/













































































































































































































































































































































































































Текст
УДК 661.4/.65+С61.8
П47
П47 Позин М. Е.
Технология минеральных солей (удо-
(удобрений, пестицидов, промышленных со-
солей, окислов и кислот), ч. I ,изд. 4-е, испр.
Л., Изд-во «Химия», 1974.
792 стр., 51 табл., 211 рис.
В монографии на современном уровне описана тех-
технология важнейших (крупнотоннажных) минеральных
солей, в том числе минеральных удобрений — фосфор-
фосфорных, азотных, калийных н других, а также некоторых
окислов и кислот (фосфорной, соляной и др.). Рассмот-
Рассмотрены свойства сырья, полупродуктов и продуктов; из-
изложены фнзико-химические основы производства; описа-
описаны, технологические схемы, режимы и аппараты.
Первая часть монографии посвящена технологии
промышленных минеральных солей и других продуктов.
Книга является практическим и справочным руко-
руководством для инженеров, работающих в химической
и смежных с ней отраслях промышленности. Она мо-
может служить учебным пособием для аспирантов и сту-
студентов старших курсоп химико-технологнческнх вузов.
Третье издание кннгн вышло в 1970 г.
31403—055
050(С1)—74
55—74
Издательство «Химия», 1974
ОГЛАВЛЕНИЕ
Предисловие к четвертому изданию 17
Предисловие к третьему изданию 17
Часть I
Глава I. Использование минеральных солей в народном хозяйстве ... 19
Минеральные соли в сельском хозяйстве 19
Минеральные удобрения . . . 19
Классификация минеральных удобрений . . 24
Минеральные яды и*другие препараты 31
Минеральные корма 34
Минеральные соли в промышленности 3.4
Развитие производства солей в СССР 36
Краткие сведения о мировом производстве удобрений и солей 40
Литература . .43
Глава II. Растворимые солн в природе и методы их добычи 45
Образование соляных залежей 45
Природные рассолы и классификация соляных озер 47
Разработка залежей ископаемых солей . 52
Горные разработки ...... ... 52
Подземное выщелачивание 53
Получение солей из рассолов и морской воды 55
Добыча самосадочных солей 55
Бассейный способ переработки рассолов 56
Литература ...... 58
Глава III. Хлористый. натрий 60
Физико-химические свойства 60
Применение 62
Сырье и методы производства 64
Каменная соль . 65
Самосадочная соль . -. 66
Получение садочной соли бассейным способом 68
Производство выварочной сбли 70
Получение соли в чренах 71
Вакуумная выварочная соль ,. . 74
Очистка рассолов 75
Конструкции аппаратов 77
Иодированная поваренная соль 82
Получение соли вымораживанием . 83
Получение поваренной соли высаливанием и перекристаллизацией ... 88
1*
Оглавление
Оглавление
Получение поваренной соли ив галитовых отходов калийных предприятий
Брикетирование и борьба со слеживаемостыо соли
Специальная (тонкая) очистка соли
Литература
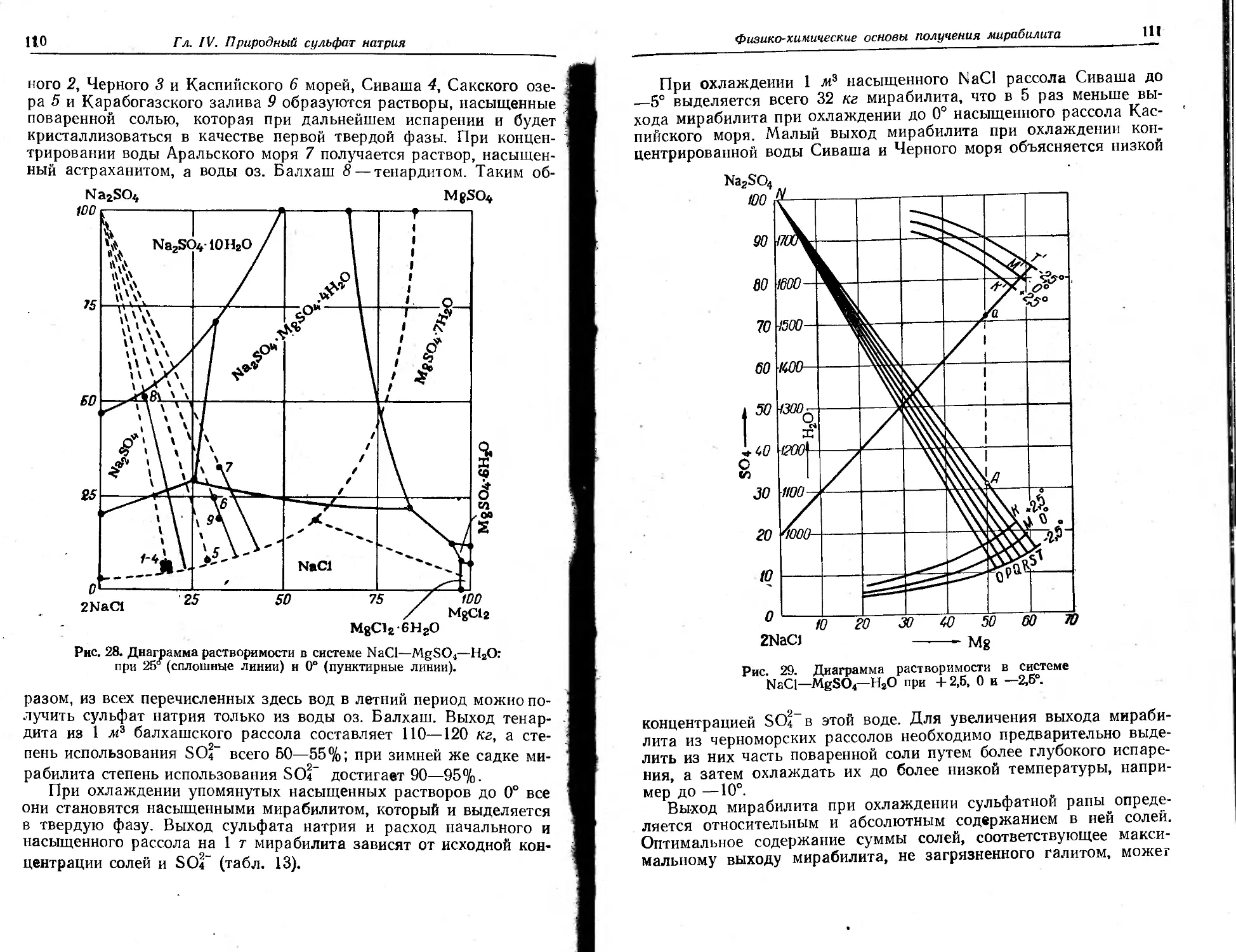
Глава IV. Природный сульфат натрии
Физико-химические свойства
Применение
Сырье .
Физико-химические основы получения мирабилита ив природных рассолов
Обезвоживание мирабилита
Обезвоживание мирабилита в естественных условиях
Заводские бпособы обезвоживания мирабилита .... , . . . .
Производство сульфата натрия на заводах СССР
Производство сульфата натрия на комбинате «Карабогазсульфат> . . .
Производство сульфата натрия на Кучукском сульфатном комбинате .
Получение сульфата натрия из твердых солевых отложении
Глауберова соль
Литература . . . ,
Глава V. Природные калийные соли
физико-химические свойства
Применение .
Сырье
Получение хлористого калия методами растворения и раздельной кри-
кристаллизации
Физико-химические основы переработки сильвинитовых руд ....
Растворение снльвииа
Кристаллизация хлористого калия .....
Схема производства хлористого калия методом растворения н кристал-
¦ лизации - ¦ . .
Переработка хартзальцевых руд
Слеживаемость хлористого калия . . . . ч
Получение хлористого калия из карналлита ...... ....
Защита оборудования от коррозии и эрозии .
Получение хлористого калия механическим обогащением калийных руд
Метод флотации
Комбинированные схемы флотационного обогащении с растворением и
кристаллизацией .
Другие способы обогащения
Получение хлористого калия при подземном выщелачивании калийных руд
Получение хлористого калия из природных рассолов
Сульфат калия .
Галургическая переработка полиминеральных руд Предкарпатья . .
' Конверсионные способы получения сульфата калия
Получение сульфатнокалийных удобрений прн флотационном обогаще-
обогащении и в комбинации с галургическими способами переработки . .
Гидротермический метод переработки калийных руд
Переработка водонерастворимых калийных руд
Прочие способы получения сульфата калия и сульфатиокаляйиых удоб-
удобрений -
Литература .
89
61
93
93
98
98
102
103
108
114
114
116
130
130
131
132
134
135
138
138
139
142
146
146
149
151
154
159
160
161
164
164
164
167
168 ¦;
170
171 '
173
173
177
179
180
182
183
184
Глава VI. Поташ 187
Физико-химические свойства 187
Применение 187
Методы производства поташа 189
Переработка золы подсолнечника 190
Переработка бардяного угля и золы .192
Получение поташа из содо-поташных растворов глиноземного производ-
производства '96
Производство поташа карбонизацией раствора едкого кали 201
Формиатный метод получения поташа 203
Триметиламиновый метод получения поташа 203
Литература . . • 204
Глава VII. Бром и его соли 206
Физико-химические свойства 206
Применение 208
Сырье ... 209
Получение брома 210
Отгонка брома паром - 210
Десорбция брома воздухом '. 213
Поглощение брома из бромо-воздушной смеси 221
Другие методы получения брома 230
Производство некоторых солей брома 231
Бромид натрия • 231
Бромид калия . . . . 233
Бромид аммония 233
Бромат калия 233
Литература 234
Глава VIII. Иод и его соли . . 236
Физико-химические свойства 237
Применение . ч . , .' 239
Сырье . . >r-.._ • 239
Получение иода 240
Извлечение иода иэ селитренной породы 240
Извлечение иода из водорослей J 241
Извлечение иода из буровых вод 242
Адсорбция иода твердыми сорбентами 243
Десорбция иода воздухом 249
Осаждение иода в виде малорастворимых солей . 253
Экстракция иода несмешивающимися с водой растворителями .... 253
Электрохимические методы извлечения иода 254
Очистка иода-сырца 255
Получение солей иода 256
Литература . 260
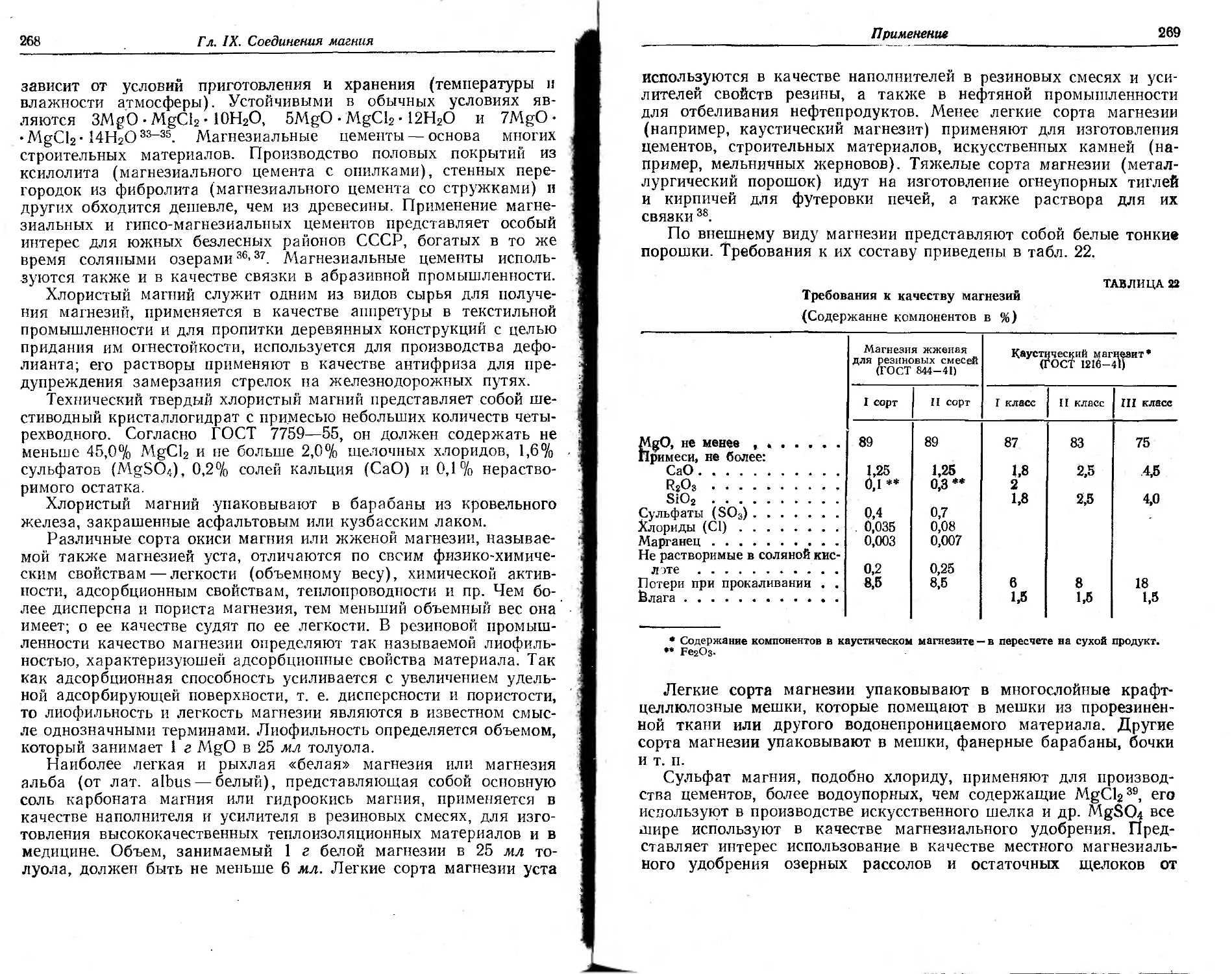
Глава IX. Соединения магнии ... 263
Физико-химические свойства 263
Применение 267
Сырье . . 270
Хлористый магний 272
Солнечная выпарка природных вод 272
Получение MgCb при переработке карналлита 274
Выпарка хлормагниевых щелоков 275
Химические способы получения хлористого магния 276
Безводный хлористый магний 278
Магнезии 281
Тяжелые формы магнезии из магнезита и доломита 281
Бикарбонатный способ получения магнезии 282
Аммиачные способы получения магнезии 287
Известковый способ получения магнезии , 287
Гидросульфидный способ получения гидроокиси магния . 296
Оглавление
Содовый способ получения легких магнезии . 296
Гидролиз и окисление хлористого магния 299
Сульфат магния и его термическое разложение 301
Получение сульфата магния 301
Термическое разложение сульфата магния %. . 303
Ньювель и совелит 304
Магниевые удобрения '• • ¦ 305
Литература 306
Глава X. Соединения бора • 311
Физико-химические свойства 311
Применение . 315
Сырье 319
Методы переработки борного сырья . . 322
Борная кислота 324
Природная борная кислота ...... 324
Получение борной кислоты из буры 324
Получение борной кислоты из магниевых боратов ........ 325
Получение борной кислоты из датолита ....... . . 335
Борат кальция . . . -. . 337
Бура 337
Получение буры из борной кислоты 338
Получение буры из боратов содовым методом 341
Переработка боратов на буру другими щелочными методами .... 343
Обезвоживание буры . . 344
Борные удобрения 344
Получение борных удобрений из отходов производства борной кислоты '344
Получен: оорных удобрений из боратового сырья L 346
Выделение бора из растворов ... ..../. 351
Перборат натрия .... I. 353
Борный ангидрид . . . . \. 356
Трехфтористый бор \ 357
Литература \358
Глава XI. Соляная кислота . . 363
Свойства хлористого водорода и соляной кислоты S63
Применение 368
Ингибированная соляная кислота 370
Получение хлористого водорода 371
Сульфатный способ . ¦ 371
Способ Гаргривса-Робинзона . 379
Синтез хлористого водорода из элементов 380
Получение хлористого водорода из хлора и водяного пара в присутствии
угля . . . 386
Хлористый водород как побочный продукт некоторых производств . . . 387
Получение чистого хлористого водорода . ..... 390
Абсорбция хлористого водорода 391
Абсорбция хлористого водорода с охлаждением 391
Адиабатическая абсорбция хлористого водорода , . 396
Жидкий хлористый водород 403
Регенерация НС1 из травильных растворов . 407
Переработка хлористого водорода в хлор . . 408
Литература . ¦ 412
Глава XII. Соли бария ................... 416
Физико-химические свойства 416
Применение 420
Сырье 424
Сульфат бария 425
Оглавление
Восстановление барита 427
Хлорид бария 434
Солянокислотный способ 434
Хлорнатриевый способ 43f
Разложение BaS раствором СаСЬ и карбонизацией 438
Хлораммониевый способ 439
Карбонатный способ 440
Хлормагниевый способ 441
Хлорный способ ¦ 443
Способы горячего хлорирования и гидрохлорирования 444
Взаимодействие ВаБСч и СаС1г - . ¦ 445
Хлоркальциевый способ . 447
Нитрат бария 451
Получение нитрата бария из BaS и азотной кислоты ' . . 451
Конверсия солей бария • 452
Карбонат бария 453
Карбонизация раствора сернистого бария 453
Получение ВаСО3 из нитратного щелока 455
Другие способы получения ВаСОз 456
Гидроокись бария . . 456
Получение гидроокиси бария из ВаСЬ и NaOH 456
Получение гидроокиси бария из сульфида бария 458
Другие способы получения гидроокиси барии 459
Окись и перекись бария 460
Литература
462
Глава XIII. Соли сульфидного ряда . . ¦-!¦.. . . 466
Физико-химические свойства . '¦. 466
Применение . . 469
Сульфид натрия . \ 470
Физико-химические основы восстановления Na2SO4 углем 470
Получение сульфида натрия восстановлением сульфата натрия углем . 477
Переработка плава в сульфид натрия . 483
Получение сульфида натрия восстановлением сульфата натрия газами 491
Другие способы получения сульфида натрия 498
Гидросульфид натрия 499
Полисульфиды . . 501
Литература 502
Глава XIV. Соли сульфитного ряда 506
Физико-химические свойства 506
Применение . 510
-ьфье ". . 513
Окисляемость сульфитных солей ." 515
Давление SO2 над растворами поглотителей 519
Бисульфит и сульфит натрия 523
Получение бисульфита и сульфита натрия из сернистого газа и щелочей 523
Получение бисульфита натрия 525
Семиводный сульфит натрия 527
Безводный сульфит натрия 529
Получение бисульфита н сульфита натрия взаимодействием SOg с рас-
раствором Na2SO4 или NaCl 532
Бисульфит и сульфит аммония 534
Пиросульфиты 535
Получение пиросульфита натрия мокрыми способами " . 535
Сухой способ получения пиросульфита натрия .... .... 537
Пиросульфит калия ". 538
Оглавление
Гидросульфит (дитиоиит) натрия 538
Производство гидросульфита натрия восстановлением сернистой кислоты
цинковой пылью 539
Другие способы получении гидросульфита натрия 543
Ронгалит 545
Тиосульфат натрия 547
Химические основы образования тиосульфата 547
Сульфатный способ 549
Полисульфидный способ 553
Сульфитный способ 555
Получение тиосульфата натрия при мышьяково-содовой очистке газов . 556
Другие способы производства .... 557
Тиосульфат аммония ... 559
Литература ¦ 560
Глава XV. Соли хрома 564
Физико-химические свойства 564
Применение 570
Сырье и методы его переработки 573
Окислительный обжиг хромитов с получением хроматов 576
Физико-химические основы окислительного обжига хромита 576
Минералогический состав хроматного спека и расчет шихты 586
Физико-химические основы выщелачивания хроматного спека 588
Физико-химические основы перевода хроматов в бихроматы 590
Методы перевода хроматов в бихроматы 590
Теория травки хроматов кислотами 591
Производство бихромата натрия 596
Получение бихромата натрия с углекислотной травкой хромата . . . 603
Бихромат калия .... 606
Хромовый ангидрид 611
Сульфаты хрома 613
Хромовые квасцы . . . . • 613
Основные сульфаты хрома^ 614
Гидроокись и окись хрома ? 615
Хроматно-серный метод получения окиси хрома 619
Хлорный хром 621
Другие методы переработки сырья и отходов, содержащих хром .... 622
Литература 625
Глава XVI. Сульфат алюминия и продукты иа его основе 632
Физико-химические свойства . 632
Применение 634
Сырье ' 636
Глины 637
Другие виды сырья 639
Производство коагулянтов " 640
Неочищенный сернокислый алюминий из каолина 641
Неочищенный сернокислый алюминий из каолииа и нефелиновой муки 642
Нефелиновый коагулянт 643
Получение очищенного сернокислого алюминия 645
Другие методы получения коагулянта и сернокислого алюминия .... 649
Получение коагулянта из золы 649
Получение коагулянта из глин и каолина методом спекания . . . '. . 650
Комбинирование производства очищенного сернокислого алюминия с сер-
нистокислотным методом разложения каолинов 651
Получение сернокислого алюминия из боксита 652
Использование отбросных травильных растворов для получения суль-
сульфата алюминия 653
Оглавление
Производство квасцов .' 653
Литература 65S
Глава XVU. Соедииения меди 661
Физико-химические свойства . 661
Применение . . 664
Сырье и способы производства медного купороса 666
Производство медного купороса из медного лома 667
Теоретические основы процесса 667
Технология процесса 670
Получение медного купороса электролизом . 675
Получение медного купороса при окислении меди хлорной, медью .... 676
Производство медного купороса из окиси меди 676
Окись меди из белого матта 676
Растворение окиси меди в серной кислоте 679
Получение медного купороса из окиси меди и сернистого газа . . . 680
Получение медного купороса сульфатизирующим обжигом белого матта 682
Получение медного купороса из окисленных медных руд 684
Производство медного купороса из колчеданных огарков ....... 685
Выщелачивание меди из огарка ... 685
Хлорирующий обжиг огарка . . ... 686
Извлечение меди из отходов медеплавильных заводов 687
Выделение меди из разбавленных растворов . . 687
Получение медного купороса из электролитных растворов медеэлектролит-
ных заводов 688
Получение других соединений меди 689
Основной сульфат меди 689
Основные карбонаты меди 690
Хлорокись меди 690
Закись меди 691
Регенерация солей меди в производстве искусственного волокна .... 691
Литература 692
Глава XVIII. Соединения железа 694
Физико-химические свойства 694
Применение 699
Получение железного купороса из травильных растворов 701
Вакуум-кристаллизационные установки 703
Получение железного купороса из колчеданных огарков 705
Сульфат окиси железа 706
Хлориды железа 707
Получение окислов железа и их гидратов 708
Литература ... . , 712
Глава XIX. Сульфат и хлорид циика 714
Физико-химические свойства 714
Применение - 718
Сырье 720
Сульфат цинка 721
Получение сульфата цинка растворением материалов, содержащих Zn и
ZnO, в серной кислоте . . ... ... 722
Получение цинкового купороса из медистой окиси цинка . . 725
Извлечение циика из колчеданных огарков хлорирующим обжигом . . . 727
Хлорид цинка " 728
Литература 729
Глава XX. Сульфат никеля -. . 731
Физико-химические свойства ' 731
Применение * 733
10
Оглавление
Получение никелевого купороса растворением никеля в серной кислоте . 734
Получение никелевого Купороса из растворов кобальтового производства 735
Получение инкелевого купороса из растворов медеэлектролитных заводов 735
Литература ... 736 ;
Глава XXI. Хлорид кальция 738
Физико-химические свойства ........ 738
Применение ... 741
Сырье ¦ 742
Получение плавленого хлорида кальция из дистиллерной жидкости содо-
содового производства 743
Получение хлорида кальция из маточного щелока хлоратного производства 745
Получение гидрооксихлорида кальция и хлорида кальция из него .... 746
Получение безводного хлорида кальция из соляной кислоты и известняка 747
Литература - 749
Глава XXII. Соединения марганца 751
Физико-химические свойства 751
Применение . ... . - 756
Сырье 759
Химическая переработка бедных марганцовых руд 761
Искусственная двуокись марганца 767
Получение двуокиси марганца электрохимическим способом 767
Производство активированного пиролюзита—ГАПа . -771
Получение активной двуокиси марганца из бедных руд 772
Получение активной двуокиси марганца термическим способом . . . 774
Сульфат марганца . . 775
Хлорид марганца . 777
Перманганат калия 778
Мажеф ¦ 785
Литература 788
Часть II
Глава XXIII. Природные фосфаты и фосфоритная мука 801
Фосфорные руды 801
Минералогический состав апатитовых и фосфоритных руд 802
Типы фосфорных руд и месторождения ... . . 804
Применение фосфатов 808
Физико-химические и механические свойства 814
Транспортировка и хранение фосфатного сырья 817
Фосфоритная мука ...... 819
Методы и продукты химической переработки фосфатов . 823
Литература ... - . ....... 824
Глава XXIV. Простой суперфосфат ...... 828
Состав и свойства суперфосфата 828
Применение 830
Методы производства • - 831
Физико-химические основы производства 832
Химизм процесса . . ... 832
Норма серной кислоты ... . . . . . 834
Механизм и скорость процесса 835
Влияние температуры 83/"
Смешение реагентов . . ....... 838
"Степень измельчения фосфатов 839
Коэффициент разложения в конце I стадии реакции 839
Разложение фосфата фосфорной кислотой (II стадия реакции) .... 840
Фосфатный комплекс суперфосфата и его фазовый состав 842
Оглавление
11
Дозревание суперфосфата 845
Особенности разложения магиийсодержащих фосфоритов 850
Нейтрализация суперфосфата твердыми добавками 853
Аммонизация суперфосфата 855
Производство суперфосфата 856
Выделение фтористых газов Ь58
Основная аппаратура и условия ее работы 859
Материалы для защиты аппаратуры от коррозии 863
•Материальный баланс производства суперфосфата 834
Технико-экономические данные S64
Получение гранулированного суперфосфата 867
Литература ... 874
Глава XXV. Производство фосфорной кислоты сернокислотным способом 881
Физико-химические свойства . . . . . 881
Применение 883
Физико-химические основы сернокислотной экстракции -фосфатов .... 884
Химизм процесса . - . 884
Скорость разложения фосфатов при сернокислотной экстракции из них
фосфорной кислоты . . 886
Кристаллизация сульфата кальция 895
. Методы сернокислотной экстракции фосфорной кислоты из фосфатов . . 904
Производство экстракционной фосфорной кислоты дигидратиым способом 908
Технико-экономические показатели 920
Концентрирование фосфорной кислоты 921
Получение концентрированной фосфорной кислоты полугидратным спо-
способом . . . 924
Литература 933
Глава XXVI. Производство фосфора и фосфорной кислоты электротермиче-
электротермическим методом ... ...... 938
Свойства фосфора и его соединения У38
Применение фосфора и термической фосфорной кислоты 940
Теоретические основы возгонки фосфора из фосфатов кальция . . 943
Производство фосфора электровозгоикой из фосфатов .... 947
Расходные коэффициенты 955
Отходы производства, их утилизация 955
Получение фосфорной кислоты 957
Литература ... 966
Глава XXVII. Концентрированные фосфорные удобрения. Двойной и обога-
обогащенный суперфосфаты . . . ...... 971
Состав и свойства 971
Применение концентрированных суперфосфатов 972
Физико-химические основы производства двойного'суперфосфата . . . 973
Химизм процесса ." " 973
Условия равновесия и кристаллизация твердых фаз ....... 974
Скорость разложения фосфатов фосфорной кислотой \ . 978
Растворение фосфата в фосфорной кислоте (без кристаллизации твердой
Фазы) 978
Разложение фосфатов (с кристаллизацией продуктов реакции) в незагу-
стевающей пульпе 982
Разложение фосфатов с образованием загустевающей пульпы ... 98Й
Производство двойного суперфосфата 9Э1
Камерный способ 992
Бескамерные (поточные) способы 997
Циклические методы получения двойного суперфосфата бескамёрным
способом _ 1004
12
Оглавление
Некоторые технико-экономические данные 1009
Обогащенный суперфосфат 1010
Литература 1013
Глава XXVIII. Концентрированные фосфорные удобрения. Преципитат (ди-
кальцийфосфат) • - • Ю17
Физико-химические свойства ... . '017
Применение 1019
Методы производства 1020
Физико-химические основы осаждения дикальцийфоефата (цреципитирова-
иия) 1024
Производство преципитата . . 1029
Кормовой преципитат : 1032
Литература ... 1035
Глава XXIX. Термические фосфаты 1038
Состав и свойства 1038
Применение 1042
Производство термических фосфатов 1043
Получение обесфторенных фосфатов . . 1043
Получение плавленых магниевых фосфатов . . 1051
Получение термощелочных фосфатов (термофосфатов) . . . . 1053
Получение метафосфата кальция 1055
Литература . 1056
Глава XXX. Промышленные продукты, содержащие фосфор 1062
Сульфиды фосфора . . 1032
Хлориды фосфора ........ \ ... 1063
Фосфиды . . . 1064
Ортофосфаты натрия и калия ...... 1065
Производство фосфатов натрия 1068
Дегидратированные фосфаты натрия 1070
Производство триполифосфата натрия и других дегидратированных
фосфатов 1076
Фосфаты аммония и двойные фосфорнокислые соли аммония .... 1085
Фосфаты кальция 1089
Литература . .... 1090
Глава XXXI. Соединения фтора .1095
Физико-химические свойстве 1095
Применение 1105
Сырье 1108
Получение фтора 1110
Фтористый водород и плавиковая кислота ... 1113
Разложение плавикового шпата и других фторидов 1113
Абсорбция и конденсация фтористого водорода 1117
Жидкий фтористый водород 1120
' Производство фтористых солей из плавиковой кислоты 1124
Криолит 1124
Фторид алюминия . 1126
Фторид натрия . . 1128
Фторид магния . 1129
Щелочные способы получения фторида натрий и криолита из плавикового
шпата . 1129
Улавливание фтористых соединений из отходящих газов и их переработка! 134
Получение кремнефтористоводородной кислоты 1136
Переработка кремнефтористоводородной кислоты в кремнефториды . 1142
Получение фтористых солей из отходящих газов 1148
Фторид натрия 1149
Фторид кальция 1158
Оглавление
13
Криолит и фтористый алюминий .... 1160
Аммиачный способ улавливания и переработки фтористых газов ... 1162
Литература 117°
Глава XXXII. Соли азотной кислоты 1178
Нитрат аммония . . 1178
Физико-химические свойства 1178
Применение П84
Сырье и методы производства 1186
Производство аммиачной селитры с выпаркой растворов 1187
Получение нитрата аммония безупарочиыми способами 1199
Получение аммиачной селитры конверсией нитратных растворов . . 1203
Удобрения на основе нитрата аммония 1204
Нитрат аммония 1209
Нитрат кальция 1210
Физико-химические свойства 1210
Применение 1211
Методы производства 1211
Нитрат и нитрит натрия 1216
Физико-химические свойства 1216
Применение 1216
Способы производства нитрата натрия 1217
Нитрат калия 1222
Физико-химические свойства 1222
Применение 1223
Способы производства нитрата калия 1223
Литература -V - 1232
Глава XXXIII. Соли аммония . 1237
Сульфат аммония 1237
Физико-химические свойства 1237
Применение > 1238
Производство сульфата аммония 1239
Хлористый аммоний 1253
Физико-химические свойства 1253
Применение 1253
Производство хлористого аммония 1254
Углекислые соли аммония 1259
Физико-химические свойства 1259
Применение 1260
Получение углекислого аммония 1262
Получение двууглекислого аммония 1263
Фосфаты аммония 1264
Физико-химические свойства 1264
Применение 1265
Производство фосфатов аммония 1266
Удобрения, содержание полифосфаты аммония 1274
Удобрения, содержащие метафосфат аммония 1274
Литература 1275
Глава XXXIV. Карбамид 1282
Физико-химические свойства , 1282
Применение и агрохимические свойства 1285
Методы производства 1287
Физико-химические основы синтеза карбамида из аммиака я двуокиси
углерода . . , 1288
Способы производства карбамида из аммиака и двуокиси углерода прямым
синтезом 1293
14
Оглавление
Способы регенерации газов дистилляции и режимы синтеза карбамида 1294
Производство карбамида по схеме с жидкостным рециклом .... 1297
Переработка растворов карбамида в готовый продукт 1300
Литература ......... 1302
Глава XXXV. Комплексные твердые сложные удобрения ...... 1307
Физико-химические основы разложения фосфатов азотной кислотой . 1308
Выделение фтора и редкоземельных элементов из азотнокислотной вы-
вытяжки 1311
Переработка раствора, полученного азотнокислотным разложением фос-
фосфатов 1314
Физико-химические основы переработки азотнокислотной вытяжки . 1314
Способы переработки азотнокислотной вытяжки 1321
Переработка азотнокислотной вытяжки в сложные удобрения .... 1325
Карбонатный способ 1329
Сульфатные способы 1331
Фосфорнокислотиый способ 1336
Нитроаммофос и нитроаммофоска 1337
Получение нитрофоски с вымораживанием части нитрата кальция . . 1339
Получение водорастворимой нитрофоски с частичной регенерацией азот-
азотной кислоты 1346
Производство сложных удобрений на баэе фосфорной кислоты .... 1347
Некоторые свойства водных систем, содержащих фосфаты, нитраты ам-
аммония, карбамид и др 1347
Способы производства 1349
Литература 1358
Глава XXXVI. Комплексные смешанные и сложно-смешанные жидкие удоб-
удобрения . . . . ... ..... 1383
Общие сведения 1363
Синергизм и антагонизм удобрений 1365
Производство смешанных удобрений 1367
Расчет состава тукосмесей . 1370
Литература ¦. 1374
Глава XXXVII. Жидкие удобрения 1375
Общие сведения 1375
Физико-химические свойства 1377
Производство жидких удобрений 1387
Схемы производства аммиачной воды и аммиакатов иа основе аммиач-
аммиачной селитры . 13Е7
Производство жидких комплексных удобрений 1350
Суспендированные жидкие комплексные удобрения 1392
Литература 1392
Глава XXXVIII. Соли мышьяка 1396
Физико-химические свойства 1396
Применение мышьяковых препаратов и технические требования к иим . 1402
Сырье .... 1405
Получение белого мышьяка обжигом мышьяковых руд 1407
Арсенит кальция 1408
Мокрый способ производства 1409
Полусухой способ производства 1410
Арсеннт натрия 1412
Парижская зелень 1413
Мышьяковая кислота 1416
Азотнокислотный способ ". . ., 14N
Оглавление
15
Другие способы получения, мышьяковой кислоты ....... 1418
Арсенат кальция 1419
Способ каталитического окисления растворов арсенита воздухом . . 1419
Получение арсеиата кальция из мышьяковой кислоты 1423
Азотнокислотный способ ..... 1423
Хлорный способ . 1425
Электрохимический способ 1426
Термический способ 1426
Получение арсената кальция из окисленных мышьяковых руд . . . 1427
Литература ' 1428
Глава XXXIX. Соли кислородных кислот хлора 1430
Физико-химические свойства 1430
Применение 1437
Производство хлорной извести 1441
Обжиг известняка 1441
Получение пушонки (гашение извести) 1442
Хлорирование пушонки 1445
Гипохлорит натрия . . 1449
Гипохлорит кальция ' 1450
Двуокись хлора 1453
Сернистокислетный способ 1454
Метанольный способ . 1456
Солянокислотный способ 1457
Хлоридный способ . : 1458
Получение двуокиси хлора из хлорита натрия 1459
Хлорит натрия 1460
Хлораты натрия, калия, кальция и магния 1461
Получение хлоратов натрия и калия электрохимическим методом . . 1461
Получение хлората калия известковым методом ... 1464
Хлорат натрия 1469
Хлораты кальция и магния 1470
Перхлораты натрия, калия и аммония и хлорная кислота 1471
Перхлорат натрия 1471
Перхлорат калия 1473
Перхлорат аммония 1473
Хлорная кислота 1474
Литература . 1476
Глава XL. Продукты высокотемпературного хлорирования руд .... 1480
Общие сведения 1480
Четыреххлористый титан 1481
Физико-химические свойства 1481
Применение . ..." 1442
Сырье . . 1483
Способы обезжелезивания ильменитового концентрата 1484
Получение четыреххлористого титана из рутилового концентрата и тита-
титанистых шлаков 1486
Получение четыреххлористого титана из других видов сырья .... 1494
Треххлористый титан 1495
Четыреххлористый кремний 1495
Физико-химические свойства 1495
Применение 1496
Получение четыреххлористого кремния 1497
Безводный хлористый алюминий 1800
Физико-химические свойства 1500
Применение . 1501
Получение безводного хлористого алюминия .......... 1501
Литература ' . . . . 1504
16
Оглавление
Глава XL1. Цианистые соединения 1509
Фиаико-химические свойства 1509
Применение 1513
Сырье 1516
Цианамид кальция 1516
Получение цианамида кальция в электрических печах 1518
Получение гранулированного цианамида кальция 1521
Другие методы получения цианамида кальция 1521
Производство цианидов натрия и калия . 1523
Получение цианистых солей из синильной кислоты 1524
Получение цианплава из цианамида кальция 1525
Фиксация азота смесью соды с углем 1525
Восстановление цианатов, полученных из аммиака и карбонатов . . 1526
Сплавление амида натрия с углем .... 1526
Сухая перегонка барды 1527
Получение цианидов из коксового газа 1528
Получение роданидов натрия и аммония из отбросных растворов мышьяко-
во-содовой очистки газов . ,. . . . 1529
Производство железисто- и железосинеродистых солей 1530
Железистосинеродистые соли 1530
Железосинеродистые соли 1531
Производство синильной кислоты контактными способами 1532
Способы синтеза синильной кислоты 1532
Производство синильной кислоты контактным окислением метана и ам-
аммиака 1535
Получение цианистого водорода из аммиака и окиси углерода (форм-
амидный способ) 1540
Получение синильной кислоты плавменным методом 1540
Литература 1543
ПРЕДИСЛОВИЕ К ЧЕТВЕРТОМУ ИЗДАНИЮ
Четвертое издание этой книги является стереотипным — в нем
лишь исправлены опечатки, обнаруженные в третьем издании, а
также данные ГОСТов в соответствии с произведенными в них но-
новыми изменениями и дополнениями.
ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
Со времени выпуска предыдущего издания этой книги не про-
прошло и десяти лет, а прогресс техники производства многих неор-
неорганических продуктов оказался настолько большим, что возникла
необходимость внести в книгу существенные дополнения и изме-
[ения. Это касается всех ее частей, но главным образом тех, в ко-
которых рассмотрена технология минеральных удобрений. Развитие
этой отрасли промышленности во всем мире, и особенно в СССР,
идет чрезвычайно быстро. Выполнение решений XXIII съезда
КПСС и последующих Пленумов ЦК КПСС обусловило рост хи-
химической промышленности, и прежде всего основной химии, как
базы, обеспечивающей непрерывное развитие сельского хозяйства.
Новые методы производства появляются быстрее, чем исчезают
старые, однако наряду с новыми заводами продолжают успешно
действовать и построенные ранее. Поэтому наиболее трудным при
подготовке третьего издания оказалось, не увеличивая и без того
значительный объем книги, дополнить ее обильными новыми све-
сведениями, сохранив при этом и весь не утративший своего значения
Материал предыдущего издания.
18
Предисловие к третьему изданию
Глава V написана Ю. Я. Каганович, главы VII и VIII —
Г. С. Клебановым, главы XXXII —XXXV и XXXVII —Л. 3. Ар-
сеньевой и В. А. Клевке, глава XXXVI — с участием Л. 3. Арсенье-
вой. Главы XXIII—XXX написаны Б. А. Копылевым, при этом ча-
частично использован материал, написанный для предыдущего изда-
издания книги А. А. Соколовским. Глава VI и стр. 1154—1157, напи-
написанные ранее А. А. Соколовским, подверглись в новом издании
лишь небольшим изменениям. Главы XXXIX—XLI и стр. 1314—1321
написаны Б. А. Копылевым. Всем этим товарищам приношу боль-
большую .благодарность.
Я благодарю также М. И. Муратову и В. А. Рябина за мате-
материалы, предоставленные для IV и XV глав книги.
М. Позин
Ленинградский технологический институт
имени Ленсовета
Май, 1969 г.
II
Гл ава I
ИСПОЛЬЗОВАНИЕ МИНЕРАЛЬНЫХ СОЛЕЙ
В НАРОДНОМ ХОЗЯЙСТВЕ
Ассортимент минеральных солей, используемых в промышлен-
промышленности, в сельском хозяйстве, в медицине и для бытовых целей,
весьма велик. Он исчисляется сотнями наименований и непрерывно
растет. Масштабы мировой добычи и производства некоторых ми-
минеральных солей достигают миллионов и даже десятков миллио-
миллионов тонн в год.
По масштабам производства и потребления первое место зани-
занимают соли и соединения натрия, фосфора, калия, азота, алюми-
алюминия, железа, меди, серы, хлора, фтора, хрома, бария и некоторые
другие. Десятки миллионов тонн минеральных удобрений и боль-
большие количества других минеральных солей используются в сель-
сельском хозяйстве.
МИНЕРАЛЬНЫЕ СОЛИ В СЕЛЬСКОМ ХОЗЯЙСТВЕ
Минеральные удобрения !~25
Минеральными удобрениями (туками) называются соли и дру-
другие продукты, содержащие элементы, необходимые для развития
растений и используемые с целью получения высоких и устойчи-
устойчивых урожаев. Основная масса применяемых удобрений вносится
в почву под посевы. Некоторые виды удобрений используют и для
внекорневого питания растений.
В образовании ткани растения, в его росте и развитии уча-
участвует большинство химических элементов (около 60). Основными
из них, образующими 90% массы сухого вещества растений, яв-
являются углерод, кислород и водород. 8—9% растительной массы
составляют: азот, фосфор, магний, сера, кальций, калий и железо.
На долю остальных элементов приходится 1—2% веса растения.
Бор, медь, марганец, цинк, иод, бром, мышьяк входят в состав
20
Гл. I. Минеральные соли в народном хозяйстве
растений в тысячных и десятитысячных долях процента, а такие
Элементы, как уран, радий, торий, — в миллионных и миллиард-
миллиардных долях процента7'в.
Основную массу кислорода, углерода и водорода растение по-
получает иа воздуха и воды, остальные элементы оно извлекает из
почвенного раствора.
Особенно важную роль в минеральном питании растения играет
азот, входящий в состав белков; последние являются основой
жтдл ткани. В растительных белках содержится 15,5—18%
азота. Азот входит и в состав хлорофилла, с помощью которого
растения усваивают углерод из находящегося в атмосфере угле-
углекислого газа и солнечную энергию. Растения извлекают азот из
минеральных солей (солей аммония и нитратов). Некоторые рас-
растения (бобовые) могут усваивать азот воздуха благодаря деятель-
деятельности развивающихся на корнях клубеньковых бактерий.
Из солей аммония азот усваивается растениями с наибольшей
легкостью; нитраты же восстанавливаются в тканях растения сна-
сначала до нитритов, затем до аммиака, перерабатываемого в амино-
аминокислоты и белки *.
Основными формами азотных удобрений являются: аммиачная
(соли аммония — сульфат, хлорид, фосфаты и др.), нитратная
(соли азотной кислоты — кальциевая, калиевая, натриевая се-
селитры), аммиачно-нитратная (NH4NOs) и амидная (карбамид
CO(NH2)s и др.). Все минеральные азотные удобрения (за исклю-
исключением двойных солей типа MeNH4PO4, например, магнийаммоний-
фосфата MgNH4PO4 • Н2О) хорошо растворимы в воде и быстро
переходят в почвенный раствор, что обеспечивает легкую усвояе-
усвояемость азота растением.
Соединения фосфора играют важную роль в дыхании и размно-
размножении растений. В пересчете на P2Os содержание фосфора в не-
некоторых частях растений достигает 1,6%. Усиление питания фос-
фосфором повышает засухоустойчивость и морозостойкость растений
и увеличивает содержание в них ценных веществ — крахмала в
картофеле, сахарозы в сахарной свекле и т. п. Восприимчивость
растением фосфорных удобрений, являющихся солями фосфорных
кислот, зависит от их растворимости и от характера почв, в пер-
первую очередь от кислотности почв. Наличие в почве значительного
запаса подвижной (усвояемой растениями) формы фосфора спо-
способствует хорошему использованию других удобрений — азотных
и калийных. Одним из методов оценки усвояемости содержащейся
в удобрении Р2О5 является растворимость фосфатных соединений
в искусственных растворах, кислотность которых близка к кислот-
кислотности почвенных растворов (стр. 30). Содержание фосфора в фос-
фосфорных удобрениях принято выражать в пересчете на Р2О5.
Большую роль в регулировании жизненных процессов, проис-
происходящих в растении, играет калий. Он улучшает водный режим
Минеральные соли в сельском хозяйстве
21
растений, способствует обмену веществ и образованию углеводов,
увеличивая, так же как и фосфор, накопление крахмала в карто-
картофеле, сахара в сахарной свекле и т. п., и еще в большей мере, чем
фосфор, повышает засухоустойчивость и морозостойкость расте-
растений. Содержание калия в сухом веществе растения достигает
4 5%. а в золе листьев 30—60%. По легкости усвоения калия
растением различают три формы его соединений: 1) содержащие
водорастворимый калий, 2) обменный калий, т. е. переходящий в
почвенный раствор в результате ионообменных процессов, и 3) не-
необменный, входящий в состав безводных силикатов, из которых
калий извлекается растением лишь частично и медленно. Содер-
Содержание калия в удобрениях выражают в пересчете на К2О.
Кальций содержится в растениях в виде солей минеральных и
органических кислот. Он способствует развитию корневой системы,
нейтрализации избыточной кислотности в клетках растений и их
устойчивости при повышенной кислотности почвы. Кальций вносят
в почву в виде фосфорнокальциевых удобрений, кальциевой се-
селитры, извести, гипса и др.
Магний содержится главным образом в зеленых частях par
стения. Он входит в состав хлорофилла (~2,7%) и фитина, спо-
способствует протеканию восстановительных процессов в растении,
образованию углеводов и переводу фосфора из минеральных в
органические соединения. Магний находится в почве главным
образом в виде силикатов и алюмосиликатов, т. е. в форме, не
усвояемой растениями. В качестве магниевых удобрений приме-
применяют доломит, магнезиальные фосфаты, содержащие магний калие-
калиевые минералы (каинит, лангбейнит) и другие соли.
Сера входит в состав белков и эфирных масел и вносится в
почву в удобрениях, содержащих сульфаты кальция, магния, ка-
калия, а иногда в виде элементарной серы, окисляемой микроорга-
микроорганизмами до серной кислоты.
Железо играет роль катализатора при образовании хлорофилла
и участвует в дыхании растений, входя в состав ферментов, регу-
регулирующих окислительно-восстановительные процессы. Ввиду до-
достаточного содержания железа в почвах соли железа в качестве
удобрений используются лишь в исключительных случаях (при
чрезмерном содержании в почве извести).
Элементы, жизненно необходимые для растения, но входящие
в его состав в ничтожных количествах (от 10~2 до 10~I2%) и иг-
играющие главным образом роль регуляторов протекающих в расте-
растении сложных процессов, носят название микроэлементов. К ним
относятся бор, марганец, медь, цинк, молибден, кобальт, иод и др.
Удобрения, содержащие эти элементы, называют микроудобре-
микроудобрениями. Значение этих удобрений исключительно велико, так как
недостаток микроэлементов, входящих в состав ферментов, вита-
витаминов, белков, гормонов, вызывает нарушение обмена веществ и
22
Гл. 1. Минеральные соли в народном хозяйстве
Минеральные соли в сельском хозяйстве
23
тяжелые заболевания растений. Особенно велико влияние микро-
микроэлементов на окислительно-восстановительные процессы, проте-
протекающие в растении, на их направление, на процессы фотосинтеза,
отток углеводов и др. Роль микроэлементов в жизненных процес-
процессах в настоящее время с успехом изучается с помощью меченых
атомов. В особую группу можно выделить ультрамикроэлементы
(содержащиеся в количествах, меньших 10~Б%), в том числе ра-
радиоактивные вещества.
Некоторые элементы, например, кальций, сера, железо, нахо-
находятся в почве в большинстве случаев в достаточном для растений
количестве. Другие же элементы, в особенности азот, фосфор, ка-
калий, имеющие наибольшее значение для питания растений, необ-
необходимо вносить в почву в виде удобрений. Питательные элементы
частично возвращаются в почву естественным путем. Так, азот,
находящийся в ткани растения в органической форме, при гниении
частично переходит в аммиачную и нитратную формы и вновь
усваивается растениями. Однако эти процессы идут медленно и
значительная часть питательных элементов в почву не возвра-
возвращается, часть их вымывается из почвы грунтовыми водами или
оказывается в форме, непригодной для усвоения растениями. По-
Поэтому запас питательных элементов в почве требуется восполнять
внесением удобрений.
Если уменьшение содержания питательных веществ в почве не
будет компенсироваться внесением удобрений, почва будет исто-
истощаться, что приведет к снижению урожайности. Это может прои-
произойти и тогда, когда в почве содержатся еще весьма большие ко-
количества необходимых для питания растений элементов, так как
урожай зависит не от общего, валового запаса их в почве, а только
от той их части, которая является усвояемой; эта часть составляет
в большинстве случаев лишь долю общего запаса.
В СССР плановая организация круговорота питательных ве-
веществ, базирующаяся на научных основах, созданных Жаном Ба-
Батистом Буссенго, А. Н. Энгельгардтом, Д. И. Менделеевым,
К. А. Тимирязевым, В. В. Докучаевым, П. А. Костычевым,
В. Р. Вильямсом, Д. Н. Прянишниковым и другими, обеспечивает
повышение плодородия почв и непрерывный рост урожаев. Опыт
передовиков сельского хозяйства, показывающих замечательные
примеры высоких урожаев, все шире внедряется в практику.
Чем выше урожайность, тем больше выносится из почвы пита-
питательных веществ. О приросте урожая и о влиянии его на вынос
питательных веществ из почвы можно судить по примерным дан-
данным, приведенным в табл. 1 и 2.
При внесении в почву полного удобрения (содержащего азот,
фосфор и калий) урожай повышается в 1,5—2 раза. В среднем
прибавка урожая от применения удобрений составляет ~40%.
Каждый рубль, затраченный на минеральные удобрения, при пра-
ТАБЛИЦА1
Вынос питательных веществ из почвы с урожаем
(в кг/га)
Питательное
вещество
N
Р2О5
к2о
При урожае
.озимой пшеницы
15 ц/га
46
22
28
30 ц/га
112
39
77
При урожае
сахарной свеклы
150 ц/га
65-85
25-28
60-70
270 ц/га
166
42
157
При урожае
кукурузы (зе-
(зеленой массы)
600 ц/га
150
70
200
ТАБЛИЦА 2
Повышение урожайности от внесения в почву фосфора, азота и калия*
Культура и вид продукции
Хлопок-сырец
Корни сахарной свеклы
Сахар » » ... • • • •
Конопля-волокно
Крахмал в клубнях картофеля . . .
Зерно озимой пшеницы
Прирост урожая, т на 1 т внесенного
N
10-14
120-160
20
5-5,6
2-2,2
120
17-18
12-25
P«O5
5-6
50-55
8-9
4-4,2
1,5-1,8
40-80
6-6,5
20-25
KjO
2
40-50
6-7
40-60
5-5,5
3-4
* Здесь приведены средние данные. Прирост урожая меняется в зависимости от харак-
характера почв, климатических условий, качества удобрений, агротехнических мероприятий и пр.
вильном их использовании дает прирост урожая в среднем на
10 руб. Следующие примерные цифры показывают, как изменяется
урожай в центнерах с 1 га от применения удобрений в условиях
надлежащей агротехники:
Без удобрений С удобрениями
Хлопок 13 33
Пшеница озимая 15 26
Сахарная свекла НО 282
Количество вносимых удобрений на 1 га посевной площади в
сельскохозяйственной практике колеблется в следующих преде-
пределах: азотные удобрения —от 30 до 300 кг N, фосфорные —от 45 до
200 кг' Р2О5, калийные — от 40 до 250 кг К2О. В последнее время
наблюдается тенденция увеличивать нормы внесения удобрений.
Микроудобрения вносятся в почву в незначительных количе-
количествах. Например, для повышения урожая волокна и семян льна на
24
Гл. 1. Минеральные соли в народном хозяйстве
заболоченных и известковых почвах на 30% достаточно внести
всего 0,5 кг бора на 1 га.
В качестве микроудобрений не обязательно использовать чис-
чистые соли микроэлементов, например, меди, марганца, цинка, бора;
они могут быть заменены природными минералами и отходами про-
промышленности. Так, в качестве источника микроэлементов могут
быть использованы пиритные огарки, содержащие медь и другие
металлы, борсодержащие отходы от производства соединений бора;
шламы от обогащения марганцовых руд и др. Распределение ма-
малых количеств микроудобрений на большие посевные площади за-
затруднительно. Поэтому микроудобрения добавляют к главным
формам удобрений еще в процессе их производства. Когда же для
питания растений микроэлементами используют чистые соли, их
предпочитают не вносить в почву, из которой извлекается лишь
незначительная их доля, а питать ими непосредственно растения
путем предпосевного пропитывания семян или опрыскивания рас-
растений растворами солей (внекорневое питание).
В СССР при большом разнообразии почвенно-климатических
условий и выращиваемых культур требуется не только большое
количество удобрений, но и широкий ассортимент их. Поэтому
азотные, фосфорные, калийные и другие удобрения изготовляются
и применяются в виде различных солей и их смесей, в которых пи-
питательный элемент находится в разных формах и количествах.
Классификация минеральных удобрений
Удобрения классифицируют по происхождению, назначению,
составу, свойствам, способам получения и др.
По происхождению удобрения разделяют на минеральные, ор-
органические, органо-минеральные и бактериальные. Минеральные
или искусственные удобрения — специально вырабатываемые на
химических предприятиях неорганические вещества, главным обра-
образом минеральные соли; однако к ним относят и некоторые органи-
органические вещества, например, карбамид. Органические удобрения
содержат питательные элементы, главным образом (но не исклю-
исключительно) в виде органических соединений, и являются обычно
продуктами естественного происхождения (навоз, фекалии, торф,
солома и др.). Органо-минеральные удобрения — смеси органиче-
органических и минеральных удобрений. Бактериальные удобрения содер-
содержат культуры бактерий, способствующих накоплению в почве
усвояемых форм питательных элементов.
По срокам внесения удобрения разделяют на основные (пред-
(предпосевные), вносимые до посева, припосевные, вносимые во время
посева (например, в рядки), и подкормки, вносимые в период раз-
развития растений.
По видам питательных элементов (табл. 3) удобрения разделя-
разделяют на азотные, фосфорные (или фосфатные), калийные (калиевые),
Минеральные соли в сельском хозяйстве
25
ТАБЛИЦА 3
Важнейшие минеральные удобрения • ¦
Название
Суперфосфат порош-
порошкообразный
Суперфосфат грану-
гранулированный
Суперфосфат обога-
1*1 f\ Т Т Т Т ? Т ТТ
щенныи
Суперфосфат двой-
двойной и тройной
Преципитат
Фосфоритная мука
Костяная мука
Фосфатшлаки (томас-
шлак или марте-
мартеновский)
Термофосфат
Плавленый фосфат
Обесфторенный фос-
фосфат
Метафосфат кальция
Аммиак жидкий
Аммиачная вода
Аммиачная селитра
(нитрат аммония)
Сульфат алшония
Сульфонитрат аммо-
аммония
Извеетково-аммиач-
ная селитра
Натриевая селитра
(нитрат натрия)
Кальциевая селитра
(известковая се-
селитра)
Хлористый аммоний
Бикарбонат аммония
Цианамид кальция
Карбамид (мочевина)
Карбамид-формаль-
дегидное
Главные компоненты
Фосфорные
Са(Н2РО4J-Н2О + ,
+ H3PO4+CaSO4
То же
То же
Са(Н2РО4J • Н2О + Н3РО4
СаНРО4 • 2Н2О
Ca5F(PO4K
Са3(РО4J + СаСО3
4СаО- Р2Об + 5СаО • Р2О6 • SiO2
Na2O-4CaO-P2O5-SiO2
4(Са, Mg)O-P2O6 +
- + 5(Са, Mg)O • Р2О6 • SiO2
ЗСаО • Р2О5 + 4СаО • Р2О6
Са(РО3J
Азотные
NH3
NH3 + Н2О
NH4NO3
(NH4JSO4
(NH4JSO4-2NH4NO8
NH4NO3 + CaCO3
NaNO3
Ca(NO3J-3H2O
NH4C1
NH4HCO3
CaCN2 + С
CC(NH2J
NHCONHCH2
Содержание
основного
питательного
вещества
14-21% Р2О6
19,5-21% Р2О5
28-32% Р2О5
38-50% Р2О6
27-46% Р2О5
16-25% Р2О5
30% Р2О5
14-20% Р2О6
20-35% Р2О6
20-35% Р2О6
20-38% Р2О5
65-70% Р2О6
82,3% N
16,5-20,5% N
34-35,0% N
20,5-21% N
25-27% N
16-20,5% N
16,1% N
13-15% N
24,5-25% N
18% N
18-23% N
42-46,6% N
33-42% N
Объемный
вес, • т/л*
1,1-1,2
1,1
1,1
0,9-1,1
0,8-0,85.
1,7-1,8
0,86
2,01-2,05
1,7
0,61
0,92-0,91
0,8-1,1
0,7-0,95
0,8-1,2
1-1,2
1,1-1,4
0,9-1,1
0,6-0,8
0,6-0,61
0,65-0,У1
• В табл. 3 приведены ориентировочные пределы объемного веса для-образцов с разной
величиной зерен и разной степени слежалости. .
26
Гл. I. Минеральные соли в народном хозяйстве
Продолжение
Название
Хлористый калий
30 и 40 % -наи калий-
калийная соль
Сильвинит молотый
Карналлит молотый
Сульфат калия
Калимаг (лангбейни-
товый)
Калимаг (шенито-
вый)
Каинит
Суперфосфат аммо-
аммонизированный
Суперфосфат двой-
двойной аммонизиро-
аммонизированный
Аммофос
Диаммофос
Нитроаммофос
Сульфоаммофос
Метафосфат калня
Калийно-аммиачная
селитра
Калиевая селитра
(нитрат калия)
Аммофоска
Нитрофоска
Нитроаммофоска
Диаммонитрофоска
Главные компоненты
Калийные
КС1
KCl + NaCl
КС1 + NaCl
КС1 • MgCl2 • 6H2O + NaCl
K2SO4
K2SO4-2MgSO4 + CaSO4+NaCl
K2SO4 + MgSO4
KC1 • MgSO4 • 3H2O + NaCl
Комплексные
CaHPO4 + NH4H2PO4 + CaSO4
CaHPO4 + NH4H2PO4
NH4H2PO4 + (NH4JHPO4
(NH4JHPO4 + NH4H2PO4
NH4NO3 + NH4H2PO4
(NH4JHPO4 + (NH4JSO4
(КРО3)„
NH4NO3 + KNO3 + NH4CI
KNO3
(NH4JHPO4 + (NH4JSO4 +
+ KNO3 + NH4Cl
NH4NO3 + (NH4JHPO4 +
+ KNO3 + NH4C1
или NH4NO3 + CaHPO4 +
+ CaCO3 + KNO3 + NH4C1
или NH4NO3 + CaHPO4 +
+(NH4JHPO4+CaSO4 • 2H2O+
+ KNO3 + NH4C1
NH4NO3+NH4H2PO4 +
+ KNO3 + NH4C1
NH4NO3+(NH4JHPO4 +
+ KNO3 + NH4C1
Содержание
ОСНОВНОГО!
питательного
вещества
50-62% К2О
30-40% К2О
12-15% К2О
12-13% К2О
48-52% К2О
19-21% К2О
28-30% К2О
8-12% К2О
1,5-3% N,
19-20% Р2О5
6-7% N,
40-50% Р2О5
11-14% N,
48-55% Р2О5
16-18% N,
46-48% Р2О5
21-22% N,
21-22% Р2О5
18-20% N,
16-20% Р2О5
57-59% Р2О6,
38-40% К2О
16% N,
26-28% К2О
13,5% N,
46,5% К2О
11 — 12% N,
11-16% Р2О6
15-20% К2О
11-20% N,
8-16% Р2О5
10-21% К2О
17^18,5% N,
17-18,5% Р2О6
17-18,5% КзО
То же
Объемный
вес, * т/л3
0,73-1,17
0,94-1,2
1,07-1,1
0,99-1,06
1,25-1,43
1,5
1
1,3-1,4
1,2-1,35
0,9-1
1,05-1,27
1-1,3
0,8-1,3
~1
Минеральные ^соли в сельском хозяйстве
27
• В табл. 3 приведены ориентировочные пределы объемного веса для образцов с разной
величиной зерен и разной степени слёжалости.
магниевые, борные и т. д. Удобрения, содержащие только ми-
микроэлементы, объединяют в общую группу микроудобрений.
По агрохимическому значению удобрения разделяют на пря-
прямые являющиеся источниками питательных элементов для ра-
растений и косвенные, служащие для мобилизации питательных
веществ из почвы путем улучшения ее физических, химиче-
химических и биологических свойств (например, для нейтрализации кис-
кислотности почвы известкованием, или для мелиорации гипсова-
гипсованием и др.).
Деление удобрений на прямые и косвенные является в извест-
известной мере условным, так как все. прямые удобрения оказывают и
косвенное действие, а элементы, входящие в состав косвенных
удобрений, в той или иной степени используются в питании рас-
растения.
Прямые минеральные удобрения могут содержать один или не-
несколько разных питательных элементов. Три главных питательных
элемента — азот, фосфор и калий — вносятся под посевы в наи-
наибольших количествах. По их содержанию удобрения разделяют на
простые, в состав которых входит только один из главных пита-
питательных элементов, и комплексные, содержащие два и более пи-
питательных элемента. По числу главных питательных элементов
комплексные удобрения называют двойными (например, типа РК)
и тройными (NPK); последние называют также полными. Удобре-
Удобрения, содержащие значительные количества питательных элементов
и мало балластных веществ, называют концентрированными, а
удобрения, все компоненты которых служат для питания расте-
растений — безбалластными. К последним относятся, например, соли,
и катион и анион которых содержат питательные элементы, такие
как KNO3, NH4NO3 и др. Концентрированные и безбалластные
удобрения обладают высокой эффективностью, а их перевозка об-
обходится дешевле, чем неконцентрированных удобрений. Так как
любой питательный элемент находится в удобрении в форме ка-
какого-либо соединения, т. е. в связи с другими элементами, в той
или иной мере используемыми растением, то, строго говоря, про-
простых удобрений нет. Применяя термины простые, двойные и трой-
тройные удобрения, имеют в виду содержание в них одного, двух или
трех главных элементов — азота, фосфора и калия или вообще
тех элементов, ради внесения которых в почву удобрение исполь-
используется; сопутствующие элементы, хотя и извлекаемые растениями,
этой классификацией не учитываются.
По конституции удобрения разделяют на простые, смешанные и
сложные. Простыми называют удобрения, содержащие только один
питательный элемент в одной форме (например, азот в NaNO3).
Смешанными удобрениями называют механические смеси удобре-
Ний, состоящие из разнородных частиц, получаемые тукосмеше-
тукосмешением. Если же удобрение, содержащее несколько питательных
28
Гл. 1, Минеральные соли в народном хозяйстве
элементов, получается в результате химической реакции в завод-
заводской аппаратуре, его называют сложным.
Сложные удобрения состоят из однородных частиц, содержа-
содержащих питательные элементы в нескольких формах. Деление удобре-
удобрений на смешанные и сложные в значительной мере условно. Сме-
Смешанные удобрения при хранении нередко становятся сложными
в результате реакций, протекающих между составляющими смесь
компонентами. Иногда называют сложно-смешанными удобрения,
получаемые в результате «мокрого тукосмешения» — смешения
твердых продуктов с жидкими (плавами, растворами) и последую-
последующего отверждения смесей, сопровождающегося перекристаллиза-
перекристаллизацией и другими процессами. (Такие удобрения за границей назы-
называют также тукосмесями мокрого смешения.)
Количества питательных веществ и их соотношения в комп-
комплексных удобрениях могут быть различными. Удобрения, в кото-
которых соотношение питательных элементов соответствует агротехни-
агротехническим требованиям (для определенной культуры, почвы и т. д.)
называют уравновешенными.
По агрегатному состоянию удобрения разделяют на твердые,
жидкие (например, аммиак, водные растворы и суспензии) и газо-
газообразные, применяемые под укрытиями (например, двуокись угле-
углерода) .
Усвоение удобрений растениями зависит от их растворимости
я от характера почв, в первую очередь от концентрации ионов во-
водорода в почвенном растворе. Например, некоторые почвы обла-
обладают свойствами, позволяющими растениям усваивать Р2О5 (хотя
и медленно) из практически не растворимого в воде трехкальцие-
вого фосфата, особенно из тонкодисперсных его разновидностей —
фосфоритной и костяной муки. Концентрация ионов водорода в
почвенном растворе, необходимая для усвоения РгО5 из разных
видов фосфорных удобрений, различна. Методом оценки усвояе-
усвояемости Р2О5 является определение растворимости фосфатных со-
соединений в искусственных растворах, кислотность которых близка
к кислотности почвенных растворов — в аммиачном растворе ци-
цитрата аммония (реактив Петермана) и в 2%-ном растворе лимон-
,ной кислоты. Для определения усвояемой РгО5 применяют также
0,05 н. раствор серной кислоты и Н-катиониты. Усвояемость кор-
кормовых фосфатов, используемых в животноводстве (см. ниже), опре-
определяют по их растворимости в 0,4%-ном растворе соляной кислоты.
По степени растворимости фосфорные удобрения разделяют на
водорастворимые, цитратнорастворимые (т. е. растворимые в ци-
цитрате аммония), лимоннорастворимые (растворимые в лимонной,
гуминовой и других слабых органических кислотах) и трудно или
нерастворимые. Почти все азотные неорганические удобрения рас-
растворимы в воде, так же как и применяемые в качестве искусствен-
искусственных удобрений соединения калия.
Минеральные соли в сельском хозяйстве
29
Водорастворимые удобрения наиболее легко усваиваются рас-
растениями, однако значительная их часть вымывается из почвы грун-
грунтовыми и дождевыми водами и исчезает непроизводительно. Из
азотных удобрений растения извлекают 50—70% N, из калийных —
40—60% КгО. Еще в меньшей степени используется фосфор, кото-
который частично переходит в форму трудно усваиваемую растениями
в результате идущих в почве химических и биологических про-
процессов.
Эффективность легко растворимых удобрений значительно воз-
возрастает при применении их в крупнокристаллическом или в гра-
гранулированном виде. При этом удобрение размещается в почве оча-
очагами и медленнее переходит в почвенный раствор, вследствие чего
уменьшаются потери питательных веществ и увеличивается ис-
использование их растениями. Повышенная эффективность гранули-
гранулированных удобрений обусловлена также возможностью их внесе-
внесения малыми дозами совместно с семенами в рядки 12.
Большим преимуществом гранулированных удобрений являются
их хорошие физические свойства — они не комкуются, не слежи-
слеживаются, не пылят. Эти удобрения можно вносить в почву с по-
помощью туковых или комбинированных сеялок, применяемых для
одновременного высева семян и минеральных удобрений 18-19, что,
помимо облегчения труда и повышения его производительности,
значительно улучшает использование удобрений.
Для создания в почве запаса питательных веществ применяют
удобрения долговременного действия. Длительно сохраняющимися
в почве фосфатами являются цитратно-, лимонно- и, особенно,
труднорастворимые фосфаты. Для создания в почве запасов азота
служат естественные и искусственные органические азотсодержа-
азотсодержащие соединения. К последним относятся, например, оксамид (ди-
амид щавелевой кислоты H2NCOCONH2), медленно разлагаю-
разлагающийся в почве с образованием NHJ и N0^ производные пири-
пиридина C5H5N; мочевино-формальдегидные высокомолекулярные
композиции — уреаформы, карбамиформы (диметилентримочевина,
триметилентетрамочевина и другие)—продукты совместной кон-
конденсации карбамида (мочевины) CO(NH2J и формальдегида
СН2О; они могут служить источником азота в почве в течение дли-
длительного времени. Для этой же цели могут служить цитратнорас-
цитратнорастворимые двойные соли типа MeNH4PO4, например, магнийаммо-
нийфосфат MgNH4PO4 • Н2О. Медленный переход любых водорас-
водорастворимых веществ в почвенный раствор может быть достигнут
также при покрытии гранул удобрения пленками из высокомоле-
высокомолекулярных соединений или при применении гранулированных удоб-
удобрений, полученных из порошков с добавками этих же соединений
(например, полиакриламида) или полимеризующихся веществ.
Исследование10 скорости растворения в воде некоторых
минеральных удобрений — NaNO3, NH4NO3, KC1, CO(NH2J —
30
Гл. I. Минеральные соли в народном хозяйстве
в условиях, аналогичных естественным, показало, что это диффу-
диффузионный процесс, характеризующийся температурными коэффи-
коэффициентами в пределах 1,1—1,8. Коэффициенты скорости растворе-
растворения для этих солей в диапазоне линейной скорости воды от 2 до
50 см]мин имеют значения от 0,1 до 0,25 см/мин. В пределах 0—30°
эти коэффициенты увеличиваются почти пропорционально повыше-
повышению температурв1. Покрытие зерен удобрения полимерной пленкой
позволяет регулировать скорость их растворения, так как послед-
последняя не зависит ни от вида удобрения, ни от скорости потока, а
определяется диффузионной проницаемостью пленки, обусловлен-
обусловленной природой полимера, его концентрацией и типом использован-
использованного растворителя. Так. скорость растворения в воде CO(NH2J.
КС1 и других солей уменьшается в 100—200 раз при покрытии их
зерен пленкой, образованной из 27%-ного раствора полистирола
в бензоле или 10%-ного раствора поливинилацетата в ацетоне, или
из 6%-ного раствора нитроцеллюлозы.
Внесение удобрений не только повышает количество усвояемых
растениями питательных веществ в почве, но влияет и на физиче-
физические, физико-химические и биологические свойства почвы, от кото-
которых также зависит ее плодородие. Одним из важных факторов
является изменение рН почвенного раствора. Внесение в почву
веществ, обладающих кислыми или щелочными свойствами, соот-
соответствующим образом влияет на реакцию почвенного раствора.
Однако вследствие неодинакового использования растениями ка-
катионов и анионов растворенных солей изменение рН может прои-
произойти и при внесении в почву нейтральных солей. Например, при
систематическом внесении в почву таких веществ как (NH.^SO^
NH4C1, почвенный раствор приобретает кислую реакцию: взамен
извлекаемых растением катионов раствор обогащается ионами во-
водорода, что приводит к накоплению в почве свободной кислоты.
Другие удобрения, например. NaNO,. Ca(NO3J. являются источни-
источниками накопления в почве ионов ОН~. Поэтому только химическая
характеристика реакции удобрений недостаточна. Они должны
различаться и по физиологическим свойствам, обусловленным не-
неодинаковой степенью использования катионов и анионов. По этому
признаку удобрения классифицируют на физиологически кислые,
физиологически щелочные и Физиологически нейтральные.
Большое значение имеют физические свойства удобрений. Во-
Водорастворимые удобрительные соли не должны быть сильно гигро-
гигроскопичными, не должны слеживаться при хранении, они должны
быть сыпучими, легко рассеваться на почву, но в то же время
сохраняться на ней в течение некоторого времени, не сдуваясь
ветром и не слишком быстро вымываясь дождевой водой. Этим
требованиям в наибольшей мере отвечают гранулированные удо-
удобрения, производство и применение которых непрерывно возра-
возрастает.
Минеральные соли в сельском хозяйстве
31
Минеральные яды и другие препараты 9-23~52>59
Использование минеральных солей в сельском хозяйстве не
ограничивается применением их в качестве удобрений. Задачей, не
менее важной, чем получение высоких урожаев, является их сохра-
сохранение. Это требует защиты растений от уничтожения и порчи вре-
вредителями (насекомыми, грызунами) и предохранения их от забо-
заболеваний. Массовое появление вредителей (саранчи, клопа-чере-
клопа-черепашки, колорадского, жука, полевых грызунов и др.) может
привести к полной гибели урожая. В США при общей годовой стои-
стоимости урожая 13 млрд. долларов потери сельскохозяйственной
продукции в некоторые годы от болезней растений оценивают в
1,75 млрд. долларов, а от вредных насекомых — в 4 млрд. долла-
долларов, т. е. они составляют более одной трети урожая9< 59. Прибли-
Приблизительно такие же потери урожая вызываются сорняками. По дру-
другим данным46, сельскому хозяйству США приносят ущерб более
6000 видов вредных насекомых, уничтожающих около 10% урожая
пищевых продуктов и прядильных культур. Там погибает ежегод-
ежегодно около 8% урожая картофеля, уничтожаемого колорадским жу-
жуком, которым заражена площадь, превышающая 6 млн. км2. Коло-
. радский жук дает в один сезон потомство, превышающее 30 млн.
особей, способных съесть больше 20 т зеленой массы картофеля.
Распространение этого вредителя идет очень быстро — за 15 лет
A859—1874 гг.) он переместился на 3200 км через весь северо-
североамериканский материк с запада на восток и заразил все карто-
картофельные поля33. Если бы не велась борьба с колорадским жуком,
он мог бы уничтожить урожай картофеля полностью.
Широкое применение в сельском хозяйстве ядовитых препара-
препаратов для борьбы с вредителями и болезнями растений позволяет
значительно (иногда в несколько раз) увеличивать урожаи и по-
повышать качество продуктов. Каждый рубль, затраченный на ядо-
ядовитые препараты при правильном применении агротехники, сохра-
сохраняет продукции, т. е. дает прирост урожая, по зерновым и овощ-
овощным культурам на 5—10 руб., по хлопку, техническим и ягодным
культурам — на 15—30 руб., по цитрусовым — на 30—40 руб.
Препараты, используемые для защиты растений, носят общее
название пестицидов. («Пестицид» в дословном переводе означает
«убивающий заразу».) Их также называют сельскохозяйственными
ядами или ядохимикатами. Ядовитые препараты, применяемые для
уничтожения вредящих растениям насекомых, называют инсекти-
инсектицидами. В их число входят овициды — средства, уничтожающие
яйца насекомых, ларвициды — вещества, уничтожающие личинок
и гусениц, афициды — уничтожающие тлей. Вещества, уничтожаю-
уничтожающие круглых червей называют нематоцидами, уничтожающие мол-
моллюсков— моллюскоцидами, уничтожающие голых слизней — лима-
Цидами, уничтожающие растительноядных клещей — акарицидами.
32
Гл. I. Минеральные соли в народном хозяйстве
Инсектициды, поражающие паразитов через наружные покровы,
называют контактными, через органы дыхания — трахейными, а
убивающие только при попадании в кишечник насекомого — ки-
кишечными. Системными или внутрирастительными называются ин-
инсектициды, проникающие через корни и листья внутрь растения и
делающие его ядовитым для паразитов.
Препараты, предназначенные для борьбы с паразитирующими
на растениях грибками и вирусными заболеваниями, называют
фунгицидами, а для борьбы с возбудителями бактериальных бо-
болезней растений — бактерицидами. Некоторые препараты являются
одновременно и инсектицидами и фунгицидами. Независимо от
этой классификации под названием инсектофунгициды часто под-
подразумевают вообще все вещества, применяемые для борьбы с вре-
вредителями и болезнями растений.
Многие препараты используются в качестве антисептических
средств для обеззараживания почв, зерна и хранилищ продукции,
а также в качестве консервирующих средств для пищевых продук-
продуктов и животного сырья. Фунгициды, используемые для обеззара-
обеззараживания семян, называют протравителями.
Для уничтожения вредных теплокровных животных приме-
-няются препараты, называемые зооцидами. Препараты, применяе-
применяемые для борьбы с грызунами, называют также родентицидами,
для борьбы с крысами — ратицидами, для борьбы с птицами —
авицидами. Отпугивающие средства, служащие для защиты людей
и животных от нападения птиц, клещей, кровососущих двукрылых
насекомых (комаров, гнуса и др.), называются репеллентами.
Средства, привлекающие полезных насекомых, называют аттрак-
тантами.
Все шире стали использовать вещества, называемые стимуля-
стимуляторами или регуляторами роста растений29-44. Это препараты, ак-
активно влияющие на физиологические процессы растительного орга-
организма. В зависимости от условий и дозы стимуляторы роста могут
по-разному влиять на растения: в малых дозах они обычно акти-
активируют рост растений, а в больших дозах замедляют или
полностью подавляют их развитие.
Стимуляторы роста используются для обработки семян и рас-
растений с целью повышения урожая, а также для получения бессе-
бессемянных плодов; для ускорения созревания плодов; для торможе-
торможения развития семян, клубней, почек и цветов, например, для пред-
предотвращения вредного влияния на них весенних заморозков или для
удлинения срока цветения декоративных растений; для задержки
прорастания картофеля и других плодов при их длительном хра-
хранении; для активирования корнеобразования при вегетативном
размножении черенками; для предотвращения преждевременного
предуборочного опадания плодов; для предуборочного удаления
листьев и высушивания растений и пр.
Минеральные соли в сельском хозяйстве
33
Применение дефолиантов, т. е. веществ, вызывающих опадение
листьев, позволяет механизировать уборку урожая хлопка, ово-
овощей и других культур; применение дессикантов, т. е. высушиваю-
высушивающих веществ, облегчает уборку корне- и клубнеплодов, например
картофеля. В качестве стимуляторов роста применяются главным
образом органические соединения, но также и некоторые мине-
минеральные.
Особенно широко применяются стимуляторы роста для унич-
уничтожения сорняков. Такие препараты называются гербицидами.
В зависимости от свойств и нормы применения гербицидов они
разделяются на гербициды сплошного действия, т. е. действующие
на все виды растений, и селективные, т. е. избирательно уничто-
уничтожающие одни виды растений и безопасные для других. Химиче-
Химическая прополка полей с использованием авиации или наземных ма-
машин позволяет в сотни раз ускорить этот трудоемкий процесс и
резко повысить производительность труда в сельском хозяйстве.
Гербициды, предназначенные для уничтожения сорных кустарни-
кустарников и деревьев, называют арборицидами, а для уничтожения водо-
водорослей — альгицидами.
Большинство минеральных гербицидов обладает сплошным
действием, т. е. уничтожает все виды растений, и непригодно для
химической прополки посевов. Они могут применяться для унич-
уничтожения растительности на невозделываемых участках, в между-
междурядьях садов и ягодников, на дорогах, парковых площадках, же-
железнодорожных путях, для подготовки земель под плантации и т. п."
и расходуются при этом в значительных дозах (до 1000 кг на
1 га), в сотни раз превышающих дозы органических гербицидов
избирательного действия, используемых для химической прополки.
Из неорганических гербицидов избирательным действием обла-
обладают, например, соли меди — нитрат, хлорид, смесь хлорида и
сульфата. Они уничтожают сорняки (преимущественно однолет-
однолетние) и не уничтожают культурные посевы (злаки) 48.
В качестве пестицидов используются многие химические веще-
вещества в твердом, жидком (растворенном) или газообразном состоя-
состоянии. Часто растения подвергают опрыскиванию суспензиями илгт
эмульсиями, а также опыливанию тонко размолотыми препарата-
препаратами — дустами, которые представляют собой смеси активно дей-
действующих веществ с инертными носителями (наполнителями). Ве-
Вещества, оказывающие токсическое действие в газообразном или
парообразном состоянии, называются фумигантами. К ним отно-
относятся и твердые вещества, выделяющие ядовитые пары (например,
Цианид кальция, выделяющий под действием атмосферного воз-
Духа пары синильной кислоты).
Вследствие биологической активности пестициды обладают то-
токсичностью по отношению к теплокровным животным и человеку.
Чо степени токсичности, определяемой средне-смертельной дозой
2 М. Е. Позин
34
Гл. 1. Минеральные соли в народном хозяйстве
на 1 кг веса животного, их условно делят на четыре группы:
1) сильнодействующие вещества, средне-смертельная доза кото-
которых меньше 50 мг/кг; 2) высокотоксичные, при дозе 50—200 мг/кг;
3) среднетоксичные, при дозе 200—1000 мг/кг и 4) малотоксичные,
при средне-смертельной дозе, превышающей 1000 мг/кгБ0.
. Катионы металлов по степени токсичности могут быть распо-
расположены в следующий ряд51:
Ag>Hg>Cu>Cr>Ni>Pb>Co>Zn>Fe>Ca
Раньше в качестве сельскохозяйственных ядов применяли глав-
главным образом неорганические вещества. В настоящее время нахо-
находят широкое применение более эффективные и менее вредные для
человека и животных органические препараты. Однако и неорга-
неорганические яды не утратили своего значения и используются в зна-
значительных количествах. Наиболее распространенными неорганиче-
неорганическими пестицидами являются соединения мышьяка, меди, бария,
серы, фтора, циана, кислородных кислот хлора.
Минеральные корма 17-53-54
Минеральные соли применяются и в качестве кормовых
средств — для подкормки скота и птицы. Добавки минеральных
солей к кормовым рационам ускоряют развитие животных, увели-
увеличивают продуктивность скотоводства и птицеводства. Для этой
цели используют поваренную соль, карбонат кальция, фосфаты
кальция —дикальцийфосфат (преципитат) и трикальцийфосфат,
соли железа, меди, цинка, кобальта и другие, а также карбамид.
Кормовые фосфаты в отличие от удобрительных не должны содер-
содержать вредного для животного организма фтора и поэтому изго-
изготовляются специально. Медь, цинк, кобальт и многие другие
элементы требуются для животного организма в ничтожных коли-
количествах, но влияют на его развитие сущестьенным образом. Так,
подкормка овец и других животных солями кобальта и меди зна-
значительно увеличивает продуктивность по мясу и шерсти.
МИНЕРАЛЬНЫЕ СОЛИ В ПРОМЫШЛЕННОСТИ55
Различные отрасли промышленности используют в своих произ-
производственных процессах большое количество разнообразных мине-
минеральных солей. Среди всех отраслей промышленности, потребляю-
потребляющих соли, особое место занимает химическая промышленность,
являющаяся не только производителем минеральных солей, но и
крупнейшим их потребителем. Здесь минеральные соли исполь-
используются в качестве сырья для получения кислот, щелочей, газов и
других солей, а также в качестве сырья и вспомогательных мате-
материалов для многих химических производств. Так, химическая про-
промышленность потребляет колоссальные количества поваренной
Минеральные соли в промышленности
35
соли для получения хлора, едкого натра, соды и других солей нат-
натрия и соединений хлора. Сульфат натрия перерабатывается в сер-
сернистый натрий и другие содержащие серу соли. В производстве
красителей применяют сернистый натрий, нитрит натрия, хлорат
калия, сульфат натрия, бихроматы калия и натрия и многие дру-
другие соли. Значительный ассортимент солей и окислов используется
в качестве катализаторов при синтезе органических продуктов,
для беспламенного горения и других процессов.
В металлургической промышленности соли используют при из-
изготовлении черных, цветных и легких металлов и изделий из них,
в качестве сырьевых материалов, флюсов, присадок, реагентов для
обработки руд и изделий, компонентов электролитов и пр.
Многие соли применяют при обработке металлов, в частности
в машиностроительной промышленности, для придания поверхно-
поверхности деталей тех или иных свойств (прочности, твердости), для за-
закалки инструментов, для пайки и сварки металлов и сплавов, для
защиты изделий от коррозии и пр.
Для шлифовки и полировки металлических изделий, а также
стекла и изделий из пластмасс абразивная промышленность изго*
товляет специальные материалы, используя для этого окислы же-
.леза (крокус), алюминия, хрома, олова, а также карбонаты каль-
кальция, магния, силикат натрия и другие соли.
В горнорудной промышленности, во флотационных процессах
также применяют некоторые соли, например, сернистый натрий,
медный и железный купоросы.
В нефтяной промышленности при бурении скважин используют
сульфат бария, а при переработке нефти — минеральные катализа-
катализаторы, например, хлорид алюминия.
¦ Различные соли применяются в электропромышленности, в
электродной, в аккумуляторной и элементной промышленности, в
гальванотехнике и пр.
Стекольная промышленность является крупным потребителем
соды и сульфата натрия — основных компонентов шихты для варки
стекла. В производстве стекла и эмалей применяются и многие
другие минеральные соединения, придающие изделиям те или
иные специальные свойства (механическую прочность, термическую
и химическую устойчивость, плотность, цвет, степень прозрачности,
электропроводность и др.).
Большие количества окислов и солей используют в производ-
производстве керамических изделий и огнеупоров, при изготовлении термо-
термоизоляционных материалов, в цементной промышленности и в про-
промышленности строительных материалов.
Потребителем разнообразных минеральных солей является тек-
текстильная промышленность, использующая их для беления тканей
и протравки их перед крашением и печатанием в качестве различ-
различных аппретур и утяжелителей.
2*
Гл. I. Минеральные соли в народном хозяйстве
Различные соли и окислы металлов используются в резиновой
промышленности в качестве наполнителей для резиновых смесей,
в качестве усилителей механической прочности резин, для ускоре-
ускорения процесса вулканизации, для придания изделию необходимого
цвета.
Окислы, карбонаты, сульфаты и другие соли применяются и
в производстве пластических масс; для изготовления взрывчатых
веществ и порохов используют нитраты, хлораты и др.
Различные соли и окислы применяются в пищевой, кожевенной,
фармацевтической и во многих других отраслях промышленности.
Чем выше технический уровень различных отраслей промыш-
промышленности, тем обширнее и разнообразнее перечень применяемых
ими солей. В увеличении ассортимента солей значительную роль
играет также постоянный рост требований к качеству изделий про-
промышленности, обусловливающий более широкое применение раз-
разнообразных химических продуктов.
Большой ассортимент химикатов, в частности солей высокой
чистоты, потребляют новые отрасли промышленности, например,
производство полупроводниковых, ядерных материалов и др.
Во многих случаях одни и те же соли применяются для разно-
разнообразных целей. В соответствии с этим устанавливаются опреде-
определенные требования к качеству солей, узаконенные государствен-
государственными общесоюзными стандартами (ГОСТ) или согласованными
между производителем и потребителем специальными техниче-
техническими условиями (ТУ), предусматривающими также определен-
определенные виды тары и методы анализов продукта.
Следует отметить общую тенденцию к повышению чистоты со-
солей и других технических продуктов, выпускаемых химической
промышленностью как за рубежом, так и в СССР.
РАЗВИТИЕ ПРОИЗВОДСТВА СОЛЕЙ В СССР561
Солевые промыслы являлись старейшими химическими произ-
производствами в России. Производство поваренной соли из природных
рассолов возникло в России еще в XII в., а с XIV в. началось
селитроварение. Кроме поваренной соли и селитры, в допетровской
Руси вырабатывался поташ, который экспортировался и в другие
страны. В небольших количествах вырабатывались купоросы и не-
некоторые минеральные краски. Выработка селитры особенно увели-
увеличилась к началу XVIII в. в связи с расширением пороходелия.
В течение XIX в. были построены заводы, производившие мине-
минеральные кислоты ^серную, соляную) и соли (сульфаты, сернистый
натрий и др.)- В 1850 г. был основан Кокшанский завод, выраба-
вырабатывавший соли хрома, медный и железный купоросы, поташ. Бон-
дюжский завод (основанный в 1868 г.) производил сернокислый
глинозем и квасцы. В 1875 г. в Петербурге был построен Тентелев-
Развитие производства солей в СССР
37
ский химический завод, по тому времени наиболее совершенный,
вырабатывавший серную, азотную и соляную кислоты и на их
основе минеральные соли—сернокислый глинозем и др. В 1896 г.
на этом заводе было освоено производство суперфосфата. В 1900 г.
в России насчитывалось 526 химических предприятий, многие из
которых вырабатывали различные минеральные соли.
Тем не менее масштабы химического производства были срав-
сравнительно небольшими. Политика царского правительства, покро-
покровительствовавшего иностранному капиталу, не способствовала раз-
развитию отечественной химической промышленности. Расширение
источников химического сырья и рост русского производства не
поощрялись, а химические товары ввозились из-за границы по по-
повышенным ценам. В нашей стране, в недрах которой скрывались
самые богатые в мире залежи солей калия, глауберовой соли и
других, добыча этих солей не производилась. Калийные соли вво-
ввозились из Германии (за исключением поташа, вырабатывавшегося
из золы растений). Глауберова соль также ввозилась, хотя некото-
некоторое количество ее производилось путем растворения и кристаллиза-
кристаллизации сульфата натрия, получавшегося при выработке соляной кис-
кислоты и переработке импортной натриевой селитры на азотную
•кислоту. Ввозились и другие соли и сырье для их производства,
в частности фосфориты, борные минералы и пр.
В 1913 г. в Россию были ввезены (в числе прочих):.
Калиевые соли 77,0
Натриевая селитра 43,4
Суперфосфат 196,7
Фосфориты 53Л
Борные минералы ."~ 3,8
На отечественных заводах в 1912 г. было выработано:
Суперфосфата 150,1
Сульфата натрия 71,0
Хлорной извести 26,3
Сернокислого глинозема и
• квасцов 19,6
Поташа 16,4
Сернистого натрия 2,9
Хлористого бария 2,1
Медного купороса 1,0
Силикатов Na и К 12,8
Сульфата аммония 11.0
Хлористого цинка ' 6,7
Железного купороса 1,7
Сульфата магния 1,0
Хромпика натриевого .... 0,9
Тиосульфата натрля 0,04
Кремнефторида натрия .... 0,04
Фторидов и HF ..... 0,02
После Октябрьской революции вместе с созданием в СССР
мощной химической промышленности получило широкое развитие
и производство минеральных солей.
В период гражданской войны большинство химических пред-
предприятий было полностью или частично разрушено. В 1920—1921 гг.
было произведено всего около 25 тыс. т разных солей. В связи.с
Гл. I. Минеральные соли в народном хозяйстве
выполнением плана ГОЭЛРО были восстановлены и построены но-
новые солевые производства на ряде химических заводов, превратив-
превратившихся в крупные предприятия (Щелковский, Чернореченский, Бон-
дюжский и др.). Были пущены новые производства сернистого нат-
натрия, тиосульфата, магнезии, квасцов, хлористого цинка и пр.
В результате широко развернувшихся геологоразведочных ра-
работ уже в годы первой пятилетки A928/1929—1932/1933 гг.) основ-
основная химическая промышленность получила возможность использо-
использовать хибинские апатиты, Соликамские калиевые соли, карабогаз-
ский сульфат и многие другие виды отечественного сырья. Это
создало предпосылки для широкого развития производства мине-
минеральных солей, в первую очередь минеральных удобрений. Наряду
с коренной реконструкцией старых предприятий в годы первой
пятилетки построили крупные химические комбинаты (Воскресен-
(Воскресенский, Невский, Константиновский, азотно-туковые заводы — Берез-
никовский, Новомосковский, Горловский, Соликамский калийный
комбинат и др.), производящие значительные количества мине-
минеральных удобрений и других солей. За первую пятилетку произ-
производство суперфосфата увеличилось почти в 4 раза, а минеральных
солей для промышленных нужд —на 30%- Заново организовали
и значительно расширили производства плавиковой кислоты и фто-
фтористых солей, борной кислоты, солей мышьяка и хрома, сернокис-
сернокислого глинозема и др.
В 1933—1937 гг. выпуск фосфорных удобрений вырос в 3,2 раза,
а азотных — в 18 раз. Всего в 1937 г. было выработано около
3,2 млн. т минеральных удобрений (в условных единицах*), в том
числе ~1,5 млн. т суперфосфата. За счет широкого использова-
использования апатитового концентрата среднее содержание усвояемого Р2О5
в суперфосфате повысилось с 14,6 до 18,5%. В эти годы также рас-
расширилось производство калийных удобрений, солей бария, солей
сульфитного ряда, медного купороса и др.
XVIII съезд ВКШб) объявил третью пятилетку A938—1942 гг.)
пятилеткой химии. За эти годы продукция химической промыш-
промышленности должна была вырасти в 2,4 раза. В первые годы пяти-
пятилетки вошли в строй многие химические предприятия, в том числе
Кемеровский и Днепродзержинский азотно-туковые комбинаты
A939 г.), Чирчикский комбинат A940 г.) и др. Производство су-
суперфосфата в 1940 г. по сравнению с 1913 г. увеличилось в 66 раз
(в натуре). К началу Великой Отечественной войны СССР распо-
располагал развитой основной химической промышленностью и мощ-
мощными сырьевыми ресурсами для ее дальнейшего роста. Были от-
* При подсчете общего баланса удобрений их весовые качества принято вы-
выражать в условных единицах: для азотных удобрений в пересчете на азот стан-
стандартного коксохимического сульфата аммония B0,5% N); для фосфатных —
в пересчете на содержание 18,7% усвояемой РгО6; для калийных — в пересчете
на 41,6% КгО.
Развитие производства солей в СССР
39
крыты новые месторождения природных солей и, минералов — фос-
фосфорных, калийных, борных и многих других.
Ущерб, нанесенный химической промышленности войной, был
быстро ликвидирован. Уже в 1944 г. продукция химической про-
промышленности превысила довоенный уровень. В последующие годы
были построены новые предприятия (Кокандский, Джамбульский,
Сумской, Северодонецкий, Руставский, Навоийский, Кейдайнский
и другие заводы и комбинаты, Березниковские и Солигорские ка-
калийные фабрики и прочие предприятия), вырабатывающие мине-
минеральные удобрения и разнообразные соли.
Резко изменилась техника производства. Были освоены непре-
непрерывные способы производства (в частности, суперфосфата), новая
мощная аппаратура, в значительной мере механизированы трудо-
трудоемкие работы, стала широко использоваться автоматизация. Стали
выпускаться новые виды удобрений и солей, в частности гранули-
гранулированные продукты (суперфосфат, аммиачная селитра и др.).
В 1954 г. производилось уже 14 видов минеральных удобрений и
16 видов неорганических ядохимикатов (а вместе с органиче-
органическими 45).
Большое значение для дальнейшего развития производства
минеральных удобрений и многих других солей имели решения
сентябрьского A953 г.) Пленума ЦК КПСС по развитию сельского
хозяйства, майского A958 г.) Пленума ЦК КПСС по ускоренному
развитию химической промышленности и утвержденные XXI съез-
съездом КПСС A959 г.) контрольные цифры развития народного хо-
хозяйства СССР на 1959—1965 гг. В соответствии с этими реше-
решениями производство минеральных удобрений в 1965 г. достигло
31,3 млн. т в условных единицах или 7,4 млн. т в виде питатель-
, ных веществ (N + Р2О5 + КгО). В 1966 г. сельскому хозяйству
были поставлены (в условных единицах): 12,9 млн. т азотных,
ТАБЛИЦА 4
Динамика роста производства минеральных удобрений в СССР
(в условных единицах)
Годы
1913
1928
1932
1937
1950
Млн. т
0,2
0,3
1,0
3,2
5,5
Годы
1955
1956
1957
1958
1959
Млн. т
9,6
10,9
11,7
12,4
12,9
Годы
1960
1961
1962
1963
1964
Млн. т
13,8
15,3
17,3
19,9
25,6
Годы
1965
1966
1967
1968
1969
Млн. т
31,3
35,8*
40,1 *•
43,43*
46,0 4*
* 8,4 млн. т питательных веществ.
** 9,4 млн. т питательных веществ.
** 10,2 млн. т питательных веществ.
** 10,6 млн. т питательных веществ.
40
Гл. I. Минеральные соли в народном хозяйстве
4,6 млн. г калийных, 8,9 млн. т фосфорных удобрений и 4,0 млн. г
фосфоритной муки61. Динамика роста производства минеральных
удобрений в нашей стране показана в табл. 4 и на рис. 1.
Директивами XXIII съезда КПСС A966 г.) было предусмотрено
дальнейшее значительное увеличение выпуска минеральных удо-
удобрений. В соответствии с постановлением ЦК КПСС и Совета Ми-
Министров СССР (май 1968 г.) общая мощность производств, вы-
вырабатывающих удобрения за
' * период с 1968 по 1972 гг., дол-
должна увеличиться на 48 млн. т.
Дальнейший рост произ-
производства минеральных удобре-
удобрений и солей должен будет со-
сопровождаться глубокими усо-
усовершенствованиями в методах
их получения, ростом произво-
производительности труда, повышени-
повышением степени использования
сырья и энергии, улучшением
качества продукции и сниже-
1960 1965 1970 нием ее стоимости. Развитие
1928
1S37
Рис. 1. Производство минеральных удо-
удобрений в СССР (в млн. г).
промышленности минеральных
удобрений должно, в частно-
частности, идти по пути увеличения
выпуска комплексных, высококонцентрированных удобрений, рас-
расширения их ассортимента. Важной задачей является освоение
новых видов сырья и строительство новых туковых предприятий
в районах значительного потребления удобрений, особенно .на
востоке страны.
Некоторым резервом поставок удобрений сельскому хозяйству
является устранение их потерь при доставке от производителей
к потребителям за счет рационализации методов транспорта, пере-
перегрузок и т. п. Эти потери пока еще чрезвычайно велики — они со-
составляют 15—20%. Их уменьшение равнозначно повышению по-
полезной мощности предприятий без капитальных затрат.
КРАТКИЕ СВЕДЕНИЯ О МИРОВОМ ПРОИЗВОДСТВЕ УДОБРЕНИИ
И СОЛЕЙ626
Производство минеральных удобрений во всем мире в 1965/66 гг.
составило 48,1 млн. т питательных веществ (N + Р2О5 + КгО) 62.
Средняя концентрация питательных веществ в удобрениях
A966 г.) —в США 37,6%, в ФРГ 31,5%. На основании данных ко-
комиссии по пищевым и сельскохозяйственным ресурсам (FAO) при
Организации Объединенных наций, учитывающих рост населения
земного шара, прогнозируется следующая мировая годовая по-
Сведения о мировом производстве удобрений и солей
41
требность в питательных веществах для растений, необходимая
для предотвращения голода во многих странах мира (в млн. т) 63:
в 1975 г. 86 млн. т C9N + 28Р2О5 + 19К2О); в 1985 г. 144 млн. т
F7N + 47Р2Об + ЗОКгО). На эти же годы общая потребность в
фосфоритной руде (в натуре) должна составить соответственно
130 и 220 млн. г, потребность в сере (все виды сырья для произ-
производства серной кислоты и др.) 53 и 93 млн. г, потребность в ам-
аммиаке 60 и 102 млн. г.
Мировое производство азотных удобрений (в пересчете на эле-
элементарный азот) составило в 1967/68 г. 24,6 млн. т, а мощность
заводов аммиака достигла 36 млн. г N64. Около трех четвертей
вырабатываемого аммиака расходуется на производство удобре-
удобрений65. Особенно значительно растут производства аммиачной се-
селитры, карбамида и комплексных азотных удобрений. Доля жид-
жидких азотных удобрений превысила 15 %N. В США потребление
жидких удобрений составило в 1964/65 г. 4,9 млн. г, т. е. 18% от
общего количества удобрений (питательных веществ); около
2 млн. г или 47% внесенного в почву азота поступило в виде
жидких удобрений, из них более 1,3 млн. т в виде безводного ам-
аммиака и более 0,5 млн. т в виде солевых растворов66-в7. Потребле-
Потребление последних превысило 1 млн. т и составило ~6% от общего
потребления тукосмесей в США 68.
Мировая добыча фосфоритов в 1967 г. составила 76,6 млн. г,
в том числе в США 35,4 млн. г и в странах Африки 19,3 млн. г71.
80% от общего количества фосфоритов добывается в США, СССР
и Марокко. Общее производство фосфорных удобрений к 1967 г.
достигло 18 млн. т Р2О5, в том числе в Америке 5,7 (в США ~5),
в Западной Европе 4,9 (из них ~25% в виде основных шлаков),
в Азии 1,5, в Океании 1,3 (в Австралии ~1), в Африке 0,6 млн. т.
В последние годы резко увеличивается производство фосфорной
кислоты, двойного суперфосфата, фосфатов аммония и непрерыв-
непрерывно снижается выпуск простого суперфосфата. Доля РгО5, выпус-
выпускаемая в виде смешанных и сложных удобрений, значительно воз-
возрастает. Производство кормовых фосфатов колеблется в пределах
2—3 млн, т в год Р2О572.
В США для производства удобрений используют около 50%
добытого фосфорита, 25% расходуют для нужд других отраслей
промышленности и 25% экспортируют. Производство простого су-
суперфосфата в США уменьшилось с 1,63 млн. т P2Os (80% от всего
PgOs) в 1952 г. до 0,9 млн. т Р2О5 B0%) в 1966 г.; в виде двойного
суперфосфата и аммофосов выпускают около 80% РгО5. В 1967 г.
мощность цехов двойного суперфосфата достигла 2,17 млн. г
РгО5, а фосфата аммония — 3,08 млн. т Р?.ОД. Число установок су-
сухого смешения удобрений достигло в США 2500, на них смешивают
более 20% всех вырабатываемых удобрений73. Мощность устано-
установок по производству фосфора к 1970 г. 720 тыс. г74. 80% всей
Гл. I. Минеральные соли в народном хозяйстве
производимой в США фосфорной кислоты полутают экстракцион-^
ным способом 74.
Мировое производство калийных солей составило в 1965 г.
13,4 млн. г К2О, в том числе в США 2,85, в ФРГ 2,38, в ГДР 1,83,
во Франции 1,81, в Канаде 1,25 млн. г. В 1966 г. во всех странах
было выработано 14,4 млн. т К2О 75. Большая часть калийных удоб-
удобрений выпускается в виде хлористого калия, содержащего50—60%
К2О. Меньше 10% от общего количества К2О выпускается в виде
сульфата калия.
Среднее потребление всех видов удобрений (сумма питатель-
питательных веществ) в кг на I га пахотной земли составляет: в Голландии
более 500, в Бельгии, ФРГ, Японии 300—400, в Англии, Франции
ТАБЛИЦА 5
Производство некоторых минеральных солей
и других продуктов в США в 1966 г.
Название
Нитрат аммония для твердых удо-
удобрений
Нитрат аммония для жидких удо-
удобрений . . . г. ,«,
Нитрат аммония для взрывчатых
веществ
Нитрат аммония
Триполифосфат натрия
Кислый пирофосфат натрия . . . .
Пирофосфат натрия
Метафосфат натрия
Тринатрийфосфат
Тетракалийпирофосфат
Фосфор элементарный
Фосфорная кислота A00% РгО5) • ¦
в том числе термическая . . -
» » » экстракционная . .
Простой суперфосфат A00% P2O6J
Двойной суперфосфат A00% Р2О6)
Фосфат аммония A00% Р2О6) . . .
Преципитат .
Сульфат алюминия
» натрия
Карбид кальция
Соляная кислота A00% НС1) . . .
Фтористоводородная кислота
A00% HF)
Азотная кислота A00% HNO3) . .
Серная кислота A00% H2SO4). . .
Тыс. т
2070*
1800*
369*
5040 **
936
24,3
103,5
78,3
52,2
51,3
6V**
4023
«73
3150
900
1360
1360
230*
963
1379
1035*
1327,5
148,5
4455*
21802,5 •
Литература
43
* 1965 г.
** 1967 г.
и некоторых других странах Западной Европы 150—200, в США
50—60. В СССР в 1967 г. среднее потребление питательных ве-
веществ составило около 50 кг/га (при общей посевной площади
207 млн. га).
В табл. 5 приведены данные о производстве некото-
некоторых минеральных солеи в~США в 1966 г.76.
ЛИТЕРАТУРА
1. Д. Н. Прянишников, Избранные сочинения, т. I, II ч III, Сельхозгиз,
1952—1953. — 2. Агрономическая химия, под ред. А. Г. Шестакова, Сельхоз-
Сельхозгиз, 1954. — 3. Д. А. Сабинин, Минеральное питание растений, Изд. АН СССР,
1940. — 4. А. В. Владимиров, Физиологические основы применения азотистых
и калийных удобрений, Сельхозгиз, 1948. — 5. А. В. Соколов, Агрохимия фос-
фосфора, Изд. АН СССР, 1950. — 6. Е. И. Ратнер, Питание растений и примене-
применение удобрений, Изд. АН СССР, 1955. — 7. Справочник агронома по удобрениям,
Сельхозгиз, 1955. — 8. Т. П. Унанянц, Минеральные удобрения, Госхимиздат,
1949. — 9. К. М. Малин, Химия и урожай, Госхимиздат, 1955.— 10. Л. А. Пен-
ч е в а, Б. А. К о п ы л е в, М. Е. П о з и н, Изв. вузов. Хим. и хим. технол., 10, 319
A967); 12, 54 A969).
//. Гранулированные удобрения, Сборник работ ВИУАА за 1950 г., 1952.—
12. Н. С. Авдонин, Гранулированные удобрения, Сельхозгнз, 1952. —
/5. М. Я. Ш к о л ь н и к, Значение микроэлементов в жизни растений и в земле-
' делии, Изд. АН СССР, 1950. — 14. М. В. К а т а л ы м о в, сб. «Исследования по
прикладной химии», Изд. АН СССР, 1955, стр. 325. — 15. О. К- Добролюб-
Ский, ЖПХ, 28, № 9, 1022 A955). — 16. М. В. Кат алым о в, Микроэлементы
и их роль в повышении урожайности, Госхимиздат, 1957. —17. Микроэлементы
В жизни растений и животных, Изд. АН СССР, 1952. —18. М. Л. Кругляков,
Машины для внесения удобрений в почву, Машгиз, 1953. —19. Н. А. Мин х,
Машины для внесения удобрений в почву^ Сельхозгиз, 1954. — 20. Техника вне-
внесения удобрений, под ред. Е. В. Бобко, Сельхозгиз, 1938.
21. Сб. «Усовершенствование техники внесения удобрений», Изд. АН УССР,
Киев, 1955. — 22. И. П. С е р д о б о л ь с к и й, Химия почвы, Изд. АН СССР,
1953. — 23. С. И. В о л ь ф к о в и ч, Н. Н. Мельников, В. И. Орлов, Хим.
пром., № 6, 321 A954). — 24. Н. Е. Пестов, Физико-химические свойства зер-
зернистых и порошкообразных химических продуктов, Изд. АН СССР, 1947.—
25. Т. П. Унанянц, Словарь по удобрениям, Госхимиздат, 1948. —
26. С. И. В о л ь ф к о в и ч, Химия в сельском хозяйстве, Изд. АН СССР, 1963. —
27. П. В. П о п о в, Справочник по ядохимикатам, Госхимиздат, 1956.—
28. Д. фрир, Химия инсектисидов и фунгисидов, ИЛ, 1948. — 29. Н. Н. Мель-
Мельников, Хим. пром., № 1, 20 A951). — 30. Ю. А. Баскаков, Н. Н. Мельни-
Мельников, Там же, № 7, 435 A955).
31. И. И. Г у н а р, М. Я. Березовский, Химические средства борьбы
с сорняками, Сельхозгиз, 1952. — 32. В. Ф. Верзилов, Н. Н. Долгополо в,
Стимуляторы роста растений и их применение в зеленом строительстве, Изд. АН
СССР, 1951. — 33. Е. G. Lodeman, The Spraying of Plants, New York. 1916.—
84. Д. А л ь г р е н, Г. К л и н г м э н, Д. Вольф. Борьба с сорными растениями,
ИЛ, 1953. — 35. L. M. Stabler, J. Agr. Food. Chem., 1, 193 A953).—
86. H. E. Д е к а т о в, Химические средства борьбы с сорной растительностью
в лесном хозяйстве, Гослесбумиздат, 1958. — 37. А. Л. Ефимов, Химический
Метод борьбы с вредителями и болезнями сельскохозяйственных растений, Сель-
Сельхозгиз, 1949.— 38. В. И. О р л о в, П. В. П о п о в, М. А. М о р о з о в а, М. Г. Г а б-
риелова, Ядохимикаты (инсектисиды и фунгисиды), Краткий справочник, Гос-
Хвмиздат, 1946. — 39. Д. Г. Хорсфолл, Фунгисиды и их действие, ИЛ, 1948.—
44
Гл. I. Минеральные соли в народном хозяйстве
40. А. Л. Е ф и м о в. Справочник по применению ядов для борьбы с вредителями
и болезнями растений, Сельхозгиз, 1956.
41. В. С. Ч у в а х и н, К. С. М у ш н и к о в а, Б. Н. Пастухов, С. Д. По-
Попов, Б. А. Герасимов, П. В. 3 а р и н г, Пособие по борьбе с вредителями и
болезнями сельскохозяйственных культур, Сельхоз1ИЗ, 1945. — 42. А. Л. Ефи-
Ефимов, И. А. К аз ас, Инсектициды и фунгициды, Сельхозгиз, 1940.— 43. А. М. Ни-
Никифоров, Борьба с вредителями и болезнями сельскохозяйственных культур,
Сельхозгиз, 1954. — 44. Н. Н. Мельников, Ю. А. Баскаков, К- С. Бока-
рев, Химия гербицидов и стимуляторов роста растений, Госхимиздат, 1954. —
45 В И Орлов, Хим. пром., № 6, 377 A956). — 46. R. С. Roark, Chem. Eng.
News, 22, № 17, 1464 A944). — 47. В. И. Орлов, Хим. пром., № ц, 23 A945).—
48. В. И. Орлов, К. А. Г ар, Там же, JYb 9, 1 A946). — 49. В. И. Орлов,
К. А. Гар, М. Г. Габриелова, Там же, № Ц, 341 A947). — 50. Л. И. Мед-
Медведь, Журн. ВХО, 9, №. 5, 561 A964).
5/. С. Н. Иванова, Там же, 9, № 5, 496 A964). — 52. Н. Н. Мельни-
Мельников, Хим. пром., № 6, 181 A950). — 53. И. С. Попов, Кормление сельско-
сельскохозяйственных животных, Сельхозгиз, 1951. — 54. М. М. Дьяков, Ю. В. Голу-
Голубе н ц е в а, Минеральное питание сельскохозяйственных животных, Сельхозгиз,
1947. — 55. А И. Шерешевский, Т. П. Унанянц, Г. Я. Бахаровский,
Химические товары, Справочник, ч. 1, Изд. «Химия», 1967. — 56. П. М. Лукья-
Лукьянов, История химических промыслов и химической промышленности России,
т. I—III, Изд. АН СССР, 1948—1951. — 57. М. А. Блох, Развитие и значение
химической промышленности, НХТИ, 1920.— 58. Н. Н. Калмыков, С. А. В а й с-
б е й н, Экономика социалистической химической промышленности, Госхимиздат,
1955. — 59. Н. Н Некрасов, Химизация в народном хозяйстве СССР, Гос-
политиздат, 1955. — 60. Н. Н. Некрасов, Экономика химической промышлен-
промышленности, Изд. «Советская наука», 1957.
61. Союз Советских Социалистических Республик. 1917—1967, Изд. «Совет-
«Советская энциклопедия», 1967, стр. 234. — 62. Bombay Market, 30, № 23, 89 A967);
Agr. Chem., 23, № 5, 48 A968). — 63. С. J. Pratt, Chem. Eng. Progr., 63, № 10,
37 A967). — 64. Nitrogen, № 51, I, 14 A968); Chem. Eng. Progr., 64, № 5, 49
A968); H. А. Симулип, Хим. пром., № 2, 106 A969). — 65. D. E. Chalkley,
W. Turner, Viland Gas Internal., 7, № ю, 49 A967). — 66. W. С Scott,
J A. Wilbanks, Chem. Eng. Progr., 63, № Ю, 58 A967). —67. Nitrogen,
№ 49, 25 A967). — 68. Ibid., № 43, 13 A966); Agr. Chem. 22, № 10, 32
A967). — 69. С. И. Вольфкович, Агрохимия, № 9, 3 A965). — 70. Г. Л. Ко-
реньков, Н. А Устинова, Горнохимическое сырье зарубежных стран, Изд.
«Химия», 1965, гл. 17.
71. Phos. a. Pot., № 32, 1 A967) № 34, 1 и 3 A968). — 72. Chem.
Age (India), 18, № 7, 482 A967); Chem. Eng. News, 46, № 37, 107A A968).—
73. T. Me Bride, J. R. Camp, World NPKS, № 7, 3 П967). — 74. Chem. Eng
News. 46, № 13, 29 A968). — 75. M. R. Freeman, Mining Ann. Rev., 80 A967).—
76. Chem. Eng. News, 43, № 36, 100 A965); 44, № 36, 72 A966).
Глава II
РАСТВОРИМЫЕ СОЛИ В ПРИРОДЕ И МЕТОДЫ
ИХ ДОБЫЧИ
Отрасль химической технологии, изучающая способы получе-
получения и первичной переработки растворимых природных солей, на-
называется галургией. Основным галургическим методом является
тепловая обработка естественных и искусственно приготовленных
водных растворов природных солей. Нагреванием, испарением и
охлаждением этих растворов, их смешением и обработкой ими ис-
ископаемых солей при определенных температурах достигают выде-
выделения тех или иных продуктов. Иногда галургические приемы со-
совмещают с более глубокой химической переработкой. Главными
продуктами галургических производств являются: хлорид и суль-
сульфат натрия; соли калия, магния, бора; бром, иод и их соли; при-
природная сода и др.
Галургия базируется на классических работах Я. Г. ВантТоф-
фа, Н. С. Курнакова и их школ по физико-химическому анализу
солевых систем. Эти работы позволили выявить условия кристал-
кристаллизации солей из рассолов. На их основе Я. Г. Вант-Гофф создал
теорию образования природных соляных залежей.
ОБРАЗОВАНИЕ СОЛЯНЫХ ЗАЛЕЖЕЙ
В земной коре и на ее поверхности наряду с залежами различ-
различных не растворимых в воде минералов находятся залежи раство-
растворимых минералов — солей, встречающихся как в виде твердых
отложений, так и в виде растворов. Эти залежи возникали на про-
протяжении многих геологических периодов жизни земли, когда созда-
создавались благоприятные для их появления геохимические, гидрогео-
гидрогеологические и климатические условия. Источником этих залежей
является морская вода, из солей которой образовались и место-
месторождения ископаемых солей, и соляные озера, и подземные рас-
рассолы ', ~ '
L
¦}
46
Гл. II. Растворимые соли и методы их добычи
При испарении морской воды, проникавшей в бессточные кот-
котловины, концентрация солей постепенно повышалась. Из насыщен-
насыщенных рассолов кристаллизовались соли, образовавшие в течение
длительного времени мощные напластования. Часто испарение
воды происходило при последовательном перемещении через не-
несколько котловин с ограниченным стоком, что приводило к обра-
образованию солевых залежей различного состава, соответствующих
составу солей, выделявшихся в разных стадиях испарения. Соле-
отложение продолжалось и в зимние периоды, при понижении тем-
температуры рассолов, что также приводило к изменению состава
кристаллических фаз.
Концентрации и соотношение солей в воде мирового океана в
разные геологические эпохи не оставались неизменными. Менялась
и последовательность кристаллизации солей. К изменению состава
первичных соляных отложений и к образованию вторичных место-
месторождений приводило размывание уже образовавшихся первичных
залежей грунтовыми водами и рассолами. Существенную роль в
этих процессах играют химические взаимодействия растворов с
окружающими их материковыми породами. Наконец, значительное
влияние на формирование солевых залежей и на последующие
изменения их оказывают тектонические явления2.
Все эти процессы, продолжающиеся и в настоящее время, при-
привели к образованию многочисленных месторождений растворимых
солей — соляных озер и их донных отложений, подземных с опле-
ний рассолов и мощных твердых залежей, состоящих из покры-
покрывающих друг друга солевых пластов различного состава. Вслед-
Вследствие осадочного происхождения твердые солевые отложения, на-
находящиеся в геологически не нарушенных районах, залегают в
виде г.ологих пластов разной толщины, измеряемой десятками и
сотнями метров и распространяющихся на значительных простран-
пространствах. Твердые хлориды натрия и калия обладают пластичностью,
вследствие чего они часто встречаются в месторождениях, имею-
имеющих форму соляных куполов, которые образовались в результате
выдавливания этих пластичных горных пород к земной поверхно-
поверхности. Такие соляные купола, обычно овальной формы, достигают
2—3 км в длину и опускаются в глубь земли на сотни и даже ты-
тысячи метров.
Главными компонентами солевых залежей являются хлоридные
и сульфатные соединения натрия, магния, калия и кальция. В твер-
твердых отложениях они встречаются в виде различных минералов —
простых и сложных солей, безводных и кристаллогидратов. Осо-
Особенно распространены: галит NaCl, мирабилит Na2SO4- 10H2O, те-
тенардит Na2SO4, эпсомит MgSO4 • 7Н2О, астраханит Na2SO4 • MgSO4 •
' 4Н2О; сильвин КС1, карналлит КС1 • MgCl2 • 6Н2О, каинит КС1 •
> MgSO4 • ЗН2О и др. Во многих отложениях находятся соли бро-
Природные рассолы и классификация соляных озер
47
ма, бора, карбонаты (например, сода NaCO3 • ЮН2О) и др. Реже
встречаются нитраты (например, NaNO3).
Солевые залежи — природные рассолы и твердые отложения —
во всех случаях содержат основные компоненты морской воды 3~6.
В морской (океанской) воде находится в среднем около 3.5% раз-
различных солей C4—38 г/л при плотности океанской воды 1,024—
1,029 г/см3). Около 78% солевой массы морской воды прихо-
приходится на долю хлорида натрия. Если бы из морской воды могли
быть извлечены все соли и распространены на площади всех мо-
морей и океанов C60 000 000 км2), то толщина такого солевого пласта
превышала бы 60 м, из которых 48 м приходилось бы на долю
NaCl. Четыре соли, связанные уравнением
2NaCl + MgSO4 5=± Na2SO4 + MgCl2
составляют 93% от веса сухого остатка океанской воды; на долю
солей калия, кальция и брома приходится около 6%, а всех осталь-
остальных соединений, в том числе газообразных и органических, око-
около 1%.
Вода океанов более концентрированная, чем вода морей, так как
последние питаются пресными водами рек*.
Запасы природных солей на территории СССР практически не-
неисчерпаемы. Распространение отложений солей натрия, калия, маг-
магния в нашей стране связано с определенными геологическими пе-
периодами, главным образом Пермского моря, простиравшегося на
нынешней территории Европейской части СССР от Белого до Кас-
Каспийского морей, а также Арало-Каспийского бассейна и др. Древ-
Древние моря, покрывавшие громадные поверхности Земли, оставили
после своего исчезновения мощные залежи ископаемых солей и
многочисленные соляные озера. По своему географическому распо-
расположению соляные месторождения образуют так называемый соля-
соляной пояс, охватывающий большую часть территории СССР, от
нижнего течения Дуная до Якутии, и переходящий в пределы
Монголии и Китая7. Озерные месторождения солей относятся к
наиболее молодым месторождениям полезных ископаемых, насчи-
насчитывающим около 8—10 тыс. лет. Многие из соляных озер образо-
образовались в современную эпоху. В то же время соляные пласты неко-
некоторых озер (например, Баскунчак, Эльтон и др.) образовались в
межледниковые эпохи и возраст их исчисляется десятками ты-:
вяч лет.
ПРИРОДНЫЕ РАССОЛЫ И КЛАССИФИКАЦИЯ СОЛЯНЫХ ОЗЕР
В современных условиях главными источниками соленакопле-
ния в СССР являются соляные озёра. Под влиянием внешних"
* Состав и концентрацию солеи в некоторых водоемах см. в табл. 12 на
етр. Ю9.
48
Гл. П. Растворимые соли и методы их добычи
факторов в них непрерывно протекают постоянные, периодические
и циклические процессы, направленные в сторону достижения рав-
равновесных состояний8. Постоянные процессы вызываются воздей-
воздействием среды на озерный рассол, которое ведет к односторонне
направленному постепенному изменению его химического состава.
Такие процессы называются процессами метаморфизации рассо-
рассолов; они сопровождаются образованием постоянных осадочных
отложений — малорастворимых продуктов метаморфизации: СаСОз,
MgCO3, CaMg(CO3h, CaSO4-2H2O и др. Периодические процессы
обусловлены многолетними колебаниями климата, а циклические —
изменением климатических факторов в течение года. Под влия-
влиянием этих процессов образуются периодические твердые фазы —
легкорастворимые соли. Периодические процессы косвенно отра-
отражаются и на метаморфизации.
Природные воды, в том числе и воды озер, по концентрации
содержащихся в них растворенных веществ (в вес.%) делятся на
три группы: пресные — меньше 0,1, солоноватые — от 0,1 до 3,5 и
соленые или соляные — больше 3,5. Вода соляных озер называется
рапой, или рассолом. Так называют и жидкую фазу соляных си-
систем.
По химическому составу воды соляных озер, как и другие ми-
минерализованные воды, разделяются на три основных типа: карбо-
карбонатный, сульфатный и хлоридный9. Принадлежность озерного рас-
раствора к тому или иному типу определяется относительным содер-
содержанием в нем главных ионов: СОз~, НСОз, SO|", СГ, Са2+, Mg2+
и Na+. Она устанавливается по так называемым коэффициентам
метаморфизации Км — характерным отношениям концентраций
(в %) главных солей в растворе. Изменение коэффициентов мета-
метаморфизации по времени года (уменьшение и возрастание) повто-
повторяется циклически в течение многих лет (годичные циклы). Изме-
Изменение величины коэффициентов метаморфизации в годичном цикле
является следствием изменения температуры и состава рапы, а из-
изменения за длительные периоды времени обусловливаются воздей-
воздействием материка на питание водоема, влиянием донных отложений,
а также биологическими процессами 10.
В качестве коэффициентов метаморфизации пользуются следую-
Na2CO3 + NaHCO3 Na2SO4 MgSO4 MgCl2
щими отношениями: — ., С(Л -; ,. cr. ; ., ni ¦; -?~~ .
Na2SO4 ' MgSO4 ' MgCl2 CaCl2
Озера карбонатного типа («содовые» озера) содержат в каче-
качестве основных компонентов Na2CO3, NaHCO3, Na2SO4 и NaCl; они
характеризуются величиной коэффициента метаморфизации
„ Na2CO3 + NaHCO3
Дм = ъг~сг\ > значение которого может изменяться в ши-
роких пределах — от 0 до оо; для этих озер Ки~ 2 4
как MgSO4 в них не содержится.
MgSO4
= «о, так
Природные рассолы и классификация соляных озер
46
Озера сульфатного типа, в которых главными компонентами
являются сульфаты и хлориды натрия и магния, разделяются на
два подтипа: сульфатнонатриевый и сульфатномагниевый (хлор-
магниевый). Первый из них не содержит MgCl2 и характеризуется
коэффициентом метаморфизации 0 <^g~<oo; здесь -д^р =
= оо. Второй содержит MgCl2, но не содержит Na2SO4, поэтому
0, а характерный для этого случая коэффициент мета-
метаморфизации М^Г| 4, называемый коэффициентом Курнакова11 /(мК,
MgSO4
мС|
g 2
_ MgSO4 .
меняется в пределах 0< ^ С1 <оо.
Для озер хлоридного типа, не содержащих сульфатов натрия и
магния. /Смк = 0; для них характерным коэффициентом является
л MgCl, ^
0<-cfci7<o°-
По величине коэффициента Курнакова соляные озера делят так-
также на озера первого и второго класса. Для океанской воды
Кмк = 0,7. Соляные озера, в которых Кык < 0.7 (от 0,7 до 0) от-
относят к первому (морскому) классу; бессульфатные озера, для ко-
' торых Кмк = 0, относят ко второму классу. При такой классифи-
классификации к сульфатным озерам относят те, в которых Кшк > 0,7.
Главными среди многих природных явлений, приводящих к ме-
метаморфизации рассолов, являются:
1) взаимодействие компонентов двух рассолов при их смеше-
смешении с образованием осадков;
2) ионообменные процессы, протекающие при участии кол-
коллоидных частиц иловых отложений, которые накапливаются на
дне озер в результате разрушения окружающих почв и горных
пород;
3) биохимические процессы, протекающие с участием микро-
микроорганизмов в рассолах и илах, содержащих органические веще-
вещества.
Рассмотрим в качестве примеров некоторые пути метаморфиза-
метаморфизации. Пресные воды, питающие озеро, несут в него Ca(HCO3h, об-
образующийся при выщелачивании известковых пород с участием
СО2 воздуха. Взаимодействие бикарбоната кальция с сульфатом
магния
MgSO4 + 2Са(НСО3J ^=± CaSO4 • 2Н2О + CaMg(CO3)
приводит к уменьшению содержания в рапе MgSO4 и может пони-
понизить коэффициент метаморфизации ..е'Г|4 До нуля, — озеро ста-
нет беосульфатным. Дальнейшее взаимодействие бикарбоната
кальция с хлоридом магния
MgCl2 + 2Са(НСО3J
CaMg(CO3)» + СаС1а + 2НаО + 2СО,
50
Гл. If. Растворимые соли и методы их добычи
приводит к появлению в рассоле СаС12. Таким образом осуществ-
осуществляется постепенный переход озер из сульфатных в хлоридные с од-
одновременным образованием осадочных пород. Питание же озера
морского типа речной водой, содержащей сульфат натрия, приво-
MgSO4
дит к увеличению отношения м С1 . т. е. к превращению озера
в сульфатное. В этом процессе большую роль играют ионообмен-
ионообменные реакции. Биохимические процессы приводят, в частности, к
восстановлению сульфатов и выделению сероводорода под влия-
влиянием СО2 воздуха; в резуль-
тате этого содержание суль-
фатов в Озере уменьшает-
ся.
Направление процессов-
метаморфизации зависит и
от климатических условий.
В сухом климате происхо-
происходит постепенное изменение
типа рассола от карбонат-
карбонатного к сульфатному и затем
к хлоридному, — это прямой
или нормальный путь мета-
метаморфизации; во влажном
климате процесс метамор-
метаморфизации идет в обратном
направлении (рис. 2) и.
Na2CO3(NaHCO3)
MgCl2
СаС12
Рис. 2. Схема главных типов природных
рассолов и путей их метаморфизации.
Для характеристики содержания в озерах других, не основных
элементов — калия, брома, бора и пр. — пользуются коэффициен-
коэффициентами, показывающими отношение концентрации этих элементов
(ионов или солей) к концентрации одной из_ основных солей или
суммы всех солей BСОлей, %). например ^ ' ¦ (умножение на
10й производят, чтобы оперировать с целыми числами). При зна-
значительном содержании К+, Вг~ и других элементов озеро соответ-
соответственно называют калийным, бромным. Так, озера, в которых
КС! • 10s ^ „„ КС1 • 10 ,
>20 и м „. > 1,
называют хлоркалиевыми.
По соотношению содержащихся в озере жидкой и твердых фаз
различают рапные, сухие и подпесочные озера. Рапные озера
сохраняют в течение всего года' поверхностный слой рапы, покры-
покрывающий илистое дно или донные отложения солей; порядок напла-
. стования солей в этих отложениях соответствует последователь-
последовательности их политермической кристаллизации, вследствие чего в верх-
верхних слоях оказываются наиболее растворимые соли12. В сухих
озерах уровень рапы находится ниже обнаженной поверхности
донных отложений, — она покрывается небольшим слоем рапы
Природные рассолы и классификация соляных озер
51
лишь во влажный период года. В подпесочных озерах уровень дон-
донной рапы в течение всего года значительно ниже поверхности твер-
твердых солевых отложений, которая обычно покрыта слоем наносов
(песка и др.); эта стадия существования соляного озера предше-
предшествует переходу в соляную залежь. В сухих и подпесочных озерах
нижние пласты соляных отложений могут содержать более рас-
растворимые соли, чем верхние.
Слой соли, выкристаллизовавшейся в озере в течение опреде-
определенного периода года, соответствующего по климатическим усло-
условиям садке этой соли (например, галита летом или мирабилита
зимой), называется новосадкой13. Слой новосадки может частично
или полностью растворяться в течение этого же года в сезон, сле-
следующий за садочным, когда рапа озера вновь окажется ненасы-
ненасыщенной этой солью. Соль, оставшаяся нерастворенной в течение
годичного цикла, слой которой сохранился в донном отложении,
называется старосадкой. Она появляется, когда донная рапа до-
достигает насыщения. Отложения старосадки характеризуются слоис-
слоистостью, соответствующей годичным циклам ее образования. Под
слоем старосадки образуется корневая соль, появляющаяся в ре-
результате вторичных процессов перекристаллизации. Корневая соль
может быть представлена минералами, образовавшимися при
взаимодействии старосадки с донной рапой в результате измене-
изменения ее состава, температуры и др. Корневая соль (соляной пол)
может состоять из галита, эпсомита, астраханита, глауберита и
мирабилита. Она играет большую роль как регулятор насыщения
рапы и садки солей. В период влажного лета в озерах без корне-
корневой соли рапа может сильно обводниться с прекращением садки —
насыщение рапы и садка соли произойдет лишь в следующем
засушливом сезоне. Это нарушает равномерную работу про-
промысла.
Озера, в которых отложения солей представлены только перио-
периодически образующейся и растворяющейся новосадкой, называются,
самосадочными. Озера же, имеющие, кроме новосадки, отложения
старосадки и корневые, называются корневыми.
В тех озерах, в которых при образовании новосадки рапа ухо-
уходит под уровень соли, ее испарение продолжается путем капилляр-
капиллярного поднятия на поверхность новосадки, хотя оно и происходит
в этом случае в 100—200 раз менее интенсивно, чем с открытой по-
поверхности озера 14.
При садке солей в озерах имеет место увлечение других солей,
относительно которых раствор не насыщен. При садке галита ко-
количество увлекаемых солей меньше, чем при садке других солей.
При совместном образовании льда и садке солей лед увлекает
около 30—40% выделившихся солей, а остальная их часть обра-
образует донные отложения. В зимних условиях в озерах Забайкалья
и Якутской АССР, а также в Северном Ледовитом океане
52
Гл. II. Растворимые соли и методы их добычи
образуются соляные выцветы на льду — «гуджир», или «ледяные
цветы». Это объясняется увеличением объема при замерзанииш.
Объектами промышленной эксплуатации являются как твердые
озерные отложения, так и рапа — поверхностная, а в сухих озерах •
подкорковая, называемая также межкристальной. !
Изучение явлений метаморфизации рассолов и законов солена- j
копления не только облегчает поиски природных солевых залежей,
но открывает возможность интенсифицировать медленные есте- ;
ственные процессы, ускорять выделение из природных растворов
полезных минералов, искусственно создавая их месторождения.
Такие методы основаны, например, на перекачке рассола мощными
насосами из одного озера в другое (с рассолом другого состава)
или рассолов из нескольких озер в одну котловину, наиболее вы-
выгодно расположенную для промышленной разработки солевых от-
отложений, образовавшихся при таком искусственном сливании при-
природных рассолов.
РАЗРАБОТКА ЗАЛЕЖЕЙ ИСКОПАЕМЫХ СОЛЕЙ
Горные разработки
Добыча солей из давно сформировавшихся твердых залежей
производится обычными горными работами. Таким путем извле-
извлекают из недр каменную соль, тенардит, сырые соли калия: сильви-
сильвинит, карналлит, каинит и др.
Когда верхний горизонт соляного пласта находится на неболь-
небольшой глубине, его разработку можно производить открытым спо-
способом. В этом случае устраивают карьер или разнос, т. е. удаляют
слой покровных пород и производят выемку соли. На крупных
разработках для этой цели используют экскаваторы, скреперы
с лебедками или с тракторной тягой, механические лопаты и дру-
другие механизмы. Обычно пласт соли предварительно разрыхляется
(дробится) механическим или взрывным способом.
Для мощных пластов, залегающих на большой глубине, откры-
открытый способ разработки непригоден. Кроме того, его недостатком
является загрязнение соли наносами пыли, а особенно заливание
карьера атмосферными осадками и почвенными водами. Поэтому
разработку пластовых месторождений ведут обычно подземным
способом путем устройства шахт и выемки соли из галерей или
камер, вырабатываемых в толще соляного пласта16. Проходка
широких ходов при закладке подземных камер осуществляется
штрекопроходческими комбайнами, врубовыми машинами; про-
проходка скважин — сбоечно-буровыми машинами 17; выемка соли и
погрузка — скреперными установками; для транспорта и подъема
соли на поверхность в крупных шахтах используют подземные
вагонеточные электропоезда и лифты м-20.
Разработка залежей ископаемых солей
Подземное выщелачивание
Грунтовые воды, проникая в залегающий в недрах пласт соли,,
размывают его — образуется естественный подземный рассол21.
Такие рассолы могут извлекаться на поверхность через колодцы
или буровые скважины и служить источниками добычи солей22.
Извлечение соли на поверхность в виде рассола проще и дешевле,
чем в твердом виде, и особенно удобно, когда эта соль подвер-
подвергается дальнейшей переработке в растворе. Поэтому в ряде слу-
случаев оказывается целесообразным осуществлять подземное выще-
выщелачивание соли из соленосной породы. Этот метод не дает воз-
возможности влиять на состав извлекаемого рассола и поэтому
широко используется пока только для извлечения из недр пова-
поваренной соли.
Подземное выщелачивание осуществляют разными способами!
систематическим орошением водой и постепенным размыванием
подземных камер в солевом пласте или затоплением камер; обра-
образующийся концентрированный рассол выкачивается насосами.
В СССР применяют более совершенный способ выщелачивания
через бурояые скважины. В скважину, закрепленную колонной
"стальных обсадных труб диаметром 150—250 мм, вставляется
труба меньшего диаметра G5—100 мм). По одной из этих труб
с помощью центробежного насоса высокого давления B0—25 ат)
в пласт соли нагнетается вода. Она растворяет соль и в виде рас-
рассола выдавливается на поверхность по другой трубе. Различают
два режима работы скважин — противоточный, когда воду подают
по наружной трубе, а рассол поднимается на поверхность по внут-
внутренней (рис. 3), и прямоточный, когда по внутренней трубе подают
воду, а рассол выдавливается по наружной трубе. Глубина сква-
скважин и давление, под которым в нее подают воду, зависят от глу-
глубины залегания пласта соли или подземного источника рассола.
Производительность скважины 10—25 м3 рассола в 1 ч. (Иногда
воду подают в скважину самотеком; в этом случае рассол, который
имеет большую плотность, не может достигнуть поверхности за
счет давления столба воды, и его откачивают глубинным насосом,
опущенным в скважину до уровня, определяемого разностью плот-
плотностей рассола и воды.)
Камера, образующаяся в пласте соли при размывании его
водой через буровую скважину, постепенно приобретает форму,
близкую к форме опрокинутого конуса, так как в результате
естественной конвекции боковая поверхность, а особенно потолок
камеры, растворяется быстрее, чем дно, покрытое насыщенным
рассолом и шламом механических примесей. Поэтому боковая по-
" верхность становится все более пологой и затем покрывается слоем
пустой породы, препятствующим дальнейшему выщелачиванию.
Интенсивность рассолообразования уменьшается, и эксплуатацию
I
54
Гл. П. Растворимые соли и методы их добычи
скважины приходится прекращать, когда образующая конуса до-
достигает угла 30—40°. В результате этого запасы месторождения
при таком способе эксплуатации используются не больше чем
на 5—15%.
Эксплуатацию скважин осуществляют также комбинированным
противоточно-прямоточным методом 23. Основная стадия здесь пря-
прямоток, когда происходит «размыв» слоя соли с образованием
большого количества рассола; при более короткой стадии работы
Рассол
V+W+V+V
+ + + +Т.
+ ++++ + + -Ш
*¦ + + + + + + + +
*:*:¦*¦>;¦>
+ + +++ + + +
+
;¦:*:¦#:¦:¦>
+v+v+++v+v
+ + + + + 4- + + +
Рис. 3. Схема выщелачивания пласта
•солн через буровую рассольную
•скважину, работающую противотоком.
Рис. 4. Схематический разрез буровой
скважины с гидрозрубом.
противотоком происходит «промывка» скважины с удалением из
нее большей части нерастворимых частиц. Продолжительность
ликла чередования направления потоков внутри скважины равна,
например, 2 ч при соотношении продолжительности режимов «раз-
«размыва» и «промывки» в пределах от 7: 1 до 3: 1.
Более совершенной является эксплуатация скважин с гидро-
гидроврубом (рис. 4) 24~26. В этом случае вместе с водой в скважину
нагнетают воздух или нефть. Вначале поддерживают уровень воды
на постоянной высоте 1—1,5 м от забоя. При этом растворение
идет только по окружности камеры, потолок же предохраняется
от действия воды тонким слоем «нерастворителя» — воздуха или
нефти. Образуется вруб — приблизительно плоская цилиндриче-
цилиндрическая камера высотой 1—1,5 м и диаметром 100 м и больше. (Бо-
(Более вероятно, что при выщелачивании гидровруба форма обра-
вующейся полости в соляной залежи соответствует форме гипер-
гиперболоида вращения.) После этого воздух или нефть выдавливают
на дневную поверхность, повышая уровень рассола, причем про-
Лолучение солей из рассолов и морской воды
55
исходит интенсивное растворение потолка камеры. Осаждение пу-
пустой породы на растворяющейся поверхности при этом исключено
и использование запасов месторождения возрастает.
Наиболее прогрессивным является ступенчатое выщелачива-
выщелачивание27, особенно для разработки пластов соли, содержащих много
нерастворимых включений. В этом случае вначале производят
размыв не в форме вруба, т. е. плоской щели, а в форме конуса»
обращенного вершиной вниз. Затем, периодически повышая уро-
уровень подвода воды и изменяя уровень отбора рассола, производят"
ступенчатое растворение соли, так что камера выщелачивания
принимает форму, близкую к цилиндру, с основанием в виде во-
воронки и сводчатой кровлей. Нерастворимые включения скапли- "
ваются в нижней части камеры. Степень использования пласта
соли резко возрастает.
Питание пласта соли водой для его размыва и откачку рас-
рассола производят и по разным скважинам — по одним подают
воду, по другим откачивают рассол. При такой групповой системе
эксплуатации скважин коэффициент извлечения соли особенно
возрастает при последовательной отработке запасов по падению
пластов и при использовании провальных воронок, образовавших-
образовавшихся в результате выщелачивания, для подачи воды к разрабатывае-
разрабатываемым верхним пластам соли28. Это позволяет сократить число во-
водоприемных скважин и значительно увеличить количество пода-
подаваемой воды.
Образующиеся при подземном выщелачивании пустоты могут
явиться причиной обрушения кровли камер — опускания и обвалов
надсолевых пород. Поэтому этот метод извлечения солей может
. использоваться лишь при достаточной прочности покровных пла-
пластов.
ПОЛУЧЕНИЕ СОЛЕИ ИЗ РАССОЛОВ И МОРСКОЙ ВОДЫ
Добыча самосадочных солей
В СССР широко развита промысловая добыча самосадочных
солей (Na'Cl, Na2SO4- 10Н2О), кристаллизующихся из рапы озер
и лиманов в результате естественного испарения воды под дей-
действием солнечного тепла (NaCl)- или в результате охлаждения
рапы в зимнее время (Na2SO4- 10H2O).
Добыча самосадочной соли производится ручным и механиче-
механическим способами. Ручной способ добычи соли требует тяжелого
физического труда. Поэтому на крупных соляных промыслах при
значительной толщине слоя соли пользуются исключительно меха-
механическими способами добычи соли: применяют скреперы, трактор-
тракторные погрузчики, бульдозеры, одно- или многоковшовые экскавато-
экскаваторы, солесосы. Солесосами пользуются при толщине слоя рапы не
менее 0,5 м. Они монтируются на железнодорожных платформах
56
Гл. П. Растворимые соли и методы их добычи
(сухопутные) и на понтонах (плавучие). Солесос (рис. 5)
представляет собой центробежный насос / с подвижной всасы-
всасывающей трубой 2, на устье которой имеется резак-разрыхлитель 3
е ножами. Разрыхлитель вращается со скоростью 36 об/мин. При
этом он разрывает слабо спаянные в пласте кристаллы соли, по-
получаются куски 5—35 мм, которые засасываются вместе с рапой
солесосом. На грохоте 7, соль отделяется от рапы и ила, стекаю-
стекающих по лотку 8 обратно в озеро. С помощью транспортера 9 —
бесконечной цепи с гребками — соль передвигается к краю грохота
Рнс. 5. Солесос:
/ — центробежный насос; 2 — подвижная всасывающая труба; 3 — резак-разрыхлитель;
4 — двигатель; 5 — нагнетательная труба; б и 8 —лоткн; 7 — грохот; 9 — транспортер.
и ссыпается в кучу или на транспортное устройство. Для всасы-
всасывания кристаллов соли в трубу требуется большая скорость дви-
движения рапы, при которой кристаллы поддерживаются во взвешен-
взвешенном состоянии. Энергичное всасывание начинается при скорости
около 2 м/сек. Часовая производительность солесосов от 30 до
116 г соли. При незначительной толщине слоя рапы, а особенно
при эксплуатации сухих озер, для сбора новосадки пользуются
скреперами с лебедками. Экскаваторы применяются для сбора
выломанной соли при толщине пласта не меньше 0,5 м, когда
ковш экскаватора не захватывает вместе с солью подстилающий
слой ила.
Бассейный способ переработки рассолов
Способ добычи соли из рапы соляных озер и лиманов с по-
помощью бассейнов, в которых происходит естественное испарение
или охлаждение рапы и кристаллизация (садка) солей, называют
бассейным. В качестве бассейнов могут быть использованы также
находящиеся близ озера естественные впадины. Грунт, в котором
находится бассейн, должен обладать очень малой фильтрующей
способностью, чтобы рассол не разбавлялся грунтовыми водами
и не просачивался в почву.
Для механизированного сбора соли с применением бульдозе-
бульдозеров, скреперов и других машин необходимо, чтобы слой соли имел
Получение солей из рассолов и морской воды
57
большую толщину, что при бассейном получении поваренной соли
практически невозможно осуществить, так как для интенсивного
испарения слой рапы в бассейне должен быть небольшим.
Высокий слой рапы в бассейне A—4 м и более) поддерживают
при зимней садке мирабилита. В этом случае рапу сульфатного
озера накачивают в садочные бассейны летом, когда концентра:
ция сульфата натрия наиболее высока. Осенью при похолодании
происходит садка мирабилита. Зимой маточный раствор сливают,
а остающийся толстый слой мирабилита @,3—1 м) собирают с
помощью механизмов и бугруют для более полного стока маточ-
маточного раствора.
Размеры бассейнов выбирают соответственно величине про-
промысла с учетом местных климатических, почвенных условий, со-
состава и концентрации рапы. При ручной ломке соли площадь,
отводимая под садку соли, разбивается на много небольших бас-
бассейнов A—4 га), имеющих форму прямоугольника. В бассейнах
больших размеров ветер развивает волны, обнажает слой соли,
что вызывает неравномерную садку. При механизированной ломке
соли перемычки между бассейнами мешают работе механизмов,
следовательно, садочные бассейны должны иметь большие раз-
размеры. Подготовительные (отстойные) и запасные бассейны вместе
занимают приблизительно такую же площадь, как и садочные,-
Перекачка рапы из бассейна в бассейн производится рапокачкой
(насосами) по желобам и канавам.
Важнейшим фактором, определяющим продуктивность бассей-
ного промысла, является испаряемость рапы29-30. Рапа испаряется
медленнее, чем вода, так как давление водяного пара над рассо-
. лом меньше, чем над водой. Отношение высоты слоя испарившейся
рапы к высоте слоя воды, испарившейся в тех же условиях, на-
называется коэффициентом испаряемости рапы. Величина коэффи-
коэффициента испаряемости, очевидно, всегда меньше единицы; она
зависит от состава и концентрации рапы, а также от климата и по-
погоды и может быть различной для одной и той же рапы, испа-
испаряющейся в разных условиях, например при разной влажности
воздуха. Коэффициент испаряемости может быть и отрицательной
величиной, что соответствует поглощению рапой влаги из воздуха.
Для крымских рассолов коэффициенты испаряемости имеют сле-
следующие средние значения:
(Вода)
1,0
1,00
1,075
0,90
1,116
0,73
1,210
0,50
1,291
0,24
1,320
0,08
Плотность рапы, г/см8 . . .
Ковффициент испаряемости
Испаряемость воды сильно меняется по месяцам года и колеб-
колеблется, например в районе Крымских промыслов, от 35 мм вод. ст.
в декабре до 300—330 мм вод. ст. в июле и августе.
58
Гл. П. Растворимые соли и методы их добычи
Литература
59
В
V
W
Скорость испарения воды с поверхности водоема площадью
более 100 ж2 может быть рассчитана по эмпирической формуле
E=*B(H-h)
где ¦ Е— скорость испарения, кг/м2 • сутки;
В — коэффициент, кг/(м2 ¦ сутки • мм рт. ст.), зависящий от
скорости ветра (рис. 6);
(Я — h) — дефицит влажности, т. е. разность между давлением
насыщенного пара при температуре поверхностного слоя
воды и давлением пара в воздухе на высоте 2 м над
водоемом, мм рт. ст.
Более точно расчет испарения воды с поверхности садочного
¦ соляного бассейна производится по тепловому балансу, учиты-
учитывающему солнечную радиацию, излу-
излучение атмосферы, тепловое излуче-
излучение водной поверхности, конвекцию
и др.31-32.
Испарение рапы и садка соли за-
замедляются выпадением атмосферных
осадков. Среднегодовое количество
осадков сильно колеблется для раз-
различных, даже сравнительно близких,
районов (например, в районе Гениче-
ска — 328,8 мм вод. ст., а в районе
Керчи — 439,1 мм вод. ст.).
Бассейны используются не только
для испарительной садки солей в
летнее время, но и для зимней садки.
При понижении температуры рапы
происходит вымораживание солей.
Так, при охлаждении концентрирован-
концентрированных растворов NaCl из них при тем-
температурах ниже +0,15° кристаллизу-
кристаллизуется дигидрат хлорида натрия NaCl •
•2Н2О, а при охлаждении сульфатных
растворов—мирабилит Na2SO4- I0H2O.
Соли, добываемые бассейным способом, более чисты, чем из-
извлекаемые из донных отложений озер, так как содержат значи-
значительно меньше ила и других механических примесей. Однако су-
существенным недостатком бассейного способа, так же как и добычи
самосадочной соли, является сезонность и зависимость от клима-
климатических явлений. О методах расчета бассейных промыслов см.33-34,
ЛИТЕРАТУРА
1. А. А. Иванов, Основы геологии и методика поисков, разведки и оценки
месторождений минеральных солей, Госгеолиздат, 1953. — 2. М. П. Фивег
Труды ВНИИГ, в. 23, 1952, стр. 3; в. 32, 1956, стр. 102. — S. Н. С. К У р на к о в,
0.1
Ш
г i б в
Скорость iempa,M/ce/(
10
Рнс. 6. Величина коэффи-
коэффициента В в зависимости от ско-
скорости ветра на высоте 4,5 м
над поверхностью озера.
В. Г. Кузнецов, А. И. Дзенс-Литовский, М. И. Равич, Соляные озера
Крыма, Изд. АН СССР, 1936. — 4. В. П. Ильинский, Труды ГИПХ, в. 40,
1948, стр. 5. — 5. Б. Л. Ронкин, Наши соляные озера и их богатства, ГОНТИ,
1931. — 6. J. Cook, Discovery, 17, № 2, 71 A956). —7. А. И. Дзеис-Литов-
ский, Труды ВНИИГ, в. 34, 1957, стр. 5. — 8. М. Г. Валяшко, Там же,
в. 23, 1952, стр. 10. — 9. М. Г. Валяшко, Там же, стр. 13. — 10. Н. С. Домб-
р о в с к а я, ИСФХА АН СССР, 22, 293 A953).
//. Экспериментальное исследование процессов метаморфизации природных
соляных вод. Труды ВНИИГ, в. 23, 1952, стр. 147. —12. О. Д. Кашкаров,
Там же, в. 32, 1956, стр. 3, 49. —13. М. Г. Валяшко, Там же, в. 23, 1952,
стр. 25. — 14. Я- И. Т ы ч и н о, М. Г. В а л я ш к о, Там же, в. 24, 1952, стр. 247. —
15. О. Д. Кашкаров, Бюлл. ВИГ, № 1—2, 13 A940). — 16. А. М. Пеньков,
А. А. В о п и л к и н, Расчет опорных целиков при добыче каменной соли, Киев,
Изд. АН УССР, 1950. — 17. С. Е. Шарапов, Н. Г. Шешунов, Хим. пром.,
№ 6, 183 A953). —18. Л. Д. Ш ев яков, Разработка месторождений полезных
ископаемых, Углетехиздат, 1953, гл. XVIII. —19. А. Н. Андреичев, Разработка
калийных и каменносоляных месторождений, Госхимиздат, ч. I, 1953; ч. II,
1954.—-20. А. Н. Андреичев, Пути механизации трудоемких работ в горно-
химической промышленности, Госхимиздат, 1953.
21. А. Е. Ходьков, Труды ВНИИГ, в. 28, 1953, стр. 3. — 22. А. И. Дзенс-
Литовский, А. А. Иванов Н. А. Корчебоков, Бюлл. ВИГ, № 6—7, 3
A939). — 23. И. А. Л е г е н ч е н к о, Хим. пром., № 8, 469 A956).— 24. П. А. К У л-
ле, Труды ВНИИГ, в. 30, 1955, стр. 3, 49.-25. П. А. Кулле, Там же, в. 20,
1°'49. —26. М. В. Сыроватко Сборник статей ВЗПИ, в. 9, 1955, стр. 28.—
27. П. С. Бобко, Труды ВНИИГ, в. 35, 1959, стр. 365, 509. — 28. П. С. Бобко,
Хкм. пром., № 2, 41 A952). — 29. О. Д. К а ш к а р о в, Труды ВНИИГ, в. 24, 1952,
стр. 229. —'80. Б. Г. Иванов, Испарение в естественных условиях, Гидромет-
издат, 1939.
31. О. Д. Кашкаров, сб. «Вопросы производства калийных удобрений»,
Изд. «Химия», 1964, стр. 79. — 32. О. Д. Кашкаров, сб. «Технология пере-
переработки природных солей и рассолов», Изд. «Химия», 1964, стр. 67, 77, —
33. W. Free Kevin, Trans. Inst. Chem. Eng., 36, № 2, 115 О&58).—
34. D. M. Myers, С W. Bony ton, J. Appl. Chem., № 4, 207 A958).
Физико-химические свойства
61
Глава III
ХЛОРИСТЫЙ НАТРИЙ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Хлористый натрий, получивший название поваренной соли, так
как с давних времен эта соль добывалась путем вываривания (вы-
(выпарки) ее из растворов, встречается в природе в виде растворов и
кристаллических отложений, главным образом каменной соли, ми-
минерала галита NaCl. Он кристаллизуется из чистых растворов
в виде правильных кубов; кристаллы, образующиеся в растворах,
содержащих примеси, могут иметь форму октаэдра или ромбиче-
ромбического додекаэдра. Плотность кристаллов при 20° 2,16 г/см3. Тем-
Температура плавления 800,8°. В плотных природных массах (в ка-
каменной соли) зерна галита обычно имеют неправильные огранения
и характеризуются мелкими размерами (от долей до нескольких
миллиметров).
В присутствии модифицирующих агентов хлористый натрий
может кристаллизоваться в форме трехмерных дендритов с объем-
объемным весом 0,7 г/см3. В таком виде он выделяется из концентриро-
концентрированных растворов хорошо растворимых в воде хлоридов или гидро-
гидроокисей, в которых растворяется лишь 1—2% NaCl, или при выпа-
выпаривании под вакуумом при медленном перемешивании рассола,
содержащего 0,05% ферроцианида щелочного металла1'2.
При температуре ниже + 0,15° соль кристаллизуется в виде
двухводного кристаллогидрата NaCl • 2Н2О (гидрогалит), выше
этой температуры — в виде безводного NaCl. Растворимость NaCl
мало изменяется с температурой. Насыщенный водный раствор
содержит (рис. 7):
NaCl, %
При —21,2° (эвтектическая
точка) 24,42
При 0° 26*28
» 20° 26,39
» 40° 26,68
NaCI, %
. При 60° 27,07
» 80° 27,55
» 100° 28,15
» 140° 29,63
» 180° 30,99
Насыщенный водный раствор под давлением 760 мм рт. ст. ки-
кипит при 108,7° (содержит 28,41% NaCl).
Чистый галит бесцветен и обладает стеклянным блеском; при-
примеси окрашивают его в различные цвета — от серого до темно-
серого (примеси глинистых веществ), в желтый, розоватый и крас-
красноватый цвет (окислы железа), в бурый (органические вещества)
и др. Галит, встречающийся одновре-
одновременно с калийными солями, имеет часто
голубую, синюю или фиолетовую окрас-
окраску, исчезающую при нагревании до 200°;
возможно, что она появляется вслед-
вследствие выделения металлического натрия
под влиянием радиоактивного излучения
калия3'4.
В зависимости от месторождения по-
поваренная соль может содержать в каче-
качестве примесей песок, породы полево-
полевошпатового и каолинитового характера,
растворимые соли щелочных и щелочно-
щелочноземельных металлов, включения маточ-
ног.о раствора и др.5. Отмечается корре-
корреляция между содержанием примесей и
размером частиц соли6. О содержании
микропримесей в поваренной соли раз-
разных месторождений см.7 Чистая соль об-
обладает весьма небольшой гигроскопич-
гигроскопичностью, увеличивающейся при наличии
некоторых примесей, например хлористо-
хлористого магния. Другие примеси, например
равномерно распределенный дисперсный
ил, несколько снижают гигроскопич-
гигроскопичность поваренной соли8.
Давление водяного пара над раствором хлористого натрия
с возрастанием его концентрации при низких температурах умень-
уменьшается незначительно (например, при 20° для 5%-ного раствора —¦
П мм рт. ст., а для насыщенного—14 мм рт. ст.), а при
высоких температурах заметно снижается (при 100° для 5%-ного
раствора — 736 мм рт. ст., а для насыщенного — 566 мм
рт. ст.)9.
Дигидрат хлористого натрия кристаллизуется в интервале от
+ 0,15 до —21,2°. При медленной кристаллизации образуются тон-
тонкие иглы, а при быстрой — хорошо ограненные и развитые моно-
клиннотпризматические кристаллы плотностью 1,6 г/см3. Объемный
вес 0,8—I т/м3. В интервале от +0,15 до —21,2° давление водяного
пара над NaCl • 2Н2О изменяется от 3,48 до 0,69 мм рт. ст., а гиг-
гигроскопическая точка — от 75,2 до 100% 1О'И.
140
120
100
60
40
20
-го\
I NaCl
аС12НгС
10
20 30
NaCl,%
40
Рис. 7. Растворимость в си-
системе NaCt-H2O.
62
Гл. III. Хлористый натрий
ПРИМЕНЕНИЕ
Поваренная соль играла большую роль в нсторин развития че-
человечества. Применение ее как приправы к пище известно с древ-
древнейших времен. В Тибете и Монголии глыбы соли использовались
в качестве денег. В древней Руси солеварение являлось широко
распространенным промыслом.
Поваренная соль с давних пор применяется также в качестве
консервирующего средства, предохраняющего от гниения, — для
засолки рыбы, мяса, овощей, кож н др. С развитием промышлен-
промышленности соль находит широкое применение в различных, производ-
производствах в качестве исходного или вспомогательного сырья12. В на-
настоящее время для пищевых целей расходуется меньше половины
мировой добычи соли. Средняя годовая норма пищевого потреб-
потребления соли на одного человека составляет 8—10 кг, а общее по-
потребление, включая промышленное, достигает в отдельных странах
25—75 кг в год. Соль идет также в пищу животных. Ее, в част-
частности, используют при заготовке силоса для скота. В качестве
корма расходуется 5—10% добываемой соли.
По способу добычи и обработки пищевая поваренная соль де-
делится на: 1) мелкокристаллическую — выварочную; 2) молотую,
разных видов (каменная, самосадочная) и различной крупности
помола; 3) немолотую — комовую (глыба), дробленку и зерновую
(ядро); 4) иодированную. Требования к качеству пищевой соли
обусловлены ГОСТ 13830—68 (табл. 6); методы испытаний см.
ГОСТ 13685—68.
ТАБЛИЦА 6
Состав пищевой соли (в %) по ГОСТ 13830—68
Применение
63
Сорт
Экстра . .
Высший . .
Первый . .
Второй . .
Содержа-
Содержание NaCl
в сухом
веществе,
не менее
99,7
98,4
97,7
97,0
Содержа-
Содержание не ра-
растворимых
в воде
веществ
в сухом
веществе,
не более
0,03
0,16
0,45
0,85
Максимальное
содержание влаги
0,1
Каменная соль 0,25
Садочная и само-
самосадочная . . 3,2
Выварочная . . 5,0
Каменная . . . 0,25
Садочная и само-
самосадочная . . 4,0
Выварочная . . 5,0
Каменная . . . 0,25
Садочная и само-
самосадочная . . 5,0
Выварочная . . 6,0
Предельное содержание
в сухом веществе
Са2+
0,02
0,35
0.5
0,65
0,01
0,05
0,1
0,25
soj~
0,16
0,8
1,2
1,5
Fe*°3
0,005
0,005
0,01
«,01
Поваренная соль не должна иметь запаха (для иодированной
соли допускается слабый запах иода). Цвет соли сорта экстра бе-
белый; для прочих сортов допускаются оттенки сероватый, желто-
желтоватый и др. Максимальное содержание Na^SO4 в пересчете на су-
сухое вещество для сорта экстра 0,2%, для других сортов 0,5%.
В соли Артемовского месторождения допускается наличие тем-
темных частиц в пределах норм иона SOij", а для Солотвинской соли
в пределах норм нерастворимого остатка. В соли «глыба» Нахиче-
ванского месторождения допускается содержание NaCl (в пере-
пересчете на сухое вещество) не менее 93% и не растворимого в воде
остатка не более 5%- В выварочной соли заводов западных обла-
областей УССР и калийных комбинатов допускается содержание ка-
калия до 0,42% (в пересчете на сухое вещество). Соль сорта экстра
должна полностью просеиваться через сито со стороной квадрат-
квадратного отверстия 0,8 мм и не менее чем на 95% через сито — 0,5 мм.
Соль молотая делится на следующие номера помолов в зави-
зависимости от просева через сита разных размеров:
Размер Количество
стороны соли,
квадратного проходящей
отверстия через сито
сита, (в %),
мм не менее
Для высшего и первого
сортов помол:
№ 0 0,8 90
№ 1 1,2 90
№ 2 2,5 90
№ 3 4,5 85
Для второго сорта помол:
№ 1 1,2 90
№ 2 2,5 90
№ 3 4.5 85
Соль «экстра» должна полностью проходить через сито со сто-
стороной квадратного отверстия 0,8 мм и на 95% через сито со сто-
стороной 0,5 мм.
Соль «дробленка» или «зерновая» выпускается с величиной
зерна до 40 мм. Глыбовая соль имеет куски весом от 3 до 50 кг.
Содержание иодида калия в иодированной соли всех сортов
25 г на 1 г соли или 0,00191% свободного иода; для стабилиза-
стабилизации KI в иодированную соль (кроме выварочной «экстра») до-
добавляют тиосульфат натрия — 250 г на 1 г соли. Отклонение этих
норм допускается до ±20%- Предельное содержание влаги в иоди-
иодированной соли 0,5%.
Техническая соль должна содержать не менее 93% NaCl. Допу-
Допустимое содержание примесей устанавливается в зависимости ог
назначения соли.
В химической промышленности NaCl является основным
сырьем для получения соды, хлора, едкого натра, хлорной извести
64
Гл. III. Хлористый натрии
Каменная соль
65
к пр.; кроме того, применяется в производстве пластмасс, в орга-
органическом синтезе и пр. Хлористый натрий используют более чем
в 1500 производствах5-13.
Хлористый натрий, имеющий дендритную структуру и объем-
объемный вес ~ 0,8 г/см3 при избыточном давлении 1,7 ат, может быть
применен в качестве пламягасящей добавки при изготовлении
безопасных аммонитов малой плотности @,9 г/см3 при избыточном
¦давлении 1,7 ат)н.
В СССР наибольшее количество поваренной соли получают,
добывая каменную и самосадочную соль. Выварочной и бассейной
соли производят незначительное количество — менее 5%- Основ-
Основным потребителем поваренной соли в США является химическая
промышленность (~70%); пищевая промышленность расходует
6—6,5% соли; для кормления скота и других сельскохозяйствен- |
ных нужд ~ 5%15~17.
•, Мировая добыча и производство поваренной соли составляли
в 1961 г. 89 млн. т, в том числе в США 23,4 млн. т.
СЫРЬЕ И МЕТОДЫ ПРОИЗВОДСТВА
Основными типами месторождений поваренной соли являются:
1) пласты и штоки каменной соли; 2) океанская и морская вода,
озерные рассолы; 3) растворы в соляных источниках и грунтовых
водах; 4) солончаки; 5) возгоны соли на кратерах вулканов и в
трещинах лавовых потоков.
Главнейшие месторождения каменной соли в Европейской ча-
части СССР расположены в отложениях бывшего Пермского моря 18.
Каменносоляные месторождения Восточной Сибири относятся
к Кембрийским отложениям. Туркменские соляные месторождения
приурочены к пограничной зоне между меловой и юрской геоло-
геологическими системами19. Соленосные отложения Северного Края \
(Котлас, Сольвычегодск и др.) связаны с различными подразделе-
подразделениями палеозоя, от девона до верхней перми 20.
Крупнейшими месторождениями каменной соли в СССР яв-
являются: Артемовско-Славянское в Украинской ССР, мощность
пластов которого превышает 200 м; Илецкое в Оренбургской обла-
области, где пласт соли разведан на глубину 1,5 км; Усольское в Ир-
Иркутской области 21 и многие другие ^ 23.
В СССР весьма распространены соляные озера, многие из ко-
которых служат источниками добычи поваренной соли ^~39. В При-
Прикаспийской низменности насчитывается до 2000 озер, из них более
200 крупных, в том числе таких, как Баскунчак и Эльтон. Котло-
Котловина оз. Баскунчак имеет овальную форму площадью около
115 км2. Запасы соли в озере за счет вод впадающих в него источ-
никпв возрастают ежегодно более чем на 800 тыс. т или на ~ 100 т
в час?6. Оз. Эльтон занимает площадь 180 км2 и является вели-
величайшим из соленых озер Европы37. В Казахской ССР количество
известных соляных озер превосходит 2500. Наибольшее количество
поваренной соли находится в морской воде (стр. 47).
Методы производства (добычи) соли разделяются на четыре
основные группы: 1) добыча каменной соли; 2) добыча самосадоч-
самосадочной соли из соляных озер; 3) добыча садочной поваренной соли
бассейным способом из морской и озерной вод; 4) получение вы-
выварочной соли путем выварки ее из естественных и искусственных
рассолов.
Для технических целей применяют каменную и самосадочную,
а для пищевых — выварочную, самосадочную и садочную соль.
Кроме того, производят специальные сорта соли — иодированную,
брикетированную и неслеживающуюся, а также соль высокой чи-
чистоты (99,9% и больше NaCl).
КАМЕННАЯ СОЛЬ
Каменная соль встречается преимущественно в виде мощных
пластов или крупных штоков и линз, толщина которых достигает
иногда сотен и тысяч метров 40~4S. Большей частью она имеет плот-
плотное компактное строение и сопровождается залежами и мощными
толщами ангидритовых, гипсовых и других пород. Поэтому ка-
каменная соль зачастую содержит различные примеси — ангидрит,
глину, а также воду, битумы, газы (табл. 7).
ТАБЛИЦА 7
Состав некоторых образцов каменной соли
Образец
Илецкая крупная . .
» - глыба . . .
Брянцевская серая .
» белая . ,
Артемовская ....
NaCl
98,94
96,93
94,42
99,02
97,7-99,6
CaSO4
0,81
0,85
0,29
0,42
1,3-0
MgCI2
0,05
0,07
0,35-0
CaClj
0,2 •
0,33
0,15-0
Нераство-
Нерастворимый
остаток
0,24
0,39
0,1 в
0,14
0,65-0
Влага
1,10
Ml
0,10
0,22
0,1-0,4
По морфологическому признаку различают следующие типы
залежей каменной соли: пластовые (например, Артемовско-Сла-
вянское месторождение), пластово-линзообразные, линзообразные
(например, в Ишимбаевском районе Башкирии), куполо- и што-
кообразиые (например, Илецкое) и гнездообразные — в виде мел-
мелких линз и блоков.
Подземную добычу каменной соли обычно ведут на глубине до
500 м, так как при больших глубинах стоимость работ значительно
увеличивается. Вследствие пластичности каменной соли44 разра-
разработку осуществляют в камерах до 30 м высотой без креплений;
3 М. Е. Позин
66
Гл. III. Хлористый натрий
длина камер достигает 500 м, ширина 20 м, при ширине целиков
8—12 м. О методах подземной разработки пластов каменной соли
см.41-50. при подземных разработках имеет большое значение учет
режима вод, природных рассолов и газов соляного месторождения.
Предложено обогащать каменную соль в электростатическом
сепараторе после предварительного опрыскивания ее раствором
смеси жирных кислот с шеллаком C:1) или раствором салици-
салициловой и фталевой кислот A:1) в органическом растворителе, ко-
который испаряется до ввода соли в сепаратор "
51
САМОСАДОЧНАЯ СОЛЬ
<
Промысловая добыча самосадочной соли составляет в СССР]
около половины добываемой соли. i
Состав рассолов соляных озер весьма различен и зависит от]
геологических, климатических, почвенных условий, а также от вре-j
мени года. Кроме того, рассол представляет собой сложный ком-]
плекс в микробиохимическом отношении52. В качестве примера.''
ь табл. 8 приведен минеральный состав рапы некоторых крымских
озер.
ТАБЛИЦА 8 i
Содержание солей в рапе некоторых крымских озер (в %)
Озеро
Тоническое (весной) .• . .
» (в сентябре). . .
Плотность
рапы,
гкм*
1,154
1,237
1,084
1,077
NaCI
15,29
13,80
8,50
7,48
KCI
0,16
0,45
0,21
0,18
MgCI2
2,74
9,43
0,92
1,22
MgSO4
1,08
3,69
0,74
0,74
CaSO4
0,20
0,33
0,39
Сумма
солей
19,52
?7.41
10,74
10,30
Поваренная соль добывается из бессульфатных озер хлорид-
ного типа, а также из сульфатных озер (стр. 49M3~56.
Кристаллизация NaCI из рапы озер хлоркальциевого типа оп-
определяется равновесием в системе CaClj—MgCl2—NaCI—Н2О 57,
политерма которой до настоящего времени изучена еще не пол-
полностью. Поле кристаллизации NaCI на изотермических диаграм-
диаграммах значительно больше полей кристаллизации других солей. На-
Например, при 25° кристаллизуется только NaCI даже из растворов,
содержащих всего 0,6 вес. % NaCI при концентрации СаС12 и MgCIa
35,6 вес.%. Концентрации (в вес.%) СаС12 и MgCl2 четверной си-
системы в насыщенных растворах NaCI могут быть определены по
данным растворимости NaCI в хорошо изученных тройных систе-
системах СаС12—NaCI—Н2О и MgCl2—NaCI—Н2О по следующим фор-
формулам:
ill ьс. „
I-1,165/ * ' с«ео|2 - k +1.165/
-IQ
Самосадочная соль
67
где С —число молей обеих солей в 100 г раствора, определяемое
по тройным системам при данной растворимости NaCI;
k и 1~ весовые количества (в %) CaClg и MgClj (по раствори-
растворимости в тройных системах при данной растворимости
NadM79.
При испарении рапы морского типа из хлорид-сульфатных озер
вначале выделяются карбонаты кальция и магния, затем гипс,
а потом поваренная соль.
Из самосадочных озер добывают как новосадку, так и старо-
садку, которая по своим качествам превосходит новосадку. Иногда
под старосадкой залегает так называемая гранатка, представляю-
представляющая собой слой рыхлой, рассыпчатой поваренной соли в виде не-
несцементированных крупных кристаллов, загрязненных илом. После
отмывки ила рапой получается соль с высокими пищевыми каче-
качествами (табл. 9J9. Гранатка обычно образуется в озерах, где со-
соляная масса целиком пропитана рапой (корневая соль отсутствует)
и является одним из основных объектов разработки. Некоторые
соляные озера, например Баскунчак, очень мелководны. В жар-
жаркое время года почти вся вода из озера испаряется, что облегчает
добычу соли60. Вывозку соли из озера производят в открытых
вагонах. Рельсовый путь укладывают непосредственно по пласту
соли.
ТАБЛИЦА 9
Состав некоторых образцов самосадочной и бассейной поваренной соли67
Соль
Крымская бассейная
Туркменская само-
самосадочная (оз. Куу-
ли)
Васкуичакская само-
самосадочная
Павлодарская само-
самосадочная
NaCI
98,16-99,17
93,7-98,3
97 7>-99,32|0-0,24]
97,91-99,22
СаС12
0,05-0.4
MgCb
CaSO4
0,3-0,95
0-1,14H,24-1,27| 0-0,!
0,48 I 0-0,85| 0-0,:
0,01-0,44|0,01 -0,43|0,03-0,24|
MgSO4
0-0,3
0,05-0,14
0,530,
Нераство-
Нерастворимый
остаток
1,19-0,5
0,2б!0,07-0.6
0,6-0,7
Добытая соль, например баскунчакская, содержит 0,5—0,6%
илистых примесей, которые придают ей грязно-желтый цвет. Про-
Промывкой дробленой соли рапой можно получить столовую (освет-
(осветленную) соль с содержанием всего 0,04—0,05% примесей. Строи-
Строительство заводов по осветлению соли более рентабельно, чем по-
постройка вакуум-выпарных заводов: капиталовложения снижаются
в 2 раза, расход топлива в 23 раза, затраты рабочей силы
в 3 разаи. Для осветления соль измельчают до размера зерен
0,8 мм и подвергают противоточной промывке в логуошере чистым
68
Гл. III. Хлористый натгрцй
Получение садочной соли бассейным способом
69
насыщенным раствором. Промытую соль с 10—12% влаги центри-''
фугируют, после чего ее влажность снижается до 4,5—б%. Затем
ее высушивают до влажности 0,2% и классифицируют не грохоте
по размерам, предусмотренным ГОСТом.
Промывку поваренной соли можно производить также в буг-
буграх, складываемых на берегу озер 62, но при этом теряются зна-
значительные количества ее63. Потери крупнокристаллической соли
в буграх при стекании с них в грунт насыщенного раствора Nad
могут быть рассчитаны по уравнениюв4:
П1 KSH
где р — потери в % от веса бугра;
/С — растворимость NaCl;
" S — площадь основания бугра;
Н — годовое количество атмосферных осадков, мм\
Y — насыпной вес соли;
V — объем бугра.
Потери мелкокристаллической соли, удерживающей влагу в ре-
результате действия капиллярных сил, зависят от влагоемкости
соли ц и могут быть вычислены по формуле в5:
где H'S' — количество испарившейся влаги;
г) — 5D—N), здесь N — номер помола соли (стр. 63).
Максимальная влагоемкость неслежавшейся соли, равная
сумме сорбционной и капиллярной влагоемкости, зависит от гра-
гранулометрического состава и от объемного веса соли в буграх, рав-
равного для неслежавшейся садочной соли 1,18—1,28 т/м3 и 1,2—
1,3 т/м3 для каменной соли (в зависимости от помола). Объемный
вес слежавшейся соли на 5—10% больше64'6в.
Содержание некоторых примесей в соли можно уменьшить пу-
путем ее измельчения и фракционирования. Например, установ-
установлено 68, что при измельчении природной поваренной соли, содер-
содержащей до 7% сульфата натрия, последний переходит главным об-
раэом в мелкие фракции, с которыми и может быть удален (до
90% от общего количества Na2SO4).
ПОЛУЧЕНИЕ САДОЧНОЙ СОЛИ БАССЕЙНЫМ СПОСОБОМ
Бассейный способ (стр. 56) получения садочной соли приме-
применяют в тех случаях, когда по гидрохимическим и другим условиям
не происходит самопроизвольной садки соли из рассола. Получе-
Получение соли бассейным способом дороже, чем добыча самосадочной
соли, так как -связано с необходимостью сооружения искусствен-
ных или приспособления естеетвенных бассейнов; кроме того, при
бассейном способе затруднена механизация процесса, и потому он
утрачивает свое значение.
Для добычи поваренной соли бассейным способом ет' 69~Т1 из не-
ненасыщенной рапы континентальных озер, из воды лиманов и при-
приморских водоемов (озер) обычно сооружают бассейны трех типов:
1) подготовительные или гипсоотстойники, 2) запасные и 3) садоч-
садочные. Вначале рапу из озера накачивают в подготовительные бас-
бассейны, где ее держат небольшим слоем 25—40 см и где она, испа-
испаряясь в жаркое время года (апрель — сентябрь), сгущается почти
до насыщения и отстаивается от механических примесей, а также
от кристаллизующихся солей: гипса и карбонатов кальция и же-
железа. Затем рапу перекачивают в запасные бассейны, где хранят
до следующей весны возможно более высоким слоем F0—60 см)
для уменьшения разбавления атмосферными осадками. (При по-
получении поваренной соли из сульфатных озер рапу перекачивают
в запасные бассейны в холодное зремя года после садки мираби-
мирабилита 72.) В конце мая или начале июня следующего года рапу из
запасных бассейнов перекачивают в садочные бассейны, где вскоре
начинается садка соли, продолжающаяся до сезона ломки (конец
августа — начало сентября). В садочных бассейнах поддерживают
толщину слоя рапы 10—16 см, а к концу сезона садки 20—25 см.
По мере испарения садочные бассейны пополняют рапой ив запас-
запасных бассейнов. К концу сезона садки пласт соли, осевшей на дно
садочного бассейна, достигает 5—6 см. Ломку (сбор) соли произ-
производят при этом ручным способом — простыми лопатами или чал-
пами (дырчатыми лопатами) — после откачки рассола обратно
в.озеро. Выломанную соль собирают в кучи на дне бассейна, а за-
затем вывозят на берег и складывают в бугры трапецеидальной
формы высотой 3—4 м для стекания маточного рассола. Сложен-
Сложенная в бугры соль содержит 8—10% рассола, в котором находятся
магнезиальные и другие примеси, придающие свежедобытой соли
горьковатый привкус. При длительном вылеживании эти примеси,
особенно соли магния и хлорид кальция, стекают с рапой, благо-
благодаря тому, что они сильно гигроскопичны и увлажняются влагой
воздуха.
Некоторые рассолы в бассейнах покрываются сверху коркой
кальциевой соли (карбоната), что замедляет процесс испарения и
кристаллизацию соли. Рассол, предварительно очищенный от
Са(НСО3)а пропусканием его через анионит и катионит, кристал-
кристаллизуется значительно быстрее неочищенного73. Для интенсифика-
интенсификации испарения рапы предложено74 устраивать в бассейнах брыз-
гальные установки, например, форсуночного типа75.
Хранить рассолы можно в больших ямах, облицованных тон-
тонкими D0 мк) политеновыми листами, — это предохраняет от раз-
разбавления атмосферными осадками и грунтовыми водами7в. ¦
70
Гл. III. Хлористый натрий
Производство выварочной соли
71
Бассейная соль имеет размеры зерен 10—15 мм. В таком виде
ее иногда отгружают. В большинстве случаев соль из бугров из-
измельчают на солемельницах — вальцовых, дезинтеграторах или
жерновых. Поваренная соль, полученная бассейным способом, со-
содержит 98—99% NaCl и примеси, зависящие от состава рапы
(табл. 9). Для получения более чистой соли ее промывают иногда
на центрифугах морской водой или неконцентрированной рапой
или предварительно очищают рапу химическими способами77.
В тех случаях, когда соль получается из сильно загрязненных рас-
. солов и содержит много примесей, ее иногда подвергают перекри-
перекристаллизации 78.
Для получения биологически полноценной поваренной соли, со-
содержащей минеральные вещества морской воды, включая микро-
микроэлементы, предложено маточный раствор после кристаллизации
NaCl испарять досуха в специальных бассейнах и образующийся
твердый остаток смешивать с NaCl. Уменьшение гигроскопичности
продукта достигается добавкой фосфата натрия Na8PO4 или кар-
карбоната натрия79.
ПРОИЗВОДСТВО ВЫВАРОЧНОЙ СОЛИ
Выварочная соль получается в результате выпаривания искус-
искусственных или естественных рассолов, добываемых из недр земли.
Такие рассолы отличаются сравнительно высокой концентрацией
NaCl и малым содержанием примесей. Для получения выварочной
соли непригодны рассолы любых поверхностных озер вследствие
высокого содержания в них кальциевых солей и других примесей.
Растворимость CaSO4 в растворах поваренной соли больше, чем
в воде. Максимум растворимости CaSO4 и СаСО3 в растворах
NaCl соответствует концентрации приблизительно 2 моль NaCl
в 1000 г воды80-81. Обычно рассол содержит (в г на 1 л):
NaCl 280-310
CaSO4 ....... 5-6
MgCl2 н MgSO4 . . 0,2-4
СаС12 0,2-0,8
Плотность рассола при 15° равна 1,19—1,20 г/см3. Высокое со-
содержание MgCl:2 в рапе не препятствует выварке из нее поварен-
поваренной соли, так как последующая промывка соли позволяет снизить
концентрацию MgCb в межкристальной жидкости и получить соль
высокого качества (стр. 113).
При выпарке рассолов морского типа (являющихся концентра-
концентратами морской воды) при температуре кипения под атмосферным
давлением после достижения насыщения кристаллизуется NaCl.
На рис. 8 показана равновесная диаграмма растворимости при
100° в водной взаимной с*истеме
2NaGl + MgSO4 - Na2SO4 + MgCl2
MgSQ,
состоящей из главных компонентов морской воды. Фигуративная
точка солевой массы жидкой фазы по мере кристаллизации NaCl
движется из начального положения /. В стабильной области кри-
кристаллизации выделяется около 70°/о NaCl, когда точка состава
жидкой фазы достигает границы полей кристаллизации NaCl и
левеита Na2SO4 • MgSO4 • 2,5Н2О в точке 2. Однако при дальней-
дальнейшем выпаривании вместо смеси галита и левеита продолжает кри-
кристаллизоваться один галит в метастабильной области (подобно
тому, как это происходит и при
солнечном испарении рассолов, ко-
когда NaCl кристаллизуется в мета-
метастабильной области без астрахани-
та Na2SO4-MgSO4-4H2O — см.
рис. 83 на стр. 272). Примерный
ход кристаллизации показан пунк-
пунктирной линией. Задержка выделе-
выделения сульфатов вследствие достаточ-
достаточно большой стойкости метастабиль-
иого состояния повышает общую
степень извлечения NaCl при кипе-
• нии раствора до 91%. При выпари-
выпаривании же обессульфаченного кон-
концентрата морской воды можно вы-
выкристаллизовать до 96% поварен-
поваренной соли82""85. Стабильные фазы
выделяются лишь при добавке боль-
большого количества затравки.
Выпаривание рассолов в заводских
30
10
2NaCl Ю 30 SO
90 MgCl2
Рис. 8. Изотерма растворимости
системы NaCl—MgSO4—Н2О при
100°.
DbindjjMDannc ра^^ши ~ „- условиях осуществляют
"либо в чренах, обогреваемых топочными газами, либо в вакуум-
выпарных аппаратах, обогреваемых паром. На чренных установ-
установках очистку рассола от примесей производят в процессе его упа-
упаривания. Соль получается в виде более крупных кристаллов, чем
при вакуумной выпарке. Для выварки соли в вакуум-выпарных
аппаратах в ряде случаев необходима предварительная очистка
рассола от кальциевых и магниевых солей.
Температуры кипения рассолов морского типа различного со-
состава могут быть определены расчетным путем. О методе расчета
см.
75
Получение соли в чренах
Это старый метод, который сохранился и до настоящего вре-
времени. Имеются чренные солеварни (варницы), действующие
с XVJ в. (солеварни в районе Соликамска и др.N9. В США чрен-
ную выварку соли осуществляют, например, на заводе в Манисти
(штат Мичиган) производительностью более 1000 т/сутки8*.я. рас-
сол, подогретый с 10—15е до 60—70°, поступает в выпарной чрен,
72
Гл. III. Хлористый натрий
175
<50
? 75
1
50
О
представляющий собой открытый прямоугольный резервуар (ско-
(сковороду), изготовленный из котельной стали толщиной 6—8 мм.
Размеры его: длина 15—20 м, ширина 8—10 м, глубина 0,4—0,5 м.
В процессе выпарки в чрене поддерживают постоянный уро-
уровень рассола 18—20 см. При нагревании рассола в чрене до 80° из
него выделяются сероводород и другие растворенные газы, а также
выпадает сульфат кальция (рис. 9). По достижении температуры
кипения A08°) происходит разложение бикарбоната кальция и
образующийся СаСО3 выделяется в
осадок; продолжается выпадение твер-
твердого CaSO4. Твердые примеси уда-
удаляются специальными гребками через
борт чрена. По достижении насыще-
насыщения (через 6—8 ч) начинает кристал-
кристаллизоваться NaCl. Магнезиальные соли
остаются в растворе, попадают в го-
готовую поваренную соль с маточным
раствором, понижая ее качество. Для
получения мелкокристаллической соли
температуру рассола в процессе кри-
кристаллизации поддерживают в преде-
пределах 90—100°. Для получения крупно-
крупнокристаллической соли температуру по-
понижают E0—60°) и выгребают соль
1—2 раза в сутки.
Соль, кристаллизующаяся в про-
процессе выпарки, механизированными
гребками выгребается через наклонный борт чрена и отжи-
отжимается на центрифугах (до влажности 3—5%) или высушивается
с сушилках.
Температуру в топке под чреном поддерживают на уровне
1000—1200°; температура газов, уходящих из последнего газохода,
350—400°. При содержании в рассоле 24—25% NaCl расходуется
0,45—0,5 т условного топлива на 1 т готовой соли; при понижении
концентрации рассола до 15—16% NaCl расход топлива возрас-
возрастает до 1,1—1,2 т/т. Среднесуточный съем соли с 1 м2 поверхности
нагрева чрена составляет 80—100 кг при исходной концентрации
рассола 300 г/л NaCl; при этом интенсивность испарения воды со-
составляет 11—12 кг в 1 ч на 1 мг поверхности нагрева чрена.
При интенсивном выпаривании раствора в чренах получается
соль с размерами зерен 0,1—0,2 мм. При снижении температуры
до 60° (для получения крупнозернистой соли) производительность
чренов уменьшается почти в 10 раз по сравнению с производитель-
производительностью при интенсивном кипении. Однако более важным считают
не интенсивность выпарки, а получение крупнозернистой соли, по-
поэтому до сих пор пользуются чренным способом вываривания
12 3 4 5
Моль CaSOi, на 1000моль HP
Рис. 9. Растворимость CaSO4
в насыщенном растворе NaCl.
Производство выварочной соли
73
соли, несмотря на его примитивность. Крупнозернистую соль
можно получить и при высокой температуре выпаривания рассола
(90—96е), для этого необходимо добавить к нему поверхностно-
активное вещество — мыла, жиры, спирты и др.88-89 @,0002% ог
веса получаемой соли). В качестве добавок предложены также
такие, как бромистый цетилпиридин90 или 0,002% Мп в виде
M11SO4 и 0,001% смеси сексвиолеата сорбитана и монолаурата
мешалка; ;;—солеииорннки; и, ю - «иуупштми, »- цпи^п
7 — транспортер; 19 — весы; 20 — ловушка; 21 — барометрический кон"
денсатор; 22 — ресивер; 23 — вакуум-насос.
полиоксиэтилен-сорбитана91. Более экономичным является брике-
брикетирование мелкокристаллической соли, полученной интенсивными
методами выварки, с последующим дроблением брикетов до зерен
требуемых размеров (стр. 91).
В зависимости от качества выпариваемых рассолов чрен оста-
останавливают на чистку через 7—12 дней. За это время в маточном
растворе накапливается много примесей, а полотно чрена покры-
покрывается накипью — плотной коркой солей (называемой в Сибири
чренным камнем или ширеем, а на Украине омокой) толщиной
7—10 см, производительность чрена сильно понижается (иногда
до 50%), и создается опасность его прогара, а расход топлива зна-
значительно возрастает.
Накипь состоит из смеси кристаллов NaCl (86—90%), неболь-
небольшого количества других растворимых солей и 5—8% нераствори-
нерастворимых осадков, главным образом сульфата кальция. Коэффициент
74
Гл. III. Хлористый натрий
Производство выварочной соли
75
теплопроводности соляной накипи равен 2—2,5 ккал/(м • ч • град),
т. е. в 25—30 раз меньше, чем стали. Очистку от накипи можно
производить механическими способами и размыванием ее струями
еоды 91.
На рис. 10 показана схема осуществляемой на Усольском за-
заводе выварки соли в круглых чренах с механизированным удале-
удалением соли 88. Выгрузка соли со дна чрена в солесборники произво-
производится при помощи скребков и проволочных щеток, укрепленных на
мешалке, вращающейся со скоростью 2—3 об/мин. Отфугованную
соль с 5—6% влаги направляют по транспортеру на склад или во
вращающуюся барабанную сушилку. Выпарной чрен изготовляют
из стальных листов толщиной 6—7 мм. Он имеет диаметр 10 ж и
высоту борта 0,5 м, сверху покрыт деревянным колпаком, снаб-
снабженным двумя вытяжными трубами высотой 10 м для отвода пара
и люками, служащими для наблюдения за работой мешалки и для
ремонта чрена. Благодаря непрерывному удалению солей со дна
чрена образование чренного камня происходит значительно мед-
медленнее, и длительность работы чрена между остановками для
чистки достигает 30 суток, т. е. в 3 раза больше, чем при выварке
соли в немеханизированных чренах.
Вакуумная выварочная соль
Вакуум-выпарка значительно рентабельнее, чем чренная, вслед-
вследствие многократного использования тепла греющего пара. Она
выгодно отличается и меньшей затратой рабочей силы. Обычно
для выварки поваренной соли применяют концентрированные рас-
рассолы и лишь в районах, очень удаленных от залежей соли, исполь-
используют для ее получения морскую воду. Вследствие того что раство-
растворимость NaCl очень мало зависит от температуры (рис. 7), соль
получают изотермической кристаллизацией при температуре кипе-
кипения насыщенного рассола, удаляя растворитель — воду. В резуль-
результате быстрого пересыщения раствора в процессе интенсивного ки-
кипения в вакуум-выпарных аппаратах образуется мелкозернистая
соль.
В процессе выпаривания рассола происходит осаждение соли
на теплопередающей поверхности — греющих трубках. Поэтому-
необходимо периодически A—2 раза в сутки) промывать аппа-
аппараты водой. Кроме того, образуется плотноструктурная накипь из
выделяющихся в осадок нерастворимых солей — сульфата и кар-
карбоната кальция и кремнекислых соединений. Пересыщение жидко-
жидкости сульфатом кальция достигается прежде всего на греющей по-
поверхности, которая и инкрустируется осадком сульфата кальция.
Сульфат кальция, кристаллизующийся в массе жидкости, образует
высокодисперсную взвесь, которая в дальнейшем также принимает
участие в образовании отложений на греющих поверхностях и
стенках аппаратов93. Возможно, что образование твердой накипи
связано также с процессами перекристаллизации сульфата каль-
кальция и перехода его при этом из одной модификации в другую. До-
Добавка к рассолу сульфитцеллюлозного щелока замедляет переход
CaSO4 • '/гНяО как в CaSO4 • 2Н2О, так и в CaSO4, что, по мнению
некоторых исследователей, должно способствовать уменьшению от-
отложения накипи64. Образование накипи вызывает необходимость
тщательной химической и механической чистки аппаратов раз
в 7—10 дней.
Для получения более чистого продукта, а главным образом для
предохранения аппаратов от накипи рассолы перед выпаркой сле-
следует очищать от ионов Са2+, Mg2+ и SOf~.
Очистка рассолов
Существуют разные способы очистки рассолов ss-B7- \) содо-
содовый, при котором в рассол добавляют соду, осаждающую Са2+ и
частично Mg2+; 2) известково-содовый, при котором в рассол од-
одновременно вводят соду и известь (известковое молоко); при этом
рассол освобождается достаточно полно и от кальциевых и от
магниевых солей; 3) известково-сульфатно-содовый, осуществляе-
осуществляемый в две фазы. В первой фазе в рассол вводят сульфат натрия
и известь, причем происходит освобождение рассола от раствори-
растворимых солей магния и кальция. Во второй фазе рассол освобо-
освобождается от гипса путем карбонизации его двуокисью углерода
(дымовым газом) или введения в него соды.
При очистке рассола этим способом идут следующие реакции!
Первая фава
Вторая фаза
Na2SO< + Са(ОНJ
MgCl2 + 2NaOH
M°So 2NOH
CaCl2 + 2Na0H
2NaOH + CO2
CaSO4 + Na2CO3
CaSO4 + 2NaOH
Mg(OH2) + 2NaCl
Mg(OHJ + Na2SO4
Ca(OHJ + 2NaCl
Na2CO3 + H2O
СаСОз + Na2SO4
После очистки рассолы отстаиваются от осадков и поступают
на выпарку. Содовый способ очистки наиболее прост, но пригоден
для рассолов, содержащих мало Mg2+, так как Mg2+ плохо оса-
осаждается содой. Очистка этим способом протекает наиболее удов-
удовлетворительно при температуре около 100е. Преимущество этого
способа — в быстроте отстаивания рассола от осадка.
Если часть соды заменить едкой щелочью, содовая очистка
даст хорошие результаты и без нагревания раосола — при пере-
перемешивании в течение 30—60 мин. Концентрация катионов Cas+ и
Mg2+ в растворе NaCl может быть снижена до 80—40 ммоль/л9В.
76
Гл. 111. Хлористый натрий
Производство выварочной соли
77
О растворимости СаСО3 и Mg(OHJ и пересыщении ими рассола
см.
99
При наличии значительных количеств Mg2+ очистку рассола
производят иэвестково-содовым или известково-сульфатно-содо-
вым способом. В обоих случаях очистку можно производить без
подогрева рассола (при 15—25е). Однако повышение температуры
рассола до 40—50° при известково-содовой очистке приводит к бо-
более быстрому отстаиванию осадков — время отстаивания сокра-
сокращается на ~25%.
Рекомендуют вводить вместе с осаждающими реагентами
0,5—2% от их веса поверхностно-активных веществ (соли алкил-
аминов с 10—18 атомами С) 10°.
Предложено много вариантов содовой и известково-содовой
очистки рассола 1Ш, а также другие методы предотвращения оса-
осаждения CaSO4 в процессе выпарки, например добавка в рассол
СаС12 в количестве, соответствующем молярному отношению
Са : SO4 > 2,4 : 1 102-108. При этом вследствие положительного зна-
значения температурного коэффициента растворимости CaSO4 в при-
присутствии СаС12 осаждение CaSO4 при выпаривании рассола не
происходит. Очистку насыщенного раствора NaCI от CaSO4 можно
осуществлять, выдерживая раствор над слоем твердой поваренной
соли при температуре выше точки перехода гипса в ангидрит
(95—10бв) '**. Для уменьшения растворимости CaSO4 можно вво-
вводить в раствор 2—6 г/л Na2SO410Б или MgSO4106. В последнем
случае Mg2+ выделяется из рассола в одной из стадий выпарива-
выпаривания в виде киверита MgSO4-7H2O, который возвращают в про-
процесс.
Наиболее совершенным, обеспечивающим хорошую очистку
при минимальном расходе соды (в 2—2,5 раза меньшем, чем по
известково-содовому способу), является известково-сульфатно-со-
довый способ, широко применяемый на современных предприя-
предприятиях 107> 108. Однако он требует наиболее продолжительного от-
отстаивания осадков. Продолжительность очистки этим способом со-
составляет 25—40 ч летом и 60—75 ч зимой В5. Ускорение отстаивания
осадков может быть достигнуто введением в рассол коагулянтов.
Представляет интерес термический способ очистки рассола от
CaSO4, основанный на уменьшении растворимости CaSO4 при на-
нагревании (рис. 9). Некоторые рассолы, например рассолы из
иркутских и славянских скважин, могут быть очищены от гипса
на 50% нагреванием их до 200° при 15 аг88'106. При отделении
CaSO^ за счет изменения его растворимости до кристаллизации
Nad и при применении обычных методов химической очистки от
клоридо§ кальция и магния, получается выварочная соль, содер-
содержащая 99,8% ftaCI11B. Об использовании осадков, получаемых
при ©чистке рассолов, см. ш
Конструкции аппаратов
При вакуум-выпарке рассолов NaCI для уменьшения отложе-
отложения накипи на греющих поверхностях применяют выпарные аппа-
аппараты ее, life—не с энергичной принудительной циркуляцией жидкости.
Циркуляция осуществляется или с помощью внутренней пропел-
пропеллерной мешалки (внутренний или встроенный насос) (рис. 11),
Упаренный I pacmSop
Рнс. 11. Выпарной аппарат
с внутренним циркуляцион-
циркуляционным насосом:
/ — насос; 2 — греющая камера;
А 4— направляющие перего-
перегородки.
Конденсат
Рис. 12. Выпарной аппарат с наружным!
циркуляционным насосом:
1 — испаритель; 2 — солеотделитель; 3 — фильтр для
соли; 4 — циркуляционный насос.
или с помощью наружного насоса (рис. 12). Скорость движения
жидкости в таких аппаратах устанавливают в пределах 1,5—
4 м/сек. При этом жидкость закипает в верхней части трубок. При
скорости меньше 1 м/сек уровень закипания смещается книзу и
вффективность от принудительной циркуляции резко падает—-
процесс идет почти так же, как и при выпаривании с естественной
78
Гл. III. Хлористый натрий
циркуляцией. Увеличение скорости циркуляции раствора больше
4 м/сек не приводит к дальнейшему повышению коэффициента
теплопередачи. Обычно рассолы, выделяющие накипь, выпаривают
при скорости циркуляции, не меньшей 2,5 м/сек.
При выпаривании морских рассолов в аппаратах с принуди-
принудительной циркуляцией интенсивность накипеобразования значитель-
значительно снижается при вынесении кипения раствора из греющих труб
в испарительную камеру, т. е. при использовании аппаратов с вы-
выносными кипятильниками 117.
Для получения при вакуум-выпарке более крупной кристалли-
кристаллической соли предложено осуществлять классификацию солевой
пульпы, выходящей из испарителя аппарата с принудительной
циркуляцией. Рассол с мелкими кристаллами, выходящий из верх-
верхней части классификатора, направляют обратно в испаритель; в
нижней части классификатора оседают крупные кристаллы 118> ш.
Другим путем является циркуляция рассола между вакуум-испа-
вакуум-испарителем, где достигается небольшое пересыщение (до 1,6 г/л), и
непрерывнодействующим кристаллизатором, работающим по прин-
принципу взвешенного слоя, где происходит снятие пересыщения и
выращивание кристаллической затравки до размера зерен
~2 лш120-121.
Для укрупнения кристаллов поваренной соли, получаемой при
выпаривании рассолов, предлагают также добавлять к растворам
карбоксиметилцеллюлозу и некоторые другие высокомолекуляр-
высокомолекулярные соединения 122>123.
В солеваренной промышленности нашли также применение кон-
конструкции выпарных аппаратов с механической очисткой теплопе-
редающей поверхности при помощи скребков или щеток. Приме-
Применение таких аппаратов целесообразно лишь для выпарки неочи-
неочищенных рассолов, содержащих значительные количества гипса.
Наряду с этими типами выпарных аппаратов для выпарки
рассола применяют и аппараты с естественной циркуляцией. Ши-
Широко распространен аппарат корзиночного типа с подвесной грею-
греющей камерой. Недостатком его, как и других аппаратов с есте-
естественной циркуляцией, являются большие габариты.
Хорошо оправдали себя в эксплуатации также вакуум-аппа-
вакуум-аппараты барометрического типа; в них питание рассолом и выгрузка
соли осуществляются так называемым открытым путем через сбор-
сборник соли, в который опущены барометрические трубы. Эти аппа-
аппараты имеют греющие камеры с горизонтальными трубками. Уро-
Уровень рассола в аппарате зависит от разрежения в нем. Поэтому
для заполнения необходимым количеством рассола, т. е. несколько
выше греющей камеры, такие аппараты в многокорпусной бата-
батарее располагаются на разной высоте, каскадом, что требует боль-
большего объема здания, чем при аппаратах других конструкций
4(рис. 13).
Производство выварочной соли
79
Очищенный рассол подогревается до 50—60° за счет тепла
конденсата из второго и третьего корпусов и поступает в общий
рассоло- и солееборник 4, откуда автоматически происходит пита-
питание вакуум-аппаратов 5—7. Кристаллизующаяся поваренная соль
поступает черве барометрическую трубу в общий сборник, откуда
транспортируется шнеком через приемник и солеизвлекатели в
центрифуги 11. При поверхности нагрева системы около 600 м2
и расходе пара A,2 ат) 4300—4400 кг/ч производительность трех-
корпусной батареи составляет 86—90 т/сутки. Для получения 1 г
Рис. 18. Схема трехкорпусной выпарной станции с аппаратами барометрич -
ского типа:
1, 15 — насосы; 9, 3 — теплообменники; 4 - солееборник; S—7 — вакуум-адпараты; 8, 13 — баро-
йетрические конденсаторы; 9 - водоотделитель, 10, 18-чакуум-иас<$еы; И — центрифуга;
14 — вакуум-регулятор; 16 — отвод конденсата.
^ухой соли расходуется 1,2 т пара, а 1 т греющего пара иснаряет
2,5 г воды. После каждых 3—4 суток работы аппараты останав-
останавливают на 1 сутки для очистки.
Для предотвращения образования накипи при выпаривении
рассолов морского типа и морской воды можно перегревать
жидкость под давлением, а затем испарять ее в испарителе при
пониженной температуре под вакуумом. Выделяющийся при испа-
испарении сульфат кальция остается суспендированным в жидкости
вместе с кристаллами соли. При концентрировании морской воды
без кристаллизации соли предложено загружать в испаритель гра-
гранулированный известняк, на котором осаждается сульфат каль-
кальция 124.
В течение длительного времени большие затруднения в произ-
производстве выварочной соли возникали иа-за коррозии аппаратуры.
80
Гл. III. Хлористый натрий
Производство выварочной
81
Даже рассолопроводы, работающие при обычной температуре, из-
изготовленные из простой стали, сильно разрушаются и требуют
частых ремонтов. Коррозия уменьшается при содержании в рас-
рассоле 0,05—0,1 г/л NaOH. Наиболее стойкими в насыщенном соля-
соляном рассоле являются такие металлы как ниобий, титан, а из
более доступных — хромоникелевые и хромоникелевомолибдено-
вые стали; из неметаллических материалов стойкими являются
текстолит (до 100°) и импрегнированный графит (до 170°) 88> 125.'2б
Современные солеваренные заводы оборудованы 4—5-корпус-
ными выпарными станциями с аппаратами из нержавеющей стали
с выносными греющими камерами и принудительной циркуляцией.
В них используют мятый пар от турбогенераторов и других ма-
машин. Для предотвращения образования накипи — инкрустации
CaSO4 — в рассол перед выпаркой вводят кристаллический CaSO4,
на поверхность которого выделяется сульфат кальция из рассола
в процессе выпарки. (Аналогично введение СаСО3 предотвращает
образование накипи из СаСО3.) Соль, отжатая на центрифугах,
имеет влажность 4—5%.
Для получения чистой соли, содержащей 99,5% NaCl, ее пос-
после фильтрации промывают чистым насыщенным рассолом. При
этом магний удаляется почти полностью, а кальций на 60—70%.
Содержание магния в промытой соли пропорционально его со-
содержанию в промывной жидкости 127. При получении поваренной
соли вакуум-выпаркой из рапы некоторых озер, например озера
Кучук, промывка соли позволяет получать высокие ее сорта даже
без предварительной очистки рассола128. После сушки во вра-
вращающихся барабанных сушилках горячим воздухом или дымо-
дымовыми газами влажность соли снижается до 0,2—0,4% или до сотых
долей процента uo-13°-lsl.
Перспективной является сушка поваренной соли в аппаратах
со взвешенным слоем 12°. В опытном аппарате поваренная соль
высушивалась от 3 до 0,03% влажности воздухом, подогретым
до 200° и поступавшим в аппарат под давлением 575 мм вод. ст.
Расход топлива был меньше, чем при сушке во вращающихся
барабанах, на 65—70% 132- ш.
- На некоторых американских заводах применяют вакуум-
фильтр-сушилку для одновременной фильтрации и сушки соли.
Отжатая от маточного раствора лепешка соли продувается на
фильтре горячим воздухом A50—160°), после чего влажность соли
снижается до 0,1%. Фильтр-сушилка при диаметре 1,8 м и ширине
барабана 0,6 м дает в сутки до 200 т соли; при ширине барабана
1,2 ж —до 380—400 т.
Высушенную соль просеивают на цилиндрических вращающихся
ситах (буратах) для отделения комков (окатов), образующихся
при высушивании влажной соли. Просеянную мелкую сухую соль
передают с помощью ленточного транспортера в расфасовочное
отделение склада. Один из барабанов ленточного транспортера
является магнитным сепаратором — в него вмонтирован электро-
электромагнит. При проходе через сепаратор из соли извлекаются могу-
могущие попасть в продукт металлические частицы.
Сухую вакуумную выварочную соль расфасовывают в картон-
картонные коробки, бумажные пакеты (по 0,5 и 1 кг) и в многослойные
крафтцеллюлозные мешки (до 50 кг).
В качестве примера приводим анализы (в %) двух образцов
выварочной соли, полученных чренной и вакуумной выпаркой из
одного и того же сырого рассола:
NeCI
Нераство-
CaCIa CaSO< MgSO4 римый Влага
остаток
Чренная соль . . 98,51 - Следы 0,03 0,50
Вакуумная соль 99,40 0,Ш - - 0,19
0,16
0.22
0,05
0,60
0,06
Для некоторых районов целесообразно комбинирование есте-
естественного концентрирования рассолов бассейным способом с по-
последующей вываркой соли из концентрированного рассола134-13S.
Таким путем, в частности, целесообразно извлекать соль из мор-
морской воды, так как полная выварка морской воды в вакуум-вы-
вакуум-выпарных аппаратах требует большого расхода тепла, и поэтому
соль получается дорогой. Тем не менее выпарку морской воды
производят. В Японии на некоторых угольных шахтах, располо-
расположенных у морского побережья, получают соль выпариванием мор-
морской воды, используя в качестве топлива низкокачественный уголь
и рудничный газ 136. При ступенчатом испарении морской воды под
вакуумом можно одновременно с поваренной солью получать ди-
QTmympoBaHHyio воду, конденсируя пары холодной воды из глу-
глубин моря 137.
Представляет интерес получение выварочной соли с использо-
использованием тепла термальных соляных источников, находящихся в
районе Камчатки и Чукотского полуострова 138.
Мелкую поваренную соль, обладающую повышенной скоростью
растворения, можно получать, высушивая раствор NaCl в распы-
распылительных сушилках при прямоточном движении снизу вверх теп-
теплоносителя и раствора, подаваемого через форсунку сжатым воз-
воздухом. Высушенная соль должна улавливаться в циклонном пы-
пылеуловителе 139.
Предложено140 получать кристаллическую поваренную соль из
рассола, выпаривая его в прямоточном аппарате, где он контак-
тируется с топочными газами при пенном режиме. Поток суспен-
суспензии NaCl в рассоле движется в восходящем направлении, если
получают мелкую (пищевую) соль и в нисходящем — при получе-
получении соли с частицами крупнее 0,165 мм. Вспененный рассол про-
проходит сепараторы для отделения насыщенного водяным паром
газа, который затем эжектируется. С пеной выводится осадок
82
Гл. III. Хлористый натрий
Получение соли вымораживанием
83
солей жесткости, образующийся при повышенной температуре под
влиянием содержащейся в топочном газе СО2. В ходе непрерыв-
непрерывной кристаллизации размеры кристаллов и скорость их роста ре-
регулируются автоматически за счет непрерывного возврата оборот-
оборотного рассола после отделения сгущенной суспензии в конической
части аппарата.
ИОДИРОВАННАЯ ПОВАРЕННАЯ СОЛЬ
Употребление иодированной поваренной соли, содержащей
примесь KI, предупреждает болезнь щитовидной железы — зоб-
зобную эндемию, развивающуюся при недостатке иода в питьевой
воде. Ежедневная потребность человека в иоде составляет 0,06—
0,1 мг.
С течением времени иодид калия окисляется с выделением бы-
быстро улетучивающегося иода. Разложение KI значительно уско-
ускоряется в присутствии влаги и в особенности окислителей. Имеются
попытки применения иодата калия, который в присутствии некото-
некоторых иодорганических соединений не разлагается в течение дли-
длительного времени ш.
Добавка в соль различных веществ, так называемых стабили-
стабилизаторов — фосфатов и карбонатов кальция или магния и других —
увеличивает стойкость иодированной соли. Лучшим стабилизато-
стабилизатором оказался тиосульфат натрия, восстанавливающий элементар-
элементарный иод до Nal. Введение тиосульфата натрия в количестве 1%
от KI или 0,00001% от веса соли позволяет хранить иодирован-
иодированную соль в течение шести месяцев без потерь иода. Без стабили-
стабилизатора KI полностью исчезает в течение 2—3 месяцев 1V2> нз. Для
предупреждения потерь иода при хранении иодированной соли
предложено также добавлять к ней активированный уголь 144.
Иодированную соль приготовляют мокрым и сухим способами.
Мокрый способ заключается в опрыскивании соли раствором KI,
содержащим тиосульфат натрия, из форсунок или капельниц. Кон-
Концентрация раствора KI должна быть такой, чтобы влажность соли
не возрастала более чем на 1%. Обычно применяют раствор, со-
содержащий 2,5—5 г/л KI. Для увеличения срока хранения иоди-
иодированной соли предложено иодировать ее опрыскиванием водным
раствором иодинола A г/л 12 и 3 г/л KI), приготовляемого из по-
поливинилового спирта и раствора иода в иодиде калия 14Б.
Иодирование производят на разных стадиях производства
соли. Например, каменную и озерную соль удобно иодировать пе-
перед ее помолом (рис. 14). В процессе дальнейшего измельчения
соли достигается равномерное распределение иодида калия в про-
продукте88. Иодирование выварочной соли мокрым способом может
быть осуществлено перед сушкой в сушильных барабанах, а также
в элеваторах или на транспортерах.
Попытка плавить иодированную соль для получения кусковой
иодированной соли показала, что при этом теряется значительное
количество иодида калияИ6. Для получения крупнокристалличе-
крупнокристаллической иодированной соли рекомендуют прибавлять KI к соляному
рассолу до бассейной кристаллизации из него NaCl U7-148.
Сухой способ иодирования соли88- 149 заключается в смешива-
смешивании сухой соли с концентратом иодида калия, представляющим
собой поваренную соль, содержа-
содержащую в 100 раз больше стабилизи-
стабилизированного KI, чем предусмотрено
нормой, т. е. 1 г на 1000 г соли. Не-
Необходимость предварительного при-
приготовления концентрата обусловле-
обусловлена невозможностью равномерного
смешения твердых веществ в соот-
соотношении 1 : 100 000. Влажность кон-
концентрата должна быть не больше
0,1%. Для смешения концентрата с
солью применяют шнековые и ба-
барабанные смесители. Более совер-
совершенными являются смесители цен-
тробежного и каскадного ти-
11ОИ . /—ленточный транспортер; 2—бункер
На ЗаВОДе фирмы Canadian Salt крупнокристаллической соли; 3-наклон-
« -г г . ныи желоб; 4 — подвод раствора иоди-
Со. НеДаВНО СТаЛИ ИСПОЛЬЗОВаТЬ стого калия с ниппелями для опрыски-
nnfitioiiunHHue пытятрпы нрппрпмд- вания соли; Я-горловина вальцового
виорационные питатели непрерыв станка; е-вальцы; 7-выход молотой
НОГО ДеЙСТВИЯ С весаМИ, ДЛЯ ВВеде- и иодированной соли.
. ния в соль (со скоростью Ш—
750 г/мин) малых количеств СаЮ4, СоСО3 и KI в смеси с алюмо-
алюмосиликатами (снижающими клейкость добавок). Эти питатели, как
сообщают ш, позволяют отказаться от предварительного смешения.
Сухой способ иодирования соли одно время применялся более
широко, чем мокрый способ, так как сухая иодированная соль бо-
более устойчива, чем влажная. Считают, однако, что после разра-
разработки метода стабилизации влажной иодированной соли целесо-
целесообразнее применять мокрый способ иодирования, отличающийся
большей простотой 88>149.
ПОЛУЧЕНИЕ СОЛИ ВЫМОРАЖИВАНИЕМ
Добыча соли из концентрированных рассолов возможна путем
кристаллизации соли при охлаждении рапы82152. Зимой, при низ-
низких температурах, из насыщенных рассолов вымерзает дигидрат
хлористого натрия NaCl-2H2O. Кристаллизация его идет тем ин-
интенсивнее, чем ниже температура, вплоть до температуры выде-
выделения криогидрата (—21,2°). Если дигидрат извлечь из рапы, то
I
84
Гл. III. Хлористый натрий
Получение соли вымораживанием
85
при повышении температуры воздуха выше +0,16° происходит его
разложение и переход в чистую поваренную соль (рис. 7).
Дигидрат хлористого натрия выделяется в зимнее время в со-
соляных источниках Якутской АССР, а также во многих других
озерах. Выделяющийся дигидрат не содержит примесей, и его вы-
вымораживание из рассола является одним из методов получения
чистой соли. При плавлении 1 т NaCl • 2НгО при 25° можно по-
получить 481,5 кг безводного хлористого натрия с выходом NaCl,
равным 77,8% "• 163.
Вымораживание соли зимой может производиться в таких же
бассейнах, как и садка соли летом. Максимальный выход безвод-
безводной соли из насыщенного рассола до-
достигает 20% от начального содержания
ее в исходном рассоле.
Однако получение поваренной соли
вымораживанием является весьма слож-
сложным, громоздким процессом, требующим
больших площадей бассейнов. При нали-
наличии дешевой электроэнергии метод ис-
искусственного вымораживания может ока-
оказаться выгодным для получения пище-
пищевой соли сорта экстра. Более эффектив-
эффективным является концентрирование морской
100
I 40
I 20
45 '-20 В°ДЫ в условиях холодного климата вы-
Температура.'С
Рис. 16. Вымораживание
льда из морской воды.
мораживанием льда.
Степень выделения льда в зависимо-
сти от температуры охлаждения пока-
8ана на рис i5e2 Охлаждением морской
воды до —17° с последующим отделе-
отделением льда можно получить рассол, содержащий 9—12% со-
солей 154,155 (по другим данным156 19%). Такой рассол после очи-
очистки от CaSC4 пригоден для выделения NaCl методом выпарива-
выпаривания. Из 1 мъ морской воды получается 30—32 кг соли 167. Качество
втой соли различно: лучшие сорта могут содержать до 97—99%
NaCl, а худшие 80—82% 1Б8. В среднем она содержит ~ 10% при-
примесей.
Непосредственная заводская выпарка морской воды требует
много различного оборудования, т. е. больших капитальных вло-
вложений и значительного расхода топлива. Для получения из мор-
морской воды, содержащей 3,5% солей (из которых 80% NaCl), 1 т
поваренной соли нужно выпарить ~45 т воды. Для этого необ-
необходимо израсходовать при использовании четырехкорпусной ва-
вакуум-выпарной батареи 3 т, а при чренной выпарке 7 т условного
топлива. С увеличением концентрации рассола количество выпа-
выпариваемой воды на 1 т соли резко уменьшается; так при концен-
концентрации рассола 8% нужно выпарить 15 т воды, а при 12% только
9 т. Поэтому предварительное вымораживание льда из морской
воды с последующей выпаркой оставшегося рассола экономически
более выгодно.
Плотность пресной воды максимальна при +3,95°, максимум
же плотности морской воды наблюдается при более низких тем-
температурах. При концентрации суммы солей 2,47% температура на-
начала выделения льда (—1,332°) становится равной температуре
максимальной плотности такого рассола (рис. 16). Более соленые
воды (океана, соляных озер) замерзают
при температурах выше температур их мак-
максимальной плотности. Например, океан-
океанская вода, соленость которой равна 3,5%,
гамерзает при —1,91°, а максимальная
плотность ее при —3,52°.
Эти свойства соленых вод имеют важ-
важное значение как для биологических, так и
для галургических процессов. В то время
как при замерзании поверхности пресного
водоема подледная вода сохраняет темпе-
температуру около +4° ввиду ее повышенной
плотности, образование льда на поверхно-
поверхности соленых вод сопровождается конвек-
конвекцией подледных рассолов из-за разности
их плотностей, пока плотность не достигает
максимума, для воды океана при —3,52°.
Это может оказаться гибельным для неко-
некоторых видов фауны и флоры, но весьма благоприятно для галур-
галургических процессов, так как циркуляция подледных рассолов и
низкая температура их максимальной плотности ускоряет охла-
охлаждение. Это приводит к увеличению степени вымораживания льда,.
укреплению рассолов и к повышению выхода солей, кристалли-
кристаллизующихся при понижении температуры. В бассейнах для зимней
садки солей температура рассолов уже через несколько суток до-
достигает средней температуры воздуха 1Б6.
На рис. 17 приведена диаграмма глубокого охлаждения мор-
морской воды. На оси абсцисс отложены значения коэффициента кон-
концентрирования К, равного отношению количества исходного рас-
раствора к количеству конечного раствора. Его величина может быть-
определена по отношению содержания иона магния, являющегося
постоянным (неизменным) компонентом. Равновесными линиям»
диаграмма разделена на несколько областей кристаллизации. Об-
Области I и II, а также IV и VIII отделены двумя линиями: пунктир-
пунктирная относится к охлаждению морской воды обычного состава,,
содержащей сульфат кальция; сплошная линия характерна дл»
морской воды, очищенной от сульфата кальция. С увеличением
концентрации растворов эти линии сближаются, так как
/ г J
Соленость воВы, %
Ри-с. 16. Температуры
замерзания (/) при наи-
наибольшей плотности BУ
морской воды.
Гл. HI. Хлористый натрий
растворимость сульфата кальция уменьшается. В растворах, на-
насыщенных хлористым натрием, растворимость сульфата кальция
столь мала, что кривые сливаются.
Как видно из диаграммы, на которой линия АВ(АВ')—путь
кристаллизации льда, а линия ВС (В'С) — мирабилита, вымерза-
вымерзание льда из морской воды начинается при —1,8°. При —7,3° вме-
вместе со льдом кристаллизуется мирабилит Na2SO4- ЮН2О, а при
—15° начинает выделяться
Коэффициент концентрирования, К сульфат кальция. При —22,4°
к этим твердым фазам при-
присоединяется NaCl-2H2O. Если
при —22,4° отделить жидкую
фазу от твердой и продолжать
ее охлаждать, то при —34°
раствор станет насыщенным
хлоридом кальция. Таким об-
образом, по мере понижения
температуры раствор обога-
обогащается хлоридами магния и
кальция, кристаллогидраты ко-
которых выделяются в осадок
при еще более глубоком охла-
охлаждении. Конечный рассол за-
замерзает при температуре око-
около —60°.
В зависимости от темпера-
температуры вымерзания льда из мор-
морских рассолов выделяющийся
лед может содержать разное
количество твердых солевых
примесей, т. е. иметь различ-
различную «соленость». До темпера-
температуры —7° образуется доста-
Получение соли вымораживанием
87
-22
Рис, 17. Диаграмма глубокого охлажде-
охлаждения морской воды.
Области кристаллизации:
/ — льда; // - льда + Na2SP4 • 10H2Oj
/// •*- льда + NagSO4 • ЮН^О + NaCI . 2H9OS
/УМБОГН;
/УМа«БО4'Г0Н2О;
V - NaCI . «HsO + Na2SO4 • 10HsO;
VI - NaCI + NasSO.4 ¦ IOH2O; VII- NaCI;
VIII — область ненасыщенных растворов.
точно чистый лед, а ниже этой температуры он все больше загряз-
загрязняется примесями. По разным источникам соленость морского
льда характеризуется величинами от 0,3 до 1%-
Из 1 т морской воды при охлаждении до начала кристаллиза-
кристаллизации мирабилита (—7,3°) вымерзает ~720 кг льда. При охлажде-
охлаждении до —16° выделяется в твердые фазы 80—85% сульфат-иона и
получается —175 кг рассола. При вымораживании до —36е из
1 т морской воды остается всего ~26 кг рассола.
В Японии и других странах концентрирование морской воды
осуществляют непосредственным вымораживанием льда1БВ. Кон-
Концентрировать морскую воду вымораживанием льда можно, раз-
•брыегивая ее при 0° в вертикальной башне, через которую цир-
циркулирует минеральное масло, с температурой —10°, В нижней
части башни накапливается'рапа плотностью 1*134 г/см\ а вверху
над маслом — лед
160
д маслига — лсд
Заслуживает внимания получение поваренной соли из морской
воды комбинированием испарения и охлаждения с одновременным
выделением мирабилита 164. Изменение состава получающихся при
этом растворов показано на рис. 18. Морскую воду (/, /°) или ее
концентраты (/, /' или /, 1"),
полученные вымораживанием
или другим методом, выпари-
выпаривают при 100° вначале до на-
насыщения (/, 1"'), а затем с
кристаллизацией NaCI (/—2).
Остающийся маточный рас-
раствор B, 2') после отделения
NaCI разбавляют водой {2,2")
и охлйждают до —10°, причем
кристаллизуется мирабилит
B—5). Маточный раствор пос-
после отделения мирабилита C,
3') выпаривают вторично при
100° для дополнительной кри-
кристаллизации NaCI C—4). Ко-
Конечный раствор состава 4, 4'
остается в небольшом количе-
количестве и может быть использо-
использован для извлечения магниевых
солей.
Важной проблемой челове-
человечества является расширение
ресурсов пресной воды, запа-
запасы которой на земном шаре
весьма ограничены. В связи с
этим интенсивно разрабаты-
разрабатываются различные методы
опреснения соленых вод1.
Помимо пресной воды при
обессоливании морской воды и других соленых вод получаются*
более концентрированные рассолы, чем исходные. Они могут слу-
служить источниками выделения NaCI рассмотренными выше спо-
способами.
Опреснение морской воды в крупных масштабах производят
различными путями. Широко применяют дистилляцию в вакуум-
испарителях 162 и вымораживание льда157- 1Б9> 16s~1^. Применяют
различные источники холода, в частности, вымораживание льда
ведут за счет испарения небольшой части воды @.5%) под ваку-
вакуумом (абсолютное давление в вакуум-аппарате 3 мм рт. ст.) ил»
2NaCI
50
W
Рис. 18. Схема переработки морской
воды и ее концентратов испарением прв
100° и охлаждением до —10°.
Гл. III. Хлористый натрий
Получение поваренной соли из галитовых отходов
120
\
за счет испарения под атмосферным давлением примешиваемых к
воде легкокипящих веществ, например: бутана, изобутана 1б6, про-
пропана, фреонов и др.168. Кристаллизующийся лед можно отделять
от остающейся воды фильтрованием, центрифугированием или
флотацией 1ег. Все шире распространяется обессоливание электро-
электродиализом 1в1 и фильтрованием под давлением через мембраны,
проницаемые для воды и почти не пропускающие растворенные
в ней соли. Мембраной служит, например, пленка из ацетатной
целлюлозы с модифицирующими добавками (перхлорат магния,
«цетон) 169. Используют и ионообменные смолы 170> т.
ПОЛУЧЕНИЕ ПОВАРЕННОЙ СОЛИ ВЫСАЛИВАНИЕМ
И ПЕРЕКРИСТАЛЛИЗАЦИЕЙ
Уделялось много внимания разработке новых способов полу-
получения поваренной соли из рассолов путем высаливания ее хло-
хлористым магнием (рис. 19) или хлористым кальциемт. Преиму-
Преимущества этих способов со-
состоят в относительной
простоте технологическо-
технологического процесса (заключаю-
(заключающегося в смешении рас-
рассолов, отделении выпав-
выпавших кристаллов соли и
сушке их), в отсутствие
расхода топлива на вы-
выпаривание рассола, в от-
отсутствие необходимости
предварительной очистки
рассола и др.
При смешении, напри-
например, 1 м3 рассола, содер-
содержащего 20,7% NaCl, 1,3%
MgSO4, 4,3% MgCl2,
73,7% Н2О (плотность
1,23 г/см3) с 1 м3 хлор-
_, .„ „ магниевого рассола, со-
Рис. 19. Ивотермы растворимости в системе cv7 cm л л ^i
NaCl-MgCl2-H2O при 10, 40 и 100°. держащего 27,6% MgCl2,
4,4% MgSO4, 1% NaCl,
€7,0% Н2О (плотность 1,26 г/см3), выход кристаллизующейся по-
поваренной соли составляет около 78 кг.
Разработаны способы перекристаллизации каменной соли, по-
позволяющие получать чистую соль более дешевым путем, чем ва-
вакуум-выпаркой. Например, каменную соль смешивают с маточным
раствором, остающимся после вторичной кристаллизации. Соле-
Солевую пульпу перемешивают острым паром, конденсация которого
приводит к растворению кристаллов соли при 100—105°. Нерас-
\
во 40 а
2С\2 на ЮОВмоль Н2О
творившаяся часть, содержащая примеси (ангидрит и др.), от-
отделяется в отстойнике, а горячий раствор направляют на кристал-
кристаллизацию в две стадии — при охлаждении его до 80°, затем до 50°.
Соль из кристаллизаторов отжимают на центрифугах и высуши-
высушивают 173.
Другой способ заключается в обработке каменной соли цир-
циркулирующим насыщенным раствором NaCl при температуре от 0
до —20°; при этом соль переходит в дигидрат, который отфиль-
отфильтровывают и нагревают. Дигидрат плавится и выделяется чистая
соль; рассол возвращают в процесс 174.
Из каменной соли, сильно загрязненной ангидритом, можно'
получить поваренную соль сорта «экстра» перекристаллизацией
из растворов хлорида кальция. Каменную соль перемешивают в
течение 30 мин с исходным раствором, содержащим 24% СаС12.
В кристаллизаторе раствор охлаждается от 112 до 20°. Выход
NaCl из 1 м3 раствора около 50 кг. Получаемая после промывки
соль содержит 99,2% NaCl. Расход СаС12 A00%) при 50-кратном
использовании составляет 11 кг на 1 т получаемой соли 175.
ПОЛУЧЕНИЕ ПОВАРЕННОЙ СОЛИ ИЗ ГАЛИТОВЫХ ОТХОДОВ
КАЛИЙНЫХ ПРЕДПРИЯТИЙ
При переработке сильвинита на хлорид калия (стр. 154) в ка-
качестве отхода (отвала) получается загрязненный примесями хло-
хлорид натрия. Количество его очень велико — до 80% от перера-
перерабатываемого сильвинита. Галитовые отходы содержат (в сухом
веществе) 92—96% NaCl, 1,2—2,5% КС1, 0,6—2% CaSO4, 0,05—
0,2% MgCb, 0,3—3% нерастворимых веществ; влажность их 5—
10% 176.
Технические сорта поваренной соли могут быть получены иа
отходов путем их промывки насыщенным раствором хлорида нат-
натрия, например на реечном классификаторе. При этом содержание
NaCl повышается до 98%), а содержание примесей снижается
(КС1 до 0,30—0,32%). Этот метод переработки отходов наиболее-
прост и дешев.
Получение более чистой пищевой соли может быть осуществ-
осуществлено растворением отходов177, химической очисткой полученного
рассола и вакуумной выпаркой его, а также флотацией отходов 178.
Последний метод имеет преимущество перед вакуум-выпаркой^
так как не требует расхода пара. Из отходов флотируют примеси,,
а не основной продукт — так называемая обратная флотация. (Воз-
(Возможна и прямая флотация в присутствии солей свинца или вис-
висмута 179.) Хотя флотация и дает продукт е высоким содержанием
NaCl (99,7%), но он загрязнен флотореагентами и имеет неудов-
неудовлетворительный внешний вид, так как представляет собой не
бесцветный (красноватый) тонкий порошок (содержание класса
0,15 мм составляет ~57%).
90
Гл. III. Хлористый натрий
Промывка галитовых отходов от переработки верхнекамских
сильвинитов по специальной технологической схеме180 позволяет
экономично получать пищевую соль хорошего качества. По этой
схеме (рис. 20) отходы смешиваются с рассолом NaCl и подвер-
подвергаются мокрой классификации на дуговых ситах 181 с целью отде-
отделения средней фракции с размером —3 + 0,5 мм, содержащей наи-
наименьшее количество примесей. Эта фракция при Ж: Т равном 0,7
Брикетирование и борьба со слеживавмостью соли
Рис. 20. Схема получения соли 1 сорта помола № 0 из галитовых отходов:
U 6 к 8 —мешалки; 2, 9 и 21 — насосы: 3, 5, 10 и ./4 —дуговые сита; 4 - планфильтр; 7 — дро- \
ойлка; A — напорный бак; 12 — контактные чаны; 13 и 15 —гидросепараторы; 16 — горизонталь-1
вай мешалка; 17 — центрифуга; 18 — транспортер; 19— отстойник; 20 — сборный бак.
доизмельчается до крупности, соответствующей помолу № 0, и пе-.
ремешивается в контактных чанах 30—40 мин. При этом раство-^j
римые примеси переходят в раствор, а затем пульпа обесшламли-
вается в гидросепараторах и на дуговом сите. Загрязненный рас- {
сол сливают, а сгущенную пульпу направляют на центрифугу.
В ней соль промывается небольшим количеством воды для оконча-
окончательного вытеснения хлорида калия. Промытую соль высушивают.
Загрязненный оборотный рассол после осветления возвращается
в процесс, но часть его удаляется для вывода из цикла хлорида
калия. При вакуум-выпарке этой части рассола из него можно по-
получать соль «экстра». Наибольшая удельная производительность
отстойной площади для оборотного рассола достигается при кон-
концентрации суспензии 5 г/л; при этом она равна 1,5 м3/(м2'Ч) при
расходе коагулянта крахмала 0,7 кг на 1 т ила, или 1,9 м&/(м2'Ч)
при таком же расходе полиакриламида 182. Получаемая соль со-
соответствует по составу первому сорту. Она содержит (в %):
NaCl 98,5—99,0 Нерастворимый оста-
CaSO4 Ор-0,6 ток 0,3-0.4
MgCh 0,02-0,07 KCI Нет
Техническая поваренная соль может быть получена и из от-
отходов флотации КС1 из сильвинита.
Содержание токсичных жирных аминов (используемых в каче-
качестве коллектора-собирателя сильвина) во флотационных отходах
составляет ~8 г/т, но в солевой фракции их практически нет, так
как они адсорбируются на глинистом шламе (в 1 т нераствори-
нерастворимой части отходов содержится ~325 г аминов). Поэтому в гото-
готовой технической соли, полученной из отходов флотации, амины от-
отсутствуют 183.
Для отмывки флотационных отходов галита от аминов пред-
предложено также обрабатывать их в классификаторах оборотным ма-
маточным раствором; из циркулирующего раствора частицы амина,
имеющие размеры 1—100 мк, отделяются фильтрованием через
несколько слоев стеклоткани ш. О приготовлении из отходов фло-
флотации сильвинита технического рассола см.185.
Для внутризаводского перемещения можно использовать гид-
гидротранспорт поваренной соли в ее насыщенном растворе с по-
последующим отделением соли в гидроциклоне. Влажность соли и»
гидроциклона 18—20% ш-
Предложена скоростная разгрузка сухой соли из опрокиды-
опрокидывающегося вагона в бетонный бассейн с рассолом, находящийся
под колеей. Пульпу (Ж:Т=1:1) перекачивают насосом в хра-
хранилище, где кристаллы отделяются от рассола, возвращаемого в
бассейн 187.
БРИКЕТИРОВАНИЕ И БОРЬБА СО СЛЕЖИВАЕМОСТЬЮ СОЛИ
Крупнокристаллическая соль, содержащая меньше 5% зерен с
размерами менее 5 мм, схватывается весьма слабо. Наибольшей
слеживаемости подвержена соль с размерами зерен менее 1,2 мм.
Слеживаемость соли резко повышается во влажном воздухе, осо-
особенно при колебаниях влажности. Сильнее слеживается соль с по-
пониженным содержанием Mg2+188.
Надежным путем предотвращения слеживаемости соли яв-
является высушивание и прокаливание ее до остаточной влажности
менее 0,1 или 0,05% (в районах со среднегодовой относительной
влажностью воздуха не выше 76%) и упаковка в сухом виде во
влагонепроницаемую тару18В.
«2
Гл. III. Хлористый натрий
Литература
93
Во многих странах, особенно в странах с влажным климатом,
вызывающим слеживание и расплывание срли, соляную пыль и
часть мелкозернистой соли брикетируют, брикетированная соль
употребляется в качестве пищевой соли. Изготовляют также кор-
кормовые брикеты для животных («лизальные камни» или «лизун-
«лизунцы») , иногда с добавкой кормовых фосфатов и микроэлементов 19°.
Брикеты, изготовленные ив соляной пыли, могут быть размолоты
на крупнозернистую соль на рифленых валках, причем количество
получаемой вновь пыли очень невелико F6,6% зерен имеют круп-
крупность более 5 мм).
Можно изготовлять легкие пористые блоки из поваренной соли
плотностью меньше 0,7 г/см3. Для этого приготовляют из кристал-
кристаллической поваренной соли и рассола (или другой жидкости) пасту
или суспензию, в которую вводят связующий агент — желатину,
алкилцеллюлозу, казеин @,1—0,5% от веса сухой соли) и насы-
насыщают ее воздухом или двуокисью углерода. Последнюю можно
генерировать непосредственно в смеси, добавляя к ней бикарбо-
бикарбонат натрия или аммония. Насыщенную воздухом кремоподобную
массу выливают в формы и высушивают при 150° ш.
Предложены различные способы предотвращения слеживаемо-
сти соли при открытом хранении на воздухе, в основном с приме-
применением разных добавок, понижающих гигроскопичность соли или
препятствующих схватыванию ее в сплошной конгломерат или
плотные комки. В качестве таких добавок предложены: водоне-
растворимые соединения марганца и железа (окись марганца и
фосфат железа или 9МпО • 4FeO • РгО8) в количествах, допусти-
допустимых при применении соли в пищу ("-0,6—0,7%I92, водораство-
водорастворимый комплексный фторид, например фторсиликат, в таком ко-
количестве, чтобы содержание фтора в соли было не больше
0,01% 193; тонкодисперсный осажденный силикат кальция или ги-
дратированная кремневая кислота с размерами частиц менее 1 мк
в количестве 0,5—5% от веса соли 194'195; алифатические амины с
числом атомов углерода от 6 до 22 (о-октиламин, дециламин) в
количестве 0,023—1,8 кг на 1 г соли, предварительно растворенные
в нефти или керосине с добавкой поверхностно-активных веществ
@,5—3%), вносимые при 65—120°1ее; и другие добавки197.198. За
границей часто в качестве таких добавок используют ферроциа-
ниды и феррицианиды E—10 ч. на 1 млн. ч. NaCl); для уменьше-
уменьшения их вымывания атмосферными осадками при хранении соли
в буграх предложено феррецианид, феррицианид или нитрило-
триацетамид предварительно обрабатывать органическим силокса-
ном @,1—5% от веса добавки), содержащим группы RR'SiO —
(где R, R'— алкильный или арильный радикалI99. Для этой же
цели предложено добавлять к рассолу при получении из него соли
поверхностным испарением моноэфир лауриновой кислоты и по-
лиоксиэтиленсорбитан 20°. Показана эффективность добавки
A1(OHS) @,005—0,1%), введенной мокрым способом — опрыски-
опрыскиванием соли растворами А1С13 @,1%) и щелочи201. Запатентован
способ получения быстрорастворяющейся в воде и неслеживаю-
щейся поваренной соли путем добавки к выпариваемому рассолу
€,0015—0,1% гексаметафосфата натрия с примесью 2—40% от
веса последнего поливалентного катиона в водорастворимой фор-
форме— в виде окислов, гидроокисей, сульфатов или хлоридов Al, Fe,
Ва, Bi, В, Cd; в получаемой соли содержится меньше 0,3% фос-
фосфата и меньше 0,0005% металла202. Для придания поваренной
соли гидрофобное™ рекомендуют обрабатывать ее пищевым жи-
жиром (меньше 1%J03 или парафином204, или жидкими неорганиче-
неорганическими галогенидами кремния, имеющими связь Si—Н, например
силикахлороформом 2М.
СПЕЦИАЛЬНАЯ (ТОНКАЯ) ОЧИСТКА СОЛИ
Соль высокой чистоты с содержанием NaCl 99,9% и больше
готовят или перекристаллизацией стандартных (продажных) сор-
сортов солиш или другими методами — промывкой и пр. Чистоту
соли можно определять измерением ее электрического сопротивле-
сопротивления207. Очистку NaCl от таких примесей, как например Na2SO4,
можно осуществлять промывкой соли соляным раствором 72>208.
Хлористый натрий с содержанием 99,97—99,98% NaCl может
быть получен перекристаллизацией NaCl • 2Н2О, а также обработ-
обработкой поваренной соли концентрированной соляной кислотой209.
Предложено очищать поваренную соль от примесей раство-
растворением ее в ненасыщенном растворе чистой соли под давлением
при 115—140° и последующей кристаллизацией NaCl из получен-
полученного насыщенного раствора, не содержащего примесей, при атмо-
атмосферном давлении210.
Представляет интерес очистка поваренной соли от органиче-
органических и бактериальных примесей плавлением. Затраты на очистку
соли этим методом, вероятно, могут быть ниже затрат на пере-
перекристаллизацию, так как затраты тепла на плавление соли зна-
значительно меньше, чем на однократное испарение воды из рас-
раствора211.
ЛИТЕРАТУРА
1. W. Е. May, Т. R. Skott, пат США 2642335, 1953; канад. пат. 497203,
1953. —2. Австрал. пат. 153929, 1953.— 3. В. Н. Дубинина, Труды ВНИИГ
в. 29, 1964, стр. 3.— 4. К. Przibram, Kali, 30 A936). —5. А. А. Иванов,
Основы геологии и методика поисков, разведки и оценки месторождений мине-
минеральных .солей, Госгеолиздат, 1953. — 6. R. Bruno, Ann. chlfnica, 56, № 11, 1287
A966). — 7. Л. Д. Панченко, Н. Д. Ивашенко, Ё. Л. Вол ченко, Труды
НИИ соляной пром., в. 2 A0), 1S59, стр. &?. — 8. Е. И. Ах умов. Е. И. Нико-
Николаева, Т. Н. Шевлякова, ЖПХ 29, № 10, 1599 A9Б6). — 9. Н. А. К у П и н а,
Труды ЛТИ им. Ленсовета, в. 40, iffi, стр. 92. —10. В. П. Ильинский,
В. Ф. Королев, Е. И. Ахумов, ЖПХ, 25, № 5, 507 A952).
94
Гл. III. Хлористый натрий
Литература
95
11. А. П. Николаевский, ИСФХА АН СССР, 4, в. 2, 217 A930).—Э|
12. М. Кучеров, Поваренная соль и ее техническое использование, Химтеорет,
1933, — /5. В. Н. Енько, Поваренная соль, Химтеорет, 1933. —14. J. Taylor,
канад. пат. 490245, 1963.— 15. Т. П. Унанянц, Хим. наука и пром., 2, № 6,
774 A957). — /б. А. И. Дзенс-Литовский, В. А. Иванов, Поваренная
соль. Требования промышленности к качеству минерального сырья, в. 77, Изд. ¦
«Недра», 1965. —17. Г. Л. Кор енько в, Н. А. Устинова, Горнохимическое
сырье зарубежных стран, Изд. «Химия», 1965. —18. И. А. Егунов, сб. «Источ-
«Источники минерального сырья для химической промышленности», т. II, 1928, стр. 213.—
19. А. А. Иванов, П. И. А л е к с е е в с к и й, Минерально-сырьевая база СССР,
вып. 36, ОНТИ, 1935. — 20. А. Е. Первухина,- Труды Сев. базы АН СССР,
в. 5, 1940.
21. П. М. Т а т а р и н о в и др., Курс нерудных месторождений, ч. I, Гор-
геолнефтеиздат, 1934. — 22. 3. А. Горелик, Залежи каменной соли в Домано-
вичском районе и перспективы поисков соли и нефти в БССР, Минск, 1947. —•
23. А. И. Дзенс-Литовский, Бюлл. ВИГ, № 1—2, 9 A940).— 24. Б. Л. Рон-
кин, Наши соляные озера и их богатства, ГНТИ, 1931. — 26. Н. С. Курнаков,
Соляная проблема и химическая промышленность, ГНТИ, 1931. — 26. М. Г. В а-
ляшко, Труды ВНИИГ, в. 23, 1952, стр. 25.-27. Г. Г. У разов, И. Н. Л е-
пешков, Труды Ин-та химии АН УзбССР, т. 2, 89 A949). — 28. А. А. II ер-
рот те, Соль, 1936. — 29. Е. В. Посохов, Минеральные богатства соляных
озер Казахстана, Алма-Ата, 1949. — 30. Я. Д Фридман, А. А. Зиновьев,
Организация солепромыслов в Киргизской ССР, Фрунзе, 1948.
31. О. И. Дейнека, Д. М. Корф, Бюлл. ВИГ, № 4—6 A939) и № 6—7
A940). — 32. Л. В. Николаев, Кулундинские соляные озера и пути их освое-
освоения, Новосибирск, 1935.—33. Н. Ф. Поярков, Автореф. канд. дисс, АН
УзбССР, Ташкент, 1955. — 34. Н. С. Курнаков, В. Г. Кузнецов,
А. И. Дзенс-Литовский, М. И. Равич, Соляные озера Крыма, Изз. АН
СССР, 1936. — 35. В. П. Блидин, ЖПХ, 24, № 10, 1096 A951).— 36. А. А. Ива-
Иванов, Природные минеральные соли, Госгеолиздат, 1951. — 37. Проблемы озера
Эльтон, под ред. Н. С. Курнакова, ч. II, Труды ВИГ, в. 16, 1939.—
38. Г. С. Клебанов, Д. М. Корф. Л. В. Е л о в с к а я. сб. «Приаральский
соляной район», Изд. АН СССР, 1937. — 39. Л. Насруллин, Нар. хоз. Ка-
Казахстана, № 4—5, 23 A940). — 40. Инструкция по применению классификации
запасов к месторождениям калийных солей и каменной соли, Госгеолиздат,
1951.
41. Я. Д. Ш е в я к о в, Разработка месторождений полезных ископаемых,
Углетехиздат, 1953. — 42. А. Н. Андреичев, Разработка калийных и каменно-
соляных месторождений, ч. I, 1953. — 43. А. Н Дегтярев, Д. П. Д у к о в л и-
нов, Хим. пром., № 2, 65 A955). — 44. Ш. А. П а н а х и, Труды Азербайджанск.
индустриального ин-та, в. 12, 1956, стр. 61 —45. А. И. Дзенс-Литовский,
Бюлл. ВИГ, № 6—7, 21 A940). — А. Н. Андреичев, Пути механизации
трудоемких работ в горнохимической промышленности, Госхимиздат, 1953. —
47. А. П. Л ы с о в, В. М. Сталин, К. А. Лохани и, Электрическая породо-
породопогрузочная машина ЭПМ-1, Углетехиздат, 1952.—48. И. Д. Косте в, Стаха-
Стахановские методы работы в бурении соли, Пищепромиздат, 1940.— 48а. П М. Ц и м-
баревич, Труды Московск. горного ин-та, № 6, 1941, стр. 36. — 49. Труды
Украинск. н.-и. ии-та соляной пром., в. 5A3), 1962.—50. И. В. Остроухое,
Там же, в. 2 ПО), 1959, стр. 34, 48.
51. Е. Mankowski, К. Lipinski, польск. пат. Б3883, 1965. — 52. П. Ка-
Кашинский, Н. Славский, Методы химического анализа соленой воды и озер-
озерной грязи, ГНТИ, 1931. — 53. А Г. Б ер гм а н, Н. П Лужная, Физико-хими-
Физико-химические основы изучения к использования соляных месторождений хлорид-суль-
хлорид-сульфатного типа, Изд. АН СССР, 1951. — 54. Н. С. Курнаков, Собрание избран-
избранных работ, т. II, ОНТИ, 1930, стр. 339.— 55. И. Н. Лепешков, Усп. хим., 21,
№ 9 1079 A952). — 55. Н. С. Курнаков, С. Ф. Жемчужный, ИСФХА АН
СССР, 1, № 1, 186 A919). — 57. В. Я. Р уд ин, Н. Л, Ярым-Агаев, ЖПХ, 30,
№ 6, 941 A957). — 68. М. К- Файзиев, ДАН УзбССР, № 3, 17 A954).—
S9. Е. И. Познер, ЖФХ, 21, № 7, 863 A947). — 60. М. К- Воротницкий,
Механизмы для добычи и погрузки соли, Пищепромиздат, 1946.
61. Н. П. Кузьмин, Труды Всесоюзн. н.-и. ин-та соляной пром., № 1 (9),
1954, стр. 4.-62. В. И. Николаев, Б. И, Степанов, Н. Е. Салит ев а,
Труды н.-и. станции НКМП КалмАССР, 1940, стр. 105. — 65. Е. И. Ах умов,
ЖПХ, 30, Ш 8, 1246 A957). — 64. Б. Ф. Вьюнов, Труды Всесоюзн. н.-и. ин-та
соляной пром., Jto 1 (9), 1954, стр. 72.— 65. Е. И. А х у м о в, Е. И. Николаева,
ЖПХ, 27, № 5, 480 A954). — 66. Е. И. А х у м о в, Л. А. Б а к к, Там же, 32, № 6,
1225 A9Б9). — 67. Б. Ю. Прасс, Руководство для мастеров-солеваров (вахте-
(вахтеров) бассейных промыслов, Пищепромиздат, 1937. —-68. Rao С. Sivaprasada,
Chem. Age (India), 16, № 6, 518 A965). — 69. Б. Ю. Прасс, С. Ф. В ере о ц-
кий, Добыча соли простейшими способами, Пищепромиздат, 1943. — 70. Н. П р е-
словски, Минно дело, 10, № 1, 68 A955).
71. М. J. Felizardo, Philippine J. Sci., 84, № 2, 177 A955). — 72. M. Pei-
sak, S. Afrik. Industr. chemist, 9, № II, 210 A955). — 73. Z. Czerwinskl,
A. Narebska, Przem. Chem., 10, № 6, 299 A954). — 74. П. А. Кулле, Труды
1ДНИСЛ, в. 2, 1940, стр. 3. — 75. Ф. А. Петрачков, М. В. Гончарова,
Труды Украинск. н.-и. ин-та соляной пром., в. 5 A3), 1962, стр. 11а.—
76 G G. Jain a. oth., Salt Res. a. Ind., 4, № 1, 10 A967). — 77. J. P. J a s s a-
walla, Chem. Age (India), 7, № 1, 53 A956). — 78. R. T. Thampy, N. R. Ku-
Ioor, D N. Daruvalla, Chem. Age (India), 5, № 3, 99 A954). — 79. Пат.
ФРГ 931168, 1955. — 80. Э. Б. Штерн ин а, Е. В. Фролова, ИСФХА АН
СССР, 21,271 A952).
81. J. D'Ans, Kali und Steinsalz, 4, № 4, 109 A965). — 82. К. Э. Гиттер-
ман, В. Ф. Королев, Труды соляной лаборатории АН СССР, в. 15. ч. 1,
1937. — 83. И. К. Пуха, Труды ВНИИГ, в. 36, 1959, стр. 322. — 84. В. П. Иль-
ииский, А. И. Данюшевская, Труды Дальневосточного филиала АН СССР.
Сер. хим., в. 2, 1956, стр. 43, 54. — 85. А. И. Данюшевская, Там же,
стр. 75. — 86. A. S. Hester, H. W. Diamond, Ird. Eng. Chem., 47, № 4, 672
.A955). —57. D. W. Koufmann, Sodium Chloride, New York—London, I960.—
«8. А. И. Зиновьев, Технология выварочной и иодированной соли, Пищепром-
Пищепромиздат, 1957. — 89. V. Jenista, Prumysl Potravin, 7, № 1, 25 A956).—
90. D. А. С г о о k s, Т. R. S с о 11, канад. пат. 498284, 1953.
91. Суги, Симидзу, Сакура и, япон. пат. 1823, 1955; Bull. Soc. Salt
Sci. Japan, 10, № 1, 27 A956). — 92. В. Н. Айзенберг, И. Н. Гладкий,
Н. Г. Пахмура, Труды Украинск. н.-и. ин-та соляной пром., в 5 A3), 1962,
стр. 115. — 9S. И. 3. Макинский, Труды Азерб. индустриального ин-та, в. 5,
1953, стр. 41. — 94. М. Б. Зеликин, А. Я. Бакулина, Н. К- Лукьянова,
Э. С. Ненно, Труды ВИСП, т. 8, 1955, стр 56.-95. А. В. В ас сер б ер г,
А. Е. Кругликов, Труды ЦНИСЛ, сб. № 1 (в. 6—8), 1941, стр. 187.—
96. А. Ф. Б о р я ч е к, Е. К. О в е ч к и н, С. С. У р а з о в с к и й, Труды ВИСП,
т. 5, 1949; стр. 71. — 97. А. А. Фурман, С. С. Шрайбман, Приготовление и
очистка рассола, Изд. «Химия», 1966. — 98. Хагино, Кобаяси, Bull. Soc.
Salt Sci. Japan, 9, № 5, 10 A955). — 99. А. Ф. Борячек, Е. Т. Громова,
О. Н. Кулагина, Труды НИОХИМ, т. 19, 1969, стр. 48. —100. Мацу но
Такэо, япон. пат 743, 1961.
101. D. W. Hengerer, канад. пат. 506130, 506131, 1954. — 102. A. Hirsch,
пат. США 2683649, 1954. — 103. Дат. пат. 77720, 1954.— 104. Ф. В. Королев,
Труды соляной лаборатории, т. 15, 1937. — 105. R. M. Hunter, R. D. Blue,
М. P. Neipert, канад. пат. 511795, 1955. — 106. А. С. Н. Arens, англ. пат.
1054594, 1954. — 107. D. Y. Gerritsen, Chem. Weekbl., 50, № 9, 159 A954).-i
108. A. E. Кругликов, Труды ВНИИГ, в. 36, 1959, стр. 244, 376. — 109. Т. Oh-
Гл. III. Хлористый натрий
kawa, япон. пат. 2279, 1953. — ПО. W. L. Badger, F. С. Standiford
Chem. Eng., 62, № 3, 173; № 4, 180 A056).
///. В. Н. Айзенберг, И. Н. Гладкий, Труды Украинск. и.-и. ин-та со-
соляной пром., в. 2 A0), 1959, стр. 87.— 112. Н. И. Тельперин, Выпарные ап-
аппараты, Госхимиздат, 1947.— 113. А. Г. Касаткин, Основные процессы и ап-
аппараты химической технологии, Госхимиздат, 1960. —114. А. Н. Плановский,
В. М. Р а м м, С. 3. Коган, Процессы и аппараты химической технологии, Изд.
«Химия», 1968. — 115. В. Ф. А ее мус, Труды ЦНИСЛ, сб. 1 (в. 6—8), 1941,
стр. 49. — 116. Кода Набуо, Plant a. Progress., 7, № 12, 23 A965).—
117. Е. М. Ковалев, В. С. Фокин, И. М. Коваль, Хим. и нефт. маш., № 10,
19 A965). — 118. Швейц. пат. 289976, 1953. — 119. Н. Domning, швед, пат.,
192439, I960. — 120. Chem. Age, 82, № 2104, 648 A959).
121. Кониси Такэо, Bull. Soc. Sea. Water Sci. Japan, 19, № 3 178
A965). —122. Голланд. пат. 88978, 1958. —123. Англ. пат. 822893, 1959.—
124. К. S u g i m о t о, Т. J a w a t а у а, япон. пат. 1266, 1954. — 125. С. С. Ш р а й б-
ман, Хим. пром., № 4, 17 A947). — 126. А. И. Кунина, Т. М. Пелен, сб.
«Коррозия и борьба с ней», № 12, Машгиз, 1952, стр. 105. — 127. Симидзу
Сатко, Chem. Techn. a. Eng., 3, № 2, 28 A959). — 128. Я. М. Хейфец, ЖПХ,
34, № 3, 473 A961). —129. П. Г. Романков, Н. Б. Рашковская, Сушка во
взвешенном состоянии, Изд. «Химия», 1968. — ISO. Food Eng., 27, № 7, 102 A955).
131. J. A. Lee, Chem. Eng, 58, № 1, 102 A951). — 132. С W. Jobes, Chem.
Eng., 61, № 1, 165 A954). —133. Ю. Я. Каганович, Хим. наука и пром., 2,
№ 6, 764 A957). — 134. П. Ф. Славнова, Соц. строительство, № 1, 42 A941,
Якутск). —135. R. H. Buchanan, Chem. Process (Australia), 13, № 4, 49
(I960). —136. J. Coal Res. Inst., Japan, 7, № 1, 7 A956). —137. L. Nisolle,
пат. ФРГ 899937, 1953. — 138. A. M. Жирмунский, Б. Я. Розен, Сообщение
Дальнезосточного филиала АН СССР, в. 6, 1954, стр. 5. — 139. В. Г. Яроцкий,
А. И. Науменко, Р. М. Самельзон, Труды ВНИИСП, в. 9 A7), 1965,
стр. 162. — 140. Англ. пат. 1091723, 1965.
141. R. Т. Thampy, N. R. Kuloor, D. N. Daruvalla, Chem. Age (In-
(India), 5, №¦ 3, 99 A954).— 142. А. И. Зиновьев, Труды ВСНИИ, № 1 (9),
1954, стр. 39. — 143. J. Tanoviceanu, Rev. igienS mlcrobiol. si epidemiol., № 2,
37 A954). — 144. W. H. En gels, канад. пат. 501309, 1954. —145. Л. Е. Олиф-
c о н, В. И. 3 а к, Л. Ф. Михайлова, авт. свид. 198353, 1966. — 146. С. P. S h а г-
da, D N Daruvalla, Chem. Age (India), 6, № 1, 91 A955).—147. Франц.
пат. 1043831, 1953. —148. Австрал. пат. 158218, 1954. —149. А. И. Зиновьев,
Труды ВСНИИ, № ] (9), 1954, стр. 66. — 150. М. М. Р а т н е р, Н. Г. П а х м у р а,
Труды Украинск. н.-и. ин-та соляной пром., в. 2 A0), 1959, стр. 78.
151. Н. Е. S ills, Chem. Process (USA), 29, № 7, ПО A966).— 152. Д. С. На-
Наде ж д и н, М. В. Гончарова, М. Е. Купличенко, Труды Украинск. н.-и.
ин-та соляной пром., в. 2 A0), 1959, стр 72. —153. В И. Николаев,
Б. И. Степанов, Труды н.-и. станции A935—1940), Изд. Местпрома, Калм.
АССР, Кануково, 1940, стр. 100. — 154. Е. И. Ахумов, ЖПХ, 29, № 4, 569
A956). —155. А. И. Данюшевская, Сообщение о научно-исследовательских
работах членов Приморского отд. ВХО им. Д. И. Менделеева, в. 3, 1957,
стр. 89. —156. В. П. Ильинский, Труды Дальневосточного филиала АН
СССР. Сер. хим., в. 2, 1956, стр. 5. — 157. J. N a k а п о, япон. пат. 2277, 1953.—
158. И. В. Кизеветтер, Труды Дальневосточного филиала АН СССР. Сер.
хим., в. 2, 1956, стр. 83. — IBs. Ayai Masao, Furusama Masoru Bull
Soc. Salt Sci. Japan, 12, № 1, 3 A9Б8). —160. T. Sakaguchi, япон. пат 2278
1953.
161. О. С. Ленчевский, Журн. ВХО, 5, № 6, 609 (I960). —162. Hud-
Hudson Т., Irvine, Chem. a. Process Eng., 42, № 9, 387 A961). — 163. Chem Eng.
News, 37, № 2, 28 A959). — 164. R. С Himes a. oth., Ind. Eng. Chem., 51, № 11,
Литература
97
1345 A959). —165 S Kiesskalt, Chem. Ingr. Techn., 38, № 2, 108, A105
A966).—166. Chem. Eng, № 12, 152 (I960).—167. К- С Himes, S. E. Mil-
Miller W H Mink, H. L. Qolring, Ind. Eng. Chem, 51, № 11, 1345 A959).—
168 E R Gi Hi land, W. K. Lewis, пат. США 3137Б54, 1956. —169. Brit.
Chem. Eng, 9, № 12, 829 A964).— 170. A. J. Weinberger, D. F. De Lapp,
Chem. Eng. Progr, 60, № 11, 56 A964).
171 В П М е л е ш к о, Н. С. А н п и л о в а, М. Н. Р о м а н о в, О. В. Чер-
в и н с к а я, ЖПХ, 33, № 11, 2481 A960). — 172. В, П. Ильинский, Н. А. В а-
рыпаев К Э. Гиттерман, Е. Шмидт, Труды Соляной лаборатории АН
СССР в. 7, 1936. —173. И. Н. Гладкий, И. Г. Кальнина, Труды ВНИИСП,
в 9 A7) 145 A965). —174. G. G. Turner, англ. пат. 1009736, 1963.—
175 И. Н. Гладкий, О. К. Журид, И. Г. Кальнина, Труды ВНИИСП,
в 9 A7) 149 A965). — 176. В. Ф. Ассмус, Э. А. Гуревич, Труды ЦНИСЛ,
сб 1 (в 6—8) 1941, стр. 281. —177. А. Г. Злобинский, М. Б. Позина,
Труды ВНИИГ, в. 49, 55 A966). — 178. Н. Н i r se, G. Sch u Iz, Freiberger Fors-
chungsh. A, № 354, 29 A965). — 179. Пат. ФРГ 875631, 1953. —180. В. В. Вязо-
в о в, Г. Н. Попов, сб. «Технология переработки природных солей и рассолов»,
Изд.' «Химия», 1964, стр. 79.
181. В. В. Вяз о вов, Г. Н. Попов, Там же, стр. 91. — 182. В. В. Вязо-
в о в, Г. Н. Попов, Там же, стр. 97. —183. Г. Н. Попов, Хим. пром., № 8,
600 A965) —184 М И. М аз ель, Н. С. Иванова, сб. «Калийные соли и
методы их переработки», Изд. ИОНХ АН БССР, 1963, стр. 60. — 185. Г. Н. П о-
п о в, Т. Н. Полторацкая, сб. «Вопросы технологии переработки галургиче-
ского сырья» Изд. «Химия». 1967, стр. 3. — 186. К- С. Бессмертный и др.,
Хим пром., № 4, 306 A966). — 187. Chem. Process. (USA), 30, № 9, 117 П967).—
188. Ц. Масудзава, Bull. Soc. Sea Water Sci. Japan, 20, № 3, 122 A966).—
189. Хкачаку, Chemistry (Japan), 14, № 1, 46 A959). — 190. Ф. А. Петрач-
к о в, Л. Д. П а н ч е н к о, А. С. С а ц ю к, Труды Украинск. н.-и. ин-та соляной
пром., в. 2 A0), 1959, стр. 62.
191. Е. G. С о о к е, англ. пат. 978712, 1962. — 192. Н. W. Diamon d, канад.
пат 504154 1954. —193. Швед. пат. 155225, 1956. — 194. Англ. пат. 735014, 1955.—
195 Пат. США 2768В98. 1956.— 196. В. Е. В а а г so n, D. Т. Oh I sen, пат. США
3*86828, 1962.—197. L. CreangS, E. Serb an, Rev. chim. (Рум.), 12, № 4,
202 A961). —198. L. Phoenix, Brit. Chem. Eng, 11, № 1, 34, 2a A966).—
199 J D Bicrhall, англ. пат. 1009196,-1963; M. P. Bhatt, D. S. D a t a r,
Salt Res. a. Ind., 5, 3—4, 57 A968). — 200. W. K. Gil key, A. C. Shy man,
пат. США 2655438, 1953.
201. Л. Б. Т о к а р с к и й, О. П. К у н и ц к а я, Т. Е. Л и ф ш и ц, Труды
ВНИИСП, в. 9 A7), 1965 стр. 157. —202. R. S. PIoss, пат. США, 3148023,
1963. — 203. Наотаке Сато, япон. пат. 2228, 1960. — 204. J. Sci. Ind. Res,
А15, № 12 539 A956). — 205. J. B. Rust, L. Spialter, пат. США 2869166,
1954. — 206. R. R. Oliver, Philippine J. Sci, 83, № 3, 245 A954). — 207. J. S u-
gi, T Ogawa, япон. пат. 2950, 1954. — 208. Инд. пат. 45907, 1953. — 209. Тоя-
ма, Bull. Soc. Salt Sci. Japan, 9, № 3, 124 A955). — 210. С. М. Hopper,
R. R. В e v e n, каиад. пат. 499538, 1954.
211. П. А. Кулле, В. Ф. Ассмус, Труды ВНИИГ, в. 24, 1952, стр. 278.
М. В. Позия
Глава IV
ПРИРОДНЫЙ СУЛЬФАТ НАТРИЯ*
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Сульфат натрия Na2SO4 встречается в природе в составе мно-
многих минералов: тенардита Na2SO4, мирабилита Na2SO4-10Н2О,
астраханита Na2SO4-MgSO4'4H2O, глауберита Na2SO4 • CaSO4,
глазерита Na2SO4-3K2SO4, беркеита 2Na2SO4- Na2CO3 и др.
Тенардит в обычных условиях представляет собой бесцветные
ромбические кристаллы с плотностью 2,68 г/см3. При нагревании
претерпевает ряд полиморфных превращений, при 890° плавится.
Природный тенардит и тенардит, полученный из горячего насыщен-
насыщенного раствора и высушенный при 110°, имеет структуру, отличную
от переплавленного тенардита. Для последнего характерны следую-
следующие фазовые переходы:
<z-Na2SO4
18о-гоо°
P-Na2SO4
240°
Y-Na2SO4
570-600°
6-Na2SO4
* При нагревании тенардита (Г), выделившегося из водных рас-
растворов, происходят следующие превращения:
570-600°
240°
Y-Na2SO4
6-Na2SO4
а при охлаждении:
Y-Na2SO4 ниже 240°» p-Na2SO4 ииже l80°
» <z-Na2SO4
Переход a-Na2SO4 в тенардит происходит при обычных усло-
условиях медленно в присутствии влаги. Полное обезвоживание сопро-
сопровождается переходом в v-Na2SO4 !~3. Со многими солями (L12SO4,
K2SO4, Na2CO3 и др.) Na2SO4 образует изоморфные твердые рас-
гворы.
• Производство сульфата натрия из поваренной соли см. в гл. XI.
Физико-химические свойства
99
Из водных растворов при температурах от 32,384 до 233° кри-
кристаллизуется безводный сульфат натрия в ромбической системе,
выше 233° — в моноклинной. Ниже 32,384° выделяются прозрачные
моноклинные кристаллы NagSCv 10Н2О— глауберовой соли или
мирабилита (от латинского слова mirabilis — удивительный). Плот-
Плотность мирабилита 1,464—1,481 г/см3. Существует также метаста-
бильный семиводный кристал-
кристаллогидрат Na2SO4 • 7Н2О.
При 32,384° мирабилит ин-
конгруэнтно плавится — раз-
разлагается на безводный сульфат
натрия и его насыщенный рас-
раствор. Растворимость Na2SO4 в
воде (рис. 21 и табл. 10) при
повышении температуры от
32,384 до ~ 120° уменьшается,
затем возрастает, а выше 233°
резко убывает, приближаясь к
й
350
300
250
200
150
100
S0
i0
4
Лед^
-w
— ti©
Naj
мои
N
1
SO4
ОКЛ.
\233'
1
Na2SOt
ромб
v
нулю при критической темпе-
температуре воды C65°) 4-6. Рас-
,—'
/
/
32fi°
/
1
10
20
3D 40
Na2SO4,%
Рис. 21. Диаграмма растворимости
в системе Na2SO4—H2O.
0 5 10 15 20 25 30 35
Температура, °С
Рис. 22. Давление дис-
диссоциации Na2SO4 • 10Н2О.
творение безводного сульфата натрия в воде сопровождается вы-
выделением тепла вследствие гидратации. Растворение мирабилита
происходит с поглощением тепла, затрачиваемого на разрушение
гидратных связей7.
На рис. 22 показана кривая давлений водяного пара над мира-
мирабилитом. Если влажность окружающего воздуха меньше давле-
давления диссоциации, кристаллы мирабилита выветриваются — покры-
покрываются слоем безводного сульфата. Давление пара насыщенных
растворов Na2SO4 при высоких температурах:
t, °C 200 250 300 350. 362
Р, кгс/см2 14,3 38 85 167 195
4*
100
Гл. IV. Природный сульфат натрия
Физико-химические свойства
101
Растворимость сульфата натрия в воде
Темпе-
Температура,
°С
-0,6
-1,2
0
10
15
20
2Б
30
32,4
Концентрация
насыщенного
раствора,
вес. %
1,96
3,85
4,5
8,2
11,7
16,1
21,9
28,8
33,2
Твердая фаза
Лед
Лед + Ш28О4-10НгО
Na2SO4-10HgO
Na2SO4 • 10Н8О +
+ Na2SO4 (ромб.)
Темпе-
Температура.
40
59
70
100
120
140
233
280
Концентрация
насыщенного
раствора,
вес. %
32,5
31,9
80,5
29,9
29,5
29,6
32.0
25,3
Твердая фаза
Na8SO4 (ромб.)
Na2SO4 (ромб.) +
+Na2SO4(MOHOpui.)
Na2SO4 (монокл.)
Растворимость сульфата натрия в насыщенных водных раство-
растворах NaCl с повышением температуры непрерывно возрастает вплоть
до температуры плавления
безводной эвтектики NaCl—
Na2SO4 (рис. 23). При этом
максимум давления пара эвто-
нических растворов тройной
NaCl
NagSO4
200
нго
Рис. 23. Политерма растворимости
в системе Na2SO4—NaCl—H8O при
температурах от 100 до 700°.
4ое boo
VC
Рис. 24. Кривые зависимости давле-
давления водяного пара от температуры
растворов, насыщенных Na2SO4, NaCl
и смесью солей (эвтоника).
системы Na2SO4—NaCl—Н2О B28—230 кгс/см2) намного (на 170—
175 кгс/см2) ниже максимума давления пара насыщенных раство-
растворов системы NaCl—НгО (рис. 24) 5.
На рис. 25 приведены изотермы растворимости в системе
Na2SO4—NaCl—Н2О. При 25° эвтонический раствор, равновесный
с Na2SO4 и NaCl, содержит 6,85% Na2SO4 и 23,14% NaCl, а в рав-
равновесии с Na2SO4 и Na2SO4 •
• 10Н2О находится раствор,
содержащий 15,61% Na2SO4
и 14,4% NaCl.
Растворимость мираби-
мирабилита в системе Na2SO4—
—NaOH—H2O при 25°
имеет минимум (рис. 26) и
затем плавный подъем к
точке превращения Na2SO4-
• 10Н2О в Na2SO4, соответ-
соответствующей содержанию в
растворе 7,73% NaOH. При
более высоких температу-
температурах, вплоть до 350°, в твер-
твердую фазу выделяется без-
безводный сульфат натрия.
Растворимость Na2SO4 при
25° при увеличении концен-
20
NaCl, бес. %
Рис. 25. Растворимость в системе
Na2SO4—NaCl—Н2О при температурах от 0
до 100°:
Г — галит; М — мирабилит; Т — тенардит.
трации NaOH от 0 до 48%
уменьшается от 21,8 до
0,45%. В системе Na2SO4—
—NaCl—NaOH—H2O рас-
растворимость Na2SO4 при 25° в том же диапазоне концентраций
NaOH (от 0 до 48%) уменьшается от 6,98 до 0,03%. При этом
растворимость NaCl в этих
растворах снижается с 22,86
до 0,95%. В системе Na2SO4—
—NaOH—Н2О в диапазоне
концентраций от 47,5 до 63,5%
NaOH при 70—200° кристалли-
кристаллизуется 3Na2SO4 • 2NaOH8. В си-
системе Na2SO4—NaOH образуют-
образуются инконгруэнтно плавящиеся
соединения вероятного состава
3Na2SO4 • 2NaOH и Na2SO4 •
• 2NaOH. Первое существует
ниже 485° и при 418° имеет по-
полиморфное превращение, вто-
второе существует ниже 316°9-10.
В растворах сульфата натрия растворяется значительное коли-
количество сульфата магния при всех температурах, вплоть до 300е.
Нагреванием этих растворов нельзя отделить Na2SO4 от MgSO4 n< lf.
4NazSO410H2O
л
\
5 Ю 1
№
2SO4
—
5 К 25 30 35 40 45 50
NaOH,%
Рис. 26. Растворимость в системе
Na2SO4—NaOH—H2O при 25°.
102
Гл. IV. Природный сульфат натрия
Сырье
103
ПРИМЕНЕНИЕ
Развитие добычи природного сульфата натрия в России в срав-
сравнительно крупных масштабах, по-видимому, относится к началу
второй половины XVIII в. и связано с использованием при варке
стекла найденной близ Барнаула глауберовой соли 13. И в настоя-
настоящее время сульфат натрия используется в стекольной промышлен-
промышленности. Наибольшие же его количества расходует целлюлозно-бу-
целлюлозно-бумажная промышленность (например, в США ~70% от общего по-
потребления сульфата натрия). Его применяют для производства
моющих средств, а также в цветной металлургии, текстильной,
кожевенной и других отраслях промышленности. Кроме того, в
медицине и ветеринарии также используют глауберову соль.
В химической промышленности сульфат натрия служит сырьем
при выработке сернистого натрия, ультрамарина, силиката натрия
и др. и. Разработаны способы переработки сульфата натрия на
серную кислоту, серу, кальцинированную и каустическую соду,
сульфат аммония и другие продукты 1Б~20. Однако в промышлен-
промышленности эти способы пока не используются, так как получение пере-
перечисленных продуктов из других видов сырья экономически вы-
выгоднее.
Основными производителями сульфата натрия являются СССР.
США, Япония, ФРГ, Канада и Франция21. В СССР, США и Канаде
значительные количества сульфата натрия получают из природных
месторождений, в США, в частности, — при комплексной перера-
переработке рапы оз. Сирлз. Сульфат натрия вырабатывают также в хи-
химической промышленности как побочный продукт многих произ-
производств. В США наибольшее количество «химического» сульфата
натрия получают из отбросных растворов производства вискозного
волокна и целлофана. Значительные количества его производят
вместе с соляной кислотой из поваренной соли (см. гл. XI), осо-
особенно в странах, не располагающих природными ресурсами суль-
сульфата натрия. Его получают как побочный продукт при производстве
бихромата натрия, фенола, борной кислоты, карбоната лития, ре-
резорцина, некоторых пигментов и проч.
