/





























































































































































































































































































































































































Текст
М. Е. ПОЗИН
ТЕХНОЛОГИЯ
МИНЕРАЛЬНЫХ СОЛЕЙ
(УДОБРЕНИЙ, ПЕСТИЦИДОВ,
ПРОМЫШЛЕННЫХ СОЛЕЙ,
ОКИСЛОВ И КИСЛОТ)
Часть II
'4-е издание, исправленное
при участии:
Л. 3. Арсеньевой, Ю. Я. Каганович, Г. С. Клебанова,
В. А. Клевке, Б. А. Копылева, А. А. Соколовского
ИЗДАТЕЛЬСТВО «ХИМИЯз
Ленинградское отделение
1974
УДК 661.4/.65Ч- 661.8
П47
П47 Позин М. Е. и др. X _
Технология минеральных солей (удо-
брений. пестицидов, промышленных со-
лей, окислов и кислот), ч. I ,изд. 4-е, испр.
Л., Изд-во «Химия», 1974.
768 стр.. 71 табл., 250 рис
В монографии на современном уровне описана тех-
нология важнейших (крупнотоннажных) минеральных
солей, в том числе минеральных удобрений — фосфор-
ных, азотных, калийных и других, а также некоторых
окислов и кислот (фосфорной, соляной и др.). Рассмот-
рены свойства сырья, полупродуктов и продуктов; из-
ложены физике химические основы производств; описа-
ны технологические схемы, режимы и аппараты.
Вторая часть монографии посвящена технологии
минеральных удобрений и пестицидов, а также фтори-
стых и цианистых соединений.
Книга является практическим и справочным руко-
водством для. инженеров, работающих в химической
и смежных с ней отраслях промышленности. Она мо-
жет служить учебным пособием для аспирантов и сту-
дентов старших курсов химико-техиологнческих вузов.,
Третье издание книги вышло в 1970 г.
п
31403—056
050 (01)—74
56—74
© Издательство «Химия», 1974
ОГЛАВЛЕНИЕ
Предисловие к четвертому изданию................................... !7
17
Предисловие к третьему изданию............................. • • •
Часть I
Глава I. Использование минеральных солей в народном хозяйстве ... 19
Глава II. Растворимые соли в природе и методы их добычи..............45
Глава III. Хлористый натрий......................................... 60
Глава IV. Природный сульфат натрия...................................98
Глава V. Природные калийные соли....................................138
Глава VI. Поташ 187
Глава VII. Бром и его соли......................................... 206
Глава VIII. Иод и его соли..........................................236
Глава IX. Соединения магния.........................................263
Глава X. Соединения бора............................................311
Глава XI. Соляная кислота.......................................... 363
Глава XII. Соли бария...............................................416
Глава XIII. Соли сульфидного ряда...................................456
Глава XIV. Соли сульфитного ряда....................................506
Глава XV. Соли хрома . 564
Г лава XVI. Сульфат алюминия и продукты на его основе...............632
Глава XVII. Соединения меди ........................................661
Г лава XVIII. Соединения железа.....................................694
Глава XIX. Сульфат и хлорид цинка...................................714
Глава XX. Сульфат никеля............................................731
Глава XXI. Хлорид кальция......................................... 738
Глава XXII. Соединения марганца.....................................751
1*
794
Оглавление
Часть II
Глава XXIII. Природные фосфаты и фосфоритная мука....................801
Фосфорные руды.....................................................801
Минералогический состав апатитовых и фосфоритных руд.............801
Типы фосфоритных руд и месторождения.............................804
Применение фосфатов .............................................808
Физико-химические и механические свойства........................814
Транспортировка и хранение фосфатного сырья .................... 817
Фосфоритная мука............................ . ................. . 819
Методы и продукты химической переработки фосфатов..................823
Литература ..........................................................824
Г лава XXIV. Простой суперфосфат.....................................828
Состав и свойства суперфосфата.......................................828
Применение ..........................................................830
Методы производства .................................................\821
Физико-химические основы производства................................832
Химизм процесса................................................... 832
Норма серной кислоты ..............................................834
Механизм и скорость процесса.......................................835
Влияние температуры................................................837
Смешение реагентов.................................................838
Степень измельчения фосфатов.......................................839
Коэффициент разложения в конце I стадии реакции.................839
Разложение фосфата фосфорной кислотой (II стадия реакции) .... 840
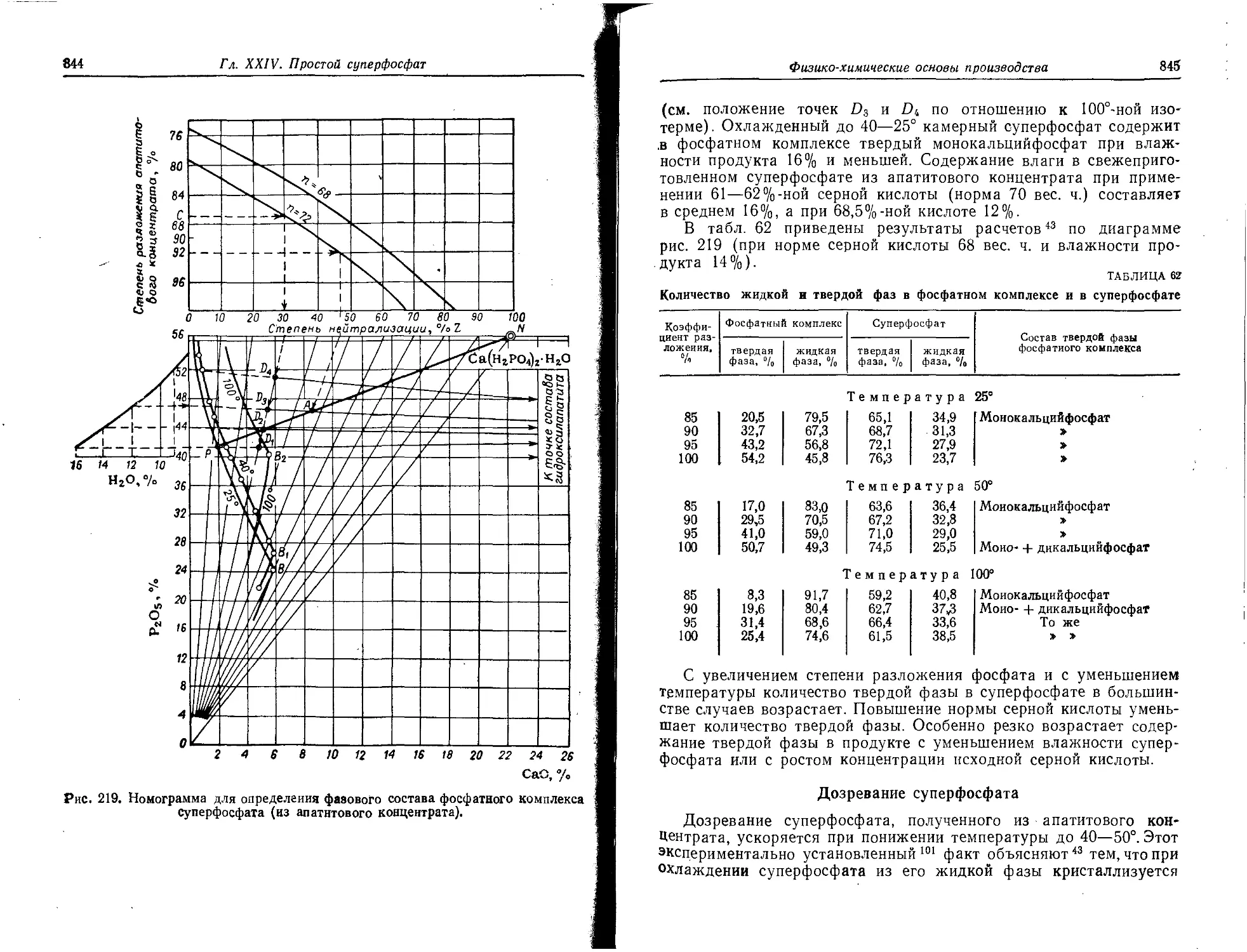
Фосфатный комплекс суперфосфата и его фазовый состав ....... 842
Дозревание суперфосфата.........................................845
Особенности разложения магнийсодержащих фосфоритов..............850
Нейтрализация суперфосфата твердыми добавками...................853
Аммонизация суперфосфата........................................855
Производство суперфосфата.........................................856
Выделение фтористых газов.....................................- . 858
Основная аппаратура и условия ее работы ....................... 859
Материалы для защиты аппаратуры от коррозии.....................863
Материальный баланс производства суперфосфата ..................864
Технико-экономические данные....................................864
Получение гранулированного суперфосфата...............................867
Литература .........................................................874
Г лава XXV. Производство фосфорной кислоты сернокислотным способом . 881
Физико-химические свойства...........................................881
Применение ....................................................... . . 883
Физико-химические основы сернокислотной экстракции фосфатов .... 884
Химизм процесса ...................................................884
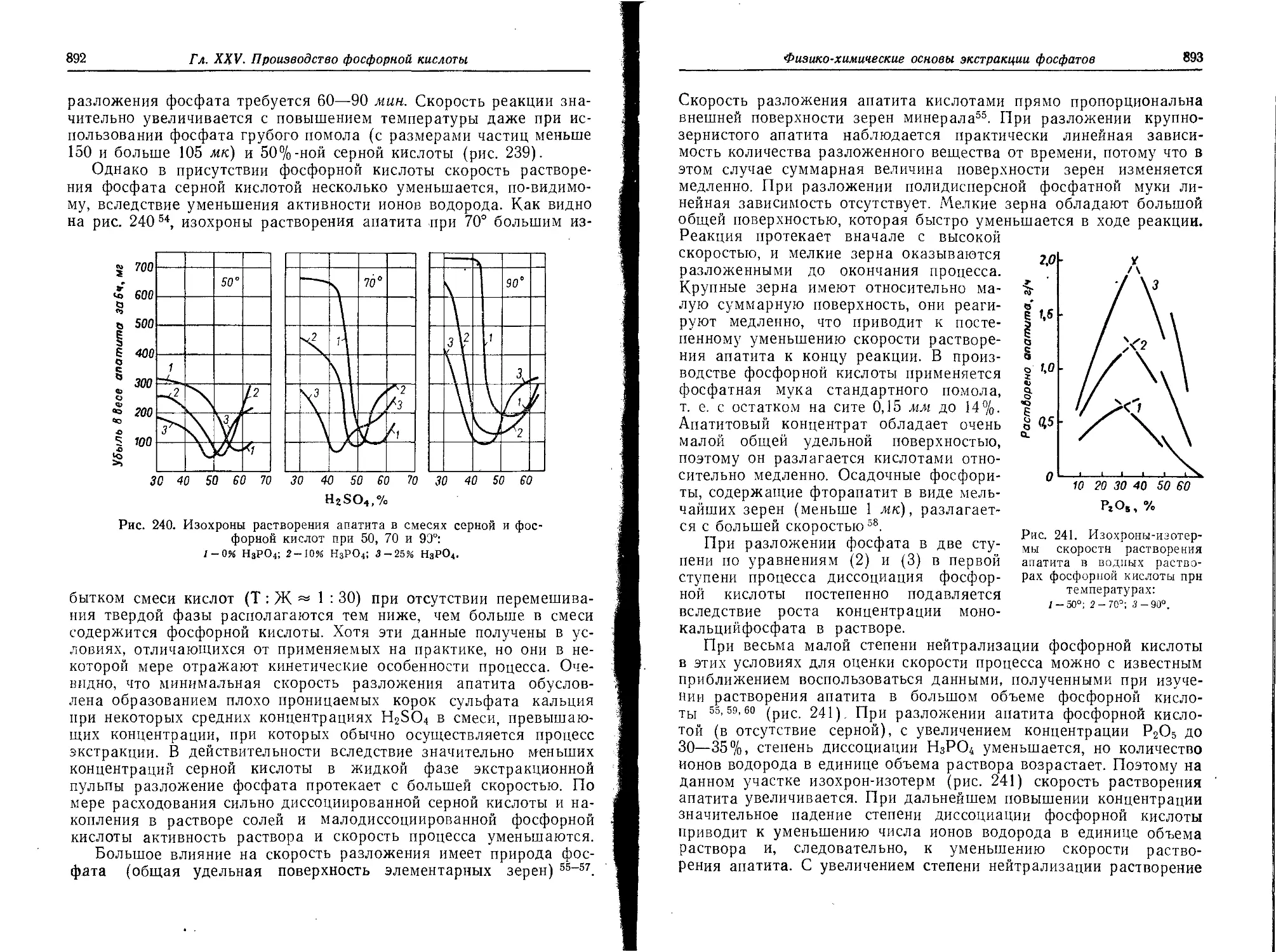
Скорость разложения фосфатов при сернокислотной экстракции из них
фосфорной кислоты . •............................................886
Кристаллизация сульфата кальция .................................. 895
Методы сернокислотной экстракции фосфорной кислоты из фосфатов . . 904
Производство экстракционной фосфорной кислоты дигидратиым способом 908
Технико-экономические показатели...................................920
Концентрирование фосфорной кислоты ............................... 921
Получение концентрированной фосфорной кислоты полугидратным спо-
собом ...............................................................924
Литература ............................................................933
Оглавление
795
Глава XXVI. Производства фосфора и фосфорной кислоты электротермиче-
ским методом ..................................938
Свойства фосфора и его соединения.................................. 938
Применение фосфора и термической фосфорной кислоты................. 940
Теоретические основы возгонки фосфора из фосфатов кальция .... 943
Производство фосфора электровозгонкой из фосфатов.................. 947
Расходные коэффициенты........................................... 955
Отходы производства, нх утилизация............................... 955
Получение фосфорной кислоты........................................ 957
Литература .......................................................... 966
Глава XXVII. Концентрированные фосфорные удобрения. Двойной и обога-
щенный суперфосфаты ....................... .... 971
Состав и свойства............................................... 971
Применение концентрированных суперфосфатов .... ... 972
Физико-химические основы производства двойного суперфосфата . . . 973
Химизм процесса.................................................. 973
Условия равновесия и кристаллизация твердых фаз.................. 974
Скорость разложения фосфатов фосфорной кислотой.................. 978
Растворение фосфата в фосфорной кислоте (без кристаллизации твердой
фазы)........................................................... 978
Разложение Фосфатов (с кристаллизацией продукта реакции) в иеза-
густевающей пульпе................................................ 982
Разложение фосфатов с образованием загустевающей пульпы . . . 988-.
Производство двойного суперфосфата................................. 991
Камерный способ................................................. 992
Бескамерные (поточные) способы................................... 997
Циклические методы получения двойного суперфосфата бескамерным
способом ..................................................... 1004
Некоторые технико-экономические данные..............................1009
Обогащенный суперфосфат.............................................1010
Литература . . .................................................... 1013
Глава XXVHI. Концентрированные фосфорные удобрения. Преципитат (ди-
кальцийфосфат) ... ..........., . ..........1017
Фнзико-химпческие свойства.........................................1017
Применение ..................................................... 1019
Методы производства................................................1020
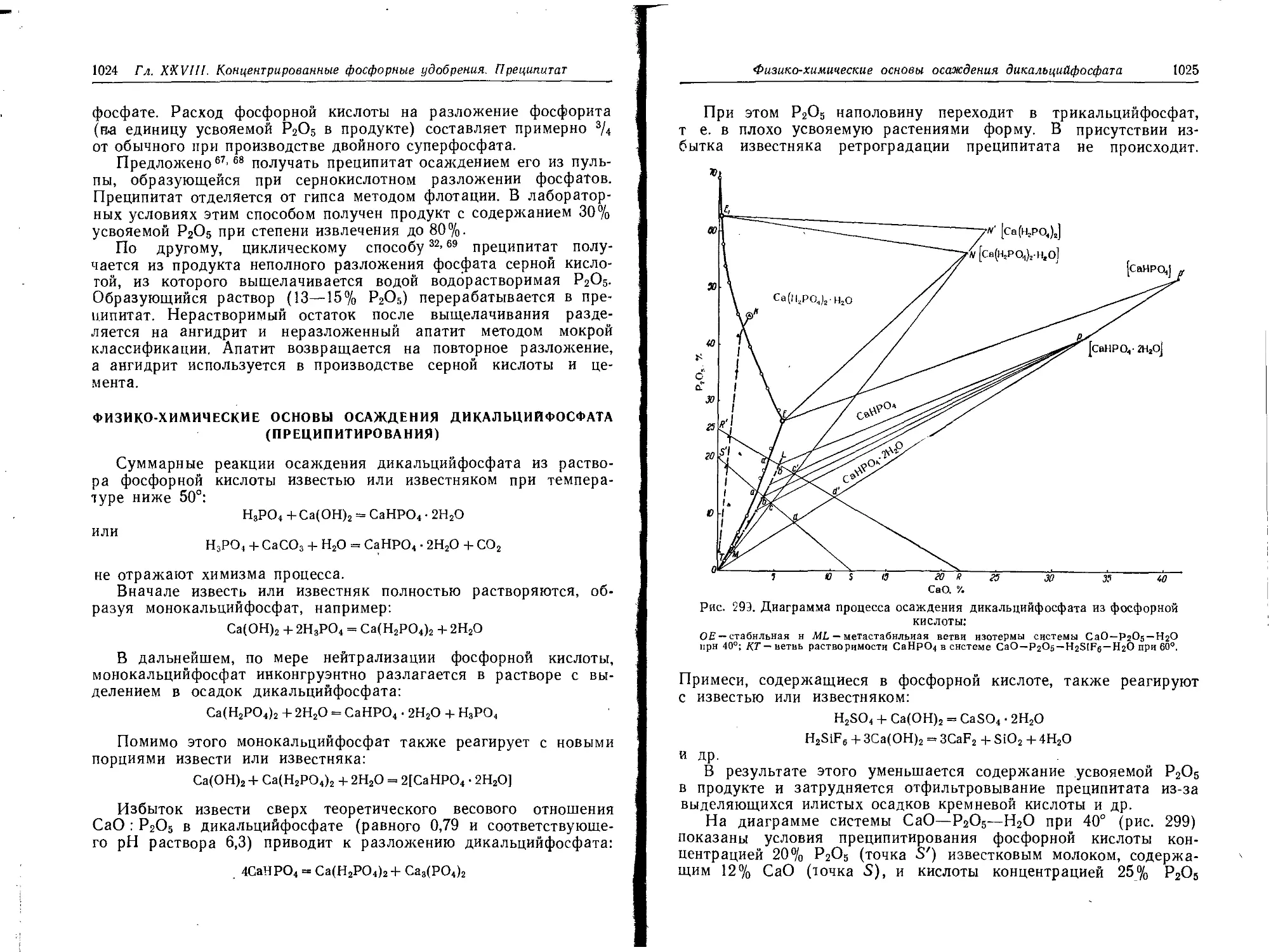
Физико-химические основы осаждения дикальцийфосфата (преципитиро-
вания).......................................................'. . Ю24
Производство преципитата . . . '.................................. 1029
Кормовой преципитат .............................................. 1032
Литература ..................................................... . Ю35
Г лава XXIX. Термические фосфаты................................... 1038
Состав и свойства................................................ 1038
Применение ................................................... 1042
Производство термических фосфатов................................. 1043
Получение обесфторенных фосфатов............................... 1043
Получение плавленых магниевых фосфатов ...................... . 1051
Получение термощелочных фосфатов (термофосфатов)................ 1053
Получение метафосфата кальция....................................1055
Литература ....................................................... Щ56
Глава XXX. Промышленные продукты, содержащие фосфор..................1062
Сульфиды фосфора...................................................1062
796
Оглавление
Хлориды фосфора.................................................. Ю63
Фосфиды .................................... ............... . • Ю54
Ортофосфаты натрия и калия ....................................... 1065
Производство фосфатов натрия................................... 1068
Дегидратированные фосфаты натрия ................................ 1070
Производство триполифосфата натрия и других дегидратированных
фосфатов .......................................................1076
Фосфаты аммония и двойные фосфорнокислые соли аммония .... 1085
Фосфаты кальция....................................................1089
Литература .......................................................• 1090
Глава XXXI. Соединения фтора ........................................1095
Физико-химические свойства....................................... 1095
Применение . ............................................... 1105 (
Сырье ..............................................................1108 \
Получение фтора............................................... . 111'
Фтористый водород и плавиковая кислота..........................111:
Разложение плавикового шпата и других фторидов...................111;
Абсорбция и конденсация фтористого водорода .....................1117
Жидкий фтористый водород......................................... ' 1120
Производство фтористых солей из плавиковой кислоты...............1124
Криолит ........................................................ 1124
Фторид алюминия..................................................1126
Фторид натрия....................................................1128
Фторид магния....................................................1129
Щелочные способы получения фторида натрия и криолита из плавикового
шпата .......................................................... 1129
Улавливание фтористых соединений из отходящих газов и их переработка 1134
Получение кремнефтористоводородной кислоты.......................1136
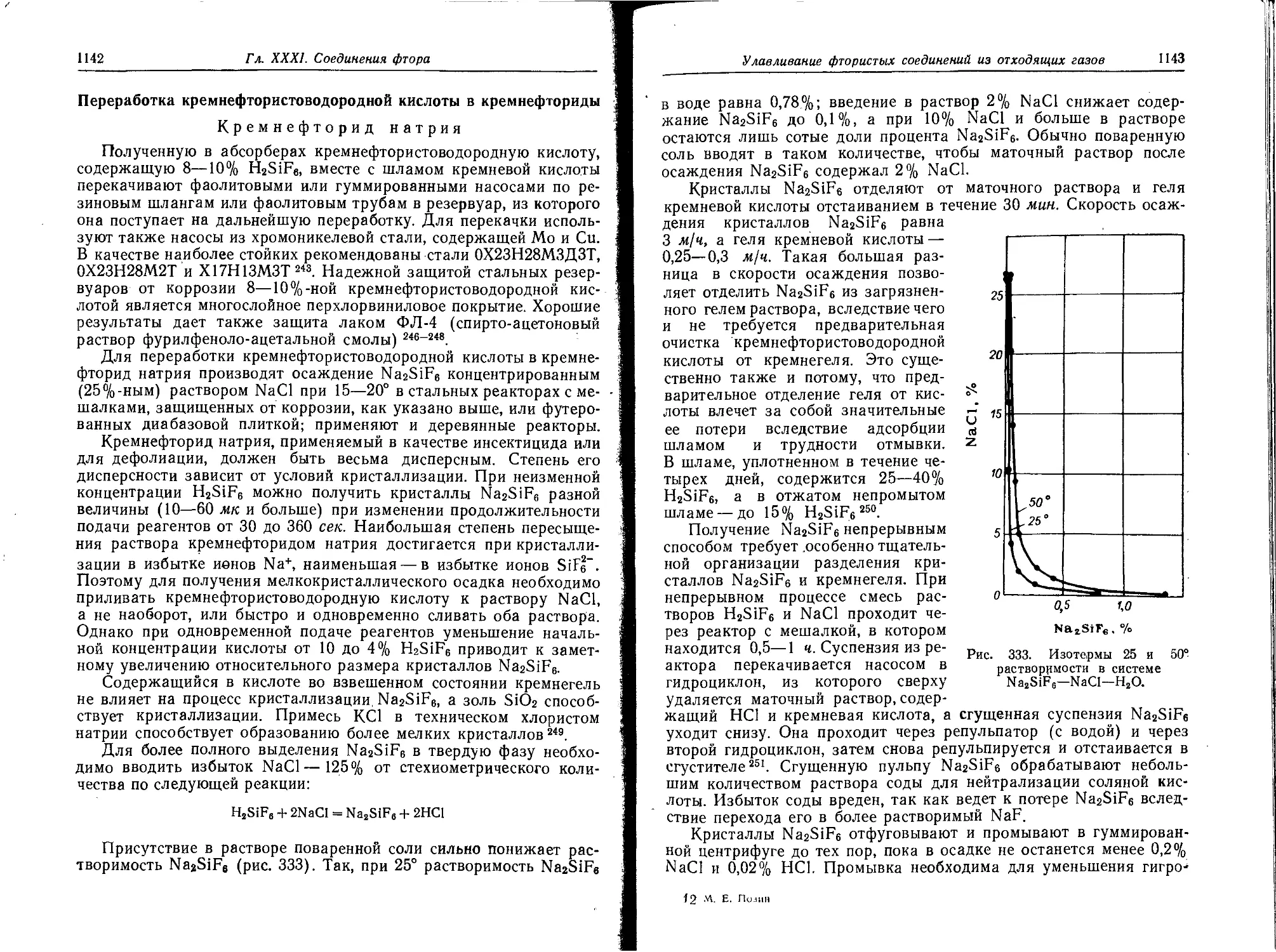
Переработка кремнефтористоводородной кислоты в кремнефториды . 1142
Получение фтористых солей из отходящих газов . .................1148
Фторид натрия ................................ . .............. 1149
Фторид кальция 1158
Криолит и фтористый алюминий ....................................1160
Аммиачный способ улавливания и переработки фтористых газов . . . 1162
Литература ..........................................................1170
Глава XXXII. Соли азотной кислоты..................................1178
Нитрат аммония.....................................................1178
Физико-химические свойства.......................................1178
Применение ......................................................1184
Сырье и методы производства......................................1186
Производство аммиачной селитры с выпаркой растворов..............1187
Получение нитрата аммония безупарочиыми способами................1199
Получение аммиачной селитры конверсией нитратных растворов . . . 1203
Удобрения на основе нитрата аммония..............................1204
Нитрит аммония ....................................................1209
Нитрат кальция .................................................. 1210
Физико-химические свойства.......................................1210
Применение ......................................................1211
Методы производства..............................................1211
Нитрат и нитрит натрия.............................................1216
Физико-химические свойства.......................................1216
Применение .................................................... 1216
Способы производства нитрата натрия..............................1217
Оглавление
797
Нитрат калия • • ........................................
Физико-химические свойства....................................
Применение ...................................................
Способы производства нитрата калия............................
Литература ................................................ • • •
Глава XXXIII. Соли аммония.........................................
Сульфат аммония ..................•...........................
Физико-химические свойства......................................
Применение .....................................................
Производство сульфата аммония...................................
Хлористый аммоний .....................................
Физико-химические свойства................................. • •
Применение .....................................................
Производство хлористого аммония.................................
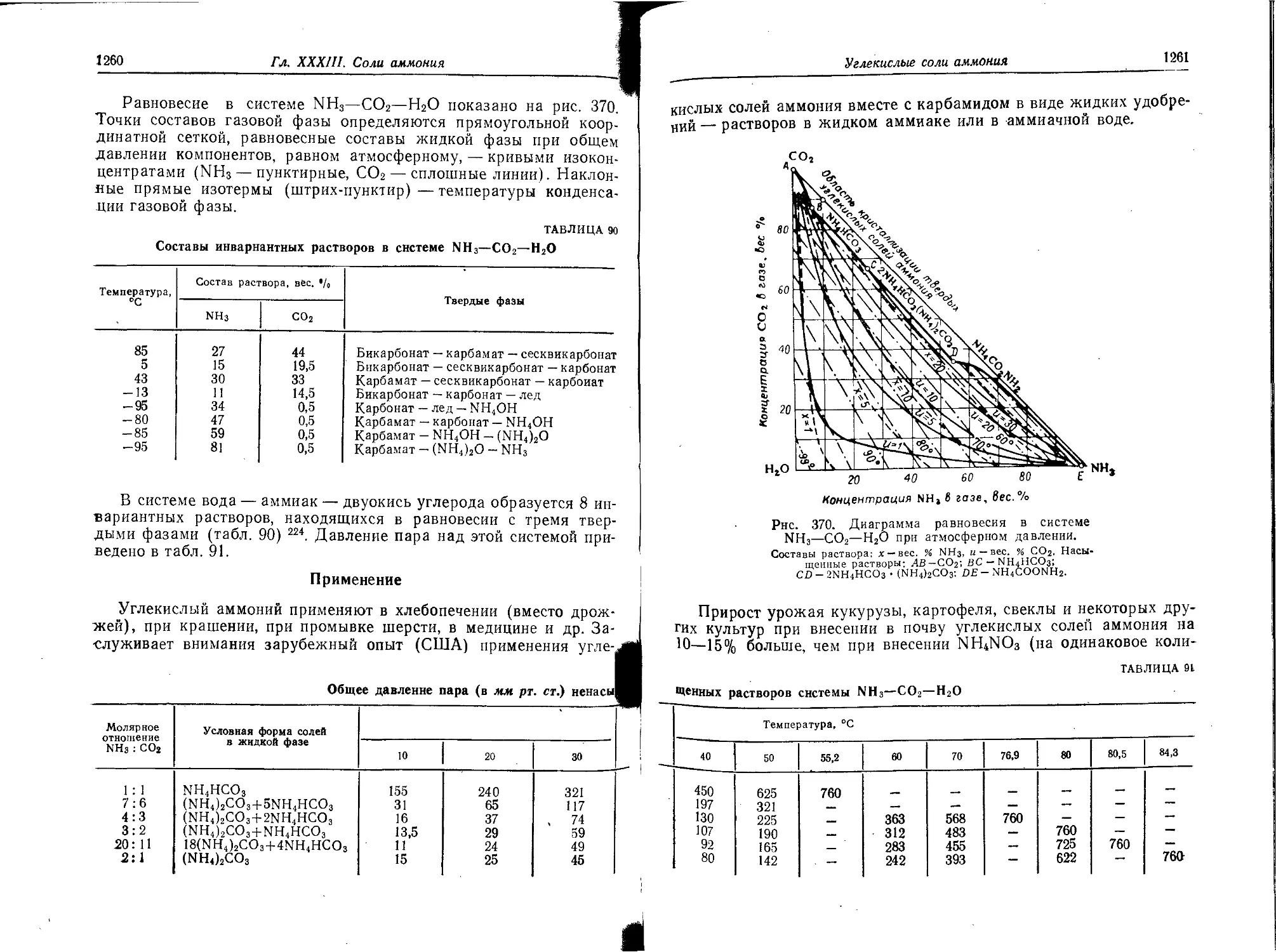
Углекислые соли аммония..........................................
Физико-химические свойства......................................
Применение .....................................................
Получение углекислого аммония ..................................
Получение двууглекислого аммония................................
Фосфаты аммония .................................................
Физико-химические свойства......................................
Применение .....................................................
Производство фосфатов аммония...................................
Удобрения, содержащие полифосфаты аммония........................
Удобрения, содержащие метафосфат аммония.........................
Литература ........................................................
1222
1222
1223
1223
1232
1237
1237
1237
1238
1239
1253
1253
1253
1254
1259
1259
1260
1262
1263
1264
1264
1265
1266
1274
1274
1275
Глава XXXIV. Карбамид ................. .............................1282
Физико-химические свойства .........................................1282
Применение и агрохимические свойства................................1285
Методы производства ................................................1287
Физико-химические основы синтеза карбамида из аммиака и двуокиси
углерода ........................,............................... 1288
Способы производства карбамида из аммиака и двуокиси углерода пря-
мым синтезом ..................................................... 1293
Способы регенерации газов дистилляции и режимы синтеза карбамида 1294
Производство карбамида по схеме с жидкостным рециклом .... 1297
Переработка растворов карбамида в готовый продукт.................1300
Литература ...........................................................1302
Глава XXXV. Комплексные твердые сложные удобрения.....................1307
Физико-химические основы разложения фосфатов азотной кислотой . . 1308
Выделение фтора и редкоземельных элементов из азотнокислотной вы-
тяжки ........................................................... I3H
Переработка раствора, полученного азотнокислотным разложением фосфа-
тов ...............................................................1314
Физико-химические основы переработки азотнокислотной вытяжки . 1314
Способы переработки азотнокислотной вытяжки...................1321
Переработка азотнокислотной вытяжки в сложные удобрения .... 1325
Карбонатный способ............................................1329
Сульфатные способы............................................1331
Фосфорнокислотный способ......................................1336
Нитроаммофос и нитроаммофоска.................................1338
798
Оглавление
Получение нитрофоски с вымораживанием части нитрата кальция . . 1339
Получение водорастворимой нитрофоски с частичной регенерацией
азотной кислоты , .... ....... 1346
Производство сложных удобрений на базе фосфорной кислоты . . . 1347
Некоторые свойства водных систем, содержащих фосфаты, нитраты
аммония, карбамид и др..........................................1347
Способы производства............................................1349
Литература ....................................................... 1358.
Г лава XXXVI. Комплексные смешанные и сложно-смешанные удобрения . 1363
Общие сведения.................................................. 1363
Синергизм и антагонизм удобрений ................................ 1365
Производство смешанных удобрений............'.....................1367
Расчет состава тукосмесей....................................... 1370
Литература ....................................................... 1374
Г лава XXXVII. Жидкие удобрения . . 1375
Общие сведения................................................ 1375
Физико-химические свойства........................................1377
Производство жидких удобрений .................................. 1387
Схемы производства аммиачной воды и аммиакатов на основе аммиач-
ной селитры................................................... 1387
Производство жидких комплексных удобрений.......................1390
Суспендированные жидкие комплексные удобрения...................1392
Литература .........................................................1392
Глава XXXVIII. Соли мышьяка ............................... . . . 1396
Физико-химические свойства....................................... 1393
Применение мышьяковых препаратов и технические требования к ним , 1402
Сырье ............................................................1405
Получение белого мышьяка обжигом мышьяковых руд...................1407
Арсенит кальция ................................................. 1408
Мокрый способ производства .................. . . . . 1409
Полусухой способ производства ................................. 1410
Арсенит натрия....................................................1412
Парижская зелень.................................................1413
Мышьяковая кислота ...............................................1416
Азотнокислотный способ..........................................1416
Другие способы получения мышьяковой кислоты.....................1418
Арсенат кальция ................................................. 1419
Способ каталитического окисления растворов арсенита воздухом . . 1419
Получение арсената кальция из мышьяковой кислоты................1423
Азотнокислотный способ..........................................1423
Хлорный способ................................................. 1425 -
Электрохимический способ.......................................1426
Термический способ .......................7.....................1426
Получение арсената кальция из окисленных мышьяковых руд . . . 1427
Литература .........................................................1428
Глава XXXIX. Соли кислородных кислот хлора..........................1430
Физико-химические свойства........................................1430
Применение ...................................................... 1437
Оглавление
799
Производство хлорной извести....................................
Обжит известняка...............................................
Получение пушоики (гашение извести)............................
Хлорирование пушонки ..........................................
Гипохлорит натрия ..............................................
Гипохлорит кальция ..................................... ......
Двуокись хлора..................................................
Сернистокислотный способ.......................................
Метанольный способ.............................................
Солянокислотный способ.........................................
Хлоридный способ...............................................
Получение двуокиси хлора из хлорита, натрия....................
Хлорит натрия..................... .............................
Хлораты натрия, калия, кальция и магния.........................
Получение хлоратов натрия и калия электрохимическим методом . .
Получение хлората калия известковым методом....................
Хлорат натрия..................................................
Хлораты кальция и магния.........................................
Перхлораты натрия, калия и аммония и хлорная кислота............
Перхлорат натрия...............................................
Перхлорат калия ...............................................
Перхлорат аммония..............................................
Хлорная кислота ...............................................
1441
1441
1442
1444
1448
1450
1453
1454
1456
1457
1458
1459
1460
1461
1461
1464
1469
1470
1471
1471
1473
1473
1474
Литература ........................................................ 1476
Глава XL. Продукты высокотемпературного хлорирования руд .... 1480
Общие сведения................................................... 1480
Четыреххлористый титан.............................................1481
Физико-химические свойства........................................1431
Применение .......................................................1482
Сырье ............................................................1483
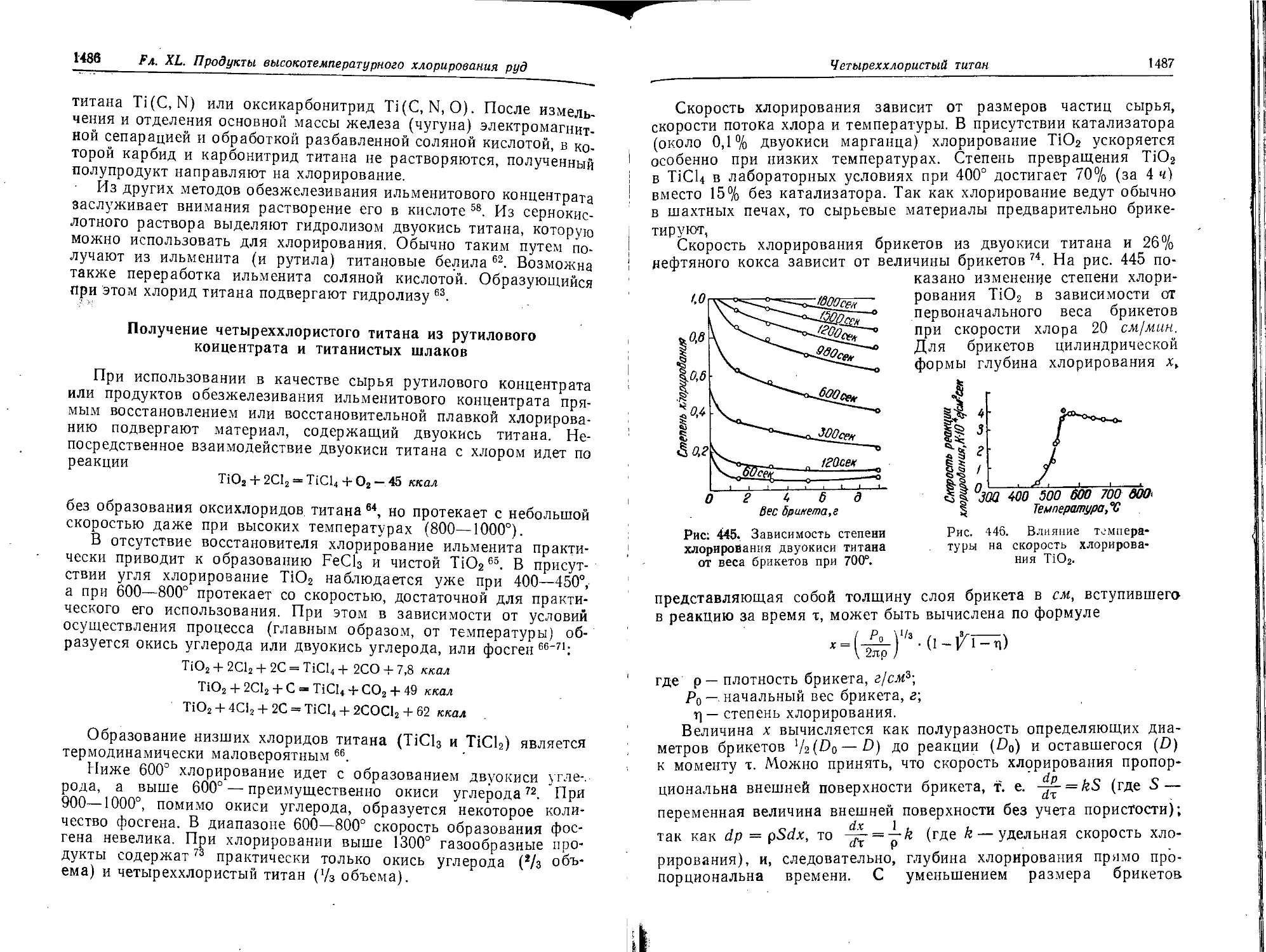
Способы обезжелезивания ильменитового концентрата.................1484
Получение четыреххлористого титана из рутилового концентрата и ти-
танистых шлаков.................................................. 1486
Получение четыреххлористого титана из других видов сырья ..... 1494
Треххлористый титан............................................_ 1495
Четыреххлористый кремний...........................................1495
Физико-химические свойства........................................1495
Применение ..................................................... 1496
Получение четыреххлористого кремния............................. 14)7
Безводный хлористый алюминий.......................................1500
Физико-химические свойства........................................1590
Применение .......................................................1501
Получение безводного хлористого алюминия..........................1501
Литература ......................................................... 1504
Г лава XL1. Цианистые соединения ....................................1509
Физико-химические свойства........................................1509
Применение ...................................................... 1513
Сырье .............................................................1516
Цианамид кальция...................................................1516
Получение цианамида кальция в электрических печах............... . 1518
Получение гранулированного цианамида кальция ................... 1521
Другие методы получения цианамида кальция , . . ................. 1521
800
Оглавление
Производство цианидов натрия и калия ............ ................
Получение цианистых солей из синильной кислоты.................
Получение цианплава из цианамида кальция.......................
Фиксация азота смесью соды с углем.............................
Восстановление цианатов, полученных из аммиака и карбонатов . .
Сплавление амида натрия с углем ...............................
Сухая перегонка барды..........................................
Получение цианидов из коксового газа...........................
Получение роданидов натрия и аммония из отбросных растворов мышья-
ково-содовой очистки газов .......................................
Производство железисто- н железосинеродистых солей................
Железистосинеродистые соли ....................................
Железосинеродистые соли......................'.................
Производство синильной кислоты контактными способами..............
Способы синтеза синильной кислоты..............................
Производство синильной кислоты контактным окислением метана и ам-
миака .........................................................
Получение цианистого водорода из аммиака и окиси углерода (форм-
амидный способ) .... ........ ......... . •
Получение синильной кислоты плазмонным методом.................
Литература ...................................................... •
Предметный указатель..............................................
1540
1540
1543
1547
Глава XXIII
ПРИРОДНЫЕ ФОСФАТЫ И ФОСФОРИТНАЯ МУКА
Фосфор является распространенным в природе элементом. Со-
держание его в земной коре (кларк) составляет 0,08—0,12% (по
весу) или около 0,07% от общего числа атомов земной коры. Вслед-
ствие высокой активности элементарного фосфора и его окислов
он образует мало- и труднорастворимые в воде минералы. Извест-
но около 120 минералов, содержащих фосфор. Из них наибольшее
промышленное значение имеют минералы апатитовой группы,1-5
входящие в состав фосфорных руд или фосфатов. Природные фос-
фаты, называемые агрономическими рудами, являются исходным
сырьем для производства соединений фосфора — элементарного
фосфора, фосфорной кислоты, минеральных удобрений, кормовых
средств, полифосфатов и т. п.
ФОСФОРНЫЕ РУДЫ
с общей формулой
Минералогический состав апатитовых и фосфоритных руд
Природные фосфорные руды разных месторождений разли-
чаются по своим физическим и химическим свойствам в зависи-
мости от минералогического состава, структуры, содержания при-
месей. Они подразделяются на два основных вида — апатитовые
и фосфоритные руды. Фосфорным веществом в обоих видах сырья
являются минералы апатитовой группые> 7 с общей формулой
ЗМ3(РО4)2 • СаХ2, где М представлен Са2+, а X — фтором, хлором,
группой ОН. Кальций, входящий в состав фосфатной части моле-
кулы, может изоморфно замещаться стронцием, редкоземельными
элементами, натрием и другими элементами; ион РО4 — ионами
SO4 и SiOl-. Помимо минералов апатитовой группы в фосфат-
ных рудах содержатся другие минералы—примеси. Наиболее рас-
пространен в природе кальцийфторапатит ЗСа3(РО4)2• CaF2 или
802
Гл. XXIII. Природные фосфаты и фосфоритная мука
Ca5(PO4)3F, а также гидроксилапатит ЗСаз(РО4)2-Са(ОН)2 или
Са5(РО4)зОН. Гидроксилапатит находится в костях животных.
В апатитовых рудах основным фосфорсодержащим минералом
является кальцийфторапатит и в небольших количествах — гидро-
ксилапатит и другие формы изоморфного замещения8. Фторапа-
тит в руде находится в виде полупрозрачных неправильных кри-
сталлов, слегка окрашенных в зеленый и желто-зеленый цвет. Зер:
на иногда имеют форму шестигранных призм. Апатиты состоят из <
крупных кристаллических частиц, характеризующихся отсутствием
полидисперсности и микропористости9’ !0.
Состав химически чистого кальцийфторапатита следующий:
42,23% Р2О5, 55,64%CaO, 3,77% фтора. Вследствие частичного изо-,
морфного замещения, природный чистый минерал содержит в сред-'
нем 40,7% Р2О5 и 2,8—3,4% фтора. Состав образцов Кольского
апатита приведен в табл. 52.
ТАБЛИЦА 52
Химический состав некоторых образцов Кольского апатита
(в вес. %)
Компоненты Образец
I II III IV
р205 40,95 40,80 38,33 40,57
AS2O5 0,00028 0,00018 0.00007 0,00016
v2o5 — 0,001 —
SiO2 0,41 0,26 1,55 0,21
TiO2 0,00 0,00 0,00 0,00
ZrO2 0,001 0,001 0,001 —
У Ln2O2 1,19 0,80 3,22 1,75
Fe2O3 0,10 0,08 0,12 0,12
A12O3 0,19 0,12 0,34 0,10
CaO 52,51 53,17 42,38 47,70
SrO 2,51 1,75 11,42 6,69
BaO 0,00 0,00 0,00 0,00
MgO 0,12 0,08 0,05 0,10
MnO 0,05 0,01 0,05 0,03
Na2O 0.14 0,30 0,13 0,36
K2O 0,08 0,08 0,07 0,04
H2O 0,15 0,19 0,28 0,10
Фтор 2,47 3,32 3,73 3,12
Хлор — 0,00 0,00 Следы
s 100,87 100.96 101,67 100,89
a* 1,04 1,39 1,57 1,31
S—a 99,83 99,57 100,10 99,58
О минералогическом составе фосфатного вещества фосфоритов
нет единого мнения. Допускают11-12, что оно состоит из трех фаз,
♦ а—количество кислорода, эквивалентное фтору в CaF^.
Фосфорные руды
803
дающих иногда твердые растворы изоморфных смесей: 1) кальций-
фторапатит (в подавляющем большинстве фосфоритов), 2) карбонат-
апатит (очень редко), 3) гидроксилапатит (в небольшом количестве
в форме изоморфной смеси фторгидроксилапатита). Предполагают
также 13~15, что фосфатное вещество фосфоритов представлено,
кроме фторапатита, франколитом (штаффелитом) и курскитом.
Молекулярное строение и свойства последних точно не определены;
это объясняется высокой дисперсностью фосфатных минералов
фосфоритов и трудностью их получения в чистом виде.
По некоторым исследованиям 16, фосфаты фосфоритных руд об-
разованы изоморфным замещением во фторапатите части фосфора
углеродом с одновременной заменой одного кислорода на ОН-
или F [хСаюРбОгдРг + г/СаюРзСО23(Р, ОН)з].
ТАБЛИЦА 53
Фосфаты группы апатита (в вес. %), входящие в состав фосфоритов
Минералы _ Формулы РгО5 СаО со2 Фтор СаО Р2О5 СО2 P2O5 F Р2О5
Фторапатит CaioPgOs^Fs 42,23 55,64 3,77 1,32 0,09
Г идроксил- апатит 42,40 55,88 — — 1,32 — —
Карбонат- апатит Са10Р5СО2з(ОН)3 39,97 63,22 4,46 — 1,58 0,12 —
Франколит СаюРб.гСо.зОгз.гРг 37,14 56,46 3,54 3,44 1,52 0,09 0,09
Курскит Са|оР4,8С1,20 22,8?2(ОН)112 34,52 56,86 5,35 3,85 1,64 0,16 0,11
В табл. 53 приведены формулы и химический состав важнейших
минералов, составляющих фосфатное вещество фосфоритов.
Фосфориты характеризуются мелкокристаллической структурой,
высокой полидисперсностыо и пористостью частиц91 10. При этом
микроструктуры фосфатного вещества различных фосфоритов
весьма разнообразны. Фосфат встречается как в виде коагулиро-
вавшего геля, близкого к аморфному, так и в явно кристаллической
форме, хотя имеются многочисленные промежуточные группы. На-
пример, в фосфоритах Каратау преобладают сравнительно крупные
И хорошо кристаллизованные частицы с размером 1—2 мк, в эстон-
ских и кингисеппских фосфоритах — частицы с размером значи-
тельно меньшим 1 мк и весьма высокой пористостью.
Апатитовые руды, кроме апатита, содержат минералы 17: нефе-
лин (Na, К) AlSiO4 • nSiO2, пироксены — эгирин NaFe(SiO3)2 и дру-
гие, титаномагнетит Fe3O4 • FeTiO- • TiO2, ильменит FeTiO3, сфен
CaTiSiO5, полевые шпаты, черную слюду, эвдиалит.
В состав фосфоритных руд входят минералы-примеси2-5’18’19:
глауконит — водный силикат типа [(R2O + RO) • R2O3 • 4SiO2 • 2Н2О],
где R2O—-Na2O и К2О, RO—MgO, СаО и FeO, R2O3—Fe2O3 и A12O3,
804 - - " ’ Гл. XXIII. Природные фосфаты и фосфоритная мука
лимонит Fe2(OH)6-Fe2O3, кальцит СаСО3, доломит CaCO3-MgCO3,
магнезиальные силикаты Mg2SiO4, каолин H2Al2Si2O8 • Н2О, пирит
FeS2, полевые шпаты, кварц, гранит и другие, а также органические
вещества. )
Количество фосфатных минералов в земной коре составляет2®
не больше 0,75%. Около 95% фосфора находится в земной коре в
виде апатитов, присутствующих почти во всех изверженных поро-
дах в чрезвычайно рассеянном состоянии и редко образующих
скопления, имеющие промышленное значение. Крупные апатитовые
месторождения—магматического происхождения13-17. Имеются
гидротермальные образования апатита, т. е. выделившиеся из го-
рячих водных растворов. Апатит образовывался также при кон-
такте магмы с известняками. Месторождения фосфоритов возник-
ли21-22 вследствие осаждения фосфатов из морской воды11-14.
Встречаются также образования фосфоритов, близких к франко-
литу, связанные с гидротермальными растворами23.
Типы фосфоритных руд и месторождения
Самыми крупными скоплениями фторапатита являются место*
рождения магматического происхождения, находящиеся в Хибин-
ской тундре (Кольский полуостров) 24-26. Здесь разведано шестг
месторождений апатито-нефелиновой руды: Кукисвумчоррское,
Юкспорское, Расвумчоррское, Саамское, Куэльпорское и Ено-Ков-
дорское, из них эксплуатируются первые три. На горе Кукисвум-
чорр мощность рудного тела достигает 200 ж. Оно состоит из верхней
богатой зоны (70—80 м) со средним содержанием в породе 29,1 %
Р2О5 и нижней бедной (120—130 м) руды, содержащей 18% Р2О5,-
Руды Юкспорского месторождения содержат от 9,5 до 21,8%
Р2О5. Вертикальная мощность рудного тела достигает 180 м.
Расвумчоррское месторождение состоит из двух участков. Сред-
невзвешенное содержание Р2О5 составляет 19,4—19,55%. Разработ-
ка ведется открытым способом (на участке Апатитовый цирк).
В начальный период эксплуатации Хибинских месторождений
добывалась руда с содержанием 32—34% Р2О5- В течение более
35 лет работы комбината «Апатит» богатая руда с разведанных
участков уже в основном выработана. В настоящее время среднее
содержание Р2О5 в добываемой руде составляет 17,5—18%. Эта
руда так же, как и богатая, легко перерабатывается флотацией
в высокосортный концентрат, содержащий не менее 39,4% Р2О5 27.
Производительность комбината «Апатит» составила в 1967 г.
8,7 млн. т концентрата, а в 1970 г. возрастет до 14,5 млн. т концен-
трата 28.
Кольский апатитовый концентрат является лучшим в мире
сырьем для производства удобрений и экспортируется в больших
количествах в страны СЭВ и другие 29-30. Промышленное значение
Фосфорные руды
805
имеет также Ено-Ковдорское месторождение апатито-магнетито-
вых РУД- После извлечения из них магнитной сепарацией железного
концентрата остаются хвосты, содержащие около 10,5% Р2О5. Из
последних дальнейшим обогащением могут быть получены концен-
траты с содержанием 25—35% Р2О5. В СССР разведано еще не-
сколько небольших залежей апатитовых руд: в Джамбульской,
Иркутской и других областях.
За рубежом31 магматические месторождения апатитовых’ руд
находятся в Северной и Центральной Швеции, где они связаны с
месторождениями фосфористых железных руд. Содержание апа-
тита в фосфатной руде составляет 8—13%• В Южно-Африканской
республике имеется месторождение апатитов контактного происхо-
ждения (от пироксеново-апатитовых пород до почти чистого
апатита с содержанием 34—40,5% Р2О5). Известны небольшие гид-
ротермальные месторождения апатитов в Канаде и в Южной Нор-
вегии. Норвежские руды содержат до 40—41,8% P2Og. Среди гид-
ротермальных месторождений апатита наибольшее значение имеют
залежи в Испании, содержащие 34,4—42,6% Р2О5. Месторождения
подобного типа в Китае (провинция Цзянси) ассоциируются с мар-
ганцовыми рудами; отгружаемая руда содержит 33% Р2О5. Круп-
ные месторождения апатитов имеются в Бразилии (Минас — Же-
райс) и в Уганде (Восточная Африка).
Запасы и добыча апатитовых руд за рубежом незначительны
по сравнению с СССР.
Осадочные фосфоритные месторождения СССР по залеганию
рудного тела подразделяются5-32 на два' типа: платформенные и
геосинклинальные.
Платформенные месторождения характеризуются распределе-
нием руды на больших площадях горизонтальным залеганием фос-
форитных слоев, разделенных прослойками пустых пород, неболь-
шой мощностью слоев (не выше 5 м).
Геосинклинальные месторождения вытянуты линейно, имеют
сложное залегание (крутые наклоны пластов и т. п.), фосфоритные
пласты мощностью до 15 м чередуются с фосфатно-кремнистыми и
фосфатно-карбонатными породами и др.
В обоих типах месторождений различают фосфориты по нх
виду; пластовые, желваковые, зернистые и ракушечные
Крупнейшие в СССР месторождения пластовых фосфоритов гео-
синклинального типа находятся в Казахстане в Джамбульской
области (горы Каратау)19-36. Наиболее важные месторождения
этого района — Чулактау, Аксайское, Джанытас, Коксу. Руды со-
держат до 26% Р2О5 и не более 2—3% полуторных окислов. На
отдельных участках встречаются зоны с содержанием в руде до
36% Р2О5. На качестве руды отрицательно сказываются примеси
доломита и силикатов магния (MgO свыше 3%). В СССР имеется
Ряд мелких неэксплуатируемых месторождений пластовых
808
Гл. XXIII. Природные фосфаты и фосфоритная мука
фосфоритов платформенного типа — Стерлитамакские,, Вольские,
хоперские и обояньрыльские.
Желваковые фосфориты залегают преимущественно в Европей-
ской части СССР и в Северо-Западном Казахстане, среди кварцево-
глауконитовых, иногда известковых песков и глин в виде желваков
(камней) разнообразной формы и цвета. Иногда желваки сцемен-
тированы известью, глиной, кварцем и фосфатами в прочные пли-
ты. Желваковые фосфориты делят на три группы: 1) песчани-
стые, 2) глауконитовые и 3) глинистые.
Песчанистые фосфориты составляют в СССР около 60% от
общих запасов фосфоритов. Наиболее крупные месторождения на-
ходятся в Актюбинской области (Ново-Украинское, Богдановское,;
Кандагачское), в Курской (Щигровское, Трухачевское, Красно-^
Полянское, Свободинское — первое эксплуатируется), в Брянской;
(Полпинское — эксплуатируется, Козелкинское, Сеннинское, Бур<
ковско-Глаженское, Толвинско-Нетвинское и др.), в Калужской
(Подбужское, Слободско-Которецкое, Бычковское, Новоселкинскоеу
Труфановское). В ряде других районов Европейской части СССР,
имеются относительно небольшие залежи песчанистых фосфоритов.
Для этих фосфоритов характерно низкое содержание Р2О5 (12—
16%, максимально 19,5—20%), большое количество нераствори-
мого остатка, т. е. не растворимых в кислоте силикатов и кремне-
зема (до 30—50%) и небольшое содержание полуторных окислов
(до 5—6%).
В глауконитовых фосфоритах преобладает примесь глауконита.
Они содержат до 24%Р2О5 (чаще 18—20%), 20—33% кремнезема
и много полуторных окислов — в желваках или плите 12—15%, а в
выветрелой руде до 20%. Типичным их представителем являются
фосфориты верхнего (рязанского) горизонта разрабатываемого
Егорьевского месторождения (Московская область).
Глинистые фосфориты наиболее богаты Р2О5 (в желваках 24—
29%). Они содержат относительно небольшое количество не рас-
творимого в кислоте остатка (3—15%), много кальцита и мало
полуторных окислов (до 8%). Фосфоритами этого типа сложены
Вятско-Камский бассейн (Кировская область) и нижний горизонт
Егорьевского месторождения. Эти крупные месторождения экс-
плуатируются.
Месторождения зернистых фосфоритов (Таджикистан, Восточ-
ная Сибирь) пока не имеют промышленного значения.
Сравнительно крупные месторождения Эстонской ССР (Маар-
ду) и Ленинградской области (Кингисеппское) образованы раку-
шечными (ободовыми) фосфоритами — это слои пород, насыщенные
фосфатизированными раковинами. Руда бедная (3—8% РгО5), но
легко обогащается, давая концентрат с содержанием до 29% Р2О5.
А4есторождения фосфатного сырья размещены на территории
СССР очень неравномерно. Наиболее благоприятно распределяется
Фосфорные руды
807
сырье для производства фосфоритной муки37. Потребляющие фос-
форитную муку центральные районы, где распространены подзоли-
стые почвы, с избытком обеспечены сырьем. Крупные фосфорит-
ные месторождения центра могут снабжать и другие районы, в
меньшей степени обеспеченные фосфатами.
В ряде крупнейших сельскохозяйственных районов страны —
Сибири, на Украине, Кавказе, Дальнем Востоке — пока не обна-
ружено промышленных запасов фосфоритов. Многочисленные ме-
сторождения Русской Платформы содержат относительно низко-
качественные фосфориты.
Весьма важной является разработка методов обогащения и ис-
пользования бедных фосфоритов многих месторождений! с целью
привлечения новых ресурсов фосфатов в производство удобрений.
Сырьевой базой для химической переработки в настоящее
время являются главным образом хибинские апатиты и частично
фосфориты Каратау.
Апатитовый концентрат перевозится на все суперфосфатные
заводы Европейской части СССР. Расстояние некоторых предприя-
тий от апатитового месторождения составляет: Винница — 2581 км,
Пермь — 2777 км, Константиновка — 2869 км, Одесса — 3019 км.
Дальние перевозки апатитового концентрата обусловливают удо-
рожание продукции. Открытие и эксплуатация мощного месторо-
ждения фосфоритов Каратау обеспечило суперфосфатные заводы
республик Средней Азии и Южного Казахстана местным сырьем и
позволило сократить перевозки в эти районы сырья или супер-
фосфата из Европейской части СССР, что дало большой экономи-
ческий эффект.
Помимо апатито-нефелиновых руд Кольского месторождения и
фосфоритов месторождения Каратау, промышленно разведанными
ресурсами сырья являются также актюбинские фосфориты и фос-
фориты Вятско-Камского месторождения.
Мало разведанные пока месторождения фосфатного сырья на
территории Восточной и Западной Сибири: Белозиминское, Бурят-
ское (Ошурковское), Сейбинское, Таштагольское и другие подго-
тавливаются для промышленного использования 33. Промышленно-
разведанные запасы фосфатного сырья в СССР обеспечивают38
потребность в нем на многие годы.
При использовании Вятско-Камского месторождения возможно
было бы уменьшить перевозки сырья на 1,3—1,5 тыс. км в районы
Средней и центральной полосы СССР, а увеличение разработки
месторождений Каратау и Актюбинского позволит вообще ликви-
дировать перевозки фосфатного сырья на расстояние свыше
3 тыс. км во все районы крупного потребления удобрений.
За рубежом наиболее крупные месторождения фосфоритов на-
ходятся в Северной Африке (Тунис, Алжир, Марокко), в США, на
островах Тихого океана и в Иордании31.
808
Гл. XXIII. Природные фосфаты и фосфоритная мука
Месторождения Северной Африки относятся к типу пластовых.
На большинстве участков фосфорит добывается открытым спосо-
бом. Руда содержит 23—33% Р2О5 и 1—2% R2O3 и используется
без обогащения. В США наибольшее значение имеют фосфориты
Флориды, содержащие от 18,4 до 34% Р2О5 и 1,5—3% R2O3. Низко-
процентные руды подвергаются обогащению.
В рудах месторождений Теннесси содержится от 16 до 23,8—
29,8% Р2О5 и до 5% R2O3. Фосфориты из месторождений в Южной
Каролине содержат около 26,6% Р2О5. У подножья Скалистых гор
(штаты Юта, Айдахо, Уайоминг и Монтана) расположены фосфо-
ритные пласты со средним содержанием Р2О5 32,1 % и R2O3 2—3%.
На Тихом океане на коралловых островах Макатеа, Науру, Океан,
Ангаур и других разрабатывают залежи гуано и месторождения
фосфоритов, образовавшиеся замещением известняков, подстилаю-
щих гуано. Качество фосфатов очень высокое (Р2О5 до 36,7—
41,2%)- Мировые геологические запасы фосфатов приблизительно
равны 475 млрд г, т. е. около 66 млрд, г Р2О5. Запасы 31>39-42 про-
мышленного значения по состоянию на 1956—1957 гг. оцениваются
во всем мире в количестве ~26 млрд т, в том числе в СССР 5,4 млрд.т
фосфоритов и 1,66 млрд, т апатитов, что составляет 27% от миро-
вых запасов. Указывают43, что мировая добыча природных фосфа-
тов составила в 1965 г. 57 млн. т (по другим данным44 —
62,5 млн. т) по сравнению с 34,8 млн. т в 1957 г. По различным
оценкам, она может в 1970 г. составить 70—115 млн. т.
В СССР добыча45 фосфоритов в 1957 г. составила 3,5 млн. т и
увеличилась по сравнению с 1955 г. на 100%.
В последующие годы добыча фосфоритов увеличивалась как за
счет расширения старых, так и открытия новых рудников по раз-
работке месторождений Каратау, Кингисеппского и др. В связи
с достижением в 1970—1976 гг. максимально допустимого объема
добычи руды и производства апатитового концентрата дальнейший
рост потребности в фосфорном сырье предполагается28 покрыть
за счет соответствующего увеличения добычи фосфоритов. В бас-
сейне Каратау годовая добыча фосфоритной руды может быть уве-
личена до 14,5 млн. т или до 3,3 млн. т Р2О5. Развитие Вятско-
Камского бассейна может обеспечить получение руды до 2,5 млн. т
Р2О3 в год. Возможно также получить до 1 млн. т Р2О5 в год за
счет введения в эксплуатацию 46 Актюбинского и Белозиминского
месторождений.
Применение фосфатов
Преобладающее количество фосфорных руд используется для
производства минеральных удобрений и в меньшей степени —для
получения элементарного фосфора и промышленных продуктов, со-
держащих фосфор, а также в металлургии черных и цветных ме-
таллов.
Фосфорные руды
809
В металлургии фосфорные руды применяются в смеси с желез-
ной рудой для выплавки в доменной печи феррофосфора и при
переработке в доменной печи малофосфористых железных руд, не
пригодных для получения нормального томасовского или литейного
фосфористого чугуна. Апатито-нефелиновые руды используются
также в литейном деле (для получения металла с повышенным со-
держанием фосфора), при получении фосфористой меди, в качестве
раскислителя при производстве фосфористой бронзы и др.
В металлургии применяются апатито-нефелиновые руды с со-
держанием не менее 28,5% Р2О5 (ОСТ 18234—39).
Основной целью переработки фосфатов с получением удобрений
является перевод не растворимых в воде и почвенных растворах
природных фосфорнокислых соединений в растворимое состояние.
В зависимости от свойств фосфатов, их минералогического и хими-
ческого составов, назначения получаемых продуктов и технико-
экономических условий 47-49 применяются различные методы их
переработки. Во всех случаях природные фосфаты подвергают
предварительно размолу и обогащению с применением методов
сухой или мокрой сепарации, обжига, флотации и др.
Наиболее распространены механические (размол), физико-ме-
ханические (флотационное обогащение) и химические методы.
Механическими и физико-механическими способами получают
также в некоторых количествах готовый продукт — фосфоритную
муку. Химическая переработка фосфатов производится с помощью
минеральных кислот, чаще всего серной, затем азотной и фосфор-
ной. Последняя, в свою очередь, является промежуточным продук-
том переработки фосфорных руд. В некоторых случаях применяют
также соляную кислоту. К химическим относятся и способы пере-
работки фосфатов при высоких температурах (1200—1800°), так
называемые термические методы (электротермическая возгонка
фосфора, термическое разложение щелочными и щелочноземель-
ными соединениями, гидротермическое обесфторивание).
Фосфоритная мука применяется в качестве удобрения на кис-
лых почвах, и, кроме того, как нейтрализующая добавка к супер-
фосфату (см. гл. XXIV). Она эффективна на торфяных почвах,
предназначенных для пастбищ и сенокосов. По масштабам воз-
можного потребления это соответствует 10% от общей потребности
в Р2О5. Для производства фосфоритной муки пригодны руды, фос-
фатное вещество которых способно растворяться в кислых почвен-
ных растворах и в слабой фосфорной кислоте. Такой способностью
не обладают апатитовые руды, пластовые фосфориты Каратау
Древнекембрийского возраста и им подобные, содержащие фосфат-
ное вещество в явно кристаллической форме50’51.
Установлено 52-53, что с уменьшением величины элементарных
зерен фосфорита или с увеличением общей их удельной поверх-
ности (стр. 839) растут химическая активность и агрономическая
810 Гл. XXIII. Природные фосфаты и фосфоритная мука
Фосфорные руды
811
ценность фосфорных руд. Для производства фосфоритной муки
используются платформенные желваковые и ракушечные фосфори-
ты Егорьевского, Вятского, Полпинского, Эстонского и других
месторождений, отвечающие указанным требованиям54"56.
Фосфоритная мука является часто местным удобрением (без
дальних перевозок). В этих условиях для ее приготовления допу-
скается применение низкокачественного сырья37, получаемого ме-
тодами простого механического обогащения руды (грохочения, от-
мывки, сепарации), а иногда даже без обогащения. При химиче-
ской, в особенности кислотной переработке фосфатов большое
значение имеет состав минералов-примесей. Наиболее вредны кис-
лотно-растворимые минералы, содержащие полуторные окислы
(нефелин, глауконит, лимонит, каолинит), а также соединения
магния (доломит и др.). Для уменьшения содержания примесей
фосфоритные руды подвергают механическому обогащению 57'58.
Первичное обогащение фосфоритов состоит в разделении руды по
крупности — более крупные фракции содержат обычно больше
Р2О5 и, соответственно, меньше примесей.
Концентраты первичного обогащения могут проходить более
глубокое вторичное обогащение методом флотации 59-63, применяе-
мым для фосфоритов Каратау, Кингисеппских и др.64-66. Однако
он еще недостаточно широко используется для обогащения фосфо-
ритов различных других месторождений.
Положительные результаты обогащения некоторых фосфоритов
получены в лабораторных условиях сочетанием флотации с магнит-
ной сепарацией, а также применением электростатических мето-
дов. Рядовой концентрат Лопатинской рудомойки, содержащий
7—10% Fe2O3 и 3—4% А120з, может быть обогащен методом маг-
нитной сепарации и флотации67 с получением флотоконцентрата
состава: 28—28,3% Р2О5, 2,2—2,4% Fe2O3, 1,1 —1,2% А12О3. Выход
продукта, пригодного для химической переработки, составляет
36—44%.
Электростатическое обогащение может оказаться экономичнее
флотации 68’69. Этим методом в лабораторных условиях получен69
из Маардусской руды с 11,2% Р2О5 концентрат, содержащий 26—
27% Р2О5 при извлечении 86—87%.
Предложены различные методы обогащения карбонатсодержа-
щих фосфоритов— прокаливанием с последующим гашением 70’7V
и удалением (отмывкой) кальция в виде гидроокисей или карбо-Д
ната (при гашении с карбонизацией), извлечением карбонатов72-7т|
при помощи растворов хлоридов или сульфатов под давлениемЯ
хлорированием хлором или хлористым водородом с последую-^
щим выщелачиванием образующихся не летучих хлоридов74
и др.
Возможно также химическое облагораживание природных фос-
фатов, предложенное первоначально для отделения апатита от
Эксплуатируемых и настоящее время месторождений Джацытас, Акса® и Чулактау.
812
Г л. XXIII. П риродные фосфаты и фосфоритная мука
ТАБЛИЦА 58
Требования к составу фосфоритов по ОСТ 10131—39
(Содержание компонентов в %)
Вятские Актю- бин- ский Егорьевские — ЛопатинскиЙ рудник Егорьев- ские — Егорьев- ский рудник
I сорт II сорт рязаиски-й горизонт портландский горизонт
I сорт II сорт
Р2О5, не менее . . . 25,3 23,5 19 22,7 22,5 19 21,7
R2O3, не более . . . 6,5 Не нор- мировано 4 Не нор- мировано 7,5 Не нор- мировано Не нор- мировано
Влага, не более . . . 10 12 9 14 8 20 • 14
нефелина растворением последнего в слабой соляной кислоте7В’ Ч
В дальнейшем было найдено, что при обработке измельченного
фосфорита Каратау разбавленной серной кислотой доломит разла-
гается с высокой скоростью, и часть магния переходит в раствор,
фосфатный же минерал растворяется в незначительной степени 77-79.
Полученный этим способом концентрат содержит 29—30% Р2О5
и лишь 0,5—1,0% СО2. Декарбонизация фосфорита Каратау может
быть также осуществлена прокаливанием80 при 1200° в течение
15—30 мин.
При обработке измельченного фосфорита хлористым водоро-
дом81-83 при 350—400° образующиеся пары FeCl3 и избыток хлори-
стого водорода охлаждают. Конденсирующийся FeCl3 отделяют, а
Химический состав фосфатного сырья важней
Марокко............................
Алжир .............................
Тунис (Гафза)......................
Египет (Сафага)....................
США —
Флорида (морские галечники) . . .
Теннесси ......................
Западные штаты ................
РаО5 FejOa А12О3
35,11 0,12 0,45
30,60 . 0,25 0,25
30,55 0,55 0,74
31,05 4,28 0,38
30-36 0,7-2,6 1,0
30-36 2,2-3,4 1,2-2,7
27-36 0,5-2,1 0,5-1,9
хлористый водород используют
уменьшает скорость растворения
снова. Прокалка при 400—800°
глауконита в кислотах7, в связи
Фосфорные руды
813
с чем этот прием может быть использован при кислотной перера-
ботке содержащих железо фосфатов (см. гл. XXV).
Прокаленный фосфорит легче измельчается и позволяет полу-
чать дальнейшей переработкой более чистые продукты84.
ТЛетод флотации широко используется для обогащения апатито-
нефелиновой руды. В результате флотации получается высококаче-
ственный апатитовый концентрат (см. табл. 54). Тонкость его по-
мола определяется требованиями главного потребителя — супер-
фосфатной промышленности85. Для повышения производительно-
сти флотационных фабрик предложена флотация руды более круп-
ного помола с разделением концентрата на стандартный и на более
крупнозернистый продукт, который тоже может найти применение
для химической переработки 86'87. Процессы обогащения апатито-
нефелиновой руды связаны с уносом апатитовой и нефелиновой
пыли с воздухом из аспирационных систем и с газами из сушиль-
ных установок. Для улавливания этой пыли в последние годы стали
применять эффективные пенные газопромыватели 88’89. После по-
вторного обогащения нефелиновых хвостов получается нефелино-
вый концентрат, содержащий 29—30% А12О3. Он комплексно пере-
рабатывается на глинозем, цемент, соду и поташ (см. гл. VI).
Апатитовый концентрат по ГОСТ 5.1188—72 должен содержать
(в пересчете на сухое вещество) не менее 39,4% Р2О5; влаги 1,0 ±
±0,5%; остатка на сите с сеткой № 016 К не более 11,5%; полутор-
ных окислов R2O3(FeO, Fe2O3, А12О3 на сухое вещество) не более
3,0% по требованию потребителей.
Требования к фосфоритам по ОСТ 10131—39 приведены в
' абл. 55.
В табл. 56 приведен химический состав фосфатного сырья не-
которых зарубежных месторождений.
ТАБЛИЦА 56
шнх зарубежных месторождений (в %)31
СаО MgO SO3 Фтор NaaO 4- К2О S1O2 Вода + органическое вещество
53,00 0,16 1,40 4,24 1,35 0,86 1,63
50,75 1,00 2,05 — — 1,65 7,25
49,68 0,99 2,46 2,86 1,81 1,64 3,69
48,74 0,35 2,70 3,21 1,04 3,81 3,24
46-50 0,1-0,6 0,2-1,5 0,1-3,9 0,1-1,1 7-10 1,6-3,0
42-49 0,02-0,3 0,6-2,7 3,2-3,8 0.3-1,0 5-9 1,4-2,0
44-52 Следы 0,3-3,1 2,9-3,9 0,3-1,4 5-17 0,7-4,7
Фосфориты Каратау, используемые в настоящее время как
промышленное сырье, должны содержать: для термической
814
Гл. XXIII. Природные фосфаты и фосфоритная мука
переработки не менее 23% Р2О5 (ГОСТ 11901—66) и для кислот-
ной— не менее 28% (МРТУ—6—12—9—66). Кроме того, в сырье,
применяемом для кислотной переработки, не должно содержаться
более 2% MgO,.3% R2O3 и 5,5% СО2.
На комбинате «Каратау» фосфатное сырье для кислотной
переработки производится как путем сухого размола высококачест-
венных руд, так и обогащением менее богатых (рядовых) фосфо-
ритных руд. При этом даже из фосфоритов со средним содержа-
нием 23,3% Р2О5 и 3,6% MgO при существующих методах обога-
щения получается флотационный концентрат, содержащий лишь
27,9% Р2О5 и 2,4% MgO. Помимо этого, обогащение фосфоритов
Каратау является дорогостоящим процессом и связано со значи-
тельными потерями сырья. Товарное извлечение Р2О5 во флотаци-
онный концентрат не превышает 63—65%, т. е. при обогащении
теряется до 35% фосфатного вещества. Хвосты обогатительной
фабрики, содержащие 16—18% Р2О5 и 4—6% MgO, не исполь-
зуются. Себестоимость 1 т Р2О5 во флотационном концентрате в
2,5—3 раза больше, чем в фосфоритной муке сухого помола из
исходной руды.
Многие природные фосфаты содержат уран92. Так, в США оса-
дочные фосфориты Флориды, Айдахо, Уайоминга, Монтаны и дру-
гих содержат до 0,018% ПзО8; Южной Каролины — от 0,001 до
0,15% U3O8; в фосфоритах Северной Африки — марокканских,
алжирских, тунисских и египетских — от 0,004 до 0,016% изО8; в
фосфоритах Тихого и Индийского океанов от 0,001 до 0,007%
U3O8; в изверженных породах — апатитах Южной Африки, Норве-
гии и других — от 0,001 до 0,03% U3O8. Природные фосфаты яв-
ляются значительным источником урана, несмотря на малое его
содержание, так как количество перерабатываемых фосфатов из-
меряется десятками миллионов тонн в год. Апатитовый концентрат
содержит в качестве полезной примеси около 0,9% лантанидов и
других редких элементов (лантан, церий, ниобий, иттрий и др.).
Физико-химические и механические свойства
Заводы фосфорных удобрений перерабатывают большое коли-
чество порошковидного фосфатного сырья — апатитового концен-
трата и фосфоритной муки Каратауского и Кингисеппского место-
рождений. Потребление фосфатного сырья на крупных заводах до-
стигает 400 000?' в год, а запас на складе до 10 Q00 т. Правиль-
ная организация транспортирования и хранения фосфатного сырья,
обеспечивающая минимальный размер капиталовложений и экс-
плуатационных расходов, зависит от выбора технических средств,
основанных на использовании физико-механических свойств сырья.
Транспортирование и особенно складская обработка мелко-
дисперсного порошковидного фосфатного сырья связаны со зна-
Фосфорные руды
815
чительным пылением. Поэтому транспортирование и хранение
сырья должны быть организованы с обеспечением нормальных
санитарных условий труда и минимальных потерь сырья.
Ниже приводятся некоторые физико-химические и механиче-
ские данные для апатитового концентрата и фосфоритной муки
КаратаУ- Сведения о фосфоритной муке, как готовом продукте,
приведены на стр. 819.
Твердость фосфоритных желваков57 находится в пределах от
2 до 5, а зерен чистого апатита равна 5 (по шкале Мооса). Тем-
пература плавления апатита 1500—1570°; стандартная теплота об-
разования при 25° равна 28 ккал/моль. Теплоемкость93 апатитового
концентрата 0,187 кал!(г•град).
Объемный вес апатитового концентрата 94-95 при небольшом
увеличении его влажности резко уменьшается. В среднем для стан-
дартного продукта объемный вес в кучах принимается 1,8 т/м3, а
на транспортерах и в малых бункерах 1,5 т/м3.
Гранулометрический анализ апатитового концентрата85 дан в
табл. 57.
ТАБЛИЦА 57
Характеристика дисперсности апатитового концентрата
Ситовый анализ Удельная поверхность фракции, см^/г Поверхность фракции
размеры частиц, мк содержание, % смеси, смУг в % от суммарной поверхности
+ 210 0,5 260 1,3 0,09
-210 +130 10,8 290 31,8 2,21
-150 +100 22,1 359 75,4 5,25
—100 + 74 14,5 481 69,7 4,88
-74 + 40 7,8 721 56,2 3,96
-40 44,3 2718 1204,0 83,61
1438,4 100,00
Поверхность наиболее тонких классов (—74 мк) достигает
80—90% от удельной поверхности всех фракций, их количество со-
ставляет 45—50% по весу. Следует различать внешнюю удельную
поверхность частиц измельченного фосфата (табл. 57) и общую
(внутреннюю) удельную поверхность элементарных зерен 52’59,
определяемую адсорбцией азота при —195°.
Фосфоритная мука месторождения Чулактау стандартного по-
мола при влажности 0,1—0,3% имеет объемный вес (в т/м3): на
ленте транспортера 1,1 —1,2, свеженасыпанная в вагоны 1,45—1,5,
после транспортирования по железной дороге до 1,6, после дли-
тельного хранения на складе 1,8—2,096. Сухая фосфоритная мука
'Улактау очень подвижна, растекается подобно жидкости,
ссыпаясь под углом 15—20°. Увлажненная до 0,75—1,5% мука
816
Гл. XXIII. Природные фосфаты и фосфоритная мука
теряет текучесть и способна слеживаться. Фосфоритная мука Чу-
лактау, в отличие от фосфоритной муки всех других месторожде-
ний, гигроскопична.
Влажность, %
Рис. 212. Зависимость
угла откоса апатито-
вого концентрата от
влажности.
Флотреагенты; I— торфя-
ная смола; 2 и 3 — жидкое
мыло. Помол: 1 п2 — гру~
бый; 3 — тонкий.
При хранении ее влажность увеличивается до
0,75—1,5°/о- Сильно увлажненная мука при-
обретает консистенцию жирной глины и после
высыхания некоторые ее разновидности пре-
вращаются в комья, требующие измельчения.
Углы покоя (естественного откоса) для
апатитового концентрата94 показаны на
рис. 212. С увеличением влажности углы
покоя возрастают. Для стандартного кон-
центрата принимается средний угол по-
коя 37°.
Сыпучесть апатитового и нефелинового
концентратов по шкале Пестова95 приведена
на рис. 213. На сыпучесть концентратов так-
же оказывает большое влияние влажность
продуктов.
Изучены также другие физико-механиче-
ские свойства апатитового концентрата и фос-
форитной муки, знание которых необхо-
димо, в частности для выбора средств их
транспортирования и хранения97. Некоторые
дополнительные свойства для апатитового
концентрата с влажностью 0,1%: коэффициент динамического тре-
ния 1,20; угол внутреннего трения 31°; коэффициент внутреннего
трения 0,68; коэффициент трения о сталь 0,58; коэффициент трения
о дерево 0.60; коэффициент трения о
резину 0,63; коэффициент трения о про-
резиненный бельтинг 0,65; начальное
сопротивление сдвигу без слеживания
6—55 кгс/м1-, плотность 3,22; коэффи-
циент истечения 0,2. При лежании апа-
титового концентрата в течение 3 суток
его свойства и значения коэффи-
циентов трения практически не из-
меняются.
Коэффициент уплотнения фосфо-
ритной муки (кроме каратауской) при
размере частиц 0,7—0,1 мм и влажно-
сти 1,5% — 1,21; угол естественного
Рнс. 213. Относительная сыпу-
честь апатитового и нефелино-
вого концентрата:
/ — нефелин; 2 — апатит.
откоса 40—45°; коэффициент трения о сталь 0,3; коэффициент тре-
ния о бетон 0,5.
Растворимость природных фосфатов в почвенных растворах к
эффективность их в качестве удобрений зависит не только от то-
нины помола и состава фосфата, но и от общей удельной поверх-
Фосфорные руды
817
нести. Хибинский апатит обладает очень низкой общей удельной
поверхностью и поэтому он малоэффективен как удобрение. Эф-
фективность муки из Кольского апатита увеличивается, если вместо
стандартной, состоящей на 80% из частиц меньше 0,15 мм, при-
менять муку более тонкого помола98 (с содержанием 80% частиц
меньше 0,06 мм).
Разные виды фосфатного сырья разлагаются кислотами с раз-
личной скоростью. Количественная характеристика химической
активности определена для некоторых видов фосфатов при разло-
жении их кислотами 99-102, а также при термическом их восстано-
влении103. Однако систематических данных о реакционной спо-
собности разных фосфатов не имеется. Реакционная способность
природных фосфатов при кислотном их разложении обычно умень-
шается после нагревания их вследствие спекания зерен и роста
кристаллов104. Разложение кислотами облегчается, если природ-
ный фосфат содержит некоторое количество двуокиси углерода.
Транспортировка и хранение фосфатного сырья
Применяемая до сих пор перевозка фосфатного сырья в кры-
тых железнодорожных вагонах общего назначения является мало
рациональной вследствие вытекания сырья в пути через неплот-
ности и пыления при загрузке и разгрузке. Это создает тяжелые
условия труда и потери сырья. Для уменьшения потерь сырья при
перевозках пол вагона выстилают бумагой и около дверей уста-
навливают деревянные или камышевые щиты. Однако при раз-
грузке бумага разрывается и щиты разрушаются, что приводит
к загрязнению сырья и необходимости его дополнительного просеи-
вания. Разгрузка фосфатного сырья из таких вагонов осущест-
вляется иногда еще вручную, а чаще электромеханическими ло-
патами с дистанционным управлением, производительностью до
30 т/ч. Применение электромеханических лопат позволило значи-
тельно уменьшить количество рабочих по сравнению с ручной раз-
грузкой, но это не исключает пыления и тяжелых условий труда.
Улучшение условий труда при разгрузке фосфатного сырья дости-
гается использованием пневматических разгрузочных установок,
аналогичных применяемым для разгрузки цемента. Производитель-
ность таких разгрузчиков, разработанных Ленинградским филиа-
лом ВНИИстройдормаша, составляет 70—100 т/ч.
Этот способ, хотя и обеспечивает беспыльную разгрузку мате-
риала, однако громоздок, требует значительного расхода энергии
1~3 квт-ч на 1 т фосфатного сырья). Для разгрузки эшелона
(маршрута) в 1500 т в течение 5 ч требуются три разгрузчика с
°бщей мощностью, равной 1350 кет.
На Самаркандском суперфосфатном заводе опробован для раз-
РУзки каратауской фосфоритной муки разгрузчик цемента типа
818
Гл. XXIII. Природные фосфаты и фосфоритная мука
С-377. Производительность его на фосфоритной муке составила!
35 т/ч, т. е. на 15 т/ч меньше, чем на цементе; расход электро-!
энергии на разгрузку сырья (без подачи на склад) составил
1,5 кет ч/т. я
Пневматические разгрузочные устройства могут быть примене!
ны на заводах, располагающих достаточными ресурсами электрии
ческой энергии в качестве временного средства при транспортире!
вании сырья в вагонах общего типа. Радикальным решением!
проблемы является транспортирование фосфатного сырья в специ||
альных саморазгружающихся вагонах бункерного типа или в пневи
матических цистернах (пневморезервуарах). ']
Саморазгружающиеся 60-тонные цельнометаллические вагоны5
бункерного типа с двухсторонней боковой выгрузкой могут быть
загружены в течение 6—7 мин и разгружены с применением ви-
браторов за 8—10 мин. При загрузке вагонов не требуется пред-
варительной подготовки (выкладывание бумагой и др.), а после =
разгрузки нет надобности в зачистке. В пути не происходит утечки!
сырья, а разгрузка вагона производится автоматически, с пневма-
тическим открыванием разгрузочных люков дистанционными при-1
способлениями. Для перевозки фосфатного сырья на небольшие
расстояния могут быть использованы пневматические цистерны. Из]
них можно производить разгрузку непосредственно в склад и]
вследствие этого они позволяют почти полностью исключить пыле-1
ние материала. I
Фосфатное сырье из железнодорожных вагонов выгружается в|
прирельсовые скреперные траншеи, расположенные по обе стороны!
железнодорожного пути. Две траншеи вмещают по 1500 т фосфат-1
ного сырья и имеют длину, позволяющую одновременно разгру!
жать четыре 50-тонных вагона общего назначения или пять 60-тон!
ных саморазгружающихся вагонов. По окончании разгрузки
железнодорожного состава траншеи опорожняются скреперами ем!
костью 1 м3, подающими сырье в устройства для отделения приме!
сей (щелевые бункера и др.) и для транспортировки на склад-1
элеваторами и ленточными конвейерами.
Хранение сырья на ряде заводов производится в старых склада!
амбарного типа, в которых вследствие значительного пыления мя
териала создаются тяжелые условия труда как на самом склад»
так и на прилегающей к нему территории. На новых заводах col
оружаются склады силосного типа, которые, хотя и требуют боль!
ших капитальных вложений, но обеспечивают улучшение условии
труда и предотвращают потери сырья. I
Силосные склады устраиваются в виде трех или шести железо!
бетонных башен внутренним диаметром 11,5 м и высотой 21,4 л!
общей вместимостью в среднем 10 000 или 20 000 т сырья. А эрг!
рование сырья в силосах производится сжатым воздухом, которы!
Фосфоритная мука
819
подводится под пористые плиты, помещенные в нижней части си-
лоса (днище).
Подача сырья из скреперных траншей в силосы и внутрицеховое
перемещение его средствами конвейерного транспорта — ленточны-
ми и винтовыми конвейерами и элеваторами — связаны с большим
числом пересыпок (перегрузок) материала и приводят к значитель-
ной запыленности помещений и потерям сырья. Аспирация обору-
дования, укрытие мест пересыпок и ленточных конвейеров не
исключает запыленности и связаны с дополнительными затратами.
Более рациональным является пневмотранспорт 106 сырья. Для
этого силосные склады оборудуются сдвоенными стационарными
камерными пневмонасосами с верхней выгрузкой и автоматиче-
ским управлением. Производительность их достигает 50 т/ч при
высоте подъема 40 м. Пневматический транспорт требует большего
расхода электроэнергии, чем конвейерный, но позволяет улучшить
условия труда и уменьшить потери сырья.
ФОСФОРИТНАЯ МУКА
влагоемкость ее 3,7%. При
Рис. 214. Рассеваемость фос-
форитной муки.
Фосфоритная мука —сухой, пылящий, тонкий порошок серого,
желтоватого, бурого и других цветов. Она не гигроскопична, не
слеживается, но рассевается механическими сеялками не доста-
точно хорошо (рис. 214). Предельная
этой влажности фосфоритная мука
почти теряет свою сыпучесть94.
Фосфоритная мука относится к
трудноусвояемым удобрениям. До-
стоинством фосфоритной муки являет-
ся длительность (в течение ряда лет)
ее действия. Фосфоритование почв —
один из важнейших приемов повыше-
ния плодородия 107~109. Для увеличе-
ния эффективности фосфоритной муки
предложена 110 ее обработка неболь-
шими дозами кислот (4—8% от веса
муки). Удобрительное действие фос-
форитной муки зависит от тонины размола. Стандартом (ГОСТ
5716—65), не распространяемым на муку Каратау, предусматри-
вается остаток на сите 0,18 мм не более 10%. Фосфоритная мука
выпускается четырех сортов: высший сорт с содержанием Р2О5 не
менее 30% (получается при флотации фосфоритов), I сорт—не
менее 25% Р2О5, II сорт — не менее 22% Р2О5 и III сорт не менее
/о Р2О5. Содержание влаги в высшем сорте не более 1,5%, в
остальных — до 3 %.
Основными производителями фосфоритной муки, применяемой в
качестве удобрения, являются Егорьевский, Брянский, Допатинский
820
Гл. XXIII. Природные фосфаты и фосфоритная мука
и другие фосфоритные рудники. Фосфоритная мука, выпускае-
мая на комбинате «Каратау» и Кингисеппском комбинате «Фос-
форит», используется в основном как сырье для производства
удобрений. 1
Производство фосфоритной муки из природного фосфата может!
быть осуществлено в зависимости от состава сырья или без его
обогащения, или путем более глубокого обогащения флотацион-
ными методами. Процесс переработки руды без ее обогащения
заключается в следующем57’9"’111""'1. Руду подвергают сначала
грубому дроблению (при размере кусков свыше 50—100 мм) обыч-
но в щековых или молотковых дробилках. Так как фосфоритная
руда, поступающая на переработку, обычно имеет влажность 7—
16%, то для дальнейшего измельчения и для получения стандарт-
ного продукта руду высушивают до содержания 2—3% влаги.
Фосфорит сушится топочными газами в противоточных барабан-
ных сушилках. Барабан, установленный под углом до 6°, делает
1—8 об/мин. Скорость прохождения газов через барабан состав-
ляет 1,5—2,0 м/сек. В зависимости от влажности руды температура
на входе в сушильный барабан поддерживается в пределах 500—
750°. Температура отходящих газов 100—120°. Сушильные бара-
баны снабжены внутри насадкой, обеспечивающей пересыпание
материала. Съем влаги с 1 м3 объема сушильного барабана 40—
60 кг/ч. Диаметр сушильных барабанов 1,2—2,8 м при длине
3,5—7 диаметров. При средней влажности руды 14% производи-
тельность сушилки диаметром 1,5 м и длиной 14 м (объем 24,8 м3)
300—350 т/сутки.
Высушенный фосфорит дробится молотковыми дробилками до
кусков размером не более 10—15 мм. Такое предварительное дроб-
ление необходимо для подготовки материала к тонкому измельче-
нию в мельницах. Для измельчения применяют шаровые мельницы
с производительностью 15—24 т/ч фосфоритной муки. На неболь-
ших установках используются мельницы кольцевого типа с про-
изводительностью 2,5—3,5 т/ч. Применение электроимпульсной
дробилки позволяет получать фосфоритную муку с размером ча-
стиц меньше 50 мк и с более высокими удобрительными свой-
ствами, чем у обычной И5.
На рис. 215 приведена схема производства фосфоритной муки,
на которой не показано крупное и мелкое дробление руды. Фос-
форитная мельница работает в замкнутом цикле с воздушным
сепаратором: измельчаемый материал из шаровой мельницы 11
непрерывно подается элеватором 12 в сепаратор 13, где крупные
частицы (крупка) отделяются от готовой муки. Последняя пере-
дается шнеками 14 в силосное хранилище, а крупка возвращается
на размол в мельницу. Запыленный воздух очищается в рукавных
фильтрах 15 или циклонах и выбрасывается вентилятором 16
в атмосферу.
Фосфоритная мука
821
Первичное обогащение руды производят путем тщательной дез-
интеграции ее и промывки. Из руды выделяют желваки, гальку
й фосфат, а глину и песок смывают водой. Первичный концентрат
Егорьевского фосфорита с размером частиц +0,5 мм содержит118
до 22% Р2О5 и является окончательным продуктом обогащения,
удовлетворяющим требованиям ГОСТ. Процесс производства фос-
форитной муки в этом случае заключается в дроблении, обогаще-
нии и сушке концентрата.
В некоторых случаях обогащение руды проводят в две стадии.
В первой — методами рудомойки выделяют первичный концентрат;
Рис. 215. Схема производства фосфоритной муки:
1 — хвостовой вентилятор; 2 — канатная дорога; 3 —бункер; 4 — загрузочная камера сушилки;
5 —сушильный барабан; 6 — выгрузочная камера сушилки; 7 —топка; 8 — элеватор; 5 —бункер;
10— питатель; 11 — шаровая мельница; /2 —элеватор для фосфоритной муки; 13 — сепаратор;
14 — шнек для фосфоритной муки; /5 —рукавный фильтр; 16 — вентилятор фильтра.
оставшиеся хвосты рудомойки, содержащие до 7,5% Р2О5, подвер-
гают во второй стадии флотации. Выход концентрата из хвостов
составляет ~38,0%. Готовый концентрат после сушки содержит
2—3% влаги. Предварительная (до флотации) магнитная обра-
ботка воды и пульпы интенсифицирует процесс и улучшает техно-
логические показатели117. Бедные оболовые руды, содержащие
5—10% Р2О5, не поддаются первичному обогащению. Их перера-
батывают флотационным методом с получением концентрата или
так называемой флотированной фосфоритной муки.
На рис. 216 показана схема производства фосфоритной муки,
получаемой флотационным обогащением фосфоритной руды. Про-
цесс осуществляют в три стадии. В первой — основной флотации —
получают промежуточный концентрат путем отделения из руды
основной массы пустой породы — хвостов. После классификации
и отстаивания хвосты (пески) откачиваются в хвостохранилище,
а жидкая среда — слив из сгустителей используется в качестве
оборотной воды на последующих стадиях процесса. Промежуточ-
ный концентрат основной флотации направляют на вторую
Методы и продукты химической переработки фосфатов
823
стадию — первую и вторую перечистные флотации. Обедненная
фракция первой перечистки возвращается на основную флотацию
в первую стадию. Обогащенная фракция первой перечистки под-
вергается второй перечистке с получением концентрата и хвостов,
направляемых на третью стадию — катионную флотацию и пере-
чистку. Концентраты второй и третьей перечисток после сгущения-
фильтруют и полученную лепешку высушивают.
Фосфоритная руда из бункера для руды через качающийся
питатель ленточным транспортером подается на молотковую дро-
билку. На транспортере устанавливают магнитный сепаратор.
Дробленая руда через ленточные весы направляется в бункер,
откуда она при помощи тарельчатого питателя дозируется в шаро-
вую мельницу мокрого помола. Сюда же поступает «хвостовой»
слив (пески) с третьей флотации, а также растворы соды и жид-
кого стекла, необходимые в процессе первой (основной) флотации.
Пульпа из шаровой мельницы насосом перекачивается в пульподе-
литель, куда подается также слив со второй стадии флотации —
обедненный продукт первой перечистки. Далее пульпа смешивается
с флотационными реагентами — керосином и талловым маслом и
поступает на первую — основную флотацию. Здесь получается
промежуточный черновой концентрат и отделяется пустая порода,
которая откачивается после классификации в хвостохранилище.
Черновой концентрат направляется на флотационные машины для
первой и второй перечистных флотаций. Концентрат анионной
флотации (второй перечистки) двухспиральным классификатором
разделяется на слив, который откачивается на сгущение, и фрак-
цию песка, которую после обработки серной кислотой направляют
на катионную флотацию и перечистку в присутствии катионного
реагента ИМ-11. В результате катионной флотации получается
отброс — «пески» и камерный продукт, который обезвоживается,
и затем присоединяется к сгущенному концентрату анионной фло-
тации. Концентраты фильтруют и высушивают в сушильном ба-
рабане.
МЕТОДЫ И ПРОДУКТЫ ХИМИЧЕСКОЙ ПЕРЕРАБОТКИ ФОСФАТОВ
Все способы химической переработки фосфатного сырья можно
разделить на две группы:
1) прямая обработка сырья с непосредственным получением
готовых продуктов;
2) разложение сырья с выделением промежуточных продук-
тов — фосфора и фосфорной кислоты, используемых для дальней-
шего производства разнообразных конечных продуктов.
Для переработки фосфатов применяют как кислотные, так и
термические методы.
иНепосредственной обработкой фосфатного сырья серной кисло-
той получают простой суперфосфат, фосфорной кислотой — двойной
2 М. Е. Позии
824
Гл. XXIII. Природные фосфаты и фосфоритная мука
(или тройной), смесью серной и фосфорной кислот — обога-
щенный (или полуторный). Все эти продукты в отличие от при-
родных фосфатов содержат фосфорные соединения, растворимые
в воде. Они служат в качестве фосфорных удобрений и разли-
чаются по содержанию усвояемой Р2О5: в простом суперфосфате
ее—14—21%, в обогащенном 28—32% и в двойном 42—48%
(до 50—55%). При прямой термической обработке фосфатов такЙ
же получают непосредственно различные фосфорные удобрения
фосфаты, используемые для технических целей — обесфторенныЯ
фосфат (кормовой и удобрительный), содержащий 23,5—38% P2OJI
плавленые магниевые фосфаты (19—21% Р2Р5), щелочные терме
фосфаты (18—28% Р2О5), а также отходы металлургии — основный
шлаки, содержащие 6—20% Р2О5. Я
Промежуточными продуктами переработки природных фосфЯ
тов являются фосфор и фосфорная кислота. Перевозка фосфоЯ
ной кислоты и, в особенности, фосфора дешевле, чем получаемый
из них продуктов, поэтому их выгоднее транспортировать заво*
дам-потребителям и там перерабатывать.
Элементарный (желтый) фосфор получают в настоящее время
исключительно электротермическим путем. При окислении фос-=
фора воздухом образуется фосфорный ангидрид, который взаимо-
действием с водой превращается в фосфорную кислоту, называе-
мую термической кислотой. Более экономичным, а поэтому и более
распространенным является производство экстракционной фосфор-
ной кислоты, получаемой экстракцией (извлечением) ее из фос-
фатов серной кислотой. При использовании для этой цели азотной
или соляной кислоты получают азотнокислотную или солянокис-
лотную вытяжку фосфатов. Последние, наряду с фосфорной кисло-
той, содержат также растворенные нитрат или хлорид кальция.
Фосфорная кислота является важнейшим полупродуктом в про-
изводстве концентрированных фосфорных и сложных удобрений,।
а также технических фосфорнокислых солей. Она служит для про-i
изводства двойного (и тройного) и обогащенного суперфосфатов,
аммофоса, аммонитрофоски, технических фосфатов кальция, нат-;
рия, аммония, полимерных и железомарганцевых фосфатов, а так-
же кормовых фосфатов (преципитата, монокальцийфосфата), до-
бавляемых в корм скоту и др.
Фосфор и фосфорный ангидрид применяют также для произ-
водства сульфидов и хлоридов фосфора, фосфорорганических
соединений и др.
ЛИТЕРАТУРА
1. М. П. Ф ивег, Труды НИУ, в. 109, 1932. — 2. Агрономические руды СССР,
Труды НИУИФ, в. 99 и 100, т. 1, 1932; в. 115 и 116, т. 2, 1934, в. 124 и 125, т. 3,
1934; в. 138, т. 4, 1937; в. 146, т, 5, 1939; в. 149, т. 6, 1941. —3. Б. М. Г им мел ь-
ф а р б, Агрономические руды, Изд. АН СССР, 1938. — 4. Н. С. Ульянов, Хим.
пром., № 7, 430 (1957). — 5. Б. М. Г и м м е л ь ф а р б, сб. «Промышленные ми-
Литература
825
неральные удобрения», Госхимиздат, 1958, стр. 167. — 6. И. Д. Борнеман-
Старынкевнч, Н. В. Белов, ДАН СССР, 19, № 4 (1938); 22, № 2 (1939);
26, Ns 8 (1940); 90, № 1 (1953). — 7. М. Л. Ч е п е л е в е ц к и й, Труды НИУИФ,
в.'137, 1937. — 8. Г. А. Манакин, Научн. зап. Одесск. политехи, ин-та, 2, № 1,
3 (1955). — 9. С. И. В о л ь ф к о в и ч, Л. Б. Г р и н ш п а н, А. Б. Ш е х т е р, ДАН
СССР, 85, 137 (1952). — /0. М. Е. К у п е р м а н, Э. В. Водзинская,
Л. Б. Гриншпан, Сообщения научно-техн. раб. НИУИФ, № 3, 63 (1957).
//.А. В. Казаков, Труды НИУИФ, в. 139, 1937; в. 145, 1939,—
/2. А. В. К а з а к о в, Проблемы советской геологии, № 7 (1937). — /3. Г. И. Бу-
шинский, Апатит, фосфорит, вивианит, Изд. АН СССР, 1952. — 14. Г. И. Бу-
ши н с к и й, Изв. АН СССР. Сер. геол., Ns 6 (1954); № 1 (1955). —/5. Р. П. О з е-
пов, Л. Б. Гриншпан, Г. И. Бушинский, Зап. Всесоюзи. Минер, об-ва,
X» 3, 303 (1956). — 16. Б. М. Масленников, Ф. А. К а в и ц к а я, ДАН СССР,
109, № 5 (1956). —/7. М. П. Фи вег, А. П. Шубин, Апатиты, ОНТИ, 1937.—
18. Я. В. Самойлов, Труды Комиссии по исследованию фосфоритов, т. 3, 4,
6, 7, 1911—1915. — 19. А. А. Ш у г и н, Труды НИУ, в. 125, 1934. — 20. А. А. С а у-
ков, Геохимия, Госгеолиздат, 1951.
21. Б. М. Г и м м е л ь ф а р б, Что такое фосфориты, где и как их искать,
Госгеолтехиздат, 1954. — 22. Б. М. Гиммельфарб, И. М. К У Р м а н, В. А. О к-
н и н а, А. И. Смирнов, Ц. И. У ф л я н д, Методика поисков фосфоритов, Гос-
химиздат, 1956.—23. К- S z р i 1 a, Arch. Mineralog, 26, № 12, 99, 1965 (1966).—
24. А. Е. Ферсман, Полезные ископаемые Кольского полуострова, Изд. АН
СССР, 1941. — 25. Апатито-нефелиновые месторождения Хибинских тундр. Труды
НИУ, в. 89, сб. 1, 1932. —26. Хибинские апатиты. Труды НИУИФ, в. 128, сб. 2,
1936. — 27. А. Н. Стрельцов, В. А. Смирнов, Ю. Я- Туров, В. Е. Тре-
тьяков, Труды ЛенНИИгипрохима, в. 1, 1967, стр. 262. — 28. Р. Пермяков,
Эконом, газ., № 24, 18 (1967). — 29. Внешняя торговля СССР за 1965 г. (стат,
обзор), Изд. «Международные отношения», 1966.—30. Т. П. У н а н я н ц, сб.
«Исследования по химии и технологии удобрений, пестицидов и солей», Изд.
«Наука», 1966, стр. 296.
31. Е. В. Орлова, Фосфориты. Справочник по минеральным ресурсам за-
рубежных стран, т. Ш, Неметаллические ископаемые, Госгеолиздат, 1948.—
32. Б. М. Г и м м е л ь ф а р б, Сборник трудов ГИГХС, в. 2, 1955. — 33. Фосфо-
ритные руды СССР. Труды НИУ, в. 24, т. 1, 1925; в. 68, т. 2, 1930; в. 72, т. 3,
1930; в. 75, т. 4, 1931; в. 78, т. 5, 1930; в. 79, т. 6, 1931; в. 85, т. 7, 1931; в. 88,
т. 8, 1932. — 34. Н. С. Ш а т с к и й, Сборник материалов совещания по осадоч-
ным породам, т. 2, Изд. АН СССР, 1955.—35. А. Н. Дегтярев, Хим. пром.,
№ 9, 313 (1953).— 36. С. И. Вольфковпч, Фосфориты Кара-Тау, Изд.
АН СССР, 1946. — 37. Т. П. У н а н я н ц, Хим. пром., Ns 6, 350 (1955).—
38. А. Н. Стрельцов, Ю. Я. Туров, В. Е. Третьяков, Г. А. Паршина,
Труды ЛенНИИгипрохим, в. 1, 1967, стр. 253.—39. Statistical Jearbook United
Nations, 1956.— 40. Г. А. Роговских, А. И. Красовская, Минеральные
Удобрения, Рынок капиталистических стран, Внешторгиздат, 1956.
41. G. R. Mansfield, Ind. Eng. Chem., 34, № 1, 9 (1942). —42. E. В. С о к-
л а ко в а, Баланс запасов полезных ископаемых СССР на 1 января 1958 г., в. 41,
Фосфориты; в. 66, Апатит, Госгеолтехиздат, 1959. — 43. Eng. a. Mining J., 167,
Ns 12, 103 (1966). 44. J. V. Beall, Mining Eng., 18, № 10, 80 (1966).—
45. А. И. Ш e p e ш e в с к и й, Требования промышленности к качеству минераль-
ного сырья, в. 19, Фосфатное сырье (апатиты и фосфориты), Госгеолтехиздат,
1959.- 45. А. Н. Стрельцов и др., Труды ЛенНИИгипрохим, в. 1, 1967,
стр. 269. — 47. С. И. Митрофанов, Селективная флотация, Изд. «Недра»,
1967.—48. Н. С. С а з ы к и н, В. Г. Скоков, Хим. пром., № 11, 871 (1966).—
49. Б. М. Г и м м е л ь ф а р б, Т. П. У н а н я н ц, Сырьевая база туковой промыш-
ленности СССР, ОНТИ, 1937. — 50. D. McConnell, Am. Min., 23, Ns 1
(1938).
2»
826
Гл. XX///. Природные фосфаты и фосфоритная мука
51, М. Л. Чепе л евецк и й, Б, М, Гиммел ьф а рб, М. Е. Куперман,
3. В. Красильникова, ДАН СССР, 119, № 1, 133 (1958). — 52. W. L. Hill,
I. Н. Caro, G. A. Wieczorek, J. Agr. Food Chem., 2, № 25, 1273 (1954). -Л
53. F. Carbona, Ind. Chim., № 463, 41 (1956). — 54. P. Laske, Landwirtscjl
10, № 2, 114 (1957). —55. I. H. С a r o, W. L. H i 11, J. Agr. Food Chem.. 4, Nsl
684 (1956). — 56. А. С. Ч e p н а в и н, Вопр. геол, агрон. руд, Изд. АН СССИ
1956, стр. 35. — 57. Справочник по удобрениям, Госхимиздат, 1933. Я
58. Л. И. С т р е м о в с к и й, Состояние фосфоритных обогатительных фабрик
пути развития обогащения фосфоритных руд Союза, Гостоптехиздат, 1940. -Я
59. В. М. Борнео в, Хим. пром., № 1, 13 (1956). — 60. Л. М. Ч е р н ы й, Там
№ 2, 82 (1957). 1
6/, М. А. Эйгелес и др., Там же, № 8, 473 (1957).—62. Л. И. С т р я
м о в с к и й, Тезисы докладов на отраслевом совещании работников основной Я
горнохимической промышленности, 1958, стр. 100. — 63. Л. И. Стр ем о векиЯ
О. А. Розанова, Хим. пром., № 5, 144 (1951).— 64. Л. И. СтремовскиИ
О. М. Кнаус, Обогащение фосфатных пуд. Труды Гос. н.-и. ин-та горнохиЯ
сырья, в. 8,. Госгортехиздат, 1962. — 65. М. И. Б а с к а к о в а, сб. «Производителя
ные силы Южного Казахстана», т. 3, Алма-Ата, Изд. «Наука», 1966, стр. 8. Я
66. Н. Т. Цапков, М. А. Орел, Хим. пром., № 5, 391 (1967). — 67. А. В. Ml
ш ь я н о в а, Ю. М. Смирнов, И. Я. Холом янский, Там же, Ns 2, 1Я
(1967). — 68. А.. И. Ангелов, В. М. Борисов, Там же, № 4, 251 (1964).—Э
69. А. И. Ангелов, Т. А. Марк, Обогащение руд, № 2 (62), 36 (1966).—j
70. J. Hoffman, В. С. Mari ache г, Mining Eng., 13, № 5, 472 (1961). J
71. C. Ben-Ari, W. J. Fuchs, англ. пат. 908137, 1962; 908021, 1962,—
72. Э. Б. Ш т е р н и и а, Е. В. Фролова, ЖПХ, 35, № 4, 751—756 (1962).—
73. Е. R. Hermann, франц, пат. 1398563, 1965. — 74. А. Н. Кето в, В. В. П е ч-
ковский, Л. П. Костин, Изв. вузов. Хим. и хим. технол., 8, № 6, 969
(1965). — 75. Третья всесоюзная конференция работников суперфосфатной про-
мышленности. Труды НИУ, в. 113, 1933, стр. 142. — 76. С. И. В о л ь ф к о в и ч,
Изв. АН СССР. ОХН, № 6, 661 (1957); № 7, 767 (1957). — 77. М. Л. Чепеле-
в е ц к и й, Е. Б. Б р у ц к у с, В. М. Р а м м, Е. В. Южная, Е. П. В е т ч и н к и и,
Сообщ. о научно-технич. раб. НИУИФ, Ns 2, 15 (1957). — 78. М. Л. Чепеле-
вецкий, Е. Б. Бруцкус, Суперфосфат, Физико-химические основы произ-,
водства, Госхимиздат, 1958. — 79. А. С. Черняк, ЖПХ, 30, № 6, 844 (1957). —|
80. А. В. С м о р о д и н ц е в, В. А. Ш а м а р и н, 3. А. Оглоблина, Хим. пром.!
Ns 4, 280 (1966). 1
8/. Л. Владимиров, Р. Герасимович, Труды НИУИФ, в. 134,
1936.—82. Л. Владимиров, Р. Герасимович, Е. Дунаевский,
Там же, в. 143, 1938, стр. 19. — 83. L. Е. Dupont, I R. Archer, W. F. К a r-
t e г, пат. CUlA 2629650. 1953. — 84. A. A. Guffey, Chem. Eng. Progr., 58,
Ns 10, 91 (1962).— 85. Л. Б. Г p и н ш п а н, сб. «Исследования по прикладной
химии», Изд. АН СССР, 1955, стр. 192.—86. Ф. Н. Белаш, Н. С. Ульянов,
Хим. пром., Ns 1, 13 (1957). — 87. Ф. Н. Белаш, А. А. Андреева, М. А. Га-1
милов, сб. «Обогащение полезных ископаемых», в. 1, Госметаллургиздат, 1958J
стр. 24. — 88. М. Е. П о з и н, Э. Я. Т а р а т, Труды ЛТИ им. Ленсовета, в. 36;
1956. — 89. М. Е. П о з и н, И. П. М у х л е н о в, Э. Я. Т а р а т, Пенные газоочи-:
стители, теплообменники и абсорберы, Госхимиздат, 1959. — 90. А. М. Дубо-
вицкий, А. И. Шерешевский, Технология минеральных удобрений, Гос-
химиздат, 1947.
91. С. К. Воскресенский, Хим. наука и пром., № 2, 129 (1956).—
92. С. И. В о л ь ф к о в и ч, Научно-техн, информ, бюлл. НИУИФ, № 4,3 (1957).—
93. В. П. П л и г у н о в, Научн. зап. Одесск. политехи, ин-та, 2, № 1 (1955).—
94. Н. Е. Песто в, Физико-химические свойства зернистых и порошкообразных
Химических продуктов. Изд. АН СССР, 1947. — 95. Н. Е. П е с т о в, Е. И. П а у т-
кина, Хим. пром., № 7, 213 (1952); сб. «Исследования по прикладной химии»,
Литература
827
Изд. АН СССР, 1955, стр. 290. — 96. В. В. Богоявленский, Хим. пром.,
№ 6, 360 (1955). — 97. Р. Л. Зенков, Тезисы н.-техн. совещания по транспор-
тированию и хранению порошковидного фосфатного сырья для производства
минеральных удобрений, ч. II, ГОСИНТИ, 1962, стр. 6, 11. — 98. Т. Lituhski,
F. G о г 1 а с h, Przem. Chem., 40, № 2, 92 (1961). — 99. Н. L. Marshall,
L F. Rader, Ind. Eng. Chem., 25, 1253 (1933). —100. M. А. Вей дерм a,
Хим. пром., № 5, 18 (1963).
/01. T. Moguchi, Т Hosoi, О. Kashimura, J. Chem. Soc. Japan. Ind.
Chem. Sect., 64, 1892 (1961). —102. W. L. Hill, Chemistry and Technology of
Fertilizers, Ed. by V. Sauchelli, N. Y.—London, 1960, p. 116.— 103. H. H. Постни-
ков, Б. В. E в з л и н а, О. В. Васильева, ЖПХ, 28, № 6, 579 (1955).—
104. J. Ando, S. М a t s u n о, Bull. Chem. Soc. Japan, 39, № 9, 1915 (1966).—
105. В. А Кононов, С. Д. Э в e н ч и к, Н. Р. Бродовский, Э. Н. Гинз-
бург, Н. Г. Нейбург, Тезисы н.-техн. совещания по транспортированию и
хранению порошковидного фосфатного сырья для производства минеральных
удобрений, вып. 1 ГОСИНТИ, 1962, стр. 1. —106. В. Иордан, Опыт экс-
плуатации пневмотранспорта апатитового концентрата. Там же, стр. 118.—
107. Ф, В. Турчин, Минеральные удобрения и их применение, Изд. АН СССР,
1943; Хим. пром., № 3, 181 (1957). — 108. А. В. Соколов, Агрохимия фосфора,
Изд. АН СССР, 1950. — 109. А. В. Соколов, Удобр. и урожай, № 2 (1956);
В. А. Тюлин, Т. Б. Беус, Хим. пром., № 7, 432 (1954). — 110. Народное хо-
зяйство СССР в 1958 году. Статистический сборник, Госстатиздат, 1959.
111. В. С. Демин, Производство фосфоритной муки, суперфосфата и крем-
иефтористого натрия, Госхимиздат, 1955. — 1/2. М. Е. П о з и н, Технология ми-
неральных удобрений и солей, Госхимиздат, 1957. —113. В. С. Вальднер,
Техника размола фосфоритов, КОИЗ, 1933. — 114. А. И. Шерешевский,
А. А. Соколовский, П. Ф. Деревицкий, Производство фосфорных
удобрений, ГОНТИ, 1938. — 115. Д. М. Дубинкин, Новые физические методы
в пищевой промышленности, Тезисы докладов, 1967, стр. 83. —116. А. Г. Ш а-
п и р о, Тезисы докладов научно-технического совещания по транспортированию
и хранению порошковидного фосфорного сырья для производства минеральных
удобрений, ч. II, 1962, стр. 40. — 117. А. В. М а ш а я н о в а, М. Я. X о л о м я н-
ский, В. Н. Классен, Хим. пром., № 9, 50 (1967).
Глава XXIV
ПРОСТОЙ СУПЕРФОСФАТ
Простым суперфосфатом называется фосфорное удобрение
получаемое разложением природных фосфатов серной кислотой
взятой в количестве, недостаточном для связывания всего кальция
в сульфат. Образующаяся при смешении кислоты с фосфатом сус
пензия по мере протекания реакций и кристаллизации из раствора
сульфата кальция постепенно загустевает и твердеет (схваты
вается, «созревает») в сплошную массу, которая затем измель
чается. Сульфат кальция остается в продукте в виде балласта
По внешнему виду простой суперфосфат представляет собой по
рошкообразный или зернистый продукт светло- или темно-серого
цвета.
СОСТАВ И СВОЙСТВА СУПЕРФОСФАТА
Суперфосфат состоит из нескольких твердых фаз и распреде
ленной между ними жидкой фазы1-э. Твердые фазы: фосфать
кальция, магния, железа, алюминия, сульфат кальция CaSC>4 (ан
гидрит) с примесью полугидрата CaSO4 • 0,5Н2О, сульфат строн
ция SrSO4 (при переработке апатитового концентрата), неразло
женные минералы, кремнегель SiO2-nH2O и др. Количество
твердых фаз составляет от 65 до 72%, в том числе 50—55% суль
фата кальция и стронция. Жидкая фаза состоит из водного рас
твора фосфорной кислоты, насыщенного монокальцийфосфатом
в качестве примесей в растворе присутствуют катионы Na+, К*
Mg2+, Al3+, Fe2+, Fe3+ и анионы SiFe”, AlFe”, FeFs", F” и др.
Качество суперфосфата оценивают по содержанию в нем усво
яемого фосфора10-17 (в пересчете на Р2О5). Усвояемая Р2О
(стр. 20) присутствует в виде соединений, растворимых в вод
[НзРО4, Са(Н2РО4)2, Mg(H2PO4)2] и в цитратном раствор^
(СаНРО4, MgHPO4, фосфаты железа и алюминия].
Состав и свойства суперфосфата
829
Простой суперфосфат содержит от 14 до 21% усвояемой Р2О5.
Его выпускают в виде следующих продуктов: 1) порошкообразный
не нейтрализованный (содержит до 5,5% свободной Р2ОВ); 2) по-
рошкообразный нейтрализованный (свободная кислотность ча-
стично нейтрализована карбонатами или фосфатами кальция);
3) гранулированный с размером зерен от 1 до 4 мм (гранулиро-
ванный продукт обычно нейтрализован); 4) аммонизированный
(свободная кислотность нейтрализована аммиаком).
Балл гигроскопичности 18-25 (по десятибалльной шкале Пе-
стова22) для суперфосфата из апатитового концентрата с влаж-
ностью 10—15% снижается от 3—5 до 1 при нейтрализации кис-
лотности известняком или аммиаком. Сушка увеличивает гигро-
скопичность (при низкой кислотности балл увеличивается до 5—7).
При одинаковых условиях гигроскопичность суперфосфата, полу-
ченного из фосфорита Каратау, на 1,5—3 балла выше, чем у апа-
титового суперфосфата, что объясняется присутствием в первом
гигроскопичного мономагнийфосфата. Суперфосфаты из фосфори-
тов Каратау обладают большой влагоемкостью24.
Нейтрализованный и высушенный суперфосфат (порошкооб-
разный и гранулированный) рекомендуется хранить в герметиче-
ской таре (например, в битумированных бумажных мешках).
Уменьшение влажности суперфосфата и содержания в нем сво-
бодной Р2ОВ повышает прочность гранул 24-26. Гранулированный
суперфосфат, полученный из апатитового концентрата, содержа-
щий 0,3—0,6% свободной Р2ОВ и 2—3% влаги, имеет гигроскопи-
ческую точку 70—80%, при которой прочность гранул высокая (из-
мельчение меньше 1%).
Слеживаемость 8’22’27 суперфосфата вызывается процессом кри-
сталлизации Са(НгРО4)2-Н2О из жидкой фазы. Охлажденный и
вызревший на складе суперфосфат, у которого процесс кристал-
лизации монокальцийфосфата закончился, а также гранулирован-
ный и нейтрализованный суперфосфат почти не слеживается.
Рассеваемость или сыпучесть 22’28-31 суперфосфата определяет
способность его высеваться сеялками. Нейтрализация свободной
кислотности и сушка суперфосфата резко улучшают его рассевае-
мость.
Объемный или насыпной вес свободно насыпанного негранули-
рованного суперфосфата из апатитового концентрата составляет
1>0—1,1 т/м3, а из фосфорита Каратау 0,8—1,0 т/м3; при хранении
в кучах высотой до 10—-И м объемный вес увеличивается до
1,25 т/м3. Насыпной вес гранулированного суперфосфата25 в не-
Уплотненном слое равен 0,85—0,87, а в уплотненном (глубина
7,3 м) 0,95—0,98 т/м3.
Угол естественного откоса для негранулированного суперфос-
фата в среднем принимается 45° и для гранулированного25
32,5—34°.
830
Г л. XXIV. Простой суперфосфат
Удельная теплоемкость32 для высушенного до постоянного веса
при 105—110° порошкообразного не нейтрализованного суперфос-
фата (из апатитового концентрата) Go100 равна 0,224 и для про-
дукта с влажностью 15,1%—0,345 кал/(г • град). Теплоемкость
гранулированного суперфосфата из апатитового концентрата со-
ставляет 0,231—0,246 кал/ (г -град) в зависимости от влажности.
Теплоемкость простого суперфосфата из галечного флоридского
фосфорита33 при 27,6° равна 0,207, а аммонизированного при 2, 3
н 4% NH3 соответственно 0,234, 0,235 и 0,248 кал//г -град).
ПРИМЕНЕНИЕ
Простой суперфосфат является наиболее дешевым и распро-
страненным фосфорным удобрением. Он (особенно нейтрализован-
ный) может быть эффективно использован под любые растения и
на любых почвах 34-40.
Значительными достоинствами обладает суперфосфат в грану-
лированном41-42 виде.
В США, Англии, Франции и других странах большая часть
суперфосфата применяется в виде смешанных удобрений. Для
приготовления тукосмесей используется аммонизированный супер-
фосфат 33-43-44.
Суперфосфат в небольших количествах употребляется в дрож-
жевой и сахарной промышленности, в качестве огнезащитного по-
крытия древесины 33-45 и др.
Требования к качеству простого суперфосфата приведены в
табл. 58.
ТАБЛИЦА 5»
Нормы стандартов на простой суперфосфат
(содержание компонентов в %)
Из апатитового концентрата по ГОСТ 8382—57 Из фосфоритов Каратау по ГОСТ 4667-49
1 сорт II сорт I сорт II сорт
Р2Ов усвояемой, не менее 20 19 16 14
Р2О6 свободной, не более 5 5 5,5 5.5
Влажность, не более 12 13 15 15
Согласно ГОСТ 5956—53, гранулированный суперфосфат из апа-
титового концентрата должен содержать усвояемой Р2О5 не менее
20,5% в I сорте и 19,5% во II. Количество свободной Р2О5 должно
быть в пределах 1—2,5%. Гранулометрический состав: количество
гранул размером 4—10 мм не более 5%, 2—4 мм не менее 74%,
1—2 мм не более 20%, меньше 1 мм не более 1%. Механическая
прочность при стандартном испытании не менее 97%.
Методы производства
831
МЕТОДЫ ПРОИЗВОДСТВА
Производство простого суперфосфата заключается в: 1) сме-
шении фосфата — апатитового концентрата или фосфоритной муки
с серной кислотой, 2) отверждении (схватывании) реакционной
массы, называемом также созреванием суперфосфата и 3) дораз-
ложении непрореагировавшего фосфата при вылеживании и до-
обработке суперфосфата на складе (дозревание).
При аммонизации и гранулировании суперфосфата вызревший
(складской) продукт подвергается дополнительной обработке.
Способы производства суперфосфата можно разделить на пе-
риодические, полунепрерывные и непрерывные 33’43’46-77.
1) В ранее широко применявшихся периодических способах
смешение фосфатной муки с серной кислотой и дальнейшее раз-
ложение фосфата (созревание в камерах) производили в перио-
дически действующих аппаратах. Для разложения фосфата и со-
зревания суперфосфата использовали 52’53’58’63’65’70’72 камеры-ва-
гоны Бескова и др.57’ 38>66.
2) В полунепрерывных способах применяют непрерывное сме-
шение фосфата с серной кислотой при периодическом созрева-
нии суперфосфата в вагонах Бескова66 или в камерах типа
Свенска70.
3) В непрерывных способах основные стадии производства осу-
ществляются в аппаратах непрерывного действия. Эти способы
наиболее совершенны, поэтому они вытесняют периодические и
полунепрерывные установки. В СССР на большинстве заводов
применяются непрерывные трехкамерные вертикальные смесители
и кольцевые вращающиеся камеры с автоматизированной подго-
товкой серной кислоты и дозировкой реагентов 33’62’63’65’67’68’70-73.
В Европе нашли применение непрерывные способы Максвелл
и Норденгрена.
По способу Норденгрена 33-62, смешение реагентов производят
в две ступени: сначала фосфат разлагают избытком серной кис-
лоты, во второй ступени дополнительно вводят фосфат, разлагае-
мый фосфорной кислотой, образовавшейся в смесителе первой сту-
пени. Созревание идет в прямоугольной камере, задняя и боковые
стенки которой неподвижны, а полом камеры служит транспортер
ИЗ стальных плит.
В США распространены непрерывные способы Бродфилда77
и Саккетта33. По первому из них смешение осуществляют в коры-
тообразном горизонтальном смесителе. Суперфосфат созревает
на движущемся вместе с боковыми стенками стальном транспор-
тере 33’50.55.57,59-63,75,76. р]0 способу Саккетта 33’64’77 разложение
происходит в цилиндрической гуммированной башне во взвешенном
состоянии. Серная кислота образует распыленную конусообраз-
НУЮ струю, которая попадает в циклоннотурбулентное облако
832 .
Гл. XXIV. Простой суперфосфат
фосфатной муки. При этом достигается быстрое и хорошее смешение
обоих реагентов. Полученную смесь дополнительно перемешивают
в горизонтальном смесителе и направляют в камеру-транспортер.
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПРОИЗВОДСТВА
Химизм процесса
В производстве суперфосфата не растворимая в воде нейтраль-
ная трехкальциевая соль ортофосфорной кислоты Са3(РО4)г,
содержащаяся в природных фосфатах в виде минерала кальцийфтор-
апатита ЗСа3(РО4)2 • CaF2, превращается в растворимые соедине-
ния, преимущественно в монокальцийфосфат Са(Н2РО4)2 и, ча-
стично, в свободную фосфорную кислоту и дикальцийфосфат
СаНРО4.
Суммарная реакция разложения апатита серной кислотой:
2Ca5(PO4)sF + 7H2SO4 + 6,5Н2О = ЗСа(Н2РО4)2 • Н2О + 7CaSO4 • 0,5Н2О + 2HF (1)
Минералы-примеси тоже реагируют с серной и фосфорной кис-
лотами 43’ 76~80.
Установлено81’82, что одновременное присутствие серной кис-
лоты и монокальцийфосфата в водном растворе при обычных для
производства температурах невозможно вследствие реакции, иду-
щей нацело слева направо:
Са(Н2РО4)2+ H2SO4 = CaSO4 + 2Н3РО4 (2)
Поэтому фторапатит и серная кислота реагируют первоначально
до образования свободной фосфорной кислоты с выделением в
твердую фазу полугидрата сульфата кальция83:
I. 7Ca5(PO4)3F + 35H2SO4 + 17,5Н2О = 21HSPO4 4-35CaSO4 • 0,5Н2О + 7HF (3)
После полного использования серной кислоты выделившаяся
фосфорная кислота реагирует с фторапатитом, образуя монокаль-
цийфосфат:
II. Ca5(PO4)3F + 7Н3РО4 + 5Н2О = 5Са(Н2РО4)2 • Н2О + HF (4)
Образующийся монокальцийфосфат вначале находится в рас-
творе, при пересыщении которого начинает кристаллизоваться. При
стехиометрическом соотношении компонентов в первой стадии реа-
гирует 70%, а во второй — остальные 30% апатита.
Первая стадия реакции начинается сразу же после смешения
реагентов и заканчивается в течение 20—40 мин при созревании
суперфосфата в камере.
По мере кристаллизации сульфата кальция реакционная масса
затвердевает в результате образования структурной сетки из мик-
рокристаллов сульфата кальция, захватывающей большие количе-
ства жидкой фазы 43’ е4.
Физико-химические основы производства
833
Этому способствует также перекристаллизация сульфата каль-
ция из полугидрата в ангидрит. При высокой температуре реак-
ционной массы в суперфосфатной камере (НО—120°) и большой
концентрации Р2О5 в жидкой фазе (42—46% в конце первой ста-
дии процесса) первоначально выделяющийся полугидрат сульфата
кальция быстро превращается в ангидрит 81-83-85-8б:
2CaSO4 • 0,5Н2О = 2CaSO4 + Н2О
Это объясняется тем, что в указанных условиях стабильной
(наименее растворимой) формой сульфата кальция является ан-
гидрит. В отличие от этого ангидрит в созревшем суперфосфате
при низких температурах (15—25°) является метастабильным, но
переход его в стабильный гипс CaSO4-2H2O в этих условиях
заторможен. Практически в суперфосфате годового вылеживания
сульфат кальция остается в виде ангидрита87.
Процесс фазового перехода полугидрата в ангидрит при взаи-
модействии серной кислоты с фосфатом начинается уже в смеси-
теле, так как при 80—90° ангидрит обладает наименьшей раствори-
мостью (стр. 896). Фазовое превращение полугидрата в ангидрит
осуществляется путем растворения метастабильного полугидрата
в жидкой фазе с образованием пересыщенного раствора и кристал-
лизации из последнего новой фазы — ангидрита. С увеличением
концентрации кислоты и температуры растворение полугидрата и
кристаллизация ангидрита ускоряются. Суперфосфатная масса
схватывается еще до полного израсходования серной кислоты,
когда образование монокальцийфосфата еще невозможно.
Вторая стадия начинается в период камерного созревания и
продолжается длительное время при хранении продукта на складе.
Примесь нефелина разлагается серной кислотой с выделением
геля кремневой кислоты:
2NaAlSiO4 + 4H2SO4 = Na2SO4 + А12(8О4)г + 2H2SiO3 + 2Н2О (5)
Образующиеся сульфаты натрия и алюминия реагируют во
второй стадии с монокальцийфосфатом:
Na2SO4 + Са(Н2РО4)2 = 2NaH2PO4 + CaSO4 (5а)
A12(SO4)3 + ЗСа(Н2РО4)2 = 2A1(H2PO4)s 4- 3CaSO4 (56)
Выделяющийся гель кремневой кислоты (содержание SiO2 в
суперфосфате составляет 0,8—1,2%) способствует схватыванию
суперфосфата6. Глауконит и другие водные алюмосиликаты и
глины, присутствующие в фосфоритах, разлагаются серной кисло-
той аналогично нефелину. Природные окислы железа также раз-
лагаются, например, по суммарной реакции:
Fe2O3 + 3H2SO4 + ЗСа(Н2РО4)2 = 3CaSO4 + 2Fe(H2PO4)3 + ЗН2О (6)
В результате разложения минералов-примесей в раствор пере-
ходят однозамещенные фосфаты натрия, калия, закисного железа,
834
Гл. XXIV. Простой суперфосфат
кислые фосфаты трехвалентного железа и алюминия. По мере
протекания второй стадии реакции и уменьшения концентрации
свободной Н3РО4 в жидкой фазе, кислые фосфаты железа и алю-
’ миния переходят в средние, малорастворимые фосфаты 88-90:
М(Н2РО4)3 + 2Н2О = МРО4 • 2Н2О + 2Н3РО4 (7)
Таким образом, конечное разложение полуторных окислов
можно представить уравнением:
(Fe, А1)2О3 + H2SO4 + Са(Н2РО4)2 = 2(Fe, AI)PO4+CaSO4 +ЗН2О (8)
Карбонаты легко разлагаются серной кислотой:
(Са, Mg)CO3+H2SO4=(Ca, Mg)SO4+СО2 + Н2О . (9)
Газообразная двуокись углерода создает пористую структуру
суперфосфата, что улучшает его физические свойства.
Выделяющиеся кремневая кислота и фтористый водород вза-
имодействуют с образованием SiF<:
4HF + H2SiO3 = SiF4 + ЗН2О
При 110—120° в смесителе и суперфосфатной камере, четырех-
фтористый кремний в присутствии паров воды стабилен и выде-
ляется в газообразном виде. Этим объясняется отсутствие в газо-
вой фазе фтористого водорода. При охлаждении газов четырех-
фтористый кремний взаимодействует с парами воды:
3SiF4 + ЗН2О = 2H2Sifs + H2SiO3
Вследствие частичного протекания этой реакции, уже в газоходе
происходит отложение белого объемистого осадка геля SiO2. При
водной абсорбции фтористых газов эта же реакция лежит в основе
получения кремнефтористоводородной кислоты в качестве побоч-
ного продукта суперфосфатного производства (см. гл. XXXI),
Соединения алюминия вступают во взаимодействие с фтористым
водородом и кремнефторид-ионом, образуя прочные комплексные;
фториды, находящиеся в числе твердых фаз суперфосфата. Be-'
роятно образование также малорастворимых солей Na2SiFs,'
KjSiFe, CaSiF6, CaF2 и NaF, особенно при охлаждении и снижении
концентрации Н3РО4 в жидкой фазе суперфосфата. J
Норма серной кислоты
Норма серной кислоты, расходуемой для разложения фосфата,
зависит от его состава.
Суммарное уравнение (1) служит для расчета стехиометриче-
ской нормы серной кислоты при переработке апатитового концент-
рата (отношение 7H2SO4: ЗР2О5 равно 686:426 = 1,61 вес. ч.
H2SO4 на 1 вес. ч. Р2О5).
Физико-химические основы производства
835
При содержании в апатитовом концентрате .39,4% Р2О5 стехио-
метрическая норма H2SO4 на 100 вес. ч. сырья составляет:
,39,4 • 1,61 = 63,4 вес. ч. С целью ускорения разложения апатита
применяется практическая норма расхода серной кислоты, равная
68—72 вес. ч. на 100 вес. ч. апатитового концентрата.
Для разложения фосфоритов предложено 43 вести расчет нормы
серной кислоты, исходя из следующих молярных соотношений:
По уравнению (1): 1 моль Р2О5 эквивалентен 2,333 моль H2SO4
» » (8): 1 » Fe2O3 или AltO3 » 1 » H2SO4
» ' » (9): I » COj » 1 » H2SO4
Механизм и скорость процесса
Концентрация HtSO«
Рис. 217. Общи! вид зависи-
мости степени разложения фос-
фата от концентрации исход-
ной сериой кислоты (нзохроиа).
Первая стадия реакции представляет собой химическое раство-
рение, осложненное осаждением на зернах фосфата плотных или
сравнительно рыхлых пористых корок сульфата кальция. Плотные
корки сильно затрудняют диффузию жидкой фазы к поверхности
фосфата, и поэтому реакция замедляется; рыхлые корки замедляют
реакцию в меньшей степени. Структу-
ра образующейся корки обусловлена
скоростью кристаллизации твердой
фазы, зависящей главным образом
от пересыщения раствора сульфатом
кальция. Поэтому скорость разложе-
ния фосфата определяется не только
активностью (концентрацией) кисло-
ты, но и степенью ее пересыщения про-
дуктами реакции. На рис. 217 пока-
зан общий вид зависимости степени
разложения фосфата за определенное
время (изохрона) от концентрации
исходной серной кислоты. С увеличе-
нием концентрации разбавленных рас-
творов (начиная от нулевой) и уменьшением концентрации креп-
ких растворов (от 100% H2SO4) активность их повышается и ско-
рость, а следовательно, и степень разложения увеличиваются. Од-
нако, начиная с некоторых концентраций кислоты (малых и
больших) возрастает пересыщение системы сульфатом кальция.
Это вызывает уменьшение скорости и степени разложения как
в области малых, так и больших концентраций кислоты. По этой
причине на кривой рис. 217 имеются два максимума. Минимум ме-
жду ними характеризует область наибольшего пересыщения рас-
гвора сульфатом кальция с образованием на зернах фосфата кор-
ки труднопроницаемой для кислоты. Положение максимумов
’Концентрация кислоты и степень разложения фосфата) зависит от
вида сырья, отношения Т : Ж, температуры и др.
836
Г л. XXIV. Простой суперфосфат
Торможение разложения фосфата кислотой может быть обус-
ловлено кристаллизацией сульфата кальция на поверхности зерен
фосфата непосредственно из диффузионного пограничного слоя91-93
и замедлением вследствие этого диффузии ионов Са2+ в массу
раствора. Наблюдениями под микроскопом найдено8, что эффект
пассивирования зерен фосфата определяется размерами и формой
образующихся кристаллов сульфата кальция. При высоких кон-
центрациях серной кислоты (выше 63%) жидкая фаза быстро
пересыщается сульфатом кальция, вследствие чего выделяется
большое количество мелких (длиной 5—7 мк и. шириной 1—2 мк)
кристаллов CaSO4-0,5H2O и CaSO4 в форме иголочек, образую-
щих налеты, которые покрывают почти всю поверхность зерен апа-
тита. Это затормаживает реакцию, в результате чего процесс про-
текает недостаточно полно и суперфосфатная масса с недостаточ-
ным количеством сульфата кальция плохо схватывается. Содер-
жащаяся в ней жидкая фаза остается на поверхности твердых
частиц и получается не рассыпчатый, а «мажущий» продукт с пло-
хими физическими свойствами. При концентрациях серной кислоты
ниже 63% жидкая фаза пересыщается в меньшей степени, поэтому
выделяются относительно большие кристаллы сульфата кальция
(10—15 мк). Они не покрывают поверхность зерен фосфата сплошным
слоем, а образуют пористую рыхлую корку, в меньшей степени за-
трудняющую диффузию кислоты к зернам. Поэтому реакция идет
быстро и получается сухой рассыпчатый продукт, так как остаю-
щаяся жидкая фаза впитывается в поры между кристаллами.
Чем меньше концентрация серной кислоты, тем крупнее полу-
чаются кристаллы; при этом кристаллы CaSO4-0,5H2O сохраняют
свою игольчатую форму, a CaSO4 образует пластинки и призмы.
Однако применение кислоты низкой концентрации (в диапазоне
левого максимума рис. 217) приводит к тому, что с ней вводится
слишком большое количество воды и вместо твердого продукта
образуется несхватывающаяся пульпа.
Образование сульфатной корки на зернах фосфата обусловлено
прилипанием94 к ним значительно меньших по размеру (шламо-
вых) кристаллов полугидрата сульфата кальция или ангидрита
под влиянием межмолекулярных95 или электростатических сил
притяжения96. Сульфатный покров образуется94 в результате при-
липания кристаллов с размерами меньше 5—10 мк. Кристаллы
крупнее 30—40 мк, близкие по размерам к зернам апатита, почти
не прилипают к их поверхности и не образуют шламового покрова.
Размеры же образующихся кристаллов сульфата кальция зависят
от условий кристаллизации, прежде всего от степени пересыщения!
жидкой фазы. При периодическом смешении фосфатной муки d
серной кислоты, в жидкой фазе в начальный момент имеется лиши
серная кислота исходной концентрации. В этих условиях для раз"
ложения апатитового концентрата должна применяться серная
Физико-химические основы производства
837
кислота концентрации 61,7—64,0%, а для фосфоритов Каратау107
применяют серную кислоту концентрацией 65—65,5%. Серную кис-
лоту такой концентрации используют потому что при более кон-
центрированной серной кислоте из-за высокой начальной скорости
процесса с самого начала реакции на зернах фосфата образуются
плотные корки сульфата кальция, замедляющие дальнейшее его
разложение. Получается медленно твердеющий суперфосфат с не-
удовлетворительными физическими свойствами97-10В.
При непрерывном питании смесителя с непрерывным выводом
смеси через перелив в смесителе находится постоянный объем
пульпы, содержащей в жидкой фазе фосфорную кислоту. Вводимая
серная кислота сразу же разбавляется жидкой фазой реакционной
пульпы, что позволяет избежать неблагоприятных условий, веду-
щих к образованию непроницаемых корок, несмотря на применение
относительно концентрированной (до 68,5—69,5%) серной кислоты.
В этих условиях обеспечиваются сравнительно высокая скорость
разложения фосфата и получение суперфосфата с хорошими физи-
ческими свойствами 91’105’106.
Концентрация серной и фосфорной кислот в жидкой фазе пуль-
пы и степень разложения фосфата в смесителе — взаимно связан-
ные величины. Экспериментально найдены43 максимальные кон-
центрации серной кислоты в жидкой фазе суперфосфатной пульпы,
при которых разложение апатита протекает практически без замет-
ного торможения реакции. Чем выше начальная концентрация сер-
ной кислоты, тем большая степень разложения апатита должна
поддерживаться в смесителе и тем меньшим должно быть отноше-
ние H2SO4 : Н3РО4 в жидкой фазе, чтобы процесс шел без образо-
вания непроницаемых корок на зернах фосфата.
Оптимальный концентрационный режим осуществляется под-
бором продолжительности пребывания пульпы в смесителе. С этой
целью изменяют полезный объем смесителя регулировкой уровня
пульпы. При начальной концентрации серной кислоты 68,5—69,5%
время пребывания пульпы в смесителе установлено 5—7 мин 43.
В условиях непрерывного перемешивания разложение фосфо-
ритов Каратау осуществляют серной кислотой такой же концен-
трации, как и апатитового концентрата.
Выбор концентрации серной кислоты зависит не только от,
способа смешения реагентов, но и от тонкости помола фосфата,
отношения Ж : Т в суперфосфатной пульпе и температуры про-
цесса 108.
Влияние температуры
Скорость реакции разложения фосфатов серной кислотой с по-
вышением температуры 92’93’108 от 50 до 90—100° и выше сильно
Возрастает. Необходимый температурный режим разложения фос-
фатов устанавливается за счет тепла реакции и теплосодержания
838
Гл. XXIV. Простой суперфосфат
фосфата и серной кислоты. Тепловой эффект реакции разложения
фторапатита в первой стадии подсчитан43 по теплотам образо-
вания:
Ca5(PO4)3F (tb.) + 5H2SO4(2,56H20) =
= 5CaSO4 (тв.) + ЗН3РО4 (4,27Н2О) + HF (г.)+ 23,3 ккал
Оптимальная температура серной кислоты для разложения апа-
титового концентрата при непрерывном смешении реагентов:
Концентрация H2SO4, °/0 . . 64’,О 65,5 67,0 68,5
Температура, °C............ 65 — 70 60 — 70 55 — 65 50 — 60
В зимнее время температура серной кислоты должна быть на
5—10° выше. При переработке фосфоритов Каратау периодическим
способом температуру серной кислоты поддерживают летом 40—50°
и зимой 50—60° 70’107. В оптимальных условиях температура пуль-,
пы, выходящей из смесителя, находится в пределах НО—115°.
В камерах температура суперфосфата достигает 115—120°. Пони-
жение температуры пульпы при указанных температурах серной
кислоты свидетельствует о недостаточном разложении фосфата.
Смешение реагентов
Работа мешалок в смесителях должна обеспечивать однород-
ность пульпы и возможно большую скорость потока жидкой фазы
по отношению к поверхности частиц фосфата. Перемешивание
ускоряет диффузионный обмен ионов в пограничном слое и объеме
раствора и повышает скорость реакции разложения фосфата.
Турбулентный режим перемешивания снижает также степень
пересыщения раствора в пограничном слое, что способствует обра-
зованию более крупных кристаллов сульфата кальция и, следова-
тельно, более проницаемых пленок на зернах фосфата. Это в свою
очередь ускоряет разложение. При неблагоприятном концентра-
ционном и температурном режимах с помощью только интенсифи-
кации смешения невозможно значительно ускорить процесс разло-
жения фосфата.
Степень измельчения фосфатов
Гранулометрический состав фосфатной муки в сильной степени
влияет на скорость ее разложения108-111. Мелкие фракции муки
разлагаются быстро. Частицы больших размеров (80—100 мк)
Физико-химические основы производства
839
разлагаются медленно. В первой стадии наиболее реакционноспо-
собные мелкие частицы фосфата в первую очередь реагируют
,с активной серной кислотой, а во второй стадии менее активная
фосфорная кислота взаимодействует с оставшимися крупными ча-
стицами с относительно низкой реакционной способностью. Устра-
нение этого явления — одна из важных задач совершенствования
технологии производства суперфосфата.
Стандарты (см. гл. XXIII) для апатитового концентрата и фос-
форитной муки Каратау допускают количество частиц размером
>150 мк не более 14%. Одновременно необходимо110-112, чтобы ко-
личество частиц размером <75 мк составляло не менее 47—48%
(см. табл. 57, стр. 815). За рубежом применяется помол фосфори-
тов с остатком 10—15% на сите 0,15 мм 48.
Скорость разложения фосфатов серной кислотой зависит от их
физической структуры, которая характеризуется общей (внутрен-
ней) удельной поверхностью элементарных зерен113’114. При одной
и той же степени измельчения фосфаты, обладающие высокораз-
витой общей удельной поверхностью (например, фосфаты Туниса
и Марокко), химически более активны, чем с низкой общей удель-
ной поверхностью (например, хибинский апатит).
Коэффициент разложения в конце I стадии реакции
При стехиометрической норме серной кислоты расчетный коэф-
фициент разложения апатита в момент окончания I стадии реак-
ции составляет 70% (уравнения 3 и 4). При любой норме серной
кислоты этот показатель рассчитывается по формуле115
М = 70—= 70-^-= 1,104п
п0 63,4
где п— фактическая норма серной кислоты в вес. ч. на 100 вес. ч.
апатита;
По-стехиометрическая норма.
Данные табл. 59 показывают, что в камере частично протекает
также и II стадия реакции. Чем выше норма серной кислоты, тем
меньше расхождение между расчетными и практическими показа-
телями для камерного- суперфосфата. В табл. 60 приведен рассчи-
танный43 состав суперфосфата в момент окончания I стадии реак-
ции при исходной концентрации серной кислоты 67% и испарении
25% введенной с кислотой воды. Практически камерный суперфос-
фат содержит меньше свободной Р2О5 вследствие частичного
протекания в камере II стадии реакции, при которой потребляется
фосфорная кислота/
840
Г л. XXIV. Простой суперфосфат
ТАБЛИЦА 59
Коэффициенты разложения апатита к концу
I стадии реакции и в камерном суперфосфате
в зависимости от нормы серной кислоты43 (в %)
Норма (серной кислоты Расчетный коэффициент разложения (К1> Практический коэффициент разложения для камерного суперфосфата
63,4 70,0 78,0
66,0 72,9 80,7
69,0 76,2 83,5
72,0 79,5 86,0
75,0 82,8 ' 88,5
78,6 86,8 90,4
83,4 92,1 92,5
ТАБЛИЦА во
Состав суперфосфата к концу 1 стадии реакции
Норма серной кислоты Выход суперфосфата CaSO4, • % H3PO4, % Р2О5 свободная, % Неразложен- иый апатит, %
68 1,91 49,1 21,4 15,5 12,7
70 1,94 49,8 21,6 15,7 11,4
72 1,97 50,4 21,9 15,9 10,0
74 2,00 51,1 22,2 16,1 8,8
78 2,05 52,5 22,8 16,5 6,5
82 2,10 53,8 23,4 17,0 4,4
86 2,15 54,9 23,9 17,3 2,3
• Сумма CaSO4+SrSO4 в пересчете
на CaSO4.
Разложение фосфата фосфорной кислотой
(II стадия реакции)
После разложения основной массы фосфата серной кислотой
оставшаяся непрореагировавшая его часть взаимодействует с На-
копившейся в системе фосфорной кислотой (II стадия). Первая
стадия — разложение фосфата серной кислотой, как указано было
выше, протекает быстро вследствие большой активности раствора
(высокой концентрации ионов водорода), а также расходования
наиболее мелких частиц фосфата. Во второй стадии апатит разла-
гается водным раствором фосфорной кислоты^ в котором все боль-
ше накапливается монокальцийфосфата. По мере увеличения сте-
Физико-химические основы производства
841
пени нейтрализации фосфорной кислоты активность раствора
уменьшается и скорость процесса замедляется 116-117. Когда жид-
кая фаза становится насыщенной фосфатами кальция (моно- и
дикальцийфосфатом), что совпадает с окончанием созревания
суперфосфатной массы в камере, скорость разложения еще боль-
ше уменьшается. Это обусловлено, помимо уменьшения активно-
сти жидкой фазы, образованием на зернах непрореагировавшего
фосфата плохо проницаемых корок, но уже не сульфата, а фосфа-
тов кальция, кристаллизующихся из пересыщенных растворов си-
стемы СаО—Р2О5—Н2О118*. Образовавшаяся в I стадии корка
сульфата кальция на зернах фосфата уже в самом начале II ста-
дии создает дополнительное сопротивление диффузии фосфорной
кислоты к реагирующей твердой поверхности. Помимо этого после
завершения I стадии непрореагировавший фосфат состоит из от-
носительно крупных частиц, что приводит к уменьшению поверхно-
сти контакта фаз. Тем не менее, в камере II стадия процесса про-
текает еще с достаточной скоростью вследствие высокой темпера-
туры и разложения фосфата мало нейтрализованными растворами
фосфорной кислоты. Но в дальнейшем при дозревании суперфос-
фата на складе процесс идет очень медленно.
Режим разложения апатита фосфорной кислотой при созрева-
нии суперфосфата вплоть до окончания камерного процесса, опре-
деляется первоначальными условиями производства, т. е. концент-
рацией и нормой серной кислоты, температурой процесса и режи-
мом смешения реагентов. Протекание процесса непосредственно в
камере обычно не регулируется. В табл. 61 приведен типичный со-
став камерного суперфосфата в зависимости от метода производ-
ства 68.
ТАБЛИЦА 61
Состав камерного суперфосфата
Метод производства Содержание, % Норма H2SO4, вес. ч. H2SO4 на 100 вес. ч. апатита Степень разложе- ния К, м Выход, ч. на I ч. апатита
Р2^5общ Р2°5усв Р2О5СВО6 н2о
Из апатитового концентрата
Периодический . . . 19,9 16,7 11,9 14,5 70,6 83,9 1,98
Полунепрерывный 20,3 17,15 12,2 13,2 71,8 84,5 1,94
Непрерывный.... 20,7 17,8 12,2 12,7 69,8 86,0 1,90
Из фосфорита Каратау (Чулактау)
Периодический
15,1 I 12,7 I 8,0 [ 12,0 I 64,0 I 84,0 I 1,82
^Подробно о скорости разложения фосфатов фосфорной кислотой см.
842
Гл. XXIV. Простой суперфосфат
Дальнейшее разложение фосфата протекает на складе, где
имеется возможность в известной степени ускорять процесс. Это
основано на изменении фазового состава фосфатного комплекса
суперфосфата "43.
Фосфатный комплекс суперфосфата и его фазовый состав
Фосфатным комплексом называют систему, состоящую из твер-
дых и растворенных фосфатов кальция, свободной фосфорной кис-
лоты и воды, т. е. всех главных компонентов системы, за вычетом
непрореагировавшего апатита, CaSO4
Рис. 218. Система
СаО—Р2О5—Н2О.
и HF. Поэтому состав фосфатного
комплекса определяется с помощью
диаграммы равновесия в системе
СаО—Р2О5—Н2О 119“125 (рис. 218). Воз-
можность применения этой диаграм-
мы для определения фазового состава
фосфатного комплекса суперфосфата,
полученного из апатитового концен-
трата, обусловлена тем, что образовав-
шийся сульфат кальция (и стронция)
находится в твердой фазе и уже не
принимает участия в химических реак-
циях. Фтористые соединения частично
улетучиваются, а частично образуют
малорастворимые соединения — крем-
нефториды, фторкомплексы алюминия
и пр. Примесей других минералов в
апатитовом концентрате всего 3%.
Поэтому без большой погрешности II
стадию процесса получения суперфос-
фата можно рассматривать как рас-
творение в фосфорной кислоте гидро-
ксилапатита Са5(РО4)3(ОН). При этом
происходит постепенная нейтрализа-
ция фосфорной кислоты. Степень ней-
трализации первого иона водорода
фосфорной кислоты определяется от-
ношением количества Р2О6, связанной
с СаО в виде монокальцийфосфата, к
общему содержанию Р2О6 в жидкой
фазе. По окончании I стадии процесса
степень нейтрализации равна нулю —жидкая фаза представлена
свободной Н5РО4. Во II стадии фосфорная кислота реагирует
с остальной частью фосфата, образуя раствор фосфорной кислоты
и монокальцийфосфата, находящийся в равновесии с твердой фа-
Физико-химические основы производства
843
зой: моно- или дикальцийфосфатом. С увеличением степени раз-
ложения фосфата степень нейтрализации возрастает, с увеличением
нормы серной кислоты она уменьшается.
В нижней части рис. 219 приведены изотермы системы
СаО—Р2О5—Н2О для 25, 40 и 100°. Пучок лучей, исходящих из
точки начала координат диаграммы, представляет шкалу степени
нейтрализации. Луч 100%-ной нейтрализации заканчивается
в точке N, отражающей состав монокальцийфосфата Са(Н2РО4)2-
•Н2О (Р2О5—56,34% и СаО — 22,22%).
В верхней части рисунка дана зависимость степени нейтрали-
зации от степени разложения апатитового концентрата при двух
нормах серной кислоты (п), а слева второй вспомогательный гра-
фик зависимости между концентрацией Р2О5 в жидкой фазе к
началу I стадии процесса и конечной влажностью готового супер-
фосфата (при норме H2SO4 72 вес. ч). На рис. 219 нанесены лучи
растворения гидроксилапатита в фосфорной кислоте различной кон-
центрации. Эти лучи направлены в точку состава Са5(РО4)3(ОН)
(Р2О3— 42,39% и СаО — 55,82%), находящуюся за пределами ди-
аграммы. Точки составов фосфатного комплекса лежат на пересе-
чении лучей растворения и степени нейтрализации. Например, при
норме серной кислоты 72 вес. ч., коэффициенте разложения апатита
92% и влажности суперфосфата 12%, состав фосфатного комп-
лекса изобразится точкой A (Р2О3— 46,2% и СаО—8,5%). Эта
точка находится в поле кристаллизации Са (Н2РО4) 2 • Н2О при
25—100°: следовательно, фосфатный комплекс суперфосфата в при-
веденном примере состоит из жидкой фазы (раствор фосфорной
кислоты и монокальцийфосфата) и монокальцийфосфата в твердой
фазе. С помощью луча кристаллизации, проведенного из точки iV
через точку А, подсчитывают, по правилу рычага, соотношение ме-
жду количествами жидкой и твердой фаз в комплексе при различ-
ных температурах. Точки пересечения луча кристаллизации с изо-
термами (например, точка Р для 25°) определяют состав жидкой
фазы при соответствующей температуре.
При норме серной кислоты 72 вес. ч. степень разложения апа-
тита в камерном суперфосфате составляет в среднем 87% (точка С
на рис. 219) и степень нейтрализации жидкой фазы равна 30%.
При влажности суперфосфата 16, 14, 12 или 9% луч степени ней-
трализации жидкой фазы суперфосфата пересечет соответствую-
щие лучи растворения в точках Оь D2, D3 или £)4. Из положения
этих точек можно заключить, что при влажности суперфосфата
16% фосфатный комплекс при 100° и выше совсем не содержит
твердого монокальцийфосфата (точка лежит в поле ненасыщен-
ных растворов), а при влажности 14% содержит его очень мало
(точка О2 лежит близко к 100°-ной изотерме). Чем меньше влаж-
ность суперфосфата, тем больше образуется кристаллического моно-
Кальцийфосфата в фосфатном комплексе камерного суперфосфата
844
Гл. XXIV. Простой суперфосфат
Рис. 219. Номограмма для определения фазового состава фосфатного комплекса
суперфосфата (из апатитового концентрата).
Физико-химические основы производства
845
(см. положение точек £)3 и О4 по отношению к 100°-ной изо-
терме). Охлажденный до 40—25° камерный суперфосфат содержит
.в фосфатном комплексе твердый монокальцийфосфат при влаж-
ности продукта 16% и меньшей. Содержание влаги в свежеприго-
товленном суперфосфате из апатитового концентрата при приме-
нении 61—62%-ной серной кислоты (норма 70 вес. ч.) составляет
в среднем 16%, а при 68,5%-ной кислоте 12%.
В табл. 62 приведены результаты расчетов43 по диаграмме
рис. 219 (при норме серной кислоты 68 вес. ч. и влажности про-
дукта 14%).
ТАБЛИЦА 62
Количество жидкой и твердой фаз в фосфатном комплексе и в суперфосфате
Коэффи- циент раз- ложения, % Фосфатный комплекс Суперфосфат Состав твердой фазы фосфатного комплекса
твердая фаза, °/о жидкая фаза, % твердая фаза, °/0 жидкая фаза, %
Температура 25°
85 20,5 79,5 65,1 34,9 Монокальцийфосфат
90 32,7 67,3 68,7 31,3
95 43,2 56,8 72,1 27,9
100 54,2 45,8 76,3 23,7
Температура 50°
85 17,0 83,0 63,6 36,4 Моиокальцийфосфат
90 29,5 70,5 67,2 32,8 »
95 41,0 59,0 71,0 29,0 »
100 50,7 49,3 74,5 25,5 Моио- + дикальцийфосфат
Температура 100°
85 8,3 91,7 59,2 40,8 Моиокальцийфосфат
90 19,6 80,4 62,7 37,3 Моио- + дикальцийфосфат
95 31,4 68,6 66,4 33,6 То же
100 25,4 74,6 61,5 38,5 » »
С увеличением степени разложения фосфата и с уменьшением
температуры количество твердой фазы в суперфосфате в большин-
стве случаев возрастает. Повышение нормы серной кислоты умень-
шает количество твердой фазы. Особенно резко возрастает содер-
жание твердой фазы в продукте с уменьшением влажности супер-
фосфата или с ростом концентрации исходной серной кислоты.
Дозревание суперфосфата
Дозревание суперфосфата, полученного из апатитового кон-
центрата, ускоряется при понижении температуры до 40—50°. Этот
экспериментально установленный 101 факт объясняют43 тем, что при
охлаждении суперфосфата из его жидкой фазы кристаллизуется
846
Г л. XXIV. Простой суперфосфат
Са (НгРО^г • Н20 и, кроме того, при этом уменьшается возмож-
ность возникновения пассивирующих корок СаНРО4. В случае
образования последних в камере, при понижении темпера-
туры суперфосфата на складе возможно их растворение. При кри-
сталлизации Са (Н2РО4)2 • Н2О происходит передвижение жидкой
фазы (микроперемешивание). Все эти факторы способствуют уско-
рению процесса.
Охлаждение суперфосфата осуществляют распылением камер-
ного продукта дисковыми, дутьевыми 126-127 или барабанными 128
разбрасывателями и периодическим
перелопачиванием суперфосфатных
куч грейферами или экскаватора-
МИ 129, 130.
Дозревание суперфосфата на
складе значительно ускоряется с
увеличением нормы серной кислоты,
используемой в I стадии процесса
(рис. 220). Это объясняется умень-
шением степени нейтрализации
жидкой фазы и началом кристалли-
зации монокальцийфосфата с обра-
зованием его корки при большей
общей степени разложения фосфа-
та. При норме серной кислоты 80—
83 вес. ч. H2SO4 на 100 вес. ч. апа-
тита степень разложения его уже
_ г,.,,, п . в камерном суперфосфате состав-
Рис. 220. Дозревание суперфос- в j ет ч-
фата из апатита в зависимости ляег 91 93/о, а при норме 83
от нормы серной кислоты. 85 вес. ч. 93—95%• Кислый супер-
фосфат содержит около 16% свобод-
ной Р2О5. При смешении его с эстонской фосфоритной мукой содер-
жание свободной кислоты достигает стандартной величины через
3 суток после нейтрализации131. Содержание усвояемой P2Os в су-
перфосфате составляет 18,7—19,2%. Недостатком производства су-
перфосфата с применением высоких норм серной кислоты является
большой расход нейтрализующих добавок (костяной или фосфо-
ритной муки, или известняка и др.) и увеличение расхода серной
кислоты на переработку апатита (в расчете иа 1 т P2Os).
Разложение фосфата избытком серной кислоты может быть
осуществлено с применением кислоты повышенной концентра-
ции33'62 (77—78% H2SO4). В этом случае используется два смеси-
теля. В первом производится смешение фосфата с избытком кис-
лоты. Образующаяся пульпа перетекает во второй смеситель, куда
подают дополнительное количество фосфата. После второго сме-
сителя пульпа подвергается непрерывному камерному вызре-
ванию.
Физико-химические основы производства
847-
Изучение8 микрогранулометрического состава суперфосфата,
приготовленного с применением кислоты разной концентрации, по-
зволило объяснить ускорение процесса образования суперфосфата
при использовании кислоты более низкой концентрации, чем при-
меняемая обычно. Показано108, что при разложении апатитового
концентрата 61,7%-ной H2SO4 (при норме 70 вес. ч. моногидрата)
в лабораторных условиях с имитацией непрерывного смешения ре-
агентов коэффициент разложения в суперфосфате после 3—7 дней
хранения достигает 93,5—95%. При еще меньшей концентрации
кислоты (40—50%-ная H2SO4) можно получить уже в камерном
суперфосфате до 90% разложения апатита. Такой суперфосфат
с влажностью 16—20% может быть использован непосредственно
для получения гранулированного продукта (стр. 867).
В ряде других исследований 132- 13е, направленных на ускорение
дозревания суперфосфата, также использовали для разложения
тонко измельченного фосфата разбавленную (40—60%) серную
кислоту с последующей сушкой продукта в барабанной сушилке.
Полученные результаты показали возможность разложения фло-
ридского фосфорита на 93% без дополнительного хранения супер-
фосфата на складе.
Ускорение дозревания суперфосфата достигается также, если
при его получении часть серной кислоты заменить на азотную 13э.
При частичной замене серной кислоты азотной образующаяся
в I стадии процесса фосфорная кислота вступает в реакцию с про-
дуктом взаимодействия апатита с азотной кислотой — нитратом
кальция. Ускорение процесса разложения апатита на II стадии
происходит, по-видимому, вследствие большей активности азотной
кислоты по сравнению с фосфорной кислотой. При норме кислоты
70 вес. ч. на 100 вес. ч. апатита достижение стандартной степени
разложения (95%) происходит при 5%-ной замене серной кислоты
за 10 суток, вместо 14—15 суток в отсутствие азотной кислоты. Су-
перфосфат имеет меньшую свободную кислотность, чем суперфос-
фат, полученный без использования азотной кислоты, и повышенное
содержание водорастворимой Р2О5; кроме того в нем содержится
0,06% азота при замене 1% серной кислоты азотной, 0,3% азота
при замене 3 и 0,5% азота при замене 5% серной кислоты.
Отрицательным явлением в производстве суперфосфата с ис-
пользованием смеси кислот является выделение азотной кислоты
в газовую фазу при варке его и вылеживании. При варке выде-
ляется от 16 до 46% азотной кислоты от вводимого количества.
Предложено 140 для предотвращения выделения окислов азота до-
бавлять к смеси кислот карбамид, амидосульфоновую кислоту или
ее производные (в количестве 0,5—1 % от веса кислоты). При этом
азотные соединения восстанавливаются до элементарного азота.
При добавлении к серной кислоте азотной кислоты (вместо
замены ею части серной кислоты) в количестве 20% сверх.
«48
Гл. XXIV. Простой суперфосфат
теоретической нормы степень разложения апатита в камерном су-
перфосфате 141 составляет 94—95%. Такой продукт можно без вы-
леживания подвергнуть нейтрализации и грануляции.
Аналогичным является процесс разложения фосфатов при до-
бавке к серной кислоте (сверх стехиометрического количества) со-
ляной кислоты 142-144. Камерный продукт высушивают. Выделяю-
щиеся при этом хлористый водород и фтористые газы поглощают
водой. Из полученного раствора осаждают кремнефторид натрия
при помощи хлорида натрия, а 16%-ный раствор соляной кислоты
возвращают на разложение фосфата. Степень разложения апатита
в камерном суперфосфате составляет 92—93%,’ а фосфорита Ка-
ратау 94—96%. Получаемый высушенный продукт не требует
•складского вызревания, а также применения нейтрализующих до-
бавок. При замене на Тайюаньском заводе (КНР) 10—15% серной
кислоты на соляную продолжительность вызревания суперфосфата,
полученного из марокканских и других фосфоритов, сократилась
до 3 суток; продукт содержит 19,2% усвояемой Р2О5145.
Из других методов, опробованных с целью ускорения дозрева-
ния суперфосфата, отметим попытки применения поверхностно-ак-
тивных веществ 146-149 анионноактивного, катионноактивного и не-
ионногенного типов, которые, однако, не дали заметных результа-
тов. Значительное ускорение доразложения фосфатов достигалось
при перемешивании суперфосфата во вращающейся трубе в тече-
ние 2 суток150, а также при разложении фосфорита в шаровой
доельнипе 151.
При оптимальных условиях производства суперфосфата из апа-
титового концентрата непрерывным методом (норма H2SO4 69—
70 вес. ч., концентрация H2SO4 68%, ее температура 55°, время сме-
щения реагентов 6 мин, время созревания в камере 1 ч) среднесу-
точный прирост43 усвояемой Р2О5 за первые 5 суток хранения су-
перфосфата при 40—50° составляет 0,3%, за следующие 5 суток
около 0,1 % и в последующие 10 суток 0,045%. Общий прирост за
20 суток достигает 2% усвояемой Р2О5. Содержание водораство-
римого фтора уменьшается 152 от 0,5—0,6% в свежем до 0,2—0,3%
в дозревшем суперфосфате при общем его неизменном содержа-
нии ~0,9%.
При обследовании режимов призводства и дообработки супер-
фосфата на различных заводах установлено 153, что лучшие резуль-
таты в камерном процессе достигаются на предприятиях, где строго
соблюдается оптимальный режим смешения реагентов, главным
образом, по температуре и концентрации серной кислоты. Пока-
зано, что в этих условиях при расходе 68—69 кг H2SO4 на 100 кг
апатита степень разложения в камерном продукте равна 84—87%.
Вылеживание суперфосфата на складе при температуре не выше
50° при трех перелопачиваниях позволяет закончить процесс дозре-
вания (степень разложения апатита 94—97%) за 10—12 суток.
Физико-химические основы производства
849
В табл. 63 приведен типичный состав дозревшего суперфосфата
(товарной продукции), полученного из апатитового концентрата
разными методами66.
ТАБЛИЦА 65
Состав товарного суперфосфата из апатитового концентрата
Метод производства Содержание, % Норма H2SO4, вес. ч. H2SO4 иа 100 вес. ч. апати- та Степень разложе- ния К, % Выход, ч. И& 1 ч. апатита
ГгОвобщ РвОбусв ^2^5св0б Н20
Периодический 20,5 18,9 5,1 12,7 70,6 92 1,92
Полунепрерывный 20,8 19,6 4,4 11,0 71,8 94 1,90
Непрерывный 21,5 20,2 8,0 9,8 69,8 94 1,83
Дозревание суперфосфата, полученного из фосфоритов, имеет
некоторые особенности, обусловленные присутствием соединений
магния (см. ниже) и больших количеств фосфатов полуторных
окислов. В процессе дозревания возникают условия для кристал-
лизации фосфатов железа и алюминия типа МРО4-2Н2О или
2МРО4 • Н8РО4 • 8Н2О, растворимость которых резко падает с умень-
шением концентрации свободной Р2О5 в жидкой фазе 84'68~90.
Средние фосфаты полуторных окислов практически не раство-
римы в воде и поэтому относительно плохо усваиваются расте-
ниями. Кислые фосфаты в нейтральной среде разлагаются:
2МРО4 • Н3РО4 • 8Н2О + aq = 2МРО4 • 2Н2О + Н3РО4 + aq
При дозревании суперфосфата, содержащего много окислов же-
леза, количество цитратнорастворимой P2‘Os возрастает, а водорас-
творимой P2Os уменьшается (ретроградация). Это объясняют154
образованием двойной соли Ca[Fe(HPO4)2]2- 6Н2О, которая рас»
творима в цитратном растворе более чем на 80%. Окись алюми-
ния, особенно в присутствии Fe2O3, вызывает образование цитрат-
нонерастворимой P2Os в суперфосфате после длительного его хра-
нения 154.
Количество водорастворимой P2Os заметно уменьшается, если
отношение —е2р л 10° в сырье превышает 7,5—8. В суперфосфате
из апатитового концентрата Feip3Q 100 в пределах 2 — 4^ ретро-
градация Р2О5 обычно не происходит. Цитратнонерастворимые
формы Р2'О5 образуются при отношении в сырье - -"-вл Д° 20.
Г-сли значительная часть полуторных окислов в сырье представ-
лена А12О3, предельно допустимая величина суммарного содержания
850
Гл. XXIV. Простой суперфосфат
полуторных окислов становится меньше1S4. Фосфориты, содер-
жащие полуторные окислы свыше указанных предельных коли-
честв, не могут эффективно использоваться для производства су-
перфосфата. Необходима глубокая очистка такого сырья (см. гл,
XXIII).
Особенности разложения магнийсодержащих фосфоритов
Каратауские, кингисеппские и некоторые другие фосфориты
загрязнены доломитом, доломитизированным известняком и маг-
незиальными силикатами. В присутствии магниевых соединений
разложение фосфоритов43, 107 протекает в три стадии:
Первая стадия заключается в образовании раствора фосфор-
ной кислоты, в который переходит также MgSO4.
I. Ca5(PO4)3F + 5H2SO4 = 5CaSO4+ ЗН3РО4 + HF
Ca, Mg(CO3)2 + 2H2SO4 = CaSO4 + MgSO4 + 2CO2 + 2H2O
Mg2SiO4 + 2H2SO4 = 2MgSO4 + SiO2 + 2H2O
Во II стадии происходит разложение части непрореагировав-
шего фосфата фосфорной кислотой с образованием мономагний-
фосфата:
II. Ca5(PO4)3F + 5MgSO4 + 7Н3РО4 = 5CaSO4 + 5Mg(H2PO4)2 + HF
Превращение сульфата магния при этом в хорошо растворимый
мономагнийфосфат, остающийся в растворе, является главной осо-
бенностью разложения фосфоритов Каратау и других, содержащих
магниевые соединения.
Третья стадия состоит в разложении остаточного апатита фос-
форной кислотой, содержащей мономагнийфосфат:
III. Ca5(PO4)3F + 7Н3РО4 = 5Са(Н2РО4)2 + HF
Ca5(PO4)3F + 2Н3РО4 - 5СаНРО4 + HF
Равновесие в конце II и на III стадиях реакции может быть
рассмотрено с помощью диаграмм систем MgO—Р2О5—Н2О и
MgO—СаО—Р2О5—Н2О 155-157. В системе MgO—Р2О5—Н2О (рис.
221) мономагнийфосфат значительно более растворим, чем моно-
кальцийфосфат в системе СаО—P2Os—Н2О. Степень нейтрализа-
ции при одинаковой концентрации Р2О5 в насыщенном растворе
выше в магниевой системе, чем в кальциевой (рис. 222).