Несмотря на то, что многие потребители используют сульфат
натрия в виде растворов, товарным продуктом является безводная
соль. Добываемый из природных источников мирабилит содержит
около 50% воды (в чистом Na2SO4-l0H2O 55,9% кристаллизацион-
кристаллизационной воды); кроме того, хотя в отличие от тенардита мирабилит
практически не гигроскопичен 22, но он плавится при невысокой тем-
температуре и из-за этого легко слеживается в монолитную массу.
Поэтому, перевозка мирабилита на большие расстояния нерента-
нерентабельна, и его подвергают обезвоживанию в естественных или в за-
заводских условиях вблизи от места добычи. В связи с этим наиболь-
наибольший интерес представляет непосредственная добыча безводной со-
соли — тенардита; его залежи, однако, сравнительно редки, глав-
главным источником природного сульфата натрия является мирабилит.
Состав и количество примесей в сульфате натрия зависят от
состава рапы, из которой образовались мирабилит или тенардит,
от условий их кристаллизации, добычи и хранения (табл. 11).
ТАБЛИЦА 11
Требования к химическому составу сульфата натрия из мирабилита
(ГОСТ 6318—68)
[Приведенные нормы (в %). за исключением влаги,
относятся к сухому веществу]
Сульфат натрия, не менее
Не растворимый в воде остаток, не более
Хлориды в пересчете на NaCl, ие более .
Сульфат кальция, не более
Железо (Fe2O3), не более
Влага, не более
Высший
сорт
99,3
0,5
0,2
0,05
0,01
0,5
I сорт
97.5
1.5
1,0
0.5
0,01
3
11 сорт
940
4.5
2,0
1.0
0,03
7
Тенардит, добываемый из донных отложений озер, должен
иметь влажность не более 15% и содержать в сухом веществе не
менее 92% Na2SO4 и не более 5% NaCl, 0,5% CaSO4) 2,5% не
растворимых в воде веществ и 0,03% окислов железа. Тенардит в
кусках разной величины перевозят навалом.
Согласно ГОСТ 5.1135—71, кристаллический сульфат натрия,
получаемый в качестве отхода в производстве вискозного шелка
(в результате взаимодействия серной кислоты с едким натром и
натриевыми солями), должен содержать не менее 99,8% Na2SO4
и не более 0,01%) хлора, 0,01% Са и Mg в пересчете на CaSO4,
0,01% железа в пересчете на Fe2(SO4K, 0,1%) ZnSO4, 0,02% нера-
нерастворимого в кислой среде остатка, 0,2% веществ, теряемых при
прокаливании, и 0,04%) воды.
Сульфат натрия перевозят навалом в полиэтиленовых мешках.
Представляет интерес холодное брикетирование сульфата натрия
с получением брикетов, пригодных к перевозке без тары. Сульфат,
подвергаемый брикетированию, должен содержать 4—5% воды.
Этого можно достигнуть смешением 93—91 вес. ч. сухого суль-
сульфата натрия с 7—9 вес. ч. мирабилита. Расходы на брикетирова-
брикетирование сульфата и последующее его измельчение с помощью дезин-
дезинтегратора не превышают стоимости бумажных мешков23-24.
СЫРЬЕ
По' морфологическому строению месторождения сульфата разт
деляются25> 26 на ископаемые тенардито-мирабилитовые и озер-
озерные; последние представляют собой периодические временные ново-
новосадки мирабилита (или тенардита) и постоянные донные корневые
Сырье
105
104
Гл. IV. Природный сульфат натрия
отложения мирабилита, тенардита, астраханита и глауберита в
виде пластов и линз под рассолами или илами озер.
Большинства месторождений сульфата натрия состоит из соче-
сочетания разных сульфатных минералов с примесью галита, гипса,
эпсомита и др.
Тенардит залегает в виде пластов, линз и желваков в донных
озерных месторождениях26. В сульфатных озерах, рапа которых
насыщена хлористым натрием, но содержит достаточное количе-
количество SO2", ежегодно осенью и зимой при температурах около 0е и
ниже выпадает мирабилит. В озерах с высококонцентрированной
рапой садка мирабилита иногда наблюдается и летом в прохлад-
прохладную погоду, при понижении температуры рапы ниже 18е. При по-
повышении температуры выпавший мирабилит вновь растворяется.
Летом же из рапы озер с весьма высокой концентрацией SO-i" про-
происходит садка тенардита (например, из рапы оз. Балхаш26). Озер-
Озерные рассолы морского происхождения A класса) представляют
практический интерес с точки зрения возможности извлечения из
них сульфата натрия, если коэффициент метаморфизации
MgSC>4 : MgCl2 больше единицы. В некоторых озерах слой оседаю-
оседающего мирабилита достигает 0,5 м и более при выходе, превышаю-
превышающем 100 кг Na2SO4 • 10Н2О из 1 м3 рассола.
В сульфатных озерах степей Казахстана и Кулунды ежегодно
в осенне-зимние месяцы выпадают миллионы тонн новосадки ми-
мирабилита, который частично садится на дно, частично же выбрасы-
выбрасывается волнами на берег. Если создаются условия, при которых не
весь мирабилит вновь растворяется в летнее время, происходит
накопление донных отложений с образованием перекристаллизо-
перекристаллизовавшегося пласта мирабилита «стеклеца» — плотного слоя с глад-
гладкой поверхностью и малой пористостью27. Встречаются месторож-
месторождения мирабилита, сцементированные песками или переслаиваю-
переслаивающиеся иловыми отложениями, которые образовались в результате
высыхания озер.
Астраханит встречается в виде донных корневых отложений в
ряде сухих озер и водоемов повышенной солености. Например,
средняя мощность пластов астраханита приаральских соляных озер
(Джаксы-Клыч, Восточного и др.) составляет 2 м2&. Встречаются
также линзы и прослойки астраханита в залежах ископаемых со-
солей. Залежи астраханита в СССР исчисляются многими десятками
миллионов тонн291.
Глауберит образует пласты в месторождениях ископаемых со-
солей и содержится в донных отложениях некоторых солевых озер.
Значительные месторождения глауберита находятся в солевых по-
породах Тянь-Шаня, в Киргизии. При взаимодействии глауберита с
водой он разлагается и сульфат натрия переходит в раствор, а
сульфат кальция остается в нерастворенном остатке324. Разрабо-
Разработан гидротермический способ получения сульфата натрия и цемен-
цемента из глауберита. При нагревании до 750—800° в присутствии во-
водяного пара глауберитовой породы, содержащей примеси галита,
кремнезема, глины и другие, осуществляется реакция35-36:
Na2SO4 • CaSO4 + 2NaCl + SiO2 + H2O = 2Na2SO4 + CaSiO3 + 2HC1
Важное значение имеют месторождения сульфатных рассолов,
пропитывающих погребенные солевые отложения. Встречаются
сульфатные рассолы карстового происхождения, образовавшиеся в
результате выщелачивания поверхностными и подземными водами
погребенных солевых пород.
Наша страна располагает богатейшими в мире месторожде-
месторождениями природного сульфата натрия в виде твердых отложений и
рапы многочисленных озер в Сибири, Казахстане и Средней Азии37.
Важнейшими месторождениями являются залив Кара-Богаз Кас-
Каспийского моря, оз. Кучук в Алтайском крае и озера Аральской
группы. Значительные количества сульфата натрия содержатся в
рапе озер Анж-Булат, Эбейты, Тениз и других, а также в ископае-
ископаемых глауберитовых породах Тянь-Шаня (Когкорка, Кетмень-Тюбе,
Джелды-Су). Практически не исчерпаемыми источниками его яв-
являются воды Каспийского38, Аральского морей и оз. Балхаш. Со-
Солевой состав этих водоемов, не имеющих связи с океаном, сильно
зависит от состава речных вод; последние же богаче сульфат-ионом,
чем океанская вода. Поэтому Каспийское и Аральское моря и
оз. Балхаш, а также генетически связанные с ними многочислен-
многочисленные озера этого обширного района, содержит значительно больше
SO4~, чем моря и озера, связанные с океаном (например, Черное
море, Крьщские озера).
Залив Кара-Богаз («Черная пасть»), расположенный на восточ-
восточном побережье Каспийского моря в районе с пустынным, засушли-
засушливым климатом, является величайшим в мире месторождением суль-
сульфатных солей. Залив, соединяющийся с морем узким проливом,
при небольшой глубине (несколько метров) имеет поверхность
18346 км2. Это громадный испарительный бассейн, в котором под
влиянием солнечного тепла в летние месяцы испаряется большое
количество воды. В результате этого уровень воды в заливе ниже,
чем в море, и происходит непрерывное пополнение залива морской
водой и накопление в нем солей. Концентрация солей в воде за-
залива в 20 раз больше, чем в море. Основными ионами являются
Na+, Mg2+. SO4~ и СГ. Запасы сульфата натрия в заливе огромны.
Они исчисляются миллиардами тонн.
На комбинате «Карабогазсульфат» получали сульфат натрия из
рапы з*алива бассейным способом путем выделения мирабилита при
зимнем охлаждении рапы и последующего естественного обез-
обезвоживания его летом 39~42. Однако уровень Каспийского моря ци-
циклически изменяется. Вследствие его понижения, начавшегося
106
Гл. IV. Природный сульфат натрия
Сырье
107
б двадцатые годы XX в., приток морской воды в залив уменьшает-
уменьшается, а концентрация рапы в заливе увеличивается (рис. 27). Так,
с 1921 до 1961 г. приток воды уменьшился с 25 до 10,5 км3/год и
глубина залива снизилась с 10 до 3,5 ж436. (Площадь высохшей
поверхности залива сейчас превышает 2000 км2). Это привело к
тому, что уже в 1939 г. рапа насытилась хлоридом натрия, а затем
сульфатом магния (эпсомитом MgSO4 • 7Н2О) и астраханитом
(Na2SO4»MgSO4-4H2O), которые и
стали осаждаться. Вследствие та-
такого обессульфачивания рапы вы-
выход мирабилита резко снизился
(как видно из рис. 27 максималь-
максимальный выход мирабилита соответ-
соответствует притоку воды в залив, рав-
равному 13—14 км3/год). Это обусло-
обусловило необходимость изменить спо-
>§ соб получения мирабилита: рапу
50 §¦ залива, направляемую в бассейны
для зимней садки, стали разбавлять
морской водой. Затем и этот способ
производства пришлось оставить,
так как вследствие продолжающе-
продолжающегося понижения уровня рапа ото-
отошла на слишком большое расстоя-
расстояние от места производства и состав
25 20 15 Ю
Приток Воды. км3/го3
Рис. 27. Изменение концентрации
NaCl и MgCl2 в рапе Карабогаз-
пийской воды.
ского залива'(сплошные'линии), ее, даже при разбавлении морской
и выхода мирабилита из 1 мь водой, стал непригоден для садки
рапы при ее охлаждении до 0° ЧИСТОГо мирабилита47-52.
E^ Повышение концентрации рапы
привело к образованию твердых со-
солевых отложений «смешанной со-
соли»— смеси галита, астраханита, эпсомита, а также гипса и глау-
глауберита, а понижение уровня рапы обнажило значительную часть
этих отложений. Разведкой дна залива установлено существова-
существование четырех солевых пластов, в том числе трех погребенных,
свидетельствующих о том, что в прошлом в заливе происходили
гидрохимические изменения, аналогичные современным. Солевые
пласты пропитаны межкристальными рассолами. Межкристальные
рассолы, запасы которых весьма велики, представляют собой
ценный источник для промышленного получения сульфата нат-
натрия 53-Б4.
Первый рассольный горизонт пропитан рапой, состав которой
близок к составу поверхностной рапы залива. Второй рассольный
горизонт расположен во втором, погребенном соляном пласте и
гипсовых песках, слагающих его кровлю. Этот пласт отделен от
первого и третьего солевых пластов практически нефильтрующими
прослойками илов. Он сложен в основном галитом и глауберитом и
обладает большой пористостью B2—34%). Мощность этого пласта
в среднем составляет 10 м при глубине залегания 5—5,5 м. Из вто-
второго рассольного горизонта в. настоящее время производят откачку
рассолов для получения сульфата натрия. Эти рассолы не являются
концентратами морской воды, а образовались в результате раство-
растворения твердых отложений глауберита и галита. Рассолы обладают
напором — их пьезометрический уровень близок к дневной поверх-
поверхности В среднем концентрация ионов (в вес. %) рассолов второго
горизонта колеблется в следующих пределах:
СГ 12,5-15
SO^~ . 2,5-8
HCOJ ...... .0,04-0,11
Na 5-8,5
Mg2+ . . 1,5-3,9
Кроме того, в них находятся (в мг/л):
К+ 2500 Вг"
В2О3
.2500
.380
350
1-10
Эти рассолы в отличие от поверхностной рапы содержат до
300 мг\л сероводорода. Их температура 15—17°.
Третий солевой пласт и пропитывающие его рассолы несколько
отличаются по минералогическому и химическому составам. В со-
состав минералов, слагающих этот пласт, кроме галита и глауберита,
входит астраханит. Пористость пласта ~ 15—25%, мощность в
среднем 3,8 м, залегание на глубине 18 м. Четвертый солевой пласт
изучен пока недостаточно.
Оз. Кучук 55~58 — крупнейшее сульфатное озеро, расположен-
расположенное в наиболее низкой части Кулундинской степи (Западная Си-
Сибирь). Площадь озера около 175 км2, средняя глубина 1,62 м. Сред-
Средние составы рапы (в вес. %):
Na2SO4 NaCl MgCfe
Зимой 0,6 16,4 5,0
Летом 3,9 16,4 4,7
Около 80% площади дна озера покрыто пластом слежавшегося
мирабилита-стеклеца толщиной 2,5—3 м. Количество его исчис-
исчисляется сотнями миллионов тонн69-60, но экономичные методы до-
добычи донного мирабилита пока отсутствуют. В засушливые годы
концентрация солей в рапе повышается настолько, что она насы-
насыщается Na2SO4 и NaCl уже при +5° и ниже этой температуры про-
происходит совместная садка мирабилита и галита.
В Приаральском районе расположено Джаксы-Клычское суль-
сульфатное месторождение, представленное группой сухих озер —
«сульфатников», образовавшихся в результате испарения водьг-
Аральского моря37-614.
108
Гл. IV. Природный сульфат натрия
В оз. Анж-Булат (Павлодарская обл.) в засушливые периоды
образуется донный пласт тенардита. Это вторичный процесс, за-
заключающийся в обезвоживании мирабилита под слоем концентри-
концентрированной рапы65. В оз. Эбейты (Омская обл.N6~69 садка мираби-
мирабилита происходит в начале осени при охлаждении рапы ниже 15—20°.
К зиме слой новосадки в центре озера достигает 20—30 см.
В качестве примера приводим средний состав (в %) рапы и
пластовой соли оз. Эбейты:
Состав рапы
Na2SO4 11,64
NaCl 13,27
MgCl2 2,39
КС1 0,04
KB - 0,032
Се(НСО8J 0,063
Состав мирабилита в пласте
Na2SO4 • IOHjjO 95,69
Na2SO4 2,00
NaCl 0,25
K2SO4 0,013
CaSO4-2H2O 0,39.
Нерастворимые примеси . . . 1,66
Состав тенардита из донных отложений сульфатных озер севе-
северо-восточного побережья Каспийского моря (в %):
Na2SO4 96-98,5 CaSO4 0,3-1,0
NaCl 0,4-1,2 Са(НСО3J 0,4-0,6
MgSO4 .' 0,2—0,5 Нерастворимые примеси . . 0,7—1,5
Следует также упомянуть тенардитовое месторождение Узун-Су,
Ходжентские соляные месторождения, сульфатные озера Гуз-Кане,
Муллалы, Денгиз-Куль, Куули и многие другие70-77.
Наиболее распространенным является бассейный метод зимней
кристаллизации мирабилита с последующим естественным или
искусственным обезвоживанием. Иногда вместо естественного зим-
зимнего вымораживания в бассейнах осуществляют заводское искус-
искусственное вымораживание мирабилита, например, на комбинате
«Карабогазсульфат» и на заводе Монэхенс в Техасе (СШАO9.
Твердые сульфатные породы разрабатывают горными методами.
При залегании пласта на большой глубине применяют шахтные
способы (например, в месторождении Узун-Су в ТуркмССР). В за-
зависимости от состава добываемой породы ее или высушивают на
воздухе, или перерабатывают заводскими методами в сульфат
натрия °"
80
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПОЛУЧЕНИЯ МИРАБИЛИТА
ИЗ ПРИРОДНЫХ РАССОЛОВ
Мирабилит можно получить из рассолов почти всех морей и
озер и тем с большим удельным выходом, чем больше коэффи-
коэффициент метаморфизации MgSO4: MgCl2. В табл. 12 приведено со-
содержание солей в воде океана, некоторых морей и соляных водое-
водоемов. Составы солей отдельных водоемов нанесены на диаграмму
растворимости в системе NaCl—MgSO4—Н2О (рис. 28)82-83. При
испарении в испарительных бассейнах воды океана /, Средизем-
Физико-химические основы получения мирабилита
109
ТАБЛИЦА 12
Состав воды океана, некоторых морей и соляных водоемов
Океан ....
Средиземное
море ....
Черное море .
Сиваш (южная
часть) . . .
Сакское озеро
Каспийское
море ....
Аральское море
Оз. Балхаш . .
Залив Кара-
Богаз (осень
. 1938 г.) ...
' Na2SO4.
Химический состав, вес. %
Са(НСО8J
0,017
0,016
0,027
0,020
0,028
0,011
0,02
0,08
CaSO4
0,12
0,13
0,068
0,38
0,31
0,09
0,164
0,09
MgSO4
MgCls
0,22
0,24
0,12
0,86
0,94
0,31
0,26
0,036
6,55
0,33
0,31
0,16
1,14
1,93
0,061
0,007
0,142*
8,05
MgBr2
KC1
NaCl
0,007
0,003
0,022
0,029
0,0008
0,07
0,06
0,04
0,27
0,36
0,013
0,018
2,80
2,84
1,45
9,61
7,76
0,81
0,56
0,143
18,74
сумма
солей
3,66
3,63
1,87
12,30
11,36
1,29
1,03
0,321
27,51
С12
0,67
0,77
0,75
0,75
0,49
5,1
37,2
1,82
ТАБЛИЦА 13
Выход мирабилита и расход воды и рассолов из разных водоемов 8!
Расход воды (в м3) для по-
получения 1 м3 раствора, на-
насыщенного NaCl:
без кристаллизации NaCl
. с кристаллизацией NaCl
Расход воды (в м3) для по-
получения 1 т мирабилита . .
Расход насыщенного рассола
(в м3) для получения 1 г
мипабилита
Выход (в кг):
мирабилита из 1 м3 насы-
насыщенного рассола ....
NaCl при получении 1 м3
насыщенного рассола .
NaCl на 1 7 мирабилита
Каспийское
море
29
182
6,28
160*
0
0
Аральское
море
45
196
4,85
230*
л
0
0
Оз.
Балхаш
125
256
2,05
490*
п
х
0
Черное
море
66
538
8,2
122,4 м
71П
1 IV
ff о
6,8
Сиваш
8Д5
69.6
8,2
122,4 ••
710
6,8
¦ При охлаждении рассола до 0°.
•» При охлаждении рассола до -10».
110
Гл. IV. Природный сульфат натрия
физико-химические основы получения мирабилита
111
ного 2, Черного 3 и Каспийского 6 морей, Сиваша 4, Сакского озе-
озера 5 и Карабогазского залива 9 образуются растворы, насыщенные
поваренной солью, которая при дальнейшем испарении и будет
кристаллизоваться в качестве первой твердой фазы. При концен-
концентрировании воды Аральского моря 7 получается раствор, насыщен-
насыщенный астраханитом, а воды оз. Балхаш 8 — тенардитом. Таким об-
2NaCl
25
SO
75
M8C12 6H2O
1DD
MgCl2
Рис. 28. Диаграмма растворимости в системе NaCl—MgSO4—Н2О:
при 25 (сплошные линии) н 0° (пунктирные линии).
разом, из всех перечисленных здесь вод в летний период можно по-
получить сульфат натрия только из воды оз. Балхаш. Выход тенар-
тенардита из 1 мъ балхашского рассола составляет 110—120 кг, а сте-
степень использования SC>4~ всего 60—55%; при зимней же садке ми-
мирабилита степень использования SO4 достигает 90—95%.
При охлаждении упомянутых насыщенных растворов до 0е все
они становятся насыщенными мирабилитом, который и выделяется
в твердую фазу. Выход сульфата натрия и расход начального и
насыщенного рассола на 1 т мирабилита зависят от исходной кон-
концентрации солей и SOl~ (табл. 13).
При охлаждении 1 м3 насыщенного NaCl рассола Сиваша до
—5е выделяется всего 32 кг мирабилита, что в 5 раз меньше вы-
выхода мирабилита при охлаждении до 0° насыщенного рассола Кас-
Каспийского моря. Малый выход мирабилита при охлаждении кон-
концентрированной воды Сиваша и Черного моря объясняется низкой
Na2SO4
2NaCl
Рис. 29. Диаграмма растворимости в системе
NaCl—MgSO4— Н2О при +2,6, 0 и —2,6°.
концентрацией SC>4~b этой воде. Для увеличения выхода мираби-
мирабилита из черноморских рассолов необходимо предварительно выде-
выделить из них часть поваренной соли путем более глубокого испаре-
испарения, а затем охлаждать их до более низкой температуры, напри-
например до —10е.
Выход мирабилита при охлаждении сульфатной рапы опреде-
определяется относительным и абсолютным содержанием в ней солей.
Оптимальное содержание суммы солей, соответствующее макси-
максимальному выходу мирабилита, не загрязненного галитом, может
112
Гл. IV. Природный сульфат натрия
быть определено графически по диаграмме растворимости в си-
системе NaCl—MgSO4—Н2О. На рис. 29 представлена такая диа-
диаграмма, составленная для карабогазской рапы83. Концентрации
SO4 и Mg выражены в индексах. На диаграмме нанесены изотермы
растворимости для +2,5, 0 и —2,5е и содержание воды в насыщен-
насыщенных растворах в молях на 100 моль солей (в верхней части диа-
диаграммы). Шесть лучей кристаллизации мирабилита, проведенных
из точки Л' до пересечения с линиями растворимости, соответ-
соответствуют следующим значениям хлормагниевого коэффициента, ха-
характеризующего весовое отношение ионов хлора и магния:
Луч NT NS NR NQ NP NO
—^ 4,0 4,2 4,4 4,6 4,3 5,0
- В качестве примера определим оптимальную концентрацию
рапы, характеризующейся коэффициентами:
Ing
= 4,0 и
р
¦' g 4
•= 2,0. Положение точки А состава рапы на диаграмме опре-
определяется индексами: SO4 — 30,8, Mg — 50,0. Проводя из точки А
вертикальную линию до пересечения в точке а с водной проекцией
луча кристаллизации,, находим оптимальное содержание воды в
рапе данного солевого состава, равное 1515 Л4/100 М солей. Прак-
Практически необходим небольшой избыток воды для предотвращения
загрязнения мирабилита галитом, так что содержание воды в рапе
перед ее охлаждением можно принять равным 1525 М/100 М со-
солей. Пересчитывая индексы на весовые количества, находим вес
100 М рапы и процентное содержание в ней солей:
SO2" . . . 30,8-96 = 2 957 г или 7,6%
С1~ . . . . 69,2-71 = 4 913 » » 12,7%
М^+ . . . 50-24,3"= 1215» » 3,1%
Na+ ... 50-46 — 2300» » 5,9%
Н2О. . . . 1525 - 18 ~ 27 450 » » 70,7%
38 835 г
100,0%
Следовательно, оптимальное и предельно допустимое содержа-
содержание суммы солей в данной рапе перед ее охлаждением для садки
мирабилита равно 29,3% •
Выход мирабилита Ъс к$) И8 1 м3 можно рассчитать по отно-
отношению длин отреэков AM : NM, пользуясь формулой
AM 100a-322d-10
NM
с -96
где 100 — индекс мирабилита по SO4; d— плотность охлаждаемой
рапы; 322 и 96 — мол. веса мирабилита и SO4; a — содержание
SO4~ в охлаждаемой рапе, %; с — индекс по SO4 охлаждаемвй
рапы. Для приведенного выше примера AM : NM = 0,142, с = 30,8,
Физико-химические основы получения мирабилита
113
а = 7,6, d = 1,25; после подстановки этих величин в формулу по-
получаем х = 150 кг/м3.
На рис. 30 показан выход мирабилита из рапы разного состава
при оптимальном содержании солей в зависимости от величины ко-
коэффициента метаморфизации рапы.
Изменение хлормагниевого коэффи-
коэффициента Cl": Mg2+ в пределах от 4
до 5 мало влияет на величину вы- 5
хода и показано на диаграмме за-
заштрихованной площадью в преде-
пределах «от» и «до»83.
При использовании рассолов с
большим содержанием MgSO4 их
разбавляют до охлаждения более
слабыми растворами или раство-
раствором поваренной соли84.
Рапа, остающаяся после кри-
кристаллизации мирабилита, содержит
большое количество MgCl2 и дру-
другие ценные компоненты и может
служить объектом дальнейшей пе-
переработки 47> 8Б~88.
Сгущенная хлормагниевая рапа
может служить материалом для по-
50 ЮО 1Щ .200
Выход мирабилита, кг/м3
уц р
состава при оптимальном содер-
содержании солей.
uivvv.1 ~..j.. Г „. рИС- 30. Выход мирабидита из
лучения хлористого магния (а из эксплуатационной рапы разного
него окиси магния и соляной кис- """"" """ «™»"о"•-""« mi">n-
лоты), брома, бора и других про-
продуктов. Рассмотрим в качестве при-
примера комплексную схему переработки Кучукской рапы. Эта рапа
после зимней садки мирабилита содержит, например (в %):
NaCl 16,52
MgCl2 4,78
MgSO« 0,69
CaSO4 ......... 0,08
Ca(HCO3J 0,066
KC1 0,029
KBr 0,045
B2O3 0,013
При вакуум-выпарке до 23% MgCl2 из рапы кристаллизуется
чистая поваренная соль, без примеси MgSO4 и CaSO4. Полученный
рассол содержит <~- 1,9 кг/м3 брома, по*сле отгонки которого из
него можно осадить хлористым кальцием полуводный гидравличе-
гидравлический гипс. При дальнейшей выпарке обессульфаченной рапы до
32% MgCl2 выделяется дополнительное количество NaCl. После
репульпации кристаллов NaCl, полученных в 1-й и 2-й стадиях
выпарки, с почти насыщенной NaCl рапой, поступающей на вы-
выпарку из рапохранилища, концентрация MgCl2 в межкристальной
жидкости снижается до 8—9%, а после центрифугирования в по-
поваренной соли остается меньше 0,6% MgCl^. При промывке ее на
114
Гл. IV. Природный сульфат натрия
Обезвоживание мирабилита
115
центрифуге водой получается соль высшего сорта, почти соответ-
соответствующая соли сорта экстра; она содержит всего по ~0,04% Mg
и Са. После отделения поваренной соли концентрированный рас-
раствор MgCl2 окончательно выпаривается до шестиводного, а затем
до двухводного хлористого магния (см. гл. IX). Последняя стадия
выпарки сопровождается частичным гидролизом MgCl2, и выде-
выделяющийся хлористый водород может быть поглощен известковым
молоком для получения хлористого кальция, частично используе-
используемого для обессульфачивания рапы.
Разработаны и другие схемы комплексного использования ма-
маточных рассолов морского типа после садки мирабилита 59. В част-
частности, путем их выпаривания при температуре кипения A26°) и
последующего охлаждения до 70° и разделения образующихся
твердых солей из них можно выделить шенит K2SO4 • MgSO4 • 6Н2О,
используемый в качестве удобрения (см. гл. V)87.
При испарении в лабораторных условиях маточных растворов,
остающихся после кристаллизации мирабилита из охлажденных
астраханитовых растворов, при 20—25° выделяется эпсомит не
только в стабильном поле его кристаллизации, но и в метаста-
бильной области, захватывающей часть поля астраханита. Напри-
Например, из 1 т астраханитового сырья оз. Куули можно получить 1 J
мирабилита и 0,3 т эпсомита S1.
Переработкой твердых отложений астраханита приаральских
озер путем водной обработки, охлаждения полученного смешан-
смешанного раствора с выделением мирабилита и с последующим его
плавлением, и выпарки маточных растворов с кристаллизацией
NaCl, можно получить из 1т астраханитовой породы 0,45 т без-
безводного сульфата натрия, 0,17 т поваренной соли и 0,18 г
MgCl2 • 6Н2О ».
ОБЕЗВОЖИВАНИЕ МИРАБИЛИТА
Наибольшее количество сульфата натрия получают обезвожи-
обезвоживанием мирабилита. При наличии ресурсов природного сульфата
натрия производство его из поваренной соли и серной кислоты
(см. гл. XI) является экономически менее целесообразным.
Различные методы обезвоживания дают продукт, сильно отли-
отличающийся по своим качествам. Например, при обезвоживании в
барабанных сушилках получается сильно комкующийся и слежи-
слеживающийся сульфат; при обезвоживании распылением — пушистый,
легкий материал с насыпным весом 450—500 кг/м3.
Обезвоживание мирабилита в естественных условиях
Выветривание мирабилита, т. е. удаление кристаллизационной
воды, происходит только, когда давление его диссоциации больше
парциального давления водяного пара в окружающей атмосфере.
Чем больше разность этих давлений, тем быстрее разлагается кри-
кристаллогидрат. На рис. 31 заштрихованная площадь соответствует
периоду выветривания, а расстояние между изохронными точками
дает понятие об относительной его интенсивности (которая зависит
и от силы ветра). Из этой диаграммы видно, что наибольшей ин-
интенсивности выветривание мирабилита в районе Кара-Богаза мо-
может достигать в июле и августе. В зимние месяцы влажность воз-
воздуха больше давления диссоциации, и находящийся на открытом
воздухе сульфат будет поглощать влагу. Это происходит иногда
и летом в ночное время, ко-
когда влажность воздуха зна-
значительно возрастает.
Естественное обезвожи-
обезвоживание мирабилита можно § g
осуществлять в штабелях | ^
или в бассейнах. В первом ^
случае мирабилит склады-
складывают на специальных пло-
площадках на берегу в штабе-
штабели высотой 30—40 см, по- „ „, v
вепхногть кптппых ттттятегть Рис- 3L ХаРактеРистика условии есте-
верхнос!ь которых^тщатель ственного обезвоживания мирабили«Ы-иа
I II Ш IV V VI VII VIIIIX X XI XII
Месяцы
но выравнивают. Обычно в промыслах
Течение Лета ПРОИСХОДИТ / — среднемесячные температуры воздуха в районе
карабогазскнх сульфатных промыслов; 2 —давле-
ПОЛНОе ВЫВетрИВаНие Мира- ние диссоциации мирабилита (построено по рис. 22);
биЛИТЗ НеДОСТатКОМ ЭТОГО **~среднемесячная абсолютная влажность воздуха.
метода является необходи-
необходимость больших площадей для штабелей мирабилита (на каждые
100 г сульфата натрия необходима площадь в 10 000 ж2). Выход
сульфата натрия с 1 м2 эксплуатируемой площади составляет
85—98 кг89-90. Этот метод в настоящее время уже не имеет про-
промышленного значения.
При бассейном методе обезвоживания 91>92 мирабилит сосредо-
сосредоточен в одном месте, благодаря чему возможна механизация про-
процесса.
По окончании зимней садки мирабилита из бассейна сбрасы-
сбрасывают (сливают) маточный раство.р с помощью системы дренаж-
дренажных устройств. Оставшийся в бассейне мирабилит слеживается
в монолитный пласт, который подвергается выветриванию в лет-
летнее время. Периодически с поверхности пласта удаляют тонкий
слой порошкообразного сульфата. Ускорению выветривания мира-
мирабилита способствует периодическое боронование (разрыхление)
верхнего горизонта пласта.
Посыпка каменным углем93 увеличивает интенсивность обез-
обезвоживания почти в 3 раза, однако снижает качество суль-
сульфата, поэтому не применяется. Предложена94 схема механизации
сбора сульфата натрия, предусматривающая распиловку пласта
116
Гл. IV. Природный сульфат натрия
Обезвоживание мирабилита
117
мирабилита циркульными пилами и вывозку кусков на берег тя-
тягачами.
Для разрыхления пласта мирабилита и интенсификации обез-
обезвоживания предлагается фрезерование его посредством вращаю-
вращающихся лопастных шнековых фрезеров и ворошение кусков при по-
помощи подметательно-уборочной машины с круглой вращающейся
щеткой. Эта же машина может производить сбор сульфата, пода-
подаваемого при помощи транспортерных лент на прицепы самораз-
саморазгружающейся конструкции. Скорость обезвоживания при работе
по этой схеме значительно увеличивается.
Таким методом может быть получен либо полностью обезво-
обезвоженный сульфат, либо частично обезвоженный, с 20—30% воды,
который может быть без особых трудностей досушен заводскими
способами (см. ниже).
Сульфат, полученный методом выветривания, содержит от 92
до 98% Na2SO4. Существенными недостатками естественного обез-
обезвоживания мирабилита являются значительные потери сульфата
натрия за счет развеивания ветром и загрязнение его пылью (пе-
(песком). Потери обусловливают повышенный расход мирабилита —
на 1 т сульфата натрия расходуется 3 т мирабилита вместо теоре-
теоретически рассчитанных 2,27 т.
Возможно осуществлять летнее обезвоживание мирабилита
с образованием тенардита в бассейнах под слоем рапы, содержа-
содержащей NaCl89~97. (Этот же процесс протекает и в естественных усло-
условиях в некоторых сульфатных озерах Кулундинской степи и др.).
Недостатком этого метода является загрязнение сульфата пова-
поваренной солью и солями магния.
Обезвоживание мирабилита в естественных условиях является
сезонным, связано с климатическими условиями, требует большой
затраты тяжелого физического труда и больших площадей.
Заводские способы обезвоживания мирабилита
Разработано и предложено много способов заводского обезво-
обезвоживания мирабилита89, в основу которых положены следующие
приемы и их комбинации: 1) плавление мирабилита, 2) выпарка
растворов в аппаратах с теплопередающей поверхностью, 3) вы-
выпарка при непосредственном соприкосновении растворов с горя-
горячими газами, 4) автоклавирование, 5) сушка воздухом и дымо-
дымовыми газами, 6) высаливание, 7) обезвоживание с применением
летучих растворителей.
Плавление мирабилита
При нагревании до температуры превращения C2,4°) мираби-
мирабилит плавится — разлагается на Na2SO4 и Н2О; освобождающаяся
кристаллизационная вода растворяет часть с)у!ьфата натрия с об-
образованием насыщенного раствора, а остальная часть его C5,8%))
остается в твердой фазе и может быть отделена отстаиванием,
центрифугированием и т. д. Можно несколько увеличить выход
сульфата натрия, нагревая расплавленную массу, так как раство-
растворимость его при этом уменьшается (рис. 21); например, при 90°
в осадке остается 45,9% Na2SO4, содержавшегося в исходном де-
кагидрате. Вследствие уменьшения растворимости Na2SO4 с по-
повышением температуры происходит зарастание плотной коркой,
сульфата теплопередающих поверхностей плавильных аппаратов98.
Этого можно избежать, осуществляя плавление мирабилита при
непосредственном соприкосновении с продуктами сгорания топ-
топлива. Однако в этом случае сульфат загрязняется золой и сажей.
Расход топлива составляет ~ 23 кг на 1 т сульфата8В.
Обычно применяют плавители мирабилита с циркулирующим
маточным щелоком, который нагревают паром в трубчатом подо-
подогревателе.
Получение сульфата натрия путем только плавления мираби-
мирабилита отличается низким коэффициентом использования сырья: рас-
расход мирабилита на 1 г безводного Na2SO4 практически достигает
4,5 т. Значительная доля сульфата сбрасывается с маточным рас-
раствором. Поэтому в современных производственных схемах плавле-
плавление мирабилита сочетается с выделением сульфата натрия из рас-
раствора разными методами — выпаркой, высаливанием, выморажи-
вымораживанием.
Выпарка растворов сульфата натрия в аппаратах
степлопередающей поверхностью
Кристаллизация сульфата натрия при выпаривании его раство-
раствора в аппаратах, обогреваемых паром, затруднена зарастанием по-
поверхности нагрева коркой сульфата. Особенно сильно инкрусти-
инкрустируются греющие поверхности при выпарке сульфатных растворов,
загрязненных примесями, вызывающими коррозию стальных кипя-
кипятильников89. Греющие поверхности выпарных аппаратов должны
изготовляться из хромистых или хромоникелевых сталей, наиболее
устойчивых в условиях выпарки сульфатного раствора.
Для уменьшения зарастания солью греющих поверхностей ре-
рекомендуется применять аппараты с интенсивной принудительной
циркуляцией раствора или с механической очисткой поверхности
нагрева (например, движущимися скребками или щетками). Не-
Несмотря на интенсивную циркуляцию C м/сек), в течение суток на-
нарастает слой накипи до 0,5 мм и коэффициент теплопередачи умень-
уменьшается с 3470 до 900 ккал/ (ж2 • ч • град). Поэтому интенсивная цир-
циркуляция не исключает необходимости частых промывок аппаратов,
что снижает производительность выпарной станции.
118
Гл. IV. Природный сульфат натрия
Обезвоживание мирабилита
119
Вследствие склонности сульфата натрия к образованию пере-
пересыщенных растворов отложение соли происходит не только в зоне
подогрева и кипения раствора, но и на всех поверхностях цирку-
циркуляционного контура. Поэтому наиболее радикальным средством
борьбы с зарастанием солью выпарных аппаратов является вы-
выпарка интенсивно циркулирующей суспензии мелких кристаллов
Na2SO4 в насыщенном растворе. В этом случае пересыщение ра-
раствора снимается главным образом за счет роста взвешенных
в жидкости кристаллов. При содержании в суспензии 5—10% твер-
твердой фазы выпаривание в опытном аппарате при скорости циркуля-
циркуляции 1,8—1,9 м/сек проходило в течение 1,5 суток без заметного
снижения коэффициента теплопередачи 10°.
Зарастание теплопередающей поверхности значительно умень-
уменьшается, если она хорошо отполирована; при этом коэффициент
теплопередачи в 1,5—2,5 раза больше101.
Подвергаемый выпариванию горячий раствор Na2SO4 обычно
используют и для плавления мирабилита. Загружаемые в плави-
тель куски мирабилита при смешении с подогретым маточным рас-
раствором «тают», рассыпаются и образующаяся суспензия безвод-
безводного сульфата натрия направляется на выпарку.
На заводе в Троне (США) рапу концентрируют в трехкорпус-
ной вакуум-выпарной батарее, а затем кристаллизуют мирабилит
при охлаждении до 5—6е. Отфильтрованный мирабилит плавят и
образующийся концентрированный раствор сульфата натрия на-
направляют в двухступенчатый испаритель, где кристаллизуется без-
безводный сульфат натрия. Кристаллы выводятся из испарителя при
60е и высушиваются в барабанной сушилке (см. ниже) при 135°.
Выпарка растворов сульфата натрия при
непосредственном соприкосновении с горячими
топочными газами
Преимущество этих методов — в отсутствие .греющих поверхно-
поверхностей, зарастающих сульфатом. Пропускание топочных газов над
зеркалом жидкости в пламенной печи дает более интенсивное вы-
выпаривание раствора и лучшее использование тепла, чем при обо-
обогреве открытого чрена через днище. Более рационально можно
осуществлять выпарку сульфатного раствора в огневой башне
(рис. 32). Такая башня из листовой стали имеет вертикальную
плоскопараллельную насадку из тонкой листовой стали. Стекаю-
Стекающий по насадке раствор выпаривается в токе идущих снизу горя-
горячих дымовых газов, поступающих из расположенной рядом топки.
Температура входящих газов около 1000°, уходящих— 100°. Плот-
Плотность орошения башни раствором 10—15 м3/ч на 1 м смоченного
периметра насадки. Производительность башни сечением 1 X 8 м
и высотой 8 л приблизительно 12 г сульфата натрия в 1 ч.
Еще более интенсивно идет процесс при использовании метода
погруженного горения 102'103, когда смесь горючего газа или распы-
распыленного жидкого топлива и воздуха подается в горелку, находя-
находящуюся под уровнем сульфатной суспензии. В этом случае тепловой
коэффициент полезного действия установки достигает 90% и боль-
больше. На рис. 33 показана схема непрерывного обезвоживания мира-
мирабилита методом погруженного горения на заводе Монэхенс в
Техасе (США) 79-89-104. Мирабилит плавят горячим маточным рас-
раствором. Насыщенный раствор Na2SO4 перекачивается в выпарной
аппарат погруженного горения 2. В горелки подают газо-воздуш-
ную смесь. Расход газа в одной горелке 52 м3/мин. Температура
факела 1300—1500е; барботирующие газы уносят испаряющуюся
воду и выбрасываются в атмосферу. Выходящая из выпарного ап-
аппарата суспензия, имеющая температуру 90° и содержащая 5%
кристаллического Na2SO4) отстаивается; осветленный маточный рас-
раствор переливают обратно в выпарной аппарат, а частично возвра-
возвращают на плавление мирабилита. Сгущенная пульпа, содержащая
~70% Na2SO4, поступает на центрифуги 4, из которых маточный
раствор возвращается на выпаривание, а сульфат с содержанием
5—6% влаги направляется в барабанную сушилку5, обогреваемую
дымовыми газами. Готовый продукт содержит 99,3—99,9% NasSO* ¦
и менее 0,01% железа. Выпарные аппараты изготовлены из стали;
внутренние стенки отполированы и после 15 лет работы не пока-
показали никаких признаков разрушенияБ9.
При погруженном выпаривании суспензий Na2SO4 в насыщен-
насыщенном растворе, содержащих 10—15 вес. % кристаллов, тепловая
нагрузка горелок достигает 175—200 тыс. ккал/ч 10Б.
Можно осуществить процесс, используя вместо погруженного
горения барботаж горячего топочного газа A000°) через выпари-
выпариваемый сульфатный раствор. Этот метод позволяет использовать
вместо газообразного, низкосортное твердое топливо, но требует
больших капитальных затрат.
Обезвоживание мирабилита в автоклавах
Этот способ основан на резком понижении растворимости суль-
сульфата натрия при высоких температурах (рис. 21). Если мирабилит
или раствор сульфата натрия нагреть в автоклаве почти до крити-
критической температуры воды, то практически весь сульфат окажется
в твердой фазе. Так как растворимость примесей с ростом темпе-
температуры увеличивается и они остаются в жидкой фазе, то сульфат
получается более чистым, чем при других методах обезвоживания
мирабилита. Этот метод, весьма экономичный в тепловом отноше-
отношении, пока не нашел практического применения из-за трудности
аппаратурного оформления.
Обезвоживание мирабилита
121
Обезвоживание мирабилита сушкой воздухом
и дымовыми газами
Низкотемпературная сушка
Низкотемпературное обезвоживание мирабилита сушкой за-
заключается в искусственном выветривании его воздухом при темпе-
температурах ниже точки плавления мирабилита C2,4°) в полочных или
барабанных сушилках. Процесс аналогичен естественному вывет-
выветриванию. Интенсивность сушки в этих условиях очень низка, про-
процесс идет медленно и требует применения аппаратуры с большими
габаритами.
Низкотемпературная сушка мирабилита воздухом в полочных
сушилках, осуществляемая периодически на сульфатном промысле
на оз. Эбейты (при благоприятных климатических условиях, не
требующих расхода топлива на подогрев воздуха), дает среднесу-
среднесуточный съем сульфата с 1 м2 сушильной площади всего 0,5 кг.
Применение вакуума при низкотемпературной сушке106 уско-
ускорило бы процесс, но не настолько, чтобы оправдать более высокую
стоимость оборудования и повышенный расход энергии.
Низкотемпературную сушку мирабилита можно осуществлять
также и дымовыми газами, разбавленными воздухом, например в
аппарате с движущейся лентой. Благодаря быстрому отводу влаги
с мощным газовым потоком возможно вести процесс при темпе-
температуре, на 10—12° превышающей температуру плавления мираби-
мирабилита, т. е. при 45°107. Интенсивность низкотемпературной сушки
значительно повышается при осуществлении процесса во взвешен-
взвешенном слое. Все же при больших масштабах производства низко-
низкотемпературная сушка неприменима.
Высокотемпературная сушка
Для интенсивного обезвоживания мирабилита сушкой необ-
необходимо осуществлять процесс при температурах, значительно пре-
превышающих температуру плавления мирабилита. Для этого ис-
используются вращающиеся барабанные сушилки. Предотвращение
схватывания и комкования за счет плавления достигается путем
смешения поступающего на сушку мирабилита с обезвоженным
сульфатом, выходящим из сушилки. Такой способ сушки назы-
называется ретурным (возвратным)—часть готового продукта цирку-
циркулирует через сушило. Кроме того, для раздавливания комков и
сдирания материала, прилипшего к стенкам, внутри сушильного
барабана помещают длинную цепь, только один конец которой
закреплен 108-109.
Технологическая схема процесса изображена на рис. 34. Смесь
мирабилита с сульфатом, подаваемая в сушилку, имеет темпера-
температуру около 30° и представляет собой зерна 1—2 мм или гранулы
Рис. 32. Схема установки обезвоживания мирабилита в огневой башне:
/ — корпус огневой башни; 2 —насадка; 3 — распределительные щелн для подачи раствора; 4 — циркуляционный насос; 5 —приемный желоб для
сульфатной суспензии со шнеком для сульфата; 5 —отстойник сульфатной суспензии-плавнтель ми^бялита; 7 —мешалка сульфатной суспен-
суспензии; S— центрифуги; 9 — возврат маточного раствора; 10 — топка; // —огневой штуцер.
Рис. 33. Схема обезвоживания мирабилита
методом погруженного горения:
/ — плавитель; 2 — выпарной аппарат погруженного горе-
иия; 3 — отстойник-сгуститель; 4 — центрифуга; 5 — бара-
баявая сушилка; 5- центробежный иасос.
Рис. 34. Схема ретурного способа обезво-
обезвоживания мирабилита в барабанной сушилке;
/ — смеситель; 2 — барабанная сушилка; 3 —шнек;
4 — элеватор; 5 — обратный шнек; е —бункер для
сульфата натрия; 7 —дымосос; S — пылеуловитель,|
122
Гл. IV. Природный сульфат натрия
размером до 1 см. Количество оборотного сульфата натрия, добав-
добавляемого к мирабилиту, должно быть таким, чтобы в смеси, посту-
поступающей на сушку, содержание воды не превышало 20—25%- При
большем содержании воды материал сильно комкуется и налипает
на стенки сушилки. Практически при содержании в мирабилите
5% гигроскопической влаги (общее содержание воды 58%) для
получения смеси, содержащей 25% воды, соотношение между ми-
мирабилитом и оборотным сульфатом равняется 1 : 1,4. При этом в
сушилку возвращается больше половины выгружаемого из нее
¦сульфата. Это требует дополнительной затраты тепла на его на-
нагревание до температуры сушки, так как сульфат натрия, имею-
имеющий при выходе из сушилки температуру около 120°, охлаждают
до возврата в сушилку.
Дымовые газы поступают в сушилку с температурой 650—850°;
температура уходящих газов 170—190°. Отходящий газ выносит из
¦сушилки до 30% Na2SO4 в виде пыли; для ее улавливания газ про-
пропускают через пылеуловитель (циклон, батарейный циклон). Ды-
Дымовые газы могут проходить через сушилку прямотоком или про-
противотоком материалу. При прямоточном режиме обеспечивается
большая скорость испарения воды в начальной стадии сушки, что
препятствует прилипанию сульфата к поверхности футеровки су-
сушильного барабана. Недостатками ретурного способа являются:
1) необходимость возврата в печь более половины получаемого
сульфата, 2) в связи с этим низкая производительность сушила,
3) необходимость шихтовки материалов с предварительным охла-
охлаждением обезвоженного сульфата во избежание образования ком-
комков при смешении и 4) образование комкующегося и пылящего
продукта, слеживающегося при хранении.
Ретурный способ сушки — один из наиболее громоздких, тре-
требующий большого расхода металла на сооружение сушилок и
транспортных устройств и большого расхода энергии и топлива.
На 1 т готового сульфата натрия расходуется до 250 кг условного
топлива G000 ккал/кг) и больше 40 квт-ч электрической энергии.
При диаметре сушила 2,8 м и длине 14 м удельный влагосъем
с 1 ж3 объема сушила составляет 35 кг/ч (при температуре входя-
входящих газов 850°, уходящих 180°).
На одном из заводов в США89-108 применялся «трехстадийный» '
•способ обезвоживания мирабилита, состоящий из трех последова-
последовательных процессов: 1) плавления мирабилита во вращающейся
печи E5°), 2) выпарки-сушки смеси плава и сульфата в трубчатой
паровой сушилке A03°) с превращением этой смеси в твердый про-
продукт, содержащий 27% воды, и 3) окончательной сушки в бара-
барабанной сушилке A40°), обогреваемой дымовыми газами. При та-
такой схеме обезвоживания расход тепла меньше, чем при сушке
¦с оборотным сульфатом. Кроме того, основная масса тепла, рас-
расходуемого при трехстадийном способе обезвоживания, получается
Обезвоживание мирабилита
123
за счет использования отработанного пара низкого давления, т. е.
обходится дешевле, чем тепло, получаемое при сжигании угля.
В Канаде на сульфатных предприятиях на оз. Чаплин (Саска-
чеван) обезвоживание мирабилита осуществляют во вращающейся
печи в комбинации с испарителем. Последний представляет собой
камеру, в которой поступающий мирабилит плавится в токе горя-
горячих газов. У днища камеры расположен быстро вращающийся
шнек с лопастями, разбрызгивающими сульфатную суспензию.
При контакте с горячим газом брызги подсыхают и получается ма-
материал с влажностью ~25%, который досушивается во вращаю-
вращающейся печи с небольшим ретуром ио. Эти методы связаны с усло-
усложнением аппаратуры.
Безретурная высокотемпературная сушка мирабилита111 в ба-
барабанной сушилке требует меньших расходов тепла и энергии на
перемещение сульфата, чем сушка с ретуром. Сущность безретур-
ного способа сушки заключается в обезвоживании мирабилита на
подушке высушенного материала (толщина слоя которого зависит
от диаметра барабана). Создание подушки достигается с помощью
специального выгрузочного устройства. Влажный продукт забра-
забрасывается на эту подушку на расстояние 4—5 м при помощи пита-
питателя, состоящего из трефеля (дозатора) и метательной лопатки.
Такой способ сушки широко используют в содовом производстве
(для кальцинации бикарбонатаI12. Процесс осуществляют в печах
с внешним обогревом. Находящаяся внутри тяжелая цепь, закре-
закрепленная с одного конца, способствует разрушению комков и хоро-
хорошему смешению свежих порций загружаемого материала и горя-
горячего высушенного продукта.
Опыт показал, что обезвоживание мирабилита в содовых печах
с безретурным питанием не сопровождается обрастанием стенок
печи сульфатной коркой. Продукт получается крупнокристалличе-
крупнокристаллический без комков с насыпным весом ~ 1,2 т/м3 и влажностью 0,05%
и меньше.
С большей интенсивностью безретурное обезвоживание мираби-
мирабилита осуществляется в сушильных барабанах с внутренним обо-
обогревом. В опытах по безретурному обезвоживанию мирабилита,
проведенных в барабане длиной 7,62 ж, диаметром 0,9 ж, устано-
установленном под углом 3°, делающим 2,5 об/мин и отапливаемом
генераторным газом, влагосъем достигал 72 кг/(м3-ч). Это при-
приблизительно в 3 раза больше влагосъема при безретурном обезво-
обезвоживании мирабилита в сушилках с внешним обогревом и прибли-
приблизительно в 2 раза больше влагосъема при ретурном обезвожива-
обезвоживании в барабанных сушилках с внутренним обогревом.
Опытом безретурной сушки сульфата натрия с повышенной
влажностью на стекольных заводах в барабанных печах с внут-
внутренним обогревом, отапливаемых генераторным газом, установ-
установлено, что если температура отходящих из печей газов не снижается
124
Гл. IV. Природный сульфат натрия
ниже 150°, то налипание сульфата на стенку барабана не проис-
происходит. Сульфат на выходе из барабана имеет при этом темпера-
температуру 90—100°. Все же периодически (каждые 1 — 1,5 ч) приходится
очищать течку и приемную часть барабана от накопившихся от-
отложений. Очистку производят ручным способом в течение 10—
12 мин. Суточная производительность печи диаметром 0,95 м и
длиной 8,25 м, делающей 4—5' об/мин, при начальной влажности
сульфата 35—40% и температуре поступающих газов 550—600°
составляет 18—20 т сульфата с влажностью 1 —1,5% Пз.
Одним из наиболее совершенных и экономически выгодных
способов обезвоживания мирабилита является безретурная сушка
во взвешенном слое в сушилках КС («кипящего слоя»I1417. Та-
Такие сушилки с выносными мазутными топками работают на ком-
комбинате «Карабогазсульфат». При площади газораспределительной
решетки 6 м2 производительность сушилки 6 т/ч обезвоженного
сульфата. Температуры: в слое около 120°; входящего газа
750—800°, отходящего газа 100—110°. Решетки чугунные с диамет-1
ром отверстий 6 мм и живым сечением 10%. При скорости газа в
слое 2—2,5 м/сек сопротивление слоя и решетки 550—600 мм вод. ст. ¦
Конечная влажность сульфата меньше 0,1%. Продукт почти цели-
целиком состоит из гранул с размерами от 0,25 до 2 мм (96%). Частиц
с размерами меньше 0,25 мм всего ~0,3%. Около 20% материала
выносится из сушилок газом, улавливается в циклоне и возвра-
возвращается в бункер для мирабилита. С отходящим газом теряется
~2% продукта.
Перед подачей в сушилку КС мирабилит следует дробить на
куски меньше 25 мм. Подачу его на «кипящий слой» рекомендуется
осуществлять с помощью пневнозабрасывателя — наклонной течки1
с воздушной струей. При площади решетки 2—7 м2 высота слоя ;
~0,7 м, а высота сепарационной зоны аппарата должна состав-
составлять 4—7 м. Газораспределительная решетка может быть щеле-
щелевой (из колосников). В верхней части аппарата должно поддер-
поддерживаться разрежение 5—8 мм вод. ст., обеспечиваемое дымо-
дымососом.
Энергетические затраты на обезвоживание мирабилита в аппа-
аппаратах КС (как и другими способами) зависят от его начальной
влажности. При начальном содержании воды 56%, температуре под
решеткой 700° и температуре слоя 120° на 1 г продукта расхо-
расходуется 140 кг мазута (с рабочей теплотворной способностью
9400 ккал/кг), 50 кет • ч электроэнергии, 4 ж3 воды, 15—20 кг пара
(на мазутную форсунку). При использовании в качестве топлива
природного газа энергозатраты на обезвоживание мирабилита во
взвешенном слое приблизительно такие же, как и при его плавле-
плавлении с последующей выпаркой сульфатного раствора, но капиталь-
капитальные вложения меньше. Пи сравнению же с барабанными сушил-
сушилками, работающими с ретуром, сушилки КС требуют на 25—30%
Обезвоживание мирабилита
125
меньше энергозатрат, в 5—7 раз меньше металла и в 2—3 раза
меньше производственной площади.
Обезвоживание мирабилита в распылительных сушилках в по-
потоке горячих газов было испытано в заводских условиях89-118, но
не нашло распространения. Для получения этим способом продук-
продукта с влажностью 0,1—0,5% температура газов в средней части
камеры не должна быть ниже 250—300°. При более высокой тем-
температуре газов увеличивается интенсивность процесса, но при тем-
температуре, превышающей точку плавления сульфата, расплавленный
сульфат проникает через кирпичные стенки камеры и инкрустирует
их наружную поверхность.
Предложены разные видоизменения обезвоживания мирабилита
с помощью распылительной сушки. Так, например, насыщенный
раствор сульфата натрия можно выпаривать в распылительной
сушилке не досуха, а получая текучую суспензию сульфата натрия,
и после отделения кристаллов его маточный раствор возвращать в
процесс"9. Можно высушивать также в распыленном состоянии и
частично обезвоженный мирабилит с содержанием менее 30% воды,
вводя его в сушилку через сопло. При такой влажности исходного
материала не происходит его налипания на стенки сушилки '20.
В СССР были проведены опыты по ретурному обезвоживанию
мирабилита в пневматической сушилке — вертикальной трубе в по-
потоке горячих топочных газов (800—850°). Газы вводились в ниж-
нижнюю часть трубы, несколько выше в нее подавали с помощью
охлаждаемого водой сопла шихту из мирабилита и сульфата на-
натрия в отношении 1 :2. Шихта высушивалась в потоке газов, пере-
перемещаясь со скоростью 10—20 м/сек, и собиралась в ловушке и ча-
частично (~10%) в циклоне121. Этот метод обезвоживания мира-
мирабилита также не использован в промышленности.
Обезвоживание мирабилита высаливанием
Извлечение сульфата натрия из его растворов может быть осу-
осуществлено методами высаливания, основанными на понижении
растворимости сульфата со смещением переходной точки в присут-
присутствии других веществ.
Наиболее дешевый высаливающий реагент — NaCl; к тому же
он часто является естественной примесью к сульфатным раство-
растворам. На рис. 35 изображена политермическая диаграмма равно-
равновесия для системы Na?SOa—NaCl—НоО (содержание Na2SO4 и
NaCl дано в cvxom остатке). Если в отсутствие NaCl точка пере-
перехода Na9SO4'ЮНоО в Na9SO4 лежит при 32.38°, то при добав-
добавке NaCl температура перехода уменьшается и становится равной
l7,Qe- когда содержание NaCl в растворе достигает 22,3%. В этой
тпойиой точке А в солевой массе эвтонического раствора 25,4%
Na2SO4 и 74,6%) NaCl, а сам раствор содержит всего 7.57%
126
Гл. IV. Природный сульфат натрия
Обезвоживание мирабилита
127
Na2SO4, в то время как в точке В в насыщенном растворе 33,2%
Na2SO4. Ниже 17,9° выделение в осадок безводного Na2SO4 не про-
происходит, а при повышении температуры поле кристаллизации
Na2SO4 несколько расширяется и степень его высаливания увели-
увеличивается. Так, при 50° в отсутствие NaCl растворимость Na2SO4
составляет 31,9%, а эвтонический раствор содержит 23,89% NaCl
и всего 5,17% Na2SO4 (в сухом остатке 17,8% Na2SO4 и 82,2%
NaCl).
Таким образом, добавка к растворам сульфата натрия NaCl
приводит к кристаллизации Na2SO4 и позволяет получить в без-
безводном виде значительную до-
лю сульфата. После отделения
Na2SO4 отстаиванием и филь-
фильтрованием остающийся маточ-;
ный раствор может быть под-
подвергнут выпариванию для
кристаллизации из него NaCl,-
возвращаемого на высалива-
высаливание. Отфильтрованный суль-
сульфат для удаления из него при-
примеси NaCl необходимо про-
промыть насыщенным раствором
Na2SO4 (в 1,5-кратном коли-
количестве к твердой фазе) 89.
Способ высаливания суль-
сульфата натрия галитом исполь-
используется на предприятии имени
32Ж
100
Na2SO4
Рис. 35. Проекционная политермическая
диаграмма равновесия в системе
Na2SO4—NaCl—H2O.
Тельмана в ГДР. Расчетные
расходные коэффициенты применительно к обезвоживанию этим
способом Аральского сульфата натрия следующие (на 1 т суль-
сульфата): галита ~0,65 т, пара 0,88 т, электроэнергии 36 кет • ч,
условного топлива 0,015 т. Этот способ получения безводного
сульфата натрия экономически оправдывается при использовании
дешевого отбросного галита, не добываемого специально для вы-
высаливания.
В качестве высаливающих реагентов для выделения безводного
сульфата натрия из растворов могут быть использованы смеси со-
солей: NaCl + КС1122, NaCl + MgSO4, а также Na2S, NaOH и др.
При использовании в качестве высаливающего средства едкого
натра вода, содержащаяся в сульфатном растворе, из которого
высаливается Na2SO4, разбавляет раствор NaOH и должна быть
удалена из него выпариванием. После выпаривания концентриро-
концентрированный раствор NaOH вновь возвращается на высаливание. Пре-
Преимущество этого способа по сравнению со способом непосредствен-
непосредственной выпарки сульфатных растворов заключается в том, что при
выпарке растворов NaOH греющая поверхность не инкрустируется
сульфатом натрия, хотя и происходит его дополнительная кристал-
кристаллизация по мере увеличения концентрации NaOH ш.
В табл. 14 приведена растворимость Na2SO4 в водных раство-
растворах едкого натра при разных температурах.
ТАБЛИЦА 14
Растворимость Na2SO4
в водных растворах NaOH
Концентрация
NaOH,
%
0
5
15
25
35
45
55
г Na2SO4 в
50°
46,8
28,4
П,7
3,7
1,9
0,6
0,3
70°
43,9
26,5
11,0
3,5
1,6
0,6
0,3
100 г НгО при
90°
42,7
24,9
10,7
3,3
1,5
0,6
0,3
100°
42,4
24,5
10,0
2,9
1,3
0,6
0,2
температуре
120°
41,8
23,5
9,6
2,6
1,3
0,6
0,2
140°
4?,0
2,2
Ш.1
2,6
1,4
0,6
0,3
По диаграмме рис. 36 можно определить степень высаливания
O из его насыщенного раствора при 60° растворами NaOH
50%
80 70 Б0
0,1 0,3 0,5 0,75 1,DD ifiS 1,50 1,75 2,00 2,25 2,5D 2.75 3,DD 3,25
Количество раствора NaOH, л/ л насыщ. раствора сульфата
Рис. 36. Степень высаливания Na2SO4 из его насыщенного
раствора при 60° растворами NaOH разной концентрации:
— концентрация исходного раствора NaOH, %, концентра-
%, концентрация NaOH в жидкой фазе получаемой суспензии
разной концентрации. При смешении 0,4—1 объема 40—50%-ного
раствора NaOH с 1 объемом насыщенного раствора сульфата на-
натрия степень высаливания Na2SO4 составляет 85—90%, а раствор
>28
Гл. IV. Природный сульфат натрия
после отделения осадка сульфата содержит 20—25% NaOH и
2,5—5% Na2SO4.
Непосредственное растворение твердого мирабилита в растворе
едкого натра приводит к образованию тонкодисперсного осадка
Na2SO4, трудно отделяемого от жидкой фазы. Лучшие результаты
получаются при предварительном расплавлении мирабилита. На-
Нагреванием мирабилита до 60° можно выделить 42% сульфата на-
натрия. При смешении отделенного от кристаллов раствора с равным
объемом 40%-ного раствора NaOH высаливается еще 52% Na2SO4.
Наконец, при выпаривании полученного разбавленного раствора
щелочи выделяются остальные 6% Na2SO4.
Для получения в процессе плавления крупнокристаллического,
хорошо фильтрующего безводного сульфата натрия плавление
необходимо вести медленно при перемешивании массы с такой ин-
интенсивностью, чтобы выделяющийся безводный Na2SO4 оставался
во взвешенном состоянии. Затем суспензия должна быть нагрета
до 70—85°. При соблюдении этих условий образуется осадок с раз-
размерами кристаллов 200—250 мк, оседающий при 85° со скоростью
8-—9,5 м/ч. В процессе высаливания крупнокристаллический, хо-
хорошо фильтрующий осадок образуется при медленном приливании
нагретого до 70—80° раствора NaOH к нагретому до этой же
температуры, интенсивно перемешиваемому раствору сульфата
натрия.
Находящиеся в сульфате натрия примеси соединений магния,
полуторных окислов, кремневой кислоты и другие образуют в ра-
растворе едкого натра гелеобразные осадки, затрудняющие отстаи-
отстаивание и фильтрацию кристаллов Na2SO4. Поэтому эти примеси
должны быть отделены до высаливания. При этом возможно по-
получение продукта, содержащего до 99,5% Na2SO4 (в пересчете на
сухое вещество).
Предложено 124 получать безводный сульфат натрия из мираби-
мирабилита нагреванием последнего в смеси с двойной солью Na2SO4'
•NH4HCO3-4H2O до 60°. Образующийся осадок Na2*SO4 отфиль-
отфильтровывают, а из фильтрата охлаждением до 20° выделяют Na2SO4-
•NH4HCO3*4H2O. Концентрированием маточного раствора, остаю-
остающегося после отделения четырехводной двойной соли, кристалли-
кристаллизуют сульфат аммония.
Обезвоживание мирабилита с применением
летучих растворителей
Для высаливания сульфата натрия из его растворов, получен-
полученных при плавлении мирабилита, могут быть использованы летучие
реагенты — аммиак, метиловый и этиловый спирты и др.125-126. По-
После отделения осадка Na2SO4 эти реагенты могут быть регенериро-
регенерированы из водного раствора отгонкой. Эти методы пока не нашли
Обезвоживание мирабилита
129
промышленного применения в связи с тем, что они связаны с поте-
потерями относительно дорогих реагентов. Однако они интересны тем,
что требуют малого расхода тепла. Так, при использовании в ка-
качестве высаливающего средства аммиака расход тепла на 1 т про-
продукта в 2 раза меньше, чем при плавлении мирабилита и выпари-
выпаривании воды из образующегося раствора.
!йз допмоссреру
ВоОа
Мирабилит
Вода 100°па техмич.
нужды
Конденсат Сульфат па упакойку
Рис. 37. Схема обезвоживания мирабилита аммиачным способом:
1 -дробилка; 2-грохот; 3-бункер; 4-реактор-аммонизатор; 5-фонарь; 6-буферный резер-
резервуар- 7 - уплотнитель суспензии; 8-диафрагменный иасос; 9— центрифуга; iO-сушнлка.
//-калорифер; 12 - вентилятор; 13- насос; 14 - дистиллер; 15 - подогреватель жидкости;
16- холодильник газа; 17 - экспанэер; /8- скруббер; /9, 20 - гидравлические регуляторы
давления; 21 - эксгаустер; жидкости и суспензии; твердые вещества!
газы и пары.
Растворимость сульфата натрия в воде в присутствии аммиака
сильно уменьшается 127-128. Поэтому при взаимодействии регене-
регенерированной аммиачно-паровой смеси, имеющей температуру 60°,
с мирабилитом, имеющим температуру 10°, выход сульфата натрия
достигает 90—95% без дополнительного нагревания или охлажде-
охлаждения реакционной смеси.
На рис. 37 показана возможная схема обезвоживания мираби-
мирабилита аммиачным способом129. Реактор по высоте разделен двумя
ложными днищами, препятствующими падению крупных кусков
мирабилита на дно. Снизу в реактор поступает аммиачно-паровая
смесь при 50—60° под давлением 20—50 мм рт. ст., а сверху —
аммонизированный раствор. Соприкасаясь с холодным мирабили-
мирабилитом, водяные пары конденсируются. Таким образом, обезвоживание
5 М. Е. Позин
130
Гл. IV. Природный сульфат натрия
сульфата натрия происходит в водном растворе аммиака. Суспен-
Суспензия Na2SO4 в водно-аммиачном растворе удаляется из нижней
части реактора через фонарь, позволяющий следить за плотностью
суспензии. После отстаивания и центрифугирования получаемый
сульфат натрия с влажностью 4—6% высушивают в барабанной
сушилке. Маточный раствор направляется на дистилляцию ам-
аммиака. Горячую жидкость, выходящую из дистиллера, направляют
в экспанзер, где образуется пар, используемый для пропарки
сульфата натрия на центрифуге. Уходящая из экспанзера отброс-
отбросная жидкость содержит всего 50—60 г/л Na2SO4.
Другим вариантом обезвоживания мирабилита с помощью ле-
летучих веществ может быть выпарка с кристаллизацией Na2SO4 и
одновременной отгонкой высаливающего агента. Так, например,
для устранения инкрустации поверхности нагрева выпарных аппа-
аппаратов при кристаллизации Na2SO4 из водного раствора предло-
предложено 130 производить кристаллизацию сульфата натрия в выпари-
выпариваемом водном растворе органической жидкости F0—90%-ном
растворе этиленгликоля или в 40—50%-ном растворе спирта). От-
Отгоняющиеся при выпарке пары органической жидкости и воды
подвергают ректификации и образующийся водный дистиллят воз-
возвращают в процесс. Кристаллы Na2SO4 выделяют из суспензии,
промывают и высушивают.
ПРОИЗВОДСТВО СУЛЬФАТА НАТРИЯ НА ЗАВОДАХ СССР
Производство сульфата натрия на комбинате «Карабогазсульфат»
Межкристальный рассол второго погребенного горизонта
(стр. 160) из скважин, расположенных в районе Кургузульской
бухты карабогазского залива, с помощью двух насосных станций
поступает на завод по чугунному трубопроводу диаметром 600 мм
в два железобетонных резервуара емкостью по 1000 мъ. Отсюда он
подается в кристаллизаторы цеха мирабилита. Охлаждение рас-
рассола осуществляют в две ступени — в предкристаллизаторах до 10°
сбросным маточным раствором, остающимся после отделения кри-
кристаллов мирабилита, и в кристаллизаторах до 0° испаряющимся
жидким аммиаком аммиачной холодильной машины. Маточный
раствор, нагревшийся в предкристаллизаторах до 8°, используют
для переохлаждения жидкого аммиака и для кондиционирования
воздуха в производственных помещениях в жаркое время года.
Кристаллизаторы представляют собой вертикальные цилиндри-
цилиндрические резервуары, снабженные внутри четырехсекционной систе-
системой трубчатых теплообменников и лопастными мешалками, а так-
также скребками для очистки поверхности трубчатки от прилипшего
мирабилита.
Производство сульфата натрия на заводах СССР
131
Параллельно работают две батареи кристаллизаторов. Каждая
состоит из двух предкристаллизаторов и пяти кристаллизаторов,
расположенных каскадом. Мирабилитовая пульпа поступает из
них самотеком через мешалку в дисковые фильтры-сгустители, за-
затем на центрифуги, откуда мирабилит ленточным транспортером
передается в цех сульфата натрия.
Плавление мирабилита осуществляется в камере плавления
циркуляционного плавителя в восходящем токе горячего щелока
(~70°), который возвращается вниз через подогреватель, где ис-
используется соковый пар вторых корпусов выпарной станции. Из
них щелок, содержащий ~30% сульфата натрия, направляется на
выпарку в двухкорпусные выпарные батареи. Греющий пар в пер-
первом корпусе имеет давление 5,5 ат. Здесь выпаривается ~50%
воды. Пульпа из первого корпуса поступает во второй, обогревае-
обогреваемый соковым паром первого. Сюда же подают маточный щелок
после центрифуг для поддержания постоянного отношения Т: Ж-
Сульфатная пульпа из вторых корпусов, пройдя самоиспарители
и сгуститель вместе со сгущенной пульпой, полученной при плавле-
плавлении мирабилита, поступает на центрифуги,
Отфугованный сульфат, содержащий 5% влаги, высушивают
топочным газом, разбавленным воздухом в аппарате КС с мазут-
мазутной топкой. Уносимая газом сульфатная пыль улавливается в цик-
циклонах и электрофильтре.
Производство<»сульфата натрия на Кучукском сульфатном
комбинате
Из рапы оз. Кучук мирабилит получают -бассейньш способом
(стр. 115), а затем его обезвоживают плавлением и выпаркой в за-
заводских условиях. В качестве садочного бассейна используют кот-
котловину высохшего оз. Селитренного, расположенного в 2,4 км от
оз. Кучук. Работу промысла ведут в двухгодичном цикле, т. е. на-
накачку рапы в садочный бассейн производят один раз в два года
D0 млн. ж3). Высота налива — 7 ж. Выход мирабилита из 1 м3
рапы 117—130 кг. В засушливые годы, в период наибольших кон-
концентраций кучукской рапы, для предотвращения совместной садки
с мирабилитом NaCl и NaCl-2H2O рапу разбавляют менее кон-
концентрированной рапой из близрасположенного оз. Кулундинского,
уровень воды в котором выше, чем в оз. Кучук.
Плавление мирабилита раствором сульфата натрия ведут при
60° в горизонтальном реакторе с рамной и шнековой мешалками.
Циркулирующий между плавителем и подогревателем раствор
содержит некоторое количество взвешенных кристаллов Na2SO4,
что предотвращает зарастание греющих поверхностей подогрева-
подогревателей, где температура раствора повышается до 100° соковым па-
паром из второго корпуса выпаркой батареи. Пульпа из плавителя
132
Гл. IV. Природный сульфат натрия
поступает на центрифуги, а слив из плавителя и фильтрат с цен-
центрифуг в первый корпус выпарной батареи. Во избежание зара-
зарастания греющих поверхностей кристаллами в циркулирующем рас-
растворе в первом корпусе поддерживают содержание твердой фазы
10—20%- Циркуляция осуществляется с помощью пропеллерных
насосов. Пульпа, поступающая на центрифугу из второго корпуса,
содержит 35% твердой фазы. Отфугованный сульфат натрия высу-
высушивают во вращающихся барабанных сушилах дымовым газом,
который затем очищается от сульфатной пыли в циклонах, муль-
мультициклонах и скрубберах.
ПОЛУЧЕНИЕ СУЛЬФАТА НАТРИЯ ИЗ ТВЕРДЫХ СОЛЕВЫХ ОТЛОЖЕНИЙ
В большинстве случаев извлечение сульфата натрия из твер-
твердых солевых отложений связано с довольно сложной их переработ-
переработкой. Сульфат натрия получают при этом наряду с другими продук-
продуктами. Только при добыче тенардита, если порода достаточно чиста,
ограничиваются ее выламыванием или извлечением из рапы (на-
(например, на оз. Джаксы-Клыч) и естественной сушкой в буграх.
Часто, однако, и тенардит подвергают перекристаллизации для
удаления примесей других солей и органических загрязнений1S1.
Наиболее крупным производством сульфата натрия из твердых
солевых отложений является комплексная переработка хартзаль-
цевых пород на заводе имени Тельмана в ГДР (стр. 159). Полу-
Получающийся при этом галитовый отвал содержит кизерит, который
отмывают. После конверсии
MgSO4 + 2NaCl = Na2SO4 + MgCl2
безводный сульфат натрия высаливают галитом.
Предложено много схем переработки на сульфат натрия астра-
ханитового сырья. Например, одна из схем заключается в раство-
растворении породы с последующим выделением мирабилита выморажи-
вымораживанием (после добавки NaCl для повышения выхода) и в обезво-
обезвоживании мирабилита выпаркой или высаливанием. Другая схема
предусматривает растворение с последующим прямым высалива-
высаливанием Na2SO4 галитом при 50° и т. д.
Разработаны способы получения сульфата натрия из глаубери-
товых пород132. При комплексной переработке полиминеральных
руд Предкарпатья возможно получение значительных количеств
сульфата натрия из лангбейнитового отвала (см. гл. V).
Запасы твердых солей в Карабогазском заливе практически
неисчерпаемы. В настоящее время обнажилась значительная часть
дна залива почти вдоль всего побережья (за исключением восточ-
восточного и части южного). Ширина прибрежной полосы солевых отло-
отложений достигает 20—30 км, а мощность пласта смешанной соли
доходит до 2,5 м. Средний состав солевых отложений в северо-за-
Получение сульфата натрия из твердых солевых отложений
133
падной части залива в районе действующего промысла комбината
«Карабогазсульфат» (в %):
NaCl 73,14 СаСО3 0,26
MgSO4 9,80 Нерастворимый остаток 0,67
Na2SO4 6,89
CaSO4 0,8
Н,О. . . . ; 8,44
Северные отложения более богаты сульфатами и содержат в
среднем (в %) 55> 1М- ш-
NaCl 58
MgSO4 20,5
Na2SO4 4,2
CaSO4 .
H2O. ,
0.7
15,6
Получение мирабилита из смеси NaCl и MgSO4 было разрабо-
разработано еще в 1929 г. применительно к Крымским соляным промыс-
промыслам 135. Разработаны и способы переработки карабогазской cMej
шанной соли («Sel mixt»). Она может быть растворена в морской
воде с последующим выделением из раствора мирабилита бассей-
ным способом или переработана на сульфат натрия заводским спо-
способом. Однако технико-экономические показатели этих способов
переработки смешанной соли несколько уступают показателям су-
существующего производственного процесса получения сульфата на-
натрия из межкристальных карабогазских рассолов. Поэтому эти
способы пока не реализуются, и ниже дается лишь их краткое опи-
описание.
Бассейный способ переработки смешанной соли на мирабилит
заключается в растворении смешанной соли в морской воде с по-
получением отношения MgSO4: NaCl, равного 0,4—0.6. Такое отноше-
отношение солей обеспечивает максимальный выход мирабилита при по-
последующем охлаждении раствора (стр. 111) в садочном бассейне.
В основу схемы заводской переработки смешанной соли 136 по-
положены также процессы растворения ее в морской воде и кристал-
кристаллизации мирабилита. При растворении смешанной соли при 20—
25° получаемый раствор должен содержать 27,5% солей (раство-
(растворимость смешанной соли при этой температуре ~30,5%). При со-
содержании в смешанной соли 80% сухих солей для растворения 1 т
соли требуется 1,9 т воды, причем образуется 2,3 ж3 рассола плот-
плотностью 1,25 г/см3. Потери рассола со шламом при отстаивании со-
составляют 2%- При охлаждении 1 ж3 осветленного рассола до 0°
образуется около 280 кг мирабилита, а до —5° около 295 кг. Ниже
—5° из рассола может выделиться также гидрогалит NaCl-2H2O.
Маточный раствор после садки мирабилита может быть использо-
использован для получения эпсомита. Отфугованный мирабилит содержит
5% влаги. При его расплавлении при 55—60° выделяется ~40%
безводного сульфата натрия или 0,15 г на 1 г мирабилита. После
отделения сульфата натрия к оставшемуся щелоку можно доба-
добавить «смешанную соль (из расчета 0,33 г на 1 г мирабилита). При
134
Гл. IV. Природный сульфат натрия
Литература
135
этом в результате высаливания выделится 0,21 т N2SO4 на 1 г
мирабилита. Маточный раствор от высаливания в количестве
0,78—0,8 т возвращают в процесс на охлаждение. Сульфат натрия,
промытый раствором от плавления мирабилита, высушивают.
Упомянем о производстве сульфата натрия из твердых отложе-
отложений Большого Соленого озера в США, которое меньше Карабо-
газского залива, но аналогично ему по характеру солей. Здесь раз-
разрабатывается береговой пласт мирабилита длиной несколько кило-
километров, толщиной 0,6—3 м и шириной 0,6—2,4 км, прикрытый
слоем песчаника. Взорванная порода, доставляемая в вагонах на
завод, содержит приблизительно 20% сульфата натрия, 20% воды
и 60% пустой породы. Измельченную руду обрабатывают горячей
водой (80—85°) и после отстаивания песка раствор упаривают.
Кристаллизующийся при выпарке сульфат натрия отделяют в от-
отстойниках, центрифугируют и высушивают во вращающейся бара-
барабанной сушилке. Маточный раствор возвращают на выпарку137.
Запатентованы138 способы очистки сульфата натрия от соеди-
соединений железа. По этим способам расплавленный сульфат нагре-
нагревают до 1200° в присутствии кремнийсодержащих материалов
(SiO2, Na2Si03) и щелочей (NaOH, Na2CO3). Затем расплав декан-
декантируют с осадка, в который переходит более 90% железа. Fe2O3
снимают также с поверхности расплава, а для более полного уда-
удаления железа охлажденный сульфат измельчают и подвергают
магнитной сепарации.
О правилах и нормах техники безопасности в производстве
природного сульфата натрия см.139.
ГЛАУБЕРОВА СОЛЬ
К чистоте десятиводного сульфата натрия, применяемого под
названием глауберовой соли для медицинских целей, предъяв-
предъявляются повышенные требования. Согласно ГОСТ 6319—52*, глау-
глауберова соль должна содержать не менее 99% Na2SO4- ЮН2О, и не
более: 0,005% не растворимых в воде веществ, 0,1% хлоридов
(NaCl), 0,0005% Fe, 0,003% солей аммония (NH4), 0,0005% тяже-
тяжелых металлов сероводородной группы (РЬ), 0,03% солей кальция.
Мышьяк, сульфиты и тиосульфата, а также гигроскопическая
влага должны отсутствовать. Реакция водного раствора должна
быть нейтральной.
Из мирабилита, содержащего большие загрязнения, глауберову
соль получают перекристаллизацией. Мирабилит растворяют в
воде при 40°. Теплый (во избежание закристаллизовывания в тру-
трубах) раствор, имеющий концентрацию 30% Na2SO4, отфильтровы-
отфильтровывают от шлама и направляют в кристаллизатор, например, бара-
барабанного типа. Кристаллическая пульпа собирается в корыто с
мешалкой A0—12 об/мин) у нижнего конца кристаллизатора и от-
отсюда поступает на центрифугу. Маточный раствор с центрифуги
используют при растворении новых порций мирабилита.
Кристаллизатор с наружным водяным охлаждением, имеющий
диаметр 0,5 м и длину 10 м, дает в сутки 13 т глауберовой соли.
Расходные коэффициенты на 1 т глауберовой соли: мирабилита
1,095 т, воды 0,215 м3, пара B ат) — 0,28 т, электроэнергии
67 кет • ч.
ЛИТЕРАТУРА
I. А. Н. Хлапова, ДАН СССР, 105, № 3, 500 A955).—5. F. С. Kracek,
J. Phys. Chem., 38, № 9, 1281 A929). —3. F. С. Kracek, R. E. Gibson, ibid.,
34, № 1, 188 A930). — 4. H. S. Dooth, R. M. Bidwell, J. Am. Chem. Soc,
72, 2570 A950). — 5. M. И. Равич, Ф. Е. Боровая, В. Я. Кеткович,
ИСФХА АН СССР, 22, 240 A953). — 6. W. L. Marshal, H. W. Wright,
С. Н. Secoy, J. Am. Chem. Educ, 31, № 1, 36 A954). —7. G. Brodale,
W. Gianque, J. Am. Chem. Soc, 80, 2042 A958). —8. В. М. Еленевская,
Автореф. канд. дисс, ИОНХ АН СССР, 1955. — 9. М. И. Р а в и ч, В. М. Еле-
невская, ИСФХА АН СССР, 25, 176 A964). — 10. А. Г. Б е р г м а н, В. А. X и т-
ров, Там же, 21, 199 A952).
II. М. Л. Гавриш, И. С. Галинкер, Зап. Харьковск. сельскохоз. ин-та,
14 E1), 29 A957). — 12. И. Н. Б о ч к а р е в а, сб. «Технология переработки при-
природных солей и рассолов», Изд. «Химия», 1964, стр. 228. —13. П. М. Лукья-
Лукьянов, История химических промыслов и химической промышленности в России,
т. Ill, Изд. АН СССР, 1951. — 14. В. М. Букштейи, Б. В. Яснопольский,
Хим. пром., № 5, 133 A948).— /5. А. И. Г орбанев, ЖПХ, 27, № 8, 804; N° 9,
921 A954). —16. Г. М. Куперман, Д. X. Гварамадзе, С. И. Джикия,
Н. П. 3 а р к у а, Труды Ин-та химии АН ГрузССР, т. II, 1953, стр. 117. —
17. Н. L. Bittrich, E. Leibnitz, J. prackt. Chem., 3, № 3—4, 126 A956).—
18. А. И. Г о р б а н е в, В. Я. Н и к о л и н а, Труды ВИСП, т. 7, 1954, стр. 5. —
19. Т а к и м о т о, X а т т а р и, J. Chem. Soc. Japan. Ind. Chem. Sec, 58, № 9,
654 A955). — 20. H. Siebourg, W. Moll, пат. ГДР 4944, 1953.
21. A. X. С пи в а к, В. Н. Белов, Хим. пром., №5,391 A968).—
22. Б. А. Б е р е м ж а н о в, С. К. Н у р л а н о в а, сб. «Химия и химическая тех-
технология», № 3—4, Изд. MB и ССО КазССР, 1965, стр. 27. — 23. Н. Е. Кири-
Кириченко, А. Ф. Б оря чек. Хим. пром., № 6, 178 A950). — 24. О. И. Пудов-
Пудовкина и др., Труды УНИХИМ, т. 7, 1958. стр. 87.— 25. А И. Дзенс-Литов-
с к и й, Труды ВНИИГ, в. 32, 1956, стр. 87.— 26. Б. А. Б е р е м ж а н о в. Реф. докл.
и сообщ. VIII Менделеевского съезда, 1, 51 A959). —27. А. И. Дзенс-Литов-
ский, Хим. пром., № 6, 1 A947). — 28. Р. А. Д ы м ш и ц, сб. «Технология пере-
переработки природных солей и рассолов», Изд. «Химия», 1964, стр. 150.—
29. В. И. Николаев, А. И. Качалов, ЖПХ, 15, № 1_2 A942); ДАН СССР.
№ 6, 57 A941). -— 50. В. И. Николаев, Л. С. Дынкина, Труды Калмыцкой
н.-и. станции, Изд. Местпрома Калм. АССР, 1940.
31. В. М. Букштейи, Р. С. Р о с к и н а, Труды ВНИИГ, в. 31, 1956.
стр. 83. — 32. В. Н. Щербина, Глауберит, глауберитовые породы и их кора
выветривания, Кирг. ФАН СССР, Фрунзе, 1952. — 33. М. А. Штерн, Труды
ВНИИГ, в. 31, 1956, стр. 99. — 34. Я. Д. Фридман, А. А. Зиновьев,
М. Д. Лапина, Двойные сульфаты натрия и кальция и переработка их при-
природных отложений, АН КиргССР, Фрунзе, 1956. — 35. В. И. Ксензенко,
С. Исмаилов, Н. А. За нем он ец, ЖПХ, 39, № 12,2622 A966).—
36. Я. Д. Фридман, В. М Одинцов, М. Д. Л о п и н а, Труды ин-та химии
АН КиргССР, в. 8, 1957, стр. 57. — 37. Б. В. Яснопольский, сб. «Сульфат
натрия в СССР», Изд. АН СССР, 1946, стр. 5,— 38. В В. Гецеу, Труды Ин-та
геологии Министерства геол. СССР, в. 6, Махачкала, 1966, стр. 154. —
136
Гл. IV. Природный сульфат натрия
Литература
137
39. В. П. Ильинский, Г. С. Клебанов, Труды Соляной лаборатории АН
СССР, т. 1. 1932; т. 4, 1934.-40. В. П. Ильинский, Г. С. Клебанов,
Я. Б. Б л ю м б е р г, Там же, т. 5, 1936.
41. Я. Б. Блюмберг, Труды Комиссии по комплексному изучению Кас-
Каспийского моря, т. 11, 1940, етр. 83. — 42. А. В. Николаев, ИСФХА АН СССР,
18, 259 A949). — 43. Г. С. Седельников, Д. В. Буйневич, сб. «Проблемы
комплексного использования минеральных богатств Кара-Богаз-Гола, Ашхабад,
1959, стр. 97.-44. Д. В. Буйневич, И. Н. Л е Пешков, Г. С. Седельни-
Седельников, В. Д. Поляков, В. К. Соловьев, П. А. Грек о.в, Л. А. Андреева,
Там же, стр. 40.—45. Г. С. Седельников, Л. С. Ефименко, В. К. Со-
Соловьев, Е. И. Азарова, Д. В. Буйневич, П. А. Греков, Изв. АН
ТуркмССР, № 3, 30 A958). — 46. Г. С. Седельников, Хим. пром., № 7, 501
A963).— 47. Я. Б. Блюмберг, Там же, № 7, 4 A947). — 48. Я. Б. Блюм-
б ер г, Там же, № 6, 342 A954). — 49. Б. Ч. Ч а р ы е в, Кара-Богаз-Гол. История
исследования и промышленного освоения, Ашхабад, 1950. — 50. А. Д. Пельш,
Труды ВНИИ Г, в. 23, 1552, стр. 118.
51. В. М. Букштейн, М. Ю. Г а р к а в и, Н. С. Гаркави, ЖПХ, 25,
№ 8, 826 A952). — 52. С. Ю. Геллер, Изв. АН СССР. Сер. географ., № ?
A954). — 53. В. А. Вахрамеева, Труды ВНИИГ, в. 32, 1956, стр. 67; в. 35,
1959, стр. 7. — 54. А. И. Д 8 ен с - Л ито век и й, М. Ю. Гаркави, Г. А. Ва-
Васильев, Л. В. Еловская, Там же, в. 35, 1959, стр. 16. — 55. В. М. Б у к-
штейи, ЖПХ, 14, № 23, 1687 A937). — 55. А. И. Гор б ан ев, Хим. пром.,
№ 9, 310 A953). — 57. А. И. Дзенс-Литовский, Наука и жизнь, № 6
A944). — 58. В. М. Букштейн, Труды ВНИИГ, в. 21, 1949, стр. 243. — ,
59. А. Д. Пельш, Хим. наука и пром., 2, № 6, 734 A957). — 60. В. М. Бук-
Букштейн, сб. «Технология переработки природных солей и рассолов», Изд. «Хи-
«Химия», 1964, стр. 3.'
61. К. А. Баллод, ЖХП, №3, 17 A939).— 62. Г. С. Клебанов,
Д. М. К о р ф, Л. В. Еловская, Труды Соляной лаборатории АН СССР, т. 12,
1937. — 63. Ф. Ф. Бадер, сб. «Исследования озер СССР», в. 6, 1934.—
64. Л. М. Гроховский, Сборник работ Мосгеолнеруд., в. 1, 1950 стр. 43.—
65. А. Ф. Горбов, ДАН СССР, 74, № 5, 975 A950). — 66. С. 3. Макаров
И. Г. Дружинин, ИСФХА АН СССР, 9, 356 A939). — 67. И. Г. Дружинин,
ДАН СССР. 31, № 1, 901 A941). — 68. И. Г. Дружинин, сб. «Сульфат натрия
в СССР», Изд. АН СССР, 1946, стр. 103. — 69. А. Б. Здановский, Труды
ВНИИГ, в. 27, 1953, стр. 193. — 70. А. И. Дзенс-Литовский, Сульфатные
месторождения СССР, Изд. АН СССР, 1947.
71. Е. В. Посохов, Изв. АН КазССР. Сер. энергет.. в. 2 A949).—
72. О. Д. Кашкаров, И. Д. Карпюк, Я. И. Тычин о, Труды ВНИИГ,
в. 23, 1952, стр. 131. — 73. М. Г. В а л я ш к о, А. А. Нечаева, Т. Б. Пале-
Палено в а, Там же, в. 24, 1952, стр. 106. — 74. А. Г. Бергман, сб. «Сульфат натрия
в СССР», Изд. АН СССР, 1946, стр. 40. — 75. С. М. Мукимов, Р. И. К а п-
к а е в а, А. Г. Б е р г м а и, Труды Ин-та химии АН УзбССР, т. 2, 1949, стр. 145.—
76. И. Н. Л е Пешков, сб. «Сульфат натрия в СССР», Изд. АН СССР, 1946,
стр. 76.-77. Г. Г. У р а з о в, И. Н. Л е п е ш к о в, Труды Ин-та химии АН
УзбССР, т. 2, 1949, стр. 89. — 78. С. М. Кореневский, Труды ВНИИГ в 35
1959, стр. 84. — 79. W. L. Weisman, R. С. Anderson, Min. Eng., 5, № 7,
711 A953). — 80. А. И. Гор бане в, В. Я. Николина, Сульфат натрия, Гос-
химиядат, 1954.
81. Я. Б. Блюмберг, Хим. пром., № 4, 209 A954). — 82. А. Д. Пельш,
сб. «Технология переработки природных солей и р.ассолов», Изд. «Химия», 1964
стр. 291. — 83. Р. С. Роскина, Труды ВНИИГ, в. 27, 1953, стр. 205 —
84. Chem. Eng., 60, № 8, 242 A953). — 85. А. Д. Пельш, Труды ВНИИГ в 24,
1952, стр. 99. — 86. А. Д. Пельш, Там же, стр. 86.-87. Н. М. Абдулга-
И и е в, сб. «Технология переработки природных солей и рассолов», Изд. «Химия»,
1964, стр. 254.-88. Я. М. Хейфец, ЖПХ, 34, № 3, 473 A961). — 89. С. 3. Ма-
Макаров Л С Иткииа, Промышленные методы обезвоживания мирабилита,
Изд. АН СССР, 1946. —S0. В. Я. Бернасовский, Вести. АН КазССР, № 12
A953).
91. В. П. Ильинский, В. М. Филиппео, ЖХП, в, № 1, 3 A929).—
92 В И. Запорожченко, Там же, № 6, 560 A935). —S3. О. Д. Кашка-
Кашкаров, Бюлл. ВИГ, К» 3 A939). — 94. Р. А. М а н дел ь, Там же, № 3, 7 A940).—
95. В. П. Ильинский, ЖПХ, 3, № 14 B0), 1135 A926). — 96. С. 3. Мака-
Макаров, Материалы к физико-химическому изучению соляных озер Кулундинской
степи, Изд. АН СССР, 1935. — 97. С. 3. Макаров, Усп. хим., 5, № 7—8. 1031
A936). —98. Г. И. Белов, И. П. Калинин, ЖХП, К» 6, 21 A940).—
99. К. Н. Шабалин, Ю- П. Каретников, ЖПХ, 24, № 1, 11 A951).—
100 Н. А. Ушатинский, С. И. Голуб, В. М. Букштейн, Хим. пром., № 6,
324 A956).
101. Л. Н. Матусевич, В. В. Ухлов, ЖПХ, 40, №> 9, 1935 A967).—
102. К. Ф. Павлов. В. А. К о пыл ев, ЖХП, № 6, 29 A938). — 103. К. А. К о-
Ье, С. W. Hauge, Power, № 8 A933). — 104. Е. W. Douglas, С. О. Ander-
Anderson, Chem. Met. Eng., № 5, 135 A941). — 105. L. Lesniewicz, Chem. stosow.,
2, № 3, 291 A958).— 106. В. И. Николаев, А. К- Сен юта, ДАН СССР, 16,
№ 4, 219 A937). — 107. А. Д. Перепелица, Новые способы комплексного энер-
энергохимического использования углей, переработки мирабилита и получения соды,
Иркутск, 1947, стр. 91. — /08." Р i ere e, Chem. Met. Eng., № 12, 718 A937).—
109 Р. А. Манд ель, Бюлл. ВИГ, № 6—7, 1939, стр. 36. —110. A A Hol-
Holland, Chem. Eng., 58, № 1, 106 A951).
111. Ю. Я. Каганович, Хим. пром., № 7, 404 A955). —112. В. Ф. Чер-
Чернов, Производство кальцинированной соды, Госхимиздат, 1956. —113. С. М. Б р е-
ховский, Стекло и керамика, № 8, 17 A949). —114. Ю. Я. Каганович,
Хим. наука и пром., 2, № 6, 764 A957). — 115. Ю. Я. Каганович, А. Г. Зло-
б и н с к и й, Хим. пром., № 5, 389 A960). — 116. Ю. Я. Каганович, А. Г. 3 л о-
б и н с к и и, сб. «Техника сушки во взвешенном слое», в. 2, изд. ЦИНТИХим-
нефтемаш, 1966, стр. 44. — 117. О. Д. Кашкаров, Труды ВНИИГ, в. 50, 1967,
стр. 102. — 118. В. Е. Грум-Гржимайло, ЖХП, № 2, 18 A925).—
119. A, Zieren, J. Rupp, пат. ФРГ 929366, 1955. — 120. Баба, Иноуэ, япон.
пат. 70, 1955.
121. А. И. Горбанев, Б. Ю. Мигулии, Труды ВИСП, т. 6, 1952,
стр. 56.-/22. В. И. Николаев, Т. И. Арнольд, ЖПХ, 15, № 3, 83
A9.42) . — 123. А. Н. Вишневский, А. Ф. Б о р я ч е к, Труды ВИСП, т. 6, 1952,
стр. 44. —124. К- О da, япон. пат. 1979, 1954. — 125. А. А. Яковкин, Изв. Рос.
ин-та прикл. хим., 1, № 2, 7 A923). — 126. А. А. Яковкин, Там же, 1, № 3, 1
A925). — 127. А. А. Яковкин, Труды ГИПХ, в. 8, 1927, стр. б. — 128. А. П. Б е-
лопольский, П. И. Александров, ЖПХ, 4, № 5, 576 A931); Труды
НИУИФ, т. 144, 1940, стр. 57.— 129. А. Н. Вишневский, А. ф. Борячек,
3. К. Зубахина, Труды ВИСП, т. 7, 1954, стр. 65. —130. R. E. Vener,
A. R. T h о m p s о n, Ind. Eng. Chem., 42, № 3, 464 A950).
131. К. К. Sapre, Chem. Age (India), 7, № 1, 55 A956).—Щ. М. А. Шт ер н.
Труды ВНИИГ, в. 31, 1956. стр. 99. —133. И. Г. Дружинин, В. И. Нико-
Николаев, И. С. Чел я дин а, А. И. Лазарева, ЖПХ, 22, № 8, 787 A949).—
134. В. И. Николаев, сб. «Сульфат натрия в СССР», Изд. АН СССР, 1946,
стр. 139. — 135. В. П. Ильинский, Г. С. Клебанов, Труды ГИПХ, в. 13,
1929, стр. 45. —Ш. Р. С. Роскина, Труды ВНИИГ, в. 31, 1956, стр. 34.—
137. В. С. Егоров, сб. «Сульфат натрия в СССР», Изд. АН СССР, 1946,
стр. 161. —138. W. Simon, G. An del finger, пат. США 8190721 и 3190722,
1965. —• 139. Правила и нормы техники безопасности и промышленной санитарии
в производстве содовых продуктов и природного сульфата натрия, Госхимиздат,
1959.
Глава V
ПРИРОДНЫЕ КАЛИЙНЫЕ СОЛИ
Важнейшими солями калия являются хлорид и сульфат и обра-
образуемые ими минералы.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Хлорид и другие галогениды калия кристаллизуются из водных
растворов по типу NaCl. Выше 0° (за исключением KF-2H2O)
кристаллизуются безводные соли. Существует кристаллогидрат
КС1 • Н2О, плавящшся при —5,30°; эвтектическая температура
КС1-Н2О + лед —9,80е1.
Плотность кристаллов КС1 1,99 г/см3; теплота плавления
6,41 ккал/моль; теплота сублимации (КС1кр—КС1Г) 53,4 ккал/моль.
Температуры плавления и кипения галогенидов калия повышаются
в ряду I —> F:
Т. плавл., °С Т. кип., °С
KI 682 1330
КВг 728 1376
КС1 768 1417
KF 856 1505
Калий также образует полигалогениды, например К1з*ЗН2О,
К1СЦ и др.
Насыщенные водные растворы галогенидов калия содержат
следующие количества растворенного вещества (в вес. %):
0° 25° 100°
KI 56,2 59,8 67,35
КВг 34,92 40,7 61,20
КС1 21,90 26,45 35.90
KF 30,70 48,90 '59,80
Диаграмма растворимости в системе КС1—NaCl—Н2О приве-
приведена на рис. 38.
Применение
139
Сульфат калия K2SO4 образует четыре полиморфных модифи-
модификации с температурами превращений 300, 350, 449 и 585°2; пла-
плавится он при 1069°. Существует ряд кислых солей сульфата ка-
калия, температура плавления которых значительно ниже, чем у
средней соли; так, например, для КзЩБС^Ь она равна 350°3.
Растворимость сульфата калия в воде при 0° — 6,71%; 25° —
10,75% и 100°—19,4%- Из водных растворов кристаллизуется без-
20 25 30 35 40
KCI, вес.%
45 50 5S
Рис» 38. Политермическая диаграмма растворимости в системе
KCl-NaCl-H2O.
водная Ьоль, а ниже 9,7е KsSO4 • Н2О; температура плавления эв-
эвтектики K2SO4 • Н2О + лед — 1,55°4.
Давление водяного пара над насыщенными растворами при
100° для КС1 — 567,8 мм рт. ст., для K2SO4— 723,8 мм рт. ст. О да-
давлении пара над растворами системы К+, Na+, Mg+|CI, SO4
см.Б-6. О давлении при высоких температурах см.7.
.2-
ПРИМЕНЕНИЕ
Калийные соли применяют главным образом в качестве мине-
минеральных удобрений. Основным видом продукции калийной промы-
промышленности является хлористый калий, около 95% которого ис-
используют в качестве минерального удобрения, а остальные 5% пе-
перерабатывают на едкое кали и другие соединения калия.
Из общего количества калийных удобрительных солей 8—10%'
вырабатывают в виде сульфата калия и двойной соли сульфатов
калия и магния (K2SO4 • MgSO4), калимагнезии, используемых
J40
Гл. V. Природные калийные соли
Применение
,141
для удобрения почв под хлорофобные культуры (табак, цитрусо-
цитрусовые и др.), качество которых ухудшается под влиянием иона хлора.
Основными производителями калийных солей до последнего
времени являлись СССР, США, ГДР, ФРГ и Франция. Начиная с
1965 г. быстро развивается калийная промышленность в Канаде
на базе Саскачеванского месторождения; предполагается, что уро-
уровень производства в Канаде в ближайшее время превысит таковой
в США. Сравнительно небольшие количества калийных солей вы-
выпускается в Испании, Израиле и Италии; ведется подготовка к ос-
освоению калийных месторождений в Конго и Эфиопии. Общая вы-
выработка калийных солей за последние десять лет возросла более
чем в 2 раза и составляет в настоящее время (в пересчете на К2О)
15 млн. т в год.
Соответственно росту производства увеличилось потребление
на единицу сельскохозяйственной площади, которое в ряде высоко-
высокоразвитых стран достигло оптимального для данного вида почв
уровня. Количество вносимых солей в пересчете на КгО в кг на
1 га сельскохозяйственной площади составляет: в США — 70,
Бельгии—120, Японии—103, ФРГ — 80. Однако во многих стра-
странах эта величина не превышает 1—1,5 кг.
В незначительном количестве в качестве удобрения используют
добываемые из недр сырые (т. е. не обогащенные) калийные соли,
содержащие около 20% КгО.
Качество хлористого калия, производимого в СССР, регламен-
регламентируется ГОСТ 4568—65; в зависимости от способа производства,
выпускается хлористый калий двух марок: К — получаемый кри-
кристаллизацией из раствора и Ф — флотационным обогащением ка-
калийных руд (табл. 15).
ТАБЛИЦА 15
Содержание хлористого калия по ГОСТ 4568—65
Хлористый калий (КС1):
в пересчете на сухое ве-
вещество в %, не менее
в пересчете на КгО в %,
не менее
Влага в %, не более . . . .
Хлористый натрий (NaCl) в пе-
пересчете на сухое вещество
в %, не более
Не растворимый в воде остаток
в перестете на сухое веще-
вещество в %, не более . . . .
Марка К
сорт
высший
99,0
0,3
0,9
0,1
первый
98,3
1,0
1.4
0,15
второй
95,0
60,0
1,0
4.5
Не
сорт
второй
95,0
60,0
1,0
4,5
нормируе
третий
92,0
58,1
1,0
7.0
гея
Марка Ф
Хлористый калий, поставляемый сельскому хозяйству, должен
быть не слеживающимся; в целях устранения слеживаемости, до-
допускается обработка его аминами или другими реагентами.
Наряду с выпуском мелкокристаллического хлористого калия
предусматривается также производство гранулированного или
крупнокристаллического продукта, получаемого кристаллизацией
из растворов и флотационным обогащением; для 3 и 2 сортов со-
содержание класса 4—2 мм должно составлять не менее 80% и
класса 1—2 мм — не более 20%.
Сульфатнокалийные удобрения в СССР выпускаются в виде
сульфата калия, калимагнезии (двойной соли сульфатов калия и
магния с примесями хлоридов калия и натрия) и калийно-магние-
вого концентрата, получаемого флотационным обогащением каи-
нито-лангбейнитовой руды.
Калимагнезия в соответствии с техническими условиями содер-
содержит в пересчете на сухое вещество: в 1 сорте КгО не менее 30%,
во 2 сорте 28%; MgO, соответственно, 10 и 8%; содержание иона
хлора не должно превышать 5% для 1 сорта, для 2 сорта — не
регламентируется. Содержание влаги для 1 и 2 сортов должно быть
не более 5%, в том числе гигроскопической — не более 2%- Ма-
Материал должен проходить через сито с отверстиями 5 мм.
Калийно-магниевый концентрат выпускают также двух сортов:
Сорт 1 Сорт 2
КгО в пересчете на обезвоженное
вещество в %, не менее 19,0 17,5
MgO в пересчете на обезвоженное
вещество в %, не менее 9,0 8,0
Влага в %, не более 4,0 4,0
Остаток на сите с отверстиями 3 мм
в %, не более 10 10
Сульфат калия, получаемый при переработке полиминеральных
руд по регламенту, разработанному ВНИИГом, выпускается 1 и
2 сортов. Содержание (в %, в пересчете на сухое вещество): КгО
для 1 сорта не менее 50, для 2 — 45; иона хлора не более 0,5 и 2,
соответственно. Влаги не более 0,5% для обоих сортов.
Хлористый калий, производимый в зарубежных странах, выпу-
выпускается с содержанием не менее 60—61 % КгО в виде так назы-
называемого, стандартного (мелкокристаллического с минимальным со-
содержанием пылевидных фракций), крупнокристаллического, со
средним размером зерен @,5—2,3 мм) и гранулированного —
@,8—3,3 мм). По специальным заказам вырабатывают мелкий
хлористый калий @,1—0,2 мм).
Хлористый, калий, производимый для технических целей, ис-
используют для получения едкого кали, хлората и перхлората калия,
применяемых в качестве отбеливающих препаратов и в производ-
. стве взрывчатых веществ, бромида и иодида калия, используемых
142
Гл. V. Природные калийные соли
в фармацевтической и фотографической промышленности, карбо-
карбоната калия, применяемого для получения специальных стекол и
глазури, силиката калия (K2Si2O5), используемого для пропитки
дерева, отбеливания тканей и других целей, цианида калия — ре-
реагента для извлечения золота из руд, перекиси калия (КОг) и про-
прочих перекисных соединений для регенерации воздуха, и других
калийных соединений. Кристаллы КС1 обладают очень высокой
прозрачностью для инфракрасных лучей, поэтому их применяют
в некоторых оптических приборах,
СЫРЬЕ
Содержание калия в земной коре составляет ~1,5%. Калий
входит в состав алюмосиликатов, слагающих многие породы, поле-
полевых шпатов, гранитов, лейцитов, гнейсов, твердых ископаемых со-
солевых отложений и рассолов морского и континентального проис-
происхождения. Составные части почв, особенно глинистые вещества,
активно удерживают (путем сорбции) калий, что, в частности,
имеет очень важное значение для жизни растений. Благодаря та-
такой способности почв вымывание калия идет сравнительно мед-
медленно, в результате чего содержание его солей в природных водах,
как правило, во много раз ниже, чем солей натрия и магния.
В табл. 16 представлены основные, наиболее распространенные
калийные минералы.
ТАБЛИЦА 16
Состав и свойства наиболее распространенных калийных минералов
Минерал
Состав
Плотность,
1,97-1,99
1,57
2,66
2,070-2,19
2,176
2,03-2,15
2,25
2,83
2,58-2,60
2,13
2,70
2,72-2,78
2,60
2,60-2,80
2,45-2,50
2,2-2,8
Твер-
Твердость
2
2-3
2
2,5
3
3-4
2,5
4-5
3
2,5-3
5-в
3,5—4
5-6
2-3
Сильвин
Карналлит
Арканит
Каинит
Бромкарнадлнт
Шенит
Леонит
Лангбейннт
Калушит
Калиборит
Глазерит ¦
Полигалит
Нефелин
Алунит
Лейцит
Глауконит
КС1
КС1 • MgCIj • 6НаО
K2SO4
КС1 • MgSO4 • ЗН2О
КВг • MgBr2 • 6Н2О
K2SO4 • MgSO4 • 6Н2О
K2SO4 • MgSO4 • 4НгО
K2SO4 • 2MgSO4
K2SO4 • CaSO4 • H2O
K2O-4MgO-llB?<V18H2O
3K,SO4 - Na2SO4
KaSO4 • MgSO4 • 2CaSO4 • 2H2O
(K, NaJO • A12O3 • 2SiO2
(K, NaJSO4-AIa(SO4K-4Al(OHK
K2O • AlaO3 • 4SiO2
[(K,NaJO + (Mg, Ca, Fe)O].(Fe, AlJO3.4SiO2-2H2O
Калийные руды определяют по преобладающему содержанию
р них тех или иных минералов. Сильвинитом называют породу,
состоящую из сильвина A0—60%) и галита B5—70%) с приме-
143
сями ангидрита, карбоната магния и глинистых веществ. Встре-
Встречаются сильвиниты с прожилками карналлита или смесь сильви-
сильвинитов с карналлитами.
Крупнейшее в мире Верхнекамское месторождение калийных
солей представлено сильвинитовыми и карналлитовыми рудами;
происхождение его связано с испарением бассейнов Пермского
моря. Примерная схема геологического разреза месторождения
района Соликамска показана на рис. 39 (по А. А. Иванову). Под
покровными породами наносного слоя (известняки, глины и анги-
ангидрит) находится слой покровной каменной соли, ниже его лежит
0 250 500 ISO Ю00 1250*
Рис. 39. Примерный геологический разрез Верхнекамского месторождения
калийных солей:
1 — наносный слой; а —известняки; 3 — сильвнннтовая порода; 4 — ангидрит; 5 —каменная соль;
6 —глины; 7 — карналлнтовая порода.
карналлитовая зона, мощность которой достигает в ряде участков
100 м. В ней пласты желтой карналлитовой породы, содержащей
50—-85% карналлита, прослаиваются каменной солью. В верхней
и нижней части карналлитовой зоны местами залегает сильвинито-
вая порода. Под карналлитовой зоной лежит основная сильвини-
товая зона, средняя мощность которой достигает 30 м. Верхний
горизонт этой зоны состоит из пестрого сильвинита — смеси кри-
кристаллов молочно-белого сильвина с серыми, голубыми и синими
кристаллами галита. Под пестрым сильвинитом залегает горизонт
красного сильвинита — смеси сургучно-красного сильвина с голу-
голубой каменной солью. Содержание КС1 изменяется в зоне пестрых
сильвинитов от 40 до 55%, в зоне красных сильвинитов от 10 до
35%. Под сильвинитовой зоной залегает мощный пласт каменной
соли. Верхняя поверхность соляной толщи располагается ниже
земной поверхности на глубинах от 100 до 350 м. Соляная залежь
имеет форму большой линзы, вытянутой в меридианальном напра-
направлении. Добыча калийных руд Верхнекамского месторождения
производится из пласта пестрых сильвинитов. Содержание КС1 в
добываемом сильвините изменяется в среднем от 23 до 30%, NaCl
144
Гл. V. Природные калийные соли
Сырье
145
от 65 до 75%, нерастворимых глинистых веществ — 0,5—3%; со-
содержание брома, находящегося в рудах в виде бромкарналлита,
составляет от 0,06 до 0,17% 8-
Одной из отличительных особенностей Верхнекамского место-
месторождения является сравнительно большое содержание газов в
виде микровключений и «свободных» газов — в порах и пустотах
породы 9-10. В состав газов входят водород, метан, азот и двуокись
углерода; горючие газы (Н2 и СН4) встречаются главным образом
в карналлитовых рудах.
В последние годы начата эксплуатация крупного месторожде-
месторождения калийных руд в Белоруссии — Старобинского (верхне-девон-
(верхне-девонские отложения). Месторождение представлено сильвинитами с не-
небольшими примесями карналлита, содержание глинистых веществ
составляет от 4—5 до 10—12%.
Предкарпатские месторождения калийных солей (эпоха мио-
миоцена) представлены рядом калиеносных линз, сложенных сульфат-
нокалийными минералами. Калийные породы переслаиваются гли-
глинистой или чистой каменной солью. Наибольшее промышленное
значение имеют Калуш-Голынское и Стебниковское месторожде-
месторождения, представленные сульфатно-хлоридными минералами; руды
сложены из каинитовой породы (галит 20—40%, каинит 35—60%,
полигалит 3—7%, глинистый материал 6—15%) и лангбейнит-каи-
нитовой породы (галит — 30%, каинит 20—30%, лангбейнит 10—
20%, сильвин 5—10%, кизерит 5—10%, глинистые материалы —
до 20%) ч-13.
Кроме перечисленных, месторождения водорастворимых калий-
калийных руд в СССР находятся в Средней Азии (Гаурдакское и Кар-
люкское), в Волго-Эмбинском районе и ряде других.
Месторождения калийных солей в ГДР и ФРГ содержат, на-
наряду с сильвином, галитом и карналлитом, значительные количе-
количества кизерита, ангидрита, а также примеси каинита, лангбейнита,
тахгидрита (CaCl2-MgCl2- 12H2O). Присутствие значительных ко-
количеств сульфатно-хлоридных минералов связано с условиями об-
образования месторождения, солеродным бассейном которого было
Цехштейновое море с относительно низкой степенью метаморфи-
зации рассола. Руды месторождения подразделяются на хартззль-
цевые и карналлитовые.
Примерный состав хартзальцевых пород (в %):
Кнзернтовый Ангидритовый
хартзальц хартзальц
Сильвии ....
Галит
Кизерит ....
Ангидрит . . .
Одно из крупнейших в мире месторождений калийных солей
находится в Канаде (Саскачеван), его эксплуатация начата в
60-х годах. Месторождение представлено сильвинитовыми и кар-
наллитовыми рудами, залегающими на значительной глуби-
глубине
и
20-24
40-60
17-28
1,5
20-23
50-61
0,5
15-20
Впервые в мировой практике на этом месторождении, наряду
с шахтным способом, организована добыча калийных солей под-
подземным выщелачиванием ls.
В США калийные месторождения сложены сильвинитовыми и
карналлитовыми рудами, в некоторых участках имеются пласты
лангбейнита (штат Нью-Мексико).
Калийные месторождения Франции и Испании представлены
сильвинитовыми и карналлитовыми рудами. В Италии перераба-
перерабатываются руды, содержащие каинит, карналлит и сильвинит.
В воде океанов и морей содержится ~0,05% калия. При
объеме воды мирового океана 1370- 106 км3 содержание в нем ка-
калия составляет примерно 7- 10м i КгО, что превышает известные
запасы месторождений калийных солей более чем в 10 миллионов
раз. Таким образом, мировой океан является, практически, неисчер-
неисчерпаемым источником солей калия. Некоторые виды водорослей
(макроцистис и др.) активно извлекают калий из морской воды,
поэтому зола этих растений может служить источником калия.
С площади моря в 400 км2 можно ежегодно собирать такое коли-
количество водорослей, при переработке которых получается более
1 млн. т К2О 16.
В последние годы значительно возросло производство калий-
калийных солей из сгущенной рапы Мертвого моря 17> 18. Калийные соли
получают при комплексной переработке рапы оз. Сирлз (США,
КалифорнияI9. Это месторождение представлено, так называе-
называемым, сухим озером — рапа пропитывает соляной пласт озера и
добывается из скважин. Пласт солей состоит из галита, глазерита
CK2SO4-Na2SO4), троны (Na2CO3 • NaHCO3 • 2Н2О) и борных
минералов. Значительное количество калийсодержащих рассолов
находится в оз. Индер (СССР), которое также относится к типу
сухих озер 20.
Источником для промышленного производства калия могут
служить полигалитовые руды, месторождения которых в СССР
находятся в Жилянском и Волго-Эмбинском районе. Промышлен-
Промышленное извлечение солей калия из полигалитов пока не производится.
Установлено, что отмытая от NaCl полигалитовая порода — хо-
хорошее минеральное удобрение.
Дополнительным источником калийных солей является пыль,
отделяемая в электрофильтрах цементных печей. В сырой шихте,
идущей на приготовление цемента, содержится 0,2—!% КгО, зна-
значительная часть которого возгоняется в процессе прокалки и вы-
выделяется при понижении температуры в электрофильтрах. Содер-
Содержание К2О в водорастворимой форме в этом продукте составляет
20—30%.
146
Гл. V. Природные калийные соли
Получение KCI методами растворения и кристаллизации
147
Переработку сильвинитовых, хартзальцевых и карналлитовых
руд на хлористый калий осуществляют:
1) Методом растворения и раздельной кристаллизации, осно-
основанной на различии температурных коэффициентов растворимости
солевых составляющих руды (этот метод называют еще тепловым
или галургическим).
2) Механическим обогащением породы, главным образом фло-
флотацией; гравитационная сепарация при обогащении калийных руд
не нашла широкого распространения.
3) Комбинацией флотационного обогащения "с растворением и J
кристаллизацией мелких фракций руды; такого рода схемы начи-
начинают широко применяться в последние годы в зарубежной прак-
практике.
4) Подземным выщелачиванием руды с последующей перера-
переработкой рассола выпаркой и кристаллизацией; этот способ приме-
применяется пока только в Канаде при переработке руды, залегающей
на большой глубине.
ПОЛУЧЕНИЕ ХЛОРИСТОГО КАЛИЯ МЕТОДАМИ РАСТВОРЕНИЯ
И РАЗДЕЛЬНОЙ КРИСТАЛЛИЗАЦИИ
Физико-химические основы переработки сильвинитовых руд
В основе получения КС1 методами растворения и кристаллиза-
кристаллизации лежат свойства системы КС1—NaCl—НгО (рис. 38). Из со-
сопоставления изотерм 25 и 100° (рис. 40) видно, что содержание
NaCl в эвтоническом растворе при понижении температуры уве- I
личивается. Фигуративная точка системы, соответствующей соста- -|
ву эвтонического раствора Еш при 100°, при охлаждении оказы-
оказывается в поле кристаллизации КС1. Следовательно, при охлажде-
охлаждении раствора, насыщенного NaCl и КС1, в осадок выпадает только
КС1. При добавлении к насыщенному раствору КС1 твердого NaCl
часть КС1 вытесняется из раствора в осадок.
При охлаждении эвтонического раствора ?100 от 100 до 25°,
вследствие выделения в осадок КС1, состав раствора будет ме-
меняться — его фигуративная точка переместится вдоль луча кри- J
сталлкзации Сп от ?100 до п. Если после отделения осадка KCI |
раствор п снова нагреть до 100°, то он окажется сильно ненасы-
ненасыщенным КС1 и лишь очень немного недонасыщенным NaCl. По- 1
этому если этим горячим раствором обработать сильвинит, то бу-
будет растворяться преимущественно КС1. (Впрочем, это зависит от
условий растворения; например, при противоточной обработке
сильвинита щелоком, когда раствор встречается с материалом,
уже почти не содержащим КС1, вначале будет растворяться боль-
больше NaCl, чем КС1, а затем будет растворяться только КС1 при
одновременном высаливании NaCl из раствора). После отделения
твердого остатка NaCl вновь будет получен горячий эвтонический
раствор ?ioo, из которого при охлаждении выделится КС1. С по-
помощью такого циклического процесса можно осуществлять разде-
разделение сильвинита на КС1 и NaCl.
Для полного выделения КС1 из сильвинита необходимо, чтобы
количество его, вводимое в цикл, соответствовало количеству цир-
циркулирующего щелока. Если принять округленно состав сильвинита
25% КС1 и 75% NaCl (точка S), то точки смесей его с маточным
раствором п расположатся на линии nS. Необходима такая смесь,
которая полностью разделялась бы на раствор Ет и твердый
NaCl. Этому условию соответ-
соответствует смесь состава К, лежа-
лежащего на пересечении линий
nS и ЕюоВ. Таким образом,
для полного растворения КС1
и получения при этом эвтони-
эвтонического раствора ?юо необхо-
необходимо обрабатывать сильвинит
таким количеством маточного
щелока состава п, чтобы от-
отношение количеств п : S рав-
равнялось отношению отрезков
SK:Kn.
Принципиальная схема пе-
переработки сильвинитовых руд
состоит из следующих основ-
основных операций:
1) выщелачивание измель-
измельченного сильвинита горячим
маточным раствором, получен-
полученным после кристаллизации КС1; при этом из сильвинита в рас-
раствор переходит KCI, a NaCl почти полностью остается в отвале;
2) отделение горячего щелока от отвала и осветление его от
мелких увлеченных твердых частиц (ил и солевой шлам);
3) охлаждение щелока, сопровождающееся кристаллизацией
КС1;
4) отделение кристаллов КС1 от маточного раствора, сушка!
5) нагревание маточного раствора, возвращаемого на выще-
выщелачивание КС1 из новых порций сильвинита.
Эта принципиальная схема лежит в основе всех производств
хлористого калия из сильвинитовых руд по методу растворения и
кристаллизации. Некоторые различия в технологических схемах
и режимах процесса вызваны главным образом изменением со-
состава сырья и применением аппаратов различных конструкций.
Практически получаемые составы твердых и жидких фаз после
выщелачивания и кристаллизации несколько отличаются от
Рис. 40. Растворимость в системе
KCI—NaCl—Н2О при 25 и 100°.
148
Гл. V. Природные калийные соли
характерных для рассмотренного выше хода процесса. Состав го-
горячего щелока после выщелачивания сильвинита отличается от
эвтонического — степень насыщения его хлористым калием, в за-
зависимости от способа выщелачивания, составляет 90—96%; по-
поэтому при охлаждении щелока вначале кристаллизуется только
NaCl. После достижения температуры, соответствующей насыще-
насыщению, начинает кристаллизоваться КС1, а выделившийся ранее
NaCl при активном перемешивании мог бы вновь раствориться,
но он обычно прикрывается кристаллами КС1 и поэтому не рас-
растворяется. Это является причиной загрязнения продукта хлори-
хлористым натрием. Так, при степени насыщения горячего щелока хло-
хлористым калием на 96% его содержание в кристаллизующейся
соли составляет 99,3%, а из щелока, насыщенного только на
90,6%, получается соль, содержащая 94,3% КС121. Это показывает
важность достижения максимальной степени насыщения горячего
щелока хлористым калием, как и использования прямоточного вы-
выщелачивания, когда в щелоке содержится меньше мелкокристал-
мелкокристаллической трудноотделяемой взвеси NaCl, образующейся в больших
количествах в результате высаливания NaCl хлористым калием'
при противоточном выщелачивании.
В производственных условиях в процессе осветления горячего
щелока он несколько охлаждается и из него кристаллизуется не-
некоторое количество NaCl, удаляющееся вместе с солевым и гли-
глинистым шламами, — происходит так называемое самоочищение го-
горячего щелока, повышается степень его насыщения хлористым
калием.
При вакуум-охлаждении щелока от 100 до 20° теоретически |
может быть испарено 12% воды без загрязнения кристаллизую-
кристаллизующегося КС1 хлористым натрием. Практически выделяющийся в ¦:
начале кристаллизации с испарением воды хлористый натрий |
прикрывается затем слоем хлористого калия и не может перейти
обратно в раствор; поэтому маточный щелок остается недонасы-
щенным по NaCl, хотя последний и находится в твердой фазе. Для
предотвращения загрязнения хлористого калия хлористым нат-
натрием к щелоку добавляют в начале кристаллизации (или перед
ней) воду — конденсат.
Если сильвинит загрязнен карналлитом, то вследствие цирку-***
ляции щелока в нем постепенно накапливается MgCl2. В этом слу-
случае щелок нужно обновлять, так как в присутствии MgCl2 раство-
растворимость КС1 уменьшается, а при концентрации MgCl2 больше
100 г/1000 г Н2О растворимость NaCl в насыщенных растворах
КС! с понижением температуры уменьшается (а не увеличивается,
как в отсутствие MgCl2). Это приводит к загрязнению КС1, кри-
кристаллизующегося при охлаждении горячего щелока, хлористым
натрием. По данным Я. М. Хейфеца, при содержании в сильвините
1,106% MgCl2 и при испарении в процессе охлаждения щелока
Получение KCI методами растворения и кристаллизации
149
4% воды содержание КС1 в продукте равно 87% при концентра-
концентрации MgGl2 в маточном растворе 137 г/1000 г Н2О и 80% при
213 г/1000 г Н2О; извлечение КС1 из сырья в первом случае 90,3,
во втором 92%.
Растворение сильвина
При растворении сильвина в оборотных щелоках требуется
получить близкий к насыщению по КС1 щелок при минимальном
содержании калия в отвале22. Необходимо также обеспечить до-
достаточно высокую интенсивность растворения. При прочих равных
условиях интенсивность растворения возрастает при увеличении
степени измельчения руды. С другой стороны, крайне нежелатель-
нежелательным является присутствие фракции —0,2 мм, так как тонкодис-
тонкодисперсные частицы, содержащие сильвин, выносятся со щелоком иэ
растворителей в виде солевого шлама и уходят в отвал.
Кроме того, появление мелких частиц руды связано с раскры-
раскрытием глинистых компонентов, обладающих весьма тонкой струк-
структурой и известной способностью к набуханию, что резко ухудшает
условия осветления щелоков. Оптимальная степень измельчения
определяется составом и структурой породы. Для верхнекамских
сильвинитов в качестве оптимального принято дробление до круп-
крупности частиц 5—0,25 мм; остаток на сите 5 мм не должен превы-
превышать 5%.
Температурный режим растворения играет чрезвычайно важ-
важную роль, поскольку при повышении температуры увеличивается
концентрация насыщения КС1 и возрастает скорость растворения.
Так как растворение проводят в аппаратах под атмосферным дав-
давлением, предельная температура горячих щелоков составляет
100—103°; этой температуре соответствует концентрация насыще-
насыщения, равная 270 г\л КС1 и 210 г\л NaCl. При понижении темпера-
температуры горячего щелока на 5—10° содержание КС1 снижается до
245—250 г/л, что вызывает соответственное уменьшение выхода
калия из единицы объема циркулирующего щелока.
Степень насыщения щелока и количество нерастворенного КС1
в отвале зависят от принятого режима растворения, которое осу-
осуществляют противотоком, прямотоком и по комбинированным схе-
схемам. При оценке эффективности различных режимов растворения-
необходимо учесть также происходящее при растворении высали-
высаливание хлористого натрия. Шламообразование отрицательно ска-
сказывается на показателях процесса в целом, так как при этом воз-
возрастают нагрузка на аппаратуру для отстаивания и обработки
шламов и потери калия с отвалом.
В режиме противотока средняя разность концентрации рабо-
рабочего раствора и концентрации насыщения значительно выше, чем
при прямотоке, что обеспечивает повышение интенсивности раство-
растворения. С другой стороны, при противотоке высаливание NaCl
L
150
Гл. V. Природные калийные соли
Получение KCI методами растворения и кристаллизации
151
значительно увеличивается. В идеальных условиях растворения
верхнекамского сильвинита скорость растворения при прямотоке в
2 раза меньше, чем при противотоке, но количество высоленного
NaCl при этом возрастает в 1,5 раза. В промышленном аппарате,
учитывая несовершенство перемешивания, эффективность противо-
точного растворения несколько снижается, главным образом в ре-
результате частичного экранирования сильвинита выделяющимися
кристаллами NaCl, но увеличение шламообразования в указан-
указанном соотношении сохраняется 23.
В большинстве случаев в качестве оптимальной выбирают ком-
комбинированную схему растворения, по которой в первом по ходу
руды растворителе движение щелока и породы происходит пря-
прямотоком, а в последующих — противотоком. По такой схеме в пер-
первый растворитель подают щелок после выщелачивания во втором
растворителе, в который поступают нагретый маточный щелок и
-слабые щелока после выщелачивания отвала в третьем раствори-
растворителе (при промывке водой); во втором растворителе происходит
дорастворение руды, передаваемой из первого растворителя. Ко-
Количество солевого шлама, выделяющегося на ! м3 осветленного
щелока при переработке верхнекамских сильвинитов, составляет
в среднем 250—260 кг и может быть снижено на 20% при подаче
части маточного щелока (~15% от общего количества) в первый
растворитель.
На большинстве современных калийных фабрик выщелачива-
выщелачивание сильвинитовых руд осуществляют в шнековых растворителях,
представляющих собой горизонтальную корытообразную мешалку
длиною до 22—24 м; перемешивание и транспортирование пульпы
•осуществляется шнековой спиралью, вращающейся на горизон-
горизонтальном валу. Расход тепла, вызванный эндотермическим эффек-
эффектом растворения КС1, вводом холодного сильвинита и отдачей
тепла в окружающую среду стенками аппарата, компенсируют
подачей пара или перегревом под давлением растворяющего ще-
щелока. В последнем случае, при вводе щелока в растворитель, про-
происходит выделение растворного пара за счет самоиспарения. Ввод
острого пара осуществляют через дюзы: в некоторых конструк-
конструкциях шнековых растворителей внутри корпуса размещены нагре-
нагревательные элементы, обогреваемые глухим паром. Последний ва- .
риант является менее удачным — трубчатки нагревательных эле-
элементов быстро выходят из строя в результате коррозии и эрозии,
кроме того, нагревательные элементы не обеспечивают быстрого
нагрева сырой соли, что снижает интенсивность растворения. Удель-
яая производительность шнековых растворителей 1,7—3 т/{м3-ч)
руды (в зависимости от ее состава).
При небольших масштабах производства применяют барабан-
барабанные растворители, в которых руда и щелок движутся параллель-
ио24. Удельная производительность барабанных растворителей
ниже, чем шнековых примерно на 30—40%, расход электроэнергии
в несколько раз выше и составляет ~ 1 кет • ч на 1 т руды (вместо
~0,25 квт-ч на 1 г в шнековых). Недостатком барабанных рас-
растворителей является также невозможность проведения противо-
точного растворения и вынос тонких фракций сильвина с щелоком.
Значительная интенсификация растворения калийных руд ожи-
ожидается при проведении процесса в условиях турбулентного по-
потока по трубам25. Этим способом осуществляют растворение от-
отвалов галита на калийных предприятиях ГДР. Время полного
растворения галита составляет ~2 мин при скорости потока в
трубе ~ 1 м/сек.
Проведены исследования, показавшие возможность растворе-
растворения галитового отвала и калийных солей в кипящем слое26.
Кристаллизация хлористого калия
На современных калийных предприятиях кристаллизацию про-
производят в многоступенчатых вакуум-кристаллизационных установ-
установках с рекуперацией тепла растворного пара. На некоторых из них
сохранились устаревшие комбинированные схемы с охлаждением
в вакуум-кристаллизаторах и башнях типа градирен. Охлаждение
в башнях происходит при самоиспарении распыленного раствора;,
выделившиеся кристаллы хлористого калия и маточный щелок
собирают внизу башни и направляют на отстойную станцию. Раз-
Разность температур между наружным воздухом и охлажденным рас-
раствором составляет летом 10—15°, а зимой 25—35°.
При охлаждении в башнях исключается возможность исполь-
использования тепла растворного пара; недостатком этого метода яв-
является также мелкокристаллическая структура получаемого про-
продукта, который вследствие этого сильно слеживается. При ком-
комбинированном охлаждении в вакуум-кристаллизаторах и башнях
кристаллы из башен имеют линейные размеры в 1,5 раза меньшие,
чем в вакуум-кристаллизаторах. Средние размеры получаемых
кристаллов колеблются в пределах 0,11—0,16 мм27.
Непрерывнодействующие многоступенчатые вакуум-кристалли-
вакуум-кристаллизационные установки обеспечивают возможность рекуперации 40—
70% тепла, затраченного на нагрев щелоков при выщелачивании
руды. Рекуперация тепла осуществляется путем нагревания ма-
маточных щелоков растворным паром в поверхностных конденсато-
конденсаторах или конденсаторах смешения. Скорость охлаждения и перепад
температур при ступенчатой кристаллизации значительно сни-
снижаются, что способствует увеличению размеров кристаллов.
При укрупнении кристаллов, наряду с улучшением товарных
качеств хлористого калия, обеспечивается повышение производи-
производительности центрифуг и сушилок за счет снижения влажности
осадка.
1Б2
Гл. V. Природные калийные соли
Получение КС1 методами растворения и кристаллизации
153
С увеличением числа ступеней охлаждения повышается сте-
степень использования тепла растворного пара, возрастает общая
площадь зеркала испарения и уменьшается перепад температур
в каждой ступени. С другой стороны, многоступенчатые вакуум-
кристаллизационные установки имеют очень большие габариты,
удельный расход металла значителен, велик также объем произ-
производственных помещений. Увеличение числа ступеней свыше 14—•
15 почти не дает дальнейшего повышения температуры нагревае-
нагреваемого щелока (рис. 41) 28. При нагревании в поверхностных конден-
конденсаторах конечная температура щелока на 7—10° ниже, чем при
юн \г /з и а
Рис. 41. Кривая выбора числа ступеней.
нагревании в конденсаторах смешения, однако качество хлористого
калия в последнем случае несколько хуже.
Влияние числа ступеней на размеры кристаллов может быть
-определено по эмпирической формуле, предложенной Вязововым:
тде d — средний размер кристаллов, мм;
N — число ступеней.
В вертикальных вакуум-кристаллизаторах обеспечивается ма-
малый унос брызг щелока с паро-воздушной смесью. Горизонталь-
Горизонтальные вакуум-кристаллизаторы с мешалками дают более равномер-
равномерное и медленное охлаждение, что способствует укрупнению кри-
кристаллов.
Присутствие в растворе взвешенных частиц шлама повышает
вероятность образования центров кристаллизации и поэтому вле-
влечет за собой уменьшение размеров кристаллов. Диспергирующее .
воздействие шламов может быть подавлено введением добавок, из
которых особенно активное влияние оказывают соли свинца, пер-
первичные алифатические амины, пектин, алыинат натрия и дру-
другие 29-31.
При эксплуатации 14-ступенчатых вакуум-кристаллизационных
установок, в которых перепад температуры в каждой ступени со-
составляет в среднем 4—5° и скорость охлаждения около 2 град/мин,
получаются кристаллы, средний размер которых не превышает
0,200 мм при значительном содержании фракции 0,150жж (80%).
Присутствие этой фракции повышает пыление продукта при скла-
складировании и внесении в почву. Для получения хорошо рассевае-
мого хлористого калия размер зерен 0,15 мм является предельным
и содержание этой фракции должно строго контролироваться.
Описаны вакуум-кристаллизационные установки с 24 ступе-
ступенями охлаждения, состоящие из 8 горизонтальных корпусов с
тремя ступенями в каждом корпусе. Первые 19 ступеней снаб-
Н Вакууп-насосу
Ританир]
Прсдунт
C/iuS
Рис. 42. Кристаллизатор „Кри-
„Кристалл" для регулируемой кри-
кристаллизации.
Рис. 43. Кристаллиза-
Кристаллизатор ДТВ для регулируе-
регулируемой кристаллизации.
жены поверхностными конденсаторами, последние 5 ступеней ра-
работают в общем цикле с конденсаторами смешения, орошаемыми
свежей водой. Увеличение числа ступеней до 24 дает некоторый
рост среднего размера кристаллов, однако содержание фракции
—0,15 мм составляет 15—30% 29.
Укрупнение кристаллов достигается при введении ретурной за-
затравки в виде кристаллов средней фракции, отделенных от основ-
основной массы на классификаторах. Наиболее благоприятные резуль-
результаты получены при введении затравки в количестве 40—70% от
выкристаллизовавшегося хлористого калия с дополнительной до-
добавкой алкиламина для подавления образования новых центров
кристаллизации 29.
В последние годы на зарубежных калийных предприятиях все
шире используется так называемая регулируемая кристаллизация,
454
Гл. V. Природные калийные соли
обеспечивающая получение крупнокристаллического продукта 32-35_
Известны два типа аппаратов Для этой цели: кристаллизаторы со
взвешенным слоем типа «Кристалл-Осло» и кристаллизаторы с
циркулирующей суспензией типа «ДТВ» с мешалкой и- всасываю-
всасывающей циркуляционной трубой. Схематическое изображение обоих
типов кристаллизаторов показано на рис. 42 и 43. В кристалли-
кристаллизаторе со взвешенным слоем снятие пересыщения происходит при
контакте раствора с массой взвешенных в потоке кристаллов, при
этом кристаллизующаяся соль отлагается на поверхности частиц
слоя. Укрупненные кристаллы непрерывно отводятся из нижней
части аппарата. Свежие порции раствора смешиваются с цир-
циркулирующим потоком и далее поступают в вакуум-испаритель,
где за счет самоиспарения образуется пересыщенный раствор,
который по центральной трубе направляется в нижнюю часть
.аппарата.
В кристаллизаторах типа «Кристалл» обеспечивается получе-
¦ние весьма крупных (до 2—3 мм) зерен хлористого калия, но
производительность их довольно низка, так как для образования
взвешенного слоя необходимо поддерживать сравнительно не-
небольшую A,5—2 см/сек) линейную скорость раствора, что требует
установки для многотоннажных производств кристаллизаторов
весьма большого диаметра. В кристаллизаторах с циркулирующей
суспензией типа «ДТВ» производительность повышается в не-
несколько раз, но размер получаемых кристаллов меньше. В настоя-
настоящее время используются 4- и 8-ступенчатые установки с диамет-
диаметром аппаратов 5 м при производительности 600 тыс. т хлористого
кадия в год38.
Схема производства хлористого калия методом
растворения и кристаллизации
Примером современного производства хлористого калия яв-
является предприятие, перерабатывающее сильвинит верхнекамского
месторождения. Схема производства изображена на рис. 44.
Дробленный до крупности 0,25—5 мм сильвинит из солемель-
ницы подают в бункеры на склад сырых солей, откуда с помощью
лоткового качающегося питателя забирают на ленточный транс-
транспортер с автоматическими весами и направляют в шнековые рас-
растворители длиной 21,5 м, диаметром 2,76 м; число оборотов
шнековой спирали 8 в минуту. Сильвинит последовательно транс-
транспортируется через два шнековых растворителя, причем первый ра-
работает по принципу параллельного тока, а второй — противотока.
Передача сильвинита из первого во второй аппарат и удаление от-
отвала из второго аппарата осуществляются наклонными элевато-
элеваторами с дырчатыми ковшами, из которых щелок сливается обратно
JB растворители. Для компенсации тепловых потерь в растворители
Рис. 44. Схема производства хлористого калия из сильвинита:
U 4, S7 и 61 — ленточные транспортеры; 2 — бункер для сильвнннта; 3 — питатель; 5 — автоматические весы; 6, н 7 10 — шнековые растворители
8, 9 vi II — ковшевые элеваторы; 12 и 19 — скребковые транспортеры; 13 — плаи-фильтр; 14 — вакуум-иасос; /5 — вакуум-котел; 16 — барометрический
конденсатор смешения; 17 н 44 — брызгоуловителн; 18 и 41 — барометрические баки; 20-резервуар для промывной воды; 21, 39, 50, 52 и 55 — на-
сосы; 22 — шестиконуспый отстойник-сгуститель; 23 — сборник солевого шлама; 24 — мешалка для глинистого шлама; 25 — отстойник-сгуститель
глинистого шлама; 26 — резервуар для промывной воды; 27 — расходный бак; 28 — вертикальный корпус вакуум-кристаллизационной устанопки;
29—34 — горизонтальные корпусы вакуум-кристаллнзационной установки; 35 — бак для хлоркалиевой пульпы; 39 — поверхностный конденсатор;
37— трубчатый подогреватель; 38 — сборник подогретого щелока; 40 — конденсатор смешения; 42 — пароструйный инжектор; 43 — сборный конден-
конденсатор смешения; 45 — дополнительный конденсатор смешения; 46 — барометрический бак для конденсата из поверхностных конденсаторов;
47 — центробежный насос для хлорка лиевой пульпы; 48 — шестиконусный сгуститель; 49 — бак для маточного щелока; 5/— мешалка для сгущенной
хлоркалиевой пульпы; 53-расходная мешалка для хлоркалиевой пульпы; 54— центрифуга; 55-бак для маточного щелока с центрифуг; 5в~барз-
банцая сушилка; 59 — топка; 60 — выгрузочная камера; 62 — дымосос; 63 — циклон-пылеудозитель,
156
Гл. V. Природные калийные соли
вводится через дюзы острый пар A,5—2 ат). Горячий маточный
щелок после вакуум-кристаллизации (растворяющий щелок), на-
нагретый до 105—115°, поступает во второй растворитель, движется
противотоком руде и вытекает в виде «среднего» щелока с плот»
ностью 1,220—1,236 г/см3, который поступает в первый раствори-
растворитель, где движется в одном направлении с сильвинитом. Выте-
Вытекающий из первого растворителя горячий (97—107°) концентри-
концентрированный щелок содержит 245—265 г/л КС1. Для окончательного
извлечения КО отвал из второго растворителя элеватором пере-
передают в третий, более короткий растворитель шнекового типа (дли-
(длиною Им), куда направляют промывные воды и фильтраты, полу-
полученные при обработке отвала и шлама на план-фильтре и при
противоточной промывке. Движение отвала и щелока в третьем
растворителе происходит также противотоком. Кроме дополни-
дополнительного извлечения КС1, в третьем растворителе обеспечивается
рекуперация тепла отвала, передающего частично свое тепло ще-
щелоку; этот щелок присоединяют к растворяющему щелоку, а отвал
элеватором передают на фильтрование. Отвал после промывки го-
горячей водой на элеваторе содержит ~15% маточного раствора.
Для уменьшения потерь хлористого калия его промывают горячей
водой на план-фильтре — фильтре непрерывного действия с гори-
горизонтальной поверхностью фильтрации; в отвале после фильтрации
содержится 4,5—6% Н2О и около 2,5% КО. Осадок сбрасывается
с план-фильтра на скребковый транспортер и удаляется из цеха.
Горячий щелок, вытекающий из первого растворителя, содер-
содержит глинистый и солевой шламы. Отдачение этих примесей осу-
осуществляют в шестиконусном отстойнике ¦— шпицкастене 22; в ка-
каждом конусе отстойника имеется мешалка A об/мин), предназна-
предназначенная для уплотнения шлама и облегчения его выгрузки.
Для увеличения скорости осветления раствора в него вводят
коагулянт, способствующий осаждению мелкодисперсных илистых
частиц. В последнее время крахмал заменяют более эффектив-
эффективными коагулянтами, главным образом полимерными вещества-
веществами36-38. Получает широкое распространение полиакриламид, рас-
расход которого в 5—6 раз меньше крахмала A00—120 г на 1 г гли-
глинистого ила), при значительно большей скорости осаждения. При
использовании подобного высокоэффективного коагулянта стано-.
вится реальной переработка калийных руд, содержащих большие
количества (до 20%) не растворимых в воде веществ.'
При осветлении щелока в отстойнике 22 происходит некоторая
классификация шлама: в первых конусах оседает главным обра-
образом солевой шлам, который возвращают в первый растворитель
или подвергают фильтрации на барабанных вакуум-фильтрах, а в
последних — илистый шлам. Отношение жидкости и твердой фазы
(Ж: Т) в выгружаемом солевом шламе не должно превышать
0,8—1. Илистый шлам из последних конусов отстойника при Ж:Т,
Получение KCI методами растворения и кристаллизации
157
равном 2—2,5, передают на противоточную промывку, состоящую
из батареи сгустителей Дорра.
Вытекающий из отстойника щелок, имеющий температуру 87—
98°, охлаждается до 17—27е в 14-ступенчатой вакуум-кристалли-
вакуум-кристаллизационной установке. Вакуум создается за счет отсоса паро-воз-
душной смеси, образующейся при самоиспарении раствора, си-
системой паровых эжекторов, установленных на конденсаторах.
Вакуум-кристаллизационная установка состоит из одного верти-
вертикального корпуса 28 (I ступень) и шести горизонтальных корпусов
29—34, имеющих 13 ступеней охлаждения (//—XIV). Горизон-
Горизонтальные корпуса имеют лопастные мешалки A6 об/мин).
Осветленный щелок из отстойника засасывается в первый кор-
корпус вакуум-кристаллизационной установки и далее перетекает по
переточным трубам вместе с образующимися кристаллами КС1 из
одной ступени в другую. Из последней XIV ступени пульпа хло-
хлористого калия самотеком по барометрической трубе сливается в
приемный бак. Растворный пар из первых девяти ступеней конден-
конденсируется в поверхностных конденсаторах, нагревая при этом ма-
маточный щелок, направляемый на растворение сильвинита. Щелок
проходит последовательно конденсаторы от IX ступени до /, на-
нагреваясь от 17—27° до 65—75°.
Дальнейшее нагревание растворяющего щелока до 113—115°
осуществляют в трубчатых подогревателях, обогреваемых паром.
Растворный пар из последних пяти ступеней вакуум-кристалли-
вакуум-кристаллизационной установки конденсируют в конденсаторах смешения,
орошаемых водой. Свежая вода подается в конденсатор XIV сту-
ступени и затем самотеком перетекает из одного конденсатора в дру-
другой до конденсатора X ступени, из которого по барометрической
трубе сливается в приемный бак. Система отсоса паровыми эжек-
эжекторами несконденсированной паро-воздушной смеси построена та-
таким образом, что паро-воздушная смесь перекачивается из по-
последней в предшествующую. Из вспомогательного конденсатора
паро-воздушную смесь отсасывают поршневыми вакуум-насосами
и через брызгоуловитель выбрасывают в атмосферу.
Конечное охлаждение щелока определяется давлением в XIV
ступени, которое, в свою очередь, зависит от температуры воды,
поступающей на конденсацию паров; поэтому режим работы ва-
вакуум-кристаллизационной установки в зимних и летних условиях
оказывается несколько отличным.
Для уменьшения кристаллизации хлористого натрия при охла-
охлаждении в процессе вакуум-испарения к раствору, поступающему
на кристаллизацию, присоединяют часть конденсата растворного
пара из первых четырех поверхностных конденсаторов.
Из сборного бака пульпу хлористого калия перекачивают в
шестиконусный отстойник 48 такой же конструкции, как отстой-
отстойник 22 для осветления горячего щелока. Осветленный холодный
158
Гл. V. Природные- калийные соли
Получение КС1 методами растворения и кристаллизации
159
маточный щелок направляют на подогрев в поверхностных конден-
конденсаторах 36, а сгущенную пульпу хлористого калия — в горизон-
горизонтальную мешалку, откуда ее перекачивают шламовыми насосам»
в расходные мешалки над автоматическими центрифугами полу-
полунепрерывного действия 54. Влажность осадка после центрифуги-
центрифугирования, в зависимости от величины кристаллов, изменяется от
5 до 8%.
Рис. 48. Схема установки для сушки хлористого калия в кипящем
слое:
J-ленточный питатель; 2 —бункер; 3-транспорте!,; 4-бункер для сухой соли;
5-печь КС; 6 — выгрузочная течка; 7-шиек для выгрузки; 8 —батарейный
- циклон;»—пенный аппарат; /0-бак для раствора соды; // — дымосос; 12 — шнек;
/3 — ленточные конвейеры; 14 — вентилятор.
Сырой хлористый калий по ленточному транспортеру направ-
направляют на сушку, которую производят в барабанных вращающихся
сушилках. Мелкий хлористый калий, увлекаемый дымовыми га-
газами, отделяют в циклонах и присоединяют к общему потоку го-
готовой продукции, направляемой на склад. Величина удельного
влагосъема при сушке равна 35—45 кг/(м3-ч), конечная влаж-
влажность хлористого калия не выше 1%.
В последние годы в СССР и в США для сушки калийных со-
солей начали применять аппараты кипящего слоя39'40. Новый спо-
способ позволил значительно повысить качество сушки благодаря
большей равномерности и степени высушивания, а также снизить
на 20—30% удельный расход топлива. Схема сушильной уста-
установки для сушки хлористого калия, производительностью 100—
120 т/ч^ представлена на рис. 45.
При сушке хлористого калия в кипящем слое продукт может
быть получен в обеспыленном виде. Это достигается или возвра-
возвратом циклонного продукта на смешение с влажной солью (такой
прием эффективен при начальной влажности не менее 8—10%),
или совмещением сушки с классификацией путем отдувки пыле-
пылевидных фракций из слоя с последующей грануляцией пыли, от-
отделяемой в циклонах.
На 1 т готового хлористого калия расходуют: около 5 т сильви-
сильвинита (в расчете на содержание в нем 22% КС1), 0,5 мгкал* пара,
30 квт-ч электроэнергии, 14 м3 воды и топлива (условного) при
сушке в барабанных сушилках— 12 кг, при сушке в аппаратах ки-
кипящего слоя — 8 кг. Количество отвала на I т продукции состав-
составляет 2,5—3 т, а мелкокристаллического солевого шлама с влаж-
влажностью 15% ~0,5 т.
Величина потерь КС1 с галитовым отвалом зависит от крупно-
крупности помола сырой руды и степени промывки отвала на план-
фильтре. Потери с отвалом заметно возрастают при увеличении
содержания фракции +5 мм в сырой руде, поэтому грануломет-
гранулометрическая характеристика последней строго контролируется.
Потери КС1 с осадком с барабанных фильтров зависят от ко-
количества выносимого солевого шлама и его состава. Для умень-
уменьшения количества еолевого шлама, образующегося в результате
высаливания мелкокристаллического NaCl при растворении, можно
часть нагретого маточного щелока направлять в первый раствори-
растворитель (стр. 156); должно быть обращено также внимание на умень-
уменьшение содержания мелких фракций в сырой руде.
Сокращение потерь КС1 с глинистыми шламами обеспечивается
поддержанием заданной плотности выгружаемых сгущенных илов
с последующей противоточной промывкой. Прочие механические
потери составляют около 1,5—2%. В процессах растворения, сгу-
сгущения, фильтрации и промывки шламов в соответствии с требова-
требованиями оптимального режима общая величина потерь составляет
8-10%.
В настоящее время разработано автоматическое регулирование
всех основных технологических операций схемы 41~43.
Переработка хартзальцевых руд
Переработка на хлористый калий хартзальцевых пород Стас-
фуртского месторождения также основана на растворении руды в
оборотном маточном растворе и кристаллизации из горячего насы-
насыщенного раствора хлористого калия 44~4б.
Некоторые различия в технологическом режиме и аппаратур-
аппаратурном оформлении процесса связаны с особенностями солевого и
• I мгкал =• 106 кал.
160
Гл. V. Природные калийные соли
Получение KCI методами растворения и кристаллизации
161
минералогического состава сырья, содержащего наряду с сильви-
сильвином и галитом значительные количества кизерита, ангидрита,
карналлита.
Растворение хартзальца ведется таким образом, чтобы воз-
возможно полнее растворить сильвин и вместе с тем предупредить
переход в раствор кизерита, для которого характерна незначи-
незначительная скорость растворения при сравнительно большой раство-
растворимости в данной системе. Для растворения обычно применяют
два или три шнековых растворителя длиною не более 14—15 м,
через которые пропускают соль последовательно, а растворяющий
щелок распределяют параллельными потоками. Примерно 30—
40% направляют в первый растворитель. Все растворители рабо-
работают прямотоком. Накопление в оборотных маточных растворах
MgCl2, неизбежное из-за наличия карналлита в перерабатывае-
перерабатываемом сырье, снижает растворяющую способность щелоков по отно-
отношению к КС1 и увеличивает относительную растворимость NaCl
(стр. 149). Это обстоятельство вызывает необходимость сброса
части маточных растворов, в результате чего общая степень из-
извлечения калия из хартзальцевой руды составляет не более 80—
82%. По этой же причине при переработке хартзальца выпускают,
в основном, соль, содержащую 40—50% КеО; получение продукта,
содержащего 60% КгО, требует специальной промывки.
Растворы, поступающие на вакуум-кристаллизацию, в зависи-
зависимости от накопления хлористого и сернокислого магния в оборот-
оборотном щелоке почти всегда являются пересыщенными по отноше-
отношению к лангбейниту или глазериту, а при понижении температу-
температуры — и к шениту или леониту. Образование двойных солей при
кристаллизации является нежелательным, поэтому их выделение
минуют, проводя процесс в метастабильной области26.
Отвал, получаемый при переработке хартзальца, содержит зна-
значительное количество кизерита. При комплексном использовании
сырья из отвала получают целый ряд продуктов: MgSO4*7H2O,
MgSO4, Na2SO4- 10Н2б, Na2SO4, K2SO4-MgSO4 и K2SO4.
Слеживаемость хлористого калия
Хлористый калий, полученный методами растворения и кри-
кристаллизации, слеживается, что вызывает значительные потери при
хранении, транспортировании и весьма затрудняет рассевание при
внесении в почву. Слеживаемость является результатом сцепления
отдельных частиц при кристаллизации КС1 из пленки насыщен-
насыщенного раствора, обволакивающей кристаллы47. Образование пленки
связано с гигроскопичностью продукта, а также с недостаточной
степенью сушки.
Один из путей уменьшения слеживаемости — глубокая сушка
до содержания влаги не выше 0,1—0,2%>- Последующее увлажне-
увлажнение при хранении, неизбежное при повышении относительной
влажности воздуха, значительно снижает эффективность глубокой
сушки.
Размеры кристаллов хлористого калия определяют степень
слеживаемости. Установлено, что нагрузка на раздавливание
спрессованных образцов, являющаяся косвенным показателем сле-
слеживаемости, составляет для частиц со средним размером 0,05 мм—
18,9 кгс/см2, 0,38 мм — 0,6 кгс/см2; частицы размером 0,7Б мм прак-
практически уже не слеживаются'; (образцы прессовались под давле-
давлением 1 кгс/см2 в течение 3 суток при подсушивании).
Для устранения слеживаемости хлористого калия вводят до-
добавку алифатических аминов в количестве 200 г на 1 г хлористого
калия. Наряду с этим, в последние годы все шире применяют гра-
гранулирование хлористого калия на валковых грануляторах 48>4S.
Получение хлористого калия из карналлита
В системе KCI—MgCl2—Н2О карналлит КС1 • MgCl2 • 6Н2О ста-
стабилен в интервале температур от —21 до +167,5° (рис. 46). Он
растворяется инконгруэнтно — при обработке ограниченным коли-
количеством воды в раствор переходит MgCl2, a KC1 остается в твердой
фазе. При испарении раствора карналлита кристаллизуется сна-
сначала КС1, а затем карналлит с растворением ранее выпавшего
КС1. Если до начала кристаллизации карналлита отделить выкри-
выкристаллизовавшийся КС], а оставшийся раствор продолжать выпа-
выпаривать или охлаждать, то в обоих случаях будет кристаллизо-
кристаллизоваться карналлит.
Природный карналлит загрязнен примесями сильвина и га-
лита. При переработке карналлита методами растворения и кри-
кристаллизации щелоки насыщены хлористым натрием, который вы-
выделяется вместе с КС1, загрязняя продукт. При кристаллизации
КС1 из горячего раствора, насыщенного КС1 и NaCl, он будет тем
чище, чем меньше в растворе хлористого магния.
Все существующие методы переработки карналлита основаны
на большей растворимости хлористого магния по сравнению с рас-
растворимостью хлористого калия и делятся на способы полного и
неполного растворения 4S> 50-52 Первые способы заключаются в
полном растворении карналлитовой руды горячим оборотным ще-
щелоком, в котором содержится значительное количество MgCl2 и
мало КС] (например, 280 г/л MgCl2, 40 г/л КС1 и 4б г/л NaCl).
При охлаждении полученного раствора большая часть хлористог©
калия кристаллизуется (с примесью NaCl). Маточный раствор вы-
выпаривают и охлаждают; при этом кристаллизуется карналлит
КС1 • MgCb • 6Н2О, называемый, в отличие от природного, искус-
искусственным. Его используют для получения магния, или для
в М. Е. Познн
162
Гл. V. Природные калийные соли
Получение КС1 методами растворения и кристаллизации
163
извлечения КС1 тем же путем, как и из природного карналлита.
В конечном щелоке содержится большое количество MgCl2.
Способы неполного растворения, при которых КС1 полностью
не растворяется, а остается частично в шламе, называют также
«шламовыми». Основными из них являются способы холодного
разложения, растворения «на конечный щелок» и «конечным ще-
щелоком».
Холодное разложение карналлита ведут обработкой его водой
или оборотным щелоком без нагревания, причем большая часть
S 10 15 20 25 Зй
SO I 60
MgCl22HjO
Рис. 46. Растворимость в системе КС1—MgCl2—H2O.
KC1 извлекается в виде тонкого шлама, взвешенного в жидкости.
После удаления галитового отвала шламовый хлористый калий
отделяют от щелока, из которого выпариванием и охлаждением
кристаллизуют искусственный карналлит, присоединяемый к при-
природному карналлиту, идущему на разложение. Остающийся рас-
раствор хлористого магния, содержащий мало примесей, перераба-
перерабатывают на твердый плавленый хлористый магний (см. гл. IX).
Растворение «на конечный щелок» или горячее шламовое раз-
разложение заключается в обработке карналлитовой руды маточным
щелоком при ~100°. При этом в раствор переходит весь MgCl2
и часть КС1 из карналлита, а остальная часть КС1 из разложен-
разложенного карналлита выделяется в виде шламового хлористого ка-
калия. После отделения галитового отвала (в котором остается и
находившийся в руде сильвин), а затем шламового хлористого ка-
лия, раствор охлаждают для кристаллизации возвращаемого в
цикл искусственного карналлита, а затем выпаривают для полу-
получения плавленого хлористого магния. Преимуществом этого спо-
способа является возможность получения без выпарки конечного ще-
щелока, богатого хлористым магнием и бедного хлористым калием.
По способу растворения «конечным щелоком» шламовому раз-
разложению подвергают предварительно полученный из природного
карналлита искусственный карналлит, мало загрязненный галитом
и потому отделение шлама КС1 от NaCl не производят. Галитовый
отвал отделяют в первой стадии, после горячей обработки при-
природного карналлита оборотным маточным щелоком, перед кри-
кристаллизацией искусственного карналлита.
Имеется много вариантов этих схем.
Способы холодного разложения отличаются тем, что в них
практически весь КС1 остается нерастворенным; в горячих же
способах КС1 полностью или частично растворяется.
Использование способов холодного разложения, как и горячих
способов неполного растворения карналлита, приводит к получе-
получению менее ценного продукта, загрязненного глинистыми и дру-
другими нерастворимыми примесями, содержащимися в сырье, однако
вполне пригодного как удобрение. Использование этих способов
оказывается целесообразным при наличии сравнительно чистого
карналлита.
Наиболее распространены способы полного растворения кар-
карналлита, они дают возможность получать наиболее чистый про-
продукт, но требуют примерно в 1,5 раза большего расхода тепла,
чем способы холодного разложения и почти в 2 раза больше тепла,
чем способ растворения «на конечный щелок». Поэтому в тех слу-
случаях, когда состав природного сырья допускает возможность их
использования, способы неполного растворения карналлита яв-
являются более экономичными.
Сложность выделения хлористого калия из карналлита значи-
значительно удорожает технологический процесс по сравнению с пере-
переработкой сильвинита. Переработка карналлита на хлористый ка-
калий более экономична, если она производится с использованием
других компонентов, входящих в состав природного карналлита,
и в первую очередь хлористого магния с целью получения соеди-
соединений магния, металлического магния, хлора, хлорпроизводных
и др.53.
В СССР хлористый калий из карналлита не вырабатывают.
Карналлитовую руду перерабатывают способом неполного (шла-
(шламового) растворения на необходимый для получения металличе-
металлического магния искусственный карналлит. Отходом в производстве
магния электролизом карналлита является калийная соль, содер-
содержащая 75—80% КС1; ее выпускают в качестве удобрения под
названием «электролит».
6*
164
Гл. V. Природные калийные соли
При переработке карналлита применяют такую же аппаратуру,
как и при переработке сильвинита. Растворение карналлита идет
с большой скоростью, поэтому его ведут и в вертикальных рас-
растворителях; по-видимому, целесообразно вести его в трубах, сов-
совмещая этот процесс с гидротранспортом.
Защита оборудования от коррозии и эрозии
Растворы и пульпы, перерабатываемые в производстве хло-
хлористого калия из калийных руд, действуют разрушающе на ап-
аппаратуру. Значительный коррозийный и эрозийный износ имеет
место в трубопроводах, растворителях, отстойниках для горячих
щелоков, насосах, ковшах элеваторов и др. Для защиты аппара-
аппаратуры применяют различного рода покрытия. Растворители покры-
покрывают диабазовой обмазкой по сетке Рабица. Вакуум-корпусы гум-
гуммируют или футеруют, например антегмитовыми плитками. Пере-
Переточные желоба, лотки, кожухи для термоизоляции изготовляют из
пластмасс или защищают обмазками на основе асбовинила (ас-
(асбест с этинолевым лаком). Металлические трубопроводы защи-
защищают фарфоровыми вкладышами на специальных цементах или
гуммируют.
ПОЛУЧЕНИЕ ХЛОРИСТОГО КАЛИЯ МЕХАНИЧЕСКИМ ОБОГАЩЕНИЕМ
КАЛИЙНЫХ РУД
Метод флотации
Этот метод переработки в настоящее время широко приме-
применяется в СССР, США и Канаде. Он основан на флотационном или
флотагравитационном разделении водорастворимых минералов
руды в среде насыщенного солевого раствора, при этом, пенным
продуктом является, как правило, сильвин или другие калийные
минералы. Основные стадии производственного процесса вклю-.
чают следующие операции:
1) сухое и мокрое измельчение руды;
2) «оттирка» глинистых примесей от частиц руды в специаль-
специальных мешалках;
3) выделение глинистых шламов путем механического обес-
шламливания в гидроциклонах и гидросепараторах:
4) обработка солевой пульпы специальными реагентами де-
депрессорами с целью подавления вспенивания шламов и сорбции
флотореагентов на поверхности глинистых частиц (наиболее рас-
распространен прием обработки шламов, сочетающий механическое
обесшламливание основной массы глинистых частиц с последую-
последующей обработкой пульпы реагентами депрессорами);
5) обработка солевой пульпы реагентами флотации, которые
подают в специальные мешалки и, частично, непосредственно во
Получение КС1 механическим обогащением калийных руд
165
флото-машины. В состав реагентов флотации входят: основной
компонент — собиратель, в качестве которого при флотации силь-
сильвина применяют водорастворимые соли алифатических аминов с
углеродной цепочкой Ci6—С20, депрессоры и деактиваторы (крем-
(кремневая кислота, полиакриламид и др.) и вспениватели (сосновое
масло, спирты и др.). Иногда дополнительно вводят реагенты для
поддержания оптимального значения рН среды и для других це-
целей;
6) основная флотация, которую осуществляют в машинах с
механическим или пневматическим перемешиванием;
7) перечистка концентрата, полученного при основной флота-
флотации с целью повышения содержания в продукте хлористого
калия;
8) контрольная флотация хвостов основной флотации для сни*
жения потерь калия с отвалом;
9) обезвоживание концентрата перечисткой флотации на
фильтрах или центрифугах и сушка;
10) обезвоживание хвостов и подача их в отвал;
11) отработка глинистых шламов, выделенных в голове про-
процесса, путем их сгущения и противоточной промывки. Для ликви-
ликвидации сброса жидких отходов необходимо обезвоживать шламы
на специальных фильтрах или центрифугах отстойного типа54-55.
Используется схема флотационного обогащения сильвинитов
с предварительным выделением глинистых примесей с помощью
пенной флотации — основной и перечистной.
В качестве реагентов пенной флотации применяют смесь кар-
боновых кислот, полученных при окислении уайт-спирита, так на-
называемого, ФР-256'57. После перечистной флотации требуется под-
подвергать глинистые шламы сгущению и прогавоточной промывке,
однако трудности, связанные с процессом разрушения пены, обра*-
зующейся при шламовой флотации, затрудняют проведение этих
операций, что приводит к значительным потерям хлористого ка-
калия с жидкой фазой. На некоторых зарубежных предприятиях пе-
переработку руды производят комбинированием флотогравитацион-
ного обогащения с пенной флотацией58'59.
С целью повышения степени извлечения калия производят тер-
термообработку хвостов, содержащих некоторое количество силь-
сильвина. Для этого нагревают галитовую пульпу до 60—70°, твердый
хлористый калий растворяется, так как раствор при повышении
температуры становится ненасыщенным КС1. Затем производят
обезвоживание хвостов и сброс их в отвал, а маточный раствор
охлаждают с выделением хлористого калия. На современных ка-
калийных фабриках охлаждение и кристаллизацию хлористого калия
при термообработке производят в вакуум-кристаллизаторах с ре-
регулируемой кристаллизацией, обеспечивающей получение крупно-
крупнокристаллического продукта54'60.
Получение КС1 механическим обогащением калийных руд
167
Существенным достижением в технологии обогащения калий-
калийных руд в последние годы является крупнозернистая флотация,
обеспечивающая получение части продукции, с размерами частиц
1—3 ммв1~6Б. Возможность флотации материала такой крупности
определяется, в первую очередь, величиной отдельных зерен силь-
сильвина и галита в руде и степенью их взаимного прорастания.
Оптимальная крупность частиц сильвина при его выделении в
пенный продукт крупнозернистой флотации составляет 3,3 мм.
Для крупнозернистой флотации потребовалась разработка спе-
специальных флотомашин66-67.
Применение крупнозернистой флотации позволило значительно
повысить качество продукции, так как хлористый калий, разме-
размерами— 1—3 мм не слеживается, не пылит и хорошо рассевается
при внесении на почву; его агрохимическая эффективность значи-
значительно выше для ряда почв и культур, чем стандартного по-
порошкообразного хлористого калия. Наряду с этим удельная про-
производительность основного оборудования — дробилок, сгустителей,
фильтров и сушилок возрастает при получении крупнозернистого
материала.
Эффективность крупнозернистой флотации в значительной сте-
степени определяется показателями операции дробления, так как
весьма важно максимально уменьшить переизмельчение руды на
стадиях сухого и мокрого помола.
Схема переработки с получением части продукции в крупно-
крупнозернистом виде изображена на рис. 47. Солевую пульпу, предва-
предварительно подвергнутую механическому обесшламливанию, разде-
разделяют по классу 1 мм. Фракции +1 мм направляют на специаль-
специальную реагентную обработку и далее на крупнозернистую флотацию.
Пенный продукт крупнозернистой флотации подвергают перечи-
перечистке, а хвосты — контрольной флотации. Переработка фракции
руды—1 мм протекает по обычной схеме.
В СССР получение хлористого калия методом флотационного
обогащения осуществляется при переработке руд Верхнекамского
и Белорусского месторождений. Используются схемы с отделением
шламов способом пенной флотации, депрессией шламов и меха-
механическим обесшламливанием руды в гидроциклонах. Показатели
процесса в значительной степени определяются содержанием шла-
шламов в руде — с повышением содержания глинистых примесей резко
возрастает расход реагентов и снижается степень извлечения
калия.
Комбинированные схемы флотационного обогащения
с растворением и кристаллизацией
Такие схемы успешно применяются на некоторых канадских
предприятиях. Установлено, что наиболее целесообразно их ис-
использование для переработки руд с повышенным содержанием
168
Гл. V. Природные калийные соли
Получение КС1 механическим обогащением калийных руд
169
иловых примесей68. При переработке по комбинированным схемам |
на растворение направляют ту часть руды, в которой концентри- j
руется большая часть илов; как правило, это наиболее мелкие
классы, выделяемые на стадии механического обесшламливания.
Применение галургического способа переработки для этой части
руды обосновано резким ухудшением структуры шламовых осад-
осадков в присутствии флотореагентов, а также повышенной вязко-
вязкостью и значительным количеством оборотных щелоков во флота-
флотационных способах переработки. По этим причинам при флотацион- J
ных способах обогащения возрастает требуемое число отстойников, 1
ухудшаются показатели по противоточной отмывке шламов и, !|
соответственно, снижается степень извлечения хлористого калия;
кроме того, с ростом содержания шламов резко возрастает рас-
расход реагентов.
В комбинированных схемах, как правило, используют регули-
регулируемую кристаллизацию, поэтому на ряде предприятий к сырью,
направляемому на растворение, присоединяют пылевидный хло-
хлористый калий, отделенный от основной массы продукции путем
рассева или воздушной классификации.
Другие способы обогащения
Наряду с флотацией и флотогравитационным разделением, обо-
обогащение калийных руд осуществляют также гравитационными ме-
методами на основе разницы в плотностях сильвина и галита. Для
повышения эффективности обогащения процесс необходимо прово-
проводить в среде с высокой плотностью, для чего применяют суспензии
тонкоизмельченных магнетита, барита, пирита и других минералов.
На одном из французских предприятий методом гравитацион-
гравитационного разделения обогащают сильвинитовые руды эльзасского ме-
месторождения, содержащие ~10% нерастворимых веществ69. Руду
измельчают до крупности —30 мм +3 мм и разделяют в статиче-
статическом сепараторе в магнетитовой суспензии. Сепаратор представляет
собой ванну, на дне которой имеются конусообразные выводы для
промежуточного продукта и хвостов. При подаче и выводе потоков
стремятся исключить завихрения; высота сепаратора сравнительно
невелика, что сокращает время контакта глины с раствором, тем
самым уменьшая разбухание и расслоение глинистых частиц; кон-
концентрат выделяют из слива. При переработке руды таким методом
получают концентрат, содержащий 65% КС1; извлечение калия со-
составляет ~90%- Гравитационное разделение в статических сепара-
сепараторах из-за небольшой разности в плотности минералов (КС1—1,98,
NaCl — 2,14 г/см3) представляет значительные трудности. Кроме
того, при разделении таким путем возможно обогащать руду толь-
только сравнительно крупного измельчения, поэтому содержание основ-
основного вещества в концентрате из-за неполного раскрытия зерен по-
получается довольно низкое.
Разделение минералов на основе центробежного ускорения в
гидроциклонах дает возможность обогащения сильвинитовых руд
сравнительно мелкого дробления и при значительном эффекте раз-
разделения70. Пульпу, приготовленную из измельченного сырого силь-
сильвинита и тяжелой магнетитовой суспензии, пропускают через
гидроциклон, в котором более тяжелый галит отбрасывается к пе-
периферии и выгружается из нижней конической части, а сильвин
концентрируется ближе к центральной части и выводится через
верхний слив гидроциклона.
На первой фазе разделения получают концентрат и промежу-
промежуточный продукт, который подвергают повторной сепарации с выда-
выдачей хвостов, содержащих минимальное количество сильвина. Верх-
Верхний продукт, образующийся при повторной сепарации, может быть
переработан флотационным методом или другими способами.
Концентрат и хвосты направляют на обезвоживание на гро-
грохоты, где отделяется основная часть тяжелой суспензии, возвращае-
возвращаемой в цикл. Дренируемый на грохоте материал содержит некоторое
количество суспензоида, который отмывают в оборотном растворе,
насыщенном по отношению к солевым составляющим породы. По-
Полученную разбавленную суспензию сгущают до требуемой плотно-
плотности и возвращают в основной процесс. Часть суспензии требуется
выводить из цикла на регенерацию для удаления окисленных ча-
частиц магнетита, присутствие которых снижает плотность разделяю-
разделяющей среды. Регенерацию осуществляют с помощью магнитного се-
сепаратора. В нем отделяют магнетит от накопившегося глинистого
шлама и окисленного суспензоида, потери которого компенсируют
добавкой свежих порций магнетита.
Все известные способы механического обогащения калийных руд
не требуют применения технологического пара (за исключением
сравнительно малого расхода на термообработку галитового от-
отвала), поэтому при строительстве перерабатывающих фабрик иск-
исключается необходимость сооружения дорогостоящих ТЭЦ. По-
Поскольку все технологические операции протекают без нагрева,
резко снижается коррозия аппаратуры и улучшаются условия труда.
Хлористый калий, получаемый таким образом, меньше слеживается
и лучше рассевается, чем получаемый путем растворения и кри-
кристаллизации.
С комбинированными методами обогащения, например флота-
флотацией и флотогравитацией или флотацией и гидросепарацией,
удачно сочетается возможность переработки части руды при более
крупном дроблении, что позволяет снизить расход электроэнергии
на измельчение породы, а также удельный расход флотореагентов.
Обогащение сильвинитовых руд может быть осуществлено элек-
электростатическим разделением сильвина и галита. Сущность метода
заключается в разделении в электрическом поле частиц, поверх-
поверхность которых заряжается разноименными зарядами.
170
Гл. V. Природные калийные соли
Обогащение таким способом предложено осуществлять либо при
предварительном нагреве руды до 400° с последующим охлажде-
охлаждением71'72, либо путем реагентной обработки руды при 100°. В ка-
качестве реагентов используют салициловую кислоту и аммиак73'74.
Разделение сильвина и галита осуществляют в сепараторах при на-
напряжении порядка 100 000 в. Электростатическое обогащение может
быть использовано для отделения нерастворимых примесей; в этом
случае руду нагревают до 120—150° и направляют на сепарацию.
Этот способ испытан на сильвинитовых и лангбейнитовых рудах75.
ПОЛУЧЕНИЕ ХЛОРИСТОГО КАЛИЯ
ПРИ ПОДЗЕМНОМ ВЫЩЕЛАЧИВАНИИ КАЛИЙНЫХ РУД
Таким способом перерабатывают калийные руды пока только на
одном предприятии в Канаде76-77; схема процесса показана иа
рис. 48.
i
Рассол из скважины
Получение хлористого калия из природных рассолов
171
Рассол из
хвостохранилища
В хВостохранилище
II продукции
12 13 н 15
Рис. 48. Схема переработки рассолов, полученных при подземном выще-
выщелачивании калийной руды:
/ — выпарные аппараты (четырехкорпусная выпарка); 2 — сгуститель; 3 — аппараты
четырехступенчатой вакуум-кристаллизации; 4— центрифуга; 5 —репульпатор; 6 — цен-
центрифуга; 7 —барабанная сушилка; 8 — циклон; 9 — гранулятор циклонной пыли; 10 — дро-
дробилка; // — грохота; 12 — гранулированный КС1; 13 — крупнозернистый KCI; 14 — средне-
зерннстый КС1; 15 — стандартный KCI.
При подземном выщелачивании через скважины получают рас-
рассол хлористых натрия и калия, который направляют на выпарку
в многокорпусную выпарную батарею. При выпарке кристалли-
кристаллизуется хлористый натрий: его отделяют от маточного раствора и
направляют в сброс. Горячий упаренный раствор, насыщенный хло-
хлористыми натрием и калием, направляют на вакуум-кристаллиза-
вакуум-кристаллизацию. Для этой цели используют четырехступенчатую установку с
регулируемой кристаллизацией Маточный раствор после кристал-
кристаллизации присоединяют к свежим порциям рассола и возвращают в
цикл выпарки. Кристаллы хлористого калия после сушки рассе-
вают с получением стандартного продукта и двух сортов крупно-
крупнокристаллического хлористого калия крупностью —2,4 +0,8 мм и
—3,3 +1,1 мм. Циклонную пыль подвергают грануляции на меха-
механических валках.
Описан ряд других способов переработки рассолов, получаемых
при подземном выщелачивании руды; так, например, предлагается
аммонизировать растворы. Растворимость хлористого калия в при-
присутствии аммиака снижается, и из раствора кристаллизуется КС1;
хлористый калий отделяют в специальных отстойниках, и на цен-
центрифуге сушат и выпускают в виде готовой продукции. Из маточ-
маточного раствора отгоняют аммиак, возвращаемый в цикл, а раствор
через теплообменник направляют в сброс77.
ПОЛУЧЕНИЕ ХЛОРИСТОГО КАЛИЯ ИЗ ПРИРОДНЫХ РАССОЛОВ
Применяемые способы извлечения калия из рассолов представ-
представляют интерес в связи с разработкой схем комплексного использо-
использования галургического сырья, особенно сгущенной морской воды,
являющейся неисчерпаемым источником солей калия и других цен-
ценных элементов.
При переработке рапы оз. Сирлз78 получают: соду, сульфат нат-
натрия, хлористый калий, сульфат калия, жидкий бром, бромиды,
очищенную буру, борную кислоту, пироборат натрия, карбонат и
фосфат лития. Выделение солей основано на выпаривании, вакуум-
кристаллизации и кристаллизации из сильно пересыщенных раство-
растворов. Выпаривание осуществляют в трехкорпусной выпарной бата,-
рее при принудительной циркуляции и противоточном движении
раствора и греющего пара. При выпарке кристаллизуются: NaCl,
Na2CO3 • Н2О, Na2CO3 • 2Na2SO4 (беркеит) и в небольших количе-
количествах Li2NaPO4. При последующей вакуум-кристаллизации полу-
получают хлористый калий. Образующийся маточный раствор является
пересыщенным по отношению к буре, но самопроизвольного выде-
выделения буры не происходит.
Рапа Мертвого моря в сравнении со сгущенной океанской во-
Дой других водоемов отличается высоким содержанием хлористого
кальция. Солевой состав рапы (в %): СаС12—14,0, NaCl — 27,8,
MgCl2 — 51,4, KCI — 4,6 и MgBr2 — 2,2; сумма солей изменяется от
23 до 33%, причем в более глубоких слоях концентрация выше, чем
172
Гл. V, Природные калийные соли
Сульфат калия
173
у поверхности. Извлечение из рапы солей осуществляют 17>18 при
испарении в системе искусственных бассейнов. Для интенсификации
испарения подкрашивают рапу небольшим количеством красите- I
лей, в результате чего интенсивность поглощения солнечных лучей
возрастает во много раз. Применяется так называемая динамиче-
динамическая бассейновая система, при которой испарение происходит при
медленном зигзагообразном течении потока рассола по ряду бас-
бассейнов.
В первом, по ходу рапы, бассейне происходит кристаллизация
поваренной соли, во втором — кристаллизация смеси карналлита
и хлористого натрия, в третьем, в самые жаркие месяцы, из ма-
маточных растворов после осадки карналлита, выделяются игольчатые
кристаллы бишофита (MgCl2* 6Н2О). В маточном растворе после
садки бишофита содержится значительное количество хлористого
кальция. Сбор карналлита производят солесосами в период садки
как только высота слоя выпавших кристаллов достигнет примерно
10 см.
Карналлит подвергается холодному разложению; образую-
образующуюся при этом смесь сильвина и галита разделяют флотационным
способом. В последние годы для переработки смеси галита и силь- |
вина используют метод растворения и кристаллизации в аппаратах
с регулируемой кристаллизацией типа ДТВ79.
В Бонневиле (США) хлористый калий получают из грунтовых \
рассолов, пропитывающих рыхлые илы, подстилающие солончаки
района Большого Соленого озера. Для извлечения рассолов со-
создана широкая дренажная система на площади около 243000 га,
глубина канав достигает 4—4,5 м. Собираемые в центральном кол-
коллекторе рассолы перекачивают в ряд испарительных бассейнов;
кристаллизация смеси сильвина и галита происходит в главном
испарительном бассейне.
Маточные рассолы сбрасывают при приближении состава рапы
к насыщению по карналлиту. Сезон испарения продолжается около
100 дней. Солевой пласт, образовавшийся в главном испаритель-
испарительном бассейне, представляет собой искусственный сильвинит
C0—35% КС1 и 65—67% NaCl). Соль собирают бульдозерами и
грейдерами, грузят на мотовозы и направляют на перерабатываю-
перерабатывающую фабрику для получения хлористого калия методом флота-
флотации80-81.
Разработана схема получения калийных солей из рапы оз. Ин-
дер. Рапу сгущают в бассейнах; вначале кристаллизуется галит, -
при этом состав рапы приближается к насыщению калийными со-
солями. Сгущенный рассол перекачивают в сборные бассейны для
хранения. При зимнем охлаждении сгущенного рассола кристалли-
кристаллизуется сильвин с примесью галита (содержит больше 85% КС1).
Дополнительное извлечение калийных солей возможно при повтор-
повторном летнем испарении и охлаждении зимой. Общее извлечение
КС1 при этом достигает 76% от содержания его в исходной рапе.
Дальнейшее испарение следует осуществлять уже в заводских усло-
условиях с получением при охлаждении карналлита.
Возможна комплексная переработка рассолов Кара-Богаз-Гола
с получением мирабилита, эпсомита, бишофита и калийных солей.
Выделение калийных солей в виде карналлита происходит при
выпарке и охлаждении обессульфаченных хлормагниевых рассо-.
лов после садки эпсомита. Карналлит может быть переработан на
хлористый калий известными способами80'82.
Выделение калия из рассолов может быть осуществлено мето-
методом прямого осаждения дипикриламином (гексанитродифенилами-
ном)83. Морскую воду обрабатывают раствором кальцийдипикрила-
мина, при этом в осадок выпадает нерастворимый калийдипикрила-
мин, который отделяют фильтрацией с промывкой. Из фильтрата
при подкислении выделяют непрореагировавший дипикриламин.
Промытый калийдипикриламин разлагают кислотой (серной, соля-
соляной, уксусной) — образуются раствор соответствующей соли калия
и осадок дипикриламина, который возвращают в цикл 80.
СУЛЬФАТ КАЛИЯ
Известны следующие пути получения сульфата калия:
1) переработкой галургическими методами — растворением и
кристаллизацией полиминеральных сульфатнокалийных руд Пред-
карпатья;
2) конверсионным и ионитным способами на основе взаимодей-
взаимодействия хлористого калия и различных сульфатных солей, например,
эпсомита, мирабилита, лангбейнита и др.;
3) комбинированием флотационного обогащения сульфатно-
калийных руд с переработкой концентрата галургическим спо-
способом;
4) при производстве соляной кислоты из серной кислоты или
сернистого газа и хлористого калия (см. гл. XI);
5) гидротермическим методом на основе взаимодействия суль-
сульфатных солей (например, гипса, сульфата натрия, эпсомита, алу-
алунитов) с хлористым калием с получением побочной соляной кис-
кислоты;
6) переработкой водонерастворимых калийных руд (полигали-
тов, лейцитов, глауконитов и др.);
Сульфатнокалийные удобрения с пониженным содержанием
КгО, получают при флотационном обогащении лангбейнитовых и
каинито-лангбейнитовых руд.
Галургическая переработка полиминеральных руд Предкарпатья
Сложность и пестрота минералогического состава и колебания
в содержании основных минералов в различных участках место-
месторождения, а также значительные примеси глинистых веществ,
174
Гл. V. Природные калийные соли
весьма осложняют переработку этого вида сырья. Существование
метастабильных равновесий в пятикомпонентной системе К+, Na+>
Mg2+|cr, SO4~, H2O еще более осложняет переработку84-85. Одна
из схем получения сульфата калия из каинитовых руд86 заклю-
заключается в растворении руды в оборотном щелоке при ~ 100—107°.
Получается раствор, насыщенный хлористым калием и лангбейни-
том; твердая фаза после выщелачивания содержит вторичный ланг-
бейнит, образовавшийся из каинита породы. Из горячего насыщен-
насыщенного раствора при охлаждении кристаллизуются КС1 и NaCl.
Маточный раствор направляют на растворение вторичного лангбей-
лангбейнита (при 60—65°). При охлаждении полученного раствора кри-
кристаллизуется шенит, который может быть переработан на сульфат.
калия. Отвал после первого растворения состоит из галита, кизе-
кизерита, полигалита с примесью лангбейнита. Галит растворяют в
холодной воде, а оставшиеся калийные соли перерабатывают на
калийное удобрение.
На Калушском химико-металлургическом комбинате осуществ-
осуществлен вариант схемы переработки полиминеральных руд, разрабо-
разработанный ВНИИГом 87-8э. Технологическая схема, предусматриваю-
предусматривающая комплексное использование сырья с получением калийных
удобрений, сульфата натрия, поваренной соли и бишофита, состоит
из четырех основных циклов:
а) производства сульфата калия;
б) производства сульфата натрия;
в) флотации нерастворенного остатка;
г) регенерации солей из избыточных щелоков.
По схеме ВНИИГ (рис. 49), породу, измельченную до —5 мм,
обрабатывают оборотным горячим щелоком при 65—70°. В про-
процессе выщелачивания в щелоке растворяются каинит, сильвин, ше-
шенит и леонит и другие легкорастворимые минералы, в отвале
остаются галит, лангбейнит, полигалит и гипс. Горячий щелок
осветляют от нерастворимых примесей и направляют на вакуум-
кристаллизационную установку для выделения шенита.
Для кристаллизации чистого шенита без примеси NaCl к ще-
щелоку добавляют маточные растворы, получаемые при последующем
разложении шенита на сульфат калия. Кристаллизация шенита
заканчивается при 20°, полученную пульпу сгущают и подвергают
фильтрации; часть маточного раствора после кристаллизации ше-
шенита возвращают в начало процесса на выщелачивание руды, дру-
другую часть подвергают выпарке для регенерации солей калия. Весь
шенит, или часть его, разлагают водой при 50е, при этом полу-
получаются сульфат калия и маточный раствор, присоединяемый к ще-
щелоку, направляемому на кристаллизацию шенита.
При высоком содержании лангбейнита в руде необходима пере-
переработка отвала, так как основная часть лангбейнита при выщела-
а)
I
3
<и
я
Я
ы
(-¦
о
«о
п)
О,
ID
о.
3
i
а.
176
Гл. V. Природные калийные соли
чивании оборотными маточными щелоками не растворяется. Извле-
Извлечение лангбейнита из отвала может быть осуществлено флотацией,
разделением в тяжелых суспензиях или отмывкой галита водой.
Последний способ наиболее прост, однако его применение связано
с необходимостью переработки больших количеств растворов хло-
хлористого натрия.
Выделенный тем или иным методом лангбейнит содержит зна-
значительные примеси полигалита. Эту смесь после сушки можно вы-
выпускать в качестве калийно-магниевого удобрения A6,0—18,0%
КгО). С целью получения концентрированных калийных удобрений
лангбейнитовый флотоконцентрат растворяют при 75—90°, щелок
охлаждают до 20° и отделяют кристаллизующийся при этом шенит
(K2SO4 • MgSO4 • 6Н2О). Разложением шенита водой при 50е полу-
получают сульфат калия F0% КгО).
Неразложенный обезвоженный шенит (K^SC^- MgSO4) также
является высококачественным бесхлорным удобрением B8—30%
КгО). Щелок, оставшийся после отделения шенита из лангбейни-
тового щелока с высокой концентрацией сульфата магния, можно
использовать для получения сульфата натрия.
Технологическая схема переработки калийных руд предусмат-
предусматривает регенерацию солей, содержащихся в выводимых из про-
процесса избыточных маточных щелоках. С этой целью щелока под-
подвергают выпарке последовательно в 3—4 стадии, выделяя после
первой стадии поваренную соль пищевых сортов, после второй и
третьей, смешанные калийные соли (каинит, карналлит), после
четвертой — бишофит (MgCb • 6Н2О) различной сортности.
Таким образом, по данной схеме получают удобрения в виде:
сульфата калия, калимагнезии I и II сорта.
Соотношение этих видов продукции определяется составом ис-
исходной руды. Для характеристики зависимости соотношения суль-
сульфата калия и калимагнезии в товарной продукции от состава
исходной руды предложен так называемый сильвинитовый коэффи-
коэффициент р, равный отношению весового содержания сильвина к каи-
каиниту в породе: при отсутствии в руде сильвина (Р=0) в результате
переработки может быть получен только шенит. При р=0,15 (по-
(порода содержит иа 1 моль каинита 0,5 моль сильвина) 2/3 продук-
продукции в расчете на КгО получают в виде сульфата калия и Уз .в
форме шенита. При эквимолярном отношении сильвина и каинита
(Р =¦ 0,30) продуктом является только сульфат калия.
Присутствие в породе лангбейнита позволяет проводить про-
процесс при значительно больших значениях р. Отношение весового
количества КС1 в сильвине и каините к общему содержанию MgSO4
во всех водорастворимых минералах породы обозначается коэффи-
коэффициентом а. При а=1,24 переработка породы на сульфат калия
возможна при значении Р=0,6, т. е. в 2 раза большем, чем для
предельного случая в отсутствие лангбейнита.
Сульфат калия
177
Соотношение между сульфатом калия и калимагнезией в гото-
готовой продукции определяется также объемом производства бишо-
фита и сульфата натрия. Если процесс ведется по варианту схемы
с максимально возможным выходом бишофита и сульфата нат-
натрия, — калимагнезия в качестве готовой продукции не произво-
производится. При сокращении производства бишофита и сульфата нат-
натрия, дол'я калимагнезии в готовой продукции возрастает.
Переработка руды с получением только бесхлорных калийных
удобрений осуществима при значении а=1,24. При а>1,24 содер-
содержание MgSO4 недостаточно для превращения КС1 в K2SO4. Если
а>1,24, КС1 при вакуум-кристаллизации выделяют в виде товар-
товарного продукта. Кристаллизация КС1 происходит из растворов, ле-
лежащих на стабильной диаграмме растворимости в поле глазерита,
однако выделение глазерита при вакуум-испарении не наблю-
наблюдается— кристаллизация КС1 происходит в метастабильной обла-
области. Примесь сульфатов в кристаллах КС1 незначительна, если
время кристаллизации не превышает 45 мин, а конечная темпера-
температура не ниже 30—28°90.
Содержание КгО в кристаллизующемся продукте, определяемое
примесью NaCl, зависит от количества воды в исходном щелоке.
Максимальное содержание КС1 (~52% КгО) получается при уве-
у
личении отношения w= экв.н2о до 14—15. При w > 15 содержа-
ние КС1 падает за счет увеличения примеси NaCl91. Расходные ко-
коэффициенты на 1 ,т КгО при получении товарной продукции с со-
содержанием 38% КгО составляют: руды 12 т, пара 5,5 т, электро-
электроэнергии 220 кет ¦ ч, топлива — 0,066 т.
Конверсионные способы получения сульфата калия
Рассмотрим конверсионный способ получения сульфата калия
на примере взаимодействия хлористого калия с эпсомитом:
2КС1 + MgSO4 ^=± MgCl2 + K2SO4
Процесс ведут в две стадии с образованием на первом этапе
шенита92. Для получения максимального выхода шенита точка С\
(рис. 50) состава исходной смеси должна лежать на луче кристал-
кристаллизации ШР, идущем из полюса шенита Ш в точку Р, положение
которой соответствует составу маточного раствора, насыщенного
шенитом, КС1 и каинитом. Раствор Р—шенитовый щелок сбрасы-
сбрасывают, а шенит обрабатывают хлоридом калия в водной среде с об-
образованием сульфата калия и маточного раствора А, насыщенного
хлоридом калия, сульфатом калия и шенитом. Этот раствор пол-
полностью используют в первой стадии конверсии и цикл, таким об-
образом, замыкается. Для получения высококачественного сульфата
178
Гл. V. Природные калийные соли
калия (~52% КгО) целесообразно использовать хлорид калия с
высоким содержанием основного вещества вз.
Разработан ряд вариантов рассмотренной схемы. Для повыше-
повышения степени использования калия проводят выпарку и охлаждение
щенитовых щелоков с выделением калийных солей в виде хлорида
калия и леонита, возвращаемых в цикл. Выпарка и охлаждение
MgSO4
Сульфат калия
179
о
2КС!
Рис. 50. Растворимость в водной системе 2КС1 + MgSO4
±=^ KSO + MCl при 25°.
маточных растворов с выделением возвратных солей могут быть
проведены также при одностадийной конверсии с прямым получе-
получением сульфата калия. При выпуске части шенита без разложения,
степень использования калия повышается, так, например, при по-
получении всей продукции в виде сульфата калия она составляет
70%, а при выпуске только товарного шенита — 87%. Повышение
степени использования калия до 87% с выработкой только суль-
сульфата калия возможно при выпарке шенитового щелока; расход
пара при этом составляет 0,45 г на 1 г К.2О.
Процесс конверсии мирабилита (или безводного сульфата) с
хлористым калием аналогичен рассмотренному выше. Конверсию
осуществляют в две стадии с образованием промежуточного соеди-
соединения—глазерита. Глазерит полностью или частично разлагают
на сульфат калия. Маточные растворы после выделения глазерита
могут быть переработаны на поваренную соль, при этом выделяются
возвратные калийные соли (смесь глазерита и хлористого калия),
которые используют в основном цикле переработки s4-95.
Промышленное получение сульфата калия на основе хлористого
калия и эпсомита осуществляется на калийных предприятиях ГДР
при комплексной переработке хартзальцевых руд45. Отвал после
лереработки руд на хлористый калий, содержащий галит, кизерит
и ангидрит, подвергают быстрой промывке водой, при этом галит,
скорость растворения которого значительно выше, чем у кизерита,
практически полностью растворяется. Затем растворяют кизерит
при 75°. При охлаждении раствора кристаллизуется эпсомит, ко-
который конвертируют с КС1 в K2SO4 в две стадии, через шенит.
Часть шенита смешивают с ангидритом, сушат и выпускают в ка-
качестве товарной продукции под названием «реформкалий»; содер-
содержание КгО в этом продукте составляет 26—30%- В товарном суль-
сульфате калия содержится после сушки 48% КгО.
Конверсионные методы получения сульфата калия в США осно-
основаны на переработке хлористого калия с лангбейнитом или беркек-
том
79
В последние годы в Канаде организовано производство суль-
сульфата калия на базе природного сульфата натрия и хлористого
калия79. При конверсии на сульфат калия астраханитовых солей
процесс можно вести через промежуточные соединения — шенит и
глазерит или их смеси.
Сульфат калия может быть получен также из стехиометриче-
ской смеси хлористого калия и сульфата аммония. Образующийся
при этом хлористый аммоний возгоняется в токе инертного газа
или водяного пара96.
Сульфат калия может быть получен ионообменным способом,
например, при пропускании горячего (90°) раствора MgSO4 через
К-катионит:
2RK + MgSO4 ^=± R2Mg+K2SO4
При охлаждении раствора до 10° из него кристаллизуется K2SO4.
Регенерацию катионита можно осуществить, обрабатывая его рас-
раствором хлористого калия:
Суммарное уравнение процесса:
2KCl + MgSO4 ^z±
Получение сульфатнокалийных удобрений при флотационном
обогащении и в комбинации с галургическими способами
переработки
Флотационное обогащение сульфатнокалийных руд осуществ-
осуществляют на базе переработки каинито-лангбейнитовой породы Стеб-
никовского месторождения. Руду, измельченную до —0,5 мм*
180
Гл. V. Природные калийные соли
подвергают флотации в присутствии коллектора — жирных кислот
(С7—С9) и депрессора — смеси кремневой кислоты и полиакрил-
амида; оптимальное значение рН среды, равное ~7, регулируют
добавкой щелочи. Пенный продукт сгущают, фильтруют и сушат в
аппаратах кипящего слоя. Хвосты флотации разделяют в гидро-
гидроциклонах — слив направляют на противоточную промывку, а пе-
сковую часть, после фильтрации, сбрасывают в отвал S7. Серьезные
трудности при осуществлении этого процесса связаны с высоким
содержанием илов в руде, в среднем 14—18%.
В ГДР разработан способ флотационного обогащения харт-
зальцевых руд. Породу измельчают до —0,4 мм; в процессе дроб-
дробления отделяют более крупные фракции, в которых содержание
К2О составляет ~13%, эта часть руды выпускается в виде товар-
товарного продукта под названием «каинит». Измельченная руда флоти-
флотируется в две стадии — на первой стадии в пенный продукт перехо-
переходит преимущественно сильвин, на второй — кизерит. После сгуще-
сгущения и фильтрации, эти продукты смешивают и сушат с получением
двух видов товарной продукции: «Катех» КгО—38^-42%, MgSO4—
10% и «Emgekali» —К2О —33—37%, MgSO4 — 14% 98.
Комбинированная схема осуществлена в последние годы в Си-
Сицилии на базе переработки каинитовых руд, содержащих кроме
каинита — карналлит, сильвин и галит. Руду измельчают до —0,6 мм
с предварительной воздушной классификацией и направляют на
отделение глинисто-солевого шлама в гидроциклонах. Слив гид-
гидроциклонов сгущают в отстойниках и подвергают противоточной
промывке с целью извлечения калия. Флотация проводится в при-
присутствии алифатических аминов со средним числом атомов С, рав-
равным 12, при расходе 120 г на \т руды; пенным продуктом яв-
является каинит. Состав щелока регулируют таким образом, чтобы
практически исключить возможность конверсии каинита в шенит
к цикле флотации. Отфильтрованный концентрат присоединяют
к части исходной руды с повышенным содержанием КгО и направ-
направляют на переработку на сульфат калия. Сульфат калия получают
в две стадии: на первой — происходит превращение каинита в ше-
шенит, на второй — разложение шенита водой на сульфат калия; ще-
щелок после этой стадии используется на первой стадии конверсии
(при превращении каинита в шенит)98.
Гидротермический метод переработки калийных руд
Метод гидротермической переработки основан на реакциях:
MgSO4 + 2KCl(NaCl) + H2O = MgO + K2SO4(Na2SO4) + 2НС1
CaSO4 (в виде гипса) + 2KCl(NaCl) + raSiO2 + Н2О =
= CaO • mSiO2 + K2SO4(Na2SO4) + 2HC1
Исследование условий гидротермической переработки калий-
калийных солей Предкарпатья показало, что при нагревании до 800° от-
Сульфат калия
181
дельных минералов, входящих в состав породы, и их смесей, про-
происходит, главным образом, дегидратация. В присутствии кисло-
кислорода воздуха и, особенно, водяного пара, конверсия солей уско-
ускоряется, особенно в том случае, когда нагреву подвергается смесь
хлоридов и сульфатов щелочных металлов; степень конверсии при
этом в 2,5—4 раза больше, чем в отсутствие водяного пара.
Это объясняется тем, что при взаимодействии с кислородом
MgCl2 разлагается с выделением хлора и образованием MgO, а в
результате гидролиза водяным паром MgCl2 также превращается
в MgO с выделением хлористого водорода. При нагревании каиниту
или смеси хлорида калия и сульфата магния реакция идет тем ин-
интенсивнее, чем больше пропускается водяного пара. В отходящем
газе содержится хлористый водород, а в остатке окись магния и
сульфат калия. Введение в реакционную смесь добавок—SiO2
(трепела), MgO и других в количестве 20—25% облегчает про-
процесс — в этих условиях реакционная масса остается рыхлой, рас-
рассыпчатой, что позволяет поднимать ее температуру до 800—900°
без превращения в жидкий плав. Это ускоряет реакцию с газовой
фазой (паром) и облегчает транспорт шихты. При 800—900° сте-
степень превращения КС1 в K2SO4 достигает 90—95%. Более высокие
температуры приводят к потерям КС1 вследствие его летучести.
Реакция с гипсом протекает при более высоких температурах,,
причем трепел является обязательным реагентом для связывания
СаО в СаО • mSiO2. Летучесть хлоридов магния и натрия в при-
присутствии трепела незначительна и не может неблагоприятно по-
повлиять на ход основной суммарной реакции 100-101.
При гидротермической переработке пород Предкарпатья про-
продуктами конверсии являются: водорастворимая солевая часть, со-
состоящая из сульфатов натрия и калия, и нерастворимая часть, со-
состоящая из трепела, окиси магния и силиката магния. Растворимые
соли выщелачивают из продуктов обжига при 100—106° в оборот-
оборотных маточных растворах, разбавленных промывными водами. Об-
Образующийся горячий раствор подвергается ступенчатому охлажде-
охлаждению в вакуум-кристаллизаторе с испарением части воды; в интер-
интервале 100—30° кристаллизуется глазерит. При охлаждении до 20°
кристаллизуется мирабилит; мирабилитовый маточный раствор
возвращают на выщелачивание. Количество оборотных щелоков на
1 т K2SO4 составляет 12—14 т. В качестве готовой продукции по
этому методу получают глазерит и сульфат натрия. Глазерит,
содержащий 40% КгО, может быть использован непосредственно-
как удобрение или- переработан на сульфат калия ш.
Подобным же гидротермическим способом можно получать суль-
сульфат калия из алунита K2SO4- A12(SO4K-4A1(OHK 103. При получе-
получении окиси алюминия из алунита сульфат калия является побочным
продуктом, однако количество его невелико, так как теоретическое
содержание K2SO4 в алуните равно 23%, а в алунитовых породах.
182
Гл. V. Природные калийные соли
еще меньше. Но если подвергать обработке водяным паром при
700° смесь алунита с хлористым калием, выход K2SO4 можно зна-
значительно увеличить за счет реакции:
6КС1 + A12(SO«K + ЗН2О = 3K2SO« + А12О3 + 6НС1
Гидротермический метод получения сульфата калия может ока-
оказаться экономичным только при условии использования на месте
производства получаемой соляной кислоты; количество ее, обра-
образующееся на 1 т КгО, составляет 6,8 т (в расчете на 27% НС1).
Переработка водонерастворимых калийных руд
Предложено много способов получения сульфата калия из по-
лигалитовых руд. Во всех способах, кроме способа, основанного на
восстановлении калийных солей до сульфидов, первоначальной
операцией является отмывка от галита и последующее прокалива-
прокаливание руды для перевода полигалита в водорастворимую форму 104.
В ряде способов предусматривается горячее выщелачивание
прокаленной руды при ~ 100°; в раствор переходят сульфаты ка-
калия и магния, в отвале остается гипс. Выделение сульфата калия и
леонита из раствора осуществляют при выпарке и вакуум-кристал-
вакуум-кристаллизации.
В других способах выщелачивание прокаленного полигалита
производят при 30е. В раствор переходит только MgSGj, твердая
фаза состоит из сингенито-гипса (K2SO4 • CaSC>4 • Н2О); ее прока-
прокаливают и выщелачивают при 100°. Сульфат калия при этом раство-
растворяется, из раствора его выделяют выпаркой и вакуум-кристалли-
вакуум-кристаллизацией.
Имеются способы, комбинирующие горячее и холодное выще-
выщелачивание прокаленного полигалита. Часть руды направляют на
горячее выщелачивание, а часть на холодное, — сопровождающееся
образованием сингенито-гипса. Процесс осуществляют при мокром
измельчении прокаленной руды в среде оборотного рассола. Полу-
Полученные кристаллы сингенито-гипса промывают, сушат и выпускают
как товарную продукцию (~25% КгО).
Наконец, существуют способы, основанные на обработке газо-
газообразным восстановителем сульфатов, содержащихся в руде. По-
Полученный при восстановлении сульфид калия хорошо растворяется
в воде, благодаря чему при холодном выщелачивании происходит
отделение калийных солей от магниевых и кальциевых. Раствор
сульфида калия подвергают карбонизации с образованием КгСО3.
Предложен способ переработки на сульфат калия лейцитовых
пород путем обжига тонкоизмельченной руды с известняком и га-
литом. Хлористый калий возгоняется; его извлекают в системе
охлаждающих и пылеулавливающих устройств.
Сульфат калия
183
В настоящее время полигалиты, а также другие водонераство-
римые калийные породы, на сульфат калия не перерабатывают, так
как технико-экономические показатели предложенных способов ока-
оказались неблагоприятными.
Целесообразна простейшая переработка полигалитовых руд —
отмывкой водой галита. Полученный при этом продукт содержит
14—15% КгО и может быть с успехом применен в качестве удоб-
удобрения. Проведены исследования отделения галита от полигалита
путем флотации.
Прочие способы получения сульфата калия и сульфатнокалийных
удобрений
Сульфат калия может быть получен восстановлением лангбей-
нита. Сырая лангбейнитовая соль, предварительно отмытая от хло-
хлористого натрия, смешивается с углем илн коксом в барабанном
смесителе (92% лангбейнита и 8% угля) и восстанавливается в-
шахтной печи в течение 4 ч при 800—900°:
K2SO4 • 2MgSO4 + 2С = K2SO4 + 2MgO + 2СО + 2SO2
Обогрев печи осуществляется газами, содержащими 95% ме-
метана, который восстанавливает SO2 до серы:
6SO2 + 4СН4 = 3S2 + 4СО + 8Н2О
Из полученного твердого остатка сульфат калия выщелачи-
выщелачивается при 100е водой. Окись магния отжимается на фильтрпрессе,
a. K2SO4 кристаллизуется из щелока при его охлаждении в отстой-
отстойниках-кристаллизаторах. Из 1 т лангбейнитовой соли получается
100 кг K2SO4 (96%), 75 кг MgO (85%)) и 20 кг серы.
Разработан способ получения сульфата калия при переработке
алунитов на глинозем и серную кислоту по восстановительной
схеме ВАМИ 105. По этой схеме при выпарке оборотных алюми-
натных растворов выделяется смесь сульфатов калия и натрия с
отношением K2SO4:Na2SO4 примерно 1 : 1. Смесь сульфатов перера-
перерабатывают на сульфат калия конверсией с хлоридом калия. Отли-
Отличием данного процесса от известных схем конверсионной перера-
переработки через глазерит является выпарка и вакуум-кристаллизация
глазеритовых маточных Щелоков с выделением возвратных солей
(глэзерита и хлористого калия) и получение из конечного щелока
пищевой соли и дополнительной порции возвратных солей. Послед-
Последняя стадия процесса протекает также при выпарке и вакуум-кри-
вакуум-кристаллизации с использованием оборотного раствора в замкнутом
цикле ш6.
При переработке на глинозем нефелинов, калийные соли выде-
выделяют из сбросного раствора 107, содержащего ионы Na+, K+, COf a
f (см. гл. VI).
184
Гл. V. Природные калийные соли
Предложен способ переработки каинитовых руд с получением
нового удобрения калушита K2SO4 • CaSO4 • Н2О 108. Процесс со-
состоит из растворения каинита и хлористого калия, обработки рас-
раствора гипсом с получением калушита и выпарки раствора после от-
отделения калушита. На первой стадии выпарки кристаллизуется
NaCl, при последующем выпаривании выделяется смесь КС1 и
NaCl, которую возвращают в начало процесса; в конечном щелоке
содержится MgCb.
Установлена возможность отделения глазерита от галита фло-
флотацией при применении в качестве флотореагента соединений типа
R — О — SO3H, где R — алкил-группа с числом атомов углерода в
цепи не ниже 5 109. Описан способ получения K2SO4 из КС1 и CaSO4
через промежуточный продукт — сингенит K2SO4 • CaSO4 • Н2О — в
водном и водо-метанольном растворе110.
ЛИТЕРАТУРА
I. Я. А. Фиалке в, В. Б. Черногоренко, ДАН СССР, 102, № 4, 739
A955).—г. М. Bernard, Jeffrey, Compt. rend., 240, № 10, 107A955); 242,
№ 21, 2512 A956) .-3. H. С. Банных, Я Е. В и л ь н я н с к и й, ЖОХ, 26, № 4,
952 A956). — 4. А. Б. Здановский, Е. Ф. Соловьева, Л. Л. Эзрохи,
Е. И. Л я х о в с к а я, Справочник экспериментальных данных по растворимости
солевых систем, т. III, Госхимиздат, 1961, стр. 1930.— 5. Л. Л. Эзрохи, Труды
ВНЙИГ, в. 31, 1956. стр. 164.— б. А. И. М у н, В. В. Т ар т ак о в ск а я, Изв.
АН КазССР. Сер. хим., № 8, 35 A955). — 7."м. И. Р а в и ч, Ф. Е. Боровая,
В. Я. Кетковнч, ИСФХА АН СССР, 22, 225 A953); 23, 258 A953).—
8. А. А. Иванов, Зап. Всероссийск, минералогическ. общества, 61, № 3
A932). — S. Ю. В. Морачевский, Бюлл. Ин-та галургии, № 5 A938). —
10. А. А. Черепенников, Газоносность солей Соликамского калийного руд-
рудника, Углетехиздат, 1949.
II. А. А. Иванов, Хим. пром., № 1—2 A946). —12. С. М. Коренеа-
ский, ДАН СССР, 88, № 6 A953). — IS. В. В Лобанова Труды ВНИИГ
в. 32, 1956, стр. 164. — /4. W. Pearson, J. Canad. Min. Metal. Bull., October
(I960). —15. Canad. Chem. Proa, June, 63 A964). —.76. E. F. Armstrong,
L. M. Mi all, Kaw Materials from the Sea (England). —17. D. E. Garrett
Chem. Eng. Progr., 10 A963). —18. Phos. a. Pot., № 27, 37 A967).—
19. J. W. High tower, Chem. Eng., 8 A961).— 20. M. Г. Валяшко Труды
ЙНИИГ, в. 23, 1953, стр. 93.
21. Ф. Ф. Вольф, В. С. Ятлов, Труды Центр, лаб. Севхимтреста, т. 1,
1929, стр. 5.-22. О. Д. Кашка ров, Труды ВНИИГ, в. 31, 1956, стр. 107.—
23. А. Б. Здановский, Там же, в. 33, 1956, стр. 185. — 24. А. Б. Зданов-
Здановский, Там же, в. 31, 1956, стр. 132. — 25. Г. А. Аксельрод, О применении
гидродинамики в кинетике растворения твердых частиц, Львов, 1955. — 26 Пат
ФРГ 956303, 1957. — 27. А. Б. Здановский, Л. Л. Эзрохи, ЖХП, № 2, 9
A948). — 28. А. Б. Нудельман, Там же, № 1, 10 A951). — 25». Н. Auten-
rieth, H. Rieke, H. Dust, Chem. Ing. Techn., № 11, 29 A957) —30 Франц
пат. 66544, !656.
31. Пат. ФРГ 108361, 1956. — 32. A. W. Bamfortt, Chem Proc. Eng 46,
№ 2, 81 A965). — 33. L. E. Garret, Ind. Eng. Chem., 53, № 8, 623 A961) —
34. В. А. Постников, ЖХП, № 11, 853 A963). — 35. J. Houghton, Chem.
Proc. Eng., 46, № 12, 639 A965).—36. V. K. LaMer, R. H. S me Hie,
P. K. Lee. J. Coll. Sci., 11, 704 A956); 1?.. 230, 566 A957). — 37. Л. И. Обрей-
Литература
185
Нова, А. К- Лившиц, сб. «Флотация», Госметаллургиздат, 1956, стр. 36,—
38. А. М. Swift, Tappi, 40, № 9 A957).—39. Ю. Я Каганович, А. Г. Зло-
О:1нский, Техника сушки во взвешенном слое, изд. ЦИНТИХИМНефтемаш,
1SJ66. — 40. Min. Eng., 18, № 10, 106 A966).
41. И. С. Лурье, Г. Г. К о л п и к о в, А. Г. 3 л о б и н с к и й, Хим. наука в
пром., 2, № 6, 767 A957). — 42. Н. И. Забродин, Б. П. Туркин, Там же, 3,
№ 1, 104 A958). — 43. Правила и нормы техники безопасности и промышленной
санитарии для проектирования, строительства и эксплуатации производства хло-
хлористого калия и обогащенного карналлита, Госхимиздат, 1961. — 44. Герм. пат.
325398, 1918; 355979, 1919; 374017, 1920; 347371, 1921; 278694, 1921. — 45. F. S е-
г о w у, Verarbeitungsmethoden der Kalirohsalze, Halle, (Saale) 1952. —
46. A. Jacob, Kaii, gewinnung und anwendung der Kalidiingesalze, Hannover,
1955. — 47. J. Ordonnaun, Annal. minerai et carb., 139, № 1 A950).—
48. Aufbereitungs Technik, № 2, 80 A967). — 49. Phos. a. Pot., № 36, 49 A968).—
50. Англ. пат. 408419, 1934; 425326, 1935; 544675, 1942; франц. пат. 799242, 1936.;
герм. пат. 687571, 1940; 650240, 1937; пат. США 2174950, 1938; К- Н. Шебалин,
сб. «Соликамские карналлиты», 1935, стр. 109.
51. А. С. Л е о н т и ч у к, сб. «Соликамские карналлиты», 1935, стр. 133. —
52. Я. С. Вильнянский, Там же, стр. 148. — 53. Я- М. Хейфец, Там же,,
стр. 25.-54. R. Noyes, Potash a. Potassium Fertilizer (USA), 1966. —
55. H. Schubert, Aufberlitungs Technik, № 6, 305 A966); № 7, 406 A967).—
56. Пат. США 2984348, 1961. — 57. Пат. США 2733809, 1960. —58. М. М. Павлю-
ченко, Э. Ф. Коршук, Калийные соли и методы их переработки, Минск,
1983. — 59. Ю. Е. Могульская, Т. В. Каплянская, Е. И. Соловьев,
сб. «Вопросы механического обогащения калийных солей», Изд. «Химия», 1966.—
60. Phos. a. Pot., № 36, 46 A968).
61. W. Wilson, Eng. Min. J, 167, № 8, 86 A966). — 62. P. Merrit, Min.
Eng., 18, № 10, 103 A966). — 63. H. С. Ульянов, И. Ф. Мещеряков,
VIII Международный конгресс по обогащению, 1968. — 64. И. Ф. Мещеряков,
Ю. В. Рябов, М. А. Подвигин, Хим. пром., № 12, 909 A968). — 66. Авт.
свид. 199125, 1967. — 66. Авт. свид. 185783, 1985.— 67. И. Ф. Мещеряков,
Цветные металлы, № 3 A968). — 68. Phos. a. Pot., № 29—32 A967).—
69. R. Burr, Rev. de l'indust. mineral., № 1 A954). — 70. В. М. Борисов,
H. А. Афанасьев, Хим. наука и пром., 1, № 2, 146 A956).
71. Пат. США 2997171, 1961. — 72. Пат. США 3052349, 1962. — 73. Пат. ФРГ
1095762, 1961. — 74. Авт. свид. 178318, 1965. — 75. Пат. США 3052349, 1965.—
76. Phos. a. Pot., № 30, 38 A967); Canad. Chem. Proc, June, 63 A964).—
77. R. A. G a s k a, R. D. G о о d e n a u g h, G. A. Stuart, Chem. Eng. Proc., Jan.
A965). 78. J. V. Hightower, Chem. Eng., 8 A961). — 79. Phos. a. Pot., № 29-
A967). — 80. А. Д. Пельш, Хим. наука и пром., 2, № 6, 734 A957).
81. Pit. a. Quarry, 47, № 6, 136 A955). — 82. А. Д. Пельш. Труды ВНИИГ,
в. 24, 1952, стр. 86. — 83. Е. F. Armstrong, L. M. Mia ! I, Raw Materials from
the Sea (England). — 84. О. Д. Кашка ров, Ю. М. Аллабердыев, Изв. АН
ТуркмССР, № 5, 84 A958); № 1, 118, № 2, 58 A959). — 85. Ю. М. Аллабер-
Аллабердыев, О. Д. Кашкаров, Изв. АН ТуркмССР, № 3, 15 A959).—
86. Д. Ленгауер, ИСФХА АН СССР, 14, 411 A941). — 87. Авт. свид. 189876,
1966. — 88. Авт. свид. 186991, 1966. — 89 Авт. свид. 184820. 1966. — 90. М. Е. П о-
зин, М. И. Муратова, ЖПХ, 30, №. 9, 1380 A957).
91. М. Е. Позин, М. И. Муратова, Там же, 1383. — ft2. H. С. Курна-
ков, Е. И. Лукьянова, Изв. АН СССР, Сер. хим., в. 1, 39 A938). —•
93. Я. Б. Влюмберг, Е. Ф. Соловьева, Труды ВНИИГ, в. 24, 1952,
стр. 77. — 94. А. И. Заславский, С. С. С и н а н и, А. А. Соколова, Изв.
АН СССР, № 1, 47 A938).— 95. Я Б. Блюмберг, Ю. Я- Каганович,
В. А. Останина, Е. Д. Ярушевич, Труды ВНИИГ в. 31, 1956, стр. 66.—
186
Литература
96. Пат. ФРГ 873838, 1953. — 97. А. А. Желнин, Л. А. О бр азид е лов а,
В. Г. Егорова, Вопросы механического обогащения калийных солей, Изд. «Хи-
«Химия», 1966. —98. Phos. a. Pot, № 21 A966). —99. G. Marullo, J. Vaccari,
Freiburg Forschung, A-267, № 39 A963).— 100. Я. А. фиал ков, сб. «Местные
минеральные удобрения УССР», в. 1, 1954. стр. 319.
101. С. Д. Ш а р г о р о д с к и й, И. О. Шор, Там же, стр. 334. —
102. С. И. Я к у б с о н: Н. К. Д а в и д е н к о, Там же, стр. 328. — 103. С. Д. Ш а р-
городский, О. И. Шор, А. С. Баранова, ЖПХ, 29, № 4, 492 A956).—
104. J. С о п 1 е у, Е. Р a t r i d g e, Соли калия из месторождения полигалитов в
Техасе и Нью-Мексико, 1944. — 105. Г. В. Л а б у т и н, Труды • ВАМИ, № 42,
1959; Авт. свид. 106048, 1955. — 106. А. Ф. Б о р я ч е к, Д. И. К У х а р е в, Труды
НИОХИМ, т. 10, 1957, стр. 67.-107. Г. С. Седельников, А. И. Лазаре-
Лазарева, Хим. пром., № 5, 401 A959). —108. Г. П. Александров, В. С. Тихо-
Тихонов, ЖПХ, 31, № 10, 1445 A958). — 109. С. А. Кузин, Там же. 12, № 3, 381
A939). — НО. Н. S с h п a b е 1, G. L i e b m а п n, W. М а у, Bergakademie, 20, № 5,
292 A968).
Глава VI
ПОТАШ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Углекислый калий, поташ, KsCO3 — белый кристаллический по-
порошок плотностью 2,43 г/см3, плавится при 896°, теплота плавле-
плавления 7800 ккал/кг-мол. Из водного раствора выше —6,2° кристал-
кристаллизуется К2СО3 • 1,5Н2О с плотностью 2,13 г/см3от — 6,2 до —36,5°
(криогидратная точка) выде- 150
ляется К2СО3 • 6Н2О. Теп-
Теплота растворения при 18°
в 400 моль воды: для К2СО3
+ 6490 ккал/кг-мол, для К2СО3-
• 1,5 Н2О - 380 ккал/кг-мол.
Насыщенный водный раст-
вор содержит при 0° 51,8,
при 15° 52,9, при 100° 60,9%
КоС03 (рис. 51). Темпера-
Температура кипения 5096 раствора
К2СО3 113,1°.
Поташ сильно гигроскопи-
гигроскопичен. Равновесная относи-
относительная влажность над
К2СО3-1,5Н2О при 25° всего
43%, на воздухе поташ расплывается, а при длительном хранении
поглощает двуокись углерода и превращается в твердый бикарбо-
бикарбонат калия КНСОз1.
ПРИМЕНЕНИЕ
Поташ используют в производстве стекла. Высококачествен-
Высококачественные сорта его применяют для изготовления медицинского, оптиче-
оптического, электровакуумного, художественного стекла, а также
¦50
! -
—.Лед
•
—
(
JO
10
W 30 40
Концентрация
50 ВО ТО
Рис. 51. Политерма системы К2СО3—H2O-
188
Гл. VI. Поташ
Методы производства поташа
189
хрусталя. Поташ употребляют для производства некоторых солей,
фармацевтических препаратов, жидкого калийного мыла, при по-
получении жидкой и твердой двуокиси углерода '•2; при крашении
и отбелке тканей, для промывания шерсти, для изготовления
печатных красок из индантреновых красителей и т. д. Его приме-
применяют для очистки промышленных газов от сероводорода по ва-
вакуум-поташному способу, особенно в коксохимической промышлен-
промышленности для очистки коксового газа3. Возможно использование по-
поташа (в смеси с содой) в качестве противопожарного средства.
Поташ используют в строительном деле в качестве морозозащит-
ной добавки в растворы и бетоны, имеющей преимущества перед
другими солями — он не вызывает коррозии, образования высолов
и проч.4. В сельском хозяйстве5-6 его применяют как щелочное
бесхлорное удобрение, однако пока в небольших количествах не
только из-за высокой стоимости, но и из-за плохих физических
свойств, обусловленных сильной гигроскопичностью. Ведутся по-
поиски способов улучшения физических свойств этого удобрения.
Пока же предпочитают транспортировать и использовать поташ в
виде жидкого удобрения — концентрированных растворов, или
предварительно перерабатывают его в бикарбонат калия7-8, об-
обладающий лучшими физическими свойствами (но почти в 1,5 раза
меньшей концентрацией калия). Бикарбонат получают карбони-
карбонизацией печным газом концентрированного горячего A30°) поташ-
поташного раствора и кристаллизацией при охлаждении.
Высокая стоимость поташа пока органичивает его применение
в строительном деле и в сельском хозяйстве, в перспективе же эти
отрасли (так же как и производство оконного, тарного и других
видов технического стекла) должны стать самыми крупными по-
потребителями поташа.
Получение поташа является старейшим химическим производ-
производством 9. На Руси оно возникло в XV в. Главнейшими потребите-
потребителями поташа были стекольное производство и мыловарение.
Сырьем для получения поташа с древнейших времен была расти-
растительная зола. Производство поташа из золы растений носило ку-
кустарный характер, и поташ мог конкурировать с другими щело-
щелочами лишь до появления дешевой соды, которую начали выпу-
выпускать с 1865 г.
Наибольшее количество поташных заводов (около 200) и наи-
наибольшая его выработка в России A7 000 т) падают на третью
четверть XIX в. Затем производство поташа сократилось. Значе-
Значение русского поташа упало также потому, что в Германии было
•создано промышленное его производство из калийных солей Стас-
фуртского месторождения 10.
К качеству поташа, вырабатываемого на современных пред-
предприятиях, предъявляются различные требования. Например, со-
согласно ГОСТ 10690—63 поташ из минерального сырья должен
содержать (в %):
Потаи! кальцинн- Поташ полутора-
рованныЙ водный
сорт I сорт 2 сорт 1 сорт 2
К2СО3, не менее 98 93 97,6* 92,5*
Натрия в пересчете на Na2CO3,
не более 0,9 4,0 0,9 6,0
Хлоридов в пересчете на хлор,
не более 0,07 1,5 0i07 2,0
Сульфатов в пересчете на SO4,~
не более 0,4 1,0 0,5 1,6
Железа в пересчете на Fe2O3,
не более 0,006 Не опреде- 0,005 Не опреде-
определяется ляется
Не растворимого в воде ос-
остатка, не более 0,1 То же 0,1 То же
' В пересчете на прокаленное вещество.
Тарой для поташа служат многослойные бумажные битумиро-
ванные или прорезиненные мешки, или мешки из пластиковой
пленки.
МЕТОДЫ ПРОИЗВОДСТВА ПОТАША
Лишь некоторые из многих способов получения поташа нашли
практическое применение п.
Один из старых способов — извлечение поташа из раститель-
растительной золы 12~17 — почти полностью потерял прежнее свое значение
вследствие недостаточной экономичности. В ряде стран (ГДР, Че-
кословакия, Польша, Италия и др.) существует производство
поташа из бардяного угля (продукта коксования упаренной па-
паточно-спиртовой барды) 13> 16>17. Бардяной уголь содержит около
60% К2СО3, но наряду с углекислым калием в нем находятся сода,
сернокислый и хлористый калий и др.
Поташ можно выделять и из золы, получаемой после выпарки
и полного сжигания паточно-спиртовой барды 18>1Э.
Производство поташа из золы растений, бардяного угля и
золы, овечьего пота имеет сейчас лишь местное значение5. В на-
настоящее время поташ вырабатывают главным образом из мине-
минерального сырья. В СССР его производят при комплексной пере-
переработке нефелинов. При этом получают глинозем, цемент и содо-
поташные растворы, перерабатываемые на соду и поташ 20-25
В небольших количествах поташ получают из озерной рапы.
За рубежом наиболее распространены способы получения по-
поташа из хлорида калия — электролитический, заключающийся в
карбонизации раствора едкого кали26, полученного электролизом
хлорида калия, и аминовый13, аналогичный аммиачному способу
получения соды, но с заменой аммиака различными аминами. Эти
190
Гл. VI. Поташ
способы дорогие, стоимость продукта высока и поэтому его выра-
вырабатывают в небольших количествах.
Хлорид калия можно переработать в поташ магнезиальным
способом Энгеля — Прехта 13- 27~29. Он заключается в карбониза-
карбонизации под давлением E—18 ат) суспензии активного карбоната
магния в растворе хлорида калия:
3(MgCO3 • ЗН2О) + 2КС1 + СО2 ^=± 2(КНСО3 • MgCO3 • 4Н2О) + MgCl2
Образующуюся двойную соль Энгеля, отделенную от маточного-
раствора, разлагают при нагревании водой или суспензией гидро-
гидроокиси магния — получают раствор поташа, перерабатываемый на
твердый продукт, и магнезию, возвращаемую в процесс. В настоя-
настоящее время этот способ, позволяющий получать весьма чистый по-
поташ, имеет ограниченное применение (в СССР не применяется)
вследствие своей сложности, связанной главным образом с обес-
обеспечением условий образования активного карбоната магния.
Для получения легко фильтрующейся соли Энгеля и более пол-
полного использования магния рекомендуют заменить СО2 бикарбо-
бикарбонатом натрия30. Вместо КС1 можно использовать содо-поташные
щелока, подвергаемые карбонизации в присутствии MgCO3 • ЗН2О 31.
Из хлорида калия можно получать поташ через кремнефторид
калия одновременно с фторидом натрия32 (см. гл. XXXI).
Поташ производят переработкой природного сернокислого ка-
калия формиатиым способом26-33. Известны и другие "¦'2 способы
получения поташа, например, способ Леблана, циана мидный, ба-
бариевый (из сульфата калия), но на практике они не исполь-
используются.
Переработка золы подсолнечника *
Зола подсолнечника 13 содержит в растворимой части от 15 до
35% К2СО3, 3,5—4,1% K2SO4 и 3,8—5,1% КС1. Не растворимый
в воде остаток составляет от 40 до 61%. Выщелачивание подсол-
подсолнечной золы осуществляют в стальных резервуарах с ложным дном
емкостью до 10 г золы по методу «настаивания». В цикле обычно
работают четыре выщелачивателя. Процесс ведут по принципу
противотока: первый аппарат, загруженный свежей золой, зали-
заливают раствором, полученным из второго, этот выщелачиватель за-
заливают раствором из третьего и т. д. Последний аппарат заливают
горячей водой. Заливку раствором каждого выщелачивателя про-
проводят в 3—4 приема. На 1 т золы расходуется около 0,7—1 мг
воды. Выщелачивание заканчивают при остаточном содержании
в сухом подзоле не более 1,5% К2СО3, что дает степень извлече-
извлечения К2СО3 около 95%. Получается концентрированный раствор
* Данные по технологическому режиму производства представлены 3. П. Ве-
ретенииковой.
Методы производства поташа
191
(плотность 1,19 г/см3) среднего состава (в %): К2СО3—15,0,
КС1 — 3,3, K2SO4—1,7. Этот раствор осветляется от взвешенных
частиц в отстойниках.
На диаграмме растворимости84 в системе КгСОв—КС1—
—K2SO4—Н2О при 75° (рис. 52) точка Н состава раствора после
выщелачивания (в пересчете на сухое вещество) лежит на вторичной
КС1.%
¦ Вточну
40 гоЬ
KjSO4.%
ВО 100
кгсо,.%
Рис. 52. Диаграмма растворимости в системе К2СО3—КС1—KaSO4—Н2О
при 76° и в системе К2СО3—КС1—Н2О при 25е (по способу вторичных
проекций36).
проекции (точка Т) в поле кристаллизации K2SO4. Вспомогатель-
Вспомогательные построения показывают, что начальный раствор Н является
ненасыщенным. В процессе выпарки этого раствора по лучу ОТ
первой кристаллизующейся фазой на участке отрезка LD является
K2SO4. На отрезке луча выпарки DK кристаллизуются одновре-
одновременно K2SO4 и КС1. В точке К выпарка должна быть прекращена
во избежание выделения также и КгСОз- 1,5Н2О. В этот момент
состав жидкой фазы определяется положением точки Е {Е'), в ко-
которой раствор насыщен тремя твердыми фазами35. Выпарку рас-
раствора заканчивают при достижении плотности 1,57—1,60 г/см3
при температуре его кипения 75—85° (давление 0,2—0,1 ат).
Наиболее чистый сульфат калия (без примеси КС1) выделяется
при выпарке до плотности 1,40—1,45 г/см3. На практике выпарен-
192
Гл. VI. Поташ
Методы производства поташа
193
ный раствор имеет следующий примерный состав (в %): К2СО3 —
50,0-ь50,6: КС1 —3,5^-4,4; K2SO4—- 0,6-=-1,0. В производственных
условиях выпаривание заканчивают несколько ранее достижения
состава тройной точки Е (?')•
Растворимость КС1 в двойной точке В системы К2СО3—КС1—
—Н2О при 25° составляет 0,97% 36- Из рис. 52 видно, что точки
состава выпаренного раствора лежат вблизи предельного луча
кристаллизации КС1 — В; следовательно, при этой температуре
возможно выделение КС1 или же смеси КС1 и КзСОз- 1,5Н2О.
Одновременно, вследствие снижения растворимости K2SO4 в чет-
четверной системе34, при охлаждении будет осаждаться в небольших
количествах и эта. соль. Выкристаллизовавшуюся смесь солей про-
промывают для уменьшения потерь КгСОз. После промывки смесь
содержит 65—70% КС1, 20—30% K2SO4 и 5—7% К2СО3. Практи-
Практически раствор поташа после охлаждения до 25—30° и кристалли-
кристаллизации солей имеет средний состав (в %): KgCO3 — 50,6; КС1— 2,2;
K2SO4—1,0 (плотность 1,56 г/см3). Раствор подогревают и выпа-
выпаривают до плотности 1,60—1,64 г/смъ, после чего его направляют
в напольные печи для прокаливания.
При переработке золы с содержанием 20% К2СО3, 4% KCI
и 2%) K2SO4 расходуют на 1 т поташа: золы — 5,125 т, пара —
4,5 т, топлива условного на прокалку — 0,24 т, электроэнергии —
75 квт-ч, воды—10 мг. При этом получается отходящих солей:
хлорида калия F5—70% КС1)—0,2 т, сульфата калия (94%
K2SO4)— 0,1 т.
Переработка бардяного угля и золы
Бардяной уголь и зола получаются выпариванием и, соответ-
соответственно, неполным или полным сжиганием барды, являющейся
отходом после сбраживания патоки свеклосахарных заводов и от-
отгонки спирта. Присутствие в бардяной золе Na2CO3, кроме КгСО3,
K2SO4 и КС1, усложняет ее переработку. Соотношение между ко-
количествами основных солей в бардяном угле и золе очень разно-
разнообразно: при колебаниях в содержании К2СО3 от 30 до 80% от-
отношение Ыа2СОз: К^СОз находится в пределах 0,03—0,50;
КС1: К2СО3 0,06—0,67- K2SO4: К2СО3 0,07—0,63.
На одной из действующих установок хг<1Т бардяной уголь из-
измельчают и выщелачивают водой при 80°. После отделения и про-
промывки шлама19 полученный раствор плотностью 1,19—1,22 г/см3
вьшарииают до плотности 1,39—1,45 г/см3, при этом выделяется
большая часть сульфата калия. Маточный раствор охлаждают до
22—25е для кристаллизации КС1. Сухое вещество жидкой фазы
содержит около 81% К2СО3, 2,5% K2SO4, 6,4% КС1 и 10% Na2CO3.
При высушивании такого раствора можно получить низкокаче-
низкокачественный 80%-ный поташ.
Для получения поташа высокого качества раствор, после от-
отделения КС1, дополнительно охлаждают для выделения двойной
соли К№СОз. Для получения 96%-ного поташа раствор охла-
охлаждают до 0°; при охлаждении до 5—6° готовый продукт может
содержать лишь 90%) КгСОз. Раствор после отделения двойной
соли дополнительно выпаривают до плотности 1,69 г/см3, при этом
Na2COs.%
HaCt%
КС1.%
Рис. 53. Диаграмма водной системы КгСОа + 2NaCl Т~*" Na2CO3 + 2KC1 при 76е
(по способу вторичных проекций35).
выделяется кристаллический поташ. К концу выпаривания тем-
температура кипения раствора достигает 140°. Концентрированный
раствор, содержащий взвесь кристаллов углекислого калия, охла-
охлаждают до 96е, благодаря чему дополнительно выделяются кри-
кристаллы поташа, затем смесь пропускают через центрифугу. Отфу-
гованный поташ высушивают. Маточный раствор плотностью
1,58—1,60 г/смъ смешивают со свежим раствором, идущим на вто-
второе выпаривание. По мере накопления в этой смеси примесей
посторонних солей она выводится из цикла и поступает в прока-
лочную печь для получения поташа пониженного качества. Поташ
высшего качества, получаемый описанным методом, содержит
7 М. Е. Позии
194
Гл. VI. Поташ
около 96% К2СО8, 0,5% Na2CO3, 0,5% K2SO4 и 1% КС1. Выделен-
Выделенную двойную соль подвергают переработке для получения низко-
низкокачественной соды G5%) и поташа пониженного качества (около
90% К2СО3).
Имеющиеся неполные данные34 по равновесию в системе
NaCl—KC1—Na2SO4—K2SO4—Na2CO3—К2СО3—Н2О показывают,
что в важных для данного процесса точках при 25—80° в жидкой
фазе содержатся лишь следы K2S0.4, поэтому без большой по-
погрешности можно пренебречь наличием в исходных растворах
K2SO4. При выпарке этих растворов сернокислый калий будет
выделяться практически полностью и в первую очередь.
Вследствие этого можно ограничиться исследованием 13 взаим-
взаимной системы K2CO3 + 2NaCl±i:Na2COs+ 2KC1 при 75° (рис. 53
и табл. 17) 34.
Система К2СО3 + 2NaCl ^ Na2CO3 + 2KCI при 75°
ТАБЛИЦА 17
f •
&
S
к
о
ь
S1
24
25
52
74
54
27
22
93
94
95
96
Состав
состав 1
О
Ч,
¦х
_
—
—
57,6
—
56,1
—
44,4
55,9
—
41,3
55,3
те
о
о
«
Z
—
31,2
—
—
—
11,13
8,48
2,14
9,81
8,2
1,65
жидко!
эаствора
G
33,3
—
—
—
18,45
2,17
_
—
17,4
3,5
2,0
О
л
2
_
27,5
—
—
17,75
—
20,9
_
—
11,82
-
фазы
. вес.
состав раствора в
счете
те
О
у
—
—
100
—
96,3
_
83,9
26,3
—
78,0
93,8
пере-
на сухое вещество
О
О
Л
Z
_
100
—
—
—
34,7
16,1
3,7
25,1
15,4
2,8
о
100
—
_
—
60.S
3,7
—
—
44,6
6,6
3,4
й
Z
100
_
_
49,1
—
65,3
_
—
30,3
-
Твердые фазы
КС1
NaCl
Na2CO3 • Н2О
К2СО3 • 1,5Н2О
NaCl + KC1
К2СО3-1,5Н2О+КС1
Na2CO3 • Н2О + NaCl
Na2CO3 • Н2О + KNaCO3
K2CO3-l,5H2O+KNaCO3
NaCl + КС1 + Na2CO3 • H2O
Na2CO3 • H2O + KNaCO3 + KCl
K2CO3 • 1,5H2O + KNaCO3+KCl
ТАБЛИЦА IS
Состав образцов растворов, полученных из бардяной золы
Точка (см.
рис. S3)
Я7(Г7)
Состав раствора, вес. %
к2со3
12,48
10,43
Na2CO3
3,41
1,85
KCl
1,44
3,70
K2SO4
0,80
3,00
Na2CO3
К2СОз
0,273
0,181
kci
K2CO3
0,115
0,354
K2SO4
КгСОз
0,064
0,288
Состав раствора в пере-
пересчете на сухое вещество
(без K2SO4)
К2СО3
72,0
65,3
Na2CO3
19,7
11,5
KCl
83
23,2
Методы производства поташа
195
Все точки состава растворов, полученных из бардяной золы и
угля (без учета содержания K2SO4), ложатся в правой части
диаграммы, несмотря на разли-
различия в соотношении солевых ком-
компонентов. Для рассмотрения
взяты два типичных раствора
(табл. 18).
При выпаривании начального
раствора Н7(Т7) при 75е первой
выделяющейся фазой (после оса-
осаждения K2SO4) будет №2СОз-
• Н2О, а затем смесь Na2CO3 • Н2О
и КС1. В точке 95 (95') в твердой
фазе появляется двойная соль
KNaCO3. Температура выпарива-
выпаривания 75е выбрана потому, что в
этих условиях отпадает после-
последующая кристаллизация солей
при более низкой температуре.
Точка состава второго рассмат-
рассматриваемого начального раствора
Н9(Тд) лежит в поле кристалли-
кристаллизации КС1. Выпадение этой соли
заканчивается вблизи переход-
переходной точки 95(95'), т. е. до начала
выделения двойной соли.
Таким образом, первый этап
выпаривания заканчивается при
приближении состава жидкой
фазы к точке 95(95'), после чего
выпавшие твердые фазы отде-
отделяют. Следующей стадией яв-
является выпаривание при 75е для
выделения двойной соли в смеси
с КС1. В точке 96 (96') выпарку
прекращают и выпавшие кри-
кристаллы KNaCO3 + KCl отделяют.
Температура 75е для этой стадии
выпаривания тоже является оп-
оптимальной, так как маточный ра-
раствор в этих условиях содержит Рис. 54. Схема производства поташа
наименьшее количество примесей из бардяной волы,
солей.
Точка 96 (96'), определяющая состав конечного маточного
раствора при 75е (табл. 17), лежит в пеле кристаллизации КС1
для 25е. Следовательно, охлаждением раствора 96 (96') до 25°
7*
196
Гл. VI. Поташ
(при небольшом разбавлении водой) можно снизить содержание в
нем КС1. После дополнительного выделения КС1 солевой состав
жидкой фазы будет: 95,5% К2СО3, 2,85% Na2CO3 и 1,65% КС1.
На основе анализа равновесных диаграмм системы предложена
и экспериментально проверена18 технологическая схема производ-
производства поташа, приведенная на рис. 54. В этой схеме использовано
свойство двойной соли растворяться в воде при 35е и выше ин-
конгруэнтно, т. е. с разложением. При соответствующем количе-
количестве воды в раствор переходит КгСО3. а большая часть Na2CO3 и
КС1 остается в осадке. Раствор карбоната калия возвращается
в процесс.
Изучен13 также вариант переработки бардяной золы, заклю-
заключающийся в выделении натрия в виде фторида. По этой комбини-
комбинированной схеме первоначальный раствор бардяной золы предва-
предварительно выпаривают с целью возможно более полного выделения
K2SO4 и КС1. После отделения смеси солей маточный раствор
смешивают с раствором фторида калия, получаемого конверсией
кремнефторида калия оборотным раствором поташа (см. гл. XXXI).
При этом частично осаждается фторид натрия. Фильтрат подвер-
подвергают вторичной выпарке для полного отделения оставшегося в
растворе фторида натрия. Дальнейшей задачей является кристал-
кристаллизация поташа; для этого осуществляют третью ступень вы-
выпарки.
Экспериментально установлена возможность получения этим
способом как высококачественного поташа (не ниже 96% КгСО3),
так и фторида натрия (в среднем 97% NaF). Степень извлечения
поташа из бардяной золы достигала 94%.
Получение поташа из содо-поташных растворов глиноземного
производства
При получении глинозема из нефелина образуются побочные
растворы (плотность 1,15—1.18 г/см3) следующего среднего со-
состава20 (в г/л): Na2CO3—157: К2СО3—62; K2SO4 (включая не-
окисленные соединения серы) — 16: собственно K2SO4— 15; R2O3 —
0,1; CI" — 0,07 Эти растворы перерабатывают на содо-поташную
смесь и на поташ. Исходные растворы содержат частично бикар-
бикарбонаты Na и К (свыше 7 г/л в пересчете на NaHCO3), наличие ко-
которых вызывало значительную коррозию стальной аппаратуры37
и загрязняло поташ окислами железа. Нейтрализация начальных
щелоков раствором NaOH резко уменьшила коррозию оборудова-
оборудования, и содержание железа в поташе снизилось с 0,06—0,14% до
0,001—0,002% 25- Поташ получают из содо-поташных щелоков по
следующей упрощенной схеме20. Производится предварительное
концентрирование щелоков в вакуум-выпарных аппаратах Кест-
нера до суммарного содержания солей 400—440 г/л (плотность
Методы производства поташа
197
1,29—1,33 г/см3). Затем ведется вторая выпарка при 130—132° до
точки насыщения раствора солями №гСО3, K2SO4 и KNaCO3 (кон-
(концентрация солей в растворе 750 г/л, плотность его 1,51—1,53 г/см3).
В процессе второй выпарки выделяется смесь из соды и сернокис-
сернокислого калия, которая отделяется на центрифугах и высушивается.
Пример анализа этой смеси: 79,06% Na2CO3, 11,27% КгСОэ (при-
(примесь из жидкой фазы), 9,28% K2SO4, 0,001% Fe2O3 и 0,31% по-
потери при прокаливании.
При расчете выпарки следует иметь в виду, что давление во-
водяного пара над растворами, содержащими К2СО3, Na2CO3 и до
б вес.% K2SO4, практически равно давлению пара над чистым
раствором КгСО3, концентрация которого равна сумме концен-
концентраций этих трех солей33.
Маточный раствор после второй выпарки смешивают с маточ-
маточным раствором после кристаллизации КгСО3-1,5^0 (см. ниже)
и выпаривают при 105—112° до точки насыщения солями K2SO4,
KNaCO3 и КгСОз- 1,5Н2О (третья выпарка). Такой раствор (плот-
(плотность 1,64 г/см3) содержит 1060 г/л КгСО3, 28,8 г/л Na2CO3,
45,2 г/л KaSCUoCii, и 6,8 г/л K2SO4.
В процессе третьей выпарки выпадает смесь KNaCO3 и K2SO4
(фактический состав: 26,2% Na2CO3, 64,6% К2СО3, 3,41% K2SO4).
Этот осадок отделяют фильтрованием, растворяют в конденсате
и возвращают на смешение с начальным щелоком.
Раствор поташа после третьей выпарки охлаждают до 45—50е
для выделения КгСО3-1,5Н2О. Поташ отжимают на центрифугах
и отгружают потребителям без сушки, а маточный раствор воз-
возвращают на третью выпарку.
Продукт имеет средний состав (в % на сухое вещество):
К2СО3 97,63
Na2CO3 0,82
K2S04o6i4 ........ 0,68
K2SO4 0,46
ci- .
А12О3
Fe2O3
Н2О .
0,07
0,043
0,002
17,22
Об автоматическом регулировании кристаллизации поташа в
однокорпусной вакуум-кристаллизационной установке см.39
Разработан способ гранулирования поташа, содержащего 5—
6% гигроскопической влаги в тарельчатом грануляторе, на кото-
который подают смесь КгСО3-1,5Н2О, мелкую и крупную фракцию
КгСОз после дробления гранул и воду в отношении 1 :0,45—
0,8:0,12—0,2. Гранулы могут быть высушены в барабанной су-
сушилке до полного обезвоживания, причем их прочность не умень-
уменьшается 40.
В циркулирующих растворах постепенно накапливаются ионы
хлора и неокисленные соединения серы. Для выделения соединений
хлора раствор поташа "эпизодически подвергают охлаждению до
температуры ниже 45е, при этом выделяется поташ, загрязненный
хлором (до 1 %), а очищенный фильтрат вновь поступает на третью
198
Гл. VI. Поташ
Методы производства поташа
199
выпарку. Единственной возможностью уменьшить загрязнение по-
поташа неокисленными соединениями серы при работе по описан-
описанной схеме является промывка поташа конденсатом. Однако про-
промывка не обеспечивает получение поташа без примесей неокис-
ленных соединений. В приведенном выше анализе поташа коли-
количество неокисленных соединений серы составляет 0,22% (на
K2SO4). Остается нерешенным еще вопрос использования промыв-
промывного раствора, содержащего после промывки поташа значитель-
значительные количества углекислого калия, загрязненного неокисленными
соединениями серы.
iatSO4 Ha2COy2Na2SO4
Рис. 85. Диаграмма водной системы КгСО3 + Na2SO4
35\
при 75° (по способу вторичных проекций 35).
В ряде исследований изучался процесс разделения содо-поташ-
ных щелоков с применением карбонизации растворов 23> 24> 41~43.
Эти способы требуют для своего осуществления аппаратуры из
нержавеющей стали. В других работах 22>2Б-33-44 разделение солей
осуществлялось на основе анализа взаимной системы К2СО3 +
+ Na2SO4^±Na2CO3 + K2SO4, изученной при 2536, 35, 50, 7534,
10045 и 150°46'47. Привлекались также данные, доказавшие су-
существование двойной соли KNaCO3 в системе КгСОз—Na2CO3—Н2О
при температуре выше 100°48-49. Равновесие в данной системе
позволяет осуществить последовательное выделение солей по схеме,
показанной на примере диаграммы для 75° (рис. 55). Точка со-
состава нейтрализованного едким натром начального раствора Н
(#') лежит в поле кристаллизации соды. Следовательно, сода бу-
будет первой твердой фазой, выпадающей в процессе изотермической
выпарки. На этом этапе сода может быть выделена наиболее
полно (свыше 80%) при 150°. В процессе дальнейшей выпарки
солевой состав жидкой фазы изменяется вдоль изотермы от точки
М (М') до точки Е (Е') и затем до точки F (F1). На этом пути
в твердую фазу выделяется смесь соды и глазерита. В точке Е
(Е1) глазерит взаимодействует с КгСО3, превращаясь в Na2CO3 и
K2SO4. По окончании этого превращения в твердую фазу выде-
выделяются сода и сернокислый калий. В точке F (F') раствор насы-
насыщен содой, сернокислым калием и двойной солью (табл. 19).
ТАБЛИЦА 19
Состав растворов в точке F(F') насыщения солями
Na2CO3 • *Н2О + KNaCO3 • #Н2О + K2SO4 при разных температурах
25
35
50
75
100
150
Состав раствора,
вес. %
«
О
о
сч
17,02
45,60
45,10
44,20
43,59
45,82
«
О
о
см
а
Z
21,00
6,70
7,22
8,14
9,18
8,95
О
«
й
0,75
Следы
»
0,31
0,74
Состав раствора
в пересчете на сухое
вещество, вес. %
О
о
сч
54,16
87,20
86,30
84,50
82,12
82,50
О
О
«ч
М
Z
43,90
12,80
13,70
15,50
17,30
16,10
О
1,94
Следы
»
0,58
1,40
Твердые фазы
Na2CO3-7H2O + Ыа2С03-КгС0з-6Н2О + K2SO4
Ыа2СОз • Н2О + Na2CO3 • К2СО3 + K2SO4
То же
» »
Na2CO3 + Na2C03-K2CO3 + K2SO4
Из данных табл. 19 следует, что K2SO4 выделится наиболее
полно при проведении второго этапа выпарки при температуре не
выше 75°, так как уже при 100° содержание K2SO4 в жидкой фазе
начинает увеличиваться. После отделения соды, загрязненной
сульфатом калия, маточный раствор F (Р), имеющий плотность
1,52 г\смг, подвергается дальнейшей выпарке. На этой третьей
стадии выпарки состав жидкой фазы изменяется вдоль изотермы
от точки F (F') до точки G ( G') (табл. 20).
ГАБЛ ИЦД 20
Состав растворов в точке О(С) насыщения солями
К2СО3 • Na СОз • #Н2О + K2SO4 + K2CO3 • 1,5Н2О при разных температурах
25
35
50
75
100
150
Состав раствора
вес. %
8
еч
48,10
49,95
52,10
56,00
59.38
66,81
О
О
л
Z
5,02
4,17
3,21
2 01
1,03
1,15
О
а
ы
0,63
Следы
»
0,07
0.25
Состав раствора
в пересчете на сухое
вещество, вес. %
8
?
89,45
92,28
94,20
96,55
98,18
97,94
О
О
CS
¦г.
9,35
7,72
5,80
3,45
1,70
1,70
О
(Л
еч
1,17
Следы
0J2
0.36
Твердые фазы
КгСОз-ЫагСОз-бНгО + K2SO4 + К2СО3-1,5Н2О
K2CO3-Na2CO3 + K2SO4 + К2СО3.1.5НгО
То же
» »
» »
Наиболее целесообразна выпарка на третьей стадии при 100°
(жидкая фаза содержит 98,18% К2СО3 в пересчете на сухое ве-
вещество, ее плотность 1,62 г/см3).
200
Гл. VI. Поташ
Исходный щелок
Нейтрализация
исходного щыт
t выпорка
{рредварилтыщ
Пары воды
_Jlapbi воды
ъ соды и суль-
т калия но сушку
X
рф)
(перваястадий
X
26ыпарнаG5")
[Вторая стадия)
Фильтрация
olfaue) C
Лары воды
3 выпарка
A00")
Фильтрация
X
Фильтрация
Дотпттепьнт
выпарка G5°)
X
Кристаллизация
поп?ви/аB50)
X
Фильтрации
Выделившуюся в осадок в процессе третьей выпарки двойную
соль, загрязненную сульфатом калия, отделяют и возвращают на
смешение с начальным щелоком. Даль-
Дальнейшая переработка маточного рас-
раствора зависит от способов борьбы с
накоплением примесей — главным об-
образом иона хлора и неокисленных со-
соединений серы. Эти соединения легко
растворимы и остаются в растворе до
завершения процесса, накапливаясь в
товарном продукте, если производст-
производственный цикл является замкнутым.
Получение поташа высокого каче-
качества из неокисленных растворов воз-
возможно посредством систематического
вывода маточного раствора после кри-
кристаллизации углекислого калия (не
замкнутый производственный цикл).
Отводимый маточный раствор высу-
высушивают для получения поташа, за-
загрязненного неокисленными соедине-
соединениями серы 25. Маточный раствор пос-
после третьей выпарки и отделения двой-
двойной соли дополнительно выпаривают
при 75—100е, в результате чего частич-
частично выделяется К2СО3-1,5Н2О (осадок
собирается в солесборнике выпарного
аппарата). Жидкую фазу охлаждают
до 25е, при этом кристаллизуется до-
дополнительное количество К2СО3 •
• 1,5Н2О. Смесь обоих осадков кристал-
кристаллического поташа промывают насы-
насыщенным раствором поташа, получен-
полученным в предыдущей операции. Промыв-
Промывка идет полнее при предварительном пе-
перемешивании осадка с частью промыв-
промывного раствора. Вторичную промывку
осуществляют непосредственно на
фильтре или центрифуге. По этой схеме
частично получается кристаллический
поташ, содержащий (в % на сухое
вещество): К2СО3—99,0, Na2CO3—0,3,
K2SO4—0,2, окисляемых соединений (в
пересчете на K2SO3)—0,05, Fe2O3—0,001.
Наашминйе-о поташа А
Промывка
Пршюгпселение
сыщеннойо
раствора
Поташ
(«о сушку)
Рис. 56. Принципиальная схема
получения чистого поташа из
содо-поташных растворов
глиноземного производства (с
применением окислительной
прокалки поташа).
Второй возможный путь, позволяющий выпускать весь поташ в
виде высококачественного продукта, — окисление соединений серы.
Методы производства поташа
201
Из испытанных25 средств окисления (перекись водорода, персуль-
персульфаты, воздух в присутствии катализаторов и окислительная про-
прокалка) наиболее эффективна окислительная прокалка поташа.
Схема получения поташа с окислительной прокалкой (рис. 56)
одинакова с другими вариантами вплоть до третьей выпарки и от-
отделения двойной соли, далее раствор высушивают и прокаливают
в течение 3 ч при 600° с доступом воздуха. В этих условиях степень
окисления соединений серы до сульфата калия достигает 96—98%.
Прокаленный поташ растворяют в воде при 25° с таким расчетом,
чтобы образовавшийся K2SO4 остался нерастворенным. Последний
отфильтровывают, а раствор выпаривают при 75° и охлаждают до
25е, как это описано выше. После промывки кристаллический по-
поташ является готовым продуктом. Маточный раствор возвращают
в процесс (замкнутый производственный цикл).
Поташ, полученный по описанной схеме, имел следующий со-
состав (в % на сухое вешество): К2СО3 —99,4, Na2CO3 — 0,2, K2SO4—
0,05, окисляемых соединений (в пересчете на K2SO3)—до 0,01,
Fe2O3 —до 0,001, Сг2О3 —0,00004, V2O5 — 0,0001. Такой поташ при-
пригоден для производства хрусталя. \
Производство поташа карбонизацией раствора едкого кали
При электролизе хлорида калия по диафрагменному способу
получается раствор, содержащий 145 г/л КОН и 180 г/л КС1. Для
отделенит КС] раствор концентрируют до 44,6—47,1 % КОН (плот-
(плотность 1,47—1,50 г/см5) и затем охлаждают. При 20—30° в растворе
остается лишь около 1 % КС1. После отделения выделившегося хло-
хлорида калия раствор разбавляют до концентрации около 30% КОН
(плотность 1,3 г/смъ) и подвергают карбонизации в колонне с на-
насадкой из керамических колец 13> 26>50. (При электролизе хлорида
калия в ванне с ртутным катодом получается раствор КОН, содер-
содержащий лишь сотые доли процента хлоридов, и его карбонизуют
без предварительной очистки.)
Запатентован51 способ двухступенчатой карбонизации концен-
концентрированного 50% раствора КОН; при этом с большей полнотой
используется СО2 из топочного газа. В I ступени в противоточном
абсорбере карбонизации газом, содержащим ~10% СО2, подвер-
подвергается циркулирующий здесь раствор смеси КОН и КгСОз. К выте-
вытекающему раствору добавляют свежий раствор КОН и 90% этого
абсорбента возвращается в абсорбер, а 10% передается во II сту-
ступень абсорбции, где точной дозировкой СО2 (например, жидкой СО2
из баллонов) достигается полная нейтрализация КОН до K2COS.
Во избежание кристаллизации К2СО3 в I ступени ведут процесс
при 40—100°, подавая в абсорбер раствор с отношением
КОН: К2СО3, равным 1 : 1, и карбонизуя здесь 90% КОН.
202
Гл. VI. Поташ
Методы производства поташа
203
Дальнейшую переработку карбонизированных растворов произ-
производят по схеме рис. 57. Раствор углекислого калия выпаривают в
трехкорпусной вакуум-выпарной батарее. Концентрированный рас-
раствор высушивают на непрерывно действующих вакуум-вальцах,
обогреваемых паром. Высушенный продукт прокаливают в вакуум-
вакуумных барабанных сушилках периодического действия с паровой ру-
рубашкой (давление пара 12 ат). Этим методом получают поташ с
К Вакуум-насосу
В атмосферу
К Вакуум-насосу
Рис. 57. Схема получения поташа карбонизацией растворов КОН:
I —карбонизационная колонна; 2 и 7—сборники; 3 — напорный бак; 4 — вакуум-выпарной аппа-
аппарат; 5 - конденсатор; 6 — барометрический ящик; 8 — вакуум-вальцы; 9— циклон; 10 к 13- транс-
транспортеры; 11 — барабанная сушилка; 12 — конусный ящик; 14 — элеватор; 15 — бункер.
содержанием 98% КгСО8 и около 1% КС1. Для выпуска более чи-
чистого поташа (по содержанию хлоридов) последний очищают пе-
перекристаллизацией.
В ГДР поташ получают также из смеси растворов, содержа-
содержащих КОН и КгСО8 (отходные растворы от производства перманга-
ната, бихромата калия и др.). Растворы карбонизуют в цилиндри-
цилиндрических сосудах и очищают от взвешенных частиц фильтрованием
на фильтрах Келли. Очищенный раствор выпаривают до выделения
кристаллов KjCOa • 1,5Н2О. Маточный раствор из солесборника
(с ложным дном) возвращают обратно в процесс. Кристаллы из
солесборника смывают сырым раствором и пульпу перекачивают
насосом на центрифугу. Поташ поступает затем в прокалочный ба-
барабан, обогреваемый газовым топливом. Газы из барабана очи-
очищают в циклоне и орошаемой водой башне, насаженной коксом.
Прокаленный поташ охлаждают в барабане, орошаемом снаружи
водой и измельчают в дезинтеграторе.
Формиатный метод получения поташа
Этот метод состоит26>31'52 в каустификации известковым моло-
молоком в присутствии окиси углерода природного промытого K2SO4.
Окись углерода образует с выделяющейся щелочью муравьино-
кислый калий:
K2SO4 + Са(ОНJ + 2СО = CaSO4 + 2HCOOK
Реакцию осуществляют в автоклаве с мешалкой при 200° (на-
(нагрев паром под давлением 32 ат). Очищенный от H2S, SO2 и СО2
генераторный газ подают в автоклав под давлением 30 ат. Время
реакции составляет 45 мин. Работают поочередно два автоклава.
Реакционная пульпа поступает в резервуар-экспанзер, где выде-
выделяется пар, используемый для подогрева смеси, идущей в автоклав.
Затем раствор декантируют, а сгущенную пульпу, содержащую
осадок CaSO4, отфильтровывают и промывают на барабанных ва-
вакуум-фильтрах. Осветленная жидкость содержит (в г/л): НСООК
165—175, СаО 6—8, K2SO4 9—11. Для отделения СаО к раствору
добавляют КгСОз, и образовавшийся углекислый кальций отде-
отделяют на фильтрпрессе. Раствор концентрируют до плотности
1,55 г/см3 в четырехкорпусной выпарной батарее, после чего его
охлаждают до 20° для выделения K2SO4, который снова используют
в производстве. Раствор выпаривают при 190° в открытых котлах
с змеевиками, обогреваемыми паром высокого давления. В котлах
получают расплав следующего состава (в %): НСООК —90;
КС1 — 0,4; К2СО3 —2,5; K2SO4 —0,1; Na2CO3 —0,3. Горячий рас-
расплав сохраняют в промежуточных сборниках с змеевиками при
температуре не ниже 160°, откуда его подают во вращающиеся печи
на кальцинацию и окисление воздухом при 800°. Образование угле-
углекислого калия происходит по реакции:
2НСООК + О2 = К2СО, + Н2О + GO8
Плав К2СО8 выгружают из печи специальным скребком. Гото-
Готовый продукт имеет следующий состав (в %): К2СО8 —98;
КС1 —0,25; K2SO4 —0,2; Na2CO8 —0,3; Fe —0,0005; не раствори-
растворимый в воде остаток — 0,01.
Триметиламиновый метод получения поташа
Этот метод13-Б3-5Б заключается в обработке раствора хлорида
калия A часть) и триметиламина D части) двуокисью углерода
под давлением и при перемешивании:
КС1 + N(CH3K + СО2 4- Н2О = КНСО3 + N(GH3K • HG1
При охлаждении выпадает кристаллический бикарбонат калия,
который легко отделяется от раствора солянокислого триметил-
204
Гл. VI. Поташ
амина. Последний снова превращают в свободный триметиламин
2[N(CH3K • НС1] + Са(ОНJ = 2N(CH3K + СаС12 + 2Н2О
а бикарбонат переводят в карбонат прокаливанием.
Предложен56-58 аналогичный процесс с применением гексаме-
тиленимина (отхода производства гексаметилендиамина) — от 1 до
1,4 части на 1 часть КС1. Концентрация водного раствора гексаме-
тиленимина 42—50%. При температуре реакции 20—40° степень
использования КС1 70—90%. После отделения бикарбоната калия
к раствору добавляют известь и регенерируют гексаметиленимин
отгонкой при 93—95°.
ЛИТЕРАТУРА
I. В. М. Р о м м, Абсорбция газов, Изд. «Химия», 1966, стр. 53, 669, 679. —
2. М. А. Л ю д к о в с к а я, С. Д. Фридман, В. А. Клевке, Хим. пром., № 5,
339 A965). — 3. Н. Н. Егоров, М. М. Дмитриев, Д. Д. Зыков,
Ю. Н. Бродский, Очистка от серы коксовального и других горючих газов,
Госметаллургиздат, 1960. — 4. И. А. Токмакова, Применение растворов и бе-
бетонов с добавкой поташа при производстве строительных работ в зимнее время,
Госстройиздат, 1963. — 5. А. А. Трипольский, Л. Г. Чернега, Хим. пром.,
№ 9, 675 A963). — 6. И. М. Стребков, Химия в сельском хозяйстве, № 4,
16 A964). —7. Chem. Week, № 2, 38 A963). —& А. В. Петербургский,
Производство и применение сложных удобрений во Франции, Сельхозгиз, 1960.—¦
9. П. М. Лукьянов, История химических промыслов и химической промышлен-
промышленности России, Изд. АН СССР, т. I, 1948; т. II. 1949; т. III, 1951. —10. В. Wae-
s е г, Chem. Ind., б, № 5, 413 A953)
II. В. Waeser, Chem. Ztg., 77, № 10/11, 323, 359 A953).— IS. П. М. Лукь-
Лукьянов, Курс химической технологии минеральных веществ, ч. II, Химтеорет,
1932. —13. Н. Д. Деркачев, Поташное производство, Химтеорет, 1932.—
14, В. О. С е д л и с, Химические товары и строительные материалы, Изд. Кубуч,
1933. —15. П. Н. Коваленко, Уч. зап. Ростовск. на Дону ун-та, 25, № 7, 99
A955). —16. П. Н. Рощии, Журн. сахарн. пром., № 4—5 A928).—
17t А. И. С к и р с т ы м о н с к и й, сб. «Пищевая промышленность СССР», № 3,
1946. — J8. А. А. Соколовский, Автореф. докт. дисс, ЛТИ им. Ленсовета,
1955. — 19. Т. И. Кузнецова, К- Л. Павлова, Труды НИИХП, в. 4, 1956,
стр. 61. — 20. И. Л. Талмуд, Хим. наука и пром., 2, № 6, 692 A957).
21. С. И. Белозуб, Тезисы докладов и сообщений на Всесоюзном совеща-
совещании работн. содовой пром., Пермь, 1958, стр. 93. — 22. А. Ф. Б о р я ч е к,
Д. И. К у х а р е в, Труды НИОХИМ т. 10, 1957, стр. 67. —23. Г. С. Седельни-
Седельников, Автореф. докт. дисс, ИОНХ АН СССР, 1954. — 24. Г. С. Седельников,
А. И. Лазарева, Хим. пром., № 5, 401 A959). — 25. А. А. Соколовский,
Т. И. Кузнецова, К- Л. Павлова, Хим. наука и пром., 2, № 4, 533 A957) .—
26. В. Ф. Чернов, Производство кальцинированной соды, Госхимиздат, 1956.—
27. Г. И. Чуфаров, В. С. Кнута рев, ЖХП, № 3, 232 A931).—
28. Н. Н. Heimann, Bull. Res. Council Israel, C5, № 1, 113 A955).— 29. Bull.
Res. Council Israel, C5, № 1, 115 A955).— 30. H. M. Bhavnagary, Chem.
Age (India), 19, № 2. 110 A968).
31. В. А. Лобанов, В. А. Шестакова, авт. свид. 209426, 1966.—
32. А. А. Соколовский, И. М. Богуславский, Т. И. Кузнецова,
Труды НИИХП, в. 2, 1955, стр. 3. — 33. И. С. Галинкер, А. С. Потерянко,
Труды ВИСП, т. 6. 1952, стр. 27.— 34. J. Е. Tee pie, The Industrial Development
Searles Lake Brines, 1929. — 35. А. А. Соколовский, ЖПХ, 29, № 5, 743
A956). — 36. W. S. В las dale, J. Am. Chem. Soc, 45, 2938, 2944 A923).—
Литература
205
37. В. Г. И и ж е ч и к, А. В. Я и у ш, Хим. ггоом., № 1, 39 A955).— 38. Д. М. Г и н 8-
бург, ЖПХ, 38, № 1, 55 A965); 42, № 5, 994 A969).— 39. М. И. Рунов,
А. С. Р о м а н е ц, В. К- Дружинин, Труды НИОХИМ, т. 19, 1969, стр. 284. —
40. К. И. Дворниченко, Е. Е. Мареиич, авт. свид. 181066, 1964; сб. «Хи-
«Химическая промышленность Украины», № 4 B6), 1966, стр. 20.
41. С. У. Горн ев, Автореф. канд. дисс, Харьковский политехи, ин-т, 1953;
Труды ВИСП, т. 9, 1956, стр. 36. — 42. Н. П. Лужная, С. Н. Косячков а,
ИСФХА АН СССР, 25, 345 A954); 26, 259 A955); 27, 358 A956); С. Н. Косяч-
Косячков а, Автореф. канд. дисс, ИОНХ АН СССР, 1954. — 43. П. С. Богоявлен-
Богоявленский, А. С. Мананникова, Сборник научных трудов Львовского гос. вете-
ринарно-зоотехнич. ин-та, № 6, 1953, стр. 376. — 44. G. Ervin, С. Е. McCart-
McCarthy, Ind. Eng. Chem., 36, № 5, 415 A944). — 46. А. Ф. Б op я чек, Н. Н. Дро-
зин, 3. К- Зубахина, М. И. Куиина, ЖПХ, 28, № 1, 100 A955).—
46. Л. С. И тки на, В. Ф. Кохов а, ИСФХА АН СССР, 26, 242 A955).—
47. Л. С. И тки на, В. Ф. Кохов а, ЖНХ, 1, № 7, 1665 A956). — 48. С. 3. Ма-
Макаров, М. П. Шульгина, Изв. АН СССР. ОХН, № 4, 510 A940).—
49. М. И. Р а в и ч, Л. С. И т к и н а, В. Ф. К о х о в а, ИСФХА АН СССР, 25, 350
A954). —50. М. X. Кишиневский, ЖПХ, 30, № 2 A957).
51. Пат. США 3254946, 1964; Англ. пат. 1050410, 1964; Норвеж. пат. 109199,
1964. — 52. Ost-Rassow, Lehrbuch der Chemischen Technologie, 1941,
S. 311. —55. Орт л и б, герм. пат. 5786, 1878; 9376, 1879; 13397, 1880. — 54. Австр.
пат. 178910, 1954. —55. Австр. пат. 178911, 1954.— 56. С. А. Крашенинников,
И. Н. Шокии, В К. Новиков, авт. свид. 186407, 1965. — 57. В. К. Нови-
Новиков. И. Н. Ш о к и н, Г. А. Крашенинников, Труды МХТИ им. Д. И. Мен-
Менделеева, в. 56, 1967, стр. 152. — 58. Л. В. К и р е е в а, И. Н. Ш о к и и, С. А. Кра-
Крашенинников, Там же, в. 60, 1969, стр. 7.
Физико-химические свойства
207
Глава VII
БРОМ И ЕГО СОЛИ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Бром Вг2 — красновато-бурая жидкость, образующая бурь'е
пары; плавится при —7,25°, кипит при 68,78°. Плотность жидкого
брома 3,187A—0,001086^) &/см3. Теплоемкость: кристаллов
0,0898 кал/г (—20,7°), жидкости 0,1071 кал/г @—57°) и пара
0,0553 кал/г (83—228°). Теплота плавления 16,14 кал/г; теплота
парообразования 44,8 кал/г. Коэффициент расширения 0,0011 на
1 градус B0—30°).
бром растворим в воде и во многих органических растворите-
растворителях— эфире, сероуглероде, четыреххлористом углероде, хлоро-
хлороформе, керосине. Растворимость брома в 95%-ной серной кислоте
при 25° 0,75%. Растворимость воды в броме при 22°0,046%. Насы-
Насыщенный водный раствор содержит при 760 мм рт. ст. 3,22—3,41%
брома в интервале 20—50°. Из насыщенного водного раствора ниже
5,84° выделяется Вг • 8Н2О.
Константа гидролиза' брома
Вг2 + Н2О = НВгО + Н+ + Вг"
[НВгО] [Н+] [Вг~]
Л [BrJ
при 0° равна 0,7.10~9, при 20° — 5,2 • 10~9 и при 36° — 11,3 • 10~9.
При нагревании бромноватистая кислота переходит в бромно-
ватую:
ЗНВгО = НВгО3-
Сульфаты и нитраты понижают растворимость брома в воде, в
хлориды и бромиды ее повышают, так как бром с ионами Вг~ и Сг
образует комплексные соединения:
Вг2 +
2Вг2 + Вг" - Вг~; Вг2 + СГ - Вг2СГ
Для разбавленных растворов бромистого калия @,01—1,0 н.)
константа равновесия для первой из этих реакций ~ 16, а для вто-
второй ~40.
Давление пара жидкого брома при 0° 66,9, при 20° 173, при 40°
392 мм рт. ст. Давление пара брома над его водными растворами
подчиняется закону Генри — Дальтона при учете количества гид-
ролизовавшегося брома или при подавлении гидролиза 2~4. Коэф-
Коэффициент распределения брома Н (равновесное отношение концен-
концентрации брома в газовой фазе к концентрации его в растворе) для
его водных растворов4 зависит от температуры:
/, °С 0
Я 0,0158
10 20 30 40 50 60
0,0287 0,0434 0,0653 0,0935 0,1275 0,1652
120
дО
Бром сильно разрушает кожу; пары его даже в малых концен-
концентрациях разрушают слизистые оболочки. Предельно допустимая
концентрация брома в воздухе
0,002 мг в 1 л; смертельная кон-
концентрация (при 30—60-минутном
воздействии) 0,05 мг в I л.
Бромистый натрий NaBr об-
образует кристаллогидраты (рис.
88). Насыщенный водный рас-
раствор кипит при 121°. NaBr-2H2O
кристаллизуется в моноклинной
системе и имеет плотность
2,18 г/см3; безводная соль кри-
кристаллизуется в кубической систе-
системе, имеет плотность 3,2 г/см3 и
плавится при 755°.
Бромистый калий КВг кри-
кристаллизуется в безводной форме
в кубической системе с плотно-
плотностью 2,76 г/смг\ температура пла-
плавления 730°.
Бромистый аммоний NH4Br
кристаллизуется в безводной фор-
форме в кубической системе с плотно-
плотностью 2,43 г/см3. При 542° возгоняет-
возгоняется, частично при этом разлагаясь.
Бромистое железо FeBr2 — кристаллы зеленого цвета, от при-
присутствия бромного железа приобретают красный или красновато-
бурый оттенок. Бромистое железо образует ряд кристаллогидратов5
(рис 59). Насыщенный раствор FeBr2 кипит при 132°. Безводная
соль кристаллизуется в гексагональной системе, имеет плотность
4,64 г/см3 и плавится при 684°; гексагидрат кристаллизуется в ром-
ромбической системе
-40.
NaBr 5HjO
) го зо 40 so
Концентрация соли, "А
№
Рис, 58. Растворимость бромидов
калия, натрия и аммония в воде.
208
Гл. VII. Бром и его соли
40 SO
FeBr2,%
Рис. 59. Политерма растворимости в системе
FeBr2—H2O.
Бромноватокислые калий КВгО3 и натрий NaBrO3 кристалли-
кристаллизуются в безводной форме, при нагревании до 370—380° разла-
разлагаются с выделением кислорода. КВгО3 образует кристаллы триго-
нальной системы с плотно-
160' ' ¦ ' ' ' ^ ' стью 3,27 г/см3, NaBrO3 —
кубической системы с плот-
плотностью 3,34 г/см3.
ПРИМЕНЕНИЕ
Элементарный бром яв-
является исходным веществом
для получения его органиче-
органических и неорганических со-
соединений. Бромиды приме-
применяют в медицине, фотокино-
фотокинотехнике, производстве неко-
некоторых красителей и фарма-
фармацевтических препаратов.
Для медицинских целей ис-
используют и многие органи-
органические соединения брома (бромалин, бромистую камфару, бром-
бромурал, ксероформ и др.). Броматы используют в аналитической хи-
химии (броматометрия) и в хлебопечении. Наибольшее количество
брома и его солей (NaBr, FeBr2) применяют для синтеза бром-
органических соединений, используемых в качестве антидетонирую-
щих добавок к моторному топливу (бромистый этил, дибромэтан
и др.), хладагентов (бромфреоны), огнетушащих средств (броми-
(бромистый метилен), фумигантов (бромистые метил, этил и др).
В США начали использовать бром для санитарной обработки
воды. Расширяется применение бромидов для получения металлов
высокой чистоты,
Технический бром выпускают трех сортов, содержащих не ме-
менее 99,9, 99,7 и 99,2% брома; ГОСТ 454—70 регламентирует нали-
наличие в нем примесей хлора, сульфатов, воды, органических и
нелетучих веществ. Перевозка и хранение жидкого брома
требует соблюдения больших предосторожностей. Жидкий
бром транспортируют в стальных баллонах емкостью 40—320 л,
футерованных свинцом, или в специальных цистернах, плакирован-
плакированных никелем или футерованных свинцом. Тарой для малых коли-
количеств брома служат толстостенные стеклянные бутыли с притер-
притертыми пробками, защищенные от света 6~9. Осушенный жидкий бром
(содержащий меньше 0,01% воды) можно транспортировать при
температуре ниже 20—25° в баллонах из углеродистой стали (Ст. 3)
и при 25—50° в баллонах из хромистой стали AX13) 10-п. Для за-
защиты нержавеющей стали рекомендуют12 добавлять в жидкий
Сырье
209
бром малые количества двуокиси азота или крепкой азотной кис-
кислоты.
В СССР основным продуктом, в виде которого выпускается
бром, является плав бромистого железа, состоящий из смеси че-
четырех- и шестиводного кристаллогидратов. Согласно ГОСТ
9814—61, плав должен содержать не менее 49% брома, не более
3% хлора (от количества брома) и 0,3% Fe^. Тарой для плава
служат стальные барабаны с двойным швом емкостью 100—130 кг.
'Продукт, более богатый бромом, — бромисто-бромное железо
(Fe3Br8 • пН2О), почти не выпускают из-за большой агрессивности
по отношению к стальной таре. Бромистое железо может транспор-
транспортироваться в обычных железнодорожных цистернах в виде кон-
концентрированных растворов (содержащих 31,3—33,6% Вг зимой и
35,2—38,1% Вг летом), если содержание Fe3+ в растворе не превы-
превышает 0,3% и в него в качестве ингибитора добавлен уротропин
@,25 %I3. При перевозке в футерованных цистернах уротропин не
добавляют, а количество бромного железа в растворе не ограничи-
ограничивается.
Мировое производство брома в настоящее время находится на
уровне 150 тыс. т в год (без СССР)?4-18.
СЫРЬЕ
Содержание брома в земной коре составляет Ы0~3 вес. %.
Бром находится в природе в виде солей. Основными источниками
Для промышленного получения брома являются: воды океанов, со-
содержащие в среднем 67 г брома в 1 м3\ некоторые рассолы соля-
соляных озер и буровые воды нефтеносных районов, содержащие
0,2—3,5 кг брома в I м3, а также маточные щелоки, получающиеся
при переработке карналлита и сильвинита, содержащие 0,6—4,5 кг
брома в 1 м3. Все эти растворы помимо бромидов обычно содержат-
СГ, SO5", НСОз, СОГ, Na+, K\ Ca2+, Mg2+, а также довольно
часто Г, В4О2~, HS~, S2", NH4+, Fe2+, Mn2+ и органические веще-
вещества. Соленость рассолов колеблется от 30 до 390 г/л, а темпера-
температура от —20° (озерные рассолы зимой) до 75° (высокотермальные
буровые воды).
Извлечение брома обычно выгоднее производить из рассолов,
более богатых бромидами. Но не только содержание брома в ис-
исходном сырье определяет экономические показатели процесса;
практически очень важными факторами являются температура, со-
соленость, щелочность и состав рассола. Так, при повышении темпе-
температуры и содержания иона SOf", а также при уменьшении соле-
солености (или при очень большой солености рассола) в процессе
извлечения брома увеличивается давление его пара над рассолом
и тем самым снижается расход пара или электроэнергии на произ-
производство. Повышение щелочности рассола увеличивает расход
210
Гл. VII. Бром и его соли-
Получение брома
211
кислоты, требующейся для подавления гидролиза брома. Неблаго-
Неблагоприятным фактором является наличие в рассолах веществ (HS~,
S2~, NH+, Fe2+, Mn2+, органические вещества и т. д.), взаимодей-
взаимодействующих с хлором, который вводится для окисления бромидов,
ибо это связано с возрастанием его расхода. Присутствие в исход-
исходном рассоле органических веществ затрудняет извлечение, снижает
выходы и загрязняет получающийся бром (из-за образования
бром- и хлорорганических веществ), особенно при отгонке паром.
ПОЛУЧЕНИЕ БРОМА
Все применяемые в промышленности способы получения брома
из природных вод основаны на окислении бромидов до элементар-
элементарного брома с последующим извлечением последнего из раствора.
Окисление бромидов обычно производят хлором или хлорной во-
водой. При наличии в исходном рассоле, помимо бромидов, еще и
иодидов извлечение брома можно осуществлять как до, так и после
извлечения иода (см. гл. VIII). При этом следует учитывать, что
если сначала извлекают иод, применяя для этого твердые сорбенты
(активированный уголь, аниониты), то последние могут поглощать
до 6% содержащегося в растворе Вг~ (количество теряемого при
этом брома зависит ог солености и состава раствора, видов окисли-
окислителей и сорбентов) 17> 18.
Извлечение элементарного брома из рассолов после окисления
производят отгонкой паром или десорбцией воздухом б> 18~24.
Отгонка брома паром I
Извлечение брома по этому способу осуществляют в колоннах
непрерывного действия при одновременном хлорировании рассола
и отгонке выделяющегося брома водяным паром. Часто применяют,
колонны системы Кубиершского — башни высотой 5,5—6 м, сло-
сложенные из толстых A8—20 см) гранитных, андезитовых или беш-
таунитовых плит и имеющие внутреннее сечение 1,5x0,8 м. Внутри
колонна разделена горизонтальными перегородками на 7—8 камер.
Для сообщения камер между собой в перегородках имеются отвер-
отверстия, перекрытые керамиковыми колпачками, образующими гид-
гидравлические затворы. Для свободного прохода газа в двух отвер-
отверстиях каждой перегородки установлены широкие керамиковые
трубы. В каждой камере горизонтально уложены керамиковые
дырчатые плиты (на расстоянии 4—5 см), служащие насадкой.
Бромсодержащий рассол, нагретый в теплообменнике и в подо-
подогревателе до 80—90°, поступает в колонну сверху.. Газообразный
хлор подают в третью снизу камеру, а в нижнюю — водяной пар
A,5—2 ат) для нагрева рассола до кипения A06—120°); пар дви-
движется по тому же пути, что и хлор. Хлор, растворяясь в рассоле,
вытесняет из его солей бром, переходящий в газовую фазу.
Наличие в исходном рассоле ионов HCOS~ и СО|~ приводит при
хлорировании (при ~100°) к образованию хлоратов:
^ + ЗС12 = СЮз + 5СГ + ЗСО2
+ ЗС12 = СЮз + 5СГ + 6СО2 + ЗН2°
Хлораты при высоких значениях рН не являются окислителями,
и затраченный на их образование хлор теряется. Для снижения
расхода хлора рассолы предварительно подкисляют25 до рН=*
=2,5—6,0 серной (иногда соляной) кислотой, вводя ее в рассоло-
провод после подогревателей или в поглотительную колонну. Выхо-
Выходящий из колонны отработанный рассол стекает в резервуар, за-
заполненный железными стружками, где оставшиеся в рассоле сво-
свободные галогены и их кислородные соединения частично (на
40—50%) связываются и превращаются в соли железа.
Для окончательного удаления свободных галогенов и предот-
предотвращения коррозии теплообменника отработанный рассол нейтра-
нейтрализуют тиосульфатом натрия:
Na2S2O3 + 4Br2 + 5Н2О = Na2SO4 + 8HBr + H2SO4
Во избежание повышения кислотности рассола в нейтрализатор
добавляют раствор соды или магнезиальное молоко (если в рас-
рассоле содержится мало SO4 — известковое молоко) до слабощелоч-
слабощелочной реакции (рН=8—10).
Пары брома, воды и остаток газообразного хлора поступают из
колонны в керамический водяной холодильник, где бром и вода
конденсируются. Содержание хлора в сыром броме составляет
3—5%. После разделения несконденсировавшийся хлор и бромная
вода C,1—3,4% Вгг) поступают обратно в колонну, а жидкий бром
рафинируется в небольшой керамической колонне с насадкой, обо-
обогреваемой горячей водой F5—75°). Содержащийся в сыром броме
хлор испаряется и поступает обратно в колонну, а частично испа-
испарившийся бром конденсируется в керамическом обратном холо-
холодильнике. Количество хлора в жидком броме зависит от темпера-
температурного режима рафинера, так как температура кипения жидкого
брома понижается от содержания в нем хлора (рис. 60J6; ее под-
поддерживают в интервале 45—54°.
Колонна Кубиершского перерабатывает 200—270 м3 рассола в
сутки на 1 м2 поперечного сечения; количество переработанного
рассола снижается с увеличением начального содержания в нем
брома, но производительность при этом повышается. В этих колон-
колоннах эффективно работает сравнительно небольшая часть объема.
Более эффективны колонны Кальтенбаха27, в которых паро-газовая
смесь переходит из камеры в камеру не по трубам, а по каналам
внутри стен, что увеличивает реакционный объем и равномерность
контакта фаз. Колонны Кальтенбаха перерабатывают около 500 м3
212
Гл. VII. Бром и его соли '
60
40
рассола в сутки на 1 м2 сечения. На американских заводах отгонку
брома из рассола паром производят в башнях с насадкой из колец
Рашига. Выход по брому при
отгонке его мало зависит от
применяемой аппаратуры и ко-
колеблется в пределах 80—95%,
в зависимости от состава сырья
и содержания в нем брома.
В сбросном рассоле, в зависи-
зависимости от конструкции колонны,-
содержится 0,05 — 0,15 кг/м3
брома (приблизительно поров-
поровну в виде Вг~ и Вг2). Иногда
вследствие забивания аппа-
аппаратуры (колонн, трубопрово-
трубопроводов и т. д.) твердыми отложе-
отложениями, а также сильного вспе-
вспенивания рассола происходит
снижение производительности
колонн. Это обычно вызывает-
вызывается наличием большого количе-
количества взвеси, пересыщенностью
поступающих рассолов и на-
f
-20
(Вг2
\
ч
\
V
ч
ч
\
\
20
W 00
СостаВ.мол.%
60
100
си
Рис. 60. Состав пара и жидкости при
температурах кипения смесей брома и
хлора:
t — жидкость; 2 — пар.
личием поверхностно-активных
веществ. Поэтому рассолы предварительно отстаивают или фильт-
фильтруют и разбавляют водой (в случае пересыщения). Иногда причи-
причиной забивания аппаратуры яв-
является выделение малораство-
малорастворимых веществ в процессе на-
нагревания, подкисления и хло-
хлорирования исходных рассолов.
Последнее в основном связано
с разрушением бикарбонатов,
образованием гипса и двуокиси
марганца, гидролизом солей
трехвалентного железа и т. д.
Когда в исходном рассоле на-
находится значительное количе-
количество органических веществ, по-
получаемый бром может содер-
I
но
по
70
/7
s
\
ч
w /.г 14 1,е w
Концентрация брома Орассжмг/м3
Рис. 61. Расход пара на, 1 т брома в за-
зависимости от его содержания в рассоле.
жать до 3,5—4% броморгани-
ческих веществ (бромоформа,
тетрабромметана, бромуксус-
ной кислоты и др.) 28-30, которые отделяют дистилляцией, иногда с
предварительной обработкой брома концентрированной серной ки-
кислотой или бромидами31.
Получение брома
213
Экономическая эффективность способа отгонки брома паром в
основном определяется расходом пара, который увеличивается с
уменьшением концентрации брома в исходном рассоле и с пониже-
понижением его температуры. На рис. 61 приводятся данные по расходу
пара на 1 т брома для рассола из Сакского озера A0—20°), из ко-
которых видно, что при концентрации брома менее I кг в I м3 расход
тепла становится столь большим, что извлечение брома этим мето-
методом оказывается экономически не целесообразным. Для получения
1 т брома в зависимости от состава исходного рассола расходуют:
хлора 0,53—1 т, серной кислоты 0—3 т; соды кальцинированной
60—200 кг, тиосульфата натрия 12—45 кг, стружек железных
10—40 кг, электроэнергии 150 — 300 квт-ч, воды пресной
100—200 м3. Расход исходного рассола составляет 106—130% ог
теоретического.
Десорбция брома воздухом
Этот способ более распространен в промышленности, чем от-
отгонка паром, так как дает возможность извлекать бром из низко-
низкопроцентного сырья, не требует сложной аппаратуры и легко может
быть автоматизирован.
Рассол
Рис. 62. Схема извлечения брома из рассолов методом воздуш-
воздушной десорбции:
/ — напорный бак для рассола; 2—регулирующее устройство для кислоты;
3 — регулирующее устройство для хлора; 4 — хлоратор; Б — десорбер; 6—брызго-
уловнтель; 7 —башня для хлорочистки; 8 — бак для хлорочистительного ра-
раствора; 9 — насос; 10 — хемосорбер; П — вентилятор для воздуха; 12 — бак
для сорбента; 13 — нейтрализатор,
Бромсодержащие рассолы подкисляют серной или соляной кис-
кислотой до рН=3—3,5, а затем в них вводят окислитель для пере-
перевода Вг~ в Вг2 (на некоторых заводах, где исходный рассол имеет
рН<^б,5, его не подкисляютK2-33.
Рассол, содержащий растворенный бром, поступает в верх-
верхнюю часть башни выдувания (десорбер, рис. 62), где стекает по
Гл. VII. Бром и его соли
Получение брома
215
ю го
Врвмя.мин
30
насадке. Навстречу рассолу из окон, находящихся у днища десорбе-
ра, поступает мощный поток воэдуха, который извлекает растворен-
растворенный бром. Полученная бромо-воздушная смесь, содержащая некото-
некоторое количество хлора и другие примеси, проходит брызгоуловитель и
хлороочистительную башню, а затем поглотительные башни (хемо-
сорберы), где бром улавливается. Отработанный воздух выбрасы-
выбрасывается хвостовым вентилятором в атмосферу или направляется
вновь в десорбционную башню. Во избежание проникновения брома
в атмосферу цеха вся аппаратура работает под вакуумом, за иск-
исключением воздуховода после хво-
хвостового вентилятора.
Подкнсление исходного рассо-
рассола и окисление бромидов хлором
осуществляют в хлораторе, пред-
представляющем собой вертикальную
асбоцементную или покрытую
эбонитом стальную трубу диа-
диаметром 250—600 мм. Кислоту вво-
вводят в нижнюю часть трубы. Для
перемешивания кислоты и рассо-
рассола над подающей кислоту труб-
трубкой устанавливают диафрагму
из кислотоупорного материала,
чем создается турбулентность по-
потока. Для облегчения регулиров-
регулировки подачи кислоты ее иногда предварительно разбавляют до кон-
концентрации 5—10%. Ввод хлора осуществляют на расстоянии 2—Ам
от ввода кислоты, обеспечивающем полное смешение кислоты с
рассолом. Газообразный хлор поступает из стальных цистерн или
ресиверов, где поддерживают давление на 0,7—1 ат выше, чем со-
создает столб жидкости в хлораторе. Если используют разбавленный
хлор, например, отходящий газ от других производств, подаваемый
под небольшим давлением, его вводят в верхнюю часть хлоратора.
При хлорировании происходит растворение хлора и взаимодействие
растворенного хлора (или продуктов его гидролиза) с составными
частями рассола.
Скорость растворения хлора практически не зависит от концен-
концентрации Вг~ в рассоле и хлор-газа34. Увеличение поверхности кон-
контакта и понижение температуры, вязкости и солености раствора
ускоряют процесс растворения хлора. Скорость взаимодействия ме-
между растворенным хлором и Вг~ велика, а с другими составными
частями рассола (что характеризуется так называемой галогено-
поглощаемостью35 рассола) относительно мала (рис. 63). При ра-
работе с теплыми и концентрированными рассолами, содержащими
большое количество С1~, скорость растворения хлора уменьшается.
Б этих случаях неполностью растворившийся хлор может попасть
Рис. 68. Изменение галогеиопогло-
щаемости Челекенской буровой воды
от времени.
в десорбционную башню и загрязнить бромо-воздушную смесь. По-
Поэтому окисление таких рассолов целесообразно вести не газообраз-
газообразным хлором, а хлорной водой36> 37. Последнюю получают, погло-
поглощая хлор холодной пресной или морской водой в асбоцементной
трубе высотой ~8 м. Хлорная вода, содержащая в 1 м3 5—7 кг
хлора, смешивается в хлораторе с бромсодержащим рассолом. При-
Применение хлорной воды снижает расход хлора на 7—20%-
При взаимодействии хлора с бромсодержащим рассолом, по-
помимо основной реакции окисления Вг~ до Вг2, идет ряд побочных
ЮО
80
;7О
160
50.
ав ав w
Экй.мора/экб. брома
1.2
Рис. 64. Зависимость степени окисления бром-ио-
бром-иона от дозировки хлора (эквиваленты) при рН ^ 4.
реакций38-43 (Вг2 + С12 = 2ВгС1; Вг2 + ЗС12 = 2ВгС13), а также ги-
гидролиз Вг2, Cl2, BrCl, BrCls и реакции комплексообразования. При
хлорировании подкисленных рассолов гидролиз брома подавляется,
а гидролиз остальных соединений идет тем в большей мере, чем
выше температура.
При введении в подкисленный бромсодержащий рассол поло-
половины теоретического количества хлора идет только реакция окисле-
окисления Вг~ до Вг2. Увеличение количества вводимого хлора до 90—•
96% приводит к частичному образованию BrCl; дальнейшее повы-
повышение количества вводимого хлора приводит к образованию
ВгС1344. По другим данным45, при окислении хлором 80—82% Вг~,
от его начального содержания в рассоле, выделяется бром с не-
небольшой примесью BrCl (до 10%) 46. Повышение дозировки хлора
приводит к взаимодействию его с бромом и образованию BrCl, при-
причем ВгС13 не образуется даже при двухкратном количестве хлора
от стехиометрического. Побочные реакции и летучесть образую-
образующихся, соединений хлора с бромом приводят к тому, что в бромо-
воздушной смеси всегда присутствует некоторое количество хлора,
а для окисления бромидов требуется вводить избыток хлора (рис.
64, 65). Зависимости, приведенные на этих рисунках, для
216
Гл. VII. Бром и его соли
¦10
различных рассолов неодинаковы, но характер кривых не меняется.
На практике окисление Вг~ ведут на 75—94% 17-47-48, затрачивая на
это 115—160% хлора от теоретического. Процесс хлорирования 45~52
контролируют или замером окислительно-восстановительного по-
потенциала рассола, поступающего из хлоратора в десорбер, или
определением содержания бром-иона в вытекающем из десорбера
отработанном рассоле.
Десорбцию брома из рассола осуществляют в деревянных, же-
железобетонных или кирпичных облицованных кислотоупорными плит-
плитками башнях, заполненных
керамическими кольцами
E0x50 мм) или хордовой
насадкой. Высота башен ко-
колеблется от 12 до 30 м; се-
сечение зависит от их произ-
производительности *. На бром-
бромных заводах СССР башни
обычно имеют высоту ~ 14 м,
высоту насадки Юл* и диа-
диаметр до 4 м.
Промышленное извлече-
извлечение брома из рассолов осу-
осуществляют при содержании
его не менее 65—70 мг/л
(океанская вода). Коэффи-
Коэффициент распределения брома
Получение брома
21Т
во во
Степень окисления В г," %
Рис. 65. Зависимость содержания хлора
в бромо-воздушной смеси (в % К сумме
Вг2 + С12) от степени окисления бром-иоиа
и рН рассола (< = 25°):
;-рН = 6; 2-рН = 5; 3-рН < 4.
между воздухом и рассолом
зависит от температуры, со-
состава и концентрации рассо-
рассола и при подавлении гидро-
гидролиза не зависит от концен-
концентрации брома89. Повышение температуры и содержания БОГ. а
также снижение концентрации рассола увеличивают коэффициент
распределения. Повышение концентрации С1~ в растворе до 5 н.
уменьшает коэффициент распределения из-за процессов комплексо-
образования, а более высокое содержание хлоридов приводит к
его повышению. На коэффициент распределения влияет и состав
катионов, но в меньшей степени, чем указанные анионы.
На рис. 66 приведена зависимость коэффициента распределения
брома от температуры для некоторых природных рассолов, состав
которых дается в табл. 21Б9.
Из океанской воды, содержащей ~ 70 г/м3 Вг~, при 18° извле-
извлекать бром не менее выгодно, чем из рассола оз. Кучук при 0° (зи-
(зимой), где содержание брома ~ 270 г/м3. Это связано с тем, что ко-
О расчете производительности десорбционных башеи ем.
53-68
ТАБЛИЦА 2J
Состав некоторых природных рассолов, содержащих бром
Состав рассола, г/л
CaSO4
1,3
Следы
4,0
4,6
2,6
СаС12
—
—
—
16,1
MgSO4
2,2
51,1
59,7
5,2
—
MgCl2
3.6
362
6,1
55,5
133
MgBr2
0,08
1,31
0,31
0,31
0,85
NaCl
29,7
24,1
145
194
104
Концен-
Концентрация
рассола, н
0,63
8,8
5,3
4,8
4.8
Океанская вода
Оз. Кургузул
Оз. Кучук (летний рассол).
Оз. Кучук (зимний рассол).
Оз. Старое
эффициент распределения брома для океанской воды при 18° равен
0,03, а для рассола оз. Кучук при 0° 0,0074, и потому равновесное
б й фазе для первого случая -равно
0.06
голг
Ю 20
Температура,"С
30
Содержание брома в газовой
0,03-70=2,1 г/м3, а для второ-
второго 0,0074-270= 2 г/м3.
Коэффициент десорбции
брома зависит от температуры,
скорости воздуха и плотности
орошения башни, он пропорци-
пропорционален скорости воздуха в сте-
степени 0,7—0,9 и плотности оро-
орошения в степени 0,3—0,5. В
производственных условиях ве-
величина его обычно колеблется
в пределах 2,0—4,8 м/чы<т и
уменьшается с повышением
температуры.
На производительность де-
сорбционной башни сильно
влияют коэффициент избытка
воздуха (т. е. отношение фак-
фактического количества воздуха
к тому его количеству, которое
требуется для получения рав-
равновесной концентрации брома) и коэффициент извлечения брома
(т. е. отношение количества брома, перешедшего в воздух, к на-
начальному количеству его в рассоле). При увеличении коэффициента
избытка воздуха возрастает движущая сила (так как снижается
концентрация брома в воздухе), что уменьшает величину необхо-
необходимой .поверхности насадки (при постоянной производительности).
С другой стороны, повышение количества воздуха (при постоянной
скорости его) потребует увеличения сечения башни, а это приведет
к уменьшению плотности орошения, снижению коэффициента
Рис. 66. Зависимость коэффициента рас-
распределения брома от температуры для
различных рассолов:
/ — океанская вода; 2 —оз. Кургузул; 3 — оз. Ку-
Кучук (летний рассол); 4-оз. Кучук (зимний рас-
рассол); 5 — оз. Старое.
218
Гл. VII. Бром и его соли
Получение брома
219
•
1
/
Off! 0JB5 090
Коэффициент извлечения бром
035 IPO
десорбции и необходимости увеличения объема насадки. Отсюда
следует, что существует определенный коэффициент избытка воз-
воздуха, при котором десорбиионная башня будет иметь минималь-
минимальный объем. Обычно работают с коэффициентом избытка воздуха,
равным 2,5—3,5. Этот коэффициент является условным, а не истин-
истинным, ибо не учитывает образования соединений брома с хлором,
которые имеют более низкое давление пара, чем бром.
Повышение коэффициента извлечения брома связано с увели-
увеличением площади насадки или с увеличением скорости воздуха. На
рис. 67 приведена зависи-
зависимость изменения относи-
относительной величины поверх-
поверхности насадки от коэффи-
коэффициента извлечения брома,
если за единицу принять
ту величину поверхности,
которая требуется при
коэффициенте извлечения,
равном 0,75. При деше-
дешевом сырье и неограничен-
неограниченных его запасах в неко-
некоторых случаях экономи-
экономически выгоднее идти на
некоторое снижение коэф-
коэффициента извлечения. На
действующих заводах ко-
коэффициент извлечения в большинстве случаев равен 0,82—
0,9547-48' 61~65. Для увеличения коэффициента извлечения на
1,5—3% предложено добавлять немного хлора к воздуху, посту-
поступающему в десорбер, — это вызывает добавочное окисление Bi6.
Другим средством, дающим возможность повысить коэффициент
извлечения (не изменяя поверхности насадки), является увеличе-
увеличение скорости воздуха, которая обычно составляет 0,5—0,6 м/сек (на
полное сечение башни), а на некоторых заводах достигает 1,2 м/сек.
Увеличение скорости воздуха создает возможность повысить плот-
плотность орошения и работать на режимах, близких к захлебыванию,
что приводит к повышению производительности башен примерно в
2 раза67 при увеличении расхода электроэнергии на 1 т брома в
1,5—1,6 раза.
Десорбция брома воздухом мсжет быть осуществлена не только
е башнях с насадкой, но и в аппаратах работающих на пенном ре-
режиме68, в частности в пенных аппаратах с провальными тарел-
тарелками69. В этих аппаратах коэффициент десорбции во много раз
больше, чем в башнях с насадкой, а объем пенного аппарата зна-
значительно меньше насадочной башни. Для этой же цели предложено
использовать аппарат со взвешенным слоем насадки70.
Рис. 67. Зависимость величины поверхности
насадки от коэффициента извлечения брома
из рассола.
В процессе десорбции брома воздухом в газовую фазу перехо-
переходят и другие летучие компоненты, содержащиеся в исходном рас-
рассоле, а потому в бромо-воздушной смеси помимо брома всегда со-
содержится хлор (в основном, в виде BrCl), двуокись углерода, во-
водяной пар, а иногда органические вещества. Содержание брома в
газовой фазе колеблется в очень широких пределах (в зависимости
от состава исходного рассола) и составляет 0,7—12 г/м3, т. е. для
извлечения 1 кг брома в заводских условиях требуется от 80 до
1500 м3 воздуха. Для уменьшения расхода воздуха стремятся под-
поддерживать повышенную температуру как рассола, так и воздуха.
Десорбцию брома при температуре рассола ниже 2—3° производить
нецелесообразно. Поддержание повышенной температуры воздуха
зимой на некоторых заводах осуществляют за счет подачи в де-
сорбционную башню отработанного воздуха, выбрасываемого хво-
хвостовым вентилятором из хемосорберов.
Содержание хлора обычно составляет 3—10% от количества
брома в воздухе и зависит от степени окисления бромида и коэф-
коэффициента извлечения, при этом чем они выше, тем больше содер-
содержится в бромо-воздушной смеси хлора. При определении содержа-
содержания хлора в последовательных порциях воздуха, пропускаемого
через хлорированный раствор, было установлено, что при дозировке
хлора 100—120% от теоретического (по отношению к содержанию
брома в рассоле) первые фракции содержат меньше хлора, чем
последние. Это объясняется меньшим давлением пара BrCl, чем
брома. При дозировке хлора более 150% от теоретического первые
фракции содержат наибольшее количество хлора, а затем оно
уменьшается. Это объясняется тем, что над раствором давление
хлора больше, чем давление брома. При дозировке хлора около
150% от теоретического содержание хлора по отношению к брому
остается практически неизменным. Это подтверждается и завод-
заводскими данными, когда при увеличении дозировки хлора при окисле-
окислении рассола выше определенного предела (при прочих равных ус-
условиях) процесс десорбции ухудшается, и содержание брома в бро-
бромо-воздушной смеси уменьшается 41-4В-47.
Содержание СО2 в бромо-вовдушной смеси зависит от чистоты
воздуха и содержания СО|~и НСО^" в исходном рассоле. При под-
кислении и хлорировании рассолов содержащиеся в них карбонаты
и бикарбонаты разрушаются, а выделяющаяся двуокись углерода
десорбируется. Молекулярное отношение СОг: Вг2 в бромо-воздуш-
бромо-воздушной смеси колеблется от 1 до 5 (иногда и больше).
. Бромо-воздушная смесь после отделения брызг и капель посту-
поступает в уловительную систему, состоящую из одной или нескольких
башен-хемосорберов. Для выбора способа улавливания брома из
газовой фазы существенно, в виде какого продукта он в дальней-
дальнейшем будет применяться. Если бром будет применяться' в виде со-
солей, то бромо-воздушную смесь следует предварительно очистить
220
Гл. VII. Бром и его соли
Получение брома
221
от хлора; при отсутствии хлороочистки, для получения кондицион-
кондиционных солей, количество брома, извлекаемого из сырья, должно быть
снижено на 15—^20% 47-48-71, и экономические показатели производ-
производства при этом ухудшаются. Если же продуктом производства яв-
является элементарный бром, то очистку бромо-воздушной смеси про-
изводить нецелесообразно, так как примеси удаляются в процессе
•его получения.
Очистку бромо-воздушной смеси от хлора производят раство-
растворами бромидов, получаемыми в хемосорберах. При питании хлоро-
очистительных аппаратов растворами бромидов щелочных и ще-
щелочноземельных металлов идут реакции: 2Вг~ + С12 = 2С1~ + Вг2,
Br" + BrCl = С1~ + Вг2. При очистке растворами бромистого же-
железа процесс усложняется — вначале Fe2+ окисляется до Fe3+ и соли
железа гидролизуются по следующим суммарным реакциям:
2Fe2+ + Вг2[С12] = 2Fe3+ + 2ВГ[СГ]
2Fe2+ + BrCl = 2Fe3+ + Вг" + СГ
12Fe2+ + 3O2 + 2Н2О = 8Fes+ + 4FeOOH
Лишь после окисления ионов Fe2+ более чем на 90% идут при-
приведенные выше реакции выделения элементарного брома "¦72
В процессе хлороочистки из бромо-воздушной смеси извлекается
80—95% содержащегося в ней хлора. Скорость абсорбции хлора
мало зависит от концентрации Вг~ в абсорбенте (если она больше
¦0,05 г-экв/л) и от плотности орошения насадочных башен @,5—
4 м/ч) 73-76. Исходные растворы для хлороочистки обычно содержат
от 100 до 400 г/л Вг~. Повышение концентрации Вг~ в этих раство-
растворах приводит к накоплению в них Вг3 и Brs, что увеличивает аг-
агрессивность среды и потери брома с отработанным раствором. Этот
раствор (содержащий менее 5 г/л Вг~ и некоторое количество Вг2)
направляют после хлорирования в десорбер для извлечения брома
•или сбрасывают в канализацию.
При хлороочистке растворами бромистого железа выделяется
•шлам, состоящий в основном из гидратированных окислов железа;
¦его накапливание ухудшает гидродинамический режим работы хло-
роочистителя77-78. Для уменьшения количества образующегося
шлама используют растворы, содержащие только Fe3+, снижают
температуру хлороочистки, уменьшают содержание кислорода в
¦бромо-воздушной смеси (работая на циркулирующем в системе воз-
воздухе). Осаждение шлама в хлороочистителе можно предотвратить,
пропуская часть циркулирующего здесь раствора через фильтр или
отстойник. Отстаивание шлама ускоряется при введении в хлоро-
очистительный раствор аммонийного мыла или полиакриламида
C5—50 л 0,025% раствора на 1 м3) — скорость осаждения дости-
достигает 5 -10~4 см/сек при степени осветления 96—97%.
Хлороочистку обычно "осуществляют в башнях из кирпича или
железобетона, футерованных кислотоупорными плитками, запол-
заполненных керамической (высота слоя 2—10 м) или деревянной хор-
хордовой насадкой, и снабженных брызгоуловителями. Трубопроводы
изготовляют из дерева, эбонитированных, стальных труб или вини-
винипласта, насосы из керамики, термосилида, эпоксида и фаолита.
Очистку бромовоздушной смеси от хлора осуществляют также в
полочной колонне, работающей на пенном режиме72'79 со скоро-
скоростью газа 1,2—1,5 м/сек. При высоте колонны 2—3 м и при содер-
содержании Вг" в орошающем растворе 20—25 г/л степень очистки до-
достигает 98—100%. Гидравлическое сопротивление колонны равно
80—90 мм вод. ст., т. е. близко к сопротивлению насадочных ба-
башен; вследствие интенсивного перемешивания в пенных аппаратах
шлам не оседает. На некоторых заводах хлороочистку осуще-
осуществляют в идущем от десорбера к хемосорберу газоходе, в кото-
который под давлением через распылитель вводят раствор бромистого
железа 8°-82; затем его улавливают в отдельном брызгоуловителе
и используют вновь. Степень очистки от хлора 50—60%.
Поглощение брома из бромо-воздушной смеси
Очищенная от хлора бромо-воздушная смесь поступает в хемо-
сорбер для улавливания брома. Улавливание брома производят
железными стружками, растворами бромидов, едких и карбонат-
карбонатных щелочей (иногда с добавлением восстановителей) и серни-
сернистым газом.
Поглощение брома железными стружками основано на реак-
реакции Fe + Br2 = FeBr2, идущей очень энергично в присутствии па-
паров воды. Образующееся бромистое железо поглощает влагу из
воздуха и переходит в раствор.
Хемосорбер — полая башня высотой 6—7 м, сложенная из беш-
таунита или кирпича, футерованного кислотоупорной плиткой.
Применяют также башни из железобетона83, ничем не футерован-
футерованные. Диаметр хемосорбера определяют исходя из скорости газа
0,25—0,4 м/сек (на полное сечение). На некотором расстоянии от
дна башни имеется деревянная или керамическая колосниковая
решетка, на которую укладывают слой железной стружки высотой
до 4 м. Верх башни закрыт деревянной крышкой, в которой имеют-
имеются люки для загрузки стружки. Бромо-воздушная смесь поступает
в верхнюю часть башни; воздух, освобожденный от брома, выхо-
выходит снизу. Обратное направление воздушного потока приводит к
накоплению в растворе ионов Fe3+ вследствие того, что более бога-
богатая бромом газовая смесь встречается с концентрированным ра-
раствором бромистого железа. Отработанный воздух поступает че-
через брызгоуловитель в вентилятор, которым выбрасывается в
атмосферу или передается в десорбционную башню, так как его
температура в хемосорбере повышается на 10—30°, Хемосорбер
?22
Гл. VII. Бром и его соли
Получение брома
223
периодически догружают железной стружкой, причем она должна
быть мало углеродистой, свободной от других металлов, не содер-
содержать масла, тряпок и других инородных включений и иметь объ-
объемный вес 300—450 ке/м3. Раньше стружку предварительно акти-
активировали травлением в слабой серной кислоте. Опыт действующих
заводов показал, что от операции травления можно отказаться,
если применять стружку, удовлетворяющую указанным выше тре-
требованиям, и если она не сильно окислена.
В процессе улавливания брома происходит окисление железа
кислородом воздуха, в результате чего образуются гидраты окис-
окислов — шлам, обволакивающий поверхность стружек. Наличие шла-
шлама приводит к ухудшению контакта между железной стружкой и
бромо-воздушным потоком и потому увеличивается проскок брома,
который обычно не превышает 1—2%. Кроме того, образование
шлама повышает сопротивление движению воздуха во всей систе-
системе, которое в конце цикла достигает 500—600 мм вод. ст., вместо
нормального 250—300 мм вод. ст. Поэтому стружки периодически
(иногда непрерывно) промывают водой или слабыми раство-
растворами бромистого железа и через определенный промежуток вре-
времени A—2 месяца) вместе со шламом выгружают. Перед выгруз-
выгрузкой шлам тщательно промывают, но даже при этом потери брома
-составляют 3—4% от веса шлама84, а иногда достигают 10% 85.
. Промывные воды используют для орошения хемосорбера или для
. хлороочистки. Раствор бромистого железа, стекающий со дна хе-
хемосорбера, содержит 400—700 г/л бром-иона и некоторое количе-
количество трехвалентного железа. Для удаления Fe3+ раствор нагревают
. или выдерживают в баках с железными стружками; при этом Fe3f
; восстанавливается:
2Fe3+ + Fe = 3Fe2+ + 45,6 ккал
Получаемый раствор бромистого железа после отстаивания вы-
выпаривают в чугунных или эмалированных чашах до тех пор, пока
.температура кипения не достигнет 132—134°, что соответствует со-
содержанию 49—51% Вг~. Затем полученный плав через сифон или
вручную сливают в стальные барабаны, в которых он застывает в
монолитную массу. Чаши обогревают топочными газами или глу-
глухим паром. Выпарку топочными газами проводят в чугунных кот-
котлах с толщиной стенек 50 мм, паровую — в стальных эмалиро-
эмалированных чашах с рубашкой. Производительность чаш определяется
паросъемом 10—15 кг/ч с 1 м2 поверхности нагрева. Превращение
раствора бромистого железа в плав возможно и выпаркой под
вакуумом 85; при этом процесс ускоряется почти в 2 раза и вслед-
вследствие более низкой температуры кипения плава E5е при вакууме
670 мм рт. ст.) уменьшается коррозия. Кроме того, этот процесс
в большей мере удовлетворяет требованиям техники безопас-
безопасности
86
Барабаны из кровельного железа, применяемые для транспор-
транспортировки бромистого железа, должны быть герметичны и в плаве не
должно быть раствора, иначе происходит окисление Fe2+ до Fe3+,
что приводит к быстрому разрушению тары и вытеканию продукта.
Получение плава бромистого железа связано с быстрым изно-
износом выпарной аппаратуры из-за образования корок основных-
солей и коррозии, вызываемой выделением НВг и НО, образую-
образующихся при гидролизе бромида и хлорида железа. В связи с труд-
трудностями, возникающими при выпарке растворов бромистого
железа, их целесообразно не выпаривая перевозить в железно-
железнодорожных цистернах после добавления ингибитора (уротропина).
Выход брома в товарный продукт при наличии, хлороочистки
составляет 75—86% от содержания его в исходном рассоле и сни-
снижается до 58—72% при отсутствии хлороочистки; потери по ста-
стадиям производства распределяются следующим образом: хлориро-
хлорирование 7—20%, десорбция 3—30% (более высокие потери в этих
двух стадиях относятся к процессам без хлороочистки); хлоро-
очистка 1—2%, улавливание 2—3%, выпарка и затаривание 1—•
2%. Для получения плава бромистого железа расходуют (в пере-
пересчете на 1 т брома): серной кислоты 0—3 т (в зависимости от ще-
щелочности рассола); хлора 0,55—0,9т (в зависимости от галогено-
поглощаемости рассола); железной стружки 0,8—1 т; электроэнер-
электроэнергии от 1000 до 10 000 кет- ч (в зависимости от применяемого сырья);
воды 20—50л3; топлива 0,6—1 т (в пересчете на уголь); кровель-
кровельного железа 50—60 кг.
Поглощение брома из бромо-воздушной смеси более целесооб-
целесообразно вести концентрированными растворами бромистого желе-
железа 87-94 в этом случае улавливание брома производят в хемосор-
берах, имеющих те же размеры и конструкцию, что и хемосорберы,
применяемые при улавливании железной стружкой, но вместо
стружки их загружают керамическими кольцами размером 50 X
X 50 мм на высоту 3—4 м. Насыщение бромом раствора броми-
бромистого железа производят при рециркуляции его (плотность ороше-
орошения насадки 1,5—3 м/ч) до тех пор, пока 80—95% Fe2+ не перейдет
в Fe3+: 2FeBr2+Br2 = 2FeBr3. После этого раствор бромного же-
железа выводят из цикла и вместо него вводят свежий раствор бро-
бромистого железа. Коэффициент абсорбции брома не зависит от кон-
концентрации брома в бромо-воздушной смеси и определяется скоро-
скоростью движения газовой фазы и химической емкостью поглотителя,
т. е. концентрацией ионов Fe2+; присутствие Fe3+ практически не
влияет (если отношение Fe3+: Fe2+ меньше 20). Для указанных
выше условий при 18е и скорости газа 0,3—1 м/сек коэффициент
абсорбции соответственно равен 45—70 м/чы.
Полученный раствор бромного железа восстанавливают желез-
железными стружками в железобетонных резервуарах, футерованных
кислотоупорными плитками, причем скорость процесса зависит от
224
Гл. VII. Бром и его соли
количества железной стружки, загруженной в аппарат, состояния
ее поверхности, отношения Fe3+ : Fe2+ в растворе, его концентра-
концентрации, а также начальной температуры. Продолжительность восста-
восстановления составляет 3—5 ч при содержании в растворе 700—800 г
Вг- в 1 л, начальной температуре 30—34° и весовом отношении
_ 7 . 1 полнота восстановления 97—99%- Во избежа-
Fe,
-стружка
раствор
ние сильного вспенивания стружка не должна быть загрязнена
маслом.
В процессе восстановления бромного железа выделяется боль-
большое количество тепла и температура раствора повышается на 40—
60° (в зависимости от концентрации). При питании хемосорбера
растворами, содержащими ~750 г Вг~ в 1 л, они становятся после
восстановления столь концентрированными, что при охлаждении
выделяется FeBr2 • 6Н2О. Для получения чистых кристаллов ра-
раствор перед охлаждением должен отстаиваться для осаждения
взвеси. Охлаждение раствора ведут при перемешивании, во избе-
избежание прилипания кристаллов к стенкам. Отделение от жидкости
производят на нутч-фильтрах.
При поглощении брома по этой схеме выход брома увеличи-
увеличивается на 2—3% (по сравнению с поглощением железной струж-
стружкой), так как уменьшаются потери со шламом, а расход стружек
снижается до 0,5 т на 1 т брома. Кроме того, можно получать до
30% кристаллического бромида железа без выпарки.
Делались попытки поглощать бром из бромо-воздушной смеси
путем распыления раствора бромида железа непосредственно в
газоходе, но из-за чрезмерных потерь вследствие брызгоуноса этот
способ не применяют95'96.
Поглощение брома из бромо-воздушной смеси растворами бро-
бромида натрия основано на образовании полибромидов (ВГд, Вг;:),
что приводит к понижению давления брома над растворами59. Та-
Такой метод абсорбции брома применяют на бромном заводе в рай-
районе Мертвого моря97, использующем бромсодержащий щелок
@,8—1 % Вг~) после извлечения калийных солей. Из бромо-воз-
бромо-воздушной смеси B00 г брома в 1 м3) бром поглощают охлажден-
охлажденным до —18° раствором бромида натрия C50 г/л). Раствор содер-
содержит после абсорбции до 35%) брома. Его отгоняют, нагревая ра-
раствор глухим паром. Регенерированный раствор NaBr возвращают
на абсорбцию. Этот способ эффективен лишь при переработке теп-
теплых природных рассолов, богатых бромом.
Поглощение брома из бромо-воздушной смеси едкими щело-
щелочами в большинстве случаев не рационально и не выгодно, так
как при относительно высоком содержании в бромо-воздушной
смеси двуокиси углерода (СО2: Вг2>1) много щелочи превра-
превращается в карбонат. Для абсорбции брома применяют растворы
Получение брома
225
соды61 и известковое молоко98, которые взаимодействуют с бро-
бромом по следующим суммарным уравнениям:
2ОН" + Вг2 = Вг" + ВгСГ + Н2О
2СОз" + Вг2 + Н2О = Вг" + ВгСГ + 2HCOJ
2HCOJ + Вг2 = Вг" + ВгО~ + 2СО2 + Н2О
Первые две реакции проходят в растворах практически пол-
полностью, а третья при 20° только на ~90%. Повышение темпера-
температуры и снижение щелочности в конце процесса приводят к тому,
что гипобромиты посте-
постепенно переходят в брома-
ты, а бикарбонаты в кар-
бонаты:
W
СО|" + Н2О + СО2
Полученные растворы,
содержащие гипоброми-
гипобромиты, броматы и бромиды,
могут быть использованы
как для получения бро-
брома, так и его солей. Вы-
Выделение брома произво-
производят путем добавления к
раствору кислоты (обыч-
(обычно серной):
10
\
V
ю
20 30
NaBr,°/o
50 60
ВгОГ+бВг +6Н+=ЗВг2+ЗН„О Рис. 68. Изотермы растворимости в системе
ВКГ+ ВГ + Ж*-Вг1 + ВД> *аВг-КаВгО3-Н2О Ч» * * * - »¦
При подкислении концентрированных бромид-броматных ра-
растворов часть брома выделяется в жидком виде и отделяется от
водного раствора декантацией. Оставшийся в водном растворе
бром отгоняют острым паром в колоннах с насадкой. Из бромид-
броматного раствора можно получить бромид натрия, а если
нужно, то и бромат натрия, различными методами. Одним из мето-
методов является выпаривание раствора с последующим его охлажде-
охлаждением (рис. 68) 10°, причем кристаллизуется до 80% бромата, со-
содержащегося в растворе. Оставшийся щелок со сравнительно
небольшим количеством бромата обрабатывают восстановителями
(например, железными стружками при кипячении), затем отде-
отделяют от нерастворимого остатка и из раствора выделяют броми-
бромистый натрий.
Для поглощения брома щелочными растворами применяют
насадочные башни из железобетона или дерева, заполненные
керамическими кольцами или хордовой насадкой. Для лучшего
Ь М. Е. Позин
228
Гл. VII. Бром и его соли
использования поглотителя раствор непрерывно циркулирует.
В процессе поглощения брома растворами соды образуется бикар-
бикарбонат натрия, который сравнительно мало растворим в бромид-1
броматном растворе (рис. 69I00. Поэтому, во избежание забивания!
насадки осадком, применяют растворы, содержащие в начале про-]
цесса только 30—50 г/л соды. Для получения более концентри-|
аНСО3
NaBr
20
60
80
NaBrOs
Рис. 69. Изотермы растворимости в системе
NaBr-NaBrO3—NaHCO3—Н2О при 25 и 35°.
рованных растворов NaBr + NaBrO3 в них можно дополнительно
вводить в процессе абсорбции твердую соду или раствор NaOH
(для перевода бикарбоната в карбонат). Степень абсорбции брома
достигает 98,5—99% 93- Расход кальцинированной соды составляет
0,75—0,8 т на 1 т брома.
Описаны методы40-100-">6 поглощения брома из бромо-воздуш-
бромо-воздушной смеси щелочными растворами, содержащими NaOH, КОН,
Са(ОНJ, Na2CO3 и др., в присутствии восстановителей — амми-
аммиака, формиатов, уротропина, карбамида — в результате чего полу-
получаются почти чистые бромиды, не содержащие кислородных соеди-
Получение брома
227
нений брома. В присутствии этих восстановителей процессы идут
по следующим суммарным уравнениям:
6ОН" + 3Br2 + 2NH3 = 6ВГ + 6Н2О + N2
2ОН" + 2Вг2 + 2НСОО" = 4Вг~ + 2Н2О + 2СО2
36ОН- + 18Вг2 + N4(CH2)e = 36Вг~ + 24Н2О + 6СО2 + 2N2
6ОН" + ЗВг2 + CO(NH2J = 6Br~ + 5Н2О + СО2 + N2
Во всех случаях при использовании щелочных абсорбентов,-со-
абсорбентов,-содержащих Na+, следует учитывать возможность образования в ка-
качестве промежуточного соединения относительно плохо раствори-
растворимого бикарбоната натрия и поддерживать такую концентрацию
раствора, чтобы избежать выделения его в осадок.
Поглощение брома известковым молоком производят в барбо-
тажных колоннах, имеющих четыре полки, на которых установ-
установлены колпаки. Жидкость перетекает с полки на полку по перелив-
переливным.трубам. Применение насадочных башен нецелесообразно, так
как образующийся карбонат кальция [за счет взаимодействия СО2
с Са(ОНJ] осаждается плотной коркой на насадке и башня зара-
зарастает. Хемосорбер орошают раствором, содержащим 5—5,5% ак-
активной окиси кальция, в который постепенно вводят 10—25%-ный
раствор аммиака. Добавление аммиака сразу в больших количе-
количествах приводит к его десорбции и образованию аэрозоля бромида
аммония. Вытекающие из хемосорбера щелока содержат 140—
170 г/л Вг". Недостатком этого метода является то, что образую-
образующийся шлам увлекает значительное количество раствора (~50%
от веса шлама) и ротор вентилятора забивается бромидом аммо-
аммония; это требует промывки, усложняет технологический процесс
и приводит к дополнительным потерям брома B—3%). Кроме
того, применение барботажных колонн вместо насадочных башен
увеличивает гидравлическое сопротивление системы на 150—
250 мм вод. ст., что повышает расход электроэнергии. Расход окиси
кальция A00%) на 1 т брома составляет 0,8—1 т и зависит от со*
держания двуокиси углерода в бромо-воздушной смеси.
Абсорбцию брома из бромо-воздушной смеси щелочными по-
поглотителями в присутствии восстановителей наиболее широко при-
применяют при получении КВг. В этом случае вследствие большой-
растворимости КНСО3 можно поддерживать высокую концентра-
концентрацию поглотителя. Раствор, вытекающий из хемосорбера, содержит
до 500—520 г/л КВг. В нем содержится также ~0,1 г-экв/л ще-
щелочи и 5—15 г/л бромата, который разрушают добавкой брома и
формалина в присутствии катализатора — ионов железа.
Поглощение брома путем взаимодействия с сернистым газом
Br2 + SO2 + 2Н2О = 2НВг + H2SO4
нашло применение на самых крупных заводах в США и Англии.
Оно может быть осуществлено двумя методами: 1) взаимодействием
228
Гл. VII. Бром и его соли
007,
в газовой фазе с образованием капель жидкости, состоящей
из смеси НВг и H2SO4, улавливаемых в брызгоуловителях; 2) аб-
абсорбцией брома водным раствором смеси НВг + H2SO4 с после-
последующим взаимодействием растворенного брома (Вг2, ВГд, Вг;:) с
сернистой кислотой в растворе. Абсорбция брома водными раство-
растворами бромидов связана с резким понижением давления его пара
из-за образования комплексных соединений (рис. 70) 59>107. Более
целесообразным является осуществление взаимодействия в газо-?
вой фазе'08, так как объем anna-i
ратуры при этом меньше, ynpo-ii
щается регулирование процесса ^
снижается расход сернистого^
газа, который составляет 0,5—]
0,6 т на 1 т брома. Взаимодейст-;
вие между сернистым газом, по-
получаемым сжиганием серы или,1
колчедана, и бромом происходит"
практически мгновенно и осуще-
осуществляется в воздухопроводе меж-
между десорбционной башней и уло-
уловителем. Концентрация сернисто-
сернистого газа практического значения
не имеет, так как он все равно
сильно разбавляется воздухом
при содержании брома в нем
0,7—12 г/л*3. Воздухопровод вы-
выполняют из кислотоустойчивого
материала, обычно из стали, по-
покрытой эбонитом, резиной и т. д.
Уловитель представляет собой
железобетонную или кирпичную
б*ашню, футерованную кислотоупорными плитками и заполненную
стеклянной ватой или керамическими кольцами. Для лучшего ула-
улавливания башня орошается (периодически или непрерывно) не-
небольшим количеством раствора (НВг + H2SO4). Башни имеют тот
же диаметр, что и десорбционные, а высоту 4—6 м. После улови-
тельной башни устанавливают брызгоуловитель, орошаемый водой
и исходным рассолом. Стекающая из брызгоуловителя жидкость
поступает в десорбционную башню. Степень улавливания брома
составляет 98,5—99%- Стекающий из уловительной башни раствор
содержит 10—20% НВг, 8—18% H2SO4 и до 1% НС1. Концентра-
Концентрация получающегося раствора зависит от содержания брома в ис-
исходном растворе и давления водяного пара над ним. Чем ниже
давление водяного пара и выше содержание брома в исходном
растворе, тем больше содержание бромистого водорода в полу-
получающейся жидкости.
50
3 4
Концентрация раапдора,г-экО
Рис. 70. Зависимость коэффициента
распределения брома от содержа-
содержания Вг~ в растворе при 0, 25 и 50°.
I
Получение брома
229
Раствор смеси кислот (НВг + H2SO4 + HC1) перерабатывают
на бром 1™-Н1 хлорированием или на бромистоводородную кис-
кислоту фракционной перегонкой. Хлорирование производят в кера-
керамических сосудах, где часть брома выделяется в жидком виде и
затем декантируется. Водный раствор, содержащий растворенный
бром, нагревают острым паром в колоннах с насадкой, Вг2 отго-
отгоняется, а отработанный раствор, содержащий НС1 и H2SO4 и не-
небольшие количества брома, стекает снизу башни. Получаемый та-
таким образом бром содержит не менее 98,5—99% Вгг. Отработан-
Отработанную смесь кислот D,6—9% НС1 и 5—10% H2SO4) используют для
подкисления исходного бромсодержащего рассола; количество от-
отработанной смеси кислот (в пересчете на 100% H2SO4), получаю-
получающейся на 1 т брома, составляет 1,6—1,8 т. При этом на 1 т брома
расходуется 0,48—0,5 т хлора и 5—8 т пара.
Для получения бромистоводородной кислоты необходимо вести
фракционную перегонку. I фракция с температурой кипения <123°
представляет собой очень разбавленный раствор соляной и броми-
бромистоводородной кислот, в котором содержится до 90% НС1 и 1,5—
2% НВг от их начального количества (поступившего на дистилля-
дистилляцию). II фракция с температурой кипения 123—126° содержит
19—22% НВг и 0 13—0,3% НС1. С этой фракцией отгоняется до
12% НВг и 8% НС1 от их начального количества. III фракция с
температурой кипения 126—127° содержит —45% НВг, 0,1% НС1
и 0,07% H2SO4; с ней отгоняется ~85% НВг от веса загружен-
загруженного. Кубовый остаток содержит 78% H2SO4, 0,02% НВг и
0,01% Вг2.
Первая фракция может быть использована для подкисления
исходного рассола, кубовый остаток которого получается до 0,9 т
(в пересчете на 100% H2SO4) на 1 т брома, может быть также ис-
использован либо для подкисления исходного рассола, либо в каче-
качестве товарной кислоты (там, где не вредна примесь НВг и Вг2).
Во II и III фракциях содержится 96—97% уловленного брома. При
отсутствии жестких требований по содержанию хлора в продукте
они могут быть смешаны, и тогда получается раствор, содержащий
НВг —40%, НС1 —0,2% и H2SO4 — 0,06%, который используется
для получения бромистых солей *.
В США бромистоводородную кислоту получают также из смеси
НВг и H2SO4, образующейся при взаимодействии брома с серой и
водой по экзотермической реакции
S + ЗВг2 + 4Н2О = 6HBr + H2SO4
идущей достаточно быстро при 90—100°; вначале сера бромируется
й S2Br2 далее реагирует с бромом и водой.
* О материалах для аппаратуры и приборах для контроля бромного произ-
производства см.4S- *•>В2- м>62- »2-н8_
230
Гл. VII. Бром и его соли
Для многих потребителей необходим элементарный бром, а
при извлечении его воздушной десорбцией получаются его соеди-
соединения. Наиболее простым методом получения жидкого брома яв-
является обработка бромид-броматной смеси кислотами (стр. 225).
В СССР жидкий бром обычно получают обработкой газообразным
хлором раствора FeBr2 C50—550 г/л Вг") в асбоцементных или
железобетонных аппаратах и последующей отгонкой его за счет
теплоты реакции и с помощью острого пара. Пары брома конден-
конденсируются в холодильнике и жидкий бром через автоматические до-
дозаторы сливается в тару. Все коммуникации изготовляются из
стеклянных труб, соединенных с помощью химически стойкой ре-
резины и фторопластовой пленки. Выход брома составляет 95—98%
от его содержания в бромистом железе11822.
Другие методы получения брома
Некоторое количество брома выделяли раньше из природных
рассолов в виде труднорастворимых органических соединений —
триброманилина, трибромфенола, тетраброманилина 123~126. Однако
это оказалось мало экономичным из-за трудности регенерации ис-
исходных органических соединений.
Были предложены методы экстрагирования брома из хлориро-
хлорированных рассолов несмешивающимися с водой органическими рас-
растворителями 126—129 Экстракция керосином, бензином, дихлорэта-
дихлорэтаном и другими приводит к их бромированию (и частично хлори-
хлорированию) и потому нерациональна. Применение для экстракции
предварительно бромированных экстрагентов (керосина) дает бо-
более благоприятные показатели, но при этом в ряде случаев наблю-
наблюдается образование стойких эмульсий, что создает большие потери
брома и экстрагента.
Извлечение брома из окисленных растворов взаимодействием с
Этиленом с целью получения дибромэтилена мало экономично, так
как из-за малых концентраций брома в рассоле образующийся
С2Н4Вг2 практически полностью растворяется и для его извлечения
требуется экстракция. Возможно извлечение брома из рассолов
осаждением хлоридом серебра130 с выходами около 90%. Весьма
небольшое применение нашел электрохимический метод получения
брома ш34.
Одним из перспективных методов является извлечение брома
из природного сырья с помощью анионитов 135-14° *. Окисленные
(хлорированные) рассолы, содержащие свободный бром, контакта-
руются с анионитом (в СССР применяют АВ-17), который после
насыщения бромом обрабатывают сернистым газом или сульфи-
сульфитом натрия для восстановления в бромид, а затем концентриро-
Производство некоторых солей брома
231
* Подробнее этот метод описан в главе VIII.
ванным раствором хлорида натрия для десорбции бромида. Полу-
Полученный раствор, отделенный от анионита, хлорируется и бром
отгоняется водяным паром при 80—90°. Пары брома конденси-
конденсируются в жидкий бром. Степень сорбции брома анионитами соста-
составляет 95—97% и не зависит от температуры (в пределах 2—50°)
и его концентрации в растворе. Насыщение ионита зависит от со-
содержания в растворе Вг~ и С1~ и составляет 0,2—1 г/г при концен-
концентрации брома в исходном растворе 0,1 — 1 кг/м3. Общий выход
брома зависит от степени окисления бромида и может достигать
80—85%.
ПРОИЗВОДСТВО НЕКОТОРЫХ СОЛЕЙ БРОМА
Бромид натрия
Бромид натрия может быть получен непосредственно из
бромо-воздушной смеси при поглощении брома растворами соды
(стр.* 225). Его также получают из растворов бромида кальция,
образующегося при улавливании брома известковым молоком в
присутствии аммиака. К раствору бромида кальция A40—170 г/л
Вг~) добавляют сульфат натрия (или содуL0-141> 142
CaBr2 + Na2SO4 - CaSO4 + 2NaBr
Пульпу перемешивают 1—2 ч, отфильтровывают сульфат каль-
пия, шелок выпаривают до плотности 1,40—1,42 г/см3 и вторично
фильтруют от осадка, выделяющегося при выпарке (CaSO,},
СаСО3). Затем щелок окончательно выпаривают до образования
густой массы, содержащей 15—20% маточного рассола. Массу по-
подают на центрифуги, где производят отделение кристаллов при
температуре выше 70е. Недостатком этого метода являются значи-
значительные потери брома B—5%) за счет увлечения раствора осад-
осадком сульфата кальция.
Небольшие количества бромида натрия получают взаимодей-
взаимодействием бромида железа с растворами соды:
FeBr2 + Na2CO3 + (л + 1) Н2О = Fe(OHJ • лН2О + 2NaBr + СО2
Благодаря наличию в растворе Fe3+, а также окислению кисло-
кислородом воздуха, в осадке всегда содержится Fe(OHK. В чугунных
котлах с мешалкой и паровой рубашкой раствор соды нагревают
почти до кипения и небольшими порциями приливают раствор
бромида железа. Из-за выделения СО2 происходит сильное вспени-
вспенивание, и потому необходимо интенсивное перемешивание реакцион-
реакционной массы при заполнении аппарата не более чем на 50%. Выде-
Выделяющиеся гидраты окислов железа отделяют от раствора, промы-
промывают разбавленными промывными водами и водой до-практически
232
Гл. VII. Бром и его соли
полного удаления Вг~. Первые порции промывных вод смешивают
с основным раствором бромида натрия и выпаривают в чугунных
эмалированных чашах. Кристаллы бромида натрия отделяют на
центрифугах, сушат, просеивают и упаковывают.
Другим широко применяемым методом получения бромида
натрия является взаимодействие брома с раствором едкого натра
(или соды) в присутствии восстановителей — формиата натрия143
или аммиака 144-14Б:
Na2CO3 + 2HCOONa + 2Br2 = 4NaBr + 3CO2 + H2O + 121,8 ккал
3Na2CO3 + 2NH3 + 3Br2 = 6NaBr + 3CO2 +3H2O + N2 + 157,4 ккал
Процесс ведут в закрытых эмалированных аппаратах, в которые
вначале заливают раствор соды (или едкого натра), добавляют к
нему восстановитель, а затем при перемешивании под слой рас-
раствора приливают бром. Ввиду большого выделения тепла и газов,
на крышке аппарата, во избежание потери брома, устанавливают
ловушки, заполненные железными стружками или орошаемые ра-
раствором соды. После окончания реакции раствор фильтруют от
взвешенных частиц, а затем перерабатывают, как было указано
выше. Для получения 1 т бромида натрия этим методом расхо-
расходуют: брома 0,8—0,о2 т, едкого натра (технического) 0,45—0,48 т,
аммиака 0,07—0,08 т или муравьиной кислоты A00%) 0,23—
0,25 т.
Одним из перспективных методов получения бромистого нат-
натрия является взаимодействие соды (или едкого натра) или хло-
хлорида натрия с бромистоводородной кислотой. Бромистоводород-
ная кислота может быть получена фракционной разгонкой смеси
HBr + H2SO4 или синтезом из элементов 146-147.
Получение бромида натрия из хлорида натрия и бромистово-
бромистоводородной кислоты
NaCI + HBr = NaBr + HCI
основано на более низкой температуре кипения азеотропной смеси
НС1—Н2О A10°), чем смеси HBr—Н2О A26°) 148. Реакцию ведут
при 110°, добавляя к твердому хлориду натрия бромистоводород-
•ную кислоту в количестве 120% от стехиометрического. В реак-
реакторе остается твердый бромид натрия, а соляная кислота отго-
отгоняется и после конденсации может быть использована для под-
кисления исходных бромсодержащих рассолов. Получающийся по
этому методу бромид натрия после промывки небольшим количе-
количеством воды содержит 0,5—0,8% NaCI. Выход по HBr составляет
96—98%. При производстве безводного бромида натрия необхо-
необходимо все операции, где имеется жидкая фаза, вести при темпера-
температуре выше 70°, иначе будет получаться дигидрат, который в даль-
дальнейшем необходимо обезвоживать,
Производство некоторых солей брома
233
Бромид калия
Получение бромида калия может быть осуществлено всеми
методами, указанными для бромида натрия. Исходным сырьем
при этом служит едкое кали или поташ. Чаще всего осуществ-
осуществляют их взаимодействие с растворами бромида железа или бро-
бромом в присутствии восстановителей. Ранее широко применявшийся
метод взаимодействия раствора едкого кали с бромом
6КОН + ЗВг2 = КВгОз + 5КВг + ЗН2О
с последующим восстановлением броматов углем при высокой тем-
температуре149 в настоящее время утратил свое значение. Получение
бромида калия несколько проще, чем бромида натрия, так как
КВг при всех температурах выделяется из растворов в безводной
форме. Для получения 1 т КВг расходуют: 0,7—0,72 т брома, 0,64—
0,58 т едкого кали, 0,06—0,065 т аммиака или 0,20—0,22 т му-
муравьиной кислоты A00 %) -
Бромид аммония
Бромид аммония получают взаимодействием концентрирован-
концентрированного раствора аммиака с бромом:
8NH3 + 3Br2 = 6NH4Br + N2 + 104,8 ккал
Для ведения этого процесса реактор вначале охлаждают, а
бром приливают небольшими порциями. Выделяющийся из рас-
раствора аммиак и образующийся дым бромида аммония улавливают
водой или слабым раствором бромида аммония. В растворе дол-
должен всегда присутствовать избыток аммиака во избежание обра-
образования взрывчатых галогенных соединений азота. Полученный
раствор выпаривают, а затем охлаждают, и выделившиеся кри-
кристаллы бромида аммония отфуговывают, сушат при 90—95е и упа-
упаковывают. На получение 1 т бромида аммония расходуют: 0,9—
0,96 т брома и 0,25—0,27 т аммиака.
Бромид аммония может быть получен при взаимодействии
растворов бромида железа и аммиака или карбоната аммония,
а также раствора бромида кальция с карбонатом аммония 150.
Бромат калия
Бромат калия можно получить взаимодействием нагретого рас-
раствора едкого кали с бромом, но при этом полезно используется
только V6 часть брома. Поэтому бромат калия получают окисле-
окислением бромида калия или брома в щелочной среде хлором ш:
6КОН + КВг + ЗС12 = КВгОз + 6КС1 + ЗН2О
12КОН + Вг2 + 5С12 = 2КВгО3 + 10КС1 + 6Н2О
Реакцию ведут в эмалированном аппарате, куда вначале за-
заливают раствор едкого кали плотностью 1,32 г/см3' (~450 г/л
234
Гл. VII. Бром и его соли
КОН) и в него постепенно приливают жидкий бром при непре-
непрерывном перемешивании. Затем в раствор пропускают хлор до
нейтральной реакции, фильтруют, а затем охлаждают до 15°. Вы-
Выделившуюся смесь КВгО3 и КС1 промывают малыми порциями
холодной воды до практически полного растворения хлорида ка-
калия. Выход по брому составляет ~90—94%.
'Бромат калия может быть получен также анодным окислением
раствора бромида калия 152> 153. Выход по току при этом составляет
80—90%, расход электроэнергии 4000—5000 квт-ч на 1 т бромата
калия.
ЛИТЕРАТУРА
1. Н. A. Liebhafsky, J. Am. Chem. Soc, 56, 1500 A934); 61, 3513 A939).—
2. Г. П. Лучинский, ЖОХ, 9, 708 A939).— 3. L. W. Winkler, Chem. Ztg.,
23, № 67, 687 A899). — 4. А. Н a n t z s с h, A. Vagt, Z. phys. Chem., 38, 705
A901). — 5. В. П. Ильинский, А. Т. Уверская, Труды ГИПХ, в 41, 1958,
стр. 112.— б. Encyclopedi of Chemical Technology, Ed. R. E. Kirk, D. E. Oth-
mer, vol. II, 1952, p. 629.-7. G. S. Haines, Ind. Eng. Chem.. 31, M> 12, 2702
A949). — 8. А. А. Попадюк, ИБП ГИПХ,* № 11—12, 51 A967) — 9. Chem.
Eng., 38, № 22. 136 (I960). — 10. В. Ф. Б о йцо в а и др. БТИ ГИПХ** № 7, 3
A958).
77. В. М. Беренблит, Т. Ф. Кур ан о в а, Там же, № 4, 23 A957).—
12. А. Bodenheifner, I. Epstein, I. Schnerb, Israel J. Chem.. 5, № 4a
145 A967). — 13. B. M. Беренблит, Т. Ф. К у р а и о в а, Труды ГИПХ в 41
1958, стр. 210. —14. Е. Ю. Левина, БТИ ГИПХ, № 16, 3 A962).—
15. Г. А. Яковкин, ИБП ГИПХ, № 2, 3 (i964); № 11—12, 4 A967).—
16. Г. Л. Кореньков, Н. А. Устинова, Горнохимическое сырье зарубежных
стран, Изд. «Химия», 1965, гл. 4. —17. Ф. Ф. Герман, БТИ ГИПХ, № 9, 25
(I960). —№. М. Л. Койфман, Г. Р. Залкинд, Р. И. Гуревич, Труды
ГИПХ, в. 48, 1965, стр. 104.-/9. Л. А. А м р о м, Д. С. С та сине в ич, Журн.
ВХО, № 1, 47 A962). — 20. Ind. Eng. Chem., № 7, 50 A964).
21. W. Miller, Bull. Bur. Mines, № 630, 447 A968). — 22. R. Korn Freiber-
gcr Fosschungsh., № 534, 53 A965).— 23. L. E. Jolles, Bromine and its Com-
Compounds, London, 1966. — 24 Inform. Chim., № 42. 37, 43, 47, 53. A966) —
25. В. П. Ильинский, В. М. Филиппе о, ЖХП, № 16, 833 A928) —
26. В. J. Karsten, Z. anorg. Chem, 53, 63 A907). — 27. М. Kaltenbach
Chim. e. Ind., 25, № 3, 1 A931). — 28. M. С/Рождественский, Л. М Б р о-
уде, ЖПХ, 10, № 4—6, 722 A937). — 29. П. Г. Данильченко, М. И. Р а в и ч,
ЖРФХО, № 59, 953 A927). — 30. М. С о d е 11, G. Norwitz, Chem. Ind K° 19
580 A957). " *
31. Пат. США 3145084, 1964. — 32. О. В. Лебедев, ИБП ГИПХ Л"» 13
72 A967). — 33. С. И. Гришко, БТИ ГИПХ, № 14, 27 A962).— 34. Л1. Е. По'
зин, М. Л. Шабашова, С. Д. Гольденберг, ЖПХ. 22, № 5, 467 A949).—
35. В. Ф. Б о й ц о в а, Г. С. Клебанов, И. П Кузьмина, ИБП ГИПХ
№ 7—8, 28 A965). —36. Н. О. Эверт, БТИ ГИПХ, № 1, 35 A955) —
37. Ф. Ф. Герман, ЖПХ, № 3, 171 A956). — 38. Н. П. Гринвуд, Усп. хим.,
22. № 4, 445 A953). — 39. Я. А. Фиал ков, Межгалоидные соединения, Изд.
АН УССР, 1958. — 40. В. И. К с е н з е н к о, Д. С. С т а с и н е в и ч, Технология
брома и иода, Госхимиздат, 1960.
41. Д. С. Стасиневич, ЖПХ, 31, № 5, 701 A958). — 42. Д. С Стаси-
иевич, Там же, № 6, 844 A958). — 43. Л. С. Лилич, ЖОХ, 22, 730 A952).—
* Иодо-бромная промышленность, Сб. обзорной информации ГИПХ.
** Бюлл. техн. информации ГИПХ по иодо-бромной промышленности.
Литература
235
44 С. И. Яворский, БТИ ГИПХ, № 1, 28 A955). — 45. О. Ф. Клочко,
В П Зобнин, Труды ГИПХ, в. 41, 1958, стр. 171.— 46. R. Magai, J. Soc.
Chem. Ind. Japan, 46, 358, 1217 A943). — 47. H. О. Эверт, М. А. Тарабри-
на ИБП ГИПХ, № 1, 4 A964). — 48. Г. Р. Залкинд, М. Л. Койфман,
Там же, № 11—12, 59 A967). — 49. R. Magai, J. Soc. Chem. Ind. Japan, 47, 33,
309 A944). — 50. J. J. Ronco, Chem. Abs., 42, № 14, 5176 A948).
51. И М Полищук, БТИ ГИПХ, № 14, 21 A962).— 52. А. Д. Кутний,
Ю. Е. Г о л ь б е р г, ИБП ГИПХ, № 1, 16; № 3—4, 70 A964).— 53. М. Е. П о з и н,
К. И Хренников, ЖПХ, № 2, 45 A948) —54. J. J. Ronco, Chem. Abs., 42,
№ 15 5623 A948). — 55. М. Е. П о з и н, ЖПХ, 29, № 9, 1336 A956).—
56 С. И. Яворский, БТИ ГИПХ, № 5, 12 A957). — 57. М. Г. Гусев, ИБП
ГИПХ, № 1, 48 A964). — 58. С. И. Яворский, Там же, № 5—6, 56 A965).—
59. Г. С. К л е б а н о в, Е. П. Б а с о в а, Э. М. В о л н я н с к а я, И. М. Г у р е в и ч,
Сборник трудов ГИПХ, в. 40, 1948, стр. 119.— 60. В. П. И л ьин ский, К- Д. Ру-
Руси н о в а, Е. Г. Дроздова, Там же, в. 41, 1958, стр. 153.
61. L. С. Steward, Ind. Eng. Chem., 4, № 26, 361 A934). —62. Manuf.
Chem. 25, № 7 305 A954). —63. R. Bock, Chem. Ind. Techn., 25, № 5, 245
A953). — 64 E Lawson, World Petrol., 25, № 4, 64 A954). — 65. Civil Eng. a.
Public. Works, 49, 577, 709 A954). — 66. О. В. Лебедев и др., ИБП ГИПХ,
№ 13, 69 A967); № 14, 61 A968). — 67. А. Е. Шноль, Хим. пром., № 9, 318
A953). — 65. М. Е. Позин Б. А. Копы л ев, Г. В. Бельченко, Изв. вузов.
Хим. и хим. технол., № 5, 142 A958).— 69. М. Г. Гусев, ИБП ГИПХ, № 7—8,
75 A965). — 70. Н. И. Гельперин, В. И. Савченко, В. 3. Гринько, Хим.
Пром., № 5, 372 A967).
71. И. Ш. Мастеровой, ИБП ГИПХ, № 2, 24 A964). — 72. М. Г. Гу-
Гусев Е. В. Мовчан, БТИ ГИПХ, № 5, 29 A957). — 73. Ф. Ф. Герм а и, ИБП
ГИПХ, № 3—4, 34 A964). — 74. Л. Д. Полякова, Там же, № 3—4, 10
A964). — 75. Е. А. Дианов, В. И. Ксензенко и др., Там же, № 3—4, 29
A964)- № 7—8, 24 A965). — 76. В. И. Ксензеико, А. Нуриев, Хим. пром.,
К» 3 183 A966). — 77. Т. К- Лукина, ИБП ГИПХ, № 3—4, 20 A964).—
78. Г. Р. Залкинд, Т. К. Лукина, Труды ГИПХ, в. 48, 1965, стр. ПО.—
79. М. Е. Позин, И. П. М у х л е н о в, Е. С. Т у м а р к и н а, Э. Я. Т а р а т, Пен-
Пенный способ обработки газов и жидкостей, Госхимиздат, 1955, стр. 231.—
80. Ф. Ф. Герман, Л. Я. Ткаченко, БТИ ГИПХ, № 10, 19 A960).
81. Ф. Ф. Герман, Там же, № 12, 22 A961). — 82. Н. П. Якубовский,
ИБП ГИПХ, № 7—8, 88 A965). — 83. Ф. Ф. Герман, ЖХП, № 2, 105 A956).—
84. Н. О. Э в е р т, Л. М. К о й ф м а н, БТИ ГИПХ, № 3,2 A956).— 85. И. Ш. М а-
стеровой, Там же, № 14, 24 A962).—86. Правила и нормы техники безопас-
безопасности и промышленной санитарии для проектирования, строительства и эксплуа-
эксплуатации производства бромистого железа по методу выдувания, Госхимиздат,
1961. — 87. В. П. Ильинский, Я- М. Сеферовичи др., Труды ГИПХ, в. 41,
1958, стр. 193 —88. М. П. Мочулкин Н. О. Эверт и др., БТИ ГИПХ, № 5,
25 A957).—«9. Н. П. Якубовский, Там же, № 6, 38 A958); № 7, 41 A958).—
90. Ф. ф. Г е р м а н, Там же, № 6, 43 A958).
91. Л. А. Кочаловский, Там же, № 6, 47 A958). — 92, Н. П. Якубов-
Якубовский, Г. Р. Залкинд, Там же, № 6, 52 A958). — 93. М. Е. П о з и н, Е. С. Т у-
маркина, ЖПХ, 23, № 4, 396 A950). — 94. В. И. Ксензенко, К. А. Еро-
Ерофеева, Хим. пром., № 3, 207; № 4, 260 A964); ИБП ГИПХ, № 7—8, 45
A965).— 95. С. Н. Титов, Н. П. Якубовский, БТИ ГИПХ, № 12, 27
A961). — 96. Ф. Ф. Герман, Там же, № 14, 30 A962). — 97. Chem. Ztg., 81,
757 A957). — 98. А. Г. Ж и в о т о в с к и й, Н. А. Б а б у ш к и н а, ЖПХ, 36, № 11,
2343 A963). — 99. Н. И. Гельперин, В. И. Ксензенко и др., Хим. пром..
Mi 11, 832 A965). —100. Г. С. Клебанов, Е. П. Басова, ЖПХ, 12, № 11,
1601 A939).
236
Гл. VII. Бром и его соли
101. М. Г. Гусев, БТИ ГИПХ, № 9, 14 A960). — 102. Л. В. Алентьева,
Там же, № 10, 15 (I960). — 103. Н. О. Эверт, М. М. Шимонова, Там же,
№ 16, 21 A962),— 104. Ф. Ф. Герман, А. М. Банвер, Там же, № 2, 1956,
стр. 10. —105. В. П. Ильинский, Я. М. Сеферович, Сборник рефератов
ГИПХ за 1945 г., 1946, стр. 48. —106. В. И. Ксензенко, Труды МИТХТ, № 8,
1958, стр. 104.— 107. М. А. Менковский, Н. А. Петров и др., ЖНХ, № 1,
1658 A956). — /05. В. П. Ильинский, В. Ф. Б о й ц о в а, Труды ГИПХ, в. 41,
1958, стр. 129. —109. В. Hart, Instruments, 20, 956 A947).—110. В. Ф. Бой-
Бойцов а, К. Д. Русинова и др., ИБП ГИПХ, № 3—4, 42 A964); № 7—8, 18
A965).
111. Г. Р. Залкинд, О. Н. Богатырева и др., ИБП ГИПХ, № 14, 49
A968). — 112. О. Ф. К л очко, В. П. Зобнин, БТИ ГИПХ, № 3, 1956, стр. 24.—
113. С. К- Афанасьев, М. А. П о р т н о в, Ю. И. Ч е п е л к и н, ЖПХ, 10, № 8,
1421 A937). —114. М. А. Портнов, С. И. Элькина, Зав. лаб., 10, № 11,
527 A945). —115. А. В. Фомичев, БТИ ГИПХ, № 6, 66 A957).—
116. И. Е. Орлов, Методы анализа рапы, буровых вод и контроль производства
иода и брома, ГОНТИ, 1939.—117. Ц. А. Аджемян, А. И. Аккерман,
Хим. пром., № 6, 14 A945). —118. Б. Д. Матюшенко, БТИ ГИПХ № 14, 32
A962).— 119. Б. П. Алешин, ИБП ГИПХ, № 11—12, 28 A967).— 120. О. В. Л е-
бедев, Н. Е. Чумичева, Там же, № 11—12, 33, 38 A967).
121. Ф. Ф. Герман, Там же, № 11—12, 47 A967). — 122. Б. П. Алешин,
Там же, № 11—12 A967). —123. А. Г. Байчиков, Л. Демидова, Е. Ефре-
Ефремов, ЖХП, № 1, 58 A934). — 124. А. Г. Б а й ч и к о в, А. Г. 3 а б р о д к и н. Там
же, № 3, 59 A934). — 125. А. Корнилов, ЖПХ, 5, 567 A932). — 126. Б. Г. П а н-
телеймонов, ЖХП, 4, № 9, 713 П927); 5, № 11—12, 484; № 21—22, 1220
A928). —127. А. Ф. Сагайдачный, Н. Захарова и др., Труды ГИПХ,
в. 18, 1933, стр. АА. —128. А. Г. Байчиков, ЖХП, № 12, 63 A934).—
t29. П. Гогоришвили, М. Каркорашвили, Труды Ин-та химии АН
ГрузССР, № 11, 1953, стр. 87. — 130. Пат. США 2622966, 1952
131. J. J. Ronco, Chem. Abs., 42, № 15, 5349 A948). —132. Япон. пат.
177354, 1949; 7008, 1951.—133. М. Schlotter, Ober electrolytische Gewinnung
Von Brom und Iod, Halle, 1907. — 134. M. H. M а ч у л к и н а, В М. Маркова
и др., Труды ГИПХ, в. 48, 1965, стр. 153. — 135. Япон. пат. 1511, 1952; 4464, 1952 —
136. Пат. США 3174828 I960. — 137. Г. Р. Залкинд, М. Л. Койфман, ИБП
ГИПХ,№ 11—12, 59 A967).—138. В. И. Ксензенко, Н. С. Гаркави, Там
Же, № 11—12, 67 A967). —139. J. Aveston, D. Everest, Chem. Ind, 14,
№ 37, 1238 A957). —140. M. Ziegler, Angew. Chem., № 8, 283 A960).
141. Авт. свид. 102805, 1954; 138231, 1961. —142. E. Я. Диан о в,
В. И. Ксензенко, Н. К- Соловьева, сб. «Проблема комплексного исполь-
использования минеральных богатств Кара-Богаз-Гола», Изд. АН ТуркмССР, Ашхабад,
1959, стр. 206. — 143. В. П. Ильинский, А. И. Ч е р т о к, С. Л. Р а х м и л е-
вич, Калий, № 4, 29 A934). — 144. М. Г. Гусев, Р. И. Аров, А. Г. Лысый,
БТИ ГИПХ, № 4, 20 A957). —145. Правила и нормы техники безопасности и
промышленной санитарии для проектирования, строительства и эксплуатации
производства бромистого калия и бромистого натрия аммиачным методом, Гос-
химиздат, 1961. —146. В. П. Ильинский, В. Ф. Бойцова и др., Сборник
трудов ГИПХ, в. 41, 1958, стр. 161. — 147. В. П. Ильинский, А. В. Попова,
БТИ ГИПХ, № 7, 25 A958). — 148. В. П. Ильинский, А. В. Попова, Труды
ГИПХ, в. 41, 1958; стр. 183. —149. В. Хютнер, Получение бромистых солей И
бромистых препаратов, Берлин, 1918. —150. Авт. свид. 106540, 1956.
151. А. Ильин, Реактивы и препараты лабораторного назначения, ГОНТИ,
1938, стр. 131, 169. —152. М. П. Мочулкин, Труды ГИПХ, в. 41, 1958,
стр. 232. — 153. А. М. Межерицкий, Я. К. Смирнова и др., ИБП ГИПХ,
№ 7—8, 82 A965).
Глава VIII
ИОД И ЕГО СОЛИ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Иод — серовато-черные кристаллы с металлическим блеском
плотностью 4,93 г/см3. Плавится он при 113,6° и кипит при 184,4°,
Теплоемкость кристаллов
0,05058+4,688-10* кал/г B5—
—113°), жидкости 0,0756 кал/г
A13,6—184°) и пара 0,0350
кал/г B5—1200°). Теплота пла-
плавления 14,85 кал/г A13,6°). Теп-
Теплота парообразования 56,9 кал/г
цри 113,6° и 39,3 кал/г при
184,4°.
Давление пара иода при
25° 0,31 мм рт. ст. и при 113,6°
90,5 мм рт. ст. Он легко возго-
возгоняется; пары имеют фиолето-
фиолетовый цвет. При конденсации па-
паров на холодной поверхности
образуются кристаллы. Рас-
20 30
Температура,°С
Рис. 71. Изменение коэффициента рас-
распределения иода между газовой фазой
и водными растворами солей и водой
в зависимости от температуры.
творимость иода в 1000 г воды
при 20° 0,28 г, 50° —0,71 г и
100° — 4,5 г. В присутствии
сульфатов растворимость иода
уменьшается, а в присутствии
I", Вг~, С1~ — увеличивается вследствие образования комплексных
полигалогеновых ионов:
Г+12=1з, ? + 12 = 1б, ВГ+12 = Вг1-
и т. п. Это сильно влияет на коэффициент распределения иода
между газовой и жидкой фазами (рис. 71), что существенно для
238
Гл. VIII. Иод и его соли
извлечения иода из растворов воздушной десорбцией1-4. Лучше,
чем в воде, иод растворяется в этиловом спирте, эфире, хлоро-
хлороформе, сероуглероде, тетрахлорметане, дихлорэтане, керосине и
других органических жидкостях, образуя окрашенные растворы.
Растворение иода в воде сопровождается гидролизом: 1г +
-+ Н2О = Н+ + I" + НЮ, константа равновесия6 которого при 20°
Сырье
239
Образующаяся иодноватистая кислота неустойчива и при по-
повышении температуры и под действием света легко перехо-
переходит в йодноватую кислоту:
ЗНЮ = ЗН+ + 2Г + Юз (этот
переход происходит легче,
чем у аналогичных соедине-
соединений хлора и брома). Поэто-
Поэтому суммарное уравнение ги-
гидролиза иода:
312 + ЗН2О = 6Н+ + 5Г + IOJ
Его константа равнове-
равновесия при 20° равна 6:
[нЧ6[г]5[ю3-]
130
ю
'о
2
NaI-2:
^^
N
~^?-
ъ^,—'
з1_ 1
,——-
70
60
501--
40
к-
= 2,45-10'
,-48
-го
о
20
Ш Ю0
Рис. 72. Растворимость Nal, NH4I, KI и
КЮ3 в воде.
Крепкой азотной кисло-
кислотой иод легко окисляется до
I2O5 при комнатной темпе-
температуре. Пятиокись иода
сильно гигроскопична и под
действием влаги воздуха пе-
переходит в Н13О8. При 280°
I2O5 необратимо разлагает-
разлагается на 12 и О2. (После про-
продолжительного нагревания
при 250е наблюдается не-
незначительная сублимация
12О5.)
Йодноватая кислота при 100—130° разлагается с образованием
ЫзО8. Дальнейшее разложение HlgOg с образованием 12О5 идет
при 195—220°6*.
Иод вызывает ожоги на коже, а его пары раздражают сли-
слизистые оболочки. Содержание иода в воздухе рабочих помещений
не должно превышать 0,001 мг/л.
Из неорганических соединений иода наибольшее значение
имеют йодистые натрий, калий, аммоний и иодноватокислый ка-
калий. Все эти соли образуют бесцветные кристаллы. Йодистый нат-
натрий Nal кристаллизуется в кубической системе с плотностью
3,67 г/см3; плавится при 651° (по другим данным при 682°). Рас-
Растворимость его в воде показана на рис. 72. NaI*2H2O кристалли-
кристаллизуется в моноклинной системе с плотностью 2,45 г/см3. Йодистый
натрий сильно гигроскопичен. При длительном хранении кристал-
кристаллы его окрашиваются в желтый цвет из-за выделения свободного
иода. Йодистый калий KI и йодистый аммоний NH4I кристалли-
кристаллизуются только в безводной форме в кубической системе. Плот-
Плотность KI 3,13 г/смг, плавится он при 723°; плотность NHJ
2,51 г/см3. Иодноватокислый калий КЮ3 образует безводные кри-
кристаллы кубической системы с плотностью 3,89 г/см3; плавится при
560е. При нагревании разлагается с выделением кислорода. Об-
Образует кислую соль КЮз'НЮз. Насыщенный водный раствор при
15° содержит 1,33% КЮ3-НЮ3.
ПРИМЕНЕНИЕ
Иод применяют в медицине в виде йодной настойки E и 10%
раствор в спирте) или раствора Лиголя (водный раствор иода,
содержащий йодистый калий) и в виде солей и органических со-
соединений (йодоформ, иодгност, сайодин, сергозин, иодол и др.).
Иод используют при синтезе органических красителей, при по-
получении титана, при изготовлении некоторых металлов высокой
степени чистоты (через иодиды). 70—75% иода перерабатывают
в йодистые соли, применяющиеся, кроме медицины, в фотографии,
лабораторной практике, в сельском хозяйстве и для иодирования
поваренной соли. Мировое производство иода составляет
~4000 г/год7-11.
Согласно ГОСТ 545—71, технический иод 1- и 2-го сортов
должен содержать соответственно не менее 99 и 97,5 % иода и не бо-
более: 0,010 и 0,015% хлора и брома, 0,1 и 0,2% органических веществ
и 0,05 и 0,15% золы. Иод упаковывают в полиэтилено-лавсановые
мешки, помещаемые в картонные барабаны или деревянные бочки.
СЫРЬЕ
Иод широко распространенный в природе, но очень рассеянный
элемент. Содержание его в земной коре составляет Ы0~4 вес.%-
Минералы, содержащие иод, встречаются редко и в очень малых
количествах. В рассеянном состоянии иод находится повсеместно.
Содержание иода в горных породах и минералах колеблется от
1,7- Юг5 до 1,2-10~4%, в природных водах от 3• 10~5 до 150 мг/л
и в воздухе 0,001—0,002 мг/м3. Наиболее богат иодом воздух при-
приморских районов, так как в морской воде содержится 0,01—
0,04 мг/л иодидов или иодатов. Иод содержится во всех
240
Гл. VIII. Иод и его соли
растительных и животных организмах, особенно в морских (неко-
(некоторые губки содержат до 8,5% иода).
Сырьем для промышленного получения иода являются некото-
некоторые природные отложения селитры, морские водоросли и буровые
воды нефтяных и газовых месторождений. Селитроносные породы,
• содержащие иод, находятся в северной части Южной Америки
(Чили, Боливия, Перу). Содержание иода в них составляет от 0,05
до 1%. Из морских водорослей особенно богаты иодом ламина-
ламинария, фукус и филлофора. В Китае эти водоросли выращивают
специально для извлечения из них иода. Содержание иода в воз-
душносухих водорослях 0,02—0,5%. Буровые воды нефтяных ме-
месторождений— основное сырье для получения иода в СССР, Япо-
Японии, США и других странах. Содержание иода в них 10—60 мг/л
(в отдельных случаях до 150 мг/л). Буровые воды, используемые
для промышленного извлечения иода, имеют сложный солевой
состав, различные соленость и термальный режим. Получение
иода выгоднее производить из вод, богатых иодом, но все сооб-
соображения, приведенные об извлечении брома, остаются в силе и
для извлечения иода (стр. 209, 216).
Иод, попутно с бурой и другими компонентами, можно извле-
извлекать из сопочных грязей, содержащих 50—200 г/м3 иода 12.
ПОЛУЧЕНИЕ ИОДА
Извлечение иода из селитренной породы13
Маточные рассолы, получающиеся при выщелачивании при-
природной натриевой селитры из породы, содержат 5—12 г/л иода
в виде иодата и иодида натрия и кальция; их обрабатывают
бисульфитом или сульфитом натрия:
2IOg + 5HSOg = 3HSO4 + 2SO^ + Н2О +12
5Г + IOg + 6HSO4 = 6SO^ + ЗН2О + 3L,
ИЛИ
2IO
+ 2HSOg
Выделяющийся в виде илистой массы иод отделяют на фильтр-
прессах. Полученный иод-сырец содержит 50—70% иода и при-
примеси Н2О, SiO2, CaSCu и др.; после его очистки получают про-
продукт, содержащий ~99% 12.
По другому методу маточные рассолы обрабатывают сульфи-
сульфитом натрия и сульфатом меди:
IOJ + 3SO|~ = Г + 3SO|~
+ Си2+=Си12
Получение иода
241
2CuI2
Выделившийся иод реагирует с избытком сульфита
12 + SO|" + Н2О = 2Г + SOf- + 2Н+
и потому практически полностью осаждается в виде малораство-
малорастворимого иодида меди. Его промывают и перерабатывают на иод
или йодистые соли
2CuI + 3MnO2 = I2 + 2CuO + Мп3О4
2Cul + 6H2SO4 + 2Fe2O3 = I2 + 2CuSO4 + 4FeSO4 + 6HgO
2CuI + K2CO3 = 2KI + Cu2O + CO2
а соединения меди переводят в сульфат и возвращают в процесс.
Выделение иода из маточных рассолов можно производить об-
обработкой их тиосульфатом натрия и серной кислотой, но этот ме-
метод применяется редко из-за высокой стоимости тиосульфата.
Извлечение иода из селитренных щелоков на действующих за-
заводах составляет всего 60—75%. Это связано с тем, что количе-
количество иода в маточных рассолах, получаемых при переработке се-
селитры, значительно превышает потребности капиталистических
стран в иоде, и многие заводы их вообще не утилизируют.
Извлечение иода из водорослей
При сжигании морских водорослей получается 18—40% золы
(от веса воздушносухой водоросли), содержащей от 0,1 до 2%
йода132. При озолении теряется до 50% иода; для уменьшения
потерь рекомендуют предварительно обрабатывать водоросли сла-
слабым раствором щелочи или известковым молоком A—2%), в
результате чего в золе остается 95—96% иода. Расход извести
A00% СаО) составляет ~6 кг на I кг иода.
Золу выщелачивают и растворы (иногда после выпарки для
выделения из них КС1, NaCl и др.) подкисляют серной кислотой
6 целью разрушения карбонатов и сульфидов; затем для выделе-
выделения свободного иода в них вводят окислитель — хлор, хлораты,
иодаты, двуокись марганца, перманганат калия и т. д. При окис-
окислении иодатом, хлором, перманганатом выделение иода происходит
быстро и образуются мелкие кристаллы (в виде темного ила),
увлекающие большое количество примесей из раствора. Хлорат
калия выделяет иод при комнатной температуре медленно A5—
20 ч) и потому образуются крупные кристаллы, легко отделяю-
отделяющиеся от раствора и увлекающие малые количества примесей.
Маточный рассол декантируют, кристаллы иода загружают в
бязевые мешки, промывают и прессуют. Полученный иод-сырец
содержит 75—90% иода, 0,5—1% хлора, 10—25% воды и —1%
солей. Оставшийся в маточном рассоле элементарный иод @,3—
0,5 г/л) адсорбируют активированным углем. Из угля его извле-
извлекают раствором каустической соды, а полученные щелока
242
Гл. VIII. Иод и его соли
смешивают с основным раствором, идущим на выделение иода.
Иод из раствора извлекают также отгонкой паром.
Извлечение йодистых солей из золы может быть осуществлено
обработкой ее спиртом (растворимость в 90%-ном спирте при
18—25° KI — 6,5 и Nal — 31%)- После отгонки спирта остаются
почти чистые иодиды. Хотя извлечение иода достигает 95—97%,
метод не нашел применения из-за относительно большого рас-
расхода спирта.
Осуществляют и комплексную переработку водорослей с ис-
использованием органической их части, применяя для этого различ-
различные методы. Первый из них — сухая перегонка при 700—800°.
Остающийся в ретортах уголь, содержащий 35—40% углерода и
60—65% золы, обрабатывают водой, подкисленной НС1, при этом
большинство солей (в том числе иодиды) переходит в раствор;
из него выделяют соли калия, а затем иод. Выход иода состав-
составляет 85—87% от его содержания в водорослях. Полученный уголь
обладает высокой адсорбционной способностью. Из 1 г сухих во-
водорослей получается 150—160 кг угля, 70—80 кг смолы, 20—25 кг
^сернокислого аммония и 50—150 м3 горючего газа.
Второй метод комплексной переработки водорослей — сбражи-
сбраживание их в присутствии фермента; в течение 15—20 суток водо-
водоросли почти полностью растворяются, а в растворе образуются
уксусная, валериановая, пропионовая кислоты и ацетон. Для вы-
выделения кислот в форме кальциевых солей к раствору добавляют
мел. Раствор отфильтровывают на вакуум-фильтрах, выпаривают
и после выделения из него органических веществ и калийных со-
солей извлекают иод. При таком способе переработки используется
~70% сухого вещества водорослей, но стоимость иода оказывается
высокой, и потому метод требует дальнейшего усовершенство-
усовершенствования.
Третий метод — обработка водорослей водой с небольшим со-
содержанием хлорной извести или гипохлорита натрия (возможно
С12, Н2О2 и др.); при этом в раствор переходит 85—90% иода,
большая часть минеральных солей, маннит и органические азотсо-
азотсодержащие вещества. Из раствора, содержащего ~0,5 г/л иода
в виде иодноватокислых солей, извлекают иод и маннит или
только иод. Из оставшихся «отбеленных» водорослей получают
агар-агар.
ИЗВЛЕЧЕНИЕ ИОДА ИЗ БУРОВЫХ ВОД
Извлечение иода из буровых вод осуществляют адсорбцией
твердыми сорбентами, десорбцией воздухом, осаждением в виде
малорастворимых солей и экстракцией несмешивающимися с во-
водой растворителями.
При одновременном извлечении из буровых вод иода и брома,
сначала обычно извлекают иод, но имеются заводы, где вначале
Извлечение иода из буровых вод
243
извлекают бром, а потом иод23-24. В последнем случае содержа-
содержащиеся в буровой воде иодиды переходят в иодаты, которые перед
извлечением иода предварительно полностью или частично вновь
восстанавливают до иодидов. В качестве восстановителя обычно
применяют сернистый газ, расход которого в несколько раз пре-
превышает теоретический, так как в сбросных рассолах бромного
производства всегда содержатся свободный бром, хлор и их вза-
взаимные соединения, которые тоже восстанавливаются. Иногда ис-
используют и другие восстановители.
Адсорбция иода твердыми сорбентами
Для адсорбции иода могут быть использованы активирован-
активированный уголь, иониты, крахмал, некоторые высокомолекулярные со-
соединения и т. д.8'13-25-31. Обычно применяют активированный
уголь и иониты, обеспечивающие высокие выходы иода.
Адсорбция иода активированным углем
К подкисленной буровой воде добавляют окислитель (нитрит
натрия, хлор, иодаты, гипохлориты и иногда хлорное железо826),
в результате чего выделяется свободный иод:
21" + 2NO2 + 4Н+ = I2 + 2NO + 2Н2О
2Г+С12 = 12 + 2СГ
< 5Г + Юз + 6Н+ = 312 + ЗН2О
2Г + СЮ~ + 2Н+ = 12 + СГ + Н2О
Выделившийся иод адсорбируют активированным углем.
Насыщенный иодом уголь, называемый в производстве «иод-
уголь», промывают водой и направляют на десорбцию иода, за-
Ш ключающуюся в нагревании иод-угля с раствором едкого натра
или сульфита натрия:
312 + 6NaOH = 5NaI + Nalb3 + ЗН2О
I2 + Na2SO3 + H2O = 2NaI + H2SO4
I2 + Na2SO3 + 2NaOH = 2NaI + Na2SO4 + H2O
Полученный щелок, содержащий иодиды и немного иодатов
(иодаты в процессе десорбции частично37 восстанавливаются),
подкисляют и обрабатывают окислителем (бертолетовой солью,
иодатами, хлором и иногда бихроматами38'39) для выделения сво-
свободного иода:
6Г + СЮз + 6Н+ = 312 + СГ + ЗН2О
6Г + Cr2Cf" + 14Н+ - 312 + 2Сг3+ + 7Н2О
244
Гл. VIII. Иод и его соли
Выделившийся кристаллический иод отделяют от раствора и
отжимают на прессах или центрифугах из титана или его спла-
сплавов40-41.
Важнейшим процессом в этом способе извлечения иода яв-
является адсорбция его активированным углем42-46. В СССР при-
применяют активированный уголь КАД, получаемый из каменного
угля, и очень редко березовый уголь БАУ. Уголь марки БАУ
имеет бблыную адсорбционную емкость, чем уголь КАД, но он ме-
менее механически прочен, имеет меньший объемный вес и стоимость
его значительно выше. Перед применением активированный уголь
обрабатывают разбавленной кислотой, что уменьшает его золь-
зольность и повышает активность4Б.
Активность угля определяют по уменьшению содержания иода
в растворе йодистого калия через 30 мин и 24 ч после внесения в
раствор навески угля. Такой метод определения активности угля
дает сильно завышенные результаты (иногда в 2—2,5 раза), так
как уменьшение содержания иода в растворе происходит не только
за счет его адсорбции, но и за счет частичного восстановления 12
до I" активированным углем. Поэтому адсорбционную способ-
способность целесообразно определять по количеству иода, извлекае-
извлекаемого из угля после обработки его раствором сульфита или едкого
натра.
Уголь КАД адсорбирует иод при температуре ниже 45°, а
БАУ — при температурах ниже 60—65°. При более высоких тем-
температурах адсорбционная емкость их сильно понижается. Увели-
Увеличение содержания иода в буровой воде повышает степень насы-
насыщения угля иодом, которая на практике для угля КАД состав-
составляет от 20 до 120 г иода на 1 л угля.
Чистота и состав применяемой буровой воды являются очень
существенными факторами, влияющими на адсорбцию. При пе-
переработке огромного количества буровой воды (для извлечения
1 т иода требуются десятки тысяч м3 буровой воды) на активи-
активированном угле осаждаются почти все взвешенные в воде при-
примеси, а также адсорбируются многие вещества, находящиеся в
растворенном виде. Это приводит к снижению адсорбционной ем-
емкости угля, усложняет десорбцию иода и ухудшает его качество.
Особенно вредные примеси — соли нафтеновых кислот и другие
органические вещества, содержащиеся в буровых водах нефтяных
скважин46-47. Буровая вода, поступающая из скважин, вначале
проходит через нефтяные ловушки, а потом отстаивается в есте-
естественных или искусственных бассейнах от механических примесей.
На некоторых заводах буровую воду после отстаивания фильт-
фильтруют через песчаные, сенные или другие быстродействующие
фильтры. При большой щелочности буровой воды ее выдерживают
в открытых водоемах 10—30 дней, в результате чего содержание
Извлечение иода из буровых вод
248
бикарбонатов уменьшается на 30—60% (иногда и на 90%):
2НСО3 = СО|~ + Н2О + СО2
Для этой же цели иногда буровую воду обрабатывают извест-
известковым молоком или обожженным доломитом; образующийся кар-
карбонат осаждается на дно бассейна.
При хранении буровой воды в естественных водоемах, на дне
которых имеются грязевые отложения, в теплое время года на-
наблюдается активная бактериальная деятельность, что приводит к
накоплению легко окисляющихся веществ (H2S, HS~ и т. п.) и по-
потому извленение иода усложняется, а расход окислителя увеличи-
увеличивается. Иногда к буровой воде добавляют сернокислый алюминий,
соли железа (окисные) или известковое молоко 48>49; содержание
нафтеновых кислот в воде уменьшается при этом иногда на 80%.
Осветленная буровая вода поступает в смеситель, где ее под-
подкисляют серной или соляной кислотой, а затем обрабатывают
окислителем. При подкислении выделяются свободные нафтеновые
кислоты, всплывающие на поверхность, и потому на некоторых
заводах после подкисления вода отстаивается, а затем уже в нее
вводят окислитель.
Расход кислоты зависит от щелочности буровой воды, содер-
содержания в ней нафтенатов и применяемого окислителя. Для окис-
окисления иодидов требуется, чтобы в буровых водах, содержащих
мало нафтенатов (чистая вода), кислотность была равной 1,1—
3 мг-экв/л, а для загрязненных нафтенатами вод 5—7 мг-экв/л.
При окислении с помощью Юз, С12, СЮ" требуется меньшая ис-
исходная кислотность, а при окислении нитритами она должна быть
повышенной.
Количество вводимого окислителя регулируют таким образом,
чтобы содержание свободного иода в буровой воде составляло
92—97% от его начального содержания (в зависимости от чи-
чистоты буровой воды и концентрации I"). Расход окислителя за-
зависит от присутствия других восстановителей (H2S, органических
веществ и др.) и для чистых вод составляет 130—200% от теоре-
теоретического (по отношению к содержанию 1~), а для загрязненных
достигает 400—900%.
После смесителей буровая вода по деревянным или асбоце-
асбоцементным желобам или трубам поступает в систему адсорберов —
деревянных или железобетонных, защищенных кислотоупорным
покрытием резервуаров с ложным дном, на котором помещается
слой активированного угля. Полезная емкость адсорбера 4—20 м3
при заполнении его объема углем на 60—80%. Адсорберы уста-
устанавливают группами по 3—4 шт., при этом осуществляется про-
противоток— через свежий уголь протекает буровая вода, прошедшая
другие адсорберы, а через наиболее насыщенный уголь —свежая
246
Гл. VIII. Иод и его соли
буровая вода. В зависимости от чистоты, солености и температуры
буровой воды, а также от качества применяемого угля скорость
протекания ее через адсорберы устанавливают от 2 до
4,5 мг1(м2'Ч), а количество свободного иода на сбросе не более
0,5—1,5 мг/л.
Насыщенный иод-уголь после промывки его водой передают
гидротранспортом или вручную на десорбцию 50~52. Для десорбции
иод-уголь загружают в стальные котлы, обогреваемые глухим или
острым паром. Вначале иод-уголь промывают горячей водой для
удаления солей и кислоты, а потом нагревают до 90—102° с 10—
1Б% раствором едкого натра. Полученный раствор сливают и
иод-уголь неоднократно промывают промывными растворами от
предыдущих операций, а затем пресной водой.
При наличии на иод-угле большого количества адсорбиро-
адсорбированных нафтеновых кислот, отмывку иода иногда производят рас-
раствором сульфита натрия, который с ними практически не взаимо-
взаимодействует53. Но так как при взаимодействии иода с сульфитом
образуется эквивалентное количество кислоты (стр. 243), то для
избежания разрушения стальной аппаратуры к раствору суль-
сульфита добавляют едкий натр или соду. В полученные после де-
десорбции растворы, содержащие 20—35 г/л иода, после отстаивания
от частиц гипса, угля и других веществ добавляют сернокислый
глинозем или закисленную серной кислотой глину54 для выделе-
выделения нафтеновых кислот. При этом осаждается шлам, с которым
теряется некоторое количество иода (при работе с загрязненными
буровыми водами потери иода достигают 5%)-
Очистку растворов от нафтеновых кислот можно осуществлять
высаливанием их поваренной солью в кислой среде. Так как в
растворах помимо иодидов содержатся иодаты, то при создании
кислой среды может выделяться свободный иод. Во избежание
его потерь иодаты в этом случае предварительно восстанавливают
сульфитом натрия.
Выделение иода из очищенного раствора ведут в кристаллиза-
кристаллизаторах, изготовленных из дерева, кирпича или стали, футерован-
футерованных кислотоупорным материалом. Раствор подкисляют и вводят
в него окислитель55. При окислении бертолетовой солью кислот-
кислотность раствора должна быть не ниже 0,7—1 н., а при применении
других окислителей значительно ниже. Применение в качестве
окислителя иодата ускоряет выделение иода в 2—3 раза по срав-
сравнению с бертолетовой солью, улучшает его качество и снижает
расход кислоты.
В маточном растворе после кристаллизации остается 0,3—
1,2 г/л свободного иода, который извлекают активированным уг-
углем. Выделившийся в кристаллизаторе иод отфильтровывают и
полученную иод-пасту, содержащую 70—85% иода, заворачивают
в матерчатые салфетки или мешки и отжимают на гидравлических
Извлечение иода из буровых вод
247
прессах под давлением 450—600 ал. Лепешки иода-сырца весом
5—10 кг упаковывают в деревянные бочки.
Этот метод получения иода-сырца трудоемок и тяжел вслед-
вследствие токсичности перерабатываемого материала56. Поэтому по-
постепенно переходят на непрерывную кристаллизацию иода57-58 и
заменяют отжим иод-пасты на прессах центрифугированием59'61
или сушкой при 52°62. Реактор-кристаллизатор непрерывного дей-
действия представляет собой вертикальный аппарат типа «труба в
трубе». В наружную трубу сверху поступают реагенты. Образую-
Образующаяся суспензия кристаллов иода поднимается по внутренней
трубе и сливается в непрерывно действующий отстойник. Сгущен-
Сгущенная суспензия подается на центрифугу. Часть маточного раствора
направляют на подкисление буровой воды. Съем иода с 1 л объ-
объема непрерывно действующего реактора составляет 2,5—3,5 кг/ч,
а в реакторе периодического действия всего 0,01—0,02 кг/ч.
Активированный уголь после десорбции из него иода, про-
промывки и замачивания в кислоте вновь возвращают на адсорбцию.
Уголь можно использовать не больше 4—6 раз, затем его подвер-
подвергают реактивации или направляют в отвал.
Реактивацию угля63 ведут при ~700° в атмосфере водяного
пара в стальных или чугунных ретортах. В процессе реактивации
органические вещества разрушаются, а гипс восстанавливается
до сульфида кальция, который затем удаляется из охлажденного
угля при обработке его разбавленной соляной кислотой. Актив-
Активность реактивированного угля составляет 35—60% от активности
свежего угля.
На некоторых заводах адсорбцию иода проводят не путем
фильтрации буровой воды через слой угля, а взмучиванием его
в резервуарах с мешалкой64. Это упрощает дозировку кислоты
и окислителя, иод-уголь получается более чистым, так как не за-
гипсовывается и не замазывается осадками (глиной, песком). При
этом также сильно уменьшается количество активированного угля,
находящегося в процессе производства, т. е. объем незавершен-
незавершенного производства. Недостатком этого способа является прерыв-
прерывность процесса, увеличение объема адсорбционной аппаратуры в
2—3 раза по сравнению с фильтрацией и большой расход электро-
электроэнергии *.
Выход иода в товарный продукт составляет 75—92%. На 1 г
иода-сырца расходуют: 4—65 т серной кислоты, 1—3,6 т нитрита
натрия, 1,1—2 т едкого натра, 0,23—0,27 т бертолетовой солиг
0,2—0,5 т сульфита натрия, 0,5—2 т сернокислого глинозема,
1,2—4 т активированного угля, 8—14 г топлива (условного); рас-
расход электроэнергии составляет 5000—8000 квт-ч. Широкие пре-
пределы значений расходных коэффициентов обусловлены различным
• О контроле йодного производства см.
248
Гл. VIII. Иод и его соли
качеством буровой воды. Для уменьшения расхода кислоты на
28—35% можно периодически через активированный уголь филь-
фильтровать отработанную буровую воду. При этом происходит ней-
нейтрализация щелочности угля и адсорбция им Н+.
Применение в качестве окислителя для буровой воды иодата
или хлора вместо нитрита натрия дает возможность снизить расход
кислоты в 2—3 раза71.
Практический интерес представляет адсорбция иода из буровых
вод углем без предварительного окисления I" до Ь72. Из сравни-
сравнительно чистой буровой воды при начальном содержании в ней кис-
кислоты 1,4—3,8 мг-экв/л адсорбция ниже 28° идет на 92—100%.
88—96% адсорбированного иода находится на угле в молекулярном
виде, хотя он и поступает в виде I"; это объясняют образованием в
активированном угле при низких значениях рН перекиси водорода
(за счет адсорбции кислорода).
Предложены электрохимический, термический и паротермиче-
ский методы десорбции иода с угля73'7*. Электрохимическая75 де-
десорбция основана на пропускании электрического тока через на-
нагретый электролит (растворы NaCl, Na2SO4 и пр.), в котором на-
находится иод-уголь; иод переходит в раствор, а затем, благодаря
повышенной температуре, возгоняется. При этом возможно достичь
100% извлечения.
Иод может быть десорбирован с иод-угля нагреванием его без
доступа воздуха при 200—400°. В зависимости от температуры, чи-
чистоты иод-угля и степени его насыщения степень десорбции состав-
составляет 72—83%. Степень десорбции может быть увеличена до 92%„
если процесс вести под вакуумом. Иод, остающийся в угле в виде
иодида, может быть отмыт водой.
Паротермический метод десорбции76-78 основан на пропускании
перегретого водяного пара через нагретый до 350—400° иод-уголь.
При этом десорбируется 55—96% иода, а остальной иод переходит
б иодид благодаря присутствию в угле восстановителей. Степень
десорбции зависит от насыщения угля иодом. При содержании иода
в угле 220 г/л степень десорбции 92—96% и расход пара ~7 /на
1 г иода, а при 50 г/л десорбируется 55% и расход пара возра-
возрастает до 76 т/т иода.
Для улучшения качества иода и освобождения его от органи-
органических примесей (без дополнительной очистки иода-сырца) пред-
предложена отгонка его паром из растворов после десорбции или из
иод-пасты79-80.
Выделение иода с помощью анионитов
В последнее время стали широко применять ионитный метод
извлечения иода из природных вод (СССР, Япония) 81~88. Этот ме-
метод в отличие от угольно-адсорбционного дает возможность пере-
перерабатывать щелочные воды (рН —8), содержащие 7—120 мг/л I",
Извлечение иода из буровых вод
249
до 150 г/л С1- и значительные количества органических веществ, и
имеющие температуру 5—60°. Вода предварительно очищается от
соединений железа и, в отдельных случаях, от органических ве-
веществ, отстаивается или фильтруется (стр.244),затем хлорируется
для перевода 1~ в 12 и пропускается через один или несколько ад»
сорберов с анионитом. Окисление хлором осуществляют, в зависи-
зависимости от экономических соображений, или после предварительного
подкисления воды серной кислотой до рН = 2—3, и тогда потери
иода на этой стадии производства составляют 1—4%, или без
подкисления, причем потери иода достигают 30—40%. Расход
хлора в первом случае составляет 120—130% от стехиометриче-
ского, во втором — 300—500 % •
В качестве анионита в Японии применяют амберлит ША-400
или IRA-900 (тип I) с основным характером, а в СССР — смолу
АВ-17 в С1~ форме. Насыщение амберлита IRA-400 иодом при
содержании в исходной воде —100 мг/л I" достигает 700—800 г
на л смолы; насыщение анионита АВ-17 при концентрации I" в
исходной воде 27—35 мг/л составляет 180—300 г/л. Потери иода
на стадии сорбции 1—7%. По окончании сорбции, для «докрепле-
ния» иод-ионита через него пропускают маточный рассол, остаю-
остающийся после выделения иода, и слабые растворы (менее 2 г/л иода)
от предшествовавшей десорбции; одновременно в них вводят
5%-ный раствор нитрита. Десорбцию ведут при 50—60°, обрабаты-
обрабатывая вначале иод-ионит раствором едкого натра A00—120 г/л) или
сульфита натрия G0—100 г/л), а затем 10% раствором хлорида
натрия; иногда применяют-смешанные растворы NaOH + NaCl или
Na2SO3+NaCl. Полученные растворы, содержащие 20—25 г/л иода,
направляют на выделение иода, а слабые (промывные) растворы —
на доукрепление иод-ионита.
После десорбции иода раствором едкого натра выделение кри-
кристаллического иода из концентратов производят подкислением их
серной кислотой, а растворы, полученные при десорбции с помощью
сульфита, хлорируют, причем содержание I" понижается в них
до 0,6 г/л. Отделение кристаллов иода, их промывку и укупорку
производят так, как описано выше (стр. 244). Потери на этой ста-
стадии составляют 1,5—2%- Общий выход иода 85—92% (при подкис-
лении воды).
На 1 т иода-сырца расходуется: 4—12,5 т 75%-ной серной кис-
кислоты, 0,7—0,75 т хлора, 0,8—0,9 г сульфита натрия A00%
Na2SO3), ~0,5 т хлорида натрия, 0,05—0,08 т нитрита натрия,
0,02—0,1 л ионита.
Десорбция иода воздухом
Извлечение иода десорбцией воздухом основано на том
же принципе, что и извлечение брома, и осуществляется в той
же аппаратуре (стр. 213), но здесь исключаются башни для
250
Гл. VIII. Иод и его соли
хлороочистки2- 69> 89~97. Для извлечения иода по этому методу не-
необходимо содержащийся в буровой воде иодид окислить в свобод-
свободный иод. Для окисления иодидов в кислой среде буровую воду
вначале подкисляют серной или соляной кислотой до рН=2—3,5.
Затем вводят окислитель — хлор или растворы гипохлоритов; ни-
нитрит натрия не применяют, так как образующиеся в процессе ре-
реакции окислы азота также десорбируются воздухом. Степень окис-
окисления обычно составляет 95—98%.
При высокой щелочности буровой воды, обусловленной нали-
наличием НСО3~, во избежание большого расхода кислоты окисление I",
в отличие от Вг~, можно вести и в щелочной среде. Растворы гипо-
гипохлоритов натрия или кальция дают более высокую степень окисле-
окисления, чем хлор. Высокощелочные воды Бакинского района (щелоч-
(щелочность 30—40 мг-эке/л), содержащие большое количество органи-
органических веществ, окисляются гипохлоритом на 89—91%, а хлором
на 60—70%. Повышение температуры уменьшает степень окисле-
окисления I", так как при этом увеличивается скорость гидролиза 12 и
Происходит разрушение бикарбонатов, а образовавшиеся карбо-
карбонаты взаимодействуют с иодом. При окислении растворами гипо-
гипохлоритов необходимо хорошее перемешивание и малая концентра-
концентрация окислителя (не более 2 г/л активного хлора), так как иначе
может происходить локальное переокисление Г до Юз- Кроме
того, окисление гипохлоритами идет сравнительно медленно—мак-
медленно—максимальная степень окисления достигается через 2—3 мин.
Окисленная буровая вода поступает в десорбцпонную башню,
где растворенный иод извлекается встречным потоком воздуха. Де-
Десорбцию осуществляют в башнях с насадкой при скорости воздуха
t),5—1 м/сек (считая на полное сечение башни), но ее можно про-
проводить и в пенных аппаратах98. Количество затрачиваемого воз-
воздуха зависит от давления пара иода над рассолом и будет тем
меньше, чем больше содержание иода в буровой воде, выше тем-
температура и ниже ее соленость (рис. 71) (или, наоборот, при очень
высокой солености).
Содержание иода в уходящем из десорбера воздухе колеблется
•от 0,05 до 0,25 мг/л, при этом расход воздуха в 1,1—1,8 раза боль-
больше теоретического (вычисленного на основании коэффициента рас-
распределения09). Степень десорбции обычно составляет 92—97%.
В получающейся иодо-воздушной смеси помимо иода содер-
содержатся и Другие вещества, обладающие заметным давлением пара
над буровой водой. При извлечении иода из кислой буровой воды в
иодо-воздушной смеси содержатся двуокись углерода (из-за раз-
разрушения СОз" и НСОз) и нафтеновые кислоты. Содержание СО2
зависит от количества карбонатов в исходной буровой воде; содер-
содержание нафтеновых кислот достигает 0,5 кг на 1 кг иода. При из-
извлечении иода из щелочной буровой воды количество СО2 и
Извлечение иода из буровых вод
251
нафтеновых кислот в ибдо-воздушной смеси относительно неве-
невелико.
Извлечение иода из иодо-воздушной смеси производят с по-
помощью сернистого газа б9-10° в присутствии паров воды:
SO2 +12 + 2Н2О = 2HI + H2SO4
Образующуюся смесь кислот улавливают в бяганях с насадкой
из керамических колец, стеклянной ваты и др. Для уменьшения
потерь иода насадка должна быть влажной и потому башни перио-
периодически или непрерывно орошаются циркулирующим раствором
смеси кислот. Плотность орошения не превышает 1 мъ/(м2-ч).
Получающаяся смесь кислот HI и H2SO4 легко окисляется кис-
кислородом воздуха с выделением свободного иода н потому во избе-
избежание его потерь расход сернистого газа увеличивают до 170—250%
от теоретического. Содержание свободного иода в циркулирующем
растворе не должно превышать 0,1 г/л. Концентрация получаю-
получающейся смеси кислот (80—120 г/л HI, 55—90 г/л H2SO4) зависит от
давления водяного пара в иодо-воздушной смеси.
Степень улавливания иода достигает 98%. Повышение темпера-
температуры и концентрации смеси HI и H2SO4 в адсорбционной башне
выше указанного предела увеличивает потери иода из-за роста дав-
давления пара йодистого водорода над раствором. Содержащиеся в
иодо-воздушной смеси нафтеновые кислоты почти не улавливаются
сорбентом и по данным, полученным на Нефтечалинском заводе,
содержание их в растворе по отношению к иоду равно 2—4: 1000,
тогда как в газовой фазе это соотношение равно 15—45:100.
В дальнейшем иод выделяют из раствора путем введения окисчи-
телей (хлора, иодата, бертолетовой соли). Выход иода составляет
95—98%. Получающиеся после выделения иода маточные рассолы,
содержащие 25—35 г/л НС1 и 80—120 г/л H2SO4, используют для
подкисления исходной буровой воды или для других целей.
Сорбент может быть также использован для получения иоди-
стоводородной кислоты или ее солей. Прямое разделение HI и
H2SO4 дистилляцией невозможно, так как при нагревании этой
смеси до 30—60° (в зависимости от концентрации раствора) идут
следующие реакции:
2HI + H2SO4 = I2 + SO2 + 2Н2О
6HI + H2SO4 = 3I2 + S + 4Н2О
8HI + H2SO4 = 4I2 + H2S + 4Н2О
Для получения чистой иодистоводородной кислоты из смеси
кислот101 последнюю предложено обрабатывать хлоридом бария,
в результате чего выделяется осадок сульфата бария, а в растворе
остается смесь НС1 и HI. Сульфат бария отфильтровывают и про-
промывают— он является побочным продуктом. Водный раствор-
НС1 и HI подвергают затем фракционной дистилляции. В первой
2Б2
Гл. VII/. Иод и его соли
Извлечение иода из буровых вод
253
фракции, кипящей ниже 125—126°, содержится практически весь
НС1, 0,002—0,04% h и 0,3—0,9% HI. Кубовый остаток имеет состав
56—56% HI, 0,1—0,2% HCI и кипит выше 126° (азеотроп HI—Н2О
кипит при 127° и содержит 57% HI). Выход HI достигает 96—98%.
Степень извлечения иода из подкисленных буровых вод состав-
составляет 77—92%, а из щелочных — 72—84 %.
Производство иода десорбцией воздухом имеет ряд преиму-
преимуществ по сравнению с угольным методом: возможность использо-
использования загрязненных и
высокощелочных буровых
вод, меньшая трудоем-
трудоемкость и простота автомати-
автоматизации процесса 6Б-69- "•1о2,
малый объем незавер-
незавершенного производства;
поэтому метод десорбции
постепенно вытесняет
угольный.
При работе на тер-
термальных водах с повы-
повышенной концентрацией
иода десорбция его воз-
воздухом является не менее
ф
V
л
\
^*e_—
—-
0 5 Ю 15
Концентрация водного раствора Н1,%
20
Рис, 73. Коэффициент распределения иода
между газовой Лазой и водным раство-
раствором HI при 18-20°.
у
эффективным методом,
чем сорбция ионообмен-
ионообменными смолами. Для низ-
низкотермальных вод с малым содержанием иода в большинстве
случаев целесообразнее использовать ионообменный метод.
Извлечение иода из природных вод десорбцией воздухом позво-
позволяет получать йодистые соли, минуя стадию получения иода. Для
этого в качестве сорбента, при улавливании иода из газовой фазы,
могут применяться или растворы соответствующих иодидов (если
они растворимы) 10°, или растворы иодистоводородиой кислоты шз.
При взаимодействии h с растворами, содержащими I", образуются
полииодиды—Ь, Ig, в результате чего давление пара иода над
раствором резко понижается (ср. рис. 73 и 71). Затем полииодиды
переводят в иодид действием сероводорода 104 или соответствую-
соответствующего основания совместно с перекисью водорода 10*:
Ц + 2ОН- + Н2О2 = ЗГ + 2Н2О + О2
Указанные схемы прямого получения йодистых солей по лабо-
лабораторным данным дают благоприятные технико-экономические по-
показатели.
Имеются указания3-106-,107 на возможность использования для
абсорбции иода из газовой фазы водных растворов тиосульфата,
сульфита натрия, щелочей, смеси сульфита с содой. Следует учи-
учитывать, что содержащиеся в иодо-воздушной смеси нафтеновые
кислоты также улавливаются растворами этих сорбентов, имею-
имеющими щелочную реакцию.
Осаждение иода в виде малорастворимых солей
Выделение иода из буровых вод возможно на основе образова-
образования малорастворимых иодидов меди (стр. 241), серебра и ртути.
Осаждение иода в виде иодида меди целесообразно вести из буро-
буровых вод, в которых содержание С1~ не превышает 4—5%, так как
при более высоких концентрациях хлоридов образуются раствори-
растворимые комплексные соединения, что приводит к увеличению потерь
меди и иода.
Извлечение иода в виде иодида серебра осуществляют по двум
схемам: или к буровой воде добавляют раствор нитрата серебра,
или буровую воду фильтруют через слой хлорида серебра. Полу-
Получаемый иодид серебра отделяют от буровой воды, промывают, а за-
затем восстанавливают железом, цинком или электрохимически до
металлического серебра. Из регенерированного серебра получают
соответствующие соли, которые вновь используют в производстве.
Остающиеся после выделения металлического серебра растворы,
содержащие иодид железа или цинка, перерабатывают в соли иода
или из них выделяют иод путем введения окислителя.
Извлечение иода из буровых вод осаждением его в виде Cul и
Agl почти всюду прекращено из-за сравнительно больших потерь
меди и серебра (и иода).
Для извлечения иода в виде иодида ртути108 к буровой воде
добавляют соли одновалентной ртути. Выделяющийся осадок гало-
галогенных соединений ртути отделяют от буровой воды и обрабаты-
обрабатывают сероводородом. Раствор HI и НС1 отфильтровывают и выде-
выделяют из него иод введением окислителя. Осадок, содержащий со-
соединения ртути, регенерируют. Выход иода достигает ~90%.
Экстракция иода несмешивающимися с водой растворителями
Извлечение иода по этому методу возможно вести как из кис-
кислых, так и щелочных вод106-112. В качестве экстрагентов могут
применяться вещества, очень мало растворимые в воде, химически
не взаимодействующие с иодом и окислителями, вводимыми для
перевода I- в 1% а также обладающие высоким коэффициентом
распределения для иода К, т. е. отношением концентрации иода в
растворителе к концентрации иода в водном растворе при рав-
равновесии 113-иб. Коэффициент распределения для одного и того же
254
Гл. VIII. Иод и его соли
экстрагента зависит от температуры, состава буровой воды, ее со-
солености и концентрации иода. Последнее связано с изменением
растворимости иода и образованием комплексных ионов 1з, Is- Для
различных экстрагентов коэффициент распределения изменяется в
широких интервалах. Так, при 20° для буровых вод Бакинского
района с плотностью 1,058—1,093 г/см3, содержащих 20—35 мг1~в
1 л и имеющих щелочность 12—16 мг-экв/л, коэффициент распре-
распределения для дихлорэтана равен 80—85, керосина 30—45 и четырех-
хлористого углерода 35—45. Для системы иод — керосин — чистая
вода /(=120.
Извлечение иода из буровой воды осуществляют в эмульгато-
эмульгаторах, в которых обе жидкости тщательно перемешиваются, а затем
разделяются отстаиванием, при этом основная масса иода перехо-
переходит в органический растворитель. Для 95—97% извлечения иода
теоретически достаточно 2—3 экстракций, осуществляемых проти-
противотоком. Например, при соотношении между буровой водой и экс-
трагентом, равном 10: 1, и при К=50 извлечение иода после одно-
однократной экстракции достигает 83,3%, после двукратной 96,8%,
после трехкратной 99,4%. Из органического растворителя иод из-
извлекают обработкой его раствором сульфита натрия, в результате
чего иод переходит в водный раствор в виде иодида, а экстрагент
после промывки водой возвращается в процесс.
При наличии в буровой воде значительных количеств органиче-
органических веществ (особенно нафтенатов) этот метод становится мало-
малорациональным, так как в процессе экстракции образуются стойкие
эмульсии, что приводит к большим потерям экстрагента и иода,
увеличению объема аппаратуры и затратам реагентов на разруше-
разрушение эмульсии.
В промышленности в качестве экстрагента применяют керосин
или его смесь с петролейным эфиром, которые при этом частично
иодируются, что приводит к потере 15—20% иода. Для уменьшения
потерь иода и снижения расхода окислителя можно последний вво-
вводить не в буровую воду, а в экстрагент. В качестве экстрагента
целесообразно применять вещества не иодирующиеся, но по стои-
стоимости и другим показателям близкие к керосину, как, например,
дихлорэтан и др.
Электрохимические методы извлечения иода
В основе этих методов лежит электролиз буровой воды, содер-
содержащей I", с выделением иода на медном или угольном ано-
анодов, ne-ii8_ pja медном аноде иод выделяется в виде иодида меди.
Выделение иода на угольном аноде может осуществляться по не-
нескольким схемам. Так, анодом может служить активированный
уголь, который и адсорбирует выделившийся иод; по другим схе-
схемам выделяющийся на аноде иод растворяется в буровой воде, а
Очистка иода-сырца
255
затем извлекается из нее активированным углем или выдуванием
воздухом. Электролитические методы не нашли промышленного
применения из-за высокого расхода электроэнергии и трудности
технологического оформления.
ОЧИСТКА ИОДА-СЫРЦА
Иод-сырец обычно содержит большее количество примесей орга-
органических веществ и минеральных солей, чем это допускается ГОСТ,
и поэтому подвергается очистке. Для отделения водорастворимых
солей лепешки иода промывают, для отделения остальных приме-
примесей иод сублимируют или обрабатывают сырец концентрированной
серной кислотой.
Наиболее широко применяемый метод очистки — сублимацию
осуществляют в стальных, чугунных или керамических ретортах,
обогреваемых топочными га-
зами. Пары иода кристалли-
кристаллизуются на стенках холодиль-
холодильников, представляющих собой
систему керамических или ас-
асбоцементных труб, охлаждае-
охлаждаемых воздухом. Для уменьше-
to
0,5
J
70°
N
50°
25°
• —.
^\
,
**- -"¦
—
*
+
'
20
Концентрация
60
100
Рис. 74. Растворимость иода в серной
кислоте.
ния влажности получающегося
иода трубы-холодильники
укладывают с небольшим на-
наклоном, а в стыках труб оста-
оставляют отверстия для стока во-
воды. Иногда иод-сырец перед
сублимацией смешивают с 8—
12% негашеной извести. Для
получения крупных кристал-
кристаллов, легко отделяемых от стенок холодильника 119, температура па-
паров иода в зоне конденсации не должна быть выше температуры
плавления иода A13,6°).
На некоторых заводах возгонку иода производят пропусканием
перегретого водяного пара (ПО—120°) через слой иода-сырца.
Пары иода и несконденсировавшиеся пары воды охлаждают в пря-
прямоточном холодильнике. Полученную пульпу, состоящую из кри-
кристаллов иода и воды, фильтруют через бязь и прессованные уголь-
угольные плитки, а затем кристаллы иода отжимают на гидравлическом
прессе. Расход пара 3,6 мгкал на 1 т иода.
Для очистки иода-сырца применяют также плавление его под
слоем серной кислоты при 120—150° или промывку его 90—93%
серной кислотой — при этом содержащиеся в сырце органические
вещества обугливаются 120>121. Использование более концентриро-
концентрированной кислоты не улучшает очистки, но увеличивает потери иода
256
Гл. VII/. Иод и его соли
из-за повышения его растворимости (рис. 74) 122. Обработанный
серной кислотой иод измельчают и промывают водой для удале-
удаления включений и серной кислоты. Потери иода составляют 2—3%, |
а расход серной кислоты 2—3 т на I т иода. Отработанную сер-
серную кислоту используют на других стадиях получения иода.
Для получения особо чистого иодаm иод-сырец возгоняют в
вакууме или перекристаллизовывают его из раствора в четырех-
хлористом углероде; захват примесей предотвращают с помощью
стеклянной ткани.
ПОЛУЧЕНИЕ СОЛЕЙ ИОДА
Получение йодистых солей прямым взаимодействием иода с рас-
растворами гидроокисей в промышленности не применяется из-за труд-
трудности отделения от иодатов. Из диаграмм растворимости в ейсте-
20
X
" /?—¦
L kio,
KI
10
20
30
40
30
60
ТО
Рис. 76. Изотермы растворимости в системе KI—КЮ3— Н2О.
мах KI—КЮз—Н2О и Nal—NaIO3— H2O (рис. 75 и 76) видно, что
в чистом виде можно выделить только иодат калия или натрия124.
30 40
Nal,%
70
Рис. 76. Изотермы растворимости в системе Nal—NalOs—Н2О.
Иодиды же всегда будут загрязнены иодатами, и для удаления
последних необходимо применять дополнительные операции.
По наиболее распространенному методу для получения иоди-
иодидов 125>126 приготавливают раствор йодисто-йодного железа Fe3ls.
Последний получают при взаимодействии железных стружек с
Получение солей иода
257
иодом в присутствии воды: 3Fe + 4I2 = Fe3I8. Затем к раствору
резЬ прибавляют растворы гидроокисей или карбонатов:
Fe3I8 + 8М0Н = Fe3O4 + 8MI + 4Н2О
Fe3I8 + 4М2СО3 = Fe3O4 + 8MI + 4СО2
где М-К, Na,NH4.
Получение иодидов ведут в чугунных или стальных реакторах с
мешалкой и паровой рубашкой. Раствор карбоната или гидроокиси
нагревают до кипения, а затем медленно прибавляют раствор иоди-
стоиодного железа. После 3—6-часового нагревания, с целью по-
получения легко фильтрующего осадка Fe3O4, последний отделяют
декантацией или фильтрованием, отжимают на фильтрпрессах и
промывают водой до практически полного извлечения иодидов.
Первые промывные воды смешивают с основным раствором и вы-
выпаривают до получения насыщенного раствора, из которого выкри-
выкристаллизовывают иодид. Кристаллы иодида центрифугируют, про-
промывают и сушат при ~ 100°. При получении иодида аммония,
последний частично разлагается и окрашивается иодом, а потому,
реакцию ведут при избытке аммиака в растворе, а окрашенные
кристаллы промывают небольшим количеством спирта или отбе-
отбеливают пропусканием через них паров аммиака. На некоторых за-4
водах вместо Fe3ls готовят раствор йодистого железа FeF-- i* ре-
реакцию ведут по уравнению:
3FeI2 + 12 + 8МОЧ = Fe3O4 + 8MI + 4Н2О
Иодиды также получают взаимодействием иодо-воздушной сме-
смеси 10°-105 (стр. 249) или иода127 со щелочными растворами в при-
присутствии перекиси водорода при 8—10° или в присутствии серо-
сероводорода:
I2 + 2MOH + H2S = 2MI + 2Н2О + S
Выход иодида (в пересчете на иод) составляет 96—98%. Кон-
Концентрации исходных растворов КОН, КгСО3 и Na2CO3, а также-
оптимальные условия кристаллизации иодидов натрия и калия мо-
могут быть установлены с помощью диаграмм растворимости в си-
системах Nal—Na2CO3—NaHCO3—H2O, KI—К2СО3—КНСО3—Н2О,
KI—КОН—КаСОз—Н2О (рис. 77—79)ш. Иодиды натрия и калия
высаливают карбонаты из растворов и в двойных и тройных эвто-
нических точках в пределах температур 0—25° содержание Nal
в солевой массе растворов составляет 98,3—99,3 мол. %, a KI—
87,7—91,1 мол.%. Для получения чистых иодидов карбонаты дол-
должны быть предварительно удалены из растворов добавкой Fe3I8
или HI. В системе Щ—КОН—Н2СО3—Н2О наибольшим высали-
высаливающим эффектом обладает КОН. Применяемые для получения KI
поташ и едкое кали всегда содержат соединения натрия A—5%),
9 М. Е. Лозин
Получение солей иода
259
которые накапливаются в производственных растворах и загряз-
загрязняют готовый продукт. Пути разделения KI и Nal могут быть
выявлены с помощью диаграммы растворимости в системе
KI—Nal—Н2О (рис. 80I29.130.
Перспективным является изготовление иодидов |25- '3i—134 взаи-
взаимодействием иодистоводородной кислоты (получаемой при извле-
извлечении иода из рапы десорбцией воздухом, стр. 249) со щелочами,
карбонатами или хлоридами:
М2СО3 + 2HI = 2MI + Н2О + СО2
Ло последней реакции можно производить и очистку иодидов
щелочных металлов от примесей хлоридов и бромидов, обрабатывая
70
60
10
Ч
Ч
ч
\
ч
s^SO'
"ч\
0°
2Н,О
Ю
30
Nal.
SO
70
Рнс 80. Растворимость в системе KI—Nal—HSO
при 0, 25 и 50°.
соли раствором HI при нагревании. Количество HI должно со-
соответствовать ~1,5 эквивалентам от содержащихся в солях Вг"
и С1-.
В лабораторных условиях разработан метод прямого получения
иодида натрия реактивной квалификации из иод-углей135. После
промывки иод-угля пресной водой из него извлекают иод раство-
раствором щелочи или сульфитно-содовым раствором. Полученный кон-
концентрат-выпаривают досуха и прокаливают при 350—450° для уда-
удаления органических веществ. Остающийся плав обрабатывают
небольшим количеством воды и получают раствор, содержащий
64-65% Nal, 0,2—0,3% NaCl, 0—1% NaBr и 0,03—0,05% Na2SO4.
9*
260
Гл. VIII. Иод и его соли
В этом растворе может содержаться и 0,02—0,03% нафтеновых
кислот, если не прокаливать остаток после сульфитно-содовой об-
обработки. Далее удаляют из раствора SC>4~ карбонатом бария и
добавляют иодистоводородную кислоту A50% от эквивалента Вг~+
+ С1~). Нагреванием до кипения A28°) отгоняют смесь кислот
(НС1 + НВг + Н1 + Н2О), после чего остается чистый иодид натрия.
Иодат калия получают по реакциям 136.
6КС1 + 312 + 10ШО3 = 6КЮ3 + 6HCI + I0NO + 2Н2О
• НЮ3 + КОН = 2КЮ3 + Н2О
Иодат калия может быть также получен раскристаллизацией
смеси К1 + КЮ3.
ЛИТЕРАТУРА
I. В. П. Ильинский, А. Н. Стрельникова, Труды ЛХФИ, в. 5, 1958,
стр. 32. — 2. Г. С. Клебанов, А. А. Гинзбург, Труды ГИПХ, в. 41, 1958,
стр. 8. — 3. R. F. Taylor, Chem. Eng. Sci., 10, № 1—2, 68 A969). — 4. F. N e le-
leva si 1, Chem. prumysl., 16, № 4, 197 A966). —5. Angelescu, Popelescu,
Z. phys. Chem., Abt., A156, 3041 A931).— 6. С. И. Яворский, Г. Р. Зал-
кинд, Труды ГИПХ, в. 48, 1965, стр. 10. — 6а. К- Selte, A. Kjekshus, Acta
chem. Scand., 22, № 10, 3309 A968). —7. Chem. Age, 94, № 2616, 660 A965).—
8. Г. А. Яковкин, ИБП ГИПХ*, № 11—12, 4 A967). — 9. Там же, № 13, 79
A967). — 10. W. M i 11 е г, Bull. Bur. Mines, № 630, 447 A965).
II. Г. Л. К о р е н ь к о в, Н. А. Устинова, Горнохимическое сырье зару-
зарубежных стран, Изд. «Химия», 1965, гл. 5. — 12. В. П. Григорович, Экономика
и культура Крыма, № 1—2 A933). — 13. Б. П. Денисович, Иод и его произ-
производство, ГОНТИ, 1938. — 14. Б. П. Пентегов, Изв. Тихоокеанской научно-про-
научно-промысловой станции, 3, 5 A929). —15. Н. Д. Аверкнев, Хим.-фарм. вестн.
№ 3—4 A936).— 16. Н. Д. Аверкиев, ЖХП, 7, № 4 A930); № 8—9 A930).—
17. А. Мамуровский, Хим. и соц. хоз., № 9 и 10 A931). —18. А. Марци-
новский, Северн, хоз. (Архангельск) № 2 A930). —19. Ф. К- Ревза, БТИ
ГИПХ**, № 4, 38 A957). — 20. В. П. Ильинский, К. И. Булгакова,
Там же, Хя 4, 42 A957).
21. Rev. prod, chim., № 1314, 546 A963). — 22. Chem. Eng., 42, № 12, 98
A964). — 23. В. П. Ильинский, К. И. Булгакова, БТИ ГИПХ, № 6, 17
A958). — 24. К. И. Булгакова, В. П. Ильинский, Труды ЛТИ им. Лен-
Ленсовета, в. 41, 1957, стр. 145. —25. А. П. Харин, ЖФХ, 30, № 8, 1776 A956).—
26. J. W. Watson, Ind. Eng. Chem., 47, № 5, 1053 AЭ55). — 27. П. В. Афа-
Афанасьев, ЖПХ, 12, № 7, 1006 A935). — 28. Б. П. Денисович, Там же, 11,
№ 1, 15 A934). — 29. О. Ю. Магидсон, Там же, 12, № 4 A935).—30. О. Ю. М а-
гидсон, Там же, 4, № 1 A927).
31. В. И. Ксензенко, Д. Е. С т а с и н е в и ч, Технология брома и иода
Госхимиздат, I960. — 32. О. И. Андреева, Б. Б. Васильев, ЖХП, 14, № 15,
1097 A937). — 33. С. В. Горбачев, И. А. Касаткина, Бюлл. Н.-и. хим.-
фарм. ин-та, № 2, 1930. —34. К Seubert, A. Dorrer, Z. anorg. Chem., 15,
339 A894). — 35. В. П. Толстяков, Сборник статей по общей химии. Изд. АН
СССР, в. 2, 1953, стр. 1249. — 36. С. И. Яворский, Г. Р. Залкинд, БТИ
ГИПХ, № 15, 26 A962); № 16, 15 A962); Изв. АН Туркм. ССР, № 2 и 3 A962);
Труды ГИПХ, в. 48, 1965, стр. 3 и 10. — 37. И. Ш. М ас те р о в о й. Б. М. Жеп-
Литература
261
* Иодо-бромная промышленность, Сб. Обзорной информации ГИПХ.
** Бюлл. техн. информации ГИПХ по нодо-бромной промышленности.
лннский БТИ ГИПХ, № 9, 25 (I960).— 38. A. Scanzo, Gazz. chim. ital., 63»
58 A933). — 39. N. Schlundt, J. Am. Chem. Sot, 17, 754 A895).— 40. Г.Г.По-
чепцов а, С. В. Тимченко, ИБП ГИПХ, № 5—6, 34 A965).
41. Г. Г. По чепцов а, С. В. Тимченко, Там же, № 5—6, 3 A965).—
42. А. Н. Алексеева, Автореф. канд. дисс, ЛТИ им. Ленсовета, 1949.—
43. А. Г. Байчиков, ЖПХ, 15, № 4, 228 A942). — 44. В. И. Верещагина,
А. И. Харин, Изв. вузов. Хим и хим. технол., 3, № 6, 1011 A960).—
45. И. Ш. Мастеровой, БТИ ГИПХ, № 13, 14 A961). — 46. С. П. Те в ос о в,
О. В. Андрейко, ДАН АзССР, № 2 A953). — 47. Ф. Ф. Герман, ИБП
ГИПХ, № 5—6, 9 A965). — 48. С. И. Яворский, К- И. Петрова, Труды
ГИПХ, в. 48, 1965, стр. 68. —49. М. А. Шенкер, Л. М. К о й ф м а н, Там же,
стр. 76; БТИ ГИПХ, № 15, 3 A962); ИБП ГИПХ, № 1. 38 A964).—
50. С. И. Яворский, В. Г. Тимофеева и др., ИБП ГИПХ, № 1, 7 A964).
51. Ф. Ф. Герман, Н. Н. Лысенко, БТИ ГИПХ, № 15, 37 A962).—
52. И. Ш. Мастеровой, Н. П. Якубовский, Там же, № 15, 42, 46
A962). — 53. С. И. Яворский, Э. Я. Фарфель, Труды ГИПХ, в. 48, 1965,
стр. 56.-54. С. И. Яворский, Е. Ф. П а ш н е в а, ИБП ГИПХ, № 5—6, 14
A965). — 55. Г. П. Третьякова, В. С. Люткевич. Там же, № 5—6, 5
A965). — 56. Правила и нормы техники безопасности и промышленной санита-
санитарии для проектирования, строительства и эксплуатации производства воды по
угольному методу, Госхимизцат, 1961. — 57. И. П. Якубовский, ИБП ГИПХ,
№ 5—6, 22 A965). — 58. Ф. Ф. Герман, Там же, № 13, 20 A967).—
59. П. В. 3 и м н и ц к и й, Там же, № 13, 25 A967); А Ф. Б о г и ч, Н. К. Г у з е в,
Там же, № 13, 27 A967), — ЬО. А. К. Гузев, Ф. Ф. Герман, Там же, Хя 5—6,
25 A965).
61. В. Н. Волховская, Ф. Ф. Герман, Там же, № 5—6. 37 A965).—
62. М. С. Го берм а н, Л. С. Котелков, Там же, № 5—6, 38 и 45 A965).—
63. А. X. Панах-3 аде, М. И. Попов, БТИ ГИПХ, № 4, 26 A959).—
64. R. Robertson, Ind. Eng. Chem., 26, 36 A934). — 65. И. Е. Орлов, Ме-
Методы анализа рапы, буровых вод и контроль производства иода и брома, ГОНТИ,
1939. — 66. В. И. Ильинский, К. И. Булгакова, Труды ГИПХ,
в. 41, 1958, стр. 92.-67. О. Ф К л ю ч к о, В. П. 3 о б н и н, Там же, стр. 171;
БТИ ГИПХ, № 3, 24 A956). — 68. В. П. 3 о б н и н, Там же, № 7, 33 A958).—
69. F. G. Sawyer, M. F. Oh man, F. E. Lusk, Ind. Eng. Chem., 41, № 8, 1547
A949). — 70. M. А. Леонов, ИБП ГИПХ, № 5—6, 20 A965).
71. Г. В. Сергеев, БТИ ГИПХ, № 1, 15 A955). — 72. В. П. Ильин-
Ильинский, К. И. Булгакова, К. Ф. Русинов а, Труды ГИПХ, в. 41, 1958,
стр. 83.-75. В. А. Пьян ко в, ЖПХ, 9, № 2, 238 A935). — 74. М. В. Т о в-
бин, О. М. Барам, Укр. хим. ж., 21, № 2, 205 A955). — 75. С. П. Тевосов,
Исследование электрохимических методов получения иода из нефтяных вод, Изд.
АН АзССР, Баку, 1959. — 76. В. П. Ильинский, К. И. Булгакова, Труды
ГИПХ, в. 41, 1958, стр. 75.-77. А. X. П а н а х - 3 а д е, Уч. зап. Азерб. ун-та,
№ 1, 31 A955); № 11, 73 A957). — 78. А. X. Панах-Заде, ИБП ГИПХ.
№ 14, 79 A968). — 79. М. И. П о п о в, БТИ ГИПХ. № 3, 17 A956).—80. Г. В. С е р-
геев, Там же, № 3, 22 A956).
81. Авт. свид. 76779, 1949; 136720, 1960; япон. пат. 1511, 1952; 6619, 1953;
11454, 1961; 11455 1961; пат. США 3177050, 1963; франц. пат. 1401113, 1964;
англ. пат. 1058429, 1964. — 82. Л. Г. Давыдов, Ю. А. Толмачева,
ЖПХ, 32, № 9, 1979 A959). — 83. V. Е. М а г i a n i, Ann. chim., № 2, 158 A961).—
84. С. И. Яворский, Э. Я. Фарфель, БТИ ГИПХ, № 12, 10 A961); № 15,
12 A962); ИБП ГИПХ, № 7—8, 5 A965); Труды ГИПХ, в. 48, 1965, стр. 36.—
85. ИБП ГИПХ, № 5—6, 68 A965). — 56. С. И. Яворский, В. И. Волкова
и др., Там же, № 11—12, 80 A967); № 13, 45 A967). — 87. С. И. Яворский,
М. С. Гоберман, Там же, № 11—12, 88, 99 A967). — 88. К- И. Булгакова,
Д. А. Гильманова и др., Труды ГИПХ, в. 48, 1965, стр. 46. — 89. Г. С. К л е-
262
Гл. VIII. Иод и его соли
б а н о в, Там же, в. 41, 1958, стр. 4. — 90. Г. С. Клебанов, А. А. Гинзбург,
БТИ ГИПХ, № 2, 14 A955).
91. Д. И. Зульфугорлы, М. И. Абдулаева, А. X. Панах-Заде
и др., Уч. зап. Азерб. ун-та, ЛЬ 4, 11 A955). — 92. С. Д. Гусейнов и др. Там
же, № 3, 17 A955). — 93. М. И. Абдулаева, Автореф. канд. дисс, Азерб.
ун-т, 1954. — 94. Т. Ragozinsky, Przem. Chem., № 9, 12, 639 A953).—
95. Ц. А. А джем ян и др., Хнм. пром., № 6, 14 A945). —96. С. И. Я в о р-
ский, В. И. Волкова и др., ИБП ГИПХ, № 11—12, 108 A967); № 13, 12
A967). — 97. Л. Д. Штутман, Там же, № 13, 8 A967). — 98. М. Е. Позин,
Б. А. К о п ы л е в, Г. В. Б е л ь ч е н к о, Изв. вузов. Хим. и хим. технол., № 5
A958). —99. С. И. Яворский, В. И. Волкова и -ф., БТИ ГИПХ, № 13 3
A961); Труды ГИПХ, в. 48, 1965, стр. 86.— 100. Г. С. К я еб а н о в. А. А. Гинз-
Гинзбург, Там же, в. 41, 1958, стр. 17.
101. Г. С. Клебанов, А. А. Гинзбург, БТИ ГИПХ, № 5, 21 A957).—
102. Э. Д. Кутний, Ю. Е. Гольберг, ИБП ГИПХ, »* 1, 16 A964).—
103. Г. С. Клебанов, А. А. Р е й х а р д, Труды ГИПХ, в. 41, 1958, стр. 40.—
104. Г. С. Клебанов, А. А. Рей хард, Там же, стр. 52. — 105. Г. С. Клеба-
Клебанов, А. А. Р е й х а р д, Там же, стр. 63. — 106. В. И. К с е н з е н к о, Е. А. Д и а-
нов. ИБП ГИПХ, № 5—6, 48 A965). — .'07. R. F. Taylor, Chem. Eng. Sci., 10,
№ 1—2, 68 A959).— 108. Z. Szabo, M Beck, Chem. Zbl!, 27, 7463 A967) —
109. G. Rovesti, Chem. Abs., 4279 A947); 9641 A950).— 110. О. Ю. Магид-
Магидсон и др., Бюлл. Н.-и. ЛХФИ, № 1—5 п930—1931).
111. О. Ю. Магидсон, ЖХП. 6. № 10, 1699 A929). — 112. Б. Г. Панте-
Пантелеймонов а, ЖПХ, Kb 9, 713 A927); № И—12, 484; № 21—22, 1220 A928).—
113. В. П. Ильинский, А Н Стрельникова, Труды ЛХФИ. в. 5, 1958,
стр. 47. — 114. А. Г. Б а й ч и к о е, ЖХП, 12, № 10, 1062 A935). — 115. М. И. К о р-
шуи, Там же, 7, № 10. 60S A930). —116. Е. И. Виноградова. ЖПХ 12,
№ 8, 1115 A939); 13, № 3. 390 A940). — 117. А. А. Ш м у к, А. П. Харин, Там
же, 10, № 2, 223 A937) — 115. Я. И. Гамзулов, Там же, 32, Х« 1, 104 A959).—
119. Е. И. Шавердова, Там же, 25, № 9, 919 A952). — 120. Н. Н. Лысенко,
ИБП ГИПХ. № 5—6. 45 A965).
121. Польск. пат 35632. — 122. Г. С. К л е б а н о в, А. В.Морозова, ЖПХ,
37, № 1, 207 A964).— 123 Ф В. Денисов. Р. Г Л е п и л и и а. Там же, 39,
№ 9 A931); № 10, 2166 A966). — 124. С. И. Яворский, БТИ ГИПХ, № 2, 21
A955). — 125. Л. С. Майофис, Химия и технология химфармпрепаратов, Изд.
«Медицина», 1964. — 126. F. Chemnitius, Chem. Z., 61. 587 608 A927).—
127. Ю. В. К а р я к и н. И. М. Ангелов, Чистые химические реактивы, Госхкм-
издат, 1955. — /2S Г. С. Клебанов, Г. Я. Пинчук ЖПХ, 39, № б, 1291
A966); 40. № 11. 2431 A967); 41, Хе 1, 94 A969). — /2Р. Г. С. Клебанов,
Г. Я Пинчук, ЖПХ, 37, Jsfe 2, 289 A964). —130. A. Hill, H. Willson,
J. В i s h о р, J. Am Chem. Soc, 55, 521 A933).
J31. А. А. Гинзбург, Н. Г. Висленева, БТИ ГИПХ, № 6, 24 A958). —
132. В. П. Ильинский, Л. И. К о к о в к и н а, Мед. пром. СССР, № 2, 20
A959) —133. Н В. Лутугина, Л. И. Коковкина, ЖПХ, 37, № 7, 1487
A965). — 134. Л. И. Кислякова. Автореф. канд. дисс, ЛХФИ, 1967; Н. В. Л у-
тугина, Л. И. Кислякова, ЖПХ, 41, № 1. 218 A969).— 135. С. И. Явор-
Яворский, Э. Я. Ф а р ф е л ь, Труды ГИПХ, в. 48, 1965, стр. 62. — 136. А. К а ц.
Реактивы и препараты лабораторного назначения, в. 1, ГОНТИ, 1938, стр. 178.
Глава IX
СОЕДИНЕНИЯ МАГНИЯ
Важнейшими соединениями магния, применяемыми в различ-
различных отраслях промышленности, являются: хлористый магний и
магнезии — окись магния и основной углекислый магний, а также
сульфат магния и некоторые другие соли.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Большинство солей магния хорошо растворяется в воде..
Ион Mg2+ придает растворам горький вкус. Галогениды магния, за
исключением MgF2, сильно гигроскопичны — на воздухе расплы-
расплываются.
Хлористый магний MgCl2 безводный плавится при 718°. В при-
присутствии следов воды «дымит» на воздухе — разлагается на НС1 и
MgO. Из водного раствора выделяются бесцветные кристаллогид-
кристаллогидраты с 1, 2, 4, 6, 8 и 12 молекулами воды (рис. 81). В интервале
температур от —3,4 до 116,7° устойчив кристаллогидрат
MgCl2-6H2O— бишофит, имеющий плотность 1,56—1,59 г/см3.
Давление водяного пара (в мм рт. ст.) над насыщенным раствором
MgCl2-6H2O: при 15°—4,11, при 30° —9,95, при 60° —44,33, при
100°—165,06. Давление пара над кристаллогидратом MgCl2-4H2O:
при 100°—12,3, при 160° — 228,6, а над системой MgCl2-6H2O —
MgCl2-4H2O: при 100° —72, при 150° — 423,5 '>2. Дигидрат стаби-
стабилен от 181,5 до 240°; при 240° он переходит в моногидрат
MgCl2-H2O, при нагревании которого выше 285° образуется основ-
основной хлорид магния Mg(OH)Cl. Выше 500° последний разлагается
на MgO и НС1.
В системе MgO—MgCl2—Н2О в пределах 0—110° и при кон-
концентрациях MgCl2, меньших 45%, образуются кристаллогидраты
оксихлорида магния MgCl2-3Mg(OHJ-8H2O и MgCl2-5Mg(OHJ.
•2Н2О, выделяющиеся ниже 100°, и MgCl2-9Mg(OHJ-5H2O и
MgCl2-2Mg(OHJ-4H2O, выделяющиеся выше 100°. Чем выше
264
Гл. IX. Соединения магния
отношение Mg(OHJ:MgCl2 в этих соединениях, тем они устойчи-
устойчивее при малых концентрациях MgCl2. Октогидраты образуются так-
также в системах MgO—СаС12—Н2О и CaO—MgCl2—Н2О3. Образую-
Образующаяся при кипячении суспензии MgO в концентрированном раство-
растворе MgCl2 основная соль MgCl2 -3Mg(OHJ-8H2O переходит при
200° в тетрагидрат и при 240° в безводную соль MgCb-3Mg(OHJ,
гоо\
50
-да,
7<?/,5*
¦-Щ
/ -I7A"
го-50° зо
4д
50
%
00
70
Рис. 81. Растворимость в системе MgCl2—Н2О:
/-MgCl2-12H2O: //-a-MgCl2-8H2O; //'-p-MgCI2.8H2O';
/// - MgCl2 • 6H2O; IV - MgCl2 • 4H2O; V- MgCI2 • 2H2O.
не разлагающуюся до 320°. Начиная с 330 [температура разложе-
разложения Mg(OHJ] до 360° происходит отщепление последних трех мо-
молекул воды 4.
При взаимодействии MgBr2 с гидроокисью магния образуются
соединения MgBr2-5Mg(OHJ-8H2O и MgBr2-3Mg(OHJ-8H2O,
изоморфные соответствующим хлоридам и устойчивые — первое в
интервале концентраций 1,6-—2,06 и второе 2,13—2,7 моль в 1000 г
раствора. Растворимость их в воде ~ 0,005 моль на 1000 г раствора.
При взаимодействии концентрированных растворов MgCl2
со щелочами возможно образование комплексного основания
{Mg[Mg(OHJ]3}(OHJ4.
Растворимость MgCl2 в водных растворах, насыщенных хлори-
хлористым водородом, 30% при 25° и 0,7% при 0°5. (О растворимости
в водных системах, содержащих MgCl2, см. 6~10)
Физико-химические свойства
265
MgCl2 образует комплексные соединения типа MtMgCb] • 6Н2О,
например, карналлит K[MgCl3] • 6Н2О или КС1 • MgCl2 • 6Н2О ".
Растворимость в системе MgCl2—КС1—Н2О приведена на стр. 162.
Разложение аммониевой соли NH4[MgCl3] • 6Н2О при нагревании
происходит без гидролиза и образования основных солей, что дает
возможность получить таким путем безводный MgCl2. В безводных
системах МС1—MgCl? (где М — щелочной металл) для К и Na
известны только соединения KMgCl3 и NaMgCl3'2.
Окись магния MgO встречается в природе в свободном состоя-
состоянии в виде минерала периклаза. Искусственная — представляет со-
собой рыхлый порошок плотностью 3,65—3,9 г/см3; плавится при
2800°. Полученная при невысоких температурах — легко раство-
растворяется в кислотах с образованием солей, соединяется с водой, об-
образуя гидрат Mg(OHJ. При сильном прокаливании становится
очень твердой и приобретает кислото- и водостойкость. Рекристал-
Рекристаллизация окиси магния при нагревании начинается выше 500°, и при
1200—1600° образуются крупные кристаллы периклаза. Поэтому
активность MgO уменьшается с повышением температуры прока-
прокаливания до 1600°. Активность материала, обожженного выше 1600°,
вновь немного возрастает, что объясняется появлением трещии
спайности13. (О равновесии в системе 2MgO^2Mg+O2 см.14)
Гидроокись магния Mg(OHJ встречается в природе в виде ми-
минералов брусита и немалита. Искусственно получается при дей-
действии щелочей на растворы солей магния в виде объемистого бе-
белого студенистого осадка; величина рН осаждения 10,5; плотность
осажденной MgCOHJ 2,36—2,40 г/см3. Она легко поглощает из
воздуха двуокись углерода. При 20° насыщенный водный раствор
содержит 0,019 г/л, а при 100° 0,04 г/л Mg(OHJ. Произведение
растворимости Mg(OH)? при 25° равно 5,5- 10~12. Вторая константя
гидролиза Mg(OHJ^Mg2"-i-2OH- равна 3 • 10~3. Давление Н2О
над Mg(OHJ при 300° равно лишь 10 мм вод. ст. Выше 500°
Mg(OHJ теряет воду и переходит в MgO. Гидроокись магния
легко растворяется в кислотах, а также в растворе NH4C1 вслед-
вследствие реакции:
Mg(OHJ + 2NH4C1 Я=
Углекислый магний MgCO3 в свободном состоянии встречается
в природе в виде минерала магнезита (а также в виде гидратов
MgCO3-3H2O и MgCO3-5H2O). Плотность магнезита 3—3,12 г/смг.
Давление диссоциации MgCO3 ч* MgO + СО2— 28.1 ккал/г-мол
равно 1 ат при 650° 15. При 25° и давлении СО2 0,0001 ат раствори-
растворимость MgCOs в воде равна ~ 0,5 мг/л, при 0,968 ат — 22 мг/л, а
при 15от — 55 мг/л. С повышением температуры растворимость
уменьшается. MgCO3 выделяется из водных растворов только, если
в них содержится большой избыток СО2; в противном случае вы-
выделяются основные соли, которые образуются и при кипячении
266
Гл. IX. Соединения магния
водного раствора MgCO3 и при взаимодействии растворов солей
магния с карбонатами щелочных металлов. Основные соли превра-
превращаются в нейтральный MgCO3 при нагревании с концентрирован-
концентрированными растворами бикарбонатов.
При насыщении двуокисью углерода водной суспензии карбо-
карбоната магния последний растворяется с образованием бикарбоната
MgCO3 + СО2 + Н2О = Mg(HCO3J
который можно рассматривать как комплексную кислоту типа
H2[Mg(CO3J]. При взаимодействии MgCO3 с карбонатами щелоч-
200
MgSO4HaO ^ MgSO4.HlO
MgSO4 4Hzo\-l//7So
cc-MgSO4 й
|S-MgSO4 7НгО
*5o! 70 20~ 30
MgSO4, вес. %
Рис. 82. Растворимость в системе MgSO4—H2O.
ных металлов образуются соли этой гипотетической кислоты — пло-
плохо растворимые двойные соли, например, K2[Mg(CO3J] • 4Н2О,
KH[Mg(CO3J]-4H2O, доломит Ca[Mg(CO3J] или СаСО3• MgCO3.
Растворимость доломита при повышенном давлении СО2 увеличи-
увеличивается б присутствии Na2SO4 |6.
При взаимодействии в растворе эквимолекулярных количеств
MgCl2 и К2СО3 или КНСО3 ниже 80°, а также при многосуточном
нагревании раствора Mg(HCO3J до 100° образуется метастабиль-
ная твердая фаза MgCO3 • ЗН2О, устойчивая при 40° и переходящая
при 60—80° в аморфный основной карбонат 5MgO • 4СО2 • пН2О
(где п=6—7). Последний разлагается при 100° с образованием
Применение
267
Mg(OHJ17-19. При более высокой температуре (~200°)
MgCO3-3H2O, а также MgCO3-4H2O превращаются в MgO20-21.
Сульфат магния образует 1, 2, 3, 4, 5, 6, 7 и 12-водные кристал-
кристаллогидраты (рис. 82). При комнатной температуре из водных рас-
растворов кристаллизуется эпсомит MgSO4 • 7Н2О, а выше 48° — са-
киит MgSO4 • 6Н2О, который в интервале 87—92° плавится инкон-
груэнтно с образованием метастабильных пяти- и четырехводной
солей. Твердый MgSO4-4H2O при 106° переходит в MgSO4-3H2O,
последний —в MgSO4-2H2O при 122—124°. От 161 до 169°MgSO4-
• 2Н2О переходит в MgSO4-H2O— кизерит. Из водных растворов
стабильный кизерит кристаллизуется при температурах выше 67,5°.
Обезвоживание кизерита наблюдается при 320—330° 22~24. По дру-
другим данным25, первые шесть молекул воды из эпсомита удаляются
при нагревании до 285°, последняя — при 420°. Чистый безводный
MgSO4 разлагается с заметной скоростью выше 1100°, причем ско-
скорость разложения увеличивается в присутствии SiO226'27. При тер-
термическом разложении MgSO4 • 7Н2О под давлением паров воды
50—200 мм рт. ст. были получены фазы с 6; 4; 2,5; 2; 1 и 0 моле-
молекулами воды28.
При взаимодействии MgO, Mg(OHJ и MgCO3 при температу-
температурах 20—100° в водной среде с водным гелем кремневой кислоты
образуется нерастворимый гидросиликат магния (сепиолит).
С кристаллическим кварцем это взаимодействие идет с трудом (при
длительном нагревании при 120° в автоклаве29).
ПРИМЕНЕНИЕ
Главными потребителями соединений магния являются произ-
производство огнеупоров, строительная и металлургическая промышлен-
промышленность.
Значительные количества соединений магния перерабатывают
на металлический магний, используемый для получения сплавов
с алюминием и другими металлами. Введение магния (~0,05%) в
чугун повышает его ковкость и сопротивление разрыву. Металли-
Металлический магний используют также для магнийтермического восста-
восстановления титана и кремния30. Магний получают восстановлением
магнезии или доломита ферросилицием или углем при ~2000° и
электролизом расплавленного безводного хлористого магния31.
В последнем случае электролитом служит или обезвоженный кар-
карналлит или безводный MgCl2 с добавками КС1 и NaCl для пони-
понижения температуры плавления и повышения электропроводности.
Безводный хлористый магний получают хлорированием окиси маг-
магния хлором, выделяющимся при электролизе32.
Хлористый магний применяют для изготовления цементов (на-
(например, цемента Сореля: 1 вес. ч. MgCl2+2 вес. ч. MgO+вода)
и на их основе различных строительных материалов. Магнезиаль-
Магнезиальный цемент состоит из оксихлоридов магния, состав которых
268
Гл. IX. Соединения магния
Применение
269
зависит от условий приготовления и хранения (температуры и
влажности атмосферы). Устойчивыми в обычных условиях яв-
являются 3MgO-MgCl2-10H2O, 5MgO • MgCl2 • 12Н2О и 7MgO •
•MgCl2- S4H2O33-35. Магнезиальные цементы — основа многих
строительных материалов. Производство половых покрытий из
ксилолита (магнезиального цемента с опилками), стенных пере-
перегородок из фибролита (магнезиального цемента со стружками) и
других обходится дешевле, чем из древесины. Применение магне-
магнезиальных и гипсо-магнезиальных цементов представляет особый
интерес для южных безлесных районов СССР, богатых в то же
время соляными озерами36'37. Магнезиальные цементы исполь-
используются также и в качестве связки в абразивной промышленности.
Хлористый магний служит одним из видов сырья для получе-
получения магнезии, применяется в качестве аппретуры в текстильной
промышленности и для пропитки деревянных конструкций с целью
придания им огнестойкости, используется для производства дефо-
дефолианта; его растворы применяют в качестве антифриза для пре-
предупреждения замерзания стрелок на железнодорожных путях.
Технический твердый хлористый магний представляет собой ше-
стиводный кристаллогидрат с примесью небольших количеств четы-
рехводного. Согласно ГОСТ 7759—55, он должен содержать не
меньше 45,0% MgCl2 и не больше 2,0% щелочных хлоридов, 1,6%
сульфатов (MgSO.-), 0,2% солей кальция (СаО) и 0,1% нераство-
нерастворимого остатка.
Хлористый магний упаковывают в барабаны из кровельного
железа, закрашенные асфальтовым или кузбасским лаком.
Различные сорта окиси магния или жженой магнезии, называе-
называемой также магнезией уста, отличаются по своим физико-химиче-
физико-химическим свойствам — легкости (объемному весу), химической актив-
активности, адсорбционным свойствам, теплопроводности и пр. Чем бо-
более дисперсна и пориста магнезия, тем меньший объемный вес она
имеет; о ее качестве судят по ее легкости. В резиновой промыш-
промышленности качество магнезии определяют так называемой лиофиль-
ностью, характеризующей адсорбционные свойства материала. Так
как адсорбционная способность усиливается с увеличением удель-
удельной адсорбирующей поверхности, т. е. дисперсности и пористости,
то лиофильность и легкость магнезии являются в известном смыс-
смысле однозначными терминами. Лиофильность определяется объемом,
который занимает 1 г MgO в 25 мл толуола.
Наиболее легкая и рыхлая «белая» магнезия или магнезия
альба (от лат. albus — белый), представляющая собой основную
соль карбоната магния или гидроокись магния, применяется в
качестве наполнителя и усилителя в резиновых смесях, для изго-
изготовления высококачественных теплоизоляционных материалов и в
медицине. Объем, занимаемый 1 г белой магнезии в 25 мл то-
толуола, должен быть не меньше 6 мл. Легкие сорта магнезии уста
используются в качестве наполнителей в резиновых смесях и уси-
усилителей свойств резины, а также в нефтяной промышленности
для отбеливания нефтепродуктов. Менее легкие сорта магнезии
(например, каустический магнезит) применяют для изготовления
цементов, строительных материалов, искусственных камней (на-
(например, мельничных жерновов). Тяжелые сорта магнезии (метал-
(металлургический порошок) идут на изготовление огнеупорных тиглей
и кирпичей для футеровки печей, а также раствора для их
связки38.
По внешнему виду магнезии представляют собой белые тонкие
порошки. Требования к их составу приведены в табл. 22.
Требования к качеству магнезии
{Содержание компонентов в %)
ТАБЛИЦА 32
MgO, не менее ,
Примеси, не более:
СаО
R8OS
SiO2
Сульфаты (SO3)
Хлориды (С1)
Марганец
Не растворимые в соляной кис
лэте
Потери при прокаливании • .
Влага
Магнезия жжеиая
для резиновых смесей
(ГОСТ 844-41)
I сорт
89
1,25
0,1**
0,4
. 0,035
0,003
0,2
8,6
II сорт
89
1,25
0,3**
0,7
0,08
0,007
0,25
8,6
Каустический магнезит*
(ГОСТ 1216-41)
I класс
87
1.8
2
1,8
6
1,5
II класс
83
2,5
2,5
8
1,5
III класс
75
4,5
4,0
18
1,5
• Содержание компонентов в каустическом магнезите — в пересчете на сухой продукт.
• FO
Легкие сорта магнезии упаковывают в многослойные крафт-
целлюлозные мешки, которые помещают в мешки из прорезинен-
прорезиненной ткани или другого водонепроницаемого материала. Другие
сорта магнезии упаковывают в мешки, фанерные барабаны, бочки
и т. п.
Сульфат магния, подобно хлориду, применяют для производ-
производства цементов, более водоупорных, чем содержащие MgClg39, его
используют в производстве искусственного шелка и др. MgSC>4 все
шире используют в качестве магнезиального удобрения. Пред-
Представляет интерес использование в качестве местного магнезиаль-
магнезиального удобрения озерных рассолов и остаточных щелоков от
270
Гл. IX. Соединения магния
соледобычи. Двойная соль (NH4JSO4-MgSO4-6H2O (соль Тут-
тона) является удобрением и фунгицидом, предупреждающим не-
некоторые болезни зерновых культур 40.
СЫРЬЕ
По своей распространенности в природе магний занимает
восьмое место — на его долю приходится 2,35% веса земной коры.
Он встречается большей частью в виде силикатов, карбонатов,
сульфатов и хлоридов. К силикатам относятся, например, оли-
оливин (Mg, FeJSi04, тальк 3MgO • 4SiO2 • Н2О, серпентин 3MgO •
•2SiO2-2H2O (серпентиниты — продукты метаморфизации олкви-
нитовых пород; волокнистой разновидностью серпентинита являет-
является асбест); к карбонатам — магнезит MgCO3 и доломит MgCOe*
•СаСО3; к сульфатам — кизерит MgSO4 • Н2О, каинит MgSO4-
•КС1:ЗН2О, лангбейнит 2MgSO4 • K2SO4, шенит K2SO4-MgSO4-
•6Н2О, эпсомит MgSO4-7H2O, сакиит MgSO4-6H2O, леонит
K2SO4 • MgSO4 • 4Н2О, полигалит K2SO4 • MgSO4 • 2CaSO4 • 2Н2О,
астраханит Na2SO4 • MgSO4 • 4Н2О; к хлоридам — бишофит MgCl2-
•6Н2О, карналлит КС1 • MgCl2 • 6Н2О.
Наиболее распространенными соединениями магния являются
вулканические образования — первичные силикатные н алюмоси-
Ликатные породы. В качестве сырья для производства соединений
магния используют обычно вторичные твердые минералы и при-
природные растворы: растворимые соли магния и их растворы (вода
морей и соляных озер), осадочные породы морского происхожде-
происхождения— доломиты, магнезиты и вторичные, преимущественно гидра-
тированные силикаты — змеевики, асбесты, тальки.
Магнезит (плотность 2,9—3,1 г!см3, твердость 4—4,5) встре-
встречается в двух разновидностях — плотной («аморфной») и кристал-
кристаллической. Плотный магнезит содержит обычно больше кремнезема
и меньше других примесей (окислов железа, алюминия, кальция),
чем кристаллический. Последний часто содержит изоморфную
смесь карбонатов Fe, Ca, Мп. В СССР имеются месторождения
магнезита на Урале, в Восточной Сибири, в Средней Азии, в Баш-
Башкирии, в районе Средней Волги и в других районах страны4|. По
американским данным, мировые запасы магнезита составляют
приблизительно 8,3 млрд. т, из которых 5 млрд. т в Китае,
2 млрд. т в Корее, 65 млн. т в США42. В США бедные магнезиты,
содержащие много примесей, обогащают флотацией и разделе-
разделением в тяжелых суспензиях. В качестве тяжелой жидкости плот-
плотностью 2,9 г/см3 применяют суспензию 1 вес. ч. молотого ферро-
ферросилиция в 2 вес. ч. воды. В этой суспензии магнезит (плотность
~3 г\ см3) тонет, а кварцит и другие примеси (плотность
~2,6 г/смг) всплывают. По этому методу работает обогатитель-
обогатительная фабрика мощностью 3000 т/сутки32'43.
Сырье
271
Природные доломиты (плотность 2—2,86 г/см3, твердость 3,5—
4), помимо основного минерала доломита CaMg(CC>3J содержат
примеси кварца, кальцита, гипса, глин и другие, придающие по-
породе различную окраску. Имеется много месторождений доломи-
доломитов в разных районах СССР.
Ценным магниевым сырьем являются ископаемые калиево-маг-
ниевые минералы44 — карналлит, каинит, лангбейнит, полигалит
и др. (см. гл. V). Морская вода, содержащая ~1,3 г/л Mg2+,—
практически неисчерпаемый сырьевой источник для производства
соединений магния45. В больших концентрациях сосредоточен Mg2+
В рапе Крымских озер, оз. Эльтон, озер Кулундинской степи, в
воде Сиваша, Каспийского моря и особенно примыкающего к нему
Кара-Богаз-Гола. Концентрация солей магния в рапе озер иногда
достигает 30%, например, в оз. Эльтон в июле-августе46-47. Рапа
этого озера представляет собой хлормагниевый щелок с малым
содержанием других солей48-49. Концентрированный хромагние-
вый щелок получается и при переработке карналлита (см. гл. V),
после извлечения из Карабогазской рапы мирабилита50'51, при
комплексной переработке рапы оз. Кучук 52>53 и др. Одним из
видов магниевого сырья является астраханит; в корневых отло-
отложениях астраханитовых озер находится эпсомит64.
Конверсия природных двойных солей позволяет искусственно
получать щелоки, содержащие соли магния55>Б6. Примером может
служить превращение карналлита в каинит:
КС1 • MgCl2 • 6Н2О + MgSO4 • 6Н2О = КС1 • MgSO4 • ЗН2О + MgCl2 + 9Н2О
Потенциальным сырьем для производства соединений магния
являются весьма распространенные в природе силикаты магния}
некоторые из них содержат больше магния, чем, например, кар-
карналлит. Наиболее распространенным силикатом магния является
оливин (Mg, FeJSi04, представляющий собой изоморфную смесь
форстерита Mg2Si04 и файялита Fe2SiO4, а также других орто-
силикатов. Под действием гидротермальных растворов оливин
разлагается с образованием серпентина. Известны большие мас-
массивы дунита — горной породы, содержащей 60—75 % оливина —
на Урале. Крупное месторождение оливина имеется на Кольском
полуострове. Волокнистая разновидность серпентинита — асбест
имеет самостоятельное техническое значение и так как содержание
его в серпентинитовой породе обычно не превышает нескольких
процентов, то при добыче асбеста на Урале, Кавказе, в Сибири
получгют значительные количества отвалов серпентинита32.
В СССР и за границей проводились опыты по получению хлори- .
стого магния обработкой серпентинитов соляной кислотой или
хлорированием при 500—1000°. Основная трудность заключается
в отделении раствора хлористого магния от образующейся при
этом кремневой кислоты. Молотый серпентин применяют в каче-
качестве медленно действующего магниевого удобрения57,
272
Гл. IX. Соединения магния
ХЛОРИСТЫЙ МАГНИЙ
Солнечная выпарка природных вод
Производство хлористого магния (MgCl2-6H2O) из морской
воды, рапы озер и лиманов осуществляют после садки солей и
извлечения брома58.
Процесс испарения морской воды и рапы соляных озер мор-
морского типа можно проследить по диаграмме (рис. 83) изотермы
25° взаимной водной си-
системы 59~63
2Nad + MgSO4 =
= rJa2SO4 + MgCl2
Эта система является
весьма сложной как по
1 количеству твердых фаз,
так и по их склонности
кристаллизоваться в ме-
тастабильном состоянии.
Метастабильные участки
мирабилита, тенардита,
галита и эпсомита вкли-
вклиниваются внутрь поля
астраханита (области,
ограниченные пунктирны-
пунктирными линиями). Состав со-
солевой массы исходной
морской воды и рассолов
большинства соляных
озер морского типа при-
приближенно можно изобра-
изобразить точкой / — в процен-
процентах ион-эквивалентов:
Mg/2 — 22,3, Na — 77,7,
Cl — 92,5, SO4/2 — 7,5.
При изотермическом ис-
испарении этого раствора
первой в твердую фазу
будет выделяться пова-
поваренная соль. Состав рас-
раствора в процессе кристал*
лизации NaCl изменяется
по лучу кристаллизации
J—2. В точке 2 состав раствора: Mg/2 —58, Na —42, Cl — 77,
SO4/2 — 23. Количество испаряемой воды в последовательных
стадиях-1 кристаллизации можно определить по водной (верх-
(верх2NaCl Ю 20 30 40 SO ВО 70 60 90 MgC\z
Рнс. 83. Изотерма растворимости водной си-
системы 2NaCl +-MgSO4 Z^. MgCl2 + Na2SO4
при 25° (сверху — водная диаграмма).
Хлористый магний
273
ней) диаграмме (рис. 83}. Если бы кристаллизовались только
стабильные фазы, то при дальнейшем испарении воды состав
раствора изменялся по отрезку кривой 2—3, что сопровожда-
сопровождалось бы совместной кристаллизацией галита и астраханита. Так
как точка 3 инконгруэнтная, то при дальнейшем выпаривании
воды в этой точке про-
произошло бы растворе-
растворение выкристаллизовавше-
выкристаллизовавшегося ранее астраханита с
выделением в осадок сме-
смеси NaCl и MgSO4-7H2O,
называемой смешанной
солью. Однако вследствие
того, что в природных
условиях имеет место дли-
длительное и устойчивое су-
существование лабильных
равновесий, при испаре-
испарении морской воды в боль-
большинстве соляных озер
астраханит не выпадает,
а вместо него, после не-
некоторого еще выделения
NaCl, начинает кристал-
кристаллизоваться смесь NaCl и
MgSO4«7H2O (ср. с про-
процессом, описанным на
стр. 71 и рис. 8.)
Состав этой смеси
изменяется по мере уве-
10 20 30 40 50 ВО Ю 80 90
2NaCl
Рис. 84. Изотерма 0° водной
34 ^=±MgCl2 + Na2SC
ной диаграммой).
сим емы
(с вид-
личения концентрации
хлористого магния. По
достижении тройной точ-
точки 4 MgSO4-7H2O теряет
вследствие обезвоживающего действия хлористого магния 1 моле-
молекулу воды, превращаясь в MgSO4 • 6Н2О.
При дальнейшем увеличении концентрации рассол достигает
конечной эвтонической точки Е с составом жидкой фазы (в моле-
молекулах на 1000 мол. воды): MgCl2—101,0; MgSO4 —9,5; Na2Cl2 —
1,1; кроме хлористого натрия и MgSO4-6H2O, начнет выделяться
бишофит MgCl2 • 6Н2О и состав рассола не изменится вплоть до
полного высыхания. Такой состав плюс некоторое количество со-
солей калия и брома будет иметь «солнечный» хлористый магний,
получаемый методом естественного испарения рапы при 25°. При
0° содержание в нем MgSO4 понизится приблизительно в 2,5 раза.
Изотерма 0° изображена на рис. 8464. (При 65° в диапазоне
274
Гл. IX. Соединения магния
6НгО
весовых концентраций 7,88—6,79% MgSO4, 7,71—0,9% MgCl2,
21,14—21,82% NaCl кристаллизуется двойной сульфат
9Na2SO4-MgSO465.) Практически рапу обычно выпаривают более
интенсивным заводским способом, используя солнечное тепло
только для начальной стадии концентрирования природных вод
до —20% MgCl266-67.
Получение MgCI2 при переработке карналлита
Независимо от способа разложения карналлита (стр. 161) хлор-
магниевые щелоки, получаемые после кристаллизации искусствен-
искусственного карналлита, могут
подвергаться выпарке на
шестиводный хлористый
магний и дальнейшему
обезвоживанию. Содер-
Содержание MgCl2 в таких ще-
щелоках обычно 28—31%.
На рис. 85 изображе-
изображена изотерма 25° для кар-
наллитовой системы 58>68.
Большую часть поверх-
поверхности кристаллизации за-
занимают поля NaCl и КС1.
Значительно меньше поле
более растворимого кар-
карналлита и меньше всего
поле бишофита. Эти че-
четыре поля сходятся в двух
тройных точках. В пер-
первой — карналлитовой с
насыщенным раствором,
имеющим состав (в
вес.%) MgCl2 —25,54,
NaCl — 2,36, КС1 — 3,29, в
равновесии находятся три
твердых фазы: NaCl, KC1
и КС1 • MgCl2 • 6Н2О. Вто-
Вторая тройная точка — би-
шофитная имеет состав
MgCl2 —35,56, NaCl —
0,32, КС1 —0,12; в этой
точке с насыщенным рас-
раствором в равновесии находятся КС1 • MgCl2 • 6Н2О, MgCl2-6H2O и
NaCl. Линия, соединяющая карналлитовую и бишофитную точки,
соответствует конечным хлормагниевым щелокам, получаемым
Хлористый магний
275
NaCl
КС!
Рис. 85. Изотерма 25° системы
NaCl—KC1—MgCl2—H2O.
,100°
б50
bo
§ 20
Твердая фаза
6Н2О
при различных способах переработки карналлитов. Чем выше при
выпарке хлормагниевых щелоков их концентрация, тем меньше
в них примесей NaCl и КС1. Обычно эти соли отделяют после
выпарки раствора до ~35% MgCl2, а затем выпарку продолжают
для получения шести-, четырех-
или двухводного хлористого маг-
магния.
Очистка карналлитового хлор-
магниевого щелока от NaCl и
КС1 может быть произведена вы-
высаливанием этих солей с по-
помощью газообразного хлористо-
хлористого водорода. КС1 высаливается
легче, чем NaCl. Уже при малых
концентрациях НС1, наряду с
NaCl и КС1 в донную фазу вы-
выпадает также карналлит. При
больших концентрациях НС1
(~300 г/л) выделяется очень чи-
чистый i%Cl2-6H2O (рис. 86). По-
Последующей продувкой раствора
воздухом можно регенерировать
Ю
1
Рис.
10 20 30 40
Концентрация НС1. Вес.%
86. Растворимость в системе
MgCl2— НС1—Н2О.
практически безводный хлористый водород69. Получение чистых
продуктов этим методом дает наилучшие результаты при сочета-
сочетании высаливания хлористым водородом с ионным обменом70.
Выпарка хлормагниевых щелоков
Независимо от способа получения щелок, содержащий MgCl2
и некоторое количество различных примесей, выпаривают. Выпа-
Выпаренный щелок разливают в тару, где он при охлаждении закри-
сталлизовывается в твердую массу — MgCl2-6H2O. Выпарка до
более высоких концентраций, связанная с повышением темпера-
температуры кипения раствора при атмосферном давлении выше 160°, не
практикуется, так как при этом MgCl2 начинает гидролизоваться
с выделением хлористого водорода.
Для выпарки хлормагниевых щелоков применяют как вакуум-
выпарные аппараты, так и аппараты, работающие по принципу
погруженного горения32-71-73. Выпарной аппарат погруженного го-
горения / (рис. 87) представляет собой чугунный или стальной фу-
футерованный цилиндр с помещенной в него горелкой 2. Горение
газообразного топлива происходит в камере сгорания 3, располо-
расположенной ниже уровня раствора. Продукты сгорания, соприкасаясь
с раствором, вызывают сильную циркуляцию его. Выпаривание
воды из раствора происходит при температуре, соответствующей
парциальному давлению водяного пара в паро-газовой. смеси, т. е.
276
Гл. IX. Соединения магния
при температуре более низкой, чем при передаче тепла через по-
поверхность нагрева. Температура отходящих газов может быть
выше температуры кипения раствора всего на 1—2°. Термический
коэффициент полезного действия весьма высок (~95%). В вы-
выпарной батарее из 3—4 последовательно включенных (по жидко-
жидкости) аппаратов хлормагниевый щелок выпаривают до концентра-
концентрации 34—35% MgCl2. Дальнейшее выпаривание до 45—57% MgClg
производят после отделения вы-
выделившихся солей и (если тре-
требуется) очистки раствора от каль-
кальция (добавлением к нему раство-
раствора MgSO,i) в аппаратах с погру-
погруженным горением или в откры-
открытых стальных футерованных ре-
резервуарах, обогреваемых паром
25—30 ат. О математическом опи-
описании выпарки хлормагниевого
щелока в аппаратах погружен-
погруженного горения см.74.
1 При розливе выпаренного ще-
щелока непосредственно в тару —
барабаны из кровельного желе-
железа — требуются большие площа-
площади для их охлаждения. Кроме
того, извлекать монолит из бара-
барабанов трудно, а сами они сильно
разрушаются вследствие корро-
коррозии, обусловливаемой действием
хлористого магния на железо.
Эти - затруднения исключаются
при получении хлористого маг-
магния в виде чешуек"
применяться деревянные
Рис. 87. Выпарной аппарат с погру-
погруженной горелкой (разрез):
/ — выпарной аппарат; 2 —горелка; 3 — ка-
камера сгорания.
случае в качестве тары могут
В этом
или фа-
фанерные ящики или бочонки, или многослойные крафтцеллюлозные
мешки. Для получения чешуйчатого хлористого магния выпарен-
выпаренный щелок (с плотностью 1,56 г/см3) кристаллизуют на вращаю-
вращающемся стальном барабане, охлаждаемом внутри водой.
Химические способы получения хлористого магния
Химическая переработка морской воды
Хлористый магний получают из морской воды также без ее
предварительного концентрирования и садки NaCl, непосредствен-
непосредственным осаждением известковым молоком гидроокиси магния
(стр. 287J и растворением ее в соляной кислоте. Осадок (кек),
Хлористый магний
277
содержащий. ~25% Mg(OHJ, смешивают с разбавленным рас-
раствором MgCb и нейтрализуют соляной кислотой. Полученный рас-
раствор, содержащий ~15% MgCl2, выпаривают. Этот способ позво-
позволяет получать Mg(OHJ, а затем и хлористый магний, не содер-
содержащие соединений бора, которые не осаждаются при больших
значениях рН. Хлористый магний, перерабатываемый на метал-
металлический магний, не должен содержать больше 0,001% бора.
Источником хлора для перевода суспензии Mg(OHJ в MgCb
может служить раствор СаС12 — дешевый отход содового произ-
производства. Суспензию Mg(OHJ в растворе СаСЬ подвергают76 кар-
карбонизации:
Mg(OHJ + СаС12 + СО2 = MgCl2 + CaCOs + Н2О
После отделения осадка получается раствор, содержащий около
10% MgCb и некоторое количество NaCl. При выпаривании рас-
раствора до 35% MgCb основная часть NaCl выкристаллизовывается
и отделяется.
Получение хлористого магния из доломита
Полностью обожженный доломит гасят водой при 95—100°.
Полученную пульпу, содержащую ~ 11 % твердой фазы, подвер-
подвергают карбонизации при 60—40°. При этом карбонизуется только
гидроокись кальция, переходящая в углекислый кальций77. Кар-
бонизованную пульпу подвергают селективной нейтрализации со-
соляной кислотой при рН = 7 — 8 во избежание растворения СаСО3.
Соляную кислоту приливают медленно, чтобы скорость нейтрали-
нейтрализации ОН" не превышала скорости растворения Mg(OHJ. После
отделения осадка отстаиванием получаемый раствор содержит
около 16% MgCl2. Часть этого раствора возвращают на взмучи-
взмучивание после первого отстаивания, а часть направляют на вторич-
вторичное отстаивание, а затем на выпарку32.
Предложено78 подвергать карбонизации при температуре ниже
40° суспензию из доломитового молока, сульфата кальция и хло-
хлорида щелочного металла (при молярном отношении MgO : CaSO4:
: MCI = 1:1:1). К концу карбонизации суспензию нагревают до
кипения. Образовавшийся СаСО3 отделяют от раствора, затем из
него выкристаллизовывают сульфат щелочного металла, а маточ-
маточный раствор, содержащий MgCl2, выпаривают.
Для получения MgCl2, не содержащего СаСЬ, рекомендуют7*
производить карбонизацию доломитового молока, полученного га-
гашением обожженного доломита раствором NH4C1, взятым в ко-
количестве, необходимом для обменного разложения половины окис-
окислов доломита. При карбонизации в осадок выделяется основная
двойная углекислая соль кальция и магния CaCO3*Mg(OHJ, a
в растворе остается MgCl2. Осадок также обрабатывают раствором
278
Гл. IX. Соединения магния
NH4C1 для перевода Mg(OHJ в MgCl2. Возможно использо-
использовать для регенерации аммиака в содовом производстве вместо из-
известкового молока доломитовое молоко, с образованием при этом
MgCb80'81. При обработке жидкости теплообменника дистилляции
доломитовым молоком вначале реагирует с NH4CI гидроокись
кальция. Гидроокись магния вступает в реакцию во вторую оче-
очередь и может прореагировать при 95° полностью лишь при отгонке
аммиака. Без отгонки NH3 реакция между Mg(OHJ и NH4C1
протекает лишь на 45—50%, идет медленно и зависит от актив-
активности окиси магния (стр. 287).
Хлористый магний из сульфатных растворов
Отбросный щелок в производстве сульфата калия, содержащий
2,9—3,2% К+, 5,5—6,5% Mg2+, 17—18% С1" и5- 12% SO;", пред-
предложено перерабатывать следующим методом82. Вначале щелок
выпаривают и кристаллизуют лангбейнит, причем удаляется до
80% SO4 . Оставшийся SO4~ осаждают хлористым кальцием, от-
отделяют CaSO4 и раствор выпаривают и выкристаллизовывают из
него карналлит и галит. Полученный раствор хлористого магния
направляют на выпарку.
В Техасе (США) хлористый магний, необходимый для произ-
производства металлического магния, получают из лангбейнитовой руды
и из доломита83. Обменным разложением лангбейнита с КС1 вы-
выделяют K2SO4. Из оставшегося раствора, содержащего около 13%
MgCl2 (при общем содержании солей ~30%), вакуум-кристалли-
вакуум-кристаллизацией извлекают карналлит, а раствор MgCl2 концентрируют в
аппаратах с погруженным горением, причем он окончательно очи-
очищается от КС1 и других солей. Обезвоженный хлористый магний
направляют на электролиз, а выделяющийся при электролизе
хлор перерабатывают в соляную кислоту, которую используют
для нейтрализации бикарбоната магния, получаемого из доломита.
Образовавшийся при этом раствор MgCl2 концентрируют и после
-обезвоживания также направляют на электролиз.
Безводный хлористый магний
Обезвоживание хлористого магния
Безводный хлористый магний, содержащий до 99% MgCl2,
можно получить84 ступенчатым обезвоживанием гексагидрата до
дигидрата с дегидратацией последнего в токе НС1 при постепен-
постепенном повышении температуры до 350—375°, с выдержками при
250 и 305°. В производственных условиях обезвоживание
¦MgCl2*6H2O осуществляют таким образом, чтобы избежать его
Хлористый магний
279
плавления в кристаллизационной воде. Обычно процесс ведут в
две стадии, удаляя 4—5 моль воды в токе горячего воздуха A00—
200°), а затем окончательно обезвоживая в токе хлористого во-
водорода при 620—650°. При выпарке раствора хлористого магния
в аппаратах с погруженным горением концентрацию раствора до-
доводят до 57% MgCl2, что соответствует MgCl2-4H2O, а обезвожи-
обезвоживание MgCl2-4H2O до MgCl2-2H2O осуществляют во вращаю-
вращающейся печи горячим воздухом. Двухводный хлористый магний
окончательно обезвоживают в токе НС1 в шахтной печи, предва-
предварительно брикетируя материал, или во вращающейся печи с внеш-
внешним обогревом. Получаемый продукт содержит до 90% MgCl2,
4—5% MgO, 0,1—0,15% Н2О, остальное — СаС12 и примеси. Для
освобождения от MgO обезвоженный продукт подвергают пере-
переплавке85-86.
Обезвоживание хлористого магния возможно с помощью кар-
карбида кальция путем внесения его в расплавленный кристаллогид-
кристаллогидрат при температуре не ниже 180°87.
Предложено обезвоживать MgCl2 добавкой к его растворам
NaCl и КС1 E0% от веса MgCl2) и кипячением смеси при 350—
400° под слоем минерального масла, плавающего на поверхности.
За 1—1,5 ч происходит полное обезвоживание88.
В США применяют метод получения безводного MgCl2 на-
нагреванием MgO с хлористым аммонием, получаемым в производ-.
стве соды76. Фирма „Dow Chemical Co" запатентовала в ряде
стран89'90 непрерывное получение безводного хлористого магния
высокой чистоты (99,5%) MgCl2) путем обработки водного рас-
раствора MgC!2 аммиаком в присутствии NH4C1 — выделяющийся
гексааммиакат хлорида магния отделяют и подвергают терми-
термическому разложению с регенерацией аммиака. Концентрацию ам-
аммиака в реакционной смеси поддерживают в пределах 35—65%
при температуре от —20 до +10°. Процесс осуществляют в си-
системе реакторов непрерывного действия по двухступенчатой схеме.
Выход гексааммиаката хлорида магния составляет 70%). Прока-
Прокаливание отфильтрованного и промытого осадка ведут при 400°
в течение 4 ч.
Известны и другие варианты, например, обезвоживание-
MgCl2-2H2O газообразным аммиаком при 200° и 2 яг в кипящем
слое; образующийся аммиакат разлагают во 2-й зоне аппарата
Ври 400° и 1 от на MgCl2 и NH391. Запатентован92 способ, по
которому 20—50%-ный раствор MgCl2 обрабатывают при 105^
хлоридом аммония, в кипящем слое при 215° в токе азота полу-
получают безводную соль NH4Cl-MgCl2 с избытком 20%) NH4CI, затем
при 480° отгоняют NH4C1 и получают безводный MgCl2. Обезво-
Обезвоживание твердого гидрата хлорида магния можно вести, нагре-
нагревая его суспензию в углеводородах с температурой кипения 110—
500°93.
i
280
Гл. IX. Соединения магния
Хлорирование окиси магния
Образование MgCl2 по реакции MgO + Cb^MgCb + V2O2
при низких температурах возможно при малых концентрациях
хлора в газовой фазе, а при высоких температурах E00—700°)
необходима большая концентрация хлора (рис. 88). Практически,
в связи с малой скоростью реакции ниже 500°, процесс ведут при
высоких температурах85-94-95. Образование MgCb ускоряется в
присутствии восстановителя, так как при этом выводится из сферы
, ' , мосы*.) ^ реакции кислород. В каче-
'оо h 1"я * ап ?- стве восстановителя приме-
применяют твердый углерод или
окись углерода:
Магнезии
281
() ()
MgCl2 (ж.) + СО (г.) + 25,7 ккал
гЛ =
боо 7оо еоо
Температура,"С
Рис. 88. Равновесие
MgO + Cl2 ^
в системе
С твердым углеродом ре-
реакция протекает с достаточ-
достаточной полнотой при 800—
900°; с окисью углерода —
при более низкой темпера-
температуре. Окись магния должна
содержать минимальное ко-
количество примесей. Особен-
Особенно вредны примеси АЬО3 и
SiO2, продукты хлорирования которых уносятся из печи с газом.
Кроме того, они частично реагируют друг с другом и с MgO, об-
образуя силикатную корку, препятствующую диффузии хлора к ча-
частицам окиси магния.
Хлорирование ведут в шахтной электрической печи32, подобной
печам, применяемым для хлорирования титанистого и другого
минерального сырья (см. рис. 448, стр. 1490). Электрический ток
подводят к насадке из угольных цилиндриков A00ХЮ0 мм), рас-
расположенных двумя рядами (верхний и нижний) в пространстве
между электродами. В печь загружают брикеты, изготовленные
из тонкоизмельченного каустического магнезита, углеродистого ма-
материала и пека. В нижней части печи имеются фурмы для подачи
хлора и летка для периодического выпуска (каждые 3—4 ч) рас-
расплавленного хлористого магния.
Для брикетирования шихты можно применять хлористый маг-
магний 96, но, по-видимому, это не имеет особых преимуществ. Хло-
Хлорирование ведут при 1000—1050° в верхней части насадки и 800—
850° — в нижней, так, чтобы выходящий MgCl2 имел 700—750°. При
более высокой температуре происходит значительное испарение
MgC!2. Производительность печи, перерабатывающей в начале
кампании 13—16 т брикетов и 9—12 т хлора в сутки, постепенно
снижается вследствие загрязнения угольной насадки окислами и
уменьшения ее электропроводности. Поэтому периодически через
1,5—6 месяцев печь останавливают для чистки от шлаков и за-
замены насадки и одновременного ремонта огнеупорной футеровки.
На производство 1 т безводного хлористого магния расходуют:
550 кет ¦ ч электроэнергии, 90 кг угольной насадки, 3 кг угольных
электродов, 800—950 кг хлора.
Представляет интерес хлорирование окиси магния, суспенди-
суспендированной в расплаве хлоридов, в присутствии восстановителя;
процесс может быть осуществлен в барботажном тарельчатом ап-
парате97-98.
Безводный хлористый магний образуется также в качестве по-
побочного продукта при восстановлении магнием титана из TiCl4".
МАГНЕЗИИ
Существует много способов производства окиси магния и дру-
других форм магнезии 10°-101. Тяжелые формы получаются при обжиге
магнезита и доломита, термическом разложении сульфата магния
и гидролизе хлорида магния. Магнезии с различной степенью
активности получают осаждением из растворов гидроокиси магния
и основных карбонатов магния и их последующей термической
обработкой.
Тяжелые формы магнезии из магнезита и доломита
Обжигом высокосортного магнезита при относительно невысо-
невысоких температурах (~700°) получают магнезию, содержащую все
примеси, имевшиеся в магнезите. Под названием «каустический
магнезит» такую магнезию используют для изготовления цементов
и строительных материалов. При высокотемпературном обжиге
магнезита A500—1800°) получается неактивная форма окиси маг-
магния, состоящая из крупных зерен периклаза, она носит название
металлургического порошка и применяется для изготовления огне-
огнеупоров. Обжиг магнезита осуществляют в шахтных, вращающихся
и механических полочных печах 32>|02, а также в печах со взвешен-
взвешенным слоем. Для получения металлургического порошка предло-
предложено103-104 обжигать во вращающихся печах при 1500—1800° и
другие магнезиальные материалы — брусит, доломит или магнезит
в смеси с небольшим количеством гидратирующихся окислов
(MgO или СаО).
Известны разные способы получения окиси магния из доломити-
зированных материалов, загрязненных силикатами и кремнеземом.
282
Гл. IX. Соединения магния
Так, предложено обжигать доломит в смеси с силикатом и не-
небольшим количеством окиси железа, улучшающей спекаемость
сырья 105. Обожженный материал затем обрабатывают водой, при-
причем гидратируется и отмывается основная масса СаО, не вошед-
вошедшей в образовавшийся при обжиге феррит. От последнего окись
магния очищают магнитной сепарацией, а затем освобождают от
остатков Са(ОНJ обработкой соляной кислотой и промывают во-
водой 106.
На заводе фирмы «Kaiser Refractories» (СШАI07 кусковой доло-
доломит отделяют от примесей песка в сепараторе, заполненном измель-
измельченной смесью ферросилиция и магнетита, через которую проду-
продувают сжатый воздух. Плотность смеси 2,72—2,76 г/см3, а доломита
2,85 г/см3; поэтому он опускается на дно и отводится, а песок и
другие более легкие примеси остаются на поверхности материала,
заполняющего сепаратор. Доломит очищают от захваченных ча-
частиц ферросилиция и магнетита в виброкамерах и промывают. За-
Затем его обжигают во вращающихся барабанных печах длиной 91 м.
При прокаливании до 1800° с добавкой 5% окислов железа полу-
получается «пережженный доломит» с большой плотностью; его измель-
измельчают и обрабатывают при 80° нефтяным битумом для предотвра-
предотвращения гидратации. Этот материал используют в качестве метал-
металлургических флюсов. Доломит, предназначенный для получения
окиси магния, прокаливают не выше, чем при 1100°. Предложено
также осуществлять избирательную кальцинацию загрязненного
силикатами и кремнеземом доломита во взвешенном слое с одно-
одновременной сепарацией продукта. Образующийся порошок окиси
магния имеет зерна меньше 0,25 мм и увлекается газовым потоком
в сборник, а более крупные зерна примесей остаются и выводятся
из зоны обжига 108.
При получении каустического магнезита освобождение от при-
примеси SiO2, присутствующей в сыром магнезите, также может быть
достигнуто в процессе обжига. При быстром нагревании магнезита
до 600—800° примесь р-модификации кварца при 575° переходит
в а-модификацию с превращением его в тончайший порошок, ко-
который легко выносится из печи проходящими через нее газами 109.
Бикарбонатный способ получения магнезии
Путем дальнейшей химической обработки продуктов обжига
магнезита и доломита можно получить из них легкие формы маг-
магнезии. Одним из методов такой обработки является перевод мало-
малоактивной окиси магния в раствор в форме бикарбоната магния с
последующим осаждением из него активной магнезии. Окись маг-
магния, полученную при обжиге магнезита, размалывают и гасят во-
водой. Образующуюся суспензию—магнезиальное молоко подвер-
подвергают карбонизации двуокисью углерода в автоклавах под давле-
Магнезии
283
нием выше 5 ст. При этом образуется раствор бикарбоната
магния
Mg(OH), + 2СО2 = Mg(HCO3)s
который после отделения от твердых примесей при дальнейшем ки-
кипячении гидролитически разлагается:
2Mg(HCO3J = MgCO3 + Mg(OHJ + ЗСО2 + HSO
Образующийся осадок основного карбоната машия отделяют
и после сушки получают в виде тончайшего порошка — магнезии
альба.
1000
900
800
700
бое
1
с
а
I
о"
и
о!
560
400
300
200
ЮО
1 1
/
7
/
/
2
3
Время
ЮО 700
Время, мин
300
Рис. 89. Термограммы диссоциа-
диссоциации карбонатов кальция и м^гияя
в атмосфере СО2 при нормальном
давлении:
/ — известняк; 2 — доломит; 3 — м агнезит.
Рис. 90. Скорость диссоциации доло-
доломита при 620°:
/ — с добавкой NaF; 2-е дебавкой NaCI;
3—без добавки.
Аналогичным путем получают магнезию и из доломита. При об-
обжиге диссоциация доломита
MgCO3 • СаСО3 = MgO + СаО ¦+- 2СО, - 74 кка i
протекает в две стадии (рис. 89). Разложение MgCO3 доломита
происходит при ~730°, т. е. на ~80° выше, чем разложение
MgCOs магнезита. Это обусловлено тепловым эффектом образова-
образования CaMg(CO3J- Опытным путем установлено отсутствие стадии
предварительного распада доломита на образующие его карбона-
карбонаты "°. При 730° доломит разлагается с образованием MgO и твер-
твердого раствора карбонатов, обедненного углекислым магнием111:
nCaMg(CO3J = (п - 1 )MgO + MgCO3 • «CaCO3 + (я - 1 )СО2
Повышением температуры до 910° достигается дальнейшая дис-
диссоциация твердого раствора:
MgCO3 • яСаСО3 = MgO + иСаО + (я + 1 )СО2
Скорость диссоциации долом.ита меньше, чем магнезита; ©на
увеличивается в присутствии 1% фторида или хлорида натрия
284
Гл. IX. Соединения магния
ц
WSO"
(рис. 90K2'112-113. Обжиг доломита ведут при 700—800° или
1100—1250° в зависимости от назначения получаемого продукта.
При неполном обжиге разлагается лишь MgCO3 и получается так
называемый полуобожженный доломит. Его можно, в частности,
получить при быстром нагревании доломита в закрытой зоне вра-
вращающейся печи до 750—800° в атмосфере СО2 в течение 15 мин и
дальнейшем охлаждении до 500° в течение 30 мин .
При гашении водой полностью обожженного доломита обра-
образуется суспензия гидроокисей магния и кальция:
(MgO + СаО) + 2Н2О = Mg(OHJ + Ca(OHJ
Скорость гидратации MgO уменьшается с повышением темпе-
температуры обжига и особенно резко выше 1300° (рис. 91); она умень-
^ шается также в присутствии
примесей СаО, Sfo2, А12О3 и
других и зависит от размеров
частиц окиси магния. При
отстаивании суспензии гидро-
гидроокись магния оказывается в
верхнем слое, что позволяет
производить этим методом гру-
грубое отделение ее от других
твердых компонентов. Неболь-
Небольшие добавки MgCl2 ускоряют
гидратацию, но при этом воз-
возможно образование оксихло-
ридов П6.
Для получения относитель-
относительно крупных кристаллов
Mg(OHJ в суспензии гаше-
гашение ведут при 95—100°, нагре-
й ф
4 в 12 16 20 24 2в 32 36
Время гидратации, сутки
Рис. 91. Скорость гидратации окиси
магния, обожженной лри разных тем-
температурах. Начальная влажность 25%;
температура 20°.
вая пульпу, содержащую ~17% твердой фазы, острым паром.
Для разделения гидроокисей магния и кальция пульпу разбавляют
до содержания твердой фазы 11%, охлаждают до 60° и подвергают
карбонизации. При этом Са(ОНJ переходит в СаСО3, a Mg(OHJ
остается без изменения:
Са(ОНJ + Mg(OHJ + СО2 = СаСО3 + Mg(OHJ + Н2О
В интервале 40—60° карбонизация гидроокиси кальция идет с
максимальной скоростью. Осаждение СаСО3 осуществляют в сталь-
стальных аппаратах под атмосферным давлением, подавая в них турбо-
газодувками газ из обжиговых печей, содержащий до 40% СО2.
Конец карбонизации гидроокиси кальция определяют по электро-
электропроводности пульпы — после окончания карбонизации Са(ОНJ
электропроводность резко увеличивается вследствие начала карбо-
яизации Mg(OHJ32. Полученную пульпу направляют на дальней-
дальнейшую карбонизацию, причем образуется раствор бикарбоната маг-
магМагнезии
285
ния. Растворимость MgO в, воде, насыщенной двуокисью углерода,
показана на рис. 92. Во избежание выделения в осадок карбоната
магния при получении бикарбоната магния температуру не подни-
поднимают выше 26°. Бикарбонатный раствор, отделенный от шлама
(СаСО3, SiO2 и др.), разлагают, нагревая его при перемешивании
до 45—50°. При этом образуется основной карбонат магния
3MgCO3 • Mg(OHJ, который высушивают и выпускают в качестве
легкой магнезии. Очистка раствора от соединений железа и мар-
марганца может быть осуществлена добавкой .
к раствору гидратированного углекислого tB
магния 117.
Скорость растворения Mg(OHJ при
карбонизации водной суспензии опреде- J- /4
ляется скоростями процессов растворения
и гидратации двуокиси углерода 118. Прак-
Практически скорость карбонизации гидроокиси "^
магния зависит от интенсивности переме- 9»
шивания суспензии и от парциального дав- 2
ления СО2 в газе. Оптимальное давление *
СО2 — 2,5—3 ат; при таком давлении рас-
растворяется 80—90% MgO и образуется пе-
пересыщенный раствор бикарбоната, содер-
содержащий 25—30 г/л MgO. Добавка к магне-
магнезиальной суспензии 25 г/л MgSO4-7H2O
облегчает растворение MgO и способствует
стабилизации пересыщенного бикарбонат-
s /5-1
12
е ¦
20 40 60 80 100
Температура, "Е
ного раствора 11В. "Средняя скорость раство- Рис< 92. РаствОрИМОсть
рения гидроокиси магния и концентрация MgO в воде, насыщен-
образующегося пересыщенного раствора ной двуокисью углерода.
Mg(HCO3J растут с увеличением концен-
концентрации MgO в суспензии до 30 г/л, а затем уменьшаются. На
степень возможного пересыщения раствора сильно влияют также
размеры частиц гидроокиси магния, зависящие от условий об-
обжига исходного магниевого минерала (прежде всего от темпе-
температуры обжига120). При карбонизации полуобожженных доломитов
наблюдается большая растворимость бикарбоната магния при ма-
малой длительности карбонизации (вследствие пересыщения раство-
раствора), но с увеличением продолжительности процесса содержание
его в растворе уменьшается, а в осадок выделяется основная угле-
углекислая соль магния, смешивающаяся с нерастворимыми состав-
составными частями доломитов121. В суспензии обожженного доломита
при обработке СО2 под давлением образуется двойная соль
Mg(HCO3J • MgCO3; при давлении 16аг в твердой фазе нахо-
находится MgCO3-3H2O, а в растворе Mg(HCO3J; при 60 ат выде-
выделяется в осадок MgCOs, а количество магния в растворе — 24 г/л
(в пересчете на MgCO3 • ЗН2ОI22.
286
Гл. IX. Соединения магния
Магнезии
287
Декарбонизация раствора бикарбоната магния с получением
основной соли ускоряется при продувке через раствор воздуха.
Однако этот воздух значительно снижает концентрацию выделяю-
выделяющейся в этом процессе двуокиси углерода, что затрудняет ее воз-
возврат на карбонизацию. Поэтому вначале разложение раствора ве-
ведут только при механическом перемешивании с использованием
выделяющегося газа. После снижения концентрации MgO до 4 г/л
через раствор начинают продувать воздух, причем выделяющийся
газ выбрасывают в атмосферу.
Для получения основного карбоната магния разложение бикар-
бонатного раствора ведут при температуре выше 45е, потому что
при 25° из пересыщенного метастабильного раствора Mg(HCO.4J
выделяется не основная соль, a MgCO3 • ЗН2О (кристаллы ромби-
ромбической системы), а при 10° —MgCO3-5H2O (кристаллы моноклин-
моноклинной системы); превращение MgCO3-5H2O в MgCO3-3H2O происхо-
происходит при 13,9° 12°-123-125. Предложено производить разложение би-
карбонатного раствора, «мгновенно» нагревая его до 100° инжекти-
инжектированием струей пара в реакционную камеру, где в мельчайших
капельках раствора образуются частицы со средним размером
~2 мк, имеющие состав 5MgO • 4СО2• Н2О, т. е. 4MgCO3-
•Mg(OHJ123. Предложено также обрабатывать раствор Mg(HCO3J
при 80—90° 2—5%-ным щелочным или канифольным мылом;
выделяющийся при этом осадок основной соли MgCO3 • Mg(OHJ.
зерна которого покрыты мылом, рекомендуется в качестве напол-
наполнителя тля резиновых смесей 126.
Карбонизацию суспензий, содержащих Mg(OHJ, с последую-
последующим разложением М?г(НСОз)9 можно использовать и для очистки
технических окиси или гидроокиси магния; в этом случае после от-
отделения примесей от раствора Mg(HCO3J его высушивают и про-
прокаливают или прокаливают полученную из него магнезию. При
энергичном перемешивании раствора бикарбоната магния с водной
суспензией гидроокиси магния ниже 50° получается карбонат
магния:
СО3J + Mg(OH)s = 2MgCO3 + 2Н2О
Для увеличения активности рекомендуется выдерживать его при
температуре ниже 27°127.
Из руд, содержащих брусит (MgO ¦ Н2О) можно извлекать
окись магния выщелачиванием раствором, насыщенным СО2 и SO2
(рН=4,5—5,5); при повышении рН полученного раствора MgHSO3
до ~9 добавкой MgO или Mg(OHJ осаждается MgSO3-6H2O,
который затем перерабатывают в окись магния с регенерацией
SO2. Часть окиси магния возвращают в процесс128.
Предложено129 получать окись магния из обожженного магне-
магнезита или брусита циклическим способом с помощью этаноламина.
Исходный материал диспергируют при 30° в течение 8 мин в про-
карбонизированном 30%-ном растворе моноэтаноламина. После
отделения от шлама на фильтре остается пересыщенный раствор
A5—17 г/л MgO). Через 45—50 мин из него начинает выделяться
чистый MgCO3, который отфильтровывают и прокаливают, а филь-
фильтрат вновь насыщают двуокисью углерода и возвращают в цикл.
Продукт содержит больше 95% MgO.
Аммиачные способы получения магнезии
В Пейнсвилле (Охайо, США) крупное производство окиси маг-
магния из доломита, содержащего 20% MgO, комбинируется с произ»
водством кальцинированной соды. Газ, получаемый при обжиге
доломита в шахтных печах и содержащий до 40% СО2, исполь-
используется в содовом производстве и для переработки гидроокиси маг-
магния. Доломитовое молоко направляют на станцию дистилляции
содового завода, где оно взаимодействует с раствором хлористого
аммония (с фильтровой жидкостью). При достаточном количестве
доломитового молока, обеспечивающем эквивалентное соотноше-
соотношение между СаО и NH4C1, гидроокись магния не вступает в реак-
реакцию, остается неизменной, а Са(ОНJ превращается в СаС12:
Mg(OH), + Са(ОНJ + 2NH4C1 = Mg(OHJ + СаС12 + 2NH3 + 2Н2О
После отгонки NH3 суспензия Mg(OHJ в растворе СаС12
уплотняется, гидроокись магния отфильтровывают, промывают и
прокаливают для превращения в окись магния76. Для получения
таким способом окиси магния, удовлетворяющей требованиям
стандарта на металлургический порошок 2-го класса, необходима
тонкая очистка известково-магнезиального молока и работа без
избытка окиси кальция 77.
По другому варианту дистиллерная жидкость, полученная при
обработке фильтровой жидкости доломитовым молоком, подвер-
подвергается карбонизации, в результате которой осаждается СаСОз,
a Mg(OHJ превращается в MgCl2:
Mg(OHJ + Ca(OHJ + 2NH4C1 + CO2 = MgCls + CaCO3 + 2NH3 + ЗНгО
Затем полученный раствор хлористого магния перерабатывают
на магнезию.
Гидроокись магния может быть получена одновременно с суль-
сульфатом аммония при обработке раствора MgSO4 аммиаком и дву-
двуокисью углерода iso—1зз
. Известковый способ получения магнезии
Этот, способ заключается в осаждении гидроокиси магния
из растворов хлористого магния известью (известковым моло-
молоком). Вследствие того, что растворимость Mg(OHJ (стр. 2651
288
Гл. IX. Соединения магния
значительно меньше, чем Са(ОНJ A,6г/л при 25°), реакция
MgCl2 + Са (ОНJ = CaCls + Mg (OHJ
идет в сторону образования Mg(OHJ. Гидроокись магния отфиль-
отфильтровывают, тщательно промывают водой и прокаливают при не-
невысоких температурах. В зависимости от режимов осаждения и
прокаливания получаются разные сорта магнезии. При обработке
известью непосредственно морской воды SO?" не осаждается
вследствие относительно высокой растворимости CaSO4, и в оса-
осадок выделяется чистая гидроокись магния, но ее концентрация
всего 3 г/л. После отстаивания в сгустителях концентрация повы-
повышается до 100 г/л, а с помощью вакуумной фильтрации она может
быть увеличена до 350 г/л134.
Трудность получения гидроокиси
магния этим способом заклю-
заключается в том, что без соблюдения
специальных условий осадок
Mg(OHJ выделяется в коллоид-
коллоидной форме, что сильно затруд-
затрудняет его отстаивание, отфильтро-
вывание и промывку. Для полу-
Магнезии
289
to-5
7
5
3-Ю'1
у
е-
&
8
2-Ю-
1 2
Время, я
чения легко фильтрующего осад-
осадка необходимо создать условия
для медленной кристаллизации
при малом пересыщении раство-
фильтрации от продолжительности
осаждения Mg (OHJ известковым
молоком.
Рис. 93. Зависимость коэффициента ра и при относительно неболь-
шом количестве центров кристал-
лизации
для замедления кристаллиза-
кристаллизации Mg(OHJ применяют сухую молотую малоактивную известь,
получаемую из соответствующих сортов известняка при особых
условиях обжига, а также известковые макароны (в ФРГ); для
этой же цели в США гашение извести производят не водой, а ма-
маточным раствором. При использовании для осаждения Mg(OHJ
сухой малоактивной извести производят очистку магнезиального
молока от непрореагировавшей извести и других твердых приме-
примесей гидромеханическими способами.
На рис. 93 показана зависимость коэффициента фильтрации
К = eL ¦ см/сек (где Q — количество см3 фильтрата, получаемого
с поверхности S см2 за т сек при напоре Н см. вод. ст. и толщине
слоя осадка / см) осадка гидроокиси магния от длительности
приливания известкового молока к раствору MgCl2135. При бы-
быстром приливании известкового молока рН раствора в зоне обра-
образования осадка больше, чем для Mg(OHJ — в этих условиях (на
кривой участок /) рост кристаллов затруднен и фильтруемость
осадка незначительная. По мере увеличения длительности прили-
приливания щелочи величина рН в зоне реакции все меньше отличается
от рН для Mg(OH)a, торможение роста кристаллов уменьшается и
коэффициент фильтрации возрастает (участок //). После того как
длительность приливания известкового молока становится столь
большой, что каждая капля щелочи полностью используется пре-
прежде чем в раствор попадает следующая капля, дальнейшее замед-
замедление приливания молока уже не влияет существенно на фильт-
фильтрующие свойства осадка (участок ///). Гидроокись магния, полу-
полученная при одночасовом приливании известкового молока, хорошо
отстаивается, через 2—4 ч после осаждения легко отмывается оя1
иона хлора и содержит 0,08—0,1% Са. После двухдневного стоя-
стояния осадок не удается отмыть от ионов хлора, а содержание каль-
кальция в нем превышает 1%. При вливании раствора магниевой соли
в щелочь частицы осадка представляют собой студенистые обра-
образования. Наоборот, при приливании щелочи к раствору магниевой
соли частицы получаются в виде плотных и прочных пластинок
с резко очерченными краями и легко отфильтровываются ш-137.
Для получения более плотного и в 5—7 раз менее загрязнен-
загрязненного окисью кальция осадка Mg(OHJ рекомендуют вводить в рас-
раствор MgCl2 (до приливания к нему щелочи) различные добавки,
например, 0,03% (ЫаРО3)б,188 или формалин139. Предварительное
подкисление морской воды соляной кислотой до рН =¦ 4 и про-
продувка ее воздухом для удаления СО2 также позволяет получить
более крупные кристаллы и, следовательно, легко отстаивающийся
осадок Mg(OHJU0. Существенное значение для образования хо-
хорошо отстаивающегося осадка имеет энергичное турбулентное пе-
перемешивание реагирующих растворов, при котором стехиометриче-
ским количеством известкового молока удается в течение 15—
20 мин при рН = 10,7—11,3 получить осадок Mg(OHJ, состоящий
из мелких хлопьев, оседающих со скоростью ~250 см/ч141.
При осаждении гидроокиси магния из разбавленных растворов,
например, из морской воды, можно применять метод наращивания
кристаллов Mg(OHJ, заключающийся в том, что осадок, получаю-
получающийся после первого осаждения в виде геля, вносится в раствор,
предназначенный для дальнейшего осаждения и т. д. После не-
нескольких наращиваний осадок приобретает зернистую структуру.
По мере увеличения числа наращиваний улучшается фильтруе-
фильтруемость осадка и уменьшается количество удерживаемой им жидкой
фазы. Практически достаточно 3—4 операций наращгзания для
приготовления осадка, пригодного для внесения в раствор MgClj
перед приливанием к нему известкового молока. Затем можно
часть выделенной из суспензии, легко отстаивающейся гидроокиси
магния переработать на продукт, а часть использовать для обра-
обработки новой порции раствора и т. д. Легкая магнезия, получаемая
методом наращивания, имеет насыпную плотность 200—300 кг/м3.
М. Е. Позив
290
Гл. IX. Соединения магния
Метод этот, однако, непригоден для переработки рассолов с высо-
высокой концентрацией MgCl2 ш- ш.
Для получения магнезии известковым способом применяют ма-
маточные растворы после кристаллизации солей из естественной
рапы, карналлитовый щелок, морскую воду и др. В зависимости
от концентрации растворов, режима осаждения, промывки и высу-
высушивания получаются различные сорта магнезии. Рапы, содержа-
содержащие ионы SO4 , предварительно обессульфачивают, добавляя к
ним раствор СаС12, получаемый после осаждения Mg(OHJ изве-
известковым молоком. При медленном добавлении (~30 мин) ра-
раствора СаС12 в перемешиваемую рапу, имеющую слабощелочную
реакцию, при 20° осадок гипса образуется в виде спаянных друз,
что обеспечивает быстрое его отделение отстаиванием 144.
Производство гидроокиси магния из морской воды известковым
способом осуществляется в США с 1928 г. Морскую воду обраба-
обрабатывают вначале небольшим количеством Са(ОНJ в аппарате с
вращающимися лопастями с целью флокуляции органических
примесей. После осветления в отстойнике воду пропускают через
песочный фильтр и направляют в реактор-отстойник, куда подают
и известковое молоко. Раствор из отстойника сбрасывают в море
а пульпу, состоящую из 12% Mg(OHJ и 88% морской воды, со-
содержащей ~3% солей, промывают противотоком в батарее де-
кантеров пресной водой. В результате промывки концентрация
Mg(OHJ в пульпе снижается до 10%, а содержание солей умень-
уменьшается до 0,05%- Часть пульпы подвергают карбонизации с даль-
дальнейшей переработкой в легкую магнезию. Другую часть пульпы
пропускают через сито с 16000 отв/см2 и выпускают в виде про-
продукта—магнезиального молока [с содержанием 9% Mg(OHJ].
Третью часть пульпы обрабатывают паром и фильтруют на бара-
барабанном вакуум-фильтре — полученную пасту, содержащую 30%
Mg(OHJ, также выпускают в виде готового продукта. Эту же па-
пасту перерабатывают в другие товарные продукты —высушиванием
ее в распылительной сушилке получают сухую гидроокись магния,
а прокаливанием ее —тяжелую окись магния145. Обжигом отфиль-
отфильтрованной гидроокиси магния при 750—800° получают легкую маг-
незию с объемным весом 0,55 кг/л. Тяжелую магнезию с объемным
весом 1,9 кг/л получают в результате продолжительного обжига
гидроокиси магния с присадкой Fe2O3 при 1700° 146. Кроме извест-
известкового молока, для осаждения гидроокиси магния применяют так-
также доломитовое молоко и обожженный доломит147.
На рис. 94 показана схема производства окиси магния из мор-
морской воды, по которой работает завод фирмы «Kaiser Refractories»
(США) 148. Профильтрованная морская вода G6 мг/мин) допол-
дополнительно фильтруется через слой обожженного доломита в трех
резервуарах 3 диаметром по 38 м. При этом удаляются растворен^
ная СО2 и бикарбонат кальция (в виде карбоната кальция) и
10*
Рис. 94. Схема производства окиси магния из морской воды на заводе Kai&er Refractories (США):
/ — бункер для обожженного доломита; 2~рез»рвуар для суспензии доломита; 3 — резервуар с доломитом; 4 — резервуар; 5 — пер"
вичный реактор; 6— вторичн-ый реактор; 7 —гребковый классификатор; S — сгуститель; 9 — промывной сгуститель: 10 — вакуум-
фильтр; .11 — шнековый пи-татель; 12 — шаговая мельница; C—печь для периклаза; 14 — U-подовая печь; 15 — бункер для окиси
магния; 16 — мельница для тонкого измельчения; 17 — смеситель; IS — шнек; 19 — элеватор; 20 — брикетный пресс; 21 — снто; 22 — бункер
для брикетов; 23 — подогреватель; 24 — печь для прокаливания брикетов; 25 — холодильник; 26 — дробилка; 27 — вентилятор;
2S — пылеуловитель.
292
Гл. IX. Соединения магния
Магнезии
293
содержание СО2 в воде снижается в 4 раза, а концентрация магния
уменьшается на 10%. Осаждение Mg(OHJ и отделение SiO2, Fe2Os
и других твердых примесей производят в двух последовательно
работающих первичных реакторах 5, в которые вводят обожжен-
обожженный доломит. Суспензия Mg(OHJ поддерживается мешалками во
взвешенном состоянии и идет в слив, а пульпа поступает во вторич-
вторичный реактор 6, где образуется добавочное количество Mg(OHJ,
возвращаемого в первичные реакторы. Шлам из вторичного реак-
реактора (SiO2, Fe2Os) направляется в гребковый классификатор 7, а
оттуда в отвал. Суспензия Mg(OHJ поступает в три параллельно
работающие сгустителя 8 диаметром 75 м. Верхний слив сгустите-
сгустителей сбрасывают в море, а шлам Mg(OHJ подвергают противоточ-
ной промывке в двух сгустителях 9 (диаметром 75 м), а пульпу,
содержащую 25% Mg(OHJ, направляют на пять барабанных
вакуум-фильтров 10 диаметром 4,2 м, длиной 5,4 м, работающих
при 500 мм рт. ст. Фильтрат возвращают в сгустители, а кек, со-
содержащий 50% Mg(OHJ, перерабатывается в магнезию. Часть
кека с добавками, понижающими температуру спекания, вводи-
вводимыми в шаровой мельнице 12, обжигают в печи 13 при 1850° в те-
течение 2,5—4 ч для получения гранул периклаза (84—97% MgO).
Основную часть кека обжигают в 11-подовой печи 14, где полу-
получается активная окись магния. Ее измельчают и выпускают в ка-
качестве готового продукта с размером частиц меньше 0,04 мм.
Часть выгружаемой из печи окиси магния перерабатывается на
зерненый продукт с размерами частиц 9 мм. Для этого ее пропу-
пропускают через шаровую мельницу, смешивают с водой в смесителе 17
и брикетируют в прессе 20 под давлением 140 ат; затем брикеты
с размерами 15 X 18 мм прокаливают во вращающейся печи в те-
течение 2 ч, охлаждают и дробят. Продукт содержит больше 98%
MgO. По описанной схеме 1 т MgO получают из 273 м3 морской
воды и 1 т обожженного доломита (содержащего 38—40% MgO).
В 1967 г. в Мексике пущен завод 149 по получению MgO из мор-
морской воды, которую сначала обрабатывают серной кислотой для
регулирования рН и суспензией Mg(OHJ из осветлителя. Затем
классифицированной известью осаждают Mg(OHJ, который отде-
отделяют в сгустителях диаметром ~ 100 м. Сгущенная суспензия про-
проходит последовательно через три противоточных декантационно-
промывных аппарата и затем обезвоживается на крупнейшем ба-
барабанном вакуум-фильтре диаметром ¦~3,5 м, длиной ~8,5 м.
Кек, содержащий 55% твердого, сушат и прокаливают в двух
12-подовых печах. Продукт содержит 98% MgO. Извлечение маг-
магния 95%.
Описан способ высушивания суспензии гидроокиси магния на
поверхности вальцов, обогреваемых паром G атI50.
Обессульфаченные рассолы морского типа могут служить ис-
источником окиси магния, предназначенной для изготовления огне-
огнеупоров. Рапу обрабатывают мелкоизмельченной негашеной из-
известью181 (или известковым или доломитовым молоком162), обра-
обрадовавшийся осадок промывают, высушивают, размалывают и обжи-
обжигают. Продукт, полученный из рапы Сиваша, содержит 95% MgO,
0,5% СаО, 1,6% ИгОз, 1,9% SiO2; изготовленные из него огнеупоры
по качеству превосходят огнеупоры, изготовленные из Саткинского
магнезита.
Раствор хлористого магния для осаждения магнезии получают
также из доломита. Обожженный при 800—900° доломит размалы-
размалывают и обрабатывают водой. Полученное доломитовое молоко кар-
бонизуют газом, выходящим из обжиговых печей. При карбониза-
карбонизации получается значительно более активная, чем доломит, смесь
карбонатов магния и кальция. Эту смесь подвергают обработке
раствором хлористого кальция при нагревании в автоклавах под
давлением около 4 ат, причем MgCO3 переходит в MgCl2 по ре-
реакции:
MgCO3 + СаС12 - MgCl2 + СаСО3
Полученный раствор хлористого магния отделяют от осадка
СаСО3 и перерабатывают в магнезию осаждением известковым
молоком. Осаждение магнезии из растворов хлористого магния
может быть произведено и доломитовым молоком, содержащим
Са(ОНJ:
MgCl2 + Mg(OHJ + Ca(OH)j = CaCls + 2Mg(OHJ
После отделения гидроокиси магния раствор, содержащий хло-
хлористый кальций, может быть использован для обработки прокар-
бонизованного доломитового молока и превращен в MgCl2. Изучен
процесс получения окиси магния из доломита и содержащих СаС12
отработанных щелоков содового производства 153. Недостатком из-
известкового способа получения магнезии является загрязнение про-
продукта значительным количеством окиси кальция. Для получения
Mg(OHJ с малым содержанием СаО следует применять свеже-
свежеприготовленное разбавленное известковое молоко при низкой тем-
температуре, а также удалять СО2 из воды и рапы. При взаимодей-
взаимодействии разбавленной рапы и доломитового молока с концентрацией
10% твердой фазы получается гидроокись магния с содержанием
4—5% СаО. Такое содержание СаО в 2—3 раза меньше, чем в
гидроокиси, полученной с помощью известкового молока. Магне-
Магнезиальный клинкер, пригодный для основных магнезиальных огне-
огнеупоров, содержащий меньше 1—2% СаО, можно получить, если
предварительно удалять из рапы ионы SOl~> обрабатывая ее до-
доломитовым молоком 154.
Отделение гидроокиси магния от непрореагировавших частиц
и примесей (СаО, S1O2) можно производить гидромеханиче-
гидромеханическими методами — отстаиванием, центрифугированием и т. д.
294
Гл. IX. Соединения магния
Предложено, например, разделять суспензию отстаиванием на две
фракции — тонкую, в которую переходит 60—70% Mg(OHJ, с со-
содержанием СаО и БЮг на 20—30% меньше, чем в исходном мате-
материале, и грубую, в которой СаО и БЮг на 30—40% больше, чем
в исходном шламе '®5. Очистку суспензии гидроокиси магния от
крупки (т. е. от непрореагировавшей и окклюдированной извести)
можно осуществлять в классификаторе непрерывного действия, а
сгущение суспензии в 7—9 раз — в отстойнике непрерывного дей-
действия. Однако при этом очень велик расход промывной воды — он
составляет ~ 150—200 м3 на 1 г твердой фазы. Уменьшения рас-
расхода промывной воды до 40—90 ms/t и сокращения площади от-
отстойников можно достичь при предварительном сгущении суспен-
суспензии в осадительной или фильтрующей центрифуге1б6.
По сравнению с отстаиванием и фильтрацией центрифугирова-
центрифугирование дает наилучшие результаты. При отношении в исходной сус-
суспензии Ж:Т, равном 10:1, удельная производительность отжим-
отжимной (осадительной) центрифуги составляет 386 кв/(ч-м2), степень
разделения 97% и влажность отжатого осадка 50%. Удельная
производительность центрифуги в ~4 раза больше производитель-
производительности барабанного фильтра 157.
Изучено электроосмотическое обезвоживание пасты гидроокиси
магния. Оно позволяет понизить влажность осадка в 3—5 раз и
довести ее до ~20%. Расход энергии на электроосмотическое обез-
обезвоживание в 15 раз меньше, чем при испарении воды157'158.
Одним из перспективных способов получения окиси магния с
малым содержанием окиси кальция является двухфазный процесс
обработки морской или озерной рапы известью или обожженным
доломитом159. Сущность способа заключается: 1) в неполном (на
85%) осаждении гидроокиси магния известью из раствора хлори-
хлористого магния и 2) в завершении взаимодействия непрореагировав-
ших MgCU и Са(ОНJ, а также СаСОз при обжиге:
Магнезии
295
= MgO + СаС12 + Н2О
+ CaCl2
Ca(OHJ + MgCl2 =
CaCO3 + MgCl2 =
После отмывки хлористого кальция из обожженного продукта
водой получается паста гидроокиси магния, содержащая (в прока-
прокаленном веществе) 97—99% MgO, 0,1—0,4% СаО, 0,5—1,8% SO3,
0,3—0,7% С1~. На рис. 95 представлена схема производства этим
способом. Обожженный доломит (или комовую известь) орошают
небольшим количеством рассола хлористого магния и гасят в ма-
машине для безотходного гашения160-161. При этом получается кру-
крупитчатая пульпа (—0,5 мм). Рассол дозируют таким образом, что-
чтобы пульпа имела достаточную текучесть, а парообразование при
гашении, производящемся при ~80°, не было чрезмерно интенсив-
интенсивным. Выходящую из гасильной машины 2 горячую пульпу
?!
о п.- 2 а
Si
разбавляют дополнительным количеством рассола (до 90% от
его общего расхода) и направляют в батарею агитаторов 5, где
она перемешивается в течение 1,5 ч, м ......
а.затем фильтруют. Осадок (кек) про- Я 1||
питывают рассолом хлористого маг- 2. 5|ш
ния в количестве 10—15% от общего
его расхода, составляющего 105—
110% от стехиометрического по отно-
отношению к извести. Кек, пропитанный
рассолом, обжигают в течение 1 ч во
вращающейся печи 8 при 800°. Отхо-
Отходящие печные газы содержат неболь-
небольшое количество НС1 (они могут быть
направлены на подкисление рапы пе-
перед извлечением из нее брома или
очищены перед выпуском в атмосферу
другим способом). Выходящий из пе-
печи спек смешивается в холодильном
барабане с раствором, полученным
при последующей промывке его водой.
Магнезиальная паста, полученная пос-
после отмывки спека от СаС12, идет на
изготовление магнезитового кирпича. ^1 «» в*
Применительно к обессульфачен-
ной рапе Сиваша разработана другая
схема 162> ш. Хорошо обожженный до-
доломит (не менее чем на 98%) гасится
небольшим количеством рапы в га-
гасильном барабане, и полученную пуль-
пульпу направляют в шаровую мельницу,
куда добавляют рапу. Здесь происхо-
происходит гидратация окиси кальция, взаи- Г°*^ТТ Г
модействие гидроокиси кальция с хло-
хлористым магнием и частичная гидра-
гидратация окиси магния из обожженного
доломита. Эти процессы завершаются
в 4-х последовательно работающих ре-
реакторах с мешалками, где происходит
формирование частиц гидроокиси маг-
магния определенной структуры и разме- I | Ч^Л!4
ров. В реакторы поступает слив из "* **
классификатора, работающего в зам-
замкнутом цикле с шаровой мельницей, а
также остальное количество рапы (из
расчета общего расхода, равного
110%, от стехиометрического по окиси
о
•К
IS К I S
il
а> s a S к
X Я и Е 4
31 в
Si!
I ЙД
L
296
Гл. IX. Соединения магния
кальция). Выходящую из последнего реактора магнезиальную
пульпу перерабатывают в пасту методами отстаивания, фильтра-
фильтрации, центрифугирования и электроосмотического обезвоживания
и затем прокаливают. Окись магния, полученная по этой схеме
из рапы Сиваша в лабораторных условиях, содержала 99,5% MgO
0,1% СаО, 0,2% SiO2, 0,1% R2O3) следы R2O.
Предложено также получать гидроокись магния из морской
воды смешением ее с продуктом неполного гашения обожженного
доломита [Са(ОНJ + MgO], предварительно очищенным воздуш- |
ной сепарацией от крупных частиц примесей СаСО3 и CaSO4. Маг-
Магнезиальную пульпу фильтруют на дисковом фильтре через ткань
из найлона 164. Запатентован 165 следующий процесс: гашение тонко
измельченного обожженного доломита водой при 80—100° в тече-
течение 2 ч, добавка к полученной суспензии рассола MgCl2 в количе-
количестве 103—110% от стехиометрического для связывания Са(ОНJ,
перемешивание 6—12 мин, гидравлическая классификация с целью
вывода из суспензии тяжелых примесей, затем фильтрование и
промывка Mg(OHJ. Предложено166 получение Mg(OHJ из обож-
обожженного доломита с размером частиц 0.1—0,6 мм, суспендирован-
суспендированного в растворе MgCl2, вести при 20° в псевдоожиженном слое без
репульпирования и фильтрования. После прокаливания Mg(OHJ
образуется продукт, содержащий 88—92,5% MgO, 2,8—4% СаО,
1,85-2,25% SiO2, 0,9-1,65% R2O3.
Гидросульфидный способ получения гидроокиси магния
Получение магнезии может быть совмещено с производством
хлористого бария из сернистого бария. При обработке BaS раство-
раствором СаС12 образуются хлорид и гидросульфид бария:
2BaS + СаС12 + 2Н2О = BaCls + Ba(HSJ + Ca(OHJ
Раствор ВаС12 и Ba(HSJ после отделения осадка Са(ОНJ
обрабатывают раствором MgCl»:
Ba(HSJ + MgCl2 + 2Н2О - Mg(OHJ + ВаС12 + 2H2S
Отфильтрованный и промытый осадок Mg(OHJ прокаливанием
лревращают в магнезию. Для получения из BaS осадка Mg(OHJ,
легко отделяемого от раствора, предварительно уменьшают рН
раствора, насыщая его сероводородом (стр. 441).
Содовый способ получения легких магнезии
Этот способ, наиболее старый, заключается в осаждении основ-
основного карбоната магния из растворимых солей магния (MgSO4,
Mg(NOsJ и MgCl2) содой. В качестве сырья используют при-
природные и промышленные растворы магниевых солей, а также маг-
магнезит.
Магнезии
297
Магнезит измельчают в шаровой мельнице до размера частиц
Q2—0,1 мм и растворяют в 28%-ной серной кислоте при нагре-
нагревании до температуры кипения острым паром. Полученный раствор
сульфата магния очищают от железа окислением Fe2+ в Fe3+ хлор-
хлорной известью и осаждением в виде Fe(OHK. Очищенный раствор
MgSO4 обрабатывают 10—12% раствором соды. Суспензию вы-
выделившегося осадка основного карбоната магния нагревают ост-
острым паром до кипения — пропаривают, после чего состав осадка
приблизительно соответствует формуле соединения, образующе-
образующегося по реакции:
4MgSO4 + 4Na2CO3 + 4Н2О = 3MgCO3 • Mg(OHJ • ЗН2О + 4Na2SO4 + СОа
Осадок отфильтровывают, промывают и сушат — получается
легкий белый порошок — магнезия альба.
Этот способ может быть использован и для получения магнезии
из озерных рассолов 167.
Осаждение основного карбоната магния из растворов хлори-
хлористого магния или карналлита производят следующим образом.
В стальной реактор с паровым барботером и мешалкой заливают
раствор соды концентрацией 130—140 г/л. При перемешивании на-
нагревают раствор до кипения и приливают к нему небольшой струей
хлормагниевый щелок или раствор карналлита A50—160 г/л
MgCl2). Температуру поддерживают выше 90°, так как при пони-
понижении температуры, так же как и при быстром сливании раство-
растворов, образуется плохо фильтрующий осадок. Подачу раствора
MgCl2 прекращают, когда в реакционной массе содержание
Na2CO3 не превышает 5 г/л при отсутствии в жидкой фазе Mg2+.
Затем суспензию кипятят в реакторе в течение 15 мин для разру-
разрушения бикарбонатов, осадок отфильтровывают и промывают на
фильтре. Промывку пасты ведут водой, подогретой до 50—60° и
содержащей не более 25 мг/л С\~ и 30 мг/л SO4~. После промывки
паста содержит —30% MgCO3 ( — 15% MgO) и не более 0,055%
С1- и 0,5% Na2CO3.
Сушку магнезии альба осуществляют в камерных сушилках на
алюминиевых противнях. Более совершенным способом является
применение сушилок ленточного типа. Сырую углекислую магне-
магнезию с фильтров подают шнеком на бесконечную ленту из прово-
проволочной сетки с отверстиями 10 мм. При помощи 16 барабанов
лента 8 раз поднимается вверх и опускается вниз (8 петель) вну-
внутри камеры сушилки, а затем вдоль нижней части сушилки дви-
движется горизонтально. За время движения магнезия высыхает в по-
потоке горячего воздуха и сбивается с сетки в бункер при помощи
специального механизма.
Сушку производят до содержания остаточной влаги меньше
0,5%. Затем магнезию размалывают в мельнице кулачково--
Ударного действия, оборудованной воздушным сепаратором и
298
Гл. IX. Соединения магния
расфасовочным бункером. Выход продукта по MgCb составляет
60—70% от теоретического, а содержание в нем основного веще-
вещества в пересчете на MgCOe должно быть не меньше 50%• На про-
производство 1 т основной углекислой магнезии расходуют: 1,47—
1,74 т MgCl2 A00%), 1J3 т кальцинированной соды (95%), 1,75 г
условного топлива, 6,2 мгкал пара, 230 квт-ч электроэнергии, 25—
50 м3 воды.
Прокаливанием основной углекислой магнезии из нее удаляют
двуокись углерода и воду и превращают ее в легкие сорта жженой
магнезии (магнезии уста) MgO. Гидратная вода удаляется из ос-
основного карбоната магния при 320°, а при 430° диссоциирует кар-
карбонат. Дегидратация гидроокиси магния также идет при 400—
420°168'169. При 500° одновременно с диссоциацией Mg(OHJ, осво-
освобождающейся из основного карбоната магния, происходит ее кар-
карбонизация двуокисью углерода, образующейся при диссоциации
карбоната. В результате получается смесь MgO и MgCOs 170. (Если
вести этот процесс под большим давлением СО2, например 50 ат,
то при 500° образуется MgCOs). Окись магния с наибольшей ак-
активностью (с удельной поверхностью ~ 100 м2/г) получается при
прокаливании основного карбоната магния и гидроокиси магния
при 500—600°. В обоих случаях активность продуктов приблизи-
приблизительно одинакова. Если прокаливание вести ниже 500 и выше 600°,
то активность магнезии, полученной из основного карбоната маг-
магния, больше, чем из гидроокиси магния при одинаковых темпера-
температурах их обработки (особенно выше 1000°).
В производственных условиях прокаливание основного карбо-
карбоната магния ведут в полочных механических печах при 700—800°.
В них материал перемещается со свода на свод сверху вниз с по-
помощью гребков, передвигающих его по сводам от периферии к
центру (на нечетных сводах, считая сверху) и от центра к периферии
(на четных сводах). Верхний свод — чугунная плита служит для
предварительной подсушки материала. Внутри кладки остальных сво-
сводов Имеются каналы для обогревающих топочных газов. В вал, на
котором вращаются гребки, подается вентилятором воздух для ох-
охлаждения металла с целью предохранения его от деформаций.
На верхнем своде содержание в пасте MgCO3 повышается от
26—32 до 36—40%. На втором своде содержание MgO в матери-
материале увеличивается до 36—40%. Окончательное разложение дости-
достигается на четвертом своде, откуда магнезия через течку поступает
в шнек и направляется на склад. Магнезиальную пыль, выносимую
из печи уходящими газами, содержащую 60—70% MgO, улавли-
улавливают в рукавных фильтрах, в электрофильтрах или в других ап-
аппаратах и возвращают на прокаливание. Для производства 1 т
жженой магнезии из раствора MgCl2 или из карналлита расхо-
расходуют: 2,55 т MgCl2 A00%), 3,05 т соды (95%), 2 т условного топ-
топлива, 6,4 мгкал пара, 335 квт-ч электроэнергии, 65 м3 воды. При
Магнезии
299
получении окиси магния содовым способом из магнезита расход-
расходные коэффициенты особенно велики: на 1 г MgO затрачивается
около 4 г серной кислоты и около 4 т соды, которые превращаются
в отбросный сульфат натрия. Этим способом пользуются только
для производства окиси магния высокой степени чистоты.
Осадки карбонатов магния, полученные из природных вод, за-
загрязнены примесями хлоридов натрия и калия. Для освобождения
от них предложено1™ промывать карбонаты магния 0,005%) раство-
раствором NH4C1; последний замещает адсорбированные хлориды нат-
натрия и калия, переходящие в раствор. При последующем прокали-
прокаливании карбонатов магния NH4C1 улетучивается и готовый продукт
содержит всего ~0,03% хлора. Расход промывной воды при этом
сокращается на 50—60%.
При получении жженой магнезии содовым способом из отброс-
отбросного раствора сульфата магния, образующегося в производстве
борной кислоты (стр. 329), необходимо предварительно осадить из
раствора бораты магния каустическим магнезитом 172.
Легкая магнезия может быть получена вместе со смешанным
удобрением из морской рапы плотностью 1,32 г/см3, остающейся
после кристаллизации NaCl и содержащей MgSO4) MgCl2 и не-
небольшие количества NaCl и КС1173. Рапу обрабатывают одновре-.
менно аммиаком и двуокисью углерода при 50° последовательно
в трех реакторах. И8 суспензии, вытекающей из каждого реактора,
отфильтровывают осадок основного карбоната магния, промывают
его водой от аммониевых солей. Фильтрат и промывные воды на-
направляют в следующий реактор. При некотором избытке карбо-
карбоната аммония извлечение магния из рапы достигает 90—95%,
@,32 кг из 1 л рапы). Высушенный продукт MgCO3-Mg(OHJ-
•ЗН2О имеет насыпную плотность 180 кг/м3. При прокаливании
этого продукта при 600—700° получается легкая магнезия с на-
насыпной плотностью 80 кг/м3. Фильтрат из третьего реактора обра-
обрабатывают гипсом, и после отделения СаСО3 выпаркой и кристал-
кристаллизацией раствора можно получить удобрение, состоящее из
NH4C1, (NH4JSO4 и КС1 и содержащее 21—22 %N и 3% К2О. Из
1 л рапы получается 0,7 кг такого удобрения.
Для получения окиси магния, имеющей повышенную совмести-
совместимость с органическими веществами, слабообожженную MgO сме-
смешивают в среде инертного органического растворителя с амином
с числом атомов С^С20, затем отфильтровывают, промывают инерт-
инертным летучим растворителем, сушат при 65—100° и измельчают.
Продукт содержит 1—25% органических веществ 174.
Гидролиз и окисление хлористого магния
Окись мапшя может быть получена И8 MgCl2 путем его гидро-
гидролиза ire-i8-'
MgCl2 + нгО ^=> MgO + 2HC1
300
Гл. IX. Соединения магния
Сульфат магния и его термическое разложение
801
и окисления174>
2MgCl2 + О2 5z± 2MgO + 2С12
При нагревании шестиводного хлористого магния до 500° вна-
вначале, после частичного обезвоживания MgCl2, образуется MgO •
• 2MgCl2 ¦ 2Н2О, а затем гидрооксихлорид магния Mg(OH)Cl:
MgCl2 + H2O 5=± Mg(OH)Cl + HCl
Выше 500—610° Mg(OH)Cl разлагается176:
Mg(OH)Cl ^z± MgO + HC1
Если вести нагревание MgCl2 в токе инертного газа и таким об-
образом удалять выделяющийся хлористый водород, хлористый маг-,
ний полностью превратится в
магнезию.
Проведение гидролиза MgCl2
с предотвращением распла-
расплавления его в кристаллизацион-
кристаллизационной воде возможно при исполь-
использовании в качестве исходного
материала магнезиального це-
цемента Сореля. При 500—600°
степень гидролиза за 1 ч со-
составляет 95—98% 184-186. Окись
магния, полученная таким пу-
путем при 600—650°, удовлетво-
удовлетворяет требованиям на каусти-
каустический магнезит, а при 1000°—
требованиям на металлургиче-
металлургический порошок для огнеупо-
огнеупоров186. В США получение
MgO гидролизом хлористого
магния осуществляют в круп-
крупных масштабах 187. Разработано проведение этого процесса распы-
распылением концентрированного раствора MgCl2 в потоке горячего
газа 188. Запатентовано189 использование для гидролиза MgCl2
реакционной камеры с фонтанирующим слоем огнеупорного инерт-
инертного материала с частицами 3—8 мм; в камеру подают раствор
или кристаллический хлорид магния, продукты реакции — MgO
и НСЛ — выносятся топочным газом и отделяются от него в ула-
улавливающих устройствах.
В процессе гидролиза хлористого магния могут образоваться
твердые растворы MgCl(Cl, ОН). При 550° давление НСЛ над гид-
рооксихлоридом достигает I at и его разложение протекает с мак-
максимальной скоростью. Выделяющийся НСЛ частично реагирует с
Mg(OH)Cl, образуя твердый раствор MgCl2 в Mg(OH)Cl. Этот
о го to во во юо
Концентрация НО в еазобои д>азе,%
Рис. 96. Поля твердых фаз в системе
MgO—HC1—Н2О:
/ -Mg(OH)CI; //-твердый раствор MgCI(CI, ОН),
цифрами обозначено содержание в нем
Mg(OH)CI в мол. %; ///-MgO. Р-суммарное
давление реакционных газов в ат.
процесс протекает на 40—45° выше температуры устойчивого со-
состояния Mg(OH)Cl E10°), т. е. при метастабильном, перегретом
состоянии Mg(OH)Cl. После исчезновения Mg(OH)Cl температура
повышается. При этом может происходить разложение твердого
раствора с образованием MgO и обогащением газовой фазы хло-
хлористым водородом, а твердого остатка хлористым магнием. На
рис 96 представлена политермическая диаграмма равновесия в си-
системе MgO—НС1—Н2О190.
При термическом разложении кристаллогидратов оксихлоридов
магния 2Mg(OHJ-MgCl2-5H2O, 3Mg(OHJ-MgCl2-8H2O, 5Mg(OHJ-
• MgCl2-8H2O и 9Mg(OHJ-MgCl2 «6H2O вначале также происхо-
происходит частичная их дегидратация, а затем процесс идет по схеме191:
nMg(OH)s • MgCl2 = HSO + (ft - 1 )H2O • nMgO + MgCl2
MgClj + HSO = MgO + 2HC1
Окись магния, полученная гидролизом MgCb, гидратируется
очень медленно. Полученная гидролизом цемента Сореля окись
магния гидратируется при 16° за 48 ч на ~5%, а при 85° за 11 ч
на -90% 192.
Окисление расплавленного хлористого магния кислородом ш
идет по реакции (рис. 88):
Для интервала температур 750—950° значение константы рав-
новесия этой реакции КР •= —оТ может быть вычислено по фор-
муле181> *•*:
1в*ГР->1-|—МБО—^2-
Для случаев, когда в жидкой фазе кроме MgCl2 присутствуют
другие хлориды, значения А и В будут:
Состав расплава Л В
KCl-3MgCl2 1,507 1183
KCl-MgCI2 0,944 1288
2KCl-MgCl2 0,103 1451
NaCl • MgCl2 G50-860)° . . . 1,799 2000
СУЛЬФАТ МАГНИЯ И ЕГО ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
Получение сульфата магния
Сульфат магния может быть получен из природных растворов
морского типа и твердых солевых отложений. Например, при ком-
комплексном использовании карабогазских рассолов можно, удалив
некоторую часть галита из маточного рассола после садки
802
Гл. IX. Соединения магния
мирабилита (см. гл. IV), кристаллизовать из него эпсомит MgSOv
• 7Н2О. Эпсомит выделяется не только в стабильном поле его кри-
кристаллизации (рис. 83), но в значительной степени B0—50% от ко-
количества, выделившегося в стабильной области) в метастабильной
области, захватывающей часть поля кристаллизации астраханита.
Из 1 т астраханитового сырья озер Куули, Джаксы-Клыч, Чумыш-
Куль могут быть получены 1 т мирабилита и 0,3 т эпсомита, а из
сырья Аральского месторождения — 0,9 т мирабилита и 0,2 т эпсо-
эпсомита195. Из сивашской рапы, выпаренной до начала выделения
эпсомита, имеющей состав 9,76% MgS04, 14,06% MgCl2, 2,94%
КС1, 5,72% NaCl, выход MgSO4 при +5° 58,2%, при —5° 75% 196.
Технически чистый эпсомит можно получить охлаждением до —5°
рассола с плотностью 1,29—1,31 г/см3, полученного испарением мор-
морской воды, с добавлением к нему 6 объемн.% морской воды. Мак-
Максимальный выход эпсомита F5—68% от MgSO4 в морской воде)
получается из раствора, содержащего 9,1% MgSO4, 14,7% MgCl2,
9,8% NaCl»97.
Обезвоживанием эпсомита получают кизерит MgSO4 • Н2О, а
затем и MgSO4. Первые шесть молекул воды из эпсомита уда-
удаляются при температуре ниже 70° (рис. 82). Кизерит получают в
качестве нерастворимого отхода при водной обработке природных
калийных солей (гл. V). Предложено производить его очистку фло-
флотацией с последующим обезвоживанием, что позволяет изготов-
изготовлять продукт, содержащий 98% MgSO41&8. Обезвоживание кизе-
кизерита во вращающихся печах, обогреваемых дымовыми газами,
практически начинается при 200° и заканчивается при 440°199.
Чистый сульфат магния может быть получен нагреванием маг-
магнезита и доломита в растворе сульфата аммония. Образуется рас-
раствор MgSO4, который после отгонки аммиака концентрируется для
осаждения CaSO4, а затем перерабатывается на MgSO4 или другие
соединения магния 20°-2о2. Предложено получать сульфат магния
обработкой водной суспензии (Ж:Т=1:1 по весу) тонкоизмель-
ченного серпентина (с размером частиц меньше 0,075 мм) кон-
концентрированной серной кислотой (плотность 1,84 г/см3). Получен-
Полученную массу разбавляют водой и фильтруют; образовавшийся рас-
раствор MgSO4 подвергают выпарке и кристаллизации203. Раствор
MgSO4 можно также получать карбонизацией суспензии окиси маг-
магния и гипса 204.
Очистку растворов сульфата магния от ионов Ni2+, Fe2+, Fe3*,
Mn2+, Ca2+, Al3+ и других, по-видимому, целесообразнее всего про-
производить осаждением этих ионов с помощью Mg(OHJ. Если ис-
использовать Са(ОНJ, то это приведет к загрязнению продукта суль-
сульфатом кальция; применение в качестве окислителя КМпО4 при ще-
щелочной обработке, так же как и реакций двойного обмена с Na2S
и КзРе(СЫN» приводит к образованию коллоидных гидрооки-
гидроокисей 205.
Сульфат магния и его термическое разложение
303
Термическое разложение сульфата магния
Сульфат магния, подобно сульфату кальция, служит сырьем для
сернокислотного производства20е. Получаемый при обжиге суль-
сульфата магния сернистый газ перерабатывается на серную кислоту,
а огарок представляет собой высококачественную окись магния,
пригодную для изготовления огнеупоров и ксилолита.
Термическое разложение MgSO4 протекает с достаточной пол-
полнотой и скоростью при 1100—1200°. При более низких темпера-
температурах, в отсутствие восстановителей и особенно при наличии ката-
каталитически действующих окислителей, например Fe2O;j, происходит
сульфатизация окиси магния газом, содержащим SO2 и Ог. Так,
в присутствии 9% Fe2O3 при соприкосновении с газом, содержа-
шим 5% QOo. при 600° MgO превращается на ~50% в MgSO4 в
течение 30 мин2сгг. В присутствии восстановителей С, S, Н2, СО,
СН. температура термического разложения MgSO4 снижается до
650°208, а в присутствии МпО даже при 500°209. Восстановление
MgSO4 углем ведут практически в интервале 700—900°. Основной
реакцией является:
2MgSO4 + С -= 2MgO + 2SO2 + COS
В результате побочных реакций, особенно при избытке угле-
углерода, SO2 частично восстанавливается до серы, a MgSO^ до MgS.
Оптимальный выход МрО достигается при молярном отношении
MgSO4: С, равном 1 : 1. В этих условиях при 800° выход МрО со-
составляет 99%, а выход серы: в SO2 — 81%, вееру—16.5%, в
MgS — 2% и остается в виде MgSO4 — 0,3% 210. Благоприятным
для увеличения выхода MgO и SO? является создание в зоне реак-
реакции окислительной атмосферы при 900—950°. При избытке кисло-
кислорода получается окись магния, практически свободная от суль-
сульфидов 2И.
Восстановление сульфата магния углем осуществляют в меха-
механических печах, применяемых для обжига колчедана 212. Могут быть
использованы барабанные и другие печи, в частности, при восста-
восстановлении окисью углерода, водородом и другими газами — печи со
взвешенным слоем материала 213. Содержание окиси магния в огар-
огарке зависит от зольности восстановителя. Воздушная классифика-
классификация огарка позволяет выделить мелкие фракции, содержащие 75—
80% MgO. Водная седиментация, а также флотация с примене-
применением мыла дают продукт с 90% MgO. Концентрат, получаемый ппи
отмучивании, содержит 89—90% MgO, 3—4% RsOs, 2—2,5% CaSO4,
2—3% SiO2 и силикатов и до 1 % MgSO4209.
Восстановление MgSO4 природным газом идет по реакции:
4MgSO4 + СН4 = 4MgO + 4SO2 + СО2 + 2Н2О
При использовании безводного MgSO4 с размерами зерен 0,2—*
0,6 мм и природного газа с содержанием 97,8% СН4 восстановление
304
Гл. IX. Соединения магния
начинается при 700—720°, а полное восстановление MgSO4 до
MgO происходит при 750—950°. В газообразных продуктах реак-
реакции, помимо S62 и СО2, находятся также S, H2S и С$?, количество
которых зависит от температуры и скорости газа214,
Предложено осуществлять восстановление MgSO4 при помощи
непрерывно циркулирующего газа, содержащего Н2 или H2S и од-
одновременно служащего теплоносителем. Реакция протекает при
большом избытке водорода или сероводорода. В первом случае из
циркулирующего газа вымывается H2S, во втором извлекается вы-
выделяющаяся серамв. Восстановление твердого сульфата магния
можно производить также в распыленном состоянии, в потоке
газа21в.
В известных условиях может представить интерес обменное
взаимодействие между MgSO4 и КС1 (или NaCl) в присутствии
водяного пара217. Реакция
MgSO4 + 2KC1 + Н2О - MgO + K2SO4 + 2НС1
протекает при ~800°. Она может иметь значение для производства
бесхлорного калиевого удобрения из КС1. Обработкой твердых
продуктов реакции водой можно разделить K2SO4 и MgO. Однако
при этом на 1 т KsO получается около 6,8 т 27%-ной соляной кис-
кислоты; для таких больших количеств кислоты трудно найти приме-
применение в районе расположения калийных фабрик.
НЬЮВЕЛЬ И СОВЕЛ ИТ
Ньювель и совелит — высококачественные теплоизоляционные
материалы, применяемые в судостроении, котлостроении и др. Они
представляют собой смесь магнезии с асбестом. Ньювель содержит
магнезию альба и 14—19% асбеста. Содержание в нем MgO (в пе-
пересчете на сухое вещество) должно быть не менее 32%, СаО не
более 2%, влаги 10—15%- Объем сухого порошкообразного мате-
материала не более 200 кг/м9, а коэффициент теплопроводности при
средней температуре 200° не должен превышать 0,07 ккал/(м • ч-
-град). Его выпускают и в виде пасты с 70% влаги. Совелит также
содержит 12—17% асбеста, но изготовляется не из магнезии альба,
а из смеси карбонатов магния и кальция218.
Порошкообразный ньювель имеет объемный вес меньше 200 кг/м3.
Изготовленная из него изоляция при разности температур 300° имеет
коэффициент теплопроводности не больше 0,08 ккал/ (м ¦ ч ¦ град),
а сопротивление разрыву не менее 1 кгс/см2 2Ш-220. Совелит в по-
порошке содержит 7—8% влаги. Объемный вес ньювеля и совелита
в изделиях (плитах) 350—450 кг/м3, коэффициент теплопроводно-
теплопроводности 0,08—0,09 ккал/(м • ч • град), временное сопротивление на из-
тиб 2—2,5 кгс/см2.
Магниевые удобрения
306
МАГНИЕВЫЕ УДОБРЕНИЯ
Магний является необходимой составной частью растений. Он
способствует образованию углеводов в растительном организме и
плодообразованию. Содержание MgO в золе большинства расте-
растений не превышает 0,2—0,5%, но иногда достигает 3°/о (в золе тур-
турнепса). Обычно в большей части почв содержится достаточное
количество магниевых минералов @,4—4% MgO), но урожай
многих культур на песчаных и супесчаных почвах значительно по-
повышается от внесения магниевых удобрений 221>222.
Простыми магниевыми удобрениями являются эпсомит, кизе-
кизерит, магнезит, дунит, серпентинит (стр. 270J23. К калиево-магние-
вым удобрениям относятся калимагнезия и калимаг, каинит, кар-
карналлит, полигалит и другие соли (см. гл. V). Доломит является
известково-магниевым удобрением. Добавляя доломит к расплав-
расплавленной аммиачной селитре, можно получить азотно-магниевое удо-
удобрение.
К фосфорно-магниевым удобрениям относятся димагнийфосфат
и магнийсиликофосфат, который получают обработкой дунита или
серпентинита слабой фосфорной кислотой. При переработке этим
методом смеси силиката магния и фосфорита получается магние-
во-кальциевый фосфат. К этому же виду удобрений относятся то-
масшлак (стр. 1041), в состав которого входит 2,5—3,2% MgO, a
также серпентиновый суперфосфат — смесь суперфосфата с дуни-
том или серпентином в количестве около 10% к весу суперфосфата.
В состав последнего удобрения, помимо обычных компонентов су-
суперфосфата, входят MgHPO4 и SiO2; содержание в нем MgO~
~4%224-225. При нейтрализации свободной кислотности суперфос-
суперфосфата (стр. 853) доломитом или магнезитом улучшаются физические
свойства суперфосфата, а количество усвояемой Р2О5, в отличие
от нейтрализации известняком или мелом, не уменьшается даже
при добавке к суперфосфату 10—20% доломита и в особенности
магнезита226.
Сложным магнезиальным удобрением является магнийаммоний-
фосфат MgNH4PO4 • Н2О. Используя это удобрение можно создать
запас азота в почве, так как он находится здесь в цитратнораство-
римой форме (другие же азотные соли водорастворимыеJ27-228.
Многосторонними удобрениями, содержащими магний, являются
также навоз и древесная зола. Удобрения, содержащие, кроме азо-
азота, фосфора и калия, также и магний, с различными соотноше-
соотношениями питательных веществ могут быть получены азотнокислотной
переработкой фосфатов, содержащих магний, например каратаус-
ких фосфоритов (см. гл. XXXIV).
В качестве магнезиально-азотного удобрения применяют также
соль Туттона MgSO4 • (NH4JSO4 • 6Н2О. В ФРГ ее получают взаи-
взаимодействием сульфата аммония с кизеритом; в СССР ее изготовляют
806
Гл. IX. Соединения магния
в небольшом количестве из отбросного сульфата аммония и магне-
магнезита. Предложены способы ее получения обработкой пульпы от-
отбросного фосфогипса (стр. 914) и обожженного доломита аммиа-
аммиаком и СОг или бикарбонатом аммония по реакции
2CaSO4 + MgO • СаСО, + 2NH4HCO3 = ЗСаСО3 + MgSO4 + (NH4)SSO4 + Н2О
с последующей кристаллизацией двойной соли из раствора226> 229.
Из других удобрений, содержащих магний, в СССР производят
ааводскими способами бормагниевое удобрение (стр. 345) и ка-
лийно-магниевые удобрения.
ЛИТЕРАТУРА
1. Н. В. Кондырев, Г. В. Березовский, Труды ВАМИ, № 11—12,
1935, стр. 29.-2. В. В. Сергеев, И. С. Качановская, ЖПХ, 32, № 8,
1671 A959). — 3. Т. Demedink, W. F. Cole, H. W. Hueber, J. Chem., 8,-
№ 2, 215 A955). — 4. Q. Wehtier, Z. ancrg. Chem., 272, № 1—4, 201 A953).—
5. А. П. Обухов, В. П. Лавров, Труды ГИПХ, в. 25, 1934, стр. 81. —
6. А. Б. Здановский, М. И. Ляховская, Р. Э. Шлеймовнч, Справоч-
Справочник по растворимости солевых систем, Госхнмиздат, т. 1, 1953 и т. II, 1954. —
7. А. Н. П е р о в а, Сообщение о научных работах Всес. хим. об-ва им. Д. И. Мен-
Менделеева, в. 2, 1955, стр. 46. — 8. Н. С. Курнаков, А. В. Николаев, Изв. АН
СССР. Сер. хим., № 2, 403 A938). — 9. Е. И. Лукьянова, В. И. Сокол,
Г. Н. Соколова, ЖНХ, 1, № 2, 208 A956). — 10. G. О. Assaron, А. В а 1-
der, J. Phys. Chem., 59, № 7, 631 A955).
//. А. И. М у н, В. Е. Т а р т а к о в с к а я, Изв. АН КазССР. Сер. хим., № 8,
35 A955). —12. Б. Ф. Марков, И. Д. Панченко, ЖОХ, 25, № 11, 2038
A9-55). — /3. П. П. Будников, ДАН УССР, № 5, 339 A950). — 14. J. Weber,
Kohasz. lapok, 2, № 4, 174 A956). — 75. Е. С г em e r, W. Allgener, Radex
Rundschau, № 2, 54 A953). — 16. О. К- Я н а тье в а, ЖНХ, 1, № 4, 751 A956).—
17. О. К. Янатьева, Г. С. Рапопорт, И. С. Рассонская, М. Б. Усти-
Устинова, ЖПХ, 34, № 2347 A961). — 18. Т. A tod a, J. Sci. Res. Inst., 47, 244 A953);
48, 173 A954); 49, 117 A955). —19. Л. Н. Щегров, В. Н. Скроботун,
А. Г. Р я д ч е н к о, сб. «Исследования в области хнмин и технологии минераль-
минеральных солей и окислов», Изд. «Наука», 1965, стр. 59. — 20. Б. В. Ефремов,
8. А. Протащик, Сборник научных работ Ин-та хнмин АН БССР, в. 5 A),
1956, стр. 58.