После окончания II стадии реакции фосфатный комплекс со-
стоит из MgO, Р2О5 и Н2О. Для этого момента составы комплекса,
рассчитанные43 для фосфорита Каратау (28% Р2О5), содержа-
щего от 3 до 14% MgO (по отношению к P2Os), при норме H2SO4
на разложение фосфатной части сырья 47,5 вес. ч. характеризуют-
ся во всех случаях ненасыщенной жидкой фазой при 50—95°. Сле-
довательно, образующийся фосфат магния остается в растворе, что
Физико-химические основы производства
851
и объясняет высокую степень нейтрализации, составляющую от
13,6 до 55,5% при рассматриваемых отношениях MgO: Р2О5 в
сырье. С увеличением степени нейтрализации фосфорной кислоты
окисью магния разложение фосфата в III стадии реакции сильно
замедляется 93’158. В связи с этим наиболее целесообразна перера-
ботка в суперфосфат обогащенных сортов фосфоритов Каратау, со-
держащих не больше 6—8%
MgO по отношению к Р2О5.
При 80° и отношении
“Я • 100 в сырье от 3 до 8
г 2^5
включительно фосфатные ком-
плексы содержат159 в твер-
дой фазе Са(Н2РО4)2 • Н2О +
+ Mg(H2PO4)2. При этом пер-
воначально кристаллизуется
СаНРО4, затем СаНРО4 рас-
творяется с одновременным
выделением Са(Н2РО4)2 Н2О.
После исчезновения СаНРО4
продолжает кристаллизовать-
ся Са(Н2РО4)2 • Н2О до насы-
щения раствора смесью двух
солей Са(Н2РО4)2 Н2О +
+ Mg(H2PO4)2.
При отношении -дИЯ • 100
г 2V>5
свыше 8 (степень разложения
88,6%) первоначально может
выделяться Са (Н2РО4)2 • Н2О
(магниевые фосфаты остают-
Рис. 221. Изотермы системы
MgO—Р2О5—Н2О при 25, 83 и 130°.
(A, Ai, Л2 — эвтонические растворы, равновес-
ные с двумя твердыми фазами).
ся в растворе). В тех же усло-
виях, но при более полном
разложений фосфата (95%)
кристаллизуется СаНРО4. Образование СаНРО4 возможно и
при менее полном разложении фосфата для сырья, содержащего
свыше 10% MgO по отношению к P2Os. Это объясняется зна-
чительным высаливающим действием фосфатов магния на фос-
фаты кальция в системе СаО—MgO—Р2О-,—Н2О. При 25° в боль-
шинстве случаев первоначально осаждается Са (Н2РО4)2 • Н2О,
а затем смесь однозамещенных фосфатов кальция и
магния.
Присутствие гигроскопичного мономагнийфосфата ухудшает
физические свойства суперфосфата из фосфоритов Каратау 160~16,4.
Образование в ряде случаев плохо проницаемых корок из кристал-
лов СаНРО4 затрудняет доразложение фосфата.
852
Гл. XX!V. Простой суперфосфат
При дозревании суперфосфата из фосфоритов Каратау с вы-
соким содержанием магниевых соединений коэффициент разложе-
ния достигает 95% при 25° в течение 45 суток83; при 80° скорость
дозревания значительно больше (особенно в течение первых 2—
3 дней), и он дозревает в 5—10 раз скорее, чем при 25°. Этим су-
перфосфат из фосфоритов Каратау отличается от апатитового су-
перфосфата. Охлаждение суперфосфата ускоряет дозревание толь-
ко при условии переработки сырья, содержащего меньше 2% MgO.
Рис. 222. Степень нейтрализации первого иоиа
водорода фосфорной кислоты в насыщенных ра-
створах систем СаО—Р2О5—Н2О и
MgO—Р2О5—Н2О при 25 и 80°.
Точки В —узловые в кальциевой системе, точки A, Aj и
Лз — узловые в магниевой системе.
Суперфосфат, получаемый из фосфорита Каратау, содержащего
27,5% P20s, 6,59% СО2 и 2,5% R2O3, разложением 68%-ной серной
кислотой при ее норме 64 вес. ч. и степени разложения 90%, имеет
следующий типичный состав: 15,6% Р2О5общ, 14,0% Р2О5Усв, 5,0%
РгОзсвоб и 10,5% Н2О. Выход его составляет 1,67 т/т. В опытах
сушки свежего суперфосфата 133 при 320—360° в течение 45 мин
степень разложения фосфорита Каратау достигала 86%. Получался
продукт, содержащий 16—17% цитратнорастворимой, около 4%
водорастворимой и 1,5% свободной Р2О5. При сушке улетучива-
лось 70% фтора, содержавшегося в свежем суперфосфате. Этими
опытами показана возможность увеличения степени разложения
фосфорита в процессе сушки свежего суперфосфата.
Переработка магнийсодержащих фосфоритов в суперфосфат
может быть осуществлена без дозревания при бескамерном его
производстве путем разложения фосфорита серной кислотой в не-
Физико-химические основы производства 853
загустевающей пульпе и сушки полученной пульпы в смеси с ре-
туром готового продукта 165’166. При норме 55%-ной серной кис-
лоты, равной 110% от стехиометрического количества, 70° и на-
чальном весовом отношении Т:Ж в пульпе 1 : 1,5, степень разло-
жения фосфорита Каратау за 60—90 мин составляет 92—95%.
В процессе сушки пульпы в смеси с ретуром готового продукта при
70—80° в течение 60 мин степень разложения фосфорита увеличи-
вается до 97—97,5%. В готовом продукте, полученном в лабора-
торных условиях из фосфорита Каратау, содержание усвояемой
формы Р2Об на 1—2% больше, чем предусмотрено ГОСТом. При
сушке суперфосфата одновременно происходит процесс грануля-
ции. Готовый продукт не требует дальнейшего дозревания. Ана-
логичным методом можно разложить апатитовый концентрат 50—
60%-ным раствором бисульфата аммония в незагустевающей пуль-
пе167. Степень разложения апатита в пульпе при 95—98° и отно-
шении Т : Ж = 1 : 2 4- 2,6 за 2 ч составляет 90,5%; после смешения
пульпы с ретуром и сушки она достигает в готовом продукте 97—
99%.
С целью повышения качества суперфосфата из фосфоритов
Каратау, разработан способ их химического обогащения168 (см.
гл. XXIII). Из облагороженного фосфорита получается суперфос-
фат с содержанием 15,5—16% усвояемой Р2О5 при коэффициенте
разложения 96—97%. Достигается экономия 8—40% серной кис-
лоты. Применение химического облагораживания фосфоритов Ка-
ратау является целесообразным способом улучшения и стабилиза-
ции качества суперфосфата, получаемого из этого сырья.
О режиме производства суперфосфата из сырья новых место-
рождений см. 168’ 169.
Нейтрализация суперфосфата твердыми добавками
Дозревший суперфосфат содержит до 5,5% свободной Р2О5.
При высевании вместе с семенами такой суперфосфат уменьшает
их всхожесть. Он замазывает высевные устройства и зависает в
бункерах механических сеялок. Высокая кислотность суперфос-
фата приводит к сравнительно быстрому разъеданию крафтцеллю-
лозной тары, вызывает коррозию туковых сеялок. Для улучшения
качества вызревший суперфосфат обрабатывают твердыми ней-
трализующими добавками 170-176. Чаще всего процесс нейтрализа-
ции сочетается с гранулированием суперфосфата (см. ниже). В ка-
честве добавок применяются костяная мука, фосфоритная мука,
известь, мел, известняк, доломит, змеевики (серпентиниты), обес-
фторенные фосфаты (см. гл. XXIX) и т. п. Наиболее целесооб-
разны добавки, содержащие фосфор, которые несколько обога-
Ща^т продукт (при внесении костяной муки или эстонского
Фосфорита снижение на 1 % концентрации свободной Р2О5 эквива-
лентно, приросту усвояемой Р2О5 в среднем на 0,42%). Нейтрали-
854
Гл. XXIV. Простой суперфосфат
зация свободной фосфорной кислоты добавками, содержащими
кальций, сводится к образованию монокальцийфосфата. Количе-
ство твердой фазы в суперфосфате при этом увеличивается и улуч-
шаются его физические свойства:
Са5(РО4)з(ОН) + 7Н3РО4 + 4Н2О = 5Са(Н2РО4)2 Н2О
СаСО3 + 2Н3РО4 = Са(Н2РО4)2 • Н2О + СО2
При значительном снижении общей кислотности суперфосфата
возможно образование малорастворимого в воде дикальцийфос-
фата:
Са(Н2РО4)2 = СаНРО4 + Н3РО4
При избыточном количестве извести образуется трикальцийфос-
фат— наступает ретроградация фосфорнокальциевых солей в труд-
ноусвояемые формы:
2СаНРО4 + Са(ОН)2 = Са3(РО4)2 • Н2О + Н2О
Плохое перемешивание реагентов может привести к местной
ретроградации Р2О5. Поэтому требуется тщательное смешение
суперфосфата с добавками. Использование добавок, содержащих
магний, в меньшей мере улучшает физические свойства суперфос-
фата из-за гигроскопичности образующегося мономагнийфосфата,
однако введение магния полезно в связи с специфическим удоб-
рительным действием этого элемента 34'36:
CaMg(CO3)2 + 4Н3РО< = Са(Н2РО4)2 + Mg(H2PO4)2 + 2СО2 + 2Н2О
Mg2SiO4 + 4Н3РО4 = 2Mg(H2PO4 )2 + H2SiO3 + Н2О
По приведенным выше уравнениям определяют коэффициенты
расхода нейтрализующих добавок на единицу свободной Р2О5
(табл. 64). Практическая норма эстонской фосфоритной муки
1,6 вес. ч., а обесфторенного фосфата 175 1,08 вес. ч. на 1 вес. ч. сво-
бодной Р2О5 в суперфосфате. С наибольшей скоростью разлагаются
измельченный известняк и обесфторенный фосфат. Эффективно
действуют также костяная мука и эстонская фосфоритная мука.
Более медленно разлагаются фосфориты Каратау и, особенно, апа-
титовый концентрат.
ТАБЛИЦА 64
Коэффициенты для расчета количества нейтрализующих добавок
Z Костяная мука Известь, мел и известняк Карбонаты Доломит, магнезиаль- ные силикаты
Молярные отношения. . . ЗР2О5: 7Р2О5 СаО : Р2О5 СО2; Р2О5 MgO : Р2О6
Коэффициенты расхода . . 0,429 0,391 0,310 0,282
Физико-химические основы производства
855
Аммонизация суперфосфата
Нейтрализация свободной кислотности суперфосфата аммиаком
является эффективным способом улучшения его качества 33'43’|77-203.
Особенно большое значение имеет этот процесс для суперфосфата
из фосфоритов Каратау 195-201’2°4. Аммонизированный суперфосфат
представляет собой сухой порошок, совершенно не гигроскопич-
ный. Введенный азот (в количестве до 4—5%) является питатель-
ным элементом. Отношение N : Р2О5 в таком продукте мало; для
азотно-фосфорных удобрений обычно требуется содержание от
0,5 до 1 и более вес. ч. азота на I вес. ч. Р2О5. Для получения
двойного удобрения на основе простого суперфосфата используют
аммонизацию с помощью аммиакатов (аммиачных растворов нит-
рата аммония, карбамида и др.) (см. гл. XXXVII).
При нейтрализации аммиаком первого иона водорода свобод-
ной Н3РО4
Н3РО4 + NH3 = NH4H2PO4 +27 ккал
вследствие выделения тепла температура увеличивается до 80°, и
суперфосфат несколько подсушивается187. При расходе аммиака
согласно этому уравнению (0,24 вес. ч. на I вес. ч. свободной Р2О3)
содержание водорастворимой Р2Об в суперфосфате не снижается.
При более глубокой аммонизации образуется диаммонийфос-
фат, который реагирует с сульфатом кальция:
(NH4)2HPO4 + CaSO4 = СаНРО4 + (NH4)2SO4
При введении 0,48 вес. ч. NH3 на единицу свободной Р2О5 и
0,24 вес. ч. NH3 на единицу Р2О3 монокальцийфосфата вероятно
полное превращение фосфатов аммония по реакциям:
Н3РО4 + CaSO4 + 2NH3 = СаНРО4 + (NH4)2SO4
Са(Н2РО4)2 • Н2О + CaSO4 + 2NH3 = 2СаНРО4 + (NH4)2S,O4
Такая глубокая аммонизация вызывает большие потери водо-
растворимой Р2Об, но содержание усвояемой Р2О5 уменьшается
незначительно. Дальнейшее увеличение расхода аммиака на еди-
ницу Р2О5 приводит к образованию трикальцийфосфата, т. е. к
ретроградации усвояемой Р2О3 180’189- 191.
В результате аммонизации суперфосфата из фосфоритов Кара-
тау202 происходит почти полное высаливание фосфатов кальция и
магния. При охлаждении до 25° кристаллизуются монокальцийфос-
фат и фосфаты магния и аммония.
Аммонизированный суперфосфат при охлаждении схваты-
вается, что объясняется изменением гидратности мономагнийфос-
фата и образованием других кристаллогидратов. Для борьбы со
слеживанием продукта на складе необходимо ускорение процессов
кристаллизации посредством сушки суперфосфата (до 3% влаги)
и его охлаждения (распыление и перелопачивание). Соли магния 3
3 М. £. Познн
856
Гл. XXIV. Простой суперфосфат
предотвращают ретроградацию лимонно- и цитратнорастворимой
Р2О5202. Добавка к апатитовому суперфосфату 3% MgSOi • 7Н2О
позволяет вводить большие количества аммиака (нейтрализация
до pH = 8,5—9,5) без уменьшения относительного содержания
усвояемой Р2О5. Концентрация NH3 в продукте достигает при
этом 11,5%. Стабилизирующее действие сульфата магния объяс-
няют205 торможением реакции образования гидроксилапатита
при взаимодействии фосфата и сульфата кальция с аммиаком.
Аммонизация суперфосфата газообразным аммиаком осущест-
вляется во вращающемся барабане с непрерывным введением ам-
миака и суперфосфата прямотоком 192. При норме аммиака около
2% к весу суперфосфата степень его поглощения составляет 97—
99%. Крупные комки суперфосфата отсеиваются на сите и измель-
чаются.
ПРОИЗВОДСТВО СУПЕРФОСФАТА
Суперфосфатный завод состоит из склада фосфатного сырья и
хранилищ серной кислоты, операционного отделения для разложе-
ния фосфата серной кислотой и склада суперфосфата, где происхо-
дит дозревание и дообработка (измельчение) продукта и иногда
его нейтрализация твердыми добавками 33’ 46-77- 206-212. в операцион-
ном отделении устанавливается также аппаратура для абсорбции
фтористых газов (см. гл. XXXI), выделяющихся при разложении
фосфата в смесителе и суперфосфатной камере.
Измельченный фосфат из железнодорожных вагонов выгру-
жается электромеханическими лопатами и с помощью различных
транспортных механизмов (скреперы, элеваторы, шнеки, ленточ-
ные транспортеры, пневмотранспортные трубы и др.) подается в
силосные склады. Во избежание зависания муки в силосах, их
стенки и днища обкладываются пористыми плитками, через кото-
рые подают сжатый воздух. Из силосов муку подают с помощью
элеватора и прямого и обратного шнеков в небольшой бункер, рас-
положенный над полуавтоматическими весами.
Серную кислоту перекачивают центробежными насосами в на-
порный бак, откуда она поступает в мерник. При периодическом
способе производства порции муки и серной кислоты смешиваются
в смесителе в течение 1—2 мин, и пульпа, с помощью клапана в
дне смесителя, спускается в камеру. В зависимости от объема ка-
меры устанавливают один или два смесителя. Количество «заме-
сов» колеблется от 50 до 130. Время загрузки камеры составляет
0,8—2,5 ч.
По окончании загрузки и созревания суперфосфат выгружают
из камеры фрезерным механизмом и направляют на склад, где он
хранится в кучах высотой до 8—10 м. При подаче на склад супер-
фосфат охлаждается распылением с помощью разбрасывателя.
Периодически (2—3 раза) его перелопачивают грейферными кра-
Sofia
3*
858 Гл. XXIV. Простой суперфосфат
нами или экскаваторами. Общее время складского хранения со-
ставляет 2—3 недели.
Дозревший суперфосфат смешивают с нейтрализующими до-
бавками во вращающемся барабане. Перед отгрузкой слежав-
шийся суперфосфат должен быть измельчен. С этой целью супер-
фосфат сначала просеивают на вибрационных грохотах, а не про-
шедшие через сито комки дробят на валковых дробилках или
дезинтеграторах.
Полунепрерывный метод отличается от периодического лишь
способом смешения реагентов. Фосфат и серная кислота дозиру-
ются с помощью питателей непрерывного действия. Так же непре-
рывно производится смешение фосфата с серной кислотой. Пульпа
непрерывно поступает в камеру, после заполнения которой пита-
ние прекращается. В дальнейшем процесс протекает так же, как
и в периодическом способе.
Производство суперфосфата непрерывным методом отличается
не только непрерывной дозировкой и смешением реагентов, но
также и вызреванием суперфосфата в непрерывно действующей
камере. На рис. 223 представлена схема непрерывной установки с
горизонтальной кольцевой вращающейся суперфосфатной камерой
(Мориц-Стандерт) 68. Кроме улучшения физико-химических усло-
вий разложения фосфата, непрерывный метод является более эко-
номичным: он требует меньшей затраты рабочей силы и меньших
капиталовложений, облегчает возможность автоматизации приготов-
ления серной кислоты нужной концентрации и дозировки обоих реа-
гентов. Значительно улучшаются условия труда. Непрерывный спо-
соб применяется и с другими суперфосфатными камерами (Брод-
филда, Максвелла, камерами с пульсирующей выгрузкой и т. д.).
По непрерывному способу Саккетта получения суперфосфата
во взвешенном состоянии 33’64174, измельченный фосфат дозируется
в горизонтальный желоб, в который нагнетается воздух. Воздуш-
ный поток увлекает фосфатную муку и вводит ее тангенциально в
цилиндрическую башню, в которой происходит разложение фос-
фата серной кислотой. Серная кислота (71%) подается в башню
насосом через распылительное сопло, образуя распыленную кону-
сообразную струю с углом 60°, встречающуюся с циклонно-турбу-
лентным облаком фосфатной муки. Достигается хорошее переме-
шивание компонентов. Пульпа падает на дно башни и поступает во
вспомогательный горизонтальный смеситель, а оттуда в камеру-
транспортер. Такая установка производит 45 т суперфосфата в час.
Выделение фтористых газов
В операционном отделении выделяются в газовую фазу в сред-
нем следующие количества фтора (в виде SiF4) по отношению к его
содержанию в фосфатах213-221: из марокканского фосфорита — 25%,
Производство суперфосфата
859
из флоридского — 21%, из теннессийского — 35,5%, из апатитового
концентрата — 40—50%. Добавка кремнегеля к фосфатам, содер-
.жащим недостаточное количество SiOg, увеличивает выход фто-
ра222. Добавка СаСОз, доломита или MgCOs в пульпу в количе-
стве ~2% от апатита, способствует увеличению выхода фтора
вследствие увеличения пористости суперфосфатной массы и интен-
сификации газовыделения 223.
Количество выделяющегося фтора зависит от температуры в
смесителе216’217: с увеличением температуры до 105° и выше выход
фтора достигает максимальной величины. С ростом концентрации
серной кислоты увеличивается концентрация Н3РО4 в жидкой фазе
суперфосфатной массы и растет43 парциальное давление пара
SiF4, в связи с чем также увеличивается степень выделения фтора.
Непрерывное смешение реагентов, позволяющее применять кон-
центрированную серную кислоту, способствует увеличению выхода
фтора. Повышенная норма серной кислоты, увеличивая степень
разложения фосфата в смесителе и камере, тоже увеличивает сте-
пень выделения фтора. Одним из условий наиболее полного выде-
ления фтора является поддержание достаточно высокого (не мень-
ше 20 мм вод. ст.) разрежения в смесителе и в камере с помощью
отсасывающего вентилятора. Причиной неполного выделения фтора
является резкое уменьшение давления пара SiF4 с понижением кон-
центрации H2SiF6 в жидкой фазе по мере вызревания суперфос-
фата. Затвердевание суперфосфата в камере также затрудняет вы-
деление газообразного четырехфтористого кремния.
Концентрация отходящих газов находится в пределах 15—35 г
фтора в 1 м3 газа. Значительные колебания в составе газа объяс-
няются подсосами воздуха.
Основная аппаратура и условия ее работы224
В современном непрерывном процессе применяют непрерывное
разбавление серной кислоты водой в смесителе с дырчатой пере-
городкой (диаметр отверстий 6—7 мм), контролируемое концентра-
томером (рис. 224). Контроль ведется по равенству плотностей кис-
лот— поступающей из смесителя и эталонной; равенство их плот-
ностей определяется с помощью дифференциального манометра по
равенству гидравлических сопротивлений при пропускании пузырь-
ков сжатого воздуха через слои кислоты равной глубины70.
Непрерывное дозирование серной кислоты может осуществ-
ляться ковшевыми, диафрагменными, сифонными и щелевыми до-
заторами70’225. Применяются также самопишущие автоматические
ротаметры с электрическим управлением и пневматическим кла-
паном для регулирования жидкостного потока 33.
Для непрерывной дозировки фосфата широко применяются ав-
томатические весовые дозаторы различных конструкций 70>22e-227.
860
Г л. XXIV. Простой суперфосфат
На рис. 225 изображена схема весового дозатора (пойдометра).
Он представляет собой ленточный транспортер, натянутый
на барабан. Рабочая часть транспортера опирается на весовой
ролик, который связан системой рычагов с весовым коромыс-
лом. Вес ленты с находящимся на ней фосфатом уравновеши-
воздух
Рис. 224. Концентратомер:
1 — освинцованный сосуд; 2 — перегородка;
3 — сосуд с эталонной кислотой; 4 —сте-
клянные трубки для подвода воздуха;
5 и 6 — сосуды для контроля количе.
ства подводимого воздуха (100 пузырьков
в минуту); 7 —дифференциальный мано-
метр.
вается специальным грузом. С по-
мощью регулирующей заслонки
устанавливают расход фосфата, по-
ступающего на ленту дозатора из
бункера, так, чтобы коромысло ве-
сов с грузом слабо колебалось около
среднего положения. При отклоне-
нии от этого положения производят
регулировку заслонкой. Системы с
механическими питателями обла-
дают большой инерционностью, что
обусловливает относительно низкую
(1—2%) точность подачи материа-
ла. Созданы двухступенчатые доза-
торы с электромагнитными вибра-
ционными и шнековыми питателя-
ми, имеющие незначительную инер-
цию и работающие с высокой точ-
ностью 227~229. Употребляются также
автоматические порционные весы, отвешивающие отдельные пор-
ции муки в течение 40—45 сек под действием электронного регу-
лятора230. Фосфоритная мука Каратау обладает большой теку-
честью, поэтому для ее дози-
рования наиболее пригодны
порционные весы и весы с
шнековым питателем 228.22Э.
В качестве непрерывных
смесителей употребляют ко-
рытообразные аппараты с
одним или двумя горизон-
тальными валами с лопат-
ками, обеспечивающими по-
ступательное движение
пульпы. Такие смесители ис-
Рис. 225. Схема весового дозатора:
J — лейточиый транспортер; 2 —барабан; 3— весо-
вой ролик; 4 — коромысло весов; 5 —груз; 6 —регу-
лирующая заслонка.
пользуются в непрерывных способах Мориц-Стандерт,- Бродфилда,
Норденгрена, Максвелл и др.
На отечественных заводах получили распространение верти-
кальные трех- или четырехкамерные смесители непрерывного дей-
ствия (рис. 226). Емкость смесителя регулируется шибером. Про-
должительность пребывания пульпы в смесителе 5—Тмин. (При
работе на Каратауском фосфорите 2—3 мин.) Мешалки в первых
Производство суперфосфата
861
двух-трех камерах вращаются с окружной скоростью 6,5—7 м/сек
и в последней камере — около 5 м/сек, при этом обеспечивается
интенсивное и хорошее смешение реагентов. Исследованием сме-
шения апатита с кислотой с применением Р32 установлено231, что
при среднем времени пребывания пульпы в смесителе 5,6 мин
отдельные части загруженного материала (особенно в верхних
слоях) быстро уходят из
смесителя. С целью удлине-
ния пути потоков необходи-
мы изменения режима сме-
шения и конструкции пере-
мешивающих устройств.
Предложены новые, усовер-
шенствованные конструкции
смесителей с дисковыми ме-
шалками 232. Из смесителя
пульпа перетекает в супер-
фосфатную камеру.
Цилиндрическая каме-
ра 68 непрерывного действия,
применяемая на суперфос-
фатных заводах СССР (рис.
227), представляет собой
вертикальный железобетон-
ный цилиндр, имеющий
стальной кожух и футеров-
ку из диабазовых плиток.
Цилиндр вращается на к ро-
ликах вокруг неподвижной
чугунной трубы, проходящей
через сальниковое уплотне-
Рис. 226. Четырехкамерный смеситель:
1 — сообщающиеся камеры; 2 — переливная коробка;
3 —съемная крышка; 4 —входное отверстие; 5 —ме-
шалка; 6 —чугунный шибер; 7 — трос ручной ле-
бедки для подъема и опускания шибера.
ние в днище цилиндра.
Вращение осуществляется с помощью электромотора через
редуктор. В течение 1,5—2,5 ч камера делает один оборот (на-
правление вращения показано на плане рис. 227 стрелкой). Желе-
зобетонная крышка камеры неподвижна. К последней подвешена
вертикальная чугунная перегородка, отделяющая зону загрузки от
зоны выгрузки. Для вырезки готового суперфосфата служит фре-
зер, вращающийся со скоростью 8—10 об/мин в направлении, про-
тивоположном вращению камеры.
Суперфосфатная пульпа из смесителя непрерывно поступает
в камеру в точке 9. Пульпа затвердевает и подходит к фрезеру
готовой для выгрузки. За один оборот фрезер срезает слой супер-
фосфата толщиной 5—25 мм. Срезанный ножами фрезера супер-
фосфат попадает на транспортер. Для уменьшения трения рас-
ширяющейся при созревании (вследствие выделения газов)
862
Гл. XXIV. Простой суперфосфат
суперфосфатной массы о неподвижную стенку центральной трубы
у наружной стенки этой трубы в зоне загрузки расположен эксцен-
трик, создающий свободный объем 233
Рис. 227. Кольцевая суперфосфатная камера непрерывного действия:
J— цилиндрический корпус камеры; 2 — центральная труба; 3 — электромотор; 4 —редуктор;
5—неподвижная крышка камеры; 6 — перегородка; 7 —фрезер; 8 — эксцентрик; 9 — место
ввода пульпы.
Плотность суперфосфатной массы, равная в начале схватыва-
ния 1,5
т/м3, уменьшается ко времени выгрузки до 1,1 т/м3. При
переработке фосфоритов, содер-
жащих карбонаты и выделяющих
при разложении двуокись угле-
рода, давление на стенки камеры
меньше, так как суперфосфатная
масса более пориста и расши-
ряется в объеме за счет сокра-
щений пор; плотность массы в ка-
мере в этом случае составляет
0,8—0,9 т/м3. Выделяющиеся при
реакции газы удаляются через
отверстие в крышке камеры в
вентиляционную трубу, по кото-
рой отсасываются в абсорбцион-
Рис. 228. Разбрасыватель суперфос- ную установку.
фата. Производительность стандарт-
ной (в СССР) камеры, имеющей
диаметр 7,1 м и высоту 2,5 м, при скорости ее вращения один обо-
рот за 2,5 ч, составляет 30 т суперфосфата в 1 ч, а при скорости
вращения один оборот за 1,5 ч — 50 т суперфосфата в 1 ч. Съем
суперфосфата с 1 л;3 объема камеры достигает 500—650 кг/ч.
Производство суперфосфата
863
При подаче на склад суперфосфат несколько охлаждают, раз-
брасывая его. Для этой цели применяют небольшой (длиной 0,7—
1 м, диаметром 0,6—0,8 м) быстро вращающийся (1000—
1400 об/мин) горизонтальный барабан с продольными лопастями
(рис. 228). Падая на наружную поверхность барабана, суперфос-
фат отбрасывается лопастями и разбивается на мелкие частицы,
которые, оседая в кучу, охлаждаются окружающим воздухом с
70—90° до 30—60°. Суперфосфат вылеживается в нескольких ку-
чах. Обычно склад оборудован грейферными кранами или другими
транспортными механизмами, с помощью которых в течение срока
хранения продукт периодически перелопачивают, т. е. пересыпают
кучи с места на место. Это способствует его охлаждению и более
быстрому дозреванию. Несмотря на это, срок вылеживания супер-
фосфата на складе составляет обычно 2—3 недели.
Для погрузки суперфосфата в железнодорожные вагоны при-
меняются механизированные погрузчики 70’234’ 235.
Материалы для защиты аппаратуры от коррозии
В производстве суперфосфата для предотвращения коррозии
серной, фосфорной и кремнефтористоводородной кислотами ши-
роко используются кислотостойкие материалы как металлические,
так и неметаллические 236"245.
Из металлических материалов применяются ферросилид и вы-
сокохромистый чугун для сернокислотных центробежных насосов
(марки КНЗ и ХНЗ), хромистый чугун для ножей суперфосфатных
фрезеров и для вентилей, хромоникелевые, хромоникельмолибдено-
вые и другие кислотоупорные стали для отдельных деталей аппа-
ратов. В небольших количествах применяются также свинец и харт-
блей (сплав свинца с сурьмой) для изготовления пробок и гнезд
клапанов, сернокислотных расходомеров, обкладки мерников и т. п.
Применяются неметаллические защитные материалы: диабазо-
вые плитки и кислотоупорный кирпич для футеровки сернокислот-
ных баков, суперфосфатных смесителей и камер, газоходов, абсорб-
ционных камер и башен; диабазовые или андезитовые замазки для
скрепления футеровочных плиток, для защитной обмазки поверх-
ностей мешалок смесителей, стенок суперфосфатных камер, кожу-
хов вентиляторов и т. д. Из кислотоупорного цемента и бетона
изготовляют отдельные части суперфосфатных камер и сборников
(стены, днища и своды камер, крышки баков). В качестве запорных
приспособлений применяются керамические краны.
Большое применение получили также резина и пластические
массы. Резина используется в качестве прокладок и уплотнений
суперфосфатных камер, для защитной обкладки турбинок венти-
ляторов и транспортерных лент для суперфосфата. Для изготов-
ления центробежных насосов (для слабой серной и кремнефтори-
864
Гл. XXIV. Простой суперфосфат
стоводородной кислот), запорных приспособлений и арматуры,
разбрызгивающих валков применяются фаолит и графолит (из-
делия на основе феноло-формальдегидной резольной смолы с
добавкой асбеста или графита), а также асбовинил. Для обклад?
ки мерников и других резервуаров используется винипласт.
О контроле производства суперфосфата и его автоматизации
см_ 12, 70, 225, 229, 246, 247.
Материальный баланс производства суперфосфата
В основу расчета 248 материального баланса (табл. 65) произ-
водства простого суперфосфата из стандартного апатитового кон-
центрата (39,4% Р20з) положены следующие показатели249:
1) норма серной кислоты 68 кг на 100 кг концентрата; 2) концент-
рация серной кислоты 68%; 3) концентрация исходной серной
кислоты (контактной)
состав суперфосфата в %:
92,5%; 4)
Камерного
Р2О5 общая 20,8
Р2О5 усвояемая 17,6
После
дозревания
на складе
21,0
19,8
Камерного
Р2О5 свободная 11,0
Влажность . . 12,0
После
дозревания
на складе
5,5
10,5
Материальный баланс
производства
апатитового
простого суперфосфата на
концентрата
ТАБЛИЦА 65 ’
1000 кг
Затрачено
кг
Получено
кг
Апатитовый концентрат
Серная кислота башен-
ная ............... .
Вода для разбавления
серной кислоты . . .
1000
895
105
2000
50,0
36,8
13,2
100,0
Газы и пар, выделившие-
ся при разложении . . .
Пары воды, выделившие-
ся при хранении супер-
фосфата на складе . . .
Товарный суперфосфат . .
106
18
1876
5,3
0,9 1
93,8 f
2000 100,0
Итого . .
%
%
Итого . .
Технико-экономические данные
Расходные коэффициенты для разных типов фосфатного сырья
приведены в табл. 66. Наименьший расход фосфата и серной кис-
лоты составляет на 1 т экспедиционного суперфосфата: 0,53—
0,55 т апатитового концентрата и 0,37—0,38 т серной кислоты;
(100%). Расходные коэффициенты на всех заводах, перерабатьН
вающих апатитовый концентрат, почти одинаковы. Между тем, се-1
Производство суперфосфата
865
ТАБЛИЦА 68
Расходные коэффициенты в производстве суперфосфата 68
На 1 m суперфосфата
На 1 иг усвояемой Р2О5
Фосфатное сырье
Апатитовый кон-
центрат (непре-
рывный способ) 39,4 20,2
Фосфорит фло-
ридский .... 33,0 20,0
Фосфорит Кара-
тау ............
с добавкой
апатитового
концентра-
та .........
0,381 0,546
0,350* 0,600 9,0
0,264 0,480 4,0
0,088
18,2
0,21
(дол-
лары)
1,885
1,750
2,500
2,700
3,000
3,410
0,600
22,8
45,0
28,0
90,2
1,06
(дол-
лары)
158
* Расход серной кислоты для флоридского фосфорита S3 вероятно занижен. Теоретиче-
ский ее расход по тем же данным 33 составляет 0,411 m H2SO4 на 1 m суперфосфата нлн
2,06 m на 1 m усвояемой Р2О5.
бестоимость суперфосфата в пересчете на 1 т усвояемой Р20.5
колеблется от 76 до 100 руб. и выше. Это объясняется тем, что
затраты на производство суперфосфата определяются главным
образом стоимостью сырья, которая составляет 89—93% себестои-
мости продукта. Стоимость же сырья — апатитового концентрата
и серной кислоты — на разных заводах различна, так как она
зависит от затрат на доставку апатитового концентрата и серного
сырья для выработки серной кислоты. Чем дальше расположен
суперфосфатный завод от источников сырья, тем дороже оказы-
вается суперфосфат (табл. 67).
Суперфосфатные заводы экономически целесообразно разме-
щать в районах потребления и подвозить к ним сырье250, расход
которого меньше количества получаемой продукции.
При производстве 1 т усвояемой P2Os из апатитового концен-
трата расходуют 4,2—4,3 т сырья и получают 4,95—5,30 т про-
дукции.
При получении суперфосфата из фосфоритов Каратау увеличи-
вается расход серной кислоты (за счет переработки карбонатов,
содержащихся в сырье), а также расход фосфата вследствие мень-
шего содержания в нем Р2О5; при переработке фосфоритов Кара-
тау расходуют 6,25—6,5 т сырья и получают 7—7,15 т суперфосфата.
866
Гл. XXIV. Простой суперфосфат
Получение гранулированного суперфосфата
867
Сравнительная калькуляция себестоимости суперфосфата (на 1 т 100% Р2О5)
№ завод ев . 2*0 о счь«сосо ОО 0 1Л О Ь? т-Г 00* о о —<* С4 92,54, 0,31 92,23 3,80 96,03
Джамбулски! цена, руб. ю '* 00 Г- _ — —- 00 со ф СО 04 СО < S§ 1 1 1 1 СО 04 со — 35 ~ о о 1 1 1 1 1
расход ф 04 о а со о _ Q 00.Ю О | | | | О О О 04 00 —• - 04 1 1 1 1 1
се ВС ВС \о сумма, руб. rj* ф rj? 1 СО О О О' 04 о со । 83,78 0,46 83,32 0,14 83,46
о 2 й § цена, РУ6- Ф Ф стз io х S «О 1 1 °- О-®- 1111 сч 0 0 СО 04 1 1 1 1 1
=г S № ИЗ ав га расход 8 । । § ооlilt ~ . о ~ 04 1 1 Г 1 1
м X S ХО S so о NOlOd ФОООЬФ «СО •“’ О 00 rj* 00 6066046 СО СО 72,85 1 0,60 72,25 4,40 76,65
скнй химко цена, руб. ю ь* 00 1О 00 08 1 < < 1 СОЬ-*^ц6 0*0 1 1 1 1 СО Ю 04 — j 22,00 1 1 1
X о и с с га расход О О ю со со о ь- о о О 00 п- ~ «66-Г оо 0,0275 1 1 1
Элементы эатржт Сырье: апатитовый концентрат (100% Р2О5), т костяная мука (100% Р2О5), т фосфоритная мука (100% Р2О5), т серная кислота (100% H2SO4),t Энергетические затраты: электроэнергия, кет ч вода, я? Зарплата с начислением Амортизация Цеховые расходы Общезаводские расходы Итого . . - * ’ Отходы фторгазов (100%), т . . . . 1 Итого себестоимость Виепроизводствеииые расходы . . . 1 Коммерческая себестоимость ....
Соответственно увеличиваются заработная плата и энергетические
затраты. При производстве суперфосфата из апатитового концен-
трата заработная плата составляет 1—2,5% и энергетические
затраты лишь 0,2—1 % 33’ 66.
Суперфосфат до сих пор является самым дешевым фосфорным
удобрением 251-254 и потому, несмотря на то, что он содержит мало
Р2О5 и много балласта, действующие заводы будут существовать
еще долго.
Дальнейшее-развитие производства суперфосфата возможно в
сочетании с получением сложно-смешанных удобрений, при ис-
пользовании отбросных растворов серной кислоты металлургиче-
ских 255 и нефтеочистных 256 заводов, а также при вовлечении в
промышленную эксплуатацию фосфоритных месторождений мест-
ного значения 257. С целью улучшения качества продукта широкое
развитие получает процесс гранулирования, а также аммонизации
суперфосфата, особенно при переработке магнийсодержащих фос-
форитов типа Каратау.
Представляет интерес 258 увеличение содержания усвояемой
Р2О5 с 19,5 до 23% в суперфосфате путем быстрого его нагрева-
ния (со скоростью 10 град!мин) до 200—220°. При этом в присут-
ствии водяных паров образуются хорошо растворимые в воде ли-
нейные полифосфаты кальция.
ПОЛУЧЕНИЕ ГРАНУЛИРОВАННОГО СУПЕРФОСФАТА
Процесс гранулирования суперфосфата 25’33’259-279 обычно объ-
единяется с нейтрализацией свободной кислотности твердыми до-
бавками, главным образом, мелом или известняком. В некоторых
случаях применяют для нейтрализации 280 феррошлаки (отходы
производства ферросплавов), содержащие 43—46% СаО, 13,5—
14% Мп и др. При хранении гранулированного суперфосфата, со-
держащего не более 0,5% водорастворимого марганца, не наблю-
дается ретроградации водорастворимой и цитратнораствори-
мой Р2О5.
Предложено281 также использовать для нейтрализации супер-
фосфата фильтровую жидкость содовых заводов, содержащую
карбонаты и бикарбонаты аммония и натрия. Содержание пита-
тельных веществ в продукте не уменьшается за счет введения
аммиака. Принятый у нас способ гранулирования состоит в окаты-
вании нейтрализованного и увлажненного суперфосфата в бара-
банном грануляторе с последующей сушкой, дроблением и рассе-
вом гранул (рис. 229). Гранулы классифицируются на грохоте по
крупности на три фракции: 1) частицы размером свыше 4 мм по-
ступают на валковую дробилку, где измельчаются и снова попа-
дают на грохот, 2) готовый продукт с размером гранул в основном
от 2 до 4 мм и 3) частицы величиной меньше 2 мм, которые
868
Гл. XXIV. Простой суперфосфат
возвращаются на смешение с исходным суперфосфатом (ретур).
Количество ретура составляет 20—30% от массы суперфосфата.
Рис. 229. Схема получения гранулированного суперфосфата:
j — бункер для суперфосфата; 2 — бункер для нейтрализующей добавки; 3 —ленточный
питатель; 4 — валковая дробилка; 5 и 9 — элеваторы; 6 — транспортер гранулятора;
7 —гранулятор; 3 —сушильный барабан; 10 — грохот; //—транспортер готового про-
дукта; 12 — дробилка; 13 и 17 — транспортеры; /4-ретурный транспортер; /5-бун-
керы для готового продукта; 16 — зашивочная машина.
' Гранулятор 70’264 представляет собой вращающийся полый ба-
рабан (рис. 230), внутри которого (на расстоянии 1 м от входа)
Рис. 230. Барабанный гранулятор:
j — вращающийся .цилиндр; 2 — бандаж; 3 —зубчатый венец привода; 4 — ролики; 5 —форсунка
для воды; б —срезающий нож; 7 —электромотор.
расположены направляющие лопасти. На входе и выходе установ-
лены подпорные кольца, обеспечивающие определенную рабочую
загрузку барабана суперфосфатом. Внутрь барабана подведена
труба с тремя форсунками для распыления воды. Для очистки сте-
нок барабана от налипающего на них суперфосфата имеется спе-
циальный нож (из диабазовых плиток).
Получение гранулированного суперфосфата
869
Вода в форсунки подается под давлением 4—5 ат. Первая
форсунка, через которую вводится 70% воды, установлена на рас-
стоянии 6,5 м от выходного отверстия гранулятора, вторая — на
расстоянии 5 м и третья — на расстоянии 1 м от выхода. Сушка
суперфосфата производится в обычных барабанных прямоточных
сушилках70.
В следующих условиях 264 гранулирование протекает с высокими
показателями: барабанный гранулятор длиной 7,5 м и диаметром
1,4 м установлен с углом наклона 2°; барабан делает 7,5 об/мин
(окружная скорость 0,51 м/сек}-, при объеме суперфосфата в бара-
бане около 3 .w3 время окатывания составляет 7 мин. Обеспечи-
вается хорошее смачивание суперфосфата водой, распыляемой в
грануляторе форсунками (под давлением 4—5 атм}. Продукт в гра-
нуляторе смачивается до содержания 15,7% Н2О (на некоторых
заводах до 17—18% Н2О). Сушильный барабан диаметром 2,2 м
и длиной 14 м работает с производительностью до 24,8 т продукта
в час при влагосъеме до 70 кг/(ч-м3), что достигается установкой
топок с площадью колосниковой решетки 0,1 м2 на 1 м3 объема ба-
рабана и отсасывающего вентилятора производительностью 600 м3
на 1 м3 объема сушильного барабана. Температура продукта в
процессе сушки не должна превышать 85°, во избежание образо-
вания нерастворимых пиро- и метафосфатов кальция. Температура
топочных газов, поступающих в сушилку, 600°, а отходящих 100—
120°. Использование в качестве топлива природного газа дает зна-
чительную экономию, позволяет повысить качество продукта 282 и
улучшить санитарные условия.
Представляет интерес применение для сушки суперфосфата
аппаратов со взвешенным слоем 283’284, в которых процесс про-
текает в 2—3 раза интенсивнее, чем в сушильном барабане.
Готовый продукт (при переработке апатитового концентрата)
содержит 19,9—20,9% усвояемой Р2О5, не выше 2,3% свободной
Р2О5 и 3,0—4,5% влаги.
Ситовая характеристика гранулированного суперфосфата:
Размер частиц, jkjk
Количество
частиц, °/о
. +4. . .
-4+2. . .
-2+1. . .
-2 . . .
2,8-3,8
82,9 - 90,8
13,8-5,0
0,5-0,4
Для уменьшения слеживаемости гранулированного суперфосфа-
та и сохранения сыпучести его охлаждают перелопачиванием на
складе, а также в процессе его перемещения. На Кедайнском хи-
мическом комбинате продукт охлаждают воздухом в специально.
870
Гл. XXIV. Простой суперфосфат
сконструированном аппарате со взвешенным слоем (рис. 231).
Гранулированный суперфосфат после сушилки поступает на на-
клонно расположенную сетчатую раму 12 по точке через затворный
питатель и скатывается в выгрузное устройство, снабженное за-
твором. В перекрестном направлении движению суперфосфата по
отверстиям в раме проходит охлаждающий воздух. Производи- ?
тельность аппарата длиной 3 м, шириной 1,4 м и высотой 2,5 ж при
поверхности сита 2,3 м составляет 13—14 т/ч продукта. В зимних
условиях при температуре поступающего воздуха —5° суперфос-
фат охлаждается до 35°, в летних — при температуре воздуха
30° — до 50—45°.
Рис. 231. Схема устройства для охлаждения гранулиро-
ванного суперфосфата конструкции Кедайнского химиче-
ского комбината:
1 — корпус; 2 — осветительный прибор; <3 — течка; 4 — питатель-затвор;
5 —контргрузы питателя-затвора; 6 — механизм для изменения угла
наклона сеточной рамы; 7 —люк; 8 — пусковой клапан; 9 — поворачи-
ваемые лопастн; /0 —затвор для отвода охлажденного продукта;
//—заслонка; /2 —сетчатая рама; 13 — смотровое окно.
В процессе сушки суперфосфата выделяются газообразные со-
единения фтора. Их количество зависит от степени нейтрализации
свободной Р2О5. Чем глубже нейтрализация, тем меньше выде-
ляется фтора, что объясняется уменьшением парциального давле-
ния пара SiF4 при понижении концентрации свободной Н3РО4 в
жидкой фазе суперфосфата 285. При указанной выше степени ней-
трализации суперфосфата выделяется до 13% фтора от его содер-
жания в суперфосфате 286. Газы из сушильного барабана проходят
циклонный пылеуловитель и поступают затем в абсорберы, оро-
шаемые водой, для улавливания газообразных фтористых соедине-
Получение гранулированного суперфосфата
871
ний. Сточные воды нейтрализуются известью или другими веще-
ствами, например нефелином 287'288.
За счет сушки выход гранулированного суперфосфата умень-
шается по сравнению с количеством вызревшего суперфосфата, не-
смотря на введение известняка для нейтрализации свободной кис-
лотности: выход простого суперфосфата на единицу апатитового
концентрата равен 1,876, а гранулированного— 1,775.
Процесс гранулирования нуждается в значительных усовершен-
ствованиях. В описанном оформлении он громоздок и содержит
серьезное противоречие. Современные непрерывные способы разло-
жения фосфатов дают возможность применять концентрированную
серную кислоту и получать суперфосфат с минимальной влажно-
стью— 10%; при гранулировании же суперфосфат увлажняют до
16—18%. Эта излишняя влага удаляется затем в сушильном бара-
бане, что связано с дополнительной затратой топлива. Для заво-
дов, выпускающих только гранулированный продукт, может пред-
ставить интерес гранулирование суперфосфата, полученного
разложением апатита кислотой пониженной концентрации
(стр. 847).
На некоторых заводах применяются грануляторы (наклонные
чаши или диски, планетарные смесители и др.), работающие без
увлажнения и сушки суперфосфата 263> 268.
Разработан метод гранулирования без увлажнения и сушки су-
перфосфата, получаемого из апатитового концентрата 266’272. В гори-
зонтальном двухвалковом смесителе в течение 2 мин производится
пластификация вызревшего суперфосфата, который затем окаты-
вается в гранулы в барабане. Гранулы припудриваются с поверх-
ности нейтрализующими добавками или аммонизируются для сни-
жения кислотности и придания им прочности. Дальнейшая обра-
ботка обычная — рассев на ситах и дробление крупной фракции.
Получены гранулы с высокой прочностью; выход продукционной
фракции —4 +1 мм составляет около 60%, и фракции —1 мм не
более 4—5%.
Некоторые из описанных выше непрерывных методов производ-
ства суперфосфата сочетаются с гранулированием 272 ’ 289' 29°. На-
пример, разложением легко разлагаемого фосфата в непрерывном
горизонтальном смесителе и сушкой суперфосфатной пульпы в су-
шильном барабане можно одновременно получить гранулирован-
ный продукт.
Этого можно также достигнуть, если гранулированию под-
вергнуть смесь суперфосфата и фосфоритной муки, к которой до-
бавлена 55—70%-ная серная кислота в количестве, необходимом
Для разложения фосфорита 201.
Получение гранулированного суперфосфата, особенно из фосфо-
ритов Каратау, целесообразно сочетать с его аммонизацией. Это
позволяет исключить расход известняка на предварительную нейт-
872
Гл. XXIV. Простой суперфосфат
рализацию суперфосфата, увлажнение его и сушку продукта. При-
менение аммонизированного гранулированного . суперфосфата
(вместо нейтрализованного известняком) имеет ряд преимуществ
в условиях среднеазиатских республик, почвы которых содержат
более 20% известняка.
Нейтрализация суперфосфата, полученного из фосфоритов ।
Каратау, аммиаком до pH = 4—4,5 не приводит к снижению усво-
яемой Р2О5. Содержание водорастворимой Р2О5 снижается лишь
на 12—18% и составляет 73—82% от усвояемой Р2О5. За счет теп-
ла, выделяющегося при аммонизации, влажность продукта сни- ’
жается на 4—5%. Содержание азота в конечном суперфосфате со- '
ставляет 0,7—1,4%.
При аммонизации суперфосфата из фосфоритов Каратау амми-
акатами аммиачной селитры или карбамида можно получить
сложное гранулированное удобрение с хорошими физическими и
агрохимическими свойствами, содержащее 20—21% питательных "
веществ, в том числе 5—6% азота 292'293.
Переработкой природного фосфата смесью серной кислоты и
сульфата аммония (бисульфатом аммония) образуется 294'295 азот-
но-фосфорное удобрение, так называемый азотированный супер-
фосфат, содержащий 6—10% N и 10,5—14% усвояемой Р2О5.
Предварительное прокаливание 296 фосфорита Каратау при 350°
ускоряет разложение его смесью серной кислоты и сульфата аммо-
ния. За 1 ч степень разложения его смесью 3 вес. ч. H2SO4 и 1 вес. ч. fl
(NH4)2SO4 достигает 99%. fl
На рис. 232 показана технологическая схема производства ам- fl
ионизированного гранулированного суперфосфата на Самарканд- •
ском суперфосфатном заводе 297.
Вызревший кислый суперфосфат после двухкратного измельче-
ния на шабмашинах (разбрасывателях) поступает в отделение
аммонизации, где с помощью распределительных транспортеров
и питателей подается в четыре аммонизационных барабана. Жид-
кий аммиак из танков-хранилищ направляется в испарительную
станцию. В испарителе, обогреваемом горячей водой, жидкий ам-
миак превращается в газ. Газообразный аммиак из испарителя
поступает в аммонизатор-гранулятор под слой суперфосфата.
Нейтрализованный продукт из аммонизатора-гранулятора по-
средством транспортеров подается на склад через разбрасыватель-
шабмашину. На складе суперфосфат в течение 3—4 суток перело-
пачивается 3—4 раза мостовым грейферным краном. Происходя-
щее при этом понижение температуры суперфосфата до его
упаковки благоприятно сказывается на его физических свойствах.
Аммонизированный суперфосфат, охлажденный в течение 3—4 су-
ток трехкратным перелопачиванием до 28—32°, не слеживается и
сохраняет полную сыпучесть в течение 4—5 месяцев. а
Получение гранулированного суперфосфата
873
Охлажденный материал краном подается в бункер, затем с по-
мощью транспортеров поступает на фракционный рассев и упа-
ковку. Крупная фракция после дробилки возвращается на рассев.
Степень использования аммиака составляет 93—96%. Выхлопные
газы из аммонизатора содержат 5—8 г/м5 NH3 и направляются на
водную абсорбцию. Образующаяся при промывке газов аммиачная
вода концентрации 2—2,5% NH3 подается для сиропки 75 %-ной
серной кислоты, используемой для получения суперфосфата.
Для получения 1 т аммонизированного гранулированного су-
перфосфата расходуется 1,02 т кислого суперфосфата и ~ 190 кг
аммиака.
Примерный состав получаемого продукта (в %): общая
Р2О5—16,0—16,8; усвояемая Р2О5—15,0—15,5; водорастворимая
Рис. 232. Схема производства аммонизированного суперфосфата:
/ — абсорбционная башня; 2 — вентилятор; 3 — аммоннзатор; 4 — транспортер; 5 — разбрасыва-
тель; 6 — грейферный кран; 7 —бункер; 8 — железнодорожный вагон; Р—мешкозашивочная
машина, 10 — автоматические весы; 11 — молотковая дробилка; 12 — грохот; 13 — элеватор.
Р2О5—11,5—12; азот 1,7—1,8; влага 7—8,5; частиц с размерами
более 4 мм — 9—11, частиц с размерами более 5 мм — 5,0—6,0;
степень разложения фосфорита в готовом продукте 91—93%. Гиг-
роскопическая точка аммонизированного суперфосфата при 25°
составляет 75—80%.
874
Гл. XXIV. Простой суперфосфат
ЛИТЕРАТУРА
1. С. И. Вольфкович, Л. В. Владимиров, Труды НИУ, в. 55 1928. —
2 Н. L. Marshall a. oth., Ind. Eng. Chem., 25, 1253 (1933). — 3. S. К r fl g e 1,
A. Ret ter, Superphosphate, № 3, 57 (1930). — 4. C. Krflgel, A. Ret ter,
ibid., № 10, 247 (1931). —5. W. L. Hill, S. B. Hendricks, Ind. Eng. Chem.,
28, 440 (1936). — 6. Л. M. Орлова, Труды НИУИФ, в. 137, 1937, стр. 87.—
7. П. Я. Крайний, А. М. Матвеенко, Научн. зап. Одесск. политехи, ин-та,
2, № 2 (1954). — 8. М. Е. П о з и н, Б. А. К о п ы л е в, В. Л. Варшавский,
Труды ЛТИ им. Ленсовета, в. 36, 1956, стр. 51. — 9. Р. Н. Иванов, ДАН
УзССР, № 8, 37 (1957). — 10. Труды НИУИФ, в. 141, 1938.
11. Р. F. Fleury, Ind. chim., 40, № 437, 351—355 (1953). —12. Ф. Н. Кель-
ма н и др. Методы анализа и контроля производства серной кислоты и фосфор-
ных удобрений, Госхимиздат, 1963. — 13. Р. X. Ошеровйч, Зав. лаб., № 7
(1952). — 14. Б. С. Кавнатская, М. Г. Мамаджанянц, Хим. пром., № 12,
373 (1952); № 8, 295 (1953). — 15. Р. X. Ошеровйч, Автореф. канд. дисс.,
НИУИФ, 1957. —16. I. R. Archer, R. Р. Thomas, Agr. Food. Chem., 4, № 7,
608—613 (1956). —17. Л. В. Владимиров, М. М. Кошелева, сб. «Иссле-
дования по прикладной химии», Изд. АН СССР, 1955, стр. 298. —
18. С. И. Вольфкович, Л. В. Владимиров, Труды НИУ, в. 55, 1928,
стр. 73. — 19. Lenglen, М i 1 h i е t, Superphosphate, № 5, 49 (1932). —
20. W Ross, R. Merz, Am. Fertilizer., № 3, 21 (1928).
21. Л. Берлин, О. Вознесенская, О. Никонова, Р. Фискина,
ЖХП, № 1, 24 (1936). — 22. Н. Е. Пестов, Физико-химические свойства зерни-
стых и порошкообразных химических продуктов. Изд. АН СССР, 1947.—
23. Н. Е. Пестов, Н И. Николаева, Хим. пром., № 12, 375 (1951).—
24. Н. Е. Пестов, А. Й. Шерешевский, Е. И. Пауткина, Там же, № 8,
297 (1953). — 25. И. Л. Гофман, Е. Е. 3 у с с е р, Д. Л. Ц ы р л и н, А. И. Ш е-
решевский, сб. «Гранулированный суперфосфат». Труды НЙУИФ, в. 157, 1955,
стр. 7. — 26. Н. Е. Пестов, Т. В. Глазова, Хим. пром., № 9, 281 (1951).—
27. Н. Е. Пестов, А. Н. Миронова, Там же, № 2 (1940),—28. A.. Meh-
ring, Ind. Eng. Chem., Anal. Ed., 1, 34 (1931).—29. Б. Евзлина, Труды
НИУЙФ, в. 143, 1938, стр. 31.—30. Н. Е. Пестов, Е. И. Па утки на, сб. «Ис-
следования по прикладной химии», Изд. АН СССР, 1955, стр. 290.
31. Д, Г. Грачев, сб. «Гранулированный суперфосфат», Труды НИУИФ,
в 157, 1955, стр. 61. — 32. В. П. Пл иг у нов, Научн. зап. Одесск. политехи,
ин-та, 2, № 1 (1955). — 33. В. Ваггаман, Фосфорная кислота, фосфаты и
фосфорные удобрения, Госхимиздат, 1957. — 34. Ф. В. Турчин, Минеральные
удобрения и их применение, Изд. АН СССР, 1943.—35. W. L. Hill, Е. I. Fox,
J Е. М i 11 i n s, Ind. Eng. Chem., № 7 (1949). — 36. А. В. Соколов, Агрохимия
фосфора, Изд. АН СССР, 1950. — 37. Меченые атомы в исследованиях питания
растений и применения удобрений. Труды совещания, Изд. АН СССР, 1955.—
38. А. В. Соколов, Удобр. и урожай, № 2 (1956).— 39. Ф. В. Турчин, Хим.
пром., № 3 (1957). — 40. А. В. Соколов, Химические проблемы земледелия
СССР, VIII Менделеевский съезд, Изд. АН СССР, 1959.
41. S. N о г d е n g г е и, Fertilizer Society Papers, № 2, 12 (1947).— 42. А. В. С о-
колов, сб. «Гранулированный суперфосфат». Труды НИУИФ, в. 157, 1955,
стр. 74, 113, 137. — 43. М. Л. Чепелевецкий, Е. Б. Бруцкус, Суперфос-
фат. Физико-химические основы производства, Госхимиздат, 1958.— 44. И. В. Ба-
бенко, Удобр. н урожай, № 11, 17 (1958).—45. А. П. Крючкова, П. И. Фи-
шер, Хим. наука и пром., 2, в. 4 (1957). — 46. С. И. Вольфкович, Усп. хим.,
25, в. 11 (1956).—47. П. В. Дыб ина, А. А. Соколовский, Сборник статей
ВЗПИ, в. 19—20, 1957, стр. 171.—48. С. К. Воскресенский, Научно-техн,
информ, бюлл. НИУИФ, Я» 1 (1957). — 49. Э. В. Брицке, Производство супер-
фосфата, Рига, 1910. — 50. L. Schucht, Die Fabrikation des Superphosphates,
Braunschweig, 1926.
Литература
875
51. А. А. Быков, Производство простого суперфосфата, Гос. научно-техи.
изд-во Украины, 1953. — 52. Н. М. Вайнман, П. П. Тюрин, Производство су-
перфосфата, Госхимиздат, 1933. — 53. А. А. Соколовский, Суперфосфаты.
Справочник по удобрениям, Госхимиздат, 1933. — 54. Везер, Русско-герм. вести,
науки и техн., № 11 (1934). — 55. A. Debaisieux, Rev. Prod. Chim., № 12—13
(1934). — 56. M. Shoe Id, Chem. Met. Eng., 41, № 4, 178 (1934). —57. Б. А. Со-
коловский, Минеральн. удобрения и инсектофунгисиды, Ns 2, 10 (1935).—
58. Н. М. Вайнман, Операционное отделение суперфосфатного производства,
ОНТИ, 1936. — 59. И. Гофман, Я. Зильберман, И. Островский, Тех-
нология суперфосфата и других фосфорных удобрений, Химтеорет, 1937.—
60. W. Packard, Superphosphate, 10, 221 (1937); 11, 7, 27, 41 (1938).
61. А. И. Шерешевский, А. А. Соколовский, П. Ф. Деревиц-
к и й, Производство фосфорных удобрений, ГОНТИ, 1938. — 62. Р. Parrish,
A. Ogilvie, Calcium Superphosphate and Compound Fertilizers, London — New
York, 1946.—63. A. M. Дубовицкий, А. И. Шерешевский, Технология
минеральных удобрений, Госхимиздат, 1947.—64. R. L. Demrnerle, W. I. Sa-
ckett, Ind. Eng. Chem., 41, Ns 7, 1306 (1949). — 65. С. И. Вольфкович,
А. П. Егоров, Д. А. Эпштейн, Общая химическая технология, т. I, Госхим-
издат, 1952. — 66. В. В. Андреев, Хим. пром., Ns 12, 370 (1952).—67. Fertilizer
Technology and Resources in the United States, New York, 1953. — 68. Д. Л. Цы p-
лин, ЖПХ, 28, № 10, 1025 (1955). — 69. I. T. Procter, Ind. Chim., 41, Ns 438,
6 (1954).—70. В. С. Демин, Производство фосфоритной муки, суперфосфата
и кремнефтористого натрия, Госхимиздат, 1955.
71. Э. Н. Гинзбург, Научно-техн, информ, бюлл. НИУИФ, № 5, 19
(1956). — 72. М. Е. Поз ин, Технология минеральных удобрений и солей, Гос-
химиздат, 1957. — 73. Д. Л. Цырлин, Исследование по производству минераль-
ных удобрений, изд. НИУИФ, 1957. — 74. В. А. Кононов, сб. «Промышленные
минеральные удобрения», Госхимиздат, 1958, стр. 185. — 75. Technologia Zwiaz-
kow Fosforowych, PWT, Warszawa, 1958. — 76. A. V. Slack, Farm. Chemie., 122,
Ns 4, 61 и Ns 5, 54 (1959). — 77. к. V. Slack, Суперфосфат, под ред. А. А. Со-
коловского, Изд. «Химия», 1969, стр. 60. — 78. С. И. Вольфкович, Л. Е. Б е р-
лин, А. Винокурова, А. Салов а, ЖХП, Ns 15—16, 1 (1931). — 79. М. Л. Ч е-
пелевецкий, Е. Б. Бруцкус, Б. Б. Евзлина, Т. А. Крюкова
Л. М. Орлова, С. С. Р у б и н о в а, Труды НИУИФ, в. 137, 1937. — 80. М. Л. Ч е-
пелевецкий, Е. Б. Бруцкус, Исследования по физико-химическим основам
производства суперфосфата, изд. НИУИФ, 1957.
81. А. П. Б е л о п о л ь с к и й, А. А. Т а п е р о в а, М. Н. Шульгина, ЖПХ,
12, Ns 1 (1939); 13, № 5 (1940); 18, Ns 9—10 (1945); 23, Ns 1 (1950).—
82. A. N. Campbell, J. W. C out is, Ind. Eng Chem., 40, Ns 8, 1295 (1948).—
83. A. S a n f о u r c h e, A. К r a p i v i n e, Bull. Soc. Chim., 53, Ns 12, 1573 (1933).—
84. M. Ч e п e л e в e ц к и й, E. Бруцкус, 3. P о д о в а, Труды НИУИФ, в. 147,
1940. — 85. Н. L. Marshall, S. В. Hendricks, W. L. Hill, Ind. Eng. Chem.,
32, 12, 1631 (1940). — 86. A. A.. Ke tel a ar, D. Heimann, Rec. trav. chim., 43,
Ns 4, 279 (1954). — 87. А. П. Белопольский, A. A. T а п ер о в а, ЖХП, 15,
№ 3, 44 (1937). — 88. E. Б. Бруцкус, Труды НИУИФ, в. 137, 1937, стр. НО.—
89. М. Л. Чепелевецкий, Т. А. Крюкова, Там же, стр. 102.—
90. Е. Б. Бруцкус, сб. «Исследования по прикладной химии»,.Изд. АН СССР,
1955, стр. 184.
91. М. Л. Ч е п е л е в е ц к и й, Е. Б. Бруцкус, 3. А. Р о д о в а, ЖХП, Ns 11,
8 и № 21, 3 (1941). — 92. Е. Б. Бруцкус, М. Л. Ч е п е л е в е ц к и й, ИСФХА
АН СССР, 20, 383 (1950). — 93. Е. В. Южная, Автореф. канд. дисс., НИУИФ,
1956. — 94. М. Е. Поз ин. Б. А. Копы л ев, И. Ф. Ефремов, В. Л. Вар-
шавский, А. С. Маркович, Коллоид, ж., 27, № 4, 593 (1965).—
95. Н. А Толстой, А. А. Спартаков, Г. И. Хилько, Там же, 22, № 6,
705 (I960). — 96. И. Ф. Ефремов, Там же, 22, № 3, 276 (I960).—
876
Гл. XXIV. Простой суперфосфат
97. С. И. Вольфкович, Л. Е. Б е р л и н, Л. Б. Грнншпан, Хибинские апа-
титы, под ред. А. Е. Ферсмана, Изд. Гостреста «Апатит», 1930, стр. 139. —
98. С. Вольфкович, Л. Берлин, А. Винокурова, А. Логинова,
А Салов а, А. Соколовский, Б. Соколовский, С. Перельман,
Н. Постников, Ю. Рабинович, Труды НИУ, в. 95, 1932. — 92. Б. А. С о-
коловский, Как работать на апатитовом сырье, Химтеорет, 1932.—
100. Л. Е. Берлин, Хибинский апатит и переработка его на суперфосфат, Гос-
химиздат, 1933.
101. Л. В. Владимиров, Е. Е. Бабкин и др., ЖХП, № 9 (1938). —
102. И. И 3 а р и н г, Там же, № 2 (1940). — 103. Н. Крючков, Я. П рже-
го д с к а я, Труды НИУИФ, в. 147, 1940. — 104. И. Л. Г о ф м а и, А. И. Сен-
гер, ЖХП, № 1 (1941).— 105. Е. Б. Бруцкус, Д. Л. Цырлин, Хим. пром.,
№ 10, 299 (1951). — 106. М. Е. П о з и н, Г. С. Григорьев, Б. А. К о п ы л е в,
А. Д. Соколова, ЖПХ, 31, № 5, 693 (1958). — 107. А. И. Шер ешевский,
ЖХП, № 7 (1940); Реф. н.-и. раб. за 1956 г., ч. I, 12, Труды НИУИФ, в. 161,
1958. — 108. М. Е. П о з и н, Б. А. К о п ы л е в, Тен Се-ден, Труды ЛТИ
им. Ленсовета, в. 36, 1956, стр. 5. —109. R. I. Nunn, Т. R. Dee, J. Food Agr., 5,
№ 6, 256 (1954). — НО. Л. Б. Г р и н ш п а н, сб. «Исследования по прикладной
химии», Изд. АН СССР, 1955, стр. 192.
111. Л. Б. Г р и н ш п а н, М. М. Кобрин, Хим. пром., № 3, 60 (1950).—
112. Д. Л. Цырлин, Реф. н.-и. раб. за 1957 г., т. I, 21, Труды НИУИФ, в. 163,
1959. —113. W. L. Hill, I. Н. Caro, G. A. Wieczorek, J. Agr. Food Chem.,
2, № 25, 1273 (1954). —114. F. С о r b о n a, Ind. chim., № 463, 41 (1956).—
115. M. Л. Чепелевецкий, ИСФХА АН СССР, 19 (1949).— 116. К. С. К Р а с-
нов, ЖПХ, 26, № 11, 1114 (1953). — 117. К. С. Краснов, Там же, 28, № 12,
1275 (1955). — 118. М. Е. Поз ин, Б. А. К о п ы л е в, Д. Ф. Жильцова, Там
же, 32, № 10, 2164 (1959). — 119. А. П. Белопольский А. А. Таперов а,
М. Т. Серебренникова, М. Н. Шульгина, ЖХП, 14, № 7, 504, 660
(1937). —120. К- L. Elmore, Т. D. Farr, Ind. Eng. Chem., 32, 580 (1940).
121. А. П. Белопольский, M. T. Серебренникова, А. В. Биле-
вич, ЖПХ, 13, 1 (1940).— 122. А. В. Казаков, Труды НИУИФ, в. 139,
1937. — 123. Д’А нс, К н ю т тер, Angew. Chem., 65, № 23 (1953). — 124. М. Л. Ч е-
п е л е в е ц к и й, Ц. С. Больц, Н. А. Василенко, С. С. Р у б и и о в а, сб. «Ис-
следования по прикладной химии», Изд. АН СССР, 1955, стр. 175.—
125. Н. I. Bassett, Chem. Soc., № 9, 2949 (1958). — 126. Н. Д. Л е в а д н ы й,
ЖХП, № 6, 45 (1940). —127. И. И. 3 а р и н г, Там же, № 2, 6 (1941).—
128. Д. Н. Шевченко, А. В. Юлиш, Хим. пром., № 1, 19 (1952).—
129. Л. Берлин, Л. Горицкая, А. Сергеева. Труды НИУИФ, в. 143,
1938, стр. 31. —130. Л. Берлин, Л. Горицкая, А. Сергеева, Там же,
в. 147, 1940.
131. Л. В. Владимиров, Н. К. Падо, С. К. Воскресенский, Сооб-
щения о научно-технических работах НИУИФ. Обмен опытом, в. 2, Госхимиздат,
1957, стр. 23. —132. G. L. Bridger, Е. С. К а р u s t a, Ind. Eng. Chem., 44,
№ 7, 1540 (1952). —133. W. Drobot, Jowa State Coll. J. Sci., 29, № 3, 405
(1955).— 134. E. E. Зуссер, Сообщения о научно-технических работах НИУИФ,
№ 2, Госхимиздат, 1957, стр. 20. —135. G. L. Bridger, I. L. Kearns, J Agr.
Food. Chem., 4, № 6, 526 (1956). — 136. G. L. Bridger, W. Drobot, ibid., 4,
№ 6, 526 (1956). — 137. R. D. Young, F. G. Heil, ibid., 5, № 9, 682 (1957). —
/38. E. E. Зуссер, Хим. пром., № 2, 177 (1959). — 139. M. Е. П о з и н,
Б. А. К о п ы л е в, Новые методы получения минеральных удобрений, Госхим-
издат, 1962. — 140. М. С е г п у, V. Lakota, I. Bartos, пат. ЧССР 115285, 1965.
141. М. Л. Варламов, И. М. К а г а н с к и й, А. М а н а к и н, Т. Ф. Том-
чик, Межвузовская республиканская конференция по химии и химической тех-
нологии, Днепропетровск, 1967, стр. 32. — 142. Е. Е. Зуссер, В. М. Р а м м,
Сообщения о иаучио-техиических работах НИУИФ, № 2, 1957, стр. 25. —
Литература
«ГГ
143. Е. Е. Зуссер, Реф. н.-и. раб. за 1956 г., ч. I, стр. 11. Труды НИУИФ,
в. 161, 1958; Реф. и.-и. раб. за 1957 г., ч. I, стр. 19. Труды НИУИФ, в. 163,
1959. — 144. А. М. Левин, Э. И. Цыпина, Сообщения о научно-технических
работах НИУИФ, № 2, стр. 21, Госхимиздат, 1957. — 145. Хаусюз Шицзе, 20,
№ 3, 121 (1966). —146. Е. I. Fox a. oth., J. Agr., Food Chem., 2, № 12, 618
(1954). — 147. E. I. Fox, I. O. Hardesty, R. Kumagai, Farm. Chem., 117,
№ 9, 43 (1954). —148. R. Kumagai, I. O. Hardesty, J. Agr. Food Chem., 3,
№ 1, 34 (1955). —149. F. A. Retzke, G. F. S a c h s e 1, R. B. Filber, ibid.,
3, № 6, 496 (1955). —150. Э. H. Гинзбург, Д. Л. Цырлин, В. H. Л а м п.
Рефераты н.-и. раб. за 1956 г., ч. I, стр. 14, Труды НИУИФ, в. 161, 1958.
151. R. R. R a u n s 1 е у, D. R. Boylan, J. Arg. Food Chem., 6, № 9, 677
(1958). — 152. Л. П. А а р е т, Я. Я. А н с о, М. А. В ей дерм а, Ю. К- Труз,
Труды Таллинск. политехи, ин-та, А, № 230, 1965, стр. 87. —153. М. Е. П о-
з и н, Б. А. Копылев, Б. Д. Г ул л ер, Хим. пром., № 9, 43 (1967).—
154. Н. L. Marschall, W. L. Hill, Ind. Eng. Chem., № 7, 1537 (1952).—
155. А. П. Белопольский, С. Я. Шпунт, M. H. Шульгина, ЖПХ, 23,
№ 8 (1.950); 24, № 4 (1951); 26, № 3 (1953); 27, № 4, 5 (1954). — 156. А. П. Б e-
л о польский, С. Я. Шпунт, М. Н. Шульгина, Там же, 27, № 5,
493 (1954). — 157. А. П. Б е л о п о л ь с к и й, С. Я. Шпунт, М. Н. Шульгина,
сб. «Исследования по прикладной химии», Изд. АН СССР, 1955, стр. 107.—
158. К. С. Краснов, Изв. вузов. Хим. и хим. технол., № 3, 100 (1958).—
159. А А. Соколовский, ЖПХ, 28, № 5, 743 (1956). — 160. А. А. Вишня-
кова, М. Н. Набиев, Изв. АН УзССР, № 11, 25 (1956).
161. А. А. Вишнякова, М. Н. Набиев, Изв. АН УзССР. Сер. хим. и.,
№ 2, 5 (1957). — 162. М. Н. Н а б и е в, А. А. Вишнякова, ДАН УзССР, № 2,
25 (1957). —163. А. А. Вишнякова, Автореф. канд. дисс.. Ин-т химии АН
УзССР, 1957. —164. А. А. Вишнякова, сб. «Материалы объединенной науч-
ной сессии по хлопководству», т. 1, Госиздат УзССР, 1958, стр. 416.—
165. М. Е. П о з и н, Б. А. Копылев, А. С е й т м а г з и м о в, авт. свид. 137934,
1961. — 166. М. Е. Поз ин, Б А. Копылев, Новые методы получения мине-
ральных удобрений, Госхимиздат, 1962, стр. 44, 75. —167. С. С. Караханяв,
Г. О. Григорян, Р. Л. М и р v м я н, Изв. АН АрмССР. Хим. н., 18, № 6, 615-
(1965); М. Л. Чепелевецкий, Е. Б. Бруцкус, В. М. Р а м м, Е. В. Юж-
ная, Е. П. В е т ч и н к и н, Там же, стр. 15. — 168. А. И. Шерешевский, Ре-
фераты н.-и. раб. за 1956 г., ч. I, стр. 8, Труды НИУИФ, в. 161, 158.—
169. Е. Б. Бруцкус, А. И. Л и ц о в а, Л. И. Кон о в а л о в а, Реф. н.-и. раб. за
1957 г., ч. I, стр. 8, Труды НИУИФ, в. 163, 1959. — 170. W. McIntire,
G. Shuey, Ind. Eng. Chem., 24, 8, 933 (1932).
171. С И. Вольфкович, H. E. Пестов, сб. «Технические сдвиги ино-
странной химической промышленности за годы экономического кризиса», 1936,
стр. 131. — 172. Р. Е. Ремен, В. И. Головкин, ЖХП, № 3, 26 (1938).—
173. Л. Берлин, О. Вознесенская, Л. Горицкая, А. Сергеева,
Труды НИУИФ, в. 147, 1940. — 174. Р. Ремен, И. Демиденко, Там же.—
175. А. И. Шерешевский, Л. С. Горицкая, Л. И. Потапова, Сооб-
щения о научно-технических работах НИУИФ, № 2, Госхимиздат, 1957, стр. 22.—
176. А И. Шерешевский, Л. И. Потапова, Рефераты н.-н. работ за
1956 г., ч. I, стр. 16, Труды НИУИФ, в. 161, 1958. —177. С. И. Вольфкович,
Л. Е Б е р л и н, И. Л. Г о ф м а н, А. А. И о н а с, Удобр. и урожай, № 6 (1930);
№ 7—8 (1930). — 178. Н. Е. Пестов, Химстрой, № 12 (1931). — 179. Л. Бер-
лин, Л. Горицкая, ЖХП, № 2 (1932). — 180. F. G. К е е n е n, Ind. Eng.
Chem., 22, 1378 (1930); 24, 44 (1932).
181. W. H. Ross, К. D. Jacob, Fixed Nitrogen, New York, 1932.—
182. Л. E. Берлин, Труды НИУ, в. 113, 1933.— /83. A. R. Merz, United States
Departament of Agriculture, Circular 185 (1931).— 184. Л. Берлин, Л. Го-
рицкая, А. Заседателева, Удобрения и иисектофунгнсиды, № 4 (1935).—
878
Гл. XXIV. Простой суперфосфат
185. L. М. White, I. О. Hardesty, W Н. Ross, Ind. Eng. Chem., 27, 562
(1935). —186. Л. Берлин, Л. Горицкая, ЖХП, № 23, 1398 (1936).—
187. I. О. Hardesty, W. Н. Ross, Ind. Eng. Chem., 29, 1283 (1937).—
188. J. В. S. Holmes, Am. Fert., 95, № 9 (1941). — 189. E. M. Harvey,
L. V. Rohner, Am. Fert., Oct, 10 (1942). — 190. R. F 1 a 11, G. Brunisholz,
S. Chapius-Gottreux, Helv. chim. acta, 34, 3, 884 (1951).
191. R. C. D a t i n, E. A. W о r t i n g t о n, G. L. P о u d r i e r, Ind. Eng. Chem.,
44, № 4, 903 (1952). —• 192. L. D. J a t e s, F. T. Nielson, G. C. Nicks, Farm.
Chem., 117, Ns 7, 38, 41, 43, 45, 47; № 8, 34, 36, 40 (1954). — 193. R. F. Knight,
Ind. chim., 41, Ns 438, 4—5 (1954). — 194. M. Gorski, H. Stupnicka, Prze-
mysl. Chem., 10, № 3, 129 (1954). —195. Л. Б. Гриншпан, А. И. Шерешев-
ский, E. M. Курочкина и др., Сообщения о научно-технических работах
НИУИФ, № 2, 18, 19 (1957). — 196. Р. Н. Иванов, Изв. АН УзССР. Сер. хим.
н., № 1, 67 (1957); Хим. пром., № 2, 79 (1957).— 197. Н. В. Бабенко, Научно-
техн. информ, бюлл. НИУИФ, Ns 9, 74 (1957). — 198. В. Д. Б а з и л е в. Тезисы
докладов на отраслевом совещании работников основной и горнохимической про-
мышленности, 1958, стр. 97. — 199. М. Н. Набиев, 3. Н. Ф а р м а н о в,
А. А. Вишнякова, Узб. хим. ж., Ns 1, 7 (1958). — 200. А. И. Шерешев-
ский, Е. М. Курочкина, Рефераты н.-и. раб. за 1956 г., ч. I, стр. 15, Труды
НИУИФ, в. 161, 1958.
201. Р. Н. Иванов, С. Я. Шпунт, Сборник аспирантских работ НИУИФ,
1959, стр. 35. — 202. Р. Е. Р е м е н, Е. И. Петрова, Реф. н.-и. работ за 1957 г.,
ч. 1, стр. 47, Труды НИУИФ, в. 163, 1959. — 203. V. W. Harwood, Agr. Chem.,
14, Ns 5, 42, 103 (1959). — 204. M. H. Набиев, А. А. Вишнякова, Аммони-
зированный суперфосфат из фосфоритов Каратау, Изд. АН УзССР, 1960. —
205. W. Kowalski, Przem. Chem., 41, Ns 10, 563 (1962). — 206. В. Головкин,
Р. Ре мен, Л. Владимиров, А. Сергеева, Труды НИУИФ, в. 143, 1938,
стр. 13.—207. В. В. Буткевич, Хим. пром., № 6, 340 (1954).—
208. Д. Л. Ц ы р л и н, Научно-техн, информ, бюлл. НИУИФ, Ns 9, 3 (1957).—
209. Д. Л. Цырлин, Тезисы докладов на отраслевом совещании работников
основной и горнохимической промышленности, 1958, стр. 53. — 210. Д. А. Пат-
рушев, М Н. Стеклова, Научно-техн, информ, бюлл. НИУИФ, № 9, 15
(1957).
211. В. В. Андреев, Тезисы докладов на отраслевом совещании работни-
ков основной и горнохимической промышленности, 1958, стр. 57. —
212. Ю. П. Иваненко, Там же, стр. 60. — 213. С. Вольфкович, Труды
НИУ, в. 55, 1928, стр. 29. -214. W. L. Hill, К. С. Beeson, J. Assoc. Offic.
Agr. Chemists, 19, 328 (1936).— 215. И. И. 3 a p и н г, ЖХП, № 12 (1938).—
216. И. И. Зар инг, Там же, № 7 (1939).—2/7. W. L. Hill, К- D. Jacob,
Mining Eng., Sec., 1, 6, Ns 10, 994 (1954).—218. R. Donald, Chem. Trade J.,
133, Ns 3467, 1197 (1953). — 219. V. C h a г г i n, Chim. Ind., 72, № 3, 524 (1954). —
220. K- A. Sherwin, Chem. Ind., Ns 41, 1274 (1956).
221. W. Augustyn, Przem. Chem., 38, № 3, 137 (1959).—222. E. Hollan-
der, P. Kerengi, Chem. prumsyl., 16, Ns 8, 478 (1966).—223. M. E. П o-
зин, Б. А. Копыле в, Г. В. Бельченко, Л. Я. Терещенко, Расчеты по
технологии неорганических веществ, Изд. «Химия», 1966. — 224. Г. С. Д у р а с с,
Бюлл. техн, информ. Гипрохима, Ns 39, 3 (1958). — 225. С. В. Кузнецов, Ав-
томатизация производства суперфосфата, Госхимиздат, 1956. — 226. Э. Н. Гинз-
бург, Хим. пром., Ns 11, 346 (1949); Ns 5, 152 (1952). — 227. В. О г и е в и ч,
Пром.-эконом, газета, Ns 137 (281), 1957. — 228. С. Д. Э венчик, В. А. Коно-
нов, Бюлл. техн. инф. Гипрохима, Ns 40—41, 3 (1958).—229. С. В. Кузне-
цов, Вестник техн, и эконом, информ., Ns 2 (7), (1958).— 230. R. В. Risk, Ind.
chim., 41, № 438, 6 (1954).
231. М К u 1 с s а г, S. О г s о s, Nehezvegyipari kutato int. kozl., 3, Ns 1—2,
51 (1966). — 232. Э. H. Гинзбург, Д. Л. Цырлин и др., Рефераты н.-и.
Литература
879
раб. за 1956 г., ч. 1, стр. 10, Труды НИУИФ, в. 161, 1958. — 233. Э. Н. Гинз-
бург, Хим. пром., № 3, 8 (1945). — 234. Ф. В. Захаров, М. Н. Волобуев,
Обмен опытом, сб. Гос. Комитета по химии, № 6 (51), 34 (1958).—
235. М. Д. Кондон и, Д. Н. Шевченко, Там же, стр. 74. — 236. К- А. По-
ляков, Ф. Б. Сломянская, К. К- Полякова, Коррозия и химически стой-
кие материалы, Госхимиздат, 1953. — 237. А. И. Р е й б м а н, М. И. Финкель-
штейн, Хим. пром., № 3, 150 (1955).— 238. К- А. Поляков, М. А. Гур-
фи и к е л ь, Коррозия и способы защиты оборудования в сернокислотной про-
мышленности, Госхимиздат, 1955. — 239. В. В. Андреев, Хим. пром., № 5,
299 (1956). — 240. И. А. Егоров, Фаолит и его применение в химической про-
мышленности, Госхимиздат, 1956.
241. А. И. Рейбман, М. И. Финкельштейн, Хим. пром., № 8, 493
(1956).—242. К. Г. Бергман, Б. И. Леви, Сообщ. о научно-техн, работах
НИУИФ, № 3, 41 (1957). — 243. Г. Г. Ш а м к о, Бюлл. техн. инф. Гипрохима,
№ 39, 36 (1958). — 244. G. В1 gе о п, Ind. chim., № 490, 130 (1958).—
245. Б. И. Л е в и, А. А. Ежова, Рефераты н.-и. раб. за 1956 г., ч. II, стр. 38,
Труды НИУИФ, в. 162, 1959.—246. Материалы совещания по автоматизации
производственных процессов в химической промышленности, ЦБТИ Мин. прибо-
ростроения и средств автоматиз. СССР, 1956.—247. С. В. Кузнецов, Бюлл.
техн, информ. Гипрохима, № 40—41, 50 (1958).— 248. Е. Hollander, Р. К е г ё-
nyi, Chem. prumysl, 17, № 2, 60 (1967); 17, № 4, 211 (1967).—249. А. А. Со-
коловский, Технология фосфорных удобрений, изд. ВЗПИ, 1954.—
250. Я. к. Иоффе, Удобр. и урожай, Ks 7, 6 (1958); Реф. н.-и. работ за 1957 г.,
ч. II, стр. 93, Труды НИУИФ, в. 164 (1959).
251, И. В. Шокальская, сб. «Финансы и кредит», в. 3, 1966, стр. 119.—
252. А. М. Левин, Э. И. Цыпина, Реф. н.-и. работ НИУИФ за 1957 г., ч. II,
стр. 94, Труды НИУИФ, в. 164, 1959. — 253. А. Н. Г о л о в а и е в а, Хим. пром.,
№ 7, 42 (430) (1958). — 254. А. М. Левин, Э. И. Цыпина, Рефераты н.-и.
раб. за 1956 г., ч. 11, стр. 89, Труды НИУИФ, в. 162, 1959.—255. V. L о go-
me г а с, Technika, 21, № 3 (1966). — 256. Ю. А. Усманов, Сборник докладов
III межобластной конференции почвоведов и агрохимиков Среднего Поволжья
и Южного Урала, Куйбышев, 1964, стр. 5. — 257. А. М. Л е в и н, Э. И. Цы-
пина, Удобр. и урожай, № 6, 26 (1959). — 258. В. И. Гл а душ ко, Тезисы
докладов на Всесоюзном совещании по конденсированным фосфатам, Алма-Ата
(1968).—259. Nordengreen, Н. Lehrecke, Am. Fert., 93, № 1, 5—7, 24,
26; № 2, 10—11, 22 (1940).— 260. I. О. Hardesty a. oth, ibid, 92, № 7
(1940).
261. I. N. Mackall, M. S h о e 1 d, Chem. Met. Eng, 47, № 2, 102 (1940).—
262. I. T. Procter, Adress before Fertilizer Society of England, 1949.—
263. A Brunn e, F. Zwanzig, M. Koch, Chem. Techn, 5, № 7, 385 (1953).—
264. Д. H. Шевченко, Хим. пром, № 3, 1 (1955).— 265. 1. S. Martinet,
Agr. Chem, 9, № 4, 46, 138, 139, 141 (1954). — 266. Д. Л. Цырлин, E. E. 3yc-
cep, Сообщения о научно-технических работах НИУИФ, № 2, Госхимиздат, 1957,
стр. 27.-267. 1. Е. Reynolds, Agr. Chem, 12, № 2, 41, 105, 107 (1957).—
268 Н. Д. Таланов, Научно-техн, информ, бюлл. НИУИФ, № 2—3, 13
(1957). — 269. Т. Р. Hignett, Agr. Chem, 12, № 1, 30, 107, 109, 111, 113
(1957). — 270. A. Spillmann, ibid, 12, № 6, 42, 113 (1957).
271 T. P. Hignett, A. V. Slack, J. Agr. Food Chem, 5, № 11,814 (1957).—
272. E. E. 3 у с с e p, Тезисы докладов на отраслевом совещании работников ос-
новной и горнохимической промышленности, 1958, стр. 86. — 273. Л. Я. Копач,
Там же, стр. 90.—274. Ф. А. Серковский, Там же, стр. 92.—
275. В. А. Ставерский, Там же, стр. 94.-276. Д. Л. Цырлин, Рефераты
н.-и. раб. за 1956 г, ч. I, стр. 16, Труды НИУИФ, в. 161, 1958.—277. А. В. Р h i 1-
1 i р s, G. C. Hicks, I. E. Jordan, T. P. Hignett, J. Agr. Food Chem, 6,
№ 6, 449 (1958). — 278. H. R u m p f, Chem. Ind. Techn., 30, № 3, 144 (1958).—
880
Гл. XXIV. Простой суперфосфат
279. Comm. Fertilizer, 96, № 5, 34 (1958). — 280. Б. С. Кавнатская,
Н. М. Школьник, Л. М. Гвоздилова, Хим. пром., № 12, 934 (1966).
281. А. И. П о с т о р о н к о, сб. «Приемы повышения урожайности с.-х. куль-
тур», Киев, Изд. «Урожай», 1967, стр. 342. — 282. И. П. Худо лей, Г. А. С к о-
р я к, Новое в суперфосфатном производстве, Изд. «Техшка», Киев, 1964, стр. 21.—
283. В. И. Гл а душно, Х1м. пром. Украши, научно-произв. сб. № 2 (25), 1966,
стр. 3.—284. И. Н. Кузьминых, Е. Л. Яхонтова, А. И. Родионов,
Изв. вузов. Хим. и хим. технол., № 3, 80 (1958).—285. А. И. Шерешевский,
Л. С. Горицкая, Л. Н. Потапова, Сообщения о научно-технических рабо-
тах НИУИФ, № 2, Госхимиздат, 1957, стр. 26. — 286. И. М. Богуславский,
Л. Д. Киоссе, Там же, стр. 54. — 287. С. И. Вольфкович, Р. Е. Р е м е н,
Там же, стр. 52. — 288. И. М. Богуславский, Тезисы докладов на отрасле-
вом совещании работников основной и горнохимической промышленности, 1958,
стр. 109. — 289. Б. А. Соколовский, Е. Н. Жучков, ЖПХ, № 6, 70
(1933).—290. I. Procter, A. Ogilvie, канад. пат. 502802, 1954.
291. Хируо Дзэнкоку, япон. пат. 16069, 1964. — 292. М. Н. Набиев,
М. Адылова, Узб. хим. ж., № 3 (1962). — 293. М. Н. Набиев, А. А. Виш-
някова, М. Адылова, Тезисы докладов на IV Всесоюзн. конференции техн,
неорганических веществ, Ташкент, 1964, стр. 99. — 294. R. С. D a t i п, пат. США
3172751, 1965. — 295. С. С. К а р а х а н я н, Г. О. Григорян, Р. Л. Миру-
м я н, Изв. АН АрмССР. Хим. наука, 18, № 5, 516 (1965). — 296. М. А. Ды-
канбаев, В. В. Тихонова, А. Б. Бектуров, Изв. АН КазССР. Сер. хим.,
№ 4, 3 (1966).—297. К- Камалов, М. Н. Набиев, Узбекск. хим. ж., № 1
(1965).
Глава XXV
ПРОИЗВОДСТВО ФОСФОРНОЙ кислоты
СЕРНОКИСЛОТНЫМ СПОСОБОМ
Фосфорная кислота является основным полупродуктом в произ-
водстве фосфорных и сложных концентрированных удобрений и
других фосфорсодержащих соединений.
Один из распространенных способов ее получения — сернокис-
лотный— заключается в разложении природных фосфатов серной
кислотой и в отделении образующейся твердой фазы — сульфата
кальция от раствора фосфорной кислоты. Он называется экстрак-
ционным, или мокрым, способом в отличие от термического
(см. гл. XXVI).
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Фосфорной кислотой упрощенно называют ортофосфорную кис-
лоту Н3РО4. Она изготовляется и применяется в виде водных рас-
творов; концентрированные растворы имеют консистенцию густого
сиропа. Безводная фосфорная кислота кристаллизуется в виде
бесцветных призматических кристаллов ромбической системы,
плавящихся при 42,35°; они легко расплываются на воздухе. При
284° и давлении 1 ат начинается дегидратация ортофосфорной кис-
лоты и превращение ее в пирофосфорную Н4Р2О7 и метафосфор-
ную НРО3 кислоты.
На рис. 233 изображена диаграмма растворимости в си-
стеме. Н3РО4—Н2О. Твердыми фазами являются: лед, Н3РО4 и
Н3РО4-0,5Н2О (или 2Н3РО4-Н2О). В системе Р2О5—НзО1-14
имеется твердая фаза Н4Р2О7— пирофосфорная кислота (стр. 942).
Безводная ортофосфорная кислота Н3РО4 содержит 72,4%
Р2О5. При ее термической дегидратации или при взаимодействии
с фосфорным ангидридом образуется полифосфорная кислота,
содержащая больше 72,4% Р2О5 и состоящая из смеси разных
фосфорных кислот: орто-, пиро- (Н4Р2О7), Триполи- (Н5РзО!0)
882
Гл. XXV. Производство фосфорной кислоты
С2,5%Н3РО4
Н3РО4 • 1/2НгО + лед 91,£%Н3РО4
,J00L—>--.-->--------1---1--1--1--1
0 20 40 SO 80 100
тетраполи- (НеРЛз). мета- (НРО3) и полиметафосфорной. Соотноше- 1
ние между этими кислотами зависит от общей концентрации Р2О5 в
растворе, а при наличии в нем полимерных метафосфорных кислот
с общей формулой (НРОз)„ — и от времени (способа получения).
Это объясняется тем, что гидратация и дегидратация метафосфор- i
ных кислот происходит
медленно. I
Кислота с концентра-
цией 70—80% РзО5 (что I
эквивалентно 96—110%- |
ной Н3РО4) называется |
суперфосфорной кисло- |
той. Пиро- и триполифос- I
форная кислоты эквива-
лентны, соответственно,
110 % - и 114 % -ной Н3РО4. |
Суперфосфорная кислота I
с концентрацией 75,6% I
Р2О5 содержит 49% Р2О5 |
в виде ортофосфорной, I
42 % —в виде пирофос-I
форной, 8% в виде три- I
полифосфорной и 1% — I
в виде тетраполифосфор- I
ной кислот. При разбав- I
лении суперфосфорной I
кислоты водой происхо- "
дит значительное выде-
ление тепла и полифос-
форные кислоты быстро
гидратируются до орто-।
фосфорной. Плотность су-1
перфосфорной кислоты I
1,92—2 г!см3. Она имеет!
при 66°) и менее активно I
действует на металлы и сплавы, чем обычная ортофосфорная кис-1
лота. Температура замерзания суперфосфорной кислоты ~0°. I
В водных растворах фосфорная кислота ионизируется. Кон-1
станты ионизации 0,1—0,01 н. фосфорной кислоты следующие: I
н3ро4 н+ + н2ро;
н2ро; н+ + нро3-
нро4“ н+ + РО3~
Теплота растворения 1 моль
1,741, в 20 моль — 4,998, в 100 моль — 5,269 кал!г-мол.
Содержание H3PO4, вес. %
i-------»-------1-----.-->
O 20 40 60
Содержание P3Os, bee. °/e
Рис. 233. Диаграмма растворимости в си-
стеме Н3РО4—Н2О.
очень большую вязкость (400—1000 сп
К, = 7,52 • 10
К2 = 6,31 • 10"’
К3 = 2,2- 10’13
Н3РО4 в 1 моль
при 25
при 25
при 18°
воды составляет
Применение
883
ТАБЛИЦА 68
Температуры кипения водных растворов фосфорной кислоты1,11
H3PO4, % Температура, °C Н3РО4, % Температура, °C Н3РО4, % Температура, °C
12,6 101,35 51,2 108,70 77,2 136,09
17,0 101,90 57,3 113,27 80,8 145,38
23,7 102,44 66,5 120,15 89,6 173,23
37,4 104,45 71,8 127,85 98,0 233,24
Вязкость8,7 растворов Н3РО4 при 75 по отношению к воде(25 )
при концентрации около 27% Н3РО4 равна 1,967, при 68% Н3РО4—
9,506, при 88,2% Н3РО4 —28,61, при 97,35% Н3РО4 — 59,24 сп.
Давление пара растворов фос-
форной кислоты ’8-12 приведено
на рис. 234 (в паре только НгО),
а температуры кипения в табл. 68.
Средняя теплоемкость13 (при
20—100°) водных растворов
Н3РО4 с концентрацией от 0 до
60% определяется уравнением:
С = 1,0109 — 0,00709%, где к —
концентрация Н3РО4, в %.
ПРИМЕНЕНИЕ
Фосфорная кислота, получае-
мая сернокислотным методом,
применяется главным образом в
производстве концентрированных
удобрений (см. гл. XXVII, XXVIII,
XXXIII). В США для этой цели
используют около 93% всей экс-
тракционной фосфорной кисло-
ты 15-21. В меньших количествах
она потребляется для получения
промышленных продуктов (см.
гл. XXX). Производство фосфор-
ной кислоты сернокислотным спо-
собом расширяется за последние
годы во многих странах, так как
Рис. 234. Давление пара растворов’
фосфорной кислоты.
увеличивается спрос на концентрированные удобрения и растет ис-
пользование побочных продуктов (соединений урана, ванадия и
фтора), выделяемых в процессе разложения фосфатов серной кис-
лотой 22-26 Имеет значение и более низкая стоимость экстракцион-
ной фосфорной кислоты по сравнению с термической (см. гл. XXVI).
884
Гл. XXV. Производство фосфорной кислоты
Состав экстракционной фосфорной кислоты зависит от приме-
няемого фосфатного сырья27-29 и условий ее производства. Кис-
лота, получаемая из апатитового концентрата дигидратным спосо-
бом (с выделением сульфата кальция в виде дигидрата или гипса
CaSO4-2H2O), содержит 25—32% Р2О5, 1,8—2,8% SO3, 0,8—1,1%
Fe2O3, 0,5—1,0% А120з, 0,2—0,4% СаО, 1,5—1,8% фтора и имеет
плотность 1,25—1,3 г/см3. При использовании фосфоритов, в состав
которых входит значительное количество нерастворимых примесей
(кремнезема, силикатов и других), образуется более разбавленная
кислота с концентрацией 20—25% Р2О5 вследствие необходимости
применения для отмывки осадка большего количества воды. При
наличии в фосфоритах соединений магния последние также пере-
ходят в кислоту, которая в этом случае содержит 1—3,5% MgO.
Содержание других примесей колеблется в тех же пределах, что и
в кислоте, полученной из апатитового концентрата. Во многих слу-
чаях получаемую разбавленную кислоту упаривают до концентра-
ции 53—55% Р2О5, необходимой для производства концентрирован-
ных фосфорных и сложных удобрений.
Концентрированную экстракционную кислоту получают также
без упарки полугидратным методом (с выделением сульфата каль-
ция в виде полугидрата CaSO4 • 0,5Н2О) и она содержит 47—48%
Р2О5, 0,9—1,1% К2О3, 0,2—0,4% SO2', 0,2—0,7% фтора, 0,1—0,2%
СаО. Ее применяют для получения кормовых фосфатов и удоб-
рений.
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ СЕРНОКИСЛОТНОЙ ЭКСТРАКЦИИ
ФОСФАТОВ
Химизм процесса
При обработке фосфата серной кислотой при повышенной тем-
пературе он разлагается с образованием раствора фосфорной кис-
лоты:
Ca6(PO4)3F + 5H2SO4 = 5CaSO4 + ЗН:РО4 + HF
Для обеспечения подвижности пульпы, образующейся при сме-
шении измельченного фосфата с серной кислотой, с целью облег-
чения перемешивания и перекачки ее весовое отношение между !
жидкой и твердой фазами (Ж: Т) поддерживают в пределах от
2,5 : 1 до 3,5 : 1. Для этого пульпа (или серная кислота, или фос-
фат) смешивается с раствором разбавления, т. е. раствором фос-
форной кислоты, получаемым при отмывке водой экстракционной
кислоты, удерживаемой осадком сульфата кальция.
Таким образом, разложение фторапатита серной кислотой про-
исходит в смеси ее с фосфорной кислотой при взаимодействии с
ионами водорода, концентрация которых зависит от соотношения
кислот. Присутствие в реакционной массе фосфорной кислоты
Физико-химические основы экстракции фосфатов
885
влияет как на активность раствора, так и на кристаллизацию суль-
фата кальция.
Процесс может быть представлен уравнением:
Ca3(PO4)3F + 5H2SO4 + nH3PO4 + aq =
= (п + 3)Н3РО4 + 5CaSO4 • рН2О + HF + aq ... (1)
В тех случаях, когда фосфат предварительно смешивают с рас-
твором разбавления, а затем добавляют серную кислоту, процесс
идет в две ступени30’31. Сначала фосфат кальция частично реаги-
рует с фосфорной кислотой
Ca5F(PO4)3 + пН3РО4 = 5Са(Н2РО4)2 + (п - 7)Н3РО4 + HF (2)
затем при добавлении серной кислоты ионы кальция осаждаются
из раствора:
Са(Н2РО4)2 + H2SO4 = CaSO4 + 2Н3РО4 (3)
При этом серная кислота также разлагает оставшийся фосфат, не
прореагировавший с фосфорной кислотой.
Одновременно с фосфатом разлагаются и другие минералы,'
входящие в состав сырья.
Карбонаты кальция, присутствующие в природных фосфатах,
разлагаются согласно суммарному уравнению:
СаСО3 + H2SO4 + nH3PO4 + aq = CaSO4 • рН2О + СО2 + nH3PO4 4- aq (4)
При разложении смесью кислот доломита, а также силикатов
магния, в раствор переходят сульфат магния и мономагнийфосфат:
2MgCa(CO3)2 + 3H2SO4 4- 2Н3РО4 F aq =
= 2CaSO4 • рН2О + MgSO4 + Mg(H2PO4)2 + 4CO2 + aq (5)
Кислоторастворимые силикаты (нефелин, глауконит, каолин,
силикаты магния) в результате взаимодействия с кислотами вы-
деляют кремневую кислоту 32'34. Последняя реагирует с фтористым
водородом, образуя SiF4
SiO2 + 4HF = S1F4 + 2Н2О
Часть S1F4 удаляется в газообразном виде, другая же превра-
щается в кремнефтористоводородную кислоту
SiF4 + 2HF = 2H2SiF6
остающуюся в растворе. Количество выделяющегося газообраз-
ного SiF4 зависит от температуры процесса и концентрации серной
и фосфорной кислот: с их увеличением возрастает парциальное
давление пара SiF4 над растворами H2SiF6 в смеси кислот 35’36, и
степень выделения фтора в газообразную фазу растет.
Кремнефтористоводородная кислота в растворе частично взаи-
модействует со щелочами (нефелина, глауконита и других кисло-
торастворимых минералов), образуя малорастворимые кремнефто-
886
Гл. XXV. П роизводство фосфорной кислоты
риды натрия или калия. При выпарке фосфорной кислоты кремне-
фтористоводородная кислота частично разлагается на SiF4 и HF.
При сернокислотной экстракции в раствор переходит до 75% фто-
ра и около 20% фтора улетучивается37.
Окислы железа и алюминия переходят в раствор и реагируют
с фосфат-ионами:
R2O3 + 2Н3РО4 = 2RPO4 + ЗН2О
(где R — ионы А1 или Fe).
Природные фосфориты, содержащие много окислов алюминия и
железа, непригодны для сернокислотной экстракции фосфорной
кислоты. Особенно вредна примесь железа. Растворимость фос-
фата железа невелика (при 70—80° растворимость его в 16—
20%-ной фосфорной кислоте составляет менее 2%) и в начале об-
разуются пересыщенные растворы, из которых постепенно выделя-
ются в осадок фосфаты железа. В результате этого часть экстра-
гированной Р2О5 теряется38. В присутствии 2—3% иона SO1~
фосфорнокислотные растворы, содержащие фосфаты железа, устой-
чивы длительное время 39-42, если они получены из природного фос-
фата, в котором весовое отношение Fe2O3: Р2О5 < 0,12. Практи-
чески для экстракции фосфорной кислоты применяют фосфа-
ты28’29, содержащие Fe2O3 не больше 8% от веса Р2О3.
Растворимость фосфатов алюминия в фосфорной кислоте зна-
чительно выше, чем фосфатов железа 43-45; количество окиси алю-
миния в фосфатном сырье большей частью меньше, чем окислов
железа. По этим причинам в условиях производства фосфорной
кислоты находящиеся в ней фосфаты алюминия обычно не осаж-
даются.
Предварительное прокаливание фосфорита при 850—1050° умень-
шает скорость растворения Fe2O3 и А12О3 в фосфорной и серной
кислотах. Фосфат же кальция при этом разлагается быстро.
Расход серной кислоты в производстве фосфорной определяется
стехиометрическим количеством, необходимым для связывания
окиси кальция и магния. Для каждого вида фосфата эксперимен-
тально находят отклонения от стехиометрической нормы, обусло-
вливаемые присутствием минералов-примесей 24- 27-29_
Скорость разложения фосфатов при сернокислотной
экстракции из них фосфорной кислоты
Исследования системы СаО—Р2О5—SO3—Н2О показали, что
фосфаты кальция и серная кислота не могут сосуществовать в рас-
творе при обычных для производства температурах, т. е реакция
(1) необратима и нацело протекает слева направо 46’47.
Гетерогенная реакция разложения апатита серной кислотой в
присутствии фосфорной сводится к взаимодействию ионов Н+ и
Физико-химические основы экстракции фосфатов
887
0,9749
HjO
Рис. 235. Плотность (в г! см?) растворов си-
стемы H2SO4—Н3РО4—Н2О (в мол. %) при
75°.
РО43" фосфата с образованием малодиссоциированной Н3РО4.
Сульфат-ионы связывают ионы кальция, и преобладающее их ко-
личество выделяется в относительно большом объеме раствора
(Т : Ж ~ 2 -г- 3 : 1) в виде малорастворимого сульфата кальция.
В этих условиях, при правильном режиме процесса, сульфат каль-
ция не осаждается на зернах растворяющегося фосфата; исклю-
чено и выделение из раствора фосфатов кальция, которые могли
бы образовать на поверхности зерен экранирующую корку.
Экспериментальное исследование скорости процесса в условиях
растворения фосфата в серной кислоте в смеси с фосфорной кисло-
той весьма затруднитель-
но из-за сложности обра-
зующейся системы.
Растворение апатита
в кислотах48 лимитирует-
ся скоростью диффузии
ионов водорода из объе-
ма раствора к поверхно-
сти частиц фосфата или
ионов кальция из погра-
ничного слоя в объем
раствора. В области вы-
соких концентраций вяз-
кость растворов фосфор-
ной кислоты значительно
увеличивается, что за-
медляет диффузию ионов
и дополнительно умень-
шает скорость растворе-
ния апатита. Таким обра-
зом, концентрация ионов водорода и вязкость раствора являются
основными факторами, определяющими скорость растворения апа-
тита в смеси серной и фосфорной кислот.
Свойства тройной системы H2SO4—Н3РО4—Н2О, компоненты
которой неограниченно растворимы во всей области концентраций
и в широком диапазоне температур, изучены недостаточно. Допу-
скают 49~50, что кислотные компоненты в этой системе образуют
ацидиевый комплекс эквимолярного состава:
h2so4 + н3ро4 [н4ро4]+ + hso;
Плотность тройных растворов сильно зависит от содержания
воды и в меньшей мере от соотношения в них кислот51’52. На
рис. 235 показана плотность растворов тройной системы при 75°.
При концентрации от 100 до 20 мол. % воды растворы с большим
содержанием H2SO4 имеют несколько большую плотность; при
4 М. Е. Позия
888
Гл. XXV. Производство фосфорной кислоты
содержании Н2О менее 20 мол.% — наоборот, — более плотными
являются растворы с меньшим содержанием H2SO4. При содержа-
нии воды около 20 мол. %
плотность изогидратных
растворов практически не
зависит от соотношения
кислот.
На изотермах вязкости
при постоянном содержа-
нии воды имеется минимум,
глубина и положение кото-
рого зависят от температу-
ры. Кривые зависимости
вязкости от концентрации
(рис. 236) указывают, что в
системе H2SO4—Н3РО4—
—НгО ассоциированные ком-
плексы, имеющиеся в двой-
Рис. 236. Вязкость (в сп) растворов си- ных системах, диссоциируют
стемы H2SO4—Н3РО4—Н2О (в мол. %) при смешении51. Имеющий-
при 75°. ся в двойной системе
H2SO4—Н2О максимум вяз-
кости, соответствующий составу H2SO4 • Н2О, обнаруживается и в
тройной системе. Добавление Н3РО4 вызывает разрушение гидрата
и в концентрированных рас-
творах с преобладанием
фосфорной над серной кис-
лотой он отсутствует полно-
стью. Изотермическая по-
верхность вязкости имеет
сложный рельеф. Область,
богатая серной кислотой, ха-
рактеризуется синклиналью
и антиклиналью: последняя
проходит в направлении
ОТ H2SO4—НгО к H2SO4 •
• Н3РО4. С повышением тем-
пературы и синклиналь и
антиклиналь исчезают и при
95° поверхность вязкости
уже почти моноклиина. При
небольших концентрациях
H2SO4 кривые вязкости
Н2О
Рис. 237. Удельная электропроводность
(в олг’-смг1) растворов системы
H2SO4—Н3РО4—Н2О (в мол. %) при 75°.
имеют монотонный характер.
На рис. 237 показаны значения удельной электропроводности
растворов тройной системы разных концентраций при 75°. Изо-
Физико-химические основы экстракции фосфатов
889
термическая поверхность удельной электропроводности имеет две
антиклинали и одну синклиналь. Антиклиналь в области разбав-
ленных растворов, по-видимому, связана с максимальной мигра-
цией ионов, а в области прилегающей к углу серной кислоты — с
оптимальными условиями передачи протона.
Давление паров воды над некоторыми растворами тройной си-
стемы определено при 60° для смесей, содержащих — 45% Р2О5
( — 62,5% Н3РО4) и от — 1 до 16% H2SO4, а для смесей, содержа-
щих — 53% Р2О5 и от 0,3 до 10% H2SO4, при 60 и 75° (табл. 69).
ТАБЛИЦА 69
Давление водяного пара иад некоторыми растворами
системы Н3РО4—H2SO4—Н2О
Температура 60° Температура 75°
состав смеси, °/0 давление пара, мм рт. ст. состав смеси, % давление пара, JHJW рт. ст.
H3PO4 H2SO4 н2О H3PO4 H2SO4 н2о
63,5 0,0 36,3 76,5
63,5 1,02 35,48 73,5
62,4 2,08 35,52 72,6
62,5 3,14 34,36 69,0
62,2 4,10 33,7 65,0
62,5 4,83 32,67 61,4
63,2 6,07 31,8 58,1
64,0 7,40 28,6 51,5
64,0 8,50 27,5 49,0
63,8 9,30 26,9 44,9
63,8 10,56 25,64 41,5
64,0 12,70 23,3 34,3
63,9 15,07 21,03 28,4
63,9 16,85 19,25 23,0
74,0 0,0 26,0 47,1 74,0 0,0 26,0 95
73,0 0,3 26,7 48,7 73,0 0,3 26,7 98,5
73,0 0,6 25,4 46,1 73,0 0,6 25,4 91,3
74,0 1,34 24,6 44,8 74,0 1,34 24,6 90,4
75,0 2,15 23,85 40,5 74,0 2,15 23,85 86,5
75,0 3,4 21,61 37,1 75,0 3,4 21,6 76,1
75,1 4,4 20,6 33,2 75,1 4,4 20,6 70,4
74,5 5,34 20,16 30,1 74,5 5,34 20,16 63,1
75,5 6,5 17,98 28,2 75,5 6,5 17,98 55,9
76,0 7,4 16,56 24,0 76,0 7,4 16,56 50,1
75,1 8,3 16,62 21,7 75,1 8,3 16,62 45,4
74,7 9,59 15,71 19,1 74,7 9,6 15,71 40,7
76,0 10,5 13,5 17,1 76,0 10,5 13,5 34,2
При 60° давление пара над смесью 62,5—64 %-ной фосфорной и
серной кислот с повышением концентрацйи серной кислоты от 0 до
16,85% H2SO4 уменьшается от 76,5 до 23 мм рт. ст. Увеличение
концентрации серной кислоты в смесях, содержащих 74—76%
4*
890
Гл. XXV. Производство фосфорной кислоты
Н3РО4, от 0 до 10,5% H2SO4, приводит к понижению давления
пара от 47 до 17 мм рт. ст. При 75° увеличение содержания серной
кислоты в растворах с концентрацией 74—76% Н3РО4 понижает
давление пара от 95 до 34,2 мм рт. ст.
Тепловые эффекты смешения и теплоемкости смесей, содержа-
щих 61,8—58% Н3РО4 и 1,03—6,5% H2SO4, характеризуются мак-
симумом, равным 0,634 ккал/ (ч • град) и соответствующим концен-
трации 4% H2SO4. Показано53, что тепловой эффект смешения
Рис. 238. Зависимость степени разложения фосфо-
рита от времени и концентрации исходной сериой
кислоты при отношении Т : Ж = 1: 4:
1 -30»/о H2SO4; 2-40% H2SO4; 3-50% H2SO4; 4 — 60% H2SO4;
5-70% H2SO4.
62 %-ной фосфорной кислоты и 94 %-ной серной с образованием
смесей, содержащих 58—62% Н3РО4 и 1—6,5% H2SO4, соответ-
ствует (в ккал/г смеси) численному значению концентрации (в %)
серной кислоты в смеси. Например, для смеси с концентрацией
1,03, 1,99% H2SO4 тепловой эффект смешения равен 1,08, 2,0 ккал/г
смеси.
Скорость разложения фосфата серной кислотой в присутствии
раствора разбавления — фосфорной кислоты, как и при производ-
стве суперфосфата, зависит не только от активности ионов водо-
рода, но и от степени пересыщения жидкой фазы продуктом реак-
ции— сульфатом кальция. Общий вид зависимости степени разло-
жения фосфата за определенное время (изохрона) от концентра-
ции серной кислоты в смеси так же как и на рис. 217 изображается
кривой, имеющей два максимума и один минимум между ними.
Так как скорость взаимодействия между фосфатом и серной кис-
лотой велика, то можно предположить, что начальная действую-
Физико-химические основы экстракции фосфатов
891
бремя,мин
Рис. 239. Изменение степени разложения
фосфорита серной кислотой во времени
при разных температурах (крупность фос-
форита — 150+105 мк).
щая концентрация серной кислоты будет определяться длительно-
стью ее подачи и объемом раствора разбавления. Например, при
подаче 100 вес. ч. моногидрата серной кислоты для разложения
100 вес. ч. апатита в объем раствора фосфорной кислоты, занимае-
мый 400 вес. ч. (начальное отношение Ж : Т = 2,5 : 1 в расчете на
сухой фосфогипс) в течение 1 ч ее начальная концентрация в смеси
при продолжительности полного смешения, равной ~ 1 мин, будет
~0,3% и всего ~ 3%, если принять (без учета взаимодействия
серной кислоты) продолжительность полного смешения, равную
10 мин. В действительности за 10 мин много кислоты прореагирует
и, следовательно, начальная
концентрация серной кисло-
ты будет во много раз
меньше.
Таким образом, разложе-
ние фосфата протекает при
взаимодействии с серной
кислотой небольшой концен-
трации, и скорость процес-
са максимальна при не-
которой оптимальной кон-
центрации серной кислоты
в смеси с фосфорной, лежа-
щей в левой части рис. 217.
. По-видимому, в области кон-
центраций серной кислоты в
смеси меньше оптимальной
разложение фосфата замед-
ляется вследствие недоста-
точной активности ионов во-
дорода, а при больших кон-
центрациях вследствие торможения реакции при экранировании
поверхности зерен фосфата коркой выделяющегося сульфата
кальция.
Непосредственных данных о скорости разложения фосфатов
смесями фосфорной кислоты, содержащими небольшие количества
(0,1—0,3%) серной кислоты, почти не имеется. Приближенно мож-
но судить о скорости процесса по данным взаимодействия фосфа-
тов с серной кислотой большей концентрации (30—50% H2SO4)
как в отсутствие, так и в присутствии в начальной смеси фосфор-
ной кислоты. При весовом отношении фосфата и кислоты (Т :Ж),
равном 1 :4, разложение подольского фосфорита, содержащего
32,8% Р2О5, с крупностью частиц меньше 53 мк при 70° раствором
серной кислоты концентрации 30% H2SO4 завершается полностью
за ~15 мин (рис. 238). С увеличением концентрации кислоты до
40% H2SO4 скорость процесса уменьшается и для полного
892
Гл. XXV. Производство фосфорной кислоты
разложения фосфата требуется 60—90 мин. Скорость реакции зна-
чительно увеличивается с повышением температуры даже при ис-
пользовании фосфата грубого помола (с размерами частиц меньше
150 и больше 105 мк) и 50%-ной серной кислоты (рис. 239).
Однако в присутствии фосфорной кислоты скорость растворе-
ния фосфата серной кислотой несколько уменьшается, по-видимо-
му, вследствие уменьшения активности ионов водорода. Как видно
на рис. 240 54, изохроны растворения апатита при 70° большим из-
Рис. 240. Изохроны растворения апатита в смесях серной и фос-
форной кислот при 50, 70 и 93°:
1-0% Н3РО4; 2-10% Н3РО4; 3-25% Н3РО4.
бытком смеси кислот (Т : Ж 1 : 30) при отсутствии перемешива-
ния твердой фазы располагаются тем ниже, чем больше в смеси
содержится фосфорной кислоты. Хотя эти данные получены в ус-
ловиях, отличающихся от применяемых на практике, но они в не-
которой мере отражают кинетические особенности процесса. Оче-
видно, что минимальная скорость разложения апатита обуслов-
лена образованием плохо проницаемых корок сульфата кальция
при некоторых средних концентрациях H2SO4 в смеси, превышаю-
щих концентрации, при которых обычно осуществляется процесс
экстракции. В действительности вследствие значительно меньших
концентраций серной кислоты в жидкой фазе экстракционной
пульпы разложение фосфата протекает с большей скоростью. По
мере расходования сильно диссоциированной серной кислоты и на-
копления в растворе солей и малодиссоциированной фосфорной
кислоты активность раствора и скорость процесса уменьшаются.
Большое влияние на скорость разложения имеет природа фос-
фата (общая удельная поверхность элементарных зерен) 55~57.
Физико-химические основы экстракции фосфатов
893
Скорость разложения апатита кислотами прямо пропорциональна
внешней поверхности зерен минерала55. При разложении крупно-
зернистого апатита наблюдается практически линейная зависи-
мость количества разложенного вещества от времени, потому что в
этом случае суммарная величина поверхности зерен изменяется
медленно. При разложении полидисперсной фосфатной муки ли-
нейная зависимость отсутствует. Мелкие зерна обладают большой
общей поверхностью, которая быстро уменьшается в ходе реакции.
Реакция протекает вначале с высокой
скоростью, и мелкие зерна оказываются
разложенными до окончания процесса.
Крупные зерна имеют относительно ма-
лую суммарную поверхность, они реаги-
руют медленно, что приводит к посте-
пенному уменьшению скорости растворе-
ния апатита к концу реакции. В произ-
водстве фосфорной кислоты применяется
фосфатная мука стандартного помола,
т. е. с остатком на сите 0,15 мм до 14%.
Апатитовый концентрат обладает очень
малой общей удельной поверхностью,
поэтому он разлагается кислотами отно-
сительно медленно. Осадочные фосфори-
ты, содержащие фторапатит в виде мель-
чайших зерен (меньше 1 мк), разлагает-
ся с большей скоростью58.
При разложении фосфата в две сту-
пени по уравнениям (2) и (3) в первой
ступени процесса диссоциация фосфор-
ной кислоты постепенно подавляется
вследствие роста концентрации моно-
Рис. 241. Изохроны-изотер-
мы скорости растворения
апатита в водных раство-
рах фосфорной кислоты при
температурах:
1 - 50°; 2 - 70°; 3 - 90°.
кальцийфосфата в растворе.
При весьма малой степени нейтрализации фосфорной кислоты
в этих условиях для оценки скорости процесса можно с известным
приближением воспользоваться данными, полученными при изуче-
нии растворения апатита в большом объеме фосфорной кисло-
ты 55'59’60 (рис. 241). При разложении апатита фосфорной кисло-
той (в отсутствие серной), с увеличением концентрации Р2О5 до
30—35%, степень диссоциации Н3РО4 уменьшается, но количество
ионов водорода в единице объема раствора возрастает. Поэтому на
данном участке изохрон-изотерм (рис. 241) скорость растворения
апатита увеличивается. При дальнейшем повышении концентрации
значительное падение степени диссоциации фосфорной кислоты
приводит к уменьшению числа ионов водорода в единице объема
раствора и, следовательно, к уменьшению скорости раство-
рения апатита. С увеличением степени нейтрализации растворение
894
Гл. XXV. Производство фосфорной кислоты
апатита замедляется. Изотермы растворения апатита в частично
нейтрализованной фосфорной кислоте аналогичны изотермам, при-
веденным на рис. 241, но средние скорости растворения уменьша-
ются с увеличением степени нейтрализации кислоты48’6,-63 (см.
также рис. 273 на стр. 980).
Скорость реакции апатита с фосфорной кислотой зависит так-
же от температуры. Температурный коэффициент скорости раство-
рения апатита в растворах фосфорной кислоты48’63 находится в
пределах от 1,31 до 1,48.
Степень разложения апатита фосфорной кислотой за одно и то
же время тем выше, чем больше температура (рйс. 242). Реакция
разложения фосфатов про-
S 0,3 1,0 CJJ 1,1)
13 8реия,ч
текает с выделением тепла
(см. гл. XXIV); тепло вы-
деляется также при разбав-
лении серной кислоты. По-
этому в условиях производ-
ства пульпа разогревается
до 75—80° и выше.
Скорость взаимодействия
с растворами кислот мине-
ралов-примесей (глаукони-
та, лимонита) 60 уменьшает-
ся по мере разложения, так
как переходящие в раствор
катионы постепенно подав-
ляют диссоциацию кислоты.
Рис. 242. Изменение степени разложения
фосфорной кислотой апатитового концен-
трата во времени при разных температурах.
Концентрация фосфорной кислоты 20% Р2О5;
Ж : Т = 20 : 1.
Прокаленный при 400—800° глауконит разлагается медленнее, чем
непрокаленный. Примесь серной кислоты в фосфорной (2—6%
SO3) практически мало влияет на скорость разложения глауко-
нита и лимонита, но способствует удержанию в растворе окислов
железа.
Разложение фосфоритов сопровождается выделением СО2, об-
разующей пену. При недостаточно интенсивном перемешивании
частицы фосфата, попадая в малоподвижную пену, образуют ко-
мочки, которые в результате взаимодействия с серной кислотой по-
крываются коркой кристаллов сульфата кальция. Это нарушает
нормальный процесс разложения фосфата. Перемешивание долж-
но обеспечить интенсивное движение верхнего слоя пены и ее по-
глощение (засасывание) пульпой в воронку, образующуюся при
вращении лопастных мешалок с окружной скоростью 4—6 м^сек.
Применяются также подавители пены. Для создания турбулент-
ного движения жидкости при перемешивании имеет значение отно-
шение Ж : Т в пульпе. Практикой установлено отношение Ж Т в
пределах от 2,5 до 3,5, которое поддерживается циркуляцией обо-
ротной фосфорной кислоты.
Физико-химические основы экстракции фосфатов
895
В производственных условиях высокая степень разложения при-
родных фосфатов (в том числе и наиболее медленно реагирующего
апатитового концентрата) достигается уже в первые 1 —1,5 ч. Од-
нако скорость разложения фосфата не является единственным
фактором, определяющим длительность процесса. Она опреде-
ляется условиями образования крупнокристаллического сульфата
кальция, обладающего хорошими фильтрующими свойствами, из
которого можно отмыть фосфорную кислоту небольшим количест-
вом воды. Это необходимо для получения кислоты максимальной
концентрации (так как промывная вода возвращается в цикл).
Практически продолжительность экстракции для разных видов
сырья и режимов колеблется в пределах 4—8 ч.
Кристаллизация сульфата кальция
В системе СаО—Р2О5—SO3—Н2О сульфат кальция в твердом
виде может существовать в трех формах: ангидрита CaSO4, полу-
гидрата CaSO4 • 0,5Н2О и дигидрата или гипса CaSO4-2H2O.
Растворимость гипса и ангидри- ,
та возрастает с увеличением кон- §
центрации кислоты до определен- 08
ной величины, а затем убывает ««
(рис. 243, 244). 46
При 80° в диапазоне концен- g-o м
граций кислоты 0—50% Р2О5 х§
стабильной формой, обладающей J” 42
наименьшей растворимостью, яв- £ 0
ляется ангидрит 46’64, а при 25° в
растворах, содержащих до ~ 30%
Р2О5, — дигидрат. Наличие при п п
г „ < . г Рис. 243. Растворимость кристалле-
ЭТИХ условиях Других форм суль- гидратов сульфата кальция в водных
фата кальция обусловлено дли- растворах фосфорной кислоты при 25°.
тельностью скрытого периода их
перехода в стабильную форму, соответствующей большей или мень-
шей устойчивостью образующегося метастабильного или пересы-
щенного раствора (по отношению к стабильной фазе). Во всех
случаях образования сульфата кальция в фосфорнокислых раство-
рах вначале выделяется полугидрат, который затем превращается
в стабильную при данных условиях форму. Это превращение про-
исходит путем растворения полугидрата и одновременной кристал-
лизации из раствора гипса или ангидрита. В системе CaSO4—
—Р2О5—Н2О гипс является стабильной формой сульфата кальция
только в растворах фосфорной кислоты, над которыми давление
водяного пара больше давления пара над твердым CaSO4-2H2O
при данной температуре.
896
Гл. XXV. Производство фосфорной кислоты
На рис. 245 приведена схема превращения кристаллогидратов
сульфата кальция в чистых растворах фосфорной кислоты в зави-
симости от их температуры и концентрации. В области выше cd
стабильной формой является ангидрит. При 80° превращение пер-
воначально выделившегося полугидрата в ангидрит происходит в
растворах, содержащих больше 33,3% Р2О5 (рис. 245). Если в рас-
творы, составы которых расположены выше cd, внести гипс, то он
сначала превращается в метастабильный полугидрат, имеющий
концентрация фоссрорной кислоты, %РгО5
Рис. 244. Растворимость кристаллогидратов сульфата
кальции в водных растворах фосфорной кислоты при
80°.
меньшую растворимость, чем гипс, а затем в ангидрит. В области ,
между кривыми cd и ab стабильной формой также является ангид-
рит, но здесь полугидрат превращается в ангидрит не непосредст-»
венно, а сначала переходит в гипс, который отличается промежу-
точной растворимостью. При 80° это происходит в растворах,
содержащих от 0 до 33,3% Р2О5 (рис. 245), что объясняется одина-
ковой растворимостью полугидрата и дигидрата при 80° и 33,3%
Р2О5. Пересечение кривых на рис. 244 отвечает сосуществованию
в растворе двух метастабильных фаз — полугидрата и дигидрата
при 80°, и кривая cd (рис. 245) представляет собой геометрическое
место точек сосуществования этих метастабильных фаз при раз-
ных температурах.
В области ниже ab стабильной формой является гипс. Кривая
ab — линия, разграничивающая растворы, равновесные со ста-
бильными твердыми фазами—ангидритом и гипсом. Она пред-
ставляет собой также геометрическое место точек сосуществова-
ния метастабильного и стабильного дигидрата. В растворах, соста-
Физико-химические основы экстракции фосфатов
897
вы которых расположены ниже кривой ab, ангидрит непосредст-
венно переходит в гипс (рис. 245), а полугидрат предварительно
претерпевает дегидратацию, переходя в ангидрит.
Превращение метастабильных фаз, связанное с уменьшением
концентрации сульфата кальция в пересыщенном (метастабиль-
ном) растворе и переходом его в стабильное состояние, зависит не
только от температуры и концентра-
ции кислоты, но и от свойств кри-
сталлогидратов. В области выше
кривой cd превращение метаста-
бильного полугидрата в стабильный
ангидрит происходит очень медлен-
но, в течение многих суток. Процесс
ускоряется с повышением темпера-
туры и увеличением концентрации
кислоты. Например, при 60° это пре-
вращение в кислоте концентрации
50% Р2О5 завершается примерно за
10—12 суток, а при 90° — за 1 сут-
ки. В кислоте, содержащей 40%
Р2О5, для перехода полугидрата в
ангидрит при 90° требуется ~2 су-
ток.
В области между кривыми cd и
ab превращение метастабильного
полугидрата в метастабильный гипс
в растворах, содержащих 10—25%
Р2О5, при 80° происходит в течение
1—8 ч ^.табл. 70). Процесс уско-
ряется с понижением температуры
и уменьшением концентрации кис-
лоты. Дальнейшее превращение гип-
са в стабильный ангидрит проте-
кает очень медленно. Образовав-
шийся в этих условиях гипс существует в метастабильной форме в
течение нескольких месяцев (при концентрации Р2О5 ниже 10%)
или нескольких суток (при 25% Р2О5).
На рис. 246 показаны изохроны скрытых периодов перехода
полугидрата в гипс и ангидрит, а на рис. 247 — изохроны кристал-
лизации гипса и ангидрита.
Присутствие в кислоте примесей влияет на растворимость кри-
сталлогидратов и в соответствии с этим изменяются устойчивость
метастабильных фаз и условия сосуществования их и стабильных
фаз. В растворах фосфорной кислоты, содержащих свободную сер-
ную кислоту или избыточное количество окиси кальция по сравне-
нию со связанной в виде CaSO4, растворимость сульфата кальция
Рис. 245. Схема превращений
кристаллогидратов сульфата каль-
ция в растворах фосфорной кисло-
ты при различных температурах:
I область. Стабильная форма —гипс;
схема превращений: полугидрат (П) —>
—> ангидрит (А) —> гипс (Г) и ангид-
рит —> гипс.
7/ область. Стабильная форма —ангид-
рит; схема превращений: полугидрат —>
—> гипс —> ангидрит и гипс —> ан-
гидрит.
III область. Стабильная форма —ангид-
рит; схема превращений: полугидрат —>
—> ангидрит и гипс —> полугидрат —>
—> ангидрит.
898
Гл. XXV. Производство фосфорной кислоты
ТАБЛИЦА 70
Продолжительность превращения полугидрата в гипс
в зависимости от температуры и концентрации
фосфорной кислоты
Температура, °C Концентра- ция, % Р2О5 Длительность превращения, ч
скрытый период кристалли- зация гипса общая продолжи- тельность
40 30 Не определено 3 3
35 1 14 15
38 12 26 38
40 60 90 150
25 0,25 1,75 2,0
60 30 2,0 3,2 5,2
35 15,0 6,4 21,4
10 0,12 1,0 1,12
80 18 1,25 1,75 3,0
25 5,0 3,0 8,0
28,5 18,0 6,5 24,5
Температура
рго5, %
Рнс. 246. Изохроны скрытых периодов перехода полугидрата в гипс
и ангидрит в фосфорнокислых растворах.
Физико-химические основы экстракции фосфатов
899
значительно уменьшается 65-68. Фосфат кальция оказывает высали-
вающее действие на сульфат кальция. При увеличении содержа-
ния свободной СаО в растворе до 1,5—2% возможна совместная
кристаллизация сульфата и фосфата кальция. Указывают также
на возможность образования 69>70 в этих условиях твердого рас-
твора CaSO4-2H2O и СаНРО4"2НгО. Увеличение концентрации
Рис. 247. Изохроны кристаллизации гипса и ангидрита в фос-
форнокислых растворах.
свободной серной кислоты в растворах фосфорной кислоты, содер-
жащих 25—35% Р2О5, при 70° приводит к уменьшению раствори-
мости как полугидрата, так и дигидрата сульфата кальция71. В
растворах фосфорной кислоты, содержащих кремнефтористоводо-
родную кислоту, растворимость сульфата кальция, наоборот, уве-
личивается.
Отношение СаО : SO3 в сульфате кальция равно 0,7. При из-
бытке СаО в растворе сверх этого отношения, а также в присутст-
вии солей магния область стабильной кристаллизации гипса рас-
ширяется 29’72-74 до концентрации жидкой фазы 35—37% Р2О5 при
температуре соответственно 80—70°. На рис. 248 изображена диа-
грамма, показывающая практическую (в пределах длительности
технологических процессов и содержащихся в растворе примесей)
900
Гл. XXV. Производство фосфорной кислоты
280Г
Е
<з
240
200
160
X CaSO4
Г20
80
40
0
Са5О4-2НгО
5 10 15 20 25 30 35 40 45 60 55 60
РЯО6,%
- CaSO4-0,5HiO
Рис. 248. Области существования кристалло-
гидратов сульфата кальция в условиях
производства фосфорной кислоты серно-
кислотным методом.
5
&
степень гидратации сульфата кальция в зависимости от темпера-
туры и концентрации производственной фосфорной кисло-
ты 25>75~77. В условиях, близких к производственным, значительно
расширяется область существования стабильного гипса и появ-
ляется достаточно широкая область существования устойчивого
полугидрата.
На практике осаждение стабильного гипса (дигидратный про-
цесс) ведется в условиях получения фосфорной кислоты с концент-
рацией до 30—32% Р2О5 при 65—80°. Из более концентрирован-
ных растворов (> 35%
Р2О5) и при повышении тем-
пературы до 90—95° оса-
ждается полугидрат, в раз-
личной степени способный
к оводнению до гипса 72-79.
Снижение температуры оса-
ждения и концентрации
фосфорной кислоты, а так-
же повышение количества
СаО или SOs в растворе
способствуют получению
быстро оводняющегося по- (
лугидрата. Присутствие j
большого количества гипса >
(затравки) тоже ускоряет .
превращение полугидрата, j
При температурах выше 100—105° и концентрации кислоты
больше ~45% Р2О5 (рис. 248) выделяется ангидрит.
Форма и размеры выделяющихся кристаллов зависят от тем-
пературы и концентрации кислоты, степени пересыщения и условий
снятия пересыщения. Установлено81 максимальное пересыщение " i
(при котором начинается самопроизвольная кристаллизация) суль- I
фатом кальция растворов системы CaSO4—Р2О5—Н2О при 25, 60 и |
80°. Кривые пересыщения фосфорнокислых растворов сульфатом I
кальция в зависимости от концентрации кислоты располагаются 1
симбатно кривым растворимости гипса и ангидрита (рис. 249). 1
Значения концентрации пересыщения растворов сульфатом каль- 1
ция не только превышают значения растворимости стабильной 1
формы, но существенно отличаются от растворимости метаста- 1
бильных форм. Например, для 45% Р2О5 при 60° концентрация
сульфата кальция в пересыщенном растворе равна ~ 0,9%, в то
время как растворимость ангидрита составляет 0,32, полугидра-
та— 0,58 и гипса — 0,69% CaSO4.
На рис. 250 представлена кривая изменения во времени кон- ]
центрации сульфата кальция в пересыщенном растворе (в расчете I
на СаО), содержащем 45,6% Р2О5 в отсутствии других посторон-
Физико-химические основы экстракции фосфатов
901
них ионов при 60°. В данных условиях стабильной твердой фазой
является ангидрит, гипс — первой метастабильной фазой, а полу-
гидрат — второй. Как видно, при переходе от первой метастабиль-
ной фазы дигидрата к другой наблюдается как бы вторичное пе-
ресыщение раствора (максимум на кривой). С увеличением содер-
жания Р2О5 в пересыщенных растворах в диапазоне от 10 до 50%
Р2О5 скорость кристаллизации сульфата кальция при 25° умень-
шается, а при 60° — увеличивается.
Размеры выделяющихся кристаллов сульфата кальция при раз-
личных условиях изучены в большей мере при образовании дигид-
большей мере при образовании дигид-
рата и в меньшей — при образова-
нии полугидрата и ангидрита.
Установлено81, что при выделении
гипса из чистых фосфорнокислых
Рис. 249. Изотермы 60°
растворимости сульфата
кальция и пересыщения
им растворов в системе
CaSO4—Н3РО4—Н2О:
( — ангидрит; 2 — полугидрат;
3 — гипс; 4 — пересыщение
сульфатом кальция.
Рис. 250. Изменение во времени
концентрации сульфата каль-
ция (в пересчете на СаО) в пере-
сыщенном растворе, содержа-
щем 45,6% Р2О5 при 60°.
растворов повышение температуры от 30 до 80° приводит к уве-
личению длины кристаллов в 10—15 раз, а ширины в 3—5 раз.
С изменением концентрации кислоты от 25 до 34% Р2О5 продоль-
ные и поперечные размеры кристаллов значительно уменьшаются.
Большое влияние на форму кристаллов оказывают примеси82~87.
Увеличение содержания СаО в реакционной пульпе вызывает
уменьшение размеров кристаллов при возрастании степени неод-
нородности их81. Некоторое уменьшение размеров кристаллов на-
блюдается при большом недостатке или избытке серной кислоты
по сравнению со стехиометрическим ее количеством. Максималь-
ные размеры кристаллов соответствуют норме серной кислоты 90 и
110% по отношению к стехиометрической. На рост кристаллов
902
Гл. XXV. Производство фосфорной кислоты
гипса оказывает влияние соотношение между концентрациями
ионов Са2+ и SOl” в растворе.
При избытке Са2+ гипс образуется в виде тончайших игл дли-
ной 20—80 мк. По мере уменьшения концентрации Са2+ и увеличе-
ния концентрации SO2- кристаллы гипса растут; при избытке
SO2- их размеры достигают 100 мк в ширину и нескольких сотен
микронов в длину. Для получения крупнокристаллического одно-
родного осадка необходимо, чтобы отношение SO3: СаО в жидкой
фазе (по весу) было по возможности постоянным, равным 1,5—3.
Оптимальным является содержание в растворе 1—2,5% SO3 и
0,35—0,75% СаО.
При небольшой концентрации SO3 примеси ионов трехвалент-
ного железа и алюминия способствуют образованию более широ-
ких и коротких (изометрических) кристаллов гипса и полугидрата.
Влияние этих примесей на размеры выделяющихся кристаллов
имеет сложный характер88. Образование кристаллов по форме,
близкой к изометрической, происходит при содержании в растворе
от ~ 0,2 до 1,5% R2O3. Это обусловливается уменьшением длины
кристаллов почти в 1,5 раза; ширина же их остается практически
неизменной.
В сочетании с ионами алюминия благоприятное влияние на
форму и размеры кристаллов сульфата кальция оказывают ионы
кремнефторида. Хорошо фильтрующий сульфат кальция обра-
зуется в присутствии 1—2% (по отношению к Р2О5—фосфата)
сульфатов магния, цинка, железа, никеля и меди 89. При наличии
в растворе большого количества кремневой кислоты гипс кристал-
лизуется в виде тончайших игл. Кроме того, выделившаяся при
экстракции фосфорной кислоты из некоторых фосфоритов кремне-
вая кислота в виде илистого осадка сильно затрудняет отделение
жидкой фазы от твердой.
О размерах кристаллов полугидрата и ангидрита, выделяю-
щихся из концентрированных растворов фосфорной кислоты при
повышенных температурах, имеются весьма ограниченные сведе-
ния. Известно,90 что полугидрат и ангидрит, выделяющиеся при
высокой температуре в фосфорнокислой среде с большим содержа-
нием Р2О5, хорошо отфильтровываются от жидкой фазы. В этих
условиях полугидрат кристаллизуется в форме небольших шести-
гранных призм размером 20—30 мк, собранных в крупные конгло-
мераты, а ангидрит — в виде широких утолщенных пластинок.
Крупные компактные конгломераты кристаллов полугидрата полу-
чены из раствора, содержащего 40% Р2О3, при небольшой концен-
трации (1,5—1,7%) SO3. Они отделяются от раствора лучше, чем
тонкие пластины или игольчатые кристаллы. Ангидрит, наоборот,
выделяется в виде более крупных кристаллов, образующих пластины
Физико-химические основы экстракции фосфатов
903
неправильной квадратной формы, из раствора концентрации
45—55% Р2О5, содержащего избыток серной кислоты (3—5% SO3).
При длительности процесса 4—6 ч из растворов, содержащих
35—43% Р2О5 при 75—80°, выделяющиеся конгломераты полу-
гидрата имеют размер от 75 до 200—300 мк; они состоят из от-
дельных мелких кристаллов ~ 10 мк. Размеры кристаллов полу-
гидрата зависят как от концентрации кислоты, так и от плотности
пульпы.91 При весовом отношении Ж : Т в пульпе в пределах от
6:1 до 10:1, концентрации кислоты — 50% Р2О5 образующиеся
при 60° кристаллы полугидрата имеют длину от 50 до 80 мк и ши-
рину от 3 до 5 мк. Размеры кристаллов полугидрата, выделяю-
щиеся при 60° из раствора фосфорной кислоты концентрации 45%
Р2О5 при Ж : Т « 4 : 1, составляют 40—50 мк по длине и 3—4 мк
по ширине.
Полученные в лабораторных условиях (периодический метод)
кристаллы полугидрата не собираются в конгломераты и имеют
игольчатую форму. Длина их превышает ширину примерно в 10 раз.
Такие кристаллы легко отфильтровываются от жидкой фазы, но
задерживают относительно большие количества кислоты, что при-
водит к значительным потерям Р2О5 при промывке.
Кристаллы полугидрата с размерами в длину до 150 мк и в ши-
рину до 15—20 мк выделяются 92 в фосфорнокислом растворе, со-
держащем — 25—33% Р2О5 и 15—25% HNO3. Такие кристаллы
отделяются фильтрованием от раствора, представляющего собой
смесь азотной ( — 50% HNO3) и концентрированной фосфорной
кислоты (—45—65% Р2О5), со скоростью в 3—6 раз большей, чем
дигидрат в производстве кислоты концентрации 30—32% Р2О5.
Образование кристаллов ангидрита связано с дегидратацией
первоначально выделяющегося полугидрата. В растворе, содержа-
щем 50% Р2О5, при 90° перекристаллизация начинается только че-
рез 6 ч и заканчивается после 30 ч (стр. 897). Дегидратация полу-
гидрата ускоряется с увеличением концентрации P2Os, а также при
наличии в растворе серной кислоты. С повышением концентрации
свободной H2SO4 в растворе скорость дегидратации полугидрата
возрастает и образующиеся кристаллы ангидрита имеют относи-
тельно небольшие размеры93. Так, в кислоте, содержащей в рас-
творе 47—50% Р2О5 и больше 5% H2SO4, дегидратация при 100°
заканчивается в течение 20—40 мин. Образующиеся кристаллы ан-
гидрита имеют прямоугольную форму и длину 15—30 мк, а ши-
рину— 6—10 мк. С понижением концентрации серной кислоты до
3% продолжительность полного превращения полугидрата при 100°
увеличивается до 80—85 мин. При этом получаются значительно
более крупные кристаллы, имеющие также прямоугольную форму,
с размерами, например, 30—40 X 6—8 мк. В этих условиях замет-
ное растворение полугидрата начинается только через —30 мин
после начала процесса, а появление первых кристаллов ангидрита —
904
Гл. XXV. Производство фосфорной кислоты
через 40 мин. Это указывает на образование раствора с относи-
тельно небольшим пересыщением ангидритом, что обусловливает
выделение более крупных кристаллов.
Дальнейшее уменьшение содержания серной кислоты в рас-
творе до 2—1 % H2SO4 связано с уменьшением нормы ее до 100 и
90% по отношению к стехиометрическому количеству (в расчете на
кальций) и накоплением в растворе ионов кальция. В этом случае
выделяются мелкие и неразвитые по форме кристаллы ангидрита, :i
хотя фазовый переход протекает очень медленно. Для полного пре- )
вращения полугидрата в ангидрит в этом случае требуется в зави-
симости от нормы кислоты от 150 до 600 мин. ’ )
С повышением температуры дегидратация полугидрата сульфа- 1
та кальция ускоряется, и соответственно уменьшаются размеры I
кристаллов ангидрита. Это, вероятно, объясняется увеличением 1
скорости растворения полугидрата при повышении температуры и
достижением в результате этого большего пересыщения раствора
сульфатом кальция. При 120° в растворе, содержащем 3—3,5%
H2SO4, продолжительность процесса меньше в 2—3 раза, чем при
100°, и составляет 30—40 мин вместо 80—120 мин. j
При увеличении содержания в пульпе жидкой фазы степень пе- 1
ресыщения раствора сульфатом кальция при прочих равных уело- 1
виях (растворимости, скорости перемешивания и др.) умень- а
шается. Это должно привести к замедлению дегидратации и уве- J
личению размеров выделяющихся кристаллов ангидрита. При
содержании в растворе ~3 — 7,5%SO4-, Ж:Т = 6:1 и 80—100° 1
выделяющиеся кристаллы ангидрита имеют размеры от 30 X 6 мк |
до 60 X 15 мк. 1
МЕТОДЫ СЕРНОКИСЛОТНОЙ ЭКСТРАКЦИИ ФОСФОРНОЙ кислоты
ИЗ ФОСФАТОВ
В зависимости от условий осуществления процесса (стр. 895) 1
различают три режима экстракции фосфорной кислоты 15>18 ’ 94-104: |
дигидратный, полугидратный и ангидритный. Сущность каждого из 1
них заключается в достаточно полном разложении фосфата, раз- 1
делении получаемой пульпы и отмывке фосфорной кислоты из I
осадка. Для успешного проведения второй операции необходимо,I
чтобы в процессе экстракции сульфат кальция выделялся в виде |
хорошо фильтрующих и легко промываемых кристаллов. Это обес- J
печивает высокую производительность фильтровальной аппарату-1
ры и получение фосфорной кислоты максимальной концентрации
(для данного режима) вследствие возможности промывки осадка <
минимальным количеством воды. Наиболее распространен диги- ;
дратный режим, в результате усовершенствования которого105-115 j
возможно в настоящее время получать при 65—80° фосфорную кис- 1
лоту с содержанием 28—32% Р2О5. Раньше этим способом полу- j
Методы сернокислотной экстракции фосфорной кислоты 905
чали кислоту концентрацией 20—27% (в зависимости от состава
фосфата). Оптимальные условия процесса достигаются за счет
поддержания необходимого температурного режима охлаждением
пульпы (барботажем воздуха или в вакуум-испарителях 1,3~116), ис-
пользования затравки для роста кристаллов (циркуляцией продук-
ционной пульпы) и применения более совершенных фильтров.
Охлаждение пульпы за счет испарения воды при продувке воздуха
или в вакуум-испарителях в то же время приводит к увеличению
концентрации P20s в жидкой фазе. Температуру можно регули-
ровать и количеством циркулирующей пульпы. Описано 117~119 по-
лучение таким путем фосфорной кислоты концентрации 30% P2Os
при 80°.
При большом количестве циркулирующей пульпы (затравки)
можно получить при температуре ниже 65° фосфорную кислоту
концентрацией до 38—40% Р2О5 с выделением гипса. Лаборатор-
ными исследованиями было установлено33, что при эквивалентных
количествах СаО и SO3 в растворе гипс выделяется и при концент-
рации кислоты 35—36% Р2О5 при 70°. Применение вакуум-фильт-
ров— карусельных, ленточных, конвейерно-лотковых и горизон-
тальных план-фильтров позволяет вести фильтрование с малым
расходом воды на промывку фосфогипса.
В практических условиях, в присутствии примесей, влияющих
на стабильность и скорость фазового перехода кристаллогидратов
сульфата кальция (рис. 248), из растворов с концентрацией боль-
ше 33—35% Р2Оэ выше 70—80° и из растворов, содержащих больше
42—45% Р2О5 выше 60°, в твердую фазу выделяется полугид-
рат. Еще большее повышение температуры и увеличение концент-
рации кислоты способствуют дегидратации полугидрата и выделе-
нию ангидрита в качестве твердой фазы, устойчивой на всем
протяжении процесса экстракции. Это наблюдается в растворах с
концентрацией 50—60% Р2О5 выше 100° и при концентрации
35—37% Р2О5 выше 120°. Таким образом, получение сернокислот-
ной экстракцией фосфатов фосфорной кислоты концентрации боль-
ше 32% Р2О5 связано с применением высокотемпературного режи-
ма и выделением в твердую фазу полугидрата или ангидрита,
которые и отделяются от жидкой фазы.
Ангидритный процесс, протекающий при температурах выше
100°, осложняется сильной коррозией аппаратуры, трудностью от-
деления мелких кристаллов, необходимостью подогревания реаген-
тов вследствие недостаточности тепла реакции для поддержания
автотермичного режима. Помимо этого большие трудности воз-
никают при отмывке фосфорной кислоты из отжатого осадка
(вследствие напряженного водного баланса) и при подготовке ан-
гидрита к транспортированию и складированию. Поэтому ангид-
ритный процесс до сих пор не вышел из стадии разработки в круп-
нолабрраторных или в небольших опытных масштабах.
906
Гл. XXV. Производство фосфорной кислоты
Получение фосфорной кислоты, содержащей 40—50% и больше j
Р2О5, при ангидритном режиме основано на предварительной об-
работке фосфата горячей фосфорной или серной кислотой или на .
использовании в качестве полупродукта двойного или простого -
суперфосфата.
При предварительном разложении фосфата горячей (135°) кон- <
центрированной фосфорной кислотой 120-122 полученную массу после ]
вызревания подвергают дальнейшей обработке смесью фосфорной
и серной кислот при высокой температуре. Образующуюся пульпу ’
охлаждают до 80° и фильтруют. Процесс осуществляют следую- ,
щим образом.
Фосфатную муку периодически смешивают 120'121 с частью обо-
ротной фосфорной кислоты (около 40% Р2О5). Образующаяся
пульпа, жидкая фаза которой содержит монокальцийфосфат и
фосфорную кислоту, поступает в обогреваемый змеевиком реактор ;
с мешалкой, оборудованной приспособлением для подавления пе- ]
ны. В реактор через определенные интервалы времени вводят смесь
серной кислоты с остальной частью оборотной фосфорной кислоты. :
Эту смесь предварительно нагревают до 135°. Пульпа затем вы-
стаивается 3—4 ч и охлаждается до 80° в чанах, которые отли- ;
чаются от реактора отсутствием змеевиков и приспособлений на ;
мешалках для уничтожения пены. Фильтрование ведут на конвей-
ерно-лотковом вакуум-фильтре по пятифильтратной схеме (четыре
противоточных промывки). Первый фильтрат (продукционная
фосфорная кислота) содержит 42—45% Р2О5, остальные фильтра-
ты соответственно 40, 35, 20 и 5% Р2О5. При переработке марок- <
канского фосфорита степень его разложения составляет 97—98% ;
и коэффициент отмывки около 98%.
При обработке серной кислотой стандартного гранулированного ’
суперфосфата возможно 28’123 получать фосфорную кислоту с 40%
Р2О5. По «клинкерному» способу 28’124, позволяющему получать"
фосфорную кислоту с концентрацией до 54% Р2О5 (в значительной
степени обесфторенную), фосфат смешивают с 98%-ной серной
кислотой и нагревают до 200—300° для образования сухого про- ;
дукта, который обрабатывают затем водой. В процессе перемеши- :
вания и нагревания улетучивается около 90% фтора, поэтому эта !
фосфорная кислота пригодна для получения кормового фосфата.
Представляет интерес 125 производство фосфорной кислоты полу-
гидратно-ангидритным способом, по которому шлам (полугидрат),
полученный в результате предварительного разложения фосфата ’
до отделения от жидкой фазы, обрабатывают серной кислотой для
образования ангидрита. Последний отделяют от жидкой фазы и
промывают водой. Жидкую фазу используют для предварительной ;
обработки фосфата.
Предложено 28,126 получать 70%-ную фосфорную кислоту (с :
концентрацией ~50% Р2О5), почти не содержащую фтористых '
Методы сернокислотной экстракции фосфорной кислоты
907
соединений. Фосфат смешивают с оборотной фосфорной кислотой
и смесь нагревают до 200°. Получается комплексный полифосфат;
который при нагревании со смесью концентрированных серной и
фосфорной кислот превращается в пирофосфорную кислоту. По-
следняя легко гидратируется до ортофосфорной кислоты. Сульфат
кальция отфильтровывают и промывают.
В отличие от ангидрита, фильтрование и промывание-полугид-
рата от фосфорной кислоты протекает быстрее примерно в 2 раза,
чем дигидрата сульфата кальция. Наряду с увеличением скорости
фильтрования осадка при этом достигается также увеличение
производительности реакционной аппаратуры в 1,5—2 раза вслед-
ствие повышения концентрации и температуры кислоты. Осуще-
ствление процесса при несколько более высоких температурах
(90—105°), т. е. на 20—25° выше, чем при выделении дигидрата,
в настоящее время не вызывает особых затруднений вследствие
наличия относительно дешевых материалов для конструирования
оборудования, стойкого при повышенных температурах в концент-
рированных растворах фосфорной кислоты.
В СССР освоено и осуществляется в промышленных масштабах
производство полугидратным методом фосфорной кислоты кон-
центрации 45—48% Р2О5 по методу, разработанному ЛТИ имени
Ленсовета, Винницким химкомбинатом и Ленниигипрохимом. В этом
направлении ведутся работы и другими организациями (НИУИФ,
Гипрохим, УПИ, Красноуральский комбинат и др.).
Разработка технических условий производства концентрирован-
ной фосфорной кислоты полугидратным способом базируется на
усовершенствованиях, достигнутых в дигидратном процессе (регу-
лирование температурного и концентрационного режимов, приме-
нение циркуляции пульпы, фильтров новых конструкций и т. п.),
а также на физико-химических особенностях выделяющегося полу-
гидрата. Полугидратный процесс отличается от дигидратного, во-
первых, условиями формирования достаточно крупных, хорошо
фильтрующих кристаллов (стр. 903), а во-вторых, режимом про-
мывания осадка вследствие малой стойкости полугидрата и гидра-
тации его в гипс в разбавленных фосфорнокислых растворах. По-
этому в различных способах полугидратного режима применяют
или промежуточную перекристаллизацию 73~75’79’80’126’127 полугид-
рата (после отделения продукционной фосфорной кислоты) в гипс,
который затем промывают водой128-131, или полугидрат выделяют
в относительно устойчивой форме, не гидратирующейся при про-
мывании в течение достаточного для производственных условий
времени и не схватывающейся при транспортировке и хранении.
Перекристаллизация полугидрата в гипс связана с дополнитель-
ной операцией и усложняет технологический процесс. Помима
этого в практических условиях не удается достигнуть полного
оводнения полугидрата в предназначенном для этого аппарате
908
Гл. XXV. Производство фосфорной кислоты
(гидраторе) и продолжающаяся гидратация полугидрата приводит
к загипсовыванию фильтровальной ткани и коммуникаций. Полу-
чение устойчивого полугидрата и непосредственное его промыва-
ние водой упрощает технологическую схему, но требует стабилиза-
ции полугидрата и применения соответствующих режимов.
ПРОИЗВОДСТВО ЭКСТРАКЦИОННОЙ ФОСФОРНОЙ кислоты
ДИГИДРАТНЫМ СПОСОБОМ
Существующие схемы дигидратного процесса различаются при-
менением или отсутствием циркуляции пульпы, распределением
реагентов между реакторами, способами охлаждения пульпы и,
наконец, методами разделения твердой и жидкой фаз и промывки
фосфогипса.
Во всех случаях основной целью является ведение процесса
без резких колебаний концентраций и температур и образование
возможно более крупных и изометрических кристаллов сульфата
кальция.
Хорошо фильтрующие осадки дигидрата сульфата кальция
в процессе экстракции получаются при следующих условиях 31> 82-84.
Степень пересыщения жидкой фазы сульфатом кальция при
разложении фосфатов должна поддерживаться не выше 0,2—0,5.
Низкая степень пересыщения достигается прежде всего непрерыв-
ным ведением процесса. Для этого реагенты непрерывно подаются
в систему и энергично перемешиваемая реакционная масса медлен-
но перетекает через серию экстракторов (4—8 шт.) или отдельные
секции многосекционного экстрактора. Чем больше относительный
реакционный объем раствора (или чем больше продолжитель-
ность взаимодействия реагентов), тем крупнее и однороднее
получаются кристаллы. Опытом установлена продолжитель-
ность пребывания пульпы в реакторах для различных типов фос-'
фатного сырья 4—7 ч 31> 85~87. 132-150. Необходимая длительность
процесса обеспечивается выбором соответствующего объема реак-
ционной аппаратуры.
Для увеличения скорости кристаллизации (снятия пересыще-
ния) и уменьшения возможности возникновения новых центров
кристаллизации (зародышей) процесс проводят в присутствии
большого количества растущих кристаллов сульфата кальция (за-
травка), что достигается посредством циркуляции — перекачивания
пульпы из последнего реактора в первый. Кратность циркуляции
не превышает 10—12 вес. ч. на единицу продукционной пульпы.
Другим важным условием является поддержание оптимальной
концентрации серной кислоты в жидкой фазе того реактора, где
происходит осаждение сульфата кальция (стр. 902). Она тем выше,
чем больше в сырье соединений полуторных окислов (табл.
7j) з1, за, 85-87,132-150 Оптимальная концентрация поддерживается
Производство экстракционной Н3РО4 дигидратным способом
909
ТАБЛИЦА 71
Концентрация H2SO4 (в пересчете на SO3) в жидкой фазе
для оптимальных условий кристаллизации гипса
Фосфатное сырье . Концентрация H2SO4 (в пере- счете на SO3), вес, °/о
схема без циркуляции схема с цирку- ляцией
Апатитовый концентрат (флотационный) .... 1,3-2,8 0,9-1,8
Вятский фосфорит, флотационный концентрат . Актюбинский (Богдановский участок) флотаци- 1,2-2,4 0,8-2,0
онный концентрат 1,2-2,4 0,8-1,8
Фосфориты Каратау (Чулактау) 2,5-3,0 —
Актюбинский (Новоукраинский участок), мытый 2,2-4,0 —
в схеме без циркуляции пульпы распределением серной кислоты
или фосфата между первыми реакторами, а по схеме с циркуля-
цией— подбором количества циркулирующей пульпы. В первом слу-
чае часть серной кислоты (иногда 10—15% от общего количества}
вводят во второй реактор, чтобы поддержать оптимальную кон-
центрацию ее в первом экстракторе. При переработке апатита со-
держание свободной H2SO4 в растворе первого экстрактора не
должно превышать 20 г/л, а в последующих экстракторах оно сни-
жается от 10 до 0 г/л. В схеме без циркуляции оптимальную кон-
центрацию серной кислоты стремятся поддерживать в первых двух
реакторах, и серную кислоту, помимо первого реактора, вводят
также в третий и четвертый реакторы. В первых реакторах проис-
ходит растворение фосфата кальция, и количество серной кислоты
в растворе уменьшается. Чем уже интервал концентраций серной
кислоты в жидкой фазе экстракторов, тем больше должна быть
кратность циркуляции31’98.
Для выращивания крупных кристаллов гипса в экстракторах
поддерживают температуру 70—75°. Это достигается за счет тепла
реакции и регулируется охлаждением.
Пульпа, вытекающая из последнего экстрактора и состоящая
из раствора фосфорной кислоты и твердой фазы—гипса, поступает
на разделение. Отделенный раствор — основной фильтрат представ-
ляет собой экстракционную кислоту. Осадок, удерживающий зна-
чительное количество раствора, промывают противотоком водой.
Отмытый осадок гипса содержит небольшие количества недоот-
мытой фосфорной кислоты — его называют фосфогипсом.
Часть основного фильтрата отводится в качестве готового про-
дукта, а остальное его количество смешивается с промывным филь-
тратом и возвращается на экстракцию в виде раствора разбавле-
ния. В зависимости от числа промывок (например, трех- или*
-910
Гл. XXV. Производство фосфорной кислоты
четырехфильтратная схема и т. д.) образуются несколько фильтра-
тов разной концентрации.
Отделение и промывание фосфогипса производят на ленточных,
конвейерно-лотковых и карусельных вакуум-фильтрах. Одно из
главных требований к фильтрам — обеспечение хорошей отмывки
гипса от фосфорной кислоты при наименьшем расходе воды. Ранее
для этой цели применяли барабанные вакуум-фильтры. При ис-
пользовании барабанных вакуум-фильтров погружного типа для
отмывки осадка его репульпируют водой и промежуточными рас-
творами. Фильтрование пульпы и промывание осадка производят
последовательно на трех вакуум-фильтрах противотоком в три сту-
пени. Это связано с образованием шести фильтратов. Но предва-
рительная репульпация осадка в течение 1—2 ч перед каждой сту-
пенью в специальном смесителе позволяет эффективно отмыть фос-
форную кислоту даже из осадков, зашламованных кремнегелем и
другими илистыми примесями. Например, при экстракции фосфор-
ной кислоты из фосфоритов Каратау степень отмывки фосфогипса
от фосфорной кислоты при ступенчатом фильтровании (шестифиль-
тратной схеме) достигает 97% при концентрации кислоты 20—25%
Р2О5, а производительность фильтров составляет 400—450 кгЦм2 • ч)
сухого фосфогипса 86-88>90’104. Барабанные вакуум-фильтры могут
быть также использованы на небольших установках при получе-
нии экстракционной фосфорной кислоты из бедных отечественных
фосфоритов 132-15°.
При переработке фосфатной руды, содержащей ~32% Р2О5,
-степень отмывки фосфогипса на барабанных вакуум-фильтрах до-
стигает18’ 105 98—99%, а выход Р2О5 в фосфорную кислоту больше
“95%. Перед вторым фильтрованием фосфогипс перемешивается со
вторым фильтратом, содержащим 15—16% Р2О5, в течение 2 ч.
При этом происходит окончательное разложение фосфата и дости-
гается равновесная концентрация сульфата кальция в жидкой фа-
•зе, что предотвращает кристаллизацию на фильтровальной ткани
и в коммуникациях. Перед третьим фильтрованием фосфогипс пе-
ремешивается с четвертым фильтратом, содержащим -~2,5% Р2О5,
в течение 1 ч с добавлением в пульпу серной кислоты до концен-
трации около 2% SO3.
До недавнего времени вращающиеся барабанные вакуум-фильт-
ры непрерывного действия 151 широко применялись в производстве
экстракционной фосфорной кислоты. Их главный недостаток — гро-
моздкость и сложность обслуживания. Попытки упрощения про-
цесса фильтрования применением двухступенчатой (четырехфиль-
тратной) схемы не увенчались успехом ,04. Помимо этого, трудность
защиты этих аппаратов от коррозии, малая поверхность фильтро-
вания привели к использованию для фильтрования фосфорной кис-
лоты новых конструкций высокопроизводительных вакуум-фильт-
ров. Технологическая схема производства значительно упрощается
912
Гл. XXV. Производство фосфорной кислоты
при применении фильтров наливного типа (ленточных, конвейерно-
лотковых или карусельных). Они позволяют совмещать основную
операцию фильтрования с процессом промывания гипса, что дает
возможность уменьшить число фильтратов. Ленточные вакуум-
фильтры27,151-155 (рис. 251) имеют сравнительно небольшую фильт-
рующую поверхность (до 10 м2) при ширине ленты 500 мм. Они
отличаются высоким съемом фильтрата и осадка с единицы фильт-
рующей поверхности в 1 ч (вследствие отложения на поверхности
фильтра в первую очередь крупных частиц осадка), легкой реге-
нерируемостью фильтрующей ткани, простотой конструкции, лег-
костью защиты от коррозии и удобством обслуживания. Недостат-
ком их является нечеткое разделение отдельных фаз фильтрования
и промывания осадка, что увеличивает расход воды на промывание
и уменьшает концентрацию продукционной фосфорной кислоты 156.
Кроме того, они имеют относительно небольшую производитель-
ность. Поэтому на заводах большой мощности требуется большое
число фильтров, занимающих значительные площади.
Применением конвейерно-лотковых фильтров достигается луч-
шее разделение промывных растворов27’ 109,118,121,157 дт0 позволяет
вести промывку с уменьшенным расходом воды. Фильтр состоит
из ряда прямоугольных лотков с бортами, смонтированными в виде
непрерывной ленты. Борта (радиальные стенки) соседних лотков
(путчей) являются общими, в результате чего образуется не-
прерывное кольцо из фильтровальных ячеек. Эти фильтры из-
готавливаются диаметром до 4 м при поверхности фильтрования
до 12 м2.
Наибольшими достоинствами обладает карусельный 114-158 ва-
куум-фильтр (рис. 252), получивший весьма широкое распростране-
ние. Он состоит из 24 отдельных лотков длиной 1,9 м, шириной
0,9 м у внутреннего конца и 1,2 ж у наружного и глубиной 0,2 м.
На их днищах уложена фильтровальная ткань. Лотки установлены
на каретках с колесиками, движущимися по круговым рельсам.
С помощью двух головок — подвижной, вращающейся вместе с
лотками, и неподвижной фильтраты отсасываются в соответствую-
щие вакуум-сборники. После прохождения зон фильтрования и про-
мывания каждый лоток порознь автоматически опрокидывается
для выгрузки лепешки фосфогипса. Фильтровальная ткань промы-
вается водой и подсушивается воздухом. Затем лоток вновь при-
нимает рабочее положение и возвращается в зону основной фильт-
рации. Цикл работы каждого лотка (путча) состоит из стадий
фильтрования, обезвоживания осадка, двух или более промываний
осадка с промежуточным обезвоживанием его, разгрузки фосфо-
гипса и промывания ткани. Во время фильтрования, промывания
и обезвоживания осадка лоток соединен с источником вакуума, во
время разгрузки осадка, сопровождаемой опрокидыванием лотка
с источником сжатого воздуха, и во время промывания — с атмо-
Производство экстракционной Н3РО4 дигидратным способом
913
сферой. Суспензия и промывная жидкость поступают равномерно
по всей длине фильтровальной перегородки лотка из специальных
дозирующих устройств. Поверхность фильтрования равна 40 и
80 м2; имеются сведения об изготовлении фильтров с поверхностью
100 м2 и более159. Благодаря тому, что отдельные лотки изолиро-
ваны друг от друга, возможно получать концентрированный, не раз-
бавленный промывной жидкостью основной фильтрат. Это позво-
ляет также производить многоступенчатую противоточную про-
мывку минимальным количеством воды.
Для фильтрования в настоящее время применяются ткани из
синтетических волокон — хлориновые и лавсановые (вместо при-
менявшихся ранее шерстяных и нитрованных шелковых и хлопча-
тобумажных тканей). Они обладают механической прочностью и
химической стойкостью в фосфорной кислоте концентрации до
45% Р2О5 и больше при 80—90°. Опробованы с хорошими резуль-
татами полиэтиленовые ткани 109. Большинство применяемых сей-
час тканей изготовлено на основе поливинилхлорида: волокно со-
виден (саран) Н4, лэйнил121, перхлорвиниловое волокно «ПЩ»,
хлорин ,60’ 161 и др. Недостатком этих тканей является то, что через
них проникают тонкие частицы; для предотвращения этого необхо-
димо применять ткани из сложных нитей.
914
. Гл. XXV. Производство фосфорной кислоты
При отделении фосфогипса на наливных вакуум-фильтрах с 1 м2
фильтрующей поверхности за 1 ч снимается 600—800 кг осадка
(в расчете на сухой). Влажность фосфогипса составляет 30—40%.
Количество получаемого фосфогипса (сухого) составляет 160% от
веса переработанного апатитового концентрата, а для других
Серная кислота
Рис. 253. Схема производства экстракционной фос-
форной кислоты (28—32% Р2О5);
/ — бункер для апатитового концентрата; 2 — ленточный
весовой дозатор; 3 — экстрактор; 4 и 10— напорные баки;
5 —дозаторы кислот; 6 — вакуум-испаритель; 7 — распреде-
литель пульпы; S — погружные насосы; 9 — барометриче-
ский конденсатор; // — лотки карусельного фильтра;
12 — вакуум-сборники; 13 — сборники фильтратов.
фосфатов зависит от сорта фосфата, степени гидратации сульфата
кальция и содержания не растворимых в кислоте примесей.
На рис. 253 изображена применяемая на отечественных заводах
схема производства фосфорной кислоты (28—32% Р2О5) из апати-
тового концентрата. Из фосфоритов Каратау и Кингисеппского
возможно получить кислоту с концентрацией 25—27% Р2О5. Раз-
ложение фосфата производится в четырехсекционном экстрак-
торе Н1>112’1,5 емкостью 600 лг3. В каждой секции установлены три
лопастных или пропеллерных мешалки. Переток пульпы через сек-
Производство экстракционной Н3РО4 дигидратным способом
915
ции осуществляется при помощи перегородок попеременно — снизу
вверх и сверху вниз. В первую секцию непрерывно поступает апа-
титовый концентрат из бункера через ленточный дозатор. Сюда же
подают оборотную фосфорную кислоту и второй — промывной —
фильтрат с фильтра, ретурную, т. е. циркуляционную пульпу из
вакуум-испарителя, и серную кислоту. Последняя может также
поступать во вторую и третью секции экстрактора. Из третьей сек-
ции часть пульпы перетекает в четвертую, откуда она идет на филь-
трацию; остальная пульпа подается погружным насосом в вакуум-
испаритель и из него направляется в первую и четвертую секции
экстрактора.
На некоторых заводах используют батарейные экстракционные
системы, состоящие из 4, 5 и более реакторов емкостью по 50—85 .и3
с пропеллерными или турбинными мешалками, вращающимися со
скоростью 400—600 об)мин.
Газы и пары удаляются из экстракторов в специальную вытяж-
ную систему.
Экстракторы, мешалки, трубопроводы защищают от коррозии —
от действия горячей фосфорной кислоты, содержащей серную и
кремнефтористоводородную кислоты и другие примеси, и от эро-
зии— истирания перемешиваемой пульпой. Для этого используют
устойчивые в указанных условиях неметаллические материа-
лы 27> 163-170. Стальные корпуса экстракторов футеруют по слою
резины или полиизобутилена кислотоупорным кирпичом или гра-
фитовыми блоками. В процессе работы поверхность футеровки по-
крывается тонким слоем осадка гипса, который защищает ее от
истирания. Валы и лопасти мешалок, а также детали вакуум-
фильтра, трубопроводы и насосы для перекачки пульпы и фосфор-
ной кислоты изготовляют из стали ЭИ-943 и для относительно сла-
бых кислот из сталей ЭИ-448 и Х18Н10Т.
Наилучшими насосами для пульпы являются погружные цент-
робежные вертикальные насосы. Во избежание инкрустации не-
доступной для чистки внутренней поверхности трубопроводов
кристаллизующимся из раствора гипсом, их изготовляют из поли-
этилена, поверхность которого не зарастает гипсом, а также из
армированной резины.
Вакуум-испаритель представляет собой резервуар, где с по-
мощью вакуум-насоса поддерживают пониженное давление. Вслед-
ствие этого поступающая в него жидкость оказывается перегретой
и она закипает, причем из нее выпаривается некоторое количество
воды. Тепло, расходуемое на испарение, отнимается от пульпы,
температура которой поэтому понижается. Таким образом, в ва-
куум-испарителе происходит одновременно охлаждение пульпы (на
5—10°, в зависимости от количества циркулирующей пульпы и раз-
ности давлений) и некоторое увеличение концентрации фосфорной
916
Тл. XXV. Производство фосфорной кислоты
кислоты за счет удаления (испарения) воды. С помощью вакуум-
испарителя температура пульпы поддерживается равной ~70°.
Соотношение между весовым количеством раствора и твердого
двухводного сульфата кальция, т. е. Ж : Т, в суспензии, находя-
щейся в экстракторе, поддерживают равным 2—2,5: 1.
Фильтрацию пульпы производят на карусельном лотковом
фильтре, где гипс отделяется и промывается по четырехфиль-
тратной схеме. Фильтраты собираются в вакуум-сборниках. Пер-
вый фильтрат Ф1 отправляется в сборник готовой продукции, а
часть его возвращается в первую секцию экстрактора. Сюда же
возвращается и второй фильтрат Ф2, полученный при промывании
осадка третьим фильтратом Ф3. Фильтрат Ф3 образуется при про-
мывании осадка четвертым фильтратом Ф4, а последний — при про-
мывании горячей водой. Промытый гипс сбрасывается с лотка в
сборник, из которого в виде суспензии удаляется в отвал или на
переработку. Концентрация Р2О5 в фильтратах: Ф1 — 30—32%,
Ф2-22—25%, Фз— И—12%, Ф4 —2,5—3,5%.
Выделяющийся в экстракторах водяной пар и газы отсасы-
ваются в абсорбер, где промываются водой для улавливания фто-
ристых соединений, затем выбрасываются в атмосферу.