21. П. И. Белькевич, Б. В. Ерофеев, Изв. АН БССР, № 4, 127
A952). — 22. J. Kamecki, S. Р а 1 е у. Roczn. chem., 29, № 2—3, 691 A955).—
23. Л. Г. Берг и др., Термография. Изд. АН СССР, 1944. — 24. R. W. Ford,
Q. В. Frost, Canad. J. Chem., 34, № 5, 591 A956). — 25. Lorant В ё 1 a, Z.
analyt. Chem., 21S, № 3, 256 A966). —Ж Е. Kowalska, Przem. chem. 12,
№ 8, 442 A956). — 27. N. J. Knopf, H. S t a u d e. Z. phys. Chem., 204, № 5—6,
265 A955). — 28 M. Lallemant, Q. Watelle-Marion, Compt. rend.. C265,
№ 12, 627 A967). — 29. N. Hast, Arkiv kerni, 9, № 4, 343 A956). — 30. W. R u t-
kowski, Pract. inst. Min-wa hutn., 8, № 2, 85 A956).
31. Г. А. Яковкин, Хим. пром., № 7, 12 A947). — 32. X. Л. Стрелец,
А. Ю. Т а й ц, Б. С. Гуляницкий, Металлургия магния, Госметаллургиздат,
I960,— 33. А. Г. Бергман, И. П. Выродов, ЖПХ, 31. № 1, 19 A958).—
34. А. Г. Бергман. И. П. Выродов, Там же, 32, № 3, 504 A959).—
35. И. П. Выродов, Там же. 34, № 6, 1208 A961). — 36. Я. М. X е й ф е ц, Со-
Соликамские карналлиты, ОНТИ, 1935.—37. Д. О. Б ер штейн, Я. Р. Красе,
Строит, пром., 6, 32 A956). — 38. Я. В. Ключаров, Н. В. Мешалкина,
Литература
807
Цемент, № 5, 14 A957). — 39. Л. Г. Берг, С. Г. Ганелина, Каустический до-
доломит, Таткнигоиздат, 1957. — 40.*Н. Keitel, франц. пат. 1024071, 1953.
41. П. М. Т а т а р и н о в, Е. И. Д в о р ш а н, Магнезит, Минерально-сырьевая
база СССР, в. 2, ОНТИ, 1935. —42. L. R. William's, Bull. Bur. Mines, №630,
537 A965). — 43. H. F. Utley, Pit a. Quarry, 46, № 12, 90 A954).—
44. И. Н. Лепешков, Хим. наука и пром., 2, № 6, 687 A956). — 45. J. Cook,
Discovery, 17, № 2, 71 A956). — 46. Б. Л. Р о н к и н, Озеро Эльтон как источник
натуральных магнезиальных солей, Госхимиздат, 1933. — 47. Проблемы озера
Эльтон, Труды ВИГ, в. 16, 1939, ч. П. — 48. А. П. П а л к и н, М. А. О п ы х т и н а.
Труды ГИПХ, в. 18, 1933, стр. 36. — 49. И. Б. Фейгельсон, А. П. Ларина,
Бюлл. Ин-та галургин, № 4—5, 42 A939). — 50. Ю. Б. Айзенберг, Строитель-
Строительные материалы Туркменистана, Ашхабад, 1951, стр. 51.
51. В. Н. Е р е м и н, Труды Научной конференции по изучению и освоению
производительных сил Сибири, т. 2, Томск, 1940, стр. 526. — 52. Я. М. Хейфец,
ЖПХ, 34, № 3, 473 A961). — 53. В. М. Букштейн, сб. «Технология перера-
переработки природных солей и рассолов», Изд. «Химия», 196.4, стр. 3. — 54. Б. И. С т е-
панов, В. И. Николаев, ДАН СССР, 27, № 3, 224 A940). —55. Н. И. Кор-
Корнилов, Изв. СФХА АН СССР, 20, 8 A950). —56. В. И. Николаев, Там же,
19, 335 A949). — 57. A. F. Burns, A. M. Smith, Agr. Chem., 20, № 9,
23 A965). — 58. А. Г. Бергман, А. П. Обухов, Труды ГИПХ, в. 25, 1934,
стр. 9.-59. Н. С. Курнаков, С. Ф. Жемчужный, ЖРФХО, 51, 28 A919);
сб. «Карабугаз и его промышленное значение», Изд. АН СССР, 1930. —
60. А. Г. Бергман, Н. П. Л у ж н а я, Физико-химические основы изучения и
использования соляных месторождений хлорид-сульфатного типа. Изд. АН
СССР, 1951.
61. А. Д. Пельш, Труды ВНИИГ, в. 21, 1949, стр. 160, 186.—
62. \. Д. Пельш, Там же, в. 27, 1953, стр. 3. — 63. М. М. Викторов, Графи-
Графические расчеты в технологии минеральных веществ, Госхимиздат, 1954,
стр. 348. — 64. А. Д. Пельш, Труды ВНИИГ, в. 27, 1953, стр. 17.—
65. Е. Ф. Соловьева, Там же, в. 31, 1956, стр. 180. —66. В. П. Ильин-
Ильинский, Труды ГИПХ, в. 40, 1948, стр. 5, 55.-67. А. Ф. С а г а й д а ч н ы й, Там
же, в. 5, 1927, стр. 18. — 68. Н. С. К У Р н а к о в, Д. П. М а н о е в, Н. А. О с о к о-
рева, Калий № 2, 25 A932). — 69. Ф. Серова, Труды ВНИИГ, в. 36, 1959,
стр. 136. — 70. В. Kassner, Bergakadernie, 7, № 4, 190 A955).
71. К. Ф- П а в л о в, Б. А. К о п ы л е в, ЖХП, № 6, 29 A938).— 72. Н. С. С п и-
рин, М. А. Бедюх, сб. «Вопросы производства калийных удобрений», 1964,
стр. 42. — 73. Н. Kurrneier, Chem. Ingr. Techn., 38, № 4, 484 A966).—
74. Л. Н. М а т и й к о, Л. В. Л о п а ч е н о к, сб. «Вопросы добычи и переработки
галургического сырья», Изд. «Химия», 1966, стр. 17. — 76. Г. С. К л е б а н о в,
В. М. Букштейн, Н. О. Эверт, Бюлл. ВИГ, № 7, 1938, стр. 24.—
76. J. M. A very, R. F. Evans, Chem. Met. Eng, 52, № 4, 94 A945).—
77. Ф. К. Михайлов, Труды НИОХИМ, т. 11, 1958, стр. 5. — 78. J. Kam let.
пат. США 2705185, 1955—79. Дат. пат. 76988, 1954.— SO. A. H. Вишнев-
Вишневский, ЖХП, 10, № 2, 23 A939).
81. Ф. К- Михайлов, Д. М. Гинзбург, Труды НИОХИМ, т. 10, i957,
стр. 39. — 82. С. Н. Fuchsmann, пат. ФРГ 944426, 1956.— S3. P. D. V. Man-
Manning, S. D. Kirkpatrick, Chern. Met. Eng., 51, № 5, 92, 142 A944).—
84. R. Manocha, Q. Sternheim, J. Sci. Ind. Res., (B—C) 15, № 7, В 375
A956). — 85. А. И. Беляев, Металлургия легких металлов, Госметаллургиздат,
1954. — 86. Е. И. Савинкова, Я- Е. В и л ь н я и с к и й, Изв. вузов. Хнм. и
хим. технол., 2, № 1, 59 A959). — 87. S. Mr о we с, польск. пат. 37688, 1955.—
88. S. M. Heins, В. Strauss, пат. США 3201208, 1963. — S9. V. J. Chris-
tensen, Q. С. Egleson, E. L. Carlson, пат. ФРГ 1183485, 1963. — 90. Нор-
Веж. пат. 105491, 1963.
308
Гл. IX. Соединения магния
Литература
30»
91. Пат. США 334726, 1965. — 92. Пат. США 3347625, 1965. —S3. Пат. США]
3345128, 1965. — 94. Т Л. Лукманова, Я. Е. Вильиянский, Изв. вузов.
Хим. и хим. технол., 7, № 3, 510 A964).— 95. Е. И. Савинкова, Т. Л. Лук-
Лукманова, Изв. вузов. Цветн. металлург. № 3, 105 A964). — 96. Ind. Chem., 21,
№ 246 351; № 247, 419 A945). — 97. А. И. Тетеревков, Я- Е. Вильнян-
ский, ЖПХ, 32, № 2, 438 A959). — 98. Я. Е. Внльнянский и др., Реф.
докл. VIII Мендел. съезда. Секции неорг. хим.. 149 A959). —95. 1. J. Кг em а,
пат. США 2668750, 1954. —100. A. Apostolide, Rev. chim., 6, №8, 425 A955).
101. И. К. Пуха, сб. «Технология переработки природных солей и рассо-
рассолов», Изд. «Химия», 1964, стр. 114. —102. А. М. Кузнецов, Производство кау-
¦ стического магнезита, Промстройиздат, 1948. — 103. V. V. Hughey, пат. США
2640759, 1953. —104. J. С. Hicks, L. W. Austin, англ. пат. 719623, 1954. —
105. F. Nadachowscki, Szklo i ceramika, 4, № 3, 99 A953).—
106. R. A. Schoenlaub, пат. США 2636809, 1963. —107. С. R. Havighorts.
S. L. Swift, Chem. Eng., 72, № 15, 150, A-l A065).— 108. F. E. Lathe, пат.
США 2694620, 1954. —109. E. Bieneck, E. S eh roth, пат. ФРГ 930738, 1955;
франц. пат. 1109716, 1956. — ПО. П. В. Г ел ьд, О. А. Ее и н, ЖПХ, 21, № 3, 240
A949).
///. А. А. Банков, Ж- русск. металлург, общ-ва, 1, 311 A913).—
112. А. А. Б а й к о в, Собрание трудов, т. V, Изд. АН СССР, 1948, стр. 49. —
113. А. А. Байков, А. С. Тумарев, Изв. АН СССР, ОТН, № 4, 565 A937).—
114. М. Hicgnet, пат. США 2637545, 1953. —//5. А. Н. Яснновский,
В. В. Гончаров, Огнеупоры, № 4—5 7 A946). —116. П. П Будников,
X. С. Воробьев, ЖПХ, 32, № 2, 253 A959). —117. G. M. Armstrong,
Н. A. Cook, пат. США 2631923, 1953. — 118. В. К. Shukla, Salt. Res. a. Ind., 5,
Ш 2, 34 A968).— /75. В. А. Клементьев, Труды ВАМИ, № 18, 1939,
стр. 11. —120. J. Horiguchi, J. Sci. Res. Inst., 47, 99, 294, 301, 319, 341 A953);
.48, 27 A954).
121. H. А. О с о к о р е в а, Труды ГИПХ, в. 25, 1934, стр. 94. — 122. J. M i 1-
bauer, J. Vlasak, Cl. Sci. math., natur. et rned., 51, 1 A953).—
123. P. И. А р а в, Л. И. Колосова, Комплексное использование ресурсов Си-
Сиваша и Перекопских озер, Киев, 1958, стр. 80. — 124. Р. И. А р а в, Е. Г. Ч у-
б а р ь, сб. «Исследования в области химии и технологии минеральных солей и
окислов», Изд. «Наука», 1965, стр. 24. —125. Р. И. Ар а в, Укр. хим. ж., 32,№5,
521 A966). —126. К. Hobold, W. Hacker, пат. ФРГ 949563, 1956.—
127. A. Tomioka, япон. пат. 1370, 1954. — 128. Т. С. Atchison, канад. пат.
497409, 1953.— 129. P. W. T rube у, J. В. Boyack, англ. пат. 965081, I960.—
130. Е. A. Prudhomme, франц. пат. 1027683, 1953.
131. Б. А. К о п ы л е в, И. Г. Л е с о х и н, А. М. Б а л я с и а я, Труды ЛТИ
нм. Ленсовета, в. 10, 1941. — 132. A. G. Wintershall, франц. пат. 1022581,
1953. — 133. Н. Keitel a. oth., англ. пат. 693108 1953; пат. ФРГ 913535, 1954 —
134. R. N. Vohra, R. N. Patel, В. К. Shukla, Chem. Age (India), 19, № 6
441 A968). —135. А. В. Николаев, ЖПХ, 20, № 3, 187 A947).—
136. Ю. А. Клячко, Н. П. Кондратюк, Зав. лаб., № 8, 912 A947).—
137. Н. П. Кондратюк, Ю. А. Клячко, Хим. наука и пром., 2, № 2, 267
A956). 138. J. Sugi, К. Shimizy, япон. пат. 1065, 1955. —139. J. Schroe-
der, К. Boron, A. Milewska, Przem. chem., 45, № 1, 14 A966).—
140. S. 1 s h i z a k a, M. T e r a d а, япон. пат. 3665, 1953.
141. G. Wehner, пат. ГДР 4400, 1953. — 142. H. С. С пир о, Хим. пром.,!
М> 5, 137 A948). — 143. Н. С. С п и р о, Труды ВНИИГ, в. 36, 1959, стр. 281.—'
144. А. Д. Пельш, Там же, в. 24, 1952, стр. 99. — 145. Chem. Eng., 54, J\ft 8, 132
A947).—146. Tidsskr. kjemi, bergves og metallargi, 13, № 5b, 83 A953).—
147. W. С Gilpin, N. Heacmen, Cloy Prod. J., 20, № 3, 33, 35 A953).—
148. С R. Havighorst, S. L. Swif t, Chem. Eng., 72, № 16, 84, A-l A965).—
149. Eng. a. Mining J., 168, № 3, 104 A967). — ISO. G. H. Weyermuller,
R. R. Kitchen, Chem. Process," 31, № 11, 39 A968).
151. Я- Д- Козин, П. Т. Данильченко, А. М. Поиизовскнй, Вестн-
АН СССР, № 11, 63 A954). — 152. Ц. Дяковска, X. Козарев, Тежка пром.
4, № 10, 45 A955).— 153. D. Marcus, J. Weiser, Rev. chim., 18, № 2, 114
A967).—154. Нага и, Есидзаки, Накаяма, J. Ceram. Assoc. Japan, 62,
№ 694, 248- № 695, 325 A954).- 155 J. A. Robertson. С. В. Miles,
D. С. L1 n t о п, пат. США 2703273, 1955. — 156. П. Г. Р о м а н к о в, Н. Б. Р а ш-
ковская, 3. А. Березовская, Инф. бюлл. ЛТИ им. Ленсовета. Аннотации
н.-и. работ, в. 1, 1956, стр. 191. —157. И. К. Пуха, Аннотации работ ВНИИГ,
№ 1, 1956, стр. 52. —158. И. К- Пуха, Труды ВНИИГ, в. 36, 1959, стр. 315.—
159. А. Д. Пельш, Там же, в. 31, 1956, стр. 3. — 160 Ю. С. 3 а я ч к о в с к и й,
Строит, пром., № 8, 13 A953).
161. Н. Д. Чалый, Механизация строительства, N° 10, 31 A953).—
162. И. К- Пуха, Труды ВНИИГ, в. 31, 1956, стр. 28. — /63. И. К. П у х а,
В. В. Т а р х о в а, Е. Я. Г р я з и н а, Н. Ф. С е д н е в а, сб. «Технология пере-
переработки природных солей и рассолов», Изд. «Химия», 1964, стр. 104. —
164 Л. L. Bradley, Eng. u. Foundryrnan, 21, № 1, 62 A956). — 165. R. A. Pat-
ton, С. В a u g h, англ. пат. 972323, 1962. — 166. В. С. С т о й н о в а, Г. Р. По-
Попов. Г. С. М о и е в, авт. евнд. НРБ 10319, 1963. — 167. В. Г. Э д и б е р, Труды
ВНИИГ, в. 36, 1959, стр. 344. —168. А. Я. В а й в а д и др., Изв. АН ЛатвССР.
Сер. хим., № 2, 163, 171 A962); № 6, 643 A965). —169. Gregg, Packer,
Wheatley, J. Chem. Soc, 1965, № 1, 46. — 170. А. С. Суслейменов, Тру-
Труды 1 совещания по термографии, Изд. АН СССР, 1955, стр. 200.
171. И. К- Пуха, Р. Э. Натинская, авт. евнд. 183724, 1964.—
172 А. А. Соколовский, Т. И. Кузнецова, К- Л. Павлова, Труды
ГНИИХП, в. 3, 1955, стр. 66.-/75. К. Seshadri, J. Gupta, J. Sci. Ind. Res.,
13, № 3, 204 A954). —174. R. A. Patton, пат. США 3211567, 1962.—
175. W. Moldenhauer, Z. anorg Chem., 51, 369 A906). —176. А. П. Обу-
Обухов, M. H. Михайлова, В. И. К о n о с о в а, А. А. Б у к в и и, Труды ГИПХ,
в. 25, 1934, стр. 84.-/77. Я- Е. Вильняиский, Е. И. Савинкова, ЖПХ,
26, № 8, 808 A953). — 178. Г. Т. К о с и ы р е в, Е. И. Савинкова, В. Н. Рас-
Рассохина, сб. «Физическая химия расплавленных солей», Изд. «Металлургия»,.
1965, стр. 331. — 179. Е. И. Савинкова, Г. Т. К о с и ы р е в, Г. А. П а р т и н а,
Цветные металлы, № 4, 56 A966). —/80. Г. Т. Коснырев, Е. И. Савин-
Савинкова, Я. Е. Вильнянский. Изв. вузов. Цветн. металлург., Ш 5, 58 A966).
181. И. Л. Резников, ЖПХ, 23, № 9, 897 A950). —182. F. Haber,
F. F1 е i s с h m a n n, Z. anorg. Chem., 51, 336 A906). — 183. В. А. Суходскнй.
А. П. Обухов, А. И. Крягова, Труды ВАМИ. № И—12, 1935, стр. 47.—
184. А. Г. Бергман, В. А. Клементьев, Е. Б. П е в з н е р, Труды ГИПХ,
в. 16, 1932, стр. 3; в. 25, 1934, стр. 20. —185. А. Г. Бергман, А. И. Ляшен-
ко, А. П. Обухов, Там же, в. 25, 1934, стр. 39. —186. А. П. Обухов,
М И. Михайлова, В. Н. Колосова, Там же, стр. 69. — 187. Chem. Eng.,
№ 8 346 A956). —188. Н. Нор ре, В. Maurer, Chem. Techn., 18, № 5, 257
A966).— 189. К- В Bengtson, L. N. Johanson, J. С. A 1 mo n d, пат. США
3251650 1963. — 190. Я. Е. Вильнянский, Е. И. Савинкова, ЖПХ, 27,
№8, 864 A955).
191. W. F. Cole, Т. Demedink, Austral. J. Chem., 8, № 2, 234 A955).—
192. В. А. Клементьев, Труды ГИПХ, в. 25, 1934, стр. 78. — 193. Ф. К- М и-
хайлов и др., Труды НИОХИМ, т. 12, 1959, стр. 59, 71. — 194. R. T such id a,
Sci. Rep. Tohoku Univ., Ser. I, 37, № 1, 9 A953).—195. В М. Букштейн.
P. С. Роскнна, Труды ВНИИГ, в. 31, 1956, стр. 83. — 196. Н. К- Давиден-
ко, Укр. хим. ж., 21, N° 6, 773 A955). — 197. В. П. Ильинский, Труды Даль-
невосточн. филиала АН СССР, в. 2, 1956, стр. 89. — 198. F. Busch. Freiberger
810
Гл. IX. Соединения магния
Forschungsh., А, № 354, 23 A965). —199. R. Burmeister, ibid., 41 A965).—
200. R. R e i s s п е г, Н. S u i d а, австр. пат. 177406, 1954.
201. H. Suida, австр. пат. 178091, 1954. — 202. Т. Весене, А. Каушпе-
дас, Научн. труды вузов ЛитССР. Хим. и хим. технол., № 6, 225 A965). —-
203. Аюкава Марнама, япод. пат. 420, 1955. — 204. Л. Г. Берг, С. Г. Г а-
нелина, Э. Ф. Губанов, ЖПХ, 32, № 11, 2676 A939). — 205. L. Rovira,
Ind minera, 12, № 143, 21 A953). — 206. A. Les, Ind. chim., 40, № 431, 167
A953). — 207. В. В. Печковский, ЖПХ, 29, № 8, 1137 A956); 30, № 11, 1579
• A957). — 208. R. Kaiser, Chem. Technlk, 6, № 4, 207 A964). — 209. А. Б. Суч-
Сучков, Б. А. Б op ок, 3. И. Морозова, ЖПХ, 32, № 6, 1382 A959).—
210. A. Beerwald, Z. anorg. Chem., 261, Nt 1—2, 52 A950).
211. В, SchStzel, Chem. Technik, 5, № 7, 394 A953). — 212. B. Schat-
zel, Ibid.. 4, № 3, 114 A952). — 213. H. A. Lehman, G. Diekers, пат.
ГДР 33284 1962. — 214. Я. П. Б е р к м а н, С. В. К у ш н и р, ДАН СССР, 104,
№ 3, 448 A965). — 215. Н. Zirngibl, пат. ГДР 5167, 1953. — 216. К. Ebner,
яат. ФРГ 887196, 1953. — 217. В. В. В я з о в о в, Хнм. наука и пром., 2, № 6,
700 (}9Б6). — 218. О. К- Янатьева, Г. С. Рапопорт, И. С. Рассонская,
М. В. Устинова, ЖПХ, 34, № 10, 2347 A961). — 219. Б. А. Шойхет,
Б. Ю. Ланге, Хим. пром., № 6, 380 A958). — 220. Б. И. Исачкин, сб. «Тер-
«Термоизоляционные материалы», № 2, 1941, стр. 9.
221. К. П. Магницкий, Магниевые удобрения, Сельхозгиз, 1952.—
222. К- П. Магницкий, Хим. пром., № 5, 131 A950). — 223. М. М. Мазаева,
Удобрение н урожай, № 11, 38 A956).— 224. Н. Е. Пестов, ЖХП, 14, № 3,
185 A937). — 225. Н. Е. Пестов, Л. Ф. Г р ев ц ев а, Труды НИУИФ, в. 129,
1936, стр. 12. — 226. В. И. Зелионкайте, И. В. Яницкий, С. В. Вайрай-
те, Труды АН ЛитССР, Сер. Б, 2, 75, 85 (Ш68). — 227. Удобрения, перев. с англ.
под ред. А. В. Петербургского, Изд. «Колос», 1965, стр. 294. — 228. В. Л. Вар-
Варшавский, Г. Я. Попова, Б. А. К о п ы л е в, М. Е. П о з н н, Труды НТК
«Оболовые фосфориты», Таллин, 1968, стр. 181. — 229. И. 3. В а л ь д штей н а о,
С. В. Вайрайте, В. И. Зелионкайте, Научные труды вузов ЛитССР. Хим.
я хим. технол., т. XI, 1968.
Гл а в а X
СОЕДИНЕНИЯ БОРА
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Бор — весьма активный элемент, не встречающийся в природе
в свободном виде. Элементарный бор восстанавливается из бор-
борного ангидрида магнием1 (и алюминием), причем образуются
также бориды магния 5~7. Более чистый бор можно получить, вос-
восстанавливая его из ВС13 металлическим цинком8, или электролизом
расплава В2О3 в криолите9. В воздухе бор загорается при 700°,
образуя борный ангидрид В2О3. Выше 900° бор соединяется с азо-
азотом, образуя нитрид бора BN, имеющий высокую тугоплавкость
(плавится под давлением выше 3000°) 10~12 При нагревании до
2500° смеси В2О3 (или В) с углем без доступа воздуха образуются
черные кристаллы карбида бора (плотностью 2,52 г/сж3I3. Карбид
бора устойчив в азотной кислоте, в хлоре и в кислороде до 1000°;
при сплавлении со щелочами в присутствии воздуха — разлагается.
Карбид бора В6С по твердости близок к алмазу, а В4С и В3С —
тверже алмаза. Бориды металлов тоже обладают большой твер-
твердостью; так, борид хрома в смеси с железом является наплавоч-
наплавочной смесью, придающей поверхности стальных изделий повышен-
повышенную износоустойчивость148. Трифторид бора BF3 образуется1-
при взаимодействии В2О3 и HF в присутствии концентрированной
серной кислоты или при нагревании до 500° тетрафторбората ба-
бария20 Ba(BF4J. Трифторид бора — газ (кипит при —100,4°, пла-
плавится при —127,1°), хорошо растворимый в концентрированных
серной, азотной, фосфорной кислотах21-22, а также в органических
растворителях. В воде разлагается, образуя борную и борофтори-
стоводородную (тетрафтороборную) кислоты:
4BF3 + ЗНгО = Н3ВО3 + 3HBF4
1 мл воды при 0° и 762 мм рт. ст. поглощает 1057 мл газообраз-
газообразного BF8, а 1 мл серной кислоты плотностью 1,85 г\см% — 50 мл
312
Гл. X. Соединения бора.
Физико-химические свойства
31»
BF3. Плотность газообразного BF3 при 760 мм рт. ст. и 20° 3,0 г/л,
г при 88° — 4,613 г/л; плотность жидкого BF3 равна 2,6999—
€,00642 Т. Давление пара BF3 выше температуры кипения вычис-
вычисляется по формуле lgP = 5,1009—889,6/7 (где Р в вг), а ниже
— 101,4° — по формуле lg Р = 8,928— 1023,5/Г (здесь Р л
мм рт. ст.J5'24.
С HF и фторидами BF3 образует борофтористоводородную кис-
кислоту HBF4 и ее соли —фторбораты, например, KBF426. Предло-
Предложены методы получения B2F426. Трихлорид бора ВС13 кипит при
+ 18°, плавится при —108°; о методах его получения см.27~29; о по-
получении ВВг3 и В13 см.30; о получении монофосфида бора ВР, ис-
используемого в качестве огнеупорного материала и полупроводника,
см.
Я—35
О свойствах и получении фосфата бора, используемого в каче-
качестве катализатора (при гидратации этилена и проч.), см.36-37.
С водородом бор образует различные гидриды бора —борово-
дороды (бораны). Последние способны образовывать многочислен-
многочисленные комплексные соединения — полигидрополибораты и др.38.
Борный ангидрид В2О3 имеет две кристаллические модифика-
модификации, плавящиеся при 294 и 465° (плотность 1,85 и 2,46 г/см3), но
обычно находится в стеклообразной форме (плотность 1,78—
1,85 г/см3; плавится при 557°); кипит при ~1860° (по другим дан-
данным при ~2200°39); гигроскопичен, с водой образует борные кис-
кислоты.
Борная (ортоборная) кислота Н3ВО3 (или В2О3 • ЗН2О) выде-
выделяется из растворов в виде бесцветных кристаллов (плотность 1,43—
1,49 г/см3). При нагревании постепенно теряет воду и переходит
(при 70—110°) в метаборную кислоту НВО2 (или В2О3-Н2О), су-
существующую в трех кристаллических формах с температурами
плавления 176, 201 и 236°. При дальнейшем прокаливании превра-
превращается в борный ангидрид В2О3. Насыщенный водный раствор
содержит при 0° 2,6%, при 21° 4,9%, при 100° 27,6% Н3ВО3. При
кипячении водных растворов борная кислота улетучивается вместе
<; парами воды. Константы диссоциации Н3ВО3:
/ 20°
(по другим данным К\
= 1,6-10~14; Н3ВО3 легко вытесняется из ее солей минераль-
минеральными кислотами, даже такими слабыми, как Н2СО3 h.H2S. Недав-
Недавними исследованиями установлено39а, что в водных растворах ион
•бората находится в форме В(ОН)~. Поэтому диссоциация борной
кислоты протекает по уравнению
В(ОНK + 2Н2О = B(OH)J + Н3О+
Тетраборная кислота Н2В4О7—более сильная; для нее К\°°— 2 • 10""
и /G>5°^2- 10~5 (по другим данным К?"^ \0~\
¦7,3-ИГ10), /СГ=1,8-1О-13 и
!5°
-13
= 5,84- Ю-10
Соли борных кислот носят общее название боратов; к ним
относятся ортобораты, метабораты и многочисленные соли поли-
полиборных кислот, имеющих общую формулу /пН3ВО3 • гсНгО или
Н3тп-2пВтО3т-п (для тетраборатов m = 4, п *= 5; для гексаборатов
m = 6, п = 7; для декаборатов m = 10, п = 12 и т. д.40).
Бура —тетраборат (пироборат) натрия Na2B4O7, кристалли-
кристаллизуется из водных растворов выше 56° в виде Na2B4O7 • 5Н2О; ниже
56° — в виде Na2B4O7 • 10Н2О. Последний образует крупные моно-
моноклинные призматические кристаллы, выветривающиеся на воздухе
(плотность 1,70 г/см3). Насыщенный водный раствор содержит при
0° 1,3%, при 100° 34,3% Na2B4O7. При прокаливании буры при
350—400° получается безводный Na2B4O7, который при 742° пла-
плавится, превращаясь в прозрачное хрупкое стекло (плотность
2,34 г/см3). При спекании буры с борной кислотой при 140—180°
получаются полибораты с отношением Na2O : В2О3 < 1 : 4 41>42. Ме-
Метабораты образуют с Н2О2 пербораты43'44. Известны истинные пер-
бораты натрия NaBO3, калия 2КВО3 • Н2О и аммония 2NH4BO3 -
• Н2О. Техническому продукту, называемому перборатом натрия,
неправильно приписывали формулу NaBO3 • 4Н2О, на самом деле
он является пергидратом метабората натрия ЫаВОг • Н2О2 • ЗН2О.
Он представляет собой бесцветные гигроскопические кристаллы,
содержит 10—10,4% активного кислорода, обладает окислитель-
окислительными свойствами. В 100 мл воды при 18° растворяется 1,17 г. Вод-
Водные растворы имеют щелочную реакцию; при действии на них раз-
разбавленных кислот выделяется перекись водорода. Выше 50° раз-
разлагается с выделением кислорода. В сухом состоянии при обыкно-
обыкновенной температуре хранится длительное время без разложения
(потеря активного кислорода ~0,1% в год). О тиоперборатах нат-
натрия и калкя см.45.
Малорастворимые бораты щелочноземельных металлов являют-
являются основными солями46> 47. Например, кальциевая соль гексабор-
ной кислоты имеет строение:
НОСаОч /Оч
ОН ОН ОН
При удвоении этой структуры получается Са2В6Оц • 5Н2О или
2СаО • ЗВ2О3 • 5Н2О — формула минерала колеманита. Если к одно-
одному из гидроксилов присоединить дополнительно молекулу воды, то
после удвоения получится формула мейергофферита Са2В6Оц • 7Н2О
или 2СаО • ЗВ2О3 • 7Н2О. При присоединении по молекуле воды к
каждому из четырех гидроксилов аниона поли борной кислоты по-
получится СаН(Н3ОLВ3О8, а после удвоения формула минерала
иньоита Са2В6Оп • 13Н2О или 2СаО • ЗВ2О3 • 13Н2О. Примером со-
солей гетероиоликислот бора (т. е. кислот, содержащих, кроме бора,
S14
Гл. X. Соединения бора
кислорода и водорода, атомы других элементов) являются бороси-
ликаты, например датолит 2СаО • В2О3 • 2SiO2 • Н2О.
При смешении борной и плавиковой кислот 48~53 образуется ком-
комплексный ион BFI тетрафторборной кислоты HBF4. Известны двой-
двойные соли, получающиеся из фтористого натрия и буры (или мета-
бората натрия или борной кислоты) и из фтористого аммония и
борной кислоты54-55. Растворимость Н3ВО3 и NaF при совместном
присутствии, т. е. в системе НзВО3 — NaF — Н2О, по сравнению
с растворимостью их в воде, резко возрастает56; например, при 25°
. . - 4,27 34,46 44,34 35,44 14,12 5,29
. . 3,86 4,19 9,85 12,63 10,68 2,70 —^
Твердые фазы NaF NaF+H3BO3 H3BO3
Применение
816
Н3ВО3 в растворе, %
NaF в растворе, % .
Это указывает на комплексообразование в растворе с появле-
появлением фторзамещенных полиборатов.
При извлечении бора аммиаком из отработанных растворов в
производстве борной кислоты образуются гексабораты аммония-
магния57. При 20—25° образуется семиводный гексаборат(ЫН4JО •
¦ MgO-3B2O3-7H2O; ниже 20° существует (NH4JO -MgO -ЗВ2О3-
- 15Н2О. Гексаборат аммония-магния гидролизуется — вода вымы-
вымывает из него борат аммония:
2(NH4JMgBeOu • aq + 9Н2О = Mg2BeOi2 •
О получении полиборатов аммония см.68.
Гидролизу подвержены и другие бораты59. В 0,02 М водном рас-
растворе, насыщенном изоамиловым спиртом, моноборат натрия гид-
гидролизуется незначительно, а пента- и тетраборат почти полностью
разлагаются до моноборатов:
NaB5O8 + 6Н2О = NaBO2 + 4Н3ВО3
^ Na2B4O7 + ЗН2О = 2NaBO2 + 2Н3ВО3
Разложение гексабората магния MgB6Oi0 начинается при кон-
концентрации меньше 0,002 М; диборат магния в 0,01 М растворе гид-
гидролизуется на 30%:
MgB2O4 + 4H2O ;fz> Mg(OHJ + 2H3BO3
Индерит Mg2B6On • 15Н2О при концентрации 0,002 М целиком
разлагается:
Mg2B6Ou + ЗН2О = 2MgB2O4 + 2Н3ВО3
Бораты кальция разлагаются легче, чем бораты магния.
При взаимодействии В2О3 с SiO2 и Р2ОБ получаются стекла.
Первичной фазой, кристаллизующейся в системе В2О3—SiO2—Р2О5
для значительной части треугольника составов, является, по-види-
по-видимому, ВРО460. Линия ликвидуса для составов с 30—70 вес.% SiO2
имеет эвтектический характер61. В этом интервале концентраций
стекла имеют малую устойчивость к атмосферной влаге.
Описаны полимеры на основе бора62'63, для которых характерна
связь бора с кислородом =В—О—В = , дающая длинные цепи и
замкнутые кольца. Эта связь сравнительно легко разрушается во-
водой. Такое же строение имеют, возможно, и полимеры метафосфата
борила (ВО)РО3, который является солью, образующейся в виде
белого порошка из НРО3 и Н3ВО3 (в присутствии концентрирован-
концентрированной уксусной кислоты), причем борная кислота проявляет в этом
случае нехарактерные для нее основные свойства.
Обнаружено64 наличие полимеров борной кислоты в ее раство-
растворах, особенно в концентрированных65. Полимеризационные свой-
свойства бора позволяют использовать некоторые его соединения
(например, арилборные кислоты) в реакциях конденсации для полу-
получения пленок и волокон66. При взаимодействии боранов с серово-
сероводородом получается высокомолекулярный полимер (BHS)n, даю-
дающий бесцветные, прозрачные пленки67. Нитрид бора образует поли-
полимеры (BN)n со слоистой структурой; они могут быть получены в
виде мягких чешуек (аналогично графиту). Стеклообразный бор-
борный ангидрид (В2О3)П относится к полимерам, имеющим простран-
пространственную структуру68. Некристаллизующийся полимерный продукт
можно также получить при выпаривании раствора тетрабората
с фосфатом и щелочью; этот продукт предложено использовать
для защитных покрытий металлов.
ПРИМЕНЕНИЕ69-88
Бор используют в ядерной физике — в связи с его способностью
поглощать нейтроны, он препятствует ядерным цепным реакциям 70.
В металлургии элементарный бор применяют для изготовления ли-
лигатур. Поверхностный слой стали, насыщенный бором, приобретает
высокие механические и антикоррозионные свойства. Такая сталь
оказывается достаточно устойчивой в смеси соляной и азотной кис-
кислот. Используют также содержащие бор сплавы алюминия, меди,
никеля, оловянные и алюминиевые бронзы и др. Способность буры
растворять окислы металлов позволяет применять ее в качестве
плавня при сварке и пайке металлов (для этой цели используют
и борат цинка).
Вследствие высокой твердости большое значение как абра-
абразивы приобрели карбиды бора. Использование нитрида бора осно-
основывается на его высокой огнеупорности (до 3000°) в нейтраль-
нейтральной или восстановительной среде. Из него изготовляют, например,
жаростойкие подставки и изоляторы для индукционных высоко-
высокочастотных печей. Бориды титана, циркония, ванадия, ниобия, тан-
тантала, молибдена, вольфрама, марганца и других тугоплавких ме-
металлов характеризуются высокими температурами плавления и
316
Гл. X. Соединения бора
твердостью71-72, положительными температурными коэффициен-
коэффициентами электросопротивления, способностью переходить в сверхпро-
водимое состояние73 и другими ценными свойствами. Это обусло-
обусловило разработку методов их получения 74-7Б. Трехфтористый бор и
его производные все шире используются как высокоактивные ката-
катализаторы в органическом синтезе, в частности в процессах пере-
переработки нефти, а также в гальванотехнике и литейном деле76.
Бура и борная кислота издавна применяются в стекольной про-
промышленности, в частности для изготовления стекловолокна, опти-
оптического стекла и химической посуды. Из применяемых для произ-
производства борного стекла боратов щелочных металлов R2O • 2В2Оз,
R2O • В2О3 и 2R2O • В2О3 (где R—Li, Na, К) наиболее прочными
в расплавах являются соединения типа R2O • В2Оз, которые улету-
улетучиваются при 1300—1400° без разложения77. В керамической про-
промышленности бура и борная кислота используются для изготовле-
изготовления глазурей. Некоторые природные борные соединения, например,
пандермит 2СаО • ЗВ2О3 • ЗН2О, в смеси с полевым шпатом приме-
применяют для пропитки керамических изделий с целью придания им
водонепроницаемости. В эмаль для металлической посуды добав-
добавляют буру для уменьшения разницы между коэффициентами тер-
термического расширения эмали и металла. В присутствии небольших
количеств В2О3 значительно уменьшается вязкость эмалевого стек-
стекла и облегчается смачивание им стали78.
В текстильной промышленности буру используют при мытье
тканей, для растворения жиров, для фиксации на тканях некото-
некоторых минеральных протрав; она заменяет мыло при очистке шелка
в процессе размотки коконов; ее примешивают к крахмалу для
придания блеска накрахмаленным тканям. Буру применяют в ко-
кожевенной и в целлюлознобумажной промышленности. В электро-
электрохимических производствах буру и борную кислоту применяют в ка-
качестве добавки к электролитам, например, при получении катод-
катодного никеля G—16 г\л Н3ВО3), в резиновой промышленности для
замедления вулканизации каучука, а также в мыловаренной и в
парфюмерной промышленности и в органическом синтезе79.
В США выпускают более 50 наименований борных соединений
неорганических и органических, в том числе полимерных. Важной
областью применения соединений бора стало производство топлив
для ракетных двигателей — боранов и их алкильных и других про-
производных, обладающих высокой теплотворной способностью80.
Из неорганических соединений бора в капиталистических стра-
странах в наибольшем количестве выпускают и потребляют различные
бораты натрия, затем борную кислоту, бораты калия, аммония,
кальция, свинца, бария и др.
Борную кислоту и буру используют для пропитки деревянных
конструкций с целью защиты их от гниения и для придания им
огнестойкости. Благодаря антисептическим свойствам борную кис-
Применение
317
лоту и буру применяют в медицине. Их используют также в ка-
качестве консервирующего средства в пищевой промышленности.
При этом следует учитывать, что соединения бора могут быть
вредны для человеческого организма при систематическом их
принятии внутрь даже в малых дозах A—2 г в сутки вызывают
быстрое, а доза 4 г — немедленное отравление81).
Буру упаковывают в бумажные мешки, а борную кислоту в
деревянные бочки или матерчатые мешки. Требования к составу
буры и борной кислоты приведены в табл. 23 и 24.
ТАБЛИЦА 23
Качество технической и пищевой буры (в %) по ГОСТ 8429—69
27 не менее
Не растворимый в воде остаток, не более
N82SO4, не более
Na2CO3 » »
Хлориды в пересчёте на хлор, не более
Тяжелые металлы сероводородной группы в пере-
пересчете на свинец, не более
Железо, не более
Мышьяк » »
Остаток после просева:
на снте 0,15 мм, ае более
» » 0,6 » » »
Качество борной кислоты (в %) по ГОСТ 2629—44
Техническая
49,5
0,2
0,4
0,4
Не нормировано
Пищевая
51,5
0.02
0.1
Отсутствует
0,005
0,01
0,004
0,001
10
Отсутствует
ТАБЛИЦА 24
HsBO3, не менее
Не растворимый в воде остаток, не более
СГ", не более "
SO2- » »
Fe » »
Тяжелые металлы группы H2S, не более.
Влага, не более
I сорт
99,5
0,005
0,001
0,008
0,001
0,001
II сорт
98,5
0.1
0.2
0,6
0.005
0,005
Техническая бура, предназначенная для экспорта по ГОСТ
10.60—71, должна содержать не менее 99,5% Na2B4O7- 10H2O и
не более: 0,004% Fe, 0,0025% Са, 0,0015% тяжелых металлов се-
сероводородной группы (РЬ), 0,0012% А1, 0,01% SO4, 0,005% хло-
хлоридов (С1).
Борную кислоту, применяемую для электротехнических кон-
конденсаторов, получают перекристаллизацией технического продук-
продукта; к ее чистоте предъявляют более высокие требования (ГОСТ
5281—50).
Перборат натрия 82, обладающий белящими свойствами, при-
применяют в качестве мягкого белителя для Отбелки шерсти, шелка,
бархата, соломы, а также для изготовления моющих средств.
818
Гл. X. Соединения бора
Окислительные свойства пербората натрия позволяют использовать
его в качестве заменителя водных растворов перекиси водорода.
В некоторых производствах источником бора служит борный
ангидрид. Например, применение его вместо борной кислоты в ка-
качестве сырья для выплавки карбида бора снижает расход электро-
электроэнергии. Бораты цинка используют в качестве люминофоров, ката-
катализаторов в органическом синтезе, для производства огнеупорных
волокнистых веществ и др.
Борсодержащие вещества являются эффективными микроудо-
микроудобрениями — среди всех микроэлементов бор имеет наибольшее зна-
значение 83~89. В большинстве почв содержание бора колеблется от 20
до 200 мг/кг, но обычно только около 5% его находятся в усвояе-
усвояемой форме90. Отсутствие или недостаток бора в почве вызывает
заболевания растений, приводящие к снижению урожая (гниль сер
дечка у сахарной и кормовой свеклы, отмирание верхушки льна
люцерны и др.). Помимо профилактического действия, бор повы-
повышает урожайность, например, внесение борных удобрений в коли-
количестве 6—9 кг/га (в пересчете на Н3ВО3) дает прирост клевера и
люцерны 0,5—1 ц/га, кормовой свеклы 4—7 ц/га, овощей 2—5 ц/га
и т. п.87- 88.
Наиболее концентрированными борными удобрениями являются
борная кислота и бура, но обычно в качестве удобрений приме-
применяют промышленные отходы, содержащие небольшие количества
бора, некоторые природные бораты или продукты их простейшей
переработки. Например, по ГОСТ 9595—61, борат магния, получае-
получаемый осаждением каустическим магнезитом из маточных растворов
от производства борной кислоты и используемый в качестве микро-
микроудобрения, должен содержать не менее 9% лимоннорастворимых
соединений бора в пересчете на Н3ВО3. Для предпосевного опудри-
вания семян применяют порошок, содержащий бор; это механиче-
механическая смесь тонкоизмельченных (меньше 0,15 мм) борной кислоты
и талька. Согласно СТУ 76—650—62, в нем должно быть 14—16%
НзВОз и менее 1% влаги.
Для получения борных удобрений целесообразно использовать
также такие виды бедного борного сырья, как сопочные грязи,
нефтяные буровые воды91, отходы от обогащения борных руд, тур-
турмалиновые руды, содержащие 2—3% В2О3, гипсы, содержащие
примеси борных минералов и др.92. Местные удобрения — зола,
торф и навоз — также имеют значение в качестве источников бора:
1 кг золы древесины содержит от 200 до 700 мг бора, 1 кг сухого
вещества навоза и торфа — около 20 мг. Небольшие количества
бора содержатся в сырых калийных солях D—8 мг/кг) и в доломи-
доломитах C—8 мг/кг). Чаще всего соединения бора добавляют к другим
удобрениям, например к суперфосфату, так как распределение
малых количеств борного удобрения на большую площадь посева
затруднительно. Соединения бора вводят и в жидкие удобрения в
Сырье
319
II
и I
виде буры или полибора 2 — частично дегидратированного пента-
или тетрабората натрия93. В США и в Канаде рекламируются бор-
борные и другие микроудобрения в виде фриттов — размолотых спла-
сплавов соединений микроэлементов со стеклом. В таком виде микро-
микроэлементы не поглощаются почвой, а медленно вымываются и по-
постепенно поступают в растения94.
СЫРЬЕ
Распространенность бора значительна — его весовой кларк для
земной коры 5-10%. Однако борные минералы (табл. 25) весьма
рассеяны, и имеется мало месторождений с высокой концентра-
концентрацией бора, представляющих практический интерес.
Турмалин — борсодержащий алюмосиликат переменного со-
состава, относящийся к первичным породам. В табл. 25 приведен его
ТАБЛИЦА 26
Некоторые борсодержащие минералы
Борацит (стасс-
фурнт)
Сульфоборит
Пннноит
Ашарит (ссай-
белеит)
Индернт
Курнаковит
Патерноит
Гидроборацит
Калиборнг
Боронатрокаль-
цит (улекснт)
Иньонит
Мейергоффернт
Колеманит (бо-
рокальцнт)
Пандермнт
(прицент)
Тннкал (бура)
Тинкалкернит
Кернит (разо-
(разорит)
Сассолин (бор-
(борная кислота)
Данбурит
Датолит
Турмалин'
Химическая формула
Твер-
Твердость
Плотность,
MgCl, • 5MgO • 7В2О3
3MgSO4 • 6MgO • 4В2О3 ....
MgO • В.О3 • ЗН2О ....
2MgO • В2О3 • Н2О или MgHBO3 ..'.'.'..
2MgO-3B2O3-15H2O
2MgO - ЗВ2О3 • 13Н2О
MgO • 4В2О3 ¦ 4Н2О ' ' '
СаО • MgO • ЗВ2О3 • 6Н2О
K2O-4MgO-llB?O3-18H,O .'
Na2O • 2СаО • 5В2О3 • 16Н2О ,
2СаО • ЗВ2О3 • 13Н2О
2СаО • ЗВ2О3 • 7Н2О
2СаО • ЗВ2О3 - 5Н2О
2СаО ¦ ЗВ2О3 ¦ ЗН2О *
Na2O - 2В2О3 • 10Н2О .
Na2O • 2В2О3 • 5Н2О ' '
Na2O • 2В2О3 • 4Н2О [ [
В2О3-ЗН2О или Н3ВО3
СаО • В2О3 • 2SiO2
2СаО • В2О3 ¦ 2SiO2 • Н2О "
Na2O • (Fe, Mg)O • 10А12О3 • 18SiO2 • 4В2О3 ¦ 5Наб
4
3-4
3-4
1,5-2
3
2-3,5
4-5
2
2
4-4,!
3-3,*
2-2,5
2,5
3
5-5,5
7-7,5
2,9-3,2
2,32-2,45
2,29
2,69
1,79
1,85
2,11
1,9-2,17
2,13
1,65-1,95
1,88
2,12
2,41-2,44
2,26-2,48
1,69-1,76
1,88
1,9
1,5
2,9-3,6
2,9-3,2
1 Пандермнту приписывают также составы 8CaO.l0B2O3.I5H20 и 5СаО.6В2О3-9Н2О.
820
Га X. Соединения бора
приблизительный состав, который выражают также приблизитель-
приблизительной формулой: (Na, Са) (R, AlN [Si6Al3B3(O, ОНKо], где R—Mg,
Fe, Mn, Li. Турмалиновые руды сопровождаются другими ценными
минералами, после обогащения которых могут быть получены тур-
турмалиновые отходы, содержащие около 3% В2Оз, 75% SiC>2, 8%
Fe2O3, 9% А12О3 и др. Обогащением этих отходов могут быть полу-
получены концентраты, содержащие до 8% В2Оз. Датолит — водный
боросиликат кальция — образовался, вероятно, в результате разру-
разрушения водой первичных пород. Датолитовые руды содержат, на-
например, 9—13% В2О3, 30—32% СаО, до 3% MgO, 3—6% А12О3,
5—8% Fe2O3 + FeO, 40—42% SiO2, до 5% СО29^96. Путем обога-
обогащения получают датолитовый концентрат: 15,5—16,5% В2О3 (вме-
(вместо 21,9% в чистом минерале), 33,8—34,5% СаО, 39,6—40,5% SiO2,
3,5—4,5% Fe2O3 + FeO и др.
Под действием воды и двуокиси углерода первичные породы
разлагаются с выделением свободной борной кислоты и ее раство-
растворимых солей, попадающих в океан, в воде которого, по данным
А. П. Виноградова, содержится бора 4,5 г/ж3. (Приводят также
цифру 3,6 - 10~4%.) По другим данным, в сухом остатке морской
воды ~0,05% В2Оз97. Зола некоторых морских растений содер-
содержит98 в 20 раз больше бора — до 1% В2О3. При испарении мор-
морской воды до эвтонической точки бор остается в растворе, в твер-
твердые фазы переходит небольшая его часть: концентрация В2Оз в
карбонатных твердых фазах составляет 0,02%, в гипсах 0,006—
—0,007%, галит кристаллизуется без бора". Бораты магния-каль-
магния-кальция, натрия-магния и другие образовались при высыхании морей,
и их месторождения расположены обычно над пластами галита и
сильвина под слоем гипса, карбонатов и других пород 46>100'1Ш.
Первично выделившиеся из морской воды преимущественно маг-
магниевые бораты в результате метаморфизации переходили в каль-
кальциевые. Растворимые бораты, такие как боронатрокальцит и дру-
Тие, явились источником накопления отложений буры, кернита и
других твердых солей, а также накопления буры и борной кислоты
в рассолах из нефтяных скважин и других буровых и минераль-
минеральных водах, а также в рапе озер. Эти рассолы, несмотря на малую
концентрацию в них бора (сотые доли процента), составляют зна-
значительные запасы борного сырья. Борная кислота содержится
парах некоторых горячих источников, куда она проникает в резуль-1
тате разложения парами воды турмалинов, находящихся в глубо-j
ких слоях почвы.
Большая часть запасов борного сырья находится в Западно»
полушарии, главным образом в США (около 90%). На долю СШЛ|
приходится 95% добычи и больше 85% мировой продукции борных
соединений. Ежегодная добыча боратов в США (в пересчете Hi
ВоОз) превышает 300 тыс. т. (По другим данным, в 1965 г. в США1
выработано 425 тыс. т борных соединений в пересчете на В2О3.)
Сырье
321
Добывают и перерабатывают только богатые бором бораты — тин-
кал, кернит, колеманит, улексит, пандермит, сассолин, борацит.
Борным сырьем в США служит также рапа оз. Сирлз. Значитель-
Значительные запасы боратов находятся в Южной Америке (в Аргентине,
Чили, Перу, Боливии), а также в Турции. Борсодержащее сырье
имеется в ФРГ, Швеции, Иране, Индии, Японии, Шри Ланка и во
франции. Общие запасы борсодержащего сырья капиталистиче-
капиталистических стран оценивается в 115—120 млн. т (в пересчете на
В2О3) 80'102-108.
В Западной Европе промышленная добыча борного сырья ве-
ведется лишь в Италии — в провинции Тоскана — извлекают борную
кислоту из паров вод горячих источников (соффионов), содержа-
содержащих несколько десятых долей процента Н3ВО3. Природные сква-
скважины дают пар с температурой около 100° и давлением 3 ат, а
пробуренные— 145° и 6 ат. Суммарный дебит скважин составляет
A965 г.) ~3000 т пара в 1 ч. Пары и воды источников, кроме бор-
борной кислоты, содержат СО2, H2S, CH4, NH3, He, Ar, Ne.
До открытия боратов в СССР буру и борную кислоту произво-
производили из импортного сырья — боронатрокальцита и кернита109.
В настоящее время используют отечественное сырье, главным об-
образом ашаритовые, гидроборацитовые, ашаритово-гидроборацито-
вые и ашарито-улекситовые руды. Наиболее богатые породы этих
руд содержат до 37% В2О3; среднее содержание В2О3 в эксплуати-
эксплуатируемых породах 17—20%, главным образом в виде ашарита. Бед-
Бедные руды, в которых преобладает гидроборацит, содержат от 1
до 6,5% В2О3 и должны подвергаться обогащению. Борным мине-
минералам сопутствуют примеси гипса, кальцита, магнезита и др. Фло-
Флотация гидроборацитовой руды дает концентрат, содержащий до
34% В2О3 при степени извлечения до 86%. Обесшламленную руду
после измельчения и классификации перемешивают с реагентами
и подвергают только основной флотации. Реагентами служат: ски-
скипидар B кг/т) и эмульсии аполярных масел, в частности керосина
G4 г/т) ш~т.
Согласно техническим условиям, боратовые руды, отгружаемые
химическим предприятием для переработки, подразделяются на
следующие виды и сорта:
Виды рул Содержание B2OS, %
I сорт II сорт III сорг
Ашаритовая Выше 27,5 22-27,5 16,5-22
Неашаритовая Выше 27,5 20-27,5 16 —20
Обычно на заводы поступает120 ашаритовая руда III сорта с
содержанием (в %): В2О3 18,9—21,2, СаО 13,2—16,8, MgO 14,1—
—21,4, А12О3 0,9—1,7, Fe2O3 0,8—1,6, SO3 4,2—6,4, СО2 4,3—5,8,
Н2О 13,9—22,2. Встречаются также боратовые руды, содержащие
значительные количества галита112-121. Такая руда может быть
II М. Е. Позин
322
Гл. X. Соединения бора
обогащена путем выщелачивания NaCl холодной водой. Так, из
образца солевого бората, содержащего 9,84% В2О3 и 45,6% NaCl,
путем промывки его холодной водой и высушивания был получен
концентрат с содержанием 44,5% В2О3 и лишь следов хлора. По-
Потери В2О3 составили при этом 2,4% 122.
Перспективным сырьем являются сопочные грязи, содержащие
до 0,5% В2О3. Для извлечения В2О3123 сопочную грязь прокали-
прокаливают в течение часа при 500° и выщелачивают водой при 80—90°.
Жидкую фазу, содержащую 0,35—0,45% В2О3, декантируют и об-
обрабатывают в течение часа при 95° окисью кальция D моль СаО
на 1 моль В2О3) — полученный продукт представляет собой бораты
кальция с 15—20% В2О3. Он может служить борным удобрением
или быть переработанным на борную кислоту сернокислотным ме-
методом.
МЕТОДЫ ПЕРЕРАБОТКИ БОРНОГО СЫРЬЯ
По условиям химической переработки борсодержащее сырье
может быть разделено на три группы:
1) легко разлагаемые минералы, главным образом осадочного
происхождения — натриевые, калиевые, натриево-кальциевые и
' магниево-кальциевые бораты (около 60 видов);
2) трудно разлагаемые минералы изверженного происхожде-
происхождения— сложные борсодержащие силикаты и алюмосиликаты (око-
(около 25 видов);
3) водные растворы, содержащие буру и борную кислоту.
В настоящее время в мировой борной промышленности приме-
применяется главным образом сырье, отнесенное к группе 1.
Методы переработки природных боратов на борную кислоту и
буру разделяют в основном на кислотные и щелочные.
Для разложения боратов могут применяться различные реа-
реагенты: кислоты — H2SO4, HNO3, НС1, Н3РО4, HF, Н2СО3(СО2),
H2SO3(SO2); щелочи и щелочные соли —NaOH, Na2CO3, NaHCO3,
Na2SO4, Na2S, Na2SO3, NH4C1, (NH4JCO3 и др. В качестве приме-
примеров ниже приведены реакции разложения ашарита некоторыми
кислотами и щелочами:
2MgO • В2О3 • Н2О + 2H2SO4 • aq = 2H3BO3 + 2MgSO4 + aq
2MgO • B2O3 • H2O + 4Н3РО4 • aq = 2H3BO3 + 2Mg(H2PO4J + aq
2BMgO • B2O3 • H2O) + ЗСО2 + aq = 4H3BO3 + 3MgCO3 • Mg(OHJ + aq
2BMgO • B2O3 • H2O) + Na2CO3 + 3CO2 + aq = Na2B4O7 + 4MgCO3+ aq
2Mgo • B2O3 • H2O + 2NaOH + aq = 2NaBO2 + 2Mg(OHJ + aq
¦ При выборе той или иной кислоты, кроме ее активности, до-
доступности и стоимости, следует учитывать и возможность использо-
использования ее магниевой соли, получаемой в качестве побочного про-
продукта.
Методы переработки борного сырья
323
Разложение некоторых боратов двуокисью углерода 124> 125 пред-
представляет экономический интерес, благодаря возможности исполь-
использования дешевой (отбросной) двуокиси углерода и связывания ее
в виде карбоната магния или кальция. Солянокислотное разложе-
разложение, так же как и использование для этой цели HF, связано с по-
потерями Н3ВО3 при кристаллизации ее из раствора и с затрудне-
затруднениями, вызываемыми коррозией аппаратуры. Это же относится и
к разложению азотной кислотой i26-128. Нагревание с раствором
карбоната и бикарбоната натрия, иногда при повышенном давле-
давлении, широко используется за границей при переработке в буру
легко разлагаемых минералов — боронатрокальцита, кернита и др.
Разработаны условия разложения гидроборацитовой и ашаритовои
руд раствором сернистого натрия129. В СССР, где главными ви-
видами промышленных борных руд являются ашарит, гидроборацит
и датолиты, применяют сернокислотную переработку боратов с по-
получением борной кислоты. Буру же вырабатывают из борной кис-
кислоты и соды, однако в ограниченных количествах, так как бура и
борная кислота почти всегда взаимозаменяемы и нет надобности
тратить средства на нейтрализацию борной кислоты. Серная кис-
кислота дает возможность вскрытия всех видов борного сырья, за
исключением трудно разложимых турмалиновых пород, которые
предварительно должны быть подвергнуты щелочно-термической
обработке. Фосфорнокислотное разложение природных боратов
магния, кальция и натрия 130 позволяет получать наряду с борной
кислотой удобрения. Так, после выделения Н3ВО3 из раствора, по-
полученного в результате разложения фосфорной кислотой датолита
и содержавшего 4,3% В2О3, 26,5% Р2О5, 5% MgO, оставался ма-
маточный раствор, в котором было 1,2% В2О3, 26,5% P2Os, 5% MgO.
В этом растворе остаются фосфаты магния и кальция, и после об-
обработки его известняком или аммиаком получаются ценные фос-
форно-магниевые и фосфорно-магниево-аммиачные удобрения, со-
содержащие бор. Обычно при кислотном разложении боратов полу-
получают пульпу, которую разделяют на шлам и раствор, идущий на
кристаллизацию борной кислоты. Если боратовое сырье содержит
много растворимой кремневой кислоты, разложение ведут так,
чтобы получить густую пульпу, и борную кислоту извлекают из
нее выщелачиванием водой; в такой пульпе кремневая кислота ко-
коагулирует и при выщелачивании не переходит в раствор.
Извлечение бората из руды, содержащей борат кальция, мож-
можно производить, например, обрабатывая ее горячим раствором
(NH4JCO3, NH4HCO3 или смесью NH3 и СО2131. Раствор отделяют
от неразложившегося остатка и образовавшегося СаСО3 и при его
охлаждении кристаллизуют борат аммония. Для регенерации NH3
и СО2 раздельно нагревают борат аммония и СаСО3 до температур
их разложения. После регенерации NH3 из раствора или кристал-
кристаллического бората аммония остается Н3ВО3.
$24
Гл. X. Соединения бора
Из реакционной массы, образующейся при разложении борато-
вой руды, борную кислоту можно извлекать спиртами (или фено-
фенолами). При этом образуются эфиры борной кислоты (например,
при использовании метанола — триметилборат). Эфир отгоняется и
при последующем его гидролизе спирт регенеруется. При охлаж-
охлаждении образовавшегося концентрированного водного раствора кри-
кристаллизуется борная кислота, а маточный раствор возвращается
в процесс.
Представляет интерес способ отгонки борной кислоты водяным
паром при 400—500° из пульпы, полученной, например, сернокис-
сернокислотным разложением боратовой руды 120. Отгонку можно вести в
аппарате с кипящим слоем материала, в этом случае пульпа пред-
предварительно гранулируется с использованием ретура. Смесь паров
4борной кислоты и воды после очистки от пыли охлаждается в кон-
конденсаторах и получаемый здесь раствор борной кислоты перера-
перерабатывается на твердый продукт выпаркой и вакуум-кристаллиза-
щией.
О технике безопасности в производстве борной кислоты см.|32,
БОРНАЯ КИСЛОТА
Природная борная кислота
В Тоскане (Италия) борную кислоту добывают примитивными
методами из фумарол — струй газов и паров, выделяющихся из
земных расщелин. Пары направляют для конденсации в бассейны
с водой. Летом, при испарении из них воды, на дно выпадают кри-
кристаллы борной кислоты. Оставшийся раствор затем концентри-
концентрируют при 85° до начала выпадения кристаллов. После этого при
медленном охлаждении раствора в течение 3—4 суток из него кри-
кристаллизуется борная кислота. Полученную «сырую» борную кис-
кислоту, содержащую 80—82% Н3ВО3, 6—8% (NH4JSO4, 3—4%
MgSO4> 1—1,5% Na2SO4 и K2SO4, примеси Fe2O3, песка, глины,
гипса и другие, подвергают перекристаллизации.
Маточный щелок после первой кристаллизации сырой борной
кислоты содержит 5—9% (NH4JSO4 и 3—5% Н3ВО3. Его возвра-
возвращают на выпаривание и после накопления в нем 15—20%
(NH4JSO4 обрабатывают щелочью; отгоняющийся аммиак погло-
поглощается серной кислотой — образуется чистый сульфат аммония.
Получение борной кислоты из буры
При обработке буры кислотами образуется борная кислота, на-
Na2B4O7 + H2SO4 + 5H3G = 4Н3ВО3 + Na2SO,
пример:
Борная кислота
325
Кроме серной и соляной кислот, можно применять и сернистую.
В последнем случае через раствор буры или через'водную суспен-
суспензию содержащего буру минерала пропускают SO2. Этот способ по-
получения борной кислоты в настоящее время в СССР не приме-
применяется.
Получение борной кислоты из магниевых боратов120> 121-124-15<>
Магниевые бораты сравнительно легко и полностью разлагают-
разлагаются серной, соляной, азотной и фосфорной кислотами. Ашарит трудно
и недостаточно полно разлагается сернистым газом, а двуокисью
углерода и растворами бикарбоната и соды ашарит практически
не разлагается. Гидроборацит сравнительно легко и в достаточной
степени разлагается сернистым газом, бикарбонатом и содой, но
труднее двуокисью углерода.
Для переработки ашаритовых пород применяют сернокислот-
сернокислотное разложение. Несмотря на то, что гидроборацитовые породы
разлагаются щелочами, последние нецелесообразно применять для
этой цели, так как в породе содержится значительное количество
сульфата кальция. При обработке содой сульфат кальция перехо-
переходит в сульфат натрия:
CaSO4 + Na2CO3 + aq = Na2SO4 + CaCO3 + aq
Это приводит к повышенному расходу соды. Получаемые ра-
растворы содержат много сульфата натрия, и после выделения из
них буры требуется затрачивать много тепла для переработки
оставшегося раствора сульфата натрия путем его выпаривания.
Поэтому гидроборацитовые породы, как и ашаритовые, и ашарито-
улекситовые, подвергаются сернокислотному разложению.
Разложение ашарита и гидроборацита серной кислотой идет
по следующим реакциям:
2MgO • В2О3 • Н2О + 2H2SO4 + aq = 2MgSO« + 2Н3ВО3 + aq
CaO • MgO • 3B2O3 • 6H2O + 2H2SO4 + aq = CaSO« + MgSO4 + 6H3BO3 + aq
Присутствующие в породе примеси, реагируя с серной кис-
кислотой, превращаются в сернокислые соли — CaSO4, MgSO.4,
A12(SO4K, Fe2(SO4K и др. Сульфат кальция практически весь пе-
переходит в осадок, что облегчает дальнейшую переработку маточ-
маточных растворов, остающихся после кристаллизации борной кислоты.
При обработке ашаритовой породы серной кислотой примеси
минералов с повышенным, против ашарита, отношением MgO : В2Оз
(в ашарите это отношение равно 1,14) разлагаются быстрее аша-
ашарита, особенно при невысоких температурах. Поэтому при опти-
оптимальных условиях, обеспечивающих хорошее разложение ашарита,
отношение MgO: В2О3 в полученных растворах соответствует от-
отношению этих компонентов в исходном сырье. Скорость разложе-
разложения ашарита (рис. 97) резко возрастает с повышением темпера-
температуры до 80°. Дальнейшее повышение температуры незначительно
326
Гл. X. Соединения бора
увеличивает скорость процесса. Величина частиц минерала в пре-
пределах от 0,2 до 0,6 мм не оказывает значительного влияния на
степень и скорость перехода В2О3 и MgO в раствор.
Кальциевые бораты разлагаются серной кислотой значи-
значительно медленнее, чем магниевые, так как на поверхности их зе-
зерен образуется корка из кристаллов сульфата кальция. В зави-
зависимости от условий разложения она имеет разную плотность упа-
упаковки и состоит из CaSO4-2H2O, p-CaSO4-0,5H2O или
CaSO4-H2SO4. Эта корка затрудняет диффузию кислоты к
поверхности растворяющегося
минерала и продуктов реакции в
раствор, что и лимитирует ско-
скорость разложения. С повышением
температуры концентрация сер-,
ной кислоты, при которой дости-
достигается максимальная скорость
разложения кальциевых боратов,
увеличивается. При сернокислот-
сернокислотном разложении ашаритовой по-
породы содержащиеся в ней при-
примеси кальциевых боратов при
размере зерен 2 мм к моменту
полного разложения ашарита
успевают разложиться в сравни-
бй 148
Борная кислота
327
80-
40
w го
30 40
Время, мин
-От
60
успеват разо
Рис. 97. Влияние продолжительности . тельн0 небольшой мере 148.
перемешивания на степень разложе- л г
ния ашарита серной кислотой. При одновременном присут-
присутствии в сырье магниевых бора-
боратов, углекислого кальция и соединений железа оказывается возмож-
возможным дозировать серную кислоту из расчета только на MgO, без
практического снижения скорости и уменьшения степени разло-
разложения боратов. Это позволяет вести процесс с уменьшенным про-
против стехиометрических количеств расходом серной кислоты за
счет уменьшения степени разложения примесей. Оптимальными
условиями являются: температура в реакторе не ниже 80° (обыч-
(обычно до 95°), количество серной кислоты 85—90% по отношению
к стехиометрической норме, рассчитаннвй по анализу сырья на
связывание MgO, CaO, Fe2O3 и А12О3 в сульфаты (за вычетом
содержащегося в сырье SO3); продолжительность перемешивания
30—60 мин; величина частиц сырья —2 мм. Серную кислоту вво-
вводят постепенно, при начальной температуре не выше 60°. По
окончании разложения пульпа должна иметь кислотность 0,2—
0,5% H2SO4. При таких условиях в раствор переходит 98—99,5%
В2О3, а степень разложения примесей значительно ниже — в рас-
растворе содержится меньше 0,4% R2O3 [в виде R2(SO4)S]. При боль-
большей кислотности пульпы сильнее разлагаются примеси, увеличи-
увеличивается расход фильтровальной ткани, затрудняется отмывка бор-
относительно
повышением температуры
ной кислоты. При недостатке кислоты пульпа может иметь щелоч-
щелочную реакцию, при этом уменьшается степень извлечения В2О3, а из
раствора могут выделяться полуторные окислы.
В отличие от ашарита гидроборацит хорошо разлагается го-
горячим содовым раствором, а при кипячении — даже водой; серной
кислотой он легко растворяется на холоду. Измельченная до тон-
тонкости меньше 0,15 мм руда разлагается за 5 мин после смешения
с кислотой больше, чем на 95%, даже при невысоких температу-
температурах. Ускорение и увеличение степени разложения
крупных зерен (— 1 мм) достигается
до 60°. Дальнейшее повышение
температуры увеличивает ско- юо
рость и степень разложения
уже в меньшей мере (рис. 98).
Отношение MgO: В2О3 в
гидробораците равно 0,19, т. е.
значительно меньше, чем в
ашарите. Если бы разложению
подвергался чистый гидробо-
гидроборацит, то получались бы рас-
растворы борной кислоты, мало
загрязненные примесями, что
уменьшило бы потери В2О3
при их отделении. Однако ги-
дроборацитовое сырье содер-
содержит большое количество при-
примесей, разлагающихся вместе
§90
3 ВО\-
I
§.70
ВО
20 3D
40 50 ВО 70
Температура, °С
ВО 90
Рис. 98. Влияние температуры на сте-
степень разложения гидроборацита (круп-
(крупность частиц ~ 1 мм):
1—5 мин; 2 —15 мин; 3 — 25 мин.
с гидроборацитом. Но гидроборацит разлагается значительно легче
других компонентов руды. Поэтому можно осуществлять разложе-
разложение неполной нормой серной кислоты B5—50% от стехиометриче-
ского); при этом борная кислота почти полностью переходит в ра-
раствор, а магниевые (не боратные) соединения разлагаются частич-
частично. Это приводит к уменьшению отношения MgO: В2О3 в растворе
по сравнению с сырьем (при учете магнийсодержащих примесей
пустой породы). В растворе, полученном при переработке гидро-
борацитовой руды, MgO : В2О3 равно 0,4—0,5.
Борную кислоту получают по следующей схеме151. Ашарито-
вую руду, содержащую —25% В2О3 и —17,5% MgO, дробят, вы-
высушивают при —400° во вращающейся барабанной печи топоч-
топочным газом, затем размалывают. Боратовую муку разлагают в си-
системе реакторов с мешалками серной кислотой в течение 1 ч при
90—95°. В 1-й реактор одновременно с мукой подают 75—78%-ную
серную кислоту, маточный раствор от кристаллизации борной
кислоты, воду и промывную воду. Пульпа из последнего реактора
поступает на ленточный вакуум-фильтр. Шлам, после промывки
на фильтре горячей водой, удаляют в отвал, он имеет влажность
328
Гл. X. Соединения бора
Борная кислота
329
~30% и содержит 1—1,5% MgO и 0,7—0,8% В2О3 (около поло-
половины этого количества в водорастворимой форме). Для улучше-
улучшения фильтрации в пульпу добавляют коагулянт — 0,5%-ный рас-
раствор полиакриламида. После контрольного фильтрования на
фильтрпрессе основной фильтрат (содержащий более 6% В2О3 и
~6%MgO) охлаждают в системе вакуум-кристаллизаторов
до 15°.
Во избежание выделения вместе с кристаллами борной кис-
кислоты полуторных окислов фильтрат, поступающий на кристалли-
кристаллизацию, должен быть слабокислым @,2—0,6% свободной H2SO4).
Кристаллы Н3ВО3 отделяют от маточного раствора на центри-
центрифуге, на которой затем промывают чистой холодной водой — кон-
конденсатом A—1,5 мъ на 1 т сырой борной кислоты). Промывную
воду возвращают в реактор. Влажность сырой борной кислоты
после промывки 5—8%. Сушку борной кислоты во вращающейся
сушилке с барабаном из нержавеющей стали осуществляют воз-
воздухом с температурой 90—100°. Более высокая температура сушки
недопустима, так как вызывает потери Н3ВО3 и частичный переход
ее в метаборную кислоту. Для получения борной кислоты повы-
повышенной чистоты (реактивной, химически чистой, медицинской и
особо чистой) ее перед сушкой подвергают перекристаллизации,
приготовляя растворением в воде при 95° 16—25%-ный раствор
и затем охлаждая его.
При переработке гидроборацитового концентрата из описанной
схемы исключается измельчение сырья и добавляется операция
нейтрализации и слабого подкисления серной кислотой фильтрата
(до кислотности 0,2—0,5%) после отделения от него шлама, так
как в этом случае фильтрат имеет слабощелочную реакцию. Кроме
того, для замыкания цикла по балансу воды необходимо осу-
осуществлять выпарку раствора (промывной воды), в противном
случае увеличатся потери В2О3.
Возможно разделение смесей Н3ВО3 и MgSO4'7H2O, а также
Na2B4O7- 10Н2О флотацией >36-137.
Борная кислота способна флотироваться без специальных реа-
реагентов. Суспензия боратовой руды в маточном растворе после от-
отделения кристаллов сульфата магния разлагается серной кислотой
при 90—95°. Шлам отделяется и промывается, промывные воды
возвращаются на разложение, а концентрированный раствор охла-
охлаждается (например, в вакуум-кристаллизаторе), причем из него
выделяется смесь кристаллов борной кислоты и сульфата магния.
При разделении этой суспензии во флотационной машине борная
кислота поступает в пенный продукт, а сульфат магния — в хвосты.
Суспензия сульфата магния сгущается и кристаллы отфуговывают-
ся или отфильтровываются; маточный раствор возвращается на
разложение руды. Пенный продукт подвергается нескольким пере-
перечисткам, а затем полученная борная кислота репульпируется в про-
промывной воде (для растворения увлеченных кристаллов сульфата
магния), отфуговывается, промывается водой и высушивается.
При разложении боратов, содержащих большие количества
MgO, серной кислотой, получаемый раствор содержит борную кис-
кислоту и сульфат магния. При возврате маточного раствора после
кристаллизации борной кислоты на разложение новых количеств
боратов раствор быстро насыщается сульфатом магния. Это делает
невозможным его дальнейшую циркуляцию в замкнутом цикле для
использования остающейся в нем борной кислоты, так как в кри-
кристаллизаторах вместе с борной кислотой начинает выделяться
MgSO4 • 7Н2О. Поэтому в цикл возвращают лишь часть маточного
раствора, а другую его часть, содержание магния в которой равно
количеству магния, извлекаемому из породы, выводят из произ-
производства.
С выводимым из цикла маточным раствором, содержащим 21—
22,5% MgSO4, 0,4—0,8% H2SO4 и 1,8—3,5% Н3ВО3, теряется часть
борной кислоты. Для того чтобы потери были наименьшими, рас-
раствор, поступающий на кристаллизацию борной кислоты, должен
содержать 20—21% MgSO4 и 10—12% Н3ВО3. Снижение концен-
концентрации MgSO4 увеличивает растворимость Н3ВО3 и количество ма-
маточного раствора, а следовательно, количество Н3ВО3 в отбросном
маточном растворе; повышение же концентрации MgSO4 может вы-
вызвать его кристаллизацию при охлаждении раствора, т. е. загряз-
загрязнение кристаллизующейся борной кислоты. Увеличение концентра-
концентрации Н3ВО3 в растворе может вызвать ее кристаллизацию еще в
процессе фильтрации пульпы, т. е. возрастание потерь В2О3.
Доля маточного раствора, возвращаемая в реактор при пере-
переработке ашаритовой руды, зависит от отношения MgO : В2О3 в
сырье — если это отношение больше единицы, маточный раствор в
процесс не возвращается; при MgO : В2О3 < 1 количество М ма-
маточного раствора, возвращаемого в процесс в кг на 100 кг сырья,
вычисляют по формуле
А-Б + п
М = -
Г-В
,— •100
где А и Б— содержание В2О3 и MgO в сырье, %;
В и Г — В2О3 и MgO в маточном растворе, %;
п — количество В2О3, возвращаемого в процесс с промыв-
промывными водами, кг на 100 кг сырья (обычно п « 1,4).
Для гидроборацитовых руд, при полном разложении бората,
важна лишь степень превращения MgO в сульфат, отношение же
MgO : B2O3 в сырье не имеет значения. Потери В2О3 с долей ма-
маточного раствора, выводимой из производственного цикла, состав-
составляют 15—30% при переработке ашаритовых руд и 2—8% — гидро-
гидроборацитовых.
Для уменьшения потерь борной кислоты путем возврата в цикл
всего маточного раствора были предложены различные методы
330
Гл. X. Соединения бора
отделения сульфата магния из циркулирующих щелоков. Вследст-
Вследствие понижения растворимости сульфата магния в присутствии сер-
серной кислоты можно вводить серную кислоту, требующуюся для
разложения сырья, не в варочный котел, а в маточный щелок после
выделения из него борной кислоты; при этом из щелока выде-
выделяются кристаллы MgSO4 • 7Н2О.
Как видно из диаграммы растворимости, в системе Н3ВО3—
MgSCU—H2SO4—Н2О при 25° (рис. 99) повышение концентрации
Рис. 99. Изотерма 25° растворимости в системе
H3BO3-MgSO4-H2SO4-H2O.
H2SO4 сильно уменьшает растворимость MgSO4; "растворимость
MgSO4 в 50%-ной серной кислоте по сравнению с растворимостью
в воде понижается почти в 10 раз.
Прн содержании в растворе 50% H2SO4 концентрация эвтони-
ческого раствора, насыщенного и MgSO4j и Н3ВО3 при 35° равна:
3,75% MgSO4 и 2,02% Н3ВО3. При высаливании сульфата магния
серной кислотой, вследствие выделения тепла разбавления серной
кислоты, температура поднимается до 60—65°. Это препятствует
одновременной кристаллизации борной кислоты, так как содержа-
содержание Н3ВО3 в эвтоническом растворе с повышением температуры
увеличивается:
45°
55°
65°
50
3,75
2,38
50
3,75
2,86
50
3,75
3,71
Состав эвтонического раствора, % 35°
H2SO4 50
MgSO4 3,75
Н3ВО3 2,02
Щелок после высаливания сульфата магния может быть раз-
разбавлен возвращаемыми в реактор промывными водами, что снизит
концентрацию в нем серной кислоты до пределов, требуемых для
Борная кислота
331
разложения бората. Одновременно снизится и концентрация ионо!
Mg2+, оставшихся в растворе, — это устрани! опасность выпадения
из раствора сульфата магния при кристаллизации борной кислоты.
Другой метод выделения сульфата магния из циркулирующего
щелока основан на уменьшении его растворимости при повышении
температуры от 70 до 180—200° (рис. 82). Это позволяет выделить
значительную долю сульфата магния из раствора, остающегося по-
после кристаллизации борной кислоты. Для этого маточный раствор
нужно сначала концентрировать выпариванием, причем содержа-
содержание в нем MgSO4 возрастет до 34—36%. Затем при нагревании до
160—165° под давлением из раствора кристаллизуется сульфат маг-
магния в виде MgSO4 • Н2О.
Маточный раствор, выводимый из производства борной кис-
кислоты, может быть переработан на теплоизоляционный материал
обработкой известковым молоком (стр. 287). Наиболее целесооб-
целесообразна переработка его на боромагниевые удобрения (см. ниже).
Использование маточного раствора для получения магнезии
или других термоизоляционных материалов 138 заслуживает вни-
внимания лишь при предварительном выделении борной кислоты. Бор
может быть извлечен из маточного раствора осаждением в виде
боратов магния окисью магния D,5—6 моль MgO на 1 моль В2Оз
в растворе); такой метод (стр. 344) требует значительно меньше
топлива, энергии и пара, чем другие способы переработки маточ-
маточных растворов ш- н0. Маточный раствор D0—50 г/л Н3ВО3) от
производства реактивных сортов борной кислоты используют так-
также для производства стирального порошка — борщелока Ul. Для
этого к раствору добавляют соду в количестве, необходимом для
образования тетрабората
4Н3ВО3 + Na2CO3 = Na2B«O7 + СО2 + 6Н2О
и для получения продукта с отношением Na2CO3: Na2B4O7
не более 10. Масса полностью затвердевает в течение 3—7 дней,
после чего ее высушивают, измельчают и расфасовывают.
А. Н. Костюковский предложил раздельное получение борной
кислоты и сульфата магния без вывода маточных растворов так
называемым методом биборации. Метод основан на обработке ма-
маточного раствора, полученного после кристаллизации борной кис-
кислоты, небольшим количеством окиси магния A4% от В2О3 в рас-
растворе). После отделения осадка, состоящего из гидратов окисей
железа и алюминия, раствор выпаривают до концентрации
10,5—11% MgO, и при охлаждении его до 15—20° кристаллизуется
сульфат магния, а В2О3 остается в растворе, который возвращают
в процесс. Недостатком этого метода является разбавление рас-
раствора, направляемого на кристаллизацию борной кислоты, в
1,3—1,5 раза и увеличение объема кристаллизаторов почти в 2
раза. Впоследствии А. А. Соколовский предложил осуществлять
332
Гл. X. Соединения бора
двухфазное раздельное получение борной кислоты и сульфата маг-*'
ния путем проведения изотермической кристаллизации борной кис-
кислоты вакуум-выпаркой при 65—75° и политермической кристалли-
кристаллизации сульфата магния. Однако и в таком варианте процесс зна-
значительно усложнен (необходимость выпарки и др.), вследствие
чего раздельное получение борной кислоты и сульфата магния не
нашло практического применения.
б точку ЮО% Н3ВО3 E6.4 %ВгО3)
MgO:B2O3*
начолыт растворе
6 6 10 12 14 16 ATid 20 22
MgO.ftr.%
26
Рис. 100. Диаграмма для расчета политермической кри-
кристаллизации борной кислоты. Точки составов кристалло-
кристаллогидратов:
Л-MgSO, . 7Н2О; ?-MgSO4 • 6Н2О; B-MgSO4 • Н2О.
- Существенное значение в производстве борной кислоты имеет
правильный режим кристаллизации Н3ВОз, базирующийся на свой-
свойствах системы НзВОз—MgSO4—Н2О >26- ш- ш. От 0 до 48° сульфат
магния в твердой фазе находится в форме MgSO4 • 7Н2О, от 48 до
68—69° — MgSO4 • 6Н2О; при более высоких температурах ста-
стабильной фазой является кизерит MgSO4 • Н2О, причем с ростом
температуры выше 68е содержание MgSO4 в эвтонических раство-
растворах уменьшается (рис. 100). Однако следует учитывать, что равно-
равновесное состояние достигается медленно (десятки часов), и в усло-
условиях производства борной кислоты, когда процесс ограничен
несколькими часами, система находится в метастабильном со-
состоянии.
Определение выхода В2О3 при кристаллизации борной кислоты
из растворов, содержащих MgSO4, можно производить графиче-
графически144 с помощью диаграммы растворимости (рис. 100), на кото-
кислота
333
0
100°
рой нанесены изотермы в пределах от и до 100" для стабильного
состояния системы, лучи кристаллизации Н3ВО3 и линии составов
начального раствора с постоянным отношением MgO: В2О3. На
этой диаграмме нанесены предельные лучи кристаллизации бор-
борной кислоты и кристаллогидратов сульфата магния, заканчиваю-
заканчивающиеся в эвтонических точках раз-
различных изотерм. Количество G (в
кг на 100 кг начального раствора)
твердой борной кислоты, кристал-
кристаллизующейся при охлаждении рас-
раствора, определяется по формуле
G= — • 100, где — — отношение
С С
'длины отрезка луча кристаллиза-
кристаллизации Н3ВО3 от точки начального
раствора до соответствующей изо-
изотермы к длине луча кристаллиза-
кристаллизации борной кислоты от точки, соот-
соответствующей 100%-ному содержа-
содержанию Н3ВО3 (или 56,4% В2О3), до
пересечения с изотермой. Выход
В2О3 (в %) (степень перехода В2Оз
из жидкой фазы в твердую) вычис-
вычисляется по формуле
56,4
а
„ = _.
[В2О3
• 100
UJ 0,5 W 09 1.1 U 0 1,Г
MgO В2о, 0 начальном раапборе
Рис. 101. Содержание В2О3 в ма-
маточных растворах (в % от содер-
содержания в начальных растворах)
и максимальные концентрации
В2О3 в начальных растворах (в %)
в зависимости от содержания MgO
при температурах кристаллизации
борной кислоты 10 и 20° (для
прямых линий — левая шкала, для
кривых —правая).
где [В2Оз]ь — начальная концентра-
концентрация В2О3 в растворе.
Например, если раствор, содер-
содержащий 5,38% В2О3 и 7,0% MgO
(MgO : B2O3 =1,3), охладить до 10°,
то 5 = 7,65 кг на 100 кг раствора и
т] = 80,1%- Как видно из рис. 100,
с увеличением концентрации В2О3 и
уменьшением отношения MgO : В2Оз количество выкристаллизовы-
выкристаллизовывающейся борной кислоты и выход В2Оз возрастают. Практически
расчет ведут не по «предельному» лучу кристаллизации, а по лучу,
пересекающему изотерму в точке, находящейся иа некотором рас-
расстоянии от эвтонической, соответствующей реальным условиям. На
рис. 101 показана зависимость содержания В2О3 в маточном рас-
растворе, выводимом из кристаллизатора, от отношения MgO: В2О3 в
начальном растворе при расчете кристаллизации по предельному
лучу при 10° A,15% В2О3 в маточном растворе) и 20° A,4% В2О3),
а также по лучу, пересекающему изотерму 20° в точке, соответст-
соответствующей содержанию 1,6% В2О3. Приведены также значения
334
Гл. X. Соединения бора
Борная кислота
335
максимальной концентрации В2О3 в начальном растворе для раз-
различных отношений MgO : В2О3.
Количество G выкристаллизовавшейся борной кислоты из 100 яг.
раствора можно также вычислить из уравнения материального
баланса кристаллизации
° 56,4-[В2Оз]м -100
где [В2О3]Я и [В2Оз]м — концентрации В2О3 в начальном и маточном
растворах.
При содержании в боратах соединений калия возможно загряз-
загрязнение борной кислоты сульфатом калия. С повышением темпера-
температуры до 90° в эвтонических растворах системы Н3ВО3—K2SO4—Н2О
E г
О 5 10 15
КНгРО4, бес.%
Рис. 102. Политерма растворимости в системе
КН2РО4-Н3ВО3-Н2О.
содержание борной кислоты увеличивается значительнее, чем суль-
сульфата калия 145-146, поэтому сернокислотное разложение боратов, со-
содержащих калий, целесообразно производить при высокой темпера-
температуре (80—90°).
При фосфорнокислотной переработке калиборатов получаемый
раствор содержит борную кислоту и фосфаты калия. В системе
Н3ВО3—КН2РО4—Н2О тройная эвтектика с составом 11,7%
КН2РО4+2,6% НзВОз+85,7% Н2О образуется при—3,3° (рис. 102).
От температуры тройной эвтектики до 40° на диаграмме состоя-
состояния имеются поля кристаллизации только исходных компонентов.
При постоянной температуре растворимость Н3ВО3 и КНгРО4 в
воде при взаимном присутствии компонентов увеличивается. С по-
повышением температуры в раствор переходит относительно больше
НзВОз, чем КН2РО4147.
О кинетике солянокислотного разложения боратов см,150,
Получение борной кислоты из датолита120> ш>ш-
Датолит 2СаО-B2O3'2SiO2-H2O и сопутствующие ему мине-
минералы (гранат ЗСаО • Fe2O3 • 3SiO2, геденбергит CaO-FeO-2SiO2,
кальцит СаСО3) не содержат магния; но в датолитовом сырье много
CaO, SiO2 и окислов железа. Датолит, плохо разлагаемый щело-
щелочами, легко разлагается кислотами; наиболее целесообразно при-
применение серной кислоты, так как кальций отделяется при этом
в виде осадка сульфата кальция CaSC>4 • 2Н2О. При обработке да-
толитового сырья серной кислотой датолит разлагается сравни-
сравнительно быстро и полно (96—100% за 60 мин при 95° и расходе на
1 т сырья 0,55 т моногидрата серной кислоты), а другие минера-
минералы— медленно и лишь частично (гранат на 12—20%, геденбер-
геденбергит на 2—4%, кальцит на ~50%). Вследствие этого в раствор
переходят значительные количества Н3ВО3 и SiO2 • д:Н2О и немного
сульфатов железа и кальция. Из-за перехода в раствор кремневой
кислоты невозможно перерабатывать датолиты по схеме, приме-
применяемой для ашаритов и гидроборацитов, так как при высокой кон-
концентрации SiO2 раствор постепенно превращается в студнеобраз-
студнеобразную массу, трудно отделимую от нерастворившегося остатка; из
этой массы трудно также извлечь и борную кислоту. Фильтрация
несколько облегчается, если добавлять в реакционную смесь ще-
щелока MgSO4 или Na2SOi, или маточный раствор после выделения
Н3ВО3153, или получать разбавленные растворы, но тогда после
отделения шлама борную кислоту необходимо перед кристаллиза-
кристаллизацией выпаривать, причем SiO2 будет выделяться в осадок или
в процессе выпарки или в процессе кристаллизации, в первом слу-
случае вызывая значительные потери Н3ВО3, а во втором — загрязняя
продукт.
Поэтому для переработки датолитов разработана другая схема
(рис. 103). Сырье разлагается серной кислотой, разбавленной на-
настолько, чтобы получилась густая пульпа. Последнюю направляют
в сушильный барабан после смешения в горизонтальных смеси-
смесителях с ретуром. При подсушке до влажности 20—26% происходит
коагуляция кремневой кислоты, перешедшей при разложении в
жидкую фазу. Подсушенный материал подвергается противоточ-
ному выщелачиванию, причем избыточную кислотность нейтрали-
нейтрализуют добавкой карбоната кальция, что приводит к удалению в оса-
Док полуторных окислов и остатка кремневой кислоты. Шлам
легко отделяется и подвергается противоточной промывке на ва-
вакуум-фильтрах (затем он может быть использован для изготов-
изготовления строительных материалов), а полученный раствор содержит
лишь борную кислоту без примесей SiO2 и окислов железа, что
позволяет получать более чистые сорта борной кислоты. После под-
кисления и контрольной фильтрации раствор направляют в ва-
вакуум-кристаллизаторы для получения борной кислоты, кристаллы
Бура
337
которой отделяют на центрифуге и там же промывают холодной
водой. Промывные воды и часть маточного раствора возвращают
для разбавления поступающей в реактор серной кислоты, другая
часть маточного раствора поступает на выщелачивание подсушен-
подсушенной пульпы, — отходы маточных растворов в этой схеме отсут-
отсутствуют. Поэтому степень использования бора превышает 90%. На
1 т борной кислоты (98,5% Н3ВО3) расходуют: 4,35 г датолитового
концентрата A6% В2О3), 2,4 т серной кислоты A00%), 0,6 т из-
известняка A00% СаСО3) и приблизительно 130 м3 воды, 8 т пара
и 800 кет ¦ ч электроэнергии; количество отбросного шлама с 50%
влаги— 12,2 т.
Извлечение бора из датолитовой руды можно осуществить, об-
обрабатывая водную суспензию прокаленного сырья двуокисью угле-
углерода 125 или двуокисью серы 155, а также гидротермическим спосо-
способом. При 1350—1400° реакция
2СаО • В2О3 • 2SiO2 • Н2О + 2Н2О = 2CaSiO3 + 2Н3ВО3
идет почти количественно — выход Н3ВО3 из зерен руды 0,1—0,5 мм
за 8—9 ч достигает 90%, но из-за медленности процесса расход
пара очень велик154.
БОРАТ КАЛЬЦИЯ
Борат кальция получают нейтрализацией борной кислоты из-
известью. В промышленных условиях для его получения используют
датолит.
Шихта из молотой датолитовой руды или концентрата и моло-
молотого известняка спекается в барабанной печи при 950—1050°; про-
продолжительность нахождения шихты в реакционной зоне 15 мин.
Датолитовый спек измельчается в валковой дробилке, гасится обо-
оборотными растворами и подвергается мокрому размолу. Получен-
Полученная пульпа (Ж : Т = 8—12 : 1) подогревается острым паром до 80°
и карбонизуется. Затем она сгущается и фильтруется. Шлам ре-
пульпируется и после вторичной карбонизации сгущается, филь-
фильтруется, промывается и сбрасывается. Вторичный фильтрат и про-
промывные воды возвращаются на мокрый помол датолитового спека
и на разбавление пульпы, а из основного фильтрата —¦ раствора
борной кислоты — известковым молоком A00 г/л СаО) осаждается
борат кальция. Суспензия бората кальция сгущается и поступает
на центрифугу. Затем борат кальция высушивается при 100—150°
в сушилке кипящего слоя.
БУРА120- 134' 157~161
Из отечественных видов сырья лишь гидроборацит легко раз-
разлагается содовым раствором и из гидроборацитового сырья мож-
можно получать щелочным методом непосредственно буру (стр. 342).
338
Гл. X. Соединения бора
Однако основным сырьем до сих пор являются ашаритовые и сме-1
шанные руды, трудно разлагаемые щелочами. Поэтому в CCCpI
буру вырабатывают из сырой технической борной кислоты варкой!
ее с содой (использовать для этой цели растворы борной кислоты]
до ее кристаллизации невозможно, так как они содержат MgSO4,|
который реагирует с содой, образуя осадок магнезии; это увели-
увеличивает расход соды и вызывает потери буры в осадке).
Возможно и непосредственное получение буры, например, из
датолитов. В автоклаве при 300° и 140—150 ат датолит в значи-
значительной мере разлагается даже водой (выход В2О3 в раствор за
1 ч составляет 19%)- В присутствии соды @,5 М раствор) разло-
разложение значительно ускоряется и выход буры по реакции
2BСаО • В2О3 • 2SiO2 • Н2О) + Na2CO3 = Na2B4O7 + 4CaSiO3 + 2Н2О + СО2
из руды, содержащей 7,5% В2О3 и измельченной до размера зе-
зерен 0,152 мм, достигает за 1 ч 92% 156.
Получение буры из борной кислоты
Процесс основан на реакции
Na2CO3 + 4Н3ВОз = Na2B4O7 + СО2 + 6Н2О
В реакторе, снабженном мешалкой, приготовляют раствор
соды, нагревают его до 95—100° и загружают борную кислоту. Во
избежание выброса раствора из-за сильного вспенивания, обуслов-
обусловленного выделением углекислого газа, борную кислоту добавляют
небольшими порциями. Соотношение между загружаемыми компо-
компонентами должно быть таким, чтобы полученный раствор содержал
14—20% Na2B4O7 и 0,5—1,0% Na2CO3. Избыток соды затрудняет в
дальнейшем отмывку буры и увеличивает количество промывных
вод; недостаток соды уменьшает выход В2О3 в буру (часть В2О3
переходит в более растворимые соли) и затрудняет ее кристалли-
аацию. На 1 т буры расходуют 0,65 т технической борной кислоты
и 0,307 т кальцинированной соды.
После 30-минутного нагревания при 100° раствор отделяют от
шлама — нерастворимых загрязнений, попавших из сырья, и на-
направляют для охлаждения от 90° до 15—20° в кристаллизаторы,
где из него кристаллизуется бура Na2B4O7 • ЮН2О. Кристаллы
буры отделяют от маточного раствора на центрифуге. Здесь же их
промывают водой. Маточный раствор и промывные воды возвра-
возвращают в реактор на растворение соды. Вследствие этого в растворе
накапливается сульфат натрия, образующийся из содержащегося
в сырой борной кислоте сульфата магния:
MgSO4 + Na2CO3 = Na2SO4 + MgCO3
На рис. 104 приведены некоторые изотермы системы Na2B4O7—
Na2SO4—Н2О (сплошные линии) 157 и изотермы 15 и 35° той же
Бура
339
системы в присутствии 1,5% Na2CO3 (пунктирные линии). Влияние
такого количества соды не существенно. Накопление же сульфата
натрия может привести к загрязнению буры мирабилитом. При 25°
в эвтоническом растворе содержится 21,9% Na2SO4. В присутствии
примеси Na2CO3 и NaCl эта концентрация несколько понижается.
Если раствор буры при кристаллизации охлаждается до 25е, то
практически недопустимо содержание в исходном растворе больше
12% Na2SO4; при этом в маточном растворе не должно быть боль-
больше 20% Na2SO4. Чем больше в растворе Na2SO4, тем выше должна
24
го
а
5
-65'
и
V
35^30°
•——
<
1
—¦' —
"- ¦—
\
\
\
— —
ft
н
^А J\
s<
Т
\
о
——.
о
\
\
\
\
\
\
^ 1 г
1
7
1 1 1
16
\
V
\ ]
\
\
\
\
-21-\
1 1
го
%
2
\
SO
«
ra2SO+
s\
3i
00
?
Рис 104. Система Na2B4O7—Na2SO«— H2O.
быть конечная температура кристаллизации буры. Следовательно,
часть маточного раствора должна выводиться из цикла. Кроме
того, в присутствии в растворе более 2,5% Na2SO4 образующиеся
кристаллы буры уменьшаются в размере157-159. Удовлетворитель-
Удовлетворительные по размерам кристаллы буры получаются, если в исходном
растворе, содержащем 1,5% Na2CO3 и 0,5% NaCl, не больше 5%
Na2SO4; маточный раствор при этом не насыщен сульфатом нат-
натрия—он содержит ~8% Na2SO4. В выводимом из цикла маточ-
маточном растворе содержится 1,3—1,7% Na2B4O7. А. А. Соколовским
разработан способ, позволяющий возвратить этот раствор в цикл
после извлечения большей части Na2SO4. Этот способ заключается
в переводе содержащегося в растворе тетрабората в более раство-
растворимый пентаборат натрия, в выпарке раствора и кристаллизации
из него сульфата натрия; обедненный сульфатом раствор, содер-
содержащий В2О3 в форме пентабората, может быть возвращен в цикл.
340
Гл. X. Соединения бора
Бура
341
На рис. 105 показана изотерма 30° системы Na2O—В2О3—Н2О.
Линия АБ — растворимость пентабората натрия Na2B10Oi6- 10H2O
или Na2O •БВгОз'10Н2О; растворимость тетрабората имеет мини-
минимум в точке В. С более щелочными растворами в равновесии нахо-
находятся метабораты натрия Na2O • В2Оз • 8Н2О и Na2O • В2О3 • 4Н2О.
Перевод тетрабората в пентаборат добавкой к раствору борной кис-
кислоты неприемлем, так как состав полученного раствора неблаго-
неблагоприятен для кристаллизации из него сульфата натрия. Этот перевод
можно осуществить добавкой к раствору такого количества серной
2
Мирабилит Щ
ПентаБсрагп\
натрия
Na,O-ZBiO3-10H,tO
Na2OB2Q,-4H2O
20 30
Na2O.%
Рис 105. Изотерма 30° системы
Na2O—В2О3—Н2О.
Борная
кислота
Рнс. 106. Изотермическая проек-
проекция B0,5°) пространственной диа-
диаграммы для системы
Na2SO4— H3BO3—Na2B4O7—H2O.
кислоты, которое требуется для нейтрализации Na2CO3 и перевода
буры в пентаборат натрия; при этом содержание Na2SO4 в растворе
увеличивается. После выпарки раствора до достижения концентра-
концентрации Na2SO4 23—24%, при охлаждении до 20° выкристаллизовы-
выкристаллизовывается мирабилит Na2SO4 • ЮН2О (рис. 106) 160. Выход сульфата
натрия E0—60%) зависит от начального состава маточного рас-
раствора и может быть увеличен до 80% и более путем повторения
операций с добавкой к вторичному маточному раствору после выде-
выделения мирабилита новых порций исходного маточного раствора.
Недостатком описанного выше процесса получения буры из бор-
борной кислоты является его периодичность. Наибольшие трудности
при переходе на непрерывный процесс вызывает кристаллизация
буры. Применяемые на заводах периодически действующие кри-
кристаллизаторы представляют собой резервуары с мешалками, охла-
охлаждаемые водой через металлические стенки или с помощью змее-
виков. Они требуют частой чистки от налипающих кристаллов -и
малопроизводительны. Попытка применения вакуум-кристаллиза-
вакуум-кристаллизаторов оказалась неудачной вследствие обильного вспенивания в-
них растворов буры и переброса пены в коммуникации. Образова-
Образование пены объясняется выделением СО2 при взаимодействии буры.
с остатками соды:
Na2B4O7 + Na2CO3 = 4NaBO2 + СО2
Метаборат натрия образуется медленно, и для полного завер-
завершения этого процесса с образованием непенящегося раствора
буры требуется длительное время. Кроме того, работа с раство-
раствором, содержащим вместо небольшого избытка соды эквивалентное-
ему количество метабората, нецелесообразна из-за ухудшения:
условий кристаллизации буры и увеличения содержания В2О3 в ма-
маточном растворе. Поэтому важной задачей является разработка
конструкции непрерывно действующего кристаллизатора с водя-
водяным охлаждением или вакуумного типа, пригодного для пеняще-
пенящегося раствора. ¦
При производстве буры из датолитового сырья для реакции
с содовым раствором можно использовать не кристаллическую-
борную кислоту, а получаемый при кислотном разложении датоли-
тов раствор борной кислоты, так как он не содержит сульфата
магния. (Таким образом исключаются стадии кристаллизации
Н3ВО3 и ее медленного растворения в содовом растворе). Оба
раствора можно одновременно подавать в реактор непрерывного
действия; после 30—60-минутного перемешивания в реакторе при
95—100° и контрольной фильтрации раствор буры должен в этом
случае подвергнуться выпарке для удаления избытка воды, после
чего может быть направлен на политермическую кристаллизацию.
Получение буры из боратов содовым методом
Получение буры из боронатрокальцита
Этот вид сырья применяется за рубежом и применялся раньше
в СССР. Извлечение буры из породы, содержащей 40—42% В2Оз,
производится путем варки ее с содой и бикарбонатом. Содержа-
Содержащаяся в боронатрокальците окись кальция переходит в нераство-
нерастворимый осадок СаСОз, а бура — в раствор по реакции:
2(Na2O • 2СаО • 5В2О3 • 16Н2О) + Na2CO3 + 4NaHCO3 =
= 5Na2B4O7 + 4СаСО3 + СО2 + 34Н2О
Варку производят в два приема. Вначале ее проводят прк
90—95° с использованием оборотных щелоков, содержащих в 1 л
130—185 г Na2B4O7 и 20—35 г соды.
342
Гл. X. Соединения бора
Бура
34Я
Полученный раствор отстаивается и отфильтровывается от не-1
растворившейся части породы и образовавшихся CaCO3HFe(OHK.
¦Отфильтрованный раствор содержит 290—325 г/л Na2B4O7 и
4—10 г/л Na2CO3. Твердый остаток в реакторе размешивают с во-
водой и переводят в автоклав для второй варки, куда добавляют
«оду и бикарбонат.
Вторую варку проводят при 120—130°, а затем раствор отделяют
ют твердого остатка, идущего после промывки в отвал; он содер-
содержит 1—3% В2О3. Раствор идет на первую варку, а промывную
воду возвращают в автоклав на вторую варку.
Из горячего концентрированного раствора буры, полученного
атосле первой варки, при охлаждении до 45—48° кристаллизуется I
<бура. Раствор не должен быть слишком концентрированным, его
плотность не должна превышать 1,15 г/см3, так как при большей
концентрации из него при температуре выше 56° будет кристалли- 1
зоваться не десятиводная, а пятиводная соль. Щелок после кри-
кристаллизации возвращают в производственный цикл, поэтому по-
постепенно в нем накапливаются примеси — до 10—15% NaCl, 2—3%
Na2SO4 и др. Время от времени щелок должен обновляться.
Получение буры из кернита (разорита)
Кернит отличается от буры только меньшим содержанием кри-
кристаллизационной воды.
В США (Калифорния) кернитовую руду (~25% В2О3) выще-
выщелачивают при 90—95° слабым раствором буры или маточным рас-
раствором после кристаллизации. Отделенный от примесей раствор
.направляют в вакуум-кристаллизатор. Отфугованный продукт вы-
высушивают в турбосушилках, состоящих из расположенных одна
над другой вращающихся тарелок. Высушиваемый материал пе- |
ресыпается с тарелки на тарелку.
Если в кернитовой породе содержатся бораты кальция, то варку
яедут в автоклавах в присутствии соды, переводящей борат каль-
кальция в борат натрия.
Получение буры из гидроборацита
Схема переработки гидроборацитового концентрата в буру за-
заключается в следующем. Концентрат смешивают в реакторе с ма-
маточным раствором и с промывными водами от промывки шлама
* буры; затем вводят смесь соды и бикарбоната натрия и осуще-
осуществляют варку 2—3 ч при 95—100°. После этого шлам отделяют
ма ленточном фильтре или фильтрпрессе, промывают горячей во-
водой и отправляют в отброс. Фильтрат (раствор буры) после кон-
контрольного фильтрпресса направляют на политермическую кристал-
кристаллизацию. Часть маточных растворов, во избежание накапливания
s цикле переходящего из сырья SOf", выводят из процесса (концен-
(концентрация SO3 в растворе не должна превышать 10%),
Получение буры из гидроборацитового концентрата можно осу-
осуществлять и по кислотно-щелочному методу, т. е. с предваритель-
предварительным получением из него борной кислоты и последующей ее обра-
обработкой содой. Потери В2О3 с выводимым из процесса маточным
раствором в обоих методах (щелочном и кислотно-щелочном) при-
приблизительно одинаковы. При учете потерь В2О3 только с маточ-
маточными растворами и при содержании в сырье 34,3% В2О3 и 7,78%
SO3 расход реагентов на 1 т В2О3 в буре составляет: при щелочном
методе (с выходом В2О3 в продукт 97,5%) соды A00%) —0,83 г
и бикарбоната натрия A00%)—0,41 т; при кислотно-щелочном
методе серной кислоты A00%) —0,76 т. Стоимость этих реагентов
в обоих методах приблизительно одинакова. Производство буры
из бората щелочным методом проще, чем кислотно-щелочным, но-
по последнему методу получается бура лучшего качества, так как
примеси отделяются при изготовлении полупродукта — 6opHofr
кислоты.
Переработка боратов на буру другими щелочными методами 11Г
Разложение боратов едким натром происходит легче, чем содой,
но он дороже, и поэтому его не используют. Для переработки аша-
ритовых боратов, плохо разлагающихся содовым раствором, пред-
предложен известково-содовый способ, при котором в растворе обра-
образуется NaOH. Боратовую муку, проходящую через сито 0,15 мм,
прокаливают в течение 2 ч при 600—650°, затем обрабатывают
3,5 ч в реакторе смесью извести и содового раствора при темпера-
температуре кипения. Пульпу фильтруют и шлам подвергают вторичному
разложению во втором реакторе, а затем в третьем, после чего
шлам, отфильтрованный и промытый горячей водой F л на 1 кг
бората), идет в отвал. В третьем реакторе шлам разлагают про^
мывными водами, во втором — фильтратом от пульпы из третьего-,
в первом — свежим раствором соды в фильтрате от пульпы из вто-
второго реактора. Фильтрат от пульпы из первого реактора перера-
перерабатывают в буру, для чего содержащийся в нем метаборат нат-
натрия переводят в тетраборат, например, путем обработки раствора-
двуокисью углерода и кипячения, и направляют на кристалли-
кристаллизацию.
Другой вариант этого способа заключается в спекании размо-
размолотой ашаритовой руды с известью при 600—700° в течение 1 ч.
Охлажденный спек разлагают в реакторах содовым раствором-
с добавкой промывных вод и маточного раствора. После фильтра-
фильтраций и промывки шлам может быть подсушен и использован в ка-
качестве борного удобрения или добавки к суперфосфату. Фильтрат
обрабатывают углекислым газом и кристаллизуют буру. По этой1
схеме выход бора в буру больше, чем при получении буры череа
борную кислоту, причем не расходуется серная кислота.
344
Гл. X. Соединения бора
Предложено несколько способов переработки боратов щелоч-
щелочным спеканием. Так, при спекании (сплавлении) бората с содой
в течение 1—3 ч при 600—1200° получается масса, из которой вод-
водным выщелачиванием извлекают в раствор метаборат; для пере-
перевода его в буру раствор обрабатывают борной кислотой или дву-
двуокисью углерода.
Бораты, содержащие кальций, могут перерабатываться на буру
путем спекания с сульфатом натрия. Например, при спекании ко-
леманита происходит следующая реакция:
2BСаО • ЗВ2О3 • 5НгО) + 3Na2SO4 = 3Na2B4O7 + 3CaSO4 + CaO + I0H2O
Бура выщелачивается из полученного спека водой и кристалли-
кристаллизуется из раствора.
Обезвоживание буры
Для получения обезвоженного продукта кристаллогидрат пред-
предварительно обезвоживают во вращающихся барабанных сушилках
и окончательно в плавильных печах. Расплавленную массу кри-
кристаллизуют на охлаждающих вальцах161.
Другой метод обезвоживания162—распыление раствора тетра-
бората натрия во вращающейся печи над непрерывно движущимся
перемешивающимся слоем безводного Na4B2O7. нагретым до
450—650°. Происходит испарение воды из капелек раствора и осе-
оседание частиц на движущийся слой; при этом продукт не агломе-
агломерируется.
Запатентован способ обезвоживания буры в две стадии163 —
•сначала жидким аммиаком в автоклаве при 20° и 8 от для получе-
получения промежуточного продукта с двумя-пятью молекулами воды на
1 молекулу соли, который затем отфильтровывают под давлением,
отдувают из него остатки аммиака воздухом и прокаливают
яри 450°.
Обезвоживание буры можно осуществить путем постепенного
¦многостадийного ее нагревания в аппарате с кипящим слоем в по-
потоке горячего воздуха при температурах ниже точки плавления.
W—90% воды удаляется в пределах 100—200°, а остальная — при
220—700°. При этом отпадает надобность в размоле продукта, ко-
который получается порошкообразным; он содержит 98—99,8%
Na2B4O7164.
БОРНЫЕ УДОБРЕНИЯ
Получение борных удобрений из отходов производства
борной кислоты132
Маточные растворы, содержащие (в %): В2О3 1,3—1,6, MgSO4
¦21—23, H2SO4 0,2—0,6, R2O3 0,2—1,2, нейтрализуют до слабощелоч-
слабощелочной реакции мелом или известняком в вертикальных резервуарах
с мешалками периодического действия. Полученную суспензию вы-
выудобрения
345
сушивают в распылительной сушилке (диаметр 5,5 м) дымовыми
газами C50—450°). Распыливание суспензии производят форсун-
форсунками, работающими на остром паре, что исключает их закупори-
закупоривание и по расходу энергии экономичнее, чем распыление сжатым
воздухом. Дымовые газы очищаются от уносимой из сушилки пыли,,
проходя через циклон и скруббер, орошаемый суспензией (маточ-
(маточным раствором). Такой способ получения бормагниевого удобре-
удобрения позволяет полностью использовать содержащиеся в маточном
растворе Н3ВО3 и MgSO4. Продукт содержит 6—8% Н3ВО3).
65—75% MgSO4, 29—17% кристаллизационной воды, CaSO4 и дру-
другие примеси и имеет небольшой объемный вес 0,2—0,3 т/л*3, зави-
зависящий от тонкости распыления высушиваемого раствора. При гру-
грубом распылении продукт получается влажным. Преимуществом
распылительной сушки маточного раствора перед выпариванием
его через теплопередающую поверхность в аппаратах с огневым
или паровым нагревом является отсутствие затрат труда на дроб-
дробление застывающей после выпарки монолитной массы, которая
к тому же содержит больше влаги, чем продукт, получаемый В;
распылительной сушилке. Однако для получения 1 т продукта тре-
требуется удалить из маточного раствора ~3 т воды. Поэтому более-
рациональна переработка маточного раствора в борное удобрение-
осаждением бора окисью магния (каустическим магнезитом) в
форме боратов магния хВ2О3 • r/MgO • 2Н2О, малорастворимых в
воде, но хорошо растворимых в 2%-ной лимонной кислоте. При
95°, норме MgO 4,5 моль на 1 моль В2О3 и 2-часовом перемешива-
перемешивании осаждается более 90% бора и в фильтрате после отделения-
осадка содержится 0,26% В2О3. Осадок промывают, высушивают
и измельчают до —2 мм. Промывные воды идут в отброс, а филь-
фильтрат может быть переработан на сульфат магния или магнезии.
Получаемый продукт содержит больше 5% лимоннорастворимого-
В2О3, 20,5% MgO (общ.), 12,5% SO3 (в виде MgSO4). Его объем-
объемный вес 0,8 т/м3 (ГОСТ 9595—61 нормирует содержание в про-
продукте только лимоннорастворимых соединений бора в пересчете
на Н3ВО3— не менее 9% и степень его измельчения — продукт
должен полностью проходить через сито с ячейками 4,5 мм).
Себестоимость 1 т В2О3 в таком удобрении значительно ниже,
чем в удобрении, получаемом путем высушивания всего маточного,
раствора.
Сырой осадок бората магния может быть использован не только
как удобрение, но возвращен в производство борной кислоты в.
виде добавки к боратовому сырью. Но в этом случае необходимо
до осаждения бората очищать маточный раствор от полуторных
окислов углекислым кальцием, а осадок бората хорошо отмывать
от MgSO4.
При обработке отбросного маточногй раствора аммиаком и
двуокисью углерода или углекислым аммонием можно получить-
346
Гл. X. Соединения бора
боросульфатаммоний и магнезию:
4Н3ВО3 + (NH4JCO3 = (NH4JB4O7 + СО2 + 6Н2О
4MgSO4 + 4(NH4JCO3 + IOHjO = 3MgCO3 • Mg(OHJ • 9H2O + 4(NH4JSO4 + CO2
Осадок основной углемагниевой соли отделяют от раствора,
промывают и высушивают, причем получается магнезия альба
3MgCO3 • Mg(OHJ • ЗН2О (стр. 297). Раствор нейтрализуют сер-
серной кислотой и высушивают — получается удобрение (боросуль-
¦фатаммоний), содержащее около 6% бора (в пересчете на В2О3),'
около 70% (NH4JSO4 и около 20% MgSO4.
Предложено также растворять в нагретом до 85° маточном рас-
растворе после кристаллизации Н3ВО3 сульфат аммония @,8 моль
¦на 1 моль MgSO4). При охлаждении раствора до 20° кристалли-
кристаллизуется двойная соль сульфатов магния и аммония (см. стр. 270,
305), содержащая после высушивания при 100—105° 36%
(NH4JSO4) 33% MgSO4, —2% Н3ВО3, 29% Н2О. Она является
удобрением с хорошими физическими свойствами. Конечный ма-
маточный раствор возвращают в цикл 165-166. После накопления в нем
Горной кислоты она может быть извлечена.
Получение борных удобрений из боратового сырья
Борные удобрения
Э4Г
Получение борных удобрений из датолитов
134
Маточные растворы из производства борной кислоты являются
весьма ограниченным источником сырья для борных удобрений;
при росте их потребления сырьем будут служить природные бора-
бораты, главным образом датолитовые руды, нуждающиеся в перера-
переработке, так как датолит не растворяется ни в воде, ни в лимонной
кислоте. Возможно получение борного удобрения из датолито-
датолитового сырья с применением аппаратуры, используемой в суперфос-
суперфосфатном производстве. С помощью серной кислоты содержащийся
в датолите ВгОз переводится в водорастворимую или усвояе-
усвояемую форму. Измельченную датолитовую руду (размер зерен
<0,25 мм) перемешивают в течение нескольких минут с 50%-ной
серной кислотой (норма кислоты 90% от стехиометрического коли-
количества в расчете на СаО); начальное отношение Т : Ж = 1. Пульпа
поступает в камеру, где происходит вызревание массы в течение
1 ч, причем вследствие экзотермичности реакций температура по-
повышается до 105—115°. После выгрузки затвердевшей массы она
представляет собой готовое удобрение с хорошими физическими
свойствами (сухость, рассыпчатость и др.) и не требует вылежи-
вылеживания на складе, так как степень разложения датолита в камере
достигает 97—99%. Датолитовое удобрение содержит 8—10%
Н3ВО3, 0,5—0,6% H2SO4, сульфат кальция, кремневую кислоту.
:На 1 г удобрения расходуется ~0,26 т H2SOi A00%),
При переработке флотационного датолитового концентрата-
A5—16% В2О3) в получаемом удобрении по нормам ГОСТ
10347—63 должно содержаться не менее 13% водорастворимой
борной кислоты; свободная кислотность в пересчете на H2SO4 не
должна превышать 1,5%.
Разработан способ получения борной кислоты из этого датоли-
датолитового удобрения. Для этого его репульпируют 15 мин при 20—30""
в оборотном щелоке, насыщенном борной кислотой. Затем пульпу
подвергают флотации. При этом борная кислота поступает в пен-
пенный продукт, а гипс и другие примеси остаются в хвостах. После
фугования и сушки пенного продукта получается 90—92%-наябор-
90—92%-наяборная кислота, а перекристаллизацией неотфугованного пенного про-
продукта можно получить стандартную техническую борную кислоту.
После отделения хвостов раствор возвращается в цикл. Хвосты
могут быть перефлотированы для получения гипсового концен-
концентрата, пригодного для переработки в строительный гипс 167.
Получение боросуперфосфата 134>168
Введение соединений бора в наиболее распространенное удоб-
удобрение — суперфосфат дает ценное удобрение — боросуперфосфат„
Его можно получать или совместным сернокислотным разложе-
разложением смеси фосфатного сырья и датолитовой муки, или добавкой"
датолитовой муки к готовому суперфосфату, или смешением супер-
суперфосфата с датолитовым борным удобрением.
Совместное разложение апатита или каратауского фосфорита
и датолитового сырья серной кислотой связано с технологическими*
трудностями. Выделяющаяся при разложении датолита кремневая
кислота обволакивает частицы апатита и препятствует их взаимо-
взаимодействию с серной и фосфорной кислотами, так что даже при дли-
длительном хранении на складе степень разложения сырья остается
недостаточно высокой. При разложении же смеси фосфорита Кара-
тау с датолитом последний разлагается недостаточно потому, что
температура в процессе хранения ниже оптимальной температуры
разложения датолита (95°). Такие же неблагоприятные условия
для разложения датолита создаются и при добавке его к вызрев-
вызревшему суперфосфату, температура которого обычно ниже 55° —
степень разложения датолита свободной фосфорной кислотой су-
суперфосфата не превышает 85%. Кроме того, для разложения
датолита требуется дополнительное хранение боросуперфосфатана
складе в течение 5 суток, т. е. необходимо увеличение размеров
суперфосфатного склада. Добавка же датолита к свежему (камер-
(камерному) суперфосфату нецелесообразна, так как ухудшает условия
доразложения апатита. Наиболее полное использование фосфат-
фосфатного и датолитового сырья достигается при смешении готового бор-
борного датолитового удобоения с суперфосфатом как. вызревшим,.
348
Гл. X. Соединения бора
-так и свежим, так как готовое датолитовое удобрение не влияет
«а доразложение фосфата. В связи с различной потребностью рас-
растений в фосфоре и боре отношения Н3ВО3: Р2О5 в боросупер-
фосфате должны составлять от 1 : 6 до 1 : 20.
Содержание Р2О5 и Н3ВО3 могут колебаться для апатитового
^оросуперфосфата в пределах от 18 до 14,6% Р2О5 и от 0,9 до 2,5%
Н3ВО3; для фосфоритового — от 13 до 11,1% Р2О5 и от 0,7 до 1,9%
"НзВО3. Боросуперфосфат обладает лучшими физическими свой-
свойствами, чем суперфосфат, а себестоимость бора в нем меньше, чем
? бормагниевом удобрении.
Получение концентрированного
боросуперфосфата
(бородвойного суперфосфата)ш-169
Датолитовая руда почти полностью разлагается слабой эк-
экстракционной фосфорной кислотой, содержащей 20—24% Р2О5
(или 27,6—33,1% Н3РС>4). При этом протекают следующие реак-
реакции:
«СаО • В2О3 ¦ 2SiO2 • Н2О + 4Н3РО4 +2Н2О = 2Ca(H2PO4J+2H3BO3+2[SiO2. H2OJ
3[СаО • FeO • 2SiO2] + 8Н3РО4 «= ЗСа(Н2РО4J + Fe3(PO4J + 6[SiO2 • Н2О]
ЗСаО • Fe2O3 • 3SiO2 + 8Н3РО4 = ЗСа(Н2РО4J + 2FePO4 + 3[SiO2 • H2OJ + ЗН2О
Оптимальные условия разложения: норма кислоты ПО—115%
от стехиометрической и 3-часовое перемешивание при 95°. Разло-
.жение датолитовой руды производят непрерывно в серии верти-
вертикальных реакторов с мешалками, соединенных перетоками
^рис. 107). Избыток фосфорной кислоты нейтрализуют добавкой
* горячей пульпе углекислого кальция. Нейтрализованная пульпа
поступает в барабанный гранулятор, где смешивается с ретуром —
-измельченным сухим материалом — в отношении ретур : пульпа,
равном 1,5. Гранулы высушивают в сушильном барабане до влаж-
влажности 2—3%. Затем материал измельчают и рассеивают на фрак- :
ции —3 +0,5 мм и —0,5 мм. Фракция —3 +0,5 мм является про-
продуктом, который в случае необходимости до упаковки нейтрали-
нейтрализуют известняком. Фракцию —0,5 мм возвращают на смешение
с пульпой. Продукт содержит 35—41% общей Р2О5, 34—40,3%
усвояемой Р2О5) 28—33,6% водорастворимой Р2О5, 6,3—8% общего
и 6—7,6% водорастворимого бора в пересчете на Н3ВО3. Влаж-
«ость продукта, определенная высушиванием при 85°, равна2—3%.
Разработан также способ получения борофосфорного удобре-
¦ния фосфорнокислотной переработкой предварительно прокален-
прокаленной данбуритовой руды или ее концентрата и смеси его с фосфо-
фосфоритом. Данбурит (CaO-B2O3-2SiO2) содержит 28% В2О3, а кон-
Борные удобрения
349
центрат 20—22% В2О3. Разложение идет в течение 1 ч при 95°,
продукт содержит до 55% питательных веществ т-т.
Изучено выделение бора из датолита в виде борнометилового
эфира. Для этого раствор датолита в серной (~52% H2SO4) или
Рис. 107. Схема производства бородвойного суперфосфата:
Л — хранилище фосфорной кислоты; 2-насос; 3-напорный бак для фосфорной кислоты;
4-дозатор кислоты; 5-реакторы: 6-питатель для датолитовой муки; 7-элеватор; 8-Пита-
8-Питатель для ретура; 9 — барабанный смеситель-гранулятор; 10- барабанная сушилка; II — пыле-
пылеуловитель; 12-вибрационный грохот; 13-дробилка; 14-питатель для известняка; М-бара-
М-барабанный нейтрализатор.
в соляной кислоте (~7% НС1) обрабатывают метиловым спиртом,
который затем регенерируют на 98—99% и снова возвращают в
процесс 172.
Производство борных удобрений методом
спекания 173~175
При спекании турмалинов и датолитов с известняком или со-
содой В2Оз переходит в усвояемую растениями форму. Из спека, по-
полученного при 1100е из турмалина F,5% В2О3 и 37,1% SiO2) и уг-
углекислого кальция A30% от стехиометрического в расчете на S1O2
турмалина), извлекается в лимоннокислую вытяжку 72,5% В2О3.
Оитимальная температура спекания турмалина с содой 900°
(рис. 108). При стехиометрическом количестве соды (по SiO2) из
содержащегося в спеке В2О3 66% извлекается в водную вытяжку
и 80,3% в лимоннокислую; при норме соды 110% от стехиометри-
стехиометрической степень извлечения В2О3 в водную вытяжку возрастает до
81,5% и в лимоннокислую до 92,3%. При спекании датолита с
СаСО3 (Ю0% от стехиометрического количества) при 1000° сте-
степень превращения В2О3 в лимоннорастворимую форму 90—98,5%;
те же результаты достигаются при спекании датолита с содой, но
при более низкой температуре (800°) (рис. 109). Спекание можно
производить с количеством соды, иа 20% меньшим стехиометриче-
350
Гл. X. Соединения бора
ского (в расчете на SiO2 датолита), однако все же использование
соды менее экономично, чем использование известняка. Спек,
после тонкого измельчения, служит удобрением.
Опыты174 спекания с известняком кварцтурмалиновой руды,
имевшей 4,26% В2О3, 61% SiO2 и 24% R2O3, дали спек, содержа-
содержащий 4,2% Н3ВО3 при степени перехода В2О3 в усвояемую форму
97—98%. Оптимальные условия спекания: норма известняка 1,32г
100
во
ео
40
s
I 20
с
0
—^
I
it/-
Mi
%
/
f
0
•о
S
а
60
40
20
<
Щ
w
к
У
N
700 800 900 1000
Темперашура,°С
Рис. 108. Влияние темпера-
температуры спекания турмалина
с содой на степень извле-
извлечения В2О3.
700 800 900
Температура ,°С
Рис. 109. Влияние темпера-
температуры спекания датолита
с содой на степень извле-
извлечения В2О;,.
на 1 т турмалина, продолжительность спекания 1,5 ч при 1000°.з
На 1 т спека расходуется 0,584 т руды и 0,77 т известняка. Таким'
ке способом можно получать удобрение, содержащее В2О3 в ли-:
аоннорастворимой форме, из данбуритового концентрата175.
Б.орные удобрения из сырья с низким
содержанием бора
Помимо турмалинов, к такому сырью относятся необогащенный;
датолит и красные и серо-зеленые глины из месторождений маг-1
ниевых боратов. Запасы этих видов сырья могут полностью удов-1
летворить потребности сельского хозяйства в боре. Эти глины]
содержат около 3% В2О3, из которых 91—93% находятся в ли-]
моннорастворимой форме и 82—93% в цитратнорастворимой фор-
форме. Поэтому эти глины после сушки и размола могут быть исполь-
использованы непосредственно в качестве борного удобрения173,
Выделение бора из растворов
351
Разработан способ получения обогащенного борного концен-
концентрата из озерной рапы, содержащей бор 176. При обработке рапы
известковым молоком получают концентрат, содержащий (в %):
Mg — 31,5 (в пересчете на Mg(OHJ—76); В2О3—8; нераствори-
нерастворимого остатка —0,3; R2O3—1,2; Са — 3; SO4—1,5; С1 — 2; Н2О,
Na и пр. — 8. Этот концентрат можно применять в качестве удоб-
удобрения. При извлечении из него бора в раствор обычными методами
и последующего осаждения известковым молоком можно получить
более богатый концентрат, содержащий 25—30% В2О3. Степень из-
извлечения бора из рассола достигает 60%. Обработкой концентрата,
содержащего 4,3—6,8% В2Оз, экстракционной фосфорной кислотой
{22—23% РгО5) были получены174 удобрения, содержащие
3,8—5,8% Н3ВО3, 30,8—42% Р2О5, 19,2—27,6% MgO; отношение
MgO : Н3ВО3 может изменяться в пределах от 6,3: 1 до 10: 1. На
получение 1 т борофосфорного удобрения расходуют: 0,3—0,48 т
концентрата D,3—6,8% В2О8) и 1,48—1,75 т фосфорной кислоты
{22,7% Р2О5).
ВЫДЕЛЕНИЕ БОРА ИЗ РАСТВОРОВ
Природные растворы, несмотря на то, что они содержат бор
в небольших концентрациях, являются мощным потенциальным
источником борного сырья, разработке методов использования ко-
которого, уделяется все большее внимание99-174~177. Трудность извле-
извлечения бора обусловлена крайне низкими концентрациями бора при
значительном содержании других веществ, мешающих его выделе-
выделению. Рапы некоторых солевых озер и некоторые буровые воды
являются концентрированными растворами хлоридов натрия, ка-
калия, магния и содержат всего около 0,02% В2Оз. В рассоле многих
озер содержится 0,01—0,06% В2Оз178. В сухом остатке морской
воды ~0,05% В2Оз97. Промышленные отходы — растворы от про-
производства борной кислоты и буры гораздо богаче бором — они со-
содержат около 1,5% В2Оз.
Выделение бора из растворов возможно или с помощью иони-
тов, или в виде труднорастворимых соединений, растворимость
которых крайне мала, меньше содержания В2©3 в природных во-
водах. Эта задача еще далеко не разрешена. Так, после осаждения
В2О3 окисью магния или кальция в растворах содержится еще
0,2—0,3% В2О3179'180. Растворимость природных боратов в воде
при 25° также выше содержания В2О3 в природных водах — в рас-
растворе калиборита, гидроборацита, иньоита и улексита содержится
0,2—0,4% В2О3 и лишь раствор ашарита содержит 0,0085%
В2О3, 181
Однако возможно выделение значительной доли бора из про-
промышленных растворов и из предварительно сконцентрированных
природных вод. Выше был описан способ осаждения бора из озер-
озерной рапы окисью кальция176, Разработан177 способ выделения
352
Гл. X. Соединения бора
бора с помощью сульфата марганца из маточных растворов от
производства борной кислоты 134-182 и растворов, получающихся
после выпарки буровых вод 120:
Na2B4O7 + MnSO4 + 5H2O = Mn(BO2J • 2H2O + Na2SO4 + 2H3BO, W
2H3BO3 + MnSO4 5=± Mn(H2BO3J + H2SO4 *
Образующуюся серную кислоту необходимо нейтрализовать
окисью кальция или магния. После обработки сульфатом мар-
марганца при 70—95° маточных растворов от производства борной
кислоты, содержавших 1,3—2% В2О3, 21—24% MgSO4, 0,2—1,2%
R2(SO4K, 0,2—0,6% H2SO4 и незначительные количества других
1 примесей, концентрация В2О3 в конечных фильтратах была
0,37—0,6% при нейтрализации окисью кальция и 0,4—0,55% при
нейтрализации окисью магния. Обработка сульфатом марганца
при 20° нейтрализованного окисью кальция концентрата буровой
воды, содержащего после выпарки и выделения бикарбоната нат-
натрия и буры 0,82% Na2B4O7 @,57% В2О3), 17,8% NaCl и 0,55%
Na2CO3, дала фильтрат с 0,3% В2О3.
Образующиеся осадки — бораты марганца — после их под-
подсушки могут быть использованы непосредственно как микроудоб-
микроудобрения, или как добавки к суперфосфату, а так же как сырье для
получения борной кислоты. Сушка этих осадков при 500° приводит
к разложению боратов марганца с образованием борной кисло-
кислоты— при выщелачивании водой в раствор переходит 70—80% В»О3
идоЗО%Мп.
Метод осаждения борной кислоты из маточных растворов
окисью магния 139' и0 дает осадок с отношением MgO : В2О3, рав-
равным 3:1. Осаждение бора в виде боратов кальция 67> "• 176 также
дает удовлетворительные результаты только для сравнительно бо-
богатых бором растворов, так как в конечной жидкой фазе содер-
содержится — 0,25% В2О3.
Совместное осаждение боратов кальция и меди позволяет вы-
выделить из раствора, содержащего 0,2—0,4% В2О3, 85—93% В2О3.
В растворе после осаждения содержится всего 0,03% B9O3, а в
осадке.22,4% В2О3> 18,3% СиО, 36,25% СаО и 23,1% Н2О. Осаж-
Осаждение должно проводиться известковым молоком, к которому до-
добавлена соль меди. Аналогичные, но несколько худшие результаты
получаются при соосаждении бората кальция с другими микро-
микроэлементами — марганцем, кобальтом 183.
Борная кислота в водных растворах образует комплексные
кислоты с винной и триоксиглутаровой кислотами. Можно выде-
выделять бор из очень разбавленных растворов, осаждая его в виде со-
солей этих кислот: боротартратов /zMe2O • тМеО • В2О3 • 2С4Н4О5 • aq
и бородитартратов пМе2О • тМеО • В^О3 • 4С4Н4О5 • aq, боротри-
Перборат натрия
353
оксиглутаратов пМе2О • тМеО • В2О3 • 2CSH6O6 • aq и диборотритри-
оксиглутаратов «Ме2О • тМеО • В2О3 • ЗС5Н6О6 • aq. Здесь Me1 —
щелочной металл или аммоний, Me11 — щелочноземельный металл,
цинк, кадмий, марганец. Некоторые из этих солей, например, боро-
дитартраты и диборотритриоксиглутараты щелочноземельных ме-
металлов, очень мало растворимы в воде, но хорошо растворимы в
кислотах, в том числе и в лимонной, и могут быть использованы
в качестве микроудобрений 184.
Форма боратов, выделяющихся из растворов, зависит от вели-
величины рН |85.
Возможно выделение борной кислоты из нефтяных вод адсорб-
адсорбционными методами — активной окисью магния и активированным
углем: 1 г угля адсорбирует из раствора борной кислоты до 15 мг
В2О3. Десорбция осуществляется кипячением суспензии адсорбен-
адсорбента в воде 69>89.
Имеющиеся в настоящее время иониты мало селективны и не-
недостаточно эффективны для выделения бора из солевых растворов.
Но они могут оказаться полезными при извлечении В2О3 из раство-
растворов борной кислоты, не содержащих солей. Так, иониты ММГ-1,
МГ, ТН, Н извлекают из раствора, содержащего 0,2% В2О3, до 90%)
борной кислоты — в растворе остается 0,02—0,03% ВгО3. Это поз-
позволяет использовать аниониты для регенерации борной кислоты из
растворов, не содержащих солей, когда невозможно осуществить
осаждение при помощи СаО и MgO. Регенерацию анионита лучше
всего производить водой или 5% раствором NaOH.
По отношению к бору наибольшей селективностью обладают
гидроксилсодержащие иониты (ПВГ, АВ-17, АВ-27, ЭДЭ-10,
ЭДЭ-10П). Ионы тетрабората поглощаются с наибольшей актив-
активностью при рН 7,5—9,5. Максимальная степень поглощения бора
в форме комплексных ионов (из растворов, содержащих компо-
компоненты, необходимые для комплексообразования) достигается при
рН 2—3,5 186- ш.
Взаимодействие Н3ВО3 с анионитами протекает очень медлен-
медленно — в отличие от сильных кислот, поглощающихся в течение не-
нескольких минут, для достижения равновесия при поглощении бор-
борной кислоты требуется 5—7 суток. Проблемой большой важности
является синтез новых ионообменных смол, пригодных для погло-
поглощения В2О3 из солевых растворов.
ПЕРБОРАТ НАТРИЯ
Перборат натрия получается43 при взаимодействии буры иле
борной кислоты с едким натром и с перекисью водорода:
Na2B4O7 • 10Н2О + 2NaOH + 4Н2О2 + Н2О = 4(NaBO2 ¦ Н2О* • ЗН2О)
Н3ВО3 + NaOH + Н2О2 + Н»О = NaBOs. HSO2 • 3HSO
12 М. Е. Позии
884
Гл. X. Соединения бора
К 30% раствору NaOH добавляют буру в количестве, превы-
превышающем стехиометрическое на 20%. В полученный раствор мета-
бората натрия NaBO2 медленно приливают 3% раствор перекиси
водорода в воде с 10—15%-ным избытком. Реакцию ведут при тем-
температуре не выше 10°. В реактор добавляют поваренную соль для
высаливания полученного пербората натрия при охлаждении до0°.
Перборат натрия отжимают на центрифуге и высушивают при не-
невысокой температуре. Выход продукта составляет 93—95%; на 1 т
пербората натрия затрачивается 0,67 i буры.
Вместо NaOH можно применять менее дорогую соду. В 5% рас-
раствор перекиси водорода при 15° вводят теоретические количества
буры и соды. Образование пербората натрия протекает по реакции:
Na2B4O7 • 10Н2О + 2Na2CO3 + 4Н2О2 + ЗН2О = 4(NaBO2 • Н2О2 • ЗН2О) + 2NaHCO,
Процесс заканчивается в течение 1 ч с использованием буры
и перекиси водорода до 68%; количество активного кислорода в
продукте 8,5%, вместо теоретических 10,38% в перборате натрия.
Усовершенствованный содовый способ получения пербората
натрия188 заключается в применении 1,5% раствора перекиси водо-
водорода (вместо 5%), с которым бура и сода взаимодействуют в при-
присутствии сульфата цинка. После перемешивания в течение
20—25 мин при 10° реакционную массу охлаждают до 5° и вводят
в нее насыщенный раствор NaCl. Выделившийся осадок отфильтро-
отфильтровывают и сушат при 40—50° в токе воздуха. Степень использования!
Н2Ог при таком способе получения пербората натрия составляет!
"86%, а буры 89,5%. На 1 т пербората натрия A00%) расходуют:!
0,25 т Н2О2, 0,69 т буры, 0,48 т соды, 0,72 т поваренной соли и1
0,05 г сульфата цинка.
Для получения пербората натрия может быть использован ма-
маточный раствор от производства борной кислоты (стр. 344). Для
осаждения из этого раствора Fe, Al, Ca, Мп и других металлов его
¦ обрабатывают при 80—85° небольшим количеством каустического
магнезита с одновременным окислением Fe2+ и Мп2+ хлорной из-
известью. После дополнительного нагревания до кипения и удаления
шлама к раствору добавляют соду для осаждения основного кар-
карбоната магния, перерабатываемого на магнезию. Остающийся
фильтрат содержит около 80% бора от количества его в маточном
растворе после производства борной кислоты. К фильтрату добав-j
ляют NaOH или Na2CO3 — образуется раствор метабората натрия.]
ГОсле охлаждения до 10° в него вводят Н2О2 G—8 кг на 1 м3\
фильтрата). Затем охлаждают до 5° или добавляют NaCl для вч
саливания пербората натрия. Кристаллический перборат отфиль-j
тровывают и высушивают при 45—60°. ]
Основным компонентом шлама, получаемого при очистке исход-]
ного маточного раствора, является борат магния. Для извлечения]
Перборат натрия
бора шлам можно обработать 5% раствором NaOH, отделенную от
осадка жидкость упарить и добавить -к фильтрату, используемому
для получения пербората.
Перборат натрия получают также сплавлением борной кислоты
с перекисью натрия и электрохимическим способом43-189-190. Элек-
Электролизу подвергают раствор, содержащий в. 1 л ~40 г Na2B4O7,
~140 г Na2GO3 и 0—40 г NaHCOs. Анод — платиновый; катод —
оловянные или стальные спиральные змеевики с охлаждающей во-
водой. Плотность тока на аноде 1500—2000 а/м2, на катоде
500—1000 а/м2. Образование пербората натрия идет через проме-
промежуточные продукты. Вначале у анода образуется перкарбонат
2СО23~ - 2е = С2О|-
который, реагируя с водой, образует перекись водорода:
C2Og~ + 2Н2О = 2НСОз + Н2О2
Имеются предположения и об ином механизме этого процесса t91.
Перекись водорода образует с боратом перборат (пергидрат бо-
бората). На катоде выделяется водород, а на аноде небольшое коли-
количество кислорода и двуокиси углерода. Эти потери СОс должны
компенсироваться периодическим введением в электролит бикарбо-
бикарбоната натрия, во избежание повышения концентрации ОН~ и усиле-
усиления их разряда. Напряжение на ванне 6—7,5 в; нагрузка иногда
достигает 11000 а. Расход энергии на 1 т продукта ~5500 квт-ч.
Выход по току 40—50%. При больших плотностях тока выход по
току увеличивается с повышением температуры. Однако темпера-
температура не должна быть выше 16°; она не должна опускаться и ниже
14°, так как пои этом вместе с перборатом натрия кристаллизуются
бура и сода. Выход резко уменьшается при загрязнении растворов
ионами тяжелых металлов, каталитически разлагающих перекись
водорода. Поэтому циркулирующий электролит очищают кипяче-
кипячением с силикагелем (8 г на 1 л раствора), лучше под давлением до
3 ат. Ввиду отсутствия в ванне диафрагмы в электролит вводят не-
немного хромовокислой соли и ализаринового масла или жидкого стек-
стекла, что препятствует восстановлению пербората на катоде. В процес-
процессе электролиза электролит пересыщается перборатом натрия, кото-
который кристаллизуется. Периодически часть суспензии кристаллов
выводят на центрифугу и отделенные кристаллы пербората натрия
высушивают. В полученном продукте 10—10,5% активного кисло-
кислорода-.
За рубежом перборат натрия выпускается в нескольких фор-
формах192. Более распространен тетрагидрат NaBO--4H2O, т. е.
NaBO2 • Н2О2 • ЗН2О, содержащий 10% активного кислорода; вы-
выпускают также NaBO2 • Н2О2 • 2Н20193; менее распространен моно-
моногидрат NaBO3-H2O, т. е. монопергидрат NaBO2-H2O2, с 15—16%'
активного кислорода, получаемый обезвоживанием тетрагидрата,
12»
356
Гл. X. Соединения бора
т
который легко теряет кристаллизационную воду. Давление пара над
NaBO2• Н2О2• ЗН2О вычисляют по уравнению lgP= 12,19—328671-'.
Производство и потребление перборатов за границей исчисляется
десятками тысяч тонн в год. Помимо пербората натрия, имеющего
наибольшее применение, известны способы получения пергидратов
боратов калия, аммония, лития, рубидия и цезия, а также истин-
истинных перборатов аммония, калия и бериллия43-194~196. Более под-
подробные сведения о свойствах и методах получения пербора-
перборатов см.43.
БОРНЫЙ АНГИДРИД
Борный ангидрид получают дегидратацией борной кислоты, на-
нагреванием ее до 400—500°189. При этом часть борной кислоты уле-
улетучивается с парами воды. При 500°, 1 ат и продолжительности
нагревания 3 ч197 дегидрата-
дегидратация протекает полностью (рис.
110), а потери B2OS состав-
составляют около 6%. Под вакуу-
вакуумом (остаточное давление
15 мм рт. ст.) можно осуще-
ствить дегидратацию Н3ВО3
при 200—250°.
Предложен способ обезво-
живания Н3ВО3 в сушильном
барабане в токе воздуха или
других газов в три последова-
последовательные стадии при 100—120°,
170—185° и 215—225°. Продол-
Продолжительность каждой стадии
1 ч. Получается порошкообраз-
порошкообразный продукт, содержащий
99% В2О3198.
Система В2О3—Н2О может
находиться в метастабильном
состоянии. В зависимости от
бор
887
50,
100 200 300
Температура^
Рис. ПО. Дегидратация борной кислоты:
1 — содержание В2О3 в плаве; 2 —потери ВзОз-
Сплошные линии — при атмосферном давлении;
пунктирные линии — при вакууме 15 мм рт. ст.
(без поглощения паров воды).
давления водяного пара и
борных кислот в газовой фазе, а также от присутствия в орто-
борной кислоте примесей других борных кислот, дегидратация
Н3ВО3 может протекать разными путями '". При общем давлении
водяного пара и борной кислоты в газовой фазе 10—15 мм рт. ст.
чистая борная кислота непосредственно переходит в борный анги-
ангидрид:
2H3BO3-=B2O3 + 3HSO
Реакция начинается при 96°. При общем давлении паров от
20 до 50 мм рт. ст. дегидратация H3BOS идет с образованием двух
промежуточн!
состава:
соединений — НВО2 и гидрата неопределенного
Н3ВО8 =- НВОа + Н2О (84 - 96°)
НВО2 = xBiO3 • i/H2O A18- 143°)
¦ f/H2O = хВ2О3 + f/H2O A45 - 150")
При общем давлении паров 66—988 мм рт. ст. дегидратация
идет через метаборную кислоту с дальнейшим образованием рас-
растворов, из которых при удалении воды получаются вязкие, пуча-
пучащиеся жидкости. Превращение HSBO2 в НВО2 при этих условиях
наблюдается от 101 до !56°, а образование растворов — от 147 до
178°. Содержание в ортоборной кислоте примеси р-формы HBOj
способствует образованию растворов при общем давлении паров от
175 до 750 мм рт. ст. В этих условиях Н3ВО3 переходит в НВО2 от
118 до 144°, а дальнейшее разложение НВО2 с образованием вяз-
вязких жидкостей идет от 179 до 194°.
ТРЕХФТОРИСТЫЙ БОР
Наиболее простой способ получения BF3 заключается в обра-
обработке концентрированной серной кислотой вмеси плавикового
шпата и борного ангидрида:
3CaF2 + В2О3 + 3H2SO4 - 2BF3 + 3GaSO4 + ЗН2О
Однако продукт, полученный этим способом, загрязнен значи-
значительным количеством SiF4, удаление которого является очень труд-
трудным. При разложении олеумом смеси флюоритового концентрата с
боратом кальция при 150—180° и 150%-ном избытке H2SO4 выход
BF3 составляет 90%; продукт содержит 2—3 мол.% SiFu200. Более
чистый продукт получается при замене плавикового шпата фторбо-
ратом натрия, аммония или калия201, содержащим меньше
кремния:
6N aBF4 + В2О3 + 6H2SO4 - 8BF3 + 6NaHSO4 + ЗН2О
Чистый фтористый бор можно получить термическим разложе-
разложением фторборатов щелочных металлов76-202. Разложение фторбо-
рата фенилдиазония идет по реакции203.
C6H5N2BF4 = C6H5F + N2 + BFS
П-ри разложении фторборатов арилдиазония 2М получается фто-
фтористый бор с содержанием лишь 0,14 мол, % SiF4. Из фторборатов
щелочных металлов наиболее легко диссоциирует LiBF4, труднее
NaBF4, очень трудно KBF4. Давление диссоциации (Р в мм рт. ст.)
может быть вычислено по формулам:
358
Гл. X. Соединения бора
Диссоциация KBF4 ускоряется добавками MgCb, CaCl2, MgSO4
или ВаС1220.
Наиболее экономичный и распространенный за рубежом способ
получения BF3 состоит из двух стадий — получения фторбората
натрия или аммония по реакциям
Na2B4O7 ¦ 10Н2О + I2HF = Na2O(BF3L + 16Н2О
6NH4HF2 + 4Н3ВО3 = 4NH3 + 1IH2O + (N^kOfBFg)*
12NH4F + 2B2O3 = I0NH3 + 5H2O + (NH4JO(BF3L
н обработки их серной кислотой:
Na2O(BF3L + 3H2SO4 = 2NaHSO4 + H2SO4 • H2O + 4BF3
Процесс осуществляют следующим образом. Буру или борную
кислоту медленно добавляют к стехиометрическому количеству
фтористоводородной кислоты или бифторида аммония. Смесь на-
нагревают и кипятят для удаления воды (и аммиака при приготовле-
приготовлении фторбората аммония). Фторборат загружают в реактор, куда
на холоду добавляют рассчитанное количество олеума, содержа-
содержащего 20% свободной SO3. При нагревании реактора выделяется
BF3, который собирают в газгольдере, а из него компрессором на-
накачивают в баллоны под давлением 140 ат76.
Трехфтористый бор можно также получить взаимодействием
' борной и фторсульфоновой кислот:
3SO3HF + Н3ВО3 = BF3 + 3H2SO4
Части аппаратуры, соприкасающиеся с BF3, изготовляют из ма-
материалов, содержащих кремнезем: стекла, керамики, фарфора; наи-
наиболее подходящим материалом является кварц.
ЛИТЕРАТУРА
1. Л. Я. Марковский, Ю. Д. Кондрате в, Г. В. Капустовская,
ЖОХ, 25, № 3, 433 A955). — 2. Л Я- Марковский. М. П. Белов, ЖПХ,
37, № 8, 1658 A964); 38, № 1. 17 A965); 40, № 4, A967). — 3. Г. В. Самсонов,
Л. Я. Марковский, А. Ф. Ж и г а ч; М. Г. В а л я ш к о, Бор, его соединения
и сплавы, Изд. АН УССР, Киев, I960. — 4. А. С. М и к у л и н с к и й, В. В. Е р е м-
кин и др., Труды УНИХИМ. в. 5, 1957, стр. 152, 166, 206, 211, 232.-
5 В. И. Михеева и др., ДАН СССР. 91, № 5, 1133 A953); ЖНХ. 2, № б,
1223, 1232, 1242, 1248 A957). — 6. А. М. Поляк и др., Труды УНИХИМ, в. 5,
1957, стр. 222, 228.-7. Л Я. Марковский. Г. В. Капустовская, ЖПХ,
35, № 4. 723 A962). — S. J. Cueilleron, Ph. Pi chat, франц. пат. 1403135,
J964. — 9. R. Monnier, P. Tissot, P. Pearson, Helv. chim. acta, 49, 67
A966).— 10. A. A. Woo d, пат. США, 3189412, 1959
//. X. Та га в а, К- Исин, япон. пат. 747, 1962. —12. В. М. Слепцов,
Г. В. Самсонов, ЖПХ, 34, № 3.501 A961).- 13. A. L i p p, Techn. Rundschau,
57, № 33, Б, 7 A965). —14. И. И. Искольдский, С. Л. Черкинская,
ЖПХ, 31, № 1, 25 A958). —15 И. И. Искольдский, А. Е. Горбунов,
Там же, 38, № 7, 1428 A965). — 16. J. L. Andrieux, S. Marion, Compt. rend.,
Литература
359
236, № 8, 805 A953). — 17. Л. Я. Марковский, Н. В. Векшииа, ЖПХ, 31,
1293 A958); 34, 16, 242, 2171 A961); 35, 30 A962).— 18 Т. И. Серебрякова,
Г. В. Самсонов, Там же, 40, № 1, 3 A967). — ii». H. S. Booth, D. R. Mar-
Martin, Boran Triffuoride and its Derivatives 1949. — 20. И. Г Рысс, Е. М. По-
Полякова, ЖОХ, 18, № 2, 286 A948).
21. M. И. В инн и к, Р. Н. КРУ г лов, Н. М. Чирков, ЖФХ, 31, № 4, 832
A957). — 22. Н. А. Е. MacKenzic a. oth., J. S. Afric. Chem. Inst., 5, 123
A951); 6, № 1, 4, 8 A953). — 23. А. В Топ ч и ев, Я. М. П о у ш к и и, С. В. 3 а-
городний, Усп. хим., 21, № 4, 422 A952). 24. И. Г. Рысс, Химия фтора и его
неорганических соединений, Госхимиздат, 1956. — 25. S. Pawl en ко, Z. anorg.
Chem., 340, № 3, 201 A965). — 26. A. L. McCloskey, H. M. Manasevit,
R. J. Brotherton, пат. США 3180707, 1961; 3180708, 1964. -27. M. В а с с а-
redda, G. F. Nencetti, Chim. e ind., 42, № 10, 1084 (I960). —Ж Е. L. Dan-
Dance, Т. Е. Drisko, E. A. Me La in, пат. ФРГ 1163300, 1959. — 29. Пат. США
3152869, 1960; 3144306, 1961; 3148030, 1961. — 30. Б. Н. Иваиов-Эмин,
Л. А. Нисельсон, И. В. Петрусевич, ЖПХ, 34, № 11, 2378 A961).
ЗУ. Г. В. Самсонов, Ю. Б. Титов, Там же, 36, № 3, 669 A963).—
32. F. Thevenot, J. Cueilleron, Bull. Soc. chim. France, 1968, № 8, 3204.—
33. J. Cueilleron, F. Thevenot, ibid., 1965, K° 2, 402. — 34. G. Gastello,
E. Biagini, S. Munari, J. Chromatogr. 20, № 3, 447 A965). — 35. Пат. ФРГ
1242580, 1963. —36. M. Baccaredda, G. F. Nencetti, R. Tartarelli,
Chim. e ind., 47, № 2, 148 A965); 48, № 1, 24, 30 A966). — 37. J. B. Moffat,
H. L. Goltz, Canad. J. Chem., 43, №. 6, 1680 A965). — 38. Сб. «Неорганические
полимеры», ИЛ, 1961, стр. 379. —39. F. Т. Greene, J. L. Margrave, J. Phys.
Chem., 70, № 7, 2112 A966). — 39a. A. Kankaanpera, P. Saloman, Acta
chem. Scand, 23, № 2, 712 A969). — 40. P. R Kemp, The Chemistry of Borates,
London, 1956.
41. К. Н. Schier, P. Wange, пат. ГДР 31302, 1962. —42. Голланд. пат.
111135, 1955. — 43. Перекись водорода и перекисные соединения, под ред.
М. Е Позина, Госхимиздат, 1951, стр. 129, 340, 381. —44. А. П. Казрагис.
А. Ю. Пр окопчик, сб. «Химия перекисных соединений», Изд. АН СССР,
1963, стр. 156. — 45. F. Chopin, P. Hagenmuller, Bull. Soc. chim. France,
1965, № 10, 3031. — 46. А. В. Николаев, Физико-химическое изучение природ-
природных боратов, Изд. АН СССР, 1947. — 47. Н. A. Lehmann, К. "S ch a a r s с ti-
timid t, I. Giinther, Z. anorg. Chem., 346, № 1, 2, 12 A966). — 48. И. Г. Рысс
ДАН СССР, 52, 421 A946). — 49. И. Г. Рысс, М М. Слуцкая, Там же, 57,
689 A947). — 50. И. Г. Рысс, М. М. Слуцкая, ЖФХ, 21, 549 A947).
51. И. Г. Рысс, М. М. Слуцкая, С. Д. П а л е в с к а я, ДАН СССР, 22,
1322 A948). — 52. А. Р. Altschuller, J. Am. Chem. Soc, 77, № 23, 6187
A955). — 53. И. Г. Рысс, А. Г. Элькеибард, ДАН СССР, 91, № 4, 865
A953). — 54. A. Bazaroff, Ber, 7, 1121, A874).— 55. Г. И. Петренко
ЖРФХО, 34, 37 A902). — 56. И. Г. Р ы с с, М. М. С л у ц к а я, Б. С. В и т у х н о в-
ская, ЖПХ, 25, № 2, 148 A952). —57. А. Д. Кешан, Е. М. Шварц, Изв
АН ЛатвССР, № 8, 107, 137 A954). — 58. G. Heller, J. lnorg a. Nucl. Chem
27, № 11, 2345 A965). — 59. E. M. Шварц, А. Ф. Иевииьш, Изв. АН
ЛатвССР, № 9, 135 A956). — 60. W. J. E n gler t, F. A. H urn m e 1, J. Soc Glass
Technol., 39, № 187, T 121 A955).
F. A. Hummel, ibid., T 113. — 62. А. А. Берлин,
|{"\Г"/"*\ t^Tl -WTT ¦¦» r-m m
A932).
61. W. F. Horn, F. A. Hummel, ibid., T 113. — 62. А. А. Берлин
В. П. Парини, Хим. наука и пром., 1, № 1, 44 A956). — 63. W. H. Zacha-
riasen, G. E. Zigler, Z. Krist. (A), 83, 354 A932). — 64. J. Edwards J
Am. Chem. Soc, 75, 6151, A953). — 65. P. J. Antikainen, Suomen Kem., 30,
№ 4, 1374 A957). — 66. Chem. Eng. News, 55, № 21, 28 A957). — 67. A. Burg
R. Wagner, J. Am. Chem. Soc, 76, 3307 A954). — 68. L. M a z e t, Chim. et ind,
64, № 4, 423 A950).— 69. А. Д. Кешан, Синтез боратов в водном растворе и
их исследование, Изд. АН ЛатвССР, 1955.-70. V. L. McKinncy, T. Rock-
360
Гл. X. Соединения бора
well, A. S. К i t z e s, W. Q. H u 11 i n g s, U. S. Atomic Energy Comm. Repts
AECD-3626, 40 A954).
71. Г. В. Самсонов, ДАН СССР, 86, № 2, 329 A952). — 72. A. Roos,
Chim. et. ind., 82, № 3, 339 A959). — 73. В. Л. Гинзбург, Сверхпроводимость
Изд. АН СССР, 1946. — 74. Г. А. М е е р с о и, Г. В. С а м с о и о в, ЖПХ, 27, № Ю,
П15 A954). — 75. Л. Я. Марковский, Н. В. Векшина, Там же, 31, № 9,
1293 A958). — 76. Г. Б у з, Д. Мартин, Химия фтористого бора и его произ-
производных, ИЛ, 1955. — 77. Н. В. Соломин, Л. В. Потемкина, ДАН СССР
96, № 1, 91 A954). — 78. Г. И. Беляев, ЖПХ, 31, № 2, 306 A958).—
79. Н. И. Г е л ь п е р и и, К. Н. С о л о п е н к о в, Хим. иаука и пром., 1, № 3,
324 A956). — 80. Г. Л. Кореиьков, Н. А. Устинова, Горнохимическое'
сырье зарубежных стран, Изд. «Химия», 1965.
81. В. Г. X л о п и и, Бор и его соединения, ОНТИ, 1919. — 82. J. R e m о п d,
Rev. Prod, chim., 57, № 1208, 451 A954). — 83. В. В. Яковлева, Борные удоб-
удобрения и их применение, Сельхозгиз, 1954. — 84. Микроэлементы в жизни растений
и животных, Изд. АН СССР, 1&52. — 85. М. В. Каталымов. Значение бора в
земледелии СССР, Сельхозгиз, 1948. — 86. Микроэлементы. Тезисы докладов Все- j
союзн. совещ. по микроэлементам, Изд. АН ЛатвССР, 1955. — 87. М. В. Кат a-j
лымов, Хим. наука и пром., 1, № 2, 155 A956). — 88. И. А. Поспелов. Бор-]
ные удобрения на подзолистых почвах СССР, Сельхозгиз, 1947. — 89. К- М. Ma-j
лин, ЖПХ, 27, № 3, 230 A954). —SO. Удобрения, пер. с англ., Изд. «Колос»,
1965.
91. И. А. X р и з м а и, Сообщения о научных работах ВХО им. Д. И. Мен-
Менделеева, в. 2, 1955. стр. 33. — fi2. H. П. Набоков, И. И. Халтурина, сб.
«Геология и ресурсы агрохимического сырья Казахстана», Алма-Ата, Изд. «Нау-
«Наука», 1965, стр. ПО.—S3. Т. R. Fischer, С. К. Mclntyre, Comm. Fertili-
Fertilizer, 94, № 6, 34 A957).—94. Т. П. Унаняиц, Вестн. техи. и эконом, ииформ.
НИИТЭХИМ, № 5 A963). — 95. И. М. К урман, сб. «Большая Эмба», т. I, Изд.
АН СССР, 1937. — 96. С. С. Смирнов, ДАН СССР, 45, № 1, 22 A944).—
97. А. Е. Ферсман, Геохимия, т. IV, Госхимиздат, 1939, стр. 51. — 98. Н. W a t-
t e n b e r g, Z. anorg. Chem., 236, 339 A938) . — 99. Т. В. Г а л а х о в с к а я, Труды
ВНИИГ, в. 52, 1967. стр. 84. —100. Сб. «Химия боратов», Изд. АН ЛатвССР,
1953.
101. Н. A. Lehman п, G. Rietz, Z. anorg. Chem., 350, № 3, 4, 168 A967).—
102. Т. П. Унанянц, Хим. иаука и пром., 2, № 6, 774 A957).— 103. М. L. Leo-
nardi, Mining Eng., 6, № 2, 203 A954). — /04. G. H. Bikler, D. С Sawener,
Ind. Eng. Chem., 49, № 3, 322 A957). — 105. Т. П. Унаняиц, Хим. пром., № 1,
69 A961). — 106. И. М. Курман, сб. «Минеральные ресурсы капиталистических
стран», Главгеолтехииздат, 1959, раздел «Бор».—107. И. С. Львова, сб. «Про-
«Производство и потребление минеральных удобрений, фосфора, фосфорной н серной
кислот и борных продуктов в зарубежных странах», изд. НИУИФ, 1964, стр. 115.—
108. W. С. Miller, Bull. Bur. Mines, № 630, 149 A965). — 109. И. Я К а б а ч-
ник, Я- Я. Лидак, Борная кислота и бура, ГНТИ, 1931. —110. Сб. «Бор и ка-
калий в Казахстане», Изд. АН СССР, 1935.
111. Сборник под ред. П. М. Татаринова, ГОНТИ, 1938. —112. А. В. Н и^
кол а ев, Изв. АН СССР. Сер. хим., № 2, 410 A938). — ИЗ. И. Н. Лепепй,
ков, ДАН СССР, 22, № 9, 591 A939).— 114. А. А. Иванов, Я. Я. Я р ж е м-1
ский, Труды ВНИИГ, в. 29, 1954, стр. 210. — 115. В. В. Герасимова, Там!
же, в. 29, 1954, стр. 215. — 116. Л. Е. Берлин, Хим. пром., № 4, 98 A949).—
И7. В. И. Классеи, Л. Д. Р ато б ы л ьс ка и, Там же, № 2, 84 A954).— \
118. И. А. Щукин, В. П. Соколов, Г. С. Александрова, А. П. Кон-|
стантинова, Обогащение борсодержащих руд, 1937. —119. В И. Классен,]
Труды ГИГХС, 1950, стр. 51. — 120. Л Е. Берлин, Производство борной кис-
кислоты, буры и борных удобрений, Госхимиздат, 1950.
Литература
121. Л. Е. Берлин, Хим. наука и пром., 2, № 6, 726 A957).—
122. Л. Е. Берлин, М. И. Лазарева, Сообщения о научно-технических ра-
работах НИУИФ, Госхимиздат, 1957, стр. 63. — 123. Т. Я. С а р у х а и я н,
И. В. Фадеева, Сообщения о научно-технических работах НИУИФ, Госхим-
Госхимиздат, 1957, стр. 64.— 124. В. Е. Грушвицкий, А. Н Ремииа, Бюлл. Ин-та
галургии, № 8, 18 A939). — 125. Ю. С. Плышевский и др., Труды УНИХИМ,
в. 7, 1958, стр. 140; в. 8, 1959, стр. 73; Реф. докладов на VIII Менделеевском
съезде, № 1, 141 A959).—126. В. Е. Грушвнцкнй, сб. «Большая Эмба»,
т. I, Изд. АН СССР, 1937, стр. 651. —127. Л. Е. Берлин, Там же, саботы
НИУИФ. —128. М В. Голошанов, ЖПХ, 26, № 3, 303 A953).—
129. А. Б. Б е к т у р о в, В. И. Антонова, Труды Ии-та хим. наук АН КазССР,
т. 1, 1957, стр. 52, 60. — 130. С. И. В о л ь ф к о в и ч, Л. Е Берлин, ДАН СССР,
43, № 6, 264 A944).
131. Франц. пат. 1391910, 1964; Пат. ГДР, 42960, 1961. —132. Правила и
нормы техники безопасности и промышленной санитарии для проектирования,
строительства и эксплуатации производств борной кислоты, буры и борных удоб-
удобрений, Госхимнздат, 1963. —133. Л. Е. Берлин, Хим. пром., № 4, 2 A949).—
134. Л. Е. Б е р л и и, ЖПХ. 28, № 8, 785 A955). — 135. Л. Е. Б е р л и н, А. А. С о-
коловский, ЖХП, 17, № 8, 21 A940). —136. А. М. Андреева, С. А. Ку-
Кузин, ЖПХ, 10, № 5, 845 A937).— 137. Л. Д. Р а т о б ы л ьс к а я. Хим. пром.,
№ 4, 234 A958). — /58. М. Р. Хай кии, Там же, № 3, 82 A947).—
139. Л. Е. Берлин, Л Ф. Ветрова, Сообщении о научно-исследовательских
работах НИУИФ, Госхимиздат, 1957, стр. 61. — 140. 1. D'Ans, К- Н. В eh rend t,
Kali u. Steinsalz, 2, N° 4, 121 A957).
141. А. И. Горячкии, Хим. пром., № 3, 82 A949). —142. Л. Е. Берлин,
Т. А. Саруханян, Там же, №3, 74 A950). — 143. Б. А. Маноле-Бежан,
Труды НИИ хим. пром.. в. 2, Росгизместпром, 1955, стр. 82; ЖПХ, 29, № 8, 1147
A956). —144. А А. Соколовский, Труды ВЗПИ, в. 2, 1952, стр. 35.—
145. А. П. Перова, ЖПХ. 27, № 12, 1275 A954). — 146. А. П. Перова, Сбор-
Сборник статей по общей химии, Изд. АН СССР, 1953, стр. 91. —147. Я. А. У гай,
ЖПХ, 23, № 5, 482 A950).— 148. А. Б. Здаиовский, В. М. Имамутди-
н о в а и др., сб. «Исследования в области химии и технологии минеральных со-
солей и окислов», Изд. «Наука», 1965, стр. 12, 17, 21. —149. Е. Л. Яхонтова,
A. Г Кузнецова, ЖПХ, 38, № 11, 2401 A965).— 150. А. Б. Здаиовский,
B. М. Имамутдинова, Там же, 36, № 8, 1675 A963); Труды ВНИИГ, в. 52,
1967, стр. 75.
151. G. Schmutzler, J. Kircheisen, Chem. Techn., 29, № 8, 468
A968). —152. Л. Е. Берлин, Е. M. Абрамова, А. И. Лазарева, Сообще-
Сообщения о научно-исследовательских работах НИУИФ, Госхимиздат, 1957, стр. 58.—
/53. G Schmutzler, J. Kircheisen, пат. ГДР 34074, 1963 и 39801, 1964.—
1S4. Е. П. Ожигов и др., сб. «Материалы по исследованию химического сырья
Дальнего Востока», Изд. АН СССР, 1958, стр. 75. —155. Е. П. Ожигов и др.,
Реф. докладов на VIII Менделеевском съезде, № 1, 140 A959). — 156. А. М. По-
Поля к, Э. М. М о р г у и о в а, Труды УНИХИМ, в. 7,1958, стр. 124. — 157. U. S b о г-
gi, E. Bovalini, L. Cappelini, Gazz. chim. ital., 54, 298 A924).—
'S8. А. Б. Здановский, Труды ВНИИГ, в. 21, 1949. — 169. D. E. Garrett,
G. P. Rosenbaum, Ind. Eng Chem., 50, № 11, 1681 A958). — 160. J. E. Teep-
• e, The Industrial Development ol Searles Lake Brines, 1929.
161. Eng. Min. J., 159, № 4, 101 A958). —162. Пат. СШД 3336103, 1962.—
163. Швейц. пат. 441247, 1964. —164. Л. Е. Берлин, М. В. Лыков, Б. Ф. Фе-
Дюшкии, А. П. Данилова, авт. свид. 219582, 1964—/05. J. Kircheisen
пат. ГДР 34859, 1964.-/E6. Q. Schmutzler, J. Kircheisen, Chem. Techn.,
20, Щ 4, 239 A968).— 167. Д. Ф. Ризе, А. С. Коробицын, Т. Ф. Куль-
Кульков а. Сборник рефератов докладов IX Менделеевского съезда, № 7, 1965.
СТР- 74. — 188. Л Е. Б е р л и н, сб. «Исследования по производству минеральных
362
Гл. X. Соединения бора
удобрений», 1957. —169. Л. Е. Берлин, Н. И. Николаева, Сообщения,
о научно-исследовательских работах НИУИФ, Госхимиздат, 1957, стр. 55. —'
170. М. Н. Н а б и е в, В. К- Дубовая, Р. А. Маннакова, Узб. хим. ж., № 2
A963).
171. Р. А. М а н н а к о в а, сб. «Физико-химические и технологические иссле-
исследования минерального сырья», Ташкент, Изд. «Наука», 1965, стр. 37.—
172. А. В. Николаев, В. Л. Богатырев, Изв. СО АН СССР. Сер. хим. н.,
№ 11, вып. 3, 150 A965).—/75. А. М. Поляк, Л. И. Девятовская, Труды
УНИХИМ, в. 4, 1957, стр. 50; А. М. Поляк, М. Е. Боронкова, Там же,
стр. 128. —174. В. Т. Микеров, Т. И. Завертяева, Сообщения о научно-
исследовательских работах НИУИФ, Госхимиздат, 1957, стр. 36.—175. Л. Е. Бер-
Берлин, Е. М. Абрамова, Б. Ф. Ф е д ю ш к и н, Сборник рефератов IX Менде-
Менделеевского съезда, № 7, 1965, стр. 16. —176. К- И. Хренников, В. И. Г у тер-
терма н, Е. Д. Я р у ш е в и ч, Л. А. Постникова, Аннотации научно-исследова-
научно-исследовательских работ ВНИИГ, сб. 1, Госхимиздат, 1956, стр. 36; сб. 2, 1958, стр. 90.—
177. Л. Е. Берлин, сб. «Химия боратов», Изд. АН ЛатвССР, 1953. —
178. Д. И. Кузнецов, ЖПХ, 13, № 9, 1332 A940). — 179. А. Г. К у р н а к о в а,
сб. «Химия боратов», Изд АН ЛатвССР, 1953. —180. А. Г Курнакова, Изв.
СФХА АН СССР, 18, 221 A949).
181. А. И. С пир яг и на, Труды ВНИИГ, в. 27, 1953, стр. 77.—
182. А. Д. Пельш. Аннотации научно-исследовательских работ ВНИИГ 1951—
1956 гг., 1958, стр. 92. —183. Е. П. Ожигов, В С. Лозинская, Сборник
рефератов докладов IX Менделеевского съезда, № 7, 1965, стр. 61. —
184. Е. М. Шварц, А. Ф. И ев и ныл, Там же, стр. 111. — 185. М. Г. В а л я ш-
ко, Г. К- Годе, ЖНХ, 5, № 6, 1316 (I960). —186. Б. П. Никольский,
Е. А. М а т е р о в а. М. Г. В а л я ш к о, Д. С. Порывкии, А. Л. Г р е к о в и ч,
М. Д. Аникеева, сб. «Современные методы анализа», Изд. «Наука», 1965,
стр. 312. — 187. А. С. Смирнов, 3. В. Архангельская, ЖПХ, 39, № 10,
2295 A966). —188. Н. Н. Дрозин, Там же, 25, № 12, 1301 A952).—
189. П. П. Ф е д о т ь е в, В. Солнышки н, Сборник исследовательских работ,
Химтеорет, 1936. стр. 104. —190. Н. Н. Дрозин, ЖПХ, 24, № 1, 86 A951).
191. Н. Е. Хомутов, М Ф. Сорокин, Л. С. Филатова, сб. «Химия
перекисных соединений», Изд. АН СССР, 1963. стр. 140. —19. G. Remond,
Rev. prod, chim, 57, № 1207, 401 A954). —193. Пат. ФРГ 1078101 и 1079603,'
1955. —194. С. Girsewald, A. Volokitin, Ber., 42, 865 A906); Chem. Ind.,
33, 100 A910)- 35, 36 A912). —195. М. И. Ц а л. Труды ЛТИ им. Ленсовета,
в. 40, 1957, стр. 53. — 196. G. В г е t s ch ne i d ег, пат. ФРГ 940348, 1956.—
197. M. С. М а к с и м е н к о, В. Н. Крылов, М. А. Липинский, Труды ЛТИ
им. Ленсовета, в. 15, 1947, стр. 91. — /S5. Л. Е. Берлин и др., авт. свид. 199120,
1966. —199. В. В. Урусов, Е. Н. Дергачева, Сообщения о научно-исследо-
научно-исследовательских работах НИУИФ, обмен опытом, в. 3, 1957, стр. 61. — 200. Г. А. Л о-
паткина, Г. Н. Богаче в, Н. Г. Бляхе р, В. Л. Закутинский, Изв. СО
АН СССР. Сер. хим. н., № 2. в. 1, 88 A968).
201. Н. S Booth, К. S. Wilson, J. Am. Chem. Soc, 57, 2273 A935).—
202- M. И. Винник, Г. В. Манелис и др., ЖНХ, 1, № 4, 628 A956).—
203. И. В. Андреева, ЖПХ, 32, № 8, 1855 A959). — 204. Г. М. Панченков,
В. М. Моисеев, Ю. А. Лебедев, ДАН СССР, 100, № 6, 1103 A955).
Глава XI
СОЛЯНАЯ КИСЛОТА
Водный раствор хлористого водорода назвали соляной кисло-
кислотой потому, что издавна его получали из поваренной соли, дей-
действуя на нее серной кислотой. Этот так называемый сульфатный
способ производства соляной
кислоты долгое время был
единственным. Затем стали по-
получать синтетический хлорис-
хлористый водород из хлора и водо-
водорода. В настоящее время оба §_
эти способа утрачивают свое В
значение, так как большие ко-
количества хлористого водорода
получаются в качестве побоч-
побочного продукта при хлорирова-
хлорировании органических веществ и в
других производствах. В связи
ру р
с этим возникла необходимость
изыскания новых путей утили-
утилизации хлористого водорода и
соляной кислоты.
Во всех случаях производ-
Ю
30 50 Г0
Темперотура,'С
Рис. 111. Общее давление паров над
соляной кислотой разных концентраций
СТВО СОЛЯНОЙ КИСЛОТЫ ' СОСТОИТ (в вес. %); (пунктирная ликия — изоба-
из двух стадий 1) получения Ра 760 мм рт. ст.).
хлористого водорода и 2) по-
поглощения (абсорбции) хлористого водорода водой. Производят
также безводный жидкий хлористый водород.
СВОЙСТВА ХЛОРИСТОГО ВОДОРОДА И СОЛЯНОЙ КИСЛОТЫ
Хлористый водород НС1 — бесцветный газ. 1 л при 0° весит
1,639 г; плавится он при —114,2°, кипит при —86,1°. Критические
температура и давление равны 51,5° и 81,6 от. Плотность жидкого
хлористого водорода 0,831 г/сж3 при 20° и 1,194 г/сж3 при —85,8°.
864
Гл. gl. Соляная кислота
Давление пара над жидким хлористым водородом:
Температура, "С.... -85,0
Давление, от 1,0
-20
14,53
0
25,46
20
41,68
40
64,52
61,6
81,6
При 30° жидкий хлористый водород растворяет меньше 0,1%
воды. Молярная теплоемкость газообразного хлористого водорода
при постоянном давлении вычисляется по формуле Ср = 6,5+0,001 Т.
1т Во влажном воздухе хлори-
gOp\:^\^i | ^У\ у/\ JC V\A^%3j стый водород образует густой
туман — мельчайшие капли со-
соляной кислоты. Вредно дей-
действует на организм, раздражая
и разрушая слизистые оболоч-
оболочки и дыхательные пути. Пре-
Предельно допустимая концентра-
концентрация HCl в воздухе рабочей зо-
зоны производственных помеще-
помещений 0,01 мг/л (С12—0,001 мг/л).
Безводный хлористый во-
водород почти не действует на
металлы, соляная же кислота
растворяет большинство метал-
металлов. В соляной кислоте устой-
устойчивы платина, золото, тантал,
ниобий, некоторые силикатные
минералы (андезит, диабаз,
кварц) и изделия (стекло, ке-
керамика, фарфор), а также эбо-
эбонит, резина, некоторые пласти-
пластические массы, например, фао-
лит, винипласт, тефлон и др.
Углеродистая сталь, нагретая
до 300—400°, и нержавеющие
стали 1Х18Н9Т и ЭИ-496, на-
нагретые до 500°, удовлетвори-
удовлетворительно устойчивы к соляной ки-
кислоте 2> 8. Окислы металлов
превращаются газообразным
хлористым водородом в хлори-
хлориды; реакции ускоряются в при-
присутствии водяного пара4.
Растворимость хлористого водорода в воде очень велика и
вильно зависит от температуры; при общем давлении 760 мм рт. ст.:
0 10 20 30 40 60 60
Свойства хлористого водорода и соляной кислоты
365
10 20 30 40 60 60 70 вО 90Ю01Ю
Температура, "С
Рис. 112. Давление НС1 иад соляной
кислотой в зависимости от температуры
н содержания HCl (в вес. %) в ра-
растворе.
Температура, СС
Число литров НС1, погло-
поглощаемых 1 л воды . . . 606,5
При парциальном давлении НС1 в газе 760 мм рт. ст. 1 л воды
при 0° растворяет 525,2 л НС1 (в растворе 46,15 вес.% НС1), при
18° — 451,2 л НС1 (в растворе 42,34 вес. % НС1). Общее давление
паров и давление НС1 над соляной кислотой приведены на рис. 111
и 112. Теплоты растворения НС1 в воде могут быть вычислены с
помощью рис. 113.
Равновесное давление НС1 над соляной кислотой понижается
при внесении в раствор CuCl, NH4C1 и повышается в присутствии
TiCl4, SnCl2, SnCl4. Предпола-
Предполагают, что в системах CuCl —
НС1-Н2О, CuCl — NH4CI— ^
-HCl —H2O, NH4C1 — HCl— I
— Н2О образуются соединения ,f
соответственно: 2CuCl • HCl, §
CuCl • 2NH4C1, NH4CI • «HCl R 5
(n—зависит от температуры)Б. |«
В системе CuCl2—HC1—Н2О ||
при неизменной температуре |,
давление пара Н2О уменьша- g-j
ется с возрастанием содержа- | ^
ния в растворе как НС1, так и | 7. /\ -1250
СиС12. Это указывает на то, что 8
в системе происходит высали- I
вание СиС12 и НС1. В системе
ZnCl2—HCl—H2O взаимодей-
взаимодействие компонентов более слож-
сложное—в области одних концент-
концентраций происходит высалива-
высаливание, в области других — всали-
/
/
Ч
/
Ч
/
\
/
\
\
\
\
\
¦ 450
400
350
200 Д
0 4 8 12 IS 20 24 2S 32 36 40
Содержание НС1, вес. %
Рис. 113. Интегральиаи S и дифферен-
дифференциальная Ф теплоты растворения газо-
газообразного НС1 в соляной кислоте.
вание отдельных компонентов6.
Для давления паров в системе НС1—Н2О характерен минимум,
соответствующий азеотропной смеси, состав которой зависит от
температуры кипения (давления). Азеотропная смесь, кипящая при
ТАБЛИЦА 2в
Концентрации азеотропных растворов в системе НС1—Н2О
473,9 442,0 411,5 385,7 361,6 338,7
Давление,
мм рт. ст.
5.0
100
200
300
400
500
600
НС1,
%
23,2
22,9
22,3
21,8
21,4
21,1
20,7
Давление,
мм рт. ст.
700
760
800
900
1000
1100
1200
НС1,
%
20,4
20,24
20,2
19,9
19,7
19,5
19,4
Давление,
мм рт. ст.
1200
1400
1600
1600
1700
1800
1900
я
19,3
19,1
19,0
18,9
18,8
18,7
18,6
Давление,
мм рт. ст.
2000
2100
2200
2300
2400
2500
НС1,
%
18,5
18,4
18,3
18,2
18,1
18,0
366
Гл. XI. Соляная кислота
110° (под давлением 760 мм рт. ст.), содержит 20,24 вес. % НС1
при 75,9—22,15%, при 66,2—23,2%, при 19,9—24,6% 7 (см. также
табл. 26).
Температуры кипения соляной кислоты при давлении;
760 мм рт. ст. приведены в табл. 27.
ТАБЛИЦА 27
Температуры кипения соляной кислоты при 760 мм рт. ст.
Свойства хлористого водорода и соляной кислоты
367
Концентрация
кислоты.
мол. % НС!
2,0
4,0
е,о
8,0
10,5
Температура
кипения.
"С
101,8
103,3
105,3
108,0
109,7
Концентрация
кислоты.
мол. % НС1
12,0
14,0
17,0
18,6
26,3
Температура
кипения.
°С
109,0
105,2
92,0
82,7
19,0
. Коэффициенты активности соляной кислоты приведены
табл. 28 (см. также8).
ТАБЛИЦА 28
Средние коэффициенты активности HCI
в водных растворах
Концентрация
НС1
в молях
иа 1 кг воды
0,01
0,1
0,5
1,0
2,0
3,0
4,0
Температура, °С
0
0,9065
0,8027
0,7761
0,8419
1,078
1,462
2,000
20
0,9052
0,7985
0,7616
0,8162
1,024
1,345
1,812
40
0,9016
0,7891
0,7432
0,7865
0,9602
60
0,8987
0,7813
0,7237
0,7541
0,9072
В системе НС1—Н2О установлено существование двух эвтектик:
при —74,7° с 23,0% НС1 и при —73,0° с 26,5% НС1. Между эвтек-
тиками находится ветвь кристаллизации конгруэнтно плавящегося
при —70° гексагидрата НС1 • 6HgO. Метастабильная эвтектика
лед —НСЬ4Н2О находится при —87,5° и содержит 24,8% НС19.
В системе НС1—Н2О существуют кристаллогидраты10-11:
НСЬ8Н2О, НСЬ4Н2О, НСЬЗН2О (tnn —24,4°), НС1 • 2Н2О
.(*пл —17,7°), НС1-Н2О (*пл —15,35°). Лед кристаллизуется из
30%-ной кислоты при —20°, из 15%-ной при —30°, из 20%-ной при
—60°, из 24%-ной при —80°.
Плотность соляной кислвты при 16°:
Концентрация НС1, % . . 10,17 20,01 30,бв 39,11
Плотность, г/см3 1,050 1,100 1,165 1,200
Удельная теплоемкость с соляной кислоты, содержащей п мо-
молей воды на 1 моль НС1 ":
п . . . 5,2 10 $0 50 100 290
с 0,660 0,749 0,Й5 0,932 0,964 0$70
Вязкость 2 н. соляной кислоты в 1,12 раза, а' 12 н. кислоты в
2,14 раза больше вязкости воды.
Растворимость хлора в соляней кислоте при 21е и парциальном
давлении хлора 760 мм рт. ст.:
Концентрация кислоты, г-же НС1/л . . 0 0,1 1.0 3,0 6,0 9,0
ббъем хлора @°, 7бО мм рт. ст.), раот-
воряющийся в 1 объеме кислоты . . 2,117 1,48 1,61 2,16 2,90 3.65
Давление хлора над его раствором в 36,3%-ной соляной кислоте
при 20° и при концентрации хлора 1,8бг/л составляет 141 мм рт. ст.,
а при 7,82 г/л— 551,8 мм рт. ст., и в этих пределах изменяется
пропорционально концентрации хлора 12.
Отношение равновесной концентрации хлора в газовой фаве к
концентрации его в 1 н. соляной кислоте (коэффициент распреде-
распределения) при 20° является величиной постоянной, равной
5,3- 105 мм рт. ст./мольные доли 13.
Число объемов @°, 760 мм рт. ст.) водорода, кислорода и дву-
двуокиси углерода, растворяющихся в 1 объеме соляной кислоты при
26°, при парциальном давлении этих газов 760 мм рт. ст.:
Концентрация кислоты, г-же НС1/л 0 0,6 12 8
Растворяется Н2 0,01754 0,0170 0,0164 0,0164 0,0146
» О2 0,02831 0,0271 0,0263 6,0346
» СО2 . . 0,759 6,738 0,732 0,72$
О растворимости НС1 в серной кислоте можно судить по дан-
данным н равновесия в системе НС1—H2SO4—Н2О (табл. 29).
В водной системе Na2SO4+2HCl-»2NaCH-H2SO4 при 25° уста-
установлено наличие поля насыщения у газообразного компонента
—НС1. В растворе с максимальной концентрацией серной кислоты,
находящейся в равновесии с NaCl и соляной кислотой при парци-
парциальном давлении НС1, равном 1 ат, содержится (в вес. %):
H2SO4 — 53,8, NaCl — 5,5, HCI — 0,8. При взаимодействии газооб-
газообразного хлористого водорода с декагидратом сульфата натрия вы-
выделяется поваренная соль и в растворе образуется серная кис-
кислота, содержащая 28% H2SO4) 18% HCI и 2,3% NaCl. Выход H2SO4
достигает 90—97% 1В.
363
Гл. XI. Соляная кислота.
Применение
369
ТАБЛИЦА S9
Равновесные концентрации в системе НС!—Н28О4— НВО
(в вес %; рНС1-в мм рт. ст.)
PHCI
H2SO4
30,60
57,36
65,76
68,96
74,66
82,97
95,82
-760
НС1
25,83
9,81
4,94
3,27
1,48
0,26
0,188
t-
РНС
H2SO4
32,5
68,1
81,75
89,23
94,03
98,45
99,6
26°
-760
НС1
18,7
2,7
0,142
0,092
0,108
0,197
0,402
PHCI
H2SO4
20
36,2
48,0
62,7
67,6
80,7
83,4
70°
-760
HCI
22,3
13,2
6,99
1,56
0,54
0,05
0,032
<-25°
PHCI
H2SO4
29,1
40,0
51,9
58,0
64,0
69,7
76,3
-230
HCI
16,8
H,2
5,6
3,3
1,5
0,4
0,1
PHCI
H2SO4
21,9
31,8
42,3
63,5
59,2
64,7
69,9
25°
-45
HCI
12,2
9,1
B,9
2,8
1,4
0,5
0,1
Газообразный HCI при парциальном давлении НС1, меньшем
0,1 мм рт. ст., быстро сорбируется смесью Na2SO4+Na2SO4 • 10Н2О,
по-видимому, с образованием твердого раствора Na2SO4 + NaCl.
Безводный Na2SO4 не взаимодействует с газообразным НС1 при
низких давлениях и начинает медленно реагировать лишь при дав-
давлении НС1, превышающем 2 ат 16.
ПРИМЕНЕНИЕ
Соляную кислоту применяют в химической промышленности
для выработки хлористых солей цинка, кальция, бария, аммония и
других и органических продуктов—анилина, дифениламина и про-
прочих, для выработки синтетического каучука (хлоропрена), краси-
красителей, для омыления жиров и масел. Соляную кислоту применяют
Лри получении гидролизного спирта и глюкозы из крахмала, в про-
производстве сахара, желатина и клея, при дублении и окраске кож,
в производстве активированного угля, при крашении тканей, для
травления металлов (снятия окислов с их поверхности) при метал-
металлообработке, в различных гидрометаллургических процессах, в
гальванопластике, в нефтедобыче для увеличения дебита скважин,
для консервирования кормов (в Японии) и т. д. Жидкий и газооб-
газообразный хлористый водород применяют для гидрохлорирования раз-
различных органических соединений с целью получения хлористого
»тила C2H5CI, хлорвинила СН2СНС1 из ацетилена, этиленхлоргид-
рина, синтетической камфоры и др.
В США производят более 1,8 мли. г в год соляной кислоты
,A00% НС1I7.
Выпускают несколько сортов технической соляной кислоты
.(табл. 30). ~~
ТАВЛИЦА 30
Требования к качеству соляной кислоты
(содержание компонентов в %)
Содержание НС1, не менее
Максимальное содержание
примесей:
серной кислоты (SO3)
серной кислоты (SO4)
железа (Fe)
мышьяка (As) ....
свободного хлора (С12)
остаток после прока-
прокаливания
ртути (Hg)
Техническая
по ГОСТ
1382-69
1-й
сорт
31,0
0.4
0,1
0,006
2-Й
сорт
27,5
0.8
03
0.01
Синтетическая техническая
по ГОСТ 857-6
марка марка
А I Б
р
А
р
Б
35,0-
38,0
0,005
0,003
0,005
0,02
31,6
0,00В
0,003
0.005
0.02
марка
31.0
0.03
0.02
0.01
0.15
по ГОСТ 5.1691-72
(аттестованная про-
продукция!
марка
А
35,0
0,005
0,003
0,0001
0,003
0,016
0,0005
марка
33,0
0,005
0,003
0,0001
0,003
0,015
0,0005
марка
В
32.0
0,008
0,008
0,0002
0,008
0,08
0,0005
Очистка разбавленных растворов соляной кислоты (до 5 М) от
соединений железа может быть произведена с помощью анионооб-
менной смолы, которую регенерируют промывкой водой 18. Очистку
концентрированной технической соляной кислоты (с концентра-
концентрацией больше 32% НС1) от ионов Fe , Fe + и SO4 — предложено про-
производить катионообменной смолой, приготовленной на основе фе-
фенола 19.
Показана возможность очищать соляную кислоту от железа
экстракцией бутилацетатом — при содержании Fe(III) 10—25 г/л
степень извлечения его превышает 99,9 °/о 20. Летучие примеси можно
выдувать из соляной кислоты воздухом.
Соляную кислоту транспортируют в стальных гуммированных
цистернах и бочках и в фаолитовых контейнерах, а также в стек-
стеклянных бутылях емкостью не более 40 л. Бутыли помещают в пле-
плетеные из прутьев корзины или деревянные обрешетки, выложенные
соломой или древесной стружкой. Хранят соляную кислоту в сталь-
стальных гуммированных резервуарах, а также в резервуарах, защищен-
защищенных' фаолитом и винипластом21. Использование гуммированных
цистерн и резервуаров сильно упрощается при осуществлении вул-
вулканизации обкладки без давления при низкой температуре22.
В отдельных случаях для транспортировки и хранения HCI, а
также для санитарных целей может представить интерес поглоще-
поглощение хлористого водорода сульфатом меди или свинца, из которых
он потом выделяется при нагревании23.
870
Гл. XI. Соляная кислота
К техническому сульфату натрия, получаемому в соляно-суль-
фатном производстве, по ГОСТ 1363—47 предъявляются следую-
следующие требования (в %):
I сорт И сорт
Na2SO4, не менее 95 91
H2SO4, не более 1,5 3,5
NaCl » » . . 1,2 3,5
Fe » » . . . 0,2 0,25
Не растворимый в воде остаток, не более ... 0,3 0,8
ИНГИБИРОВАННАЯ СОЛЯНАЯ КИСЛОТА
Некоторые органические вещества, добавленные к соляной кис-
кислоте, сильно тормозят растворение ею металлов. Такие вещества
называются ингибиторами243. Чем концентрированнее кислота,
тем с меньшей скоростью она растворяет металл в присутствии не-
незначительного количества ингибитора, но тем более стойким дол-
должно быть вещество, применяемое в качестве ингибитора. К числу
распространенных относятся ингибиторы ПБ — полимеры бутил-
имина. Тормозящее действие ингибиторов объясняется адсорбцией
органического вещества на поверхности .металла, в результате ко-
которой образуется защитный слой, повышающий перенапряжение
выделения водорода на металле. Большинство ингибиторов прояв-
проявляет свое защитное действие ниже 70—80°30. Представляют инте-
интерес ингибиторы, стойкие при более высоких температурах (до
100—110°); к ним относятся, например, уротропин, акридин, «пе-
нореагент» ПР-1 и др.31 Так как стойкость ингибиторов в концен-
концентрированной и горячей кислоте невелика, то ингибированную кис-
кислоту выпускают с концентрацией 19—20% НС1 в летнее время и
до 25% НС1 зимой. Она содержит 0,8—1 % ингибитора ПБ-б и
0,01—0,015% соли "мышьяка (в пересчете на As), усиливающей
действие ингибитора. Содержание свободного хлора не должно
превышать 0,1%, а солей железа — 0,01%. Цвет кислоты — светло-
коричневый. Процесс приготовления ингибированной кислоты за-
заключается в следующем: кислоту разбавляют водой, добавляют
раствор Na3As03 или AsCl3 и смешивают с раствором ингибитора
ъ кислоте.
Ингибированная кислота растворяет сталь в десятки и в сотни
раз медленнее, чем неингибированная. Средняя скорость растворе-
растворения стали в ингибированной кислоте не превышает 0,15 е/(м2-ч),
что соответствует уменьшению толщины стенок резервуара всего на
0,16 мм в год. Однако пары ингибированной кислоты разрушают
не защищенную от коррозии сталь. Ингибиторы не оказывают за-
защитного действия также на линии раздела фаз (жидкой и газо-
газовой) ; относительно менее устойчивы и сварные швы. В этом отно-
отношении гуммированные емкости имеют преимущества.
Применение ингибированной кислоты особенно удобно, когда
оно не связано с необходимостью растворять металлы; в последних
Получение хлористого водорода
371
случаях ингибитор предварительно должен быть разрушен на-
нагреванием кислоты. В процессах травления ингибированная
кислота легко (с такой же скоростью, как и неингибированная)
очищает поверхность черного металла от ржавчины, окалины, на-
накипи и других отложений, растворимых в соляной кислоте; в от-
отсутствие ингибитора этот процесс может сопровождаться разруше-
разрушением металла, особенно в местах клепанных и сварных швов изде-
изделий из углеродистых и некоторых легированных сталей.
Ингибированную соляную кислоту перевозят в стальных желез-
железнодорожных цистернах и в автоцистернах, окрашенных изнутри
химически стойкой эмалью ХСЭ-93 в три слоя и лаком ХСЛ-93 в
два слоя. Ими же окрашивают хранилища ингибированной кис-
кислоты.
ПОЛУЧЕНИЕ ХЛОРИСТОГО ВОДОРОДА
Сульфатный способ
Сырье
Сырьем для получения хлористого водорода и сульфата натрия
служат поваренная соль (обычно измельченная каменная соль —
бузун) и купоросное масло — 92—93%-ная серная кислота. Менее
концентрированную серную кислоту не применяют, так как в этом
случае хлористый водород был бы чрезмерно разбавлен парами
воды, что затруднило бы получение концентрированной соляной
кислоты. Применение крупнозернистой выварочной соли предпо-
предпочтительнее вследствие ее пористости — она легко пропитывается
кислотой с образованием однородной массы. Однако выварочная
соль содержит переменное количество влаги, что затрудняет дози-
дозировку сырья и регулирование температурного режима печей. Ка-
Каменная соль характеризуется постоянной влажностью, но она бо-
более загрязнена примесями CaSO4, РегОз и другими (см. гл. III),
переходящими в сульфат натрия. Помимо этого, применение камен-
каменной соли связано с необходимостью ее измельчения и более интен-
интенсивного перемешивания с серной кислотой 34.
Физико-химические основы взаимодействия
хлористого натрия с серной кислотой
Реакция между хлористым натрием и серной кислотой
2NaCl + H2SO4 = Na2SO4 + 2НС1
— эндотермическая. При применении серной кислоты концентра-
концентрацией меньше 100% расход тепла на реакцию увеличивается за счет
дегидратации серной кислоты. В табл. 31 приведены значения теп-
теплового эффекта АИ и изменения изобаj ого потенциала AZ для
372
Гл. XI. Соляная кислота
некоторых реакций взаимодействия NaCl с серной кислотой и кис-
кислыми сульфатами натрия при различных температурах88.
ТАБЛИЦА 31
Значения ЛЯ и AZ реакций взаимодействия хлористого натрия с серной
кислотой и кислыми сульфатами натрия
Получение хлористого водорода
373
№
1
2
8
4
5
6
Реакция
2NaCl + H2SO4 (ж., 100%)~Na2SO4 + 2HCl (г.)
2NaCl + H2SO4 (ж., 93%) =
- Na2SO4 + 2НС1 (г., 0,83 ат) + 0,41Н2О (г., 0,17 ат)
NaCl + H2SO4 (ж., 100%) =
= NaHSO4 + HCl (г., 1 ат)
NaCl + H2SO4 (ж., 93%) = NaHSO4 +
+ НС1 (г., 0,71 от) + 0,41Н2О (г., 0,29 ат)
NaCl + 2NaHSO4 = Na8H(SO4J + НС1 (г.)
NaCl+Na3H(SO4J = 2Na2SO4 + HCl (г.)
t, °c
25
227
25
100
227
327
25
186
25
186
25
138
144
186
2Б
127
160
227
270
AH,
кал/моль.
12 130
9 615
19 560
18 370
16 080
. 18 960
-950
-2Б80
6 400
4100
10 450
9840
9 800
9 580
15710
15 240
15 260
15 440
15 690
Ъ-Z.
кал/моль
-3 120
-12 820
-290
-Б 190
-13 020
-18 670
-6 990
-9 960
-4 000
—9 150
2 930
170
0
-950
4860
1270
120
-2 230
-3760
Взаимодействие хлористого натрия с серной кислотой начи-
начинается даже при 0° с выделением в газовую фазу почти безводного
НС1, но быстро прекращается; жидкая фаза представляет собой |
раствор образовавшегося сульфата натрия в серной кислоте. При
нагревании реакция возобновляется — одновременно с хлористым
водородом удаляется водяной пар вследствие дегидратации серной
кислоты. Обезвоживание жидкой фазы облегчается тем, что рас-
растворение образующегося сульфата натрия в серной кислоте повы-
повышает равновесное давление водяного пара.
В системе Na2SO4--H2SO4—Н2О при 25° существуют следую-
следующие твердые фазы: Na2SO4 • !0Н2О, Na2SO4 • 4,5Н2О, Na2SO4,
NasH(SO4J> NaHSO4-H2O, NaHSO4 и NaH3(SO4J-H2O. При 0°
существуют эти же фазы и H2SO4-H2O; NaH3(SO4J • Н2О нахо-
находится в метастабильном состоянии, имеется метастабильная фаза
Na2SO4-5,5H2O36. Однако поскольку реакцию с хлористым нат-|
рием ведут при высокой температуре и используют концентриро-
концентрированную серную кислоту, содержащую очень мало воды, вскоре
удаляющейся в газовую фазу, практически процессы идут в без-
безводной системе. В зависимости от температуры реакционной массы
образующийся сульфат натрия может полностью оставаться в
жидкой фазе или частично кристаллизоваться из нее в виде кис-
кислых солей: 2Na2SO4-9H2SO4, Na2SO4 • 2H2SO4, Na2SO4 • H2SO4 (би-
(бисульфат или гидросульфат натрия NaHSO4) и 3Na2SO4 • H2SO4
[тринатрийгидросульфат Na3H(SO4J]. Нейтральный сульфат кри-
кристаллизуется только в заключительной стадии процесса37.
На рис. 114 дана диаграмма растворимости в двойной системе
H2SO4—Na2SO437. Молярные проценты Na2SO4 (верхняя шкала)
численно равны степени превращения NaCl в Na2SO4. Раствори-
Растворимость сульфата натрия в серной кислоте сильно возрастает, начи-
начиная от 4% (вес.) при 0° до 59% при 186°. При повышении темпе-
температуры до 270° растворимость увеличивается до 67%.
Во избежание преждевременного затвердевания реакционной
массы необходимо, чтобы температура на каждой стадии процесса
была не ниже той, при которой исчезает жидкая фаза. Реакция в
сульфатных печах протекает с высокой интенсивностью, если тем-
температурные условия не допускают значительной кристаллизации
промежуточных кислых сульфатов натрия. Так, если успела проре-
прореагировать третья часть хлористого натрия, а температура при этом
еще не достигла 109°, вся масса затвердевает в смесь кристаллов
NaCl и Na2SO4 • 2H2SO4. Когда в некоторой зоне печи, где темпе-
температура ниже 186°, NaCl прореагировал на 50%, там будут нахо-
находиться в твердом состоянии хлористый натрий и бисульфат нат-
натрия— жидкая фаза будет отсутствовать. Если NaCl прореагировал
на 75% и температура будет ниже 270°, реакционная масса пол-
полностью затвердевает, образуя конгломерат, состоящий из остатка
NaCl и NasH(SO4J. Последний особенно часто образуется в суль-
сульфатных печах. Выше 270° тринатрийгидросульфат инконгруэнтно
плавится, и освобождающаяся серная кислота реагирует с остав-
оставшимся хлористым натрием —¦ появляется кристаллический ней-
нейтральный сульфат натрий.
ТАБЛИЦА 32
Примерное содержание NaCl и Na2SO4
в реакционной массе при различной степени
разложения NaCl
NaCl, вес. К
25,2
22,0
9,3
Б,7
2,8
Na2SO4, вес. К
38,3 ..
61,0
81,7
86,7
90,7
Степень
разложения NaCl,
%
55,6
69,6
88,0
92,6
96,4
374
Гл. XI- Соляная кислота
Изменение содержания NaCl и Na2SO4 в реакционной массе и
степень разложения хлористого натрия по мере взаимодействия его
с серной кислотой показаны в табл. 32 37.
Реакции, рассмотренные в табл. 31, являются обратимыми, т.
газообразный хлористый водород может вытеснять серную кислот
из нейтрального и кислых сульфатов. Равновесное давление НС1
мол. %
О Ю 20 30 40 50 60 70 8090100
В 50
ЬОО
ййП
иоо
&J50
Jt dUV
«J OKfi
t200
«;
*- 150
100
60
0
1 /
/
—I—
4
с
С/
a
о
t~
a
Ss
IT
k
щ
1
i
i
/
/,
о*
in
Ж
CO
w-
z,
1
/
1
1
Г 2/u-
46"
ей
Мол.%
SD 75
О 10 20 30 40 50 60 70 SO 30100
H2SO4 вес. % Na2SO4
Рис 114. Политерма растворимости в си-
системе H2SO4—Na2SO4.
BD ВО K2SO*
Вес.%
Рио. ПБ. Политерма раство-
растворимости в системе
H2SO4—K2SO4.
системе NaCl+2NaHSO4 (реакция б) вычисляется по формуле
lgP =. — 2241,27м+5,37; оно равно при 25° 0,007 ат, при 138° 0,83 ат,
при 144° 1 ат, при 186° 3,09 ат. Равновесное давление НС1 в си-
системе NaCl—Na3H(SO4J (реакция 6):
t, °С
26
Р, ат 27-10"
127 160 227 270
0,202 0,87 9,4 32,6
Получение НС1 взаимодействием H2SO4 с КС1, в отличие от
NaCl, требует несколько более высоких температур (на 100—150°)
для своего завершения. На рис. 115 приведена политерма системы
H2SO4 — K2SO438'39. Для полного разложения остатков жидкой^
фазы в этой системе требуется нагреть ее до 700—750°.
Получение хлористого водорода
376
Сульфатные печи
40-48
Реакцию между поваренной солью и серной кислотой осуществ-
осуществляют в механических или во вращающихся печах. Механическая
сульфатная печь (рис. 116) представляет собой муфель, под (чаша)
12 и свод // которого сложены из фасонных огнеупорных и кисло-
кислотостойких шамотных плит. Под муфелем и над ним расположены
дымоходы. Толщина плит пода 120 мм; свод муфеля для облегче-
облегчения передачи тепла к реакционной массе делают более тонким —
30 мм. Внутреннюю кладку печи и топки выполняют из шамотного,
а наружную — из простого кирпича. Под печью расположен привод
мешалки. Наиболее распространены печи с диаметром муфеля
4,25 м; длина и ширина печи равны 5,7 м, высота 5,2 м; поверх-
поверхность нагрева свода 14 м2, реакционный объем 1,8 м3. Поваренная
соль через загрузочную воронку непрерывно поступает в центр
муфеля с помощью шнекового питателя 8. .Туда же по трубе по-
подается купоросное масло. Оно поступает в укрепленный на головке
вала и вращающийся вместе с ним распределитель 13 кислоты —
«гусек», имеющий форму чайника, через носик которого сливается
в муфель. Перемешивание поваренной соли и кислоты и передви-
передвижение реакционной массы по поду муфеля от центра к периферии
производится укрепленной на чугунном валу 15 мешалкой с че-
четырьмя плечами, расположенными крестообразно, на каждом из
которых имеется по два (всего восемь) термосилидовых или чугун-
чугунных гребков с керамическими41 или карборундовыми наконечни-
наконечниками (ножами, зубьямиL2. Вращение вала @,75—1,25 об/мин)
производится через зубчатую передачу электромотором мощностью
4,5 кет при диаметре муфеля 4,25 м и 6,8 кет при диаметре муфеля
5,5 м.
Обогрев муфеля, температуру в котором поддерживают в пре-
пределах 500—550°, производят дымовыми газами, имеющими темпе-
температуру 950—1100°. Газы поступают из топки /, примыкающей к
муфельной печи. Дымовые газы уходят из-под чаши муфеля с тем-
температурой ~700° через два борова. Образовавшийся в муфеле
сульфат натрия попадает через люк 17 в холодильно-размольный
барабан 18 или в трубчатую шаровую мельницу с водяной рубаш-
рубашкой. Измельченный и охлажденный сульфат натрия ссыпается на
транспортер (или в вагонетку) и отправляется на склад. При нор-
нормальном режиме сульфат натрия перед выходом из печи имеет
лимоняо-желтый цвет, в отличие от сероватого цвета, характерного
Для «сырого» сульфата, образующегося при низкой температуре
в муфеле. Уходящий из муфеля хлористоводородный газ имеет
температуру 375—400°. Теоретический состав этого газа при ис-
использовании для разложения поваренной соли 93%-ной серной
Кислоты — 83% НС1 и 17% водяного пара; в сухом же газе дол-
должно содержаться 100% НС1. Однако вследствие того, что внутри
37в
Гл. XI. Соляная кислота
Jt3L
Дымовые газы
Рис. 116. Механическая сульфатная печь (вертикальные разрезы):
1 —шахтная топка; 2 —загрузочная коробка для топлива; 3-колосшщовая решетка;
4 — зольнНк; 5 —топочный порог; 6 — канал для вторичного воздухе! 7 — загрузочная
воронка для соли; 8 — загрузочный шнек для соли; 9-солевая труба; 10-нислотнай
труба; 11 — свод Муфеля; 12 ~ под муфеля; 13 — питатель-распределитель кислоты («гусек»
нли «чайник»); 14*-головка ^ешалки; 75 — вал мешалки; 16 — коронная шестерня при-
привода мешалки; 17 — люк для выхода сульфата натрия; 18 - холодильно«размольный
барабан для сульфата; 19 — труба для отвода хлористого водороде.
Получение хлористого водорода
377
муфеля поддерживают вакуум 1—2 мм вод. ст. (во избежание про-
проникновения газа в атмосферу цеха), происходит подсос в муфель
воздуха и дымовых газов и фактическая концентрация НС1 в сухом
газе колеблется на уровне 30—40%. При хорошем уплотнении му-
муфеля концентрация НС1 в газе может быть повышена до 50% и
выше43. Газ из сульфатной печи загрязнен парами серной кислоты,
сульфатной пылью, летучими соединениями мышьяка, переходя-
переходящими из серной кислоты.
Производительность сульфатной механической печи зависит от
размеров муфеля, от числа оборотов вала, температуры в муфеле
и других условий режима работы. При диаметре муфеля 5,5 м печь
дает до 12—14 т, а при диаметре муфеля 4,25 м 9—Юг сульфата
натрия в сутки. Выработка сульфата натрия составляет 0,59—0,64 г
на 1 т 27,5 °/о соляной кислоты. Суточная производительность 1 м2
пода печи достигает 500—540 кг перерабатываемой поваренной
соли. При замене шамотного свода муфеля карборундовым, имею-
имеющим большую теплопроводность, производительность сульфатной
печи возрастает примерно на 30% 44 (по другим данным, на
35—40%). Аналогичные результаты дает и свод, выполненный из
ферросилициевых плит.
Механические сульфатные печи работают в очень жестких усло-
условиях, — детали их подвергаются действию агрессивной среды при
высокой температуре. Это вызывает необходимость их ежегодной
остановки на ремонт.
Муфельный свод работает до 10 лет4Б. Под муфеля перести-
перестилают чаще — он подвергается механическому износу под влиянием
работы гребков и периодических чисток печи от сульфата натрия
(через 2,5—3 месяца) и может служить без ремонта до трех лет46.
Недостаточно хорошие условия теплопередачи через стенку му-
муфеля механической печи не обеспечивают интенсивного протекания
последней стадии процесса — превращения тринатрийгидросуль-
фата в сульфат, требующего высокой температуры. Интенсивно
протекает лишь образование тринатрийгидросульфата, не требую-
требующее температуры выше 200—250°. Процесс можно значительно ин-
интенсифицировать, осуществляя в механической печи только первую
его стадию и перенеся вторую в трубчатую вращающуюся печь с
противоточным нагревом массы непосредственно дымовыми газами
до 600—700°. При этом 70—75% НС1 будет выдаваться из муфель-
муфельной печи в виде газа высокой концентрации (выше 50%), а осталь-
остальные. 25—30%—из вращающейся печи, разбавленные продуктами
горения топлива 37. Впервые вращающаяся печь была использована
в соляно-сульфатном производстве именно для второй стадии про-
процесса п.
Получение сульфата натрия и хлористого водорода в одну ста-
стадию во вращающихся трубчатых печах с внутренним нагревом ре-
реакционной массы топочными газами47 осложнено образованием
3Z8
Гл. XI. Соляная кислота
в зоне печи, где реакционная масса имеет температуру 186—270°,
налипающего на стенки твердого тринатрийгидросульфата. Силь-
Сильный прогрев и хорошее перемешивание массы в той зоне печи, где
степень превращения повышается от 50 до 75%, устраняет это на-
налипание. Хлористый водород из вращающихся печей сильно раз-
разбавлен топочными газами — концентрация НС1 в газе около 5%,
что, однако, достаточно для получения стандартной кислоты.
Преимуществом вращающихся печей является их большая про-
производительность— при работе на 93%-ной серной кислоте печь,
имеющая длину 12 м и диаметр 1,5 м, выдает 25 т сульфата в сут-
сутки. Площадь печного цеха, оборудованного вращающимися печами,
в 3—3,5 раза меньше площади цеха равной производительности,
оборудованного механическими муфельными печами.
В США в производстве сульфата натрия и соляной кислоты
получила сравнительно большое распространение вращающаяся
печь типа Лоури. На одном конце эта печь имеет откатывающуюся
нефтяную или газовую топку, на другом, холодном, конце произво-
производится загрузка сырья — поваренной соли и бисульфата натрия.
Холодная часть печи служит для смешения сырья и отделена от
горячей части чугунной пластиной, в центре которой имеется от-
отверстие @,6 м) для прохода газов и прорези у периферии для про-
прохода реакционной массы; в этой части печи выделяется около 25%
хлористого водорода. Реакция протекает в основном при высокой]
температуре в горячей части печи, что предотвращает налипаний
больших количеств материала на стенки печи. Для получения ка]
чественного, т. е. рыхлого и пористого сульфата натрия темпера |
туру в горячем конце печи поддерживают выше 600°, но ниже точку
плавления сульфата, т. е. 890°4S.
На выработку 1 г 27,5%-ной соляной кислоты и соответствую]
щего количества сульфата натрия затрачивается: 0,47—0,53 т солу
(97% NaCl), 0,40—0,42 г серной кислоты (93% H2SO4); pacxo.d
условного топлива G000 ккал/кг) в механических муфельных ne-J
Чах 0,19—0,22 г (во вращающихся печах 0,12 т), электроэнергии-
~43 кет • ч, воды ~ 14 мъ.
При переработке в механических сульфатных печах хлористого
калия на 1 т 100%-ного НС1 в виде соляной кислоты получают 1,22 т
К2О в виде сульфата калия; при этом расходуется: 2,21 г хлори-'
стого калия (95% КС1), 1,62 г серной кислоты (98% H2SO4), 0,665 г
мазута, 0,67 мекал пара, 105 кет -ц электроэнергии. При пере-
переработке хлористого калия происходит более сильная коррозия
печей (муфеля), чем при работе с NaCl.
Предложен видоизмененный сульфатный способ получения НС1.
Он заключается в смешении при температуре ниже 150° NaCl с
H2SO4, например, в шнековом смесителе с греющей рубашкой, в
молярном отношении 2:1с целью получения эквимолекулярной
смеси NaCl и NaHSC>4, образование которой заканчивается в
Получение хлористого водорода
879
течение 5 мин. Затем эту смесь вводят на слой расплавленного
сульфата натрия толщиной 1—1,2 м, находящийся в вертикальной
печи, где температура (до 1100°) поддерживается с помощью элек-
электрической дуги. Хлористый водород непрерывно отводят сверху, а
снизу выгружают Na2SC>4 и гранулируют его. Тепло, выделяющееся
при охлаждении плава Na2SO4, используется для проведения пер-
первой стадии процесса — образования бисульфата натрия49.
Способ Гаргривса-Робинзона
По этому способу60 сульфат натрия и малоконцентрированный
хлористый водород (8—10% НС1) получают из поваренной соли,
содержащей ~0,2% окиси железа, являющейся катализатором,
по суммарной реакции:
4NaCl (тв.)-+ 2Н2О (г.) + 2SO2 (г.) + О2 (г.) = 2Na2SO4 (тв.) + 4НС1 (г.) + 99 кшл
В начале XX в. этот способ довольно широко применяли в За-
Западной Европе, особенно в Англии. По этому способу увлажнен-
увлажненную noBapenHvio соль с добавкой небольшого количества сульфата
железа и глины прессуют для получения пористых брикетов с
соломой и опилками. Последние потом выгорают, образуя пори-
пористую массу. Просушенные брикеты загружают в несколько чугун-
чугунных котлов — камер, имеющих высоту 4—5 м, диаметр 5—6 м н
емкость 50 т каждая, обогреваемых снаружи топочными газами.
Через камеры последовательно проходит смесь сернистого газа с
воздухом и водяным паром. Реакционная масса прогревается до*
350°, затем начинается выделение тепла экзотермической реакции
и дальше процесс протекает самостоятельно. Процесс ведется при
оптимальной температуре в наиболее горячей зоне около 540° и
проходит очень медленно — в течение 15—20 суток; выход суль-
сульфата достигает 90—97%. Значительного повышения температуры
сверх оптимальной не допускают, так как уже при 640° в системе
NaCl—Na2SC>4 появляется эвтектический расплав, содержащий
32,6% NaCl. Корка спекшейся массы вокруг расплава препят-
препятствует проникновению газов к непрореагировавшей соли и в этом
случае из камеры вместе с сульфатом натрия выгружают глыбы,
содержащие много NaCl. Периодически из камер выгружается
сульфат и они вновь загружаются брикетами соли. При этом осу-
осуществляется противоток — свежая газовая смесь поступает в ка-
камеру с наиболее отработан-ной массой и выходит из вновь загру-
загруженной камеры.
Процесс очень громоздок. Экономичность этого способа опре-
определялась дешевизной сырья — использованием больших количеств
отбросных сернистых газов медеплавильных и других метал-
металлургических заводов. В настоящее время этот способ почти не
880
Гл. XI. Соляная кислота
применяется. В некоторых случаях его используют для получения
сульфата калия Б1.
Раньше считали52, что образование сульфата натрия при суль-
фатизации хлористого натрия влажной смесью сернистого газа
с воздухом идет через сульфит натрия
502 + Н2О + 2NaCl = Na2SO3 + 2HC1
¦быстро окисляющийся кислородом воздуха в сульфат, и предпо-
предполагали, что образование сульфита лимитирует общую скорость
процесса. Позднее было доказано63, что вначале происходит окис-
окисление SOfi в БОз, который переводит хлорид в сульфат:
503 + Н2О + 2NaCl = Na2SO4 + 2HCI
Это позволяет значительно интенсифицировать процесс путем
добавки к поваренной соли катализаторов, ускоряющих окисление
§Ог- Лучшим катализатором в этих условиях является окись же-
железа. Так, добавка к NaCl 1% колчеданного огарка сокращает
длительность процесса с 15—20 суток до 1—1,5 ч при 95—96%-ном
выходе сульфата натрия. Проведение процесса с увеличенным ко-
количеством катализатора позволяет значительно рационализиро-
рационализировать старый способ, заменив громоздкую аппаратуру более про-
производительной. В условиях СССР, располагающего большими ко-
количествами отбросных сернистых газов, такой модернизованный
¦способ может оказаться экономичным для получения сульфата
калия, являющегося ценным удобрением, качество которого не
снижается от содержания в нем небольших количеств соединений
железа.
Этот процесс может быть осуществлен в печи со взвешенным
слоем. Горячие газы, содержащие SCb, Ог и Н2О, пропускают че-
через слой хлорида со скоростью 0,3—1,5 м/сек, достаточной для
уноса частиц образующегося сульфата калия. Уносимую сульфат-
йую пыль отделяют от газа, содержащего НС1, до его охлаждения,
.а часть газа возвращают в нижнюю зону печи. При 600° степень
превращения КС1 в K2SO4 достигает 98% 54-Б5. Может представить
¦интерес и видоизменение этого процесса при осуществлении его
в отсутствие водяного пара
2КС1 +SO2 + О2 =-KjSO4 + С12
с получением хлора или хлористого сульфурила56.
Синтез хлористого водорода из элементов
Синтез из элементов дает концентрированный хлористоводо-
хлористоводородный газ, содержащий 80—90% и больше НС1, легко поддаю-
поддающийся сжижению, а поглощение его дистиллированной водой поз-
позволяет получать чистую реактивную кислоту, концентрация кото-
Полученив хлористого водорода
38!
рой при необходимости может достигать 38% 87-в0. Реакция взаи-
взаимодействия хлора с водородом50
Н2 + Cl2 ^=± 2HC1 + 44 ккал
обратима. Константа равновесия реакции
J .2
Ри2 • Pci2
(где/? — парциальные давления компонентов) вычисляется по
уравнению
QRR4
lg Яр = -^г1 - 0,533 lg T + 2,42
где Т—абсолютная температура.
По другим данным61:
18
-0,44 lg T + 2,16
Обозначив через х долю диссоциированного НС1, получим для
константы равновесия реакции выражение
„ 4A-*)=
17« 2,51-10
.427 1,12-10
727 1,34-10
Г7
>—в
с помощью которого вычислены следующие значения х\
t, -с х t,°cx
1227 6,10- ИГ4
1727 0,41-Ю
2227 1,30 • 10~2
Таким образом, распад НС1 на элементы становится заметным
лишь при очень высоких температурах — выше 1500°.
Теплота реакции синтеза, равная при 0° 44 ккал, несколько уве-
увеличивается с повышением температуры:
qt = 44+ 0,001 К ккал
При адиабатическом сгорании стехиометрической смеси хлора
и водорода, имеющей температуру 0°, теоретическая температура
факела пламени равна -~2500°. Практически, вследствие неко-
некоторой диссоциации НС1, температура пламени снижается при-
примерно до 2400°. Избыток в газовой смеси одного из компонентов —
хлора или водорода — еще несколько понижает температуру го-
горения.
При обычной температуре, в отсутствие сильных световых лу-
лучей, реакция образования НС1 из элементов идет очень медленно.
При нагревании смеси хлора и водорода или под действием яркого
света происходит взрыв вследствие цепной реакции62:
С1+Н2=НС1+Н;
и т. д.
S82
Гл. XI. Соляная кислота
Цепная реакция могла бы прерваться в результате таких ре-
реакций: Н + С1 = НС1; С1 + С1 - С12; Н + Н = Н2. Однако вероят-
вероятность этих реакций очень мала, так как концентрация атомов
ничтожно мала по сравнению с концентрацией молекул.
Присутствие кислорода замедляет соединение хлора с водоро-
водородом. По-видимому, имеет место обрыв цепи реакций вследствие
соединения кислорода с атомарным водородом63. Скорость реак-
реакции обратно пропорциональна концентрации кислорода в газовой
смеси; в отсутствие кислорода реакция практически протекает
мгновенно. Зависимость скорости фотохимической реакции от кон-
концентрации компонентов может быть выражена эмпирической фор-
формулой Тона
d [HC1] [С12]2 [Н2]
dx =Л " [О2]{[Н2]+0,1[С12]}
где К — константа скорости реакции, зависящая от. интенсивности
возбуждающего светового луча' или теплового импульса.
Реакции, возбуждаемые световым или тепловым импульсом, но
скорость протекания которых в дальнейшем, в отличие от чисто
фотохимических реакций, зависит от температуры, называются
темновыми.
Считают, что определяющей стадией рассматриваемого тем-
нового процесса является реакция С1 + Н2 = НС1 4- Н. Если обо-
обозначить константу скорости этой реакции К\, а константу диссо-
диссоциации молекул хлора /Ср, то константа скорости образования НС1,
равная К = 'А/Ср • Ки может быть вычислена по эмпирической
формуле64:
/С=1,18-102е
35 370
RT
Скорость реакции зависит также от присутствия катализато-
катализаторов — влаги, твердых пористых тел (губчатой платины, древесного
угля) и некоторых минеральных веществ (кварца, глины и т. п.).
Абсолютно сухие хлор и водород не взаимодействуют между со-
собой. Присутствие следов влаги ускоряет реакцию столь интенсив-
интенсивно, что она может произойти со взрывом. Повышение содержания
влаги сверх 5- 10~5 уже не отражается на скорости реакции.
В производственных установках осуществляется спокойное, не
взрывное горение водорода в токе хлора. Водород подается с из-
избытком 5—10%, что позволяет полностью использовать более цен-
ценный хлор и получить незагрязненную хлором соляную кислоту.
Возможно, однако, вести процесс и при подаче стехиометрических
количеств водорода и хлора или очень малого избытка водорода
(до 1 %); при этом получаемый хлористый водород содержит до
1% С12 и 2% Н2. Очистку газа от хлора можно осуществить, про-
пропуская его через активированный уголь при 450° с объемной ско-
Получение хлористого водорода
ТАБЛИЦА 33
J
Пределы взрывных концентрации
в газовых смесях (в объемн
Температура,
°С
-20
0
20
50
100
4-91,5
3,5-92
3-92,5
3-93
. 3-93
Н2 + о2
4-96
4-96
4-96
4-96
4-97
водорода
. %)
Н2 + воздух
4-72
4-73
4-76
8,7-76
3-80
ростью 260 м3/ч на 1 ж3 угля6Б. Избыток водорода выше 20%
опасен, так как может привести к взрыву в процессе абсорбции
НС1 вследствие образования гремучей сме-
смеси. В табл. 33 приведены пределы взры-
ваемости смесей водорода с хлором, кис-
кислородом и с воздухом (при исходном об-
общем давлении 0,5—1,5 ат) 66.
Температура самовоспламенения смеси
хлора с водородом в воздухе равна 240°.
Сжигание смеси хлора и водорода про-
производится в печах различных конструкций,
представляющих собой небольшие каме-
камеры из огнеупорного кирпича, плавленого
кварца, графита или из металла. Неотъем-
Неотъемлемой частью печей являются горелки для
сжигания водорода в токе хлора. Устрой-
Устройство горелки показано на рис. 117. Она
состоит из двух стальных трубок, вмонти-
рованных в стальной патрон. Хлор подает-
подается по внутренней трубке, водород — по на-
наружной. В наружной трубке горелки укреп-
укреплен жаростойкий динасовый наконечник 4.
Газы, сгорающие в горелке, образуют
длинный факел пламени, температура
которого понижается по длине печи
от 1800—2000° до 800—1200°. Регулировка
подачи газов производится с помощью рота-
ротаметров или шайб с дифференциальными ма-
манометрами и контролируется по анализу от-
отходящего газа.
Простейшими печами являются полые кирпичные камеры, фу-
футерованные внутри огнеупорным кирпичом. При внутренних раз-
размерах камеры: ширина 0,61 м, длина 2,16 ж, высота 0,71 ж, объем
ее составляет 0,94 м3 и производительность — от 3,5 до 7,5 т соляной
Рис. 117. Горелка:
1~трубка для ввода хлора;
2 — трубка для ввода водо-
водорода; з — патрон; 4 — динасо-
динасовый наконечник.
384
Гл. XI. Соляная кислота
кислоты в сутки. Иногда реакционные камеры таких печей запол-
заполняются контактным материалом — графитом, коксом, кварцем,
огнеупорной глиной и т. п.
Несколько большее распространение получили вертикальные
печи, монтируемые из кварцевых труб. Кирпичные и кварцевые
печи работают под небольшим вакуумом во избежание проник-
проникновения хлористого водорода в атмосферу цеха. Недостатком
этих печей является отсутствие условий для хорошего отвода
тепла, вследствие чего газ уходит из них с очень высокой темпера-
температурой.
Наиболее совершенными являются печи, построенные из спе-
специальных или простых сталей, которые достаточно устойчивы, не-
несмотря на содержание в хлористоводородном газе влаги, так как
температура газа выше точки росы. Металлические печи устраи-
устраиваются, как и кварцевые, в форме вертикальных труб, но обла-
обладают значительно большей производительностью 1 м3 объема, так
как отвод реакционного тепла из них через наружные стенки идет
во много раз интенсивнее, чем через стенки печей из силикатных
материалов. В ряде случаев стальные печи снабжаются сна-
снаружи водяными кожухами и охлаждаются проточной водой. Это
позволяет дать на них значительно большую нагрузку. Стальная
печь при диаметре 0,25 м и высоте 2,5 м дает от 6 до 25 т соляной
кислоты в сутки (в зависимости от способа отвода тепла). Кроме
того, стальные печи могут работать под давлением, что увеличи-
увеличивает концентрацию НС1 в газе, вследствие устранения подсосов
воздухаБ9. При водяном охлаждении температура воды в кожухе
стальной печи не должна быть ниже 90—95°, т. е. ниже точки росы
газа, во избежание конденсации из него влаги на стенке печи679.
Выходящий газ обычно имеед температуру не ниже 400°. При со-
содержании в газе ~5°/о влаги не следует допускать дальнейшее
его охлаждение в отводных железных трубах ниже 200°. При не-
необходимости охлаждать газ до 30—60° его транспортируют по
трубам из пропитанного фенольной смолой графита. При необхо-
необходимости получить сухой хлористоводородный гае его сушат в
стальной насадочной колонне концентрированной серной кислотой,
причем он охлаждается до ~250°59.
Опубликованы патенты, предусматривающие использование по-
подаваемого в печь избыточного водорода из отбросного газа после
абсорбции НС1, путем инжектирования этого rasa свежим водо-
водородом в добавочную печь70.
В СССР применяют двухконусные печи с естественным воздуш-
воздушным охлаждением (рис. 118). Благодаря тому, что форма печи
близка к форме факела пламени, обеспечивается равномерная
тепловая нагрузка стенок печи. Корпус печи сварен из 8-милли-
8-миллиметровой листовой стали (Ст. 3). Нижняя цилиндрическая часть
корпуса и съемное днище футерованы огнеупорным кирпичом.
Получение хлористого водорода
383
ФИ00/6
Верхний конец печи прикрыт зажатой между фланцами предохра-
предохранительной мембраной из листа паранита или асбеста, которая вы-
выталкивается в случае внезапного взрыва газовой смеси.
Водород, освобожденный от влаги охлаждением его до 25—
30°, последовательно проходит гравийный пламягаситель, водоот-
водоотделитель, гравийный искрогаситель, обратный клапан и поступает
в печь. Скорость подачи газов в печь составляет, в
зависимости от нагрузки, 40—120 мъ/ч хлора и
43—130 м3/ч водорода, что почти в 1,5 раза выше
скоростей газовых потоков в цилиндрической печи.
Давление хлора и водорода в подводящих трубо-
трубопроводах должно быть не меньше 800 мм рт. ст.
В печи поддерживают давление около 600 мм вод. ст.
Температура наружной стенки печи достигает
400°, а температура отходящего газа в газопро-
газопроводе перед абсорбционной колонной (см. ниже)
должна быть не больше 250°. Автоматическая ре-
регулировка подачи газов обеспечивает нужное со-
соотношение между хлором и водородом, поступаю-
поступающими в горелку. Помимо этого, процесс контроли-
контролируют по цвету пламени, который должен быть
молочно-белым. В случае избытка хлора, что не-
недопустимо, цвет пламени приобретает зеленоватый
оттенок, при большом избытке водорода — го-
голубой.
Технические газы, используемые для синтеза
НС1, должны быть по возможности чистыми. Кон-
Концентрация водорода не должна быть ниже 95%, а
концентрация хлора — не ниже 90%. В получае-
получаемом хлористоводородном газе содержание НО не
должно быть меньше 80%. Производительность
описанной печи достигает 10 т 100%-ного хлори-
хлористого водорода в сутки. Расход газов на 1 т
31%) -ной соляной кислоты составляет: хлора
A00%-ного) 0,305—0,312 г; водорода A00%-ного)
ПО—112 мъ; расход электроэнергии 35 квт-ч, воды
20 м3, пара 0,014 мгкал.
Представляет интерес осуществление синтеза
НС1 из элементов в графитовой горелке, погру-
погруженной в соляную кислоту, находящуюся в сталь-
стальном 'резервуаре, футерованном кислотоупорным
Рис. 118. Схема
стельной печи
для синтеза
хлористого во-
водорода:
1 — корпус печи;
2~ гспелка; 3 — за-
запальный люк;
4 — смотровый
штуцер с кварце-
кварцевым стеклом;
5—il't цер для от-
отвода > лэристого
•водорода. 6 — пре-
дай
#ите^ы{ый
лист; 7 — опорная
К-нструкцнр.
кирпичом по слою резины. Температуру кислоты поддерживают
несколько ниже равновесной, соответствующей кипению кислоты
при данном парциальном давлении НС1 в газе. Теплота горения
и растворения отводится путем циркуляции кислоты через во?
Д*цюй холодильник. Непрореагировавшие водород и хлор перед
. Е. Позин
386
Гл. XI. Соляная кислота
отводом из аппарата разбавляют воздухом во избежание образо-
образования взрывчатой смеси. Недостатком этого способа является
большой расход воды на охлаждение кислоты59.
Получение хлористого водорода из хлора и водяного пара
в присутствии угля
При пропускании смеси хлора с водяным паром через слой
раскаленного угля образуется хлористый водород по реакции:
2С12 + 2Н2О(пар) + С = 4НС1 + СО2 + 67 ккал
Ввиду большой экзотермичности реакции она протекает без
внешнего подвода теплап-72. В отходящих газах содержится
65—78% НС1, 15—18% СО2, 2—4% СО. При выходе из реакци-
реакционной зоны газы могут содер-
содержать также небольшие коли-
количества фосгена (СООг), кото-
который затем, гидролизуясь во-
водяным паром, превращается в
СО2 и НО. С повышением
температуры степень превра-
превращения С12 в НС1 увеличивает-
увеличивается. Она зависит также от сор-
сорта угля и от наличия в нем
примесей, являющихся ката-
катализаторами, например окиси
железа. Как видно из рис. 11973,
для достижения 95—97%-ного
160
200 300 400 500
Температура,'С
Рис. 119. Зависимость степени превра-
щения С12 в НС1 от температуры:
i-древесный уголь; 2-кокс; з-древесный
ью окнси желе
ванный уголь.
600 выхода НС1 при работе с ак-
активированным углем, с коксом
и с древесным углем, содержа-
содержащим примесь Fe2O3, в реакци-
реакционной зоне должна поддержи-
- I».
р
уголь с примесью окнси железа; 4 — активиро- »«иь1.и lcmucjJdiypd tw 4OV ,
й б
p
при работе с древесным углем
550—600°.
Расход кокса или древесного угля составляет 10—11% от веса
получаемого хлористого водорода. Процесс осуществляют в ге-
генераторах шахтного типа, построенных из стали и футерованных
внутри кислотоупорным и огнестойким кирпичом.
По сравнению с прямым синтезом НС1 из элементов, угольный
метод обладает крупным недостатком — здесь получается менее
концентрированный хлористый водород, причем не сухой, а с при-
примесью вдуваемого в генератор водяного пара. Это вызывает кор-
коррозию металлических деталей генератора, в частности загрузоч-
загрузочного устройства. Поэтому в СССР угольный метод не приме-
применяется.
Получение хлористого водорода
887
Хлористый водород как побочный продукт некоторых
производств
Хлористый водород образуется при хлорировании многих ор-
органических соединений. Этот источник хлористого водорода с раз-
развитием промышленности органического синтеза приобрел главен-
главенствующее значение. Например, в 1965 г. в США было выпущено
1179000 т соляной кислоты A00% НС1), причем ~80% этого ко-
количества было получено в виде отхода от производства органи-
органических хлоропродуктов74. Наибольшие количества хлориетого во-
водорода образуются при реакциях заместительного хлорирования
и гидрохлорирования. Примером может служить получение НС1
при производстве хлорбензола из бензола по реакции:
С12 + С6Н6 = С6Н5С1 + НС1
Еще одним примером, имеющим большое перспективное зна-
значение, может служить хлорирование метана и других алифатиче-
алифатических углеводородов. Хлористый водород образуется в результате
цепной реакции 76: С12 = Cl + Cl; C1 + СН4 = СН3 + НС1; СН8 +
+ С1 = СН3С1 и т. д.
В подобных процессах в большинстве случаев получается кон-
концентрированный хлористый водород. Иногда он значительно раз-
разбавлен воздухом и почти всегда загрязнен органическими веще-
веществами и хлором.
Хлористый водород образуется и при реакциях конденсации в
присутствии А1С1з и РС13 (конденсация фталевого ангидрида с
толуолом и др.), при реакциях фосгенирования и проч. В этих
процессах выделяются небольшие количества НС1, и установки
для его улавливания очень компактны. Так, например, установка
производительностью около 1 т соляной кислоты в сутки занимает
всего 20 м2 площади76.
При сульфохлорировании углеводородов образуется смесь SO2,
НС1 и SO2CI2. Из этой смеси возможно выделить НС1, если до-
добавить к ней хлор в количестве, эквивалентном количеству SO2,
и пропустить ее через активированный уголь (при 20е). При этом
образуется хлористый сульфурил SO2CI2, который отделяют кон-
конденсацией. Затем хлористый водород для очистки от SO2 и SO2CI2
промывают концентрированной серной кислотой 77.
Хлористый сульфурил также может служить концентрирован-
концентрированным источником соляной кислоты (одновременно и серной кис-
кислоты) , так как под действием воды он разлагается на смесь этих
кислот:
SOjGlj + 2Н2О = H2SO4 + 2HC1
При ограниченном количестве воды НС1 выделяется в газо-
газообразной форме. Так как хлористый сульфурлл — жидкость, не
действующая на сталь, то его легко транспортировать в стальных
888
Гл. XI. Соляная кислота
бочках или цистернах к потребителям, нуждающимся в серной и
соляной кислотах.
Предложены различные способы очистки хлористого водорода
от летучих органических веществ. Например, можно, добавив к
загрязненному хлористоводородному газу хлор, а затем водород и
воздух или водяной пар, сжечь такую хлороводородную смесь;
при этом органические примеси также сгорят и останется чистый
хлористый водород78-79.
Другим методом очистки хлористого водорода от органических
примесей является его промывка в скруббере концентрированной
или разбавленной соляной кислотой. В последнем случае кислота
концентрируется за счет хлористого водорода из газа. Очищенный
газ поступает во второй скруббер, где поглощается с образова-
образованием соляной кислоты. Промывную кислоту из первого скруббера
направляют на регенерацию для выделения примесей. Если тем-
температура кипения этих примесей выше температуры кипения азео-
тропйЪй смеси НС1—НгО, то вначале отгоняют хлористый водород,
затем азеотропную смесь. В противном случае вначале отгоняют
примеси с некоторым количеством НС1 и оставляют в жидкой
фазе чистую азеотропную смесь, а выделившийся загрязненный
хлористый водород очищают от примесей поглощением водой с
последующей фракционированной перегонкой полученного рас-
раствора .
Хлористый водород является побочным продуктом и в ряде
процессов неорганической технологии, например, при гидролизе
хлористого магния с целью получения окиси магния (стр. 299),
при некоторых способах конверсии хлоридов в нитраты азотной
кислотой, при переработке хлористого калия и полиминеральных
калийных руд (см. гл. V) на бесхлорные удобрения и пр. На не-
некоторых заводах в США при производстве металлического магния
из хлористого магния в электролизеры загружают не полностью
обезвоженный хлористый магний (приблизительно MgCl2- 1,25Н2О).
На этих заводах 40% хлора выделяется в виде хлористого водо-
водорода, улавливаемого водой с образованием слабой соляной кис-
Лоты81.
Возможно каталитическое хлорирование сероводорода
Н28 +-С12 - S + 2НС1
на катализаторе из окиси алюминия, при объемном соотношении
СЬ : H2S 0,9—0,95. Максимальный выход серы (90%) достигается
при 400° и объемной скорости 20 м3/ч на 1 м3 катализатора82.
Хлористый водород может быть получен при регенерации ам-
аммиака в производстве соды по некоторым новым схемам, например,
путем обработки твердого хлористого аммония расплавленным
бисульфатом натрия 8з. В первой стадии процесс осуществляется
при 220—270°, .причем в газовую.фазу, выделяется НС1; во второй
Получение хлористого водорода
стадии при 330—380° происходит диссоциация образовавшейся в
первой стадии двойной соли с выделением в газовую фазу ам-
аммиака, а оставшийся плав бисульфата натрия возвращают в про-
процесс. Таким образом бисульфат натрия является средой, с по-
помощью которой удается разделить аммиак и хлористый водород,
образующиеся при диссоциации хлористого аммония. Давление
паров (в мм рт. ст.) над NH4C1 в пределах 100—400° может быть
вычислено по уравнению:
0,05223-83486
Оно равно при 209,8е—10 мм рт. ст., 245° — 40 мм рт. ст.,
271,5°—100 мм рт. ст., 316,5° —400 мм рт. ст. и при 337,8° —
760 мм рт. ст. Давление паров NH3 над (NH4JSO4 равно:
2,33 мм рт. ст. при 225°, 10,44 мм рт. ст. при 255°, 22,26 мм рт. ст.
при 280° и 50,84 мм рт. ст. при 300°.
Другой вариант этого способа предусматривает переработку
растворов хлористого аммония с использованием в качестве теп-
теплоносителя органической жидкости с высокой температурой ки-
кипения, пары которой стойки до 500°84. Исходный раствор NH4C1
смешивают с нагретым теплоносителем, в результате чего про-
происходит выпаривание и образуется безводная пульпа, содержащая
20—25% NH4C1 и 80—75% теплоносителя. Пульпу смешивают с
расплавленным бисульфатом натрия и отгоняют из смеси НС1,
затем NH3. Хлористый водород сушат в башне, орошаемой серной
кислотой, и пропускают через низкотемпературный холодильник
для конденсации паров теплоносителя, который, так же как и би-
бисульфат натрия, непрерывно циркулирует в системе.
Еще один способ регенерации аммиака из хлористого аммония
в содовом производстве с получением при этом хлористого водо-
водорода заключается в следующем 85. Сухой хлористый аммоний раз-
разлагают при 335—350° и полученную смесь газов NH3. и НС1 про-
пропускают через нагретый до 500° платиновый или железоокисный
катализатор для разложения аммиака на водород и азот. После
отмывки следов аммиака фосфорной кислотой из газовой смеси
выделяют НС1 сжижением его под давлением 200 ат, а оставшуюся
азото-водородную смесь очищают от следов НС1 окисью кальция
и направляют на синтез аммиака. В результате этого кругового
процесса для производства соды требуются только NaCl и СО2 и
не. образуется никаких отбросов; на 1 г соды получается 0,7 т НС1
(~2г соляной кислоты).
Получение хлористого водорода из КС1 при переработке его
на бесхлорное удобрение — K2SO4 возможно не только по способу
Гаргривса — Робинзона (стр. 379), но и другим путем. При раз-
разложении хлористого калия большим избытком концентрированной
^серной кислоты при сравнительно низкой температуре (~80°)
390
Гл. XI. Соляная кислота
выделяется концентрированный и достаточно сухой хлористый во-
водород, а образовавшиеся кислые сульфаты калия направляются на
разложение фосфата86. Аналогичный способ разработан87 и при-
применительно к хлористому аммонию — с образованием НС1 и би-
бисульфата аммония, который затем используется для разложения
фосфата с целью получения азотно-фосфорного удобрения. В раз-
различных металлургических процессах, в которых никелево-медные,
оловянные и другие руды подвергаются хлорированию в присут-
- ствии водяного пара, выделяется хлористый водород и в качестве
побочного продукта может быть получена соляная кислота.
Хлористый водород образуется при взаимодействии хлоридов
щелочных или щелочноземельных металлов с глиноземистым или
силикатным материалом в присутствии водяного пара при 500—
800°88,89 j^ горючему газу добавляют пар D0%) и обрабатывают
им шихту во взвешенном слое 90.
В связи с проблемой утилизации отбросного хлористого каль-
кальция от производства соды установлена возможность получения
соляной кислоты гидролизом СаС12. При 728° и давлении водя-
водяного пара 1 ат в системе
СаС12 + Н2О
СаО + 2НС1
давление НС1 равно 6,2- 10~4аг, а в системе
СаС12 + Н2О + SiO2 з=± CaSiO3
давление НС1 равно 0,35 ат. При гидролизе Cafcl2-6H2O в смеси -\
с глиной A :2,5 по весу) при 700° и 10-кратном избытке пара вы- ,
ход НС1 составляет 80—90%, а концентрация получаемой кислоты ¦ |
25—28%. Подсчитано, что расход пара на 1 г соляной кислоты со-
составит — 0,25 г91.
Наконец, следует упомянуть о возможности получения газа, |
содержащего 85,7% НС1, взаимодействием сухого хлора с аммиа-
аммиаком при 800° в присутствии СаС12 как катализатора 92:
Реакция происходит самопроизвольно и полностью, но нельзя
допускать смешения СЬ и NH3 при низких температурах, так как
это приводит к образованию сильно взрывчатого треххлористого
азота.
Получение чистого хлористого водорода
Для получения реактивной соляной кислоты в настоящее вре-
время пользуются наиболее чистым хлористым водородом, синтези-
синтезируемым из элементов. При малых масштабах производства доста-
достаточно чистый хлористый водород может быть получен из техни-
технической соляной кислоты, даже основательно загрязненной нелету-
нелетучими примесями, по способу Газенклевера. Этот способ применяли >
Абсорбция хлористого водорода
391
раньше для получения реактивной кислоты. Он заключается во
вливании струи соляной кислоты в концентрированную серную
кислоту (купоросное масло), которая связывает воду, освобождая
выделяющийся хлористый водород. Получающуюся разбавленную
серную кислоту использовали в различных производствах, а хло-
хлористый водород поглощали дистиллированной водой; образую-
образующаяся соляная кислота содержит всего ~0,0015% H2SO493.
Чистую соляную кислоту C8,2% НС1) предложено получать
десорбцией НС1 при 110° из технической кислоты C7% НС1), к
которой добавляют 0,01—0,1% триэтаноламина, связывающего
примеси соединений Fe, Си, А1 и Мп в прочные комплексы, отхо-
отходящие с кубовым остатком B8,2% НС1). Отгоняемый хлористый
водород проходит через насадочную колонну, в середину которой
подают пар, получаемый из дистиллированной воды; пары из
верха колонны пропускают через конденсатор, из которого отби-
отбирают чистую кислоту94.
Чистый хлористый водород получают также, выдувая его воз-
воздухом из технической соляной кислоты. Непосредственно из газа
сульфатных печей получают реактивную соляную кислоту после
очистки этого газа от сернокислотного тумана, SO2, соединений
мышьяка, железа и других загрязнений95. Такую очистку осуще-
осуществляют сначала, промывая газ соляной кислотой, затем пропуская
его через несколько поглотителей с активированным углем и вновь
промывая чистой соляной кислотой.
АБСОРБЦИЯ ХЛОРИСТОГО ВОДОРОДА
Абсорбцию хлористого водорода осуществляют двумя способа-
способами— с отводом и без отвода тепла, выделяющегося при гидрата-
гидратации НС1. В качестве материалов для абсорбционной аппаратуры
используют пропитанную смолой древесину, естественные кисло-
кислотоупорные камни (гранит, песчаник), кислотоупорные керамику и
эмаль, плавленый кварц, графит, графолит и антегмит (АТМ-1 —
теплопроводный материал из графита и феноло-формальдегидной
смолы) и некоторые виды пластических масс (фаолит, винипласт,
полиэтилены, фторопласты, найлон и др.), абсорберы из которых
отличаются легкостью и малыми габаритами9602. Для изготовле-
изготовления кислотопроводов, запорных приспособлений, насосов и т. п.
применяют железокремниймолибденовый сплав (антихлор,
МФ-15) »03, 104_
Абсорбция хлористого водорода с охлаждением
Раньше считали, что для получения концентрированной соля-
соляной кислоты газ, содержащий хлористый водород, перед абсорб-
«щней должен быть обязательно охлажден и сам процесс абсорбции
392
Гл. XI. Соляная кислота
должен вестись при возможно низкой температуре—из абсорбе-
абсорберов должно отводиться тепло, выделяющееся при растворении НС1
в воде. Это убеждение было основано на опасении, что при недо-
недостаточном отводе тепла из абсорберов жидкость в них закипит и
при этом невозможно будет получить кислоту, содержащую более
20,24% НС1, т. е. более концентрированную, чем постоянно кипя-
кипящая смесь хлористого водорода с водой.
Действительно, пользуясь рис. 113 (стр. 365), можно подсчи-
подсчитать, что, например, при получении из НС1 и Н2О 20%-ной соля-
соляной кислоты выделяется 89 ккал на 1 кг раствора. Этого тепла
вполне достаточно, чтобы образующаяся кислота закипела, так
как при ее теплоемкости "-'0,7 ккал/(кг • град) этого тепла хва-
хватило бы для повышения температуры на 127°.
В соответствии с этим абсорбционные установки сооружены
таким образом, чтобы обеспечить наиболее полный отвод тепла
гидратации НС1. Обычно газ вначале охлаждают в длинных га-
газопроводах и керамических холодильниках. Охлаждение совме-
совмещают с осушкой газа, что дает возможность получать более кон-
концентрированную соляную кислоту. Осушку производят в башнях
с насадкой, где конденсируются пары H2SO4 и влага и задержи-
задерживается сульфатная пыль, уносимая из сульфатных печей. В дру-
других случаях газ в башнях высушивают концентрированной серной
кислотой.
Синтетический хлористый водород по выходе из печей синтеза
охлаждают в холодильнике из графолитовых или кварцевых труб.
При охлаждении до 38—40° образуется некоторое количество кон-
конденсата— 36%-ной соляной кислоты105. Охлажденный газ направ-
направляется на абсорбцию водой. Абсорбцию газа осуществляют в фао-
литовых, керамических или футерованных стальных башнях с на-
насадкой. Газ проходит последовательно две башни. Вторую башню
орошают водой, из первой отбирают готовую соляную кислоту.1
Газ по выходе из последней башни выбрасывается в атмосферу:
керамическим вентилятором. При абсорбции газа из сульфатных
печей содержащийся в нем сернокислотный туман проходит через
всю установку и частично конденсируется лишь под действием
центробежной силы хвостового вентилятора, из которого выпу-
выпускается небольшое количество кислоты. Кислота циркулирует в
каждой башне с помощью насосов — центробежных керамических
и др. Тепло, выделяющееся при абсорбции, отводится с помощью
установленных под башнями керамических змеевиков или графо-
графолитовых холодильников, по которым циркулирует кислота. Змее-
Змеевики опущены в резервуары с проточной холодной водой. Для
поглощения хлористого водорода водой значительное распро-
распространение, особенно за рубежом, получили установки, оборудо-
оборудованные кварцевыми и графолитовыми холодильниками и абсор-
абсорберами 106.
Абсорбция хлористого водорода
393
Кварцевые холодильники (рис. 120) представляют собой S-об-
разные трубы (называемые иногда «интегралами») длиной 2 м
с внутренним диаметром 200 мм. Холодильники соединяются по-
последовательно в вертикальной плоскости и орошаются сверху
(снаружи) водой. Абсорберы (рис. 121) соединяются, как холо-
холодильники, и также орошаются снаружи водой. В верхний абсорбер
подается вода (слабая соляная кислота), переливающаяся в на-
находящийся под ним, и т. д.
У сливного конца абсорбера име-
имеется порог, определяющий уро-
уровень жидкости в абсорбере. Газ
впускается в нижний абсорбер и
движется противотоком жидко-
жидкости. На верхней поверхности аб-
абсорбера имеется пережим, спо-
.л .,. ъ \*\
Рис. 120. Кварцевые холодильники. Рис. 121. Кварцевый абсорбер (внешний
вид и разрез).
собствующий лучшему соприкосновению газа с жидкостью. Такие
же абсорберы изготовляются и из графолита.
На рис. 122 изображена схема установки для охлаждения и
абсорбции синтетического хлористого водорода. Газ из печи син-
синтеза, имеющий температуру около 1000°, проходя по газоходу /,
остывает до ~200°, затем входит сверху в секцию холодильни-
холодильников 2. Из последних с температурой около 100° он поступает снизу
в секцию абсорберов 3, после которых промывается водой в не-
небольшой керамической башне 4 с насадкой. Конденсат из холо-
холодильников смешивается с продукционной кислотой, содержащей
32—35% НС1.
Большим преимуществом кварцевых и графолитовых абсорб-
абсорбционных установок является компактность, обусловленная ин-
интенсивным охлаждением. Помимо этого, в кварцевых абсорберах
394
Гл. XI. Соляная кислота
получается чистая кислота, не загрязненная примесями, извлекае-
извлекаемыми из керамики, в частности железом. Недостатком их является
высокая стоимость и малая механическая прочность. Так как ско-
скорость абсорбции НС1 водой очень велика, то число абсорберов, из
которых монтируют абсорбционную установку, определяют, ис-
исходя из условий, необходимых для полного отвода тепла гидра-
гидратации НС1. Для производства 7 г соляной кислоты в сутки доста-
достаточна поглотительная установка из 13 кварцевых холодильников
Вода
Абсорбция хлористого водорода
395
Рис. 125. Схема установки охлаждения н абсорбции хлористого
водорода:
/ — газоход; 2 — холодильники; 3 — абсорберы; 4 — хвостовая башня; 5 —венти-
—вентилятор; 6 — сливная труба; 7 —сливной штуцер для кислоты; Я —слнвная труба
для конденсата.
и 11 абсорберов. Используют и кварцевые абсорберы, имеющие
форму вертикальной трубы — скруббера с насадкой, собранного
из отдельных кварцевых частей66.
Вполне стойкими по отношению к соляной кислоте вплоть до
330° являются тантал и ниобий. Эти металлы хорошо проводят
тепло и легко поддаются механической обработке. В абсорбере из
вертикальной танталовой трубы диаметром 150 мм и высотой 2 м,
помешенной в стальной кожух для охлаждения водой, можно по-
поглотить более 225 кг НС1 в час. Однако тантал (так же как и ни-
ниобий) является дорогим металлом и поэтому танталовые абсор-
абсорберы для хлористого водорода используют пока, по-видимому, на
очень немногих заграничных предприятиях. Но имеются указания,
что, благодаря простоте устройства танталовых абсорберов и боль-
большой продолжительности их службы, капиталовложения и эксплуа-
эксплуатационные расходы на 1 кг 100%-ного хлористого водорода при
использовании танталовых абсорберов ниже, чем при использова-
нии абсорберов из других материалов 107. Описана установка не-
небольшой мощности для получения соляной кислоты из синтетиче-
синтетического хлористого водорода, в которой
из тантала выполнены лишь немногие
детали абсорберов67. Выходящий из
печи синтеза хлористый водород охла-
охлаждают сначала воздухом в стальных
трубах, потом в трубах из графита,
пропитанного фенольными смолами.
Затем по гуммированной стальной
трубе газ поступает' в двухступенча-
двухступенчатый абсорбер (рис. 123). Первая сту-
ступень абсорбера представляет собой
трубу из тонкого гофрированного лис-
листа тантала, помещенную в стальной
кожух — водяную рубашку. Газ здесь
идет прямотоком со слабой соляной
кислотой, поступающей из второй сту-
ступени. Остаток газа, не поглощенный
в первой ступени, поступает во вто-
вторую ступень — керамический абсор-
абсорбер, также снабженный танталовой
трубой с водяным охлаждением и
насадкой. Во второй ступени осуще-
осуществляется противоток газа с посту-
поступающей сверху дистиллированной во-
водой. Отходящий газ разбавляют
воздухом для снижения концентрации
водорода ниже опасного предела и
выпускают в атмосферу.
Абсорбция хлористого водорода из
разбавленных газов, содержащих вла-
влагу (например, из отходящих газов от
выпарки хлористого магния), затруд-
затрудняется образованием тонкодисперс-
тонкодисперсного тумана соляной кислоты, кото-
который абсорбируется с большим трудом.
Разработан метод акустической коа-
коагуляции аэрозоля соляной кислоты из
таких газов, полнота которой, при по-
повышении их относительной влажности
до 97%, достигает 94 %108.
Из . отбросных газов можно извле-
извлекать НС1, пропуская их через колонну с синтетическим цеолитом.
Адсорбционная емкость его в среднем равна 6 г НС1 на 100 г
Долита. Стойкость цеолита к НС1 значительно возрастает после
Рис. 123. Двухступенчатый
абсорбер хлористого водорода:
/ — трубопровод для охлаждающей
воды; 2 — трубопровод для дистил-
дистиллированной воды; 3 н 5 — гофриро-
гофрированные танталовые трубки; 4 — ввод
хлористого водорода в первую сту-
ступень абсорбера; 6 —вывод абгаза из
второй ступени абсорбера; 7—водя-
7—водяная рубашка; 8 — керамический
скруббер (вторая ступень абсорбера);
S —стальной кожух первой ступени
абсорбера; ю — трубопровод для
выпуска кислоты.
Гл. XI. Соляная кислота
Абсорбция хлористого водорода
897
его обработки водным раствором хлорида цинка илн кальция. Де-
Десорбцию НС1 осуществляют при нагревании до 300—800° или про-
дувкои сухим воздухом ша.
Адиабатическая абсорбция хлористого водорода
Наиболее совершенным является способ абсорбции хлористого
водорода без отвода тепла, называемый также способом Гаспа-
ряна110-111, доказавшего ошибочность мнения о невозможности по-
получить при этом соляную кислоту концентрированнее 20,24%.
Принцип, лежащий в основе способа, заключается в следующем.
Если пропускать в воду хлористый водород, то за счет теплоты
растворения температура образующейся соляной кислоты начнет
повышаться, что приведет к увеличению давления ее паров. Когда
общее давление пара станет равным внешнему, например, атмо-
атмосферному давлению, жидкость закипит и начнется интенсивное ис-
испарение воды. Затрата тепла на испарение воды в адиабатических
условиях вызовет снижение температуры, кипящей кислоты и по-
поэтому концентрация ее, являющаяся функцией температуры, нач-
начнет при дальнейшей подаче хлористого водорода возрастать.
На рис. 111 приведены кривые общего давления пара над со-
соляной кислотой разных концентраций. Пересечение кривых с
пунктирной линией соответствует точкам кипения под атмосфер-
атмосферным давлением G60 мм рт. ст.). С понижением температуры ки-
кипящей кислоты ее концентрация, начиная от 20,24%, может и воз-
возрастать и убывать. Это же видно из рис. 124. При общем давле-
давлении 700 мм рт. ст. азеотропная смесь кипит при 105,8° и содержит
20,4% НС1. При более низкой концентрации кислоты ее пары обо-
обогащены водяным паром, а при более высокой — хлористым водо-
водородом. Охлаждение кислоты азеотропного состава вследствие ее
испарения при абсорбции, т. е. переход от изотермы ta3, например,
к t\, может привести к образованию как более разбавленной (точ-
(точка Л), так и более концентрированной (точка В) кислоты. Возра-
Возрастание или убывание концентрации кислоты будет зависеть от со-
состава газовой фазы. Если парциальное давление НС1 в газе пре-
превышает давление НС1 над постоянно кипящей смесью, то концен-
концентрация кислоты должна возрастать. Поэтому при подаче хлори-
хлористого водорода кипящая в адиабатических условиях кислота будет
концентрироваться.
По мере укрепления соляной кислоты содержание НС1 в ухо-
уходящих из абсорбера парах также будет повышаться. На рис. 125
показано, например, изменение давления паров Н2О и НС1 над
соляной кислотой, получаемой по мере насыщения воды с началь-
начальной температурой 30° при общем давлении 700 мм рт. ст. 100%-ным
хлористым водородом без охлаждения абсорбера. Пока концен-
концентрация кислоты невелика, в паровую фазу переходит, главным
образом, вода, и раствор концентрируется как вследствие про-
продолжающегося поглощения хлористого водорода, так и вследствие
но
t,
too
60
¦70
'60
so
JO
го\-
f
IS*
r
f.
-/-
/
1/
Л
/
-\
\
\
s %.
1
1
. 1
В Г"***
— 1
1
\
1
1
!
\
¦
I
1 .
I
i
\ i
\!
1
i
\ i
\
1
i
i
^^
4
к
\
\
ч
4
ч
4
\
\
t
ч
\
\
\
\
0,1
02 OJ Д4 05 0.B 0,7 08 09
HC1. мм. доли
Рнс. 124. Кривые состав — температура кипения системы
НС1—Н2О при различных давлениях.
Цифры на изобарах:
Р¦ — Рнс, + Рн q вл» рт. ст. (масштаб абсцисс вправо от пунк-
пунктирной линии в 2 раза меньше).
удаления воды. При достижении концентрации кислоты 14% НС1
(молярная концентрация НС1 в жидкости х — 0,072) и темпера-
температуры 102,5° раствор закипит. Дальнейшее насыщение раствора
898
Гл. XI. Соляная кислота
Абсорбция хлористого водорода
399
хлористым водородом будет идти при постоянном давлении паров
над кислотой, равном 700 мм рт. ст., но по мере возрастания кон-
концентрации кислоты парциальное давление водяного пара над ней
будет уменьшаться, а парциальное давление хлористого водоро-
водорода — увеличиваться. Поэтому при получении концентрированной
кислоты в одноступенчатом абсорбере большая часть хлористого
водорода останется неабсорбированной.
700
у
/
/
л
/|
1
\
\
\
/
1
/
X
\
\
0
i:
Г
Е 3
L
/
1
/
1
0,050.072 0J0 D.IS 0.20
концентрация HG1 в жидкости, мо/ш
пгз
Рис. 125. Изменение давления паров НС1 (кри-
(кривая /) и Н2О (кривая 2) в процессе насыще-
насыщения воды хлористым водородом (начальная
температура воды 30°, давление 700 мм рт. ст.).
10 15 Z0 25 30 ЗЛ
Концентрация соляной кислоты, J
Ввс.%
Рис. 126. Число теоретиче-
теоретических тарелок, обгспечивэю-
щее получение соляной
кислоты заданной концен- ,
трации.
• Наиболее рационально процесс горячей абсорбции может быть
осуществлен в тарельчатой или насадочной абсорбционной ко-
колонне. Для получения стандартной соляной кислоты, содержащей
27,5% НС1, требуются всего 4 теоретические тарелки (рис. 126),
а 31% НС1 — 5. На практике применяются не тарельчатые, а на-
садочные колонны, более простые и удобные в эксплуатации.
Понижение температуры, подаваемой на верх абсорбера воды,
немного уменьшает количество уходящего из него водяного пара,
а следовательно, и потери уносимого им хлористого водорода. Не
должна быть слишком высокой и температура поступающего на'
абсорбцию газа. Увеличение содержания в газе инертных компо-
компонентов также влияет на абсорбцию благоприятно (несмотря на
понижение концентрации НС1 в газе), так как понижает темпера-
температуру в абсорбере. Но высота абсорбера должна быть при этом
больше, так как увеличивается высота слоя насадки, эквивалент-
эквивалентная одной теоретической тарелке. Если поступающий на абсорб-
абсорбцию хлористый водород содержит водяные пары, то максималь-
максимальная концентрация получаемой соляной кислоты одинакова с кон-
концентрацией кислоты, конденсирующейся при охлаждении газа до
точки росы. Если хлористый водород разбавлен инертным газом
(воздухом), то максимальная концентрация соляной кислоты бу-
будет соответствовать концентрации кислоты, находящейся в равно-
равновесии с поступающим на абсорбцию газом. Эту концентрацию
можно определить по рис. 112 или вычислить по формуле112
с = @,304/ — 0,0016) р0'15, где с — концентрация кислоты, г НС1
на 1 г Н2О; tf —температура кислоты, °С; р— парциальное давле-
давление НС1, мм рт. ст. Так, при содержании в поступающем на аб-
абсорбцию газе 30%) НС1 (парциальное давление НС1 —228 мм
рт. ст.) максимальная концентрация соляной кислоты при 80° бу-
будет 28,5%, при 60° ¦—31,8%, для 50%-ного газа — соответственно
30,5 и 33,3%.
Производственные колонны для абсорбции хлористого водо-
водорода без охлаждения изготовляют из фаолита и заполняют насад-
насадкой из керамических колец B5X25 мм). Широко применяют также
абсорбционные колонны из графитовых блоков, пропитанных тер-
термореактивными смолами с последующей термообработкой). При
диаметре колонны 0,45 м и высоте 6,4 м в ней можно получить из
синтетического хлористого водорода до 30 г в сутки 31%-ной со-
соляной кислоты. Температура поступающего в колонну газа не
должна превышать 250°, а концентрация НС1 в нем должна быть
не меньше 80%). Воду подают в колонну через фаолитовый ороси-
ороситель. Вытекающая из колонны соляная кислота должна иметь тем-
температуру не выше 60° и плотность 1,155—1,160 г/см3 при 20°. Она
поступает в оросительный холодильник из графолитовых трубок
(диаметром 78/102 мм, длиной 3,6 м) и течет по трубкам, орошае-
орошаемым снаружи холодной водой. Охлажденная до 40° соляная кис-
кислота направляется в хранилище готовой продукции, например,
стальные, футерованные диабазовой плиткой резервуары. При по-
повышении температуры выходящей из колонны кислоты уменьшают
количество подаваемой воды, так как это указывает на избыток'
воды. При понижении температуры ^зеличивают расход воды на
орошение, во избежание получения слишком концентрированной
кислоты и увеличения потерь НС1 с газом.
Уходящая из верхней части абсорбционной колонны паро-га-
зовая смесь содержит незначительное количество НС1. Перед вы-
выбросом в атмосферу содержание НС1 в ней снижают до санитар-
санитарной нормы, для чего ее пропускают через промывную, также фао-
литовую колонну диаметром 0,6 м и высотой 4 м, с насадкой из
керамических колец, орошаемую водой. Стекающая из промыв-
промывной колонны кислая вода, содержащая всего ~1,5% НС1, не ис-
пвльзуется и является отбросом производства. Перед спуском в
400
Гл. XI. Соляная кислота
Абсорбция хлористого водорода
401
канализацию ее нейтрализуют известняком. Иногда нейтрализа-1
¦цию осуществляют в промывной башне, орошая ее раствором соды.
Имеется опыт40-113 горячей абсорбции газа (выходящего из |
сульфатных печей и содержащего 30—50% НС1) в системе из ше-
шести последовательно соединенных фаолитовых барботажных аб-1
сорберов, расположенных каскадом.
Аналитический расчет адиабатической
абсорбции HCl111-114
Состав жидкой и паровой фаз, а также температурный режим
на каждой тарелке (сечении колонны) можно определить из по-
постоянства в адиабатических условиях для любого сечения аппа-
аппарата разности теплосодержаний AQ поднимающихся в колонне |
паров и нисходящей жидкости. Значение AQ определяется из ма-
материального и теплового потоков между (п + 1)-й и n-й тарел-
тарелками (счет тарелок снизу вверх):
Уп-Хп+, [ \~уп
здесь At— разность температур между (л + 1)-йн л-й тарелками;.
Ь — количество воды в получаемой кислоте, кг-мол/ч;
у и х— молярные концентрации НС1 в газовой и жидкой фа-
фазах;
гй — молярная теплота сжижения НС1 (тепло конденсации
и диссоциации), ккал/моль;
гв — молярная теплота конденсации H2O, ккал/моль;
са и св—молярные теплоемкости НС] и Н2О, ккал/моль-град.
Пусть, например, производительность абсорбера 365 кг/ч НС1,
температура поступающего газа 50°, начальная температура воды
36° и давление в колонне 700 мм рт. ст. При получении 30%-ной
кислоты @,174 мольных долей) температура вытекающей кислоты
будет 88,3° (рис. 127), а концентрация НС1 в парах, поднимаю-
поднимающихся с первой тарелки, т. е. равновесных с вытекающей жидко-
жидкостью, у\ = 0,725 мольных долей. Величина AQ, т. е. разность ме-
между теплосодержаниями Q поступающего газа и q отбираемой
кислоты: AQ = Q — q= 16500-10 4-7-10-50 —88,3A8 • 47,3 +
4-7-10) = 87300 ккал/ч [здесь: 16500 ккал/моль — средняя моляр-
молярная теплота сжижения (абсорбции) НС1; 10 моль/ч — количество
абсорбируемого НС1; 7 ккал/моль — молярная теплоемкость НС1;
18 ккал/моль — молярная теплоемкость воды; 47,3 >
10A-0,174)
0,174
количество воды в отбираемой кислоте, моль/ч]. Подставляя зна-
значение AQ и других известных величин в приведенное выше урав-
уравнение, получим при п =* 1, т. е. для первой тарелки:
473A-0,725),,
Это уравнение содержит два неизвестных х2 и /2 и ввиду от-
отсутствия аналитического выражения функции t «* f(x) (рис. 127)
решается методом подбора. Прих2 ¦"= 0,146 /2 = 101,5 . Первый мно-
множитель правой части уравнения —' с17^5-о 14б'— =3,29 выражает
количество воды (моль/ч), уносимой с первой тарелки. Произве-
Произведение этого множителя на величину - _'^ 7^ , т. е. 3,29 • 2,64 = 8,68,
1__
87 300 =
0,1 0,2 0,3 0,4. 05 00 0,7 OJB
Мол доли НС1
Рис. 127. Определение состава жидкости и
пара в системе НС1—Н2О по диаграмме х, у, t
при давлении 700 мм рт. ст.
представляет количество уходящего с 1-й тарелки газообразного
хлористого водорода (моль/ч). Следовательно, на 1-й тарелке аб-
абсорбируется 10—8,68 = 1,32 моль/ч или 13,2% от поступающего
на абсорбцию НС1. По температуре t2 = 101,5° определяем содер-
содержание НС1 в парах над второй тарелкой: у2 = 0,41. Далее, анало-
аналогично приведенному выше, определяем: t3 = 104,3°, хъ = 0,093, ys =
= 0,043, /4 = 97,7°, х4 = 0,011, #4~0. Таким образом, требуется
колонна с четырьмя теоретическими тарелками, причем на первой
абсорбируется 13,2% от общего количества НС1, на второй —
29,8%, на третьей — 50% и на четвертой — 7%.
Графический расчет адиабатической
абсорбции HCl11*-116
Схема диаграммы /, / — х, у, энтальпия — состав, для двухфаз-
двухфазной трехкомпонентной системы (поглощаемый компонент, рас-
растворитель и инертный газ) изображена на рис. 128. Диаграмма
построена для постоянного приведенного давления
402
Гл. XI. Соляная кислота
Жидкий хлористый водород
403
где
Р — общее давление;
р° — давление инертного газа;
рА и рв - давление паров поглощаемого компонента и раствори-
растворителя.
Линия АВ — энтальпия i жидкости при кипении в зависимости
' от ее состава х, линия CD — энтальпия / пара состава у при кон-
конденсации. Эти линии ограничивают двухфазную область ACDB и
пересекающуюся изотермами, например MN. Жидкость состава х
в точке М находится в равновесии с паром состава у в точке N.
И
-/1
Шдкость М
D
В
О х у 1
х,у '-
Рис. 128. Схема диаграммы эн-
энтальпия — состав.
Рнс. 129. Определение числа тео-
теоретических тарелок на диаграмме
энтальпия — состав.
Если заданы количества поступающего на абсорбцию газа V\
(без инертного компонента) и уходящей с абсорбции жидкости Lu
а также их составы и температуры, то для определения числа тео-
теоретических тарелок противоточного абсорбера при помощи диа-
диаграммы /, i — х, у необходимо сначала найти координаты хТ и ip
точки Р, называемой полюсом: хр*=-д, h = ~^- Здесь разности
A = Vn — Ln; В = Vnyn — L-nXn, C=VnIn — Lnin являются по-
постоянными для любой тарелки (сечения) аппарата, следователь-
следовательно и для места ввода газа в аппарат, для которого легко опре-
определяются их значения, так как все параметры с индексом п = 1
известны.
Через полюс Р и точку начального состояния газа НA\,у\)
проводят прямую (рис. 129), которая пересекает линию кипения
раствора в точке А. Эта точка характеризует состояние жидкости,
уходящей с первой (нижней) тарелки. Через точку А проводят
изотерму АВ'. Точкой В' определяется состав газа ух, уходящего с
1-й тарелки и находящегося в равновесии с жидкостью состава Х\.
Затем из точки В' проводят луч В'Р к полюсу Р, пересекающий ли-
линию кипения раствора в точке В. Точка В определяет состояние
жидкости Х2, уходящей со 2-й тарелки и равновесной с газом со-
состава У2- Последний определяется по изотерме ВС. Из точки С
снова проводят луч к полюсу Р и т. д. Построение продолжают до
тех пор, пока луч, проведенный через полюс, не пройдет через точ-
точку N', соответствующую конечному составу газа ук, или через точ-
точку N, соответствующую начальному составу поступающей на аб-
абсорбцию жидкости хИ. Число изотерм между начальной и конечной
точками построения диаграммы равно числу теоретических тарелок.
На рис. 130 показано графическое определение числа теорети-
теоретических тарелок на диаграмме /, i — х, у для абсорбции хлористого
водорода при концентрации получаемой соляной кислоты 31%
(xv = 0,31), температуре поступающего газа 50°, температуре воды
t = 30° и общем давлении 760 мм рт. ст. Построение выполнено для
случая, когда абсорбции подвергается чистый НС1, не содержащий
инертных газов и водяного пара, т. е. Уп=\, и хлористый водород
полностью поглощается, т. е. (/к=0. Так как ун=1 и хк=0,31 изве-
известны, то направление первого луча к полюсу определяется при со-
соединении этих точек прямой линией. Кроме того, имеем ук=0 и
*н=0, что обусловливает хр=0. Для определения ip необходимо
продолжить линию ys — хк до пересечения с осью ординат, это
дает значение ip =—100 ккал/кг. Как видно из построения, тре-
требуются четыре теоретические тарелки. На диаграмме видно также
распределение температур по тарелкам (снизу вверх): 82, 96, 108,2
и 103,5°. Количество тепла, которое необходимо отвести в дефлег-
дефлегматоре: —ip — tB *= 100 + 30= 130 ккал на 1 кг добавляемой
воды или 130A —0,31) = 90 ккал на 1 кг соляной кислоты.
ЖИДКИЙ ХЛОРИСТЫЙ ВОДОРОД
На рис. 131 приведена схема производства жидкого хлористого
водорода, содержащего 99,9% НС1. Горячий газ, содержащий
94—95% НС1, охлаждается в кварцевом холодильнике, в котором
конденсируется небольшое количество соляной кислоты. Эта соля-
соляная кислота образуется из воды, получившейся в печи синтеза из
кислорода, загрязняющего хлор. Затем газ осушают концентриро-
концентрированной серной кислотой.
Вся аппаратура, расположенная после кварцевого холодильника
и соприкасающаяся с влажным газом, изготовляется из сплавов,
стойких в присутствии влажного НС1 (хромоникелевые и меднони-
келевые сплавы с добавкой молибдена и вольфрама, алюминиевые
бронзы и др.). В сухом хлористом водороде железо не подвержено
коррозии. Поэтому всю аппаратуру и трубопроводы, расположен-
расположенные после сушильной башни, изготовляют из стали.
HOI, tec %
Жидкий хлористый водород
405
Рис. 130. Графическое определение числа теоре-
теоретических тарелок для адиабатической абсорбции
хлористого водорода на диаграмме энтальпия —
состав.
Сухой газ сжимают до 60 ат и охлаждают в холодильнике —
змеевике, погруженном в воду. Образующийся жидкий НС1 со-
собирается в сборнике, откуда разливается в тару — стальные бал-
баллоны.
Помимо сернокислотной сушки предложено много других спо-
способов получения безводного хлористоводородного газа, пригодного
для ожижения, из разбавленных газов или из растворов (соляной
кислоты). Из газов, образующихся при разложении NaCl серной
кислотой, можно путем абсорбции и последующей десорбции НС1
Рис. 131. Схема производства жидкого хлористого водорода:
/ — печь для синтеза HCI; 2 — газовый холодильник; 3 — сепаратор конденсата соляной
кислоты; 4 — ведтилятор; 5 —сушильная башня; 6 —насос; 7 —резервуар для серной
кислоты; 8 — компрессор; 9 — конденсатор; 10 — сборник жидкого хлористого водорода;
11 — баллон для жидкого хлористого водорода; 12 — показатель уровня; 13 — уравнитель-
уравнительная трубка.
получить газ, содержащий больше 99% НС1. Абсорбентом служит
соляная киелста азеотропного состава; поглощенный ею хлори-
хлористый водород отгоняется при дистилляции в виде почти 100%-ного
НС1, а оставшийся азеотропный раствор охлаждают и вновь воз-
возвращают на абсорбцию НС1 из печного газа 117- 118.
Другой способ119 заключается в выпаривании соляной кислоты,
содержащей больше 20% НС1, под давлением ~15 ат. Пары
охлаждают до —10, —18°, причем конденсируется концентрирован-
концентрированная соляная кислота, а в паровой фазе остается безводный хлори-
хлористый водород. Можно осуществлять процесс и не используя повы-
повышенное давление, проводя его в две стадии в двух ректификацион-
ректификационных колоннах. В первой колонне из соляной кислоты отгоняется
НС1 при нормальном давлении; остающаяся азеотропная смесь
разделяется во второй колонне при пониженном давлении на водя-
водяной пар и более концентрированную кислоту, из которой вновь
406
Гл. XI. Соляная кислота
Регенерация НС1 из травильных растворов
407
можно отогнать НС1 при нормальном давлении. Этот процесс
можно осуществлять непрерывно и циклически, используя тепло
уходящего из первой колонны хлористого водорода для подогрева
кислоты, подаваемой на разделение во вторую колонну 120~123.
С целью получения безводного хлористого водорода из соляной
кислоты, содержащей больше НС1, чем в азеотропной смеси, реко-
рекомендуют также применять для обезвоживания концентрированный
раствор гигроскопической соли, например, 30—60%-ный раствор
СаС12. Процесс солевой экстрактивной ректификации можно осу-
осуществлять в отгонной колонне, в верхнюю часть которой поступает
исходная соляная кислота, а в нижнюю — греющий пар. На уровне,
где соляная кислота достигает азеотропного состава, в колонну
вводится горячий раствор соли. Нижняя часть колонны является
исчерпывающей, а безводный хлористый водород отводится из
верха колонны. Вытекающий из колонны разбавленный раствор
соли выпаривают и возвращают в колонну 124~126. Тепло, выноси-
выносимое уходящим хлористым водородом, можно использовать для по-
подогрева поступающей в колонну соляной кислоты. Подобный же
¦способ с применением MgCl2 разработан для получения чистого
НС1 из абгазов, образующихся при термическом разложении хло-
хлорида магния 127.
Предварительное концентрирование хлористого водорода из
слабых водных растворов может быть осуществлено и экстрак-
экстракционными методами. Предложено, например, после предваритель-
предварительного концентрирования слабой соляной кислоты выпариванием, до
содержания в ней 10% НС1, экстрагировать НС1 пентанолом или
изоамиловым спиртом. Регенерацию спирта, т. е. выделение из него
.концентрированного водного раствора (~30% НС1) производят
при помощи бензола, разделяя затем спирт и бензол разгонкой;
•степень регенерации спирта достигает 98—99% 12S-
Для получения концентрированного хлористого водорода из без-
безводной смеси с другими газами рекомендуется обрабатывать газ
хлористым или бромистым тетраметиламмонием (CHsbNCl или
(CH3LNBr. При соприкосновении газа с этими реагентами в тече-
течение 10—100 сек при 60° образуются продукты присоединения НС1,
которые легко разлагаются при нагревании до 135—150° с выде-
выделением концентрированного хлористого водорода и с регенера-
регенерацией поглотителя. Поглощение НС1 может осуществляться как
при пропускании газа через неподвижный или подвижный слой
реагентов, так и через их суспензию в инертной жидкости, напри-
например, в четыреххлористом углероде, 1,2,4-трихлорбензоле и др.1М
Сульфаты тяжелых металлов (Pb, Cu, Cd, Ag, Hg, Sn, Bi, Sb, Ti) и
некоторые фосфаты также поглощают хлористый водород из раз-
разбавленного газа и при нагревании выделяют чистый хлористый во-
водородсс.
РЕГЕНЕРАЦИЯ НС1 ИЗ ТРАВИЛЬНЫХ РАСТВОРОВ
Использование соляной кислоты для травления стали все уве-
увеличивается, так как по эффективности удаления окалины соляная
кислота в 5 раз превосходит серную, а регенерация солянокислых
травильных растворов легче сернокислых. При солянокислотном
травлении ~0,4% железа переходит в раствор, содержащий в
конце процесса около 10% НС1 и 15% FeCl2.
В США, Канаде, Англии и в других странах регенерацию
травильного раствора осуществляют, распыляя его в печи, обогре-
обогреваемой горелками. Капли раствора испаряются и образующиеся
твердые частицы FeCl2 в нижней части печи при 300—800° превра-
превращаются в НС1 и Fe2O3. Окись железа выгружают из печи, а обжи-
обжиговый газ освобождается от частиц Fe2O3 в циклоне и проходит
через абсорбционную колонну, где образуется 20%-ная соляная
кислота, возвращаемая на травление. Обжиговый газ после цик-
циклона содержит еще 3—10% Fe2O3 от количества, образовавшегося
в печи. Для того чтобы эта часть окиси железа не растворялась в
соляной кислоте в абсорбере и не возвращалась бы в травильные
ванны в виде FeCb, газ между циклоном и абсорбером проходит
через промывную колонну, орошаемую частью травильного раство-
раствора, направляемого в обжиговую печь. При этом раствор насы-
насыщается хлористым водородом и частично упаривается, а улов-
уловленная им Fe2O3 превращается в FeCl3. Используют также про-
промывную насадочную и абсорбционную колонны, объединенные
в один агрегат. Установка работает под разрежением, создавае-
создаваемым хвостовым вентилятором, выбрасывающим абгаз в атмо-
атмосферу 130> ш.
Канадская фирма Stelco предложила аппарат, в котором про-
продукты разложения регенерируемого травильного раствора отде-
отделены от продуктов сгорания топлива. Раствор разбрызгивают на
крышку вращающегося цилиндра, под которым сжигают газ.
Стенки цилиндра погружены в проточную воду, образующую ги-
гидрозатвор. В результате мгновенного испарения травильного рас-
твоса и разложения при 480—540° FeCl2 превращается в НС1 и
Fe2O3, которая непрерывно удаляется скребком с крышки цилин-
цилиндра. Удельная производительность установки 0,6 м3/(м2-ч). HCI
извлекается из газов с образованием соляной кислоты, возвра-
щаемоей на травление. Такой аппарат в 10 раз ниже распылитель-
распылительной печи с одинаковой производительностью |32.
Описана установка ш, в которой часть обработанного травиль-
травильного раствора упаривается и поступает в реактор с кипящим слоем
песка, где при 800° в присутствии кислорода воздуха идет конвер-
конверсия FeClj в Fe2O3. После очистки отходящего газа в циклоне or
частиц Fe2O3 его промывают в адиабатическом абсорбере второй
частью травильного раствора для извлечения НС1.
408
Гл. XI. Соляная кислота
Описаны несколько вариантов метода регенерации НС1, заклю-
заключающегося в кристаллизации FeCl2 при охлаждении травильного
раствора. Кристаллы отделяют на центрифуге и разлагают в печи
на Fe2O3 и НС1 ш.
Предложен также способ регенерации, заключающийся в окис-
окислении содержащегося в травильном растворе FeCb в FeCU возду-
воздухом под давлением или хлором. Затем раствор обрабатывают в
насосе-смесителе раствором трибутилфосфата в керосине. Смесь
поступает в отстойную колонну, в которой водная фаза, содержа-
содержащая НС1, собирается внизу и возвращается на травление, а орга-
органическая фаза, содержащая FeCls, отводится сверху. FeCU экстра-
экстрагируют из нее водой, нейтрализуют известью и сбрасывают, а ре-
регенерированную органическую фазу возвращают на обработку
окисленного травильного раствора '"
135
ПЕРЕРАБОТКА ХЛОРИСТОГО ВОДОРОДА В ХЛОР
Все большие количества хлористого водорода получаются в
виде отхода от других производств. Наряду с этим непрерывно
растет потребность в хлоре для органического синтеза. Поэтому
Созданию рациональных методов переработки хлористого водорода
в хлор уделяют значительное внимание. До сих пор, однако, отсут-
отсутствуют методы регенерации хлора из НС1, которые были бы эко-
экономичнее получения хлора электролизом поваренной соли.
В прошлом веке, до развития промышленного электролиза хло-
хлоридов, хлор получали из НС1 по способу Вельдона DНС1 + МпО2=
= МпС12+ 2Н2О + С12) ш, а затем по способу Дикона DНС1 + О2=
= 2НгО + 2С12). Впоследствии эти способы были полностью вы-
вытеснены электрохимическими, но в настоящее время химические
способы получения хлора вновь возрождаются на новой техниче-
технической базе. Все они основаны на прямом или косвенном окислении
НС1 (или хлоридов), причем наиболее распространенным окисли-
окислителем является кислород воздуха 137-]зв.
Равновесие реакции Дикона 50> IS6
4НС1 + О2 ^=± 2Н2О (г.) + 2С12 + 27,3 ккал
смещается в сторону хлора при пониженных температурах и повы-
повышенных давлениях и может рассматриваться как совместное рав-
равновесие систем: 2НС1 ^С12 + Н2 и 2Н2 + О2 ^ 2Н?О.
Константа равновесия реакции Дикона может быть вычислена
по формуле:
„2
/>hci ¦ Po2
5750
- 2,136 lg T - 0,000857Г + 0,683 • 1O~V2 + 0,296
или по упрощенной, но достаточно точной для технических расче-
расчетов формуле lg/С = 6034 T~l — 6,972.
Переработка хлористого водорода в хлор
40»
Значения igK:
t,°C....
25
+ 13,28
4Б0 600
+1,5 -0,05
650 1984
-0.4 -4,3
Константа равновесия в наибольшей мере зависит от парци-
парциального давления НС1 в равновесной газовой смеси, так как эта
величина входит в уравнение изотермы реакции в четвертой сте-
степени. В меньшей мере влияет давление водяного пара и совсем
мало парциальное давление кислорода, так что выходы реакции
при сжигании НС1 в кислороде и в воздухе отличаются незначи-
незначительно. Например, при 430° смесь из 40 объемн. % НС1 и 60 объ-
емн. % воздуха дает в состоянии равновесия 70—71% превраще-
превращения. Если освободить эту смесь от азота, то начальный ее состав
будет 76,1% НС1 и 23,9% О2, а равновесная степень превращения
вследствие увеличения парциального давления НС1 повысится до
76,1%. Если же при прежней начальной концентрации НС1 D0%)
повысить соответственно парциальное давление кислорода, то вы-
выход возрастет только до 72%- Если из равновесной газовой смеси
(полученной из 40% НС1 и 60% воздуха), степень превращения в
которой 70—71%, удалить водяной пар, то степень превращения
возрастет до 82,5—85%. Увеличение выхода реакции за счет повы-
повышения давления НС1 или удаления водяного пара связано со зна-
значительными техническими осложнениями, поэтому стремятся по-
повысить выход, понижая температуру окисления НС1. Это связано
с необходимостью использования катализаторов, так как без них
реакция при низких температурах идет чрезвычайно медленно.
В диконовском процессе в качестве катализатора использовали
хлорную медь, 50%-ным раствором которой (иногда с добавкой
NaCl) пропитывали пористый керамический носитель. Оптималь-
Оптимальная температура реакции на таком катализаторе была обычно в
пределах 430—490°. Этот катализатор легко отравляется соедине-
соединениями мышьяка, с которыми образует неактивный арсенат меди, а
также двуокисью и трехокисыо серы. Присутствие в газе даже не-
небольших количеств паров серной кислоты вызывает резкое умень-
уменьшение выхода хлора в результате последовательно идущих ре-
реакций:
H2SO4 = SO2 + У2О2 + Н2О
SO Cl 2HQ 2H':i HO
Cl2 + Н2О = '/2О2 + 2НС1
Таким образом, серная кислота является катализатором, спо-
способствующим обратному превращению С1г в НС1. Поэтому хлори-
хлористоводородный газ до окисления на медном катализаторе должен
подвергаться тщательной очистке от примесей, снижающих выход
хлора.
Установка Дикона состояла из подогревателя газа, газового
фильтра и контактного аппарата — стального цилиндрического
L
410
Гл. XI. Соляная кислота
кожуха, внутри которого находились два концентрически располо-
расположенных керамических цилиндра с отверстиями; кольцевое про-
пространство между ними заполнено катализатором. Хлористый водо-
водород окисляли воздухом, поэтому хлор получался разбавленным.
В контактный аппарат подавали смесь, содержавшую 25 объемн.%
НС1 и 75 объемн.% воздуха (~16% О2), а газ, выходивший из
аппарата, содержал около 8% С12, 9% НС1, 8% водяного пара и
75% воздуха. Такой газ, после отмывки из него НС1 и осушки
серной кислотой, использовали обычно для получения хлорной из-
извести.
Реставрация процесса Дикона в настоящее время базируется
иа окислении хлористого водорода не воздухом, а кислородом, что
позволяет получать концентрированный хлор при применении вы-
высокоактивных катализаторов. Образующуюся хлорокислородную
смесь отмывают от остатков НС1 последовательно 36- и 20%-ной
соляной кислотой и осушают серной кислотой. Затем хлор сжи-
сжижают, а кислород возвращают в процесс. Отделение хлора от кис-
кислорода производят также, поглощая хлор под давлением 8 ат хло-
хлористой серой, которую затем регенерируют, получая 100%-ный
хлор:
C12+S2C12 ^± S2C14
Используют низкотемпературные катализаторы, например, дву-
хлористую медь, активированную солями редкоземельных метал-
металлов, что дает возможность вести процесс даже при 100° и поэтому
резко увеличить степень превращения НС1 в СЬ 137-ш. На окисно-
хромовом катализаторе сжигание НС1 в кислороде производят при
340—480° ио. Описано применение при 250—420° катализатора из
смеси V2O5 с пиросульфатами щелочных металлов и активаторами
на силикагеле U1. Изучены механизм и кинетика этого процесса и
установлены оптимальные условия его осуществления ш< 143, в ча-
частности в псевдоожиженном слое144.
Окисление хлористого водорода кислородом производят также
с помощью расплавленной смеси FeCl3 + KC1 в две стадии, осу-
осуществляемые в отдельных реакторах 137. В первом реакторе проис-
происходит окисление хлорного железа с образованием хлора:
2FeCl3 + 1 '/А = Fe2O3 + 3C1S
Во втором реакторе хлорное железо регенерируется из окиси
железа хлористым водородом:
Fe2Os + 6НС1 - 2FeCls 4- ЗНаО
Хлористый калий добавляют для уменьшения давления пара
хлорного железа. Этот процесс предложено145> ш осуществлять
также в одном аппарате, в котором контактная масса, состоящая
из РегОз, КС1 и хлорида меди, кобальта или никеля, нанесенных
Переработка хлористого водорода в хлор
41»
на инертный носитель, перемещается сверху вниз аппарата.
Вверху аппарата она проходит горячую зону хлорирования, где
регОз превращается в FeCb, взаимодействуя с НС1, находящимся
в потоке идущего снизу вверх газа. Затем контактная масса опу-
опускается в зону охлаждения, где под действием кислорода обра-
образуется элементарный хлор, a FeCl3 переходит в Fe2O3. Окисленная
контактная масса снова возвращается в зону хлорирования.
Подобное же косвенное окисление НС1 в С1г осуществляется
по схеме:
2НС1 + MgO = MgCl2 + Н2О
Представляет интерес сочетание этого процесса с регенерацией
аммиака окисью магния в производстве соды (стр. 287). Предло-
Предложено одновременно получать хлор и серную кислоту, пропуская
через ванадиевый катализатор при 400—600° газ, содержащий
НС1, Ог и большой избыток SO2. Затем из газа конденсируют
H2SO4 и HSO3C1 и абсорбируют SO3 серной кислотой — хлор ос-
остается в газовой фазе. HSO3CI гидролизуется и выделяющийся
НС1 возвращают в процесс 147.
Окисление С1~ с целью получения С1г можно осуществлять и
и электролизом соляной кислоты или получаемых из нее хлоридов
металлов. В последнем случае регенерацию хлора можно комбини-
комбинировать с извлечением из руд чистых металлов, в частности порош-
порошкообразных. Промышленный электролиз соляной кислоты 136'148; 149
был освоен в небольших масштабах в США еще в 30-х годах.
В Германии во время второй мировой войны работала опытная
установка по электролизу соляной кислоты. В США получены удо-
удовлетворительные результаты на полупромышленной установке по
электролизу СиС12, оборудованной ванной мощностью 4000 о.
Электролиз водного раствора СиС12 ведут при 80°, при плотности
тока 11 а/дм2 и напряжении на ванне 1,8 в, с графитовыми анодом
и катодом:
2СиС12 = 2СиС1 + С12
Двухлористая медь легко регенерируется при обработке смеси
CuCl и соляной кислоты воздухом в абсорбционной колонне при
83—87°:
2СиС1 + 2НС1 + '/jOj = 2CuCl2 + Н2О
Для установки производительностью 35 т хлора в сутки на 1 т
Хлора требуется затратить 1600 квт-ч постоянного тока и
155 квт-ч переменного, т. е. расход энергии на 1 т хлора прибли-
приблизительно в 2 раза меньше, чем при электролизе раствора хлори-
хлористое натрия '*7>1Я0. Помимо меди, в качестве «переносчика» при
412
Гл. XI. Соляная кислота
электрохимической регенерации хлора могут быть использованы
любые другие поливалентные металлы — Fe, Cr, Mn, Ni и пр.
Напомним, что хлор может быть получен и окислением хлори-
хлоридов. Например, при взаимодействии NaCl и SO3 идут реакции:
2NaCl + 2SO3 = 2NaSO3Cl
2NaSO3Cl = Cl2 + SO2 + Na2SO«
Распад NaSO3Cl происходит при 275°. Смесь газов SO2 и Cl2
можно разделить, поглощая хлор S2C12 или СС14 или подвергая ее
ректификации, в результате которой получается азеотропная смесь,
содержащая 88 мол. % С12 и 12 мол. % SO2. Азеотропную смесь '
можно далее разделить, переводя SO? в SO?C12 и отделяя избыточ-
избыточный хлор, a SO2C12 разлагая при 200° на SO2 и С12, которые до-
добавляют к смеси, направляемой на ректификацию.
Хлор можно получить и окислением хлорида или хлористого
водорода азотной кислотой, а также двуокисью азота (см. гл.
XXXII);
ЗНС1 + HNO3 = С12 + NOC1 + 2Н2О
Разложение хлористого нитрозила может быть достигнуто его
окислением:
2NOC1 + О2 = 2NO2 + С12
Предложено, например, окислять NOC1 75%-ной азотной кис-
кислотой 181:
2NOC1 + 4HNO3 = С12 + 6NO2 + 2Н2О
Смесь хлора и двуокиси азота разделяют, перерабатывая NO2
в слабую азотную кислоту, используемую затем для окисления
НС1 в первой стадии процесса с образованием С12 и NOC1. Основ-
Основным затруднением при осуществлении этого процесса в промыш-
промышленных масштабах является устранение коррозии. В качестве ма-
материалов для аппаратуры применяют керамику, стекло, свинец,
никель, пластмассы. По этому методу в США в 1952—1953 гг.
работала установка производительностью 75 т хлора в сутки|37.
Разработан циклический способ получения хлора окислением
хлористого водорода азотной кислотой без образования хлори-
хлористого нитрозила по реакции:
2НС1 + 2ШО3 = С12 + 2NO2 + 2Н2О
Процесс идет в жидкой фазе при 80°, выход хлора достигает
100%, NO2 получается в жидком виде182.
ЛИТЕРАТУРА
1 М Е Позин Технология минеральных удобрений и солей, Госхимиздат,
1957 'гл. XIII. — 2. X. Л. Цейтлин, ЖПХ, 21, № 1, 35 A948). —3. X. Л. Цейт-
лии С М Бабицкая, Там же, 31, № 1, 84 A958). — 4. А. ВоиПё, С h а-
' Ь ё г о и х, Compt. rend., 242, № 25, 2947 A956). — 5. И. С. М о Р о з о в, Изв. Г.ГГР
Литература
413
ОХН, № 1, 37 A946). —б. Л. С. Л илич, В. И. Тимофеев, Вестн. ЛГУ, № 10,
Сер. физ. и хим., в. 2, 68 A956). —7. М. С. Вревский, Работы по теории
растворов, Изд. АН СССР, 1953. —8. А. К. Coving ton, J. E. Prul, J. Chem.
Soc, 1957, 1930. — 9. G. V u 111 a r d, Compt. rend., 241, № 19, 1308 A955).—
W. M. А. Клочко, М. Ш. Курбаиов, ИСФХА АН СССР, 24, 237 A954).
11. H. А. Лоури, Соляная кислота и сульфат натрия, Химтеорет, 1934.—
12. И. И. Ангелов, М. Ф. Володина, Труды ИРЕА, в. 20, 1951, стр. 190.—
13. М. Е. П о з и н, Труды ЛТИ им. Ленсовета, в. 20, 1951, стр. 3.— 14. А. Б. 3 д а-
н о в с к и й, Е. И. Л я х о в с к а я, Р. Э. Ш л е й м о в и ч, Справочник по раствори-
растворимости солевых систем, т. 1, Госхимиздат, 1953, стр. 27. —15. Д. М. Корф,
A. П. Шатровская, ЖОХ, 10, № 13, 1231 A940).—16. S. W. Benson,
R. L. Richardson, J. Am. Chem. Soc, 77, № 16, 4206 A965).— 17. Chem. Eng.
News, 44, № 36, 72A A966). — 18. F. X. McGarvey, пат. США 2695875, 1954.—
19. G. M obi us, пат. ГДР 7361, 1954. — 20. П. Ь. Д ы б и н а, ЖПХ, 33, № ю,
2184 A960).
21. S. Schein, Chem. Met. Eng., 35, № 11, 673 A928). — 22. И. В. Бирю-
Бирюков, Хим. пром., № 4, 113 A952). — 23. Пат. США 2234738, 1941. — 24. И. Н. Пу-
Путилова, С. А. Балезин, В. П. Бараний к, Защита металлов от разъеда-
разъедания кислотами, Госхимиздат, 1945.— 25. И. Н. Путилова, С. А. Балезин,
B. П. Баранник, Ингибиторы коррозии металлов, Госхимиздат, 1958.—
26. В. П. Б а р а и н и к, И. Н. Путилова, Хим. пром., 266 A948).— 27. С. А. Б а-
л е з и н, В. П. Б а р а и н и к, И. Н. Путилова, Применение ингибиторов кис-
кислотной коррозии, Госхимиздат, 1948.—Ж X. Л. Цейтлин. Хим. пром., № 7,
199 A951). — 29. В. П. Баранник, И. Н. Путилова, Там же, № 4, 103
A949). — 30. С. А. Балезин, Уч. зап. МГПИ, в. 76, 5 A953).
31. И. П. А н о щ е н к о, ЖПХ, 30, № 3, 393; № 4, 523 A957).— 32. В. А. X и т-
ров, В. П. Задорожный, Там же, 37, № 3, 574 A964). —33. Т. Д. Колпа-
кова, В. П. Баранник, Хим. пром., М> 7. 596 (I960). — 34. А. И. Горба-
нев, В. Я. Николина, Сульфат натрия, Госхимиздат, 1954. — 35. Я. Е. В и л ь-
нянский, 3. Л. Персии, Труды УНИХИМ, в. 4, 1957, стр. 83.—
36. Е. И. Лукьянова, Сборник статей по общей химии, т. 1, Изд. АН СССР,
1953, стр. 106. — 37. Я- Е. В ильн я некий, 3. Л. Персиц, Хим. пром № 6,
19 A946). — 38. J. Kendall, M. L. London, J. Am. Chem. Soc, 42, 2131
A920). — 39. H. С. Б а н н ы х, Автореф. канд. дисс, Свердловск, 1951; Н. С. Б а н-
ных, Я- Е. Вильнянский, Труды УНИХИМ, в. 7, 1958, стр. 94.—
40. И. М. Вассерман, Производство минеральных солей, Госхимиздат, 1982,
гл. II.
41. С. А. Жихаревич, С. Д. Лукова, ЖПХ, 9, № 7, 1230 A936).—
42. С. В. Лозовой, Хим. пром., № 4, 244 A957). — 43. Я. Е. Вильиянский,
В. П. 3 а б и я к о, Там же, № 1, 22 A945). — 44. А. М. Г и н с т л и н г. Хим. маш.,
№ 2, 3 A939); ЖХП, № 9, 31 A939).— 45. М. И. Ц а л, Я. Е. Клейман, Хим.
пром., № 4, 120 A951). — 46. Н. В. Гвоздев, Производство соляной кислоты и
сульфата, Химтеорет, 1933. — 47. Н. В. Гвоздев, А. М. Г и н с т л и н г
Д. М. Зильбермаи, ЖХП, 17, № 8, 34 A940). — 48. Хим. пром., № 7 21*
A944). — 49. Франц. пат. 1397699, 1964; пат. США 3363977, 1963. — 50. П. Па-
Паскаль, Синтез и катализ в основной химической промышленности ГОНТИ
1938.
51. Chem. Eng., № 5, 132 A954). — 52. В. Neumann, H. Kunz, Z. ang
Chem., 42, № 47, 1085 A929). — 53. С. И. Вольфкович, Ф. Г. Марголис
Изв. АН СССР, ОХН, № 5, 322 A942). — 54. Г. В. Л а бути н. А М Маль-
Мальцев, Труды ВАМИ, № 40, 1957. —55. С. Cannon, пат. США 2706144, 2706145,
1955. — 56. .С. М. Вольфкович, Ф. Г. Марголис, ДАН СССР, 51, № 1, 23
A943). — 57. В. Г. Флейшман, Хим. пром., № 7, 411 A957).— 58. Л В. Гант-
ман, Е. П.Шведов, Там же, №4,19 A947). —59. А. Н. Maude, Chem Eng
Progr., 44. № 3, 179 A948). — 60. A. H. Maude, Chem. Ind., № 9,-348 A942).
414
Гл. XI. Соляная кислота
61. С. Д. Бесков, Техно-химические расчеты, Госхимиздат, 1950, стр. 246.—
62. A. W. Umland, J. Electrochem. Soc, 101. № 12, 626 П954). — 63. А. И.Роз-
лове кий, ЖФХ, 29, № 1, гЗ A955). — 64. А. И. Розловский, ДАН СССР,
92, № 3, 621 A953). — 65. Суэла, Асайо, япон. пат. 6561, 1954. — 66. J. Van
Dlest, R. De G r u f, Ind. chim. beige, 30, № 11, 1195 A965). — 67. Ind. chemist,
№ 314. 115 A951); № 333, 451 A952). — 68. W. Kunzen. W. Rasche, пат.
ФРГ 923844, 1955. — 69. Франц. пат. 1070017, 1954.-70. A. Czernotzky,
пат. ГДР 7039, 1954; 10779, 1955.
71. W. Hirschkind, Ind. Eng. Chem., 17, № 10, 1071 A925).— 72. А. А, Ч й-
жик, ЖХП, № 9, 37 A932). — 73 В. Neumann, R. Domke, Z. ang. Chem.,
№ 11, 368 A926). — 74. Б. Д. Мельник, Хим. пррм., № 7, 548 A966).—
75. H. О. Pr itch a r d, J. Am. Chem. Soc, 76, № 4, 1201 A954). — 76. Я- С. Де-
миховский, Пром. орг. хим., 7, № 2, 107 A940). — 77. A. Weidemann,
В. Bruzs, пат. ГДР 4698, 1953.— 78. U. Kopsch, англ. пат. 695063. 1954 —
79. О. Sly, австрал. пат. 159181, 1954. — 80. Н. С. Wohlers, пат. США
2730194, 1956.
81. Chem. Met. Eng., 52, № 4, 130 A945). — 82. М. Н. Kh u dk a r, M. R a ki-
boddowla, Chem Ind. № 5, 128 A954). — 83. На Пег, англ. пат. 716754,
1954; франц. пат. 1107573, 1956.— 84. С. F. Р г u 11 о n, W. S. М i 11 е г, L. С. Н i r ri-
rile г, пат. США 2735749, 1956. — 85. К. Herod, польск. пат. 37981, 1955.—
86. Б. Д. Мельник, Хим. пром., № 6, 330 A957). — 87. М. Г. Манвелян,
Г. О. Григорян, С. С. Караханян, Изв. АН АрмССР. ОХН, 17, № 5, 573
A964); 18, № 1, 92 и № 5, 516, 521 A965). — 88. Е. Berl, H. Standinger,
Z. ang. Chem., 43, № 46, 1006 A930).— 89. Англ. пат. 478851, 1938. — 90. Франц.
пат. 1072599, 1954.
91. Ф. К. Михайлов, Л. М. В о л о в а, Л. С. Коваленко, Хим. пром.,
№ 8, 595 A964). — 92. $. G. Osborn, пат. США 2126803, 1338.-95. В. Анд-
Андреев, Г. Креймер, ЖХП, № 2, 146 A927).— 94. Пат. ЧССР 118613, 1965.—
95 М. Н. Мерлис, ЖПХ, № 17, 1043 A936). — 96. W. Sobievanski, Chemik,
7, № 2, 34 A954). — 97. S. H. Friedmann, Chem. Eng., 69, № 14, 133 A962).—
98. А. В. Горяинова, Журн. ВХО, 10, № 1. 58 A965). — 99. А. П. Прозо-
Прозоров, Я- Е. Нусинов, И. К. Шмелев, Хим. пром., № 2, ЮЗ A955> —
100. И. Я. К л и н о в, Дерево, как материал для химической аппаратуры, Гос-
Госхимиздат, 1956.
101. И. Я. Клинов, Хим. наука и пром., № 4, 492 A958). — 102. И. А. Его-
Егоров, Фаолит и его применение в химической промышленности, Госхимиздат,
1956. — 103. К. А. Поляков, Ф. Б. С л ом ян ска я, К. К. Полякова, Кор-
Коррозия и химически стойкие материалы. Госхимиздат, 1953. — 104. И. М. Корни-
Корнилов, Хим. наука и пром., № 2, 23*8 A957).— 105. С. С. Brumbaugh, А. В. Т i 11-
mann, R. С. Sutter, Ind. Eng. Chem., 41, № 10, 2165 A949).— 106. W. Peck,
Ind. Chem., 14, № 166, 445 A938). — 107. F. Hunter, Ind. Eng. Chem., 30, № 11,
1214 1938). — 108. M. Л. Варламов, К. К. Беленавичус, ЖПХ, 36, № 4,
697 A963).—109. R. W. Haisty, J. W Ross, пат. США. 3197942, 1964.—
110. A. M. Г а с п а р я н, Абгорбция хлоррводорода в колонных аппаратах, изд.
Ереванск. политехи, ин-та, 1Э40; А. М. Г а сп ар ян. Труды Ереванского поли-
политехнического ии-та, в 1, 1941.
111. В. Haase, H. Koch, Chem. Techn.. 20, № 12. 738 A968) —
112. В. Н. Ш у л ь ц. Расчеты по технологии соляной кислоты и сульфата, ОНТИ,
1934. — 113. П. С. А р т а м о и о в, В. Н. Извеков, Хим. пром., № 3, 13 A946).—
114. В. М. Рамм, Абсорбция газов, Изд. «Химия», 1966, стр. 275, 281, 731, 732.—
115. В. М. Рамм, Абсорбционные процессы в химической промышленности, Гос-
Госхимиздат, 1951, стр. 124. 314. — 116. G. Hellemanns, H. Seibring, Chem.
Ingr. Techn.. 38, № 10, 1046 A966). — 117. W. Bey, швейц. пат. 288158, 1953. —
118. R. С. Sutter, пат. США 3165453, 1959. —119. С. С. Brumbaugh, пат.
США, 2665240, 1954. — 120. Фииск. пат. 26930, 1954.
Литература
415
121. Франц. пат. 1040338, 1953. — 122. С. G e r d e s, С. Н a m m a n n пат
ГДР 878487, 1953. —123. Т. Riehm, канад. пат. 514290, 1955. —124 Н Р Ro-
Roberts, канад. пат. 490323, 1953. — 125. R. Vie we g, Chem. Techn., 12, № 14, 188
A960). —126. О. В. Т ы in e ц к а я, И. М. Г р и н ш т е й н, Труды Всесоюзн. н.-н.
ин-та гидролизной и сульфитноспиртовой пром т 14 1965 стр 225 280
127. J. Synowies, Ind. chim. beige, 33, № 4, 351 A968). — 128. E. D. Cri t ten-
den, A. N. H i kso n, Ind. Eng. Chem., 46, № 2, 265 A954). — 129. D. H. Camp-
fa e 11, пат. США 2717199, 1955.— 130 Sheet Metal Ind., 43, № 473, 718 A966).
131. A. Hake, Blech., 13, № 5, 264 A966).— 132. Canad. Chem. Process., 49,
№ 7, 93 A965). — 133. Chem. Age (India). 17, № 8, 635 A966). — 134. A. Hake
Sheet Metal Ind., 42, № 461, 696 A965). —135 Фраиц. пат.' 1417037, 1964.—
136. В. В. Стендер, Электролитическое производство хлора и щелочей Хим-
теорет, 1935. — 137. Tekn. tidskr., 83, № 32, 653 A953). — 138. F. Wolf F'Riin-
ge, R. Korn, Z. anorg. Chem., 304, № 1, 2, 48 (I960). —139. Галланд. пат
112095, 1960; пат. ГДР 33098, 1961; пат. США 3201201, 1962; англ пат 993496
1963. — 140. R. В a n n е г, Т. Р е г г i п, пат. США 2678259, 1954.
141. Пат. ФРГ 1240830, 1961. —/42. А. А. Солошенко, Я. Е Вильнян-
ский, ЖПХ, 37, № 3, 535 A964); Кинетика и катализ, V, № 5 881 A964) —
143. A. A. Jones, H. Bliss, С. A. Walker, J. Am. Inst. Chem. Eng., 12, № 2,
260 A966). —Ш K- H. Fleurke, Trans. Inst. Chem. Eng., 46, № 2- Chem Eng
№ 216, 41 A968).- 145. D. J. Pye, W. J. Joseph, пат. ФРГ 888386 1953 -
146. A. Abbey, англ. пат. 689370, 1953. — 147. R. Bauwens J Molliere
франц. пат. 1430215, 1965. —148. W. Gardiner, Chem. Eng' 54, № 1 100
^V.nTJ49- ?¦ Gallone. G- Mess пег, Electrochem. Technol., 3, № 11, 12
321 A965). -150. С. Р. Robert 8, Chem. Eng. Progr., 46, 456 A950).
151. W. J. Cong don, S. W. Grossmann, пат. США 2665195, 1954.-^
152. Chem. Age, 88, № 2246, 131 A962).
Глава XII
СОЛИ БАРИЯ
Важнейшими промышленными солями бария являются хлорид,
карбонат и сульфат. Выпускают также гидроокись, окись и пере-
перекись бария, нитрат, хромат, манганат, хлорат и другие соли, а так-
также сульфид бария, который служит полупродуктом для производ-
производства других соединений бария.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Окись бария ВаО обычно получается в виде аморфной, пори-
пористой массы с плотностью 4,73—5,46 г/см3; могут быть получены
кубические кристаллы с плотностью 5,72 г/см3. Плавится при 1923°.
Довольно хорошо растворяется в расплавленных хлоридах и суль-
сульфатах щелочных металлов, не вступая с ними в реакции обмена;
так, например, растворимость ВаО при 1200° в NaCl 5,33, а в
Na2SO4 7,58 мол. % 1- Окись бария энергично соединяется с волой
ВаО + Н2О = Ва(ОНJ + 22,3 ккал
образуя гидроокись бария — белый, аморфный порошок с плотно-
плотностью 4,5 г/см3. При 20° в воде растворяется 3,75%, а при 80°
53,17% Ва(ОНJ; в присутствии хлоридов и нитратов щелочных
металлов растворимость увеличивается, а в присутствии гидрооки-
гидроокисей — уменьшается.
Из водных растворов системы Ва(ОНJ—Н2О кристаллизуется
Ва(ОНJ-8Н2О с плотностью 1,66 г/см3, конгруэнтно плавящийся
при 78,3°. Растворение Ва(ОНJ-8Н2О в воде идет с поглощением
тепла (—15,2 шал/моль). Из концентрированных растворов, содер-
содержащих 57—62,2% Ва(ОНJ, кристаллизуется Ва(ОН2) • ЗН2О; кон-
концентрации 62,2% соответствует перитектическая точка при 88°. При
более высоких концентрациях выделяется Ва(ОНJ-Н2О, имеющий
точку полиморфного превращения при 185° и перитектического пе-
перехода в безводный Ва(ОНJ при 199° (концентрация в точке пе-
Физико-химические свойства
417
рехода 88,2% Ва(ОНJ). Безводная гидроокись бария при 250° пре-
претерпевает полиморфное превращение и плавится без разложения
при 408°2. При нагревании до 900—1000° получается безводная
окись бария3.
Перекись бария4 ВаО2 — белый порошок с плотностью
4,96 г/см3, весьма устойчивый в сухом виде. Технический продукт
содержит 80—90% ВаО2 и окрашен соединениями железа в желто-
желтоватый или зеленоватый цвет. Перекись бария обладает парамаг-
парамагнитными свойствами. При нагревании с углем действует как депо-
деполяризатор, при этом протекает реакция:
2ВаО2 + С = 2ВаО + СО2
Элемент BaOj — уголь развивает электродвижущую силу, рав-
равную 1 в. При нагревании выше 700° ВаО2 переходит в тестообраз-
тестообразную массу и разлагается по обратимой реакции:
2ВаО2 ^=± 2ВаО + О2-34,2 ккал
Эта реакция ускоряется в присутствии влаги, гидроокиси бария
и других веществ.
Многочисленные литературные данные4 о давлении диссоциа-
диссоциации ВаО2 при разных температурах весьма противоречивы. Веро-
Вероятно, равновесное давление кислорода равно 1 ат приблизительно
при 840°.
Перекись бария является сильным окислителем. Она окисляет
SO2 в сульфат (на холоду), N2O и NO в нитрат (при нагревании),
разлагает сухой хлористый водород с выделением С12, С12О и даже
О3. Окисление органических веществ может сопровождаться взры-
взрывом и воспламенением. С водой ВаО2 образует сначала ВаО2»8Н2О,
затем В а (ОН) 2 и Н2О2. При действии перекиси водорода на слабо-
слабощелочные растворы бариевых солей ниже 30° или на октагидрат
перекиси бария (при 0°) образуются белые кристаллы ВаО2 • 2Н2О2.
Выше 30° образуется ВаО2 • Н2О2. В системе Ва(ОНJ—Н2О2—Н2О
в пределах температур от —10 до +50°, помимо октогидрата пе-
перекиси бария ВаО2 • 8Н2О, выделяющегося только при температу-
температурах, близких к 0°, и малых концентрациях Н2О2, в твердых фазах
существуют дипероксигидрат ВаО2 • 2Н2О и монопероксигиДрат
ВаО2 • Н2О5. Установлена возможность получения надперекиси ба-
бария Ва(О2J при взаимодействии перекиси бария с кислородом под
давлением больше 3000 ат6.
Хлористый барий ВаС12 • 2Н2О кристаллизуется из водных рас-
растворов с двумя молекулами воды в виде бесцветных пластинок с
плотностью 3,05 г/см». Выше 113° теряет кристаллизационную
воду. Безводный имеет плотность 3,86 г/см3 и плавится при 962*.
Насыщенный водный раствор содержит при 0° — 24,0%, при
100°—37,0% ВаС12. Водные растворы, содержащие ВаС12 и
Вв(ОНJ, обладают хорошими буферными свойствами в облает^
А А М. Е. Позин
418
Гл. XII. Соли бария
20
8? 20
10
\JTBb
ДВа(
ч\
]\
¦зВа
:ы
70
s
гн2<
\
№
рН —13,62-=-13,82 7. В присутствии НС1 и особенно CaCfe раство-
растворимость ВаС12 в воде резко уменьшается (рис. 132)8.
Изучены свойства растворов ВаС12 в расплавленных солях —•
нитратах9, хлоридах и других10. В системе ВаС12—СаС12, имею-
имеющей эвтектику с 35 мол. % ВаС12 при 605°, обнаружено соединение
ВаС12-СаС12с^л632°и.
В системе ВаС12—КС1—NaCl существует двойная соль ВаС12«
¦ 2КС112.
Нитрат бария Ba(NO3J представляет собой бесцветные окта-
адрические кристаллы плотностью 3,21 г/см3, плавящиеся при 592°.
При нагревании разлагается с образова-
образованием вначале нитрита бария, затем окиси
бария. Насыщенный водный раствор при 0°
содержит 4,8%, при 20° —7,9%, при 100°—
25,5% Ba(NO3J. В присутствии нитрата
и хлорида кальция растворимость Ba(NO3)a
значительно понижается. Составы эвто-
нических растворов при 100° (в мол.%):
для системы Ba(NO3J—Ca(NO3J—Н2О
меньше 0,1 Ba(NO3J и больше 99,9
Ca(NO3J, количество воды 140,9 М на
100 М солей; для системы Ba(NO3J—
ВаС12—Н2О 28,5 Ba(NO3J и 71,5 ВаС12, ко-
количество воды 1320 М на 100 М солей. Вы-
Высаливающее действие СаС12 объясняется,
по-видимому, его гидратацией 13- и.
Карбонат бария ВаСО3 имеет три .модификации: ниже 811° —
а-модификация, кристаллы ромбической системы (витерит) с плот-
плотностью 4,3 г/см3, выше 811° стабильна гексагональная модифика-
модификация, выше 982° — кристаллы кубической системы. Карбонат бария
устойчив при достаточно высоких температурах. Давление диссо-
диссоциации при 1200° равно 92 мм рт. ст., при 1400° ~ 1 ат. Несколько
ниже 1400° ВаСО3 начинает спекаться; плавится при 1740°. В воде
почти не растворим — при 18° в 1 л воды растворяется ~0,02 г
ВаСО3. Растворимость его повышается в присутствии угольной кис-
кислоты или солей аммония, а особенно в концентрированных рас-
растворах Na2SO4, MgSO4, ZnSO4, Ca(NO3J, CaCl2, K2CO3, MgCl2.
В водной системе ВаСО3 + Na2SO4 +* BaSO4 4- Na2CO3, при 33° и
оптимальном отношении ВаСО3: Na2SO4, равном 11:5, после 7-ча-
7-часового перемешивания выход Na2CO3 в виде 5—6%-ного раствора
достигает 89% 15- В системе BaSO4 — К2СО3 — Н2О при всех тем-
температурах стабильной парой солей является ВаСО3 и K2SO4. Опи-
Описаны условия превращения ВаСО3 в окись, феррит и другие
соединения бария 16. При нагревании смеси ВаСО3 и SiO2 взаимо-
взаимодействие начинается выше 700° с образованием метасиликата ба-
бария, а. выше 800°— ортосиликата. При 1000° и избытке ВаСО3 на-
10 го 30
СаС12,%
Рис. 132. Растворимость
ВаС1г в воде в присут-
присутствии СаС12.
Физико-химические свойства
419
ряду с ортосиликатом образуется трехбариевый силикат. В смесях,
богатых SiO2, метасиликат образуется только выше 1000°". Взаи-
Взаимодействие эквимолекулярных количеств ВаСО3 и Т1О2 начинается
выше 400° с образованием BaTiO3. В интервале 1150—1250° обра-
образуется тетрагональная модификация BaTiO3. Титанат бария обла-
обладает сегнето-электрическими свойствами в широком диапазоне тем-
температур и в этом отношении имеет ряд преимуществ перед другими
сегнето-электриками. Монокристаллы титан ата бария могут быть
получены из расплавленных систем титаната бария и фторидов,
хлоридов, сульфатов, силикатов, карбонатов и других соединений
щелочных металлов; титанат бария плохо растворим в расплавах
большинства галогенидов щелочных металлов, за исключением
фторидов натрия и калия 18> 19.
Сульфат бария BaSO4 имеет плотность 4,5 г/см5, при ~1150°
претерпевает полиморфное превращение, при ~1400° начинает за-
заметно диссоциировать, плавится при 1580°. Искусственно осажден-
осажденный в виде белого, тонкого, почти аморфного порошка называется
бланфиксом. Почти совершенно не растворим в воде20 — насыщен-
насыщенный водный раствор при 18° содержит 0,00023% BaSO4. Раство-
Растворяется в расплавленных щелочах, в концентрированной серной
кислоте с образованием кислого сернокислого бария, немного рас-
растворяется в кипящей соляной кислоте. В системе BaSO4 — NaCl
наблюдается полная смешиваемость в жидком состоянии при от-
отсутствии соединений и твердых растворов; в эвтектике при 741° со-
содержится 33,3 вес.% BaSO421. При длительном кипячении измель-
измельченного барита в растворе соды образуется двойная соль сульфата^
и карбоната бария.
Сульфид бария BaS при кристаллизации из расплавленного со-
состояния образуется в виде кристаллов кубической формы с плот-
плотностью 4,25 г/см3. Технический продукт представляет собой пори-
пористую массу серого или черного цвета (цвет зависит от количества
примеси угля). При растворении в воде BaS гидролизуется с об-
образованием гидроокиси и гидросульфида бария, т. е. ионов ОН~
и HS-:
2BaS + 2Н2О = Ba(HSJ + Ba(OHJ
Из водного раствора может быть выкристаллизован BaS • 6Н2О.'
Кислоты, в том числе и СО2 воздуха, выделяют из BaS сероводо-
сероводород. При нагревании с концентрированными кислотами BaS раз-
разлагается с выделением SO2 и S. В чистом виде сернистый барий
обладает люминофорными свойствами.
При взаимодействии раствора Ва(ОНJ с сероводородом обра-
образуются полисульфиды бария с примесью небольших количеств тио-.
сульфата, сульфида и сульфита. Полисульфиды образуются также
при растворении в воде смеси тонко размолотых плава сернистого,
бария и серы. Образовавшиеся вначале низшие полисульфиды,.
14*
420
Гл. XII. Соли бария
реагируя с серой, переходят в высшие — BaS-Ss, BaS«S422. Поли-
Полисульфиды бария достаточно устойчивы: температуры плавления
BaS2 —925°, BaS3 —554°.
Фторид бария BaF2 имеет плотность 4,83 г/см3 и плавится при
1280°. При 18° в 1 л воды растворяется 1,63 г BaF2.
Кремнефторид бария BaSiF6 получают взаимодействием рас-
растворов кремнефтористоводородной кислоты и хлорида бария, а так-
также по реакции 23
BaS + H2SiF6 = BaSiF6 + H2S
при небольшом избытке H2SiF6 во избежание образования BaF2 по
реакции:
BaSiF6 + 2BaS + 2H2O = 3BaF2 + SiO2 + 2H2S
При 25° в 1 л воды растворяется 0,25 г BaSiF6.
Все растворимые соединения бария ядовиты. Смертельная доза
~0,8 г бариевой соли. Средство первой помощи — прием внутрь
разбавленного раствора сульфата магния. О технике безопасности
в производстве бариевых солей см.24.
ПРИМЕНЕНИЕ
Хлорид бария применяется в качестве яда для борьбы с вреди-
вредителями полевых, огородных и садовых культур, в частности для
уничтожения свекловичного долгоносика25. Его применяют также
в керамической промышленности, в текстильном производстве, для
изготовления некоторых минеральных красок, для очистки котель-
котельной воды и рассолов от иона SOr, при получении некоторых ред-
редких металлов (например, радия) в качестве соосадителя при выде-
выделении их солей из растворов и т. д.
Нитрат бария применяют для получения окиси и перекиси ба-
бария, в пиротехнике при изготовлении осветительных составов (Ва
окрашивает пламя в желтовато-зеленый цвет), в производстве не-
некоторых взрывчатых веществ. Системы, содержащие нитрат бария
и хлориды натрия и калия, представляют интерес в качестве низко-
низкотемпературных соляных ванн для термической обработки метал-
металлов
26
Карбонат бария используют для изготовления карбюризаторов,
применяемых для науглероживания (цементации) стальных изде-
изделий при высоких температурах. Они представляют собой зерна дре-
древесного угля или активированного каменноугольного полукокса
(ГОСТ 5535—50), покрытые слоем ВаСО3; содержание последнего
10—25%. Карбонат бария применяют в керамической промышлен-
промышленности, в производстве оптического стекла, эмалей, а также в радио-
радиоламповом производстве для покрытия катодов27'*8. Карбонат ба-
бария используют также в качестве зооцида — для борьбы с грызу-
грызунами. "
Применение
421
Окись, гидроокиси и перекись бария производятся в небольших
количествах. Окись бария добавляют к материалам, из которых
изготовляют сердечники электромагнитов. Перекись бария приме-
применяют для изготовления запалов, используемых при термитной свар-
сварке. Гидроокись бария в некоторых странах употребляют в сахар-
сахарной промышленности для выделения сахара из мелассы.
Природный сульфат бария — барит служит в СССР основным
сырьем для производства всех солей бария. Тонкоизмельченный
барит используют как утяжелитель глинистых растворов при глу-
глубоком бурении нефтяных и газовых скважин. В США для этой
цели используют около 80% потребляемого барита29-30. Помимо
этого, его применяют для производства бланфикса и литопона, а
также при изготовлении специальных сортов стекла, эмалей, гла-
глазури и др. Бланфикс и лучшие сорта барита используют в качестве
наполнителя при производстве высших сортов бумаги, при произ-
производстве резины, пластмасс, при изготовлении аппретуры в тек-
текстильной промышленности, для получения огне- и кислотоупорных
красок и замазок, в электротехнической и парфюмерной промыш-
промышленности, а также в рентгенотехнике. Литопон —эквимолекулярная
смесь BaSO4 и ZnS, осаждающаяся при сливании растворов BaS
и ZnSO4, имеет наибольшее значение как стойкая против действия
щелочей и сероводорода белая краска 81>32. Однако производство
литопона резко сокращается — его вытесняют титановые пигменты,
имеющие лучшую укрывистость, светостойкость и др.
Сульфид бария является промежуточным продуктом при изго-
изготовлении соединений бария из барита и применяется в кожевенной
промышленности, а также при изготовлении инсектофунгицидов,
в особенности сольбара — полисульфидов бария. Сольбар получают
смешением размолотого до тонкости частиц меньше 0,1 мм плава
сернистого бария с молотой серой. Сольбар, в отличие от извест-
ково-серных отваров, обладает транспортабельностью и незначи-
незначительной фитоцидностью33. Фунгицидное и инсектицидное действие
сольбара (и других полисульфидов) обусловлено не только поли-
полисульфидами, но и тонкодисперсной элементарной серой, являю-
являющейся продуктом их разложения. Сернистый барий особой чистоты
используют при изготовлении люминофоров.
Фтористый барий является антисептиком для древесины и ин-
инсектицидом. Более широкое распространение в качестве инсекти-
инсектицида получил кремнефтористый барий, в отличие от BaF2 не ожи-
ожигающий растения.
Хлорат бария применяют в пиротехнике и в производстве взрыв-
взрывчатых веществ, перхлорат бария — в качестве обезвоживающего
средства, ацетат бария — при крашении тканей, манганат бария —
в малярном деле и проч.
Титанат и ферриты бария, обладающие сегнето-электрическими
ввойствами, применяют в радиоэлектронике. . Как тугоплавкий
422
Гл. XII. Соли бария
Применение
423
полупроводниковый материал представляет интерес дисилицид ба-
бария BaSi234.
Требования к качеству важнейших технических солей бария
приведены в табл. 34 и 35 (в %).
ТАБЛИЦА 35
Требования к качеству сернокислого бария
(содержание компонентов в %)
Содержание сернокислого бария:
в пересчете на сухое веще-
вещество, не менее
в натуральном продукте, не
менее
Содержание влаги, не более . .
Потери при прокаливании, не
более
Содержание примесей в пере-
пересчете на сухое вещество, не
более:
железа
хлоридов в пересчете на хлор
CaSO4
водорастворимых сульфатов
(S0«) .
SiO2
Для баритования
(ГОСТ 5694—68)
марка А
98,5
69
30
0,002*
0,04
0,1
марка Б
98
68.6
30
-
0,006 *
0,05
0,1
Аккумуля-
Аккумуляторного
(ГОСТ 11380—65)
98,5
0,2
1,0
0,01 **
0,04
0,5
0,4
* В пересчете на Fe2O3.
•* В пересчете на Fe.
Сульфат бария, предназначенный для аккумуляторной промыш-
промышленности, не должен содержать примесей соединений тяжелых ме-
металлов, за исключением свинца, и должен иметь определенную
дисперсность: общий вес частиц крупнее 5 мк не должен превы-
превышать 20%, а вес частиц размером 1 мк и менее допускается в пре»
делах 10—40%; остаток после просева на сите № 016 не должен
превышать 1%. Наличие твердых комков не допускается. Насып-
Насыпная плотность продукта 0,73 г/сма Для некоторых потребителей из-
изготовляется продукт с повышенной дисперсностью C0—40% веса
составляют частицы меньше 1 мк).
Бланфикс, выпускаемый в виде пасты для баритования бумаг,
должен иметь белизну не менее 97% (для марки А) и 96% (для
марки Б) от белизны эталона, не содержать веществ, восстанавли-
восстанавливающих КМпО4, сульфидов, сульфатов и тиосульфатов, за исклю-
исключением тех случаев, когда его выпускают подкрашенным в сирене-
сиреневый, голубой, кремовый цвета. Продукт должен удовлетворять
' ТАБЛИЦА 34
Требования к качеству технических соединений бария
(содержание компонентов в %)
s „ . „ Углекислый Азотнокислый барий „ ,
Хлористый барий йяпий * — Гидрат окиси барий
(ГОСТ 742-72) (ГОСТ 2149-65) (ГОСТ 1713-72) «ГОСТ 10848-72)
——————^——^— со знаком ~^~~~~~~~~~~^^~~~~
В1сортЙ 1-й сорт 2-й сорт В"о™тИЙ 1-й СОРТ ""сорт^ '"й СОРТ (ГОсПл468-72) 1-й сорт 2-й сорт 3-Й сорт
Основного вещества, /
не менее 98,5** 96,5** 95** 99,0 97,5 99,5 99,2 -99,4 95.53* 93,03* 88,03*
Примесей, не более: ¦»
гигроскопической
влаги .... 1,0 3,0 3.8 0,5 1,5 0,2 0,3 0,3. — — —
нерастворимого
остатка ... 0,1** 0,154* 0,34* 0,1 5* 0,25* 0,054* 0.154* 0,1 4* 0Л05* 0,15s* 0,36*
железа (Fe) . . 0,002 0,003 0,03 0,005 0,008 0,001 0,005 0,001 0,003 0,005 0,03
хлоридов. . . . 0,3б* 0,4б* 0,8б* 0,087* 0,127* 0,058* 0,18* 0,18* 0.057* 0.087* 0,107*
сульфатов (SO4) — — — 0,05 0,12 — — — — — —
сульфидов . . . 0.059* 0,109* 0,39* 0,01'°* 0,0310* — — — См. ГОСТ —
серы общей (SO*) — — — 0,06 0,21 — — — — — —
ВаСО3 — — , — — — — — — 1,5 1,7 2,5
кальция, магния
и стронция в
пересчете на Са — — — 0,2 0,4 — — — 0,10"* 0,15й* 0,3 м*
натрия — — — — — — — — 0,03 — —
Реакция водного рас-
раствора — — — — — Нейтральная — — —
• Показатели для углекислого бария даны в.пересчете иа сухое вещество.
** ВаС1г-2Н2О.
8* Ва(ОНJ-8Н2О.
4* В воде.
5* В соляной кислоте.
6* Прочие хлориды, кроме BaClj, в пересчете на CaClg.
7* В пересчете на хлор,
8* В пересчете на ВаС12.
в* В пересчете на BaS.
Ю* В пересчете на S.
424
Гл. XII. Соли бария
Сульфат бария
425
требованиям предусмотренного ГОСТ испытания на дисперсность;
остаток после просева на сетке № 008К не должен превышать
0,01%. Для марки А количество металлических вкраплений по ба-
баритовому слою на площади 9 X 12 см не должно превышать шести.
СЫРЬЕ
Атомный кларк бария 0,005%. Он входит в состав силикатных
и других пород, но сырьем для получения соединений бария слу-
служат только минералы барит или тяжелый шпат BaSO4 и витерит
ВаСОз. Мировая добыча барита в 1966 г. составила 3,44 млн. г35.
Витерит более ценное сырье, чем барит, так как легко растворяет-
растворяется в кислотах, что значительно упрощает производственный про-
процесс. Однако месторождения витерита встречаются значительно
реже и менее богаты, чем месторождения барита. Крупные залежи
витерита имеются только в Англии. В СССР основным видом ба-
бариевого сырья является барит. Месторождение витерита имеется
в Туркменской ССР; добываемый здесь витерит после сортировки
и отборки содержит 75—80% ВаСО336.
Наибольшее промышленное значение имеют месторождения ба-
барита, происхождение которых связано с магматическими изверже-
извержениями. В таких месторождениях барит является одним из основ-
основных минералов жилы и сопровождается кварцем, флюоритом, кар-
карбонатами и сульфидами тяжелых металлов. Барит встречается в
виде вторичных скоплений в осадочных породах в форме желва-
желваков, прожилок, иногда пластов. Соединения бария адсорбируются
отрицательно заряженными частицами гидроокислов марганца и
поэтому являются спутниками коллоидальных марганцовых руд.
Например, чиатурские марганцовые руды содержат до 1,5% окиси
бария.
Наиболее мощные месторождения барита сосредоточены в За-
Закавказье (Кутаисское, Ганджинское, Шамхорское, Катар-Коварт-
ское, Актальское и др.), где содержание BaSO4 в рудах иногда
доходит до 97%, в Средней Азии и в примыкающих к ней районах
Западной Сибири. Имеются также месторождения BaSO4 на Урале.
На Алтае барит встречается в месторождениях полиметаллических
или свинцово-цинковых руд, при выделении из которых сульфидов
свинца, цинка, меди, железа, серебра получается баритовый кон-
концентрат (Змеиногорское, Лениногорское, Белоусовское, Березов-
ское, Салаирское и другие месторождения) 37. Сортировка руды
Змеиногорского месторождения дает барит с содержанием 98%
•BaSO4;'8.
Помимо промывки, грохочения и сортировки на классифика-
классификационных столах широко применяют флотационное обогащение ба-
баритовых руд. Отделение барита от пирита возможно только фло-
флотацией, причем содержание барита в пирите может быть снижено
от Ю до 1 % 39. Для разделения смесей барита и флюорита с суль-
сульфидами необходима трехступенчатая селективная флотация в при-
присутствии лаурилсульфоната с добавками растворимого стекла и
лимонной кислоты. Вначале отделяют сульфиды, затем барит и,
наконец, флюорит. Окислы, силикаты, карбонаты и кварц остаются
в отходах40. Предложено очищать тяжелый шпат от CaF2 кипяче-
кипячением измельченного сырья в концентрированном растворе Aids4I.
В Западной Европе в последнее время для отбеливания барита и
удаления сульфидов железа стали применять подкисление (обыч-
(обычно одновременно с дроблением).
В зависимости от применяемых методов первичной обработки
руды получают кусковой, молотый барит или баритовый концен-
концентрат; все эти виды сырья выпускают четырех сортов (табл. 36).
ТАБЛИЦА Зв
Требования к качеству баритового сырья
(Содержание компонентов в сухом веществе в % по ГОСТ 4682 — 49)
Высший
сорт
95
1,5
¦ 0,5
0,3
3
1
I
№ 1
I сорт
90
2,5
1.5
1.0
5
^йтральиа'
№3
II сорт
85 s
4.0
3.0
1.0
5
2
№5
III сорт
80
Не нормировано
1.0
10
Не нормировано
BaSO4, не менее
SiO2, не более
Fe2O3 » »
Вещества, растворимые в воде,
не более
Влага в баритовом концен-
концентрате, не более
Влага в молотом барите, не
более
Реакция водной вытяжкн . .
Цвет по шкале белого цвета,
не выше 1
Молотый барит всех сортов по гранулометрическому составу
подразделяют на класс А (наполнитель), для которого остаток на
сите 0,085 мм должен быть не более 1% и на сите 0,15 мм — не
более 0,1%, и на класс Б (утяжелитель), для которого остаток на
сите 0,085 мм должен быть не более 5%.
Лучшие сор*та кускового барита и высококачественный моло-
молотый барит применяют в лакокрасочной промышленности. Для про-
производства солей бария используют обычно кусковой барит I и II
сортов и баритовый концентрат.42
СУЛЬФАТ БАРИЯ
Искусственный сульфат бария изготовляют в виде тонкодис-
тонкодисперсного порошка следующими способами:
1 1) осаждением из водных растворов солей бария серной кисло-
т*й или сернокислыми солями;
426
Гл. XII. Соли бария
Восстановление барита
427
2) обработкой природного барита различными способами.
Сульфат бария получается также в качестве побочного про-|
дукта при сульфатной очистке соляных рассолов, при полученш
перекиси водорода разложением перекиси бария серной кислотор
(или другими кислотами с последующей обработкой продуктов
реакции серной кислотой); его можно получать из отходов очистки
от сульфатов дистиллерной жидкости содового производства43.
Для осаждения BaSO4 из растворов часто используют различ-|
ные отбросные растворы и промывные воды, получаемые при прс
изводстве соединений бария и содержащие растворимые соли
BaS, BaCl2, Ba(NO3J и др. Лучшие сорта сульфата бария полу-|
чают обычно из специально приготовленных растворов хлорида
бария путем осаждения серной кислотой или сульфатом натрия
по реакциям:
ВаС12 + H2SO4 = BaSO4 + 2НС1
ВаС12 + Na2SO4 = BaSO4 + 2NaCl
При осаждении из горячих растворов при низких значениях pf
(т. е. при осаждении серной кислотой) получается менее дисперс|
ный кристаллический осадок, легко оседающий на дно аппарата)
Осаждение из растворов с высоким значением рН (например, ^
растворов BaS или из растворов нейтральных солей с помощьь
Na2SO4), особенно при невысоких температурах, приводит к обра{
зованию очень медленно оседающих осадков. Подбором соответ!
ствующих условий осаждения можно получить осадки любой сте|
пени дисперсности, вплоть до коллоидных44.
Осадки BaSO4, отделенные от жидкости, подвергают тщатель!
ной промывке, иногда неоднократному кипячению в воде и пропа-
риванию с целью удаления примесей, особенно С1~ и других, спо-1
собных окрашивать продукт. Затем осадки отжимают на фильтр-
прессах и выпускают в виде пасты, содержащей 20—30% водьь
Если требуется сухой продукт, то промытый осадок отжимают ни
гидравлическом прессе и затем сушат при ПО—120°, а иногда про!
каливают при 300—350°. После сушки и прокалки скомковавшийсЯ
продукт размалывают на вальцовой мельнице.
Для получения осадка BaSO4, легко отмываемого от окклюди-1
рованного SO?", рекомендуют45 производить осаждение при бы-.'
стром размешивании непрерывно поступающих в реакционный:\
аппарат растворов ВаС1г и Na2SO4, причем в реакционной зоне
молярная концентрация SOi~ должна быть все время больше,
чем Ва2+. Осажденный BaSO4 можно получать в процессе каусти-'
фикации Na2SO4 с помощью раствора Ва(ОНJ или Ва(ОН) (HS).'I
Выход NaOH в виде 10%-ного раствора достигает 87% 46- Предло-j
жено47 получать BaSO4 со свойствами, характерными для блан-з
фикса, путем тончайшего размола тяжелого шпата (лишь ~%]
помола с размером частиц больше 4 мк). Очищенный BaSO4 мокег)
быть получен из природного барита обработкой тонкоизмельчен-
ного минерала серной или соляной кислотой с целью удаления из
него примесей. Так, при обработке барита олеумом BaSO4 раство-
растворяется, а затем, после отделения нерастворившегося остатка, вы-
выделяется при разбавлении раствора водой. При обработке природ-
природного барита соляной кислотой в раствор переходят соединения
алюминия, железа, кальция, а в осадке остаются BaSO4 и ЭЮг.
Для такой обработки целесообразно использовать богатые бариты.
Так, из минерала, содержащего 95—98% BaSO4, 1—2% SiO2 и
1—3% СаО, А12Оз, Fe2O3, после двухкратной обработки соляной
кислотой с плотностью 1,19 г/см3, разбавленной 1 : 10, в течение
2 ч при кипячении с последующей отмывкой ионов хлора водой
был получен продукт, содержащий 99% BaSO4 и 1% SiO2. Устано-
Установлено, что примесь инертного SiO2 не препятствует использованию
продукта для рентгеноскопии43.
Очистку природного барита можно производить также раство-
растворением его в расплавленных солях — NaCl или СаС12; после от-
отстаивания примесей и декантации расплава его охлаждают, и по-
после обработки застывшей массы водой получают чистый BaSO449.
Таким путем можно получить BaSO4, пригодный для бумажной и
аккумуляторной промышленности, лишь из первосортной барито-
баритовой руды60.
ВОССТАНОВЛЕНИЕ БАРИТА
Для перевода практически не растворимого ни в воде, ни в кис-
кислотах барита в растворимое соединение его восстанавливают в
сернистый барий. В дальнейшем сернистый барий легко перера-
перерабатывают на любые соединения бария. Восстановление барита
ведут почти исключительно в трубчатых вращающихся печах с по-
помощью угля и природного или генераторного газа.
Несмотря на то, что шихта состоит из барита и угля, BaSO4
восстанавливается главным образом окисью углерода:
BaSO4 + 4СО = BaS + 4СО2
Двуокись углерода восстанавливается углеродом до СО по
реакции СО2 -г С^2СО. Выше 1000° в составе равновесной газо-
газовой смеси в присутствии углерода СО2 практически отсутствует.
Поэтому восстановление барита выше 900—1000° протекает по
суммарной реакции
BaSO4 + 4С = BaS + 4CO - 138,5 ккал A)
а при 600—800° по суммарной реакции:
BaSO4 + 2С= BaS+ 2СО2-56,1 ккал
B)
Вследствие эндотермичности этих реакций необходим подвод
тепла, для чего сжигают в печи природный или генераторный газ,
428
Гл. XII. Соли бария
Восстановление барита
429
компоненты которого (СН4, Н2, СО) при неполном их сгорании
также восстанавливают барит.
С точки зрения расхода угля и затраты тепла выгоднее вести
процесс восстановления при низких температурах, по реакции B),
но так как он при этом протекает медленно, то предпочитают осу-
осуществлять его по реакции A), идущей значительно быстрее, и для
этого температуру в «горячем» конце печи поддерживают выше
1000°.
В трубчатых печах осуществляется противоток восстанавливае-
восстанавливаемого материала и греющих газов. Поэтому практически в газах,
г.
Рис. 133. Восстановление барита камен-
каменным углем при разных температурах:
1 — кислоторастворимые солн баркя 2 — водо-
водорастворимые соли бария.
700 750 600 650 900 050
ТЬмпература^С
Рис. 134. Зависимость восстано-
восстановления барита от вида восстано-^
вителя:
/—древесный уголь; 2 — кокс; 3— антра-J
цит; 4 — каменный уголь.
уходящих из «холодного» конца печи, соприкасающихся с шихтой,
богатой углеродом, СО почти не содержится, а содержание СО2
достигает 20—25%. Этому способствует также соответствующее
регулирование подачи в горелку печи воздуха, необходимого для[
сжигания газа. Таким образом, суммарной реакцией процесса вос-
восстановления является реакция B). При интенсивных режимах
обжига, с целью увеличения температуры факела горящего газа,
работают с недостатком воздуха; в этом случае СО полностью не|
сгорает, и в отходящих газах содержится иногда до 5% СО.
Скорость восстановления барита сильно зависит от темпера-I
туры (рис. 133) и сорта твердого углеродистого восстановителя
(рис. 134). В лабораторных опытах, результаты которых приве-
приведены на рис. 133 и 134, использован барит, содержащий (в %):
ВаО —64,98, SO3 —34,13, СаО — 0,26, Fe2O3 — 0,06, SiO2 —0,65,
Н2О — 0,08. Восстановители содержали: высушенный древесный
уголь—18,8% летучих и 3,8% золы; кокс—1% летучих, 0,6% вла-
влаги, 9,2% золы; антрацит — 5,4% летучих, 2,6% влаги, 2,7% зольш
каменный уголь — 27,4% летучих, 1,2% влаги, 7,5% золы. Дисперс-
Дисперсность барита и восстановителей — 0,13 мм. Количество восстано-
восстановителя брали с 60—55%-ным избытком по углероду 51>Б2. Как вид-
видно, каменный уголь — более активный восстановитель, чем древес-
древесный уголь, кокс и антрацит. Поэтому предпочитают употреблять
каменный уголь, например марки ПЖ, несмотря на то, что расход
его больше, чем тощих углей (например, антрацита), содержащих
мало летучих веществ. При невысоких температурах скорость вос-
восстановления барита увеличивается в присутствии карбонатов нат-
натрия и калия, особенно Na$CO353.
Барит восстанавливается вначале до
BaSO4 + СО = BaSO3 + СО2
а лишь затем до BaS — в получаемом плаве сернистого бария со-
содержится некоторое количество ВаБОз. Выше 600° сульфит бария
с большой скоростью диссоциирует54-57
BaSO3
ВаО + SOjj
поэтому содержание его в плаве незначительно. Но с еще большей
скоростью ВаО реагирует с парами элементарной серы, образо-
образовавшейся в результате восстановления SO2
2ВаО + P/2S2 = 2BaS + SO2
поэтому ВаО в плаве не обнаруживается. BaSO3 образуется также
по реакции:
BaS
+ H2O = BaSOj + H2S
Может иметь место и обратное окисление BaS сернистым га-
газом до BaSO4S8:
BaS + 2SO2 = BaSO4 + S2
Частичное взаимодействие BaSO3 и BaS с СОг
BaSO3 + COS = ВаСОз + SO2
BaS + 4CO2 = BaCO3 + SO2 + 3CO
приводит к появлению в плаве ВаСО3, а взаимодействие ВаЭОз с
S — к появлению тиосульфата
BaSO3+ '/282=638203
Содержащиеся в барите примеси (SiO2, AI2O3, CaF2, Fe2Os)-
выше 1000° вступают в реакции, затрудняют процесс восстановле-
восстановления-и снижают выход BaS. Поэтому бедные бариты следует пред-
предварительно обогащать. Кремнезем образует с сульфатом бария
430
Гл. XII. Соли бария
мета-, орто- и трехбариевый силикаты и способствует появлению
в газовой фазе SOg и S:
BaSO4 + 8Ю2 = BaSiO3 + SO3
2BaSO4 + BaSiO3 = Ba3Si05 + 2SO3
SO3 + CO = SO2 + CO2
Ортосиликат и трехбариевый силикат при выщелачивании пла-
плава водой разлагаются с образованием гидроокиси и метасиликата
бария:
Ba2SiO4 + Н2О = BaSiO3 + Ba(OHJ
Ba3Si05 + 2Н2О = BaSiO3 + 2Ba(OHJ
Ю0
§60
I50
При выщелачивании BaS из плава водой нерастворимые при-
примеси BaSO3, ВаСО3 и силикатов бария являются вредными, так
как ведут к потерям бария в остаю-
остающемся после выщелачивания шламе.
При кислотном выщелачивании плава
барий из этих кислоторастворимых
соединений также переходит в раствор.
Окислы алюминия и железа в про-
процессе восстановления барита реагиру-
реагируют с сульфидом и сульфатом бария,
| образуя алюминаты и ферриты бария
„ 10 к Ю & 26 30 34 36 42 (ВаО-А12О3 и BaO-Fe2O3). При взаи-
СодержжЬугля бшите,% модействии окиси железа с сульфидом
бария образуется также тиоферрит
Рис. 135. Зависимость степени BaFe2S4. Эти побочные реакции так-
также приводят к уменьшению выхода
растворимых бариевых соединений.
Если, однако, выщелачивание плава
водой вести не по обычной противот-4"-
ной схеме, а при повышенных температуре и давлении, то выход
бария в раствор повышается на 10—15% за счет разложения сили-
силикатов, алюминатов и ферритов. Для дополнительного извлечения
бария предложено шлам после водного выщелачивания BaS спе-
спекать с твердым СаС12 при 300° и затем нагревать в течение 0,6—1 ч
при 600—800°, после чего из охлажденного спека водой извлекается
ВаС1259. Для обезвреживания отбросного шлама его предложено
обрабатывать хлорной известью 60.
На рис. 135 показано изменение степени перехода BaSO4 в BaS
при восстановлении природного барита и содержания BaS в плаве
в зависимости от количества каменного угля в шихте. Эти дан-
в плаве (крнвая 2) от коли-
количества угля в шихте.
Восстановление барита
431
ные61 получены для барита, содержавшего 81,5% BaSO4, 0,4%
СаО, 4,2% SiO2, 5,2% R2O3, 0,2% CaF2, и малозольного каменного
угля, подвергавшихся прокаливанию при 950° в течение 3 ч. При
оптимальном составе шихты B2% угля) достигается выход BaS
~97% и плав содержит ~67% BaS. Увеличение содержания SiOg
в барите до 12,6% при снижении BaSO4 до 77,66% приводит в тех
же условиях к уменьшению выхода BaS до 81,4% и содержания
BaS в плаве до 40,2%. Аналогичные результаты получаются,
когда шихта содержит 9,7% Fe2O3 и 73% BaSO4.
Для предотвращения образования силикатов бария при ис-
использовании баритов, содержащих много S1C>2, к шихте можно до-
добавить известь или известняк. При этом SiO2 связывается в не ра-
растворимый в воде силикат кальция. Это имеет смысл лишь в том
случае, если плав подвергается водному выщелачиванию, так как
при кислотном выщелачивании это лишь увеличивает расход кис-
кислоты на связывание окиси кальция.
Присутствие в шихте примеси известняка ускоряет процесс вос-
восстановления, так как выделяющаяся при разложении карбоната
двуокись углерода способствует перемешиванию шихты, а окись
кальция препятствует сплавлению шихты легкоплавкими приме-
примесями, находящимися в барите. В результате увеличивается степень
восстановления барита. Кроме того, кальций заменяет часть бария
при взаимодействии с примесями, находящимися в природном ба-
барите. При восстановлении баритов, в состав которых входит кар-
карбонат бария, целесообразно добавлять в шихту CaSO4, это повы-
повышает содержание серы в газовой фазе и способствует переходу
карбоната бария в сульфид, т. е. увеличивает содержание водо-
водорастворимых соединений бария в плаве62.
Производственный процесс получения плава сернистого бария
сводится к следующему. Измельченные барит (до крупности 0,2—
0,3 мм, иногда 4—5 мм) и уголь (до крупности 2—3 мм) посту-
поступают через автоматические дозаторы в барабанный или лопастной
смеситель. Тонкое измельчение барита не обязательно, так как
крупнозернистый барит при температуре восстановления растрес-
растрескивается и распадается на частицы меньше 0,5 мм, а основная
масса их имеет размеры 100—300 mk6S-m. Шихта поступает в бун-
бункер, откуда через автоматический питатель, например, тарельча-
тарельчатого типа, подается в печь для восстановления. Трубчатая вра-
вращающаяся печь, расположенная с уклоном 3—6°, делает от 1 до
5 об/мин. В зависимости от производительности (до 40—50 г ба-
барита в сутки) устанавливают печи длиной до 40 м с внутренним
диаметром до 1.5—2 м. Стальной корпус печи футерован внутри
двумя слоями шамотного кирпича. Нижняя торцевая часть бара-
барабана печи введена в неподвижную каретку; в ней имеется горелка,
через которую подают газообразное топливо и воэдух. Из бара-
барабана печи в каретку поступает и выгружается из нее раскаленный
432
Гл. XII. Соли бария
Восстановление барита
433
плав сернистого бария. Противоположный конец печи входит в
пыльную камеру.
С этой же стороны в печь введена течка от тарельчатого пита-
питателя для шихты. Течка охлаждается водяной рубашкой. Иногда
вместо газа для обогрева печи используют пылевидное твердое
топливо (уголь) или жидкое топливо (мазут). При температуре
в реакционной зоне вблизи факела горящего газа 1050—1100° про-
продолжительность прохождения шихты через печь составляет 1,5—
2 ч. Повышением температуры можно значительно сократить дли-
длительность восстановления и повысить производительность печи
(это потребует соответствующего увеличения числа оборотов
печи). Отходящие газы выносят из печи большое количество пыли,
которая улавливается в пыльных камерах, циклонах или электро-
электрофильтрах и возвращается на приготовление шихты. Тепло газов
утилизируется в паровых или водяных котлах. Раскаленный плав
выходит из печи в виде сыпучей массы, состоящей из пористых
гранул величиной с горошину. Он проходит через холо„ильник—
вращающийся стальной барабан, снаружи охлаждающийся возду-
воздухом или орошаемый водой. Нагретый воздух из холодильника по-
поступает в печь. По остывании плав имеет черный или темно-серый
цвет. Он содержит 63—75% BaS (водорастворимый барий),
15—20% кислоторастворимых соединений бария (BaSO3, ВаСО3,
ВаВЮз) и примеси, в состав которых входит и некоторое количе-
количество непрореагировавшего угля.
Во влажном воздухе плав сернистого бария карбонизуется и
выделяет сероводород по реакции BaS + CO2+H2O = BaCO3+H2S,
в результате чего содержание водорастворимого бария пони-
понижается. Кроме того, BaS постепенно окисляется кислородом воз-
воздуха. Поэтому в случае необходимости транспортирования за пре-
пределы завода плав укупоривают в деревянные бочки.
Восстановление баритового концентрата, имеющего высокую
дисперсность (средний размер частиц ~40 мк, 10% меньше
10 мк) и, особенно, содержащего меньше 92% BaSO4, труднее, чем
измельченного кускового барита, так как шихта при этом сильно
спекается, приплавляется к стенкам печи, а газ уносит много ба-
баритовой пыли (до 10%). Опыт восстановления шихты из барито-
баритового концентрата (93—94% BaSO4) и кокса в соотношении 4 : 1 во
вращающейся печи диаметром 2,2 м, длиной 20 м с рабочим объе-
объемом 50 м3 и поверхностью нагрева ПО м2 при степени заполнения
барабана 6—9% и температуре в передней части 1050—1100° по-
показал, что степень восстановления в среднем равна 92% и дости-
достигает 95%, а нагрузка печи 35—40 кг/(м3-ч), т. е. почти такая же,
как при восстановлении молотого барита 41 кг/(мг-ч). Плав выхо-.:
дит из печи в виде гранул 2—15 мм. Отмечено, что материал дви- '<
жется в печи концентрическими слоями — мелкая фракция нахо-
находится в ядре потока и оказывается плохо восстановленной. Расчет
показывает, что расход тепла на восстановление BaSO4 составляет
приблизительно 32%, на испарение и перегрев пара 4%, с отходя-
отходящими газами 36%, с плавом 10%, теряется излучением 15% и в
результате химического недожога 4—5% 65. Предварительная гра-
грануляция баритового концентрата в зерна 3—5 мм с добавкой свя-
связующих уменьшает пылеунос из печи.
Заводскими опытами показана целесо-
целесообразность гранулирования шихты из
флотационного барита и коксика (с
размером зерен меньше 3 мм) с добав-
кой в качестве связующего 8%-ного
раствора сульфитного щелока (8—10%
от веса шихты) 66. Благодаря пористо-
пористости гранулированного барита его вос-
восстановление идет быстрее, чем дробле-
дробленого, и равная степень восстановления
достигается при температуре на ~ 100°
более низкой. Исследовано восстанов-
восстановление барита электротермическим спо-
способом в лабораторной однофазной пе-
печи (d = 300 мм) мощностью 25 кет67,
Большие преимущества имеет вос-
восстановление барита газами. При этом
получается чистый плав, не загрязнен-
загрязненный углем и золой, что значительно
облегчает его дальнейшую перера-
переработку.
При взаимодействии BaSO4 с СО
в интервале 700—1000° образуется
главным образом BaS и лишь немного
600
Температура,"?
Рис. 136. Зависимость степени
восстановления BaSO4 водо-
водородом (зерна барита меньше
0,3 мм) и метаном (зерна ба-
барита меньше 0,5 мм) ст темпе-
температуры при продолжит гльностл
процесса 1 ч в лабораторных
условиях.
ВаСО3. Скорость восстановления барита окисью углерода прибли-
вительно равна скорости восстановления коксом. Равновесие ре-
реакции
BaSO4 (тв.) + 4СО (г.) ^±. BaS (тв.) + 4СО2 (г.)
в отсутствие свободного углерода сильно сдвинуто в сторону обпа-
зования BaS даже при высоких температурах. Так, при 1200° в
равновесной газовой смеси 91% СО2 [lg Кв - lg (Pco2: Pqo) =a
8
Восстановление BaSO4 водородом ниже 700° идет очень мед-
медленно, при 850—900° — быстро. Восстановле'.яе BaSO4 метаном
идет медленнее, чем водородом, и требует более высоких темпера-
температур (рис. 136) 69. Начальная скорость восстановления BaSO4 водо-
водородом или окисью углерода растет пропорционально давлению
гава (изучено до 600 мм рт. ст.); энергия активации этих реакций
434
Гл. XII. Соли бария
Хлорид бария
435
составляет соответственно 47 000 и 56 000 кал/моль 70. Для реакции
BaSO4 (тв.) + 4Н2 (г.) = BaS (тв.) + 4Н2О (г.)
в интервале 800-1000° lgftp.= 5,447+- т
Перспективно восстановление барита природным газом в печах I
с кипящим слоем. При 900° за 30—40 мин степень восстановления |
достигает 94%, а степень использования восстановителя (газа)
32—36% 72. По другим данным73, при восстановлении барита про-|
дуктами неполного сгорания природного газа (~13% Н2 и 10%
СО) в течение 70—80 мин при 800—900° и высоте кипящего слоя|
1 м получается «плав», содержащий 62—64% BaS и 10—12%
ВаСОз- Степень использования газов в процессе восстановления:)
СО — 74% и Н2 — 52%. Гранулометрический состав плава прибли-J
зительно такой же, как исходного барита.
ХЛОРИД БАРИЯ
Из многих способов получения хлорида бария74 наибольшее
промышленное распространение получили солянокислотный и
хлоркальциевый способы. Хлорнатриевый, хлораммониевый, хлор-
магниевый, хлорный и другие способы получения хлорида бария
не имеют широкого применения, хотя некоторые из них в опреде-
определенных условиях используются или могут быть эффективно ис-
использованы.
Солянокислотный способ
. В основе этого способа лежит реакция:
BaS + 2НС1 = ВаС12 + H2S
Охлажденный плав сернистого бария размалывают в шаровой
или трубчатой мельнице и направляют на разложение соляной ки-
кислотой. При размоле плава в мельницах мокрого помола выгру-
выгружаемый из печи плав не охлаждается, а поступает по желобу в
мельницу, в которую подается вода или слабый щелок б количе-
количестве, достаточном для образования густой шламообразной массы.
При этом отпадает необходимость в улавливании вредной пыли и
уменьшаются потери от окисления плава воздухом.
Разложение плава соляной кислотой производят в стальных!
реакторах, футерованных кислотоупорным кирпичом или диабазо-
диабазовой плиткой в два слоя. Подачу плава в соляную кислоту произ-
производят в течение 1,5—2 ч при перемешивании реакционной массы
до тех пор, пока раствор не приобретет щелочную реакцию, при
содержании 18—20 г избыточного BaS в 1 \г. Это приводит к оса-
осаждению из раствора примеси железа
FeCl2 + BaS = FeS + BaCl,
и других тяжелых металлов.
Для полного разложения кислоторастворимых соединений
(BaSO3, ВаСОз, силикатов бария) и для того чтобы выделяю-
выделяющаяся при разложении силикатов кремневая кислота не перехо-
переходила в раствор, используют соляную кислоту с концентрацией не
ниже 13% НС1, лучше 15% НС1. Применение более концентриро-
концентрированной кислоты нежелательно, так как это способствует выделе-
выделению элементарной серы, теряемой с отвалом.
По окончании разложения в реактор добавляют горячие про-
промывные воды для разжижения реакционной массы с целью уско-
ускорения отстаивания раствора от нерастворимых компонентов плава
и в особенности от тонкодисперсного илистого осадка сернистого
железа. После отстаивания осадка раствор декантируют, остав-
оставшийся шлам, содержащий до 5% ВаСЬ • 2Н?О, промывают горячей
водой (~80°). Полученные при промывке шлама слабые растворы
хлористого бария используют для следующих промывок, для раз-
разжижения реакционной массы перед отстаиванием и для мокрого
помола плава сернистого бария. Щлам после промывки смывают
струей воды и отправляют по желобу в отвал.
Запатентована76 непрерывно действующая установка для раз-
разложения раствора BaS соляной кислотой, состоящая из двух по-
последовательно включенных реакторов. Первый из них представляет,
собой трубу, в которой смешиваются растворы, а второй — резер-
резервуар, разделенный на две камеры вертикальной перегородкой, не
доходящей до крышки; реакционная смесь поступает в нижнюю
зону одной камеры, а удаляется из другой.
Отделение щелоков от шлама и промывку его производят те ре-
реже на центрифугах, добавляя предварительно к пульпе древесные
опилки, что облегчает отжим илистого осадка. Отделенный от
шлама щелок для освобождения от взвешенных частиц пропу-
пропускают через фильтр-пресс и направляют в стальной закрытый .ре-
.резервуар с мешалкой, где щелок нейтрализуют соляной кислою.-.
Для удаления растворенного сероводорода щелок продувают ост-
острым паром. Полученный чистый раствор, содержащий 260—300 г/л
ВаС12, смешивают с маточными щелоками, оставшимися после
кристаллизации, и подвергают выпариванию до концентрации
400—600 г/л ВаС12, а затем при охлаждении до 25—35е кристал-
кристаллизуют ВаС12-2Н2О. Иногда выпаривают только маточные ще-
щелоки, которые затем сливают с чистым раствором, полученным
после выщелачивания, и смешанный щелок направляют на кри-
кристаллизацию. На современных заводах солянокислотное разложе-
разложение пульпы сульфида бария ведут непрерывным способом в кас-
каскаде реакторов. Затем шлам отделяют с помощью автоматических
436
Гл. XII. Соли бария
Хлорид бария
437
5 10
15 20 25
ВаС1г,вес.Уо
30
фильтрпрессов76. Выпарку растворов хлористого бария осуществ-
осуществляют в многокорпусных вакуум-выпарных аппаратах. Температура
кипения раствора в первом корпусе равна 105—106°; в послед-
последнем— около 65°. Иногда выпаривание в вакуум-аппаратах совме-
совмещают с кристаллизацией.
Кристаллизацию часто осуществляют в непрерывно действую-
действующих кристаллизаторах, имеющих форму желоба с двойными стен-
стенками, между которыми циркулирует охлаждающая вода. Длина
желоба 16 м, ширина 1,5 м, высота 0,5 м. Раствор течет по желобу
противотоком охлаждающей воде. Выпадающие на дно желоба
кристаллы передвигаются с помощью механических гребков к
концу кристаллизатора и сваливаются
в приемник, из которого посту-
поступают на центрифугу. Для кристалли-
кристаллизации применяют также барабанные
кристаллизаторы с воздушным охлаж-
охлаждением.
Отжатый на центрифугах от маточ-
маточного раствора кристаллический хлори-
хлористый барий промывают водой. Кри-
Кристаллы, выгружаемые из центрифуги,
содержат обычно не более 4% гигро-
гигроскопической влаги и укупориваются в
бочки. Иногда продукт подвергают
сушке горячим воздухом (ПО—120°)
в барабанных вращающихся сушилках для понижения содержа-
содержания гигроскопической влаги до 1%.
При получении хлористого бария описанным солянокислотным
способом на 1 г 97%-ного ВаС12-2Н2О расходуют: 1,25—1,35 т ба-
барита A00% BaSO4), 0,4—0,5 т реакционного угля G000 ккал/кг)
и 1,4—1,6 т 27,5%-ной соляной кислоты. В качестве отхода на
каждую тонну хлористого бария из реактора удаляется 0,11—
0,13 т сероводорода, который перерабатывают на элементарную
серу, сернистый газ (серную кислоту), гидросульфид или тиосуль-
тиосульфат натрия и пр. Вследствие ядовитости сероводорода должны
быть обеспечены условия, препятствующие его проникновению в
производственные помещения; предельно допустимая концентра-
концентрация H2S в воздухе рабочей зоны 0,01 мг/л.
Выделение кристаллов хлористого бария из щелока может быть
произведено высаливанием путем добавления к щелоку соляной
кислоты. Растворимость ВаС12 в воде при 25° равна 27,1%. В рас-
растворе, содержащем 21,66% НС1, растворимость его понижается до
0,1% (рис. 137). При смешении равных объемов ~27%-ного рас-
раствора НС1 и насыщенного при 25° раствора ВаС12 выкристалли-
выкристаллизуется около 86% соли. При повышении температуры эффектив-
эффективность высаливания увеличивается. Для уменьшения содержания
Рис. 137. Растворимость в ci.-
стеме ВаС12— НС1—Н2О.
кислоты в кристаллическом осадке кислоту следует добавлять мед-
медленно при 60°. Затем кристаллы отмывают от кислоты двухкратной
промывкой нейтральным раствором хлористого бария. Оставшийся
после высаливания кислый щелок (соляную кислоту с примесью-
ВаС12) направляют на разложение плава сернистого бария.
Хлорнатриевый способ
В солянокислотном способе получения хлористого бария соля-
соляная кислота может быть частично заменена поваренной солью779.
При этом разложение плава сернистого бария производится умень-
уменьшенным в 2 раза количеством
соляной кислоты по реакции:
2BaS + 2HC1 = ВаС12 + Ba(HSJ
Из полученного гидросуль*
фидного щелока — раствора
гидросульфида и хлорида ба-
бария — хлооид бария высали-
высаливается поваренной солью:
ВаС12 + Ba(HSJ + 2NaCl + 2H2O =¦
= 2ВаС12 - 2Н2О + 2NaHS
Одновременно получается
гидросульфид натрия без за-
затраты едкого натра. NaHSf
При обработке водной сус-
суспензии плава сульфида бария,
нагретой до 70°, рассчитан-
рассчитанным количеством соляной кис-
кислоты можно получить гидро-
гидросульфидный щелок любой концентрации вплоть до насыщенного
хлоридом бария раствора. Несмотря на уменьшенное количество
соляной кислоты, растворение плава происходит быстро, так как
равновесие
н2о
Рис. 138. Схема диаграммы раствори-
растворимости для системы Ba(HSJ + 2NaCl=»
¦=BaCl2 + 2NaHS при 20°.
BaS (тв.)
BaS (раств.) <--
Ba(HSJ+Ba(OHJ
сдвинуто вправо вследствие нейтрализации ионов ОН соляной
кислотой.
На схеме диаграммы растворимости водной системы Ba(HSJ +j
. ¦+ 2NaCl = ВаС12 + 2NaHS при 20° (рис. 138) видно, что поле кри-
кристаллизации хлористого бария занимает большую часть квадрата
и что точка Е\ совместного существования NaCl, Ba(HSJ и
ВаС12'2Н2О является инконгруэнтной. Наибольший выход кри-
кристаллического хлористого бария получится в случае кристаллиза-
кристаллизации по лучу DEi. Фигуративные точки комплексов, получающихся
438
Гл. XII. Соли бария
Хлорид бария
439
смешением Ba(HSJ и NaCl, лежат на диагонали АС. Следова-
Следовательно, наивыгоднейший состав исходной смеси Ba(HSJ и NaCl
будет изображаться точкой гр, лежащей на пересечении диагонали
и луча кристаллизации. Введение в систему хлористого бария сдви-
сдвинет положение точки исходной смеси к точке D и путь кристалли-
кристаллизации удлинится, что является признаком увеличения выхода кри-
кристаллов.
Если процесс вести, применяя растворы, не содержащие ВаС12,
т. е. при начальном составе системы, изображаемом точкой -ф, то,
согласно диаграмме, молярное соотношение 2NaCl: Ba(HSJ равно
0,54:0,46 (при содержании в исходном растворе 9,5 моль воды на
1 моль суммы солей). Для устранения совместной кристаллизации
NaCl и Ba(HSJ конец пути кристаллизации не должен доходить
до точки Еи оканчиваясь вблизи ее, например, в точке S.
Для получения раствора состава точки -ф необходимо исходить
из 35% раствора гидросульфида бария. Даже насыщенный серо-
сероводородом при 100° раствор сульфида бария не даст раствора гид-
гидросульфида такой концентрации. Однако такой раствор, содержа-
содержащий 35% Ba(HSJ при 90°, можно получить, если выщелачивать
плав сульфида бария не водой, а раствором гидросульфида бария".
Необходимый же для выщелачивания раствор гидросульфида ба-
бария можно получить, насыщая раствор BaS сероводородом.
Для соблюдения оптимального соотношения необходимо ввести
ъ 1 кг 35% раствора гидросульфида бария 0,234 кг поваренной
соли. При этом выход ВаС12 • 2Н2О в твердую фазу при охлажде-
охлаждении смеси до 20° достигает 72,6%.
Сероводород, необходимый для предварительного получения
раствора гидросульфида бария, может быть получен, например, по
реакции
NaHS + NaHCO3 = Na2CO3 + H2S
из. маточного раствора после кристаллизации хлористого бария.
Остающийся в этом растворе ВаС12 B7,4% от начального коли-
количества) можно предварительно осаждать содой, образующейся по
указанной реакции, что приводит к получению второго продукта —
углекислого бария. Из 1 г барита по такой схеме можно получить
0,34 г хлористого и 0,36 г углекислого бария.
Разложение BaS раствором СаС12 и карбонизацией
Разработаны методы получения гидросульфидного щелока и из
него хлористого бария и без применения (или с очень малым рас-
расходом) соляной кислоты 80>8I. Так, например, можно производить
высаливание ВаС12-2Н2О (до 70%) при 30—40° 32% раствором.
СаС12 из гидросульфидного щелока [около 28% ВаС12 + Ba(HSJ],
получаемого при выщелачивании плава сернистого бария 10% рас-
раствором СаС12 при 60—100°. Остающийся маточный раствор исполь-
используют для выщелачивания новых порций плава.
Можно также производить полукарбонизацию суспензии суль-
сульфида бария. При этом по реакции
2BaS + Н2О + СО2 = ВаСО3 4- Ba(HSJ
получаются карбонат и гидросульфид бария в равных количествах.
Однако образующийся раствор Ba(HSJ имеет низкую концентра-
концентрацию и требует выпарки. При многократном, например, трехкрат-
трехкратном, выщелачивании плава и карбонизации суспензии, с использо-
использованием растворов для повторного выщелачивания плава можно по-
получить раствор с содержанием 27% Ba(HSJ. Такая концентрация
достаточна для высаливания хлорида бария поваренной солью с
выходом не меньше 75%. Остальные 25% бария можно осадить
в виде карбоната или сульфата81.
Способ получения хлористого бария карбонизацией углекислым
газом раствора, содержащего эквивалентные количества BaS и
СаС12
BaS + СаС12 + СО2 + Н2О = ВаС12 + СаСО3 + H2S
не нашел применения из-за больших потерь бария в форме ВаСО3,
образующегося наряду с СаСО3 (по-видимому, в виде двойного
соединения — барито-кальцита). Этого можно избежать, если под-
подвергнуть карбонизации щелок, получаемый после выщелачивания
плава сернистого бария раствором хлористого кальция и отделения
выпавшего осадка гидрата окиси кальция24:
2BaS 4- 2СаС12 + 2Н2О = ВаС12 + Ba(HSJ + СаС12 + Са(ОНJ
ВаС12 + Ba(HSJ + СаС12 + Н2О + СО2 = 2ВаС12 + СаСО3 + 2H2S
При содержании в растворе избытка СаС12 [150% от стехиомр-
трического по отношению к Ba(HSJ] потери бария в шламе СаСО3
весьма невелики. Из получаемого раствора, содержащего ВаС12 и
некоторое количество СаС12, после выпарки кристаллизуют ВаС12 •
• 2Н2О. По этой схеме расход СО2 на карбонизацию в 2 pasa мень-
меньше, чем при. карбонизации щелока без отделения Са(ОН)а.
Хлораммониевый способ
Производство хлористого бария может быть экономично орга-
организовано в комбинации с регенерацией аммиака из хлористого ам-
аммония, содержащегося в жидкости теплообменника дистилляции
содовых заводов82"84:
BaS + 2NH4C1 = ВаС12 + 2NH3 + H2S
При этом для образования хлористого бария используется от-
отбросный ион хлора содового производства (вместо ценных мате-
материалов— НС1 или СаС12) и, кроме того, аммиак регенерируется
440
Гл. XII. Соли бария
Хлорид бария
441
с помощью BaS, а не Са(ОНJ. В результате этого затраты на про-
производство ВаС12 оказываются меньше на 20—30%.
Попутно, кроме ВаС12, могут быть получены и другие товарные
продукты — сульфид аммония, гидросульфид натрия, сера, серная
кислота. Это позволяет комплексно использовать сырье, применяе-
применяемое для получения хлористого бария и соды. Однако масштабы
потребления хлористого бария значительно меньше масштабов со-
содового производства.
Жидкость теплообменника дистилляции имеет состав (в %):
NaCl — 5,8, Na2SO4 — 0,2, NH4C1— 14, (NH4JCO3 — 0,24, свобод-
свободного NH3~2 и остальное вода. Предварительно в специальной ко-
колонне отгоняют из жидкости свободный аммиак. Взаимодействие
плава BaS с жидкостью теплообменника дистилляции должно про-
производиться при температуре не ниже 95° и при 5—7%-ном избытке
BaS от стехиометрического количества. Это обеспечивает полную
отгонку аммиака и сероводорода и получение раствора, не содер-
содержащего ионов HS", присутствие которых снижает степень исполь-
использования бария и приводит к загрязнению продукта. Отгонка
связанного аммиака этим методом, происходит примерно в 5 раз
медленнее, чем при обработке жидкости известковым молоком. Ос-
Осветленный раствор, содержащий около 17% ВаС12, 5,5—6% NaCl и
0,6% СаС12, направляют на вакуум-выпарку, в процессе которой
происходит кристаллизация хлористого бария, завершающаяся при
охлаждении выпаренного раствора до 20°. Выпаренный раствор,
направляемый на охлаждение, содержит (в %): ВаС12—13,7,
NaCl — 20,9, СаС12 — 0,12. Маточный раствор, полученный после
кристаллизации охлаждением, содержит приблизительно 3,6%
ВаС12, 25% NaCl и 2,4% СаС12. ¦->
Для возврата аммиака в содовое производство отгоняемая па-
¦ро-газовая смесь должна быть промыта раствором NaOH, причем
•сероводород связывается в гидросульфид.
Карбонатный способ
_Этот способ получения хлористого бария является по существу
¦видоизменением солянокислотного способа. Он заключается в рас-
растворении в соляной кислоте природного витерита или ВаСО3< осаж-
осажденного из раствора BaS (или других растворов, содержащих Ва2+):
BaS + Н2О + СО2 = ВаСО3 + H2S
ВаСО3 + 2НС1 = ВаС12 + Н2О + СО2
При использовании природного ВаСО3 получение из него хлори-
хлористого бария отличается большой простотой, дающей преимущества
перед другими способами. При необходимости получать углекис-
углекислый барий осаждением из BaS экономичность метода сильно по-
понижается. Его преимуществом перед описанным выше солянокис-
лотным методом является лишь то, что продукт получается более
чистым, так как при осаждении ВаСО3 все растворимые примеси,
содержавшиеся в плаве сернистого бария, остаются в растворе и
растворению в соляной кислоте подвергают чистый ВаСО3.
Хлормагниевый способ24
При взаимодействии BaS с раствором MgCl2 при нагреваний
идет реакция:
BaS + MgCl2 + 2H2O = ВаС12 + Mg(OHJ + H2S
Используя отход калийной промышленности — щелока хлори-
хлористого магния, можно при этом получить три ценных продукта —
' хлористый барий, сероводород и гидроокись магния, которая может
быть переработана на окись магния. Эта реакция, однако, долго не
могла найти промышленного применения из-за трудности отстаива-
отстаивания, фильтрации и промывки получающегося коллоидального осад-
осадка гидроокиси магния.
Вследствие гидролиза BaS в водных растворах при сливании
растворов BaS и MgCl2 протекает реакция:
2BaS + MgCl2 + 2H2O = Ba(HSJ + ВаС12 + Mg(OHJ
Наличие в растворе значительного количества ионов ОН" ведет
к мгновенному выделению нерастворимого, вязкого, не отстаиваю-
отстаивающегося осадка Mg(OHJ. По мере его образования происходит
дальнейшее гидролитическое расщепление BaS до полного его ис-
исчезновения. Активность ионов ОН~ в растворе определяется про-
произведением растворимости [Mg2+][OH~]2= ^Смв<он),. Так как Кн,о =
= [НЦОН-], то
^Mg(OHJ |н+]2
[Mg2+] =-
<Н2О
Принимая, что [Mg2+] = fMg2+CMg2+ (где fMg2+ — коэффициент
активности, а СМ2+ — концентрация ионов магния), имеем:
К
Mg(OHJ
[нЧ2
Mg2+
Приняв для упрощения fMg2+ = 1 и логарифмируя, получим:
"
14;
При 20°/Сме(онь ¦= 1,2 • 10 " и /Сн2о= Ю 14; подставив эти зна-
значения в последнее уравнение и меняя знаки для перехода к вели-
величине рН, имеем
2H-17.08
442
Гл. XII. Соли бария
Хлорид бария
443
где pMg2+— логарифм концентрации ионов магния (в г-ионах/л),
взятый с отрицательным знаком.
Пользуясь этим равенством, легко вычислить максимально воз-
возможное содержание Mg2+ (в г/л) в растворах с различным значе-"
нием рН при насыщении гидроокисью:
рН 8 9 10 11 12
Содержание Mg2+, г/л . . . . 2,92-102 2,92 2,92 • 10~2 2,92 • 10"~4 2,92- 1(Гв
Эти данные показывают, что при рН < 9 раствор может содер-
содержать достаточное количество Mg2+, не выделяя его в виде гидро-
гидроокиси. Понижение рН среды или, что то же, уменьшение количества
ионов ОН" может быть достигнуто путем предварительного насы-
насыщения сероводородом раствора сернистого бария для превращения
его в Ba(HSJ. При приливании к такому раствору MgCl2 осадка
не образуется. При постепенном нагревании раствора, содержа-
содержащего Ba(HSJ и MgCl2, в силу повышения равновесного давления
H2S последний медленно удаляется; одновременно с этим концен-
концентрация ОН~ медленно нарастает и происходит выделение хорошо
Отстаивающегося и отфильтровываемого осадка гидроокиси магния.
Вместо раствора сернистого бария, получаемого при выщела-
выщелачивании плава сернистого бария водой, можно пользоваться гид-
гидросульфидным щелоком, образующимся при выщелачивании плава
BaS раствором СаС12 по реакции:
2BaS + СаС12 + 2Н2О = Ba(HSJ + ВаС12 + Са(ОН)г
Образующийся при этом Са(ОНJ выпадает в осадок и его
отделяют, а раствор насыщают сероводородом до требуемой вели-
величины рН и сливают с раствором хлористого магния. Выщелачива-
Выщелачивание плава сернистого бария растворами СаС12 позволяет получить
растворы с высокой концентрацией бария. Плав сернистого бария
подвергают выщелачиванию при 65° 10% раствором СаС12, количе-
количество которого берут с таким расчетом, чтобы перевести весь BaS,
содержащийся в плаве, в Ba(HSJ и обеспечить добавочное извле-
извлечение бария из карбоната и силиката бария в виде ВаС12. Для этой
цели шлам, оставшийся после выщелачивания, дополнительно раз-
разваривают при 100°.
Отстоявшийся щелок, содержащий 12—13% Ba(HSJ, 15—16%
ВаС12 и 0,5—1,5% СаС12, поступает на реакцию с MgCl2 в аппарат
колонного типа. В верхней части колонны происходит донасыщение
стекающего вниз щелока поднимающимся кверху отходящим серо-
сероводородом с доведением рН раствора до 8—8,5. Ниже поступает
30—35%-ный раствор MgCl2 в количестве 125—150% от стехиомет-
рического по отношению к Ba(HSJ. При нагревании раствора по
мере прохождения его вниз по колонне от 60 до 100° происходит
постепенное удаление H2S и выделение Mg(OHJ. Остатки H2S
удаляют кипячением раствора в нижней части колонны при 103—
104°. Горячий раствор ВаС12 с осадком Mg(OHJ поступает на от-
отстаивание и фильтрацию на вакуум-фильтрах. Прозрачный рас-
раствор ВаС12 идет на вакуум-выпарку и кристаллизацию, а маточные
щелоки после кристаллизации возвращают в производство.
Получение раствора хлористого бария менее высокой концен-
концентрации, чем по описанному способу, осуществляют по более про-
простой схеме. Раствор сернистого бария в воде насыщают сероводо-
сероводородом. К полученному гидросульфидному раствору добавляют
хлормагниевый щелок. При нагревании этой смеси из нее удаляет-
удаляется сероводород, частично возвращаемый вновь на насыщение ис-
исходного раствора BaS, и выделяется осадок Mg(OHJ. После отде-
отделения осадка остается раствор ВаС12. Расходные коэффициенты
на 1 т 97%-ного ВаС12-2Н2О составляют: 1,14 т барита A00%
BaSO4), 0,29 т СаС12 A00%), 0,19 т MgCl2 A00%) и 0,34 т реак-
реакционного угля G000 ккал/кг); в качестве побочных продуктов по-
получают 0,13 т сероводорода A00%) и 0,08 г MgO A00%).
Хлорный способ
Этот способ заключается в обработке газообразным хлором
раствора, полученного водным выщелачиванием плава сернистого
бария85"87. Суммарная реакция процесса имеет вид:
l2=BaCl2
Вследствие гидролиза сернистого бария и хлора образуются
промежуточные соединения: Ва(ОНJ, Ba(HSJ, HC1, НС1О,
Ва(СЮJ. Помимо этого, в процессе хлорирования протекают мно-
многочисленные побочные реакции, ведущие к образованию некоторых
количеств BaSO4, BaS2O3 и других соединений, что понижает вы-
выход хлористого бария и серы. Наибольшие выходы ВаС12 и свобод-
свободной серы достигаются при хлорировании раствора, содержащего
около 170 г/л BaS, при 85—90°. При этом выход ВаС12 равен 9^ °V,
а серы —90%. Необходимую температуру поддерживают теплом,
выделяющимся в процессе хлорирования (при начальной темпера-
температуре раствора 90°).
Избыток хлора ведет к образованию большего количества
BaSO4, уменьшая выход ВаС12 и серы:
ВаС12 + S + ЗС12 + 4Н2О = BaSO4 + 8HC1
Кроме того, перехлорирование щелока до кислой реакции вы-
вызывает коррозию стальных аппаратов и трубопроводов, применяе-
применяемых в дальнейших стадиях переработки раствора хлористого бария.
Поэтому хлорирование ведется до нейтральной или слабощелочной
реакций (до обесцвечивания щелока, имеющего желто-зеленый
Цвет)-. Содержание BaS в прохлорированном щелоке не должно
превышать 0,5—1 г/л.
•444
Гл. XII. Соли бария
Хлорид бария
445
Хлорирование ведут в керамических резервуарах или в башнях,
или в другой аппаратуре, устойчивой по отношению к влажному
хлору. Прохлорированные щелоки продувают воздухом для удале-
удаления растворенных газов (сероводорода, хлора) и направляют на
•отстаивание от серы. При содержании в них более 0,5% BaS по-
последний осаждают раствором хлорного железа в виде FeS. Щелок,
¦отстоявшийся от тонкого осадка серы и сернистого железа, под-
подвергают переработке на кристаллический хлористый барий (вы-
(выпариванию, кристаллизации, сушке) теми же методами, как и в
других способах, описанных выше. Отвал, оставшийся после вод-
водного выщелачивания плава сернистого бария, обрабатывают соля-
соляной кислотой для извлечения бария, содержащегося в виде кисло-
торастворимых соединений, и промывают водой. Полученные слабые
растворы хлористого бария присоетиняют к основному раствору
поступающему на хлорирование. На производство 1 т 97%-ного
ВаС12-2Н2О расходуют: 1,25—1,35 т барита A00% BaSO4), 0,31 —
€,32 т хлора A00%), 0,1—0.12 т соляной кислоты B7,5% НС1),
¦0,4—0,5 т реакционного угля G000 ккал/кг); в качестве побочного
продукта получается 0,12 т серы A00 %).
Способы горячего хлорирования и гидрохлорирования
Разработаны способы получения хлористого бария из сухого
плава сернистого бария действием на него газообразного хлора
или хлористой серы при высоких температурах:
BaS + С12 = BaCI2 + S
BaS + S2C12 = ВаС12 + 3S
Применение этих способов затрудняется сильной коррозией
¦аппаратуры хлором и хлористой серой85"87.
Показана возможность получения ВаС12 хлорированием BaSO-
¦в расплаве хлоридов, например, в расплаве ВаСЬ, в присутствии
•восстановителя (кокса). Растворенный в хлориде сульфат бария
¦вначале восстанавливается до BaSO3 или до BaS и эти продукты
восстановления хлорируются. Процесс идет с достаточной интен-
интенсивностью уже при 800°. Можно получить плав, содержащий до
"94% ВаС1288. На одном из индийских заводов хлористый барий
-получают пропусканием хлора или хлористого водорода через рас-
раскаленную до 600° шихту, состоящую из барита и кокса. Попутным
продуктом является сера89. Взаимодействие барита с хлористым
. on оо
'водородом
90-92
BaSO4 + 2HC1 = ВаС12 + Н2О + SO2 +
Т>езко ускоряется выше 600°, особенно при осуществлении его во
взвешенном слое и с увеличением концентрации НС1 в применяе-
применяемом газе.
Взаимодействие BaSO4 и CaClg
Константа равновесия реакции
BaSO4+CaCl2 ^± CaSO4+BaQ2
в водной среде определяется отношением произведений раствори-
мости BaSO4 и CaSO4: Кр = ПРВа5О4: ПРСа5О4 = С^ : ССа2+. При
26° ПРВа5о4 = 9,9 • 10~",ПРСа5о4 = 6,3- 10~5и#р~1,6. 10 6. Таким
образом, в водном растворе при не очень больших концентрациях
СаС12 и низких температурах концентрация ионов бария ничтожно
мала по сравнению с концентрацией ионов кальция и равновесие
реакции сдвинуто влево.
Но если обрабатывать BaSO4 концентрированным водным рас-
раствором СаС12при повышенных температурах^эта реакция протекает
слева направо. Это обусловлено уменьшением растворимости
CaSO4 и увеличением растворимости BaSO4 при повышении тем-
температуры, а также малой растворимостью CaSO4 и ВаС12 в концен-
концентрированных растворах СаС12, имеющих высокую температуру
кипения. Кроме того, с повышением температуры быстро возра-
возрастает активность СаСЬ. Поэтому, обрабатывая BaSO4 концентри-
концентрированным раствором СаС12, можно полечить ВаС12. Так. после
2—3-часовой выдержки при 170° выход ВаС12 составляет —90%.
Он возрастает с увеличением избытка СаС12 и уменьшением коли-
количества «оды в реакционной смеси. Поэтому извлечь ВаС12 из реак-
реакционной смеси выщелачиванием подой невозможно — реакция прой-
пройдет в обратном направлении. Предложено экстрагировать ВаС12
из этой смеси метанолом93. При сплавлении безводной смеси BaSO4
и СаС12 сульфат кальция образуется в неактивной, слабо растип-
римой форме94. Это позволяет осуществить выщелачивание ВаС12
не только метанолом, но и водой, которая успевает извлечь BaClg
раньше, чем неактивный CaSO4 вступит в реакцию.
Возможность обменного разложения между расплавленными
BaSO* и СаС12 в условиях, когда реакция идет с достаточной для
практических целей скоростью, определяется равновесием:
BaSO4 (ж.) + СаС12 (ж.) «= CaSO4 (ж.) + ВаС12 (ж.)
г, fCaSO4l ГВаС121
Константа равновесия этой реакци Kv — TgaSOTTCaCrT' ГДе Ве"
личины в скобках — активности. Ввиду отсутствия для этой систе-
системы данных о степенях ионизации и о коэффициентах активности,
можно допустить, что активности пропорциональны молярным до-
долям и распада на ионы не происходит95. Молярные доли N в рав-
равновесной системе при степени превращения BaSO4 в ВаС12, равной
*J*h при исходном составе смеси щ молей BaSO4 и п2 молей СаС12
446
Гл. XII. Соли бария
будут:
N
BaS04
Щ{\-х)
n,
n2 — ntx m
N
BaCIj"
N,
CaSO4 =
Подставив эти значения в выражение для Kv и обозначив отно-
отношение CaCfe: BaSO4B исходной смеси, т. е. п2: Пь через г0, получим:
X2
Ар
Отсюда:
(\-х)(п2-п1х) A-х)(го-х)
2(/СР-1)
Значения /СР можно вычислить из уравнений изменения изобар-
изобарного потенциала:
Если принять для всех компонентов, участвующих в реакции,
жидкое состояние в качестве стандартного, то уравнения функций
ЛЯ = f(T) и Д2° = f(T) будут такими:
ДЯ = 9777 - 2.73Г + 3,68 • 10~3Г2 + 1,57 • Ю^
- AZ° = 9777 - 6,297" lg T + 3,68 • 10~3Г2 - 0,785 • 10бГ~! + 12.75Г
При 1144° К (871° С) —AZ° = 7129 кал и Kv = 22,9. Это дает при
г0 = 1 значение х = 0,823 и при г0 = 2 д; = 0,966. Таким образом,
при 870° возможно превращение BaSO4 в ВаС12 на 82,3% в стехио-
метрической смеси и на 96,6% при двукратном избытке СаС12. Эти
вычисленные величины хорошо подтверждаются эксперименталь-
экспериментальными данными93. При 870° степень превращения BaSO4 в ВаС12
при г0 = 1 оказалась равной 82,9%, а при г0 = 298,2%.
, Присутствие в смеси BaSO4 и безводного СаС12 плавней (NaCl,
КС1 и пр.) значительно ускоряет реакцию при низких температу-
температурах. Так, синтетический BaSO4, взаимодействуя с эквивалентным
количеством СаС12, при 500° за 28 мин превращается в ВаС12 на
48%, а в присутствии в смеси 9 вес.% NaCl — на 79%, причем
NaCl не является хлорирующим агентом. Аналогичные результаты
получаются и с природным баритом. Интенсифицирующее действие
плавня проявляется в интервале температур, в котором присутст-
присутствие плавня вызывает появление жидкой фазы, т. е. 500—600°. Ниж-
Нижний предел этого интервала E00°) соответствует эвтектике NaCl
и СаС12 E05°, 51.5 мол. % СаСЬ), ;а затем тройной эвтектике, тем-
температура которой на 5—10° ниже, а верхний предел F05°) — эвтек-
эвтектике СаС12 и ВаС12. При использовании в качестве плавня Ca(NO3J
жидкая фаза появляется уже при 409°. Оптимальным является не-
небольшое количество добавки, обеспечивающее появление в реак-
Хлорид бария
447
ционной смеси всего 10—15 вес. % жидкой фазы, что позволяет
вести процесс при относительно низкой температуре, сохраняя сы-
сыпучесть реакционной массы. При более высоких температурах до-
добавка плавня не имеет смысла, так как жидкая фаза появляется и
в смесях, не содержащих плавня. Так, уже при 600° в отсутствие
плавня степень превращения в лабораторных условиях при раз-
разных молярных соотношениях CaClj : BaSO4 достигает следующих
величин:
CaCl2:BaSO4 1,6 1,3 1,0 0,9 0,8 0,7 0,6
f за 13 мин 87,3 83,9 78,7 76,7 70,3 66,3 55,4
Степень превращения: | ^ ^ мш 958 92>д 892 86K g^, 698 602
Степень превращения за 43 мин, при соотношениях СаС12:
: BaSO4. равных 0,8—0,6, достигает теоретических пределов.
Хлоркальциевый способ
На практике рассмотренную выше реакцию
BaSO4 + СаС12
CaSO4 + ВаС12
осуществляют, прокаливая компоненты в присутствии восстанови-
восстановителя — угля для восстановления сульфатов в сульфиды. А так как
растворимость BaS значительно больше, чем CaS, растворимость
которого при 20° всего около 0,2 г/л, то при выщелачивании плава
обратная реакция не идет.
Взаимодействие BaSO4 с СаС12 и углем, лежащее в основе хлор-
кальциевого способа получения ВаС12, известного также под наз-
названием способа Дюфло, является сложным процессом, состоящим
из многих реакций, протекающих с разной скоростью. Наряду с
основными реакциями обменного разложения BaSO4 и СаС12 и вос-
восстановления CaSO4
BaSO4 + СаС12 = CaSO4 + ВаС12 + 7,8 ккал
CaSO4 + ЗС = CaS + 2CO + СО2 - 80,2 ккал
BaSO4 + ЗС + СаС12 = ВаС12 + CaS + 2CO + СО2 - 72,4 ккал
идут приводящие к тем же результатам реакции восстановления
BaSO4 и обменного разложения BaS и СаС12:
BaSO4 + 4С = BaS + 4CO - 138,3 ккал
BaS + СаС12 = ВаС12 + CaS + 24,9 ккал
BaSO4 + 4С + СаС12 = ВаС12 + CaS + 4CO - 113,4 ккал
Степень перехода BaSO4 в ВаС12 и скорость процесса зависят
от качества барита, от дисперсности компонентов и состава шихты,
от температуры и др. 96~98- Примеси, содержащиеся в низкосорт-
низкосортном сырье, образуют побочные продукты, обволакивающие зерна
BaSO4- и препятствующие взаимодействию их с хлористым каль-
кальцием и углем. Поэтому степень перехода BaSO4 в ВаС12 возрастает
448
Гл. XII. Соли бария
с увеличением содержания BaSCU в барите и с ростом моляр-
молярного отношения СаСЬ: BaSO4. Повышение температуры до 1100°
резко интенсифицирует процесс. Дальнейшее повышение темпера-
температуры до 1200° приводит к некоторому снижению выхода хлористого
бария, очевидно вследствие значительного роста доли побочных
реакций. По этой же причине уменьшается степень перехода BaSO4
е ВаС12 при значительном увеличении длительности прокалки ших-
шихты. Применение мелкоизмельченных барита и угля (—0,15 мм)
приводит к увеличению выхода ВаС12, сокращению длительности
процесса и, следовательно, к увеличению производительности пе-
печей. (Однако высказывалось мнение, что измельчение барита не
обязательно, так как кусковой барит при нагревании до ~250° рас-
растрескивается и рассыпается — см. стр. 431.) Консистенция плава
обусловлена главным образом содержанием в нем ВаС12 и СаС1а.
При переработке барита, содержащего 95% BaSO4, подвижная
масса, легко выгружаемая из печи, получается при 10%-ном из-
избытке СаС12 (выход ВаС12 91%). Из низкокачественных руд, содер-
содержащих 57—79% BaSO4, можно получить достаточно подвижный
плав лишь при применении большого избытка (~2О°/о) хлористого
кальция. Выход хлористого бария при этом составляет 88—91%.
Однако плавы, полученные из низкосортных баритов, отличаются
после охлаждения повышенной плотностью и труднее поддаются
выщелачиванию. Кроме того, содержание в них большого количе-
количества СаСЬ приводит к увеличению потерь ВаСЬ с отбросным ма-
маточным раствором при кристаллизации. В общем же, примеси, со-
содержащиеся в сырье, значительно меньше влияют на процесс вос-
восстановления барита в присутствии СаС12, чем без него.
Схема получения хлористого бария хлоркальциевым способом
изображена на рис. 139. Прокаливание реакционной смеси ведется
в периодически действующих коротких барабанных вращающихся
печах (тамбурах), расположенных горизонтально. Топливом слу-
служит мазут, подаваемый в печь форсункой. Длина печи 6—7 м, вну-
внутренний диаметр 1,5 м; толщина огнеупорной футеровки 250 мм;
число оборотов печи— 1,6 в 1 мин. Полезный объем печи 3,6—5 ж3.
В средней части барабана имеется люк, через который произво-
производится загрузка шихты и выгрузка плава. Для загрузки барабан
поворачивают люком кверху, для выгрузки —люком книзу.
В печь загружают 1,5—2 т барита, измельченного до 0,5—5 мм,
и уголь в количестве 13—15% (в пересчете на углерод) от веса
барита. Загрузка длится 10 мин, после чего люк закрывают; печь
приводится во вращение и из резервуара, обогреваемого отходя-
отходящими из печи газами, через трубу, входящую в заднюю горловину
печи, в течение 30—40 мин подают концентрированный раствор
хлористого кальция, содержащий 800—900 г/л СаС12. Обогрев ре-
вервуара с раствором хлористого кальция требуется во избежание
кристаллизации последнего. Количество хлористого кальция (в пе-
15 М. В. Позин
Рис. 139. Схема получения хлористого бария хлоркальциевым способом:
/ — щековая дробилка для барита; 2 — элеватор; 3 —бункер для дробленого барита; 4 — труб-
трубчатая мельница для барита; 5 —бункер для молотого барита; 6 — бункер для угля; 7 —авто-
—автоматические весы для барита; S — автоматические весы для угля; 9 — смеситель; ю — вращаю-
вращающаяся печь (тамбур); //—резервуар для мазута; 12 — котел для раствора хлористого кальция;
13 — вагонетка с тиглем; 14 — щековая дробилка для плава; 15 — выщелачиватель плава;
16 — центрифуга для отделения шлама; 17 — резервуар-отстойник для раствора хлористого
бария; 18 — центробежные насосы; 19-резервуар для раствора хлористого бария, поступаю-
поступающего на выпарку; 20 — вакуум-выпарная батарея; 21 — греющая камера; 22 — барометрический
конденсатор; 23 — брызгоуловитель; 24 — кристаллизатор хлористого бария; 25 — вентилятор
для подачи охлаждающего воздуха в кристаллизатор; 26 — центрифуга для отделения кри-
«таллов хлористого бария; V — сборник маточного щелока; 28 — бункер для хлористого бария;
39 — сушилка.
450
Гл. XII. Соли бария
ресчете на 100%), загружаемого в печь, составляет 48—49% от
веса барита. Требуемый раствор хлористого кальция получают
выпариванием в вакуум-выпарных аппаратах дистиллерной жид-
жидкости содового производства (содержащей ~ 100 г/л СаС12) или
маточных щелоков, получаемых в качестве отхода в производстве
бертолетовой соли (содержащих ~400 г/л СаСЬ).
В течение 1,5—2 ч после загрузки температуру в печи поддер-
поддерживают на уровне 770—780° — происходит испарение влаги и
шихта превращается в расплавленную массу. Затем температуру
повышают до 900—950°—расплавленная масса сильно пенится;
через 1—1,5 ч реакция заканчивается. К концу процесса темпера-
температуру повышают до 1000—1100°, происходит усадка массы, она ста-
становится однородной. Температура отходящих газов колеблется
от 200 до 800° (в среднем 300—400°). Длительность одной опера-
операции плавки 4—5 ч. Она зависит от количества загруженной шихты,
качества сырья, тонкости его помола. Передержка плава в печи,
помимо снижения содержания в нем ВаС12, приводит к его загу-
стеванию, что может вызвать козлообразование и затрудняет по-
последующее выщелачивание плава.
Когда плав готов, вращение печи прекращают, поворачивают
ее люком книзу и в течение 5 мин сливают плав в находящийся
под печью плоский чугунный резервуар («паром») или в пере-
передвижные тигли, ковши, установленные на вагонетках. Здесь плав
застывает в течение 1,5—3 ч (в зависимости от его качества и тем-
температуры плава и окружающего воздуха). Плав содержит 55—
60% ВаС12, 4—12% СаС12, около 7% CaS, около 3% BaSO4, около
0,5% BaS, 20—25% других примесей — непрореагировавшего угля
F—8%), ВаСОз, FeS, SiO2 и проч. Выход хлористого бария в
плаве составляет около 90% от теоретического по отношению к
загруженному бариту. Остывший плав разбивают на куски, дро-
дробят на щековой дробилке до крупности меньше 3 см к направляют
на выщелачивание.
Выщелачивание плава производят горячей водой F0—80°). По-
Повышенную температуру в процессе выщелачивания поддерживают
острым паром. Применяют выщелачиватели различных типов —
резервуары с мешалками на вертикальном валу, желобы с лопаст-
лопастными мешалками на горизонтальном валу, барабанные вращаю-
вращающиеся выщелачиватели и др.
После выщелачивания раствор отделяют от шлама на центрифу-
центрифугах. Шлам промывают водой и выбрасывают в отвал. Он содержит
более 20%) CaS, 8—<10% BaSO4 и другие нерастворимые веще-
вещества, а также 18—«20% влаги и около 1%, ВаС12. Количество от-
отвала составляет 0,8—1,1 т на 1 т готового продукта. Промывные
воды с центрифуг смешивают с основным раствором. Раствор, со-
содержащий 300—350 г/л ВаС12, отстаивается от мути и поступает
на выпарку и кристаллизацию хлористого бария.
Нитрат бария
451
Маточные щелоки после кристаллизации хлористого бария со-
содержат в среднем 80—120 г/л ВаС12 и 100—150 г/л СаСЬ. Их при-
присоединяют к раствору, идущему на выпарку. Постепенно содержа-
содержание в них СаС12 повышается, а содержание ВаС1г понижается, так
как хлористый кальций обладает большей растворимостью и выса-
высаливает ВаС12 (рис. 132). Когда содержание ВаС12 в маточных ще-
щелоках снижается до 25—50 г/л ВаС12-2Н2О, они выводятся из ци-
цикла и выбрасываются.
На 1 г хлористого бария (94%) ВаС12-2Н2О), получаемого
хлоркальциевым способом,расходуется 1,07—-1,08 т барита A00%),
0,35—0,38 т топлива G000 ккал/кг), 0,53—0,70 т хлористого каль-
кальция A00%), 2,7—2,8 мгкал пара, 54 квт-ч электроэнергии.
Для осуществления непрерывного выщелачивания плава пред-
предложено его предварительно отверждать на охлаждаемой поверх-
поверхности в тонком слое ".
Преимуществом хлоркальциевого метода получения хлористого
бария перед солянокислотным является большая простота процесса,
так как в одну стадию обжига получается плав хлористого бария,
причем различные примеси — соединения железа, SiO2, CaS и дру-
другие при выщелачивании не переходят в раствор, что исключает
необходимость специальной очистки раствора.
Серьезным недостатком хлоркальциевого метода является при-
применение печей периодического действия. Попытки осуществить про-
процесс в непрерывно действующих вращающихся печах пока не дали
положительных результатов в связи с сильным приплавлением
шихты к футеровке печи. Другим недостатком хлоркальциевого ме-
метода является потеря серы с уходящим в отвал сернистым кальцием.
НИТРАТ БАРИЯ
Из способов получения нитрата бария 10° наибольшее техниче-
техническое значение имеют:
1) разложение сернистого бария азотной кислотой;
2) обменное разложение солей бария и нитратов.
Получение нитрата бария из BaS и азотной кислоты
Процесс получения Ba(NO3b из плава сернистого бария и азот-
азотной кислоты аналогичен солянокислому разложению сернистого
бария. Здесь, однако, должна применяться аппаратура из специаль-
специальных материалов, устойчивых в присутствии азотной кислоты и
окислов азота. Для разложения раствора, полученного после вод-
водного выщелачивания плава сернистого бария (~ 125 г/л BaS), ис-
используют разбавленную азотную кислоту C0—35% HNO3). При-
Применение концентрированной кислоты приводит к большим потерям
окислов азота в результате восстановления HNO3 сероводородом:
BaS -f 2HNO3 = Ba(NO3J + H2S
0 2HNO j + 3H2S = 4H2O + 2NO + 33»
15V
452
Гл. XII. Соли бария
При этом выделяется элементарная сера, загрязняющая полу-
получаемый продукт, так как ее трудно отделить от раствора.
Исходные растворы смешивают в стальном цилиндрическом ре-
реакторе, футерованном кислотоупорными плитками, снабженном ме-
мешалкой A20 об/мин). Компоненты дозируют так, чтобы в выходя-
выходящем из реактора щелоке имелся избыток кислотности 1—5 г/л
HNO3. Далее в «разварнике» с помощью острого пара при темпе-
температуре не ниже 95° производят отдувку из кислого щелока серо-
сероводорода и коагуляцию выделившейся в твердую фазу серы. Затем
производят нейтрализацию кислотности щелока при 95—97°, доба-
добавляя к нему сульфидный раствор (BaS) при перемешиваний до
•избыточной щелочности 0,16—0,5 г/л BaS. Большее повышение ще-
щелочности и понижение температуры раствора вызывают накопле-
накопление в растворе H2S, т. е. Ba(HSJ, чем создаются условия для раз-
разложения нитрата бария:
*' 2Ba(HSJ + Ba(NO3J + H2S = 2NO + Ва(ОНJ + 2BaS + 2Н2О + 3S
Нейтрализованный щелок осветляется в отстойнике Дорра при
температуре не ниже 90°. Шлам, содержащий элементарную серу,
сульфит бария и другие примеси, идет в отвал. Осветленный щелок
выпаривают в двухкорпусном вакуум-аппарате. Выделившуюся во
втором корпусе соль через солесборник спускают на центрифугу.
Маточный раствор выводят из цикла при накоплении в нем болыйе
3 г/л примесей, содержащих серу. Выгружаемые из центрифуги
кристаллы азотнокислого бария имеют влажность <—' 1,5%. При вы-
выработке продукта I сорта, содержащего меньше 0,5% влаги, соль
сушат воздухом (90—95°) в барабанной сушилке. На производ-
производство I т продукта расходуют: 1,7—1,8 т барита II сорта (в пере-
пересчете на 100% BaSO4), 0,5 т азотной кислоты (в расчете на НМОз),
0,4—0.42 г условного топлива, 0,18 т каустической соды (92%),
137 ж3 воды, 4,5 мгкол пара, 23,5 кет-ч электроэнергии, —4500 м*
газообразного топлива. На каждую тонну продукта получается
0,5 т шдамов; шл_ам из отстойников содержит 4% BaS, 10—15%
ВаСОз, 15—20% BaSO4, 10—15% С. 30—40% золы; шлам из реак-
реакторного отделения представляет собой серу, смоченную раствором
Ba(NO3J.
Конверсия солей .бария
Поэтому способу концентрированный раствор хлористого бария
при 80—90° обрабатывают NaNO3 йЛи KNO3:
ВаСЬ + 2NaNO3 = Ba(NO3J + 2NaCl
При охлаждении раствора до 30—35° из него кристаллизуется
Ва(ЫОз)г. который отделяют йа фильтре, промывйют и высуши-
высушивают.
Для получения нитрата бария, содержащего только 0,1—0,3%
хлоридов, необходимо продукт Первой кристаллй&ацйи перекри-
Карбонат бария
453
2NH4NO3
2NH4CJ
сталлизовывать. Этот способ отличается простотой, возможностью
использования аппаратуры из стали (Ст. 3) и малым расходом
пара и электроэнергии. Однако он связан с применением дорогого
сырья — ВаС12 и NaNO3.
Для конверсии хлорида бария в нитрат может быть использо-
использован нитрат аммония 101:
ВаС12 + 2NH4NO3 = Ba(NO3J + 2NH4C1
На рис. 140 изображена диаграмма растворимости в этой си-
системе при 25°. Аналогичный вид имеют диаграммы для 0 и 100°.
При сливании насыщенных рас-
растворов NH4NO3 и ВаС12 при 25°
кристаллизуется около 65%
ВаA\ЮзJ. Если выпаривать при
100° раствор, содержащий 2 моль
NH4NO3 на 1 моль ВаС12, до на-
насыщения NH4C1, выход Ва(ЫОз)г
составит ~73%.
Для обменного разложения
можно использовать также вод-
водную систему:
ВаСО3 + Ca(NO3J => СаСО3 + Ba(NO3J
Вследствие меньшей раствори-
растворимости СаСОз по сравнению с
ВаСО3 реакция идет слева на-
направо.
Нитрат, хлорид и другие чи-
чистые водорастворимые соли бария
fia(NO3J
/Ш7
74395/95
1871
186
ВаС1г
Рис. 140. Растворимость в водной сис-
системе BaCI2 + 2NH4NO8»=Ba(NO3J +
+ 2NH4CI при 25°. (Цифрами отме-
отмечены моль Н2О на 100 моль солей.)
могут быть изготовлены из рас-
раствора, получаемого при выщела-
выщелачивании плава BaS, пропуска-
пропусканием его при 50—70° через сильно
кислый катионит в Na+- или К+-форме (или сильно основной ани-
онит). Для извлечения 100%) бария катионит не доводят до пол-
полного насыщения и регенерируют раствором соли натрия или калия
с соответствующим анионом, добавляя к нему и маточный раствор,
остающийся после кристаллизации соли бария. Из раствора, со-
содержащего сульфиды щелочных металлов, выделяют соответствую-
соответствующие сульфиды, а маточный раствор возвращают на выщелачива-
выщелачивание плава BaS I02.
КАРБОНАТ БАРИЯ
Карбонизация раствора сернистого бария
Наиболее распространенный способ получения технического
Углекислого бария основан на карбонизации раствора сернистого
A60—170 г/л BaS) при 30—40° двуокисью углерода,
464
Гл. XII. Соли бария
содержащейся в газах, отходящих из известково-обжигательных
печей, из печей в производстве сернистого бария и др. В применяе-
применяемых для этой цели печных газах не должно содержаться заметных
количеств кислорода во избежание окисления сернистого бария, а
содержание СО2 желательно не ниже 20%.
Ввиду того, что сернистый барий в растворе гидролизован
2BaS + 2Н2О = Ва(ОНJ + Ba(HSJ
при карбонизации идут две последовательные реакции. Вначале
карбонизуется Ва(ОНJ:
Ва(ОНJ + СО2 = ВаСО3 + Н2О
При дальнейшей карбонизации выделяется сероводород по ре-
реакции:
Ba(HSJ + СО2 + Н2О = ВаСО3 + 2H2S
Таким образом, процесс идет по суммарному уравнению:
BaS + СО2 + Н2О = ВаСО3
Печной газ проходит последовательно через 3—4 цилиндриче-
цилиндрических резервуара с коническим днищем, где барботирует через
предварительно отстоявшийся от шлама раствор сернистого бария.
Это обеспечивает хорошее использование содержащейся в газе
двуокиси углерода. Вместе с СО2 газ приносит в последующий
карбонизатор H2S, выделившийся в предыдущем по ходу газа ап-
аппарате. H2S поглошается сульфидом бария с образованием гидро-
гидросульфида:
BaS + H2S = Ba(HSJ
В дальнейшем гидросульфид бария карбонизуется. По оконча-
окончании реакции в первом по ходу газа аппарате его отключают и
дают отстояться образовавшемуся осадку ВаСО3. Затем жидкость
сливают и направляют на выщелачивание плава сернистого бария,
а осадок промывают несколько раз горячей водой, отжимают на
фильтрах (а иногда под прессом) и сушат.
Получаемый таким путем углекислый барий всегда, даже при
хорошей промывке, содержит небольшое количество сульфидов и
серы. С целью получения продукта, не содержащего серы, осаж-
осажденный ВаСО3 смешивают с водой, добавляют небольшие коли-
количества щелочей (NaOH, NH4OH, Na2CO3, NaHCO3 и т. п.) и нагре-
нагревают до кипения. При этом образуются сульфиды, легко отмывае-
отмываемые от осадка горячей водой. Более полная очистка осадка ВаСО3
от сульфидной серы достигается при нагревании его в слабых
растворах щелочей до 140—150° в автоклавах. Общее содержание
серы становится при этом ниже 0,2% в пересчете на BaSOt.
Карбонат бария
455
Осаждение ВаСОз из растворов BaS производят также содой
по реакции:
Ва(ОНJ + Ba(HS)8 + 2Na2CO3 = 2ВаСО3 + 2Na2S + 2Н2О
Для получения более чистого осадка следует к 15—16% рас-
раствору соды, нагретому до 70—80°, медленно приливать 11—15%
раствор BaS, не допуская полного израсходования соды (избыток
соды должен равняться 1—2%). Полученный осадок BaCOs легко
отстаивается и промывается.
Получение ВаСО3 из нитратного щелока
Для производства ВаСО3 используют, например, раствор, со-
содержащий 140—160 г/л Ba(NO3J — осветленный нитратный щелок
из производства азотнокислого бария. К щелоку, после контроль-
контрольной фильтрации на вакуум-фильтре, добавляют раствор NaOH
с концентрацией 500 г/л до свободной щелочности 1—2 г/л (в пере-
пересчете на NaOH). Подщелачивание ведут при 60—70° в резервуаре
с змеевиками для подогрева паром и барботером для перемеши-
перемешивания раствора воздухом. Затем нитратный раствор обезврежи-
обезвреживают (удаляют из него сернистые соединения), медленно добавляя
раствор гипохлорита натрия A20 г/л активного хлора) до появ-
появления свободного активного хлора, определяемого пробой на иодо-.
крахмальную бумажку. При этом идут следующие реакции:
BaS + 4NaClO = BaSO4 + 4NaCl
S + 3NaClO + H2O = H2SO4 + 3NaCl
H2SO4 + Ba(NO3J = BaSO4 + 2HNO3
После отстаивания раствора от BaSO4 и других примесей его
приливают к перемешиваемому, подогретому до 80—90° раствору
соды B00 г/л Na2CO3). Конец реакции
Ba(NO3J + Na2CO3 = BaCO3 + 2NaNO3
устанавливают по щелочности реакционной смеси — около 0,5 г/л
(в пересчете на ОН"). По окончании осаждения карбоната бария
продолжают перемешивание при 85—90° в течение 10—15 мин,
затем дают осадку ВаСО3 отстояться в течение 20—30 мин. Рас*
твор NaNO3 декантируют и отправляют на выпаривание, а осадок
промывают на вакуум-фильтре горячей водой (80—909) от нитрата
натрия и ионов хлора. Затем осадок разваривают при 80—90°, от-
отфильтровывают, окончательно промывают, сушат и укупоривают.
На 1 т продукта (в пересчете на 100%) ВаСО3) расходуют: 0,59—
0,6 г кальцинированной соды (95% Na2CO3), 2,4 т барита A00%),
0,675—0,71 г азотной кислоты A00%), 0,22—0,25 т едкого натра
(S2%), 0,02—0,026 т гипохлорита натрия A00% активного хлора).
456
Гл. XII: Соли бария
Другие способы получения ВаСО3
При небольших масштабах производства для осаждения угле-
углекислого бария используют производственные растворы хлористого
бария, обрабатывая их содой:
ВаС12 + Na2CO3 = BaCO3 + 2NaCl
Обычно раствор содержит 180—200 г/л ВаС12. Его предвари-j
тельно очищают от ионов кальция и железа обработкой раствором..
NaOH или промывными водами, получаемыми при производстве*
гидроокиси бария, содержащими 30—40 г/л Ва(ОН)г. Для осаж-'
дения берут раствор соды, содержащий 180—-200 г/л №2СО3. Про-;
цесс ведут так же, как и при осаждении ВаСО3 из Ba(NO3J. Про-
Промывку осадка на фильтре горячей водой ведут до содержания
в нем С1~ не больше 0,05%- На 1 т 100%-ного углекислого бария
расходуется 1,276—1,29 т ВаС12-2Н2О A00%), 0,553—0,575 т каль-;
цинированной соды (95%), 0,011т едкого натра (92%), 100—
120 кет 'Ч электроэнергии, ~3 мгкал пара, ~100лг3 воды.
Существуют и другие способы получения ВаСО3, например.
осаждение его из растворов BaS или ВаС12 углекислым аммонием,
или разваривание BaSO4 в концентрированном растворе углекис-;
лого калия под давлением 5 ат.
При перемешивании 12—15 мин суспензии BaSO4 в 20% рас-г
творе №2СОз (с небольшой добавкой NaOH) в автоклаве при 8—'\
10 ат и 200° выход ВаСО3 достигает 90 %103.
Возможно получение ВаСО3 взаимодействием BaSO4 и
ь среде расплавленного NaCl104.
ГИДРООКИСЬ БАРИЯ
Получение гидроокиси бария из ВаС12 и NaOH
Реакцию обменного разложения
ВаС12 + 2NaOH = Ва(ОНJ + 2NaCl
осуществляют в цилиндрических обменниках-кристаллизаторах из|
нержавеющей стали, снабженных водяной рубашкой для охлажде-j
¦ ния и мешалкой D5 об/мин). В аппарат загружают раствор хлори-
хлористого бария, имеющий концентрацию 365 г/л ВаС12 и темпера-]
туру 40°, и затем, при непрерывном размешивании, спускают из]
мерника раствор едкого натра F50 г/л) с 18%-ным избытком про-'
гив стехиометрического количества. Массу размешивают 1 ч и за-ч
тем пускают в рубашку воду для охлаждения. После охлаждения'
до 25° (продолжающегося 12—15 ч) массу спускают на фильтр для
отделения кристаллической гидроокиси бария Ва(ОНJ • 8Н2О.
Продукт промывают на фильтре водой до содержания хлора
Гидроокись бария
457
30
го
15
to
2NaCl
\
Ыиаодныиpai
\
\
TlBOff
меньше 0,09% и отжимают под вакуумом. Степень перехода бария
в продукт составляет ~72%. Маточный раствор, содержащий
(в г/л): NaCl 180—200, Ва(ОНJ до 30 и NaOH 30—60, а также
промывные воды C0—40 г/л NaCl и 20—25 г/л Ва(ОНJ) исполь-
используют для растворения хлористого
бария, направляемого для оса-
осаждения углекислого бария.
На 1 г технического продукта
расходуют: 0,92 т хлористого ба-
бария (95%), 0,37 г жидкой каусти-
каустической соды (92%)), 0,085 т каль-
кальцинированной соды (для исполь-
использования бария из маточного
раствора и промывных вод),
1,4\ мгкал пара, 50 ж3 воды,
57 кет • ч электроэнергии; в каче-
качестве побочного продукта полу-
получается 0,15 т ВаСОз A00%). Как
показали исследования105, опи-
описанный здесь производственный
режим не является оптимальным
и может быть улучшен.
В системе ВаС12—NaOH—Н2О
при 30° могут присутствовать
следующие твердые фазы:
ВаС12 • 2Н2О, ВаС1 (ОН) • 2Н2О,
Ва(ОНJ-8Н2О, Ва(ОНJ-ЗН2О,
Ва(ОНJ-Н2О, NaCl и NaOH •
•Н2О (рис. 141). Наибольший
выход Ва (ОНJ • 8Н2О получается
Ва(ОН), 8НгО
Z
,! NaOH
(ОН),
Ва
Эк6и8а/1енгпы.дш единицы
Рис. 141. Диаграмма системы
BaC!2-NaOH-H2O при 30°.
из раствора, содержащего экви-
эквимолекулярные количества ВаС12
и NaOH (точка В) при заверше-
завершении кристаллизации в точке А и
при водном числе исходного рас-
раствора 22—23. Описанным же выше
производственным условиям соответствуют водное число 16,4
и 18%-ный избыток NaOH. Хотя избыток NaOH и увеличивает
очень немного выход гидроокиси бария, но влечет значительное
увеличение расхода щелочи, а уменьшенное водное число приво-
приводит к загрязнению осадка примесями, отмывка от которых влечет
за собой потери гидроокиси бария. Исходный раствор оптималь-
оптимального состава при использовании жидкой каустической соды с кон-
концентрацией 650 г/л NaOH получается при добавлении ее к 22%
раствору ВаС12. Дозировать NaOH следует с избытком ~1% во
избежание потерь ВаС12. Если, кроме этого, после отделения основ-
458
Гл. XII. Соли бария
ной Массы кристаллов Ва(ОНJ-8Н2О при 25° центрифугирова-
центрифугированием, охладить маточный раствор до —5° и выделившиеся допол-
дополнительно кристаллы вместе с основной их массой перекристалли-
перекристаллизовать растворением в воде при 80° и охлаждением до 25°, то
можно получить технический продукт со значительно меньшими
затратами сырья на 1т: 0.785 т хлористого бария (95%), 0,265 т
жидкой каустической соды (92%), 0,027 т кальцинированной соды
(95%), 0,85 т пара, 2,2 т конденсата, ~20 м3 воды, 100 квт-ч элек-
электроэнергии; в качестве побочного продукта получается всего 47 кг
ВаСОз A00%I05-
О расчете степени использования бария и NaOH см.106.
Разработан метод ступенчатой вакуум-кристаллизации <
Ва(ОНJ-8Н2О достаточно крупных размеров из горячего рас-]
твора G5—80°), полученного взаимодействием 40%) раствора-
NaOH с 19—20% раствором ВаС12-2Н2О. При расходе воды на'
"промывку кристаллов 1—1,5 кг на 1 кг продукта содержание*
в нем С1- меньше 0,05% 107.
Возможно непрерывное осаждение Ва(ОНJ*8Н2О 42—44%)
раствором NaOH без подогрева исходного раствора ВаС12 B4—
28% ВаС12'2Н2О) при скорости потока последнего не более 6 л/ч
на 1 л рабочего объема аппарата. Избыток щелочи в реакционной :
смеси —10%, а ее температура 20—25°, но при 5—6° достигается
наибольший выход продукта. Такой «холодный» способ повышает
производительность аппаратуры 103.
Запатентован109 способ дегидратации продукта горячим воз-
воздухом A00—225°) в распылительной сушилке, куда подают рас-
расплав Ва(ОНJ-8Н2О или раствор Ва(ОНJ. Для уменьшения со-
содержания ВаСО3 в продукте воздух рекомендуют очищать от СО2.
Гидроокись бария
459
Получение гидроокиси бария из сульфида бария
BaS
Гидроокись бария можно получить обработкой щелока
марганцовыми рудами или отходами, содержащими МпОг. При
этом идут реакции
Ba(OH)(HS) + 2MnO2 =. Ва(ОНJ + Мп2О3 + S
Ba(OH)(HS) + 1,5МпО2 = Ва(ОНJ + Ч2Ш3О4 + S
Ba(OH)(HS) + МпО2 = Ва(ОНJ + МпО + S
2Ba(OH)(HS) + МпО2 = 2Ва(ОНJ + MnS + S
A)
B)
C)
D)
константы равновесия которых при 25°: Ki = Ю35, /С2 = 1034,
Д'з = 1021 и Ка = 109. Следовательно, равновесие этих реакций
резко сдвинуто вправо и процесс обессеривания щелока должен
идти практически до конца. Действительно, при осуществлении
противоточной обработки марганцовой руды щелоком B7—69 г/л
BaS) в аппаратах с мешалками при 25—40° в течение 60 мин, при
молярном отношении МпО2 : Ва от 4 до 2,4, выход Ва(ОНJ соста-
составляет около 99%)- Увеличение количества МпО2 против приведен-
приведенной выше оптимальной величины приводит к снижению выхода,
вероятно вследствие протекания реакции:
Ва(ОНJ + МпО2 = ВаМпОз + Н2О
Раствор Ва(ОН)г получается разбавленным и до кристаллиза-
кристаллизации его требуется выпаривать110'111.
Гидроокись бария может быть получена (с выходом 95—98%)
взаимодействием водной суспензии ZnO и BaS. После отделения
ZnS, охлаждая раствор, выделяют Ва(ОНJ • 8Н2О 112. Лучшие ре-
результаты дает осаждение гидроокиси бария с помощью Си2О —
осадок Cu2S меньше по объему и фильтруется в 10—20 раз быст-
быстрее, чем ZnS, поэтому меньше потери раствора Ва(ОНJ, кроме
того он менее загрязнен ш.
Запатентован ш способ изготовления гидроокиси бария непо-
непосредственной ее кристаллизацией из раствора, получаемого выще-
выщелачиванием плава BaS, при охлаждении до 13°. При двухкратной
промывке кристаллов насыщенным раствором Ва(ОНJ при 22°
(при Т: Ж = 1 : 1 по весу) получается продукт с отношением
Ba(OHJ:Ba(HSJ = 48 : 1.
Другие способы получения гидроокиси бария
Гидроокись бария может быть получена из осажденного кар-
карбоната бария и тонкоизмельченного (—10 000 отв/смг) кремнезема
любого сорта, например кварцевого песка. При спекании в течение
1 ч этих материалов при 1100° в присутствии 1—2% ускоряющих
процесс добавок (марганцовый шлам или свежеосажденная окись
железа) образуется трехбариевый силикат ш:
ЗВаСОз + SiO2 = Ba3SiO5 + ЗСО2
Его подвергают 3—4-минутной обработке водой при 80°:
Ba3SiO5 + 2Н2О = 2Ва(ОНJ + BaSiO3
Полученный после выщелачивания раствор перерабатывают на
кристаллическую гидроокись бария, а метасиликат бария возвра-
возвращают на спекание с карбонатом бария, причем также образуется
трехбариевый силикат:
BaSiO3 + 2ВаСО3 - Ba3Si05 + 2СО2
Выход продукта достигает 90—95%) 116.
Гидроокись бария можно получать одновременно с литопоном 3.
Фначала получают цинкат бария взаимодействием раствора
460
Гл. XII. Соли бария
сернистого бария с суспензированной в нем окисью цинка при
80—90°j
2BaS + 3ZnO = 2ZnS + 2BaO • ZnO
2BaS + 4ZnO = 2ZnS + 2BaO ¦ 2ZnO
Затем в реакционную массу добавляют новую порцию BaS и
сернокислый барий — происходит осаждение литопона:
2ВаО • ZnO + BaS + BaSO4 + ЗН2О = ЗВа(ОНJ + ZnS + BaSO,
После отделения литопона гидроокись бария выделяют из рас-
раствора охлаждением 117.
Предложено получать гидроокись бария прокаливанием смеси
тонкоизмельченных барита и пирита (FeS2: BaSC>4=2—5) при
750—950° с пропусканием через шихту водяного пара. При обра-
обработке полученного огарка водой образуется раствор Ва(ОНJ. Ре-
Рекомендуется вести выщелачивание под давлением при 150°ш.
Разработаны способы получения гидроокиси бария из щелоков
BaS 119 или из растворов хлорида, нитрата или ацетата бария ш
с помощью сильнокислотных (К+- или Ыа+-форма) или сильно-
сильноосновных (ОН~-форма) ионитов, которые регенерируют раство-
растворами едких щелочей. Для сорбции бария из сульфидных раство-
растворов рекомендуется фильтровать его через катионит КУ-2 при 60°
и элюировать его при этой же температуре раствором NaOH; этим
способом можно получить продукт, содержащий больше 99% гид-
гидроокиси бария ш.
Разработан способ получения гидроокиси бария электролизом
раствора ВаС12. При концентрации исходного раствора ВаС12
330—335 г/л и катодной плотности тока до 2500 а/м2 образуется
амальгама с концентрацией 0,1—0,15% Ва; при температуре в раз-
лагателе больше 80° получается раствор, содержащий 300 г/л
Ва(ОНJ. Напряжение на ванне 5 в; катод — ртутный; выход
Ва(ОН)г по току превышает 90%) 122.
ОКИСЬ И ПЕРЕКИСЬ БАРИЯ
Окись бария получают прокаливанием азотнокислого или угле-
углекислого бария в тигельных или в муфельных печах различных кон-
конструкций (углекислый барий прокаливают также и в других печах,
например шахтного типа).
Реакция
2Ba(NO3J = 2ВаО + 4NO + ЗО2
протекает при нагревании до 1000—1050°. После нескольких часов
прокаливания получается пористая масса, содержащая более 96%)
ВаC. Выделяющиеся сильно разбавленные воздухом окислы азота
улавливают щелочными растворами. При прокаливании Ba(NO3J
в стальных тиглях высотой 0,5 м и диаметром 0,4—0,5 м (слегка
Окись и перекись бария
461
конической формы) на внутреннюю и наружную поверхность тиг-
тиглей наносят слой шамотной обмазки. Тигли закрыты крышкой с от-
отверстием для выхода газов. Нагрев ведут медленно, в течение 30—
35 ч, во избежание бурного выделения газов и уноса материала из
тиглей. Затем печь охлаждают 10—11 ч и выгружают тигли. После
дополнительного охлаждения из них выбивают окись бария, кото-
которую помещают в герметически закрываемые стальные ящики для
предотвращения карбонизации на воздухе.
Разложение ВаСО3 на ВаО и СО2 начинается выше 900°. Для
полного разложения требуется накаливать углекислый барий до
1400°. Снижение температуры в печи приводит к обратному обра-
образованию ВаСО3 из продуктов его диссоциации. Частичное оплавле-
оплавление поверхности прокаливаемых кусков материала также сильно
затрудняет процесс. Эти трудности устраняются при прокалива-
прокаливании карбоната бария в смеси с углем. Вследствие того, что угле-
углерод восстанавливает СО2 до СО реакция протекает по схеме:
ВаСОа + С = ВаО + 2СО
Отсутствие в газовой фазе СО2 исключает обратный ход реак-
реакции, что дает возможность вести ее при температурах ниже 1200°
и избежать оплавления материала 123. Для этого процесса предло-
предложено применять аппаратуру из карборунда с присадкой корунда ш.
Предложено также осуществлять процесс в псевдоожиженном
слое 125.
Получение перекиси бария основано на способности окиси ба-
бария поглощать кислород при не очень высоких температурах
(стр. 417). Выше 900° перекись бария целиком разлагается на
окись бария даже в атмосфере чистого кислорода. Интенсивное
поглощение кислорода окисью бария идет при 450—700°. Процесс
осуществляют нагреванием окиси бария в муфельных печах до
этих температур в струе воздуха или путем нагнетания кислорода
под давлением 2—3 ат в герметически закрытую реторту из нержа-
нержавеющей стали, снабженную электрообогревом. В реторту, предва-
предварительно нагретую до 200°, быстро загружают свежеприготовлен-
свежеприготовленную окись бария, содержащую не меньше 96%) ВаО. Затем реторту
закрывают, продувают для удаления воздуха и нагнетают в нее
кислород, очищенный от СО2 и влаги. В дальнейшем через каждые
1,5—2 ч производят продувку реторты, удаляя через продувочный
кран накапливающиеся в процессе поглощения кислорода инерт-
инертные газы (примеси в кислороде). После достижения ~700° элек-
электрообогрев выключают и температуру поддерживают на этом
уровне за счет тепла реакции около 4 ч. Когда процесс близок
К "завершению, температура начинает снижаться, и при достиже-
достижении 600—580° включают подогрев. После 4-часовой выдержки элек-
"Врообогрев прекращают и массу охлаждают при подаче кислорода
462
Гл. XII. Соли бария
до 350—380°. Затем прекращают подачу кислорода и выгружают
продукт в подставленный под реторту барабан из оцинкованной
стали. Продукт размалывают в шаровой мельнице и просеивают
,A600отв/ам2). Отсев содержит 83—87% ВаО2 и идет в отход
A12кг на 1 т продукта I сорта). Продукт содержит более 90%
ВаО2. Выход ВаО2 из ВаО составляет ~92% от теоретического.
Для получения 1 т продукта расходуют (при производстве окиси
бария прокаливанием азотнокислого бария в тиглях): 2,2 т азотно-
азотнокислого бария (99%), 645 м3 кислорода, 7,05 т условного топлива
и 1500 кет -ч электроэнергии.
Описанный способ является малопроизиодительным и громозд-
громоздким. Предложены способы получения ВаО2 окислением кислородом
или воздухом при 400—700° расплавленной гидроокиси бария. Раз-
Разработан также способ окисления твердой, нерасплавленной гидро-
гидроокиси 126. Процесс заключается в обезвоживании октагидрата гид-
гидроокиси бария и окислении Ва(ОНJ при 550—600°. Окисление
идет быстро и получаемый продукт содержит 93—95% ВаО2.
Можно получать ВаО2 через дипероксигидрат ВаО2 • 2Н2О2, обра-
образующийся при действии Н2О2 на Ва(ЫОзJ в аммиачной среде при
обычной температуре. При этом сначала образуется гидроокись,
которая сразу же реагирует с перекисью водорода. После высуши-
высушивания при ПО—115° получается продукт, содержащий 92—95%
ВаО2 5-127.
Предложено получать из Ва(ОНJ и Н2О2 водную суспензию
ВаО2-8Н2О и затем обезвоживать перекись бария, нагревая сус-
суспензию выше 55°, отфильтровывать ВаО2 и высушивать при ПО—
150°128.
ЛИТЕРАТУРА
/. Н. К. Воскресенская, Г. Н. Кащеев, ИСФХА, 25, 168 A954).—
2. М Mich and, Compt. rend., C262, № 14, 1143 A966). — 3. Д. А. Дымчи-
шин, Производство бариевых солей, ГОНТИ, 1938.—4. Перекись водорода и
перекисные соединения, под ред. М. Е Познна, Госхимиздат, 1951.—
5. Н К- Григорьева, Т. И. Арнольд, С. 3. Макаров, сб. «Химия пере-
кисных соединений». Изд. АН СССР, 1963, стр. 94.-6. И. П. Во льнов,
А. Н. Ш ату нин а, ДАН СССР, ПО, ЛЬ 1, 87 A956). —7. М. Brook, Chemist-
Analyst, 42, № 2, 28 A953). — 8. А. М. Голуб, Г. М. Кил им ник, ЖПХ, 29,
№ I i7 A956). — ?. J. Doucet, N. Bizouard, Compt. rend., 242, № 17, 2137
A956). — 10. И. Н. Беляев, М. Л. Шолохович, ЖПХ, 25, № 8, 818 A952).
// П. П. Будников, П. А. Володин, С. Г. Тресвятский, Укр.
хим. ж., 22, № 3, 292 A956). —12. Г. А. Б уха лов а, Е. С. Ягубьяы, Изв.
вузов Хнм. и хим. технол., 3, № 5, 783 A960). —13. Л. Н. Успенская,
А. Г. Бергмаи, ЖОХ, 25, № 7, 1277 A955). —14. Л. Н. Успенская,
Н. П. Глушкова, А. Г. Бергман, Там же, 25, № 9, 1658 A955). —¦
15. Г. М. К у п е р м а н, Д. X. Г в а р а м а д з е, С. И. Д ж и к и я, Н. П. 3 а р«
к у а, Труды Ин-та химии АН ГрузССР, т. 11 1953, стр. 117. — 16. A. Paoloni,
J. electr. Ind. electrochimi, 58, 55, 82 A949). — 17. Э. К. Келлер, В. Б. Глуш-
Глушкова, ЖНХ, 1, № 10, 2283 A956). — 18. Н. Н. Беляев, М. Л. Шолохович,-
ЖПХ, 25, № 6, 657 A952). — /?. М. Л. Шолохович, Н. Н. Беляев, ЖОХ,
Литература
463
24, № 2, 218 A954). — 20. К. Н. Leiser, Z. anorg. Chem., 335, № 5, 6, 225
A965).
21. J. Bye, J. Holder, ВЫ! Soc. chim. France, 1953, № 4," 399.—
22. M. Г. Габр нелова, ЖХП, № 10. 38 A939).— 23. А. И. Заславский,
Сборник статей к 20-летню ГИПХ, Госхимиздат, 1939, стр. 87. — 24. Правила и
нормы техники безопасности и промышленной санитарии для проектирования,
строительства и эксплуатации производств сернистого бария, хлористого бария
и гидроокиси бария, Госхимиздат, 1963. — 25. М. Г. Габриеле в а, М. А. Мо-
Морозова, Производство неорганических ядохимикатов, Изд. «Химия», 1964,
гл. 5. — 26. В. П. Радищев, Научно-иссл. работы хим. ин-тов и лаб. АН
СССР за 1940 г., Изд. АН СССР, 1941, стр. 21. — 27. А. А. Иванов, Электро-
Электровакуумная технология, Госэнергоиздат, 1944. — 28. Г. Герман, С. Вагенер,
Оксидный катод, Гостехнздат, 1960. — 29. Г. Л. Кореньков, Н. А. Устиио-
в а, Горнохимическое сырье зарубежных стран, Изд. «Химия», 1965, гл. 2. —
30. R. W. Lewis, Bull. Bur. Mines, № 630, 91 A965).
31. E. Ф. Беленький. И. В. Р и с к и и. Химия и технология пигментов,
Госхимиздат, 1960.—32. Е. Ф. Беленький, Производство минеральных кра-
красок и лаков, ГНТИ, 1931. — 33. В. И. Орлов, К. А. Г а р. Хим. пром., № 9, 3
A946). — 34. В. С. Нешпор, В. Л. Юпко, ЖПХ, 36, № 5, 1139 A963).—
35. Mining Ann. Rev., 81, 83 A967).—36. А. И. Майоров, ЖХП, № 7, 13
A940). — 37. Ю. В. Морачевскнй, Труды ГИПХ, в. 37, 1946, стр. 120.—
38. П. Д. Набоков, Хим. пром., № 8, 20 A950). — 39. J. A. Caddiek, Mining
Mag., 90, № 5, 280 A954).— 40. L. Stragiotti, Ind. mineraria, 5, № 8, 451
A954).
41. F. Vogel, пат. ГДР, 10091, 1953. —42. Ф. И. Стригунов, Цвета.
металлы, № 2, 18 A968). — 43. Н. Ш. Сафиуллин, Сборник научно-техн.
информ. материалов НИОХИМ, в. 1, 1960, стр. 74.—44. U. Franc a is. англ.
пат. 724272, 1955. — 45. G. M. Hood, пат. США 2685499, 1954.-4E. Г. М. Ку-
Купе р м а н, С. И. Д ж и к и я, Н. П. 3 а р к у а, Труды Ин-та химии АН ГрузССР,
т. 11, 1953, стр. 127. — 47. R. Albert, пат. ФРГ 907102, 1954.— 48. И. С. Ша-
Шапиро, Хим. пром., № 12, 14 A944). — 49. Н. S. Boots, E. F. Pollard,
М. J. Rentschler, Ind. Eng. Chem., № 10, 1981 A948). — 50. О. И. Пудов-
Пудовкина, Э. М. Моргунова, Труды УНИХИМ, в. 3, 1955, стр. 130.
51. Н. Ф. Юшкевич, П. М. Спиридонов, Минеральное сырье и его
переработка, 1, № 5, 374 A926). — 52. П. В. Дыбина, Технология минераль-
минеральных солей, Госхимиздат, 1949, гл. III.—53. В. В. Печковский, С. А. А м и-
рова, А Н. Кет о в. Уч. зап. Пермского гос. ун-та, 17, в. 1, 45 A960).—
54. А. Н. Кетов, В. В. Печковский, Хим. и хим. технол., № 4, 667 A958).—
55. В. В. Печковский, А. Н. Кетов, Труды Пермского политехи, ин-та,
№ 10, 1961, стр. 3; ЖПХ, 33, № 8, 1719 (I960). — 56. С. А. А мир о в а,
В. В. Печковский, А. Н. Кетов, Там же, стр. 33. — 57. А. Ф. Ложкин,
В. В. Печковский, Н. Л. Субочева, Там же, стр. 57.-58. В. В. Печ-
Печковский, Там же, 32, № 10, 2189 A959). — 59. Польск. пат. 48579, 1963.—
60. Н. Ш. Сафиуллини др., авт. свид. 179287, 1965.
61. W. Steinmetzer, H. Plischke, Chem. Techn., 9, № 1, 38 A957).—
62. А. Ф. Ложкин, В. В. Печковский, Н. Л. Субочева, Уч. зап.
Пермск. гос. ун-та, 17, в. 1, 55 (I960). —63. И. Н. Аверко-Антонович,
Труды Казанск. хим.-техн. ин-та, № 7, 1.938, стр. 3. — 64 L. L e p i n s k i Che-
mik, 18, № 1, 23 A965). — 65. Ф. И. Стригунов, Н. Ш. Сафиуллин,
3. Ф. Гаврилова, Хим. пром., № 3, 227 A967); Труды НИОХИМ, т. 19, Изд.
«Химия», 1969, стр. 123. — 66. В. И. М у м л а д з е, Тезисы докладов VI Всесоюз-
Всесоюзной конференции по технологии неорганических веществ, Тбилиси, 1968,
Стр. 117. — 67. Г. М. К у п е р м а н, С. И. Д ж и к и я, Труды Тбилисск. хим.
ин-та АН ГрузССР, т. 3, 1938, стр. 125. —68. Н. Ф. Юшкевич, П. М. Спи-
Р*донов, Минеральное сырье и его переработка, 2, № 12, 803 A927).~»
464
Гл. XII. Соли бария
69. В. П. Ильинский, Г. С. Клебанов, Труды соляной лаб. АН СССР,
в. 2, 1932. — 70. В. А. Шушунов, Г. И. Садовников, Б. Я. Андреев,
ЖПХ, 28, № 8, 1472 A954).
71. R. V. Culver, С. J. Hamdorf, J. Appl. Chem., 5, № 8, 383 A955).—
72. Н. Ш. С а ф и у л л и н, Э. Ф. Г а в р и л о в a, XiM. пром. Гнформ. иаук техи.
зб., № 3 B3), 32 A965). — 73. В. Н. Чазов, Л. К. Уд а лов, О. Б. Клеба-
Клебанов, Хим пром., № 2, 113 A966). — 74. М. Е. Позин. Технология минераль-
минеральных удобрений и солей, Госхимиздат, 1957, гл. IX. — 75. Польск. пат. 53368,
1965, — 76. Г. А. Ткач и др.. Труды НИОХИМ, т. 19, 1969, стр. 269.—
77. М. К. Гольдберг, ЖХП, № 18, 1115 A936). — 78. Д. В. Безуглый,
Ф. М. Куца ков, Там же, № 8, 37 A939).— 79. В. М. Какабадзе, И. Г. Б у-
чукури. Там же, № 8, 240 A950). —SO. Г. Н. Купер май, С. Н. Джикия,
Труды Тбилисск. хим. ин-та АН ГрузССР, т. 7, 1944, стр. 117.
81. В. М. Какабадзе, Я. Г. Бучукури, Хнм. пром.. № 1, 16 A951).—
82. Ф. К. Михайлов, Там же, № 7, 393 A956). — 83. В. Ф. Чернов. Про-
Производство кальцинированной соды, Госхимиздат, 1956, стр. 300.— 84. Д. М. Гинз-
Гинзбург, Н П. Красильникова, С. С. Де мирская. Труды НИОХИМ,
т. 10, 1957, стр 146. — 85. Ф. Н. Строков, Труды ГИПХ, в. 11, 1928,
стр. 167. —86. Ф. Н. Строков, Там же, в. 10, 1928, стр. 36. —87. В. М. Гри-
невич. ЖПХ, 14, № 1, 63 A941). — 88. В. В. Печковский, А. Л. С о ф р о-
нов, ЖПХ, 34, № 10, 2153 A966). —<?5. S. S. Bratnagar, S. Parthasa-
rathy, A. L. Sundara Rao, J. Sci. Ind. Res. (India), № 3, 108 A944).—
90. Франц. пат. 64928, 1928.
91. M О. X a p м а д а р я н, К. И. Б р о д о в н ч, Укр. хим. ж., 8, 1 A933). —
92. Я. Г. Бучукури, Сообщение АН ГрузССР, 10, № 6, 329 A949).—
93. R. N. Schreve, R. К. То пег, Ind. Eng. Chem., 32, № 4, 568 A940).—
94. R. N. Schreve, M. F. Wiegant. ibid., 35, № 5, 608 A943). — 95. P. Вен-
нер. Термохимические расчеты, ИЛ, 1950, стр. 196. — 96. О. И. Пудовкина,
Е. И. Толстикова, Труды УНИХИМ, в. 3, 1955, стр. 133.— 97. A.M. Гинст-
линг, А. Д. Волков, ЖПХ, 33, № 8, 1700 (I960). — 98. Ф. И. Стригунов,
Хим. пром., № 11, 856 A968). — 99. Ф. И. Стригунов, авт. свид. 215908,
1966. —100. М. А. Миниович, Соли азотной кислоты (нитраты), Госхимиз-
Госхимиздат, 1946, стр. 87.
101. А. Б. Здаиовский, Бюлл. ВИГ, № 3, 1940; Труды ВНИИГ, в. 21,
1949, стр 348. — 102. J. P. Leaver, M. Т. Pope. англ. пат. 1017341, 1963 —
103. Е. Zirngiebl, пат. ФРГ 1177122, 1963. — 104. Н. S. Booth, E. F. Pol-
Pollard, Ind. Eng. Chem., 40, № 10, 1983 A948).— 105. M Б. Зеликиы, С. С. Де-
мирская, Тоуды НИОХИМ, т. 10, 1957, стр. 126. — 106. В. И. Панов и др..
Хим. пром., № 11, 849 A968). —107. Н. Ш. Сафиуллин, С. К- Соляник,
сб. «Химическая промышленность Украины», № 6 C0), 1966, стр. 8.—
108. В. Л. Сокол, Е. А. Риф, А. В. Б р о м б е р г, Л. И. Ушакова, Труды
ИРЕА, в. 27, 1965, стр. 365. —109. S. К- May, R. J. Sinnott, англ пат.
1000301. 1962. — 110. В. М. Какабадзе, П. И. Гагнидзе, ДАН СССР, 105,
№ 5, 1053 A955).
111. П. И. Гагиидзе, Автореф. каид. дисс, Грузинск. политехи, ин-т,
Тбилиси, 1955. — 112. В. М. Какабадзе, Т. А. Иванова, И. П. Чача-
нидзе, Реф. докл. VIII Менделеевского съезда, № 1, 145 A959). —113. R. Scho-
ber, пат. ФРГ 1181188, 1963. —.Ш. J. G. Marshall, A. E. Oates, англ. пат.
980813, 1962. — 115. В. Б. Глушкова, Э. К. Келлер, ЖНХ, 2, № 5, 1001
A957). —116. В. М. Какабадзе, Г. Д. Чачанидзе, ЖПХ, 26, № 8, 787
A953). —117. В. М. Какабадзе, Г. Д. Чачанидзе, Труды Грузинск. по-
политехи, ин-та, № 28, 1953, стр. 3. — 118. F. W. J a h п, пат. США 2724639, 1956. —
119. J. G. Marshall, A. E. Oates, N. A. McDonald, аигл пат. 992793,
1961. —120. 3. P. Leaves, M. Т. Pope, англ. пат. 1013711, 1963.
i
Литература
465
121. Л. Л. К о ш а к о ш в и л и, Я. Г. Бучукури, В. Д. Э р и с т а в и, Те-
висы докладов VI Всесоюзной конференции по технологии неорганических ве-
веществ, Тбилиси, 1968, стр. 54.-/22. Л. А. И сэров, Труды НИОХИМ, т. 10,
1969, стр. 155. —123. С. N. Satterfield, F. Feakes, Chem. Eng. J., 5, № 1,
122 A959). —124. Kfisberg, пат. ФРГ 881494, 1953. —125. W. H. Kahn,
J. С. S ind linger, франц. пат. 1076381, 1954. — 126. Л. А. И с аров, сб. «Хи-
«Химия перекисных соединений», Изд. АН СССР, 1963, стр. 101. —127. С. 3. Ма-
Макаров, Н. К. Григорьева, ЖПХ, 32, № 10, 2184 A959). — /25. Э. М. Мит»
к е в и ч, И. П. К н и г а в к о, В. В. Ч е с н о к о в а, сб. «Химическая промышлен-
промышленность Украины», № 6 D2), 1968, стр. 17,
Глава XIII
СОЛИ СУЛЬФИДНОГО РЯДА
К солям сульфидного ряда относятся производные сероводо-
сероводорода (сульфиды), полисернистых водородов (полисульфиды),
' а также оксисульфиды типа RO • RS. Они широко распространены
в природе и являются сырьем для получения цветных металлов и
серной кислоты. К ним относятся сульфиды меди, свинца, серебра,
цинка, никеля, кобальта, железа, мышьяка, сурьмы, ртути и др. *.
Природные сульфиды не растворимы в воде. Важнейшими суль-
сульфидными продуктами, получаемыми заводскими способами или об-
образующимися при очистке промышленных газов от сероводорода,
являются растворимые соединения щелочных металлов. В этой
главе рассматриваются производства сернистого натрия, гидро-
гидросульфида натрия и некоторых полисульфидов.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Сернистый натрий Na2S в безводном состоянии имеет плот-
плотность 1,86 г/см3, плавится при 1180°2. Сильно гигроскопичен. На-
Насыщенный водный раствор содержит при 18° 15,3%, при 90° 36,4%
Na2S. Из водных растворов ниже 48° кристаллизуется Na2S • 9Н2О,
выше 48° — Na2S • 6Н2О. Водные растворы имеют сильно щелоч-
щелочную реакцию вследствие гидролиза:
Na2S + H2O ^з± NaOH + NaHS
В 0,1 н. растворе при 25° степень гидролиза Na2S составляет
86,5%. Дальнейший гидролиз по схеме NaHS + H2O^NaOH + H2S
практически не идет; в 0,1 н. растворе при 25° NaHS гидролизуется
лишь на 0,12%.
Гидросульфид (сульфгидрат) натрия NaHS устойчив только в
растворах. Кислород воздуха медленно окисляет эти растворы, ве-
вероятно, по следующей схеме3:
HS" + О2 = HSOJ; 2HSOJ = S2O§~ + H2O; 2HSOJ + О2 - 2HSO3
Физико-химические свойства
467
Образование тиосульфата идет быстрее, чем сульфита, поэтому
главным продуктом окисления является тиосульфат. Кроме того,
продуктами окисления являются также сульфат и полисульфиды.
Сульфид натрия окисляется медленнее гидросульфида; механизм
его окисления сложнее — помимо тиосульфата, сульфита и суль-
сульфата образуются политионовые кислоты, преимущественно тритио-
новая (см. гл. XIV). При нагревании растворов NaHS переходит
в Na2S и выделяется сероводород:
2NaHS ^± Na2S + H2S
Растворимость NaHS в воде при 20° равна 42% 4.
Сульфид аммония устойчив при температуре ниже —16°, вышь
этой температуры разлагается:
(NH4JS = NH3 + NH4HS
Поэтому при взаимодействии газообразных NH3 и H2S без
охлаждения образуется гидросульфид аммония, устойчивый до'
~32°. Давление диссоциации твердого NH4HS (с выделением эк-
эквимолекулярной смеси NH3 и H2S) равно:
Температура, °С 4,2 9,5 22 25 32,6 >44,4
Давление, мм рт. ст 132 175 410 501 772 1560
О системе NH3—H2S—Н2О см.'5-6, а о получении (NH4JS см.7.
Гидросульфиды натрия или калия образуются при поглощении
сероводорода8 из промышленных или природных газов водными
растворами почти всех применяемых поглотителей — карбонатов,
фосфатов, фенолятов натрия или калия, солей аминокислот (на-
(например, диметиламиноуксусного калия):
Na2CO3 + H2S ^=± NaHCO3 + NaHS
K3PO4 + H2S 5=± K2HPO4 + KHS
C6H5ONa + H2S ^=t C6H5OH + NaHS
(CH3JNCH2COOK + H2S ^± (CH3JNCH2COOH + KHS
При регенерации растворов выделяется сероводород, который
может быть использован.
При поглощении сероводорода водными растворами идут сле-
следующие равновесные процессы:
H2S(r.) ^=± H2S(pacTB.) 5=*: H++HS"
Левое из этих равновесий зависит от парциального давления
H2S в газе и от температуры, а правое, также зависящее от тем-
температуры, устанавливается в соответствии с константой диссоциа-
диссоциации /? =
[H+][HSi
-1-7
щ S1 . При 25° ft =1,2-10 '. Для хорошего погло-
поглощения сероводорода нужно, чтобы концентрация H2S в растворе
била малой, что достигается при значительной диссоциации, когда
468
Гл. XIII. Соли сульфидного ряда
отношение [HS~]: [HjS] велико. Это осуществляется в растворе
с малой концентрацией ионов Н+. Если, например [HS~]: [H2S] =
= К : [Н+] = 100, то [Н+] = 1,2 • 10~9, что соответствует значению
рН«*9. Приблизительно такой порядок значений рН и имеют рас-
растворы упомянутых выше поглотителей. Еще лучшими поглотите-
поглотителями были бы растворы с большими значениями рН, однако по
этой же причине регенерация таких растворов с выделением погло-
поглощенного сероводорода была бы затруднена. Хорошо поглощают
сероводород и легко отдают его при регенерации вещества, для
реакции которых с H2S изменение изобарного потенциала AZlw
находится в пределах от +15 до —19.
Константы равновесия для систем H2S—К2СО3—КНСО3—
KHS—HsO (I) и H2S—Na2CO3—NaHCO3—NaHS—H2O (II) при
25° io, п.
____ [KHCOsHKHS] ^007i г-мол
1 [K2CO3] pH'2S ~° ' л - мм рт. dr. '
• Применение.
469
_
[Na2CO3
_
г-мол
л-мм рт. ст.
Их зависимость от температуры в пределах 25—60°:
2800 -3,2; • - 25°°
Равновесное давление сероводорода над раствором для этих си-
систем:
pLs = 0,714(/ + 43)C2-88 и PKS
где С — концентрация раствора, н.
Полисульфидными называются соединения типа M2Sn или
M2S • Sn_i, так как предполагается, что атомы серы здесь связаны
с сульфидом. В водных растворах полисульфиды гидролизуются 12,
например:
2 (S • S4J" + 6Н2О = 2S2O|" + 6H2S
Способность к гидролизу понижается с уменьшением числа ато-
атомов серы в молекуле полисульфида, вероятно вследствие уменьше-
уменьшения при этом степени диссоциации13. Полисульфиды в твердом
вкде сильно гигроскопичны и во влажном воздухе окисляются
с выделением свободной серы
s2r+i,5o2=s2ol-+sn_2
и далее
В присутствии двуокиси углерода полисульфиды наряду с се-
серой рыделяют также сероводород:
sz~ + со2+н2о = со|~+h2s + sn_,
Полисульфиды образуются при взаимодействии сульфидов с се-
серой, а также при медленном окислении кислородом воздуха гид-
гидросульфидных растворов, например:
4MHS + О2 = 2Н2О + 2M2S2
Для натрия известны полисульфиды с числом атомов серы да
я = 5. Как видно из рис. 142, в системе NagS—S .полисульфиды мо-
Na2S
900^°
50
60 70
Сера. Sec.%
ВО
Рис. 142^, Система Na2S—S.
гут существовать до 920° (по новейшим данным2 до 1180°); выше
этой температуры существует только моносульфид натрия. Для
калия известны полисульфиды с числом атомов серы до 6. Поли-
Полисульфиды кальция менее устойчивы, чем полисульфиды натрия и
калия и сохраняются лучше всего в растворах, но известны и су-
сухие препараты полисульфидов кальция. Еще менее стабильны
полисульфиды аммония.
ПРИМЕНЕНИЕ
Сульфид натрия служит сырьем для производства сернистых
красителей, применяется в текстильной промышленности при кра-
крашении этими красителями хлопчатобумажных тканей, в кожевен-
кожевенной промышленности для удаления волоса со шкур, в производстве
красок. Его используют также для получения тиосульфата и гид-
гидросульфида натрия и во флотационных,процессах, в частности при
флотации цинковой обманки и руд, содержащих железо, цинк и
470
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
471
свинец. Описаны опыты по замене сульфидом натрия едкого натра
при химической обработке глинистых растворов для выщелачива-
выщелачивания гуминовых веществ из бурых углей14. Сульфид натрия яв-
является полупродуктом в некоторых способах получения соды и
едкого натоа из сульфата натрия.
Технический сернистый натрий выпускают в виде плавленого
продукта в железных барабанах или в чешуированном виде в поли-
полиэтиленовых мешках, вложенных в бумажные. Согласно ГОСТ
596—70, он должен содержать (в %):
Высший 1-й 2-й
сорт сорт сорт
Сернистого натрия (Na2S), не менее ... 70 66 63
Не растворимых в воде веществ, не бо-
более 0.2 0.5 1,5
Железа (Fe), не более 0,08 0,15 0.45
' Гидросульфид натрия применяют главным образом в производ-
производстве искусственного шелка, а также в кожевенной промышленно-
промышленности. Его выпускают в виде раствора, содержащего 22% NaHS и
не более 3% Na2S и транспортируют в стальных цистернах, кон-
контейнерах и бочках.
Полисульфиды также используют в кожевенной промышленно-
промышленности и при флотации руд и особенно в качестве средств борьбы
с вредителями растений 15>16. Наиболее распространенным поли-
полисульфидным инсектофунгицидом является известково-серный от-
отвар, содержащий полисульфиды кальция; реже применяются по-
полисульфиды натрия или калия, называемые иногда серной печенью,
а также аммония и бария, б том числе и сольбар (стр. 421).
Токсичность полисульфидов объясняется действием свободной
серы, выделяющейся при разложении препаратов на воздухе. По-
Полисульфиды натрия нашли также широкое применение в производ-
производстве полимеров, используемых для получения полисульфидных кау-
чуков (резинит, тионол и др.).
СУЛЬФИД НАТРИЯ
Сульфид натрия получают, главным образом, восстановлением
сульфата натрия углем. Полученный плав содержит, помимо Na2S,
значительные количества примесей, для освобождения от которых
¦его подвергают выщелачиванию. Образовавшийся раствор серни-
сернистого натрия отделяют от непрореагировавшего угля и других не-
нерастворимых веществ и подвергают выпариванию, в процессе ко-
которого происходит очистка раствора от растворимых соединений.
Концентрированный раствор разливают в барабаны, где он засты-
Бает в продукт, называемый плавленым сернистым натрием.
Физико-химические основы восстановления Na2SO4 углем
Восстановление Na2SO4 осуществляют чаще всего в коротких
горизонтальных барабанных вращающихся печах периодического
действия, в которых смесь сульфата с углем нагревается топоч-
топочными газами до 1200°. При этом проходят следующие суммарные
реакции:
Na2SO4 (тв.) + 2С (тв.) = Na2S (тв.) + 2СО2 (г.) — 53,8 ккал/г-мол A).
Na2SO4(TB.) + 4C(TB.) = Na2S(TB.) + 4CO(r.)-136,2 « B)
Na2SO4 (тв.) + 4СО (г.) = Na2S (тв.) + 4СО, (г.) - 28.6 « C).
Значительная масса сульфата натрия восстанавливается, ви-
видимо, по реакции A). Образующаяся при этом СО2, реагируя
с углеродом, превращается в СО, так как выше 800° равновесие
в системе СОо+Сч^СО — 41,2 ккал/г-мол сдвинуто вправо. Неко-
Некоторое количество сульфата восстанавливается окисью углерода по
реакции (ЗI7, а образующаяся двуокись углерода, реагируя с угле-
углеродом, вновь превращается в СО; в итоге это приводит к частич-
частичному протеканию процесса по реакции B). Окись углерода и часть
реакционного угля при содержании в топочных газах кислорода
сгорают до СО2; поэтому газы, выходящие из печей, содержат
много СО2 и мало СО, а газы из плохо герметизированных печей
могут совсем не содержать СО. Однако это не доказывает, что-
восстановление идет только по реакции A). То, что оно идет и no-
реакциям B) и C) и сопровождается простым выгоранием угля,,
подтверждается расходом угля, который в заводских условиях зна-
значительно превышает количество, требующееся теоретически по ре-
реакции A), т. е. 0,17 т углерода на 1 т сульфата натрия A00%).
Механизм восстановления сульфата натрия достаточно сложен.
В первой стадии восстановления Na2SO4 переходит в Na2SO3..
В дальнейшем протекает реакция диспропорционирования 18~23
O4 D).
идущая и в отсутствие восстановителей; она суммирует следующие
процессы:
Na2SO3 = Na2O + SO2 E>
2Na2SO3 + SO2 = 2Na2SO4 + '/2S2 F>
Na2O + '/2S2 = Na2S + '/2O2 G\
Na2SO3 + '/2O2 = Na2SO4 ' (8>
В присутствии же восстановителей образование серы из SO?
особенно облегчается, так как реакции SO2 + 2CO •= i/2S2 + 2СОг
и SO2 + С = 4/г52 + СО2 идут со значительной скоростью, что-
ускоряет и реакции E) и G).
Восстановление сульфата натрия начинается при температурах
ниже температуры его плавления (890°) в основном окисью угле-
углерода и .другими компонентами газовой фазы; но в производствен-
производственных условиях, при применении в Качестве восстановителя сравни-
сравнительно крупнозернистого каменного угля, интенсивный процесс вос-
восстановления твердым углеродом протекает лишь после появления.
472
Гл. XIII. Соли сульфидного ряда
жидкой фазы, смачивающей поверхность частиц угля. Наобо-
Наоборот, восстановление газами замедляется при плавлении шихты, так
как жидкая фаза затрудняет доступ газа внутрь реакционной
массы — газ может проникнуть в нее только растворяясь и мед-
медленно диффундируя. Процесс может быть разбит на три периода
(рис. 143). Период / начинается после загрузки шихты в печь. Он
характеризуется нагреванием и постепенным плавлением сульфата
натрия, сопровождающимся нарастанием скорости процесса.
Основной период // характери-
характеризуется «кипением» плава, т. е.
бурным выделением газа. Этот
то'
1
/
/
/
1
о
эо too
время.ыин
150
Рис. 143. Кривая нарастания со-
содержания Na2S в плаве.
Рис. 144. Диаграмма плавкости в си-
системе Na2SO4—Na2S.
период, когда плав остается жидким, соответствует наибольшей
скорости процесса. Период ///, наступающий к концу процесса,
характеризуется загустеванием шихты и снижением скорости нара-
нарастания количества ЫагБ в связи с уменьшением концентрации
Na2SO4 в жидкой фазе.
Если в шихте имеются примеси (сульфаты и сульфиды щелоч-
щелочных и щелочно-земельных металлов и др.), играющие роль плав-
плавней, расплавляющих часть сульфата натрия ниже 890°, то интен-
интенсивность процесса возрастает уже при низких температурах. Роль
такого плавня выполняет и ЫагБ. Вследствие появления его, хоть
и в небольшом количестве в начале процесса (а также из остаю-
остающегося на стенках печи плава после выпуска предыдущей за-
загрузки), жидкая фаза эвтектического состава (рис. 144) образуется
уже при 665 ±10°24 и в дальнейшем реакция сильно интенсифици-
интенсифицируется.
На рис. 144 изображена диаграмма плавкости в системе
Na2SO4—Na2S. Эвтектическая смесь, имеющая температуру плав-
плавления около 665°*, содержит приблизительно 57% Na2SO4 и 43%
Na2S (точка Е). Проследим ход процесса восстановления в изо-
изотермических условиях при 950°. Состав жидкой фазы будет не-
* По разным данным температура этой эвтектики находится в пределах
€65—740°.
Сульфид натрия
473
прерывно изменяться по пунктирной прямой ас до пересечения ее
с кривой плавкости BE в точке с. В этой точке начнется выделение
в твердую фазу сернистого натрия, т. е. начнется загустевание
плава. В дальнейшем состав жидкой фазы будет оставаться по-
постоянным (точка с). Количество жидкой фазы этого состава будет
непрерывно уменьшаться, а количество твердой фазы — возрас-
возрастать. При полном восстановлении исчезнет весь сульфат натрия и"
вместе с ним исчезнет вся жидкая фаза — плав затвердеет.
Плав в печи обычно несколько перегрет. Вследствие высокой
температуры топочных газов по мере выделения Na2S в твердую-
фазу температура плава не остается постоянной, а непрерывно,
повышается. Изменение температуры и состава жидкой фазы со-
совершается вдоль кривой плавкости BE. Если плав выгружается
при 1050°, то изменение температуры жидкой фазы идет вдоль
отрезка cd до точки d.
Для уменьшения вязкости плава, предотвращения его налипа-
налипания на стенки печи и для облегчения выгрузки плава из печи не-
необходимо, чтобы он обладал достаточной подвижностью. С этой
целью в печи поддерживают высокую температуру A100—1200°),
Если в конце процесса температура в печи выше температуры за-
затвердевания Na2S, то сульфат натрия может быть восстановлен:
полностью. В этом случае плав имеет достаточную текучесть для
выпуска его из печи. При более низких температурах часть Ыаг&
находится в плаве в твердом виде и для того, чтобы плав содер-
содержал достаточное количество жидкой фазы и не был слишком гу-
густым, необходимо оставлять часть сульфата невосстановленной..
Количество невосстановленного сульфата должно быть тем.
больше, чем ниже температура в печи.
Кроме сульфата и сульфида натрия в плаве содержатся сода
и метасиликат натрия (см. ниже), которые также находятся в.
жидкой фазе. Содержание сульфата в выгружаемом плаве может
быть тем меньше, чем больше в нем соды и метасиликата, которые
заменяют часть сульфата в жидкости, обеспечивающей подвиж-
подвижность плава.
Избыток угля в шихте значительно увеличивает вязкость плава
и сильно уменьшает его теплопроводность. Реакция восстановления
сульфата протекает очень быстро, в течение нескольких минут или
даже долей минуты. Общая же продолжительность процесса, из-
измеряемая часами, определяется длительностью прогрева шихты..
Чем больше угля в шихте, тем медленнее она прогревается, тем
больше продолжительность пребывания плава в печи и тем ниже
производительность печи. Уголь должен быть пористым и мало-
малозольным (содержать не более 15% золы) во избежание чрезмерного
загрязнения плава и уменьшения выхода продукта. Лучшим вос-
восстановителем является древесный уголь, обладающий большей по-
пористостью, однако в производстве пользуются более дешевым.
474
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
47»
рядовым каменным углем. Торф восстанавливает сульфат натрия
в 2 раза быстрее, чем уголь, но для этого требуется брикетировать
шихту, а расход торфа в 2 раза больше, чем угля25.
Вредными примесями в применяемом для восстановления тех-
техническом сульфате натрия являются кислотность и повышенное
содержание NaCl, а в природном — повышенная влажность, NaCl
и нерастворимые вещества. При повышенной влажности сульфата
затрудняется приготовление шихты и усиливается разрушение фу-
футеровки восстановительных печей. Поэтому сульфат, содержащий
больше 5% влаги, подвергают сушке.
При соотношении в шихте Na2SO4: С, равном 3 : 1, при 850°
ъ лабораторных условиях 95%-ный выход Na2S достигается за
2—3 мин, причем часть этого времени затрачивается на прогрев
реакционной массы до температуры реакции; при 1100° выход в
88,6% был достигнут за 40 се/с. 26~34-Излишняя продолжительность
процесса в производственных условиях, связанная с необходи-
необходимостью прогрева больших масс шихты, ухудшает результаты, так
как возникают побочные реакции, при которых расходуется уже
.образовавшийся сульфид натрия.
Продолжительность процесса восстановления в заводских усло-
условиях зависит также от интенсивности перемешивания шихты, со-
состава и температуры греющих газов и т. п. Она колеблется в пре-
пределах от 1 до 2,5 ч. При этом степень восстановления сульфата
составляет 80—85%. При крупном угле получается плав с мень-
меньшим содержанием Na2S и с большим содержанием примесей.
Помимо основных реакций, ведущих к образованию Na2S, про-
протекают побочные процессы, в результате которых в плаве обнару-
обнаруживаются некоторые количества Na2SO3, Na2CO3, Na2Si03, Na2S2O3
И др. Na2SO3 появляется в результате неполного восстановления
сульфата; ЫагСО3 — в результате взаимодействия Na2S с СОг и
водяным паром, имеющимся в топочных газах:
Na2S + СО2 + Н2О = Na2CO3 + H2S
Образование соды термодинамически возможно85 и по реак-
реакциям
Na2SO4 + СО = Na2CO3 + SO2
Na2SO4 + ЗСО = Na2CO3 + 2СО2 + VaS2
а в присутствии водорода, образующегося из влаги шихты и топ-
топлива, по реакциям:
Na2SO4 + Н2 + ЗСО = Na2CO3 + H2S + 2СО2
Na2SO4 + 2Н2 + 2СО =¦ Na2CO3 + H2S + Н2О + СО2
В результате этих реакций содержание соды в плаве посте-
постепенно возрастает, особенно при недостатке угля в шихте. Однако
к концу процесса количество ее в плаве уменьшается. Приведен-
Приведенные реакции обусловливают потерю серы в газах. В результате
взаимодействия SO2 с СО, а также с H2S образуется элементарная
сера. В газах обнаруживаются также небольшие количества COS.
Газы, интенсивно выделяющиеся из плава в процессе восста-
восстановления, изолируют его от непосредственного соприкосновения
с топочными газами. В период дозревания плава, когда интенсив-
интенсивное выделение из него газов прекращается и соприкосновение с то-
топочными газами облегчается, при наличии подсосов в печь воз-
воздуха, а также кислорода в топочном газе, может происходить
частичное выгорание серы из плава с одновременным образова-
образованием соды:
2Na2S + ЗО2 = 2Na2O + 2SO2
2Na2O + 2СО2 = 2Na2CO3
2Na2S + 3O2 + 2CO2 = 2Na2CO3 + 2SO2
Окисление сульфидной серы кислородом воздуха до SO2 идет,,
по-видимому, с образованием в качестве промежуточного продукта
моноокиси серы SO36; цепная реакция
2SO = SO2 + S; S + О2 = SO + О; ...
обусловливает образование атомарного кислорода, что способ-
способствует окислению ЫагБ.
Потеря серы в виде газообразных соединений приводит при пе-
передержке плава в печи к уменьшению содержания в нем Na2S.
Появление в газовой фазе БОг обусловливает возможность реак-
реакции:
Na2S + SO2 + H2O = Na2SO3 + H2S
Содержащийся в плаве метасиликат натрия образуется из соды
и кремнезема, находящегося в золе реакционного угля или попа-
попадающего в шихту в виде загрязняющего сырье песка, а также из,
футеровки печи:
Na2CO3 + SiO2 = Na2Si03 + CO2
Чем больше кремнезема в шихте, тем меньше выход сернистого
натрия и тем больше в плаве метасиликата натрия и содыет. Это
можно объяснить тем, что, помимо образования Na2SiO3 из соды,
кремнезем взаимодействует с первичным продуктом восстановлен
Ния — сульфитом:
Na2SO3 + SiO2 - Na2Si03 + SO2
Обнаруживаемый при анализе плава тиосульфат натрия обра-
образуется, в качестве быстро исчезающего промежуточного продукта,
По реакции
Na2SO3 + ViS2 ¦= Na2S2O8
476
Гл. XIII. Соли сульфидного ряда
за счет сульфита и серы, возникающих в процессе восстановления.
Кроме того, он, вероятно, образуется при растворении плава в
воде за счет взаимодействия с имеющейся в плаве свободной серой
<в присутствии кислорода воздуха:
2Na2S + 2S + ЗО2 - 2Na2SaO3
Помимо примесей, образовавшихся в результате побочных ре-
реакций, плав содержит остатки неизрасходованного реакционного
угля и минеральные вещества из угля (золу), а также примеси из
сульфата. Обычно в плаве содержится: 70—76% Na2S, до 2%
Na2Si03, 0,5—2,5% Na2SO3, 1—3% Na2S2O3) 5—13% Na2CO3, до
15% нерастворимых минеральных веществ и до 8% углерода (не-
(невыгоревший уголь). Сульфат натрия в плаве из механических пе-
печей обычно отсутствует или содержится в количестве, меньшем
1—1,5%, а содержание NaCl зависит от количества его в исход-
исходном сульфате. Чем меньше золы содержится в реакционном угле,
тем более высокопроцентным получается плав. Из-за примеси угля
,плав имеет черный цвет.
Помимо сульфата натрия, восстановлению с целью получения
/сернистого натрия могут подвергаться сульфит и тиосульфат на-
натрия. Заводы, производящие анилиновые красители, получают в
.^качестве отхода от производств фенола, бета-нафтола и других
;-значительные количества загрязненного сульфита натрия, содер-
содержащего до 80% Na2SO3 (в сухом веществе) и примеси Na2SO4
и Na2S2Os. Этот сульфит используют, примешивая его к сульфатно-
угольной шихте в производстве сульфида натрия. Сырьем для про-
производства сульфида натрия может служить также тиосульфат на-
натрия, являющийся побочным продуктом при очистке газов от
.сероводорода мышьяково-содовым способом (стр. 556). Восстано-
Восстановление технического тиосульфата углеродом протекает с образо-
образованием сульфида натрия и элементарной серы
Сульфид натрия
477
,или
2Na2S2O3.+ ЗС = 2Na2S + ЗСО2 + S2
2Na2S2O3 + 6С = 2Na2S + 6СО + S2
Для получения моносульфида требуется осуществлять процесс
выше 920° (рис. 142); при 700—800° получается сульфид натрия,
.содержащий некоторые количества полисульфида. Обстоятель-
Обстоятельством, затрудняющим осуществление этого процесса, является не-
.обходимость извлечения серы из газообразных продуктов реак-
реакции — образующаяся при восстановлении тиосульфата сера в при-
.сутствии угля и вносимой с тиосульфатом влаги большей частью
превращается в сероводород, частично в другие газообразные со-
соединения (COS и CS2), частично остается в элементарном виде38.
Получение сульфида натрия восстановлением сульфата натрия
углем
Если сульфид натрия получают из природного сульфата, влаж-
влажность которого иногда достигает 15—18%, то его подвергают
сушке разбавленными воздухом топочными газами во вращаю-
вращающейся барабанной сушилке до влажности меньше 5%. Затем его
измельчают в дезинтеграторе до размера зерен 4 мм и меньше и
смешивают, например, в двухвальком лопастном смесителе с из-
измельченным 39 и просеянным углем. Весовое отношение твердого
углерода к Na2SO4 в шихте составляет 0,25—0,27.
Рис. 145. Внешний вид вращающейся печи перио-
периодического действия с камерой горячего выщела-
выщелачивания.
На рис. 145 и 146 показана реакционная печь. Для загрузки
шихты и слива плава в барабане печи имеется люк, крышка кото-
которого с внутренней стороны футерована. Тепло уходящих из печи
газов, имеющих высокую температуру (выше 900°), используют для
обогрева котлов, в которых концентрируют щелок сернистого на-
тр'ия, полученный после выщелачивания плава. Барабаны печей
изготовляют из 10—14-миллиметровой стали, они имеют диаметр
2—3 м, длину 5—8 м. Например, печь с диаметром барабана 3,1 м
и длиной 5,5 м имеет суточную производительность ~ 17 т плава,
содержащего 70—79% Na2S, при расходе газа на отопление до
2000 м3/ч (при нормальных условиях); барабан печи вращается со
скоростью 2,6 об/мин электромотором мощностью 25 кет.
Для футеровки печей сернистого натрия следует применять као-
линитовый или очень плотный высокоглиноземистый шамотный
кирпич горячего прессования, содержащий больше 50% глинозема,
478
Гл. XIII. Соли сульфидного ряда
имеющий пористость 14—17%, с временным сопротивлением
сжатию не ниже 250 кгс/см2 и дополнительной усадкой при 1400°
не больше 0,5% 4а3. Используют также форстеритовый кирпич,
основным веществом которого является Mg2Si04 и хромомагнези-
товый кирпич. Предложены и другие материалы, показавшие до-
достаточную устойчивость к расплавленному сернистому натрию при
лабораторных испытаниях44 — хромитовый огнеупор на глинистой.
300
Сульфид натрия
479
-2000—Л
Рис 146. Продольный разрез вращающейся печи периодического действия
с камерой горячего выщелачивания плава:
1 — барабан печи (диаметр 2500 мм); 2 - опорный ролик; 3 - привод печи; 4 - камера для гаше-
гашения н выщелачивания плава; 5-газовая горелка; 6 — пыльная камера; 7 —газовая горелка
(подтопок).
связке, высокоглиноземистые огнеупоры, изготовленные из ко-
корунда, двуокиси титана и глины. В строго восстановительной ат-
атмосфере возможно применение углеродистой (графитовой) футе-
футеровки с гарниссажем на ее внутренней поверхности45.
При повернутом кверху люком барабане печи производят за-
загрузку печи шихтой через люк в течение 5—7 мин. Затем люк за-
закрывают, приводят печь во вращение и в течение 30—40 мин на-
нагревают реакционную массу до 850—900°. При этом происходит
частичное выгорание угля и полное расплавление сульфата. При
дальнейшем повышении температуры до 1000—1200° реакционная
масса вспенивается (вследствие выделения газов) и увеличивается
в объеме. Во избежание перелива массы через горловины в топку
и в пыльную камеру, в печь забрасывают через люк пыльной ка-
камеры 2—3 лопаты мелкого угля или прекращают на некоторое
время вращение печи. По прекращении вспенивания наступает пе-
период интенсивного «кипения» массы, продолжающийся 10—15 мин.
При этом масса быстро уменьшается в объеме, загустевает и при-
прилипает к внутренней поверхности печи. Дальнейшим нагреванием
массу доводят до жидкокашеобразной консистенции, после чего
плав готовят к сливу. Слив реакционной массы производят в те-
течение 15—30 мин через надеваемый на люк ограничитель — диа-
диафрагму диаметром 100—130 мм. При жидком плаве пользуются
диафрагмой 60—70 мм. Плав спускают небольшими порциями в
камеру гашения, куда предварительно заливают нагретый до ки-
кипения слабый щелок.
В качестве реакционного угля для восстановления сульфата
натрия чаще применяют жирный уголь с зольностью не больше
12% и содержанием летучих 30—33%. Активность этого угля
выше, чем тощих углей, а летучие, выделяясь, способствуют пере-
перемешиванию реакционной массы и, сгорая, ускоряют ее прогрев;
в результате производительность печей, оборудованных угольными
топками (не дающими очень высокой температуры), при работе на
жирном реакционном угле оказывается больше. Но при обогреве
вращающихся печей газовым топливом или мазутом легко дости-
достигается высокая температура входящих в печь газов A250—1270°)
и поэтому в качестве реакционного угля можно применять более
дешевые виды топлива, например антрацитовую мелочь. При этом
реакция протекает без существенного вспенивания массы и оказы-
оказывается возможным увеличить количество единовременно загру-
загружаемой в печь шихты. Плав получается однородным (жидким), не
налипающим на стенки. Расход антрацитового угля на 15—20%
меньше, чем жирного угля м. Недостатком антрацитового угля яв-
является высокая абразивность его золы.
При отоплении механических печей мазутом форсунки лучше
располагать в специальных камерах сжигания мазута, так как
температура факела слишком высока (больше 1500°), и это приво-
приводит к быстрому разрушению футеровки перегретым плавом серни-
сернистого натрия. Лучше, чтобы температура поступающего в печь го-
горячего газа была не больше 1300°.
В коротких печах (~5л) съем плава с 1 м3 полезного объема
достигает 0,8—0,9 т в сутки, в то время как в длинных печах
G—8 м) — немногим более 0,5 т. Это объясняется большей про-
продолжительностью плавки в длинных печах, необходимой для за-
завершения реакции в конце печи, противоположном вводу горячих
газов. В коротких печах реакционная масса прогревается быстрее
и равномерней, и продолжительность одной операции плавки
(включая время загрузки шихты и выгрузки плава) составляет
~2,5 ч, вместо 4 ч в печах с длиной барабана 7—8 м.
Механические вращающиеся печи периодического действия зна-
значительно совершеннее, чем применявшиеся ранее для восстановле-
480
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
481
ния сульфата натрия ручные пламенные подовые печи47-54, однако
и они имеют существенные недостатки. Основные из них — невоз-
невозможность выпускать и выщелачивать плав непрерывным способом
и низкий тепловой коэффициент полезного действия вследствие
того, что барабан печи короткий и газы уходят из него с очень
высокой температурой (900—950°). Эти недостатки могли бы быть
устранены применением вращающихся печей непрерывного дей-
действия, что, однако, связано с рядом пока еще непреодоленных за-
затруднений.
Во вращающихся печах непрерывного действия тепло могло бы
быть использовано лучше, так как шихта и греющие газы дви-
движутся противотоком и уходя-
уходящие газы имеют температуру
600—750°. Во избежание коль-
цеобразования шихту требует-
требуется составлять на неспекаю-
щемся угле. При этом сокра-
сокращается и продолжительность
восстановления. Чем больше
угля в шихте, тем больше вяз-
ч
?
>
«о
п
/
7 16 19 20 21 22 23 24 25 2S 3
Не тб.уелерода на /00нгШ2ЗОь
Рис 147. Зависимость необходимого
времени пребывания в печи от содер-
содержания твердого углерода в шихте
(уголь ПЖ).
кость плава и необходимое для
восстановления время пребы-
пребывания его в печи (рис. 147).
Съем плава с 1 м3 внутрен-
внутреннего объема печи составляет
1,2—1,3 т в сутки31. Выпуск
плава из печи производится в
зоне с наиболее высокой температурой, на небольшом расстоянии
от нижнего конца печи, где имеется специальное звено с несколь-
несколькими отверстиями, через которые при вращении печи плав выте-
вытекает в приемник или в камеру выщелачивания. Как показал
опыт, несмотря на высокую температуру в реакционной зоне
печи непрерывного действия (до 1250°), оказывается трудным
избежать кольцеобразования, т. е. налипания шихты на футеровку
в средней зоне печи. Это нарушает устойчивость ее работы. По-
Поэтому, а также из-за быстрого разрушения футеровки в наиболее
горячей зоне восстановление сульфата натрия в таких печах не
получило распространения. Снижения температуры восстановления
с сохранением при этом подвижности реакционной массы во вра-
вращающейся печи непрерывного действия можно было бы достичь
добавкой в тонкодисперсную шихту порошкообразных инертных
наполнителей, в присутствии которых жидкая фаза распределяется
в большом количестве твердого материала и реакционная масса
остается сыпучей. Наполнителем может служить, в частности, и
значительный избыток реакционного у1ля55-57. Добавка в шихту
извести резко повышает скорость реакции восстановления суль-
фата углем при низких температурах (ниже 750°) вследствие уда-
удаления СО2 из сферы реакции в виде твердого углекислого каль-
кальция
58
Для получения плава сернистого натрия используют также не-
непрерывно действующие шахтные печи59>"°. Восстановлению в них
подвергают природный кусковой сульфат натрия. Шихта из кокса
и сульфата натрия, содержащего
2,5—25% Н2О, поступает в печь че-
через бункер и питатель (рис. 148).
Количество кокса в шихте — 50—
55% от веса натурального сульфата.
. Можно восстанавливать также бри-
брикеты, изготовленные из увлажнен-
увлажненного сульфата или из его смеси с
реакционным каменным углем. Печь
представляет собой цилиндрическую
шахту высотой 6 м с внутренним
диаметром 1,2 м. Нижняя часть пе-
печи с подом является горном; он опи-
опирается на колеса и при ремонте пе-
печи откатывается в сторону. В горне
имеются три летки для выпуска пла-
плава в стальные тигли, вмещающие
около 80 кг плава. Летки и нижняя
часть шахты печи — кессон, т. е.
зоны наиболее высоких температур,
имеют снаружи водяные рубашки
для отвода тепла. В зоне наиболее
высоких температур (горн и кессон)
печь футерована хромомагнезито-
вым кирпичом, в остальной части —
шамотным кирпичом. Непосредст-
Непосредственно над горном расположен фур-
фурменный пояс с отверстиями, через
которые в печь засасывается воздух, необходимый для горения кок-
кокса; количество воздуха B000—2800 м3/ч) регулируют таким обра-
образом, чтобы в печном газе было не больше 1,5% кислорода. Печной
газ отсасывается из печи в стальную башню, где он очищается от
пыли и охлаждается водой, причем из него конденсируется водяной
пар. Охлажденный газ отсасывают из башни эжектором (гидро-
(гидроавтоматом), создающим разрежение в колошниковой части печи
400—800 мм вод. ст.61 Рабочей жидкостью в эжекторе является
вода с примесью известкового молока для нейтрализации компо-
компонентов газа, содержащих серу. Вода, циркулирующая с помощью
центробежного насоса, перед возвратом в эжектор частично обно-
обновляется с выводом части загрязнившейся жидкости.
16 М. Е. Позин
Рнс. 148. Схема установки шахт-
шахтной печи для восстановления суль-
сульфата натрия:
1 — бункер для шихты; 2—питатель;
в —шахта печи; 4 — горн; 5 —тигель;
в—башня для охлаждения печного
газа; 7 —гидравлический затвор под
башней; 8 — гидроавтомат; 9 — газоводо-
газоводоотделитель; 10 — брызгоуловитель;
// — насос.
482
Гл. XIII. Соли сульфидного ряда
Описанная шахтная печь может выпускать в сутки больше
25 т плава, содержащего 67% Na2S. Однако практически, вслед-
вследствие больших подсосов воздуха (~50%) и недостаточности дутья,
такие печи дают 20—22 т плава в сутки с содержанием всего 60%
Na2S. Количество же примесей, образующихся в результате окис-
окисления Na2S кислородом, проникающим в печь с подсасываемым
. воздухом, в особенности сульфата натрия, значительно выше (на
5—6% и больше), чем в плаве из механических печей. При этом
расходные коэффициенты в пересчете на 1 т 63%-ного плава со-
составляют: 2 т натурального сульфата натрия, 0,7 т кокса, 0,015 т
извести, 300 кет • ч электроэнергии.
Для приготовления брикетов сульфат натрия увлажняют до со-
содержания 10—-15% Н2О. Увлажнение порошкообразного сульфата
сопровождается выделением тепла гидратации. При увлажнении
всего лишь с 3 до 10% этого тепла достаточно чтобы в адиабати-
адиабатических условиях температура сульфата повысилась на 31,1°. Это
вызывает необходимость отводить тепло в процессе увлажне-
увлажнения, не допуская перегревания продукта выше точки превращения
C2,4°), а практически, поддерживая температуру при гидратации
на уровне 23—28°. При хранении гидратированного сульфата или
брикетов летом их также следует предохранять от нагревания окру-
окружающим теплым воздухом. Охлаждение в процессе гидратации
можно осуществлять в аппарате с псевдоожиженным слоем в по-
потоке воздуха (если требуется — предварительно охлажденногоN2.
Брикетирование осуществляют на непрерывно действующем валь-
вальцевом прессе. По выходе из пресса брикеты для созревания (упро-
(упрочнения) вылеживаются 12—18 ч в системе небольших подвижных
бункеров, объединенных в рампы, оборудованные транспортными
механизмами.
Разработан способ низкотемпературного восстановления суль-
сульфата натрия в виде брикетов из шихты, содержащей Na2SO4 и спе-
спекающийся каменный уголь (Na2SO4 : С не больше 2) или торф (при
отношении торфа к сульфату больше 1,6). Наибольшую прочность
имеют сульфатно-угольные брикеты с примесью 10% торфа в ка-
качестве связующей добавки. Восстановление при 750—800° идет без
видимого плавления брикетов. Поэтому оно может быть осуществ-
осуществлено в непрерывно действующей вращающейся печи, а также в
шахтной печи. В опытах на модельной вращающейся печи, при
продолжительности процесса 40—50 мин был получен полупродукт,
содержавший до 75% Na2S21-63.
При наличии дешевой электроэнергии восстановление сульфата
натрия углем можно осуществлять в электрических печах64-65.
В последнее время в промышленность внедряются высокоэф-
высокоэффективные печи циклонного типа. В них восстановление порошко-
порошкообразного сульфата натрия, смешанного с тонкоизмельченным ре-
реакционным углем, происходит при 1400° за короткое время в
Сульфид натрия
483
пленке расплавленной шихты, омываемой быстро движущимся га-
газом. Шихта из бункеров вводится струей сжатого воздуха в цик-
циклонную камеру тангенциально со скоростью 20—25 м/сек. Туда же
подается жидкое или газообразное топливо. Воздух для сжигания
топлива и поддержания интенсивного вращательного движения
вводится тангенциально со скоростью 100—120 м/сек. За 10 мин
степень восстановления сульфата достигает 95—97%- Съем плава
с 1 м3 объема циклонной печи значительно выше, чем в механиче-
механической и шахтной печах, а циклонный эффект в сочетании с жидкой
пленкой расплава дает высвкую степень улавливания пыли в са-
самой печи. Испытание циклона с водяным охлаждением стенок по-
показало, что стойкость гарниссажа (слоя плава, застывшего на
стенках) настолько велика, что, вероятно, возможно будет обой-
обойтись без специальной футеровки печи. Так как циклонная печь
работает непрерывно, то облегчается проведение в непрерывно дей-
действующей аппаратуре последующих операций переработки плава.
Плав стекает по стенке камеры через диафрагму в копильник, где
завершается восстановление («дозревание») и откуда он непре-
непрерывно удаляется через леткувв>67.
Переработка плава в сульфид натрия
Выщелачивание сульфида натрия из плава холодным спосо-
способом, т. е. после его охлаждения и затвердевания теперь почти везде
прекращено и заменено более рациональным горячим выщелачи-
выщелачиванием.
При холодном выщелачивании плав из печей выливается (из
ручных печей — выгружается) в котелки (тигли, бадейки и т. п.),
закрываемые затем крышками, застывает в них и охлаждается
10—12 ч. Высказывалось мнение80' 68>69, что в процессе охлаждения
плава содержание в нем NagS уменьшается вследствие протекания
побочных реакций. В действительности это уменьшение происходит
лишь в поверхностном слое горячего плава, соприкасающемся с
воздухом — Na2S окисляется в Na2SO,t. Поэтому выгрузка из ко-
котелков еще не остывшего плава приводит к потерям Na2S.
Слитки застывшего плава выгружают из котелков опрокидыва-
опрокидыванием и разбивают ручным способом. Затем плав дробят вначале
на щековых, затем на валковых дробилках и направляют на выще-
выщелачивание. Для выщелачивания твердого плава применяй^ резер-
резервуары с мешалками, дырчатые вертикальные барабаны, вращаю-
вращающиеся на вертикальном валу в неподвижных цилиндрических ре-
резервуарах 47, неподвижные и вращающиеся автоклавы, в которых
процесс осуществляется под давлением пара 5—6 ат и др. Выще-
Выщелачивание производят горячими щелоками сначала более, затем
менее концентрированными, постепенно укрепляя их, и горячей во-
водой, которой окончательно промывают шлам перед его удалением
16*
484
Гл. XIII. Соли сульфидного ряда
из выщелачивателей. При методическом (последовательном) вы-
выщелачивании получают щелоки четырех-пяти концентраций:
Первый щелок (крепкий) — плотность при 50—80° 1,26—
Второй » (средний)— » » » 1,22 —
Третий » (слабый) — » » » 1,14—
Четвертый » » » » 1,08—
Пятый » » » » 1,02-
,32 г/см3
,24 »
,16 »
,10 »
,04 »
Четвертый и пятый щелоки получают при промывке шлама во-
водой.
В системе Na2S—Na2SO4—H2O 7°-72 при 30—90° с увеличением
концентрации Na2S и с ростом температуры растворимость Na2SC>4
уменьшается (табл. 37). При температуре не ниже 80—90° воз-
возможно получить щелок, содержащий 32,5—35,5% Na2S и меньше
0,5—0,8% Na2SO470. В интервале температур 90—140° содержание
Na?S в насыщенном растворе растет от 35,8 до 47,2%, а концентра-
концентрация Na2SO4 уменьшается с 0,81 до 0,20% 71- Это указывает на целе-
целесообразность осуществления выщелачивания плава при темпера-
температуре выше 100°.
таблица з
_ Растворимость Na2SO4 в водных растворах Na2S (вес. %O0
(Жирным шрифтом обозначены составы эвтонических растворов)
Сульфид натрия
485
30°
Na2S
6,00
10,31
12,24
13.15
15,10
17,31
20,00
Na2SO4
28,80
21,00
!3,60
10,76
7,05
6,40
5,85
40°
Na2S
,60
6,43
10.01
12,82
16,00
17,62
21,02
Na2SO4
32,50
21,22
18,40
13,02
8,20
5,61
4,50
60»
Na2S
5,60
9,00
12,41
18,01
23,71
28,69
29,92
Ma2SO4
31,01
18,02
14,42
8,91
6,30
4,39
2,00
80°
Na2S
6,11
13,62
20,71
27,22
30,61
32,40
33,50
Na2s04
?0,19
14,03
7,05
3,59
2,32
0,92
0,80
90°
Na2S
9,91
13,62
20,39
28,19
32,21
35,51
36,42
Na2SO4
30,00
12,02
6,29
2,59
0,92
0,72
0,50
Горячее выщелачивание жидкого плава осуществляют сразу
после выпуска его из печи без промежуточного охлаждения и за-
затвердевания. Это дает экономию в расходе топлива, электроэнер-
электроэнергии и значительно повышает производительность труда, так как
устраняет громоздкие операции розлива, охлаждения, дробления
и перегрузки плава, создающие к тому же тяжелые условия труда
вследствие обильного тепло- и газовыделения. При горячем выще-
выщелачивании плава используется его физическое тепло, вследствие
чего почти в 2 раза сокращается расход пара на подогрев щелоков
и уменьшаются потери МагБ, вызываемые окислением кислородом
и карбонизацией СО2 воздуха. Сокращаются производственные
площади, особенно за счет того, что отпадает необходимость иметь
большие площадки для охлаждения слитков плава. Сокращаются
количество оборотных щелоков и емкости для их хранения.
Горячее выщелачивание плава позволяет получить щелок высо-
высокой концентрации, не требующий выпаривания. После фильтрации
такого щелока при 190—200° через металлическую сетку для отде-
отделения от непрореагировавшего угля и- других твердых примесей
щелок мог бы быть разлит по барабанам, где он застынет в гото-
готовый продукт. Этот прием, однако, не получил промышленного рас-
распространения из-за большой вязкости щелока и возможности кри-
кристаллизации его при охлаждении в процессе фильтрации, что за-
затрудняет отделение от него нерастворимого шлама. По этим же
причинам не получило осуществления и предложение предвари-
предварительно (до выщелачивания) фильтровать через металлическую
сетку под вакуумом горячий жидкий плав69. Вследствие этого по-
получают щелок, содержащий в 1 л 300—320 г Na2S. После отделе-
отделения от него шлама отстаиванием и с помощью фильтров или цент-
центрифуг щелок выпаривают и разливают в тару (барабаны), где он
и застывает.
В цехах, оборудованных периодически действующими вращаю-
вращающимися печами, горячее выщелачивание плава осуществляют по-
после окончания плавки — печь останавливают и выпускают через
разгрузочное отверстие жидкий плав в камеру гашения или горя-
горячего выщелачивания46-50. Камера представляет собой стальную
сварную коробку емкостью около 10 ж3, снабженную паровыми
барботерами и вытяжными трубами для отвода паров воды. Пере-
Перемешивание пульпы производится мешалкой или циркуляционным
¦насосом. Камера смонтирована в одном блоке с вращающейся
печью и обхватывает печь поясом (рис. 145 и 146); она устанавли-
устанавливается под печью так, что плав не может быть слит за* пределы
камеры. Перед выпуском плава из печи в камеру до половины ее
объема D—5 м3) заливают средний щелок A5—20% Na2S) и на-
нагревают его острым паром до 100—105° (лучше почти до кипения).
Затем постепенно сливают плав из печи — происходит гашение и
выщелачивание щелоком плава, имеющего температуру около
1000°, сопровождающееся интенсивным парообразованием, так что
по окончании операции объем полученной пульпы на 15—20%
меньше объема исходного щелока (при гашении 1 т плава из ще-
щелока испаряется 650—700 кг воды). После окончания слива содер-
содержимое камеры перемешивают 15—20 мин при одновременном
подогреве для завершения выщелачивания Na2S. Завершение выще-
выщелачивания можно производить и в специальном агитаторе. Затем
полученный щелок, содержащий 28—32% Ma2S, отделяют от шлама
на центрифуге или нутч-фильтре или отстаиванием. Соотношение
между количествами среднего щелока и плава регулируют с уче-
учетом их состава так, чтобы крепкий щелок не был слишком кон-
концентрированным. Оптимальной концентрацией крепкрго щелока -
486
Гл. XIII. Соли сульфидного ряда
при современном аппаратурном оформлении процесса, по-види-
по-видимому, следует считать ~30% Na2S; плотность такого щелока
~ 1,35 г/см3, температура кипения ~ 125°, а температура кристал-
кристаллизации выше 70° G0° —для чистого 30% раствора Na2S). При
более высокой концентрации затрудняется дальнейшая перера-
переработка щелока, так как вследствие значительной его вязкости за-
замедляется отстаивание шлама и создается опасность выпадения
кристаллического сернистого натрия в процессе отделения шлама
вследствие некоторого охлаждения щелока. Если, однако, обеспе-
обеспечить сохранение высокой температуры крепкого щелока в процессе
отделения шлама, которое производить не отстаиванием, а прину-
принудительным способом, например центрифугированием, то оптималь-
оптимальная концентрация крепкого щелока окажется значительно выше.
Следует, однако, учитывать, что в суспензии из выщелачивателя
отношение Ж: Т колеблется от 20 : 1 до 45 : 1. Для разделения этой
суспензии применение нутч-фильтров и вакуум-фильтров затруд-
затруднено вследствие того, что шлам, получаемый после горячего выще-
выщелачивания плава, имеет очень тонкую структуру и забивает поры
фильтрующих тканей. Использование центрифуг для таких разбав-
разбавленных суспензий также нецелесообразно. Поэтому лучше освет-
осветлять крепкий щелок отстаиванием, например, в сгустителях типа
Дорра, а сгущенный шлам окончательно отжимать и промывать на
центрифуге, куда он может быть подан диафрагмовым насосом.
При промывке шлама на центрифуге или путем репульпации
слабым щелоком F0—80 г/л Na2S) получают средний щелок, ко-
который используют вновь для горячего выщелачивания плава. Вто-
Вторую и третью промывки шлама производят горячей водой, получая
растворы, содержащие соответственно 60—80 и 25—40 г/л Na2S.
Промывные воды смешивают с частью крепкого щелока, получая
средний щелок для выщелачивания плава. Удаляемый в отвал
промытый шлам представляет собой илоподобную массу, содержа-
содержащую непрореагировавший уголь, золу, сульфиды тяжелых метал-
металлов (главным образом FeS) и влагу, в которой растворены соли.
Его состав в пересчете на сухое вещество: 10—30% угля, 0,3—0,8%
Na2S, 0,6—1,5% Na2S2O3, 0,7— 1 % Na2SO3, 0,2—0,8% Na2SO4, 0,4—
0,6% Na2COs; остальное — нерастворимые минеральные соедине-
соединения. Влажность шлама, отделенного отстаиванием ~50%, а цент-
рифугирован-ием — 30—35 %.
Применение метода горячего выщелачивания плава в комби-
комбинации с периодически действующими печами связано с некоторыми
затруднениями. Большие порции жидкого плава нельзя сразу на-
направлять в воду (в щелок), так как это вызывает мгновенное паро-
парообразование и разбрызгивание, сопровождающиеся хлопками и
ударами, подобными взрывам. Поэтому выпуск плава производят
медленно, через ограничительные шайбы — это неизбежно приво-
приводит к уменьшению производительности печи. Для более спокойного
Сульфид натрия
487
гашения плава концентрация щелока, поступающего в гаеитель,
должна быть не ниже 20% Na2S. Заливать в камеру перед выпу-
выпуском в нее плава лучше только небольшую часть щелока, а основ-
основное его количество подавать одновременно с плавом через систему
трубок, создающих в камере жидкостную завесу (сетку); проходя
через нее, плав гасится и гранулируется, а затем уже выщелачи-
выщелачивается. При горячем выщелачивании плава из печи непрерывного
действия легче организовать процесс так, чтобы плав, выпускаемый
из печи тонкой струей, сначала гасился малым количеством ще-
щелока и лишь затем смешивался с основной его массой. Можно так-
также выливать плав на вращающийся стальной барабан, частично
погруженный нижней своей частью в pe-зервуар со щелоком. Струя
плава растекается по верхней части барабана и застывает на нем
в виде тонкой пленки, легко растворяющейся при вращении бара-
барабана. При этом тепло плава используется так же, как и в камере
гашения.
Упомянутые выше затруднения при горячем выщелачивании
плава из печей периодического действия послужили основанием
для предложений73-74 производить выщелачивание плава, частично
охлажденного в изложницах. По этому способу изложницы, имею-
имеющие отверстия в боковых стенках, с несколько охлажденным пла-
плавом, застывшим с поверхности и у стенок, с помощью мостового
крана переносятся от печи и устанавливаются на поддон, распо-
расположенный в верхней части выщелачивателя — резервуара с мешал-
мешалкой. Плав орошается слабым щелоком, а образующийся крепкий
щелок стекает с поддона в расположенный рядом с выщелачива-
телем отстойник. В процессе выщелачивания щелок, соприкасаю-
соприкасающийся с плавом, кипит, но он отделен от раскаленного (жидкого)
плава коркой застывшего плава, поэтому выщелачивание происхо-
происходит спокойно, без хлопков. Скорость выщелачивания плава умень-
уменьшается при увеличении содержания в нем (или в исходном суль-
сульфате натрия) хлористого натрия. Слиток плава весом 170—450 кг,
полученный из сульфата, содержащего меньше 3% NaCl, выщела-
выщелачивается полностью за 1—1,5 ч1ь.
Состав получаемого крепкого щелока зависит от качества
сырья и условий восстановления и выщелачивания. Обычно он со-
содержит 28—30% Na2S, 1—6% Na2CO3, 0,5—1% Na2SO4, 0,2—1%
Na2SO3, 0,5—1,5% Na2S2O3 и имеет плотность 1,26—1,32 г/см3 при
70°. Если щелок не подвергался фильтрации, то его осветляют от
взвешенных твердых частиц отстаиванием в стальных цилиндриче-
цилиндрических резервуарах большой емкости с коническим днищем. Отстой-
Отстойники покрыты снаружи тепловой изоляцией и снабжены паровыми ¦
змеевиками для периодического подогрева, чтобы температура ще-
щелока не снижалась ниже 70° во избежание кристаллизации серни-
сернистого натрия. Профильтрованный или отстоявшийся щелок на-
направляют на выпарку.
488
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
489
При выпаривании чистого раствора Na2S под атмосферным да-
давлением температура его кипения повышается до 186,5 . При этой
температуре появляются кристаллы, после чего она понижается
до 184,5° и в процессе дальнейшей кристаллизации остается неиз-|
менной7Б.
Выпарку технического крепкого щелока производят в откры-1
тых чугунных котлах, обогреваемых газами, отходящими из вос-
восстановительных печей. Часто выпаривание ведут в две стадии, в|
последовательно работающих котлах. Предварительное выпарива-
выпаривание производится до ~40% Na2S, когда температура кипения до-
достигает 135°. По мере испарения воды котлы доливают щелоком.
При окончательном выпаривании температура щелока повышается'
до 175—185°, что соответствует концентрации 63—65% Na2S. Более
рациональной является выпарная установка из 4 котлов, через ко-
которые щелок проходит непрерывным потоком и упаренный сли-
сливается в сборник готового щелока, откуда разливается в тару.
Размеры котлов разные, например диаметр 2 м, высота 2,5 м.
Наиболее стойким материалом для выпарной аппаратуры, осо-
особенно при высоких концентрациях Na2S, являются высокохроми-
высокохромистые и хромоникелевые стали76, например, следующие (по сни-
снижающейся стойкости): Х28, Х28НА, Х25Т, Х25Н4Т, 0Х21Н5Т,
1Х21Н5Т, 0Х21Н6М2Т, 1Х18Н10Т, Х17Н13М2Т, Х17Г9АН,
Х14П4НЗТ, XI7, Х1377. Предложено также применять котлы из
стали (Ст. 3), выложенные цинком78. На современных установках
удаляющийся из котлов в процессе выпарки водяной пар отводят
в водяной конденсатор; получаемая здесь теплая вода исполь-
используется в производстве (например, для промывки шлама).
В процессе выпаривания из щелока выделяются осадки солей,
Содержащие главным образом Na2CO3, а также Na2S2O3, Na2SO3,
Na2SC>4. Эти соли кристаллизуются в результате сильного уменьше-
уменьшения их растворимости в концентрированных растворах Na2S. Осад-
"ки раньше вычерпывали дырчатыми черпаками, и так как они содер-
содержали значительные количества (до 65%) Na2S — их направляли
в выщелачиватели. Осадки, вычерпывание которых является тяже-
тяжелой и трудоемкой операцией, образуют на стенках котлов корку,
ухудшающую теплопередачу. Помимо ухудшения использования
тепла, это ведет к тому, что стальные котлы прогорают, а чугун-
чугунные лопаются. Во избежание этого котлы снабжают мешал-
мешалками — выделяющиеся осадки остаются взмученными в щелоке,
но продукт содержит больше примесей79. Периодически корку,
образовавшуюся на стенке котла, растворяют развариванием ее
в воде.
Продолжительность выпарки щелока на разных заводах раз-
различна и зависит от концентрации поступающего на выпарку рас-
раствора, размеров котла, температуры греющих газов и т. д.
При наличии специальной топки, при емкости котла 7—8 т готового
продукта и при работе в две стадии с доливкой продукцион-
продукционного котла продолжительность одного цикла выпарки равна
35—40 ч.
На новых предприятиях предусмотрена выпарка крепкого ще-
щелока до 50% Na2S в вакуум-выпарных аппаратах с выносной
греющей камерой и принудительной циркуляцией (с последующей
выпаркой до 65% в уварочных котлах). Наибольшее значение ко-
коэффициента теплопередачи в вакуум-аппаратах /(=1000—
1100 ккал/ (м2 ¦ ч • град) достигается при следующих условиях: ва-
вакуум в сепараторе г = 550—600 мм рт. ст., температура греющего
пара tn = 127—128°, температура кипения щелока tK = 114—122°.
Если аппарат выполнен из углеродистой стали, то для замедления
коррозии лучше вести выпарку до 55% Na2S (при tn = 138—143°,
г = 500—550 мм рт. ст. и tK= 123—130°, К = 800—900 ккал/(м2-чХ
Хград). Опытами на модели вакуум-выпарного аппарата показана
возможность одностадийной упарки щелока от 30 до 65—72%
Na2S, что позволит отказаться от выпарных котлов; в качестве оп-
оптимальных рекомендуются: /к = 164—167° (пар с избыточным да-
давлением 6—6,5 ат), г = 500—550 мм рт. ст., tK = 145—152°, при этом
/(=800—1000 ккал/(м2 • ч • град). Существенной инкрустации грею-
греющей поверхности не наблюдалось.80
Щелок, упаренный до 63—65% Na2S, разливают в барабаны из
кровельного железа, в которых он застывает (закристаллизовы-
вается) при естественном охлаждении в течение 24 ч в сплошную
монолитную массу, после чего крышки барабанов запаиваются.
Емкость барабана 160—170 кг продукта. Разливка щелока в бара-
барабаны производится через спускной штуцер по короткому сталь-
стальному желобу или с помощью вертикального центробежного насоса,
смонтированного на одной оси с электромотором. Такой насос с
помощью тали опускают сверху в котел, погружают в жидкость и
выкачивают ее в разливной желоб.
Продукт — плавленый сернистый натрий — обычно содержит
63—68% Na2S, 3—5% Na2CO3, 0,5—1% Na2SO4, 0,5—1,5%
Na2S2O3+Na2SO3, 0,2—3% NaCl, 0,1—0,2% А120з+Ре20з, 0,3—
0,4%) SiO2, 0,5—1,5% не растворимого в воде остатка (в том числе
до 1% углерода) и 20—30% Н2О. В отдельных случаях содержа-
содержание соды в продукте может достигать 10%). Находящаяся в про-
продукте вода связана" с Ma2S в виде кристаллогидрата Na2S-2H2O,
содержание которого достигает 90—95%.
Для получения более чистого сернистого натрия крепкий щелок
может быть подвергнут перед выпаркой обработке твердым пла-
плавом (или раствором) сернистого бария. При этом щелок не только
очищается от примесей карбоната, сульфата, сульфита и тиосуль-
тиосульфата, образующих с BaS не растворимые в воде соли бария, но в
нем значительно повышается концентрация Na2S (за счет обра-
образующегося из BaS), что приводит к уменьшению количества
490
Гл. XIII. Соли сульфидного ряда
выпариваемой воды и соответствующему уменьшению расхода топ-
топлива на выпарку.
Выход сернистого натрия по отношению к израсходованному
сульфату в цехах, оборудованных механическими печами периоди-
периодического действия с горячим выщелачиванием плава, составляет
75—80% от теоретического. Потери распределяются приблизитель-
приблизительно так: при шихтовке ~0,5%, от уноса пыли из печи ~ 1%, от по-
побочных реакций при восстановлении 7—10%, при выщелачивании
и хранении щелоков 6—8%, со шламом из выщелачивателей за
счет недостаточной отмывки шлама 0,2—0,3%, с осадками солей
при выпарке 1—2%, при разливе готового продукта в барабаны и
другие механические потери 1,5—3%. При этом расходные коэффи-
коэффициенты на 1 т стандартного продукта составляют: 1,35—1,5 т суль-
сульфата натрия A00% Na2SO4), 0,5—0,6 г реакционного угля
G000 ккал), 0,3—0,4 т топлива G000 ккал), 0,4—0,6 т пара, 3—7 ж3
воды, 100—200 кет -ч электроэнергии.
Затраты на сырье составляют ~50% себестоимости сернистого
натрия, в то время как энергетические затраты—15%, зарплата
с начислениями—13%, амортизация — 2% и цеховые расходы —
20%- Поэтому дальнейшее снижение себестоимости должно идти
главным образом по пути сокращения расхода сырья (уменьшение
потерь) и затрат на его перевозку81. Осуществление непрерывного
воссстановления сульфата натрия в сочетании с горячим выщела-
выщелачиванием плава и возвратом избыточного угля в производственный
процесс может привести к дальнейшему снижению расходных ко-
коэффициентов по сульфату и по реакционному углю45.
Технический сернистый натрий можно выпускать не только в
виде сплавленной монолитной массы, но и в виде мелких чешуек,
получаемых при срезании с наружной поверхности вращающегося
горизонтального барабана застывшего на ней слоя расплавленного
сернистого натрия. Ванна с расплавленным продуктом, в которую
опущена нижняя часть барабана, должна иметь температуру ПО—
120°. Температура охлаждающей воды, подаваемой внутрь бара-
барабана, 39—40°. Потери сернистого натрия в этом процессе незначи-
незначительны ( — 0,1—0,2%).
Гранулированный сернистый натрий можно получить охлажде-
охлаждением на бесконечной металлической ленте расплавленного техни-
технического продукта, подаваемого на ленту в виде капель. Для уско-
ускорения затвердевания капель и сокращения длины ленты между
местами загрузки и выгрузки ленту можно обдувать воздухом.
В этом процессе потери сернистого натрия несколько больше, чем
при получении чешуйчатого продукта82.
При охлаждении крепкого щелока, содержащего 28—30% Na2S,
получаемого после выщелачивания плава, из него выделяются кри-
кристаллы Na2S • 9Н2О, слегка окрашенные в желтый цвет примесью
сернистого железа. Прибавление к щелоку небольшого количества
Сульфид натрия
491
цианистого натрия дает возможность получить прозрачные бес-
бесцветные кристаллы83. Те же результаты достигаются добавкой
цинкового купороса A0 г на \ л щелока) —сернистое железо бы-
быстро осаждается вместе с сернистым цинком84. Кристаллический
сернистый натрий содержит в 2 раза меньше Na2S C1—32%), чем
плавленый, но он менее гигроскопичен.
Путем плавления кристаллов Na2S'9Н2О при температуре выше
60° и выдерживания расплава при 5—55° можно получить кри-
кристаллы Na2S • 6Н2О. Обезвоживание кристаллического сернистого
натрия (Na2S«9H2O) при 150° под вакуумом позволяет подучить
продукт, содержащий 95—98% Na2S.
Безводный сернистый натрий получается путем восстановления
Na2SO4 водородом (см. ниже). Его можно также получать обра-
обработкой при 600—850° соды или ее смеси с твердым едким натром,
сероводородом или сероокисью углерода в среде восстанавливаю-
восстанавливающих газов.
Получение сульфида натрия восстановлением
сульфата натрия газами
Восстановление сульфата натрия не твердым углем, а газооб-
газообразными восстановителями (природным, генераторным и другими
газами) по суммарным реакциям
Na2SO4 + СН4 = Na2S + СО2 + 2Н2О
Na2SO4 + 4СО = Na2S + 4СО2
Na2SO4 + 4Н2 = Na2S + 4Н2О
дает возможность сразу получать продукт, не загрязненный углем,
и устраняет необходимость громоздких операций по переработке
плава — выщелачивания, отделения шлама, выпаривания щелоков.
Помимо этого, газ является более дешевым восстановителем, чем
твердое топливо.
Выход Na2S при восстановлении сульфата натрия водородом и
водяным газом при 800° в течение 1 ч составляет ~85% от теоре-
теоретического. Восстановление окисью углерода идет медленнее, чем
водородом, а метаном еще медленнее85. Отсутствие конструкций
печей, в которых можно было бы осуществлять достаточно полное
восстановление газом жидкой реакционной массы — расплавленно-
расплавленного сульфата, затрудняло организацию этого процесса в заводских
масштабах. Восстановление сульфата натрия газами при низкой
температуре, исключающей возникновение жидкой фазы, протекает
сравнительно медленно86.
Из многих газовых восстановителей — окиси углерода, водо-
Рода, метана, генераторного и природного газа — в настоящее вре-
время для получения Na2S в промышленном масштабе используется
лишь водород. Это объясняется тем, что в присутствии примеси
492
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
493
небольших количеств катализаторов (меньше 0,2% соединений же-
железа, никеля, меди и др.) сульфат натрия восстанавливается водо-
водородом в сернистый натрий при температурах более низких, чем
температуры плавления сульфата и его эвтектических смесей. Вос-
Восстановление начинается при 500° и протекает с достаточной ско-
ростью при 600°, что позволяет избежать оплавление материала.
Получается безводный продукт, содержащий больше 95% Na2S,
называемый сульфиграном (сульфид гранулированный) 27>87. С по-
повышением содержания водяного пара в газе равновесие основной
реакции
Na2SO, + 4Н2 ^=± Na2S + Н2О
сдвигается влево и ее скорость резко уменьшается. При 20% Н2О
в газе и 630° довести реакцию до конца невозможно. Кроме того,
повышенное содержание водяного пара ускоряет побочную реак-.
цию, приводящую к потере серы в виде H2S:
Na2S + 2H2O ^z± 2NaOH + H2S
Поэтому если осуществляется рециркуляция водорода, подавае-
подаваемого в восстановительную печь в большом избытке, то по выходе
газа из печи водяной пар должен быть сконденсирован в процессе
рекуперации тепла (см. рис. 149).
Восстановление сульфата натрия природным газом или мета-
метаном, вероятно, связано с диссоциацией последнего, происходящей
при высоких температурах (800—1000°). Разложение метана уско-
ускоряется в присутствии древесного угля, кокса, железа, никеля
и др. Выход Na2S при 600° незначительный, сильно возрастает
выше 750°, при 800° за 3 ч достигает —88%, а при 850° — 95%. Бо-
Более высокая температура приводит к загрязнению продукта угле-
углеродом, выделяющимся при разложении метана, а более длитель-
длительное нагревание — к образованию побочных продуктов. При восста-
восстановлении не выше- 850° содержание углерода в продукте меньше
1 % 88-89.
Восстановление сульфата натрия окисью углерода начинается
около 700°, уже при 730° протекает со значительной скоростью, а
при 850° практически заканчивается в течение 25 мин. Процесс со-
сопровождается побочными реакциями, приводящими к образованию
соды и серы ё0:
3Na2SO4 + Na2S + 8СО = 4Na2CO3 + 4СО2 + 4S
В присутствии водяного пара образование соды из побочного
процесса превращается в основной; восстановление окисью угле-
углерода идет по реакции
Na2SO4 + 4СО + Н2О = Na2CO3 + ЗСО2 + H2S
а восстановление полуводяным газом по реакциям
Na2SO4 + Н2 + ЗСО = Na2COs + H2S + 2СО2
Na2SO4 + 2Н2 + 2СО = Na2CO8 + H2S + СО2 + Н2О
причем с увеличением парциального давления водяного пара пред-
предотвращается плавление материала и ускоряется образование соды.
Полуводяной газ при 630° и /?НгО»=0,35 ат за 4 ч восстанавли-
восстанавливает сульфат на 97,2% с выходом 2Na2CO3 92,7%. Это послужило
Рис. 149. Схема производства сернистого натрия восстановлением суль-
сульфата натрия водородом во вращающейся печи:
/ — бункер для сульфата; 2 —печь с вращающимся подом для предварительной про-
прокалки сульфата; S—пэчь для восстановления сульфата; 4 — циклон; 5 — теплообмен-
теплообменник; 6 — скруббер с керамич^ков насадкой для охлаждения газа и конденсации водяного
пара; 7 и 5 — скрубберы со стальной насадкой для поглощения H2S и осушки газа;
9 — газодувка; W — подогреватель водорода.
основанием для разработки метода получения соды восстановле-
восстановлением сульфата натрия газами91-94.
Скорость восстановления сульфата натрия в сульфид газами
растет с увеличением расхода газа и дисперсности частиц Na2SO4,
а при относительно низких температурах также в присутствии ка-
катализаторов— при высоких температурах скорость реакции стано-
становится настолько большой, что роль катализаторов оказывается
несущественной. Однако, если относить скорость реакции не к еди-
единице поверхности раздела фаз, а ко всей массе сульфата, то уско-
ускорение реакции с повышением температуры идет лишь до некоторого
предела. По мере оплавления частиц реакционная поверхность
Уменьшается и процесс замедляется. Так, восстановление водородом
пРи температурах выше 650° дает меньшие выходы- Na2S, чем
494
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
495
при более низкой температуре (полное восстановление при 650е I
достигается лишь за 15 ч). Выше 800* наблюдается полное сплав-
сплавление реакционной массы. Предотвратить отрицательное влияние
плавления материала на ускорение процесса с повышением темпе-
температуры можно, если в начале процесса поддерживать 600—610° и
повышать температуру до 700—710° только после того, как сте-
степень восстановления превысит ~50%, когда содержание Na2S в
реакционной смеси будет больше, чем в эвтектике (стр. 472), и
температура плавления начнет возрастать. Таким способом за 8 ч
можно получить продукт, содержащий 96% Na2S85. Уменьшение
оплавления материала достигается также при предварительном!
гранулировании или брикетировании сульфата. Предложено до-|
бавлять к сульфату натриевую соль алифатической кислоты с|
целью осуществления процесса во вращающейся печи без спека-|
ния с получением зерненого продукта 95.
Изучено восстановление брикетов сульфата натрия газами пи-|
ролиза нефти в стационарном и в псевдоожиженном слое. В усло-|
виях восстановления сульфатных брикетов при 700—725° в шахт-1
ной печи основными восстановителями в этих газах являются водо-1
род и олефины. Парафиновые углеводороды участвуют в восста-1
новлении в меньшей степени. В присутствии соединений железа|
скорость восстановления увеличивается96.
При промышленном восстановлении сульфата натрия водоро-1
дом в качестве катализатора используют обычно железо. При co-J
держании в сульфате всего лишь 0,07—0,08% Fe скорость процесс?
уже достаточно велика при 600—650°87. Для равномерного распре-1
деления катализатора его следует вводить в сульфат в виде вод-|
ного раствора какой-либо соли, например, FeSO4.
Предложено также получать сульфат, легко воестанавливае-1
мый водородом при 700° во вращающейся печи, смачиванием смеси|
сульфата с колчеданным огарком 35%-ной серной кислотой A,5 кг
огарка и 20.к« кислоты на 100 т сульфата) и последующим брике-
брикетированием 9Г.
На рис. 149 показана принципиальная схема производства сер-|
нистого натрия восстановлением сульфата натрия водородом на|
заводе в Леверкузене98. Восстановление осуществляется в горизон-
горизонтальной вращающейся A об/мин) печи длиной 20 м с наружный
диаметром барабана 3,1 м. Концы барабана имеют форму усечен-j
ных конусов с диаметром малых оснований 1,6 м, что обеспечивает
толщину слоя перемещающегося в печи материала 0,75 м. Печу
футерована шамотным кирпичом по слою теплоизоляции из стек]
лянной ваты. Герметичное уплотнение между барабаном и неподН
рижяыми головками печи достигается с помощью лабиринтов, за-]
Полненных под давлением вязкой (хлорнафталиновой) смазкой]
Нагрузка и выгрузка материала из печи производится с помощью
торцовых шнеков. Сульфат натрия из сульфатных печей, размоло-!
тый до тонкости 1—2 мм, для освобождения от серной кислоты об-
обрабатывается небольшим количеством сульфита натрия и для
окончательного удаления H2SO4 прокаливается при 600° в обогре-
обогреваемых коксовым газом печах с вращающимся подом. Затем он
с температурой 400—450° поступает в восстановительную печь. Ка-
Катализатор — пыль из электрофильтров колчеданных печей — добав-
добавляется в сульфат перед прокалкой. Водород, поступающий из про-
производства хлора, подается в восстановительную печь нагретым до
820° в трубчатом подогревателе из жароупорной стали, где сжи-
сжигается коксовый газ. Количество водорода G20 мъ\ч) в 6—8 раз
превышает теоретическое. Газ, выходящий из печи, очищают в ци-
циклоне от сульфатной пыли, возвращаемой в прокалочную печь, и
охлаждают в теплообменниках с 530 до 150—170°, затем для даль-
дальнейшего охлаждения и конденсации водяного пара промывают хо-
холодной водой, а для поглощения H2S и осушки 35 и 50% раство-
растворами NaOH. После этого вместе со свежим водородом циркули-
циркулирующий водород через теплообменники (в которых подогревается
до 400°) и через нагреватель возвращается в печь. В системе под-
поддерживается избыточное давление в несколько сотых долей ат.
Сернистый натрий выходит из печи в виде кусков 10—15 см с тем-
температурой 750° и охлаждается в стальном барабане, а затем в
шнеке с водяной рубашкой. В этих аппаратах и в элеваторе между
ними продукт находится в атмосфере азота. Затем перед тариро-
тарированием его измельчают в молотковой дробилке до размера кусков
5 мм. Готовый сульфигран содержит 95% Na2S, 2% Fe2O3, 0,5—1%
Na2CO3, 1% NaCl и 1—1,5% Na2S2O3 и Na2SO3. На 1 т сульфи-
грана (в пересчете на 98% Na2S) расходуют 1,85 т сульфата нат-
натрия, 1350 м3 водорода, 820 кет ¦ ч электроэнергии и ~ 1300 м3 коксо-
коксового (или генераторного) газа.
На другом заводе (в Вольфене) восстановление сульфата нат-
натрия водородом производят в шахтных печах 45'8В.97. Горячий суль-
сульфат из механических сульфатных печей охлаждается в холодиль-
холодильнике до 40—50°, а затем в наклонном шнеке, где он смачивается
водой C—4%); после этого его брикетируют. Сульфат должен со-
содержать 0,2—0,3% Fe. Брикеты овальной формы размером 40—
50 мм с плотностью 1,8—2 г/см3 для усиления прочности вылежи-
вылеживаются на складе 24 ч. Затем через сито для отсева мелочи и пыли
брикеты поступают в отапливаемую генераторным газом наклон-
наклонную вращающуюся печь, где нагреваются до 550—600°, причем из
них удаляется влага, а содержание NaCl за счет реакции с H2SO4
(бисульфатом) снижается до 0,1% (большее содержание NaCl не-
нежелательно). Нагретые брикеты поступают через питатель в шахт-
нУю восстановительную печь производительностью 7—8 т сульфи-
гРана в- сутки. Шахта печи цилиндрической формы футерована
Шамотом. Между футеровкой и стальным кожухом кизельгуровая
изоляция. Шахта разделена вертикальной шамотной стойкой на
496
Гл. XIII. Соли сульфидного ряда
Сульфид натрия
497
три вертикальные секции, к каждой из которых подводится водо-1
род. Водород поступает с хлорного завода; он подогревается до|
400° в теплообменниках за счет тепла уходящего из печи газа, а|
затем до 700—710° в электрическом подогревателе, в котором со-1
противлением служат графитовые шары диаметром 40 мм. Подо-1
греватель представляет собой футерованную камеру с электро-1
дами, присоединенными к однофазному трансформатору F000 а\
60 в). Водород, выходящий из печи с температурой 560°, очищается
от сульфатной пыли в циклоне, охлаждается в теплообменниках дс
~ 150°, затем в водяном скруббере до 25—30°, где освобождается!
от основной массы водяного пара. После этого, вместе со свежей
долей водорода, с помощью компрессора газ возвращается в цикл.|
Он должен содержать не меньше 97% Н2 и не больше 30 г водя-
водяного пара в 1 м3; количество проходящего через печь водорода
5 раз больше теоретического. Выгрузка сернистого натрия из печи|
производится с помощью вращающегося стола A об/ч). Продукт
представляет собой гранулы серо-коричневого цвета и содержит
96—97% Na2S, 0,4% Na2S2O3, 0,7% Na2SO3, 0,36% NaHS, 0.4%|
Na2CO3, 0,4—0,6% Fe2C>3, 1,4% нерастворимых веществ. На 1 т про-
продукта расходуется 1,9 т сульфата натрия, 1320 мг водорода, 720 \
генераторного газа, — 1000 квт-ч электроэнергии, 60 м3 воды (пс
другим данным, 15 м3) и 15 чел.-часов рабсилы.
Недостатком этой схемы является необходимость гранулирова-1
ния сульфата натрия, что, помимо затрат на этот процесс, приво-
приводит к уменьшению реакционной поверхности материала, к замед-|
лению восстановления (продолжительность пребывания материале
в печи достигает 20 ч) и увеличению необходимых размеров печи]
Восстановление сульфата газами может быть значительнс
интенсифицировано и усовершенствовано при осуществлении егс
в циклонных печах", а также в печах со взвешенным слое»
(рис. 150). Здесь можно подвергать восстановлению сульфат
размерами зерен меньше 2 мм. Имеется много предложений
осуществлению восстановления сульфата натрия во взвешенно!*
слое, направленных главным образом к предотвращению агломе-|
рации частиц 4Б-100~105. Так, например, для этого предложено
реакционное пространство, заполненное в основном сульфидом!
натрия, вводить сульфат в таком количестве, чтобы его содержа]
ние в массе составляло 5—10%- Это позволяет вести процесс без
спекания частиц при постоянной и оптимальной температуре, пре
вышающей эвтектическую, находящейся в пределах 675—7501
(в зависимости от вида катализатора). При размере зерен суль-1
фата 0,2—0,3 мм в присутствии 1% железного катализатора, дли-1
тельность восстановления водородом составляет 8 мин. Рекомен-|
дуют поддерживать температуру стенок печи ниже или равной
температуре реакционной смеси, чтобы исключить частичное шлако-1
вание, особенно на стенках. Это достигается подачей в печь суль-1
фата и газа, предварительно нагретых до температуры, превы-
превышающей реакционную.
Одним из существенных преимуществ восстановления суль-
сульфата натрия водородом является прямое получение продукта с со-
содержанием 96—98% Na2S без необходимости производить перера-
переработку плава, дающую к тому же продукт, содержащий только 65%
Na2S, перевозка которого требует повышенных транспортных рас-
расходов. Однако этот способ имеет и недостатки: большой расход
водорода; необходимость организации установки для получения
Рис. 150. Принципиальная схема производства сернистого натрия
восстановлением сульфата натрия во взвешенном слое:
1 — бункер для сульфата; 2 — автоматический питатель сульфата; 3 — шнек;
4 — реактор-восстаиовительная печь со взвешенным слоем сульфата; 5 —холо-
—холодильный барабан для охлаждения сернистого натрия; 6 —газовый теплообмен-
теплообменник; 7 — электрофильтр для улавливания пыли, уносимой водородом из реактора;
8 —водяной скруббер для охлаждения водорода и конденсации водяного пара;
9 — скруббер для отмывки водорода от H2S; 10 — газодувка; // — нагреватель
водорода.
азота, требующегося для промывки аппаратуры перед пуском и
остановкой, во избежание образования гремучей смеси; необходи-
необходимость применения сульфата, не содержащего хлористого натрия
(понижающего температуру появления жидкой фазы в системе
Na2SO4—Na2S). В связи с большой стоимостью водорода использо-
использование этого способа рационально пока лишь в районах с избыточ-
избыточным балансом водорода, особенно при наличии там природного
сульфата.
Более перспективным, и потому заслуживающим дальнейшей
разработки и производственного освоения, является восстановле-
восстановление сульфата натрия природным газом и газами, содержащими
СО — отходящими из производств фосфора, карбида кальция
и др., — а также коксовым газом. Помимо того, что эти газы де-
дешевле водорода, а вернее, именно поэтому, можно не возвращать
в цикл непрореагировавшую часть газа, а использовать ее в каче-
качестве топлива; это значительно упростит технологическую схему и
снизит капитальные и эксплуатационные затраты.
498
Гл. XIII. Соли сульфидного ряда
Другие способы получения сульфида натрия
Сравнительно малые количества сернистого натрия полу-
получаются не восстановлением сульфата натрия, а другими способами.
При улавливании раствором едкого натра отбросного сероводо-
сероводорода 106'107, получающегося, например, при производстве хлори-
хлористого бария, сероуглерода, при очистке нефти, или сероводорода,
извлеченного из коксового и других промышленных газов, обра-
образуется гидросульфид натрия (см. ниже) или сернистый натрий по
реакции:
2NaOH + H2S = Na2S + 2Н2О
Вместо едкого натра для поглощения сероводорода можно
пользоваться известковым молоком с примесью соды:
Na2CO3 + Са(ОНJ + H2S = Na2S + СаСО, + 2Н2О
Сернистый натрий получается при сливании растворов гидро-
гидросульфида и едкого натра:
NaHS + NaOH - Na2S + Н2О
Na2S получается также путем переработки содержащих Са$
отвалов, образующихся в производстве соды из сульфата натрия,
известняка и угля или в производстве хлористого бария хлоркаль-
циевым методом. При разваривании этих отвалов в растворе, со-
содержащем соду и сульфат натрия, происходит гидролиз CaS по
уравнению:
2CaS + 2Н2О = Ca(HSJ + Ca(OHJ
Са(ОНJ каустифицирует соду по реакции:
Са(ОНJ + NagCOa = 2NaOH + CaCO8
а в результате реакции
Ca(HSJ + Na2SO4 = CaSO4 + 2NaHS
получается гидросульфид натрия, образующий с едким натром
NajS. Общее уравнение процесса имеет вид:
2CaS + Na2CO3 + Na2SO4 = CaSO4 + СаСО, + 2Na2S
Полученный раствор сернистого натрия отделяют от осадков и
выпаривают.
Предложено производить Na2S одновременно с сульфитом нат-
натрия взаимодействием серы с кипящим 50% раствором едкого на-
натра. Нагревание продолжают пока температура кипения раствора
не достигнет МО—150°. При этом Na2S остается в растворе, а суль-
сульфит натрия выделяется в твердом виде и после охлаждения смеси
до 120—110° Na2SC>3 отделяют центрифугированием. Затем раствор
охлаждают до 20—25°, отделяют выделившийся кристаллический
сернистый натрий, а маточный раствор возвращают в процесс108.
Гидросульфид натрия
499
Так как восстановление сернокислого бария в промышленных
условиях осуществляется с меньшими трудностями, чем восстано-
восстановление сернокислого натрия, то сернистый натрий можно получать
обработкой плава сернистого бария раствором сернокислого нат-
натрия по реакции:
BaS + Na2SO4 = BaSO4 + Na2S
Осадок сернокислого бария, отделенный от раствора Na2S, мо-
может быть вновь возвращен на восстановление109. Сернистый нат-
натрий получают также в качестве побочного продукта при производ-
производстве углекислого бария по реакции:
BaS + Na2CO3 - Na2S + ВаСО3
Раствор Na2S, остающийся после отделения осадка ВаСО3, вы-
выпаривают.
При обработке смеси поваренной соли с углем концентрирован-
концентрированной серной кислотой в механических муфельных сульфатных печах
или в пламенных печах при 650° получается плав, содержащий
больше 60—62% Na2S u0. Процесс идет по схеме:
2NaCl + 2С + H2SO4 = 2НС! + Na2S + 2СО2
Здесь совмещаются две реакции: образование сульфата натрия
и хлористого водорода и восстановление сульфата углем. Таким
образом, можно получать сернистый натрий непосредственно из
поваренной соли, минуя стадию предварительного производства
сульфата. Выделившийся при этом хлористый водород, как и при
получении сульфата, поглощается водой с образованием соляной
кислоты.
Описано получение ЫагЭ, основанное на разложении амаль-
амальгамы натрия, образующейся при электролизе поваренной соли с
ртутным катодом, раствором полисульфида натрия или серой по
реакциям:
6Na + Na2S4 = 4Na2S; 2Na + S = Na2S
При обработке амальгамы, содержащей 0,2% Na, раствором
состава: 10—11% NaaS и 11—12% полисульфидной серы, полу-
получается раствор, содержащий 29—30% Na2S и 0,2—0,3% Na2S2Qs,
который без дальнейшей переработки используют в производстве
красителей98,
ГИДРОСУЛЬФИД НАТРИЯ
Гидросульфид натрия получают улавливанием сероводорода
При промывке содержащих сероводород промышленных газов рас-
раствором едкого натра8
H2S + NaOH = NaHS + НаО
BOO
Гл. XIII. Соли сульфидного ряда
Полисульфиды
501
или раствором сернистого натрия:
H2S + Na2S = 2NaHS
Промывку содержащих сероводород газов производят в баш-
башнях с насадкой, орошаемой сверху поглощающим раствором.
Концентрация сероводорода в промываемом газе не имеет боль-
большого значения. От нее зависят размеры абсорбционной башни. Что
касается конечной концентрации получаемого раствора гидросуль-
гидросульфида, то при рециркуляции поглощаемого раствора она мало за-
зависит от концентрации H2S в газе. Это позволяет улавливать серо-
сероводород из газов, содержащих десятые доли процента H2S, и полу-
получать при этом растворы, содержащие до 30% NaHS. Если, однако,
промываемые газы содержат много влаги, а концентрация серово-
сероводорода в них невелика, то это ведет к значительному разбавлению
раствора конденсирующейся из газа водой.
При поглощении щелочью сероводорода из промышленных га-
газов (например, из коксового газа) из них вымываются и некоторые
другие примеси, загрязняющие раствор, в частности цианистые
соединения
HCN + NaOH = NaCN + Н,@
и сероуглерод:
CS2 + 2NaHS = Na2CS3 + H2S
Na2CS3 + 3H2O = Na2CO3 + 3H2S
При содержании в газах кислорода растворы легко екисляются
2NaHS + 2О2 = Na2S2O3 + Н2О
а цианистые соединения переходят в роданистые:
2NaCN + 2H2S + О2 = 2Na NCS + 2Н2О
Промышленные газы, из которых улавливается сероводород,
обычно содержат СО2, а технический едкий натр — соду. Поэтому
растворы гидросульфида, получаемые промывкой газов раствором
едкого натра, содержат некоторое количество NaHCO3. Раствор
гидросульфида, не содержащий бикарбоната, может быть получен,
если вести поглощение сероводорода известковым молоком по ре-
реакции
Са(ОН>2 + 2H2S = Ca(HS>2 + 2Н2О
с последующим обменным разложением содой:
Ca(HS)j + Na2CO3 - 2NaHS + СаСО3
При этом содержащаяся в rase двуокись углерода связывается
известью.
При поглощении сероводорода известковым молоком можно по-
получить почти насыщенный раствор гидросульфида кальция, кото-
который в ряде случаев может с успехом применяться вместо гидро-
гидросульфида натрия. При обработке этого раствора сухой содой полу-
получаются растворы, содержащие до 385 г/л NaHS.
Более концентрированный продукт, содержащий свыше 50°/о
NaHS, получается при действии сероводорода на твердый плавле-
плавленый сернистый натрий при 80—90е. При этом масса плавится к
образуется раствор гидросульфида.
Описано111 получение гидросульфида действием серной кислоты
на Na2S по реакции:
2Na2S + H»SO« = 2NaHS + Na2SO4
Гидросульфид и сульфид натрия содержатся в щелоках — отхо-
отходах нефтеперерабатывающих заводов. Перед сбросом эти щелоки
необходимо обезвреживать. Разработан метод регенерации NaOH
из этих щелоков с выделением из них серы с помощью окисли-
окислителя— двуокиси марганца по реакциям:
NaHS + MnO2 = NaOH + MnO + S
Na2S + MnO2 + Н2О = 2NaOH + MnO + S
При отношении Na2S : Mn2O, равном 1 : 2,5, и 30° процесс за-
завершается за 2 ч. Из отработанной марганцовой руды серу можно-
извлечь перегретым паром или органическим экстрагентом П2.
Растворы NaHS используют для получения чистого сероводо-
сероводорода по реакции:
2NaHS + H2SO4 = Na2SO4 + 2H2S
Разложение раствора NaHS можно производить стехиометриче-
ским количеством серной кислоты с доведением рН отработанного-
раствора до 8—3. Процесс сопровождается выделением неболь-
небольшого количества элементарной серы (до 3%), причем, чем выше
рН, тем меньше выделяется серы113.
ПОЛИСУЛЬФИДЫ
Полисульфиды натрия, вследствие сильной гигроскопичности их
кристаллов, обычно производят и применяют в виде растворов,,
имеющих желто-бурый цвет. Их можно получать разными спосо-
способами114; наиболее распространенным является растворение тонко-
измельченной серы (серного цвета) в растворе щелочи:
6NaOH + 2nS = 2Na2Sn_, + Na2S2O3 + 3H2O
Во избежание окисления нагревание суспензии серы в растворе
Щелочи можно вести в атмосфере азота. Образующиеся полисуль-
Фиды содержат от 2 до 4,5 атомов серы. Присутствие тиосульфата
натрия обычно не мешает использованию полисульфидного рас-
раствора, в частности и в процессах полимеризации. Если полисуль-
полисульфидный раствор предназначен для использования в качестве
Й02
Гл. XIII. Соли сульфидного ряда
инсектофунгицида, его готовят 8—10-кратным разбавлением воде
суспензии, полученной из серы и 20% раствора NaOH, взятых в В'
совом соотношении 0,35: 1.
Дисульфид натрия можно получить взаимодействием серы с г
рячим раствором сернистого натрия
или с раствором, содержащим NaOH и NaHS:
NaOH + NaHS + S = Na?S2 + H2O
Получаемые этими способами растворы содержат дисульфид
или дисульфид и сульфид натрия; они получаются быстрее, чем
при ризварке элементарной серы в растворе едкого натра, требую-
требующей длительного кипячения суспензии.
Чистые полисульфиды натрия получают из элементов:
2Na + 2S = Na2S2
В качестве реакционной среды используют спирт или жидкий
аммиак 115.
Полисульфиды калия (и твердые полисульфиды натрия) полу-
получают сплавлением серы с углекислым калием (или содой). Полу-
Получаемая «серная печень» имеет вид комьев желто-бурого цвета. Она
хорошо растворима в воде, и поэтому ее иногда называют «раство-
«растворимой серой».
Полисульфиды кальция изготовляют продолжительным кипя-
кипячением тонконзмельченной серы в известковом молоке. Весовой
состав суспензии — S : СаО : Н?О=10 : 5 : 85. Полученный раствор
отстаивается от осадка извести и применяется под названием изве-
стково-серного отвара. В его состав входят тетрасульфид и пен-
тасульфид кальция15. Сухие препараты полисульфндов кальция
получают выпариванием концентрированного известково-серного
отвара, добавляя к нему в качестве стабилизатора сахар1б, или
сушкой отвара в распылительной сушилке. Эти препараты содер-
содержат 64—67% полисульфидов кальция, 23—28% CaS2O8, 5—11% S
я 0,3—'1,1% СаСОз. О полисульфидах бария (сольбаре) см. стр. 421.
ЛИТЕРАТУРА
1. Минералы СССР, т. II, под редакцией А. Г. Бетехтииа, Изд. АН
СССР, 1940. — 2. Е. J. Kohemeyer, G. Lohrke, Z. anorg. Chem., B281,
H. 1—2 A956).— 3. С. А. Щукарев, Е. М. Киреева-Тузулахова, ЖОЧ,
1, № 8—9, 1126 A931).— 4. Д. В. Безуглый, Ф. М. Куцаков, ЖХП, № 8,
37 A939). —5. В. М. Оратовский, А. М. Гамольский, Н. Н. Климе н-
жо, ЖПХ, 37, № 11, 2392 A964).— 6. Д. М. Гинзбург, Н С. Пикулина,
В. П. Литвин, ЖПХ, Там же, 38, № 9, 2117 A965); 39, № 10, 2371 A966).—
7. М С. Л и т в и н е н к о, М. Б. Хват, Н. А. Г у р е в и ч. Кокс и химия, № 8,
<51 A965).—в. В. М. Р а м м, Абсорбционные процессы в химической промыш-
Литература
60S
ленности, Госхимиздат, 1951, стр. 255. — 9. М. С. Л и т в и и е н к о, С. П. Л у н-
пин, ЖПХ, 29, № 4, 543 A966). —10. М. С. Литвиненко, Там же, 25, № 5„
516 A952).
//. М. С. Л и т в и и е н к о, Там же, 25, № 7, 696 A952). — 12. Е. S с h u I e к,
Е Кб г os, L. Ma r os, Acta chim. Acad. Sci. Hung., 10, № 1—3, 291 A956).—
13. G. R. Levi, V. Riganti, Ann. chimica, 46, № 1—3, 174 A956).—
14. И. Л. Багбанлы, M. М. Гуревнч, А. К. Мискарли, Труды Инсти-
Института химии АН АзССР, в. 13, 1954, стр. 114. — 1Б. А. Л. Ефимов, Химический
метод борьбы с вредителями и болезнями сельскохозяйственных растений, Сель-
хозгиз, 1949. — 16. Д. Ф р и р, Химия инсектисидов и фунгисидов, ИЛ, 1948. —
17. П. П. Будников, М. И. Некрич, Хим. пром., № 7, 18 A955).—
18. F. Foerster, К. К u b е 1, Z. anorg. Chem., 139, 261 A924).— 19. А. П. Б у н-
тин, Т. М. Сосинатров, Труды лим.-металлург. ин-та, Зап.-Сиб. филиала
АН СССР, № 5, 1951, стр. ST. —20. А. Б. Бектуров, В. В. Тихонов; Труды
Ин-та хим. наук АН КазССР, т. 1, 1957, стр. 20.