Технологический выход P20s, т. е. степень перехода Р2О5 из
сырья в фосфорную кислоту (без учета механических потерь) со-
ставляет 95—96% для апатита и колеблется в пределах 71—94%
для разных фосфоритов. Его определяют по общему содержанию
Р2О5 в отбросном фосфогипсе
КвЫХ 100
[Р2О5 общ] Г -100
[Р2О5 фосфата]
%
где в квадратных скобках дано процентное содержание общей-
Р2О5 в фосфогипсе и Р2О5 в исходном фосфате (в пересчете на су-
хое вещество). а Г — гипсовое число, т. е. выход сухого фосфо-
гипса на единицу фосфата.
Технологический выход обычно на 2—3% ниже коэффициента
извлечения Р2О5 в раствор (Кизвл).- Это объясняется недостаточ-
ной отмывкой фосфогипса от раствора фосфорной кислоты.
Коэффициент извлечения Рг'Об в раствор (в %) определяется
по содержанию водонерастворимой Р2О5 в фосфогипсе:
{[Р2О5 общ] [Р2О5 водораств]} Г - 100
Кизвл - 100------;-------------=---------%
1^2^5 фосфата!
Для различных фосфатов /СИзвл находится в пределах 95—
99%.
Коэффициент отмывки фосфогипса (в %) характеризует потери
водорастворимой Р2О5 в отбросном фосфогипсе:
Котм = 100 —
[Р2О5 водораств] Т* 100* 100
[РгО5 фосфата] Кизвл
%
Производство экстракционной Н3РО4 дигид ратным способом
917
В оптимальных условиях он составляет 97—99%. Очевидно, что
технологический выход определяется, в свою очередь, коэффициен-
тами извлечения Р2О5 в раствор и отмывки фосфорной кислоты из
фосфогипса
If __ ^ИЗВЛ^ОТМ о/
Л вых----100-- 0
Количество воды, необходимое для отмывки фосфогипса, опре-
деляется 171 в соответствии с рассчитанными или установленными
экспериментально исходными показателями 98,128 с использованием
уравнения материального баланса, которое для 100 вес. ч. фосфата
имеет вид:
100 + S + W = Ф + Ю0Г + Гк + G
где S — расход серной кислоты исходной концентрации; W — рас-
ход воды на промывку фосфогипса; Ф — количество продукцион-
ной фосфорной кислоты; Г — гипсовое число; — содержание
жидкой фазы в отбросном промытом фосфогипсе; G — количество
выделяющихся газов (SiF4 и СО2) и водяного пара.
Количество продукционной фосфорной кислоты определяется
в зависимости от ее концентрации с учетом установленного опыт-
ным путем коэффициента выхода Р2О5:
[Р2О5 фосфата)
ф = ip о Г ''вых
[Н2и5 ф. К-ты!
где [Р2О5Ф.к-ты] — концентрация Р2О5 в фосфорной кислоте, %.
Количество выделяющихся газов и водяного пара G при отсут-
ствии опытных данных можно вычислить из баланса процесса раз-
ложения фосфата
I00 + S+ R = F + G
где R — количество раствора разбавления для поддержания задан-
ного отношения Ж : Т в пульпе;
F — количество пульпы
F = 100/ (n+ 1)
п — заданное отношение Ж ’• Т в пульпе.
Баланс фильтрации и промывки фосфогипса составляется по
экспериментальным данным171.
Увеличение степени отмывки фосфорной кислоты из фосфогипса
достигается при использовании для экстракции фосфатов серной
кислоты с концентрацией 93% H2SO4 (вместо 75%-ной). Это позво-
ляет улучшить баланс воды в технологическом процессе и ввести
большее количество воды для промывания гипса. Благодаря этому
уменьшаются потери фосфорной кислоты с фосфогипсом, идущим
в отвал, и облегчаются процессы фильтрации и промывки. Однако
увеличение концентрации серной кислоты не изменяет концентрации
918
Гл. XXV. Производство фосфорной кислоты
получаемой фосфорной кислоты, которая предопределяется
оптимальными условиями кристаллизации гипса, рассмотренными
выше.
Экстракционная фосфорная кислота, полученная дигидратным
способом из апатитового концентрата, содержит 0,8—1,5% фтора
в виде H2SiF6, так как в раствор переходит до 75% фтора, нахо-
дящегося в природном фосфате. Очистка кислоты от фтора может
быть произведена осаждением FUSiFe солями натрия, калия, ба-
рия37, 144> 145> 172~174. Обычно в раствор вводят 30—40 г NaCl на 1 л
фосфорной кислоты. Образующийся по реакции
H2SiFe + 2NaCl = Na2SiFe + 2НС1
плохо растворимый кремнефторид натрия выделяется в осадок
в форме хорошо осаждающихся и фильтрующих шестигранных
кристаллов размером около 15 мк. Его отделяют от раствора сна-
чала отстаиванием, а затем центрифугированием или фильтрова-
нием. Таким образом выделяется 75—85% фтора и содержание его
в фосфорной кислоте снижается до 0,2—0,3%. В промытом и вы-
сушенном осадке кремнефторида содержится до 97% Na2SiF6.
Фосфорная кислота, обесфторенная хлоридом натрия, вызывает
более сильную коррозию аппаратуры, особенно при повышенной
температуре. Поэтому при необходимости последующего концент-
рирования кислоты выпаркой обесфторивание осуществляют с по-
мощью соды или фосфата натрия. При осаждении содой вводят
20—22 г Ыа2СОз на 1 л кислоты (130—150% от стехиометрического
количества).
Очистку фосфорной кислоты, полученной из каратауских или
кингиссепских фосфоритов, от соединений магния можно произве-
сти 175 выпариванием ее до концентрации 37—39% Р2О5 при 80—90°
в смеси с кремнефтористоводородной кислотой, добавляемой в ко-
личестве, необходимом для образования MgSiF6. При 20° из упа-
ренной массы выделяется MgSiF6, в результате чего отношение
MgO : Р2О5 в фосфорной кислоте не превышает 3%.
При сернокислотной экстракции апатитового концентрата в рас-
твор переходит ~20% содержащихся в нем соединений редкозе-
мельных элементов 24>176. Они могут быть выделены осаждением
в виде фосфатов при частичном усреднении фосфорной кислоты.
Большее извлечение редкоземельных элементов из апатита (до
70%) достигается при соляно- или азотнокислотном его разложе-
нии.
Из фосфорной кислоты, полученной из фосфоритов, содержа-
щих до 1 % V2O5, ванадий может быть осажден раствором желе-
зистосинеродистого натрия 177 или в виде фосфорнованадиевой кис-
лоты при добавлении окислителей, например, хлоратов и персуль-
фатов 28,
Производство экстракционной Н3РО4 дигидратным способом
919
В процессе сернокислотной экстракции природных фосфатов
(месторождений США, Северной и Южной Америки и других), со-
держащих от 0,001 до 0,15% Us'Os 178-182 в раствор переходит от
70 до 90% урана. Из фосфорной кислоты уран извлекают разными
методами 24-28’182-207. Например, экстракцией слабым (1—2%-
ным) раствором алкилпирофосфата в керосине по схеме
U4+ + 2R2P2O7H2 = (R2P2O7)2U + 4Н+
После разделения фаз уран осаждают плавиковой кислотой
(R2P2O7)2U + 4HF - UF4 + 2R2P2O7H2
Растворитель возвращается в процесс.
Уран извлекают также из раствора с применением ионообмен-
ных смол; непосредственным осаждением U(H2PO4)4 с помощью
восстановителей — гидросульфита натрия Na2S2O4, или формальде-
гвдсульфоксилата NaHSO2 • НСНО • 2Н2О205 и др.
Фосфогипс содержит небольшие количества (0,3—0,5%) недоот-
мытой фосфорной кислоты и поэтому может быть использован в
качестве удобрения, но лишь в районах, близких к месту его получе-
ния, так как перевозка такого низкопроцентного удобрения не эко-
номична. Фосфогипс может применяться для гипсования солонцо-
вых почв или перерабатываться в штукатурный алебастр и литые
строительные детали 208’209. Термическим разложением в составе
цементной шихты его можно превратить в цементный клинкер и
в сернистый газ 210-217, а из последнего получить серную кислоту.
Таким путем возможно регенерировать серную кислоту, затрачен-
ную на разложение фосфата. Фосфогипс может также служить
источником сульфат-иона (взамен серной кислоты) при конверсии
его аммиаком и двуокисью углерода в сульфат аммония. Этот про-
цесс представляется перспективным в сочетании с производством
сложных удобрений азотно-сернокислотным и азотно-сульфатным
методами (стр. 1331). Представляет также интерес высокотемпе-
ратурная обработка фосфогипса в смеси с каолином и природными
фосфатами кальция и алюминия, с получением силикофосфорного
и алюмофосфорного удобрения212.
Перед отправкой фосфогипса к месту использования его необ-
ходимо высушить для улучшения транспортабельности (и для пре-
дотвращения смерзания при перевозке и хранении в зимнее время)
до содержания влаги меньше 3% и измельчить скомковавшийся
при сушке материал до порошкообразного состояния. Это услож-
няет его утилизацию. Пока ни в СССР, ни за рубежом фосфогипс
не используют — он является отходом производства. Влияет и ши-
рокая распространенность природного сульфата кальция, перера-
батывать который проще, чем фосфогипс.
5 М.. Е. Позин
920
Гл. XXV. Производство фосфорной кислоты
Технико-экономические показатели
Расходные коэффициенты сырья на производство экстракцион-
ной фосфорной кислоты зависят от применяемого фосфата и усло-
вий процесса. Например, на одном из заводов на 1 т Р2О5 в гото-
вой кислоте расход апатитового концентрата составляет 2,64 т, и
при норме серной кислоты 91,5 кг на 100 кг фосфата расход ее ра-
вен 2,41 т (в пересчете на моногидрат). Расходные коэффициенты
при переработке разных фосфоритов сильно колеблются: примерно
от 3,3 до 6 т фосфорита и от 2,6 до 4,1 т серной кислоты. Расход-
ные коэффициенты при переработке фосфоритов больше, чем при
переработке апатита: по фосфату в 1,5—2,3 раза,’по Р2О5, содер-
жащейся в фосфате, в 1,02—1,27 раза; по серной кислоте в 1,2—-
1,7 раза (в зависимости от количества примесей на 1 вес. ч. Р2О5
в фосфате).
Стоимость сырья в общих затратах на производство фосфор-
ной кислоты экстракционным способом составляет 70—80%. В за-
висимости от стоимости сырья, а также степени его использования
себестоимость экстракционной фосфорной кислоты в пересчете на
ТАБЛИЦА 72
Сравнительная калькуляция себестоимости 1 т Р2О5 в экстракционной
фосфорной кислоте из апатитового концентрата
Воскресенский завод Красиоуральский завод
Элементы затрат расход цена руб. сумма руб. расход цеиа руб. сумма руб.
Сырье: апатитовый концен- трат (39,4% Р206) серная кислота (100% H2SO4) Вспомогательные матери- алы Отходы 2,64 2,44 12,97 15,05 32,25 36,73 2,71 2,577 14,50 13,80 58,00 35,60 1,64 -1,36
Всего за вычетом отходов Энергетические затраты электроэнергия, квт • ч пар, мгк вода, jm3 сжатый воздух, тм3 . . Зарплата с начислениями Амортизация Цеховые расходы Общезаводские расходы . . 128 40,3 0,461 0,01465 0,0064 2,89 70,98 1,88 0,26 1,33 1,28 3,55 4,45 2,59 132 0,164 47,2 0,0138 4,75 0,0013 93,88 1,82: 0,78 0,61 2,08 3,64 4,5 1,34
Итого себестоимость . . . 86,32 108,68
Производство экстракционной Н3РО4 дигид ратным способом
921
1 т Р2О5 колеблется в широких пределах от 85—90 до ПО руб. и
выше.
В качестве примера ниже приведена калькуляция себестоимо-
сти экстракционной фосфорной кислоты (в пересчете на 1 т Р2О5)
для Воскресенского и Красноуральского заводов (табл. 72).
Показатели процесса зачастую ухудшаются из-за отклонения
параметров процесса от оптимального режима вследствие трудно-
сти ручного регулирования.
При автоматизации 28’218-222 технологического процесса годовой
расход серной кислоты уменьшился на 4,8%, а апатитового кон-
центрата на 5,1%.
Концентрирование фосфорной кислоты
Для получения концентрированных фосфорных и сложных удоб-
рений требуется фосфорная кислота, содержащая 37—55% Р2О5.
Так как в процессе сернокислотной экстракции фосфатов дигид-
ратным способом такая фосфорная кислота не образуется, ее по-
лучают выпариванием более слабой.
При нагревании водных растворов фосфорной кислоты любых
концентраций (вплоть до 98% Н3РО4) в газовую фазу выделяется
только водяной пар. Поэтому теоретически фосфорная кислота мо-
жет быть выпарена до очень высоких концентраций. Практически
выпарка осложняется коррозией материалов аппаратуры под дей-
ствием горячей фосфорной кислоты, которая сильно увеличивается
с повышением концентрации кислоты. Это требует применения вы-
сококачественных кислото- и термостойких материалов для изго-
товления оборудования выпарных установок. Выпарка затрудняет-
ся также выделением осадков, состоящих из примесей, загрязняю-
щих кислоту (сульфата кальция, кремнефторидов и др.) вследствие
уменьшения растворимости некоторых из них в концентрированной
кислоте. Осадки инкрустируют греющие поверхности, ухудшая
теплопередачу.
Примеси, находящиеся в фосфорной кислоте, ограничивают в
некоторых случаях предельную ее концентрацию после выпарки.
Например, фосфорная кислота, получаемая из необогащенных фос-
форитов Каратау, может быть сконцентрирована только до содер-
жания 37—38% Р2О5. При более высокой концентрации кислота
загустевает. Это объясняется 223 выделением MgSiFe • 6Н2О, кото-
рый выше 80° диссоциирует 224:
MgSiFe = SiF4 + MgF2
Тонкодисперсный фтористый магний увеличивает вязкость фос-
форной кислоты.
С увеличением концентрации экстракционной фосфорной кис-
лоты значительно возрастает давление пара растворенной в ней
кремнефтористоводородной кислоты 36-225. Значительная ее часть
5»
822
Гл. XXV. Производство фосфорной кислоты
выделяется в газовую фазу в виде смеси SiP4 + 2HF. При выпарке
кислоты до 33—35% РгО5 удаляется 25—35% фтора, а до 55—57%
Р2О5 — около 85% фтора, содержащегося в фосфорной кислоте.
Выпарку экстракционной фосфорной кислоты производят в ап-
паратах, в которых тепло передается через греющую поверхность,
обогреваемую паром (или другими теплоносителями), и в аппара-
тах с непосредственным нагреванием кислоты топочными газами.
В первом случае применяют однокорпусные вакуум-выпарные ап-
параты с выносной греющей камерой 32> 109,226-228. Корпус аппарата
гуммирован, нагревательные трубки — графитовые. Употребляются
также выпарные аппараты других конструкций, например, аппа-
раты с вертикальными свинцовыми трубами28, пленочные вакуум-
выпарные аппараты114 и др.
Для уменьшения инкрустации греющих трубок процесс осуще-
ствляют по циркуляционной схеме 229, т. е. концентрированная фос-
форная кислота частично возвращается на смешение с исходной
слабой кислотой. Количество выпаренной кислоты берут такое,
чтобы концентрация смеси была лишь немного ниже, чем в готовой
кислоте. Растворимость примесей в такой кислоте значительно
меньше, чем в исходной. Поэтому при смешении содержащиеся
в слабой кислоте примеси кристаллизуются. Их отделяют в отстой-
нике и осветвленную кислоту направляют в выпарной аппарат.
Меньшую часть выходящей из выпарного аппарата кислоты отво-
дят как готовый продукт, а большую часть возвращают в цикл на
смешение со слабой экстракционной кислотой. Применение рецир-
куляции выпаренной кислоты с предварительным отстаиванием
примесей значительно уменьшает отложения солей в нагреватель-
ной камере. Небольшая накипь один раз в 7—10 дней растворяется
кипящей водой или содовым раствором. Осадок из нижней части
отстойника откачивается шламовым насосом в экстракционное
отделение. Недостатком этого метода является необходимость цир-
куляции большого количества выпаренной фосфорной кислоты
(кратность циркуляции составляет 100—150 по отношению к про-
дукту) .
Выпарку фосфорной кислоты непосредственным нагреванием то-
почными газами осуществляют в барботажных концентраторах —
камерах из кислотоупорного материала. В них выпаривание произ-
водится при барботаже через поверхностный слой кислоты горячих
топочных газов. При этом выделяющиеся осадки твердых солей
остаются во взвешенном состоянии и выносятся из аппарата вме-
сте с кислотой, которая затем очищается отстаиванием.
На рис. 254 приведена схема установки для выпаривания фос-
форной кислоты в однокамерном барботажном концентраторе. То-
почные газы вводятся со скоростью 90—ПО м’.сек; их температура'
на входе колеблется в пределах 650—900° и на выходе 90—110°;
температура кислоты на несколько градусов ниже температуры
924
Гл. XXV. Производство фосфорной кислоты
отходящих газов; на 1 кг испаряемой воды расходуется 730—
790 ккал (при средней теплоте сгорания мазута 9,5 тыс. ккал!кг).
Влагосъем с 1 м2 зеркала испарения выпарной камеры около
400 кг/ч; полезное использование тепла горения топлива превы-
шает 80% 2181230’231. В выпаренной кислоте содержится 52—56%
Р2О5, 0,2—0,4% фтора, 3,5—5% SO3. В газе, выходящем из кон-
центратора, содержится при нормальных условиях от 2,5—3 до
8—9 г/м3 фтористых соединений (в пересчете на фтор). Фтористые
газы поглощаются водой в абсорберах с образованием раствора
HzSiFe.
Особенно интенсивно протекает выпарка фосфорной кислоты в
концентраторе с погруженным горением природного газа 28’118’232-234.
Аппарат гуммирован и футерован. На испарение 1 кг воды расхо-
дуется ~720 ккал. Горение газа вызывает пульсацию жидкости,
благодаря чему внутри камеры создается циркуляция, предотвра-
щающая осаждение твердых частиц. Выпаривание фосфорной кис-
лоты (30% Р2О5) до концентрации 50% Р2О5 производится в две
стадии. Для предварительной выпарки используется тепло отхо-
дящих газов второй стадии.
Уходящий из барботажных концентратов и аппаратов с по-
груженным горением дымовой газ уносит значительное количество
тумана фосфорной кислоты, который должен улавливаться в элек-
трофильтрах.
Выпаривание фосфорной кислоты непосредственным нагрева-
нием производят также в безнасадочных скрубберах-башнях, в ко-
торых навстречу горячему газу, поступающему из топки, падают
капли выпариваемой кислоты. Использование роторного разбрыз-
гивателя, образующего крупные капли, препятствует появлению
кислотного тумана. Многократная циркуляция выпариваемой кис-
лоты в скрубберах предотвращает накопление в них осадка. Этот
спосо-б выпарки требует большей затраты топлива и энергии, чем в
аппаратах с погруженным горением.
При получении в барботажном концентраторе 1 т кислоты кон-
центрации 54% Р2О5 расходуется, по данным Красноуральского
медеплавильного комбината: 2,227 т слабой фосфорной кислоты
(в пересчете на 25% Р2О5), 0,097 г мазута, 45,8 квт-ч электро-
энергии. Цеховая себестоимость 1 т кислоты концентрации 54%
Р2О5 равна 67,89 руб.
В расчете на 1 т Р2О5, находящейся в кислоте, это составит
125,54 руб. Следовательно, упаривание кислоты с 25 до 54% P2Os
обходится в ~17 руб. в расчете на 1 т P2Os.
ПОЛУЧЕНИЕ КОНЦЕНТРИРОВАННОЙ ФОСФОРНОЙ КИСЛОТЫ
ПОЛУГИДРАТНЫМ СПОСОБОМ
Получение концентрированной экстракционной фосфорной кис-
лоты (без упарки) полугидратным способом отрабатывается в на-
Получение концентрированной Н3РО4 полугидратным способом
925
стоящее время в ряде стран в разных вариантах на крупных опыт-
но-промышленных установках. Их различия обусловлены выделе-
нием полугидрата с разной стабильностью и способностью к гидра-,
тации, что отражается на условиях отмывки фосфорной кислоты из
осадка, его подготовке к транспортировке и хранению. В зависи-
мости от этого экстракцию фосфорной кислоты полугидратным спо-
собом проводят или с промежуточной гидратацией полугидрата
(полугидратно-дигидратный процесс) или без гидратации его
(стр. 907).
вода
Рис. 255. Схема получения фосфорной кислоты полу-
гидратио-дигидратным способом:
/ — вакуум-холодильник полугидратной пульпы; 2— фильтр полу-
гидрата; 3 — смеситель полугидрата, второго фильтрата и сер-
ной кислоты; 4 — вакуум-испаритель гипсовой пульпы; 5 —по-
гружной насос; 6 — сборник гипсовой пульпы; 7 —гидратор;
3 —сборник полугидратной пульпы; Р —экстрактор (полугидрат-
ный); 10 — фильтр.
На рис. 255 приведена схема полугидратно-дигидратного про-
цесса 127 получения фосфорной кислоты концентрации 42,5% Р2О5,
разработанная фирмой «Singmaster and Braier» (США). Разложе-
ние фосфата производится в первом экстракторе(полугидратной)
при 95° и недостатке серной кислоты по отношению к стехиомет-
рическому ее количеству. Фильтрованием пульпы получают продук-
ционную фосфорную кислоту, а отжатый осадок полугидрата, не
отмывая из него фосфорную кислоту, смешивают с серной кисло-
той и гидратируют в гипс. Получаемый при этом маточный рас-
твор — разбавленная кислота возвращается на разложение фосфата.
В первый экстрактор подают фосфоритную муку и часть необ-
ходимой серной кислоты. Другая часть кислоты добавляется в
926
Гл. XXV. Производство фосфорной кислоты
вакуумный холодильник, откуда она вместе с охлажденной цирку-
лирующей пульпой поступает в экстрактор через сборник-питатель
полугидратной пульпы. В вакуум-холодильник подается часть про- 1
Аукционной кислоты — основного фильтрата, полученного после от- |
деления полугидрата. Другая часть его отводится в качестве про- 1
дукции. Непосредственно в экстрактор вводится также разбавлен- |
ная кислота — первый фильтрат, полученный после отделения 1
гипса, который вместе с частью продукционной кислоты составляет I
раствор разбавления. В этом фильтрате содержится до 10% H2SO4. |
Таким образом, серная кислота, используемая для разложения 1
фосфата подается в экстрактор тремя потоками — непосредственно ]
с циркулирующей пульпой, из вакуум-холодильника и с разба-
вленной кислотой, полученной после перекристаллизации полу-
гидрата в гипс. Пульпа из первого экстрактора погружным насо- 1
сом откачивается в вакуум-холодильник, где она охлаждается 1
на 12—15°.
Холодильник снабжен турбинной мешалкой, обеспечивающей |
хорошее перемешивание потоков и циркуляцию пульпы в отдель- |
ных частях холодильника. Горячая пульпа подается через сопло |
в нижней части холодильника и охлаждается не только за счет |
испарения, но и теплообменом с охлажденными циркулирующими s
потоками. Дальнейшее выделение в твердую фазу полугидрата и 1
кремнефторидов в результате охлаждения массы в холодильнике j
происходит в объеме пульпы и они откладываются на поверхности |
имеющихся кристаллов. Это позволяет предотвратить образование |
твердых отложений на стенках холодильника и охлаждать пульпу 1
на 10—15°, т. е. примерно в 2—3 раза интенсивнее, чем в обычных )
вакуум-испарителях.
Охлажденная пульпа с температурой 80—85° через сборник j
фильтра частично возвращается в экстрактор (циркулирующая) и 1
частично направляется на фильтрование (или центрифугирование) 1
для отделения продукционной кислоты. Некоторое количество ее з
направляют в вакуумный холодильник с целью разбавления посту- I
пающей туда концентрированной серной кислоты и получения не- |
обходимого отношения Ж: Т в пульпе. Скорость фильтрования ]
пульпы весьма большая. На фильтрпрессе процесс продолжается 1
~2 мин. ;
Отжатый на фильтрпрессе (или центрифуге) осадок, состоящий !
из полугидрата сульфата кальция и содержащий значительное ко- 1
личество фосфорной кислоты, направляют в аппаратуру гидрата- J
ции (перекристаллизации полугидрата в гипс). Одновременно в 1
процессе гидратации происходит регенерация фосфорной кислоты,
поступающей с неотмытым осадком полугидрата. Она возвра- (
щается на разложение фосфата в виде первого фильтрата, полу- ,
чаемого при отфильтровывании образовавшихся кристаллов гипса j
от жидкой фазы.
Получение концентрированной Н3РО4 полугидратным способом
997
Гидратация полугидрата производится в гидраторе, снабженном
мешалкой и погружным насосом. В него поступает лепешка полу-
гидрата, первый фильтрат, полученный после отделения гипса, и
некоторое количество серной кислоты Жидкая фаза пульпы в
гидраторе содержит 20—25% Р2О5 и 5—10% H2SO4. Температуру
пульпы в гидраторе поддерживают равной 75—77° и регулируют
ее при помощи циркулирующей части пульпы, охлаждаемой в гип*
совом вакуум-холодильнике. Благодаря большому разбавлению
пульпы (Ж:Т) и высокой концентрации серной кислоты создаются
условия для выделения гипса в виде крупных ромбических кристал-
лов с размерами до 100—-150 мк, а иногда и до 500 мк. При этом
почти полностью исключается сокристаллизация с гипсом дикаль-
цийфосфата, что обусловливает уменьшение потерь Р2О5 с отмы-
тым фосфогипсом.
Пульпа из гидратора погружным насосом перекачивается в
вакуум-холодильник, откуда часть (циркулирующая) охлажденной
пульпы возвращается в гидратор. Оставшееся ее количество на-
правляется на фильтрацию и промывку водой (по двухфильтрат-
ной схеме).
В холодильнике за счет испарения некоторого количества воды
отводится теплота разбавления серной кислоты, поступающей на
гидратацию, а также тепло, выделяющееся при перекристалли-
зации полугидрата сульфата кальция в гипс. Фильтрование ди-
гидратной пульпы производят на ленточном (или карусельном)
фильтре при 65—68°. Вследствие выделения при гидратации полу-
гидрата крупных хорошо фильтрующих кристаллов гипса фильтро-
вание пульпы происходит с большой скоростью. Длительность
всего цикла—основной фильтрации с одной или двумя противо-
точными промывками — составляет ~ 1 мин. При этом образуется
лепешка толщиной 45—50 мм.
В отмытом фосфогипсе содержится от 0,3 до 0,7% общей Р2О5,
в том числе 0,2—0,5% водорастворимой Р2О5, 0,1—0,2% Р2О5 рас-
творимой в цитратном растворе и 0,1—0,3% нерастворимой Р2О5.
Степень извлечения Р2О5 в кислоту составляет 97—98%.
Недостатками этого процесса является образование неустойчи-
вого полугидрата сульфата кальция вследствие чего невозможна
отмывка его от фосфорной кислоты, а также очистка водой филь-
тровальной ткани или перегородки. Химическая их регенерация
также исключается из-за отсутствия пригодного растворителя По-
этому представляется трудно осуществимой длительная кампания
фильтровального материала фильтров или центрифуг. Помимо
этого получаемая фосфорная кислота загрязнена монокальцийфос-
фатом и имеет недостаточно высокую концентрацию (~42,5°/о
P2OS), что в некоторой степени ограничивает области ее приме-
нения.
928 Гл. XXV. Производство фосфорной кислоты
Более перспективен процесс, основанный на выделении полу-
гидрата, не гидратирующегося при промывке водой и в дальней-
шем в течение достаточного времени необходимого для транспорти-
ровки. Для успешного осуществления процесса требуется помимо
стабильности полугидрата образование его в виде крупных хо-
рошо фильтрующих компактных конгломератов, легко отмываемых
от фосфорной кислоты небольшим количеством воды.
Рис. 256. Схема производства экстракционной кислоты полугидратным
методом по способу ЛТИ — Винницкого химкомбината — Ленниигипрохима:
1 — эл ^ватор; 2 — бункер; 3 — питатель; 4 — шнек; 5 — дозатор; 6 — наборный бак; 7 — сбор-
ник фильтрата; 8 — экстрактор; Р —ленточный вакуум-фильтр; 10 — вакуум-приемннк;
11 - репульпатор; 12 -сборник; 13 — промежуточный бак; 14 - вакуум-испаритель.
На рис. 256 представлена схема производства концентрирован-
ной фосфорной кислоты прямым полугидратным методом, т. е.
без перекристаллизации полугидрата в гипс, разработанная в
СССР ЛТИ имени Ленсовета, Винницким химическим комбинатом
и Леннигипрохимом.
Отработка способа и создание промышленного производства
концентрированной фосфорной кислоты были осуществлены в спе-
циальном опытно-промышленном цехе, проектирование которого
было начато Ленниигипрохимом в 1963 г.
Этот способ заключается в практически полном разложении
апатита в избытке фосфорной кислоты и в обработке полученной
пульпы (представляющей собой суспензию монокальцийфосфата
в растворе фосфорной кислоты, насыщенном монокальциифосфа-
Получение концентрированной Н3РО4 полугидратным способом
929
том) серной кислотой при регулируемой кристаллизации полугид-
рата сульфата кальция.
Разложение апатитового концентрата производится в 3,5—4-
кратном избытке фосфорной кислоты от стехиометрического коли-
чества, необходимого для образования монокальцийфосфата, при
90—95° в течение 1,2—1,7 ч. Требуемый избыток фосфорной кис-
лоты, содержащей 45—48% Р2О5, создается введением в реактор
первого фильтрата — части продукционной кислоты и фосфорно-
кислотно-полугидратной пульпы. Степень разложения апатита со-
ставляет 98,5—99%.
С целью удаления переходящего в раствор иона SiF6 к апа-
титу добавляют немного соды; в результате этого около 50% со-
держащегося в апатите фтора осаждается в виде кремнефторида
натрия. Около 35% фтора выделяется в форме газообразных со-
единений.
Образующаяся в первой стадии монокальцийфосфатная пульпа
подается на вторую стадию — обработку серной кислотой для экс-
тракции фосфорной кислоты с выделением полугидрата сульфата
кальция в виде изометричных компактных друз, хорошо фильтрую-
щих и достаточно стабильных при промывке водой. Серная кис-
лота, необходимая для взаимодействия с монокальцийфосфатом,
подается в смеси со вторым фильтратом, содержащим 34—35%
Р2О5 в виде так называемого раствора регенерации. Разложение
монокальцийфосфата серной кислотой и кристаллизация полугид-
рата сульфата кальция при 95—100° происходит почти мгновенно
(в пределах времени, необходимого для смешения потоков). В жид-
кой фазе пульпы поддерживается «нулевой» и «минусовый» серно-
кислотный режим (практически отсутствует свободная серная кис-
лота). Отношение между жидкой и твердой фазами в пульпе со-
ставляет от 2,5 : 1 до 3 : 1.
При указанном технологическом режиме полугидрат выде-
ляется в виде шарообразных изометрических конгломератов диа-
метром до 100 мк.
Прореагировавшую пульпу, представляющую собой суспензию
полугидрата сульфата кальция в растворе фосфорной кислоты кон-
центрации 45—50% Р2О5, частично возвращают на стадию разло-
жения фосфата, частично отводят на фильтрацию. Предварительно
Доследнюю часть пульпы можно направить в гидроциклон для сгу-
щения до отношения Ж:Т = 2:1 и одновременной классификации
полугидрата. Фильтрация и промывка полугидрата сульфата каль-
ция осуществляются на наливном (ленточном или карусельном)
фильтре по четырехстадийной схеме (одна основная фильтрация и
три промывки). Получаемый первый фильтрат частично отводится
в виде продукции, а остальная масса его подается на разложение
апатита. Промытый полугидратный осадок удаляется любыми ви-
дами транспорта.
930
Гл. XXV. Производство фосфорной кислоты
Степень отмывки из осадка фосфорной кислоты при расходе
~ 800 кг воды на 1т апатита при вакууме 0,45—0,5 ат достигает
98%. Влажность осадка после последней стадии промывки равна
17,5%; во влажном осадке после первой зоны фильтрации содер-
жится 30—35% жидкой фазы. Вода подается нагретой до 60°.
Продолжительность фильтрования и промывания составляет
1,2—1,5 мин, а производительность фильтра достигает 1300—1500 кг
сухого осадка с 1 м2 в 1 ч. Распределение поверхности фильтра по
вонам (в % от общей фильтрующей поверхности) составляет: в
первой зоне 50—60, второй — 20—30, третьей и четвертой — по 10.
При фильтрации и промывке осадка фильтровальная ткань
(лавсан на шелку) не гипсуется и срок ее службы, достигающий
10—12 суток определяется механической прочностью. На второй
и третьей зонах фильтра откладываются незначительные количе-
ства осадка (в основном из кремнефторидов), которые легко рас-
творяются при периодической (1 раз в 7—10 дней) промывке си-
стемы горячей водой.
Промытый осадок полугидрата сульфата кальция устойчив при
хранении: он не схватывается в монолитную массу в течение весь-
ма длительного времени в закрытом помещении цеха, на открытой
площадке и на шламовом поле. Значительная часть осадка исполь-
зуется для гипсования почв.
Получаемая фосфорная кислота, содержащая 45—48% Р2О3,
0,1—0,2%. СаО, 0,5—0,7% фтора, 0,2—0,4% SOtn 1,2% R2O3, ис-
пользуется для производства кормовых фосфатов и удобрений.
Описанный процесс отличается высокими технико-экономиче-
скими показателями. На производство фосфорной кислоты расхо-
дуется в расчете на 1 т Р2О3:2,63 т апатитового концентрата и
2,36 т серной кислоты (моногидрат). Съем Р2О3 с реакторов обеих
стадий составляет 29—30 кг с 1 м3 в 1 ч. Необходимая поверх-
ность фильтрации в расчете на 1 т/ч Р2О3 в продукционной кислоте
составляет 2,8—2,85 .и2. Кратность циркуляции пульпы, определяе-
мая отношением количества пульпы, возвращаемой на разложение
апатита и отбираемой на фильтрацию составляет ~2: 1. На
рис. 257 приведен общий материальный баланс процесса на 1 т
апатитового концентрата.
Объем реакционной аппаратуры, необходимый для стадии экс-
тракции фосфорной кислоты меньше в 2,5 раза, чем для разложе-
ния апатита.
По сравнению с дигидратным производство концентрированной
фосфорной кислоты полугидратным методом позволяет увеличить
производительность реакционной аппаратуры в 1,5—1,6 раза, филь-
трующего оборудования не менее чем в 2 раза по съему Р2О3 с 1 м2,
съем фтора, в зависимости от температуры и метода обесфторива-
ния, до 30—60% от содержания его в фосфатном сырье и умень-
Получение концентрированной Н3РО4 полугидратным способом
931
шить на 70—100 кг на 1т РгО5 расход серной кислоты, а также
массу отбросного сульфатного осадка на 20—25%.
В СССР и за рубежом находятся в стадии опытной разработки
и другие способы осуществления процесса полугидратным мето-
Рис. 257. Схема материальных потоков производства концентри-
рованной экстракционной фосфорной кислоты полугидратным мето-
дом по способу ЛТИ — Винницкого химкомбината — Ленннигипро-
хима (цифрами обозначены количества в кг на 1000 кг апатитового
концентрата; количество добавляемой соды не учитывается).
дом, основанные на несколько иных принципах (НИУИФ — Гипро-
хим, Красноуральский медеплавильный комбинат и другие в СССР,
фирма «Fisons Fertiliser ltd» в Англии и др.).
На рис. 258 показана схема 131 получения концентрированной
кислоты прямым полугидратным методом по способу, разработан-
ному фирмой «Fisons Fertiliser ltd». Выделение в втом случае в
процессе экстракции достаточно стабильного полугидрата дости*
гается при определенной температуре и концентрации кислоты
регулированием степени пересыщения жидкой фазы сульфатом
932
Гл. XXV. Производство фосфорной кислоты
кальция, а также количества кристаллической затравки. При тем-
пературе и концентрации кислоты, соответствующим большей рас-
творимости гипса, чем полугидрата (т. е. в условиях промывки
осадка) стабильность полугидрата тем больше, чем больше степень
пересыщения раствора сульфатом кальция. Это обусловливает осу-
ществление экстракции и промывки с получением жидкой фазы,
сильно пересыщенной сульфатом кальция. Помимо этого, в твердой
фазе, а также на стенках аппаратов не должно быть отложений
гипса, так как гидратация полугидрата в указанных условиях резко
ускоряется в присутствии затравки кристаллов гипса. Стабильность
срорная кислота
Рис. 258. Схема получения фосфорной кислоты полу-
гидратным методом по способу Fisons:
/ — первый реактор; 2 — второй реактор; 3 — сборник для питаю-
щего фильтрата; 4 —фильтр; 5 —сборники фильтрата.
полугидрата в разбавленных растворах при относительно пони-
женных температурах (55—65°) также возрастает с увеличением
размеров его частиц, так как замедляется их растворение в жид-
кой фазе, как предварительного этапа, предшествующего фазовому
переходу. Поэтому экстракцию необходимо осуществлять в усло-
виях, обеспечивающих максимальный рост кристаллов полугид-
рата в реакционном объеме. Это достигается ступенчатой подачей
серной кислоты и осаждением сульфата кальция в несколько ста-
дий. Обычно для этого необходимо минимально два экстрактора
(или две ступени). При осуществлении процесса в одну стадию
(в одном реакторе без перегородок) невозможно получить кри-
сталлы полугидрата с удовлетворительными размерами. Наилуч-
шие результаты получены при использовании четырех аппаратов
или одного четырехсекционного реактора. При этом количество сер-
ной кислоты по аппаратам (или секциям) регулируют так, чтобы
в первых двух осаждалось около 30—40% сульфата кальция от
общего его количества, а остальное количество в последних сек-
циях. Разложение фосфорита и выделение полугидрата протекает
при 85—110°. Степень использования Р2О5 фосфата зависит от
концентрации получаемой кислоты и изменяется от 97—98%
Литература
933
до 92—94% при получении кислоты, содержащей от 40 до 55%
Р2О5.
ЛИТЕРАТУРА
1. А. И. Гельбштейн, М. И. Темкин, ЖОХ, 23, в. 8, 1278 (1953).—
2. К. И. Загвоздкин, Ю. М. Рабинович, Н. А. Барилко, ЖПХ, 18,
№ 1, 29 (1940). — 3. R. N. Bell, Ind. Eng. Chem., 40, 8, 1464 (1948).—
4. L. F Andrieth, R. N. Bell, Inorganic Syntheses, 3, 85 (1950).—
5. M. А. Клочко, M. Ш. Курбанов, ИСФХА АН СССР, 24, 252
(1954).—6. С. И. Скляренко, И. В. Смирнов, ЖФХ, 25, № 1, 24
(1951). — 7. Н. Д. Литвинов, Т. А. Крюкова, Е. А. Курочкина, ЖПХ,
7. № 7, 1121 (1934). — 8. И. А. Каблуков, К- И. Загвоздкин, Труды
НИУ, в. ПО, 1933. — 9. К. Загвоздкин, Труды НИУИФ, в. 143, 1938,
Стр. 19. —10. Е. Н. Brown, С. D. Whitt, Ind. Eng. Chem., 44, № 3, 615
(1952).
11. Э. В. Б р и ц к е, Н. Е. Пестов, Труды НИУ, в. 59, 1929.—
12. Р. Kharabanda, Ind. Chem., 32, № 374, 120 (1956). — 13. М. М. Попов,
Н. Н. Феодосьев, С. М. Скуратов, Труды НИУ, в. ПО, 1933.—
14. Н. Basset, J. Chem. Soc., 1958, № 9, 2949. —15. И. Л. Гофман, Хим.
Йром., № 2, 119 (1955). — 16. I. Gordon, Chem. Week., 71, № 5, 26 (1952).—
17. Chem Eng., 60, № 3, 196 (1953). — 18. Statistical Yearbook United Nations,
1956.— /9. W. L. Hill, Ind. Eng. Chem., 44, № 7, 1526 (1952). — 20. Ibid, 46,
№ 6, 1121 (1954).
21. Chem. Age (India), 18, № 7, 482 (1967). —22. С. И. Вольфкович,
Усп. хим., 25, в. 11, 1309 (1956).—23. С. И. Вольфкович, Научно-техн, ин-
форм. бюлл. НИУИФ, № 4 (1957). — 24. П. В. Дыбина, А. А. Соколов-
ский, Сборник статей ВЗПИ, в. 19—20, 1957, стр. 171. — 25. В. Ваггаман,
Фосфорная кислота, фосфаты и фосфорные удобрения, Госхимиздат, 1957.—
26. С. Н. Young, Military Eng, 50, № 335, 165 (1958).—27. Технология фос-
форной кислоты, двойного суперфосфата и фосфатов аммония, под ред.
С. Вольфковича, Труды НИУИФ, в. 153, 1940. — 28. С. К. Воскресенский,
ЖПХ, 27, № 7, 699 (1954). — 29. С. К. Воскресенский, Хим. наука и пром,
1, № 2, 129 (1956). — 30. А. А. Соколовский, ЖПХ, № 11 (1932); № 10
(1933); № 2 (1936).
31. С. И. Вольфкович, С. К- Воскресенский, А. А. Соколов-
ский, Р. Е. Ремен, М. М. Кобрин, Труды НИУИФ, в. 153, 1940, стр. 12.—
32. Е. Бруцкус, Там же, в. 134, 1936.—33. Л. М. Орлова, Там же, в. 137,
1937, стр. 87. — 34. В. Н. Свешникова, М. Н. Ляшенко, ЖНХ, 1, вып. 3,
532 (1956).—35. М. Л. Ч е п е л е в е ц к и й, Е. Б. Бруцкус, Исследования по
физико-химическим основам производства суперфосфата, изд. НИУИФ, 1957. —
36. М. Л. Ч е п е л е в е ц к и й, Е. Б. Б’руцкуС, Суперфосфат, Физико-химиче-
ские основы производства, Госхимиздат, 1958. — 37. А. А. Соколовский,
М. В. Слад ков а, И. Л. Гофман, Труды НИУИФ, в. 153, 1940, стр. III.—
38. А. А. Соколовский, М. М. Кобрин, А. А. И о н а с с, С. К. Милова-
нова, Там же, стр. 54.— 39. Е. Б. Бруцкус, Труды НИУИФ, в. 137, 1937",
стр. ПО. — 40. М. Л. Ч е п е л е в е ц к и й, Т. А. Крюкова, Там же, в. 137,
1937, стр. 102.
41. Е. Бруцкус, М. Чепелевецкий, Труды НИУИФ, Там же, в. 147,
1940, стр. 24. — 42. Д. Хейфец, Химизация соц. земледелия, № 5,3 (1936).—
43. Е. Бруцкус, Труды НИУИФ, в. 143, 1938, стр. 19. — 44. Ё. Б. Бруц-
кус, сб. «Исследования по прикладной химии», Изд. АН СССР, 1955, стр. 184.—
45. I. С. Brosheer, F. A. Lenfesty, I. F. Anderson, J. Am. Chem. Soo.,
?6, № 23, 5951 (1954).—46. А. П. Белопольский, А. А. Таперов J,
934
Гл. XXV. Производство фосфорной кислоты
М. Н. Шульгина, ЖПХ, 12, 1 (1939); 13, 5 (1940); 18, 9—10 (1945); 23, 1
(1950).— 47. A. N. Campbell. J. W. Coutts, Ind. Eng. Chem., 40, № 8, 1295
(1948).-48. К. С. Краснов, ЖПХ, 28, № 12, 1275 (1955). —49. Д. И. Бе-
ло цк и й, М. Ф. Хохол, ЖНХ, 5, 708 (1960).—50 С. А. Щ у к а р е в,
Т. Г. Б а л и ч е в а, К- Я- Б о р ч а, Там же, 8, 1437 (1963).
51. В. И. Владимирова, Б. А. Конышев, В. П. М а ш о в е ц. Тезисы
докладов НТК ЛТИ им. Ленсовета, «Химия», 1965, стр. 57.—52. В. И. Влади-
мирова, В. П. Машовец, ЖПХ, 39, № 9, 1936 (1966). — 53. М. В. Ж д а н о-
в а, Р. Ю. 3 и н ю к, Б. А. К о п ы л е в, Тезисы докладов НТК ЛТИ им. Ленсове-
та, 1968, стр. 15. — 54. Е. Б. Б р у ц к у с, М. Л. Ч е п е л е в е ц к и й, ИСФХА АН
СССР, 20, 383 (1950).— 55. М. Л. Ч е п е л е в е ц к и й, Труды НИУИФ, в. 137, 1937,
стр. 10. — 56. W. L. Hill, I. Н. Саго, G. A. Wieczorek, J.. Agr. Food. Chem.,
2, № 25, 1273 (1954).—57. F. Carbona, Ind. chim., № 463, 41 (1956).—
58. M. Л. Ч e п e л e в e ц к и й, С. С. Р у б и н о в а, Труды НИУИФ, в. 137, 1937,
стр. 36, 59, 66.—59. М. Е. Позин, Б. А. Ко пылев, Тен С е - д е н, Труды
ЛТИ им. Ленсовета, в. 36, 1956, стр. 22. — 60. М. Л. Ч е п е л е в е ц к и й,
Е. Б. Б р у цк у с, К. С. Краснов, Е. В. Южная, ЖНХ, 1, № 7, 1512 (1956).
61. К. С. Краснов, ЖПХ, № 11, 1114 (1953). — 62. К- С. Краснов,
Изв. вузов. Хим. и хим. технол., № 3, 100 (1958); ЖПХ, 31, № 3, 345 (1958).—
63. Е. В. Южная, Автореф. канд. дисс., НИУИФ, 1956.—64. X. Б ел о поль-
ский, А. Таперов а, М. Шульгина, Труды НИУИФ, в. 143, 1938,
стр. 16. — 65. Е. Б. Бру цк у с, О. И. Курт ев а, Сообщения о научно-техни-
ческих работах НИУИФ, № 2, Госхимиздат, 1957; Реф. н.-и. раб. за 1956 г.,
ч. I, стр. 3, Труды НИУИФ, в. 161, 1958.—66. О. И. Куртева, Е. Б. Бруц-
кус, Сборник аспирантских работ, изд. НИУИФ, 1959, стр. 15. — 67. О. И. Кур-
тева, Е. Б. Бруцкус, Реф. и.-и. раб. за 1957 г., ч. I, стр. 16, Труды НИУИФ
в. 163, 1595.—68. Е. В. Хамский, М. Л. Чепелевецкий, ЖПХ, № 5,
948 (1959). — 69. Campbel, Cootts, Ind. Chem., 40, № 7, 1295 (1948).—
70. M. Л. Чепелевецкий, Е. В. Хамский, ЖПХ, 32, Ns 5, 948 (1959).
71. О. И. Куртева, Е. Б. Бруцкус, Там же, 34, № 1, 8 (1961).—
72. Е. В. Хамский, Автореф. канд. дисс., НИУИФ, 1955. — 73. Е. В. Хам-
ский, М. Л. Чепелевецкий, ЖПХ, 31, № 7, 976 (1958).—74. М. Л. Че-
пелевецкий, Н. К. Падо, Реф. н.-и. раб. за 1957 г., ч. I, стр. 14, Труды
НИУИФ, 1959, стр. 163, —75. Н. Lehrecke, Chem. Fabr., Ns 50, 505 (I933);'Me-
tallborse, Ns 57 (1933). — 76. A. Sanfourche, Ind. chim., Ns 244, 331 (1934);
№ 245, 406 (1934). — 77. H. Tiedemann, W. Gundelach, Chem. Fabr.,
Ns 49 (1933). — 78. S. G. Nordengreen, пат. США 2002547, 1935.—
79. С. К. Воскресенский, А. А. И о и а с с, Труды НИУИФ, в. 153, 1940,
стр. 92. — 80. М. Е. П о з и н, Б. А. Копы л ев, Я. К. Ядацкая, Изв. вузов.
Хим. и хим. технол., 9, Ns 2, 171 (1966).
81. Т. В. Любченко, Б. А. Копы лев, М. Е. П о з и н, ЖПХ, 40, № 10,
2225 (1967). — 82. М. Л. Чепелевецкий, Б. Б. Евзлина, Труды НИУИФ,
В. 137, 1937, стр. 127.—83. М. Л. Чепелевецкий, С. Рубинов а. Там же,
В. 147, 1940, стр. 24. — 84. Е. R. McCartney, J. Coll. Sci., 13, № 4, 383
(1958).—85. С. К. Милованова. Автореф. канд. дисс., НИУИФ, 1955.—
86. С. К- Воскресенский, С. К- Милованова, Сообщения о научно-тех-
нических работах НИУИФ, Ns 2, Госхимиздат, 1957, стр. 9. — 87. С. К- Мило-
ванова, Научно-техн, информ, бюлл. НИУИФ, № 9, 49 (1957). — 89. Б. А. Ко-
пы л е в, Т. В. Любченко, М. Е- П о з и н, ЖПХ, 42, № 11, 2431 (1969).—
89. Е. II е i n е г t h, пат. ФРГ 923845, 1955.—90. С. К. Воскресенский,
А. А. И о н а с с, Труды НИУИФ, в. 153, 1940, стр. 92, 94.
91. М. Е. П о з и н, Б. А. Копы л ев, В. Л. Варшавский, И. Пинтер,
ЖПХ, 34, 2384 (1961).—92. М. Е. Поз ин, Б. А. Ко пыл ев, Б. А. Дмит-
ревский, Неопубл, материалы.—93. М. Е. П о з и н, Б. А. Ко пыл ев,
Б. Л. Варшавский, Неопубл, материалы. — 94. X. А. Соколовский,
Литература
935
ЖХП, № 11 (1932); № 10 (1933); № 2 (1936). — 95. И. Гофман, Я. Зиль-
берман, И. Островский, Технология суперфосфата и других фосфорных
удобрений, Химтеорет, 1937. — 96. Н. М. Вайнман, Д. К- Панарин,
А. А. Соколовский, Производство преципитата, ГОНТИ, 1937.—
97. А. И. Шерешевский, А. А. Соколовский, П. Ф. Деревицкий,
Производство фосфорных удобрений, ГОНТИ, 1938. — 98. А. М. Дубовиц-
кий, А. И. Шерешевский, Технология минеральных удобрений, Госхим-
издат, 1947. — 99. А. А. Соколовский, Технология фосфорных удобрений
(учебное пособие), изд. ВЗПИ, 1954. —100. М. Е. Позин, Технология мине-
ральных удобрений и солей, Госхимиздат, 1957.
101. А. А. Соколовский, Технология минеральных удобрений, йзд.
«Химия», 1966. — 102. С. К- Воскресенский, Научно-техн, информ, бюлл.
НИУИФ, № 1, 1957. — 103. С. К. Воскресенский, Тезисы докладов на от-
раслевом совещании работников основной и горнохимической промышленности,
1958, стр. 65. — 104. С. Д. Э в е н ч и к, В. А. Коионов, Там же, стр. 72; Бюлл.
техн, информ. Гипрохима, № 40—41, 3 (1958). — 105. W. С. Weber, Chem. Met.
Eng., № 12 (1932). — 106. I. Atwell, Ind. Eng. Chem., 41, № 7, 1318 (1949).—
107. Ind. Chem., 27, № 317, 256 (1951). — 108. I. I. Porter, I. F r i s k e n, Chem.
Trade J., 132, № 3426, 249 (1953); Farm. Chem., 118, № 11, 38, 43, 70 (1953). —
109. G. C. Inskeep. W. R. Fortr, W. C. Weber, Ind. Eng. Chem., 48, № 10,
1804 (1956). — 110. W. R. Horn, Agr. Chem., 12, № 1, 39 (1957).
111. Chem. Eng. News, 35, № 4, 59 (1957); J. Agr. Food Chem., 5, № 3, 161
(1957). —//2. G. Burnet, J. Agr. Food Chem., 5, № 4, 258 (1957). — 113. Ab-
страл. пат. 164898, 1955. — 114. R. I. McNally, Mining Eng., 8, № 10, 1017
(1956). —115. Хим. и хим. технол., № 11, 42 (1958). — 116. С. К. Воскресен-
ский, И. Л. Гофман, С. К. Милованова, Р. М. Серебряная, Реф.
н.-и. раб. за 1957 г., ч. 1, стр. 13, Труды НИУИФ, в. 163, 1959. — 117. Chem. а.
Process Eng., 34, № 11, 353 (1953). —118. Chem. Age, 69, № 1784, 593 (1953);
Chem. Trade J. a. Chem. Eng., 133, № 3459, 689 (1953). — 119. Engineer, 196,
Ns 5096, 409 (1953). — 120. S. Nordengreen, Ind. chim., 38, № 411, 278
(1951).
121. S. Nordengreen, R. Francia. P. Nordengreen, ibid., 43,
№ 464, 80 (1956). —/22. V. S a u c h e 11 i, Agr. Chem., 11, № 8, 51 (1956); 11,
№ 10, 69, 71 (1956). —123. M. S h о e 1 d, пат. США 2384773, 1945 —
124. CL C. Legal, I. N. Pryor, T. O. Tongue, P. L. V e 11 m a n n, Ind. Eng.
Chem., 49, № 3, I, 334 (1957). — 125. B. W. Hammaren, R. V. Town end,
канад. пат. 497336, 1953. — 126. I. H. Coleman, пат. США 2384813, 1945;
2384814, 1945.— /27. R. Chelminski, R. L. Somerville, Chem. Eng.
Progr., 62, № 5, 108 (1966). — 128. Неопубликованные материалы ЛТИ им. Леи-
ровета.—129. J. Е. Do ver port, J. G. Get sin ger, F. K- Carrol, Ind.
Eng. Chem. Design a. Development, 4, № 1, 84 (1965). —130. Chem. Age (India),
16, Ns 12, 1080, 1081 (1965).
131. N. Robinson, Phos. a. Pot., № 31, 23 (1967); Brit. Chem. Eng., 12,
Ns 11 (1967). —132. С. Воскресенский, С. Милованова, Труды
НИУИФ, в. 134, 1936. — 133. М. М. Кобрин, Там же. — 134. А. А. И о н а с с,
М. М. Кобрин, ЖХП, 13, Ns 5, 271 (1936). —/55. А. И о н а с с, Труды
НИУИФ, в. 134, 1936. —136. М. Дорниш, М. Сладкова, Там же.—
137. С. К. Воскресенский, С. К. Милованова, ЖПХ, 13, Ns 13, 770;
Ns 14, 850 (1936).—J38. С. Воскресенский, А. Ионасс, Труды НИУИФ,
в. 143, 1938, стр. 13. —139. М. Сладкова и др., Там же, стр. 14. —140.
С. Воскресенский, С. Милованова, Б. Розенберг, Труды НИУИФ,
в. 143, 1938.
141. С. Воскресенский. А. Ионасс, Там же, стр. 15.—
142. А. Ионасс, Р. Ошерович, Там же, стр. 16. —143. М. Сладкова,
Там же, в. 147, 1940, стр. 23. — 144. А. А. Соколовский, М. Кобрин,
936
Литература
М. Сладко в а, Там же, стр. 27. — 145. А. Соколовский, М. Сусаров,
Там же. —146. к. Соколовский, М. Сладкова, Там же, стр. 22.—
147. С. К. Воскресенский, Там же, в. 153, 1940, стр. 43. — 148. С. К. Вос-
кресенский, С. К- Милованова, А. А. Ионасс, Там же, стр. 65,—
149. С. И. Вольфкович, С. К. Воскресенский, Н. И. Крючков,
Р. Е. Р е м е н, Э. И. С в и д е р с к и й, Г. М. С т р о н г и н, Там же, стр. 143.—
150. М. В. Сладкова, Труды НИУИФ, в. 153, 1940, стр. 103.
151. 3. Б. Канторович, Машины химической промышленности, т. I, Маш-
п.з, 1957. — 152. Э. Н. Гинзбург, Хим. пром., № 3, 79 (1951).— 153.
Э. Н. Гинзбург, Научно-техн, информ, бюлл. НИУИФ, № 2—3, 3 (1957).—
154. Э. Н. Гинзбург, И. Л. Гофман, Б. И. 3 е н з и и о в, С. К. Милова-
нова, Р. М. Серебряная, Реф. н.-и. раб. за 1956 г., .ч. 1, стр. 18, Труды
НИУИФ, в. 161, 1958; Реф. н.-и. раб. за 1957 г., ч. 1, стр. 17, Труды НИУИФ,
в. 162, 1959. — 155. Э. Н. Гинзбург, И. Л. Гофман, С. К- Милованова,
Хим. пром., № 5, 73 (443) (1959). —156. П. Я- Крайний, М. Л. Варламов,
А. Г. Большаков, Э. К. Л о п а т т о, Л. С. Ф ош ко, Научн. зап. Одесои.
политехи, ин-та, 2, № 2, 4 (1954). —157. Chem. Eng. News, 34, 2452, May 14-
(1956). —/58. Chem. Eng., 61, № 6, 128, 296, 298 (1954). —159. В. А. Жужи-
ков, Фильтрование, Изд. «Химия», 1968. — 160. Г. А. Максудов, И. И. Бу-
лыгин, Сообщ. о научно-технич. работах НИУИФ, № 1, 1957, стр. 16.
161. Г. А. Максудов, И. И. Булыгин, А. А. Ежова, Реф. н.-и. раб.
за 1956 г., ч. II, стр. 40, Труды НИУИФ, в. 162, 1959; Реф. н.-и. раб. за 1957 г.,
ч. 1, стр. 84, Труды НИУИФ, в. 163, 1959. — 162. В. В. К а ф а р о в, В. Д. П ci-
до й м а, Хим. пром., № 2, 86 (1957). — 163. W. С. Weber, Chem. Met. Eng.,
39, № 10 (1932). — 164. И. Зарин г, А. Логинова, К- Петрова, ТрудЙ
НИУИФ, в. 147, 1940, стр. 42. — 165. Chem. Eng., № 10, 109 (1948).—
166. I. I. Porter, G. G. Lowriston, Chem. Trade J., 133, № 3465, 1073-
(1953); Ind. chim., 41, № 438, 14 (1954). —167. Л. А. Кузнецов, Б. И. Леви,
Сообщ. о научно-техн, работах НИУИФ, № 3, 1957, стр. 37.— 168. К- Г. Берг-
ман, Б. И. Л е в и, Там же, стр. 41. — 169. К- Г. Бергман, И. И. Булыгин,
Б. И. Леви, А. А. Ежова, Реф. н.-и. раб. за 1956 г., ч. 2, стр. 39, Труды
НИУИФ, в. 162, 1959; Реф. н.-и. раб. за 1957 г., ч. I, стр. 79, 82, Труды НИУИФ,
в. 163, 1959. — 170. G. Bi geon, Ind. chim., № 490, 130 (1958).
171. M. E. П о з и н, Б. А. К о п ы л е в, Г. В. Бельченко, Л. Я. Тере-
щенко, Расчеты по технологии неорганических веществ, Изд. «Химия», 1967.—
172. И. Л. Гофман, Труды НИУИФ, в. 147, 1940, стр. 23; в. 153, 1940,
стр. 122. — 173. М. Л. Чепелевецкий, Ц. С. Больц, ЖПХ, 10, № 7, 1186
(1937). — 174. Ц. Больц, Труды НИУИФ, в. 147, 1940, стр. 23.—
175. С. Я. Шпунт, С. К- Воскресенский и др., Сообщ. о научно-техн,
работах НИУИФ, № 2, 1957, стр. 14; Хим. наука и пром., № 2 (1957); Реф. н.-и.
раб. за 1957 г., ч. 1, стр. 11, Труды НИУИФ, в. 163, 1959. — 176. W М a z g a i,
Chem. Technik, 9, № 6, 350 (1957). —/77. F. L a i s t, пат. США 1544911, 1925. —
178. C. Arambdurg, J. Orcel, Compt. rend., 233, 1635 (1951).—
179. T. M. Ware, Mining Congress J., 39, № 2, 52 (1953).— 180. C. F. David-
son, D. Atkin, Geol. Survey (Gr. Britain) a. Museum, Atomic Energy Div.,
Rept. № 124, 11 (1952); Chem. Abs., 48, № 3, 1209 (1954).
181. Chem. Eng., 63, № 5, 120 (1956).-—182. И. Л. Гофман, Хим. пром.,
№ 1, 48 (1956). — 183. F. Hecht, M. G e r h о 1 d, Mikrochemie ver. Mikrochim.
Acta, 35, 359 (1950); Chem. Abs., 44, № 17, 7712 (1950). — 184. V. E. McKelvey,
I. M. Nelson, Econ. Geol.. 45, 35 (1950).-— 185. Chem. Eng., 58, № 12, 108
(1951). — 186. R. G. Gibbs, Chem. Eng. News, 29, №49,5112 (1951). — /87. Chem.
Week., 69, № 22, 21 (1951). —/88. I. Gordon, ibid., 71, № 5, 26 (1952).—
189. A. A. G u n t z, Compt. rend., 234, 868 (1952). — 190. Ch. В i z a r d, ibid.,
236, 1587 (1953).
Литература
937
191. М. Е. Thompson, US Geol. Survey Bull., № 988-D, 45 (1953); Chem.
Abs., 47, № II, 5321 (1953). — 192. R. T. R u n n e 1 s, I. A. S c h 1 e i c h e r, H. S. V a n
N о r t w i c k, State Geol. Survey Kansas Bull., № 102, 93 (1953); Chem. Abs., 48,
№ 1, 88 (1954). —193. Chem. Eng. News, 31, 4540 (1953). —194. C. F. Dela-
ney, С. M. Matthews, I. H. Poole, Sci. Proc. Roy. Dublin Soc., 26, 165
(1953); Chem. Abs., 48, № 8, 4386 (1954). — 195. H. Curtis, Chem. a. Process
Eng., 34, № 8, 251 (1953). —196. I. M. Schreyer, C. F. Baes, Anal. Chem.,
25, 644 (1953); J. Am. Chem. Soc., 76, № 2, 354 (1954). —197. A. Z enoble,
H. Sa Ivan, V. Ziegler, Compt. rend., 238, 1720 (1954). —198. R. M. Max-
ham, US Geol. Survey, Circ. № 230, 4 (1954); Chem. Abs., 48, № 17, 9874
(1954). —199. H. Wedow, US Geol. Survey, Circ. № 316, 9 (1954); Chem. Abs.,
48, № 15, 8705 (1954).— 200. Ind. chim., 41, № 438, 32 (1954).
201. Chem. Eng., 61, № 6, 118 (1954). —202. Farm. Chem., 117, № 7, 64
(1954). —203. J. Agr. Food Chem., I, № 4, 292 (1954).— 204. P. С. Лонг,
Д. А. Эллис, P. Г. Б e й л о с, Химия ядерного горючего, Госхимиздат, 1956.—
205. R. F. S t е d m a n, Chem. Ind., № 6, 150 (1957) — 206. B. F. G r e e n, O. W. A 1-
len, D. E. Tynan, Ind. Eng. Chem.. 49, № 4, 628 (1957). — 207. R. Heiser,
Jowa Eng., 58, № 6, 36 (1958)—208. P. Си м а н о в с к а я, Труды НИУИФ, в 143,
1938, стр. 23. — 209. Р. Симановская, Там же, в. 147, 1940, стр. 25.—
210. Р. Э. Симановская, Автореф. канд. дисс., НИУИФ, 1946.
211. I. М. Stinson, С. Е. Mu mm a, Ind. Chem., 46, № 3, 453 (1954).—
212. R. F. Knight, Ind. chim., 43, № 464, 77 (1956).—2/3. P. Э. Симановская,
3 В. Водзинская, Сообщения о научно-технических работах НИУИФ, № 3,
1957, стр. 26.—214. Р. Э. Симановская, 3. Ф. Коротова, Там же,
стр. 30.—215. Р. Э. Симановская, 3. Ф. Коротова, Там же, стр. 31.—
2/6’. С. Я. Шпунт, 3. И. Гусева, Там же, стр. 34.—2/7. Сб. «Гипс и фосфо-
гипс», под ред. С. И. Вольфковича, Труды НИУИФ, в. 160, 1958.—218. С. К- В о с-
кресенский, Р. М. Серебряная, Сообщения о научно-исследовательских
работах НИУИФ, № 2, 1957, стр. 8. — 219. Ф. Н. Кельм ан и др., Методы ана-
лиза и контроля производства серной кислоты и фосфорных удобрений, Гос-
химиздат, 1963. — 220. С. В. Кузнецов, Автоматизация производства супер-
фосфата, Госхимиздат, 1956.
22/. Сб. «Автоматизация химических и коксохимических производств», Гос-
металлургиздат, 1958.—2/2. Ю. Г. Бондаренко, Состояние и перспективы
развития работ по автоматизации производств фосфорных удобрений, Северодо-
нецк, 1967,— 223. С. Я. Шпунт, Ф. Е. М о с т о в и ч, ЖПХ, 30, № 3, 386 (1957) .—
224. В. С. Я т л о в, Е. Н. Пинаевская, ЖОХ, 18, № 17 (1938). — 225. И. И. 3 а-
р и н г, ЖХП, 13, № 5, 266 (1936). — 226. И. Л. Гофман, К- И. Макарьин,
Химстрой, № 2 и 3 (1935). — 227. И. Гофман, Труды НИУИФ, в. 134, 1936,—
228. И. Гофман, Там же, в. 143, 1938, стр. 18. — 229. W. С. Weber, пат.
США 2091898, 1937. — 230. С. К. Милованова, Научн.-техн. бюлл. НИУИФ,
№ 9, 60 (1957).
231. И. Н. Кузьминых, В. Н. Кочетков, Труды Московск. хим.-технол.
ин-та им. Д. И. Менделеева, в. 24, 1957, стр. 441. — 232. К. Ф- Павлов,
Б. А. К о п ы л е в, ЖХП, № 6, 29 (1938). — 233. А. Г. Амелин, Производство
серной кислоты, Госхимиздат, 1956.—234. П. Г. Удыма, Вести, техн, и эконом,
информ., № 2 (7), 39 (1958)..
Глава XXVI
ПРОИЗВОДСТВО ФОСФОРА И ФОСФОРНОЙ кислоты
ЭЛЕКТРОТЕРМИЧЕСКИМ МЕТОДОМ
СВОЙСТВА ФОСФОРА И ЕГО СОЕДИНЕНИЯ
Фосфор существует в нескольких модификациях; основными
являются: белый (а и Р), фиолетовый и черный1-5 фосфор. При
электровозгонке из природных фосфатов получается белый фосфор
(ядовит, на воздухе легко самовоспламеняется). Стеклообразный
фосфор6 в виде рентгеноаморфной массы представляет собой тем-
но-серое хрупкое вещество (плотностью 2,26 г/см3).
Белый фосфор имеет плотность 1,8 г/см3, плавится при 44,1°,
кипит при 280,5°; теплота плавления 0,156 ккал/ (г-атом), теплота
испарения 4,0 ккал) (г-атом). Теплоемкость (Ср) при 25° равна
5,55 кал/(мольград). Ниже —77° a-модификация белого фосфора
превращается в p-модификацию также белого фосфора. В жидком
и парообразном состоянии до 800° фосфор четырехатомен (Р4),
выше этой температуры начинается распад на двухатомный (Р2).
Плотность расплавленного фосфора при 100° 1,7 г/см\ при темпе-
ратуре кипения 1,5г/с.«3.
Давление пара белого фосфора:
i°, С...... 20 50 80 100 200 281
Р, мм рт. ст. . . . 0,02 0,28 1,39 3,62 266 760
Белый фосфор обладает специфическим запахом, светится в
темноте. В воде практически не растворим. Растворим хорошо в
сероуглероде, бензоле, толуоле и др. Под действием света приобре-
тает желто-бурую окраску (вследствие образования незначитель-
ных количеств красного фосфора), поэтому в технике его обычно
называют желтым.
Во избежание окисления и самовоспламенения на воздухе его
хранят под водой. При нагревании без доступа воздуха до 270—
300° белый фосфор переходит в красный, являющийся полимером
Свойства фосфора и его соединения
93»
белого. Процесс ускоряется7 при смешении белого фосфора с крас-
ным, служащим активатором полимеризации.
Красный фосфор имеет плотность 2,1—2,2 г)смъ, на воздухе не-
загорается, не ядовит, не растворяется в сероуглероде, бензоле и
других растворителях, в которых растворим белый фосфор. При
нагревании возгоняется. Пары красного фосфора идентичны парам
белого; при их конденсации получается белый фосфор. Однако ис-
парение фосфора сопровождается полимеризацией молекул пара
с образованием красной модификации. Вследствие меньшего дав-
ления пара над красным фосфором, чем над жидким, процесс про-
текает в сторону образования все большего количества красного-
фосфора. Поэтому при длительном нагревании можно получить,
красный фосфор из жидкого, не прибегая к высоким давлениям.
При высоких температурах и очень высоких давлениях образуется
черный фосфор.
При сжигании фосфора образуется пятиокись фосфора — фос-
форный ангидрид Р2О5. Теплота реакции окисления фосфора кис-
лородом до фосфорного ангидрида 360 ккал!г-мол Р2О5. Молярная
теплоемкость Р2О5 (Ср) 24,1 кал! (моль • град).
При обычной температуре фосфорный ангидрид представляет
собой белое кристаллическое вещество. Он гигроскопичен и яв-
ляется одним из наиболее интенсивных осушителей газов. При 359°’
возгоняется. Пары фосфорного ангидрида полимеризованы и отве-
чают по своему составу димеру пятиокиси, т. е. имеют удвоенную-
молекулу Р4О10. Фосфорный ангидрид известен в виде нескольких
модификаций: летучая имеет структуру димера Р4О10, нелетучие-
обладают координационным строением. Диаграмма состояния
включает два расплава и несколько твердых фаз, так как нелету»
чие модификации имеют различные кристаллические решетки. Тех-
нический продукт состоит из смеси летучей и нелетучих модифика-
ций, остающихся после отгона Р4О10 в виде твердой, спекшейся'
массы.
Разные модификации имеют плотность от 2,28 до 3,05 г/см*.
Давление пара летучей модификации при 316,5°—207 мм рт. ст.,
при 358,7° — 760 мм рт. ст. Превращение в нелетучие модификации
идет при 300—360°8. Температура плавления 563° (под давлением),,
теплота плавления 8,2 кал)г-мол. Существует стеклообразная моди-
фикация фосфорного ангидрида, имеющая значительно меньше»
давление пара и возгоняющаяся при более высоких температурах.
При гидратации фосфорного ангидрида последовательно обра-
зуется ряд фосфорных кислот. Вначале фосфорный ангидрид пере-
ходит в тётраметафосфорную кислоту Н4Р4О12, затем в тетра-
полифосфорную НеР4О1з; последняя, гидратируясь, образует смесь
ОрТОфосфорНОЙ Н3РО4 И ТрИПОЛИфосфОрНОЙ Н5Р3ОЮ кислот. При
дальнейшей гидратации триполифосфорная кислота, отщепляя
Н3РО4, переходит в пирофосфорную Н4Р2О7, которая присоединяя
940
Гл. XXV/. Производство фосфора и фосфорной кислоты
воду, дает ортофосфорную кислоту, представляющую собой конеч-
ный продукт гидратации фосфорного ангидрида. При 284° орто-
фосфорная кислота начинает дегидратироваться с превращением
в первичные продукты гидратации фосфорного ангидрида — пиро-
фосфорную Н4Р2О7 и метафосфорную НРОз кислоты.
Фосфорный ангидрид является промежуточным продуктом в
производстве фосфорной кислоты. Свойства фосфорной кислоты,
а также суперфосфорной кислоты9’10 приведены выше (стр. 881).
ПРИМЕНЕНИЕ ФОСФОРА И ТЕРМИЧЕСКОЙ ФОСФОРНОЙ КИСЛОТЫ
Белый (или желтый) фосфор служит для получения фосфор-
ного ангидрида, фосфорных кислот и других разнообразных неор-
ганических и органических соединений фосфора. В оборонном деле
он применяется в качестве дымообразующего и зажигательного
средства, для изготовления трассирующих пуль. В значительных
количествах используются его производные: хлористый фосфор —
в производстве пластификаторов, инсектицидов, продуктов органи-
ческого синтеза; сульфид фосфора — в спичечной промышленности
и для изготовления смазочных средств; фосфиды цинка и каль-
ция— для военных целей, для изготовления средств сигнализации
и в производстве пестицидов. Он является также полупродуктом
для получения красного фосфора. Фосфор высокой чистоты, содер-
жащий около 1 • 1СГ4% примесей, применяют в электронике и для
изготовления полупроводников11, фосфидов бора, алюминия и ред-
ких металлов. Его можно получить очисткой технического желтого
фосфора разными методами: фильтрованием и промыванием рас-
твором щелочи11, ионообменной очисткой и промыванием водой12,
зонной плавкой 13, дистилляцией 14’15 и другими 16>17. Желтый фос-
фор транспортируют в стальных цистернах без нижнего слива, в
стальных бочках и в банках из оцинкованного железа под слоем
воды или незамерзающего раствора (хлорида цинка или натрия).
Красный фосфор потребляется спичечной промышленностью —
он является основным компонентом смеси, наносимой на боковые
поверхности спичечных коробок. Из него изготовляют фосфиды
.алюминия и меди, применяемые в металлургии, сельском хозяйстве
и для получения сварочных средств.
Фосфорный ангидрид, являющийся полупродуктом для получе-
ния фосфорной кислоты, применяется также как самый энергич-
ный осушитель среди всех остальных веществ, употребляемых для
осушки18. Остаточное давление водяного пара над фосфорным
ангидридом составляет при 25° лишь 0,00001 мм рт. ст. При взаи-
модействии с водой на 1 г-мол Р2О5 выделяется 34 ккал. Исполь-
зуется он и как реагент при производстве эфиров фосфорной кис-
лоты и в нефтяной промышленности при очистке дистиллятов неф-
ти и получении продуктов с увеличенным октановым числом 19.
Применение фосфора и термической фосфорной кислоты
941
Большая часть фосфора перерабатывается в фосфорную кис-
лоту, называемую термической, в соответствии со способом ее по-
лучения.
Преимущество термического способа получения фосфорной кис-
лоты состоит в возможности производства кислоты любой концен-
трации (вплоть до 100% Р2О5) и высокой степени чистоты при ис-
пользовании любых фосфатов, в том числе и низкокачественных
без их предварительного обогащения. Термическая ^фосфорная кис-
лота используется в основном для получения фосфорнокислых со-
лей и в меньшей степени — в производстве концентрированных
удобрений.
Термическая суперфосфорная кислота20 начинает применяться
для получения более концентрированных, чем обычно, удобрений:
жидких, двойного суперфосфата, гранулированных смешанных (без
сушки) и др.
Термическая фосфорная кислота расходуется на изготовление
многих солей, потребляемых различными отраслями промышлен-
ности— пищевой, сахарной, керамической, стекольной, текстильной
и др.20. В пищевой промышленности она употребляется при изгото-
влении напитков для придания им кислого вкуса, при очистке са-
харных сиропов в производстве сахара-рафинада. Присутствие
фосфорной кислоты в пищевых продуктах признается полезным, так
как соединения фосфора играют важную роль в питательном ра-
ционе человека. Она применяется для приготовления зубных це-
ментов 19, фосфатных вяжущих веществ — фосфатных цементов и
связок, отличающихся жаростойкостью, специальными электротер-
мическими и теплофизическими свойствами, и используемых для
новой техники, а также в качестве средств защиты от радиации21.
Фосфорная кислота применяется 19’22’23 также при получении
активированного угля для пропитки спичечной соломки (нетлею-
щие спички), для удаления накипи в паровых котлах, при изготов-
лении тканей с огнезащитной пропиткой, кинопленок и фотореак-
тивов и т. д. Она является составной частью препаратов, исполь-
зуемых для защиты металлов от коррозии и катализатором
в процессах дегидрирования, полимеризации и алкилирования уг-
леводородов.
Большое промышленное значение имеют мета- и полифосфаты
натрия, а также ортофосфаты: три- и динатрийфосфат, применяе-
мые для умягчения воды, для борьбы с котельной накипью, для
изготовления различных моющих средств без затраты пищевых
жиров, в производстве фармацевтических препаратов, в фотогра-
фии, для регулирования вязкости буровых жидкостей, как утяже-
лители шелка и для многих других целей.
Фосфаты аммония используются в качестве противопожарных
средств, для культивирования дрожжей и др. Фосфаты марганца,
железа и другие применяются для защиты металлов от коррозии.
«42
Гл. XXVI. Производство фосфора и фосфорной кислоты
Рис. 259. Система Р2О5—Н2О.
Точки плавления: L\ — 2Н3РО4 • Н2О;
L2 — H3PO4: L3 — H4P2O7.
Фосфаты кальция, помимо их значения как удобрения, исполь-
зуются в бумажной промышленности, в качестве хлебопекарных
препаратов, для изготовления зубных порошков, как добавка к
столовой соли, к корму скота и проч.
Мировая выработка элементарного фосфора увеличилась с
520 тыс. т24-37 в 1960 г. до 750 тыс. т в 1965 г., а в 1970 г. ожи-
дается увеличение его производства до 1250 тыс. т38-41. В 1965 г.
•90% мирового выпуска фосфора было переработано на фосфорную
кислоту и лишь 10% в другие продукты 38’39. При этом 50% произ-
веденного фосфора в виде термической кислоты было использо-
вано для получения триполифос-
фата и тетрапирофосфатов нат-
рия, 18% — для выработки дру-
гих технических фосфорных солей
и на прочие нужды. Остальное
количество фосфора (22% от об-
щего выпуска) также в виде тер-
мической кислоты было израсхо-
довано на производство мине-
ральных удобрений и кормовых
фосфатов*
Согласно ГОСТ 8986—59, фос-
фор желтый (белый) I и II сор-
тов должен содержать соответ-
ственно не менее 99,9 и 99,7%
фосфора и не более 0,1 и 0,3% ве-
ществ, не растворимых в CS2.
Красный фосфор должен со-
держать (ГОСТ 8655—57) желто-
го фосфора не больше 0,005% в
1-м сорте и 0,03% во 2-м сорте и
иметь температуру воспламенения
не ниже 270 °C.
По ГОСТ 10678—63 термиче-
ская фосфорная кислота (техни-
ческая I и II сортов) должна со-
держать не менее 73% Н3РО4, хлоридов (на хлор) не более 0,05%,
тяжелых металлов, кроме железа (в расчете на свинец), не более
0,008% в I сорте и 0,03% во II, мышьяка не более 0,008% в I сорте и
0,01% во II. В I сорте допускается содержание сульфатов (на SO4)
до 0,25% и железа0,05% при отсутствии взвешенных частиц, во II—
количество сульфатов и железа не нормируется. Содержание других
примесей в термической кислоте (РЬ, фтор, SiOj) незначительно и
составляет сотые или тысячные доли процента. Состав термической
фосфорной кислоты для производства удобрений не нормирован.
Пищевая фосфорная кислота, согласно ГОСТ 10678—63, дол-
Основы возгонки фосфора из фосфатов кальция
943
жна содержать: фосфорной кислоты — не менее 70%; сульфатов
(в пересчете на SO4) — не более 0,02%; хлоридов (в пересчете на
С1)—не более 0,02%; мышьяка — не более 0,0003%; железа — не
более 0,01%; взвешенные частицы должны отсутствовать.
Для получения пищевой фосфорной кислоты используется тер-
мическая фосфорная кислота, которая очищается от мышьяка и
тяжелых металлов с помощью сероводорода или сернистого нат-
рия и др.19’ 42~44.
Выделение твердой фосфорной кислоты является эффективным
способом ее очистки от всех примесей. Данные 19’45’46 по раствори-
мости ортофосфорной кислоты (рис. 259) позволяют определять
условия (температуру и концентрацию) для кристаллизации кри-
сталлогидрата НзРО4-0,5Н2О или безводной Н3РО4 (кристалли-
зуется в ромбической системе).
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ВОЗГОНКИ ФОСФОРА
ИЗ ФОСФАТОВ КАЛЬЦИЯ
Суммарная реакция восстановления трикальцийфосфата угле-
родом выражается уравнением 47’48:
Са3(РО4)2 + 5С = Р2 + 5СО + ЗСаО(—418,9 ккал)49 (—413,0 ккал)50 (1)
Реакция начинается (без флюсов) при 1100° и при 1400° завер-
шается в течение 1 ч (рис.
260) 51-60.
С увеличением коли-
чества углерода степень
восстановления трикаль-
цийфосфата растет (рис.
261 ) 49’61. При высоких
температурах (1500—
1550°) влияние избытка
углерода менее заметно52.
Большое значение имеет
природа углерода. Чем
мельче кристаллики гра-
фита, тем выше его
химическая актив-
ность51’52’61’62. По возра-
станию восстановитель-
Рис. 260. Влияние температуры иа степень
восстановления фосфатов кальция (в течение
60 мин):
/—синтетический Саз(РО4)2; 2 —синтетический
Са5(рО4)зР; 3 — апатито-нефелииовая руда; 4 —вятский
фосфорит; 5 —фосфорит Каратау; 6 — синтетический
Са4Р2О9.
ной способности углеро-
дистые материалы распо-
лагаются в ряд: кокс неф-
тяной, графит, антрацит,
кокс металлургический,
древесный уголь.
На практике возгонку фосфора осуществляют с введением
в шихту кремнезема в качестве флюса. Исследовалось также
944
Гл. XXVI. Производство фосфора и фосфорной кислоты
применение глинозема и алюмосиликатов60. Влияние флюсов объяс-
няется тем, что они, связывая окись кальция, смещают равновесие
реакции в сторону образования фосфора. Процесс восстановления
в этих случаях может быть представлен следующими уравнениями:
Са3(РО4)2+5С + 2SiO2 = Р2 + 5СО + Ca3Si2O7(—368,9 ккал)49 (—348,8 ккал)50 (2)
Ca3(PO4)2 + 5С + ЗА12О3= Р2 + 5СО+3(А12О3 • СаО) (—386,0 ккал)50 (3)
В присутствии кремнезема восстановление идет при более низ-
ких температурах49-61: от 1000 до 1200—1300°. Установлено52'63
что при восстановлении трикальцийфосфата образуется преимуще-
ственно трикальцийдиси-
Рис. 261. Влияние избытка углерода на уве-
личение степени восстановления трикальций-
фосфата. Продолжительность нагрева 60 мин:
J -Са3(РО4)2 + хС, 1400°; 2 - Са3(РО4)2 + хС + 3 SiO2,
1300°; 3 - Са3(РО4)2 + хС + 2 SiO2, 1300°; 4-Са3(РО4)2+
+ xC + 3SiO2, 1200°.
ликат Ca3Si2O7, другие же
силикаты, в частности ме-
тасиликат CaSiO3, могут
образовываться в резуль-
тате вторичных реакций.
Кремнезем, введенный
в шихту в количестве
большем, чем нужно для
образования трикальций-
дисиликата и не прореаги-
ровавший с последним —
возгоняется, загрязняя
фосфор64. Степень вос-
становления трикальций-
фосфата возрастает с
увеличением отношения
SiC>2:CaO в шихте. Влия-
ние SiC>2 становится мало
заметным (особенно при температурах выше 1300°) при количе-
стве SiO2, большем чем в трикальцийдисиликате49-61.
В присутствии S1O2 облегчается также удаление из печи изве-
сти, которая в этом случае выпускается в виде легкоплавкого шла-
ка— силиката кальция. На рис. 262 видно, что температура плав-
ления шлака зависит от соотношения в нем SiO2: СаО. О составе
шлака судят по показателю кислотности—отношению весового со-
держания S1O2 и СаО. Например, для метасиликата кальция, пла-
вящегося при 1540° и содержащего 51,7% SiO2 и 48,3% СаО, пока-
затель кислотности равен 1,07. Шлаки с меньшим показателем кис-
лотности, содержащие больше СаО, чем метасиликат, называются
основными, а шлаки с большим показателем кислотности кислыми.
Основные и кислые шлаки, показатель кислотности которых
меньше или больше 1,07, плавятся при температурах более низких,
чем метасиликат кальция.
Глинозем и алюмосиликаты также положительно влияют на
восстановление трикальцийфосфата, но в меньшей степени, чем
Основы возгонки фосфора из фосфатов кальция
945
кремнезем 61’65-67. Для восстановления имеет значение и природа
флюсов. Аморфные двуокись кремния или глинозем дают при про-
чих равных условиях более высокую степень восстановления, чем
кристаллические 61,66-69. Щелочные соли повышают степень восста-
новления трикальцийфосфата углеродом, особенно при низких тем-
пературах 65,70-73.
Приведенные выше суммарные уравнения реакции восстанов-
ления трикальцийфосфата углеродом не дают представления о
механизме процесса, взгляды на кото-
рый различны74-91.
Установлено, что при термическом
получении фосфора из шихты апа-
тита и углерода в присутствии SiO2,
последняя не может вытеснять Р4Ою
из апатита даже при 1400°. При тем-
пературе выше 1000° апатит реагирует
с SiO2, образуя CaSiO3 и Са3(РО4)2 с
выделением Н2О и SiF4. Затем
Са3(РО4)2 восстанавливается углеро-
дом (легче, чем апатит), а избыток
окиси кальция реагирует с SiO2, обра-
зуя легкоплавкий шлак.
Восстановление фосфатов водоро-
дом, природным газом и другими га-
зообразными реагентами протекает
значительно медленнее, чем твердым
углеродом. Показано92, что в присут-
ствии природного газа при температу-
рах до 1250° (до оплавления шихты)
Рис. 262. Диаграмма плавкости
в системе SiO2—СаО.
восстановление идет преимущественно углеродом, отлагающемся
при пиролизе метана на поверхности и в порах фосфорита; при тем-
пературах выше 1300° — водородом, образующимся при пиролизе
метана. Для промышленных процессов, протекающих при 1400—
1450°, продувание шихты метаном нецелесообразно, так как вос-
становление за счет водорода идет медленнее, чем металлургиче-
ским коксом. В то же время выделившийся при пиролизе метана
углерод обладает большей восстанавливающей способностью, чем
кокс. Взаимодействие природных фосфатов с газообразными вос-
становителями ускоряется в присутствии солей натрия и магния,
снижающих температуру плавления шихты93. Из щелочных солей
наиболее активно действует94 добавка Na2SO4. Максимальное влия-
ние флюсов наблюдается при температуре плавления шихты
(- 1300°).
Восстановление фосфатов газами связано с необходимостью
применения большого избытка восстановителя (20—30-кратного
количества) и громоздкой аппаратуры95.
«48
Гл. XXVI. Производство фосфора и фосфорной кислоты
Скорость реакции восстановления трикальцийфосфата углеро-
дом лимитируется диффузией реагирующих компонентов 52-60'96.
Опытные данные подтверждают, что факторы, ускоряющие диффу-
зию в твердых телах, повышают степень восстановления трикаль-
цийфосфата углеродом. Степень восстановления возрастает с умень-
шением размера частиц шихты. Особенно показательно положи-
тельное влияние брикетирования шихты. Скорость диффузии, а
следовательно и восстановления, растет с увеличением температуры,
©ведение в шихту флюсов, образующих полиэвтектические смеси,
ускоряет процесс. Температура превращения кремнезема в менее
плотные модификации совпадает с начальной температурой вос-
становления трикальцийфосфата в присутствии кремнезема (900—
1000°). Ускорение диффузии может быть объяснено внутрикристал-
лическими превращениями. Взаимодействие между фосфатом и
коксом в присутствии кварца, сопровождающееся кристаллохими-
ческими превращениями с образованием соединений промежуточ-
ных степеней окисления фосфора, протекает главным образом
в расплаве90. Только до его появления идет прямое восстановление
фосфата окисью углерода и углеродом в результате диффузии в
твердых фазах.
В процессе возгонки протекают побочные реакции. При темпе-
ратуре выше 1600° и большом избытке углерода происходит обра-
зование фосфида и карбида кальция: •
Са3(РО4)2 + 8С = С.а3Р2 4- 8СО
Са3Р2 + 6С = ЗСаС2 + Р2
Содержащаяся в фосфатах окись железа связывает часть фос-
фора в фосфиды железа, например:
Fe2O3 + ЗС = 2Fe + ЗСО
4Fe + P2 = 2Fe2P
Они выпускаются из печи в расплавленном виде и застывают
в чугунноподобную массу — феррофосфор.
Существенное значение имеет возгонка щелочей и выделение
фтористых соединений:
2CaF2 + SiO2 = 2СаО + SiF4
Четырехфтористый кремний при конденсации фосфора погло-
щается водой с выделением кремневой кислоты, которая загряз-
няет продукт. Содержащиеся в сырье сульфиды и сульфаты обра-
зуют сероводород. Идет также разложение карбонатов; выделяю-
щаяся при этом двуокись углерода восстанавливается до окиси.
Имеющаяся в шихте влага реагирует с фосфором, образуя фосфо-
ристый водород и фосфористую кислоту, что снижает выход фос-
фора. Для уменьшения потерь фосфора сырье подвергается иногда
предварительной прокалке.
Производство фосфора злектровозгонкой из фосфатов
947
ПРОИЗВОДСТВО ФОСФОРА ЗЛЕКТРОВОЗГОНКОЙ ИЗ ФОСФАТОВ
Возгонка фосфора в печи типа доменной не получила промыш-
ленного развития, несмотря на большое количество исследователь-
ских и опытных работ 97-107. Это объясняется сложностью процес-
са, высокими капитальными затратами и большей стоимостью фос-
фора, чем получаемого злектровозгонкой 108. Интерес представляет
применение плазменных печей для возгонки фосфора. Но это тре-
бует еще значительных изысканий. В настоящее время для воз-
гонки фосфора применяются электрические печи 4'14'25,109-140
с угольными или графитовыми электродами, погруженными в
шихту. Нагревание происходит от пламени электрической дуги, воз-
никающей между электродами, и за счет сопротивления самой
шихты.
Современные печи большой мощности работают на основной
шихте, т. е. при весовом отношении S1O2: СаО, равном 0,8—1,0
(см. выше). Этим достигается значительно меньшая возгонка крем-
ния, чем при работе на кислой шихте. Если фосфат содержит мень-
ше SiO2, чем это необходимо, в шихту вводят чистый кварцевый
песок (освобожденный от пыли) или кварцит. Фосфатные руды, со-
держащие избыток SiO2, обычно не применяются, потому что та-
кие руды бедны фосфором и корректировка их состава добавле-
нием СаО еще больше уменьшает количество Р2О5 в шихте. При
расчете шихты учитывается также содержание в сырье окислов
алюминия, щелочей и магния 141>142.
Углерод вводится в виде каменноугольного кокса или антра-
цита в количестве 110—120% по отношению к теоретической нор-
ме 143’144, рассчитанной для восстановления Р2О5 до фосфора, ЕегОз
до железа и СО2 до СО. Шихту составляют из кускового мате-
риала. Размеры кусков фосфорита и кварцита от 5 до 50 мм, гра-
нулированного порошкообразного фосфорита от 5 до 25 мм, антра-
цита или кокса 3—25 мм. Более мелкие частицы фосфата отсеи-
вают и укрупняют различными способами. Порошкообразную
шихту тоже превращают в кусковой материал. В зависимости от
свойств исходного сырья применяются следующие способы подго-
товки ШИХТЫ 24’144.
1) гранулирование с последующим обжигом (этот способ наи-
более пригоден для глинистых фосфоритов);
2) агломерация при высоких температурах;
3) спекание смеси тонкоизмельченного фосфорита и кокса;
4) брикетирование шихты. По этому способу осуществляют
смешение измельченного фосфата и связующего, например жид-
кого стекла 133 или глины 138, с последующим формованием брике-
тов и прокалкой последних при температуре около 750°. Необхо-
димость тесного соприкосновения реагентов делает целесообраз-
ным введение в брикеты измельченного кокса или угля 52>5®.
948
Г л. XXVI. Производство фосфора и фосфорной кислоты
Для прокалки и сушки компонентов шихты используются отхо-
дящие газы установки (СО). Предварительное прокаливание фос-
форита производят с целью дегидратации, декарбонизации, а так-
же разложения органического вещества фосфоритов. Это приво-
дит к уменьшению расхода электроэнергии 145.
Рис. 263. Трехфазная электрическая печь для возгонки фосфора с тре-
угольным расположением электродов;
/«-кожух печи; 2 — летка для выпуска шлака; 3 — электродержатель с набивным
самоспекающимся электродом; 4 — бункеры с течками для компонентов шнхты;
5 — трансформатор; 6 — шины; 7 —штуцер для вывода газа.
Возгонка фосфора осуществляется в трехфазных печах, рабо-
тающих на переменном токе.
Мощность современных печей составляет 35—50 тыс. кет и
больше148. Ведутся работы по интенсификации действующих и со-
зданию новых печей мощностью 70 и 100 тыс. кет.
Печи изготовляют из огнеупорного и кислотоупорного кирпича
с наружным стальным кожухом для обеспечения герметичности.
Под и стенки реакционной зоны выполняются из угольных блоков
(рис. 263 и 264). Диаметр ванны печей достигает 8,5 м (при диа-
метре кожуха .10,5 м). Они выпускают до 60 т фосфора в сутки.
950 Гл. XXVI. Производство фосфора и фосфорной кислоты
При использовании мощных печей расход энергии на производство
фосфора сокращается147. Так, в печах мощностью до 5 тыс. кет
расход электроэнергии на 1 т фосфора составляет 17,5—18 тыс.
кет • ч, в печах мощностью 25—50 тыс. кет расход энергии сни-
жается до 13—15 тыс. кет • ч148. Использование мощных печей вы-
годнее также и вследствие уменьшения удельных капитальных за-
трат на сооружения и эксплуатационных расходов. Печи средней
мощности питаются переменным током напряжением 170—260 в.
Повышение напряжения позволяет повысить мощность печи и уве-
личить ее производительность. Печи мощностью 35—50 тыс. кет
работают на напряжении 300—500 в.
В трехфазных печах электроды располагают в ряд или тре-
угольником. В первом случае печи имеют эллиптическое или пря-
моугольное сечение, во втором — цилиндрическую форму или фор-
му равностороннего треугольника с закругленными вершинами. г
К каждому из трех электродов подключен свой трансформатор 1
для преобразования тока высокого напряжения, нагрузку которого
можно регулировать. Чаще всего применяются угольные электроды
диаметром 800—1500 мм. Электроды закреплены в электродержа-
телях, охлаждаемых через рубашку в нижней части водой. Ток
проходит по токонесущим шинам к токонесущему кольцу, оттуда :
через охлаждаемые водой медные кабели к контактным плитам, i
удерживающим электроды. По мере сгорания электрод опускают. ;
В верхней его части наваривают новые цилиндрические звенья, 1
заполняемые электродной массой, которая спекается по мере опу-
скания электрода (самоспекающиеся электроды).
На рис. 265 приведена схема опытной вращающейся печи для ’!
возгонки фосфора мощностью 7500 кет25. 12e>|29. Внутренний диа- i
метр вращающегося тигля составляет около 5 м. Скорость враще-
ния от 1 оборота за 15 ч до 1 оборота за 100 ч. Электроды графи-
товые диаметром 600 мм. Печь работает с напряжением до 325 в.
Благодаря вращению печи создается возможность плавить не толь-
ко кусковую руду, но и не агломерированные мелкие фосфаты, что
удешевляет производство. На этой печи достигнута экономия в
расходе электроэнергии на 11%. При этом срок службы футеровки
увеличивается в 3—4 раза.
Для восстановления фосфата и расплавления шихты послед-
няя в реакционной зоне нагревается до 1500—1600°. Это обеспечи-
вает понижение вязкости шлака и легкий выпуск его через летки,
находящиеся в стенке печи против электродов. Расстояние от
электродов до стенок должно быть таким, чтобы температура у .
стенок была ниже реакционной. Температура выходящих из печи
газов колеблется в пределах 300—500°. Процесс в печи идет
непрерывно (с периодической загрузкой шихты и выпуском
шлака).
Производство фосфора злектровозгонкой из фосфатов
951
Днище и стены печи до уровня шлака футерованы угольными
блоками. Выше стены выложены огнеупорным кирпичом. Сталь-
ной свод печи также футерован. Продолжительность службы фу-
теровки зависит от качества угольных блоков, соблюдения техно-
логического режима и соответствия размеров печи ее мощности.
При нарушениях технологического режима износ футеровки на-
блюдается уже через 5—6 месяцев. Короткий межремонтный про-
Рис. 265. Вращающаяся электрическая печь для
возгонки фосфора:
/ — гидравлический затвор (расплавленный свинец); 2 —вра-
щающийся тигель; 3 — неподвижная крышка; 4 —графитовый
электрод; 5— вал тигля; 6 — угольный блок; 7 —летка для
шлака; 6 —летка для феррофосфора; Р —графитовый блок;
/(/ — гибкие кабели; // — центральная загрузочная труба;
12— боковая загрузочная труба; 13 — огнеупорный кирпич.
бег электропечей создает напряженную обстановку и приводит
к значительным убыткам. Рабочий период между остановками
электропечей на капитальный ремонт футеровки должен быть 5—
10 лет.
Особенностью печей для возгонки фосфора является их полная
герметичность. В печи поддерживается избыточное давление газов
порядка 30—60 мм вод. ст.; это предотвращает возможность под-
соса воздуха, что привело бы к сгоранию фосфора.
Печные газы содержат по весу около 25% фосфора; остальные
75% —окись углерода с примесью H2S, СО2, N2, РНз, SiF4, водя-
ного пара и др.
6 И. Е. Поэин
952
Гл. XXVI. Производство фосфора и фосфорной кислоты
Технологическая схема электровозгонки приведена на рис. 266.
Для загрузки печи установлено несколько бункеров, что облегчает
равномерное ее заполнение. Дозировка компонентов шихты — фос-
форита, кокса и кварца — производится раздельно и смесь посту-
пает в бункеры. Последние герметически закрываются крышками,
Рис. 266. Схема производства фосфора:
/ — печь? // — электрофильтр ; /// — конденсационная установка; / — загрузочный бункер;
2 —дозатор; 3 —система разгрузки; 4 — кожух; 5 —подводка тока; 6 — труба факельного сжи-
гания; 7 —вентилятор отходящих газов; 8 — газопровод; 9 — холодильная установка; /0—башня
(холодная); // — башня (горячая); /2—приемник для хранения фосфора; /3 —шнеки для транс-
портировки пыли; Д/ —камеры для сбора пыли; /5—летка для выпуска феррофосфора;
16 — труба для вывода газов; 17— электрод; 73 — трансформатор.
а в нижнюю их часть непрерывно подается инертный газ — азот
или дымовой. Это предохраняет от выделения в атмосферу цеха
печного газа. При заполнении бункеров инертный газ выделяется
наружу, а при закрытых крышках он поступает в печь. Феррофос-
фор сливается из печи один раз в сутки, а шлак через каждые
3—4 ч.
Феррофосфор из печи через летку сливается в ковш, стоящий
на железнодорожной платформе, и отвозится на розлив в излож-
ницы, из которых извлекается после застывания. Шлак сливают
Производство фосфора электровозгонкой из фосфатов
953
через две другие летки (расположенные немного выше) по желобу
в разборные чугунные кокили, установленные на железнодорожной
платформе, и отвозят для переработки в изделия. Если шлак не
используется, его выпускают из летки в грануляционную мель-
ницу, куда подают воду. Измельченный шлак отвозится в гон-
долах.
или 20—25 вес.°/о
Уробень ,
боды 10
5
Желтый rf- ~ ~
гроссрор
Выходящий из печи газ, содержащий 220—330 г фосфора в 1 м3
при нормальных условиях (4—6 объемн.%
Р4, 65—85% СО, 2—4% СО2, 0,5—1%
SiF4) поступает в электрофильтры, где
из него выделяется уносимая из печи
пыль (шихта). Электрофильтры рабо-
тают под напряжением 40—60 тыс. в.
Во избежание конденсации фосфора
температуру в электрофильтрах под-
держивают на уровне 250—300°. С этой
целью электрофильтры и газоход ме-
жду ними заключены в футерованные
кирпичом кожухи, внутри которых
циркулирует горячий топочный газ, ‘
подаваемый теплоизолированным вен-
тилятором. Газ получают сжиганием
в топке части печного газа после вы-
деления из него фосфора. Температура
в электрофильтрах регулируется авто-
Конденсат
Рис. 267. Конденсатор фосфо-
ра с вращающимися дисками:
/ — орошение охлаждающей водой;
2 —штуцер для входа и выхода
газов; 3 —колпак; 4 — чаша с водой;
5 — соединительная труба: 6 — пере-
лив для воды; 7—крышка к чаше;
8 — впускное отверстие для пара;
9 — чаша с паровой рубашкой;
10 — разбрызгивающие диски;
И — ящик.
матически — при недостатке тепла сжи-
гается дополнительное количество га-
за, а избыток его выбрасывается в
трубу. Пыль, оседающая в газоходе
между печью и первым электрофильт-
ром, сбрасывается шнеком в бункер
под электрофильтром. Выгружаемая
из бункеров электрофильтров пыль от-
возится в отвал.
Из электрофильтров газ поступает в конденсаторы фосфора.
Конденсация фосфора достигается в результате промывки газа
водой и охлаждения его при этом до 57—60°149. В качестве конден-
саторов используют орошаемые циркулирующей водой полые
стальные башни, цилиндрические конденсаторы с внутренними пе-
регородками или полками, а также, при не очень больших мощно-
стях производства, аппараты с вращающимися дисками. Цирку-
лирующая в них вода постепенно подкисляется, поэтому ее нейтра-
лизуют содой, поддерживая pH = 5,5—6,0. Степень конденсации
фосфора достигает 99%.
На рис. 267 приведена схема конденсатора с вращающимися
дисками182. Аппарат состоит из колпака, охлаждаемого снаружи
6*
954
Гл. XXVI. Производство фосфора и фосфорной кислоты
водой, и нижней чаши, обогреваемой через паровую рубашку.
Внутри корпуса вращаются (со скоростью 500—700 об/мин) в про-
тивоположных направлениях два валика о дисками, погруженными
частично в воду. Происходит тонкое распыление воды, образующей
плотную завесу. Газ проходит последовательно через несколько
таких промывателей.
Уходящий из конденсаторов газ содержит до 85 объемн.°/о
СО, 0,05% фосфора (потери), 0,2—0,4% РНз, 0,5—1% H2S и дру-
гие примеси. Этот газ должен быть очищен от Р, H2S и РН3 для
использования окиси углерода в синтезах или же применяться без
очистки как топливо. Часть газа сжигают для подогрева электро-
фильтров.
Сконденсировавшийся жидкий фосфор собирается под водой
в чашах-сборниках, непосредственно присоединенных к конденса-
торам. Сборники, а иногда и газоходы, изготовляются из алюми-
ниевой бронзы и имеют рубашки для обогрева горячей водой, —
это предохраняет жидкий фосфор от затвердевания. Из сборников
фосфор сифонируется в отстойник, а из него через мерник — мон-
тежю — в обогреваемый водой резервуар на складе. Отсюда горя-
чей водой, накачиваемой насосом через подогреватель, жидкий
фосфор передавливают по обогреваемым водой трубам в цех фос-
форной кислоты, а для отправки потребителю — в железнодорож-
ные цистерны, снабженные обогревающими змеевиками и предва-
рительно частично залитые водой.
В конденсаторах и во всех сборниках жидкий фосфор нахо-
дится под слоем горячей воды с температурой 60°. При сливе фос-
фора освобождающийся объем замещается водой, а при заполне-
нии фосфором избыток воды сливается в сборник кислой воды, из
которого насосом через напорный бак вновь перекачивается в ап-
параты.
В отстойнике жидкий фосфор расслаивается с выделением шла-
ма. В шлам переходят примеси — неуловленная в электрофильтрах
пыль, сажа, кремневая кислота, образовавшаяся при гидролизе со-
держащегося в газах фтористого кремния:
3SiF4 + 4H2O = 2H2SiF6+SiO2-2H2O
Образующаяся по этой реакции кремнефтористоводородная кисло-
та растворяется в воде и подкисляет ее. Шлам из отстойника пе-
риодически сливается через сифон в специальный резервуар. В су-
хом веществе шлама содержится до 50% фосфора; его направляют
в цех фосфорной кислоты, где сжигают.
Для отделения примесей вместо отстаивания применяют также
фильтрацию сконденсированного фосфора на закрытых дисковых
фильтрах, работающих под давлением150.
Производство фосфора злектровозгонкой из фосфатов
955
Вследствие огнеопасности, взрывоопасности и вредности произ-
водства фосфора, все процессы, связанные с выпуском из -печи и
удалением шлака и феррофосфора, загрузкой электродной массы
механизируются и автоматизируются 151.
Расходные коэффициенты
Расход компонентов шихты зависит от состава природного фос-
фата, углеродистого и силикатного сырья. Последнее в ряде слу-
чаев совсем не расходуется. Ниже приводятся пределы расхода
сырья и материалов на 1 т фосфора: природный фосфат 7—10 т,
кокс 1,4—1,6 т, кварцит до 3 т, электродная масса 0,015—0,04 т,
инертный газ 200—500 л3, вода до 500 л3, пар 2—3 т.
Количество побочных продуктов: газ (содержащий СО) до
4000 лР, феррофосфор 0,1—0,5 т, шлак 7,5—11 т, пыль из электро-
фильтров 0,05—0,35 т.
Расход электроэнергии на 1 кг фосфора составляет 14—
18 кет • ч— он зависит от мощности печей (см. стр. 950). Выход
фосфора достигает 92%.
Отходы производства, их утилизация
Ценным отходом производства элементарного фосфора являет-
ся высококалорийный газ, содержащий 80—85% окиси углерода.
Последняя частично используется как топливо в самом производ-
стве, частично же газ может быть использован для других нужд
(химические синтезы).
Полезным побочным продуктом возгонки является феррофос-
фор, сплав фосфидов железа Fe2P и FesP, количество которого за-
висит от содержания окислов железа в исходной руде. Получаю-
щийся феррофосфор имеет примерный состав (при переработке
апатитового концентрата): около 72% Fe, 24—26% Р, 0,5—1,5%
Si, 0,5—0,8% V, 0,8—1,2% Мп, 0,4—0,6% Сг, 0,8—1,1% Ti. Его
используют главным образом в металлургической промышленности
как присадку в литейном производстве, как раскислитель, а также
как защитный материал 152 от радиоактивного излучения.
Силикатный шлак состоит из 38—43% SiO2, 2—5% А120з, 44—
48% СаО, 0,5-1,5% MgO, 0,5-1% Fe2O3, 0,5-3% Р2О5. Он может
быть использован для изготовления литых изделий, плиток для до-
рожного строительства, шлаковых цементов и кирпича, шлаковой
ваты или пемзы для теплоизоляции и т. д.
Представляет интерес одновременное получение желтого фос-
фора и глиноземистого цемента153 или цементного клинкера154.
В первом случае восстановлению подвергают шихту из апатито-
вого концентрата, титаноглиноземистого шлака, глины и коксика.
956
Гл. XXVI. Производство фосфора и фосфорной кислоты
Во втором случае процесс разработан в лабораторных условиях
применительно к фосфориту Каратау |54’155. Вначале шихту прока-
ливают при 1100° во вращающейся печи, а затем в ретортной или
электропечи, где происходит возгонка фосфора и окончательное
формирование клинкерных материалов.
При очистке газов возгонки в электрофильтрах собирается
пыль156, которая содержит до 22% усвояемой Р2О5 и иногда до
15% К2О. Такая пыль может быть использована в качестве удоб-
рения. Шлам, получающийся при очистке жидкого фосфора в от-
стойниках, как указано выше, сжигается в отделении фосфорной
кислоты.
Считают 157, что наличие фосфора в шламе связано с адсорб-
цией его гелеобразным кремнеземом. В результате захвата неко-
торого количества свободного фосфора образуются макрострук-
туры фосфорного шлама. Предложено 158 вязкий шлам диспергиро-
вать 25%-ным раствором NaOH (в количестве 1,5—2 вес. ч. на
6000 вес. ч. шлама). После длительного перемешивания шлама,
суспендированного в горячей воде, возможно центрифугированием
отделить жидкий фосфор чистотой 92% Р при выходе 93%. Для
уменьшения вязкости суспензии к шламу предварительно доба-
вляют аммонийлигносульфонат 159.
В табл. 73 приведен примерный материальный баланс получе-
ния фосфора в электрической печи 132 при переработке апатитового
концентрата с выходом продукта около 92%. В * * *
ТАБЛИЦА 73
Материальный баланс производства фосфора
Введено кг % Получено кг %
Фосфат (апатитовый концентрат в виде агломерата, содержа- щий 30% Р2О5, 40% СаО, 7% SiO2) Кварц. . . Кокс 8 250 3 000 1 500 64,7 23,5 11,8 Фосфор желтый Феррофосфор Окись углерода и другие газы Кремнистый шлак .... Пыль из электрофильтров Шлам сухой 1 000 150 3 000 8 150 350 100 7,8 1,2 23,5 64,0 2,7 0,8
Итого .... 12 750 100,0 Итого .... 12 750 ЮО.СГ
В балансе (табл. 73) не учтены некоторые составляющие: вво-
димый инертный газ, сгоревшие электроды, водяной пар и др.
При использовании фосфорита, содержащего 23,15% Р2Ов, по-
тери фосфора достигают 13% 15°.
Получение фосфорной кислоты
957
ПОЛУЧЕНИЕ ФОСФОРНОЙ КИСЛОТЫ
Производство термической фосфорной кислоты возможно дву-
мя методами: одно- и двухступенчатым 24’151-159.
При одноступенчатом или непрерывном методе печные газы,
содержащие элементарный фосфор и окись углерода, поступают
в камеры сгорания, где фосфор окисляется до фосфорного ангид-
рида кислородом воздуха, газы охлаждаются, гидратируются и
пропускаются через электрофильтры для улавливания фосфорной
кислоты. По двухступенчатому методу фосфор сначала конденси-
руется, а затем сжигается и Р2О5 гидратируется до фосфорной
кислоты.
Несмотря на кажущиеся преимущества одноступенчатого не-
прерывного метода, наиболее широко распространен двухступен-
чатый способ, так как последним получается более чистая фосфор-
ная кислота. Кроме того, в этом случае возможно при необходи-
мости выпускать в качестве продукта также желтый, фосфор и
эффективно использовать окись углерода, являющуюся побочным
продуктом. Затраты на оборудование ниже, чем при одноступен-
чатом способе24, так как в последнем улавливание фосфорной кис-
лоты из образующихся больших объемов газа требует громоздкой
аппаратуры. Расход электроэнергии, воды и воздуха при односту-
пенчатом методе также значительно больше.
Первоначальной стадией процесса получения фосфорной кис-
лоты является окисление фосфора:
Р4 + 5О2 = Р4О10
По одноступенчатому способу окисляется не только фосфор, но
и окись углерода:
Р4 + ЮСО + 10О2 = р4о10 + 10СО2
При этом выделяется большое количество тепла, которое прак-
тически не может быть использовано из-за коррозионного дейст-
вия фосфорного ангидрида, содержащегося в продуктах горения.
Требуется охлаждение газов в камере горения, предупреждающее
быстрый износ аппаратуры и обеспечивающее полное поглощение
фосфорного ангидрида.
Вследствие высокой температуры газа (800—1000°) при взаи-
модействии фосфорного ангидрида с водой первоначально обра-
зуется метафосфорная кислота НРО3 (в парообразном состоянии):
Р4О10 + 2Н2О = 4НРО3
Последняя при гидратации превращается в туманообразную
ортофосфорную кислоту (через ряд промежуточных соединений):
НРО3 + Н2О = Н3РО4
Во избежание образования низших окислов фосфора, переходя-
щих при гидратации в фосфористую и фосфорноватистую кислоты,
сжигание фосфора производится в двухкратном избытке воздуха.
958
Гл. X.X.VI. Производство фосфора и фосфорной кислоты
С целью использования окиси углерода, содержащейся в газах
возгонки, исследована возможность избирательного окисления фос-
фора 105’,67. Установлено, что при поддержании в окислительной
камере 450—540° и подаче 120—130% воздуха от теоретического
количества, необходимого для окисления фосфора, можно окислить
практически весь фосфор до Р2О5, а окись углерода окисляется
только на 5—7%. Эти исследования направлены к эффективному
применению одноступенчатого способа получения фосфорной кис-
лоты, хотя при этом она получается менее чистой.
Представляет интерес прямое получение пятиокиси фосфора пу-
тем окисления фосфора воздухом в процессе восстановления фос-
фора в смеси с углем и кварцем в пламенной печи 170 при темпера-
туре 1200—1600°, развиваемой за счет сгорания образующихся фос-
фора и окиси углерода. Другой возможный путь — возгонка Р2О3
из смеси фосфата и SiO2. Равновесное давление Р4О10 над смесью
Са3(РО4)2 и SiO2 при 1700° К равно 7,7- 10~7ат. Из смеси, содер-
жащей 3 моль SiO2 на 1 моль Са3(РО4)2 при 1650° и давлении
1О'3 мм рт. ст. выход Р2О5 в лабораторных условиях составил
99,5% 171.
По двухступенчатому способу производства газы возгонки очи-
щаются от пыли, затем фосфор конденсируется в виде жидкости
(температура выше точки плавления — около 60°), а окись угле-
рода может быть использована в качестве топлива. Фосфор сжи-
гается в камере сгорания, фосфорный ангидрид охлаждается во-
дой и гидратируется в башнях172. Фосфорная кислота, полученная
двухступенчатым способом, содержит 85—86% Н3РО4.
Этим же методом получают некоторые количества суперфос-
форной кислоты, содержащей 105—115% Н3РО4 (76—82% Р2О5).
Для этого фосфорный ангидрид, образующийся при сжигании эле-
ментарного фосфора, поглощают ограниченным количеством воды.
По другому способу в обычную термическую фосфорную кислоту
вводят твердый фосфорный ангидрид до получения кислоты тре-
буемой концентрации.
Ведутся работы по одновременному получению фосфорной кис-
лоты и водорода, который может быть использован, например в
синтезе аммиака241168’169. Этот процесс заключается в окислении
фосфора паром при температуре от 500—600° до 700—800° в при-
сутствии катализатора (платина, палладий или фосфаты алюми-
ния, титана, циркония, активированные платиной, палладием):
Р4 + 10Н2О = Р4О10 + 10Н2
При молярном соотношении водяной пар : фосфор, равном 18:1,
и температуре 700—800° на опытной установке удавалось переве-
сти в фосфорную кислоту до 93,8% фосфора и получить отходя-
щие газы с содержанием 99,8% водорода.
Получение фосфорной кислоты
959
Основной особенностью в производстве термической фосфорной
кислоты является отвод большого количества тепла, выделяемого
при сжигании фосфора. В зависимости от метода отвода тепла
различают циркуляционный и испарительный способы производ-
ства термической фосфорной (и полифосфорной) кислоты, возник-
шие одновременно в 30-х годах и подвергшиеся в дальнейшем зна-
чительному усовершенствованию. В первом случае основное тепло
отводится с циркулирующей кислотой (и снимается водой в холо-
дильниках) и около 20% с отходящими газами. Во втором случае
тепло снимается в основном за счет испарения воды и частично
отводится с отходящими газами. Ввиду низкой теплоемкости фос-
форной кислоты при циркуляционном способе на 1 т сжигаемого
фосфора-Необходимо подавать до 400—450 т кислоты. По-
этому нормальное осуществление процесса зависит от работы кис-
лотных насосов, холодильников кислоты, горячих кислотопроводов
и связано с ремонтом этих узлов. В системах, работающих по ис-
парительному принципу нет надобности в сооружении громоздкого
кислотно-холодильного хозяйства, но усложняются требования к
конструкционным материалам для изготовления аппаратуры. В свя-
зи с этим более распространенными являются циркуляционные си-
стемы, достигшие большой производительности. Совершенствова-
ние испарительного способа связано с применением материалов
(сталей), устойчивых при высоких температурах по отношению к
корродирующему и термическому действию фосфорного факела.
Разработано173 в полузаводских условиях производство кис-
лоты с комбинированным отводом тепла как за счет теплообмена
между горячими газами и водой через стенки аппарата, так и за
счет испарения воды. В теплообменно-испарительной системе бла-
годаря большой разности температур факела и охлаждающей воды
достигается высокий теплосъем при меньших габаритах основной
аппаратуры, чем в циркуляционной системе одной и той же про-
изводительности. При этом 70% тепла снимается за счет наруж-
ного охлаждения металлических стенок аппаратуры и только 5—
10% за счет испарения воды.
В циркуляционных системах сжигание фосфора и гидратацию
фосфорного ангидрида производят или в одном аппарате или (для
лучшего распределения теплоотвода) в отдельных аппаратах.
На рис. 268 представлена технологическая схема получения
термической фосфорной кислоты с применением одного аппарата
(башни) для сжигания фосфора и поглощения фосфорного ангид-
рида. Под давлением горячей воды из расходного резервуара фос-
фор передавливается по трубопроводу к шести-семи форсункам,
охлаждаемым водой, расположенным в горизонтальной решетке,
перекрывающей башню-камеру сжигания; форсунки изготовляют
из кислотоупорной хромоникелевой стали или хромистого чугуна.
Корпус башни изготовлен из стали, внутри гуммирован и по слою
960
Гл. XXVI. Производство фосфора и фосфорной кислоты
резины футерован кислотоупорными плитками. При высоте башни
10 м, диаметре вверху 2,75 м, внизу 2,35 м в ней можно перерабо-
тать до 12 т фосфора в сутки, что соответствует 37,5 т1сутки,
100%-ной фосфорной кислоты.
Фосфор распыляется поступающим в форсунки воздухом под
давлением до 6 ат я и сгорает, образуя факелы, обращенные вниз.
Количество сжатого (первичного) воздуха составляет ~7% от
общего количества, необходимого для окисления фосфора. Осталь-
ное количество воздуха (вторичного) подается по патрубку в цен-
Расплавлен-
ный cpoctpop
Воздух
Рис. 268. Схема производства термической фосфорной кислоты:
1 ~ форсунки; 2 — башня-камера сжигания; 3 — электрофильтр; 4 — вентилятор;
5 — сборник для кислоты; 6 — насос; 7 — оросительный холодильник.
тре крышки башни. Вторичный воздух, благодаря особой конструк-
ции патрубка, поступает внутрь башни спиралеобразно. Этим обес-
печивается лучший контакт воздуха с разбрызгиваемым фосфором
и в некоторой мере заглушается шум работающих форсунок.
У верхнего края башни имеется «воротник» — кольцевой желоб,
куда подается охлажденная фосфорная кислота (при 40—60°) и
часть воды, необходимой для образования фосфорной кислоты цз
сжигаемого фосфора. Разбавленная водой в верхнем кольцевом
желобе холодная фосфорная кислота переливается через его внут-
ренний край и стекает в виде пленки, которая предохраняет внут-
реннюю поверхность башни от воздействия высоких температур.
Равномерному омыванию кислотой стенок способствует немного
конусообразная форма башни. Кислота, стекающая по стенкам,
поглощает некоторое количество фосфорного ангидрида и упари-
Получение фосфорной кислоты
961
вается за счет тепла, выделяющегося при сжигании фосфора. Ос-
тальное количество фосфорного ангидрида поглощается стекающей
кислотой в нижней части башни, куда также подается дополни-
тельная часть воды. Фосфорный ангидрид проходит через жидкост-
ную завесу, создаваемую форсунками, расположенными в середине
башни. Количество воды, подаваемой в башню, регулируется так,
чтобы вытекающая из башни кислота имела концентрацию 88—90%
Н3РО4.
Газ, охлажденный до 80—100° циркулирующей кислотой и за
счет испарения воды из нижней части башни поступает в пластин-
чатый электрофильтр, где улавливается содержащаяся в нем ту-
манообразная фосфорная кислота, а затем гуммированным венти-
лятором выбрасывается в атмосферу. Стальная, футерованная кис-
лотоупорным кирпичом камера электрофильтра имеет длину 11,5 л,
ширину Зли высоту 5,7 м. Внутренние детали электрофильтра из-
готовлены из нержавеющей стали. Для вывода кислоты дно элект-
рофильтра имеет уклон к середине. Вытекающая из электрофильтра
кислота содержит 75—77% Н3РО4, т. е. меньше, чем из башни.
Концентрация получаемой в электрофильтре кислоты тем меньше,
чем ниже температура газа в нем, так как при низкой температуре
конденсируется большее количество водяного пара.
Кислота из башни, имеющая температуру 80—85°, и кислота из
электрофильтров стекают по желобам в стальной гуммированный
и футерованный кислотоупорным кирпичом сборник, где они сме-
шиваются, образуя 80—85%-ную фосфорную кислоту. Эта кислота
подается насосом из нержавеющей стали в оросительный холо-
дильник также из нержавеющей стали, где она охлаждается водой
до 30—40°. Часть кислоты направляется на склад готовой продук-
ции, а основная ее масса в качестве циркулирующей кислоты по-
ступает на орошение башни. Циркулирующая кислота в башне
поглощает все тепло реакции (на каждый кг сжигаемого фосфора
выделяется 6530 ккал). Но температура кислоты на выходе не
должна превышать 85°, так как при более высоких температурах
она сильно разрушает материал оборудования.
При сжигании фосфора и гидратации фосфорного ангидрида
в отдельных аппаратах — в башнях (камерах) сжигания и гидра-
тации возникает возможность постепенного охлаждения фосфорных
газов и уменьшения вследствие этого образования туманообразной
фосфорной кислоты. Это упрощает отделение фосфорной кислоты
из газовой фазы и позволяет уменьшить объем электрофильтров
или вовсе их исключить. Фосфор сжигают (рис. 269) в камере
(башне) сжигания, орошаемой оборотной фосфорной кислотой. Бла-
годаря охлаждению газов циркулирующей фосфорной кислотой тем-
пература их на выходе из башни составляет ~ 145—160°. При
этом часть образующегося фосфорного ангидрида поглощается
кислотой, подаваемой из башни гидратации. Из башни сжигания
962
Гл. XXVI. Производство фосфора и фосфорной кислоты
отводится продукционная кислота. Оставшаяся масса газов из ка-
меры (башни) сжигания поступает в башню гидратации, где ула-
вливается остальная часть фосфорного ангидрида с образованием
фосфорной кислоты. Башня гидратации орошается оборотной раз-
бавленной фосфорной кислотой. Непрореагировавшие газы из ба-
шни гидратации с температурой 50—60° поступают в электрические
фильтры для окончательной их очистки перед выбросом в атмо-
сферу.
В некоторых системах раздельное сжигание фосфора и гидра-
тацию фосфорного ангидрида сочетают в одной башне, но на раз-
ной высоте. В верхней части ее производят сжигание фосфора с
охлаждением образующихся газов орошаемой охлажденной кисло-
той (см. выше). В этом случае орошение башни кислотой осуще-
ствляется двумя потоками. Часть кислоты (в количестве 300—
320 м3/ч) при 60° поступает сверху, а остальное ее количество
(320—350 м3/ч) вбрызгивается форсунками в середину башни.
Отходящие газы, содержащие значительные количества непо-
глощенной Р2О5 и тумана фосфорной кислоты, можно улавливать
в скрубберах Вентури более дешевых, чем электрофильтры.
В нижнюю наклонную часть трубы Вентури форсункой вбрыз-
гивается фосфорная кислота концентрации 75% Н3РО4. В верти-
кальной части трубы газ с помощью форсунок промывается свежей
водой. Вбрызгивание воды производится с целью выравнивания
концентрации кислоты и снижения температуры газа от 100° до
80—90°. Это необходимо для защиты трубопроводов от коррозии.
Вбрызгиваемая в узком сечении разбавленная фосфорная кис-
лота в расширяющейся части трубы Вентури образует влажную
завесу, которая действует как преграда. Пройдя через завесу газ
насыщается влагой. Одновременно при адиабатическом сжатии
газа происходит конденсация паров воды, причем частички Р2О5 и
Н3РО4 в газе действуют как центры конденсации. Частички, окру-
женные оболочкой воды, утяжеляются и могут поэтому в дальней-
шем легко отделяться от газовой фазы. Выделение жидкой фазы
из газового потока производится частично в переходной трубе из
скруббера Вентури и, в основном, в циклоне.
Вытекающая кислота собирается в сборник 45 %-ной (по Р2О5)
кислоты, откуда она погружным насосом подается в горловину
трубы Вентури для орошения газа. Избыток кислоты отводится в
сборник башенной кислоты.
Газодувка, создающая разрежение в системе, сблокирована с
насосами для воды, подаваемой на передавливание фосфора на
складе фосфора. При неисправности газодувки выключаются и на-
сосы, вследствие чего прекращается подача фосфора на башню.
Отходящий газ после циклона, пройдя каплеотделитель, выбра-
сывается в атмосферу. Температура газа, отходящего из башни,
должна быть ниже 105°.
Рио. 269. Схема производства фосфорной кислоты термическим способом:
1 — погружной насос; 2 —холодильник; 3 — бачок-расходомер; 4 — башия сжигания; 5 — башня
гидратации; 6 — электрофильтр.
В атмосферу
Тепло с 65*/»-ной
кислотой из башни
гидратации ?
Тепло реакции
Тепло с Воздухом С продукционной 75%-ной
146130 * ',Л
Тепло с tpoccpo
ром 32400
ния
Кислотой 494700
С отходящими
газами 4227200
Холодильник! щей
51588160
Потери
20000
18062400
13641100
37947060
37947060Х>
С
Рис. 270. Схема тепловых потоков (в ккал/ч) в башне сжигания
(производительностью 3000 кг/ч фосфора).
Башня ^циркулирующей
сжига- ~~~'о-ной кислотой
964
Гл. XXVI. Производство фосфора и фосфорной кислоты
При использовании для очистки газов скрубберов Вентури
часть нерастворимых примесей загрязняет кислоту. Очистка кис-
лоты производится фильтрацией через фильтровальное полотно,
на которое наносится слой кизельгура в количестве 1 кг на 1 м2
фильтрующей поверхности. В кислоту добавляется 200 г кизель-
гура на 1 м3 фильтруемой кислоты. Кизельгур связывает загряз-
нения и сохраняет пористость намытого слоя.
Отфильтрованная кислота стекает в сборник чистой кислоты и
Насосами подается на склад готовой продукции. Склад кислоты со-
стоит из гуммированных сборников емкостью 200 м3, снабженных
змеевиками для подогрева кислоты и погружными насосами. От-
грузка фосфорной кислоты в железнодорожные цистерны из не-
ржавеющей стали или гуммированные осуществляется по трубо-
проводу из склада к устройству для налива. Передача кислоты
Производится погружными насосами, установленными в хранили-
щах кислоты.
Ниже приведен материальный баланс производства фосфорной
Кислоты на 1 т фосфора (без учета циркулирующих потоков):
Приход Кг Расход Кг
1. Фосфор . . . 1000 1. Продукционная фосфорная
2. Воздух . . . 10 370 кислота (75% Н3РО4) . . . 4 194
в том числе: в том числе:
02 . . . 2467 Н3РО4 3 146
n2 ....... . . . 7 953 Н2О 1 048
8. Вода . . . 2915 2. Отходящий газ 10 086
в том числе: Ра 1 133
n2 7 953
Н2О 1 000
3. Примеси * 5
Всего.................... 14 285
Всего................ . . 14285
• Примеси, содержащиеся в жидком фосфоре, отдельно ие выводятся, Они поступают
частично в продукцию, частично могут скапливаться в виде шлама.
Количество оборотной (циркулирующей) кислоты на 1 т сжи-
гаемого фосфора составляет в башне сжигания —400—450 г, а в
башне гидратации —50—60 т.
На рис. 270 представлено графическое изображение тепловых
Потоков в башне сжигания.
При отсутствии циркуляции кислот (в испарительных схемах)
отвод тепла сжигания фосфора производится вначале охлажде-
нием водой стенок башни, а затем в холодильнике газов. Оконча-
тельное охлаждение газа и поглощение фосфорного ангидрида про-
исходит в башне гидратации за счет некоторого испарения впрыс-
киваемой в нее воды. Фосфор сжигают в прямоугольных или
цилиндрических камерах-башнях сжигания, изготовленных из гра-
Получение фосфорной кислоты
965
фитовых блоков и имеющих поддон из нержавеющей стали. Они
имеют24 диаметр 4,4 м и высоту 10,7 м. Снаружи камера охлаж*
дается стекающей по ее поверхности водой. В результате охлаж-
дения наружным теплообменом температура газов снижается до
~800° (от температуры факела, достигающей 2000°). До подачи
в башню гидратации газы дополнительно охлаждают до 180—200°
в холодильнике, расположенном ниже камеры сжигания. Холо-
дильник представляет собой резервуар из угольных плит с распо-
ложенными внутри горизонтальными графитовыми трубами174
(диаметром 7,5 см), по которым циркулирует охлаждающая вода.
В холодильник вытекает также из поддона камеры сжигания мета-
фосфорная кислота, образующаяся в некотором количестве из фос-
форного ангидрида при высокой температуре. Резервуар холодиль-
ника дополнительно охлаждается водой также снаружи. Здесь газ,
выходящий из камеры сжигания с температурой 800°, охлаждается
до 180° и затем поступает в башню гидратации, орошаемую водой,
подаваемой через распылители.
Башни гидратации (диаметром 2,4 м и высотой 10,8 ж) изготов-
ляются из кислотоупорного кирпича без наружного кожуха и футе-
руются угольными плитками 175. На разной высоте в башни через
распылители вводится вода. В ней ~55% Р2О5 превращается в
фосфорную кислоту, причем газ охлаждается до 100°. Остальное
количество фосфорного ангидрида и туманообразная фосфорная
кислота улавливаются в электрофильтре.
На рис. 271 приведена технологическая схема и схема контроля
и регулирования процесса.
По сравнению с циркуляционными испарительные схемы имеют
ряд преимуществ: уменьшаются эксплуатационные расходы и
удельные капиталовложения, сокращается расход нержавеющих
сталей и облегчается получение кислот с более широким диапазо-
ном концентраций (в том числе и суперфосфорной кислоты). На
сооружение теплообменно-испарительной системы, разработанной
в НЙУИФе 173, требуется меньше в 2,5—6 раз нержавеющей стали
и в 2—2,5 раза капитальных вложений, чем на циркуляционную
систему. При этом уменьшаются также и эксплуатационные рас-
ходы. Аппаратура системы изготовляется из нержавеющей стали
и не футеруется. Внутренняя поверхность аппаратов защищается
от коррозии пламенем и горячими газами пленкой кислоты.
При сжигании фосфора в производстве фосфорной кислоты сте-
пень его использования составляет 98%. При этом на производство
термической фосфорной кислоты (в пересчете на 100%-ную Н3РО4)
расходуется 0,32—0,33 т желтого фосфора.
В производстве фосфорной кислоты термическими способами
около 92% общей стоимости продукта составляет стоимость фос-
фора, поэтому стоимость термической фосфорной кислоты в основ-
ном определяется затратами на электровозгонку фосфора.
966 Гл. XXVI. Производство фосфора и фосфорной кислоты
По сравнению с сернокислотным методом, получение фосфор-
ной кислоты одной и той же концентрации (85% Н3РО4) электро-
термическим способом обходится, как правило, дороже24.
В связи с этим перспективным является развитие комплексных
способов переработки, например, окисление фосфора водяным
паром с одновременным получением водорода 168>169, электровоз-
гонка фосфора из кремнеземистых фосфатов с использованием
Рис. 271. Схема производства термической фосфорной кислоты и размещения
контролирующих и регулирующих приборов:
/ — расходомер воздуха; 2 — регистратор температуры; 3 — регистратор температуры и термо-
регулятор; 4 —форсунка; 5 —камера сжигания; 6 —диафрагменный водомер; 7 — регулирующая
заслонка; 5 —воздуходувка для подачи вторичного воздуха; 9 — холодильник для газа;
1Q* башня гидратации; // — электрофильтр; 12 — дифференциальный манометр; /3 — регулятор
давления; /4*-хвостовой вентилятор; /5 —прибор для дистанционного управления; 16 — газоход
для подачи газа в выхлопную трубу.
шлаков на шлакопортландский цемент60, получение фосфора из
апатитовой руды и бокситов с одновременным выпуском глинозе-
мистого цемента 60’176’177, переработка апатита и нефелина с полу-
чением шлаков, пригодных для извлечения из них окиси алюми-
ния 60 и др. Перечисленные способы в той или иной степени изу-
чены. Описан60 процесс одновременного производства фосфора и
карбида кальция.
ЛИТЕРАТУРА
I, А. Финдлей, Правило фаз и его применение, ГОНТИ, 1932. — 2. В. И. Н и-
К о л а е в, ЖРФХО, 6, 1045 (1928).—3. К- Р a t z, Abhandl. Dtsch. Akad. Wiss.
Berlin KI- Chem. Biol, und Geol., № 7, 93, 1955 (1957).—4. H. H. Знамен-
ский, А. П Шубников, Производство желтого и красного фосфора, ОНТИ,
1938. — 5. Н. Д. Таланов, Т. А. Мишакова, Реф. н.-и, раб. за 1957 г., ч. I,
Литература
967
стр. 26, Труды НИУИФ, в. 163, 1959. — 6. R. С. Ellis, Inorgan. Chem., 2, № 1,
22 (1963). — 7. Н. Д. Таланов, авт. свид. 135473, 1960.—8. К. И- Загвозд-
кин, Н. А. Барилко, ЖПХ, 13, № 1, 29 (1940). — 9. М. М. S tri plin,
D. McK night, G. Н. Me gar, J. Agr. Food. Chem., 6,- № 4, 298 (1958).—
10. J. Agr. Food. Chem., 7, № 4, 233 (1959).
11. R. G. Brautigam, Compounds Semiconduct., v. 1, N. Y. — London,
1962, p. 79. —12. H. Д. Таланов, Г. В. Астахова, Т. А. Локтюхина, авт.
свид. 150536, 1963. — 13. J. К г е m е г, Н. К г i b 1 е, Chem. Ingr., Techn., 36, № 9,
957, А 1439, А 1443 (1964). — 14. R. G. В г a n t i g a m, пат. США 3033653, 1962. —
15. F. V Williams, пат. США 3039854, 3053637, 3056660, 1962. — 16. J. Kre-
mer, Н. Krible, F. R о d i s, пат. ФРГ 1143794, 1963. — 17. Н. F. Н a d a m о v-
sky, J. Kaltermann, пат. ГДР 31317, 1965. — 18. S. Glixelli, К. Bora-
tynsky, Z. anorg. Chem., 235, № 3, 225 (1938). — 19. В. Ваггаман, Фосфор-
ная кислота, фосфаты и фосфорные удобрения, Госхимиздат, 1957. — 20. A. L. Ни-
hti, Р. A Gartaganis, Canad. J. Chem., 34, 785 (1956).
21. С. Л. Г о л ы и к о - В о л ь ф с о н, М. М. Сычев, Л. Г. Суд а к ас,
Л. И. С к о б л о, Химические основы технологии и применения фосфатных свя-
зок и покрытий, Изд. «Химия», 1968. — 22. И. Л. Гофман, Хим. наука и пром.,
2, № 6, 706 (1957). — 23. A. S. Michaels, Р. М. Williams, К. В. В a n d о 1 f,
Ind. Eng. Chem., 50, № 6, 889 (1958). — 24. С. H. All, Ind. Eng. Chem., 44, № 7,
1520 (1952). —25. W. L. Hill, ibid., 44, № 7, 1526 (1952).—25. J. Gordon,
Chem. Weekblad., 71, № 5, 26 (1952). —27. Chem. Eng., 60, № 3, 196 (1953).—
28. H A. Curtis, J. Electrochem. Soc., 100, № 4, 81C (1953). — 29. Ind. Eng.
Chem., 46, № 6, 1121 (1954). —5(7. И. Л. Гофман, Хим. пром, № 2, 119
(1955).
31. E. С. К a p u s t a, Ind. Eng. Chem., № 1, 42A—44A (1956).—32. Commerc.
Fertilizer, 92, № 6, 76, 78 (1956). — 33. W. 1. Riley, Chem. Eng. News, 34, №44,
4312 (1956). — 34. С. И. Вольфкович, Усп. хим., 25, № 11, 1309 (1956).—
35. G. Bixler, I. Work, R. L a 11 i g, Ind. Eng. Chem., 48, № 1, 2 (1956). —
36. Scholl a. oth. Farm. Chemicals, 120, № 2, 53 (1957). — 37. H. H. Постни-
ков, Хим. пром., № 6, 381 (1958). — 38. A. H. Стрельцов, Ю. Я. Туров,
В. Е Третьяков, Г. А. Паршина, Труды Ленниигипрохима, в. 1, 1967,
стр. 248. —39. Н. KISkner, Chem. Ind., 18, № 11, 682 (1966).— 40. Phos. a.
Pot., № 25, 10 (1966).
41. H. M. Постников, M. Г. Френкель, Журн. ВХО, 7, № 5, 500
(1962). — 42. E. E. 3 у с с e p, ЖХП, 13, № 9, 536 (1936). — 43. Г. Люстринг-
сгаузер, Труды НИУИФ, в. 143, 1938, стр. II. — 44. Фреден, И о н а ш к у,
Rev. chim., 5, № 12, 619 (1954).— 45. W. Н. R о s s a. oth., Ind. Eng. Chem., 17, 1087
(1925). — 46. H. Basset, J. Chem. Soc., 1958, № 9, 2949.—47. H. E. Пестов,
Удобр. и урожай, № 6 (1930); № 10 (1931).—48. L. Wohler, Ann. Phys. Chem.,
17, № 1, 178 (1829). —49. W. H. Ross, A. L. M e h г i n g, R. M. Jones, Ind.
Eng. Chem., 16, № 6, 563 (1924).—50. К. M a t i g n о n, Chem. Ind., 31, № 6,
1268 (1934).
5/. H. Frank, H. Fiildner, Z. anorg. Chem., 204, 97 (1932).—
52. H. H. Постников, сб. «Исследования no прикладной химии», Изд. АН
СССР, 1955, стр. 67. — 53. Н. Н. Постников, Б. Б. Е в з л и н а, О. В. В а-
с и л ь е в а, ЖПХ, 27, № 6 (1955). — 54. Н. Н. П о с т н и к о в, А. Д. М и х а й л и и.
Сообщения о иаучио-технических работах НИУИФ, № 2, 1957, стр. 46.—
55. Н. Н. Постников, ЖПХ, 31, № 9, 1281 (1958). — 56. А. И. Климович,
Изв. вузов. Хим. и хим. технол., № 6, 71 (1958).—57. Н. Н. Постников,
А. Д. М и х а й л и н, ДАН СССР, 120, № 2, 378 (1958). — 58. А. Д. М и х а й л и н,
Н. Н. Постников, Реф. н.-и. раб. за 1956 г., ч. 1, стр. 25, Труды НИУИФ,
в. 161, 1958. — 59. А. Д. Михайлин, Автореф. канд. дисс., НИУИФ, 1957.—
60. Н. Н. Постников, Автореф. докт. дисс., Ин-т металлургии АН СССР в
НИУИФ, 1959.
968
Гл. XXVI. Производство фосфора и фосфорной кислоты
61. К. Jakob, D. Reinolds, W. Hill, Ind. Eng. Chem., 20, 11, 1204
(1928); 21, 1126 (1929). — 62. Л. Я. Марковский, Труды ГИПХ, в. 34, 1940,
стр. 69.—63. Н. Н. Постников, Б. Б. Евзлина, О. В. Васильева, Хим.
пром., № 1, 8 (1951). — 64. Н. Н. Постников, Л. П. Б е л о т е л о в, Мин.
удобр. и инсектофунгис., № 3, 11 (1935). — 65. Н. Н. Постников, Ю. М. Ра-
б и н о в и ч, Удобр. и урожай, № 11—12,911 (1930); ЖПХ, № 21—22, 1 (1931).—
66. А. П. Любан, Анализ явлений доменного процесса, ч. I, Госметаллургиздат,.
1955.—67. Э. В. Брицке, Н. Е. Пестов, Труды НИУ, в. 59, 1929.—
68. А. П. Любан, Металлург, 2, № 2, 54 (1936). — 69. И. Е. Шихуцкий,
ЖПХ, № 18, 979 (1928). — 70. Н. Е. Пестов, А. Е. С д о б н о в, Там же, № 2.
129 (1931).
71. К. И. 3 а г в о з д к и н, Там же, 7, № 3, 455 (1934). — 72. П. М. Л у к ь я*
иов, Общий курс электротермии, изд. МХТИ им. Д. И. Менделеева, 1940. —
73. А. И. Шерешевский, сб. «Производство удобрений за границей», № 3.
1932, стр. 55.—74. Е. A u b е г t i n, L. В oblique, Bull. Soc. chim., 2, 9, 335-
(1868). — 75. H. M о i s s о n, Z. Electrochem., 5, 515 (1899). — 76. A. Reneult,
ibid., 5, № 48, 332 (1899). — 77. W. Hempel, Z. anorg. Chem., 18, 132.
401 (1905). — 78. A. L a s s i e r, Orig. Comm. 8 Intern. Congr. of Appl. Chem., 2,
175 (1912). — 79. O. Nielsen, Ferrum, 10, 97 (1913). — 80. К. M a t i g n о n,
Chim. Ind., 5 (1929).
81. Э. В. Брицке, H. E. Пестов, H. H. Постников, Мин. сырье и
цветы, металлы, № 4, 375 (1929).—82. К. И. Загвоздки н, ЖХП, 12, № о,
597 (1935). — 83. Э. В. Брицке, Б. К. Веселовский, Изв. АН СССР, № 4,
479 (1937). — 84. Б. Ф. Ерофеев, ЖФХ, 828 (1937). — 85. К. Загвоздки н,
С. Больц, ЖПХ, 11, 1548 (1938). — 86. Е. Бабкин, К- Загвоздки н.
Ю. Рабинович, Труды НИУИФ, в. 134, 1936. — 87. Е. Е. Бабкин, К. И. 3 а-
гвоздкин, Ю. М. Рабинович, Труды НИУИФ, Там же, в. 143, 1938,
стр. 134.—88. G. Т г б m е 1, Н. Harkort, W. Hot op, Z. anorg. Chem 256,
4 (1948). — 89. П. В. Г e л ь д, В. Г. Власов, Н. Н. Серебренников, ЖПХ,
25, № 2, 121 (1952). — 90. Д. А. Патрушев, А. С. М и к у л и н с к и й, Там же,
33, № 4, 774 (1960); Труды УНИХИМ, в. 5, 1957, стр. 236.
91. F. Wodtcke, Z. Chem., 1, № 1, 15 (1960).—92. А. А. Анисонян,
И. Р. Шебло, ЖПХ, 37, № 3, 482 (1964).— 93. А. Б. Бектуров, В. В. Ти-
хо н о в, В. К. Э с и н, Труды Ин-та хим. наук АН КазССР, т. 10, 1964,
стр. 94.-94. А. А. Анисонян, И. Р. Шебло, ЖПХ, 37, № 11, 23 (1964).—
95. В. Tendaj, Przem. Chem., 43, № 11, 596 (1964). — 96. Н. Н. Постников,
Хил. пром., № 9, 260 (1952). — 97. С. В г i s о п, англ. пат. 3515, 1868.—
98. W. Н. Waggaman, С. R. Wagner, Ind. Eng. Chem., 10, 353 (1918).—
99. W H. W a g g a m a n, T. В. T u r 1 e у, Chem. Met. Eng., 23, 25, 1057 (1920).—
100. А. И. Шерешевский, Удобр. и урожай, № 11—12, 1082 (1931).
‘ 101. W. Н. Waggaman, Ind. Eng. Chem., 24, 983 (1932). — 102. H. W. E as-
terwood, Chem. Met. Eng., 40, 283 (1933).— 103. P. H. Royster a oth.,
Techn. Bull., 543, US Dept. Agr. (1937). — 104. А. Малец, Г. Лютринсгау-
зер Труды НИУИФ, в. 143, 1938, стр. 9. —105. Т. Р. Hignett, Chem. Eng.
Progr., 44, № 10, 753; № 11, 821; № 12, 895 (1948). — 106. W. H. Waggamaft,
R. E. Bell, Ind. Eng. Chem., 42, № 2, 283 (1950). — 107 H. A. Curtis, Chem.
Met. Eng., 42, № 6, 299, 320 (1935). — 108. H. W. Waggaman, R. E. Bell,
Ind. Eng. Chem., 42, № 2, 276 (1950). —109. W. H. Ross a. oth., ibid., 9, 26
(1917); 16, 563 (1924). — HO- I. N. Carothers, ibid., 10, 35 (1918).
111. С. И. В о л ь ф к о в и ч, Техн.-эконом, вести., № 8—9 (1925). — 112. Е. U г-
b a i n, Chem. Ind., 17, 4, 536 (1927). — 113. В. G. К 1 u g h, Chem. Met. Eng., №11
(1929).— 114. H. H. Постников, Ю. M. Рабинович, ЖПХ, 1, № 21—22
(1931); Удобр. и урожай, № 11—12, 911 (1930). — 115. В. G. К 1 u g h, Ind. Eng.
Chem., 4, 371 (1932). — 116. С. И. Вольфкович и др., Труды НИУ, в. 95,
1932, стр. 42. —117. Н. Е. Пестов, А. И. Шерешевский, Усп. хим., 3,
Литература
969
№ 4, 610 (1934). — 118. Н. Н. Постников, Л. П. Бел отелов, Мин. удобр.
и инсектофунгис., № 3, 11 (1935). — 119. Н. A. Curtis a. oth., Chem. Met. Eng.,
42, № 6, 320 (1935); 43, № 8, 408; № 11, 647; № 12 (1936); 45, № 3, 116; № 10,
-536 (1938). — 120. И. Гофман, Я. Зильберман, И. Островский, Тех-
нология суперфосфата и других фосфорных удобрений, Химтеорет, 1937.
121. Н. Постников, И. Орлов, Н. Крючков, А. Белотелое, Р. Ре-
мен, Труды НИУИФ, в. 143, 1938, стр. 9. —122. R. Н Newton, Chem. Met.
Eng., 45, № 7, 374 (1938). — 123. А. И. Шерешевский, сб. «XXV лет работы
НИУИФ», 1946, стр. 74. — 124. 1. Н. Walthall, Proc. Eng. Soc., 32, № 4, 74
(1946). —125. T. El let sen, Trans. Electrochem. Soc., 89, 307 (1946); ам. пат.
2300355, 1942. — 126. Ritter, Chem. Ind. Techn., № 22, 253 (1950). —127. TVA,
Chem. Eng. Rept, № 4 (1950). — 128. M. M. S tri pl in a. oth., Chem. Eng., 58,
№ 7, 108 (1951). — 129. I. R. C a 11 a h a m, Chem. Eng., 58, № 4, 102 (1951).—
130. Л. Я. Марковский, А. Л. Оршанский, В. П. Прянишников,
Химическая электротермия, Госхимиздат, 1952.
131. Т. М. Ware, Mining Congress J., 39, № 2, 62 (1953).—132. F. В. Shep-
herd, Chem. Ind., № 45, 1324 (1956). —133. G. Born, Chem. Techn., 8, № 9,
538 (1956). — 134. M. E. Позин, Технология минеральных удобрений и солей,
Госхимиздат, 1957. — 135. Technologia Zwiozkow Fosforowych, PWT, Warszawa,
1958. — 136. H. H. Постников, И. И. Абличенков, С. С. Андрушко,
Реф. н.-и. раб. за 1956 г., ч. 1, стр. 26, Труды НИУИФ, в. 161, 1958. —
137. В. М. Medyn ski, Przemysl. Chem., 13, № 7, 373 (1957).—
138. H. H. Постников, Г. Г. Н о з а д з е, Реф. и.-и. раб. за 1957 г., ч. 1, стр. 27,
Труды НИУИФ, в. 163, 1959. — 139. С. И. Вольфкович, 3. А. Роговин,
Ю. П. Руденко, И. В. Ш м а н е н к о в, Общая химическая технология, т. II,
Госхимиздат, 1959. — 140. G. В г е i 1, Chem. Ingr. Techn., 35, Аз 8, 549 (1963).
141. H. Постников, Химстрой, № 8 (1932). — 142. А. С. Мик ул ин-,
с к и й, Д. А. Патрушев, Труды УНИХИМ, в. 5, 1937, стр. 263. — 143. Д. А. П а-
т р у ш е в, Д. Ш. Литвак, Там же, в. 8., 1959, стр. 163. —144. А. П. Боль-
шаков, В. Ф. Осипов, А. И. 3 а и к и н а, Труды Ленниигипрохима, в. 1, 1967,
стр. 8. — 145. J. С. Barber, Е. С. Marks, J. Metals, 14, № 12, 902 (1962).—
146. Д. А. Патрушев, Труды УНИХИМ, в. 5, 1957, стр. 252, 281. — 147. G. В г е- '
i 1, К. Schlosser, Elektrochem. Z., 13, № 12, 327 (1961). — 148. G. S c h m a d t,
Chem. Techn., 17, № 3, 157 (1965). —149. К- Загвоздки н, Труды НИУИФ,
в. 143, 1938, стр. 11. — 150. Т. Е. Me in ho Id, E. N. Taylor, Chem. Process,
26, № 2, 21 (1963).
151. R. E. L e M a y, J. К. M e t с a 11 e, Chem. Eng. Progr., 60, № 12, 60
(1964). — 152. R. Sevin, J. four electr. et inds electrochim., 66, № 8, 271
(1961). —153. И. И. Абличенков, H. H. Постников, Хим. пром., № 6,
431 (1964). — 154. M. Н. Набиев, С. Т. Т а х т а х о д ж а е в, Б. И. Нудель-
м а н, А. М. Попова, А. И. Кузнецова, Узб. хим. ж., № 3, 5 (1965).—
155. Т. А. Атакузиев, Г. М. Пузикова, Д. Т. И р и г б а е в а, Б. И. Н у-
дельман, С. Т. Тахтоходжаев, М. И. Набиев, Тезисы докладов VI Все-
союзной конференции по технологии неорганических веществ и минеральных удо-
брений, Тбилиси, 1968, стр. 79. —156. Н. Н. Постников, ЖПХ, № 1, 65
(1960). —157. Д. А. Патрушев, А. Г. Полубоярцев, Журн. ВХО, 9, № 2,
235 (1964); Хим. пром., № 7, 499 (1964).— 158. J. С. Barber, G. Н. М е g а г,
Т. S. Sloan, пат. США 3113839 и 3084029, 1963. — 159. J. С. В а г b е г, G. Н. М е-
g а г, Т. S. Sloan, пат. США 313660, 1964. — 160. В. А. Ершов, М. И. По-
борцев, Е. А. Качанова, П. П. Логинов, Труды Ленниигипрохима, в. 1,
1967, стр. 24.
161. Т. I. Swann, Ind. Eng. Chem., 14, 630 (1922).— 162. W H. W a g g a-
m a n a. oth., US Dep. Agr. Bull. 1179 (1923).— 163. Lilienroth, ам. пат. 1594372,
1926; 1605960, 1926; 1673691, 1928. — 164. Э. Брицке, Н. Пестов, Н. Пост-
ников, Мии. сырье, № 4 (1929). —165. Н. Н. Постников, И. И. Орлов,
970 Гл. XXVI. Производство фосфора и фосфорной кислоты
Н. И. Крючков, ЖХП, № 10, 728 (1937).— 166. L. Н. Almond, Н. К- Stein-
fa i s s, Chem. Eng., 10, 105 (1948). —167. M. M. Strip! in, TVA, Chem. Eng.
Rep., № 2 (1948). —168. I. F. Schultz, G. Tarbutton, Ind. Eng. Chem., 42,
№ 8, 1608 (1950). —169. L. B. Hein a. oth., ibid., 42, № 8, 1616 (1950).—
170. H. Ruter, E. Cherdcon, G. Bayer, пат. ФРГ 1150958, 1964.
171. С. И. Вольфкович, Р. Г. Аз и ев, ДАН СССР, 162, № 6, 1310'
(1965). —172. Н. Литвинов, А. Мельникова, Труды НИУИФ, в. 134,
1936. — 173. М. Г. Френкель, Тезисы докладов VI Всесоюзной конференции по
технологии неорганических веществ и минеральных удобрений, Тбилиси, 1968,
стр. 163. — 174, Л. И. Ломбер г. Материалы по обмену опытом, НИУИФ,
1959. — 175. А. Белотелое, Труды НИУИФ, в. 143, 1938, стр. 10.—
176. Н. Н. Постников, И. И. Абличенков, И. А. Иноземцева, Реф.
н.-и. раб. за 1956 г., ч. 1, 26, Труды НИУИФ, в. 161, 1958. — 1-77. Б. Л. Гутер-
ман, А. М. Левин, Реф. н.-н. раб. за 1957 г., ч. 2, стр. 103, Труды НИУИФ,
в. 164, 1959.
Глава XXVII
КОНЦЕНТРИРОВАННЫЕ ФОСФОРНЫЕ УДОБРЕНИЯ.
ДВОЙНОЙ И ОБОГАЩЕННЫЙ СУПЕРФОСФАТЫ
Двойным (тройным) суперфосфатом называется концентриро-
ванное фосфорное удобрение, получаемое разложением природных
фосфатов фосфорной кислотой. По внешнему виду он похож на
простой суперфосфат, но содержит в 2—3 раза больше усвояемой
Р2О5.
Обогащенный суперфосфат производится разложением фосфа-
тов смесью фосфорной и серной кислот и по содержанию Р2Ов за-
нимает промежуточное положение между простым и двойным.
СОСТАВ И СВОЙСТВА
Фазовый состав двойного и обогащенного суперфосфатов мало-
отличается от простого (см. гл. XXIV); обогащенный суперфосфат
содержит меньше сульфатов кальция и стронция, а в двойном су-
перфосфате сульфаты присутствуют лишь при разложении фосфа-
тов фосфорной кислотой, загрязненной ионом SO?'.
В зависимости от сырья и условий производства в двойном су-
перфосфате содержится: 45—56% общей Р2О5, 42—55% усвояемой
Р2О5, 38—54% водорастворимой Р2О5 и 1,5—2,5% свободной
Р2О51-6. Двойной суперфосфат, содержащий 50—55% усвояемой
Р2О5, называется также тройным.
Содержание цитратнорастворимой P20s в двойном суперфос-
фате зависит преимущественно от состава фосфата и в меньшей
степени от состава фосфорной кислоты. Более существенно влияют
примеси фосфорной кислоты на относительное содержание водо-
растворимой Р2О5. Обогащенный суперфосфат содержит 22—34%
усвояемой Р2О5. Состав обогащенного суперфосфата зависит от
степени замены серной кислоты фосфорной; при 20—30%-ной за-
мене содержание общей Р2О5 составляет2,7 соответственно 28 и
32%, при этом отношение усвояемой и общей Р2О5 равно 99 и 98%.
972
Гл. XXVII. Концентрированные фосфорные удобрения
Концентрированные суперфосфаты выпускаются в гранулиро-
ванном и аммонизированном виде8.
Гигроскопичность двойного суперфосфата (табл. 74), так же
как и простого, зависит от содержания свободной фосфорной кис-
лоты и влажности продукта. При большой свободной кислотности
(4—6% Р2О5 и выше) суперфосфат обладает тем £олее высокой
гигроскопичностью, чем меньше содержится в нем влаги. Поэтому
сушка «кислых» суперфосфатов целесообразна только до такой
влажности, при которой их гигроскопическая точка соответствует
средней относительной влажности воздуха той местности, где су-
перфосфат хранится или применяется. Нейтрализация свободной
кислотности до 1,5—2% Р2О5 позволяет высушивать двойной су-
перфосфат до стабильной влажности 2—4%. Рассеваемость 10 двой-
ного нейтрализованного гранулированного суперфосфата очень вы-
сокая (около 9 баллов по шкале Пестова при влажности от 3 до
20%).
ТАБЛИЦА 74
Гигроскопичность двойного суперфосфата
{Образец Содержа- ние ^2^5 своб, % Влаж- ность, % Гигроскопиче- ская точка, % относитель- ной влажно- сти при 25° Балл по шкале гигроско- пичности
Из вятского фосфорита (флотацион- ный концентрат) Из апатитового концентрата, аммо- 4,6 Нет 12,3 70,0 4,0
низированный 7,8 85,6 0,9 ,
Объемный вес двойного суперфосфата из апатитового концен-
трата11 в камере равен 1 т/м3, после вызревания в свободно насы-
панном состоянии — 0,9 т/м3.
Вызревший двойной суперфосфат из апатитового концентрата
имеет угол естественного откоса 11 в пределах 40—43°.
ПРИМЕНЕНИЕ КОНЦЕНТРИРОВАННЫХ СУПЕРФОСФАТОВ
Двойной и обогащенный суперфосфаты обладают такой же
агрохимической эффективностью как и простой суперфосфат при
внесении равных количеств усвояемой Р2О512~1е. Главное преиму-
щество концентрированных суперфосфатов заключается в относи-
тельно меньшем количестве балласта. Это сокращает затраты на
транспортирование и хранение питательного вещества (Р2О5),
уменьшает расход тары, снижает затраты на внесение удобрения
в почву. Поэтому применение обогащенного и, особенно, двойного
.•суперфосфата экономически эффективнее, чем простого суперфос-
фата. Большое значение имеет возможность при производстве
Физико-химические основы производства
973
двойного суперфосфата переработки местного фосфатного сырья,
которое не пригодно для получения простого суперфосфата высо-
кого качества.
Перечисленные преимущества двойного суперфосфата объяс-
няют причины непрерывного увеличения его производства 17~27. :
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПРОИЗВОДСТВА ДВОЙНОГО
СУПЕРФОСФАТА
Химизм процесса
Фторапатит взаимодействует с фосфорной кислотой по ре-
акции:
Ca5(PO4)3F + 7Н3РО4 + 5Н2О = 5Са(Н2РО4)2 • Н2О + HF (1 >
Фтористый водород вступает в реакцию с кремневой кислотой,
выделяющейся в результате разложения кислоторастворимых си-
ликатов, при этом образуются кремнефтористоводородная кислота
и четырехфтористый кремний. Кремнефтористоводородная кислота
превращается в малорастворимые кремнефториды кальция, натрия
и калия. Четырехфтористый кремний частично выделяется в газо-
образном состоянии. Степень его выделения растет с увеличением
температуры и концентрации Р2О5 в жидкой фазе.
Карбонаты также разлагаются фосфорной кислотой:
СаСО3 + 2Н3РО4 = Са(Н2РО4)2 • Н2О + СО2 (2>
Присутствующая в фосфорной кислоте примесь серной кислоты
превращается в сульфат кальция (а также в сульфат стронция при
переработке хибинского апатита).
Вследствие разложения минералов-примесей, содержащих
окислы железа и алюминия, образуются средние фосфаты полу-
торных окислов:
(Al, Fe)2O3+2Н3РО4 + НаО = 2(А1, Fe)PO4 • 2Н2О (3>
Подсчет нормы термической фосфорной кислоты на единицу
фосфата производится по уравнениям (1), (2) и (3).
При разложении апатитового концентрата расчет может быть
упрощен п. Количество фосфорной кислоты определяют только по
уравнению (1), согласно которому на 1 вес. ч. Р2О5 в апатите тре-
буется 2,33 вес. ч. Р2О5 в кислоте. Поэтому норму фосфорной кис-
лоты п на 100 вес. ч. апатита подсчитывают по формуле
[Р2О5 своб] .
где f — принимаемая доля от стехиометрической нормы; [Р2О5ап] —
содержание Р2О5 в апатитовом концентрате, %; [РгОбсвоб] —
«74
Гл. XXVII. Концентрированные фосфорные удобрения
«одержание в фосфорной кислоте свободной Р2О5 (в %), определяе-
мой титрованием.
Фосфорная кислота, полученная сернокислотной экстракцией,
частично нейтрализована примесями катионов, содержит также сво-
бодную серную и кремнефтористоводородную кислоты. В этом слу-
чае норму фосфорной кислоты подсчитывают по концентрации
ионов водорода 6> 28. Она определяется титрованием щелочью с ин-
дикаторами метиловым желтым или бромкрезоловым зеленым,
точки перехода которых соответствуют нейтрализации первого иона
водорода фосфорной кислоты и обоих ионов водорода серной и
кремнефтористоводородной кислот.
Стехиометрическую норму фосфорной кислоты п на 100 вес. ч.
фосфорита подсчитывают по формуле
Юо(2“+-2£__2^\
< 56 145 142 /
ге== d
где а, Ь, с — содержание в фосфате СаО, (Al, Fe)2O3 и Р2О3, %;
d — концентрация ионов водорода, %; 56 — молекулярный вес СаО;
145 — молекулярный вес (Al, Fe)2O3 из расчета среднего содержа-
ния 75% Fe2O3 и 25% А12О3; 142 — молекулярный вес Р2О5.
Условия равновесия и кристаллизация твердых фаз
На рис. 272 в нижней части нанесены две изотермы (75 и
100°)29-31 и политермическая кривая составов эвтонических раство-
ров Eo°Ei52° тройной системы СаО—Р2О5—Н2О, а также кривые
ATw, KL и КТ— проекции изотермы (60°) четверной системы
•СаО—Р2О5—CaSiF6—Н2О на плоскость СаО—Р2О5 32. '
В верхней части рис. 272 приведена зависимость степени ней-
трализации Z первого иона водорода Н3РО4 от коэффициента раз-
ложения К апатитового концентрата при стехиометрической и из-
быточной (110%) норме Н3РО4, рассчитанная по формуле29
ЗЗЗАК
АК + 72,5п
где А — содержание Р2О5 в апатитовом концентрате, % (39,4%);
п — норма Н3РО4, кг на 100 кг апатитового концентрата (при сте-
хиометрической норме п = 126,2 кг, при норме 110% от стехиомет-
рической п = 138,8 кг Н3РО4). Линии с постоянными значениями
•степени нейтрализации нанесены также в нижней части диаграм-
мы (лучи, исходящие из точки начала координат). Луч 100%-ной
степени нейтрализации проходит через точку N состава монокаль-
цийфосфата. Поэтому все точки систем (составы комплексов), об-
разуемых при смешении фосфорной кислоты разных концентраций
и гидроксилапатита, взятых в стехиометрических количествах, ле-
жат на луче ON. При норме 110% они располагаются на луче сте-
пени нейтрализации 93,8%.
Физико-химические основы производства
975
При периодическом смешении реагентов растворение фосфата
начинается в ненейтрализованной фосфорной кислоте и продол-
жается при все возрастающей концентрации в растворе ионов каль-
ция. После насыщения жидкой фазы фосфатами кальция дальней-
шее разложение фосфата сопровождается кристаллизацией твер-
дой фазы
СаО. %
Рнс. 272. Номограмма для определения условий равновесия в системе
апатит — фосфорная кислота — вода.
При непрерывном смешении фосфата с фосфорной кислотой
оба компонента поступают в смеситель, где находится перемеши-
ваемая пульпа, и поэтому вначале разложение фосфата происхо-
дит в жидкой фазе постоянного состава, содержащей фосфорную
кислоту и растворимые продукты реакций, т. е. в растворе, не на-
сыщенном фосфатами кальция. Состав жидкой фазы пульпы зави-
сит от соотношения между количествами реагентов и от продол-
жительности реакции в смесителе. В дальнейшем разложение фос-
фата протекает (в реакторах) с изменением состава жидкой фазы
976
Гл. XXV11. Концентрированные фосфорные удобрения
и после достижения насыщения раствора фосфатами кальция со-
провождается кристаллизацией твердой фазы.
Таким образом, процесс состоит из двух стадий — растворения
фосфата вначале в растворах фосфорной кислоты ненасыщенных,
а затем — насыщенных фосфатами кальция 33~41. Эти стадии раз-
личаются по своим равновесным39, а также кинетическим40усло-
виям.
На рис. 272 момент насыщения раствора определяется точками
пересечения луча растворения гидроксилапатита (например, AR в
кислоте концентрации 25% Р2О5) с ветвями изотерм (точки а и ai
при 75 и 100° для указанного примера). Степень разложения фос-
фата (гидроксилапатита) для отдельных точек на кривых раство-
римости определяется по проходящей через них линии степени ней-
трализации (лучу растворения) при помощи верхней части диа-
граммы. Пример графического определения коэффициента раз-
ложения фосфорной кислотой концентрации 40% PsOsnpn 100° (для
узловой точки при 100°) показан тонким пунктиром. Из положения
узловых точек Е видно, что степень нейтрализации жидкой фазы,
а следовательно, и коэффициенты разложения в этих точках близки
к максимуму для каждой температуры. Для графического опреде-
ления начальных концентраций фосфорной кислоты, обеспечиваю- '
щих насыщение раствора в узловых точках Е, проводятся лучи рас-
творения гидроксилапатита через точки Е до пересечения с орди-
натой (точки А, В, С, F соответственно для 40, 75, 100 и 115°).
Наибольшее разложение апатита в равновесных условиях с обра-
зованием насыщенного раствора наблюдается при более низких
температурах и концентрациях фосфорной кислоты (табл. 75). Уве-
ТАБЛИЦА 75
Условия максимального разложения апатита в момент
насыщения жидкой фазы при различных температурах
Температура, °C Начальная концентрация H3PO4, % Р2О5 Степень нейтрализации в узловых точках, % Степень разложения, %
при стехио- метрической норме Н3РО4 при норме Н3РО4 110% от стехио- метрической
25 21,0 60,9 52,5 57,5
40 24,7 55,3 46,5 50,5
50,7 27,6 50,0 41,5 45,5
60 30,2 45,7 37,5 41,0
75 33,6 41,7 33,5 36,5
100 40,0 34,4 27,0 29,5
115 43,7 32,8 25,7 28,0
60* 45,2) 15,7 11,0 12,5
* Для точки К в присутствии в растворе 0,74% фтора.
Физико-химические основы производства
977
личение нормы фосфорной кислоты (до 110% от стехиометриче-
ской) несколько повышает коэффициент разложения апатита.
Примесь в фосфорной кислоте кремнефтористоводородной кислоты
расширяет границы существования дикальцийфосфата и резко сни-
жает его растворимость32 (рис. 272). Поэтому начальная концен-
трация фосфорной кислоты, необходимая для образования насы-
щенного раствора в узловых точках, с ростом содержания фтора
значительно увеличивается, одновременно уменьшаются степень
нейтрализации и коэффициент разложения (например, точка К на
кривой КЕбо°).
Образующиеся растворы, соответствующие по своему составу
узловым точкам, находятся в равновесии с твердыми моно- и ди-
кальцийфосфатом. В практических условиях апатит разлагают фос-
форной кислотой концентрации 45—55% РгО5 с образованием в
первой стадии насыщенных растворов, равновесных с одним твер-
дым монокальцийфосфатом. Точки составов этих растворов распо-
ложены на верхних кривых диаграммы системы (например, точки
г и Г1 при 75 и 100°). Степень разложения фосфата в момент насы-
щения жидкой фазы составляет всего 5—7%.
Дальнейшее разложение фосфата во второй стадии происходит
при взаимодействии его с насыщенным раствором и сопровождается
выделением твердой фазы. Данные о равновесии в системе могут
быть использованы для выбора условий кристаллизации твердой
фазы того или иного состава. При образовании в первой стадии
насыщенных эвтонических растворов, отвечающих по своим соста-
вам узловым точкам, дальнейшее разложение фосфата (если оно
возможно было бы по кинетическим условиям) сопряжено с выде-
лением в твердую фазу вначале смеси моно- и дикальцийфосфата,
а к концу процесса — одного дикальцийфосфата. В отличие от
этого при растворении апатита в фосфорной кислоте концентрации,
например, 55% Р2О5 (луч растворения DR на рис. 272) после на-
сыщения жидкой фазы (точки гиг, при 75 и 100°) точки состава
фосфатного комплекса сначала попадают в поле кристаллизации
Са(Н2РО4)2-НгО (например, точки е и et при 75 и 100°). Состав
жидкой фазы изменяется вдоль изотерм системы (для рассматри-
ваемого примера от точек г или п до узловых точек £). Последо-
вательное растворение апатита протекает при постепенном умень-
шении концентрации Р2О5. После достижения раствором узловых
точек в твердую фазу начинает выделяться смесь Са(Н2РО4)2 • Н2О
и СаНРО4. В точках s или Si (в зависимости от нормы Н3РО4) в
равновесных условиях разложение апатита полностью заканчи-
вается. Степень нейтрализации жидкой фазы по мере выделения
твердой фазы на ветви монокальцийфосфата повышается, а на
ветви дикальцийфосфата — уменьшается. Повышение температуры
уменьшает степень нейтрализации на ветвях дикальцийфосфата
увеличивает ее на ветвях монокальцийфосфата.
078
Гл. XXVII. Концентрированные фосфорные удобрения
Скорость разложения фосфатов фосфорной кислотой
Скорость взаимодействия фосфатов и достигаемая степень их
разложения за определенный промежуток времени зависит в пер-
вую очередь от количества применяемой для разложения фосфор-
ной кислоты и образующегося фазового состава системы. В соот-
ветствии с этим различают:
1. Растворение фосфата в большом количестве кислоты, до-
статочном для образования гомогенного (не считая нерастворимых
примесей) раствора.
2. Взаимодействие фосфата с фосфорной кислотой в незагусте-
вающей пульпе, когда количество выделившихся кристаллов про-
дукта реакции относительно невелико по сравнению с количеством
жидкой фазы. Это происходит при растворении фосфата в разбав-
ленной или в избытке концентрированной фосфорной кислоты по
сравнению со стехиометрическим ее количеством, необходимом для
образования монокальцийфосфата, но недостаточном для образо-
вания гомогенного раствора. Процесс протекает вначале в нена-
сыщенном растворе, затем при достижении насыщения образуются
пересыщенные растворы, из которых кристаллизуется твердый
продукт.
3. Взаимодействие фосфата с концентрированной фосфорной
кислотой (50—55% Р2О5), взятой в количестве, равном или не-
сколько большем (на 5—10%) стехиометрического количества в
расчете на образование монокальцийфосфата. В начальный пе-
риод— до образования насыщенного раствора — вся масса фос-
форной кислоты реагирует с небольшой частью фосфата. Весовое
отношение между кислотой и реагирующим фосфатом Ж : Т соста-
вляет ~20 -ь 50: 1 (в зависимости от концентрации и темпера-
туры кислоты), затем в течение некоторого времени твердая фаза
выделяется в небольшом количестве с образованием подвижной —-
текучей пульпы. По мере дальнейшего разложения фосфата и уве-
личения количества твердой фазы масса (загустевающая пульпа)
постепенно густеет, пока не схватится в сплошной твердый монолит.
Растворение фосфата в фосфорной кислоте
(без кристаллизации твердой фазы)
Скорость разложения фосфата фосфорной кислотой с образова-
нием гомогенного раствора определяется законами диффузионных
процессов: она пропорциональна концентрации ионов водорода,
реагирующей поверхности, интенсивности перемешивания и т. д.