21. В. А. Никитин, Т. И. Кун и и, Журн.'ВХО, 5, № 3, 350 (I960).—
22. И. Н. Севрюков, Е. М. Сергиевская, ЖПХ, № 1, 54 A961) —
23. А. А. Фотиев, Там же, 35, № 11, 2402 A962).—24. С. J. N у m а п,
Т. D. O'Brien, Ind. Eng. Chem., 39, № 8, 1019 A947). — 25. Т. И. К у и и н,
В. А. Никитин, Изв. вузов. Хим. и хим. технол., № 5, 61 A958).—
26. В. И. М и н а е в. Изв. Томск, технол нн-та, 44, в. 1, 11 A923).— 27. Е. А. Ш и-
лов, П П. Будииков, Изв. Ив.-Вознесенск. полнтехн. ин-та, 10, 57 A927).—
28. П. П. Будников, А. Н. Сысоев, Укр. xiM. ж., 3, № 2, 113 A928) —
29. П. П. Будников, ДАН СССР, 1, № 6, 330 A934).— 30. С. К. Чирков,
ЖХП, №6, 9 A938).
3/. А. А. Порет, Труды ГИПХ, в. 35, 1941, стр. 22. —32. М. Е. Познн
Хим. пром., № 12, 10 A944). —33. А. Г. Репа, Е. П. Данильченко, ЖПХ,
24. № 1, 20 A951).— 34. А. П. Бунтин, Т. М. С о с и п а т р о в. Труды Хим.-
металлург. ин-та Зап.-Сиб. филиала АН СССР, № 8, 1954, стр. 39.— 35. Р. В. М и-
ролюбов, Ф. А. Кульков, М. А. Серебренникова, Хнм. пром., № 7,
206 A950). — 36. Н. П. Диев, А. И. Окунев, В. В. Падучев, В. В. Топо-
Топорова, В. С. Мокроносов, ДАН СССР, 107, № 2, 273 A956) —
37. Н. С. Лапшни, Хим. пром., № 4, 122 A951). — 38. 3. С. Банных,
Е. М. Полякова, Труды УНИХИМ, в. 4, 1957, стр. 64. — 39. Т. И. Куннн
В. А. Никитин, Труды ИХТИ, в. 5, 1956, стр. 85; в. 7. 1958, стр. 61.—
40. Б. В. Федосеев, К- Л. Добрусин, Л. И. Шапиро, Хим. пром., № 12,
377 A949).
41. С. В. Глебов, Труды ГИПХ, в. 35, 1941, стр. 52.-42. П. П. Будни-
Будников. В. И. Ендовнцкий, ДАН УССР, № 4, 35 A940).— 43. Н. С. Лап-
Лапшин, Хим. пром., № 7, 16 A948).— 44. Л. В. Юма нова, М. С. Герма ид-
зе. Труды УНИХИМ, в. 2, 1954, стр. 187. — 45. К. И. Макарьин, Труды
Московск. инж.-экон. ин-та, в. 1, 1954, стр. 83. — 46. В. П. Филиппов
И. М. Вейсбаид, Хим. наука н пром., 2, № 6, 752 A957). — 47. М. Е. По-
зин, Технология минеральных удобрений и солей, Госхимиздат, 1957, гл XI —
«о- И. Шейнберг, Химстрон. № 7, 207 A929). — 49. П. В. Дыбина, Тех-
Технология минеральных солей, Госхлмнздат, 1949.—50. И. М. В а ее ер м а н.
Производство минеральных солей, Госхимиздат. 1962.
51. Л. О. Черкасский, Сернистый натр, Свердловск, 1931.— 52. А. Н По-
Пономарев, Хнм. пром., № 5, 146 A949). — 53. Н. Н. Чер ейская. Там же,
¦I* 7, 193 A951). — 54. М. Д. Лнберман, Тезисы докладов на отраслевом
совещании работников основной и горнохимической промышленности, 1958
стр. 129.^55. Г. Я. Тарасов, А. С. Изотова, ЖПХ, 6, № 4. 673 A933) —
°?- G. A. Pirs, Brit. Abs., 1, 410 A946).-57. Ю. П. Никольская, Труды
лим.-металлург. ин-та Зап.-Снб. филиал АН СССР, в. 1, хим., 1949, стр 61
oe- J- F. White, A. H. White, Ind. Eng. Chem., 28, № 2, 244 A936)! —
504
Гл. XIII. Соли сульфидного ряда
69. О. И. Пудовкина, Тезнсы докладов на отраслевом совещании работни-
работников основной и горнохимической промышленности, 1958, стр. 127; О. И. Пудов-
Пудовкина и др., Труды УНИХИМ, в. 7, 1958, стр. 87, 91.— 60. И. П. Щербак
и др., авт. свид. 194066, 1965.
61. П. В. Бурдуков, Цветные металлы, № 1, 47 A951). — 62. В. Ф. Вол-
Волков и др.. Хим. пром., № 9, 687 A967). — 63. Т. И. Кунин, В. А. Никитин
Изв. вузов. Хим. и хим. технол., 1, № 3, 93; № 5, 61 A958); 3, № 2, 324 (I960)-'
Труды Ивановского хнм.-технол. ин-та, в. 5, 1956, стр. 85; в. 7, стр. 61 и 64. —
€4. В. П. Ильинский, Г. Я. Тарасов, О. Я. К а л а н и, Труды ГИПХ,
в. 16, 1932, стр. 89. — 65. В. П. Ильинский, Труды соляной лаборатории
АН СССР, в. 2, 1932.—66. Ф. А. Кульков, Тезисы докладов на отраслевом
¦совещании работников основной и горнохимической промышленности, 1958
стр. 125. —67. В. Г. Брексон, Труды УНИХИМ, в. 14, 1967, стр. 87; сб.'
«Циклонные энерготехнологические процессы и установки», изд. ЦНИИинформ.
Мин. цвет, мет., 1967, стр. 264. — 68. Л. Черкасский, Техник на производ-
производстве. № 14, 140 A932). — 69. В. Заболоцкий, Б. П. Полетаев, Труды
Днепропетровск, химико-технол. ин-та, т. 2, в. 2, 1941, стр. 58. — 70. Б. А. Б е-
Ф ем ж а нов, Г. Г. Пронина, Уч. зап. Казахск. ун-та, 22, 30 A956).
71. Б. А. Беремжанов, Изв. АН КазССР. Сер. хим., в. 10, 104 A956).—
72. А. И. Мун, Там же, в. 10, 22 A956). — 73. Ф. А. Кульков. Е. М. Поля-
Полякова, Е. И. Толстикова, Труды УНИХИМ, в. 3, 1955, стр. 74. —
74. Т. И. Кунин, Н. С. Лапшин, В. А. Н и к и т и н, Труды ИХТИ, в. 7,
1958, стр. 64.-75. Ю. П. Каретников, В. Н. Тарасова, К- П. Ж и п и-
лева, ЖПХ, 34, № 3, 682 A961). — 76. А. И. Аккерман, Труды ГИПХ,
в. 35, 1941, стр. 60. — 77. Л. А. Полуборцева, А. А. Рейфер. Т. М. М а н-
торова, И. Г. Воликова, 3. Ф. Истрина, Хим. пром., № 3, 230 A966). —
18. S. Nakaya, япон. пат. 1866, 1953. — 79. П. Михайленко, Н. Три лев.
Производство сернистого натрия, Химтеорет, 1933.-5G. Ю. С. Шмелев,
И. В. Костарева, Ф. И. Китаева Труды УНИХИМ. в. 19, 1967, стр. 84.
81. Н. Н. Ч ер ей ска я. Труды Московск. инж.-экон. ии-та, в. 1, 1954,
стр. 75.-82. Т. И. Куиин, Н. С. Лапшин, Хим. пром., № 3. 9 A948).—
S3. И. Г. П о в а р н и н. Производство сернистого натра на малых установках,
Гизмегтпром, 1946. — 84. Раб. химик, № 10, 14 A934).— 85. В. П, Ильинский,
Г. Я. Тарасов, А. А, Гинзбург, Труды ГИПХ, в. 16, 1932, стр. 80.—
-86. R. Ley, Chem. Ztg., № 85, 859 A934).— 87. С. J. Nyraan, T. D. O'Brien,
Ind. Eng. Chem., 39, № 8, 1019 A947). — 88. Д. Е. Днонисьев, ЖПХ, 9, №8,
1378 A936). — 89. А. И. Горбанев, В. Я. Николнна, Сульфат иатрня, Гос-
химиздат, 1954. — 90. Okuno, Masumi, Fukuyoma, J. Soc. Chem. Ind.,
Japan, 36, № 8, 466B A933).
91. А. И. Горбанев, ЖПХ, 27, № 8, 804 A954). — 92. А. И. Горбанев,
Там же, 28, № 9, 921 A954). — 93. А. И. Горбанев, Там же, № 10, 1033
A954). — 94. В. В. Чнчков, А. П. Шурыгин, Хнм. пром., № 12, 898
A965). —95. К- Wintersberger, G. Kondela, пат. ФРГ 878197, 1953. —
¦96. В. Т. Кунин, В. А. Никитин, И. П. К и р и л л о в, Изв. вузов. Хим. и
хим. технол., 8, № 6, 974 A965); 9, № 4, 609 A966); 10, № 7, 792 A967).—
97. Zirngibl, пат. ГДР 6592, 1954.— 98 L'ind. chim., 36, № 379, 25 A949).—
$9. А. П. Шурыгин, В. В. Ч и ч к о в, сб. «Циклонные энерготехнологические
•процессы», Цветметннформация. 1966. — 100. G. Kondela, К. Wintersber-
Wintersberger, пат. США 2719076, 1955.
101. Франц. пат. 1031456, 1954. — 102. Франц. пат. 1080224, 1954.—
103. Англ. пат. 738702, 1955. — 104. К. Wintersberger, G. Kondela, пат.
¦ФРГ 344605, 1956. — 105. Я. Циборовский, С. Вронский, ЖПХ, 32, №3,
473 A959). —106. Г. Джамбов, Г. Папуджиков, В. Николова, Химия
и индустрия (болг.), 38, № 1, 31 A966). — 107. L. Eckert, F. Wolf, M. F a r-
kas, Chem. Techa, 18, № 5, 293 A966). —108. G. N. Terziev, пат. США
Литература
5H5
9705187 1955—109. В. П. Ильинский, Г. С. Клебанов, ТруДы Соляной
лаборатории АН СССР, в. 2, 1932. —110. Ульященко, Раб. химик, № 10,
10 A934).
111 С Bandrowska, Przem. Chem., 45, № 5, 271 A966). —112. B.C. Пе-
Пеина Б. А. Старцев, ЖПХ, 38, № 6, 1212 A965).-113. Т. Д. Авер-
И П Баки на, Л. И. Булычева, Труды УНИХИМ, в. 14, 1967,
Ft J S Jk Id
'
I
21
у, ру
l37 —114 Е М Fettes, J. S. Jorczak, Ind. Eng. Chem., 42, №
ti I
l37 114 Е М Fette, g
2217 A950). — 115. D. M. Jost, H. Russell, Systematic Inorganic Chemistry,
N. Y., 1944, p. 152.
Физико-химические евойства
БОТ
Глава XIV
СОЛИ СУЛЬФИТНОГО РЯДА
К солям сульфитного ряда относятся соли кислот, образован-
образованиых комплексными ионами:
с П2- о П2-
sol" (и hsos~), s2o|-, sso!~,s2o2-
т. е. сернистой (сульфиты и бисульфиты), тио-
¦сернистой (тиосульфиты), серноватистой или тиосерной (тиосуль-
фаты), гидросернистой (гидросульфиты или дитиониты), пиросер-
нистой (пиросульфиты или мета бисульфиты), дитионовой (дитио-
наты) и политионовых (политионаты).
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
При растворении SO2 в воде образуется сернистая кислота
H2SO3, которая в свободном состоянии выделена только в форме
кристаллогидрата H2SO3 • 6Н2О. Большинство ее средних солей,
сульфиты (за исключением сульфитов щелочных металлов), в воде
не растворимы. Бисульфиты устойчивы только в растворах; при их
выпаривании в твердую фазу выделяются пиросульфиты, вслед-
вследствие равновесия такого типа: 2NaHSO34*Na2S2O5+H2O. Пиросер-
нистая кислота H2S2O5 не получена. В сернистой кислоте и боль-
НОч
шинстве ее солей сера четырехвалентна ^S = O; допускают
но/
нох .о
нАо
с шестивалентной серой. Сульфиты являются сильными восстано-
восстановителями. В водных растворах ион SOf~ окисляется даже кисло-
кислородом воздуха, но медленно: 2SOi + Ог -j2So!~. Процесс этот
каталитический, сопровождающийся цепной реакцией; ничтожные
количества ионов тяжелых металлов резко ускоряют процесс, а
лекоторые органические добавки (например, спирты) — замедляют.
также существование в некоторых случаях структуры
Сульфиты присоединяют серу е образованием тиосульфатов:
)f~ + S = БгОз". При действии на раствор тиосульфата кислотами
эта реакция идет в обратном направлении с выделением серы и
H2SC>3, так как H2S2C>3 в свободном состоянии неустойчива.
В ионе тиосульфата один атом серы шестивалентный, а другой
двухвалентный, чем объясняются сильные восстановительные свой-
свойства тиосульфатов. Сильные окислители окисляют S2O3~ до суль-
сульфата SOl", слабые — до тетратионата S^". Так, например, иод
окисляет S2C>3~ до тетратионата в водном растворе при окисли-
окислительном потенциале иода меньше 540 мв; в спирто-водных рас-
растворах при потенциале иода, большем 540 мв, окисление идет
до сульфата и серы, а при потенциале иода больше 650 мв, когда
становится возможным окисление свободной серы, продуктом окис-
окисления является только сульфат'.
При взаимодействии водных растворов SOs и H2S происходит
образование тиосерной и политионовых кислот, причем первичным
продуктом, вероятно, является не полученная в свободном состоя-
состоянии тиосернистая кислота H2S2O2. Это взаимодействие можно
представить следующим рядом реакций:
SO2 + Н2О = H2SOS
H2S2O2 + H2S *> 3S + 2H2O
H2SO3 + S = HjS2Os
HjSjOj, + 2H2SOj = H2S4Oe + 2H2O
H2S4Oe + H2SO3 - H3SsOe + H2S2OS
HsS4Oe + H2S = 2H2SaO3 + S
H2S2O2 + 2H2S2O, = H2SBOe + 2H2O
H2S6O6 = H3S5O6 + S и т. д.
При этом образуются политионовые кислоты H2SnO6 с п —
= 3—6. Ион дитионовой кислоты БгОб" может быть получен окис-
окислением водного раствора SO2, например, двуокисью марганца. По-
Политионовые кислоты мало устойчивы, существуют только в рас-
растворах в сильно диссоциированном виде, но образуют хорошо кри-
кристаллизующиеся соли, легко растворимые в воде. В политионовых
кислотах атомы серы образуют цепочку НО—SO2—S—S—S—-
—SO2—ОН; центральные атомы серы двухвалентные, поэтому поли-
политионаты легко окисляются. Политионовые кислоты (в частности,,
с п > 6) могут образовываться и в результате разложения не вы-
леленной в свободном состоянии сульфоксиловой кислоты S(OHJ-
или H2SO2, играющей роль промежуточного продукта, например 2:
2H2SO2 ^r± H2S2O3 + Н2О
H2SO2 + 2H2S2O3 = H2SB®8 + 2Н2О
H2S6O6+'(/j-5)H2SaO3 ^±
«08
Гл. XIV. Соли сульфитного ряда
Гидросернистая кислота H2SO4 очень неустойчива; в водных
растворах идет реакция диспропорционирования:
2H2S2O4 + Н2О = 2H2SO3 + H2S2O3
Однако эта кислота образует достаточно устойчивые, но легко
¦окисляемые кристаллические соли — гидросульфиты.
Приведенные выше соображения о строении соединений суль-
сульфитного ряда вероятны, но не бесспорны; химическая структура
этих соединений окончательно не уста-
установлена и, как и их свойства, является
предметом многочисленных исследова-
исследований 3-4. Из соединений сульфитного
ряда наибольшее значение имеют
сульфит и бисульфит натрия, тиосуль-
тиосульфат и гидросульфит натрия, а также
ронгалит5.
Сульфит натрия кристаллизуется
из водных растворов ниже 33,4° в виде
Na28O3 • 7Н2О, а при более высоких
температурах — в безводной форме
(рис. 151). Давление водяного пара
в системе Na2SO3-7H2O,:<*~Na2SO3+
+ 7Н2О (г.):
100
90
ВО
70
•3D
W
10
/
/
\
Л
\
Р, мм рт.
ст.
. 15
.8,76
20
12,99
25
18,59
30
26,83
10
20
30
Рис. 151. Растворимость в си-
системе Na2SO3—Н2О.
Насыщенный раствор бисульфита
натрия содержит при 15°—39,2, при
97,2° —49,1% Na2S2O5.
Сульфит аммония кристаллизуется
из водных растворов в форме моноги-
моногидрата (NH4JSO3-H2O. Температура
полной диссоциации сульфита аммо-
аммония при 760 мм рт. ст. 160—180°. Пиросульфитом аммония
(NH4JS2O5 насыщен раствор, содержащий при 20° 76%, при 60°
«6% NH4HSO3 (рис. 152, 153) 6-12.
Растворимость в воде MgSO3 при 23° равна 0,6%, а при содер-
содержании в растворе 400 г/л Mg(HSO3J растворимость MgSC>3 уве-
увеличивается до 1,6% B0 г/лI3. О растворимости в системах
MgSCy- MgSCy-H2O, Mg(HSO3J—MgSO4—H2O и MgSO3—
Mg(HSO3J—Н2Осм.14.
Сульфит кальция кристаллизуется из водного раствора в виде
CaSO3-0,5H2O16. Растворимость в воде при 18° равна 0,0043%.
Тиосульфат натрия Na2S2O3 • 5Н2О плавится при 48е и перехо-
переходит в дигидрат Na2SoO3 • 2Н2О. Очень хорошо растворим в воде:
в насыщенном растворе при 0° 34,4%, при 100° 72,7% Na2S2O316-
Физико-химические свойства
509
Ю W 30 id 50 W W
Рис. 152. Растворимость в системе
(NH4hSOs—NH4HSO3—H2O.
<NH4|2SO3
ВО 70 60 SO iO 30 20
— (NHJ2SO4,%
Рис. 153. Изотерма 30° системы
(NH4JSOS—NH4HSOS—(NH4JSO4—H2O. (Концентрации даны
в вес. %; цифры на изогидрических линиях — содержание
воды в насыщенном растворе в % от веса сухих солей.)
Z10
Гл. XIV. Соли сульфитного ряда
В присутствий железа17 образуются окрашенные соединения —
комплексный ион [FeS2O3l+, а также возможно [Fe (ОН) 2 • FeS2O3]2+.
Тиосульфат аммония (ЙН^гЗгОз кристаллизуется в безводной
форме; насыщенный раствор содержит при —10е 60,3%, при 60°
69,4% (NH4JS2O3. Сильно гигроскопичен (гигроскопическая точка
~64%). Концентрированные растворы выше 50° разлагаются с вы-
делением серы.
Гидросульфит (дитионит) натрия Na2S2O4 ниже 52° кристалли-
кристаллизуется в виде Na2S2O4 • 2Н2О. В форме кристаллогидрата и в вод-
водных растворах гидросульфит натрия в присутствии кислорода (воз-
(воздуха) весьма неустойчив. При 0° насыщенный водный раствор
содержит 19,3%, при 60е — 29,4% Na2S20.4. С повышением темпера-
температуры растворимость увеличивается, но точно она не установлена
вследствие разложения Na2S2O4. Добавка NaCl в количестве 25%
от веса насыщенного раствора понижает растворимость ЫагБгОф
в 3 раза. Плотность технического гидросульфита 2,3—2,4 г/см3, на-
насыпная плотность 1,2 г/см5. При нагревании до 250е гидросульфит
натрия самовоспламеняется и горит.
Ронгалит (формальдегидсульфоксилат натрия) NaHSO2*
• СН2О • 2Н2О — натриевая соль ронгалитовой кислоты, в свобод-
свободном состоянии не существующей. При 63е плавится в кристаллиза-
кристаллизационной воде, выше 120° р'азлагается. В I л холодной воды рас-
растворяется 500 г ронгалита.
ПРИМЕНЕНИЕ
Применение солей сульфитного ряда в разных отраслях техники
основано главным образом на их восстановительных свойствах.
Сульфит натрия применяют в кожевенной и в фото-кинопромыш-
фото-кинопромышленности, в фармацевтической промышленности и в медицине,
а также во многих производствах химической промышленно-
промышленности, в частности в производстве искусственного волокна, являю-
являющемся основным потребителем сульфита. Сульфит натрия исполь-
используют в качестве реагента при флотации руд цветных металлов, в
анилокрасочной и парфюмерной промышленности, при деаэрации
котловой воды 18 и в качестве добавки к цианистым электролитам
в гальванических ваннах 1в. Бисульфит натрия применяют в коже-
кенной промышленности, в пищевой промышленнести в качестве
консервирующего средства, в текстильной промышленности при
белении и крашении тканей и т. п. Белеяие с помощью бисульфит-
ных растворов (или с помощью SO2, H2SO3) основано на присоеди-
присоединении SC>2 или 5Оз~ к органическим красящим веществам с обра-
образованием бесцветных продуктов и применяется для материалов
(шерсти, шелка, соломы), разрушающихся при белении сильными
окислителями20. Бисульфит аммония используют в производстве
капролактама, в синтезе дубителей, для варки целлюлозы и др.
Применение
511
В бумажной промышленности для получения целлюлозы из
древесины по сульфитному способу применяют большие коли-
количества сульфитной кислоты — раствора бисульфита кальция
•Ca(HSO3J с избытком SO2, получаемого взаимодействием SO2 с
известняком и водой. Обычно раствор содержит 2,7-—3,5% SO2, из
них 52—65% в свободном состоянии, и 0,8—1,3% СаО. За грани-
границей наметились тенденции к замене в производстве целлюлозы би-
бисульфита кальция бисульфитом аммония, что повышает произво-
производительность завода на ~ 10% вследствие увеличения выхода цел-
целлюлозы из древесины и сокращения продолжительности варки;
кроме того, улучшается качество целлюлозы и сокращается время
ее отбелки и расход белящих материалов. Бисульфит аммония ис-
используют также в качестве источника 100%-ного SO2. Растворы
сульфита и бисульфита аммония (а также натрия) могут быть
использованы как эффективные поглотители при очистке газов от
малых количеств сероводорода21.
Пиросульфит натрия, называемый также метабисульфитом, при-
применяют для тех же целей, что и бисульфит натрия. Он является
эффективным консерватором зерна, зеленых кормов и кормовых
промышленных отходов.
Тиосульфат натрия часто неправильно называют гипосульфи-
гипосульфитом. Его применяют в фото-кинопромышленности в качестве закре-
закрепителя. В текстильной промышленности его используют для унич-
уничтожения остатков активного хлора после хлорного беления тканей
(и поэтому называют антихлором). Тиосульфат натрия используют
также в кожевенной промышленности, в медицине и ветеринарии,
в целлюлозно-бумажной, фармацевтической, химической промыш-
промышленности.
Гидросульфит натрия применяют в текстильной промышлен-
промышленности и в пищевой — для обесцвечивания сахара, патоки и др.-
Ронгалит-—сильный восстановитель. Его применяют для от-
отбелки шерстяных тканей, сахара, жиров, мыла и т. п. В текстиль-
текстильной промышленности с его помощью получают на тканях вытра-
вочные узоры. Он обесцвечивает почти все красители, не действуя
на волокно, в том числе и на хлопчатобумажное.
Требования к качеству технических продуктов, установленные
ГОСТами, приведены в табл. 38.
Сульфит, тиосульфат и пиросульфит натрия упаковывают в по-
полиэтиленовые мешки, помещаемые в фанерные, картонные бара-
барабаны или в деревянные бочки, или в многослойные бумажные
мешки; гидросульфит натрия — в герметически закрываемые же-
железные барабаны, которые для предохранения от порчи вставляют
в фанерные барабаны.
Бисульфиты натрия и аммония выпускают в виде растворов,
окрашенных примесями в светло-желтый цвет. Раствор бисульфита
натрия должен содержать (ГОСТ 902—68) не менее 22,5% SO2,
Сырье
513
связанного в виде NaHS03, т. е. не менее 36,5% NaHS03. Несвя-
Несвязанный S02 должен отсутствовать. Содержание Na2SO3 (в пере-
пересчете на S02) должно быть не больше 1,0%, соединений железа
(FeO)—не больше 0,01%- Плотность этого раствора 1,31 г/см\
Раствор бисульфита аммония содержит ~820 г/л NH4HSO4, плот-
плотность его при 20° не более 1,35 г/см3, а кислотность в пересчете на
H2SO4 должна быть не меньше 3 г/л. Бисульфиты натрия и аммо-
аммония перевозят в гуммированных цистернах, а также в стальных
цистернах и бочках.
В США выпускают22 разные сорта гидросульфитов и сульфо-
ксилатов: гидросульфит натрия, содержащий ~90% Na2S2O4 и не-
немного стабилизатора — соды или Na2S2O3, гидросульфит натрия
с добавкой соды, смесь гидросульфита натрия с Na4P2O7 в от-
отношении 2:1; формальдегид-сульфоксилат натрия (ронгалит),
Н-формальдегид-сульфоксилат цинка Zn(HSO2• CN2OJ, Н-ацет-
альдегид-сульфоксилат цинка (дектролин) ZnS02 • СН3СНО.
СЫРЬЕ
Основным сырьем для производства разнообразных солей суль-
сульфитного ряда является сернистый ангидрид, получаемый при об-
обжиге серного колчедана, серы и других видов серного сырья. Газ,
отбираемый из колчеданных печей, содержит 7—12% SO2. Значи-
Значительно более экономичным является использование отходящих га-
газов, содержащих S02, в частности выхлопных газов из производ-
производства серной кислоты контактным методом, содержащих 0,3—0,8%
SO2. Основными компонентами этих газов являются азот и кисло-
кислород. Промывка их щелочными растворами, поглощающими SO2,
позволяет обезвреживать газы, выбрасываемые в атмосферу, и од-
одновременно получать ценные продукты — сульфит, бисульфит нат-
натрия и др. Возможно использование газов с любой концентрацией
SO2, даже и очень малой, например получаемых при сжигании
топлив дымовых газов, содержащих десятые и даже сотые доли
процента SO2. Однако утилизация SO2 из отбросных газов цвет-
цветной металлургии и работающих на твердом топливе электростан-
электростанций осложнена необходимостью охлаждения и особенно очистки
этих газов. Это не всегда оказывается экономичным и, например
в Англии, имеется несколько установок, на которых очищают ды-
дымовые газы от SO2 известью или известняком, но получающиеся
растворы бисульфита кальция выбрасывают. В СССР известковый
способ был испытан для очистки дымовых газов электростанций23.
Его рентабельность зависит от того, будет ли возможно рациональ-
рациональное использование, например для строительных целей или в про-
производстве целлюлозы24, получающегося шлама, состоящего из
CaSO3 • 0,5Н2О, CaSO4-2H2O и золы и содержащего 50% влаги.
Водные суспензии некоторых сортов известняков .с размерами
17 М. Е. Позии
ТАБЛИЦА 38" w
Требования к качеству сульфитных солей (содержание компонентов в %) *°
Сульфит натрия
ПИнатпия*ИТ ГВДн°тоияФИТ Тиосульфат натрия
„ натрил натрия /тч/ч^ч-г^'г».! л i^«\
кР??"^™ес™й безводный (ГОСТ 5644-66) (ГОСТ 11683-66) (ГОСТ 246-67) <гост 244-68)
A UL.1 yUo"~~bbJ ¦
фотографический фотогра фический
____________ техни- ____________ техни- , _ , .. фотографы- техни-
технический чески* марка А марка Б сорт 1 сорт 2 ческий ческий '
А Б А Б
: _ , Р
Основное ве- >^
щество, не ' 5
менее . . . 96 * 93* 91* 97 94 92 946* 916* 90 85 99.0-100,17* 98,07* >
Примеси, ие 9
более |
нераствори-
нерастворимые в воде 0,01 0,02 0,1 0,01 0,02 0,08 0,05 0,1 0,5 1,0 0,04 0,05 «
железо. . 0,005**0,005" 0,03 ** 0,0053* 0,0053* 0,0053* 0.0053* 0,0153* 0,1 ** 0,154* 0,0023* 0.0033* |_
щелочь а с
пересчете 3
на <3
Na2CO3-10H2O 0,3 0,6 2,5 0,1 0,4 0,7 - - - - - - °
Na2S ... — — — Отсутствие — — 0*2 0,4 Отсутствие 'g
S2O|"" . . 0,01 0,01 - 0,005 0,02 0,02 - -¦ ' - - - S1
тяжелые
металлы - 0,001s* 0,002s* _____ -
мыщьяк ,— — _ — -— 0,0001 0,0001 — — — -
цинк ... - _л _ - _ _ _ _ 0,2 8,4 - -
63 1«11бЗИ8«еМяа„^%лиФад?)ГР^И.^СмИЙ? nTHOf7?b*aT «атрия повышенного катеств» по ГОСТ 5.1189-72 должен содержать
таллоа ос^дпемы^српоноппп^^'.(PM^rfj'r"^* ™ б°Лее 0Ш% Fe' W% не Растворимых в воде веществ, 0,001 % тяжелых ме-
^У^У^^ю^Ф^иЫЬ S " ВеЩеСТВ "е раСТВ°РИМЫХ в ам«°™«вь,х растворах; требуется отсутствие при-
* Na2SO3-7H2O. " В пересчете на FeO. 3* В пересчете на Fe. "* Сумма Fe2O3 н А12О3. 5* В пересчете на РЬ. 6* Общее содевж
нне SO2 в пересчете на Na2S2Os. Na2S2O3-5H2O.
514
Гл. XIV. Соли сульфитного ряда
Окисляемость сульфитных солей
515
частиц меньше 0,09 мм поглощают SO2 с такой же скоростью, как
известково-магнезиальное молоко. При размерах частиц ~0,2 мм
скорость абсорбции уменьшается на 10—15%. Доломитизирован-
ные известняки абсорбируют SO2 медленнее, так как СаСО3 • MgCO3
имеет меньшую реакционную способность по отношению к SO2,
чем СаСОз25.
Предложены и в разной степени разработаны многие методы
извлечения SO2 из газов с помощью абсорбентов — водных рас-
растворов и суспензий химически активных поглотителей, таких как:
известь (известковый метод), известняк, окись магния (магнези-
(магнезитовый метод), сульфит аммония (аммиачный метод), окись цинка
(цинковый метод), сульфит натрия и окись цинка (содо-цинковый
метод), ксилидин, фосфаты, нефелин, основной сульфат алюминия,
основной сульфат хрома и другие, а также каталитические методы,
основанные на поглощении SO2 и окислении БОз~ в SO4~ в вод-
водном растворе кислородом в присутствии ионов Mn, Fe, Си и дру-
других металлов6-26-29. Подавляющее большинство этих методов
очистки газов от SO2 связано с образованием сульфитов и бисуль-
бисульфитов, причем наиболее эффективными являются циклические ме-
методы, при которых абсорбция SO2 чередуется с регенерацией аб-
абсорбента десорбцией или другими способами. В СССР эксплуати-
эксплуатируется аммиачный метод очистки дымового газа ТЭЦ30-35. Он
основан на равновесии:
=± 2NH4HSOS
Рабочий раствор содержит сульфит и бисульфит аммония. Этот
раствор поглощает SO2 из дымовых газов при ~30°, а при нагре-
нагревании до кипения выделяет чистый SO2 с примесью водяного пара,
который легко конденсируется. Степень отгонки SO2 зависит от мо-
молярного отношения SO2: NH8 в исходном растворе, от плотности
орошения колонны и температуры кипения раствора в ее нижней
части. Из 1 м3 раствора бисульфита аммония можно выделить ма-
максимально 140 кг SO2; теоретический расход пара на отгонку будет
при этом равен 1 т на 1 т SO2. При выделении 100 кг/м3 SO2 тео-
теоретический расход пара 1,5 т/т SO2; практический расход пара
1,9—2,7 т/т SO236-40.
Аммиачный метод используется также на металлургическом за-
заводе в Трейле (Канада), где очищают 8500 mz/muh газа с содержа-
содержанием 0,75% SOo; после абсорбции в газе остается 0,1% SO2. Полу-
Получаемый раствор бисульфита аммония B40 г/л серы) перерабаты-
перерабатывают с помощью серной кислоты на сульфат аммония и 100%-ный
SO241. Этот вариант в ряде случаев можно предпочесть, так как
циклический аммиачный метод с регенерацией поглотителя имеет
существенные недостатки — окисление SO^ до SO2", а также
саморазложение раствора вследствие его термодинамической
неустойчивости с образованием SOi~H элементарной серы:
4HSOg - S3O*f + SO2." + 2Н2О
S3°6~ + Н2° = SO4~ + S2°3~ + 2H+
S2O2- + 2HSOJ = 2SO^- + 2S + H2O
Сера, реагируя с бисульфитом, дает тиосульфат, образующий
с бисульфитом еще большие количества SO4 и серы и т. д. По-
Поэтому необходимо выделять из циркулирующего раствора сульфат
аммония и выводить часть раствора из цикла во избежание на-
накопления тиосульфата, компенсируя эти потери добавкой свежего
аммиака. Эти вредные процессы усиливаются содержащимися в
газе примесями — пылью окислов металлов, соединениями селена,
мышьяка, что обусловливает необходимость тонкой очистки газа,
еще больше усложняющей и удорожающей процесс. Поэтому ра-
рационально дальнейшее усовершенствование этого метода, которое
может идти как по пути изыскания мощных ингибиторов, заторма-
затормаживающих реакции окисления, и сокращения длительности про-
процесса применением высокоинтенсивной аппаратуры 82, так и по пути
выбора оптимальных соотношений между количеством регенери-
регенерируемого и перерабатываемого на другие продукты поглотителя.
Заслуживает внимания вариант аммиачного метода с термиче-
термической диссоциацией получающегося твердого сульфата аммония для
регенерации аммиака42. Сульфат аммония разлагают при нагре-
нагревании до 200—300°. а образующийся бисульфат взаимодействует
с сульфит-бисульфитом аммония, причем регенерируется SO2:
(NH4JSO4 = NH, (г.) + NH4HSO4
3NHS + 2SO2 + 2Н2О = NH4HSO3 + (NH4JSQS
NH4HSOS + (NH4JSOS +3NH4HSO4 = 2SO2 + 3(NH4JSO4 + 2H2O
ОКИСЛЯЕМОСТЬ СУЛЬФИТНЫХ СОЛЕЙ
Вследствие легкой окисляемости сульфитных солей (в особенно-
особенности солей аммония) получаемые продукты — бисульфит, сульфит —
содержат некоторые количества сульфата. Процесс окисления
продолжается и при хранении продуктов. При хранении кри-
кристаллического сульфита в нем иногда накапливается до 20% суль-
сульфата, что делает продукт нестандартным. Крупнокристаллический
сУльфит лучше противостоит окисляющему влиянию атмосферы
вследствие меньшей поверхности кристаллов, приходящейся на еди-
единицу веса. Его окисление усиливается во влажной атмосфере и под
Действием света, а также в случае использования в производстве
^од загрязнений ионами металлов6-43-45. В сухой атмосфере
7HO теряет кристаллизационную воду и переходит в
516
Гл. XIV. Соли сульфитного ряда
безводный Na2SO3, но не окисляется. На этом основании рекомен-
рекомендуется изготовлять безводный продукт и хранить его в условиях,
препятствующих гидратации46.
В растворе окислительные процессы происходят, по-видимому,
в результате цепной реакции. Начальными центрами цепи, воз-
возможно, являются одновалентные ионы SOJ, образующиеся из
ионов SOl" в результате потери ими отрицательного заряда (на-
(например, под действием ультрафиолетовых лучей). При этом об-
образуется гипотетическая монотионовая кислота HSO3. В дальней-
дальнейшем цепная реакция идет по следующей схеме:
|~ + ОН" + 2Н+
HSO3 + O2 + H2O +
OH + SO^ + H+ = HSO3 + он~ и т- Д-
, Обрыв цепи в отсутствие кислорода происходит в результате
димеризации 2SO3 = S2O6~— образуется стабильная дитионовая
кислота.
Образование сульфата в бисульфите в процессе его изготовле-
изготовления тем меньше, чем больше концентрация SO2 в газовой смеси,
так как на количественную сторону окисления влияет главным об-
образом длительность процесса. Наиболее сильное окисление проис-
происходит при рН раствора 7—9, т. е. при регенерации раствора и уда-
удалении SO2. Этому способствует также повышение температуры.
В диапазоне 25—100° скорость разложения возрастает в 25 раз.
Значение рН для чистого раствора бисульфита аммония по разным
ранным находится в пределах 2,2—4,7; вероятное значение 3,547-48.
Об окислении сульфитов кальция и магния и бисульфита магния
см. «¦50.
Окисление сульфита кислородом при рН = 5 — 8 протекает, как
реакция второго порядка, а при меньших и больших значениях
рН — как реакция первого порядка. В присутствии сильных окис-
окислителей, например двуокиси марганца, окисление сульфита значи-
значительно ускоряется, причем максимум скорости наблюдается в кис-
кислой среде, при рН = 4б1~54. При поглощении кислорода раствором
сульфит-бисульфита аммония окисляются и SOl н НЭОз, но пре-
преимущественно бисульфит. Несмотря на это, содержание его в рас-
растворе не изменяется, а содержание сульфита уменьшается. Это
объясняется тем, что образующийся при окислении HSOJ ион HSOZ,
являясь более сильной кислотой, реагирует с сульфитом, вновь
образуя бисульфит, так что из раствора исчезают только ионы
БОз~- В диапазоне изменения содержания бисульфита 63—100%
от суммы сульфита и бисульфита скорость окисления раствора
возрастает приблизительно в 6 раз. Когда в растворе сверх бисуль-
бисульфита содержится свободная сернистая кислота, т. е. значение рН
раствора понижено, скорость окисления резко падает до нуля. Ско-
Окисляемость сульфитных солей
517
рость поглощения кислорода раствором снльфит-бисульфита аммо-
аммония [Go,, г/(л*2 •'«)], эквивалентная скорости образования сульфата
аммония, может быть вычислена по формуле
где S —содержание БОг в сульфите и бисульфите, г-мол/л;
С —содержание NHB в сульфите и бисульфите в пределах
7,5—3 г-мол/л, при постоянном общем содержании NH3 в
растворе в виде сульфита, бисульфита, сульфата, тиосуль-
тиосульфата и тритиоиата, равном 8 г-мол/л.
Для С<3 г-мол/л:
Эти формулы выведены55 для поверхностной абсорбции кисло-
кислорода сравнительно концентрированными растворами и отличаются
о 2 « в 8
Кониептрашю NHSeblt ,г-иол/л
Рис. 154, Изохроны скоростей поверхностного
поглощения кислорода воздуха растворами
сульфит-бисульфита аммония при 20—22° и при
начальном CJC 07808
от кинетических закономерностей, наблюдаемых для разбавленных
растворов в иных гидродинамических условиях569. По мере уве-
увеличения общей концентрации солей скорость поглощения кисло-
кислорода вначале возрастает, затем убывает (рис. 154). Максимум
наблюдается при общей концентрации солей 2—3 г-мол/л, но сме-
смещается в сторону больших значений этой концентрации с увеличе-
увеличением относительного количества сульфата (изохроны 2 и 3 для
больших значений времени располагаются правее, чем /). Наличие
максимума, очевидно, объясняется тем, что в разбавленных рас-
растворах процесс окисления лимитируется скоростью реакции в рас-
растворе, а в концентрированных — скоростью абсорбции кислорода
из воздуха. С увеличением парциального давления кислорода ско-
Рость окисления возрастает60, но абсолютный прирост концентра-
концентрации SO4~ в растворе зависит от того, какая стадия лимитирует
общую скорость процесса. Так, в пенном аппарате скорость
518
Гл. XIV. Соли сульфитного ряда
поглощения SO2 в 10—15 раз больше, чем в насадочном скруббере,
а скорость поглощения кислорода — в 2—3 раза. В итоге степень
окисления раствора в пенном аппарате в 5 раз меньше, чем в на-
насадочном скруббере61.
В результате обобщения производственных данных предло-
предложена 62 эмпирическая формула для определения скорости окисле-
окисления растворов сульфит-бисульфита аммония, натрия, кальция, маг-
магния в процессе извлечения SO2 из газов в скрубберах с насадкой
(хордовой, реечной или из керамических колец):
°*<г'щ>(т
//Здесь Go2 — скорость поглощения кислорода, г/(м2-ч), отнесенная
к 1 м2 поверхности насадки; t — температура, °С; Q — плотность
орошения, м3/(м2-ч); у— плотность раствора, кг/м3; ц — вязкость
раствора, кг-сек/м2; S/C — среднее молярное отношение содержа-
содержания SOo в жидкой фазе к содержанию связанного с ним основания.
Это уравнение получено для следующих пределов изменения
условий: Q — от 52 до 2, t — от 50 до 25°, у — от 1 до 1,3, ц — от
1,1 до 3, S/C — от 0,7 до 0,9, скорости газа в полном сечении скруб-
скруббера— от 0,3 до 3 м/сек, исходной концентрации SO2 в газе — от
0,06 до 0,35 объемн.%, содержание кислорода в газе-—от 8 до 19
объемн.%, общего содержания растворенного SO2 (в виде суль-
фит-бисульфитных солей) —от 0,016 до 7 моль/л, рН раствора —
от 4,0 до 6,0.
Окислительные процессы могут быть подавлены введением в
производственные растворы отрицательных катализаторов — ве-
веществ, каталитически препятствующих окислению, антиокислите-
антиокислителей или ингибиторов. Механизм ингибиторного действия объяс-
объясняют образованием комплексов из молекул реагента и ингибитора.
Так, при ингибировании спиртами процесса окисления SO2 возду-
воздухом образуются комплексные соединения между SO2 и спиртом.
По эффективности ингибиторного действия фенолов на реакцию
окисления слабых водных растворов NaHSO3 при 40—60° их можно
расположить в следующий ряд: гидрохинон > пирогаллол > р-наф-
тол > а-нафтол > пирокатехин > резорцин > фенол > флороглю-
цин. Особенно эффективными антиокислителями являются диме-
тилпарафенилендиамин и парафенилендиамин — они предохраняют
от окисления даже при концентрации 1 :200 0006!-65. Следы окис-
окислов азота сильно ухудшают действие ингибиторов 66.
Накопление сульфата в производственных растворах при обра-
обработке их сернистым газом происходит не только за счет их окисле-
окисления, но и вследствие поглощения БОз, обычно содержащегося в
сернистом газе.
Давление SO2 над растворами поглотителей
519
ДАВЛЕНИЕ SO2 НАД РАСТВОРАМИ ПОГЛОТИТЕЛЕЙ
В основе образования соединений сульфитного ряда лежит про-
процесс растворения SO2 в воде. В системе Н2О—SO2 устанавли-
устанавливаются следующие равновесия:
H2O + SO2 ^=? H2SO3 ^r
Для первого из этих равновесий давление SO2 над раствором
где Н — константа Генри;
[H2SO3J — концентрация недиссоциированных молекул H2SO3.
Для двух других равновесий
[h+][hso3|
[H2SO3]
= 1,3 . Iff"
j
[HSO3]
3,11 . 1(Г7
(КГ"-3,36- И
°
1). Приводят также67 значение
6,28 • 10~8. Так как общая концентрация SO2 в растворе
SO] [HSO] [SOI"]
Jill ^^ J ЙЛ ^r Л %^ » ^ ЪЛ Ik Л L^^4& ^^ ^^
S = [H2SO3] + [HSO3] + [SOI"],
то
. _„с [h+F
Pso2
у
у
У
fit
*у
'//
¦^Лг,
21
1
У*
S
И"
у
—ч
= HS-
Следовательно, равновесное §.*
давление р50г над раствором I'
(рис. 155) * зависит от общей
концентрации S02 в растворе S
и от температуры, определяю-
определяющей величины Н, Ki и /Си, а
также от кислотности раство- ЩЗО
ра — чем меньше рН, тем дав- 20
ление SO2 больше. Поэтому в
тех случаях, когда поглощение
SO2 из газов сопровождается
последующей регенерацией по-
поглотительного раствора нагре-
нагреванием, необходимо выбирать поглотительные растворы с такими
значениями рН, для которых равновесное давление SO2 мало при
15—25° и резко повышается при нагревании до 60—100°. Это рас-
растворы солей слабых кислот или слабых оснований, обладающие
буферными свойствами — сульфит-бисульфитов аммония и натрия,
фосфатов натрия, основных сульфатов алюминия и хрома и др.
* См. также •»¦и.
20 30 40 50 60 70 60 90100
Температура,"С
Рис. 155. Давление SOS иад водными
растворами.
520
Гл. XIV. Соли сульфитного ряда
В системе SO2—NH3—H2O равновесие устанавливается в ре-
результате следующих реакций:
SO2(r.) + H2O -^=± SO2-H2O
NH3(r.) + H2O :
NH3 • Н2О
NHj+OH"
Н2О
При 25° в системе в заметных количествах присутствуют сле-
сле67
дующие ионы и молекулы
67
При рН < 4,2 S02.H2O, HSO-, NH+
4,2-7,0 HSOJf, SO*", NH+
» »
» » 7,0-9,5
» » > 9,5
HSO3
-, NH+, NH3.H2O
^, NH3.H2O, OH~
На рис. 156 и 157 показана изменение давлений SO2 и NH3, от-
отнесенных к единице концентрации NH3, над растворами суль-
1.0
^so2
Рис 156. Давления SO2 и NHS (в мм рт. ст.) иад раство-
растворами (NH4JSOS и NH4HSOS, отнесенные к единице концен-
концентрации NH3 в растворе.
(Сплошные линии — ps0^. пунктирные — PNHS-)
фит-бисульфита аммония. Здесь Cso2 — общее содержание SO2 в
растворе сульфита и бисульфита и Cnhs — содержание NH3 в
г-мол/\00г-мол воды. Для раствора сульфита аммония отношение
Cso2/Cnhs = 0.5, а для бисульфита Cso2/Cnh3 = 1- Парциальные да-
давления SO2 и NH3 над раствором сульфит-бисульфита аммония
Давление SO2 над растворами поглотителей
могут быть вычислены по формулам:
Psot'
BCSO2-CNHsJ О3-51"
S4S6
CNHS ~ CSO2
мм рт. ст.
2CSo2-(
'NHs
мм рт. ст.
где T — абсолютная температура6-70-78.
Эти приближенные формулы выведены71 из равновесий
^± 2HSOg
и могут применяться для растворов любых концентраций, имею-
имеющих практическое значение, — погрешность этих формул обычно
меньше 10%, но сильно возрастает при больших значениях ps0 .
Давление SO2 (в мм рт. ст.) над растворами сульфит-бисуль-
сульфит-бисульфита аммония может быть также вычислено по эмпирическим фор-
формулам ""
72.
»gPso2 = 20.54l'g-
JNHs
Сспп
2443
2697
+ 9,997 при
- 0,87 -
В присутствии сульфата в растворе сульфит-бисульфита аммо-
аммония, парциальное давление SO2 над раствором возрастает73.0 дав-
давлении §Ог над растворами сульфит-бисульфита магния в присут-
присутствии MgSO4 см.74-75.
На рис. 158 приведены изотермы равновесного давления SO2
над раствором Na2SO3+NaHSO3. На оси абсцисс отложена степень
превращения Na2SC>3 в NaHSO3 по мере насыщения раствора SO2,
т. е. величина Cso2/CNa- Значения ps02 (в мм рт. ст.) отнесены к
единице концентрации Na(CNaHCso2B г-мол/ЮО г-мол воды) и
должны умножаться на CNa. Диаграмма построена по данным для
CNa = 8, что соответствует ~20% SO2 при насыщении раствора га-
газом до образования NaHSO3.
На рис. 159 приведена зависимость давления SO2 над раствора-
растворами бисульфита кальция при 25°, которые могут существовать только
в области между линиями АВ и АС, т. е. в присутствии свободной
SO2. Линия АВ — растворимость SO2 в воде, линия АС — раство-
растворимость сульфита кальция. Давления SO2 над растворами
^a(HSO3J, насыщенными CaSO3, показаны на рис. 16079-80. В от-
отличие от бисульфита кальция растворы бисульфита магния могут
9
м-
«а-
Оз~
.(Л S
К го
8 КЗ
Бисульфит и сульфит натрия
523
существовать в отсутствие избытка SO213> 81; в этом отношении
растворы бисульфита цинка подобны растворам бисульфита каль-
кальция ^ 83. О давлении SO2 над растворами основного сульфата алю-
А
у
/
//
//
из
%
/
у
/
/
<,
k/
/
/
/
{
#/
/
/
-3
у
ч
/
У
/
у
с
/
/
//
50
Содержание Sty в растворе ,г/ЮОг
Рис. 160. Давление SO2 над растворами Ca(HSO3J,
насыщенными CaSO3.
Линии одинакового содержания Са в растворе:
/ — 0,62; г-1,24; 3-1,86; 4-2,48 г/100 г воды.
миния, основного сульфата хрома, фосфатов натрия, ксилидина
см.6-78. О составе водных растворов SO2 при небольших концент-
концентрациях и различных рН см.84.
БИСУЛЬФИТ И СУЛЬФИТ НАТРИЯ
Получение бисульфита и сульфита натрия из сернистого газа
и щелочей
Теория процесса
При поглощении SO2 раствором едкого натра в зависимости от
степени его насыщения образуется сульфит или бисульфит натрия.
Обычно поглощение сернистого газа производят более дешевым
раствором соды. При этом вначале образуются бикарбонат и суль-
сульфит натрия по реакции:
2Na2CO3 + SO2 + H2O = 2Na HCO3 + Na2SO3 A)
Затем происходит постепенное разрушение бикарбоната, уда-
удаляется углекислый газ и получается раствор сульфита натрия:
B)
При последующем поглощении SO2 раствором сульфита обра-
образуется бисульфит:
is + H2O=2NaHSO4
C)
B24
Гл. XIV. Соли сульфитного ряда
26
Степень нейтрализации, %
50 73 Si
-г
Суммарными уравнениями реакций образования сульфита и би- -jj
сульфита натрия будут:
Na2CO3 + SO2 = Na2SO3 + СО2
Na2CO3 + 2SO2 + H2O = 2NaHSO3 + CO2
Изменение состава содового раствора в процессе поглощения
им SO28S показано на рис. 161 [степень нейтрализации (в %) для
исходного раствора соды рав-
равна нулю, для готового раствора
бисульфита—100]. Как видно
из диаграммы, в растворе про-
происходит накопление бикарбо-
бикарбоната, концентрация которого
достигает максимума, когда
поглощено около 25% от об-
общего количества SO2, требую-
требующегося для превращения соды
в бисульфит натрия. Реакция
A) заканчивается раньше, чем
реакция B). После исчезнове-
исчезновения всей соды содержание би-
бикарбоната натрия в растворе
еще очень велико и идет посте-
постепенное его снижение за счет
образования сульфита. Когда
поглощено 50% от общего ко-
количества SC>2, реакция B) за-
заканчивается,— в растворе име-
имеется только сульфит натрия.
1
Бисульфит и сульфит натрия
525
0 0,2 0,4 0,6 0,8 ф
ДпинцСысота) аппарата по нопраблению
дбиженип жидкости, доли единицы
Рис. 161. Изменение состава содового
раствора в процессе поглощения SO2:
i-Na2CO3; 2-NaHCO3; 3 - SO2 сульфитный;
4—SO2 бисульфитлый.
После этого начинается обра-
образование бисульфита натрия по
реакции C) — концентрация
сульфита падает.
Если абсорбция SO2 осуще-
осуществляется в башне с насадкой,
орошаемой содовым раство-
раствором, то реакция A) заканчивается в зоне, соответствующей при-
приблизительно 30%, а реакция B) —40% верхнего объема насадки.
Нижние 60% объема насадки соответствуют зоне образования би-
бисульфита по реакции C). Количество SC>2, поглощаемое при обра-
образовании сульфита в 40% верхнего объема башни, равно количеству
SO2, поглощаемому при образовании бисульфита в 60% нижнего
объема башни. Следовательно, скорость поглощения SC>2 изме-
изменяется по высоте аппарата. Скорость увеличения содержания об-
общего SO2 (сульфитного и бисульфитного) в растворе вначале уве-
увеличивается, достигает максимума, когда в растворе содержится
только Na2SO3, а к концу процесса уменьшается вследствие уве-
увеличения равновесного давления SO2 над раствором по мер|
его бисульфитизации, т. е. перехода сульфита в бисульфит
(рис. 158).
При насыщении раствора сернистым газом его плотность уве-
увеличивается за счет поглощения SO2 и за счет испарения из рас-
раствора воды, так как при незначительном содержании SO2 в газе,
поступающем на абсорбцию, через аппарат проходит большой
объем газа, уносящего много влаги. Так, при работе на 7—8%-ном
газе, при температуре раствора 50—70°, его плотность увеличи-
увеличивается от 1,24—1,25 г/'см3 для исходного раствора соды и до 1,34—
1,36 г/см8 для раствора бисульфита. При 3%-ной концентрации SO2
в газе, поступающем на абсорбцию, плотность раствора возрастает
от 1,19 до 1,36 г/см3. Чем ниже концентрация SO2 в исходном газе,
тем меньше должна быть концентрация исходного раствора соды
для получения раствора бисульфита стандартной концентрации.
При концентрации соды в исходном растворе 20—23% максималь-
максимальная концентрация бикарбоната достигает 9—11%. максимальная
концентрация сульфита 24—28% A2—14% сульфитного SO2). Во
избежание выпадения в абсорбционных аппаратах осадков
NaHCO3 и Na2SO3 при концентрации соды в исходном растворе
около 30% и при работе на 7—8%-ном газе, необходимо поддер-
поддерживать температуру раствора не ниже 60° по крайней мере в пер-
первой стадии процесса или в пер,вой по ходу раствора зоне аппарата.
При абсорбции SO2 из отбросных газов малой концентрации,
ввиду сильного возрастания при этом количества испаряющейся
воды и применения содового раствора меньшей концентрации, в
абсорберах может поддерживаться температура ниже 606. При со-
соблюдении режима процесса абсорбции можно получать готовые
растворы бисульфита, более концентрированные (около 27% SO2),
чем предусмотренные стандартом B2,5% SO2), что дает экономию
при транспорте, хранении и проч.
Получение бисульфита натрия
Получение бисульфита натрия из отходящих газов контактных
сернокислотных заводов производят поглощением SO2 из этих газов
в башнях с насадкой. Материал башен — свинец, пластмасса или
Дерево; в последнем случае применяют хордовую насадку. Обычно
газ проходит последовательно через две башни, в каждой из ко-
которых с помощью кислотоупорных центробежных насосов осущест-
осуществляется циркуляция раствора. Из первой по ходу газа башни выво-
выводят часть готового раствора бисульфита, а убыль раствора попол-
пополняют раствором из второй башни. Свежий раствор соды добавляют
к раствору, циркулирующему во второй башне. Таким образом,
баш орошаются не раствором соды, а сульфит-бисульфитным
526
Гл. XIV. Соли сульфитного ряба
Бисульфит и сульфит натрия
527
щелоком, что предотвращает выделение в твердую фазу бикарбо-
бикарбоната натрия.
На каждую тонну моногидрата серной кислоты, вырабатываем
мой на сернокислотном заводе, можно получить 0,15—0,18 г стан-
стандартного раствора бисульфита натрия. Впрочем, его количество
тем меньше, чем лучше работает контактный завод (чем выше сте-
степень контактирования).
При использовании газов ватержакетных печей6 газы, имею-
имеющие температуру ~250°, предварительно промывают от пыли и
охлаждают до 30° водой в футерованной башне с насадкой, затем
пропускают через брызгоуловитель — деревянную башню с насад-
насадкой, после чего направляют в деревянную поглотительную башню с
хордовой насадкой, орошаемую вначале 22—23% раствором соды
при 35—40°. Получается раствор, содержащий 23% SO2 C8%
NaHSO3). Количество его 2,5—3 г в сутки при размерах башни:
диаметр 1,3 м, высота 8 м, поверхность насадки 380 м2. Продолжи-
Продолжительность одного цикла около 6 ч, причем в течение первых 4 ч SO2
поглощается практически полностью, а затем, после того как со-
содержание SO2 в растворе достигает ~16%, появляется проскок
SO2, резко увеличивающийся. Более рациональна описанная выше
непрерывная схема производства. По-видимому, вследствие недо-
недостаточной очистки газа от пыли на описанной установке около 10%
от пропускаемого SO2 окисляется до Na2SO4. При получении би-
бисульфита из газа колчеданных печей, содержащего, кроме SO2, де-
десятые доли процента SO3, горячий печной газ, после очистки от
огарковой пыли в электрофильтрах необходимо промывать серной
кислотой для извлечения SO3. При работе по такой схеме на 1 г
стандартного раствора бисульфита расходуют: 208 кг соды A00%).
240 кг SO2) 20 кг серной кислоты A00%), 21 квт-ч электроэнергии,
0,18 мгкал пара, 18 ж3 воды86.
Бисульфит натрия может быть получен с помощью ионообмен-
ников 87, причем этот процесс сочетается с очисткой газа от H2S.
Ионообменная смола должна содержать активный водород и яв-
являться кислотой более слабой, чем сернистая, но более сильной,
чем H2S. Сероводород извлекают из газа раствором соды и полу-
полученный раствор гидросульфида натрия обрабатывают смолой, при-
причем выделяется концентрированный сероводород, который окис-
окисляют до SO2. Взаимодействием последнего с образовавшейся на-
натриевой солью ионообменной смолы получают раствор бисульфита
при одновременной регенерации смолы:
2H2S + Na2CO3 = 2NaHS + Н2О + СО2
H2S + 1' /2О2 = SO2 + Н2О
RNa + SO2 + H2O = NaHSOs + RH
Семиводный сульфит натрия
Нейтрализация раствора соды сернистым газом приводит к об-
образованию сравнительно разбавленных растворов сульфита. Это
усложняет процесс кристаллизации. Поэтому кристаллический
Na2SO4 • 7Н2О получают из концентрированного раствора, обра-
образующегося при нейтрализации содой раствора бисульфита натрия,
содержащего 45—47% NaHSO3:
2NaHSO3 + Na2CO3 = 2Na2SO3 + H2O + CO2
Во избежание выделения NaHCO3 реакцию ведут в кислой сре-
среде, приливая раствор соды к раствору бисульфита. Концентрации
растворов должны быть такими, чтобы получаемый раствор суль-
сульфита был близок к насыщению при 38—40°. Приливание раствора
соды к бисульфиту ведут медленно вследствие сильного вспенивания
массы выделяющейся двуокисью углерода. Теплоту нейтрализации
отводят с помощью находящегося в реакторе водяного змеевика,
поддерживая 38—40°. После нейтрализации раствор должен иметь
нейтральную или слабокислую реакцию. Он отстаивается и на-
направляется в кристаллизаторы. При кристаллизации сульфита в
барабанных кристаллизаторах с воздушным охлаждением, имею-
имеющих длину 12—20 м, диаметр 0,75—0,9 м и вращающихся со ско-
скоростью 8—12 об/мин, съем кристаллов с 1 мв полного объема кри-
кристаллизатора составляет 0,5—0,75 т в сутки, в зависимости от тем-
температуры воздуха и температуры и концентрации поступающего на
кристаллизацию раствора. (Для этого процесса очевидно рацио-
рациональнее использовать более пооизводительные вакуум-кристалли-
вакуум-кристаллизаторы.) Кристаллы Na2SO3 • 7Н2О отжимают иа центрифуге, а ма-
маточный сульфитный раствор возвращают на растворение соды или
направляют на поглощение SO2 для получения раствора бисуль-
бисульфита. На 1 т семиводного сульфита натрия расходуют: 0,26—0,27 т
S(\ и 0,44—0,46 г кальцинированной соды.
Запатентован способ непосредственного получения твердого
сульфита натрия обработкой концентрированного раствора би-
бисульфита твердой содой при 50°, после чего раствор выливают в
форму, в которой он затвердевает; продукт содержит 52,6% Na2SO3,
44 1% Н2О и 2,3% Na2SO488.
Некоторая экономия соды в производстве сульфита может быть
Достигнута при переработке бисульфита с помощью СаСО3 — мела
или известняка. Этот процесс, возможно, представит интерес для
очистки отработанных и дымовых газов с частичной регенерацией
SO2 или_ выпуском двух продуктов — сульфитов натрия и кальция.
По этому способу89 вначале SO2 поглощают из газов раствором
сульфита и бисульфита натрия (аммония). Затем раствор переме-
перемешивают' с известняковой пульпой (СаСО3: Н2О = 5,2) при
40—70°:
2NaHSO3 + СаСО3 + Н2О = Na2SO3 + CaSO3 • 2Н2О + СО2
528
Гл. XIV. Соли сульфитного ряда
Значительные количества сульфита натрия получаются в каче-
качестве отхода при производстве фенола. Фенол изготовляют из бен-
золсульфокислоты, которую вначале нейтрализуют раствором
сульфита натрия. Полученный 50% раствор бензолсульфокислсго
натрия сплавляют с едким натром при 300—315° в стальных кот-
котлах с огневым обогревом, причем образуется фенолят натрия и
сульфит натрия. Плав гасится водой и из него выщелачивается
фенолят натрия, а большая часть менее растворимого сульфита
натрия остается в твердом виде и отфильтровывается. Раствор фе-
фенолята натрия нейтрализуют сернистым газом, который выделяется
при обработке сульфокислоты сульфитом. При этом образуются
фенол и раствор сульфита натрия, который вновь возвращают на
нейтрализацию сульфокислоты. Таким образом, весь процесс вы-
выражается реакциями:
Бисульфит и сульфит натрия
529
2C6H5SO3H + Na2SO3 =
2C6H;SO3Na + 4NaOH =
2C6H6ONa + SO2 + H2O =
¦ 2CeH5SO3Na + SO2 + H2O
¦¦ 2C6HsONa + 2Na2SO3 + 2H2O
2C6H5OH + Na2SO3
2C6H5SO3H + 4NaOH = 2C6H5OH + 2Na2SO3 + 2H2O
Сульфит натрия, отфильтрованный от раствора фенолята нат-
натрия, выпускают в качестве товарного технического продукта.
Сульфит натрия можно получить, насыщая сероводородом сус-
суспензию Fe(OHK в 5% растворе Na2S:
6H2S + 6Fe(OHK = 6FeS + 15H2O + l,5O2
Na2S + l,5O2 = Na2SO,
Процесс следует вести при 10—20°, так как в горячем растворе
выделяется сера:
3Na2S + 2Fe(OH)s = 2FeS + S + 6NaOH
Отделенное от раствора сульфита сернистое железо может
быть регенерировано в H2S и Fe(OHK. Из 1 г Na2S A00%) этим
методом можно получить 1.16 г Na2SO3 или 2,32 т Na2SO3-7H2O90.
Окисление сульфида натрия в сульфит может быть осуществлено
и воздухом. Для этого смесь концентрированного раствора Na2S с
инертным компонентом (например, продуктом), представляющую
собой хлопьевидную рыхлую массу, продувают при перемешивании
влажным воздухом с температурой 120—150°91.
Запатентован способ получения сульфита натрия из отбросного
сульфидного щелока целлюлозных заводов; содержащийся в ще-
щелоке Na2S окисляют кислородом (воздухом) при 156—180° и дав-
давлении 5,5—8,5 ат92.
Безводный сульфит натрия
Безводный сульфит натрия весьма устойчив против окисления.
При хранении в течение нескольких месяцев содержание Na2SO3 в
продукте практически не изменяется. Кроме того, егоперевозка де-
дешевле, чем Na2SO3 • 7Н2О, так как он содержит больше SO2 и
имеет больший насыпной вес A,2—1,3, вместо 0,8—0,9 г/ж3 для
Na2SO3-7H2O).
Безводный сульфит натрия можно получить кристаллизацией из
концентрированного раствора при 95—100° или плавлением
Na2SO4 • 7Н2О. В последнем случае получается минимальное коли-
количество маточных растворов. При плавлении Na2SO3 • 7Н2О полу-
получаемую кристаллическую пульпу выдерживают в обогреваемом
паром реакторе в течение 30 мин для образования более крупных
кристаллов, затем повышают температуру до 75—80° (так как при
повышении температуры растворимость Na2SO3 уменьшается) и
выпускают пульпу на центрифугу. Отфугованный продукт высуши-
высушивают при 75—80°. Этот метод44, однако, не используется в про-
промышленности вследствие его громоздкости (двойная кристаллиза-
кристаллизация, малая производительность реактора, периодичность процесса).
Поэтому безводный Na2SO3 получают преимущественно кристалли-
кристаллизацией из насыщенного раствора, приготовленного при темпера-
температуре ~35°, соответствующей наибольшей растворимости соли.
Раствор очищают от железа, нейтрализуя его кислотность содой и
подщелачивая едким натром до содержания 25—28 г/л NaOH и
отделяя шлам после отстаивания. Затем раствор нагревают при
перемешивании в течение 1 ч до 95—100°, причем из него кристал-
кристаллизуется около '/в части содержащегося в нем Na2SO3. Кристалли-
Кристаллическую пульпу разделяют на нутч-фильтре, который служит и су-
сушилкой. Маточный раствор возвращают в цикл (на растворение
соды), а кристаллы, после промывки на фильтре спиртом, сушат
воздухом, нагретым в паровом калорифере до 80—90°. При сушке
300 кг сульфита в течение 1,5 ч выгружаемая соль содержит 0,5—
1% влаги. Ее подвергают дополнительному досушиванию в сушил-
сушилке полочного типа. Спирт применяют для ускорения сушки с це-
целью уменьшения окисления продукта, но при добавке к нему анти-
антиокислителей (стр. 518) от промывки спиртом можно отказаться.
На производство 1 т безводного сульфита натрия этим способом
расходуется: 3,76 т* бисульфита натрия C6,6%), 0,133 г кальци-
кальцинированной соды A00%), 0,185 т** каустической соды A00%),
76 л этилового спирта (96%), 190 квт-ч электроэнергии, 2 мгкал
пара, 300 м3 воды86.
* Эта величина, приведенная по практическим заводским данным, в 2 раза
больше теоретической, что объясняется, по-видимому, неправильным учетом
Количества маточного раствора, возвращаемого на растворение соды.
** Этот расход может быть снижен путем частичной замены каустической
соды кальцинированной.
530
Гл. XIV. Соли сульфитного ряда
Бисульфит и сульфит натрия
531
Запатентован 93 способ сушки безводного сульфита натрия воз-
воздухом в псевдоожиженном слое при температуре ниже 120°. В свя-
связи с быстрым высыханием продукта значительного образования
Na2SO4 не происходит.
Гипрохимом разработана типовая схема производства сульфита
натрия из отработанных газов контактных сернокислотных систем
(рис. 162). SO2 из газов извлекается в башне циркулирующим
Рис. 162. Схема производства безводного сульфита натрия:
1 — сульфитная башня; 2 - циклон-брызгоуловитель; 3 — сборник дляяульфит-бисуль-
фитного раствора; 4 — распределительный бачок для сульфит-бйсульфнтйЬго ра-
раствора; 5 — расходный бак для сульфит-бисульфитнрго раствора! б — питательный
бак для сульфит-содового раствора; 7 —расходный бак для сульфит-содового ра-
раствора; 8 — нейтрализатор; 9 — обезвоживатель; 10 — центрифуга; Ц, 13 — бункеры;
12 — автоматические весы; 14 — тара; 15 — растворитель соды; 16 — фильтрпресс;
17 — сборник фильтрованного сульфит-содового раствора; 18 — колонна разложения;
19 — сборник раствора сульфата натрия; 20 — насос.
сульфит-бисульфитным раствором, избыток которого выводится из
цикла и нейтрализуется сульфитно-содовым раствором при 33° в
аппарате с мешалкой и паровым змеевиком. Нейтрализованный
сульфитный раствор нагревается в обезвоживателе — резервуаре с
мешалкой, снабженном паровой рубашкой, до 90—95°. (Образую-
(Образующаяся сульфитная пульпа передается на центрифугу, с которой
отжатые кристаллы с влажностью 3—6% выгружаются в бункер,
затем поступают на автоматические весы и тарируются. Маточный
раствор направляют на растворение соды. В связи с накоплением
в нем сульфата натрия, часть его выводится из цикла и заменяется
соответствующим количеством воды. Выводимый раствор обраба-
обрабатывают в колонне разложения серной кислотой; выделяющийся при
этом SO2 отводят в контактную систему, а образующийся раствор,
содержащий ~25% Na2SO4 — в кремнефтористое отделение супер-
суперфосфатного цеха, где он используется взамен N а С1 (стр. 1142). Сте-
Степень улавливания SO2 ~90%; доля SO2, удаляемая в виде отво-
отводимого маточного раствора, достигает 20—23%. Переработку полу-
получаемого при абсорбции SC>2 раствора, т. е. нейтрализацию, обез-
обезвоживание и отжим кристаллов, можно осуществлять в аппаратах
периодического действия и небольшой емкости при малой мощ-
мощности установок B—4 т\'сутки). При производительности более
10 т/сутки сульфита целесообразно осуществление процесса по не-
непрерывной схеме.
Раствор сульфита натрия для производства безводной (и семи-
водной) соли можно получать нейтрализацией бисульфита извест-
известковым молоком94:
2NaHSO3+ Са(ОНJ = Na2SO3 + CaSO3 • 2Н2О
После отделения осадка CaSO3 • 2Н2О отстаиванием или фильт-
фильтрацией получается раствор, содержащий до 23% Na2SO3. Во избе-
избежание потерь Na2SO3 со шламом температура раствора до его от-
отделения должна быть в пределах 25—70°, в которых растворимость
Na2SO3 относительно высока.
Безводный Na2SO3 выделяется при смешении растворов каусти-
каустической соды с концентрацией больше 35% NaOH и бисульфита
с концентрацией больше 35% NaHSO3 в молярном соотношении
NaOH : NaHSO3>2 : 1 при температуре выше 70°. Выход продукта
в расчете на бисульфит натрия превышает 80%. Если содержание
железа в растворах не больше 25 мг/л, то в образующемся осадке
сульфита содержится 2—Змг Fe на 100 г соли. Такое содержание
железа не вызывает окрашивания продукта и не препятствует его
использованию в фототехнике ss.
Безводный сульфит натрия можно получать «сухим» способом,
насыщая твердую соду сернистым газом. Реакция /
Na2CO3 + SO2 = Na2CO3 + СО2
идет лишь в присутствии влаги, поэтому газ должен быть увлаж-
увлажнен (~8% Н2О). Наилучшим образом процесс осуществляется в
аппарате с кипящим слоем при 120—170°. При более низких
температурах образуется пиросульфит натрия (стр. 537), а выше
200° — главным образом сульфат натрия и сера (в результате раз-
разложения пиросульфита)
2Na2S2O5 = 2Na2SO4 + SO2 + S
В интервале 120—170° пиросульфит разлагается по реакции
Na2S2O5 = Na2SO3 + SO2
v
и получаемый продукт содержит больше 90% Na2SO3 и незначи-
незначительное количество Na2SO4 (до 3%) и Na2CO3 (до 8%)96'97,
532
Гл. XIV. Соли сульфитного ряда
Получение бисульфита и сульфита натрия взаимодействием SO2
с раствором Na2SO4 или NaCl
Делались попытки замены содовых продуктов в производстве
бисульфита натрия сульфатом натрия или хлористым натрием.
Взаимодействие SO2 с Na2SO4 идет в присутствии извести. При
этом вначале образуется суспензия мелкокристаллического осадка
сульфита кальция
Са(ОНJ + SO2 = CaSO3 + Н2О
который при дальнейшем поглощении SO2 переходит в раствори-
растворимый бисульфит кальция, вступающий в реакцию с NajSC^ с выде-
выделением в осадок гипса. Общее уравнение процесса:
Na2SO4 + Са(ОНJ + 2SO2 + 2Н2О = 2NaHSO3 + CaSO4. 2Н2О
'Его осуществление возможно лишь для разбавленных раство-
растворов, содержащих меньше 17% Na2SO4, с получением раствора би-
бисульфита натрия, содержащего до 15% SO2. Получение же стан-
стандартного раствора бисульфита B2,5% SO2) требует, чтобы в ис-
исходном растворе было не меньше 25% Na2SO4, а при такой
концентрации происходит загустевание реакционной массы, за-
затрудняющее дальнейшее ее взаимодействие с SO2 и отделение осад-
осадка от раствора. Эти трудности устраняются при двухступенчатом
осуществлении процесса S8. В первой стадии известково-сульфатная
суспензия, содержащая в жидкой фазе 17% Na2SO4, насыщается
сернистым газом с отводом реакционного тепла для поддержания
30—35°. В течение 2—3 ч достигается 95—100%-ное превращение
сульфата по реакциям:
Са(ОНJ + 2SO2 = Ca(HSO3J
Ca(HSO3J + Na2SO4 + 2H2O = 2NaIISO3 + CaSO4 • 2H2O
- • Реакционная пульпа, содержащая крупнокристаллический осадок
гипса, остается подвижной. Во второй стадии в пульпу, кристаллы
гипса в которой являются хорошей основой для дальнейшей кри-
кристаллизации, вводят вторую порцию сульфата в количестве, рас-
рассчитанном на закрепление раствора бисульфита до концентрации
22—23% SO2. Затем продолжают насыщение пульпы сернистым
газом при непрерывной подаче в реактор сухой извести или
пушонки A моль СаО на 1 моль введенного во второй порции
Na2SO4). Загрузка извести производится со скоростью, обеспечи-
обеспечивающей слабокислую реакцию массы, т. е. присутствие свободной
H2SO3, что препятствует появлению CaSO3 и ведет к образованию
Ca(HSO3J (рис. 159). Процесс завершается без загустевания
пульпы. Осадок гипса может быть отделен от раствора бисульфита
натрия отстаиванием и фильтрацией на вакуум-фильтрах. В первой
ступени известь может быть заменена эквимолекулярным количе-
Висульфит и сульфит натрия
63»
ством сульфита кальция, являющегося отходом некоторых произ-
производств.
Способ получения сульфита натрия из поваренной соли, серни-
сернистого газа и аммиака" используется в Канаде 10°. В основе этого
способа лежит реакция
2NaCl + (NH4JSO3= Na2SO3 + 2NH4Cl
Равновесие в этой системе при 60—85° сдвинуто таким обра-
зом, что наибольшую площадь занимает поле кристаллизации
Na2SO3 (рис. 163). Поэтому мелкокристаллический хлористый нат-
натрий при внесении в горячий раствор сульфита аммония будет легко
и в значительных количествах растворяться, а в осадок перейдет
2NaCl
Na2SO3
NaCl
NaHSO3
Рис. 163. Изотерма раствори-
растворимости водной системы 2NaCl+
+(NH4JSO3 ^=± Na2SO3+
+2NH4C1 при 60D.
NH4HSO
Рис. 164. Изотерма раствор»
мости водной системы NaCl 4-
+NH4HSO3 ^=± NaHSO3+
+NH4C1 при 25°.
безводный сульфит натрия. После отделения этого осадка в рас-
растворе останется хлористый аммоний. Кристаллизация NH4C1 до-,
стигается насыщением раствора сернистым газом с целью перевода
оставшегося сульфита в бисульфит и охлаждением раствора до 25°,
Как видно из рис. 164, поле кристаллизации NH4C1 при этом зна-
значительно увеличивается. Это позволяет подобрать такие концент-
концентрации раствора, при которых удается произвести кристаллизацию-
NH4C1. Оставшийся раствор, содержащий некоторое количество
Mi4Cl и бисульфита натрия, насыщается аммиаком, причем би-
бисульфит вновь превращается в сульфит, раствор нагревается и
вновь обрабатывается NaCl и т. д. Таким образом, можно осущест-
осуществить замкнутый цикл с периодической кристаллизацией Na2SO3 и
NH4C1. '
Конечный щелок, получаемый после кристаллизации хлористого,
аммония,содержит приблизительно 7% NHJ, 12-13% СГ, 19%
534
Гл. XIV. Соли сульфитного ряда
Пиросульфиты
535
Он насыщается аммиаком до содержания 0,5% свободного
NH3, — получается так называемый исходный раствор. При опти-
оптимальных концентрациях растворов, позволяющих вести замкнутый
процесс, в исходном растворе должно содержаться 10,5% NHJ
(при этом в нем содержится 17—18% SO3~, 4 — 5% Na+). Этот
раствор обрабатывается стехиометрическим количеством NaCl.
Выход Na2SO3 в одном цикле составляет около 55% по отноше-
отношению к SOl", содержащемуся в исходном растворе; выход NH4C1 —-
около 38% по отношению к связанному аммиаку.
При изучении растворимости в четверной системе NaCl—
(NH4JSO3—НгО при 20 и 60° помимо исходных компонентов были
обнаружены и другие твердые фазы, вероятно твердые растворы
присутствовавшего в качестве примеси Na2SO4 в Na2SO3101. При
осуществлении циклической схемы конечный щелок (раствор после
кристаллизации NH4C1) должен периодически очищаться от на-
накапливающегося в нем сульфата (например, осаждением - SO4~
хлористым кальцием).
БИСУЛЬФИТ И СУЛЬФИТ АММОНИЯ
Концентрированные растворы бисульфита аммония содержат
70—75% NH4HSO3; в ряде случаев их целесообразно использовать
в качестве источника 100%-ного SO2 (см. стр. 514). Бисульфит
аммония получают насыщением водного раствора аммиака серии-
•стым ангидридом, содержащимся в хорошо очищенных выхлопном
м производственном газах контактных сернокислотных систем.
Выхлопной газ из контактной системы проходит последова-
последовательно через две башни с насадкой — сульфит-бисульфитную и
•сульфитную, каждая из которых работает на замкнутом цикле
орошения. Сульфит-бисульфитный раствор поступает на циркуля-
циркуляцию в абсорбер распыливающего действия, куда подается крепкий
газ. В абсорбере крепкого газа сульфит-бисульфитный раствор
превращается в концентрированный бисульфитный раствор, насы-
насыщаясь очищенным сернистым газом, отбираемым после компрессо-
компрессоров контактного сернокислотного цеха. Газ из этого абсорбера воз-
возвращается в контактную систему. Соотношение между сульфитом
и бисульфитом в растворе сульфитной башни поддерживают
равным 0,3 (по весу), в абсорбере крепкого газа циркулирует рас-
раствор, состоящий только из бисульфита аммония; в сульфит-бисуль-
-фитной башне устанавливается среднее соотношение между суль-
сульфитом и бисульфитом. Часть раствора бисульфита аммония, цир-
циркулирующего через абсорбер крепкого газа, выводится из цикла в
качестве продукта. Этот раствор пропускают через центрифугу для
отделения взвешенных в нем кристаллов сульфата аммония, кото-
который является побочным продуктом производства. Для производ-
производства 1 т бисульфита аммония (в пересчете на 100%-ный) расхо-
расходуется 30 000 нм3 выхлопного газа с содержанием 0,3% SO2,
2370 нм3 крепкого газа G,5% SO2), 0,226 г аммиака в виде амми-
аммиачной воды с концентрацией не ниже 18% NH3, 124 квт-ч электро-
электроэнергии и 1 м3 воды. В качестве отхода получается 0,068 т сульфа-
сульфата аммония (в пересчете на 100%-ный).
Кристаллический моногидрат сульфита аммония (NH4JSO3-
• Н2О можно получить, высаливая его газообразным аммиаком из
раствора сульфита аммония. Маточный раствор после разбавления
служит для абсорбции SO2, которую ведут так, чтобы в растворе
оставался свободный аммиак, не превращая его полностью в суль-
сульфит аммония; затем раствор возвращают в процесс на поглощение
аммиака102. Описан способ получения (NH4JSO3 при взаимодей-
взаимодействии пирита с кислородом (воздухом) и аммиаком в присутствии
добавки NaCl; при 500° и двухчасовой обработке выход сульфита
составляет 82—87% 103.
ПИРОСУЛЬФИТЫ
При растворении в воде пиросульфита (метабисульфита) нат-
натрия образуется раствор бисульфита натрия. Следовательно, во
всех случаях применения эти продукты взаимозаменимы. Однако
пиросульфит является более ценным веществом, так как он полу-
получается в виде сухой соли Na2S2OE, выгодно отличающейся от би-
бисульфита своей транспортабельностью.
Существует несколько способов получения пиросульфита нат-
рия — так называемые мокрые и сухой методы. Основой всех мок-
мокрых методов является кристаллизация пиросульфита и растворов,
достигаемая: 1) выпариванием растворов бисульфита: 2) насыще-
насыщением сернистым газом сульфитной пульпы — суспензии твердого
сульфита натрия в его насыщенном водном растворе; 3) насыще-
насыщением сернистым газом пульпы, получаемой смешением кальцини-
кальцинированной соды с бисульфитом. Сухой метод заключается в
обработке сернистым газом увлажненной кальцинированной соды-
или бикарбоната натрия.
Получение пиросульфита натрия мокрыми способами
Метод получения пиросульфита натрия выпариванием раствора-
бисульфита требует расхода топлива; кроме того, при выпаривании-
раствора вместе с водяным паром улетучивается 20—26%' SO2.
В результате реакции
2NaHSO3 = Na2SO3 + Н2О + SO2
в растворе появляется сульфит, сильно загрязняющий кристалли-
кристаллизующийся пиросульфит. Хотя фракционированной кристаллиза-
536
Гл. XIV. Соли сульфитного ряда
Пиросульфиты
537
цией можно получить часть продукта, содержащего до 93 %*
Na2S2O5, но с очень низким выходом — 25—30%.
Получение пиросульфита натрия насыщением сернистым газом
сульфитной пульпы производят следующим образом. В реактор
заливают маточный бисульфитный раствор, оставшийся после
кристаллизации пиросульфита, и загружают кристаллический
Na2SO3 • 7Н2О. Пульпу нагревают паром до 40° и подвергают об-
обработке сернистым газом из колчеданных печей, подаваемым в
аппарат через барботер. По мере поглощения SO2 сульфит раство-
растворяется, превращаясь в бисульфит, и одновременно из раствора
кристаллизуется пиросульфит:
N a2SO3 + SO2 = Na2S2O5
По окончании обработки газом пульпу охлаждают. При этом
в осадок переходит дополнительно некоторое количество пиросуль-
пиросульфита натрия. Кристаллы пиросульфита отделяют от маточного
раствора на центрифуге и сушат воздухом при 70—80°, так как
влажный пиросульфит легко окисляется. В связи с тем, что с
Na2SO3 • 7Н2О в процесс вводится большое количество воды, объем
получаемого маточного раствора значительно превышает количе-
количество раствора, загружаемого в реактор. Поэтому часть маточного
раствора (раствор бисульфита) выводят из производственного про-
процесса. Этим обусловлены высокие расходные коэффициенты: на
1 т пиросульфита расходуется до 7 т семиводного сульфита нат-
натрия и до 1 т 100%-ного SO2.
Наиболее рациональным является насыщение сернистым газом
содово-бисульфитной пульпы, получаемой введением твердой каль-
кальцинированной соды в насыщенный раствор бисульфита натрия (ма-
(маточный раствор после кристаллизации пиросульфита) 104>105. При
этом идут следующие процессы:
NaHSO3 + Na2CO3 = Na2SO3 + NaHCO3
NaHSO3 + NaHCO3 = Na2SO3 + CO2 + H2O
Na2SO3 + SO2 + H2O = 2NaHSO3
Так как исходный маточный раствор бисульфита является насы-
насыщенным, а температурный коэффициент растворимости пиросуль-
пиросульфита натрия в пределах производственных температур B5—40°)
невелик, то при последовательном или одновременном проведении
процессов нейтрализации раствора содой и насыщения реакцион-
реакционной массы сернистым газом происходит кристаллизация пиро-
пиросульфита. Оптимальная температура процесса 35—45°. Концентпа-
ция поступающего в абсорбер сернистого газа может быть любой.
Даже при содержании в газе десятых долей процента SO? при до-
достаточных размерах абсорбционной колонны поглощение SO2 будет
практически полным. Оптимальное весовое соотношение между ко-
количествами соды и маточного бисульфитного раствора при приго-
приготовлении исходной пульпы равно 1 : 4. Это соответствует 40—50%
соды от стехиометрического количества, требующегося для полной
нейтрализации всего бисульфита. Загрузка соды в большем коли-
количестве приводит к сильному загустеванию пиросульфитной пульпы,
что затрудняет ее движение в аппаратах и трубах. В пульпу необ-
необходимо добавлять некоторое количество воды для компенсации
потерь воды в виде маточного раствора, смачивающего кристаллы
выводимого из цикла пиросульфита и в виде паров, уносимых га-
газом, уходящим из абсорбера.
Производство значительных количеств пиросульфита натрия
рационально лишь непрерывным способом. При этом поглоще-
поглощение SO2 можно осуществлять, например, в механических абсорбе-
абсорберах с последующим разделением пиросульфитной пульпы в непре-
рывнодействующих центрифугах; сушку пиросульфита во избежа-
избежании его разложения следует производить в течение очень малого
времени, измеряемого секундами, например, распыляя продукт в
потоке теплого воздуха.
Об опыте насыщения сернистым газом сульфит-бисульфитной
пульпы см.ш6, а о получении пиросульфита натрия сульфитно-
известковым способом см.107
Наибольшей стойкостью в средах, перерабатываемых
при производстве пиросульфита, обладают стали 1Х18Н9Т,
0Х23Н28МЗДЗТ, X17HI3M2T и 0Х21Н6М2Т 108.
Сухой способ получения пиросульфита натрия 109-ш
Этот способ основан на реакции:
Na2CO3 + 2SO2 = Na2S2O5 + СО2
Вначале образуется сульфит натрия8, который при дальнейшем
поглощении SO2 переходит в пиросульфит. если температура под-
поддерживается на уровне ~60° (не выше 80° — см. стр. 531) 97. Со-
Совершенно сухие вещества — сода и сернистый газ — не вступают
во взаимодействие. Наилучший выход пиросульфита достигается
при влажности соды 14—16%, т. е. соответствующей составу
Na2CO3-H2O, содержащему 14,52% воды. Сода, увлажненная до
такой степени и растертая для уничтожения образовавшихся при
этом комков, остается сухим, сыпучим, легко подвижным материа-
материалом. При взаимодействии с сернистым газом образуется продукт,
содержащий 90—94% Na2S2O5 и несколько процентов влаги. При
более значительном увлажнении соды B—3 моль Н2О на 1 моль
^та2СО3) выход пиросульфита не понижается, но продукт содержит
больше влаги. Вместо моногидрата соды может применяться би-
бикарбонат натрия.
Сернистый газ должен быть также увлажнен. Сухой газ подсу-
подсушивает соду быстрее, чем образуется пиросульфит. Чем меньше
содержится в газе SO2, тем большей должна быть его .влажность.
538
Гл. XIV. Соли сульфитного ряда
Гидросульфит (дитионит) натрия
539
При пользовании отбросными сернистыми газами, содержащими
0,4—0,5% SO2, относительная влажность их должна быть не
ниже 20%.
При содержании в газе кислорода к соде должен примеши-
примешиваться в незначительном количестве ингибитор, препятствующий
окислению продукта в сульфат, например диметилпарафенилендиа-
мин @,05%). Количество сульфата в продукте зависит от содер-
содержания кислорода в газе, от количества и рода ингибитора и т. п.
В продукте содержится также незначительное количество непро-
реагировавшей соды, которая при растворении продукта в воде
реагирует с бисульфитом, образуя сульфит. Для реакции между
твердой содой и сернистым газом могут применяться аппараты не-
непрерывного действия шнекового или полочного типа и аппараты с
кипящим слоем llz.
Пиросульфит калия
Пиросульфит калия в небольших количествах получают113 на-
насыщением 35—40% раствора К2СО3 сернистым газом, содержащим
6—7,5% SO2. Температуру раствора поддерживают на уровне 55—
60° за счет теплоты газа и теплоты нейтрализации; к концу про-
процесса раствор охлаждают водой для кристаллизации пиросульфита
калия. Отжатые на нутч-фильтре кристаллы содержат 20—25%
влаги. Их сушат в чугунных эмалированных чашах, имеющих шту-
штуцеры для ввода и вывода газа. Сушильным агентом служит серни-
сернистый газ, содержащий 6—7,5% SO2 и не больше 0,03% влаги.
Температура газа 80—90°, так как выше 100° возможно частичное
разложение верхнего слоя продукта. Выходящий газ имеет темпе-
температуру около 50°. Пиросульфит натрия также сушат сернистым
газом.
ГИДРОСУЛЬФИТ (ДИТИОНИТ) НАТРИЯ
Получение гидросульфита натрия связано с большими трудно-
трудностями, обусловленными чрезвычайной нестойкостью этого продукта
в двухводной форме и во влажном состоянии, особенно в присутст-
присутствии кислорода воздуха вследствие окисления по реакциям
2Na2S2O4 + О2 + 2Н2О = 4NaHSO3
или
Na2S2O4 + О2 + Н2О = NaHSO4 + NaHSO3
Гидросульфит разлагается и в отсутствие окислителя п4. В ней-
нейтральном растворе он сравнительно быстро распадается на тио-
тиосульфат и бисульфит
2Na8S2O4 + Н2О = Na2S2O3 + 2NaHSO3
причем в качестве промежуточного соединения образуется сульф-
оксиловая кислота H2SO2 или в слабощелочной среде сульфокси-
лат Na2SO2. В щелочной среде образуются тиосульфат и сульфит:
2Na2S2O4 + 2NaOH = Na2S2O3 + 2Na2SO3 + H2O
При избытке щелочи гидросульфит количественно взаимодейст-
взаимодействует с тиосульфатом (так же как и с полисульфидом):
Na2S2O, + Na2S2O3 + 4NaOH = 3Na2SO3 + Na2S + 2K О
Поэтому в сильнощелочном растворе, при 6—9-кратном избытке
щелочи по отношению к гидросульфиту, он медленно разлагается
по суммарной реакции:
3Na2S2O4 + 6NaOH = 5Na2SO3 + Na2S + 3H2O
Производство гидросульфита натрия восстановлением сернистой
кислоты цинковой пылью
Наиболее распространенный промышленный способ получения
гидросульфита натрия основан на восстановлении сернистой кис-
кислоты цинковой пылью. Суспензия цинковой пыли в воде обрабаты-
обрабатывается 100%-ным сернистым газом; при этом образуется раствор
гидросульфита цинка:
Zn + гНг
ZnS2O4 + 2Н2О
Полученный раствор смешивают с раствором соды:
ZnS2O4 + Na2COa= Na2S2O4 + ZnCO8
После отделения нерастворимого углекислого цинка фильтро-
фильтрованием гидросульфит натрия высаливают из раствора поваренной
солью в форме Na2S2O4 • 2Н2О. Ввиду сильной неустойчивости двух-
водного гидросульфита его подвергают дегидратации и затем су-
сушат. Продукт выпускают в виде безводного Na2S2O4, который в от-
отсутствие влаги выдерживает длительное хранение.
Цинковая пыль должна просеиваться через сита, имеющие
3600—4900 отверстий на 1 см2, и содержать не менее 95% актив-
активного цинка (определяемого иодометрическим способом) и не более
5% ZnO и 0,05% тяжелых металлов. Водная суспензия цинковой
пыли должна содержать 25% цинка (по весу). Суспензия насы-
насыщается сернистым газом в стальном реакторе с мешалкой и змее-
змеевиком для охлаждения при непрерывном перемешивании при 40—
50° в течение 1—2,5 чш. Более быстрое и более медленное насы-
насыщение ведет к разложению гидросульфита цинка (который еще
более нестоек, чем гидросульфит натрия), в первом случае вслед-
вследствие перекисления раствора сернистым газом, во втором — вслед-
вследствие чрезмерного затягивания процесса. Необходимый для
540
Гл. XIV. Соли сульфитного ряда
процесса 100%-ный SO2 получают разложением бисульфита кон-
концентрированной серной кислотой (купоросным маслом) в «газовом»
аппарате, представляющем собой стальной котел, футерованный
диабазовой плиткой, со стальной крышкой. О конце процесса гази-
газирования судят по изменению цвета суспензии гидросульфита цин-
цинка, который из серого переходит в светло-желтый. Получение
100%-ного SC>2 разложением бисульфита натрия серной кислотой
является нерациональным методом. Более выгодно производить
обогащение сернистого газа до 100%-ного с помощью циклических
методов (стр. 514).
Полученный раствор гидросульфита цинка вливают в стехио-
метрическое количество 22,5% раствора соды (плотность 1,24 г/см3),
загруженного в стальной аппарат с мешалкой и змеевиком для
охлаждения. Для получения крупнокристаллического, легко отфиль-
отфильтровываемого осадка углекислого цинка, реакционную массу пере-
перемешивают 30 мин при 25—35°. При более высокой температуре об-
образуется липкий, плохо фильтрующий осадок основного углекис-
углекислого цинка. В полученном растворе должно быть не меньше 170 г/л
Na2S2O4 и избыток №2СОз 5—35 г/л. В осадке содержится около
5% Na2S^O4B виде малорастворимой двойной соли Na2S2O4-гпБгО^
что приводит к потерям гидросульфита с углекислым цинком. От-
Отфильтрованный осадок промывают сначала слабым 3% раствором
гидросульфита натрия (промывной еодой от предыдущей опера-
операции) в количестве 25% от объема фильтрата, а затем водой. Пер-
Первую промывную воду добавляют к фильтрату; вторую промывную
воду (содержащую ~3% Na2S2O4) используют для промывки
осадка в последующих операциях. Для стабилизации раствора гид-
гидросульфита натрия путем увеличения рН раствора в приемник
фильтрата предварительно заливают 35% раствор едкого натра
в количестве 5% от объема фильтрата. Раствор должен содержать
55—65 г/л NaOH и не меньше 120 г/л Na2S2O4.
Раствор гидросульфита (фильтрат, смешанный с первой про-
промывной водой), содержащий около 16% Na2S2O4, передается в ре-
резервуар с мешалкой и змеевиком для охлаждения, где произво-
производится высаливание при понижении температуры от 33—36° до 20—
25°. При непрерывном перемешивании раствора в него вносится
поваренная соль в количестве 28—30 кг на 100 л раствора. Во из-
.бежание выделения мелких кристаллов Na2S2O4 • 2Н2О соль добав-
добавляют к раствору небольшими' порциями. После 45 мин перемеши-
перемешивания основная масса гидросульфита оказывается в суспензии
в виде относительно крупных кристаллов Na2S2O4 • 2Н2О; маточ-
маточный раствор, содержащий 12—16% NaCl, 1,5—2% NaOH, про-
продукты разложения гидросульфита (сульфит, сульфат, тиосульфат)
и от 1 до 25 г/л Na2S2O4 (в системе NaCl—Na2S2O4—Н2О эвтони-
ческий раствор при 25° содержит 1,65% Na2S2O4), декантируется
и сливается в отброс (содержащийся в нем тиосульфат натрия
Гидросульфит (дитионит) натрия
541
может быть выделен и выпущен в качестве побочного продукта
производства).
Кристаллы, содержащие небольшое количество маточного рас-
раствора, быстро нагреваются с помощью паровой рубашки при мед-
медленном вращении мешалки до температуры выше 52°, являющейся
точкой перехода дигидрата в безводную соль. При нагревании
до 58—62° дегидратация заканчивается в течение 15—30 мин. Без-
Безводный гидросульфит отделяют от горячего маточного раствора,
содержащего около 3,4% Na2S2O4; при охлаждении этого раствора
до 25—20° из него можно выкристаллизовать Na2S2'O4 • 2Н2О в ко-
количестве 4 % от дигидрата, полученного в основном процессе
Дегидратированный гидросульфит натрия с целью удаления
межкристальной жидкости подвергают двукратной промывке эти-
этиловым спиртом — сначала 80%-ным, затем 96%-ным — и сушат
в течение 1,5—2,5 ч под вакуумом в токе воздуха, входящего с
температурой 80—100° ( в замкнутом цикле с промежуточной кон-
конденсацией паров спирта). Скомковавшийся при сушке продукт раз-
размалывают и укупоривают в тару—металлические барабаны. Го-
Готовый продукт представляет собой порошок сероватого цвета,
содержащий 80—90% Na2S2O4. Вследствие больших потерь от раз-
разложения общий выход гидросульфита составляет 30—40% от тео-
теоретического по израсходованному количеству цинковой пыли. Овод-
йенный этиловый спирт регенерируют ректификацией. Расходные
коэффициенты на производство 1 т гидросульфита натрия этим
способом составляют: 0,78 т цинковой пыли A00%), 1,94 г каль-
кальцинированной соды A00%), 0,51 т каустической соды A00%),
2,3 т серной кислоты A00%), 8,0 г бисульфита натрия B2,5%),
2,2 т поваренной соли A00%), 133 л этилового спирта (96%),
440 кет • ч электроэнергии, 2,65 мгкал пара, 330 м2 воды. В каче-
качестве отходов на 1 т продукта получаются: 9 г сырого углекислого
цинка, содержащего 20—30% общего цинка, 15—20% общего СО3,
0,35—0,4% железа, 35—45% воды; 0,12 т не вступившего в реак-
реакцию металлического цинка; 8,5 т раствора из газового аппарата,
содержащего смесь сульфата и бисульфата натрия A:2); 5,5 м*
холодного маточного раствора после высаливания; 1,75 ж8 горя-
горячего маточного раствора после дегидратации гидросульфита.
Более совершенный и современный способ освобождения от ос-
остатков влаги после дегидратации гидросульфита натрия заклю-
заключается в следующем. В центрифугу, где производят отделение кри-
кристаллов дегидратированного гидросульфита натрия от маточного
раствора, подают СО2, которая превращает содержащийся в меж-
межкристальной жидкости NaOH в Na2CO3. Образующийся карбонат
натрия связывает оставшуюся в отфугованной лепешке влагу и
превращается в кристаллогидрат — это приводит к исчезновению
свободной влаги в кристаллах гидросульфита натрия. Приме-
Примешанный к продукту карбонат натрия помимо этого является и
542
Гл. XIV. Соли сульфитного ряда
стабилизатором гидросульфита. Продукт, выгруженный из центри-
центрифуги, сушат в вакуум-сушилке при 80е, затем охлаждают, просеи-
просеивают и укупоривают.
Для высаливания гидросульфита можно вместо NaCl приме-
применять Na2CO3, NaOH или Na2SO3; тогда холодный маточный рас-
раствор не будет содержать NaCl и обработкой сернистым газом его
можно превратить в бисульфит натрия, что значительно понизит
затраты на сырье.
Для уменьшения потерь гидросульфита натрия от разложения
в процессе его производства предложено116 изолировать производ-
производственные растворы, содержащие гидросульфит, от действия атмос-
атмосферного воздуха, сохраняя их под тонким слоем A0—20 мм)
бензола. При отделении кристаллов гидросульфита от маточного
раствора они также остаются под слоем бензола, а при сушке про-
продукта бензол отгоняется, так же как и спирт. При этом выход про-
продукта может быть повышен до 50—60 % 94.
За границей имеются установки, производящие до 900 г в ме-
месяц гидросульфита и выпускающие в качестве побочного продукта
500—600 т окиси цинка ш. Для получения ZnO пигментной квали-
квалификации предложено перед дальнейшей переработкой очищать рас-
раствор ZnS2O4 от тяжелых металлов (железа) катионитом в форме
цинковой соли118. Предложено также вместо цинковой пыли ис-
использовать гранулированный сплав, содержащий цинк и 1—3,5%
натрия119. Для приготовления гидросульфита цинка применяют
медные реакторы, а для высаливания гидросульфита натрия — эма-
эмалированные котлы с рубашкой, в которой циркулирует, в зависи-
зависимости от условий процесса, холодная или горячая вода 12°. Дегидра-
Дегидратацию гидросульфита предложено осуществлять парами органиче-
органических жидкостей, температура кипения которых немного выше
температуры дегидратации (СН3ОН, С2Н5ОН, СС14, С6Н6) 121. Для
предотвращения загрязнения гидросульфита цинка примесями, со-
содержащимися в цинковой пыли, газирование суспензии рекомен-
рекомендуют вести до достижения установленного заранее определенного
значения окислительно-восстановительного потенциала 122. Предло-
Предложены также добавки к гидросульфиту натрия с целью уменьшения
его разложения на воздухе — жидкие эфиры__полученные из спир-
спиртов с пятью и более атомами углерода 123.
Для снижения потерь гидросульфита натрия от разложения при
сушке, необходимо предотвратить гидратацию дегидратированного
Na2S2O4 при его фильтровании и промывке, для чего эти процессы
следует осуществлять при температурах выше температуры пре-
превращения двухводного вещества в безводное, не допуская охла-
охлаждения фильтруемой суспензии. Можно также вести промывку го-
горячим спиртом, при этом получается продукт, содержащий больше
98% Na2S2O4124. Сушка в атмосфере СО2 также предохраняет про-
продукт от разложения 125.
Гидросульфит (дитионит) натрия
543
Другие способы получения гидросульфита натрия
Гидросульфит натрия может быть получен восстановлением
раствора бисульфита натрия различными восстановителями — цин-
цинковой пылью, порошкообразным железом или алюминием, амаль-
амальгамами цинка или натрия, муравьинокислым натрием или аммо-
аммонием и др. Наиболее распространенным восстановителем является
цинковая пыль. Применяется концентрированный раствор бисуль-
бисульфита, содержащий 47% NaHSO3. Реакция протекает при 15—25°
по уравнению:
4NaHSO3 + Zn = Na2S2O4 + Na2SO3 + ZnSO3 + 2H2O
При одновременном насыщении сернистым газом реакция идет
по уравнению:
2NaHSO3 + SO2 + Zn = Na2S2O4 + ZnSO3 + H2O
В отличие от предыдущей реакции, натрий используется пол-
полностью и получаются более концентрированные растворы гидро-
гидросульфита. При этом образуются также нерастворимые двойные
соли гидросульфита натрия и сульфита цинка, сульфита натрия и
сульфита цинка. Для разрушения двойных солей реакционную
смесь обрабатывают известковым молоком. При этом образуются
осадки Zn(OHJ и CaSO3, а в растворе остается гидросульфит
натрия, который перерабатывают по способу, описанному выше.
При использовании в качестве восстановителя порошкообраз-
порошкообразного железа (электролитического, губчатого) наилучшие резуль-
результаты получаются, когда ведут восстановление при 15° раствора,
содержащего около 25% NaHSO3 и 1—1,3% свободной H2SO3, что
соответствует рН=2,4—2,45 126. Для поддержания такой кислотно-
кислотности в процессе восстановления реакционную массу подпитывают
сернистым газом в количестве 50—65% от стехиометрического
по реакции:
2NaHSO3 + SO2 + Fe = Na2S2O4 + FeSO3 + H2O
Получаемый раствор содержит 15—17% Na2S2O4. Лучшим спо-
способом очистки его от примеси растворимых соединений железа
является обработка его гашеной известью-пушонкой при 30-минут-
30-минутном перемешивании. Вследствие каустификации сульфита известью
раствор содержит 1,3—1,4% NaOH; поэтому отпадает необходи-
необходимость в специальной добавке едкого натра для стабилизации рас-
раствора. Осадок сульфита железа, отделенный от раствтэра гидро-
гидросульфита, может быть разложен серной кислотой с целью возвра-
возвращения SO2 в производственный процесс.
Представляют интерес электрохимические способы получе-
получения гидросульфита натрия прямым восстановлением иона HSO3
на твердом катоде или восстановлением амальгамой натрия,
544
Гл. XIV. Соли сульфитного ряда
получаемой электролизом в ванне с ртутным катодом ш-128. Нор-
Нормальный потенциал окислительно-восстановительной реакции
Ронгалит
645
равен —0,009 в 129.
Электролиз водных растворов SO2 на твердом катоде очень
сложен. При рН=0,6—6,2 образуется H2S2O4. При рН < 0,6 на
поверхности катода образуются желто-коричневые маслообразные
сульфаны, а в электролизере появляется тестообразная масса —
смесь сульфанов и серы. Первоначальным продуктом восстанов-
восстановления при этом также является H2S2O4, которая превращается
в сульфан и SO2. В зависимости от молярного отношения SCb и
H2S2O4 состав сульфанов различен, например, H2S33—H2S,?.
H2S,0-H2S12 и др. 13°.
Восстановление бисульфита натрия в гидросульфит на твер-
твердом катоде происходит в кислой среде, в которой гидросульфит
весьма нестоек. Это приводит к серьезным затруднениям: электро-
электролиз приходится вести при высокой объемной концентрации тока
(т. е. при малом объеме католита), при небольшой катодной плот-
плотности тока и низкой температуре. Так как процесс образования
гидросульфита сопровождается одновременным образованием ще-
щелочи, необходимо непрерывно вводить в катодное пространство
газообразный SO2, разбавленный инертным газом во избежание
перекисления раствора в местах введения газа. Анолитом служит
раствор сульфита натрия. Наиболее целесообразно применение
проточного электролизера с диафрагмой, который дает возмож-
возможность получать растворы, содержащие до 180 г Na2S2O4 в 1 л.
Лучшими материалами для катода являются платина, золото, мо-
молибден, серебро, свинец, никель. На этих материалах водород вы-
выделяется с большим перенапряжением (потенциал восстановления
HSO.3 равен —0,163 е, потенциал выделения водорода —0,157 е).
Однако некоторые из этих металлов каталитически ускоряют даль-
дальнейшее восстановление гидросульфита до тиосульфата:
S2O^ + 2Н+ + 2е = S2Oi*- + Н2О
При серебряных, титановых и свинцовых катодах потери от
этого процесса минимальны.
Имеется много патентов на использование в качестве восстано-
восстановителя амальгамы натрия. Необходимость обеспечения при этом
тесного и быстрого контакта амальгамы с раствором NaHSOs и
раствора с газообразным SO2 связана с довольно сложным кон-
конструктивным оформлением реакционных аппаратов, снабженных
мешалками с большим числом оборотов. Для интенсивного разме-
размешивания предложен, например, аппарат в форме барабана с бы-
быстро вращающимся полым валом, через который вводят SO2 и на
наружной поверхности которого имеются щетки для разбрызгива-
разбрызгивания амальгамы 13<-132.
Раствор бисульфита натрия (NaHSO3 : Na2SO3 = 10: 1) разла-
разлагают, перемешивая его с амальгамой натрия при 40°. Выделив-
Выделившиеся кристаллы Na2S2O4 • 2Н2О отфильтровывают и обезвожи-
обезвоживают быстрым нагреванием до 60—65°, промывают спиртом и суш-
сушкой в вакууме. Выход продукта по SO2 70—75%, но он содержит
до 0,002% ртути и поэтому не может применяться в фотографии.
Предложены способы получения гидросульфита натрия и дру-
других щелочных металлов восстановлением жидким или газообраз-
газообразным SO2 их боргидридов при температурах от —70 до 70°133' ш.
РОНГАЛИТ
Ронгалит, или сульфоксилатформальдегид ЫаНЭОг • СН2О •
•2НгО, * получают при взаимодействии бисульфита натрия с цин-
цинковой пылью в присутствии формалина:
NaHSO3 + СН2О + Zn + Н2О = Zn(OHJ + NaHSOj • СН2О
При смешении бисульфита с цинковой пылью с большой ско-
скоростью идет реакция
4NaHSO3 + Zn = Na2S2O4 + ZnSO3 + Na2SO3 + 2H2O
и частично протекают реакции:
2ZnSO3 + Zn + 2H2O = ZnS2O4 + 2Zn(OH)j
ZnS2O4 + Na2SO3 = Na2S2O4 + ZnSO3
При введении в смесь формалина образуется ронгалит
Na2S2O4 + СН2О + Н2О = NaHSO2 • СН2О + NaHSO3 \
а выделяющийся при этом бисульфит снова вступает в реакцию
с цинком, но частично (весьма медленно) соединяется с формали-
формалином:
NaHSO3 + CH2O=NaHSO3 • СН2О
Для производства ронгалита 13Б>136 используют раствор бисуль-
бисульфита, цинковую пыль и раствор формалина. Лучшие выходы по-
получаются при последовательном вводе в реактор воды, цинковой
пыли, бисульфита и затем, приблизительно через 10 мин, форма-
формалина. При этом уменьшается количество образующегося бисульфит-
* Ронгалитом называюттакжегидросульфитформальдегидНа252О4 • 2СН2О • 2Н2О;
18 М. Е. Позив
646
Гл. XIV. Соли сульфитного ряда
формальдегида NaHSO3-CH2O, который восстанавливается мед-
медленно. Оптимальные условия: температура процесса 90—95°,
содержание SO2 в растворе бисульфита 15—20%- Количество фор-
формалина должно быть стехиометрическим. Цинковую пыль вводят
с большим избытком; ее расход достигает иногда 200—205% от
теоретического, причем степень восстановления бисульфита равна
~85%. При смешении реагентов в реакторе с мешалкой восста-
восстановление идет вначале с большой скоростью, но затем резко за-
замедляется вследствие экранирования реагирующей поверхности
частиц цинка образующейся гидроокисью цинка. Этим и объяс-
объясняется большой расход цинковой пыли. Скорость, а следовательно,
и степень восстановления возрастают при увеличении дисперсно-
дисперсности цинковой пыли. При использовании пыли со средним разме-
размером частиц 22 мк (95%) в количестве 130% от теоретического при
оптимальных условиях за 3 ч степень восстановления NaHSO3 рав-
равнялась ~40% и степень использования цинка ~30%; при среднем
же размере частиц 4 мк (92%) степень восстановления увеличи-
увеличилась до 81%, а степень использования цинка до 63%. Для ускоре-
ускорения процесса предложено137 осуществлять его в вибрационных
мельницах с цинковыми шарами, где создаются условия для уда-
удаления Zn(OHJ с поверхности частиц цинка. Здесь при загрузке
цинковой пыли в количестве 110—120% от теоретического за 3 ч
степень восстановления NaHSO3 равна 92—95%. Так как при этом
расходуется и цинк, из которого изготовлены шары, общий расход
цинка составляет 150—160% от теоретического. Но изготовление
цинковых шаров дешевле, чем цинковый пыли. Замена цинковых
шаров стальными невозможна, так как при этом степень восста-
восстановления не превышает 20%.
Безводный ронгалит весьма устойчив, но во влажной атмосфере
постепенно разлагается. Двухводный ронгалит разлагается с вы-
выделением кристаллизационной воды, которая далее еще больше
ускоряет разложение. Разбавленные растворы разлагаются значи-
значительно быстрее концентрированных, так как уменьшение величины
рН ускоряет разложение 138>139. Гигроскопическая точка двухвод-
ного ронгалита равна 60%. Давление водяного пара над двухвод-
>ium ронгалитом можно подсчитать по приближенной формулеи0
2125
lgP = 8,26 j- (здесь Р в мм рт. ст.). В атмосфере^ насыщенной
водяными парами, двухводный ронгалит разлагается со скоростью
в ~6 раз большей, чем в сухой среде.
Отход от производства ронгалита, так называемая окшара, со-
содержит гидроокись и окись цинка с примесью соединений серы.
Ее перерабатывают в цинковые белила. Разработан электрохими-
электрохимический способ регенерации цинка из окшары; его получают в фор-
форме губки, которая по активности в процессе производства ронга-
ронгалита не уступает цинковой пыли 141«
Тиосульфат натрия
847
ТИОСУЛЬФАТ НАТРИЯ
Химические основы образования тиосульфата
Существует много способов получения тиосульфата натрия, в
основе которых лежат разные реакции. Производственное значение
имеют главным образом следующие:
сульфитный способ
Na2S2O3
гидросульфидный способ
2NaHS + 4NaHSO3 = 3Na2S2O3 + 8H2O
сульфидный способ
2Na2S + Na2CO3 + 4SO2 = 3Na2S2O3 + CO2
сероводородный способ
2H2S + 2Na2SO3 + 2NaHSO3 = 3Na2S2O3 + 3H2O
дисульфидный (полисульфидный)
2N a2S2 + 6NaHSO3 = 5Na2S2O3 + 3H2O
или, в общем виде:
2Na2Sn + 6NaHSO3 + B« - 4) Na2SO3
1) Na2S2O3 + 3H2O
Кроме того, тиосульфат натрия получается в качестве побоч-
побочного продукта в производстве гидросульфита и при очистке про-
промышленных газов от серы. Его можно получать также сульфат-
сульфатным способом, используя Na2SO4. Из методов, уже утративших
промышленное значение, следует упомянуть действие смеси серни-
сернистого газа и кислорода (воздуха) на раствор сернистого натрия
BNa2S + 2SO2+O;> = 2Na2S2O3) и окисление сернистого кальция
(отвала от производства соды) кислородом воздуха в тиосульфат
кальция с последующим обменным разложением с сульфатом
натрия.
Механизм образования тиосульфата был предметом многочис-
многочисленных исследований Высказывались предположения, что в по-
построении сулыЬоксиловой, тиосерной и политионовых кислот уча-
участвует моноокись серы:
H2SO2 H2S2O3 H2S4Oe H2S-,O6
SO-H2O 2SO-H2O 3SO-SO2-H20 5SO-H2O
На -основании этого следовало бы считать, что ангидридом
тиосерной кислоты является димер (SOJ. Присутствие в газооб-
газообразной моноокиси серы ассоциированных молекул, в частности
(SOJ> было установлено спектрографически142. Однако изучение
Рефракции содержащих серу соединений143 приводит к выводу, что
18*
548
Гл. XIV. Соли сульфитного ряда
тиосульфат является продуктом сочетания сульфита с атомарной
серой. Это согласуется со взглядами Менделеева, считавшего тио-
серную кислоту продуктом присоединения серы к сернистой кис-
кислоте, подобно серной кислоте — продукту присоединения кислорода
к сернистой кислоте144.
На этом основании механизм образования тиосульфата при
получении его гидросульфидным способом представляется следую-
следующим 145. Вначале гидросульфид, взаимодействуя с бисульфитом,
образует в качестве промежуточного продукта тиосернистую кис-
кислоту:
Ионы водорода, появляющиеся вследствие диссоциации бисуль-
бисульфита
8HSO3
вступают в реакцию с новым количеством гидросульфида и с тио-
сернистой кислотой, образуя элементарную серу:
HS" + S2O|- + ЗН+ = 3S + 2Н2О
Выделяющаяся сера в активном состоянии реагирует с сульфи*
том, образуя тиосульфат:
Суммарное уравнение процесса:
2HS" + 4HSOJ = 8S2Ol"+8Н2О
При отношении HS~ : HSO^, равном 1 :2, в результате проме-
промежуточных реакций образуются эквивалентные количества сульфита
и серы, связывающиеся в тиосульфат, чем исключается появление
побочных продуктов.
При получении тиосульфата сульфидным способом14в-ш по об-
общему уравнению
2S2" + СО*" + 4SO2 = 3S2O^ + СО2
вначале образуются сульфит и гидросульфид:
+ 8О2 + Н2О = SOl~ + H2S
Гидросульфид, реагируя с БОг, также превращается в сульфит:
' 2HS" + SO2 + Н2О = SO|" + 2H2S
Тиосульфат натрия
Б49
Выделяющийся сероводород, взаимодействуя с сернистым га-
газом, образует тиосернистую кислоту, которая далее окисляет серо-
сероводород до серы:
H2S + SO2 + Н2О = H2S2O2 + Н2О
H2S + H2S2O2 = 3S + 2H2O
И, как и в предыдущем способе, тиосульфат образуется в ре-
результате реакции сульфита с серой:
Наибольший выход тиосульфата достигается при отношении
исходных реагентов ЫагСО3: Na2S, равном 1 :2, когда образуются
эквивалентные количества сульфита и серы.
Образование тиосульфата натрия при взаимодействии сульфита
.натрия с серой является частным случаем целого класса реакций.
Подобно тиосульфатам получаются селеносульфаты при взаимо-
взаимодействии серы или селена с сульфитом щелочного металла, аммо-
аммония или кальция И8.
При образовании тиосульфата (так же как и полисульфидов)
имеет важное значение гидролиз серы как первичная стадия про-
процесса. Установлено149, что при кипячении порошка серы с водой
гидролиз идет по реакции:
2S + 2Н2О = H2S + H2SO,
Вследствие крайней неустойчивости сульфоксиловая кислота
разлагается при рН < 7 по реакции
2H2SO2 = SO2 + S+2H2O
а при рН > 7 по реакции:
2H2SO2
S2O?f + Н2О + 2Н+
Нагревание серы в щелочном растворе в токе азота приводит
к образованию полисульфидов:
12S + 6ОН~ = 2(S • S^2"-)- S2Off + ЗН2О
При этом вначале, очевидно, образуется сульфид, связываю-
связывающий затем свободную серу. Максимальное количество серы, свя-
вываемое Na2S, вероятно, соответствует образованию Na2S5, что
следует из опытов по диализу. С течением времени идет дальней-
дальнейший гидролиз полисульфида:
2(S • S4)
2~
2S2O?f + 6H2S
Сульфидный способ
Этим способом в СССР производили раньше значительные ко-
количества тиосульфата натрия. В настоящее время его производят
Другими способами. Применявшаяся и описанная ниже схема
550
Гл. XIV. Соли сульфитного ряда
?
3
производства является несовершенной. Использование этого спо-
способа в отдельных случаях может оказаться рациональным при
более современном аппаратурном оформлении.
Схема производства тиосульфата натрия сульфидным способом
показана на рис. 165. Исходный щелок, содержащий 120—130 г/л
Na2S и 60—80 г/л ЫагСОз, готовили растворением соды в щелоке
сернистого натрия или по-
получали при растворении
осадков, образующихся в
производстве сернистого
натрия и извлекавшихся
из выпарных котлов (стр.
488). Щелок насыщали
6—7%-ным сернистым га-
газом в керамической аб-
абсорбционной башне, наса-
насаженной керамическими
кольцами. Щелок, стекая
по насадке навстречу га-
газовому потоку, в нижней
части башни поглощает
SO2, а в верхней — H2S
(стр. 548). Башня должна
иметь высоту, достаточ-
достаточную для полного поглоще-
поглощения сероводорода F м при
диаметре 1 м). Макси-
Максимального выхода тиосуль-
тиосульфата достигали при плот-
плотности орошения насадки
~ 1 msI(m2 • ч); уменьше-
уменьшение плотности орошения
от 6 до 1м3/(м2-ч) уве-
увеличивало выход от 63 до
84%, дальнейшее умень-
уменьшение интенсивности оро-
Тиосульфат натрия
551
Рис.
165. Схема производства тиосульфата
натрия сульфидным способом:
*/ — резервуар для щелока сернистого натрия; 2 — на-
напорный бак для щелока; 3 - бак для горячей воды;
4 — абсорбционная башня; 5 — сборник тиосульфатного
щелока; 6 ~ фильтрпресс: Г-сборник фильтрован-
фильтрованного щелока; 8 — напорный бак; 9 — подварочный ко-
котел; 10- уварочный котел; // - колодильник; 12- кри-
кристаллизатор; 13 — центрифуга; 14 — выщелачиватель
шлама.
шения приводило к сни-
снижению выхода. После
8—10-дневной непрерывной работы башню промывали горячей
водой, так как насадка забивалась шламом, вносимым со щело-
щелоком, и осадком кремневой кислоты, содержащейся в щелоке сер-
сернистого натрия в виде силиката натрия и выделяющейся. из ще-
щелочного раствора во время его нейтрализации сернистым газом. Из
башни вытекал разбавленный тиосульфатный щелок B50—300 г/^г
Na2S2Os), нагретый вследствие экзотермичности реакции до 35—45 .
Щелок после фильтрации на фильтрпреосе поступал на выпарку.
Выпарку производили в открытых котлах с паровым обогревом в
две стадии. В первой стадии щелок выпаривали до концентрации
/~450 г/л Na2S2O3. Конечная концентрация Na2S2O3 в щелоке на
разных заводах была в пределах 48—57% G00—900 г/л). Щелок
отстаивали от осадка, содержащего Na2SO3, Na2SO4, NaCl, Na2S2O3,
и сливали. Чем выше конечная концентрация щелока, тем больше
выделяется осадка и тем больше теряется с ним тиосульфата. Оса-
Осадок собирали и перерабатывали отдельно разваркои в горячей воде
с целью растворения тиосульфата. Полученные при разварке рас-
растворы присоединяли к щелоку, вытекающему из башни. При пред-
предварительном выпаривании щелока в подварочном котле (первая
стадия) его кипятили 28 ч при 101—104°, затем подвергали от-
отстаиванию от осадка в течение 8 ч. Окончательное выпаривание
вели также 28 ч при 104—110°. После вторичного отстаивания от
осадка в течение 2 ч выпаренный щелок охлаждали до 60—62° в
течение 2—2,5 ч в холодильнике, а затем направляли в кристалли-
кристаллизаторы. Выделившиеся при охлаждении щелока до 20—30е кри-
кристаллы Na2S2O3-5H2O сортировали на три сорта (реактивный, фо-
фото, технический) и каждый сорт в отдельности отделяли от рас-
раствора на центрифуге. Маточный раствор возвращали на выпарку.
Растворимость как сульфита, так и сульфата натрия значи-
значительно снижается при увеличении концентрации тиосульфата (рис.
166). Так как растворимости сульфата и сульфита отличаются не
сильно, можно выразить условно суммарную концентрацию Na2SO4
и Na2SO3 в щелоке, поступающем на выпарку, через концентра-
концентрацию Na2SO3. Затем, сопоставляя растворимости в системе
Na2S2O3—Na2SO3—Н2О при температуре выпарки и при конечной
температуре кристаллизации, можно определить состав получае-
получаемого тиосульфата. При этом содержание в нем Na2SO4 и Na2SO?
будет выражено через Na2SO3.
На рис. 167 даны изотермы растворимости в системе Na2S2O3—
Na2SO3—Н2О при 80 и 25° (изотерма 80° расположена выше изо-
изотермы 25°, потому что растворимость сульфита натрия, как и суль-
сульфата, с повышением температуры несколько понижается). Состав
исходного щелока изображается точкой т\. Выпариваемой при 80°
Щелок вскоре насыщается сульфитом (точка т2), после чего при
Дальнейшем выпаривании выделяется осадок безводного сульфита,
а состав раствора изменяется вдоль изотермы от т2 по направле-
направлению к эвтонической точке Ет. Допустим, что выпарка прекращает-
прекращается, когда точка системы достигла т3, а состав раствора изобра-
изображается .точкой п. После отделения выпавшего осадка Na2SO3 ос-
оставшаяся новая система (п) охлаждается до 25°. При этом точка
п оказывается в области аЬЕъь, соответствующей совместной кри-
кристаллизации Na2S2O3-5H2O и Na2SO3-7H2O. Поэтому при охлаж-
охлаждении до 25° состав раствора окажется в эвтонической точке ?25>
а состав' выделившихся кристаллов изобразится точкой /с. Таким
Б52
Гл. XIV. Соли сульфитного ряда
образом, кристаллы будут состоять из тиосульфата с небольшой
примесью сульфита (и сульфата). Соотношение между тиосульфа-
тиосульфатом и примесью определяется отношением отрезков ак: Ьк. При ме-
менее глубокой выпарке примеси будет меньше или даже она будет
совершенно отсутствовать (когда точка п окажется в поле кри-
кристаллизации Na2S2O3 • 5Н2О), однако уменьшится и выход тиосуль-
тиосульфата при кристаллизации.
Тиосульфат натрия
553
NazS2Of
Рис. 166. Изотерма 25° растворимости в системе
Na2S2O3—Na2SO3—Na2SO4—H2O (концентрации растворов
выражены в г на 100 г воды).
Поля кристаллизации:
/- Na2SO4 • 10Н2О; // - Na2SO4 • 7Н2О; /// - Na2SO4 • 10Н2О+
+ Na2S2O3 • 5Н2О (смешанные кристаллы); IV — Na2S2O3 • 5Н2О.
Количество выпариваемой воды может быть сокращено в 1,5—
2 раза, если применять исходный щелок с содержанием не 120—
130, а 250—270 г/л Na2S и разбавлять его до концентрации 120 г/л
маточными щелоками от кристаллизации тиосульфата, вместо того
чтобы направлять их на выпарку. Присутствие тиосульфата в ще-
щелоке не мешает протеканию основной реакции.
На 1 т товарного тиосульфата, получавшегося по описанной
выше схеме, расходовали 0,56 г 62,5%-ного Na2S в виде щелока,
0,68 г 45%-ного серного колчедана для получения необходимого
количества сернистого газа и 0,05 т кальцинированной соды (в рас-
расчете на 95%-ную).
При получении тиосульфата в результате побочных реакций
образуются политионаты, разлагающиеся с течением времени с вы-
выделением серы. Это приводит к пожелтению кристаллов тиосуль-
тиосульфата иногда уже при нх кристаллизации, иногда при хранении.
fNa2S2O3-5H,O)b,
NazS2O3
Na2SO3
Рис. 167. Схема определения состава получаемого тиосульфата натрия
по изотермам равновесия в системе Na2S2O3—Na2SO3—Н2О.
Предотвратить это можно ш добавкой соды в горячий щелок перед
его фильтрованием (~3 кг на 1 г готового продукта):
Na2S3Oe + Na2CO3 = Na2S2O3 + Na2SO4 + CO2
Полисульфидный способ
Для -приготовления раствора полисульфида Na2S2 используют
горячий раствор сернистого натрия G0°), обычно щелок, получае-
получаемый при выщелачивании плава сернистого натрия. Этот щелок
имеет плотность 1,264 г/см3 и приблизительно следующий состав
(в %): 20 —Na2S, 2-Na2S2O3( 2,5-Na2CO3) 1,6 — Na2SO<. В нем
654
/V»4 XIV. Соли сульфитного ряда
растворяют стехиометрическое количество молотой комовой или
fa3OBoft серы. Применение тонкоизмельченной серы нежелательно,
так как при этом увеличивается выделение сероводорода. К рас-
раствору полисульфида прибавляют медленно (во избежание силь-
сильного его вспенивания и выброса) раствор бисульфита натрия до
нейтральной реакции. Получение полисульфида и нейтрализа-
нейтрализацию его бисульфитом осуществляют в стальном варочном котле,
снабженном паровым змеевиком и мешалкой, вращающейся со
скоростью 30 об/мин. Полученный разбавленный раствор тио-
тиосульфата фильтруют и перерабатывают на кристаллический про-,
дукт теми же приемами, которые применяются в сульфидном,
методе.
Степень использования сырья в производстве тиосульфата по-1
лисульфидным методом составляет: сернистого натрия 84—85%,.|
бисульфита ~92%, серы ~90%. На 1 г тиосульфата натрия рас-
расходуют: 0,235 г сернистого натрия F2%), 0,056 г комовой серы,.
1,45 г бисульфита натрия, 3,5 г пара, 2,5 мъ воды, 40 кет ¦ ч электро-
электроэнергии. Так как сырье (сернистый натрий, бисульфит) вводится '
в производство в виде водных растворов, то на каждую тонну вы-1
пускаемого продукта необходимо выпаривать ~870 кг воды—по-
воды—почти половину количества воды, содержащейся в слабом растворе
тиосульфата. Однако производство тиосульфата полисульфидным]
методом может быть организовано по замкнутой циклической схе- j
ме, позволяющей полностью устранить из производственного про-1
цесса выпарку тиосульфатного щелока 150. По этой схеме раство-1
рение соды для приготовления бисульфита должно вестись не в'
воде, а в оборотном маточном растворе от кристаллизации тио-:
сульфата. Полученный тиосульфатно-содовый раствор обрабаты-!
вают обычным способом сернистым газом, после чего тиосульфат-!
но-бисульфитный щелок направляют на реакцию с полисульфидом]
натрия. При этом образуется концентрированный раствор тио|
сульфата, который после фильтрования можно направлять непо|
средственно на кристаллизацию.
Несмотря на отсутствие выпарки, при работе по этой схеме иЛ
раствора тиосульфата также выделяются загрязняющие его при!
меси в виде осадков, отделяемых фильтрованием перед кристалли*
зацией. В тиосульфатном маточном растворе, почти независимо от
его концентрации, можно растворить значительное количество со-
соды (~150 г/л). В процессе обработки сернистым газом сода пере-
переходит в бисульфит, растворимость которого в присутствии тиосуль-
тиосульфата также очень высока и который высаливает из раствора суль-
сульфат, являющийся постоянной примесью в маточном растворе.
Когда после реакции бисульфита с полисульфидом образуется
тиосульфат, то, переходя в раствор, он высаливает то же количе-
количество примесей (Na2SO3, Na2SO4, NaCl), которое выделяется и при
выпарке раствора до той же концентрации тиосульфата.
Тиосульфат натрия
555
Сульфитный способ
Сульфитный способ получения тиосульфата натрия заключает-
заключается в растворении тонкоизмельченной серы в горячем растворе
сульфита натрия. Эта реакция протекает с малой скоростью осо-
особенно вначале, когда идет медленное смачивание серы; для уско-
ускорения этого процесса рекомендуют добавлять в раствор катионно-
активные вещества — бромиды амил-, октил-, децил-, додецил-, тет-
радецил-, бензил- и цетилпиридина1Б1 и другие, или производить
предварительное измельчение серы в смеси с маточным раствором
тиосульфата 152.
Варку раствора сульфита с серой производят в чугунном реак-
реакторе с мешалкой и паровой рубашкой. В реактор загружают воду,
безводный сульфит, 40—41% раствор NaOH D л на 100 л воды)
для предотвращения разложения сульфита и серу. Варку ведут
3—5 ч при 90—100°. К концу варки плотность раствора достигает
1,57 г/см3 и содержание сульфита в нем снижается до I —1,5%. По-
Полученный раствор имеет щелочную реакцию и усредняется раство-
раствором бисульфита натрия, к которому предварительно добавляют
кальцинированную соду C кг на 50л раствора бисульфата).Усред-
бисульфата).Усреднение продолжается 1 ч при 60—80°. Его производят для пере-
перевода избыточной щелочи и образующихся в процессе варки суль-
сульфида и дисульфида натрия в сульфит и тиосульфат:
NaOH + NaHSO3 = Na2SO3 + Н2О
2Na2S + 6NaHSO3 = 3Na2S2O3 + 2Na2SO3 + 3H2O
2Na2S2 + 6NaHSO, = 5Na2S2Os 4- 3H2O
Присутствие некоторого количества Na2SO3 в растворе необ-
необходимо как стабилизатора для создания определенного значения
рН, а также для перевода политионатов в устойчивый тритионат и
тиосульфат:
S5O26-
2S2Of"
Усредненный раствор отфильтровывают от примесей и направ-
направляют на кристаллизацию.
Сульфитный способ получения тиосульфата отличается боль-
большой простотой и высокой степенью использования сырья. Для про-
производства 1 г тиосульфата натрия расходуют 0,61 т сульфита нат-
Рия сорта «фото» (в расчете на 90%-нь1й), 0,2 т серы A00%),
4,01 т кальцинированной соды A00%), 0,04 г каустической соды
\Щ%) и 0,4 т бисульфита натрия B2,5%).
868
Гл. XIV. Соли сульфитного ряда
Получение тиосульфата натрия при мышьяково-содовой
очистке газов
В процессе очистки от сероводорода коксового и других газов
мышьяково-содовым методом, помимо основной реакции
Na3AsS3O + H2S = Na3AsS4 + Н2О
протекают побочные реакции, в частности взаимодействие серово-
сероводорода со щелочами, приводящее к образованию NaHS. При реге-
регенерации рабочего раствора кислородом воздуха тиомышьяковый
натрий переходит в тиооксимышьяковый натрий с выделением
серы
2Na3AsS4 + О2 = 2Na3AsS3O + 2S
Тиосульфат натрия
557
а гидросульфит окисляется в тиосульфат:
2NaHS + 2О2 = Na2S2O3 + Н2О
Помимо тиосульфата в растворе накапливаются также сульфат
и роданид натрия (образующийся из содержащихся в очищаемом
газе цианистых соединений). Содержание в рабочем растворе тио-
тиосульфата больше 250 г/л и сульфата натрия больше 40 г/л заметно
снижает поглотительную способность мышьяково-содового рас-
раствора, часть которого поэтому должна выводиться из цикла и за-
заменяться свежим. Из отработанного раствора выделяют тиосуль-
тиосульфат натрия. Для этого часть регенерированного раствора, после
отделения от него серы на вакуум-фильтре, выпаривают в вакуум-
выпарном аппарате до плотности 1,48—1,62 г/см3. Образующийся
при выпарке осадок, содержащий в частности Na2SO4, отделяют
отстаиванием или на друк-фильтре и раствор охлаждают в шнеко-
вом кристаллизаторе до 25—30°. Выделившиеся кристаллы тио-
тиосульфата натрия отделяют от маточного раствора центрифугиро-
центрифугированием и выпускают в качестве технического продукта 153.
Чистый тиосульфат натрия (сорт «фото») получают перекри-
перекристаллизацией технического продукта. Для этого приготовляют рас-
раствор тиосульфата плотностью 1,45 г/см3 при нагревании до 65—80°
и обрабатывают его 3—4 ч двуокисью углерода для осаждения
сернистых соединений мышьяка. После отделения осадка из рас-
раствора выкристаллизовывают чистый тиосульфат. Если технический
продукт загрязнен роданистыми соединениями, то для получения
из него фотографического тиосульфата необходима двойная пере-
перекристаллизация 154—156_ д0 перекристаллизации в продукте содер-
содержится 94-98% Na2S2O3-5H2O, 2-4,5% NaCNS, 0,17-1,7%
NaHCO3. 0,03—0,17% As2O3; после первой перекристаллизации
97—98,5% Na2S2O3 • 5Н2О, 0,2—1,45% NaCNS, 0-0,4% NaHCO3 и
0,015—0,05% As2O3; после второй перекристаллизации 99,2—99,7%
Na2S2O, • 5Н2О, от следов до 0,03% As2O3, NaCNS и NaHCO3 от-
отсутствуют.
В процессе выпарки раствора тиосульфата содержащиеся в нем
иолитионаты могут быть разложены сульфидом натрия; оптималь-
оптимальными для такого разложения являются температура 45—56° и
Другие способы производства
Среди других способов получения тиосульфата прежде всего*
следует обратить внимание на сероводородный способ, так как он,
в отличие от описанных выше, не требует в качестве сырья ценных
материалов — сернистого натрия и серы, а использует сероводород
любой концентрации, который является отбросом многих произ-
производств. Путем насыщения в абсорбционной башне сернистым га-
газом раствора соды подготовляется исходный сульфит-бисульфит-
ный раствор, содержащий около 220 г/л Na2SO3 и 180 г/л NaHSOe-
Этот раствор направляют в другую башню, где происходит абсорб-
абсорбция сероводорода. Вытекающий из башни раствор тиосульфата
фильтруют и направляют, как обычно, на выпарку и кристалли-
кристаллизацию.
Важным условием проведения реакции поглощения сероводо-
сероводорода является эквимолекулярное соотношение Na2SO3 : NaHSO3 в
исходном растворе. Оптимальное значение рН раствора равно 6,3.
Без заметного понижения выхода допустимы колебания лишь на
0,05 рН в ту и другую сторону. При значительном отклонении от
эквимолекулярного соотношения (больше 10%) выход тиосуль-
тиосульфата сильно падает, а содержание в растворе примесей (главным
образом политионатов) увеличивается. Оптимальная температура
реакции 20—30°.
Сероводородный способ также возможно осуществить без вы-
выпарки. Для этого соду растворяют в тиосульфатном маточном рас-
растворе и полученный раствор обрабатывают сернистым газом до
образования раствора, содержащего сульфит и бисульфит в экви-
эквимолекулярном соотношении.
Сульфатный метод позволяет получать тиосульфат без затраты
соды или продуктов, из нее изготовляемых (сульфита, бисуль-
бисульфита) 158. В основе его лежит реакция:
Na2SO4 + СаО + S + SO2 = Na2S2O3 + CaSO4
В раствор сульфата натрия вводят известь и серу и суспензию
перемешивают при 70—80° в течение 3 ч. Затем реакционную массу
обрабатывают сернистым газом при температуре не выше 65° во
избежание потери серы в виде сероводорода. Вначале, вероятно, об-
образуются сульфид, полисульфид и тиосульфат кальция, которые
вступают в обменное разложение е сульфатом натрия, давая тио-
тиосульфат натрия, а также сульфид и полисульфид натрия. Послед*-
ние, взаимодействуя с сернистым газом, Также переходят в тио-
тиосульфат. Помимо Ътого сульфат натрия частично каустифицируется
538
Гл. XIV. Соли сульфитного ряда
известью с образованием едкого натра и гипса. Едкий натр, реа-
реагируя с сернистым газом, образует сульфит, переходящий в при-
присутствии серы в тиосульфат. ¦
Источником тиосульфата могут служить сточные воды произ-
водств некоторых органических продуктов 159-160. Так, сточные воды
производства тиокарбанилида содержат 19—20% Na2S2O3, 4—5%
NaHS, 11—12% Na2SO3 и 2—3% органических веществ. Их смеши-
смешивают с раствором щелочи в варочном котле, снабженном рамно?
мешалкой, змеевиком и барботером. В котел вводят серу и желез-
железный купорос (для обесцвечивания), перемешивают реакционную
массу Зчи продувают через нее при 94—96° в течение 3 ч воздух.]
По окончании окисления раствор отфильтровывают от примесей
и направляют на кристаллизацию тиосульфата натрия.
Фильтраты из производства сернистых красителей содержат
18—27% Na2S2O3161; очисткой их от примесей, выпариванием и
кристаллизацией можно получить тиосульфат сорта «фото».
При взаимодействии сульфита натрия с пента- и тетратионатом
натрия образуются тиосульфат, а также тритионат:
Na2S6O6 + 2Na2SO3 = 2Na2S2O3 + Na2SsO6
Na2S4O6 + Na2SO3 = Na2S2O3 + Na2S3O6
При кипячении раствора 1 —1,5 ч в присутствии сульфита три-
тритионат почти полностью разлагается с образованием тиосуль-
тиосульфата 162. В этих условиях тиосульфат образуется в результате гид-
гидролиза тритионата, а сульфит натрия служит буфером, защищаю-
защищающим ионы S2O3~ от дальнейшего распада под влиянием ионов Н+.
В некоторых иностранных патентах предложено получать тио-
тиосульфат натрия распылением расплавленного натрия в атмосфере
SO2 с помощью вращающегося диска или сопла, через которое по-
подается струя SOa, или взаимодействием амальгамы натрия с раз-
разными растворами.
Обычный товарный продукт, пятиводный тиосульфат натрия
Na2S2O3 • 5Н2О, содержит 36,3% кристаллизационной воды. Безвод-
Безводный тиосульфат натрия вырабатывается пока в небольших коли-
количествах нагреванием пятиводной соли до полного обезвоживания.
При этом вследствие сильных местных перегревов происходит зна-
значительное разложение тиосульфата с выделением элементарной
серы. Поэтому при растворении безводного тиосульфата, получен-^
ного таким методом, образуются мутные растворы. я
Обезвоживание без разложения может быть произведено163 пла(
влением пятиводной соли при 48° в аппарате открытого типа или
в вакуум-аппарате. Расплав выпаривают до содержания в нел
78—82% Na2S2Os и выливают на холодильные вальцы, где он за-
застывает в прозрачную, стекловидную массу, состав которой соот-1
ветствует примерно двухводному тиосульфату. Застывшую соль i
виде чешуек направляют на вторую стадию обезвоживания в су-
Тиосульфат аммония
559
шилку, где ее сначала подсушивают при температуре не выше 62е
(во избежание плавления); затем температуру сушки поднимают
до 80—100° и продукт досушивают до безводного состояния.
Безводные или малогидратированные тиосульфата натрия и
аммония можно получать обработкой их растворов органическими
веществами, хорошо растворимыми в воде, но не образующими с
водой азеотропных смесей и не растворяющих тиосульфатов. Если
обрабатывать пятиводный тиосульфат метиловым спиртом, нагре-
нагретым до 60°, то при охлаждении массы до 30° будет выкристалли-
выкристаллизовываться малогидратированный тиосульфат, который затем мо-
может быть окончательно обезвожен сушкой в вакууме при 50°. Ме-
Метиловый спирт после отделения тиосульфата можно подвергнуть
дистилляции.
ТИОСУЛЬФАТ АММОНИЯ
Промышленные способы получения тиосульфата аммония анало-
аналогичны способам получения тиосульфата натрия или сводятся к об-
обменным реакциям между тиосульфатом натрия и солями аммония;
однако во всех случаях продукт образуется более загрязненным.
Чистый продукт получается по сложному и дорогому способу, в
основе которого лежит обменная реакция между тиосульфатом
кальция и карбонатом аммония.
Высококачественный тиосульфат аммония может быть получен
при действии концентрированного сероводорода при невысокой
температуре на раствор сульфит-бисульфита аммония по реакции:
2NH4HSO3 + 2(NH4JSO3 + 2H2S = 3(NH4)s,S2O3 + 3H2O
Выпарка растворов тиосульфата аммония, во избежание его
разложения с образованием сульфата и серы, должна произво-
производиться в вакууме при температуре ниже 80°164.
Тиосульфат (как и сульфат) аммония можно получить, пропу-
пропуская воздух при 150—160° и давлении 20—30 ат через содержащую
сероводород аммиачную воду коксовых и газовых заводов. Про-
Процесс ускоряется в присутствии катализаторов — переносчиков кис-
кислорода— солей меди, кобальта, никеля и других или активирован-
активированного угля 165'166.
Тиосульфат аммония можно также получать обработкой
раствора сульфита аммония элементарной серой в присутствии сво-
свободного аммиака, который предохраняет сульфит от разложения
при повышенной температуре (выше 80°) и высаливает часть тио-
тиосульфата. Маточный раствор после отделения этой части высолен-
высоленного тиосульфата возвращают в процесс. Для его ускорения тем-
температуру после начала реакции повышают до ПО—130° 167.
Запатентован способ получения тиосульфата аммония насы-
насыщением NH3 и SO2 раствора, получаемого смешением в отноше-
отношении 1:5—10 65% раствора полисульфида аммония и раствора,
660
Гл. XIV. Соли сульфитного ряда
содержащего 800 г/л (ЫН^гЭгОз. Насыщение газами ведут при из-
избытке аммиака и при температуре не выше 80°. Затем, охлаждая
раствор, выкристаллизовывают тиосульфат аммония, а маточный
раствор возвращают в процесс168. Запатентован способ предотвра-
предотвращения разложения безводного тиосульфата аммония добавкой к
нему F,01—10%) аммонийной соли слабой кислоты (угольной,
фосфорной, фталевой, лимонной) 169.
При производстве солей, рассмотренных в настоящей главе, че-
через неплотности в аппаратах в воздух рабочих помещений могут
проникать сернистый газ и сероводород. Эти газы вредны для здо-1
ровья. Поэтому аппараты должны быть герметичными или соеди-1
няться с вентиляционным устройством. Предельно допустимыми f
концентрациями у рабочего места являются: для сернистого анги-J
дрида 0,02 мг, для сероводорода 0,01 мг в 1 л воздуха.
ЛИТЕРАТУРА
1. П. А. Ходунов, Автореф. канд. дисс, ВММА, 1950. — 2. Э. И. Па-*
цаускас, Автореф. канд. дисс, АН ЛитССР, Вильнюс, 1952. — 3. N К о п о-
р i k, Monatsch. chem., 84, № 6, 1243 A953) .—4. R. H. Sahasrabudney
Current Sci., 22, № 3, 74 A953). — 5. Я- И. 3 и л ь б е р м а н, Труды ГИПХ, в. 35,
1941, стр. 3. — 6. 3. П. Розеикноп, Извлечение двуокиси серы из газов, Гос-
химиздат, 1952. —7. Н. А. Василенко, ЖПХ, 21, Mb 9, 917 A948)- 22, № 4
337 A949); 23, № 5, 472 A950); 26, № 6, 650 A953). — 8. Н. А. Василенко,'
Научно-техн. информ. бюлл. НИУИФ, в. 5—6, 105 A957). — 9. Е Тег res
Е. Hahn, Gas u. Wasserfach., 70, 309, 339, 363, 389 A927). —10. F. Ischika-
wa, H. Hagisawa, Bull. Inst. Phys. Chem. Res. (Tokyo), 10, 166 A931).
//. F. Ischikawa, T. Murooka, Sci. Rep. Tohoky, Imp. Univ., 22, 201
A933). —12. И. Н. Кузьминых, Е. Л. Яхонтов а, В. Г. Воробьева
Труды МХТИ, в. 22, 1956, стр. 202. — 18. Е. М. Я к и м е ц, М. С. А р х и п о в а!
Труды УНИХИМ, в. 1, 1954, стр. №. — 14. В. А. П и н а е в, ЖПХ, 37, № 6,
1361 A964). — 16. А. Н. Кет о в, В. В. Печковский, Там же, 31, № 12 1783
A958). — 16. Н. С. Mel, Z. Z. Hugus, M. W. L a t i m e r, J. Am. Chem.'Soc,
78, № 9, 1822 'A956). — П. J. Das, D. Patnak, J. Indian Chem. Soc 33, № 4,
243 A956). —18. M. Darrin, Can. Chem. Proc. Ind., 33, № 6, 512 A949) —
19. S. Haas, Metal. Finish, 49, № 4, 64 A951). — 20. Польск. пат. 35331, 1953.
21. Я. Зильбермаи, Л. А. Запутряева, Труды ГИПХ, в. 39, 1947,
стр. 3. — 22. G. В г е а к 1 у, J. S t а г к i e, Am. Dyest Rep., 38, № 22, 775 A949). —
23. Н. Г. 3 а л о г и н, Труды совещания по очистке промышленных газов, Гос-
металлургиздат, 1941. — 24. А. Г. Павлович, авт. свид. 58179, 1940.—
25. Н. П. Слотвииский-Сидак, Автореф. каид. дисс, МХТИ,' i953. —
26. Н. Г. Залогии, С. М. Шухер, Очистка дымовых газов, Госэиергоизаат,
1954.— 27. Chem. Eng., 66, Mb 12, 147 A959). — 28. R. Dermerdonk Cli-m
Ing. Techn., 37, № 11, 1136 A965).— 29. D. H enrich, Staub, 26, Mb 10, 429
A965). — 30. А. П. Андрианов, М. И. Левин, М. А. Гурфинкель Хим.
пром., Mb 11, 339 A950).
31. А. П. Андрианов, В. А. Чертков, Хим. пром., № 7, 394 A954).-|
32. Б. А. Чертков, Г. Е. Аристов, Д. Л. П у х л и н а, Там же № 1 11
A956).— S3. Б. А. Чертков, ЖПХ, 32, № 8, 1695 A959); Кокс и химия № II
48 A959). — 34. Б. А. Чертков, Д. Л. Пухлина, ЖПХ, 33, № 1, 5 A960) --Г
35. Б. А. Чертков, Там же, 37, № 11, 2437 A964).— 36. Б А Чертко1
Хны. пром., № 1, 44 A964). — 37. Б. А. Чертков и др., Там же Я° 1 ~
¦ Литература
561
П964) —38 Б. А. Чертков, Там же, № 9, 685 A966). — 39. Л. С. Гордеев,
Б А Чертков, Там же, № 2, 129 A967). —4ft И. Н. Кузьминых,
Д. М. Попов, Б. И. Горбачев, Там же, № 2, 128 A960).
41 R A. King, Ind. Eng. Chem., 42, Mb 11, 2241 A950). -42. H. В. Се-
Седов Научно-техн. информ. бюлл. НИУИФ, в. 5-6, 79 A957). -43. Я- Зиль-
бер'ман П Иванов, ЖПХ, 13, Mb 4, 541 A940). — 44. В. Фридман,
Н Тимкин, Хим. пром., № 5, 25 A946). -45. S. J. Pirt, D. S Callow,
W A. Gillett, Chem. Ind., № 23, 730 A957).— 46. А. Б. Пакшвер,
T И Kvhhh А. В Куконкова, Хим. пром., № 7, 209 A950). —
47 Б.' А. Черт'ков, ЖПХ. 32, Mb 8. 1695, 1707 A959); № 12. 2609 A959).—
48 Б А Чертков, Д. Л. Пухлина, Г. И. Пекарева, Там же, № 6.
1385 'A959) — 49. Б. А. Чертков, Там же, 33, № 8, 1708; М> 10, 2165
(I960). — 50. С. А. Сапотиицкий, Н. В. Глущенко, Там же, 35, Mb 10,
2191 A962).
51 А М Кагановский, П. Н. Таран, Укр. хем. ж., 21, № 4, 472
П955)' — 52 D Winkelmann, Z. Elektrochem., 59, Mb 9, 891 A955).—
53 С Н Barron, H. A. O'Hern, Chem. Eng. Sci., 21, Mb 5, 397 A966).—
54 К / A Waal, ibid., № 6—7, 559.-55. Б. А. Чертков. ЖПХ. 30,
Mb' ю' 1496 A957). — 56. M. Terres, A. Heinsen. G. W. F., 994 A929).—
57 F S W a r t m a n U. S. Dep. Inter. Bfiro Mines Rep. Inv., Mb 3339, 47
A937) — 58 E С Fuller. R. H. Grist, J. Am. Chem Soc, 63, Mb 6, 1644
A941) —59 С И. Вольфкович, Д. Л. Цырлин, ЖПХ, Mb 18, 1323
A929). — 60. Б. А. Чертков, ЖПХ, 32, Mb 1, 78 A959).
61 Б А Чертков, Там же, 32, № 5, 952, 960 A959); Вестн. техи. и экон.
ииф Mb 5 A0), 7 A958). — 62. Б. А. Чертков, ЖПХ, 34, Mb 4, 771 A961).—
63. Т. Д. Авербух, Хим. пром., Mb 11, 1 A948). — 64. Л. И. Каштанов,
А Б Лукьянов, Труды Московск. инж.-экои. ин-та. Химия и химические
производства, Mb 1, 1954, стр. 135. — 65. М И. Дорыкииа. Там же. стр. 143.—
66 М Г Манвелян и др., ЖПХ, 34, Mb 4, 934 A961). — 67. W. D. Scott,
J Z McCartney, Ind. Chem. Fundament., 6, Mb 1, 40 A967). — 68. J. Vo
solsobe, A. Simecek, J. Michalek, B. Kadlec, Chem. prumysl., 16, Mb7,
401 A965). — 69. Т. К. Джабагин, Д. К- Рой. П. А. Семенов, Хим. пром.,
Mb 11 870 A963). — 70. Н. F. John stone, P. Leppla, J. Am. Chem. Soc,
Mb 56,'2233 A934).
71 H F Johnstone a. oth., Ind. Eng. Chem., 27, № 5, 587 A935); 27,
№ 6, 659 A935); 29, Mb 3, 286 A937); 29, Mb 12, 1396 A937); 30, Mb 1, 101 A938);
32, № 8 1037 A940). — 72. P. А. Б е р д я и с к а я, С. М. Г о л я и д, Б. А. Черт-
Чертков ЖПХ 32, № 9, 1930 A959). — 73. Б. А. Чертков, Н. С. Добромыс-
лов'а, Там же 37, Mb 8, 1718 A964). — 74. В. А. П и и а е в. Там же, 36, Mb 10,
Si 16 A963) —75. В. А. Пинаев, Н. П. Пителина, Там же, 38, № 8, 1652
A965) —76. И. Н. Шокин. И. М. Егоркин, М. И. Хайтович, ЖХП,
№ 7 11 A938) —77. В. М. Р а м м. Абсорбция газов, Изд. «Химия», 1966.—
78. Т. Д Авербух, Труды УНИХИМ, в. 1, 1954, стр. 92.-79. F. H. Con-
Conrad, W L. Beuschle'in, Paper Trade J., 105, Mb 4, 37 A937); J. Am. Chem.
Soc, 56, 2554 A934). — 80. И. H. Кузьминых, М. Д. Бабушкина, ЖПХ,
29, Mb 10, 1488 A956)'.
81. И. Н. Кузьминых, М. Д. Бабушкина, Там же, 30, Мг 3, 466
П957).->-в2 И. Н. Кузьминых, А. Г. Кузнецова, Там же, 27, Mb 8
A954). — 83. И. Л. Пейсахов, В. Д. Карамзина, Там же, 32, № 1, 70
A959). — 84. Г. П. Архипов а, И. Е. Флис, К- П. Мищенко, Там же,
37. Mb 10, 2306 A964); 38, № 7, 1494 A965). — 85. М. Е. Позин, Хим. пром.,
№ 6, 14 A944). — 86. И. М. Вассермаи, Производство минеральных солей,
Госхимиздат, 1954. — 87. К. R. Gray, H. L. Crosby, пат. США 2656249,
•953. —88. Япои. пат. 8176, 1954. —89. R. Griessbach, H. Hohmann, пат.
562
Гл. XIV. Соли сульфитного ряда
ГДР 7483, 1954.— 90. В. И. Николаев, Т. И. Арнольд, ЖПХ, 16, № 7—8
A943).
91. R. S. Aries, A. Poll а к, пат. ФРГ 900335, 1953; каиад. пат. 504058 |
1954; швейц. пат. 297007, 1954. —92. 1. Е. Dreenawalt, пат. США 3165378,
1961. — 95. W. Spormann, J. Heinke, пат. ФРГ 1152386, 1962.—
94. И. Л. Гофман, Р. Е. Ошерович, Хим. пром., № 12, 13 A944).—
95. Е. Р а е г s с h, пат. ГДР 5657, 1954. — 96. Т. И. К у н и н, В. С. Епифанов,
Изв. вузов. Хим. и хим. технол., 4, № 6, 992 A961); 5, № 5, 770 и № 6, 864 |
A962). — 97. И. Г. Бляхер, М. М. В а й с б е й н, И. С. Черный, авт. свид. I
197536, 1966. —98. И. Л. Гофман, Р. Е. Ошерович, Хим. пром., № 2—3,
24 A944). — 99. Я. И. Зильберман, П. Т. Иванов, В. М. Фридман,
Сборник статей к 25-летию ГИПХ, Госхимиздат, 1944, стр. 223, 236. — |
100. J. Cook, Chem. Trade J., № 3029 A945).
101. J. A. L a b a s с h, G. R. L u s b y, Canad. J. Chem., № 5, 774, 787 j
A955). — 102. D. A. Skinner, пат. США 2666687, 1954. —103. N. Bhuiyan,
Pakistan J. Sci. Res., 16, № 2, 74 A964). —104. Я- И. Зильберман,
В. M. Фридман, Труды ГИПХ, в. 39, 1947, стр. 52. — 105. L. К г a u s, M. R и-
ску, В. Hayek, пат. ЧССР 115249, 1963. — 106. И. Г. Бляхер, Т. Д. А вер-
вербу х, М. М. Вайсбейн, Н. И. Кротов а, Труды УНИХИМ, в. 14, 1967,
Стр. 25. —107. Л. С. Ефименко, А. Аширова, А. Атаджанова, Изв.
АН ТуркмССР. Сер. физ.-техн., хим. и геол. наук, № 2, 24 A963).—
108. Е. С. Л е ц к и х, Н. В. Б о й к о в а, Е. Я. Зверева, Хим. пром., № 4,
309 A967). —109. Е. М. Якимец, Л. М. Гольдвассер, ЖХП, № б|
A934). —110. Л. К. Ахрап-Симонова, Я. И. Зильберман, К- Ф- Пав-|
лов, Труды ГИПХ, в. 35, 1941, стр. 38; ЖПХ, 12, № 3, 346 A939).
Ш. 3. С. Б а иных, Е. М. Полякова, Труды УНИХИМ, в. 8, 1959,1
стр. 91, 101. —112. И. Г. Бляхер, А. Г. Л ар юшки на, ЖПХ, 35, № 3, 503!
A962). — 118. М. А. Черняк, И. А. Генкин, Хим. пром., № 6, 178 A950).—
114. Е. М. Маршак, Хим. иаука и пром., 2, № 4, 524 A957). — 115. Т. И. Ку-
нин, Ю. А. Лобанов, Изв. вузов. Хим. и хим. технол., 8, № 6, 921 A965).—
116. Я. И. Зильберман, И. Л. Хмельницкая, В. р. Чудинов,
А. Ф. Павлова, Труды ГИПХ, в. 37, 1946, стр. 21. —117. Chem. Trade J.,
119, № 3086, 35 A946). — 118. V. L. Hansley, L. F. Moormeier, пат. США j
3216791, 1962. —119. V. L. Hansley, L. F. Moormeier, S. Schott, пат.
США 3205038, 1961. —120. Ind. Chemist a. Chem. Manufact., 29, № 343, 3551
A953).
121. Франц. пат. 1065095, 1954. —122. Франц. пат. 1024671, 1953. — |
123. Англ. пат. 695375, 1953. — 124. Ю. А. Лобанов. В. А. Ш о р м а н о в,
Т. И. Кунин, Изв. вузов. Хим. и хим. технол., 9, № 1, 10 A966). —• I
125. И. Б. 3 и л ь б е р б е р г, М. И. С т у л ь, М. И. Галкин, Б. И. Ястре-
Ястребов, авт. свид. 175047, 1964. — 126. Я- И. Зильберман, Н. Г. Маркова»
Труды ГИПХ, в. 37, 1946, стр. 5. —127. В. Г. Хомяков, В. П. Маш овец,
Л. Л. Кузьмин, Технология электрохимических производств, Госхимиздат,
1946. -128. В. К. В ей дин, Труды НИОХИМ, т. 19, 1969, стр. 176.—
129. R. Jellinek, Elektrochem. angew. phys. Chem., 17, 157 A911)'.—
ISO. F. Feher, E. S с h 1 i e p, H. Weber, Z. Elektrochem., 57, № 10, 916 A953).
131. Норвеж. пат. 84483, 1954.—132. A. Wurbs, Chem. Age (India), 16,
№ 10, 825 A965). —133. J. A. D у а п, Т. P. Whaley, пат. США 3226185,
1961. — 134. J. H. Muril, пат. США 3216790, 1962. — /55. В. Ф. Постников,
Т. И. Кунин, ЖПХ, 13, №2,185A940). —136. В. Ф. Постников, Т. И. Ку-
Кунин, Труды Ивановск. химико-технол. ин-та, в. 3, 1949, стр. 126. —137. Т. И. Ку-|
нин, М. А. Власюк, Изв. вузов. Хим. и хим. технол., 4, № 4, 636 A961).
138. Т. И. Кунин, Труды Ивановск. химико-технол. ин-та, в. 5, 1956, стр. 90. — I
139. Т. И. Кунин, ЖПХ, 21, № 6, 685 A948).— 140. Т. И. Кунин, М. А. В л а-1
сю к. Труды Ивановск. химико-технол. ин-та, в. 5, 1956, стр. 97.
Литература
663
141. В. В. Буданов, Т. И. К у к и н, Г. И. Куракина, Изв. вузов. Хим.
и хим технол., 10, № 5, 546 A967). — 142. Schenk, Р 1 a t z, Z. anorg. Chem.,
222, 177 A935). —143. А А Гринберг, ЖПХ, 21, № 5, 425 A948).—
144 Д И. Менделеев, Основы химии, т. II, изд. 13, 1947, стр. 538.—
145 И М Вассерман, Труды ИРЕА, в 20, 1951, стр. 176.—146. Я. И. Зиль-
Зильберман, К. Г. Круглякова, Н. Д. Черткова, ЖПХ, № 9, 6 A941).—
147 Франц пат. 1077539, 1954. — 148. Е. Schulek, Е. Кбгбз, L. Maros,
Ada chim. Acad. Sci. Hung., 10, № 1—3, 291 A956). — 149. Г. П. Л у ч и н с к и й,
М И Попова, ЖПХ, 12, № 11, 1664 A939). —150. Я. И. Зильберман,
. № 11, 21 A940).
151. G. I. P. Levenson, J. Appl. Chem., 4, № 1, 13 A954). — 152. Т. Г. Ах-
метов Л И. Кузнецов-Фетисов, Труды Казанск. хим.-технол. ин-та,
в. 34, 1965, стр. 18; з. 36, 1967, стр. 26; авт. свид. 172729, 1964; 181059. 1965. —
153. И. Я А з б е л ь, Мышьяково-содовая очистка газа от сероводорода, Госхим-
Госхимиздат, 1956. — 154. Н. Н. Поляков, А. Ф. Ложкин, ЖПХ, № 4—5, 55
/1940) —155. Н. Н. Поляков, А. В. Гранжан, Хим. пром., № 7, 199
A950). —156. А. Б. Телепнева, Т. Д. Авербух, Труды УНИХИМ, в. 8,
1959, стр. 122. —157. Т. Г. Ахметов, Л. И. Кузнецов-Фетисов, Труды
Казанск. хим.-технол. ин-та, в. 34, 1965, стр. 25. — 158. Г. П. Л у ч и н с к и й,
Труды ГИПХ, в 35, 1941, стр. 35. — 159. П. В. Д ы б и н а, Технология мине-
минеральных солей, Госхимиздат, 1949, стр. 166. — 160. Р. И. Л е в е н з о н, В. В. К а-
фаров. ПОХ, 7, № 8, 439 A940).
161. П. В. Дыбина, ЖХП, № 1, 58 A940). — 162. И. Л. Хмельницкая,
Труды ГИПХ, в. 37, 1946, стр. 64.-/65. Я. И. Зильберман, В. М. Ф р и д-
ман, Там же, в. 39, 1947, стр. М. — 164. Я. И. Зильберман, В. М. Фрид-
Фридман, Сборник статей к 25-летию ГИПХ, 1944, стр. 203. —165. Англ. пат. 71867Б,
1954. —166. Пат. ФРГ 902973, 1954; 925408, 1955. —167. Норвеж. пат. 81235.
1953; дат. пат. 76777, 1953. —168. W. Muller, пат. ФРГ 1184330. 1963.—
169. Пат. США 3350168, 1963.
Физико-химические свойства
565-
Глава XV
СОЛИ ХРОМА
Из соединений хрома наибольшее промышленное значение при-
приобрели натриевые и калиевые соли хромовых кислот, в особен-
особенности соли двухромовой кислоты — хромпики — Na2Cr2O7-2H2O и
К2СГ2О7, а также хромовые квасцы и основные сульфаты хрома
(сухие хромовые дубители), хлорный хром, окись хрома и хромо-
хромовый ангидрид. Современная технология этих продуктов разрабаты-
разрабатывалась в СССР главным образом в Уральском научно-исследова-
научно-исследовательском химическом институте и на предприятиях и наиболее
полно отражена в монографии '.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Хром образует с кислородом окислы СгО, СггОз, СгСЬ, CrOs и
перекиси СгО4, СгСу, из них наибольшее практическое значение
имеют Сг2О3 и СгО3 и их соединения.
Закись хрома СгО черного и красного цвета, не растворима в
воде и в разбавленных кислотах. Гидрат закиси хрома Сг(ОН)з
желто-коричневого цвета, разлагается в воде и при нагревании;
растворим в концентрированных кислотах.
Окись хрома Сг2Оз зеленого цвета кристаллизуется в гексаго-
гексагональной системе, имеет плотность 5,21 г/см3, плавится при 2435°2.
Температура кристаллизации окиси хрома несколько изменяется
в зависимости от условий ее получения3. Состав гидратов окиси
хрома окончательно не установлен. По разным данным гидраты
окиси хрома могут содержать от одной до девяти молекул воды, в
зависимости от условий получения4. Разноречивы и данные о тем-
температурных интервалах существования отдельных гидратов 6~10.
Так, например, по одним данным8 в системе Сг2О3—Н2О сущест-
существуют три стабильные фазы: Сг(ОНK, СгООН и а-Сг2О3; граница
между Сг(ОНK и СгООН лежит при ~55°, а между СгООН и|
СО при ~540° и мало смещается при повышении давления.
По другим данным9 стабильной является и Сг2О3 • 5Н2О, устойчи-
устойчивая между 68 и 85—95°, а Сг(ОНK устойчива до 150°, СгООН об-
образуется при 320—360° под давлением 320—350 ат. Данные дру-
других авторов отличаются от приведенных. Прокаленная окись хрома
в воде не растворима и плохо растворяется в кислотах. Гидроокись
хрома растворяется в кислотах с образованием солей трехвалент-
трехвалентного хрома, а с сильными основаниями образует хромиты:
Сг(ОНK + 3NaOH = Cr(ONaK + ЗН2О
Зеленая или фиолетовая окраска солей трехвалентного хрома,
их кристаллическая форма и свойства зависят от возникновения
тех или иных комплексных структур. Например, гексагидрат хлор-
хлорного хрома СгС13 • 6Н2О имеет две структуры — зеленую
[СгС12(Н2ОL]С1 -2Н2О и фиолетовую (называемую также серой)
[Сг(Н2О)б]С1зп'12. Безводный хлорный хром образует красно-фио-
красно-фиолетовые кристаллы (tnsi 1152°), легко возгоняющиеся в токе хлора;
начинает реагировать с кислородом при 350е (СгС12 — при 404°);
при 514° восстанавливается водородом до СгС12; перегретым водя-
водяным паром при 350—450° разлагается на Сг2О3 и НС113. Раствори-
Растворимость СгС13 в воде очень мала (почти не растворим), но увеличи-
увеличивается в присутствии следов СгС12. Растворимость зеленого
СгСЬ • 6Н2О при 15е около 130 е в 100 г воды. Гидролиз СгС13 в
водных растворах приводит к образованию при 20—30е Сг(ОН)С12,
а при 40—50° Сг(ОНJС1 ">'15 • О Na3CrCl6, KsCrCl6, К3Сг2С19 см.le.
Сульфат хрома, получаемый растворением гидроокиси хрома в
серной кислоте, выделяется из раствора в виде темно-фиолетовых
кристаллов Cr2(SO4K- 18Н2О 17>18. Они плавятся при 80—85е с от-
отщеплением 10 молекул воды; остальные 8 молекул воды удаля-
удаляются при ПО—115°. Сообщают также19, что Cr2(SO4)s- 18H2O при
нагревании отщепляет только 16 молекул воды, при дальнейшем
же обезвоживании выделяется SO3 и образуется основная соль
Cr2O(SO4J устойчивая в интервале температур 460—640°. По дру-
другим данным 20 фиолетовый сульфат хрома, полученный введением
эквивалентного количества HsSO4 в охлажденный раствор
Cr(NO3K, имеет состав Cr2(SO4K • 15Н2О; при 90—95° он перехо-
переходит в девятиводный, при 120° — в шестиводный кристаллогидрат, а
полная дегидратация протекает при 400° и не сопровождается гид-
гидролизом. Разложение обезвоженного сульфата до Сг2О3 протекает
при 700—735°.
Зеленая модификация сульфата Сг3+ получается при восстанов-
восстановлении GrO3 в сернокислом растворе. При этом обычно образуется
пересыщенный раствор, превращающийся при выпаривании в тем-
темно-зеленую 21 вязкую массу, а после высушивания ее в стекловид-
стекловидное вещество состава Cr2(SO4K • 8Н2О. Эта соль теряет 2,6 моле-
кулы воды при 120—125°, остальная вода удаляется только при
440—445°18. Описаны22 кислые сульфаты трехвалентного хрома
566
Гл. XV. Соли х$ома
3Cr2(SO4K-SO3, Cr2(SO4K-SO3, 3Cr2(SO4)8-2H2SO4- 3H2O cepo-
эеленого цвета, не растворимые в воде, выделяющиеся в мелкокри-
мелкокристаллическом виде при кипячении с серной кислотой бихроматов К,
NH4 и окиси хрома.
Хромовые квасцы — двойные соли сульфата трехвалентного
хрома и сульфатов Na, К, NH4 и других — имеют состав
M5SO4-Сг2($О4)з-24Н2О. Растворимость квасцов в воде резко
возрастает с повышением температуры. Они кристаллизуются в
виде темно-фиолетовых октаэдров. Водные растворы также имеют
фиолетовую окраску, но при нагревании они становятся зелеными
вследствие диссоциации двойных солей (для хромово-калиевых
квасцов выше 78е). При выпаривании растворов выделяется
аморфная зеленая масса. Калиевые квасцы имеют плотность
1,84 г/ал*3, плавятся при 89е; в 1 л насыщенного водного раствора
содержится при 26° 243,9 г K2SO4- Cr2(SO4K -24Н2О (или 1?.5,1 г
безводной соли). Аммониевые квасцы имеют плотность 1,72 еГсмъ,
разлагаются при 100е; растворимость при 26е 21,2 г в 100 е воды.
Описано получение некоторых аммиакатов трехвалентного хро-
хрома - [Cr(NH?)eJ(8O4)?- 2H2SO4 • ЗН2О, [Cr(NH3)e]2(SO4)8 • H2SO4>
[Cr(NHs)e]2(SO4K-5H2O, [Cr(NH8MH2O](NO8)8, [Cr(NH8MH2O]Cl8.
• CuCl2. Последний, в отличие от нитратного комплекса, устойчив
при хранении. Осажденная аммиаком из хромовых квасцов гидро-
гидроокись хрома при нагревании в течение 1 ч до 40—45е образует рас-
раствор аммиаката 23.
В системе Cr2O3—SiO2 установлено существование соединения
Сг2О3 • 3SiO2, плавящегося без разложения при 1995°; имеются две
эвтектики — при 1750е F5 мол. % Сг2О3) и при 1680е E мол. %
Сг2О3) 24.
Хромовый ангидрид СгО3 красного цвета кристаллизуется в
ромбической сингонии, имеет плотность 2,7 е/см3, при 170° незна-
незначительно разлагается в твердом состоянии, в связи с чем плавится
при непостоянной температуре в пределах 180—202°; расплавлен-
расплавленный склонен к переохлаждению и затвердевает около 170°8. Яв-
Является сильным окислителем. Растворимость в воде при 0°61,7%,
при 100° 67,46% СгО325. При растворении в воде образует хромо-
хромовую кислоту Н2СгО4, в свободном виде неизвестную. В системе
СгОз—H2SO4—Н2О существуют твердые фазы СгО3 и СгО8 • SO3 2б.
При растворении СгО3 в разбавленной серной кислоте (до
~30% H2SO4) протекают процессы полимеризации с образова-
образованием иона Сг2О|" и ассоциации с образованием иона НСг2О7 и не-
Диссоциированнбй двухромовой кислоты:
2НСгСГ ^=± Сг2О2
Cr2O2" + H+ ^i
f+ H+ ^zt H2Cr2O7
Физико-химические свойства
567
Н2О
В растворе, содержащем от 30 до 55% H2SO4, кроме двухро-
двухромовой кислоты, образуется, по-видимому, также хромосерная кис-
кислота
27, Й.
CrO3 + H2SO4 +± H2CrSO7
Предполагают, что в растворах с концентрацией больше 55°/о
H2SO4 идет дальнейшее присоединение серной кислоты к хромосер-
ной кислоте
H2CrSO7 + H2SO4 ^~± CrO2(HSO4J + Н2О
дальнейшая полимеризация
2Н2Сг2О7 + H2SO4 ^=±: Н2Сг4О,з + H2SO4 • Н2О
H2CrSO7 + H2Cr2Or ^z± H2Cr3SO,3 + H2O
и дегидратация хромосерной кислоты:
2H2CrSO7 + H2SO4 ^z± 2(СгО3 ¦ SO3) + H2SO4 • 2H2O
В растворах, содержащих H2SO4 • 2Н2О, т. е. при концентрации,
большей 73% H2SO4, хромовый ангидрид неустойчив. Цвет таких
растворов постепенно из оранжевого переходит в зеленый. Вообще
при взаимодействии с сильными кислотами при нагревании СгО3
образует зеленые или фиолетовые соли этих кислот, например
Cr2(SO4K. При нагревании водного раствора хромовой кислоты до
300—325° в закрытом сосуде (при равновесном давлении кислорода
50—200 ат) она разлагается с образованием осадков СгО2 и
HCrO2 2S.
При термическом разложении хромового ангидрида при нор-
нормальном давлении образуются: декахромат хрома Сг2(Сг10О31)з =¦
= СгО2,90в, бихромат хрома Сг2(Сг2О7)з = CrO2i625 и монохромат
хрома Сг2(Сг04K = Сг02,4о, а также твердые растворы в пределах
от СгО3 до СгО2,98 и от CrOi,54 до CrOi^o- Под давлением обра-
образуется также двуокись хрома СгО28'30-31. О перекисях и перекис-
ных соединениях хрома см.32-34.
Оксихлорид хрома СгО2С12, или хлористый хромил, — темно-
красная жидкость с плотностью 1,911 г/см3 при 20е, плавится при
'—96,5е. Давление пара в (ммрт.ст.): 10 при 13,8°, 100 при 58е, 400
при 95,2°, 760 при 117,1° (tK). Водой, а также на свету и при нагре-
нагревании разлагается. Образуется при взаимодействии газообразного
НС1 и СгОз или при обработке концентрированной серной кисло-
кислотой смеси бихромата и хлорида.
В системе CrO3—HF—Н2О при 0° существуют две твердые фа-
фазы— СгО3 и CrO2F235. Последний плавится (под давлением)
при 31,6°.
Соли хромовой кислоты имеют желтую окраску, двухромовой —
оранжево-красную. Концентрированные растворы Na2Cr04 и
КгСгО4 имеют рН *» 9, Na2Cr2O7 и К2Сг2О7 — около 4 36. Кислая
Реакция двухромовокислых солей обусловлена взаимодействием
568
Гл. XV. Соли хрома
иона Сг2О7 с.водой с образованием ионов Н+ и СгО4~, причем уста
навливается равновесие:
f z± 2НСЮ4 ^=
Na2Cr2O,
Физико-химические свойства
2
При прибавлении к раствору какой-либо кислоты (ионов Н+)
равновесие сдвигается влево; при прибавлении какой-либо щелочи
(ионов ОН~) — вправо. Это по-
позволяет переводить монохроматы
в бихроматы и наоборот путем
изменения рН раствора. В кислой
среде бихроматы являются силь-
сильными окислителями; при их вос-
восстановлении образуются соедине-
соединения трехвалентного хрома.
Хромат (монохромат) натрия
Na2Cr04 кристаллизуется из вод-
водных растворов до 19,52° в виде
моноклинных сильно гигроскопич-
гигроскопичных (расплывающихся на воз-
воздухе) кристаллов декагидрата
Na2Cr04 • 10Н2О с плотностью
1,483 г/см3; в пределах от 19,52 до
26,6° B5,9°) кристаллизуется гек-
сагидрат Na2Cr04- 6Н2О, от 26,6
до 62,8° — Na2Cr04 • 4Н2О. Выше
62,8° образуются ромбические
кристаллы безводного a-Na2Cr04,
переходящие при 413° в гексаго-
,00 нальные p-Na2Cr04, плавящиеся
при 792°. Растворимость хромата
натрия в воде 24,2% при 0°,
44,3% при 20° и 56,1% при 100°
(рис. 168). В системе Na2CrO4—
Na2SO4—Н2О при 15° твердая фа-
Na2CrQ«
Na2CrO4-4HjO
Na*CrO4
20 40 €0 80
Температура, °С
Рис. 168. Растворимость хроматов и
бихроматов в воде.
за представляет собой непрерывный ряд смешанных кристаллов
декагидратов хромата и сульфата натрия, а при 25° кристалли-
кристаллизуются твердый раствор Na2SO4-10Н2О—Na2CrO4-6H2O, безвод-
безводный Na2SO4 и Na2CrO4-6H2O (рис. 169) 37-38. В системе Na2Cr04—
Na2CO3—Н2О при 35° кристаллизуется Na2CrO4-4H2O, Na2CO3-
• Н2О и твердый раствор беркеитового типа, образуемый двойной
солью 2Na2CrO4-Na2CO3 и исходными безводными солями39.
Хромат калия К2СгО4 кристаллизуется безводным. Ниже 669°
устойчива ромбическая модификация, выше этой температуры —
гексагональная. Плотность при 18° 2,73 г/см3; плавится при 975°.
Растворимость в воде 37,1% при 0°, 45,2% при 100° (рис. 168).
В системе К2Сг04—Кг8О4—НгО при 25° выделяется одна твердая
фаза изоморфных солей. В системе К2СгО4—Na2CrO4—Н2О
при 25° выделяются три твердые фазы: Na2CrO4 • 6Н2О,
ЗК2Сг04 • Na2CrO4 (хромовый глазерит) и К2Сг04. Хромовый гла-
зерит, в отличие от сернокислого, образует двусторонний твердый
раствор с пределами концентраций 3,16К : Na и 2.39К : Na. В си-
системе КгСгО4—Na2SO4—Н2О при 25° имеется пять полей кристал-
кристаллизации: 1) непрерывного твердого раствора хромата и сульфата
калия, 2) сложного тетрагенного твердого раствора глазерита,
3) твердого раствора Na2SO4 • 10Н2О—Na2Cr04 • 6Н2О, 4) безвод-
безводного Na2SO4 и 5) Na2Cr04 •
. 6Н2О 38-40. При 208—220° К2Сг04 #г
взаимодействует с расплавлен-
расплавленным СгО3 с образованием КгСг2О7. И
При 270—280° К2Сг04 реагирует
с продуктом разложения СгО3 —
бихроматом хрома:
//
///
= ЗК2Сг207 + Сг2(СгО4)з
ю
50-
20 30 40
Na2CrO4 ,0ec.%
При ЭТОМ ВЫХОД К2Сг207 ПО рт m Изот растворимости
обеим реакциям составляет 80— в системе Na2CrO4-Na2SO4-H2O
85%. Для полного превращения при 25°:
ХрОМата В бихромат ТребуетСЯ ИЗ- /-твердые растворы Na2SO4.10H2O а
быток СгОз против стехиометри- Na2»?iVfa"c?o"^2oS°4!
ческого количества 4l. О свойствах
безводных систем, содержащих хроматы и гидроокиси, сульфаты,
нитраты, фториды натрия и калия, см. 42~45.
Хромат аммония (NH4JCr04 выделяется из водных растворов
в форме моноклинных кристаллов; плотность при 12°— 1,917 г/см8.
Выше 180° и в горячих водных растворах разлагается. Раствори-
Растворимость в воде при 30° 28,8%.
Хромат кальция существует в пяти модификациях: СаСгО4,
СаСгО4 • 0,5Н2О, СаСгО4-Н2О, а- и р-СаСгО4-2Н2О. Наиболее
» растворим в воде а-дигидрат A4,75% СаСгО4 при 0°), наименее-
растворим безводный хромат кальция D,3% СаСгО4 при 0°). С уве-
увеличением температуры их растворимость несколько уменьшается.
Безводный хромат кальция образуется выше 400°, двухводные гид-
гидраты — ниже 25°.
Бихромат натрия Na2Cr20? кристаллизуется выше 84,6° в без-
безводном виде, а ниже этой температуры — с двумя молекулами во-
Ды (Na2Cr2O7 • 2Н2О) в виде моноклинных призм или тонких игл»,
имеющих плотность 2,35 г/см3. Сильно гигроскопичен, на воздухе-
¦ Расплывается. Безводный плавится при 357°; при 400° разлагается
с выделением кислорода. Растворимость Na2Cr2O7 в воде при 20е*
73,18%, при 100° 91,43%. Растворимость в присутствии Na2SO4 и*
С1 см.46. Существуют три- и тетрахроматы натрия Na2Cr3Oi0 ц
670
Гл. XV. Соли хрома
Применение
571
Na2Cr4Oi3, которые плавятся (соответственно) при 232 и 168° и раз-
разлагаются при 337 и 320°47.
Бихромат калия К2СГ2О7 кристаллизуется из водного раствора
безводным в триклинической системе, негигроскопичен, плотность
при 25° 2,68 г/см3. Существует в трех модификациях — устойчивая
при обычной температуре модификация А превращается при 269°
в модификацию С с увеличением объема на 5,2%; последняя при
охлаждении переходит в метастабильную при обычной температуре
модификацию В, которая при нагревании до 255° переходит в С с
уменьшением объема на 0,1 %48. Плавится при 398°.
Бихромат калия до 800° практически не разлагается. Изополи-
хроматы К2СГ3О10 и К2СГ4О13 плавятся соответственно при 243 и
210° с одновременным разложением на К2СГ2О7 и СгОз, при даль-
дальнейшем нагревании разлагается СгО3 49. Растворимость КгСг2О7 в
воде при 0° 4,3%, при 20° 11,7%, при 100° 50,2%. Составы эвтони-
ческих растворов в системе К2СГ2О7—K2SO4—Н2О50-Б1: при 20°
6,24% К2Сг2О7 и 8,23% K2SO4, при 40° 14,40% К2СГ2О7 и 8,20%
K2SO4. О растворимости в системе К2СГ2О7—КС1—Н2О см.52. О си-
системах (NH4hCr2O7— (NH4JSO4—Н2О и (NH4JCr207—NaCl—H2O
см.53-54.
Бихромат аммония (NH4JCr207 кристаллизуется из водного
раствора в виде дигидрата и безводной соли. Плотность при 25°
.2,15 г/см3. При нагревании до 185° разлагается. Растворимость
в воде при 0° 15,16%, при 20° 26,67%, при 100° 60,89% (NH4JCr2O7.
Обладает способностью к внутримолекулярному горению. Темпе-
.ратура вспышки 240°. В простейшем виде механизм горения пред-
представляется совокупностью следующих реакций55:
(NH4JCr207 = 2NH3 + Н2О (пар) + 2СЮ3 - 64,5 ккал
2СгО3 = Сг2О3 + 1,5О2 — 5,6 ккал
2NH3 + 1,5О2 = N2 + ЗН2О (пар) + 151,3 ккал
Бихромат кальция СаСг2О7 кристаллизуется из водного рас-
раствора от 0 до 10° в виде гексагидрата СаСг2О7 • 6Н2О, при 20—40°
в виде СаСг2С>7 • 5Н2О, при 50—60° в виде СаСг2О7 -41^0; выше
70° вследствие гидролиза
СаСг2О7 + Н2О ^=± СаСгО4 + Н2Сг04
кристаллизуется смесь бихромата и монохромата кальция56. Рас-
Растворимость в воде при 0° 53,47%, при 20° 57,87%, при 60° 65,54%,
при 100° 72,7% СаСг2О7. Бихромат кальция гигроскопичен и на
воздухе расплывается. О растворимости в системе СаСггО?—
К2Сг2О7—Н2О см.57-68.
ПРИМЕНЕНИЕ
Хром и его соединения широко используются в различных от-
отраслях промышленности59. Металлургической промышленностью
выпускаются специальные хромистые стали, отличающиеся высо-
высокой механической прочностью, жаростойкостью и антикоррозион-
антикоррозионными свойствами. Хром входит также в состав сплавов — алюми-
алюминиевых, кобальтовых, титановых и др.60. Получаемый восстановле-
восстановлением окиси хрома углем в атмосфере водорода карбид хрома
Сг3Сг служит для изготовления химически и термически стойких
металлокерамических сплавов (керметов), а также режущих и на-
наплавочных твердых сплавов 61>62. Соединения хрома используются
для получения хромомагнезитовых и других огнеупоров, применяе-
применяемых в металлургических печах.
Окись хрома служит сырьем для получения металлического
хрома (обычно алюмотермическим путем), карбида хрома, шлифо-
шлифовальных паст и красок, стойких к свету, огню и кислороду воз пуха.
Ее применяют также для окрашивания стекла и керамики. Окись
хрома является компонентом весьма часто применяемых в неорга-
неорганическом и особенно органическом синтезе хромовых катализато-
катализаторов (для дегидрогенизации алифатических углеводородов, арома-
т°йзации парафиновых углеводородов, гидрирования и крекинга,,
конверсии нефтяных газов и проч., а также для реакций изотоп-
изотопного обмена) 8>63. Для производства металлического хрома в по-
последнее время все шире стали использовать в качестве полупро-
полупродукта хлорный хром.
Хромовые квасцы, основной сульфат хрома, хромпики и неко-
некоторые другие соединения хрома используются в качестве дубиль-
дубильных веществ в кожевенной промышленности и в качестве протрав
и пропитывающих средств при крашении, печатании тканей в тек-
текстильной промышленности.
Хромпики служат сырьем для производства хромовых красок—¦
желтого крона РЬСгО4, красного крона PbCrO,f Pb(OHJ, оранже-
оранжевого крона (смеси средней и основной солей хромата свинца), жел-
желтых хромовых солей цинка и др. Соли хромовых кислот дают жел-
желтые, оранжевые, красные и коричневые краски; соли окиси
хрома — зеленые; синие и черные краски получаются из соедине-
соединений хрома и железа.
Хромпики используют также при получении некоторых органи-
органических красителей и для приготовления катализаторов. Хромпики
применяют в жировой, спичечной, бумажной промышленности, в
электротехнике, в пиротехнике, в фотографии и в других отраслях
техники, а также в сельском хозяйстве (для протравливания се-
семян). Из хромпиков в наибольших количествах используют более
Дешевый бихромат натрия.
Соединения шестивалентного хрома (хромовый ангидрид,
хпомпики, хлористый хромил) используют в качестве сильных
окислителей, в частности во многих процессах органического син-
синтеза— при получении ализарина, сахарина, бензойной кислоты,
антрахинона, гидрохинона, в производстве резин, пористых пла-
пластиков.
572
Гл. XV. Соли хрома
Хромовый ангидрид используют при получении синтетического
каучука, органических красителей, для очистки жиров и масел.
Его водные растворы применяют для травления и гальваническо-
гальванического хромирования металлов, для получения цветных пленок на по-
поверхности сплавов, для электролитической полировки металлов,
для электрохимической обработки режущего инструмента. Элек-
Электролизом растворов хромовой кислоты получают металлический
хром высокой степени чистоты.
Хромат натрия является хорошим ингибитором коррозии стали
водой, содержащей до 1 % NaCl64.
Технический бихромат натрия выпускают в виде безводного
кристаллического или гранулированного, водного гранулирован-
гранулированного, а также плавленого продуктов, которые должны удовлетво-
удовлетворять требованиям ГОСТ 2651—70 (табл. 39). Технический бихро-
бихромат калия по ГОСТ 2652—71 и ГОСТ 5.991—71 выпускают выс-
высшего и первого сортов, содержащих соответственно (в %):
К2СГ2О7 не менее 99,7 и 99,3; сульфатов (SO4) не белее 0,02 и
€,08; хлоридов (С1) не более 0,07 и 0,02; не растворимого в воде
остатка не более 0,03 и 0,1; влаги не более 0,05 и 0,1.
ТАБЛИЦА 39
Состав технического бихромата натрия (по ГОСТ 2651—70)
(содержание компонентов в %)
Хромовый ангидрид, не
менее
или Na2Cr2O7, не менее
Сульфаты (SO4), не более
Хлориды (С1), не более .
}К.е Г1езо, не более . . . .
Не растворимый в воде
остаток, не более . . .
1>Н водного раствора
Кристаллический
безводный
высший
сорт *
75,5
98,9
0,1
0.2
0.0Э2
0.01
I сорт
75,0
98,25
0.2
0,4
0,003
0.02
Гранули-
Гранулированный
безвод-
безводный
74,0
0,5
0,6
0,005
Гранулированный
водный
I сорт 1 II сорт
72,5
0,5
0,6
0.005
ода I о.оз
Не ниже 3,5
71
0,7
0,7
0,005
0,05
Плавле-
Плавленый
70
0,7
0,7
0,005
0.05
* Этот сорт соответствует также регламенту по ГОСТ 5.1493—72 на аттестованный
продукт.
Технический хромовый ангидрит, согласно ГОСТ 2548—62,
выпускается в виде чешуек или гранул (не более 3 мм) и должен
содержать в высшем, 1- и 2-м сортах, соответственно, не менее
99,5, 99,0, 98,5% СгО3 и не более 0,03, 0,06, 0,10% не растворимых
в воде веществ и 0,10, 0,20, 0,40% SO4. К качеству аттестованно-
аттестованного продукта ГОСТ 5.1599—72 предъявляют следующие требова-
требования: содержание не менее 99,6% СгО3 и не более: 0,02% не раст-
растворимых в воде веществ, 0,06% SO* и 0,005% натрия.
Требования к качеству технической окиси хрома представ-
представлены в табл. 40. Помимо приведенных в ней, ГОСТ предусматри-
предусматривает следующие показатели: содержание цветных металлов
Сырье и методы его переработки
573
Состав технической окиси хрома (по ГОСТ 2912—6в)
(содержание компонентов в %)
ТАБЛИЦА 40
Показатели
Общий хром (Сг2О3),
не менее
Соли хрома-VI
(СгОз), не более . .
Сера (S), не более .
Железо (FeO), не
более
Углерод (С), не бо-
более
Влага, не более . .
Не растворимые в водв|
вещества, не более
в том числе-суль-
числе-сульфаты (SO4), не
более ....
рН водной вы-
вытяжки ....
«Металлургическая»
«Пигментная>
«Часовая»
сорт
ОХМ-1
98,0
0,5
0,015
0,15
0,05
0,15
ОХМ-2
98,0
0,5
0,03
0,20
0,10
0,15
Не нормировано
ОХП-1
ОХП-2
99,0
0,02
99,0
0,05
Не нормировано
0,15
0,3
0,04
6—8
0,15
0,8
0,08
6-8
ОХЧ-1
98,0
ОХЧ-2
98,0
Не нормировано
0,15
0,5
0,15
. 0,5
Не нормировано
в'ОХМ-1; удельную поверхность и класс чистоты поверхности по-
после полирования для ОХЧ; рН водной вытяжки, красящую способ-
способность, цвет и оттенок, укрывистость и крупность зерен для ОХП.
В окиси хрома ОХМ-2, получаемой из хромового ангидрида, приме-
применяемой в других производствах (не для выплавки металлического
хрома), допускается общее содержание серы (S) не более 0,06%.
Тарой для хромпиков, хромового ангидрида и окиси хрома
служат стальные барабаны или полиэтиленовые мешки.
СЫРЬЕ И МЕТОДЫ ЕГО ПЕРЕРАБОТКИ
Сырьем для производства соединений хрома служит хромит или
хромистый железняк — минерал состава FeO-СггОз или FefCK^b,
являющийся нерастворимой солью хромистой кислоты НСгО2. Он
относится к группе шпинелей с общей формулой RnO • RniO3. В чи-
чистом виде хромит, содержащий, согласно приведенной формуле,
около 68% окиси хрома, встречается крайне редко. Природный ми-
НеРал, представляет собой различного вида хромшпинелиды с об-
Щей формулой (Fe, Mg)O-(Cr, Al, FeJO365. Их можно рассмат-
'Ривать как изоморфные твердые растворы солей: Fe(CrO2J,
^e(FeO?J, Mg(CrO2b и Mg(AlO2h- Помимо хромшпинелидов в
РУДе имеются примеси, других пород (серпентина, талька и проч.N6,
574
Гл. XV. Соли хрома
так что содержание Сг2О3 обычно составляет 35—57%- Содержа-
Содержание других компонентов: FeO + Fe2O3 15—25%, А12О3 7—20%
MgO 5—18%, SiO2 3—8%, CaO 1—3%. Бедная руда, содержащая
меньше 35% Сг2О3, называется железо-хромовой. На химические
заводы хромит поступает в виде кусковой руды, а также в виде
концентрата, получаемого обогащением бедных руд67-68 (табл. 41).
Марки и сорта хромитовой руды
ТАБЛИЦА 41
Марка
I
II
III
iv.
Наименование руды
Маложелезистая
Маложелвзнстая
Средиежелезнстая
Железистая
Хромитовые концентраты
Сорт
I
II
III
IV
V
VI
VII
VIII
IX
X
XI
XII
XII
XIII
XIV
XV
Содержание,
Сг2Оз
60
56-59
50-55
45-49
40-44
60
55—59
50—54
46-49
40—45
40—44
38-39
35-37
32-34
35-39
32—34
40-60
S1O2
6
6
6
6
6
6
6
6
6
6
8.5
8,5
8.5
8,5
11,0
11,0
и
GaO
МММ
—
3.0
3,0
3,0
3,0
(Сг8О3\
\ FeO /среди
3
3
3
2.8
2,8
2.8
2.8
2,8
2.3
23
23
23
1,8
1,8
Нормальная влажность руд б%, концентратов — 8%. По харак-
характеру подготовки руды подразделяют на четыре класса:
Класс Метод подготовки Размер кусков, мм
1 Дробленые и грохочение 10—75 (мельче 10 мм не более 10%)
2 Грохоченые мелкне До 10 (крупнее 10 мм не более 5%)
3 Рядовые (не дробленые и не
грохоченые) До 250
4 Концентраты
По содержанию Сг2О3 хромиты (руды) делят также на следуй'
щие классы69:
Сг2О3, К
Весьма высокохро.мистый Выше 54
Высокохромиетый 50—54
Среднехромистый 38—50
Низкохромистый 32—37
Обогатительный 5—38
Сырье и методы его переработки
575
Требования к составу хромитовой руды, применяемой для про-
производства магнезито-хромитовых огнеупоров, указаны в табл. 42.
Требования к хромнтовой руде (по ГОСТ 10154—62)
ТАБЛИЦА 42
Содержание, вес. %
Сг2О3. не менее
SiO2. не более
СаО, не более
Железа (Fe2O3), не более
Марка
К-пшс
50
5,5
1
14
к-мхс
50
5,5
1
14
с-мхс
36
6
1.6
В СССР имеются богатейшие запасы высококачественных хро-
хромитов. Месторождения хромовых руд находятся в Казахской ССР
б Актюбинской области65> 66 и на всем протяжении Уральского
хребта: на Северном Урале — Сарановское, Гологорское, Ключев-
ское, на Южном Урале — Верблюжьегорское, Халиловское и др.
Имеются также месторождения хромовых руд в Закавказье704.
Промышленные месторождения хромитов имеются в Индии, в
Югославии, в Румынии и в других странах. Наиболее крупные
месторождения хромитов имеются в Южно-Африканской Респу-
Республике, в Южной Родезии, в Турции и на Филиппинских островах,
где сосредоточено ~90% всех достоверных запасов хромитов в
капиталистических и некоторых других зарубежных странах. США
обладают небольшими запасами низкосортных хромовых руд
(8,1 тыс. тOБ. В 1953 г. импорт хромовой руды в США превысил
2 млн. г, а в 1962 г. снизился до ~ 1,3 млн. т. В 1962 г. в США из
^общего количества потребленных хромитов 52% израсходовано в
металлургии, 32% в керамической промышленности и 16% перера-
переработано в химической промышленности главным образом в бихро-
мат натрия, из которого затем получают другие соединения. В США
40% хромовых химикатов потребляют в виде пигментов, 18%—в
виде дубильных веществ, 18% в виде хромового ангидрида76.
Богатые хромиты подвергают плавке в электропечах для полу-
получения феррохрома (~60% Сг), применяемого в качестве присадки
пРи выплавке хромистых сталей. При получении феррохрома про-
¦ Исходит восстановление хромита углеродом:
FeCr2O4 + С = Fe + СО + Сг2О8
2Сг+ЗСО
576
Гл. XV. Соли хрома
В процессе восстановления, помимо сплавов Fe—Сг—С, обра-
образуются карбиды хрома (СГ4С, Сг7С3, СгзС2O7-78.
Спеканием хромитов с окислами кальция, магния, алюминия
получают хромитовые или хромомагнезитовые огнеупоры, приме-
применяемые в качестве футеровочных материалов для металлургиче-
металлургических и других печей. Для изготовления этих огнеупоров обычно
используют маложелезистые и малокремнеземистые хромиты, со-
содержащие не более 20% FeO и 7% SiO2. Связкой служит глинозе-
глиноземистый цемент, содержащий однокальциевый алюминат СаСЬ
• АЬОз- Температура плавления СаО-А12О3 1600°, а СаО-Сг2Оз
2090°.
В системе СаО • Сг2О3—СаО • А12О3 образуется ряд твердых рас-
растворов (линия ликвидуса не имеет экстремумов). Хромомагнезито-
вый кирпич плавится при 2000—2050° 79~81.
Сырьем для производства соединений хрома могут служить
феррохром — полупродукт, получаемый из хромитовых руд метал-
металлургической промышленностью, вырабатывающей хромистые стали,
а также шлаки, получаемые в производстве феррохрома и хроми-
хромистых сталей.
Получение солей окиси хрома путем кислотного выщелачивания
хромита не имеет пока промышленного значения. Даже в присут-
присутствии сильных окислителей не удается быстро достичь хорошего
разложения руды. Щелочное разложение хромита, сопровождаю-
сопровождающееся окислением, позволяет получать растворимые соединения
шестивалентного хрома с большим выходом. Этот метод и приме-
применяется в промышленности. Из хромита получают хроматы и би-
хроматы; соединения же трехвалентного хрома изготовляют их
восстановлением. (В последнее время, однако, некоторое распро-
распространение получает электрохимическое окисление трехвалентного
хрома.)
Главным продуктом в производстве солей хрома является би-
хромат натрия. Для его получения тонкоизмельченный хромит под-
подвергают окислительному обжигу в смеси с содой и окисью кальция
в виде доломита или известняка. При этом образуется монохромат
чатрия, который выщелачивают из прокаленной шихты (спека) и
переводят в бихромат натрия введением в раствор кислоты.
ОКИСЛИТЕЛЬНЫЙ ОБЖИГ ХРОМИТОВ С ПОЛУЧЕНИЕМ ХРОМАТОВ
Физико-химические основы окислительного обжига хромита
Химизм процесса
Химизм процесса окислительного обжига хромита не может
быть описан одной или несколькими простыми реакциями. Шихта,
составленная из хромита, соды и доломита вместе с газовой фазой,.
с которой она взаимодействует, является очень сложной многокрМ-
Окислительный обжиг хромитов с получением хромате
577
понентной системой. В ней при высоких температурах помимо га-
газовой и жидкой фаз имеется большое число твердых фаз, состав
которых зависит от режима обжига (стр. 586). Условно окисли-
окислительную прокалку хромита в присутствии соды можно представить
уравнениями реакций:
4(?еО • Cr2CV + 8Na2CO3 + 7О2 = 8Na2Ci O4 + 2Fe2O3 + 8СО2
4(MgO • Cr2O3) + 8Na2CO3 + 6О2 = 8Ma2CrO4 + 4MgO + 8СО2
Одновременно идут реакции, в результате которых появляются
многие другие соединения (см. ниже), в первую очередь ферриты,
алюминаты, силикаты натрия и кальция и хроматы кальция.
Окись железа, находящаяся в шихте в свободном состоянии и
в составе хромшпинелида, взаимодействует с Na2O, СаО, MgO с
образованием ферритов и алюмоферритов. Образуется также фер-
ратAУ) натрия
Na2O • Fe2O3 + 3Na2CO3 +¦ 0,^Oa = 2Na4FtO, + 3CO2
который растворяется в жидкой фазе прокаливаемой массы и оки-
окисляет хром(III); это интенсифицирует окислительный обжиг, так
как, расходуясь на окисление хромита, многократно регенерируясь,
феррат служит переносчиком кислорода из газовой фазы к поверх-
поверхности зерен хромита82'83.
Окисление Сг(III) в Cr(VI) идет очевидно последовательно
через Cr(IV) и Cr(V). Наличие в спеке хроматов(У) натрия и
кальция установлено84; они растворены в жидкой фазе шихты и
участвуют в передаче кислорода — образуясь в результате окисле-
окисления хромита (III) хроматом (VI), они затем окисляются газообраз-
газообразным кислородом до хромата (VI).
Образование хромата из хромита является эндотермическим
процессом, требующим затраты значительных количеств тепла.
Чем более высок температурный уровень прокаливания шихты и
чем больший избыток кислорода в печи, тем быстрее идет оки-
окисление.
' Взаимодействие хромита с содой и окисью
кальция
Окислительный обжиг производят в трубчатых вращающихся
печах при температурах, достигающих 1100—1200°. Хромат натрия
появляется при нагревании шихты еще при сравнительно низких
температурах и образует с содой эвтектический расплав, содержа-
содержаний 62,5% Na2CrO.j. Появление жидкой фазы в системе Na2CrO4—
Na2CO3 возможно уже при 655° 85. При дальнейшем нагревании ре-
реакционной массы жидкая фаза обогащается содой, причем состав
Расплава вначале изменяется от эвтектической точки вдоль линии
ликвидуса по направлению к точке плавления соды (рис. 170). Но;
*9 М. Е. Позии
578
Гл. XV. Соли хрома
одновременно с возрастающей скоростью происходит образование
хромата натрия, замещающего соду в жидкой фазе. Сода расхо-
расходуется не только на основную реакцию, но и на реакции с другими
компонентами шихты — на образование феррита, алюмината и си-
силиката натрия (см. ниже), которые затем в свою очередь реаги-
реагируют с хромитом, известью и кислородом, давая хромат натрия и
кальциевые неплавкие соединения. До 1000° скорость образования
хромата натрия больше скорости взаимодействия соды с другими
окислами (SiO2, А12Оз, Fe2O3)86. По мере исчезновения соды из
жидкой фазы происходит растворе-
растворенное ние новых количеств соды. В ре-
результате этих процессов твердая
сода исчезает задолго до достиже-
достижения шихтой температуры плавления
соды. На рис. 170 схематически изо*
бражено измерение состава жидкой
фазы по мере повышения темпера-
температуры; момент исчезновения твердой
соды выражен изломом кривой в
точке А.
Диаграмма условно не учиты-
учитывает возможность растворения в
жидкой фазе других компонентов
«55е
73?"
NatCOj
Рис. 170. Схема изменения со-
состава жидкой фазы.
прокаливаемой шихты. Поэтому на диаграмме жидкая фаза в кон-
конце процесса прокаливания состоит из чистого хромата натрия. На
самом деле в жидкой фазе содержится некоторое количество хо-
хорошо растворимого в хромате натрия хромата кальция — при 1000°
. . растворимость хромата кальция в хромате натрия достигает 80%.
Практически, однако, в расплаве растворено небольшое количе-
количество хромата кальция, так как растворенные ионы кальция всту-
. пают в реакции, приводящие к образованию нерастворимых каль-
кальциевых соединений — силикатов, ферритов, алюминатов и др. Толь-
Только при большом недостатке соды в шихте возможно появление
значительного количества хромата кальция в жидкой фазе.
На рис. 171 приведена диаграмма плавкости системы СаСгО4+
4-Na2CO3 = Na2CrO4+CaCO3 при невысоких температурах и в ат-
атмосфере углекислого газа, когда еще не происходит разложение
углекислого кальция86. В этих условиях система является трехком-
понентной. (Составы даны в молярных процентах.) В равно-
равновесии с расплавом находятся следующие твердые фазы: СаСгСч.
СаСО3, Na2Ca(CO3J и твердые растворы тЫа2СО3'пСаСОз и
/МагСгСч • gCaCrO4. На диаграмму нанесены изотермы и обозна-
обозначены температурные минимумы (точки плавления двойных и трой-
тройных эвтектик) и максимумы. Как видим, появление жидкой фазы
в прокалочной печи возможно уже при 645°. По мере повышения
температуры состав жидкой фазы меняется. Одновременно меняет-
Окислительный обжиг хромитов с получением хроматов
57»
ся и состав реагирующих твердых фаз, диссоциирует карбонат
кальция. В системе Na2CrO4—СаСгО4 имеется максимум при 812°,
обязанный образованию твердых растворов; двойная эвтектика
содержит 48,4 мол. % СаСгО4, плавится при 740° и состоит из
твердого раствора (Na2, Ca)CrO4 и чистого хромата кальция87.
Наиболее легкоплавкие смеси расположены вдоль пограничных ли-
линий, соединяющих точки тройных эвтектик F68, 645 и 648°) и
точку двойной эвтектики соды и хромата натрия F55°). Из этих
смесей образуется жидкая фаза в шихте, содержащей соду, ниже
Na2CO3
855°
50
— CO3.%
75
W0
СаСО3
Рис. 171. Диаграмма плавкости системы
CaCrO4+Na2CO3 = Na2CrO4 + CaCO3.
700е. При прокаливании бессодовых (известковых) шихт образова-
образование хромата кальция ниже 800° идет медленно88, а при более высо-
высоких температурах образуются хромито-хроматы, снижающие вы-
выход хромата (см. ниже).
В промежуточных стадиях обжига в жидкой фазе содержится
' также некоторое количество силиката натрия. Na2Cr04 и Na2Si03
образуют эвтектику при 770°, содержащую 10% Na2Si0s. Однако
содержание силиката натрия в жидкой фазе не может быть боль-
большим, так как хромитовая шихта составляется из материалов с ма-^
лым содержанием силикатов, и кроме того, силикат натрия, по-"
Добно алюминату и ферриту натрия, образуется только в качестве
промежуточного продукта. Анионы кремневой кислоты довольно
быстро извлекаются из жидкой фазы катионами кальция с обра-
_ зованием нерастворимых и труднорастворимых силикатов, заме-
Щаясь анионами хромата. Только при большом избытке соды в
щихте возможно значительное содержание силиката натрия в рас-
расплаве 85.
19*
880
Гл. XV. Соли хрома
Примеси, содержащиеся в хромитовой руде, не влияют суще-
существенно на предельную степень окисления окиси хрома. От при-
присутствия окислов кремния, алюминия и железа степень окисления
Сг2О3 не уменьшается, если прокаливаемая шихта содержит окись
кальция в количестве, достаточном для связывания кислотных оки-
окислов. Следует однако отметить, что примеси влияют на скорость
окисления хрома — снижение содержания Сг2О3, т. е. повышение
содержания примесей (SiO2, Fe2O3, A12O3) приводит к увеличению
продолжительности обжига хромитовых шихт в производственных
условиях (см. стр. 584).
Наполнители в содо во-хромитной шихте
Составные части шихты и кислород газовой фазы образуют ге-
гетерогенную систему. Скорость реакции в такой системе зависит от
развития межфазной поверхности. Поэтому большое значени
имеет тонкость помола хромита. То обстоятельство, что сода нах
дится в жидкой фазе, значительно ускоряет процесс, — в этих ус-
условиях частицы хромита смачиваются расплавом, содержащи
соду, и реакционная поверхность увеличивается во много раз. Пра
вда, при наличии жидкой фазы, обволакивающей твердую частицу,
газообразный компонент должен диффундировать через пленку
жидкости. Тем не менее наличие ограниченного количества жидкой
фазы сильно ускоряет процесс. При большом же количестве жид-
жидкой фазы происходит спекание шихты, налипание ее на стенки
печи, что мешает ее продвижению, затрудняет доступ кислорода к
реагирующим частицам руды из-за слипания их в крупные комья.
В результате этого процесс сильно замедляется, и выход хромата
понижается89. При относительно низких температурах, когда коли-
количество расплава в шихте невелико, происходит, по-видимому, непо-
непосредственное окисление окиси хрома газообразным кислородом;
выше же 900° окисление идет с участием расплава, в котором воз-
возрастает содержание реакционно-способных частиц—ионов кисло-
кислорода, образующихся в результате диссоциации карбоната и хро-
хромата натрия и при посредстве промежуточных веществ — ферра-
таAУ) и, вероятно, перекиси натрия. При большем содержании
¦окиси железа в шихте ее положительное влияние, как передатчика
кислорода через ферратAУ) натрия уменьшается и не компенси-
компенсирует ухудшение процесса вследствие уменьшения концентрации ос-
основных реагентов — Сг2О3 и Na2CO3; при этом затрудняется и об-
образование феррата натрия90.
Твердые составные части шихты хорошо смачиваются образую-
образующейся в процессе прокалки жидкой фазой89. Помимо поверхност-
поверхностного смачивания, под влиянием капиллярных сил происходит по-
почти полное заполнение пор между отдельными частицами матери-
материалов. В разных стадиях обжига твердые фазы удерживают Olf^
Окислительный обжиг хромитов с получением хроматов
581
0,35 г расплава на 1 г твердого вещества. Во избежание спекания
реакционной массы в шихту необходимо добавлять какие-либо
тонкоизмельченные вещества (их обычно называют наполните-
наполнителями, хотя они и не являются инертными компонентами) в таком
количестве, чтобы слой жидкой фазы на поверхности твердых ча-
частиц был очень тонким 91~93. В реальных соотношениях компонен-
компонентов в хромитовой шихте толщина жидкой пленки на поверхности
твердых частиц не превышает 1 мк. В этих условиях шихта остает-
остается практически сухой, рассыпчатой, легко подвижной, не спекается
и обладает большой газопроницаемостью. Однако при этом по-
поверхность добавки удерживает часть жидкой фазы и использова-
использование последней может быть обеспечено только при условии хоро-
хорошего перемешивания шихты, что и осуществляется в реакционных
прокалочных печах.
Таким образом, добавка наполнителя к шихте приводит, с од-
одной стороны, к положительному эффекту, уменьшая толщину жид-
жидкой пленки на поверхности частиц, а с другой — к отрицательному,
устраняя часть жидкой фазы с реакционной поверхности хромита.
Очевидно, что существует оптимальное соотношение между коли-
количеством хромитовой руды и наполнителем, при котором полезный
эффект от введения наполнителя оказывается максимальным. Мак-
Максимальное окисление наблюдается при содержании в шихте 16,2—
16,5% Сг2О3. С повышением температуры диапазон концентраций
Сг2О3 в шихте, при которых достигается максимальное окисление,
расширяется. При 1200° степень окисления практически одинакова
для шихт, содержащих от 12 до 19,5% окиси хрома94'95.
Среди доступных реальных наполнителей отсутствуют такие
твердые вещества, которые в условиях высокотемпературного про-
прокаливания хромитовой шихты были бы инертными ко всем компо-
компонентам шихты. Это обусловливает протекание побочных реакций,
сопровождающих основную реакцию образования монохромата
натрия. Из ряда возможных наполнителей — CaO, CaCO3, MgO,
MgCO3, Fe2C>3, A12O3, SiO2 —¦ наилучшим из простых является из-
известковый (жженая известь, мел или известняк), а из ком-
комбинированных, как показал производственный опыт, доломит
(MgCO3 • СаСО3) и его смесь с известняком 95~97. Положительная
роль извести заключается в том, что окись кальция вытесняет
окись натрия из феррита, алюмината и силиката натрия, образо-
образовавшихся из компонентов шихты; освобождающаяся же окись нат-
Рия образует хромат натрия, выход которого поэтому увеличи-
увеличивается. Но вместе с тем работа с доломитовым и известковым на-
наполнителями предопределяет и появление в прокаливаемой шихте
, хромата кальция. Хромат магния практически не образуется, так
как это соединение разлагается выше 500°, и около 90% MgO,
содержащейся в прокаленной шихте, находится в свободном со-
состоянии. Давление диссоциации MgCrO4, равное 1 ат, достигается
582
Гл. XV. Соли хрома
уже при ~535°; [igP (в мм рт.ст) = 23,271 — 16488/Г]. Термиче-
Термическое разложение MgCrO4 сопровождается образованием вначале
неустойчивого 2MgO • Сг2Оз, постепенно разлагающегося на MgO
и MgO • Сг2Оз98. Вследствие высокого давления диссоциации при
сравнительно низких температурах MgCrO4 не может находиться
в прокаленной шихте. Для полного же разложения СаСгО4 тре-
требуется очень высокая температура. Чистый хромат кальция при
нагревании на воздухе начинает заметно разлагаться при темпера-
температуре около 1000° с выделением кислорода. В присутствии 0,1 моль
СаО разложение ускоряется уже при 800°. Однако даже при на-
нагревании в течение 4 ч при 1300° хромат кальция полностью не
разлагается". В заводских условиях максимальная температура
прокалки шихты обычно не превышает 1200° и содержание СаСгО4
в прокаленной шихте составляет 5—8% от получаемого хромата
натрия.
Хроматы кальция образуются из окиси хрома и окиси кальция
при нагревании их на воздухе при 600—1000°. При молярном отно-
отношении СаО: Сг2О3 от 2 до 3 вся окись хрома практически окис-
окисляется уже при 600—70СР за 2 ч и переходит в СаСгО4 и оксихро-
миты кальция. Оксихромиты также интенсивно окисляются. При
600—700° за 2 ч монохромит СаО • Сг2О3 окисляется на 2/3, давая
смесь 2СаСгО4 + Сг2О3, двухкальциевый оксихромит 2СаО • Сг2ОЕ
окисляется полностью с образованием СаСгО4, а трехкальциевый
оксихромит 3CaO-Cr2Os окисляется на 75% с образованием
ЗСаО • 2СгО3100-105.
Хромат кальция выше 1000° диссоциирует с образованием хро-
мито-хроматов95-1М- Ш6, например, по реакции
12СаСгО4 = 3(СаО • Сг2О3) + 9СаО • 4СгО3 • Сг2О3 + 6О2
В присутствии свободной извести хромито-хромат ©бразуется
по реакциям:
3MgCr2O4 + 9СаО + ЗО2 = 9СаО • 4СгО3 • Сг2О3 + 3MgO
3FeCr2G4 + 9СаО + 3,75О2 = 9СаО • 4СгО3 • Сг2О3 + l,5Fe2O3
Из других хромито-хроматов кальция в прокаленной шихте
вероятно присутствие ЗСаО-2СгО3-2Сг2О3.
При не очень высоких температурах хромито-хроматы окис-
окисляются кислородом с образованием СаСгО4, например
9СаО • 4СгО3 • Сг2О3 + 1,5О2 = 6СаСгО4 + ЗСаО
Окись кальция расходуется также на образование клинкерных
минералов — алюмоферритов (браунмиллерита 4СаО • А12Оз • ЁегОз
и др.), алюминатов кальция—12СаО-7А12О3 (которому раньше
приписывали формулу 5СаО-ЗА12О3) и других, дикальциевого си-
силиката р-2СаО • SiO2, феррита кальция 2СаО • Fe2O3. Количе-
Количество ее должно быть достаточным для связывания А12О3, Fe2O3,
SiO2.
Окислительный обжиг хромитов с получением хромитов
583
При прокаливании шихты, содержащей соду, она реагирует
с хромит-хроматами даже при 600°. Это послужило основанием для
предложения способа переработки хромита, заключающегося в
предварительном получении хромит-хромата кальция при 1000°
с последующей обработкой полупродукта содой при 600—700°.
В этом способе исключается необходимость в наполнителе (окиси
магния, доломите), а следовательно, уменьшается количество от-
отбросов, которое при возврате окиси кальция в процесс становится
незначительным Ш7.
При применении в качестве наполнителя доломита количество
остающегося в прокаленной шихте хромата кальция значительно
меньше, чем при известковой шихте. Кроме того, доломитовая
шихта меньше спекается. Оплавление шихты при замене доломита
карбонатом кальция связано с образованием карбонатной эвтек-
эвтектики86. Преимуществом доломита, который содержит 52—54%
СаСОз и 30—40% MgCO3, является то, что диссоциация карбона'
тов здесь происходит по всей реакционной зоне печи, в темпера-
температурном интервале от 650 до 1100° Ш8-109. При нагревании доломита
в пределах 650—750° происходит выделение СО2 из MgCO3, а в
пределах 800—1100° — из СаСО3. Это обеспечивает сравнительно
равномерное выделение СО2 из шихты по всей длине печи. Ско-
Скорость разложения известняка и магнезита определяется диффу-
диффузионной кинетикой и зависит от скорости прохождения газа через
слой карбоната. При увеличении скорости газа до 7—8 м/сек ско-
скорость разложения карбонатов возрастает, затем процесс контроли-
контролируется химической кинетикой и дальнейшее увеличение скорости
газа не вызывает интенсификации разложения. Скорость же раз-
разложения доломита практически не зависит от скорости газа, так
как процесс контролируется не диффузионной, а химической кине-
кинетикой уже при малых скоростях газа в конвективном потоке110.
Выделяющийся углекислый газ, пронизывая шихту снизу вверх,
производит ее перемешивание и обеспечивает ее рыхлость, что
облегчает доступ кислорода к нижним слоям реагирующей массы.
Лабораторными опытами установлено, что еще лучшим напол-
наполнителем, чем карбонат кальция и доломит, является карбонат
бария1И.
Как уже указывалось выше, максимальный полезный эффект
от введения в шихту наполнителя, дающий наибольший выход во-
водорастворимых хроматов, соответствует определенному оптималь-
оптимальному содержанию наполнителя в шихте95. На рис. 172 показана
кривая зависимости выхода водорастворимых хроматов от соотно-
соотношения между Na2CO3 и СаО, при содержании в шихте 15% Сг2Оз
(в виде 34,9%-ной руды), при прокаливании в течение 30 мин
при 1000°. Максимальный выход соответствует соотношению
Na2CO3: СаО, равному 60:40. Увеличение общего содержания
Сг2О3 в шихте с 16 до 20% нли частичная замена доломита окисью
584
Гл. XV. Соли хрома
или карбонатом магния не влияет на окисление, если имеется
достаточно СаО для связывания кислотных окислов. При недо-
недостатке СаО степень окисления хромита снижается. То же отно-
относится и к влиянию нерудных компонентов хромита, содержащих,
кроме MgO, главным образом окислы алюминия, железа и крем-
кремния, — изменение их количества не влияет на предельную степень
окисления хромита, если содержание СаО достаточно для свя-
связывания кислотных окислов 112,
а продолжительность прокалки
велика. В промышленных ус-
условиях влияние их заметно.
Следует отметить, что в ших-
шихтах, содержащих мало соды и
много извести, обнаруживает-
обнаруживается повышенный против теоре-
теоретического выход водораствори-
водорастворимых хроматов. Пунктирная
прямая AN (рис. 172) показы-
показывает стехиометрическое коли-
количество соды, необходимое для
перевода окиси хрома в хро-
Зд/ мат натрия. Точка В соответ-
СаО ствует количеству соды, равно-
равному 100% от стехиометрическо-
Рис. 172. Кривая выхода водораствори- го. Кривая выхода в правой
мых хроматов в присутствии СаО. части диаграммы лежит выше
стехиометрической прямой AN.
Как видно из расположения кривой, степень использования соды
в присутствии СаО (или СаСО3) может достичь — 140%. Это свя-
связано с переходом хромат-иона в раствор не только в виде хромата
(гатрия, но и в виде некоторого количества хромата кальция. Рас-
Растворимость безводного хромата кальция в воде равна 4,3% при 0°,
1,11% при 50е и 0,42% при 100°.
При выщелачивании прокаленной шихты большим количеством
воды в раствор переходит заметное количество СаСгО4, что и дает
повышенную цифру выхода. При применении неизвестковых напол-
наполнителей кривая выхода растворимого хромата лежит всегда ниже
стехиометрической линии AN.
Выход хроматов зависит не только от состава нерудной части
шихты, но, естественно, и от состава руды 95. Различные руды окис-
окисляются с разной скоростью. Быстрее окисляются руды с малым
содержанием А1 и Fe2+. Бедные руды, содержащие меньше 30%
СггОз, дают наибольший выход хроматов при прокаливании в
смеси с одной содой. Частичная замена ее в шихте карбонатами
кальция и магния снижает выход. Степень окисления хрома г>
шихте, содержащей повышенные количества SiO2, меньше, чем для
Окислительный обжиг хромитов с получением хроматов
585
Na2CO3
50 Вес-%
USD
BSD*
железистой руды или руды, не содержащей каких-либо примесей.
Помимо химического состава на реакционную способность разных
хромитовых руд влияют и их физико-химические свойства 89>93- нз-п5_
Процесс образования хромата из хромита является эндотерми-
эндотермическим и требует затраты значительных количеств тепла И6. Чем
более высок температурный уровень прокаливания шихты, тем
быстрее идет окисление. На рис. 173 показана зависимость степени
окисления Сг2О3 в СгО3 от про-
продолжительности прокаливания
при разных температурах при из-
известковой (пунктирные линии) и
доломитовой шихте (сплошные
линииI17.
Применявшийся для опытов
хромит содержал: 44,61% Cr2Os,
24,28% FeO, 9,86% А12О3, 4,04%
SiO2; обожженная известь содер-
содержала: 90% СаО, 6,43% MgO и
2,04% А120з+Ре20з. Исходные
вещества смешивали в соотноше-
соотношении хромит : известь : сода, рав-
равном 1:1:0,64. При составлении
доломитовой шихты применялся
обожженный доломит, содержав-
содержавший 65,04% СаО, 29,7% MgO и
4,2% SiO2.
Из диаграммы видно, что
равная степень окисления при
высоких температурах достигает-
достигается на доломитовой шихте быстрее,
чем на известковой. Работа на
12 3 4 5 6?
Продолжительность пронали-
бания, v
Рис. 173. Зависимость степени окисле*
ния хромита в хромат от продолжи-
продолжительности прокаливания шихты,
доломитовой шихте дает более
благоприятные результаты и в отношении степени перехода СгО$
в раствор при последующем выщелачивании прокаленной шихты.
Скорость и степень окисления хромитовой шихты зависят от
интенсивности поглощения кислорода из газовой фазы, в частно-
частности от концентрации кислорода в газе. При обжиге хромита в
смеси с содой и доломитом содержание кислорода в выходящем
из печи газе должно быть не ниже 7,5—10% 118.
Скорость процесса зависит также от активности образующейся
"ри диссоциации карбонатов окиси кальция. Наличие примесей в
карбонатных наполнителях приводит к образованию неактивной
. извести. Если в карбонатном сырье содержится меньше 3% при-
примесей, то заметное количество неактивной окиси кальция обра-
образуется лишь при температурах выше 1350°119. О кинетике окисле-
окисления хромитовых шихт см. также99- 12°-1М.
586
Гл. XV. Сот хрома
Окислительный обжиг хромитов с получением хроматов
587
Минералогический состав хроматного спека и расчет шихты
Минералогический состав хроматного спека зависит от коли-
количества соды в шихте, количества окиси кальция, введенной в шихту
с доломитом, и от содержания А12О3, Fe2O3 и других компонентов
в исходной руде. Кроме того, состав спека зависит от температуры
обжига, хотя, по-видимому, равновесие в системе не достигается.
Он зависит также от скорости охлаждения прокаленной массы;
при быстром охлаждении в спеке могут присутствовать метаста-
бильные фазы. По этим причинам минералогический состав спека
не является однозначным и сведения о нем, полученные в разных
исследованиях, не идентичны. Несомненно85- 87-123, что в спеке на-
находится C5—40%) твердый раствор Na2Cr04—СаСгО4 с содержа-
содержанием ~80% Na2Cr04. Установлено также присутствие сле-
следующих соединений: p-2CaO-SiO2, 12CaO • 7А12О3, 2CaO-Fe2O3r
4СаО • А12О3 • Fe2O3, в чистом виде или в виде твердых растворов.
В спеке содержатся также хромшпинелиды, хромит(III) и хро-
хромат (V) кальция и алюминат натрия, неразложенные карбонаты
кальция и натрия. Большая часть окиси магния находится в спеке
в свободном состоянии, а около 10% в виде твердого раствора
с 4СаО • А12О3 • Fe2O3123. Возможно, что в спеке присутствует также
магниевый браунмиллерит 4СаО • 2MgO • А12О3 • Fe2O3 m.
Практикой эксплуатации хроматных печей установлена неце-
нецелесообразность введения в шихту стехиометрического количества
соды, так как степень использования соды не выше степени окис-
окисления хромита. Вследствие этого количество соды в шихте рассчи-
рассчитывают по формуле
[Na2CO3] = 1,395А;[Сг2Оз]
где [Ыа2СОзЗ'— кг соды на 100 кг руды; [Сг2О3] — вес.% Сг2Оз
в руде; 1,395 — стехиометрический коэффициент; к — степень окис-
окисления.
Для расчета количества СаО, необходимого для составления
шихты, предложены различные полуэмпирические формулы, исхо-
исходящие из возможного образования тех или иных соединений в про-
прокаленном спеке96-Ш6-ш7-113> 123~127. При переработке хромитов, со-
содержащих 47—56% Сг2О3, рекомендуются' формулы (для доло-
доломитовых шихт):
при [А12О3]: [Fe2O3]>0,64
[СаО] = l,88[SiO2] + 0,91[А12Оз] + 0,82[FesO3] + 0,26[СГ!О3]
при [Al2O3]:[Fe2O3]<0,64
[СаО] = l,88[SiO2] + 1,Ю[А12О3] + 0,70[FesO3] + 0,26[Сг2О,]
Здесь [СаО] в кг на 100 кг руды; [SiO2], [A12O3], [Fe2O3],
{Сг2О3] — вес.% окисла в руде. . -
В промышленных печах степень окисления хромита умень-
уменьшается с понижением содержания Сг2О3 в шихте и с увеличением
их загрузки шихтой127. Так, для шихты, содержащей 16,5% Сг2О3,
изменение степени окисления (А%) в зависимости от загрузки
(С кг/ч) можно представить эмпирической формулой:
А =130,4- 0,01125С
Условная производительность печи Р, т. е. количество Сг2О3
(в кг), окислившееся в печи в течение 1 ч, при содержании Сг2О3
в шихте В%, будет:
Р = ABC -10-4
Например, для шихты с содержанием 16,5% Сг2О3:
Р = С A30,4-0,01125С) • 16,5 • КГ4
Из этого выражения видно, что условная производительность Р
возрастает с увеличением нагрузки, хотя при этом степень окисле-
окисления уменьшается. В зависимости от конструкции печи и режима
ее работы существует оптимальная концентрация окиси хрома в
шихте, при которой достигается максимальная степень окисле-
окисления93- 106' из. На наших заводах она принята равной 16,5—17,5%
Сг2О3.
Для определения производительности печи в зависимости от её
размеров предложена эмпирическая формула 128
Р = 3,5A00-ЛH-45/I'5/.
здесь Р — производительность печи по хроматным щелокам, кг/ч
Na2Cr207 • 2Н2О; D — внутренний диаметр печи, м; L — длина
печи,м.
В заводской практике делались попытки полной и частичной
замены щелочного компонента шихты — соды более дешевым —
сульфатом натрия 129. Эти попытки не привели к удовлетворитель-
удовлетворительным результатам — выходы хромата оказывались более низкими,
а худшие физические свойства прокаленной шихты (спекаемость
и вязкость при обжиге, твердость при выщелачивании) затрудняли
проведение производственных процессов. Описаны и опыты замены
соды на Na2SO4 или NaCl, давшие положительные результаты.
Рекомендуется оптимальное соотношение в шихте (мол.)
Cr2O3: Na2SO4: СаО, равное 1: 1,8: 4,4, или Сг2О3: NaCl: СаО, рав-
равное 1 : 3",6 : 4,4. Температура прокаливания 900—950°, т. е. ниже,
чем для содовой шихты. Степень окисления Сг2О3 85—90% 1S0,
Установлена возможность замены соды нефелином114-131.
Добавка наполнителей к хромитовой шихте наряду с их поло-
положительным влиянием приводит к возрастанию расходов на
588
Гл. XV. Соли хрома
Физико-химические основы выщелачивания спека
589
транспортировку, размол и увеличивает количество неиспользуемых
отходов 132. Поэтому предлагаются различные видоизменения схемы
окислительной прокалки хромитов. Одно из них рассмотрено выше
(стр. 583). Другое заключается в осуществлении процесса в три
стадии с уменьшенным количеством наполнителя (завод в Левер-
кузене). В первой стадии прокалке подвергают шихту, составлен-
составленную из руды и 25% стехиометрически необходимой соды. Полу-
Полученный после выщелачивания спека шлам с добавкой еще 25%
соды прокаливают во второй стадии. Образовавшийся спек снова
выщелачивают и оставшийся шлам после подсушки смешивают
с остальными 50% соды и половинным количеством доломита
(в расчете на первоначальное количество руды) и вновь прокали-
прокаливают (третья стадия). При такой схеме расход доломитао'мень-
шается в 2 раза, а степень окисления хромита повышается ш.
Описан опыт прокалки хромитовых шихт в модели печи со
взвешенным слоем 134. Хромито-содово-доломитовую шихту с влаж-
влажностью 18% гранулировали 3 мин в тарельчатом грануляторе и
гранулы C—5 мм) прокаливали в цилиндрической печи с двумя
слоями на решетках из жароупорных труб, охлаждаемых воздухом.
Топочный газ поступал под нижний слой с температурой 1150° со
скоростью 3,5 м/сек. При степени окисления 90% интенсивность
по шихте равнялась 500 кг/(м2-ч). Достаточно большая степень
окисления была достигнута и на содово-известковой шихте. Фир-
Фирмой „ Chemische Fabrik Galvachrom S. А." (ФРГ) разработан метод
окисления в кипящем слое хромито-содовой шихты при 600—
1200°135.
О прокалке хромитовых шихт в кольцевой печи см.136.
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ВЫЩЕЛАЧИВАНИЯ
ХРОМАТНОГО СПЕКА
При обработке прокаленной шихты — хроматного спека водой
из него выщелачиваются твердый раствор хроматов натрия и каль-
кальция, алюминат натрия и сода. Другие твердые составные части
спека под действием воды превращаются в новые минеральные
фазы, остающиеся в шламе или взаимодействующие с компонен-
компонентами системы. Так, в результате медленной гидратации и гидролиза
силикатов кальция образуются гидросиликаты кальция и в раствор
переходит гидроокись кальция, которая реагируя с ионами СОз
или Mg2+ образует нерастворимые СаСО3 или Mg(OHJ — это
ускоряет гидролиз. Аналогично этому при взаимодействии с водой
алюминатов кальция образуются гидроалюминаты кальция и гид-
гидроокись алюминия. Вследствие гидролиза клинкерных минералов,
главным образом алюмоферритов, происходит деградация водо-
водорастворимых хроматов с образованием гидрохромалюмината каль-
кальция ЗСаО-А12Оз-СаСгО4-12Н2О137-141. например, по схематиче-
схематическому уравнению:
4СаО • А]2О3 • Fe2O3 + Na2Cr04 + 16H2O = ЗСаО • AJjO3 • СаСгО4 • 12Н2О +
+ 2Fe(OHK + 2NaOH
В результате этого потери хрома в шламе могут быть значи-
значительными, так как в хроматных спеках содержится до 20% браун-
миллерита И2.
Во избежание потери хромового ангидрида в виде малораство-
малорастворимых соединений — основных хромито-хроматов (стр. 582) и гид-
рохроматалюмината кальция — в процессе водного выщелачивания
суспензию хроматного спека обрабатывают углекислым газом. При
сатурации (карбонизации) суспензии идут реакции, приводящие
к образованию СаСО3 и А1(ОНK и способствующие переходу хро-
хромата в раствор:
Са(ОНJ + СО2 = СаСО3 + Н2О
Na2O • AljOs + СО2 + ЗН2О = Na2CO3 + 2А1(ОНK
Са(ОНJ + Na2CO3 = СаСО, + 2NaOH
CaCrO4 + Na2CO3 = Na2CrO4 + CaCO3
В начальной стадии сатурации в результате гидролиза браун-
миллерита и алюминатов кальция могут образоваться гидроалю-
гидроалюминаты кальция, а также карбоалюминаты кальция, например, по
уравнению:
4СаО • А12О3 • Fe2O3 + СО2 + 14Н2О = ЗСаО • А12О3 • СаСО3 • 11Н2О + 2Fe(OH)8
При дальнейшей сатурации эти соединения разрушаются угле-
углекислым газом
ЗС О- А! О,-6 12О i-ЧСО. = ЗСаСОа + 2А1(ОЧK + ЗНгО
ЗСаО • А12О3 ¦СаСО, •11Н2О + ЗСО2 = 4СаСО3 + 4AJ(OHK + 8Н2О
что полезно, так как предотвращает в дальнейших стадиях пере-
переработки хроматной пульпы гидролиз этих соединений с образова-
образованием нерастворимых гидрохромалюминатов кальция.
Более полное извлечение хрома в раствор в результате обра-
обработки пульпы углекислым газом сводится, в основном, к следую-
следующим процессам 141:
2ОН- + СО2 = СО2- + Н2О
СаСгО4 + СО2," = СЮ2" + СаСО3
ЗСаО' • А12О3 • СаСгО4 • 12Н2О + Na2CQ3 + ЗСО2 - NasCrO4 + 2A1 (ОНK +
+ 4СаСО, + 9Н2О
Таким образом при сатурации гидролиз минералов, входящих
в состав хроматных спеков, направляется в сторону образования
590
Гл. XV. Соли хрома
соединений, не содержащих хрома. После отделения этих соедине-
соединений в виде шлама, получается монохроматный щелок.
Установлено143, что потери хрома со шламом можно снизить,
добавляя к поступающему на выщелачивание спека оборотному
раствору хромата натрия метасиликат натрия.
Физико-химические основы перевода хроматов в бихроматы
Б9Ь
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ
.144
ПЕРЕВОДА ХРОМАТОВ В БИХРОМАТЫ1
Методы перевода хроматов в бихроматы
Этот процесс, носящий в промышленной практике название
«травки» монохроматных щелоков, осуществляют с помощью сер-
серной кислоты:
2Na2CrO4 + H2SO4 = Na2Cr207 + Na2SO4 +- H2O
При этом более 60 7о затраченной на производство соды пере-
переводят в менее ценный продукт — сульфат натрия, что является
существенным недостатком этого метода. Обычно в сульфате
остается 2—3% хромпика; это увеличивает потери хрома и обусло-
обусловливает необходимость очистки сульфата для его использования.
В производстве хромового ангидрида (стр. 611) травку монохро-
матного щелока ведут бисульфатом натрия.
Предложен ряд других методов травки. Из них наибольшего
внимания заслуживает углекислотный 145>Н6. Он заключается в об-
обработке монохроматных щелоков углекислым газом под давлением.
Выделяющийся в осадок бикарбонат натрия
2Na8Cr04 + 2СО2 + Н2О ^±. Na2Cr2O7 + 2NaHCOs
может быть использован взамен соды при прокалке хромита. Это
обстоятельство, а также то, что угольная кислота дешевле серной,
делает углекислотный метод травки весьма экономичным. Его при-
применяют на некоторых зарубежных заводах.
Травка монохроматных щелоков плавиковой кислотой позво-
позволила бы совместить производство хромпика с производством фто-
фтористых солей, например
2Na2CrO4 + 2HF = Na2Cr2O7 + 2NaF + Н2О
2Na2CrO4 + NaA102 + 6HF = Na2Cr207 + Na3AlFe + 3H2O
И Т. П.
При этом исключается расход серной кислоты в производстве
хромпика и соды в производстве фтористых солей. Образующийся
при травке хроматного щелока плавиковой кислотой или фтористо-
фтористоводородным газом труднорастворимый фтористый натрий промы-
промывают на фильтре водой. При содержании в нем 5—7% NaCr2O7 его
предложено использовать для получения антисептика («уралита»),
состоящего из NaF, Na2Cr207, и динитрофенола, предназначенного
для пропитки деревянных шпал и телеграфных столбов147.
Аммиачно-углекислый метод травки 148-149-, аналогичный амми»
ачно-содовому процессу, заключается в обработке монохроматных
щелоков аммиаком и углекислым газом (при обычном давлении):
2Na2CrO4 + 2NH3 + 2CO2 + 2H2O ^=± 2NaHCO3 + (NH4JCr04 + Na2CrO4
Затем путем кипячения хроматный раствор переводят в бихро*
матный с одновременной регенерацией аммиака:
(NH4JCrO4 + Na2CrO4 = Na2Cr20T + 2NH3 + Н2О
Степень перехода хромата в бихромат составляет 90—92%, и
получение стандартного продукта возможно при дополнительной
обработке щелока серной кислотой |33. Этот метод используется в
Чехословакии.
Перевод хроматов в бихроматы можно осуществлять и электро-
электрохимическим путем 150; при этом на аноде выделяется кислород, а
иа катоде водород:
2Н2О ^=±. 2Н+ + 2ОН";
Выход по току для насыщенных растворов хромата натрия до-
достигает 92—95% при 15—25° и 75% при 50—60°.
Разработан также хлорный метод травки, состоящий в обра-
обработке хроматного раствора при 70—80° газообразным хлором, при-
причем наряду с бихроматом образуется хлорат натрия:
6Na2CrO4 + ЗС12 = 3Na2Cr2O7 + 5NaCl + NaClO3
Для разделения солей может быть использована их различная
растворимость.
Хлорный метод может быть совмещен с электролизом: при
электролизе совместного раствора монохромата и хлорида обра»
зуется бихромат, а в качестве побочных продуктов получаются,
хлорат и едкая щелочь.
Наиболее экономичен, по-видимому, метод травки плавиковой
кислотой: он может дать наивысшую эффективность капиталовло-
капиталовложений и лучшую производительность труда, не сопряжен с боль-
большими капитальными затратами, что приводит к меньшей стоимости
продукции. Применение для травки отбросной двуокиси углерода
приводит к значительной экономии соды, но требуе^г больших капи-
капиталовложений в оборудование.
Теория травки хроматов кислотами144
Перевод монохроматов в бихроматы основан на равновесии:
j~ + 2Н+ 5=± 2НСЮ7 ^=± Ст2О27~ + Н2О
В результате гидролиза в растворе монохромата содержат-
содержатся ионы CrOiT, HCrOJ и ОН". При введении в этот раствор
592
Гл. XV. Соли хрома
Физико-химические основы перевода хроматов в бихроматы
593
кислоты НА в нем появляются также ионы Н+ и А". Очевидно, си-
система будет стремиться к состоянию равновесия, определяемому
в общем случае константами равновесия протекающих между
ионами обратимых реакций и растворимостями компонентов. При
расчете равновесий между хроматами и кислотами можно допу-
допустить, что практически соли как сильные электролиты полностью
диссоциированы и что в растворе отсутствуют гидробихроматные
ионы HCr2Of.
При условии, что все компоненты системы хорошо растворимы,
т. е. не выпадают в твердую фазу, как, например, бывает при
травке серной кислотой, ионный состав и степень перехода опреде-
определяются следующими уравнениями:
константа диссоциации травящей кислоты:
A)
1X1 ~ [НА]
константа диссоциации гидрохроматного иона:
_ [нЧ[сгоГ]
Д'~ [НСгО4-]
константа гидролиза бихроматного иона:
[Сг2оГ]
B)
C)
Значения К% и /Сз пока точно не установлены. Приводятся зна-
значения: для К* от 0,9-10 до 1,26- Ю, для К% от 0.9Ы0-2 до
3,03-10~2 (при 25°).
' Если обозначить начальную молярную концентрацию кислоты а
и начальнуюконцентрацию хромата с, то
D)
E)
а=[НА] + [А~]
с = [СгО^-] + [НСгО;] + 2 [Cr2Of-]
Условие электронейтральности раствора (концентрацией Н+ и
ОН~ можно пренебречь) отвечает уравнению
[М+] = 2 [OrOj-] + 2 [Сг2О|-] + [НСгО;] + [А'] - [СгО^] + с + [А~]
откуда, учитывая, что [М+] = 2с, получаем:
Степень перехода:
FI
-[СгО*1 i
Совместное решение уравнений A — 7) приводит к выражению
2KUl-xJ(a-cxJ
(I - х) (а - сх) = К22К3сх3
(8)
из которого можно определять х методом последовательного при-
приближения или графически.
Допущение отсутствия бихроматных ионов в растворе (прене-
(пренебречь ими можно уже при величинах Кз, больших единицы) при-
приводит к более упрощенному урав-
уравнению для степени перехода
Щ
(а + с) - V Ki (с - аJ
(9)
из которого видно, что степень
перехода практически не зависит
от концентраций, а лишь от мо-
молярного отношения кислоты и
хромата.
В практически важном случае,
когда а = с, уравнение (9) после
преобразований дает:
' = (Ю)
Щ
\щ
1«*
а
о.г\
1 +
Z 3 4 5 6 7 в 9 Ю И
А2
д7~ Рис. 174. Зависимость степени пере-
перехода хромата в бихромат от коя-
На рис. 174 нанесены кривые, станты диссотцыиапц™ „TL™efl кисло"
показывающие зависимость сте-
степени перехода х монохромата в -/- i = i
бихромат от —lg K\, т. е. от кон-
константы диссоциации травящей кислоты для —= 1 и Ki= Ю~т (кри-
(криты при а = с:
-tf2 = 810~T;
н Ь\ =0.05 с.
вая /) и 8 • 10 '(кривая 2).
Если травку производят хорошо растворимой кислотой, но с
образованием малорастворимой соли, выпадающей в твердую фазу
(например, при травке плавиковой кислотой), то ионный состав
и степень перехода определяются уравнениями A) и B) (нали-
(наличием Сг2О?" можно пренебречь) и уравнением, характеризующим
растворимость МА. По закону действующих масс равновесие
MA ^=± M* + A~
(Твердая фаза) (Раствор)
определяется константой (произведение растворимости):
594
Гл. XV. Соли хрома
В первом приближении можно принять линейную зависимость
растворимости МА в хроматно-бихроматных растворах от степени
перехода. По значениям для крайних точек можно найти
[ А"] = -J-5— = А. + (ь - —) х
[М+] 2с \ 2с I
[А~] = 6,4-6^ (
или
где #1 = -к растворимость МА в хроматном, Ь — в бихроматном
растворе, а Ъ2 = Ъ — Ъх.
Уравнение E) в данном случае будет иметь вид
[НА] + [А~] = A-#)а A2)
где у — степень перехода кислоты в виде соли МА в твердую фазу.
Так как [М+] = 2с — уа, то из условия электронейтральности
раствора следует:
Совместное решение уравнений приводит к квадратному урав-
уравнению:
Если
¦ 0 (растворимость МА постоянна) и а = с, то
2/CiC + K2bj - VKjbi {Кфх + 4/Clc)
A5);
На рис. 174 нанесена кривая 5 степени перехода для а =
Ь\ = 0,05с и Кч — 10. При травке монохроматного щелока плави-
плавиковой кислотой можно принять К\ = Ю-3 и Ь\ = 0,05с, при этом
уравнение A5) даст х = 0,998.
Если травку производят малорастворимой кислотой (например
угольной), то можно принять [НА] = const:
К, [НА]
] У, [НА] A-х
[HCrOj] [А"] [НСГО4] (bi+b9x)x
При Ь2 =¦= 0
откуда
[НА]
[НА]
A6)
Физико-химические основы перевода хроматов в бихроматы
595
Пока насыщение раствора солью МА еще не достигнуто, можно
принять:
[] []
Тогда
/Ci[HA](l-x)
г= сх*
- ft, [НА] + У К\ [НА]2 + 4KjK2c [НА]
2К2с
A7)
Если применяемая для травки кислота (или ангидрид) газо-
газообразна, то в результате наличия равновесия
[НА]газ +=*: [НА]р.р
на степень перехода должно влиять давление. Если для первого
приближения допустить, что система подчиняется закону Генри, то
[НА]=/Ср
где р — парциальное давление кислоты или ее ангидрида в газовой
фазе.
Подставив это значение [НА] в уравнения A6) и A7), можно
найти зависимость х от давления. При наличии МА в твердой
фазе
при отсутствии МА в твердой фазе
- КхКр +
Из уравнения A8) следует
х = -
2К2с
1
1+-
A9)
B0)
откуда видно, что с повышением давления степень перехода моно-
монохромата в бихромат растет.
Влияние температуры сказывается, главным образом, на изме-
нении растворимости НА и МА, так как отношение -^- можно при*
нять не зависящим от температуры. Вследствие того, что раство-
растворимость газов сильно уменьшается с повышением температуры,
а растворимость солей обычно увеличивается, то, как видно из
Уравнения B0), степень перехода с повышением температуры дбл-
нша уменьшаться.
Когда все компоненты хорошо растворимы, степень перехода х,
как мы видели выше, не зависит от концентрации. Иначе обстоит
Дело при малорастворимой кислоте. Так как Ь\ сильно уменьшается
5 ростом концентрации F, = -^-), то, как видно из уравнения A8),
596
Гл. XV. Соли хрома
значение х должно существенно повышаться с ростом концентра-
концентрации при наличии МА в твердой фазе (изменением [НА] с концен-
концентрацией можно пренебречь). Наоборот, при отсутствии МА в твер-
твердой фазе величина х уменьшается с ростом концентрации, как это
видно из уравнения A9).
Что касается влияния «силы» травящей кислоты, то на рис. 162
видно, что с уменьшением константы диссоциации кислоты до
/(j = 10~4 степень перехода снижается с 1 до 0,97—0,99; при
К\ = Ю~5 х = 0,91—0,97; с дальнейшим снижением К\ ДО 10~9
значение х падает по S-образной кривой до 0,09—0,2 и асимптоти-
асимптотически приближается к нулю.
ПРОИЗВОДСТВО БИХРОМАТА НАТРИЯ151
Схемы производства бихромата натрия отличаются аппаратур-
аппаратурным оформлением. Одна из них изображена на рис. 175. Хромит
E1—53% Сг2О3) и доломит подвергают вначале грубому дробле-
дроблению в щековых дробилках до 40—50 мм, и, в случае их повышен-
повышенной влажности, высушивают дымовыми газами в барабанных су-
сушилках до влажности менее 1%. Затем их измельчают в шаровых
мельницах до тонкости, соответствующей остатку 1—2% при про-
просеивании через сито с 4900 отверстиями на 1 см2 92> 133- isi-i56_ rj0.
мимо доломита в шихту подают пыль, улавливаемую в электро-
электрофильтрах из газов прокалочных печей, выносящих до 10% шихты,
и пыль от размольных механизмов, улавливаемую в рукавных
фильтрах, а также высушенный шлам после второй фильтрации
(см. ниже), полученный в процессе дальнейшей переработки про-
прокаленного спека A5—20% доломита заменяют шламом). Хромит,
соду и наполнители смешивают в шнеках, затем в двухвальных
лопастных смесителях. (Можно производить смешение в кипящем
слое 154.)
Для прокаливания хромитовои шихты служат барабанные вра-
вращающиеся печи, установленные с уклоном 3—6°, делающие 0,7—
1,5 об/мин. Шихта проходит через печь в течение ~3 ч.
Противотоком шихте движутся греющие газы, получающиеся
сжиганием пылевидного угля, или газа, или нефти. Количество
кислорода, подаваемого в печь с воздухом, должно быть достаточ-
достаточным не только для сжигания топлива, но и для окисления хромита.
Оно должно в 3 раза превышать стехиометрическое количество,
необходимое для окисления окиси хрома, — отходящие газы долж-
должны содержать не меньше 7,5% О2155-156. Топливо должно быть
малосернистое во избежание понижения степени окисления вслед-
вследствие взаимодействия хромата натрия с двуокисью серы:
2Na2CrO4 + 2SO2 = Cr2O3 + 2Na2SO4 + 0,5O2
При использовании низкокалорийного топлива требуется по-
подогревать подаваемый в печь воздух. Температуру в нижней части_
TiiiaislUjjeii
;r_. и ^ га
о а ев ?
^г чч „«= з ь г я g " к •-
:ililli!si!:SI
«И1В
И1
|грШ&8-|3|
598
Гл. XV. Соли хрома
лечи поддерживают в пределах 1100—1200°; температура отходя-
отходящих газов 500—750°. Тепло этих газов используется под паровыми
котлами, снабжающими паром дальнейшие стадии производства.
Пыль, уносимая газами из печи, улавливается в электрофильтрах,
в которые газ поступает с температурой 250—300°.
Газы из электрофильтров вентилятором (дымососом) частично
выбрасываются в дымовую трубу, частично подаются в сатуратор
для карбонизации шлама, получающегося в результате выщела-
выщелачивания прокаленной шихты, а часть газов смешивается со струей
воздуха, подающей в печь угольную пыль. Смешение печного газа
•с воздухом, необходимым для подачи в печь пылевидного топлива
и для его сжигания (или для сжигания газообразного или жидкого
топлива), позволяет регулировать температуру в печи.
Нормальная работа печи в значительной мере зависит от усло-
условий, предотвращающих образование «козлов» или настылей157.
Важно поддерживать постоянный состав шихты, влияющий на по-
появление в печи жидкой фазы. Вредным является наличие большого
количества жидкой фазы в начале процесса (приблизительно в се-
середине печи) и уменьшение ее по мере завершения химических
реакций. Чтобы избежать больших количеств расплава на проме-
промежуточных стадиях процесса предложено в шихтах, составленных
на богатой руде, заменять значительную часть соды карбонатом
кальция, т. е. работать на «малосодовой» шихте. Примеси в шихте
м топливе окислов железа, кремнезема и глинозема образуют с со-
содой вязкий и липкий плав. Поэтому при отоплении печей газом,
вместо угольной пыли, образование настылей уменьшается.
Уменьшение вязкости хромитовой шихты может быть также
достигнуто добавкой разрыхляющих материалов — древесных опи-
опилок, рисовой шелухи и др.
Уменьшению настылеобразования способствует применение фу-
футеровки, химически инертной к компонентам прокаливаемой шихты,
например из глиноземистого, хромитового или хромомагнезитового
огнеупоров.
Применение гранулированной или брикетированной шихты по-
позволяет полностью исключить налипание частиц на футеровку и
устранить кольцеобразование. Гранулирование шихты легко до-
достигается окатыванием предварительно увлажненной шихты в ба-
барабане или на наклонно расположенном круглом диске с невысо-
невысокими бортами 157-158.
Существенным является не перегружать печь. Перегрузка печи
приводит к увеличению длительности прогрева шихты, к уменьше-
шию степени окисления хромита, к увеличению расхода топлива и
к опасности «закозления» печи 159.
Производительность печи, имеющей длину 44 м и внутренний
диаметр 2,5 м, составляет 160—190 т шихты в сутки при степени
окисления Сг2О3 в СгО3, равной 89—91%. В печи, имеющей длину.
Производство бихромата натрия
59»
55 м и внутренний диаметр 2,3 м, реакционная зона больше
и при той же производительности степень окисления достигает
92-93%.
О материальном и тепловом балансах прокалочной печи
см.
160
При содержании в исходной шихте 16—17,5% Сг2О3 концентра-
концентрация общего хрома в прокаленной массе в пересчете на СгОз со-,
ставляет 26—30%, причем доля СгО3 в водорастворимых соединен
ниях равна 88—90%. Выходящий из печи в виде гранул прока-,
ленный спек, имеющий температуру 800—900°, проходит через-
барабанный воздушный холодильник, измельчается в вальцовых
дробилках и поступает в шаровые мельницы мокрого помола, где:.
происходит гашение, размол и выщелачивание хроматного спека.
Мельница представляет собой вращающийся стальной барабан,,
выложенный панцырными плитами, внутри которого находятся-
стальные измельчающие шары. Для гашения и выщелачивания
спека применяют слабые щелоки, содержащие 20—50 г/л Na2CrOt>.
получающиеся при промывке на фильтрах горячей водой шлама^
идущего в отвал. В мельнице образуется пульпа (Т:Ж=1:3),
жидкая фаза которой является раствором монохромата натрия.;,
Температура пульпы 80—85°.
Пульпа из мельницы поступает в сатуратор, где происходит
насыщение ее углекислым газом.
Применяют барботажные и барабанные сатураторы. Последние-
менее эффективны — они представляют собой вращающийся гори-
горизонтальный цилиндрический стальной барабан длиной 20 м и диа-
диаметром 1,6 м, снабженный внутри продольными полками из швел-
швеллеров, которые при вращении зачерпывают шламообразную массу
и по мере поднятия вверх разбрызгивают ее. Концы сатуратора-
соединены с загрузочной и разгрузочной головками, через которые,
производится ввод и вывод пульпы и сатурационного газа. Головки-
установлены на роликовых тележках и откатываются во время;
чистки сатуратора.
, Карбонизация пульпы производится очищенным в электрофиль-
электрофильтрах дымовым газом из прокалочной печи, содержащим 10—16%.
СО2. Использование углекислого газа в сатураторе составляет
25—35%. Пульпу разделяют на барабанных вакуум-фильтрах. По-
Получаемый здесь монохроматный, или так называемый желтый'
«крепкий» щелок первой фильтрации содержит 230—270 г/л
Na2Cr04 и 6—10 г/л Na2CC>3. Шлам на фильтре промывают горя-
горячей водой или подогретым до 80° фильтратом второй фильтрации.
При промывке получают слабый щелок, содержащий 80—85 г/л
Na2Cr04, который смешивают с промывной водой второй фильтра-
фильтрации и используют для выщелачивания прокаленной шихты в мель-
мельнице мокрого помола. Средняя производительность 1 м2 фильтрую-
Щей поверхности составляет 100 кг шлама в час.
€00
Гл. XV. Соли хрома
В качестве фильтрующего материала используют капроновую
ткань, но она служит не более 2—3 суток, так как засоряется. При-
Причиной засорения является главным образом выделение кальцита
из раствора монохромата натрия. Промывка капроновой ткани
слабой соляной кислотой позволяет в несколько раз увеличить
срок ее службы. Работы, проведенные в СССР и за рубежом, по-
показывают, что непрерывная регенерация капроновой фильтрующей
ткани может быть осуществлена при применении фильтров со схо-
сходящим полотном 161.
Шлам с первых фильтров смывается водой на вторые (диско-
(дисковые) вакуум-фильтры, откуда после промывки смывается в шламо-
шламовые пруды. Промытый шлам, уходящий в отвал, содержит1б2 28—
35% СаО, 26—33% MgO, 6—10% Fe2O3, 5—8% А12О3, 3—6% SiO2,
5—8% Сг2Озобщ, 1% С; в нем содержится 1—1,5% водораствори-
водорастворимого СгОз и 1—1,2% кислоторастворимого СгОз. Со шламом те-
теряется 15—17% хрома исходной руды. Показана возможность
уменьшения этих потерь путем автоклавного окисления трехва-
трехвалентного хрома кислородом воздуха в процессе выщелачивания
•спека при добавке соды, что способствует полному связыванию
содержащейся в спеке окиси кальция в карбонат и повышает вы-
выход хрома в раствор 163-164. Одной из важных проблем хроматного
производства является утилизация шлама, накапливающегося в
•очень больших количествах. Частично этот шлам может быть ис-
использован взамен части наполнителя (доломита) при составлении
хромитовой шихты 165'1ба. Его можно использовать для строитель-
строительства дорог в качестве добавки при укреплении грунтов 167. Шлам,
получаемый при переработке известковых (бездоломитовых) шихт,
т. е. шлам с пониженным содержанием MgO, после прокалки с уг-
углем для восстановления Cr(VI) до Сг(Ш), может быть использо-
использован для получения силикатного кирпича (искусственного камня) и
¦портланд-пемента ч*8-170.
¦ Желтый хроматный щелок подвергается «подтравке» — предва-
предварительному травлению серной кислотой с целью нейтрализации
щелочности и разложения оставшегося в растворе алюмината нат-
натрия:
2NaOH + H2SO4 = Na2SO4 + 2Н*О
Na,CO3 + H2SO4 = Na2SO4 + H2O + CO,
2NaA10, + HjSO4 + 2H2O = 2A1(OHK + Na2SO4
Полное превращение алюмината в гидроокись алюминия дости-
достигается при рН = 8,21Т|. Подкисление щелока производится 73—
77%-ной серной кислотой при интенсивном перемешивании паром
до начала покраснения щелока так, чтобы в растворе образовалось
не более 10 г/л Na2Cr207 • 2Н^О. Предварительно щелок нагревают
острым паром до 75—80°, так как при этом получается осадок
гидроокиси алюминия, более плотный и легче отстаивающийся.
Производство бихромата натрия
601
80
2 20
Монохроматный щелок фильтруется и отстаивается от осадка
гидроокиси алюминия. Осадок после отжатая и промывки водой
имеет вид пастообразной желто-зеленоватой массы и содержит
10—18% А12Оз, до 3% СгОз, 0,5—9% SiO2, 2—16% СаО, 1—6%
MgO и до 85% воды. Он носит название хромаль и идет в отброс»
но может быть использован в кожевенном производстве или пере-
переработан на алюмогель, пригодный в качестве адсорбента для осуш-
осушки воздуха, очистки масел и
т. п.172. Алюмогель из хромаля
имеет следующий примерный
состав (в % на сухое веще-
вещество): 63,6 —А12О3, 8,5 —SiO2,
7,7 —Сг2О3, 9,4 —СаО, 8,9 —
MgO, 0,8 —SO4~. Для получе-
получения 1 т хроматного щелока, со-
содержащего 195 г/л Na2Cr04,
расходуют: 202 кг хромовой
руды E0% Сг2О3), 288 кг до-
ломита C0% СаО), 71 кг со- и ™
ды (95% Na2CO3), 7,4 кг сер-
ной кислоты A00% H2SO4),
512 м3 генераторного газа,
0,11.2 мгкал пара, 8,5 м3 воды.
Выход хрома в монохромат-
монохроматный щелок составляет 75—81 %.
Потери распределяются следу-
следующим образом: с неразложен-
ным хромитом 10—14%, со
шламом 6—10%, потери пои
размоле и пылеунос 1,5—3%.
Отстоявшийся монохроматный щелок подвергают травлению —
обработке 73—77%-ной серной кислотой для перевода монохро-
монохромата в бихромат. После травки в раствор добавляют гипохлориг
кальция (или хлорную известь) для окисления хрома, содержаще-
' гося в хроми-хроматах. Полученный раствор бихромата натрия
(«красный» щелок) выпаривают в две стадии в многокорпусных
вакуум-выпарных батареях. После первой выпарки до концентра-
концентрации Na2Cr207 600—660 г/л отделяют на центрифуге выпавшие кри-
кристаллы безводного сульфата натрия и щелок упаривают вторично-
до ь-пниентраттии Na2Cr207 1100—1350 г/л.
На рис. 176 приведена политермическая диаграмма раствори-
растворимости в" системе Na2Cr207—Na2SO4—Н2О 173. Поверхность ACHF
# отвечает растворам, насыщенным Na2Cr207 • 2Н2О; поверхность-
FHMN— растворам, насыщенным Na2Cr207; поверхность CBDE —
Na2SO4 • 10Н2О; поверхность EDPMH—Na2SO4. При испарении
воды из раствора, в котором молярное отношение . Na2Cr207:.
Рис. 176. Политерма растворимости
в системе Na2Cr2O7—Na2SO4—Н2О.
602
Гл. XV. Соли хрома
: Na2SO4 равно 1:1, будет происходить кристаллизация Na2SO4.
При повышенных температурах луч, проведенный в плоскости изо-
изотермического сечения диаграммы из начала координат, пересекает
линию насыщения безводным сульфатом натрия, например при
98° — в точке К. При дальнейшем испарении воды, по мере выде-
выделения в осадок Na2SO4 точка состава раствора будет перемещаться
по этой линии (КМ) в направлении к эвтонической точке М, в ко-
которой раствор окажется насыщенным обеими солями. Так как в эв-
тоническом растворе содержание сульфата натрия весьма мало, то
в процессе травки и последующей выпарки в осадок выделяется
почти весь сульфат натрия. В осадок переходит также некоторое
количество гидроокисей алюминия и железа.
Сульфат натрия подсушивают во вращающейся барабанной
сушилке и используют как товарный продукт, содержащий в I сорте
не меньше 78% Na2SO4 и не больше 1% Na2Cr207 • 2Н2О и во
II сорте — не меньше 55% Na2SO4 и не больше4% Na2Cr207 • 2Н2О.
На 1 г бихромата натрия получается до 700 кг сульфата.
Крепкий бихроматный щелок ( ~ 1100 г/л СгОз) после отстаива-
отстаивания перерабатывается на твердый продукт. Для этого его увари-
уваривают в однокорпусных вакуум-выпарных аппаратах или в откры-
открытых плавильных котлах, снабженных паровыми змеевиками и ме-
мешалкой, до концентрации 67,5—69% Сг2О3 и более. В таком плаве
содержится 0,4—0,7% SOf" и столько же С1". Для получения плав-
плавленого продукта плав разливают в стальные барабаны, где он пол-
полностью застывает (закристаллизовывается) при охлаждении на
воздухе, после чего барабаны укупоривают. Из плава изготовляют
также гранулированный и чешуйчатый продукты. Гранулирование
плава осуществляют в двухвальном лопастном грануляторе, дей-
действующем периодически. Освоен и непрерывный способ получе-
получения гранулированного продукта из бихроматного раствора
A100 г/л СгОз) распылением его на кипящий слой гранул в по-
потоке топочных газов 174. Небольшую часть бихромата натрия вы-
выпускают в кристаллическом виде. Для этого охлаждают горячий
красный щелок, выпаренный до концентрации 1200—1300 г/л СгОз.
Кристаллы отделяют на центрифуге, а маточный раствор присоеди-
присоединяют к щелоку, идущему на выпарку 175>176.
Причиной загрязнения бихромата натрия небольшим количе-
количеством сульфата (и хлорида) натрия является недостаточно полное
отделение наиболее мелкой фракции этих солей при отстаивании
бихроматного щелока. При получении кристаллического продукта
содержание этих примесей может быть незначительным при хоро-
хорошем отстаивании бихроматного раствора перед его кристаллиза-
цией. При плохой очистке этого раствора значительная часть взве-
взвешенного сульфата натрия может быть отделена от более крупных
кристаллов бихромата натрия классификацией кристаллической
пульпы на ситах. Наконец незначительное разбавление бихромат-
Производство бихромата натрия
60S
ного раствора перед кристаллизацией (от 1120 до 1070 г/л СгО3)
может обеспечить растворение примесей, а охлаждение раствора
при кристаллизации до 35° — предотвратить их выпадение из рас-
раствора в процессе кристаллизации177.
Отделение бихромата натрия от менее растворимого сульфата
натрия рекомендуют производить высушиванием раствора, содер-
содержащего обе эти соли, например, в распылительной сушилке, с по-
последующим выщелачиванием водой бихромата и отделением суль-
сульфата натрия на центрифуге. Вторичное высушивание полученного-
концентрированного раствора бихромата натрия в распылительной
сушилке дает сыпучий, не пылящий и легкорастворяющийся про-
продукт 130. Эти операции, однако, значительно усложнят и удорожат
производство.
Предложено178 также отделять от бихроматного раствора суль-
сульфат натюия и примеси добавлением к щелоку после травки серной
кислотой крепкого паствора бихр_омата с повышением концентра-
концентрации до 800 г/л NaRCr207 ¦ 2Н2О. После отфильтровывания осадка
щелок упаривают.
На 1 г плавленого бихромата натоия F7,1% СгО3) расхо-
расходуется 1,38—1,41 т хромита E0% Cr9Os^ 2,0—2,15 т доломита,
0,93—0,95 т кальцинированной соды (95% Na2CO3), 0,32—0,40 г
серной кислоты A00%) и 0.005—0,01 т гипохлорита кальция. Рас-
Расход условного топлива G000 ккал/кг) составляет 0,78—0,95 т,.
электроэнергии 400—500 кет • ч, пара 6—8 т, воды 20—40 т.
Получение бихромата натрия с углекислотной травкой хромат»
При травке раствора хромата натрия угольной кислотой сте-
степень перехода хромата в бихромат по реакции
2Na2CrO4 + 2СО2 + Н2О
Na2Cr2O7 + 2NaHCO3
зависит от температуры, концентрации оаствора хоомата натрии
Ji давления двуокиси углерода. С понижением температуры увели-
увеличивается растворимость СО? и уменьшается растворимость NaHCOs,
что способствует протеканию реакции слева направо. В то же-
время с понижением температуры уменьшается растворимость хро-
хромата натоип, что может повлиять неблагоприятно на степень пере-
перехода 179. Степень превращения ч бихромат насыщенного раствора
хромата, содержащего 16,98% Сг, при 23° под давлением СО2 4 ат
Достигает 66% за 5 ч. В ненасыщенном растворе A4,3% Сг) сте-
степень превращения под давлением 8 ат достигает 65% за 2 ч.
Повышение давления СО2 до 40 ат приводит к сравнительно*
^большому увеличению выхода бихромата. При содержании в
'00 мл раствора 16,6 г Сг и 10° степень превращения за 8—12 ч со-
составляет под давлением 10 ат ~68%, а при 40 ат ~83%.
604
Гл. XV. Соли хрома
С повышением температуры до 30° степень превращения умень-
уменьшается до 50% при 10 ат и до ~72% при 40 ат. С увеличением
концентрации хрома в растворе степень превращения возрастает
в большей мере при небольших давлениях и в меньшей мере при
¦повышенных давлениях180.
Остающийся в растворе при однократной травке угольной кис-
кислотой непрореагировавший хромат натрия B5—35% от общего
количества хрома) может быть переведен в бихромат при дотравке
60
«30
60°
10
Ю 20
30 40 50
Na2Cr2O7,%
60
70 60
Рис. 177. Растворимость в системе
Na2CrO4 - Na2Cr2O7 - Н2О.
раствора серной кислотой (после отделения бикарбоната). Для со-
сокращения расхода серной кислоты предложено травленый раствор
выпарить и выкристаллизовать из него при охлаждении часть хро-
хромата. При этом доля бихроматного хрома в растворе может повы-
повыситься до 91%, а расход серной кислоты сократиться в 2,5—3,5
раза ш. Это объясняется тем, что при кристаллизации хромата из
насыщенных растворов, содержание бихромата в которых мало,
происходит обогащение их бихроматом (рис. 177). Так, при 10°
¦эвтонический раствор содержит 6,73% Na2Cr04 и 55,7% Na2Cr207,
¦т. е. относительное содержание в растворе хрома в виде Na2CrC>4
равно 8,89%, а в виде Na2Cr207— 91,11%. При выпаривании рас-
раствора до концентрации 82% Na2Cr207, соответствующей темпера-
температуре кипения его ~ 145° практически весь хромат удаляется из
раствора, а остающийся насыщенный раствор бихромата при ох-
охлаждении застывает в твердый продукт 182.
Преимущества комбинированной травки угольной и серной кис-
кислотами по сравнению с сернокислотной' травкой заключаются в
уменьшении расхода серной кислоты почти в 5 раз, соответствен-
соответственном сокращении количества образующегося сульфата натрия и no.-i
Производство бихромата натрия
605
терь с ним хромпика, в двукратном уменьшении расхода соды
вследствие возврата бикарбоната в процесс.
Возможна и полная замена серной кислоты угольной при двух-
стадийном осуществлении процесса травки с промежуточным от-
отделением бикарбоната натрия. По такой схеме травка хромата
угольной кислотой в крупных заводских масштабах осуществлена
1-я стадия
насыщения
Рис. 178. Схема производства бихромата натрия с углекислотной травкой
монохром атного щелока:
/ — мельница для размола руды; 2 — шихтосмешеиие; 3 — печь для обжига извести; 4 — коль-
кольцевая печь для обжига шихты; 5 —приемный бак; 6 — мельница мокрого помола; 7 —фильтр;
8 — сгуститель; 9 — трехстадийная выпарка; 10 — двухстадийная выпарка; и — травочиые аато-
клавы; 12 — компрессор.
i
в Германии в 30-х годах. На рис. 178 показана схема производ-
производства бихромата натрия с углекислотной травкой 132>133-183. Про-
Прокалку шихты с известковым наполнителем осуществляют в кольце-
кольцевых печах. В этих печах с горизонтальным вращающимся подом
исключается налипание спека на стенки печи. Благодаря быстрому
прогреву шихты и отсутствию непрерывных перевалок сохраняется
пористая, структура спека. Это обусловливает возможность приме-
применения извести вместо доломита и, следовательно, снижение рас-
¦ хода наполнителя. В этом случае расход наполнителя составляет
0,755 т на 1 т бихромата натрия вместо 1 т/т при трехстадийной
прокалке (стр. 588) и вместо 2 т/т при одностадийной прокалке
Шихты.
606
Гл. XV. Соли хрома
Травку раствора хромата натрия производят под давлением
15—16 ат в две стадии смешанным газом (из известково-обжига-
тельных печей и из печей кальцинации бикарбоната), содержащим
50% СО2. В автоклавы первой стадии карбонизации поступает рас-
раствор при 70° с содержанием 820 г/л хромата натрия. В процессе
карбонизации раствор охлаждается до 20° и хромат переходит в
бихромат на 86—87%. Затем раствор отделяют от выделившегося
бикарбоната на центрифуге и выпаривают до концентрации 1300 г/л
в пересчете на хромат натрия; выделившийся частично в осадок
хромат натрия отделяют отстаиванием. Щелок подвергают второй
карбонизации, причем температура его снижается от 70 до 30°, а
степень перехода хромата в бихромат достигает 94—96%. Вторая
стадия карбонизации завершается в течение 6 ч, причем повыше-
повышение давления СО2 сверх 6 ат на выходе бихромата практически
не отражается. Выделившийся бикарбонат также отделяют на
центрифуге, а раствор поступает на дальнейшую переработку. Би-
Бикарбонат натрия промывают водой, кальцинируют и полученную
«желтую» соду (содержащую ~ 1 % Na2Cr04) возвращают на ших-
шихтовку с хромитом. Расход соды при этом методе производства со-
составляет 500 кг на / т хромпика вместо 920 кг при травке серной
кислотой.
БИХРОМАТ КАЛИЯ
Калиевый хромпик может быть получен путем поташного вскры-
вскрытия хромитов, т. е. аналогично натриевому хромпику, но с приме-
применением поташа взамен соды. Этот способ в настоящее время не
применяется, так как рациональнее пользоваться более дешевыми
источниками калия, чем поташ, а именно хлоридом или сульфатом
калия, получая калиевый хромпик обменным разложением этих ка-
аиевых солей с бихроматом натрия по реакциям:
Na2Cr2O7 + 2KC1 = K2Cr2O7 + 2NaCl
Na2Cr2O7 + K2SO4 = K2Cr2O7 + Na2SO,
Поташный способ вскрытия хромитов может, однако, найти
применение в сочетании с травкой хромата угольной кислотой с
возвратом поташа в шихту, по схеме:
4К2СО3 + 2(FeO • Сг2О3) + 3.5О2 = 4K2Cr04 + Fe2O3 + 4CO,
4К2СгО4 + 4СО2 + 2Н2О = 4КНСО3 + 2К2Сг2О7
4КНСО3 = 2К2СО3 + 2Н2О + 2СО2
При содержании в шихте 17,55% Сг2О3, стехиометрического ко-
количества поташа, 5%-ного избытка СаО (в виде доломита) сте-
степень окисления хромита за 60 мин составила в лабораторных усло-
условиях ~95% 184. Увеличение продолжительности прокаливания до
2 ч повышает степень окисления на 1—2%. Уменьшение содержа-
содержания К2СО3 в шихте до 50% от стехиометрического количества, не,-
Бихромат калия
607
обходимого для образования хромата, обусловливает понижение
степени окисления лишь на ~4%.
Описано 132> 133> 135-178-185 получение бихромата калия на заводе
в Биттерфельде по щелочному способу спеканием хромита с едким
кали:
2Сг2О3 + 8КОН + ЗО2 = 4К2СгО4 + 4Н2О
Тонкоизмельченный хромит G—10% остатка на сите с отвер-
отверстиями 0,044 мм) смешивают с 50% раствором КОН, не содержа-
содержащим больших количеств КС1. Полученную пульпу подвергают мок-
мокрому спеканию в течение 9—10 ч при 400—500° в печи, обогревае-
обогреваемой водородным пламенем. Обогрев печи водородным пламенем
исключает образование К2СОз, который налипает на стенки печи.
Плав выщелачивают раствором бикарбоната калия. Получаемый
раствор, содержащий 150—220 г/л К2ОО4 и 120—185 г/л К2СО3,
выпаривают до концентрации 670 г/л КгСО3. Выделившийся при
этом в осадок хромат калия растворяют в воде и подвергают трав-
травке двуокисью углерода под избыточным давлением 4 ат.
2К2СгО4 + 2СО2 + Н2О ^± К2Сг2О7 + 2КНСО3
Осадок бихромата калия отделяют, промывают и высушивают,
а раствор бикарбоната калия направляют на выщелачивание плава.
Раствор поташа, полученный после отделения хромата калия, очи-
очищают от остатков хромата, выпаривают и выпускают в виде то-
товарного продукта. Степень использования хрома по этому методу
составляет 92—96%, т. е. выше, чем по другим методам, и мало
зависит от качества руды. На 1 т бихромата калия F7,1 % СгО3)
расходуется около 2000 м3 водорода и получается 1,2—2 т поташа.
В СССР бихромат калия производят обменным разложением
бихромата натрия с хлоридом калия, более дешевым, чем сульфат
калия. Помимо этого, сульфаты натрия и калия образуют глазе-
ритовые твердые растворы в широком интервале концентраций, что
осложняет осуществление процесса 132>186. Процесс может быть
также осуществлен с применением сильвинита, т. е. смеси КС1 и
* NaCl. Однако в этом случае несколько уменьшается выход КгСг2Ог
и возрастает количество воды, которое необходимо выпарить 187.
На рис. 179 изображена диаграмма растворимости в системе
Na2Cr207 + 2KC1 = К2Сг207 + 2NaCl при 25 и 100°188-191. При
этих температурах стабильной парой солей являются КгСг2О7 и
NaCl; эти соли обладают и наибольшими полями кристаллизации.
Поле кристаллизации К2Сг207при низких температурах сильно уве-
увеличено, поэтому обменное разложение Ыа2СггО7 и КС1 можно было
бы проводить при обычной температуре B0—25°), причем в твер-
твердую фазу выделялся бы КгСг2О7. Однако при этом значительные
количества КгСг2О7 оставались бы в маточном растворе, содержа-
содержащем NaCl. С целью более полного разделения этих солей обменное
608
Гл. XV. Соли хрома
Вихромат калия
609
разложение лучше проводить при повышенной температуре, когда
при соответствующем соотношении компонентов в твердую фазу
будет удаляться NaCl. После отделения осадка NaCl путем охла-
охлаждения щелока из него можно выкристаллизовать К2СГ2О7.
В качестве примера рассчитаем выход К2СГ2О7 при 25° из экви-
эквимолекулярной смеси, содержащей (в г-ионах): 0,5— К, 0,5 — Na,
0,5 — С1 и 0,5 —Сг2О7 (точка А на рис. 179). Этому составу соот-
соответствует содержание воды в насыщенном растворе26 М на 1 г-мол
2КС\0,1 0,2 0,3 Ofi 0,5 0,6 0,7 0,8 0,9К2СггО7
0
2NaC10,S D,B 0,7 0,6 0,5 Ofi 0,3 0,2 0J Na2Cr2O7
Рис. 179. Изображение циклических методов получе-
получения бихромата калия на диаграмме система
Na2Cr207 + 2KCl ^± К2Сг207 + 2NaCl.
солей. Весовой состав раствора (в г): 29,25 — NaCl, 73,55 — К2СГ2О7
и 468 — Н2О. Он может быть получен растворением 37,28 г КС1 и
65,5 Na2Cr2O7 в 468 г Н2О и будет содержать 11,5% Na2Cr207-
(Аналогично, в насыщенном растворе точки А— при 100° 29,9%
Na2Cr207.)
При смешении указанных количеств солей с меньшим количе-
количеством воды, например с 8,5 моль или 153 г Н2О, из системы будет
выкристаллизовываться бихромат калия, а состав жидкой фазы
будет изменяться по лучу кристаллизации КгСг2О7 — Л до точки
В, которая соответствует насыщенному раствору, содержащему
8,5 моль воды, 0,876 моль NaCl и 0,124 моль К2СГ2О7. Если обозна-
обозначим количество этого раствора у, а х — количество выкристаллизо-
выкристаллизовавшегося на участке А — В К2СГ2О7, то уравнение материального
баланса будет:
0,5K2Cr2O7 + 0,5NaCl = *К2Сг,О7 + у @,876NaCl + 0,124К2Сг2О7)
Учитывая, что количество NaCl в растворе не изменяется, т. е.
0,5 NaCl = у • 0,876 NaCl, находим у - 6,571 и х = 0,429. Следова-
0 429
тельно, выход К2Сг207 в твердую фазу составит-^- • 100 = 85,8%.
Это максимальный выход К2СГ2О7, достигаемый при 25° из опти-
оптимальной (стехиометрической) смеси исходных солей при исключе-
исключении опасности загрязнения продукта вследствие кристаллизации
NaCl, начинающейся в точке С.
Дополнительное извлечение КгСггО? из маточного раствора
можно осуществить путем его концентрирования, например, при
100°. Раствор состава JB при 100° находится в- поле кристаллизации
NaCl. Поэтому при выпаривании из раствора будет кристаллизо-
кристаллизоваться NaCl, а состав раствора будет меняться по линии NaCl — В
по направлению к точке D, по достижении которой выпаривание
следует прекратить, во избежание выделения наряду с NaCl также
К2Сг207. Состав раствора в точке D (в молях): 0,609 — NaCl,
0,391 — K2Cr2C>7 и 5,67 — Н2О. По уравнениям материальных ба-
балансов для точки D легко найти что будет испарено 6,7 моль воды
(из 8,5 моль) и выделится 0,683 моль NaCl, или 78% от содержа-
содержащегося в исходном растворе @,876 моль). При охлаждении маточ-
маточного раствора (после отделения NaCl) до 25° возможно дополни-
дополнительно выделить 0,305 моль К2СГ2О7, или 78% от содержащегося
в растворе. Общий выход К2Сг207 в результате двухступенчатой
кристаллизации будет 85,8 + -—~0 ' ' = 96,9% .
Циклическое получение бихромата калия обменным разложе-
разложением бихромата натрия и хлорида калия может быть осуществлено
разными путями. Например, по циклу К—G — L — К исходный
раствор, соответствующий по составу точке К при 100°, охлаждают
до 25°. Во избежание выделения NaCl маточный раствор должен
иметь состав точки G; при этом из первоначального раствора вы-
выделяется 0,346 г-же бихромата калия на 1 г-же суммы солей. Ма-
Маточный раствор нагревают и выпаривают при 100° до получения
раствора состава L. При этом выделяется в осадок 0,346 г-же
NaCl на 1 г-же суммы солей. К отделенному от NaCl раствору до-
добавляют 0,692 г-же смеси состава А. В результате снова полу-
получается 1 г-же исходного раствора К и цикл возобновляется.
Возможно также получение бихромата калия по циклу Е —
*—G—И—Е. Однако в этом случае выход бихромата меньше и
требуется удалять при выпарке раствора большее количество
воды, но аппаратурное оформление процесса проще.
20 М. Е. Позин
610
Гл. XV. Соли хрома
Выход бихромата по обоим циклам легко определить графиче-
графически. Прямые отрезки ЕЕХ и ККи параллельные прямой G —
— Na2Cr2O7, образующиеся при пересечении со стороной квадрата
К2СГ2О7—Na2Cr207 в точках Ei и Ки соответствуют (в масштабе)
количеству бихромата калия, кристаллизующегося при охлажде-
охлаждении растворов Е и К до точки G.
Практически реакция между Na2Cr2'O7 и КС1 проводится в реак-
реакторе, куда предварительно заливают промывные воды и маточный
раствор и выпаривают их до плотности 1,37—1,38 г/см3. Затем
в реактор добавляют раствор бихромата натрия, содержащий не
менее 925 г/л CrOs, и в горячий раствор (~105°), содержащий
260—280 г/л СгО3, вводят эквивалентное количество КС1. Раствор
слегка подкисляют серной кислотой для перевода примеси хромата
в бихромат и окисляют хроми-хроматы хлорной известью. После
отстаивания горячего раствора от выделившегося NaCl осветлен-
осветленный раствор направляют в кристаллизаторы, где при охлаждении
до 30-—35° из него выделяется К2СГ2О7. В процессе кристаллизации
раствор несколько раз разбавляют водой от промывки кристаллов
К2&2О7, чтобы избежать выпадения NaCl вместе с К2СГ2О7. Выход
бихромата калия по хрому достигает 96% 188> 189-
На получение 1 т бихромата калия (98% К2Сг207) расходуется:
1,004 т бихромата натрия A00%), 0,55 т хлорида калия (95%),
0,017 т серной кислоты A00%), до 0,001 т хлорной извести, 5,2 т
пара, 300 кет • ч электроэнергии.
Другой путь разделения солей заключается в выпаривании ще-
щелока после обменного разложения, причем NaCl выделяется в оса-
осадок; его отфильтровывают и промывают. Промывные воды возвра-
возвращают в производство. Отфильтрованный раствор К2СГ2О7 концент-
концентрируют (до 500—525 г/л КгСгг'О?) и после отстаивания направляют
на кристаллизацию в стационарные или вращающиеся кристалли-
кристаллизаторы. Кристаллы КгСг2О7 подсушивают при 40—50°.
Кристаллизация бихромата калия (так же как и бихромата
натрия) сопровождается инкрустацией охлаждающих поверхностей
выделяющимися кристаллами. Для устранения или уменьшения
инкрустации требуется осуществлять интенсивное перемешивание
раствора, перемещающее процесс образования и роста кристаллов
с охлаждающих поверхностей в массу раствора, и обеспечить ма-
малую разность температур между раствором и охлаждающей водой.
Наиболее легко это достигается в кристаллизаторах непрерывного
действия 192. Использование вакуум-кристаллизаторов приводит к
получению мелких кристаллов, как и при чрезмерно интенсивном
перемешивании раствора в механических кристаллизаторах. Кри-
Кристаллизация бихромата в аппаратах с охлаждением и перемеши-
перемешиванием раствора барботирующим воздухом исключает инкруста-
инкрустацию, дает возможность использовать для сооружения кристалли-
кристаллизатора или его облицовки нетеплопроводные, но химически стойкие
Хромовый ангидрид
611
материалы-—пластмассы, резину и др. Однако расход энергии
в барботажных кристаллизаторах в несколько раз больше, чем
в вакуум-кристаллизаторах'""
193
ХРОМОВЫЙ АНГИДРИД
Применяемые методы получения хромового ангидрида основаны
на обработке хромпиков концентрированной серной кислотой:
Na2Cr2O7 + 2H2SO4 = 2CrO3 + 2NaHSO4 + H2O
В стальной реактор с мешалкой и наружным электрообогревом
загружают плав бихромата натрия F0—65% СгО3) и добавляют
92—93%-ную серную кислоту (несколько меньше стехиометриче-
ского количества). Реакционную массу нагревают в течение 1,5—
2 ч, при этом вначале она вспенивается в результате интенсивного
удаления водяного пара, с которым уходят и образующиеся (из
содержащейся в исходном бихромате примеси NaCl) газы — С12.
НС1 и СгО2С1г; отходящий газ для улавливания этих веществ про-
промывают в скрубберах раствором соды. По окончании вспенивания
добавляют остальное количество серной кислоты (до ~105% от
стехиометрического) и поднимают температуру в реакторе немно-
немного выше 200°. Это обусловлено температурами плавления бисуль-
бисульфата натрия A86°) и хромового ангидрида (~200°). Нагревание
массы выше 205° приводит к быстрому разложению СгО3 с выде-
выделением кислорода и образованием соединений СгО3 с Сг2О38-194.
Реакция образования СгО3 на самом деле сложнее, чем приве-
приведенная выше, так как в системе Na2'0—СгО3—SO3—Н2О могут су-
существовать при разных температурах, помимо представленных в
уравнении, и другие соединения, например, Na2SO4, Na3H(SO4J«
•Н2О, Na3H(SO4J177. Кроме того, в небольшой мере идут побоч-
побочные реакции диссоциации СгО3 и образования сульфата хрома
и его двойных соединений с сульфатом натрия и серной кислотой.
Соединения трехвалентного хрома появляются вследствие реакций:
2Na2Cr207 + 10H,SO4 = 2Cr2(SO4K + 4NaHSO4 + 8H2O + 3O2
4CrO3 + 6H2SO4 = 2Cr2(SO4K + 6H2O + 3O2
Сульфат хрома образует с серной кислотой нерастворимые без-
безводные кислые сульфаты хрома 2Cr2(SO4K • H2SO4, Cr2(SO4b-
•H2SO4 и другие, а также соли Na[Cr(SO4J] и Na3[Cr(SO4K] и
кислые соли xNa2SO4 • #Cr2(SO4K • zH2SO4.
После завершения реакции плав сливают в отстойник, снабжен-
снабженный наружными электроподогревателями. Вследствие разности
плотностей расплавленных хромового ангидрида и бисульфата
натрия (при 200° плотность СгО3 —2,30 г/смг, плотность NaHSO4
~2,05 г/см3) и незначительной взаимной растворимости этих ве-
Ществ, они расслаиваются. Скапливающийся в низу отстойника
20*
612
Гл. XV. Соли хрома
Сульфаты хрома
613
жидкий хромовый ангидрид направляют на охлаждаемые изнутри
водой полые барабаны — вальцы, на поверхности которых он за-
застывает и снимается ножами в виде чешуйчатого продукта, кото-
который укупоривают в стальные барабаны. После слива хромового
ангидрида из отстойника выпускают бисульфат натрия — его ис-
используют для травки хромата натрия, которую осуществляют в
этом случае здесь же, в цехе хромового ангидрида ввиду трудно-
трудности транспорта расплавленного бисульфата в бихроматный цех.
Из 1 т хромового ангидрида получают более 1,5 т бисульфата нат-
натрия. Он содержит ~3,5% СгО8, 2% Сг2О3 и 32—33% титруемой
H2SO4.
Полученный продукт содержит более 99% СгОв. Использование
хрома составляет ~96%. На 1 г хромового ангидрида (98% СгО3)
расходуется: 1,52 т бихромата натрия F7,1% СгО3) и 1,29 т серной
кислоты A00%).
Применение бихроматного плава с содержанием 68—70% СгОз
(вместо 63%) позволяет сократить продолжительность плавки на
20—40%, а замена купоросного масла олеумом A03—104% H2SO4)
приводит к сокращению продолжительности процесса в 3 раза 195.
Предложено производить очистку хромового ангидрида добав-
добавкой к нему кристаллического бихромата и его насыщенного вод-
водного раствора и нагреванием массы до плавления. При этом плэб
¦расслаивается, — в нижний слой переходит чистый СгО3, а в верх-
верхний — загрязнения 196.
Из общего количества Сг2О3, содержащейся в бисульфате, до
0,5% (абсол.) находится в форме водорастворимых соединений.
Можно уменьшить их содержание (до 0,05% Сг2О3) выдержива-
выдерживанием бисульфата в смеси с 15% Na2S'O4 (для предотвращения раз-
разложения СгО3) при 185° в течение 1 ч. При этом разлагается лишь
5—!О°/о от содержавшегося в бисульфате СгО3. а Сг2О3 почти пол-
полностью переходит в нерастворимые соединения, которые могут
<шть отделены при приготовлении раствора бисульфата 197. При ис-
использовании бисульфата для травки желтого щелока содержа-
содержащиеся в нем нерастворимые примеси отделяются после травки вме-
вместе с выделяющимся сульфатом натрия. Оставшиеся в щелоке во-
водорастворимые соединения Сг3+ окисляют гипохлоритом 198''".
За рубежом запатентован и осуществлен, так же как и в СССР,
непрерывный способ производства хромового ангидрида200-201. Би-
хромат и серная кислота смешиваются и взаимодействуют, про-
проходя последовательно шнековые аппараты, в последнем из кото-
которых поддерживают температуру выше точки плавления СгО3, за-
затем плав разделяется в непрерывно действующем отстойнике; из
него хромовый ангидрид поступает на охлаждаемые вальцы для
получения чешуйчатого продукта, а бисульфат используется для
травки хроматного щелока. Непрерывный способ обеспечивает воз-
возможность полной автоматизации производства.
Хромовый ангидрид может быть получен из монохроматных
(желтых) щелоков через хромат кальция по следующей схеме:
хромат кальция осаждают обработкой хроматного щелока хлори-
хлоридом кальция при кипячении; отфильтрованный и промытый хромат
кальция разлагают серной кислотой, причем образуются гипс и
раствор СгО3; гипс отделяют на фильтре и раствор выпаривают
для получения твердого СгО3202. Материалом для выпарного ап-
аппарата может служить титан.
Описано много других путей получения СгО3 (например, разло-
разложение хромата натрия азотной кислотой 195), но они пока не имеют
промышленного значения.
СУЛЬФАТЫ ХРОМА
Сульфаты хрома применяются главным образом в качестве ду-
дубителей203. Раньше для этой цели вырабатывали хромовые квасцы,
которые использовались для приготовления дубильных соков, те-
теперь они заменены готовыми сухими хромовыми дубителями — ос-
основными сульфатами хрома, содержащими к тому же больше Сг2О3.
Хромовые квасцы
Хромовые квасцы могут быть получены любым методом, при
котором в растворе образуется смесь сульфата хрома и сульфата
натрия, калия или аммония. Так, например, квасцы могут быть
получены растворением феррохрома в серной кислоте с последую-
последующим отделением от железа и введением в раствор сернокислого
хрома сульфата щелочного металла.
Промышленное производство хромовых квасцов 155 основано на
восстановлении хромпиков в водном растворе серной кислоты.
Восстановление производится органическими веществами, которые,
окисляясь, полностью переходят в углекислый газ и воду, не за-
загрязняя квасцы продуктами своего окисления. В качестве восста-
, новителей могут применяться щавелевая кислота, спирт, сахар,
патока, ржаная или пшеничная мука, древесные опилки, каменно-
каменноугольная смола и т. п.
Реакция образования квасцов выражается схематическим урав-
уравнением
2Na2Cr2O7 + 8H2SO4 + 40Н2О + ЗС = 2 [Na2SO4 • Cr2(SO4K • 24Н2О] + ЗСО2
и протекает со значительным выделением тепла. В аппарат зали-
заливают бихроматный щелок, содержащий 1100—1200 г]л Na2Cr2O7, a
¦ затем 6.5—75%-ную серную кислоту. Количество кислоты коррек-
корректируют по анализу массы в конце процесса. После заливки кис-
кислоты при непрерывном перемешивании в реактор очень медленно
вводят восстановитель, например каменноугольную смолу. При
614
Гл. XV. Соли хрома
Гидроокись и окись хрома
615
этом масса разогревается, сильно вспенивается вследствие выде-
выделения СО2 и увеличивается в объеме. Поэтому полезная емкость
аппарата составляет лишь около 30% его объема.
Температура реакционной массы не должна превышать 60—65е,
так как перегретая масса после выпуска из реактора застывает
чрезмерно долго.
Вследствие того что квасцы кристаллизуются с 24 молекулами
воды, твердый продукт может быть получен простым охлаждением
реакционной массы. Поэтому по окончании реакции масса выпус-
выпускается из аппарата непосредственно в тару — деревянные бочки, в
которых и застывает в твердый продукт — хромо-натриевые квас-
квасцы. На 1 г 100%-ных хромо-натриевых квасцов расходуется 0,3 т
100%-ного бихромата натрия, 0,41 т 100%-ной серной кислоты и
несколько десятков килограммов восстановителя (в зависимости
от его состава).
Хромо-калиевые квасцы производятся (из бихромата калия)
тем же методом, как и хромо-натриевые. Разница лишь в том, что
хромо-калиевые квасцы образуются не в виде загустевающей
массы, а кристаллизуются из раствора204. Они лучше по качеству,
чем хромо-натриевые квасцы, так как являются более чистым про-
продуктом, но стоят дороже.
Хромовые квасцы можно получать, пропуская сернистый газ
через охлажденный до 0° насыщенный раствор бихромата.
Изучена распылительная сушка водных растворов хромовых
квасцов подогретым воздухом при 95 и 120°. Расход тепла составил
~2700 ккал на 1 кг удаленной влаги и "-3900 ккал на 1 кг сухого
продукта205. Уменьшение удельного расхода тепла возможно при
использовании в качестве сушильного агента смеси воздуха с то-
топочными газами.
Сернокислый раствор хромо-аммониевых квасцов используют в
качестве электролита в производстве металлического хрома206.
Для получения хромо-аммониевых квасцов восстанавливают хро-
хромат натрия сернистым газом и отделяют кристаллизацией сульфат
натрия или растворяют в серной кислоте (отработанном электро-
электролите) феррохром и очищают раствор от сульфата железа.
Основные сульфаты хрома
Основной сульфат хрома получают так же как и хромовые
квасцы — восстановлением раствора бихромата натрия органиче-
органическими веществами или сернистым ангидридом и его производными.
Восстановление органическими веществами ведут в кислом рас-
растворе. Например, при использовании мелассы, реакция идет по
схеме:
8Na2Cr2O7 + 24H2SO4 + С„Н22О„ = 16Cr(OH)SO4 + 8Na2SO4 + 12СО2 + 27Н2О
Полученный раствор выпаривают до концентрации 300 г/л
Сг2О3, затем высушивают. Продукт содержит до 30% Сг2О3207. Его
основность зависит от количества вводимой в процесс серной кис-
кислоты. Поэтому, а также из-за явлений гидролиза и других факти-
фактическая формула основного сульфата хрома отличается от формулы
Cr(OH)SO4, согласно которой его основность равна 33,3% 208.
В США выпускают продукты с основностью от 33 до 52% при со-
содержании Сг2О3 около 25%.
Пока менее распространенным, но более совершенным методом
является восстановление бихромата двуокисью серы (а также со-
солями сернистой и тиосерной кислот). Восстановление по реакции
Na2Cr2O7 + 3SO2 + Н2О = 2Cr(OH)SO4 + Na2SO4
идет в отсутствие свободной серной кислоты и дает продукт с ос-
основностью 33,3%. После отдувки избыточного сернистого газа
острым паром полученный концентрированный раствор высуши-
высушивают в распылительной сушилке.
При восстановлении монохромата натрия по реакции
2Na2Cr04 + 3SO2 + 2H2O = Cr2(OHLSO4 + 2Na2SO4
можно получить продукт с основностью 66,7%, а при использова-
использовании смеси бихромата и монохромата получаются сульфаты хрома
с основностью 40—45%, представляющие наибольший интерес как
дубители209.
Основные сульфаты хрома, так же как и хромовые квасцы, мо-
могут быть получены из феррохрома210. Предложены, но не нашли
широкого распространения методы кислотного вскрытия хроми-
хромитов21115. Существенным осложнением этих процессов является не-
необходимость отделения железа216 и других примесей.
ГИДРООКИСЬ И ОКИСЬ ХРОМА
Гидроокись хрома обычно получают из бихромата натрия. Рас-
Раствор Na2Cr207, нагретый до 60°, подкисляют соляной кислотой и
затем вводят отдельными порциями смесь, состоящую из форма-
формалина C2—35%) и соляной кислоты (плотность 1,19 г/см3). Полу-
Получается раствор хлорного хрома, который обрабатывают избытком
аммиака для осаждения гидроокиси хрома. Осадок, после отделе-
отделения от маточного раствора и промывки водой для удаления С1~,
содержит 40—60% воды и подвергается высушиванию. В зависи-
зависимости от условий осаждения и высушивания получаются разные
гидраты окиси хрома: Сг2О3 • 7Н2О, Сг2О3 • 5Н2О, Сг2О3 • 4Н2О
и др. 8.
Окись хрома, предназначенную для полировальных паст и для
Малярных целей, получают дегидратацией гидратов окиси хрома217
или прокаливанием смеси бихромата калия или натрия с серой или
616
Гл. XV. Соли хрома
углем131 (древесными опилками) при 700—800°. Восстановление
протекает по суммарным реакциям:
K2Cr2O7 + S = K2SO4 + Cr,O3
К2Сг2О7 + 2С = К2СО3 + СО + Сг2О3
Процесс осуществляется при первоначальном нагревании смеси
и затем протекает бурно без подвода тепла. Прокаленную массу
по остывании измельчают и обрабатывают водой, или сразу гасят
водой, которая отмывает от окиси хрома растворимые соли. Отмы-
Отмытую окись хрома высушивают и иногда прокаливают при 700—800°
для выжигания органических примесей, а затем, после охлажде-
охлаждения, вновь промывают водой при 50—60°. При получении окиси
хрома из бихромата натрия, серы и древесных опилок (с золь-
зольностью 7,4%) или древесного угля (с зольностью 5,5%) наиболь-
наибольший выход Сг2О3, равный 96,6%, получается при шихте состава
Na2Cr207: S : уголь = 15:2:3. В продукте содержится —82%
Сг2О3. В отсутствие серы выход Сг2О3 не превышает 87% 218.
Прокаливание бихромата калия с 9—11% древесного угля от
веса бихромата осуществляют в футерованных камерах, а также
на непрерывно движущейся ленте, заключенной в стальном ко-
кожухе. Часть кожуха футерована внутри огнеупорным кирпичом,
накаливаемым снаружи топочными газами. При движении через
раскаленную футерованную часть шихта воспламеняется и в ней
идет реакция образования окиси хрома. Выделяющаяся окись
углерода сгорает в этой части печи за счет засасываемого воздуха.
Образующаяся окись хрома сгружается у конца ленты через спе-
о 191149 O1Q
циальныи люк; ее отмывают от поташа и высушивают 1Л1> "".-*1а.
Восстановление бихромата калия углем может быть совмещено'¦
с получением бихромата калия поташным способом (стр. 606), с
использованием получающегося поташа для приготовления хроми-
товой шихты. Расход поташа в этом случае определяется лишь ве-
величиной производственных потерь 184.
Аналогичным образом процесс можно осуществлять, применяя
вместо бихромата хромат натрия. В этом случае образование оки-
окиси хрома идет через метахромит натрия ЫаСгОг, содержание кото-
которого в спеке достигает 10—15%. При выщелачивании спека мета-
метахромит натрия гидролизуется с образованием едкого натра и гид-,
роокиси хрома. Это приводит к необходимости повторной прокалки,
выщелоченной массы с последующей отмывкой продукта от обра-'
зовавшегося монохромата натрия 220.
Содержание свободного и карбонатного углерода в окиси хрома,;
полученной восстановлением К2Сг207 древесным углем, составляет}
0,3—0,5%. Для получения металлургической окиси хрома, содер-
содержащей не более 0,15% углерода, производят его выжигание при-
650—700° в электрических вращающихся печах. Этот процесс про-
протекает крайне медленно из-за плохой теплопроводности материала^
Гидроокись и окись хрома
617
и малого содержания выжигаемого углерода. Процесс возможно
осуществить при более низкой температуре с применением окисли-
окислителя, например NH4NO3 или СгО3221. В присутствии 10% NH4NO3
(от веса Сг2О8). выгорание углерода за 1 ч при 350° протекает на
85—90%.
Удаление углерода происходит в результате следующих реак-
реакций:
2NH4NO3 + С = СО2 + 2N2 + 4Н2О
2NH4NO3 + К2СО3 = 2NH3 + СО2 + Н2О + 2KNO3
Образовавшийся нитрат калия реагирует с окисью хрома, да-
давая хромат (и, возможно, бихромат):
Cr2O3 + 4KNO3 = 2K2Cr04 + N2O3 + 2NO2
Непрореагировавший нитрат аммония разлагается (стр. 1183).
Окись хрома может быть получена восстановлением твердого
бихромата или хромата газообразными восстановителями — водо-
водородом222'223, окисью углерода224 и другими, например в печи со
взвешенным слоем22S. Запатентованы методы восстановления хро-
матов газами (Н2, СО) в растворах под давлением 150—
200 аг226^228. Окись хрома получается также обработкой хлорного
хрома при 500° кислородом воздуха229-230, или водяным паром231. .
Чистую окись хрома можно получать прокаливанием гидро-
гидроокиси хрома, осажденной из солей трехвалентного хрома аммиа-
аммиаком или едкими щелочами, а также при нагревании бихромата
аммония или смеси бихромата калия или натрия с хлоридом или
сульфатом аммония. В основе последних процессов лежит реакция:
(NH4JCr2O7 = Cr2O3 + N2 + 4Н2О + 123 ккал
Образование окиси хрома происходит, очевидно, за счет вос-
восстановления Сг6+ до Сг3+ аммиаком, входящим в состав бихромата
аммония.
Непосредственное восстановление бихромата натрия аммиаком
* идет при 300° до хроми-хромата натрия. Хромат натрия при этой
температуре не реагирует с аммиаком. При 700° бихромат и хро-
хромат натрия восстанавливаются аммиаком до хромитов:
Na2Cr2O7 + 2NH3 = Na2O • Cr2O3 + ЗН2О + N2
2Na2Cr04 + 2NH3 = 2Na2O • Cr2O3 + 3H2O + N2
Однако гидролиз хромитов натрия протекает с трудом и при
выщелачивании их большим количеством горячей воды удается пе-
- ревести в раствор не более 30% щелочи232.
Окись хрома может быть получена восстановлением щелочного
раствора хромата натрия сернистым газом233-234. При этом осаж-
осаждается хромат хрома (хроми-хромат) 2Сг2О3 • СгО3 • л:Н2О, который
618
Гл. XV. Соли хрома
Гидроокись и окись хрома
619
при дальнейшем прокаливании переходит в окись хрома. Послед-
Последний процесс основан на том, что при нагревании СгО3 выделяет
кислород и переходит в Сг2О3:
2BСг2О3 • СгО3 • *НгО) = 5Сг2О3 + 1,5О2 + 2хН2О
Взаимодействие бёсщелочного раствора хромата с двуокисью
серы при рН = 9,2 протекает с частичным образованием бихро-
мата:
2 + хН2О = 2Сг2О3 • СЮ3 • *Н2О + 6SO2~ |
Максимальная степень восстановления хромата достигается при
рН = 5,3; она равна ~57%, а максимальная степень осаждения
хрома 71,4%. При дальнейшем введении SO2 ион Сг2О72~ восста-
восстанавливается до '/з-основного сульфата хрома CrOHSO4, переходя-
переходящего затем в нейтральный сульфито-сульфат хрома. Одновременно
может происходить растворение ранее выделившегося осадка хро-
хромата хрома, также переходящего в сульфито-сульфат хрома.
При восстановлении сернистым газом кипящего щелочного рас-
раствора монохромата натрия (с добавкой Na2CO3 или NaOH) дости-
достигается практически полное осаждение гидроокиси хрома (при по-
понижении рН до 5,3) по реакции
2СгО2~ + COf- + 3SO2 + *Н2О = Сг2О3 • *Н2О + 3SO^~ + СО2
или по реакции:
" + 2ОН~ + 3SO2 + (х
1)Н2О = Сг2О3 • хН2О + 3SO'f
Окись хрома получается после прокаливания отфильтрованной
и отмытой гидроокиси; маточный раствор может быть переработан
на товарный сульфат натрия.
Помимо способов, кратко рассмотренных выше, окись хрома
можно получать прокаливанием хлоридов хрома23Е или основных
сульфатов хрома или карбонизацией их в водных суспензиях с
последующим прокаливанием образующейся гидроокиси хрома23в
и другими путями. В СССР в настоящее время окись хрома полу-
получают главным образом двумя способами — термическим разложе-
разложением хромового ангидрида и восстановлением хромата натрия в
растворе серой и тиосульфатом (хроматно-серный метод).
Термическое разложение хромового ангидрида 131 позволяет по-
получать тонкодисперсную окись хрома для шлифовальных матери-
материалов и для лакокрасочной промышленности. В результате прока-
прокаливания расплавленного хромового ангидрида при 850—900° окись
хрома образуется по реакции:
2СгО3 = Сг2О8 + 1,БО2
Однако при этом появляются промежуточные продукты (стр.
667), разлагающиеся по мере повышения температуры по схеме8:
285-300° 340-370°
Выше 800" .
—> СгО2,40
По более поздним данным 236:
СгО3(тв.) -^-°> СгО3(ж.) —
СгО2,в2(тв.)
330-370°
СгО2.«(тв.)
СгО,,5(тв.)
Полученный спек гасят водой (промывными растворами) и
систематически промывают (для извлечения растворимых веществ,
главным образом сульфата натрия). В хромовом ангидриде содер-
содержатся примеси соединений SO?" — бисульфата натрия и кислой
соли сульфата хрома %Na2SO4 • #Cr2(SO4K • zH2SO4 (стр. 566). Кис-
Кислый сульфат хрома образуется и при прокаливании в результате
взаимодействия окиси хрома с бисульфатом. Во избежание загряз-
загрязнения окиси хрома нерастворимым кислым сульфатом хрома
предложено238 добавлять к хромовому ангидриду соду, которая
переводит и бисульфат натрия и сульфат хрома в сульфат натрия:
2NaHSO4 + Na2CO3 = 2Na2SO4 + Н2О + СО2
Cr2(SO4J + 3Na2CO3 = 3Na2SO4 + Cr2O3 + 3CO2
Отфильтрованную после выщелачивания и промытую пасту
окиси хрома высушивают в барабанной сушилке с наружным обо-
обогревом. Окись хрома, получаемая термическим методом, содержит
99—99,5% Сг2О3, 0,01—0,015% S, -0,03% С, 0,02—0,07% FeO,
0,03—0,07% Н2О.
Хроматно-серный метод получения окиси хрома
Этим методом, заключающимся в восстановлении водного рас-
раствора хромата натрия серой200, производят наибольшее количе-
количество окиси хрома, главным образом, для переработки на металли-
металлический хром. Преимущество метода — использование сравнительно
дешевого сырья—раствора монохромата натрия и, вследствие
этого, низкая себестоимость продукта (на 13—17% ниже себестои-
себестоимости окиси хрома, полученной термическим разложением хромо-
хромового ангидрида200; см. также239).
Щелочной раствор монохромата натрия, содержащий 110—
140 г/л СгО3 и около 10 г/л щелочи (в пересчете на NaOH), обра-
обрабатывается в каскаде реакторов при кипячении A03°) тонкоиз-
мельченной @,2 мм) серой, добавляемой в виде суспензии в части
ароматного щелока, приготовленной в шаровой мельнице. По дру-
другому варианту восстановление осуществляют в автоклавах рас-
расплавленной серой.
620
Гл. XV. Соли хрома
Хлорный хром
«21
Вследствие гидролиза серы в горячем щелочном растворе обра-
образуются ионы (S-SnJ" и другие (см. гл. XIV), которые восстанав-
восстанавливают шестивалентный хром до трехвалентного, а сера превра-
превращается главным образом в S2O3~- Общее уравнение реакции вос-
восстановления:
4Na2CrO4 + 6S + Bх + 1)Н2О = 2(Сг2О„
+ 2NaOH
Выделившийся осадок гидроокиси хрома отфильтровывают на
барабанных вакуум-фильтрах, промывают и прокаливают для по-
получения окиси хрома в барабанной печи при ~1100° в потоке га-
газов, полученных сжиганием топлива в избытке воздуха для созда-
создания окислительной атмосферы. При этом примеси сернистых соеди-
соединений и щелочи превращаются в водорастворимые соли Na2SO4 и
Na2Cr04. Для их извлечения выходящий из печи спек окиси хрома
выщелачивают, затем несколько раз промывают на фильтрах с
промежуточными репульпациями и высушивают в барабанной су-
сушилке с наружным обогревом.
Щелочной раствор тиосульфата натрия, полученный после отде-
отделения гидроокиси, перерабатывают с помощью серной кислоты на
сульфат натрия и серу по реакции:
3Na2S2O3 + H2SO4 = 3Na2SO4 + 4S + H2O
Более рациональным является использование этого раствора
для восстановления хромата натрия по реакции24043:
10Na2CrO4 + 3Na2S2O3 + 7H2SO4 + Bx- 7)H2O =
= 2BСг2О3 • СгО3 • хН2О) + 13Na2SO4
При этом окись хрома получают по так называемой упрощен-
упрощенной схеме хроматно-серного метода, заключающейся в двухстадий-
ном восстановлении монохромата — сначала часть щелочного рас-
раствора монохромата восстанавливают жидкой серой в автоклаве, <
затем к полученной суспензии окиси хрома в щелочном тиосуль-'
фатном растворе добавляют остальную часть монохром атн ого рас-
раствора и серную кислоту и смесь кипятят. После этого дополни-
дополнительно обрабатывают раствор в автоклавах при 150°. Отфильтро-
Отфильтрованный осадок хроми-хроматов прокаливают, при этом он превра-
превращается в окись хрома, не содержащую адсорбированной щелочи,
так как процесс восстановления завершается в кислой среде. Эта
окись хрома не требует дальнейшей обработки и выпускается в
качестве готового продукта. Раствор, оставшийся после отделения
хроми-хроматов, перерабатывают на товарный сульфат натрия.
В упрощенном варианте хроматно-серного метода отсутствует ряд
операций, имеющихся в основном варианте —выщелачивание,
фильтрование с репульпациями и сушка окиси хрома.
На производство 1 г окиси хрома хроматно-серным методом
расходуется 2,02 т монохроматного щелока F7,1% Cr2Os), 0,85 г.
серы и 0,06 т кальцинированной соды, а по упрощенному варианту
2,05 т монохроматного щелока, 0,175 т серы, 1 т серной кислоты
A00%) и 0,02 т кальцинированной соды. Получаемая окись хрома
содержит 98—99% Сг2О3, меньше 0,01% S, 0,03% С, -0,1% FeO,
до 0,08% Н2О.
хлорный хром
В связи с трудностью получения металлического хрома непо-
непосредственно из железистой хромитовой руды, ее сначала перераба-
перерабатывают на соединения хрома, которые (главным образом Сг2Оз)
и служат сырьем для металлургов. Одним из перспективных видов
такого сырья является безводный хлорный хром СгС1з — он обра-
образуется при хлорировании газообразным хлором хромитов, в част-
частности низкопроцентных, в присутствии восстановителя при 800—
jqOo°244-247 г]рИ хлорировании брикетов руды с каменным углем,
например в шахтной печи, образуется СгС13 наряду с другими ле-
летучими и нелетучими хлоридами: FeCl3, A1C13, SiCl4, MgCl2 и др.
При небольшом избытке хлора низшие хлориды хрома и железа
не образуются. В отсутствие восстановителя хлорируется только
закись железа. Летучие хлориды могут быть разделены дробной
конденсацией вследствие различного давления их пара.
Хлорирование при 900—1000° обеспечивает практически полное
извлечение хрома из руды в виде СгС18. Восстановителем является
главным образом окись углерода. В указанном интервале темпе-
температур при размерах частиц угля меньше 0,15 мм, а частиц руды
меньше 0,07 мм, скорость процесса лимитируется реакцией газифи-
газификации углерода. Если частицы руды крупнее 0,15 мм, лимитирую-
лимитирующей становится реакция хлорирования. В последнем случае резко
возрастает расход углерода, так как отношение СО : СО2 в отходя-
отходящем газе увеличивается 248>249.
Удаление железа из хромитовой руды или концентрата позво-
позволяет использовать их для непосредственной переработки в хром
металлургическими методами. Такое обогащение хромита может
быть достигнуто селективным хлорированием в присутствии огра-
ограниченного количества углерода248-250'251. Входящие в состав руды
окислы могут быть расположены в следующий ряд по уменьшению
своего сродства к хлору (в присутствии углерода при 800—1000°):
MgO, Fe2O3, FeO, СггОз, А12О3, S1O2. Окислы железа хлорируются
легче окиси хрома и потому, если в шихте содержится лишь такое
количество углерода, которое необходимо для связывания кисло-
кислорода окислов железа, то в результате хлорирования почти пол-
полностью удаляется железо и только небольшая доля хрома (менее
Ш отн. %), которая может быть извлечена из газовой фазы в виде
• СгС13 его фракционной конденсацией. Например, при лаборатор-
лабораторном хлорировании руды, содержавшей 51,6—55,0% Cr2Og, 1,6—
Ю,3% Fe2O3, 3,1-9,9% FeO, 13,7-17,4% MgO, 8,8—8,4% A12O3,
622
Гл. XV. Соли хрома
4,4—5,9,% SiO2 и до 1,1% СаО, получался продукт — безжелези-
безжелезистый хромовый концентрат, в котором было 60—64% Сг2О3 и
меньше 0,2% железа; содержание углерода не превышало 0,02%,
а хлора —2,3% 250.
Хлорный хром можно получать хлорированием феррохро-
феррохрома 2Б2-256. При хлорировании углеродистого феррохрома избытком
хлора при 900—1000° образуются газообразные СгСЬ и FeCb. Без
избытка хлора образуется также жидкий FeCl2. Процесс может
быть осуществлен автотермично. Хлорирование можно вести и в
среде расплавленных хлоридов (Na, К, Mg, Ca, Ba, FeJ53-2S7. Для
хлорирования хромита и феррохромита может быть использован
также хлористый водород258'2Б9.
ДРУГИЕ МЕТОДЫ ПЕРЕРАБОТКИ СЫРЬЯ И ОТХОДОВ, СОДЕРЖАЩИХ
ХРОМ
Несмотря на то что в промышленности получила распростране-
распространение окислительная прокалка хромита с целью переработки его в
соли шести- и трехвалентого хрома, в отдельных случаях могут
представить интерес и другие методы переработки хромовых руд.
Следует также отметить, что щелочная прокалка хромита с окис-
. лением его кислородом воздуха экономична лишь при использова-
использовании высококачественных руд с малым содержанием кремнезема.
Низкокачественную хромитовую руду предложено перерабаты-
перерабатывать кислотным методом, заключающимся в следующем. Руду, из-
измельченную до размера частиц меньше 75 мк, растворяют в 75%-
ной серной кислоте при 140—160° в присутствии СгО3 A0% от
веса руды), который служит катализатором. Степень разложения
руды за 4 ч составляет 90%. Образующийся раствор зеленого
цвета разбавляют водой, отфильтровывают от неразложенного хро-
хромита и кремнезема и после дополнительного разбавления до кон-
концентрации 45 г/л Сг2Оз подвергают электрохимическому окисле-
окислению. Электролиз осуществляют в ванне с диафрагмой и с элек-
электродами из двуокиси свинца при плотности тока ~ 2 а/дм2 н
напряжении 3,2 в. Степень окисления раствора составляет 95%, вы-
выход по току 50—55%. Полученный раствор красного цвета выпа-
выпаривают до концентрации 750 г/л SO42~, причем из него выделяется
85—90% содержащегося в нем сульфата железа и 60—80% суль-
сульфата алюминия. После отделения сульфатов фильтрат выпари-
выпаривают до концентрации, соответствующей температуре кипения 150°.
Охлаждением выпаренного раствора из него выкристаллизовывают
СгО3. Отфильтрованный и промытый продукт содержит до 95%
СгОз (или до 99% СгОз в сухом веществе). Фильтрат, содержащий
значительное количество СгОз и H2SO4, возвращают на разложе-
разложение руды. Степень извлечения хрома из руды, содержавшей 37 7%
Сг2О3, 12,5% FeO, 26,8% А12О3, 18,8% MgO и 4,6% SiO2, на опыт-
опытном заводе составляла 75% 260.
Другие методы переработки сырья и отходов
623
Представляет интерес применение пористых сорбентов для раз-
разделения соединений хрома и железа при помощи ферроцианида ка-
калия, как осадителя, и древесного активированного угля в качестве
сорбента (носителяJ61.
Хромовый ангидрид, полученный кислотным методом из бедных
руд, может быть использован без дальнейшей очистки для произ-
производства дубильных солей, хроматов, бихроматов и красок. При до-
дополнительной перекристаллизации можно получить продукт, при-
пригодный для хромирования.
Изучение выщелачивания хромита серной кислотой в присут-
присутствии К2Сг207 в качестве окислителя показало, что растворение
определяется окислением Fe2+ в Fe3+ и зависит от отношения содер-
содержащегося в руде общего количества FeO к количеству FeO, свя-
связанному с Сг2О3. Для высокосортной руды, содержавшей 43,95%
Сг2О8 и 18,7% FeO (FeOoBm : FeOxp=0,9), извлечение хрома из
руды достигло 94,5%; из низкосортной руды, содержавшей 37,41%
Сг2О3 и 11,66% FeO (FeOoBm : FeOxp = 0,66), было извлечено около
46% хрома268.
Предложено перерабатывать сырье, содержащее хром, при по-
помощи кислого сульфата аммония. При этом в раствор переходят
Cr2(SO4K, (NH4JSO4 и сульфаты других металлов. После отделе-
отделения нерастворимого остатка из раствора осаждают аммиаком в
отдельности гидроокиси металлов и Сг(ОНK. Последний раство-
растворяют в серной кислоте для получения Cr2(SO4K- Из фильтрата,
после отделения Сг(ОНK кристаллизуют (NH4JSO4, который нагре-
нагреванием до 370° переводят в NH4HSO4, возвращаемый в процесс263.
Представляет интерес получение соединений трехвалентного
хрома, а также металлического хрома из феррохрома, получаемого
непосредственно при плавке руды. Предложено перерабатывать
феррохром в Na2Cr04 и СаСгО4 путем определенной дозировки в
шихте соды и окиси кальция. Соду вводят в шихту в количестве,
необходимом для превращения половины хрома, содержащегося в
сырье, в хромат натрия. Окись кальция вводят в количестве, боль-
большем, чем это требуется для связывания остальной части хрома в
хромат кальция. Процесс ведут с тонкоизмельченным феррохро-
феррохромом, прокаливая шихту при температурах, близких, но не превы-
превышающих температуру плавления феррохрома. Продукт обжига
содержит Na2Cr04 и СаСгО4 приблизительно в одинаковых моле-
молекулярных количествах264.
Хромат и хромит кальция могут служить исходным материалом
для производства металлического хрома алюмино- и силикатер-
мическими методами94. В связи с этим патентовались и разраба-
• тывались способы их получения из хроматного щелока, а также из'
хромита и из феррохрома 265-267
На заводах анилино-красочной промышленности, широко
использующих бихроматы в качестве окислителей; получают
624
Гл. XV. Соли хрома
Литература
в качестве отходов производства значительное количество кислых
фильтратов, содержащих сульфат хрома (~ 100 г/л Сг2О3 и 350 г/л
H2SO4). Из этих растворов сульфата хрома регенерируют электро-
электролизом двухромовую кислоту с возвратом ее в производство в смеси
с серной кислотой. Электролиз ведут в ванне с диафрагмой и со
свинцовым анодом. На поверхности анода образуется двуокись
свинца, играющая роль передатчика кислорода. Электролиз ведут
при плотности тока на аноде 120—300 а/м2 и температуре 30—50°.
Напряжение на ванне около 3,5 в. Выход по току 85—90%. Малые
плотности тока на аноде и большие плотности тока на катоде при-
приводят к высоким выходам по току. С повышением плотности тока
на катоде уменьшается восстановление. На 1 т окиси хрома расхо-
расходуется около 3400 кет - ч268.
Электрохимическое окисление трехвалентного хрома в концен-
концентрированных растворах хромо-натриевых квасцов изучено в преде-
пределах концентраций 40—170 г/л Сг3+ и 45—850 г/л H2SO4269. Оно мо-
может быть осуществлено р электролизере с диафрагмой или без
диафрагмы со свинцовыми электродами. Наибольшие выходы по
току (~91%) получаются при содержании в исходном растворе
около 80 г/л Сг3+ и 55 г/л H2SO4, при плотности тока 3 а/дм2 и тем-
температуре 55°. Напряжение на ванне 3,5—4 в; степень окисления
Сг3+ в Сг6+ 90—95%- Об электрохимическом окислении сульфата
хрома в разбавленных растворах см. ^о-2?2.
Электролиз отработанных растворов соединений хрома, содер-
содержащих ионы Cr2O?~, CrOl", Cr3+ и другие, с применением желез-
железных электродов в ванне без диафрагмы в присутствии веществ, де-
деполяризующих железный анод (например, NaCl), приводит к вы-
выделению из электролита нерастворимого осадка, соответствующего
по составу хромиту железа Fe(CrO2J. При плотности тока на
аноде 10—12 а/дм2, напряжении 10—15 в и температуре электро-
электролита 20—30° хромпиковый раствор с содержанием 2 г/л Сг2Оз, алю-
мо-хромовый с содержанием ~10 г/л K2AlCr(SO4L и хромовый с
содержанием 11 г/л Cr2(S04b порознь и в смеси из равных весо-
весовых количеств осветляются полностью при электролизе в течение
10—12 мин. Образующийся на дне электролизера осадок желто-
бурого цвета содержит до 30—35% Сг2О3. Этот метод может ока-
оказаться пригодным для извлечения хрома из сточных вод кожевен-
кожевенной промышленности, гальванотехнических производств и др.273.
Предложены различные методы регенерации отработанных рас-
растворов после хромирования. Так, осаждение ионов загрязняющих
металлов можно осуществить добавкой раствора едкой щелочи.
После нагревания до кипения раствор хромата отделяют от
осадка 274.
Очистка отработанных хромовых растворов после анодной об-
обработки алюминия, хромирования металлов или снятия медных
Покрытий с регенерацией хромовой кислоты может быть осущест-
осуществлена с помощью ионообменной смолы типа сульфированных поли-
полимеров поливинилароматических соединений276.
Большинство соединений хрома вредны, а соединения шести-
шестивалентного хрома ядовиты, и работа с ними требует тщательного
соблюдения условий, обеспечивающих сохранение здоровья рабо-
работающих. Проникая в кожу через незначительные повреждения, со-
соединения хромовых кислот разрушают белковые вещества клеток,
вызывают язвы. Особенно сильному разрушению подвержены сли-
слизистые оболочки. Вдыхание пыли и испарений, содержащих соеди-
соединения хрома, приводит к поражению слизистой оболочки носа и к
прободению его хрящевой перегородки.
Важнейшим мероприятием, обеспечивающим безопасность ра-
работы, является устранение выделения в атмосферу цеха пыли из
мельниц, смесителей, прокалочных печей и паров, содержащих
соединения хрома. Это достигается герметизацией аппаратов и ис-
использованием санитарной вентиляции275. Сточные воды должны
подвергаться очистке от соединений хрома 277. В качестве средств
индивидуальной защиты должны применяться респираторы, за-
защитные очки, резиновая обувь, перчатки, передники. Соблюдение
правил техники безопасности, а также правил личной гигиены и
использование профилактических средств (смазывание кожи рук
умягчающими мазями и др.) вполне предохраняют работающих от
поражений.
ЛИТЕРАТУРА
I. Т. Д. А в е р б у х, П. Г. Павлов, Технология соединений хрома, Изд.
«Химия», 1967. — 2. М. Y. U d у, Chromium, v. I, Chemistry of Chromium and its
Compounds, N. Y. — London, 1956.— 3. Т. В. Роде, Труды 1-го Совещания по
термографии, Казань, 1953, Изд. АН СССР, 1955, стр. 154. —4. М. С. Ковель,
Труды Уральск, политехи, ин-та, № 152, 1966, стр. 5.—5. С И Дьячков-
Дьячковский, В. К. Рубцова, ЖОХ, 10, № 4, 380 A940). —б. С. И. Дьячков-
Дьячковский, В. М. Офицеров, Там же, 11, № 5—6, 371 A941). —7. М W S h а-
ter, R. Roy, Z. anorg. Chem., 276, № 5—6, 275 A954).— 8. Т. В. Роде, Авто-
реф. докт. дисс, 1ЮНХ АН СССР, 1956; Кислородные соединения хрома и
хромовые катализаторы, Изд. АН СССР, 1962.—Р. М. А. Миниович, ЖХП,
№ 7, 34 A939). —10. Г. Грассер, Краткий курс хромового производства,
* Гизлегпром, 1934.
II. А. А. Гринберг, Введение в химию комплексных соединений, Госхим-
нздат, 1951, стр. 231. —12. А. В. Памфилов, Н. Н. Гуменюк, ЖОХ, 23,
№ 7, 1065 A953). — IS. В. В. Печковский, С. А. Алперова и др ЖАХ
9, № 9, 2059 A964). —14. S. N. Tewari, С. Chosh, Kolloid Z., 138, № 2 87
A954). — /5. S. N. Tewari, S. Ghosh, Z. anorg. Chem., 279, № з_4, 225
A955). — 16. И. В. Васильева, А. И. Ефимов, Б. 3. Питеримов, ЖНХ,
9, № 3, 754 A964). —17. Н. Н. Пучков а, Автореф. канд. дисс, Черновицк.
гос. ун-т, 1954. — 18. А. В. Памфилов, Н. Н. Пучкова, ЖОХ 26, № 4,
955 A956). —19. Lor ant В ё 1 a, Z. analyt. Chem., 219, № 3, 256 A966) —
• 20. И. Д. Музыка, О. И. Шор, ЖОХ, 26, № 5, 1277 A956).
21. А В. Памфилов, А. И. Л о п у ш и н с к а я, А. М. Белая, Укр хим
ж., 30, № 2, 173 A964).— 22. О. Quadrat, V. В alien sky, J. Fafe, Sbornlk
yysoke skoly chemicko-technologicke, Praze, 1957. — 23. Morry, J. Chem. Soc.
626
Гл. XV. Соли хрома
Литература
627
Japan. Pure Chem. Sec, 74, № 4, 253 A953). — 24. В. Ф. Смачная, П. Я. Саль-
да у, Зап. ЛГИ нм. Г. В. Плеханова, 32, № 3; 313 A956). — 25. W. H. Hart-
Hartford, Ind. Eng. Chem., 41, № 9, 1933 A949). —26. А. В. Ра ко век и и,.
Д. Н. Тарасенков, ЖРФХО, 60, 7 A929). — 27. В. Н. Васильева,.
Автореф. канд. дисс. Ивановск. хим.-технол. ин-т, Иваново, 1955. — 28. К. Б. Я Ц и-
ыирский, В. Н. Васильева, ЖНХ, 1, № 5, 984 A956). — 29. В. J. T h a-
m e r, R. M. D о п g I a s s, E. Staritsky, J. Am. Chem. Soc, 79, № 3, 547
A957).—SO. С. М. Ария, С. А. Щукарев, В. Б. Глушкова, ЖОХ, 28„
№ 8, 1241 A953).
31. О. G 1 е m s e r, U. H a u s с h i I d, F. T г и р е 1, Naturwissenshaften, 40,.
№ 11, 317 A953). — 32. Перекись водорода и перекисные соединения, под ред.
М. Е. Позина, Госхимиздат, 1951. — 33. R. С. R a i, S. Prakash, Z. anorg.
Chem., 275, № 1—3. 94 A954).— 34. R. C. R a i, J. Indian Chem. Soc, 34, № 1.
59 A957).— 35. H. С. Николаев, Ю. Н. Буслаев, ИСФХА ИОНХ АН
СССР 26, 270 A955). — 36. Н. J. Kaufmann, W. В. L a u d e r, R. К. К е р-
ner, Ind. Eng. Chem.. 32, № з, 423 A940).— 37. Isakichi Takenchi, Me-
Memoirs of the College of Science Kyoto Imper. Univ., 1, № 5, 249 A915).—
38. И. Г. Дружинин, Изотерма растворимости 25° и твердые растворы чет-
четверной системы: сульфаты — хроматы калия и натрия, Изд. АН СССР, 1938. —
39. И. Г. Дружинин, Изв. АН СССР.- Отд. мат. и ест. наук. Сер. хим.,
№ 5—6, 1141 A938).— 40. С. 3. Макаров, И. Г. Дружинин, Там же.
№ 6, 1291 A937).
41. В. И. С п и ц ы н, Н. С. Афонский, В. И. Цир ельников, ЖНХР
5, № 9, 1970 (I960).— 42. А. Г. Бергман, В. А. X и т р о в, ИСФХА АН СССР,
24, 204 A954).— 43. И. С. Рассонская, ДАН СССР. 78, № 2, 279 A951).—
44. И С. Рассонская, А. Г. Бергман, ЖОХ, 23, № 1. 7 A953).—
45. Е. П. Дергунов, А. Г. Бергман, ИСФХА АН ГССР, 21, 184 A952).—
46. А И. Жуков, В. М. Шутова, Труды УНИХИМ, в. 1, 1954, стр. 29.—
47. В И. Спицы н, Н. С. Афонский, ЖНХ, 8, № 10, 2412 A963).—
48. J. Jaffray, A. Labary. Compt. rend.. 242, № И, 1421 A956).—
49. В. И. Спицыи, Н. С. Афонский, В. И. Цирельн иков, ЖНХ, 5, №7,
1505 (I960). — 50. А. В. Раковский, А. В. Бабаева, Труды ИРЕА, в. 11.
1931, стр. 15.
51. П. С. Богоявленский. ЖОХ, 22, № 4, 551 A952). — 52. П. С. Бо-
Богоявленский, Изв. Воронежск. гос. пед. ич-та, 5. в. 2, 49 A939).—
53. А. Д. Шевелева, ЖНХ, 1, № 8, 1883 A956).— 54. Я. И. Герасимов,
Труды ИРЕА, в. 11, 1931, стр. 34.— 55. А. А. Шидловский, С. А. Оран-
Оранжерее в. ЖПХ, 26, № 1, 25 A953). —56. Д. Н. Тарасенков, 3. И. Ко-
нопкина. Там же, 27, № 2, 198 A954).— 57. 3. И. Коиопкина, Там же,
29, .У» 1, 22 A956). — 58. А. Я- Д е й г, ЖНХ, 5, № 2, 503 (I960).— 59. Р. Н е П-
kel, Bayer-Ber., № 17, 24 A966). — 60. М. Y. Lid у. Chromium, v. II, Metallurgy
of Chromium and its Allays, N. Y. — London, 1956.
61. Г. В. Самсонов, Я. С. У м а н с к и й. Твердые соединения тугоплав-
тугоплавких металлов. Госметаллургиздат, 1957. — 62. Т. Я. Ко со л а по в а, Г. В Сам-
Самсонов. ЖПХ. 32, № 1, 55 A959) —63. W. Weller, S. E. F о 11 z, Z. phys.
Chem., 5, № 1—2. 100 A955).— 64. M. Dsrrin, Ind. Eng. Chem., 38, № 4, 368
A946). — 65. M. Ш Кац, Труды УНИХИМ. в. 12. 1966, стр. 25.— 66. С. М. 3 у б-
к о в, Б. В. К а р л ы ш е в. Э. Н. Юсупова, сб. «Металлургия, обогащение,
огнеупоры», Изд. АН КазССР, 1960. — 67. С. А. Сысолятин, Труды Ин-тэ
УРАЛМЕХАНОБР, в. 12, 1965, стр. 89.-68. Л. И. К р у п и н, Там же,
стр. 531.—69. С. С. Горланов. Требования промышленности к качеству ми-
минерального сырья. Хромит, в. 15, Госгортехиздат, 1963. — 70. Сб. «Хромиты
СССР», т. 1, Изд АН СССР, 1937; т. 2, 1940.
71. Н. А. Дюкалов, Хромит, 1931. —72. Г. И. Б о г а ч е в, Тоуды
УНИХИМ, в. 7, 1958, стр. 18. — 73. Н. В. Володомонов, Хромит, Мине-
Минерально-сырьевая база СССР, в. 8, ОНТИ. 1935. — 74. Ф. Ф. В о л ь ф, Е. Н П и-
наевская, ЖХП, № 10, 949 A931). — 75. Г. Л. Кореньков, Н. А. Усти-
Устинова Борнохимическое сырье зарубежных стран, Изд. «Химия», 1965, стр. 330.—
76. R. W. Но Hi day, Bull. Bur. Mines, № 630, 211 A965). — 77. П. В. Гель д.
О. А. Есин, ЖПХ, 23, № 12, 1260 A950)— 78. П. В. Гельд, О. А. Есин,
Там же. 23, № 12, 1271 A950). — 79. Ф. И. Васенин, Там же, 12, № 5. 651
A939). — 80. Я В. Ключ ар о в, С. М. Зубаков, Труды ЛТИ им. Ленсовета,
в 34. 1955. стр. 32.
81 В. А М о л е в а, Вопросы петрографии и минералогии, т. 2, Изд. АН
СССР, 1953, стр. 350. — 82. С. А. Паюсов, Я. Е. Вильнянский, Труды
УНИХИМ, в. 12, 1966, стр. 14; в. 16, 1967, стр. 41; Труды Уральск политехи,
ин-та. № 152. 1966, стр. 37; ЖПХ. 40, № 4. 805 A967); 42, № 9, 1974 A969).—
S3. Я. Е. Вильнянский. О. И. Пудовкина, ЖПХ, 22, № 7. 683
A949). — 84. В. В. Тельпиш, Я. Е. Вильнянский. ЖПХ, 42, № 5, 1180 и
№ 10, 2337 A969). — 85. Я. Е. Вильняискнй, О. И. Пудовкина. ЖПХ,
20, № 8. 794 A947). — 86. Я. Е. Вильнянский, О. И. Пудовкина, Там
же, 21, № 12, 1242 A948). — 87. Я. Е. Вильнянский, О. И. Пудовкина,
ЖОХ, 18, № 6, 1033 A948). — 88. В. А. Рябин, Труды УНИХИМ. в. 12, 1966,
стр. 35. — 89. Т. Д. А в е р б v х, М. А. С е р е б о е н н и к о в а, Н. Д. М а с л о в а.
Труды УНИХИМ. в. 1 1954. стр. 3: в. 3, 1955. стр 45; в. 7, 1958, стр. 23,—
90. Ю. И Кротов, Я. Е. Вильнянский. ЖПХ, 38, № 6, 1206 A965).
91. Н. Ф. Юшкевнч, ЖХП, 1, № 3, 51а A925). — 92. Л. И. Попова,
Там же, 2, № 6 A2), 465 A926). —93. М. В. К и р е е в а, ЖПХ, 31, № 10, 1484
A958); Труды конференции по усовершенствованию технологии производств
хромовых и фтористых солей, Госхимизпат. 1959, стр. 38. — 94. Н. С. М а с а л о-
вич, Г. Н. Богачов, Труды УНИХИМ. в. 12. 1966. стр. 75.-95. С. К. Чир-
Чирков, ЖПХ, 13, М> 4, 521, 528 A940): ЖХП. № 4—5; 41; № 7. 39 A939)- № 3,
29 П940).-96. Н. И X а й д у ко в, ЖХП, № 11, 38 A939).- 97. Ф. Ф Вольф,
Л И. Попова. Сборник оабот по химической технологии, минеральных ве-
веществ, в. 1, Свердловск, 1929, стр. 109.—98. И. Г Рысс, Р. Г. Урнцкая,
ДАН СССР, 4, 213 A934).— 99. М. Raman Nayar, H. E. Watson,
J. J. Sudboroagh, J. Ind. Inst. Sci.. 7, № 4, 53 A924).—700. V T. Atha-
vale, S. K. Jatkar, J. Indian Inst. Sci., 20A, № $. 55 A937); 21A, 199, 159,
179, 273 A938).
101. W. F. Ford, W. J. Rees, Trans. Brit. Ceram. Soc. 45, 125 A946)- 47,
№ 6, 207 A948); 48, № 8, 291 A949).— 102. W. F Ford, J. White, Trans. Brit.
Ceram. Soc, 48, № 11, 417 A949). — 103. Я. В. Ключ аров. В. Г Егер ЖПХ,
35, № 9, 1916 A962); Огнеупоры, № 3, 126 A963).— 104. Ф. И. Васенин
ЖПХ, 21, № 5, 429 A948).—105. П. Д. Пяти коп. Там же, 34, № 4, 766
A961).— 106. М. В. Киреева, Автореф канд. дисс, УПИ. Свердловск, 1959.—
*107. М. В. Киреева, А. А. Солошенко, ЖПХ, 33, № 1. 43 (I960) —
108. R. J. Harker, О. F. Tuttle, Am. J. Sci.. 253, № 3. 274 A955) —
' 109. M. В. Киреева, Т. В. С т р а т о н о в а. Труды УНИХИМ, в 16 1967,
стр. 142. —110. А. П. Любан, В. Г. Манчинский, ЖПХ, 25, № 8 808
A954).
А. Макарова,
4. А. Се-
111. И. Я. Федосеев, А. А. Перемышлева, М. а. макг
Труды Вороиежск. ун-та, т. 40, 1956, стр 35. —112. Т. Д А в ер бух, М. А. Се-
Серебренникова. Н. Д. Маслова, ЖПХ, 29, № 4. 498 A956); 31, № 6, 835
A958). — ИЗ. М. В. Киреева и др., Труды УНИХИМ. в. 8, 1959, стр. 5, 13;
сб. «Исследования в области химии и технологии минеральных солей и окислов»,
' Изд. «Наука», 1965, стр. 101; Труды УНИХИМ, в. 12, 1966, стр. 3 и 8.—
114. В А. Рябин, Автореф. канд. дисс., УПИ, Свердловск, 1962. —
115. Н. Р. Кузнецов, В. А. Р я б и и, В. В. Тельпнш, ЖПХ, 36, № 12. 2754
A963). —116. Я. Е. Вильияиский, Л. А. Боровских, Труды УНИХИМ,
628
Га. XV. Соли хрома
Литература
629
в. 7, 1958 стр. 3. — 117. Н. Ф. Юшкевич, А. Л. У раз о в, ЖХП 4,№ 5
387 A927) —118. О. И. Пудовкина, Н. А. Гущина, ЖПХ, 24, № 11, 1136
A951).—ПР. Н, П. Табунщиков, В. В. Тимошенко, Труды ВИСП т. 6,
1952, стр. 5; т. 7, 1954, стр. 44 —120. М. Bogitsh. Compt. rend., 178, № 14,
2254 A924).
121 Ю И Уатенко Е. А. К л и м к о в и ч, Труды Днепропетровск. ХТИ,
т 5 1956'стр 156. —122. С. А. Паюсов, Я. Е. Вильияиский, Труды
УНЙХИМ, в 16, 1967, стр. 30. — 123. О. И. Пудовкина, М. В. Киреева,
ЖПХ 29, № 6, 828 A956); 32, № 3, 499 A959). —124. Н. И. Хаидуков,
А. Э 3 а я р н ы й, Е. П. П о д т ы м ч е н к о, Труды УНИХИМ, в. 4, 19&7, стр 24.—
126 М В Киреева, А. А. Солошенко, ЖПХ, 33, № 2, 337 (I960).—
126. М. В. Киреева, Э. П. Клюки на, Там же, 34, № 6, 1348 A961) —
127. Г Н Богачов, Н. С. Банных, М. Я. Попильскии, Труды УНИХИМ,
в 4 1957 стр 3 — 128. М. Я. П о п и л ь с к и й, Л. И. Д е в я т о в с к а я, М. Т. А в-
д е е в а, Там же, в. 12, 1966, стр. 56. — 129. А. А. Ф е д о р о в, Хим. пром., № 10,
22 A945). —130. F. Schatzler, К. Plassmeyer, Chemiker-Ztg. Chem.
Apparat., 90, № 19, 661 A966).
131. В А Рябин, ЖПХ, 38, № 7, 1600 A965). —132. Г. Н. Богачов,
Хим наука н пром., 2, № 6, 714 A957).— 133. P. Diltey, Chem. Techn., 459
A950) — 134 Г Н Б о г а ч о в, В. М. П а в л о в, В. И. Ч е р н ы х, Э. П. К л ю-
кина, Хим. пром., № 9, 655 A861). — 135. С. Bandrowska, Chemik 21 № 9,
328 A968) —136. В М. Павлов, Г. А. Богачов и др., Труды УНИХИМ,
в 5 1963 стр 72. — 137. В. А. Рябин, А. А. П о т о р о ч и н, П. С. Ремпель,
И А Леонтьева, Там же, в. 12, 1966, стр. 43. —138. В. А. Рябин,
А А П о т о р о ч и н, Там же, стр. 50. — 139. Н. П. Левченко, В. А. Рябин,
П С. Р е м п е л ь, Там же, стр. 53. — 140. В. А. Р я б н н и др., Изв. АН КазССР.
Сер. хим., № 2 A967).
141. Б В. Миролюбов, В. А. Рябин и др., Труды УНИХИМ, в. 14,
1967, стр 66.-/42. Б. В. Миролюбов, В. А. Рябин и др.. Там же, в. 16,
1967' стр 75—143. Б В Миролюбов, В. А. Рябии и др., Там же,
стр 82.— '144. Т Д. Авербух, Хим. пром., № 11, 13 A946).— 145. Я. Р. Гольд-
штейн ЖХП № 7 A3), 564 A926); № 6—7, 495 A927). — 146. В. Neumann,
С. Е k s s n e r, Angew. Chem., 43, 440 A930). — 147. И. Г. Р ы с с, С. С. О р л о в,
ЖХП № 4 53 A933). — 148. Ф. Ф. Вольф, Л. И. Попова, Труды Центр, лаб.
сев хим треста, в. 1, Свердловск, 1929, стр. 97. —149. Е. Н. Пинаевская,
Н Л Антошки на и др., ЖПХ, 37, № 5, 989; № 12, 2563 A964).—
150. И. Г. Щ е р б а к о в, О. Е с и н, Z. Elektrochern., 32, 396 A926).
151. Д. А. Дымчишин, Производство хромовых солей, Госхимиздат,
1934. — /52. М. В. Киреева, Л. А. Дунаевская, С. И. Ремпель, сб.
«Исследования в области химии и технологии минеральных солей и окислов»,
Изд «Наука», 1965, стр. 105. —153. М. В. Киреева, Л. А. Дунаевская,
ЖПХ, 37, № 1, 204 A964).— 154. В. М. Павлов, И. И. Шншко, Хим. пром.,
№ 1Г 781 A961). — 155. П. Л. Мясников, О. И. Пудовкина, Э. М. Мор-
Моргунова, Труды УНИХИМ, в. 7, 1958, стр. 32. —156. О. И. Пудовкина,
М. Я Попильскии, Э. М. Моргунова, Н. В. Горбунова, Там же,
СТр 41 —157 Т Д Авербух, Л. В. Юманова, А. С. Микулинскнй,
Труды УНИХИМ, в. 1, 1954, стр. 36. — 158. П. М. Лукьянов, ЖХП, № 1, Ц
A924).— 159. Л. М. Стрибман, Там же, № 7, 17 A939). — 160. В. С. Ш е р-
м а н, Р. Н. С у х а н о в а, Труды УНИХИМ, в. 14, 1967, стр. 175.
161 Л Г Ш и р и н к и н, Л. С. С к а т и н а, В. М. Павлов, Труды
УНИХИМ,' в. 12, 1966, стр. 82, 89. 93. — 162. В. А. Рябин, А. А. Шибаева,
Там же в 12 1966, стр. 157. — /53. М. Я. Попильскии, Р. А. Горохова,
Ю. К. К а си ль и др.. Там же, в. 16, 1967, стр. 23, 191-— 164. М. В. Киреева,
Р А Горохова, Г. С. Краснова и др., Там же, стр. 201. — 165. Б. А. Р я-
бнн. Я. Е. Вильняиский, Там же, стр. 107. —166 В. А. Рябин и др.,
Там же, стр. 123. — 167. Н. Г. Ех лаков а, Там же, в. 12, 1966, стр. 151.—
168. Я. Е. В и л ь н я н с к и и, авт. свид. 18247, 1929. — 169. В. А. Рябин и др.,
Труды УНИХИМ, в. 14. 1967, стр. 103. —170. В. А. Пьячев, М. Ф. Чебуков,
Изв. вузов. Хнм. и хим. технол., 8, № 1, 118 A965).
171. Д. И. Мильман, Ю. Б. Смирнов, Хим пром., № 7, 540 A965).—
172. Г. К. М а г а р ш а к, В. Н. Тарасова, Б. П. Волгин, Труды УНИХИМ»
в. 1, 1954, стр. 55.— 173. А. В. Раковский, Е. А. Никитина, Труды ИРЕА,
т. 2, 1931, стр. 5. — 174. В. Ф. Волков и др., Хим. пром., № 6, 450 A966); № 6,
449 A968). —175. В. А. Одиицов, Л. Н. Матусевич, Там же, № 9, 687
A965). —176. Л. Н. Матусевич и др., Там же, № 12, 927 A966).—
177. А. А. Солошенко, Ю. К Касиль, М Я Попильскии Труды
УНИХИМ, в. 16, 1967, стр. 110. —178. Н. S chafer, P. Henkel, M. Weist,
пат. ФРГ 1205508. 1962. —179. Н. Ф Юшкевич, М. Н Левин, ЖХП, 2,
№ 3 (9), 247 A925); № 4—5 A0—11), 329 A926). — 180. G. Agde. К- Е. Fet-
Fetter, Angew. Chem., 48, № 5, 92 A935).
181. Н. Ф, Юшкевич, В. А. Каржавин, И. Н. Шокин, ЖХП № 12
A8), 9&-1; № 14 B0), 1119 A926). —182. Н. Ф. Юшкевич, И. Н. Шокин,
Там же, № з, 204 A927). — 183. О. Nydegger, Chimia, 4, № 12, 288 A950).—
184. Г. Н. Богачов, Е. М. Полякова, М. А. Грязных, Труды УНИХИМ,
в. 4, 1957, стр. 13.— 185. Р. В. Миролюбов, Труды конференции по усовер-
усовершенствованию технологии производств хромовых и фтористых солей, Госхим-
Госхимиздат, 1959, стр. 42. — 186. Л. А. Боровских, Я. Е. Вильнянский, Там же,
стр. 46.-/87. В. С. Ятлов, ЖПХ, 2, № 5, 561 A929). — /88. J. В. Robert-
Robertson, J. Soc. Chem. Ind., 43, № 47, 334T A924). —189. Г. Н. Богачов, Хим.
пром., № 5, 144 A952). —190. 3. С. Банных, Г. А. Лопатки на, Труды
УНИХИМ, в. 7, 1958, стр. 50.
191. Я. Е. Вильнянский, 3. С. Банных, Там же, в. 8, 1959, стр. 46.—
192. Ю. П. Каретников, Г. Н. Богачов, Г. Ф Фефелова Труды
УНИХИМ, в. 1, 1954, стр. 85. —193. В. М. Павлов, Там же, в 5, 1957;.
стр. 291. —194. И. Г. Рысс, А. И. Зелянская, Acta phys., 8, № 5, 623
A938). — 195. Т. Д. Авербух, Труды Конференции по усовершенствованию тех-
технологических производств хромовых и фтористых солей, Госхимиздат, 1959,
стр. 23. — 196. Австрал. пат. 157986, 1954; 164665, 1955. —197. М. А. Остро-
у м о в, Автореф. каид. дисс, Уральск, политехи, ин-т, Свердловск, 1953. —
/98. М. Я. Попильскии, Труды Конференции по усовершенствованию тех-
технологических производств хромовых и фтористых солей, Госхимиздат 1959,
стр. П.—199. М. Я. Попильскии и др., Труды УНИХИМ, в. 16 1967,
стр. 23. — 200. Пат. США 1872588, 1873889, 1932; герм. пат. 565156, 1932 и 742797,
1943; пат. ФРГ 1203748, 1961; пат. ЧССР 101948, 1961; англ. пат 959745,
1964.
201. А. М. Поляк, Журн. ВХО, 7, № 1, 56 A962). — 202. И Г Рысс,
*А. Э. Заярный, А. И. Зелянская, ЖПХ, 14, № 1,46 A941).— 203. А. Н. М и-
, х а й л о в. Химия дубящих веществ и процессов дубления, Гизлегпром 1963.
гл. \\\. —204. С. К. Чирков, ЖПХ, 10, № 17—18, 1257 A937).—
205. Н. И. Г е л ь п е р и н, Н. Г. К р о х и н, К. И. Б о г а ч е в а, Труды Всесоюзн.
н.-и. ин-та синтет. и натур, душистых веществ, № 2, 1954, стр. 165. — 206. А Сал-
Салли, Хром, Госметаллургиздат, 1958. — 207. Ф. Б Бланше, авт свид 149178,
1962. — 208. И. Н. Головастиков, ЖПХ, 29, № 4, 553 П956) —
209. С. Д. К а ц, авт. свид. 61020, 62191, 1942. — 210. Г. Д. М а з а л е ц к и й Труды
УНИХИМ, в. 12, 1966, стр. 269.
211. П. Чекии, ЖПХ, № 6, 565 A926). — 212. К. И. Лосев, К А Махо-
- нин, Там же, И, № 12, 1564 A938).— 213. Геры. пат. 369816, 1923- 390602 1924;
444798, 1927. — 214. W. J. Biermann, M. Heinrichs. Canad. J. Chem 38,
№ 9, 1449 (I960). — 215. H. F: Johnstone, R. W. Derbyshire, Ind. Eng-
Chem., 34, 280 A942). — 216. Д. П. Зосныович, К. Б. КладннцкаЯ,
€30
Гл. XV. Соли хрома
Н. Д. Иванова, ЖПХ, 36, № 2, 333 A963). — 217. Е. Ф. Беленький,
И. В Р и с к и н, Химия и технология пигментов, Госхимиздат, 1960. —
218. D. R. Dhingra, S. N. К а р о s г, V. Paul, J. Sci. Soc, 2, 21 A953).—
219. О. И. Пудовкина, В. М. Павлов, М. Я. Попильский, Н. В. Г а р-
к у н о в а, Труды УНИХИМ, в. 8, 1959, стр. 53.— 220. Н. С. Б а н н ы х, В. Ф. Б е з-
р у к о в а, Труды УНИХИМ, в. 3, 1955, стр. 52.
221. к М Поляк М Т. Серебренникова, М. Я. Попильский,
Там же, в. 4, 1957, стр. 25; в. 7, 1958, стр. 46.-222. В. М. Власенко,
Л А Кухарь М. Г Розенфельд, М. Т. Русо в. Хим. пром., № 8, 955
A962).— 223. Англ. пат. 748610, 749097, 1956; пэт. США 2854313, 1958.—
224. Р. Манд ель. Новости техники, № 21, 37 A938).— 225. J. С. Kalbach,
канад пат. 512959, 1955.—226. А. Н. Торокии, Т. Д. Авербух, Труды
УНИХИМ, в. 12, 1966, стр. 219. — 227. Е. А. Н a h п, Е. Р е t е г s, Canad. J. Chem.,
39, № i( 162 A961). — 228. В. Н. Ипатьев и др., Вег., 59, № 7, 1418 A926)
и 60, № 8, 1980 A927)- Bull. Soc. chim. France, 39, 1384 A926); Compt. rend., 183,
505 A926). —229. Л. Г. Булатова, Я. Е. Вильнянский. Труды УНИХИМ,
в. 8, 1959, стр. 41; в. 12, 1966, стр. 267. — 230. Л. И. Девятовская,
Я. Е. Вильнянский, Там же, в. 8, 1959, стр. 23.
231. В. П. Иванцов, Труды Томск, ун-та, т. 154, 1962, стр. 32, 42.—
232. А. М. П о л я «, Л. И. Д е в я т о в с к а я, Труды УНИХИМ, в. 4, 1957, стр. 80.—
233. Я. Е. Вильнянский, Н. С. Банных, Хим. пром., № 10, 19 A945).—
234. Я. Е. Вильнянский, Л. А. Боровских, М. С. К о в е л ь, Журн. ВХО,
8, № 1, 116 A963); ЖПХ, 39, № 10, 2266 A966); Труды Уральск, политехи, ин-та,
№ 152, 1966, стр. 13, 28. — 235. Л. Г. Булатова, Я. Е. В и л ь н я и с к и й, Труды
УНИХИМ, в. 16, 1967, стр. 129. —236. Г. Д. Мозалецкий, Р. С. Байта-
к о в а, Там же, стр. 3. — 237. R. R i с h e I m i, M. Zaffitte, Compt. rend., C265,
№ 10, 541 A967). — 238. M. Я. Попильский, авт. свид. 77540, 1949.—
239. С. Л. К р а п и в н е р, Труды конференции по усовершенствованию техно-
технологических производств хромовых и фтористых солей, Госхимиздат, 1959,
стр. 55. — 240. Б. П. Середа, Труды УНИХИМ, в. 12, 1966, стр. 191.
241. Б. П. Середа, 3. А. Самоделкииа, Там же, стр. 199.—
242. Б. Н. Середа, 3. А. С а м о д е л к и н а, А. Г. Никулин, А. М. 3 а г у-
д а е в, Там же, стр. 208. — 243. Б. П. Середа, М. Я. Попильский и др.,
Труды УНИХИМ, в. 16, 1967, стр. 146, 152, 164. —244. В. С. Ятлов, А. В. По-
Попова, ЖПХ, 6, № 6, 1049 A933).— 245. С. G. M a i e r, Sponge Chromium,
U. S. Bur. Mines Bull., № 436, 109 A942). — 246. E. A. Pokorny, Extraction
and Refining of Rarer Metals, London, 1957, p. 34 — 247. R. L. A n n i s, R. G. В a n-
n e r, J. G. К о u r i 1 o, F. S. M a h n e, F. S. P e r r i n, Mines Magazine, 49, № 8,
21 A959). — 248. И. А. Магидсон, Г. В. Карсанов, М. И. Герасимова,
Т. В. Калмыкова, ЖПХ, 34, № 5, 953; 11, 2391 A961). — 249. И. А. Матид-
с о н и др., сб «Металлургия и химическая промышленность Казахстана», № 4,
1961, стр. 15; ЖПХ, 36, № 10, 2132 A963).— 250. Т. С. Шибнева, Г. Ф. Ф е-
фелова, Труды УНИХИМ, в. 12, 1966, стр. 174.
257. Т. С. Шибнева, С. П. Сол яков, А. В. Покровский, Н. И. Син-
Синцов, Г. Ф. Ф е ф е л о в а, Труды Ключевского заЕода ферросплавов. Металло-
Металлотермия, № 2, 1965. — 252. Л. И. Девятовская, Я. Е Вильнянский,
Труды УНИХИМ, в. 8, 1959, стр. 23; в. 12, 1966, стр. 255; сб. «Металлургия и
химическая промышленность Казахстана», № 3, 1961, стр. 59. — 253. Т. Е. Ш и б-
нева, Г. Ф. Фефелова, А. В. Покровский, Труды УНИХИМ, в. 12, 1966,
стр. 262. — 254. Л. Г. Булатова, Я. Е. Вильнянский, Там же, стр. 267. —
255. А. А. Солощенко, Я. Е. Вильнянский, Л. Г. Раевская, Г. В. С а й-
ф у л л и н а, Там же, в. 16, 1967, стр. 61.— 256. Л. И. Д е в я т о в с к а и, С. П. С о-
л я к о в, Т. А. Я к и м е ц, Т. Н. Колосова, Там же, стр. 89. — 257. И. В. В а-
с и л ь е в а, М. П. С у с а р е в, И. В. К р и в о у с о в а, сб. «Исследования в об-
области химии и технологии минеральных солей и окислов». Изд. «Наука», 1965,
Литература
631
стр. 139. —258. А. А. Солошенко, Я. Е. Вильнянский, ЖПХ 37, № 3
535 A964).—259. А. И. Тетеревков, Я. Е. Вильнянский, Труды УНИХИМ^
в. 16, 1967, стр. 52, 116; сб. «Металлургия и химическаи промышленность Казах-
Казахстана», № 5, 1961, стр. 58. — 260. J. L. Clay, J. F. Ре arse D H T r e t h e--
wey, J. Soc. Chem. Ind., 69, № 9, 275 A950).
261. И. Д. Музыка, В. А. Молодцов а, ЖПХ, 32, № 9, 1936 A959).—
262. Накадзава, J. Mining Inst. Japan, 72, № 814, 207 A956).—
263. N. H. Brundin, швед. пат. 142749, 1953. — 264. M. Y. Udy пат ФРГ
919287, 1954. — 265. E. M. Полякова и др., Труды УНИХИМ. в 12' 1966
стр. 162, 284.-266. А. М. Поляк, М. А. Серебренникова и др., Там же^
ггр. 170, 184. — 267. Е. Н. Пинаевская и др., Там же, в. 8, 1959,'
стр. 19; ЖПХ, 34, № 8, 1722 A961).— 268. В. Г. Хомяков, В. П. Машовец,
Л. Л. К у з ь м и н, Технология электрохимических производств, Госхимиздат 1949
стр. 406.— 269. Б. И Томилов, ЖПХ, 30, № 12, 1785 A957).— 270. V. Е ngel-*
hardt, Handbuch der technishen Elektrochemie, Leipzig, 1939, S. 133.
271. R. H. McKee, S. T. Leo, Ind. Eng. Chem., 12, № i, i6 Л920) —
272. H. С. Дроздов, ЖОХ, 3, № 3, 345 A933). — 273. А. Ф. Богоявлен-
Богоявленский, M. И. Гаркав и, Г. 3. Афанасьев, К. Ш. Шамсутдинов Уч
Зап. Каз. ун-та, 113, кн. 8, 23 A953). — 274. Н. Cuppers, пат. ГДР 6402'
1954. — 275. R. L. Costa, пат. США 2733204, 1956. — 276. Правила и нормы тех-
техники безопасности промышленной санитарии для строительства и эксплуатации
предприятий производства хромовых соединений, Госхимиздат, 1961.—
277. 3. А. Филиппова, В. Ф. Плехоткин, Техн.-экон. информ. НИИТЭХИМ
Охрана труда и техника безопасности, № 6, 17 A966)
Физико-химические свойства
635
Глава XVI
СУЛЬФАТ АЛЮМИНИЯ И.ПРОДУКТЫ НА ЕГО ОСНОВЕ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Сульфат алюминия A12(SO4K безводный представляет собой
•белые кристаллы, имеющие плотность 2,71 г/см3. Растворимость
при 0е равна 31,2, при 100° 89 г в 100 г воды. Из водного раствора
кристаллизуются бесцветные моноклинные кристаллы Al2(SO4K-
•18Н2О, имеющие плотность 1,69 г/см3 A7°) и обезвоживающиеся
при 86,5°.
Алюминиевые квасцы имеют общую формулу M2SO4 -AbCSC^)з*
-24Н2О или MA1(SO4J- 12Н2О.
Алюмо-аммониевые квасцы NH4A1(SO4J- 12Н2О — бесцветные
кристаллы с плотностью 1,64 г/см3, плавятся при 93,5° без разло-
разложения. При нагревании расплавленной массы до 120° отщепляются
10 молекул воды, а при 200° происходит полное обезвоживание.
Растворимость при 0° равна 2,6 г, при 80° 35,2 г NH4Al(SO4J-
• 12Н2О в 100 г воды.
Алюмо-калиевые квасцы KAl(SO4J* 12НгО — бесцветные кри-
кристаллы с плотностью 1,76 г/см3, плавятся при 92е. Растворимость
при 0° — 2,96, при 20° —5,75, при 70° —35,9, при 90°— 109. при
100°—154 г KA1(SO4J в 100 г воды. Безводный сульфат алюми-
алюминия-калия KA1(SO4J гигроскопичен, на воздухе расплывается,
имеет плотность 2,75 г/см3.
Алюмо-натриевые квасцы NaAl(SO4J- 12Н2О — бесцветные кри-
кристаллы с плотностью 1,675 г/см3, плавятся при 61°. Растворимость
при 0° —37,4, при 20° —40,8. при 40° —44,3 г NaAl(SO4J в 100 г
воды. В системе Na2SO4—A12(SO4K—Н2О при 0° эвтонический рас-
раствор состава (в вес.%): 3,74 —Na2SO4 и 21,58 —A12(SO4K нахо-
находится в равновесии с твердыми фазами: Na2SO4 и инконгруэнтно
растворимых квасцов Na2SO4 • А12EО4)з*24Н26. Точке перехода
соответствует состав раствора 1% Na2SO4 и 27,12% A12(SO4K, рав-
равновесный с твердыми фазами Na2SO4-A12(SO4K«24H2O и
A12(SO4K-18H2O1.
При взаимодействии сульфата алюминия с содержащимися в
естественных водах бикарбонатами кальция и магния образуется
гидроокись алюминия, которая является коагулирующим агентом.
В результате гидролиза2'3 сульфата алюминия из водных раство-
растворов вначале выделяются промежуточные основные соли, а затем"
гидроокись алюминия:
A12(SO4:8 + 2Н2О = A12(SO4J(OHJ + H2SO4
A12(SO4J(OHJ + 2Н2О = A12(SO4)(OHL + H2SO4
A12(SO4)(OHL + 2H2O = 2A1(OHK + H2SO4
A12(SO4K + 6H2O = 2A1(OHK + 3H2SO4
Гидролиз A12(SO4K даже при высоких температурах в интер-
интервале 200—300° (при повышенном давлении) протекает с малой
скоростью и не превышает
50%. При добавке 15 г/л
Na2SO4 гидролиз значитель-
но ускоряется. В получаю-
щемся основном сульфате
алюминия содержится до
3% Na2O.
По другим данным4,
продуктом гидролиза
Al2(SO4K при повышенных
температурах и давлении ~
является основной сульфат <
состава ЗА12О3 • 4SO3 • 7Н2О,
выход которого возрастает
при увеличении давления до рис
70 ат и продолжительности
процесса до 30 мин 5.
Основной сульфат А12Оз'45Оз-ЗН2О изоморфен с NH4A1(SO4J
• и, вероятно, имеет структуру, отвечающую формуле [A1(SO4J]OH3.
Соединение 3A12O3-4SO3-9H2O переходит при определенных усло-
условиях в ЗА12О3 • 4SO3 • (NH4) 2O • 6Н2О6.
Основные соединения алюминия образуют истинные или колло-
коллоидные растворы в воде7, весьма нестойкие. С течением времени
или при повышении температуры они разлагаются с образованием
осадка переменного состава8, превращающегося в устойчивый
комплексный ион [А1зс(ОН)г;(Н2О)г]<3:1:-^+ В системе А1(ОНK—
A12(SO4K — Н2О (рис. 180) 9 образуются твердые фазы: 4А12Оз*
•3SO3-24H2O — при разбавлении водой, так называемый гидроли-
гидролитический осадок (с 75% щелочности), ЗАЬОз^БОз-ЗШгО — рас-
растворимый осадок, медленно выделяющийся ниже 56° (с 56,6%•!
10 EO 30 40 50 6ff
Alz{SO^K, g иа /DCs H2O
180. Растворимость в системе
A1(OHK-A12(SO4K-H2O.
«34
Гл. XVI. Сульфат алюминия и продукты на его основе
щелочности), А12О3- SO3-3,5H2O — нерастворимый осадок, выде-
выделяющийся при кипячении раствора F6,7% щелочности), и ЗА12О3-
-5SO3-R2O- 10Н2О (где R—Na, К, NH4) — откладывающийся в
виде накипи при кипячении в присутствии щелочных солей E5,6%
щелочности). Под щелочностью здесь имеется в виду отношение
количества алюминия в виде А1(ОНK к общему его количеству.
Стойкость растворов основного сульфата алюминия зависит от спо-
способа их приготовления. Наиболее стойкие растворы образуются
при обработке растворов сульфата алюминия водной суспензией
мела или известняка при кипячении.
Начало разложения A12(SO4K при нагревании наблюдается при
530°. Заканчивается оно при 860°, причем продуктом разложения
является у-АЬОз10. Процесс интенсифицируется добавкой 5—10%
Fe2O3 или Сг2О3 и особенно в восстановительной среде, например
в потоке окиси углерода11. По другим данным12, разложение суль-
сульфата алюминия при нагревании идет по следующим стадиям:
До 610° A12(SO4K=SO,+A12O(SO4J
» 810° А12О(8О4J=О2+А12О(8ОЛ2
» '910° A|2O(SO3J=2SO2+A12O3
Термохимическое разложение сульфата алюминия представляет
интерес для переработки алунита 13. Образующийся при этом гли-
глинозем обладает высокой активностью и способностью к спеканию,
вследствие большой дисперсности и дефектности кристаллови.
Аналогично протекает и термическое разложение сульфитов алю-
алюминия 1Б.
При обжиге во взвешенном слое обезвоживание основных алю-
мо-аммониевых квасцов происходит при 460—520°, а выделение
окислов серы при нагревании до ~1100°. При 1000° в течение 5,5-
минутного обжига получается продукт, содержащий 90% А12О3,
а в течение 12-минутного — 95% А12О3. Максимальная концентра-
концентрация окислов серы в лабораторных условиях была 14,5% 16-
Продукты термического разложения различных солей алюми-
алюминия отличаются разной удельной поверхностью и активностью.
Продукт разложения A12(SC>4K при 400—700° имеет удельную по-
поверхность 20—25 м2/г, продукт разложения А1С!3 в тех же усло-
условиях—80—160 м2/г, а продукт разложения A1(NO3K—100—
170ж2/г17.
ПРИМЕНЕНИЕ
Сульфат алюминия является наиболее распространенным коа-
коагулянтом, применяемым в водоочистке для обработки питьевых и
промышленных вод, и используется в ряде других отраслей тех-
техники. Коагулирующие свойства A12(SO4K обусловлены образова-
образованием коллоидной гидроокиси алюминия и основных сульфатов в
результате гидролиза. В процессе коагуляции А1(ОНK коллоидные
Применение
635
частицы примесей, находящиеся в воде, захватываются и выде-
выделяются вместе с А1(ОНK в виде студенистых хлопьев. После хими-
химической обработки воду фильтруют.
Помимо водоочистки сернокислый глинозем применяется в
больших количествах в целлюлозно-бумажной промышленности
для проклейки бумаги и других целей; его используют в текстиль-
текстильной промышленности в качестве протравы при крашении хлопчато-
хлопчатобумажных, шерстяных и шелковых тканей, при дублении кож, для
консервирования дерева, в производстве древесно-волокнистых
плит, в промышленности искусственных волокон и др.
Алюмо-калиевые квасцы применяют в мясной, кожевенной, тек-
текстильной промышленности, в меньших количествах — в меховой,
фармацевтической и проч. 18>19. Алюмо-аммониевые квасцы служат
сырьем в производстве камней синтетического корунда.
Основной сульфат алюминия может быть использован для обо-
обогащения бедных сернистых газов (см. гл. XIV) 20.
Предложено использование сульфата алюминия и квасцов в ка-
качестве фунгицидов для зеленых растений в виде 0,5—2%-ного рас-
раствора при рН 3,5—4 21.
В США ежегодно производят около 1 млн. т сульфата алюми-
алюминия 22.
По химическому составу сульфат алюминия и квасцы должны
удовлетворять требованиям, приведенным в табл. 43.
ТАБЛИЦА 4*
Требования к химическому составу технических сернокислого алюминия
и квасцов
(содержание компонентов в %)
А12О3, не менее . .
H2SO4 (своб.), не бо-
более
Железо (Fe2O3), не
более
As2O3, не более . .
Нерастворимый оста-
остаток, не более. . .
Технический сернокислый алюминий
очищен-
очищенный
(ГОСТ
5.740-71)
16,3
Отсут-
Отсутствие
0,02
0.001
0.3
очищенный
(ГОСТ 12966-67)
I сорт II еорт III сорт
15,0
0,05
0,04
0,003
0,5
14,5
0,10
0,10
0,003
0,7
13,5
0,10
1.5
0,003
1,0
неочи-
неочищенный
(ГОСТ
5155-49)
9
2
0,8
0,003
23
Квасцы алюмокалне-
вые технические
(ГОСТ 15028-69)
высший
сорт
10,60
0,0015
0,03
сорт
10.50
0,002
0,035
II сорт
10,30
0,035-
0,1
Технический сернокислый алюминий очищенный, получаемый
из гидроокиси алюминия или из обогащенного каолина имеет вид
Плотных кусков мелкокристаллического строения белого цвета с
зеленоватым оттенком, сообщаемым примесью солей ¦ закисного
636
Гл. XVI. Сульфат алюминия и продукты на его основе
Сырье
637
железа (FeSO4). При длительном хранении на воздухе куски
сульфата алюминия окрашиваются с поверхности в желтоватый
цвет из-за окисления примеси закисного железа в окисное.
Неочищенный сернокислый алюминий, получаемый обработкой
серной кислотой каолина с последующей добавкой нефелинового
концентрата (ГОСТ 5155—49), содержит кроме сульфата алюми-
алюминия также квасцы, примеси других сульфатов и нерастворимых
веществ. По внешнему виду — это плотные куски кристаллического
строения неопределенной формы, серого цвета.
Иногда под названием коагулянтов выпускают и другие про-
продукты, содержащие в качестве основного вещества, помимо суль-
сульфата алюминия, и сульфат трехвалентного железа, обладающий
таким же коагулирующим действием, как и сульфат алюминия.
Поэтому для производства коагулянтов можно применять глины и
другие виды сырья, содержащие соединения железа, в частности,
колчеданный огарок.
Коагулянты и нечищенный сернокислый алюминий более деше-
дешевые, но худшие продукты, чем очищенный сернокислый алюминий,
так как содержат много нерастворимых примесей и меньше А12Оз.
Удаление примесей из водоочистительной аппаратуры требует до-
дополнительных затрат. Недостатком сернокислого алюминия яв-
является его способность слеживаться при хранении. Наряду с твер-
твердым сульфатом алюминия используют «жидкий коагулянт» — рас-
раствор сульфата алюминия, содержащий 8,3% А12О3. Такой раствор *
дешев, он устойчив при низких температурах (кристаллизация на-
начинается ниже —20е), его легко применять23.
Взамен сульфата алюминия в качестве коагулянта предложено
употреблять оксихлориды алюминия, в частности пентаоксихлорид
А12(ОНMС1. В воде он диссоциирует на А12(ОН)д и СГ. Его полу-
получают действием NaOH на А1С1з или обработкой А1(ОНK слабой
соляной кислотой. После упаривания раствора и сушки продукт
содержит 80% А12(ОНMС1, т. е. более 40% А12О3, что намного
больше, чем в сернокислом алюминии237.
СЫРЬЕ
Сырьем для производства сернокислого алюминия28.29 во мно-
многих странах служит наиболее дешевый и доступный вид природ-
природного материала — глина. Разложение глины производят серной
кислотой, которая растворяет содержащуюся в глине окись алю-
алюминия по реакции:
А12О3 + 3H2SO4 = A12(SO4K + ЗН2О
В СССР сернокислый алюминий получают из каолина, а также
растворением в серной кислоте гидроокиси алюминия, вырабаты-
вырабатываемой в больших количествах на заводах цветной металлургии.
¦Сернокислый алюминий или содержащие его коагулянты получают
и из других глиноземистых материалов — нефелина (стр. 639),
уртита, кианита (А12О3-SiO2), а также из высококремнистых бок-
•еитов и золы от сжигания углей.
Глины
Основными минералогическими компонентами глин являются30
водные алюмосиликаты: каолинит Al2O3-2SiO2-2H2O, галлуазит
А12О3- 2SiO2-2H2O + aq, аллофан А12О3-SiO2 + aq, пирофиллит
А12О3 • 4SiO2 • 4Н2О, монтмориллонит А12Оз • 4SiO2 • Н2О + aq, се-
серицит K2O-3Al2O3-6SiO2-2H2O или KAl2[Si3AlOi0](OHJ и их кол-
коллоидные аналоги. Некоторые глины в основной своей части образо-
образованы слюдистыми минералами. Помимо этого, в глинах встречаются
полевые шпаты, кальцит, доломит, гипс, рутил, турмалин, глау-
глауконит, роговые обманки и другие минералы, а также иногда и
¦свободный глинозем в виде диаспора или гиббеита, соединения
ванадия и редких элементов.
Каолинитовые глины или каолины в основном состоят из вод-
водных алюмосиликатов и кремнезема. Другие минералы обычно при-
присутствуют в ничтожных количествах. Из водных алюмосиликатов
в каолин входит в основном каолинит в кристаллическом состоянии
или в виде геля, а также диккит и накрит, имеющие тот же
состав А12Оз • 2SiO2 • 2Н2О, но отличающиеся рентгенографической
характеристикой. В небольших количествах содержатся галлуазит,
аллофан, пирофиллит и монтмориллонит. Кремнезем присутствует
в каолинах главным образом в форме кварца; встречаются также
опал и халцедон. Примеси в каолинах представляют собой поле-
полевые шпаты, слюды, железосодержащие минералы (лимонит, гема-
гематит, магнетит, сидерит), а также ильменит, рутил, кальцит, доло-
доломит, гипс и др.
Сырье, используемое для производства сульфата алюминия,
должно содержать значительное количество глинозема и легко раз-
разлагаться кислотами. Таким требованиям удовлетворяют нефели-
нефелиновые породы и каолинитовые глины. Эти глины содержат 15—40%
А12О3, 55—75% SiO2, 1—2% Fe3+ и 0,5—1% Fe2+. Обычно исполь-
используют глины, содержащие после обезвоживания 30—40% А12Оз.-
Бокситы, хотя и содержат большие количества глинозема, труднее
разлагаются кислотами. Трудно разлагаются кислотами и глины,,
содержащие значительные количества полевошпатовых пород. Для
решения вопроса о пригодности того или иного сорта глины обычно
производят пробную варку сернокислого алюминия.
Месторождения каолинитовых глин весьма распространены,
хотя не все они достаточно изучены в качественном и технологиче-
технологическом отношении. Наиболее богаты разведанными месторождениями
каолинов Украина, затем Урал31.
638
Гл. XVI. Сульфат алюминия и продукты на его основе
Сырье
639
Многочисленные месторождения огнеупорных глин, богатых
каолинитом, также могут быть использованы как источники сырья
для производства сульфата алюминия.
Обжиг глин
При обработке серной кислотой непрокаленной глины, даже
при продолжительном кипячении, в раствор переходит лишь часть
окиси алюминия. Выход А12О3 в раствор различен для разных
сортов глин, но редко превышает 60% (при 5-часовом кипячении),
а обычно значительно ниже. Для облегчения процесса растворения
окиси алюминия в кислоте раздробленную глину подвергают об-
обжигу при 700—800°. Обжиг обычно производят во вращающихся
трубчатых печах, в которых глина нагревается газами, получен-
полученными сжиганием топлива при большом избытке воздуха.
После прокаливания глин степень извлечения из них А12О3.
значительно повышается и достигает для некоторых сортов 99%
при варке с 63—65%-ной серной кислотой в течение 6—8 ч при
115°. Коагулянт, полученный аналогичным путем из суворовских
глин Подмосковного района, содержащих 38—43% А12О3, 1,4—
2,75% Fe2O3 и 44—45% SiO2, имеет 0,1—0,2% железа и может
быть использован не только для очистки воды, но и бумажной про-
проза, зз
мышленностью
По данным разных исследователей, обезвоживание каолинов " I
при прокаливании наблюдается при различных температурах в
пределах 500—600°34, а обезвоживание боровичских глин —даже
при 400°35. Термический анализ показывает, что обезвоживание
каолинов и глин происходит в интервале 400—500°36.
При нагревании выше 500° молекула каолинита распадается:
А12О3 - 2SiO2 • 2Н2О = А12О3 + 2SiO2 + 2Н2О
Вначале, при частичной потере воды образуется водный мета-
каолинит Al2O3-2SiO2-(l—п) Н2О, в дальнейшем переходящий в
безводный метакаолинит. При более длительном прокаливании он
диссоциирует на А12О3 и SiO2, которые остаются в аморфных фор-
формах37-38. Поэтому обожженная глина резко отличается от необож-
необожженной легкостью растворения А12О3 в кислотах. В действитель-
действительности процессы, протекающие при нагревании каолина, весьма
сложны. Продуктом эндотермической реакции при 500—550° яв-
является аморфное вещество, близкое по своим свойствам к алло-
фану и аллофаноидам, представляющим собой механические смеси
из гелей SiO2 и А12О3, с переменным их отношением, образовав-
образовавшиеся в результате совместной коагуляции39.
Кривые, приведенные на рис. 181, по данным А. М. Соколова,
показывают влияние температуры прокаливания каолина в тече- *
ние 1 и 5 ч на потерю в весе вследствие удаления воды (пунктир-
(пунктирные линии) и на долю А1гО3, переходящую в раствор при после-
последующей обработке прокаленного каолина соляной кислотой (сплош-
(сплошные линии). Чем выше температура прокаливания, тем сильнее
разрушается каолин, теряя воду, и тем большая доля окиси алю-
алюминия растворяется в кислоте. Наилучший эффект достигается при
700—800°, даже при сравнительно непродолжительном обжиге —
около 1 ч. Прокаленная обезвоженная глина обладает наибольшей
пористостью, что также спо-
способствует ускорению раство-
растворения А12О3 при кислотной
обработке 40~42.
Выше 800° образуются
новые кристаллические фа-
фазы— Y~AbO3 и нераствори-
нерастворимые соединения А1гО3 и
SiO2, в частности силлима-
силлиманит А12О3 -SIO2, муллит
ЗА12О3 • 2SiO2 и др.4°-43. По-
Поэтому температура обжига 5.
не должна превышать 800е. *"
При нагревании глин
кроме воды удаляются и
другие летучие вещества —
СО2, SO3 и т. п.
При обжиге содержа-
SDO
600 700 800
Темпершпурв,вС
900 1000
Рис. 181. Влияние температуры прокали-
прокаливания каолина на потерю в весе (пунктир-
(пунктирные линии) и количество А12О3, растворяю-
растворяющееся в кислоте (сплошные линии).
щаяся в глине закись желе-
железа окисляется кислородом,
находящимся в топочных
газах, и переходит в Fe3O4,
которая плохо растворима в серной кислоте. При обработке про*
каленной глины кислотой небольшие количества железа все же
переходят в раствор. Выделение железа из раствора в виде
Fe(OHK достигается путем так называемой основной варки, в
конце которой, благодаря избытку глины, рН раствора повышается
до такого предела, когда железо осаждается в виде гидроокиси.
Соли окисного железа гидролизуются легче, чем соли алюминия:
рН осаждения гидроокиси железа около 2, а гидроокиси алюми-
алюминия 4,1. Это позволяет выделить железо из раствора сульфата алю-
алюминия при достижении в конце варки незначительной кислотности
раствора.
Другие виды сырья
Различные сорта сернокислого алюминия, в частности коагу-
коагулянты, могут быть получены из золы некоторых углей, содержа»
щих много А12Ов — иногда до 35%, а также из сланцев4*.
640
Гл. XVI. Сульфат алюминия и продукты на его основе
Хорошим сырьем для получения коагулянта и квасцов является
нефелин (К, NaJO- Al2O3-2SiO2. Крупнейшие залежи нефелина
находятся на Кольском полуострове, где он входит в состав сиени-
сиенитов и образует сплошные массы вместе с апатитом. Нефелиновые
породы залегают в Ильменских горах в районе Миасса, в Каслин-
Каслинском районе Урала, в районе Жданова, в Восточной Сибири, в
Узбекистане и др.31 -
Средний состав нефелинового концентрата, получаемого при
разделении апатито-нефелиновой породы Кольского полуострова
(в %):
SiO2 43,22 ТЮ2 0,37 Na2O 12,00
AI2O3 29,00 CaO 1,70 КаО 7,12
Fe2O3 3,00 P2O5 0,65 Потери при про-
FeO 0,80 MgO 0,80 каливанни . . . 1,26
Нефелиновый концентрат представляет собой тонкий материал,
90% которого проходит через сито 1300 отв/см2 и около 40% — ,
через сито 6400 отв/см2.
¦ Особый интерес представляют встречающиеся в природе основ-
основные алюминиевые квасцы K2SO4- A12(SO4K-4A1(OHK, относя-
относящиеся по своему минералогическому характеру к алунитам. В их
состав вместо калия или наряду с ним может входить и натрий.
Примерный химический состав алунитовой породы, содержащей
50—55% алунита (в %): 20—22 А12О3) 3—4 Fe2O3, 35—40 SiO2,
1,5—2 Na2O, 2,5—3 К2О, 20—21 SO3, 6—7 Н2О45.
В результате обжига алунитовой породы при 500—550° про-
происходит дегидратация и разложение молекулы алунита- с образо-
образованием безводных алюминиевых квасцов и у-А12О3:
K2SO4 • А12( SO4K • 4А1(ОНK = K2SO4 • AI2(SO4K + 2А12О3 + 6Н2О
После выщелачивания обожженной породы получают раствор
квасцов, содержащий 28 г/л A12(SO4K, подвергаемый дальнейшей
переработке13' 46~48. При выщелачивании с добавкой серной кис-
кислоты в течение 24 ч при 90—95° можно повысить концентрацию
раствора до —60 г/л A12(SO4K49.
При водном выщелачивании алунита, обожженного при 800°
до прекращения выделения сернистых газов, _ образуется раствор
сульфатов щелочных металлов, окись алюминия остается в шламе.
Обработка этого шлама серной кислотой позволяет получить
A12(SO4K-18H2O50.
ПРОИЗВОДСТВО КОАГУЛЯНТОВ
Наиболее простым и наиболее старым способом получения не-
неочищенного сернокислого алюминия является варка непрокален-
ного, но подсушенного каолина с серной кислотой. Степень превра-
превращения А12О3 глины в сульфат не превышает 70—80%. Усовершен-
Производство коагулянтов
641
ствованием этого метода явились разложение каолина избытком
серной кислоты для повышения степени извлечения А12О3 и ней-
нейтрализация избыточной кислоты нефелином. Успешное применение
нефелина в качестве добавки к каолину послужило основанием для
прбизводства нефелинового коагулянта из одного нефелина (без
каолина).
Получающиеся по всем этим способам продукты — неочищенг
ный сернокислый алюминий или коагулянты — после варки за-
затвердевают и не подвергаются дополнительной переработке. Они
содержат все примеси сырья, в частности и нерастворившиеся ве-
вещества. Для получения очищенного сернокислого алюминия про-
производят отделение нерастворимых примесей, что значительно
усложняет производственный процесс.
Неочищенный сернокислый алюминий из каолина
Неочищенный сернокислый алюминий раньше получали2-51 из
необожженной каолинитовой глины, которую подвергали сушке;
в пламенной печи при 300—400°. При этом химически связанная
вода удаляется лишь частично. После сушки охлажденный до
50—80° каолин разлагали в варочном котле 65—67%-ной серной
кислотой 6—8 ч при 105—110°. По окончании варки массу, содер-
содержавшую не более 6—8% свободной серной кислоты спускали в
зрельники, где количество свободной кислоты понижалось до 2—
2,5%, затем спускали на кристаллизационный стол (лоток)—пло-
(лоток)—плоскую прямоугольную плиту с бортами высотой 0,25—0,5 м, выло-
выложенную кислотоупорными плитками. На столе масса в результате
охлаждения закристаллизовывалась — полностью затвердевала, по-
после чего ее выбивали вручную с помощью лома и отправляли на
склад в виде кусков весом 15—20 кг. Для получения 1 т неочи-
неочищенного сернокислого алюминия по этому способу расходовали:
0,42 т каолина (с 15% влаги), и 0,32 т серной кислоты A00%),'
0,2—0,3 г пара и 6—6,2 кет • ч электроэнергии.
При использовании каолина, прокаленного при 700—800°, раз-
разложение в варочных котлах завершалось, и надобности в дозре-
дозревании массы в зрельниках не было. Были предложены различные
способы механизации процесса кристаллизации сернокислого алю-
алюминия — путем распыления массы до ее застывания, кристаллиза-
кристаллизации на вращающихся барабанах с внутренним охлаждением или
в шнеках, в вагонетках с откидными полыми стенками, охлаждае-
охлаждаемыми водой, на пластинчатых бортовых транспортерах, в ковше-
ковшевых конвейерах и др. Рациональный способ механизации удаления
•застывшего глинозема удалось разработать при получении глино-
глинозема с определенной структурой, при которой остывший и за-
затвердевший продукт легче извлекается из кристаллизаторов. Это
достигается разложением каолина избытком серной кислоты,
21 М. Е. Поэнн
642
Гл. XVI. Сульфат алюминия и продукты на его основе
нейтрализуемой в дальнейшем нефелином. Автоматически дейст-
действующая машина для снятия сернокислого алюминия с плоских кри-
кристаллизационных столов представляет собой сдвоенную пластинча-
пластинчатую цепь с ножами и гребками. Беспрерывно перемещаясь попе-
поперек кристаллизатора, имеющего размеры 9 X 2,7 м, она срезаег
материал слой за слоем и сбрасывает его гребками на ленточный
транспортер, расположенный вдоль кристаллизатора52.
Процесс получения неочищенного сернокислого алюминия из
каолина, требующий сушки каолина и вызревания продукта, яв-
является малоинтенсивным и громоздким. Более рационально про-
производство коагулянта из смеси каолина и нефелина или из одного
нефелина.
J Неочищенный сернокислый алюминий из каолина
и нефелиновой муки
¦• Сущность метода заключается в почти полном разложении сы-
сырого природного каолина избыточным количеством серной кислоты
в течение короткого времени и нейтрализации избытка кислоты не-
нефелиновой мукой:
А12О3 • 2SiO2 • 2Н2О + 3H2SO4 = A12(SO4K + 5H2O + 2SiO2
(Na, KJO ¦ А12О3 • 2SiO2 + 4H2SO4 = (Na, KJSO4 • A12(SO4K + 4H8O + 2SiO2
Повышение температуры реагирующей массы за счет взаимо-
взаимодействия кислоты с водой каолина и добавляемой водой также спо-
собствует более быстрому и полному разложению каолина. Обра-
Образующиеся при разложении нефелина квасцы сообщают продукту
способность схватываться в плотную прочную массу. Скорость
схватывания и прочность массы тем больше, чем больше избыток
кислоты и соответствующий расход нефелиновой муки на ее ней-
нейтрализацию.
Степень извлечения глинозема в раствор из сырого каолина
зависит от сорта глины, концентрации и нормы кислоты. Отмучен-
Отмученный положский каолин (содержащий 35% А12О3 и 47,5% SiO2),
высушенный и измельченный до величины зерен 0,5—1 мм, раз-
разлагается с наибольшей скоростью в избытке 54—55% -ной серной
кислоты. Обработка каолина двойной нормой кислоты (по отноше-
отношению к стехиометрической) в течение 2 ч при 105—120° переводит
в раствор около 85% исходного глинозема63. При использовании
концентрированной кислоты реакция замедляется вследствие обра-
образования на зернах корок труднорастворимого безводного сульфата
алюминия или кислых сульфатов и увеличения сопротивления диф-
диффузии.
Процесс может быть также осуществлен путем подачи купорос»
ного масла в водную суспензию каолина (Ж:Т = 3:2).
Производство коагулянтов
643
При получении неочищенного сернокислого алюминия из као-
каолина и нефелиновой муки5I в варочный котел — стальной цилиндр
g диабазовой футеровкой, снабженный мешалкой, — загружают
воду и купоросное масло для разбавления кислоты до 75—76%
H2SO4. Затем загружают каолин и производят варку массы в те-
течение 15—35 мин при непрерывном перемешивании. Температуру
100—110° поддерживают подачей в котел острого пара. По окон-
окончании варки массу, содержащую 4,5—7,5% А12О3 и 20—25% сво-
свободной кислоты, разбавляют водой и вводят в котел в течение
15 мин отдельными порциями (по 5—10 кг) нефелин. Реакционную
массу перемешивают еще 2—3 мин, затем быстро, во избежание
схватывания, сливают на кристаллизационный стол.
Степень перехода А12О3 в сернокислый алюминий составляет из
каолина 65—75%, а из нефелина около 90%. Для получения 1 г
продукта по этому методу расходуют: 0,17 т каолина (с 15% вла-
влаги), 0,18 т нефелиновой муки сухой (или 0,12 т А12О3 в каолине
и нефелиновой муке), 0,31 т серной кислоты A00%), 0,21 т пара,
2,5 квт-ч электроэнергии и 0,12 м3 воды.
Нефелиновый коагулянт
При смешении нефелинового концентрата с башенной серной
кислотой без последующего разбавления водой смесь быстро за-
загустевает, так как находящаяся в ней вода связывается с образо-
образовавшимися солями в твердые кристаллогидраты. Это сопровож-
сопровождается сильным повышением температуры, вызывающим значитель-
значительное парообразование, что приводит к резкому увеличению объема
смеси, которая превращается в твердую пористую массу, легко
рассыпающуюся в порошок. Этот продукт, состоящий из смеси
калиевых, натриевых квасцов, SiO2 и прочих примесей, находив-
находившихся в нефелине и образовавшихся при обработке его серной
• кислотой, называется нефелиновым коагулянтом. Его правильней
было бы назвать неочищенным нефелиновым коагулянтом в отли-
отличие от очищенного нефелинового коагулянта, которым является
смесь квасцов, полученная кристаллизацией раствора после отде-
отделения от него кремнеземистого осадка.
Температура реакции, количество испарившейся воды, выход и
свойства коагулянта зависят от концентрации исходной кислоты53.
В продукте, полученном при разложении нефелина 63—84,5%-
ной кислотой, обнаружен бисульфат алюминия. Это объясняется
неполной нейтрализацией серной кислоты:
2(R2O • А12О3 • 2SiO2) + 8HjSO4 + 8Н2О = A12(SO4K • 3H2SO« - 12Н2О + 2RHSO4 +
+ 2SiO2 + R2O • A12O3 • 2SiO2
Наличие в коагулянте гигроскопичных кислых солей обусловли-
обусловливает поглощение им влаги из воздуха. В результате бйоднения
2>1*
644
Гл. XV/. Сульфат алюминия и продукты на его основе
продукта происходит дальнейшее разложение непрореагировавшего
нефелина. Этот процесс «дозревания» протекает на воздухе мед-
медленно, около 12 суток, вследствие покрытия зерен непрореагировав-
непрореагировавшего нефелина кристаллами коагулянта. При растворении кристал-
кристаллов в воде процесс дальнейшего разложения ускоряется и завер-
завершается в холодной воде в течение часа, а в горячей — в течение
5 мин. Таким образом, замедление взаимодействия нефелина с кон-
концентрированной серной кислотой (выше 63% H2SC>4) объясняется
недостатком воды в жидкой фазе53. С наибольшей скоростью не-
нефелин разлагается 47—73%-ной серной кислотой (рис. 182)в*.
IIIя
а
го
Получение очищенного сернокислого алюминия
645
~W W 50 60 10
Концентрация серной кислоты,!
ВО
Рис. 182. Влияние концентрации серной кислоты на ско-
скорость разложения нефелина.
Получение неочищенного нефелинового коагулянта было пред-
предложено производить смешением нефелинового концентрата с ба-
башенной серной кислотой в котлах с мешалками и выливанием по-
полученной Пульпы до ее загустевания в аппараты для «созревания»,
т. е. затвердевания массы. Твердая масса подвергается измельче-
измельчению. При смешении нефелина с 92% серной кислотой реакция
идет очень медленно и незагустевшая пульпа может легко перете-
перетекать в желоб со шнеком, куда добавляется вода для разбавления
кислоты. После этого реакция идет очень быстро, и масса, интен-
интенсивно перемешиваемая шнеком и передвигаемая им вдоль аппа-
аппарата., быстро затвердевает, превращаясь в мелкие зерна.
Рекомендована следующая схема производства нефелинового
коагулянта55. Смешение купоросного масла и нефелинового кон-
концентрата производят в двух смесителях, снабженных трехлопаст-
трехлопастными мешалками и соединенных последовательно. В один из сме-
смесителей подают непрерывно кислоту и нефелиновый концентрат.
Образующаяся пульпа перетекает во второй смеситель, откуда вы-
выходит из нижней части его через гидравлический затвор в ковше-
ковшевой дозатор. В выходящей пульпе должно содержаться от 1,5 до
4% ивбыточнои серной кислоты (в зависимости от качества нефе-
нефелина). Под избыточной понимают кислоту, содержащуюся в пульпе
сверх того количества, которое может прореагировать к концу про-
Лесса при гидратации. Из ковшевого дозатора пульпа поступает в
шнек-реактор, куда добавляют воду иа расчета разбавления кие-
лоты до 70—73% H2SO4. Продолжительность пребывания массы в
шнеке-реакторе составляет 28—30 сек и степень разложения нефе-
нефелина за это время достигает 85—88%. Из реактора сухая рассып-
рассыпчатая масса с температурой 80—100 поступает на склад, где про-
происходит дозревание и охлаждение продукта в течение 2—4 суток.
На производство этим методом 1 т нефелинового коагулянта тре-
требуется: 0,32 г нефелиновой муки (до 1% влаги) или 0,105 т А12Оз
A00%), 0,378г серной кислоты A00%).
ПОЛУЧЕНИЕ ОЧИЩЕННОГО СЕРНОКИСЛОГО АЛЮМИНИЯ
При производстве очищенного сернокислого алюминия раство-
растворением в серной кислоте гидроокиси алюминия (или окиси алю-
алюминия) процесс осуществляют, например, следующим способом.
В реакционный котел (стальной резервуар, футерованный кислото-
кислотоупорным кирпичом по слою диабазовой плитки) одновременно за-
загружают гидроокись алюминия, серную кислоту и воду в прибли-
приблизительно стехиометрическом соотношении, соответствующем содер-
содержанию в продукте ~90% Al^SC^b- 18H2O и ~10% свободной
воды. Перемешивание ведут острым паром, поддерживая темпе-
температуру на уровне ПО—120°, и заканчивают его через 20—30 мин,
когда количество свободной серной кислоты в пробе реакционной
м.аесы станет меньше 0,1%. Реакционную массу, содержащую
13,5—15% А12Оз (в виде сульфата алюминия), для ускорения по-
последующей кристаллизации охлаждают в реакторе до 95°, проду-
продувая через нее в течение 10 мин воздух. Затем ее сливают на кри-
кристаллизационный стол, оборудованный автоматической машиной
для срезки застывшего продукта (стр. 642). Кристаллизация плава
на столе продолжается ~50 мин и столько же времени занимает
извлечение продукта из кристаллизатора, имеющего площадь 32—
34 м2 (емкость ~6 г). Расход материалов на 1 т продукта со-
.ставляет: 0,142 т гидроокиси алюминия (в пересчете на А12О3)
и 0,40 т серной кислоты A00%).
Кристаллизацию ведут также на охлаждаемой изнутри наруж-
наружной поверхности горизонтального вращающегося барабана — на
холодильных или кристаллизационных вальцах. Барабан частично
погружен в находящийся в поддоне плав, имеющий температуру
бб—100е. Кристаллизация на вальцах облегчает условия труда,
обеспечивает непрерывный режим производства, улучшает товар-
товарные свойства продукта. Снимаемый с вальцев чешуйчатый про-
продукт, содержащий 13,5—14% А12Оз, при хранении слеживается.
Неслеживающийся продукт получают, повышая содержание А12Оз
до 15,3—16,8% A5,3% соответствует концентрации А12Оз в кри-
кристаллогидрате Al2(SO4b- 18H2O). При длине барабана вальцев
2,2 м и диаметре 1,8 м (поверхность теплообмена 12,4 м2), при вы-
выпуске продукта с содержанием 13,5—14% А126з, число оборотов
646
Гл. XVI. Сульфат алюминия и продукты на его основе
барабана составляет 4,3 в минуту и средняя рабочая производи-
производительность вальцев равна 2,4 т/ч; при выпуске продукта, содержа-
содержащего 15,3—15,8% А12О3, барабан делает 1—1,2 об/мин и произво-
производительность снижается до ~ 1 т/ч.
Для получения неслеживающегося продукта предложено5Ба
также смешивать пульпу гидроокиси алюминия с 60%-ной серной
кислотой, взятой в количестве 95—97% от стехиометрического и
образующийся раствор с температурой 100° направлять для кри-
кристаллизации на холодильные вальцы. Продукт содержит примесь
основной соли.
ЗапатентованБ6 непрерывный способ получения сульфата алю-
алюминия, в котором водная суспензия А1(ОНK и серная кислота в
стехиометрическом отношении подаются с большой скоростью до-
дозирующими насосами в смесительные форсунки реактора, в кото-
котором масса находится не менее 30 сек. Затем она охлаждается до,
температуры ниже 100° в проточном холодильнике и продавлива-
продавливается через сопла или прорези для образования мелкогранулиро-
ванного продукта.
Предложено обрабатывать гидроокись алюминия серной кис-
кислотой во вращающихся автоклавах при 145—165° в течение 5—
20 мин с последующим завершением реакции во вращающейся
печи при 175—500°; из печи выходит обезвоженный, гранули-
гранулированный сульфат алюминия, легко транспортируемый и дози-
дозируемый.
Производство сульфата алюминия из каолина заключается в
следующем *7-б8. Обогащенный мокрым способом каолин дробят
(а мелочь гранулируют) в частицы 2—7 мм, обжигают при 750—
800°, затем обрабатывают в реакторе циркулирующей через слой
крупки серной кислотой для выщелачивания А12Оз. Процесс начи-
начинают при 70°, отводя от раствора тепло реакции, а затем подни-
поднимают температуру до 104—105°, т. е. выше температуры плавления
в кристаллизационной воде сульфата алюминия, содержащего
13,5% А12О3. Полученный плав перекачивают насосами из реак-
реактора в сборник, откуда он поступает на кристаллизационные валь-
вальцы. Получаемый здесь чешуйчатый продукт, содержащий 13,5%
А12О3. транспортируется на склад. На 1 т сернокислого алюминия
A3,5% А12О3) расходуется 0,6 т каолина мокрого обогащения (при
15% влажности) и 0,43 т купоросного масла. После слива из реак-
реактора плава сульфата алюминия, в оставшемся сиштофе задержи-
задерживается до 40% продукта, который извлекают трехкратной система-
систематической промывдой, направляя полученный водный . раствор,
содержащий ~б,8% А1263, на разбавление купоросного масла; по-
получаемая кислота с концентрацией 31% Н2$О4 поступает в реак-
реактор. Промытый сиштоф, содержащий 0,4% А12О3, удаляется гидро-
гидротранспортом, отделяется от воды и иейольвуется для производств?»
цемента.
Получение очищенного сернокислого алюминия
647
Для кристаллизации сульфата алюминия применяют также от-
открытые плоские металлические ванны с двойным днищем, где
циркулирует охлаждающая вода и вращающиеся барабанные холо-
холодильники, в которых плав гранулируется. Непрерывную кристалли-
кристаллизацию с получением стекловидного сульфата алюминия рацио-
рационально осуществлять на движущейся конвейерной ленте из нержа-
нержавеющей стали (или на горизонтальном диске). В этом случае на
кристаллизацию подают раствор (плав), содержащий 17% А12'Оз,
и в него вводят в качестве затравки измельченный сульфат алюми-
алюминия. Раствор с температурой 130° поступает на ленту, охлажден-
охлажденную до 10—30° и смоченную водой. Нижняя поверхность ленты
охлаждается водой (первые 32 м ленты рекомендуют охлаждать
водой, подогретой до 70°). Раствор начинает загустевать при 110°.
При толщине слоя 5—10 мм его верхняя поверхность отвердевает
через 4—6 мин. При проходе ленты вокруг вала твердый слой суль-
сульфата трескается и ссыпается. Затем его охлаждают воздухом и
измельчают. Лента длиной 60 м и шириной 0,72 м имеет произво-
производительность 3 т сульфата алюминия в час. Этот способ, как и при-
применение кристаллизационных вальцев, позволяет полностью механи-
механизировать процесс, снизив затраты на оборудование, требует малых
площадей для его размещения59.
Запатентовано60 и воздушное охлаждение ленты с помощью не-
нескольких сопел; скорость воздушной струи 40—50 м/сек. В плав
предварительно добавляют 2% твердой пыли сульфата алюминия
в качестве затравки. В плаве содержится 17,3% А12О3; его темпе-
температура 110°. При длине ленты 18 м, ширине 0,8 м, толщине слоя
плава 19 мм и скорости движения ленты 3,25 м/мин, ее производи-
производительность равна 2,2 т/ч.
Возможно61 использование прорезиненной ленты; в этом.слу-
этом.случае плав перед подачей на ленту охлаждают до 83—88°.
Старый способ получения сернокислого алюминия из глин
в СССР утратил свое значение. Он заключался в следующем.
Обожженную и размолотую до тонкости 1—2 мм глину варили
с 65—70%-ной серной кислотой 4—8 ч при кипячении до получения
нейтрального раствора сульфата алюминия, что достигалось до-
добавлением в котел избытка глины. В конце варки содержание сво-
свободной серной кислоты в растворе не превышало 0,3% г/л. Рас-
Раствор содержал около 45% A12(SO4K- Из такого раствора уже при
90° начинает кристаллизоваться A12(SO4K' 18H2O.
Во избежание кристаллизации сульфата алюминия при даль-
дальнейшей переработке раствор по окончании варки разбавляли водой
до плотности 1,2—1,25 г/см3. Получение такого раствора сразу,
путем применения более разбавленной кислоты, нерационально,
так как' извлечение А12О3 из глины при этом ухудшается. После
разбавления раствор сульфата алюминия отфильтровывали на
фильтрпрессе с деревянными рамами (применение ¦ чугунного
648
Га, XVI. Сульфат алюминия и продукты на его основе
Другие методы получения коагулянта и пр.
649
фильтрпресса привело бы к загрязнению раствора железом). Оса-
Осадок на фильтрпрессе промывали водой и удаляли в отвал. Промыв-
Промывные воды возвращали в производство — на разбавление концентри-
концентрированного раствора после варки. Профильтрованный раствор со-
содержал еще значительное количество мути. После отстаивания его
дополнительно фильтровали через контрольный фильтрпресс и вы-
выпаривали до плотности 1,53—1,58 г/см3. Эта плотность соответ-
соответствует концентрации, при которой раствор при охлаждении пол-
полностью закристаллизовывается. Кристаллизацию производили на
кристаллизационных столах. Застывший продукт разламывали на
куски и отправляли на склад. Расход серной кислоты (моногид-
(моногидрата) на 1 т стандартного продукта A3,5% А12О3) составлял
0,44—0,5 т, а расход прокаленной глины 0,5—0,6 г.
Суммарная потеря А12О3 во всех стадиях производства дохо-
доходила иногда до 40%. Столь высокие потери окиси алюминия объ-
объясняются главным образом низкой степенью извлечения А12Оз из
глины при ее основной варке, когда процесс ведется почти до ней-
нейтральной реакции, при которой переход А12Оз в раствор прекра-
прекращается. Нерастворенная в кислоте А12О3 остается при этом втвер-
„ дом отвале.
Потери АЬОз значительно меньше при двух последовательных
варках — кислой и основной, когда нерастворившаяся глина после
основной варки, отделенная от раствора, подвергается кислой вар-
варке с добавкой к ней кислоты и порции свежей глины. Раствор
с кислой варки при этом направляется на основную варку с до-
дополнительным количеством глины, добавляемым для нейтрализа-
нейтрализации кислоты.
В дальнейшем были предложены пути рационализации отдель-
отдельных стадий этого процесса 62~67: кальцинация глины, смоченной во-
водой и небольшим количеством серной кислоты; более тонкий раз-
размол обожженной глины; систематическое выщелачивание окиси
алюминия из глин кислотой повышающейся концентрации; исполь-
использование для фильтрации слоя листовой целлюлозы; замена фильт-
фильтров отстойниками; замена выпарных котлов пламенными ванными
печами и аппаратами с погруженным горением; замена кристалли-
кристаллизационных столов конвейерным ковшевым кристаллизатором, хо-
холодильными вальцами, распылительной сушкой и др.
Предложено получать кристаллический сульфат алюминия вы-
высокой чистоты выдерживанием в специальном сборнике пульпы,
полученной при выпарке раствора, до ее охлаждения, с целью об-
образования более крупных частиц примесей. После отделения при-
примесей массу охлаждают для кристаллизации68.
Был предложен способ получения сернокислого алюминия без
выпарки раствора, заключающийся в том, что обожженная глина,
раздробленная на зерна величиной 4—7 мм, выщелачивается сер-
серной кислотой69. Раствор после выщелачивания сразу застывает в
твердый стандартный продукт. Выщелачивание должно вестись при
100° путем заливки крупки горячей кислотой такой концентрации
D5—47%), чтобы сразу получался продукт с содержанием 13,5%
А12О3. После завершения реакции полученный концентрированный
раствор сливают через ложное дно, и в аппарат заливают свежую
кислоту. Эти операции повторяют до тех пор, пока не будет достиг-
достигнуто почти полное извлечение А12Оз из глины. По окончании выще-
выщелачивания шлам в том же аппарате промывают водой и промыв-
промывную воду используют для разбавления концентрированной серной
30
to
? 20
10
SO 40
50
GO 70
Рис. 183. Растворимость сульфата алюминия в серной
кислоте при 25°.
кислоты. Полное извлечение кислоторастворимого АЬОз дости-
достигается за 20—25 ч. Расчетная производительность 1 м3 выщелачи-
вателя 1,5—2 г стандартного продукта в сутки.
Растворимость сульфата алюминия в присутствии серной кис-
кислоты сильно понижается (рис. 183). Поэтому в некоторых случаях
может представить интерес выделение кристаллического сульфата
алюминия из растворов высаливанием концентрированной серной
кислотой. Имеется указание70, что при этом получается продукт
•с малым содержанием железа.
Предложено много способов очистки растворов сульфата алю-
алюминия и квасцов от железа71-72. Большинство из них основано на
добавке окислителей (МпО2, Н2'О2, С12, Вг2 и др.) для перевода
Fe2+ в Fe3+ с последующим выделением Fe3+ в виде нерастворимых
соединений (например, Fe(OHK, Fe4[Fe(CNN]3) или экстракцией
органическими растворителями.
ДРУГИЕ МЕТОДЫ ПОЛУЧЕНИЯ КОАГУЛЯНТА И СЕРНОКИСЛОГО
АЛЮМИНИЯ
Получение коагулянта из золы
Коагулянт может быть получен из золы некоторых углей, со-
содержащей много А12Оз- В золе подмосковных углей, например,
имеется 25—35% А12О8, 8—15% Fe2O3, 43—60% SiO2. Из такой
650
Гл. XVI. Сульфат алюминия и продукты на его основе
золы получают коагулянт, содержащий 8—10% R2O3. Извлечение
А12Оз из золы, однако, ниже F0—80%), чем из специально обож-
обожженной глины, так как при сгорании топлива развиваются значи-
значительно более высокие температуры и часть окиси алюминия в золе
связывается в форме алюмосиликатов и других соединений, не рас-
растворяющихся в серной кислоте735.
Получение коагулянта из глин и каолина методом спекания
При варке неочищенного сернокислого алюминия или коагу-
коагулянта из сырой глины в ряде случаев получаются сравнительно
низкие выходы А12О3 и низкое содержание А12О3 в продукте при
повышенной его кислотности, что связано с содержанием в глинах
полевошпатовых пород. В этом отношении большими преимуще-
преимуществами обладает способ получения коагулянта из сырых глин спе-
спеканием с серной кислотой, дающий возможность получать коагу-
коагулянт с более высоким содержанием А12О3 и лучшим выходом его,
не содержащий свободной кислоты.
По этому способу сырую глину смешивают с 75%-ной серной
кислотой, взятой в количестве 50—70% от того, которое требуется
для связывания в сернокислые соли А12О3, Fe2O3, CaO, MgO, Na2O,
К2О. Смешение производят в глиномялках или в аппаратах шнеко-
вого типа. Затем смесь прокаливают в течение 2—5 ч в подовых
или вращающихся печах при 300—500°. Получается рассыпчатый
продукт, содержащий до 15% R2O3. На 1 т такого продукта рас-
расходуют: около 1 т низкосортной сырой глины (содержащей 20%
А12О3 и 12% Fe2O3) и 0,48 т серной кислоты A00%-ной).
Помимо того, что метод спекания позволяет получать коагулянт
из необожженной низкосортной глины, он имеет и другие преиму-
преимущества по сравнению с методом варки коагулянта из глины. К ним
относятся возможность легкого осуществления процесса непрерыв-
непрерывным способом в шнеке или вращающейся печи, получение рассып-
рассыпчатого продукта и возможность получать продукт с любой свобод-
свободной кислотностью, вплоть до отсутствия кислотности, что имеет
большое значение для ряда потребителей, например для бумажной
промышленности. Полученный этим методом из каолина или белых
глин неочищенный коагулянт, не содержащий свободной серной
кислоты, пригоден для частичной замены в бумажной промышлен-
промышленности более дорогого очищенного сернокислого алюминия76.
Спек, изготовленный описанным выше способом, можно под-
подвергнуть выщелачиванию; после фильтрования получается- сравни-
сравнительно чистый раствор сульфата алюминия, содержащий лишь
небольшое количество соединений железа и сульфатов щелочных
металлов. Вследствие термической диссоциации Fe2(SO4)s в печи
основная масса железа в виде окиси железа переходит в шлам
вместе с кремнеземом и отделяется при фильтровании 77,
Другие методы получения коагулянта и пр.
651
Спеканием можно получить сульфат алюминия и из каолинов.
В СССР разработан метод двухстадийной сульфатизации каоли-
каолинов, заключающийся в спекании их с серной кислотой B0% нор-
нормы) при 600° и в последующем выщелачивании спека серной кис-
кислотой при температуре кипения 78.
Комбинирование производства очищенного сернокислого
алюминия с сернистокислотным методом разложения каолинов
Производство очищенного сернокислого алюминия может быть
организовано с использованием алюминиевого концентрата, полу-
получаемого при предварительном обогащении каолина или глин.
Одним из таких методов обогащения является разложение кремне-
кремнеземистых соединений алюминия сернистым газом. Представляет
интерес использование при этом отбросного сернистого газа метал-
металлургической и химической промышленности и получение высоко-
высококонцентрированного газа при гидролизе сульфита алюминия.
При взаимодействии окиси алюминия с водным раствором дву-
двуокиси серы протекают реакции:
А12Оз + 6H2SO3 = 2A1(HSO3K + ЗН2О
А12О3 + 3H2SO3 = A12(SO3K + ЗН2О
Процесс идет в основном с образованием бисульфита алюминия
(при 20° константы диссоциации H2SO3 следующие; Ki *- 2 • 10~2,
Я2=1-Ю-7).
Разложение каолина сернистой кислотой в интервале 25—65°
при атмосферном давлении протекает медленно. Степень извлече-
извлечения А12О3 в раствор в течение 5—7 ч не превышает 8,5%. В этих
условиях степень извлечения окиси железа составляет ~22,5%.
Повышение давления и температуры способствует резкому уве-
увеличению скорости разложения каолина и степени перевода А12О3
в водорастворимое состояние. При 20 ат и 170° степень перехода
А12О3 в раствор из водной суспензии каолина с отношением по весу
" Т:Ж, равным 1 : 10, достигает за 8—10 ч 95—96%- Опыты прово-
проводились с каолином предварительно прокаленным в течение 1 ч
/при 600°, измельченным до полного прохождения через сито с от-
отверстиями 0,25 мм и имевшим состав (в %): 38,10 — А12О3; 47,11 —
SiO2; 1,61—FejOg; 1,10 —CaO; 0,40 — TiO2, 0,60 —MgO, 11,18 —
потери при прокаливании78'80.
После отделения нерастворимого осадка, представляющего со-
собой в основном кремнезем, раствор сульфита и бисульфита алюми-
алюминия подвергают гидролизу:
A12(SO3K + 6Н2О = 2А1(ОНK + ЗН2О + 3SO2
Al(HSO3K + 3H2O = А1(ОНK + ЗН2О + 3SOa
В действительности в результате гидролиза образуютоя
основные сернистокислые соли алюминия различного состава»
652
Гл. XVI. Сульфат алюминия и продукты на его основе
A1(OH).(HSO3J, A1(OHJ(HSO3), A1(OH)SO3, A12(OHLSO3 и др.
Некоторые из образующихся основных солей трудно растворимы
в сернистой кислоте и не подвергаются дальнейшему гидролизу.
Вследствие этого продукт гидролиза состоит из гидроокиси и ос-
основных солей алюминия и может служить прекрасным сырьем для
получения сернокислого алюминия, попутно с обогащением серни-
сернистого газа.
Процесс упрощается, если раствор сульфит-бисульфита алюми-
алюминия, отделенный от примесей, обработать серной кислотой. При
этом образуется довольно концентрированный раствор сульфата
алюминия и почти чистая двуокись серы:
A12(SO3K + 3H2SO4 = A12(SO4K + 3H,0 + 3SO2
Этот процесс аналогичен переработке сульфит-бисульфитных
растворов в процессе очистки газов от двуокиси серы по аммиач-
аммиачному способу (стр. 520).
Следует, однако, отметить, что для переработки каолина опи-
описанный метод является мало перспективным вследствие малой ско-
скорости взаимодействия каолина с сернистой кислотой. Более пер-
перспективным является получение по этому способу квасцов из нефе- •
лина (стр. 655).
Получение сернокислого алюминия из боксита
Разложение боксита производят81 в стальном реакторе, футеро-
футерованном кислотоупорными плитками или покрытом стекловидной
эмалью. В реактор вначале загружают 93%-ную серную кислоту,
которую разбавляют слабыми растворами от промывки шлама и
нагревают до кипения. Боксит вводят в реактор периодически не-
небольшими порциями во избежание обильного вспенивания. Варку
осуществляют при ~ 135°. По окончании варки пульпа поступает
в отстойники, где раствор отделяется от нерастворимых примесей.
Осветленный раствор или выпаривают с получением твердого про-
продукта, или используют непосредственно.
В последнем случае, когда установка для получения раствора
сульфата алюминия соединена с системой очистки воды, продол-
продолжительность реакции и объем реактора меньше, чем в случае по-
получения раствора при стехиометрическом соотношении реагентов,
предназначенного для переработки в твердый продукт. При за-
загрузке в раствор боксита, 93%-ной кислоты и воды приблизительно
в весовом соотношении 1:2:1 уже в течение 3-минутного переме-
перемешивания массы без внешнего подогрева достигается 92% извлече-
извлечение А12О382.
Сообщают и другие режимы варки боксита с серной кислотой,
например, перемешивание в течение 30—60 мин при 150—200°83
или осуществление этого процесса в шаровых мельницах м.
Производство квасцов
653
Описана85 сравнительно недавно построенная в США уста-
установка для получения из боксита 120 т/сутки 17%-ного сульфата
алюминия. Измельченный боксит разлагают в футерованном кис-
кислотоупорным кирпичом котле с диаметром 6,1 л и глубиной 5,5 м.
В него заливают 22 мг воды, затем 16 м3 90%-ной серной кислоты
и, поддерживая острым паром температуру, равной 118°, посте-
постепенно засыпают 19 т боксита. Перед окончанием реакции добав-
добавляют холодную воду для понижения температуры до 82°. В течение
суток проводят две варки. После отстаивания в котле жидкость и
шлам фильтруют отдельно. Фильтрование производят на барабан-
барабанном вакуум-фильтре, применяя в качестве вспомогательного филь-
фильтрующего материала диатомит. Фильтрат, содержащий 8,3% А12О3,
выпаривают в течение 4 ч до 17% А12О3 в футерованном стальном
ревервуаре с паровым змеевиком и направляют на кристаллиза-
кристаллизационный стол с поверхностью 93 м2. Затвердевший материал дро-
дробят на куски и выдерживают в бункере 24 ч, что способствует бо-
более легкому последующему измельчению в дезинтеграторе.
Использование отбросных травильных растворов для получения
сульфата алюминия
Запатентован86.87 способ получения сульфата алюминия с ути-
утилизацией травильных растворов, содержащих до 10% H2SO4 и
~25% FeSO4. Эти растворы упаривают, продувают воздухом для
окисления Fe2+ в Fe3+ и обрабатывают ими глиноземсодержащий
материал (глину и др.) в автоклаве при 100—350° и давлении
200—350 ат. При этом в растворе Fe2(SO4K заменяется на
A12(SO4K, а в шламе остаются SiO2 и Fe(OHK. Профильтрован-
Профильтрованный раствор подкисляют и обрабатывают'сернистым газом для
восстановления небольшой примеси Fe3+ в Fe2+, после чего из него
кристаллизуют сульфат закиси железа, а оставшийся раствор суль-
сульфата алюминия перерабатывают в твердый продукт. Очистка рас-
' твора сульфата алюминия от FeSO4 достигается при нагревании
раствора выше 50° (до температуры кипения)—это приводит к
/Настолько значительному выделению в осадок сульфата закиси же-
железа, что получаемый раствор может быть переработан в продукт,
пригодный в качестве коагулянта для сточных вод. Осуществляя
вторую стадию кристаллизации сульфата железа при температуре
ниже 50°, получают чистый сульфат алюминия 88.
ПРОИЗВОДСТВО КВАСЦОВ
Для получения алюмо-калиевых квасцов KaSO^ A12(SO4K«
•24Н2О к концентрированному горячему раствору сульфата алю-
алюминия, содержащему 8% А12О3, добавляют сульфат калия и пере-
перемешивают в течение 1 ч при 80—90°. Затем раствор направляют на
654
Гл. XVI. Сульфат алюминия и продукты на его основе
Производство квасцов
655
кристаллизацию, например, в вакуум-кристаллизационную уста-
установку. Кристаллы квасцов отделяют на центрифуге и промывают
водой. Маточный раствор и промывную воду используют, возвра-
возвращая их на разбавление раствора сульфата алюминия перед до-
добавкой к нему сульфата калия, пока содержание примеси сульфата
железа в них не повысится до 1,1% Fe2O3. После этого их направ-
направляют на разбавление купоросного масла, используемого для варки
сульфата алюминия67.
Алюмо-калиевые квасцы могут быть получены при видоизме-
видоизменении процесса производства очищенного сульфата алюминия.
Вначале производят варку каолина с серной кислотой. По оконча-
окончании нейтрализации серной кислоты в реактор добавляют сульфат
натрия из расчета получения натриевых квасцов. Последние, вслед-
вследствие своей большой растворимости, находятся в растворе. После
разбавления раствора до плотности 1,33 г/см3 его отделяют от
осадка кремнезема, охлаждают и смешивают с насыщенным рас-
раствором хлорида калия. При этом в осадок выделяются алюмо-ка-
алюмо-калиевые квасцы, плохо растворимые при невысокой температуре.
В маточном растворе после отделения кристаллов алюмо-калиевых
квасцов остаются растворимые примеси — соединения железа и
хлорид натрия89.
В настоящее время алюмо-калиевые квасцы производят по сле-
следующему варианту этого способа. Полученный из пульпы гидро-
гидроокиси алюминия и серной кислоты раствор сульфата алюминия,
разбавленный водой до концентрации ~29% A12(SO4K, смеши-
смешивают со стехиометрическим количеством 15% раствора Na2SO4 и
с 25% раствором КС1. Последний загружается с 10%-ным избыт-
избытком для высаливания квасцов, образующихся по реакции:
A12(SO4K + 2KC1 + Na2SO4 = 2KA1(SO4J + 2NaCl
Реакционную смесь нагревают до 70е, затем раствор, содержа-
содержащий 18,5—19% KAl(SO4b, пропускают через фильтрпресс для
очистки от шлама и направляют в четырехкорпусную вакуум-кри-
вакуум-кристаллизационную установку. Остаточное давление в 4-м корпусе —
7 мм рт. ст., что позволяет снизить температуру раствора до 10°.
При такой температуре насыщенный раствор содержит ~4%
KA1(SO4J- Кристаллизуется ~86% квасцов. Кристаллы поддер-
поддерживаются во взвешенном состоянии при помощи мешалок, кото-
которыми снабжены вакуум-кристаллизаторы; отношение Ж: Т в чет-
четвертом корпусе равно 2: 1. Отсюда пульпа направляется на цент-
центрифугу, маточный раствор возвращается на производство сульфата
алюминия, а кристаллы транспортируются в бункер готовой про-
продукции II сорта. Для получения продукта I сорта отфугованные
кристаллы высушивают в барабанной сушилке горячим воздухом
A20—130°). На 1 т продукта расходуют: 0,14 г гидроокиси алюми-
алюминия A00%), 0,23 т хлорида калия A00%), 0,192 т сульфата натрия
A00%), 0,40 т серной кислоты A00%), 0,015 т кальцинированной
соды (используемой для нейтрализации свободной кислотности
раствора сульфата натрия), 168 кет• ч электроэнергии, ~60 м3
воды, 1,27 мгк пара.
Хорошим сырьем для получения квасцов является нефелин. При
взаимодействии нефелина с серной кислотой образуется смесь ка-
калиевых и натриевых квасцов по реакции:
(К. NaJO • А12О3 • 2SiO2 + 4H2SO4 + 20H2O -
= (К, NaJSO4 • A12(SO4K • 24Н2О + 2SiO2
Этот процесс осуществляют следующим образом. В кипящую
40—50%-ную серную кислоту постепенно всыпают нефелин. Рас-
Раствор по мере загустевания разбавляют водой. После полной ней-
нейтрализации кислоты раствор некоторое время кипятят, затем раз-
разбавляют водой до плотности 1,16—1,23 г/см3 и отфильтровывают от
шлама — кремневой кислоты. Затем раствор подвергают выпари-
выпариванию и охлаждению в лотках, причем он полностью закристалли-
зовывается в квасцы. Недостатком этого способа является плохая
фильтруемость пульпы, полученной после варки. Это объясняется
присутствием геля кремневой кислоты. Часть шлама проходит с
раствором через фильтр, вследствие чего готовый продукт иногда
содержит нерастворимого остатка больше, чем допускается стан-
стандартом.
Для получения легко отделяемого на фильтре осадка разложе-
разложение нефелина необходимо вести 74—76%-ной серной кислотой при
повышенной температуре54. В этих условиях происходит дегидра-
дегидратация кремневой кислоты, вследствие чего она не переходит в кол-
коллоидную форму и не затрудняет фильтрацию. Кроме того, при бо-
более низкой концентрации кислоты разложение идет слишком бы-
быстро (рис. 182), что может затруднить осуществление процесса.
При концентрации кислоты выше 76% реакция резко замедляется.
Кислота указанной концентрации берется в количестве 83—88%
' от теоретического. Температура смеси к концу процесса повы-
повышается до 140°. По окончании реакции получается рыхлая, пори-
пористая масса, из которой квасцы легко выщелачиваются горячей во-
водой. Оптимальное соотношение воды и нефелина при выщелачи-
выщелачивании квасцов составляет 2: 1.
Ввиду высокой температуры в реакционном аппарате, возни-
возникающей вследствие сильной экзотермичности реакции, выделяю-
выделяющаяся кремневая кислота находится в коагулированном виде. Од-
Однако, ввиду того что реакция не проходит количественно и продол-
продолжается при выщелачивании квасцов, в растворе обнаруживается
и некоторое количество золя кремневой кислоты. Несмотря на это,
раствор легко отфильтровывается — в 7—10 раз быстрее, чем при
разложении нефелина 40—50%-ной серной кислотой. Разложение
Нефелинового концентрата кислотой, созревание массы и ее
656
Гл. XVI. Сульфат алюминия и продукты на его основе
выщелачивание можно производить в одном аппарате — варочном
котле, снабженном подъемной мешалкой. Как только после смеше-
смешения нефелина с кислотой, продолжающегося 1—2 мин, температура
начинает повышаться, мешалка должна быть поднята. После со-
созревания массы в течение 14—15 мин в котел подают горячую про-
промывную воду для выщелачивания растворимых солей. Выщелачи-
Выщелачивание производят 20—25 мин при постепенном опускании мешалки.
Полученный раствор содержит около 8% KA1(SO4J, 23%
NaAl(SO4)» и 1% сульфатов закисного и окисного железа. Для
того чтобы железо содержалось в
основном в виде сульфата окиси
(что требуется при применении ква-
квасцов для очистки воды), необходимо
к серной кислоте, идущей на разло-
разложение нефелина, добавлять неболь-
бое количество окислителя — азот-
азотной кислоты.
Полученный раствор квасцов мо-
может быть подвергнут выпарке до
концентрации А12Оз 10,5—12,5%, по-
после чего при охлаждении получает-
получается готовый твердый продукт. При
выпарке раствора имеет место силь-
сильный гидролиз квасцов, в результате
которого образуется осадок основ-
основных солей и кислотность раствора
повышается. Во избежание получе-
получения продукта с кислотностью выше
допускаемой @,1%) в конце выпар-
выпарки необходимо производить нейтра-
нейтрализацию раствора гидроокиси алю-
алюминия нефелином или содой. Примерный состав получаемого таким
способом продукта следующий: 40,2% Al2(S04b A2% А12О3), 1,7%
fe2(SO4K @,7% Fe2O3), 11,3% Na2SO4, 4,6% K2SO4, 40,4% кри-
кристаллизационной воды и 1,8% примесей.
Калиевые квасцы являются значительно более ценным продук-
продуктом, чем натриевые, так как в отличие от последних они путем пе-
перекристаллизации могут быть получены в очень чистом виде, что
важно для многих потребителей. Поэтому раздельное получение
калиевых и натриевых квасцов более рационально, чем получение
смешанных квасцов путем прямой выпарки раствора. Раствори-
Растворимость калиевых квасцов резко уменьшается по мере увеличе-
увеличения концентрации натриевых квасцов и понижения температуры
(рис, 184). Путем охлаждения раствора, полученного сернокислот-
сернокислотным разложением нефелина, до 30е можно выделить в твердую
фазу до 90% калиевых квасцов. Оставшийся раствор содержащий
16
,2на Ю0гНгО
Рис. 184. Изотермы раствори-
растворимости KA1(SO4J в растворах
NaAl(SO4JB0, 80, 40 и 50°).
Производство квасцов
637
в основном натриевые квасцы, может быть переработай на твер-
твердый продукт выпаркой и охлаждением.
Из раствора квасцов, полученного из нефелина, можно выде-
выделить до 90% алюминия в виде калиевых квасцов путем обработки
натриевых квасцов хлоридом калия по реакции:
NaAl(SO4J + КС! ^=± KAl(SO4J + NaCl
Получение квасцов из нефелина можно осуществить разложе-
разложением водной суспензии неЛелиновой муки сернистым газом80. При
этом образуется раствор бисульфитов:
(К, NaJO • А12О3 • 2SiO2 + 8SO2 + 4H2O ^±
3 + K;HSO3 + 2Al(HSO3K
В отличие от сернистокислотного разложения каолина, разло-
разложение нефелина идет с практически достаточной скоростью при
обычных температуре и давлении. При отношении Т : Ж в пульпе,
равном 1 : 5, степень извлечения А12О3 из нефелиновой муки в рас-
раствор при 20° за 3 ч составляет около 90%. Для получения хорошо
отфильтровывающегося осадка SiO2 с предотвращением гелеобра-
зования взаимодействие двуокиси серы из отбросных газов с вод*
ной суспензией нефелина следует осуществлять ступенчато, с посте-
постепенным увеличением количества жидкой фазы путем разбавления
водой или оборотными растворами91. В начале процесса величина
Т:Ж должна быть равной 1:1, а в конце 1 :5. Раствор, получен-
полученный после отделения кремнезема, может быть переработан в ква-
квасцы обработкой его серной кислотой с последующим выпарива-
выпариванием и кристаллизацией. В качестве побочного продукта обра-
образуется почти 100%-ная двуокись серы.
Очистку растворов алюмо-калиевых квасцов от железа можно
осуществлять с помощью материалов, содержащих глинозем, на-
например дегидратированной алунитовой породой и др. При этом в
результате гидролиза железо-калиевых квасцов образуются труд-
труднорастворимые основные железо-калиевые квасцы:
1 3[K2SO4 • Fe2(SO4K] + 14Н2О ^=±
^=± K2SO4 • 3Fe2O3 • 4SO3 • 9Н2О + 5H2SO4 + 2K2SO4
Нейтрализация образующейся свободной серной кислоты ма-
материалом, содержащим глинозем, способствует сдвигу реакции ги-
гидролиза в сторону нерастворимых основных соединений железа92.
Получение не содержащих железа алюмо-аммониевых квасцов
из загрязненных железом растворов сульфата алюминия можно
осуществить осаждением их сульфатом аммония. К раствору суль-
сульфата алюминия добавляют вначале раствор бисульфата аммония,
содержащий ~26% SO2, нагревают смесь до 60—706 и выдержи-
выдерживают при этой температуре 30—40 мин. При этом содержащийся в
658
Гл. XVI. Сульфат алюминия и продукты на его основе
Литература
659
растворе сульфат окиси железа восстанавливается в сульфат за-
закиси железа. Это восстановление необходимо осуществить потому,
что сульфат окиси железа образует с сульфатом аммония изомор-
изоморфные с алюмо-аммониевыми квасцами железо-аммониевые квасцы,
совместно выделяющиеся из раствора.
После восстановления бисульфитом аммония к охлажденному
раствору сульфата алюминия, содержащему ~20—25% A12(SO4K,
прибавляют раствор сульфата аммония с концентрацией ~35%
(NH4JSO4. Для более полного осаждения алюмо-аммониевых ква-
квасцов берут 10—12%-ный избыток сульфат аммония по отношению
к стехиометрическому количеству согласно уравнению:
A12(SO4)S t- (NH4JSO4 + 24Н2О = (NH4JSO4 • A12(SO4K • 24H2O
Вследствие экзотермичности реакции раствор разогревается до
50—60°. Его охлаждают до 15—20° с целью увеличения выхода
квасцов. Отделенные кристаллы промывают и высушивают.
Этот способ переработки сульфата алюминия представляет, в
частности, интерес при получении из него гидроокиси, а затем
окиси алюминия. В этом случае алюмо-аммониевые квасцы вто-
вторично перекристаллизовывают и затем обрабатывают их водным
раствором аммиака для осаждения гидроокиси алюминия, а обра-
образующийся раствор сульфата аммония возвращают в процесс93'94.
ЛИТЕРАТУРА
1. J. A.. S k u г а 1 i s, Н. А. Н а г а п, R. М а 1 е е п у, J. Am. Chem. Soc., 76,
№ 5, 1450 A954). — 2. П. В. Дыбина, Технология минеральных солей, Госхим-
издат, 1949.— 3. П. В. Дыбина, Сборник статей ВЗПИ, № 3, 1953, стр. 37.—
4. S. Bretsznajder, J. Boczar, J. Piskorski, J. Porowski, Przem.
chem., 11,№2, 89 A955).— 5. S. Bretsznajder, W. Kawecki, Poczn. Chem.,
29, № 2—3, 287 A955). — 6. E. А. Крогиус, Научный ежегодник за 1954 г.
Саратовск. ун-та, Саратов, 1955, стр. 534. — 7. Chem. Ind., 44, № 2, 171 A939).—
8. Танабэ, J. Pharm. Soc. Japan, 75, № 8, 954 A955). — 9. Т. Д. А в ер б ух,
И. В. С т р е ж н е в, Н. П. Б а к и н а. Хим. пром., № 6, 6 A948). — 10. А, К. Т р о-
фимов, ДАН СССР, 104, № 3, 427 A955).
//. В. В. Печковскнй, А. Н. Кетов, ЖПХ, 30, № 10, 1506 A957).—
12. Lorant В el a, Z. analyt. Chem., 219, № 3, 256 A966). — 13. И. Л. Баг-
банлы, А К. Посадовская, Изв. АН АзССР 1, 13 A955). —14. Г В Ку-
к о л е в, Е. Н. Л е в е, ЖПХ, 28, № 9, 909 A955). — 15. П. В. Д ы б и н а, Там же,
37, № 8, 1711 A959). —16. L. Lesniewicz, S. Bretsznajder et al., Przem.
chem., 12, № 7, 371, 378 A956). —17. B. Vrnelik, M. Mathien, M. Prettre,
Compt. rend., 242, № 6, 773 A956).— 18. M. А. Миниович, Хим. пром., № 7,
19 A944). — 19. Там же, № 3, 22 A946). — 20. S. В г е t s z п a j d e r, Przem. chem.,
10, № 5 250- № 6, 295 A954); 11, № 3, 115 A955); 12, № 1, 33 A956); Roczn.
chem., 29, № 2—3, 227 A955).
21. Англ. пат. 784706, 1953. —22. Chem. Eng. News, 44, № 36, 72 A A966).—
23. Ю. П. Вейцер, Журн. ВХО, 5, № 6, 628 (I960). — 24. Э. А. Левицкий,
Хим. пром. № 7 557 (I960). — 25. Э. А. Левицкий, В. Н Максимов,
И. Ю. М а р ч ен к о, ДАН СССР, 139, № 4; 141, № 4 A961). — 26. Э. А. Л е в и ц-
. кий, Б. М. Щепачев, Изв. вузов. Цвета, металлург., № 2 A96L).—
27. Б. М. Щепачев, Труды НИОХИМ, т. 19, 1969, стр. 133.— 28. В. М. Куз-
Кузнецов, Производство сернокислого глинозема, ОНТИ, 1932. — 29. G. Schmutz-
ler, Chem. Technik, 17, № 12, 720 A965). — 30. П. М. Татарине в, С. Ф. Ма-
лявкин, А. Н. Гейслер, Курс нерудных месторождений, ч. II, ОНТИ, 1935.
31. Ю. В. М о р а ч е в с к н й, Труды ГИПХ, в. 37, 1946, стр. 120. —
32. П. В. Д ы б и н а, Хим. пром., № 5, 15 A945).— 33. В. К. П е р ш к е, Н. Н. Л а-
шнн, ЖХП, № 14—15, 954 A930). — 34. Технология керамики н огнеупоров, под
ред. П. П. Будникрва, Госстройиздат, 1955.—35. Г. Г. Уразов, Н. И. Влода-
вец, Изв. Ин-та фнз.-хим. анализа, 3, № 2 A927). — 36. П. В. Д ы б н н а, Сбор-
Сборник статей ВЗПИ, № 6, 49 A954). — 87. А. С. Гинзберг, Хр. С. Никогосян,
А. В. Ч и т а е в, Труды Института прикладной минералогии и металлургии, в. 22,
1926, стр. 5. — 38. Э. К- Келлер, А И Леонов, Усп. хим., 22, № 3, 334
A953).— 39. Д. С. Белянкин, ИСФХА АН СССР, 19, 51 A949).— 40. А. М. С о-
. колов, Изв. СПБ технол. ин-та, 22, 1 A913).
41. Н Видено в, Л. Димитрова, Г. Монев, Тежка пром., 4, № 6, 33
A955). — 42. P. Murray, J. White, Clay Minerals Bull., 2, № 13, 255 A955).—
43. А. И. Августиннк, Л. В. Козловский, ЖПХ, 25, № 3, 265 (I952).—
44. Chem. Ind., 14, № 6, 313 A962). — 45. В. А. М азе ль, Производство глино-
глинозема, Госметаллургиздат, 1955. — 46. Н. Н. Николаев, Водоснабж. и сантех-
сантехника, № 3, 54 A938). — 47. Г. Кутерман, Труды Ин-та химии АН ГрузССР,
в. 14, 1958, стр. 203. — 48. Т. В. Панцулая, М. С. Мерабишвнли,
Т. Н. Бегиашвили, Труды Кавказ, ин-та минерального сырья. Сер. технол.,
в. 6 (X), 1965, стр. 107. — 49. И. Л. Багбанлы, Научно-нссл. работы хим.
институтов и лаборат. АН СССР за 1941—1943 гг., 1945, стр. 300. — 50. И. Баг-
Багбанлы, Труды Ин-та химии АН АзССР, в. 15, 1956, стр. 5.
51. И. М. Вассерман, Производство минеральных солей, Госхимиздат,
1962.-55. К. В. Иванов, Хнм. пром., № 1, 48 A955). — 53. С. И. Савчук,
Автореф. канд. дисс, НИУИФ, 1955. — 54. С. С. Марков, Труды ГИПХ, в. 34,
1940, стр. 22 — 55. Н. К. Саракуз, Хнм. пром., № 6, 361 A955).— 55а, Ф. К. Ми-
Михайлов, Ю. В. Ласточкин, А. Д. Соколова, В. А. Немо в, авт. свид
223077, 1967. — 56. Австр. пат. 213375, 1961. — 57. Б. Д. Мельник, ЖПХ, 10,
№ 10, 2165 A967). — 58. П. В. Дыбина, А. А. Соловьева, Ю. И. Вишняк,
Расчеты по технологии неорганических веществ, Изд. «Высшая школа», 1967,
стр. 415. — 59. Пат. ФРГ 946436, 1956; австр. пат. 154214, 1953; франц пат
t220251, 1959. — 60. Пат. ГДР 36653, 1964, пат. США 3011878, 1961.
61. Пат. ФРГ, 1146042, 1963. — 62. И. Л. Багбанлы, X. Л. Зейпалова
Труды Ин-та химии АН АзССР, в. 13, 1954, стр. 104. —63. Н. Kretzschmar,
пат. ФРГ 930871, 1955. — 64. В. W. Hammaren, P. A. F1 о г i о, R. V. Tow-
•nend, канад. пат. 492396, 1953. — 65. S. Kojima, япон. пат. 2627, 1953,—
66 L. Lesniewicz, S. Bretsznajder, J. Lis, J. Piskorski, Przem.
chem., 12, № 10, 566 A956). — 67. Кувата, Сугахара, япон. пат. 318, 1955 —
й8. Н. Kretzschmar, пат. ФРГ 920964 1954.— 69. С. С Марков С О Рез-
Резникова. Труды ГИПХ, в. 39, 1947, стр. 69.-70. Н. W. Schmidt, пат ФРГ
886597, 1953.
71. Г. Б. Ш а х т а х т и и с к и й, А. Н. X а л и л о в, Азерб. хим. ж., № 1 109
A967). — 72. Пат. США 3141733, 1961; 3148024, 1962; 3211521, 1962; 3254948
1963. — 73. П. В. Дыбина, Хим. пром., № 4, 19 A948). — 74. П. В Дыбина,
Там же, № 8, 16 A944). — 75. П. В. Дыбина, Труды МХТИ, в 11, 1947,
стр. 61. — 76. Г. К. Магаршак, Хим. пром., № 3, 18 A946). — 77. А. Воз-
ницка, Przem. chem., 42, № 11, 649 A963). — 78. Л. В. Гладушко, В. С. С а-
жин, А. К. Запольский, Хим. пром. Украины, № 6 A967); № 7 A968); сб.
«Исследодания по комплексной переработке нефелиновых пород и каолинов
Украины на глинозем и другие продукты», Изд. АН УССР, 1968. — 79. П. В. Д ы-
Оина, Хим. пром., № 3. 9 П953). — 80. П. В. Дыбина. Сборник статей ВЗПИ,
в- 2, 1952, стр. 28.
660
Гл. XVI. Сульфат алюминия и продукты на его основе
81. Chem Ens., 57, № 12, 172 (I960). — 82. П. $. At. Commission 73, 747
A962) — 83 Пат ФРГ 1126894, 1962. — 84. Пат. США 30?$228, 1963. — 85. Chem.
Й1Е-, 68, № 6, 132 A961). —S6. Пат. США 3078146, 1963. — «7. Франц. пат.
1347556 1963, —«А Польск. пат. 44574, 1961. — 89. П. М. Токарскйй, ЖХП,
h 6, 57 A940). —SO. И. И. Искольдский, В. В. Громой, К проблеме
окиси алюминия, ОНТИ, 1934.
91. 3 П Розенкноп, авт. свид. 65934; Бюлл. изобрет., К» 2, Ц A946).—
92. Б. К. Шварцман, Автореф. канд. дйсс, ВАМИ, 1949.— 93. Л. Г. К 6 6-
ленц, Получение окиси и металлического алюминия из глин БобриковсКого
месторождения подмосковного каменноугольного бассейна, изд. Моехимэнерго-
строй, 1932. — 54. Польск. пат. 46752, 1961.
Глава XV11
СОЕДИНЕНИЯ МЕДИ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Медь легко соединяется с кислородом при нагревании '•2, обра-
8уя окислы Си2О и СиО. Давление кислорода над СиО при 450°'
равно 6,963-Ю5, при 1000°—118, а при 1070° —458 мм рт. ст.
Давление кислорода над Си2О при 450° равно 7,561 • 10~26 мм рт. ст*
и 760 мм рт. ст. при 1935°.
Закись меди Си2О образует красные кристаллы кубической син-
гонии с плотностью 6,0 г/см3, плавящиеся при 1235°. Окись меди
СиО черного или коричнево-черного цвета кристаллизуется в три-
триклинической и кубической сингонии; плотность 6,4—6,45 г/смК
Окислы меди в воде не растворимы.
При взаимодействии растворов солей меди с основаниями об-
образуются осадки зеленого или коричневого цвета. Считают, что-
осадки зеленого цвета являются гидратами гидроокисей, а корич-
коричневого— гидратами окислов меди3. Гидрат закиси меди СиОН
образует желтые кристаллы с плотностью 3,4 г/см3, не растворим
в воде, дегидратируется при 360°. Гидрат окиси меди Си(ОНJ-
окрашен в цвета от синего до желтого, имеет плотность 3,4 г/см\
при нагревании обезвоживается с образованием окиси меди. В хо-
холодной воде плохо растворим, в горячей воде разлагается. Произ-
йедение растворимости гидратированной окиси меди, соответст-
соответствующее равновесию
СиО • пН2О + Н2О ^~± Си2+ + 2ОН~ + яН2О
при 25—28° равно4 1,5 • Ю0; по другим данным 5 ПРСц<он>2 =•
=4,8-100, ПРси(ОН)!.нго=2,0- 10~19. Гидрат окиси меди раство-
растворяется в водных растворах аммиака с образованием темно-синего
комплекса [Cu(NH3L](OHJ5. Аналогичные комплексы образуют
и соли меди. Гидроокись меди растворяется также в водных рас-
растворах NaOH. С солями меди неорганических и сильных органиче-
органических кислот гидроокись меди образует зелено-желтые основные
соли типа CuSO4-3Cu(OHJ6 или CuSO4-2Cu(OHJ7.
€62
Гл. XVII. Соединения меди
Карбонат одновалентной меди С112СО3 имеет желтый цвет и
плотность 4,4 г/см3. Карбонат двухвалентной меди СиСО3 не вы-
Делен в свободном состоянии, но существует в виде основных солей.
Основной карбонат меди—азурит (медная лазурь) 2CuCO3-Cu(OHJ
образует моноклинные кристаллы синего цвета с плотностью
8,88 г/см3; разлагается при нагревании до 220°. Основной карбонат
меди — малахит CuCO3-Cu(OHJ образует моноклинные кристал-
кристаллы темно-зеленого цвета, свежеосажденный окрашен в голубой
цвет; плотность 4,0 г/см3. Эти основные соли не растворимы в хо-
холодной воде (растворимость малахита
0,0008%, азурита 0,006%), а в горячей
воде разлагаются.
Хлорид одновалентной меди Си2С12
выделяется в виде кубических кри-
кристаллов белого цвета с плотностью
3,53 г/см3, плавится при 422°, кипит
при 1366°; при 0° в 100 г воды раство-
растворяется 0,0062 г Си2С12. Растворим в
соляной кислоте и аммиаке с образо-
образованием Н[СиС1]2 и [Cu(NH3J]Cl.
Хлорид двухвалентной меди
СиС12 — коричнево-желтый порошок с
плотностью 3,05 г/см3, плавится при
498°, при 993° разлагается с образова-
образованием Си2С12. Из водных растворов
выделяются: ниже 15° СиС12 • 4Н2О, ни-
ниже 26° СиС12-ЗН2О, ниже 42° СиС12-
• 2Н2О (ромбические кристаллы сине-
синего цвета) и до 117° CuCI2 • Н2О. Насы-
Насыщенный водный раствор содержит
при 0° 40,7%, при 101,8° 52,8%
СиС12.
Основной хлорид меди СиС12 • 2СиО • 4Н2О сине-зеленого цвета
не растворим в воде; при нагревании до 140° отщепляет ЗН2О. Из-
Известны и другие основные хлориды меди: ЗСиО • СиС12-Н2О,
ЗСиО ¦ СиС12 • 4Н2О (минерал атакомит), ЗСиО • СиС12 • 5Н2О,
4СиО • СиС12 • 6Н2О, 4СиО • СиС12 • 8Н2О и др.8-9.
В концентрированных водных растворах СиС12 образуется ком-
комплексное соединение [CuClJCu, которое не диссоциирует на ионы
и обусловливает зеленую окраску растворов. При их разбавлении
происходит разложение:
50 60 70 80 90
Температура ,°С
Рис. 185. Давление водяного
пара над кристаллогидратами
сульфата меди:
I- CuSO4 ¦ 5Н2О ^=±
5=±Г CuSO4 ¦ ЗН2О + 2Н2О;
2-CuSO4 • ЗН2О 7~>"
4=2 CuSO4 ¦ Н2О +2НгО;
3 - CuSO4 ¦ 5Н2О ~j~*~
5=2 CuSO4 ¦ Н2О + 4Н2О.
Физико-химические свойства
663
[CuCl4]Cu
2Си2++4СГ
С повышением температуры равновесие этой реакции сдвигается
влево ш. Ионы Си+ и Си2+ образуют комплексы и с другими хлрри-
дами, например кальция, ртути, цинка — СиС12 • СаСЬ, СиС12 •
.HgCl2, CuCl2-ZnCl2n-12.
Пятиводный кристаллогидрат . сульфата меди CuSO4-5H2O
(хальконтит), называемый медным (синим) купоросом, образует
асимметричные ярко-синие кристаллы триклиноэдрическои системы
с плотностью 2,29 г/см3. При нагревании он плавится при 110° с
потерей части кристаллизационной воды и переходит в трехводный
(голубого цвета) и одноводный (белого цвета) сульфат меди. Выше
258° образуется безводный
сульфат меди белого цвета,
сильно гигроскопичный. При
819—860° CuSO4 разлагается
по реакции
2CuSO4 = SO3 + CuO • CuSO4
а при 897—934° полностью дис-
диссоциирует на CuO и SO313~15.
Давление водяного пара над
кристаллогидратами сульфата
меди показано на рис. 185.
При обычной температуре кри-
кристаллы медного купороса на
воздухе не выветриваются 16.
Насыщенный водный рас-
раствор медного купороса содер-
содержит при 0°— 12,9%, при 20° —
17,4%, при 55° —26,9%, при
100° —42,4% CuSO4. Безвари-
Безвариантное равновесие CuSO4 •
• 5Н2О + CuSO4 • ЗН2О + рас-
раствор + пар существует при 96°
.и 540 мм рт. ст. Раствори-
Растворимость медного купороса в при-
присутствии свободной серной кис-
кислоты понижается (рис. 186); в растворе образуется179 комплекс-
комплексный ион [Cu(SO4J]2~. При повышенных температурах из кислых
растворов кристаллизуется CuSO4 • ЗН2О (в области, лежащей
правее и выше пунктирной линии).
В системе CuSO4 — FeSO4 — H2SO4 — H2O в интервале 27—95°
и диапазоне концентраций H2SO4 от 7 до 37 г/л и FeSO4 от 10 до
300 г/л твердые фазы представляют собой три типа твердых рас-
растворов (Си, Fe) SO4 • 5Н2О с содержанием FeSO4 (в вес. %): 0—2,6,
2,7—4,7 и 8,5—9,2. С повышением температуры растворимость твер-
твердого раствора уменьшается, а содержание FeSO4 в нем возрастает.
В растворах с увеличенной кислотностью повышается содержание
FeSO4 в кристаллах, но понижается их растворимость20.
кии
750
700
650
600
г/л
О &сл
*3 ЬПП
$350
ХЛ ?Л1?
^ 250
200
150
100
50
\
\
ч
К
s
ч,
\
S
\
\
\
ч
\
ч.
ч,
\
\
s
ч
\
\
\
ч
\
ч
ч
ч
>
10*
ч
\
?s
\
N
но
ч
70'
ч
\
ч^
ч,.
S
ч|
^ч
ч
ч,
ч^
ч
О Ч,
<ч,
ч^
"ч.
s
"•ч
\,
ч
ч
ч_
О 50 iOO /50 гОО 260 300350 МО </5О S00S51T
Нонцентрация H2SO4, г/л
Рнс. 186. Изотермы растворимости мед-
ного купороса в присутствии свободной"
серной кислоты.
«64
Гл. XVII. Соединения меди
Применение
665
ПРИМЕНЕНИЕ
Из соединений меди в наибольших количествах применяется
медный купорос. Его используют в гальванических элементах в
качестве электролита, в гальванотехнике, для консервирования де-
дерева, для изготовления некоторых минеральных красок, в произ-
производстве искусственного волокна и при обогащении руд.
Растворимые соединения меди ядовиты. Этим обусловлено ши-
широкое использование в сельском хозяйстве препаратов, содержа-
содержащих медь21. Преимущественное применение находят сульфат меди,
основной сульфат меди, хлорокись меди, основные карбонаты меди,
закись и окись меди. Препараты меди, предназначенные для опры-
опрыскивания, содержат 16—55%, а иногда до 80% Си. За границей
выпускают также медные препараты с повышенной фунгицидной
активностью в форме высокодисперсных веществ под общим на-
названием «коллоидная медь». Например, препарат «Duphar Colloi-
Colloidal Copper» представляет собой пасту, состоящую из коллоидной
хлорокиси меди (с размерами частиц 0,01 мк) и поверхностно-ак-
поверхностно-активного вещества. По рекламным данным, этот препарат, содержа-
содержащий 27% Си, дает экономию меди по сравнению с сульфатом меди
в 4 раза, по сравнению с хлорокисью меди — в 3 раза и по срав*
нению с закисью меди — почти в 2 раза. Выпускают также мине-
минерально-масляную эмульсию—пасту, с 40% меди в виде одной из
солей. Соединения меди входят также в комбинированные фунги-
фунгициды, содержащие коллоидную или молотую серу E—25% Си и
40—50% S). Соединения меди добавляют к органическим фунги-
фунгицидам, заменителям медных, для усиления их эффективности и
придания универсальности действия22.23. Закись меди используют
как компонент красок для подводной части морских судов с целью
защиты от обрастания водорослями.
Среди неорганических веществ медный купорос является одним
из наиболее эффективных препаратов для борьбы с болезнями
плодовых деревьев, виноградников и других растений, и из всех
медных препаратов применяется в СССР в наибольших количе-
количествах. Чаще всего его используют в смеси с известью или другими
наполнителями. Смесь водного раствора медного купороса с из-
известью A кг CuSO4-5H2O и 0,75 кг свежегашеной извести на 100 л
воды) известна под названием бордосской жидкости, представляю-,
щей собой водную суспензию основного сульфата меди ЗСи(.ОНJ*
• CuSCu и CaSO4. Основная соль полностью разрушается в щелоч»
ной среде. Для образования суспензии стойкой основной соли мо-
молярное отношение СиО : СаО должно быть равным 1 : 0,75, а ве-
весовое отношение 1:0,53, или весовое отношение СиО: Са(ОНJ,
равным 1 :0,7. В связи с частичной карбонизацией извести, при
хранении и перевозках, при изготовлении бордосской жидкости
принимают весовое отношение медного купороса к извести 1:0,-75.
При смешении раствора медного купороса с раствором соды
получается бургундская жидкость — суспензия основного карбо-
карбоната меди 3Cu(OHJ-2CuCO3. Преимуществом ее перед бордос*
ской жидкостью является хорошая прилипаемость и отсутствие
комков, забивающих распылительные устройства. Медный купорос
применяют для изготовления парижской зелени (см. гл. XXXVI).
Описано24-25 применение медного купороса для борьбы с чрез-
чрезмерным развитием водной растительности в водохранилищах, при-
придающей воде запах и привкус.
Требования к качеству медного купороса представлены в табл. 44^
ТАБЛИЦА 4*
Состав медного
CuSO4 • 5Н2О, не менее
в пересчете на Си. не менее .
Железо (Fe), не более
Свободная H2SO4, не более . .
Не растворимый в воде остаток,
не более
Мышьяк (As), не более ....
купороса (в
По ГОСТ
5.1688-72
(со знаком
качества)
«2
3
«
и
99,0
26,19
0,035
0.25
0,05
0,005
&
а
>,
и«
о Я
95,0
24,17
0,03
0,25
0.04
0,001
%)
Марка А
сорт I
I
о
>>
и
98
24.9
0,06
0,25
0,1
0.015
<ь
в
>>
и«
о 3
К 53
94
23,9
0,03
0,25
0,05
0,015
о.
8
94
23,9
0.1
0,25
0,1
0,015
1
92
23,4
0,3
0,25
0,4
0,03
Марка Б
96
0,02
0,25
0,05
0,015-
Медный купорос I и II сортов марки А предназначается для
сельского хозяйства, III сорта — для обогатительных предприятий,
марки Б — для предприятий искусственного волокна. Тарой для
. медного купороса служат деревянные бочки, фанерные барабаны
или ящики с бумажными или полиэтиленовыми вкладышами, а
также двойные полиэтиленовые и джутовые мешки и четырехслой-
'ные бумажные мешки.
Все большее значение приобретает хлорокись меди, которая со-
содержит 55—56% Си и имеет ряд преимуществ по сравнению с мед-
медным купоросом. Расход меди при применении хлорокиси на 30%
меньше, чем при употреблении медного купороса и при этом нет
надобности в расходовании извести, необходимой для изготовления
из медного купороса бордосской жидкости. Помимо этого хлор-
хлорокись меди и препараты на ее основе оказывают меньшее ожигаю-
ожигающее действие на растения26.
Согласно ГОСТ 13200—67, 90%-ный смачивающийся порошок:
хлорокиси меди, представляющий собой смесь хлорокиси меди,.
концентрата сульфитно-спиртовой барды и декстрина, применяю-
«66
Та. XVП. Соединения меди
щийся в виде водных суспензий в качестве фунгицида, должен со-
содержать 51 ± 2% Си и не более 0,8% С1~ и 2% влаги.
Медь является одним из микроэлементов, необходимых для
жизни всех растений и животных. В качестве медных микроудоб-
микроудобрений могут быть использованы низкопроцентные руды, содержа- 1
щие медь, а также пиритные огарки °7
Производство медного купороса из медного лома
667
27
СЫРЬЕ И СПОСОБЫ ПРОИЗВОДСТВА МЕДНОГО КУПОРОСА
Основным сырьем для получения медного купороса служат сер-
•ная кислота и медь: медный лом или отходы металлообрабатываю-
металлообрабатывающей промышленности — стружка, опилки и т. п., а также отходы
или полупродукты металлургии меди — белый матт и окись меди,
ватержакетная пыль, шлаковые отходы, электролитные растворы
медеэлектролитных заводов, цементная медь, извлекаемая из руд-
рудничных вод и из колчеданных огарков и др.
Важным видом сырья для получения солей меди является ва-
ватержакетная пыль, представляющая собой тонкий порошок и со-
содержащая 0,5—5% Си в форме сульфида и сульфата, 40—50% Fe,
3—5% А12Оз, 3—6% Zn, до 15% S, 7—10% SiO2 и др. Перспектив-
Перспективным видом сырья являются шлаковые отходы медеплавильных за-
заводов, накапливаемые в течение многих лет в виде отбросов. Медь
в этих отходах содержится в окисной, сульфидной и силикатной
формах, а также в форме металла и ферритов. Примерный состав
шлаковых отходов следующий: 2—7% Си, 5—7% Fe2O3, 15—25%
A12OS, 45—50% SiO2, 1—5% CaO, 5—10% MgO, 1—3% S и 1—2%
прочих примесей.
Некоторый интерес в качестве сырья для производства медного
купороса представляют рудничные воды, образующиеся при вы-
выщелачивании отвалов атмосферными осадками и содержащие до
¦0,7 г/л меди28-29.
Большим резервом сырья для производства солей меди яв-
являются накапливаемые массы огарков от обжига колчедана на сер-
сернокислотных заводах. В старых огарках от сжигания рядового кол-
колчедана содержится до 1,5% меди в виде CuSO4, CuSO3, CuO, Cu2S,
CuS, CuFeS2. Необходимость извлечения соединений меди из огар-
огарков диктуется условиями их использования металлургической
промышленностью в качестве заменителя железной руды.
Указанные виды сырья в основном перерабатывают в медный
купорос, который, помимо непосредственного употребления, служит
также исходным материалом для получения всех других солей
меди.
Способы производства медного купороса различают главным
образом по видам применяемого сырья:
1) из медного лома и отходов меди (стружки, высечки, прово-
проволоки, опилок и т. п.) с окислением меди кислородом воздуха,'элек-
воздуха,'электролизом или раствором хлорной меди;
2) из окиси меди, получаемой из белого матта;
3) из окиси меди и сернистого газа;
4) из окисленных медных руд, содержащих незначительное ко-
количество меди, переработка которых на металлическую медь плав-
плавкой в печах является неэкономичной;
5) из колчеданных огарков и других отходов;
6) из отбросных электролитных растворов медеэлектролитных.
заводов.
За рубежом основными производителями медного купороса яв»
ляются Франция и Италия, где в качестве сырья используют глав-
главным образом медный лом и окисленные руды. В отличие от этого»
в США используют в основном электролитные щелоки, из которых,
производят больше половины ввех солей и препаратов меди.
ПРОИЗВОДСТВО МЕДНОГО КУПОРОСА ИЗ МЕДНОГО ЛОМА
Теоретические основы процесса
В отсутствие окислителей, в частности кислорода воздуха, в.
разбавленной серной кислоте медь практически не растворяется.
Она с достаточной скоростью растворяется в горячей концентриро-
концентрированной серной кислоте, но осуществлять этот процесс нерациональ-
нерационально, так как при этом половина затрачиваемой кислоты восстанав-
восстанавливается до SO2, окисляя медь в окись меди, которая и раство-
растворяется в серной кислоте, образуя медный купорос. Схема этого-
процесса может быть выражена следующими уравнениями реак-
реакций:
Си + H2SO4 = CuO + Н2О + SO2
CuO + H2SO4 = CuSO4 + Н2О
Си + 2H2SO4 = CuSO4 + 2Н2О + SO2
С целью экономии серной кислоты окисление меди производят-
кислородом воздуха одновременно с процессом «натравки», т. е.
растворения в серной кислоте. Медный лом предварительно пере-
переплавляют для рафинирования (очистки от примесей Fe, Zn, A1, РЬ-
и др.) и придания ему формы, удобной для растворения — пусто-
пустотелых гранул, обладающих большой поверхностью, что ускоряет
растворение в кислоте в 5—10 раз.
Очистка и грануляция медного лома
Чистая медь плавится при 1084°, а в присутствии примесей —
при более низкой температуре. Примеси летучих металлов и окис-
окислов — металлический цинк, трехокиси мышьяка и сурьмы — уда-
удаляются при нагревании меди до ее расплавления. При расплавле-
расплавлении медь окисляется до закиси меди, устойчивой выше 1100°. За-
Закись меди накапливается на поверхности расплавленной меди*
€68
Гл. XVI1. Соединение меди
Производство медного купороса из медного лома
в твердом (до 1200°) и в жидком (выше 1235°) виде и частично
растворяется в меди, а затем вступает во взаимодействие с приме-
примесями, например:
Cu2O + Fe = FeO + 2Cu
По мере расходования растворенной закиси меди новые ее ко-
количества переходят с поверхности в раствор, и медь подвергается
дальнейшему окислению.
Образующиеся окислы железа, магния, кальция и других ме-
металлов не растворимы в меди и переходят в шлак, всплывающий
на поверхность металла. Вследствие взаимодействия закиси меди
¦й некоторыми окислами (например, с окисью железа с образова-
образованием феррита меди) часть ее также переходит в шлак и содержа-
содержание в нем СигО достигает 30—40%.
После окисления, ошлакования примесей металлов и удаления
шлака температуру в печи немного снижают с целью окисления
присутствующей в меди полусернистой меди:
Cu2S + 2Cu2O ^±. 6Cu + SO2
Эта реакция протекает бурно, и выделяющаяся двуокись серы
увлекает брызги меди с образованием «медного дождя» («кипение»
массы).
В производстве медного купороса дальнейшая очистка меди не
требуется, а присутствие в ней кислорода и двуокиси серы необхо-
необходимо для получения пористых и пузыристых гранул. Растворимость
газов в расплавленной меди возрастает с повышением темпера-
температуры. В твердой меди, нагретой даже до температуры плавления,
растворимость газов незначительная. Процесс гранулирования
с получением пузыристой и пористой меди основан на быстром вы-
выделении газов при внезапном охлаждении и затвердевании рас-
расплавленной меди. Это осуществляется выливанием ее тонкой стру-
¦ей в холодную воду.
Серы, содержащейся в меди, обычно недостаточно для образо-
образования полых гранул. Поэтому в период «кипения» расплаве в него
добавляют некоторое количество полусернистой меди или комовой
серы A—1,6%). Образующаяся при этом двуокись серы раство-
растворяется в меди, а при ее грануляции выделяется и раздувает капли
меди в пустотелые шарики с тонкими стенками.
Растворение меди в серной кислоте (натравка)
При взаимодействии гранул меди с разбавленным раствором
серной кислоты, содержащим также сульфат меди, в присутствии
воздуха, кислород воздуха растворяется в кислоте, диффундирует
к поверхности меди и окисляет ее до закиси меди:
4Cu+O2 =
Закись меди растворяется в серной кислоте:
Си2О + H2SO4 = Cu2SO4 + Н2О
Образующийся сульфат вакиси меди легко окисляется в суль-
сульфат окиси меди:
2Cu2SO4 + 2H2SO4 + О2 - 4CuSO4 + 2Н2О
Общая скорость процесса лимитируется наиболее медленной
его стадией — окислением меди до закиси меди. Это объясняется
малой растворимостью кислорода и медленной его диффузией
к поверхности гранул меди. Процесс значительно ускоряется, когда
в растворе уже присутствует медный купорос. ,В результате депо-
деполяризации
CuSO4 восстанавливается медью до CU2SO4, а затем CU2SO4 вновь
окисляется растворенным кислородом до CuSO4. Таким образом,
медный купорос играет роль переносчика кислорода.
В присутствии металлической меди в растворе медного купороса
может находиться лишь ничтожное количество одновалентной меди.
Константа равновесия реакции Си2++Сиз^2Си+ при 25° /С={Си+]2:
[Си2+}=0,62-10. В растворе, содержащем Б0 е/л H2SO4 и 32 е/л
Си в виде CuSO4, имеется только ~0,022 г/л одновалентной меди,
т. е. меньше 0,1% от общего ее количества30-32.
Повышение температуры, как и в других случаях, ускоряет
химические реакции, но вызывает уменьшение растворимости кис-
кислорода, что замедляет окисление. Поэтому в натравочной башне
поддерживают температуру не выше 80—85°. При этом на окисле-
окисление меди используется приблизительно 'А кислорода, поступающего
в башню с воздухом, расход которого составляет около 1000 нм9
на 1 г медного купороса.
Растворимость кислорода уменьшается с ростом концентрации
CuSO4 в растворе. Поэтому при повышении концентрации C11SO4
скорость растворения меди сначала увеличивается за счет катали-
каталитического действия CuSO4, а затем уменьшается вследствие недо*
статка кислорода. Максимум скорости растворения наблюдается
при концентрации 120 г/л CuSO4 (для раствора, содержащего
~ ПО г/л H2SO4) 33-84. Но даже при содержании в растворе 300 г/л
CuSO4 скорость растворения меди в 1,6 раза больше, чем в отсут-
отсутствие медного купороса34. С увеличением концентрации серной кис-
кислоты растворимость кислорода в ней уменьшается, но усиливаются
ее окислительные свойства. Поэтому повышение кислотности
раствора вызывает не очень большое уменьшение скорости рас-
растворения меди — всего на 10% при повышении концентрации
H2SO4 e 2,5 до 20% 33. Растворение меди значительно ускоряется
670
Гл. XVII. Соединения меди
Производство медного купороса из медного лома
671
в присутствии в растворе ионов железа вследствие деполяризации
2Cu + 4Fe3+ = 2Cu2+ + 4Fe2+
Ионы Fe2+ вновь окисляются в Fe3+ и служат, таким образом,
катализатором процесса. Доля растворяющейся меди под дей-
действием ионов Fe3+ в растворе, содержащем —110 г/л H2SO4, 60 г/л
CuSO4 и 20—22 г/л FeSO4, составляет около 60% от всего количе-
количества меди, перешедшей в раствор34.
Ионы железа попадают в циркулирующий при растворении меди
раствор с серной кислотой и вследствие растворения оставшихся
в меди примесей. Содержание сульфатов железа в растворе непре-
непрерывно возрастает и достигает иногда 70 г/л и более. Вследствие
этого при кристаллизации медного купороса выделяется также и
сульфат железа, загрязняющий продукт (см. ниже). Поэтому, ко-
когда концентрация железа в растворе становится столь большой,
что создается опасность получения нестандартного по содержанию
железа медного купороса, раствор полностью выводят из обра-
обращения.
Существенным является обеспечение равномерного орошения
(смачивания) гранул меди раствором. В местах, плохо орошаемых
кислотой, образовавшаяся окисная пленка растворяется непол-
неполностью, появляется основной сульфат меди CuSO4-2Cu(OHJ, ко-
который вследствие малой своей растворимости кристаллизуется из
раствора и цементирует при этом гранулы и шлам.
Технология процесса
Производство медного купороса из медного лома делится на три
стадии: 1) получение гранулированной меди; 2) получение рас-
раствора сульфата меди; 3) кристаллизация и сушка медного купо-
купороса.
Получение гранулированной меди
Медный лом («тяжелую» медь) плавят в медеплавильной печи.
Проволоку, стружку, высечку и т. п. («легкую» медь) перед пода-
подачей в печь брикетируют33. Плавку лома ведут обычно в пламен-
пламенных печах из огнеупорного шамотного кирпича, отапливаемых ма-
мазутом 35.
Плавка меди в печи продолжается, в зависимости от количе-
количества примесей, 4,5—6 ч. После удаления шлака б «кипящую» медь
забрасывают серу, затем ее выпускают тонкой струей в воду, на-
находящуюся в гранулировочном бассейне. Он представляет собой
бетонированную яму высотой 1,6 ж и диаметром 2,5 м. В бассейн
помещают стальную корзину с дырчатыми стенками высотой 1 м
и диаметром 1,6 м; в последней собираются гранулы. При подъеме
корзины с гранулированной медью вода стекает через отверстия
в стенках корзины. Образующиеся гранулы имеют диаметр
5—15 мм. Вес 1 л гранул не должен превышать 2 кг. 1 кг таких
гранул имеет поверхность до 1500 см2.
«Натравка» и получение медного купороса
Гранулированную медь загружают в натравочную башню
(рис. 187), высота которой около 6 м, диаметр 2,5 м. Башня изго-
изготовлена из листовой стали, внутри футерована кислотоупорным
кирпичом и диабазовыми плитками. На высоте 0,5—0,9 м от дна
в башне имеется ложное днище, лежащее на колосниковой решетке
В атмосферу
3
В атмосферу
А
В атмосферу
б
Парс - воздушная
смесь
Рис. 187. Схема производства медного купороса нз гранулированной медн:
1 и 12 — ковшевые элеваторы; 2 — натравочная башня; 3 —бак с постоянным уровнем;
4 — сборник маточного раствора; 5 —расходный бак для серной кислоты; 6 и 7 —центро-
—центробежные насосы; 8 — вращающийся кристаллизатор; 9 и 14 — вентиляторы; 10 — сборник
с мешалкой; 11 — центрифуга; 13 — сушилка; 15 — нагреватель воздуха; 16 — разгрузоч-
разгрузочный бункер.
из стальных балок, опаянных свинцом. На ложном днище нахо-
находится слой меди, высоту которого поддерживают периодическими
загрузками на уровне 0,25 м от крышки башни. Под крышкой по-
помещена турбинка, с помощью которой медь непрерывно орошается
смесью серной кислоты с маточным раствором. Количество нахо-
находящейся в башне меди составляет 22—28 т.
В башне происходит одновременно окисление и растворение
меди. Эти процессы идут с выделением тепла, достаточным для
повышения температуры до необходимого уровня, т. е. до 70—85°.
Для окисления меди в башню под колосниковую решетку вдувают
воздух в смеси с паром. Пар подают для нагревания воздуха. Вду-
Вдувание холодного воздуха вызвало бы охлаждение щелока и выде»
ление из него кристаллов медного купороса, что привело бы к за-
кристаллизовыванию нижнего слоя гранулированной меди. Пода-
Подачей пара регулируют и температуру в башне. Уходящая из нее
паро-воздушная смесь выбрасывается в атмосферу.- С 1 м3
672
Гл. XVII. Соединения меди
натравочной башни можно получить в сутки более 1,3 т медного
купороса.
Орошающий щелок имеет 55—60° и содержит 20—30%
CuSO4-5H?O и 12—19% свободной H2SO4. Оптимальная плотность
орошения натравочной башни, равная 1,5—2,1 м3/(м2-ч), обеспечи-
обеспечивает образование на поверхности медных гранул очень тонкой
жидкостной пленки, через которую кислород диффундирует к меди
с достаточной скоростью. При большей плотности орошения
[4—5м3(м2'Ч)] происходит снижение производительности башни
вследствие замедления диффузии кислорода через толстые жидко*
стные слои. При этом снижение производительности башни про-
происходит после кратковременного ее возрастания, башня как бы
«вымывается».
Жидкость подают в башню при переменном режиме, создавае-
создаваемом или периодическим изменением вращения разбрызгивателя
в обратном направлении или периодическим выключением и вклю-
включением орошения через каждые 10—15 мин (при непрерывной по-
подаче горячего воздуха). Это приводит к возникновению по всему
объему башни миграционных участков окисления и растворения,
в результате чего растворение меди идет в 1,5—2 раза быстрее,
чем при постоянном орошении башни36-37.
Вытекающий из натравочной башни горячий щелок G4—76°)
представляет собой почти насыщенный раствор медного купороса —
он содержит 42—49% CuSO4-5H2O и 4—6% свободной H2SO4.
Этот щелок подают центробежным насосом из хромоникелевой
стали во вращающийся кристаллизатор непрерывного действия
с воздушным охлаждением раствора. Смесь кристаллов медного ку-
купороса с маточным раствором через сборник с мешалкой поступает
в центрифугу из нержавеющей стали, где кристаллы, отжатые ог
маточного раствора, промываются водой. На центрифугирование
поступает пульпа с соотношением Т: Ж от 1 :2 до 1 : 1,5. Отфуго-
ванный продукт, содержащий 4—6 %влаги и 0,15—0,2% кислоты38,
высушивают в барабанной сушилке воздухом при 90—Л00°. Маточ-
Маточный раствор и промывную воду после смешения с серной кислотой,
возвращают в производственный цикл.
В маточном растворе происходит постепенное накопление при-
примесей, все больше загрязняющих продукт. Содержащийся в мед-
медном купоросе сульфат никеля можно удалить с достаточной пол-
полнотой при однократной перекристаллизации. Для удаления FeSO4
необходима многократная перекристаллизация. Получение медного j
купороса с содержанием 99,9% CuSO4-5H2O однократной пере-
перекристаллизацией из раствора, насыщенного при 70°, возможно при. |
содержании в нем не более 0,3% NiSO4 и не более 0,15% FeSO439-
Если в растворе больше 40 г/л FeSO4, то количество железа в
продукте больше 0,4%, т. е. выше нормы, допускаемой ГОСТ для
продукта III сорта (рис. 188). Из растворов, содержащих больше
Производство медного купороса из медного лома
673
100—120 г\ FeSO4, выделяются смешанные кристаллы железного
и медного купоросов с характерной сине-зеленой окраской40.
Содержание железа в кристаллах медного купороса можно
уменьшить предварительным окислением Fe2+ в Fe3*. Окислителем
может служить воздух (длительный барботаж), азотная кислота,
перекись водорода и др. Степень очистки повышается в 2—4 раза
при добавке к раствору незначительного количества HF (плавико-
(плавиковой кислоты), что приводит к образованию фторидных комплексов
л
V
i
- »
— -
— —
— '¦
- -
2
-Т"
— »
100
80
О ВО
0,4 0,8 1,2 1,6 2,0 2,4 2.в 3,2 3.6 4,0 4,4 4,8
Fe в кристаллах CuSQ* ¦ 5H,O,V«
Рнс 188. Зависимость между содержанием FeSO4 в рас-
растворе медного купороса и содержанием Fe в кристаллах
CuSO4 • 5НгО при разных концентрациях раствора.
Содержание CuSO4 ¦ 5НгО в растворе (в г/л): 1 — 400; 2 — 500; 3 — 550;
4-606.
Fe3+41. Установлено также, что при усилении перемешивания в
процессе кристаллизации получаются кристаллы с меньшим со*
держанием железа, но и размеры их уменьшаются. Присутствие
ионов никеля также уменьшает размеры кристаллов, а мышьяка-—
'увеличивает42.
На производство 1 т кристаллического медного купороса рас-
расходуют: 0,27—0,29 т металлической меди и 0,39—0,40 т серной кис-
лоты A00%).
На заводе имени Войкова общие затраты тепла на производство
медного купороса составляли43 0,76мгкал на 1 т продукта. Расход
тепла распределяется следующим образом. В натравочную башню
через инжекторы вводится 47% тепла, на подогрев воздуха в
калориферах сушильного агрегата затрачивается 26% тепла и
27% тепла расходуется на подогрев раствора в сборниках, на
разогрев мазута в цистернах и т. д. Как видно из теплового ба-
баланса натравочной башни (табл. 45), для обеспечения необходи-
необходимого ее температурного режима подвода тепла извне в йиде
пара не требуется. Количество тепла, выводимого с паро-воздушцой
22 М. Е. Позии
674
/Ж XVII. Соединения меди
смесью, больше тепла, вводимого с паром вследствие дополнитель-
дополнительного парообразования, обусловленного выделением тепла реакций.
Поэтому вместо паро-воздушной смеси можно вдувать в башню
теплый воздух из кристаллизатора с добавкой 20—25% пара от
обычного количества, при температуре смеси, исключающей за-
кристаллизовывание нижнего слоя гранул в башне.
ТАБЛИЦА 45
Тепловой баланс иатравочной башни
(на 1 т 100%-ного CuSO4-5H2O)
Приход тепла, тыс. ккал
От химических реакций
С орошающим раствором . . . .
С паро-воздушной смесью . . .
Всего . .
304
134
362
800
Расход тепла, тыс. ккал .
С паро-воздушной смесью . . ,
На нагрев загружаемых гра-
гранул и содержащейся в них
В окружающую среду
Всего . .
510
ITS
3,2
11,8
800
Ввод пара в натравочную башню может быть и вовсе исключен
при осуществлении процесса с рециркуляцией паро-воздушной
смеси. Отходящую из башни паро-воздушную смесь с температурой
~80° направляют при помощи вентилятора из нержавеющей стали
под ложное дно башии. Необходимое для реакций окисления коли-
количество воздуха подсасывается через штуцер в нижней части башни.
Избыток паро-воздушной смеси отводят через специальную трубу
из верхней части башни. При осуществлении процесса по такой
схеме возможно введение в цикл газообразного кислорода, что
значительно интенсифицирует растворение меди.
Отходом производства медного купороса являются илы, скап-
скапливающиеся в резервуарах с производственными растворами. Ко-
Количество илов составляет 1—2% от перерабатываемой меди. Со-
Состав их различен; они могут содержать до 8,5% Ag2O, до 5% Bi2O3,
0,05—0,1% Аи, Pt, Pd. Такие илы могут быть переработаны гид-
гидрометаллургическими методами для извлечения из них ценных
металлов 44.
Предложено получать медный купорос из натравочного шелока
добавкой к нему серной кислоты (башенной, купоросного масла,
олеума или SO3) до содержания свободной H2SO4 60% й более.
При этом быстро осаждается мелкокристаллический белый безвод-
безводный сульфат меди, примеси же остаются в растворе. CuSO4 отфу-
говывают и растворяют в чистом маточном растворе медного купо-
купороса, из которого кристаллизуется CuSO4 • 5Н2О. Кислый щелок
после осаждения безводного CuSO4 возвращается на растворение
меди. После накопления в нем значительного количества ценных
Получение медного купороса электролизом
675^
примесей (никель, цинк, серебро и пр.) их можно извлечь. Преиму-
Преимущество этого способа — в простой и быстрой кристаллизации мед-
медного купороса без затраты тепла и холода и высокой чистоте про-
продукта 4Б.
Можно вообще отказаться от выпуска пятиводного сульфата
меди и выпускать безводный продукт, концентрация меди в кото-
котором больше C9,8% вместо 25,5% в CuSO4-5H2O). Производство
и транспорт его будут дешевле, хотя он и потребует более тща-
тщательной упаковки из-за гигроскопичности. Впрочем, даже при не-
небрежной упаковке на поверхности белого порошка появится лишь
синеватая окраска вследствие гидратации влагой воздуха, но это
не ухудшит качества продукта, который предназначен для раство-
растворения в воде. Однако во избежание слеживания упаковка должна
быть герметичной.
Очисткя сточных вод, сбрасываемых в водоемы из производств
медного купороса и других медных солей, от ионов меди может
быть осуществлена на 70—90% с помощью сульфата алюминия.
Выделяющаяся при гидролизе сульфата алюминия гидроокись алю-
алюминия адсорбирует ионы меди 46.
ПОЛУЧЕНИЕ МЕДНОГО КУПОРОСА ЭЛЕКТРОЛИЗОМ
Может представить интерес электрохимическое окисление меди
с растворением ее в серной кислоте. При проведении электролиза
с растворимым медным анодом в растворе любой соли щелочного
металла получающаяся на аноде медная соль, реагируя с образую-
образующейся на катоде щелочью, дает гидроокись меди с одновременной
регенерацией электролита.
Можно получить электролизом и непосредственно раствор мед-
медного купороса, осушествляя процесс в ванне, в которой анод, нахо-"
дящийся на дне ванны, состоит из спрессованных или сплавленных
обрезков меди. Чеоез полый катод, помещенный сверху, подается
•серная кислота. Движением раствора от катода к аноду не до-
п\-скается нежелательное в данном случае осаждение меди на'
катоде.
Пои проведении электролиза с растворимым медным анодом
R оаствоое сульфата натрия в ванне с диафрагмой можно одновре-
одновременно получать медный купорос и едкий натр. Особый интерес это
может представить при применении ртутного катода с получением
из образовавшейся амальгамы натрия концентрированной щелочи.
Анодная жидкость, кроме медного купороса, будет содержать гуль-
фат натрия, однако медный купорос и сульфат натрия могут быть
легко отделены друг от друга (как известно, трудность разделения
серной кислоты и сульфата натрия является одним из сложных
вопросов в проблеме электролиза сульфата натрия). Таким обра-
образом, этот способ позволяет получать щелочь и медный купорос без
затраты кислоты.
876
Гл. XVII. Соединения меди
Производство медного купороса из окиси меди
677
ПОЛУЧЕНИЕ МЕДНОГО КУПОРОСА ПРИ ОКИСЛЕНИИ МЕДИ
ХЛОРНОЙ МЕДЬЮ
Этот метод основан на образовании хлористой меди из хлорной
и металлической меди:
Си + ОиС1, = 2СиС1
(Хлористую медь получают также хлорированием цементной меди 1
в растворе поваренной соли.) Хлористую медь окисляют воздухом 1
с образованием оксихлорида меди:
6СиС1 + 1,5О? + ЗН,0 = 3[Cu(OHJ ¦ CuCl,]
Оксихлорид растворяют в серной кислоте, в результате чего
образуется раствор сульфата меди и регенерируется хлорная медь:
3[Си(ОНJ • СиС12] + 3H,SO4 = 3CuSO4 + 3CuCl2 + 6H2O
Получение оксихлорида меди осуществляют в бетонном баке,
куда загружают медь и заливают раствор хлорной меди. После
этого продувают массу воздухом, пока вся металлическая медь не
перейдет в нерастворимый оксихлорид. После отстаивания и декан-
декантации пульпу растворяют при нагревании в серной кислоте. При
охлаждении раствора из него кристаллизуется CuSC>4-5HjO. Ма-
Маточный раствор возвращают в процесс.
Этим способом можно получать также хлорокись меди
(стр. 690L7.
ПРОИЗВОДСТВО МЕДНОГО КУПОРОСА ИЗ ОКИСИ МЕДИ
До распространения способа получения медного купороса из
медного лома в натравочных башнях медный лом предварительно
окисляли в печах в окись меди, которую затем перерабатывали в
медный купорос.
В настоящее время медный лом перерабатывают в медный ку-
купорос только методом «натравки», а производство медного купо-
купороса растворением окиси меди в серной кислоте базируется на
окиси меди, получаемой из полупродуктов и отходов медеплавиль-
медеплавильных заводов.
Окись меди из белого матта
' Белый матт образуется при извлечении меди из сульфидных
руд в результате дальнейшей переработки штейна, состоящего из
сульфидов меди и железа и получающегося после первой плавки
сырья с отделением пустой породы. При добавке к штейну кварца
и продувке воздухом сульфид железа окисляется и переходит в
силикат. После удаления шлака остается полусернистая медь,
имеющая в изломе серебристый белый цвет, поэтому ее называют
белым металлом или белым маттом.
Белый матт получается в виде плит толщиной 6—8 см. Он со-
содержит, кроме Cu2S, до 10% металлической меди и 0,5—3% же-
железа; общее содержание меди 75—78%- Он служит для получения
черновой, а затем рафинированной меди.
Лол завода
Рис. 189. Обжиговая печь (разрез):
I — неподвижный под; 2 — подвижный под; 3 — гребки; 4 — верхний свод.
Для переработки на медный купорос белый матт измельчают и
подвергают обжигу в печах, с целью окисления сульфида в окись
меди. Для обжига используют печи разных конструкций. Разрез
одной из них показан на рис. 189. Печь имеет четыре пода, из ко-
которых два неподвижны, а два, находящиеся между ними, вра-
Щаются вокруг предполагаемой вертикальной оси, совпадающей
с осью печи. Все металлические части печи вынесены наружу. Под-
Подвижные поды 2 опираются на ролики и опоясаны зубчатыми
678
Гл., XVII: Соединения меди
Производство медного купороса из окиси меди
679
кольцами, с помощью которых приводятся во вращение. Поды вы-
выложены в форме пологих сводов из кислотоупорного кирпича. В
каждый свод при кладке печи вставляются гребки 3, расположен-
расположенные таким образом, что при вращении подвижных подов материал
перемещается по сводам от периферии к центру или в обратном на-
направлении и пересыпается со свода на свод. Разогрев печи про-
производится топочным или генераторным газом, поступающим под
нижний свод печи. Наиболее распространены печи с диаметром
4,5 м. Производительность печи составляет 6—7 т обожженного
матта в сутки при продолжительности пребывания материала в
печи 10—12 ч. Обжиг белого матта ведут с добавкой 1,75—2%
угля, обеспечивающего снижение температуры воспламенения суль-
сульфида меди, что ускоряет его окисление. Для предотвращения спе-
спекания в шихту добавляют до 15% измельченного «нагара». (На-
(Нагар — комочки спекшегося, плохо обожженного белого матта, отде-
отделяемые при просеивании обожженного матта). Температурный
режим в печи устанавливается за счет тепла, выделяющегося при
горении белого матта и угля. Обычно температуру поддерживают
в следующих пределах: 670—700° на первом поде (сверху), 740—
760° на втором, 650—675° на третьем и 450—475° на четвертом33.
При обжиге белый матт превращается в окись меди по реак-
реакции:
Cu2S + 2О2 = 2CuO + SO2
Небольшая доля сернистого газа, в связи с присутствием в бе-
белом матте железа, каталитически окисляется до БОз, который
сульфатизирует окись меди. Поэтому в продукте обжига белого
матта, помимо основного компонента — окиси меди, а также остат-
остатков сульфида, содержится некоторое количество CuSO4. С учетом
этого общая реакция окисления белого матта может быть записана
так48-49:
2Cu?S + 4,5О2 = 2CuO + CuO • CuSO4 + SO2
При недостатке кислорода или при плохом перемешивании мо-
может образоваться' некоторое количество, закиси меди:
2Cu2S + ЗО2 =' 2Cu2O + 2SO2
Закись меди растворяется в серной кислоте хуже, чем- окись,
поэтому наличие ее в обожженном матте (огарке) нежелательно.
Обожженный продукт содержит 87—90% СнО и 8—10% Cu2S или
70—72% Си и 2—2,5% S. В нем несколько меньше меди, чем
в исходном белом матте, что объясняется загрязнением продукта
нагаром и золой угля. Основная масса серы уходит из обжиговой
печи в виде сернистого газа, содержащего 1,5—2% SO2, 0,5—1%
СО2, 15—17% O2i имеющего температуру 250—300°.
Растворение окиси меди в серной кислоте
Продукт обжига белого матта, огарок — окись меди — просеи-
просеивают для отделения спекшихся комочков — «нагара» и подают в
варочный чан для растворения в серной кислоте (рис. 190) 33. От-
Отсеянный «нагар» после измельчения возвращают б печь, добавляя
его к идущему на обжиг белому матту. Варочный чан изготовляют
из андезитовых плит с внутренней свинцовой футеровкой. Исполь-
Используют также чаны из нержавеющей стали, выложенные кислото-
кислотоупорным кирпичом и футерованные внутри листовым свинцом тол-
Серноя
кислота д w
s~\ В атмосферу
1S •'
„Нагар напоВторнов
измельчение
Рис. 190. Схема производства медного купороса из белого матта:
1 и 3 —дробилки; 2 —шихта; 4 — элеватор; 5 —обжиговая печь; 6 — сита; 7 — электрический
тельфер; 8 — варочный чан; 9 — напорный бак; 10 — сборник; 11 — промежуточный бак; 12 — иасос;
13 — питательный бак; 14 — кристаллизатор; 15 — вентилятор; 16 — сборник пульпы; 17 — сборник
маточных растворов; 18 — центрифуга. ¦. ..
щиной 5 мм. Вначале в чан загружают маточный раствор, содер-
содержащий 28% сульфата меди, затем серную кислоту до получения
раствора с концентрацией 15—20% H2SO4. Массу подогревают до
кипения острым паром, подаваемым через опущенные в раствор
свинцовые трубы. В кипящий раствор загружают огарок неболь-
. шими порциями в течение 30—40 мин при перемешивании массы
острым паром. Растворение ведут до образования раствора, содер-
содержащего 43% CuSO4 и 3—4% H2SO4.
I Окись меди легко растворяется в серной кислоте. Содержа-
Содержащиеся в огарке металлическая медь и неокислившийся белый матт
(Cu2S) практически не растворяются в серной кислоте и образуют
нерастворимый шлам. В шлам частично переходит и плохо раство-
растворяющаяся в серной кислоте закись меди. По окончании варки от-
отстоявшийся раствор направляют на кристаллизацию. В зависимо-
зависимости от качества обжига белого матта очистку реакционного чана
от шлама производят или после каждой варки или после 3—4 ва-
варок. В сухом веществе шлама содержится ~50% меди, а также
некоторые количества золота и серебра, зависящие от содержания
их в исходной руде. Этот шлам возвращают для переработки на
медеплавильные заводы. • :
680
Гл. XVII. Соединения меди
Производство медного купороса ия окиси меди
681
Получение медного купороса из окиси меди и сернистого газа
Этот способ производства медного купороса50 является весьма
экономичным. Однако применение его целесообразно главным об-
образом в районах расположения медеплавильных заводов, где
имеется соответствующее сырье — окись меди и отбросный серни-
сернистый газ.
В связи с этим особый интерес приобретает получение медного
купороса из белого матта. При окислительном обжиге белый матт
превращается в окись меди. Выделяющийся при этом сернистый
газ рационально использовать для превращения полученной окиси
меди в медный купорос. Недостающее количество SO2 может быть
пополнено за счет сернистых газов медеплавильных печей. Таким
образом, белый матт может быть переработан на медный купорос
без затраты серной кислоты и с полным использованием его ком-
компонентов — меди и серы.
Способ производства медного купороса из окиси меди и серни-
сернистого газа основан на взаимодействии при 85—95° суспензии окиси
меди в водном растворе медного купороса со слабым сернистым
газом, содержащим SO2 и кислород. Отбросный сернистый газ, в
случае необходимости, должен разбавляться воздухом. Это уско-
ускоряет процесс, так как концентрация SO2 в газе не имеет сущест-
существенного значения, а увеличение содержания кислорода ускоряет
реакцию.
Образование медного купороса происходит в результате двух
независимо идущих процессов. Первый из них заключается в том,
что сернистый газ в присутствии каталитически действующих ионов
меди окисляется кислородом в серную кислоту:
2SO2 + О2 + 2Н2О = 2H2SO4
Образовавшаяся кислота растворяет окись меди, причем полу-
получается медный купорос:
H2SO4 + CuO = CuSO4 + Н2О
Второй, параллельно идущий процесс заключается в частичном
восстановлении сернистым газом двухвалентной (окисной) меди
в одновалентную (закисную) с образованием плохо растворимой
в воде соли Шевреля51 — комплексной окисно-закисной соли сер-
сернистой кислоты Cu(CuSO3J- 2Н2О или CuSO3-Cu2SO3-2Н2О:
3CuSO4 + 3H2SO3 + ЗН2О = CuSO3 • Cu2SO3 • 2Н2О + 4H2SO4
Эта соль в отсутствие кислорода при кипячении суспензии разла-
разлагается с выделением закиси меди:
3(CuSO3 • Cu2SO3 • 2Н2О) = CuSO4 + 2Cu,O + 5SO2
Однако под действием сернистого газа и кислорода в резуль-
результате дальнейшего образования серной кислоты закись меди снова
переходит в раствор, и осадок соли Шевреля постепенно исчезает
из суспензии, также превращаясь в медный купорос:
CuSO3 • Cu2SO3 • 2Н2О + SO2 + 2О2 = 3CuSO4 + 2Н2О
Окисление соли Шевреля при действии SO2 и О2 протекает с
образованием вначале основного сульфата меди:
2(CuSO3 • Cu2SO3 • 2Н2О) + ЗО2 = 2Си(ОНJ • CuSO4 + 3CuSO4 + 2Н2О
Эта реакция идет с большей скоростью, чем образование сер-
серной кислоты под каталитическим влиянием ионов меди. По мере
накопления H2SO4 основной сульфат меди переходит в раствор:
2Си(ОНJ • CuSO4 + 2H2SO4 = 3CuSO4 + 4Н2О
В результате этих процессов из суспензии исчезают все твер-
твердые фазы —и CuO и CuSO3-Cu2SO3-2H2O и 2Cu(OHJ-CuSO4 —
и суспензия превращается в раствор медного купороса. Таким об-
образом, в общем процессы сводятся к окислению четырехвалентной
серы (SO2) в шестивалентную и могут быть выражены суммарным
уравнением:
2CuO + 2SO2 + О2 = 2CuSO4
Растворимость соли Шевреля возрастает с повышением темпе-
температуры и содержания в растворе CuSO4. При 20° растворимость
этой соли в воде равна 0,042%, а при 60° — 0,14%. В 30% рас-
растворе CuSO4-5H2O при 20° растворимость повышается до 0,1%,
а при 60° — до 0,379%. Поэтому, будучи суспендирована в рас-
растворе медного купороса, комплексная соль окисляется быстрее,
чем в водной суспензии. Следовательно, для приготовления исход-
исходной суспензии окиси меди целесообразно брать не воду, а раствор
медного купороса.
Скорость окисления соли Шевреля возрастает с уменьшением
концентрации SO2 в газе. Последнее объясняется, вероятно, тем,
что в газовых смесях с высоким содержанием SO2 количество кис-
кислорода недостаточно для окисления. При содержании в газе 1—4%
SO2 и температуре 95° соль Шевреля окисляется полностью за
15—20 мин. Однако длительность процесса увеличивается за счет
времени, необходимого для предварительного растворения окиси
меди и образования соли Шевреля. При 95° и достаточном содер-
содержании кислорода в газе (при объемном отношении О2: SO2 > 4)
степень использования меди за 1 ч составляет 94—97%, а за 1,5 ч
больше 99%.
Тгхнологическая схема производства медного купороса этим
способом весьма проста. Окись меди суспендируют в маточном рас-
растворе, оставшемся после кристаллизации медного купороса, суспен-
суспензию нагревают до 85—95° и насыщают отбросным сернистым
682
• Гл. XVJI. Соединения меди
газом, разбавленным воздухом. Из полученного раствора при охла-
охлаждении до 20° кристаллизуется медный купорос. Кристаллы отжи-
отжимают на центрифуге, и маточный раствор возвращают в процесс.
ПОЛУЧЕНИЕ МЕДНОГО КУПОРОСА СУЛЬФАТИЗИРУЮЩИМ ОБЖИГОМ
БЕЛОГО МАТТА
Существенным недостатком способа получения медного купо-
купороса из белого матта путем его окислительного обжига и после-
последующего растворения полученной окиси меди в серной кислоте
является то, что основное количество серы, содержащейся в белом
матте, не используется. Между тем за счет этой серы теоретически
возможно было бы перевести в медный купорос 50% меди, находя-
находящейся в белом матте, и тем самым снизить в 2 раза расход серной
кислоты при последующей обработке продукта обжига. С этой
целью белый матт должен подвергаться не простому окислитель-
окислительному, а сульфатизирующему обжигу, т. е. длительной прокалке при
сравнительно невысоких температурах D00—500°) при достаточном
избытке кислорода. В этих условиях реакции
28О2 + О2 ^ 2SO3
CuO + SO3 ^=t CuSO4
смещены направо и 60—70% сульфидной серы переходят в суль-
сульфатную, что соответствует превращению 30—35% меди в сульфат
меди. Для обработки продукта обжига расходуется в 1,5 раза мень-
меньше серной кислоты, чем при простом окислительном (не сульфати*
зирующем) обжиге, а общее использование меди достигает 90%.
Механизм образования сульфата меди при сульфатизирующем
окислении белого матта можно представить следующими элемен-
элементарными реакциями 52. Часть сульфида непосредственно окисляется
в сульфат:
С и-2 3 + 2,5О2 = Си SO4 + CuO
Наряду с этим происходит окисление сульфида меди с образо-
образованием двуокиси серы и окиси меди:
Cu2S + 1,бО2 = Cu2O + SO2
Ж 2CuO
Окись меди далее реагирует с серным ангидридом, образую-
образующимся при каталитическом окислении SC>2 в присутствии содержа-
содержащейся в белом матте окиси железа, а частично аульфатизируется
по реакциям
CuO + SO2 = CuSO3
4CuSO3 = 3CuSO4 + CuS
с последующим окислением образующегося CuS. Для успешной
сульфатизации белого матта необходимо обеспечить достаточно
Получение медного купороса обжигом белого матта
683
высокую концентрацию кислорода в газовой фазе. Этого можно до-
достигнуть или использованием обогащенного кислородом воздуха
или применением добавок, обладающих высоким равновесным дав-
давлением кислорода в температурных условиях обжига.
Степень перехода серы в газовую фазу в виде двуокиси серы
значительно возрастает при добавке окиси меди (рис. 191) 53. Зави-
Зависимость степени перехода сульфидной серы в сульфатную от тем-
температуры выражается кривой с максимумом (рис. 192), что объяс-
объясняется разницей в скоростях образования и разложения сульфата
too ¦
600
Рис. 191. Степень перехода серы
в газовую фазу при окислении
сульфида меди в зависимости от
температуры:
1 — без добавки CuO; 2 — при добавке
к сульфиду меди 10% CuO; 3 — то же
при 2596 CuO.
iOO 500 600 700
Темперотура,°С
Рис. 192. Влияние темпера-
температуры на переход Cu2S в
CuSO4 при разном содер-
содержании окиси меди в смеси.
Содержание CuO в смеси (в %)•
1-0; 2-10: 3-25; 4-50.
меди (и промежуточных продуктов) при различных температурах;
Степень окисления сульфида меди при 450° в течение 60 мин в от-
отсутствие добавки составляет 29,5%. С увеличением температуры
выше 450° в этих условиях степень перехода сульфидной серы в
сульфатную резко падает и при 750—800° практически равна нулю.
В присутствии добавок окиси меди увеличивается степень пере-
перехода сульфидной серы в сульфатную, а температура, отвечающая
максимуму сульфатообразования, сдвигается в сторону более вы-
высоких температур. При добавке 25% CuO в интервале 500—550°
за 60 мин 35—40% сульфидной серы переходит в сульфатную, а
общее количество окислившейся серы достигает 90—95% 58.
Наиболее интенсивно сульфатизирующий обжиг сульфидов и
окислов меди идет в кипящем слое, особенно при предварительном
мелком измельчении материала Б4>5Б.
684
Гл. XVII. Соединения меди
Производства медного купороса из колчеданных огарков
685
Изучена сульфашзация Cu2S крепкой серной кислотой. До
300° она идет по реакциям:
Cu2S + 2H2SO4 = CuS + CuSO4 + SO2 + 2H2O
CuS + 2H2SO4 = CuSO4 + SO2 + S + 2H2O
S + 2H2SO4 = 3SO2 + 2H2O
Наибольший выход CuSO4 достигается при 200°. При высоких
температурах частично улетучивается серная кислота и в резуль-
результате взаимодействия CuSO4 с Cu2S образуется Cu2SO4, а затем
Си2ОБ6.
Изучены условия превращения Cu2S в CuSO4 путем автоклав-
автоклавного выщелачивания белого матта слабой серной кислотой в при-
присутствии кислорода. При этом параллельно идут следующие реак-
реакции:
Cu2S + 2,5O2 + H2SO4 = 2CuSO4-l-H2O A)
Cu2S + O2 + 2H2SO4 = 2CuSO4 + S + 2H2O B)
Скорость растворения Cu2S пропорциональна давлению кисло-
кислорода в степени 0,5. Для преимущественного (на 93%) осуществле-
осуществления процесса по реакции A), т. е. с полным использованием серы
и с меньшим расходом серной кислоты, оптимальными условиями
являются: давление кислорода ~4 ат, концентрация серной кис-
кислоты 0,01 моль/л, температура 140°57.
Аналогичным способом можно получать медный купорос из
водной суспензии халькопиритного концентрата (Т: Ж = 1:3)
с добавкой СаО или СаСО3 для регулирования гидролиза образую-
образующегося сульфата железа и выделения H2SO468.
ПОЛУЧЕНИЕ МЕДНОГО КУПОРОСА ИЗ ОКИСЛЕННЫХ МЕДНЫХ РУД
Из общих запасов медных руд 10—15% составляют руды,
содержащие медь в окисленной форме. При незначительном содер-
содержании меди в руде извлечение металлической меди методами плав-
плавки оказывается зачастую неэкономичным. В таких случаях целесо-
целесообразно перерабатывать эти руды гидрометаллургическими мето-
методами, заключающимися в извлечении (выщелачивании) меди
каким-либо растворителем. Содержащаяся в окисленных рудах
окись меди хорошо растворяется в серной кислоте. Полученные
разбавленные растворы сульфата меди или выпаривают, или выде-
выделяют из них медь цементацией (стр.687). Из выпаренных раство-
растворов получают кристаллизацией медный купорос, а цементную медь
также Перерабатывают в медный купорос.
Выщелачивание измельченной руды 10—15%-ной серной кисло-
кислотой осуществляют в растворителе, снабженном мешалкой,- куда
предварительно заливают разбавленную кислоту, подогревают ее
глухим паром до 50—80°, а затем загружают руду в таком коли-
количестве, чтобы отношение Т : Ж в пульпе составляло 1 : 3. Раство-
Растворение ведут при перемешивании и 60—70° в течение 30—40 мин.
При содержании в руде 3—5% Си выщелачивание ведут 3—5%-
ной серной кислотой в течение ~ 2 ч.
Очистку раствора медного купороса перед его дальнейшей пе-
переработкой от примесей сульфатов железа, алюминия и других
можно осуществлять с помощью известняка и обожженной медной
руды, пропуская его затем через гравийный фильтр для отделения
образовавшихся осадков34> 5В-60.
ПРОИЗВОДСТВО МЕДНОГО КУПОРОСА ИЗ КОЛЧЕДАННЫХ ОГАРКОВ
Медь в колчеданных огарках содержится в виде различных
соединений — CuSO4, CuSOs, Cu2O, CuO, Cu2S, CuS, CuFeS2. Это
затрудняет полное ее извлечение каким-либо одним простым ме-
методом.
Сульфат и сульфит меди легко выщелачиваются водой, а окись
меди — разбавленной серной кислотой. Сульфиды меди, в виде
которых находится до 20% меди огарков, не растворяются в воде
и в растворах серной кислоты, но переходят в раствор при нагре-
нагревании в результате взаимодействия с сульфатом (или хлоридом)
трехвалентного железа:
CuS + Fe2(SO4K = CuSO4 + 2FeSO4 + S
Cu2S + 2Fe2(SO4K = 2CuSO4 + 4FeSO4 + S
Этим же способом извлекают медь из сульфидных медных
руд61. Интенсивность выщелачивания увеличивается при бакте-
бактериальном окислении сульфидов 62. Из получаемого раствора мед-
медного купороса удаляют железо окислением Fe2+ в Fe3+ пиролюзи-
пиролюзитом или кислородом воздуха и обработкой известняком:
Fe2(SO4K + ЗСаСОз + ЗН2О = 2Fe(OHJ + 3CaSO4 + ЗСО2
Халькопирит с помощью сульфата железа растворить нельзя.
Его можно превратить в растворимые соединения хлорированием:
2CuFeS2 + 7С1г = 2CuCl2 + 2FeCl3 + 2S2C1jj
Наиболее разработанными способами получения медного купо-
купороса из огарков и являются водно-кислотное выщелачивание63,
применяемое на некоторых заводах, и хлорирующий обжиг в при-
присутствии NaCl.
Выщелачивание меди из огарка
При выщелачивании огарка 1% раствором серной кислоты на
холоду в течение 1 ч при отношении Т: Ж, равном 1 :3, переходит
в раствор от 60 до 80% меди. В результате шестикратного выще-
выщелачивания могут быть получены щелоки, содержащие 15—18 г/а
685
Гл. XVII. Соединения меди
меди. Эти щелоки загрязнены соединениями железа E—6 г/л Fe),
которые удаляются из раствора осаждением мелом, после их окис-
окисления бертолетовой солью или хлором. Выпариванием очищенного
щелока до концентрации меди 82 г/л и последующей кристаллиза-
кристаллизацией можно получить медный купорос. Без предварительной очи-
очистки щелока от железа получается продукт, содержащий 2—2,5%
Fe. Непосредственное получение фунгицидных препаратов из ще-
щелока может оказаться более экономичным, чем переработка его
на медный купорос64.
Извлечение водо- и кислоторастворимых соединений меди из
огарка можно осуществлять, орошая свалку огарка подкисленной
водой C—5% H2SO4). Для сбора образующегося раствора, содер-
содержащего 5—15 г/л CuSO4-5H2O, необходим колодец, откуда рас-
раствор откачивается на цементацию65. Аналогичным образом можно
производить выщелачивание огарка, находящегося в плоских пря-
прямоугольных ямах, имеющих большую площадь. Загрузку и вы-
выгрузку огарка можно осуществлять при этом с помощью мостового
крана, движущегося по опорам, уложенным вдоль краев ямы.
Хлорирующий обжиг огарка
Хлорирующий обжиг огарка заключается в обработке его хло-
хлористым натрием при высокой температуре с целью превращения
нерастворимых сульфидов меди в водорастворимые соединения66:
CuS + 2NaCl + 2O2 = CuCl2 + Na2SO4
К огарку добавляют 4—5% NaCl (в зависимости от содержа-
содержания в нем меди), смесь измельчают на вальцовой мельнице до
размера частиц не более 2 'мм и обжигают в механической колче-
колчеданной печи при 550—600°. Развитие требуемой температуры обес-
обеспечивается сжиганием генераторного газа или другого горючего.
В качестве горючего можно применять серу или серный колчедан.
В этом случае шихту составляют с соблюдением молярного отно-
отношения Си : 3S : 6NaCl. Огарок состоит из окислов железа и
CuCl2, CuCl, CuO, CuSO4 и Na2SO4. Часть хлорида натрия реаги-
реагирует с серой и водяными парами, образуя хлористый водород. В
отходящих газах, помимо продуктов сгорания топлива и хлори-
хлористого водорода, содержится некоторое количество Cl2, SO2, SO3,
водяных паров и мышьяковистых соединений. Газы из печи посту-
поступают в башню, орошаемую водой. Здесь получается 10%-ная со-
соляная кислота с примесью серной кислоты и хлора. Ее используют
для выщелачивания обожженного огарка.
Медноколчеданные огарки, содержащие 0,5—1% меди, можно
хлорировать газообразным хлором67. Систематическое выщелачи-
выщелачивание растворимых соединений меди из обожженного огарка ведут
при 20—25°. Общая продолжительность выщелачивания состав-
составляет 4—5 дней36,
Производство медного купоросд из колчеданных огарков
687
Извлечение меди из отходов медеплавильных заводов-
К отходам производства меди относятся: ватержакетная пыль,
шлаки, рудничные отвалы и др. Эти материалы содержат, как и
колчеданные огарки, различные соединения меди и перерабаты-
перерабатываются аналогичными методами — непосредственным выщелачива-
выщелачиванием или выщелачиванием после сульфатизирующего или хлори-
хлорирующего обжига. Полученные растворы подвергают цементации.
При наличии отбросного тепла их можно выпаривать для кристал-
кристаллизации из них медного купороса.
Водное выщелачивание ватержакетной пыли при 76—80° позво-
позволяет извлечь из нее в течение 4 ч от 20 до 70% меди в раствор
в виде сульфата меди. При последующей обработке остатков от
выщелачивания водой в течение 4 ч 4%-ной серной кислотой в рас-
раствор переходит еще 15—30% меди68.
Выщелачиванием шлаков 5%-ной серной кислотой при 90—
95° и Т : Ж, равном 1 : 8, с продувкой воздухом удается извлечь
в раствор только 50—53% меди. Путем предварительного сульфа-
сульфатизирующего обжига шлака в смеси с 50% -ной серной кислотой
при Т : Ж, равном 1 : 1, и 600° в течение 1 ч можно увеличить из-
извлечение меди в раствор до 80% 69.
В рудничных отвалах обычно содержится 1—2% меди. Она мо-
может быть извлечена в раствор теми же методами, как и из других
отходов, а также обработкой водной пульпы сернистым газом *.
Степень извлечения меди достигает при этом 98% в течение 1 ч,
если предварительно произвести хлорирующий обжиг материала
при 300°, с добавкой к нему 20% NaCl.
Выделение меди из разбавленных растворов
При извлечении меди из колчеданных огарков, отходов меде-
медеплавильных заводов, рудничных отвалов, а также из окисленных
• медных руд получаются разбавленные растворы медного купороса
(или хлорной меди). Рудничные воды, образующиеся на медных
рудниках в результате медленного окисления сернистой меди кис-
кислородом воздуха, также представляют собой слабый раствор мед-
медного купороса. Так как концентрирование таких слабых растворов
не экономично, медь выделяют из них цементацией70-71. Этот про-
процесс заключается в вытеснении меди из растворов железными
стружками и железным ломом:
Cu2+ + Fe = Fe2+ + Си
Электродный потенциал меди значительно выше, чем железа —
в М растворах, содержащих ионы Си2+ или Fe^+, при обычной
температуре и давлении водорода 1 ат он равен для Си +0,34 в,
Для Fe —0,44 в. Поэтому железо вытесняет медь из 'раствора в виде
Тонкого металлического шлама, называемого цементной медью.
683
Гл. XVII. Соединения меди
Цементацию осуществляют в стальном футерованном или освин-
освинцованном баке, куда загружают очищенный от грязи и ржавчины
железный лом. Затем в бак подают разбавленный раствор суль-
сульфата меди. Для полноты осаждения меди раствор ке должен
содержать значительных количеств серной кислоты. Оптималь-
Оптимальная концентрация серной кислоты равна ~0,05% или около
5 • 1(Н г-мол/л 72. При такой кислотности практически не происхо-
происходит растворения железа серной кислотой и обеспечивается наибо-
наиболее полное удаление меди из раствора, до содержания Си2+
~5 • 10~6 г-ионов/л73.
Образующийся в результате цементации разбавленный раствор
сульфата железа спускают в канализацию, а в реактор заливают
другую порцию исходного раствора, содержащего медь. Обработку
бдной и той же загрузки железа проводят 10—12 раз. После этого
оставшееся железо удаляют и выгружают осевшую на дно цемент-
цементную медь, которую затем промывают от частиц железа 10—15%-
вой серной кислотой при непрерывном перемешивании. По удале-
йии железа медь промывают водой до полной отмывки от серной
кислоты. Промытая цементная медь получается в виде пасты крас-
красновато-бурого цвета; она содержит 65—70% Си, до 35% влаги и
около 1% примесей и перерабатывается в медный купорос теми же
методами, что и медный лом. Дисперсность цементной меди возра-
возрастает с увеличением рН раствора и при уменьшении концентрации
в нем CuSO4 и С1~74. Цементацию меди можно осуществлять и в
псевдоожиженном слое железных гранул. Разработан способ из-
извлечения цементной меди флотацией78. Порошкообразную медь
можно получить из кислых растворов солей меди, добавляя к ним
растворимые в воде полисахариды (~1%) и обрабатывая газооб-
газообразным восстановителем под давлением, например, водородом при
30 ат и 140°76.
Медь может быть извлечена из разбавленных растворов C11SO4
обработкой их слабой аммиачной водой. При этом образуется оса-
осадок Cu(OHJ'CuSO4, который после отделения от раствора можно
растворить на фильтре серной кислотой для получения медного
купороса. Если в растворе присутствуют, кроме меди, ионы железа
и никеля (например, при переработке полиметаллических руд),
возможно ступенчатое осаждение их аммиаком при нейтрализации
раствора последовательно до рН = 3, затем 4,5 и б77-78.
Разработаны методы извлечения меди из разбавленных рас-
растворов экстракцией органическими растворителями79"*1.
ПОЛУЧЕНИЕ МЕДНОГО КУПОРОСА ИЗ ЭЛЕКТРОЛИТНЫХ РАСТВОРОВ
МЕДЕЭЛЕКТРОЛИТНЫХ ЗАВОДОВ
При электрохимическом рафинировании меди применяют-элект-
применяют-электролит, содержащий в 1 л 30—45 г меди в виде сульфата и около
200 г свободной серной кислоты. Помимо этого, в электролите лрн-
Получение других соединений меди
689
сутствуют примеси NiSO4, FeSO4, As^O3, ZnSO4, CaSO4 и др.
В связи с накоплением этих примесей и переходом в раствор меди
часть электролита должна выводиться из процесса. Наиболее эко-
экономичным способом утилизации выводимого из цикла электролит-
электролитного раствора является переработка его на медный купорос, так
. как стоимость последнего выше, чем стоимость затраченных на
его производство меди и серной кислоты82.
Поэтому находящуюся в выведенном электролите серную кис-,
лоту нейтрализуют материалами, содержащими медь, — катодным
скрапом, стружкой, гранулированной катодной и анодной медью,
шлаком анодных печей и т. п. Нейтрализацию производят при цир-
циркуляции раствора через слой материала, содержащего медь, загру-
загруженного в резервуары, называемые окислителями. В них вдувают
воздух, требующийся для окисления меди в процессе ее растворе-
растворения в серной кислоте. Здесь происходит тот же процесс, что и опи-
описанный выше, идущий в натравочных башнях. Температуру рас-
раствора поддерживают около 70—80° нагреванием через паровые
змеевики или острым паром. После снижения содержания свобод-
свободной кислоты в растворе до 0,5%, для чего обычно требуется от 12
до 24 ч, раствор поступает на выпаривание, причем из него ча-
частично выделяются соли железа и кальция. Затем раствор охла-
охлаждается в кристаллизаторах, где из него выделяются кристаллы
медного купороса. Из оставшегося маточного раствора извлекают
никелевый купорос, загрязненный сульфатом меди.
Содержащиеся в меди примеси при рафинировании переходят
в шлам, в котором находятся значительные количества меди. Из-
Извлечение меди из шлама производят выщелачиванием серной кис-
кислотой при 85° с продувкой воздухом. Несмотря на длительность
этой операции (до 18 ч), в шламе остается 8—10% меди. Для уско-
ускорения этого процесса предложено применять в качестве интенсив-
интенсивного окислителя меди персульфат аммония83.
ПОЛУЧЕНИЕ ДРУГИХ СОЕДИНЕНИИ МЕДИ
I Основной сульфат меди
Основной сульфат меди можно применять в качестве фунгицида
вместо бордосской жидкости. Так как он не растворим в воде, то
для опрыскивания растений его используют в виде водных суспен-
суспензий. Для получения смачивающегося порошка к продукту добав-
добавляют порошкообразный сульфитный щелок, а для удерживания
его на листьях — декстрин.
Основной сульфат меди получают осаждением содой и мелом
из раствора медного купороса по реакциям:
4CuSO4 + 3NatCO3 + C + m)H20 = 3Cu(OHJ • CuSO4 • mHsO + 3Na2SO4 + 3CO»
4CuSO4 + 3CaCCV+ (9 + m) H»O
690
Гл.' XVII. Соединения меди
Регенерация солей меди .
691
Мел дешевле соды, но при осаждении мелом образующийся
нерастворимый сульфат кальция разбавляет продукт. Поэтому для
получения препарата, содержащего 27—29% меди, необходимо 4б%
меди из раствора CuSO4 осадить содой, а остальные 60% — моло-
молотым мелом. Осаждение производят при 80—85° из раствора, содер-
содержащего ~ 160 г/л CuSO4 и 5—10 г/л H2SO4. Отфильтрованный оса-
осадок с влажностью 25—35% поступает в барабанную сушилку, где
высушивается топочными газами при 150° до влажности 3%. Затем
в" шаровой мельнице производят его размол и смешение с сульфит-
сульфитным щелоком и декстрином 84'8Б.
Запатентован способ получения основного сульфата меди
CuSO4 • 3Cu(OHJ- 5Н2О медленным осаждением из раствора, со-
содержащего 50—60 г/л CuSO4, аммиаком или аммиачной водой. При
рН в конце процесса 7—8 выход меди в осадок 100%- Отфильтро-
Отфильтрованный и промытый осадок высушивают при температуре ниже
150°. Гранулометрический состав продукта зависит от концентра-
концентрации реагентов и продолжительности осаждения. Примером может
служить продукт, в котором 80% частиц меньше 5 мк, 18% —от 5
до 8 Мк и 2%—больше 8л1К86.
Основные карбонаты меди
Основные карбонаты меди получают взаимодействием раство-
растворов медного купороса и соды. В зависимости от температуры и
другиК условий осаждения образуется или голубая соль
2СиСО3 • Си (ОН) 2 или зеленая СиСО3 • Си (ОН2). При смешении
растворов вначале выделяется студенистый осадок голубого цвета,
переходящий затем в кристаллический, имеющий состав СиСО3 •
• Си(ОНJ -х НгО, зеленого цвета. Для получения продукта такого
состава исходные растворы с концентрацией ~1 м должны сме-
смешиваться в отношении Ыа2СОз: CuSO4, равном 1,1:1; реакцион-
реакционная масса должна умеренно перемешиваться в течение длитель-
длительного времени для старения осадка и перехода студня в кристалли-
кристаллическое СОСТОЯНИе 63- 87-89
Хлорокись меди
Изразличных основных соединений хлорной меди — оксихлори-
дов (стр. 662) наибольшее значение имеет соль ЗСиО • СиС12 - 4Н2О
или ЗСи(ОНJ • СиС12 • Н2О, которая известна под названием хлор-
окиси меди90'91. Ее получают осаждением из растворов хлорной
меди известковым молоком или мелом:
. 4СиС12 + ЗСа(ОНJ + Н2О = ЗСиО • СиС12 • 4Н2О + ЗСаС12
ИЛИ
4СиС12 + ЗСаСОз + 4Н2О = ЗСиО • СиС12 • 4Н2О + ЗСаС12 + ЗСО2 .
Для получения хлорокиси меди могут быть использованы также
растворы хлористой меди, которые окисляют воздухом при 45—50е,
По этому способу получается соединение СиО • СиСЬ или
ЗСиО • СиСЬ • пН2О (в зависимости от длительности окисления 92).
Процесс может быть осуществлен путем взаимодействия металлу
ческой меди с хлорной медью при продувке воздухом (стр. 676).
В частности для этой цели могут быть использованы растворы, со-
содержащие медь, полученные при выщелачивании хлорированных
пиритных огарков.
Хлорокись меди можно также получить взаимодействием ме-
металлической меди с водным раствором хлоридов кальция и аммо-
аммония, через который продувают воздух. Выделяющийся осадок хлор-
хлорокиси меди отделяют фильтрацией93.
Для переработки медного скрапа в хлорокись меди его обраба-
обрабатывают раствором CuSO4 и NaCl при 80°. Медь растворяется в ре-
результате восстановления Си2+ в Си+. Затем окислением раствора
воздухом при 30—40° осаждают основной хлорид меди. При этом
содержание меди в растворе уменьшается с 60 до 2 г/л 94.
Закись меди
Закись меди получают электролизом раствора поваренной соли
с медными катодами и анодами95. На катоде выделяется водород,
образуются ионы ОН~, на аноде медь растворяется с образованием
ионов Си+. В результате вторичного взаимодействия
2Си+ + 2ОНГ = Си2О + Н2О
осаждается порошкообразная безводная закись меди. Процесс ве-
ведут при температуре не ниже 70° во избежание образования гид-
гидрата закиси меди. Напряжение на ванне ~4 в, катодная и анод-
анодная плотности тока 200 с/ж2. Выход по току 99%, расход энергии
1600 кет- ч на 1 т закиси меди. Материал электролизера — дерево.
Закись меди промывают водой, затем спиртом и высушивают
• в вакууме.
/ РЕГЕНЕРАЦИЯ СОЛЕИ МЕДИ В ПРОИЗВОДСТВЕ
ИСКУССТВЕННОГО ВОЛОКНА
При производстве медно-аммиачного штапельного волокна на
1 кг волокна расходуют 0,5—0,6 кг медного купороса (без учета
регенерации его). До 60% израсходованной меди извлекается из
волокна при отделочных операциях в виде раствора CuSO4, кото-
который используют для приготовления солей и гидроокиси меди. Около
7з меди удаляется в промышленную канализацию с раствором пря-
прядильной ванны.
Регенерация меди (и аммиака) может производиться двумя
способами:
ГА. XVII. Соединения меди
1) кипячением отработанного раствора прядильной ванны под
вакуумом — при этом медь выделяется в виде окиси меде, обра-
образующей медный шлам;
2) фильтрацией раствора через Н-ионитовый материал — в ре-
результате ионного обмена медь оседает на ионите, а раетвор на-
направляется на отгонку аммиака.
Первый способ, позволяющий одновременно регенерировать,
медь и аммиак, наиболее эффективен при переработке растворов
из прядильных ванн, второй — при извлечении меди из сточных и
промывных вод96.
Предложено извлекать медь из отработанного медно-аммиач-
ного раствора осаждением ее в виде основного сульфата меди
после подкисления раствора серной кислотой и доведения вели-
величины рН до 6—797.
ЛИТЕРАТУРА
I. J. Р. В a u r, D. W. В г i d g e s, W. M F a s s e 11, J. Electroetem. Soc, 103,
№ 5, 273 A956). — 2. A. Goswami, J. N. Trehan, Trans. Faraday Soc, 52»
№ 3, 358 A956).— 3. С Ott, Compt. rend., 238, № 10, 1111 A954).— 4. С. С. S i r-
car, B. Prasad, J. Indian Chem. Soc, 33, № 5, 361 A956).— 5. M. И. Архи-
Архипов, A. Б. Пакшвер, Н. И. Под бор нова, ЖПХ, 23, № 6, 650 A951).—
6. J. G a n t h i e r, Bull. Soc. chim. France, 1956, № 4, 661. — 7. E, Ш. Г а н с л и н а,
ЖПХ, 37, № 6, 1358 A964).—8. Д. Фрир, Химия инсектисидов и фунгисидов,
ИЛ, 1948. — 9. L. W а 11 е г - L ё v у, А.-М. G о г е о u d, Compt. rend., С265, № 28.
1314 A967). — 10. Е. В. К и с е л е б а, ЖФХ, 27, № 3, 443 A953).
II. W. Savelberg, Chem. Ztg., 77, № 10, 317 A953).—12. J. Ouzo, Chem.
zvesti, 11, №2, 107 A957).— 13. A. H. Кетов, В. В. П ечко век ий, Б. Н. Вар-
ский, Н. П. Старков, ЖПХ, 34, № 3, 517 A961). — 14. В. В. Печковский,
А. Н. Кетов, Труды Пермск. политехи, ин-та, № 10, 1961, стр. 3. — 15. L 6 г а п t
Be la, Z. analyt. Chem., 219, № 3, 256 A966). —16. С B. Frost, R A. Camp-
Campbell, Canad. J. Chem., 31, № 2, 107 A953). — 17. Я. А. Ф и а лко в, 3. А. Шек а,
ЖНХ, 1, № 6, 1238 A956). —18. R. Nasanen, Suomen kemisti lenti, 26,
№ 5—6, 67 A953). — /». S. Fronaens, Acta chem. Scand., 8, № 7, 1174 A954).—
20. H. Д. Мазовер, ЖПХ, 26, № 6, 612 A953).
21. С. Н. Иванова, Журн. ВХО, 9, № 5, 496 A964). — 22. В. И. Орлов,
Хим. пром., № 6, 377 A956).— 23. В. И. О р л о в, М. А. М о р о з о в а, М. Н. К р V-
тицкая, Журн. ВХО, 5, № 3, 270 (I960).— 24. R. L. Derby, D. W. Graham,
Proc. Am. Soc. Civil Eng., 79, № 203, 1 A953). — 25. Water Works Eng., 106,
№ 8, 709 A953). — 26. С. И. Вольфкович, Н. Н. Мельников, В. И. Ор-
Орлов, Хим. пром., № 6, 321 A954). — 27. М. В. Ката л ым о в, сб. «Исследования
по прикладной химии», 1955, стр. 325. — 28. Н. И. Хита ров, ЖХП. J4 1, 50
A937). — 29. А. И. Соловьев, Там же, № 8, 779 A935). — 30. В. Г. Хомя-
Хомяков, В. П. М а ш о в е ц, Л. Л. Кузьмин, Технология электрохимических про-
производств, Госхимиздат, 1949.
31. М. Р о u r b a i х, Thermodynamics of Dylute Aqueous Solutions, London,
1950. — 32. H. H. Милютин, ЖПХ, 34, № 4, 848 A961). — 33. И. М. Вассер-
м а н, Производство минеральных солей, Госхимиздат, 1954. — 34. А. И. К и н е в-
ский, ЖПХ, 28, № 10, 1088, 1113 A955). — 35. П. В. Дыбина, Технология
минеральных солей, Госхимиздат, 1949. — 36. А. И. Киневский, ЖПХ, 33,
№ 3, 608 (I960). — 37. Б. Д. Степии, И. А. Гильденблат, С. А. Грин-
штейн, Хим. пром., № 3, 175 A957).— 38. Б. Д. Степин, Там же М 6, 376
A954).— 39. Н. Eckstein, A. Lissner, Chem. Technik, 6, № 1, №* A954).—
40, А. Ф. Горбатова, Б. С. Кавиатская, Хим. пром., Ке 11, 344 A949),
1
Литература
693
41. Авт. свид. 149412, 1962. — 42. Л. М. М а ту се в и ч, Цветные металлы, 31,
№ 9. 32 A958); 32, № 11, 37 A959); 33, № 9, 4б (I960). — 43. Б. Д. Степин,
И А. Гильденблат, С. А. Гринштейн, Хим. пром., № 5, 303 A956).—
44. R. Stratta, Chim. Ind., 21, № 11, 612 A939).— 45. Fogel, Chem. Ztg., 81.
K» 21, 705 A957). —46. Б. А. Скопинцев, М. Т. Голубев а, ЖПХ, 12, № 6,
813 A939).— 47. В. В. Михайлов, ЖХП, № 4—5, 91 A940).— 48. H. П.Диев,
Ю В. Карякин, ЖПХ, 12, № 5, 655 A939).— 49. Д. М. Чижиков,
Г. С. Френц, Б. Я. Трацевицкая, Изв. АН СССР ОТН, № 4, 523 A953).—
50. Я. М. Песин, М. Л. Шабашова, ЖПХ, 23, 278, 350, 461 A950).
51. Е. П. Ожигов, Сообщ. Дальиевост. фил. АН СССР, № 6, 10 A954).—
52. В. И. Смирнов, А. И. Тихонов, Обжиг медных руд и концентратов, Гес-
металлургиздат, 1958. — 53. В. В. Печковский, ЖПХ, № 8, 802 A955); 32.
№ 8, 1857 A959). — 54. А. Бабенко, В. Смирнов, Труды Уральск, политехи,
ин-та, № 73, 1958, стр. 259.-55. Кришна Мурти, J. Sci. Ind. Res. (India),
20D, № 5, 175 A961). —56. А. П. Сиурников, В. Ф. Ларин, сб. «Исследова-
«Исследования в области химии и технологии минеральных солей и окислов», Изд. «Наука»,
1965, стр. 161. — 57. Г. Н. Доброхотов, Е. В. Майорова, ЖПХ, 36, № 10,
2148 A963).— 58. Авт. свид 158871, 1963. — 59. Т. В. 3 а б о л о т ски й, С. М. Б е-
зяйко, ЖХП, № 22, 18 A941).— 60. Т. В. Заболотский, СМ. Безяйко,
Там же, № 29, 13 A941).
61. Н. Veltman, S. Pellegrini, V. N. MacKiw, J. Metals, 19, № 2,
2! П967). — 62. F. H. Fitsh, С J. V. Da vies, Sulphur, № 61, 19 A965).—
63. И. Н. Померанцев, Д. Э. Соркииа, Труды НИУИФ, в. 123, 1935,
стр. 144. — 64. К. Sun gun, Demir ve celik, 4, № 2, 25 A955). — 65. П. В. Ды-
Дыбина, ЖХП, № 12, 42 A938). — 66. С. А. Амирова, А. М. Поляк, Труды
УНИХИМ, в. 7, 1958, стр. 217.— 67. Д. М. Чижиков, Ш. Ш Марголина,
ЖХП, № 8, 811 A935). — 68. Н. Н. Ефремов, М. М. Наркевич, сб. «Ра-
«Работы по химической технологии минеральных веществ», Свердловск, 1929,
стр. 125. —69. П. В. Дыбина, Хим. пром., № 3, 79 A947); № 9, 276 A949);
№ 12, 367 A950). — 70. Mining Eng., 18, № 6, 70 A966).
71. А. И. Г о д е с, В. Н. Пантелеев, В. А. П е х о в и ч, сб. «Порошко-
«Порошковая металлургия», Рига, 1958, стр. 58. — 72. И. Н. П л а к с и н, Н. А. Суворов-
Суворовская, Цветные металлы, № 3, 37 A948). — 73. И. Л. Агафонов, ЖПХ, 29,
№ 2, 295 A956). — 74. В. Д. Самыгин и др., Цветные металлы, № 12 36
A968). — 75. С. М. Гуревич и др., авт. свид. 192708, 1963.— 76. Япон. пат
5650, 1964. — 77. Н. S u w а. К- М i h а г а, япон. пат. 5175, 1953. — 78. М у к о я м а,
япон. пат. 854, 1955. — 79. D. W. Agers a. oth., Mining Eng., 17, № 12, 76
A965). — 80. Япон. пат. 9674, 1962.
81. М. Olteanu, J. Mo 1 do van, Rev. chim. (рум.) 17, № 1, 20 A966).—
82. В. А. Аюлицкий, А. К. Шарова, ЖХП, № 8, 591 (937).— 83. А. Г. Лот-
Лотка рев, В. С. Калеватова, ЖПХ, 21, № 9, 954 A948). — 84. М. Моро-
Морозова, Н. Кольцов, Н. Трушвина, Е. Лазарева, Сборник работ НИУИФ
в. 167, I960, стр. 151. —J^. С. Decroly, M. Ghodsi, Ind. chim. belg., 82, № 3,
265 A967).— 86. Франц. пат. 1124612, 1956. — 87. С. Т. Hsu, J. Appl. Chem., 6,
№ 2, 84 A956). —вв. В. И. Орлов, П. В. Попов, М. А. Морозова,
М. Г. Габриелова, Ядохимикаты (иисектисиды и фунгисиды), Госхимиздат,
1946. — 89. А. Л. Ефимов, Химические методы борьбы с вредителями и болез-
болезнями сельскохозяйственных растений, Сельхозгиз, 1949. — 90. М. А. Морозова,
Н. С. К о л ь ц о в, Сборник работ НИУИФ, в. 167, 1960, стр. 133.
91. М. Ф. Зубов, Там же, стр. 146.— 92. D. Lazarov, G. Bliznakov,
Z. phys. Chem. (DDR), 233, № 3, 4, 255 A966). —S3. P. K- Anderson, пат,
США 2655432, 1953. — 94. E. Irmisch, пат. ГДР 34170, 1964. — 95. H. П. Фе-
Д о т ь е в и др., Прикладная электрохимия, Госхимиздат, 1962, стр. 462. —
*& П. В. Михайлов, Л. Е. О к у н е в, Вестн. техн. и экон. информ., № 5, 34
A958). — 97. Е. М и п е k a t а, англ. пат. 728520, 1955.
Физико-химические свойства
30
[го
Глава XVIII
СОЕДИНЕНИЯ ЖЕЛЕЗА
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Сульфат закиси железа, ферросульфат, FeSO4 в пределах от
— 1,82 до 90° кристаллизуется из водных растворов в форме
FeSO4 • 7Н2О, FeSO4-4H2O и FeSO4 • Н2О (рис. 193) \ Семиводный
сульфат закиси железа, называемый в технике железным (зеле-
(зеленым) купоросом, кристаллизуется в интервале от —1,82 до 56,8°
в форме моноклинных кристаллов
голубовато-зеленого цвета с плот-
плотностью 1,899 г /см3 A5°). На возду-
воздухе выветривается, окрашиваясь в
желтый цвет, вследствие образова-
образования с поверхности Fe(OH)SO4. Че-
тырехводный кристаллогидрат с
плотностью 2,2 г/смг, окрашенный в
зеленый цвет, стабилен от 56,8 до
64,0°; при других температурах он
может выделяться в метастабиль-
ном состоянии. Выше 64° кристал-
кристаллизуется FeSO4 • Н2О белого цвета.
При нагревании FeSO4 • 7Н2О он те-
теряет шесть молекул воды. Обез-
Обезвоживание FeSO4 •Нг'О сопровождается разложением с выделением
SO2 и образованием основного сульфата железа; прокаливание же
на воздухе сопровождается окислением Fe (II) в Fe (III) с образо-
образованием Fe2O(SO4J2. Концентрация насыщенных водных раство-
растворов: в криогидратной точке (—1,821°) — 14,91%, при 20° —21,01%,
при 56,7°—35,06%, при 64° —35,57%, при 90,1° — 27,15% FeSO4.
Давление диссоциации одноводного сульфата закиси железа
FeSO4 • Н2О =f± FeSO4 + Н2О составляет 0,4 мм рт. ст. при 50°,
0,8 мм рт. ст. при 70°, 3,8 мм рт. ст. при 90°. В обычных условиях
FeSO4
4НгО
Чо
го 40 so
Темперагпура,°С
ВО 100
Рис. 193. Политерма растворимости
в системе FeSO4—Н2О.
FeSO4 • Н2О полностью обезвоживается при 300°, частично разла-
разлагаясь при этом.
Растворимость сульфата закиси железа уменьшается в присут-
присутствии свободной серной кислоты, которая высаливает кристалло-
кристаллогидраты FeSO4 из растворов (рис. 194) •. Уменьшается также и пре-
предельная концентрация раствора, соответствующая точке перехода
FeSO4 • 7Н2О в FeSO4 • Н2О. В системе FeSO4—H2SO4—Н2О при 50°"
существуют области кристаллизации стабильных FeSO4'7H2O,.
5 Ю 15 20 25 30 35 40
H8SO4,%
Рис. 194. Растворимость FeSO4 • 7М2О в зависи-
зависимости от содерлЦЙия в растворе свободной
H2SC>4 при различных температурах.
FeS'O4-4H2O, FeSO4-H2O и FeSO4 и метастабильных
FeSO4 • 7Н2О и FeSO4'4H2O. При концентрациях H2SO4, лежащих
за стабильной областью существования гепта- и тетрагидрата, и»
пересыщенных растворов в первую очередь выделяется метаста-
бильный гептагидрат, который затем превращается в метастабиль-
ный тетрагидрат с большей скоростью, чем последний переходит
в стабильный моногидрат. При б6° гептагидрат не образуется, а
при 70 и 90° не выделяется также и тетрагидрат.
В интервале 50—90° при концентрации H2SO4 меньше 27,2%
температурный коэффициент растворимости FeSO4 • Н2О отрицате-
отрицателен, а при более высокой концентрации серной кислоты — положи-
. телен (в пределах 50—70° температурный коэффициент снова ста-
становится отрицательным при концентрациях выше 76% H2SO4).
При высоких температурах растворимость FeSO4 в серной кислоте
Резко уменьшается. Так, при 200—240° в растворе, содержащем
Гл. XVIII. Соединения железа
Физико-химические свойства
~91% HjSO4, находится всего 0,2—0,3% FeSO4. При этом FeSO4
частично переходит в Fe2(SO4K в результате реакции:
2FeSO4 + 2H2SO4 = Fe2(SO4K + SO2 + 2H2O
Растворимость Fe2(SO4)s в концентрированной серной кислоте
до 200° немногим превышает 0,1% 3.
Сульфат окиси железа, феррисульфат, Fe2(SO4K кристалли-
кристаллизуется из водных растворов в форме 12, 10, 9, 7, 6 и 3-водных кри-
кристаллогидратов. Десятиводный кристаллогидрат и безводная соль
встречаются в природе в виде минералов квенстетита и коквим-
сбита. Желтые кристаллы девятиводной соли Fe2(SO4K • 9Н2О
имеют плотность 2,1 г/см3. Ромбические кристаллы безводной соли
имеют плотность 3,1 г/см3 A8°). При 20° в 100 г воды может рас-
раствориться 440 г Fe2(SO4K • 9Н2О. В водных растворах сульфат
окиси железа сильно гидролизуется, особенно при повышенных
температурах. Он очень гигроскопичен, на воздухе расплывается.
С сульфатами щелочных металлов и аммония он образует двойные
соли M[Fe(SO4J]-12Н2О, из которых наиболее распространены
железные квасцы NH4[Fe(SO4J]- 12H2O.
Давление диссоциации сульфата окиси железа по реакции
Fe2(SO4h^Fe2O3 + 3SO3 равно при 614° 70 мм рт. ст., при 650°
149 мм рт. ст.
Давление диссоциации сульфата закиси железа по реакции
2FeSO4 ^ Fej,O3 + SO2 + SO3 равно при 614° 254 мм рт. ст., при
650° 600 мм рт. ст.
В отсутствие кислорода FeSO4 разлагается с достаточно боль-
большой интенсивностью и полнотой при 700°. В токе воздуха его раз-
разложение значительно ускоряется в присутствии восстановителей —
угля и пирита. При взаимодействии FeSO4 с FeS или FeS2 G00—
800°) образуются Fe3O4 и SO24. Смесь из 82,3% FeSO4 • Н2О, 15,2%
FeS2 и 2,5% угля разлагается в токе воздуха при 600° за 40—
60 мин на 98%- С увеличением содержания кислорода в газовой
фазе скорость разложения FeSO4 уменьшается в отсутствие вос-
восстановителей (колчедана, угля), но возрастает в их присутствии.
Это объясняется тем, что восстановители, сдвигая реакцию
SO3 +± SO2 + '/гОг вправо, ускоряют тем самым диссоциацию
FeSC^50. О термическом разложении сульфата закиси железа во
взвешенном слое см. и.
Сульфат окиси железа в присутствии углерода восстанавли-
восстанавливается до сульфата закиси железа
Fe2(SO4K + С - 2FeSO4 + СО2 + SO2
а сульфат закиси железа при не очень высоких температурах!
только частично разлагается до закиси железа:
2FeSO4 +• С = 2FeO + СО2 + 2SO2
Степень превращения Fej(SO4K в FeSO4 возрастает с увеличе-
увеличением температуры, длительности взаимодействия и поверхности
частиц угля. При 300° и диаметре частиц угля меньше 3 мм сте-
степень превращения за 60 мин составляет 53%, за 90 мин — 76%, за
120 мин — 90% 12.
На отличии свойств сульфатов железа (давление диссоциации,
растворимость) от сульфатов других металлов основано обогаще-
обогащение руд и концентратов в цветной металлургии сульфатиза-
цией 13- м. Материал, гранулированный с серной кислотой, подвер-
подвергают сульфатизирующему обжигу во взвешенном слое. Прн этом
железо переходит в нерастворимые соединения, и при последую-
последующем выщелачивании получают растворы, в которых отношение
Fe к Со, Си, Zn и другим ценным металлам в десятки раз меньше,
чем в исходных материалах.
В присутствии воздуха или водяного пара FeSO4 конвертирует
хлориды щелочных металлов в сульфаты 1В.
В присутствии кислорода в водном растворе происходит по-
постепенное окисление FeSO4. В присутствии серной кислоты и с по-
повышением ее концентрации скорость окисления ферросульфата
в феррисульфат значительно уменьшается16. Окисление FeSO4
ускоряется при пропускании через раствор газа, содержащего SO2
и О217. При этом происходит образование Fe2(SO4K:
2FeSO4 + O2 + SO2 = Fe2(SO4K
В свою очередь, Fe2(SO4K восстанавливается двуокисью серы
до FeSO4 с одновременным образованием свободной «ерной кис-
кислоты:
Fe2(SO4K + SO2 + 2Н2О = 2FeSO4
Образующийся по последней реакции Fe2+ вновь окисляется в
Fe3+. В зависимости от условий взаимодействие FeSO4 с SO2 мо-
может быть направлено в сторону образования Fe2(SO4K илиН25О4.
На этом основан каталитический метод окисления SO2 в водном
растворе в присутствии ионов железа. При 80°, концентрации SO2
в газе 3% (а также и при меньших концентрациях), отношении
SO2: Ог, равном 1 :4, и содержании Fe2+ в растворе 30 г/л можно
получать растворы, содержащие 20—25% H2SO4 13> 19.