По мере нейтрализации свободной кислоты и ионов кальция
вплоть до образования насыщенного раствора скорость процесса
уменьшается. Однако вследствие значительного избытка кислоты
по отношению к реагирующему фосфату и абсолютно малой сте-
пени нейтрализации кислоты (табл. 76) скорость разложения фос-
Физико-химические основы производства
979
фата в этих условиях достаточно велика даже при использовании
кислоты с относительно небольшой концентрацией. На рис. 242
показана зависимость времени от степени разложения крупнозер-
нистого отобранного апатитового концентрата фосфорной кислотой
концентрации 20% Р2О5 при отношении Т:Ж=1:20. Как видно,
процесс вначале идет весьма быстро, а затем замедляется. В на-
чальной стадии повышение температуры увеличивает скорость раз-
ложения и общую степень разложения. При 80° апатитовый кон-
центрат полностью переходит в раствор за ~3ч.
ТАБЛИЦА 76
Весовые отношения фосфорной кислоты и апатита
(Ж : Т), необходимые для образования насыщенных
растворов
Концен- трация кислоты, % Р2О5 Значения Ж Т по весу при температуре в °C
25 40 50,7 80
20 9,60 10,50 11,80 13,40
25 9,42 8,61 9,38 17,40
30 12,10 10,75 9,32 10,20
35 16,13 13,62 11,66 9,15
40 24,11 19,04 15,94 10,81
45 36,96 29,26 22,32 14,63
46 48,66 34,90 26,71 15,22
48 53,00 40,32 30,30 17,25
50 70,00 53,77 36,77 19,67
Температурный коэффициент скорости растворения апатита до
достижения насыщения в относительно разбавленных растворах
фосфорной кислоты находится в пределах от 1,3 до 1,5, а в раство-
рах42 содержащих 51,5—53,5% Р2О5 — от 1,68 до 1,73.
Скорость разложения фосфорита Каратау в большом объеме
фосфорной кислоты (200 мл кислоты на 10 г фосфорита) значи-
тельно больше43, чем апатита и также увеличивается с повыше-
нием температуры. При 60° уже через 30 мин степень разложения
фосфорита кислотой концентрации 29% Р2О5 (40% Н3РО4) состав-
ляет 99,86%, тогда как при 20° она достигает 98,32% лишь за 1,5 ч.
Температурные коэффициенты скорости разложения фосфорита в
диапазоне концентрации кислоты от 7,2 до 22,0% Р2О5 изменяются
соответственно от 1,28 до 1,34.
Предварительная нейтрализация фосфорной кислоты соедине-
ниями магния приводит к резкому уменьшению скорости разложе-
ния фосфата36. В растворе фосфорной кислоты, нейтрализованной
карбонатом магния на 30 и 50%, скорость разложения апатита при
40° и Т: Ж= 1 30 уменьшается в 3 и 14 раз 36 по сравнению с не-
нейтрализэванной кислотой.
980
Гл. XXVII. Концентрированные фосфорные удобрения
При определенных условиях растворения апатита в большом
объеме кислоты, когда состав жидкой фазы мало изменяется вслед-
ствие незначительной степени разложения (например, при отсут-
ствии перемешивания твердой фазы) можно пренебречь концен-
трацией образующихся продуктов (соединений кальция) в рас-
Рис. 273. Сопоставление изохрон растворения апатита
в частично нейтрализованных растворах фосфорной
кислоты (при различной степени нейтрализации Z)
С изотермой системы СаО—Р2О5—Н2О при 40“
(кривая АВС).
творе. В этом случае скорость растворения фосфата пропорцио-
нальна растворимости окиси кальция в фосфорной кислоте и кри-
вая изменения скорости растворения от концентрации кислоты (изо-
хрона растворения) соответствует диаграмме растворимости 34’35’38
системы СаО—Р2О5—Н2О (рис. 273).
Как видно, скорость разложения апатита в этих условиях34, Зб,
отвечающих также начальному моменту растворения (когда "В
Физико-химические основы производства
981
растворе еще нет продукта реакции), значительно уменьшается по
мере нейтрализации первого иона водорода фосфорной кислоты
(рис. 273). Это замедляет достижение равновесных коэффициентов
разложения апатита.
Несмотря на большой избыток жидкой фазы (Т :Ж=1 : 30), сте-
пень разложения апатита в указанных условиях в растворах, рав-
новесных с дикальцийфосфатом, не превышает за 5—6 ч 0,5—0,7%
при 40—75° и 2—3% при 100°. В растворах, равновесных с моно-
кальцийфосфатом, максимальные значения степени разложения
апатита за 5—6 ч достигают 20—30% при 45—60°.
Концентрация
кислоты, % РгО5
Рис. 274. Часовые изотер-
мы-изохроны разложения
апатита ненасыщенными
растворами фосфорной
кислоты при разных темпе-
ратурах:
/ — 40°; 2—50,7°; 3-80°.
Рис. 275. Зависимость актив-
ности ионов водорода (кри-
вая 1) и вязкости (кривая 2)
насыщенных растворов системы
СаО—Р2О5—Н2О от концен-
трации фосфорной кислоты
при 40°.
На рис. 274 показаны44 изохроны-изотермы разложения апа-
тита в условиях интенсивного перемешивания (1000 об/мин) рас-
творами фосфорной кислоты разных концентраций при весовом
отношении кислоты к апатиту (табл. 76), превышающем на 15—
25% теоретическое (равновесное) отношение, вычисленное при раз-
ных температурах по диаграмме СаО——;Н2О. В этих усло-
виях заведомо не достигается насыщение растворов. Из располо-
жения кривых на рис. 274 видно, что степень растворения апатита
за 1 ч возрастает при увеличении концентрации исходной кислоты
лишь до определенного предела (до ~46% Р2О5). При повышении
концентрации кислоты до 48 и 50% Р2О5 степень разложения резко
уменьшается. Это объясняется, по-видимому, изменением активно-
сти ионов водорода и вязкости растворов кислоты (рис. 275).
В диапазоне концентраций фосфорной кислоты от 26 до 46% P2Os
982
Гл. XXV11. Концентрированные фосфорные удобрения
активность ионов водорода возрастает в большей мере, чем вяз-
кость и скорость разложения увеличивается. При содержании в
растворе 48—50% Р2О5 и больше скорость разложения умень-
шается вследствие того, что торможение процесса в результате рез-
кого возрастания вязкости раствора превалирует над ускорением
его под влиянием незначительного увеличения активности ионов
водорода.
Разложение фосфатов (с кристаллизацией продукта реакции)
в незагустевающей пульпе
Разложение фосфатов с кристаллизацией продукта реакции
представляет собой вторую стадию процесса, протекающую после
насыщения жидкой фазы системы растворимыми фосфатами каль-
СаО, %
Рис. 276. Схема разложения апатита в насыщен-
ных растворах системы СаО—Р2О5—Н2О.
ция. Это возможно при уменьшенных значениях Ж Т по сравне-
нию с указанными в табл. 76. Дальнейшее разложение связано с
образованием пересыщенных растворов, которые, в результате
кристаллизации из них твердой фазы, переходят в равновесное
состояние. Получаемая при этом вторичная жидкая фаза имеет
меньшую концентрацию, чем первоначальный раствор.
На рис. 276 показано схематически по ступеням разложение
апатита в насыщенных растворах38. Последовательные процессы
Физико-химические основы производства
983
пересыщения и кристаллизации изображены в виде зигзагообраз-
ных кривых б В при 100° и rg при 25°. Полностью процесс разло-
жения фосфата при 25° до образования фазового комплекса со-
става D может быть представлен ломаной линией aDg, т. е. движе-
нием по лучу растворения aD и по лучу кристаллизации Dg.
В незагустевающей пульпе выделяющиеся кристаллы продукта
Рис. 277. Растворимость (а) и концентрации
пересыщения (б) в системе СаО—Р2О5—Н2О при
разных температурах:
/-25°; 2-40°; 3-50°; 4-75°; 5-100°.
друга и последние (при достаточном количестве жидкой фазы и
интенсивном перемешивании) не покрываются изолирующей (шла-
мовой) коркой вплоть до полного их растворения. При этих усло-
виях скорость процесса определяется, главным образом, скоростью
кристаллизации твердой фазы из пересыщенного раствора, так как
пересыщение жидкой фазы достигается быстро и является след-
ствием разности скоростей растворения и кристаллизации. В свою
очередь, чем больше пересыщение раствора, тем больше движу-
щая сила и скорость кристаллизации, и тем меньше размеры выде-
ляющихся кристаллов.
Наибольшие пересыщения наблюдаются в концентрированных
растворах вследствие высокой их вязкости, а также, хотя и в
7 М. Е. Позин
984
Гл. XXVII. Концентрированные фосфорные удобрения
меньшей мере, в разбавленных растворах, по-видимому, вследствие
меньшей вероятности образования в них кристаллических зароды-
шей (рис. 277 и табл. 77) 45. Повышение температуры приводит к
уменьшению пересыщения концентрированных растворов, увеличе-
нию его в сильно разбавленных растворах и практически мало
сказывается на пересыщении растворов средних концентраций.
Скорость кристаллизации фосфатов кальция из пересыщенных
растворов зависит не только от пересыщения, но и от диффузион-
ных условий, определяемых концентрацией, вязкостью, температу-
рой кислоты, перемешиванием и т. п.
ТАБЛИЦА 77
Растворимость и пересыщение СаО в фосфорнокислотных растворах (%)
Содержание Р2О5 в растворе, %
55 50 45 40 35 20 15 10 5
При 26° Растворимость . . . 0,42 0,73 1,48 2,31 3,41 5,08 4,10 2,92 1,59
Пересыщение , . . 1,10 1,45 1,95 2,78 3,90 5,80 4,75 3,55 2,00
абсолютное . . 0,68 0,72 0,47 0,47 0,49 0,72 0,65 0,65 0,41
относительное, % 162 98 32 20 14,5 14 16 21,5 25,5
При 40° Растворимость . . . 0,57 1,04 1,85 2,79 3,93 4,51 2,21 1,40
Пересыщение .... 1,28 1,71 2,35 3,26 4,77 5,44 4,55 3,40 1,85
абсолютное . . . 0,71 0,67 0,50 0,47 0,83 0,93 1,04 0,79 0,45
относительное, % 125 64 27 17 14 20,7 29,5 30 32
При 50,7° Растворимость . . . 0,90 Ь47 2,27 3,42 4,38 4,19 3,22 2,30 2,31
Пересыщение .... 1,51 2,08 2,85 3,78 4,90 5,05 4,10 3,00 1,80
абсолютное . . . 0,61 0,61 0,58 0,39 0,52 0,86 0,88 0,70 0,49
относительное. % 68 42 25 10,5 12 20,8 27,5 30,5 37
При 75° Растворимость . . . 1,52 2,21 3,17 4,23 3,68 2,78 1,95 1,06
Пересыщение .... . 2,02 2,90 3,85 4,92 — 4,40 3,50 2,57 1,60
абсолютное . . . 0,50 0,69 0,68 0,69 — 0,72 0,77 0,62 0,54
относительное, % 33 31 21 16 — 19,5 25 31,5 51
При 100° Растворимость . . . 2,52 3,37 4,37 5,53 3,22 2,44 1,66 0,85
Пересыщение .... 2,70 3,62 4,72 5,85 — 3,88 3,10 2,24 1,55
абсолютное . . . 0,18 0,25 0,35 0,32 — 0,66 0,66 0,58 0,70
относительное, % 7 7 8 6 — 20 27 34 71
При 25° пересыщенные растворы являются достаточно устойчи-
выми и не разлагаются в течение длительного времени. Из раство-
ров, содержащих 34—39% Р2О5, кристаллизация монокальцийфос-
фата протекает в течение нескольких суток. С повышением темпе-
ратуры увеличивается средняя скорость (рис. 278) и достигаемая
Физико-химические основы производства
985
степень кристаллизации за определенное время (рис. 279). Из пе-
ресыщенного раствора, содержащего 55,5% РгО5, монокальций-
фосфат при 25° начинает выделяться через 5 мин. В дальнейшем,
по мере образования зародышей скорость кристаллизации увели-
чивается, достигает максимальной величины и затем уменьшается
вследствие понижения концентрации монокальцийфосфата в пере-
сыщенном растворе. С по-
М0- 5
вышением температуры от
25 до 100° увеличивается
скорость кристаллизации и
максимальное ее значение
достигается быстрее. Анало-
гичное изменение скорости
кристаллизации наблюдает-
ся и из растворов других
концентраций. Это связано с
240 360
Время,мин
Рис. 278. Изменение во времени средней ско-
рости кристаллизации монокальцийфосфата из
пересыщенных растворов, содержащих
55,5% Р2О5 при разных температурах:
1 - 25°; 2 - 40°; 3-50°; 4 - 75°; 5-100°.
Время,мин
Рис. 279. Изменение во вре-
мени степени кристаллизации
монокальцнйфосфата из пере-
сыщенных растворов, содер-
жащих 40% Р2О5 при разных
температурах:
1 -40; 2 — 50; 3-75.
уменьшением вязкости раствора при повышении температуры и об-
легчением образования зародышей кристаллов из пересыщенных
растворов.
Наибольшая скорость растворения апатита в насыщенных рас-
творах наблюдается при оптимальной концентрации кислоты. По
экспериментальным данным34 она равна 48,3% Р2О5, при 40°,
45,9% Р2О5 при 75° и 47% Р2О5 при 100°.
При разложении фосфата в незагустевающей пульпе кристал-
лизация твердой фазы значительно ускоряется. В каждый после-
дующий момент реагенты поступают в ранее прореагировавшую
массу, содержащую кристаллы, являющиеся затравкой. В зависи-
мости от условий (соотношения между кислотой и фосфатом, кон-
центрации и температуры кислоты и др,) изменяется степень
986
Гл. XXVII. Концентрированные фосфорные удобрения
разложения фосфата в отдельные стадии, т. е. при растворении его
до начала кристаллизации (к моменту образования пересыщен-
ного раствора) и в последующем при кристаллизации продукта
реакции.
ТАБЛИЦА 78
Степень разложения апатита фосфорной кислотой при образовании
пересыщенных растворов (до начала кристаллизации)
Концентрация фосфорной КИСЛОТЫ, % Р2О5 Темпера- тура, °C Степень разложения (в %) прн нормах кислоты (в % от стехиометрического количества)
200 300 400 . 500
50 32,9 49,2 65,8 82,0
40 60 38,5 57,6 76,8 96,0
70 42,0 62,8 83,5 —
50 22,5 33,6 44,9 55,0
45 60 26,5 39,7 52,8 66,4
70 29,5 44,5 59,1 74,0
50 14,7 22,2 29,5 37,0
50 60 17,1 25,6 34,1 42,8
70 19,8 29,8 39,5 49,6
50 9,7 14,6 19,4 24,3
55 60 11,6 17,4 23,2 29,0
70 12,5 18,8 25,1 31,4
В табл. 78 приведены вычисленные значения степени разложе-
ния апатита фосфорной кислотой при образовании пересыщенных
растворов (до начала кристаллизации монокальцийфосфата).
С увеличением концентрации и понижением температуры раствора
степень разложения апатита к моменту завершения первой стадии
уменьшается. Соответственно увеличивается минимальное количе-
ство фосфорной кислоты, необходимое для полного его разложения
без кристаллизации продукта реакции (табл. 79).
ТАБЛИЦА 79
Минимальное количество фосфорной кислоты,
необходимое для разложения 100 вес. ч. апатита
с образованием пересыщенных растворов
(до кристаллизации СаН2РО4-Н2О)
Темпе- ратура, °C Минимальное количество фосфорной кислоты (в вес. ч.) при концентрации Р2О5 (в °/д)
40 45 50 55
50 1495 1945 2670 3680
60 1295 165 J 2300 3090
70 1170 1480 1982 2824
Скорость разложения апатита термической фосфорной кислотой'
изучена 45 в зависимости от концентрации ее (от 40 до 55% Р2О5)
Физико-химические основы производства
987
и нормы (от 200 до 500% по сравнению со стехиометрическим коли-
чеством) в диапазоне температур от 50 до 70°. Изменение степени
разложения апатита при разных нормах кислоты показано на
рис. 280. С увеличением концентрации фосфорной кислоты от 40
Время, мин
Рис. 280. Степень разложения
апатита при 60° при разных нор-
мах фосфорной кислоты концен-
трации 45% Р2О5. Норма кислоты
в % от стехиометрического коли-
чества:
7 — 200; 2-300; 3 - 400; 4-500.
£ 1 80-
н
Н60-
и s
4(?L
—1----1-----1----1___L— I
20 40 60 80 100 120
Время,мин
Рис. 281. Изменение степени раз-
ложения апатита во времени при
разных концентрациях фосфорной
кислоты. Норма кислоты 400%;
температура 50°. Начальная кон-
центрация фосфорной кислоты в
% PjOs1
/-40; 2-45; 3-50; 4-55.
Время, мин
Рис. 282. Степень разложения апа-
тита во времени при разных тем-
пературах. Начальная концентра-
ция фосфорной кислоты 45% Р2О5;
норма 400%; температура в °C:
/ — 50; 2-60; 3-70.
до 55% Р2О5 при норме 200% степень разложения апатита при
50° за 120 мин повышается от 59,3 до 74,6%. В этом же диапазоне
концентраций кислоты при норме ее, равной 400% от стехиометри-
ческого количества, степень разло-
жения апатита возрастает лишь при
повышении концентраций фосфор-
ной кислоты от 40 до 50% Р2О5
(рис. 281).
При повышении температуры от
50 до 70° разложение апатита уве-
личивается в тем большей степени,
чем больше норма кислоты. При
норме кислоты 400 и 500% от сте-
хиометрической повышение темпе-
ратуры от 50 до 60° приводит к уве-
личению степени разложения апа-
тита за одно и то же время от 86,3
и 86,9 до 96,1 и 98,5%. Наиболее
заметно степень разложения апати-
та увеличивается при повышении температуры от 50 до 60° и не-
значительно при изменении температуры от 60 до 70° (рис. 282).
Применение термической фосфорной кислоты концентрации
40,5—41% Р2О5 в количестве ~700—450% от стехиометрической
нормы (при степени нейтрализации жидкой фазы в конце процесса
988
Гл. XXVII. Концентрированные фосфорные удобрения
20—30%) приводит при 100°48 к разложению апатита за 1 ч на
88,1%—при норме кислоты, равной 450%, на 93%—при норме
538% и на 91,8%—при норме кислоты 694% от стехиометриче-
ской. При степени нейтрализации кислоты 35 и 40%, достигаемой
при уменьшении нормы кислоты, разложение апатита кислотой
концентрации 42% РгО5 в тех же условиях составляет 78,6 и 72%.
Разложение фосфатов с образованием загустевающей пульпы
При уменьшении нормы кислоты до стехиометрической вели-
чины и увеличении ее концентрации до 50% Р2О5 и-больше весовое
отношение между фосфатом и кислотой Т : Ж составляет ~ 1 :2.
В этих условиях разложение фосфата в первой стадии (до образо-
вания насыщенного раствора) протекает в небольшой степени и
в течение очень короткого времени (5—10 лшм) 4l. В дальнейшем
начинается кристаллизация новой твердой фазы и по мере израс-
ходования кислоты количество жидкой фазы уменьшается, а коли-
чество твердой фазы увеличивается. В результате этого пульпа
постепенно густеет и, наконец, полностью загустевает (схваты-
вается), как и при получении простого суперфосфата. Кристалли-
зация новой твердой фазы и схватывание пульпы происходит тем
быстрее, чем выше концентрация исходной фосфорной кислоты.
Выделяющиеся кристаллы продукта реакции тесно соприкасаются
с непрореагировавшими зернами фосфата и откладываются на их
поверхности. Образующаяся корка по мере ее формирования ока-
зывает все большее сопротивление диффузии ионов водорода.
Вследствие этого скорость разложения фосфата определяется все
нарастающим сопротивлением твердой фазы41. Суммарное влияние
условий протекания процесса в первой стадии и в период образо-
вания корки на зернах фосфата предопределяет скорость разложе-
ния в загустевающей пульпе.
Известно 33-38, что в случае торможения процесса сопротивле-
нием образующейся корки скорость его изменяется по уравнению
прямой в координатах х/у—’/т, где у — количество вещества, про-
реагировавшего к моменту времени т. Как видно на рис. 283, раз-
ложение апатита фосфорной кислотой в загустевающей пульпе
характеризуется в целом значительно более сложной зависи-
мостью. Однако, если исключить первый период (10—20 лшм), ко-
гда разложение протекает в ненасыщенных растворах и в незагу-
стевающей пульпе, то в дальнейшем процесс описывается указан-
ной зависимостью.
В отличие от апатитового концентрата разложение фосфорита
Каратау в течение длительного времени (от 5 до 150 мин) хорошо
описывается прямолинейной зависимостью в указанных координа-
тах (рис. 284). Это, очевидно, объясняется тем, что при наличии
в растворе соединений магния насыщение раствора и кристаллиза-
Физико-химические основы производства
989
ция высаливаемой соли — монокальцийфосфата наступает значи-
тельно раньше момента отбора пробы для первого определения.
Изменение степени разложения фосфата во времени в загусте-
вающей пульпе определяется совместным влиянием активности
ионов водорода раствора и свойств кристаллизующейся твердой
фазы. Поэтому в течение некоторого времени, пока корка на зер-
нах фосфата еще является рыхлой и достаточно проницаемой, про-
Рис. 283. Зависи-
мость между 1/у
и 1/т при разло-
жении апатита фос-
форной кислотой:
у — степень разложе-
ния (в %) апатита кис-
лотой концентрации
59% Р2О5; т —время
в мин.
Рис. 284. Зависимость
между 1/у и 1/т при
разложении фосфо-
рита Каратау фосфор-
ной кислотой концен-
трации 52,5% Р2О5:
// — степень разложения,
%; т — время в мин.
цесс протекает с заметной скоростью, а в дальнейшем, когда корка
увеличивается по своей толщине и становится плотной, сильно за-
медляется.
Разложение апатитового концентрата стехиометрической нор-
мой фосфорной кислоты при 20° (рис. 285) ускоряется по мере
увеличения концентрации кислоты от 13,6 до 53,6% Р2О5, но замед-
ляется при использовании более концентрированной кислоты40. По-
видимому, повышенное значение оптимальной концентрации фос-
форной кислоты при разложении фосфата небольшим ее количе-
ством объясняется42 уменьшением концентрации кислоты в начале
взаимодействия реагентов до величины, при которой фосфат раз-
лагается насыщенными растворами с наибольшей скоростью (см.
выше). При использовании в этом случае менее концентрирован-
ной кислоты (например, 40—45% Р2О5) снижение концентрации
жидкой фазы в начале процесса произошло бы до величины, при
которой апатит разлагается с меньшей скоростью.
В течение первых 10—20 мин с начала реакции степень разло-
жения мало зависит от нормы кислоты в пределах 50—130% от
990
Гл. XXVII. Концентрированные фосфорные удобрения
стехиометрической, необходимой для образования монокальций-
фосфата (рис. 286). Это объясняется тем, что в первый момент
реагирует только поверхностный слой зерен, поэтому при всех ука-
занных нормах кислоты имеется большой ее избыток. Некоторое
увеличение нормы кислоты позволяет осуществить процесс с не-
сколько большей скоростью в течение более длительного проме-
жутка времени, по крайней мере в течение первых часов ьзаимо-
Рис. 285. Изменение коэффициента разложения апати-
тового концентрата во времени при стехиометрической
норме фосфорной кислоты и температуре 20°.
Концентрация кислоты (в % Р2О5): / — 13,6; 2 — 21,0; 3 — 36,4;
4 - 64,8; 5 - 45,6, b - 55,8; 7 - 51,5; 8 - 53,6.
действия (рис. 286). Но увеличение нормы кислоты больше 110%
от стехиометрического количества не влияет на степень разложе-
ния в продукте после двухнедельного его вылеживания49.
Существенное повышение степени разложения фосфата наблю-
дается при изменении температуры от 20 до 40° (рис. 287). Даль-
нейшее повышение температуры до 70—80° приводит к незначи-
тельному увеличению 6’34’50 степени разложения апатита. Это объ-
ясняется тем, что при температурах выше 40° уже через очень ко-
роткий промежуток времени от начала реакции выделяется значи-
тельное количество кристаллов монокальцийфосфата, создающих
большое сопротивление диффузии кислоты. В результате этого ско-
рость процесса при температурах выше 40° определяется диффу-
зионным сопротивлением твердой корки продукта реакции, которое
мало или вовсе не зависит от температуры. Практически и при
растворении апатита в большом количестве насыщенного раствора
Производство двойного суперфосфата
991
(в отсутствие перемешивания твердой фазы) также наблюдается
независимость скорости процесса от температуры34. По-видимому,
при отсутствии интенсивного отвода образующихся солей в общую
массу системы они отлагаются в непосредственной близости от
места их возникновения, т. е. у поверхности зерен фосфата. Сле-
дует также учесть и резкое уменьшение концентрации ионов водо-
рода с повышением температуры кислоты одной и той же концен-
трации, вследствие понижения диссоциации кислоты. При этом
Рис. 286. Степень разложения
апатита фосфорной кислотой
прн 20° в зависимости от нормы
кислоты (в % от стехиометри-
ческого количества):
/ — 50; 2-75; 3-90; 4-100; 5-110;
J — 120, 7-130.
Рис. 287. Степень разложения
апатита фосфорной кислотой кон-
центрации 53,6% Р2О5 прй стехио-
метрической норме кислоты и
разных температурах:
/—20; 2-40; 3-50; 4-60; 5-80;
5 — 100.
скорость реакции между ионами водорода и фосфатом с увеличе-
нием температуры возрастает в меньшей мере (или в такой же
степени), чем уменьшение концентрации ионов34. Таким образом,
не только диффузионные, но и кинетические условия осуществле-
ния процесса предопределяют незначительное влияние повышения
температуры на его скорость.
На скорость разложения фосфата фосфорной кислотой, как и
при разложении серной кислотой (см. гл. XXIV), влияют интен-
сивность перемешивания, гранулометрический состав и общая
(внутренняя) поверхность элементарных зерен фосфата.
ПРОИЗВОДСТВО ДВОЙНОГО СУПЕРФОСФАТА
Производство двойного суперфосфата осуществляется двумя
способами46: 1) камерным (подобно простому суперфосфату) и
2) бескамерным (поточным).
В камерном способе используется концентрированная фос-
форная кислота: термическая или экстракционная (последняя
992
Гл. XXVII. Концентрированные фосфорные удобрения
предварительно выпаривается до 45—55% Р2О5). Схема производ-
ства двойного суперфосфата камерным способом почти тожде-
ственна схеме получения простого суперфосфата. Камерный двой-
ной суперфосфат, так же как и простой, вылеживается и дообра-
батывается на складе.
Бескамерный способ5’47 в настоящее время осуществляется
преимущественно следующим образом. Измельченный легко раз-
лагаемый фосфат обрабатывается экстракционной фосфорной кис-
лотой концентрации 28—39% Р2О5. Жидкую пульпу смешивают с
ретурным продуктом и направляют в сушильный барабан. Высу-
шенные гранулы сортируют на три фракции: мелкую (ретур),
среднюю (товарный продукт) и крупную (поступает на дробление
и снова на рассев). Новейшие заводы двойного суперфосфата по-
строены по бескамерному способу; стоимость производства обоими
методами практически одинакова.
Камерный способ
Для камерного способа с применением экстракционной фосфор-
ной кислоты была определена ее оптимальная концентрация в 45—
50% Р2О5 для разложения фосфоритов и 50—55% для апатито-
вого концентрата®-п>51-63. При разложении фосфоритов применяют
стехиометрическую норму кислоты, а для апатитового концентрата
105—110% от стехиометрической нормы, рассчитанной по первому
иону водорода. На рис. 288 представлена технологическая схема
производства двойного суперфосфата камерным способом с полу-
чением гранулированного продукта.
Апатитовый концентрат смешивается с концентрированной фос-
форной кислотой (например, 54% Р2О5). Образующаяся пульпа
поступает в цилиндрическую камеру непрерывного действия, где
происходит вызревание массы. Вызревший камерный продукт по-
сле дозревания на складе, нейтрализуют, гранулируют и высуши-
вают.
При взаимодействии апатитового концентрата с концентриро-
ванной фосфорной кислотой в процессе вызревания в камере обра-
зуется очень твердая, монолитная масса; ее выгрузка из камеры и
дообработка на складе вызывает большие затруднения. Поэтому
перед подачей пульпы из смесителя в суперфосфатную камеру к
ней добавляют небольшое количество (до 3% от веса апатита)
молотого известняка11’59. Вследствие выделения из известняка
двуокиси углерода схватившаяся в камере суперфосфатная масса
(«пирог») обладает большей рыхлостью и пористостью и легче
выгружается с помощью фрезера.
Размягчению двойного суперфосфата способствуют некоторые
неионогенные вещества (например, алкилфениловый эфир поли-
П роизводство двойного суперфосфата
993
этиленгликоля и другие),
введенные в очень малых ко-
личествах (0,1 — 1 кг на 1 г).
Для производства двой-
ного суперфосфата камер-
ным способом применяют не
только цилиндрические вра-
щающиеся камеры, но также
ленточные транспортеры с
бортами 54, 55. Применение
ленточных транспортеров
оказалось возможным вслед-
ствие быстрого затвердева-
ния пульпы. Варианты не-
прерывных схем камерного
способа различаются аппа-
ратурой для смешения фос-
фата с фосфорной кисло-
той. Применяются лопаст-
ные смесители различных
типов с продолжительно-
стью смешения 1,5—3 мин.
В США разработан 4>50’65-76
конический смеситель, где
фосфат попадает в вихревой
поток, образуемый тангенци-
ально поступающей кисло-
той (рис. 289). Благодаря
этому возможно использо-
вать в процессе64 фосфор-
ную кислоту повышенной
концентрации, содержащую
55,5—56% Р2О5. При полу-
чении 30 т суперфосфата в
час время пребывания пуль-
пы в смесителе не превы-
шает 2 сек. Опыты 77’78 раз-
ложения фосфорита в ко-
ническом смесителе супер-
фосфорной кислотой (см.
гл. XXVI) показали возмож-
ность получения двойного су-
перфосфата с содержанием
до 55% усвояемой Р2О5.
При разложении фосфо-
ритов Теннесси и флоридских
994
Гл. XXVII. Концентрированные фосфорные удобрения
Рис. 289. Непрерывно-
действующий конический
смеситель:
/ — сопла для кислоты, соз-
дающие вихревое движение
вдоль стенки смесителя;
2 — желоб для подачи фосфат-
ной муки; 3 — патрубок для
выгрузки суперфосфатной
пульпы.
термической фосфорной кислотой, содержащей до 56,5% Р2О5,
при 38—71° образуется быстро схватывающаяся пульпа, которая
затвердевает на ленточном транспортере за 45 сек.. Лента транспор-
тера (шириной 0,91 м и длиной 11,3 м) движется со скоростью
13 м/мин. При этом производительность установки достигает 40—
42 т суперфосфата в час. При работе по
этой схеме с упаренной экстракционной
фосфорной кислотой (50% Р2О5) затверде-
вание пульпы происходит значительно ме-
дленнее (до 15 мин) -, на ленту вводят часть
сухого порошкообразного суперфосфата,
являющегося подстилкой для пульпы. При
применении суперфосфатных камер про-
должительность пребывания в них пульпы
колеблется в пределах 15—90 мин. Наибо-
лее медленно затвердевает пульпа, полу-
ченная разложением апатитового концент-
рата.
Во Франции осуществлен вариант ка-
мерного способа, по которому смешение
фосфата и выпаренной фосфорной кислоты
производится в небольшом по объему сме-
сителе, снабженном очень быстроходной
мешалкой. Последняя засасывает воздух и
эмульгирует пульпу, что придает схватив-
шемуся продукту рыхлую структуру. За-
тем пульпа поступает на ленточный транс-
портер, на котором она затвердевает.
Производство двойного суперфосфата
камерным способом во всех случаях связа-
но с необходимостью окончательного до-
разложения фосфата и дообработки, охла-
ждения и перелопачивания продукта на
складе в течение 2—3 недель и более.
Охлаждение двойного суперфосфата (как
и простого) при хранении его на складе способствует доразложе-
нию апатита 34. При хранении двойного суперфосфата, полученного
из относительно высококачественных фосфоритов, заметно повы-
шается содержание усвояемой Р2О5, количество же водораствори-
мой Р2О5 не изменяется. Содержание окислов железа в сырье
свыше 7,5—8% по отношению к Р2О5 приводит к ретроградации во-
дорастворимой Р2О5. В продукте из апатитового концентрата при
вызревании увеличивается содержание как усвояемой, так и водо-
растворимой P2O5 6’56"58.
С помощью радиоактивного фосфора изучено64 распределение
Р2О5 в двойном суперфосфате. Найдено, что при применении фос-
Производство двойного суперфосфата
995
форной кислоты с содержанием 54,5% Р2О5 в водной вытяжке су-
перфосфата содержалось 96—98% кислотной Р2О5 и 20—100%
фосфоритной Р2О5 (тем больше, чем выше норма кислоты). Следо-
вательно, при разложении фосфорита лишь часть его переходит в
монокальцийфосфат, остальная же его часть превращается в цит-
ратнорастворимую форму, не содержащую кислотной Р2О5; цит-
ратнорастворимая часть содержит также фосфаты полуторных
окислов, образованные кислотной и фосфоритной Р2О5.
На действующих в СССР предприятиях на производство двой-
ного суперфосфата (в расчете на 1 т усвояемой Р2О5) из апатито-
вого концентрата камерным способом с применением упаренной
экстракционной фосфорной кислоты (включая затраты на произ-
водство кислоты) расходуется 1,13 т Р2О5 фосфата, 1,94 т серной
кислоты (моногидрата), 0,14 т известняка (100%), 0,14 т условного
топлива, 340 кет-ч электроэнергии и 54 ж3 воды.
При использовании термической фосфорной кислоты на 1 т
Р2О5 в двойном суперфосфате расходуется апатитового концен-
трата 0,35 т (в расчете на 100% РгОб) и кислоты 0,775—0,80 т (в
расчете на 100% Р2О5), 0,36 т известняка (100% СаСО3), 0,78т
условного топлива и (с учетом затрат на производство фосфорной
кислоты) ~7000 квт-ч электроэнергии и 148 м3 воды.
Большим недостатком камерного способа, как и в производстве
простого суперфосфата, является необходимость длительного
складского вылеживания и дообработки продукта. Затвердевание
суперфосфатной массы в камере происходит, когда степень разло-
жения апатита достигает уже 60%. Дальнейшее разложение фос-
фата в схватившейся массе протекает весьма медленно и ведется
в течение длительного времени. При этом даже через 30 суток ко-
нечная степень разложения апатита в продукте не превышает
80—85%. Помимо недостаточного использования фосфата, значи-
тельных затрат на сооружение громоздких складов, расхода труда
и энергии на периодическое перемешивание продукта, при склад-
ском дозревании суперфосфата выделяются загрязняющие атмо-
сферу фтористые газы, уловить которые практически невозможно.
Другой недостаток этого способа заключается в необходимости
применения концентрированной фосфорной кислоты. Поэтому при-
ходится предварительно выпаривать экстракционную фосфорную
кислоту или использовать более дорогую термическую кислоту.
Разложение апатитового концентрата при камерном способе
производства двойного суперфосфата можно значительно интенси-
фицировать, заменив небольшую часть фосфорной кислоты (15
экв.%) азотной45 или соляной кислотой. Последние, в отличие от
фосфорной кислоты, сильно диссоциированы и поэтому легко раз-
лагают апатит, например:
Ca5F(PO4)3 + IOHNO3 = 3Ca(NO3)2 + ЗН3РО4 + HF
996
Гл. XXVII. Концентрированные фосфорные удобрения
В системе СаО — Р20Б — М20Б + Н20 (стр. 1318) нитрат каль-
ция стабилен только при небольших температурах и концентра-
циях Р2ОБ. Так, при 25° он стабилен в растворах, содержащих
меньше 36% Р2ОБ, а при 50° — меньше 29%. Поэтому при повы-
шенной температуре, достигаемой в камере, нитрат кальция раз-
лагается фосфорной кислотой по реакции:
Ca(NO3)2 + 2Н3РО4 = Са(Н2РО4)2 + 2HNO3
При этом азотная кислота регенерируется и вновь вступает в
реакцию с апатитом. При разложении апатитового концентрата
фосфорной кислотой с концентрацией 53—55% Р2ОБ с заменой
15 экв.% ее 50%-ной азотной кислотой и при суммарной норме
кислот 105% степень разложения апатита в камерном продукте
достигает 75%, а после 3-суточного вылеживания 97—98% (вместо
80% после 20—30-суточного вылеживания при обычном камерном
получении продукта без добавки азотной кислоты). Примерный
состав продукта после 3-суточного дозревания: общей Р2ОБ—
43,6%, водорастворимой Р2ОБ — 43,3%, свободной Р2ОБ — 3,8%,
N — 0,97%. Содержащийся в продукте азот в форме нитрата каль-
ция является полезным компонентом удобрения. Вследствие высо-
кой степени разложения апатита его расход на 1 т усвояемой Р2ОБ
значительно сокращается, что заметно снижает себестоимость про-
дукта. Для получения двойного суперфосфата этим способом мо-
жно приспособить действующие заводы простого суперфосфата,
если обеспечить их фосфорной кислотой.
Вылежавшийся двойной суперфосфат подвергается измельче-
нию и затем гранулированию одним из способов, применяемых и
для простого суперфосфата.
При аммонизации двойного суперфосфата, полученного из апа-
титового концентрата79, до содержания 13,2% NH3 при 42,8%
Р2ОБусв отношение Р2ОБусв : Р2ОБ общ остается постоянным; оно не
изменяется и при значительно большем количестве вводимого ам-
миака ( количество водорастворимой Р2ОБ уменьшалось от 95 до
68%). Указанное количество аммиака является предельным лишь
с точки зрения стабильности продукта.
На опытной установке TVA (Tennesy Valey Authority, США)
был разработан процесс, объединяющий аммонизацию с гранули-
рованием8-80. Аммонизирующий продукт и кислота вводились под
перекатывающийся слой смеси в непрерывном барабанном аммо-
низаторе. Гранулировались смеси с малым содержанием влаги;
тепло реакций аммонизации использовалось для сушки продукта.
Этим методом производились смешанные удобрения, одно из них
'(12—12—12) содержало в своем составе двойной суперфосфат.
Такой продукт, при условии малого содержания влаги (1,3%), по-
казал очень небольшое превращение Р2ОБ в цитратнонераствори-
мую форму даже после хранения в течение 90 суток при 82°,
П роизводство двойного суперфосфата
997
Бескамерные (поточные) способы
При производстве двойного суперфосфата по так называемым
поточным схемам81-92 фосфат обрабатывают фосфорной кислотой
и полученную пульпу подвергают выпариванию (сушке) в отсут-
ствие или в присутствии ретура готового продукта. Процесс осу-
ществляют в цепи аппаратов непрерывного действия, вплоть до
выпуска гранулированного продукта. В этом случае не требуется
созревания продукта и отпадает надобность в камерах, вследствие
чего метод получил название «бескамерного». Нет нужды и в дли-
тельном складском дозревании продукта.
Возможность применения в бескамерных методах слабой фос-
форной кислоты, содержащей 25% Р2О5, позволяет использовать
экстракционную кислоту и из каратауских и других магнийсодер-
жащих (и загрязненных другими примесями) фосфоритов, которую
трудно выпаривать из-за выделения при этом чрезмерно большого
количества содержащихся в ней примесей. Удаление воды происхо-
дит на стадии сушки продукта. Производство двойного суперфос-
фата бескамерным способом по поточной схеме можно осущест-
вить разложением фосфата стехиометрическим (или несколько
большим) количеством одной фосфорной кислоты или фосфорной
кислотой в присутствии рециркулирующей азотной или соляной
кислоты, или смесью серной и азотной (или соляной) кислот.
Бескамерный фосфорнокислотный способ
(без рецикла)
В настоящее время в промышленности бескамерным способом
получают двойной суперфосфат разложением фосфата фосфор-
ной кислотой. В первоначальном варианте29-58-60 бескамерный
способ пытались осуществить непосредственным нагреванием сме-
си слабой фосфорной кислоты и фосфата во вращающейся печи
(трубокамере). В таком виде он не получил развития из-за малой
степени разложения фосфата и аппаратурных трудностей. В даль-
нейшем он был видоизменен и усовершенствован, что позволило
осуществить производство двойного суперфосфата по непрерывной
поточной схеме. Технологические и аппаратурные условия разли-
чаются в зависимости от концентрации фосфорной кислоты.
На рис. 290 представлена схема производства гранулирован-
ного двойного суперфосфата бескамерным способом с примене-
нием концентрированной (упаренной) экстракционной фосфорной
кислоты. Фосфоритная мука разлагается экстракционной фосфор-
ной кислотой, выпаренной до концентрации 39% Р2О3 в трех сме-
сителях при обогреве острым паром до 80—100°. Полученную
пульпу смешивают в грануляторах с мелкой фракцией готового
продукта (ретур). Гранулятор представляет собой наклонный
998
Гл. XXVII. Концентрированные фосфорные удобрения
стальной корытообразный смеситель с двумя валами, на которые
насажены перемешивающие лопатки. Смесь, выходящая из грану-
лятора, высушивается в прямоточном вращающемся сушильном
барабане поступающими внутрь топочными газами. Продукт на-
гревается до 95—100° (на выходе) и высушивается др содержания
2—3% влаги. Фосфорит в смесителях разлагается на 80—85%.
В сушилке процесс продолжается, и степень разложения фосфата
по выходе из барабана достигает 94%. Перегрев суперфосфата 93
до 105° и выше приводит к все увеличивающейся потере водорас-
творимой и усвояемой Р2О5.
Рис. 290. Схема производства гранулированного двойного суперфосфата
«бескамерным» способом с применением упаренной кислоты (39°/0 Р2О5):
/ — бункер для фосфата; 2 — дозатор фосфата; 3 — смеситель; 4 — грануляторы; 5 — топка;
6 —сушильный барабан: 7 —циклон; 3 —скруббер; 9— молотковая дробилка; 10 — грохот.
При использовании неупаренной экстракционной фосфорной
кислоты схема производства (рис. 291) почти не отличается от
применяемой при работе с упаренной кислотой. Но в этом случае
несколько снижается степень разложения фосфорита при смеше-
нии реагентов и в конечном продукте, и увеличивается количество
ретура, необходимого для образования достаточно сухого и сыпу-
чего материала, не налипающего на стенки транспортных устройств
сушилки. При смешении фосфата с фосфорной кислотой концен-
трацией 28—32% Р2О5 при 50—95° он разлагается на 50—65%
(в зависимости от нормы кислоты и др.) в течение сравнительно
короткого времени. Процесс протекает с образованием незагусте-
вающей пульпы при относительно высокой скорости (стр. 982).
Для достижения максимально возможного разложения фосфата
фосфорной кислотой относительно низкой концентрации применяют
легко разложимые фосфориты (кингисеппский, и др.).
г
1000
Гл. XXVII. Концентрированные фосфорные удобрения
Сушка двойного суперфосфата, применяемая при бескамерном
способе, способствует дальнейшему разложению фосфата. При
удалении влаги состав фосфатного комплекса перемещается по лу-
чам выпарки (совпадающим с лучами степени нейтрализации) в
область, где кристаллизуется моиокальцийфосфат, и степень ней-
трализации жидкой фазы уменьшается (рис. 272). Благодаря
этому увеличивается ак-
тивность раствора, что и
'благоприятствует ускоре-
нию процесса.
В .высушенном продук-
те, полученном с примене-
нием упаренной экстрак-
ционной кислоты, сте-
пень разложения фосфата
достигает 94% (см. вы-
ше); продукт содержит не
больше 2—3% свободной
Р2О5 и не требует нейтра-
лизации. Его рассеивают
на вибрационном грохоте.
Мелкую фракцию и до-
полнительно измельчен-
ную крупную фракцию
направляют в качестве
ретура в грануляторы для
смешения с пульпой. Сред-
нюю фракцию, соответ-
ствующую по крупности
зерна требованиям к продукту, отправляют на склад как готовую
продукцию (рис. 290). При использовании для разложения фосфо-
рита неупаренной экстракционной фосфорной кислоты часть сред-
ней фракции направляется в нейтрализационный барабан
(рис. 291) на нейтрализацию молотым известняком и затем на
склад готовой продукции. Остальная часть средней фракции сме-
шивается с мелкой и с предварительно раздробленной крупной
фракциями и эта смесь используется в качестве ретура.
Недостатком бескамерного способа получения двойного супер-
фосфата является необходимость в большом количестве ретура
(от 3—4 до 8—9-кратного в зависимости от концентрации кисло-
ты). Это приводит к излишним затратам тепла, значительном за-
тратам энергии при эксплуатации и металла при сооружении про-
изводства, а также к усложнению технологического процесса.
Количество ретура можно уменьшить (или вовсе устранить)
высушиванием большей части (около 90%) пульпы в распылитель-
ной сушилке 63’94 в потоке теплого газа (рис. 292). Сухой продукт
Производство двойного суперфосфата
1001
из распылительной сушилки смешивают с остальной частью пульпы
и подают в сушильный барабан.
При высушивании распылением всего количества пульпы про-
дукт подвергают последующему гранулированию, так как он по-
лучается мелкозернистым. Перед гранулированием его смачи-
вают для пластификации небольшим количеством невысушенной
пульпы.
Одним из прогрессивных способов высушивания пульпы яв-
ляется сушка во взвешенном (кипящем) слое с одновременной
грануляцией продукта. Для этой цели при-
меняются аппараты разных конструкций.
На рис. 293 представлена схема аппарата,
не имеющего решетки для поддержания
взвешенного слоя. Его нижняя часть вы-
полнена в виде пневматической форсуний,
через которую поступает пульпа и горя-
чий воздух. Внизу конусной части аппара-
та при больших скоростях воздуха проис-
ходит частичная сушка распыленной пуль-
пы. По мере увеличения сечения аппарата
скорость воздуха уменьшается и находя-
щиеся внутри него гранулы образуют взве-
шенный слой. Подсушенная распыленная
пульпа в виде мелких частиц оседает на
гранулах, размеры которых за счет этого
увеличиваются. В центральной части аппа-
рата образуется фонтанирующий поток
гранул, которые отбрасываются к стенкам
и около них движутся книзу. Вследствие
t Отработанный
воздух
Суспензий
Рис. 293. Схема аппа-
рата для высушивания и
грануляции пульпы во
взвешенном (кипящем)
слое.
отвода части гранул через перелив уста-
навливается стационарный режим, соответ-
ствующий определенной средней величине
гранул (с довольно узким диапазоном раз-
меров) и определенной высоте слоя в аппа-
рате. Скорость формирования гранул регулируют изменением ско-
рости подачи пульпы.
В СССР двойной суперфосфат бескамерным способом произво-
дят разложением кингисеппского фосфорита неупаренной экстрак-
ционной фосфорной кислотой, получаемой из апатитового концен-
трата. Часть образующейся пульпы высушивают в распылитель-
ной сушилке и высушенный продукт в смеси с оставшейся пульпой
гранулируют и нейтрализуют молотым мелом или известняком.
Используемая фосфорная кислота содержит не менее 29,5%
Р2О5, 1,2% СаО, 1,7% SO3 и 2,3% фтора. В качестве фосфорит-
ной муки применяют кингисеппский флотационный концентрат
1002
Гл. XXVII. Концентрированные фосфорные удобрения
(высший сорт), содержащий (согласно МРТУ— 12—12—66) 28%
Р2О5, 43,3% СаО, 2,5% MgO, 3% R2O3, 4,9% СО2, 2,76% фтора и
1,5% Н2О. Тонина помола муки определяется по количеству остат-
ка на сите с отверстиями 0,18 мм, которого должно быть не более
20%.
Двойной суперфосфат выпускают, согласно МРТУ—Б—08—
25—66, трех сортов, различающихся по минимальному содержа-
нию усвояемой P20s’. в первом сорте — 46%, втором — 44% и в
третьем — 42%. Остальные показатели одинаковые для всех сор-
тов. Так, отношение водорастворимой и усвояемой P20s состав-
ляет 90%, свободная кислотность 5% Р2О5, содержание поверхно-
стной свободной кислоты 2,5% P20s, прочность гранул 97%. По
своему гранулометрическому составу продукт должен состоять из
частиц размером от 1 до 4 мм не меньше, чем на 90%, частиц с
размерами менее 1 мм — 5% и частиц с размерами более 4 мм —
5%.
На производство двойного суперфосфата (в расчете на 1 т
100% Р2О5) расходуется: 0,87 т фосфорной кислоты (100% Р2О5),
0,283 т фосфоритной муки (100% Р20б), 0,064% т молотого мела
(100 %СаО), 0,242 т условного топлива, 16,7 м3 воды, 191 квт^ч
электроэнергии, 470 м3 сжатого воздуха.
Применение описанного бескамерного способа производства
двойного суперфосфата имеет ряд ограничений. Это обусловлено
необходимостью использования в процессе легко разложимых
фосфатов (фосфоритов) и относительно чистой фосфорной кис-
лоты небольшой концентрации. Таким образом, бескамерный
способ в неизмененном виде не может быть осуществлен с приме-
нением апатитового концентрата, концентрированной фосфорной
кислоты (что позволило бы уменьшить затраты на сушку пульпы) и
загрязненной фосфорной кислоты, полученной из бедных фосфори-
тов Каратау, кингисеппских и других.
Для достижения достаточно высокой степени разложения
трудноразложимого апатитового концентрата предложено37’83
добавлять к пульпе из апатита и фосфорной кислоты (32—38%’
Р20в) немного серной кислоты (93% H2SO4). Последняя осаж-
дает часть кальция в виде сульфата кальция, степень нейтрали-
зации раствора снижается, и разложение апатита улучшается.
Затем пульпу смешивают с ретуром (гранулируют) и высуши-
вают. Продукт содержит 42—43% усвояемой Р2О5.
Непосредственное использование в бескамерном способе труд-
но разложимого апатитового концентрата связано с увеличением
концентрации и количества фосфорной кислоты (сверхстехиомет-
рического) и необходимостью возврата (рециркуляции) или ней-
трализации избытка ее. Применение стехиометрического коли-
чества концентрированной фосфорной кислоты (экстракционной
53,6% Р2О5 или термической 56% Р2О5) возможно в бескамерном
Производство двойного суперфосфата
1005
способе, если разложение фосфата фосфорной кислотой и грану-
лирование произвести одновременно во вращающемся барабан-
ном смесителе-грануляторе95, но полученный продукт требует
вызревания. Достигнутая степень разложения галечного флорид-
ского фосфорита после суточного вылеживания составила
89—90%, которая возросла в течение двух недель до 93—96%.
При наличии дешевой фосфорной кислоты двойной суперфос-
фат (или продукт типа двойного суперфосфата) может быть по-
лучен аналогичным образом нейтрализацией кислоты известня-
ком и высушиванием образующейся пульпы.
Фосфорную кислоту, полученную из фосфорита Каратау и со-
держащую 22—32% общей Р2О5 при 18—25% свободной Р2О5,
смешивали с сухим измельченным известняком96. Количество
известняка составляло 120—130% от нормы, рассчитанной на
нейтрализацию свободной Р2О5 до монокальцийфосфата. Смесь
высушивали вместе с ретурным сухим продуктом и рассеивали.
В полученной товарной фракции — продукте содержалось
45—47% усвояемой РгО5 в том числе около 35% в водораствори-
мой форме.
Предложено получать двойной суперфосфат бескамерным спо-
собом через преципитатную пульпу. Первоначально этот метод
был изучен97 применительно к получению удобрения типа двой-
ного суперфосфата из отработанных растворов фосфорной кис-
лоты, содержащих 16% Р2О5 и много примесей. В дальнейшем
он был разработан в лабораторных условиях как самостоятель-
ный способ переработки некоторых бедных фосфоритов в концен-
трированные фосфорные удобрения981". Из части фосфорной
кислоты, полученной сернокислотным разложением фосфорита,
осаждают преципитат. После отстаивания или фильтрации пуль-
пы преципитат обрабатывают второй частью фосфорной кислоты.
При этом дикальцийфосфат переходит в монокальцийфосфат.
Конверсия дикальцийфосфата в монокальцийфосфат осуществ-
ляется без нагревания. Пульпу монокальцийфосфата высушивают.
Так как для преципитирования можно использовать слабые рас-
творы фосфорной кислоты, существенно облегчается водный баланс
на стадии отмывки фосфогипса.
Из фосфорита Каратау, содержащего 25,65% Р2О5, 40,4% СаО,
3,2% MgO, 3,0% R2O3 2,6% фтора, 8,45% СО2, в лаборатории был
получен двойной суперфосфат следующего состава: 55—56% об-
щей Р2О5, 50—54% усвояемой Р2О5, 48—55% водорастворимой
Р2О5. Аналогичный продукт получен при переработке кингисепп-
ского фосфорита, содержавшего 25,7% Р2О5, 2,0% MgO, 3,3%
R2O3. Недостатком способа является повышенный расход серной
кислоты. Практически расход серной кислоты на 1 т Р2О5 в двой-
ном суперфосфате будет таким же, как на 1 т Р2О5 в экстракцион-
ной фосфорной кислоте.
1004
Гл. XXVII. Концентрированные фосфорные удобрения
Циклические методы получения двойного суперфосфата
бескамерным способом
Получение двойного суперфосфата из апатитового концентрата
циклическим бескамерным способом основано на разложении апа-
тита и кристаллизации монокальцийфосфата в незагустевающей
пульпе. Растворение апатита производят или в избытке концен-
трированной (термической) фосфорной кислоты или стехиометри-
ческим количеством фосфорной кислоты в присутствии азотной
(или соляной) кислоты. В первом случае в процессе рециркули-
рует маточный раствор, т. е. избыточная фосфорная кислота, насы-
щенная монокальцийфосфатом; во втором случае — избыточная
азотная (или соляная) кислота. В обоих случаях выделяющийся
в твердую фазу монокальцийфосфат отделяют от жидкой фазы,
нейтрализуют содержащуюся в нем свободную кислоту. После ней-
трализации его гранулируют и высушивают. Полученный продукт
представляет собой почти чистый одноводный (или с некоторой
примесью безводного) монокальцийфосфат и содержит до 55—57%
Р2О5, т. е. примерно в 3 раза больше, чем в простом суперфосфате,
поэтому его называют также тройным суперфосфатом.
Получение монокальцийфосфата разложением
апатитового концентрата термической
фосфорной кислотой
Получение двойного суперфосфата бескамерным способом с
циркуляцией маточного раствора фосфорной кислоты, насыщенной
монокальцийфосфатом, основывается на его кристаллизации из
пересыщенных растворов (стр. 982). Апатитовый концентрат, тер-
мическая фосфорная кислота концентрации 53—55% Р2О5 и цир-
кулирующий маточный раствор поступают в первый реактор. Ве-
совое соотношение между жидкими материалами и твердым (апа-
титом) составляет ~7—8: 1, а к концу процесса при образовании
монокальцийфосфата отношение Ж:Т равно ~2,5—3,0; 1. Прита-
ком отношении масса не схватывается. Это обеспечивает
разложение апатита и кристаллизацию монокальцийфосфата при
60—90° в незагустевающей пульпе с относительно большой ско-
ростью. К концу разложения (через 1,5—2 ч), достигаемому во
втором реакторе, пульпа состоит из фосфорной кислоты, насыщен-
ной монокальцийфосфатом, кристаллов одноводного монокальций-
фосфата и незначительного количества непрореагировавшего фос-
фата. Пульпу направляют на отстаивание. Сгущенную массу раз-
деляют фильтрацией. Полученный твердый монокальцийфосфат —
сырой двойной суперфосфат перерабатывают гранулированием в
конечный продукт. Маточный раствор с фильтра вместе со сливом
из сгустителя возвращают в реактор.
Производство двойного суперфосфата 1005
Технологический режим разложения апатитового концентрата
термической фосфорной кислотой установлен45-в результате экспе-
риментального его изучения в лабораторных и модельных усло-
виях. С увеличением отношения Ж: Т в пульпе степень разложе-
ния апатита возрастает. При 60° в течение 2 ч с увеличением Ж: Т
по весу от 4: 1 до 8: 1 степень разложения апатита при содержа-
нии в жидкой фазе 45% свободной Р2О5 повышается от 76,7 до
96,1%. С повышением температуры от 50 до 70° степень разложе-
ния апатита за 2 ч при весовом отношении Ж : Т, равном ~6 : 1,
увеличивается от 76,8 до 96,1 %.
Двойной суперфосфат, полученный разложением апатитового-
концентрата термической фосфорной кислотой в незагустевающей
пульпе, содержит 53—54% общей P2Os, 52,5—53% усвояемой Р2О5
и 52—52,5% водорастворимой Р2О5.
В сыром не нейтрализованном продукте содержится 18—20%
свободной Р2О5 при влажности его 25—40%. Выделяющиеся кри-
сталлы одноводного монокальцийфосфата имеют длину 120—
140 мк и ширину 60—80 мк. Увеличение отношения Ж:Т в пульпе
или повышение температуры способствуют увеличению размеров
кристаллов. Отделение монокальцийфосфата от маточного рас-
твора не представляет трудностей.
Другой способ получения монокальцийфосфата из апатитового
концентрата и термической фосфорной кислоты 100’101 заключается
в следующем. Разложение апатита осуществляют при 100—115°
смесью фосфорной кислоты и оборотного маточного раствора с
образованием насыщенного или немного недонасыщенного рас-
твора, т. е. без выделения монокальцийфосфата. Кристаллиза-
ция последнего протекает при некотором упаривании полученного
раствора и последующем его охлаждении. Выделившийся мо-
нокальцийфосфат отделяют от раствора и обрабатывают до
конечного продукта. Маточный раствор возвращают в про-
цесс.
Установлено 45, что оптимальные результаты достигаются если
разложение вести при 115° с образованием почти насыщенного рас-
твора, содержащего 47% Р2О5 и 4,7 СаО (степень нейтрализации
раствора равна ~25%), а упаренный раствор охладить до 40°.
При этом концентрация исходной фосфорной кислоты составляет
33,5% Р2О5, конечного раствора после упарки — 55,3% Р2О5 и
5,4% СаО, а маточного раствора после охлаждения — 55,0% Р2О5
и 0,5% СаО. Весовое отношение Ж'.Т в пульпе после упаривания
3,4: 1. Процесс осуществляется при использовании Р2О5 (от све-
жей кислоты и циркулирующего маточного раствора) в количестве-
574 % от стехиометрической нормы. На 1 т сухого продукта рас-
ходуется 429 кг апатита и 1588 кг исходной фосфорной кислоты
(33,5% Р2О5), циркулирует 3130 кг маточного раствора и испа-
ряется 767 кг воды.
1006
Гл. XXVII. Концентрированные фосфорные удобрения
Получение двойного суперфосфата разложением
апатита смесью фосфорной и циркулирующей
азотной (илисолянойилидругойлетучейкислоты)
Выше (стр. 995) была указана возможность интенсификации
производства двойного суперфосфата камерным способом при ча-
стичной замене фосфорной кислоты азотной (или соляной) кисло-
той. Этот же прием еще с большей эффективностью (в отношении
полноты использования сырья и скорости процесса) может быть
применен и для производства двойного суперфосфата бескамерным
способом. Процесс основан на переработке (высушивании) рас-
твора, содержащего эквивалентные количества фосфорной кислоты
и кальциевой соли летучей кислоты, необходимые для образования
монокальцийфосфата, например:
2Н3РО4 + Ca(NO3)2 = Са(Н2РО4)2 + 2HNO3
2Н3РО4 + СаС12 = Са(Н2РО4)2 + 2НС1
Помимо азотной или соляной кислоты могут быть использованы
другие летучие кислоты, в частности, фтористоводородная и крем-
нефтористоводородная кислоты.
При перемешивании апатита, экстракционной фосфорной и азот-
ной (или другой летучей) кислоты образуется раствор, содержа-
щий эквивалентные количества фосфорной кислоты и соответствую-
щей кальциевой соли. При высушивании его с ретуром готового
продукта (монокальцийфосфатом) азотная (или другая летучая)
кислота регенерируется. Ее пары улавливаются в абсорбере пуль-
пой из апатита и фосфорной кислоты, причем одновременно идет
разложение апатита. Этим методом получается из апатитового
концентрата и невыпаренной экстракционной фосфорной кислоты
продукт, содержащий до 55—60% Р2О5 (в зависимости от условий
сушки) при высокой степени использования сырья. Процесс изу-
чен в лабораторных условиях с применением соляной7, азотной43
и других летучих кислот (см. ниже). Технологическая схема про-
цесса мало зависит от применяемой циркулирующей кислоты.
На рис. 294 представлена схема получения гранулированного
двойного суперфосфата из апатитового концентрата и невыпарен-
ной экстракционной фосфорной кислотой с циркуляцией азотной
кислоты. Потери азотной кислоты можно компенсировать, добав-
ляя ее в смеситель. Вместо этого можно вводить туда нитрат нат-
рия, который реагирует с образующейся в процессе разложения
апатита кремнефтористоводородной кислотой. При этом выде-
ляется плохорастворимый кремнефторид натрия и некоторое коли-
чество азотной кислоты, достаточное для компенсации потерь. При
использовании кремнефтористоводородной кислоты в качестве цир-
Производство двойного суперфосфата
1007
кулирующего реагента 102’103 не требуется введения этой кислоты
со стороны, так как производственные потери компенсируются за
счет фтора, содержащегося в сырье. Сущность процесса заклю-
чается в образовании при экстракции, наряду с фосфорной кисло-
той, также кремнефторида кальция, из которого затем регенери-
руется кремнефтористоводородная кислота. Разложение апатита
протекает при 70—80° по реакции:
Ca5(PO4)3F + 5H2SiF6 = 5CaSiF6 + ЗН3РО4 + HF
Рис. 294. Схема производства гранулированного двойного суперфосфата из апа-
титового концентрата и фосфорной кислоты бескамерным способом с циркуля-
цией азотной кислоты:
1 — напорный бак для пульпы; 2 —бункер для ретура; 3 и 8 — дозаторы; 4 —грохот; 5 —дро-
балка; 8—абсорбер азотной кислоты; 7 —бункер; Р—насос для пульпы; 10 — смеситель-сборник
пульпы; и — никлон-пылеуловитель; 12 — дробилка; 13 — сушильный барабан; 14 — смеситель-
граиулятор.
При этом около 70% образовавшегося кремнефторида кальция
выделяется в твердую фазу в виде CaSiF6 • 2Н2О, которую отфиль-
тровывают. Остающийся раствор содержит кремнефторид кальция
и фосфорную кислоту в эквивалентных количествах, необходимых
для образования монокальцийфосфата.
При обезвоживании этого раствора образуется монокальций-
фосфат, содержащий около 10% свободной Р2О5:
ЗН3РО4 + l,5CaSIF6= 1,5Са(Н2РО4)2 + 1.5SiF4+3HF
1008
Гл. XXVII. Концентрированные фосфорные удобрения
Отделенный от раствора осадок кремнефторида разлагают при
900—1000° в присутствии кремнезема и водяного пара (в две или
три стадии):
CaSiF6 + 2Н2О = СаО + SiF4 + 2HF + Н2О
В отличие от отхода — фосфогипса, получаемого при сернокис-
лотной экстракции фосфатов, окись кальция может быть исполь-
зована как побочный продукт в ряде производств.
Выделившуюся при разложении кремнефторида и при обезво-
живании раствора эквимолекулярную смесь SiF4 и HF поглощают
в скрубберах водой. Абсорбция фтористых газов может быть осу-
ществлена в аппарате для разложения апатита, т. е. совмещена с
процессом экстракции. Образующаяся при поглощении фтористых
газов кремнефтористоводородная кислота возвращается в процесс.
Однако в промышленности этот способ не применяют из-за
ряда технических трудностей, связанных с циркуляцией больших
потоков агрессивных веществ и необходимостью разложения крем-
нефторида кальция при высокой температуре 104.
Получение двойного суперфосфата циклическим методом мо-
жет' быть также осуществлено с применением циркулирующей
фтористоводородной кислоты 105. При разложении апатита смесью
фосфорной и плавиковой кислот образуется пульпа из фосфорной
кислоты и фторида кальция:
Cas(PO4)3F + 7Н3РО4 + 9HF = 5CaF2 + ЮН3РО4
После смешения реагентов в течение ~30 мин суспензию вы-
сушивают при 220—240° с получением монокальцийфосфата:
CaF2 + 2Н3РО4 = Са(Н2РО4)2 + 2HF
Выделившийся фтористый водород, содержащий некоторую
примесь четырехфтористого кремния, абсорбируют водой и возвра-
щают в процесс. Полученный безводный монокальцийфосфат со-
держит 57—58% общей Р2О5, в том числе 97—98% в водораство-
римой форме. Повышением температуры сушки до 280° может
быть получен кислый пирофосфат кальция (СаН2Р2О7).
Получение двойного суперфосфата с циркуляцией азотной кис-
лоты, по-видимому, целесообразно осуществлять комбинированием
процесса с экстракцией фосфорной кислоты из фосфатов. Напри-
мер, при обработке апатитового концентрата смесью серной и
азотной кислот в соответствующем отношении можно получить
эквимолекулярный раствор фосфорной кислоты и нитрата
кальция:
Ca5(PO4)3F + 3,5H2SO4 + 3HNO3 = 3,5CaSO4 + 1,5Ca(NO3)2 + 3H3PO4 + HF
Некоторые технико-экономические данные
1009
Непосредственная обработка апатита смесью концентрирован-
ной серной и 50%-ной азотной кислоты приводит к образованию
густой трудно фильтрующейся суспензии. Разработаны условия 106
получения крупных хорошо фильтрующих кристаллов сульфата
кальция при осуществлении процесса с возвратом на разложение
части получаемого фильтрата. Если отделить осадок сульфата
кальция, а оставшийся раствор высушить с небольшим количе-
ством ретура (1,25—1,5 вес. ч. на 1 вес. ч. раствора), то получится
гранулированный двойной суперфосфат, содержащий 58—61% во-
дорастворимой Р2О5, в том числе 4—5% свободной. При сушке
азотная кислота полностью регенерируется и возвращается в про-
цесс, так как давление пара в системе
Ca(NO3)2 + 2Н3РО4 = Са(Н2РО4)2 + 2HNO3
уже при 107° равно атмосферному. Преимущество этого способа
заключается в быстром и практически полном разложении апати-
тового концентрата, а, следовательно, в уменьшении расхода сырья
и снижении себестоимости продукта.
Аналогичный процесс исследован с применением циркулирую-
щей соляной кислоты 107~109. Фосфат разлагается смесью серной и
соляной кислот. Образовавшийся раствор после отделения гипса
нагревается с выделением монокальцийфосфата и хлористого во-
дорода, который поглощается водой и возвращается в процесс.
НЕКОТОРЫЕ ТЕХНИКО-ЭКОНОМИЧЕСКИЕ ДАННЫЕ
При получении двойного суперфосфата камерным или поточ-
ным способом расход фосфата и экстракционной или термической
фосфорной кислоты (в пересчете на Р2О5) колеблется в пределах
1,07—1,15.
Расход серной кислоты в производстве экстракционной фос-
форной кислоты (см. гл. XXV) зависит от качества сырья. Практи-
чески, в большинстве случаев, расход моногидрата серной кислоты
на 1 т усвояемой Р2О5 в двойном суперфосфате составляет около
2 т. Значительно выше расход серной кислоты (2,6 т) при перера-
ботке необогащенных фосфоритов Каратау. Обогащение этого сырья
позволит резко снизить расход серной кислоты.
Стоимость фосфорной кислоты во всех случаях является основ-
ной составляющей в себестоимости двойного суперфосфата4-110.
Большая стоимость электроэнергии, расходуемой в производстве
термической фосфорной кислоты, определяет значительно более
высокую стоимость 1 т усвояемой Р2О5 в полученном из нее двой-
ном суперфосфате.
Экономическая эффективность двойного суперфосфата (по
сравнению с простым суперфосфатом) определяется более высо-
кой концентрацией усвояемой Р2О5111-115. Заводская себестоимость
1010 Гл. XXVII. Концентрированные фосфорные удобрения
1 т усвояемой Р2О5 в двойном суперфосфате из апатитового кон-
центрата в одинаковых условиях выше, чем в простом гранули-
рованном суперфосфате. Однако если учесть издержки на транс-
портирование удобрения и внесение его в почву, изготовление
двойного суперфосфата целесообразнее при протяженности желез-
нодорожных перевозок более 1000—1200 км, независимо от протя-
женности последующих автоперевозок. При перевозках по железной
дороге менее чем на 800—1000 км более целесообразно выпускать
простой суперфосфат113. Низкие качественные показатели в произ-
водстве простого суперфосфата из необогащенных фосфоритов
Каратау делают экономически целесообразным получение из
этого сырья двойного суперфосфата (вместо простого) во всех
случаях.
Удельная доля основных составляющих калькуляции себестои-
мости простого и двойного суперфосфата характеризуется следую-
щими показателями113:
В простом супер-
фосфате, °/о
Стоимость сырья.......... 84,0
Стоимость переработки , . 16,0
В двойном супер-
фосфате, °/о
69,5
30,5
Сравнительно высокая доля стоимости переработки в произ-
водстве двойного суперфосфата указывает на необходимость упро-
щения технологических схем, уменьшения энергетических затрат и
автоматизации производства.
ОБОГАЩЕННЫЙ СУПЕРФОСФАТ
Обогащенный суперфосфат выпускают в отдельных случаях в
цехах простого суперфосфата, стремясь облагородить выпускае-
мый продукт; с этой целью часть серной кислоты заменяют фос-
форной. Однако его производят в весьма ограниченных количест-
вах, предпочитая расходовать фосфорную кислоту для получения
более концентрированного двойного суперфосфата.
При использовании неупаренной экстракционной фосфорной
кислоты, содержащей 30—32% Р2О5, и башенной серной кислоты,
(75—76% H2SO4) получают обогащенный суперфосфат из апати-
тового концентрата при замене 10—16% (по весу) серной кислоты
фосфорной. Суммарная норма кислот составляет 71—73 вес. ч.
на 100 вес. ч. апатита, а содержание воды в смеси находится в
пределах 34—38%. После складского дозревания и обычной до-
обработки экспедиционный продукт содержит 23—24% усвояе-
мой Р2О5.
По своим физико-химическим особенностям производство обо-
гащенного суперфосфата, осуществляемое взаимодействием фос-
фата со смесью серной и фосфорной кислот, аналогично получе-
нию простого суперфосфата. Соотношение между кислотами дол-
Обогащенный суперфосфат
1011
жно быть таким, чтобы обеспечивались схватывание пульпы и вы-
сокая скорость разложения фосфата. Исследования получения
обогащенного суперфосфата, начатые еще в двадцатых годах в
СССР 116’117 и в США58, возобновились в последнее время2’8'118’119.
Большинство из них направлено к установлению условий изготов-
ления обогащенного суперфосфата в обычной аппаратуре заводов
простого суперфосфата. Указывают118’119, что из флоридского фос-
форита может быть получен обогащенный суперфосфат с примене-
нием смеси кислот в относительно большом диапазоне их соотно-
шений. Для получения схватившейся массы необходимо регули-
рование содержания воды в системе и температуры в более узких
пределах, чем это допустимо в производстве простого суперфос-
фата. В то же время отмечают 12°, что, регулируя продолжитель-
ность смешения марроканского фосфорита с серной и фосфорной
кислотами, можно получить обогащенный суперфосфат при любом
соотношении кислот. Это однако невозможно при использовании
Кольского апатитового концентрата.
Экспериментально изучены 121 составы смесей в системе
H2SO4—Н3РО4—Н2О, образующие с апатитовым концентратом
схватывающиеся пульпы. Образование загустевающей пульпы
происходит при тем большем содержании воды в системе, чем
больше отношение в ней содержания серной и фосфорной кис-
лот и зависит от степени взаимодействия реагентов. В табл. 80
ТАБЛИЦА ’0
Степень разложения апатита смесью серной и фосфорной кислот
разного состава через 1 ч после смешения
Весовое отношение H2SO4 : Н3РО4 (разрез на рис. 295) Состав исходной кислоты, °/о Содержание Р2О5 в жид- кой фазе к концу 1 стадии, °/0 Степень разложения апатита, %
н2о H2SO4 H3PO4 [н+]
4,5 : 1 11,6 72,3 16,1 1,64 61,0 43,9 *
(разрез I) 25,4 61,0 13,6 1,38 53,5 54,5*
27,8 59,0 13,2 1,34 44,6 49,5*
33,4 54,5 12,1 1,50 42,3 73,8
2: 1 15,2 56,5 28,3 1,44 58,7 38,8*
(разрез II) 20,2 53,0 26,8 1,36 54,4 38,0*
. 29,5 46,6 23,9 1,19 46,1 69,0
32,0 45,2 22,8 1,16 44,6 69,5
1,17:1 18,0 44,1 37,9 1,29 56,5 30,0*
(разрез III) 25,9 39,9 34,2 1,66 50,5 62,1
31,7 37,0 31,3 1,08 46,2 65,2
38,4 33,2 28,4 0,97 40,2 62,5
0,75: I 20,5 34,1 45,4 1,17 55,7 45,0*
(разрез IV) 27,2 31,0 41,5 1,07 49,5 60,1
33,2 28.8 38,0 0,98 45,6 61,9
* Масса не схватилась.
1012
Гл. XXVII. Концентрированные фосфорные удобрения
приведены значения степени разложения апатита в свежем супер-
фосфате (через 1ч после смешения реагентов), полученном с ис-
пользованием смесей с разным отношением серной и фосфорной
кислот при норме смеси кислот, равной 110% от стехиометрической.
Как видно, степень разложения апатита смесью кислот в ка-
мерном процессе, зависящая от концентрации серной' и фосфор-
ной кислот, определяется в основном активностью среды и кри-
сталлизацией твердой фазы. На рис. 295 показаны наименьшие
н2о
H2SO4 I II Ш IV V VI Н3РО«
Рис. 295. Область концентраций растворов системы
H2SO4—Н3РО4—Н2О (/—2), образующих с апатитовым
концентратом схватывающиеся пульпы.
(кривая 1) и наибольшие (кривая 2) концентрации растворов в
смеси в зависимости от соотношения H2SO4: Н3РО4, при которых
происходит схватывание реакционной массы. При чрезмерно ма-
лом (15—20%) и чрезмерно большом содержании (выше 55%) сер-
ной кислоты в первоначальной смеси кислот степень разложения
апатита уменьшается вследствие уменьшения активности ионов
водорода в первом случае и неблагоприятных условий для кри-
сталлизации продукта реакции (сульфата кальция) — во втором.
Область образования загустевающей пульпы с достаточно высо-
кой степенью разложения апатита при любых соотношениях кис-
лот, использованных в опытах, характеризуется концентрацией
ионов водорода в пределах 0,9—1,2 вес.%.
Литература
1013
Концентрация в системе воды и фосфорной кислоты также
обусловливает возможность образования схватывающейся пульпы.
Содержание воды в системе является одним из критериев схваты-
вания массы, полученной с применением той или иной смеси кис-
лот. Допустимые пределы содержания воды, в которых происхо-
дит схватывание пульпы, изменяются в зависимости от соотноше-
ния H2SO4 и Н3РО4. При отношении этих кислот, близком к 1 : 1
(по весу), схватывание массы происходит при содержании в си-
стеме воды от ~24 до 43%; при больших и меньших соотношениях
допустимое содержание воды (рис. 295) изменяется в сторону
уменьшения (на величину от 16 до 7 абс.%).
Применение для разложения апатита смеси кислот того или
иного состава приводит к образованию в конце первой стадии
(после израсходования всей серной кислоты) жидкой фазы, со-
держащей разное количество фосфорной кислоты. В табл. 80 обо-
значены знаком * составы кислот, образующих с апатитом несхва-
тывающиеся массы. Образование загустевающей пульпы проис-
ходит лишь при определенной концентрации Р2О5 в жидкой фазе
к концу первой стадии (началу второй стадии процесса). Она
имеет различные значения в зависимости от соотношения серной
и фосфорной кислот, находящихся в исследованных условиях в
диапазоне 40,2—50,5%.
Продукт с хорошими физическими свойствами образуется в
лабораторных условиях при использовании серной кислоты кон-
центрации 75% H2SO4 и фосфорной, содержащей 45% Р2О5, с от-
ношением НгБСХ: Н3РО4 (по весу) от 2,1 до 0,75. При этом степень
разложения в вызревшем суперфосфате через 14 суток составляет
92—93%. Вызревший продукт содержит в зависимости от отно-
шения серной и фосфорной кислот в смеси от 26 до 33% усвояе-
мой P2OS.
ЛИТЕРАТУРА
1. С. К. Воскресенский, ЖПХ, 27, № 7, 699 (1954); Хим. наука и пром.,
№ 2, 129 (1956). — 2. И. Л. Гофман, Хим. пром., № 1, 48 (1956). — 3. G. С. I п-
skeep, W. R. Fort, W. С. Weber, Ind. Eng. Chem., 48, № 10, 1804 (1956).—
4. В. В а г г а м а н, Фосфорная кислота, фосфаты и фосфорные удобрения, Гос-
химиздат, 1957.—5. I. I. Porter, I. Frisken, Farm. Chem., 116, № II, 38, 43,
70 (1953); Chem. Trade J. a. Chem. Eng., 132, № 3426, 249 (1953).— б. С. К. Вос-
кресенский, С. К- Милованова, P. E. Ремен, Труды НИУИФ, в. 153,
1940. — 7. G. L. Bridger, Tennessee Valley Authority, Chem. Eng. Rep., 5
(1949).—8. L. B. Hein, G. C. Hicks, I. C. Sil verberg, L. F. S e a t z, J.
Agr. Food Chem., 4, № 4, 318 (1956).—9. H. E. Пестов, Физико-химические
свойства зернистых и порошкообразных химических продуктов. Изд. АН СССР,
1947. — 10. Н. Е. П е с т о в, Е. И. Па у тки на, сб. «Исследования по прикладной
химии», Изд. АН СССР, 1955, стр. 290.
И. Е. Б. Бруцкус, Д. Л. Ц ы р л и н, Хим. пром., № 7, 394 (1956).—
12. ф. В. Т у р ч и и, Минеральные удобрения и их применение, Изд. АН СССР,
1943. —13. А. В. Соколов, Агрохимия фосфора, Изд. АН СССР, 1950.—
1014
Гл. XXVII. Концентрированные фосфорные удобрения
14. Н. Т. Rogers, R. W. Pearson, L. Е. Е n s m i n g е г, Soil and Fertilizer
Phosphorus, v. IV (Agronomy), 1953. —15. А. В. Соколов, Удобр. и урожай,
№ 2 (1956). — 16. Ф. В. Т у р ч и н, Хим. пром., № 3, 181 (1957). — 17. Д. Л. Ц ы р-
л и н, ЖПХ, 31, № 6, 817 (1958).— 18. С. И. Вольфкович, Усп. хим., 25,
№ 11, 1309 (1956). —19. С. И. Вольфкович, Вопросы геол, агрон. руд, Изд.
АН СССР, 1956, стр. 7. — 20. Statistical Yearbook United Nations, 1956.
21. Г. A. P о г о в с к и x, А. И. Красовская, Минеральные, удобрения,
Рынок капиталистических стран, Виешторгиздат, 1956. — 22. С. И. Вольфко-
вич, Научно-техи. информ, бюлл. НИУИФ, № 4 (1957). — 23. С. К. Воскре-
сенский, Там же, № 1. — 24. П. В. Д ы б и и а, А. А. С о к о л о в с к и й, Сбор-
ник статей ВЗПИ, в. 19—20, 1957, стр. 171.—25. Т. П. У н а и я н ц, Бюлл. техи.
эконом, информ. Всесоюзн. ин-та научн. и техн, информ., № 6, 89 (1958); Удобр.
и урожай, № 9, 53 (1959). — 26. Chem. Ind., № 1, 3 (1958).’—27. Я. И. Иоффе,
Вестиик техн, и экоиом. информ., № 5 (10), 54 (1958). — 28. А. А. С о коло в-
ский, Технология фосфорных удобрений, изд. ВЗПИ, 1954. — 29. М. Л. Чепе-
левецкий, ИСФХА АН СССР, 19, (1949). — 30. К. L. Elmore, Т. D. Farr,
Ind. Eng. Chem., 32, 580 (1940).
31. H. Bassett, J. Chem. Soc., 1958, 9, 2949.— 32. А. В. Русадзе,
С. Я. Шпунт, Сборник аспирантских работ НИУИФ, 1959, стр. 3; Реф. н.-и.
раб. за 1957 г., ч. 1, стр. 7, Труды НИУИФ, в. 163, 1959. — 33. М. Л. Чеиеле-
вецкий, Там же, в. 137, 1937, стр. 10. — 34. К- С. Краснов, ЖПХ, 24, № 11,
1114 (1953); 28, № 12, 1275 (1955); 30, № 1, 25 и № 8, 1121 (1957); 31, № 3,
345 (1958); Изв. вузов. Хим. и хим. технол., № 3, 100 (1958). — 35. М. Л. Че-
пелевецкий, Е. Б. Бруцкус, К- С. Краснов, Е. В. Южная, ЖНХ, 1,
№ 7, 1512 (1956). — 36. Е. В. Южная, Автореф. каид. дисс., НИУИФ, 1956.—
37. Е. Б. Бруцкус, М. Л. Чепелевецкий, Л. И. Коновалова,
А И. Л и цо в а, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 10, Труды НИУИФ, в. 163,
1959.—38. М. Л. Чепелевецкий, Е. Б. Бруцкус, Суперфосфат, Физико-
химические основы производства, Госхимиздат, 1958. — 39. А. А. Со кол о в-
с к и й, Сборник технической информации Гипрохима, № 5, 1959, стр. 3.—
40. М. Е. П о з и н, Б. А. К о п ы л е в, Д. Ф. Жильцова, ЖПХ, 32, № 3, 509;
№ 4, 710; № 10, 2164; № 11, 2377 (1959).
41. Д Ф. Жильцова, Автореф. каяд. дисс., ЛТИ им. Ленсовета, 1959.—
42. М. Е. Поз и и, Б. А. Копылев, Д. Ф. Жильцова, ЖПХ, № 10, 2164
(1959).—43. К Г. Бектуров, С. Г. Трофимова, Изв. АН КазССР, 72,
63 (1949).— 44. М. Е. Поз ин, Б. А. Копылев, И. П. Кыршев, ЖПХ, 36,
№ 6, 1175 (1963). — 45. М. Е. П о з и н, Б. А. К о п ы л е в. Новые методы полу-
чения минеральных удобрений, Госхимиздат, 1962. — 46. Chem. Age, 68, № 1751,
201 (1953).— 47. Н. Н. Meyers, пат. США 1475959, 1923,— 48. Н. В. Игна-
това, А. А. Соколовский, сб. «Исследования по химии и технологии удоб-
рений, пестицидов и солей», Изд. «Наука», 1966, стр. 170. — 49. А. В. Philips,
R. D. J о u n g, J. S. Lewis, F. G. Heil, J. Agr. Food. Chem., 6, № 8, 584
(1958). — 50. H. L. Marshall, L. Rader, K- Jacob, Ind. Eng. Chem., 25,
11, 1253 (1933).
51. Л. E. Берлин и др., Труды НИУИФ, в. 109, 1932; ЖПХ, № 10, 37, 45
(1933). — 52. Р. Ремен, Там же, в. 134, 1936. — 53. И. И. Орлов, ЖПХ, 13,
№ 7, 441 (1936).—54. Н. Постников, И. Орлов, Н. Крючков, А. Бе-
лотелое, Р. Ремен, Труды НИУИФ, в. 143, 1938.—55. С. Воскресен-
ский, С. Милованова, Там же, в. 147, 1940, стр. 28, 29.—56. С. Воскре-
сенский, С. Милованова, Д. Цырлин, Н. Благовещенская,
Там же, в. 147, 1940. — 57. Е. Б. Бруцкус, С. К- Милованова, Сообщение
о научно-технических работах НИУИФ, № 2, 1957, стр. 17. — 58. К- A. Jacob,
Ind. Eng. Chem., № 1, 14 (1931).—59. С. И. Вольфкович, Л. Е. Берлин,
ЖХР, № 14, 1 (1931). — 60. С. И. Вольфкович, Л. Е. Берлин, В. Т. Ми-
керов, Удобр. и урожай, № 11—12, 1079 (1931).
Литература
1013
61. Л. Е. Берлин, Тезисы докладов на отраслевом совещании работников
основной и горнохимической промышленности, 1958, стр. 71. — 62. Л. Е. Бер-
лин, Е М. Абрамова, Т. И. Завертяева, Реф. н.-и. раб. за 1957 г.,
ч. 1, стр. 36, Труды НИУИФ, в. 163, 1959.—63. С. К- Воскресенский, Те-
зисы докладов на отраслевом совещании работников основной и гориохимиче-
ской промышленности, 1958, стр. 65. — 64. Chem. Eng., 58, № 5, 208 (1951).—
65. Н. A. Curtis, Chem. Met. Eng., 42, 488 (1935). — 66. W. L. Hill,
S. B. Hendricks, Ind. Eng. Chem., 28, 440 (1936). — 67. H. A. Curtis a. oth.,
Chem. Met. Eng., 1, 43, 583, Nov.; 2, 43, 647, Dec. (1936). —68. R. H. Newton,
R. L. Cop son, Ind. Eng. Chem., 28, 1182 (1936). — 69. R. L. Cop son,
R. H. Newton, I. D. Lindsay, ibid., 28, 923 (1936). —10. R. L. Cop son
a. oth., Ind. Eng. Chem., 29, 175 (1937).
71. H. L. Marshall, W. L. Hill, ibid, 32, 1128 (1940). —72. G. L. Brid-
ger, R. B. Burt, W. W. Cerf, ibid, 37, 9, 829 (1945). — 73. G. L. Bridger,
R. A. Wilson, R. B. Bert, ibid., 39, № 10, 1265 (1947). —74. G. L. Bridger,
I. W. Markey, ibid, 45, № 2, 418 (1953). — 75. E. J. Fox, H. E. Batson,
A. V. Breen, J. Agr. Food Chem, 2, № 12, 618 (1954). —76. W. L. Hill,
H. E. Batson, W. M. Hoffman, J. Assoc. Offic. Agr. Chem, 38, № 4, 874
(1955). — 77. M. M. S triplin, D. Me Knight, G. H. Me gar, J. Agr. Food
Chem, 6, № 4, 298 (1958). — 78. G. L. Ter men, I. Sil verberg, Farm. Chem,
121, № 6, 27, 31 (1958). — 79. Л. Берлин, Л. Горицкая, ЖХП, № 10, 21
(1933).— 80. W. W. Harwood, Agr. Chem, 14, № 5, 42, 103 (1959).
81. E. L. Lar is on, Ind. Eng. Chem, № 12, 1172 (1929). —82. И. Гоф-
ман, Я- Зильберман, И. Островский, Технология суперфосфата и
других фосфорных удобрений, Химтеорет, 1937. — 83. А. И. Шерешевский,
А. А. Соколовский, П. Ф. Деревицкий, Производство фосфорных удоб-
рений, ГОНТИ, 1938. — 84. А. И. Дубовицкий, А. И. Шерешевский,
Технология минеральных удобрений, Госхимиздат, 1947. — 85. Р. Parrish,
A. Ogilvie, Calcium Superphosphate and Compound Fertilizers, 1946. — 86. Ind.
Chem, 27, № 317, 256 (1951). —87. Chem. Age, 64, № 1660, 701 (1951); Chem.
Trade J, 128, № 3335, 1017 (1951); Engineering, 171, № 4449, 526; № 4450, 560;
№ 4451, 592 (1951). —88. P. A. Retzke, G. F. S a c h s e 1, R. B. Filbert, Agr.
Chem, 9, № 5, 37, 115, 117, 119 (1954). —89. Farm. Chem, 116, № 3, 49 (1953)';
Ind. Chem, 30, № 357, 488 (1954). — 90. R. I. McNally, Mining Eng., 8, № 10,
1017 (1956).
91. M. E. П о з и и, Технология минеральных удобрений, Изд. «Химия»,
1965. — 92. Technologia Zwiazkow Fosforowych, PWT, Warszawa, 1958.—
93. E. E. Зуссер, Хим. пром., № 2, 85 (1959). — 94. С. Д. Э венчик,
В. А. Кононов, Бюлл. техн, информ, Гипрохима, № 40—41, 3 (1958).—
95. А- В. Phillips, R. D. Young, I. S. Lewis, F. G. Heil, J. Agr. Food
Chem, 6, Xs 8, 584 (1958). — 96. E. E. Зуссер, Сообщение о научно-техниче-
ских работах НИУИФ, Xs 2, 1957, стр. 13. — 97. М. С. В о ль д штейн,
А. Н. Д о х о л о в а, Вести, техн, и эконом, информ, в. 2, изд. НИИТЭХИМ,
1964. — 98. М. Е. Позин, М. С. Вольдштейн, Д. Ф. Жильцова,
Р. Т. П е т р о в а, * Хим. пром, Хэ 10, 1 (1967). — 99. Д. Ф. Жильцова,
М. Е. П о з и н, Р. Т. Петрова, Ободовые фосфориты как сырье для химиче-
ской промышленности, Труды НТК, Таллин, 1968, стр. 129. —100. А. А. Соко-
ловский, Сборник технической информации Гипрохима, в. 5, 1959, стр. 3. -
101. А. А. Соколовский, Изв. вузов СССР. Хим. и хим. техиол, 6, 3
(1963). — 102. В. А. Кононов, С.Д- Эвенчик, Журн. ВХО, 6, Xs 1, 6 (1961).—
103. К. И. Макарин, Изв. вузов. Хим. и хим. технол. Xs 3, 440 (1963).—
104. С. Я. Шпунт, Хим. пром. Xs 1, 28 (1970). —105. С. Я. Шпунт,
А. Н. Архипова, 3. Л. Ленева, 3. И. Гусева, Хим. и пром. Xs 10
(1965). —106. М. И. Позин, Б. А. Копылев, Г. А. Морозова, ЖПХ, 42,
Xs 6, 1215 (1969). —107. Б. Д. Мельник, Там же, Хэ 1, 5 (1957).—
108. Р. Е. Ремен, Е. И. Петрова, Реф. н.-и. раб. за 1957 г, ч. 1, стр. 48;
8 М. Е. Позин
1016
Литература
Труды НИУИФ, в. 163, 1959. — 109. G. Т. Gad re, J. Sci. Ind. Res., 12, № 4,
174 (1953). — 110. R. E. Bell, W. H. Waggaman, Ind. Eng. Chem., 42, 276,
283 (1950).
111. A. M. Левин, Э. И. Цыпина, Удобр. и урожай, № 6, 26 (1959).—
112. А. М. Левин, Э. И. Цыпина, Я- А. Иоффе, Б. С. Розенберг,
Научно-техн, информ, бюлл. НИУИФ, № 1-2-Р (1957). — 113. В. А. Коионов,
С. Д. Э в е и ч и к, Бюлл. техн, информ. Гипрохима, № 40—41, 40 (1958).—
114. Я- А. Иоффе, Удобр. и урожай, № 7, 6 (1958). — 115. А. С. Шапиро,
Вести, техн, и эконом, информ., № 3 (8), 13 (1958). — 116. С. И. Вольфко-
вич, Труды НИУ, в. 16, 1923. —117. С. И. Вольфкович, В. П. Камзол-
кии и др., Там же, в. 67, 1929. —118. Е. I. Fox, W. L. Hill, Ind. Eng. Chem.,
44, № 7, 1532 (1952). —119. L. D. dates, F. T. N i e 1 s s о n, E. I. Fox,
R. M. Magness, ibid., 45, № 3, 681 (1953). — 120. U. -Lamin, W. Wo from,
Chem. Techn., 17, № 2 (1965).
121. Б. А. Ко пылев, M. В. Жданова, M. E. Поз ин, ЖПХ, 41, № 7,
1402 (1968).
Глава XXVIII
КОНЦЕНТРИРОВАННЫЕ ФОСФОРНЫЕ УДОБРЕНИЯ.
ПРЕЦИПИТАТ(ДИКАЛЬЦИЙФОСФАТ)
Преципитатом называют фосфорное удобрение, содержащее до
40% Р2О5 в цитратнорастворимой форме; основной его компо-
нент— двухводный дикальцийфосфат СаНРО4-2Н2О. Преципитат
получают в виде порошка белого (или кремового) цвета осажде-
нием (преципитированием) из фосфорнокислых растворов извест-
ковым молоком или водной суспензией измельченного известняка
и последующими отфильтровыванием, сушкой и измельчением.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Дикальцийфосфат может существовать в виде монетита
СаНРО4 (триклинные кристаллы с плотностью 2,89 г/см3) и бру-
шита СаНРО4-2Н2О (моноклинные кристаллы с плотностью
2,31 г/см3).
В системе СаО—P2Os—Н2О ниже 36° стабилен брушит, при бо-
лее высокой температуре — монетит1-5. При 40—50° брушит осаж-
дается как метастабильная фаза. При более высокой температуре
выделяется безводная соль6-9.
На рис. 296 показана растворимость 10 метастабильной фазы
СаНРО4-2Н2О и стабильной СаНРО4 при 40°, а на рис. 297 ско-
рость полиморфного превращения СаНРО4 • 2Н2О —> СаНРО4 при
этой же температуре. Метастабильный брушит превращается в
стабильную безводную соль тем скорее, чем выше концентрация
Р2О5 в жидкой фазе. Потеря кристаллизационной воды при фазо-
вом превращении брушита в растворах вызывает заметное (до
50 отн. %) уменьшение количества усвояемой (цитратнораствори-
мой) Р2О5 в продукте 10’ п.
8*
1018 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
При очень малых концентрациях Р2О5 (pH раствора от 6,4 до
8,0 и выше) в твердой фазе системы СаО—Р2О8—Н2О появляются
более основные фосфаты (рис.
298)3,5’12, с чем также связано по-
нижение усвояемости продукта.
Температура и скорость дегид-
ратации брушита при сушке зави-
сит от давления, величины кристал-
Рис. 296. Растворимость
метастабильиой фазы
СаНРО4 • 2Н2О и стабиль-
ной фазы СаНРО4 в системе
СаО—Р2О5—Н2О при 40°.
297. Фазовое превращение
безводную соль
в зависимости от концентра-
Рис.
СаНРО4 • 2Н2О в
при 40°
ции Р2О5 в жидкой фазе, равновес-
ной с двуводной солью.
Концентрация Р2Од; 7 — 18%; 2—16%;
3-14%; 4 — 12%; 5-10%.
лов, способов осаждения
и
т. д., поэтому имеющиеся данные раз-
норечивы 10' 13“16. Под атмосфер-
ным давлением заметное обезво-
живание брушита наблюдается при
нагревании в течение 1 ч до 100°
и выше10. Дегидратация брушита
сушкой снижает содержание усвояе-
мой Р2О5 только на 10 отн. %,
т. е. в меньшей степени, чем при.
фазовом превращении через рас-
твор.
Нагревание дикальцийфосфата до
175° и выше вызывает отщепление
конституционной воды с образова-
нием неусвояемого пирофосфата
кальция 13. Технический преципитат
(кормовой и удобрительный) со-
держит от 35 до 50% Р2О5 общей и от 33 до 48% РгО8
усвояемой (цитратнорастворимой). При переработке фосфоритов
400
300
200
СаНРО4-2НгО
а
о
100
50
30
10
/Саэ(РО4)д
1 .73Са3(РО4)гСа(он),
' i 1
152050 200 400600 1000 1200
СаО, мг/л
1г
Рис. 298. Система
СаО—Р2О6—Н2О при 25° в области
низких концентраций3.
Применение
1019
Каратау преципитат содержит от 3 до 8% водорастворимой
Р2О5.
Преципитат обладает малой гигроскопичностью (при 7,5%
влажности гигроскопическая точка равна 84%, балл по шкале Пе-
стова 1,2)17. Он хорошо рассевается только при очень небольшой
(до 1%) влажности; при влажности 9% преципитат совершенно
не способен рассеваться (табл. 81).
Объемный вес его18 равен 0,86—
0,87 т/л3.
Димагнийфосфат с 2% влаги не ги-
гроскопичен даже при 100%-ной отно-
сительной влажности воздуха, поэтому
его присутствие в преципитате улуч-
шает физические свойства продукта.
ТАБЛИЦА 81
Рассеваемость и угол откоса
преципитата
Влаж- Рассе- ваемость Угол
ность, % по шкале Пестова откоса
1,0 5,9 44° 15'
ПРИМЕНЕНИЕ 5,0 1,5 40°1(У
7,0 1,1 46°5(Х
В качестве удобрения преципитат обладает многими достоинствами 19~23: 9,0 0 47°5(У
высокая концентрация усвояемой P2Os,
одинаковая с суперфосфатом агрохимическая эффективность на
всех типах почв и для всех культур, уменьшение почвенной кис-
лотности при его применении на кислых почвах. Преципитат нейт-
рален, поэтому не разрушает тару. Он пригоден для получения сме-
шанных и сложных удобрений. Сравнительно высокая стоимость
преципитата ограничивает его применение как удобрения. Фосфор-
ную кислоту, получаемую термическим способом или сернокислот-
ной экстракцией фосфатов, выгоднее перерабатывать в двойной
суперфосфат, производство которого проще, не требует затраты из-
вести и громоздких операций для ее приготовления. Стоимость ус-
вояемой Р2О5 в двойном суперфосфате меньше примерно в 2,2 раза,
чем в преципитате. Поэтому преципитат получают обычно лишь из
фосфорнокислых растворов, являющихся отходами других произ-
водств 24-32.
Технический преципитат в различной степени загрязнен приме-
сями фосфатов, магния, железа и алюминия, сульфатами и карбо-
натами кальция, кремнефторидами и др. Фосфаты полуторных
окислов, присутствующие в удобрительном преципитате, примерно
в 2 раза менее эффективны, чем дикальцийфосфат, хотя они и рас-
творяются в цитратном растворе 19. Поэтому необходима очистка
фосфатного сырья от полуторных окислов.
Согласно ГОСТ 1175—41, удобрительный преципитат, получае-
мый на основе сернокислотного разложения природных фосфатов,
должен содержать не менее 31% (в I сорте) и 27% (во II сорте)
цитратнорастворимой Р2О5 и не более 10% влаги; остаток на сите
2 мм не более 1,5%.
1020 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
Преципитат применяют в СССР и за рубежом в небольших ко-
личествах в качестве кормового средства, как минеральную до-
бавку к корму сельскохозяйственных животных24. В кормовом
преципитате должны отсутствовать вредные для здоровья живот-
ных примеси. В частности, содержание мышьяка не должно пре-
вышать 0,001% и фтора —0,2—0,3%.
Мировое производство преципитата 33’34 составляет около
500 тыс. т в год.
МЕТОДЫ ПРОИЗВОДСТВА
Производство преципитата состоит из двух "стадий 36-49: 1 ) по-
лучения раствора, содержащего фосфорную кислоту, и 2) осаж-
дения дикальцийфосфата (преципитирование).
Исходным раствором для получения преципитата служит фос-
форная кислота и различные фосфорнокислотные растворы, напри-
мер, образующиеся в качестве отхода в производстве желатины на
костеобрабатывающих заводах. Перед переработкой органиче-
ского вещества обезжиренных костей в желатин их обрабатывают
2—5 %-ной соляной кислотой для растворения основного вещества
кости — трикальцийфосфата. Образующийся при этом фосфорно-
кислый раствор, содержащий монокальцийфосфат и хлорид каль-
ция, подвергают преципитированию.
Небольшие количества преципитата вырабатывают на некото-
рых суперфосфатных заводах50 из отбросных солянокислых рас-
творов с концентрацией 4—4,5% НС1, получаемых при перера-
ботке кремнефтористоводородной кислоты в кремнефторид натрия
(стр. 1142). Образуется разбавленный фосфорнокислый раствор
Ca5(PO4)3F + 10НС1 = ЗН3РО4 + 5СаС12 + HF
который преципитируют. Содержащийся в растворе хлорид каль-
ция не участвует в образовании осадка дикальцийфосфата, но
высаливает его из раствора. При получении фосфорнокислых рас-
творов из фторсодержащего сырья перед преципитированием из
раствора осаждают кремнефторид натрия. При переработке апати-
тового концентрата целесообразно выделение также фосфатов ред-
ких земель.
Различия в методах осаждения преципитата зависят преиму-'
щественно от концентрации Р2О5 в исходных растворах, от вида
осадителя — суспензии известняка, известкового молока или же по-
следовательно того и другого, — а также от назначения готового
продукта. Схемы осаждения кормового преципитата определяются
составом начальных растворов. Из чистых растворов (из очи-
щенной фосфорной кислоты, полученной электротермическим спо-
собом, или из отходящих растворов производства пищевой жела-
тины) дикальцийфосфат осаждается в одну ступень. Экстракцион-
ную фосфорную кислоту нейтрализуют в две ступени: в первой
Методы производства
1021
осаждается дикальцийфосфат, загрязненный фосфатами полутор-
ных окислов и другими примесями, во второй — более чистый про-
дукт. Удобрительный преципитат осаждают в одну ступень суспен-
зией известняка, известковым молоком или последовательно тем
и другим.
Концентрация РгО5 в исходном растворе определяет необходи-
мость предварительной декантации преципитатной пульпы, воз-
можность прямого ее фильтрования или же непосредственного вы-
сушивания с ретурным продуктом.
Предложены разные способы переработки преципитатной пуль-
пы или смеси фосфорной кислоты с известняком непосредственной
сушкой ее без предварительного фильтрования. Преципитатную
пульпу, полученную 51>52 взаимодействием фосфорной кислоты с
суспензией известняка в количестве 80—90% от стехиометрической
нормы СаО на образование СаНРО4, смешивали с ретуром сухого
продукта до содержания 30—32% влаги и высушивали при темпе-
ратуре не выше 90°. Готовый продукт из обесфторенной кислоты с
концентрацией 22% Р2О5, полученной из фосфорита Каратау, со-
держал около 33% усвояемой Р2О5, в том числе 8% водораствори-
мой Р2О5. Из актюбинского фосфорита подобным методом полу-
чали продукт с содержанием до 42% усвояемой Р2О5, в том числе
12—20% водорастворимой Р2О5.
При непосредственном образовании преципитата из фосфата и
серной кислоты по реакции
Ca5(PO4)3F + 2H2SO4 = 2CaSO4 + ЗСаНРО4 + HF
расход кислоты меньше, чем при получении не только экстракци-
онной кислоты, но и простого суперфосфата. В этом случае стои-
мость единицы усвояемой Р2О5 в преципитате была бы ниже, чем
в других удобрениях. Однако реакция образования преципитата по
указанному уравнению протекает в очень малой степени и практи-
чески в прямом виде неосуществима. В связи с этим предложены
различные циклические методы, позволяющие в конечном итоге
получить преципитат с указанным расходом серной кислоты. В ка-
честве циркулирующих реагентов могут быть использованы как
легко летучие кислоты — азотная или соляная, так и фосфорная
кислота. При этом в большинстве методов исключается также опе-
рация преципитирования и, следовательно, расход извести или из-
вестняка и другие затраты.
Получение преципитата сернокислотным разложением фосфатов
с циркуляцией азотной (или соляной) кислоты аналогично получе-
нию таким методом двойного суперфосфата (стр. 1008). Процесс
может быть представлен53 следующей схемой. Разложение фос-
фата (апатита) осуществляют смесью серной и циркулирующей
азотной кислоты:
Ca3(PO4)3F + 2H2SO4 + 6HNO3 = 2CaSO4 + 3Ca(NO3)2 + 3H3PO4 + HF
1022 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
При этом расход серной кислоты меньше на ~40% (2 моль
вместо 3,5 моль на 1 моль апатита), чем в производстве простого
суперфосфата. После отделения сульфата кальция раствор смеши-
вают с ретуром готового продукта и высушивают. При этом обра-
зуется преципитат, а выделяющиеся пары азотной кислоты конден-
сируются и возвращаются в процесс в виде раствора:
Ca(NO3)2 + Н3РО4 = СаНРО4 + 2HNO3
В лабораторных условиях образование дикальцийфосфата из
раствора, смешанного с ретуром (0,75—1 вес. ч. дикальцийфосфата
на 1 вес. ч. раствора) при 120—150° практически заканчивается в
течение 30 мин. Полученный продукт содержит 1,5—3,5% влаги,
43—50% общей Р2О5, в том числе 40—45% Р2О5, растворимой в
4 %-ном растворе соляной кислоты.
Аналогично при использовании циркулирующей соляной кисло-
ты 54>55 получают вначале раствор с эквимолекулярным содержа-
нием Н3РО4 и СаС12, из которого затем удаляют хлористый водо-
род при температуре около 128°:
Н3РО4 + СаС12 = 2НС1 + СаНРО4
Хлористый водород поглощают водой и образующуюся соля-
ную кислоту возвращают на разложение фосфата. В этом процессе
количество циркулирующей соляной кислоты может быть умень-
шено примерно в 2 раза, если вместо раствора хлорида кальция
в фосфорной кислоте подвергнуть разложению монокальцийхлор-
фосфат — СаСЛНгРС^ или СаС1г • Са(Н2РО4)2 в виде двух- или од-
новодной соли при 225—300° в присутствии водяного пара:
СаС1Н2РО4 = СаНРО4 + НС1
Монокальцийхлорфосфат выделяется в твердую фазу разло-
жением фосфатов при 25° концентрированной соляной кисло-
той 56-6°, содержащей больше 20% НС1:
Ca5(PO4)3F + 7НС1 = ЗСаС1Н2РО4 + 2СаС12 + HF
При этом в маточном растворе остается хлорид кальция и со-
ляная кислота полностью не регенерируется. Осуществление про-
цесса в замкнутом цикле возможно при разложении природного
фосфата циркулирующим газообразным хлористым водородом с
добавкой фосфорной или серной кислоты59 по схемам:
пСаО • Р2О6 + пНС1 + (п - 2)Н3РО4 + ЗН2О = пСаС1Н2РО4 • Н2О
ИЛИ
пСаО • Р2О5 + 2НС1 + (п - 2)H2SO4 + (5 - п)Н2О =
= 2СаС1Н2РО4 • Н2О + (it - 2)CaSO4
где п — молярное отношение СаО : Р2О5 в природном фосфате.
Методы производства
П)?з
Образовавшийся монокальцийхлорфосфат разлагают при на-
гревании, при этом хлористый водород почти полностью регене-
рируется.
В этих способах на превращение фторапатита в дикальций-
фосфат теоретически расходуется в 2,5 раза меньше ионов водо-
рода кислот, чем по обычной схеме, и совсем не расходуется из-
весть и известняк. Необходимость термической обработки при
температурах, превышающих 100°, вероятно, затруднит получение
удобрительного преципитата удовлетворительного качества по со-
держанию цитратнорастворимой Р2О554. Для кормового продукта
это не имеет большого значения.
Более сложным представляется получение дикальцийфосфата
преципитированием фосфорной кислоты природным фосфатом
2Н3РО4 + Ca5(PO4)3F = 5СаНРО4 + HF
или с использованием фосфорной кислоты, образующейся (реге-
нерируемой) при реакции гидролиза монокальцийфосфата:
Са(Н2РО4)2 • Н2О + Н2О = СаНР04 • 2Н2О + Н3РО4
Продукт, содержащий СаНРО4, можно получить путем прямо-
го взаимодействия 61 фосфорита, фосфорной кислоты и воды в ав-
токлаве под давлением 7 ат при 200°.
Для разложения фосфорита фосфорной кислотой, образую-
щейся при гидролизе монокальцийфосфата 62 в дикальцийфосфат63,
последний отделяют фильтрованием от разбавленной фосфорной
кислоты, которую концентрируют и используют для разложения
фосфорита.
В других работах 64-66 фосфорит смешивали с монокальций-
фосфатом и водой или же с фосфорной кислотой и водой (в этом
случае соотношение такое, чтобы только часть фосфорита превра-
щалась в монокальцийфосфат). Смесь нагревали сначала в аппа-
рате с обратным холодильником де температуры около 110° для
ускорения гидролиза монокальцийфосфата, а затем в открытом
аппарате до 185°. Образовавшаяся при этом в результате гидро-
лиза фосфорная кислота концентрировалась и реагировала € фос-
форитом. Источником монокальцийфосфата может служить про-
стой или, лучше, двойной суперфосфат. Для более полного раз-
ложения фосфорита рекомендуется повторение циклов процесса
путем увлажнения полученного первоначально и измельченного
продукта. Содержание усвояемой Р2О5 в продукте достигает 98%
по отношению к общей Р2О5. В зависимости от соотношения меж-
ду фосфоритом и монокальцийфосфатом (или кислотой) усвояе-
мая Р2О5 может частично находиться в водорастворимой форме
за счет неполного использования монокальцийфосфата (или кис-
лоты). Концентрация усвояемой Р2О5 в продукте, полученном с при-
менением фосфорной кислоты, близка к таковой в двойном супер-
1024 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
фосфате. Расход фосфорной кислоты на разложение фосфорита
(на единицу усвояемой Р2О5 в продукте) составляет примерно 3/4
от обычного при производстве двойного суперфосфата.
Предложено 67'68 получать преципитат осаждением его из пуль-
пы, образующейся при сернокислотном разложении фосфатов.
Преципитат отделяется от гипса методом флотации. В лаборатор-
ных условиях этим способом получен продукт с содержанием 30%
усвояемой Р2О5 при степени извлечения до 80%.
По другому, циклическому способу32> 69 преципитат полу-
чается из продукта неполного разложения фосфата серной кисло-
той, из которого выщелачивается водой водорастворимая Р2О5.
Образующийся раствор (13—15% Р2О5) перерабатывается в пре-
ципитат. Нерастворимый остаток после выщелачивания разде-
ляется на ангидрит и неразложенный апатит методом мокрой
классификации. Апатит возвращается на повторное разложение,
а ангидрит используется в производстве серной кислоты и це-
мента.
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ОСАЖДЕНИЯ ДИКАЛЬЦИЙФОСФАТА
(ПРЕЦИПИТИРОВАНИЯ)
Суммарные реакции осаждения дикальцийфосфата из раство-
ра фосфорной кислоты известью или известняком при темпера-
туре ниже 50°:
Н3РО4 +Са(ОН)2 = СаНРО4 • 2Н2О
или
Н3РО4 + СаСО3 + Н2О = СаНРО4 • 2Н2О + СО2
не отражают химизма процесса.
Вначале известь или известняк полностью растворяются, об-
разуя моиокальцийфосфат, например:
Са(ОН)2 + 2Н3РО4 = Са(Н2РО4)2 + 2Н2О
В дальнейшем, по мере нейтрализации фосфорной кислоты,
моиокальцийфосфат инконгруэнтно разлагается в растворе с вы-
делением в осадок дикальцийфосфата:
Са(Н2РО4)2 + 2Н2О = СаНРО4 2Н2О + Н3РО4
Помимо этого моиокальцийфосфат также реагирует с новыми
порциями извести или известняка:
Са(ОН)2 + Са(Н2РО4)2 + 2Н2О = 2[СаНРО4 • 2Н2О]
Избыток извести сверх теоретического весового отношения
СаО : Р2О5 в дикальцийфосфате (равного 0,79 и соответствующе-
го pH раствора 6,3) приводит к разложению дикальцийфосфата:
4СаНРО4 = Са(Н2РО4)2+Са3(РО4)2
Физико-химические основы осаждения дикальцийфосфата
1025
При этом Р2О5 наполовину переходит в трикальцийфосфат,
т е. в плохо усвояемую растениями форму. В присутствии из-
бытка известняка ретроградации преципитата не происходит.
Рис. 29Э. Диаграмма процесса осаждения дикальцийфосфата из фосфорной
кислоты:
ОЕ — стабильная н ML — метастабнльиая ветви изотермы системы СаО—Р2О5 — Н2О
при 40°; кг-ветвь растворимости СаНрО4 в системе СаО—Р2О5—HaSIFg—Н2О при 60°.
Примеси, содержащиеся в фосфорной кислоте, также реагируют
с известью или известняком:
H2SO4 + Са(ОН)2 = CaSO4 • 2Н2О
H2SiF6 + ЗСа(ОН)2 = 3CaF2 + SiO2 + 4Н2О
и др.
В результате этого уменьшается содержание усвояемой Р2О5
в продукте и затрудняется отфильтровывание преципитата из-за
выделяющихся илистых осадков кремневой кислоты и др.
На диаграмме системы СаО—Р2О5—Н2О при 40° (рис. 299)
показаны условия преципитирования фосфорной кислоты кон-
центрацией 20% Р2О5 (точка S') известковым молоком, содержа-
щим 12% СаО (ючка S), и кислоты концентрацией 25% Р2О5
1026 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
(точка R') суспензией известняка с отношением Ж: Т, равным
1,5 : 1, и концентрацией СаО около 22% (точка R). При преципи-
тировании состав системы изменяется вдоль соединительных пря-
мых SS' и RR'.
В равновесных условиях, при нейтрализации кислоты до обра-
зования насыщенного раствора в точках а и а', жидкая фаза,
состоящая из свободной Н3РО4 и Са(Н2РО4)2, должна стать на-
сыщенной по отношению к дикальцийфосфату СаНРС^. Практи-
чески насыщение наступает позже, в точках b и Ь' на метаста-
бильной ветви ML растворимости твердой фазы СаНРО4"2Н2О.
Для ее кристаллизации необходимо образование растворов, пере-
сыщенных метастабильной фазой. Интенсивное образование ди-
кальцийфосфата, по-видимому, начинается в момент полной
нейтрализации первого иона водорода фосфорной кислоты (точ-
ки с и с'), т. е. при растворении половинного количества окиси
кальция, затрачиваемой на осаждение преципитата.
Как указывалось выше, кристаллизация дикальцийфосфата
происходит, по-видимому, в результате разложения монокальций-
фосфата из-за инконгруэнтности состава равновесных его раство-
ров по отношению к составу твердых фаз * (луч растворения моно-
кальцийфосфата ONN' проходит вне поля кристаллизации этой
соли):
Са(Н2РО4)2 + 2Н2О = СаНРО4 • 2Н2О + Н3РО4
В указанных точках степень разложения монокальцийфосфа-
та70-72 составляет соответственно 32,6 и 36,1%. Образование сво-
бодной фосфорной кислоты в момент спонтанной кристаллизации
дикальцийфосфата и вызывает интенсивное разложение оставше-
гося карбоната, не прореагировавшего к этому моменту.
Изучение системы СаО—Р2О5—H2SiF6—Н2О при 60° пока-
зало73, что растворимость дикальцийфосфата резко снижается под
влиянием образующегося CaF2 (см. изотерму КТ на рис. 299). В
связи с этим преципитирование экстракционной необесфторенной
фосфорной кислоты протекает, вероятно, в условиях более высо-
ких пересыщений.
Фосфорная кислота, получаемая сернокислотным методом из
необогащенных фосфоритов Каратау, содержит до 15% MgO по
отношению к Р2О5. Осаждение дикальцийфосфата в данном слу-
чае должно быть рассмотрено с помощью диаграммы системы
MgO—СаО—Р2О5—Н2О 74. При норме СаО 0,71 вес. ч. на 1 вес. ч.
Р2О5 (90% от стехиометрической®6) фосфаты магния остаются в
жидкой фазе. Только применение избытка СаО в виде известко-
вого молока позволяет почти полностью осадить Р2О5 (частично,
в виде димагнийфосфата).
Реакция нейтрализации фосфорной кислоты известью (в виде
известкового молока) протекает с очень высокой скоростью. При
Фивико-химические основы осаждения дикалъцийфосфата
1027
смешении фосфорной кислоты с известковым молоком создается
пересыщение раствора в тонком слое вокруг частиц извести, что
вызывает осаждение фосфатов кальция в виде корок, замедляю-
щих дальнейший процесс. Медленное введение известкового мо-
лока, высокая дисперсность частиц извести и интенсивное пе-
ремешивание уменьшает опасность таких «местных» пересы-
щений.
Скорость осаждения преципитата карбонатами определяется
скоростью разложения последних фосфорной кислотой. Природ-
ные известняки, начиная от меловидных до мрамороподобных по-
род, обладают различной плотностью. Более плотные разновид-
ности требуют для своего разложения наиболее тонкого измельче-
ния (до 0,075—0,060 мм), менее плотные породы измельчают до
величины частиц 0,10—0,075 мм. Осажденный мел и мел-рухляк
реагируют с наибольшей скоростью. Фосфорная кислота концент-
рации 10k--15% Р2О5 разлагает тонко измельченный известняк со-
ответственно на 80—90% в течение 10—15 мин. После нейтрали-
зации фосфорной кислоты до pH = 3,8—4,0, разложение карбо-
ната резко замедляется. Частичная нейтрализация катионами
фосфорной кислоты, полученной сернокислотной экстракцией
низкокачественных фосфоритов, значительно уменьшает скорость
разложения известняка. Фосфорнокислые растворы, полученные
взаимодействием соляной кислоты и фосфоритов, разлагают изве-
стняк с большей скоростью чем фосфорная кислота той же кон-
центрации, полученная сернокислотным способом.
Нейтрализация фосфорной кислоты известняком осложняется
обильным выделением двуокиси углерода с образованием в слое
пены очень мелких кристаллов дикальцийфосфата. Найдены75-77
способы, предотвращающие бурное вспенивание пульпы. При
периодическом преципитировании суспензия известняка должна
вводиться медленно, синхронно с разложением карбоната. Для
уменьшения степени пересыщения раствора целесообразно после
введения около 70% известняка в течение некоторого времени
прекратить его подачу и затем продолжать осаждение. При непре-
рывном процессе следует вводить в первый реактор всю фосфор-
ную кислоту и не больше 70% известняка, во втором реакторе
производить только перемешивание пульпы и заканчивать осажде-
ние в третьем реакторе, вводя в него остальное количество суспен-
зии известняка. Такой же режим необходим и при осаждении пре-
ципитата известковым молоком. Несоблюдение этого приводит к
осаждению мелких (5—50 мк) кристаллов дикальцийфосфата. При
раздельном введении суспензии известняка выделяются крупные
кристаллы дикальцийфосфата длиной до 100—150 мк и до 50 мк
шириной, большей частью собранные в конгломераты. При раз-
дельном введении известкового молока образуются кристаллы при-
мерно таких же размеров, но более тонкие. Очень мелкие кристалллы
1028 Гл. XXVIII- Концентрированные фосфорные удобрения. Преципитат
образуются под влиянием примесей экстракционной фосфорной кис-
лоты 78.
В конце преципитирования процесс затормаживается образо-
ванием на частицах карбоната и, особенно, извести непроницае-
мой корки кристаллов дикальцийфосфата. По условиям равнове-
сия в системе СаО—Р2О5—Н2О фосфорная кислота должна осаж-
даться почти полностью. Практически же с помощью известняка
нейтрализовать всю фосфорную кислоту оказывается невозмож-
ным, так как скорость разложения карбоната в конце процесса
Количество введенной Са (ОН)г, % от
стехиометрической нормы
Рис. 300. Изменение pH раствора в процессе преци-
питирования фосфорной кислоты.
крайне мала. Причиной этого, помимо образования экранирующей
корки с большим диффузионным сопротивлением, является и поч-
ти 100%-ная нейтрализация первого иона водорода в жидкой фазе.
Остаточное содержание P2Os в растворе может быть доведено
обычно до 0,2—0,3%. Большая степень осаждения Р2О5 известко-
вым молоком достигается только при условии высокой дисперсно-
сти частиц извести (не крупнее 0,15 мм). Более крупные частицы
извести покрываются непроницаемой коркой, вследствие этого
жидкая фаза может содержать до 3% Р2О5, несмотря на присут-
ствие в твердой фазе свободной извести. Для получения высоко-
дисперсного известкового молока требуется производить гашение
извести трехкратным количеством горячей воды79. Помимо этого
необходима классификация известкового молока для выделения ча-
стиц размером свыше 0,15 мм. Избыток свободной извести в твер-
дой фазе может привести к образованию более основных фосфа-
тов кальция (трикальцийфосфата и гидроксилапатита), плохо
усвояемых растениями.
Изменение pH раствора и концентрации в нем Р2О5 в зависи-
мости от количества введенной извести показано на рис. 300.
Производство преципитата
1029
Кристаллизация дикальцийфосфата начинается после прибав-
ления 50% извести от стехиометрического количества (см. выше),
когда pH раствора становится равным 3—3,2. При последующем
введении извести величина pH постепенно возрастает примерно до
4,8, а затем даже при добавке незначительного количества извести
величина pH резко увеличивается, что вызывает образование три-
кальцийфосфата.
При pH около 4,8 первый ион водорода фосфорной кислоты
полностью нейтрализуется. В этой точке происходит резкий пере-
лом кривой pH, поэтому именно в данной области осаждение нуж-
но вести очень осторожно, чтобы не ввести избыток извести. Рез-
кий перелом кривой изменения pH позволяет контролировать ко-
нец преципитирования, который определяют по индикатору
метиловому красному, изменяющему окраску до желто-оранжевой;
более удобен смешанный индикатор (метиловый красный и ме-
тиловый синий) окраска которого изменяется от фиолетовой до
светло-зеленой 80.
ПРОИЗВОДСТВО ПРЕЦИПИТАТА
Технологический процесс и схема преципитирования опреде-
ляются в основном концентрацией фосфорнокислых растворов.
Обычно преципитатную пульпу подвергают фильтрованию без
предварительного ее отстаивания. При использовании чрезмерно
разбавленных растворов осуществляют декантацию преципитат-
ной пульпы перед ее фильтрованием 39> 40’ 45> 46-81-33.
Известковое молоко должно содержать 50—100 г/л СаО и при
фильтрации через сито с 1600 отв/см2 не давать остатка более
2 г/л. Суспензия известняка должна приготовляться из высокока-
чественного известняка, содержащего не менее 95% СаСО3. Сус-
пензию готовят с отношением Ж Т от 2 : 1 до 1 : 1 (обычно 1,5: 1).
Плотность известняковой суспензии с отношением Ж:Т= 1,5:1
составляет ~1,35 г/см3.
При применении известкового молока процесс осаждения длит-
ся 1,5—2 ч; осаждение суспензией известняка протекает дольше,
в течение 4—7,5 ч. Однако производительность реакторов при этом
не уменьшается, так как суспензия известняка имеет более высо-
кую концентрацию (в расчете на СаО).
Преципитат, осажденный известняком хорошо отстаивается,
фильтруется и промывается. Известняк дешевле извести, при его
использовании нет опасности введения избытка свободной СаО,
что ведет к потере цитратнорастворимой Р2О5. Поэтому примене-
ние суспензии известняка имеет преимущество перед известковым
молоком.
Обработка обесфторенного раствора известняком позволяет
осадить 90—92% Р2О5. Затем осаждение может быть завершено
известковым молоком. Возможность большего или меньшего
1030 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
использования известняка вместо извести 75’77 зависит от концентра-
ции Р2О5 в исходном фосфорнокислом растворе и от степени нейт-
рализации ее примесями. При преципитировании фосфорной кисло-
ты, полученной из фосфорита Каратау, установлена51 возмож-
ность введения в процесс 65—70% СаО в виде известняка. Вна-
чале преципитирование ведут суспензией известняка с отноше-
нием Ж : Т = 1 : 1 при 55—58° в течение 4,5 ч. При этом известняк
Н,РО«
I Суспензия
Г-кСаСО3(Ж-Г-2>)
,lif I Известковое молоко
I и I
_ < - ' I —I 1 I
5
1а
ha разбавление ------1—
О 7
На гашение
ибвести
111 суспензии
шЛ с>со>
° b=bJ-04
Фильтрат^ (соАО,2-ОгЗ%РгО^
f В отброс
*
§
Р
Крупные частицы извести
Рис. 301. Схема одноступенчатого непрерывного преципи-
тирования известняком и известью (без декантации филь-
трата):
1 — преципитатор для реакции с известняком; 2 — преципитатор для
реакции с известковым молоком; 3 — сборник преципитатной пульпы;
4, 8, /2—центробежные насосы; 5 —барабаииый вакуум-фильтр;
—шаровая мельница для мокрого размола известняка; 7 — сборник
суспензии известняка; 9 —аппарат для гашения извести; 10 — класси-
фикатор известкового молока; // — сборник известкового молока.
разлагается на 95—98%. Затем пульпу обрабатывают при 35—40°
известковым молоком в течение 1,5 ч. Общая степень осаждения
Р2О5 в обеих стадиях составляет 95—97%.
Если исходная кислота достаточно чиста (например, получен-
ная из апатитового концентрата), то возможно ее преципитирова-
ние одним известняком75 При этом подучается продукт высокого
качества, содержащий 40—44% Р2О5 в цитратнорастворимой фор-
ме (в пересчете на сухое вещество). Отделенный от преципитата
фильтрат, содержащий остаток Р2О5 (0,2—0,3%), частично можно
использовать для приготовления известкового молока и известня-
ковой суспензии. Остальная часть его должна сбрасываться для
сбалансирования воды в процессе и для отвода примесей (напри-
мер, хлорида кальция).
На рис. 301 приведена одна из схем 51175,77, 84-89 получения пре-
ципитата непрерывным способом. Преципитатная пульпа перете-
кает последовательно через шесть реакторов-преципитаторов. В
первых четырех фосфорная кислота осаждается известняком, а в
Производство преципитата
1031
последних двух — известковым молоком. Распределение суспен-
зии известняка между первым и третьим преципитаторами позво-
ляет предотвратить бурное вспенивание пульпы и осадить преци-
питат с удовлетворительными фильтрующими свойствами. Устра-
нение пенообразования возможно также применением различных
пеногасителей90, например, нефти, растительных масел и т. д.
Ступенчатая подача известкового молока обеспечивает возмож-
ность лучшего регулирования процесса на второй стадии осажде-
ния и уменьшение ретроградации усвояемой Р2О5 в продукте.
После завершения операции осаждения преципитат отделяется
на фильтре и направляется на сушку. Возможно высушивание
пульпы после ее сгущения декантацией (без фильтрования). Суш-
ка должна производиться в условиях, исключающих дегидратацию
двухводного дикальцийфосфата (см. выше) и образование соеди-
нений (СаНРО4 и СазРгО?), содержащих Р2О5 в неусвояемой фор-
ме. Поэтому во избежание потери содержания цитратнораствори-
мой Р2О5 сушку преципитата ведут таким образом, чтобы темпе-
ратура к концу процесса была как можно ниже и не превышала
100°.
Преципитат, осажденный известняком, может быть высушен
при 80—90? до содержания общей влаги около 6% без опасения
ретроградации цитратнорастворимой Р2О5 (при осаждении изве-
стью необходимо при сушке оставлять 10% общей влаги).
Сушка осуществляется в прямоточных барабанных сушилках
топочными газами, смешиваемыми с воздухом для понижения их
температуры до 550—600°. Так как материал, поступающий в су-
шилку, имеет большую' влажность, то он не нагревается выше
температуры кипения воды. Продолжительность сушки 30—40 мин.
Влажность готового продукта не должна превышать 10%. Содер-
жание общей P2OS в готовом преципитате составляет 35—45%
(в зависимости от состава исходного фосфорнокислого раствора);
содержание цитратнорастворимой Р2О5 на 2—3% меньше, чем
общей.
На производство преципитата, содержащего 40,7% РгО3 из фос-
форной кислоты, полученной из апатитового концентрата, тре-
буется 91 (в расчете на 1 т усвояемой Р2О3): 3,0 т апатитового кон-
центрата (39,4% Р2О5), 2,79 т серной кислоты (100% H2SO4),
1,84 т известняка (100% СаСО3), 0,35 т условного топлива и
600 кет • ч электроэнергии.
Производство преципитата как удобрения из экстракционной
фосфорной кислоты по технико-экономическим показателям не мо-
жет конкурировать с производством простого или двойного супер-
фосфата 92-95.
Значительное удешевление преципитата возможно при исполь-
зовании отбросных растворов 94’95, например, от производства же-
латины (так называемых мецерационных щелоков) и других, или
1032 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
при комбинировании производства преципитата с другими процес-
сами, например, с гидролизом древесины концентрированной сер-
ной кислотой для получения глюкозы, спирта, дрожжей и пр.
В этом случае получаемый после обработки древесины кислый
раствор используют для разложения природного фосфата; отде-
ленную от фосфогипса жидкость обрабатывают известью: в осадок
выделяется дикальцийфосфат, а фильтрат, содержащий сахара,
подвергают дальнейшей переработке.
При использовании отходов производства желатины96 мецера-
ционный щелок, содержащий около 3,5% Р20з, осаждается при
40—30° по непрерывной схеме зольной жидкостью, содержащей
0,7% СаО. Преципитирование заканчивается при pH = 4,5—5,0
в последнем преципитаторе. Пульпа с отношением Ж: Т, равным
35:1, сгущается до Ж:Т=10:1 и фильтруется на барабанном
вакуум-фильтре (производительность фильтра 150 кг/(л2-ч) су-
хого преципитата). Фильтрат сбрасывается в канализацию. Высу-
шенный продукт при общей влажности 17% (около 2% влаги
гигроскопической) содержит 32,0% общей Р2О5 и 30,9% усвояе-
мой Р2О5. Изучено97 осаждение преципитата известковым молоком
из бросовых растворов гидрометаллургических производств, со-
держащих 19—40 г/л Р2О5. Преципитат содержит около 30% об-
щей Р2О5, 21,6% усвояемой Р2О5, 13% SO3, 7,2% А120з, 6,5% FeO,
13,2% Fe2O3 и другие примеси.
КОРМОВОЙ ПРЕЦИПИТАТ
Кормовой преципитат, наряду с другими фосфатами и мине-
ральными солями, применяют для подкормки скота и птицы. Он
отличается от удобрительного отсутствием вредных для животных
примесей — соединений фтора, мышьяка и др. Согласно ГОСТ
10566—63, в кормовых фосфатах содержание вредных примесей не
должно превышать (в %): 0,012 As, 0,2 фтора и 0,008 тяжелых
металлов (кроме железа) в пересчете на свинец. Поэтому приме-
няемые для производства кормового преципитата фосфорнокислые
растворы необходимо предварительно очистить. Например, при
сероводородной очистке термической фосфорной кислоты98-100 воз-
можно получить преципитат, не содержащий свинца и мышьяка,
при концентрации в нем лишь 0,022% фтора (в пересчете на
сухое вещество). Термическая кислота, полученная из фосфоритов
Каратау, может быть использована без дополнительной очистки
для выработки кормового преципитата. Преципитирование доста-
точно чистой термической фосфорной кислоты можно осуществлять
в одну ступень по обычной схеме с применением суспензии извест-
няка или известкового молока. При этом в первый преципитатор
вводят всю фосфорную кислоту и 70—75% СаО от общего ее ко-
Кормовой преципитат
1033
личества; остальное количество СаО вводят в третий преципитатор.
Применение известняка, более целесообразное, чем извести, по
технико-экономическим соображениям, возможно, однако, при усло-
вии биологической его доброкачественности. Получаемый продукт
содержит до 50—52% P2Os в цитратнорастворимой форме (в пере-
счете на сухое вещество) при общей влажности (включая кристал-
лизационную влагу) после сушки 14—17%.
Из термической фосфорной кислоты можно также получать
кормовой преципитат смешением ее с известняком и ретуром гото-
вого продукта и высушивания полученной массы. Кормовой пре-
ципитат можно получать и из отходов производства желатины и
экстракционной фосфорной кислоты. В первом случае осуществ-
ляют биологическую очистку растворов и биологический контроль
готового продукта96. Во втором случае кислоту очищают от при-
месей как вредных (фториды и др.), так и балластных, загряз-
няющих продукт и снижающих концентрацию Р2О5 в нем (суль-
фаты, силикаты и др.). В большинстве случаев для очистки кислоты
применяют частичное ее преципитирование. При этом в осадок
выделяется удобрительный преципитат, содержащий соосажден-
ные с ним примеси, в том числе и фтористые соединения (если они
не отделены заранее). После его отделения оставшийся фосфорно-
кислый раствор подвергают вторичному преципитированию, полу-
чая более чистый кормовой преципитат.
На рис. 302 показана схема такого двухступенчатого процесса
получения удобрительного и кормового преципитата из экстрак-
ционной фосфорной кислоты, производимой сернокислотным раз-
ложением апатитового концентрата 101>102. В I фазе расходуется
70—75% известняка от теоретического количества и осаждается
40—65% Р2О5. Одновременно с преципитатом выделяется в осадок
основная масса загрязнений (фтористые соединения, сульфаты,
полуторные окислы и редкие земли). Во II фазе расходуется
остальное количество известняка и осаждается кормовой преци-
питат. После высушивания он содержит 42—45% усвояемой Р2О5,
меньше 0,2% фтора, 0,7% SO3, 0,5% Fe2O3 и следы мышьяка.
Оставшийся после преципитирования раствор направляется вна-
чале на разбавление суспензии известняка для утилизации содер-
жащегося в нем незначительного количества Р2О5, а затем после
сгущения этой суспензии слив, содержащий Р2О5, удаляется как
отброс. Это в особенности необходимо, когда используются раз-
бавленные растворы в целях сбалансирования воды.
При использовании фосфорной кислоты, полученной соляно-
кислотной экстракцией фосфатов 103’104, преципитат отмывают во-
дой на фильтрах от хлорида кальция. После 1 фазы преципитиро-
вания удобрительный продукт содержит 46% усвояемой Р2О5,
0,7% хлора и 0,9% фтора (в пересчете на сухое вещество). Кор-
мовой преципитат, полученный во II фазе, содержит (в пересчете
1034 Гл. XXVIII. Концентрированные фосфорные удобрения. Преципитат
на сухое вещество) 45,7—45,9% усвояемой Р2О5, 0,1—0,13% фтора
и от следов до 2% хлора.
Экстракционная фосфорная кислота, полученная с применением
контактной серной кислоты, почти не содержит примесей свинца
и мышьяка. В этом случае после обесфторивания кислоты (выпа-
риванием или осаждением) из нее можно получать кормовой пре-
ципитат так же, как и из термической кислоты, смешением с из-
вестняком и высушиванием с ретуром готового продукта.
8
NaCl
Осветленная фосфорная кислота
Суспензия
известняка
из мельницы
H3PQ,
Суспензия известняка
"I I фаза преци-
'"'-чц * с ' 1 цитирования
Растбор NaCl(3%)
3 Кремнефторид на сушку
, ,, питирования j
Удобрительный преципитат
Слив б отброс
6
Фильтрат // фазы
на сушку
И (раза пре-
Суспензия ципитцрования
известняка Кормовой npet
таггГнб сушку
4
Рис. 302. Схема производства удобрительного и кормового преципитата двух-
ступенчатым способом:
/ — реакторы для осаждения кремнефторида натрия; 2 —сгуститель для отстаивания кремне-
фторида; 3 — центрифуга для кремнефторида; 4 — прецнпитатор 1-й ступени осаждения; 5 — бара-
банный вакуум-фильтр для удобрительного преципитата; d —прецнпитатор 2-й ступени оса-
ждения; 7 —барабанный вакуум-фильтр для кормового преципитата; 8—смеситель для разба-
вления суспензии известняка; 9 — промежуточный сборник суспензии; 10 — сгуститель для
слива избытка жидкости из суспензии; //—сборник суспензии известняка.
Представляют интерес предложенные53 способы получения кор-
мового преципитата из простого суперфосфата, являющегося бо-
лее дешевым источником Р2О5, чем экстракционная и тем более
термическая фосфорная кислота. При обработке суперфосфата
водой из него можно извлечь практически полностью водораство^
римые фосфорные соединения. После отделения твердого остатка —
сульфата кальция полученную вытяжку можно переработать пол-
ностью в кормовой преципитат (предварительно обесфторив ее)
или в кормовой и удобрительный (в две фазы).
Обесфторивание суперфосфатной вытяжки целесообразно осу-
ществлять в процессе выщелачивания суперфосфата, добавляя к
его водной суспензии хлорид калия. При этом значительная доля
фтора осаждается в виде кремнефторида калия и после отделения
j
Н
Литература
1035
твердых фае отношение Р2О5: F (по весу) превышает 130—150,
что позволяет при одноступенчатом преципитировании получить
продукт, содержащий не больше 0,2% фтора. Для облегчения обес
фторивания и уменьшения расхода хлорида калия следует исполь-
зовать длительно вылежавшийся на складе суперфосфат. По мере
вылеживания вместе с ростом степени разложения фторапатита
уменьшается содержание в нем водорастворимых соединений
фтора — частично вследствие удаления его летучих соединений,
частично вследствие образования труднорастворимых фторком-
плексов алюминия. Из суперфосфата, хранящегося больше двух
месяцев, может быть получена водная вытяжка, в которой отно-
шение Р2О5 : F будет настолько большим, что специального ее обес-
фторивания не потребуется. Такая вытяжка может быть сразу
подвергнута преципитированию. Ее можно использовать и непо-
средственно для смачивания кормов после нейтрализации избы-
точной кислотности, например, известняком; при этом образуется
суспензия, содержащая полезные фосфаты как в жидкой, так и в
твердой фазах.
Кормовой преципитат является еще более дорогим продуктом
(в расчете на Р2О5), чем удобрительный. Несмотря на это, при-
менение его в животноводстве дает значительный экономический
эффект, так как увеличение стоимости получаемой дополнительной
продукции значительно превышает затраты на производство пре-
ципитата. Этим и объясняется преимущественное развитие произ-
водства преципитата для нужд животноводства. Преципитат же
для удобрительных целей получают преимущественно из отброс-
ных растворов других производств.
ЛИТЕРАТУРА
1. Н. Bassett, Z. anorg. Chem., 53, 34 (1907); 59, 1—55 (1908); J. Chem.
Soc., London, 111, 620 (1917). — 2. А. Казаков, Труды НИУИФ, в. 134, 1936.—
3. А. В. Казаков, Там же, в. 139, 1937. — 4. К- L. Elmore, Т. D. Farr, Ind.
Eng. Chem., 32, 580 (1940).—5. H. Bassett, J. Chem. Soc., 1958, № 9, 2949.—
6. К. H. Швецов, Отчет об опытах по химии, переработке фосфоритов, в. 5,
1915, стр. 23. — 7. Э. В. Брицке, С. С. Драгунов, Труды НИУ, в. 55, 1928;
ЖХП, 4, № 1, 49 (1927).—8. Там же, в. 67, 1929, стр. 108. — 9. К. К. Апуш-
кин, Л. М. Гуревич, Удобр. и урожай, № 9—10, 740 (1930). —/8. М. Л. Че-
пелевецкий, Ц. С. Больц, Н. А. Василенко, С. С. Рубикова,
сб. «Исследования по прикладной химии». Изд. АН СССР, 1955, стр. 175.
11. С. С. Драгунов, Удобр. и урожай, № 3, 269 (1930). —12. А. В. Ка-
заков, Труды НИУИФ, в. 145, 1939. —13. С. С. Драгунов, Удобр. и урожай,
№ 5, 409 (1930). —14. Н. Larson, Ind. Eng. Chem., An. Ed., 7, 401 (1935.) —
15. С. И. Вольфкович, В. В. Урусов, Изв. АН СССР. ОХН. № 4, 341
(1951). —16. А. В о u 11 ё, М. D. Dupont, Compt. rend., 226, 1617 (1948); 240,
№ 8, 860 (1955); 241, № 1, 42 (1955).— 17. Н. Е. Пестов, Физико-химические
свойства зернистых и порошкообразных продуктов, Изд. АН СССР, 1947. —
18. Н. Е. Пестов, Удобр. и урожай, № 9 (1931); Справочник по удобрениям,
Госхимиздат, 1933, стр. 414, —19. Ф. Перитурии, М. Курахтанов, Труды
1036
Литература
НИУИФ, в. 134, 1936, стр. 78. — 20. Ф. В. Тур ч и и, Минеральные удобрения
и их применение, Изд. АН СССР, 1943.
21. А. В. Соколов, Агрохимия фосфора, Изд. АН СССР, 1950. —
22. А. В. Соколов, Удобр. и урожай, № 2 (1956).—23. Д. В. Харьков,
С. М. Гуревич, К- П. Магницкий, Т. С. Мельникова, Сообщение
ю научно-технических работах НИУИФ, № 2, 1957, стр. 77. — 24. И. Л. Го ф-
м а н, Хим. пром., № 1, 48 (1956). — 25. Ind. chim., 41, № 439, 42; № 440, 76
(1954).—26. С. И. Вольфкович, Усп. хим., 25, № 11, 1309 (1956). —
21. С. И. Вольфкович, Вопросы геолого-агрономических руд, Изд. АН СССР,
1956, стр. 7. — 28. П. В. Д ы б и н а, А. А. Соколовский, Сборник статей
ВЗПИ, в. 19—20, 1957, стр. 171. — 29. С. К- Воскресенский, Научио-техн.
информ, бюлл. НИУИФ, в. 1, 1957, стр. 27. — 30. С. И. Вольфкович,
Там же, Xs 4.
31. В. В а г г а м а н, Фосфорная кислота, фосфаты и фосфорные удобрения,
Госхимиздат, 1957.—32. М. Е. Позин, Б. А. Копылев, авт. свид. 121455,
1958.—33. Я. А. Иоффе, Вести, техн, и эконом, информ., № 5 (10), 54
(1958). — 34. Т. П. У н а н я н ц, Бюлл. техн.-экоиом. информ. АН СССР, № 6, 89
(1958). — 35. Контрольные цифры развития народного хозяйства СССР на 1959—
1965 годы, Госполитиздат, 1959.— 36. С. Вольфкович, С. Перельман,
Мии. сырье, Xs 3 (1930). — 37. Л. В. Владимиров, Производство преципи-
тата, Госхимиздат, 1932. — 38. А. Соколовский, Преципитат. Справочник по
удобрениям, Госхимиздат, 1933, стр. 249.—39. И. Миркин, И. Никонова,
С. Перельман, ЖХП, Xs 3 (1933). — 40. Хим. реф. ж., Х° 6 (1935).
41. И. Гофман, И. Зар инг, Труды НИУИФ, в. 134, 1936. —
42. Н. М Вайнман, Д. К. Панарии, А. Соколовский, Производство
преципитата, ОНТИ, 1937. — 43. И. Гофман, Я. Зильберман, И. Остров-
ский, Технология суперфосфата и других фосфорных удобрений, Химтеорет,
1937.—44. А. И. Шерешевский, А. А. Соколовский, П. Ф. Дерев>иц-
к ий, Производство фосфорных удобрений, ГОНТИ, 1938. — 45. С. Вольфко-
вич, А. Логинова, ЖПХ, 17, Xs 7, 381 (1944); ДАН СССР, 44, Х° 4, 168
(1944), — 46. С. Вольфкович, А. Логинова, А. Соколовский, ЖПХ,
№ 3, 1 (1945).—41. А. М. Дубовицкий, А. И. Шерешевский, Техноло-
гия минеральных удобрений, Госхимиздат, 1947.—48. М. Е. Позин,
Технология минеральных удобрений, Изд. «Химия», 1965.—49. Technolo-
gia Zwiazkow Fosforowych, PWT, Warszawa, 1958. — 50. И. M. Богуслав-
ский, С. Б. Казакова, И. И. Усачева, Реф. н.-и. раб. за 1956 г., ч. 1,
стр. 43, Труды НИУИФ, в. 161, 1959.
51. С. К. Воскресенский, С. К. Милованова, Л. Н. Архипова,
Сообщения о научно-технических работах НИУИФ, Xs 2, 1957, стр. 11.—
52. С. Воскресенский, С. Милованова, Труды НИУИФ, в. 147, 1940,
стр. 29. — 53. М. Е. Позин, Б. А. Копыле в, Новые методы получения мине-
ральных удобрений, Госхимиздат, 1962, стр. 183. — 54. Б. Д. Мельник. Хим.
пром., № 1, 5 (1957). — 55. Р. Е. Ремен, Е. И. Петрова, Реф. н.-и. раб. за
1957 г., ч. 1, стр. 48, Труды НИУИФ, в. 163, 1959. — 56. Е. I. F о х, К- G. С 1 а г к
Ind. Eng. Chem., Хе 6, 701 (1938). — 57. L. Walter-Levy, P. Maarten de
Wolff, I. P. Wincent, Compt. rend., 240, Xs 3, 308 (1955).—58. L. Wal-
ter-Levy, ibid., 241, Xs 18, 1207 (1955).— 59. E. I. Fox, R. G. Clark, Ind.
Eng. Chem., Ind. Ed., 35, 12, 1264 (1943).—60. И. Л. Гофман, Хим. пром..
Xs 6, 27 (1945).
61. T. W. Zbornik, пат. США 2361444, 1944. — 62. I. H. Walthall, Fer-
tilizer Technology and Resources, ed. K- D. Jacob, New York, 1953, 236—241.—
63. M. E. Позин, Б. А. Копы лев, Д. Ф. Жильцова, ЖПХ, 32, Хе 4, 710
(1959). — 64. G, L. Bridger, Т. I. Horzella, К. Н. Lin, Agr. Food. Chem.,
4, Xs 4, 331 (1956); J о w a, State College Press, 134 (1955). — 65. D. R. Boy-
land, Commerc. Fertilizer, 94, Xs 5, 24, 27, 32 (1957). —66. E. Б. Бруцкус
Литература
1037
И. Л. Гофман, К. С. Зотова, Реф. н.-и. раб. за 1957 г., ч. I, стр. 21; Труды
НИУИФ, в. 163, 1959. — 67. М. Е. Позин, Б. А. Копылев, В. Л. Варшав-
ский, Изв. вузов. Хим. и хим. технол., 2, № 3, 412 (1959); авт. свид. 107487,
1957. — 68. М. Е. Позин, Б. А. Копылев, Р. Ю. Зинюк, Труды ЛТИ им.
Ленсовета, в. 58, 1959, стр. 59. — 69. М. Е. Позин, Б. А. Копылев,
Д. Ф. Жильцова, ЖПХ, 32, № 3, 509 (1959). — 70. К. П. Б е л о п о л ь с к и й,
А. А. Таперов а, М. Т. Серебренникова, М. Н. Шульгина, ЖХП, 14,
7, 504 (1937).
71. А. А. Соколовский, Технология фосфорных удобрений, изд. ВЗПИ,
1954. — 72. М. Л. Чепелевецкий, Е. Б. Бруцкус, Суперфосфат, Физико-
химические основы производства, Госхимиздат, 1958. — 73. С. Я. Шпунт,
А. В. Русадзе, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 7, Труды НИУИФ, в. 163,
1959; Сборник аспирантских работ, НИУИФ, 1959, стр. 3. — 74. К. П. Бело-
польский, С. Я- Шпунт, М. Н. Шульгина, ЖПХ, 23, № 8 (1950); 24,
№ 4 (1951); 26, № 3 (1953); сб. «Исследования по прикладной химии», Изд. АН
СССР, 1955, стр. 107. — 75. А. Соколовский, М. Сладкова, Труды
НИУИФ, в 147, 1940, стр. 26. — 76. А. С о к о л о в с к и й, М. Кобрин,
М. Сладкова, Там же, стр. 27. — 77. А. Соколовский, М Сус аров,
Там же. — 78. М. Чепелевецкий, Ц. Больц, Там же, стр. 28.—
79. А. М. Кузнецов, ЖХП, № 19 (1937). — 80. М. Д о р н и ш, М. Слад-
кова, Труды НИУИФ, в. 134, 1936, стр. 65.
81. И. Г о ф м а и, А. Ионасс, в. 134, 1936. — 82. G. Т. G a,d г е, I. С u р t а,
J. Sci. Ind. Res., 12, № 4, 171 (1953). —83. N. В. Patel, ibid., В 12, № 8, 373
(1953).—84. Л. Владимиров, Удобр. и урожай, № 11—12 (1930).—
85. Л. М. Гуревич, Труды НИУ, в. 109, 1932.—86. М. Сладкова и др.,
в. 143, 1938, стр. 21. —87. А. А. Соколовский, Там же, стр. 22.-88. С. Вос-
кресенский, М. Кобрии, Там же, стр. 20.—89. А.. А. Соколовский,
Юбилейная научная конференция НИУИФ, Тезисы докладов на фосфатно-туко-
вой секции, 1940, стр. 24. — 90. М. Кобрин, Труды НИУИФ, в. 147, 1940,
стр. 28.
91. С. Д. Э в е н ч и к, В. А. Кононов, Тезисы докладов на отраслевом
совещании работников основной и гориохимической промышленности, 1958,
стр. 72; Бюлл. техн, информ. Гипрохима, № 40—41, 3 (1958).— 92. В. А. Коно-
нов, С. Д. Э в е и ч и к, Бюлл. техи. информ. Гипрохима, № 40—41, 40 (1958).—
93. Я. А. Иоффе, Удобр. и урожай, № 7, 6 (1958). — 94. С. К. Воскресен-
ский, Хим. наука и пром., № 2, 129 (1956).—95. С. К. Воскресенский,
Тезисы докладов на отраслевом совещании работников основной и горнохими-
ческой промышленности, 1958, стр. 65. — 96. С. К- Милованова, А. Н. Д о-
х о л о в а, Получение удобрения типа преципитата из отходов производства же-
латины, изд. НИУИФ, 1958. — 97. С. Черняк, Цвета, металлы, № 3, 58
(1957). — 98. М. Кобрин, М. Сладкова, Г. Гуткина, А Григорьева,
Труды НИУИФ, в. 147, 1940, стр. 28.-99. Е. Е. Зуссер, ЖХП, 13, № 9, 536
(1936). — 100. Г. Лютринсгаузер, Труды НИУИФ, в. 143, 1938, стр. 11.
101. А. Соколовский, М. Сладкова и др., Там же, стр. 21.—
102. Н. Крючков, Н. Благовещенская, Там же, в. 147, 1940, стр. 28.—
103. С. С. Перельман, И. Н. Никонова, ЖХП, № 12 (1932). —104. Ind.
Chem., 33, № 391, 457 (1957).
Глава XXIX
ТЕРМИЧЕСКИЕ ФОСФАТЫ
Термическими фосфатами называют продукты, получаемые
спеканием при высоких температурах природных фосфатов со ще-
лочами (содой, смесью сульфатов с углем) или сплавлением их
с кварцем, известняком, силикатами магния, щелочными соедине-
ниями и др. К ним относятся обесфторенные фосфаты, плавленые
магниевые и термощелочные (термофосфаты), а также основные
металлургические шлакц, богатые фосфором, и метафосфат
кальция.
В образующихся веществах Р2О5 находится в усвояемых расте-
ниями лимонно- и цитратнорастворимой формах. Поэтому продукты
термической обработки фосфатов, называемой также их щелочным
разложением, в измельченном виде являются хорошими удобре-
ниями, в особенности на кислых почвах. Их применение, помимо
использования содержащегося в них фосфорного ангидрида, дает
дополнительный эффект, аналогичный известкованию почвы. Они
негигроскопичны’, не слеживаются и содержат от 20 до 35%
общей Р2О5, в том числе 90—98% в усвояемой форме.
Продукты спекания природных фосфатов со щелочами назы-
вают термофосфатами, а продукты сплавления их с различными
добавками — плавлеными фосфатами.
СОСТАВ И СВОЙСТВА
При термической обработке природного фосфата, в особенно-
сти в присутствии добавок, происходит разрушение кристалли-
ческой решетки апатита с выделением фтористых соединений в
газовую фазу и образованием в твердой фазе- прежде всего три-
калъцийфосфата ЗСаО • Р2О5, а затем других веществ. Трикальций-
фосфат существует в двух модификациях2 — аморфная а и кри-
сталлическая р, точка перехода которых равна 1180°. Ниже этой
температуры стабильна неусвояемая кристаллическая р-модифи-
Состав и свойства
103»
кация, выше — стабильна усвояемая (лимонно- и цитратнораство-
римая) a-модификация. Предотвращение перехода аморфной а-
формы в кристаллическую ₽ достигается быстрым охлаждением
массы (закаливанием ее).
В присутствии кремневой кислоты и соды одна молекула СаО
в трикальцийфосфате замещается на Na2O, а освобожденная окись
кальция образует с SiO2 ортосиликат кальция:
ЗСа3(РО4)2 4- SiO2 4- 2Na2CO3 == 2[Na2O • 2СаО • Р2О3] 4- 2СаО • SiO2 4- 2СО2
Фосфаты и силикаты кальция (содержащиеся в природных фос-
фатах или получаемые при взаимодействии со щелочами) образуют
силикофосфаты 5CaO-P2Os-SiO2 или Na2O"4CaO’P2O5- SiO2.
При избытке СаО и недостатке или отсутствии SiO2 возможно
образование тетракальцийфосфата 4СаО-Р2Ов. Все эти соединения
также растворимы в лимонной кислоте и цитрате аммония, и по-
этому содержащаяся в них Р2ОВ усваивается растениями.
Обесфторенные фосфаты, получаемые из апатитового концен-
трата и песка, содержат сс-трикальцийфосфат, хорошо растворимый
в 2%-ной лимонной кислоте3. Примесь p-модификации уменьшает
количество лимоннорастворимой Р2О5 в продукте. Растворимость
Р2О5 обесфторенных фосфатов в нейтральном растворе лимонно-
кислого аммония (более соответствующем по своим свойствам
почвенному раствору) составляет 70—20% от растворимости в ли-
монной кислоте. Обесфторенные фосфаты, полученные из каратау-
ских фосфоритов и известняка, представляют собой твердые рас-
творы переменного состава: силикокарнотита 5СаО-Р2ОВ-SiO2,
тетракальцийфосфата 4СаО • Р2О5 или нагелылмитита 7СаО-
• Р2О5 • 2SiO2, в зависимости от соотношения компонентов в ших-
те3. Удобрения с преобладающим содержанием силикокарнотита
обладают хорошей растворимостью как в лимонной кислоте, так
и в нейтральном растворе лимоннокислого аммония.
В зависимости от состава фосфата и количества добавляемого
кремнезема (от 2 до 50% от веса фосфата) или известняка
(~40% от веса высококремнеземистых фосфоритов) обесфторен-
ные фосфаты содержат4-9: 20—39% общей Р2О5, 19—36% лимон-
норастворимой Р2О5, 28—55% СаО, 2,7—48,7% SiO2, 0,01—0,3%
фтора. Они могут содержать примеси: неразложенный фторапатит,
свободную окись кальция (при недостатке кремнезема на образо-
вание ортосиликата кальция), ортосиликаты магния (при достаточ-
ном количестве кремнезема), периклаз MgO (при недостатке
кремнезема и наличии свободной СаО), шпинелиды типа
MgO-AI2Os, ферросиликаты, кварц и др. Их сорбционная влаго-
емкость не превышает 1,14% при относительной влажности воз-
духа 90%. Насыпной вес колеблется от 1 до 1,82 г{см3 в зависимо-
сти от влажности и высоты слоя. Сыпучесть плохая; наилучшей
1040
Гл. XXIX. Термические фосфаты
сыпучестью обладает продукт, полученный из фосфорита Чулак-
тау с добавкой 60% известняка. Рассеваемость обесфторенного
фосфата из апатитового концентрата малоудовлетворительна и
расценивается в 4,75 балла по шкале Пестова при влажности
0,02—0,14%. Продукты из фосфоритов Чулактау рассеиваются зна-
чительно лучше (7 баллов при 0,02% влаги), но балл рассеваемо-
сти падает до 5 при увеличении влажности до 0,15%.
Плавленые магниевые фосфаты, получаемые сплавлением при-
родных фосфатов с магнезиальными силикатами или доломитом
(при содержании в фосфатах значительных количеств ВЮг), со-
стоят из фосфатов кальция и силикатов магния.'Содержание фтора
не оказывает большого влияния на усвояемость Р2О58’11. Они
содержат8"11: 19,5—22,5% общей Р2О5, в том числе 19—21% ли-
моннорастворимой Р2О5, 9—14% MgO, ~30% СаО, 23% БЮг,
7,5% R2O3 и 1,8% фтора.
Плавленые магниевые фосфаты после измельчения обладают
очень хорошими физическими свойствами 12.
Термощелочные фосфаты (термофосфаты) получают спеканием
измельченных природных фосфатов с щелочными солями и мине-
ралами (сода, поташ, сульфаты и бисульфаты натрия и калия,
содовые шлаки после обессеривания чугуна, нефелины, лейциты
и т. д.). Термофосфаты обычно имеют различные торговые назва-
ния (рёхлинг-фосфат, ренания-фосфат, супертомасин, палатиа, фос-
фаль и др.).
Фосфатное вещество большинства термофосфатов представлено
ренанитом Na2O • 2СаО • Р2О5 или CaNaPO4, хорошо растворимым
в 2%-ной лимонной кислоте, а также ортосиликатом кальция,
дающим твердые растворы с кальцийнатрийфосфатом. Фтор свя-
зывается в виде фторида натрия. Исключение составляет «фос-
фаль», получаемый во Франции термической обработкой сене-
гальского алюмо-кальциевого фосфорита и содержащий цитратно-
растворимый кайьцийалюминийфосфат (34% Р2О5 общей и 26%
цитратнорастворимой)13114.
Вместо щелочных солей применяют также кизерит MgSO4-H2O
(отход от обогащения калийных солей). Состав получаемого про-
дукта: 18—22% Р2О5 (около 95% Р2О5 цитратнорастворимой)
28—30% СаО, 12—14% MgO, 28—30% SO4, а также микроэле-
менты Мп, В, Сг, Си и Zn ’С Примеры анализов некоторых термо-
фосфатов даны в табл. 82.
Основные металлургические шлаки являются побочными про-
дуктами при получении стали по томасовскому или основному
мартеновскому способу. Они соответственно называются томас-
шлаком и основным мартеновским шлаком. Эти удобрения содер-
жат фосфат, растворимый в 2%-ной лимонной кислоте19’20. Про-
изводство удобрений из затвердевших шлаков состоит в их дроб-
лении и измельчении.
Состав и свойства
1041
ТАБЛИЦА 82
Состав термофосфатов (в %)4, 16—18
Продукт Р2О5 (общая) Р2О5 (лнмои- нораст- воримая) СаО SiO2 Na2O A12O3 ЕегОз Фтор
Фосфат: а) из апатитового концентрата ' н соды 29,1 28,0 40,1 10,5 15,4 1,5 0,7 2,2
б) нз фосфоритов Кюрасао и Кон- стантина в смеси с содой 31,2 28,0 43,1 9,8 13,4 0,6 0,3 1,3
Рёхлинг-фосфат из фос- форита Флориды и содовых шлаков . . . 19,8 18,6* 32,0 19,8 12,9 4,1 5,1 0,3
* P2Os, растворимая в растворе
лимоннокислого аммония.
Состав фосфатного вещества шлаков твердо не установлен.
Предполагают, что эффективным компонентом томасшлака яв-
ляется тетракальцийфосфат (хильгенштокит) 4СаО-Р2Ов или
Са4Р2О9 2,~23. Считают также, что Р2ОВ в томасшлаке находится
в виде силикокарнотита и Са4Р2О9 • CaSiO3 и что часть Р2ОВ свя-
зана в виде основного фосфата железа24. В настоящее время пола-
гают, что томасшлак содержит и другие силикофосфаты кальция
и железа, например, стедит 3(ЗСаО • Р2ОВ)- 2СаО -(2СаО • SiO2),
томасит бСаО • Р2О5 • 2FeO • SiO2.
Химический состав основного томасовского шлака колеблется
в широких пределах (в %) 25: х
Р2О5................
SiO2................
СаО.................
Fe2C3...............
11-23
3-13
38-59
6-25
МпО2................. 1—6
А12О3.............0,2-3,7
MgO.................. 2-8
S..................0,2-11.4
Мартеновский основной шлак содержит от 7 до 14% Р2О5 (из
них 85% в лимоннорастворимой форме)26. Менее растворимый
мартеновский шлак получают при переделе чугуна с большой до-
бавкой плавикового шпата CaF2 в качестве флюса 27.
Химически чистый метафосфат кальция СаО-Р2Ов или Са(РО3)2
содержит 71,7% Р2ОВ, технический продукт — до 65% усвояемой
Р2ОВ. Высокая концентрация питательного элемента является глав-
ным достоинством этого удобрения. Метафосфат кальция раство-
рим в нейтральном растворе лимоннокислого аммония. Под воз-
действием воды он медленно превращается в водорастворимый
моиокальцийфосфат28.
1042
Гл. XXIX. Термические фосфаты
Не гигроскопичный продукт с максимальной растворимостью
в растворе лимоннокислого аммония получается при молярном
отношении Р2ОВ: СаО, близком к единице 29’30. Гигроскопичность
удобрения с высоким относительным содержанием Р2О5 значитель-
но уменьшается добавлением измельченного известняка28’31.
Состав метафосфата кальция, получаемого из бурого фосфо-
рита Теннесси*: 64,6% Р2О5, 24,5% СаО, 4,3% SiO2, 3,7% R2O3,
0,2% фтора.
ПРИМЕНЕНИЕ
Термические фосфаты можно применять в качестве удобрений
на всех типах почв 5’12’32-37.
В отношении прибавки урожая все виды термических фосфатов
не уступают суперфосфату при применении на кислых подзолистых
почвах. На черноземах и сероземах обесфторенные фосфаты, тер-
мофосфаты и томасшлак дают несколько (72—87%) меньшую при-
бавку урожая. Эффективность плавленых магниевых фосфатов,
растворимых в лимоннокислом аммонии, близка к суперфосфату
и на нейтральных и щелочных почвах.
Усвояемость термических фосфатов в сильной степени зависит
от тонкости помола 4 Наиболее эффективна фракция, пределы
дисперсности частиц которой соответствуют размерам отверстий
сит 0,075—0,105 мм.
Обесфторенные фосфаты применяются в качестве минеральной
добавки в корм животных5’9’38-42 и как нейтрализующая добавка
к суперфосфату.
Согласно ГОСТ 10516—63, обесфторенный фосфат, получаемый
спеканием апатитового концентрата с небольшими добавками
песка в присутствии паров воды, должен содержать не менее:
36% Р2ОВ, растворимой в 0,4%-ном растворе соляной кислоты,
48% СаО и не более: 0,2% фтора и 0,01% металлического железа
с размером частиц более 0,5 мм. По внешнему виду он представ-
ляет собой тонкоизмельченный порошок серого или светло-корич-
невого цвета.
Термические фосфаты (за исключением металлургических
шлаков) получают и применяют в небольших количествах.
В СССР основные металлургические шлаки получают при пе-
реработке фосфористых железных руд керченского, кустанайского,
восточносибирского (ангарского) и других месторождений43.
Наличие свободных оснований не позволяет применять терми-
ческие фосфаты для смешивания с аммиачными удобрениями, по-
этому в США, где весьма распространены смешанные удобрения,
содержащие азот, производство термических фосфатов всех типов
развивается медленно35.
Термические фосфаты являются источником обеспечения сель-
ского хозяйства усвояемым фосфором при уменьшенных капиталь-
Производство термических фосфатов
1043
ных и эксплуатационных затратах на производство по сравнению
с суперфосфатом 43-47. Из них наиболее экономичными являются
томасовские и мартеновские фосфатные шлаки42. Низкая стоимость
производства допускает их перевозку на большие расстояния
(до 1500 км) при содержании 11 —12% усвояемой Р2О5.
ПРОИЗВОДСТВО ТЕРМИЧЕСКИХ ФОСФАТОВ 4’ 8’ 47-91
Получение обесфторенных фосфатов
Наиболее быстро и полно обесфториваются фосфаты при тер-
мическом разложении их в присутствии водяного пара. Гидротер-
мическая обработка фторапатита при 1400—1450° приводит вна-
чале к изоморфному замещению ионов фтора гидроксил-
ионами92-101:
Ca10(PO4)6F2 + 2Н2О = Ca10(PO4)e(OH)2 + 2HF
Способность ионов фтора и гидроксила изоморфно замещать
друг друга объясняется близостью их ионных радиусов. Теплота
этой реакции98 составляет 62,3 ккал/г-мол. Однако гидроксилапа-
тит не является конечным продуктом реакции 98’100 вследствие его
разложения на три- и тетракальцийфосфат:
CaJ0(PO4)e(OH)2 = 2Са3(РО4)2 + Са4Р2О9 + Н2О
Трикальцийфосфат выделяется при температуре обесфторива-
ния в виде «-модификации, которая при быстром охлаждении
водой (закалка) может быть сохранена в метастабильном состоя-
нии и при обычных температурах.
При обесфторивании фосфатов в шихту вводят кремнезем
(от 2 до 50%), принимающий участие в разложении гидроксил-
апатита:
Ca10(PO4)e(OH)2 + 0,5SiO2 = ЗСа3(РО4)2 + 0,5Ca2SiO4 + Н2О
В суммарном виде процесс может быть представлен уравнением:
Ca10(PO4)sF2 + Н2О + 0,5SiO2 = ЗСа3(РО4)2 + 0,5Ca2SiO4 + 2HF
В этих условиях основной фазой продукта, получаемого из
апатитового концентрата, также является а-трикальцийфосфат3’98.
В присутствии кремнезема температура перехода a-формы три-
кальцийфосфата в р снижается и скорость этого перехода на-
столько уменьшается, что продукт не теряет своих ценных свойств
даже при медленном охлаждении плава на воздухе. Процесс же
обесфторивания значительно ускоряется, так как кремнезем спо-
собствует разрушению кристаллической структуры апатита. В за-
висимости от количества кремнезема в системе СаО—Р2О5—
—SiO2 'о2-1 и образуются силикаты и силикофосфаты разного
состава, обладающие различными температурами плавления.
1044
Гл. XXIX. Термические фосфаты
В соответствии с этим обесфторенные фосфаты можно получать
двумя способами — плавлением в шахтных и других печах или спе-
Рис. 303. Фазовая диаграмма системы
СаО—SiO2—ЗСаО • Р2О5:
С— СаО; S—S1O2; Р —Р2О5; Sc — снликокарнотит;
а —область обесфторениых фосфатов, получаемых
спеканием прн содержании в шнхте 45—50% крем-
незема; б — область обесфторениых фосфатов,
получаемых плавлением прн содержании в шихте
20—25% кремнезема; в —область томасшлаков;
г —область обесфторениых фосфатов, получаемы»
спеканием при малом (2—5%) количестве кремне-
зема в шихте.
канием шихты во вращаю-
щихся печах.
При получении обесфто-
ренного фосфата по способу
плавления в шихту вводят от
20 до 25% кремнезема,
так как шихта такого соста-
ва плавится при наиболее
низкой температуре. В этом
случае фазовый состав полу-
чаемого продукта характе-
ризуется первично выделив-
шимся а-трикальцийфосфа-
том и стекловидной массой,
состоящей из твердого рас-
твора SiO2 и кальциевого
силиката СаО • SiO2 (об-
ласть б на рис. 303, 304).
Для спекания необхо-
димо, во избежание появле-
ния заметных количеств
жидкой фазы, чтобы коли-
чество кремнезема в шихте
было либо очень большим,
либо очень малым. В США
вводят в шихту до 50%
кремнезема от общего веса,
т. е. смешивают приблизи-
тельно равные количества
фосфорита и песка. В этом
случае получается продукт,
содержащий только около
20% лимоннорастворимой
р2о5.
При большом (45—50%)
содержании кремнезема в
шихте (способ спекания)
второй главный фазовый
компонент представлен кри-
стобалитом SiO2 (рис. 303 —
область а)3’96-110.
Процесс спекания осуществляют и при малом (2—5%) коли-
честве кремнезема в шихте, тогда образуется наиболее концентри-
рованный по Р2О5 продукт. Полученные в этих условиях обесфто-
Производство термических фосфатов
1045
ренные фосфаты тоже находятся в области метастабильного а-три-
кальцийфосфата, что обусловливает их растворимость в 2%-ной
лимонной кислоте. Кварц присутствует в незначительных коли-
чествах 3> 96.
Вследствие хорошей растворимости силикокарнотита предло-
жено110 спекать шихту, содержащую 5—11,5% SiO2. В процессе
РгО5, бес. “/<>
10»,5 20 30 40 50 GO 70 80 90 100
05% СаО SiOt,0ec.%
Рис. 304. Разрез фазовой диаграммы системы СаО—SiO2—ЗСаО • Р2О5 по линии
35% Р2О5 —SiO2:
а —область получения обесфторенного фосфата спеканием при содержании в шихте
5—11,5% S1O2; б —область получения обесфторениых фосфатов плавлением при содержании
в шихте 20—25% SiO2.
охлаждения до 1300° и ниже выделяются две фазы — силикокар-
нотит и тетракальцийфосфат 4СаО • Р2О5, при этом главным ком-
понентом является силикокарнотит. При увеличении количества
SiO2 в шихте до 11,5% получается чистый силикокарнотит (об-
ласть г на рис. 303 и область а на рис. 304).
Гидротермическое обесфторивание спеканием фосфоритов Ка-
ратау протекает наиболее полно при введении в шихту известняка
в количестве 40—60 вес. ч. на 100 вес. ч. фосфорита 3>96-98. Это
1046
Гл. XXIX. Термические фосфаты
объясняется тем, что каратауские и другие фосфориты, содержа-
щие много примесей, образуют низкотемпературные эвтектики.
Введение известняка в шихту уменьшает содержание жидкой фазы
в прокаливаемой масве, в результате чего она сохраняет свою
сыпучесть и не налипает на стенки печи. Но получаемый продукт
имеет пониженное содержание Р2О5. В этих условиях получаются
продукты, силикофосфатная часть которых состоит из стабильных
силикокарнотита и нагелыпмитита3’96. Преобладание первого из
них обеспечивает хорошую растворимость таких фосфатов в ци-
тратном растворе.
Механизм процесса обесфторивания фосфоритов Каратау тре-
бует дальнейшего изучения в связи с тем, что обнаружена98 воз-
можность одинаково высокой степени выделения фтора как с
введением водяного пара, так и без него.
Исследованоэз’97’98 влияние небольших (0,5%) добавок окис-
лов алюминия и магния на распад гидроксилапатита. Окись маг-
ния способствует этой реакции, как и- кремнезем; окись алюминия
тормозит процесс распада.
Одновременное воздействие водяного пара и кремнезема зна-
чительно ускоряет процесс образования растворимых в лимонной
кислоте силикофосфатов92. Скорость взаимодействия фторапатита
с кремнеземом в начальный период реакции пропорциональна ко-
личеству введенного кремнезема. В дальнейшем реакции лимити-
руются диффузионными процессами8’112. Скорость обесфторивания
апатита (и его смесей с кремнеземом) водяным паром пропорцио-
нальна парциальному давлению воды в газах8.
Впервые обесфторивание фосфатов было осуществлено по спо-
собу плавления в шахтных печах (рис. 305)4-53 высотой 12,9 м и
внутренним диаметром 2,4 м, производительностью около 75 т гото-
вого продукта в сутки. По этому способу используют брикетирован-
ную шихту, содержащую 20—25% песка. Печь обогревают нефтью
до 1400—1480° (на 50—150° выше точки плавления шихты)99.
В бетонном желобе, выложенном кирпичом, куда под давлением
7 ат вбрызгивают воду, расплав охлаждается и одновременно
гранулируется. Затем его высушивают и измельчают. На 1 т гото-
вого продукта расходуется: 1,12 т шихты, 210 л нефти на обогрев
печи, 20 л нефти на сушку продукта, 20000 нм3 воздуха, 17 м3 во-
ды на охлаждение, 145 кет • ч электроэнергии. Получаемый в США
обесфторенный фосфат содержит: 26—30% Р2О5, 38—40% СаО,
20—25% SiO2 и 0,1—0,2% фтора.