В гидрометаллургии цинка окисление Fe2+ производят при его
концентрации в растворе всего ~2 г/л Fe2+ (в виде FeSO4) и при
рН « 5—5,2 в присутствии твердой фазы из продуктов гидролиза,
окисленных соединений железа. В этом случае значительную роль
,в процессе окисления Fe2T играет адсорбция кислорода осадками
окисных соединений железа20.
Запатентованы методы бактериального окисления солей двух-
двухвалентного железа в кислых растворах21.
Гл. XVIII. Соединения железа
Применение
693
В водных растворах ион Fe8+ может гидролизоваться пр сле-
следующим схемам22:
железо образует комплексы типа [FeClJ- и [FeCl6]2~. В солянокис-
солянокислых растворах, насыщенных хлоридом щелочного металла, ион
29
находится только в виде FeCl3 и FeCl4
Fe8+ + 2H2O ^rj
2Fe3+ + 2H2O ^r± Fe2(OH)|+ + 2H+
В'соответствии с этим из растворов солей трехвалентного же-
железа выделяются разные основные соли 23- 24.
Железо-аммонийные квасцы NH4Fe(SO4J* 12H2O — фиолето-
фиолетовые кристаллы октаэдрической формы с плотностью 1,71 г/см3.
Плавятся при 40°. Растворимость в 100 г воды равна при 25° 124 г,
при 100° 400 г.
Хлористое железо FeCl2 образуется в виде зеленовато-серых
гексагональных кристаллов с плотностью 2,98 г/см3; на воздухе,
они желтеют вследствие окисления. Плавится при 672°, кипит при
1026° G60 мм рт. ст.). Давление пара при 700° 10 мм рт. ст., при
842° 100 мм рт. ст. В паре при температуре кипения находятся
FeCl2 и Fe2Cl4, а выше 1500° только FeCl2. Образует кристалло-
кристаллогидраты с 1,2, 4 и 6 молекулами воды. Температура превращения
FeCl2-2H2O в FeCl2-4H2O (голубовато-зеленые моноклинные кри-
кристаллы с плотностью 1,93 г/см3) 76,5°, температура превращения
FeCl2-4H2O в FeCl2-6H2O равна 12,3°. В насыщенном водном рас-
растворе при 20° содержится 40,5%, при 117,5° (/J 50,3% FeCl2.
Водные растворы FeCl2 слабо гидролизованы. С хлоридами щелоч-
щелочных металлов FeCl2 образует три типа комплексов: M[FeCl3],
M2[FeCl4] и M4[FeCl6], например, KFeCl3¦ 2Н2О, K3Na{FeCl6].
Хлорное железо FeCl3 — сильно гигроскопичные коричневато-
черные кристаллы гексагональной формы с плотностью 2,8 г/см3.
Плавится при 309°, кипит при 319° G60 мм рт. ст.). Давление пара
при 194° 1 мм рт. ст., при 235,5° 10 мм рт. ст., при 272,5° 100 мм
рт. ст., при 298° 400 мм рт. ст. Пары при 400° состоят из Fe2Cl6, при
750°—из FeCl3. Выше 500° под разрежением FeCl3 разлагается на.
FeCl2 и хлор. Хлорное железо образует ряд сильно гигроскопиче-
гигроскопических кристаллогидратов, расплывающихся на воздухе, из которых
конгруэнтно плавятся коричнево-желтые кристаллы FeCl3 • 6Н2О
при 37°, FeCl3 • 3,5Н2О при 32,5°, FeCl3 • 2,5Н2О при 56°, FeCl3 -2H2O
при 73,5°. Ниже 0 ± 2е существует плавящийся инконгруэнтио
FeCl-j-ЮН2О25. Криогидратная точка водного раствора —5,5°. На-
Насыщенный водный раствор содержит при 20° 47,9% FeCl3.
В разбавленных растворах FeCl3 гидролизуется до Fe(OHJ+
(константа гидролиза при 20° равна 24,8- 10~4), а в очень разбав-
разбавленных (Ю-3—10 М) до Fe(OHJ ' . Из разбавленных и концен-
концентрированных растворов выделяются основные соли — оксихлориды,
например 7Ге2О3-РеС1з-21Н2О, 8Fe2O3-FeCl8- 16H2O28. Хлорное
Окислы железа в воде не растворимы. Закись железа FeO (вюс-
тит) имеет черный цвет и плотность 5,7 г/см3. Закись-окись железа
Fe3O4 (магнетит) также черного цвета, плотность 5,2 г/см3. Окись
железа Fe2O3 (гематит) красного цвета, плотность 5,24 г/см3. Тем-
Температура диссоциации FeO (р0 = 0,21 ат) равна 2500°. Давление
О» над FeO при 750° равно 1,602 • 10~1Г мм рт. ст., а при 950° —
1,49- 10~п мм рт. ст.
Окись железа диссоциирует при не очень высоких температурах
(] 350—1500°) с образованием твердых растворов Fe2O3—Fe3O4 и
Fe3O4. Давление О2 над Fe2O3 при 427° равно 2,241 • 10~26 мм рт. ст.
Закись и закись-окись железа при нагревании (в присутствии ноч-
духа) переходят в окись железа. Давление О2 над Fe3O4 при 725°
равно 4.25- 10~17 мм рт. ст.
Гидрат закиси железа Fe(OHJ имеет зеленоватый цвет и плот-
плотность 3,4 г/см3. Растворимость при 18° 0.00015 г в 100 г воды. Рас-
Растворяется в кислотах и в растворе NH4C1. При 150—9пп° пола-
полагается30-31:
4Fe(Om2 = Fe + Fe3O4 + 4Н2О
Гидрат окиси железа Fe(OHK красно-коричневого цвета, плот-
плотность 3,4—3,9 г/см*. При нагревании до 500° отщепляет 1,5 Н2О.
Растворимость при 18° 0,000048 г в 100 г воды.
ПРИМЕНЕНИЕ
Железный купорос, являющийся контактным ядом, используют
в сельском хозяйстве для борьбы с вредителями садов и слизнями.
Его применяют также для уничтожения мхов, лишайников и гриб-
грибных спор, которые он убивает уже при концентрации 0,14%. По
своим фунгицидным свойствам железный купорос в 10 раз слабее
медного купороса32'33. Железный купорос используют и для пита-
питания растений. Железо необходимо растениям как катализатор для
образования хлорофилла. При недостатке железа растения забо-
заболевают хлорозом, и листья теряют зеленую окраску. Помимо этого,
железо входит в состав многих окислительных ферментов и играет
большую роль в дыхании растений.
Железный купорос применяют в текстильной промышленности,
для изготовления чернил, в фотографии и др. Наряду с окислами
желрза его используют для приготовления минеральных красок.
' Отбросные растворы сульфатов железа можно переработать в
изоляционный материал — феррон или ферри-гипс, представляю-
представляющий собой смесь гидратов окислов железя и гипса с наполнителем
^ материал пригоден для тепловой изоляции химической
Гл. XVIII. Соединения железа
Получение железного купороса ш травильных растворов 701
аппаратуры, из него могут быть изготовлены и строительные пли-
плиты 34. В США значительные количества сульфата окиси железа
применяют для травления металла; выпускаемый для этой цели
препарат «Феррисул» содержит 90% Fe2(SO4K и примеси — 0,5—
2% FeSO4, 3% Fe2O3 (нерастворимой), 2% SiO2, 1,5% A12(SO4K,
— 3% свободной H2SO4, 0,5% CaSO4 и др.35.
Сульфат окиси железа, а также хлорное железо используют в
качестве коагулянтов при очистке воды. Хлорное железо является
хорошим коагулянтом в щелочной среде. За рубежом его приме-
применяют также для очистки вод, содержащих серу, сульфиты и суль-
сульфиды, и как добавку при сушке осадков, отделяемых фильтрацией
при очистке канализационных вод. Применяемое для этой цели
хлорное железо выпускают под названием «перхлорид железа» в
баллонах в виде 41% раствора и разбавляют перед использова-
использованием36. Для очистки сточных аммиачных вод на небольших газо-
газовых заводах рекомендуют применять железный купорос37. Его же,
так же как и хлорное железо, можно использовать для очистки
промышленных газов от сероводорода с получением при этом эле-
элементарной серы 38. Сульфат окиси железа может применяться в ка-
качестве протравы при крашении шерсти и в качестве среды для обо-
обогащения многосернистых углей. Кислые растворы сульфата окиси
железа используют как окислительную среду при извлечении по-
полезных компонентов из руд39.
Окислы и гидраты окислов железа служат пигментами. Гидро- i
окись железа обладает сорбщюнными свойствами, которые исполь- '
зуются в гидрометаллургии при очистке растворов от примесей
некоторых катионов. Ферромагнитные окислы железа — магнетит,
у-окись железа, а также ферриты применяют в электротехнике, в
частности в производстве магнитных звуковых лент40.
Технический железный купорос, согласно ГОСТ 6981—54, дол-
должен содержать (в %):
Марка А Марка Б
FeSO4, не менее 53,0 47,0
Свободной серной кислоты,
не более 0,25 1,0
Нерастворимого остатка, не
более 0,4 1,0
Продукт упаковывают в деревянные бочки, фанерные барабаны
или деревянные ящики.
Аккумуляторный железный купорос, получаемый перекристал-
перекристаллизацией технического продукта, должен содержать не менее
19,5% железа и не более (в % от Fe): 0,1—Мп, 0,02 —Са, 0,03-
Сг, 0,03 — Ti, 0,02 —V, 0,02 —А1, 0,03 —Mg и 0,04 Си; сумма при-|
месей в продукте (без Мп и Си) не должна превышать 0,03%.
Хлорное железо безводное, получаемое хлорированием при вы-[
сокой температуре, выпускают 1 и 2 сорта. По ГОСТ 11159,-65,1
оии должны содержать соответственно не менее 97 и 95% b
и не более 1 и 2% FeCb; не растворимых в воде примесей в про-
продукте 1 сорта должно быть не более 2%. Тарой для хлорного же-
железа служат стальные барабаны, покрытые внутри лаком этиноль.
ПОЛУЧЕНИЕ ЖЕЛЕЗНОГО КУПОРОСА ИЗ ТРАВИЛЬНЫХ РАСТВОРОВ
Способ получения железного купороса специальным растворе-
растворением железа в серной кислоте в настоящее время утратил свое
значение в связи с тем, что на металлообрабатывающих заводах в
числе отходов производства имеются большие количества травиль-
травильных растворов, которые легко могут быть переработаны на желез-
железный купорос. Травильные растворы получают при обработке (тра-
(травлении) серной кислотой стальных изделий с целью удаления с
них окалины (окислов железа) перед дальнейшей обработкой по-
поверхности. Для травления применяют разбавленную 20—25%-ную
серную кислоту. Отбросные травильные растворы содержат глав-
главным образом железный купорос A5—20% FeSO4) и несколько
процентов свободной серной кислоты B—10%).
С развитием металлургии и металлообработки до современных
масштабов проблема утилизации отбросных травильных растворов
¦стала очень острой. Удаление больших количеств кислых жидко-
жидкостей в естественные водоемы невозможно. Очистка сточных тра-
травильных растворов нейтрализацией их известью представляет боль-
большие трудности вследствие образования объемистых трудно отстаи-
отстаивающихся осадков. Объем осадка сульфата кальция и гидрата
закиси железа в 20—25 раз превышает объем расходуемой на
травление металла серной кислоты (в расчете на моногидрат).
В связи с этим на многих заводах утилизируют травильные рас-
растворы, выделяя из них сульфат железа кристаллизацией с получе-
получением товарного продукта — железного купороса, при одновремен-
одновременном возврате остающейся маточной жидкости — серной кислоты в
травильные ванны. Однако потребность в железном купоросе пока
значительно меньше, чем количество его, которое можно получить
при полной утилизации травильных растворов, поэтому эта проб-
проблема остается нерешенной. Эти трудности однако постепенно пре-
преодолеваются. Описан способ обработки отбросного травильного
раствора водной суспензией Са(ОНJ при 90°, позволяющей полу-
получить быстро отстаивающийся осадок, состоящий из смеси Fe3O4
и гипса, которую разделяют магнитной сепарацией*'•42.
Железный купорос является также побочным продуктом при
, производстве двуокиси титана из титановых руд. На некоторых
заводах' за рубежом железный купорос используют в качестве
вторичного сырья для получения серной кислоты, примешивая его
(~20%) к поступающему на обжиг серному колчедану.-
702
Гл. XVIII. Соединения железа'
Получение железного купороса из травильных растворов
703
Сульфат железа содержится и в сточных жидкостях некоторых
других производств, например в отработанных красящих раство-
растворах, из которых он также может быть регенерирован43.
• Кристаллический железный купорос может быть выделен из
травильных растворов при охлаждении их до —5°, —10° или вы-
выпаркой с последующей кристаллизацией при охлаждении до 20—
25°. Можно также производить высаливание железного купороса
из травильной жидкости серной кислотой (см. рис. 194) и после
отделения кристаллического осадка возвращать маточный раствор
на травление железа.
Обычно выделение железного купороса из травильных раство-
растворов производят охлаждением их воздухом или водой, или в ваку-
вакуум-кристаллизационных установках. Предназначенную для трав-
травления металла серную кислоту часто используют при этом предва-
предварительно в качестве высаливающего средства. Вымораживание
железного купороса при охлаждении травильного раствора холо-
холодильным рассолом или выпаривание его с последующим вымора-
вымораживанием 44 являются менее экономичными способами.
Высаливание железного купороса можно также производить
ацетоном и бутиловым спиртом. При смешении 1 л ацетона с 1 л
травильного раствора можно выкристаллизовать до 85% желез-
железного купороса 45-46.
Заслуживает внимания предложение47 регенерировать травиль-
травильные растворы их нагреванием. При этом сульфат железа, раство-
растворимость которого с повышением температуры выше 64° умень-
уменьшается, выделяется в виде FeSCvH2O, а маточный раствор воз-
возвращается на травление. Из раствора, насыщенного FeSO4-H2O,
можно выделять это соединение в твердую фазу высаливанием,
добавляя при 65—85° FeSO4-7H2O48.
Если выделение железного купороса производится охлажде-
охлаждением отработанного травильного раствора, содержащего а%
FeSO4, а в остающемся маточном растворе b% FeSO4, то из
1000 кг травильного раствора будет получено 1,83хкг FeSO4-7H2O
или х кг FeSO4, причем
(а -6) -1000
100-1,836
IOjc
а степень извлечения FeSO4 из травильного раствора будет %.
Если производить охлаждение отработанного травильного рас-
раствора с одновременным введением в кристаллизатор серной кис-
кислоты в количестве m кг на 1000 кг травильного раствора, то:
_ (я-6)- 1000-fern
Х~ 100-1,836
Так; если отработанный травильный раствор, имеющий плот-
плотность 1,275 г(см3, содержащий 300 г/л или 23,5% FeSO4 и 1—2%
H2SO4, охладить до ~30°, он станет насыщенным (см. рис. 194,
табл. 46). При дальнейшем охлаждении до 3—4° концентрация
маточного раствора станет равной 14% FeSO4. При этом из 1 т
травильного раствора будет получено 234 кг FeSO4-7H2O, а сте-
степень извлечения FeSO4 составит 54%- Если же охладить раствор
только до 30°, но с одновременной добавкой в кристаллизатор
0,25 т 75%-ной серной кислоты на 1 т отработанного травильного
раствора для повышения кислотности маточного раствора до
~ 16% B00 г/л H2SO4), то из 1 г травильного раствора будет полу-
получено 75 кг FeSO4-7H2O, а степень извлечения FeSO4 составит в од-
одном цикле 17,5%. Но при высаливании серной кислотой с одно-
одновременным охлаждением до 3—4° выход железного купороса из
1 т травильного раствора составит ~290 кг, а степень извлечения
FeSO4 в одном цикле ~67%. При работе на замкнутом цикле,
т. е. с возвратом всего маточного раствора на травление, весь об-
образующийся сульфат железа может быть выделен в виде желез-,
ного,купороса.
ТАБЛИЦА 46
Растворимость FeSO4 в водных растворах серной кислоты
Темпера
0
5
10
20 '
25
30
Растворимоста
0
13,6
15,2
17,2
20,9
23,0
24,8
2
12,9
14.4
16,1
19,7
21,4
23,1
FeSO4 (в
4
12,0
13,5
15,2
18,4
20,4
22,0
%) при содержании
7
10,8
12,0
13,7
16,8
18,6
20,5
. 10
9,5
10,7
12,3
15,7
17,3
19,2
в растворе
15
7,2
8,0
10,1
13,3
15,0
16,9
H2SO4 (в
20
5,5
6,5
8,2
11,2
12,8
14,8
К)
' 35 "
3.4
4.7
6,6
9,6
11,4
13.2
Воздушное охлаждение травильного раствора, предварительно
отстоявшегося в течение 2—3 ч от окалины и других примесей, осу-
осуществляют иногда в установленных вне здания под навесом ба-
баках-кристаллизаторах 49. Кристаллы железного купороса оседают
на стенках кристаллизаторов и на опущенных в них щитах. Сте-
Степень извлечения FeSO4 составляет 48%. Перемешивание раствора
способствует его охлаждению и ускоряет кристаллизацию. Воздуш-
Воздушное охлаждение протекает медленно и может быть использовано
лишь на установках малой производительности. Поэтому во мно-
многих случаях осуществляют кристаллизацию с применением водя-
водяного охлаждения 50>51.
Вакуум-кристаллизационные установки
' Наиболее эффективным методом выделения сульфата железа
из травильных растворов является вакуум-кристаллизация50. При
остаточном давлении в кристаллизаторе 25—10 мм рт. ст. раствор,
704
Гл. XVIII. Соединения железа
Получение железного купороса из огарков
705
кипит при 5—10°. Охлаждение раствора достигается за счет его
самоиспарения. Вакуум создается в аппаратах при помощи паро-
пароструйных эжекторов, в которых смешиваются пар высокого и низ-
низкого давлений. Применением многоступенчатых эжекторов повы-
повышается их коэффициент полезного действия и значительно сокра-1
щается расход пара. Другим способом сокращения расхода пара ¦
является ступенчатое испа-
испарение раствора при разных
температурах.
В растворе, поступаю-
поступающем иа кристаллизацию из
травильных ванн с темпе-
температурой 63—65°, содер-
содержится ~ 10% H2SO4 и 18%
FeSO4. В кристаллизатор
добавляют серную кислоту
в количестве, необходимом
для получения маточного
раствора с концентрацией
— 23% H2SO4. Получающий-
Получающийся маточный раствор со-
со97% FSO
Рис. 195. Вакуум-кристаллизационная уста-
установка непрерывного действия:
1 — мерник отработанного травильного раствора5 ДерЖИТ 6,9—7% FeSO^ При
2-мерник серной кислоты; 3 - вакуум-кристалли; „ i _ пя-»ппЛ,РЛ,пй us
затор; 4-главный эжектор первой ступени сжатия- ЭТОМ на 1 1 расходуемой Hd
Л-главный конденсатор; 6, 7 и 8 - эжекторы вто- травление 76%-НОЙ СвОНОЙ
рой, третьей и четвертой ступеней сжатия; 9 — про г А —
межуточный сдвоенный конденсатор; /0-баро- КИСЛОТЫ ПОЛучаСГСЯ О,/ —
метрический сборник; И -водоотделитель; 12-кои- QC т -гпяпипкнпгп пягтпппя
деисационный горшок; 13 - иасос; М-сборник °'° ' ПРАВИЛЬНОГО pdCTBOpa,
кристаллической пульпы; /5 —центрифуга. ИЗ КОТОрОГО ИСПарЯвТСЯ При
кристаллизации 0,73 г воды.
Теоретический выход железного купороса на 1 г перерабатывае-
перерабатываемой кислоты равен 2,16 г. Вследствие потерь раствора с металлом,
с парами воды и других, практический выход составляет 1,8—2 г.
Вакуум-кристаллизационные установки периодического дейст-
действия позволяют выдерживать раствор в кристаллизаторе в течение
любого времени. Их используют в травильных цехах небольшой
производительности при расходе на травление металла до-2000 т
76%-ной серной кислоты в год. Каждый кристаллизатор рассчиты-
рассчитывают на производительность, соответствующую годовому потреб-
потреблению 500—750 т 76%)-ной серной кислоты. Расход пара на эжек-
цию составляет 0,825 г на 1 г железного купороса.
При непрерывном процессе вакуум-кристаллизации расход па-
пара значительно сокращается. Он зависит от числа корпусов уста-
установки. В четырехкорпусной установке он составляет ~0,5 т на
1 т железного купороса. Однако установки непрерывного действия
более «ложны в эксплуатации. На рис. 195 показана схема одной
из таких установок. Охлаждение раствора происходит в четырех
последовательно соединенных кристаллизаторах. Кристаллизаторы
снабжены якорными или пропеллерными мешалками. Аппараты
изготовляют из нержавеющей стали, их внутренняя поверхность
гуммируется для защиты от коррозии. Однако защитный слой ре-
резины сравнительно быстро изнашивается; эрозия вызывается пере-
перемешиванием.
Смесь отработанного травильного раствора и свежей серной
кислоты охлаждается в первом корпусе от 60 до 40°, во втором —
до 30°, в третьем — до 22° и в четвертом — до 10°. Пары из пер-
первого и второго корпусов поступают в главный конденсатор, разде-
разделенный по высоте на две части, работающие под разным давле-
давлением D5 и 34 мм рт. ст.), что позволяет эффективней использовать
охлаждающую воду. В нижнюю часть конденсатора входит пар
из первого корпуса, в верхнюю — из второго. Пары из третьего и
четвертого корпусов вначале сжимаются до 34 мм рт. ст. в первой
ступени сжатия в двух параллельно работающих эжекторах, а
затем идут в верхнюю часть главного конденсатора. Несконденси-
ровавшаяся часть пара, выходящая из основного конденсатора,
после сжатия эжекторами второй ступени направляется последо-
последовательно на промежуточную конденсацию, затем на сжатие в
третью ступень, на дополнительную конденсацию и, наконец, после
сжатия в четвертой ступени выбрасывается в атмосферу. Выходя-
Выходящая из четвертого корпуса кристаллизационной установки пульпа
поступает на центрифугирование для отделения железного купо-
купороса от маточного раствора.
За рубежом работают несколько установок, на которых выде-
выделенный из отработанных травильных растворов железный купорос
подвергается термохимическому разложению; получающийся сер-
сернистый газ перерабатывают в серную кислоту.
Отработанные травильные растворы предложено52-53 перераба-
перерабатывать в азотно-железистые удобрения аммонизацией их аммиа-
аммиаком или аммиачной водой. В результате реакции
FeSO4 + 2Н2О + 2NH3 = (NH4JSO4 + Fe(OHJ
получается суспензия, которая может содержать до 300 г/л суль-
сульфата аммония. Травильный раствор можно предварительно ис-
использовать для улавливания окислов азота из выхлопных ннтроз-
ных газов, а затем его аммонизировать.
Описан способ обработки кислого травильного раствора коксо-
коксовым газом, содержащим ЫНз- Ион железа при этом окисляется
кислородом с образованием магнетита, который отфильтровы-
отфильтровывают— остается раствор сульфата аммония54.
ПОЛУЧЕНИЕ ЖЕЛЕЗНОГО КУПОРОСА ИЗ КОЛЧЕДАННЫХ ОГАРКОВ
Для этой цели лучше использовать колчеданный огарок, охлаж-
охлажденный после обжига колчедана без доступа воздуха 4С, обладаю-
обладающий магнитными свойствами и содержащий некоторое количество
М. Е. flojHH
7C6
Гл. XVIII. Соединения железа
сульфида. Его примерный состав: 46% Fe2Os, 26% FeO и 10—16%
FeS. При обработке его серной кислотой образуются сульфаты за-
закиси и окиси железа:
FeO + H2SO4 = FeSO4 + Н2О
Fe2O3 + 3H2SO4 = Fe2(SO4K + 3H2O
Наряду с этим образующийся при разложении сульфида серо-
сероводород восстанавливает сульфат окиси железа:
Fe2(SO4K + H2S = 2FeSO4 + H2SO4 + S
При недостатке сульфида для восстановления сульфата окиси
железа применяют металлическое железо:
Fe2(SO4K + Fe =
Разложение огарка серной кислотой проводят в котле с мешал-
мешалкой в течение 5—10 мин. Перемешивание прекращают и массу вы-
выдерживают в реакторе еще 30 мин. За это время она превра-
превращается в твердый пористый продукт — частично оводненный суль-
сульфат закиси железа. Его растворяют в воде и раствор после
отстаивания направляют на кристаллизацию семиводного желез-
железного купороса. Для получения 1 т железного купороса расходуют:
0,3—0,35 т колчеданного огарка, 0,45—0,5 г серной кислоты A00%),
0,13—0,14 т металлического железа и 1,5 т пара.
Изучено взаимодействие сульфидов железа с серной кислотой.
Сульфатизация FeS с выделением H2S протекает бурно уже при
60—60°, сульфатизация FeS2 начинается при 200° и идет медленно
с выделением SO2 и S; для полного превращения FeS2 при 300°
необходим большой избыток кислоты55.
Железный купорос может быть получен из колчеданного огарка
сульфатизацией его в присутствии воды сернистым газом 56:
2Fe2O3 + 6H2SO3 + ЗО2 = 2Fe2(SO4K + 6Н2О
Fe2(SO4K + SO2 + 2Н2О = 2FeSO4 + 2H2SO4
СУЛЬФАТ ОКИСИ ЖЕЛЕЗА
Сульфат окиси железа получают обычно растворением окиси
железа в 76—80%-ной серной кислоте.
Растворы сульфата окиси железа могут быть получены окисле-
окислением серного колчедана азотной кислотой57-58:
2FeS2 + 10HNOS = Fe2(SO4K + H2SO4 + 4H2O + 10NO
Железный коагулянт (стр. 706) с низким содержанием суль-
сульфата закиси железа и свободной серной кислоты предложено по-
получать обработкой колчеданного огарка горячей серной кислотой.
Просеянны-й огарок предварительно должен быть обработан кон-
концентрированной азотной кислотой (в количестве 8—10 кг на, 1 г
Хлориды железа
70?
коагулянта). Окисленный огарок перемешивают и выдерживают
перед варкой коагулянта в течение 5—6 ч59.
Основной сульфат окиси железа может быть получен в про-
процессе переработки концентрата сульфидов металлов, состоящего
главным образом из сульфида железа и содержащего сульфиды
меди, никеля, кобальта. Водную пульпу концентрата с 10—40%
твердой фазы перемешивают 1—3 ч в автоклаве при 220—240° и
давлении 30—100 ат в присутствии кислорода. При этом сульфиды
Си, Ni, Co окисляются в растворимые сульфаты, а железо осаж-
осаждается в виде основного сульфата 60.
ХЛОРИДЫ ЖЕЛЕЗА
Самый простой способ получения хлоридов железа — это рас-
растворение металлического железа, закиси или окиси железа в соля-
соляной кислоте. Раствор FeCl2 получается также при травлении сталь-
стальных изделий соляной кислотой. Хлорное железо можно получить
из хлористого хлорированием суспензии или раствора FeCl2 или
окислением его кислородом воздуха. Получаемый раствор FeCU
выпаривают до концентрации, при которой он при остывании за-
затвердевает в кристаллический продукт FeClg • 6Н2О. Такой продукт
получается и в качестве отхода от некоторых производств, в част-
частности в производстве брома в процессе очистки бромо-воздушной
смеси от примеси хлора раствором бромида железа.
Прямое получение хлоридов железа утратило свое значение в
связи с развитием производства различных продуктов (TiCl4, AICI3
и др.) горячим хлорированием руд (см. гл. XL) с образованием
значительных количеств хлоридов железа, являющихся побочными
продуктами. В связи с ограниченностью потребности в них возни-
возникает необходимость дальнейшей их переработки с целью регене-.
рации хлора.
За границей все большее распространение получает изготовле-
"ние безводного хлорного железа FeCl3 горячим хлорированием
окиси железа или железной руды в присутствии восстановителя61.
Хлористое железо получается при обработке материала, содер-
содержащего окись железа, смесью равных объемов хлористого водо-
водорода и водорода. Температура твердой фазы повышается в печи
за счет тепла реакции от 260 до 650° 62> 63. Описано получение без-
безводного хлористого железа по реакции
2FeCl3 + С6Н,С1 = 2FeCl2 + СвН4С12 + НС1
при 140° в стальном реакторе (футерованном стеклом) с обратным
холодильником; в атмосфере азота выход 99,6% 64.
Получаемая при хлорировании никелевой руды в присутствии
восстановителя смесь FeCl2 и NiCl2 может быть разделена пропу-
пропусканием через металлическое железо при 1200—1300°. При этом
23*
T08
Гл. XVIII. Соединения железа
Получение окислов железа и их гидратов
709'
хлористый никель восстанавливается до металла и единственным
газообразным продуктом является хлористое железо65.
Предложены разные способы регенерации хлора или хлори-
хлористого водорода из отбросных хлоридов железа. При взаимодей-
взаимодействии хлорного железа с воздухом при 600—800° образуются окись
железа и хлор 66. Выделение хлорида железа из отработанного со-
солянокислого травильного раствора можно производить высалива-
высаливанием его хлористым водородом. Образующийся при этом соляно-
кислотный раствор используется для травления металла67-68 (см.
гл. XI).
Запатентован69 способ регенерации сернокислого травильного
раствора насыщением его хлористым водородом (после упарива-
упаривания). Образующаяся по реакции
FeSO4 + 2HC1 = FeCl2 + H2SO<
-серная кислота возвращается на травление, a FeCl2 подвергается
гидротер.мической обработке для превращения в окислы железа и
хлористый водород, возвращаемый на регенерацию травильного
раствора. Гидролиз хлористого железа водяным паром по реакции
3FeCl2 + 4Н2О = Fe3O4 + 6НС1 + Н2 + 75,44 ккал
целесообразно проводить при 450—650° (при 300° в равновесной
газовой фазе содержится всего ~I% HC1). При 650° скорость
гидролиза очень велика и реакция практически завершается за
15—20 мин70.
Описана регенерация солянокислых травильных растворов
с помощью жидкого анионообменника 71 и ионообменных смол72.
О других способах регенерации солянокислых травильных раство-
растворов см. 73- 74.
Хлорное железо может быть извлечено из солянокислых рас-
растворов (с целью их очистки от железа) некоторыми сложными
эфирами и кетонами, в частности трибутилфосфатом 75 и диэтило-
вым эфиром. Сущность механизма экстракции заключается в из-
изменении диэлектрической проницаемости водной фазы под влия-
влиянием кислот или солей, в результате чего работа отрыва молекул
гидратированного хлорного железа становится равной работе
сольватации их эфиром. На основании этого высказано мнение,
что появление в эфирной фазе HFeCl4 является вторичным про-
процессом, не имеющим, вопреки установившимся взглядам, непосред-
непосредственного отношения к механизму реакции "
76
ПОЛУЧЕНИЕ ОКИСЛОВ ЖЕЛЕЗА И ИХ ГИДРАТОВ
При окислении свежеосажденного гидрата закиси железа кис-
кислородом воздуха образуются не растворимые в воде основные соли
окиси железа и некоторые формы гидратированной окиси железа.
Они применяются в качестве пигментов, называемых марсами.
В зависимости от относительного содержания Fe2O3, SO3, H2O, от
температуры осаждения и от рН среды, в которой происходит окис-
окисление гидрата закиси железа, окраска марсов изменяется от жел-
желтой до красной. Осаждение гидрата закиси железа производят
обработкой раствора FeSO4 B00—250 г/л) 10% раствором соды
или 10%-ной суспензией мела в воде. Осаждение следует вести на
холоду, при рН, равном в конце процесса 5,5—5,8. Окисление про-
производят при этом же значении рН с постепенным добавлением
осадителя по .мере снижения рН в процессе окисления. При сус-
пендировании гидрата закиси железа в воде, т. е. в отсутствие ма-
маточного раствора, окисление протекает значительно быстрее77.
Из гидратов окиси железа наибольшее значение имеет моно-
моногидрат окиси железа FeO • ОН или Fe2O3 • Н2О, применяемый в ка-
качестве желтого пигмента. Он имеет очень яркий и чистый охряно-
желтый цвет с оттенками от лимонно-желтого до оранжевого.
Выше 150—200° желтая окись железа обезвоживается и переходит
в красную окись железа. При 275—300° этот процесс протекает
весьма быстро.
Желтую окись железа получают в производственных условиях
путем окисления воздухом металлического железа, погруженного
в зародышевую суспензию — раствор сернокислого (или хлори-
хлористого) железа, содержащий взвесь свежеосажденной гидроокиси
железа. Процесс ведут при 65—70° в течение 48 ч и более. Жел-
Желтую окись железа получают также в качестве побочного продукта
в процессе образования ароматических аминов восстановлением
нитросоединений металлическим железом (в частности, при полу-
получении анилина) в присутствии солей Al, Cr, Sn и некоторых других
элементов 78.
Желтый гидрат окиси железа Fe(OHK или Fe2O3 • ЗН2О (жел-
(желтый марс) получают или взаимодействием хлористого железа
с мелом и окислением осадка воздухом или непосредственным
окислением углекислого железа воздухом.
При осаждении хлористого железа и окислении образующегося
осадка протекает реакция:
2FeC!2 + 2СаСО3 + ЗН2О + 0,5О2 = 2Fe(OHK + 2СаС12 + 2СО2
Для ускорения окисления к раствору хлористого железа кон-
концентрацией 50—100 г/л FeCl2 добавляют 3—5% NaNO2 или ZnCl2-
Мел вводят в количестве 100—120% от веса FeCl2. Затем через
массу пропускают воздух. Образующийся осадок промывают, филь-
фильтруют и сушат при температуре не выше 80°.
По другому .методу желтый гидрат окиси железа получают пу-
путем предварительного выделения углекислого железа взаимодей-
взаимодействием FeSO4 и Na2CO3 и последующего его окисления:
2FeSO4 + 2Na2CO3 = 2FeCO3 + 2Na2SO4
2FeCO3 + лН2О + 0,5O2 = Fe2O3 • nH2O + 2CO2
710
Гл. XVIII. Соединения железа
Окисление ведут или воздухом при 20—25° в течение 10—12 ч
или раствором бертолетовой соли при 50—60°. В последнем случае
окисление заканчивается в течение 3—4 ч.
Окись железа — красный железоокисный пигмент — в зависи-
зависимости от условий получения может иметь различные оттенки — от
оранжево-красного до синеватого и даже фиолетово-красного.
Красную окись железа получают либо прокаливанием железного
купороса или гидрата окиси железа, либо из колчеданных огарков.
Разложение железного купороса при 650—700° идет до конца
с образованием окиси железа:
2FeSO« = Fe2O3 + SO2 + SO3
При более низких температурах D00—600°) продуктами раз-
разложения являются основные сернокислые соли Ре2Оз • mSO3 • яН2О.
Для получения окиси железа железный купорос вначале обезво-
обезвоживают в барабанных сушилках воздухом, нагретым до 250—300°,
а затем прокаливают в муфельных или пламенных печах. При
сушке с ретуром (возвратом части обезвоженного сульфата) воз-
возможно применять воздух, нагретый до 350°.
На современных предприятиях красные железоокисные пиг-
пигменты получают, просасывая через перемещающийся на конвейере
слой железного купороса горячий топочный газ и перегретый во-
водяной пар. При этом последовательно идут процессы дегидратации,
окисления и разложения сульфата до окиси железа. Таким же спо-
способом готовят желтые железоокисные пигменты, но с другим ре-
режимом прокаливания 79. Чем выше температура прокаливания, тем
меньше в продукте примеси сульфата. Но прокаливание при 850°
и выше вызывает наряду с уменьшением содержания сульфата
резкое снижение удельной поверхности окиси железа за счет спе-
спекания частиц — размеры частиц увеличиваются до ~3000 А. При
более низкой температуре получается продукт с частицами раз-
размером 150—200А80.
Природная окись железа — железный сурик — является одним
из важнейших пигментов. Его производство базируется главным
образом на красковых рудах Криворожского месторождения. Пиг-
Пигментные свойства (перетираемость, цвет, коррозионная стойкость)
зависят от степени измельчения и содержания окиси железа. Вы-
Высококачественный сурик получают путем термической обработки
природного при 600° и микроизмельчении на пароструйных мель-
мельницах 7S.
Для получения окиси железа из колчеданных огарков их под-
подвергают классификации для выделения наименее загрязненных
фракций. Последние размалывают и промывают водой для удале-
удаления примесей. После высушивания продукт содержит 90% РЮ
6% SiO2, 2,5% А12О3 и имеет темный фиолетово-красный цвет.
Получение окислов железа и их гидратов
711
Получение закиси-окиси железа РезО4 основано на взаимодей-
взаимодействии гидратов окиси и закиси железа:
2Fe(OHK + Fe(OHJ = FeO • Fe2O3 + 4H2O
Обычно исходят из гидрата закиси железа, который подвергают
окислению. Образующийся гидрат окиси железа при этом вступает
во взаимодействие с еще не окислившимся гидратом закиси же-
железа. Источником гидрата закиси железа может служить углекис-
углекислое железо, которое гидролизуется при нагревании:
FeCO, t- H2O = Fe(OHJ + СО2
Углекислое железо получают взаимодействием раствора же-
железного купороса (концентрацией 200—300 г\л FeSO4 • 7Н2О) и
раствора соды (концентрацией 100—150 г/л). Образующуюся сус-
суспензию нагревают до 80—90° и затем через нее пропускают воз-
воздух. Окисление длится 20—30 ч 81.
Окись-закись железа Fe3O4, пригодную для приготовления масс,
применяемых в магнитной звукозаписи, можно получить осажде-
осаждением Fe(OHJ при обработке раствора FeSO4 10 н. раствором
NaOH с последующим окислением Fe(OHJ водным раствором
NaNO382. Fe3O4 получают также восстановлением окиси железа
водородом и другими восстановителями83. Магнетит и окись же-
железа с магнитными свойствами получают окислением металличе-
металлического железа или термическим разложением карбоната или окса-
лата железа.40-84, или термической обработкой окиси железа выше
600° для изменения ее кристаллической структуры; обычно при
этом материал подвергают последовательному восстановлению до
Fe3O4 и окислению до Fe2O385.
Окись железа может быть получена из растворов солей двух-
двухвалентного железа при окислении газообразными окислителями
под давлением. Предварительно понижая рН раствора до 2—6,
¦ отделяют примеси, затем раствор подщелачивают до рН = 9,6,
подогревают его до 95° и обрабатывают окислителем под давле-
давлением ~2 ат. После промывки и сушки образовавшегося осадкз
Fe(OHK получается темно-коричневая с красным оттенком окись
железа 86.
Окись железа Fe2O3 можно также получить при гидролизе FeCl3
с небольшим избытком пара при 400—800° на контактной поверх-
поверхности частиц огнеупорного материала (из Fe2O3, SiO2, A12O3 и др.),
находящихся во взвешенном состоянии87.
Предложено перерабатывать железный купорос на гидроокись
железа и твердый сульфат аммония без выпаривания. Для этого
железный купорос FeSO4 • 7Н2О окисляют кислородом воздуха
в оксисульфат Fe2O(SO4J. Последний загружают в насыщенный
Раствор сульфата аммония и обрабатывают суспензию газообраз-
газообразным аммиаком в молярном отношении 1 : 1 88.
712
Гл. XVIII. Соединения железа
Железоокисный пигмент коричневого цвета, пригодный для ма-
малярной краски, может быть получен из пыли, накапливающейся
в электрофильтрах при очистке сернистого газа колчеданных пе-
печей. Для этого пыль, представляющую собой тонкодисперсную
окись железа, содержащую разные примеси, обрабатывают слабой
аммиачной водой или маточным раствором аммонийных солей для
перевода находящихся в ней сульфатов в гидраты 89- 90.
ЛИТЕРАТУРА
I. А. П Белопольский, С. Я Шпунт и др., ЖПХ, 14, 716 A941); 21,
781, 794 A948)- 23, № 2 A950).—2. Lorant Bel a, Z. analyt. Chem., 219, № 3,
266 A966). — 3. Г А. Захарченко, В. А. Цицорин, ЖПХ, 22, № 7, 703
A949); 24, № 7, 779 A951). — 4. Н. Ш. Сафиулин, Там же, 32, № 10, 2178
A959).—5. М Е. Познн, А. М. Гинстлинг, Там же, 23, № 11, 1149 и
№ 12, 1245 A950); 24, № 2, 134 A951).— 6. С. А. Амитова, В. В. Печков-
ский, Б С Камеко, А Ф. Степанова, Уч. зап. Пермск. гос. ун-та, 17, 61
(I960). —7. В В Печковский, ЖПХ, 23, 2613 A959). — 8. В. В. Печков-
Печковский, А. Г. Звездин, С. В. Островский, Там же, 36, № 7, 1454 A963).—
9. В. В. Печковский, А. Н. Кетов, Труды Пермск. политехи, ин-та, № 10,
1961, стр. 3. — 10. С. А. Амирова, В. В. Печковский, А. Н. Кетов,
Гам же, стр. 33.
II. i. Dankiewicz, J. Kopczynski, W. Stelmachowski, Przem.
Chem., 10, N° 3. 121 A954). — 12. С. А. Гордон, М. А. Менковский, ЖНХ,
2, № 1, 30 A957). —13. Л. С. Гецкин, А. Г. Батюк, П. П. Цыб, Цветные
металлы, 7, 23 A957). — 14. В. В. Печковский, Н. Л. Су б оч ев а, ЖПХ,
32, № 8, 1857 A959). — 15. С. Д. Ш а р го р о д ски й, Р. И. Заславская,
Укр. хем. ж., 21, № 6, 694 A955). —16. А. П. Богуславский, В. В. Уру-
Урусов, ЖПХ 21, № 9, 903 A948). —17. И. Н. Кузьминых, Хим. пром., № 10,
24 A945). —18 Н. Е. К eyes, Chem. Met. Eng., 57, № 5, 126 A946).—
19. M E П о з и н, И. П. М у х л е н о в, Л. С. В а с и л е с к у, ЖПХ, 28, № 6, 573;
№ 7, 681 A955). — 20. Л. С. Гецкин, В. Д. Пономарев, Там же, 29, № 7,
981 A956).
21. Англ пат 1056262 1963. —22. В. О. A. Hedstrom, Arkiv kemi, 6,
№ 1—2, 1 A953). — 23. Б. А. Рязанов, ЖОХ, 24, № 9, 1477 A954).—
24 Н В Шишкин, И Г Шубцова, Уч. зап. Саратовск. ун-та, 34, 29
A954). — 25. W. F. Link, J. Phys. Chem., 60, № 1, 91 A956). — 26. А. Н. Ива-
»т л г» f~* TJ Л тт л иг гт ТТ ГТлТТ'ТТП TTF г ЛД /"\^">?/"ЛТЭ Г*1Т п _V nl/Q IT TX^f TtllillinnOQDQ 1Э V/
D. O. Jordan, J. Chem. Soc, 1953, № 5, 1435.
net, J. phys. Chem., 60, № 11, 1569 A956).
31. F. J. Shipko, D. L. Douglas, ibid., 1519, 1522. —32. В. И. Орлов,
П. В. Попов, М А. Морозова, М. Г. Г а б р и е л о в а, Ядохимикаты (ин-
еектисиды и фунгисиды), Госхимиздат, 1946.— 33. А. Л. Ефимов, Химический
метод борьбы с вредителями и болезнями сельскохозяйственных растений, Сель-
хозгиз 1949 —34. И. И Отваги н, Хим. пром., № 8, 19 A945). — 35. J. О. Р е г-
clval С P. Dyer, M. H. Taylor, Ind. Eng. Chem., № 12 A941). — 36. R. L e-
v i e 1, Ind. Chim., 4, № 6, 499, 45 A959). — 37. J. P г с h 1 i k, L. H 1 i n a k, D. К a r-
takova, Paliva, 35, № 6, 157 A955). — 38. Г. П. Григорьев, Д. Г. Трабер,
Труды ЛТИ целлюлозно-бумажной промышленности, № 3, 1955, стр. 107. —
39. P. H. Johnson, Mining Eng., 17, № 8, 64 A965).— 40. R. Robl, Ang.
Chem., 70, № 12, 367 A958).
Литература
713
41. R. K. Rathmelle, пат. США 3261655, 1962. — 42. Sulphur, № 70, 33
A967). — 43. Ив. Радулова, П. Брайкова, Лека пром., 5, № 1, 41
(J956). — 44. П. В. Дыбина, Технология минеральных солей Госхимиздат,
1949. — 45. Chem. Met. Eng., 1003 A943). — 46. Е. Б. Конюхова, И Н. Ш о-
кин, А. Г. Кузнецова, Труды МХТИ, в. 56, 1967, стр. 120, 202; в 60, 1969,
стр. 36, 40.— 47. R. Haarmann, пат. ФРГ 918428, 1954. — 48. Голланд пат.
111813, 1958. — 49. Н. И. Шефтель, Сталь, № 7, 655 A954). — 50. Н. Ф. Сери-
Сериков, Там же, 651.
51. Sulphur, № 75, 30 A968). — 52. С. Н. Ганз, А. И. Лукьяннца,
Л. А. Бельчина, ЖПХ, 37, № 7, 1606 A964).— 53. J. 111 i n i с z, Probl.
projekt. huln. i przem. maszyn., 16, № 5, 155 A968). — 54. К. Тамаки и др.,
«Фудзи Сэйтецу Гихо. Techn. Rep. Fuji Iron and Steel Co», 14, № 3, 370
A965). — 55. А. П. Снурников, В. Ф. Ларин, ЖПХ. 39, № 12, 2634 A966).—
56. Ц з я н Ж у н - г у а н, Л э и Ц з ы - ц н, Л э й, X у н - к а н, Хуасюэ Шицзе,
№ 10, 475 A955). — 57. С. А. Г о р д о н, М. А. М е н к о в с к и и, Сборник научных
работ Московск. горного ин-та, в. 1, 1957, стр. 61. — 58. В. С Каминский,
С. Я. Л ейтке, Хим. пром., № 2, 138 (I960). — 59. А. И. Эттингер, 3. Я. Г о-
родишер, Д. М. Корф, Н. И. Самарин, авт. свид. 80463, 1949; Бюлл.
изобрет., № 3, 1941 A950). — 60. W. R. McCormic, пат. США 2718455, 1955.
61. J. Heu be I, Bull. Soc. chim. France, 1954, № 11—12, 1473.-52. M. E. Gra-
Graham, A. S. Henderson, J. P. Whitehouse, пат. США 2665191. — 63. Австр
пат. 153627, 1953. — 64. К- L. Piggott, A. F. Reid, P. С. Wailes, Austral.
J. Appl. Sci., 15, № 3, 186 A964). — 65. M. E. Graham, E. А. В e i d 1 e г, пат
США 2677594, 1954.— 66. R. H. Sawyer, пат. США 2642339, 1953. — 67. Metal
Progress, 64, № 4, 134, 136 A953). — 68. O. Runther, англ пат 703142 1954 —
69. Пат. США 2785957, 1955. — 70. М. В. Ионин и др. ЖПХ 3, №'l2 2651
(I960); 35, № 4, 900 A962).
71. Н. Wiedmann, Wasser, Luft u. Betrieb, 10, № 3, 154, 159, 183 A966).—
72. X. Tamura a. oth., J. Chem. Soc. Japan. Ind. Chem. Soc, 69, № 5 1018
A966). —73. А. Наке, Sheet Metal Ind., 42, № 461. 696 A965); 43, № 474 718-
A966); Metallurgie et constr. mec, 97, № 7, 553 A965). — 74. G. Diez P N i e d-
ner, Ind.-Anz., 87, №56, 1303 A965). — 75. О. Д. Лях, И. А Шека А И Пер-
Перфильев, ЖПХ, 39, № 8, 1799 A966). — 76. Е. И. Денисов. А С Халонин
Там же, 38, № 2, 295 A965). — 77. Я. М. Пес и н, М. А. Ш т е р н Е М Горо-
Городецкий, Хим. пром., № 6, II A948). — 78. И. Рискин Т Великосла-
винская, ЖПХ, 19, 149, 263, 271 A946). — 79. М. А. Штерн, И. С. Ша-
Шапиро, Лакокрасочные материалы и их применение, № 5 28 A967).
S0. Н. М. Сорокина, И. Г. Бляхер, Л. Н. Калит и на, ЖПХ, 38, № 12
•2844 A965).
81. Е. Ф. Беленький, И. В. Р и с к и и, Химия и технология пигментов
1осхимиздат, I960. — 82. P. P. Zaponni, англ. пат. 701433, 1953 —
*5- В. П. Рождественский, В. Г. Косягин, Л. М. Г а с а н о в а, Труды
Саратовск. ин-та «Гипрониигаз», № 5, 1966, стр. 343. — 84. W. Wolski,
A- Kwiatkowski, Przem. chem, 46, № 7, 407 A967). — 85. Япон пат 11733*
I960; 10307, 1961. — 55. С. С. Clarke, пат. США 2656282, 1953. — 87. L Ree-
Reeve, англ. пат. 701797, 1954. — 88. F. LimmerhakI, чехосл. пат 84026 1955 —
°9- А. А. Коринфский, Хим. пром., № 10—11, 35 A944). — 90. Правила я
нормы техники безопасности и промышленной санитарии для строительства и
Эксплуатации производств железоокисных пигментов, Госхимиздат, 1962.
Физико-химические свойства
715
Глава XIX
СУЛЬФАТ И ХЛОРИД ЦИНКА
Соединения цинка имеют большое значение в металлургиче-
металлургической, лакокрасочной и химической промышленности. Важнейшими
из них являются цинковый купорос и хлорид цинка. Другие со-
соединения — окись и гидроокись, сульфид цинка и прочие — играют
роль сырья, полупродуктов и продуктов в ряде производств. Здесь
рассмотрены некоторые свойства главнейших соединений цинка и
технология цинкового купороса и хлорида цинка.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Окись цинка ZnO — порошок белого цвета с плотностью
5,42 г/см3. При нагревании ZnO принимает желтую окраску. Выше
1100° начинает возгоняться. Плавится под давлением 52 ат при
2000°. Восстановление ZnO углеродом и окисью углерода начи-
начинается при температуре возгонки металлического цинка (910—
920°) и протекает полностью при 1200—1300°.
Окись и гидроокись цинка Zn(OHJ легко растворяются в кис-
кислотах. Zn(OHJ растворяется в водных растворах аммиака, при-
причем ее растворимость возрастает с увеличением концентрации NHj
вследствие образования комплексных гидроокисей [Zn(NH3J](.OHJ',
При взаимодействии 1 н. растворов ZnSO4 и NaOH при 20J
выделяется осадок основной соли ZnSO4 • 3Zn(OHJ- xH2O, мед-
медленно реагирующий со щелочью. Выше 68° осаждается Zn(OHJ2.
При растворении Zn(OHJ в разбавленных растворах NaOH обра-
образуются ионы HZnO2 и ZnO2". При 25° первая константа кислот-
кислотной диссоциации3 Zn(OHJ[Zn(OHJ(TB.) = H+ +HZnOj] равнЕ
1,2-10~17. Вторая константа диссоциации [Zn(OHJ (тв.) - 2Н+
+ ZnO2~]paBHa 2,2 • Kh30. Соответственно, константа диссоциации'
HZnO2[HZnOi = H+ + ZnO2~] равна 1,83-103. При нагревании
Zn(OHJ начинает разлагаться на ZnO и Н2О уже при 35°, ,но ин«
тенсивно процесс идет при 100—160° и заканчивается при 200°4.
По другим данным5, температура разложения Zn(OHJ 130°.
Сульфид цинка ZnS имеет плотность около 4 г/см3. Он не пла-
плавится, но с другими сульфидами (Cu2S, FeS, PbS и пр.) образует
легкоплавкие штейны 6. При нагревании ZnS сублимируется. Так,
например, при 1000° за 30 мин потеря в весе составляет 0,4%- При
давлении 10~4 мм рт. ст. за 1 ч улетучивается ~23% ZnS при 900°
и 78% при 975°. В присутствии восстановителей наряду с суб-
сублимацией имеет место также восстановление сульфида цинка. Ско-
Скорость сублимации уменьшается, а скорость восстановления увели-
увеличивается с повышением давления окиси углерода. В водородной
среде при повышении давления Н2 возрастают и скорость субли-
сублимации и скорость восстановления. Это связано с большей ско-
росты© диффузии молекул сульфида от твердой поверхности в га-
газовый объем. Давление пара над сульфидом цинка (в мм рт. ст.)
вычисляют по формуле 7 lg Р = —11 300/Г + 7,9.
При окислении кислородом воздуха суспензий искусственного
сульфида ZnS (так же как и сульфидов других металлов и неко-
некоторых природных сульфидных минералов) в растворах едкого
натра скорость перехода сульфидной серы в ион SOl~ ниже 100°
мала, но резко возрастает в пределах 150—250° 8.
Сульфат цинка ZnSO4 кристаллизуется из водных растворов
в пределах от —5,8° (криогидратная точка 27,87% ZnSO4) до 38,8°
в виде бесцветных ромбических кристаллов цинкового купороса
2nSO4 • 7Н2О. Между 7,6 и 24,9° образуется также метастабильная
моноклинная модификация ZnSO4-7H2O, а между 11,4 и 38,8° —
метастабильные моноклинные кристаллы ZnSO4-6H2O, стабиль-
стабильные от 38,8 до 70°. Ниже 11,4° метастабильная соль ZnSO4>6H2O
необратимо переходит в моноклинную форму ZnSO4 • 7Н2О. Выше
70° кристаллизуется ZnSO4 • Н2О. Насыщенный водный раствор
содержит при 0° —29,4%, при 25° — 36,7%, при 75° — 40,9%, при
99° — 37,7% ZnSO4. Водные растворы ZnSO4, не содержащие сво-
свободной кислоты, иногда мутнеют вследствие выделения осадка ос-
основного сульфата цинка 3Zn(OHJ-ZnSO4 • 4Н2О 9<10.
В системе ZnSO4—H2SO4—Н2О при 20° и общей концентрации
сульфата и кислоты 0,25—0,5 г-мол/л образуются п комплексные
ионы [Zn(SO4b]2~. На рис. 196 представлена диаграмма раствори-
растворимости в этой системе 10, позволяющая рассчитать кристаллизацию
ZnSO4 • 7Н2О.
Из 100 кг исходного раствора кристаллизуется
?1о
— 1, loX
ZnSO4 илн 1,78л: кг ZnSO4-7H2Q
где А а а — содержание ZnSO4 в растворе до и после кристалли-
кристаллизации, вес.%- О плотности, вязкости и электропроводности серно-
сернокислых растворов сульфата цинка см.12.
716
Гл. XIX. Сульфат и хлорид цинка
Физико-химические свойства
В системе ZnSO4—(NH4JSO4—Н20 образуется двойная соль.
При 50° поле ее кристаллизации больше, чем при 35°. При нагре-
нагревании до 360° эта соль полностью обезвоживается, а выше 370°
разлагается9'13. В системе ZnSO4— (NH4JSO4— Na2SO4—H2O при
35° могут существовать твердые фазы: ZnSO4 • (NH4JSO4 -6H2O
(наименее растворимая), ZnSO4 ¦ Na2SO4> 4H2O, Na2SO4-(NH4JSO4'
•4Н2О, ZnSO4-7H2O, Na2SO4 и (NH4JSO4. В системе ZnSO4—
Na2SO4—Н2О образуются двусторонние ограниченные твердые рас-
растворы ZnSO4- Na2SO4 -4H2O с су-
сульфатами натрия и цинка 14. В си-
системе ZnSO4—CuSO4—FeSO4—
—H2SO4—H2O в интервале 25—60°
образуются твердые растворы
(Си, Fe, Zn)SO4-5H2O, содержа-
содержащие от 0 до 4% ZnSO4 и от 0 до
2% FeSO4. Растворимость твердо-
твердого раствора возрастает с повыше-
повышением температуры и уменьшается
с ростом концентрации ZnSO4 в
жидкой фазе. Содержание ZnSCU
и FeSO4 в твердом растворе воз-
возрастает при повышении темпера-
температуры и при увеличении концентра-
концентрации ZnSO4 в жидкой фазе 15.
В системе ZnSO4—Na2SO4
установлено существование двух
твердых растворов на основе а-
и p-Na2SO4 и соединений 3ZnSO4-
•Na2SO4, ZnSO4 • Na2SO4, ZnSO4«
• 2Na2SO4 и ZnSO4 . 3Na2SO4 16.
В системе ZnSO4—K2SO4 образуются два соединения: 2ZnSO4 •
с ^л = 487° и ZnSO4-K2SO4 с *пл = 470°. При 510° и
717
/о го
H2SO4,%
Рис. 196. Растворимость в системе
ZnSO4—H2SO4—H2O. Твердые фазы:
ниже асй — ZnSO4 • 7Н2О; в области
abc - ZnSO4 • 6Н2О; выше bed—
ZnSO4 • Н2О.
^K2SO4 с *пл 487 и ZnSO4K24 пл р
60 мол.% Кг$О4 образуется твердый раствор. Во взаимной системе
ZnSO4—КС1 обнаружены три гетероионных конгруэнтно плавя-
плавящихся соединения: цинковый каинит ZnSO4-KCl с tV4 =¦= 485°,
2ZnCl2 • K2SO4 с *пл = 450° и КС1 • K2SO4 • 4 ZnCl2 с /пл = 350°. В си-
стеме ZnSO4—NaCl в широком интервале температур вплоть до
комнатной устойчивы ZnCl2-2NaCl, ZnSO4-Na2SO4 и 3ZnSO4-
• Na2SO4. В системе ZnSO4—ZnCl2 образуется соединение ZnSO4 •
•ZnCl2, существующее, по-видимому, в узком интервале концентра-
концентраций (от ~ 10 до 30 мол. % ZnSO4) и плавящееся инконгруэнтно
при 437°. С увеличением концентрации ZnSO4 это соединение рас-
распадается с образованием 3ZnSO4- ZnCl2. В тройной взаимной си-
системе под влиянием третьего компонента ZnSO4-ZnCl2 становится
более устойчивым 16. В системе Na, К, Zn || SO*, C1 понижение тем-
температуры кристаллизации достигает 282° 17. .
Разложение сульфата цинка в струе сухого воздуха наблю-
наблюдается уже при 600° и идет с образованием промежуточного про-
продукта ZnSO4-ZnO18. (По другим данным, его состав 2ZnSO4 •
. ZnO 19~21.) Интенсивное разложение с образованием ZnO и SO2
идет при 800—900°. Разложение ускоряется при добавке 1—2%
Fe2Os и при 900° в течение 15 мин достигает ~80%. Присутствие
SO2 в исходном газе (при отсутствии катализаторов, способствую-
способствующих превращению SOj в SO3) не влияет на скорость разложения
100,
znci2- н2
2ZnClj-3H
2ZnC12-5H,O
У10 20 30 40' 50 60 70 80 90 ZnCls
—-Вес У.
Рис. 197, Система ZnCl2— H2O.
сульфата цинка. В присутствии Fe2O3 скорость разложения ZnSC>4
ниже 700° уменьшается, а выше 800° увеличивается с ростом кон-
концентрации SO2 в исходном газе, что связано с условиями равнове-
' СИЯ SO2 + V2O2 3F* SOg 22.
Хлорид цинка ZnCl2 существует в двух формах — ас/пл = 315°
и р с 4Л =¦ 326°23; кипит при 730°. На рис. 197 показана политерма
растворимости ZnClj в воде. Из концентрированных растворов
выше 28° ZnCl2 кристаллизуется в безводной форме, при низких
температурах — с 1, l'/г, 2'/2, 3 и 4 молекулами воды. Насыщенный
раствор содержит при 25° 81,2%, при 100е — 86% ZnCl2. В системе
ZnCl2—NH3 образуются комплексы: ZnCl2 • VeNH3, ZnCl2 • У2]ЧНз,
ZnCl2-NHs и ZnCl2 • 1,5NH3. Первый из них наиболее прочен; он
Дистиллируется без разложения при 590—680°. Комплекс ZnCl2»
-1/2NH3 метастабилен. ZnCl2*NH3 плавится при 164°, а сплавы
ZnCl2 с ZnCl2'NH3 и ZnCl2*l,5NH3 характеризуются еще более
низкими температурами плавления, а также малым давлением
Диссоциации, Поэтому они могут быть использованы в качестве
718
Гл. XIX. Сульфат и хлорид цинка
Применение
719
теплоносителей24-25. Плавление смеси моноаммиаката хлорида цин-
цинка с металлическим цинком B:1) при 280—470° до прекращения
выделения водорода с последующим охлаждением плава, обработ-
обработкой водой и сушкой дает моноамид хлорида цинка ZnClNH2 (он мо-
может найти применение в производстве неорганических полимеровJ6.
Известны гидрооксихлориды цинка Zn5(OH)8Cl2 • Н2О и ZnOHCl.
Первый из них при нагревании в пределах 110—147° теряет кри-
кристаллизационную воду, а выше 167° разлагается на ZnO, ZnCl2 и
Н2О. Второй разлагается на эти же компоненты выше 227°5.
; ПРИМЕНЕНИЕ
Сульфат цинка применяется в текстильной промышленности,
в гальванотехнике, для изготовления минеральных красок, в част-
частности литопона — смеси эквимолекулярных количеств ZnS и
BaSO4, и проч. Окись цинка используют для изготовления цинко-
цинковых белил, в производстве резиновых изделий, в фармацевтиче-
фармацевтической промышленности. Сульфат и хлорид цинка служат для кон-
консервирования древесины. Хлорид цинка во многих случаях
заменяет сульфат. Его используют также в качестве водоотнимаю-
щего средства. Расплавленный хлорид цинка применяют в каче-
качестве катализатора гидрокрекинга каменного угля и его экстрак-
экстрактов
27
Сульфат цинка является полупродуктом при производстве ме-
металлического цинка гидроэлектрометаллургическим методом, ко-
которым производят приблизительно одну треть мировой продукции
цинка. По этому методу цинк получают электролизом раствора
ZnSO428, приготовленного растворением в серной кислоте окиси
цинка — продукта обжига цинковой руды. Регенерированную при
электролизе серную кислоту (~1,5 кг на 1 кг осажденного цинка)
возвращают на растворение окиси цинка —• выщелачивание
спека 29. Сульфат цинка используют также в качестве электролита
при цинковании изделий из черных металлов.
Цинк является микроэлементом, необходимым для питания
растений. Содержание цинка в сухом веществе растений составляет
5—30 мг/кг, а в некоторых культурах доходит до 45—50 (торох,
вика) и даже до 60—70 (плоды томата) мг/кг. Содержание цинка
в урожае ячменя составляет 70—75 г на 1 го, а в урожае сахар-
сахарной свеклы — до 120 г на 1 га. Отсутствие или недостаток цинка
приводит к замедлению роста и заболеванию растений, выражаю-
выражающемуся в уменьшении размера листьев и в обесцвечивании их.
• Восполнение недостатка в цинке производят внесением цинковых
удобрений в почву и опрыскиванием растений растворами цинко-
цинковых солей. В качестве цинковых удобрений могут быть использо-
использованы различные отходы промышленности, содержащие цинк30-31.
Препараты цинка применяют также для борьбы с болезнями рас-
растений (цинеб — цинковую соль этилен-б«с-дитиокарбаминовой кис-
кислоты, цирам — цинковую соль диметилдитиокарбаминовой кислоты
й Др.) 82. Фунгицидным действием обладают основные хроматы
цинка, а арсенаты цинка могут служить антисептиками для древе-
древесины. Фосфид цинка используют в качестве зооцида 83.
Согласно ГОСТ 8723—58, в цинковом купоросе должно содер-
содержаться (в %):
I сорт II сорт
Цинка, не менее 22,5 21,8
Железа, в пересчете на FeO,
не более 0,02 0,1
Серной кислоты свободной,
не более 0,05 0,1
Веществ, не растворимых в
воде, не более 0,04 0,3
Хлоридов, в пересчете на С1, •
не более 0,2 0,3
Марганца, в пересчете на МпО,
не более 0,04 0,2
Медн, свинца, кадмия и ни-
никеля в пересчете на РЬ, не
более 0,01 0,03
При содержании в цинковсш купоросе меньше 7 молекул воды
©тношение количества примесей к цинку не должно быть больше,
ТАБЛИЦА 47
Требования к качеству хлористого цинка по ГОСТ 7345—68
(содержание компонентов в %)
Внешний вип ...
Содержание:
хлористого цинка,
не менее ....
железа, не более .
сульфатов, не более
остатка, не раство-
растворимого в соляной
кислоте, не более
свинца, не более . .
меди, не более . .
мышьяка, не более
свободных кислот .
величина рН, не ме-
менее
Марка А
Пластинки
серого цвета
толщиной
1—2 мм
97
0,3
0,05
0,1
и
—
Марка В
1-й сорт
Бесцветный
желтый
прозрачный
49
0,008
Отсутствие
0,01
0,002
0,002
0,001
3.7
2-й сорт
или светло-
раствор
допускается
муть
47
0,16
0,02
0,1
—
3.7
Марка В
Мутный
раствор
бурого
цвета
40
20
10
Отсутствие
720
Гл. XIX. Сульфат и хлорид цинка
чем в семиводном продукте. Цинковый купорос упаковывают в по-
полиэтиленовые и бумажные мешки, фанерные барабаны и деревян-
деревянные бочки. Растворы цинкового купороса перекачивают по сталь-
стальным трубам, но присутствие примеси медного купороса вызывает
сильную их коррозию.
Хлорид цинка выпускают в виде растворов (табл. 47) и в виде
твердого пластинчатого продукта—• затвердевшего плава. (Раньше
плав выпускали в виде монолитной массы, застывшей в таре.)
Твердый продукт упаковывают в герметические стальные или кар-
картонные барабаны с вкладышами из полиэтилена; загрузку в тару
производят в среде сухого азота или воздуха. Растворы хлористого
цинка перевозят в стальных цистернах и бочках, а также в поли-
полиэтиленовых канистрах и стеклянных бутылях.
СЫРЬЕ
Сырьем для производства цинкового купороса служат серная
кислота и металлический цинк (пуссьера, отходы переработки
лома цветных металлов и пр.), цинковая зола, являющаяся отхо-
отходом при оцинковке металлов, а также сульфидные руды. Пуссьера
представляет собой цинковую пыль — отход, образующийся при
дистилляции цинка в ретортных печах в процессе конденсации суб-
сублимата. Получение цинковой пыли объясняют образованием
пленки ZnO, которая обволакивает мельчайшие капельки цинка,
препятствуя их слиянию34. Пуссьера содержит 60—75% цинка
в виде металла и окиси, уголь, поваренную соль и другие примеси.
В отходах от переработки лома цветных металлов содержится
45—65% Zn. Лучшим сырьем для выработки сульфата цинка яв-
являются поддувальные отходы производства цинковых белил. Они
состоят почти целиком из окиси цинка, содержат незначительные
количества силиката цинка и металлического цинка и легко реаги-
реагируют с серной кислотой.
Главными рудами, используемыми для выплавки металличе-
металлического цинка, служат цинковая обманка (сфалерит) ZnS, силикат
цинка (каламин, галмей) Zr^SiCVb^O или Zn4(Si207) (ОНЬ> Н2О,
цинковый шпат (смитсонит) ZnCO3, а в последнее время и мате-
материалы, содержащие цинк в форме феррита (ZnO-Fe2O3). Мине-
Минералы цинка встречаются обычно в полиметаллических рудах со-
совместно с соединениями свинца, серебра, меди. Цинковые руды
обогащают флотацией35.
•Сырьем для получения хлорида цинка служат соляная кислота
и те же материалы, содержащие цинк, которые используют для
получения цинкового купороса, в частности отход текстильной про-
промышленности— окшара, содержащая до 20% ZnS, ~20% Na2SC>4,
!~20% Na2COs и 10% Н2О.
Сырьем для получения окиси цинка могут служить разные
щшкеодержащие отходы химической, медеплавильной и металло-
Сульфат цинка
721
обрабатывающей промышленности. Эти отходы представляют со-
собой в большинстве случаев шламы, содержащие цинк в виде ме-
металлического цинка, окиси, хлорида, сульфата или карбоната. Для
получения окиси цинка из отхода карбоната цинка, образующегося
в производстве гидросульфита натрия, его необходимо предвари-
предварительно отмыть от водорастворимых солей (Na2SO4, Na2SO3 и др.),
содержание которых доходит до 15%. Промытый карбонат цинка,
содержащий до 65% влаги, подсушивают до влажности 25—27%
и прокаливают при 500—600°. При использовании шламов, содер-
содержащих металлический цинк, их целесообразно обрабатывать соля-
соляной кислотой и из полученного раствора ZnCl2 осаждать содой
ZnCO3, который затем прокаливать для получения окиси цинка 36.
Потенциальным источником цинкового сырья являются колче-
колчеданные огарки. Целесообразность извлечения цинка из огарков
определяется не только использованием его для производства со-
соединений цинка, но и необходимостью удаления цинка до перера-
переработки огарков в металлургии37. Цинк из колчеданных огарков мо-.
жет быть извлечен в виде сульфата цинка или в виде хлорида
цинка (при хлорирующем обжиге огарка).
СУЛЬФАТ ЦИНКА
Наиболее распространенным способом производства цинкового
купороса является растворение в серной кислоте различных мате-
материалов, содержащих цинк и окись цинка. Примеси меди, железа,
никеля, свинца, олова и другие обусловливают необходимость спе-
специальной очистки растворов.
Сульфат цинка может быть получен в качестве побочного про-
продукта при улавливании отбросных сернистых газов цинковым ме-
методом 38, из полиметаллических руд обработкой их раствором суль-
сульфата окиси железа, а также окислительным обжигом цинковой
обманки в муфельных и других печах при температуре около 600°.
Перекаливание материала приводит, вследствие диссоциации
ZnSO4, к образованию ZnO. Из обожженной массы сульфат цинка
выщелачивают водой и выкристаллизовывают из раствора цинко-
цинковый купорос.
С большей легкостью сулыЬат цинка получается при сульфати-
зирующем обжиге цинковой обманки в атмосфере сернистых га-
газов, а также при сульфатизации сернистым газом окиси
Цинка 39-42.
Сульфид цинка легко сульфатизируется серной кислотой по
реакции:
ZnS + H2SO4 = ZnSO4 + H2S
Выделяющийся сероводород окисляется до серьп
H2S + H2SO4 = SOS + 2Н2О + S
722
Гл. XIX. Сульфат и хлорид цинка
При 160—200° степень сульфатизации цинка достигает 97% 43.
При более высоких температурах она уменьшается за счет улету-
улетучивания серной кислоты, а выше 600° за счет реакции:
3ZnSO4 + ZnS = 4ZnO + 4SO2
Водная суспензия ZnS (флотационного концентрата цинковой
обманки) может быть превращена в раствор ZnSO4 окислением
кислородом при давлении 10 ат и температуре ~200° в автоклаве
в присутствии CuSO4 в качестве катализатора (Си :Zn = 1 : 10).
Процесс описывается уравнениями:
ZnS + C11SO4 = ZnSO4 + CuS
CuS + 2O2 = CuSO4
Менее, чем за 2 ч выход цинка в раствор достигает 98% (в от-
отсутствие C11SO4 — только 40%) *4.
Получение сульфата цинка растворением материалов, содержащих
Zn и ZnO, в серной кислоте
Цинковое сырье, содержащее цинк и окись цинка, размалы-
размалывают в шаровой мельнице, а затем растворяют в 18—25%-ной сер-
серной кислоте4Б. Растворение производят в резервуарах, футерован-
футерованных кислотоупорным материалом и снабженных мешалками.
Тепло, выделяющееся при предварительном разбавлении серной
кислоты до указанной концентрации, а также тепло экзотермиче-
экзотермических реакций
ZnO + H2SO4 = ZnSO4 + Н2О (ж.) + 2БД ккал
Zn + H2SO4 = ZnSO4 + Н2 + 40,0 ккал
обеспечивает повышение температуры реакционной массы до 80—
100° без дополнительного подогрева. При содержании в сырье зна-
значительного количества металлического цинка процесс сопрово-
сопровождается интенсивным ценообразованием вследствие выделения
водорода. Реакционные резервуары должны быть сообщены с силь-
сильной вытяжной вентиляцией, предотвращающей накопление-водо-
накопление-водорода в атмосфере цеха.
Для ускорения нейтрализации серной кислоты, которая замед-
замедляется к концу процесса, в реактор загружают избыток цинкового
сырья. Образующиеся при этом основные соли цинка разрушаются
впоследствии добавкой серной кислоты после удаления раствора.
По окончании реакции, когда содержание свободной серной кис-
кислоты в растворе понизится до 1—2 г/л, раствору дают отстояться
и фильтруют его через барабанный вакуум-фильтр, а в реактор
с остатком сырья заливают новую порцию серной кислоты и до-
добавляют цинковое сырье. При содержании в сырье значительных
количеств свинца и олова, не переходящих при сернокислотной
Сульфат цинка
723
переработке в раствор, твердый остаток после промывки направ-
направляют на переработку для извлечения этих металлов.
При содержании в сырье олова образуются плохо фильтрую-
фильтрующие осадки. Поэтому считают целесообразным46 при переработке
отходов, содержащих олово, отделять нерастворившийся остаток
не фильтрацией, а отстаиванием. При разбавлении суспензии до
плотности 1,2—1,21 г/см3, что соответствует содержанию в рас-
растворе 200—250 г/л ZnSO4, отстаивание идет с достаточной
скоростью (рис. 198). Полное от-
отстаивание достигается только в
нейтральной среде. При отстаи-
отстаивании кислых растворов в сливе
содержится муть. При продол-
продолжительности отстаивания 10—
15 ч получается пульпа с отно-
отношением Т : Ж, в пределах от 1 : 1,2
1 : 1,5.
(В
16
В
\
\
\
\
V
\
\
•
W 1,19 1& 1,23 1,2S 1,27 1,29 1,31
Плотность при 60,"г/см3
Рис. 198. Влияние плотности суспен-
суспензии на скорость ее осаждения.
ДО
Профильтрованный раствор
содержит 400—420 г/л ZnSO4 и
примеси FeSO4, C11SO4, CdSO4,
NiSO4, количества которых за-
зависят от содержания соответст-
соответствующих металлов в сырье. Очи-
Очистку раствора цинкового купоро-
купороса от этих примесей производят
в несколько приемов. Вначале
очищают раствор от железа, до-
добавляя окислитель для перевода
Fe2+ в Fe3+. В качестве окисли-
окислителя обычно применяют раствор
гипохлорита натрия. После окис-
окисления железо осаждают известью в виде основных солей окиси
железа. Избыток извести вреден, так как он осаждает также ос-
основные соли цинка. Произведения растворимости: Zn(OHJ
A,80 - 10~14) и Fe(OHJ(l,64 • 10~14) почти равны; произведение
растворимости Fe(OHK значительно меньше A,1-Ю-36).
После выделения в осадок соединений железа раствор кипятят
Для разрушения небольшого избытка гипохлорита натрия и от-
отфильтровывают от осадка. Осадок этой второй фильтрации, как и
осадок от предыдущей, первой фильтрации, промывают на фильтре
водой для извлечения из него ZnSO4; промывные воды используют
Для разбавления серной кислоты в реакторе.
После очистки от железа раствор поступает на очистку от Си2+,
Cd2+. Эту очистку производят путем интенсивного перемеши-
перемешивания раствора с добавленной к нему цинковой пылью в течение
4-—6 ч. Потенциал у Zn более электроотрицательный, чем у Си, Ni
724
Гл. XIX. Сульфат и хлорид цинка
Сульфат цинка
725
и Cd. Поэтому металлический цинк вытесняет эти металлы из рас-
растворов их солей по реакциям:
Zn + C11SO4 =- Си + ZnSO« + 49,9 ккал
Zn + NiSO4=-Ni + ZnSO4 + 20,9 »
Zn + Cd SO4 - Cd + ZnSO4 + 12,5 »
Наиболее полно и быстро протекает цементация меди. Выделе-
Выделение из раствора никеля и кадмия сопряжено со значительными
трудностями вследствие того, что они близко стоят к цинку в ряду
напряжений. При повышенной температуре и интенсивном пере-
перемешивании быстро протекает обратный процесс — окисления и
растворения кадмия. В то же время для достижения полноты це-
цементации никеля требуются большой избыток цинковой пыли и
высокая температура 47.
Применяемую для цементации цинковуш пыль обрабатывают
предварительно небольшим количеством слабой серной кислоты
для удаления с поверхности металла тонкого слоя окиси. Выпав-
Выпавшие в виде тонкого шлама металлы отделяют от раствора на
фильтре, после чего раствор подвергают вторичной очистке от же-
железа, которое в небольших количествах содержится в цинковой
пыли. Профильтрованный после вторичной очистки раствор содер-
содержит 300—350 г/л ZnSO4 и до 10 г/л С1".
Полученный раствор цинкового купороса содержит некоторое
количество основных солей сульфата цинка. Для их разрушения
в очищенный раствор цинкового купороса добавляют немного сер-
серной кислоты. После этого раствор направляют на кристаллизацию
цинкового купороса или используют для производства литопона и
других продуктов48. Сушку цинкового купороса можно вести во
взвешенном слое. На изготовление 1 г цинкового купороса расхо-
расходуют: 0,47 г цинка (в пересчете на металл) и 0,72 т серной кис-
кислоты A00%).
Из твердого остатка, получающегося при растворении цинка
в серной кислоте, извлекают свинец и олово49.
Наибольшее количество сульфата цинка изготовляют сейчас из
маломедистой пыли, получаемой при вторичной переработке цвет-
цветных металлов, содержащей 65—70% цинка, 1—6% олова, 7—10%
свинца и ~0,6% меди. Суспензию пыли в оборотном растворе под-
подвергают «нейтральному» выщелачиванию — обработке серной кис-
кислотой при 90° и при продувке барботирующим воздухом. Количе-
Количество добавляемой кислоты недостаточно для извлечения из сырья
всей окиси цинка и в раствор переходит ~90% ZnO. Избыток
окиси цинка и окисление Fe2+ в Fe3+ кислородом воздуха обеспе-
обеспечивают хорошую очистку раствора от железа. Величину рН регу-
регулируют так. чтобы избежать осаждения меди, перешедшей в рас-
раствор из сырья.
После «нейтрального» выщелачивания пульпа сгущается в от-
отстойниках, и сгущенная пульпа поступает на «кислое» выщелачи-
выщелачивание, а слив проходит контрольную фильтрацию через фильтр-
пресс и очищается от меди цементацией цинковой пылью при
50—70° в аппарате с мешалкой. После отделения цементной меди
на фильтрпрессе полученный раствор содержит 130—140 г/л цинка
в виде ZnSO< и направляется на выпарку в двухкорпусную вы-
выпарную батарею, работающую под давлением. Давление в I и
II корпусах составляет соответственно 3 и 1,5 ат, и температуры
кипения раствора в них 140 и 120°. Из II корпуса выводится ймесь
насыщенного раствора и взвешенных в ней кристаллов ZnSO4 • Н2О,
причем общая концентрация соответствует отношению ZnSO4 : Н2О,
равному 1 : 5—6. Суспензию охлаждают до полного отверждения
на поверхности барабана, внутри которого циркулирует вода, в
получают чешуйчатый продукт, состоящий из ZnSO4 • 7Н2О с при-
примесью ZnSO4 • Н2О.
«Кислое» выщелачивание пульпы, сгущенной после «нейтраль-
«нейтрального» выщелачивания, осуществляют серной кислотой также при
нагреве до 90° и с продувкой — перемешиванием воздухом. При
этом окись цинка практически полностью переходит в раствор, со-
содержащий ~100 г/л Zn и 1 г/л H2SO4. После фильтрации на
фильтрпрессе этот оборотный раствор используют для приготов-
приготовления суспензии исходной пыли. Отделенные на фильтре евинцово-
оловянные кеки направляют на дальнейшую переработку.
Получение цинкового купороса из медистой окиси цинка
К неиспользуемым отходам в производстве вторичной меди
принадлежит так называемая медистая окись цинка, содержащая
35—45% ZnO, до 10% закисной и окисной меди, до 20% SiO2. Ее
не используют для выплавки меди из-за образования вязких шла-
шлаков и настылей, а для выработки цинкового купороса — из-за вы-
высокого содержания меди.
Получение цинкового купороса из медистой окиси цинка воз-
возможно следующим способом 50, основанным на переводе большей
части окиси цинка в раствор, а окиси меди в осадок в результате
реакции:
CuSO4 + ZnO - ZnSO4 + CuO
Очевидно, реакция протекает через стадию гидратации окиси цин-
цинка с образованием гидроокиси меди.
Процесс осуществляют в две стадии, проводимые в разных
реакторах. В первом реакторе происходит образование конечного
Раствора, содержащего 45—47% ZnSO4, направляемого на кри-
кристаллизацию. В этот реактор поступает содержащий медь ней-
нейтральный 37% раствор из второго реактора, цинковое сырье в
726
Гл. XIX. Сульфат и хлорид цинка
Сульфат цинка
727
40—80% от общего количества серной кислоты, необходимого д
сульфатизации ZnO, PbO и СаО (без учета расхода H2SO4 на
взаимодействие с CuO, FeO, Fe2O3, которые остаются в кеке в виде
окислов). Твердый остаток (промежуточный кек) из первого реак-
реактора обрабатывают серной кислотой во втором реакторе, где из
него извлекается остаток окиси цинка и растворяется некоторое
количество окиси меди. Оптимальная температура во втором реак-
реакторе 70°. Основная масса меди,
поступающей в сырье выводится;
из второго реактора с конечным
кеком, который промывают водой,
Рис. 199. Влияние длитель-
длительности процесса на очистку
растзоров ZnSO4 от меди
при 50 и 70°.
0 0,5 1,0 1,5 2,0 2,5 3,0
Время, ч
Рис. 200. Содержание меди
в кристаллах ZnSO4 в за-
зависимости от длительности
первого выщелачивания при
50, 60 и 70°.
для уменьшения потерь растворимого сульфата цинка. Промыв-
Промывную воду возвращают во второй реактор. Содержание цинка
в остаточном кеке колеблется в пределах 6—8%, выход кека
~55% от веса сухой исходной пыли. Степень извлечения ZnO со-
составляет 90—92,5% •
Взаимодействие окиси цинка с сульфатом меди замедляется
с увеличением концентрации ZnSO4 в исходном растворе. Так, при
повышении концентрации с 31 до 36% ZnSO4 продолжительность
процесса увеличивается в 2 раза. Таким способом можно получить
раствор ZnSO4, содержащий всего 0,05 г\л меди, без затраты ме-
металлического цинка на очистку раствора (рис. 199 и 200).
Удаление железа из раствора основано на предварительном
окислении Fe2+ в Fe3+ и взаимодействии сульфата окиси железа
с окисью цинка:
Fe2(SO4K + 3ZnO + 3H2O = 2Fe(OHK + 3ZnSO4
Окисление можно производить барботирующим через раствор
воздухом; кислород воздуха в нейтральной среде энергично окис-
окисляет Fe2+. На рис. 201 показана зависимость содержания железа
в кристаллах цинкового купороса от содержания его в исходном
растворе. Кристаллы, полученные из
растворов с одинаковым содержанием
железа и меди, загрязнены железом
приблизительно в 10 раз больше, чем
медью. Это обусловливает необходи-
необходимость хорошей очистки растворов цин-
цинкового купороса от железа для полу-
получения продукта высокого качества.
0А\
¦ад
.0,1
О 0,01 0,02 0,03
Содержание Fe Вкристаллах,%
Рис. 201. Влияние содержа-
содержания
pax
цинка.
Извлечение цинка из колчеданных
огарков хлорирующим обжигом
Представляет интерес извлечение
цинка из старых колчеданных огар-
огарков, скопившихся на сернокислотных
заводах, в тех случаях, когда они со-
содержат значительные количества цин-
цинка. При хлорирующем обжиге этих
огарков и последующем выщелачива-
выщелачивании цинк извлекается в раствор в ви-
виде смеси сульфата и хлорида. Разделение их связано со значитель-
значительными трудностями, поэтому получаемый раствор используют, глав-
главным образом, для производства литопона 49.
Используемые колчеданные огарки содержат ~ 9,5% Zn и
5—6% S. Их смешивают с необожженным колчеданом, поваренной
солью и с огарком, не содержащим цинка, так, чтобы шихта со-
содержала 4,5—5% Zn и 4—4,5% S. Смесь составляют приблизи-
_ тельно из 40% огарка, содержащего цинк, 40 % использованного
' огарка (без цинка), 3,57о колчедана и 16,5% поваренной соли. Об-
Обжиг производят при 550° в механической полочной печи. Из печи
выходит «плав», содержащий 4,8—5,1% общего цинка, 4,2—4,7%
водорастворимого цинка, 13,4—15,2% С1 (в расчете на NaCl),
0,01—0,03% водорастворимого железа и сульфат натрия. При вы-
выщелачивании «плава» в раствор переходят ZnSO4, ZnCl2 и Na2SO4.
Получаемый раствор содержит (в г/л): 100—130 Zn, 300—400
Na2SO4 и 270—380 С1 (в пересчете на NaCl). После выморажива-
вымораживания сульфата натрия этот раствор используют для осаждения ли-
литопона смешением его с раствором сульфида бария:
ZnSO4 + ZnCl8 + 2BaS = 2ZnS + BaSO4 + BaCl2
В связи с тем, что в растворе имеется не только ZnSO4, но и
ZnCl2, литопои получается с повышенным содержанием ZnS, Для
728
Гл. XIX. Сульфат и хлорид цинка
Литература
729
устранения этого к осадку добавляют тонкоизмельченный отбе-
отбеленный барит. Раствор хлорида бария, остающийся после отделе-
яйя литопона, перерабатывают на кристаллический хлорид бария
(выпаркой и кристаллизацией) или на бланфикс — осаждением
BaSO4 сульфатом натрия.
ХЛОРИД ЦИНКА
Наиболее распространенный способ получения хлорида цинка
заключается в обработке цинковых отбросов технической 27—
28%-ной соляной кислотой. Обработку ведут в резервуарах, в ко-
которые загружают цинковые отбросы и заливают кислоту. Переме-
Перемешивание осуществляют сжатым воздухом или с помощью меша-
мешалок. Иногда несколько резервуаров располагают террасообразно,
« жидкость перетекает из одного в другой, причем концентрация
ZnCl2 постепенно повышается до 45%- Количество кислоты берут
•из расчета получения раствора ZnCl2, не содержащего свободной
кислоты.
Находящиеся в сырье примеси соединений свинца, S1O2 и дру-
тие при растворении в соляной кислоте образуют осадки, отклады-
откладывающиеся на зернах растворяющегося материала, содержащего
цинк, в виде непроницаемой корки. Предотвратить это можно пу-
путем предварительного окислительного обжига измельченных цин-
цинковых отходов при 300—500°61.
Реакции растворения цинка и окиси цинка экзотермичны и про-
протекают весьма энергично, но общая продолжительность процесса
достигает 10 ч и более, так как к концу он замедляется. При рас-
растворении выделяются большие количества водорода:
Zn + 2НС1 = ZnCl2 + Н2
Реакционные резервуары обычно устанавливают вне здания,
Под навесом, и водород удаляется в атмосферу.
Полученные растворы хлорида цинка подвергают очистке от
Экелеза, которую производят с помощью окислителей, не загряз-
загрязняющих раствор значительными количествами продуктов окисле-
окисления. В качестве окислителей применяют газообразный хлор, берто-
бертолетову соль и чаще всего наиболее дешевый реагент — хлорную
известь. В последнем случае в раствор переходит СаС12. Окисное
железо осаждают из раствора при нейтрализации кислотности, на-
например добавкой извести. Обычно известь вводят одновременно
•С хлорной известью. Раствор отстаивается от осадка и нераствори-
нерастворимой мути — песка и других примесей, введенных с сырьем, и де-
декантируется, а иногда фильтруется.
При применении в качестве сырья окшары необходимости в
очистке раствора нет, так как в окшаре большинство примесей на-
находится в виде растворимых солей, которые выщелачиваются горя-
горячей водой перед растворением окшары в соляной кислоте. ,
Для получения плавленого хлорида цинка раствор выпаривают
в чугунных котлах, обогреваемых топочными газами. Температура
раствора по мере повышения его концентрации поднимается. Про-
Процесс уварки сопровождается загустеванием массы при 190—280°
вследствие выделения твердой фазы; при дальнейшем подъеме тем-
температуры масса вновь становится жидкой. При подъеме темпера-
температуры до 310—340° можно получить продукт, содержащий всего
_~1,2% воды52. Без применения вакуума удалить всю воду невоз-
невозможно, так как для этого пришлось бы поднять температуру выше
400°, что сопровождалось бы значительным гидролизом ZnCl2. Гид-
Гидролиз имеет место и при менее высоких температурах; для его
уменьшения в процессе выпаривания в раствор добавляют немного
соляной кислоты. Выделяющийся при выпаривании хлористый во-
водород разъедает стенки чугунного котла, и продукт загрязняется
хлористым железом.
В связи с трудностью полного удаления воды и увеличением
"расхода топлива на последней стадии обезвоживания выпарку за-
заканчивают при сравнительно низкой температуре B20—250°). При
этом в готовом плаве остается 9—12% воды. Такой плав заливают
в железные барабаны, где он при охлаждении затвердевает, после
чего барабаны запаивают.
На 1 г 45% раствора хлорида цинка расходуют 0,23—0,25 т ме-
металлического цинка (96%-ного) или соответствующее количество
материала, содержащего цинк, и 0,9—1 т 27,5%-ной соляной кис-
кислоты. На 1 т плавленого хлорида цинка расходуют 2,4 т 45% рас-
раствора ZnCb и около 0,5 г угля G000 ккал/кг).
ЛИТЕРАТУРА
I- М. И. Архипов, А. Б. Пакшвер, Н. И. Подборнова, ЖПХ, 23.
№ 6, 650 A951). — 2. Н. Keifiig, H. Reimers. Chem.-Ztg. Chem. Apparat., 86,
№ 14, 488 A962). — 3 3. W. Fulton, D. F. Swinehart, J. Am. Chem. Soc
.76, № 3, 864 A954). — 4. В. В. Печковский, Л. П. Костин, А. Г. Звез-
дин, Труды Пермск. политехи, ин-та, № 10, 1961, стр. 155. — 5. О. К. S r i v a s-
tava, E. A. Sec с о, Canad. J. Chem., 45, № 6, 579 A967). —б. Е. Про, Цинк
и кадмий, ГОНТИ, 1931. —7. Б. Д. А в е р б у х, Е А. Ветреико Г. И Ч у-
фаров, ЖПХ, 32, № 6, 1221 A959). — 8. В. Г. Трои ев, Научио-иссл. работы
химических ин-тов и лабораторий АН СССР за 1941—1943 гг.. Изд. АН СССР
1945. — 9. А. С. Карнаухов, В. Г. Шевчук, ДАН СССР. 90. № 2, 191
A953). —10. И. Н. Кузьминых, С. Л. Любалина, Хим пром., № 12 358
A950).
П. Я. А. Фиалков, 3. А. Шека, ЖНХ, 1, № 6, 1238 A956).—
'2. В. М. Чайковская, Г. Ф. Афанасьев, Г. Н. Знаменский, ЖПХ
36, № 6, 1355 A963). —13. В. Г. Шевчук, И. Г. Дружинин, Изв. вузов
хим. и хим. технол., № 2, 25 A958). — 14. В Г. Шевчук И Н Лепешков
ЖНХ, 1, № 8, 1888 A956). — 15. Н. Д. Мазовер, ЖПХ, 25, № 7, 756 A953) —
F- Н. Н. Евсеева, А. Г. Бергман, ИСФХА АН СССР, 21, 208 A952); 22,
162 A953); 24, 162 A954). —17. Н. П. Лужная, Научно-иссл. работы химиче-
химических ин-тов и лабораторий АН СССР за 1941—1943 гг., Изд. АН СССР, 1945,
СтР. 71. —18. В. В. Печковский, А. Н. Кетов, Уч. зап. Пермск. гос. унта.
730
Гл. XIX. Сульфат и хлорид цинка
17, №' 1, 15 A960); Труды Пермск. политехи, ин-та, № 10, 1961, стр. 3.—
19. С. Д. Шаргородский, Укр. хим. ж., 15, 332 A949).— 20. Б. И. Скач
ков, Цветные металлы, № 11, 25 A968).
21. Е. В. М а р г у л и с, В. Д. Пономарев, Изв. АН КазССР. Сер. метал-
металлург., обогащения и огнеупоров, № 3 F) A960).—22. В. В. Печковский,
ЖПХ, 31, № 8, 1139 A958).— 23. J. Besson, P. Grange, Compt. rend., 238,
№ 20, 2002 A954).— 24. М. И. Равич, Е. В. Фролова, Научио-иссл. работы
химических ин-тов и лабораторий АН СССР за 1941—1943 гг.. Изд. АН СССР
1945. — 25. 3. К. Зубахина, Труды НИОХИМ, т. 19, 1969, стр. 105. — -
26. Л. М. Во лов а, В. И. Остроушко, авт. свид. 199129, 1966.—
27. С. W. Zielke a. oth, Ind. Eng. Chem., № 2, 158 A966). — 28. А. А. С a-
л и н, М. Е. С ы р о е ш к и н, Электролиз сернокислого цинка, Госметаллургиздат,
1959. — 29. С. А. Т и т о в, Выщелачивание и электролиз цинка, Госметаллургиз-
Госметаллургиздат, 1943. — 30. М. В. В. К а т а л ы м о в, сб. «Исследования по прикладной хи-
химии», Изд. АН СССР, 1955, стр. 325.
31. С. И. В о л ь ф к о в и ч, сб. «Микроэлементы в жизни растений и жи-'
вотных», Изд. АН СССР, 1952. — 32. С. Н. Иванова, Журн. ВХО, 9, № 5,
496 A964). — 33. С. И. Вольфкович, Т. М. Стронгин, Р. Е. Рем ей,
К. Е. Писарев, А. И. Шишкина, Труды НИУИФ, в. 167, 1960, стр. 5.—
34. Г. Г. У р а з о в, Л. Р. Э д е л ь с о н. Материалы по металлургии цветных ме-
металлов, Кубуч, 1932. — 35. М. R е у, P. R a f f i n o t, Rev. ind. minerale, 35,
№ 605, 134 A954). — 36. В. А. Куракин, ЖХП, № 13, 25 A941).—
37. Б. П. Волгин, Е. М. Л я п у с т и н а. Труды УНИХИМ, в. I, 1954, стр. 133.—
38. И. Л. П е й с а х о в, сб. «Обогащение и очистка газов цветной металлургии»,
Госметаллургиздат, 1953. — 39. С. И. Вольфкович, Ф. Г. Марголис Изв
АН СССР. ОХН, 5, 322 A942); ЖПХ, 28, № 5, 453 A955).— 40. В. И. Смир-
Смирнов, А. И. Тихонов, Обжиг медных руд и концентратов, Госметаллуогиздат,
1958.
41. В. В. Печковский, ЖПХ, 30, № 11, 1579 A957); 32, № 5, 966
A959). — 42. В. В. Печковский, Уч. зап. Пермск. гос. ун-та, 17, № 1, 91
(I960).—43. А. П. Сну р ников, В. Ф. Ларин, сб. «Исследования в области
химии и технологии минеральных солей и окислов», Изд. «Наука», 1965,
стр. 97.-44. Пат. ФРГ 1192167, I960. — 45. В. Л. Щукин, Производство лито-
литопона. ГОНТИ, 1931.— 46. Е. В. Санадзе, Хим. пром., № 2, 51 A950).—
47. Е. Schwarz-Bergkampf, Berg und Huttenmann, Monatschr., 98, № 1, 17
A953). — 48. Г. М. Ракович, С. Ф. Матвеева, Бюлл. Центр, ин-та информ.
цвет, металлург., № 3, 23 A956).— 49 Е. Ф. Беленький, И. В. Р и скин,
Химия и технология пигментов, Госхимиздат, 1960. — 50. И. Н. Кузьминых.
С. Л. Л ю б а л и н а, ЖПХ, 25, № 3, 257 A952).
51. Н. Н. Ефремов, А. М. Р о з е н б е р г, сб. «Работы по химической тех-
технологии минеральных веществ», Свердловск, 1929, стр. 171, 185.—52. Н. Н. Е ф-
р е м о в; Е. М. Я к и м е ц, сб. «Работы по химической технологии минеральных
веществ», Свердловск, 1929, стр. 153.
Глава XX
СУЛЬФАТ НИКЕЛЯ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Никель образует два основных окисла: закись никеля NiO и
окись никеля №2О3 и соответствующие им гидроокиси. Гидрат за-
закиси никеля Ni(OHJ образуется в виде светло-зеленого осадка при
действии щелочей на растворы солей двухвалентного никеля. При
нагревании теряет воду и переходит в закись никеля серо-зеленого
цвета. Степень гидратации является важным свойством гидрата
закиси никеля, особенно в связи с применением его для изготовле-
изготовления анодной массы щелочных аккумуляторов — степенью гидрата-
гидратации определяется структура и электрохимические свойства анодной
массы. Вода в гидрате имеет две формы связи — молекулярную и
гидроксильную. По мере обезвоживания возрастает удельная по-
поверхность и пористость гидрата1. Гидрат окиси никеля Ni(OHK,
имеющий черно-бурый цвет, образуется при окислении Ni(OHJ.
В водных аммиачных растворах растворимость Ni(OHJ при 20°
равна 2,35 г/л Ni при концентрации 36,4 г/л NH3 и 14,5 г/л Ni при
193 г/л NH3 2.
При взаимодействии 0,2 н. раствора NiCl2 с 2 н. щелочными
смесями, содержащими NaOH и менее 50% Na2CO3, образуются
суспензии гидрата закиси никеля, характеризующиеся интенсив-
интенсивным уменьшением рН раствора (на 1—1,8 за 24 ч). Старение осад-
осадка сопровождается уменьшением количества щелочи в растворе и
превращением карбоната в бикарбонат за счет обмена ионов ОН~
раствора на ионы СОз"\ адсорбированные осадком3. О старении
основных карбонатов никеля см. 4~7.
Основной карбонат никеля является промежуточным продуктом
при аммиачном выщелачивании никелевых руд. Предварительно
РУды подвергают восстановительному обжигу. Выщелачивание
производят водным раствором аммиака, содержащим карбонат
аммония, в присутствии кислорода. После выщелачивания из
732
Гл. XX. Сульфат никеля
раствора отгоняют аммиак, в результате чего осаждается основной
карбонат никеля, который подвергают обжигу8.
С окисью углерода никель образует жидкое летучее соедине-
соединение— тетракарбонил никеля №(СОL, которое при нагревании раз-
разлагается с выделением металлического никеля. На этом основан
один из способов извлечения никеля из некоторых продуктов пер-
первичной переработки руд, в которых основная масса никеля нахо-
находится уже в виде металла.
Сульфат никеля NiSO4 кристаллизуется из водных растворов
при температурах от —3,15 до 31,5° в виде NiSO4-7H2O — никеле-
никелевого купороса, кристаллы которого имеют форму ромбических
призм изумрудно-зеленого цвета. При выветривании купороса на
воздухе, а также при кристаллизации из водных растворов при
температуре от 31,5 до 53,6° образуется cc-NiSO4 • 6Н2О синеватого
цвета; в интервале 53,6—99° кристаллизуется |3-NiSO4-6H2O зеле-
зеленого цвета. При более высоких температурах образуются метаста-
бильные кристаллогидраты с меньшим числом молекул воды (от
5 до 2) и стабильный NiSO4 • Н2О с точно не установленными тем-
температурами превращения вследствие затруднений, обусловленных
образованием сильно пересыщенных растворов. Выше 280° сульфат
никеля обезвоживается. N1SO4 устойчив до 800°, а выше этой тем-
температуры разлагается с выделением SO39. Насыщенный водный
раствор содержит при 0° —21,4%, при 99° —43,4 NiSO4.
Обычно при кристаллизации охлаждением насыщенного рас-
раствора вначале выделяется шестиводный сульфат никеля, а затем
образуется некоторое количество семиводного. Соотношение между
ними зависит от температуры начала и конца кристаллизации, а
также от скорости перехода одной формы сульфата в другую. При
быстрой кристаллизации из концентрированных растворов полу-
получается продукт приблизительного состава N1SO4 • 6,2Н2О, при мед»
ленной кристаллизации — NiSO4 • 6,5Н2010.
При взаимодействии растворов сульфата никеля и едкой ще-
щелочи выделяются осадки основных сульфатов никеля, состав кото-
которых зависит от концентрации растворов. При добавлении 0,1 н.
раствора NaOH к растворам NiSCli с концентрацией, меньшей 0,1 М,
образуется осадок гидрата закиси никеля; в интервале концен-
концентраций А, 1—\М NiSO4 осаждается 3NiSO4-4Ni(OHJ, а при еше
больших концентрациях NiSO4 вначале выделяется 3NiSO4-
• 2Ni(OHJ, постепенно переходящий в 3NiSO4-4Ni(OHJn. По
другим данным12, при приливании раствора NaOH к раствору
NiSO4 образуется осадок NiSO4-4Ni(OHJ, а при обратном по-
порядке сливания растворов — NiSO4-5Ni(OHJ. Четырехосновиой
сульфат никеля неустойчив в воде и растворах NaOH и переходит
в"№(ОНJ.
В системе NiSO4—H2SO4-—Н2О существуют13 комплексные
ионы [Ni(SO4J]2". Никельаммонийсульфат Ni(NH4J(SO4J,обра-
i
Применение
733
зуется при подкислении серной кислотой аммиачного раствора суль-
сульфата никеля от рН = 7,1 -*¦ 8 до рН =» 4 при 55—60°. В присутствии
в растворе также кобальта при снижении рН до 6,2 выделяется
почти весь никель (практически свободный от кобальта) в виде
Ni(NH4J(SO4J. При снижении рН оставшегося раствора до 4 вы-
выделяется остальной никель и почти весь кобальт в "виде
Ni(NH4J(SO4J и Co(NH4J(SO4J14-
В системе NiSO4—COSO4—Н2О образуются смешанные кри-
кристаллы; при 20° в ряду смешанных кристаллов имеется разрыв 15.
В системе NiSO4—Na2SO4 образуются твердые растворы в ши-
широкой области со стороны сульфата натрия и три двойные соли:
3Na2SO4 • N1SO4, Na2SO4 • N1SO4 и Na2SO4 • 3NiSO4. Имеются две эв-
эвтектики: при 41% NiSO4 —671° и при 55% NiSO4 — 700°16.
ПРИМЕНЕНИЕ
Никелевый купорос применяют главным образом в гальвано-
гальванотехнике при никелировании металлов, для изготовления аккумуля-
аккумуляторов, для производства твердых сплавов, в жировой и парфюмер-
парфюмерной промышленности, для изготовления катализаторов и пр.
В смеси с другими препаратами его используют в качестве фун-
фунгицида ".
Технический никель сернокислый выпускают трех марок.
НС-0 для производства реактивов, НС-1 с Государственным зна-
знаком качества и НС-1 для никелирования, аккумуляторной, жиро-
жировой и парфюмерной промышленности. Согласно ГОСТ 2665—73,
сульфат никеля должен содержать не менее 20,6% никеля вместе
с кобальтом, а при отсутствии последнего не менее 20,3% никеля.
Допустимые количества примесей указаны в табл. 48.
ТАБЛИЦА 48
Максимально допустимые количества примесей в техническом
сульфате никеля, %
Марка
НС-0
НС-1 с Гос. зн.
кач.
НС-1
Со
0.2
—
РЬ
0,001
0,001
0,002
Zn
0,004
—
—
Fe
0,001
0.002
0.003
Си
0,001
0,002
0,003
Са
0,08
0,2
0,2
Mg
0.03
0.03
0,03
CI
0,01
0.1
0.1
МЫЙ
таток
I Q.O
0,03
0.05
0.05
В сульфате никеля марки НС-0 содержание Са, Mg, Na и К в
сумме не должно превышать 0,3%; в продукте для аккумуляторной
промышленности должно быть меньше 0,02% магния, а для никели-
никелирования— меньше 0,004% цинка.
734
Гл. XX. Сульфат никеля
ПОЛУЧЕНИЕ НИКЕЛЕВОГО КУПОРОСА РАСТВОРЕНИЕМ НИКЕЛЯ
В СЕРНОЙ КИСЛОТЕ
Небольшие количества технического сульфата никеля изготов-
изготовляют растворением металлического никеля в серной кислоте, к ко-
которой добавлено немного азотной кислоты. На 1 кг металлического
никеля берут 5 кг 33—35% -ной серной кислоты и 120—150 г азот-
азотной кислоты A00%). Реакция идет при нагревании паром до 60°.
Полученный раствор сульфата никеля, отделенный от нерастворив-
шегося остатка, в случае необходимости подвергают очистке от
примесей железа и меди. Медь может быть вытеснена из раствора
порошком металлического никеля, а железо после этого выде-
выделяется в виде гидрата окиси действием черного гидрата никеля
(смесь гидратов 2, 3 и 4-валентного никеля) или зеленого гидрата
никеля (основной карбонат никеля) в присутствии кислорода воз-
воздуха. Полученный раствор сульфата никеля подвергают кристал-
кристаллизации обычными способами.
Примеси железа и меди могут быть отделены от раствора
NiSO4 ступенчатым осаждением гидроокисей аммиаком с отделе-
отделением осадка гидроокиси железа при рН=3 и гидроокиси меди при
рН=4,5. Осаждение гидроокисей облегчается при введении перед
каждой ступенью нейтрализации затравки соответствующей гидро-
гидроокиси 18. Разработан метод очистки раствора NiSO4 от примесей
железа и меди противоточной экстракцией; экстрагентом является
Ni-мыло 19.
Очистка раствора NiSO4 от ионов цинка может быть осущест-
осуществлена анионообменным методом с применением смолы — вофатита
MD. Предварительно ее обрабатывают сероводородом для насы-
насыщения ионами S2"; при обработке под давлением 19 ат концентра-
концентрация серы достигает 0,58 г на 100 мл смолы. В результате ионного
обмена при рН = 3,2 протекает реакция:
S-вофатит + Zn2+ + SO4" - ZnS + SQff + вофатит
При этом 80—90% выделяющегося сульфида цинка задержи-
задерживается в свободном объеме ионообменной колонны, равном 60—
70% от объема смолы. Остальное количество ZnS улавливается не-
неполярным фильтром — вофатитом F20.
Ионообменные смолы могут быть также использованы для
очистки раствора NiSO4, полученного из отработанных катализато-
катализаторов. Предварительно смолу обрабатывают щелочью. При пропу-
пропускании через такую смолу кислого водного раствора, содержащего
ионы Fe3+ и Ni2+, в результате ионного обмена рН раствора повы-
Получение никелевого купороса
735
шается до 3—5 и из раствора выделяется только железо. Наиболее
эффективно Fe3+ отделяется при рН=4,5. Такая величина рН до-
достигается при обработке щелочных смол раствором NH4C121,
Очистка растворов NiSO4 при помоши ионообменных смол яв-
является, по-видимому, наиболее эффективной.
ПОЛУЧЕНИЕ НИКЕЛЕВОГО КУПОРОСА ИЗ РАСТВОРОВ
КОБАЛЬТОВОГО ПРОИЗВОДСТВА
Наибольшее количество сульфата никеля получают сейчас из
никелевых сульфатных растворов кобальтового производства, со-
содержащих ~30 г/л Ni. Они образуются при растворении в серной
кислоте зеленых гидратов, получаемых при электролитическом рас-
растворении никель-кобальтовых анодов или при растворении кобаль-
кобальтового шпур-штейна в атмосфере кислорода под давлением (в ав-
автоклаве).
Из сульфатного раствора, являющегося отходом кобальтового
производства, осаждают раствором соды карбонат никеля. Реак-
Реакционную смесь перемешивают воздухом в аппаратах Пачука и на-
нагревают паром до 80°. Полученная суспензия отстаивается и верх-
верхний слив удаляют в бассейн сточных вод, а из сгущенной пульпы
отделяют карбонат никеля на дисковом вакуум-фильтре. Осадок,
снятый с фильтра, имеет влажность 70%; его репульпируют с во-
водой в мешалке и повторно отфильтровывают на барабанном ва-
вакуум-фильтре. С последнего промытый влажный осадок направ-
направляют в аппараты Пачука, где его растворяют при 80—85° в кон-
концентрированной серной кислоте (купоросном масле) для получения
раствора NiSO4, содержащего 160 г/л Ni. Этот раствор очищают
от примесей железа, меди и кобальта добавкой влажного карбо-
карбоната никеля. После отделения осадков-примесей на фильтрпрессе
раствор выпаривают в вакуум-выпарной батарее до содержания
260—270 г/л Ni и направляют на кристаллизацию никелевого купо-
купороса в механические или вакуум-кристаллизаторы, где раствор
охлаждается до 17°. Кристаллы NiSO4-7H2O отделяют на центри-
центрифугах и с гигроскопической влажностью 3—5% укупоривают или
предварительно брикетируют на гидравлических прессах в блоки.
Часть маточного раствора, содержащего 120—140 г/л Ni, добав-
добавляют к раствору, получаемому после растворения карбоната ни-
никеля в серной кислоте, другую часть присоединяют к исходному
раствору, поступающему из кобальтового производства.
ПОЛУЧЕНИЕ НИКЕЛЕВОГО КУПОРОСА ИЗ РАСТВОРОВ
МЕДЕЭЛЕКТРОЛИТНЫХ ЗАВОДОВ
При электролитическом рафинировании меди на медеэлектро-
литных заводах сульфат никеля также получают в качестве по-
побочного продукта. При электролитическом растворении медных
736
Гл. XX. Сульфат никеля
анодов и осаждении меди на катоде электролитом служит раствор
медного купороса (стр. 688). При рафинировании меди некоторые*
содержащиеся в ней примеси почти полностью переходят в раствор,
другие — в шлам, а третьи — частично в шлам, частично в раствор.
Полностью переходят в раствор металлы, более электроположи-
электроположительные, чем медь. К ним относятся никель, цинк и железо. Эти ме-
металлы не осаждаются на катоде и постепенно накапливаются в
растворе, что приводит к уменьшению растворимости сульфата
меди и к ухудшению условий электролиза. Для поддержания в
электролите минимальной концентрации примесей часть раствора
периодически выводят из цикла электролиза и взамен добавляют
к электролиту серную кислоту. Выведенный раствор подвергают
регенерации.
При растворении 250 г анодов в сутки, содержащих 0,1% Ni,
в раствор переходит 250 кг Ni и необходимо ежесуточно выводить
из цикла 10 м3 раствора при содержании в нем 25 г/л № или 25 ж3
при концентрации 10 г/л Ni. Обычно раствор, отбираемый для ре-
регенерации, содержит (в г/л): 40 — Си, 180 — H2SO4, 15—Ni, 3 —
Fe, 7 — As и др. Его вначале нейтрализуют медным скрапом в на-
травочной башне при продувке воздухом (стр. 671) и после вы-
выпарки кристаллизуют при охлаждении медный купорос. Маточ-
Маточный раствор подвергают дополнительной выпарке с последующей
кристаллизацией уже загрязненного медного купороса, который
очищают перекристаллизацией. Конечный маточный раствор со-
содержит ~50 г/л Си, 40 г/л H2SO4 и 50 г/л Ni. Его подвергают
электролизу в ваннах со свинцовыми анодами, причем медь осаж-
осаждается на катодах с большим количеством примесей и отправ-
отправляется затем в переплавку вместе с черновой медью. Отработан-
Отработанный электролит содержит до 120 г/л H2SO4, 2 г/л Си и 55 г/л Ni.
Его выпаривают и охлаждают для кристаллизации никелевого ку-
купороса. Оставшийся маточный раствор вторично выпаривают и
подвергают кристаллизации и, при надобности, второй маточный
раствор подвергают аналогичной обработке. При первой кристал-
кристаллизации получают достаточно чистый никелевый купорос, а при
следующих операциях кристаллизуется загрязненный продукт, ко-
который подвергают перекристаллизации. Конечный раствор после
выпарки до 1200 г/л H2SO4 и очистки от примесей возвращают на
электролиз меди22.
ЛИТЕРАТУРА
1. Н. Г. Клюки на, Г. М. Кудряшова, ЖПХ, 36, № 3, 495 A963).—
2. М И. Архипов, А. Б Пакшвер, Н. И. Подборнова, Там же,23, №6,
650 A950). —3. О. Bagno, Compt. rend., 236, № 12, 1275 A953).—
4. И. М. В а ее ер м а н, X. 3. Брайнина, ЖПХ, 31, № 11, 1617 A958).—
5. И. М. В а с с е р м а н, Там же, 32, № 9, 1959 A959). — 6. И. М. В а с с е р м а н,
Е. А. Фомина, X. 3. Брайнина, Там же, 32, № И, 2619 A959). —
7. И. М. Вассерман, Е. А. Фомина, Там же, 34, № 1, 90 A961).—
Литература
737
'
Госметаллургаздат.
1238 A956) — 14. F. A. Forward, канад. пат. 511442 1955 —15 О С Гп
болева Наук зап. Льв1вськ. ун-та, 34, 56 A955) -16 К А Боль
ЖуГВВХО 9 4А°^ Ж 2<Sf I Ш ™ -П- С" ? Иванова,
Журн. ВХО, 9, J\s 5, 496 A964). —18. My ко ям а, япон. пат 854 1955 —
19. Н И. Гельперин и др., ЖПХ, 34, № 8, 1728 A966)- сб «Процессы жил
костной экстракции и хемосорбции», Изд. «Химия» 1966 Рстр 29* -
20. К. Eckstein, Bergakademie, 7, № ю, 483 A955)
12 М966?УВИ А 1°ПИЬйаВа' К°баяси- J- Oil Chem. Soc. Japan, 3. № Б,
дат 1958 Цейдлер. Металлургия меди и никеля, Госметаллургиз-
V224 М. Е. Поэвн
Физико-химические свойства
739
Глава XXI
ХЛОРИД КАЛЬЦИЯ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Хлорид кальция СаС12 образует белые кристаллы кубической
формы с плотностью 2,51 г/см3. Плавится при 772°. Сильно гигро-
гигроскопичен на воздухе расплывается. Равновесная относительная
влажность воздуха над СаС12-2Н2О при 20° равна 22 %, при 50°—
17%, а над СаС12-6Н2О при 10° —38%, при 20° —32,3%, при
24,5°'—31%. Высушивание гранулированным хлоридом кальция
при 25° позволяет понизить влажность газа до 0,14—0,25 г воды
в 1 м3. Практическая влагоемкость хлорида кальция при 25° (по-
(поверхностная) :
Относительная влажность газа, % .... 36 60 70 80 85 90 95
Влагоемкость, кг/кг 1,0 1.6 2,0 2,8 3,5 5,0 8,4
Сведения о растворимости и кристаллогидратах СаС12 даны
в табл. 49 и на рис. 202. О вязкости водных растворов СаС12 см.'.
Температуры кипения растворов СаС12:
СаС12, % 40,8 50 58 76
гкип, -С ., 120 130 140 175
С аммиаком СаС12 образует двойные соединения CaCl2-4NH3,
CaCl2-8NH3.
При повышенных температурах СаС12 разлагается кислородом
воздуха и парами воды по реакциям
СаС12 + 0,5О2 ^± СаО + С12
СаС12 + Н2О ^r± CaO + 2HC1
а в присутствии SiO2 или А12Оз идут реакции:
2CECI2+SiO2 + O2 ^± 2CaO-SiO2 + 2Cl2
ЗСаСB+А12О3+1,5О2 ^± ЗСаО • A12OS + ЗС12
Заметное разложение СаС12 в сухом и влажном воздухе идет
выше 830°, в присутствии SiO2 — выше 720°, в присутствии А12Оз —
выше 800°2.3.
Растворимость СаС12 в воде
ТАБЛИЦА 49
Температура,
°С
—49,8
—25
0
20
30
29,5
29,2
40
45,3
50
75
100
125
150
175,5
200
260
300
Концентрация СаС12
в растворе, %
30,5
33,4
37,3
42,7
50,0
52,4
53,0
53,6
56,5
56,9
59,1
61,3
63,7
67,2
74,9
75,7
77,7
80
Твердые фазы
Лед + СаС12 • 6Н2О
СаС12 • 6Н2О
То же
» »
СаС12 • 6Н2О + а-СаС12 • 4Н2О
СаС12 • 6Н2О + Р-СаС12 • 4Н2О (мет).
СаС12 • 6Н2О + Y-CaCl2 • 4Н2О (мет.>
а-СаС12 • 4Н2О
а-СаС12 • 4Н2О + СаС12 • 2Н2О
СаС12 • 2Н2О
То же
» »
» »
» »
СаС12 • 2Н2О + СаС12 • Н2О
СаС12 • Н2О
СаС12 ¦ Н2О + СаС12
СаС12
Известен субхлорид кальция СаС12/устойчивый при температуре
выше 800°, образующийся при нагревании СаС12 с металлическим
кальцием. Субхлорид кальция окрашен в
J00 i , 1 1 а красно-фиолетовый цвет. Он может быть
получен при комнатной температуре в
250 j ... | ) | л метастабильном состоянии при быстром.
переохлаждении.
200
t
Щюо
50
CaCl2
а:-СаС1г-4Ь
СаС1г-6Н2О^
CaCl
¦2Н2О
I
СаС12
,Н2О
/
1
60
,40
-50
20 40 ВО
СаС12, Вес. %
Рис. 202. Растворимость
еистеме СаС12—Н2О.
20
-20
ВО
Са(ОН),
?СаС12-ЗСа(ОН)г-12
Лед
СаС1г-6Н2О
/О
50 60
Рис.
20 30 40
СаС1г,еес.%
203. Растворимость в системе
СаС12—Са(ОНJ—Н2О.
В системе СаС12—Са(ОН)г—Н2О (рис. 203) в интервале 5—75°"
кристаллизуются 8 твердых фаз: Са(ОНJ, СаС12- Са(ОНJ- Н2О,
740
Гл. XXI. Хлорид кальция
СаС12-ЗСа(ОНJ-12Н2О, СаС12.2Н2О, а-, р- и у-СаС12-4Н2О и
СаС12 • 6Н2О '. Гидрооксихлорид кальция СаС12 • ЗСа (ОНJ • 12Н2О'
кристаллизуется в виде прозрачных игольчатых кристаллов ромби-
ромбической сингонии. В воде и при температуре выше 50° разлагается
с образованием раствора СаС12 и Са(ОНJ.
В солянокислых растворах растворимость СаС12 уменьшается
и тем в большей степени, чем ниже температура. При 0° в растворе
с содержанием 27,98% НС1 растворяется 9,1% СаС12 (вместо
37,30% в воде), при 15° и содержании в растворе 26,20% НС1 рас-
растворяется 21,3% СаС12 (вместо 41,2% в воде). В системе НС1—
СаС12—Н2О при 15° кристаллизуются две твердые фазы: СаС12-
-2Н2О и СаС12-6Н2О, при 25° —три твердые фазы: СаС12-2Н2О,
СаС12 • 4Н2О, СаС12 -6Н2О. Хлорид кальция высаливает NaCl из
его водных растворов, особенно при пониженных температурах.
Растворимость NaCl в системе СаС12—NaCl—Н2О (при 0°) непре-
непрерывно уменьшается по мере растворения СаС12 (от 26,23% в воде
до 0,62% в растворе, содержащем 34,87% СаС12). При более высо-
высоких температурах, помимо NaCl, кристаллизуются также СаС12 •
- 6Н2О или СаС12 • 2Н2О; при —5, —23° кристаллизуются твердые
фазы: лед. NaCl • 2Н2О, NaCl и СаС12 • 6Н2О.
В системе СаС12—NaCl—Н2О из метастабильных растворов при
концентрации NaCl до 0,5 мол. % могут выделяться а-, |3- и y-моди-
фикации СаС12-4Н2О. В системе СаС12—НС1—Н2О существуют,
наряду со стабильными, также метастабильные области выделения
всех трех модификаций тетрагидрата СаС12 и СаС12 • 2Н2О. Мета-
Метастабильные растворы характеризуются большими значениями вяз-
вязкости и плотности 5< О давлении пара в системах СаС12—NaCl—
Н2О, СаС12—КС1—Н2О и СаС12—НС1—Н2О см.6.7.
В системе СаС12—MgCl2—Н2О в интервале температур от 0 до
170° кристаллизуются MgCl2-6H2O, СаС12-6Н2О, 2MgCl2-CaC!2-
- 12Н2О, СаС12-4Н2О (стабильная и метастабильная), СаС12-2Н2О,
MgCl2-2CaCl2-12H2O, MgCl2 • 2СаС12 • 6Н2О и MgCl2-6H2O. Наи-
Наименьшую растворимость при 55° имеет инконгруэнтно растворяю-
растворяющаяся двойная соль MgCl2-2CaCl2-12Н2О. По сравнению с изо-
изотермой 25° ветвь кристаллизации этой соли при, 55° больше
приблизительно в 5 раз8. В безводной системе СаС12—MgCl2 эвтек-
эвтектической точке соответствуют 51% СаС12 и 611°.
В водной системе Са2+, Mg2+, К+|[С1" между 18 и 93° с увели-
увеличением температуры возрастает поле кристаллизации СаС12«
- 2MgCl2 • 12Н2О, появляющееся при 22°, и уменьшаются поля кри-
кристаллизации СаС12 • 6Н2О и MgCl2 • 6Н2О. Увеличивается также
поле кристаллизации СаС12 • КС1 за счет полей СаС12 • 2Н2О и
карналлита. При 92° начинает кристаллизоваться 2СаС!2 • MgCl2 •
- 6Н2О, поле кристаллизации которого возрастает при дальнейшем
повышении температуры за счет полей СаСЬ -MgCla* 12H2O и
СаС1
2 '
2Н2О!
Применение
741
ПРИМЕНЕНИЕ
Хлорид кальция используют в производстве хлорида бария, не-
некоторых красителей, для коагуляции латекса, в химико-фармацев-
химико-фармацевтической промышленности, при обработке сточных вод, в систе-
системах для кондиционирования воздуха, при экстракции масел и др.10.
В связи с большой гигроскопичностью его часто применяют в ка-
качестве осушителя газов и жидкостей. Его применяют также для
получения металлического кальция электролизом, для производ-
производства кальциевых сплавов и баббитов.
Низкие температуры замерзания водных растворов хлорида
кальция позволяют применять его в качестве хладоносителя в хо-
холодильном деле и в качестве антифриза для двигателей внутрен-
внутреннего сгорания в авиации, автомобильном транспорте, для борьбы
с гололедицей, для предотвращения смерзаемости угля и руд при
транспортировании в зимнее время и др. Существенным недостат-
недостатком его является сильное коррозирующее действие на металлы,
которое уменьшается при введении в раствор хлорида кальция
окислителей — хроматов или бихроматов "• 12. Коррозия умень-
уменьшается и в присутствии ионов магния. Раствор СаС12 -f MgCl2
также служит жидкостью с низкой температурой замерзания.
Его приготовляют как из твердых солей, так и смешивая, на-
например, конечный карналлитовый щелок (стр. 161) с предва-
предварительно упаренной дистиллерной жидкостью содового производ-
производства 13.
За рубежом масштабы применения хлорида кальция весьма
велики вследствие использования его для обеспыливания грунто-
грунтовых и щебеночных дорог и при строительстве дорог. Периодиче-
Периодическое C—4 раза за летний сезон) нанесение раствора СаС12 устра-
устраняет пылеобразование. При строительстве дорог СаС12 используют
в качестве одного из компонентов цементо-грунта — подстилаю-
подстилающего слоя дороги. При строительстве 1 км силикатированной до-
дороги расходуют ~3г 75% раствора СаС12.
В присутствии СаС12 ускоряется твердение бетона и увеличи-
увеличивается морозостойкость строительных растворов. Некоторое рас-
распространение в последние годы хлорид кальция получил в качестве
добавки к шихте при производстве кирпича-сырца на заводах с
естественной сушкой.
Хлорид кальция можно применять для предотвращения пыле-
ния угля, в частности для предотвращения появления взрывоопас-
взрывоопасной угольной пыли в шахтах, а также в качестве гербицида для
уничтожения сорняков на железнодорожных путях.
Хлорид кальция может быть использован для мелиорации со-
солонцовых почв с большей эффективностью, чем гипс. Возможно
также использование его раствора в качестве тяжелой жидкости
при углеобогащении.
24 М Е. Позин
742
Гл. XXI. Хлорид кальция
Хлорид кальция выпускают в виде раствора, плавленого про-
продукта (порошкообразного, чешуйчатого или гранулированного),
представляющего собой смесь двух- и четырехводного кристалло-
кристаллогидратов, и в виде кальцинированного, обезвоженного продукта
(порошкообразного или гранулированного) (табл. 50).
ТАБЛИЦА 50
Требования к качеству хлорида кальция (содержание компонентов в %)
Получение плавленого СаСЬ из дистиллерной жидкости
743
СаС12, не менее . . .
MgCl2, не более . . .
Прочие хлориды
(NaCl), не более . .
Сульфаты (SO4), не
более
Железо (Fe), не бо-
более
Не растворимый в
воде остаток, не
более .......
КСЮ3> не более . . .
Влага, не более . . .
Кальцинированный Плавленый | Жидкий
по ГОСТ
5.831-71
[со знаком
качества)
96,5
0.5
1,0
0,05
0,004
0,1
3,0
по ГОСТ 450-70
1-й
сорт
96
0,5
2-й сорт
90
0,6
Не нормируется
0,06
0.004
Не нормн
руется
Не норми
руется
1-й
сорт
2-й сорт
76
0,3
2,0
0.2
0,02
0,2 0,6 0,2
Не нормируется
Н е
67
0.5
Не нормн
руется
0,3
0,05
0,5 0,03
2,7 Не норми-
нормируется
нормируется
1-й сорт
38
0,2
2.5
2-й сорт
32
0,3
3,0
Не нормируется
0,01
Не норми
руется
0,15
1.2
Кильцинированный и плавленый хлорид кальция упа-
упаковывают в металлические барабаны, в полиэтиленовые или бу-
бумажные пятислойные битумированные мешки. Предложено пере-
перевозить его в железнодорожных цистернах, загружаемых горячим
концентрированным раствором СаС12, затвердевающим при есте-
естественном охлаждении. Для разгрузки в цистерну необходимо по-
подать воду или разбавленный раствор в количестве, требующемся
для получения стандартного раствора 14.
СЫРЬЕ
Сырьем для производства плавленого хлорида кальция служат
промышленные отходы растворов хлорида кальция, получающиеся
в больших количествах на содовых заводах, при производстве хло-
хлората калия и др.
Дистиллерная жидкость, получающаяся в производстве соды
по аммиачному способу при регенерации аммиака по реакции
2NH4C1 + Са(ОНJ = СаС12 + 2Н2О + 2NH3
имеет плотность 1,12—1,13 г/см3 и содержит 9,2—11,3% СаС12,
4,7—5% NaCl и небольшие количества растворенных гипса, гидро-
гидроокиси кальция и аммиака в смеси с взвешенным осадком. Послед-
Последний состоит из СаСОз, Са(ОНJ и CaSO4 • 2ШО; количество осадка
равно 20—25 ке/м3. На каждую тонну производимой соды .выбра-
.выбрасывается в дистиллерной жидкости более 1 г СаС12 и 0,5—0,6 т
<ТоП
В производстве бертолетовой соли после кристаллизации
остается маточный щелок, содержащий 400—500 г/л СаС12, по 10—
20 г/л КСЮз и КС1 и незначительные количества других примесей.
Этот щелок является отходом производства и выпускается в виде
товарного продукта, применяемого для разных целей, в частности
для производства хлорида бария (см. гл. XII). Путем его выпари-
выпаривания получают твердый (плавленый) хлорид кальция.
Для производства хлорида кальция сырьем служат также со-
соляная кислота и высококачественный известняк, содержащий
малые количества примесей R2OS, MgO, SiO2, S и P.
В некоторых случаях хлорид кальция B2—25% раствор) полу-
получают из хлормагниевого рассола, обрабатывая его гашеной из-
известью и отделяя на фильтре осадок гидроокиси магния 15.
Хлорид кальция образуется при хлорировании окиси кальция
выше 600° 16, а также при хлорировании сульфата кальция в среде
расплавленного СаС12 в присутствии восстановителя выше 800° 17>18.
Эти способы получения СаС12 в промышленности не применяются.
ПОЛУЧЕНИЕ ПЛАВЛЕНОГО ХЛОРИДА КАЛЬЦИЯ
из дистиллерной жидкости содового производства
Получение товарного хлорида кальция из дистиллерной жид-
жидкости содового производства 19>20 заключается в последовательном
выпаривании дистиллерной жидкости от концентрации ~10%
СаС12 до 67%, требуемой условиями ГОСТ на плавленый продукт.
Выпарку ведут в одиночных или батарейных плавильных котлах,
обогреваемых топочными газами, в вакуум-выпарных аппаратах,
обогреваемым паром, в распылительных сушилках и др.
Отстоявшуюся дистиллерную жидкость можно подвергнуть кар-
карбонизации и обработке хлоридом бария с последующим отстаива-
отстаиванием для очистки жидкости от растворенных в ней извести и гипса.
При наличии в растворе ионов SO4 может произойти загипсовыва-
ние греющих поверхностей выпарных аппаратов. Очистка раствора
с помощью ВаС12 приводит, однако, к увеличению производствен-
производственных издержек и не позволяет использовать выделяющуюся при
выпарке поваренную соль в качестве пищевого продукта. Поэтому
карбонизацию дистиллерной жидкости и обработку ее хлоридом
бария часто не производят.
При выпаривании не очищенной от ионов SO|~ и ОН" дистил-
дистиллерной .жидкости в выпарном аппарате с естественной цирку-
циркуляцией коэффициент теплопередачи за 92 ч снижается на -~40%
от своего первоначального значения. [Опыт был выполнен с ди-
дистиллерной жидкостью следующего состава (в г/л): 122,7 — СаС12,
24*
744
Гл. XXI. Хлорид калоция
Получение СаС12 из щелока хлоратного производства
745
62,84 — NaCl, 0,93 —CaSO4> 1,61 —Са(ОНJ; плотность 1,134 г/сж3.]
Выпаривание с принудительной циркуляцией при скорости жидко-'
сти в трубах ~3,5 м/сек и при добавке кристаллической подкладки
в виде гипса C—5% от веса жидкости) не предотвращает инкру-
инкрустирования греющих поверхностей сульфатом кальция. Однако про-
производительность аппарата при этом увеличивается в ~3 раза, а
продолжительность работы до чистки в ~2 раза21. Интенсивность
зарастания греющей поверхности и твердость образующейся корки
сульфатов (состоящей из гипса, полугидрата сульфата кальция
или ангидрита) сильно зависит от температурного режима выпарки.
На некоторых содовых заводах выпаривают неочищенную дистил-
лерную жидкость, останавливая аппарат для удаления инкруста-
инкрустаций после 25—35 суток непрерывной работы.
Осветленную дистиллерную жидкость выпаривают обычно в мно-
многокорпусных выпарных аппаратах. По достижении концентрации
~40% СаС12 выделяется в осадок почти вся содержащаяся в жид-
жидкости поваренная соль. Она может быть возвращена в производ-
производство соды при условии тщательной отмывки от СаС12 (во избежа-
избежание увеличения расхода соды на стадии предварительной очистки
рассола NaCl). Отфугованная от маточного раствора, промытая и
высушенная поваренная соль очень чиста и пригодна для пищевых
целей (если дистиллерная жидкость предварительно не обрабаты-
обрабатывалась хлоридом бария). Попутное получение чистой пищевой по-
поваренной соли является важным условием рентабельности произ-
производства хлорида кальция 22~2*.
Концентрированный раствор СаС12 после отделения поварен-
поваренной соли продолжают выпаривать до концентрации 67% СаС12
обычно в плавильных аппаратах непрерывного действия до повы-
повышения его температуры кипения до 175°. Затем жидкость разли-
разливают в барабаны, где она застывает в плавленый продукт. Для по-
получения чешуйчатого продукта плав выпускают на поверхность
охлаждаемого барабана.
Другим вариантом получения твердого хлорида кальция из дис-
тиллерной жидкости является следующий 2Б>26. Осветленную ди-
дистиллерную жидкость выпаривают до концентрации 40%) СаСЬ,
отделяют осадок NaCl, а раствор нейтрализуют соляной кислотой
и добавляют к нему хлорную известь для окисления S2dt> SO3-
и Fe2+. После перемешивания добавляют Са(ОНJ или NaOH, от-
отделяют осадок, а фильтрат упаривают до концентрации 52% СаС12.
Затем, после охлаждения до 50°, отстаиванием и фильтрованием
удаляют выделившиеся кристаллы NaCl, а раствор высушивают
в распылительной сушилке, где получается продукт I сорта.
Разработан способ обезвоживания хлорида кальция азеотроп-
ной дистилляцией с помощью фракции нефти, кипящей в пределах
160—260°27. Нефть после регенерации можно возвращать на ди-
дистилляцию — при этом продукт в меньшей мере окрашен в желтый
цвет. Обезвоженный хлорид кальция содержит меньше 0,1% Н2О.
На дистилляцию 50%-ного исходного материала подают ~4/сз
нефти, а на дистилляцию 75%-ного — 3 кг нефти на 1кг безвод-
безводного СаСЬ. При таком способе обезвоживания коррозия аппара-
аппаратуры и расход тепла меньше, чем при выпарке раствора СаС12.
Хлорид кальция может быть получен в результате регенерации
аммиака из хлорида аммония мелом сухим способом:
2NH4C1 + СаСОз - СаС1, + 2NH3 + СО2 + Н,О
Реакция протекает по схеме:
NH4C1 ^rj
2НС1 + СаСОз =
* NH3 + HC1
. СаС12+СО2 + Н2О
При 200—225° процесс лимитируется скоростью реакции вза-
взаимодействия газообразного хлористого водорода и СаСОз; при
350° процесс лимитируется диффузией28. Опыты в модели шахт-
шахтной печи показали29, что при пропускании возогнанного хлорида
аммония через слой кускового мела C—7 мм) при 420—450° полу-
получается продукт, содержащий 80—85% СаС12. Промышленное осу-
осуществление этого процесса затруднено сильной коррозией и обра-
образованием настылей. Кроме того, при использовании стальной
аппаратуры, теряется до 30%) аммиака вследствие его каталитиче-
каталитического разложения. В керамической и эмалированной аппаратуре
потери аммиака невелики.
ПОЛУЧЕНИЕ ХЛОРИДА КАЛЬЦИЯ ИЗ МАТОЧНОГО ЩЕЛОКА
ХЛОРАТНОГО ПРОИЗВОДСТВА
Получение плавленого хлорида кальция из маточного щелока
хлоратного производства, содержащего в 4—5 раз больше СаС12,
чем дистиллерная жидкость, является значительно более эконо-
экономичным. Здесь, однако, идет более сильная коррозия вследствие
примеси хлората. Процесс осуществляется аналогично получению
хлористого магния из хлормагниевых щелоков (стр. 275), т.е. пу-
путем выпаривания в чугунных котлах, обогреваемых топочными га-
газами. Иногда выпаривание ведут в стальных котлах, в стенках ко-
которых заделаны стальные змеевики; по змеевикам циркулирует
перегретая вода или другой теплоноситель. Выпаривание ведут до
тех пор, пока температура кипения жидкости не поднимается до
165—175°. При этом концентрация щелока достигает 67—75%
СаС12, после чего его чешуируют на холодильном барабане или
сливают в тару, где он застывает в плав, состоящий из смеси
СаС12 • 2Н2О и СаС12 • 4Н2О.
746
Гл. XXI. Хлорид кальция
Получение CaClj из соляной кислоты и известняка
7*7
ПОЛУЧЕНИЕ ГИДРООКСИХЛОРИДА КАЛЬЦИЯ И ХЛОРИДА КАЛЬЦИЯ
ИЗ НЕГО
Гидрооксихлорид кальция СаС12-ЗСа(ОНJ' 12Н2О образуется
при смешении в стехиометрическом отношении хлорида кальция,
молотой извести и воды. Его можно выделить из дистиллерной
жидкости без выпарки ее или на определенной стадии ее выпари-
выпаривания. Он может быть использован непосредственно, например
в строительной технике в качестве добавки, ускоряющей твердение
бетона 18-30, или переработан на хлорид кальция31.
Из водного ~10% раствора СаС12 прибавлением сухой гаше-
гашеной извести можно выделить при 0—1° в виде гидрооксихлорид а
кальция до ~26% СаС12. При этом образуется хорошо кристалли-
кристаллизующийся осадок. В маточном растворе, количество которого со-
составляет ~80% от исходных количеств раствора СаС12 и Са(ОНJ,
содержится 6—7% СаС12 и ~0,1% Са(ОНJ. Из дистиллерной
жидкости, предварительно выпаренной до содержания 21—22%
СаС12, при этих же условиях выделяется до 65% СаС12. Для полу-
получения кристаллов в хорошо фильтрующейся форме в этом случае
необходимо применять вместо сухой гидроокиси кальция извест-
известковое молоко. При этом осаждение следует производить, добавляя
раствор СаС12 к подогретому до 50—60° известковому молоку при
постоянном перемешивании. Затем массу охлаждают до ~ 1° для
получения максимального выхода гидрооксихлорида. В жидкой
фазе, количество которой ~50% от исходной смеси, после кристал-
кристаллизации гидрооксихлорида остается 7—8% СаС12 и ~0,15%
Са(ОНJ.
На рис. 203 видно, что при обработке СаС12-ЗСа(ОНJ-12Н2О
водой при 50—60° он разлагается с образованием растворов, со-
содержащих до 32% СаС12 и чистого Са(ОНJ. Для получения 30%
раствора СаС12 при 55° необходимо обработать гидрооксихлорид
небольшим количеством воды — 5—6% от веса гидрооксихлорида.
Однако при этом получается плохо фильтрующаяся масса. Лучшие
результаты достигаются при разложении гидрооксихлорида не-
несколько большим количеством воды — приблизительно 14% от €го
веса. В этом случае в полученном растворе содержится 24—25%
СаС12.
Изложенное свидетельствует, что переработка гидрооксихло-
гидрооксихлорида кальция, получаемого из дистиллерной жидкости, не может
быть экономичной и поэтому она не применяется.
Использование более концентрированных (чем дистйллерная
жидкость) растворов СаС12 для получения гидрооксихлорида каль-
кальция дает существенные преимущества. Так, если применять рас-
раствор с концентрацией 25—35% СаС12, то можно получить высокий
Выход продукта при охлаждении реакционной массы до 20° (вме-
(вместо 0е).
ПОЛУЧЕНИЕ БЕЗВОДНОГО ХЛОРИДА КАЛЬЦИЯ
из соляной кислоты и известняка
Получение хлорида кальция этим методом заключается в рас-
растворении известняка в соляной кислоте, в очистке образующегося
«сырого» (неочищенного) раствора СаС12 от примесей и в обезво
живании его. Продукт получается более чистым, чем из отходящих
жидкостей содового или хлоратного производства.
Растворение известняка (куски не больше 50 мм) производят
в стальных баках, футерованных двумя слоями диабазовой плитки.
В нижней части растворителя имеется решетка иэ диабазовых
плиток, поддерживающая загружаемый известняк. Соляную кис-
кислоту, разбавленную до 14% НС1, подают из напорного бака. Обра-
Образующийся раствор СаС12, вытекающий из растворителя через шту-
штуцер в нижней его части по винипластовой трубе, должен содержать
не больше 14 г/л свободной кислоты. Этого достигают, поддержи-
поддерживая определенную высоту слоя известняка.
Выделяющиеся из растворителей газы, содержащие СО2 и НС1,
протягиваются вентилятором через керамическую башню, запол-
заполненную известняком и орошаемую разбавленным раствором хло-
хлорида кальция. Вытекающий из башни раствор, содержащий 300—
350 г/л СаС12, примешивают к основному раствору.
Получающийся сырой раствор, содержащий 450—600 г/л СаС12,
очищают от примесей соединений Fe, Mg, A1 и SO4~. Очистку про-
производят в стальном, футерованном диабазовой плиткой реакторе
с пропеллерной мешалкой C0 об/мин). Вначале раствор очищают
от сульфатов. В реактор заливают ~ 10 м3 сырого раствора и вво-
вводят в него в сухом виде при перемешивании ~ 15 кг хлористого
бария. Осаждение сульфата бария заканчивается в течение 20—
25 мин. Затем раствор подогревают острым паром до 70—75° и
добавляют к нему известь-пушонку для осаждения гидроокисей
железа, магния и алюминия. После 40—50-минутного отстаивания
раствор профильтровывают. Количество примесей в нем не должно
превышать: 0,003 г/л Fe, 0,03 г/л SOf~, 0,025 г/л Mg. Раствор со-
содержит немного Са(ОНJ (в пересчете на СаО 2,8—3,5 г/л). Для
получения безводного продукта в распылительной сушилке раствор
должен иметь нейтральную или слабощелочную реакцию: при зна-
значительной щелочности раствор вспенивается, что затрудняет ра-
работу разбрызгивающей форсунки. Нейтрализацию избыточной ще-
щелочности осуществляют, добавляя при перемешивании соляную кис-
кислоту в сборник очищенного раствора. Затем очищенный раствор
проходит через пенный аппарат (см. ниже), где его концентрация
повышается до 700 г/л СаС12, и поступает на обезвоживание.
Обезвоживание хлорида кальция производят, распыляя раствор
в потоке горячего газа. Стальная сушильная башня изнутри выло-
выложена листом из стали 1Х18Н10Т и имеет диаметр 5,5 и высоту
748
Гл. XXI. Хлорид кальция
11 ж с коническими верхней и нижней частями. Вокруг нижнего
основания верхнего конуса с наружной стороны расположен желоб v
высотой около 0,5 м, из которого раствор СаС12 подается в фор-
форсунку. Распыление раствора производят предварительно высушен-
высушенным сжатым воздухом C ат). В верхнюю часть сушильной башни
поступает топочный газ, получаемый сжиганием природного газа,
разбавленный воздухом для понижения его тем-
температуры до 500°. Распыленный раствор и горя-
горячий газ движутся в сушилке прямотоком сверху
вниз. При этом вода из раствора выпаривается
и образуется почти безводный продукт в виде
11 сухого порошка. Часть СаС12 оседает внизу баш- Ц
У™ ни и скапливается в конусном бункере. Большая \
часть продукта уносится потоком воздуха и улав-
улавливается в двух параллельно работающих цикло-
циклонах. Продукт выгружают как из циклонов, так |
и из башни и упаковывают в барабаны из оцин-
оцинкованного железа.
Газ, выходящий из циклонов, уносит значи-
значительное количество хлорида кальцияB—2,5г/ж3).
Для его улавливания применяют32 пенные аппа-
аппараты, служащие одновременно и пылеуловите-
пылеуловителями и утилизаторами тепла газа; это тепло ис-
используется для выпарки раствора перед его пос-
поступлением в сушильную башню. Пенные аппараты,
применяемые в разных отраслях промышленно-
промышленности, позволяют осуществлять очистку газов,
ливнвя и сливная абсорбцию, теплопередачу и другие процес-
>бки! /—ёвЬя газа;
¦ сы, происходящие при контакте газов с
жидкостями с весьма большой интенсив-
интенсивностью 33-34.
Пенный аппарат (рис. 204), диаметром 2,2 м
^-смотровое отвер- и высотой 4,1 м, имеет горизонтально располо-
женную решетку с отверстиями 6 мм. Живое се-
сечение перфорированной части решетки составляет 21%. Раствор
подается на решетку с одной стороны, течет по ней в виде слоя
пены и отводится с решетки с другой стороны. Газ поступает в ап-
аппарат снизу и, пройдя через отверстия решетки, вспенивает на-
находящуюся на ней жидкость. При скорости газа в полном сече-
сечении аппарата 1,5—3,5 м/сек создается высокая турбулентность
газо-жидкостной системы, обеспечивающая интенсивное протека-
протекание процессов в этой системе, в данном случае улавливания пыли
хлорида кальция и теплообмена. Схема установки для очистки
газа от хлорида кальция показана на рис. 205. Очищенный раствор
СаС12 предварительно проходит через пенный аппарат. Выходя-
Выходящий из него подогретый и выпаренный раствор концентрацией
Литература
749
Рис. 204. Пенный
аппарат с ловуш-
ловушкой:
1 — корпус аппарата)
2 — решетка; §-^ ввод
жидкости; 4 - ОТВОД
жидкости; б — порог;
коробки:
8 —вывод газа; $ — от-
отбойная решетка!
10 — от борный зонт;
// — ловушка; 12 — от-
отвод утечной жидко-
жидкости; 13 — выхлоп газа;
СаС12 до 700 г/л подают в сушильную башню. Выпарку раствора
осуществляют отработанным газом, температура которого сни-
снижается от 150—160° до 80—90°. Улавливание брызг раствора, уно-
уносимых газом из пенного аппарата,
производится
вушке.
Твердый
в специальной ло-
гранулированный Воздух
"V
хлорид кальция можно получать "V
смешением в барабане порошко- у
образных отходов безводного и ча-
частично обезвоженного СаС12 с не-
непрерывно распыляемым раство-
раствором, содержащим более 50%
СаС12. Смешение производят в по-
потоке газа с температурой 200—
500°. Если температура массы
150—180°, то в результате высу-
высушивания массы при такой темпе-
температуре получается продукт, содер-
содержащий от 3 до 13% Н2О. Даль-
Дальнейшей сушкой при 250—500°
получают безводный продукт.
В зависимости от требований к
продукту производят дробление и
рассеивание массы, полученной
при первой или второй сушке, с
возвратом отходов в производст-
производственный цикл35'36. Запатентован способ гранулирования СаС12-Н2О
пропусканием расплава через узкое сопло под давлением, превышаю-
превышающим в 1,4—2,9 раз давление насыщенного пара над расплавом"
Рис. 205. Схема установки для
очистки воздуха от хлорида кальция:
/ - сушильная башня; 2 - пеиный аппарат;
3 —сборник раствора; 4 — промежуточный
ваш Я — насос.
37
ЛИТЕРАТУРА
1. Л Л Эзрохи, Труды ВНИИГ, в. 2, 1967, стр. S, — 2. 9. И. К у ж а х м е-
тов, Г. И. Гнатышенко, ЖПХ, 39, № 6, 1266 A966). — 3. Ф. К. Михай-
Михайлов Л М Волов а, Л С. Коваленко, Хим. пром., № 8, 696 A964).—
4. С. 3.'Макаров, И. И. Вольнов, ИСФХА АН СССР, 26, 320 A954).—
5. И Г Дружинин А И Шепелев, Труды Ин-та химии АН КиргССР,
№ 7,' 1956, стр. 3. — 6. А. И. Мун, ЖОХ, 26, № 4, 1021 A956). — 7. Л. С. Л я-
лич, М. Д. Аники ев а, Вестн. ЛГУ, № 22, сер. физ. и хим., в. 4, 97 A956).—
8. А. П Перова, Сообщения о научных работах ВХО им. Д. И. Менделеева,
в 2 1955 стр 46—5 G. О. A s s a r s s о n, A. Balder, J. Phys. Chem., S9»
№ 7, 631 'A955). 10. M. Boivin, Chim. et ind., 97, № 9, 1339 A967).
// И Л Розенфельд, Замедлители коррозии в нейтральных средах,
Изд 2ШСССР 1953. — 12. 3. И. Конопкина, ЖПХ, 31, Mb 9, 1310 A958).—
W. Пат. ГДР 32362, 1963.—14. Н, D. Loss, канад. пат. 495340, 1953.—
15. А. Ё. К р у г л и к о в и др., Тезисы доклада на VI Всесоюзной конференции
по технологии неорганических веществ, Тбилиси, 1968, стр. 248. —16. А. Н. К§-
тов, В. В. П е ч к о в с к и и, Л. П. Костин, сб. «Исследования в области хийии
750
Гл. XXI. Хлорид кальция
и технологии минеральных солей и окислов». Изд. «Наука», 1965, стр. 202.—
17. В. В. Печковский А. Л. Софронов, ЖПХ, 40, № 12, 2711 A967).—
/8. П. П. Будников,' Э. И. Креч, Там же, 14, № 6, 747, 765 A941).—
19. В Л. Рыдник Хим. пром., № 8, 253 A953). — 20. Д. С. Бодров, Труды
НИОХИМ, т. И, 1958, стр. 27.
21. В. В. Тимошенко, Там же, стр. 251. — 22. В. Ф. Чернов, Произ-
Производство кальцинированной соды, Госхимиздат, 1956. — 23. А. Ф. Б о р я ч е к,
А. Н. Вишневский, П. Г. Власов, А. Я. Бакулииа, Труды НИОХИМ,
т. 13, 1961, стр. 10. — 24. Ф. К. Михайлов, Хим. пром., № 7, 391 A956).—
25, В. В. Тимошенко, Э. Ф. Овчинникова, XiM. пром. Украши, № 2
№2), 25 A965). — 26. Р. Г. Пушкар, Там же, № 1 B5), 6 A966). —j
St. Е. Pischinger, Z. Szufarski, Przem. Chem., 38, № 7, 426 A959). —I
28. M. К. Шелудько, А. И. Черников, Т. А. Жиляева, 29, № 5, 708|
(J966). — 29. Ф. К. Михайлов, В. И. Панов, В. С. Еремеев, Труды!
НИОХИМ, т. 12, 1959, гтр. 90. — 30. В. Ф. Журавлев, Химия вяжущих ве-|
ществ, Госхимиздат, 1951.
S1. И. И. Вольнов, Е. И. Латышева, ЖПХ, 80, № 7, 977 A957).—
82. Ь- И. Штейнберг, Обмен опытом ГК по химии при Совете Министров
СССР, № 6 E1), 7 A958); Бюлл, по обмену опытом в азотной пром., № 11, 5
A9б9).—33. М. Е. Позин И. П. Мухленов, Е. С. Ту Маркина,
Э. Я. Т а р а т, Пенный способ обработки газов и жидкостей, Госхимиздат,
.1955. — 34. М. Е. Позин, И. П. М у х л е к о в, Э. Я. Т а р а т. Пенные газоочи-
газоочистители, теплообменники и абсорберы, Госхимиздат, 1959. — 35. W. R. В е п n e t,
N L. Carmouche, пат. США 2646343, 1953. — 35. A. Wilcox Le Roy,
А. С. S peer, пат. США 3250593, 1964. — 37. Япон. пат. 21128, 1962.
Глава XX 11
СОЕДИНЕНИЯ МАРГАНЦА
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Марганец образует ряд окислов: закись МпО, окись Мп2О3,
двуокись МпО2, марганцоватистый ангидрид Mn2Os *, марганцови-
марганцовистый ангидрид МпОз * и марганцовый ангидрид Мп2О7. Окислы и
гидроокислы марганца' являются важнейшими марганцовыми ми-
минералами (табл. 51).
ТАБЛИЦА 61
Природные кислородные соединения марганца
Минерал
Пиролюзит и полианит или рамс-
деллит
Вернадит
Курнакит или браунит
Манганит и гроутит
Манганозит
Пирохроит и бекстремит
Родохрозит или марганцовый шпат
Родонит
Гаусманит
Гидрогаусманит
Биксбиит
Псиломеланы или воды
Химическая формула
MnO2 (P и у)
МпО2 • пН2О
Мп2О3(а и Р)
8(Мп, FeJO3-Mn8iO,
Мп2О3 • Н2О (а и Р)
МпО
МпО • Н2О (а и В)
МпСО3
MnSiO3
Мп3О4(а, Р и v)
Мп3О4 • пН2О (а и Р)
(Mn, FeJO3
*RO • г/МпО2 ¦ zH2O, где
R—Mn, K2, Ba, Ca и др.
Плотность ••
4,75—5
3,0—3,2
4,7—4,9
4,2-4,4
3,4—3,6
3,5—3,7
4,7—4,8
2.3-4.7
В состав природного пиролюзита входит 1—3% воды; полианит
безводен. Пиролюзит растворяется в соляной кислоте с выделением
* Мп2ОБ и МпО3 в свободном состоянии ие выделены; существуют их соли
Гипоманганаты (Мп5+) и манганаты (Мп6*).
** Плотности минералов относятся к плотным разностям.
752
Гл. XXII. Соединения марганца
хлора и в серной кислоте в присутствии восстановителя, а также
в сернистой кислоте; азотная кислота взаимодействует с пиролю-
пиролюзитом медленно. Природный манганит не разлагается азотной и
разбавленной серной кислотами, но медленно растворяется в сер-
сернистой кислоте; искусственно приготовленный Мп2О3 • Н2О разла-
разлагается разбавленными кислотами. Гаусманит реагирует с мине-
минеральными кислотами, образуя смесь солей двух- и трехвалентного
марганца. Браунит представляет собой марганцовую соль мета-
марганцоватистой кислоты МпО(ОНJ, имеющую структурную
/°\
формулу О=Мп Мп. Вследствие присутствия изоморфного
ХК
MnSiO3 в минерале содержится до 15% SIO2.
Псиломелан относится к гелеобразным марганцовым рудам не-
неопределенного состава. Его можно рассматривать как твердые рас-
растворы манганита, пиролюзита, полианита и, возможно, других
окислов марганца друг в друге и воды в них, содержащие также
соли метамарганцоватистой МпО (ОН) 2 и ортомарганцоватистой
Мп(ОНL кислот. По химическим свойствам он аналогичен водной
двуокиси марганца, быстро растворяется в сернистой кислоте. Со-
Содержит 60—80% МпО2, 8—25% МпО, 3—4% Н2О. Псиломеланы
имеют разные названия в зависимости от преобладающего состава
входящей в минерал группы RO: псиломелан (МпО), криптомелан
(К2О), рансьеит (СаО), романешит (ВаО) и т. д.
Родохрозит или марганцовый шпат МпСОз, имеющий окраску
от розовой до красной, встречается обычно в изоморфных смесях
с карбонатами кальция, магния, железа, кобальта, цинка и в смеси
с Мп8Юз; основному минералу сопутствуют продукты его разло-
разложения— пиролюзит, псиломелан и др. Разложение синтетического
карбоната марганца при нагревании с образованием МпО может
быть обнаружено при 70°, со значительной скоростью он разла-
разлагается при 200—300°2, а равновесное давление СО2, равное 1 ат,
достигается при 632°. Значения этих температур различны для
разных образцов природного минерала. При 200 — 300° в токе
воздуха МпСОз окисляется последовательно до Мпз'О4, Мп2Оз и
МпО2.
Закись марганца МпО — основной окисел серо-зеленого цвета,
<пл=!785°, на воздухе окисляется, при растворении в кислотах
образует соли двухвалентного марганца слабо-розовой окраски.
Розовая окраска кристаллогидратов и растворов солей Мп2+ обус-
обусловлена ионом [Мп(Н2ОL]2+. Безводный сульфат марганца MnSO4
бесцветен, с водой образует стабильные и метастабильные кристал-
кристаллогидраты с 1, 4 (только метастабильный), 5 и 7 молекулами
воды3. Полностью обезвоживается выше 280°; безводный плавится
при 700°. Термически более устойчив, чем сульфаты Fe, Co, №. По
другим данным4, MnSO4-5H2O при нагревании одновременно с по-
Фиаико-химичвские свойства
753
терей последней молекулы воды' окисляется кислородом воздуха
с образованием Mn2O(SO4J. С повышением температуры раство-
растворимость MnSO4 в воде уменьшается: насыщенный водный раствор
содержит при 10° —37,4%, при 60° —35,2%, при 100° —26,5%
MnSO4; выше 200° она приближается к нулю. Хлорид марганца
ниже 58° кристаллизуется из водного раствора в форме
МпС12 • 4Н2О, выше этой температуры МпС12 • 2Н2О; насыщенный
раствор содержит при 25° —43,6%, при 100° —53,7% МпС12. При
нагревании тетрагидрат хло-
хлорида марганца теряет пер-
первую молекулу воды при 120°,
вторую — при 150°, а две по-
последние — при 220°; полное
обезвоживание сопровож-
сопровождается незначительным гид-
гидролизом A—1,5%)Б-МпС12
разлагается перегретым во-
водяным паром на МпО и НС1
при 350—450°, с кислородом
начинает реагировать при
500°; ниже температуры
плавления F50°) водородом
практически не восстанав-
200
№0 13DO
Температура, "С
ливается. Нитрат марганца
кристаллизуется в виде
Mn(NO3J-6H2O ниже 24°, а
Рис. 206. Изобара диссоциации МпО2.
выше этой температуры Mn(NOsJ«
ЗН2О; в насыщенном растворе при 0° — 50,5%, при 35,5 — 76,8%
Mn(NO3J. Растворимость в воде двойной соли NH4MnPO4 • Н2О
при 20° с уменьшением рН от 8,0 до 4,0 увеличивается от 1,43-10~*
до 7,99 • 10~4 г-мол/л6. Из растворов солей Мп2+ Мп(ОНJ оса-
осаждается при рН = 8,7.
Окись марганца Мп2О3 бурого цвета, обладает слабоосновными
свойствами. Соли трехвалентного марганца в растворах устойчивы
лишь в присутствии свободных кислот; гидролизуясь, они дают
осадок гидратированной окиси марганца или соли двухвалентного
марганца и гидратированную двуокись марганца.
Двуокись марганца МпО2 — амфотерный окисел; она образует
с кислотами соли четырехвалентного марганца, легко разлагаю-
разлагающиеся водой с выделением гидратированной двуокиси марганца,
а как кислотный окисел — соли марганцоватистой кислоты, манга-
ниты. Двуокись марганца 7 представляет собой мелкокристалличе-
мелкокристаллическое вещество черного цвета, не растворимое в воде. В зависимости
от способа образования МпО2 существует в различных модифика-
модификациях, отличающихся физическими свойствами. При нагревании
МпО2 отщепляет кислород и переходит последовательно в окись
марганца Мп2О3, Мп3О4 и, наконец, в закись марганца МпО.
7Б4
Гл. XXII. Соединения марганца
Физико-химические свойства
755
Температуры перехода МпО2 в МпгО3 несколько различны для раз-
разных модификаций МпО2. Однако общий вид изобары диссоциации
мало меняется и остается подобным, изображенному на рис. 206.
Переход МпОг в Мп2О3 и другие окислы происходит с образованием
твердых растворов окислов. Выше 1350° существует только МпО.
Термический распад пиролюзита и манганита может быть пред-
представлен схемами8:
МпО-ОН
Манганит
Р-МпО2
Пиролюзит
260°.
560°
р-Мп2О3
Р-Враунит
940°
3
a-Mn2O3
а-Браунит
-> МпО2)
Пиролюзит
Р-Мп3О4
Гаусмаиит
600°
1180° .
Y-Mn3O4
Гаусманнт
940°
р-Мп2О3 > р-Мп3О4
Р-Враунит
Гаусманнт
*4
0
-4
М-12
-16
0,4 06 08 W U? 1,4 1,6 1,6
На рис. 207 приведены значения равновесных давлений диссо-
диссоциации (lgpo)p.nn окислов марганца1-9 (см. также 10-12). Линии/,
2, 3 и 4 делят диаграмму на поля
устойчивости отдельных фаз —
металлического марганца, закиси
марганца, гаусманита, трехокиси
марганца и двуокиси марганца.
Соединения шести- и семива-
семивалентного марганца обладают кис-
кислотными свойствами. Они обра-
образуют, соответственно, марганцови-
стокислые соли — манганаты и
марганцовокислые соли—перман-
ганаты. Марганцовистая кислота
Н2Мп04 неустойчива и распа-
распадается на марганцовую кислоту
Рис. 207. Зависимость lgpOa от HMnO4 и МпО2. Манганаты в
4- при диссоциации окислов мар- кислых растворах разлагаются с
„„„„„ ' образованием соединений четы-
ГИНЦа. А
По реакциям: Рех" и семивалентного марган-
ца — в осадок выделяется дву-
двуокись марганца, а в растворе
образуется перманганат. Пер-
Перманганат калия КМпО4 представ-
представляет собой темно-пурпуровые с фиолетовым оттенком, почти чер-
черные кристаллы ромбической формы. Насыщенный водный раствор
содержит при 0° —2,75%, при 20° —6%, при 65° —20,0% КМпО4.
При охлаждении растворов NaMnO4 с концентрацией, меньшей
41,4%, кристаллизуется лед, в пределах концентраций 41,4—
75,2% — NaMnO4-3H2O с /Пл = 68,7°13. В щелочных растворах
перманганат разлагается
1 - 2МпО2 = Мп2Оз + V» О'
2 - ЗМп2О3 - 2МпяО4 +'/, (
S — МпзО* = ЗМпО + '/« О2
причем разложение может идти и глубже. Соединения с ионами
МпО4" имеют зеленую окраску. Начальная скорость восстановле-
восстановления перманганата калия концентрированными растворами КОН
@,9—13,8 М) при 18° очень велика, затем скорость реакции умень-
уменьшается и стремится к нулю в области степеней окисления, соответ-
соответствующих четырех-, пятивалентному марганцу. В этой области
реакцию ускоряют свет и ультрафиолетовые лучи, кислород за-
замедляет ее. Предполагают14", что восстановление КМпО4 проходит
через три стадии:
очень быстро
более медленно
1) МпО 4 + ОН" = МпО|~ + ОН
MnOJ + ОН = MnOf" + Н+ + О
2) МпО^" + ОН" = МпО|" + ОН
МпО4~ + ОН = МпО^~ + Н* + О
3) Переход МпО^" в МпО2
При малых концентрациях КОН возможны реакции распада
ионов:
^- + МпО2 + 4О1Г (~ Ш КОН)
+ 4ОН~
^-+ 2Н2О + О2
+ 2Н2О ^1± 2М11О4 + МпО2 + 4ОН~ ( ~ 0,0 Ш КОН)
Восстановление ионами гидроксила Мп7+ ->- Мп6+ (при 0 и 20°)
и Мп6+->Мп5+ (при —10 и 0°) является процессом половинного
порядка, а дальнейший процесс Мп5+ -> Мп4+ (при 20—30е) пер-
первого порядка. Энергии активации этих процессов равны соответ-
соответственно 9,3; 11,2 и 23 ккал/моль 15.
При нагревании выше 200° КМпО4 разлагается с потерей кис-
кислорода по реакции:
2КМпО4 = К2МпО4 + МпО2 + О2
а также по реакции 12:
ЗКМпО4 = К3МпО4 + 2МпО2 + 2О2
Термическое разложение быстро ускоряется с повышением тем-
температуры и в присутствии металлического алюминия. Так, при 350°
чистый КМпО4 разлагается в ~3 раза быстрее, а в присутствии
16,5% порошкообразного алюминия в ~5 раз быстрее, чем при
265°. Добавка алюминия уменьшает время достижения максималь-
максимальной скорости разложения 16- |7.
Перманганаты и .манганаты восстанавливаются в щелочном
растворе до гипоманганатов. Так, КзМпО4 получается при нагре-
нагревании до 700° К2Мп04 или КМпО4 в смеси с 4,0—45% раствором
КОН (при отношении К: Мп, равном 3:2). Гипоманганат калия
может быть также получен выпариванием концентрированного
756
Гл. XXII. Соединения марганца
раствора КОН со свежеосажденной МпО2 (отношение К: Мп равно
3,3—3,6) с последующим нагреванием в атмосфере кислорода до
750—800° и выдерживанием при этой температуре 8 ч |8. Кристаллы
Na3Mn04 • ЮН2О голубого цвета выделяются при обработке ще-
щелочных растворов NaMnO4 и Na2Mn04 восстановителями, напри-
например Na2SO3.
Взаимодействием КМпО4 с H2SO4 на холоду может быть полу-
получен высший окисел марганца Мп2О719. Это маслянистая жидкость
со специфическим запахом, зеленая в отраженном и темно-синяя
в проходящем свете, с температурой плавления +5,9° и плотностью
при 20° 2,396 г/см3. При 55° начинает разлагаться на а-Мп2О3 и
кислород, быстро разогреваясь, и при ~90° взрывает; скорость де-
детонации ~400 м/сек; чувствительность к удару такая же, как
у гремучей ртути. При взаимодействии с влагой воздуха Мп2О7
разлагается до МпО2 с выделением О2 и О3, но при отсутствии
влаги может сохраняться в закрытом сосуде при —10° длительное
время. Мп2О7 — сильнейший окислитель, например окисляет NaCl
HNal floNaClO3HNaIO320.
Соединения марганца, особенно Мп2+, являются ядами, вызы-
вызывающими хроническое отравление. Систематическое вдыхание
пыли марганцовых соединений может вызвать нервное расстрой-
расстройство, длящееся годами, истощение и смерть. Предельно допусти-
допустимая концентрация пыли марганцовых соединений (в пересчете на
МпО2) в воздухе рабочих помещений 0,0003 мг/л.
ПРИМЕНЕНИЕ
Больше 90% мировой добычи марганцовых руд потребляется
металлургической промышленностью21. Они идут на изготовление
ферромарганца F0—90% Мп), зеркального чугуна, силико-мар-
ганца, фосфо-марганца и др. В доменном процессе марганцем
обессеривают чугун. Марганец служит легирующей добавкой при
получении чугуна повышенной прочности и, особенно, твердых ста-
сталей, из которых большое значение приобрели молибденово-марган-
цовые. Марганец используют и в производстве сплавов на основе
цветных металлов — меди (например, манганин), алюминия
(дуралюмин) и др. Металлургическая промышленность использует
богатые марганцем руды с минимальным содержанием SiO2 и фос-
фосфора.
Использование в химической технике пиролюзита и искусствен-
искусственной двуокиси марганца основывается в значительной мере на хо-,
роших адсорбционных свойствах этих веществ7. Они применяются
для изготовления промышленных противогазов, в качестве депо-
деполяризаторов в гальванических элементах22, в качестве низкотем-[
пературных катализаторов для некоторых химических процессов.
Таким катализатором является, например, используемый в проти-!
.вогазах для поглощения окиси углерода гопкалит — смесь, содер-
Применение
757
жащая 50—60% МпО2 и окиси меди, серебра или кобальта. Пер-
манганат серебра AgMnO4 является основной составной частью
катализаторов, служащих для окисления СО в СО2. В чистом виде
он обладает низкими каталитическими свойствами, но весьма эф-
эффективен при нанесении на металлические окислы и в отличие от
гопкалита не отравляется парами воды, и поэтому не требует пред-
предварительной осушки отравленного воздуха23.
Двуокись марганца хорошо поглощает пары ртути; при про-
промывке водной суспензией МпО2 (рН = 2—5) в двухполочном пен-
пенном аппарате из газа улавливается до 99% содержащихся в нем
ртутных паров. Количество поглощенной ртути достигает 20% от
веса сухой МпО2 в суспензии. Ртуть может быть извлечена, а МпО2
регенерирована прокаливанием при 500° в течение 2 ч24. Восста-
Восстановленную пероксидную марганцовую руду предложено использо-
использовать в качестве катализатора для селективного окисления примеси
СО в азото-водородной смеси, применяемой для синтеза ам-
аммиака 2Б. Из окислов марганца с добавкой окислов редкоземель-
редкоземельных элементов получают монокристаллы, обладающие пьезоэлек-
пьезоэлектрическими и ферроэлектрическими свойствами.
Марганцовые руды и двуокись марганца применяются в произ-
производстве стекла, в керамической промышленности для изготовления
глазури, для придания изделиям пурпурного или коричневого от-
оттенка, а также для приготовления фиолетово-черной эмалевой
краски и эмалирования железных изделий. Соединения марганца
широко используются в лакокрасочной промышленности для изго-
изготовления сиккативов и в качестве красящих пигментов (углекис-
(углекислый марганец — марганцовый белый, окись марганца — марган-
марганцовый зеленый, метафосфат марганца — марганцовый фиолетовый
и двуокись марганца — марганцовый черный). Предложено ис-
использовать марганцовые руды и получающиеся при их обогащении
шламы для очистки газов от SOl6, от сероводорода с получением
серы 27, для обессеривания сульфида натрия с получением каусти-
каустической соды28. Восстановленную пероксидную руду (МпО) исполь-
используют для очистки азота от примеси кислорода — при 250—500°
достигается очистка 10~4—-10~S29. Искусственную двуокись мар-
марганца, изготовленную электрохимическим способом и специальной
обработкой пиролюзита (ГАП — гипховский активированный пи-
пиролюзит), используют главным образом в химических источниках
тока, как обладающую хорошими деполяризующими свойствами.
Емкость гальванических элементов, изготовленных на ГАПе, на
15—20% выше, чем элементов, изготовленных на электролитиче-
электролитической двуокиси марганца. Особенно ценным свойствам элементов,
изготовленных на ГАПе, является их сохранность в течение дли-
длительного времени — до двух лет.
Техническими условиями установлены следующие требования
к качеству продуктов.
758
Гл. XXII. Соединения марганца
Активированный пиролюзит для элементной промышленности
(МРТУ 6—09 № 2939—66) выпускают в зернах; он должен со-
держать (в %):
МпО2, не менее ; 70
Мп2О3, не более 15
Fe2O3, не более |,5
№, не более , ; 0,03
Со, не более . . .' 0ДО4
Си, не более 0.03
Сульфатов (SO4). не более 0.7
Влаги, не более 4,0
Не растворимого в соляной кислоте остатка, не более . . 12,0
Однако более важными показателями, чем химический состав
активированного пиролюзита, являются его электрохимические
свойства при работе в качестве деполяризатора в элементах.
В условиях стандартного испыта1 чя электродвижущая сила эле-
элемента 336Х должна быть в пределах 1,74—1,84 в, а емкость не
(Лиже 1,12 а-ч [при разряде на 117 ом на элемент до 1 в (анодный
режим)], при этом допускаются отклонения от указанного содер-
содержания МпО2.
Электролитическая двуокись марганца должна содержать не
менее 90% МпО2 и не более: 8% Мп2О3, 4% гигроскопической
влаги, 0,6% сульфатов (SO3), 1,5% Fe2Os; величина рН — не ме-
менее 3,6. Через сито № 021 должно проходить 95% материала, че-
через сито № 007 — не менее 65 %.
Двуокись марганца и активированный пиролюзит упаковывают
в прорезиненные или крафтцеллюлозные мешки, в стальные ба-
барабаны (лакированные внутри) или в плотные деревянные бочки,
выложенные бумагой.
Перманганат калия, мировое производство которого состав-
составляет 30—40 тыс. т в год 30, применяют главным образом в каче-
качестве окислителя, в частности, для беления тканей, жиров, масел.
Применение в медицине также основано на его сильных окисли-
окислительных свойствах. Для этих же целей применяют и перманга-
перманганат натрия, но в ограниченных количествах из-за трудности полу-
получения в чистом виде и сильной гигроскопичности. Са(МпО2J-5Н2О
применяют для обезвреживания питьевой воды. КМпО4 известен
как хороший дизенсектор3i и дезодоратор, используемый в ус-
установках для кондиционирования воздуха32. Согласно ГОСТ
5777—71, перманганат калия 1-, 2- и 3-го сортов должен содер-
содержать, соответственно, не менее 99,0, 98,0 и 95,0% КМпО4 и не бо-
более: 0,3, 0,75 и 1,0% двуокиси марганца, 0,05, 0,2 и 0,3% сульфа-
сульфатов в пересчете на SO4 и 0,5, 0,5 и 2,0% воды. Тарой для перман-
ганата калия служат стальные барабаны и банки, которые за-
запаиваются или завальцовываются.
Сырье
759
Хлорид марганца МпС12*4Н2О используют в текстильном про-
производстве, для окраски кирпичей при их отжиге, добавляют в
электролиты аккумуляторов; безводную соль применяют для очист-
очистки магния и его сплавов, для получения антидетонаторов, вводи-
вводимых в моторное топливо.
Марганец является одним из эффективных микроэлементов,
требующихся для развития растений. Его вводят в состав удобре-
удобрений, например, суперфосфатов, в виде разных соединений. В Гру-
Грузии в качестве микроудобрения широко используют марганцовый
шлам — отход от обогащения чиатурской марганцовой руды.
Разработано33 получение марганцового микроудобрения из
шлама, содержащего 77,5% МпО2, 23% МпО и 5% органических
веществ. Шлам смешивают с древесными опилками и обрабаты-
обрабатывают 98%-ной серной кислотой — получается спекшаяся рассыпча-
рассыпчатая масса, содержащая до 60% MnSO4, которую разбавляют зем-
земляной пылью или песком. При обработке водой этого продукта
получаемый раствор имеет рН = 3—4. Опрыскивание листвы рас-
растений 0,25—0,12% раствором MnSO4 • Н2О или опыление их по-
порошком сульфата марганца более экономично, чем внесение мар-
марганцовых солей в почву, где они под влиянием биологических фак-
факторов окисляются и переходят в нерастворимое состояние34.
Препарат, называемый .мажефом (марганец — железо — фос-
фосфор), является смесью кристаллогидратов монофосфатов (диги-
дроортофосфатов) марганца Мп(Н2РО4J и железа Fe(H2PO4J и
свободной фосфорной кислоты. По внешнему виду это мелкокри-
мелкокристаллический порошок белого цвета, который, согласно ГОСТ
6193—52, должен содержать 46—52% РгО5, не менее 14% Мп,
0,3—3% Fe и не более: 0,07% водорастворимых сульфатов (в пере-
пересчете на SO3), 0,06% СаО, 6% не растворимых в воде веществ и
19% влаги. Мажеф применяют для фосфатирования стали — нане-
нанесения защитного покрытия, предохраняющего сталь от коррозии.
Общая кислотность мажефа, выраженная в количестве милли-
миллилитров 0,1 н. раствора NaOH, требующихся для титрования на 0,3 г
препарата, должна быть не меньше 25. Прт погружении стального
изделия в нагретый до 98° раствор мажефа происходит растворение
железа с выделением водорода, а на поверхности металла оса-
осаждается плотный, прочный и не растворимый в воде кристалличе-
кристаллический защитный слой фосфорнокислых солей серо-черного цвета.
Когда этот слой достигает определенной толщины, дальнейшее
растворение железа прекращается (стр. 785).
СЫРЬЕ
В земной коре содержится 0,09% Мп — в 50 раз меньше, чем
Fe, но больше, чем любого другого тяжелого металлаЗБ. Марганец
встречается только в связанном виде, главным образом в марган-
марганцовых и железных рудах осадочного происхождения и в малых
760
Гл. XXII. Соединения марганца
Химическая переработка бедных марганцовых руд
761
концентрациях в минеральных и морских водах, и во всех расти-
растительных и животных тканях. Марганцовые руды подразделяют на*
три класса: марганцовые — при содержании Мп > 40%, Fe < 10%;
железомарганцовые —5—40% Мп, 10—35% Fe; марганцовистые"
железные руды при содержании Мп<5%- Запасы марганцовых
руд СССР значительно превосходят запасы во всех других странах
мира36. Важнейшими месторождениями высококачественных мар-
марганцовых руд являются Чиатурское в Грузии и Никопольское на
Украине37-38. Чиатурская пероксидная руда представлена пиролю-
пиролюзитом, псиломеланом, манганитом, браунитом и содержит 60—70%
МпО2. Основная примесь — кремнезем. Концентрат, получаемый
после мокрого обогащения руды, содержит до 96% МпО2. Для
нужд химической промышленности потребляется сырье, содержа-
содержащее не меньше 70% МпО2. Чиатурские карбонатные марганцовые
руды представлены манганокальцитами и кальциевыми родохрози-
родохрозитами. Обогащенная руда содержит более 22% Мп. При нагрева-
нагревании руды сначала диссоциирует МпСО3, затем СаСО3. Это может
быть использовано в некоторых методах обогащения карбонатных
руд39.
Разрабатываются методы химического обогащения инфильтра-
ционных руд Чиатурского месторождения (кремнистых пород, про-
пропитанных гидроокисью марганца) — восстановительным обжигом
с последующей обработкой серной кислотой, а также хлорирова-
хлорированием 40.
Никопольская руда несколько менее высокого качества, чем
Чиатурская, — содержание МпО2 в ней на 15—20% ниже, а со-
содержание SiO2 доходит до 15%. Она представлена пиролюзитом
и псиломеланом.
Крупные месторождения марганцовых руд осадочного харак-
характера, кроме СССР, находятся в Индии, Бразилии и Гане; в других
странах месторождения значительно меньшей мощности с рудами
невысокого качества.
Для промышленности Урала и Сибири представляет интерес
использование местных бедных марганцовых руд21, содержащих
26—28% Мп, 2-6% Fe, 30—40% SiO2, 0,05—0,1% S. Марганцовые
руды Полуночного и некоторых других месторождений "Урала со-
состоят из разных типов оксидных руд — пиролюзитовых, псиломе-
лановых и др. В некоторых образцах полуночной руды содержится .
До 0,02% молибдена41. Концентрат, полученный обогащением пи-
пиролюзитовых руд Полуночного месторождения, как и чиатурская
пиролюзитовая руда, может быть использован для непосредствен-
чого изготовления элементов или активированной двуокиси мар-
марганца.
При мокром обогащении марганцовых руд из-за размыва мяг-
мягких минералов образуются шламы, представляющие собой тонкие
фракции руд. Эти шламы трудно осаждаются и поэтому в значи-
значительной мере теряются со сбросными водами. Шламы содержат
18—30% Мп, и поэтому их использование имеет существенное эко-
экономическое и санитарное значение42. Установлена возможность
ускорения осаждения шламов с помощью сенсибилизаторов 43. Пе-
Переработка шламов, как и бедных марганцовых руд, возможна
только химическим путем, так как эффективный метод флотации
окисных марганцовых минералов пока не найден44. Шламы и бед-
бедные марганцовые руды применяют в качестве удобрений.
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА БЕДНЫХ МАРГАНЦОВЫХ РУД
Разработаны химические методы переработки бедных марган-
марганцовых руд и шламов. Из них наибольшего внимания заслуживает
сернистокислотный способ извлечения марганца, а также обжиг
руды в смеси с пиритом или сульфатизация ее сернистым газом.
Получаемый этими способами сульфат марганца служит полупро-
полупродуктом, перерабатываемым в электролитическую двуокись мар-
марганца и в другие соединения. Разложение бедных марганцовых
руд сернистым газом с последующим пиролизом сульфата мар-
марганца осуществлено на нескольких крупных заводах в США46.
При насыщении сернистым газом водной суспензии пиролюзи-
товой руды или шлама46 вначале образуется дитионат марганца
MnO2 + 2SO2 = MnS2O6
который с течением времени, и в особенности при повышении тем-
температуры, разлагается с образованием сульфата марганца:
MnS2O6 = MnSO4 + SO2
Увеличению количества MnSO4 способствуют побочные реак-
реакции, например окисление SO2 в H2SO4 в присутствии ионов мар-
марганца и взаимодействие МпО2 с дитионатом в присутствии SO2:
MnO2 + MnSsOu = 2MnSO4
При неизменной продолжительности процесса изменение весо-
весового отношения Т : Ж от 1 : 2 до 1 : 10 не влияет на количество
переходящего в раствор марганца, т. е. последнее не зависит от
разбавления суспензии. Но при концентрированных суспензиях ,
уменьшается степень перехода в раствор железа и других нежела-
нежелательных примесей, в особенности фосфора; однако такие суспензии *
труднее отстаиваются и фильтруются. С увеличением концентра-
концентрации сернистого газа степень перехода марганца в раствор возра-
возрастает. При тонкости помола руды 900 отв/см2, при Т : Ж = 1:10,
отношении Мп : SO2, приблизительно равном 1 : 3, концентрации
SO2 ~14% и температуре 80° в течение 90 мин достигается
94—95 % -ное извлечение марганца в раствор 47~49
25 М. Е. Позии
762
Гл. XXII. Соединения марганца
Переработка манганитных руд, содержащих окись марганца,
может быть осуществлена комбинированным методом50'м. Вна-4
чале руду обрабатывают разбавленной серной кислотой:
Мп2О3 + H2SO4 = MnSO4 + MnO2 + Н2О
Выделившийся осадок двуокиси марганца далее может быть
переработан в сульфат марганца сернистым газом.
При насыщении сернистым газом суспензии манганитной руды
(без предварительной сернокислотной обработки) образуются
сульфит и сульфат или дитионат марганца:
Мп2О3 f 2SO2 = MnSO, t- MnSO4
Мп2Ол + 3SO2 = MnSOj + MnS2Oe
Так как дитионат разлагается до сульфата, а сульфит окис-
окисляется содержащимся в газе кислородом (или кислородом воз-
воздуха), то в результате получается раствор сульфата марганца. Од-
Однако манганитные руды реагируют с сернистым газам значительно
медленнее пиролюзитных. Увеличение дисперсности ускоряет про-
процесс, лимитируемый, по-видимому, растворением руды, причем
содержание в растворе дитионата увеличивается, а сульфата отно-
относительно уменьшается. Это объясняется разностью в скоростях
растворения руды и разложения дитионата. В отличие от пиролю-
зитной руды при разбавлении суспензии из манганитной руды (от
Т : Ж = 1 : 2 до 1 : 10) степень извлечения марганца увеличивается
за счет роста количества растворенной двуокиси серы. Повыше-
Повышение концентрации SO2 в газе и в растворе ускоряет разложение
дитионата. При обработке водной суспензии манганитной руды
(Т:Ж= 1 : 10) сернистым газом A6,2—17,6% SO2) при избыточ-
избыточном отношении Mn : SO2, равном 1 : 1,9, и температуре 80° в тече-
течение 3 ч достигается 90%-ное извлечение марганца в раствор.
При обработке сернистым газом водной суспензии карбонатной
марганцовой руды вначале также образуется сульфит, который
содержащимся в газе кислородом или воздухом окисляется в
сульфат:
МпСО3 + SO2 = MnSO, + СО2 ¦
MnSO3 + V2O2 = MnSO4
Дитионат образуется в очень малых количествах, которыми
можно пренебречь. При 18—20° взаимодействие SO2 с суспензией
карбонатной руды идет весьма .медленно. Ускорение процесса воз-
возможно путем предварительного обжига сырья 13- 52. Для некоторых
типов карбонатных руд отмечается 46 достаточно интенсивное раз-
разложение и при ~20°, а при 80° была достигнута высокая степень
извлечения марганца. Наилучшие результаты получены при Т : Ж.
равном 1:10. Для руды, имевшей состав (в %): 24,32 — МпО
(Мп— 18,86), 30,81—СаО, 1,77 — MgO, 4,16 —SiO2, 2,66 — AkO*
Химическая переработка бедных марганцовых руд
763
0,89 —Fe2Os, 1,45 —SO3, 0,55 — Р2О5, 1,68 —влаги, 34,08 —потери
при прокаливании и тонкость помола 10 000 отв/см2, двукратная
обработка избытком 14—15%-ного сернистого газа (Mn : SO2, рав-
равном 1 :2,6) при 80е в течение 50 мин привела к почти 100%-ному
извлечению марганца. Образец другой руды, мало отличавшейся
по составу B1,28 —МпО, 33,18 —СаО, 2,68 —MgO и т.д.), но с
другой структурой разлагался при тех же условиях значительно
медленнее — 95%-ное извлечение марганца достигалось за ~1,5ч.
Поэтому схема и условия переработки карбонатных руд могут
быть различными для разных их типов.
Сульфатизация двуокиси марганца может быть произведена
е помощью сульфата закиси железа или пирита
2MnO2 + 2FeSO4 = 2MnSO4 + Fe2O3 + '/гОа
4MnO2 + 2FeS2 + 5,5O2 = 4MnSO4 + Fe2O3
т. е. без применения серной кислоты или даже сернистого газа.
Смесь пиролюзитной руды с сульфатом железа или пиритом под-
подвергается обжигу и из полученного спека выщелачивается MnSOv
В опвятах 53 обжига смеси искусственной двуокиси марганца с суль-
сульфатом железа при 550—600° в течение 2—3 ч степень извлечения
марганца в раствор была 98—99%, содержание марганца в выще-
выщелоченных остатках 0,7—1,2%. При обжиге с пиритом выход мар-
марганца ниже 89—90%, но следует учитывать, что пирит, например
флотационный колчедан, дешевле сульфата железа. Представляет
интерес дальнейшая разработка этого процесса.
Изучена сульфатизация соединений марганца сернистььм газом
при повышенной температуре64. Сульфатизация Мп2О3 протекает
с заметной скоростью уже при 450°, резко увеличивается в интер-
интервале 450—600° и затем до 750° практически не изменяется. Повы-
Повышение концентрации SO2 в газе увеличивает скорость сульфатиза-
сульфатизации Мп2О3 (рис. 208). Аналогично идет процесс взаимодействия
с SO2 природной карбонатной марганцовой руды, но в этом случае
скорость сульфатизации возрастает во всем интервале температур
и особенно резко выше 600° (рис. 209).
Обогащение бедных окисных марганцовых руд возможно суль-
сульфидным способом — при использовании их для очистки промыш-
промышленных газов от сероводорода55. Образующийся сульфид мар-
марганца можно перевести в сульфат сульфатизирующим обжигом s6~s8
или обработкой серной кислотой (в последнем случае регенери-
регенерируется концентрированный сероводород).
Алюмосиликатные марганцовые руды разлагают путем восста-
восстановительного обжига в присутствии CaSO4 или СаСО3. Использо-
Использование СаСОа предотвращает выделение H2S при последующем
выщелачивании59. Возможно извлечение марганца из бедных
руд. в том числе и карбонатных, обжигом с добавкой аммоний-
аммонийных солей— (NH4hSO4 или NH4C160-61. При обжиге Чиатурской
25*
764
Гл. XXII. Соединения марганца
марганцовой карбонатной руды в псевдоожиженном слое практи-
практически полная диссоциация карбоната достигается уже при 750°62.
Низкосортную марганцовую руду можно обогатить, подвергнув ее
после восстановительного обжига при 900—1000° и охлаждения
гидро- или магнитной сепарации63. Запатентован64 способ получе-
получения окиси марганца, пригодной для производства ферромарганца
\30
500
600 700
Рис. 208. Зависимость сте-
степени сульфатизации Мп2О»
за 30 мин от температуры
и концентрации SO2 в газе.
Цифры на кривых — концен-
концентрация SO2 в %.
" 500 600 700
Температура,°С
Рис. 209. Зависимость сте-
степени сульфатизации при-
природной карбонатной мар-
марганцовой руды за 30 мин
от температуры и концен-
концентрации SO2 в газе. Цифры
на кривых — концентрация
SO2 в газе в %.
восстановлением сульфата марганца пиритом при обжиге этой
смеси в псевдоожиженном слое или во вращающейся печи при
700—800° в отсутствие воздуха.
Из растворов, образующихся при извлечении марганца из руд,
можно получить концентрат высокого качества и с хорошим выхо-
выходом при осаждении его в виде МпСО3. Осаждение можно вести,
например, раствором соды периодическим или непрерывным спо-
способом 65. В последнем случае раствор, содержащий Мп2+, и раствор
соды подают в нижнюю часть реактора, а суспензию МпСОз отби-
отбирают из верхней. Суспензия циркулирует, так что перемешивание
ее при температуре выше 55° и при рН 5,7—7,2 F—6,9) продол-
продолжается 3—5 ч. Это необходимо для того, чтобы плотность кристал-
кристаллов МпСОз достигла величины-1—1,3 г/см3. Суспензия из реактора
Химическая переработка бедных марганцовых руд
765
поступает в сгуститель, затем отделяется от маточного раствора на
фильтре; осадок промывают и высушивают.
Извлечение марганца из бедных карбонатных руд может быть
осуществлено углекислотным методом, заключающимся в обра-
обработке водной суспензии руды двуокисью углерода под давле-
давлением66. Давление СО2 должно быть таким, чтобы карбонат мар-
марганца полностью перешел в раствор в виде бикарбоната, а количе-
количество воды, взятой для приготовления суспензии, должно быть до-
достаточным, чтобы после снижения давления до 1 ат, при обратном
выделении в осадок МпСО3, перешедшие также в раствор бикар-
бикарбонаты Са, Fe и другие в основном остались в растворе. Давление
СО2 и минимальное количество воды могут быть вычислены для
руды определенного состава с помощью следующих констант рав-
равновесий при 25°: *
п2+ + СО2," К, = [Mr
МпСО3
FeCO3 = Fe!
СаСО3<
¦2+
СО2"
[н2со3][со2-]
[Н2СОз1
= 8,1 -103
/со.
3,37 • 10'
v-2
Ввиду малой растворимости компонентов можно вместо актив-
активностей воспользоваться молярными концентрациями. Константы
диссоциации Н2СО3: *
[н+][нсог1 . [н+]Гсо2-1
Я, = i—Lt У. _ 4,52 • 10; Ки = ^-г1 Р- = 5,59 • 10""
[Н2СО3] Л" [НСОз]
Вследствие малой диссоциации иона бикарбоната можно счи-
считать, что все ионы карбоната превратятся в ионы бикарбоната.
Тогда:
A)
/со2
[Са2+][НСО3]2_
/со2
= 5,76-10"
1>320
B)
C)
• Разными авторами приводятся также следующие значения констант:
5.10-» и 1 • Ю-10; Kf - 2.5. НГ11; fff = 4,52 • 10"9; Kf - 4,45 • 10 ;
766
Гл. XXII. Соединения марганца
Например, если руда содержит 32,9% МпСО3, 23,0% FeCO3 и
28,6% СаСОз, то в 1 кг руды 2,86 моль МпСОз, 1,98 моль FeCO3 й
2,86 моль СаСО3. Обозначим х— число кг воды на 1 кг руды, не-
необходимое для растворения всего СаСО3, т. е. 2,86 моль Са2+, при
/?«),= 1 аг. Тогда [Са2+] = ^- и [НСО;] = ^, где Sra — число
молей Мп2+ + Fe2+ + Ca2+, остающихся в растворе, из которых чи-
число молей Мп2+ обозначим у, а число молей Fe2+ — z. Подставив
эти значения в уравнения A) — C) и решив их, находим t/=0,299,
2 = 0,0125 и х = 478 кг. Давление СО2, необходимое для полного
растворения 2,86 моль Мп2+ в 478 кг воды, можно рассчитать из
системы уравнений A) и B). Обозначим количество растворяю-
растворяющихся молей Fe2+ в I кг воды через г. Тогда:
: 1,38 • Ю
= 5,76-КГ9
2,86 Г
478 |/
,г.
т
, ( 2,86
4 478
,/2,86
4 478
о2
+
2,86
478
2,86
478
i.YP
"л
/со2
Решив эту систему уравнений, находим г=2,5-10~4, т. е. вместе
с МпСОз растворится 2,5-10- 478=0,119 моль FeCO3, и /со2=
= 29,7 ат. Так как коэффициент активности СО2 при 25° =«82%, то
необходимое давление СО2 равно 36 ат. Полученный из 1 кг руды
раствор, отделенный от нерастворившегося шлама, будет содер-
содержать 2,86 моль МпСОз, 2,86 моль СаСО3 и 0,119 моль FeCO3 в
виде бикарбонатов. После понижения давления до 1 ат из рас-
раствора выделится в осадок 2,86—0,299=2,56 моль МпСОз и 0,119—
—0,0125 = 0,107 моль FeCOe. Кальций полностью останется в рас-
2 56
творе. Степень извлечения из руды марганца составит -^ • 100 =
= 89,5%, а железа всего
1,Уо
100 = 5,4%.
1,Уо
Чиатурский флотационный марганцовый карбонатный концен-
концентрат перерабатывается азотнокислотным способом в гидрат закиси
марганца, который служит полупродуктом для получения электро-
электролитической двуокиси марганца и других продуктов. Примерный
состав сырья (в %): МпО —21,6, Мп62 —2,5, СаО—10,5, MgO —
1,6, FesO8—1,6, AI2O3 —2,0, Р2О5 —0,4, СО2 —28, SiO2—16,8,
Н2О — 20, прочих примесей—1,5. Концентрат обрабатывают азот-
азотной кислотой и из вытяжки осаждают газообразным аммиаком
при рН = 4—4,5 Fe, A1 и Р в виде FePO4, A1PO4, MnHPO4, Fe(OHK
в присутствии добавки полиакриламида. После отделения осадка
примесей из раствора, содержащего нитраты Мп, Са и Mg, осаж-
осаждают аммиаком при рН = 8,5 Мп(ОНJ. Вследствие образования в
растворе комплекса [Mn(NH3)n](NO3J степень осаждения мар-
Искусственная двуокись марганца
767
ганца составляет только 80—85%. После отфильтровывания осадка
Мп(ОНJ этот комплекс можно разрушить добавкой раствора кар-
карбоната аммония
[Mn(NHj)n](NOsJ + (NH4JCO3 =
+ «NH3 + 2NH4NO3
или окисляя его в течение 2—3 ч воздухом при рН = 8—9:
2[Mn(NH3)n](NO3J + 2Н2О + '/2О2 = Mn2O3 + 2NH4NO3 + (я - 2) NH3
Отфильтрованная и промытая паста Мп(ОНJ содержит 28—
32% Мп, 1—2% МпО2, меньше 0,5% СаО + MgO, меньше 0,3%
азота, 50—60% свободной воды. Прокаливанием гидрата закиси
марганца при 700° получают марганцовый концентрат, содержа-
содержащий 62—65% Мп и меньше 1% СаО + MgO. Его используют в
производстве перманганата калия.
Шламы, отделяемые от растворов после осаждения примесей
и разрушения комплекса [Mn(NH3)n](NO3J, высушиваются и ис-
используются в качестве микроудобрений, а оставшийся раствор
NH4NO3 может быть переработан в твердую аммиачную селитру.
Общие потери азота в описанном процессе в виде окислов азота
(образующихся вследствие частичного восстановления азотной
кислоты) и аммиака достигают 5—8%.
ИСКУССТВЕННАЯ ДВУОКИСЬ МАРГАНЦА
Получение двуокиси марганца электрохимическим способом
Чистая крупнокристаллическая двуокись марганца (у-модифи-
кация, пригодная для элементной промышленности) может быть
.получена электролизом растворов солей марганца. Электролиз хло-
хлорида марганца67> 68 можно вести с помощью свинцовых или графи-
тированных угольных электродов или электродов из металлических
сплавов на основе титана. Хотя продукт, получаемый электроли-
электролизом хлорида марганца, легче отмывается от электролита, чем при
электролизе сульфата марганца, в настоящее время принят способ,
в котором электролитом является сернокислый марганец7'69"1.
Для приготовления раствора электролита природная руда — пи-
ролкэзит восстанавливается до закиси марганца по реакциям:
2МпО2 + С = 2МпО + СО2
МпО2 + С - МпО + СО
С этой целью шихту, состоящую из 90—85% пиролюзита и 10—
15% угля, нагревают до 600—800° в ретортных или вращающихся
печах. Восстановление МпО2 в МпО достигает 90%. Процесс проте-
протекает с образованием промежуточных окислов Mn2Os, MnsO4 72-?4.
Восстановление может производиться в печах с кипящим слоем
промышленными газами. Степень восстановления при этом дости-
достигает 96—99%. Уменьшение содержания в газе Н2 и увеличение СО
768
Гл. XXII. Соединения марганца
Искусственная двуокись марганца
769
способствует устранению слипания зерен руды в процессе обжи-
обжига 75- 76. Материал, выгружаемый из печи, во избежание окисления
на воздухе охлаждается в герметических барабанах.
Охлажденную техническую закись марганца измельчают до тон-
тонкости, соответствующей ситу с 1600 отв/см2, и подвергают выщела-
выщелачиванию отработанным раствором электролита, поступающим из
электролитических ванн и содержащим серную кислоту. Выщелачи-
Выщелачивание производится в резервуарах с мешалками при интенсивном
перемешивании и при нагревании пульпы до 70—80°. Систематиче-
Систематическое выщелачивание неизмельченной технической закиси марганца
может производиться также в диффузионных батареях — высоких
цилиндрических резервуарах, заполненных зернами твердого мате-
материала, через которые циркулирует нагретый раствор электролита.
Этот способ является более громоздким.
При выщелачивании происходит растворение закиси марганца
в серной кислоте и образуется сульфат марганца:
MnO + H2SO4 = MnSO4 + Н2О
Одновременно с серной кислотой реагируют примеси: СаО,
MgO, AI2O3, Fe2O3 и FeO, образуя соответствующие сульфаты. Так
как в технической закиси марганца содержится некоторое количе-
количество двуокиси марганца, то закисное железо окисляется в окисное:
2FeSO4 + 2H2SO4 + МпО2 = MnSO4 + Fe2(SO4K + 2Н2О
После полной нейтрализации серной кислоты вследствие гидро-
гидролиза сульфатов в осадок выделяются гидраты окиси железа и алю-
алюминия. Таким образом, в полученном растворе из указанных при-
примесей остается лишь небольшое количество сульфата магния.
После последующего подкисления серной кислотой раствор элек-
электролита должен содержать 300—350 г/л MnSO4 и 200—180 г/л
H2SO4.
Необходимый для электролиза раствор сульфата марганца мо-
может быть получен из окисных руд, содержащих МпО2, восстанов-
восстановлением ее в кислых растворах порошкообразным ферромарганцем
G5—80% Мп и 8—15% FeO7. При перемешивании измельченных
руды и ферромарганца (частицы меньше 0,25 мм) с циркулирую-
циркулирующим электролитом (H2SO4 + MnSO4) и с добавляемой к нему сер-
серной кислотой МпО2 восстанавливается двухвалентным железом
с образованием MnSO4 и Fe2(SO4K по реакции, приведенной выше.
Растворяющийся ферромарганец вновь восстанавливает Fe3+ до
Fe2+ по реакциям:
Fe2(SO4K + Mn = 2FeSO4 + MnSO4
Fe2(SO4K + Fe = 3FeSO4
Образовавшееся двухвалентное железо снова вступает в реак-
реакцию с МлОг. Таким образом, процесс идет за счет растворения
ферромарганца. Его можно вести без подогревания, или при
65—75° 78>79 в течение 4—12 ч. Затем, повышая рН до 6—6,5 до-
добавлением ферромарганца или Са(ОНJ, осаждают гидроокись
железа, которая окклюдирует и другие примеси (Са, К, Na, As
и проч.). После отделения осадка и подкисления серной кислотой
раствор направляют на электролиз.
Очистку раствора MnSO4 от ионов кальция предложено80 осу-
осуществлять, добавляя эквивалентное количество оксалата или F"
(NH4F) при одновременном окислении Fez+ (воздухом) и осажде-
осаждении Fe(OH)s.
Электролиз ведется при 20—25° в ваннах, футерованных свин-
свинцом или винипластом, со свинцовыми электродами, при напряже-
напряжении на ванне 3—3,5 в и при плотности тока на аноде 500 а/ж2 и на
катоде 1000—1200 а/м2. В ваннах большой мощности целесооб-
целесообразна установка охлаждаемых анодов. В результате окисления на
аноде двухвалентного марганца образуется двуокись марганца; на
катоде выделяется водород. Суммарное уравнение процесса имеет
вид:
MnSO4 + 2Н2О = MnO2 + H2SO4 + Н2
При 18° нормальный потенциал окисления Мп2+ —» Мп3+ равен
+ 1,511 в, а для Мп3+—»Мп4+ +1,642 в, т. е. на аноде наиболее
легко образуется Мп3+. В сильнокислом растворе образование
Мп4+, требующее более высокого потенциала, идет после заверше-
завершения превращения Мп2+ в Мп3+. В слабокислом растворе механизм
процесса иной — имеет место равновесие 2Мп3+ч=*=Мп2+ + Мп4+, ко-
которое сдвигается вправо вследствие гидролиза
Mn(SO4J + 2Н2О = MnO2 + 2H2SO4
так что образование МпО2 происходит без окисления' Мп3+ на
аноде, где окисляется лишь Мп2+. Поэтому выход МпО2 по току
зависит от кислотности раствора, которая увеличивается в про-
процессе электролиза по мере уменьшения концентрации Мп2+. В силь-
сильнокислом растворе, где концентрация Мп3+ и Мп4+ велика, частич-
частично идет их обратное восстановление на катоде, доходящее до 25%.
В слабокислом растворе анод покрывается рыхлой коркой двуокиси
марганца, образующейся здесь в результате гидролиза, затруд-
затрудняющей доступ к аноду Мп2+ и понижающей перенапряжение вы-
выделения кислорода, вследствие чего его образование усиливается.
Уменьшение концентрации Мп2+ также снижает выход по току.
Поэтому электролит, отбираемый из ванн и направляемый вновь
на выщелачивание закиси марганца, содержит 50—60 г/л MnSO4
и до 450 г/л H2SO4. Сопоставление с начальным составом электро-
электролита показывает, что окисление на аноде достигает 80—85%.
Потери от катодного восстановления можно уменьшить приме-
применением графитовых катодов и совсем устранить, закрывая ка
77C
Гл. XXII. Соединения марганца
Искусственная двуокись марганца
771
диафрагмой из ткани, стойкой в кислом электролите. Однако диа-
диафрагма засоряется двуокисью марганца, что усложняет ее исполь-
использование. Электролит необходимо очищать от примесей железа,
меди, кобальта и других, сильно понижающих выход по току. Так,
в присутствии всего 0,05 г/л Fe или Си выход по току падает при-
приблизительно на 30%. Рекомендуются порошковые (угольные) ка-
катоды 81.
Исследованием процесса электролиза проточного раствора хи-
химически чистых MnSO4 и H2SO4 в ванне со стальными катодами,
анодами из сплава РЬ с 4% Sb и хлопчатобумажной диафрагмой
установлено82, что выход по току снижался от 95,5 до 39,4% при
повышении анодной плотности тока от 418 до 2520 а/м2 и возра-
возрастал от 39,4 до 79,1% с увеличением температуры анолита от 26,5
до 73°. Максимальный выход по току был достигнут при содержа-
содержании в электролите 207 г/л MnSO4.
Раствор электролита выпускают из ванны вместе с осадком
двуокиси марганца на освинцованную или гуммированную центри-
центрифугу. Здесь осадок отделяют и промывают. В связи с хорошей
адсорбционной способностью двуокиси марганца отмывка ее от
электролитов идет с трудом и требует большого расхода воды.
После отмывки двуокись марганца высушивают при температуре
не выше 100° во избежание ухудшения ее адсорбционных свойств.
Расход энергии на производство 1 т МпО2 составляет около
2000 кет • ч.
По другому варианту, получение электролитической двуокиси
марганца ведут в ваннах с графитированными угольными электро-
электродами 82. Сырьем служит паста гидрата закиси марганца (стр. 767),
которую растворяют в серной кислоте (в кислом отработанном
электролите) с добавкой перекиси водорода для восстановления
МпО2 по реакции:
МпО2 + H2SO4 + Н2О2 = MnSO4 + 2Н2О + О2
После отделения фильтрацией шлама (состоящего главным об-
образом из CaSO4) получают электролит, содержащий 100 г/л MnSO4,
20 г/л H2SO4 и меньше 0,03 г/л Fe и 0,03 г/л Си. Электролиз ведут
при 85—95°, при анодной плотности тока 100 а/м2 и катодной
ЮО—140 а/м2 и напряжении 2,5—3 в. Выход по току 90—93%;
расход электроэнергии 2600 кет- ч на 1 т продукта. Использование
анодов одноразовое (продолжительность электролиза 800—1000 ч),
а катодов — до капитального ремонта (~5000 ч). Образующаяся
в процессе электролиза МпО2 осаждается плотным слоем на ано-
анодах — графитированных угольных стержнях, укомплектованных
в анодных рамах. По окончании электролиза эти рамы с осадком
промывают и демонтируют, затем их комплектуют новыми элек-
электродами и загружают в ванны. Расход электродов доставляет
300 кг на 1 г МпО2. Осадок, снятый с анодов, дробят, размалы-
размалывают, промывают водой, высушивают и упаковывают. В продукте
содержится небольшое количество графита (менее 4%).
Производство активированного пиролюзита—ГАПа
Для получения ГАПа используют наиболее высококачествен-
высококачественные сорта пиролюзита, содержащие 80—90% МпО283. Пиролюзит
обжигают во вращающихся печах при 800—950°, причем двуокись
¦марганца переходит в низшие окислы, преимущественно в Мп3Оз.
О качестве обожженного материала судят по коэффициенту вос-
восстановления, т. е. по молярному отношению МпО: МпО2. Жела-
Желательно, чтобы коэффициент восстановления был равен единице,
что соответствует восстановлению МпО2 до МпгО3. Практически
коэффициент восстановления колеблется от 0,96 до 1,1.
Обожженную руду выщелачивают 17—18%-ной серной кисло-
кислотой при температуре около 80°. Серная кислота взаимодействует
с окисью марганца по реакции:
Мп2О3 + H2SO4 = MnO2 + MnSO4 + Н2О
Образующийся сульфат марганца переходит в раствор, а в
твердой фазе остается двуокись марганца.
Как показала практика, выщелачивание наиболее рационально
вести в аппаратах периодического действия. В бак, снабженный
ложным дном, загружают сначала слой более крупного материала,
а потом обожженную руду с размером зерен 0,5—0,6 мм; затем
заливают горячую серную кислоту. После некоторого выстаивания
рбразовавшийся раствор сульфата марганца сливают, а бак зали-
заливают новой порцией кислоты. Таким образом осуществляют
25—30 заливов кислоты, после чего аналогичным образом мате-
материал промывают горячей водой. Остаточную кислотность нейтра-
нейтрализуют слабым раствором аммиака. Выщелачивание вначале идет
быстро: раствор выдерживают в баке около 30 мин, а к концу —
до 4 ч. Выщелачивание может производиться и полунепрерывным
путем, при циркуляции кислоты с помощью центробежного на-
насоса. Чем полнее извлекается из материала окись марганца, тем
более высокого качества получается ГАП. После промывки водой
и нейтрализации продукт высушивают при температурах не выше
100°. Более высокие температуры сушки понижают его активность.
Отходом при производстве ГАПа является раствор сульфата
марганца. Его используют в качестве электролита в производстве
электролитической двуокиси марганца. Из него может быть также
получен электролизом металлический марганец. Наконец, он мо-
может быть переработан на кристаллический сульфат марганца или
на карбонат марганца, из которого затем получают другие соеди^
нения, в частности реактивы.
772
Гл. ХХМ. Соединения марганца
Искусственная двуокись марганца
775
Получение активной двуокиси марганца
из бедных руд
Качество активных марганцовых материалов, как деполяриза-
деполяризаторов, зависит от их дисперсности и малой упорядоченности ре-
решетки. Некоторые бедные руды, например чиатурские пористые
руды, обладают высокой дисперсностью и путем их химической' и
термической обработки можно получить84 активные марганцовые
материалы, пригодные для применения в элементной промышлен-
промышленности. Так, при обработке этих руд серной кислотой или вначале
щелочью, а затем серной кислотой получаются препараты, не усту-
'пающие по своей активности искусственной двуокиси марганца.
Изучено разложение карбонатных руд окислами азота. Получае-
Получаемый нитрат марганца может быть подвергнут пиролизу8Б или
электролитическому разложению86, в последнем случае на аноде
образуется очень чистая и активная двуокись марганца, при рас-
расходе энергии 2,5 кет • ч на 1 кг Мп.
Азотнокислотная переработка бедных марганцовых руд осуще-
осуществлялась на опытной установке87 производительностью 450 кг/ч.
Руду, измельченную до частиц размером 0,25 мм, обжигают в вос-
восстановительной печи, обрабатывают азотной кислотой и получен-
полученный раствор нитрата марганца после отделения нерастворимого
остатка выпаривают при 116° до концентрации 65% твердого
вещества. Затем его разлагают в обогреваемой паром двухвальцо-
вой сушилке, причем для получения у-МпО2, идущей на изготовле-
изготовление сухих элементов, температура разложения не должна превы-
превышать 350°. Выделяющиеся пары HNO3 и нитрозные газы воз-
возвращают в процесс, а МпО2 размалывают, промывают и сушат.
Разложение раствора нитрата марганца ведут также во вращаю-
вращающейся пламенной печи, причем из газов регенерируется до 95%
азотной кислоты88. Запатентован способ переработки нитрата мар-
марганца в двуокись, названную е-МпО2, которую можно применять в
качестве катализатора окисления или деполяризатора. Он заклю-
заключается в барботировании через слой расплавленной соли смеси
азота (до 40%) и водяного пара при 130°. После охлаждения из
реакционной массы отмывают остаток нитрата 89. Окисление Мп2+
(нитрат или сульфат) можно производить с помощью КМпО4 в
сильно кислой среде при 80°; после отмывки и сушки при 30° про-
продукт имеет поверхность 180ж2/г, а при 60° — 350 м2/г; отношение
Мп : О равно 1,94—1,9590. Некоторые виды сырья можно обраба-
обрабатывать азотной кислотой без предварительного их обжига, причем
такой переработке могут подвергаться руды с большим содержа-
содержанием соединений железа, которые в этих условиях не растворяют-
растворяются ei, 92 в США, в связи с отсутствием высококачественных мар-
марганцовых руд, считают целесообразным использовать и обогащен-
обогащенные, содержащие марганец шлаки доменных печей.
Предложено93 регенерировать МпОг из растворов, содержащих
MnSO4 и (NH4JSO4, получающихся при окислении двуокисы®
марганца органических соединений. Растворы обрабатывают из-
известковым молоком, выделяющийся аммиак улавливают, а осадок
Мп(ОНJ растворяют в азотной кислоте. После отделения гипса
раствор выпаривают до получения сухого нитрата марганца, из
которого нагреванием до 200—300° получают МпО2.
Двуокись марганца может быть получена из низкосортных
марганцовых руд солянокислотным способом. Максимальное из-
извлечение марганца концентрированной соляной кислотой из пред-
предварительно обожженной при 800° руды достигается в течение
20 мин. При содержании в руде 36,6% Мп и 30,4% Fe в раствор
перешло почти 70% марганца и остаток содержал 55,54% Fe и
9,25% Мп. Гидролиз полученного раствора хлорида марганца при
10-кратном разбавлении его водой в присутствии воздуха дает
осадок продукта, содержащего 85% МпО2 и практически не содер-
содержащего железа94. Получение МпО2 из различных марганцовых
материалов через хлорид марганца привлекает все большее вни-
внимание. Описан способ получения МпО2 окислением хлором в ще-
щелочной среде осадка Мп(ОН)г, полученного обработкой раствора
МпС12 содой 95. Запатентован катионитный способ извлечения Мп2+
из сернокислого раствора с регенерацией катионита соляной кис-
кислотой, кристаллизацией хлорида марганца высаливанием его хло-
хлористым водородом, термическим разложением осадка на МпО и
НС1 и с последующим окислением МпО кислородом воздуха; про-
продукт содержит 55% Мп96. Двуокись марганца, пригодную для де-
поляризяторов в сухих батареях, можно получить обработкой
солянокислой вытяжки из восстановленного природного пиролю-
пиролюзита, содержащей 5—25% Мп в виде хлоридов и 0,5—5% свобод-
свободного НС1, щелочным раствором гипохлорита при охлаждении.
После выделения осадка МпО2 рН смеси должен быть в пределах
2—5. Осадок промывают и выдерживают длительное время в
97
воде
Предложено 98 перерабатывать MnSO4 в МпС12 эквимолекуляр-
эквимолекулярным количеством СаС12 и после отделения осадка CaSO4 обраба-
обрабатывать раствор МпС12 большим количеством Са(ОНJ. В получен-
полученную суспензию Мп(ОНJ и избытка Са(ОНJ в растворе СаСЬ
пропускают воздух (кислород), для перевода Мп2+ в Мп4+. Для
завершения окисления суспензию хлорируют и образующуюся дву-
двуокись марганца отделяют от раствора СаС12.
В присутствии водяного пара при ~600° твердый хлорид март
ганца гидролизуется с образованием окислов
2МпС12 + ЗН2О
ЗМпС12 + 4Н2О
Мп2О3 + 4НС1 + Н2
Мп3О4 + 6НС1
774
Гл. XXII. Соединения марганца
Сульфат марганца
775
или, с большей скоростью, в присутствии паро-воздушной смеси:
4МпС12 + 4Н2О + О2 ^=± 2Мп2О3 + 8НС1
6МпС12 + 6Н2О + О2 ^=± 2Мп3О4 + 12НС1
При 600° степень превращения в паровой среде в течение 2,5 ч
достигает 73%, а в паро-воздушной среде за 2 ч 99%. С повыше-
повышением температуры скорость гидролиза увеличивается. Получаемые
продукты являются смесью р-Мп2О3 и р-Мп3О4".
Возможна переработка марганцовых руд с помощью аммиач-
аммиачных растворов26. Руды, содержащие МпО, могут обрабатываться
~18%-ной аммиачной водой с ~2% сульфата, хлорида, нитрата,
карбоната или карбамата аммония. Образуется раствор комплекс-
комплексной марганцово-аммиачной соли с концентрацией в пересчете на
Мп(ОНJ ~ Ю г/л. Выщелачивание ускоряется в присутствии вос-
восстановителей, например (NH4JS (в количестве 3—15 кг на I т
руды). Из отделенного раствора с помощью СО2 осаждают МпСО3
при температуре выше 60°, поддерживая молярное отношение
СО2: Мп в растворе больше 1,75. Нагреванием в течение несколь-
нескольких часов при 150—315° в воздухе или в кислороде полученный
МпСОз переводят в МпО2 шо.
Получение активной двуокиси марганца термическим способом
Этот способ проще и экономичнее химических способов. Он за-
заключается в непосредственном окислении манганитных руд 101 при
275—300° в течение 20—30 мин:
Мп2О3 • Н2О + У2О2 = 2МпО2 + Н2О
i
Скорость окисления и активность окисленного продукта зави-
зависят от физических свойств руды, содержания в ней марганца и
других компонентов, в особенности щелочных соединений. Послед-
Последние являются катализаторами, ускоряющими окисление окиси мар-
марганца 102. В отличие от манганитной руды искусственная окись
марганца в отсутствие катализаторов окисляется до МпО2 очень
медленно, но при добавке к ней 4—6% NaOH или КОН при 400°
в течение 2 ч степень окисления достигает 85—90% 103. Интенсив-
Интенсивность процесса зависит и от условий приготовления Мп2О3.
Для получения высококачественного продукта термическим
способом необходимо выбирать пригодные для этой цели руды.
Сравнительная легкость перехода природных манганитных руд
в руды пиролюзитного типа с активными свойствами позволяет
широко использовать термический способ для переработки бедных
марганцовых руд.
Для получения двуокиси марганца реактивной чистоты лучшим
считается 104 метод термического разложения осажденного МпСОз
i < последующим окислением воздухом образовавшейся МпО в при-
присутствии добавки — азотной кислоты для предотвращения диссо-
диссоциации МпО2. Отжатый на центрифуге осадок, содержащий
~70% МпСО3, сушат при 200°, затем смачивают азотной кислотой
и подвергают окислительному обжигу при 450—500° до полного
удаления окислов азота.
СУЛЬФАТ МАРГАНЦА
Сульфат марганца получают способами, рассмотренными выше,
из природной двуокиси марганца (стр. 767) и из бедных руд
(стр. 761). В связи с возрастающей потребностью в сульфате мар-
марганца как микроудобрении и для переработки его в другие соеди-
соединения изыскиваются и другие способы его производства. Многие
из них также основываются на предварительном восстановитель-
восстановительном обжиге природной руды, например аммиачный способ, в кото-
котором выщелачивание восстановленной руды производится сульфа-
сульфатом аммония 21> 105. Другие способы основаны на обработке серной
кислотой марганцовой карбонатной руды50> 106.
Серную кислоту, образующуюся в результате побочной реакции
окисления SO2 в H2SO4 под каталитическим влиянием ионов мар-
марганца, нейтрализуют добавкой к суспензии карбонатной руды
A0—15% от начального количества руды). При этом облегчаются
отстаивание и фильтрация пульпы. Очистка раствора достигается
его подщелачиванием до рН = 7—8, причем железо, фосфор и
другие примеси осаждаются. После отстаивания и фильтрации
осадок с фильтра, промытый водой, направляют в отброс, промыв-
промывные воды — на приготовление пульпы, а раствор MnSO4 — на
дальнейшую переработку.
Запатентован способ получения MnSO4 из карбонатной руды
обработкой ее разбавленной серной кислотой (отношение
Мп : H2SO4, равное 1 : 2,1, Ж : Т «* 3) в присутствии катализатора
KI10?. Процесс ведут при рН = 2—3, температуре 80—90° и при
'вдувании воздуха почти до полного выщелачивания марганца. За-
Затем горячую пульпу фильтруют, раствор MnSO4 направляют на
переработку, а осадок промывают холодной водой и промывную
воду возвращают на разложение руды.
Растворение карбонатной руды ускоряется в присутствии окис-
ной руды (пиролюзита) 108.
Когда сульфат марганца является промежуточным продуктом
производства, его раствор перерабатывают электролизом, осажде-
осаждением марганца в виде карбоната или гидроокиси и т. д. Термиче-
Термическая диссоциация твердого сульфата марганца позволяет регенери-
регенерировать из него серу и окислы марганца. Добавка к обжигаемому
сульфату марганца 5—10% угля ускоряет процесс его разложе-
разложения. Приемлемые для практических целей скорости разложения
MnSO4 в атмосфере воздуха достигаются при 850°, азота — при
776
Гл. XXII. Соединения марганца
800°, причем твердые продукты обжига состоят почти целиком из
MnsO4, а газовая фаза содержит серу в виде SO2 (содержание SO3
меньше 3% от суммы SO2 + SOSI09.
Выделение кристаллического сульфата марганца из его рас-
раствора осуществляется выпариванием и кристаллизацией или выса-
высаливанием концентрированной серной кислотой.
Ввиду того, что растворимость сульфата марганца с повыше-
повышением температуры в интервале от 27 до 100° понижается, кристал-
кристаллизация охлаждением концентрированного раствора оказывается
невозможной. Изотермическая кри-
кристаллизация при выпарке из раствора
воды, т. е. выпарка «на кристалл» не-
несколько затруднена, потому что на
греющих поверхностях выпарного ап-
аппарата нарастает толстый слой соли,
сильно снижающий коэффициент теп-
теплопередачи Кроме того, при выпарке
образуются чрезвычайно мелкие кри-
кристаллы MnSO4 • Н2О, отделение кото-
которых на фильтре от маточного раство-
раствора идет крайне медленно.
Для избежания этих трудностей
кристаллизацию сульфата марганца
можно осуществлять нагреванием рас-
раствора ио. Растворимость сульфата мар-
марганца выше 100° резко снижается
(рис. 210); в твердой фазе находится
одноводная соль MnSO4 • Н2О. Если,
например, исходный раствор содержит
20% MnSO4) то при нагревании его до
140 160
Температура, "С
Рис. 210. Растворимость MnSO4 160° в твердую фазу выделится 86%.
в воде при 120-200°. а при 180° —94% от общего количе-
количества MnSO4.
Выделение сульфата марганца может быть осуществлено на-
нагреванием раствора до 160е в автоклаве острым паром, имеющим
давление 7—8 ат. Достаточно 15-минутной выдержки при этой
температуре, чтобы в маточном растворе осталось 3,8% MnSO4.
Затем маточный раствор спускают через фильтр, находящийся
внутри автоклава, и в автоклав загружают новую порцию свежего
раствора. После двух- или трехкратной кристаллизации соль вы-
выгружают. Наиболее подходящим материалом для автоклава яв-
является медь или бронза.
Маточный раствор от кристаллизации может быть употреблен
для разбавления серной кислоты, идущей в производство ГАПа,
вследствие чего конечный выход сульфата марганца будет бли-
близок к 100%. Так как в исходном растворе сульфата марганца со-
Хлорид марганца
777
держится около 1 % MgSO4, то в маточном растворе будет накап-
ливаться сульфат магния. Присутствие последнего благоприят-
благоприятствует кристаллизации MnSO4 • Н2О, так как понижает его рас-
растворимость. Часть маточного раствора должна периодически из
цикла выводиться.
Растворимость сульфата марганца в присутствии серной кис-
кислоты сильно падает. Так, при 25° при содержании в растворе
40,8% H2SO4 растворимость MnSO4 составляет 3,8%, при 62%
H2SO4— 0,5%. При концентрации H2SO4 ниже 62% твердой фазой
является MnSO4 • Н2О, при более высокой концентрации — MnSOt
При еще более высокой концентрации и низкой температуре в
твердую фазу выделяется кислая соль MnSO4 • H2SO4 • Н2О.
На основании этого Д. М. Корфом предложена следующая
схема извлечения сульфата марганца из раствора. К раствору
добавляют серную кислоту до концентрации 40% H2SO4. При этом
выпадает в осадок около 80% сульфата марганца. Отделенный
осадок, содержащий до 40% маточного раствора, промывают рас-
раствором сульфата марганца. Растворы от промывки поступают на
высаливание серной кислотой. Полученная соль содержит около
68% MnSO4 и 0,3—0,6% H2SO4. Недостатком этого способа яв-
является большой расход серной кислоты, примерно в 3 раза превы-
превышающий расход ее на производство ГАПа. Получающаяся после
высаливания слабая серная кислота лишь частично может быть
использована для производства ГАПа, что и затрудняет реализа-
реализацию этого способа.
ХЛОРИД МАРГАНЦА
Хлорид марганца производят растворением в соляной кислоте
марганцовой руды, пиролюзита ш, карбоната или гидроокиси мар-
марганца, а также закиси марганца, получаемой предварительным
восстановительным обжигом руды (стр. 767). Двуокись марганца
растворяется по реакции Уэльдона
МпО2 + 4НС1 = МпС12 + С12 + 2Н2О
применявшейся раньше для регенерации хлора из соляной кис-
кислоты. С меньшим расходом кислоты и без потери хлора идет рас-
растворение закиси марганца:
МпО + 2ИС1 = МпС12 + Н2О
Наиболее чистый раствор получается при растворении в соля-
соляной кислоте предварительно осажденных из растворов гидрата за-
закиси или карбоната марганца. При обработке соляной кислотой
сырой или восстановленной руды в раствор переходят примеси
тяжелых и других металлов. Если производить «основное» выще-
выщелачивание, т. е. при избытке руды, то перешедшие вначале в рас-
раствор железо и алюминий к концу процесса осаждаются в виде
778
Гл. XXII. Соединения марганца
гидроокисей. Обычно очистку слабокислых растворов МпС12 от Fe,
А1, Си, №, Со и других примесей производят, нейтрализуя их ам-
аммиаком. Тяжелые металлы можно выделять в виде сульфидов.
Очищенный раствор МпС12 выпаривают до насыщения, а затем,
при охлаждении до 20°, кристаллизуется МпС12 • 4Н2О. Дополни-
Дополнительную кристаллизацию можно осуществлять высаливанием со-
соляной кислотой с возвратом маточного раствора на обработку
сырья.
Описанm способ получения тонкодисперсного кристалличе-
кристаллического МпС12-4Н2О из раствора в одну стадию — сушкой в фонта-
фонтанирующем слое.
Предложено перерабатывать в МпС12 отбросный шлам от про-
производства КМпО4 (стр. 783). С этой целью его сначала промывают
10%-ной соляной кислотой, а затем обрабатывают 30%-ной. В ре-
результате экзотермической реакции и нагревания до 90° выделяется
газообразный хлор и 95% МпО2 превращается в МпС12. Раствор
нейтрализуют карбонатом марганца, фильтруют и высушивают в
распылительной сушилке воздухом при 150°. Продукт содержит.
82% МпС12 "в.
Раствор МпС12 может быть получен взаимодействием MnSO4
с СаС12 или обработкой раствором СаС12 твердого карбоната мар-
марганца.
Исследование реакции
МпСО3(кр.) + СаС!2(раств.) ^г* МпС12 (раств.) + СаСО3 (кр.)
выполнено114 с синтетическим углекислым марганцем МпСОз*
• 0,5Н2О. С повышением концентрации СаС12 равновесная концен-
концентрация МпС12 резко возрастает: для 100° при концентрации СаС12
5,5 и 7,5 г-мол/л концентрация МпС12 равна соответственно 79,2 и
146,5 г/л\ для 200° при концентрации СаС12 4 и 5 г-мол/л кон-
концентрация МпС12 соответственно 98,8 и 162 г/л. Смещение равно-
равновесия реакции в концентрированных растворах СаС12 в сторону
образования МпС12 объясняется образованием прочных гидратов
Са2+. При концентрации СаС12 4—5 г-мол/л при 100° за 8 мин 65%
MnCOg превращается в МпС12, за ХЪмин — 85%, за 60 мин — 90%.
Эта реакция первого порядка. Лимитирующей стадией является
образование промежуточного соединения — гидрооксикарбоната
марганца Мп(ОН)(НСО3) за- счет присоединения молекул воды
к карбонату марганца. Изучены также равновесия в водных115 и '
безводных116 системах, содержащих хлориды Мп, К и Na.
Хлорид марганца можно получать117 хлорированием газооб-
газообразным хлором в присутствии восстановителя пористых чиатур-
ских руд, кристаллического пиролюзита или оксидной марганцо-
марганцовой руды при 550—600°. После охлаждения хлорированной руды
МпС12 выщелачивается водой и выкристаллизовывается. Отделе-
Отделение примесей железа достигается фильтрованием раствора через
Перманганат калия
779
толстый слой активированного пиролюзита с диаметром частиц
1—1,5 мм или добавлением к раствору сырой карбонатной марган-
марганцовой руды с последующим отфильтровыванием осадка.
Во время второй мировой войны в США значительные количе-
количества хлорида марганца готовили из окиси, полученной сжиганием
пирофорной смеси марганцовой руды (содержащей МпО2) с фер-
ферромарганцем и древесными опилками118. Ферромарганец служил
восстановителем, опилки обеспечивали необходимую пористость и
восстановительную атмосферу в слое горящей шихты. Спекшийся
материал измельчали и растворяли в соляной кислоте, раствор
МпС12 очищали от железа и других примесей и перерабатывали
в кристаллический продукт.
Безводный хлорид марганца получают из кристаллов
МпС12-4Н2О. При 58° тетрагидрат плавится. Поэтому обезвожива-
обезвоживание лучше всего производить, смешивая МпС12 • 4Н2О с большим
количеством ретура — безводного МпС12 и нагревая смесь в бара-
барабанной сушилке выше 220° или осуществляя этот процесс в аппа-
аппарате с кипящим слоем. Безводный МпС12 можно получить хлори-
хлорированием марганца или ферромарганца газообразным хлором ш.
Разработана технологическая схема извлечения марганца из руд
и шлаков хлорированием их при 900° и фракционной конденсацией
из отходящего из реакционной печи газа МпС12 при 550° и FeCl3
при 50" 12°. Предложен способ121 избирательного хлорирования
марганца обработкой материала, содержащего больше железа, чем
марганца, при 150—480° газообразным хлористым водородом
с примесью такого количества водорода, чтобы в отходящем газе
обнаруживались его следы. Процесс ведут до превращения ~50%
марганца в хлорид и достижения соотношения MnCl2: FeCl2, рав-
равного по крайней мере 1 : 1.
ПЕРМАНГАНАТ КАЛИЯ
Перманганат калия получают разложением МпО2 едким кали
и разложением ферромарганца едким кали и электролизом 30. Наи-
Наиболее распространено щелочное разложение пиролюзита с полу-
получением манганатного плава. На старых установках его осуще-
осуществляют в обогреваемых топочным газом котлах, на современных
установках — во вращающихся тарельчатых печах и в других не-
непрерывно действующих аппаратах.
При щелочном разложении производство перманганата калия
осуществляется в две стадии. В первой стадии получается манга-
натный плав, содержащий К2Мп04; во второй стадии манганат
окисляется в перманганат.
Получение манганата.в виде манганатного плава достигают
сплавлением пиролюзита с едким кали в присутствии воздуха:
2МпО2 + 4КОН + О2 = 2К2Мп04 + 2Н2О
780
Гл. XXII. Соединения марганца
Тонко размолотый в шаровой мельнице высокосортный пиро-
пиролюзит и 50% раствор КОН сплавляют при 200—270°. Более высо-
высокие температуры ведут к разрушению уже образовавшегося ман-
ганата с выделением кислорода. Разложение КгМпСХ при
475—960° в атмосфере кислорода или азота 30-122 протекает в ос-
основном по реакции
ЗК2Мп04 = 2К3Мп04 + МпО2 + О2
и небольшое количество манганата (8—10%) разлагается по ре-
реакции:
2К2Мп04 - 2К2Мп03 + О,
Получаемая по первой реакции двуокись марганца теряет часть
кислорода и фактически находится в плаве в виде вещества с со-
составом MnOi]8_l,75-
При получении манганатного плава в плоских чугунных кот-
котлах, обогреваемых снизу топочными газами и снабженных мешал-
мешалками скребкового типа, делающими до 30 об/мин, котлы эти обыч-
обычно открыты для облегчения доступа воздуха; над ними устанавли-
устанавливают вентиляционные вытяжные колпаки. В нагретый котел
вначале загружают пиролюзит и влажную двуокись марганца, по-
полученную во второй стадии процесса при выщелачивании манга-
манганатного плава. Материал подсушивается, затем к нему добавляют
небольшими порциями 50% раствор КОН. Общее количество за-
загружаемой в ¦ котел щелочи соответствует весовому отношению
МпО2: КОН, равному I : 1,45. Иногда смешение пиролюзита с рас-
раствором едкого кали производят в специальных смесителях, после
чего GMecb загружают в прокалочные котлы. Операция плавки
продолжается около суток при непрерывном перемешивании. Плав
имеет вид мелких комочков. Процесс протекает медленно, так как
окисление двуокиси марганца в манганат происходит преимуще-
преимущественно на поверхности этих комочков; внутренняя их часть почти
не окисляется. Поэтому выход манганата в лучшем случае дости-
достигает 60%; получаемый плав содержит до 30—35% К2Мп04, около
25% КОН, значительное количество МпО2, КгСО3 и другие при-
примеси.
Находящиеся в пиролюзите примеси влияют на физические
свойства плава — Fe2Os действует как отощающий материал и не
является помехой, а А12О3 и SiO2 образуют с КОН растворимые
(низкоплавкие) соединения, что приводит к увеличению липкости
плава. Добавка извести не устраняет появления этих соединений 30.
Иногда плавку производят в закрытых котлах, в которые вду-
вдувается воздух, в два приема, с промежуточным измельчением плава
в шаровых мельницах для устранения комков и ускорения про-
процесса окисления. Процесс плавки в котлах является периодическим
и поэтому весьма трудоемким.
Перманганат калия
781
Вследствие малого содержания манганата в получаемом плаве
при дальнейшей переработке его в перманганат теряются значи-
значительные количества едкого кали (расход 200% от теоретического).
и<манганата (расход 150% от теоретического).
При использовании для получения манганатного плава вра-
вращающихся барабанных печей, в них подают смесь молотого пиро-
пиролюзита и 85%-ного едкого кали при 250°, причем суспензия по-
поступает на нагретый до 350° гранулят. При этом смесь спекается
не контактируясь со стенками печи. Применяют печи с внутренним
обогревом, имеющие, например, кольцевую горелку для сжигания
газообразного топлива, а в центре пламени — сопло для подачи
суспензии 123. Из такой печи плав-гранулят для завершения реак-
реакции направляют в другую печь — «печь дожигания», через кото-
которую он перемещается при 140—250° в течение не более 4 ч. Эту
печь обогревают газами, отходящими с первой стадии, содержа-
содержащими 8—30 объемн.% О2 и 10—35 объемн.% Н2О. Вращающиеся
печи позволяют получать манганатный плав более высокого каче-
качества, чем в прокалочных котлах.
Манганатный плав повышенного качества может быть также
получен следующим способом. Размолотый пиролюзит смешивают
с расплавленной 75—85%-ной щелочью и полученную смесь гра-
гранулируют на валках. Гранулированный манганитный плав сушат
при 160—180°, т. е. при температуре ниже температуры его раз-
размягчения. Такая сушка обеспечивает однородность плава. После
этого плав окисляют воздухом, причем манганит почти полностью
переходит в манганат. Полученный таким способом плав содержит
60—65% К2Мп04, 12—13% МпО2 и 8—9% КОН + К2СО3. Вслед-
Вследствие высокого содержания манганата и малого содержания ще-
щелочей, дальнейшая переработка такого плава в перманганат зна-
значительно облегчается, а расход сырья и топлива уменьшается.
Другим вариантом является подача суспензии пиролшзита в
80%-ном едком кали на наружную поверхность вращающихся в
разные стороны вальцов, обогреваемых изнутри топочным газом.
Время пребывания материала на вальцах при 350—400° состав-
составляет 1 мин. Плав соскабливается ножами. Производительность
вальцов ~50 кг/(м2-ч); промышленные агрегаты с поверхностью
5 м2 дают до 1000 т в год KMnO430. По одному из патентов 124, про-
процесс ведут в три стадии. Сначала с помощью дисков и направлен-
направленной по касательной к ним воздушной струи суспензию пиролюзита
в едком кали наносят на вальцы, нагретые до 450°, где происхо-
происходит сушка материала. Для инициирования реакции на вальцы на-
набрызгивают воду в то место, где заканчивается сушка. Вторая ста-
стадия заключается в измельчении плава, частично состоящего из
манганата до размера частиц 0,05—0,1 мм. Третью стадию — даль-
дальнейшее окисление плава, осуществляют при 210° в печи с кипящим
слоем материала, где ен контактируется с кислородом и парами
782
Гл. XXII. Соединения марганца
воды. При длине вальцов 5л и диаметре 0,8 м получают в сутки 1
39,5 г плава, содержащего 35% К2Мп04. На получение 16,72 т/сут- |
ки КгМпО4 расходуют 10000 м3 воздуха и 1,5 г водяного пара.
Так как спекание смеси пиролюзита со щелочью не требует
длительного времени, то его можно осуществлять в распылитель-
распылительной башне, в потоке горячего газа.
Манганат может быть получен из пиролюзита электрохимиче-
электрохимическим способом при применении в качестве электролита расплав-
расплавленного едкого кали, в котором пиролюзит находится во взве-
взвешенном состоянии. Электролиз должен вестись при 195—200°. Вы-
Выход не превышает 60% от теоретического. Большой избыток едкого
кали в получаемом полупродукте затрудняет дальнейшее электро-
электрохимическое окисление КгМпО4 в КМпО4.
Превращение манганата в перманганат происходит уже при
простом кипячении водного раствора по реакции:
ЗК2Мп04 + 2Н2О = 2KMnO4 + MnO2 + 4KOH
Процесс значительно ускоряется при обработке раствора угле-
углекислым газом
ЗК2МпО4 + 2СО2 = 2КМпО4 + МпО2 + 2К2СО3
однако получаемый при этом карбонат калия требуется подвергать
каустификации известью для регенерации едкого кали. Получение
перманганата таким способом оказывается невыгодным, так как
значительная доля манганата превращается в двуокись марганца.
Окисление манганата хлором по реакции
2К2Мп04 + С12 = 2KMnO4 + 2KC1
также невыгодно, так как регенерация едкого кали из хлорида
калия, например электролизом, является дорогим процессом.
В настоящее время перевод манганата в перманганат осуще- '¦]
ствляется обычно электрохимическим окислением. При этом на ~*
аноде образуется перманганат
Перманганат калия
783
а на катоде едкая щелочь и водород:
Процессы, происходящие в электролизере, могут быть схемати-
схематически выражены суммарным уравнением:
2К2Мп04 + 2Н2О = 2КМпО4 + 2КОН + Нг
Манганатный плав выщелачивается в резервуарах с мешал- 1
ками маточным щелоком, полученным после электролиза. Раство- t\
рение манганата при 70° продолжается 1—1,5 ч. Отстоявшийся А
раствор направляется на электролиз, а шлам поступает на бара-
барабанные вакуумфильтры, там отделяется от раствора и потом воз-
возвращается на получение манганатного плава. Шлам содержит
35—50% МпО2 (не прореагировавшего при получении манганата)
и другие примеси, перешедшие из пиролюзита. Периодически, при
значительном накоплении этих примесей, шлам выбрасывается.
Электролиз производится в ваннах, представляющих собой же-
железный цилиндрический резервуар с коническим днищем, по кото-
которому уложен змеевик; с помощью этого змеевика регулируют тем-
температуру в ванне, впуская в него греющий пар или охлаждающую
воду. Ванна снабжена мешалкой и спускным краном. Железные
аноды расположены внутри ванны в виде нескольких концентриче-
концентрических цилиндров на расстоянии 100 мм друг от друга. Применяет
также никелевые аноды. Между анодами находятся катоды—же-
катоды—железные стержни диаметром 20—25 мм. Общая поверхность като-
катодов приблизительно в 10 раз меньше поверхности анодов, что
уменьшает потери от катодного восстановления. Плотность тока
на аноде 60—70 а/м2, на катоде ~700 а/м2. Анодные и катодные
пластины опираются на стеклянные или фарфоровые изоляторы.
Диаметр ванны 1,3—1,4 м, высота цилиндрической части 0,7—0,8 ж,
конической части — 0,5 м. В ванну вмещается 900—1000 л рас-
раствора электролита. Электролиз ведется при 60°. Напряжение на
ванну в начале электролиза составляет ~2,7 в, нагрузка 1400—
1600 а. В конце электролиза напряжение возрастает до 3 в, а сила
тока несколько падает. Ванны работают сериями, по несколько
штук. Число ванн в серии определяется характеристикой генера-
генератора постоянного тока. Расход энергии на 1 т КМпО4 составляет
700 кет • ч.
Электролиз ведут без диафрагмы, так как она засоряется дву-
двуокисью марганца, небольшое количество которой образуется при
электролизе. Поэтому выход по току зависит главным образом от
•степени обратного восстановления перманганата на катоде. Боль-
Большая щелочность электролита препятствует использованию добавок
для образования на катоде защитной пленки. Уменьшению выхода
по току способствует также выделение на аноде кислорода и об-
обратный переход КМпО4 в КгМпО^ из-за высокой конпентпаиии
щелочи:
4КМпО4 + 4КОН = 4К2МпО4 + 2Н2О + О2
Эта реакция каталитически ускоряется присутствующей в элек-
электролите двуокисью марганца. Увеличению выхода по току способ-
способствует низкая анодная плотность тока и искусственное перемеши'
вание электролита, уменьшающее концентрационную поляризацию
на аноде; при перемешивании в прианодном слое создается более
высокая концентрация КгМпО4, понижается анодный потенциал и
вследствие этого уменьшается выделение кислорода 125.
784
Гл. XXII. Соединения марганца
Мажеф
785
Выход по току и степень окисления увеличиваются при элек-
электролизе насыщенного раствора КгМпО4 в присутствии кристаллов.
Такой раствор содержит приблизительно 180 г/л К2М.ПО4, 30—
40 г/л КМпО4, 150 г/л КОН и 50 г/л К2СО3. Электролиз продол-
продолжается несколько часов, пока концентрация КгМпО4 не упадет до
15—30 г/л. Образующийся КМпО4 плохо растворим и частично
выделяется в виде кристаллов. По окончании электролиза раствор
электролита вместе с кристаллами перманганата калия поступает
в стальные холодильники с мешалками, охлаждаемые с помощью
водяных рубашек. Здесь происходит окончательная кристаллиза-
кристаллизация перманганата калия. Выпавшие кристаллы отделяют на цен-
центрифуге и промывают водой; маточные щелоки и промывные воды
возвращают на выщелачивание манганатного плава. Примерный
состав маточного щелока: 23 г/л КМпО4, 16 г\л К2МПО4, 210 г/л
КОН, 60 г/л К2СО3.
После промывки на центрифуге и высушивания получается за-
загрязненный технический перманганат калия, содержащий 80—95%
КМпО4) примеси МпО2, КгМпО4, сульфатов, поташа и щелочи. Для
получения чистого продукта кристаллы, промытые на центрифуге,
подвергаются перекристаллизации, для чего их растворяют в воде
при 85° и раствор охлаждают. Выделившиеся кристаллы отфуго-
вывают и сушат.
Если необходимое для производства едкое кали получают кау-
стификацией поташа известью, то расход основных материалов на
1 т перманганата калия приблизительно составляет: пиролюзита
A00% МпО2) — 0,8 т, поташа A00%) — 0.85 т и извести A00%
СаО) — 0,7 т.
Часть маточного щелока после кристаллизации перманганата
калия во избежание чрезмерного накопления примесей необходимо
выводить из цикла. Он содержит, кроме перманганата и щелочей,
алюминаты, ванадаты и др. Его можно каустифицировать известью
[СаО или Са(ОН)г] и, отделив осадок, возвратить раствор на вы-
выщелачивание манганата126. Можно утилизировать маточный ще-
щелок восстановив КМпО4 и К2М.ПО4 37%-ным растворам формалина
до МпО2; оставшийся после отделения МпО2 раствор КОН и КгСО3
при нейтрализации азотной кислотой позволяет получить калий-
калийную селитру 3 сорта 127.
Возможно непосредственное получение перманганата калия
анодным растворением марганца в щелочном электролите, содер-
содержащем КОН или КгСОз, при электролизе с анодами из ферромар-
ферромарганца, с ~70% Мп и 1—6% углерода. Процесс идет по общему
уравнению:
2Мп + 2ОН~ + 6Н2О = 2МПО4 + 7Н2
При содержании в аноде меньше 44% Мп перманганат не об-
образуется. Катод может быть из меди, стойкой в щелочном перман-
ганатнам растворе. Электролиз может осуществляться без диа-
диафрагмы или с диафрагмой из асбестового полотна; в последнем
случае катодное восстановление уменьшается и выход по току
больше. Лучшая температура электролита 16—18°. Повышение
температуры приводит к увеличению степени превращения пер-
перманганата в манганат. Электролит должен содержать 20—30%,
КОН или К2СО3. Электролизу препятствует образующаяся на фер-
ромарганцовом аноде оксидная пленка, повышающая потенциал*
особенно при малой концентрации щелочи в электролите. При при-
применении анодов из силикамарганца пассивирующая пленка обра-
образуется только при малых концентрациях электролита и высоких
плотностях тока. Слишком высокие концентрации электролита при-
приводят к появлению растворимых соединений железа, образующихся
при повышенном потенциале.
Оптимальная анодная плотность тока при использовании в ка-
качестве электролита раствора, содержащего 300 г/л К2СО3, 16—
18 а/дм2, а при 200—250 г/л КОН —30—40 а/дм2. Выход по току
не превышает 50%, а выход продукта (степень перехода раство-
растворившегося марганца в перманганат) 80—85%; расход энергии
12 кет • ч на 1 кг КМпО4. Продукт электролиза — КМпО4 полу-
получается в виде мелких кристаллов, смешанных с большим количе-
количеством электролитического шлама. Электролит охлаждают, отде-
отделяют от осадка на барабанном вакуум-фильтре и центрифуге и
возвращают в процесс. Осадок обрабатывают горячей водой для
извлечения КМпО4, который затем выделяют кристаллизацией128^
Фильтрование горячей G0—90°) электролизной пульпы для отде-
отделения шлама перед кристаллизацией перманганата позволяет по-
получить весьма чистый продукт (до 99,7% КМпО4), но пока не при-
применяется из-за отсутствия устойчивого фильтрующего материала 129_
МАЖЕФ
Для фосфатирования стальных изделий предложены и приме-
применяются разные препараты: порошок Паркера130, дигофат, мажеф
и др. Они представляют собой смеси фосфатов марганца и фос-
фосфатов железа. Железо-марганцовые фосфатные препараты имеют
преимущество перед марганцовыми, так как дают покрытия, более-
устойчивые против коррозии, и требуют меньшей длительности фос-
фосфатирования. Вредными примесями в фосфатных препаратах яв-
являются соли алюминия, мышьяка, свинца131. Наибольшее примене-
применение получили препараты, состоящие из монофосфатов марганца и
железа (мажеф), с соотношением между ними 10: 1 н- 15: 1 132.
Предложен также продукт взаимодействия феррофосфата с МпО2
при молярном отношении в исходной смеси Мп : Fe, равном 5:8 и.
11:4 133.
786
Гл. XXII. Соединения марганца
Для приготовления разных фосфатов может быть использована
фосфорная кислота или суперфосфат. Mn(H2PO4h может быть по-
получен из апатита, серной кислоты и марганцовых шламов 134. Ди-
гофатам назван дигидроортофосфат марганца, который можно
получить из фосфорной кислоты и сульфата марганца или восста-
восстановленного пиролюзита. По этому способу дигофат раньше изго-
изготовлялся в заводском масштабе 135, но он содержал свободную сер-
серную кислоту, а иногда большой избыток фосфорной кислоты, что
ухудшало антикоррозионные
свойства получаемого с его по-
помощью защитного покрытия136.
В Институте металлов был разра-
разработан способ получения дигофа-
та из фосфорной кислоты и фер-
ферромарганца. В настоящее время в
СССР основным препаратом для
фосфатирования служит мажеф.
При фосфатировании проис-
происходит разложение хорошо рас-
растворимых фосфатов марганца на
более основные фосфаты
Мп(Н2РО4J ^=± МпНРО4 + Н3РО4
ЗМпНРО4 ^=± Мп3(РО4J + Н3РО4
Рис. 211. Стабильная изотерма 100°
системы МпО—Р2О5—Н2О.
кристаллизующиеся как на по-
поверхности металла в виде плен-
пленки, так и в растворе (шламI37-14'.
Появление фосфатной пленки на металле является вторичным
процессом, которому предшествует растворение металла и связан-
связанное с этим изменение состава слоя раствора, прилегающего к анод-
анодным и катодным участкам поверхности металла. Пленка трудно-
труднорастворимых фосфатов покрывает поверхность металла и защищает
его от коррозии. Она достаточно стойка против влияния атмосфер-
атмосферных осадков, но не защищает железа даже в слабокислой среде и
в растворах солей. Но фосфатная пленка служит хорошим грунтом
для последующего нанесения органических защитных и декоратив-
декоративных покрытий — лаков, красок, смол и т. д. Состав таких пленок
много изучался, но окончательно не установлен. Для выяснения
состава осадков, выделяющихся из водных растворов фосфатов
марганца, можно воспользоваться диаграммой растворимости в си-
системе МпО—Р2О5—Н2О (рис. 211) ш- из. При 100° в твердую фазу
сначала выделяется метастабильный трехводный кристаллогидрат
МпНРО4 • ЗН2О, который переходит полностью в стабильную при
этой температуре безводную соль лишь в течение нескольких су-
суток. Так как процесс фосфатирования длится всего 40—60 мин, то
метастабильный МпНРО4 • ЗН2О не успевает разложиться и, таким
Мажеф
787
образом, входит в состав фосфатной пленки. Другим ее компонен-
компонентом является безводный дифосфат марганца МпНРО4 Образова-
Образование в пленке Мпз(РО4J, по условиям кристаллизации в этой си-
системе, является маловероятным.
В чистом монофосфате марганца Мп(Н2РО4J отношение МпО :
:Р2О5 равно 0,5 (рис. 211). В применяемом же для фосфатирова-
фосфатирования при 100° ~3%-ном насыщенном растворе монофосфата мар-
марганца отношение МпО : Р2О5 должно быть —0,3, т. е. препарат
для фосфатирования должен состоять из смеси Мп(Н2РО4J • 2Н2О
и свободной фосфорной кислоты, для того чтобы он полностью
растворился в воде при 100° без разложения при приготовлении
— 3%-ного раствора.
Для ускорения фосфатирования предложено применять при
70—80° раствор, содержащий железо-марганцовые фосфаты (на-
(например, мажеф) 30—35 г/л и нитрат цинка 50—70 г/л. В присут-
присутствии последнего пленка образуется в 10—12 раз быстрее 144. Фос-
фатирование может вестись и холодным способом, при этом не на-
нагретый раствор мажефа должен иметь повышенную концентра-
концентрацию A00—200 г/л).
Марганцовый препарат для фосфатирования сталей мажеф, со-
содержащий монофосфаты марганца и железа, может быть получен
несколькими способами:
1) растворением в избытке горячей (90°) фосфорной кислоты
закиси марганца, восстановленной углем из пиролюзита, в присут-
присутствии железа; полученный раствор отфильтровывают от нераство-
нерастворимого остатка, выпаривают до плотности 1,67—1,69 г/см3 и охла-
охлаждают для кристаллизации монофосфата марганца с примесью
монофосфата железа;
2) растворением в фосфорной кислоте ферромарганца с после-
последующей выпаркой раствора и кристаллизацией соли;
3) получением кислых фосфатов марганца и железа из сульфа-
сульфатов марганца и железа и тринатрийфосфата.
Первый способ дает продукт, загрязненный растворимыми
в фосфорной кислоте примесями, содержащимися в пиролюзите.
Второй, как и первый, связан с необходимостью выпарки фосфор-
фосфорнокислых растворов, которая осложняется отсутствием материа-
материалов, достаточно устойчивых по отношению к горячей фосфорной
кислоте. Третий 145 не обладает этими недостатками. По этому спо-
способу смешивают растворы сульфата марганца, сульфата закиси
железа и тринатрийфосфата при 60—70°. В результате протекают
реакции:
3MnSO4 + 2NasPO4 = Mns(PO4J + 3Na2SO4
3FeSO4 + 2Na3PO4 = Fe3(PO4J + 3Na2SO4
Образующиеся фосфорнокислые соли марганца и железа вы-
выделяются • в объемистый осадок. Осадок отфильтровывают на
788
Гл. XXII. Соединения марганца
вакуум-фильтрах и промывают водой до полного удаления серно-
сернокислых солей, а затем отжимают под гидравлическим прессом от
избыточной влаги и высушивают при температуре до 300°. Для
осаждения фосфатов марганца и железа вместо тринатрийфосфата
можно использовать динатрийфосфат.
Полученный порошок, содержащий 30—35% Мп и 2—3% Fe,
смешивают г. фосфорной кислотой, количество которой должно
быть достаточным для перевода тризамещенных фосфатов в одно-
замещенные:
Мп3(РО4J + 4Н3РО4 = ЗМп(Н2РО4J
Fe3(PO4J + 4Н3РО4 = 3Fe(H2PO4J
Частично происходит образование МпНРС>4 по уравнению:
Мп3(РО4J + Мп(Н2РО4J = 4МпНРО«
Ввиду экзотермичности этих реакций смесь быстро разогре-
разогревается, а по охлаждении застывает в однородную мелкокристалли-
мелкокристаллическую массу, которую высушивают при температуре не выше 80—
100° и размалывают. Полученный препарат содержит 17—19%
Мп, 1,5—2,5% Fe, 49—52% Р2О5.
По минеральному составу мажеф представляет порошок, со-
состоящий в основном из монофосфата марганца, небольших коли-
количеств фосфатов железа и дифосфата марганца.
Несколько лучшими качествами обладает препарат, получае-
получаемый растворением фосфорнокислого марганца и железа в избытке
горячей фосфорной кислоты с последующей кристаллизацией из
раствора монофосфатов марганца и железа. Такой препарат содер-
содержит меньшее количество нерастворимого остатка.
ЛИТЕРАТУРА
1. Е. Я. Роде,
Кислородные соединения марганца, ^Изд. АН СССР, 1952. —
2. В. В. Печковский, А. Г. 3 вез дин, Уч. зап. Пер'мск. гос. ун-та, 17, в. 1,
35 (I960). —3. J. Н. К repel k а, В. Rejha, Coll. Czech. Chem. Comm., 5, 67
A953); A. Seidell, Solubil. Inorg. a. Metalorg. Сотр., 1, 1940. — 4. L6rant
Bel a, Z. analyt. Chem., 219, № 3, 256 A966) —5. В. В. Печковский,
¦С А. Амирова, Н. И Воробьев, Т. В. Островская. ЖНХ, 9, № 9,
2059 A964). —б. I. P. Saraswat, Т. R. Kalra, J. Indian Chem. Soc, 44, № 6,
481 A967). —7. E. В. Алексеевский, Активная двуокись марганца, Химтео-
рет, 1937. — 8. Е Я Роде, Г Г Цуринов, Научно-иссл. работы химических
ин-тов и лабораторий АН СССР, Изд. АН СССР, 1945, стр. 50. — 9. Н. Ulich,
Z Elektrochem., 45, 521 A939).— 10. М. 011 о Earl, J. Electrochem. Soc, 112,
№4, 367 A965).
11.1 Matsushima, W. J. Thoburn, Canad. J. Chem., 43, № 26, 1723
A965). —12. H Peters, K--H. Radeke, L. Till, Z. anorg. Chem.. 346, № 1.
2. 1 A966). — 13. J. С White, R. R M i 11 e r, J. Am. Chem. Soc, 75, № 13,
3282 A953) . — 14. Б. Ежовская-Тжебятовская, Я. Навойская,
M. Вронская, Бюлл. Польск. Акад. наук, отд. III, 1, № 7, 314 A953).—
15. Б. Ежовская-Тжебятовская, Я Навойская, М. Вронская,
Там же, отд. III. 2, № 9, 451 A954); Roczh. chem., 29, № 2—3, 259 A955).—
Литература
789
16.
наук,
I Е. С. Осиновик, П. И. Белькевич, Изв. АН БССР Сео *из -техн
1Ук, № 1, 127 A956).-17. Е. И. Шмук, Автореф. канд дисс Йис?итуГго-
рючих ископаемых АН СССР, 1952. —18. R. Scholder D F i <s r h p r HWa-
terstradt, Z. anorg. Chem., 277, № 5, 234 A954). — 19 ' A S i m о n F Fi-
Fiji e r, Z. Elektrochem., 38, 137 A932). — 20. O. Hemser, H Schroder Z
anorg. Chem., 271, № 5—6, 293 A953).
21. Ю. В. Клименко, А. П. К в а с к о в. Химическое обогащение марган-
марганцовых руд, Госметаллургиздат, 1944. — 22. С. С. Марков, АвтопеФ аокт дисс.
ГИПХ. 1951.-23. М. Katz, S. Hal pern, Ind. Eng. Chem 42 »2 343
A950). — 24. Ф. А. Петрачков, Н. С. Жигулина, Т. Т Готьмаиова
Хим. пром., № 4, 301 A964). — 25. В. Т. Чагунава, Э. Р. Дзнеладзе,
Л. И. Твасалия, Тезисы докладов V Всесоюзной конференции по ТНВ и ми-
минеральным удобрениям, Киев, 1966, стр 192 — 26. В. Т. Ч а г у н а в а, Р А. Д и-
дидзе, Там же, стр. 193. — 27. Я. Я. До донов, Л. Д. Бор зова' В С Ко-
Колосова, В. С. Покаевская, ЖПХ, 32, № 11, 2373 A959). — 28.' В. М. Ка-
кабадзе, Т. А. Иванова, Труды Груз, политехи, ин-та, № 5 D0), 1955,
стр. 30. — 29. В. М. К а к а б а д з е, В. Т. Чагунава и др., Тезнсы докладов V
Всесоюзной конференции по ТНВ и минеральным удобрениям, Киев, 1966,
стр. 83. — 30. W. Baronius, J. Ma г с у, Chem. Techn., 18, № 12, 723 A966).
31. А. Л. Ефимов, И. А. К а з а с, Инсектициды и фунгициды, Сельхозгиз,
1940—32. Н. Beduneau, Rev. prod, chim., 68, № 1339, 539, 547 A965).—
33. E. Feill, J. Rabska, Biul. inform. Inst. Przem. Drobnego, 2, № 2. 1
A955). — 34. S. Trocme, G. Barbier, Agr. Chem., 11, № 5, 35 A956).—
35. А. Г. Бет ex тин. Промышленные марганцовые руды СССР, Изд. АН СССР,
1946.—36. В. И. Вернадский, СМ. Курбатов, Земные силикаты, алюмо-
алюмосиликаты и их аналоги, ОНТИ, 1937. — 37. А. Г. Бетехтин, Чиатурское мар-
марганцовое месторождение и его промышленная характеристика, ОНТИ, 1936. —
35. М. Ю. Морачевский, Труды ГИПХ, в. 37, 1946, стр. 123. — 39. Е. Я. Ро-
Роде, Г. Г. Цуринов, Научно-иссл. работы химических ин-тов и лабораторий АН
СССР, Изд АН СССР, 1945, стр. 51.— 40. В М. Какабадзе, Я. Г. Бучу-
кур и н др., Тезисы докладов VI Всесоюзной конференции по технологии не-
неорганических веществ, Тбилиси, 1968, стр. 49.
41. Е. Я. Роде, Научно-исследовательские работы химических ин-тов и ла-
лабораторий АН СССР, Изд. АН СССР, 1945, стр. 53. — 42. Д. И. Э р и с т а в и,
Д. Н. Барнабашвили, Е. С. Джапаридзе, Научно-исследовательские
работы химических нн-тов и лабораторий АН СССР за 1939 г., Изд. АН СССР,
1940, стр. 107. — 43 Б. С. Канделаки, И. Д. Гавберидзе, И. Р. Д о-
л и д з е. Там же, стр. 106. — 44. Обогащение марганцовых руд. Груз. фил. АН
"СССР, Хим. институт, 1940. — 45. Н. С. Weber, С. Н. Davenport, Trans. Am.
Inst. Chem. Eng., 39, 585 A943). — 46. В. М. Какабадзе, ЖПХ, 24, № з, 252;
№ 7 681 A951); Хим. пром., № 6, 174 A953). -47. С. Е. Wood, С. L. Walf-
red, E. P. Barrett a. oth., U. S. Bur. Mines Rept. Invest., № 3609, 30 A942).—
48. A. P. Herring, S. F. R a v i t z, Trans. Soc. Mining Eng. A1ME, 232, № 3,
191 A965). — 49. В. М. Какабадзе, Аннотации докладов на III Научно-техни-
Научно-технической конференции Грузинского индустриального института, 1940, стр. 56. —
50. В. М. Какабадзе, Изв. Груз, полнтехн. ин-та, № 2, A6) 29 A945).
51. Р. И. Агладзе и др., сб. «Исследования по переработке марганцевого
и топливного сырья Грузии», Тбилиси, 1967, стр. 63. - 52. К. М. С а л д а д з е,
Труды МХТИ, № 6, 1940, стр. 88. —53. Н. П. Д и е в, М. И. К очн е в, В. В. Па-
дучев, Г. Я Сн ридзе ЖПХ, 27, № 4, 356 A954).— 54. В. В. Печков-
Печковский, Там же, 29, № 7, 997 A956); 32, № 12, 2613 A959). — 55. Н. В. Небие-
ридзе, Я. Г. Бучукури, Тезисы докладов V Всесоюзной конференции по
ТНВ и минеральным удобрениям, Киев, 1966 стр 113. — 56. В. М. Какабад-
Какабадзе, Т. В. Панцулая, Журн. ВХО, 5, № 4, 471 (I960).— 57. Я. Г Бучукури,
В. Т. Чагунава, Т. В. Панцулаи, Сообщ. АН ГрузССР, 17, № 8, 703
790
Гл. XXII. Соединения марганца
A956) . — 58. Т. В. П а н ц у л а я, Я. Г. Б у ч у к у р и, Труды Груз, полигехн. ин-
та, т. 5, 1956, стр. 92. — 59. I. Moldovan et al. Rev. chim. (рум.), 18, № 7,
399 A967), — 60. Jl M Вучурович и др., Гласник хем друштва Београд
(сербо-хорв.), 29, № 5—6 A964).
61. И Петков Т. Николо в, Годишник хим.-технол. инст. (болг.), 11»
№4, 161 A964—1965).- 62. Н. В. Небиеридзе, В. М. Какабадзе, Я-Г.Бу-
чукури, Т А Иванова, Сообщ. АН ГрузССР, 48, № 2, 335 A967).—
63. Пат ПНР 49946, 1963. — 64. Н. С. Fuller, пат. США 3301634. 1963. —
65 Пат США 326752, 1963. — 66. Kelley, Anderson, Bur. Mines Bull., 384
A935). — 67. В. П. Ильинский, Н. П. Лапин, ЖПХ, 4, № 6, 757 A931).—
68. Л. Н. Джапаридзе, Л. И. Б а ц а н а д з е, Р. В. Ч а г у н а в а, Тезисы
докладов VI Всесоюзной конференции по технологии неорганических веществ,
Тбилиси, 1968, стр. 86. — 69. С. С. Марков, Сборник статей к 20-летию ГИПХ,
1939, стр. 74. — 70. С. А. 3 а р е ц к и й, Там же, стр. 174.
71. Р И Агладзе Бюлл. ВХО. № 5, 41 A940); № 5, 29 A941); Метал-
Металлург № 9 A17), 15 A939).— 72. В. А. Козлов. В. Г. Власов, ЖПХ, 32,
№ 3 523 A959). — 73. А. Ф. Ложкин, Н. Л. С у б о ч е в а, НДВШ, Хим. и хим.
технол., 2, 204 A958). — 74. А. Ф. Ложкин, Т. В. Сычева, В. Г. Сумен-
ков, Уч. зап. Пермск. гос. ун-та, 17, в. 1, 97 A960). — 75. Р. И. Агладзе,
Э. Д. Ч х и х в а д з е, сб. «Исследования по химической переработке руд», Тби-
Тбилиси, 1966, стр. 81. — 76. А. Ф. Ложкин, Изв. вузов. Хим. и хим. технол., 11,
№ 8 908 A968). — 77. Р. И. Агладзе, В. Ю. Минднн, авт. свид. 185863,
1963.'— 78. J. Samonides, пат. США 3227520, 1962. — 79. Франц. пат. 1448939,
1965. — 80. Пат. ГДР 45351, 1965.
81. W. To mas si, Z.-D о m i n i с z a k, Przem. chem., 43, № 11, 608 A964).—
82. H. K. Chakrabarti, T. Banerjee, J. Sci. Ind. Res.. В 12, № 5, 211
A953). —53. С. С. Марков, Труды ГИПХ, в. 37, 1946, стр. 154. —84. Е. Я. Ро-
Роде, А. П. Лазарева, Г. Г. Ц у р и н о в, Научно-исследовательские работы хи-
химических ин-тов и лабораторий АН СССР, Изд. АН СССР, 1945, стр. 52.—
85 В М Какабадзе, "Бюлл. ВХО, № 4, 32 A940). — 86. R. S. Dean,
A L. Fox, A. E. Beck, U. S. Bur. Mines Rept. Invest., № 3626, 63 A942).—
87. Chem. Eng., 61, № 10, 122, 124 A954). — 88. R. Freitag, Sprechsaal Kera-
mic, Glas, Email, 89, № 2, 32 A956). — 89. Франц. пат. 1443907, 1965.—
90. О. G 1 e m s e r, G. О a 11 о w, пат. ФРГ 1157590, 1960.
91. S. С. Rao, J. Electrochem. Soc. (India), 15, № 4, 105 A966).— 92. Пат.
ЧССР 114767, 1964; япон. пат. 736, 1962. —S3. К. Marx, M. Schulze, пат.
ФРГ '752326 1954. —94. Prasad N. S. Krishna, J. Indian Inst. Sci., A 36,
№ 1, 19 A954). — 95. A. Esme, L'Ind. chim., 34, № 361, 150 A947). 96. J. Pet-
Иска V Bastecky, пат ЧССР 114779, 1964. — 97. Пат. ФРГ 1205067.
1960 — 98. Франц. пат. 1041511, 1953. — 99. Г.И.Зворыкина, ЖПХ, 30, № 10,
1515 П957) —100. R. S. Dean, A. L. Fox, пат. ФРГ 922882, 1955, англ. пат.
690134, 1953.
!01 Е Роде Т Роде Физико-химическое изучение марганцовых руд и
минералов^ Изд АН СССР, 1937, стр. 70. — 102. J. Ka t о, Т. М a t s u h a s h i, J.
Soc. Chem. Ind. Japan, 32, № 11, 313 A929). — 103 В. М. Какабадзе, ЖПХ,
20 № 7 670 A947). — 104. Д. М. К о р ф, А. А. М а з у р е н. Хим. пром.. № 5,
361 П961).—105. Е Leaver, U. S. Bur of Mines, № 6770, 107 A934).—
106 P И Агладзе Горный ж., № 12, 39 A939). —107. Пат. ВНР 152894.
1965. — 108. Е. Maleczky, S. Fekete, Т. Bodacsoni, A. Horvath, Ba-
nyasz Kutato int. kozl. (венг.), 10, № 1, 2, 211 A965—1966). — 109. В. В. Печ-
ковский, ЖПХ. 28, № 3, 237 A955). — ПО. С. С. Марков, Сборник статей
к 25-летию ГИПХ. 1944. стр. 177.
Ш Technical Field Information Agency Rep., № 757 A947).— 112. Ю. Г. Кли-
Клименко, M. И. Рабинович, сб. «Тепло-массообмен», Киев, 1968, стр. 118. ——
Литература
791
ИЗ. Н. Маг су, W. Wiedner, пат. ГДР 40748, 1964—//4 п лд птя„пп-
СКН^ сб «Исследования по прикладной химии»" ИздАН %СР1955
?Л f-T'l5- Л- А- °1еР°в. ТРУДЬ. Воронежск. ун-та в 28 195Я сто 24 -
116. А. Г. Бергман, Е. И. Вонашек, Научно-исп „ к ' ' Р' „м
тов и лабораторий АН СССР за 1941-1943 ггГиз^Гшссср' йТТГ 67
117. Е. Я. Роде, И. С. Морозов, Там же, стр 54 - ШР п' гшГ^704"Й
1945. -119. Англ. пат. 680710, 1952. -120. A. /cochin V ШрДкеj'
Metals, 19, № 4, 28 A967). гаш' w- L t" a Ike, J.
tnn 12L L P- Whitehouse, M. E. Graham пат США 9WRR14 IQ44
П964?" S^OidRr> "о Wat?/SMradt' Z" аП°Ге- СЬетША277?5S'i-i П2
A9b4). — 123. W. В а г о n l u s, H. M a r с у, пат. ГДР 40745 1964 124 A I a n-
ge, W. D r esc her, пат. ГДР 32784, 1962. -125 В. Г Хо я К7в в П W
шовец, Л. Л. Кузьмин, Технология электрохимических пооизволств Гос
химизда^ .949, стр. 399/26. М. В С a/us, пат. "ша TS" 961 -
U7. Р. И. Агладзе, В. В. Чепурин, сб. «Исследования в области эчектпо-
химин и радиационной химии», Тбилиси 1965 стр 121 -128 R Г Ln о
швилн, Автореф. канд. дисс, АН ГрузССР; 1956.-129 Э И Бахтадзь
ше3ДпЫ Т«°нК„ЛаД03.^й ВСеС°^Н°Й к„°"ФДРен«ии по технологии неорганических в?
ществ, Тбилиси, 1968, стр. 230. —130. Parker, герм. пат. 463778, 1926
/fcJf' A; of nC °J °п ° В С1И Й' 3VM' КУлагина, ЖПХ, 18, № 7-8, 412
A945). —132. В. И. Вульфсон, Г. П. Рабинович Корпозия и борьба с
Т? ^/'F A936)--Ш- W. H. Woodstock, пат! СшТ1б57922 1953-
/« м-,^^ЛуКЬЯНОВ> К" М- СалДадзе, ЖПХ, 17, № 10, 19 A940)' —
135. И. И. Кукушкин, Там же, 13, № 5, 279 A936).-136. А. 3. Ривкинд
гггр шч?аЛЬЯкН4г; РУ?Й о11 K0HJePeH«™ по коррозии металлов прн АН
мяп, ' шкйСТ1\?, ^ м- 5- с- Лопату хин, Фосфатирование металлов,
nSST1 9о'^ ,лДпМ- 1°РФ' R R Самарин, ЖПХ, 15, № 1, 38
) п5п<" ,^ „В; R вУЛьфсон, Коррозия и борьба с ней, 5, № 1—2 7
A939). — 140. Д. М. К о р ф, Там же, стр. 85.
141 \Лё R Ло° Д,а Н е Р' 3аЩитные пленки на металлах, Изд. Харьковск гос
У?оло\ ЬГ 1\ ¦ А" Таперов а, М. М. Исаева, ЖПХ, 22, № 4 342
S1 А)-~7* 3- М- В- Голощапов. ЖНХ, 1, № 11, 2633 A956) -
/ L?- ¦ Хаин- Коррозия и борьба с ней, 5, № 1—2 A939); ЖПХ 18, № 4—5
A945); 19, № 5—6 A946); Труды Центрального н.-и. нн-та технологии судо-
судостроения, в. 15, 1957, стр. 3; ЖПХ. 32, № 11, 2463, 2473; № 12, 2662 A959)- 33
^7'!?2 A9е°)-—145- Б- Д- Мельник, авт. свид. 46914, 1936; ЖХП,' 22,