Показана113 целесообразность применения твердого топлива
(кокса) для получения плавленых фосфатов в шахтной печи.
При использовании топлива, богатого водородом, например,
нефти, мазута, специальной подачи водяного пара не требуется,
так как достаточное его количество содержится в продуктах го-
рения (больше 10%).
Производство термических фосфатов
1047
Обесфторивание различного фосфатного сырья методом плав-
ления может быть значительно интенсифицировано применением
циклонных печей ,14-116, широко используемых в энергетике для
сжигания топлива. Удельная нагрузка их достигает 2000—
3000 кгс/(м3.- ч) обесфторенного фосфата.
Методом плавления в циклонных печах, комбинированных с
паровыми котлами, так называемых энерго-технологических агре-
гатах, рационально вести обесфторивание при 1500—1600° легко-
плавких каратауских и других фосфоритов117-118, содержащих
Рис. 305. Схема производства обесфторенного фосфата в шахтной печи
способом плавления:
2 —шахтная печь; 2 —желоб для охлаждения расплава водой; <? —склад охлажденного
продукта; 4 —грохот; 5 — барабанная сушилка; d —молотковая дробилка; 7—пылеуло-
внтель; 8— абсорбционная башня для улавливания фтора.
большое количество примесей. Жидкое или газообразное топливо
и нагретый воздух вводят в печь-циклон тангенциально (рис. 306).
Фосфоритную муку подают таким образом, что она попадает на
стенки, где плавится и стекает вниз. По выходе из печи плав быст-
ро охлаждают водой, причем образуются мелкие стекловидные
гранулы. Гранулированный плав высушивают и размалывают.
Тепло отходящих из печи газов, составляющее 60—70% от общего
количества подведенного к циклонной камере, используют в па-
ровом котле-утилизаторе и в нагревателе поступающего в печь
воздуха. Затем газы, охлажденные до 200—300°, очищают от пыли
в электрофильтре и направляют в абсорбционную систему для
улавливания HF и SiF4.
Полученный таким способом обесфторенный фосфат из кара-
тауской фосфоритной муки содержит 28—30% усвояемой Р2О5 и
меньше 0,1% фтора. Степень выделения фтора достигает 96—98%.
9 М. Е. Позин
1048
Гл. XXIX. Термические фосфаты
Для переработки тугоплавких фосфатов (апатитового концен-
трата, эстонского фосфорита и др.) в циклонных печах требуются
слишком высокие температуры (~1700°).
Спекание шихты, содержащей 42—45% кремнезема 5-53, осу-
ществляют в трубчатой печи типа цементной с подачей шихты в
виде пульпы, содержащей 35—45% воды. Фосфат и песок (каж-
дый в отдельности) измельчают в мокрой трубчатой мельнице до
прохождения 85% частиц через сито 0,074 мм. Шихту готовят сме-
шением пульпы измельченного фосфата и песка в баках с мешал-
ками. Трубчатые печи диаметром 2 м и длиной 41 м обогревают
нефтью. Скорость вращения — один оборот за 66—108 сек. В конец
Рис. 306. Схема циклонной энерго-технологнческой уста-
новки:
/ — плавильный циклон; 2 —сепаратор шлака; 3 — радиационный
котел (камера охлаждения); 4 — охлаждающий элемент; 5 - паропере-
греватель; 6 — экономайзер (водяной); 7 — подогреватель воздуха.
печи вводят струю воды для охлаждения продукта (закалка). Ре-
акционная зона печи (зона горения) составляет 9 л. В этой зоне
поддерживают 1480—1600°, время пребывания в ней шихты 20—
30 мин. Печь футерована в горячем конце (24 м) огнеупорным
кирпичом, содержащим около 70% глинозема. Остальная часть
печи футерована высококачественным огнеупорным кирпичом
(толщиной 15 см). Клинкер выходит из печи в виде частиц серова-
то-зеленого цвета размером 1,6—12,7 мм. Его измельчают на вал-
ковой мельнице до прохождения 60% частиц через сито 0,074 мм.
Производство термических фосфатов
1049
В СССР осуществляют обесфторивание апатитового концентра-
та спеканием шихты, содержащей около 2% кремнезема.
Технологическая схема процесса изображена на рис. 307. Ших-
ту, смоченную водой до влажности 12—14%, подвергают спеканию
во вращающейся печи119 при 1450—1550°. Печь отапливают мазу-
том. Газы, выходящие из печи при 400—800°, несколько охлаж-
дают в полых скрубберах, вбрызгивая в них воду, затем очищают
е электрофильтре от выносимой из печи пыли и направляют в аб-
сорбционную установку для улавливания фтористых соединений.
Уловленную пыль смешивают с поступающей в печь шихтой. По-
лученный в печи клинкер после охлаждения измельчают. Тонкость
Шихта
Фильтрованная вода
Рнс. 307. Схема производства обесфторенного фосфата нз апатитового концен-
трата:
/ — элеватор; 2 — бункер; <? —шнековый питатель; 4 —шнековый смеситель; 5 — увлажнитель;
6 — электрофильтр; 7 — скруббер; 8 — вращающаяся печь; 9 — шнек; 10 — вентилятор; 11 — пластин-
чатый транспортер; 12— грохот; 13 — дробилка; 14 — питатель; /5 —шаровая мельница; 16 — абсор-
бер для фтористых газов; 17— брызгоуловнтель; 18 — автоматические весы; 19 — зашивочная
машина; 20 — рукавный фильтр.
его помола должна быть такой, чтобы остаток на сите 0,15 мм
был не больше 10%. При условиях оптимального режима 8-88-,2°:
температуре обжига 1450—1550°, времени пребывания в зоне горе-
ния около 20лшн, содержании водяных паров в газовой фазе 10—
12%, степень обесфторивания достигает 94—96%. Продукт содер-
жит 121 ~36% лимоннорастворимой Р2О5 (больше 38% РаО5, рас-
творимой в 0,4 %-ной соляной кислоте) и меньше .0,2 % фтора. Фтор
выделяется в виде смеси HF и SiF4 в различных соотношениях.
Концентрация фтора в отходящих газах составляет -~8—9 г/м3.
Значительный унос пыли из печи усложняет производство. Приме-
9*
1050
Гл. XXIX. Термические фосфаты
нение мокрого питания, а также гранулирование шихты118 сни-
жает пылеунос.
Оптимальными условиями обесфторивания фосфоритов Кара-
тау 8’90 по методу спекания являются: применение шихты, состоя-
щей из 100 вес. ч. фосфорита и 40 вес. ч. известняка (поддержи-
вают молярные отношения СаО : Р2ОЙ: SiO2 в пределах соответст-
венно 5—6:1:1—1,5), температура обжига 1430—1450°, продол-
жительность обжига 20—30 мин, содержание водяных паров в газе
10—12%. Степень обесфторивания достигает 95—97%. Концентра-
ция фтора в газе .0,5—1 г/м3 в виде смеси HF и SiF4.
Предложено для увеличения степени обесфторивания и сниже-
ния температуры обжига смешать апатит перед обработкой с рас-
творимым стеклом 122 плотностью 1,09—1,1 в количестве 70—90 л
на 1 т апатита. Указывают, что добавки к природным фосфатам
небольших количеств фосфорной кислоты 123 или кислого фосфата
натрия124 или соды и фосфорной кислоты125> 126, или сульфата
натрия также позволяют осуществить обесфторивание при 1300—
1400°. Обесфторивание оболовых фосфоритов можно осущест-
вить127 при 1200—1250° в присутствии фосфорной кислоты в коли-
честве, необходимом для уменьшения соотношения СаО : Р2О5 в
смеси до 3.
При добавке к природному фосфату (до обжига) мазута или
керосина увеличивается пористость реакционной массы, что спо-
собствует превращению Р2О5 в усвояемую форму при более низ-
ких температурах, а введение фосфорной кислоты повышает проч-
ность гранул 128.
Показана 129 возможность обесфторивания апатитового концент-
рата в псевдоожиженном слое при 1550—1600° без добавки крем-
незема. Удельная нагрузка печи в лабораторных условиях дости-
гает 170 кгс/(м3 • ч). Процесс ускоряется и температура может
быть снижена до 1500—1520° при добавке к апатиту фосфорной
или кремнефтористоводородной кислоты (~2 вес. % на 100 вес. %
апатитового концентрата). Образующийся продукт содержит 37—
38% P2Os, растворимой в 0,4 %-ной соляной кислоте, и меньше
0,2% фтора.
Исследовано130 обесфторивание фосфорита Каратау в смеси с
сульфатом кальция, SiO2 и древесным углем. Твердофазным про-
дуктом реакции являются силикофосфаты кальция. Вначале про-
цесс необходимо вести при температуре не выше 1250°, так как
шихта из фосфорита и CaSO4 плавится при 1260—1280°. Затем
после образования силикатов кальция температуру можно поднять
до 1450°.
Установлена131 возможность гидротермического обесфторива-
ния (без добавок) флотационных концентратов, содержащих 27—
.30% Р2О5, полученных из оболовых фосфоритов. Изучена 132 ско-
рость протекания процесса в кипящем слое.
П роизводство термических фосфатов
1051
Выполнены133 лабораторные исследования режима обесфтори-
вания вурнарских фосфоритов, из которых по способу спекания
получен продукт с содержанием 26,2% лимоннорастворимой Р2О5
и 0,3% фтора,
К обесфторенным фосфатам относится также кормовой три-
кальцийфосфат, образующийся при прокаливании при 1200° вы-
зревшего суперфосфата во вращающейся трубчатой печи, обо-
греваемой топочными газами противотоком9> 134-139. Большая часть
фтора (98—99%) улетучивается, а монокальцийфосфат и сульфат
кальция разлагаются:
Са(Н2РО4)2 -F 2CaSO4 = Са2(РО4)2 4- 2SO2 4- О2 4- 2Н2О
Предложен140 способ обесфторивания фосфатов хлорирующим
обжигом. Процесс протекает в обогреваемой снаружи реторте при
760—1200° в присутствии хлорирующих агентов (НО, NH4CI, С1г)
при соотношении Cl: F > 10. Продукт содержит 0,5% фтора.
Обесфторенные фосфаты по себестоимости 1 т усвояемой Р2О5
занимают промежуточное положение между простым и двойным
суперфосфатами. Более высокое содержание Р2О5, чем в простом
суперфосфате, делает обесфторенные фосфаты экономически эф-
фективным удобрением при дальних его перевозках к местам по-
требления.
Получение плавленых магниевых фосфатов
Плавленые фосфаты получают сплавлением природных фосфа-
тов с флюсами (известняком, кварцитом и др.), образующими
низкоплавкие смеси. Процесс осуществляют при 1400—1500° в
мартеновских или шахтных печах. Вытекающий из печи плав под-
вергают грануляции, быстро охлаждая его водой, — это сохра-
няет в продукте высокое содержание лимоннорастворимой Р2О5.
Гранулированный материал высушивают и измельчают.
Применение в качестве добавок серпентинов, змеевиков (по-
роды, содержащие водный силикат магния) и других магниевых
минералов позволяет снизить температуру плавления фосфатов
до 1250—1350°. Преимуществом получаемых при этом магниевых
плавленых фосфатов является также повышенное содержание
Р2О5 (так как атомный вес магния меньше, чем кальция) и при-
сутствие полезного для растений магния.
Исследования процесса сплавления природных фосфатов с
магниевыми силикатами не обнаружили химического взаимодейст-
вия между исходными компонентами 8’10’ ш-153. Магний также не
встраивается в кристаллические решетки силикофосфатов каль-
ция. Действие магния проявляется в основном в образовании лег-
коплавких магниевых силикатов, препятствующих кристаллизации
1052
Гл. XXIX. Термические фосфаты
фторапатита 154 и способствующих уменьшению вязкости образую-
щихся расплавов.
Превращение фосфатов в растворимое (в 2%-ной лимонной
кислоте) состояние связано с образованием аморфного стекловид-
ного расплава.
Установлено155, что в системе RO—А12О3—SiO2—Р2О5 наимень-
шей кристаллизационной способностью обладают стекла, в кото-
рых молекулярное отношение MgO и СаО равно 2. При их кри-
сталлизации выделяются фосфаты кальция и алюминия. Закалка
расплава (быстрое охлаждение) является необходимым условием
высокой растворимости P2Og. Чем больше скорость охлаждения,
которая достигается уменьшением диаметра капель расплава,
соприкасающихся с водой, тем больше растворимость Р2О5. Умень-
шению диаметра капель способствует снижение вязкости рас-
плава 156-158 посредством значительного его перегрева (примерно
на 150° выше температуры плавления). Поэтому на практике плав-
ление шихты ведут при 1450—1500°. Медленное охлаждение рас-
плава (особенно ниже 750°, т. е. температуры превращения рас-
творимой Р2О5 в нерастворимую форму), значительно снижает
растворимость Р2О5.
При образовании плавленых магнезиальных фосфатов в газо-
вую фазу выделяется не более 10% фтора. Поэтому при расстек-
ловывании аморфного продукта, содержащего значительные коли-
чества фтора, возможно образование апатитовой структуры10, так-
же снижающей растворимость Р2О5. Оптимальные соотношения
основных компонентов: на 1 моль Р2О5 3—5 моль СаО, 2—1 моль
MgO и SiO2. Температура расплава на выходе из печи 1350—1400°.
Размеры гранул после охлаждения расплава 1—3 мм.
Получение плавленых магнезиальных фосфатов позволяет
эффективно использовать низкокачественные фосфаты без затраты
кислот или щелочей4- 53-55> 159-168.
Плавление можно проводить в электропечах, пламенных и цик-
лонных печах 4> м'169-172.
При осуществлении процесса в трехфазной дуговой печи расход
электроэнергии составляет 850 кет • ч на 1т продукта. Нижнюю
часть кожуха печи охлаждают водой, поэтому на внутренней стен-
ке образуется защитный твердый слой магниевого фосфата. Выше
уровня плава печь футеруют огнеупорным кирпичом. Пол печи по-
крыт утрамбованной углеродистой пастой. Плавка длится 1—2 ч.
Из 1 т шихты получается 0,9—0,95 т готового продукта. Плав
охлаждают- и гранулируют струей воды 173, подаваемой под давле-
нием. После обезвоживания гранул (дренирование и сушка) их
измельчают до размеров частиц не выше 0,1 мм.
В опытно-промышленных масштабах получают плавленый фос-
фат в шахтной печи из смеси Кольского апатитового концентрата,
вьетнамского апатита и серпентина 174. Печь отапливают коксом.
Производство термических фосфатов
1053
Показано175, что кролевецкий фосфат полностью разлагается
в присутствии карбоната магния уже при 900° в течение ~ 1,5 ч.
В полученном продукте 99% от общего количества Р2О5 нахо-
дится в лимоннорастворимой форме. Легко текучий плав из бед-
ных болгарских фосфоритов, содержащих 10,8% Р2О5, 41,1% СаО,
4,75% R2O3, 14% SiO2 и 1% фтора, получается 176 при спекании их
в смеси с апатитовым концентратом в соотношении 2:3с добав-
кой ~3% SiO2 при 1475—1500°.
Плавленые магнезиальные фосфаты, несмотря на меньшую
концентрацию усвояемой Р2О5, по себестоимости питательного эле-
мента на месте потребления способны конкурировать с простым
суперфосфатом и с обесфторенными фосфатами, особенно если
учитывать агрохимическую эффективность не только Р2О5, но
и MgO42.
Получение термощелочных фосфатов (термофосфатов)
Щелочное разложение фосфатов отличается простотой техно-
логического процесса и позволяет обойтись без затраты кислот, не-
обходимых при других способах получения фосфорных удобрений,
а также дает возможность использовать без обогащения низкока-
чественное сырье, содержащее сравнительно мало Р2О5 и значи-
тельные примеси R2O3.
Химизм образования лимоннорастворимого фосфата — ренани-
та при спекании природных фосфатов с содой 17>50'177-193 может
быть представлен уравнением:
Ca5(PO4)3F + 2Na2CO3 + SiO2 = 3CaNaPO4 + Ca2SiO4 + 2CO2 + NaF
Количество соды определяется содержанием в сырье Р2О5 и
фтора, количество кремнезема рассчитывается на связывание из-
быточной (по отношению к составу ренанита) окиси кальция.
Обычно норма соды находится в пределах 1,2—1,25 моль на
1 моль Р2О5.
Производство термофосфата, осуществляемое в промышленном
масштабе на ряде предприятий 41 56,194-190, заключается в спекании
смеси измельченного природного фосфата и соды во вращаю-
щихся печах при 1100—1250° н в последующем охлаждении, дроб-
лении и размоле полученного клинкера до частиц размером
0,15 мм.
В ФРГ производится фосфат «ренания» из северо-африканских
фосфоритов, фосфоритов Кюрасао, хибинского апатитового кон-
центрата и др. Фосфат и песок высушивают, смешивают и измель-
чают в трубчатой мельнице. На 100 вес. ч. фосфата вводят
11,5 вес. ч. песка, содержащего 92% SiO2. Шихту и соду дозируют
автоматическими весами и смешивают в смесительных шнеках.
1054
Гл. XXIX. Термические фосфаты
Состав смеси для северо-африканского фосфорита: 10—11% SiO2)
17% Ыа2СОз. Увлажненную до 10% влаги шихту спекают в бара-
банных печах, обогреваемых пылевидным топливом, при 1100—
1250°. Печь длиной 35 м и диаметром 1,7 м, производительностью
4—5 т клинкера в час делает один оборот за 55—80 сек. Темпе-
ратура отходящих газов 400°. Расход угля около 15% от веса про-
дукта. Газами увлекается пыль в количестве 4—10% от веса ших-
ты, улавливаемая электрофильтрами. В горячую зону печи вводят
воду (1,9 л/мин). Это способствует некоторому обесфториванию
продукта, однако в клинкере остается еще много фтора (в преде-
лах 1,25—2,5%). Горячий клинкер охлаждают в’холодильнике до
400—600°, дробят до величины кусков около 25 мм и измельчают
до остатка на сите 0,088 мм не более 15%.
Термофосфат может быть также получен спеканием фосфатов
с сульфатами натрия и магния, с астраханитом и с другими ще-
лочными солями и минералами и сульфатом кальция 199~216.
Разложение апатита сульфатом натрия ведут в присутствии
восстановителя — угля. Процесс протекает в две стадии 199. Перво-
начально происходит сгорание большей части углерода и восста-
новление сульфата натрия до сульфида. Затем сульфид натрия
реагирует с апатитом:
Ca5(PO4)3F + 2Na2S = 3CaNaPO4 + 2CaS + NaF
При охлаждении продукта на воздухе возможно окисление
сульфида до сульфата и обратное превращение кальцийнатрий-
фосфата в апатит. Так как обратное превращение P2Os наиболее
полно протекает при 600—900°, то целесообразно быстрое охлаж-
дение клинкера до температуры не выше 400ь.
Апатит превращается в лимоннорастворимую форму на 85—
90% при 1100—1200°; шихта содержит на 100 вес. ч. апатитового
концентрата 60 вес. ч. сульфата натрия и 30 вес. ч. углерода;
продолжительность процесса 30 мин.
При спекании фосфатов с сульфатом магния в течение 30 мин
при температуре около 1200° тоже образуются лимоннораствори-
мые кальциймагниевые фосфаты и сульфат кальция.
Изучено217'218 получение термофосфата взаимодействием апа-
тита с кремнеземом и сульфатами кальция и магния с одновре-
менным выделением двуокиси серы. Полное разложение апатита и
сульфата кальция достигается при 1350—1400° (при содержании
40% апатита в исходной смеси). При сплавлении фосфорита Кара-
тау или апатита с астраханитом (Na2SO4 • MgSO4 • 4Н2О) обра-
зуется продукт, состоящий из анизотропных зерен CaNaPO4 и
стеклообразного вещества. Максимальная степень разложения фос-
фата достигается при содержании в шихте 3 моль углерода, не-
обходимого для восстановления сульфатов астраханита219’22°.
Производство термических фосфатов
1055
Для получения термофосфата могут применяться нефелины,
лейциты и другие щелочные силикаты. Большое значение имеет
возможность переработки на термофосфат необогащенной апатито-
нефелиновой руды, применение которой позволит уменьшить рас-
ход щелочных солей 181-216.
Получение метафосфата кальция
Метафосфат кальция может быть получен обезвоживанием
однозамещенных ортофосфатов при 275—300°:
Са(Н2РО4)2 • Н2О = Са(РО3)2 + ЗН2О
На этом основаны разные способы получения метафосфата
обработкой природного фосфата серной или фосфорной кислотами
или их смесью221 и прокаливанием пульпы при 350—400°. Вместо
серной кислоты можно также применить газообразный серный
ангидрид в присутствии 2% влаги и осуществить процесс непосред-
ственно при высокой температуре 222. При обработке полигалита
экстракционной фосфорной кислотой с последующим выпарива
нием и прокаливанием пульпы образуется смесь метафосфатов
калия, кальция и магния 223.
Другой способ заключается во взаимодействии фосфорного ан-
гидрида с трех- или двухзамещенными ортофосфатами при 1000—
1200°:
Са3(РО4)2 + 2Р2О5 = ЗСа(РО3)2
Этот способ, предложенный Э. В. Брицке 224, получил промыш-
ленное применение в США 225-229. Процесс осуществляется в шахт-
ной печи, загружаемой кусковым или агломерированным фосфо-
ритом. Через слой фосфорита проходят горячие газы, полученные
при сжигании фосфора в камере сгорания, расположенной у осно-
вания печи. Образующийся метафосфат кальция в расплавленном
состоянии стекает вниз и периодически выводится из печи. После
быстрого охлаждения расплава водой он дробится и измельчается.
Примеси, находящиеся в природном фосфате, время от времени
выводят из печи в виде жидкого шлака. Отходящие газы про-
пускают через скрубберы для поглощения соединений фтора.
Аналогично метафосфат кальция образуется при взаимодейст-
вии паров метафосфорной кислоты с известью или с три- или
дикальцийфосфатами.
Получение метафосфата кальция осложняется тем, что обыч-
ные огнеупорные материалы для футеровки печи оказались недо-
статочно устойчивыми. Сложной задачей является также подбор
материала для изготовления колосниковой решетки, на которой
удерживается фосфатная шихта в печи. Наружное охлаждение
кожуха печи, а также устройство колосников из охлаждаемых
1056
Гл. XXIX. Термические фосфаты
водой труб, защищенных рубашками из графита, удлиняет срок
службы печи.
Метафосфат кальция в США используют для приготовле-
ния высококонцентрированных смешанных фосфорных уДОбре-
НИЙ 43, 230, 231
Непосредственное применение метафосфата кальция в качестве
удобрения ограничено его низкой растворимостью в воде, а в про-
изводстве смешанных удобрений — из-за невозможности аммони-
зации и необходимости тонкого измельчения. Поэтому метафосфат
предварительно подвергают гидратации, при которой большая
часть Р2О5 переходит в водорастворимую форму, хорошо погло-
щающую аммиак. Гидратацию ведут в горячей воде (104—110°),
к которой добавляют небольшое количество кислоты 232. Пульпу
после гидратации направляют на аммонизацию. Возможно также
сочетание гидратации и аммонизации в одной операции 233, заклю-
чающейся в обработке метафосфата (с размерами частиц менее
0,15 мм) 18—20%-ной аммиачной водой под давлением ~7 аг
при 60—80°. Гидратацию метафосфата можно также осуществить
в щелочной среде 234, содержащей ~1 вес. ч. КОН, NaOH, Ыа2СОз
и др. на 10 вес. ч. метафосфата. Указывают, что добавка SiOa
(в отличие от добавки глинозема) увеличивает растворимость ме-
тафосфата кальция 235.
Предложено231 получать обогащенный суперфосфат взаимодей-
ствием метафосфата кальция, измельченного природного фосфата,
серной кислоты и воды. При смешении этих компонентов происхо-
дит гидратация метафосфата кальция до монокальцийортофос-
фата, который затем реагирует с серной кислотой, образуя фосфор-
ную кислоту. Дальнейший процесс разложения фосфата выделив-
шейся фосфорной кислотой осуществляется в стадии дозревания.
Получается удобрение с содержанием до 30% Р2О5 усвояемой.
ЛИТЕРАТУРА
1. Н. Е. Пестов, Е. И. П а у т к и н а, Сообщение о научно-технических ра-
ботах НИУИФ, № 2, 1957, стр. 44. — 2. Э. В. Б р и ц к е, В. К- Веселовский,
Изв. АН СССР. ОТН, № 4, 479 (1937). —3. Н. Н. Постников, М. Г. Френ-
кель, Б. Б. Е в з л и н а, А. И. Смирнов, В. И. Плотникова, ЖПХ, 31,
1453 (1958). — 4. В. Ваггаман, Фосфорная кислота, фосфаты и фосфорные
удобрения, Госхимиздат, 1957. — 5. W. Т. Whitney, С. A. Hollingsworth,
Ind. Eng. Chem, 41, № 7, 1325 (1949). — 6. T. P. Hignett, T. N. Hubbuch,
ibid, 38, № 12, 1208 (1946). — 7. I. H. Walthall, Fertilizers Technology and
Resources in US, 205, N. Y, 1953. — 8. С. И. Вольфкович, H. H. Постни-
ков, В. В. Илларионов, А. А. И о н а с с, Р. Е. Р е м е н, Термические про-
цессы переработки фосфатов на удобрения, изд. НИУИФ, 1957. — 9. И. Л. Гоф-
ман, Хим. наука и пром, 2, № 6, 706 (1957). — 10. В. Бобровницкий,
Г. Пенионжек, Г. Шредер, Przem. Chem, 12, № 7, 392 (1956).
. 11. R. W. Moulton, Chem. Eng, 56, № 8, 102 (1949). — 12. А. В. Соко-
лов, T. Д. К о р и ц к а я, сб. «Исследования по прикладной химии», Изд. АН
СССР, 1955, стр. 313. — 13. L. М о г i с е a u, Ind. chim, 40, № 431, 161 (1953);
Литература
1057
41, № 439, 42 (1954). —14. Н. Huber, Chem. Ztg., Chem. Apparatus № 3, 76;
№ 4, 109 (1959) — 15. Ind. Chem., № 439, 42 (1954); № 440, 76 (1954)- Ind. Eng.
Chem., 45, № 3, 646 (1953). — 16. I. R. Hawes, F. M. Lea, US Dept. Com.,
Office Techn. Services PB — 18913; Brit. Intelligence Objectives Sub. Commitee
Final Report, 94 (1945). — 17. W. H. M a с 1 e n a n, 1. Angus, С. H. Corbett,
BIOS Final Rept., № 1744 (1946). — 18. G. Behhen, Ind. chim., 38, № 411, 283
(1951). —/9. P. Wagner, Chem. Ztg., 19, № 63, 1419 (1895). — 20. I. B. Lind-
sey, Mass. Exp. St. 22nd Annual Rept., part 1, 74, 1910.
21. G. Hilgen stock, Stahl u. Eisen, 9, 498 (1883). — 22. H. W. Wiley,
W. H. Krug, J. Analyt. Chem., 5, 685 (1891). — 23. I. Fritsch, Manufacture of
Chemical Manures, London, 1920. — 24. P. Wagner, Diingungsfragen, 1, 28
(1896). — 25. A E. M a у e r, Lehrbuch Agrikultur Chemie, 1924.—26. E. I. Rus-
sell, Trans. Faraday Soc., 16, pt 2, 263 (1921). —27. J. E. Stead, Trans. Fara-
day Soc., 16, pt 2, 305 (1921).—28. G. L. Frear a. oth., Ind. Eng. Chem., 36,
835 (1944). — 29. H. A. Curtis a. oth., Chem. Met. Eng., 44, № 3, 140
(1937).— 30. H. A. Curtis a. oth., ibid., 45, № 6, 318 (1938).
31. W. H. McIntire a. oth., Ind. Eng. Chem., 29, 224 (1937).—
32. Ф. В. T у p ч и н, Минеральные удобрения и их применение, Изд. АН СССР,
1943.— 33. W, Н. McIntire a. oth., Soil Sci., 57, 423 (1944). —34. А. В. Со-
колов, Агрохимия фосфора, Изд. АН СССР, 1950.—35. К. D. J а с о b, Plant
Food J., 8, № 1, 2 (1954).—36. А. В. Соколов, Т. Д. К о р и ц к а я, Удобр. и
урожай, № 11, 26 (1958). —37. Д. В. Харьков, С. М. Гуревич, К- Л. Маг-
ницкий, Т. С. Мельникова, Сообщение о научно-технических работах
НИУИФ, № 2, 1957, стр. 77. — 38. В. F. Barrentin a. oth., Nutrition, 27, 35
(1944).—39. N. R. Ellis a. oth., J. Assoc. Offic. Agr. Chemists, 28, 129 (1945).—
40. H. P. Bird, J. P. Mattingly a. oth., ibid., № 1, 1181 (1945).
41. D. E. Williams a. oth., Ind. Eng. Chem., 38, № 6, 651 (1946).—
42. Am. Fert., 114, № 5, 39 (1951).—43. A. M. Левнн, А. В. Соколов,
T. П. У н а н я н ц, Э. И. Цыпина, А. С. Шапиро, Научно-техн, информ,
бюлл. НИУИФ, № 8—9р (1956). — 44. Я. А. Иоффе, Удобр. и урожай. № 7,
6 (1958). — 45. Ф. В. Турчин, Там же, № 8, 7 (1958). — 46. С. Д.-Эвенчик,
В. А. Кононов, Бюлл. техн, информ. Гипрохима, № 40—41, 40 (1958).—
47. Т. И. У н а н я н ц, Бюлл. техн.-эконом, информ. АН СССР, № 6, 89 (1958).—
48. С. Вольфкович, Томасшлак, Справочник по удобрениям, Госхимиздат,
1933, стр. 251.—49. А. Соколовский, Там же, стр. 253. — 50. И. Гофман,
Я. Зильберман, И. Островский, Технология суперфосфата и других
фосфорных удобрений, Химтеорет, 1937.
51. И. Л. Гофман, Хим. пром., № 6, 27 (1945).—52. А. М. Дубовиц-
кий, А. И. Шерешевский, Технология минеральных удобрений, Госхим-
издат, 1947. — 53. И. Гофман, Хим. пром., № 1, 48 (1956). — 54. И. Л. Гоф-
ман, Там же, № 7, 52 (1956).—55. С. И. Вольфкович, Усп. хнм., 25,
№ 11, 1309 (1956).—56. Н. Н. Постников, А. А. И о н а с с, Хим. наука и
пром., 1, № 2, 150 (1956). — 57. М. Е. Поз ин, Технология минеральных удоб-
рений и солей, Госхимиздат, 1957. — 58. С. И. Вольфкович, Научно-техн, ин-
форм. бюлл. НИУИФ, № 4 (1957). — 59. П. В. Д ы б и н а, А. А. Соколов-
с к и й, Сборник статей ВЗПИ, в. 19—20, 1957, стр. 171. — 60. С. Д. Э в е н ч и к,
В. А. Кононов, Бюлл. техн, информ. Гипрохима, Xs 40—41, 3 (1958).
61. С. Вольфкович, Н. Постников, В. Илларионов, А. Ионасс,
Р. Р е м е н, сб. «Промышленность минеральных удобрений», Госхимиздат, 1958,
стр. 210. — 52. Ионеску Эмиль, Там же, стр. 239. — 63. В. К а ч е р, Там же,
стр. 232. — 64. А. Г. П ф е ф ф е р, Там же, стр. 228. — 65. В. Ризе ль, Там же,
стр. 222. — 66. Technologia Zwiazkow Fosforowych, RWT, Warszawa, 1958.—
67. С. И. Вольфкович, H. H. П о с т н и к о в, А. А. Ионасс, В. В. Илла-
рионов, Р. Е. Ремен, VIII Менделеевский съезд, Секц. неорг. хим. и технол.,
Изд. АН СССР, 1959, стр. 134. — 68. А. К. Бектуров, Там же, стр. 137,—
1058
Гл. XXIX. Термические фосфаты
69. J. К u u s к, Acta et Commont Univ. Tartu, A. XXI (1930). — 70. P. Cald-
well, пат. США 1902932, 1933.
71. H. A Curtis, пат. США 2044774, 1936. — 72. D. Reynolds, К. Jacob,
L. Rader, Ind. Eng. Chem., 26, 4, 406 (1934). —73. D. S. Reynolds
K. D. Jacob, H. L. Marshall, L. F. Rader, ibid., 27, 87 (1935).—
74. H. L. M a r s h al 1, D. S. Reynolds, K. D. Jacob, L. F. Rader, ibid., 27,
255 (1935); 28, 678 (1936). —75. K- D. J а с о b, D. S. Reynolds, H. L. Mar-
shall, Am. Inst. Min. Met. Eng’es. Techn. Publ, 695 (1936). — 76. А. Шере-
шевский, H. Пестов, С. Кремер, Труды НИУИФ, в. 134, 1936; сб. «Ис-
следования по прикладной химии», Изд. АН СССР, 1955, стр. 207. —
77. С. St. Jacques, Ind. Eng. Chem., News Ed., 15, 29 (1937). — 78. H. A. Cur-
tis, R. L. С о p s о n, H. Earl, K- Pole, R. Gordon, Ind. Eng. Chem., 29,
Ns 7, 766 (1937). — 79. T. F. Bally, пат. США 2121776, 1938, — 80. E. Lu sc her,
пат. США 2220575, 1940.
81. F. К a e s s, герм. пат. 699313, 1940. — 82. H. Franck, F. К a e s s, герм,
пат. 676016 (1939); 689581 (1940). — S3. F. К a e s s, I. Roeder, герм. пат. 712557,
1941, — 84. К. L. Elmore, E. T. Huffman, W. W. Wolf, Ind. Eng. Chem.,
34, 40 (1942). — 85. E. J. M a u s t, пат. США 2446978, 1948. — 86. E. J. M a u s t,
C. A. Hollingsworth, пат. США 2478200, 1949. — 87. I. А. Brabson,
I. P. Smith, A. Do г row, J. Assoc. Offic. Agr. Chemists, 33, 457 (1950).—
88. I. A. Hedvall, H. O. G e г n a n d t, I. A k e s s о n, Trans. Charmers Univ.
Technol. (Gotheburg), 100, 3 (1950); Chem. Abs., 45, № 9, 4007 (1951).—
89. С. И. Вольфкович, H. H. Постников, А. А. Ионасс, P. E. P ем e h,
Сообщение о научно-технических работах НИУИФ, № 2, 35 (1957).—
90. С. И. Вольфкович, Н. Н. Постников, А. А. Ионасс, Р. Е. Ре-
мен, Там же, 37.
91. С. И. Вольфкович, В. В. Илларионов, А. А. Ионасс,
А. А. М а л ы й, Р. Е. Ремен, Гидротермическая переработка фосфатов на удоб-
рения и кормовые средства, Изд. «Химия», 1964. —92. С. И. Вольфкович,
В. В. Илларионов, Р. Е. Р е м е н, Хим. пром., № 4, 203 (1954).—
93. С. И. Вольфкович, В. В. Илларионов, Р. Е. Р е м е н, ДАН СССР,
97, № 4, 715 (1954).—94. F. Trombe, М. F б е х, Bull. Soc. chim. France, 10,
1282 (1954). — 95. H. H. Постников, сб. «Исследования по прикладной хи-
мии», Изд. АН СССР, 1955, стр. 67. — 96. Н. Н. П о с т н и к о в, М. Г, Френ-
кель, Б. Б. Евзлина, О. В. Васильева, Сообщение о научно-технических
работах НИУИФ, № 2, 41 (1957). — 97. С. И. Вольфкович, В. В. Илла-
рионов, Р. Е. Ремен, Хим, пром., Ns 8, 469 (1957). — 98. С. И. Вольфко-
вич, В. В. Илларионов, Р. Е. Ремен, 3. Г. Кулагина, Сообщение
о научно-технических работах НИУИФ, № 2, 38 (1957). — 99. А. А. Ионасс,
Т. А. Алимова, Там же, стр. 42 —100. С. И. Вольфкович, В. В. Илла-
рионов, Р. Е. Ремен, Реф. н.-и. раб. за 1956 г., ч. 1, стр. 35, Труды НИУИФ,
в. 161, 1958; Реф. н.-и. раб. -за 1957 г., ч. 1, стр. 40; Труды НИУИФ,
в. 163, 1959.
101. Н. Н. Постников, Б. Б. Евзлина, М. Генкель, Реф. н.-и. раб.
за 1956 г., ч. 1, стр. 22; Труды НИУИФ, в. 161, 1958. — 102. F. КбгЬег,
G. Trommel, Z. Elektrochem., 38, 678 (1932). —103. М. А. В redig
Н.Н. Franck, Н. Fuldner, ibid., 38, 158 (1932); 38, 959 (1933).— 104. G. Trom-
mel, Stahl u. Eisen, 63, 21 (1943). —105. G. Trommel, H. Harkort,
W. H о t о p, Z. anorg. Chem., 256, № 4—5 (1948). — 106. F. Trombe, M. F 6 e x,
Compt. rend., 236, № 11, 1167 (1953). —107. П. С. Мамыкин, С. Г. Златкин,
ЖФХ, 9, 3, 392 (1937). — 108. Д. С. Белянкин, H. А. Торопов, В. В. Л a-
п и и, Физико-химические системы силикатной технологии, Промстройиздат,
1949. — 109. R. L. Barett, М. С. С a u g h е у, Am. mineralogist, 27, 10, 680
(1942). — ПО. 1. Вега k, Przern. Chem., 12, № 9, 478 (1956).
Литература
1059
111. С. И. Вольфкович, В. В. Илларионов, Р. П. Озеров,
Р. Е. ремен, ЖПХ, 33, № 3, 524 (1960). —112. Н. Н. Постников и др.,
Реф. н.-и. раб. за 1957 г., ч. 1; Труды НИУИФ, в. 163, 1959. — 113. А. А. И о н а с с,
Р. Е. Ремен; Б. А. Гесс, А. М. Чернышев, Реф. н.-и. раб. за 1956 г., ч. 1,
стр. 23; Труды НИУИФ, в. 161, 1958. —114. П. Ф. Деревицкий и’др., авт.
свид. 113811, 1957; Обмен опытом. Сб. Гос. к-та по химии, № 10 (55), 75
(1958). —115. С. И. Вольфкович. Н. Н. Постников, А. А. Ионасс,
Р. Е. Ремен, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 41; Труды НИУИФ, в. 163,
1959. — 116. С. И. Вольфкович, Н. Н. Постников, А. А. Ионасс,
Р. Е. Ремен, Т. Н.. Ягодина, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 52; Труды
НИУИФ, в. 163 (1959); Реф. н.-и. раб. за 1956 г., ч. 1, стр. 19; Труды НИУИФ,
в. 161, 1958. — 117. А. А. Ионасс, Л. Н. С и д е л ь к о в с к и й, сб. «Исследо-
вания по химии и технологии удобрений, пестицидов, солей». Изд. «Наука»,
1966, стр. 182. — 118. С. И. Вольфкович, А. А. Ионасс, Е. Б. Мельни-
ков, Р. Е. Ре м е н, Л. Н. С и д е л ь к о в с к и й, Ю. В. Троян кин, А. П. Шу-
рыгин, Т. Н. Ягодина, Хим. пром., № 6, 394 (1961). — 119. В. Б. Земель-
ман, Труды НИУИФ, в. 205, 1964, стр. 58; ЖПХ. 37, № 1, 41 (1964).—
120. Г. М. И л ь и н, Труды НИУИФ, в. 208, 1965, стр. 133.
121. С. И. Вольфкович, А. А. Ионасс, Р. Е. Ремен, Л. Н. С и д е л fa-
ко в с к и й, В. Н. Щевелев, ЖПХ, 38, № 1, 3 (1965). — 122. М. С. Г о л ь д е р-
б и т е р, М. Е. Г и л л е р, И. М. К и л о ч и ц к и й, П. И. С а в ч е н к о в, В. Д. В а-
сильченко, авт. свид. 150845, 1962. — 123. Д. Андо, С. Мацу но, J. Chem.
Soc. Japan. Ind. Chem. Sec., 67, № 6, 880 (1964). — 124. Д. E. T упан, пат. США
3058804, 1962. —125. C. A. Hollingsworth, W. A. Kirkland, пат. США
2995436; С. A. Hollingsworth, пат. США 2995437, 1961; австрал. пат. 242653,
1963. —126. С. И. Вол ьфкович, М. Е. Гиллер, М. С. Гольденбитер,
А. А. Ионасс, И. М. Килочицкий, Р. Е. Ремен. Хим. пром., № 6, 18
(1965). — 127. М. А. Вейдерма, сб. «Исследования по химии и технологии
удобрений, пестицидов, солей», Изд. «Наука», 1966, стр. 201. — 128. С. И. Вольф-
кович, А. И. Окунев, В. И. Пирогов, авт. свид. 171393, 1965.—
129. С. И. Вольфкович, Г. Лоренц, В. А. Жукова, Л. Н. Сидель-
ковский. В. Л. Руссо, Т. Н. Ягодина, Хим. пром., № 6, 459 (1965).—
130. А X. Бронников, Тезисы докладов на Всесоюзн. совещании по фосфа-
там, Изд. «Недра», 1966. стр. 13.
131. М. А. Вейдерма, С. И. Вольфкович, ЖПХ, 37, № 5, 937
(1964). — 132. М. А. Вейдерма, С. И. Вольфкович, Хим. пром., № 8, 687
(1964). — 133. С. И. Вольфкович, Р. Е. Р е м е и, Е. И. Петрова, Реф.
н.-п. раб. за 1957 г., ч. 1, стр. 38; Труды НИУИФ, в. 163, 1959. —
134. Е. Н. Wight, D. L. Anderson, пат. США 2234511, 1941. — 135. М. S h о-
eld, пат. США 2288112, 1942; 2328884, 1954; US Reissued Pat. 22500, 1944,—
136. С. Butt, пат. США 2360197, 1944; 2442969, 1948. —137. N. С. Rockwood,
Rock Products, 47, № 7, 44 (1944). — 138. К. Elmore, L. Kelly, пат. США
2368649, 1945. — 139. E. I. F о x, W. L. Hill, K. D. Jacob, D. S. Reynolds,
Ind. Eng. Chem., 38, № 3, 329 (1946). —140. C. A. Hollingsworth, пат.
США 2726928, 1955.
141. H. E. Пестов, Л. Ф. Гревцева, Труды НИУИФ, в. 129, 1936,
стр. 12. — 142. А. Шерешевский, Е. Бабкин, Там же, в. 143, 1938, стр. 12. —
143. I. Н. Walthall, G. L. Bridger, Ind. Eng. Chem., 35, № 7, 774 (1943).—
144. К. И. Загвоздки н, Н. А. Барилко, ЖПХ, 20, № 6, 502 (1947).—
145. S Gericke, Angew. Chem., 56, № 21—22, 149; № 41—42, 287 (1943); 57,
№ 1, 23 (1944); 60, № 4, 98 (1948); 61, № 10, 410 (1949); 62, № 8, 193 (1950). —
146. S. Nagai, I. Kawasumi, T. Nakasawa, J. Electrochem. Soc. Japan,
18, № 6, 192 (1950); 18, № 8, 261 (1950); 19. 26 (1951); Chem. Abs., 45, № 18,
8178 (1951). —147. S. Nagai, I. Ando, J. Chem. Soc. Japan. Ind. Chem., Sec.,
54, № 8, 495 (1951). — 148. S. Nagai, I. Ando, M. Sakamaki, H. Vasui,
J. Chem. Soc. Japan. Ind. Chem. Sec., 56, № 2, 62 (1953). — 149. Э. В. Брицке,
1060
Гл. XXIX. Термические фосфаты
А. А. Ионасс, сб. «Исследования по прикладной химии», Изд. АН СССР, 1955,
стр. 58. —150. I. Ando, J. Chem. Soc. Japan. Ind. Chem. Sec., 61, № 3, 310
(1958).
151. I. Ando, Bull. Chem. Soc. Japan, 31, № 2, 201 (1958). — 152. T. S a t a,
ibid., № 4, 408 (1958).— 153. Г. Г. H о з а д з e, Сообщение АН ГрузССР, 20,
№ 5, 541 (1958). — 154. А. И. Соклаков, С. И. Вольфкович,
В В. Илларионов, Р. Е. Ремен, ЖПХ, 35, № 7, 1405 (1962).—
155. Р. Я. Б е р з и н ь, У. Я. Сед мал, А. Я- В а й в а л д, Изв. АН ЛатвССР,
Сер. хим., № 6, 663 (1963). — 156. R. К i у о u г а, Т. S a t a, J. Chem. Soc. Japan.
Ind. Chem. Sec., 56, № 9, 666, 668 (1953). — 157. R. Ki у our a, T. S a t a, ibid.,
56, № 10, 748 (1953). — 158. H. H. Постников, Г. Г. H о з а д з e, Сообщение
о научно-технических работах НИУИФ, № 2, 43 (1957). —159. Jashido, Juke-
to, J. Chem. Soc. Japan, 63, 439 (1942). —160. Chem. Trade J., № 3073, 797
(1946).
161. P. Caldwell, Chem. Eng., 53, № 11, 212 (1946); 54, № 5, 214 (1947). —
162. Chem. Trade J., № 3200, 392 (1948); № 3206, 556 (1948). — 163. W. L. Hill,
F. N. Ward, W. H. A r m i g e r, K. D. Jacob, J. Assoc. Offic. Agr. Chemists,
31, № 2, 381 (1948). —164. S. Nagai, I. Kawasumi, T. Nakasawa, J.
Electrochem. Soc. Japan, 18, № 5, 155 (1950). — 165. R. Ki у our a, T. S a t a)
J. Chem. Soc. Japan. Ind. Chem. Sec., № 9, 668 (1953); № 10, 718, 750 (1953).—
166. Huang-Tha-Ho, Farm. Chem., № I, 41, 46, 49 (1953). —167. Chem.
Process. Eng., 35, № 7, 213 (1954). —168. Fert. Feed Stuffs J., 41, № 5, 175
(1954). — 169. W. I. Gromberg, Rock Products, 51, № 10, 108 (1948).—
170. K. Elmore, L. Kelly, Eng. Mining J., 150, 98 (1949).
171. A. Cuzuki, O. Miydzaki, M. T о m i k i, M. Koga, J. Chem. Soc.
Japan. Ind. Chem. Sec., 57, № 4, 261, 263 (1954); № 5, 350 (1954).—
172. А. А. И о н а с с, P. E. Ремен, T. H. Ягодина, Реф. н.-и. раб. за 1957 г.,
ч. I, стр. 50; Труды НИУИФ, в. 163, 1959. — 173. L. Kaser, чехосл. пат. 102459,
1962.— 174. J. Dankiewich, К- Przylgcko-Grochowiczowa, М. D а п-
kiewicz, A. Choroszcynski, L. Chojnacki, Przem. Chem., 41, № 2, 77
(1962). — 175. А. В. Гринева, В. Ф. П а к о в с к а я, Научный ежегодник Одес-
ского ун-та. Хнм. ф-т, в. 2, Одесса, 1961, стр. 36. — 176. Д. Г. Иванов,
И. П. К ы р ш е в, Т. Трен да фи лов, Хим. и индустрия, № 2, 49 (1962).—
177. Д. Н. Прянишников, Из результатов вегетационных опытов и лабора-
торных работ, 6, 292 (1911). — 178. A. Messerschmidt, Z.anorg. Chem., 35,
537 (1922). — 179. С. И. Вольфкович, С. С. Перельман, ЖХП, 7, № 31—
33, 1880 (1930); Удобр. и урожай, № 7—8, 570 (1930). — 180. С. И. Вольфко-
вич и др., сб. «Хибинские апатиты», т. II, 1932; Труды НИУИФ, в. 95, 1932.
181. А. Шерешевский, С. Кремер, Там же, в. 134, 1936. — 182. А. М е s-
serschmidt, Angew. Chem., H14, 197 (1938). — 183. А. Б. Б e к т у p о в, Ис-
следование химии и химической технологии термофосфатов, Изд. АН КазССР,
1947.— 184. R. Schober, Angew. Chem., 63, № 3, 71 (1951). —185. Л. А. Ка-
душкина, А. Б. Бектуров, Изв. КазССР. Сер. хим., № 5, 107 (1953) —
186. К. Doer ff el, Chem. Technik, 5, № 3, 139 (1953). — 187. F. Tro mb e,
M. Foex, Bull. Soc. chim. France, 1954, № 10, 1282. — 188. Huang-Tha-Ho,
J. Agr. Food Chem., 2, № 8, 487 (1954). —189. K. Akerman a. oth., Przem.
Chem., 10, № 9, 460 (1954). — 190. K- Akerman, K- Lasiewicz, H. Za-
wadzka, ibid., 10, № 9, 465 (1954).
191. В. Бобровницкий, 3. Свенцкий, Билл. Польск. АН, отд. 3,
3, № 4, 231 (1955). — 192. I. I. S. Cornes, New Zealand J. Sci. Technol., В 37,
№ 1, 78 (1955). —193. P. В lane, Chim. analyt, 37, № 2, 57 (1955).—
194. K. D. Jacob, W. Z. Hill, H. L. Marshall, D. S. Reynolds, US Dept.
Agr. Tech. Bull., 364 (1933). — 195. Chem. Trade J., 118, № 3069, 341 (1946).—
196. E. M. Crow ter, F. M. Lea, Chem. Trade J., 118, № 3073, 797 (1946). —
Литература
1061
197. Chem. Age, № 1509, 801 (1948)’. —/98. W. L. Hill, Agr. Chem., 5, № 12,
55 (1950). —199. Ф. Ф. Замятина, Автореф. канд. дисс., НИУИФ, 1955.—
200. G. Т. G a d г е, J. Sci. Ind. Res., 13, № 1 В, 46 (1954).
201. А. Б. Б e к т у p о в, Ю. К. У в а л и е в, Изв. АН КазССР. Сер. хим.,
№ 10, 3 (1956). — 202. А. Б. Бектуров, С. И. Калмыков, Труды Ин-та
хим. наук АН КазССР, в. 1, 1957, стр. 42.—203. А. X. Бронников, Изв. ву-
зов. Хнм. и хим. технол., № 3, 86 (1958). — 204. Е. I о n е s с u, A. Wohl, Rev.
chim., 10, № 1, 17 (1959). — 205. A. Wohl, ibid., 8, № 4, 234 (1957).—
206. E. Grydlik e. a., Przem. Chem., 40, № 6, 341 (1957).—207. S. N a g a i,
T. Miharu, J. Chem. Soc. Japan. Ind. Chem. Sec., 46, 1152 (1943); Chem. Abs.,
42, 6483 (1948). — 208. N. Jayaraman, Proc. Natl Inst. Sci. India, 9, 147
(1943); Chem. Abs., 42, 7910 (1948). — 209. S. N a g a i, I. Kawazumi, J. Chem.
Soc. Japan. Ind. Chem. Sec., 52, 127 (1949); Chem. Abs., 45, № 4, 1713 (1951).—
210. S. N a g a i, Okada, T. S a t a, J. Chem. Soc. Japan. Ind. Chem. Sec., 45,
№ 9, 3977 (1951).
211. B. Schatzel, Chem. Techn., 3, № 5, 138 (1951).—212. G. L. Brid-
ger, D. R. В о у 1 a n d, Ind. Eng. Chem., 45, № 3, 646 (1953). — 213. А. Б. Бек-
туров, В. А. Тимофеева, Изв. АН КазССР. Сер. хим., № 5, 102 (1953).—
214. D. R. В о у 1 a n d, М. A. Larson, J. Agr. Food Chem., 5, № 2, 104 (1957). —
215. M. A. Larson, D. R. В о у 1 a n d, ibid., 7, № 6, 408 (1959).—
216. С. И. Вольфкович, H. H. Постников, А. А. Ионасс, P. E. Ремен,
Реф. н.-и. раб. за 1957 г.„ ч. 1, стр. 38; Труды НИУИФ, в. 163, 1959. —
217. W. Bobrownick i, Т. Pieniazek, Chem. stosow, 4, № 3—4, 359
(1960).—218. W. Bobrownick i, T. Pieniazek, польск. пат. 46877, 1963.—
219. А. Б. Бектуров, Ю. А. Покровская, С. И. Калмыков,- Изв. АН
КазССР. Сер. хим., в. 2 (16), 21 (1960). — 220. А. Б. Бектуров, В. В. Тихо-
нов, С. И. Калмыков, сб. «Циклонные плавильные энерго-технологические
процессы», Госметаллургиздат, 1963, стр. 94.
221. А. . Б. Бектуров, А. К. Ильясова, Р. А. Г е с к и н а, авт.
свид. 179284, 1966. — 222. Англ. пат. 873426, 1961. — 223. А. Б. Бектуров,
В. В. Тихонов, В. К. Э с и к, Изв. АН КазССР. Сер. хим. наук, № 4, 3
(1965). — 224. Э. В. Брицке, Н. Е. Пестов, Труды НИУ, в. 59, 1929, стр. 131;
Удобр. и урожай, Ns 2, 67 (1929).— 225. Э. В. Брицке, С. С. Драгунов, Мин.
сырье и цвета, металлург., Ns 4, 387 (1929). — 226. V. Pristoupil, пат. США
1925644 и 1925645, 1933.— 227. G. L. F г е а г, L. Н. Hull, Ind. Eng. Chem., 33,
1560 (1941). —228. H. A. Curtis, Chem. Eng. Report, Ns 1, TVA, 14, 35 (1948).—
229. G, R. Pole, A. W. В e i п 1 i c h, Bull. Am. Cer. Soc., 20, 229, July (1941).—
230. I. E. Seymour, Comm. Fertilizer, 91, Ns 2, 19 (1955); Farm. Chem., 118,
№ 9, 38 (1955); Agr. Chem., Ns 9, 86, 129 (1955).
231. A. B. Phillips, R. D. Y о u n g, I. S. Lewis, I. S i 1 v e r b e r g, J. Agr.
Food Chem., 5, № 11, 839 (1957).—232. R. D. J о u n g, F. G. H e 1 e, A. B. Phil-
lips, J. Agr. Food Chem., 9, Ns 1, 4 (1961).—233. M. R. Siegel, J. G. Geit-
singer, H. C. Mann, ibid., 10, № 1, 72 (1962).—234. J. E. Segmour, пат. США
2978312, 1961. —235. E. H. Brown, J. R. Lehr, J. Agr. Food Chem., 12, № 3,
201 (1964).
Глава XXX
ПРОМЫШЛЕННЫЕ ПРОДУКТЫ, СОДЕРЖАЩИЕ ФОСФОР
К неорганическим соединениям фосфора, применяемым в раз-
ных отраслях промышленности (стр. 940), относятся сульфиды и
хлориды фосфора, фосфиды, фосфорный ангидрид и фосфорная
кислота, ортофосфаты и дегидратированные фосфаты натрия и
калия, фосфаты аммония и двойные фосфорнокислые соли аммо-
ния, фосфаты кальция и магния, фосфаты цинка, марганца, меди,
серебра, железа, алюминия, кобальта, церия и т. д.1-5
Свойства и способы производства фосфорной кислоты и фос-
форного ангидрида описаны выше (стр. 940). Ниже приводятся
сведения о некоторых других соединениях фосфора, имеющих
наибольшее промышленное значение.
СУЛЬФИДЫ ФОСФОРА
Фосфор полуторасернистый, или сесквисульфид фосфора, P4S3
и фосфор пятисернистый P2S5 получают взаимодействием желтого
фосфора и расплавленной серы в атмосфере углекислого или
инертного газа при температуре выше 250°. Полуторасернистый
фосфор имеет плотность 2,03 г]с.м?, кристаллизуется в ромбической
системе, плавится при 178°, воспламеняется при 80—89°. Он при-
меняется в качестве составной части смеси, которой покрывается
терка спичечных коробок.
Пятисернистый фосфор (плотность 2,03 zfcAfi, расплывается на
воздухе, плавится при 276°) служит в качестве полупродукта для
некоторых органических синтезов, например, крезиловых аэрофло-
тов (флотационный реагент) и соединений, добавляемых к сма-
зочным маслам для повышения их стойкости6. Согласно
ГОСТ 7200—54, содержание серы и фосфора в сумме должно быть
fi продукте 1-го сорта не менее 98%, 2-го сорта — не менее 96,5%,
Хлориды фосфора
1063
при отношении серы к фосфору 2,5—2,7 : 1; количество не раствори-
мого в соляной кислоте остатка — не более 0,3% для 1-го сорта
и 0,6% для 2-го сорта.
Помимо P4S3 и P4S10 (P2S5) известны также P4Ss и P4S77-9.
По другим данным 10, на кривой ликвидуса системы Р — S отме-
чены максимумы, отвечающие соединениям P4S10, P4S7, P4S3 и P4S2.
ХЛОРИДЫ ФОСФОРА
В технике нашли применение6 треххлористый фосфор РС1з, пя-
тихлористый фосфор PCI5 и хлорокись фосфора РОС13. Известны
и другие хлориды фосфора, например, дифосфортетрахлорид
Р2С14П.
Треххлористый фосфор образуется при действии газообразного
хлора на расплавленный фосфор в виде подвижной бесцветной
жидкости плотностью 1,57 г!см3, дымящей на воздухе и кипящей
при 76°.
Технический продукт содержит 99—97,5% треххлористого фос-
фора и 2,5—5% хлорокиси фосфора.
Для получения треххлористого фосфора непрерывным способом
предложено 12’13 обрабатывать газообразным хлором раствор фос-
фора в треххлористом фосфоре при 70—80°. Жидкий фосфор по-
дают насосом с помощью воды в емкость с растворителем, откуда
раствор фосфора в РС13 направляют в хлоратор. Температуру
регулируют, поддерживая концентрацию фосфора в растворе, рав-
ной 5—12%. Выделяющиеся пары РС13 конденсируют и часть кон-
денсата возвращают в процесс.
Треххлористый фосфор высокой чистоты, содержащий до
2 -10 4% примесей, можно получить, используя или исходные про-
дукты высокого качества14, или ректификацией сырого продук-
та 15, образующегося при хлорировании раствора фосфора в РС13.
Пятихлористый фосфор получают в твердом виде (плотность
1,6 г/см3) пропусканием хлора через треххлористый фосфор.
Хлорокись фосфора представляет собой бесцветную дымящую
жидкость плотностью 1,68 г/см3, кипящую при 107°. В системах
РС13—РОС13 и РС15—РОС13 образуются эвтектические смеси16.
Хлорокись фосфора получается взаимодействием хлора со смесью
метафосфата кальция или природных фосфатов 17 и угля или окис-
лением треххлористого фосфора кислородом.
Предложено 18 окисление треххлористого фосфора проводить в
присутствии 0,003—0,03% органической перекиси при 100—200°
и давлении 5—20 ат. Процесс осуществляют в системе последо-
вательно соединенных охлаждаемых снаружи труб. Изучена 19 за-
висимость выхода различных продуктов при взаимодействии смеси
кислорода и хлора с расплавленным фосфором в присутствии
хлорокиси фосфора.
1064
Гл. XXX. Промышленные продукты, содержащие фосфор
Хлориды фосфора содержат подвижные атомы хлора, легко
вступающие в химические реакции. В связи с этим они исполь-
зуются для получения хлоропроизводных углеводородов, спиртов
и др.
Указывают20, что взаимодействием оксихлорида фосфора и
аммиака можно получить удобрение замедленного действия.
ФОСФИДЫ
Известны21 фосфиды многих металлов, находящие разнообраз-
ное применение.
Фосфид железа Fe2P (плотность 6,56 г/с.и3) является побочным
продуктом при электровозгонке фосфора из железосодержащих
фосфоритов, выпускаемым под названием феррофосфор. Он может
специально получаться в доменной печи22. Применяется в метал-
лургической промышленности при производстве стали и чугуна
с повышенным количеством фосфора. Феррофосфор содержит около
20% фосфора, до 6% марганца, 4—8% кремния, до 0,5% серы.
Предложена23'27 переработка феррофосфора в тринатрийфосфат
путем спекания его в присутствии избытка воздуха с содой, с по-
следующим выщелачиванием горячей водой. Разработан28 процесс
переработки феррофосфора в высокопроцентное железо и фосфат-
ный шлак, годный для использования в качестве удобрения
или кормового средства. Он заключается в сплавлении фйр-
рофосфора в электрической печи с кремнеземистой железной
РУДОЙ.
Фосфид цинка 7п3Рг (плотность 4,55 г/см5, кристаллизуется
в кубической системе) получают сплавлением фосфора с металли-
ческим цинком и измельчением плава в шаровых мельницах до
остатка на сите с сеткой № 009 не более 17%. Разработан способ
его получения из фосфорной кислоты и окиси цинка29. В соответ-
ствии с ГОСТ 13081—67 содержание фосфидного фосфора в фос-
фиде цинка должно быть не менее 20%. Применяется в качестве
яда для борьбы с грызунами.
Фосфид кальция Са3Р2 (плотность 2,24 г 1см3) получают при
взаимодействии паров фосфора с накаленной известью.
Процесс может быть осуществлен также с применением крас-
ного фосфора30, ’ загружаемого в нижнюю обогреваемую часть
реактора. В верхней его части помещают известь, которая взаимо-
действует с парами фосфора, поступающими снизу. Фосфид кальция
применяется в смеси с карбидом кальция в качестве наполнителя
сигнальных буев, действие которых основано на самозажигании
фосфористого водорода, образующегося при взаимодействии фос-
фида кальция с водой.
Ортофосфаты натрия и калия
1065
Взаимодействием фосфида кальция с порошкообразными ме-
таллами или их хлоридами могут быть получены фосфиды титана,
ванадия, ниобия, тантала и др.31
Фосфид меди CujP применяется как раскислитель при про-
изводстве бронз в виде продукта с содержанием 9—15% Р (плот-
ность 6,59—6,75 г/см3). Получается из красного фосфора и рас-
плавленной меди в графитовых тиглях; при этом степень исполь-
зования фосфора невелика. Представляет интерес непосредственное
получение СазР в электрических печах из апатита, глины, квар-
цита, угля и металлической меди 32>33.
Фосфид алюминия А1Р — порошкообразное кристаллическое
вещество зеленоватого цвета применяется для борьбы с амбарными
грызунами и хлебными вредителями. Содержит 30—40% активного
фосфора. Хранится в герметической таре. Производится34 пропу-
сканием паров фосфора над нагретым алюминием или нагреванием
до 400—500° смеси тонкого (50 мк) алюминиевого порошка с крас-
ным фосфором в бескислородной среде. Реакция экзотермическая
и протекает в дальнейшем без подвода тепла.
Рекомендуют35 добавлять в смесь алюминиевого порошка и
красного фосфора карбонаты аммония, позволяющие снизить тем-
пературу реакции, вследствие чего взаимодействие между реаген-
тами протекает более спокойно.
Фосфид алюминия может быть также получен путем замеще-
ния34’36— взаимодействием металлического алюминия с фосфидом
цинка. Процесс проводят при 700—1000° по стехиометрическим ко-
личествам реагентов37.
О фосфиде бора см. в гл. X.
ОРТОФОСФАТЫ НАТРИЯ И КАЛИЯ
Растворимость 38-44 фосфатов натрия, калия и аммония в воде
приведена на рис. 308. Однозамещенные фосфаты натрия более
растворимы, чем двухзамещенные, а последние более растворимы,
чем трехзамещенные. Области кристаллизации кристаллогидратов
фосфатов натрия в системе Na2O—P2Os—Н2О при 25° показаны
на рис. 309.
Однозамещенные фосфаты натрия и калия растворяются в
воде почти без разложения. 0,05 н. раствор NaH2PO4 имеет
pH ~ 4,6 вследствие частичной диссоциации иона Н2РО4.
Двух- и трехзамещенные фосфаты натрия и калия в водном
растворе гидролизуются по схемам: .
Na2HPO4 + Н2О = NaH2PO4 + NaOH
Na3PO4 + Н2О = Na2HPO4 + NaOH
Рис. 308. Растворимость ортофосфатов натрия, калия и аммония.
Рис. 309. Система Na2O—Р2О6—Н2О при 25°.
Ортофосфаты натрия и калия
1067
При 25° 0,04 М раствор динатрийфосфата имеет pH = 9,7,
а 0,1 М раствор тринатрийфосфата—12,5 (степень гидролиза
34%) 45.
Изучена растворимость К2НРО4 в растворах фосфорной кис-
лоты46 при 25°. В системе K2SO4—Н3РО4—НгО образуется двой-
ная соль47 KHSO4 • КНгРО4.
Моно- и динатрийфосфат (так же, как и фосфаты аммония)
используют вместе с другими веществами (бура, борная кислота,
силикат натрия и др.) для пропитки тканей и дерева с целью
придания им огнестойкости. При повышенных температурах фос-
фаты разлагаются:
2NaH2PO4 — Na2H2P2O7 -I- Н2О
2Na2HPO4 • 12Н2О = Na4P2O7 + 25Н2О
(NH4)2HPO4 = НРО3 + 2NH3 + H2O
Образующиеся при этом пиро- или метафосфаты являются легко-
плавкими веществами, покрывающими нагреваемую поверхность
тонкой пленкой, которая и защищает ее от воспламенения. Фос-
фаты натрия в огнезащитных составах более предпочтительны по
сравнению с фосфатами аммония, так как при выделении аммиака
повышается кислотность фосфата, что способствует разрушению
ткани и изменению цвета ее окраски.
Ортофосфаты натрия и калия используются в производстве
фармацевтических препаратов, в медицине, фотографии и гальва-
нопластике.
Основные области применения ди- и тринатрийфосфата: вну-
трикотловая обработка воды для предупреждения образования
накипи или ее удаления, умягчение и обработка воды для питания
котлов и других целей 1’2-48’49. Фосфаты натрия реагируют с со-
лями жесткости с образованием нерастворимых солей, кото-
рые выделяются в виде хлопьев, легко удаляемых по мере на-
копления.
В настоящее время тринатрийфосфат теряет свое прежнее зна-
чение как составная часть средств для стирки и мытья, в связи
с применением более эффективных дегидратированных фосфатов
(стр. 1074).
Динатрийфосфат употребляется для культивирования дрож-
жей и в процессах брожения, в текстильной промышленности для
обработки смесей шерсти, хлопка и синтетического волокна перед
крашением и в качестве утяжелителя шелка, в стекольной и кера-
мической промышленности (фосфатные стекла, фарфоровые эмали
и глазури), в производстве красителей и пигментов (диспергатор),
как минеральная подкормка для скота, как эмульгатор при про-
изводстве сыров и т. д. Для выращивания дрожжевых культур
применяется также монокалийфосфат.
1068
Гл. XXX. Промышленные продукты, содержащие фосфор
В СССР выпускают несколько сортов динатрийфосфата, кото-
рые должны удовлетворять следующим требованиям (в %):
По ГОСТ 5.679-70 (аттестованный продукт) По ГОСТ 451-41
I сорт 11 сорт III сорт
Na2HPO4 • 12Н2О, не менее .... 98 96 92 88
Сульфаты (SO3), не более 0,1 0,1 1,0 2,0
Железо, не более 0,01 0,02 — —.
Нитраты (NO3), не более 0,003 0,003 —• —
Тяжелые металлы, осаждаемые
сероводородом, не более .... 0,002 0,002. — —
Мышьяк, не более 0,001 0,001 — —
Хлориды (С1), не более 0,5 0,07 —
Не растворимые в воде, не более 0,01 0,02 —• —
По ГОСТ 201—58 в тринатрийфосфате должно быть не менее
23,7% РО4 и не более 0,1% не растворимых в воде веществ.
Производство фосфатов натрия
Фосфаты натрия получают преимущественно из термической
фосфорной кислоты50. Разработаны и используются также способы
производства чистых солей из фосфорной кислоты, полученной сер-
нокислотным методом, а также из суперфосфата.
Мононатрийфосфат получают нейтрализацией 25%-ной фосфор-
ной кислоты раствором кальцинированной соды. Выделившиеся
примеси отфильтровывают, раствор выпаривают до плотности
1,5 а/сж3 и кристаллизуют NaH2PO4 *2И2О. Из более концентриро-
ванных растворов может быть выкристаллизован NaH2PO4'H2O.
При использовании кислоты, содержащей больше 40% Н3РО4, ре-
комендуют 51 компоненты подавать в нейтрализатор попеременно.
Вначале нейтрализацией кислоты содой получают раствор
Na2HPO4, в который затем вносят избыток Na2CO3, после чего до-
бавляют необходимое количество кислоты. Нейтрализацию прово-
дят при 85—95°. Для удаления ионов SO4~ из экстракционной
(сырой) фосфорной кислоты предложено52 добавлять к ней каль-
цийсодержащие вещества (СаО, СаСО3, двойной суперфосфат) и
произвести последующее катионирование кислоты. Нейтрализа-
цией содой или поташом кислоты, очищенной от сульфат-ионов,
можно получить концентрированные растворы фосфатов натрия
или калия. Для получения безводной соли мононатрийфосфат
должен быть нагрет до 100°. Раствор мононатрийфосфата можно
гранулировать в распылительной сушилке.
Получение фосфатов натрия из суперфосфата основано на об-
работке его раствором сульфата натрия 53. При этом в жидкой фа-
зе образуется мононатрийфосфат:
Са(Н2РО4)2 + Н3РО4 + Na2SO4 + 2Н2О = 2NaH2PO4 + Н3РО4 + CaSO4 • 2Н2О
Ортофосфаты натрия и калия
1069
Кислый раствор мононатрийфосфата отфильтровывают от не-
растворимого остатка суперфосфата и от вновь выделившегося
сульфата кальция и затем перерабатывают как описано выше.
Ди- и тринатрийфосфат производят 53-55 нейтрализацией фос-
форной кислоты в две ступени: сначала кальцинированной содой
до динатрийфосфата, а затем едким натром до тринатрийфосфата;
Н3РО4 + Na2CO3 = Na2HPO4 + СО2 + Н2О
Na2HPO4 + NaOH = Na3PO4 + H2O
При производстве динатрийфосфата для полноты разложения
соды применяют небольшой избыток фосфорной кислоты, который
нейтрализуется затем маточным раствором. Реакторы нагревают
паром до 98—100°. Раствор динатрийфосфата после отделения вы-
делившегося осадка перерабатывают на кристаллический динат-
рийфосфат. Чистый динатрийфосфат (безводный, двух- или семи-
водный, в зависимости от температуры) выделяется при взаимо-
действии с содой технически чистого NH4H2PO4, полученного из
экстракционной фосфорной кислоты 56. Соду и моноаммонийфосфат
подают в раствор Г4агНРО4, насыщенный при температуре реак-
ции. При этом в газовую фазу выделяются NH3 и СО2, а в твердую
фазу кристаллический Na2HPO4.
Для получения тринатрийфосфата раствор динатрийфосфата
нейтрализуют едким натром, снова осветляют и направляют на
кристаллизацию. За счет тепла нейтрализации температура рас-
твора поднимается до 112° (температура кипения). Если исходная
фосфорная кислота имела концентрацию 25—29% Р2О5 (экст-
ракционная), растворы ди- или тринатрийфосфата до кристалли-
зации из них соли охлаждением предварительно выпаривают57
При применении концентрированной (около 45% РаО5) термической
фосфорной кислоты растворы не выпаривают. После охлаждения
нейтрализованных растворов до ~30° ди- или тринатрийфосфат
кристаллизуются в виде 12-водных кристаллогидратов. Их отде-
ляют на центрифугах и высушивают. Двенадцати водный кристал-
логидрат динатрийфосфата плавится в собственной кристаллиза-
ционной воде при 60°, а тринатрийфосфата при 70°. Это ослож-
няет высушивание продукта без выделения кристаллизационной
воды. Более просто процесс осуществляется при получении рас-
творов динатрийфосфата концентрации 19,8% и тринатрийфосфата
18,7% Р2О5, при охлаждении которых до 60° они полностью затвер-
девают в распылительной башне в гранулированный продукт 58 или
на охлаждаемых вальцах в чешуйчатый продукт. Для уменьшения
слеживаемости тринатрийфосфат дополнительно охлаждают воз-
духом в шнеках или вращающихся барабанах.
Предложено 59 получать Na3PO4 добавкой к исходным моно- и
динатрийфосфатам едкого натра, соды или основных солей и
последующей термической обработкой смеси. При смешении
1070
Гл. XXX. Промышленные продукты, содержащие фосфор
порошкообразного динатрийфосфата и жидкого технического кау-
стика образуется паста, которую можно переработать в обогревае-
мом паром аппарате типа вальцовой сушилки60. Откладываю-
щийся на поверхности вальцев тринатрийфосфат срезается ножом
и охлаждается.
При переработке в тринатрийфосфат экстракционной фосфорной
кислоты необходимо предварительно выделить из нее кремнефто-
рид натрия с помощью соды 61>62:
H2SiFe 4- Na2CO3 » Na2SiFg 4- СО2 4- Н2О
После отделения кремнефторида первый ион водорода фосфор-
ной кислоты нейтрализуют раствором соды по непрерывной схеме
при pH = 4,2—4,4. В этих условиях осаждаются фосфаты железа
и алюминия в форме легко фильтрующихся осадков63. При перио-
дическом способе усреднения фосфорной кислоты в первой сту-
пени до pH = 8,8—9,0 получаются плохо фильтрующиеся илистые
осадки. Дальнейшую переработку растворов осуществляют по опи-
санной выше схеме. Из экстракционной фосфорной кислоты (из
апатитового концентрата) получают тринатрийфосфат с содержа-
нием 19—19,2% Р2О5, 0,6—0,8% SO5. 0,1—0,4% NaOH и около
0,2% фтора. Качество этого продукта несколько хуже, чем при пе-
реработке термической фосфорной кислоты.
При обработке фосфата алюминия раствором едкого натра или
спеканием с содой образуется алюминат натрия и тринатрийфос-
фат. Последний выделяется из щелочного раствора в твердую
фазу. Аналогичным образом получают алюминатный раствор и
тринатрийфосфат щелочным выщелачиванием содово-алюминатно-
го спека. Этот процесс известен как метод получения А120з из руд,
содержащих алюминий в виде вивианита и других фосфорных со-
единений.
Электролизом раствора сульфата натрия и экстракционной
фосфорной кислоты 64'65 можно получить тринатрийфосфат и сер-
ную кислоту.
ДЕГИДРАТИРОВАННЫЕ ФОСФАТЫ НАТРИЯ
При дегидратации ортофосфатов натрия образуются различ-
ные конденсированные фосфаты 2-66-78 со структурой 66-68, анало-
гичной силикатам, так как координационное число как кремния,
так и фосфора по отношению к кислороду всегда равно четырем.
Один из четырех атомов кислорода связан с пятивалентным ато-
мом фосфора двойной связью и поэтому не может быть связую-
щим звеном между двумя атомами фосфора. В зависимости от
строения конденсированные фосфаты относят к метафосфатам или
полифосфатам.
Метафосфаты имеют общую формулу Мп[РОз]п, например,
при п, равном 3 или 4, три- или тетраметафосфаты. Структура ме-
Дегидратированные фосфаты натрия
1071
тафосфатов характеризуется образованием кольцеобразных анио-
нов79, например, для триметафосфата Na3[P3O9]:
(X yONa
o' О
NaOx| ЬО
Р Р
cZ Xo/^ONa
У солей с кольцеобразными анионами отношение заряда ка-
тионов к количеству атомов фосфора строго равно 1:1.
Известен двойной метафосфат натрия и кальция Na^CaPsOi»
с предполагаемым строением80:
На рис. 310 схематически показаны условия образования раз-
личных продуктов и их превращений68 при нагревании на воздухе
дигидрата однозамещенного фосфата натрия. Выше 160° обра-
зуется пирофосфат, который при температуре выше 240° переходит
в стабильную форму триметафосфата Na3P3O9 и частично в соль
Маддреля [низкотемпературную (NaPO3)n(I) и высокотемператур-
ную (NaPO3)n(II) формы]. Соль Маддреля формы (NaPO3)„(II)
при температуре около 400° переходит необратимо в триметафос-
фат На3РзОэ(1). Последний при 625° образует расплав (NaPO3)x.
При медленном охлаждении расплава получается триметафосфат
как стабильной Ыа3РзО9(1), так и нестабильных форм Na3P3O9(H)
и Па3Р3О9(Ш). При быстром охлаждении расплава метафосфата
образуется стекловидная соль Грэма. Последняя ниже 600° пере-
ходит в стабильный триметафосфат, а в присутствии затравки в
натриевую соль Курроля (ПаРОз)ж(А). Под действием влаги или
механической обработки из формы А соли Курроля образуется
форма В, которая при 380—450° переходит в триметафосфат. Обе
эти формы могут быть получены также из расплава метафосфата.
Форма А соли Курроля при охлаждении переходит в (NaPO3)n(II).
Полифосфаты имеют общую формулу Мп+гРпОзп+i или
М„Н2РпОзп+ь где п может быть равно от 1 до ~ 10е. Полифос-
фаты характеризуются тем, что их анионы состоят из неразветвлен-
ных цепей, представляющих собой (РО4) — тетраэдры, связанные
1072
Гл. XXX. Промышленные продукты, содержащие фосфор
•е
о
•е
о
к
с
o0f3<
ofiSS-OOS
(NaPO3)r (расплав) (закалка) > I (Napo3^ (стекло) |
Рис. 310. Образование различных продуктов й их превращения при нагревании на воздухе Naf^POi • 2Н2О
(основные продукты помещены в прямоугольники).
Дегидратированные фосфаты натрия
1073
друг с другом атомами кислорада. Пример триполифосфата с не-
разветвленной цепью — Na5[P3Oi0]:
ООО
II II II
NaO—Р—О—Р—О—Р—ONa
I I I
ONa ONa ONa
Отношение заряда катионов к количеству атомов фосфора прибли-
жается к 1 : 1 с возрастанием длины цепи.
Широко известный стекловидный фосфат, неправильно назы-
ваемый гексаметафосфатом (соль Грэма), вследствие цепного
строения относится к типу полифосфатов (а не метафосфатов, как
считали ранее).
Конденсированные фосфаты, имеющие разветвленную цепь, со-
держат один или несколько агомов фосфора, которые связаны че-
рез атомы кислорода с тремя другими атомами фосфора, на-
пример:
ООО
II II II
МО—Р—О—Р—О—Р—ОМ
I I I
ООО
I I I
МО—Р—О—Р—О—Р—ом
II II II
ООО
Вследствие своей разветвленной структуры они получили назва-
ние ячеистых фосфатов (изомета- и изополифосфаты). К ним отно-
сится и летучая модификация фосфорного ангидрида [РЮюЬ, где
п может иметь значение от 1 до весьма большого числа. Изомета-
и изополифосфаты имеют такой же состав, что и мета- и полифос-
фаты. Эти фосфаты при обычной температуре быстро гидролизуют-
ся в нейтральном щелочном растворе с образованием нормальных
полифосфатов. Гидролиз происходит в местах разветвления цепей,
где все три связующие атома кислорода соединены с атомами фос-
фора ковалентной связью.
Среди метафосфатов наиболее известен триметафосфат натрия
Na3[P3O9]. Метафосфаты являются солями очень сильных кислот
(их водные растворы имеют нейтральную реакцию). В воде хорошо
растворимы только щелочные соли. Безводный триметафосфат на-
трия устойчив при нагревании почти до точки плавления. Осталь-
ные метафосфаты при высоких температурах переходят в смеси
полифосфатов путем гидролитического расщепления кольцеобраз-
ных анионов. В нейтральном водном растворе метафосфаты устой-
чивы при комнатной температуре, а в кислой среде при темпера-
туре выше ~60° они быстро гидролизуются через промежуточные
1074
Гл. XXX. Промышленные продукты, содержащие фосфор
ступени до ортофосфата. В щелочных растворах они разлагаются |
до смеси полифосфатов (три- и тетрафосфатов). |
Полифосфаты и относящаяся к этой группе не растворимая в 1
воде соль Маддреля [NaPO3]n • Н2О являются солями кислот, обра- 1
зующих ряд, представленный в табл. 83 67. Цепи полифосфорных |
кислот имеют у каждого атома фосфора сильнокислую группу ОН |
и, кроме того, на концах цепи по две слабокислых ОН-группы. Со- 5
ответственно этому растворы полифосфатов, полученных из пер- 1
вичных ортофосфатов, дают слабокислую реакцию, а растворы их |
«нейтральных солей» — слабощелочную реакцию. Слабокислые, ней- ’
тральные и щелочные растворы полифосфатов прй обычной темпе- (
ратуре являются стойкими; выше 60°, особенно в кислой среде, они ;
гидролизуются. ?
ТАБЛИЦА 83
Полифосфорные кислоты Нп+2Р„О3п+1
Кислота Количество групп ОН
сильно- кислых слабо- кислых
Моноортофосфорная Н3РО4 1 2
Дипирофосфорная Н4Р2О7 2 2
Трнфосфорная Н6Р3О]0 3 2
Тетрафосфорная HeP40i3 4 2
Полифосфорная кислота, соответствующая соли Маддреля, Нп+2РпО3„+1 п 2
Наиболее важным свойством полифосфатов натрия, на котором
основано их широкое практическое применение, является способ-
ность связывать кальций и магний, умягчая тем самым воду81-87.
Эта способность полифосфатов объясняется тем, что они обладают
свойствами ионообменников88. Триполифосфат Na5PsOi0 с солями
жесткости образует соль Ca2NaP3Oi0, выделяющуюся в осадок при
достаточной концентрации ионов Са2+ в растворе. Он способен свя-
зать 10—11% кальция или 6,4% магния (от своего веса). Стекло-
образные фосфаты могут связывать 12—18% кальция или 2,9—
3,8% магния.
Другим свойством дегидратированных фосфатов (особенно три-
полифосфата) является способность пептизировать суспензии и
снижать их вязкость, вследствие чего они используются при флота-
ции руд89. Добавка во флотационную пульпу ~1 кг/т гексамета-
фосфата натрия увеличивает содержание ценного компонента в кон-
центрате при перечистке примерно в 2 раза90. В присутствии поли-
фосфатов натрия органические поверхностно-активные вещества
значительно улучшают свою моющую способность. В связи с ука-
занными выше свойствами триполифосфат и стекловидный фосфат
сейчас широко применяют для устранения солей жесткости из
Дегидратированные фосфаты натрия
1075
воды, идущей для питания силовых установок, и во всех других
случаях, когда необходимо предотвратить выделение осадков. Сте-
кловидные фосфаты используют непосредственно при стирке белья
и поверхностной чистке одежды. Триполифосфат успешно приме-
няют как составную часть (до 85%) большинства моющих смесей.
Тетранатрийфосфат добавляют к обычному мылу. Дегидратирован-
ные фосфаты применяют как средство торможения коррозии ме-
талла 91> 92, для удаления жира и масла из хлопка, шерсти и шелка,
как катализаторы ё процессах дегидрирования, алкилирования и
полимеризации углеводородов, как нефтеочистные агенты, для регу-
лирования вязкости бурового шлама, в процессах флотации, как
активные диспергаторы красок и минеральных суспензий (каоли-
нов, известняка и др.), как эмульгаторы в производстве сыров,
в гальванопластике и при электролизе, в производстве синтетиче-
ского каучука, в кожевенной промышленности, при отбелке соломы,
для стабилизации растворов пергидроля, для улучшения фильтруе-
мости вискозных растворов, для промотирования роста кристал-
лов93 и др. Пирофосфат натрия используют как реагент при обога-
щении природных серных руд. Кислый пирофосфат натрия приме-
няют преимущественно в качестве составной части хлебопекарных
порошков. Тринатрийпирофосфат Na3HP2O7 вследствие нейтраль-
ной реакции (pH = 7) наиболее приемлем в производстве колбас и
других мясных изделий.
Триполифосфат натрия должен отвечать требованиям, пред-
ставленным в табл. 84.
ТАБЛИЦА 84
Требования к качеству триполифосфата натрия
Из термической кислоты ПО ГОСТ 13493-68 Из экстракционной кислоты
1-го сорта 2-го сорта 1-го сорта 2-го сорта
Внешний вид .... Белый порошок Порошок бе- лого цвета с желтоватым или .сероватым оттенком Белый порошок Белый порошок с желтоватым или сероватым оттенком
Содержание P2OS 56,5 53,5 51,5
в %, не менее . . Содержание Na5P3O10 54,0
в %, не менее . . Содержание нераст- 92,0 90,0 75,0 60,0
воримого Ма5РзОю 12,0
в %, не более . . 10,0 12,0 20,0
pH 1 % -ного раствора 9,3-9,8 9,3-9,8
в пределах .... Не норми- ровано Не нормиро- вано
1076
Гл. XXX. Промышленные продукты, содержащие фосфор
Производство триполифосфата натрия и других
дегидратированных фосфатов
Для производства триполифосфата натрия используют как тер-
мическую кислоту, так и экстракционную кислоту при условии ее
очистки86. Фосфорную кислоту нейтрализуют содой до получения
раствора с отношением 5Na2O:3P2O5:
ЗН3РО4 + 2,5Na2CO3 + nH2O = 2Na2HPO4 + NaH2PO4 + (п + 2,5)Н2О + 2,5СО2
Раствор выпаривают, и смесь сухих солей подвергают дегидра-
тации при 340—400°. Это обеспечивает получение низкотемператур-
ной модификации Na5P3Oio, устойчивой ниже 450°, которая в отли-
чие от высокотемпературной модификации, устойчивой выше 450°,
не слеживается во влажной среде и не комкуется при растворении
в воде94. Для стабилизации низкотемпературной формы триполи-
фосфата предложено95 применять добавки небольших количеств
(до 1%) карбамида, азотной кислоты, нитрата аммония и др. Эти
добавки способствуют также более полному превращению орто-
фосфатов в триполифосфат и придают белизну продукту.
Указывают96, что добавка к неполным веществам 1—2%
NH4NO3 действует каталитически на образование Ма5Р30ю, проте-
кающее при более низкой температуре. В результате этого полу-
чается низкотемпературная модификация Ма5Р30ю, отличающаяся
хорошей растворимостью97.
Ортофосфаты NaH2PO4 и Na2HPO4 при прокаливании предва-
рительно превращаются в пиро- и метафосфаты98. Например,
2Na2HPO4 + NaH2PO4 — Na4P2O7 + NaPO3 + 2H2O
Образование промежуточных соединений протекает с макси-
мальной скоростью при 185—220°. Триполифосфат натрия частично
образуется при быстрой первичной дегидратации ортофосфатов до
230°. Основная же его масса получается по реакции между пиро-
и метафосфатами:
Na4P2O7 + NaPO3 = Na5P3O10
Она протекает с максимальной скоростью при 290—310° и идет
с достаточной полнотой только при условии очень тесного контакта
между частицами.
Дегидратация ортофосфатов с образованием триполифосфата
завершается в течение 15—30 мин.
Выпаривание раствора ортофосфатов натрия с получением су-
хих солей (сушку) и дегидратацию (прокаливание) их осуществ-
ляют как раздельно, так и в одном совмещенном аппарате. По-
следний способ получил значительное распространение. Для раз-
дельной сушки предварительно упаренного в трехкорпусном
выпарном аппарате раствора применяют99 полые вращающиеся
вальцы, обогреваемые паром давлением до 5—8 ат, или распыли-
Дегидратированные фосфаты натрия
1077
тельные башни. Дегидратацию сухих фосфатов осуществляют во
вращающихся барабанных печах. Последние могут быть исполь-
зованы для совмещенной сушки исходного раствора и кальцина-
ции ортофосфатов. В этом случае во избежание налипания мате-
риала на стенки барабана упаренный раствор, содержащий
~32% Р2О5, смешивают с ретуром готового продукта 86-100’101
(в соотношении по весу 3 -е- 3,5 : 1 при влажности смеси 10—12%)
или распыливают раствор на слой сухого материала 102.
Предложено 103 получать триполифосфат во вращающемся ба-
рабане, разделенном кольцевой перегородкой на две зоны: в пер-
вой раствор, вводимый в барабан посредством распыливающих
форсунок, высушивается, а во второй протекают дальнейшие про-
цессы. Барабан обогревается изнутри с помощью газовых или неф-
тяных горелок.
Более совершенными являются распылительные сушилки 104-107.
При производительности завода 7500 т триполифосфата в год рас-
пылительная сушилка 108 представляет собой цилиндрическую баш-
ню диаметром 6,6 ж и высотой 17 м. Раствор ортофосфатов подается
под давлением и разбрызгивается в потоке горячих газов, посту-
пающих в верхнюю часть башни с температурой 400—700° и ухо-
дящих с температурой 100—200°. Из сушилки продукт с влажно-
стью менее 5% поступает для прокаливания во вращающуюся печь
или в неподвижную печь, снабженную горизонтальными лопастны-
ми мешалками,* служащими для передвижения материала. Прока-
ливание сухих ортофосфатов может быть также осуществлено в-
распылительной сушилке совместно с сушкой раствора. Перспектив-
ным представляется проведение сушки и дегидратации ортофосфа-
тов в печи со взвешенным слоем 2> 109. Но это связано с трудностями
регулирования гранулирования продукта, так как вследствие агло-
мерации первоначальных частиц в крупные агрегаты происходит
нарушение и затухание взвешивания слоя 110.
В СССР триполифосфат производят двумя способами: 1) в со-
вмещенном процессе с применением сушильно-прокалочной рас-
пылительной печи и 2) с раздельной сушкой раствора в распыли-
тельной сушилке и прокаливанием сухих ортофосфатов в кальци-
наторе специальной конструкции — турбокальцинаторе.
При совмещенном процессе111*112 сушка раствора, прокалива-
ние сухих ортофосфатов и охлаждение продукта происходят в
одном аппарате — сушильно-прокалочной печи.
Сушильно-прокалочная печь (рис. 311) представляет собой ци-
линдрическую башню, футерованную изнутри огнеупорным кир-
пичом. В нижней части печь имеет две прокалочные и две холо-
дильных тарелки; верхняя прокалочная тарелка изготовляется из
чугуна или жаропрочной стали, а нижняя — из обычной стали,
футерованной шамотным кирпичом, а холодильные — из обычной
1078
Гл. XXX. Промышленные продукты, содержащие фосфор
стали. Распыление раствора осуществляется пневматическими
форсунками под давлением 5—7 ат. Горячие газы, полученные
сжиганием газа или мазута с температурой 800—900°, поступают
Рис. 311. Совмещенная сушильно-прокалочная печь:
2 — башня; 2 — желоб; 3 —коллектор для пара; 4—форсунка;
5 и 6 —прокалочные полки; 7 —конус; 3 —шлицы: 9 —скребки;
10 и ZZ —полки охлаждения; 12 — приводной механизм;
Z3 —вал; 14 — боров.
в нижнюю часть печи из кольцевого борова через шлицы. Основ-
ная масса газа выходит в полость между прокалочными полками,
а часть газа поступает в зону над верхней тарелкой.
Сухой и частично прокаленный материал, оседающий на верх-
ней тарелке, перемещается скребками к центру и ссыпается -на
вторую прокалочную полку. Скребки и вал выполняются полыми;
Дегидратированные фосфаты натрия
1079
для их охлаждения через них продувают воздух. Со второй прока-
лочной полки материал с температурой ~400° поступает на холо-
дильные тарелки, по которым пропускают охлаждающую воду. На
выходе из башни продукт имеет температуру 30—4Сг. Топочные
газы выводятся с температурой 140—200°.
На рис. 312 показана схема производства триполифосфата нат-
рия в совмещенном процессе с применением распылительной су-
шильно-прокалочной печи. Исходный раствор перед сушкой упа-
ривают в выпарном аппарате с принудительной циркуляцией. Упа-
ренный до 60%-ной концентрации (~30—32% РгО5) раствор
Рис. 312. Схема производства триполифосфата натрия с применением
совмещенной сушильно-прокалочной печи:
/ — выпарной аппарат; 2 — скруббер; 3 — брызгоуловитель; 4 — дымосос; 5 — циклон;
6 н 12 — бункеры; 7 —мигалка; 8 — сушнльно-прокалочная печь', 9 — напорный бак;
10 — классификатор; //—дробилка; 13 — вентилятор; 14 — шнек; /5 —топка; 16 — сбор-
ник; /7 —насос; 18 — сборник раствора; 19 — фильтр; 20 — реактор; 21 — бункер для
соды; 22 — бак для фосфорной кислоты.
насосом подают через напорный бак в распределительный желоб
печи, где происходит выпаривание и сушка раствора, дегидратация
ортофосфатов и охлаждение триполифосфата. Отработанные газы
вместе с пылью направляются в батарейный циклон для очистки от
пыли. Уловленная пыль собирается в бункере, откуда она через
две мигалки возвращается на первую прокалочную полку печи.
Выходящие из циклона газы дополнительно очищаются от пыли в
скруббере, орошаемом разбавленным раствором и, пройдя циклон-
ный брызгоуловитель, выбрасываются в атмосферу.
Полученный в печи готовый продукт после грубого дробления
пневмотранспортом подается в сепаратор, откуда крупные частицы
поступают на дополнительное измельчение в мельницы тонкого
помола. Мелкий порошковидный материал улавливается в цикло-
нах и подается в бункер готового продукта.
Ю М. Е. Познн
1080
Гл. XXX. Промышленные продукты, содержащие фосфор
Недостатками совмещенного процесса являются частое забива-
ние тракта пылеочистки и трудности регулирования температур-
ного и технологического режимов сушки и прокалки продукта. При
резком повышении температуры в прокалочной зоне качество про-
дукта снижается. В сушильно-прокалочных печах с комбинирован-
ным вводом (верхним и нижним) горячего газа 112~114 процесс
сушки протекает более интенсивно. Однако полностью недостатки,
присущие совмещенному осуществлению процесса, не устраня-
ются.
Продукт более высокого качества образуется при раздельном
получении смеси сухих солей и последующем их прокаливании в
специально предназначенных для этого аппаратах. По этой схе-
ме115 для сушки раствора применяется распылительная сушилка,
обеспечивающая получение тонкодисперсного порошка, который
легко и быстро дегидратируется.
Прокаливание тонкодисперсной смеси солей эквимолекуляр-
ного состава производится в турбокальцинаторе, представляющем
собой цилиндрическую стационарную камеру, в которую вмонти-
ровано большое количество вращающихся тарелок и турбин для
интенсивного перемешивания горячих газов и обрабатываемого
твердого материала. Этим достигается хороший контакт между
продуктом и греющим газом и исключаются местные перегревы,
ухудшающие качество продукта.
Помимо условий дегидратации качество продукта зависит от
режима нейтрализации кислоты и ее состава. При отношении мо-
лярных концентраций в нейтрализованном растворе Na2O и Р2О5
меньше или больше 1,67 качество продукта снижается. Для точ-
ного поддержания этого отношения предпочитают вести нейтрали-
зацию периодически или полунепрерывно; этим достигается более
тонкая дозировка реагентов и корректировка состава конечного
раствора. На заводе в РНР116 процесс осуществляется непрерыв-
ным путем. Нейтрализацию кислоты содой ведут в три стадии
(в трех реакторах). В первом реакторе получают мононатрийфос-
фат, во втором — динатрийфосфат и, наконец, в третьем реакторе
раствор динатрийфосфата обрабатывают автоматически дозируе-
мым количеством фосфорной кислоты до образования эквимолеку-
лярной смеси NaH2PO4 и 2Na2HPO4.
Триполифосфат натрия (как и другие дегидратированные фос-
фаты), получаемый из экстракционной фосфорной кислоты, хуже
по качеству, чем триполифосфат, полученный из термической кис-
лоты. Это объясняется тем, что содержащиеся в экстракционной
кислоте примеси не только разбавляют конечный продукт, но и
действуют как каталитические пассиваторы на процесс дегидра-
тации.
При использовании экстракционной фосфорной кислоты про-
цесс осуществляют следующим образом 117. Кислоту, содержащую
Дегидратированные фосфаты натрия
1081
(в %): ~20 Р2О5) 0,4—0,6 SO3 и 1,5—1,8 фтора, нейтрализуют со-
дой до pH = 7. Выделяющийся шлам, состоящий в основном из
фосфатов полуторных металлов и кремнефторида натрия, отделяют,
а раствор упаривают в распылительной сушилке. При этом содер-
жащиеся в растворе моно- и дифосфат натрия (в молярном от-
ношении 2:1) дегидратируются при 400° за счет тепла отходящих
газов. Продукт содержит: 54,3—55% РгО5 (из них 53,2—54,3% в
полиформе и 0,4—0,9% в ортоформе), 2,8—3,5% Na2SO4 и 1—1,5%
NaF.
В другом случае118 используют экстракционную кислоту боль-
шей концентрации ( — 30% Р2О5) и содержащую большее коли-
чество сульфатов (1,8—2,0% SO3); ее предварительно очищают от
SO|". Кислоту состава: 30,2%Р2О3, 1,8% SO3, 0,2% СаО, 0,7%
фтора, 0,4% хлора, 0,3% А12О3, 0,3% MgO и 0,3% Fe2O3 обрабаты-
вают вначале известняком, а затем концентрированным раствором
соды. После отделения гипса и кремнефторида натрия к раствору
добавляют ВаСО3 и нейтрализуют его содой с последующей пере-
работкой в конечный продукт.
Полученный в процессе очистки экстракционной фосфорной
кислоты шлам, состоящий в основном из фосфатов железа и алю-
миния, используют в производстве гранулированного суперфос-
фата117. Комплексное удобрение предложено119 получать при сме-
шении шлама с нитратом аммония и калийной солью.
Для получения из экстракционной кислоты триполифосфата
такого же качества как из термической, необходима тонкая очист-
ка ее от примесей. Особенно вредной примесью являются соедине-
ния фтора. Содержание основного вещества в продукте — триполи-
фосфате натрия — обратно пропорционально содержанию фтора в
кислоте.
Предварительное обесфторивание кислоты осаждением из нее
кремнефторида натрия 120 соответствующим количеством соды при-
водит к осложнению последующих стадий процесса. Кислоту после
отделения кремнефторида натрия подвергают дальнейшей нейтра-
лизации содой до образования моно- и диортофосфатов натрия.
Выделяющиеся при этом фосфаты полуторных окислов значитель-
но хуже фильтруются, чем при одновременном соосаждении крем-
нефторида натрия. Кроме того, остаточное содержание фтора в
кислоте достаточно велико в соответствии с растворимостью в ней
кремнефторида натрия (например, -—0,5% Na2SiF6 в кислоте кон-
центрации 22—24% Р2О3). В присутствии солей алюминия и же-
леза содержание фтора в конечном нейтрализованном растворе
увеличивается вследствие образования фторалюминатов и крио-
литов. Помимо этого при повышенной температуре кремнефторид
натрия реагирует с содой:
Na2SiFe + 4Na2CO3 + 2Н2О = 6NaF + SiO2 + 4NaHCO-,
ID*
1082
Гл. XXX. Промышленные продукты, содержащие фосфор
Образующийся фторид натрия обладает значительно большей
растворимостью, чем кремнефторид.
Установлено121, что наименьшая растворимость фтористых со-
единений (0,04—0,05% фтора) наблюдается при нейтрализации
содой экстракционной фосфорной кислоты до pH = 4,5—5,0. При
исходной концентрации в кислоте 17—24% Р2О5 это соответствует
полной нейтрализации первого и 20—30% второго ионов водорода
Н3РО4. Одновременно происходит полное выделение в твердую
фазу соединений полуторных окислов. Осуществляя нейтрализа-
цию экстракционной фосфорной кислоты в две стадии с промежу-
точным отделением примесей, возможно получить -триполифосфат
высокого качества.
Для очистки раствора от сульфат-иона обычно применяют
карбонат бария.
Пищевые продукты можно получить из экстракционной кис-
лоты, очищенной с помощью алифатических растворителей и акти-
вированного угля 122. Получаемая этим способом пищевая фосфор-
ная кислота дешевле термической.
Указывают116, что при тщательной очистке фосфорной кислоты
в условиях трехстадийной непрерывной нейтрализации экстрак-
ционной фосфорной кислоты с двухкратным отделением осадков
(после нейтрализации и затем после упарки раствора) получаемый
триполифосфат содержит: 91—93% Na5P3Oi0, 55—56,8% Р2О5,
3—4% Na2SO4, 0,005% Fe и 0,003% As. Плотность его равна
0,5—0,75 г!смй, а pH 1%-ного раствора триполифосфата состав-
ляет 9,5—9,6.
Пищевой гексагидрат триполифосфата натрия 86> 123 получают
из технического продукта, растворяя его при 60—70° в воде. По-
лученный при этом 17—20%-ный раствор На5РзОю отделяют от
нерастворимых примесей. Вакуум-кристаллизацией при 60°
(с целью предотвращения гидролиза) выделяют из раствора кри-
сталлы чистого Na5P3Oi0 • 6Н2О, которые отфуговывают от маточ-
ной жидкости и высушивают.
Во избежание перехода триполифосфата в пиро- или ортофор-
мы предложено 124 сушку гексагидрата Na5P3Oio • 6Н2О производить
горячими газами, содержащими водяной пар при парциальном
давлении последнего около 200 мм рт. ст. и относительной влаж-
ности менее 40%. Этого же можно достичь 125 путем перегрева гек-
сагидрата при повышенном давлении с последующим высушива-
нием его при атмосферном давлении в распылительном аппарате
или сушкой водяным паром 126, перегретым до 250—450°.
Стекловидный фосфат натрия (соль Грэма) получают нагрева-
нием и расплавлением при 620—700° мононатрийфосфата с по-
следующим очень быстрым охлаждением плава 127>128. Для получе-
ния продукта в виде тонких чешуек расплав выливают между вра-
щающимися охлаждаемыми вальцами. Продукт содержит ~ 69,6 %
Дегидратированные фосфаты натрия
1083
Р2О5. Его раствор имеет pH около 5,2. Свойства продукта (содер-
жание основного вещества, устойчивость и степень полимериза-
ции) зависят от температуры плавления и скорости кристаллизации
плава. В периодических условиях не обеспечивается постоянство
режимных параметров как при нейтрализации кислоты содой с по-
лучением мононатрийфосфата, так и при полимеризации и кри-
сталлизации плава.
Представляет интерес разработанная в Ленниигипрохиме 129 не-
прерывная схема получения стекловидного фосфата. Нейтрали-,
зацию фосфорной кислоты содой осуществляют непрерывным
путем при автоматическом регулировании pH среды, температуры,,
времени пребывания раствора в реакторе, сигнализации уровня в
расходных емкостях. Плавление мононатрийфосфата и полимериза-
ция осуществляется в отражательной печи непрерывного действия.
Получаемый продукт отличается повышенной растворимостью и ак-
тивностью.
Разработан (А. А. Соколовский) способ получения стекловид-
ного фосфата натрия из суперфосфата через раствор мононатрий-
фосфата, получаемый обменным разложением монокальцийфос-
фата с сульфатом натрия. Процесс можно осуществить в шахтной
печи с регулированием температуры в зонах испарения, прокали-
вания и полимеризации 13°.
Описана131 установка для получения стекловидного фосфата
расплавлением NaH2PO4 при 800° в электрической печи с двумя
угольными электродами, через которые пропускается переменный
ток 200 в.
Предложено132 получение стекловидного фосфата во вращаю-
щейся трубчатой печи из смеси, состоящей из 0,5—1,5 моль
ПаНгРО4, 1,5—0,5 моль КН2РО4 и 0,1—0,45 моль ПагНРО4, которую
нагревают до 430—480° и затем быстро охлаждают. В такой же
печи возможно получение триметафосфата щелочных металлов.
Мононатрийфосфат смешивают с 13—60% КН2РО4 и с ретурным
триметафосфатом, и смесь нагревают до 360—420°.
Представляет интерес непосредственное получение мета- (и Три-
поли-) фосфата из фосфора и соды 133 в камерах для сжигания
фосфора, минуя операции приготовления исходных растворов и их
упаривание. При 600—700° образуются твердые продукты с отно-
шением Na2O : Р2О5 от 1 : 1 до 1 :2, в том числе и 5 :3:
Р4>+ 5О2 + 0,5xNa2CO3 = (0,5xNa2O) • 2Р2О5 + 0,5хСО2
Пирофосфат натрия получают дегидратацией динатрийфосфата
при 350—400°:
2Na2HPO4 = Na4P2O7 + Н2О
В присутствии небольших количеств NaH2PO4 снижается темпе-
ратура и увеличивается скорость конденсации динатрийфосфата 134.
1084
Гл. XXX. Промышленные продукты, содержащие фосфор
Скорость образования пирофосфата зависит также от молярного
отношения \агО : Р2О5135. Она имеет минимальную величину
при 170° при отношении ИагО : Р2О5, равном 1,5, вследствие образо-
вания соли NaH2PO4 • НагНРО4, устойчивой до температуры пре-
вращения ее в пирофосфат. Добавка ~1°/о NH4NO3 к весу безвод-
ного NaHPO4 ускоряет примерно в 2 раза реакцию образования
пирофосфата при 300°136.
Пирофосфат натрия можно получать в такой же аппаратуре,
как и триполифосфат137 с соответствующим изменением режима
по нейтрализации кислоты, упарке раствора и температуре прока-
ливания. Для перехода с производства одного продукта на дру-
гой требуется ~4 ч. Термическую фосфорную кислоту (70—80%)
нейтрализуют кальцинированной содой до рН = 8,8—9,0. Раствор
кипятят для удаления СОг, выпаривают до концентрации 48—50%
Ыа2НРО4 и смешивают с 3—4 вес. ч. оборотного продукта (до со-
держания влаги в смеси 10—12%). Смесь нагревается топочными
газами до 360—400° во вращающемся барабане. Спекшиеся’ча-
стицы размером до 10 мм после охлаждения измельчают. Безвод-
ный пирофосфат натрия содержит 52—53,5% общей Р2О5, в том
числе 50,5—52% в пиро-форме и 0,9% в орто-форме.
Аналогичным методом получают пирофосфат и из экстракцион-
ной фосфорной кислоты 138.
Пищевой пирофосфат натрия получают перекристаллизацией
технического пирофосфата86. Водный раствор, содержащий ~22%
Na4P2O7, полученный растворением технической соли в воде или
оборотном маточном растворе, фильтруют при 70°. Из осветлен-
ного раствора охлаждением до 13—15° выделяют десятиводные
кристаллы пирофосфата натрия.
Кислый пирофосфат натрия ЫагНгРгО? получают осторожной
дегидратацией мононатрийфосфата при 225—250° или обработкой
при более высокой температуре, но в течение очень короткого вре-
мени 139> 14°:
2NaH2PO4 = Na2H2P2O7 + Н2О
Выше 250° образуется не растворимая в воде соль Маддреля,
ниже 220° сильно снижается степень конверсии ортофосфата в пи-
рофосфат. Готовый продукт содержит до 63,9% Р2О5. Его раствор
имеет pH около 4,2.
Тринатрийпирофосфат получают123-141 взаимодействием тетра-
натрийфосфата с соляной кислотой:
Na4P2O7 + НС1 = Na3HP2O7 + NaCl
. Исходный технический пирофосфат натрия растворяют при
20—25° в воде и фильтруют нерастворимые примеси. Из осветлен-
ного раствора, содержащего 20—22% Na4P2O7, охлаждением до
10° выделяют Na4P2O7- ЮН2О.
Фосфаты аммония
1085
После отделения кристаллов на центрифуге их обрабатывают
соляной кислотой концентрацией 35% НС1 при 35° в реакторе,
снабженном мешалкой и рубашкой, в которую, по мере необходи-
мости, подают пар или воду. В полученном растворе концентрация
солей должна быть не более 35%. При охлаждении рассолом до
0—1 % из него выделяют кристаллы \азНР2О7 • 9НгО. Их отделяют
центрифугированием от маточного раствора (хлористого натрия).
В полученном продукте содержится; 31—33% Р2О5, 0,005—0,01 %
не растворимых в воде веществ, 0,5—0,6% хлоридов (в расчете на
хлор), менее 0,15% SO3, менее 0,01% Fe.
ФОСФАТЫ АММОНИЯ И ДВОЙНЫЕ ФОСФОРНОКИСЛЫЕ
СОЛИ АММОНИЯ
Растворимость фосфатов аммония 142'143 приведена на рис. 308.
Некоторые свойства фосфатов аммония и двойных фосфорнокис-
лых солей аммония представлены в табл. 85.
ТАБЛИЦА 85
Некоторые свойства фосфатов аммония и двойных фосфорнокислых солей
аммония
Формула Давление паров NH3, мм рт. ст. pH 0,1 М раствора Плот- ность, г1см* Кристаллическая система
100° 125°
(NH4)H2PO4 0,00 0,05 4,4 1,80 Тетраэдрическая
(NH4)2HPO4 5,0 30,0 8,0 1,62 Моноклинная
(NH4)3PO4 — — 9,4 —
(NH4)3PO4 -3H2O . . . 643 1177 — —
Na(NH4)HPO4 • 4H2O . . — — — —
Mg(NH4)PO4 • 6H2O . . 207,1 * — — 1,7 Ромбическая
» При 80°.
Вследствие неустойчивости триаммонийфосфат производят и
применяют в небольших количествах. Моно- и диаммонийфосфаты
в технических целях (помимо удобрений) употребляют преимуще-
ственно в производстве огнезащитных средств (антипиренов). Фос-
фаты аммония высокой чистоты, получаемые из термической фос-
форной кислоты, не содержащие примесей, используются главным
образом в пищевой и фармацевтической промышленности.
Изучаются 144 свойства и способы получения таких фосфорно-
кислых солей аммония, как Mg(NH4)PO4, Zn(NH4)PO4 и
Fe(NH4)PO4. Вследствие плохой растворимости в воде эти соли мо-
гут применяться в качестве антипиренов в составе пластических
масс и красок и для пропитки древесины и тканей, употребляемых в
условиях воздействия воды. Железные (а также алюминиевые)
1086
Гл. XXX. Промышленные продукты, содержащие фосфор
соли могут применяться в составе красок для предохранения ме-
таллических сооружений от коррозии.
Технические фосфаты аммония используют в качестве питатель-
ной среды для культивирования дрожжей, в производстве нетлею-
щих спичек, для проклейки слюды, в производстве изоляционного
материала — миканита и т. д. Технический моноаммонийфосфат
содержит ~60% Р2О5, 15% NH3 и 0,15% железа (в пересчете на
сухое вещество) и примерно 8% влаги. По ГОСТ 8515—57, диам-
монийфосфат марки А (из термической фосфорной кислоты) дол-
жен содержать не менее 50,5% Р2О5 и 22,4% NH3 при максималь-
ной влажности 6% и марки Б (из экстракционной фосфорной кис-
лоты)— не менее 48,5% Р2О5 и 21,5% NH3, при влажности не более
8%. В продукте для пищевой промышленности должно быть не
более 0,005% мышьяка и 0,3% фтора.
Моно- и диаммонийфосфат в отличие от производства их
смеси (аммофоса) получают из термической фосфорной кислоты
(не выше 77% )145. Насыщение фосфорной кислоты аммиаком ве-
дут для получения моноаммонийфосфата при 112—113°, при этом
аммиак не теряется. Маточный раствор после отделения кристал-
лов на центрифуге возвращают на смешение с фосфорной кислотой.
Отфугованные кристаллы моноаммонийфосфата, содержащие
3—4% влаги, высушивают при температуре ~100°. При получении
диаммонийфосфата, во избежание потерь аммиака, температура
насыщения не должна превышать 80—90°. Поэтому нейтрализа-
цию ведут в две ступени: в первой вводится 60% общей нормы
аммиака при температуре до 110°, затем пульпу охлаждают и вто-
рично насыщают при указанной предельной температуре. Для со-
хранения подвижности пульпы на 1 кг 77%-ной фосфорной кис-
лоты необходимо добавить 3 кг оборотного маточного раствора.
Кристаллы диаммонийфосфата менее крупные, чем моноаммоний-
фосфата, после фугования они содержат 6—10% влаги. Продукт
высушивают при 60—70°.
При получении диаммонийфосфата из термической фосфорной
кислоты (75—85%) применяется и одноступенчатое усреднение
при непрерывном введении кис-лоты и газообразного аммиака в
сатуратор, содержащий маточный раствор1’2. Необходимая темпе-
ратура (60—70°) поддерживается за счет значительного испарения
воды. Кислота нейтрализуется до pH = 5,8. В сатураторе начи-
нается кристаллизация диаммонийфосфата. Известен 148>147 одно-
ступенчатый способ усреднения термической фосфорной кислоты в
вакуум-кристаллизаторе до pH = 5,8—6,0 при 60°. Процесс можно
также проводить в горизонтальном трубчатом реакторе, в который
впрыскивается фосфорная кислота и аммиачная вода или подается
газообразный аммиак148.
Разработан 149~15* и осуществлен способ ступенчатого получе-
ния моно- и диаммонийфосфата из экстракционной фосфорной кис-
Фосфаты аммония
1087
лоты (22—25% Р2О5). Фосфорную кислоту вначале для очистки от
примесей обесфторивают содой и усредняют аммиаком при по-
стоянной величине pH = 4—4,5 (непрерывный процесс). Выделяю-
щийся при этом осадок фосфатов железа и алюминия, соединений
кальция и фтора хорошо отстаивается в сгустителях непрерывного
действия. Сгущенную пульпу отфильтровывают или отфуговывают
и осадок промывают водой. Высушенный осадок содержит около
35% Р2О5 и 5% NH3 в усвояемой форме и может быть использо-
ван как удобрение. Очищенный раствор моноаммонийфосфата вы-
паривают до концентрации 34—36% Р2О5 и охлаждают в кристал-
лизаторах до 18—20°. Выделившиеся кристаллы отделяют на цен-
трифуге, а маточный раствор возвращают на нейтрализацию или
выпарку.
При получении диаммонийфосфата выпаренный раствор перед
кристаллизацией донейтрализовывают аммиаком при охлаждении
(температура не выше 80°) в реакторе второй ступени до рН~8.
Затем раствор диаммонийфосфата направляют на кристаллиза-
цию, центрифугирование и сушку.
Для предупреждения накопления примесей в маточном раство-
ре, тормозящих рост кристаллов (NH4)2HPO4, рекомендуют152
часть маточного раствора выводить из системы в таком количестве,
чтобы циркулирующий раствор содержал в весовых отношениях
R2O3: РгО5<0,1, фтор : Р2О5 = 0,1 и SO3: Р2О5~0,3. Отводимый
раствор высушивают с получением побочного продукта, содержа-
щего 15—20% N и 35—45% Р2О5 в водной и цитратнорастворимой
формах. Его можно использовать как удобрение.
Сушку диаммонийфосфата производят при температуре не
выше 60°, во избежание потери им аммиака и перехода в моно-
аммонийфосфат. Сушку же моноаммонийфосфата можно вести при
температуре до 100—110°.
При аммонизации моноаммонийфосфата, содержащего 10—15%
влаги, безводным аммиаком получается практически безводный
диаммонийфосфат 153 вследствие испарения воды за счет тепла ре-
акции.
Предложено 154> 155 получать диаммонийфосфат нейтрализацией
выпаренного до 26% Р2О5 раствора первой ступени в вакуум-кри-
сталлизаторах при pH = 6 и 60°. В этих условиях теряется не боль-
ше 2,5% аммиака.
Представляет интерес выделение магнийаммонийфосфата при
нейтрализации аммиаком экстракционной фосфорной кислоты, со-
держащей фосфат магния (например, при получении ее из кара-
тауских или кингиссепских фосфоритов). В первой стадии нейтра-
лизации производится очистка кислоты при pH=4—4,5, а при
рН = 6—6,5 осаждают магнийаммонийфосфат. Оставшийся раствор
непосредственно или при дополнительной нейтрализации перера-
батывают в фосфаты аммония.
1088
Гл. XXX. Промышленные продукты, содержащие фосфор
В системе MgO—Р2О5—NH3—Н2О 156 при 75° выделяется в
твердую фазу одноводный магнийаммонийфосфат MgNH4₽O4 •
•Н2О. Он является значительно более устойчивым, чем шестивод-
ный гидрат, который кристаллизуется при 20—30°.
В настоящее время небольшие количества магнийаммонийфос-
фата получают из фосфорной кислоты и магниевых соединений.
В качестве последних можно использовать хлористый магний 157,
гидроокись магния 158 и др. Процесс заключается в смешении кис-
лоты с магниевой солью при нагревании и в насыщении раствора
аммиаком при одновременной кристаллизации одноводного маг-
нийаммонийфосфата. После отделения выделившихся кристаллов
от раствора их промывают водой и сушат при 105—110°.
Возможно получение безводного диаммонийфосфата из азот-
нокислотной вытяжки фосфатов159. Нейтрализацией вытяжки ам-
миаком до pH = 3,8—4 при 80—90° выделяют основную массу
примесей. Из оставшегося раствора охлаждением до 18—20° кри-
сталлизуют NH4H2PO4 и при последующей нейтрализации при 45°
до pH = 8 выделяется (NH4)2HPO4 • СаНРО4. Из отделенного филь-*
трата нейтрализацией до насыщения получают (МН4)зРО4 • ЗН2О.
Последний смешивают с NH4H2PO4, удаляют в центрифуге выде-
лившуюся воду и получают безводный (NH4)2HPO4.
Большое внимание уделяется разработке методов улавлива-
ния аммиака из коксового газа растворами фосфорной кислоты
или моноаммонийфосфата с получением фосфатов аммония, в ча-
стности, диаммонийфосфата 160’161. Равновесное содержание СО2 и
H2S в растворах моноаммонийфосфата в присутствии фосфорной
кислоты невелико 162. Поэтому предварительная очистка коксового
газа от них не требуется.
При оптимальных условиях: температуре 50—55° и соотноше-
нии NH3: Н3РО4, равном 2, выделяются крупные кристаллы диам-
монийфосфата с размерами более 0,5 мм163. Органические примеси,
содержащиеся в газе, не влияют на размеры кристаллов. В то же
время примеси цианистых соединений железа и, в особенности,
алюминия резко уменьшают их величину. Поэтому при использо-
вании экстракционной кислоты они должны быть выделены из
раствора (как и в обычном процессе) до кристаллизации диаммо-
нийфосфата.
С целью максимального поглощения аммиака при небольшом
парциальном давлении его в газе целесообразно проводить про-
цесс в многосекционном аппарате164 с получением раствора, со-
держащего до 500—600 г)л (NH4)2HPO4. При двухступенчатой ней-
трализации экстракционной фосфорной кислоты с промежуточным
выделением шлама продукт по своему составу не отличается от
диаммонийфосфата, полученного с применением термической фос-
форной кислоты 165.
Фосфаты кальция
1089
ФОСФАТЫ КАЛЬЦИЯ
Свойства системы СаО—Р2О5—НгО рассмотрены (гл. XXIV,
XXVII, XXIX). Промышленное применение имеют моно-, ди- и три-
кальцийфосфат, осажденный гидроксилапатит и пирофосфат
кальция.
Моиокальцийфосфат в виде безводной или одноводной соли ис-
пользуется для приготовления хлебопекарных порошков. Действие
монокальцийфбсфата (а также кислого пирофосфата натрия) осно-
вано на его реакции с бикарбонатом натрия, начинающейся в хо-
лодном тесте и завершающейся во время выпечки
ЗСа(Н2РО4)2 • Н2О + 8NaHCO3 = 8СО2 + Са3(РО4)2 + 4Na2HPO4 + 11Н2О
Выделяющаяся при этом СОг вызывает подъем теста. Моно-
кальцийфосфат часто примешивают к муке в количестве 0,25—0,75%
для устранения в хлебопекарных изделиях нежелательной щелоч-
ности, возникающей при употреблении соды и для изготовления
саморазрыхляющейся муки. Моиокальцийфосфат в смеси с дру-
гими веществами (бромат калия, поваренная соль, сернокислый
аммоний, крахмал) широко применяют для улучшения качества
дрожжевого теста (стимулирование роста дрожжевых клеток) '.
Примесь фосфата кальция в муке признается полезной для орга-
низма человека.
Моногидрат монокальцийфосфата получают нейтрализацией
фосфорной кислоты (~75% Н3РО4) негашеной известью при ин-
тенсивном перемешивании. Известь вводят с такой скоростью, что-
бы поддерживалась температура 75—110°. Получаются сухие ком-
ки, которые затем окончательно высушивают и измельчают. Про-
дукт обычно содержит небольшое количество дикальцийфосфата.
Кристаллический моиокальцийфосфат получают 166 путем одно-
временного введения извести и концентрированной фосфорной кис-
лоты в раствор ~ 50 % -ной Н3РО4 при температуре не ниже 80°.
Кристаллизация заканчивается при охлаждении реакционной смеси,
после чего моиокальцийфосфат отделяется на центрифуге от кис-
лого маточного раствора и тщательно промывается. Следы фос-
форной кислоты нейтрализуют известью. Предложен способ 167, по
которому 75%-ную фосфорную кислоту и известковое молоко (5%
избытка) плотностью 1,1 г/см3 смешивают и полученную пульпу
высушивают в распылительной сушилке. Получается сыпучий про-
дукт с примесью дикальцийфосфата.
Безводный моиокальцийфосфат гигроскопичен, поэтому необхо-
димо покрытие частиц защитной пленкой 168. Его получают ступен-
чатой нейтрализацией фосфорной кислоты известью при 140—170°
с добавлением в пульпу соединений калия. Смесь нагревают затем
до 200—220°, вследствие чего поверхность кристаллов покрывается
тонкой пленкой пирофосфата калия 169> 17°.
1090
Гл. XXX. Промышленные продукты, содержащие фосфор
Та же цель достигается образованием на частицах монокаль-
цийфосфата пленок пирофосфата алюминия 171.
Трикальцийфосфат природный, в виде костяного угля и костя-
ной золы, применяется для подкормки скота и птиц.
Кормовой трикальцийфосфат в виде измельченной кости содер-
жит около 33% Р2О5, 45% окиси кальция, 0,6% азота, 0,4% жи-
ров, 4,0% минеральных примесей, 0,15% железа, 1,5% влаги. Про-
дукт подвергается бактериологической проверке.
Трикальцийфосфат употребляется, кроме того, для очистки са-
харных сиропов в производстве сахара-рафинада, в производстве
керамики и стекла, для изготовления зубных паст и порошков, он
входит в состав абразивов для полировки и шлифовки металлов,
служит для удаления фтора из воды, для кондиционирования по-
варенной соли, применяется в медицине и др. Трикальцийфосфат,
содержащий диспергированные окислы металлов, используется в
качестве катализатора дегидрирования бутана и бутена 172.
Пирофосфат кальция, как и трикальцийфосфат, применяется в’
сравнительно небольших количествах в качестве мягкого абразива
для полировки металлов и в зубных порошках и пастах. Его полу-
чают 173 прокаливанием дигидрата дикальцийфосфата при 900°.
Трикальцийфосфат получают осаждением из разбавленной
фосфорной кислоты известковым молоком. Продукт по своему со-
ставу близок к гидроксилапатиту.
При нейтрализации на холоду разбавленной фосфорной кис-
лоты известковым молоком выделяется двухводный дикальцийфос-
фат, гидролизующийся с образованием Са(Н2РО4)2 и трикальций-
фосфата. Избыток гидроокиси кальция нейтрализует дикальций-
фосфат вначале до неустойчивого соединения с весовым отноше-
нием Са : Р ~ 1,8 и затем до трикальцийфосфата. Из горячих рас-
творов, нагретых до 90°, выделяется гидроксилапатит 174. В зависи-
мости от pH среды и содержания примесей Mg2+ или Fe2+ выде-
ляется безводный или гидратированный трикальцийфосфат ,7S. При
pH = 6,95 образуется безводная соль, при больших pH — гидрат.
В присутствии примесей и при pH меньше 4 выделяется только
безводный трикальцийфосфат.
Изучено176 взаимодействие метафосфата кальция с окисью
кальция при 900° с образованием трикальцийфосфата. Процесс
протекает через промежуточные соединения Са2Р2О7 и Са4Р20э и
связан с длительным прокаливанием.
ЛИТЕРАТУРА
1. В. В а г г а м а н, Фосфорная кислота, фосфаты и фосфорные удобрения,
Госхимиздат, 1957.—2. И. Л. Гофман, Хим. наука и пром., 2, № 6, 706
(1957). — 3. Comm. Fertilizer, 92, № 6, 76 (1956).—4. W. И. Hill, Ind. Eng.
Chem., 44, № 7, 1526 (1952). —5. Chem. Statistics Handbook Manufact. Chemists
Assoc. In., 1955. — 6. G. Perot, Chim. et ind., 87, № 1, 78 (1962). —7. A. R. Pi-
Литература
1091
tochelli, L. F. Audrieth, J. Am. Chem. Soc., 81, № 17, 4458 (1959).—
8. H. Falins, Naturwiss, 50, № 1, 126 (1963). — 9. D. T. Dixon, F. W. Ein-
stein, B. R. Penfold, Acta cryst., 18, № 2, 221 (1965). — 10. R. F б r t h m a n n,
A. Schneider, Naturwiss., 52, № 13, 390, 391 (1965).
11. A. A. Sandoval, H. C. Moser, Inorgan. Chem., 2, № 1, 27 (1963).—
12. P. Re in shagen, K. L i m p e 11, H. Schmidt, пат. ФРГ 1104931, 1961,—
13. Ю. В. Мухин, А. П. Шубников, Б. И. Матвеев, Н. Ф. Крутов,
Б. Я. Либман, А. Д. Логинов, В. В. П о з д н е в, 3. X. М а р к а з е н,
В. К- Горбунов, авт. свид. 146298, 1962. — 14. Н. Д. Таланов, И. Н. С м и р-
н о в, Труды по химии и химической технологии в. 2(18), Горький, 1963, стр. 220.—
15. W. К о с h m a n n, W. Marx, J. Diehlmann, R. Schumann, пат. ГДР
22997, 1962.-/6. Б. А. Войтович, ЖНХ, 6, № 8, 1914 (1961). —/7. I. Lie-
big, G. Wehner, Н. Krausenecker, К. Schwalm, Chem. Techn., 12, № 5,
254 (1960). —18. В. Кохмани, Р. Шуман н, авт. свид. 137502, 1961.—
19. G. Miiller-Schiedmayer, М. Harnisch, Z. anorg. Chem., 333, № 4—
6, 260 (1964). — 20. С. Узки, Т. Какадзахи, Rept. Govt. Chem. Ind. Res. Inst.
Tokyo, 57, № 3, 137, 15 (1962).
21. Г. В. Самсонов, Л. Л. В e p e й к и и а, Фосфиды, Изд. АН УССР,
Киев, 1961.—22. А. Малец, Труды НИУИФ, в. 143, 1938, стр. 10.—23. Пат.
США 1909996, 1933; 1936307, 1934, — 24. Н. W. Е asterwood, Chem. Met. Eng.,
№ 6 (1933). — 25. А. Малец, Ф. Мордяшева, Г. Лютринсгаузер,
Т. Маркина, Труды НИУИФ, в. 134, 1936, стр. 48. — 26. Я- И. М и х а й л е н к о,
А. П. Крешков, ЖПХ, № 5, 346 (1937). —27. Пат. ГДР 6004, 1954.—
28. I. М. Potts a. oth., J. Electrochem. Soc., 105, № 3, 148 (1958).— 29. Р. Е.Ре-
м е н, А. Ю. К и з а с, Л. М. Лапина, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 41;
Труды НИУИФ, в. 163, 1959.—30. V. Arvamythan, R. Srinivasan,
N. Meenakshisundaram, Chem. Age (India), 17, № 6, 476, 477 (1966).
31. R. L. Ripley, J. Less-Common Metals, 4, № 6, 493 (1962).—
32. H. H. Постников, Г. Г. H о з а д з e, О. В. Васильева, Реф. н.-и. раб.
за 1956 г., ч. 1, стр. 24, Труды НИУИФ, в. 161, 1958. — 33. Н. Н. Постников,
Г. Г. Нозадзе, О. В. Васильева, Л. А. Бекасова, Реф. н.-и. раб. за
1957 г. ч. 1, стр. 25, Труды НИУИФ, в. 163, 1959.—34. X. R a b е n a u, Compound
Semiconduct., v. 1, N. Y„ 1963, p. 181.—35. W. Stachura, S. Weychert,
польск. пат. 42876, 1960.—36. С. C. Wang, М. L a h е е г и d d i n, L. H. S p i-
n a r, J. Inorg. a. Nucl. Chem., 25, № 3, 326, 327 (1963).—37. A. Addamiano,
пат. США 3008805, 1961.— 38. В. Wendrow, A. Kobe Kenneth, Chem.
Rev., 54, Ns 6, 891 (1954). — 39. К. Сулайманкулов и др., Сборник научных
работ Киргизского гос. ун-та, № 1, 1954, стр. 21. — 40. А. Бугу б а ев, Б. И м а-
накумов, М. Жакипов, Там же, стр. 31.
41. М. И. Р а в и ч, Л. Г. Щербакова, ИСФХА АН СССР, 26, 248 (1955).—
42. Булле, Домине-Бер ж, Морен, Compt. rend., 241, № 24, 1772 (1955).—
43. R. J ary, Ann. chim., 2, janv.-fevr., 58 (1957).—44. Справочник химика, т. Ill,
Изд. «Химия», 1964, стр. 215.—45. Б. П. На деинский, Теоретические обосно-
вания и расчеты в аналитической химии, Изд. «Советская наука», 1956. —
46. М. Ebert, A. Muck, Coll. Czech. Chem. Comm., 28, № 1, 257 (1963).—
47. В. H. Ламп, М. Л. Чепелевецкий, Труды, НИУИФ, 1963, стр. 9.—
48. С. Schwartz, С. I. М u п t е г, Ind. Eng. Chem.,34, 32 (1942). —49. Ф. И. Б а-
л а н, Водоподготовка, Гос9нергоиздат, 1958. — 50. Н. Е. Пест ов, ЖХП, № 3
6 (1932).
51. К- Beltz, F. Ro dis, пат. ФРГ 1086678, 1964.— 52. Кавасэ Акио,
япои. пат. 14909, 1963. — 53. А. М. Дубовицкий, А. И. Шерешевский,
Технология минеральных удобрений, Госхимиздат, 1947.:—54. F. D. Snell, Ind.
Eng. Chem., 23, 5, 470 (1931). — 55. А. И. Крым, Б. Н. Ю р ы г и н, Производ-
ство и применение тринатрийфосфата, ОНТИ, 1936.—56. Пат. ФРГ 114514,
1963. — 57. И. Л. Гофман, Реф. н.-и. раб. за 1957 г., ч. 1, 17; Труды НИУИФ,’
1092
Гл. XXX. Промышленные продукты, содержащие фосфор
в. 163, 1959. —58. Т. F. Meinhold, Т. W. S с h i е b, Chem. Process, 20, № 11,
48, 50 (1957). — 59. I. В d lint, G. L a d о г, венг. пат. 1480062, 1961.—
60. Г. M. Стр онг ин, А. С. Г у н д а р и н, авт. свид. 150826, 1962.
61. И. Л. Гофман, авт. свид. 52922, 1937, — 62. Пат. США 2271712, 1942. —
63. И. Л. Гофман, С. К. Милованова, авт. свид. 62810, 1943.—
64. М. Л. Ч е п е л е в е цк и й, Е. В. Южная, Реф. и.-и. раб. за 1956 г., ч. I,
стр. 8; Труды НИУИФ, в. 161, 1958. —65. М. Л. Ч е п е л е в е цк и й, Е. Б. Бруц-
кус, А. И. Л и ц о в а, Реф. н.-и. раб. за 1957 г., ч. 1, стр. 18; Труды НИУИФ,
в. 163, 1959. — 66. Ю. В. Ходаков, ЖНХ, 1, № 3, 368 (1956).— 67. Э. Тило,
ЖПХ, 29, № 11, 1621 (1956).— 68. Б. А. П р о д а н, Л. И. П р о д а н, Н. Ф. Ер-
мол е и к о, Триполифосфаты и их применение, Изд. «Наука и техника», Минск,
1969. — 69. Е. I. Griffith, J. Am. Chem. Soc., 78, № 16, 3867 (1956).—
70. О. P f r e n g 1 e, F e 11 e, S e i f e n, Anstrichmittel, 58, № 2, 81 (1956).
71. C. F. Callis, J. van W a z e г, I. S. Metcalf, J. Am. Chem. Soc., 77,
№ 6, 1471 (1955). — 72. Mo pa, ibid., 75, № 23, 5794 (1953).— 73. M. E. П о з и н,
Б. А. Ко пылев, Хим. наука и пром., 2, № 6, 677 (1957). — 74. I. R. vanWa-
z е г, Е. I. Griffith,!. Am. Chem. Soc., 77, № 23, 6140 (1955). — 75.1. F. McCul-
lough, I. R. van W a z e r, E. I. Griffith, ibid., 78, № 18, 4528 (1956).
76. А. В. Памфилов, H. M. Домбровский, Уч. зап. Черновицк. ун-та,
в. 21, 1956, стр. 36.— 77. R. Klement, R. Popp, Naturwiss., 44, № 11, 327
(1957). — 78. G. W. Morey, J. Am. Chem. Soc., 80, № 4, 775 (1958).—
79. M. H. Omdik, Acta cryst., 18, № 2, 226 (1965). — 80. E. J. Griffith, Inor-
gan. Chem., 1, № 4, 962 (1962).
81. A. X. Бронников, ЖПХ, 12, № 9, 1287 (1939). — 82. Morgen,
Swoope, Ind. Eng. Chem., 35, № 7, 821 (1943). — 83. G. Corsaro, Am. Perfu-
mer, 48, № 8, 64 (1946).—84. R. N. Bell, Ind. Eng. Chem., 39, № 2, 136 (1947).—
85. I. Green, ibid., 42, № 8, 1542 (1950).— 86. В. С. H о f f о г d, ibid., 46, № 9,
1938 (1954). — 87. И. Л. Г о ф м а и, Н. В. Трутнева, Сообщение о научно-
технических работах НИУИФ, № 2, 1957, стр. 28. — 88. Е. Thilo, A. Sonntag,
К Н. R a 11 а у, Z. anorg. Chem., 283, 365 (1956). — 89. J. W. U у о п s, R. Р. L а п g-
guth, Quart. Colorado School Mines, 56, № 3/2, 557 (1961).—90. О. К- Щер-
баков, Труды Н.-и. и проектного ин-та обогащен, и механ. обработки полезн.
ископаемых Уралмеханобр, в. 11, 1964, стр. 25.
91. Pl. Inclei 11, Ann. Univ. Ferrara, sez., 5, 1, Suppl. 59. Discuss., 69
(1956). — 92. H. К er st, R. G. Dalbke, Proc. Internat. Water. Conf., 22, 9
(1961), Discuss., 22. — 93. M у p о т а н и, Bull. Soc. Salt Sci., Japan, 10, № 5,
219 (1956).—94. Ваи Везер, Фосфор и его соединения, ч. I, ИЛ, 1962,—
95. Австр. пат. 176547, 1953. — 96. L. Uh lit, L. D г а р а 1, Sb. Vedec. praci. Vysd-
ke skoly Chem. technol. Pardubice, № 1—2, 63, 1962 (1963). — 97. B. Bigot,
пат. США 3094382, 1963. — 98. H. .M. Домбровский, ЖНХ, 7, № 1, 95, 104
(1962). — 99. Хим. пром, за рубежом, № 5 (68), 84 (1968). — 100. Пат. ГДР 6312,
1954.
101. Хим. наука и пром., № 2, 237 (1956). — 102. Chem. Eng., 61, № 2, 132,
320 (1954). — 103. Пат. США 2419147, 1947. — 104. Chem. Eng., 61, № 12, 132, 320
(1954). —105. И. Л. Гофман и др., Реф., и.-и. раб. за 1956 г., ч. 1, стр. 4;
Труды НИУИФ, в. 161, 1958. — 106. F. R о d i s, G. H a г 11 a p p, пат. ФРГ 1097421,
1961, — 107. К. Hermes, К. Schill, О. Pfrengle, пат. ФРГ 976323, 1963 —
108. Т. F. Meinhold, Т. W. Shieb, Chem. Process, 20, № 11, 48, 50 (1957). —
109. A. Ko eb пег, R. H. Edwards, англ. пат. 970727, 1964. —110. M. В. Лы-
ков, Д. Н. Шевченко, И. П. Ху до лей, Хим. пром., № 9, 628 (1961).
111. М. В. Лыков, Д. Н. Шевченко, Там же, № 3, 258 (1959).—
112. М. В. Лыков, Б. Н. Леоичик, Распылительные сушилки, Изд. «Маши-
ностроение», 1966. — 113. Пат. ФРГ 1029808, 1958. — 114. И. Л. Г о ф м а н, К. С. 3 о-
т о в а, М. Г. Лысенко, сб. «Исследования по химии н технологии удобрений,
Литература
1093
пестицидов и солей», Изд. «Наука», 1966, стр. 142. — 115. Р. А. Садовская,
В. Я. Рот а нин а, Т. Р. Сувырина, сб. «Исследования в области химиче-
ской электротермии», Труды Ленниигипрохима, в. 1, 1967, стр. 283. —116. Rev.
chem., № 3, 17 (1964). — 117. И. Л. Гофман, М. В. Лыков, И. П. Худо-
лей, Хим. пром , № 9, 620 (1961). — 118. А. Т а 1 m i, Е. Herman, В. Р е s k i п,
Bull. Res. Council. Israel, 41, № 2, 235 (1962). —119. E. К о r d i к, J. Sule,
чехосл. пат. 107645, 1963. — 120. И. Л. Гофман, С. К- Милованова, авт.
свид. 62810, 1943.
121. Л. А. С а р к и ц, М. Е. П о з и н, Б. А. Копы л ев, Р. Ю. 3 и н ю к,
сб. «Новые исследования по технологии минеральных удобрений», Изд. .«Химия»,
1970. — 122. Т. М. Найшуллер, Н. Н. Ромашова, Хим. пром, за рубежом,
№ 5(65), 30 (1968).— 123. К. С. Зотова, И. Л. Гофман, Пром, удобр.
и серной кислоты, НИТЭХИМ, № 4, 37 (1966). — 124. W. О. Groves, пат.
США 3063801, 1962. — 125. М. В. Лыков, К. П. Деревщикова, И. Л. Гоф-
ман, авт. свид. 160165, 1964. — 126. F. R о d i s, J. Krause, К. Beltz, пат.
ФРГ 1007748, 1961. — 127. А. Бронников, В. Постников, ЖПХ, 11, № 9,
1295 (1938). — 128 М. Ralcsewa-Minsev, Е. Banyai, Madyar Kem. Fo-
lyoirat, 56, 201 (1950). — 129. Л. H. P e у т о в и ч, Е. А. Ф о м и н а, Ю. И. Г е р ж-
бер, Л. А. Леонтьева, Межвуз. респ. конф, по хим. и хим. технол., Днепро-
петровск, 1966, стр. 17. —130. М. П. По тоскуев, Сборник научных трудов
Ивановского энергетического ин-та, в. 10, ч. 2, 1962, стр. 115.
131. Япои. пат. 217, 1953; Chem. Abs., 48, № 2, 964• (1954). —132. Пат. ФРГ
877594, 1953; Zbl„ 125, № 1, 168 962 (1954); Пат. ФРГ 876399, 1959.—
133. Н. Н. Постников, Н. Д. Таланов, Труды Всесоюзн. отрасл. совещ.
работников фосфорной промышленности, 1968, стр. 53. — 134. Н. М. Домбров-
ский, ЖНХ, 5, № 8, 1699 (1960). —135. Н. М. Домбровский, Там же, 7,
№ 6, 1360 (1962).— 136. Z. J h 1 i f, О. D г а р а 1 a v a, Sb. Vedec. praci Vysone
Skoly chem.-technol. Pardubica, Praha, 2, 115 (1961). —/37. И. Л. Гофман,
H. В. Трутнева, Сообщения о научно-технических работах НИУИФ, № 2,
1957, стр. 30. —138. И. А. Гофман, М. В. Лыков, И. П. X у д о л е й, Хим.
пром., № 9, 620 (1961). —/39. Е. N. Hetzel, G. Е. Taylor, пат. США
2408258, 1946. — 140. С. R. М с С u 11 о и g h, пат. США 2021012, 1935.
141. И. Л. Гофман, Н. С. Зотова, И. П. X у д о л е Й, Д. Н. Шев-
ченко, авт. свид. 175493, 1965, Бюлл. изобрет., № 20, 19 (1965). —142. А. К а-
дыралиев, К. Файзундаев и др., Сборник научных работ студ. Киргиз-
ского гос. ун-та, № 1, 1954, стр. 39.— 143. К. С. Чернова, ИСФХА АН
СССР, 15 (1947). — 144. С. И. Вольфкович, Р. Е. Р е м е и, сб. «Исследования
по прикладной химии», Изд. АН СССР, 1955, стр. 149. — 145. А. А. Ионасс,
М. М. Кобрин, Труды НИУИФ, в. 153, 1940, стр. 193. — 146. Н. L. Thomson
a. oth., Ind. Eng. Chem., 41, № 3, 485 (1949). —147. H. L. Thomson a. oth,
ibid., 42, № 10, 2176 (1950). —/48. J. J. Marino, Meeh. Eng., 84, № 1, 34
(1962). — 149. И. Л. Гофман, Труды НИУИФ, в. 143, 1938, стр. 26.—
150. И. Л. Гофман, Там же, в. 153, 1940, стр 202; ЖХП, № 2 (1939).
151. И. Л. Го ф май, авт. свид. 92149, 1950. —/52. Австр. пат. 239762,
1965.-/53. Пат. США 2999006, 1961, —/54. Е. С. Houston, L. D. Jates,
R. L. Ha up schild, J. Agr. Food Chem., 3, № 1, 43 (1955). — /55. I. G. Get-
singer, E. C. Houston, F. P. Achorn, ibid., 5, № 6, 433 (1957).—
156. T. Ф. Абашкина, E. Б. Бруцкус, Труды НИУИФ, в. 208, 1965, стр. 42,—
/57. Т. Д. Корицкая, Р. Е. Р е м е н, Журн. ВХО, 7, № 5, 520 (1962).—
158. С. К а су н о, Р. Т а к а ч и, Ц, Сабо, япон. пат. 1774, 1960. — 159. W. М а г-
gaj, J. Kwiecien, К. Baran, W. Biclecki, польск. пат. 47630, 1963,—
160. Е. J. Н е 1 m, Е. V. S с h u 11 е, пат. США 2946655, 1960.
161. W. G. Schulze, Blast Furnace, Coke Oven a. Row Mater. Comm Iron,
a. Steel Div. Metallurg. Soc. Am. Inst. Mining, Metallurg, a Petrol. Eng. Proc.,
v. 20, N. Y., 188, 1961, Discuss. 221, —/52. H. Г. Бунаков, Г. Д. Харлам-
1094 Гл. XXX. Промышленные продукты, содержащие фосфор
п о в и ч, ЖПХ, 38, № 9, 1915 (1965). — 163. Г. Д. X а р л а м п о в и ч, Р. И. Куд-
ряшова, Кокс и химия, № 11, 38 (1961).— 164. Г. Д. Харлампович,
М. В. Г о ф м а и, Р. И. Кудряшова, В. Е. Привалов, Там же, № 10, 9
(1964). —165. Г. Д. Харлампович, Р. И. Кудряшова, Там же, № 2,
23 (1965). — 166. С. Н. Mi Hi gran, пат. США 2121208, 1938. — 167. С. Т. W h i-
tier, пат. США 1442318, 1923; Е. В. Thornton, пат. США 1689697, 1928;
I. Carothers, Р. Logue, пат. США 1818114, 1931. — 168. R. Т. Cochran,
J. Techn. Acad. Sci., 17, July (1942). —169. 1. Schlaeger, пат. США 2160232
и 2160233, 1939, — 170. W. H. Knox e. а., пат. США 2160700, 2160701, 1939;
2462104, 1949.
171. Пат. США 2631102, 1953; канад. пат. 513755, 1955. —/72. R. S. В о и-
m a n, L. J. Piasecky, пат. США 3149081, 1964. — 173. St. Pierre, J. Am.
Chem. Soc., 77, № 8, 2197 (1955). — 174. L. Winand, Bull. She. rog. Sci. Liege,
30, № 11—12, 512 (1961). —175. J. Leroux, T. Baratali, G. Montel, Compt.
rend., 256, № 6, 1312 (1963). —176. С. И. Вольфкович, В. В. Илларио-
нов, Р. Е. Ремен, А. И. Со кл а ко в, ЖПХ, 35, № 6, 1165 (1962).
Глава XXXI
СОЕДИНЕНИЯ ФТОРА
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Свободный фтор F2 представляет собой почти бесцветный
(слегка зеленовато-желтый) газ, имеющий при нормальных усло-
виях плотность 1,696 г/л. Температура плавления фтора —219,6°,
температура кипения (при 760 мм рт. ст.), —188,1°, критическая
температура —129°, критическое давление 55 ат. Давление насы-
щенного пара жидкого фтора в интервале от —219,6° (1,7 мм рт. ст.)
до —183,7° (1220лм<рт. ст.) можно вычислить по формуле:
lg Р = 7,08718 - 357,2587"' - 1,3155 • 10|37~8
Фтор является самым электроотрицательным элементом. Потен-
циал нормального электрода для фтора равен 2,85 в. Фтор обла-
дает чрезвычайно большой реакционной способностью и соеди-
няется непосредственно со всеми элементами, за исключением
инертных газов и азота. Вследствие образования плотной защит-
ной пленки нелетучего фторида некоторые металлы (железо, медь,
магний, никель) в отсутствие воды весьма устойчивы против дей-
ствия фтора '.
Фтор энергично реагирует с большинством химических соеди-
нений других элементов, а также почти со всеми органическими
веществами. С окисью и двуокисью углерода фтор не взаимодей-
ствует. Воду он разлагает:
F2 + Н2О = 2HF + '/2О2
По этой реакции предложено осуществлять конверсию фтора
перегретым выше 260° водяным паром во фтористый водород 2. При
действии фтора на воду при обычной температуре вытесняемые
атомы кислорода частично окисляют воду до Н2О2 и фтор до окиси
фтора F2O; окись фтора — сильно ядовитый бесцветный газ с за-
пахом, похожим на запах озона. Известны и другие фториды кис-
лорода 3’4.
1096
Гл. XXXI. Соединения фтора
Трифторид хлора CIF3 — при обычных условиях газ, медленно
гидролизуется водой с образованием HF, Cl2, О2, С1О2, C1O2F, C1F,
CIO3F, количества которых зависят от соотношения C1F3:H2O5’6.
ТАБЛИЦА 86
Свойства насыщенного пара HF
t, °C р, мм рт. ст. Плотность пара, г/л Фактор ассоциации Теплота испарения, * кал/молъ
0 363,8 2,015 4,717 1257
10 536 Д 2,521 4.148 1445
ня 773,2 3,170 3,743 1616
30 1093 3,979 3,438 1777
40 1516 4,976 3,203 Х925
50 2069 6,197 3,015 2063
60 2778 7,645 2,861 2194
70 3677 9,390 2,734 2312
80 4801 11,44 2,625 2427
90 6191 13,85 2,534 2530 .
100 7891 16,64 2,453 2631
105 8872 18,20 2,417 2678
* С учетом фактора ассоциации; для неассоциированного
6,15 ккал/моль
Фтористый водород HF бесцветен, кипит (при 760 мм рт. ст,)
при 19,9°, плавится при —83,1°; критические точки7: температура
188 ± 3°, давление 66,2 ± 3,5 кгс/см2, плотность 0,29 + 0,03 г/см3.
Рис. 313. Средний молекулярный вес
(сплошная линия) и давление насыщен-
ного пара (пунктирная линия) фтори-
стого водорода.
при температуре кипения. Жидкий
В газообразном, в жидком
состоянии, а также в водных
растворах молекулы HF ас-
социированы 8>9 (рис. 313).
Для температур от Одо 105°
давление насыщенного пара
(табл. 86) HF (в мм рт. ст.)
вычисляется10 по фор-
муле:
lg Р = 8,38 - 1952,6/(355,5 + f)~'
Плотность жидкого фто-
ристого водорода убывает
почти прямолинейно от
1,21 г/см3 при температуре
плавления до 0,0959 г/см3
фтористый водород является
энергичным растворителем. В нем хорошо растворяются фториды
щелочных металлов, несколько в меньшей мере фториды щелочно-
земельных и других металлов. Растворенная в нем вода становится
сильным электролитом вследствие диссоциации по схеме
Н2О + 2HF н3о+ + hf;
Физико-химические свойства
1097
в основе которой, как и при диссоциации других электролитов в
жидком фтористом водороде, лежит реакция и:
2HF Н+ + HF2
Безводный фтористый водород является более сильной кисло-
той, чем в водных растворах, диссоциация в которых идет по той
же схеме (HF Н++ F~ и F~ + HF HF2)1Z. С фторидами ще-
лочных металлов HF образует кристаллические соединения типа
MF • nHF; например, для KF п = 2, 3 и 4 13.
Рис. 314. Концентрация HF
в газовой фазе над плавико-
вой кислотой.
lg Р
2.7
2,5
2.3
Л’
1.9
1,7
1,5
’,3
1.1
0,3
0,7
2,8 2.9 3,0 3,1 3,2 3,3 3,4 3,5 3,6 3,7 3,8
у-108
Рис. 315. Давление пара HF (Р, мм рт. ст.)
иад плавиковой кислотой разных кон-
центраций.
Фтористый водород хорошо растворим в воде; его водные рас-
творы называют фтористоводородной или плавиковой кислотой.
Равновесные концентрации HF в газовой фазе над плавиковой
кислотой приведены на рис. 314 14 и 315 15.
О давлении пара Н2О в системе HF—Н2О и давлении HF и
Н2О в системе HF—H2SO4—Н2О см.15’16.
Температуры замерзания плавиковой кислоты приведены на
рис. 316 (в присутствии примеси H2SO4 и H2SiFe см.17). Темпера-
туры кипения в системе HF—Н2О и равновесные составы пара при
атмосферном давлении приведены на рис. 317. Эти данные отно-
сятся к растворам плавиковой кислоты, содержащим 0,1—1,8%
1098
Гл. XXXI. Соединения фтора
H2SiF6. Как видно из диаграммы, в системе HF—Н2О имеется
азеотропная смесь, содержащая 37,5% HF и кипящая при 109°
(760 мм рт. ст.)18. По другим данным19, составы азеотропных сме-
сей и точки их кипения: 43,2% HF и 111° или 35,4% HF и 120°;
такие расхождения, очевидно, вызваны трудностью очистки плави-
ковой кислоты от FKSiFe- Более точным, вероятно, является состав
азеотропной смеси с концентрацией HF 38,26%, кипящей при 112°
под давлением 750,2 мм рт.ст.1-20. На рис. 318 и 319 приведены
Концентрация HF, мол
Рис. 316. Диаграмма плавкости
системы HF—Н2О.
Рис. 317. Температуры кипения и со-
ставы пара в системе HF—Н2О при
760 мм рт. ст.:
/ — состав жидкости; 2 — состав пара.
температуры кипения и составы пара тройных смесей HF—
—H2SO4—Н2О в области концентраций, имеющих практическое
значение для получения безводного фтористого водорода.
Сухой фтористый водород не реагирует с большинством эле-
ментов и их окислов, но энергично вступает в реакции в присут-
ствии влаги. Плавиковая кислота разрушает стекло и силикаты,
так как легко вступает во взаимодействие с окисью кремния:
4HF + SiO2 = SiF4+2H2O. Некоторые металлы, например свинец и
медь, относительно устойчивы в плавиковой кислоте вследствие
защитного действия пленки продуктов реакции. Более устойчи-
выми являются резина, эбонит и некоторые пластмассы.
Плавиковая кислота растворяет клетчатку и потому разрушает
кожу, вызывая ожоги. Даже ожоги слабой кислотой, вначале не
замеченные, могут вызвать глубокое поражение через несколько
часов. Поэтому обращение с плавиковой кислотой и фтористым
водородом требует особой осторожности.
Н2О,%
Рис. 318. Температура кипения в системе HF—H2SO4—Нг0
при атмосферном давлении.
Рис. 319. Содержание HF (в %) в паре иад кипящими
растворами в системе HF—H2SO4—Н2О при атмо-
сферном давлении.
1100
Гл. XXXI. Соединения фтора
Кислые фториды щелочных металлов типа MHF2 являются бо-;
лее слабыми кислотами, чем HF, а соединения других металлов,:
например A1F3-3HF или H3A1F6, сильнее, чем HF. Бифторид нат-;
рия NaHF2 имеет плотность 2,08 г/см\ растворимость в воде при
0° 2,28%, при 40° 4,92% zl. Давление диссоциации NaHF2’
(в мм рт. ст.) определяется по формуле22: 1g Р = —3830/7 + 9,97
или (в ат для интервала 273—371° К) по формуле23: 1пР =
Концентрация NH4F, бес. %
Рис. 320. Изотермы растворимости в си-
стеме NH4F—NH4HF2—Н2О при 0, 15,
25, 50 и 75°.
=—3940/7+6,677. NaF легко
поглощает HF из разбавлен-
ного газа при 100° и выделяет
его в виде концентрированного
HF при 250°. Бифторид калия
KHF2 имеет плотность 2,35
г/см\ существует в двух фор-
мах с температурой перехода
195—196°. Давление диссоциа-
ции KHF2 (в мм рт. ст.)
определяется по формуле:
1g Р =—4000/7 + 8,574. Насы-
щенный водный раствор содер-
жит при 0°—18,63%, при 100°—
50,5% KHF2. В системе KF—
HF—Н2О 21 образуются поли-
гидрофториды калия KF • 4HF,
KF-3HF, 2KF-5HF, KF-2HF,
идентичные двойным соедине-
ниям в системе KF—HF *.
В системе NH4F—HF обра-
зуются соединения NH3 • nHF,
где п — 2, 3 и 424. О парциаль-
ном давлении NH3 и HF в
этой системе см.25. На рис. 320 приведены изотермы растворимости
в системе NH4F—NH4HF2—Н2О 2б. Эвтонический раствор в системе
NH4HF2—NH4C1—Н2О при 25° содержит 36,75% NH4HF2, 10,51%’
NH4C1 и 52,74% Н2О27>28.
При взаимодействии HF с SO3 или HSO3C1 образуется фтор-
сульфоновая кислота HSO3F:
SO3 + HF = HSO3F
HSO3C1 + HF = HSO3F + HC1
Фторсульфоновая кислота — бесцветная жидкость с плотностью
при 18° 1,740 г]см\ замерзает при —87,3°, кипит при 162,6°. Без-
водная фторсульфоновая кислота диссоциирует на ионы Н+ и
SO3F_ и лишь в небольшой степени на HSO3 и F-, а также на
H2F+ и S2O6F“. Хорошо растворяет большинство фторидов 629.
Физико-химические свойства
1101
Фторид натрия NaF имеет плотность 2,79 г/см3-, плавится при
995°, выше температуры плавления имеет значительную лету-
честь30. Насыщенный водный раствор содержит при 0° 3,95%,
при 94° 4,73% NaF. Теплота растворения NaF в воде при 25°
0,213 ккал/моль. При хранении фтористый натрий слеживается
(фториды щелочных металлов, в том числе и NaF, гигроскопичны).
Фторид калия KF имеет плотность 2,48 г/см3, плавится при
856°. В отличие от NaF хорошо растворим в воде: насыщенный
раствор содержит при 20° 47,75%, при 100° 59,83%, при 140°60,5%,
при 500° 71% KF. Из водных растворов выделяются кристаллогид-
раты: KF • 4Н2О (от —21,8 до 17,7°), KF • 2Н2О (17,7—45°); выше
45° кристаллизуется безводный KF. О системах NaF—NaOH и
KF—КОН см.31, а о системах NaF—KF и др.32 (в системе NaF—KF
эвтектика находится при 710°, 60% KF).
Фторид аммония NH4F имеет плотность 1,315 г/см3, возгоняется
без плавления. Насыщенный водный раствор содержит при 20°
45% NH4F. Ниже —16,8° кристаллизуется NH4F • Н2О. Фторид
аммония является единственным неорганическим соединением, для
которого обнаружены смешанные кристаллы со льдом; они содер-
жат до 10% NH4F33"35.
Фториды щелочноземельных металлов имеют следующую рас-
творимость в воде при 18° (в г/л): MgF2 —0,0874, SrF2 —0,1173,
BaF2— 1,605. Произведение растворимости CaF2 при 18° 3,4-10-11-,
при 18° и pH = 7 растворимость CaF2 равна 0,000205 г-мол/л36.
Фторид кальция имеет плотность 3,18 г/см3, плавится при 1418°37.
Фторид магния имеет плотность 3,0 г/см3, температуру плавления
1396° (по другим данным 38 1256°). Выше 900° MgF2 интенсивно
гидролизуется водяным паром38.
В системе CaF2—SiO2 эвтектика находится при 1240° (47%
SiO2)39. О системе CaF2—СаО—Р2О3 см.40.
Фторид алюминия A1F3 имеет плотность 3,07 г/см3. Давление
пара A1F3 при 1094° 31 мм рт. ст., при 1251° 614 мм рт. ст. и при
1294° 760 мм рт. ст. При 800—1000° (в вакууме при 650—700°) BF3
взаимодействует с алюминием, образуя монофторид (А1Р)Ж, сущест-
вующий только в газообразном состоянии и разрушающийся при
конденсации:— (AlF)x—»2А1+ A1F3. Полученный сухим путем без-
водный A1F3 в воде практически не растворим; полученный из рас-
творов кристаллогидрат немного растворим — насыщенный раствор
при 0° содержит 0,127 %, при 20° 0,498%, при 50° 0,805% и при 102°
2,42% A1F3. При 20° в 10%-ном растворе HF растворяется
2,4—2,84% A1F3, а в 15% растворе HF, 2,87—3,1% A1F341. В рас-
творах сильно гидролизован; кристаллизуется обычно в виде
A1F3 ЗН2О. Известны и другие кристаллогидраты: A1F3 • 9Н2О,
A1F3 • 3,5Н2О, A1F3 • Н2О, A1F3 • 0,5Н2О '. Чем выше степень обезво-
живания A1F3, тем меньше его гигроскопичность42.
1102
Гл. XXXI. Соединения фтора
Криолит Na3AlF6 или 3NaF-AlF3 является комплексной солью
гексафторалюминиевой кислоты H3[A1F6J. Название криолит (в пе-
реводе с греческого означает «ледяной камень») дано вследствие
внешнего сходства со льдом природного минерала, применяемого
в технике наряду с искусственным. Криолит плавится при 1011°.
Предполагают, что в расплаве криолита комплексный анион [A1F6]3-
частично диссоциирует на [A1F4]~ и F-. Возможно, что этот процесс
происходит и ниже точки плавления, что объясняет высокую элек-
тропроводность криолита при температурах ниже точки плавления
и малую величину скачка электропроводности при его плавлении43.
В системе NaF—A1F3 помимо инконгруэнтно плавящегося криолита
Na3AlF6 существуют инконгруэнтно плавящийся хиолит Na5Al3F14
(5NaF • 3A1F3) и тетрафторалюминат натрия NaAIF4 (выше 480°)44.
В системе NaF—A1F3—Н2О при 25° могут существовать две
твердые фазы — при концентрации NaF меньше 1,4% конгруэнтно
растворяющаяся NanAl4F23 или 3Na3AlF6 • 2NaF • A1F3, а при кон-
центрации NaF больше 1,4% инконгруэнтно растворяющийся крио-
лит Na3AlF6 45. По другим данным46, из водных растворов выде-
ляются кристаллогидраты криолита Na3AlF6 • 0,5Н2О и хиолита
Na3Al3F14 • Н2О. Для обезвоживания этих фторалюминатов их
нужно нагревать до 300°. Вследствие образования твердых раство-
ров в промышленной практике из водных растворов не удается по-
лучить криолит, соответствующий формуле Na3AlF6 — наибольшее
значение криолитового модуля (т. е. отношения NaF: A1F3) в этом
случае равно 2,75—2,847-49. Растворимость криолита в воде при 25°
равна 0,417 г, а при 16°—0,35 г в 1000 г раствора. О равновесии в
системе NH4F—A1F3—Н2О см.50.
Четырехфтористый кремний SiF4 — бесцветный газ (критиче-
ские температура —14,15°, давление 36,7 ат), при охлаждении пе-
реходит непосредственно в твердую фазу. Твердый возгоняется при
—94,8° (под давлением 1318 мм рт. ст. плавится при —90,2°, а под
давлением 1520 мм рт. ст. — при —70°). Давление сублимации
твердого SiF4 (в мм рт. ст.) вычисляется по формуле: lg Р =
=—6100/4,577’+10,382. Фторид кремния восстанавливается желе-
зом, легко реагирует с окислами металлов; хорошо поглощается
водой (265 объемов на 1 объем воды), гидролизуясь при этом и
образуя кремнефтористоводородную и кремневую кислоты:
3SiF4 + (n + 2)Н2О = 2H2SiF6 + SiO2 • nH2O
Этот процесс идет столь интенсивно, что ему не препятствует
присутствие в растворе других кислот (например до 50% H2SO4) 51.
Гидролиз SiF4 водяным паром при температурах до 600—800°
протекает в незначительной степени.
Комплексная кремнефтористоводородная кислота H2SiFe обра-
зуется также при растворении SiF4 в плавиковой кислоте:
2HF + SiF4 5=+ H2SiFe
Физико-химические свойства
1105
В жидком сухом фтористом водороде SiF4 не растворяется, но
чем больше влаги в жидком фтористом водороде, тем больше рас-
творимость в нем SiF4. При концентрации HF больше 96% раство-
римость SiF4 меньше 0,05% (рис. 321) 52. Высокая концентрация
ионов водорода в растворах H2SiFe объясняется смещением вправо
равновесия диссоциации HF (2HF^2H+ + 2F_) вследствие связыва-
%Н*
Рис. 321. Растворимость SiF4 во фто-
ристоводородной кислоте при 15°
(в вес. %).
Рис. 322. Равновесные давле-
ния SiF4 и HF над водными
растворами H2SiF6 при 60 и 80°.
ния F~ в комплексный ион SiFg (2F + SiF^pacTB.) SiF| ). Кон-
станта равновесия гидролиза
SiFg- + (п + 2) Н2О 4Н+ + 6F“ + SiO2 • пН2О
К = [H+]4[F’]6/[SiF|-] при 20°, по разным данным *>53, находится в
пределах 4-10-28—НО-27. Водные растворы кремнефтористоводо-
родной кислоты содержат смесь гексафторкремневой H2SiF6 и пен-
тафторкремневой HSiF5 кислот; последняя образуется вследствие
2- 54
гидролиза иона SiFe
Кремнефтористоводородная кислота выделяется из концентри-
рованных водных растворов в виде бесцветных кристаллов H2SiF6-
•2Н2О, плавящихся при 19°. В парах H2SiF6 сильно диссоцииро-
вана на HF и SiF4. Газовая фаза, находящаяся в равновесии с
растворами H2SiF5, инконгруэнтна растворам — с повышением кон-
центрации кислоты отношение SiF4: HF в газовой фазе растет.
При температуре кипения в парах над кислотой с концентра-
цией H2SiF6 больше 13,3% преобладает SiF4, при меньших кон-
центрациях— HF (по сравнению со стехиометрическим в моле-
куле HaSiFe)55. На рис. 322 приведены парциальные равновесные
1104
Гл. лХл1. Соединения фтора
Рис.
гидролиза SiFg-
Концентрация раствора, мояь/л
323. Зависимость степени
от концентрации
раствора при 11°.
давления SiF4 и HF над растворами H2SiF6 56. По прежним дан- 5
ним57, общее содержание фтора в газовой фазе, находящейся в :
равновесии с растворами H2SiF6 концентрацией 2,31; 7,52; 9,34 и .
12,69%, равно соответственно (в г/м3): 0,0049, 0,0246, 0,0508 и;
0,0914 при 50° и 0,01, 0,0408, 0,0841 и 0,1104 при 70°. О давлении :
паров над концентрированными растворами H2SiFe (больше 20%) :
см.58. По более поздним данным59, над кислотой с концентрацией
19,39%, 29,35% и 37,32% H2SiFe парциальное давление SiF4 в га- :
зовой фазе соответственно соста-
вляет (в мм рт. ст.): при 25° 0,002,
0,013 и 0,093; при 50° 0,035, 0,10 и
1,2; при 75° 0,17, 0,92 и 9,7.
Давление диссоциации Na2SiF6
на NaF и SiF4 при 480° 8 мм рт. ст.,
при 522° 46 мм рт. ст., при более вы-
соких температурах оно резко воз-
растает, но точно не установлено'
(данные разных исследователей
сильно расходятся; приводится, на-
пример, величина 786 мм рт. ст. при
~580°). Растворимость Na2SiFe в
воде при 0° 0,53%, при 20° 0,66%,
при 60° 1,49%, при 80° 1,95%, при
100° 2,45%. В плавиковой кислоте
растворимость немного больше (при
0° в 25,6% растворе HF растворяется 1,0% Na2SiF6, при 20 в 21%
растворе HF 1,07—1,22% Na2SiF6, при 50° в 26,5% растворе HF
1,42% Na2SiF6)41-60. В водных растворах Na2SiF6 гидролизуется,
особенно значительно в слабых растворах (рис. 323); pH гидроли-
зованного раствора составляет 3,5—4,0. Скорость разложения SiF|~
в щелочном растворе по реакции
SiF|“ + 4ОН~ = SiO2 + 6F" + 2Н2О
прямо пропорциональна концентрации SiFe” и не зависит от кон-
центрации щелочи; константа скорости разложения (К в сект1) мо-
жет быть вычислена по формуле: 1g К =—4248/Т+13,033
Кремнефторид кальция кристаллизуется в виде CaSiF6-2H2O,
обезвоживается выше 140°, при 365° разлагается на CaF2 и SiF4;
растворимость CaSiFe в воде при 22° 105 г/л. При растворении эта
соль частично гидролизуется с выделением CaF2 и SiF4; при повы-
шении концентрации раствора гидролиз уменьшается61.
Растворимости других кремнефторидов: MgSiFe при 0° 20,85%,
при 20° 23,53%; ZnSiF6 при 0° 33,76%, при 20° 35,16%. Кремнефто-
рид бария обладает очень малой растворимостью в воде — при 25°
Применение
1105
насыщенный водный раствор содержит 0,025% BaSiF6. О раствори-
мости CaSiF6 • 2Н2О в водных растворах H2SiF6 см.б2.
Соединения фтора с серой63 — фториды серы (S2F2, SF2, SF4,.
S2F10, SFe), а также тиофториды (SOF2, SOC1F) и сульфурилфто-
риды (SO2F2, SO2C1F, SO2BrF) в обычных условиях (20° и 760 мм
рт. ст.) представляют собой бесцветные газообразные вещества или
легко кипящие жидкости (S2Fi0 С /кип 29° и SO2BrF С /кип 40°).
Фторсульфинаты щелочных металлов (KSO2F, NaSO2F) устойчивы
до 150°84. Из этой группы веществ наибольшее значение имеют гек-
сафторид серы SFe и фторсульфоновая кислота HSO3F (/кип 165,5°).
Гексафторид серы имеет плотность 6,6 г/л (0°; 753,5 мм рт.
ст.) 65, /кр=45,5°, РКр = 36,8 ат86 или 37,113 ат67, возгоняется при
—63,8°; является одним из наиболее инертных веществ — не разла-
гается при нагревании до 800°, не реагирует со щелочами, с раска-
ленными медью, окисью меди и обладает крайне малой раствори-
мостью в воде (при 0,1° 0,0147 объемов, при 14,9° 0,0076 объемов на
1 объем воды) — меньше растворимости любого другого газа.
ПРИМЕНЕНИЕ
Свободный фтор используют для получения некоторых фтори-
дов, например гексафторида урана UFe с целью разделения изото-
пов U235 и U238, гексафторида серы, используемого в качестве изо-
лирующей среды в высоковольтных кабелях, конденсаторах и
других электрических устройствах, фторидов хрома и др. Фтор ис-
пользуют также для получения фторированных невоспламеняющихся
углеводородов 68-69 с высокими температурами кипения и большими
плотностями, употребляемых в качестве теплоносителей. Фтор, его
галогениды и окислы, в частности окись фтора F2O, могут служить
окислителями ракетных топлив 7°’71. Фтор выпускают в баллонах
под давлением ~30 ат. Более удобным в обращении и в перевозке
как носитель фтора является CIF3 ®8,72.
Жидкий фтористый водород применяют в качестве растворителя
спиртов, альдегидов, эфиров и катализатора73 для процессов поли-
меризации, изомеризации и алкилирования, в частности при син-
тезе высокооктановых моторных топлив. Для этих же целей в ряде
случаев применяют и фторсульфоновую кислоту и гексафторфос-
форную кислоту. Значительные количества безводного газообраз-
ного и жидкого HF применяют для получения фторзамещенных ор-
ганических соединений — фторуглеродов, используемых в качестве
теплоносителей, диэлектриков, средств огнетушенйя, термоустойчи-
вых смазочных веществ, а также для изготовления термо- и хими-
чески стойких пластических масс — фторопластов, — в частности
тетрафторэтилена (тефлона) и проч.; хлорсодержащие фторугле-
роды, называемые фреонами, получили широкое распространение
в качестве рабочих тел в холодильных машинах. Безводный HF
1105
Гл. XXXI. Соединения фтора
используют для обработки хлоридов металлов с целью получения
TiF4, Zrp4, NbF5, TaF5, VF4 и др. Высшие фториды кобальта C0F3,
марганца MnF3, церия CeF4, свинца PbF4 и серебра AgF2 также
служат катализаторами при фторировании углеводородов. При син-
тезе многих органических соединений, в частности каучуков, фар-
мацевтических препаратов и других, в качестве катализатора ис-
пользуют фтористый бор; в других случаях катализаторами служат
фториды сурьмы, ртути, цинка. Фтористый водород используют для
извлечения урана из фосфатов и для получения фторидов ура-
на 74-83, а плавиковая кислота может быть применена для селек-
тивного извлечения металлов из полиметаллических руд (напри-
мер, ниобия и тантала из цирконпирохлоровых концентратов84).
Значительное количество солей фтора используется в металлур-
гии. В США около 70% добываемого плавикового шпата (CaF2)
расходуют в качестве флюса в мартеновских и электрических пе-
чах. В качестве флюса при производстве магниевых сплавов и при
термической обработке режущего инструмента используют фторид
магния. Криолит, фториды алюминия, натрия, лития применяются
в производстве алюминия. Фторид бериллия и его двойная соль с
фторидом натрия используются в производстве бериллия. Фториды
натрия, калия, аммония входят в состав легкоплавких смесей, ис-
пользуемых при извлечении различных металлов из их соедине-
ний 85. Плавиковую кислоту применяют для очистки чугунных от-
ливок от формовочного песка.
В гальванических процессах используют фторборную кислоту
(при электролитическом платинировании, при изготовлении алюми-
ниевых рефлекторов), кремнефтористоводородную кислоту (при
рафинировании свинца и свинцевании, при хромировании) 85-87.
Кислые фториды, в особенности бифторид калия, используют
главным образом для получения элементарного фтора и безводного
фтористого водорода. Смесь бифторидов натрия и калия может
быть использована в качестве флюса для пайки металлов 88. Флюсы
для пайки серебром содержат фторид калия или фторборат ка-
лия89. Бифторид аммония и плавиковую кислоту используют в про-
изводстве ламп накаливания90.
Минеральные соединения фтора нашли широкое применение в
промышленности строительных материалов и в керамической про-
мышленности91. При изготовлении керамики используют фториды
натрия, лития, меди, бериллия, бария, стронция, цинка, алюминия
и некоторые кремнефториды. Для ускорения варки стекла и для по-
лучения опаловых и матовых стекол, непрозрачных эмалей исполь-
зуют плавиковый шпат и кремнефторид натрия. Он же служит ми-
нерализатором, ускоряющим клинкерообразование в производстве
цемента, так же как MgF2 39 и другие фториды и кремнефториды.
Для матирования стекла применяют плавиковую кислоту и фтори-
стый аммоний. Для флюатирования поверхности каменных зданий
Применение
1107
и сооружений с целью предохранения их от преждевременного раз-
рушения применяют кремнефториды магния и цинка80, а железобе-
тонные сооружения можно флюатировать также 3,5—7% раствором
H2SiF6 92. Сухой газообразный фтористый кремний начали приме-
нять для обработки бетонных изделий под давлением 4—6 ат. При
этом в результате реакции SiF4 с Са(ОН)2 образуются CaF2 и гель
кремневой кислоты — увеличиваются объемный вес, механическая
прочность (почти в 2,5 раза) и химическая стойкость бетона в агрес-
сивных средах 80. Кремнефторид натрия входит в состав кислото-
упорных замазок93’94.
Чистый, бесцветный плавиковый шпат, называемый оптическим
шпатом, применяют в оптических приборах. Для этой же цели ис-
пользуют искусственные кристаллы CaF2 и LiF85.
Из плавленого под давлением MgF2 изготовляют окошки, про-
пускающие ИК-излучение, и окна космических аппаратов95. Оса-
жденный фтористый кальций особо высокой дисперсности и пори-
стой структуры применяют при изготовлении люминофоров и в ка-
честве теплостойкого наполнителя для резины.
В текстильном производстве используют в качестве протрав фто-
риды сурьмы, хрома. При мытье тканей, для удаления остатков ще-
лочи и разрушения непрореагировавших отбеливающих препаратов
пользуются кремнефторидом натрия. Кремнефторид натрия исполь-
зуют при флотации пирита в качестве подавителя пустой породы и
регулятора среды. Фторид натрия применяют в производстве про-
теиновых клеев; наряду с кремнефторидом натрия его используют
для фторирования питьевой воды 90.
Растворимые фториды и кремнефториды ядовиты 96 — этим об-
условлено их использование в качестве инсектофунгицидов и анти-
септиков. Фторид натрия и кремнефториды натрия и бария исполь-
зуют для борьбы с вредителями сахарной свеклы, льна, овощных
и других культур, а также в качестве зооцидов, как и фторацетат
бария. Кремнефторид натрия в смеси с цианамидом кальция (1:5
или 2 : 3) 97 используют в качестве дефолианта для предуборочного
удаления листьев хлопчатника. Фториды и кремнефториды широко
применяются для антисептирования древесины. Для консервирова-
ния железнодорожных шпал, телеграфных столбов и деревянных
конструкций, помимо давно используемого фторида натрия, реко-
мендуются фториды калия, аммония, цинка; кремнефториды нат-
рия, магния и цинка лучше проникают в древесину, чем фториды,
и не дают осадка с известью и солями кальция, поэтому их можно
применять для консервирования древесины, соприкасающейся со
штукатуркой 98-101.
Техническую фтористоводородную кислоту, согласно ГОСТ
2567—54, выпускают с содержанием не менее 40% HF и не более
0,1% H2SiF6 и 0,05% H2SO4. Тарой для нее служат эбонитовые
баки, полиэтиленовые баллоны и гуммированные цистерны.
1108
Гл. XXXI. Соединения фтора
Безводный фтористый водород, согласно ГОСТ 14022—68, дол-
жен содержать (соответственно в сорте 1 и 2): не менее 99,9 и
99,8% HF и не более 0,05 и 0,08% влаги, 0,01 и 0,015% SO2, 0,02 и
0,05% H2SO4, 0,02 и 0,05% H2SiF6. Его перевозят в стальных ци-
стернах, бочках и баллонах.
Кремнефтористоводородная кислота выпускается в виде водного
раствора, замутненного гелем кремневой кислоты; она содержит
8—10% H2SiFe.
К качеству технического фторида натрия, согласно ГОСТ
2871—67, предъявляются следующие требования:
Сорт I • Сорт II
(в о/о) (в Vi
NaF, не менее........................ 95,0 80,0
Na2CO3, не более...................... 0,5 —
Сульфаты (в пересчете на Na2SO4), не
более................................. 0,5 3
Не растворимые в воде, не более ... 2,0 —
Влага, не более....................... 0,5 2,0
К качеству технического кремнефторида натрия, согласно ГОСТ
87—66, предъявляются следующие требования:
Na2SiFe, не менее.................
Свободные кислоты (в пересчете на
НС1), не более..................
Влага, не более...................
Остаток на сите № 0063, не более
Сорт высший IB •/.) 98 Сорт 1-й (В °/о) 95 Сорт 2-й (в °/а> 93
0,1 0,15 0,15
1,0 1,0 1,0
15 15 15
Фторид алюминия, согласно ГОСТ 10017—62, должен содер-
жать (в пересчете на сухое вещество) не менее 64,5% фтора, 31,5%
алюминия и не более 0,5% SiO2 + Fe2O3, 1,3% SO4; влажность не
должна превышать 6,5%. При сушке продукта в печах с внутрен-
ним обогревом допускается содержание фтора не менее 61 %, а
влаги не более 4%. Экспортный продукт по ГОСТ 10.58—71 дол-
жен содержать в сухом веществе не менее 65% F и 31% А1 и не
более 0,35% SiO2 + Fe2O3, 0,8% SO4; влажность его должна быть
меньше 3,5%.
Криолит выпускают двух марок: К-1 и К-2; согласно
ГОСТ 10561—63, к нему предъявляются следующие требования:
К-1 К-2
(в %) (в %)
Фтор, не менее .... 54 54
Алюминий, не менее . . 13 13
Натрий, не более ... 30 30
SiO2, не более .... 0,9 1,5
К-i к-’
(в %) (в %)
Fe2O3, не более .... 0,1 0,1
Сульфаты (в пересчете
иа SO4), не более . . 1,2 1.2
Влага, не более .... 0,8 1,0
СЫРЬЕ
Содержание фтора в земной коре да 0,08% по весу, т. е. больше,
чем хлора 109.
Сырье
1109
Наиболее распространен фтор в виде содержащих фтор фосфо-
ритов и апатитов (стр. 811). Фтор входит в состав руд некоторых
металлов, природных вод103, животных и растительных организ-
мов 13. Повышенное содержание фтора в питьевых водах вызывает
заболевание эндемией — «крапчатостью» эмали зубов.
Основными видами сырья для промышленного производства
фтора н его соединений являются содержащие SiF4 и HF отходя-
щие газы заводрв, вырабатывающих фосфорные удобрения, и
природный фторид кальция — плавиковый шпат или флюорит
CaF2, месторождения которого имеются в Приморском крае, в За-
байкалье, в Средней Азии, на Урале, в районе Амдермы и др.104.
Крупные запасы плавикового шпата находятся в США, Мексике,
Канаде, а также в Испании, Франции, Италии, Англии, ФРГ —
в них добывают около 97% от общего количества плавикового
шпата, добываемого в капиталистическом мире. Примерные за-
пасы плавиковошпатовых руд (с содержанием более 35% CaF2) во
всех капиталистических странах на конец 1961 г. составляли
60 млн. т. В то же время запасы фтора в фосфатах оценивались в-
900—1300 млн. т, что эквивалентно 1700—2500 млн. т CaF2 105-106.
Плавиковый шпат в чистом виде бесцветен, но обычно сопрово-
ждается примесями кварца, кальцита, иногда барита, сульфидами
свинца, цинка и других тяжелых металлов и окрашен в белый, зе-
леный, желтый, розовый, синий и фиолетовый цвета107. Особенно
нежелательной примесью является кварцит, так как при производ-
стве фторидов он связывает часть фтора в кремнефториды.
Обычно добываемый плавиковый шпат подвергают обогаще-
нию 108. Большей частью применяют комбинированные методы.
В ФРГ очистку флюорита от барита производят гидромеханиче-
ским и флотационным методами, при этом получают концентрат,
содержащий до 98% CaF2. В США отделяют флюорит от кальцита
осаждением в тяжелых суспензиях и флотацией; вначале в тяже-
лой суспензии обрабатывают частицы размером 2 мм и получают
концентрат с 90% CaF2, затем его размалывают до 0,2—0,3 мм и
флотируют, получая продукт с 98% CaF2109.
Изучено обезвоживание пульпы флотационного концентрата в
псевдоожиженном слое инертного материала при 600° — концен-
трат уносится газом и должен улавливаться пылеуловителями (ци-
клонами и др.)110. При обогащении низкосортного плавикового
шпата в тяжелых суспензиях (плотность 2,9 sfcMz) утяжелителем
служит главным образом ферросилиций, расход которого соста-
вляет 0,2—0,5 кг на 1т руды (потери при регенерации суспензии
на магнитных сепараторах)80-1П-112. Некоторые сорта плавикового
шпата могут подвергаться термическому обогащению, основанному
на том, что при нагревании до 500—800° минерал растрескивается
на мелкие кусочки, в то время как пустая порода не подвергается
растрескиванию. После прокалки материал рассеивают.
1110
Гл. XXXI. Соединения фтора
Предложен метод химического обогащения плавикового шпа-
та— очистка его от S1O2 обработкой плавиковой кислотой. Обра-
зующийся раствор H2SiFe отделяют от осадка CaF2 и подвергают
гидролизу выпариванием при 600—800°. Продуктами гидролиза
являются кремнегель (мельчайший белый порошок с плотностью
0,05—0,16 г)см? и удельной поверхностью 200 л2/г) и фтористый
водород, который улавливают водой и возвращают в процесс 1|3 ~116.
Согласно ГОСТ 7618—70, плавиковый шпат выпускают следую-
щих марок:
ФР-55, ФР-40, ФР-30, ФР-20
ФК-95А, ФК-95Б, ФК-92, ФК-85, ФК-75, ФК-65
ФГ-95А, ФГ-95Б, ФГ-92, ФГ-85, ФГ-75, ФГМ-75, ФГ-65
ФФ-97А, ФФ-97Б, ФФ-95А, ФФ-95Б, ФФ-92
ФО-95 А, ФО-95Б, ФО-92, ФО-85
Буквами ФР, ФК, ФГ, ФФ и ФО обозначены, соответственно,
флюорит рядовой (руда), кусковой сортированный, гравитацион-
ный концентрат, флотационный концентрат и окатыши обожжен-
ные; число указывает содержание CaF2 (%); буквами А и Б отме-
чено, соответственно, допускаемое пониженное и повышенное коли-
чество примеси SiO2, а буквой М — мелкая разновидность. Помимо
SiO2 ГОСТ регламентирует содержание карбоната кальция, серы,
фосфора и гранулометрический состав.
ПОЛУЧЕНИЕ ФТОРА
Промышленные методы получения фтора основаны на электро-
лизе расплава, состоящего из KF и HF 117~119. При составе электро-
лита KF + 12HF электролиз ведется при температуре от —30 до
—50°; при составе электролита KF HF температура электролиза
выше 250°. Она определяется физическими свойствами электро-
лита. По-видимому, наиболее удобным является состав электро-
лита KF • 2HF, который расплавляется ниже 100°, а электролиз
ведется при 90—120° при непрерывной подаче фтористоводород-
ного газа в электролизер 751120.
На аноде выделяется фтор, на катоде — водород. Для корпуса
ванны, диафрагмы, катода и других элементов конструкции при-
меняют сталь и красную медь; аноды — угольные или графитовые.
Для отвода выделяющегося тепла ванну охлаждают снаружи водя-
ной рубашкой. В анодном газе содержится до 4—8% HF. После
отделения HF получается газ примерного состава: 99% F2, 0,12—
0,14% СО2, 0,3—0,5% О2 и 0,1—0,4% N2121’122.
Было предложено много конструкций ванн для получения фто-
ра 128-127 Одна из них показана на рис. 324. Ванна имеет длину
1,81 м, ширину 0,46 м и высоту 0,61 м; работает при нагрузке
750—1500 а, что соответствует плотности тока 1075—2150 а/м2.
Получение фтора
111!
Рабочая поверхность анодов около 0,7 м2. Содержание HF в элек-
тролите 37—40%; температура электролита 95—115°. Выход по
току достигает 95%; он зависит от качества фтористоводородного
газа и чистоты бифторида калия, который не должен содержать
заметных количеств НгО, SOl-, SiFe- и др. При снижении концен-
трации HF в электролите ниже 37% начинает кристаллизоваться
KF, и возрастает сопротивление электролита. Улучшение смачивае-
мости анода электролитом дости-
гается при добавке к нему 1—1,5%
LiF 128.
Вследствие большой реакцион-
ной способности фтор обычно пере-
рабатывают сразу по выходе его из
электролизера. В случае необходи-
мости транспортировать фтор пред-
почитают сжижать путем глубокого
охлаждения жидким азотом или
кислородом. Сжижение фтора при
более высоких температурах под
давлением затруднено сильной кор-
розией движущихся частей компрес-
соров. В связи с этим представляет
интерес применение для сжатия
фтора диафрагменного четырехсту-
пенчатого компрессора 129.
Для транспорта фтора приме-
няют стальные и никелевые балло-
ны iso-132 Образующаяся на внут-
ренней поверхности баллона пленка
из фтористой соли препятствует
дальнейшей коррозии, признаки ко-
торой не обнаруживаются даже по-
сле годового хранения фтора 133. Для
транспорта под атмосферным дав-
лением применяют стальные трубы,
Рис. 324. Схематический разрез
электролизера для получения
фтора:
/ — выход водорода; 2 — выход фтора;
3 —крышка; 4 — перегородка, к которой
прикрепляется диафрагма; 5 — уровень
электролита; 6 — анод; 7 — катод; 8 — кор-
пус электролизера; 9 —диафрагма из
сетки; 10 — водяная рубашка; // — угло-
вое железо; 12— фарфоровый изолятор;
13 — опоры из швеллерного железа.
но при этом следует опасаться увлажнения фтора или загряз-
нения его органическими примесями. При повышенном давлении
более пригодны трубы из никеля или монель-металла 134.
Разработана конструкция автоцистерны для перевозки фтора и
резервуара для его хранения. Это три горизонтальных стальных
цилиндра, находящиеся один в другом. Во внутреннем цилиндре
содержится жидкий фтор (—188°) под атмосферным давлением,
между стенками внутреннего и среднего цилиндров находится
жидкий азот (—196°) также под атмосферным давлением, а коль-
цевое пространство между средним и наружным цилиндрами
заполнено тепловой изоляцией. Жидкий азот предотвращает
11 М. Е. Поэин
1112
Гл. XXXI. Соединения фтора
испарение фтора, наружная изоляция уменьшает потери азота.
Перекачку жидкого фтора производят, передавливая его сжатым
гелием 135.
Отходящие газы после сжижения или использования фтора
перед выбросом в атмосферу необходимо очищать от остатков фтора
промывкой раствором щелочи, например по следующей схеме136.
Газ отмывают от фтора в поглотительной колонне с насадкой,
орошаемой 5—15% раствором NaOH с температурой 40—65°; при
такой концентрации NaOH идут только следующие реакции
F2 + 2NaOH = 2NaF + ’/2О2 + Н2О; F2 + Н2О = 2HF + ‘/2О2;
HF +NaOH = NaF+ Н2О
и чрезвычайно ядовитая окись фтора не образуется. Вытекающий
из колонны раствор, содержащий NaF, перекачивают насосом в
регенерационный бак, где его обрабатывают известковым молоком.
При этом регенерируется едкий натр (по реакции 2NaF +
+ Ca(OH)2 = CaF2 + 2NaOH) и после отстаивания суспензии CaF2
осветленный раствор через теплообменники возвращается на аб-
сорбцию фтора. При такой схеме расходуется вместо едкого натра
более дешевый продукт — известь — и предотвращается накопле-
ние в системе NaF, который вследствие малой растворимости мог
бы выделяться в осадок и забивать насадку абсорбера и трубы.
Расход извести -~4,5 кг на 1 кг улавливаемого фтора.
Необходимый для производства фтора бифторид калия готовят
смешением рассчитанных количеств плавиковой кислоты и поташа
или едкого кали. К полученному раствору добавляют этиловый
спирт для высаливания бифторида, который затем высушивают.
Так как с исходными растворами вводится вода, которая также
выделяется и при реакции
4HF + 2КОН - 2(KF • HF) + 2Н2О
то для более полного использования реагентов необходимо произ-
водить выпаривание раствора до начала кристаллизации при 90—
100°, а затем разгонку водно-спиртовой смеси для регенерации
спирта и вывода из системы воды. Можно, однако, осуществлять
процесс без выпарки раствора и без разгонки водно-спиртовой
смеси по следующей схеме 114. В абсорбционной колонне, орошае-
мой спиртом, поглощают фтористый водород, получая 30—40%
раствор HF в спирте. Едкое кали растворяют в оборотном спирте,
и после отделения от этого раствора водно-солевого слоя смеши-
вают спиртовые растворы КОН и HF. Выпавший бифторид калия
отделяют центрифугированием и высушивают при 100°. Маточный
раствор спирта, а также конденсат из паров спирта, уловленных
при сушке бифторида, возвращают в цикл для приготовления
исходных растворов.
Фтористый водород и плавиковая кислота
1113
ФТОРИСТЫЙ ВОДОРОД И ПЛАВИКОВАЯ КИСЛОТА
Разложение плавикового шпата и других фторидов
Получение фтористого водорода основано на обработке тонко-
измельченного плавикового шпата 90—92%-ной серной кислотой:
CaF2 + H2SO4 = 2HF + CaSO4
При низких температурах в этой системе возможно образование
метастабильного кислого сульфата кальция CaSCh • 3H2SO4, кото-
рый около 80° инконгруэнтно плавится с образованием CaSO4 •
• H2SO4137. Последний устойчив до 200°; выше этой температуры
выделяется CaSCh. Поэтому реакцию осуществляют при 220—280°.
При более низких температурах процесс осложняется также обра-
зованием фторсульфоновой кислоты и обволакиванием частиц
флюорита плохопроницаемой коркой сульфата кальция.
Одновременно протекают побочные реакции — разложение
серной кислотой СаСО3 и других примесей и образование SiF4:
SiO2 + 4 HF = SiF4 + 2Н2О
Плавиковый шпат подвергают, в случае необходимости, сушке,
а затем дроблению на щековых, конических и валковых дробилках
и измельчению в шаровых мельницах до тонкости, при которой на
сите с 3000 отв]см2 остается не более 10% материала.
Измельченный шпат смешивают с 90%-ной серной кислотой в
смесителях-питателях непрерывного действия. Так как при смеше-
нии начинается реакция, которая ведет к загустеванию и затвер-
деванию смеси вследствие удаления HF и образования CaSO4, то
продолжительность пребывания массы в смесителе не должна быть
большой.
Наиболее рациональным, обеспечивающим равномерную дози-
ровку компонентов, является смеситель шнекового типа, в котором
продолжительность пребывания массы составляет всего 5—6 мин.
Чем меньше содержится CaFs в натуральном плавиковом шпате,
тем меньше требуется на его разложение серной кислоты и тем
гуще получается масса в смесителе. При этом для сохранения ее
текучести следует применять менее концентрированную кислоту.
Однако применение серной кислоты, содержащей меньше 88%
H2SO4, вызывает образование кольцевых настылей в печи. Избы-
ток серной кислоты приводит к увеличению ее расхода и также
затрудняет работу печи. Недостаток же серной кислоты приводит
к неполному использованию CaF2. Поэтому весьма существенной
является точная дозировка материалов, которую рассчитывают по
анализу сырья. Из смесителя-питателя масса поступает в печь.
Для получения HF применяют барабанные вращающиеся печи
непрерывного действия 138-139. Барабан из котельной стали толщи-
ной 12—16 мм заключен в кирпичную кладку и обогревается
11*
1114
Гл. XXXI. Соединения фтора
снаружи газом. Масса из смесителя поступает в печь через отвер-
стие в неподвижной загрузочной головке (снабженной также па-
трубком для отвода газа) и по мере протекания процесса превра-
щается в рассыпчатый сухой материал, который удаляют через на-
клонный желоб в задней крышке печи. Реакция протекает наиболее
интенсивно в передней части печи. Эта часть барабана печи защи-
щена от коррозии вставленной внутрь броней из малоуглеродистой
стали толщиной 14 мм. Срок службы брони 2—3 месяца, а брониро-
ванного барабана — 1 год. Броня из стали ЭИ-533 (Х23Н23МЗДЗ)
является в 10 раз более стойкой, а из стали ЭИ-529 (Х23Н28МЗДЗТ)—
в 22 раза 14°.
Достаточная для завершения реакции продолжительность пре-
бывания реакционной массы в печи составляет 55—60 мин при
температуре в передней части печи 160—180°, а в задней 220—280°.
Более высокие температуры хотя и ускоряют процесс, но нежела-
тельны, так как приводят к повышенному содержанию паров сер-
ной кислоты в уходящем из печи газе, который имеет температуру
120—140° и содержит до 80% HF. Производительность печи с диа-
метром барабана 1,9 м и длиной 12 м составляет ~ 10 т в сутки
плавиковой кислоты (в пересчете на 100% HF). Выгружаемый из
печей материал содержит более 80% CaSO4, 2—6% CaF2 и 10—
12% свободной серной кислоты. Обычно этот материал является
отходом производства. Перед сбросом в отвал его целесообразно
нейтрализовать. Применение для этой цели суспендированного в
воде тонкоразмолотого известняка, даже при значительном его из-
бытке, не дает полной нейтрализации серной кислоты вследствие
покрытия зерен СаСОз коркой сульфата 141. Предложено нейтрали-
зовать отбросный сульфат кальция доменным шлаком (15%) при
их совместном размоле до размера частиц 90 мк с последующей
добавкой в смесителе СаО или Са(ОН)2 (1% от веса CaSO4)142.
Применяются и более мощные печи с внутренним обогревом
(диаметром 2 и 2,2 л и длиной 22 и 20 м). В печах диаметром
8 м и длиной 50 м в передней части (со стороны загрузки реаген-
тов) барабан футерован кислотоупорным кирпичом, в средней
части — угольными блоками, а со стороны выгрузки — шамотным
кирпичом. На разгрузочном конце имеется порог для увеличения
времени пребывания реакционной массы в печи. Печи с внутрен-
ним обогревом имеют более продолжительный срок службы, ббль-
шую интенсивность (почти в 2 раза) и работают с меньшим рас-
ходом греющего газа (в 1,5 раза), чем печи с наружным обогре-
вом. Но они дают разбавленный газ, что приводит к большему
расходу электроэнергии на его транспорт (в ~2 раза), работают
с повышенным расходом серной кислоты, а в газе содержится до
20 г/м& SO2143. При прямоточном обогреве этих печей можно пони-
8ить содержание CaF2 до 3% и серной кислоты до 10% в отбросе
из печи и снизить его температуру до 130—140°144.
Фтористый водород и плавиковая кислота
1115
Во избежание проникновения газов в атмосферу цеха через не-
плотности, печи работают под небольшим разрежением (2—3 мм
вод. ст.), создаваемым вентилятором, просасывающим уходящие
из печи газы через абсорбционную установку. Вследствие этого в
печи подсасывается значительное количество воздуха.
Газы, уходящие из вращающихся барабанных печей с внутрен-
ним обогревом, имеют температуру 210—250° и вследствие раз-
бавления продуктами горения топлива содержат 15—23 объемн.%
HF. Для очистки газов от туманообразной серной кислоты их
пропускают через колонку (грязевик), заполненную кусками кокса
и древесного угля. При охлаждении газов содержащийся в них
SiF4 реагирует с HF и превращается в H2SiF6 по реакции: SiF4 +
+2HF=H2SiF6.
Наиболее полная очистка газов от серной кислоты (до 90%) с
наименьшими потерями HF и H2SiF6 в конденсирующейся на
коксе серной кислоте достигается при поддержании в очистителе
высокой температуры. С этой целью газоход от печи до коксовой
колонки покрывают тепловой изоляцией. Очищенный газ с темпе-
ратурой 75—90° направляют на абсорбцию водой для получения
плавиковой кислоты.
Для получения высококонцентрированного HF-газа разложение
плавикового шпата производят в неподвижных ретортных печах
периодического и непрерывного действия 145. Ретортные печи непре-
рывного действия имеют обычно два неподвижных, расположенных
один над другим горизонтальных барабана, внутри которых реак-
ционная масса перемещается с помощью лопастей, насаженных на
вращающийся горизонтальный вал. Барабаны обогреваются сна-
ружи генераторным газом. Масса из смесителя поступает в верх-
ний барабан, движется к его концу, где проваливается в нижний
барабан, в котором продвигается в противоположном направле-
нии и поступает в барабанный холодильник.
Опубликованы патенты, по которым разложение CaF2 серной
кислотой ведут сначала при 80—180° во вращающейся печи 146 или
в грануляторе 147, а затем завершают процесс термообработкой при
250—300° (по другим заявкам148, — до 1000°); в первой стадии вы-
деляется концентрированный HF, а из газа второй стадии конден-
сируют серную кислоту с примесью HF, которую направляют на
разложение плавикового шпата. Нагревание реакционной смеси
и регулировку температуры в первой стадии предложено осуще-
ствлять за счет экзотермического эффекта взаимодействия H2SO4
с парами воды149. Запатентовано осуществление противоточного
процесса в реакторе, разделенном на две зоны — испарительную и
реакционную. Из первой во вторую поступает смесь паров H2SO4,
SO3, Н2О и HF и контактнруется с движущимся слоем CaFj при
100—300°; HF выводят из этой зоны. Твердый остаток переме-
щается затем до выгрузки через испарительную зону, где его
1116
Гл. XXXI. Соединения фтора
опрыскивают серной кислотой и куда вводят смесь паров SO3, НгО
и HF, присоединяющуюся к испаряющейся здесь H2SO4. Эта смесь
паров поступает в реакционную зону 15°.
Предложено также предварительно готовить суспензию CaF2 в
концентрированной серной кислоте (105—115% от стехиометриче-
ского количества) при 5—25° и подавать ее на нагретый до 120—
200° слой CaSO4; для повышения устойчивости суспензии в нее
следует вводить HF или HSO3F (5—10% от веса H2SO4) 151.
Описано получение HF из плавикового шпата и концентриро-
ванной серной кислоты в печи со взвешенным слоем. Реакционную
смесь с добавкой инертного вещества (ангидрита-и др.) приготов-
ляют при комнатной или слегка повышенной температуре, гранули-
руют, формуют или измельчают и загружают в печь. Псевдоожи-
жение смеси достигается при помощи газообразного фтористого
водорода 152. По другому варианту153, смесь CaF2 и H2SO4 пред-
ч ложено подавать на псевдоожиженный слой, состоящий из частиц
CaSCh с размерами от 10 до 1000 мк. Высота слоя 0,9—1,8 м, тем-
пература 200—300°; псевдоожиженный слой создается и нагре-
вается поступающим снизу газом. Уходящий газ, содержащий HF,
очищают в другом аппарате с псевдоожиженным слоем из CaSO4.
Предложено154 разлагать СаРг не серной, а термической фос-
форной кислотой. При 250° за 3 ч выход HF достигает 90%. В твер-
дой фазе образуется кислый пирофосфат кальция, который можно
использовать в качестве удобрения.
Помимо кислотного разложения представляет интерес гидро-
термическое разложение плавикового шпата или синтетического
CaF2 при 1000—1250° по реакции 155-156:
CaF2 + H2O СаО + 2HF
Природный крупнокристаллический минерал разлагается па-
ром значительно медленнее, чем синтетический CaF2. Процесс за-
медляется также образованием на реагирующих с паром зернах
покрывающей корки продукта реакции — извести. Последнее устра-
няется добавкой 30% SiC>2 Для связывания извести в двухкаль-
циевый силикат. При этом в газовую фазу помимо HF выделяется
немного SiF4, а скорость реакции повышается в несколько раз и,
например, при 1250° за 15—30 мин степень превращения достигает
85—90%.
Изучено получение HF действием серной кислоты на NaF. При
стехиометрическом соотношении реагентов для практически пол-
ного разложения NaF требуется нагрев реакционной массы до 500°.
Она получается подвижной при концентрации серной кислоты не
больше 75% H2SO4. Даже при избытке кислоты и двухчасовом
нагреве в сульфате натрия остаются десятые доли процента
фтора 157.
Фтористый водород и плавиковая кислота
1117
Запатентован способ получения HF из фторидов щелочных ме-
таллов взаимодействием их с водным раствором NH4HF2 при 25°
в течение 2 ч 158. Безводный фтористый водород может быть полу-
чен из водных растворов H2SiFe. Для этого сначала из них осаж-
дают аммиаком кремневую кислоту. Затем к полученному раствору
NH4F добавляют избыток (60%) концентрированной серной кис-
лоты и выделяют HF фракционной перегонкой, а из остатка извле-
кают (NH4)2SO4,159. По другому патенту, добавкой MgO к раствору
NH4F осаждают MgF2, причем выделяющийся аммиак вновь
используют для обработки H2SiFe; действуя на MgF2 концентриро-
ванной H2SO4, получают HF 16°. Запатентован способ использова-
ния теплоты гидратации олеума для проведения реакции сернокис-
лотного разложения фторидов с целью получения HF161. (О полу-
чении HF из H2SiF6 см. стр. 1149, а через фторид и бифторид
аммония — стр. 1167).
В качестве побочного продукта фтористый водород образуется
при пламенном газофазном гидролизе SiFe по реакции
SiF4+2H2O SiO2 + 4HF
осуществляемой с целью получения тонкодисперсного кремнезема,
имеющего удельную поверхность до 200 м2/г и используемого в ка-
честве высокоактивного сорбента и наполнителя (аэросил, аэро-
гель) 162. Реакция эта обратима (lg/Cp — — + 5,547). При
1000—1200° степень превращения SiF4 в стехиометрической смеси
0,43—0,51, при 4-кратном избытке Н2О 0,93—0,98, а при 7,5-крат-
ном— около 1. Для реакции можно использовать как концентри-
рованный SiF4, TaK и разбавленный отходящий газ суперфосфат-
ного производства. Газ смешивают с углеводородом (метан,
пропан) и сжигают в горелке из высокоглиноземистого шамота.
При чрезмерной влажности газа ее частично удаляют (например,
сернокислотной осушкой). Содержание кислорода в смеси, по-
ступающей в горелку, должно быть не ниже 16% — при необхо-
димости это достигают добавкой воздуха. Фильтрованием через
пористый материал реакционные газы освобождают от SiO2, после
чего из них может быть извлечен фтористый водород.
Абсорбция и конденсация фтористого водорода
Абсорбция HF производится последовательно в нескольких
свинцовых или пластмассовых башнях диаметром 2 м, высотой
6—8 м, заполненных коксом, угольными кольцами или деревян-
ными рейками. Если используют стальные башни, то их гуммируют
и футеруют в два слоя угольными блоками на бакелитовой за-
мазке. Последняя башня орошается водой, и получаемая слабая
кислота передается на орошение предыдущих башен противотоком
газу. Из первой по ходу газа башни вытекает продукционная
1118
Гл. XXXI. Соединения фтора
кислота. Для просасывания газов через систему за последней аб-
сорбционной башней устанавливают гуммированный вентилятор.
Такой вентилятор не может дать большого разрежения, и иногда,
для уменьшения сопротивления системы, башни включают парал-
лельно, попарно. С этой же целью иногда применяют полые
башни, без насадки, в которых абсорбция идет на поверхности
падающих капель жидкости. С газом, уходящим из абсорбционной
установки, теряется менее 1 % фтора. Для обезвреживания этого
газа его промывают раствором соды.
Кислота циркулирует с помощью эбонитовых насосов. Иногда ее
охлаждают в змеевиковых холодильниках; это позволяет получать
более концентрированную кислоту. Как видно из рис. 314 (стр. 1097),
для получения стандартной плавиковой кислоты, содержащей 40%
HF, при 25° требуется, чтобы концентрация HF в газе, поступаю-
щем на абсорбцию, была выше 7 мг/л, а при 50° — выше 30 мг/л.
Основное количество получаемой кислоты обычно сразу же пе-
рерабатывают на фтористые соли. В этом случае не стремятся
получать концентрированную кислоту. Орошение башен водой ре-
гулируют таким образом, чтобы в продукционной кислоте содер-
жалось 28—30% HF.
На получение 1 т 100%-иого HF в плавиковой кислоте расхо-
дуется: 2,55 т плавикового шпата (100% СаРг), 3,75 т серной кис-
лоты (100% H2SO4).
В абсорбционных башнях, помимо поглощения HF, происходит
конденсация H2SiF6. Количество HaSiFe в «грязной» плавиковой
кислоте, вытекающей из абсорбционной установки, зависит от со-
держания SiC>2 в плавиковом шпате и колеблется от 2 до 10%.
Очистку плавиковой кислоты от H2SiF6, дающую побочный про-
дукт— кремнефторид натрия, производят обработкой ее содой в
стальных гуммированных чанах с лопастными мешалками. Обра-
зующийся по реакции
H2SiF6 -Ь Na2CO3 — Na2SiF3 -Ь Н2О -Ь СО2
плохорастворимый кремнефторид натрия выделяется в осадок, ко-
торый отделяют отстаиванием или отфильтровывают на вакуум-
фильтре и промывают водой. Промывную воду добавляют к плави-
ковой кислоте, идущей на переработку во фтористые соли.
Применение избытка соды для осаждения Na2SiF6 недопустимо,
так как при этом нейтрализуется часть HF и получаемый NaF
также осаждается, а содержащаяся в плавиковой кислоте серная
кислота (0,5—1,5%) переводится в Na2SO4:
2HF + Na2CO3 = 2NaF + Н2О + СО2
H2SO4 4- Na2CO3 = Na2SO4 -Ь H2O + СО2
При применении же стехиометрического количества соды, тре-
буемого для очистки от H2SiF6, эти реакции не имеют значения, так
Фтористый водород и плавиковая кислота
1119
как растворимость NaaSiFe меньше, чем растворимость NaF и
Na2SO4, и потому имеющаяся в растворе кремнефтористоводород-
ная кислота осаждает Na2SiFe из этих солей по реакциям:
2NaF + H2SiF6 = Na2SiF6 + 2HF
Na2SO4 + H2SiF6 = Na2SiF6 + H2SO4
В очищенной кислоте содержится немного H2SO4, NaF и неко-
торое количество Na2SiF6, зависящее от концентрации HF и тем-
пературы (рис. 325)135; содержание Na2SiFe несколько больше
соответствующего его растворимости.
Заводы, перерабатывающие плавиковую кислоту в криолит, «те-
ряют» значительную часть фтора, выпуская его в виде кремнефто-
рида или фторида натрия, по-
лучаемых в качестве побочных
продуктов в процессе очистки
«грязной» плавиковой кислоты.
Чем меньше содержится SiO2 в
исходном плавиковом шпате, тем
меньше эти «потери». Впрочем,
получаемую при очистке пульпу
кремнефторида натрия можно
также переработать сначала в
NaF, а затем взаимодействием
NaF с A1F3 в криолит (стр. 1160)163.
Для получения плавиковой
кислоты, не содержащей I^SiFe
HF б очищенной кислоте, %
Рис. 325. Растворимость Na2SiF4
в плавиковой кислоте.
или Na2SiF6, предложено очищать печной газ от SiF4 до абсорбции
HF. Очистка от SiF4 может быть осуществлена и сухим способом,
при использовании в качестве поглотителей кислых фторидов ка-
лия, натрия или бария, образующих кремнефториды, стойкие при
температуре газа, выходящего из печи94.
Предложен 164 способ очистки газообразного HF от SiF4 в про-
цессе абсорбции. Для этого газ нагревают до 150—500° и затем
направляют в абсорбер, где за счет вносимого газом тепла идет
разделение и концентрирование получаемых кислот — из нижней
части колонны отбирают концентрированный раствор H2SiF6, а из
средней 25—38%-ную плавиковую кислоту. Уносимая газом вода
конденсируется в дефлегматоре. Для очистки технической плави-
ковой кислоты от летучих примесей (I^SiFe, SO2) рекомендуют 168
подвергать ее дистилляции, сопровождаемой продувкой воздухом
или азотом под давлением 2,5 ат, при температуре в кубе 48°.
Расход воздуха 25 кг на 1 кг HF, потери HF с отходящим га-
зом 1%.
Описана136 опытно-промышленная установка для получения
80%-ной плавиковой кислоты двухступенчатой конденсацией HF
из печного газа. В первой ступени конденсируются пары воды,
1120
Гл. XXXI. Соединения фтора
H2SO4, H2SiF6 и часть HF — конденсат отводится в виде «грязе-
вой кислоты». Во второй ступени в трубчатом противоточном теп-
лообменнике конденсируется кислота, содержащая 80% HF. Не-
сконденсировавшаяся часть HF поступает на водную абсорбцию.
50 во 10 80 90 100
Содержание HF в газе, %
Рис. 326. Выход жидкого фто-
ристого водорода в зависи-
мости от концентрации газа.
Жидкий фтористый водород
Для получения жидкого фтористого водорода167 необходимо,
чтобы газы, уходящие из печи, были высококонцентрированными
и не содержали значительных количеств SiF4. С этой целью при-
меняют в качестве сырья отборный плавиковый шпат или концент-
рат с содержанием 96—97% CaF2 и
с минимальным количеством SiO2.
Концентрированный газ по выходе
из печи охлаждается в поверхностных
холодильниках сначала водой, а за-
тем холодильным рассолом, причем из
него конденсируется часть HF в жид-
кий, почти безводный продукт. Остав-
шийся газ направляют в абсорбцион-
ную установку для получения плави-
ковой кислоты или улавливают 96—
100%-ной серной кислотой, которую
направляют затем на разложение шпа-
168
На рис. 326 показан выход жидкого
фтористого водорода в зависимости от
концентрации газа, при охлаждении его от 80 до —15°. Выход
резко падает с понижением концентрации газа и при 54% дости-
гает нуля. Из газа, содержащего меньше 54% HF, можно сконден-
сировать жидкий фтористый водород лишь при температурах более
низких, чем —15°, что не рационально.
Присутствие в газе даже небольших количеств воды в значи-
тельной степени ухудшает выход жидкого фтористого водорода,
так как при этом часть продукта выводится в виде плавиковой
кислоты. Содержание же воды в газе зависит от наличия примесей
в плавиковом шпате, разлагающихся с выделением воды:
СаСОз + H2SO4 = CaSO4 -Ь СО2 4- Н2О
R2O3 + 3H2SO4 = R2(SO4)s + ЗН2О
SiO2-f-4HF = SiF4 + 2Н2О и др.
Поэтому при производстве жидкого фтористого водорода качество
шпата имеет особенно важное значение. Рекомендуют до разло-
жения плавикового шпата серной кислотой подвергать его окисли-
тельному прокаливанию при 480—590° в псевдоожиженном слое169
или обрабатывать плавиковой кислотой, причем удаляются карбо-
Фтористый водород и плавиковая кислота
1121
наты и SiO217°. Освобождение фтористоводородного газа от влаги
осушкой затруднительно. Обычно применяемые в технике осуши-
тели — серная кислота, безводный сульфат натрия, хлорид кальция
и другие — в данном случае малоудобны, так как HF вступает с
ними в химические реакции. С концентрированной серной кислотой
HF образует некоторое количество фторсульфоновой кислоты
h2so4 + hf hso3f + h2o
с сульфатом натрия образуется, по-видимому, соль этой кислоты,
а из хлорида кальция фтористый водород вытесняет НС1. Поэтому
наилучшим способом отделения воды является дробная конденса-
ция фтористого водорода из газа.
Так как фтористый водород смешивается с водой во всех отно-
шениях, образуя так называемые высококонцентрированные плави-
ковые кислоты, содержащие 80% HF и больше, то одним из спо-
собов получения жидкого фтористого водорода является метод
Рис. 327. Схема производства безводного фтористого водорода:
/ — резервуар для серной кислоты; 2— воздушный клапан; 3 — насос; 4 — вагонетка для плави-
кового шпата; 5 —элеватор; 6 — печь; 7 —дымовая труба; 8 — шнек; 9 — бункер для шпата;
/0 —напорный бак для серной кислоты; // — ротаметр; 12 — пылеуловитель; 13 — скруббер для
промывки газа серной кислотой; 14— абсорбционная колонна, выпускающая 80%-иую плави-
ковую кислоту; /5 —холодильник для кислоты; 16 — абсорбционная колонна, выпускающая
60%-ную плавиковую кислоту; 17 — холодильник; 18— абсорбционная колонна, выпускающая
30%-ную плавиковую кислоту; 19 — приемник для 80%-иой плавиковой кислоты; 20—вентиля-
тор; 21 — ректификационная колонна; 22 —дефлегматор.
ректификации таких кислот171. В этом случае в кубе ректифика-
ционной колонны остается постоянно кипящая смесь (стр. 1098),
а концентрированный, почти безводный фтористый водород, ухо-
дящий из колонны, может быть легко сконденсирован в жидкий
продукт.
На рис. 327 показана схема производства безводного фтори-
стого водорода 172. Плавиковый шпат, содержащий не менее 97%
CaF2 и не больше 1 % SiO2 и 1 % СаСОз, проходящий через сито
6400 отв/см2, поступает через автоматический питатель в печь, где
разлагается при 230° 99 %-ной серной кислотой. Цилиндрическая
печь из мягкой стали диаметром 1,82 м, длиной 12,2 м, с наклоном
1 :48, делает 1 об1мин. Сульфат кальция выгружается непрерывно.
Газ промывается в скруббере серной кислотой и поступает в
две параллельно работающие стальные абсорбционные колонны,
где образуется 80%-ная плавиковая кислота, затем проходит
1122
Гл. XXXI. Соединения фтора
последовательно еще две пластмассовые абсорбционные колонны,
в которых образуется 60% - и 30%-ная плавиковая кислота. Цирку-
лирующие кислоты охлаждаются в рассольных холодильниках. Пе-
ред выпуском в атмосферу газ промывают щелочью в колонне с
коксовой насадкой. 80%-ная плавиковая кислота подвергается
ректификации в медной колпачковой колонне; отбираемый ди-
стиллят представляет собой товарный продукт — жидкий фтори-
стый водород, а кубовый остаток — 60%-ная плавиковая кислота —
поступает в стальные, гуммированные неопреном резервуары и от-
туда на питание абсорбционных колонн, где укрепляется до
80% HF.
Для удаления из жидкого фтористого водорода небольших коли-
честв воды, которые невозможно отделить простой дистилляцией,
его обрабатывают 90—100%-ной серной кислотой в экстрактивно-
дистилляционной колонне. В верхнюю часть колонны подают сер-
ную кислоту, в середину колонны — жидкий HF. Избыточное дав-
ление в колонне 0,35—0,7 ат\ температура вверху 27—50°, внизу
150—180°. С верха колонны отводится безводный HF, а снизу —
80%-ная серная кислота 173’174. Повышенная температура внизу
колонны и разбавление серной кислоты обусловливают гидролиз
образующейся в небольшом количестве фторсульфоновой кислоты.
Безводный фтористый водород можно получить ступенчатым
гидролизом жидкой фторсульфоновой кислоты:
HSO3F + Н2О = H2SO4 + HF
Количество воды, добавляемой на каждой ступени, должно
быть меньшим, чем необходимо для полного разложения HSO3F175.
Фтористый водород обезвоживают также, пропуская его через
раскаленный кокс 176’177. При этом кокс окисляется водяным па-
ром— выше 900° практически до окиси углерода. До 1200° взаимо-
действие HF с углеродом и продуктами его окисления водяным
паром термодинамически невозможно. Образующиеся СО и На
разбавляют фтористый водород, однако, так как температура кон-
денсации под атмосферным давлением фтористого водорода
( + 19,9°) значительно выше, чем окиси углерода (—192°) и водо-
рода (—253°), то при охлаждении газовой смеси получается жидкий
фтористый водород. Выход его в зависимости от концентрации HF
(мономера) в исходном газе и температуры конденсации приведен
в табл. 87. Это расчетные данные 178, полученные при допущении,
что фактор ассоциации газообразного HF равен 4; на самом деле
величина фактора ассоциации зависит от температуры (см.
табл. 86, стр. 1096), поэтому действительные степени конденсации
HF будут несколько отличаться от приведенных.
Так как давление водяного пара над концентрированной плави-
ковой кислотой (более 40% HF) ниже 10° ничтожно мало15, то при
конденсации HF из влажного газа вся вода практически переходит
Фтористый водород и плавиковая кислота
1123
ТАБЛИЦА 87
Расчетный выход жидкого фтористого водорода при конденсации (в %)
Концентрация HF в исходном газе, мел. % мономера Температура конденсации, °C
0 -10 -20 -30 -40 —50 -60 -70 -80
5 . — 17,5 62,0
10 — — — — — —. 23,7 63,1 83,4
16 — — — — — 15,5 55,1 80,5 9X0
23 — — — — 1,3 45,0 71,0 86,5 93,5
31 — — — — 35,6 63,5 80,7 90,6 95,8
40 — — — 22,0 57,3 76,0 87,0 93,8 97,4
51 — — 11,4 49,6 72,4 84,7 91,5 96,1 98,0
64 — 3,4 48,1 70,3 83,8 91,0 95,0 97,8 98,9
80 9,6 57,3 77,0 87,0 93,0 96,1 97,8 98,2 99,5
в конденсат. Чем ниже температура конденсации, тем больше
выход плавиковой кислоты и, следовательно, тем больше ее кон-
центрация. Увеличение же влажности исходного газа, естественно,
приводит к уменьшению концентрации HF в конденсате, но выход
его при этом возрастает 178.
Запатентованы способы179-181 получения безводного HF экс-
тракцией его из водных растворов органическими растворителями
(алкиламины, бутанол, циклогексанол, метилизобутилкетон, метил-
изопропилкетон, гексанол и др.) с последующей дистилляцией
экстракта для разделения его на безводный HF и азеотропную
смесь (HF + НгО), возвращаемую на экстракцию. Из водной фазы
дистилляцией также получают азеотропную смесь. Изучен меха-
низм экстракции HF трибутилфосфатом 182.
Предложено извлекать HF из газовой смеси раствором фенола
в органическом растворителе, например CCI4. Фенол образует с
HF неустойчивый комплекс, разрушающийся при 100—150° с пол-
ным выделением безводного HF. Его конденсируют вместе с рас-
творителем, причем конденсат расслаивается с образованием 96—
97%-ного фтористого водорода и растворителя. Последний возвра-
щается на смешение с фенолом 183.
Безводный HF можно получать, разлагая при нагревании (300—
500°) кислые фториды щелочных металлов. Например, высушивая
при 100—120° пульпу из смеси гранул NaF и 25—35%-ной плави-
ковой кислоты, можно испарить воду и получить сухой бифторид
натрия. При его прокаливании выделяется 100% HF и регенери-
руется NaF184. Прокаливание рекомендуют вести в аппарате из
алюминия или его сплавов 185. Безводный NaHFs можно получить и
контактируя с измельченным NaF при 125—150° испаренную пла-
виковую кислоту18в. Запатентован способ получения для этой же
цели бифторида калия путем экстракции HF из разбавленной
1124
Гл. XXXI. Соединения фтора
плавиковой кислоты вторичным лауриламином или его раствором
в углеводороде и добавки к экстракту KF для полного осаждения
KHF2187. О получении безводного HF через NH4HF2 см. стр. 1167.
ПРОИЗВОДСТВО ФТОРИСТЫХ СОЛЕЙ из плавиковой кислоты
Очищенную от H2SiF3 плавиковую кислоту, содержащую в каче-
стве примесей небольшие количества серной кислоты, Na2SiF6 и
NaF, перерабатывают на криолит, фторид алюминия и фторид нат-
рия путем осаждения этих плохорастворимых продуктов соответ-
вующими реагентами 135.
Криолит
Для получения криолита в плавиковой кислоте вначале раство-
ряют гидроокись алюминия:
А1(0Н)3 + 6HF = H3A1F6 + ЗН2О
Раствор фторалюминиевой кислоты H3A1F3 нейтрализуют содой:
2H3A1F6 -Ь ЗМа2СОз — 2Na3AlFg 4- ЗСО2 4- ЗН2О
Образующийся криолит выделяется в осадок.
Фторид натрия, содержащийся в виде примеси в плавиковой
кислоте, также превращается в криолит:
3NaF + H3A1F6 = Na3AlF6 4- 3HF
При избытке соды примесь Na2SiFe переходит в NaF:
Na2SiFg 4- 2Na2CO3 = 6NaF 4- 2CO2 4- SiO2
Для получения чистого криолита стандартного качества
(стр. 1108) необходимо, чтобы плавиковая кислота содержала ма-
лые количества примесей — H2SiF6, Na2SiF6 и H2SO4. Обычно «чи-
стая» плавиковая кислота, идущая на приготовление криолита, со-
держит до 25% HF, 0,7—0,8% H2SiF6 и около 1,5% H2SO4. Примеси
в криолит попадают также из гидроокиси алюминия, который по
техническим условиям не должен содержать более 0,3% SiO2, 0,6%
Na2O и 0,1% Fe2O3.
Получение криолита осуществляют 188 в стальных резервуарах,
футерованных угольными плитками и снабженных мешалками.
Процесс ведут непрерывно в 3—4 соединенных последовательно и
расположенных каскадом резервуарах. Кислоту и пульпу гидро-
окиси алюминия вводят через дозаторы в первый резервуар, содо-
вый раствор — в третий. Гидроокись алюминия дозируют из рас-
чета связывания 55% HF плавиковой кислоты. Нейтрализацию
фторалюминиевой кислоты в третьем резервуаре ведут так, чтобы
в маточном растворе осталось 2—3 г]л свободной HF во избежание
перевода примеси Na2SiF6 в NaF и SiO2 и перехода в осадок при-
меси железа, которое при меньшей кислотности выделяется вместе
Производство фтористых солей из плавиковой кислоты
1123
с криолитом в виде Fe(OH)3. Отстоявшийся криолит отфильтровы-
вают и промывают водой на барабанном вакуум-фильтре. Снимае-
мую с фильтра пасту криолита, содержащую 15—20% влаги,
высушивают в стальной барабанной сушилке при 130—140°. На
производство 1 т криолита расходуют: 0,69—0,71 т HF в виде пла-
виковой кислоты, 0,29—0,30 т А12О3 и 0,8—0,9 т Na2CO3.
Для получения криолита могут быть использованы промывные
воды и другие отбросные жидкости, содержащие NaF. При обра-
ботке их горячим свежеприготовленным раствором А1(ОН)3 в пла-
виковой кислоте 99% фтора осаждается в виде криолита, содержа-
щего 90—98% Na3AlF6189.
Криолит предложено получать также обработкой смеси А1(ОН)3
и Na2CO3 газообразным HF при 300° в горизонтальном вращаю-
щемся барабане 19°.
При приливании раствора хлористого алюминия к раствору по-
варенной соли и плавиковой кислоты образуется криолит и выде-
ляется соляная кислота:
А1С13 + 6HF + 3NaCl - Na3AlFe + 6НС1
Этот метод применяют при переработке алюминиевых шлаков
(«алюминиевой золки») растворением их в соляной кислоте. Выде-
лившуюся соляную кислоту возвращают в процесс.
Изучено191 образование криолита при взаимодействии каоли-
нита с плавиковой кислотой и с кислыми растворами NaF. Процесс
идет согласно уравнению:
Al2Si2O,(OH)4 + 24F' + 14Н3О+ = 2A1F8- + 2S1F*" + 23Н2О
В нейтральном растворе (pH ~5) вместо кремнефторида обра-
зуется гидроокись кремния:
Al2Si2O5(OH)4 + 12F" + 6Н3О+ = 2A1F|- + 2S1(OH)4 + 7Н2О
По-видимому, и в кислых растворах коллоидная гидроокись
кремния является промежуточной фазой при образовании кремне-
фторида.
Исследована возможность получения криолита борфтористово-
дородным методом 192. Сначала получают раствор HBF4 обработкой
CaF2 серной и избытком борной кислоты при 95° в присутствии
Na2SO4 для снижения растворимости CaSO4. Оптимальная концент-
рация Н3ВО3 ПО—120 г/л. К отделенному от сульфата кальция
раствору HBF4 добавляют гидроокись алюминия и соду, отделяют
осадок криолита, а маточный раствор с избытком Н3ВО3 возвра-
щают в цикл. Вследствие накопления примесей в циклическом про-
цессе при 8-кратном обороте маточного раствора извлечение фтора
снижается до 64—73%. Потери Н3ВО3 составляют 2—4% от веса
получаемого криолита.
. 1126
Гл. XXXI. Соединения фтора
Применение вместо порошкообразного гранулированного крио-
лита при электролитическом получении алюминия снижает потери
фтора в этом процессе. Гранулирование производят на тарельча-
том грануляторе из снимаемой с фильтра криолитовой пасты, влаж-
ность которой должна быть ~19%. Оптимальная температура по-
следующего обезвоживания гранул во вращающейся барабанной
печи ~ 600°193.
Фторид алюминия
Для получения фторида алюминия плавиковую кислоту нейтра-
лизуют гидроокисью алюминия:
А 1(ОН)3 + 3HF = A1F3 + ЗН2О
Находящиеся в растворе примеси NaF и NajSiFe переходят в
результате реакций
3NaF + AlF3 = Na3AlFe
3Na2SiFe + 4A1F3 = 2Na3AlFe + Al2(SiFe)3
в криолит, который выделяется в осадок вместе с A1F3. Так как об-
разующийся по последней реакции A12(SiFe)3 хорошо растворим, то
он также взаимодействует с плавиковой кислотой и превращается
в плохорастворимый AlFj:
AI2(Si Fe)3 + 6HF = 2A1F3 + 3H2SiFe
В результате этих реакций содержащиеся в плавиковой кислоте
соли натрия переходят в криолит, а взамен Na2SiF6 в растворе по-
является эквивалентное количество HaSiFg.
Растворение гидроокиси алюминия производят в предварительно
нагретой до 90—95° плавиковой кислоте. Массу перемешивают при
этой температуре в реакторе с мешалкой в течение часа. Вследствие
склонности фторида алюминия образовывать пересыщенные рас-
творы кристаллизацию ведут также из горячих растворов при 90°,
когда степень пересыщения минимальная. Продолжительность кри-
сталлизации около 2,5 ч. Фторид алюминия выделяется в виде
АШз-ЗНгО. Так как растворимость его больше, чем криолита, то
он кристаллизуется относительно медленнее, и кристаллы получа-
ются крупнее. Для получения более крупных, быстрее осаждаю-
щихся кристаллов реакционную смесь нагревают и вводят в нее за-
травку—небольшое количество кристаллов (суспензии), получен-
ных в предыдущей операции. Иногда затравку получают так: в
реакционную смесь вводят струю холодной воды (небольшое коли-
чество); резкое охлаждение части раствора вызывает выделение
некоторого количества мелких кристаллов, которые и служат за-
травкой.
Так как часть воды из реакционной смеси связывается при кри-
сталлизации A1F3 • ЗН2О, то пульпа значительно загустевает и осаж-
Производство фтористых солей из плавиковой кислоты
1127
дение кристаллов происходит медленно. Для ускорения процесса
применяют плавиковую кислоту, содержащую не больше 9—10%
HF. После некоторого охлаждения пульпы отстоявшиеся кристаллы
отделяют от маточного раствора на фильтре, промывают и высу-
шивают. На получение 1 т фторида алюминия расходуют: 0,8—
0,83 т HF в виде плавиковой кислоты и 0,71—0,72 т AI2O3.
Поступающая на сушку паста фторида алюминия содержит 25—
30% A1F3, 30—35% кристаллизационной воды и 35—40% маточного
раствора. Свободная влага из маточного раствора при сушке уда-
ляется в первую очередь. При нагревании до 300° AIF3 • ЗН2р те-
ряет большую часть кристаллизационной воды и превращается в
A1F3 • 0,5Н2О. Дальнейшее удаление кри-
сталлизационной воды происходит очень
медленно при 350—400°. Однако при этом
невозможно полностью удалить воду, и
окончательное обезвоживание ведется
при 500—550°. При этой температуре под
действием имеющегося в прокалочной
печи (сушилке) водяного пара происхо-
дит частичное разложение продукта и за-
грязнение его А12О3 194, 195:
2( A1F3 • 0,5Н2О) + 2Н2О = А12О3 + 6HF
Предложено вести сушку в псевдо-
ожиженном слое в две стадии — сначала
сушка при 200—300°, когда разложение
когда скорости гидролиза замедлена благодаря малому содержа-
нию влаги, кратковременное досушивание при 600°. Таким
путем можно получить продукт с влажностью меньше 1 % и содер-
жанием фтора больше 66%, при потере фтора на обеих стадиях
сушки 1,5—2% 196.
В маточном растворе после кристаллизации остается значитель-
ное количество фторида алюминия. Растворимость его в воде и
слабокислых растворах невелика (рис. 328), но вследствие склон-
ности к образованию пересыщенных растворов содержание AIF3
в маточном растворе составляет 50—60 г/л. Кроме того, в растворе
остается до 15—30 г/л HF и немного других примесей — соединений
железа, кремнезема, сульфатов. Маточный раствор используют для
получения из содержащегося в нем A1F3 криолита путем внесения
в него кристаллического NaF.
Предложено получать A1F3 из криолита или хиолита обработкой
их при 200° (под давлением) смесью растворов алюминиевой соли
(например, А1С13) и минеральной кислоты197. Запатентованы спосо-
бы получения фторида алюминия из железисто-глиноземной руды
(боксита) обработкой ее стехиометрическим количеством плавико-
вой или кремнефтористоводородной кислоты или их смеси при
Рис. 328. Растворимость
фторида алюминии в сла-
бых растворах HF.
еще не велико, а затем,
1128
Гл. XXXI. Соединения фтора
35—90°198. Описано получение A1F3 взаимодействием А1(ОН)3 с
подогретым выше 70° газообразным фтористым водородом во взве-
шенном слое, разделенном на три зоны; реакция протекает в сред-
ней зоне при 400—650°
Запатентован способ получения фторида алюминия нагреванием
в течение 2 ч при 80° смеси безводного А1С13 и NaF в среде ССЦ.
Затем отделяют непрореагировавший ССЦ, твердый продукт про-
мывают метанолом, потом водой и высушивают при 200° 20°.
Фторид натрия
Для получения NaF производят полную нейтрализацию техни-
ческой плавиковой кислоты содой; при этом нейтрализуются и при-
меси других кислот:
2HF + N а2СО3 = 2N aF + Н2О + СО2
H2SiFg -ЬNа2СО3 == Na2SiF3 -Ь Н2О -Ь СО2
H2SO4 + Na2CO3 = Na2SO4 + Н2О + СО2
15 30 45 во 75 во
Время, мин
Рис. 329. Скорость разло-
жения кремнефторида на-
трия в содовом растворе
(150,7 t/л Na2CO3).
При введении соды
После нейтрализации кислот образовавшийся кремнефтористый
натрий при повышенной температуре разлагается содой:
Na2SiFe + 4Na2CO3 + 2H2O =
= 6NaF + SiO2+ 4NaHCO3
а затем бикарбонатом:
N a2SiFe + 4N aHCO3 = 6N aF + SiO2 + 2H2O + 4CO2
При достаточном прогреве бикарбо-
нат разрушается:
2N аНСОз = N а2СО3 + Н2О + СО2
Зависимость скорости разложения
Na2SiF3 содой от температуры показана
на рис. 329. При содержании в растворе
150,7 г/л Na2CO3 после нагревания реак-
ционной смеси при 90—95° в течение часа
Na2SiF6 оказывается разложенным прак-
тически полностью 135.
1 раствор «грязной» плавиковой кислоты
в первую очередь нейтрализуется H2SiF6 и образующийся Na2SiF6
выделяется в осадок. При разложении кристаллов Na2SiF6 после
нейтрализации раствора на их поверхности отлагается пленка крем -
невой кислоты, затрудняющая доступ соды, и разложение идет
очень медленно даже при очень интенсивном перемешивании. Во
избежание этого процесс ведут таким образом, чтобы реакционная
масса была все время щелочной; в этих условиях Na2SiF6 вообще
не образуется. В реактор загружают только меньшую часть пла-
Щелочные способы получения NaF и криолита
1129
виковой кислоты и всю соду. Количество загружаемой кислоты
должно быть достаточным для того, чтобы реакционная масса была
подвижной. Затем в щелочной раствор добавляют остальную,
основную массу плавиковой кислоты. При таком порядке загрузки
кремнефторид натрия в осадок не выделяется, а полностью разла-
гается содой.
Ввиду малой растворимости, образующийся NaF выделяется в
виде кристаллического осадка. Чем больше содержится H2SiF6 в ис-
ходной плавиковой кислоте, тем больше получаемый NaF загрязнен
примесью SiO2. Осадок NaF отфильтровывают, промывают от ма-
точного раствора для удаления растворимых примесей и высуши-
вают. На получение 1 т фторида натрия расходуют: 0,45—0,46 т HF
в виде плавиковой кислоты и 1,08—1,10 т соды.
Представляет интерес для получения фторида натрия (и других
фторидов) использование ионного обмена201. Так, фторид натрия
может быть получен при обработке плавиковой кислотой катиони-
та в натриевой форме:
HF 4- RNa = NaF+RH
Аналогично KF образуется при пропускании раствора NaF через
калиевый катионит:
NaF 4- RK = KF + RNa
Фторид магния
Фторид магния можно получать осаждением F~ из водных рас-
творов солями магния или спеканием обезвоженного MgSO4 с NaF
при 600—650°. В последнем случае после выщелачивания плава во-
дой или слабой серной кислотой, промывки его и сушки при 105°
выход MgF2 достигает 94—96%. На 1 т MgF2 затрачивают 4,7 т
MgSO4-7H2O и 1,6 т NaF 202. (Об аммиачном способе получения
MgF2 см. стр. 1169).
ЩЕЛОЧНЫЕ СПОСОБЫ ПОЛУЧЕНИЯ ФТОРИДА НАТРИЯ
И КРИОЛИТА ИЗ ПЛАВИКОВОГО ШПАТА
Фторид натрия может быть получен по щелочному способу 203-207
спеканием плавикового шпата с содой и кремнеземом:
CaF2 + N а2СО3 + SiO2 == 2NaF+ CaSiO3 4- СО2
Реакция между CaF2 и Na2CO3
CaF2 4- Na2CO3 = 2NaF 4- СаО 4- СО2
идет и в отсутствие SiO2, но добавка его к шихте необходима для
того, чтобы связать СаО в нерастворимый силикат во избежание
обратной реакции между СаО и NaF и образования CaF2 при вы-
щелачивании NaF из полученного спека. (Заметим, что эвтектика в
ИЗО
Гл. XXXI. Соединения фтора
системе CaF2—СаО содержит 76,5 мол. % CaF2 и плавится при
1360° 2°5).
Для щелочного способа можно использовать низкосортный пла-
виковый шпат, содержащий 70—75% CaF2, непригодный для
переработки кислым способом. Щелочной способ не связан с выде-
лением вредного фтористого водорода в атмосферу цеха и с корро-
зией им аппаратуры.
Так как шихта из плавикового шпата, соды и кварца расплав-
ляется при температуре более низкой, чем идет образование NaF,
то это затрудняет спекание во вращающейся печи. Этого можно
избежать, если ввести в шихту не кварц, а аморфный кремнезем
(инфузорную землю, трепел), — получается не плав, а сухой спек,
что дает возможность вести процесс во вращающейся печи.
Эвтектика в системе CaF2—Na2CO3 содержит 45,2 мол. %
(36 вес.%) CaF2 и 54,8 мол.% (64 вес.%) Na2CO3 и плавится при
555 ± 5°. В присутствии небольших количеств (до 5%) SiO2, Fe2O3,
Na2SiO3, Na2O • Fe2O3 температура плавления эвтектической смеси
снижается до 500—520°. Заметное же образование NaF в смеси
CaF2 и Na2CO3 наблюдается лишь выше 900°. В противополож-
ность этому смеси, содержащие эквимолекулярное количество ак-
тивного кремнезема, например CaF2+Na2CO3+SiO2,CaF2+Na2SiO3,
a Ta^eCaF2 + Na2SiO3-|-Na2CO3(B молярном отношении 1:1: 0,05),
расплавляются при 900 ± 5°, а образование NaF в них протекает
интенсивно уже при 600°. Предотвращение появления жидкого
плава в печи при введении в шихту активной SiO2 объясняется
предварительным частичным связыванием соды, а также адсорб-
цией жидкой фазы высокоразвитой поверхностью добавки.
Максимальный выход NaF, достигаемый при спекани эквимо-
лекулярной смеси CaF24-Na2CO3-|-SiO2, составляет 85—87% 20в. Это
объясняется тем, что 10—15% Na2CO3 и соответствующее количе-
ство SiO2 расходуются на образование нерастворимых натриево-
кальциевых силикатов: Na2O-CaO-SiO2, Na2O-2CaO-3SiO2 и
Na2O • ЗСаО • 6SiO2. Выше 900° количество образующихся силика-
тов резко возрастает. Кроме того,- при наличии в исходном сырье
СаСО3, образующаяся из карбоната окись кальция не полностью
связывается кремнеземом, остается в свободном состоянии и при
выщелачивании спека реагирует с NaF, переводя его в нераство-
римый CaF2. Для увеличения выхода NaF соду следует дозировать
с учетом связывания в алюминат натрия содержащейся в шихте
гидроокиси алюминия. Содержание СаСО3 в плавиковом шпате
должно быть не больше 10—15%.
При составлении шихты из просушенных и тонкоизмельченных
плавикового шпата, трепела (содержащего 80—85% SiO2) и соды,
с соблюдением следующих молярных соотношений:
Na2CO3 _ CaF2+CaO
CaF2 + А12О3 + Fe2O3 Fe2O3 + SiO2 ’
Щелочные способы получения NaF и криолита
1131
при спекании шихты при 850° в течение 1 ч можно достичь выхода
NaF 93—94% для высокосортного шпата (90—95% CaF2) и
88—89% при низкосортном сырье (~75% CaF2).
Избыток соды в шихте повышает выход фтора в растворимое со-
стояние в процессе спекания, ио увеличивает потери фтора при вы-
щелачивании спека, так как непрореагировавшая сода при выщела-
чивании способствует обратному переходу NaF в CaF2:
CaSiO3 + Na2CO3 = Na2SiO3 + CaCO3
2NaF + CaCO3 = CaF2 + Na2CO3
Во избежание плавления спека спекание лучше производить при
температуре не выше 825°. При нагревании шихты до 450° начи-
нается образование силиката натрия:
Na2CO3 + SiO2 “ Na2SiO3 + СО2
При дальнейшем нагревании силикат натрия реагирует с CaF2:
Na2SiO3 + CaF2 - 2NaF + CaSiO3
Аналогично идет процесс при наличии в шихте окиси железа 206 г
Na2CO3 +Fe2O3 = Na2O • Fe2O3 4-СО2
Na2O • Fe2O3 + CaF2 = 2NaF + CaO • Fe2O3
При этом реакция c Fe2O3 протекает при температуре на
50—100° более высокой, чем с SiO2 и с меньшими потерями щелочи.
Фторид натрия извлекается из спека выщелачиванием водой при
50—55°. Одновременно из спека извлекаются и растворимые при-
меси — сода, алюминат натрия и другие, мало влияющие на рас-
творимость NaF, которая в пределах 15—100° составляет всего
около 4%. При простом выщелачивании с отношением Ж: Т, рав-
ным 10: 1, извлечение фтора достигает 70—75% от общего его ко-
личества в спеке, а получаемый раствор содержит ~25 г/л NaF.
Для получения почти насыщенных растворов (37—39 г/л NaF) не-
обходимо измельчить спек до размера зерен 0,2 мм и выщелачивать
его в течение 30—60 мин при интенсивном репульпировании.
В процессе выщелачивания может происходить взаимодействие
между NaF и СаО, а также с алюминатом натрия с образованием
нерастворимых CaF2 и Na3AlFe:
2NaF+CaO + H2O—>CaF2+2NaOH
6NaF + NaAlO2 + 2H2O Na3AlFe + 4NaOH
Это приводит к «вторичным» потерям NaF. Побочные реакции
возникают вследствие гидролиза натриево-калиевых силикатов, со-
держащихся в спеке, приводящего к образованию Са(ОН)2, гидро-
лиза алюмината натрия с выделением А1(ОН)3, а также при нали-
чии в спеке свободной окиси кальция — продукта разложения
1132
Гл. XXXI. Соединения фтора
СаСОз. Уменьшения гидролиза силикатов можно достигнуть увели-
чением крупности зерен спека до 0,2 мм, снижением температуры и
сокращением длительности выщелачивания (при интенсивном ре-
пульпировании) до 30—60 мин. Кроме того, для уменьшения гидро-
лиза алюмината натрия необходимо ввести в раствор щелочь
в количестве, обеспечивающем каустический модуль (т. е.
Na2O : AI2O3), равный 3,0. При этих условиях можно получить почти
насыщенный раствор NaF (37—39 г/л) с высоким выходом
(90-95%).
Остающийся после выщелачивания шлам после промывки вы-
брасывают, а раствор NaF мог бы быть подвергнут выпариванию
с целью переработки на кристаллический продукт. Но так как этот
раствор содержит меньше 4% NaF, то выпаривание его не эконо-
мично. Более выгодно перерабатывать этот раствор на криолит
смешением с раствором алюмината натрия и карбонизацией полу-
ченной смеси. Образующийся по реакции
6NaF +NaAlO2 +2СО2 = Na3AIFe + 2Na2CO3
криолит отфильтровывают, промывают и высушивают.
В процессе карбонизации, вероятно, идут реакции гидролиза
алюмината натрия, образования фторида алюминия и последую-
щего криолитообразования:
NaA102 + 2H20 Al(OH)3 + NaOH
3NaF+Al(OH)3 AlF3+3NaOH
Введение в раствор двуокиси углерода способствует протеканию
этих реакций слева направо. Взаимодействие NaF и A1F3 характе-
ризуется образованием вначале метастабильного в этих условиях
тетрафтор алюмината NaAlF4-H2O, который затем превращается
в устойчивый комплекс №цА14р23 • НгО. Степень этого превращения
возрастает с уменьшением в растворе отношения A1F3: NaF, с уве-
личением продолжительности взаимодействия и с повышением тем-
пературы. Выбор условий карбонизации в значительной мере обу-
словлен возможностью образования натриевого алюмокарбоната
Na2O-А12О3-2СО2-пН2О, что приводит к повышенному содержанию
в криолите трудноотмываемой щелочи и получению некондицион-
ного продукта. Натриевый алюмокарбонат образуется при большой
скорости карбонизации и температуре раствора выше 60° при на-
личии в исходном алюминатно-фторнатриевом растворе избытка
алюмината натрия по сравнению с необходимым для получения
NanAl4F23-H2O. Однако при осуществлении процесса при темпера-
туре ниже 50—60° образуется медленно отстаивающийся и плохо
отфильтровывающийся криолит. Поэтому карбонизацию алюми-
натно-фторнатриевых растворов следует вести при 60—80°. При
этом, в отсутствие избытка алюмината натрия в исходном растворе
Щелочные способы получения NaF и криолита
1133
и при постепенном снижении скорости карбонизации с выкручива-
нием пульпы под конец процесса, можно полностью избежать обра-
зования натриевого алюмокарбоната и, следовательно, получить
кондиционный криолит.
Получение криолита карбонизацией алюминатно-фторнатрие-
вых растворов изучено также применительно к раствору NaF, полу-
ченному из кремнефтористых газов (стр. 1151) или из Na2SiF6.
Рекомендованы следующие оптимальные условия 208. Исходный рас-
твор NaF берут с 10%-ным избытком по отношению к стехиометри-
ческому количеству (в расчете на А120з), а раствор алюмината
натрия должен быть свежеприготовленным с модулем 1,6—1,8. Эти
растворы необходимо подавать в реактор одновременно при непре-
рывной карбонизации, причем величина pH среды должна быть
9,8—10,2, что соответствует отношению ' в реакционной массе
NaHCO3: Na2CO3, равному 0,2—0,3. К концу процесса это отноше-
ние доводят до 1,'что обеспечивает полное выделение криолита из
раствора. При таком осаждении криолит хорошо отфильтровы-
вается. Образующийся раствор Na2CO3 возвращают в процесс
после предварительного выпаривания. На производство 1 т крио-
лита по этому методу из кремнефтористых газов требуется (по
опытным данным): 0,275 т А120з, 0,268 г Na2O, 1,064 г Na2CO3 (для
перевода Na2SiF6 в NaF), 0,324 т СО2 (100%) и необходимо выпа-
рить 1,551 т воды.
Предложен вариант описанного способа, совмещающий получе-
ние криолита с производством окиси алюминия из кремнистого бок-
сита 207 Шихту составляют из боксита, плавикового шпата и соды
с таким расчетом, чтобы в получаемом спеке содержание алюми-
ната натрия было значительно выше, чем требуется для получения
криолита. Спек размалывают и выщелачивают. Фторид натрия и
алюминат натрия переходят в раствор, который после обескремни-
вания подвергают ступенчатой карбонизации. В первой стадии кар-
бонизации выделяется гидрат окиси алюминия. Его оставляют
в растворе в количестве лишь немного большем, чем это требуется
для получения криолита. После отделения выделившегося А1(ОН)3
раствор вновь карбонизуют, причем в осадок выделяется криолит.
Способ этот, однако, сложен и мог бы представить некоторый инте-
рес лишь для получения криолитизированного глинозема.
В некоторых условиях может представить интерес спекание пла-
викового шпата с природным сульфатом натрия и углем
CaF2 + Na2SO4 + 4С = 2NaF + CaS + 4СО
с целью получения фторида натрия и переработки его на крио-
лит 209.
Гидротермальный способ получения NaF заключается во вза-
имодействии CaF2 с концентрированным алюминатным раствором
1134
Г л. XXXI. Соединения фтора
при 205—220° (под давлением 12—13 ат); образуются NaF и
Са(ОН)2207:
CaF2 + 2NaOH = 2NaF + Са(ОН)2
При понижении температуры до 150° гидроокись кальция пере-
ходит в нерастворимый алюминат кальция ЗСаО-А12О3-6Н2О. Фто-
рид натрия извлекают из осадка выщелачиванием водой. Процесс
можно осуществить в автоклаве с применением алюминатного рас-
твора, содержащего около 300 г/л каустической Na2O и имеющего
каустический модуль 2—4. При этом выход NaF за 2—3 ч достигает
90% и возрастает с увеличением каустического модуля раствора и
отношения количества раствора к обрабатываемому CaF2. Этот
метод может представить интерес в сочетании с получением окиси
алюминия из бокситов по способу Байера. Для этого к бокситу до-
бавляют некоторое количество концентрата плавикового шпата
(вместо обычной добавки извести). При выделении А1(ОН)3 обра-
зующийся NaF остается в растворе и выпадает вместе с содой лишь
в процессе регенерации раствора, т. е. при его выпаривании. При
обработке осадка водой сода растворяется, a NaF остается в твер-
дом виде. При добавке к бокситу 4—7% CaF2 выход NaF состав-
ляет 95—98%, причем степень извлечения А12О3 не снижается.
УЛАВЛИВАНИЕ ФТОРИСТЫХ СОЕДИНЕНИИ ИЗ ОТХОДЯЩИХ
ГАЗОВ И ИХ ПЕРЕРАБОТКА
Отходящие фтористые газы являются основным источником
фтора для производства его соединений. Это, главным образом,
газы, образующиеся при переработке природных фосфатов. Боль-
шие масштабы производства фосфорных удобрений обеспечивают
возможность попутного получения значительных количеств фтори-
стых солей.
При кислотной переработке фосфатов в удобрения скорость и
полнота выделения фтористых соединений в газовую фазу опреде-
ляется соотношением различных форм комплексообразователей.
Первоначально образуются фтористые комплексы растворенных
полуторных окислов (RF2), которые при наличии кремниевых соеди-
нений переходят в летучий кремнефторид. При недостатке разла-
гающихся кремниевых соединений в газовую фазу выделяется фто-
ристый водород. В этом случае для повышения выхода фтора
в газовую фазу целесообразно добавлять в реакционную массу
гидратированную кремневую кислоту,-получаемую, в частности, при
абсорбции фтористых газов210*213. Взаимодействие SiO2 с HF наи-
более полно протекает в системах с преобладанием фосфорной кис-
лоты, однако степень выделения SiF4 уменьшается при замене сер-
ной кислоты на фосфорную, так как уменьшается парциальное дав-
ление SiF4 над этими растворами214.
Улавливание фтористых соединений из отходящих газов
1135
При переработке природных фосфатов на суперфосфат около
40% содержащегося в них фтора выделяется в газовую фазу в виде
SiF4 и туманообразной HjSiFe и отсасывается из аппаратуры с вен-
тиляционными газами215. Концентрация фтора в газах, уходящих
из смесителя и камеры созревания, невелика (15—35 г/м3). Эти
газы направляют в абсорбционную установку для улавливания из
них фтора.
При сушке гранулированного суперфосфата выделяется Ю—17%
фтора, оставшегося в суперфосфате; степень выделения фтора воз-
растает с повышением температуры и длительности сушки. При
экстракции фосфорной кислоты в ней остается 78—80% фтора, вно-
симого с апатитом, 15—17% его переходит в фосфогипс, а в газы
удаляется всего 3—5% фтора. При выпаривании экстракционной
кислоты в газовую фазу выделяется 80—90% содержащегося в ней
фтора. При производстве двойного суперфосфата камерным спосо-
бом из смесителей выделяется всего 1—3% фтора, а при сушке
двойного суперфосфата, получаемого поточным методом, — 50—60%
фтора от его общего содержания в фосфате и фосфорной кис-
лоте216'217. При гидротермическом обесфторивании фосфатов фтор
удаляется из них почти полностью (93—97 %).
Количество фтора, извлекаемое в газовую фазу при разложении
природных фосфатов серной кислотой, зависит не только от его со-
держания в фосфате, но и от многих других факторов. Оно тем
выше: 1) чем меньше в фосфате окислов щелочных металлов и по-
луторных окислов, связывающих фтор в виде фторидов, остающихся
в суперфосфате; 2) чем выше температура серной кислоты и супер-
фосфатной массы, так как при повышении температуры количества
H2SiF6, остающейся в суперфосфате, уменьшается; 3) чем выше
концентрация серной кислоты, так как это вызывает соответствую-
щее увеличение концентрации фосфорной кислоты в жидкой фазе
в начале разложения, а возрастание концентрации фосфорной кис-
лоты в свою очередь связано с увеличением равновесного давления
паров SiF4, т. е. с интенсификацией выделения фтора в газо-
вую фазу.
Помимо этого увеличению выхода фтора в газовую фазу спо-
собствует повышенная норма серной кислоты, интенсификация
перемешивания, увеличение разрежения в системе и. др.
Как и в газах из производства простого суперфосфата, в газах
при производстве двойного суперфосфата, а также экстракционной
фосфорной кислоты содержится в основном SiF4. При сушке грану-
лированного суперфосфата и выпарке экстракционной фосфорной
кислоты фтор выделяется в виде эквивалентной смеси SiF4 + 2HF.
При гидротермическом обесфторивании фосфатов в газах также
находится смесь SiF4 + HF с преобладанием HF, а при хлорирую-
щем обжиге фосфатов — в основном HF. Газы из аппаратов
экстракции фосфорной кислоты содержат ~2,5 г/м2, фтора, а при
1136
Гл. XXXI. Соединения фтора
охлаждении пульпы в экстракторах воздухом 0,18—0,34 г/л3 (при
нормальных условиях). Газы из барабанных концентраторов фос-
форной кислоты содержат 8,5—9 г/л3 фтора, а после разбавления
их воздухом перед электрофильтром -~3 г/л<3. Концентрация фтора
в газе из смесителей упаренной фосфорной кислоты, апатита и фос-
форитной муки в производстве двойного суперфосфата камерным
способом всего 0,16 г/л3218. Фтористые газы, содержащие SiF4 и HF,
могут быть также получены из азотнокислотной вытяжки фосфатов
при обработке ее паром 120—140° 218-227.
Получение кремнефтористоводородной кислоты
Абсорбция фтористых газов в полых камерах
Газ, поступающий на абсорбцию из аппаратов производства
простого суперфосфата, должен иметь температуру не ниже 65°,
так как при более низкой температуре SiF4 гидролизуется находя-
щейся в газе влагой
3SiF4 + 4Н2О = 2H2SiF6 + SiO2 2Н2О
и выделяющаяся при этом гелеобразная кремневая кислота будет
оседать в газоходе в виде шлама, пропитанного кремнефтористо-
водородной кислотой, что приведет к необходимости частой чистки
газоходов.
Водная абсорбция фтористых газов 227 практически сводится
к поглощению туманообразной EhSiFe, образовавшейся в резуль-
тате гидролиза SiF4, и на суперфосфатных заводах обычно про-
изводится в две ступени. Сначала газ промывают в механических
абсорберах — камерах с разбрызгивающими валками, где улавли-
вается 80—90% фтора, затем в полых башнях, орошаемых водой,
где поглощается остальная часть фтористых соединений. Башни
орошают водой и образовавшийся здесь слабый раствор кремне-
фтористоводородной кислоты подают на орошение камер. Раньше
камеры строили из кислотоупорного кирпича и футеровали
диабазовыми плитами. Теперь на многих заводах установлены
стальные механические абсорберы, гуммированные или футерован-
ные кислотоупорными плитками по слою полиизобутилена или
асбестового картона на кислотоупорном цементе.
Нижняя часть кирпичных камер и башен выполняется из кислото-
упорного бетона и служит приемником для кремнефтористоводо-
родной кислоты. Применения абсорберов с насадкой в данном слу-
чае избегают из-за возможности засорения ее кремневой кислотой.
Устройство кирпичной абсорбционной камеры показано на
рис. 330. Ее наружные габариты: длина 10—18 м, высота 5—6 м и
ширина 1,5—2 м; объем 36 м3 и больше.
Улавливание фтористых соединений из отходящих газов
1137
Внутри она разделена перегородками на несколько отделений,
через которые газ проходит последовательно и промывается жид-
костью, распыляемой мелкими брызгами с помощью вращающихся
Рис. 330. Абсорбциониая камера.
разбрызгивателей. Более современный стальной механический аб-
сорбер показан на рис. 331. При равной производительности он
имеет в 1,5 раза меньший объем.
Рис. 331. Механический абсорбер.
Разбрызгиватель состоит из вала с лопастями, проходящего по-
перек отделения камеры на расстоянии нескольких сантиметров от
зеркала жидкости, так что лопасти погружены в жидкость на глу-
бину 2—5 см. Разбрызгиватели вращаются со скоростью
350—400 об!мин. Интенсивность разбрызгивания жидкости регули-
руется поднятием или опусканием валов с помощью подвижных
подшипников. Вода или слабая кремнефтористоводородная кис-
лота подается в камеру с конца, противоположного входу газа, и
1138
Гл. XXXI. Соединения фтора
движется противотоком ему. Из переднего конца вытекает кремне-
фтористоводородная кислота вместе со шламом кремневой кислоты,
частично оседающим на дно камеры, работу которой поэтому пе-
риодически прерывают для очистки от осевшего геля.
Качество работы абсорбционных камер в значительной мере за-
висит от конструкции разбрызгивающих валков и их установки.
Лучшие результаты получаются при валках с диаметром 350 мм,
имеющих четыре лопасти, загнутые вперед по направлению враще-
ния; загиб лопастей по кругу около 50 мм, выходной угол 55°. При
Рис. 332. Разбрызгивающий валок (поперечное сечение):
слева — незащищенный, справа — с железобетонной защитой.
вращении валка лопасть зачерпывает жидкость. При глубине по-
гружения ~3 см оптимальная скорость вращения валка 420—
450 об/мин. На некоторых заводах валки защищены от коррозии
обмазкой из армированного бетона (рис. 332). Чаще их изготов-
ляют из стали ЭИ-629, из текстолита или фаолита, иногда из де-
рева. При высоте камеры над осью валка 2 м объемный коэффи-
циент абсорбции составляет 2000—3400 ч-1; использование более
высоких камер, обычно применяемых на заводах, не обосновано,
так как пространство выше 2 м над осью валка является бесполез-
ным 228.
Применение в качестве абсорберов полых башен круглого или
квадратного сечения, имеющих полезную высоту до 10 м и орошае-
мых с помощью форсунок более удобно, чем камер с механиче-
скими разбрызгивателями, так как башни имеют меньшую площадь
основания и поэтому их легче очищать от осевшей кремневой
кислоты.
Для лучшегб улавливания H2SiFe обычно Газ пропускают после-
довательно через несколько (2—3) абсорберов, работающих с про-
тивоточной схемой орошения. Из первого по ходу газа абсорбера
вытекает кислота, содержащая 8—10% HaSiFg. Объем абсорбцион-
Улавливание фтористых соединений из отходящих газов
1139
ной системы определяется производительностью суперфосфатного
завода из расчета 0,5 м3 на 1 т суперфосфата в час. Степень улав-
ливания H2SiF6 достигает 98—99%. Отходящие из абсорбционной
установки газы содержат 0,1—0,2 г/м3 фтора, а на некоторых за*
водах 0,04—0,05 г/м3-, они выбрасываются вентилятором в атмо-
сферу. На суперфосфатных заводах на каждую тонну суперфос-
фата вентилятор должен отсасывать 250—300 м3 фтористых газов.
Вентилятор, устанавливаемый в конце системы, раньше изготов-
ляли из просмоленного дерева или из железобетона с чугунным или
деревянным ротором, покрытым диабазовой обмазкой. Теперь
используют вентиляторы с чугунным ротором производительностью
25000—30000 мъ/ч, нижняя часть корпуса которых железобетон-
ная, верхняя — стальная; внутри они покрыты диабазовой обмаз-
кой или асбовинилом 229.
Фтористые газы, выделяющиеся при сушке гранулированного
суперфосфата, поглощаются с меньшей полнотой (чем газы из сме-
сителей и суперфосфорных камер), так как значительная часть со-
единений фтора находится в этих газах в виде трудно улавливае-
мого аэрозоля 23°. Аэрозоль образуется в результате охлаждения
горячих газов, сопровождающегося конденсацией пара и образова-
нием стойкого тумана, в котором сосредоточена основная часть
фтористых соединений; они переходят в жидкую фазу аэрозоля, так
как давление их пара над разбавленным раствором H2SiF6 неве-
лико. При 50—70° в газе (при равновесии) содержится лишь
0,02—0,04 г/м3 фтора 60. Очистку этого газа от тумана можно осу-
ществлять в электрофильтрах231. Разработан акустический метод
коагуляции такого аэрозоля 232: при озвучиваний в течение 3 сек
(155 децибел, 16,5 кгц) степень очистки возрастает до 80—95%
(от 40—70% без озвучивания) —содержание фтора в газе после
акустической коагуляции снижается от 0,18 до ~0,025г/м3. При
этом увеличение общего влагосодержания в газе от 15 до 120 г/м3
приводит к возрастанию степени очистки от 73 до 95%.
Запатентован способ, по которому с целью достижения высоких
степеней абсорбции фтористых соединений (85—95%) газ без ох-
лаждения промывается в скруббере циркулирующим раствором,
H2SiF6, имеющим приблизительно ту же температуру, что. и газ.
Благодаря этому не происходит конденсации воды и образования
тумана, a SiF4 и HF абсорбируются 232.
Абсорбция фтористых газов в аппаратах
других конструкций
Поглощение фтористых газов, как хорошо растворимых газов,
может быть осуществлено в абсорберах разных конструкций при
соблюдении условий, предотвращающих их засорение кремне-
гелем 230>237. Этим объясняется преимущественное использование
Ц40
Гл. XXXI. Соединения фтора
камер (механических абсорберов) и полых башен для улавливания
газов в производстве простого суперфосфата. При абсорбции газов,
образующихся в процессе выпарки экстракционной фтористой кис-
лоты, содержащих эквимолекулярную смесь 2HF + SiF4, избыточ-
ной кремневой кислоты не образуется; поэтому для улавливания
этих газов можно использовать аппараты любых конструкций. На
заводе Des Plaines Chemical Со в Моррисе (Иллинойс, США) фто-
ристый газ, образующийся при вакуум-выпарке экстракционной
фосфорной кислоты, поглощается в барометрическом конденсаторе
(температура 21—65°, давление 35—260 мм рт. ст.) кремнефтори-
стоводородной кислотой, причем ее концентрация увеличивается
с 15 до 25% H2SiF6 238. Ниже приводятся краткие результаты
испытаний абсорбции фтористых газов в аппаратах разных ти-
пов 239~247.
Объемный коэффициент абсорбции газов, выделяющихся при
выпарке фосфорной кислоты, содержащих эквимолекулярную смесь
2HF + SiF4, в колонне с насадкой из колец размером 25 X 25 мм,
при плотности орошения 3,6 м3/(м2-ч) и скорости газа ~ 1,5 м/сек
составляет ~7000 ч-1. Гидравлическое сопротивление при этом до-
вольно велико — около 160 мм вод. ст. на 1 м высоты слоя насадки.
В колонне с хордовой насадкой в диапазоне скорости газа
0,6—1,6 м/сек, коэффициент абсорбции этих газов меняется в пре-
делах 2750—6400 ч-1, пропорционально скорости газа в степени 1,4.
От плотности орошения [6,7—15,3 л3/(л2-ч)] в этих условиях коэф-
фициент абсорбции не зависит. Гидравлическое сопротивление хор-
довой насадки высотой 1 м при скорости газа 1 м/сек—'&ммвод. ст.,
а при скорости газа 1,5 м/сек — до 25 мм вод. ст.
Башни с хордовой насадкой могут быть использованы также для
водной абсорбции четырехфтористого кремния. При равномерном и
непрерывном орошении отложения геля на поверхности насадки не
происходит. Перерыв в орошении больше 10 мин недопустим. На
Невском химическом заводе такие башни с успехом используются
в качестве второй ступени улавливания фторгазов после механиче-
ских абсорберов с валками. Две последовательно включенные
башни работают первая противотоком, вторая — прямотоком;
содержание фтора в газе снижается от 1—7 до 0,01—0,2 г/нм3.
Чистку башен производят периодически, во время капитального
ремонта.
В колонне с плоско-параллельной насадкой (пакеты из жестких
перхлорвиниловых или текстолитовых пластин) коэффициент аб-
сорбции фтористых газов достигает 1600—4500 ч-1 при линейной
скорости газа 2 м/сек (оптимальная скорость газа 3—5 м/сек).
Испытания абсорбции фтористых газов (SiF4 и 2HF + SiF4)
в тарельчатых аппаратах при пенном режиме показали их высокую
эффективность. При линейной скорости газа в общем сечении аппа-
рата с колпачковой тарелкой ~2 м/сек коэффициент абсорбции
Улавливание фтористых соединений из отходящих газов
1141
эквимолекулярной смеси 2HF + SiF4 составляет 17000—19000 м/ч
(в расчете на площадь основания колпака). Степень поглощения на
одной тарелке 82,5%. Таким образом, поглощение на 97% может
быть достигнуто в аппарате с двумя колпачковыми тарелками, при
гидравлическом сопротивлении ~ 150 мм вод. ст. 237. В аппарате
с ситчатыми тарелками без переливных устройств, работающем на
пенном режиме при полном провале жидкости через отверстия та-
релок. при скорости газа 1,5 м/сек, достаточно 3 тарелок 239 для сни-
жения концентрации фтора в газе до 0,02 г/м3. Гидравлическое со-
противление каждой тарелки 25—30 мм вод. ст. Отверстия тарелок
кремнегелем не забиваются.
Коэффициент абсорбции фтористых газов в трубе Вентури в ра-
счете на узкое сечение трубы достигает 300 000—350 000 м/сек; в ра-
счете на весь объем, занимаемый жидкостью и газом, коэффициент
абсорбции равен ~6000 ч~г, т. е. превышает всего в ~2 раза
значение коэффициента абсорбции в валковой камере. Для из-
влечения фтора из газов суперфосфатного производства требуется
установка из восьми циклонных скрубберов Вентури (труб Вентури
с циклонными сепараторами); их изготовляют из мягкой стали и
футеруют неопреном. Степень извлечения SiF4 при промывке
газа 15%-ной кремнефтористоводородной кислотой достигает
90—98% 238.
Опыт абсорбции фтористых газов, выделяющихся при обесфто-
рировании фосфатов, в аппарате Вентури бесфорсуночного типа по-
казал, что степень поглощения фтора в одной ступени достигает
84—96% при гидравлическом сопротивлении аппарата 100—
130 мм вод. ст.241.
В качестве абсорбера фтористых газов испытан аппарат распы-
ливающего типа, представляющий собой полый вертикальный ци-
линдр с одной или несколькими поперечными распределительными
обращенными книзу конусообразными перегородками, имеющими
широкие отверстия в центре. Газ, поступающий сверху, проходит
через отверстие конуса со скоростью 20—25 м/сек и распыливает
стекающую жидкость. Степень абсорбции водой и 5%-ным раство-
ром соды (в двухконусном аппарате) ~95% при гидравлическом
сопротивлении менее 140 мм вод. ст. 242.
Описано использование для улавливания фтористых газов аб-
сорбера с плавающей насадкой (полые пластмассовые шары, под-
держиваемые внутри башни вертикальным потоком газа во взве-
шенном состоянии между нижней опорной и верхней ограничиваю-
щей решетками). При высоте насадки (в неподвижном состоянии)
0,3 м и скорости газа 2,5 м/сек степень улавливания HF ~90%,
а гидравлическое сопротивление 120—130 мм вод. ст.243.
Запатентован способ одновременной очистки и концентрирова-
ния разбавленных растворов кремнефтористоводородной кислоты
дистилляцией в смеси с 65 %-ной фосфорной кислотой 244.
1142
Гл. XXXI. Соединения фтора
Переработка кремнефтористоводородной кислоты в кремнефториды
Кремнефторид натрия
Полученную в абсорберах кремнефтористоводородную кислоту,
содержащую 8—10% H2SiFe, вместе с шламом кремневой кислоты
перекачивают фаолитовыми или гуммированными насосами по ре-
зиновым шлангам или фаолитовым трубам в резервуар, из которого
она поступает на дальнейшую переработку. Для перекачки исполь-
зуют также насосы из хромоникелевой стали, содержащей Мо и Си.
В качестве наиболее стойких рекомендованы стали 0Х23Н28МЗДЗТ,
0Х23Н28М2Т и X17H13M3T 243. Надежной защитой стальных резер-
вуаров от коррозии 8—10%-ной кремнефтористоводородной кис-
лотой является многослойное перхлорвиниловое покрытие. Хорошие
результаты дает также защита лаком ФЛ-4 (спирто-ацетоновый
раствор фурилфеноло-ацетальной смолы) 246-248.
Для переработки кремнефтористоводородной кислоты в кремне-
фторид натрия производят осаждение Na2SiF6 концентрированным
(25%-ным) раствором NaCl при 15—20° в стальных реакторах с ме-
шалками, защищенных от коррозии, как указано выше, или футеро-
ванных диабазовой плиткой; применяют и деревянные реакторы.
Кремнефторид натрия, применяемый в качестве инсектицида или
для дефолиации, должен быть весьма дисперсным. Степень его
дисперсности зависит от условий кристаллизации. При неизменной
концентрации H2SiF6 можно получить кристаллы Na2SiF6 разной
величины (10—60 мк и больше) при изменении продолжительности
подачи реагентов от 30 до 360 сек. Наибольшая степень пересыще-
ния раствора кремнефторидом натрия достигается при кристалли-
зации в избытке ионов Na+, наименьшая — в избытке ионов SiFe--
Поэтому для получения мелкокристаллического осадка необходимо
приливать кремнефтористоводородную кислоту к раствору NaCl,
а не наоборот, или быстро и одновременно сливать оба раствора.
Однако при одновременной подаче реагентов уменьшение началь-
ной концентрации кислоты от 10 до 4% H2SiF6 приводит к замет-
ному увеличению относительного размера кристаллов Na2SiF6.
Содержащийся в кислоте во взвешенном состоянии кремнегель
не влияет на процесс кристаллизации, Na2SiF6, а золь SiO2 способ-
ствует кристаллизации. Примесь КС1 в техническом хлористом
натрии способствует образованию более мелких кристаллов 249.
Для более полного выделения Na2SiF6 в твердую фазу необхо-
димо вводить избыток NaCl—125% от стехиометрического коли-
чества по следующей реакции:
H2SiFe + 2NaCl = Na2SiFe + 2НС1
Присутствие в растворе поваренной соли сильно понижает рас-
творимость Na2SiFe (рис. 333). Так, при 25° растворимость Na2SiFe
Улавливание фтористых соединений из отходящих газов
1143
Рис. 333. Изотермы 25 и 50°
растворимости в системе
Na2SiFe—NaCl—Н2О.
в воде равна 0,78%; введение в раствор 2% NaCl снижает содер-
жание Na2SiF6 до 0,1%, а при 10% NaCl и больше в растворе
остаются лишь сотые доли процента Na2SiF6. Обычно поваренную
соль вводят в таком количестве, чтобы маточный раствор после
осаждения Na2SiF6 содержал 2% NaCl.
Кристаллы NajSiFe отделяют от маточного раствора и геля
кремневой кислоты отстаиванием в течение 30 мин. Скорость осаж-
дения кристаллов Na2SiFe равна
3 м)ч, а геля кремневой кислоты —
0,25—0,3 м)ч. Такая большая раз-
ница в скорости осаждения позво-
ляет отделить Na2SiF6 из загрязнен-
ного гелем раствора, вследствие чего
и не требуется предварительная
очистка кремнефтористоводородной
кислоты от кремнегеля. Это суще-
ственно также и потому, что пред-
варительное отделение геля от кис-
лоты влечет за собой значительные
ее потери вследствие адсорбции
шламом и трудности отмывки.
В шламе, уплотненном в течение че-
тырех дней, содержится 25—40%
H2SiFe, а в отжатом непромытом
шламе — до 15% H2SiF6 250.
Получение Na2SiFe непрерывным
способом требует .особенно тщатель-
ной организации разделения кри-
сталлов NasSiFe и кремнегеля. При
непрерывном процессе смесь рас-
творов H2SiFe и NaCl проходит че-
рез реактор с мешалкой, в котором
находится 0,5—1 ч. Суспензия из ре-
актора перекачивается насосом в
гидроциклон, из которого сверху
удаляется маточный раствор, содер-
жащий НО и кремневая кислота, а сгущенная суспензия Na2SiFe
уходит снизу. Она проходит через репульпатор (с водой) и через
второй гидроциклон, затем снова репульпируется и отстаивается в
сгустителе251. Сгущенную пульпу Na2SiFe обрабатывают неболь-
шим количеством раствора соды для нейтрализации соляной кис-
лоты. Избыток соды вреден, так как ведет к потере NasSiFg вслед-
ствие перехода его в более растворимый NaF.
Кристаллы Na2SiFe отфуговывают и промывают в гуммирован-
ной центрифуге до тех пор, пока в осадке не останется менее 0,2%
NaCl и 0,02% НС1. Промывка необходима для уменьшения гигро-
12 М. Е- Позпн
1144
Гл. XXXI. Соединения фтора
скопичности и слеживаемости продукта 252. Промывную воду
используют для растворения поваренной соли. Кристаллы, снятые с
центрифуги и содержащие 7—13% влаги, высушивают до влаж-
ности менее 1%. Сушку следует вести при температуре не выше
400°, так как при дальнейшем повышении температуры резко воз-
растает давление диссоциации NazSiFe (стр. 1104). Сушка произ-
водится в прямоточных вращающихся барабанных сушилках. При-
меняют также неподвижные барабанные сушилки, в которых ма-
териал перемещается шнеком и высушивается в токе горячего воз-
духа, нагреваемого с помощью калориферов; снаружи они обогре-
ваются топочными газами, которые затем поступают в калориферы
для нагрева воздуха. Используют сушилки и других типов.
Так как при сушке продукт комкуется, то, в соответствии с тре-
бованиями потребителя, высушенный продукт подвергают очень
^тонкому помолу — до величины частиц 15—30 лк (требуется, что-
"6ы остаток на сите с отверстиями 0,053 мм был не более 15%).
; Размол производят в дисмембраторе; готовый продукт с требуе-
мой тонкостью помола отделяется с помощью воздушного сепара-
тора. На некоторых заводах применяют мельницы вертикального
типа также с пневматической классификацией. Эффективность этих
мельниц значительно увеличивается при предварительной воздуш-
ной сепарации сухого кремнефторида натрия, так как при этом бо-
лее 50% продукта отбирается в виде мелкой фракции помимо
мельницы253. „
• Описана сушка кремнефторида натрия в псевдоожиженном
слое горячим воздухом, подогретым до 350° газовым топливом.
Влажность материала понижается с 15 до 0,2%. Температура воз-
духа на выходе из сушилки 200°. Пылевидный продукт не требует
размола — 90% его улавливается в циклонах, остальные 10% в ме-
шочных фильтрах. Во избежание конденсации влаги воздух по всей
трассе не должен охлаждаться ниже точки росы (53—55°) 254.
Газы из сушил, особенно топочные газы из вращающихся су-
шил, а также воздух, отсасываемый из размольных установок, со-
держат кремнефторидную пыль и немного НС1. Для обезврежива-
ния перед выбросом в атмосферу эти газы следует промывать во-
дой, например, в небольшом барботере. На производство 1 т
NajSiFe расходуют: 0,725 т H2SiF6, 1,09 т технической поваренной
соли, ~380 м3 воды, ~ 175 кет • ч электроэнергии, 0,066 т топлива.
Обычно выход готового кремнефторида натрия не превышает
7—9 кг на 1 т суперфосфата. При сокращении потерь производства
он может быть повышен, так как при получении суперфосфата из
апатитового концентрата в газовую фазу выделяется 5,5—6 кг
фтора, что соответствует 9,6—10,4 кг Na2SiF6.
В США утилизация фтора в виде кремнефторидов при произ-
водстве простого и двойного суперфосфатов составляет всего 17%.
Улавливание фтористых соединений из отходящих газов
1145
Это объясняется нерентабельностью улавливания фтористых газов
на заводах малой мощности, на которых в США производят боль-
шую часть фосфатных удобрений 255.
Отходом производства кремнефторида натрия является слабый
раствор соляной кислоты, содержащий 3—4% НО, 2% NaCl и
около 0,15% Na2SiFe. Использование такого раствора затруднитель-
но; обычно его нейтрализуют известью и спускают в канализацию.
Для получения более концентрированной соляной кислоты (10—
16% НС1) ее можно укреплять путем возврата части кислоты,
остающейся после осаждения Na2SiFe поваренной солью, обратно в
абсорбционную систему.
Отбросный раствор может быть нейтрализован и одновременно
обесфторен обработкой нефелином. Весь фтор при этом переходит
в осадок, который может быть использован в производстве стекла,
эмалей, цемента, кислотостойких замазок, антисептической пасты
для защиты древесины от гниения и огня или для получения фтори-
стых солей 258. Этот слабый и грязный раствор соляной кислоты
может быть использован как добавка к кислоте при разложении
апатитового концентрата в производстве удобрений (стр. 848). При
обработке этим раствором апатитового концентрата выделяется
фосфорная кислота с концентрацией ~2% Р2Об; после ее нейтра-
лизации мелом получается преципитат, содержащий 27—30%
Р2О5 80.
Абсорбция фтористых газов раствором соды исключает необхо-
димость нейтрализации отбросного раствора, но из-за дефицит-
ности соды используется для получения Na2SiF6 редко.
Для получения взамен слабого отбросного раствора НС1 товар-
ной соляной кислоты можно производить осаждение Na2SiFe из
кремнефтористоводородной кислоты 35% раствором бисульфата
натрия, получаемым разложением поваренной соли серной кисло-
той. Выделяющийся хлористый водород поглощается водой, а ма-
точный раствор после отделения кремнефторида натрия, содержа-
щий ~11% H2SO4 и ~2% Na2SiF6, может быть использован как
добавка к серной кислоте в суперфосфатном производстве. На 1 т
кремнефторида натрия получается ~ 1,4 т стандартной соляной
кислоты 257.
Другим отходом производства является кремнегель, количество
которого при улавливании фтористых газов суперфосфатного про-
изводства составляет ~20% от веса кремнефтористоводородной
кислоты или 1,6—1,8 кг на 1 т суперфосфата. Этот кремнегель на-
зывают белаксом (в отличие от более дисперсной и активной белой
сажи). После промывания, нейтрализации известью, мелом, амми-
аком или содой и высушивания полученный белакс, содержащий в
зависимости от способа нейтрализации 37—45% CaF2, или 32—
35% Na2SiF6, или (при повышенном расходе соды) 40—45% NaF,
может быть использован как добавка к стекольным и цементным
1146
Гл. XXXI. Соединения фтора
шихтам или как теплоизоляционный материал [коэффициент тепло-
проводности 0,045 ккал) (м-ч- град) ]80’258.
Белакс значительно отличается от кремнегеля, получаемого в
щелочной среде, например от белой сажи, изготовляемой из сили-
ката натрия углекислотным методом или из кремнефтористоводо-
родной кислоты аммиачным методом (стр. 1168). Белая сажа яв-
ляется активным сорбентом; применяется, в частности, в качестве
ингредиента резиновых смесей взамен обыкновенной черной (угле-
родистой) сажи при изготовлении цветных резиновых изделий, от-
куда и получила свое название. Белакс же имеет значительно мень-
шую активность, но все же его можно использовать как наполни-
тель в производстве резинового линолеума — для этого он должен
быть нейтрализован и не содержать значительного количества сое-
динений фтора 259.
Как показал опыт работы Винницкого химического комбина-
та 26°, предварительная очистка кремнефтористоводородной кисло-
ты от кремневой на центрифугах позволяет значительно повысить
качество получаемых из нее кремнефторидов и фторидов. Осадок
кремнегеля, промытый на центрифуге теплой водой, нейтрализован-
ный слабым содовым раствором, подается в виде суспензии в рас-
пылительную сушилку, где высушивается в потоке топочных газов
от сжигания природного газа. Полученный таким способом белакс
применяется как наполнитель в производстве микропористой рези-
ны, однако он хуже белой сажи. Он имеет пластинчатую струк-
туру261 и размеры частиц 100—200 ммк, в то время как белая сажа
БС-50 состоит из частиц 20—100 ммк. В лабораторных условиях
при моделировании переработки H2S1F6 в A1F3 (стр. 1161) был по-
лучен более активный белакс, подобный белой саже. Для этого ре-
комендуют использовать профильтрованную кислоту с концентра-
цией не более 9% H2SiF6 262>263.
При азбтнокислотцом разложении фосфатов освобождение полу-
чающейся вытяжки от H2SiF6 может быть осуществлено добавкой
к ней раствора NaNO3. Максимальная степень осаждения дости-
гается при 300—400 %-ном избытке NaNO3 от стехиометрического
количества 264’265. Растворимость Na2SiF6 в водных растворах HNO3
значительно больше, чем в растворах НС1, H2SO« и Н3РО4. Наибо-
лее полно Na2SiF6 осаждается с помощью NaCl 266.
Описано 267 действующее в Китае производство Na2SiF6 из пла-
викового шпата, серной кислоты, кремнезема и поваренной соли.
Вначале получают кремнефтористоводородную кислоту
3CaF2 + SiO2 + 3H2SO4 = H2SiFe + 3CaSO4 + 2H2O
которую затем обрабатывают поваренной солью. Применяется от-
бросная серная кислота (30—40% H2SO4) от производства органи-
ческих веществ; источником SiO2 служит молотая зола шелухи зла-
ков, содержащая больше 70% SiO2. Для производства 1 т 72%-
Улавливание фтористых соединений из отходящих газов
1147
ного Na2SiF6 расходуют: 1,86т плавикового шпата (93% CaF2),
2,3т серной кислоты (100% H2SO4), 0,615т золы (с 40% влаги),
1,1 г технической поваренной соли.
Кремнефторид аммония
Кремнефторид аммония получают нейтрализацией кремнефтори-
стоводородной кислоты аммиаком или 25 %-ной аммиачной водой.
При выпаривании нейтрализованного раствора образуется продукт,
содержащий 85—86% (NH4)2SiF6. При концентрации H2SiF6 в
исходной кислоте 12% на 1 г товарного продукта приходится вы-
паривать ~6,5 г воды.
Возможна организация этого производства и без выпарки рас-
твора 288. Для этого поглощение SiF4 из отходящих газов ведут обо-
ротным водным раствором кремнефторида аммония, затем получен-
ный кислый раствор нейтрализуют 25%-ной аммиачной водой:
SiF4 + (2 + га)Н2О + (NH4)2SiFe = 2H2SiFe + SiO2 • гаН2О + (NH4)2SiFe
2H2SiFe + SiO2 -nHjO + (NH4)2SiFe + 4NH4OH = 3(NH4)2SiFe + SiO2 • raH2O + 4H2O
Степень оборачиваемости раствора (NH4)2SiF6 определяется
растворимостью этой соли в растворе H2SiF6. При 25° в воде рас-
творяется 18,75% (NH4)2SiF6, в присутствии 6,67% H2SiF6 раство-
римость (NH4)2SiF6 уменьшается до 16,31%, а в 13% растворе
H2SiF6 до 13,75%. Практически раствор, поступающий на абсорб-
цию SiF4, ведущуюся при 55°, должен содержать 17—18%
(NH4)2SiF6. После нейтрализации кислого раствора до остаточной
кислотности 0,5—0,8% и охлаждения до 25° отфильтровывают вы-
делившийся осадок кремнефторида аммония, который содержит
85—86% (NH4)2SiF6. Если кремнегель отделить от кислого рас-
твора до его нейтрализации, продукт будет содержать 95—97%
(NH4)2SiF6. На 1 т 100%-ного (NH4)2SiF6 связывается 640кг фто-
ра в виде SiF4 и расходуется 190 кг аммиака. Преимуществом
этого процесса (по сравнению с производством Na2SiF6 с примене-
нием NaCl) является отсутствие кислых сточных вод.
Кремнефториды калия, магния, кальция,
цинка и алюминия
Кремнефторид калия получают обработкой кремнефтористо-
водородной кислоты хлористым калием и при очистке плавиковой
кислоты поташом 269.
Кремнефториды магния, кальция и цинка могут быть получены
при обработке природного или каустического магнезита, молотого
известняка или извести и окиси цинка при 50—60° в течение 2—
2,5 ч технической кремнефтористоводородной кислотой (8—10%
1148
Гл. XXXI. Соединения фтора
H2SiFe) с 1,5—2%-ным ее избытком к стехиометрическим количест-
вам. При этом получаются растворы, содержащие приблизительно:
11,5% MgSiFe, или 15% CaSiF6, или 14,5% ZnSiF6. Использование
концентрированной кислоты (~20% H2SiFe) позволяет получить
почти насыщенные растворы солей. После фильтрации растворы
выпаривают при 80—90°, так как при более высокой температуре
возможно разложение солей 27°. При охлаждении выпаренных рас-
творов выделяются осадки кристаллогидратов MgSiF6 • 6Н2О, или
CaSiF6’2H2O, или ZnSiFe • 6Н2О, содержащие после отфуговывания
маточного раствора около 98% основного вещества 271’272.
Эти кремнефториды могут быть получены и при непосредствен-
ном улавливании SiF4 из газов увлажненными порошками (напри-
мер, MgCO3) или суспензиями 273’274.
Кремнефторид магния MgSiFe • 6Н2О можно получить в качестве
побочного продукта при очистке экстракционной фосфорной кисло-
ты от ион-а магния путем добавки к ней H2SiF6 275.
Предложено производить обезвоживание раствора кремнефто-
рида магния, полученного путем нейтрализации кремнефтористово-
дородной кислоты окисью магния, в распылительной сушилке с по-
лучением кристаллического продукта. При этом целесообразно
вести выпарку раствора с последующей кристаллизацией
MgSiFe • 6Н2О, а не до полного обезвоживания. Для этого необхо-
димо выпарить раствор до концентрации 55—60% MgSiF6 • 6Н2О.
Обезвоживание раствора ведут при 65°, во избежание разложения
продукта 276.
Кремнефторид алюминия 237 получается при взаимодействии кис-
лой соли фторида алюминия с кремнегелем. Для этого растворяют
гидрат окиси алюминия в нагретой до 80° 15 %-ной плавиковой
кислоте и небольшими порциями добавляют силикагель:
2А1(ОН)3 + 12HF = 2A1F3 • 3HF + 6Н2О
2A1F3 • 3HF + 6HF + 3SiO2 = Al2(SiFe)3 + 6Н2О
После охлаждения из раствора выделяется желатинообразный
осадок кремнефторида алюминия. Он может быть получен также
при введении силикагеля в смесь плавиковой и кремнефтористово-
дородной кислот:
2А1(ОН)3 + 12HF + H2SiFe + 2SiO2 = Al2(SiFe)3 + ЮН2О
ПОЛУЧЕНИЕ ФТОРИСТЫХ СОЛЕИ ИЗ ОТХОДЯЩИХ ГАЗОВ
Потребность в кремнефториде натрия все более ограничивается
в связи с появлением новых эффективных дефолиантов. Поэтому
возникает необходимость его переработки на другие продукты или
прямого их получения из фтористых газов 278.
Для получения HF, потребность в котором все возрастает, не-
обходимо сначала переработать кремнефторид в CaF2 или NaF с
отделением SiO2. Далее фториды могут быть разложены серной
Получение фтористых солей из отходящих газов
1149
кислотой. Так, полное разложение NaF серной кислотой достигается
при нагревании стехиометрической смеси до 500°; получаемый при
этом сульфат натрия содержит, однако, 0,2—0,3% HF и до 0,7%
NaF, что ограничивает области его применения 279.
Фтористый водород можно получить дегидратацией водного рас-
твора H2SiF6 93—98,5%-ной серной кислотой при 125° в течение 3—
4 мин. При этом основная часть HF поглощается серной кислотой,
а в газовую фазу выделяется SiF4, который поглощают в абсор-
бере разбавленным раствором H2SiFe. Образующийся здесь 12—
25%-ный раствор H2SiF6 возвращают в процесс. Десорбцию HF
(80—98%) из раствора в серной кислоте (70—80% H2SO4) произво-
дят под вакуумом или добавляя концентрированную серную кисло-
ту и нагревая до 200°. Более полному извлечению HF способствует
продувка горячей серной кислоты воздухом- или инертным газом
(например, гексаном и др.). Из газа HF выделяют конденсацией
или водной абсорбцией получают плавиковую кислоту 28°.
Разработан двухстадийный цикличный способ получения HF из
фторсодержащих газов или из H2SiF6, полученной водной абсорб-
цией этих газов. В первой стадии спекают Na2SiF6 с Na2SiOs и
Ре20з, получая фторсодержащий спек. Во второй стадии его гидро-
лизуют перегретым паром, в результате чего выделяется HF, а спек
обесфторивается:
6NaF • Fe2O3 • 3SiO2 + ЗН2О = 6HF + 3Na2O • Fe2O3 • 3SiO2
Часть обесфторенного спека (одну треть) выщелачивают водой;
образующийся щелочной раствор обрабатывают кремнефтористо-
водородной кислотой или поглощают им отходящие фтористые
газы, получая Na2SiF6. Остальную часть (две трети) обесфторен-
ного спека смешивают с Na2SiFe и возвращают на спекание281.
Представляет интерес поглощение HF из газов высокотемпера-
турной переработки фосфатов при помощи NaF при ~ 100°. Обра-
зующийся бифторид натрия разлагается прй 250° с выделением
100%-ного HF 282.
В настоящее время наиболее целесообразной является перера-
ботка отходящих фтористых газов во фториды натрия, алюминия и
в криолит для использования их в производстве алюминия. Это воз-
можно осуществить как при непосредственном улавливании фтори-
стых газов, так и переработкой предварительно полученных H2SiFe
или Na2SlFe283>284.
Фторид натрия
Термическое р аз ложение Na2SiF6
При Нагревании в лабораторных условиях в течение 1 ч в му-
фельной печи или 20 мин во вращающейся печи при QQQ----6200 сте-
пень разложения Na2SiFe по реакции
Na2SiFe = 2NaF + SiF4
1150
Гл. XXXI. Соединения фтора
достигает 98—100% 285- Выделяющийся SiF4 может быть вновь по-
глощен водой и переработан на Na2SiFe— на этом основана цикли-
ческая схема производства NaF термическим методом 286. Продукт
содержит 85—88% NaF, до 1,5% Na2SiF6 и 8,5—10% не раствори-
мого в воде остатка, состоящего в основном из SiO2. Расход кремне-
фторида натрия при однократном использовании его составляет
2,34 т на 1 т фторида натрия, а при замкнутом процессе е использо-
ванием выделяющихся газов — 0,83 т (с учетом 10% потерь отвеса
сырья, загружаемого в печь; при прокаливании гранулированного
Na2SiF4 потери от уноса пыли значительно уменьшаются). Концен-
трация SiF4 в газе, получающемся при разложении Na2SiFe, со-
ставляет 2,5—8,6 объемн. %; с увеличением концентрации SiF4 раз-
ложение замедляется и качество продукта ухудшается. При
улавливании этого вторичного SiF4 можно получить чистую крем-
нефтористоводородную кислоту с концентрацией 36—39% H2SiF6,
которая может быть возвращена в производственный цикл или пе-
реработана в кремнефториды.
С одо в о-термичес к ий способ
Прокаливание Na2SiFe в присутствии соды
Na2SiF6 = 2NaF + SiF4
SiF4 + 2Na2CO3 = 4NaF + SiO2 + 2CO2
♦ Na2SiFe + 2Na2CO3 = 6NaF + SiO2 + 2CO2
позволяет упростить производство NaF, так как отпадает надоб-
ность в переработке SiF4. В отходящих газах содержится лишь не-
значительное количество (0,04%) SiF4.
Шихта из 47% кремнефторида натрия и 53% соды 287 увлаж-
няется до содержания 25% воды с целью понижения температуры
реакции в печи и уменьшения уноса материалов с газами. Шихта
нагревается до 600° в трубчатой печи топочными газами, движущи-
мися противотоком материалу. Зона максимальной температуры
580—600° находится на расстоянии 2 м от выгрузочного конца печи.
Печь длиной 10 м, Диаметром 0,9 м при 1,3 об!мин имеет произво-
дительность 250—300 т продукта в месяц. Продукт охлаждают в
шнековом водяном холодильнике, размалывают и расфасовывают
в фанерные барабаны. Он содержит 70—74% NaF вместо 85—88%
при бессодовом разложении. Более низкое качество продукта вслед-
ствие повышенного содержания в нем SiO2, так же как и необ-
ходимость расходования соды, являются недостатками метода.
Расходные коэффициенты на 1 т 70%-ного фторида натрия по по-
лузаводским данным: 0,675 т соды (100% Na2CO3), 0,61 т (с учетом
потерь до 10%)кремнефторида натрия, 0,16 т условного топлива,
180 кет • ч электроэнергии, 40 м3 воды.
Получение фтористых солей из отходящих газов
1151
Содовый способ
Этот способ получения NaF из отходящих газов, заключающий-
ся в промывке их содовым раствором, наиболее прост. Абсорбция
SiF4 по реакции
SiF4 + 2Na2CO3 + 2Н2О = 4NaF + SiO2 • 2H2O + 2CO2
и осаждение NaF осуществляются в одной операции. Однако при
этом получается смесь NaF с SiO2-2H2O, полное разделение кото-
рой затруднительно (громоздко и дорого).
По этому методу можно получить и раствор NaF, практически
свободный от кремневой кислоты 208, абсорбцией кремнефтористых
газов столь разбавленным содовым раствором, чтобы образующийся
NaF полностью в нем растворялся. В растворах, содержащих ме-
нее 1% Na2CO3, растворимость NaF при 20—100° практически рав-
на его растворимости в воде (~4%). В более концентрированных
растворах, содержащих 10—28% Na2CO3, растворимость NaF вука-
занном интервале температур ~1%.
Суспензию кремнегеля в растворе NaF разделяют отстаиванием
и центрифугированием. Промывные воды от промывки шлама ис-
пользуют для растворения соды. Шлам можно и не промывать, так
как раствор разбавленный и потери NaF в шламе не очень велики.
Аналогичным способом можно получать не содержащие крем-
ния растворы NaF из Na2SiFe или из технического фторида натрия,
загрязненного соединениями кремния.
Эффективное отделение кремневой кислоты от раствора NaF до-
стигается добавкой реагента (с 50—100%-ным избытком от стехио-
метрического), содержащего в 1 л 20 г ZnO, 40 мл аммиачной воды
и 40 г (ПН4)2СОз. При кипячении раствора аммиак улетучивается.
Очищенный таким способом раствор NaF может быть переработан
в криолит, содержащий всего 0,03% SiO2 288.
Содово-суспензионный способ
Этот способ заключается в использовании для реакции взаимо-
действия с раствором соды суспензии кремнефторида натрия, что
значительно сокращает объемы реакционных растворов 289’290. Про-
дукт содержит 70—75% NaF и 20—25% SiO2.
Этот способ получения NaF осуществляли в аппаратуре кремне-
фтористого отделения Винницкого химического комбинта следую-
щим образом 286’291. В нагретую до 70—80° пульпу Na2SiFe вводили
постепенно при интенсивном перемешивании сухую кальцинирован-
ную соду с избытком 1—2% к стехиометрическому количеству. Со-
отношение Ж:Т в реакционной массе 2—2,5: 1. После получасо-
вого нагрева острым паром и проверки завершения реакции (по
малиновой окраске фенолфталеина) горячую суспензию центри-
фугировали. Маточный раствор, содержащий до 3,5% NaF,
1152
Гл. XXXI. Соединения фтора
возвращали в процесс. Продукт после центрифугирования направ- |
ляли на сушку, которая затруднена его высокой влажностью (20— 1
25%) и присутствием большого количества гидратированной дву- |
окиси кремния. i
В дальнейшем этот процесс был усовершенствован 2S2-296. Уста- <
новлена зависимость его скорости и качества продукта от порядка ;
сливания и интенсивности перемешивания реагентов (стр. 1129). =
Хорошее перемешивание не только выравнивает концентрации реа- |
гентов, но и предотвращает в некоторой мере обволакивание крем- !
невой кислотой кристаллов Na2SiF6, что ускоряет их растворение. ’
Оптимальная окружная скорость перемешивания 1,2—1,8 м!сек.
При большей скорости масса сильно вспенивается, что осложняет
ведение процесса.
При внесении Na2SiF6 в содовый раствор скорость процесса почти
в 4 раза больше, чем при введении содового раствора в пульпу.
Но при этом происходит интенсивное пенообразование. Поэтому
вначале всю пульпу кремнефторида натрия вводят в часть содо-
вого раствора, равную одной трети от общего количества, а затем
постепенно при перемешивании добавляют в массу остальное коли-
чество раствора.
На рис. 334 изображена схема получения фторида натрия из
кремнефторида натрия, по которой работал Одесский суперфос-
фатный завод до замены этого производства производством крио-
лита. Эта схема позволяет вырабатывать каждый из продуктов
(NaF и Na2SiFe) в отдельности или совместно. Исходным сырьем
является кремнефтористоводородная кислота, полученная водной
абсорбцией отходящих фтористых газов. Кремнефторид натрия полу-
чают в стальных, футерованных диабазовой плиткой мешалках-ре-
акторах 5 емкостью по 2,7 м3, куда подают кислоту из стального,
футерованного плиткой мерника 2 (емкостью 1,9 м3) и раствор
поваренной соли из стального мерника 4 (емкостью 1,5 м3). Полу-
ченная пульпа кремнефторида натрия может быть непосредственно
переработана в товарный продукт, для чего твердую фазу отде-
ляют от жидкости на центрифуге 7 и направляют на сушку, размол
и расфасовку. Взаимодействие пульпы Na2SiFe с содовым раство-
ром осуществляют в стальных реакторах 6 с мешалками емкостью
по 2,4 м3, обогреваемых острым или глухим паром; в последнем
случае они снабжены паровыми рубашками. Обогрев глухим паром
значительно уменьшает объем маточных растворов. В реактор за-
ливают половину требуемого количества насыщенного раствора со-
ды, нагревают его до 60—80° и загружают полную порцию пульпы
кремнефторида натрия, а затем постепенно добавляют остальное
количество содового раствора. По окончании реакции (через 40—
45 мин) пульпу спускают при перемешивании в центрифуги 7. Ма-
точный раствор направляют через стальной сборник 9 в содорас-
творитель 10. Отфугованный продукт высушивают в шнековой су-
Получение фтористых солей из отходящих газов
1153
шилке. За один цикл (40—45 мин) в аппаратах указанных объемов
получают 300 кг продукта. Производительность шнековой сушилки
6—8т/сутки. Получаемый продукт содержит 72—75% NaF, 2,5—
3,5% влаги, 16—19% нерастворимого остатка (ЗЮг); остаток на
сите № 05 0,2—0,6%. Капитальные затраты на дооборудование
кремнефтористого отделения для выпуска фторида натрия во много
раз меньше затрат на организацию производства фторида натрия
содово-термическим способом.
в канализацию
Рис. 334. Схема реакторного отделения производства кремне-
фторида натрия и переработки его на фторид натрия содово-
суспензионным способом:
1 — сборник кремнефтористоводородной кислоты; 2 —мерник кремнефто-
рнстоводородной кислоты; 3 —растворитель поваренной соли; <4 —мерник
насыщенного раствора поваренной соли; 5 — реактор для получения
NaaSiFg; 6 — реактор для получения NaF; 7 — центрифуга; 3 —мерник на-
сыщенного раствора соды; 9 —сборник маточного раствора NaF; /О —рас-
творитель соды; П —отстойник для сбросного раствора.
Представляет интерес отделение кремневой кислоты от NaF с
помощью гидроциклона 296. Разделение в гидроциклоне тем легче,
чем больше отличаются по размерам частицы фтористого натрия
и кремневой кислоты. Наибольшая разница в размерах этих ча-
стиц достигается за счет укрупнения кристаллов NaF в процессе-
нейтрализации кремнефтористоводородной кислоты содой, в част-
ности за счет сравнительно медленного перемешивания реакцион-
ной массы294. В пульпе, поступающей в гидроциклон, отношение
Ж : Т должно быть равно 6—8, для чего концентрация исходной
пульпы NaaSiFe должна быть 100—80 кг/м3. При диаметре гидро-
циклона 45 мм его производительность 4—4,5 м3/ч пульпы, что
1154
Гл. XXXI. Соединения фтора
соответствует 7—10 т] сутки продукта с содержанием до 90% NaF.
Потери NaF со сливом меньше 8—10% от количества, поступаю-
щего на обогащение. Повторное пропускание слива через гидро-
циклон позволяет извлечь основную часть NaF.
Описан способ флотационного разделения NaF и БЮг 297.
Освобождение фтористого натрия от SiO2 может быть достиг-
нуто при обработке щелочью — образуется раствор жидкого стекла,
который отделяется от нерастворяющегося NaF. Получающийся про-
дукт содержит 94—99% NaF. Необходимость утилизации отхода —
раствора жидкого стекла осложняет организацию этого процесса80.
Поташный способ
Поташный способ 298.299 производства NaF заключается в кон-
версии кремнефторида калия оборотным раствором поташа, разде-
лении образовавшегося раствора фторида калия и выделившейся
кремневой кислоты, конверсии фторида калия содой с осаждением
NaF и отделении от растворимых калиевых соединений осадка NaF.
Исходный кремнефторид калия может быть получен осаждением
хлористым калием из раствора кремнефтористоводородной кислоты
H2SiF6+ 2КС1 = K2SiF6 + 2НС1 (1)
или абсорбцией газообразного четырехфтористого кремния оборот-
ным раствором поташа:
3SiF< + 2K2CO3 = 2K2SiF6+ 2СО2 + SiO2 (2)
Дальнейшие стадии процесса в обоих случаях характеризуются
следующими уравнениями:
K2SiF6 + 2К2СОз = 6KF + SiO2 + 2СО2 (3)
6KF + 3Na2CO3 ЗК2СО3 + 6NaF (4)
Весь раствор углекислого калия, образующийся в процессе кон-
версии фтористого калия, полностью возвращается на реакции (2)
и (3); если для получения кремнефторида применяется хлористый
калий, ’/з этого раствора должна быть переработана на товарный
поташ.
По обоим вариантам образуется осадок аморфной кремневой
кислоты, который после промывки, сушки и разрыхления является
’побочным продуктом — белой сажей 300.
Разложение кремнефторида калия раствором поташа протекает
с наибольшей скоростью при температуре кипения смеси (103—
113°). Кремнефторид при этом разлагался практически нацело при
небольшом (5%) избытке углекислого калия сверх теоретического
количества301. При начальной концентрации в растворе 10—13,5%
К2СО3 кремнефторид разлагается в течение 1 ч. С увеличением
Получение фтористых солей из отходящих газов
1155
концентрации раствора поташа продолжительность взаимодействия
увеличивается, например, при 21% К2СО3 реакция длится 3 ч, а
при 30% К2СО3 —4 ч.
Осаждаемый в описанных условиях гель кремневой кислоты
фильтрует с высокой скоростью и легко промывается. На полуав-
Рис. 335. Диаграмма взаимной водной системы 2KF + Na2CO3
SNaF + KjCOj при 75° в прямоугольных координатах (разрез по
оси NaF—К2СО3 и области в координатах 2NaF—К2СО3—2KF):
7 — разрез через поверхность насыщения NaF ирн отношении К2СО3 : KF = 2,2;
II — то же при KsCOs : KF — 3,4.
тематической горизонтальной центрифуге достигнута производи-
тельность в 470 кг/ч по влажному (44%) осадку. С целью полного
осаждения кремневой кислоты из раствора KF необходимо охла-
ждение реакционной смеси до 25°. Отфильтрованный после этого
раствор содержит лишь 0,05—0,06% SiOa.
На рис. 335 показана часть диаграммы взаимной системы
2KF+Na2CO3=ps:2NaF-|-К2СО3 при 75°301 >302. Для рассматриваемого
1156
Гл. XXXI. Соединения фтора
процесса осаждения NaF имеют значение разрезы диаграммы этой
системы через поверхность насыщения NaF при различном соотно-
шении в растворе К2СО3 й KF. Экспериментально определены два
таких разреза для 75° при отношениях К2СО3: KF, равных 2,2 и 3,4
(кривые I и II). Они показывают, что по мере увеличения концен-
трации K2CO3 + KF происходит полное высаливание NaF до начала
кристаллизации КдСОз- 1,5Н2О. Это позволяет построить технологи-
ческий процесс так, чтобы путем выпарки раствора поташа в при-
сутствии фтористого калия практически полностью выделялся фто-
ристый натрий.
KjSlF,
бода от лроныВ^и NaF
Вода
8
Охлаждение
пульпы
яг
1<Рш»тообаниа\ Растбор
пульпы |
Вода
НПройыдна |
шлама SiO;|
Шлам SjO2
СОа
II азаза
юбани»
а/мовЪ1аТ
Пары бобы |
|Суш»а NaF
Калоцинировенно»
сооо
Растбо
ни» COl
Фторид
натри»
(таборный)
lodt'
Осодон NaF I ФигоДаиие
[столяр/ NaF |
Пары боды
Кристаллы Na Г
(Промыбяа
сталяоб f
Маточный растбор
(К^О, ♦ KF)
бобо
Кальцинация 1 Пары боды
поташа
Поташ
( таборный )
бода
| / жмбедсия
I
&
5
J
Рис. 336. Схема производства фторида натрия и поташа нз кремнефторида
калия и соды.
Один из вариантов схемы производства фторида натрия поташ-
ным способом приведен на рис. 336. Конверсия кремнефторида ка-
лия осуществляется в обогреваемых паром реакторах с мешалкой
взаимодействием с оборотным раствором, содержащим около 39%
К2СО3 и 11,5% KF. Этот раствор образуется в процессе первой вы-
парки смеси, состоящей из фильтрата фторида калия и раствора
от промывки поташа. В процессе первой конверсии получается рас-
твор с содержанием до 23% KF и до 0,05% SiO2 при практически
полном (98,8—99%) разложении кремнефторида. Выделение кри-
сталлов NaF производится в два приема: в процессе второй кон-
версии и при первой выпарке смеси растворов. Фильтрат, не
содержащий NaF, выпаривается вторично для выделения кристалли-
ческого поташа К2СО3 • 1,5Н2О. По этой схеме на 1 т фтористого
натрия получается около 0,5 т поташа (прокаленного). На это коли-
чество продукции расходуется 1,04 т кремнефторида калия (свлаж-
Получение фтористых солей из отходящих газов
1157
ностью 10%) и 1,35 т кальцинированной соды при расходе пара на
выпаривание 5,5 т. Получаемый фтористый натрий содержит (на
сухое вещество): 98% NaF, 1% KF, 1% К2СО3 и 0,05% SiO2.
Запатентован 303 более простой метод. KF получают.из раствора
H2SiFe по реакции:
H2SiFB + ЗК2СО3 = 6KF + SiO2 + Н2О + ЗСО2
Для получения осадка SiO2, легкоотделяемого фильтрацией,
реакцию ведут при 50—100° с избытком (до 150% от стехиометриче-
ского количества) К2СО3. Отфильтрованный и упаренный раствор
Рис. 337. Схема производства фторида натрия из газообразного четырех-
фтористого кремния и соды.
обрабатывают содой для получения NaF [по реакции (4)]; после
Отделения кристаллов NaF раствор К2СО3 возвращают в процесс.
По другому варианту 304 фтористые газы (SiF4 или смесь SiF4+
+ HF) поглощаются в абсорбере оборотным раствором поташа
(рис. 337). Суспензия из абсорбера поступает в систему из несколь-
ких обогреваемых паром реакторов, где протекает процесс первой
конверсии. После отделения на центрифуге двуокиси кремния рас-
твор фтористого калия (18—20% KF) выпаривают до концентрации
около 40% KF. Выпаренный раствор поступает на взаимодействие
с содовой суспензией (40% Na2CO3) при 85—90°; выделяющийся
при этом осадок NaF отфуговывают, а раствор поташа, содержа-
щий 30—35% К2СО3, возвращают на поглощение фтористых га-
зов. По этой схеме на 1 т фтористого натрия расходуется 1,38 т
1158
Гл. XXXI. Соединения фтора
кальцинированной соды, 0,47 т фтора (в виде газов), 0,06 т карбо-
ната калия (потери) и 2,56 т пара. Потери карбоната калия могут
восполняться введением в процесс кремнефторида калия.
Поташный способ экономичнее, чем способ производства фтори-
стого натрия из плавикового шпата и серной кислоты. Его достоин-
ством является получение очень чистого продукта.
Фторид кальция *
Синтетический фторид кальция 80-304’305 отличается от природ-
ного необогащенного плавикового шпата тонкодисперсной структу-
рой. При взаимодействии H2SiF6 с СаСОз или СаО вначале обра-
зуется кремнефторид кальция, затем фторид кальция.
H2SiF6 + СаСО3 = CaSiF6 4- Н2О 4- СО2
CaSiFg 4- 2СаСО3 =» 3CaF2 4- SiO2 4- 2СО2
H2SiFB 4- ЗСаСОз = 3CaF2 4- SiO2 4- ЗСО2 4- Н2О
При использовании карбоната кальция образуется густая пена.
Хотя реакция протекает с выделением тепла, но для завершения
разложения CaSiF6 требуется подогрев смеси до 70—80° и дли-
тельное перемешивание (2—Зч). При исходной концентрации
H2SiF6 8—14% оптимальное весовое отношение СаСО3: H2SiF6 рав-
hq 2,5. При использовании 12%-ной кремнефтористоводородной кис-
лоты в результате реакции образуется суспензия с весовым отноше-
нием Т : Ж, равным 1 : 4. Осадок CaF2 получается тонкодисперсный
и трудно отстаивающийся. Фильтруемость осадка зависит от соот-
ношения HF и H2SiF6 в кислоте и вида используемого карбоната
кальция, который во всех случаях должен быть тонкоизмельчен-
ным. Если в жидкости много HF, для нейтрализации следует при-
менять молотый известняк, так как применение мела в этом случае
приводит к образованию очень тонкого, плохо фильтрующегося
осадка. Чем больше в смеси H2SiFe, тем легче фильтруется осадок,
полученный с применением мела. При отношении HF : SiF4, мень-
Щем 1 : 12, уже возможно использование мела 306, позволяющего до-
стичь полной нейтрализации кислоты.
Для улучшения отстаивания и фильтрации тонкодисперсного
CaF2 предложено добавлять в реакционную массу при его осажде-
нии олеиновую кислоту (1—2,5%). Олеиновая кислота может быть
использована многократно, так как она не вступает в реакцию и
при. перемешивании реакционной массы всплывает наверх 307.
При непосредственном поглощении отбросных фтористых газов
меловой суспензией можно нейтрализовать практически весь карбо-
нат, оставляя в суспензии ~0,5 г)л СаСОз с целью предотвращения
проскока фтористых газов. Коэффициент абсорбции SiF4 меловой
* Об аммиачном способе получения CaF2 см. стр. 1168.
Получение фтористых солей из отходящих газов
1159
суспензией больше, чем водой, и составляет в башне с хордовой на-
садкой235 ~ 10 000 ч-1.
Сгущение осажденного фторида кальция можно осуществить на
центрифуге отстойного типа. В отфугованной массе остается до
60% влаги. После высушивания получается тонкодисперсный про-
дукт с величиной частиц около 5 мк и меньше, являющийся смесью
кристаллического CaF2 и аморфной SiO2. В нем содержится ~63%
CaF2, 24—30% S.iO2, 10% СаСОз и 3% прочих примесей, количество
и состав которых зависят от качества известняка или мела. При
использовании для осаждения свежеобожженной извести выход
CaF2 составляет по фтору 85—92%, по окиси кальция 74—87%.
Фтористый кальций, содержащий больше 94% CaF2, получается
при термической диссоциации CaSiFe при 380—400°:
CaSiFB = CaF2 + SiF4
Реакция нейтрализации H2SiF6
HjSiFa + СаО = CaSiFB + Н2О
протекает нацело даже на холоду, причем основная масса CaSiF6
выделяется в осадок, а из фильтрата, содержащего 12—15%
CaSiF6, можно выделить добавочное количество этой соли выпари-
ванием. Маточный раствор может быть использован для поглоще-
ния SiF4.
Можно получать CaF2 из H2SiF6 при помощи СаС12 по схеме:
H2SiFB + СаС12 = CaSiFB + 2НС1
CaSiFB + 2СаО = 3CaF2 + SiO2
2НС1 + СаО - СаС12 + Н2О
В результате нейтрализации соляной кислоты вновь образуется
СаС12, который возвращают в процесс.
Предложено получать фтористый кальций поглощением фтори-
стых газов растворами фторидов щелочных металлов и последую-
щей конверсией образовавшихся кремнефторидов едкой щелочью и
каустификацией раствора фторида гидроокисью кальция 308:
SiF4+2MF = M2SiFB
M2SiFB + 4МОН = 6MF + S i(OH)4
4MF + 2Ca(OH)2 = 2CaF2 + 4MOH
В этом процессе щелочь не расходуется (за исключением по-
терь); часть получаемого во второй стадии раствора фторида воз-
вращают на абсорбцию, а часть направляют на каустификацию.
Каустификация идет с большой скоростью и практически заканчи-
вается в течение 1 ч. Конверсия кремнефторида также идет с доста-
точной для практических целей скоростью и полнотой. Получаемые
шламы хорошо отстаиваются. Лабораторные испытания показали,
1160
Гл. XXXI. Соединения фтора
что этим способом может быть получен фтористый кальций хоро-
шего качества.
Однако при наличии в газах, кроме SiF4, также HF и H2SiF6
осадок CaFa, образующийся при применении Са(ОН)2, получается
слишком тонкий, трудно отделяемый от жидкости (как при осажде-
нии CaF2 из смеси кислот мелом). В связи с этим этот способ не
применим для прямого улавливания фтористых газов суперфосфат-
ных заводов. Считают, что он может быть использован для перера-
ботки на CaF2 газов с малым содержанием HF. Впрочем этот спо-
соб не имеет преимуществ перед переработкой на CaF2 кремнефто-
ристоводородной кислоты без получения из нее Na2SiF6.
Описана крупная заводская установка в США для улавливания
известняком HF из газов, выделяющихся в производстве обесфто-
ренного плавленого фосфата. После очистки от пыли газы с темпе-
ратурой 100—450° пропускают через башню со слоем известняка
2,5—3 м (размер кусков 6—40 мм). При концентрации HF, соответ-
ствующей 0,15—6 г/м3 фтора, степень поглощения 95—96%. Перио-
дически часть известняка (~40%) заменяют свежим. Фторид каль-
ция накапливается в мелочи, содержащей 80—95% CaF2. Крупные
куски, не прошедшие через сито с отверстиями 3,33 мм, содержат
лишь 20—40% CaF2 и после отделения от мелочи их возвращают
в башню 309-312. Изучена зависимость степени поглощения HF и ско-
рости процесса от свойств известняка, температуры и пр.313.
Криолит и фтористый алюминий
Карбонатный способ получения криолита заключается в карбо-
низации смеси растворов NaF и алюмината натрия 288’314:
12NaF + Na2O • А12О3 + 4СО2 = 2Na3AlFe + 4Na2CO3
Na2CO3 + СО2 + Н2О = 2NaHCO3
Раствор фторида натрия получают абсорбцией фтористых газов
5% раствором соды (или другими способами, рассмотренными вы-
ше). Осадок кремнегеля отделяют от раствора NaF центрифугиро-
ванием. Алюминатный раствор с содержанием 8% А.12О3 и 8% Na2O
приготовляют растворением гидроокиси алюминия в 50% растворе
NaOH и последующим его разбавлением. Растворы NaF и NaAlO2
смешивают при одновременной их подаче в реактор и карбониза-
ции.
Для образования хорошо фильтрующего криолита величина pH
раствора должна быть 10,2—10,7. Карбонизацию ведут газом, со-
держащим 12—15% СО2, до достижения отношения Na2CO3:
: NаНСОз, равного 1. Осадок криолита отделяют от раствора в сгу-
стителе, затем на фильтре промывают и высушивают. Маточный
раствор и промывные воды возвращают на абсорбцию (на приго-
товление содового раствора).
Получение фтористых солей из отходящих газов
1161
Более рациональным является кислотный способ УНИ-
ХИМ 277>315-316, по которому вначале получают из H2SiF6 фтористый
алюминий, затем криолит. Для этого в полученной из фтористых
газов кремнефтористоводородной кислоте, содержащей 12,5—
13,5% H2SiFe и кремнегель, нагретой до 85°, растворяют в течение
15-минутного перемешивания гидрат окиси алюминия:
H2SiF6 + 2А1(ОН)з = 2A1F3 + SiO2 • 2Н2О + 2Н2О
Это взаимодействие идет в две стадии:
3H2SiF6 + 2А1(ОН)з = Al2(SiF6)3 + 6Н2О
Al2(SiF6)3 + 12Н2О = 2A1F3 + 3SiO2 • 2Н2О + 12HF
Образующийся по последней реакции HF не выделяется, а ней-
трализуется гидроокисью алюминия:
12HF + 4А1(ОН)3 = 4A1F3 + 12Н2О
Образование в качестве промежуточного продукта кремнефто-
ристого алюминия завершается при 50—90° в течение 1—3 мин,
разложение же его в растворе и образование AIF3 идет в десятки
раз медленнее317.
Благодаря способности AIF3 образовывать стойкие пересыщен-
ные растворы из них возможно отделить SiO2 и выпустить его в
виде активного наполнителя. Для получения достаточно хорошо
фильтрующего осадка кремнегеля свободная кислотность пульпы
после завершения реакции должна быть ~5 г/л H2SiF6. Активность
кремнегеля возрастает с увеличением скорости введения гидрата
окиси алюминия и интенсивности перемешивания318. При перера-
ботке кислоты с концентрацией до 8% H2SiF6 может быть получен
аморфный кремнезем с малым содержанием кристаллической фазы,
имеющей удельную поверхность -~50 м2/г (БС-50) 319’ 32°. Получе-
ние такого кремнезема из кислоты большей концентрации затруд-
нительно. Содержание в кремнеземе до 0,3% фтора и до 15% ги-
дроокиси алюминия существенно не влияет на его усиливающие
свойства.
Раствор после отделения от него фильтрованием кремнегеля со-
держит до 14% A1F3 и остается пересыщенным. На холоду и без
затравки он не кристаллизуется в течение длительного времени.
Для кристаллизации A1F3-3H2O в раствор вводят затравку и пере-
мешивают при 80—90° в течение 4 ч. Активную затравку можно по-
лучать с помощью ультразвука 321>322. Первые кристаллы выделяю-
щегося фторида алюминия представляют собой твердый раствор
A1F3-3H2O и A1F3-3,5H2O лишь затем образуется устойчивый три-
гидрат 323. Его кристаллизация ускоряется с увеличением концен-
трации исходного раствора 324. Нагревая тригидрат до 500°, полу-
чают безводный фтористый алюминий. Выше 500° начинается
1162
Гл. XXXI. Соединения фтора
Рис. 338. Растворимость AIF3
в кремнефтористоводородной
кислоте при 25 и 50°.
заметный гидролиз AIF3 с потерей HF 325>326. Маточный раствор
после кристаллизации фтористого алюминия содержит 0,13—0,35%
H2SiFe и 1,94—2,35% A1F3 (рис. 338). Его можно использовать для
абсорбции фтористых газов суперфосфатного производства без
опасения кристаллизации фтористого алюминия. Разработан не-
прерывный способ производства фторида алюминия из кремне-
фтористоводородной кислоты и гидроокиси алюминия 327.
Для изготовления криолита раствор фтористого алюминия
(7,5—8,5%) смешивают с раствором NaF (3,5—4%), причем, если
эти растворы загрязнены кремнеземом, то их рекомендуется сна-
чала подкислить до' pH = 3,5; тогда
при их сливании будет кристаллизо-
ваться криолит, свободный от кремне-
зема328. Этот способ получения крио-
лита связан с образованием больших
количеств маточного раствора (из-за
малой растворимости NaF) и, поэто-
му, с большими потерями.
Лучшим вариантом является изго-
товление криолита из кристаллическо-
го фтористого натрия или его пульпы
и пересыщенного раствора фтористого
алюминия — при этом количество ма-
точного раствора резко сокращается,
а фильтруемость осадка улучшается; модуль получаемого продукта
близок к трем 45’ 329-332 Наибольшее влияние на фильтруемость
осадка оказывает величина pH раствора, которая должна быть в
пределах 2,8—5,0. Для получения кондиционного по содержанию
SiO2 криолита (ГОСТ 10561—63, стр. 1108) необходимо, чтобы рас-
твор фторида алюминия содержал не больше 4 г/л H2SiF6 333. Если
исходная кремнефтористоводородная кислота содержит примеси
соединений и фосфора и кальция, то фильтрующие свойства осадка
получаемого криолита ухудшаются по сравнению со свойствами
осадка, образующегося в присутствии лишь одного из этих элемен-
тов. Наибольшее замедление фильтрации наблюдается при содер-
жании в исходной кислоте по 0,3% Р20з и СаО. При охлаждении до
50° раствора A1F3, полученного из такой кислоты выделяется оса-
док Ca(AlF4)2-5,5H2O 334. Непрерывный способ получения криолита
из суспензии кристаллов фторида натрия и раствора фторида алю-
миния имеет наилучшие технико-экономические показатели 335’336.
АММИАЧНЫЙ СПОСОБ УЛАВЛИВАНИЯ И ПЕРЕРАБОТКИ
ФТОРИСТЫХ ГАЗОВ
Представляет интерес получение из отбросных фтористых газов
или из продуктов их улавливания фтористого аммония, легко пере-
рабатываемого в другие фтористые соединения 337’338.
Аммиачный способ улавливания и переработки фторгазов
1163
Растворами фторида аммония можно пользоваться для получе-
ния почти всех ныне применяемых фтористых солей, причем полу-
ченные аммиачным методом они содержат меньшее количество при-
месей. Этот метод позволяет избежать затраты соды, применяемой
для получения некоторых фтористых солей. Кроме того, при этом
методе не имеет места частичный выход фтора в виде относительно-
менее ценного кремнефтористого натрия, являющегося побочным
продуктом при получении фтористых солей сернокислотным разло-
жением плавикового шпата. Это позволяет с успехом перерабаты-
вать не только отходящие фтористые газы, но и низкосортный не-
обогащенный шпат, содержащий 40—50% CaF2, с предварительным
получением из него кремнефтористоводородной кислоты 339. Повы-
шенное содержание кремнезема в шпате при этом не только не
является нежелательным, но, наоборот, дает возможность увели-
чить выход побочного продукта — белой сажи.
При нейтрализации аммиаком или аммиачной водой кремнефто-
ристоводородной кислоты, полученной из отходящих фтористых га-
зов, а также неочищенной плавиковой кислоты, получающейся при
кислотной переработке плавикового шпата, идут следующие ре-
акции:
hf + nh3 = nh4f
H2SiFB + 2NH3 = (NH4)2SiFB
(NH4)2SiFB + 4NH3 + (zi + 2)H2O *= 6NH4F 4- SiO2 • лН2О
При непосредственном улавливании фтористых газов аммиачной
водой
SiF4 + 4NH4OH + aq = 4NH4F + SiO2 • nH2O
четырехфтористый кремний легко поглощается практически пол-
ностью. При абсорбции SiF4 6—10% раствором аммиака образую-
щаяся кремневая кислота хорошо отстаивается и сравнительно
легко отделяется от жидкости. Более слабые или боле₽ крепкие
растворы аммиака дают или хлопьевидный, трудно фильтрую-
щий, или студневидный осадок 340. Лучшим вариантом является
абсорбция фтористых газов 8—9%-ным раствором NH4F с после-
дующей нейтрализацией образующегося кислого раствора [18—20%,
(NH4)2SiF6 + 2% H2SiF6] аммиаком или аммиачной водой 257.
Выделившийся осадок аморфной кремневой кислоты после про-
мывки, сушки и разрыхления выпускается в виде белой сажи.
После отделения осадка кремневой кислоты остаются весьма чи-
стые растворы фторида аммония, которые и перерабатываются на
различные фтористые соли.
Фторид аммония хорошо растворим в воде. При нагревании су-
хой соли и ее растворов происходит диссоциация с выделением ам-
миака и образованием бифторида аммония согласно уравнению:
2NH4F = NH4F-HF + NH3
1164
Гл. XXXI. Соединения фтора
Кремнефторид аммония также хорошо растворим в воде и при
взаимодействии с аммиаком разлагается согласно уравнению реак-
ции, приведенному выше. Эта реакция обратима и подбором coot- j
ветствующих условий ее можно проводить практически до конца j
как в одном, так и в другом направлении. При 15—20° и небольшом j
избытке аммиака (4—7 г/л) получаются почти чистые растворы
фторида аммония, а кремнезем опять переходит в раствор.
Это позволяет перерабатывать на ценную белую сажу, обладаю-
щую хорошими адсорбционными свойствами, любой кремнеземи-
стый материал. Процесс сводится к обработке кварцевого песка и ।
других кремнеземистых материалов раствором фторида аммония 3
при нагревании или предварительно полученным из него бифтори- J
дом аммония. При этом выделяется аммиак. Процесс идет через ’
следующие промежуточные реакции:
2NH4F = NH4F.HF + NH3
4NH4F ..HF + SiO2 = 4NH4F + SiF4 + 2H2O
SiF4 + 2NH4F = (NH4)2SiF6
Рекомендуются 257 следующие условия производства. Кварцевый
лесок, просеянный через сито 0,5 мм, промывают водой, потом сла-
бой соляной кислотой (для удаления карбонатов и соединений
лолуторных окислов) и снова водой. Затем его обрабатывают при
80° и перемешивании раствором бифторида аммония с концентра-
цией 30% (лучше более 50%). Получающийся раствор кремнефто-
рида аммония отфильтровывают от неразложившегося остатка и
насыщают при 15—20° до слабо щелочной реакции аммиаком, вы-
делившимся в первой стадии процесса. При этом ЗЮг выделяется
•обратно в твердую фазу, но уже в виде геля, который после про-
мывки и сушки суспензии в распылительной сушилке обладает
ценными активными свойствами. Раствор фтористого аммония
вновь возвращается в цикл.
Переработка раствора фторида аммония на фтористые соли осу-
ществляется следующими способами.
Непосредственным выпариванием этого раствора до насыщен-
ного состояния и последующей кристаллизацией может быть полу-
чен твердый фторид аммония. Так как при выпаривании идет
частичная диссоциация NH4F с образованием кислой соли и удале-
нием аммиака, то выделяющийся аммиак улавливают и возвра-
щают в выпариваемый раствор к началу кристаллизации соли. Это
обеспечивает выделение в осадок средней соли, которую отделяют
и высушивают при 35—40°. Маточный раствор возвращают на вы-
паривание. При выпаривании раствора NH4F без возврата NH3 по-
лучается плав, состоящий из смеси NH4HF2 (70—75%) и NH4F
(16—20%), который при охлаждении на холодильных вальцах пре-
вращается в твердый чешуйчатый продукт — фторид-бифторид ам-
Аммиачный способ улавливания и переработки фторгазов
1163’
мония. Он сильно гигроскопичен, на воздухе расплывается. Аппа-
ратура для выпарки раствора NH4F изготовляется из стали ЭИ-448-
или из графитопласта.
Криолит можно получить, прибавляя к нагретому раствору
NH4F алюминат натрия, по реакции:
6NH4F + Al(ONa)3 = Na3AlF6 + 6NH3 + 3H2O
Выделяющийся аммиак улавливают и возвращают в цикл, я
криолит отфильтровывают от маточного раствора и сушат. Изменяя
модуль алюмината (т. е. отношение Na2O к А12О3),' можно получать
или криолит (3NaF-AlF3) или хиолит (5NaF-3AlF3).
Криолит может быть выделен и из кислого раствора NH4F
(pH ~ 5,5), который смешивают без нагревания с раствором алю-
миниевой и натриевой соли:
12NH4F + 3Na2SO4 + A12(SO4)3 = 2Na3AlF6 + 6(NH4)2SO4
Полученный криолит отфильтровывают, промывают и сушат.
Из раствора (NH4)2SO4 регенерируют аммиак281.
Фторид натрия можно осадить из растворов NH4F:
1) 2NH4F + Na2CO3 = 2NaF + 2NH3 + H2O + CO2
2) 2NH4F + Na2SO4 = 2NaF + (NH4)2SO4
3) NH4F +NaCl = NaF+ NH4C1
Наиболее прост первый вариант, так как в нем сразу регенери-
руется почти весь аммиак, который может быть возвращен в цикл.
Однако применение для осаждения NaF относительно более доро-
гой соды и получение трудно используемого маточного раствора,,
содержащего около 4% NaF, снижают ценность этого метода.
По второму варианту наряду с осадком NaF образуется раствор-
сульфата аммония, содержащий до 40 г/л NaF. Избыток сульфата
натрия не высаливает NaF, так как образуется двойная соль
Na2SO4 • 2NaF. Выделить оставшийся в растворе фтор можно в виде-
криолита добавлением к раствору сульфата алюминия:
12NaF + A12(SO4)3 = 2Na3AlF6 + 3Na2SO4
Это усложняет производство фторида натрия, но позволяет по-
лучать криолит.
Наиболее целесообразным является третий вариант, так как из-
бытком поваренной соли можно выделить из раствора почти весь
фторид натрия, выход которого из раствора достигает 95% (при
стехиометрическом количестве NaCl выход NaF 60%, при 25%-ном
избытке выход 70% и при 100%-ном избытке выход 96%). Выход.
NaF возрастает также с увеличением содержания аммиака в рас-
творе и достигает максимальной величины при 12% NH3. В этом:
случае и при 50% избытке NaCl выход NaF ~90% 34°. После от-
деления кристаллов NaF аммиак может быть регенерирован из;
1166
Гл. XXXI. Соединения фтора
маточного раствора хлористого аммония обычным способом — об-
работкой известковым молоком при нагревании.
Фторид алюминия может быть получен нагреванием раствора
фторида аммония с гидроокисью алюминия в течение 6—8 ч. При
этом осаждается крупнокристаллический, легко фильтрующий ам-
мониевый криолит (NH4)3A1F6 или 3NH4F-A1F3. Так как в газовую
фазу вследствие диссоциации NH4F удаляется аммиак и в растворе
образуется бифторид аммония, то реакция, видимо, проходит по
уравнению:
3NH4HF2 + А1(ОН)3 = (NH4)3A1FB + ЗН2О .
Осадок отфильтровывают и прокаливают при 450—500°. При
этой температуре происходит разложение комплексной соли с об-
разованием A1F3. Выделившиеся газы (NH3 4- HF) могут быть
уловлены и возвращены в производство.
Фторид алюминия получается при нагревании окиси алюминия
с бифторидом аммония выше 130° по реакции
А12О3 + 3NH4HF2 = 2A1F3 + 3NH3 + ЗН2О
которая идет в две стадии. Вначале с экзотермическим эффектом
происходит нейтрализация кислого фторида аммония окисью алю-
миния
А12О3 + 6NH4HF2 = 2AIF3 + 6NH4F + ЗН2О
причем в газовую фазу выделяется водяной пар и лишь небольшое
количество аммиака. При дальнейшем нагревании окись алюминия
реагирует с фторидом аммония:
А12О3 + 6NH4F = 2A1F3 + 6NH3 + ЗН2О
В качестве побочных промежуточных продуктов образуются ам-
мониевый криолит (NH4)3A1F6 и комплексная соль NH4A1F4, кото-
рые выше 440° полностью разлагаются. При этом в газовую фазу
удаляется и некоторое количество NH4F341.
Описан циклический способ производства A1F3 342. Вначале по-
лучают тетрафторборную кислоту HBF4 разложением плавикового
шпата раствором, содержащим 216 г/л H2SO4, ПО г/л Н3ВО3 и
50 г/л (NH4)2SO4 при нагревании в течение 6 ч. После охлаждения
и нейтрализации избытка серной кислоты известняком тетрафтор-
борную кислоту отфильтровывают от осадка сульфата кальция и
обрабатывают гидроокисью алюминия и аммиаком (или карбона-
том аммония); при этом выделяется в осадок двойная соль
NH4F-A1F3. Эту соль отделяют от раствора Н3ВО3, возвращаемого
в цикл, и нагревают в смеси с А12О3 или А1(ОН)3 до ~350° (по
другим данным80, до 450—500°); образуется A1F3 и аммиак, кото-
рый возвращают в процесс.
Аммиачный способ улавливания и переработки фторгазов
1167
В отличие от 3NH4F-A1F3, устойчивого до 100°, который выде-
ляется в осадок при введении раствора A1F3 в избыток раствора
NH4F, образование NH4F-A1Fs происходит при введении рассчитан-
ного количества NH3 в раствор А1(ОН)3 в плавиковой кислоте.
Двойная соль NH4F-A1F3 может быть получена и обработкой рас-
твора NH4F гидроокисью алюминия. Если к водной суспензии
NH4F-A1F3, нагретой до 90°, добавлять постепенно NaF, образуется
осадок ЫазА1Р6. Промытый криолит имеет примерный состав: 46%
A1F3, 53,6% NaF, 0,5% А12О3-ЗН2О 343.
Получаемые из отходящих фтористых газов фторид и бифторид
аммония могут служить полупродуктами для производства концен-
трированного фтористого водорода. Их можно разлагать серной
кислотой по реакциям:
nh4f + h2so4=nh4hso4 + hf
NH4HF2 + H2SO4 = NH4HSO4 + 2HF
По второй реакции расход аммиака и серной кислоты, а также
количество побочного продукта — бисульфата аммония — в 2 раза
меньше, чем по первой. При разложении кристаллического бифто-
рида аммония 93—98 %-ной серной кислотой при 200—250° выход
HF составляет 93—96%, а содержание фтора в бисульфате аммо-
ния 0,5—1% 344-345. Безводный HF выделяется из твердых фторида
или бифторида аммония при действии на них хлористым водоро-
дом; в твердой фазе остается хлорид аммония 346.
Большой интерес представляет термический метод, не требую-
щий расхода реагентов и не дающий побочных продуктов или от-
ходов. Он заключается в получении из фторидов аммония бифто-
рида натрия и его термическом разложении 347. Раствор фторида
аммония выпаривают, при этом получается бифторид аммония:
2NH4F = NH4HF2+NH3
Аммиак вновь используется для получения NH4F из фтористых
газов, а раствор бифторида аммония обрабатывают твердым NaF:
NH4HF2 + NaF = NaHF2 + NH4F
Маточный раствор NH4F возвращается на получение бифторидэ-
аммония, a NaHF2 разлагают при 300°;
NaHF2 = NaF+HF
Фторид натрия вновь используют для обработки NH4HF2 348.
Предложено также34э-350 подвергать термическому разложению
непосредственно NH4HF2. При этом вначале получают смесь HF и
NH3, затем ее пропускают через никелевый катализатор при
700—800° для разложения NH3 на N2 и Н2. Из полученной смеси
1 IF, N2 и Н2 конденсируют HF.
1168
Гл. XXXI. Соединения фтора
Аммиачный метод позволяет получать и осажденный, в част- fl
ности весьма дисперсный и пористый фторид кальция 351-352. При fl
нейтрализации полученной из отходящих фтористых газов (или fl
другим путем) кремнефтористоводородной кислоты молотым мелом fl
образуется осадок смеси CaF2 и SiO2: fl
HgSiFjj + ЗСаСО3 = 3CaF2 + SiO2 -F H2O 4- 3CO2 fl
Для освобождения от SiO2 влажный осадок обрабатывают рас- 1
плавом фторид-бифторида аммония: fl
SiO2 + 4NH4HF2 = (NH4)2SiF6 + 2NH4F + 2H2O I
Оставшийся CaF2 отфильтровывают, промывают и высушивают, fl
а маточный раствор усредняют аммиаком для перехода кремнефто-
рида во фторид аммония: fl
(NH4)2SiF6 + 4NH3 + 2Н2О - 6NH4F + SiO2 |
Выделившаяся двуокись кремния после промывки и сушки яв- 1
ляется побочным продуктом — белой сажей, а раствор NH4F и про- .1
мывная вода упариваются. При этом получается возвращаемый 1
л процесс плав фторид-бифторида аммония: 1
2NH4F = NH4HF2 + NH, I
Аммиак улавливается водой и также снова используется. В этом 1
процессе для получения легко отделяемого на фильтре осадка 1
•CaF2 + SiO2 следует добавлять кремнефтористоводородную кис- I
лоту к меловой пульпе, тогда производительность фильтра дости- 1
гает 500—700 кг/(м2-ч) сухого вещества. При обратном порядке Я
смешения реагентов она резко уменьшается [до 30 кг/(м2-ч)]. Она |
значительно уменьшается [до 180 кг/(м2-ч)] и при использовании 1
осажденного карбоната кальция. Степень обескремнивания осадка |
фторид-бифторидом аммония при 70° достигает 98%. Производи- |
тельность фильтрации обескремненного CaF2 45—50 кг/(м2-ч), но |
юна возрастает до 360 кг/(м2-ч) сухого вещества при иснользова-
нии для получения смеси CaF2 + SiO2 осажденного карбоната
кальция. 1
По другому варианту CaF2 можно изготовить по следующей 1
схеме. Отходящие фтористые газы улавливают оборотным раство- 1
ром NH4F, который затем нейтрализуют аммиаком. Осадок крем- j
невой кислоты отделяют, промывают, высушивают и выпускают j
л виде белой сажи, а раствор NH4F упаривают для получения j
плава бифторида и фторида аммония; выделяющийся при этом ам- ?
лшак возвращается в процесс. Плав вводится в меловую пульпу [
^концентрация которой 1 : 3) для осаждения CaF2: i
2NH4HF2 + СаСОз = CaF2 + 2NH4F + CO2 + H2O
Аммиачный способ улавливания и переработки фторгазов
116»
Осадок CaF2 отфильтровывают, промывают и высушивают. Рас-
твор NH4F возвращается в процесс; промывная вода идет на при-
готовление меловой пульпы. При использовании для осаждения.
CaF2 пульпы из осажденного карбоната кальция производитель-
ность фильтрации достигает 1300 кг/(м2-ч), а при работе с сепари-
рованным мелом — всего 100 кг/(м2 • ч). Содержание SiO2 в гото-
вом продукте 0,3—0,5%.
Вместо СаСОз для осаждения CaF2 из раствора NH4F можно-
использовать СаО, а также CaSO4. В последнем случае идет кон-
версия:
2NH4F + CaSO4 = CaF2 4- (NH4)2SO4
При использовании фосфогипса степень конверсии по фтору
достигает 95—97%, в продукте содержится 88—90% CaF2,
~6% CaSO4, 0,5—0,6% SiO2. Производительность фильтрация.
~ 150 кг/(м2- ч) сухого вещества; влажность осадка ~48%. Остаю-
щийся после отделения CaF2 раствор [20—22% (NH4)2SO4] может-
быть переработан в побочный продукт —сульфат аммония.
Фторид кальция может быть получен взаимодействием раство-
ров NH4F и СаС12. Раствор NH4F (~135 г/л) с небольшой ско-
ростью (в виде брызг) вводят в кипящий раствор СаС12 (60—
300 г/л), взятый с 5—25%-ным избытком по отношению к фториду
и предварительно подкисленный соляной кислотой до содержания
0,05—0,25% НС1. Осадок отфильтровывают и промывают на цен-
трифуге, сушат и размалывают образовавшиеся после сушки
комки 353.
Фторид кальция, полученный из раствора NH4F осаждением’
карбонатной суспензией состоит из плотных кристаллов размером
1,8—3,2 мк-, при осаждении окисью кальция и хлористым кальцием-
получаются пористые кристаллы с размером 0,01—0,1 мк 282.
Фторид магния также получают аммиачным способом 257-354.
Он заключается в обработке кремнефтористоводородной кислоты
при 70—75° окисью магния (каустическим магнезитом) и в после-
дующем отделении кремнегеля с помощью бифторида аммония..
При растворении MgO вначале образуется MgSiFe
MgO 4- H2SiFe = MgSiFe + H2O
который при дальнейшей нейтрализации гидролизуется:
3MgSiF6 4- 2Н2О = 3MgF2 4- SiO2 4- 2H2SiFg
В результате процесс идет по суммарной реакции:
3MgO 4- H2SiF6 = 3MgF2 4- SiO2 4- H2O
Для разделения твердых продуктов этой реакции — MgF2 ш
SiO2 — их обрабатывают при 70—75° раствором фторид-бифторида.
аммония. При этом кремнегель растворяется:
4NH4HF2 4- SiO2 = (NH4)2SiF6 4- 2NH4F 4- H2p
1170
Гл. XXXI. Соединения фтора
Присутствующая в растворе свободная H2SiFe также превра-
щается в кремнефторид аммония:
2H2SiF6 + 6NH4F + SiO2 = 3(NH4)2SiF6 + 2Н2О
Осадок MgF2 отделяют от раствора на фильтре, промывают,
репульпируют в воде и высушивают в распылительной сушилке.
При этом исключается последующий размол. Продукт содержит
83—85% MgF2 и 2—4% CaF2. Раствор, содержащий 16—18%
(NH4)2SiF6 вновь перерабатывают в фторид-бифторид аммония.
При получении MgF2 описанным аммиачным Способом двойные
фториды магния и аммония не образуются. Гидратированная соль
(NH4)2MgF4-2H2O получается при взаимодействии MgF2 с концен-
трированным раствором NH4F. Эта соль при 110° дегидратируется,
при 215° разлагается с выделением одной молекулы NH4F, а при
285° распадается на NH4F и MgF2. Безводные соли NH4MgF3 и
(NH4)2MgF4 можно получить сплавлением MgF4 и NH4F (при
180°) 355.
ЛИТЕРАТУРА
1. И. Г. Р ы с с, Химия фтора и его неорганических соединений, Госхимиздат,
1956. — 2. С. R. Schmitt, S. Н. Smiley, пат. США 2706676, 1955,—
3. A. G. Streng, А. V. Grosse, J. Am. Chem. Soc., 88, № 1, 169 (1966).—
4. J. J. Turne r, Endeavor, 27, № 100, 42 (1968). — 5. R. В 0 u g о n, M. Carles,
J. Aubert, Compt. rend., C265, № 23, 179 (1967). — 6. H. С. Николаев,
В. Ф. С у x о в e p x о в, Ю. Д. Шишков, И. Ф. Аленчикова, Химия галоид-
ных соединений фтора, Изд. «Наука», 1968. — 7. Е. U. F г а п с k, W. S р а 11 h о f f,
Z. phys. Chem., 8, № 3—4, 255 (1956). — 8. W. Strohmeier, G. Briegleb,
Z. Elektrochem., 57, № 8, 662 (1953). — 9. Дж. X. Саймонс, сб. «Фтор и его
соединения», т. I, ИЛ, 1953, стр. 192. —10. R. L. Jarry, W. Davis, J. phys.
Chem., 57, 600 (1953).
11. E. Darmvis, J. Chim. phys. et phys.-chim. biol., 53, № 5, 445 (1956).—
12. A. J. Ellis, J. Chem. Soc., 1963, № 9, 4300. —13. С. А. Вознесенский,
Химия фтора, ОНТИ, 1937. —14. Н. И. Хай дуков, 3. 3. Липецкая,
А. А. Богиоваров, ЖПХ, 9, № 3, 439 (1936). —15. Е. М. Полякова,
А. И. Зелинская, Труды УНИХИМ, в. 1, 1954, стр. 64. —16. Р. A. Mun ter,
О. Т. А е р 1 i, R. А. К о s s a t г, Ind. Eng. Chem., № 7, 1504 (1949). — 17. E. H. П и-
ц a e вс кая, Труды УНИХИМ, в. 1, 1954, стр. 75. —18. Е. М. Поляков а,
В. С. Ятлов, Там же, стр. 68. —19. К. Fredenhagen, М. Wellmann, 2.
phys. Chem., 162, 454 (1932). — 20. Р. A. Mun ter, О. Т. Aepli, R. A. Kos-
satz, Ind. Eng. Chem., 39, № 3, 427 (1947).
21. И. В. Тана наев, ЖОХ, И, № 4, 267 (1941); ЖПХ, 11, № 2, 214 (1938),
ИСФХА ИОНХ АН СССР, 14, 353, 364 (1941).— 22. J. F. Froning,
М. К. Richards, Т. W. Stricklin, S. G. Т u г п b и 11, Ind. Eng. Chem., 39,
№ 3, 275 (1947). —23. A. R. Miller, J. Phys. Chem., 71, № 4, 1144 (1967).—
24. И. А. Семерикова, А. Ф. Алабышев, ЖФХ, 34, № 6, 1341 (1962).—
25. Ю. И. Юсова, А. Ф. Алабышев, Там же, 37, № 8, 1870 (1963).—
26. Ю. Д. Некрасов, Э. М. Рычкова, М. М. Субботина, ЖНХ, 12, № 1,
192 (1967). —27. А. К. Жданов, ЖНХ, 1, № 9, 2024 (1956). — 28. S. Р. Buet-
tner, A. W. Jacke, Inorg. Chem., 2, 19 (1963). — 29. A. A. Woolf, J. Chem.
Soc., 1955, 433. — 30. H. А. Торопов, M. M. Сычев, 3. Д. Алексеева,
Труды ЛТИ им. Ленсовета, в. 34, 1955, стр. 23.
Литература
1171
31. С. Ruby, Compt. rend., С264, № 14, 1172 (1967). —32. J. L. Holm,
Acta chem. Scand., 19, № 3, 638 (1965). — 33. В. С. Ятлов, Е. М. Полякова,
ЖОХ, 15, 724 (1945).— 34. R. Brill, S. Zaromb, Nature, 173, № 4398, 316
(1954).—35. S. Zaromb, R. Brill, J. Chem. Phys., 24, № 4, 895 (1956)'.—
36. Л. M. Михеева, А. В. Новоселова, P. Биктимиров, ЖНХ, 1, Ns 3,
499 (1956). — 37. L. Hillert., Acta chem. Scand., 18, № 10, 2411 (1964).—
38. H. К о j i m a, S. G. W h i t e w a g, C. R. Masson, Canad. J. Chem., 46, Ns 18,
2968 (1968). — 39. M. M. Сычев, Труды ЛTH им. Ленсовета, в. 34, 1955,
стр. 29.—40. J. Berak, Pocz. Chem. № 1, 23, 69 (1961).
41. Н. С. Николаев, Н. А. Иванов, С. Г. К а л т ы п и н, ЖПХ, 9, № 7,
1183 (1936). — 42. Г. Н. Богачов, Ю. Г. Мясников, Цветные металлы,
№ 12, 31 (1967). — 43. G. J. Landon, A. R. Ubbelohde, Proc. Roy. Soc.,
A 240, № 1221, 160 (1957).—44. M. M. Be тюков, сб. «Физическая химия рас-
плавов солей и шлаков», Госметаллургиздат, 1962, стр. 77. — 45. И. В. Т а н а-
наев, Ю. Л. Лельчук, ДАН СССР, 41, Ns 3, 118 (1943); ЖАХ, 2, Ns 2, 93
(1947). — 46. Ю. А. Козлов, Н. В. Белова, И. А. Леонтьева, Г. Н. Бо-
гачов, сб. «Исследования в области химии и технологии минеральных солей и
окислов», Изд. «Наука», 1965, стр. 119.—47. В. С. Ятлов, Е. Н. П и н р е в-
ская, ЖОХ, 19, 24 (1949). — 48. В. С. Ятлов, Там же, 7, 2439 (1937).—
49. В. И. Михеев, Рентгенографический определитель минералов, Госгеолтех-
издат, 1957. — 50. А. В. Новоселова, ЖОХ, 10, 1547 (1940).
51. Ф. Т. Парамонов, А. Г. Амелин, Сборник аспирантских работ
НИУИФ, 1963, стр. 77, —52. Н. С. Николаев, ИСФХА АН СССР, 25, 375
(1954). —53. И. Г. Рысс, Н. П. Бакина, ДАН ССР, 2, 19 (1936).—
54. Л. Н. Архипова, С. Я- Шпунт, Труды НИУИФ, в. 208, 1965, стр. 88.—
55. Е. Baur, Вег., 36, 4209 (1903); Z. phys.’Chem., 48, 483 (1904).— 56. В. В. И л-
ларионов, 3. Г. Смирнова, К. П. Князева, ЖПХ, 36, № 2, 237 (1963). —
57. В. В. Илларионов, Т. И. Соколова, 3. Г. Кулагина, К. П. Кня-
зева, Сообщения о научно-технических работах НИУИФ, в. 2, 1957, стр. 48.—
58. A. L. Whynes, Trans. Inst. Chem. Eng., 34, 117 (1956). — 59. Г. И. Шиш-
кин, Г. Н. Богачов, Труды УНИХИМ, в. 17, 1968, стр. 104, 108.—50. М. Ч е-
пелевецкий, Ц. Бальц, ЖПХ, 10, № 7, 1183 (1937).
61. Л. И. Архипова, С. Я. Шпунт, Труды НИУИФ, в. 208, 1965,
стр. 69. — 52. Л. И. Архипова, С. Я. Шпунт, Там же, в. 208, 1965, стр. 55.—
63. Н. С. Miller, J. F. Gall, Ind. Eng. Chem., 42, Ns 11, 2223 (1950).—
64. F. Seel, H. Jonas, L. Riehl, J. Langer, Angew. Chem., 67, Ns 1, 32
(1955). —55. W. C. Schumb, Ind. Eng. Chem., 39, № 3, 421 (1947).—
56. К. E. Me К о r m a c k, W. G. Schneider, Canad. J. Chem., 29, 699, 845
(1951). — 57. H. C. Miller, L. S. W e r d e 11 i, J. F. Gall, Ind. Eng. Chem.,
43, 1126 (1951). —68. Riki, Technique, 28, № 5, 353 (1953). — 69. Г. H. Бога-
чов, Журн. ВХО, 7, № 1, 39 (1962). — 70. Я. М. П а у ш к и н, Химический состав
и свойства реактивных топлив, Изд. АН СССР, 1958.
71. М. Caron, Ind. chim., 55, № 617, 521 (1968). —72. Chem. Trade J., 119,
№ 3092, 217 (1946). — 73. Дж. X. С а й м о н с, сб. «Фтор и его соединения»,
т. 1, ИЛ, 1953, стр. 223, 336. — 74. Сб. «Химия фтора», под ред. И. Л. Кнунянца,
№ 1—3, ГИЛ, 1948. — 75. В. Михайлов, Хим. пром., № 9, 271 (1947).—
76. И. Л. Кнунянц, О. В. Кильдишева, Усп. хим., 15, № 6, 685 (1946).—
77. В. Бокемюллер, Органические соединения фтора, Оборонгиз, 1939.—
78. J. Н. Simons, Ind. Eng. Chem., 39, Ns 3, 238 (1947). — 79. Г. А. Я к о в к и н,
Хим. пром., № 7, 12 (1947). — 80. М. Г. Габриелов а, Хим. наука и пром., 2,
Ns 6, 722 (1957).
81. А. А. Берлин, Хим. пром., Ns 10, 21 (1947). —82. А. В. Топчиев,
Я. М. П а у ш к и н, С. В. 3 а в г о р од с к и й, Усп. хим., 21, № 4, 422 (1952). —
83. А. Н. Stuewl, Chem. Eng. News, 36, Ns 51, 34, 57 (1958). — 84. И. П. Со-
рокин, Ю. П. Кольцов, сб. «Исследования в области химии и технологии
1172
Гл. XXXI. Соединения фтора
минеральных солей и окислов». Изд. «Наука», 1965, стр. 115. — 85. С. С. Fin-
ger, F. Н. Reed, Chem. Ind., 64, № 1, 51 (1949). — 86. П. Беляев, Электроли-
тическое свинцевание, Госхимиздат, 1933. — 87. К. Gebauer, К. Sommer,
Metalloberflache, 1, № 2, 25 (1947). — 88. М. С. Липман, авт. свид. 69981,
1947,— 89. Е. Н. Пин а ев ск ая, 3. А. Чазова, Труды УНИХИМ, в. 1, 1954,
стр. 77. — 90. Chem. Ind., 18, № 1, 21 (1966).
91. В. А. Вейль, сб. «Фтор и его соединения», т. 1, ИЛ, 1953, стр. 475.—
92. В. Москвин, Новости техники, № 1, 33 (1940). — 93. Н. В. Надененко,
Фтор и его соединения, ОНТИ, 1935.—94. А. А. Чижик, Г. В. Уставщи-
ков а, Сб. статей к 25-летию ГИПХ, 1944, стр. 186. — 95. Пат. США 3294878,
1961; 3365271, 1966. — 96. Правила и нормы техники безопасности и промышлен-
ной санитарии для строительства и эксплуатации производства криолита, фто-
ристого алюминия и побочных продуктов, Госхимиздат, 1962. — 97. В. И. Ор-
лов, М. А. Морозова, М. Н. Крутицкая, Журн. ВХО, 5, № 3, 268
(I960).—98. А. Л. Ефимов, И. А. Каза с, Инсектициды и фуигициды, Сель-
хозгиз, 1940. — РР. С. Hempel, Chem. Eng. News, 27, № 34, 2420 (1949).—
100. H. M. Мельников, Хим. пром., № 6, 181 (1950).
101. Б. К- Фенюк и др., Грызуны и борьба с ними, в. 4, Саратов, 1955,
стр. 157. — 102. А. Е. Ф е р с м а и, Геохимия, т. 1, Госхимиздат, 1933, стр. 146,
150. —103. Е. Н. Егорова, Бюлл. Ин-та галургии, № 4—5, 25 (1940).—
104. Ю. В. Морачевский, Труды ГИПХ, в. 37, 1946, стр. 120. —105. Г. Л. К о-
реиьков, Н. А. Устинова, Гориохимическое сырье зарубежных стран, Изд.
«Химия», 1965, гл. 9. —106. А. И. Шерешевский, Плавиковый шпат (флюо-
рит). Требования промышленности к качеству минерального сырья, в. 8, Госгеол-
техиздат, 1960. — 107. Жури. ВХО, 5, № 1, 85, 1960. —108. БНТИ М-ва геол.
СССР, сер. «Изуч. веществ состава минер, сырья и технол. обогащ. руд», № 6,
1968. —109. W. К. Finn, Z. Erzbergbau u. Metallhiittenwesen, 6, № 1, 13
(1953). — ПО. К- И. Филиппова, В. А. Сине г рибо в, В. И. Бочкарев,
Хим. пром., № 7, 520 (1967).
111. L. Stragistti, Ind. mineraria, 5, № 8, 451 (1954). —7/2. Chem. Pro-
cess,, 11, № 12, 6 (1965). —113. W. H. McIntire, J. Assoc. Offic. Agr. Chem.,
38, № 4, 913 (1955). — 114. G. E. E n g e 1 so n, R. N. S e с о r d, франц, пат. 107485,
1954; мексик. пат. 55239, 1955; пат. США 2631083, 1953. — 115. G. L. Cabot,
австрал. пат. 164397, 1955. —116. Англ. пат. 728095, 1955. —117. Д. X. Кэди,
сб. «Фтор и его соединения», т. 1, ИЛ, 1953, стр. 251. —118. J. М. Siegmund,
Chem. Eng. Progr., 63, № 6, 88 (1967). —119. H. П. Галкин, А. Б. Крути-
ков, Технология фтора, Атомиздат, 1968. —120. А. А. Чижик, Труды ГИПХ,
в. 37, 1946, стр. 87.
121. R. Murray, S. Osborne, М. Kircher, Ind. Eng, Chem., 39, № 3,
249 (1947). —122. К. Matiasovsky, M. Gregor, Chem. prumysl., 9, № 8,
393 (1959). —123. G. H. Cady, D. A. Rogers, C. A. Carlson, Ind. Eng.
Chem., 34, 4, 443 (1942). —124. R. Downing, A. Benning, F. Downing,
R. McHorness, M. Richards, T. Tomkowit, ibid., 39, № 3, 259 (1947). —
125. R. Fowler, W. Burford, H. Anderson, J. Hamilton, C. Weber,
ibid., 39, № 3, 266 (1947). —126. W. Schamb, R. Young, K- Radimer, ibid.,
39, № 3, 244 (1947). —/27. Y. Gall, H. Miller, ibid., 262. —/28. I. T. Pink-
ston, ibid., 255. — 129. S. Osborne, M. Brau degee, ibid., 273. — 130. J. F r o-
ning, M. Richards, T. Stricklin g, S. Turnbull, ibid., 39, № 3, 275
(1947).
131. W. Bockemiiller, Angew. Chem., 53, 37/38, 419 (1940). — 132. H. War-
tenberg, Z. anorg. Chem., 242, H. 4, 406 (1939). —/33. H. Priest, A. Gros-
se, Ind. Eng. Chem., 39, № 3, 28 (1947). —/34. R. Landau, R. Rosen, ibid
281, —/35. Chem. Eng., 66, № 19, 78, 80 (1959). — 136. R. Landau, R. Rosen
Ind. Eng. Chem., 40, № 8, 1389 (1948). —/37. С. А. Амирова, С. В. Остров-
ский, Тезисы докладов VI Всесоюзной конференции по технологии иеорганиче-
Литература
1173
ских веществ и минеральных удобрений, Тбилиси, 1968, стр. 222. —138. Г. Н. Б о-
г а ч о в, С. Ю. Г у з ь. Производство криолита, фтористого алюминия и фтори-
стого натрия, Госметаллургиздат, 1940. —139. С. Ю. Гузь, Р. Г. Баранов-
ская, Производство криолита, фтористого алюминия и фтористого натрия. Изд.
«Металлургия», 1964. — 140. Э. И. Антоновская, Ю. Н. В и л ь к. Хим. пром.,
№ 5, 429 (1960).
141. Г. Н. Богачов, Н. П. Б а к и и а, Труды УНИХИМ, в. 7, 1958,
стр. 108. —142. К. Sch.aupp, G. Broja, пат. ФРГ, 1176628, 1962.—
143. А. В. Кириченко, Труды конференции по усовершенствованию техноло-
гии хромовых и фтористых солей, Госхимиздат, 1959, стр. 66. —144. А. П. Ма-
лова, Там же, стр. 71, — 145. I. Duldner, С. Trabaika, Rev. chim. (рум.),
15, № 11, 668 (1964). —146. Б. П. Зверев, А. Л. Г о л ьд и и о в, А. В. С у ш и и-
цева, А. А. Ш а б а л и и, авт. свид. 167833, 1963. — 147. Фраиц. пат. 1406063,
1964. —148. М. А. Скребков и др., авт. свид. 176263, 1963. —149. Бельг, пат.
634648, 1963, — 150. Франц, пат. 1372500, 1963; пат. США, 3278265, 1965.
151. S. С. Carson, аигл. пат. 1050374, 1965. —152. W. Faber, пат. ФРГ
939867, 1956. —153. М. А. Михайлов, сб. «Материалы по исследованию хими-
ческого сырья Дальнего Востока», Изд. АН СССР, 1958, стр. 61; Реф. докл.
VIII Менделеевского съезда, № 1, 1959, стр. 152. —154. D. Р. Burkhardt,
пат. США 3207579, 1961. —155. А. Л. Климов и др., авт. свид. 178796, 1965,—
156. Г. Н. Богачов, Н. П. Голубчеико, Труды УНИХИМ, в. 5, 1957,
стр. 288. —157. Н. П. Г а л к и и, В. А. Шубин, А. С. Крылов, Хим. пром.
№ 3, 190 (1963). — 158. Англ. пат. 953943, 1962, — 159. Австр. пат. 236340, 1963.—
160. Пат. ФРГ 1182212, 1962.
161. Швейц, пат. 417546, 1963. —162. Я. Я- Лаукевиц и др., сб. «Иссле-
дования по химии и технологии удобрений, пестицидов, солей», Изд. «Наука»,
1966, стр. 256. —163. Л. А. Нега нов а, Г. Н. Богачов, Труды УНИХИМ,
в. 17, 1968, стр. 3. —164. Пат. США 3256062, 1962, —165. Швейц, пат. 391671,
1961. — 166. Г. Н. Богачов, Г. М. Б о к а с т о в, Ю. А. Козлов, Хим. пром.,
№ 1, 59 (1968). — 167. А. А. Чижик, Е. А. Алябииа, Труды ГИПХ, в. 37,
1946, стр. 71, — 168. Чехосл. пат. 116456, 1946, — 169. Пат. США 3160473, 1961,—
170. Пат. ФРГ 1171879, 1959.
171. Chem. Eng., 66, № 19, 82, 84 (1959). —/72. Ind. Chem., 24, № 287, 801
(1948). —/73. W. A. Shire, пат. США 2661319. —174. J., E. Friden,
W A. Shire, пат. США 2661266, 1953. —/75. W. F. Mitchel, J. A. Grant-
Mackey, пат. США 2702233, 1955. —/75. Фраиц. пат. 1167191, 1958.—
177. Н. П. Галкин, В. А. Шубин, А. С. Крылов, Хим. пром., № 10, 750
(1962). —178. Н. П. Галкин, В. А. Шубин, А. С. Крылов, А. Д. Сенатов,
Там же, № 9, 686 (1963). —179. W. Н. Hardwick, Р. F. Wace, Chem. Progr.
Eng., 46, № 6, 283 (1965). — 180. Пат. США 3271273, 1963.
181. Франц, пат. 1477742, 1966. —182. С. С. Коровин, Ю. И. Кольцов,
А. М. Резник, И. А. Апраксин, ЖНХ, 11, № 1, 180 (1966). —183. А. П.Лы-j
сенко, В. Г. Плюсиии, Г. Н. Якунина, М. И. Зеленцова, авт. свид.1-
194063, 1965, — 184. Пат. США 3140152, 1962,— 185. Англ. пат. 1050405, 1964.—,
186. Пат. США 3157469, 1962. —/87. Фраиц. пат. 1350760, 1962. —/88. А. И. Бе-,
л я ев, Металлургия легких металлов, Госметаллургиздат, 1954. — 189. Фраиц?
пат. 1108320, 1956. — 190. I. Manet oshi, япои. пат, 1065, 1953.
191. В. Semmens, А. В. Meggy, J. Appl. Chem., 16, № 4, 122, 125
(1966). —192. G. Singh, В. С. К a r, NML Techn. J., 7, № 3, 21 (1965).—
193. H. А. А и т о in к и и а, А. А. В о л ь б е р г, Г. Н. Богачов, А. С. Л у ч в и н.
Труды УНИХИМ, в. 17, 1968, стр. 63. —194. J. К. Bradley, Chem. Ind., 32,
1027 (I960). — 195. Австр. пат. А1254, I960,— 196. И. И. Ш и ш к о, Ю. Г. М я с-
ников, В. Ф. Волков, В. А. Веселков, Хим. пром., № 6, 465 (1967).—
197, A. Harder, R. Shulze, пат. ГДР 4701, 1953. —198. Аигл. пат. 735156,
1174
Гл. XXXI. Соединения фтора
1955; австрал. пат. 164900, 1955. — 199. A. J. Edwards, канад. пат. 505199,
1954. — 200. Япон. пат. 28567, 1961.
201. М. М. С е и я в и н, Хим. .наука и пром., 2, № 6, 754 (1957).—
202. Г. Н. Б о г а ч о в, Г. А. Л о п а т к и н а, М. А. Грозных, авт. свид. 192187,
1963. — 203. Н. П. Сажин, ЖХП, № 3, 36 (1930). — 204. Ф. Н. Строков,
Труды ВАМИ, т. 20, 1940, стр. 93. — 205. П. П. Будников, С. Г. Т р е с в я т-
с к и й, ДАН СССР, 89, № 3, 479 (1953). — 206. К. Е. М а и о й л о в, М. Н. С м и р-
н о в, Труды ВАМИ, т. 22, 1940, стр. 98, 109, 120. — 207. Г. В. Лабутин,
Н. А. Иванов, Г. С. Морозов, Там же, 20, 1940, стр. 80, 103.—
208. С. И. Вольфкович, Т. И. Соколова, 3. Г. Кулагина-Смирно-
ва, К- П. Князева, ЖПХ, 31, № 7, 970 (1958). — 209. Р. Li-Chi, V. Wen-
Hsing, J. Chem. Eng. China, 4, 261 (1938).—210. M. E. Позин, Б. А. Копы-
лев, P. Ю. 3 и н ю к, Изв. вузов. Хим. и хим. технол., 61, № 1, 98 (1963).
211. Р. Ю. 3 и н ю к, Б. А. Копылев, М. Е. Позин, Хим. пром., № 1, 37
(1967). — 212. L. D. Н а п d, J. М. Р о 11 s, А. V. S t а с k, J. Agr. Food Chem., 11,
44 (1963). — 213. E. Hollander, P. Kerenyi, Chem priimysl, № 8, 478
(1966). — 214. M. E. Позин, Б. А. Копылев, P. Ю. 3 и н ю к, ЖПХ, 37, № 1,
9 (1964). — 2/5. М. Л. Чепелевецкий, Е. Б. Бруцкус, Суперфосфат, Гос-
химиздат, 1958, гл. X. — 216. М. Г. Г а б р и е л о в а, А. Н. Семенов, Э. Я. П а-
рылис, В. Г. Никиташ, Хим. пром., № 12, 924 (1965). — 217. S. Atkin,
Е. Р е 1 i 11 i, P. A. Vila, J. H e g e d u s, Ind. Eng. Chem., 53, № 9, 705 (1961). —
218. И. H. Кузьминых, E. Л. Яхонтова, E. И. Ермакова, Научно-техн,
информ, бюлл. НИУИФ, № 9, 22 (1958). — 219. А. И. Шерешевский,
Л. С. Горицкая, Л. Н. Потапова, Сообщения о научно-технологических
работах НИУИФ, Обмен опытом, № 2, 1957, стр. 26.—220. С. И. Вольфко-
в й ч„ Н. Н. Постников, А. А. Ионасс, Р. Е. Ремен, Там же, стр. 35.
221. И. М. Богуславский, Л. Д. Киоссе, Там же, стр. 54.—
222. М. Plusje, канад. пат. 511785, 1955. — 223. С. A. Hollingsworth, пат.
США 2726928, 1955, — 224. R. Donald, Chem. Trade J., 133, № 3467 (1953).—
225. V. Charrin, Chim. e. ind., 72, № 3, 524 (1954).—226. K- A. Sherwin,
Chem. Ind., № 41, 1274 (1956).—227. A. L. Whynes, Trans. Inst. Chem. Eng.,
34, № 2, 117 (1956).—228. И. M. Богуславский, В. M. P а м м, В. И. Мэ-
тр о з о в, Сообщения о научно-технических работах НИУИФ, Обмен опытом,
№ 6—7, 1958, стр. 10.—229. М. Г. Габриелов а, М. А. Морозова, Произ-
водство неорганических ядохимикатов, Изд. «Химия», 1964. — 230. К. Е. Lunde,
Ind. Eng. Chem., 50, № 3, 293 (1958).
231. M. Л. Вармамов, Е. Л. Кричевская и др., ЖПХ, 34, № 1, 78
(1961); Научи, зап. Одесск. политехи, ин-та, 40, 62 (1962).—232. Chem. Age,
90, № 2306, 401 (1963).—233. К- A. Cherwin, Ind. chim., 41, № 438, 7 (1954).—
234. Ibid., 52, № 580, 321 (1965). —235. И. M. Богуславский, В. М. Р а м м,
Сообщения о научно-технических работах НИУИФ, Обмен опытом, № 6—7,
1958, стр. 41. — 236. Я- В. Шварцштейн, В. М. Р а м м, Там же, стр. 55.—
237. В. И. Матрозов, В. М. Р а м м, Г. М. Юсова, Там же, стр. 102, 168. —
238. Agr. Chem., 15, № 4, 70 (I960). — 239. И. Н. Кузьминых, Е. Л. Яхон-
това, Е, И. Ермакова, Научио-техн. информ, бюлл. НИУИФ, № 9, 1958,
стр. 22.—240. И. М. Богуславский, В. М. Рамм, Сообщения о научно-
технических работах НИУИФ, Обмен опытом, № 2, 1957, стр. 49.
241. В. М Р а м м, 3. В. Чагина, Е. К. Ревазов, сб. «Исследования по
химии и технологии удобрений, пестицидов, солей», Изд. «Наука», 1966, стр. 225.—
242. А'. Н. Терновская, Е. К. Ревазов, Е. П. Кольцова, Там же,
стр. 218.—243. A. W. К i е 1 b а с k, Chem. Eng. Progr. Symp. Ser., 57, № 35, 51
(1961).—244. Австр. пат. 258250, 1965. — 245. E. В Уваров, Б. А. Копылов,
Хим. пром., № 1, 44 (1968). — 246. А. И. Рейбман, М. И. Финкельштейн,
Там же, № 8, 493 (1956).—247. К. Г. Бергман, И. И. Бульчин, Мате-
риалы по обмену опытом НИУИФ, № 10, 45 (1955). — 248. К. Г. Бергман,
Литература
1175
Б. И. Леви, Сообщения о научно-технических работах НИУИФ, Обмен опытом,
№ 3, 1957, стр. 4.—249. И. М. Богуславский, Хим. пром., № 6, 18 (1948);
№ 16 292 (1949). — 250. А. Д. Соколова, А. 3. Брук, Материалы по обмену
опытом НИУИФ, № 8—9, 30 (1955).
251. S. Ambrus, J. Szamosi, Maguar Kern, lapja, 20, № 7—8, 436
(1965). — 252. К. А. Гар, H. E. Пестов, Хим. пром., № 5, 15 (1946).—
253. И. Я. Завьялов, Там же, № 6, 17 (1948)—254. Р. Jaravete, Rev.
chim. (рум.), 15, № 12, 731 (1964). — 255. W. L. Hill, К. D. Jacob, Mining
Eng., Sec. 1, 6, № 10, 994 (1954). — 256. С. И. Вольфкович, P. E. Ремен,
Сообщения о научно.-технических работах НИУИФ, Обмен опытом, № 2, 1957,
стр. 52.-257. И. М. Богуславский, И. Н. Усачева, Техн, ииформ.
НИУИФ, в. 1, 1958, стр. 4. — 258. М. Г. Габриелов а, Сообщения о научно-
технических работах НИУИФ, Обмен опытом, № 2, 1957, стр. 53. — 259. М. Г. Г а-
бриелова, В. Ф. Левин, О. П. Субботина, Хим. пром., № 6, 417 (1963).—
260. И. П. Ху до л ей, Использование отходов суперфосфатного производства,
Киев, 1968.
261. М. Г. Габриел ов а, В. Ф. Левин, Хим. пром., № 6, 433 (1961).—
262. Н. И. Смирдова, А. В. Скоричеико, С. С. Иванчев, Хим. пром.,
№ 3, 229 (1967). — 263. Н. А. Бердышева, Ю. Н. Тюрнн, М. А. Агеева,
ЖПХ, 40, № 2, 429 (1967). — 264. М. Н. Набиев, Ю. И. Шакиров, ДАН
УзбССР, № 8, 25 (1954).— 265. М. Н. Набиев, Азотнокислотное разложение
фосфатов АН УзбССР, Ташкент, 1957. — 266. Б. И. П о л я к о в, М. Н. Набиев,
В. М. Беглов, Узб. хим. ж., № 4, 12(1966); ДАН УзбССР, № 10, 30 (1966).—
267. Чэнь Цзы-цаюнь, Ян Чжи-Юаиь, Линь Каи-фу, Хуасиэ синцзе,
№ 4, 148 (1955). — 268. И. М. Богуславский, С. Б. Казакова, Экспресс-
информация НИУИФ, № 4, 4 (1958). — 269. М. А. Ко лен ко в а, А. И. Лай-
нер, А. Г. Мотина, Изв. вузов. Хим. и хим. технол., 5, № 1, 115 (1962).—
270. Т В. Островская, С. А. Амирова, Изв. вузов. Хим. и хим. технол.,
10, № 4, 371 (1967).
271. М. Г. Габриелов а, сб. «Исследования по прикладной химии», Изд.
АН СССР, 1955, стр. 244. — 272. Г. А. Лопаткина, Т. Н. Колосова,
М. А. Грозных, ЖПХ, 41, № 7, 1608 (1968). —273. Швейц, пат. 289677,
1953. — 274. J. Kaptanoglu, Demir ve Celik, 2, № 8, 182 (1953). —275. W. Ko-
walski, O. Komi nek, R. John, Przem. Chem., 44, № 2, 71 (1965).—
276. И. И. Заринг, ЖХП, № 6, 26 (1940). —277. В. Щербакова, ЖПХ, 10,
№ 8, 1402 (1937). — 278. Е. Д. Файнберг, О. Е. Эдельштейн, Хим. пром.,
№ 2, 24 (1959). — 279. Г. Н. Богачов, Н. П. Г о л у б ч е н ко. Труды УНИХИМ,
в. 5, 1957, стр. 288,—280. Пат. США 3218124—3218129, 1962; пат. США 3257167, 1963.
281. А. Г. Павлович-Волковыский, С. X. Зариицкий, Вести,
техн, и эконом, информ. НИИТЭХИМ, № 21, 21 (I960). —282. G. Tarbutton,
Т. D. Farr, Т. М. Jones, Н. Т. Lewis, Ind. Eng. Chem., 50, № 10, 1525
(1958).—283. Г. H. Богачов, Труды конференции по усовершенствованию тех-
нологии производства хромовых и фтористых солей, 1959, стр. 73. —
284. М. Г. Г а б р и е л о в а, О. П. Субботина, Н. Д. Томилова, сб. «Ис-
следования по химии и технологии удобрений, пестицидов, солей», Изд. «Наука»,
1966, стр. 246. — 285. С. И. Вольфкович, Труды НИУ, в. 55, 1928; Мнн. сырье.
4 (1929). — 286. С. И. Вольфкович, М. Г. Габриелов а, Хим. пром., № 3,
72 (1951). — 287. Е. М. Малышева, М. Г. Г а б р и е л о в а, Научио-техн.
информ, бюлл. НИУИФ, № 5-р, 39 (1956).—288. J. Lobo, V. V. D a d а р е, Ind.
J. Technol., 4, № 10, 307 (1966). — 289. И. М. Б о г у с л а в с к и й, М. Г. Г а б р и е-
л о в а, сб. «Исследования по производству минеральных удобрений», Госхимиз-
дат, 1957, стр. 109. — 290. Г. А. Лопаткина, Труды УНИХИМ, в. 8, 1959,
стр. 62.
291. М. Г. Габриелов а, Р. Е. Ремен, Сообщения о научно-исследова-
тельских работах НИУИФ, Обмен опытом, № 2, 50 (1957). — 292. Г. Я. Елагин,
13 М. Е. Позин
1176
Гл. XXXI. Соединения фтора
Б. С. Кавнатская, Хим. пром., № 3, 166 (1956). — 293. Г. Я. Елагин,
Б С. Кавнатская, П. М. Школьник, Научно-техн, информ, бюлл. НИУИФ,
№ 5-р, 45 (1956). — 294. А. С. Коробицын, Г. Н. Богачов, Д. Ф. Ризе,
Труды УНИХИМ, в. 17, 1968, стр. 112,’ 118; ЖПХ, 42, № 3, 700 (1969).—
295. Г. А. Лопатина, Г. Н. Богачов, Н. С. В о л е й к о, ЖПХ, 35, № 10,
2180 (1962). — 296. Л. И. Аг а рев и др., Труды УНИХИМ, в. 7, 1959, стр. 111.— -
297. Д. ф. Ризе, А. С. Коробицын, Хим. пром., № 2, 127 (1969).—
298. W. Siegel, пат. США 1581819, 1925; англ. пат. 263623, 1926 и 554126,
1942. —299. Chem. Trade J., 113, № 2936, 197 (1943).— 300. Г. А. Блох,
И. Е. Неймар к, П. И. Балабкин, М. И. Пржебыльский, Хим. пром.,
№ 7, 209 (1953).
301. А. А. Соколовский, И. М. Богуславский, Т. И. Кузнецо-
в а, Труды НИИХП, в. 2, 1955. — 302. А. А. Соколовский, Автореф. докт.
дисс., ЛТИ им. Ленсовета, 1955.—303. Пат. ФРГ 1133347, 1959.—
304. С. И. Вольфкович, М. Г. Габриелов а, ЖПХ, 25, № 4, 345 (1952). —
305. М. Г. Габриелов а, Сообщения о научно-технических работах НИУИФ,
Обмен опытом, № 2, 1957, стр. 51. — 306. И. М. Богуславский, С. Б. Каза-
кова, Н. А. Усачева, Там же, стр. 45. — 307. С. X. Зарницкий,
А. Г. Павлович-Волковыский, Вести, техн, и эконом, информ.
НИИТЭХИМ, № 10, 23 (I960).— 308. М. Б. 3 е л и к и н, А. Я. Бакулина,
Труды НИОХИМ, т. 10, 1957, стр. 111.—309. И. Л. Гофман, Хим. пром., № 7,
436 (1956). — 310. Т. Р. High nett, М. R. Siegel, Ind. Eng. Chem., 41, № 11,
2493 (1949).
311. J. H. Walthall, Fertilizers Technology and Resources in US, N. Y.,
1953, p. 205. — 312. N. Gilbert, T. A. Hobbs, W. D. Sandberg, Chem. Eng.
Progr., 49, № 3, 120 (1953).— 313. B. Zawadzki, J. Szaufer, Przem. Chem.,
38, № 5, 304 (1959). — 314. M. H. С м и p н о в, Труды ВАМИ, в. 22, 1940, стр. 92.—
315. 3. А Ч а з о в а, А. С. Королева, Рефераты докладов на VIII Менделеев-
ском съезде, № 1, 1959, стр. 153. — 316. М. Т. Серебренникова, А. Н. Чет-
верикова, Труды УНИХИМ, в. 8, 1959, стр. 58.—317. Л Д. С к р ы л е в,
ЖПХ, 39, № 1, 58 (1966). — 318. Л. Д. Скрылев, Там же, 41, № 1, 3 (1968).—
319. Л. Д. Скрылев, Там же, 39, № 2, 305 (1966).—320. Г. Н. Богачов,
3. Н. Чазова, Труды УНИХИМ, в. 17, 1968, стр. 45.
321. Ю. Н. Тюрин, ЖПХ, 38, № 5, 1126 (1965). —322. Н. А. Бердыше-
ва, Ю. Н. Т ю р и и, В. С. Агеева, Труды УНИХИМ, в. 17, 1968, стр. 19.—
323. А. У. Антоневич, Я. Е. Вильняхский, И. А. Юдин, Там же,
стр. 24. — 324. Е. Н. Пинаевская, Г. М. Бокасто в, Там же, стр. 13 —
325. A. S с h m i d t, Chem. Ing. Techn., 39, № 9—10, 521 (1967). —326. Ю. Г. M я c-
ников, Г. H. Богачов, Труды УНИХИМ, в. 17, 1968, стр. 27. — 327. Г. Н. Б о-
г а ч о в, 3. А. Чазова, Н. П. О к у н ц о в а, Там же, стр. 32, 39. — 328. L. N о-
tarbartolo, канад. пат. 505661, 1954. — 329. Ю. А. Козлов, ЖПХ, 37, № 3,
470 (1964). — 330. Ю. А. Козлов, Г. Н. Богачов, Там же, 39, № 5, 1183
(1966).
331. Пат. ГДР 41603, 1964. —332. Ю. А. К о з л о в, Г. Н. Б о г а ч о в,. Труды
УНИХИМ, в. 17, 1968, стр. 71.—333. Ю. А. Козлов, А. М. Загудаев.
Г. Н. Богачов, Там же, стр. 77, — 334. И. Н. Горшкова, 3. А. Чазова,
3. И. Шишкина, Г. Н. Богачов, Там же, стр. 82.—335. Г. Н. Богачов
и др., Там же, стр. 92. —336. Л. А. Н е г а н о в а, С. П, П р о к у ш е в а, Там же,
стр. 96.—337. А. А. Чнжик, Сборник статей к 20-летию ГИПХ, 1939, стр. 56,—
338. F. G. Heil, R. D. J о u n g, J. J. S t и m р е, J. Agr. a. Food Chem., 9, № 6,
457 (1961). — 339. W. Augustyn, Przem. Chem., 38, № 3, 137 (1959). — 340.
А. И. П о з и г у н, Труды Одесск. ин-та, сб. хим. фак., т. 3, 1953, стр. 107.
341. W. Augustyn, L. Firlus, К- Frackowiak, Przem. Chem., 40, № 5,
225 (1961).—342. H. W. Heiser, F. C. F г а г у, канад. пат. 494180, 1953.—
343. Пат. ГДР 3396, 1954. —344. Ю. Д. Некрасов, С. С. Марков, Труды
Литература
1177
ГИПХ, в. 40, 1960, стр. 324. — 345. И. М. Богуславский, С. И. Вольфко-
вич, С. Б. Казаков, Н. С. Богданова, Хим. пром., № 7, 450 (1961).—
346. Н. С. Николаев, Е. Г. Ипполитов, ЖНХ, 6, № 4, 1001 (1961).—
347. Ю. Д. Некрасов, С. С. Марков, Труды ГИПХ, в. 46, 1960, стр. 309,
315. — 348. П. П. Белова, Ю. Д. Некрасов, ЖНХ, 11, № 7, 1723 (1966). —
349. Пат. США 3316060, 1964. — 350. Н. П. Г а л ь и н, В. А. Ш у б и н, А. С. К р ы-
л о в, А. Д. Сенатов, Хим. пром., № 10, 752 (1963).
351. И. М. Богуславский, С. Б. Казакова, М. М. Вии ник,
Н. С. Богданова, сб. «Исследования по химии и технологии удобрений, пе-
стицидов, солей», Изд. «Наука», 1966, стр. 231. — 352. М. Г. Габриелова,
Журн. ВХО, 6, № 3, 352 (1961). — 353. J. F. R о s s, Н. W. S 1 о у е г, пат.
США 2653857, 1953; швед. пат. 140783, 1953; пат. ФРГ 925344, 1955.-
354. М. Г. Габриелова, Е. А. Малышева. И. П. Хд/ долей, Д. Н. Шев-
ченко, Б. К. Черный, Хим. пром., № 12, 914 (1967). — 355. Р. Ch аг pin,
N. Roux, J. Ehrets matin, Compt. rend., C267, № 6, 484 (1968).
13'
Глава XXXII
СОЛИ АЗОТНОЙ кислоты
НИТРАТ АММОНИЯ
Физико-химические свойства
Нитрат аммония, или аммиачная селитра, NH4NO3, белое кри-
сталлическое вещество 1-6, содержащее 35% азота в аммиачной и
в нитратной формах. Нитрат аммония плавится при 169,6°; его
теплота плавления 16,2 ккал!кг, теплоемкость при 20—28°
0,422 ккал/(кг-град).
Растворимость нитрата аммония показана на рис. 339, а темпе-
ратурные интервалы стабильности разных кристаллических форм —
в табл. 88 7.
ТАБЛИЦА 88
Кристаллические формы аммиачной селитры
Условные обозначения крист аллических форм Системы Температурные интервалы стабильного существования, °C Плотность, Теплота превраще- ния, кал/г
I Кубическая 169,6-125,2 16,75
II Тригональная(«-ромбо- эдрическая) .... 125,2 - 84,2 1,69 12,24
III Моноклинная (р-ромбо- эдрическая) .... 84,2- 32,3 1,66 4,17
IV Ромбическая От 32,3 до -16,9 1,726 4,9Э
V Тетрагональная .... Ниже —16,9 1,725 1,6
Обнаружено существование точки перехода также в интервале
44—57°5. Превращения одной кристаллической формы аммиачной
Нитрат аммония
1179
селитры в другую часто сопровождаются явлениями переохлажде-
ния или перегрева. Например, при быстром охлаждении от 125,2°
до температуры ниже 32° модификация II может превращаться не-
посредственно в форму IV без образования кристаллической фор-
мы III. В присутствии примесей (сульфаты аммония, калия, каль-
ция, магния) температура превращения может понижаться8-16.
Примеси разных ионов изменяют также форму кристаллов
NH4NO317~19. Повышение давления изменяет температуры превра-
щений: так, при 800 ат температура превращения NH4NO3 (///)**
«г NH4NO3 (IV) -повышается до 60,8°20.
Давление пара над нитратом аммония (в мм рт. ст.) при различ-
ных температурах: 76° — 0,0024; 100° — 0,0154; 123° — 0,0738;
160° —0,958; 170°—1,40;
200° —6,31; 240° —33,4.
Удельная теплоемкость Ср
(в кал/г-град) водных раство-
ров нитрата аммония для тем-
ператур 16—52° составляет
для концентрации 9,18% —
0,929; 28,5% — 0,7227; 47,1 % —
0,697; 63,9%—0,61; 88% —
0,47; 92%—0,45; 96%—0,43;
98,5% - 0,42.
Аммиачная селитра весьма
Рис. 339. Растворимость нитрата аммо'
ния в воде.
гигроскопична. Пользуясь но-
мограммой (рис. 340), можно
видеть, что, например, при 30°
давление пара над насыщен-
ным раствором аммиачной селитры равно ~ 18,5 мм рт. ст., а ги-
гроскопическая точка равна ~60%; при относительной влажности
воздуха больше 60% NH4NO3 будет увлажняться. Нитрат
аммония в сухие периоды года сыреет при хранении в Северо-
Западных областях СССР, а в течение года — повсюду, за
исключением районов Средней Азии21. Гигроскопичность нитрата
аммония и скорость поглощения влаги воздуха уменьшаются
при смешении или сплавлении с другими веществами (на-
пример, с сульфатом аммония), если давление водяного пара над
насыщенным раствором обеих солей больше давления водяного
пара над насыщенным раствором нитрата аммония. Эффективным
средством для предотвращения увлажнения (и подсыхания) ам-
миачной селитры является упаковка соли в плотную, хорошо гер-
метизированную тару, например в пятислойные битумированные
мешки.
' Угол естественного откоса нитрата аммония при относительной
влажности воздуха 50—70% и температуре 10—-30° составляем
36—37°.
% ‘ютлреор
QUUDOHMDl/g UDH9l/dLUn00HWQ
ujjwd nn ‘odou oeoHtfgog annai/goff
Нитрат аммония 1181
Слеживаемость и меры ее предотвращения
Аммиачная селитра обладает способностью сильно слежи-
ваться 22-37. Этому благоприятствует сравнительно большая рас-
творимость аммиачной селитры в воде, высокий температурный ко-
эффициент растворимости, гигроскопичность соли и полиморфные
превращения. При остывании горячей аммиачной селитры в таре,
а также при ее длительном хранении, когда изменяется ее темпера-
тура (нагрев и охлаждение), происходит кристаллизация аммиач-
ной селитры из маточного раствора. Кристаллы, выделившиеся из
раствора, находящегося на поверхности частиц, связывают смеж-
ные частицы; в результате этого селитра слеживается. Кроме того,
при охлаждении горячей соли в мешках селитра претерпевает мо-
дификационное превращение при 32,3° с перестройкой кристалличе-
ской решетки; при этом слеживаемости способствует давление, раз-
виваемое весом вышележащей массы соли31.
По другим данным 32, полиморфное превращение NH4NO3(IV)^
^NH4NO3(III) ведет к разрыхлению структуры и само по себе
не может являться причиной слеживаемости аммиачной се-
литры.
Обработка аммиачной селитры гидрофобными веществами
уменьшает ее слеживаемость. Однако распространения этот метод
борьбы со слеживаемостью селитры не получил.
Уменьшение слеживаемости достигается припудриванием частиц
соли порошкообразными минеральными добавками — фосфоритной
или костяной мукой, талькмагнезитом, золой, гипсом, каолином
и др. Одни из этих добавок только уменьшают активную поверх-
ность частиц, другие обладают также адсорбционными свойствами.
Влияние неорганических добавок на уменьшение слеживаемости
аммиачной селитры в основном определяется: 1) понижением со-
держания свободной влаги в частицах, 2) понижением гигроскопи-
ческой точки соли, что ведет к уменьшению количества испарив-
шейся межчастичной влаги, 3) ослаблением связи между кристал-
лами и 4) изменением удельного объема маточного раствора13.
Однако припудривающие добавки постепенно мигрируют с поверх-
ности во внутрь частиц, и способность последних адсорбировать и
десорбировать влагу вновь восстанавливается. Кроме того, дей-
ствие добавок, обладающих адсорбционными свойствами, ограни-
чено их емкостью в отношении влаги. Таким образом, эффект от
припудривающих добавок можно рассматривать как временный26.
В качестве одной из лучших припудривающих добавок для устра-
нения слеживаемости аммиачной селитры (а также карбамида и
сложных удобрений) рекомендуют добавку из силиката магния и
аммония (Аттакот) 27,40-42.
Для уменьшения слеживаемости предложено использовать при
получении аммиачной селитры очень малые добавки красящих
1182
Гл. XXXII. Соли азотной кислоты
веществ (0,01—0,03% от веса соли), которые влияют на форму
образующихся кристаллов — придают им хрупкость. Эти добавки
не увеличивают взрывоопасность аммиачной селитры. Тем не менее
красители полностью не устраняют слеживаемости селитры и эф-
фективность их заметно понижается с повышением количества по-
глощенной влаги. В качестве добавок рекомендуют также поверх-
ностно-активные вещества 28, но употребление их ограничено срав-
нительно высокой стоимостью.
Изменение формы частиц — переход от мелкокристаллической
селитры к гранулированной — значительно способствует пониже-
нию слеживаемости соли. Насыпной вес гранулированной аммиач-
ной селитры колеблется в пределах 0,8—1,2 т/м6 в зависимости от
плотности упаковки.
Одним из заслуживающих внимания мероприятий по уменьше-
нию слеживаемости аммиачной селитры, особенно гранулирован-
ной, является также ее охлаждение перед затариванием 2. В гер-
метизированной таре охлажденная ниже 30° аммиачная селитра
долгое время сохраняется в сыпучем состоянии27.
В нашей отечественной практике по борьбе со слеживаемостью
аммиачной селитры нашли широкое применение кондиционирую-
щие добавки — азотнокислые соли кальция и магния, получаемые
растворением в азотной кислоте доломита, а также продукты азот-
нокислотного разложения фосфатов — раствор фосфоритной муки
(РФМ) или апатитового концентрата (РАП). Их вводят в раствор
нитрата аммония до его кристаллизации4’38. Гранулированная ам-
миачная селитра с добавками 0,4—0,6% нитратов кальция и маг-
ния практически не слеживается в течение 3—4 месяцев хранения
в разных климатических условиях зэ. В присутствии этих добавок
уменьшается растворимость нитрата аммония, следовательно при
охлаждении или подсушивании выделяется меньшее количество
кристаллов соли из ее насыщенного раствора, находящегося на по-
верхности кристаллов. Добавки способствуют перемещению влаги
с поверхности внутрь частиц, что также уменьшает слеживае-
мость 12’13. Кроме того, добавки влияют на температуру полиморф-
ных превращений аммиачной селитры и уменьшают давление пара
насыщенного раствора NH4NO3. Все это способствует уменьшению
слеживаемости аммиачной селитры.
В присутствии добавок изменяется и форма кристаллов. Так, до-
бавка нитрата магния, повышающая вязкость раствора, способ-
ствует кристаллизации NH4NO3 в виде дендритов. Кристаллы такой
формы хрупки и не способны прочно цементировать ранее образо-
вавшиеся кристаллы. Хорошо влияет на улучшение физико-химиче-
ских свойств аммиачной селитры и повышает эффективность ее как
азотного удобрения добавка полиакриламида 43. Менее всего под-
вержена слеживаемости амйиачная селитра в виде гранул со сред-
ним размером зерен 1—3 мм, полученная из плава концентрацией
Нитрат аммония
1183
более 99% NH4NO3 с применением кондиционирующих добавок и
охлажденная перед упаковкой в тару до температуры ниже
32 3° 36, 37, 44-55.
Термическое разложение нитрата аммония,
его огне- и взрывоопасность
Длительный опыт производства и применения аммиачной се-
литры показал, что при соблюдении установленных правил аммиач-
ная селитра безопасна 57~66. Чистая аммиачная селитра не чувстви-
тельна к толчкам, ударам или трению. Однако при определенных
условиях нитрат аммония обладает взрывчатыми свойствами. На
этом основании его используют и как сырье в производстве амми-
ачно-селитренных взрывчатых веществ. Они взрывают только от
детонатора. Взрывы чистой аммиачной селитры могут быть вы-
званы в основном или воздействием детонаторов, или термическим
разложением соли в замкнутом пространстве.
Взрывоопасность нитрата аммония возрастает в присутствии
минеральных кислот.и легко окисляющихся материалов, таких как
органические вещества и некоторые металлы, особенно в порошко-
образном состоянии (например, алюминий, цинк, свинец, сурьма,
висмут, никель, медь, кадмий). В большинстве случаев в присут-
ствии этих металлов (особенно кадмия и меди) образуется неустой-
чивый, легко разлагающийся нитрит аммония.
При увеличении размера частиц и повышении влажности взры-
воопасность аммиачной селитры значительно уменьшается. Влаж-
ная соль, содержащая более 3% воды, не взрывает даже при
взрыве детонатора 58’5Э.
При нагревании нитрат аммония начинает разлагаться согласно
уравнению:
NH4NO3 = NH3+HNO3 —41,7 ккал
Это разложение становится заметным выше 150°, но, даже при
165°, потеря в весе аммиачной селитры не превосходит 6% за сутки.
При более высоких температурах нитрат аммония разлагается ин-
тенсивно по следующим реакциям 67:
при 200—270’
NH4NO3 = N2O + 2Н2О + 8,8 ккал
при быстром нагревании до высокой температуры
NH4NO3 = N2 + 2Н2О + 1 /зО2 + 28,5 ккал
^Теплоты этих реакций даны для 18° и для газообразного состояния
продуктов реакции.) Последнее уравнение соответствует взрывному
разложению NH4NO3. Термическое разложение NH4NO3 может про-
исходить одновременно по нескольким реакциям, причем одна из
них может доминировать над другими. Термический распад азотной
кислоты обусловливает появление в газообразных продуктах
1184
Гл. ХХХ11. Соли азотной кислоты
разложения аммиачной селитры NO и NO2. По-видимому, выделяю-
щиеся в результате термического распада азотной кислоты NO2 и
Н2О являются катализаторами дальнейшего разложения NH4NO3e8.
Термическое разложение расплавленной аммиачной селитры уско-
ряется также в присутствии соединений Сг6+, Сг3+, Сг2+ и др. 69. Та-
ким образом, чистую аммиачную селитру следует безусловно от-
нести к классу потенциально взрывчатых веществ.
Нитрат аммония, хранящийся в открытых складах, не взрывает-
ся даже в случае сильного пожара. Пожары же аммиачной селит-
ры, которые имели место в закрытых помещениях, например, в ко-
рабельных трюмах, контейнерах и т. п., кончались, как правило,
сильным взрывом. Предполагают, что термическое разложение ни-
трата аммония при атмосферном давлении протекает иначе, чем
под повышенным давлением, при котором скорость разложения мо-
жет быть большей и быстро образуются большие объемы газооб-
разных продуктов. Было показано 64 существование «предельного»
давления (около 6 ат) после достижения которого при соответ-
ствующей температуре наступает взрывное разложение аммиачной
селитры.
С другой стороны, легкую воспламеняемость и взрываемость
аммиачной селитры, находящейся в непроветриваемых закрытых
помещениях, можно объяснить не повышением общего давления,
что является вторичной причиной, а накоплением продуктов мед-
ленного разложения селитры. Самопроизвольное разложение ам-
миачной селитры в присутствии способных окисляться, например,
органических веществ является автокаталитическим. Такое разло-
жение может привести к воспламенению и взрыву. Автокатализ вы-
зывается главным образом образующейся при разложении NH4NO3
двуокисью азота, а также, но в меньшей мере, водяным паром. По-
следнее обстоятельство указывает на недопустимость тушения вос-
пламенившейся селитры водяным паром.
Стабилизаторами, предотвращающими самопроизвольное разло-
жение аммиачной селитры, могут быть вещества, связывающие
образующиеся при ее разложении азотную кислоту и NO2, или вы-
деляющие при взаимодействии с NH4NO3 аммиак. Последний ней-
трализует азотную кислоту и восстанавливает окислы азота до
элементарного азота. Стабилизаторами являются, например, карб-
амид (0,05—0,1% от веса селитры)70-73, карбонат кальция или маг-
ния (5%), хлориды, уротропин и др.67.
Применение
Аммиачная селитра является одним из наиболее эффективных
азотных удобрений. Впервые в чистом виде в качестве удобрения
ее стали применять в СССР. Большое содержание азота позволяет
осуществлять ее перевозки на значительные расстояния с меньшими
Нитрат аммония
1185
затратами на тонну азота, чем при перевозках других азотных удо-
брений (за исключением карбамида). Аммиачная селитра дешевле,
чем другие азотные удобрения 74’75. Относительная стоимость азота
в азотных удобрениях характеризуется следующими условными по-
казателями:
В аммиачной селитре............ 1
» сульфате аммония............. 1,3
» кальциевой селитре........... 1,5
Аммиачная селитра обладает потенциальной (физиологической)
кислотностью. Физиологически нейтрализованную аммиачную се-
литру получают сплавлением ее с известняком, доломитом и дру-
гими материалами76. Взрывоопасность и слеживаемость аммиачной
селитры сдерживали ее производство в капиталистических странах.
Лишь в послевоенный период, на основе успешного опыта СССР,
вначале в США, а затем и в других странах использование аммиач-
ной селитры в качестве азотного удобрения получило широкое раз-
витие.
Аммиачную селитру применяют для изготовления взрывчатых
веществ — аммонитов (смесей аммиачной селитры с органическими
материалами — древесная, жмыховая и другая мука с добавкой ни-
тропродуктов), аммоналов (смесей, содержащих алюминиевый по-
рошок) и др. Для этих целей выпускают водоустойчивую се-
литру 77т79.
Состав аммиачной селитры приведен в табл. 89.
ТАБЛИЦА 89
Состав аммиачной селитры (в %)
Нитратного й аммиачного азота в сухом
веществе в пересчете:
на NH4NO3, не менее................
на азот, не менее .................
Добавок в сухом веществе:
фосфатов (PjOr), не меиее .......
нлн нитратов Са н Mg (СаО), не менее
Влаги, ие более ......................
Не растворимых веществ:
в воде, не более ..................
в соляной кислоте, не более .......
Гранул
в пределах 1—3 мм, не меиее........
мельче 1 мм, не более..............
Жирных кислот и парафина..............
Железа.................................
Кислотность (на HNO3), не более.......
По ГОСТ 2-65 По ГОСТ 14702-69 Водоустойчивая
марка А марка кристал- лическая гранули- рованная
99,5 34,8 97,7 34,2 99,0 99,0
Отсутствие Отсутствие 0,5 ОЛ 0,3 0,4 0,8 0,9
0,05 Не о п р е д Н е О е л я е т с я I р е д е л я 0,01 е т с я 0,01
90 6 0,3-0,4 0,06-0,09 0,07 0,3-0,4 0,06-0,09 0,07
* Предприятиям, применяющим фосфорсодержащие добавки, разрешается вырабатывать
аммиачную селитру марки В с содержанием NH4NO3 не меиее 962$, азота не менее 33,62$«
1186
Гл. XXXII. Соли азотной кислоты
При упаковке селитры температура должна быть не выше 50°.
Ее упаковывают в битумированные бумажные мешки (трех--------
пятислойные), а также и в полиэтиленовые мешки80. Аммиачная
селитра марки Б, используемая в сельском хозяйстве и промышлен-
ности, должна быть рассыпчатой. Рассыпчатость определяют путем
однократного сбрасывания любых пяти мешков с селитрой на пол
плашмя с высоты 1 м с последующим рассевом на сите 5 лиг.
За время рассева селитра должна полностью пройти через сито;
допускается остаток на сите отдельных комков легко измельчае-
мых рукой.
Сырье и методы производства
Теплота нейтрализации
ккал/г-мол NH4NO3
Рис. 341. Теплота нейтрализации
азотной кислоты газообразным ам-
миаком (при 1 ат н 18°).
Производство аммиачной селитры состоит из нейтрализации
азотной кислоты газообразным аммиаком81-84 и кристаллизации
продукта. Аммиак не должен содержать более 1% влаги; в нем не
допускается присутствие масла.
Азотную кислоту берут концентрацией более 45% HNO3 85; со-
держание окислов азота в ней не должно превышать 0,1 %. Для по-
лучения аммиачной селитры мо-
гут быть использованы также от-
ходы аммиачного производства—
например, аммиачная вода и
танковые и продувочные газы,от-
водимые из хранилищ жидкого
аммиака и получаемые при про-
дувках систем синтеза аммиака.
Состав танковых газов: 45—70%
NH3, 55—30% H2 + N2 (со следа-
ми метана и аргона); состав про-
дувочных газов: 7,5—9% NH3,
92,5—91 % H2 + N2 (со следами
метана и аргона). f
Кроме того, для производства
аммиачной селитры используются
также газы дистилляции с производства карбамида; их примерный
состав: 55—57% NH3, 18—24% СО2, 15—20% Н2О86.
Тепловой эффект реакции NH3(r.)+НМО3(ж.)-> NH4NO3 со-
ставляет 35,46 кка.л1г-мол. При производстве аммиачной селитры
обычно применяют 45—58%-ную кислоту. В этом случае тепловой
эффект реакции нейтрализации соответственно уменьшается на
величину теплоты разбавления азотной кислоты водой и на вели-
чину теплоты растворения аммиачной селитры (рис. 341). При ра-
циональном использовании выделяющегося тепла нейтрализации
можно получить за счет испарения воды концентрированные рас-
творы и даже плав аммиачной селитры (рис. 342)87.
Нитрат аммония
1187
В соответствии с этим различают
аммиачной селитры с последующим
ваемый многостадийный процесс) и
дийный или безупарочный про-
цесс).
Для выбора рациональной схе-
мы нейтрализации в СССР были
проверены четыре принципиально
различные схемы получения аммиач-
ной селитры с использованием теп-
ла нейтрализации88-101:
1) установки, работающие при
атмосферном давлении (избыточное
давление сокового пара 0,15—
0,2 ат);
2) установки с вакуум-испарите-
лем;
3) установки, работающие под
давлением, с однократным исполь-
зованием тепла сокового пара;
4) установки, работающие под
давлением, с двукратным использо-
ванием тепла сокового пара (по-
лучение концентрированного плава).
В промышленной практике на-
шли широкое применение как наи-
более эффективные установки, рабо-
тающие при атмосферном давлении,
трализации и частично установки с в
схемы с получением раствора
выпариванием его (так назы-
: получением плава (односта-
Рис. 342. Концентрация раство-
ров NH4NO3, получаемых при ис-
пользовании тепла реакции, в за-
висимости от концентрации азот-
ной кислоты (потери тепла учтены
в размере 3%).
Температуры азотной кислоты н ам-
миака: ;-70°; 2-50°; 3-50° (HNO3) н
20° (NH3k 4-20°.
с использованием тепла ней-
акуум-испарителем.
Производство аммиачной селитры с выпаркой растворов
Получение аммиачной селитры по этому методу состоит из сле-
дующих основных стадий:
1) получение раствора аммиачной селитры нейтрализацией
азотной кислоты аммиаком;
2) выпаривание раствора аммиачной селитры до состояния
плава;
3) кристаллизация соли из плава;
4) сушка и охлаждение соли;
5) упаковка.
Получение раствора нитрата аммония
Процесс нейтрализации осуществляют в нейтрализаторе, позво-
ляющем использовать тепло реакции для частичного выпаривания
раствора. Он получил наименование аппарата ИТН (использование
1188
Г л. XXXII. Соли азотной кислоты
Рис. 343. Нейтрализационный ап-
парат ИТН:
/ — цилиндрический сосуд; .2—внутрен-
ний цилиндр; 3 —устройство для подачи
азотной кислоты; 4 —устройство для по-
дачи аммиака: $ —штуцер для опорож-
нения аппарата; 6 — сепаратор; 7 —гидра-
влический затвор.
тепла нейтрализации). Этот аппарат (рис. 343) представляет собой
цилиндрический сосуд 1, внутри которого помещен цилиндр 2. Про-
странство во внутреннем цилиндре составляет нейтрализационную
часть аппарата, кольцевое пространство между внешними стенками
аппарата и внутренним цилиндром является испарительной частью
нейтрализатора. Аппарат выпол-
няется из нержавеющей стали мар-
ки 1Х18Н9Т.
Раствор аммиачной селитры, об-
разовавшийся при. взаимодействии
аммиака и азотной кислоты, пере-
ливается через верхний край вну-
треннего цилиндра в испарительную
часть аппарата, в которой частично
упаривается за счет тепла, выделив-
шегося при нейтрализации. Упарен-
ный раствор вытекает из аппарата
через гидравлический затвор 7 и се-
паратор 6. Выделяющийся при упа-
ривании соковый пар удаляется че-
рез верхний штуцер. Спускной шту-
цер 5 служит для опорожнения его
при ремонте и внутреннем осмотре.
Нейтрализацию проводят при 115—
120° и некотором избыточном давле-
нии (0,15—0,2 ат) для того, чтобы
образующийся в аппарате соковый
пар можно было использовать как
греющий агент. Производительность
аппарата составляет 250—400 т/сут-
ки в пересчете на 100%-ный
NH4NO3.
В аппарате, изображенном на рис. 344, нейтрализационная ка-
мера имеет снизу отверстия, благодаря чему раствор более интен-
сивно циркулирует в реакционном пространстве, и, следовательно,
достигается лучшее взаимодействие реагентов, что обусловливает
повышение производительности аппарата до 700 т/сутки и боль-
ше 90.
Процесс нейтрализации можно проводить в слабокислой или в
слабощелочной среде. Однако для уменьшения потерь связанного
азота (в виде NH3, HNO3, NH4NO3 или NO3) с соковым паром вы-
годнее вести процесс в слабокислой среде, так как при избытке
азотной кислоты давление пара HNO3 над растворами аммиачной
селитры значительно меньше, чем давление NH3 при его избытке в
растворе 102. Так, при изменении содержания NH3 в растворе от 0,1
до 0,5 г/л потери аммиака с соковым паром возрастают с 2 до 10 кг
Нитрат аммония
1189
NH,
Рис. 344, Нейтрализационный ап-
парат ИТН с циркуляцией раствора
(обозначения те же, что и на
рис. 343) .
(или с 1,6 до 8 кг азота) на 1 т аммиачной селитры. При нейтрали-
зации же в слабокислой среде, даже если содержание HNO3 в рас-
творе изменится с 0,2 до 1 г/л, потери азотной кислоты с соковым
паром возрастут с 0,4 кг только до 1 кг (или с 0,1 г до 0,25 кг азо-
та) на 1 т аммиачной селитры3.
В некоторых случаях для уменьшения коррозии аппаратуры
предпочитают вести нейтрализацию в слабощелочной среде. В на-
стоящее время на основании боль-
шого промышленного опыта уста-
новлено, что химическая стойкость
нержавеющей стали при слабокис-
лом и слабощелочном режимах ра-
боты примерно одинакова.
При ручной регулировке процес-
са нейтрализации очень трудно под-
держивать такой режим, при кото-
ром получаются наименьшие поте-
ри азота 91. С введением в промыш-
ленную практику автоматическо-
го регулирования нейтрализацию
удается проводить при неизменном
содержании NH3 или HNO3 в рас-
творе. Например, легко поддержи-
вать заданную концентрацию NH3
в растворе 0,1 г/л и меньше. Газо-
образный NH3 подают в нейтрали-
затор под давлением 2,5—3,5 ат. По
выходе из нейтрализатора раствор
аммиачной селитры подвергают до-
нейтрализации добавлением соот-
ветственно NH3 или HNO3. Этим
предотвращается коррозия выпар-
ных аппаратов в случае кислых ще-
локов и уменьшаются потери азота
в случае щелочной среды. Далее ще-
локи центробежным насосом передают на выпаривание. При высо-
кой точности автоматического дозирования компонентов нейтрали-
зацию можно вести в нейтральной среде; в этом случае надобность
в донейтрализаторе отпадает.
Нейтрализацию HNO3 аммиаком с получением раствора ни-
трата аммония можно осуществлять в аппарате колонного типа.
В нижней части колонны располагаются концентрические цилин-
дрические сосуды, являющиеся нейтрализаторами и испарителями.
Во внутреннем сосуде помещаются горизонтально две группы со-
пел для NH3 и HNO3, таким образом, чтобы жидкость получила
вращательное движение.
1190
Г л. XXXII. Соли азотной кислоты
Для создания турбулентности у верхнего края этого сосуда по-
мещаются направляющие лопатки. В средней и верхней частях ко-
лонны помещается теплообменная и испарительная аппара-
тура 103,1М.
Разработана конструкция нейтрализатора типа насадочной ко-
лонны высотой 5 м с разбрызгивающим устройством и с использо-
ванием реакционного тепла. Вследствие высокой интенсивности про-
цесса достигается значительное уменьшение удельного объема
нейтрализационного аппарата (приблизительно в 7,5 раз по сравне-
нию с аппаратом ИТН). Этот аппарат пригоден и для переработки
танковых газов и газов дистилляции карбамидного производства105.
При осуществлении нейтрализации в скрубберном или в тарель-
чатом аппарате под пониженным давлением горячий раствор ам-
миачной селитры направляют затем в вакуум-испаритель, в кото
ром вследствие пониженного давления раствор аммиачной селитры
оказывается перегретым и начинает кипеть при температурах, соот-
ветствующих разрежению в аппарате. При этом часть воды испа-
ряется и концентрация раствора повышается.
При использовании 45%-ной кислоты с температурой 50° и ам-
миака с температурой 70° концентрация получаемого раствора
аммиачной селитры составляет ~63%, т. е. на 1 т аммиачной се-
литры выпаривается около 400 кг воды. Обычно потери аммиака на
1 т NH4NO3 составляют 2—2,5 кг, потери азотной кислоты 7—7,5 кг
и больше. Одним из важнейших факторов, повлиявших на сниже-
ние потерь в процессе нейтрализации, явился перевод аппаратов
нейтрализации на автоматическое регулирование 106.
Выпаривание растворов нитрата аммония107
Обычно получаемые в аппаратах ИТН растворы аммиачной се-
литры, содержащие 60—80% NH4NO3, упаривают до состояния
плава. Конечная концентрация плава зависит от способа кристал-
лизации. При кристаллизации в грануляционных башнях раствор
аммиачной селитры обычно упаривают до 98—98,5% NH4NO3, а
для получения селитры высокого качества — 99,5—99,8% NH4NO3;
при кристаллизации на охлаждающих вальцах — до 96—97%; в
случае кристаллизации аммиачной селитры в кристаллизаторах ча-
шечного типа концентрацию плава доводят до 94,5—95% NH4NO3.
Температура кипения 95%-ного плава аммиачной селитры при ат-
мосферном давлении равна 176°; 96,89% раствор NH4NO3 кипит
уже при 196°. Высококонцентрированный плав аммиачной селитры
при 185° и выше начинает разлагаться с выделением тепла. По-
этому при получении высококонцентрированных плавов аммиачной
селитры, чувствительных к действию высоких температур, выпари-
вание растворов следует проводить в вакууме, что позволяет значи-
тельно снизить температуру кипения раствора. Кроме того, для
Нитрат аммония
1191
уменьшения термического разложения NH4NO3 при выпаривании
предложено вводить в раствор или в плав карбамид в количестве
0,1—1% от веса аммиачной селитры 108-110
Выпаривание растворов аммиачной селитры проводят в много-
корпусных вакуум-выпарных аппаратах с использованием вторич-
ного пара, причем применяют двух- и трехступенчатые схемы вы-
парки.
При применении 54—56 %-ной кислоты в аппарате ИТН полу-
чается 80—82% раствор аммиачной селитры. Вследствие высокой
температуры кипения такого концентрированного раствора исполь-
зовать соковый пар из аппарата ИТН для последующего выпари-
вания раствора не представляется возможным. В этом случае для
выпаривания применяют только свежий пар при давлении не более
9 ат. На 1 т аммиачной селитры, вырабатываемой в час, при выпа-
ривании раствора от 64 до 98,5% NH4NO3 требуется 30—40 м2
греющей поверхности.
При трехступенчатом выпаривании раствора селитры достигает-
ся некоторая экономия свежего пара по сравнению-с двухступенча-
той выпаркой. При этом выпарной аппарат II ступени работает при
давлении 1,15—1,2 ат и обогревается паром 9 ат. Соковый пар из
этого аппарата присоединяется к соковому пару, получаемому в ап-
парате ИТН, и используется как греющий агент в выпарном аппа-
рате I ступени.
Окончательная выпарка до плава производится в аппарате
III ступени под разрежением 500—550 мм рт. ст.
Пои использовании для нейтрализации 55 %-ной кислоты вме-
сто 45—47%-ной для получения плава аммиачной селитры можно
ограничиться одноступенчатой выпаркой, вместо двух- или трех-
ступенчатой. Такая схема (нейтрализация—одноступенчатая вы-
парка— грануляция) более экономична как по капиталовложениям,
так и по эксплуатационным расходам110.
Для получения высококачественной аммиачной селитры, практи-
чески не слеживающейся, изготовляют высококонцентрированный
плав (99,5—99,8%). По-лучение последнего ведут в пленочных аппа-
ратах с падающей или восходящей пленкой ,!2. В этом случае плав
аммиачной селитры после выпарки II или III ступени концентрации
97—98% поступает в верхнюю часть пленочного аппарата на труб-
ную решетку и равномерно распределяется по внутренней поверх-
ности теплообменных трубок. В нижнюю часть аппарата подается
нагретый до 175—180° воздух, движущийся вверх по трубкам, на-
встречу стекающему вниз в виде пленки плаву. Греющий пар (13—
14 ат) подается в межтрубное пространство выпарного аппарата.
Продуваемый через трубки аппарата горячий воздух обеспечивает
необходимый массообмен за счет разности давлений паров воды
над плавом и в сухом воздухе. Увлажненный воздух выбрасывается
в атмосферу. Вытекающий из аппарата плав пропускается через
1192
Г л. XXXII. Соли азотной кислоты
фильтр и поступает в разбрызгиватель грануляционной башни. )
Высококонцентрированный плав аммиачной селитры можно по-:
лучать также в роторном пленочном испарителе, сочетающем высо- J
неэффективный процесс теплообмена с малым временем пребывания ‘
продукта в аппарате, что существенно для переработки вещества
с относительно низкой температурой термического разложения.
Роторные пленочные испарители обладают незначительным гидра-
влическим сопротивлением — это позволяет вести процесс при ;
малом остаточном давлении. Наиболее широкое распространение'5
получили роторные испарители систем: «Лува» (Швейцария), «Сам-
бай» (ФРГ), «Сако» (США). Роторные испарители отличаются
типом ротора и способом распределения жидкостной пленки на теп-
лообменной поверхности. Лопасти ротора могут быть жестко за-
креплены на валу, образуя небольшой зазор с корпусом, или шар-
нирно закреплены на валу и скользить по поверхности ротора; в
других типах ротор распределяет жидкость на поверхности корпуса
под действием центробежных сил. Фирма «Кальтенбах» (Фран-
ция) получает 99,5—99,7%-ный плав нитрата аммония, выпаривая
95%-ный раствор в роторном выпарном аппарате типа «Luwa —
Kaltenbach», обогреваемом снизу горячим воздухом81.
Выпаривание раствора нитрата аммония до высокой концентра-
ции можно вести в аппаратах пластинчатого типа с паровой ру-
башкой. Аппарат рассчитан для работы при температуре 232°. Дав-
ление в рубашке 17,5 ат, в испарительной части аппарата 0,18 ат 118
Кристаллизация нитрата аммония
В зависимости от способа кристаллизации аммиачную селитру ;
можно получать в виде мелкокристаллической или в виде агрегатов
кристаллов, плотно связанных друг с другом в форме чешуек или
гранул. В настоящее время в СССР промышленность производит
для сельского хозяйства аммиачную селитру в гранулированном
виде и небольшое количество чешуйчатой селитры. Мелкокристал-
лическую селитру используют исключительно для технических це-
лей.
Высококонцентрированный плав аммиачной селитры при незна-
чительном охлаждении быстро затвердевает. При кристаллизации
плава происходят превращения кристаллических модификаций со-
ли, протекающие с выделением тепла *>2>6. Если, например, в кри-
сталлизатор поступает 92—93%-ный плав при температуре выше
126°, а температура соли на выходе из кристаллизатора меньше 32°,
то количество тепла, выделяющегося при переходе соли из расплав-
ленного состояния в кристаллическую модификацию IV с промежу-
точными превращениями в модификации I, II и III, составляет
38,15 кал!г. Кроме того, при повышении концентрации плава в про-
Нитрат аммония
1193
цессе кристаллизации также выделяется тепло в количестве, соот-
ветствующем теплоте растворения NH4NO3. Общего количества
тепла, выделяющегося при кристаллизации и расходующегося на
испарение содержащейся в плаве воды, достаточно, чтобы из 92,5—
94%-ного плава аммиачной селитры, имеющего температуру выше
126°, получить почти сухую соль (с влажностью 0,1—0,2%). Од-
нако превращения кристаллических модификаций селитры проис-
ходят медленно, и поэтому кристаллизация с использованием вы-
деляющегося тепла требует громоздкой и малопроизводительной
аппаратуры. При таком способе кристаллизации получается мелко-
кристаллическая аммиачная селитра114.
Кристаллизацию аммиачной селитры с использованием тепла
плава и тепла кристаллизации для удаления содержащейся в плаве
влаги осуществляют в чашечных кристаллизаторах Кестнера ”5, в
продуваемых холодным или горячим воздухом шнековых кристал-
лизаторах е рубашками водяного охлаждения116 и во вращаю-
щихся горизонтальных грануляторах барабанного типа, в которых
плав разбрызгивается в противоточном потоке воздуха над слоем
ретура 1171118.
В крупных промышленных установках, вырабатывающих ам-
миачную селитру для нужд сельского хозяйства, в настоящее время
применяют непрерывные способы кристаллизации в грануляцион-
ных башнях или на охлаждающих вальцах. При кристаллизации на
поверхности охлаждающих вальцов тепло кристаллизации исполь-
зуется неполностью. Пленка плава закристаллизовавшейся соли
разделяется на отдельные агрегаты кристаллов в виде чешуек, пло-
хо поддающихся сушке. По гранулометрическому составу чешуйки
очень неоднородны и потому менее удобны для рассева при внесе-
нии в почву, чем гранулированная соль.
Обычно при кристаллизации аммиачной селитры на охлаждаю-
щих вальцах концентрация поступающего плава составляет 97—
97,5%; температура плава 140°. При понижении концентрации пла-
ва до 94,5—95,5% и медленном отводе тепла от поверхности вальца
(температура отходящей воды при этом должна составлять 70—
75°) можно получать мелкокристаллическую соль. В этом случае
сравнительно влажная горячая масса кристаллов, снятая ножом
с поверхности вальцов, далее поступает в сушильный барабан, в
котором постепенно подсушивается за счет тепла кристаллизации
соли и горячего воздуха, продуваемого над солью. Получается мел-
кокристаллическая соль с влажностью около 1 %.
Для получения аммиачной селитры в гранулированном виде
кристаллизацию ее из плава проводят в башнях, имеющих цилин-
дрическую или прямоугольную форму с конусным разгрузочным
днищем. Высота их различна: от 15 до 100 м119. Башни изготов-
ляют из железобетона, из алюминия, из стали и алюминия. В СССР
грануляционные башни изготовляют из бетона или из кирпича и
1191
Гл. ХХХ11. Соли азотной кислоты
футеруют их кислотоупорным кирпичом на диабазовой замазке; вы-
сота башен 35 м, диаметр 16 м.
Конусное днище башни выполняется из бетона или из углероди-
стой стали. Для удобства чистки конуса в случае налипания соли
на его внутреннюю поверхность конус изготовляют в виде отдель-
ных секций, между которыми имеются отверстия.
Плав аммиачной селитры распределяется по сечению башни с
помощью вращающегося разбрызгивателя — конической корзины с
отверстиями 12°. Однако брызги распределяются по сечению башни
неравномерно, что является существенным недостатком такого типа
гранулятора 121. Падающие капли плава охлаждаются встречным
потоком холодного воздуха и кристаллизуются в виде гранул.
В нижней части конуса башни размещается аппарат для доохла-
ждения гранул в кипящем слое 1221 123. Затем гранулы через ниж-
нее отверстие диаметром 1,2 м поступают на транспортер, подаю-
щий аммиачную селитру на упаковку.
Воздух входит в башню через отверстия, расположенные по ок-
ружности внизу цилиндрической части башни. Часть воздуха по-
дается вентиляторами в доохладитель с кипящим слоем. Для со-
здания тяги воздуха в специальных вытяжных трубах помещены
вентиляторы (по два вентилятора на каждую башню) производи-
тельностью до 100 000 н.ч3/ч. Режим грануляции: концентрация пла-
ва 98,3—98,5% NH4NO3, температура плава перед грануляцией не
ниже 160°, число оборотов гранулятора 450 об/мин (при размере
отверстий на верхнем поясе гранулятора не больше 1,9 мм). По-
дача воздуха для охлаждения гранул 8000—10000 мэ/ч на 1 т
NH4NO3124.
На выходе из башни воздух имеет 35—45°. Температура гранул,
выгружаемых из башни, колеблется от 85—90 до 60° и зависит от
температуры воздуха на входе в башню, его количества и, в мень-
шей мере, от нагрузки, с которой работает грануляционная башня.
Например, при количестве воздуха 100 000 м3/ч с температурой на
входе в башню 14—15° изменение нагрузки на башню с 20 до
10 т]ч плава снижает температуру гранул на выходе из башни с 76
до 72°; при снижении же температуры воздуха на входе в башню
до 4 и 2° температура гранул на выходе из башни при той же на-
грузке уменьшается соответственно до 63—59°. Понижение темпе-
ратуры гранул, выходящих из башни, например до 50—60°, яв-
ляется очень желательным 125, так как при упаковке в мешки грану-
лированной селитры с температурой выше 65° в последующем
значительно снижается рассыпчатость гранул. Соль, загруженная
в мешки с температурой ниже 55°, сохраняет рассыпчатость.
В грануляционной башне гранулы слегка подсушиваются. Их
влажность на 0,1—0,15% меньше влажности поступающего в баш-
ню плава. Это объясняется тем, что даже при 100%-ной относитель-
ной влажности поступающего в башню воздуха, давление водяного
Нитрат аммония
1195
пара над горячими гранулами больше парциального давления вла-
ги в воздухе.
Гранулирование аммиачной селитры возможно и безбашенным
способом, например пропусканием горячей кристаллической селит-
ры через экструзионную машину с механическим срезыванием в ней
гранул 126.
Предложено также грануляцию осуществлять в грануляторе та-
рельчатого типа. Тарельчатый гранулятор имеет следующие раз-
меры: диаметр тарелки 4,5 м, высота борта тарелки 30 см, угол
наклона тарелки 45°, скорость вращения тарелки 14 об/мин. Во
вращающуюся тарелку на слой соли поступают 44—75% раствор
NH4NO3 в аммиаке и 55—95%-пая азотная кислота. При этом про-
исходит образование дополнительного количества NH4NO3 и испа-
рение влаги. Одновременно происходит и классификация гранул.
Необходимое количество тепла для испарения влаги регулируется
подогревом самой тарелки и исходных реагентов 127.
Испытывалась грануляция NH4NO3 в инертной жидкости !28.
Широко распространен процесс кристаллизации- и охлаждения
гранул в кипящем слое 122>129~133. Предлагается также разбрызги-
вать в реакторе с кипящим слоем гранул горячий раствор NH4NOj
концентрацией 50—95%; снизу поступает горячий воздух134.
Важной задачей является разработка мощных агрегатов произ-
водства нитрата аммония, например, с производительностью 1400—
1500 т/сутки. Расчеты показывают, что удельные капитальные вло-
жения при сооружении таких агрегатов примерно на 25% ниже, чем
при средней мощности действующих в настоящее время. В связи
с этим необходима разработка новых более производительных гра-
нуляторов. Опыт показывает, что при замене существующих цен-
тробежных грануляторов статическими и использовании в нижней
части башни «кипящего слоя» можно значительно увеличить произ-
водительность существующих грануляционных башен и улучшить
гранулометрический состав готового продукта.
Сушка нитрата аммония135-136
Удаление влаги из аммиачной селитры методом конвективной
сушки является длительным процессом. Чем крупнее частицы соли,
тем труднее ее высушить. Мелкокристаллическая и порошкообраз-
ная соль имеет большую поверхность по сравнению с гранулирован-
ной или с чешуйчатой солью и поэтому подсыхает значительно бы-
стрее.
При сушке гранулированной или чешуйчатой аммиачной селит-
ры соль находится в сушилке от 40 мин до 1 ч. В этих условиях
Удаляется не более 40% влаги, содержащейся в поступающей в су-
шилку аммиачной селитре (например, влажность снижается с 1,5
1196
Г л. XXXII. Соли азотной кислоты
до 0,8% при температуре соли 70—75°). При этом удаляется глав-
ным образом влага, находящаяся на поверхности гранул или че-
шуек соли. Вторая и третья стадии сушки (продвижение влаги, на-|
ходящейся внутри частиц соли, к поверхности за счет капиллярных
сил и диффузии) в промышленных условиях почти не протекают,'
так как для полного удаления влаги требуется не менее 7—8
Гранулы NH4NO3, покрытые диатомовой землей (в количестве 3—|
4% от веса продукта), при сушке с влажностью от 2,5—3 до 0,2%
высыхают в 3 раза быстрее, чем непокрытые гранулы 137.
Рис. 345. Схема сушки аммиачной селитры охлажденным воздухом:
/—сушильный (охлаждающий) барабан; 2 — центробежный вентилятор для
циркуляции воздуха; 5 —скруббер; -/ — испаритель жидкого аммиака; 5 —центро-
бежный насос.
Сушку аммиачной селитры осуществляют воздухом, нагретым
до 105—110° (горячая сушка), или охлажденным воздухом с одно-
временным охлаждением соли (холодная сушка).
Горячей сушке обычно подвергают аммиачную селитру, содер-
жащую после кристаллизации 2—3% влаги, например чешуйчатую
селитру, получаемую при кристаллизации на охлаждающих валь-
цах.
На рис. 345 показана схема «холодной» сушки аммиачной се-
литры2. Гранулированная или чешуйчатая селитра, содержащая
1,5—2,5% влаги, поступает в сушилку 1 при 70—90°. Противото-
ком соли в сушильный барабан подается воздух, предварительно
охлажденный до —5, —8° в скруббере 3. Скруббер орошается цир-
кулирующим 35—45% раствором NH4NO3, имеющим температуру
—10°. Предварительное охлаждение раствора производится испа-
ряющимся при —20° жидким аммиаком. По мере повышения кон-
центрации охлаждающего раствора вледствие поглощения им
пыли аммиачной селитры, выносимой воздухом из сушильного бара-
бана, часть раствора выводят из цикла и направляют на выпарива-
ние, а к остающемуся раствору добавляют воду. Расход воздуха на
сушку 1 т NH4NO3 составляет около 4000 кг. Соль, поступающая
Нитрат аммония
1197
с влажностью 1,6%, охлаждается до 25—30° и высушивается до со-
держания 1—0,9 % влаги. После сушки горячим воздухом содер-
жание влаги в готовой селитре на 0,1—0,2% меньше, чем после «хо-
лодной» сушки.
Представляет интерес двухступенчатая сушка аммиачной се-
литры— вначале подогретым воздухом, а затем сушка и охлажде-
ние продукта охлажденным воздухом. Это позволяет использовать
преимущества горячей и холодной сушки. Двухступенчатая сушка
дает возможность выпускать чешуйчатую аммиа'чную селитру с хо-
рошими физическими свойствами без применения добавок138. Пер-
спективно осуществление сушки и охлаждения аммиачной селитры
в кипящем слое; это значительно интенсифицирует процессы.
На рис. 346 представлена технологическая схема производства
аммиачной селитры 139-142. Азотная кислота из склада поступает в
напорный бак 1, затем в нейтрализатор ИТН 5 через подогрева-
тель 2, в котором нагревается до ~50° конденсатом сокового пара
из выпарки I ступени 9. Газообразный аммиак подается в нейтра-
лизатор под постоянным давлением 2,5—3,5 ат. Вначале он прохо-
дит отделитель-испаритель жидкого аммиака 3 и подогреватель 4,
где нагревается до 50—70° вторичным паром (1,2 ат) из расшири-
теля конденсата 30. Из нейтрализатора ИТН 5 раствор аммиачной
селитры поступает в сборник 6, где он донейтрализовывается газо-
образным аммиаком до нейтральной реакции и перекачивается в
напорный бак 8, из которого направляется на выпарку I ступени 9.
В I ступени раствор выпаривается под вакуумом ~600 мм рт. ст.
до концентрации 80—82% NH4NO3. Греющим паром здесь служит
соковый пар из сепаратора 7 аппарата ИТН и пар (1,2 ат) из рас-
ширителя конденсата 30, получаемый при снижении давления кон-
денсата II ступени выпарки 143.
Обычно для I ступени выпарки, т. е. для упаривания растворов
невысокой концентрации, используют вертикальный выпарной ап-
парат пленочного типа из нержавеющей стали марки 1Х18Н9Т.
Раствор из выпарного аппарата I ступени поступает в сборник 10, где
в него вводятся добавки (продукты азотнокислотного разложения
доломита или фосфатов) и затем насосом подается в напорный бак
13 выпарки II ступени 14. Здесь раствор выпаривается под вакуу-
мом 500—550 мм рт. ст. до 98—98,5% NH4NO3. Теплоносителем яв-
ляется свежий пар (9—13 ат). Для выпарки II ступени до послед-
него времени применялись трубчатые горизонтальные аппараты.
Установлена возможность использования для II ступени более эф-
фективных выпарных аппаратов пленочного типа.
Выходящая из аппарата 14 паро-жидкостная эмульсия разде-
ляется в сепараторах 15 и 16, откуда соковый пар направляется в
конденсатор 17. Тепло этого пара может быть использовано для
подогрева раствора, идущего на выпарку,44. Двойная сепарация
уменьшает потери селитры 145’146.
КЗОЭОН DQQQ
Нитрат аммония
1199
Раствор, выходящий из второго сепаратора 16, имеет меньшую
концентрацию, чем после первого сепаратора 15 и потому вместе
с раствором после I ступени выпарки возвращается на выпаривание
в аппарат II ступени 14. Плав из выпарки II ступени через гидрав-
лический затвор 18, желоб 19 и напорный бачок 20 поступает в вы-
парной аппарат III ступени 21. В напорный бачок 20 подается газо-
образный аммиак, который нейтрализует избыточную кислотность
плава, образовавшуюся вследствие частичного гидролиза аммиач-
ной селитры в процессе выпаривания. В III ступени выпарки плав
концентрируется до 99,5—99,7% NH4NO3 и затем через желоб 22 и
бачок 23 подается на грануляцию. Третья ступень выпарки осуще-
ствляется в аппарате с падающей пленкой с одновременной про-
дувкой горячим воздухом (~180°). Из выпарного аппарата воз-
дух удаляется в атмосферу.
Грануляция аммиачной селитры осуществляется в грануляцион-
ной башне 26 в потоке воздуха, идущего снизу вверх. Плав раз-
брызгивается с помощью гранулятора 2.7. В нижней части конуса
башни помещается охладительный аппарат 28 с двумя кипящими
слоями, в котором гранулы доохлаждаются в начале атмосферным
воздухом (первый кипящий слой) и затем воздухом предваритель-
но охлажденным в аммиачном холодильнике 29. Охлажденная ам-
миачная селитра (25—30°) поступает на упаковку и в склад.
Для производства 1 т гранулированной аммиачной селитры
(100% NH4NO3) расходуется 0,213 т аммиака, 0,800 т азотной кис-
лоты (100% HNO3) и при работе на 47—49%-ной кислоте 0,3 мгкал
пара и 15 квт-ч электроэнергии, а при работе на 56—57%-ной кис-
лоте— 0,11 мгкал пара и 3,8 квт-ч электроэнергии 147-150.
Получение нитрата аммония безупарочными способами
Способ получения аммиачной селитры, известный как способ
Стенгеля, реализован в промышленном масштабе в США 150-153.
Он основан на быстром взаимодействии в реакторе предварительно
подогретых в теплообменниках 58 %-ной азотной кислоты и аммиа-
ка (рис. 347). Отделенный от паро-газовой смеси в сепараторе плав
NH4NO3 стекает в аппарат, предназначенный для продувки плава
воздухом с целью уменьшения содержания в нем влаги от 2 до
0,2%. Плав после продувки его воздухом имеет температуру 193—
205°. Затем его направляют на кристаллизацию, которая происхо-
дит на наружной поверхности движущейся, изнутри охлаждаемой
водой, ленты из нержавеющей стали (ленточный кристаллизатор),
или в грануляционной башне.
Преимуществом метода Стенгеля является уменьшение капи-
тальных затрат при строительстве цехов аммиачной селитры, по-
скольку отпадает необходимость сооружения выпарных установок.
Кроме того, удельный объем реактора-нейтрализатора здесь в 10—
16 раз меньше, чем при использовании аппаратов ИТН. Однако при
1200
Гл. ХХХП. Соли азотной кислоты
работе по безупарочному методу потери аммиачной селитры и азот-
ной кислоты несколько больше вследствие высокой температуры в
зоне реакции (выше 205°), что способствует их разложению 154. Для
уменьшения этих потерь соковый пар из сепаратора подвергают
тщательной промывке азотной кислотой в двух последовательно
установленных скрубберах, что усложняет схему. На 1 т аммиач-
ной селитры расходуется 0,221 т аммиака и 0,820 т азотной кислоты
(100%).
Рис. 347. Схема установки для производства гранулированного нитрата
аммония безупарочным методом:
1 —испаритель аммиака; 2 —бак азотной кислоты; 3 —насос для перекачки азотной
Кислоты; 4 — подогреватель азотной кислоты; 5 — нагреватель азотной кислоты;
о и 7 — нагреватели аммиака; 3 —реактор (нейтрализатор); 9 — центробежный сепара-
тор; 10 —скруббер для продувки плава воздухом; 11 — нагреватель продувочного воз-
духа и конденсатор сокового пара; /2 —скруобер для улавливания NH4NO3 и NH3 нэ
сокового пара; 13 — насос для циркуляции раствора, орошающего скруббер; 14 — кон-
денсатор для конечной конденсации соковых паров; 15 — распределительный бачок для
плава; 16 — кристаллизатор — охлаждаемая водой лента; 17— бак — сборник воды после
охлаждаемой ленты и питатель для орошения скруббера; 18 — размольное устройство;
IP —дисковый питатель; 20— элеватор; 21 — вибрационные сита; 22 — готовый продукт,
поступающий на припудривание; 23 —барабан для припудривания; 24 — бункер для
атомизнрованной земли; 25 — питатель; 26 — устройства для подготовки атомиэирован-
ной земли; 27 — бункер для готового продукта; 28 — весы; 29 — зашивочная машина;
30 — транспортер.
В СССР разработан вариант безупарочного способа получения
аммиачной селитры по следующей схеме 155-157. Газообразный ам-
миак подают в цех под давлением не ниже 4,3 ат и перед поступ-
лением в реактор нагревают до 155° паром (9 ат). Азотная кислота
поступает также под давлением 4,3 ат и нагревается до 155°, про-
ходя последовательно два подогревателя 158. В первом подогрева-
теле из нержавеющей стали для подогрева кислоты до 85° исполь-
зуют соковый пар, во втором подогревателе подогрев производят
свежим паром (9 ат). Второй подогреватель должен выполняться
из тантала или других стойких к горячей азотной кислоте материа-
лов. Подогретые аммиак и азотная кислота поступают в верхнюю
часть реактора, представляющего собой цилиндрический аппарат
(диаметр 630 мм и высота 4 м), заполненный насадкой из металли-
ческих колец на высоту 3,8 м.
Нитрат аммония
1201
Процесс образования аммиачной селитры идет под давлением
3,5 ат при температуре 210—235°. Вследствие короткого времени
пребывания продуктов реакции в реакторе (0,46 сек) разложения
аммиачной селитры не происходит 159. Образующаяся под воздей-
ствием тепла реакции паро-жидкостная эмульсия удаляется из ниж-
ней части реактора и поступает в сепаратор. При этом происходит
NH4NO3, вес. °/°
Рис. 348. Давление пара HNO3 в системе HNO3—NH4NO3—Н2О при 30°:
Л —поле кристаллизации NH4NO3, числа на изобарах — давление в мм рт. ст.
снижение давления с 3,5 до 1,2 ат, причем за счет самоиспарения
концентрации плава с 96,5% повышается до 97,5%.
Соковый пар из сепаратора проходит ловушку и направляется
в Водяной конденсатор. Часть сокового пара используют для подо-
грева азотной кислоты до 85°.
Из сепаратора плав может быть направлен в вакуум-испари-
тель, где за счет снижения давления от I до 0,27 ат (вакуум
550 мм рт. ст.) концентрация плава повышается с 97,5 до 98,7%.
Нейтрализацию плава и смешение его с кондиционирующими
1202
Г л. XXXII. Соли азотной кислоты
добавками можно производить как до, так и после вакуум-испари-
теля. Далее плав поступает в сборник и насосом перекачивается в
гранулятор.
Безупарочный способ получения аммиачной селитры по сравне-
нию со способом, использующим одноступенчатую выпарку, требует
Рис. 349. Общее давление паров HNO3 и Н2О в системе
HNO3—NH4NO3—Н2О при 30°:
А — поле кристаллизации NH4NO3, числа на изобарах—давление в мм рт. ст.
на ~30% меньшей затраты нержавеющей стали для изготовления
аппаратов и на — 34% меньшего расхода пара.
На рис. 348 и 349 приведены давления пара HNO3 и общие дав-
ления паров HNO3 и НгО над растворами в системе HNO3—
—NH4NO3—Н2О при 30°. Вследствие взаимодействия HNO3 и
NH4NO3 в жидкой фазе с образованием комплексов, в области по-
вышенных концентраций HNO3 давление пара HNO3 над раство-
рами меньше, чем в отсутствие NH4NO3, а в системе HNO3—
NH4NO3—NO—NO2—Н2О равновесная концентрация HNO3 в жид-
кой фазе больше. Поэтому при абсорбции нитрозного газа водным
Нитрат аммония ' 1203
раствором NH4NO3 можно получить более крепкую азотную кис-
лоту, чем при абсорбции водой. Нейтрализация концентрированного
раствора HNO3 и NH4NO3 аммиаком приводит к образованию вы-
сокопроцентного плава аммиачной селитры без выпарки. Это поз-
волило разработать следующую схему производства аммиачной се-
литры 160’ 161.
Полученный окислением аммиака нитрозный газ со степенью
окисленности 90—95% поглощается в 40-полочной абсорбционной
колонне, орошаемой 60%-ным раствором NH4NO3; 10 нижних таре-
лок колонны снабжены охлаждающими змеевиками. Из колонны
вытекает раствор, содержащий 40% NH4NO3, 40% HNO3 и 20%
НгО, т. е. раствор NH4NO3 в ~ 65%-ной азотной кислоте. Этот рас-
твор обрабатывается в нейтрализаторе аммиаком, подогретым до
100°, причем образуется плав, содержащий 95—96% NH4NO3.
Часть этого плава разбавляется водой (конденсатом газовых холо-
дильников) до 60% и возвращается в абсорбционную колонну, дру-
гая часть доупаривается до 98—99,5%, гранулируется и выпускает-
ся в качестве продукта. Такой способ производства аммиачной
селитры позволяет сократить расход пара на выпарку, не требует ка-
питальных затрат на выпарное оборудование. Он может быть осу-
ществлен на стандартном оборудовании цехов аммиачной селитры
и слабой азотной кислоты. Вследствие взаимодействия окислов азо-
та с NH4NO3 на стадии абсорбции происходит незначительная (при
30°) дефиксация азота; общие потери азота в описанном процессе
не превышают 1 —1,5%.
Получение аммиачной селитры конверсией нитратных растворов
В ряде производств получаются растворы нитратов кальция и
магния, переработка которых в твердые продукты не представ-
ляется целесообразной. Это относится также к растворам нитрата
кальция, получающегося при азотнокислотном разложении фосфа-
тов (см. гл. XXXV).
В качестве примера можно привести раствор следующего со-
става (в г/л): 41,8 СаО, 23,9 MgO, 3,7 МпО, 1,27 SO4, 80 МОбщ,
22,0 NaMM (NH4NO3).
Переработку таких растворов целесообразно осуществлять кон-
версией нитратов карбонатом аммония:
Ca(NO3)2 + (NH4)2CO3 = СаСОз + 2NH4NO3
Образующийся раствор упаривают и кристаллизуют NH4NO3.
Использование для этого газообразных NH3 и СОг связано с труд-
ностью регулирования реакции конверсии и значительными поте-
рями двуокиси углерода 162. При использовании раствора (МН4)2СОз
концентрацией 27% (125—135% от стехиометрического количества)
и подогреве исходных растворов до 60° конверсия с образованием
1204
Гл. XXXII. Соли азотной кислоты
кристаллических осадков карбонатов практически завершается
в течение 30 мин163. Основной фильтрат содержит 200—250 г/J
NH4NO3, 8—10 г/л NH3, 5—10 г/л СО2, 0,5—1,5 г/л Mg. Промытый
карбонатный шлам содержит 60—65% влаги и 1,2—1,5% общего
азота (во влажном). Содержание связанного азота в карбонатном
шламе в пересчете на аммиачную селитру составляет 8—10%|
NH4NO3. Часть связанного азота присутствует в шламе в виде двой!
ной соли MgCO3-(NH4)2CO3-4H2O, плохо растворимой в воде и
в растворах аммиачной селитры. Карбонатный шлам может быта
использован в качестве удобрения или обработан известковым мо|
локом с последующей отгонкой аммиака: I
MgCO3 • (NH4)2CO3 • 4I-I2O + Са(ОН)2 = 2NH3 + 6Н2О +MgCO3 + СаСО3 I
Применение промывных вод и части основного фильтрата для
приготовления раствора (NH4)2CO3 позволяет повысить концентра!
цию растворов аммиачной селитры на 3%, что приводит к сниже!
нию расхода греющего пара для выпарки на ~16,5% 164. I
Переходящие в раствор нитрата аммония примеси солей магния!
кальция, а также солей тяжелых металлов вызывают вспенивани!
при выпаривании раствора. Примеси солей магния могут быть вы!
делены в значительном количестве из основного фильтрата избыт!
ком (NH4)2CO3165. Выпаривание раствора до плава (96—98°/|
NH4NO3) с предотвращением пенообразования осуществляют путем
предварительной выпарки раствора до концентрации 55—65 °/(|
NH4NO3 и подкисления выпаренного раствора азотной кислотой!
нейтрализуемой далее газообразным аммиаком до слабощелочной
реакции 166. I
Примеси других солей, остающиеся в растворах (после очист!
ки), улучшают физические свойства готового продукта, но обра|
зуют отложения осадков на греющих поверхностях выпарных аппа!
ратов. Основным компонентом, входящим в состав осадка, является
сульфат магния. Его можно выделить при обработке раствора ди|
аммонийфосфатом 167. I
Удобрения на основе нитрата аммония I
Известко во - амми ачна я селитра I
Сплав нитрата аммония с известняком — известково-аммиачная
селитра получила широкое распространение в странах Западной
Европы. При длительном внесении в почву аммиачной селитры про-
исходит ее подкисление вследствие физиологической кислотности
аммиачных удобрений и протекающих в почве процессов нитрифи-
кации — окисления аммиака в азотную кислоту под действием
микроорганизмов почвы ies-i7i_ Поэтому аммиачную селитру реко|
мендуют применять на кислых почвах после их известкования 172|
Нитрат аммония
1205
Более удобным является применение известково-аммиачной се-
литры.
Известково-аммиачную селитру выпускают со следующими бесо-
выми соотношениями NH4NO3 : СаСО3 — во Франции 80 : 20; в ГДР
65 : 35; в США, Польше 60 : 40; в Англии 53 : 47 173.
Наилучшими физическими свойствами обладают смеси, содер-
жащие 60% нитрата аммония и 40% известняка 174; при этом соот-
ношении в смеси содержится 20,5% азота. В известково-аммиачной
селитре обычно содержится ~1% нитрата кальция. Вследствие
этого она более гигроскопична, чем чистая аммиачная селитра. Гиг-
роскопическая точка известково-аммиачной селитры на 2—3% ниже
гигроскопической точки аммиачной селитры. Но слеживаемость ее
в 2,4—3 раза меньше слеживаемости чистой аммиачной селит-
ры 19’ 175“178. Была исследована 179 взрывоопасность известково-амми-
ачной селитры; при содержании в ней азота меньше 22% она без-
опасна, продукт, содержащий 26% N взрывоопасен.
Известково-аммиачную селитру (НАС) получают смешением
94—95%-ного плава нитрата аммония с тонкоизмельченным извест-
няком и гранулированием полученной смеси в грануляционной
башне или в шнеках-грануляторах 139’ 141> 142’ 18°-i82.
При смешении известняка с плавом аммиачной селитры проис-
ходит частичное образование Са(ИО3)г (0,4—1,2%):
2NH4NO3 + СаСО3 = (NH4)2CO3 + Ca(NO3)2
Карбонат аммония диссоциирует на СО2 и NH3, вследствие чего
теряется некоторое количество аммиака — обычно 0,3—0,8% от со-
держания азота в плаве аммиачной селитры; это соответствует
0,8—2,1 кг NH3 на 1 т готового продукта (60 : 40). Потери аммиака
резко возрастают с увеличением продолжительности смешения 183.
Например, при 10-минутном смешении они составляют 0,32%, при
30-минутном — 0,82%, при 60-минутном—1,47% от общего содер-
жания азота в смеси. Так же резко возрастают потери аммиака
с ростом температуры сплавления. При 135° потери увеличиваются
в 2 раза, а при 145° в 5 раз по сравнению с потерями при 125°
(0,17%). Выделение NH3 из известково-аммиачной селитры умень-
шается с увеличением содержания в ней Ca(NO3)2 184.
Оптимальными условиями сплавления аммиачной селитры с из-
вестняком являются: концентрация плава 94—95,5%, степень из-
мельчения известняка 0,12 мм, соотношение исходных веществ
60:40, температура 125—135°, продолжительность смешения
Ю мин 171’ 185~187.
Смешение известняковой муки с плавом аммиачной селитры
производят в двухвальном смесительном шнеке, валы которого вра-
щаются со скоростью около 40 об)мин. Корпус шнека выполняют из
хромоникелевого чугуна (2,8% хрома, 1,5% никеля), крышку и
валы — из нержавеющей стали. При длине корпуса шнека 4,5 м,
1206
Гл. XXXII. Соли азотной кислоты
ширине 0,8 м и высоте 1 м его суточная производительност!
400—600 т известково-аммиачной селитры. Чтобы избежать закри-
сталлизовывания плава, шнек снабжен паровой рубашкой (давле-
ние греющего пара 3,5 ат, поверхность нагрева 6,5 .и2). Недостатков!
конструкции шнекового смесителя является необходимость часто!
смены сальника вала, который работает в тяжелых условиях (срав-
нительно высокая температура смешения и большая вязкость пере-
мешиваемой массы). Плав после смешения по обогреваемым парок
трубам направляют на грануляцию в грануляционные башни, по-
добные применяемым для грануляции аммиачной селитры. Грануль
выгружают из башни при помощи скребкового’сбрасывателя 115 нг
транспортеры, которыми они подаются в отделение обработки. Тем
пература гранул на выходе из башни 70—80°.
Продукт подвергают дроблению и рассеву для получения гранул
стандартного размера (2—5 мм), затем охлаждают воздухом до
25—35°, припудривают известняком, или кремнеземом, каолином
и т. п. 188-191 и направляют в склад192.
Удельный расход воздуха, на грануляцию 1 т продукта состав
ляет 5100—8750 м3)ч. На производство 1 т известково-аммиачной
селитры затрачивают 0,133 т аммиака, 0,485 т азотной кислоты
(100%), 0,475 т известняка (с учетом расхода на припудривание)
0,2 т пара (9 ат), 65 квт-ч электроэнергии, 42 000 ккал холода,
18 .и3 воды, 0,5 м3 сжатого воздуха.
Запатентован способ получения известково-аммиачной селитры
с пониженным содержанием Ca(NO3)2 (0,02—0,04%), заключаю
щийся в 30-минутном смешении плава NH4NO3 и известняка (или
доломита) при 180—210° с добавкой 0,1 — 1% кремнефторида нат
рия, калия или аммония 193.
Известково-аммиачную селитру можно получать взаимодей
ствием Са (NO3)2-4H2O с карбонатом аммония или с газообразными
NH3 + СО2 194-195.
Сульфат-нитрат аммония
Впервые механическую смесь, а также сплав нитрата аммония
с сульфатом аммония стали изготовлять в Германии; в СССР это
удобрение распространения не получило.
В сульфат-нитрате аммония содержится (в пересчете на сухое
вещество) 25,5—26,5% N, кислотность (в пересчете на H2SO4) не
превышает 0,3%, а влажность 2,5%.
Преимуществами этого вида удобрения (по сравнению с состав-
ляющими его компонентами) являются: меньшая гигроскопичность
по сравнению с аммиачной селитрой, большее содержание азота по
сравнению с сульфатом аммония и уменьшение расхода «балласт-
ной» серной кислоты на единицу азота по сравнению с расходом
в производстве сульфата аммония.
Нитрат аммония
1207
При смешении или сплавлении сульфата с нитратом аммония
образуется двойная соль (NH4)2SO4-2NH4NO3, содержащая при-
месь избыточного компонента (сульфата или нитрата).
На рис. 350 даны изотермы растворимости в системе NH4NO3—
—(NH4)2SO4—Н2О при 0, 30, 70 и 100°. Пунктирные линии, прохо-
дящие через эвтонические точки а2, а3, щ и bt, b2, Ь3, bt, отделяют
области существования отдельных
система трехкомпонентная, то при
условии ее инвариантности наи-
большее число фаз в ней может
быть равно пяти, из которых три—
твердые. Следовательно, все три
твердые фазы в присутствии жид-
кой и парообразной фаз могут
существовать одновременно лишь
при одной определенной темпера-
туре, концентрации и давлении.
Как видно из диаграммы, эта
температура не находится в пре-
делах от 0 до 100°. В этом интер-
вале температур в присутствии
жидкой фазы совместно могут
существовать только две твердые
фазы — двойная соль и нитрат
аммония или двойная соль и
сульфат аммония.
Чем выше содержание в суль-
фат-нитрате сульфата аммония,
тем меньше его гигроскопичность,
слеживаемость и взрывоопас-
ность. В сельском хозяйстве ши-
Рис. 350. Изотермы растворимости
в системе NH4NO3— (NH4)2SO4—Н2О
при 0, 30, 70 и 100°.
роко применяется продукт, содержащий 65—67% сульфата и
33—35% нитрата аммония, получаемый простым смешением этих
солей. Перед поступлением в смеситель соли отсеиваются от круп-
ных слежалых кусков, подвергающихся дроблению, и нагревают-
ся— сульфат аммония до 120°, а нитрат — до 40—60°.
Более совершенным способом получения сульфат-нитрата аммо-
ния является смешение сухого сульфата аммония с 83% раствором
нитрата аммония. Смешение производят в шнековых аппаратах, из
которых выходит масса с содержанием около 6% влаги. Эта масса
измельчается между двумя чугунными валами с шипами, высуши-
вается до влажности 0,1—0,2% в барабанной сушилке и рассеи-
вается. Фракция с величиной кусков больше 4 мм подвергается
размолу и возвращается на сито, а фракция с частицами менее
0,8 мм возвращается в смеситель. Можно получить сульфат-нитрат
аммония нейтрализацией смеси серной и азотной кислот. В реакт—т-
14 М. Е. Позин
1208
Гл. XXXII. Соли азотной кислоты
сатуратор, снабженный мешалкой, одновременно подают 50%-ную
азотную кислоту, 68%-ную серную кислоту и аммиак. Вследствие
экзотермичности реакции нейтрализации температура в сатураторе
достигает 145—150° и тепла хватает для испарения воды, вводимой
с кислотами. Отходящие из сатуратора газы (аммиак) и пары
воды поступают в ректификационную колонну, откуда аммиак воз-
вращается в сатуратор. Непрерывно вытекающий из реактора плав
содержит 2,5—3% свободной кислоты. Избыточная кислотность
плава снижается до 0,1—0,3% введением в него аммиака в доней-
трализаторе. Из донейтрализатора плав, содержащий 97—98%
сухой соли и 2—3% влаги, направляют на кристаллизацию. Кри-
сталлизацию осуществляют на поверхности охлаждающего бара-
бана 197.
, Калийно-амми ачная селитра
Калийно-аммиачная селитра, выпускаемая в довольно значи-
тельном количестве в ряде зарубежных стран, содержит 16—16,5%
N и 25% КгО. Ее изготовляют следующими способами: 1) механи-
ческим смешением сухих или увлажненных нитрата аммония и хло-
рида калия; 2) совместным выпариванием растворов нитрата аммо-
ния и хлорида калия; 3) введением в концентрированный раствор
или плав аммиачной селитры тонкоизмельченного хлорида калия
с последующим гранулированием плава в грануляционных башнях.
Последний способ представляет наибольший интерес, так как по-
зволяет получить более однородное по составу удобрение.
При получении калийно-аммиачной селитры по любому спо-
собу протекает в той или иной мере реакция обменного разложения:
КС1+NH4NO3 = KNO3 + NH4C1
Получаемый продукт содержит тесно сросшиеся кристаллы
KNO3 и NH4C1 198. В результате образования KNO3 — почти не гиг-
роскопичной соли, — калийно-аммиачная селитра имеет лучшие фи-
зические свойства, чем чистый нитрат аммония.
В зависимости от условий приготовления калийно-аммиачная
селитра содержит различное количество KNO3. При механическом
смешении увлажненных компонентов (2% влаги) образуется около
7% KNO3, в случае совместного выпаривания растворов нитрата
аммония и хлорида калия реакция образования KNO3 протекает
почти нацело.
При смешении плава аммиачной селитры, состоящего из
45 вес. ч. нитрата аммония и 5 вес. ч. воды, с 55 вес. ч. размолотого
хлорида калия, около 88% нитрата аммония превращается в KNO3.
При этом получается сухое рассыпчатое удобрение, которое не сле-
живается и легко рассевается. В условиях смешения горячего плава
Нитрит аммония
1209
с хлоридом калия возможны потери азота; при 130° потери состав-
ляют 0,3% от общего веса смеси.
На некоторых заводах кристаллизацию калийно-аммиачной се-
литры из плава проводят на поверхности охлаждаемого вращающе-
гося барабана. Далее продукт размалывают на сравнительно круп-
ные частицы (3—4 мм) и сушат. В США испытан метод продавли-
вания горячего плава в формовочной машине через отверстия
диаметром 3 мм. При этом получается шнур, который охлаждают
воздухом на сетчатом конвейере и сушат. Затем его разрезают на
частицы длиной 3—4 мм.
НИТРИТ АММОНИЯ
Как и большинство солей азотистой кислоты нитрит аммония
NH4NO2 термически не стоек и в твердом виде сохраняется только
при обыкновенной температуре (20—25°) 199. Загрязненный нитрит
аммония очень не стоек и постепенно разлагается даже при 20°.
Разложение его при 50° по уравнению
NH4NO2 = N2 ч-2Н2О (г.) ч-51,6 ккал
идет с большой скоростью [скорость детонации (NH4NO2) прибли-
женно равна 4000 м/сек]. Нитрит аммония гигроскопичен. В 100 г
насыщенного водного раствора нитрита аммония содержится при
—11° 50, при 19° 64,3, при 33,5° 75 г соли. Насыщенные растворы
нитрита аммония во избежание сильного разложения хранят при
10—15°.
Нитрит аммония имеет ограниченное применение. Раствор
NH4NO2 используют для получения гидроксиламинсульфата в про-
изводстве капролактама. Предложено получать из NH4NO2 водные
растворы NH4NO3 разложением NH4NO2 азотной кислотой в рас-
пыленном состоянии 200. Но это предложение вряд ли найдет прак-
тическое применение.
Производственные растворы нитрита аммония приготавли-
ваются абсорбцией окислов азота аммиачной водой или раствором
карбоната аммония:
NO + NO2 + (NH4)2CO3 = 2NH4NO2 + СО2
2NO2 + (NH4)2CO3 = NH4NO2 + NH4NO3 + CO2
Для получения раствора NH4NO2 с малым содержанием в нем
NH4NO3 необходимо, чтобы степень окисленности нитрозного газа,
поступающего в абсорбер, не превышала 35—37%.
Для уменьшения разложения нитритный раствор в процессе аб-
сорбции нитрозных газов охлаждают до 0—4°. Абсорбцию можно
осуществлять как при атмосферном так и повышенном давле-
нии201.202. Раствор нитрита аммония, предназначенный для произ-
водства гидроксиламинсульфата, имеет следующий примерный со-
став (в г/л): NH4NO2 150—200; NH4NO3 до 20; (NH4)2CO3 20—30:
своб. NH3 до 6; плотность раствора 1,08 г/см3.
14*
1210
Гл. XXXII. Соли азотной кислоты
Разработан способ получения нитрита аммония взаимодей-
ствием NaNOs с NH4CO2NH2 в среде жидкого аммиака при темпе-
ратуре ниже 30° 203.
НИТРАТ КАЛЬЦИЯ204
Физико-химические свойства
Рис. 351. Растворимость в системе
Ca(NO3)2-H2O.
Нитрат кальция (известковая или кальциевая селитра) — бес-
цветное вещество, образующее несколько кристаллогидратов
(рис. 351). При обычной температуре из раствора кристаллизуется
стабильная a-форма тетрагидрата Са (NO3)2 • 4Н2О. Растворимость
тетрагидрата характеризуется
кривой АВС. Максимум на
этой кривой в точке В соответ-
ствует плавлению тетрагидра-
та при 42,7°. Линия А'С' —
растворимость метастабильной
p-формы тетрагидрата. Кри-
вая CDE — растворимость
Ca(NO3)2-3H2O с максимумом
в точке D при температуре
плавления тригидрата 51,1°.
Линия EF — растворимость
метастабильного бигидрата
Ca(NO3)2-2H2O. Линия EG —
растворимость безводной соли,
почти не увеличивающаяся с
повышением температуры. Точ-
ка G соответствует температу-
ре кипения насыщенного рас-
твора (151°), содержащего
78,4% Ca(NO3)2. Безводная
соль плавится при 561°, однако
уже при 500° начинается ее
разложение с потерей кислорода и образованием нитрита кальция,
который далее распадается на СаО и NO2.
Как безводная соль, так и кристаллогидраты нитрата кальция
сильно гигроскопичны и на воздухе расплываются. Давление пара
над 50% раствором азотнокислого кальция 205’206 в диапазоне темпе-
ратур от 70 до 110° возрастает от 135 до 730 мм рт. ст., а над 75%
раствором в диапазоне 90—140° возрастает от 90 до 740 мм рт/ст.
Раствор с концентрацией 77,9% Ca(NO3)2 кипит при 143,3° под
нормальным давлением и при 117° под давлением 300 мм рт. ст. '.
Вязкости растворов кальциевой селитры 1 при различных тем-
пературах (в сп) : 40%-ного раствора — 2,05 (50°) и 0,96 (100°);
Нитрат кальция
12П
60%-ного — 7,73 (60°) и 3,71 (100°); 80%-ного—105,62 (90°) и
51,65 (110°).
Вязкости растворов кальциевой селитры с 5%-ной добавкой
МНдМОз: при концентрации Ca(NO3)2 41,8%—3,22 (50°) и 1,22
(90°); при концентрации Са(ЫО3)г 60% — 10,12 (50°) и. 1,80 (130°);
при концентрации Ca(NO3)2 79%—46,0 (120°) и 30,4 (130°).
Применение
Кальциевая селитра является универсальным физиологически
щелочным удобрением, пригодным для всех почв и прежде всего
для почв нечерноземной полосы с недостаточным содержанием
кальция 205. Однако, вследствие малого содержания азота (15,5%),
перевозка ее на дальние расстояния невыгодна.
Большим недостатком кальциевой селитры является также ее
высокая гигроскопичность.
Согласно МРТУ 6-03 195—67, кальциевую селитру для удобре-
ния выпускают в виде чешуйчатого продукта, светло-коричневого
цвета, содержащего (в пересчете на безводное вещество) не менее
17,5% общего азота; содержание аммиачной селитры .4—7%; со-
держание влаги (общей) не должно превышать 14%.
С целью улучшения агрохимических свойств допускается по-
верхностная обработка кальциевой селитры углекислым кальцием
(до 6%), или парафинистым мазутом (до 2%). Кальциевую се-
литру хранят в закрытых сухих складах в битумированных бумаж-
ных мешках.
Методы производства 207-211
Известны следующие промышленные методы производства
кальциевой селитры:
1) прямое взаимодействие мела или известняка с азотной кис-
лотой;
2) азотнокислотная экстракция фосфатов (см гл. XXXV) 212’213;
3) поглощение нитрозных газов известковым молоком с после-
дующей инверсией полученных растворов азотной кислотой;
4) взаимодействие окиси кальция с двуокисью азота при 300—
400°— так называемый сухой способ, имеющий ограниченное при-
менение.
Производство нитрата кальция из
известняка и азотной кислоты
На диаграмме рис. 352 изображено равновесие в системе
Ca(NO3)2—HNO3—Н2О при 25°. Точки S2, S3 и Х4 соответствуют
Ди-, три- и тетрагидрату Ca(NO3)2- Области существования
этих кристаллогидратов находятся в пределах заштрихованных
1212
Гл. XXXII. Соли азотной кислоты
криволинейных треугольников: в треугольнике SJA^N' — тетрагид-
рат, в треугольнике —• тригидрат, в треугольнике S2MN —
дигидрат. Эти области ограничены кривой N'MrMN растворимости
кристаллогидратов в кислых растворах и линиями перехода кри-
сталлогидратов. Ниже и левее линии растворимости находится об-
ласть ненасыщенных кислых растворов.
Для получения нитрата кильция применяется обычно слабая
азотная кислота. При нейтрализации известняком 30 %-ной (А) или
Рис. 352. Равновесие в системе
Ca(NO3)2—HNO3—Н2О при 25°.
18%-ной (А') азотиой кислоты изменение состава раствора по мере
нейтрализации изображается прямыми АВ и А'В'. Эти прямые не
пересекают кривой насыщения N'MjMN. Следовательно, твердую
соль можно получить лишь после выпаривания нейтрализованного
раствора.
Допустим, что в результате нейтрализации 30%-иой кислоты из-
вестняком получен слабокислый раствор, состав которого изобра-
жается точкой Ро. При выпаривании этого раствора фигуративная
точка системы, из которой удаляется вода, будет передвигаться
вдоль прямого луча РоР. На этом луче интервал Р]Р соответствует
содержанию в системе приблизительно 75—78% Ca(NO3)2. Если
состав раствора после выпаривания изображается точкой Р, то при
его охлаждении до 25° выкристаллизуется смесь ди- и тригидрата.
Состав этой смеси изображается точкой S, а состав маточного
раствора — точкой М.
Если же выпаривание произведено до точки то после
охлаждения до 25° будет получена смесь кристаллов три- и тетра-
Нитрат кальция
1213
гидрата состава Si; состав маточного раствора в этом случае изо-
бразится точкой При выпаривании до точки Р3 после охлажде-
ния будут получены кристаллы тригидрата Ca(NO3)2 • ЗН2О.
Нейтрализацию кускового известняка или мела осуществляют
обычно в башнях циркулирующей 40—48 %-ной азотной кислотой.
Кислый раствор кальциевой селитры, вытекающий из башни, про-
ходит отстойники для отделения от частиц песка и других нерас-
творимых примесей и снова подается на орошение башни.
Когда остаточная кислотность раствора понизится до 1,5—1,9%
HNO3, его отводят на донейтрализацию аммиаком или известью —
пушонкой. При применении аммиака в кальциевой селитре обра-
зуется некоторое количество нитрата аммония, в присутствии ко-
торого облегчается процесс кристализации Ca(NO3)2 из плава.
Башни, донейтрализатор, отстойники и насосы изготовляются
из нержавеющей стали марки 1Х18Н9Т. Для фильтрования обра-
зующихся вязких растворов Ca(NO3)2 обычно применяют фильтр-
прессы. Отфильтрованный раствор, содержащий ~49% Са(МО3)г
и 3% NH4NO3, выпаривают под вакуумом до 75—82% Ca(NO3)2.
Получаемый плав кальциевой селитры кристаллизуют на охлаж-
дающих вальцах или в грануляционной башне.
В СССР кальциевую селитру производят не из азотной кислоты,
а из отбросных нитрозных газов, что дает более дешевый продукт.
Производство нитрата кальция из нитрозных
ГаЗОВ 210» 211, 214, 215
При промывке нитрозных газов известковым молоком обра-
зуется щелок, содержащий нитрит кальция с примесью некоторого
количества нитрата кальция:
Са(ОН)2 + NO + NOj = Ca(NO2)2 + Н2О
2Са(ОН)2 + 4NO2 = Ca(NO3)2 + Ca(NO2)2 + 2Н2О
Абсорбцию нитрозных газов известковым молоком осуществ-
ляют в башнях циркулирующим раствором, в котором поддержи-
вают избыточную щелочность до 30 г/л добавкой к нему известко-
вого молока, содержащего 100—130 г/л СаО или сухой извести,
что позволяет получать более концентрированный щелок. Темпера-
тура щелока, поступающего на орошение, 30—35°. Из хвостовых
газов улавливается ие менее 92% окислов азота.
Вытекающий из абсорбционных башен щелок подвергается'
инверсии азотной кислотой с целью перевода нитрита в нитрат:
3Ca(NO2)2 + 4HNO3 = 3Ca(NOs)2 + 2Н2О + 4NO
Выделяющиеся при инверсии окислы азота возвращают в аб-
сорбционную систему производства слабой азотной кислоты. Ин-
версию проводят при обогреве острым паром и при интенсивном
перемешивании раствора сжатым воздухом (рис. 353).
1214
Г л. XXXII. Соли азотной кислоты
В инвертированном растворе Ca(NO3)2 остается избыток азот
ной кислоты, которую нейтрализуют аммиаком. При этом обра
зуется некоторое количество аммиачной селитры, что в данном слу
чае является полезным (стр. 1215). Далее раствор фильтруют, по
догревают до 60—70° и направляют на выпаривание в одно-
многокорпусные выпарные установки. Обычно применяют выпар
ные аппараты вертикального типа с внутренней или
греющей камерой.
и
ВЫНОСНОЙ
Рис. 353. Схема производства кальциевой селитры методом щелочного
поглощения нитрозных газов:
/ — поглотительная башня; 2 — сборник нитрит-нитратного раствора; 3 — центробежный
насос; 4 —инвертор; 5 — донейтрализатор; 6 — фильтрпресс; 7 —напорный бачок для
отфильтрованного раствора; S —выпарной аппарат; 9 —барометрический конденсатор;
10 — сборник выпаренного раствора кальциевой селитры; 11 — сборник плава кальцие-
вой селитры; 12 — барометрический ящик; 13 — охлаждающие вальцы.
При одноступенчатой схеме выпарки (однокорпусная
ка) выпаривание проводится при следующих условиях:
установ
5
Давление греющего пара, ат..........’.................
Разрежение в аппарате, мм рт. ст......................
Температура плава на выходе из аппарата, °C....... .
Температура сокового пара, °C ........................
Состав раствора, поступающего на выпарку............; .
Состав плава на выходе из аппарата ...................
500
120
105
25% Ca(NO3)2
1,4% NH4NO3
77-82% Ca(NO
5-6% NH4NO3
В случае применения трехступенчатой выпарки первый
и вто
рой выпарные аппараты работают непрерывно, третий
чески. Режим работы трехкорпусной установки
периоди
характеризуется
следующими показателями:
Концентрация раствора, %
на входе в аппарат ........................
на выходе из аппарата......................
Давление греющего пара, ат.....................
Давление сокового пара, ат.....................
Температура кипения раствора, °C..............
I П АП
корпус корпус корпус
25 36 ' 60
36 60 86
6 1,5 6
1,5 0,2 0,2
118-120 78-80 110-11
Нитрат кальция
1215
Кристаллизация является наиболее сложным процессом в про-
изводстве кальциевой селитры 214-217_ Механизм образования кри-
сталлов кальциевой селитры до настоящего времени недостаточно
выяснен. Структура образующихся кристаллов соли очень раз-
лична: иногда при кристаллизации получаются твердые, легко от-
деляющиеся от охлаждающей поверхности агрегаты кристаллов, в
других случаях образуется вязкий сиропообразный плав, не засты-
вающий в твердую корочку. Даже при сильном переохлаждении
плава кальциевой селитры, в некоторых случаях кристаллизация
может еще не наступить. Чтобы вызвать кристаллизацию, в плав
вносят затравку — кристаллы кальциевой селитры, однако кристал-
лизация при этом не всегда проходит одинаково. Большое влияние
на процесс кристаллизации кальциевой селитры оказывает аммиач-
ная селитра, в присутствии которой скорость кристаллизации
Ca(NO3)2 увеличивается в 1,5—2 раза с одновременным повыше-
нием температуры кристаллизации на 50°. Чем больше содержится
аммиачной селитры в растворе, тем лучше идет кристаллизация.
Обычно добавка NH4NO3 составляет 5—6% от веса кальциевой се-
литры.
Установлено 208, что при содержании 5% аммиачной селитры
72%-ный плав кальциевой селитры (плотность 1,72 г/см3) на охлаж-
дающих вальцах совсем не кристаллизуется; плав, содержащий
73—82% Ca(NO3)2 + NH4NO3 (плотность 1,76— 1,88 г/сЛ3), кри-
сталлизуется хорошо, 83%-ный плав (плотность свыше 1,88 г/см3)
кристаллизуется плохо. Примеси нитратов железа и алюминия
почти не оказывают влияния на скорость кристаллизации, в при-
сутствии же силикатов и нитрата натрия кристаллизация затруд-
няется — получаются липкие и плохо затвердевающие кристаллы
соли.
Кристаллизацию кальциевой селитры с добавкой аммиачной
селитры на охлаждающих вальцах проводят при 90°; при этом
большая часть соли кристаллизуется в виде двухводного гидрата.
Температуру плава в корыте вальцов поддерживают около 110°.
Перед загрузкой в тару кальциевую селитру следует охладить по
крайней мере до 30°, так как горячая соль склонна к слипанию,
что ухудшает ее рассеваемость. Охлаждение кальциевой селитры
производят в барабане, через который продувают охлажденный
воздух. Схема охлаждения воздуха такая же, как и при «холод-
ной» сушке аммиачной селитры (стр. 1196).
Для производства 1 т кальциевой селитры [82% Ca(NO3)2]
расходуется 3,3 т 25%-ного раствора Ca(NO3)2, 2 т пара (6 ат),
75 м3 воды, 23,5 кет • ч электроэнергии.
Разработан способ гранулирования кальциевой селитры (а так-
же карбамида и других плавов), рекомендуемый вместо охлаж-
дения ее на поверхности барабана или разбрызгивания в башне
ь потоке воздуха. Этот способ заключается в кристаллизации
1216
Гл. XXXII. Соли азотной кислоты
капель раствора Ca(NO3)2, разбрызгиваемых в минеральном мас-
ле, содержащем зародыши кристаллов, с последующим отделением
гранул от масла центрифугированием 218. -
Предложено также упарку растворов, грануляцию и охлажде-
ние нитрата кальция производить в одном аппарате с кипящим
слоем 21Э.
НИТРАТ И НИТРИТ НАТРИЯ204
Физико-химические свойства
Нитрат натрия NaNO3 (натриевая селитра) представляет со-
бой бесцветные, прозрачные кристаллы, имеющие плотность
Рис. 354. Политерма растворимости в системе
NaNO3—NaNO2—Н2О.
2,257 г/см3. Температура
плавления 309,5°, выше
380° он разлагается на
нитрит натрия и кисло-
род.
Нитрит натрия NaNO2
представляет собой бес-
цветные или слегка жел-
товатые кристаллы плот-
ностью 2,17 г/сл3; темпе-
ратура плавления 271°.
Растворимость NaNO3 в
воде (в г/100 г Н2О):
при 0° — 70,7; 20° — 88;
120° —213; 180° —383.
Температура кипения на-
сыщенного раствора при
736 мм рт. ст. равна 119°.
Растворимость NaNO2 в
воде (в а/100 г Н2О):
при 0° —73; 20° — 81,8;
100°—163; 128° — 219,5. Температура кипения насыщенного рас-
твора при 761,5 мм рт. ст. 128°. Политерма растворимости в систе-
ме NaNO3—NaNO2—Н2О представлена на рис. 354.
Применение
Нитрат натрия — ценное удобрение, содержащее 16,47% азота.
Это физиологически щелочное удобрение, поэтому применение его
наиболее целесообразно на кислых почвах. Нитрат натрия находит
также применение в пищевой, стекольной и металлообрабатываю-
щей промышленности, входит в состав взрывчатых веществ, ракет-
ных топлив и пиротехнических смесей 220.
Нитрат и нитрит натрия
1217
Нитрит натрия используют в производстве органических краси-
телей, в пищевой, текстильной, резиновой промышленности и в ме-
дицине. NaNO2 — ядовитое вещество, и при производстве NaNO2 и
обращении с ним следует соблюдать меры предосторожности.
(В последнее время большое значение приобрел также нитрит
аммония, применяемый для получения гидроксиламина — одного
из исходных материалов в производстве капролактама. Нитрит
аммония образуется при поглощении окислов азота в щелочной
среде в присутствии ионов NH4 или взаимодействием окислов азо-
та с аммиаком в газовой фазе221).
Нитрат натрия выпускают двух сортов (ГОСТ 828—68): в про-
дукте 1-го сорта должно быть не менее 99,5% NaNO3 (в пересчете
на сухое вещество) и не более 1,0% влаги; в продукте 2-го сор-
та — не менее 99% NaNO3 и не более 1,8% влаги; в продукте 1-го
сорта должно быть не более: 0,05% не растворимых в воде веществ,
0,5% хлоридов (NaCl), 0,02% окисляемых веществ (NaNO2).
Нитрит натрия выпускают также двух сортов (ГОСТ 6194—69)г
в продукте l-ro сорта содержание NaNO2 должно быть не менее
98,5%, NaNO3—не более 1,0%, а не растворимого в воде остат-
ка— не более 0,03% (в сухом веществе); влаги не более 1,8%; про-
дукт 2-го сорта должен содержать не менее 97% NaNO2, не более
2,3% NaNO3, 0,07% нерастворимого остатка, 2,5% влаги. Кроме
того, выпускается аттестованный продукт (ГОСТ 5.1077—71), ко-
торый должен содержать в сухом веществе не менее 98,7 % NaNO2
и не более 1,0% NaNO3 и 0,03% не растворимого в воде остатка;
содержание влаги не должно превышать 1,4%.
Способы производства нитрата натрия
Нитрат натрия добывают из природных залежей и производят
заводскими способами. В СССР имеются природные месторожде-
ния нитрата натрия в Средней Азии, на Кавказе, в Крыму, однако
они не имеют промышленного значения 222-223.
Промышленное месторождение натриевой селитры имеется в
Чили (Южная Америка), где и производится ее добыча.
Содержание NaNO3 в селитроносной земле чилийского место-
рождения сильно колеблется. Самый богатый селитроносный слой,
называемый калише, имеющий толщину 0,25—1,5 м, залегающий на
глубине 0,5—2,5 м от поверхности земли, содержит обычно от 15 до
65%, а иногда и до 95% NaNO3. Типичной является порода, содер-
жащая около 18% NaNO3. Помимо NaNO3 в породе содержатся
различные количества NaCl, KNO3, Na2SO4, MgSO4, CaSO4 и др.
Извлечение селитры из калише осуществляется противоточным
горячим выщелачиванием и последующей кристаллизацией при
охлаждении почти насыщенного раствора. Получаемый продукт со-
держит 94—96% NaNO3 и примеси других солей. Более чистую
(рафинированную) селитру, содержащую 99—99,5% NaNOs, полу-
чают перекристаллизацией.
1218
Гл. XXXII. Соли азотной кислоты
Заводские способы получения нитрата натрия основаны на аб-
сорбции окислов азота раствором соды, или на обменном разложе-
нии других нитратов с соединениями натрия, или на катионном об-
мене.
Запатентован 224 способ получения NaNO3 из NaCl и азотной
кислоты (аналогичный рассмотренному ниже способу получения
KNO3 — см. стр. 1229).
Наиболее простой способ получения нитрата натрия — нейтрали-
зация азотной кислоты содой или едким натром в промышленности
не используется из-за неэкономичности. Наиболее распространен-
ный промышленный способ основан на абсорбции щелочами окис-
лов азота из выхлопных нитрозных газов производства азотной
кислоты.
Получение нитрата натрия абсорбцией
окислов азота щелочами 225-230
При абсорбции окислов азота щелочами протекают следующие
реакции:
2NaOH + NO + NO2 = 2NaNO2 + H2O
2NaOH + 2NO2 = NaNO3 + NaNO2 + H2O
ИЛИ
Na2CO3 + NO + NO2 = 2NaNO2 + CO2
Na2CO3 + 2NO2 = NaNO3 + NaNO2 + CO2
Получаемые в результате щелочной абсорбции нитрозных газов
растворы (нитрит-нитратные щелоки) перерабатывают в нитрат
натрия (рис. 355). Соотношение между нитратом и нитритом нат-
рия в получаемых щелоках зависит от температуры и степени окис-
ленности нитрозных газов 228. Примерный состав «сырых» нитрит-
нитратных растворов (после щелочного поглощения): 250—400 г/л
NaNOs", 50—80 г/л NaNOj; 3—5 г/л NaHCO3 и ЫагСОз; 2—4 г/л
NaCl; 850—820 г/л Н2О.
Отфильтрованный на фильтрпрессе раствор поступает в аппа-
рат-инвертор периодического или непрерывного действия, в кото-
ром происходит превращение (инверсия) нитрита в нитрат натрия
под действием азотной кислоты:
3NaNO2 + 2HNO3 = 3NaNO3 + 2NO + Н2О
Образующиеся в результате инверсии окислы азота удаляются
из раствора нагнетаемым в инвертор воздухом и направляются на
переработку в абсорбционное отделение цеха слабой азотной кис-
лоты. По окончании процесса инверсии раствор нейтрализуют со-
довым раствором. Вновь полученный раствор, содержащий 40—50%
NaNOs и имеющий температуру 40—60°, направляют на выпари-
вание.
1220
Гл. XXXI1. Соли азотной кислоты
Предложено инверсию нитритных щелоков проводить в пенном
аппарате — процесс протекает с большой скоростью и степень ин-
версии достигает 99,9% 231.
Для выпаривания растворов натриевой селитры обычно приме-
няют двухкорпусную вакуум-выпарную установку. Здесь получают
суспензию, в которой содержание NaNO3 составляет ~75%, при
этом в растворе содержится 62% NaNO3, остальное количество со-
ли находится во взвешенном состоянии в виде кристаллов. Эта сус-
пензия из выпарного аппарата поступает в шнековый кристаллиза-
тор с водяной рубашкой, в котором при охлаждении от 90—93° до
40—45° происходит дальнейшая кристаллизация’ соли. Кристаллы
NaNO3 отделяют от раствора в центрифуге и высушивают в су-
шильном барабане 232. Маточный раствор из центрифуги присоеди-
няют к инвертированному раствору, направляемому на выпари-
вание.
Для получения 1 т натриевой селитры (100% NaNO3) расхо-
дуют: 0,455 т азотной кислоты (100%), 0,03 т соды, 2,5 т пара
(8 ат), 65 м'А воды, 120 кет • ч электроэнергии. В качестве отхода
производства получают (на 1 т NaNO3) 0,38 т окислов азота (в пе-
ресчете на HNO3), направляемых на абсорбцию.
Конверсионный способ получения нитрата натрия
Нитрат натрия может быть получен методами обменного разло-
жения по следующим реакциям:
Ca(NO3)2 + Na2SO4 = 2NaNO3 + CaSO4
Ca(NO3)2 + 2NaCl = 2NaNO3 + CaCl2
NH4NO3 + NaCl = NaNO3 + NH4C1
2NH4NO3 + Na2CO3 = 2NaNO3 + (NH4)2CO3 и др.
Каждая из этих реакций имеет свои специфические особенно-
сти. Так, при проведении обменного разложения между нитратом
кальция и сульфатом натрия необходимо вводить в процесс избы-
ток нитрата кальция, который частично попадает в продукт и де-
лает его гигроскопичным. Выделение в осадок гипса приводит к
сильному загустеванию реакционной массы; для того чтобы она
оставалась достаточно подвижной, необходимо разбавлять ее рас-
твором нитрата натрия после отделения от него гипса, т. е. возвра-
щать часть раствора в реактор. Скорость обменного разложения
зависит от температуры. Так, в данном случае реакция идет при
50° значительно быстрее, чем при 70°; в течение 1 ч при 50° конвер-
сия достигает 96%, а при 70° только 11%. Для получения крупно-
кристаллического, легко отфильтровываемого осадка гипса необхо-
димо вводить затравку, добавляя в реактор кристаллический гипс
и т. д.
Нитрат и нитрит натрия
1221
Производство нитра та натрия методом
катионного обмена
Представляет интерес разработка новых способов производства
нитратов калия и натрия, не требующих затраты щелочей. К таким
способам относится получение нитрата натрия из NaCl и окислов
азота, аналогичное получению калиевой селитры (стр. 1229), а так-
же метод катионного обмена 233’234.
Процесс получения натриевой (или калиевой) селитры методом
катионного обмена состоит из трех основных стадий:
1) получение раствора Ca(NO3)2;
2) катионный обмен и регенерация катионита (обмен цонов Саг+
на ионы Na+);
3) выпаривание раствора NaNO3, кристаллизация соли, центри-
фугирование, сушка и упаковка.
. Реакции, протекающие в первых двух стадиях процесса, могут
быть схематически выражены следующими уравнениями:
Ca(NO3)2 + (катионит)\а2 < 2NaNO3 + (катионит)Са
(катнонит)Са + 2NaCl < (катнонит)№2 + СаС12
При этом происходит непрерывное чередование процессов кон-
версии кальциевой селитры и регенерации катионита кальция рас-
творами хлорида натрия (или хлорида калия).
Промежуточными стадиями этого процесса являются: промыв-
ка водой катионита, выпарка и кристаллизация конвертированных
растворов.
Описываемый метод нашел промышленное применение в Норве-
гии. Растворы кальциевой селитры пропускают через ряд катионо-
обменников, в которые загружен цеолит (природный минерал типа
полевого шпата). Для регенерации цеолита используют морскую
воду. Образующийся раствор натриевой селитры концентрируют
выпариванием и далее кристаллизуют соль с последующей пере-
кристаллизацией (для получения более чистого продукта). Гото-
вый продукт используют как удобрение.
Получение нитрита натрия
Одним из способов получения нитрита натрия является его вы-
деление из нитрит-нитратного щелока до инверсии. В этом случае
процесс абсорбции окислов азота ведут таким образом, чтобы по-
лучить щелок по возможности с большим содержанием нитрита и
с меньшим содержанием нитрата. Этого можно достигнуть умень-
шением содержания кислорода в газе, поступающем на абсорбцию,
чтобы к моменту подхода газа к щелочным башням степень окис-
ления содержащихся в нем окислов азота соответствовала эквимо-
лекулярной смеси NO + NO2.
1222
Гл. XXXII. Соли азотной кислоты
Нитрит натрия можно получить также из нитрит-нитратных рас-
творов, образующихся при поглощении окислов азота раствором
соды по обычному режиму, но при небольшом изменении схемы
процесса переработки растворов. В этом случае «сырой» нитрит-ни-
тратный щелок нагревают до 80—90° .для перевода растворимых
примесей — бикарбонатов магния и кальция — в нерастворимые
карбонаты. Затем щелок профильтровывают или осветляют и на-
правляют в выпарную установку, подобную используемой для вы-
паривания инвертированного раствора. Выпаривание ведут при
118—125° 235. В выпарной установке содержание NaNO2 в растворе
доводится до ~63°/о.
Далее при охлаждении раствора до 40—45° в кристаллизаторе
часть нитрита натрия кристаллизуется. Полное выделение нитрита
натрия в одном цикле кристаллизации невозможно, что видно из
диаграммы растворимости в системе NaNO3—NaNO2—Н2О
(стр. 1216). Эвтонические растворы при всех температурах содержат
значительные количества NaNO2. Кристаллический осадок NaNO2
отделяют на центрифугах от раствора и промывают паровым кон-
денсатом для отмывки ионов хлора, которые содержатся в воде.
Затем нитрит натрия высушивают в сушильном барабане нагре-
тым до 100° воздухом. Готовый продукт содержит менее 1,5% влаги
Маточный раствор, содержащий после отделения кристаллов
нитрита натрия, приблизительно 500 г/л NaNO2 и 200 г/л NaNO3.
вместе с промывными водами направляется в инвертор, где нитрит
натрия окисляется в нитрат. Дальнейшая переработка инвертиро-
ванного раствора проводится по описанной выше схеме (см.
рис. 355).
Выход нитрита натрия и его качество зависят главным образом
от отношения нитрита к нитрату в «сыром» щелоке. Качество про-
дукта улучшается, если в процессе получения «сырого» нитрит-нит-
ратного щелока используют не техническую воду, а паровой кон-
денсат (для растворения соды и др.).
НИТРАТ КАЛИЯ204
Физико-химические свойства 1
Нитрат калия KNO3 (калиевая селитра) представляет собой без- I
водную кристаллическую соль белого цвета (иногда с желтовато- )
сероватым оттенком), плотностью 2,11 г!см3, плавящуюся при 334°. j
Выше 338° разлагается на нитрит калия и кислород. Известны две |
кристаллические модификации KNO3: при низких температурах об-
разуются кристаллы ромбической формы, при более высоких темпе- |
ратурах — ромбоэдрические кристаллы 236. Растворимость KNO3 в 1
воде (в г/100 г Н2О): при 20° —31,5; 30° — 45,6; 40° —63,9; 60°— I
109,9; 114°—312. I
Нитрат калия
1223
В растворах азотной кислоты растворимость KNO3 с увеличе-
нием концентрации кислоты понижается, достигает минимума, за-
тем возрастает; минимальная растворимость KNO3 при 50° равна
24,91% при содержании в растворе 27,63% HNO3 и 47,46% Н2О 237.
Применение
Нитрат калия используют для производства порохов, в пиро-
технике, в пищевой и стекольной промышленности и как удобре-
ние238. К преимуществам'нитрата калия по сравнению со многими
другими удобрениями относятся: отсутствие балласта, что особенно
важно при дальних перевозках и малая гигроскопичность.
' Согласно ГОСТ 1949—65, к качеству калиевой селитры предъ-
являются-следующие требования (в %):
Сорт 1-й Сорт 2-Й Сорт 3-й
KNO3 (в пересчете на сухой продукт), не менее 99,8 99,5 98,0
Влага, не более 0,1 02 2,0
Хлористые соли (в пересчете на NaCl), не более 0,03 0,1 Не норми-
Углекислые соли (в пересчете иа К2СО3). не более 0,02 0.04 руется То же
Не растворимый в воде остаток, не более 0,03 0,03 » »
Не растворимый в соляной кислоте про- каленный остаток, не более 0,035 0,02 > »
Окисляемые вешества, не более 0,01 Не яорми- » »
Соли кальция и магния (в пересчете на кальций), не более 0,02 - руется 0,02 > » >
Калиевая селитра, предназначаемая для производства порохов,
должна содержать солей кальция и магния не более 0,002%.
Качество аттестованного продукта регламентировано ГОСТ
5.1138—71; он должен содержать в сухом веществе не менее 99,9%
KNO3; содержание примесей не должно превышать: влаги — 0,08%,
хлоридов (NaCl)—0,017%, карбонатов (К2СО3)—0,01%, не рас-
творимого в воде остатка — 0,01%, не растворимого в соляной кис-
лоте прокаленного остатка — 0,004%, веществ, окисляемых мар-
ганцевокислым калием (в пересчете на KNO2) — 0,01%, солей Са
и Mg (в пересчете на Са) — 0,002%.
Способы производства нитрата калия
Нитрат калия в природе встречается в виде небольших зале-
жей. Искусственным способом, известным с давних времен, калие-
вую селитру получали в так называемых селитряницах из компо-
стов, в которые входили навоз, зола, известь, хворост и др. В ре-
зультате биохимических процессов с течением времени в таких
компостах образовывалась калиевая селитра, которую выщелачи-
вали водой и подвергали кристаллизации; при этом получался
сравнительно чистый продукт.
1224
Гл. XXXII. Соли азотной кислоты
Получение нитрата калия нейтрализацией щелочей азотной кис-
лотой вследствие необходимости затраты дорогого сырья — едкого
кали или поташа и азотной кислоты осуществляют редко. Для ней-
трализации берется 30—35% раствор КОН и 50%-ная кислота или
сухой поташ, содержащий 85—87% К2СО3 и около 5% КНСОз, и
25—30%-ная азотная кислота. Полученный раствор, содержащий
около 30% KNO3, выпаривают при 110—120°, отфильтровывают от
примесей на фильтрпрессе и направляют на кристаллизацию. Кри-
сталлы отфуговывают и высушивают. •
Способ получения нитрата калия абсорбцией калиевыми щело-
чами нитрозных газов также применяется в ограниченных масшта-
бах из-за дефицитности едкого кали и особенно поташа. Процесс
этот аналогичен описанному выше процессу получения нитрата нат-
рия абсорбцией нитрозных газов содой. При подаче на абсорбцию
раствора едкого кали отбираемый из первой абсорбционной башни
щелок содержит 350—400 г/л KNO2, 80—100 г/л KNO3 и 2—3 г/л
КОН. Инверсия проводится при 70—80° с избытком азотной кис-
лоты до 30 г/л
Наибольшее промышленное распространение имеет конверсион-
ный способ получения нитрата калия. Представляет интерес полу-
чение его из окислов азота и хлористого калия.
Нитрат калия (так же как и натрия) получают с помощью ка-
тионного обмена (см. стр. 1221).
Производство нитрата калия конверсионным
способом 239-244
Этот способ основан на обменном разложении NaNO3 и КС1:
NaNO3 + КС1 = NaCl + KNO3
Обменное разложение с целью получения нитрата калия может
осуществляться и между другими нитратами и хлоридом, сульфа-
том или карбонатом калия, например:
NH4NO3 + КС1 = KNO3 4- NH4CI
Однако в промышленности используют пока только первую
реакцию.
На рис. 356 изображена диаграмма растворимости в системе
КС1 + NaNO3 NaCl + KNO3 при 5, 25, 50 и 100° 242. При низких
температурах поле кристаллизации KNO3 занимает большую часть
площади квадрата. Если приготовить раствор эквимолекулярной
смеси КО и NaNO3 при 100°, то фигуративная точка а солевой
массы.такого раствора, лежащая на пересечении диагоналей ква-
драта, окажется в поле кристаллизации NaCl. При выпаривании
из этого раствора воды при 100°, когда будет достигнуто насыще-
ние, начнется кристаллизация NaCl, и состав солевой массы рас-
Нитрат калия
1225
твора будет изменяться по линии ab. В точке b раствор станет на-
сыщенным также и КС1. Если выделившиеся к этому моменту
кристаллы NaCl отделить и затем охладить раствор, например до
5°, то точка b окажется в поле кристаллизации KNO3; эта соль и
будет выделяться в осадок при охлаждении, причем состав рас-
твора будет меняться по линии Ьс.
Так как расстояние между точками а и b невелико, то при вы-
паривании воды из раствора, содержащего эквимолекулярные ко-
личества NaNO3 и КС1, в осадок выделяется лишь небольшое коли-
чество NaCl, и раствор вскоре
становится насыщенным также и
хлоридом калия. Это уменьшает
и выход кристаллического KNO3
при охлаждении раствора. Чтобы
увеличить количество отделяемо-
го NaCl и повысить выход KNO3,
как видно из диаграммы, следует
вводить в исходный раствор из-
быток NaNO3. Наибольший вы-
ход получается, если к концу вы-
деления NaCl раствор насыщен
тремя солями —NaCl, КС1 и KNO3,
т. е. солевая масса его изобра-
жается точкой Е2. Тогда, после
отделения выделившегося NaCl,
кристаллизация KNO3 при охлаж-
дении раствора идет по наиболее
длинному пути E2d, что обеспечи-
вает наибольший выход продукта.
Рис. 356. Изотермы растворимости
в системе NaNO3 + KCl KNO3+
+ NaCl при 5, 25, 50 и 100°.
Для производства калиевой селитры конверсионным способом
используют растворы нитрата натрия, получаемые инвертированием
растворов, образующихся при щелочной абсорбции выхлопных
нитрозных газов. Средний состав инвертированных растворов нат-
риевой селитры (в г/л): 450—500 NaNO3; до 4 NaHCO3+Na2CO3;
до 1 NaNO2; 1 NaCl + Na2SO4.
Хлорид калия, применяемый для обменного разложения, содер-
жит 95—98% КС 1.
Раствор натриевой селитры предварительно фильтруют, выпа-
ривают до содержания NaNO3 600—700 г/л и затем направляют на
обменное разложение (рис. 357), которое проводят в стальном
реакторе /, обогреваемом паром. В реактор постепенно вводят из-
мельченный хлорид калия или раствор его в слабых горячих щело-
ках, содержащих нитрат натрия. Избыток NaNO3 сверх стехио-
метрического количества составляет 90—120 г/л.
Для ускорения процесса конверсии и предотвращения забива-
ния реактора осадком хлорида натрия в нижнюю часть аппарата
1226
Гл. XXXII. Соли азотной кислоты
подают сжатый воздух или перемешивают раствор механической
мешалкой. Благодаря интенсивному перемешиванию почти весь об-
разующийся хлорид натрия находится в реакторе во взвешенном
состоянии.
Процесс конверсии продолжается около 4 ч. В начале реакции
температура раствора в аппарате 80—90°, к концу процесса ее по-
вышают до 119—122°. Для уменьшения пенообразования в реактор
Рис. 357. Схема получения KNO3 конверсионным способом:
1—реактор; 2—фильтр; 3— приемный бак растворов нитрата калия; 4—центробежный насос;
5—напорный бак; 6 н 10—кристаллизаторы; 7 н 12—центрифуги; 8—сборник для первого
маточного раствора; 9—аппарат для «распарки»;// — фильтрпресс: 13 — сушильный барабан.
добавляют небольшое количество минерального масла. Поваренную
соль отделяют от раствора на тканевых фильтрах 2, работающих
под давлением 4 ат; плотность раствора перед спуском на фильтры
должна составлять 1,54—1,6 г/смъ. Поваренную соль, содержащую
после фильтрации 15—20% KNO3, промывают на фильтре холодной
водой, при этом содержание KNO3 в шламе снижается до 1—3%.
Удаляемый из аппарата хлорид натрия можно использовать для
технических целей. Раствор нитрата калия и промывные воды на-
правляют в приемные баки 3. Во избежание кристаллизации KNO3
температуру в баках поддерживают в пределах 90—105° при по-
мощи парового обогрева. Для разложения примесей (Na2CO3,
NaNO2) в раствор нитрата калия вводят небольшое количество
нитрата аммония; при этом протекают следующие реакции:
Na£CO3 + 2NH4NO3 = (NH4)2CO3 + 2NaNO,
NaNO2 + NH4NO3 = NH4NO2 + NaNO3
Нитрат калия
1227
Образующиеся аммонийные соли разлагаются с выделением
газов, которые удаляются в атмосферу:
(NH4)2CO3 = 2NH, + СО2 + Н2О
NH4NO2 = N2 + 2Н2О
Горячий раствор нитрата калия и промывные воды направляют
в кристаллизатор 6. Здесь при охлаждении раствора до 25—30° вы-
деляются кристаллы KNO3 (первая кристаллизация). Кристалли-
ческий продукт после отделения на центрифуге 7 от маточного
раствора, возвращаемого в реактор, содержит 94—96% KNO3 и до
6% NaCl.
Для получения более чистой калиевой селитры (1-го сорта) кри-
сталлический нитрат калия после первой кристаллизаций обычно1
промывают холодной водой, растворяют в паровом конденсате
(«распарка») и подвергают повторной кристаллизации. Распарку
проводят в аппарате 9, куда подают острый пар. Концентрирован-
ный раствор KNO3, получаемый при растворении соли в паровом
конденсате, отфильтровывают на фильтрпрессе 11 245 и направляют
в кристаллизатор 10. Содержание NaCl в растворе перед второй
кристаллизацией не должно превышать 1—2%. После отделения от
маточного раствора на центрифуге 12 кристаллический нитрат ка-
лия сушат горячим воздухом (105—110°) во вращающемся бара-
бане 13232. Маточные растворы, получаемые после вторичной кри-
сталлизации KNO3, используют для получения калиевой селитры
2-го сорта.
На получение 1 т нитрата калия конверсионным способом рас-
ходуют: 0,93 т нитрата натрия, 0,92 т хлорида калия, 0,05 т нитрата
аммония, 11 т пара; 20 м2 воды, 50 кет -ч электроэнергии.
В циклическом процессе кристаллизация NaCl осуществляется
во время выпаривания из системы воды при постоянном давлении
и при переменной температуре. Описание и расчет реального
цикла наиболее точно могут быть произведены с помощью ком-
бинации изотермического и изобарического сечений диаграммы
(рис. 358) 2461247. Для обеспечения возможности точного контроля и
регулирования необходимо поддерживать неизменный режим про-
цесса, что возможно лишь при работе по определенному циклу. На
рис. 358 представлен пример оптимального цикла (В. Я. Рудин,
Н. А. Ярым-Агаев) для случая, когда кристаллизация KNO3 завер-
шается при 50°. Точка а на изотермическом сечении характеризует
состав маточного раствора после кристаллизации KNO3 на отрез-
ке Ьа в процессе охлаждения. В начале кристаллизации солевому
составу раствора соответствует точка Ь. Перед кристаллизацией,
к нему добавляют такие количества воды, чтобы он насытился
хлоридом натрия лишь при заданной температуре конца кри-
сталлизации (50°). Раствор b получается в результате выпарива-
ния воды и кристаллизации NaCl из кипящего раствора с. Точка с
1228
Гл. XXXII. Соли азотной кислоты
принадлежит к изобарическому сечению диаграммы и лежит на
луче выпаривания ВсЬ. Исходный раствор для выпаривания с по-
лучается при смешении маточного раствора а с эквимолекулярной
смесью d КС1 и NaNO3. Таким образом цикл осуществляется по
треугольнику cba. Чем выше солевой коэффициент цикла (т. е. от-
ношение массы полученного KNO3 к массе выпаренной воды), тем
меньше расход энергии на выпаривание. Чем выше конечная тем-
пература кристаллизации KNO3, тем меньше затраты на охлажде-
ние раствора. Наиболее экономичными являются циклы с темпера-
Рис. 358. Оптимальный цикл
конверсии KCl + NaNO3 =
=KNO3 + NaCl в диаграм-
ме с изотермическим (50°)
и изобарическим (1 ат) се-
чениями.
Рис. 359. Изотермы раствори-
мости в системе NH4NO3 +
+ КС1 КИОз + ИН4С1 при
О и 100°.
дурными интервалами от точки кипения до конечной температуры
кристаллизации в пределах 50—25°. При этом для луча упарива-
ния ВЬ оптимальные отношения К+: NO3 находятся в пределах
0,69—0,96 — они обеспечивают сравнительно высокие солевые коэф-
фициенты и небольшие объемы циркулирующих растворов.
Представляет интерес получение нитрата калия обменным раз-
ложением хлорида калия и нитрата аммония 248’249.
В качестве побочного продукта в этом случае получается хло-
рид аммония, т. е. продукт более ценный, чем поваренная соль, об-
разующаяся при обменном разложении хлорида калия и нитрата
натрия.
Диаграмма растворимости в системе NH4NO3 + КС1 = KNO3 +'
+ NH4C1 (рис. 359) показывает, что поле кристаллизации KNO3
•сильно увеличивается при понижении температуры. Поэтому легко
подобрать условия, обеспечивающие кристаллизацию большей
части KNO3 при охлаждении концентрированного раствора, полу-
ченного смешением NH4NO3 и К.С1, Использование этого способа
Нитрат калия
122»
затрудняется необходимостью после выделения KNO3 выпаривать
раствор хлорида аммония, интенсивно разрушающий выпарные
аппараты, кроме того, потребность в хлориде аммония относи-
тельно невелика; как удобрение хлорид аммония имеет весьма'
ограниченное применение.
Получение нитрата калия из хлорида калия
и азотной кислоты или окислов азота
Этот способ, не нашедший пока широкого распространения
в промышленности вследствие трудностей, связанных главным об-
разом со значительной коррозией аппаратуры, представляет, од-
нако, интерес, так как не требует затраты дефицитных щелочей и
большого расхода пара 236-243.
Взаимодействие хлорида калия с азотной кислотой или окис-
- лами азота идет по следующим схемам:
KC1 + HNO3 = KNO3 + HC1 (1>
3HC1 + HNO3 = NOC1 + C12+2H2O (2>
2КС1 +3NO2 + Н2О = 2KNO3 + 2НС1 + NO (3)
НС1 + 2NO2 = HNO3 + NOCI (4>
Реакция (1) идет слева направо при сравнительно низких тем-
пературах (25—60°). Реакция (2) легко обратима, начинается при
низких температурах; при 100° равновесие сдвинуто почти нацело
в сторону NOC1 и CI2-
Образованию хлористого нитрозила способствует повышение
концентрации кислот в растворе. При большой концентрации кис-
лот и высокой температуре давление паров НС1 и HNO3 над рас-
твором увеличивается, что приводит к образованию больших коли-
честв хлористого нитрозила и хлора. При применении 30—40%-ной
азотной кислоты и температуре ниже 60° потери азота в виде хло-
ристого нитрозила невелики, и хлор накапливается в растворе
в виде НС1. При охлаждении раствора из него выделяется значи-
тельная доля KNO3, а маточный раствор может быть возвращен
в цикл. В дальнейшем, при накоплении значительных количеств
соляной кислоты, перед возвратом раствора в процесс необходимо'
отгонять из него часть хлористого водорода. Отгоняемые пары кон-
денсируются в виде соляной кислоты.
Нитрат калия можно получать из хлорида калия и азотной кис-
лоты 224'251 и при повышенной температуре по схеме, изображенной
на рис. 360. В стальной реактор 1, футерованный диабазовыми
плитками, подают азотную кислоту, маточный раствор от предыду-
щей операции и загружают твердый хлорид калия. Реакционный
раствор перемешивают сжатым воздухом и нагревают острым паром
До 75—85°. Вначале реакция протекает бурно, затем замедляется.
Образующиеся газы и водяные пары направляют воздушным
1230
Гл. XXXII. Соли азотной кислоты
эжектором в абсорбер. С газами удаляется и часть хлористого
водорода. По окончании реакции раствор, содержащий в среднем
520 г/л KNO3, 35—65 г/л HNO3 и 120—140 г/л НС1, поступает на
кристаллизацию. Выделяющийся при охлаждении раствора до
25—30° нитрат калия отделяют на центрифуге 5, промывают и вы-
сушивают. Маточный раствор в смеси с промывными водами ча-
стично возвращают в реактор. Приблизительный состав возвра-
щаемого раствора: 90—110 г/л KNO3, 20—40 г/л HNO3 и 70—80 г/л
Рис. 360. Схема получения нитрата калия из хлористого калия
и азотной кислоты:
/ — реактор; 2 — центробежный насос; 5 —сборник маточных растворов;
4 —кристаллизатор; 5 —центрифуга; 6 — сушильный барабан; 7— холодильник
для воздуха; 8 — компрессор.
НС1. Часть маточного раствора нейтрализуют раствором едкого
кали и направляют на выпаривание (на схеме не показано), из
выпаренного раствора кристаллизуют нитрат калия, который может
быть использован в качестве продукта 3-то сорта.
Выход нитрата калия по азотной кислоте при использовании
части маточного раствора составляет ~70%. На производство 1 т
продукта расходуется только 4 т пара вместо 11 т по методу кон-
версии КС1 и NaNO3.
Получение KNO3 по реакции (1) можно осуществить с помощью
жидких экстрагентов — бутилового, изоаминового спиртов и т. п. —
с последующей их регенерацией 252.
Интересен метод получения KNO3, основанный на осуществле-
нии топохимической реакции между твердым хлоридом калия и
газообразной или жидкой двуокисью азота:
КС1 + 2NO2 = KNO3+ NOC1
Нитрат калия
1231
Эта реакция каталитически ускоряется в присутствии ничтожных
количеств воды и идет с большой скоростью при низких температу-
рах (—10, —12°). Скорость реакции, лимитируемая скоростью диф-
фузии, возрастает при применении тонкоизмельченного хлорида
калия и при увеличении скорости жидкого или газового потока,
омывающего твердые частицы.
Реакция с жидкой двуокисью азота может проводиться в авто-
клаве, а с газообразной — в шаровых или трубчатых мельницах.
Себестоимость калиевой селитры, полученной взаимодействием
жидких окислов азота с хлоридом калия, должна быть приблизи-
тельно такой же, как при получении ее методом катионного об-
мена 253,254. (Последний метод, однако, дает более чистый нитрат
калия.)
Получение нитратов калия и натрия взаимодействием хлоридов,
с NO2 или с азотной кислотой является одним из экономичных
путей производства этих продуктов при условии использования
хлора, выделяющегося в газовую фазу в виде С12, НС1 и NOC1.
Особенно важно использование хлористого нитрозила, так как
в противном случае потеря содержащегося в нем азота делает про-
изводство нерентабельным Хлористый нитрозил может быть окис-
лен до NO2 и С12 кислородом воздуха в присутствии концентриро-
ванной азотной кислоты или в присутствии катализаторов: МпОг,
РегОз и др. Хлористый нитрозил может быть использован также
для хлорирования окислов и других веществ; освобождающаяся
при этом NO может быть переработана в азотную кислоту. Суще-
ствуют и другие методы переработки хлористого нитрозила. В по-
следнее время интерес к этому способу привлекает внимание еще
и потому, что хлористый нитрозил, ранее не находивший примене-
ния, может быть использован для получения полупродуктов, при-
меняемых в производстве полиамидных смол.
Разработаны также способы получения KNO3 из КС1 и азотной
кислоты без образования NOC1. Например, в среде расплавленного
KNO3 (или других солей) в отличие от водной среды, реакция идет
согласно уравнению:
КС1 + 2HNO3 = KNO3 + ‘/гС12 + NO2 + Н2О
Вследствие того, что в систему с азотной кислотой! вводится
вода, точка плавления KNO3 понижена и процесс осуществляется
при 200—250° при давлении ~20 ат. Часть плава возвращают на
растворение в нем КС! перед обработкой азотной кислотой, а дру-
гую (меньшую) продувают азотом для удаления газов, гранули-
руют и выпускают в качестве продукта. Из отходящего газа извле-
кают конденсацией и сорбцией хлор и двуокись азота, которую на-
правляют на производство азотной кислоты 255.
Другой способ заключается в осуществлении реакции 25в:
2КС1 + 2HNO3 + '/2О2 = 2KNO3 + С12 + Н2О
1232
Гл. XXXII. Соли азотной кислоты
ЛИТЕРАТУРА
1. В. А. Клевке, Л. М. К о н т о р о в и ч, Труды ГИАП, в, 5, 1956, стр, 71;
в. 7, 1957, стр. 33. — 2. В. А. Клевке, Н. П. Поляков, Л. 3. Арсеньева,
Технология азотных удобрений, Госхимиздат, 1963,—3. В. А. Клевке,
Я- Д. Хаскина, ЖХП, № 7, 408 (1936). — 4. М. Н Набиев, Азотнокислотная
переработка фосфатов, Изд. АН УзбССР, 1957. — 5. С, И. Вольфкович,
Т. В. Глазова, Изв. АН СССР. ОХН, № 4, 314 (1943); ДАН СССР, 41, № 8,
346 (1943). — 6. D. Н. Booth, V. С. V i и g а г d, J. Appl, Chem., 17, № 3, 86
(1967). — 7. Нагатани Масанори, Ямадзоэ Нобору, Сэияма Тэ-
цуро, J. Chem. Soc. Japan. Ind. Chem. Sec., 67, № 9, 1342. A80 (1964).—
8. R. Tierney er, Z. Kristallogr., 97, 386 (1937). — 9. D. N. Campbell,
A. R. Campbelle, Canad. J. Res., 24, № 4, 93 (1946). —10. A. M. Дубовиц-
кий, Ф. Г. Марголис, T. В. Глазова, ЖПХ, 27, № 2, 93; № 4, 368 (1954).
11. Т. В. Заболоцкий, К. Н. Швецова, Хим. пром., № 6, 171 (1953).—
12. А. Л. Ш и е е р с о н, В. А. Клевке, М. А. М и н и о в и ч, Труды ГИАП, в. 5,
1956, стр. 283; ЖПХ, 28, № 5, 682 (1956). — 13. А. Л. Ш и е е р с о н, В. А. Клев-
ке, Труды ГИАП, в. 6, 1956, стр. 252. —14. I. Whetstone, Ind. Eng. Chem.,
44, № 11, 2663 (1952). —15. A. M. Дубовицкий, Ф. Г. Марголис, Хим.
пром., № 12, 363 (1951). — 16. В. В. Болдырев, Л. А. Алексеенко, ЖПХ,
№ 9, 1316 (1956). —17. Т. В. Заболоцкий. В. К. Нелюбин, ДАН СССР,
75, № 2, 215 (1950). — 18. S. К- G h о s s, V. К. Srinivasa, Technology (India),
2, № 1, 3 (1965). — 19. Нагатани Масанори, J. Chem. Soc. Japan. Ind.
Chem. Soc., 68, Ns 3, 424, A 23 (1965). — 20. И. А. Леско в ич, ДАН СССР,
85, № 3, 595 (1952).
21. Н. Е. Пестов, Физико-химические свойства зернистых и порошкообраз-
ных химических продуктов, Изд. АН СССР, 1947. — 22. И. Р. Кричевский,
Л. М. Конторович, М. Бергау з, ЖХП, № 7, 56 (1933). — 23. А. М. Ду-
бовицкий, Ф. Г. Марголис, Хим. пром., № 5, 136 (1947). — 24. W. Н. R о s s,
I. В. Adams, Ind. Eng. Chem., 36, Ns 12, 1088 (1944). — 25. H. П. Курин,
Хим. пром., Ns 5, 129; Ns 7, 193 (1953). — 26. P. Delfosse, Fertil. Feeding
Stuffs J., 45, 333 (1956). —27. J. Agr. Food. Chem., 7, Ns 5, 36 (1959).—
28. M. H a s h i k a w a a. oth., J. Chem. Soc. Japan. Ind. Chem. Sec., 62, № 1, 38
(1959). — 29. В. А. Клевке, Хим. пром., № 5, 26 (1945). — 30. В. К. Федоро-
ва, Д. В. Гер нет, А. Н. Машковский, Там же, № 5, 296 (1954).
31. Ф. В. Турчин, В. И. Соколова, Хим. пром., № 2, 68 (1955).—
32. Б. В. Ерофеев, Н. И. Мицкевич, ЖПХ, 31, № 12, 1805 (1958).—
33. Ф. Г. Марголис, А. М. Дубовицкий, Т. В. Глазова, Хим. пром..
Ns 5, 138 (1951). —34. В. А. К л е в к е, Н. К- Це л ь м, Там же, Ns 5, 139 (1947) —
35. Т. В. Заболоцкий, ЖПХ, 33, № И, 1127 (1950). —36. A. Kiss, Mag.
Kern. Lapja, 20, Ns 6, 295 (1965). — 37. Б. В. Хамский и др., ЖПХ, 36, № 12,
2620, 2631 (1963), — 38. Опыт Чирчикского электрохимкомбината, ИНТИ, 1953’
Госхнмиздат, 1957. — 39. М. А. М и н и о в н ч, П. В. С и ч к о в, Н. К. Ц е л ь м',
Труды ГИАП, т. 10, 1959, стр. 285.—40. Франц, пат. 1464873, 1966.
41. Пат. США 3190775, 1965,— 42. Пат. США 3230038, 1966. — 43. П. А. В л а-
с ю к, П. Ф. Ж а б и ц к и й, В. Н. М у с и ч, Доклады ВАСХНИЛ, № 5, 5 (1968) .—
44. Г. Моне в, М. Кръстева, О. Василева, Т. Андонова, Химия и Ин-
дустрия (Бъелг.), 38, Ns 6, 266 (1966). — 45. F. Micek, J. Nyvlt, Chem. pru-
mysl, 16, Ns 10, 584 (1966). — 46. Канадзава Такафуми, К а г а к у Когё
Chem. Ind. (Japan), 17, Ns 9, 885 (1966). — 47. В. Л. Максимов, Е. М. Алек-
сандрова, Н. С. Торочешников, Т. Г. Антонова, Э. М. Серебрен-
никова, О. А. Мартынова, Труды МХТИ им. Д. И. Менделеева,. в. 54,
1967, стр. 96. —48. А. И. Санникова, Н. С. Торочешников, Там же
в. 56, 1967, стр. 190. — 49. И. Я. К У з н е цо в, С. Н. Г а н з, Е. И. Доброволь-
ский, Н. И. Д з ю б а н, сб. «Химическая технология», в. 8, Изд ХГУ, 1967.
стр. 158. — 50. Nitrogen, № 46, 33 (1967).
Литература
1233
51. Z. Lyson, Chernik (польск.), 20, № 4, 115 (1967). — 52. С. H. Гана
Е. И. Кузнецов, Е. И. Добровольский, М. И. Шахова, Изв. вузов.
Хим. и хим. технол., 9, № 6, 924 (1966). — 53. Е. И. Добровольский,
С. Н. Га н з, И. Е. Кузнецов, А. У. О ш у р к о, сб. «Химическая промышлен-
ность Украины», № 4, (34), 1967, стр. 4. — 54. С. Н. Г а н з, И. Е. Кузнецов,
Г. И. В и л е с о в, М. И. Шахова, Е. И. Добровольский, А. П. Г л о з-
м а н, Н. П. Кузь, сб. «Химия и технология минеральных удобрений», Ташкент,
Изд. ФАН, 1966, стр. 128. — 55. М. Gregor, V. J е s е п a k, D. В i с е к, Chem.
Prfimysl, 14, № 12, 619 (1964). — 56. К. М. Бялко, Огнеопасные и взрывчатые
свойства азотнокислого аммония, изд. ВНИТО химиков, 1937. — 57. В. А. А с со-
нов, Свойства и технология взрывчатых материалов, Химтеорет, 1938.—
58. Н. Ф. Б о с т а н ж о г л о, Б. В. Росси, Аммиачно-селитренные взрывчатые
вещества, Оборонгиз, 1940. — 59. Г. Каст, Л. Метц, Химическое исследование
взрывчатых и воспламенительных веществ, Госхимиздат, 1934. — 60 Г. А. А б и н-
дер, ЖХП, № 22, 1351 (1936).
61. I. Е. Jacobson, Meehan. Eng., 12, 1054 (1947). — 62. G. Feick,
R. M. Hainer, J. Am. Chem. Soc., 76, № 22, 5860 (1954). — 63. В. А. Клевке,
-Д. Ю. Гамбург, Хим. пром., № 7, 22 (1950). — 64. С. Павлнковский,
А. Каминский, Przem. Chem., № 4, 220 (1957). — 65. R. Smith, Trans. Fara-
day Soc., 53, № 10, 1341 (1957).— 66. M. A. Cock, E. L. Talbot, Ind. Eng.
Chem., 43, № 5, 1098 (1951). — 67. Б. Ю. P о з м а н, О термической стойкости
аммиачной селитры, изд. ЛИИВТ, 1957, — 68. Б. Ю. Розман и др., ЖХП,
31, № 7, 1101 (1958). — 69. А. А. Шидловский, Изв. вузов. Хим. и хим,
технол., № 3, 105 (1958). — 70. Авт. свид. 191498, 1967.
71. S. Varma, D. К- Sen, Technology (India), 2, № 1, 43 (1965).—
72. Я. И. Кильман, Безоп. труда в пром., № 11, 41 (1964).—73. Англ,
пат. 940807, 1963.— 74. В. А. Клевке, ЖПХ, 30, Ns 12, 1725 (1957). —75. За
увеличение выпуска азотных удобрений. Бюллетень по обмену опытом ГИАП,
№ 3—4, ОБТИ ГИАП, 1957. — 76. ф. В. Турчин, Хим. пром., № 12, 354
(1950). — 77. Авт. свид. 197535, 1967. — 78. А. П. Ер ж, К. Г. Ильин, Химия и
технология минеральных удобрений, Ташкент, Изд. ФАН, 1966, стр. 124. —
79. Япон. пат. 25352, 1963. — 80. Энка бинару то порима (япон.), 5, №8, 1 (1965).
81. Э. Н. Вагин а, Производство аммиачной селитры, под ред. В. А. Клевке,
изд. ШАП, 1967. — 82. В. Н. Богуславский, Производство аммиачной се-
литры, Киев, Изд. «Техника», 1967. — 83. A. Voinea, Нитрат аммония, Бухарест,
1963. — 84. М. А. М и н и о в и ч, Производство аммиачной селитры, Изд. «Хи-
мия», 1968. — 85. М. С. Витухновская, ЖПХ, 38, № 5. 967 (1965).—
86. Польск. пат. 47813, 1963.— 87. Англ. пат. 1002939, 1965.— 88. А. М. Мур-
зин, В. А. Клевке, ЖХП, № 13, 897 (1937).— 89. Л. Эпштейн, М. Вик-
торов, Там же, № 4, 249 (1937). — 90. Я. И. Кильман, 3. А. Сафронова,
М. К. Авилова, Бюллетень по обмену опытом в азотной промышленности,
ГИАП, № 5, 1957, стр. 42.
91. А. Г. Феоктистов, Хим. пром., № 8, 496 (1955). — 92. А. М. Дубо-
вицкий, А. В. Кудрявцев, Производство аммиачной селитры, ГОНТИ.
1938.—93. А. М. Дубовицкий, Я. И. Кильман, Технология аммиачной
селитры, Госхимиздат, 1949.—94. G. F а u s е г, Chem. Metall. Eng., № 9, 590
(1930). — 95. G. F a u s e r, ibid., № 8, 456 (1931). — 96. M. А. Миниович,
Солн азотной кислоты (нитраты), Госхимиздат, 1946. — 97. А. Н. Пла нов-
ей и й, В. М. Р а м м, С. 3. Каган, Процессы и аппараты химической техно-
логии, Госхимиздат, 1955. — 98. В. И. Леви, Н. А. Василенко, ЖПХ, 24,
7, 698 (1951). — 99. Н. Ф. Алексеев, Хим. пром., № 1, 17 (1953).—
'06- Д. Ю. Гамбург, Там же, № 10, 311 (1952).
101. Я. И. Кильман, Узб. ж., № 1, 23 (1965). — 102. В. В. К а ф а р о в,
G Л. Ах паз а ров а, Хим. пром., № 1, 15 (1965). —103. Польск. пат. 49867,
1965. —104. Англ. пат. 1002939, 1965. — 105. Я. И. Кильман, Узб. хим. ж.,
1234
Гл. XXXII. Соли азотной кислоты
№ 2, 9 (1965). — 106. И. А. И х л о в, В. В. Васильев, Г. С. Храпунов, Хим.
•яром., № 5, 286 (1954). —107. А. Бруштейн, Упаривание растворов аммиач-
ной селитры, Госхимиздат, 1959. — 108. Б. Розман, Л. Бородкин, авт. свид.
117370, 1958. — 109. С. J. С 1 г d 1 е г, Nitrogen, № 49 (1967). — 110. Brit. Chem.
jEng., 7, № 7, 508 (1962).
111. Chem. Eng., 59, Ns 8, 215 (1952). —112. M. E. Иванов, Я. И. К и л fa-
ман, Узб. хим. ж., № 1, 69 (1967). — 113. G. Weyermuller, Chem. Process
(USA), 29, № 8, 66, 70 (1966). —114. Япон. пат. 10208, 1965, —115. М. Е. По-
зин, Технология минеральных солей, Госхимиздат, 1961.— 116. W. С. S е е m а п,
I. W. McCamy, Е. С. Н о n s t о n, Ind. Eng. Chem., 44, № 8, 1912 (1952). —
117. Nitrogen, № 49, 32, 34 (1967). — 118. Англ. пат. 968751, 1964; 990487, 1965. —
119. Nitrogen, № 28, 22 (1964). — 120. С. Ф. Хох л о. в, А. Ф. Головко,
Я. И. Стримовский, сб. «Химическая промышленность Украины», № 2 (38),
1968, стр. 43
121. М. 3. Ц в и к, М. Н. Набиев, Н. У. Р и з а е в, К. В. Меренков,
В. С. В ы з г о, Узб. хим. ж., № 2, 50 (1967). — 122. Chem. Process Eng., 49,
№ 4, 69 (1968). — 123. Франц, пат. 83386, 1964. — 124. Н. В. Мещеряков,
Н. Н. Артемьева, Труды ГИАП, т. 8, 1957, стр. 194. —125. А. И. Бру-
штейн, Хим. пром., № 4, 200 (1954). — 126. Р. Мильвицкий, авт. свид.
117660, 1958. — 127. Пат. США 3117020, 1964; 3211522, 1965, —128. Австр. пат.
.245619, 1963. —129. Е. А. Казакова и др., Хим. пром., № 5, 330 (1962).—
130. Е. А. Казакова и др., Там же, № 2, 132 (1960).
131. Н. А. Шахова, А. И. Рычков, Хим. пром., № 11, 839 (1962).—
132. Н. А. Шахова, Л. Бахтин, А. А. Соколовский, Хим. пром., № 8,
.594 (1965). — 133. П. Г. Романков, Н. Б. Рашковская, Сушка во взве-
шенном состоянии, Изд. «Химия», 1968, гл. IV. —134. Швец. пат. 431576. —
135. D. К г е v е 1 е п, И о f t i z е г, J. Soc. Chem. Ind., 11, № 2, 568 (1949).—
136. Пат. США 3127249, 1964. — 137. J. E. Q a r b i e 1 s о n, J. D. Foster,
•G. Burnet, J. Agr. Food Chem., 14, № 3, 271 (1966). —138. Я- И. Кильман,
.Л. A. Ax Назаров, Бюллетень по обмену опытом в азотной промышлен-
ности, ГИАП, в. 9, 1958, стр. 19.—139. Brit. Chem. Eng., 11, № 3 (1966).—
140. Nitrogen, № 46, 36 (1967),
141. S. К о c i e r z, Przem. Chem., 46, № 9, 502 (1967). — 142. Бэссацу кояаку
когё, 10, № 5, 206, 208 (1966). — 143. А. И. Матковский, В. А. Мелентьев,
П. Я. Стариков, Хим. пром., № 8, 483 (1955). — 144. И. С. Шипунов,
М. И. С а нж ар о в, Там же, № 2, 18 (1948). — 145. Я. И. Кильман,
В А, Клевке, Труды ГИАП, т. 7, 1957, стр. 213. — 146. Я. И. Кильман,
Бюллетень по обмену опытом в азотной промышленности, ГИАП, в. 9, 1958,
.стр. 27. — 147. Нормы техн, проектир., МХП ОБТИ ГИАП, 1957. —148. R. Е. D а-
v i s, J. О. Hardesty, Ind. Eng. Chem., 37, № 1, 59 (1945). — 149. Пути сниже-
ния капитальных вложений, ОБТИ ГИАП, 1958. — 150. I. I. Dorsey, Ind. Eng.
• Chem., 47, № 1, 11 (1955).
151. Petroleum Processing, September, 1957. — 152. A. S. Hester, Ind Eng.
•Chem., 46, № 4, 622 (1954). —153. Chem. Eng. News, № 35, 4192 (1956).—
154. Я. И. Кильман, Журн. ВХО, 9, № 5, 591 (1964). — 155. Я- И. К и л ь м а н,
В. Клевке, В. Соболева, авт. свид. 123150, 1959. —156. Я. И. Кильман,
И. В. Гриднев, Н. Н. Артемьева, М. Ф. Тавобил ов, Хим. пром., № 5,
.361 (1967). — 157. Я- И. Кильман, Узб. хим. ж., № 3, 64 (1966). — 158.
Я. И. Кильман, Там же, № 2, 9 (1965). — 159. Я. И. Кильман, Вести; техн,
и эконом, информ. Н-и. ин-та техн.-экон, исслед. Гос. Ком-та хим. пром, при
Госплане СССР, в. 7, 12 (1964). — 160. М. Е. Позин, Г. В. Бельченко,
Н. н. Т р е у ш е н к о, В. П. Панов, Р. П. Дмитревская, сб. «Массообмен-
ные процессы химической технологии», вып. 3, Изд. «Химия», 1968, стр. 100, 101;
Тезисы докладов НТК ЛТИ им. Ленсовета, Секция ТНВ, 1967, стр. 20 и 1968,
-стр. 18; Тезисы докладов VI Всесоюзн. конф, по ТНВ, Тбилиси, 1968, стр. 136
Литература
1235
161. М. Е. Позин, Г. В. Бельченко, Н. Н. Т р е у щ е н к о, сб. «Массо-
обменные процессы химической технологии», вып. 4, Изд. «Химия», 1969,
стр. 174. —162. Я. И. Кильман, В. А. Клевке, Труды ГИАП, т. 9, 1959,
стр. 376. —163. Я. И. Кильман, Там же, т. 8, 1957, стр. 173; т. 9, 1959,
стр. 385; т. 10, 317, 1959, стр. 322. — 164. Я- И. Кильман, И. П. Кузь,
Н. Е. Ветров, М. Н. Алексеева, Там же, т. 8, 1957, стр. 164.—
165. Я И. Кильман, Там же, т. 7, 1957, стр. 219. —166. Я. И. Кильман,
М. М. Коростелева, Е. Т. Шевченко, Там же, т. 10, 1959, стр. 306.—
167. Я. И. Кильман, 3. М. Сафронова, М. К. Авилова, Там же,
стр. 337. — 168. А. Ф. Иванов, Хим. наука и пром., № 2, 213 (1956).—
169. П. А. Баранов, ЖХП, № 10, 59 (1934). — 170. Ф. В. Турчин, Там же,
№ 1, 22 (1940).
171. Л. 3. Арсеньева, Материалы по обоснованию выбора схемы произ-
водства известково-аммиачной селитры, ОБТИ ГИАП, 1959. — 172. С. И. Вольф-
кович, И. И. Мельников, В. И. Орлов, Хим. пром., № 6, '321 (1954).—
173. FC1 News, 5, № 6, 2 (1967). — 174. М. S i s i г, М. Samaddar, Н. Roy,
Technology (India), 2, № 1,7 (1965). —175. S. Pawlikowski, S. Szymonik,
Przem Chem., 38, № 1, 39 (1959). — 176. M. A. M и н и о в и ч, Труды ГИАП,
т. 8, 1957, стр. 178. — 177. N. D i m i t r i g e v i c, G. S i v i r i c a, Hem. ind. (сербо-
хорват.), 19, № 10, 261 (1965). — 178. S. Varma, N. P. M i s r a, Technology
(India), 3, № 3, 121 (1966). — 179. S. Varma, Technology, 1, № 2, 15 (1964).—
180. Франц, пат. 1479319, 1967.
181. Франц, пат. 1347177, 1963. —/82. Пат. ФРГ 1024099, 1964,— 183. М. N о-
v a k, Chem. priimysl. № 5, 255 (1966). — 184. S. Varma, N. P. M i s r a, Tech-
nology (India), 3, № 1, 43 (1966). —185. Я- И. Кильман, В. А Клевке,
Труды ГИАП, т. 6, 1956, стр. 239. — 186. Я- И. Кильман, Там же, т. 5, 1956,
стр. 292. — 187. С. И. Вольфкович, Р. Е. Ремен, Труды НИУИФ, в. 46,
1927. — 188. \. J е s е п a k, J. Н г i с, J. Р е t г о v i с, Chem. priimysl, 15, № 11,
644 (1965). — 189. J. Kraft, ibid., 14, № 8, 393 (1964). — 190. P. R. R a m a s u fa-
ram on y, N. S. Monsy, Fertil. News, 9, № 6, 22 (1964).
191. A. S. Kiss, Magyar Кёт lapja, 20, № 6, 295 (1965). —192. Chem.
Process (USA), 29, Xs 4, 120 (1966). — 193. Пат. США 3351454, 1964. — 194. Пат.
ЧССР, 115397, 1965. — 195. Phos. a. Pot. № 24, 25 (1966). — 196. J. N ё m e e ё к,
J. V i к, Chem. priimysl, 15, № 10, 616 (1965). — 197. Англ. пат. 1049782, 1966.—
198. J. Silverberg a. oth., j. Agr. Food Chem., 6, X» 6, 442 (1958).—
199. M. Л. Варламов, E. И. С т a p о с e л ь с к и й, Г. А. М о н а к и н, ЖПХ,
41, № 1, 47 (1968).—200. Польск. пат. 50892 и 51176, 1966.
201. J. Brandner a oth., J. Chem. Eng. Data, 7, Xs 2, 227 (1962).—
202. Франц, пат. 1363086, 1964. — 203. Япон. пат. 18212, 1965. — 204. Справочник
азотчика, т. 2, Изд. «Химия», 1969, — 205. К. D. Jacob, W, S h о 1 1, Comm. Ferti-
lizer, 91, Xs ЗА, 94, 99 (1955). — 206. Д. Н. Прянишников, Избранные сочи-
нения, т. 1, Агрохимия, Сельхозгиз, 1952.— 207. W. Hennel, Chim. Ind., 29,
786, 1042 (1933). — 208. Н. А. Яцута, Л. М. Конторович, В. А. Клевке
ЖХП, 13, Xs 21, 1289 (1936).— 209. С. С. Перельман, Л. М. Конторо-
в^ич, Там же, Xs 6 (1940). — 210. А. Животовский, Химстрой, Xs 3, 158
211. И. А. Миркин, Т. В. Глазова, Труды НИУИФ, в. 92, 1932.—
212. Phos. a. Pot., Xs 24, 25 (1966). — 213. J. N ё m е с ё k, J. V е k, Chem. priimysl,
'о, Xs 16, 616 (1965).—214. Я- И. Кильман, В. А. Клевке, Труды ГИАП,
! т- 3, 1954, стр. 139. — 215. В. Е. Горфункель, Я. И. Кильман, Там же,
стр. 166.—216. А. В. Шубников, Основы кристаллографии, Изд. АН СССР
I 1947.-2/7. Пат. ГДР 50587, 1966. — 2/8. Chem. Age, 83, Xs 2116, 201 (1960). —
. 19. В. И. Г л а д у ш к о, П. Т. Шевченко, сб. «Химия и технология минералы
1236
Гл. XXXII. Соли азотной кислоты
ных удобрений», Ташкент, Изд. ФАН, 1966, стр. 226. — 220. В. D. Boni
Р. W. М. Jacobs, J. Chetn. Soc., А, 1966, № 9, 1265. я
221. S. Pawlikowski, S. Bistro n, Przem. Chem., 37, № 12, 771
(1958). — 222. С. H. Селяков, Нитратно-хлоридные солончаки и месторожде-
ния селитры Средней Азии, Труды Почв, ин-та им. В. В. Докучаева, т. 21, в. 2,
Изд. АН СССР, 1941.—223. Некрич, Спивак, Труды ХХТИ, № 6, 1947,
стр. 159. — 224. Австр. пат. 254918, 1964. — 225. В. Ф. Гогин, М. А. М и н и о-
вич, Химстрой, № 9, 2470 (1933).— 226. В. И. Николаев, ЖРФХО, 59,
№ 7—8 685 (1927); ЖПХ, 3, № 5, 653 (1930). — 227. А. Животовский,
Химстрой, № 7, 38 (1934). — 228. В. И. Атрощенко, Е. Г. Седышева,
Труды ХПИ, т. 1, № 1, 1952. — 229. И. Р. Кричевский, Л. М. Контор о-
вич, ЖХП, № 2, 139 (1935).—230. Т. П. Воронов, Бюллетень по обмену
опытом в азотной промышленности, № 9, 1958, стр. 10.
231. Ив. Кръстев, Изв. Ин-та общ. и неорган. хим., 5, 111 (1967).—
232. Польск. пат. 50990, 1966. — 233. Д. И. Рябчиков, Хим. наука и пром., 1,
№ 5, 554 (1956). — 234. А. Б. Пашков, В. С. Титов, Хим. пром., № 5, 10
(1958). — 235. В. А. Демидкин, Там же, № 9 , 277 (1950).—236. Д. А. Эп-
штейн, И. Н. Никонова, сб. «Исследования по прикладной химии», Изд.
АН СССР, 1955, стр. 50. — 237. Е. И. А х у м о в, ЖПХ, 27, № 12, 1324 (1954).—
238. Q. Т. G a d г е, Fertil. News, 14, № 2, 31(1969). — 239. М. А. Мнниович,
В. Ф. Гогин, Химстрой, № 3, 121 (1934). — 240. М. А. М и н и о в и ч, И. М. Ли-
би н с о н, ЖХП, № 7, 680 (1935).
241. М. А. М и н и о в и ч, М. С. Комаровский, Хим. маш., № 3 (1935). —
242. Е. J а п е с k е, Z. angew. Chem., 41, № 33, 916 (1928). — 243. В. И. Нико-
лаев, ЖПХ, 3, № 5, 653 (1930). — 244. В. Я- Рудин, Н. Л. Ярым-Агаев,
Там же, 39, № 10, 2227 (1966). — 245. G. Grosse, W. Rieger, Z. W i s s, Techn.
Hochsehull Chem. Leuna Merseburg, 9, № 3, 269 (1967). — 246. H. Л. Ярым-
Агаев, В. Я- Рудин, Т. А. Цейтлеиок, ЖНХ, 10, № 4, 976 (1965).—
247. В. Я. Рудин, Н. Л. Ярым-Агаев, ЖПХ, 40, № 6, 1204 (1967).—
248. Аронова, Лунская, Калий, № 2, 24 (1933). — 249. И. Р. Кричев-
ский, Э. А. Гольдман, Z. anorg. Chem., 218, № 3, 253 (1934).—
250. М. L. Spealman, Chem. Eng., № 23, 200 (1965).
251. Э. M. Верховская, И. Н. Шокин, А. Г. Кузнецова, Труды
МХТИ им. Д. И. Менделеева, в. 51, 1966, стр. 124. — 252. Э. М. Верховская,
И. Н. Шокин, А. Г. Кузнецова, Там же, в. 60, 1969, стр. 18.—
253. В. А. Клевке, Н. П. Поляков, Л. 3. Арсеньева, Технология азотных
удобрений, Госхимиздат, 1956. — 254. М. М. С е н я в и н, Хим. наука и пром.,
2, № 6, 754 (1957). — 255. R. W. Р f е i f f е г, V. J. D i F г а п с о, L. F. А 1 b г i g t h,
J. Agr. Food. Chem.,15, № 6, 949 (1967). — 256. Chem. Eng., 74, № 24, 118 (1967),
Глава XXXIII
СОЛИ АММОНИЯ
СУЛЬФАТ АММОНИЯ
Физико-химические свойства
Сульфат аммония (NH4)2SO4— бесцветные кристаллы ромбиче-
ской формы с плотностью 1,769 г)см3. Технический сульфат аммо-
ния имеет серовато-желтоватый оттенок. При нагревании сульфат
аммония разлагается с потерей аммиака, превращаясь в кислые
соли. Давление NH3 над сульфатом аммония равно при 205°
Рис. 361. Растворимость (NH4)2SO4 в воде.
°,5 мм рт. ст., при 300° 50,8 ммрт. ст. *. При 513° он полностью раз-
лагается на NH3 и H2SO4. При растворении 1 г-экв соли в 1 л воды
поглощается при 19,6° около 1 ккал тепла. При кристаллизации
сУльфата аммония из раствора выделяется 2,6 кал/г-мол. Раство-
римость (NH4)2SO4 в воде показана на рис. 3612>3. В присутствии
аммиака растворимость сульфата аммония значительно снижается.
Температура кипения насыщенного водного раствора (NH4)2SO4
при 760 мм рт. ст. равна 108,9°.
1238
Гл. aXXIII. Соли аммония
Применение ;
Промышленное использование сульфата аммония весьма огра
ничено, — небольшие его количества потребляют в производств
аккумуляторов и в медицине. Очень большие количества сульфат:
аммония применяют в сельском хозяйстве в качестве азотного удоб
рения. Оно содержит 20,5—21% азота (химически чисты,
(NH4)2SO4 содержит 21,2% N).
Объем производства сульфата аммония в капиталистических
странах составлял в 1960 г. приблизительно 30% от общего объема
производства азотных удобрений (в пересчете'на азот). /Мировое
производство сульфата аммония в 1960 г. равнялось 15 млн. т. Осо
бенно велика доля сульфата аммония в производстве азотных
удобрений в Японии, Италии, Великобритании, США и Индии 4-15
В СССР доля сульфата аммония в общем балансе азотных удо
брений (N) составляла в 1967 г. ~8,5%.
Сульфат аммония физиологически кислое удобрение. Для устра
нения его подкисляющего действия требуется вносить в почву
в 1,7 раза больше СаСО3, чем при пользовании аммиачной се
литрой.
По своим физическим свойствам сульфат аммония лучше амми
ачной селитры, он не огнеопасен, меньше слеживается и обладает
значительно меньшей гигроскопичностью. Равновесная относитель
ная влажность воздуха над насыщенным раствором сульфата ам
мопия при 30° равна 79,2% (для аммиачной селитры 59,4%) 16-18
Сульфат аммония должен иметь крупнокристаллическую структуру
и содержать наименьшее количество влаги и смолистых приме"
сей 19~32.
Сульфат аммония (для удобрения), выпускаемый на коксо
химических предприятиях, должен удовлетворять
бованиям (ГОСТ 9097—65):
следующим тре
Азот (иа сухое вещество), не менее . .
Свободная серная кислота, не более . .
Влага, не более ....................
Фракция более 0,25 мм, не менее , . .
Высший сорт (в %) 1-й сорт (В %) 2-й сорт (в %)
21,0 20,8 20,8
0,025 0,05 0,05
0,2 0,3 0,3
85 70 Не норми-
руется
Сульфат аммония упаковывают в мешки и отгружают в крытых
железнодорожных вагонах; допускается (по согласованию с потре
бителем) отгрузка навалом в крытых железнодорожных вагонах
Сульфат аммония аккумуляторный получают из синтетического
аммиака и аккумуляторной серной кислоты. Его выпускают двух
сортов и используют в производстве свинцовых аккумуляторов
Согласно ГОСТ 894—41, в обоих сортах должно содержаться
(в пересчете на сухое вещество) не менее 99,05% (NH^SCh и не
Сульфат аммония
1239
более 0,3% кислоты (в пересчете на H2SO4). В продукте допу-
скаются незначительные примеси хлоридов, железа, мышьяка и
марганца. Продукт упаковывают в деревянные бочки.
Сульфат аммония очищенный, изготовленный из синтетического
аммиака и серной кислоты, согласно ГОСТ 10873—64, должен со-
держать не менее 21% азота (в сухом веществе) и не более: 1%
влаги, 0,15% свободной серной кислоты, 0,002% хлоридов, 0,015%
Fe, 0,00005% As, 0,00005% Мп, 0,015% не растворимого в воде
остатка, 0,001% азотной и азотистой кислот, 0,005% роданидов
(SCN), 0,0005% тяжелых металлов сероводородной группы (РЬ) и
0,02% фосфатов (РО4).
Производство сульфата аммония
Для производства сульфата аммония используют аммиак, полу-
чающийся как побочный продукт при сухой перегонке каменного
угля в коксохимической и газовой промышленности, а также син-
тетический аммиак 33-48.
В СССР сульфат аммония-—удобрение производят только из
аммиака, содержащегося в коксовом газе. Синтетический аммиак
предпочитают перерабатывать в аммиачную селитру, являющуюся
более концентрированным азотным удобрением, стоимость еди-
ницы азота в котором ниже, чем в сульфате аммония.
Основными промышленными способами производства сульфата
аммония являются следующие 49-5°:
1) поглощение серной кислотой аммиака, содержащегося в газе
коксовых печей 51-61;
2) нейтрализация серной кислоты газообразным синтетическим
аммиаком 62> 63;
3) обработка гипса растворами карбоната аммония 64-74;
4) переработка растворов, являющихся отходом производства
капролактама 75-77.
Кроме этих основных широко применяемых способов, сульфат
аммония может быть получен окислением сульфита аммония, об-
разующегося из NH3 и SO2 (стр. 534). Сульфит аммония окис-
ляется под давлением 2—3 ат 82—98%-ным кислородом в сульфат
аммония. Получаемый 40% раствор (NH4)2SO4 выпаривают и кри-
сталлизуют.
Для производства 1 т сульфата аммония требуется 100—106 м3
кислорода (100%) и 80—90 квт-ч электроэнергии 78-82.
Разработан способ производства сульфата аммония из дымовых
газов электростанций (и выхлопных газов сернокислотных заво-
дов). Газообразный аммиак вводят в горячие дымовые газы между
экономайзером и подогревателем воздуха. Аммиак связывает со-
держащиеся в газе окислы серы в сульфит, бисульфит, сульфат,
тиосульфат аммония. Эти соли улавливают из газового потока-
15 м. г. г? - ин
1240
Гл. ХХХ111. Соли аммония
вместе с золой в электрофильтрах и после отделения золы пере-
рабатывают в сульфат аммония 83-88.
В СССР в НИУИФ разработан способ, дающий возможность
одновременно получать из природного сульфата натрия два цен-
ных продукта — соду и сульфат аммония. Он основан на реакции:
Na2SO4 + 2СО2 + 2NH3 + 2Н2О = 2NaHCO3 + (NH4)2SO4
Процесс аналогичен аммиачному способу получения соды из по-
варенной соли, с той лишь разницей, что здесь вместо хлорида
аммония образуется сульфат аммония, который, после отделения
ют кристаллов бикарбоната, извлекается из раствора путем его
выпаривания и охлаждения. Использование сырья здесь значи-
тельно выше: натрий используется не на 60—70%, а на 95—97%;
кроме того, отсутствуют отходы загрязненных растворов хлорида
Кальция, получаемые при регенерации аммиака из хлорида аммо-
ния и сливаемые в «белые моря» 89-91.
- Разработаны способы получения сульфата аммония из отходов
производства метилметакрилата 92, из кислого гудрона 93’94, из про-
мывных вод от травления металлов94, из сточных вод96 и др.97.
Физико-химические основы получения сульфата
аммония нейтрализацией серной кислоты
аммиаком
Сульфат амм'ония получается по реакции
2NH3(r.) + H2SO4 = (NH4)2SO4 + 66,9 ккал
путем нейтрализации газообразного аммиака серной кислотой. При
мокром способе производства продукт кристаллизуется из пересы-
щенных растворов, при сухом — осуществляется нейтрализация
мелких брызг серной кислоты в газообразном аммиаке.
При насыщении аммиаком 75—78%-ной серной кислоты в реак-
ционном аппарате выделяется значительное количество тепла, до-
статочное для нагрева реакционной смеси до температуры кипения
и для испарения из нее значительного количества воды. Башенная
серная кислота, часто применяемая в этом производстве, содержит
сотые доли процента окислов азота в виде нитрозилсерной кис-
лоты. В непрерывном производственном процессе башенную кис-
лоту смешивают с реакционным раствором; при этом она разбав-
ляется и нагревается за счет тепла гидратации и нейтрализации
аммиаком. Вследствие этого происходит денитрация с выделением
окислов азота в газовую фазу по реакции:
2SOsNH + Н2О = 2H2SO4 + NO + NO2
При больших масштабах производства сульфата аммония коли-
чество выделяющихся окислов азота велико, несмотря на незначи-
Сульфат аммония
1241
тельное содержание их в серной кислоте. Поэтому должны прини-
маться меры к тому, чтобы эти газы, вредно действующие на чело-
веческий организм, не проникали в атмосферу цеха.
Наиболее целесообразно производить денитрацию серной кис-
лоты до нейтрализации ее аммиаком, так как в противном случае
некоторое количество аммиака теряется вследствие следующей
реакции:
NO + NO2 + 2NH3 = 2N2 + ЗН2О'
Обычно и осуществляют йредварительную денитрацию башен-
ной кислоты, разбавляя ее водой или маточным раствором суль-
фата аммония, причем концентрация кислоты, направляемой на
нейтрализацию, снижается на 8—10%.
Рис. 362. Изотермы растворимости в системе
(NH4)2SO4—H2SO4— Н2О при 10, 30, 50, 70 и 90°-
Из примесей, находящихся в башенной серной кислоте (около
0,2%), наиболее вредное влияние в процессе получения сульфата
аммония оказывают сульфаты железа и алюминия 98. При нейтра-
лизации кислоты из этих солей образуются коллоидные гидроокиси
железа и алюминия
(Fe, А1)2 (SO4)3 + 6NH3 + 6Н2О = 2 (Fe, Al) (ОН)3+ 3 (NH4)2 SO4
обволакивающие кристаллы сульфата аммония и тормозящие их
Рост. Устранение этого явления достигается путем неполной ней-
трализации кислоты — в непрерывно действующих реакторах под-
держивают кислую реакцию среды.
На рис. 362 изображены изотермы растворимости в системе
(HH4)2SO4—H2SO4—Н2О " 10°. В этой системе в твердой фазе
1242
Гл. XXXIII. Соли аммония
могут существовать различные кислые соли. Отдельные участки
кривых соответствуют насыщению раствора: 1—(NH4)2SO4; 2—
4(NH4)2SO4-H2SO4; 3 — 3(NH4)2SO4-H2SO4; 4— (NH4)2SO4-H2SO4;
5—(NH4)2SO4-3H2SO4. Поле кристаллизации (NH4)2SO4 лежит
в области систем, содержащих небольшие количества серной кис-
лоты — aiExc при 10° и а2Е2с при 90°. Во избежание выпадения кис-
лых солей содержание H2SO4 в жидкой фазе системы должно быть
меньше, чем в точках Е, т. е. меньше 11,08% при 10° или 19,77%
при 90°. Хотя в процессе нейтрализации реакционная масса имеет
высокую температуру, но при последующем отделении кристаллов
она охлаждается, поэтому при выборе состава реакционного рас-
твора необходимо исходить из положения поля кристаллизации
сульфата аммония при невысоких температурах. Практически
кислотность раствора поддерживают на уровне 6—7% свободной
H2SO4, что исключает возможность кристаллизации кислых солей.
Кристаллы сульфата аммония, выделяющиеся из такого кислого
раствора, сравнительно мелки, что несколько затрудняет их отфиль-
тровывание и отмывку от маточного раствора. Для получения бо-
лее крупных кристаллов содержание свободной кислоты в растворе
должно было бы быть в пределах 9—15 г H2SO4 в 1 л. При еще
меньшей кислотности раствора опять получаются мелкие кри-
сталлы21’101. Как при высокой, так и при очень незначительной
кислотности раствора сульфат аммония образует пересыщенные
растворы, что ведет к появлению мелких кристаллов. Указанная же
кислотность соответствует условиям, при которых кристаллизация
идет медленнее, а кристаллы получаются крупнее 102. Однако ра-
бота с такой кислотностью допустима лишь при незначительном со-
держании в растворе веществ, образующих коллоидные осадки 103.
Вредное влияние коллоидных осадков при кристаллизации из
слабокислых растворов может быть устранено путем осаждения со-
единений железа и алюминия добавкой небольших количеств супер-
фосфата, фосфорной кислоты и других реагентов 26> 28’29’ 104> ‘Ч
Величина кристаллов увеличивается также с возрастанием про-
должительности их роста. В непрерывно действующих аппаратах
интенсивное перемешивание кристаллической суспензии инертными!
газами — воздухом или водяным паром — препятствует осаждению!
кристаллов плотным слоем на дно аппарата, где их рост прекраН
щается. Замедление процесса кристаллизации достигается также
при двухступенчатой нейтрализации—в первой ступени полу-
чается кислый насыщенный раствор сульфата аммония, который
после охлаждения донасыщается аммиаком во второй ступени.!
Равномернее распределение аммиака по всему объему нейтрали-
зуемой массы обеспечивает равномерное распределение тепла и за-
рождающихся центров кристаллизации 10®> 107.
При отжиме образовавшихся кристаллов сульфата аммония на
центрифугах в нем остается часть маточного раствора (до 1,5%
Сульфат аммония
124Й
влаги). При охлаждении соли продолжается ее кристаллизация из
оставшегося маточного раствора, что приводит к срастанию поверх-
ностей отдельных кристаллов, и вся масса цементируется в сплош-
ную глыбу. Во избежание этого после отжима от маточного рас-
твора соль следует промывать на центрифуге водой. При промывке
вода растворяет мелкие кристаллы, и масса становится более рых-
лой. Иногда для уменьшения кислотности продукта его промывают
аммиачной водой. Когда центрифугированию подвергается соль,
имеющая щелочную реакцию, т. е. содержащая свободный аммиак,
промывку лучше производить горячей водой, так как выделив-
шиеся в этом случае гидроокиси железа и алюминия, забивающие
поры между кристаллами, коагулируют в более крупный осадок и
промывка облегчается.
С целью улучшения физических свойств готового продукта и
уменьшения слеживаемости его подвергают сушке 108’ 109. Сушку
сульфата аммония проводят различными способами — на вибро-
транспортере и в барабанных сушилках. В последнее время в свя-
зи с повышенными требованиями к влажности продукта (влаж-
ность должна быть <1%) сушку стали осуществлять в псевдоожи-
женном слое110-116. Кроме того, испытана высокоэффективная
сушка в вихревой камере, дающая продукт с влажностью 0,1—
0,6% 117> и в вертикальной трубе, в которой удается высушить про-
дукт до содержания влаги 0,02—0,08% 118. Сушится сульфат аммо-
ния воздухом, предварительно нагретым паром до ИО—130°.
Производство сульфата аммония из аммиака
. коксового газа
Содержащийся в коксовом газе аммиак (7—10 г/м3) можно пе-
рерабатывать в сульфат аммония тремя способами: косвенным, пря-
мым и полупрямым119^134.
Косвенный способ состоит в следующем: коксовый газ подвер-
гают охлаждению в газовых холодильниках для выделения из него
смолы и водяных паров. Оставшийся в газе аммиак поглощают во-
дой в аммиачных скрубберах. Затем из аммиачной и надсмольной
воды, образовавшейся вследствие расслоения смолы и сконденсиро-
вавшихся водяных паров, в дистилляционных колоннах отгоняют
аммиак, который связывают серной кислотой с образованием суль-
фата аммония. Этот метод требует больших капиталовложений на
сооружение аммиачных скрубберов, а также большого расхода
электроэнергии воды и пара. Кроме того, он связан с большими по-
терями аммиака. Вследствие своей нерентабельности этот способ в
СССР не применяется.
В прямом способе поглощение аммиака серной кислотой с обра-
зованием сульфата аммония производится непосредственно из го-
рячего коксового газа, температура которого превышает темпера-
туру конденсации содержащихся в газе водяных паров.
1244
Гл. XXXIII. Соли аммония
Обычно температуру поддерживают на уровне 110°. Этот способ
также не применяется в нашей промышленности из-за сложной тех-
нологии, а также потому, что не позволяет улавливать ценные пири-
диновые основания.
Полупрямой способ как наиболее экономичный получил самое
широкое применение в коксохимической промышленности. Сущ-
ность этого способа состоит в том, что вначале из коксового газа
выделяют смолу и водяные пары путем его охлаждения. Образо-
вавшаяся жидкая фаза расслаивается на два слоя: нижний — смо-
лу и верхний — надсмольную воду, в которой частично растворен
аммиак.
Надсмольную воду подвергают обработке для выделения из нее
аммиака, который затем направляют на поглощение серной кисло-
той вместе с очищенным от смолы коксовым газом, содержащим
оставшееся количество аммиака 16’ 135~137.
Для выделения из надсмольной воды аммиака ее подвергают
дистилляции. При нагревании до 100° острым паром из нее выде-
ляется растворенный аммиак и происходит разложение части ам-
монийных солей, образовавшихся при взаимодействии аммиака кок-
сового газа с содержащимися в нем примесями — двуокисью угле-
рода, сероводородом, серным и сернистым ангидридом, хлористым
водородом, цианистыми соединениями и др. Такие аммонийные
соли, как (NH4)2CO3, NH4HCO3, (NH^S, легко разлагаются при
нагревании с выделением аммиака. Для разложения остальных
аммонийных солей надсмольную воду обрабатывают в дистилля-
ционной колонне известковым молоком, например:
(NH4)2 SO4 + Са (ОН)2 = 2NH3 + 2Н2О + CaSO4
2NH4NCS + Са (ОН)2 = 2NH3 + 2Н2О + Са (NCS)2
Поглощение аммиака из коксового газа можно производить в
сатураторах (сатураторный метод) или в скрубберах (бессатура-
торный метод) 138. До последнего времени переработку аммиака
коксового газа в сульфат аммония производили в сатураторах, в
которых совмещены процессы поглощения аммиака серной кисло-
той и кристаллизации сульфата аммония. Совмещение этих двух
процессов в одном аппарате не позволяет поддерживать техноло-
гический режим, который бы являлся оптимальным одновременно
для обоих процессов, т. е. обеспечивал бы более полное поглощение
аммиака из коксового газа и способствовал бы образованию круп-
нокристаллического сульфата аммония. Было установлено, что наи-
более рационально процессы поглощения и кристаллизации прово-
дить раздельно — поглощение аммиака вести в скрубберах, а
кристаллизацию сульфата аммония на кристаллизационных уста-
новках (бессатураторный метод). По этому методу теперь рабо-
тают многие заводы и получают крупнокристаллический продукт
с хорошими физическими свойствами.
Сульфат аммония
1245
На рис. 363 представлена схема производства сульфата аммо-
ния из аммиака коксового газа сатураторным методом 16.
Коксовый газ, охлажденный в холодильниках до 25—30° и очи-
щенный от смолы, подогревают паром в решофере 2 до 60—80° и
направляют в сатуратор 3. Перед сатуратором к коксовому газу
примешивают паро-аммиачную смесь, полученную при переработке
надсмольной воды.
Сатуратор (рис. 364) представляет собой цилиндрический аппа-
рат с коническим днищем, выполненный из углеродистой стали и
Рис. 363. Схема производства сульфата аммония из аммиака коксового газа
сатураторным методом:
1 — напорный бак для серной кислоты; 2 — подогреватель газа (решофер); 3 — сатуратор;
4-барботажная труба; 5 —кислотная ловушка; 6—8 —насосы; 7 —циркуляционный бак;
S — кристаллоприемиик; 10 — приемный сосуд; 11 — центрифуга; 12 — внбрацнрнно-сушильйый
транспортер; 13 — резервный сборник.
футерованный внутри кислотоупорными плитками. Он снабжен
выносной кислотной ловушкой. Применяются также сатураторы с
внутренней ловушкой, а также с несколькими барботажными тру-
бами и с принудительной циркуляцией, создаваемой мешалкой про-
пеллерного типа, вмонтированной в конус сатуратора 136’ >39"149.
Сатуратор заполнен насыщенным (маточным) раствором суль-
фата аммония, в который непрерывно дозируется 78 %-ная сернай
кислота.
Коксовый газ подается через барботажное устройство 4. Рас-
пределение газа происходит по тангенциальному направлению, ко-
торое приводит раствор во вращательное движение, способствуя
лучшему контакту и перемешиванию газа с раствором. Образую-
щийся сульфат аммония выделяется в виде кристаллов, осаждаю-
щихся на дно сатуратора >23’125’130’ >50-155. в низу сатуратора нахо-
дится перемешивающее сопло (ажитатор). Циркуляция раствора^
1246
Гл. ХХХШ. Соли аммония
подаваемого насосом в сопло ажитатора, также улучшает переме-
шивание.
Выходящий из сатуратора газ, содержащий водяной пар, осво-
бождается от брызг и направляется на дальнейшее использование.
Рис. 364. Сатуратор с одной центральной барботажной
трубой и выносной кислотной ловушкой:
1 — штуцер для входа газа; 2 — штуцер для выхода газа; 3 — цен-
тральная барботажная труба; 4 — барботажный зонт; 5 —труба
для подвода кислоты; 6— штуцер для перетока маточного рас-
твора в циркуляционную кастрюлю; 7 —труба для возврата рас-
твора из кастрюли обратных токов; 8 — труба для выдачи кри-
сталлов сульфата; 5— труба для подачи раствора в перемеши-
вающее сопло; 10 — перемешивающее сопло.
Концентрация аммиака в газе, выходящем из сатуратора, должна
быть не более 0,02—0,03 г/м3.
Равномерная концентрация кислоты в растворе поддерживается
непрерывной циркуляцией маточного раствора при помощи центро-
бежного насоса 6. Пульпа, состоящая из кристаллов сульфата ам-
мония и маточного раствора, непрерывно удаляется со'дна сатура-
тора и центробежным насосом 8 передается в кристаллоприемник9.
Сульфат аммония
1247
Маточный раствор из кристаллоприемника возвращается в сатура-
тор через приемный сосуд 10, а кристаллы поступают в центрифугу
непрерывного действия 11 для полного отделения от маточного рас-
твора. Сульфат аммония, содержащий после центрифугирования
около 2% влаги, при хранении слеживается, слипаясь в комки и
глыбы. При содержании влаги менее 0,2% сульфат аммония прак-
тически не слеживается. Поэтому после центрифуги сульфат ам-
мония поступает на вибрационно-сушильный транспортер 12,
где он высушивается в токе горячего воздуха, подогретого до
110—130°.
Для сушки сульфата аммония применяют также другие су-
шилки 108-118> 156-159.
Для получения 1 т (NH4)2SO4, содержащего 20,5% азота, необ-
ходимо израсходовать 0,75—0,76 т серной кислоты (100%), 0,26—
0,27 т аммиака, 2,7—6 т пара, 8 ле3 воды и 25—30 квт-ч электро-
энергии.
Для уменьшения слеживаемости предложено покрывать кри-
сталлы сульфата аммония красителем 160-162, или вводить добав-
ки25-29. Уменьшить слеживаемость сульфата аммония можно так-
же, выпуская его в гранулированном виде 163-168.
По бессатураторному методу поглощение аммиака проводят в
скрубберах. Последние применяют как с насадкой (керамические
кольца или деревянная хордовая насадка), так и без насадки (с
брызгалами). Орошение скрубберов производится раствором суль-
фата аммония, содержащим 5—6% свободной серной кислоты. Тем-
пературу поддерживают в пределах 47—55°.
Из скрубберов насыщенный раствор сульфата аммония посту-
пает на кристаллизацию. Иногда раствор вначале упаривают, за-
тем охлаждают и из полученного пересыщенного раствора произ-
водят кристаллизацию 169. В других случаях кристаллизацию про-
водят под вакуумом (600—700 мм вод. ст.) с одновременным кон-
центрированием раствора.
Разработана новая конструкция сатуратора брызгального типа
с кристаллизационной ванной (рис. 365). Расположение брызгал
(одного во входном штуцере газа и двух в самом скруббере на раз-
ных уровнях) обеспечивает почти полное улавливание аммиака из
газа.
Кристаллизатор «Кристалл» английской фирмы «Пауэр-Газ»
(рис. 366) состоит из испарительной части 1 и контейнера суспен-
зии 3. Из испарительной части насыщенный маточный раствор по
трубе 2 поступает в контейнер, где происходит рост и классифика-
ция кристаллов. Крупные кристаллы оседают на дно, откуда отво-
дятся на фильтрацию. Маточный раствор с мелкими кристаллами
Из верхней части контейнера вместе со свежим раствором циркуля-
ционным насосом 4 через подогреватель 5 возвращается в испари-
тельную часть, где раствор концентрируется за счет испарения
1248
Гл. XXXIII. Соли аммония
части воды. По данным фирмы, в этом кристаллизаторе 94—98%
получающихся кристаллов имеют размеры 0,42—1,68 мм 170>171.
На рис. 367 приведена бессатураторная схема английской фир-
мы «Саймонс-Карвэ». Коксовый газ поступает в безнасадочный
скруббер 2, снабженный брызгалами 3, где улавливается аммиак.
Скруббер разделен на две ступени. Первая ступень (нижняя часть
скруббера) орошается маточным раствором, содержащим 1 % из-
Маточный
раствор
раствор
Газ
\3
Маточный
раствор
Маточный
раствор
Маточный
раствор
Газ
Пульпа кристаллов
н насосу
Рис. 365. Сатуратор с брызгалами
и кристаллизационной ванной:
J— сатуратор; 2 — кристаллизационная ван-
на; 5—циркуляционная кастрюля; 4 — брыз-
гали; 5 — перегородка.
Рис. 366. Кристаллиза*
тор «Кристалл»:
1 — испаритель; 2— централь-
ная труба; 3 — контейнер
суспензии; 4 — циркуляцион-
ный насос; 5 — подогрева-
тель; 6 — штуцер для ввода
свежего раствора
быточной серной кислоты, вторая — (верхняя часть скруббера) оро-
шается маточным раствори, содержащим 10—12% избыточной сер-
ной кислоты. Коксовый газ из абсорбера проходит ловушку 1 и на-
правляется на дальнейшую переработку.
Серная кислота и вода поступают в сборник 4, из которого на-’
сосом 5 осуществляется циркуляция раствора в верхней части
скруббера. Маточный раствор, вытекающий из нижней части скруб-
бера, содержащий 40% сульфата аммония, поступает в сборник 11
и насосом 12 подается на орошение нижней части скруббера.
В сборник 11 поступают также раствор из верхней части скруб-
бера от насоса 5 и маточный раствор из центрифуги 7. Для обеспе-
чения хорошей циркуляции в скруббере насосы 5 и 12 имеют боль-
шую производительность (по 220—240 м3/ч).
Сульфат аммония
1249
Часть раствора из нижней зоны скруббера отбирается в сбор-
ник 10 и насосом 9 подается на сгущение в выпарной аппарат 6.
Выпарка осуществляется под вакуумом 650—680 мм рт. ст., обра-
зовавшиеся кристаллы опускаются в коническую нижнюю часть,
где они поддерживаются длительное время во взвешенном состоя-
нии путем подачи свежего раствора в низ конуса. По данным фир-
мы, при такой системе кристаллизации не менее 60% кристаллов
получаются с размером более 0,5 мм.
Рис. 367. Схема производства сульфата аммония из аммиака коксового газа
беесатураторным методом:
/“-ловушка; 2—абсорбер брызгального типа; 3^ брызгали; 4, 10 и 11 —сборники; 5, 8, 9
и 12 — центробежные насосы; d—выпарной аппарат; 7 —центрифуга.
Пульпа из выпарного аппарата центробежным насосом 8 по-
дается для фильтрации на центрифугу 7. Затем соль сушится, упа-
ковывается и направляется в склад I3S.
Проведены опыты гранулирования кристаллического сульфата
аммония с добавкой примерно 25% фосфоритной муки. Получае-
мые гранулы достаточно прочны 172.
Производство сульфата аммония
из синтетического аммиака
Сульфат аммония из синтетического аммиака производят двумя
способами: мокрым и сухим. Мокрый способ 173-175 сходен со спосо-
бом получения сульфата аммония из аммиака коксового газа.
1250
Гл. XXXIII. Соли аммония
Нейтрализацию серной кислоты аммиаком проводят в сатураторах,
заполненных кислым насыщенным раствором сульфата аммония
(избыточная кислотность раствора 4—7%). 75—78%-ную серную
кислоту и аммиак непрерывно подают в сатуратор и для предупре-
ждения чрезмерного загустевания пульпы вводят в нее некоторое
количество воды, что компенсирует испарение воды за счет реакци-
онного тепла и тепла от разбавления серной кислоты (с 78 до 7%).
В сатураторе поддерживают температуру около 110°. Смесь кри-
сталлов сульфата аммония и маточного раствора из сатуратора
поступает для разделения на центрифугу. Маточный раствор воз-
вращают в сатуратор, а кристаллы высушивают. Пары воды, ухо-
дящие из сатуратора, содержат 12—15 г/м3 аммиака. Для его улав-
ливания пары промывают серной кислотой, поступающей затем в
сатуратор. При использовании синтетического аммиака в сатуратор
вводится значительно меньший объем газа, чем при производстве
сульфата аммония из аммиака коксового газа. Это позволяет умень-
шить объем сатуратора по сравнению с применяемым в коксохими-
ческой промышленности. Расходы сырья и энергетические затраты
мало отличаются от таковых в производстве сульфата аммония из
аммиака коксового газа.
При сухом способе производства сульфата аммония 16-49'50-"-
,?б-179 распыленная с помощью вращающегося диска серная кислота
приводится в соприкосновение с газообразным аммиаком в сталь-
ной камере. За счет теплоты реакции температура повышается до
200—220°, почти вся вносимая с кислотой влага испаряется и в ка-
мере образуется готовый продукт в виде сухого мелкокристалличе-
ского, легко сыпучего и очень пылящего порошка (вес 1 м3 0,38—
0,475 т). Осевший на дно камеры продукт удаляется с помощью
вращающегося скребка.
При получении сульфата аммония сухим способом на 1 т про-
дукта, содержащего 21% азота (в пересчете на сухое вещество),
расходуют 0,25—0,26 т NH3 (100%), 0,75 т серной кислоты (100%),
1 м3 воды и 18 квт-ч электроэнергии.
Получение сульфата аммония из гипса 1
По этому способу, в основе которого лежит реакция 1
CaSO4 + (NH4)2 СО3 = (NH4)2 SO4 + СаСО3
сульфат аммония может быть получен без применения серной кис-
лоты. В качестве исходного сырья используют природный
гипс 180-185 и ангидрит186; может быть использован и фосфогипс —
отход производства фосфорной кислоты сернокислотной экстрак-
цией 187-189, также может быть использован гипс, осажденный из
морской воды 71. Этот способ широко применяется для промышлен-
ного производства сульфата аммония в Индии и некоторых других
Сульфат аммония
1251
странахв-13’64’б5’72~74. Качество гипса оказывает большое влияние
на технологический процесс и на качество готового продукта. При-
сутствие глинистых веществ, соединений железа, алюминия и дру-
гих примесей затрудняет фильтрацию и промывку шлама
(СаСОз) 66’68’70. Гипс, используемый для производства сульфата
аммония, должен содержать не менее 97—97,5% CaSO4 (в расчете
на безводное вещество).
Из гипса сульфат аммония может производиться двумя мето-
дами: жидкостным и газовым. При жидкостном методе пульпу тон-
коизмельченного гипса в воде обрабатывают 25—33% раствором
карбоната аммония; при газовом методе обработку пульпы гипса
производят газообразным аммиаком и двуокисью углерода.
Газовый метод проще жидкостного (не требуется предваритель-
ное приготовление карбоната аммония), но имеет недостатки, кото-
рые заставили отказаться от него и перейти на жидкостный метод.
Недостатками газового метода являются: периодичность процесса и
большой расход двуокиси углерода. При газовом методе образуют-
ся мелкие игольчатые кристаллы СаСО3, которые значительно хуже
фильтруют и промываются, чем крупные пластинчатые кристал-
лы, образующиеся при жидкостном методе. Время, необходимое для
проведения реакции при газовом методе, требуется значительно
большее, чем при жидкостном 190. Существенным недостатком газо-
вого метода является также то, что для отвода реакционного тепла
необходимо устанавливать холодильники для охлаждения пульпы в
реакторах и требуется сернокислотная промывка отходящих газов
из реакторов. При жидкостном методе отвод реакционного тепла
осуществляют циркуляцией через холодильники чистого раствора
карбоната аммония, и необходимости в сернокислотной промывке
газов нет.
Предложен также метод получения сульфата аммония путем
смешения в твердом виде 150 вес. ч. (NH4)2CO3 и 150 вес. ч. CaSO4
с добавлением 40 вес. ч. воды. Вся эта масса хорошо перемеши-
вается и для прохождения реакции выдерживается в течение 4—
5 суток при обыкновенной температуре. Затем производится клас-
сификация готового продукта67.
На рис. 368 показана схема производства сульфата аммония из
гипса жидкостным методом. Измельченный гипс (30% остатка на
сите 10 000 отв/см2) пневмотранспортом подают в мешалку 2, где
он смешивается с 25—33% раствором карбоната аммония, предва-
рительно нагретым в подогревателе 1 до 50—55°. Вначале с рас-
твором смешивают около половины требуемого количества гипса и
смесь направляют в первый реактор 4. Из реактора пульпу через
напорный бак 5 возвращают в мешалку 2, куда вводят остальное
количество гипса и смесь перекачивают во второй реактор 7. При
таком способе введения гипсовой муки в раствор образуются круп-
ные, легко фильтрующие и промывающиеся кристаллы СаСОз.
1252
Гл. XXXIII. Соли аммония
Время, необходимое для
(NHJjCOs, составляет 6—8 ч.
полного взаимодействия CaSO4 и
Температура в реакторе 50—60°.
Пульпу из реактора, состоящую
из раствора сульфата аммония и
осадка СаСО3, насосом перекачи-
вают на фильтрпресс 9.
Для получения крупных кри-
сталлов СаСО3 предложено ис-
пользовать реактор, имеющий
форму колонны, с коническим
днищем, заполненный раствором
(NH4)2CO3. Внутри колонны, в
нижней ее части смонтирован во-
дяной змеевиковый холодильник,
предназначенный для отвода ре-
акционного тепла. Газообразные
NH3 и СО2 подаются в низ колон-
ны. Пульпа гипса поступает в се-
редину колонны. Температура в
колонне поддерживается 40—50°.
Крупные кристаллы СаСО3 отво-
дятся из нижней части колонны
на фильтрацию, а мелкие кри-
сталлы удаляются из зоны реак-
ции через перелив в отстойник.
Кристаллы, полученные при такой
системе кристаллизации, отфиль-
тровываются в 2,6 раза быстрее,
чем полученные в обычных реак-
торах67.
Раствор сульфата аммония
(после отделения от него карбо-
натного шлама), содержащий
40—42% (NH4)2SO4 и около 1,5%
(NH4)2CO3, направляется на вы-
паривание. Выпарку проводят в
2-корпусном выпарном аппарате
12 и 13 из хромоникелевомолибде-
новой стали марки Х18Н12М2Т.
Первый корпус (обогревается па-
ром 2—3 ат) работает под давле-
нием 1,2 ат; температура раство-
ра 105—110°; концентрация выхо-
дящего раствора 48—50%. Во
втором корпусе выпарка произво-
дится под вакуумом 600—700 мм
Хлористый аммоний
1253
рт. ст. Выпарку ведут до получения пульпы, содержащей 55—60%
кристаллов и 40—45% насыщенного раствора (NH4)2SO4.
Кристаллизацию сульфата аммония из растворов осуществляют
также в 6—10-ступенчатых вакуум-кристаллизаторах при 40—45° и
вакууме 700—720 мм рт. ст.
Из выпарного аппарата пульпу по желобу 16 направляют в цен-
трифугу 17. Маточный раствор возвращают в цикл, а кристаллы на-
правляют на сушку. Применяются центрифуги непрерывного или
полунепрерывного действия, у которых все части, соприкасающиеся
с сульфатом аммония, выполняются из нержавеющей стали. Сушку
производят подогретым до НО—130° воздухом или дымовыми га-
зами с температурой около 200°. Высушивают сульфат аммония от
начальной влажности 2—2,5% до 0,1—0,3%.
Для получения 1 т (NH4)2SO4, содержащего 20,5% азота (в пе-
ресчете на сухое вещество), расходуют: 1,13 т гипса (100%
CaSO4), 0,74 т карбоната аммония (100%), 1,4 т пара, 25 м3 воды,
65 кет-ч электроэнергии и 71,5 кг условного топлива. В качестве
побочного продукта на 1 т сульфата аммония получается 760 кг
СаСОз (в пересчете на сухое вещество) в виде тонкого порошка,
который может быть использован для приготовления известковб-
аммиачной селитры или преципитата, для известкования почвы или
производства различных строительных материалов.
ХЛОРИСТЫЙ АММОНИЙ
Физико-химические свойства
Хлористый аммоний NH4C1 — бесцветные кристаллы кубической
системы с плотностью 1,53 г/см3191_ При нагревании возгоняется без
плавления, диссоциируя на аммиак и хлористый водород, причем
давление паров равно при 210° 10 мм рт. ст., при 310° 341,3 мм рт.
ст., при 337,8° 760 мм рт. ст., при 350° 1,4 ат и при 451° 10 ат192. На-
сыщенный водный раствор содержит при 0° — 23,0%, при 100° —
43,6%, при 116,0° (температура кипения)—46,5% NH4C1 193.
Применение
Хлористый аммоний применяют при пайке металлов, цинкова-
нии, лужении, в текстильной промышленности, при производстве
фармацевтических препаратов, а также для наполнения гальвани-
ческих элементов и т. д. Хлористый аммоний может быть также ис-
пользован для получения из него аммиака и хлора. Процесс разло-
жения NH4C1 на NH3 и С12 проводится в присутствии катализатора
При 400—500°. Выход по NH3 —98,8%, по С12 —85% 194’19S.
Как удобрение хлористый аммоний имеет весьма незначительное
применение, что объясняется отрицательным действием иона хлора
'1254
Гл. XXXIII. Соли аммония
на некоторые культуры, в особенности при накоплении хлора в поч-
ве в результате неоднократного внесения этого удобрения.
Наблюдается значительный рост производства и потребления
хлористого аммония в Японии. Например, в 1954 г. там произвели
61 тыс. т, а в 1963 г. 574 тыс. т NH4C1 196.
Хлористый аммоний технический (нашатырь), согласно ГОСТ
2210—51, выпускают двух сортов, которые должны соответствовать
следующим требованиям (в %):
1-й сорт 2й сорт
NH4CI, не менее .99,5 99,0
NaCl, не более 0,05 0,1
Углекислых н двууглекислых солей (в пересчете на NH4HCO3), не более . . Железа, не более 0,02 0,04
0,003 0,01
Тяжелых металлов, не более Мышьяка, не более 0,0005 0,0025
Отсутствие 0,001
Влаги, не более 1,0 1,5
Не растворимых в воде веществ, не более . . . 0,02 0,05
Хлористый аммоний упаковывают в четырехслойные бумажные
мешки, деревянные ящики и деревянные бочки. Для улучшения фи-
зических свойств хлористого аммония и уменьшения слеживаемо-
- СТИ ВВОДЯТСЯ ДОбаВКИ И ПрИМеНЯЮТСЯ ПОКрЫТИЯ 197~200.
Производство хлористого аммония
Основными способами производства хлористого аммония яв-
ляются 201’ 202:
1) взаимодействие NH3 и НС1;
2) выделение из фильтровой жидкости, получаемой в производ-
стве соды;
3) обменное разложение хлоридов с солями аммония.
Получение хлористого аммония из хлористого
водорода и аммиака
Получение хлористого аммония по реакции
HC1 + NH3 = NH4C1
осуществляют как жидкофазным, так и газофазным способами203.
При смешении газообразных НС1 и NH3 хлористый аммоний обра-
зуется в виде дыма — чрезвычайно мелких частиц, чему способ-
ствует, в частности, выделение большого количества тепла при ре-
акции. Осаждение дымообразного хлористого аммония связано со
значительными трудностями. Предложен способ получения зерни-
стого NH4CI из газообразных NH3 и НС1 путем конденсации хлори-
стого аммония в псевдоожиженном слое 204. Для получения более
крупных кристаллов NH4C1 предлагается газообразные NH3 и НС1
Хлористый аммоний
1255
вводить в поток воздуха, который служит псевдоожижающим
агентом. Процесс проводится при атмосферном давлении и 160—
200° 205.
Нейтрализация водного раствора НС1 аммиаком или водного
раствора аммиака хлористым водородом привела бы к необходимо-
сти последующего выпаривания раствора для выделения хлори-
стого аммония в кристаллическом виде, что осложняется сильной
коррозией выпарных аппаратов и требует затраты тепла. Наиболее
целесообразно проведение реакции между аммиаком и хлористым
водородом, растворенными в такой жидкости, которая ,не раство-
ряла бы образующийся хлористый аммоний. В качестве такой жид-
кости используют насыщенный водный раствор хлористого аммо-
ния 206, в котором в достаточной мере растворяются NH3 и НС1,
например при 90° растворимость NH3 в насыщенном растворе
NH4C1 составляет 3,19%, растворимость НС1 — 8,2%.
Синтетический хлористый водород, охлажденный до 30—50°, по-
глощают раствором хлористого аммония, например в барботажном
абсорбере, представляющем собой фаолитовую колонну с пятью
колпачковыми тарелками. Во избежание образования взрывоопас-
ного треххлористого азота хлористый водород не должен содержать
хлора, а температура раствора хлористого аммония должна быть
не ниже 70°.
Насыщенный хлористым водородом раствор хлористого аммония,
имеющий температуру 105—110°, выводится из абсорбера в дере-
вянный бакелитированный реактор, где нейтрализуется газообраз-
ным аммиаком до содержания свободного NH3 около 3 г/л. Обра-
зующаяся в реакторе суспензия хлористого аммония отстаивается,
и кристаллы отделяются на центрифуге из нержавеющей стали; ма-
точный раствор снова возвращается на орошение абсорбера.
На получение 1 т хлористого аммония по этому способу затра-
чивается 0,43 т аммиака, 0,75 т хлора и 240 м3 водорода.
Получение хлористого аммония из фильтровой
жидкости содового производства
При получении соды аммиачным способом по реакции
NaCl + NH3 + СО2 + Н2О = NaHCO3 + NH4C1
после отделения бикарбоната натрия остается так называемая
фильтровая жидкость, в 1 л которой содержится 170—180 г NH4CI,
70—80 г NaCl и небольшие количества карбонатов — NH4HCO3,
(НН4)2СОз — и других примесей.
Из фильтровой жидкости регенерируют аммиак, возвращаемый
в содовый процесс. Часть ее на некоторых заводах используют для
получения хлористого аммония 207-21(). Хлористый аммоний выде-
ляют выпариванием фильтровой жидкости в вакуум-выпарных
1256
Гл. XXXIII. Соли аммония
аппаратах211-213. Предварительно жидкость нагревают с целью реге-
нерации из нее аммиака и двуокиси углерода, содержащихся в фор-
ме карбоната и бикарбоната аммония. Разделение оставшихся со-
лей — NaCl и NH4C1 — осуществляется в процессе выпаривания
раствора и основано на различной растворимости этих солей. Рас-
творимость NaCl меняется с температурой мало, растворимость
NH4CI меняется значительно.
На рис. 369 приведена политермическая диаграмма раствори-
мости в системе NH4C1—NaCl—Н2О. Если, например, исходная
фильтровая жидкость содержит 175,0 г/л (15,5%) NH4C1 и
Рис. 369. Пути кристаллизации NH4C1 на диаграмме растворимости в си-
стеме NH4CI—NaCl—Н2О.
76,6г/л (6,8%) NaCl, то состав ее изобразится на диаграмме точ-
кой М. В процессе выпаривания она передвигается по лучу испаре-
ния PN по направлению к точке N. До сравнительно высоких кон-
центраций и температур этот луч проходит в поле кристаллизации
NH4CI. Если выпаривание произвести до точки N, лежащей на изо-
терме 80°, то раствор станет насыщенным NaCl. При дальнейшем
выпаривании воды при 80° из раствора будет кристаллизоваться
NaCl, пока раствор не станет эвтоническим (точка Л). После отде-
ления NaCl состав системы (оставшегося раствора) изображается
точкой А. При охлаждении этого раствора, например до 50°, из него
будет кристаллизоваться NH4C1, так как точка А лежит в поле кри-
сталлизации NH4CI, ограниченном при 50° изотермой BS, осью абс-
цисс ST и лучом ВТ, проведенным из эвтонической точки В к точке
Т твердого NH4C1. В процессе кристаллизации NH4C1 точка состава
раствора перемещается по отрезку луча кристаллизации А К, и ког-
да раствор охладится до 50°, его состав изобразится точкой К.
Охлаждение лучше осуществлять в многоступенчатых вакуум-
кристаллизаторах, в которых происходит дополнительное испаре-
ние воды, и за счет этого выход твердого NH4C1 увеличивается. Так,
если охлаждение до 50° производится в вакуум-кристаллизаторе, то
Хлористый аммоний
1257
NH4CI дополнительно выделяется в процессе испарения воды при
этой температуре, причем точка состава раствора перемещается по
отрезку изотермы КВ. В точке В раствор снова становится эвтони-
ческим, и дальнейшее испарение из него воды при 50° недопустимо.
Однако в следующей ступени вакуум-кристаллизатора раствор со-
става В может быть охлажден, например, до 35°. При этом вновь бу-
дет кристаллизоваться NH4C1, так как точка В находится внутри
поля кристаллизации этой соли при 35° (для того чтобы ограни-
чить это поле следует провести луч из точки Т в точку С). В ре-
зультате кристаллизации, обусловленной охлаждением, точка рас-
твора переместится из В в L, а испарением — в эвтоническую
точку С. Так как самоиспарение воды и вызванное им охлаждение
происходит в корпусах вакуум-кристаллизатора одновременно, то
изменение состава раствора на самом деле идет не по ломаным ли-
ниям АКВ или BLC, а по некоторым кривым (на диаграмме не по-
казаны) .
Для упрощения производства фильтровую жидкость можно
охладить, не отделяя хлористый натрий, выкристаллизовавшийся
в конце ее выпаривания. Так как точка N лежит з поле кристалли-
зации NH4CI при 50°, то при охлаждении системы до этой темпера-
туры . кристаллы NaCl растворятся, a NH4C1 выкристаллизуется.
Однако количество его (выход) будет меньше, чем в случае пред-
варительного отделения части NaCl.
Кристаллы NH4CI отделяют от маточной жидкости на центри-
фуге. Отжатый хлористый аммоний содержит ~3% NaCl и до 5%
влаги.
Аппаратуру для переработки растворов, содержащих хлористый
аммоний, изготовляют из кислотоупорных материалов. Из металлов
(и сплавов) наиболее стойким в кипящих растворах является ти-
тан214. Сушку хлористого аммония ведут при температурах не вы-
ше 70° во избежание его диссоциации.
Хлористый аммоний можно выделить из фильтровой жидкости
вымораживанием, охлаждая жидкость, предварительно насыщен-
ную аммиаком, двуокисью углерода и поваренной солью. Этот спо-
соб не требует расхода пара на выпарку, а главное его преимуще-
ство — значительное уменьшение коррозии аппаратуры при пони-
женных температурах215-217.
Хлористый аммоний может быть получен одновременно с содой
(бикарбонатом натрия) и легко выделен из раствора и без при-
менения выпаривания или вымораживания. Для этого в содовый
процесс вводят третью соль, например NaNOs218, (NH4)2SO4 219~221,
высаливающую хлористый аммоний.
При насыщении раствора NaNO3 аммиаком и двуокисью угле-
рода образуются бикарбонат натрия и нитрат аммония:
NaNO3 + NH3 + СО2 + Н2О = NaHCO3 + NH4NO3
1258
Гл. ХХХШ. Соли аммония
После отделения осадка бикарбоната натрия в оставшийся рас-
твор вводят поваренную соль:
NH4NO3 + NaCl = NH4CI + NaNO3
Образующийся по этой реакции NH4C1 выделяется в осадок и
после его отделения регенерированный раствор NaNO3 снова воз-
вращают на насыщение аммиаком и двуокисью углерода. Отжатый
на центрифуге NH4C1 содержит до 5% влаги, до 1% NaHCO3 и ни-
чтожные количества NaCl.
Вообще, наиболее экономичным является метод производства
хлористого аммония высаливанием его из фильтровой жидкости
твердым хлористым натрием. Этот процесс может быть организо-
ван, когда содовый завод использует в качестве сырья не рассол, а
твердую каменную соль.
При получении хлористого аммония, пригодного для производ-
ства сухих элементов, предложено производить предварительную
очистку раствора, содержащего NH4C1, с помощью смол 222 или друя
гими способами 223. fl
Получение хлористого аммония обменным Я
разложением хлоридов с солями аммония219'220'229!
Конверсия аммонийных солей хлоридами базируется на реак-
циях обменного разложения, например 225>226:
(NH4)2SO4 + 2NaCl(2KCl) = 2NH4Cl + Na2SO4(KgSO4) J
nh4no3 + kci = nh4ci + kno3 fl
(NH4)2SO4 + CaCl2 = 2NH4Cl + CaSO4 fl
Оптимальные условия проведения реакций (температуры и коня
центрации) определяются по соответствующим диаграммам раствоЯ
римости. Во всех приведенных реакциях из образующейся пары со-1
лей хлористый аммоний остается в растворе, а вторая соль выде-1
ляется в осадок. После ее отделения хлористый аммоний можно 1
извлечь из раствора кристаллизацией при охлаждении или выпари- 1
ванием. Аппаратуру для переработки растворов, содержащих хло- ]
ристый аммоний, изготовляют из углеродистой стали и футеруют |
кислотоупорными плитками. Сушку хлористого аммония ведут не 1
выше 60—70° во избежание его диссоциации. |
Хлористый аммоний, получаемый конверсией хлористого калия ’
с аммиачной селитрой, имеет следующий состав (в %): NH4C1 —
88,9, KNO3 —5,2, NH4NO3 —3,2, NaNO3 —0,7, Н2О — 2,0. На 1 т
хлористого аммония, получаемого этим методом, расходуется: 1,6 т
хлористого калия (2-го сорта), 1,7 т аммиачной селитры (100%
NH4NO3), 0,2 т аммиака (100% NH3), 5,4 т пара (6 ат), 76,6 м3 i
воды и 183,5 квт-ч электроэнергии.
Углекислые соли аммония
1259
В качестве дополнительных продуктов на 1 т NH4CI получается
1,75 т азотнокислого калия и 0,52 т сложного удобрения, содержа-
щего.азот и калий; возврат аммиака составляет 0,18 т.
Описанный метод не получил пока промышленного распростра-
нения вследствие недостаточной экономичности — он требует зна-
чительной затраты дорогостоящего сырья.
УГЛЕКИСЛЫЕ СОЛИ АММОНИЯ
К углекислым солям аммония относятся: нейтральный карбонат
аммония, безводный и одноводный (NH4)2CO3-Н2О; «полуторная»
соль, или сесквикарбонат (NH4)2CO3-2NH4HCO3; бикарбонат аммо-
ния NH4HCO3; карбаминовокислый аммоний NH4COONH2 (карба-
мат аммония).
Физико-химические свойства
Карбонат аммония — бесцветные кристаллы с сильным запахом
аммиака, теряющие на воздухе свой блеск и переходящие я
NH4HCO3, который, в свою очередь, также подвергается разложе-
нию. В водных растворах карбонат аммония существует только при
низких температурах: при нагревании водного раствора до 70° вы-
деляются NH3 и СО2, а при комнатной температуре водный раствор1
разлагается с образованием кислой соли:
(NH4)2 СО3 = NH3 + NH4HCO3
Одновременно происходит разложение соли с выделением воды
и с образованием карбаминовокислого аммония:
(NH4)2 СО3 = NH4COONH2 + Н2О
Сухая соль при 58° полностью улетучивается, разлагаясь на
NH3, СО2 и Н2О.
Технический углекислый аммоний представляет собой смесь кар-
боната, бикарбоната и карбаминовокислого аммония; обычно он со-
держит 29—32% NH3, около 56% СО2 и 12—15% Н2О. В 100 г во-
ды растворяется при 15° приблизительно 25 г, а при 65° около 67 а
технического углекислого аммония.
Бикарбонат аммония — белые ромбические кристаллы с плот-
ностью 1,57 г/см3. Он более стоек, чем карбонат аммония. На воз-
духе медленно разлагается. Декарбонизация водного раствора би-
карбоната аммония по реакции
2NH4HCO3 = (NHJ2 СО3 + Н2О + СО2
медленно идет при 20°, а при нагревании раствора резко уско-
ряется.
Насыщенный водный раствор бикарбоната аммония содержит?
при 0°—10,9%; при 20°—17,5%; при 60° —37,2% соли 227.
1260
Гл. XXXIII. Соли аммония
Равновесие в системе NH3—СО2—Н2О показано на рис. 370.
Точки составов газовой фазы определяются прямоугольной коор-
динатной сеткой, равновесные составы жидкой фазы при общем
давлении компонентов, равном атмосферному, — кривыми изокон-
центратами (NH3— пунктирные, СО2— сплошные линии). Наклон-
ные прямые изотермы (штрих-пунктир) — температуры конденса-
ции газовой фазы.
ТАБЛИЦА 90
Составы инвариантных растворов в системе NH3—СО2—Н2О
Температура, °C Состав раствора, в&с. ®/о Твердые фазы
NH3 со2
85 27 44 Бикарбонат — карбамат — сесквикарбонат
5 15 19,5 Бикарбонат — сесквикарбонат — карбонат
43 30 33 Карбамат — сесквикарбонат — карбонат
-13 11 14,5 Бикарбонат — карбонат — лед
-95 34 0,5 Карбонат — лед — NH4OH
-80 47 0,5 Карбамат — карбонат — NH4OH
-85 59 0,5 Карбамат — NH4<JH — (NH4)2O
-95 81 0,5 Карбамат — (NH4)2O — NH3
В системе вода — аммиак — двуокись углерода образуется 8 ин-
вариантных растворов, находящихся в равновесии с тремя твер-
дыми фазами (табл. 90 ) 224. Давление пара над этой системой при-
ведено в табл. 91.
Применение
Углекислый аммоний применяют в хлебопечении (вместо дрож-
жей), при крашении, при промывке шерсти, в медицине и др. За-
служивает внимания зарубежный опыт (США) применения угле-
Общее давление пара (в л<лс рт. ст.) ненасы|
Молярное отношение NH3 : СО2 Условная форма солей в жидкой фазе
10 20 30
1 : 1 NH4HCO3 155 240 321
7:6 (NH4)2CO3+5NH4HCO3 31 65 117
4:3 (NH4)2CO3+2NH4HCO3 16 37 , 74
3:2 (NH4)2CO3+NH4HCO3 13,5 29 59
20: 11 18(NH4)2CO3+4NH4HCO3 11 24 49
2:1 (NH4)2CO3 15 25 45
Углекислые соли аммония
1261
кислых солей аммония вместе с карбамидом в виде жидких удобре-
ний— растворов в жидком аммиаке или в аммиачной воде.
Рнс. 370. Диаграмма равновесия в системе
NH3—СО2—Н2О при атмосферном давлении.
Составы раствора: х —вес. % NH3, и— вес. % СО2. Насы-
щенные растворы: ЛВ—СО2; ВС — NH4HCO3;
CD - 2NH4HCO3 • (NH4)2CO3: DE - NH4COONH2.
Прирост урожая кукурузы, картофеля, свеклы и некоторых дру-
гих культур при внесении в почву углекислых солен аммония на
Ю—15% больше, чем при внесении NH4NO3 (на одинаковое коли-
ТАВЛИЦА 91
•Ценных растворов системы NH3—СО2—Н2О
Температура, °C
40 -— 50 55,2 60 70 76,9 80 80,5 84,3
450 625 760 ___ — — —
J97 321 — — — — — —
130 225 363 568 760 — — —
107 190 312 483 760 — —
92 165 283 455 725 760 —
80 142 • — 242 393 — 622 —• 760
1262
Г л. XXXIII. Соли аммония
чество азота) 228. Бикарбонат аммония дешевле аммиачной селитры,
яо его существенным недостатком является значительное разложе-
ние. В обычных условиях NH4HCO3 теряет при хранении в сутки
~0,3% своего веса, т. е. при двухмесячном хранении потери состав-
ляют ~ 18%. Специально обработанный NH4HCO3 теряет за 90 дней
хранения 3,5—10% веса 229. По другим данным 230, потери бикар-
боната аммония от улетучивания на воздухе при 20° составляют
за 5 суток около 4%, за 10 суток около 9%, а при 30° соответ-
ственно 10 и 22%.
Кристаллический продукт, выпускаемый под названием «угле-
аммонийные соли», согласно ГОСТ 9325—60, должен содержать не
меньше 20,6% NH3; остаток после прокаливания не должен превы-
шать 0,05%.
Углекислый аммоний для пищевых целей (ОСТ 10199—39) дол-
жен содержать не менее 28 и не более 35% NH3; остаток при про-
каливании не более 0,2%. Должны отсутствовать соли тяжелых
металлов, мышьяка, соляной кислоты и органические примеси.
Углекислый аммоний упаковывают в водонепроницаемые мешки
(полиэтиленовые, бумажные битумированные или из прорезиненной
ткани) 231’232.
Для большей устойчивости углекислого аммония предлагается
его упаковывать в мешки из поливинилхлорида, которые затем по-
мещать в стальные коробки 233.
Получение углекислого аммония Л
Углекислый аммоний может быть получен насыщением углеки"
лым газом аммиачной воды или одновременным поглощением во-
дой NH3 и СО2, а также взаимодействием газообразных аммиака
и двуокиси углерода в присутствии паров воды 233. Обычно исполь-
зуют комбинацию этих приемов. Вначале получают водный раствор
углекислого аммония, из которого отгоняют NH3 и СО2. Взаимодей-
ствием последних получают твердый продукт.
Получение водного раствора карбоната аммония с концентра-
цией 25—27% осуществляют абсорбцией водой углекислого газа и
газообразного аммиака в двух последовательно работающих абсор-
берах— карбонизаторе и промывном скруббере, орошаемых цирку-
лирующей жидкостью (противотоком движению газов). В качестве
источника двуокиси углерода используют газ известково-обжига-
тельных печей. В первый карбонизатор поступает двуокись угле-
рода и 80% аммиака от общего его расхода. К газам, поступаю-
щим из первого во второй карбонизатор, добавляют остальные 20%
аммиака. Далее газы пропускают через промывной скруббер, оро-
шаемый слегка подкисленным 10—15% раствором аммиачной се-
литры, и выводят в атмосферу. Тепло, выделяющееся в карбониза-
торах при абсорбции, отводят охлаждением циркулирующего рас-
Углекислые соли аммония
1263
твора (с 50 до 35°) в водяных холодильниках. Этот же процесс
предлагается проводить с минимальными потерями аммиака в од-
ной вертикальной колонне, что сокращает затраты на оборудова-
ние 234.
Раствор карбоната аммония, отбираемый из первого карбониза-
тора, передают на дистилляцию. Выходящие из дистилляционной
колонны двуокись углерода, аммиак и пары воды при 60° посту-
пают в сублимационные камеры, в которых происходит образова-
ние сырого углекислого аммония. Для получения товарного про-
дукта проводят повторную возгонку сырого карбоната аммония.
Предварительное получение раствора карбоната аммония яв-
ляется необходимым главным образом для коксохимических заво-
дов, так как позволяет связывать аммиак коксового газа без за-
траты серной кислоты с получением вместо сульфата аммония кар-
боната аммония.
При использовании синтетического или другого газообразного-
аммиака в концентрированном виде и двуокиси углерода и пароя
воды получают непосредственно кристаллический карбонат аммо-
ния. Процесс проводят в герметически закрытой реакционной ка-
мере, выложенной алюминием и охлаждаемой холодной водой, ко-
торая циркулирует в полых алюминиевых трубах, расположенных,
в верхней части камеры. Образующаяся в камерах твердая кри-
сталлическая соль представляет собой смесь карбоната, бикарбо-
ната и карбаминовокислого аммония. Продукт периодически вы-
гружают из камеры и упаковывают 235’236.
Получение двууглекислого аммония
Двууглекислый аммоний получают абсорбцией газообразного
аммиака и двуокиси углерода раствором карбоната аммония в са-
тураторах с отводом реакционного тепла. Вначале в сатуратор, за-
полненный раствором карбоната аммония, подают углекислый газ;
при этом происходит реакция:
(NH4)2 СО3 (раств.) + СО2 (г.) + Н2О — 2NH4HCO3 + 10,1 ккал
По мере насыщения раствора двуокисью углерода начинается
выделение кристаллов бикарбоната аммония. Осевшие кристаллы
инжектируются углекислым газом в отстойник-декантатор и далее
поступают на центрифугу для отделения от маточного раствора. Из-
Центрифуги соль поступает на укупорку 237.
Маточный раствор бикарбоната аммония из декантатора напра-
вляют в аммиачный абсорбер для насыщения аммиаком:
NH4HCO3 + NH3 — (NH4)2 СО3 -f-11,0 ккал
Отвод реакционного тепла в скруббере производят с помощью
змеевиковых холодильников. Образующийся раствор карбоната
аммония вновь поступает в сатуратор.
1264
Гл. XXXIII. Соли аммония
Представляет интерес совмещение процессов получения углеам-
монийных солей и очистки азото-водородной смеси в производстве
аммиака. Это может значительно сократить стоимость производ-
ства 238.
ФОСФАТЫ АММОНИЯ
Физико-химические свойства
Фосфатами аммония обычно называют соли ортофосфорной
жислоты — моноаммонийфосфат NH4H2PO4, . диаммонийфосфат
(NH4)2HPO4 и триаммонийфосфат (МН4)зРО4. Наиболее устойчи-
Н3РО4,%
Рис. 371. Изотермы растворимости, си-
стеме NH3—Н3РО4—Н2О при 0, 25, 50
и 75°.
вым соединением является
моноаммонийфосфат, при
нагревании которого до
100—Н0° не наблюдается
потерь аммиака. Диаммо-
нийфосфат при 70° теряет
аммиак и переходит в моно-
аммонийфосфат:
(NH4)2 НРО4 = NH4H2PO4 + NH3
Триаммонийфосфат раз-
лагается на воздухе уже при
30—40°.
Давление диссоциации
при 100° над NH4H2PO4 рав-
но практически нулю, над
(NH4)2HPO4 — 5 мм рт. ст.
и над (NH4)sPO4 — 643 мм
рт. ст. При 125° давление
NH3 над этими солями воз-
растает соответственно до
0,05, 30 и 1177 мм рт. ст.
При 20° в 100 г воды
растворяется: NH4H2PO4
40,3 г, (NH4)2HPO4 71,0 г,
(МН4)зРС>4 17,7 г. Раствори-
мость в системе NH3—
Н3РО4—Н2О показана на
рис. 37 1 239~244.
Значения pH 0,1 молярного раствора равны: для NH4H2PO4—
4,4, для (NH4)2HPO4 — 8,0 и для (NH4)3РО4 — 9,4.
Моно- и диаммонийфосфаты малогигроскопичны. Гигроскопи-
ческая точка NH4H2PO4 при 50° равна 88%, а при 15° — 97%.
При смешении моноаммонийфосфата с такими веществами, как
Фосфаты аммония
1265
Са(Н2РО4)2-Н20, (NH4)2SO4, NH4NO3, NH4C1 и СО(NH2)2, полу-
чаются удобрительные смеси, обладающие хорошими физическими
свойствами, с низкой гигроскопичностью и не слеживающиеся при
хранении 245-246.
При смешении моноаммонийфосфата с Са(ЫОз)2 в присутствии
небольших количеств воды протекает обменная реакция:
Са (NO3)2 • 4Н2О + 2NH4H2PO4 = 2NH4NO3 + Са (Н2РО4)2 • Н2О + ЗН2О
' Эта смесь обладает большой гигроскопичностью.
При смешении диаммонийфосфата и хлористого калия с суль-
фатом аммония, суперфосфатом или с моноаммонийфосфатом полу-
чаются смеси с хорошими физическими свойствами. При смешении
с нитратом аммония или карбамидом полученная смесь при хране-
нии во влажном воздухе плохо рассевается 239- 247-249.
Система NH4NO3—NH4H2PO4—Н2О представляет большой прак-
тический интерес в связи с расширяющимся производством слож-
ного удобрения типа нитроаммофоски. Совместная растворимость
компонентов данной системы была исследована для 100—200° 25°.
В связи с целесообразностью извлечения NH3 из коксового газа
растворами фосфатов аммония изучалось равновесие в системе
Н3РО4—NH3—СО2—H2S—Н2О 251.
В системе NH4H2PO4—NaNOs—Н2О в диапазоне температур 0—
110° кристаллизуются NH4H2PO4, NaH2PO4-2H2O, NaNOs, лед 252.
Все большее внимание уделяется изучению свойств и технологии
полифосфатов аммония, получаемых аммонизацией суперфосфор-
ной кислоты 253.
Полифосфаты аммония представляют собой смесь аммонийных
солей орто-, пиро-, Триполи- и других полифосфорных кислот 254-259.
Полифосфаты аммония являются стойкими соединениями и хорошо
растворимыми в воде 260-261, что важно для использования их в ка-
честве удобрений. Содержание питательных веществ в них колеб-
лется в пределах: N — 13—22% и Р2О5 — 54—68%. О свойствах по-
лифосфатов и стабильности кристаллических фаз в системе NH3—
—Н4Р2О7—Н2О при 25° и в системе NH3—Н5РзО10—Н2О при 0 и
25° — СМ. 262-26-».
Применение
Из ортофосфатов аммония промышленностью производятся толь-
ко моно- и диаммонийфосфат; триаммонийфосфат ввиду его нестой-
кости не изготовляют. Производят также полифосфаты аммония.
Наиболее широкое применение как фосфаты аммония, так и по-
Лифосфаты аммония нашли в сельском хозяйстве в качестве удоб-
рения. Они содержат два основных питательных элемента — азот
и фосфор — в водорастворимой форме. Фосфаты аммония приме-
няют также в виде компонентов комплексных удобрений и для по-
учения жидких удобрений 265~274.
1266
Гл. XXXIII. Соли аммония
Фосфаты аммония являются высококонцентрированным удобрен
Пнем благодаря большому содержанию питательных веществ, а
именно- (в вес. %): I
Р2О3 NHs I
Моноаммонийфосфат NH4H2PO4......................61,7 14,3
Диаммонийфосфат (NH4)2HPO4 .................... 53,8 25,8
Пирофосфат аммония (NH4)2H2P2O7.................67,0 16,0
» » (NH4)3HP2O7 . ............. 62,0 22,3
Триполифосфат аммония (NH4)3H2P3Oio.............68,9 16,5
Отношения N : Р2О5 составляют для NH4H2PO4 1 : 5 и для
(ЫН4)гНРО4 1 :2,5. В диаммонийфосфате это соотношение являет-
ся более благоприятным, однако вследствие своей меньшей устой-
чивости, чем моноаммонийфосфат, один диаммонийфосфат в каче-
стве удобрения не применяется, а используют его смесь с моноам-
монийфосфатом. Такую смесь называют аммофосом.
Для увеличения отношения азота к фосфору к смеси фосфатов
аммония добавляют какое-либо азотное удобрение, например суль-
фат аммония. В этом случае смесь называют сульфоаммофо-
сом 275-277.
Фосфаты аммония применяют также в пищевой и фармацевти-
ческой промышленности; их используют и в качестве антипире-
нов— для пропитки тканей, дерева и строительных материалов
с целью придания им огнестойкости.
В последнее время благодаря тому, что были разработаны спе-
циальные методы выращивания крупных кристаллов моноаммоний-
фосфата, последние нашли применение для изготовления осцилля-
торов высокой частоты, используемых в электронной технике 6-278.
Технический диаммонийфосфат, согласно ГОСТ 8515—57, выпу-
скают двух марок — А и Б, — в которых должно содержаться, со-
ответственно, не менее 50,5 и 48,5% Р2О5 и не более: 22,4 и 21,5%
NH3, 6 и 8% влаги.
Производство фосфатов аммония
Сырьем для производства фосфатов аммония являются аммиак
и ортофосфорная кислота, как экстракционная, так и термиче-
ская 276, 277, 279-288.
Фосфорная кислота нейтрализуется по реакциям:
H3PO4 + NH3=NH4H2PO4 -Ц
H3PO4 + 2NH3 = (NH4)2HPO4 fl
H3PO4 + 3NH3=(NH4)3PO4
Как видно из рис. 371, наибольший выход твердого монофос-
фата аммония достигается при осуществлении процесса по лучу АВ.
При нейтрализации экстракционной кислоты, содержащей 35—
Фосфаты аммония
1267
40% Н3РО4, выход кристаллов даже при 25° небольшой; поэтому
предпочитают использовать предварительно выпаренную кислоту.
При нейтрализации термической фосфорной кислоты (75% Н3РО4)
состав системы соответствует точке С и количество образующейся
твердой фазы весьма велико даже при температуре массы выше 75°.
Этому способствует и испарение части воды за счет тепла реакции.
При применении с целью получения аммофоса экстракционной
фосфорной кислоты, загрязненной примесями, в процессе нейтрали-
зации ее аммиаком, по достижении pH = 4—5,5, выделяются в оса-
док фосфаты железа и алюминия. Выделяется также гипс. Эти ве-
щества осаждаются в форме кристиллогидратов (Са8О4-2НгО,
реРО4-2НгО, Fe2(SO4) 3 • 9Н2О и т. п.), что связано с удалением из
раствора некоторого количества воды 289-293.
Для того чтобы получаемая при нейтрализации пульпа не была
слишком густой (это затрудняет поглощение ею аммиака), концен-
трация исходной фосфорной кислоты не должна быть слишком вы-
сокой. Чем больше в кислоте примесей железа и алюминия, тем ее
концентрация должна быть ниже для того, чтобы компенсировать
убыль воды, переходящей в твердую фазу с осадками, образующи-
мися из примесей. Обычно исходная экстракционная фосфорная
кислота, полученная из флотированных фосфоритов, берется с кон-
центрацией 23—26% Р2О5, а более чистая кислота из апатитового
концентрата — 26—30 % Р2О5.
Фосфаты аммония, получаемые из термической фосфорной кис-
лоты, обладают высокой чистотой, не содержат примесей и исполь-
зуются в основном в пищевой, фармацевтической промышленности
или для других технических целей. Концентрация термической кис-
лоты не должна быть выше 77% Н3РО4.
В настоящее время фосфаты аммония производят несколькими
способами, которые отличаются между собой условиями нейтрали-
зации кислоты и процессом кристаллизации готового продук-
та 294-296
При использовании термической фосфорной кислоты процесс мо-
жет быть осуществлен по сатураторной схеме 297 и по схеме с кри-
сталлизацией в вакуум-кристаллизационной установке 298.
При нейтрализации экстракционной фосфорной кислоты, загряз-
ненной примесями, выделяющиеся в осадок фосфаты железа и
алюминия, гипс и другие примеси остаются в готовом продукте, за-
грязняют его и снижают содержание основных компонентов. Для
получения более чистого продукта из экстракционной фосфорной
кислоты процесс нейтрализации ведут в две ступени. Сущность
Двухступенчатой нейтрализации состоит в том, что в первой сту-
пени неупаренная кислота нейтрализуется до рН = 4—4,5. При этом
в осадок выделяется большая часть примесей, которые зат(»м отде-
ляют от основного раствора фильтрацией. Отфильтрованный осадок
сУшат до содержания 5—6% влаги, и он может быть использован
1268
Гл. XXXIII. Соли аммония
для удобрительных целей, как аммофос 2-го сорта. Он содержит
около 5% NH3 и 30—35% Р2О5 в усвояемой форме.
Иногда его примешивают к готовому продукту — аммофосу. Очи-
щенный раствор подвергают дополнительной нейтрализации (2-я
ступень) после упаривания299-303. После нейтрализации кислоты до
моноаммонийфосфата осуществляется охлаждение пульпы для до-
полнительной кристаллизации; затем кристаллы отфуговывают и
сушат, маточный раствор возвращают в реактор.
Раствор, содержащий в основном моноаммонийфосфат, выпари-
вают под вакуумом до концентрации 34—36% Р2О5. Выпарка очи-
щенного и частично нейтрализованного раствора значительно про-
ще, чем предварительная выпарка фосфорной кислоты, вследствие
отсутствия отложений на греющих элементах выпарных аппаратов
и меньшей коррозии. Нейтрализацию очищенного раствора с полу-
чением частично диаммонийфосфата осуществляют непрерывным
способом последовательно в нескольких реакторах (обычно 3—
4 ) 276. В процессе нейтрализации за счет выделения тепла реакции
масса разогревается до 100—110°. Часть тепла расходуется на ис-
парение влаги. В реакторы вводят аммиак из расчета образования
10—20% диаммонийфосфата. Так как диаммонийфосфат лучше
растворим, чем моноаммонийфосфат, то чрезмерного загустева-
ния пульпы не происходит — она остается достаточно подвиж-
ной.
Из последнего реактора пульпа поступает в шнековый смеси-
тель, где ее смешивают с частью высушенного продукта. Из сме-
сителя массу направляют в барабанную вращающуюся сушилку,
затем измельчают и просеивают. Большую часть ее возвращают
в смеситель, а меньшую отправляют на склад 304.
Полученный продукт — аммофос — представляет собой смесь
моно- и диаммонийфосфата 305~307.
Этим способом возможно также получить предназначаемые для
промышленных целей моно- и диаммонийфосфат в отдельности.
Моноаммонийфосфат получают охлаждением выпаренного очи-
щенного раствора до 18—20°. Выпавшие кристаллы отделяют на
центрифуге и после этого высушивают. Маточный раствор возвра-
щают в производственный процесс.
Получение диаммонийфосфата следует вести в две ступени; так
как при подаче всего количества аммиака в одной ступени темпера-
тура поднимается очень высоко и получается слишком густая пуль-
па, вследствие чего имеют место потери аммиака. Поэтому после
реакции в первой ступени пульпа охлаждается, а затем поступает
на реакцию второй ступени, кристаллизацию, центрифугирование и
сушку.
Для получения диаммонийфосфата выпаренный раствор моно-
аммонийфосфата дополнительно насыщают аммиаком в реакторе
второй ступени до pH около 8. Во избежание потери аммиака на-
Фосфаты аммония
1269
сыщение ведут при температуре ниже 80°. Затем раствор диаммо-
нийфосфата направляют на кристаллизацию, центрифугирование и
сушку 308-31Ч
Для получения более прочных кристаллов диаммонийфосфата
предлагается кристаллизацию вести в присутствии неорганических
добавок — сульфатов или хлоридов 303’312’313.
Сушку диаммонийфосфата производят при температуре не выше
60° во избежание потери им аммиака и перехода в моноаммоний-
фосфат. Сушку же моноаммонийфосфата можно вести при темпера-
туре до 100—110°.
Фосфаты аммония можно получать и другими способами, на-
пример действием аммиака на раствор монокальцийфосфата или
на водную суспензию дикальцийфосфата (т. е. используя суперфос-
фат) 314
ЗСа (Н2РО4)2 + 8NH3 = Са3 (РО4)2 + 4 (NH4)2 НРО4
ЗСаНРО4 + 2NH3 = Са3 (РО4)2 + (NH4)2HPO4
или взаимодействием хлористого аммония с фосфорной кислотой
в присутствии бутилового и изоамилового спиртов. Степень пре-
вращения достигает 85% 315-318.
Процесс нейтрализации фосфорной кислоты можно также ве-
сти путем распыления Н3РО4 с помощью дискового распылителя в
башне в атмосфере аммиака, который поглощается каплями кис-
лоты 319-323.
Изучался процесс получения азотно-фосфорного удобрения в
конусно-цилиндрическом аппарате с псевдоожиженным слоем, в
котором были совмещены процессы нейтрализации кислоты ам-
миаком, выпарки, грануляции и сушки. Подогретая кислота распы-
ляется с помощью форсунки в поток поступающей непосредственно
под решетку подогретой аммиачно-воздушной смеси 324. Исследова-
лась возможность получения фосфатов аммония с высоким содер-
жанием азота за счет частичного образования (NH4)3PO4 325.
Разработан способ получения кристаллического NH4H2PO4 ней-
трализацией экстракционной фосфорной кислоты (49% Н3РО4) при
^=2,1 ат и 165—170° с последующим распылением плава в башне
высотой 15 м 325>327. Полученный кристаллический NH4H2PO4 может
быть подвергнут грануляции или использован для получения ком-
плексных удобрений 328-331
Фосфаты аммония можно получать также путем воздействия
фосфорсодержащих компонентов на сульфат аммония 332. Наблю-
дается тенденция замены серной кислоты фосфорной в процессе
Улавливания аммиака из коксового газа с получением моно- и ди-
аммонийфосфатов. Этот процесс может быть осуществлен как по
старой сатураторной схеме, так и по бессатураторной схеме с
Вакуум-выпаркой 327> 333~3к\
1270
Гл. XXXIII. Соли аммония
Предложено также использовать раствор моноаммонийфосфата
для извлечения аммиака из коксового газа. При этом образуется
диаммонийфосфат:
NH4H2PO4 + NH3 (NH4)2HPO4
Этот процесс эффективен тем, что раствор NH4H2PO4 поглощает
только один аммиак 341’342. В НИУИФ разработан способ для про-
мышленного использования отбросных сернистых газов цветной
металлургии с получением аммофоса. Этот метод основан на по-
глощении SO2 из газов аммиаком с последующим разложением
получаемых при этом растворов сульфит-бисульфита аммония
фосфорной кислотой:
SO2 + 2NH3 + Н2О = (NH4)2 SO3
SO2 + (NH4)2 SO3 + Н2О = 2NH4HSO3
Н3РО4 + nh4hso3 = nh4h2po4 + so2 + H2O
H3PO4 + 2NH4HSO3 =* (NH4)2HPO4 + 2SO2 + 2H2O
Получающуюся аммофосную пульпу перерабатывают в аммофос
по схеме с сушкой в распылительной сушилке. Удаляемый из пуль-
пы сернистый ангидрид может быть использован для получения
серной кислоты 343>344.
Схема производства аммофоса с применением распылительной
сушки пульпы 345, при которой не требуется предварительной вы-
парки и очистки фосфорной кислоты, изображена на рис. 372. Экс-
тракционная фосфорная кислота (22—28% Р2О5) нейтрализуется
аммиаком непрерывным способом последовательно в нескольких
реакторах 2. Вытекающая из последнего реактора пульпа посту-
пает в распылительную сушилку 7. Сушка в распылительной
сушилке производится дымовыми газами с температурой 650°, по-
лучающимися в топке при сжигании газообразного топлива. Выхо-
дящие из сушилки дымовые газы имеют температуру 100° и прохо-
дят для очистки от пыли батарейный циклон 8.
Высушенный порошкообразный аммофос непрерывно дозируется
в шнек-гранулятор 12, куда одновременно поступает также мелкая
фракция готового продукта и пульпа аммофоса. Из шнека-грануля-
тора гранулированный аммофос поступает в сушилку барабанного
типа 13. Сушку осуществляют дымовыми газами (500°) из топки 14.
Высушенные гранулы рассеивают.
Фракцию с размером зерен больше 4 мм растворяют в фосфор-
ной кислоте, идущей на аммонизацию; мелкую фракцию с части-
цами меньше 1 мм направляют в гранулятор; фракцию с.частицами
Фосфаты аммония
1271
1—4 мм выпускают в качестве готового продукта. Продукт содер-
жит 51% усвояемой Р2О5 и 11,5% N при использовании
ционной фосфорной кислоты,
полученной из апатитового
концентрата, и 47% усвояемой
P2Os и 10,7% N при использо-
вании кислоты, полученной из
фосфоритов Каратау.
О других вариантах полу-
чения ортофосфатов аммония
с другим аппаратурным офор-
млением см. 346“348.
Получение аммофоса из
фосфатных руд, содержащих
соединения магния (фосфориты
Каратау, Кингисеппские и др.),
DUJOi/ЭПН VDH
-dadjoodt tton
-nonhxDdujjxg
экстрак-
goi/nj DU
watyodu ntigowoj
осложнено выделением в про-
цессе аммонизации фосфорной
кислоты осадка магнийаммо-
нийфосфата MgNH4PO4 • Н2О.
Он присоединяется к осадкам
фосфатов полуторных окислов
и это приводит к загустеванию
реакционной пульпы. Путем
ступенчатой нейтрализации
фосфорной кислоты газообраз-
ным аммиаком можно после-
довательно выделить в твер-
дую фазу полуторные окислы
и магнийаммонийфосфат 349’35°.
После их отделения и выпар-
ки оставшегося чистого рас-
твора можно получить круп-
нокристаллический (или гра-
нулированный) фосфат аммо-
ния. По такой схеме можно
эффективно перерабатывать
Даже очень бедные и плохо
обогащаемые флотацией фос-
фориты, содержащие, напри-
мер, меньше 20% Р2О5 и боль-
Ще4% MgO. Отфильтрован-
ный и высушенный магний-
аммонийфосфат может приме-
няться в качестве самостоя
тельного азотно-фосфорномаг-
Рис. 372. Схема производства аммофоса:
Г —напорный бак для фосфорной кислоты; 2 - реактор; .3 - вентилятор; 4 — сборник; 5 - центробежный насос; 6 - напорный бак
для раствора фосфатов аммония; 7 распылительная сушилка; 3 — батарейный циклон для улавливания пыли; 9 — шнек; 10— эле-
ватор, //—бункер; /2 — шнек-гранулятор; 13— сушильный барабан; 14 — топка; 15 — двухснтный грохот; 16 — сборник с мешалкой;
/7 — транспортер.
16 М. Е. Позин
1272
Гл. XXXIII. Соли аммония
ниевого удобрения, содержащего азот в цитратнорастворимой фор-
ме, или же в смеси с аммофосом.
Для получения хорошо фильтрующих осадков аммонизацию ве-
дут при 80° сначала до pH 3,0—3,2, затем, после отделения осадка
фосфатов железа и аммония, добавляют к фильтрату соду или сульЙ
фат натрия с целью его обесфторивания. Это обеспечивает образе®
вание хорошо фильтрующего осадка магнийаммонийфосфата npfl
последующей аммонизации до pH 6—6,5. Осадок кремнефторида
натрия, образующийся при обеефторивании раствора, может быть
отфильтрован вместе с магнийаммонийфосфатом. Высушенный при
95—100° магнийаммонийфосфат содержит, например, 45,4% Р2О5,
17,5% MgO и 12,7% NH3.
При переработке описанным способом образца недообогащен-
ного Кингисеппского фосфорита, содержащего 19,65% Р2О5, 4,28%
MgO, 1,36% R2O3 и 1,52% фтора, основные компоненты распреде-
лялись следующим образом. В неотмытый осадок фосфатов полу-
торных окислов перешло 50,8% Р2О5 от его исходного количества
в фосфорной кислоте, из них 40,4% в виде водорастворимого фос-
фата аммония и 10,4% в виде цитратнорастворимых соединений.
В неотмытый осадок магнийаммонийфосфата перешло 17,4% Р2О5
и в очищенном растворе осталось 31,8% Р2О5. Следовательно, при
смешении фосфатов полуторных окислов с готовым продуктом —
аммофосом в него перейдет 82,6% Р2О5, из которых 87,4% -'-в во-
дорастворимой форме (в виде фосфатов аммония) и 12,6% —в ци-
тратнорастворимой. С магнийаммонийфосфатом удаляется 73,7 %
MgO от исходного количества, остальное переходит в аммофос в
виде цитратнорастворимого димагнийфосфата (8,65% MgO) и во-
дорастворимых кремнефторида и сульфата магния (16,65% MgO).
В аммофос переходит 90,8% израсходованного аммиака (из них
87,4% в виде фосфатов аммония), остальные 9,2% NH3 — в магний-
аммонийфосфат.
Разложение природного фосфата серной кислотой можно вести
с применением в качестве реакционной среды кислого раствора мо-
ноаммонийфосфата. При этом образуется осадок гипса, фильтрую-
щий лучше, чем при разложении в отсутствие иона аммония, и
производительность фильтров значительно увеличивается. Улуч-
шается на 1—1,5% и степень отмывки кислоты от осадка. Реак-
ционный раствор после разложения фосфата становится более кис-
лым, чем до разложения, и, после отделения гипса, его нейтрали-
зуют аммиаком для перевода вновь образовавшейся кислоты
в моноаммонийфосфат. Затем часть этого раствора вместе с ча-
стью суспензии гипса возвращают в первый сернокислотный экс-
трактор, а остальную часть после аммонизации до требуемой сте-
пени высушивают для получения аммофоса.
Аммофос может быть получен непосредственно из природных
фосфатов без предварительной их переработки в фосфорную кис-
Фосфат аммония
1273
лоту. Этот способ351 заключается в разложении фосфата концен-
трированным раствором фтористого аммония по реакции:
2Ca5F (РО4)3 + 18NH4F = 10CaF2 + 6 (NH4)3_X НХРО4 + 6xNH3
При 95—102° и трех-четырехкратном избытке 35—45%-ного рас-
твора NH4F против стехиометрического количества апатитовый кон-
центрат разлагается в течение 2—6 ч практически полностью. По-
сле отделения осадка CaF2 разделение содержащихся в растворе
фосфатов аммония от избытка фторида аммония можно осущест-
вить или выпаркой с кристаллизацией, или введением в раствор со-
единений алюминия для осаждения гексафторалюмината аммо-
ния— аммониевого криолита 3NH4F- AIF3 352. В последнем случае
остается раствор, из которого получается аммофос, содержащий,
например, ~58% Р2О5 и ~ 15% N. Из криолита легко регенери-
руется NH4F. Из CaF2 фтористый аммоний может быть регенери-
рован гидротермическим (стр. 1116) или гидрохимическими спосо-
бами, например, конверсией в СаСО3 и др. Сырьем для получения
аммофоса по этой схеме являются только природный фосфат и
аммиак — расход кислотного реагента (серной кислоты) полностью
исключается. Потери фтора при регенерации его из фторада каль-
ция и криолита, а также с раствором фосфатов аммония компен-
сируются за счет фтора, содержащегося в природном фосфате.
Остальная часть этого фтора может быть выпущена в виде фтори-
дов— CaF2, AIF3 (см. ниже) или других. Отсутствует неорганизо-
ванное выделение фтористых газов, что улучшает санитарные усло-
вия производства; уменьшается потребность в специальных кор-
розионноустойчивых материалах, так как разложение идет в ней-
тральной среде — величина pH конечного раствора 6,7—6,9.
Фосфаты полуторных окислов, так же как и фосфат кальция,
легко разлагаются фторидом аммония; при этом образуются фос-
фаты аммония и криолиты по реакции:
RPO4 • aq + 6NH4F = 3NH4F • RF3 + (NH4)3_X HXPO4 + xNH3 + aq
Поэтому этот способ позволяет вовлечь в производство удобре-
ний природные фосфаты и фосфорные промышленные отходы с
большим содержанием полуторных окислов, что расширяет сырье-
вую базу туковой промышленности.
При получении аммофоса по обычной схеме — с предваритель-
ным сернокислотным разложением природного фосфата — введе-
ние в реакционную пульпу NH4F увеличивает использование сырья
(Р2О5) за счет выделения полуторных окислов в форме криоли-
тов 353. Это позволяет и в этом процессе применять сырье е повы-
шенным содержанием полуторных окислов.
Этим же методом можно получать фторид алюминия с пони-
женным содержанием Р20з через гексафторалюминат аммония из
газов суперфосфатного производства.
16*
1274
Гл. XXXIII. Соли аммония
УДОБРЕНИЯ, СОДЕРЖАЩИЕ ПОЛИФОСФАТЫ АММОНИЯ
Получение полифосфатов аммония основано на аммонизации су-
перфосфорной кислоты (74—85% Р2О5) газообразным аммиаком3®4.
При этом получаются комплексные двойные удобрения, содержа-
щие до 80% усвояемых N + P2O5. Процесс проводится при 180—
190° и давлении 2,8—7 йт 355-358. Аммонизацию суперфосфорной
кислоты нужно вести при условиях, обеспечивающих содержание
в конечном продукте до 76—84% пирофосфатов общей формулы
(NH4)xH4_xP2O7, которые обладают лучшей растворимостью, чем
продукты с высоким содержанием полифосфатов 359“ 362.
Фирмой TVA предложена следующая схема получения полифос-
фатов аммония. Взаимодействие суперфосфорной кислоты с ам-
миаком осуществляется в двтоклаве из нержавеющей стали под да-
влением 2,75—3,5 ат. Автоклав снабжен мешалкой и охлаждающим
змеевиком для отвода тепла реакции. Плав из автоклава гранули-
руют в готовый продукт обычными способами 363’364. Эта же фирма
разработала способ производства полифосфатов аммония непо-
средственно из экстракционной фосфорной кислоты. Процесс ней-
трализации фосфорной кислоты проводят в две ступени в реакто-
рах, снабженных мешалками. Кислота и газообразный аммиак дви-
жутся противотоком. Для предупреждения затвердевания плава в
реакторах поддерживается высокая температура: в первом 155° и
во втором — 246°. Плав из второго реактора вместе с ретуром по-
дается в шнековый гранулятор. При концентрации исходной фос-
форной кислоты 55% Р2О5 получается продукт состава N : Р2О5 =
= 12 : 60, содержащий 50% Р2О5 в полифосфорной форме и осталь-
ной в ортоформе 365'367. |
УДОБРЕНИЯ, СОДЕРЖАЩИЕ МЕТАФОСФАТ АММОНИЯ ]
С недавнего времени все большее внимание стали уделять раз-
работке способов получения высококонцентрированного азотно-
фосфорного удобрения, содержащего около 17% N и 80% Р2О5,
путем взаимодействия аммиака с парами фосфорного ангидрида,
получаемыми сжиганием паров фосфора в сухом воздухе 368’369.
Твердый продукт выделяют из потока избыточного воздуха в элек-
трофильтре. Избыток воздуха необходим для поддержания в реак-
ционной камере температуры около 315°, так как при более высо-
кой температуре образуется стекловидный продукт с малым содер-
жанием азота.
В этих условиях взаимодействие аммиака с фосфорным ангид-
ридом идет по реакции:
Р4О10 + 4NH3 = 4HNPO (ОН) + 2Н2О
Выделившаяся вода гидролизует половину образовавшейся
метафосфиновой кислоты, в результате чего получается метафос-
фат аммония:
2HNPO (ОН) + 2Н2О = 2NH4PO3
Литература
1275
Суммарной реакцией процесса является:
Р4О10 + 4NH3 = 2HNPO (ОН) + 2NH4PO3
При избытке воды метафосфиновая кислота гидролизуется пол-
ностью, а при избытке аммиака образуется ее аммонийная соль
(NHPOONH4). Практически продуктом реакции является смесь
метафосфата аммония, метафосфиновой кислоты и ее аммонийной
соли, причем не менее половины азота содержится в аммиачной
форме, а другая половина — в имидной (группе NH). Эта часть
азота усваивается значительно медленнее, чем аммиачная. Поэтому
такое удобрение может представить интерес, как позволяющее
создать в почве запас усвояемого азота. Продукт представляет со-
бой пушистый белый малогигроскопичный порошок с объемным
весом 320—480 кг/м3. При хранении он несколько уплотняется.
ЛИТЕРАТУРА
1. Н. Седов, Научно-техн, информ, бюлл. НИУИФ, № 5—6, 79 (1957).—
2. J. Myl, D. Konbova, Chem. prumysL, 14, № 1, 38 (1964).—3. В. А. Ше-
стаков, С. С. Финкер, Т. М. Ч е р е м и ч к и н а, Кокс и химия, № 12, 37
(1965). — 4. J. Bell, Coke е. Gas, 12, № 134, 253; № 135, 277 (1950).—
5. L’ind. chim., 45, № 486, 32 (1958). — 6. Ind. Eng. Chem., 43, № 2, 45 A
(1951). — 7. I. R i s I, Chem. Proc. Ind., 34, № 12, 972 (1950). — 8. L’ind. chim.,
37, № 407, 180 (1951). — 9. Ind. Chem., 28, 327, 161 (1952). — 10. Chem. Eng.,
59, № 6, 242 (1952).
11. Chem. Age, 66, № 1702, 310 (1952). —/2. Chem. a. Proc. Eng., 37, № 12,
419 (1956). — 13. R. Sop or a, Compressed Air Mag., 64, № 6, 25 (1959).—
14. S. S. Sirur, V. K. Saolapurkar, Fertilizer News, 8, № 5, 27 (1963).—
15. S. A. Lou rich, Agr. Chem., 18, № 9, 54, 175, 177 (1963). — 16. В. А. Клев-
ке, H. H. Поляков и др., Технология азотных удобрений, Госхимиздат,
1956. — 17. Coke a. smokless-Fuel. Age, 8, № 87, 169 (1946). — 18. G. Е 111 е,
Vertilizer. Soc. Eng. Proc., 5, 47 (1949); L’ind. chim., 38, № 402, 16 (1951).—
19. А. П. Б p о н ш т e й н, В. Г. Л о п а р е в, В. В. С ы р е й щ и к о в, А. М. Ж а р-
нох, Кокс и химия, № 7, 42 (1961).—20. М. Я. Финкель, А. И. Т о л о ч к о,
₽. И. Меламед, Стандартизация, № 11, 38 (1961).
21. А. А. Р о д и л о в с к и й, Кокс и химия, № 1, 39 (1962).— 22. Г. П. Коп-
тев, Там же, № 5, 38 (1962). —23. М. И. Люкимсон, В. Е. Коз ель, Там
же, № 1, 37 (1963). — 24. Пат. ФРГ 1124930, 1962, —25. Англ. пат. 917567,
1963. — 26. Япон. пат. 3660, 1963. — 27. Т. Весен е, А. Каушнедас, Хим.
и хим. технол., № 5, 111 (1964). — 28. М. С. Литвиненко, А. И. Б р о д о-
вич, Л. А. Тумаркин, М. Я. Финкель и др., Кокс и химия, № 10, 22
(1964). — 29. Япон. пат. 17003, 1963. — 30. Пат. ФРГ 1171407, 1964.
31. А. Н. Туманов, М. К- Горлов, Н. А. Ю ф е р о в а, сб. «Химические
продукты коксования углей Востока СССР», в. 4, 1967, стр. 55.—32. Г. Н. Ле-
бедева, Л. А. Шашмурина, В. П. Хлопцев, В. Г. Лопарев, Там же,
Авт- свид. 138239, 1961, — 34. Канада Тацуо, Никкакё гэппо,
4, № 11, 637 (1961).—35. Саотомэ Хаятоси, Chem. Ind. Econ., 9, № 2,
04 (1962).—35. Качаку Кэйдзай, ibid., 10, № 3, 267 (1963). —37. Пат,
nocl 3035899, 1962. — 38. G. Schmutzler, Chem. Techn., 17, № 12, 720
,v5!-—39. J. Cleunett, Meeh. a. Chem. Eng., Trans. Inst. Eng. (Austral.).
54 MOfio144 (1965)-—40- Nitrogen, № 39, 34 (1966); Chem. Eng. Progr., 64, № 5.
1276
Гл. XXXIII. Соли аммония
41. Япон. пат. 10496, 1966. — 42. С. Н. Л а з о р и н, Е. Я. Стеценко, Про-
изводство сульфата аммония на коксохимических заводах, Изд, «Металлургия»,
1965. — 43. Справочник азотчика, т. 2, Изд. «Химия», 1969, стр. 193.— 44. J. N е 1-
8 е г, J. Jugmann, Sb. praci. Ped. fax. Ostrav6, № 6, 49 (1967).— 45. И. A. Мы-
кольннков, H. А. Гурьянов, В. К. Качаев, Т. Н. Бобровникова,
Кокс и химия, № 12, 46 (1967). — 46. Ито Сюит и, Миямото Рок уро,
Кочё есуй, № 109, 24 (1967).—47. Авт. свид. 202423, 1967. — 48. Н. В а н г н и ц,
Кокс и химия, № 2, 49 (1968).—49. Технология азотных удобрений, под ред.
С. И. Вольфковича, ОНТИ, 1935. — 50. В. Г. Постоловский, Производство
сульфата аммония из синтетического аммиака, ОНТИ, 1935.
51. Пат. США 3006725, 1961.—52. Г. Н. Лебедева, Л. А. Бурмист-
ре н к о, И. В. Колодяжный, Е. Г. Каплина, О. Г. Полануер,
И. П. Кулик, Кокс и химия, № 9, 42 (1963). — 53. Т. С. Калганов,
Т. Л. Савицкая, А. П. Шевченко, Там же, № 1, 33 (1953).—54. Авт.
свид. 161705, 1964.—55. Д. С. Петренко, Е. Б. Иванов, Кокс и химия,
№ 4, 43 (1965). — 56. Д. Д. Ай вен ко, Там же, № 9, 42 (1965). — 57: Пат.
США 3180713, 1965. — 58. К. А. Белов, Кокс и химия, № 1, 35 (1966).—
59. М. Я. Ф и н к е л ь,' Ю. И. Р е з у н е н к о, И. М. Давыденко, В. И. К а-
лашников, Т. Е. Гимельштейн, А. И. Шевченко, В. И. Ильяшен-
ко, М. С. К о м а р о в с к и й, № 4, 30 (1966). — 60. Авт. свид. 176573, 1965.
61. J. Karbowski, Koks, smola, gaz, 11, № 6, 235 (1966).—62. Япон.
пат. 3169, 1961. — 63. Англ. пат. 883313, 1961. — 64. R. Stahl, Chim. et Ind., 86,
№ 2, 123 (1961). — 65. S. V e d a r a m a n, Proc. Natl Acad. Sci. (India), A31,
№ 1, 48 (1961).—66. A. K. Roy, К. C. Banerji, M. К. В a r d h a n,
К R. ChaKravorty, J. Sci. Ind. Res., D 20, № 10, 361 (1961).—67. Англ,
пат. 886848, 1962. — 68. Англ. пат. 888031, 1962. — 69. А. К. Roy, В. В. Roy,
A. Mukherjee, N. S. R. A n j a n е g u 1 и, Indian J. Technol., 1, № 6, 258
(1963). — 70. Англ. пат. 937304, 1963. ’
71. S. К. R a m а п, S. R а о, D 1 w a k а г, Res. a. Ind., 9, № 7, 187 (1964).—
72. Inform, chim., № 43, 71 (1966). — 73. Nitrogen, № 46, 21 (1967). — 74. Chem.
Age (India), 18, № 3, 173 (1967). — 75. Япон. пат. 13466, 1964. — 76. Авт. свид.
183723, 1966. — 77. Япон. пат. 8529, 1967.— 78. К. King, Ind. Eng. Chem, 42,
№ 11, 2241 (1950). — 79. Shoichiro Hori, Shikazo Sai bar a, Rept.
Tokyo, Ind. Testing Lab., 37, 1 (1942).—80. Франц, пат. 1290193, 1962.
81. Япон. пат. 16862, 1963.— 82. Пат. ФРГ 1187593, 1965.—83. Chem. Age,
82, № 2102, 571 (1959).—84. Англ. пат. 903822, 1962.—85. Франц, пат. 1303816,
1962.—86. Англ. пат. 1031724, 1966. — 87. Г. О. Григорян, Р. М. Кирако-
сян, Айкакан кимнакан амсагир (Арм. хим. ж.), 20, № 2, 164 (1967). — 88. Ру-
мын. пат. 43128, 1965. — 89. А. М. Дубовицкий, И. М. Эпштейн, Произ-
водство сульфата аммония, ГОНТИ, 1938. — 90. Труды НИУИФ, в. 144, 1940.
91. I. Rupp, пат. ФРГ 889, 290, 1953. —92. Англ. пат. 1059241, 1965,—
93. Пат. США 3013860, 1961. — 94. К. А. Иванов, В, А. К о д а н а ш в и л и,
Труды Грузинского политехи, ин-та, № 5 (85), 1962, стр. 101. — 95. Авт. свид.
148035, 1962. — 96. Авт. свид. 186990, 1966. — 97. Япон. пат. 20172, 1965.—
98. Д. Д. Т а г а н о в и ч, Автореф. канд. дисс., Томский политехнический ин-т,
1954.-99. Н. А. Василенко, ЖПХ, 21, № 9, 31 (1948); 22, № 4, 337 (1949);
23, № 5, 472 (1950). — 100. М. Е. П о з и н, Технология минеральных удобрений и
солей, Госхимиздат, 1957.
101. М. А. Рабкин, А. П. Романенко, М. Е. Ковальская, ЖПХ,
24, № 12, 1257 (1951). — 102. К. П. Попов, В. Ф. Соколов, Кокс и химия,
№ 8, 35 (1961). — 103. J. N у v 11, V. V 5 с 1 a v fl, Chem. prtimysl (чешек.), 12.
№ 4, 170 (1962). — 104. Япон. пат. 3660, 1963. — 105. Япон. пат. 16863, 1963.—
106. Д. С. Петренко, Л. М. Арабова, Кокс и химия, № 2, 22 (1967).—
107. М. Б. К а р ц ы п е л ь, И. М. Ханин, Н. И. В а н ж а, О. Н. Бакун, сб.
Литература
1277
«Химическая технология», в. 7, 1967, стр. 33. — 108. С. Ю. С и п о в и ч, Кокс
и химия, № 8, 42 (1961). —109. К- П. Попов, Там же, № 5, 43 (1963).—
НО. М. Д- Кузнецов, И. Л. Непомнящий, П. Л. Новицкий,
3. Г. Л и н н а я, Там же, № 8, 39 (1961).
Ill Н. Ф. Симонов, Ю. П. К о с ь к о в, Там же, № 11, 30 (1964).—
112. М Д. Кузнецов, П. Л. Новицкий, Там же, № ТО, 55 (1965).—
ИЗ. О Г. Нели па, Там же, № 4, 37 (1966). — 114. М. Д. Кузнецов,
П. Л. Новицкий, Металлургия и коксохимия, Ns 5, 130 (1966).—
115. Н. Д. Кручинина, Г. Л. Грошев, С. М. Д о п о в, Г. В. Рыбин,
В. Г. Михеев, Хим. пром., № 10, 737 (1967). — 116. А. Д. Алексеев,
В. М В а т ю г и н, Ю. А. Янович, Изв. Томск, политехи, ин-та, 148, 115
(1967). — 117. П. 3. Ш у б е к о, С. К. Хмелевой, И. В. Колодяжный,
Кокс и химия, № 1, 38 (1963). — 118. Чжу Бао-кан, Хуасюэ шнцзе (кит.),
19, № 5, 232 (1965). — 119. Ind. Eng. Chem., 49, № 2, 57А —58 (1957).—
120. Chem. Eng., 59, Ns 6, 242 (1952).
121. Ibid., 57, № 6, 229 (1950). — 122. J. Bell, The Gas World, 80, № 3394,
Coking Section, 105 (1949). — 123. W. В о u 1 i n, J. Usines a. Gas, 76, Ns 1, 4
(1952). — 124. J. V. van D о u b u r g, Chem. Eng., 54, № 8, 93 (1947).—
125. G. Higson, Chem. Ind. № 26, 492 (1950); № 36, 750 (1951).—
126. W. К1 e m p t, Ber. Ges. Kohlentechn., 5, 401 (1950).—127. Ch. Bragg,
Petrol. Process., 59, Ns 6, 177 (1952) . —/28. L’ind. chim., 34, Ns 365, 232 (1947).—
129. Gas World, Coking Section, 60, № 2840, 12 (1939). — 130. W. Mantel,
H. Hansen, Brennstolf—,Chem., 33, Ns 5/6, 69 (1952).
131. Coke a. Gas, 21, № 242, 261 (1959). — 132. C. Otto, пат. США 2509520,
1950. — 133. W. Tiddy, пат. США 2511306—7, 1950. — 134. Франц, пат. 999342,
1952.— 135. К. А. Белов, Улавливание химических продуктов коксования, Гос-
химиздат, 1948, стр. 96. — 136. И. Е. Коробчанский, М. Д. Кузнецов,
Расчеты аппаратуры для улавливания химических продуктов коксования, Гос-
химиздат, 1952, стр. 107. — 137. А. Гомельский, Аппаратчики коксохимиче-
ских производств, Госметаллургиздат, 1953, гл. V. — 138. Д. С. Петренко,
Производство сульфата аммония, Изд. «Металлургия», 1966.— 139. М. С. Лит-
виненко, Ж. ам. техн., 21, Ns 3, 191 (1944). — 140. Англ. пат. 647742, 1947.
141. Пат. ФРГ 1081870, 1960. — 142. Пат. ФРГ 1056593, 1961, —143. Пат.
ФРГ, 1101380, 1961. —144. Е. Я. Стеценко, Кокс и химия, Ns 7, 41 (1963).—
145. Япон. пат. 2449, 1958. — 146. Г. П. Коптев, М. И. Сор к ин, Кокс и хи-
мия, Ns 10, 37 (1963). — 147. Франц, пат. 1352367, 1964. — 148. Япон. пат. 8770,
1964.-/49. Франц, пат. 1352367, 1964, — 150. J. Manning, L’ind. chim., 34,
Ns 362, 181 (1947).
151. G. Berkhoff, Chem. Met. Eng., № 7, 366 (1937). — 152. W. К 1 e m p t,
Brennstoff — Chem., 33, Ns 7/8, 114 (1952). —/53. Chem. Eng., 60, Ns 1, 338
(1953). — 154. Пат. США 2516420, 1950.— /55. Chem. Eng., 62, Ns 1, 236 (1955). —
156. J. Buchham, R. Moulton, Chem. Eng. Progr., 51, Ns 3 126 (1955).—
157. Ind. Chem., 29, Ns 346, 535 (1953). —/58. E. К a g a t e, K. Akizama, Coal
har (Japan), 6, 394 (1954); P. Remy-Gennete Bull. Soc. chim. France, 1953,
11/12, 1042.— 159. Япон. пат. 177414, 1949. — 160. Франц, пат. 984768, 1951.
М/. Пат. США 2616788, 19521— 162. Канад, пат. 477870, 1951. —/53. Пат
уША. 2600753, 1952. — 164. Пат. США 2631084, 1953.— /55. Nitrogen, Ns 45, 33
1кяЬ~166- Авт- свид. 192766, 1967. —/57. Япон. пат. 12726, 1966 —
19fiRM' Turlure- Bull- Hais coker, № 13, 15 (1963). —169. Пат. ФРГ 1004141,
хгоТ/7®' Кокубо Р ё, Сосаки Снкэкаузу, Chem. Eng. (Japan), 28,
№ 2,138(1964).
172 17а' Bamforth, Chem. Proc. Eng., 46, Ns 2, 81 (1965).—
СТПл a ш м У Р и н а и ДР-- Кокс и химия, № 10, 33 (1962). — 173. Пат.
2656248, 1953.-/74. Пат. США 2653077, 1958, —175. Пат. США 2659659.
1278
Гл. XXXIII. Соли аммония
1953. —176. Пат. США 2524341, 1950. —177. A. Bam forth, Chem. Proc. Enu.. .»
35, № 4 (1954); Corrosion Techn., 1, № 2, 31 (1954). — 178. К. S u g i b a у a s h i, fl
Chem. Eng. (Japan), 3, № 1, 16 (1954). — 179. A. Ward, Ind. Chem., 24, № 286, Я
722 (1948). — 180. Genie Civill, 129, 14 (1952). Я
181. К. Seshadri, J. Gupta, J. Sci. Ind. Res. (India), 13, 788 (1954). — Я
182. Ind. Eng. Chem., 49, № 2, 57A (1957). —/83. Франц, пат. 973296, 1951. —Я
184. Франц, пат. 55464, 1952. — 185. Франц, пат. 1011508, 1952. —186. G. В. С о г- |Н
dell, Ind. Eng. Chem. Proc. Design a. Developm., 7, № 2, 278 (1968). — 187. Воль ф-Я
кович, В. Камзолкин, А. Соколовский, Труды НИУ, в. 64, 1929; ЖХП, Ч
№ 13 и 14 (1929). —188. С. И. Вольфкович, А. А. Соколовский, ЖХП, !
№ 4 (1932). —189. А. А. Соколовский, Труды НИУ, в. 101, 1932.—
190. С. Г а н з, С. Лейбович, И. Г о р б м а н, ЖПХ, 32, № 5, 975 (1959).—
1191. Е. А. Рашковская, Е. И. Черненькая, ЖПХ, 40, № 2, 301
" (1967). —2192. S. Kaminski, Przem. Chem., 45, № 8, 431 (1966). -i!93. H. Па-
нови др., ЖПХ, 35, № 4 (1962). — 194. Франц, пат. 1395701, 1965. — 195'. Франц,
пат. 84833, 1965. —196. Токуда Сигэру, Качачу кэйдзай, Chem. Ind.
Econ., 12, № 4, 359 (1965). — 197. К- Дворниченко, Е. М. Зубова, Хим.
пром., № 1, 51 (1967). — 198. Швейц, пат. 401935, 1966. —199. Япон. пат. 25209,
1965. — 200. Пат. ГДР 39660, 1965.
4201. И. Р. Кричевский, В. А. Клевке, ЖХП, № 12, 17 (1932).—
Z202. Е. Ja песке, Z. angew. Chem., № 33, 916 (1928). —>203. В. Г. Флейш-
м а н, Хим. пром., № 7, 414 (1957). -6204. В. Г. Шляхтов, В. В. С т р е л ь- 1
ц о в, Изв. вузов. Хим. и хим. технол., 11, № 2, 232 (1968); 12, Ns 1 (1969).—
$205. Авт. свид. 191497, 1967. -^206. Л. И. 3 л о т и н, М. К- Голубцова, Хим. !
пром., № 7, 414 (1957). — 207. Ф. К. Михайлов, Е. А. Рашковская,
Е. И. Черненькая, ЖПХ, 40, Ns 6. 1173 (1967).— 208. Англ. пат. 970943,
1964. — 209. Япон. пат. 2852, 1964.— 2/0. Япон. пат. 7968, 1958.
211. В. Л. Р ы д н и к, Хим. пром., Ns 8, 253 (1953).— 212. Ф. К. Михай-
лов, Там же, Ns 7, 393 (1956).—213. В. Ф. Чернов, Производство кальци-
нированной соды, Госхимиздат, 1956. —214. Ц. Л. Д р у х и др., Труды НИОХИМ,
т 19, 1969, стр. 160. — 215. Япон. пат. 10209, 1965. — 216. Авт. свид. 202085,
1967.— 2/7. Look Japan, 10, Ns 105, 2 (1965).—2/8. М. К- Шелудько, сб.
. «Химия и технология минеральных удобрений», Ташкент, Изд. ФАН, 1966,
стр. 299. — 219. И. Р. Кричевский, Е. Гольдман, Z. anorg. Chem., 218,
253 (1934). —220. W. Gluud, Z. angew. Chem., 43 (1930).
221. M. К. Шелудько, H. Ф. Кулиш, Труды Днепропетровского ХТИ,
в. 5, 1956, стр. 201. — 222. Япон. пат. 1100, 1966. — 223. М. М. К У ш н и р,
В. Н. Васильков, сб. «Химическая промышленность Украины», Ns 1 (37), ।
1968, стр. 14. —224. Е. Janecke, Z. Elektrochem., 35, Ns 6, 332 (1929).—
225. Япон. пат. 1336, 1966. — 226. М. Нише в, М. Кръестева, Д. Дим и- ,
т р о в, Химия и индустрия (Бълг.), 36, № 9, 347 (1964). — 227. Е. А. Р а ш к о в- \
ска я, Е. И. Черненькая, ЖПХ, 40, Ns 2, 301 (1967). — 228. С. Н. Г а н з,
Рефераты докладов на секции агроном, химии и удобрений VIII Менделеевского
съезда, 1959, стр. 16. — 229. И. П. У с ю к и н, Там же, стр. 13. — 230. И. Теле-
то в, Г. Горштейн, Н. Ткаченко, ЖХП, Ns 10, 53 (1933).
231. И. П. У с ю к и н, Хим. пром., Ns 6, 457 (1962). — 232. И. П. У с ю к и
И. П. Кармазин, Б. Н. Бобров, А. П. Уварова, А. В. Р е з н и к о в|^В
А. А. Черенкова, Хим. пром., Ns 5, 355 (1966). — 233. S. Р a w 1 i к о w s k i^H
S. Aniol, S. Bistro n, A. Chomiakow, S. Szymonik, ^Magyar kern, lapfa^fl
21, Ns 2, 92 (1966). — 234. Пат. США 3310367, 1967. — 235. В. M. Р а м м, КарбоЯ
нат аммония — абсорбция газов, Изд. «Химия», 1966.—236. С. Н. Г а н з, ЖПХ.^Ч
33, Ns 6 (1960); 34, Ns 6 (1961). — 237. И. А. Шахова, А. И. Рычков, ,
Е. В. Дмитренко, Хим. пром., № 11, 783 (1961). — 238. С. Н. Г а н з, I
Г. И. Васильев, Л. В. Лопатин, Изв. вузов. Хим. и хнм. технол., 2, Ns 6,
Литература
1279
913 (1959). — 239. В. Ваггаман, Фосфорная кислота, фосфаты и фосфорные
удобрения, Госхимиздат, 1957. — 240. Б. А. Муромцев, Калий, Ns 1, 36 (1937).
241. Б. А. Муромцев, Л. А. Назарова, Изв. АН СССР. Сер. Хим., № 1,
177 (1938). — 242. С. И. Вольфкович, Л. Е. Берлин, Б. М. М а н ц е в,
ЖПХ 5, № 1, 1 (1932). — 243. J. С. В г о s h е е г, J. F. Anderson, J. Am.
Chem.’ Soc., 68, № 4, 902 (1946). — 244. К. Чернова, ИСФХА АН СССР, 15,
112 (1947). — 245. С. S. Huey, Н. V. Tartar, J. Am. Chem. Soc., 62, 26
(1940).—246. A. R. Merz, W. H. Fry, J. О. H о r d e s t y, J. R. Adams, Ind.
Eng Chem., 25, 136 (1933).—247. H. L. Thompson, P. Miller a oth., ibid.,
41, 458 (1949).— 248. К. C. Beeson, ibid., 29, 705 (1937). — 249. E. L. L a r i-
so'n, ibid., 1176. — 250. С. Д. Фридман, И. H. Поляков, Л. С. С к у м,
Р. Я. К и р и н д а с о в а, Хим. пром., № 3, 46 (1967).
251. Н. Г. Бунаков, Г. Д. Харлампович, ЖПХ, 38, № 9, 1915
(1965). —252. С. Я. Шпунт, Там же, 20, № 7, 685 (1947); 30, № 7, 985
>1957). —255. М. М. Striplin, Chem. Eng., 68, № 19, 160 (1961). — 254. R. С о а-
t е s, G. Woodward, J. Chem. Soc., 1964, 1780. — 265. D. Corbridge, E. L o-
wa/ibid., 493 (1954). — 256. F. К a s p a r e k, Acta Vniv. Palackianac Olomeic.,
Fac. Rerum Nate, 9, 217 (1962).—257. I. Kobayashi, Nippon Kagaku Zasshi,
83, 132 (1963). —258. E. Thilo, H. G r u n z e, Z. anorg. Chem., 281,262 (1955).—
259. J. R. Van W a z e r, Phosphorus a. Its Compounds, N, Y., 1958, p. 619.—
260. A. W. Frazier, J. P. Smith, J. R. Lehr, J. Agr. Food Chem., 13, Ns 4,
316 (1965).
261. G. O. G u e r r a n t, D. E. Brown, 13, № 6, 493 (1965). — 262. A. W. F r a-
zier, J. P. Smith, J. R. Lehr, ibid., 13, № 4, 316 (1965). — 263. Harau С ё-
umupo J. Chem. Soc. Japan. Ind. Chem. Sec., 69, № 11, 2077, A117 (1966).—
264. T. D. Farr, J. D. Fleming, J. D. Hatfield, J. Chem. Eng. Data, 12,
№ 1, 141 (1967). — 265. G. L. Terman, J. Sil verb erg, Farm. Chem., 121,
№ 6, 27, 31 (1958); Хим. и хим. технол., № 2 (1959).—266. J. Silverberg,
Hoffmeister, № 2 (1959). — 267. M. Man, L. C r i s t e s c u, A. Popescu, A. N e-
ciu, Rev. chim., 14, № 7, 375 (1963).—258. Пат. США 3005696, 1961, — 269. Япон.
пат., 9118, 1961. — 270. Б. Д. Мельник, Хим. пром., № 1, 5 (1957).
271, Т. X и г н е т, Г. Хикс, Дж. Джордан, Chem. a. Chem. Techno!., 12,
22 (1957). — 272, V. N. К a s t u г i г a n g а л, S. N. Sastri, Fertilizer News, 8,
№ 11, 68 (1963). — 273. Эбихара Иосиро, Кояаку Когё, Chem. Ind., 12,
№ 11, 980 (1961). — 274. J. M. Potts, D. W. R i n d t, A. V. S 1 a k, Chem. Eng.
Progr., 58, № 9, 89 (1962). — 275. L. Kramer, Chem. Ind., 40, № 7 (1948).—
276. J. Atwell, Ind. Eng. Chem., 41, 1318 (1949). — 277. A. M. Дубовиц-
кий, А. И. Шерешевский, Технология минеральных удобрений, Госхимиз-
дат, 1947. — 278. Г. Д. Харлампович, П. М. Поздеев, Хим. пром., № 6,
26 (1967). — 279. W. S. Kramer, Ind. Eng. Chem., 28, 1470 (1936).—
280. W. H. Ross, A. R. Merz, К. D. Jacob, ibid., 21, 286 (1929).
281. W. C. Weber, Chem. Met. Eng., 72 (1933).—282. E. C. Hons ton
284°th ’ 1 Agr Food Chem- 3’ № 43 (1955).— 283. Ibid., № 5, 374 (1955).—
Э- И. Цыпина, Ю. А. 3 а б е л е т и н с к и й, Вести, техн, и эконом,
ииформ., НИИТЭХИМ, Ns 4 (1964).—285 В. В. Краснушкин, Хим. пром.,
9o73’,J49 (1964). —285. С. И Вольфкович, Природа, № 10, 75 (1961).—
р В- Гофман, Журн. ВХО, № 1, (1960). — 288. Г. Д. Харлампович,
„л И, К у д р я ш о в а, Кокс и химия, № 2 (1964). — 289. Япон. пат. 7106, 1965.—
• С. К. Воскресенский, Хим. наука и пром., 1, № 2 (1956).
С- К- Воскресенский, Исследования в области концентрированных
т₽хиРНЫх Удобрений, НИУИФ, 1957. — 292. С. К- Милованова, Научно-
информ, бюлл., № 9 (1957).— 293. С. К- Воскресенский, С. К- Ми-
и Нова’ Обмен опытом, НИУИФ, № 2 (1957). — 294. Дж. Бернет, Хим.
м технол., Ns 1, 31 (1958). — 295. Гарн Вальтер, Бюлл. техн, информ
1280
Гл. XXXIII. Соли аммония
Гипрохима, № 38 (1958). — 296. Экспресс-информ. ВИНИТИ. Сер. хим. и технол..
неорг. веществ, № 17 (1964). — 297. И. Л. Гофман, Хим. наука и пром., 2,
№ 6, 711 (1957). — 298. Дж. Гетзингер, Хим. и хим. технол., № 4, 26
(1958). — 299. Е. С. Houston, ibid., № 1, 54 (1956).—300. Хаустов, Бюлл.
техн, информ. Гипрохима (1959).
301. ВИНИТИ, Экспресс-информ. Сер. хим. и технол. неорг. веществ, № 22
(1961). — 302. N. Р. С h о р е у, Экспресс-информ. ВИНИТИ. Сер. хим. и технол.
неорг. веществ, № 25 (1962). — 303. J. I. Marino, Meeh. Eng., 84, № 1, 34
(1962). — 304. Brit. Chem. Eng., 12, № 1, Suppl. (1967). — 305. Япон. пат. 13467,
1964 — 306. Пат. США 3153574, 1964. — 307. Пат. США 3155490, 1964. —
308. Авт. свид. 199847, 1967.—309. Дай Юань-фа, Чэнь Цин-юй, Хуагун
Сюэбао, № 3, 145 (1965). — 310. К. N. S у n g h a b, Fertilizer News, 13, № 2,
32 (1968).
311. Австр. пат. 239762, 1965.—3/2. Авт. свид. 189875, 1967. — 313. Г. Д. Хар-
ла м п о в и ч. Р. И. Кудряшова, Кокс и химия, № 11, 38 (1961).—314. Пат.
США 3245777, 1966.—315. А. Н. Соловьева, И. Н. Шокин, Е. Л. Яхон-
това, сб. «Химия и технология», в. 6, 1967, стр. 206. — 316. А. Н. Соловьева,
И. Н. Шокин, Е. Л. Яхонтова, Труды Московского хим.-технол. ин-та
им. Д. И. Менделеева, в. 51, 1966, стр. 128.—317. Япон. пат,. 16348, 1965.—
318. Япон. пат. 3412, 1964.—319. Brit. Chem. Eng., 11, № 3 (1966).—
320. L. G. Nilsson, Phos. a. Pot., № 24, 22 (1966).
321. Synowies Jerzy, Przem. Chem., 45, № 10, 547 (1966).—322. Brit.
Chem. Eng., 12, № 11, Suppl. (1967). —323. Пат. США 3310371, 1967. -
324. В. С. В ы з г о, А. И. Павлова, М. Н. Набиев, Узб. хим. ж., № 4,
5 (1965). — 325. Франц, пат. 1372022, 1964.—326. Brit. Chem. Eng., 12, № 1,
Supl. (1967).—327. Г. Д. X а р л а м п о в и ч, П. М. Поздеев, В. С. Ю ш и н а.
Н. Г. Бунаков, Г. Н. Лебедева, К.’ Н. Разумов, В. М. Kara со в,
О. П. Хлебников, Г. Н. Воронин, Кокс и химия, № 2, 37 (1968) —
328. Chem. Eng., 73, № 26, 58 (1966). — 329. Англ. пат. 1032628, 1966, — 330. Англ,
пат. 1035483, 1966.
331. Англ. пат. 1042132, 1966.—332. Авт. свид. 203642, 1967.—333. В. М. Ка-
та с о в, О. П. Хлебников, И. П. Белоусов, А. Ф. Тимофеев, Г. В Во-
ронин, И. М. Трощипенко, Н. Г. Бунаков, Г. Н. Лебедева,
Г. Д. Харлампович, Кокс и химия, № 12, 39 (1967). — 334. G. D. С h ат-
lampowicz, N. G. В u n а к о w, G. N. Lebiediewa, W. М. К a g a s о w,
О. Р. С h 1 е b n i к о w, Koks, Smola, Gaz, 12, № 11, 286 (1967).—335. Англ,
пат. 934436, 1963. — 336. Англ. пат. 996209, 1965. — 337. Г. Д. Харлампович,
Кокс и химия, № 10, 10 (1964). —338. W. G. Schulze, Eng. Proc., 20, 188
(1961).—339. Пат. США 2970888, 1961. — 340. В. В. Краснушкин, Хим.
пром., № 3, 149 (1964).
341. Г. Д. Харлампович и др., Кокс и химия, № 2, 34 (1962).—
342. М. В. Гофман и др., Журн. ВХО, 5, № 1, 38 (I960). — 343. Н. А. Ва-
силенко, 3. П. Розенкноп, Рефераты научно-исследовательских . работ,
НИУИФ, в. 163, 1959, стр. 6. — 344. Н. А. Василенко, М М. Чернобаев а,
С. Л. Реутова, Хим. пром., № 6, 30 (1961). — 345. С. К. Милованова,
В. Е. М о с т о в и ч, Рефераты научно-исследовательских работ НИУИФ, в. 101,
1958. — 346. Austral. Chem. Process a. Eng., 18, № 10, 32 (1965). — 347. Англ,
пат. 997881, 1965.—348. Пат. ГДР 28902, 1964.—349. М Е. П о з и н, Б. А. К °"
пылев, Г. Я. Попова, В. Л. Варшавский, ЖПХ. 41, № 9, 1877 (1968);
42, № 6, 1209 (1969). — 350. В. Л. Варшавский, Г. Я. Попова, Б. А. Ко-
пы л е в, М. Е. П о з и н, Труды НТК «Оболовые фосфориты как сырье для хи-
мической промышленности», Таллин, 1968, стр. 181.
351. М. Е. П о з и н, Р. Ю. 3 и н ю к, И. Н. Куприянова, авт. свид. 214550,
1967; Хим. пром., № 3, 196 (1969). — 352. М. Е. П о з и н, Р. Ю. Зиню к,
Литература
1281
И. Н- Куприянова, ЖПХ, 41, № 11, 2352 (1968).— 353. И. Е. П о з и н,
р Ю. Зиню к, И. Н. Куприянова, авт. свид. 223822, 1967. — 354. Phos. а.
Pot., № 24, 30 (1966). — 355. Пат. США 3228752, 1966. — 356. Пат. США 3171733,
1965. — 357. Япон. пат. 24032, 1965. —358. Англ. пат. 1001984, 1965. — 359. Пат.
США 3264085, 1966. — 360. Пат; США 3226222, 1965.
36!. Young Donald С., пат. США 3243279, 1966. —362. Япон. пат. 15603
1962. — 363. Chem. Process, 9, № 3, 8 (1963). — 364. J. G. Get sin ger,
M. R. Siegel, H. C. Mann, J. Agr. Food Chem., 10, № 4, 341 (1962) —
365. Chem. Eng. News, 43, № 39, 63 (1965). — 366. Пат. США 3044851, 1962.
367. Уэда Сиро, Ояма Кэйда и, Кома Кэйд з и, J. Chem. Soc. Japan.
Ind. Chem. Sec., 66, № 5, 586 (1963).—368. Дж. Стинсон и др., Хим. и хим.
технол., № И, 44 (1956).—369. J. С. Driskell a. oth., J. Agr. a. Food Chem,
7, № 9, 618 (1959).
Глава XXXIV
КАРБАМИД
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Карбамид ССЦМНгЦ— диамид угольной кислоты (мочевина)
в чистом виде представляет собой бесцветные кристаллы, не имею-
щие запаха, с плотностью 1,335 г/см3 и температурой плавления
132,4°. Технический карбамид — белые или желтоватые кристаллы,
имеющие форму иглообразных ромбических призм; его насыпной
вес колеблется в пределах 0,52—0,64 т)м3 (в зависимости от влаж-
ности). В чистом карбамиде содержится 46,6% азота или, в пере-
счете на аммиак, 56,7%.
Сухой карбамид при нагревании под атмосферным давлением
выше температуры плавления разлагается. Механизм его разложе-
ния предполагается следующий. Вначале карбамид изомеризуется
в цианат аммония, который диссоциирует на циановую кислоту и
аммиак:
CO(NH2)2 —► NH1NCO —> HNCO4-NH3
Циановая кислота, взаимодействуя с карбамидом, образует
биурет:
HNCO + СО (NH2)2 = nh2conhconh2
При более сильном нагревании биурет разлагается на аммиак
и циановую кислоту, полимеризующуюся в циануровую кислоту
(НМСО)з, которая образует с выделившимся аммиаком амиды и
имиды циануровой кислоты ’~8. В присутствии избытка аммиака
разложение карбамида приостанавливается 9. Добавление NH4NO3
к карбамиду приводит к весьма существенной стабилизации послед-
него, тем большей, чем больше содержится нитрата в смеси 10. На-
сыщенный раствор карбамида в воде при 20° содержит 51,83%, при
60° — 71,88%, при 120° —95% CO(NH2)2. Выше 130° в водном рас-
творе карбамид разлагается на аммиак и двуокись углерода. Тем-
пературы кипения водных растворов карбамида различной концен-
Физико-химические свойства
1283
трации при различных давлениях могут быть определены по номо-
грамме на рис. 373. Линия, ограничивающая на номограмме сетку
кривых, — кривая растворимости. Эвтектика соответствует темпера-
туре —12° и концентрации 32% CO(NH2)2- Косые линии, идущие
сверху вниз, — величины плотности растворов, а кривые, идущие
слева направо, — давления насыщенного пара над растворами.
Рис. 373. Свойства водных растворов карбамида (номограмма Фрежака),
Карбамид хорошо растворяется в спирте и в аммиаке. С аммиа-
ком образует соединение (ЬШгЬСО • NH3, содержащее 77,9% кар-
бамида и 22,1% аммиака. С повышением температуры раствори-
мость карбамида в аммиаке значительно повышается; выше 30°
растворимость карбамида в жидком аммиаке больше, чем в воде.
Давление паров аммиака над растворами карбамида в аммиаке
значительно ниже, чем над жидким аммиаком.
На рис. 374 приведена диаграмма состояния системы карба-
мид—аммиак—вода и кривые давления пара насыщенных раство-
ров ”,
Карбамид вступает в реакцию с кислотами, давая солеобраз-
ные соединения. Например, малорастворимый в воде нитрат кар-
бамида NH2CONH2-HNO3 кристаллизуется в виде моноклинных
призм, плавящихся при 163° (при нагревании разлагается со
1284
. Гл. XXXIV. Карбамид
взрывом), фосфат карбамида NH2CONH2-H3PO4 образует ромби-
ческие кристаллы с температурой плавления 117,5°. Фосфат карба-
мида хорошо растворяется в воде 12, но при этом полностью диссо-
циирует.
С солями карбамид образует комплексные соединения. Большой
интерес представляют, в частности, те из них, в которых оба компо-
нента являются удобрениями, например Са(ЫОз)2-4СО(ЫН2)2 и
NH4C1-CO(NH2)213’14.
Рнс. 374. Диаграмма состояния системы кар-
бамид — аммиак — вода (в мол. %) с кривыми
давления пара насыщенных растворов (тонкие
линии).
При взаимодействии карбамида с монокальцийфосфатом обра-
зуются фосфат карбамида и дикальцийфосфат |5>16
Са (Н2РО4)2 • Н2О + СО (NН2)2 = СО (NН2)2 • Н3РО4 + СаНРО4 + Н2О
При нагревании водных растворов карбамида под атмосферным
давлением выше 80° в результате гидратации карбамид переходит
в карбамат аммония:
СО (N Н2)2 + Н2О = N H2COON Н4
Карбамат аммония (соль карбаминовой кислоты NH2COOH)
плавится при 145—150°. В присутствии карбамида, воды и аммо-
нийных солей температура плавления карбамата аммония сни-
жается.
При растворении в воде карбамат аммония частично гидрати-
руется с образованием карбоната
NH2COONH4+H2O (NH4)2COs
Применение и агрохимические свойства
1285
который в последующем превращается в бикарбонат аммония, рас-
падающийся в свою очередь на аммиак и двуокись углерода:
(NH4)2 СОз = NH4HCO3 + NH3
NH4HCO3 = СО2 + Н2О + NH3
В разбавленных растворах карбамат аммония почти полностью
переходит в карбонат. Степень гидролиза карбамата аммония зна-
чительно понижается в присутствии ам-
миака.
На рис. 375 показана, растворимость
в системе NH2COONH4—Н2О9’17’18. Ни-
же 60° растворы карбамата аммония со-
держат примеси других аммонийных со-
лей. От —13 до +5° карбамат аммония
находится в растворе совместно с карбо-
натом, а от +5 до +60° с комплексной
солью (МН4)2СОз-2МН4НСОз — сескви-
карбонатом аммония.
В интервале температур от 0 до 118°
растворимость карбамата аммония в
жидком аммиаке незначительна. В при-
сутствии карбамида его растворимость в
аммиаке повышается.
ПРИМЕНЕНИЕ И АГРОХИМИЧЕСКИЕ
СВОЙСТВА
Карбамид широко применяют как в
сельском хозяйстве, так и в промышлен-
ности. Он является высококонцентриро-
Концентрация карбамата
аммония, бес. %
Рнс. 375. Растворимость в
системе карбамат аммо-
ния — вода.
ванным безбалластным азотным удобрением. По сравнению с дру-
гими азотными удобрениями (кроме NH3) карбамид содержит наи-
большее количество азота. Азот карбамида очень легко усваивается
растениями и по усвояемости равноценен азоту, содержащемуся в
фосфатах аммония 19~26.
По своим физическим свойствам карбамид как удобрение также
имеет преимущества перед аммиачной селитрой. Он не взрыво-
опасен, менее гигроскопичен и не так сильно слеживается. Гигро-
скопическая точка для карбамида при 20° равна 80% (для NH4NO3
Ь7%) 27-31.
Давление водяного пара при 20° над насыщенными растворами
карбамида и NH4NO3 равно соответственно 14,05 и 11,74 мм рт. ст.32.
Попадая в почву, карбамид под действием влаги очень быстро
Превращается в карбонат аммония, который далее нитрифици-
руется. Поэтому на кислых почвах карбамид вначале оказывает
нейтрализующее действие, а затем начинает действовать анало-
ично аммиачной селитре, т. е. подкисляет почву. При внекорневой
1286
Гл. XXXIV. Карбамид
подкормке растений карбамид безопасен, так как не вызывает
ожогов листьев, если в нем содержится не больше 0,25% биурета33.
В последнее время карбамид начали широко употреблять в
виде водных растворов с добавлением нитрата аммония, аммиака
и других азотсодержащих соединений 34-36. Например, в США почти
половина карбамида, расходуемого в качестве удобрения, исполь-
зуется в виде растворов. Примерный состав удобрительных рас-
творов (в вес. %):
I и I II
Свободный аммиак . . 26 36,8 Карбамид............. 12 32,5
Нитрат аммония ... 50 — Вода 12 30,7
Содержание азота в таких растворах составляет 44—45,5% 37.
Представляют интерес карбамидно-формальдегидные удобре-
ния (КФУ), получаемые конденсацией карбамида с формальдеги-
дом. КФУ относятся к концентрированным удобрениям. Содержа-
ние азота в них доходит до 40%. Азот в КФУ находится в трудно-
растворимой форме, поэтому этот вид удобрения характеризуется
медленной отдачей азота растениям в течение длительного вре-
мени. В настоящее время в ряде стран, также и в СССР, создают-
ся промышленные установки по производству КФУ 38-55-
В животноводстве карбамид применяют для откормки скота —
в качестве добавки к корму, содержащему мало белков и много
углеводов. В сычуге жвачных животных микроорганизмы превра-
щают амидный азот карбамида в белковые соединения 56-6°.
В промышленности карбамид применяют для приготовления
карбамидно-формальдегидных смол, пластических масс, клеев, фар-
мацевтических препаратов, для депарафинизации смазочных масел,
для полимеров, перерабатываемых в волокна (японская полимоче-
вина Урилон), в текстильной и бумажной промышленности, в про-
изводстве красителей и моющих средств. Его применяют также в
деревообрабатывающей и кожевенной промышленности61-65.
Согласно ГОСТ 2081—63, карбамид для промышленного приме-
нения должен содержать не менее 46,3% азота (в сухом веществе).
Его выпускают в мелкокристаллическом виде двух марок — А и
Б — с содержанием, соответственно, не более: 0,2 и 0,8% биурета,
0,005 и 0,015% свободного аммиака, 0,003 и 0,02% SO4, 0,02% не
растворимых в воде веществ, 0,2 и 1,0% влаги; для марки Б -- не
более 0,005% железа в пересчете на Fe2O3.
Сельскому хозяйству поставляют гранулированный карбамид с
содержанием не менее 46,0% азота (в сухом веществе) и не бо-
лее 1,0% биурета. Его выпускают двух фракций с размером гра-
нул: 1 — не менее 90% от 0,2 до 1,0 мм и 2 — не менее 90% от 1,0
до 2,5 мм. Во фракции 1 гранул с размером более 3 мм допускает-
ся не более 5%; гранулы этой фракции для уменьшения слеживае-
мости могут быть покрыты жиром животного происхождения
(0,05% по весу) или другими добавками.
Методы производства
1287
Карбамид упаковывают в бумажные шестислойные мешки и
в деревянные сухотарные бочки, выложенные парафинированной
или оберточной бумагой. Изучается возможность перевозки карб-
амида навалом 66 и хранения в силосах большой емкости (30 000 г)
с кондиционированием воздуха 67.
Производство карбамида непрерывно увеличивается как
в СССР, так и за рубежом. В 1954 г. мировая мощность установок
карбамида составляла 317 тыс. т, в 1969 г. она превысила 13 млн. т.
Одновременно растет производительность промышленного агрегата,
которая увеличилась с 45—135 (1950 г.) до 450 т/сутки 38’ 68~102.
МЕТОДЫ ПРОИЗВОДСТВА
Разработано несколько способов производства карбамида.
Карбамид может быть получен из аммиака и циановой кислоты
CONH + NH3=CO(NH2)2
и взаимодействием фосгена и аммиака, с одновременным получе-
нием хлорида аммония:
СОС12 + 2NH3 = CO(NH2)2 + 2НС1
HC1+NH3=NH4C1
При взаимодействии аммиака с сероокисью углерода обра-
зуется тиокарбамат аммония, который при 100°, разлагаясь, обра-
зует карбамид и сероводород:
2NH3 + COS = NH2COSNH4
NH2COSNH4 = CO (NH2)2 + H2S
Цианамидный способ получения карбамида основан на разло-
жении цианамида кальция двуокисью углерода с образованием
цианамида:
CaCN2 + Н2О + СО2 = CNNH2 + СаСО3
В кислой среде при легком нагревании цианамид присоединяет
воду, образуя карбамид:
CNNH2 + H2O = CO(NH2)2
Однако эти способы не нашли промышленного применения
вследствие своей нерентабельности и трудности осуществления.
В последнее время изучался способ производства карбамида
из окиси углерода, серы и аммиака 891 103’112. Разработан способ по-
лучения карбамида обработкой газообразным аммиаком газов пи-
ролиза кислого гудрона, содержащих окись углерода и серу 113.
Проводились также исследования фотохимических реакций взаимо-
действия окиси углерода с аммиаком 114. Рассматривается вопрос
получения синтез-газа для производства аммиака с промежуточ-
ным получением карбамида 113
1288
Гл. XXXIV. Карбамид
В настоящее время карбамид в промышленном масштабе произ-
водят только прямым синтезом из аммиака и двуокиси углерода.
(Этот синтез впервые был осуществлен Базаровым в 1870 г. И6)
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ СИНТЕЗА КАРБАМИДА
ИЗ АММИАКА И ДВУОКИСИ УГЛЕРОДА
Синтез карбамида из аммиака и двуокиси углерода протекает
в две стадии. В первой стадии происходит непосредственное соеди-
нение реагентов с образованием карбамата аммония:
2NH3 + CO2 NH2COONH4 + 38,0 ккал
Во второй стадии карбамат аммония отщепляет воду и превра-
щается в карбамид:
NH2COONH4 ^z± NH2CONH2 + H2O-6,8 ккал
Процесс протекает с образованием двух фаз: газообразной
(NHs, СОг, Н2О) и жидкой, состоящей из расплавленных солей
(карбамата аммония, карбамида, аммонийных солей) и воды.
Рис. 376. Диаграмма состоянии системы карбамид—
карбамат аммонии—аммиак (в вес. %).
Установлено 117-12°, что карбамид образуется только в жидкой
фазе, т. е. когда карбамат аммония находится в расплавленном
состоянии. При нагревании твердого карбамата аммония образова-
ние карбамида идет очень медленно.
Физико-химические основы синтеза карбамида из NH3 и С02
1289
На рис. 376 приведена диаграмма растворимости системы кар-
бамид—карбамат аммония—аммиак. На ней ограничены три поля
кристаллизации — CO(NH2)2, CO(NH2)2-NH3 и NH2CO2NH4, и об-
ласть расслоения, в которой система состоит из двух трехкомпо-
нентных жидкостей; точки составов этих жидкостей находятся на
пересечении соответствующей изотермы с кривой, ограничивающей
область расслоения.
Карбамат аммония обладает высоким давлением паров. Из-за
этого синтез карбамида ведут под высоким давлением (200—300 ат).
Рис. 377. Равновесное дав-
ление паров над плавом
карбамида, получаемым при
его синтезе при молярном
соотношении NH3: СО2 = 2.
Плотность загрузки в г/см$:
/-0,93-1,1; 2-0,676; 3-0,5;
4-0,3.
Концентрация СО(ПНг)г,бес.%
Рис. 378. Диаграмма плавкости
системы карбамат аммония —кар-
бамид.
На рис. 377 показано изменение давления паров над плавом карба-
мида в зависимости от температуры синтеза 12|> 122. Это давление
зависит не только от температуры, но и от состава системы, в част-
ности от соотношения компонентов NH3: СО2 в исходной смеси.
„° резко увеличивается при возрастании содержания СО2 в исход-
ной смеси сверх стехиометрического соотношения. Избыток же NH3
не приводит к такому сильному возрастанию давления. При из-
ытке аммиака давление понижается при добавлении к системе
ОДЫ. Помимо температуры и состава системы на величину рав-
весного давления влияет степень заполнения реакционного аппа-
та жидкой фазой (плотность загрузки) —чем она больше, тем
больше давление 123-125.
бли К0Р°СТь образования карбамата аммония увеличивается при-
тельно пропорционально квадрату давления. При атмосферном
1290
Гл. XXXIV. Карбамид
давлении и при невысокой температуре образование карбамата
аммония протекает очень медленно, а при 100 ат и 150° проходит
практически мгновенно 126.
Температура плавления карбамата аммония находится в пре-
делах 145—150°, следовательно, процесс образования карбамида
должен был бы протекать при температурах выше 150°. Однако он
идет и ниже этой температуры, так как по мере нагревания карба-
мат аммония отщепляет воду, которая снижает его температуру
Рис. 379. Зависимость выхода карбамида от
продолжительности дегидратации карбамата
аммония при разных температурах (в долях от
теоретического).
плавления. Так, при содержании в расплаве 9,2% воды темпера-
тура плавления NH2COOiNH4 снижается до 140°, при 14,7% воды —
до 130°. При этом увеличивается объем жидкой фазы.
Под действием воды часть карбамата переходит в карбонат
аммония и затем в бикарбонат, которые в свою очередь также по-
нижают температуру плавления карбамата, что вызывает увеличе-
ние скорости реакции образования карбамида.
На рис. 378 видно, насколько сильно понижается температура
плавления NH2COONH4 в присутствии CO(NH2)2- Эвтектическая
смесь, содержащая 51% NH2COONH4 и 49% CO(NH2)2, плавится
при 98°.
Процесс образования карбамида в зависимости от продолжи-
тельности нагревания карбамата аммония при разных температу-
рах показан на рис. 379 1191 126-129. Скорость образования и выход
карбамида тем больше, чем выше температура. Выше 180° кривые
выходов проходят через максимум — при дальнейшем увеличении
продолжительности нагревания реакционной смеси степень превра-
щения карбамата в карбамид уменьшается. Это объясняется уси-
лением роли побочных реакций. Кроме того, выше 200° резко усй-
Физико-химические основы синтеза карбамида из NHa и COs
1291
ливается коррозия аппаратуры реакционной смесью. Поэтому син-
тез карбамида ведут при температурах не выше 200°.
Образующаяся вода в начале процесса увеличивает степень-
конверсии карбамата аммония в карбамид, так как способствует
появлению жидкой фазы. По мере накопления воды реакция за-
медляется вследствие сдвига равновесия. По этой причине выход
карбамида, достигнув известного предела, далее не увеличивается
(рис. 379). На рис. 380 показано уменьшение выхода карбамида
при добавке воды к карбамату аммония 121.
Число молей боды на 1 моль
карбамата аммония
Рис. 380. Влияние добавки
воды к карбамату аммония на
равновесный выход карбамида
(по СО2) при 160°.
Давление, ат
Рис. 381. Влияние давле-
ния на степень конверсии
карбамата аммония в карб-
амид.
Избыточное количество СО2 не оказывает влияния на выход,
карбамида 130, но концентрация ее оказывает значительное влия-
ние 131. Чем выше концентрация СО2 в исходном газе, тем выше
степень конверсии. Обычно источником СО2 служит экспанзерный
газ — отход от производства аммиака. Содержащиеся в углекис-
лом газе примеси (Н2, СО, N2, О2 и другие) уменьшают парциаль-
ное давление аммиака и, следовательно, его растворимость в жид-
кой фазе. Так, например, если при начальном содержании СО2:
в исходном газе 98—99% степень конверсии составляет 65—66%,
при содержании СО2 85—86% степень конверсии, при прочих рав-
ных условиях, снижается до 45%. Рост степени конверсии с повы-
шением общего давления в системе синтеза показан на рис. 381.
Гак как карбамид образуется только в жидкой фазе, то чем
больше степень заполнения ею аппарата (плотность загрузки), тем
больше в нем жидкой фазы и больше равновесное давление над
ней газовой фазы (см. выше) — это увеличивает выход карба-
1292
Гл. XXXIV. Карбамид
Наибольшее влияние на увеличение выхода карбамида оказы-
вает избыток аммиака в исходной смеси против стехиометриче-
Рис. 382. Влияние из-
бытка аммиака на равно-
весный выход карбамида
(по средним данным для
185—195°, 185-320 ат и
плотности загрузки
0,7 г!см3).
ского количества 9- 119’ 132-137, определяемого
молярным соотношением NH3 : СО2 = 2
(рис. 382). Избыточный аммиак уменьшает
вредное действие выделившейся в процессе
воды — он связывает воду и тем самым
сдвигает равновесие реакции превращения
карбамата аммония в карбамид в сторону
последнего. По этой . же причине замед-
ляется гидролиз карбамата аммония с об-
разованием побочных продуктов. Кроме
того, избыточный аммиак уменьшает кор-
розию аппаратуры.
Для определения равновесного выхода
карбамида может быть использована номо-
грамма Фрежака (рис. 383). Например,
при 180°, отношении NH3: СО2=5,3 и до-
бавке 1,6 моль воды на 1 моль СО2 равно-
весный выход карбамида по СО2 будет 0,58;
при тех же условиях, но без добавки воды
он равен 0,8.
При выборе условий, обеспечивающих высокий выход карба-
мида, следует учитывать, что избыточный аммиак или должен быть
Рис. 383. Номограмма для определения равновесного вы-
хода карбамида (по СО2).
возвращен в цикл или переработан в другие продукты, например
в аммиачную селитру. Рециркуляция аммиака связана с возраста-
Способы производства карбамида из NH3 и СОг
1293
нием капитальных и эксплуатационных расходов, приводящим
к удорожанию карбамида. При переработке избыточного аммиака
в аммиачную селитру количество получаемой аммиачной селитры
превышает количество вырабатываемого карбамида — оно изме-
няется в зависимости от величины избытка аммиака и метода ди-
стилляции плава карбамида (одно- или двухступенчатая дистил-
ляция).
СПОСОБЫ ПРОИЗВОДСТВА КАРБАМИДА ИЗ АММИАКА
И ДВУОКИСИ УГЛЕРОДА ПРЯМЫМ СИНТЕЗОМ
Прямой синтез карбамида из аммиака и двуокиси углерода со-
стоит из нескольких стадий: взаимодействия NH3 и СО2 (синтез),
дистилляции продуктов синтеза и переработки растворов карб-
амида в готовый продукт.
Применяемые способы производства карбамида мало отли-
чаются условиями процесса синтеза и подразделяются главным об-
разом по методам использования газов дистилляции. При осуще-
ствлении процесса без возврата избыточного аммиака из газов
дистилляции (без рециркуляции), его перерабатывают в другие
продукты: нитрат аммония, сульфат аммония, аммонийные соли
или в аммиачную воду.
В способах с рециркуляцией аммиака, не превращенного в карб-
амид, его выделяют из газов дистилляции плава и возвращают
в цикл. Дистилляцию плава карбамида производят в одну или
в две ступени.
При одноступенчатой дистилляции плав из колонны синтеза
дросселируется до давления 1,2 ат и подвергается, дистилляции
при 70°. Отогнанный аммиак и продукты разложения аммонийных
солей направляются на переработку в аммиачную селитру или
другие продукты. Раствор карбамида выпаривают и кристалли-
зуют 138-140.
Расходные коэффициенты на 1 т карбамида составляют:
При производстве
гранулированного
карбамида
с двухступенчатой
дистилляцией плава
При производстве
кристаллического
карбамида
с одноступенчатой
дистилляцией плава
Аммиак (100%), т.................. .
Двуокись углерода, т..................
Пар, т................................
Вода, м3 .............................
Электроэнергия, кет • ч...............
Отход
Аммиак на производство аммиачной се-
Литры, т . .
1,54
1,2
3,0
130
400
2,30
1,2
3,6
136
513
0,94
1,67
В табл. 92 приводятся данные о количестве получаемой ам-
миачной селитры из аммиака, отходящего от синтеза карбамида.
1294
Гл. XXXIV. Карбамид
ТАБЛИЦА 92
Количество аммиачной селитры, получаемой в зависимости от избытка
аммиака при синтезе карбамида и схемы дистилляции
Избыток NHs сверх стехио- метрического. Степень конверсии карбамата аммония Одноступенчатая дистилляция плава карбамида Двухступенчатая дистилляция плава карбамида
получается степень отгоики аммиака, “/о получается
% в карбамид, % аммиачной селитры, т/т карбамида аммиачной селитры, т/т карбамида
0 53 2,35
50 65 3,5 16,0 3,2
100 70,3 4,9 55,0 2,85
150 72,5 6,54 68,5 2,75
200 74,0 8,2 76,0 2,67
300 76,0 11,4 84,0 2,58 ш
При двухступенчатой дистилляции плава карбамида большая
масть аммиака может быть возвращена в цикл. С увеличением
избытка аммиака при синтезе карбамида возрастает доля отгоняе-
мого аммиака в 1-й ступени дистилляции, и количество аммиака,
перерабатываемого в аммиачную селитру, уменьшается. Это позво-
ляет применить в процессе синтеза больший избыток аммиака, чем
в процессе с одноступенчатой дистилляцией плава.
Способ производства карбамида с полным возвратом в цикл
газов дистилляции является наиболее совершенным. Особенное
значение этот способ имеет, когда необходимо перерабатывать весь
аммиак в карбамид без выпуска других азотсодержащих продук-
тов 1, 61, 103, 111, 141-178
Способы регенерации газов дистилляции и режимы
синтеза карбамида
Существует несколько способов регенерации газов дистилля-
ции с целью их рециркуляции: 1) горячее компримирование, 2) по-
глощение газов минеральным маслом, 3) избирательная абсорбция
.аммиака и двуокиси углерода (газовый рецикл) ,21>|38*140.179-18»,
4) возврат в цикл NH3 и СО2 в виде водных растворов аммонийных
солей (жидкостный рецикл). По способу горячего компримирова-
ния190’191 смесь NH3 и СО2 сжимается при 175—210°, т. е. в усло-
виях, исключающих образование твердого карбамата аммония 192.
Этот метод широкого применения в промышленности не получил
из-за трудностей, возникших при компрессии смеси газов при вы-
соких температурах и малой экономичности 193-195, несМ0тря на вы-
сокое использование аммиака (до 93%).
Способы производства карбамида из NH3 и С02
1295
фирма «Chemico» США предлагает рециркуляцию NH3 и СО2
осуществлять в горячем состоянии, разлагая карбамат аммония по
стадиям: при 70, 14—28 и 1 ат. Выделившиеся газы дожимаются
до давления 210 ат, при котором осуществляется синтез. Подогрев
газов происходит за счет адиабатической компрессии. Избыток
тепла в процессе синтеза используется для получения пара 196>197.
При поглощении смеси NH3 и СО2 инертным минеральным
маслом (способ Пешине) 186- 198-201 образуется суспензия карбамата
аммония, которая поступает в колонну синтеза. Процесс синтеза
проводится при 210 ат и 180°. Степень превращения NH3 и СОг
в карбамид составляет 40—50%. По этому способу в 1955 г.
в США пущены две крупные установки 202’203. Он характеризуется
небольшими эксплуатационными расходами и меньшей коррозией
аппаратуры 204-208. в дальнейшем фирмой Пешине совместно с фир-
мой Грейс введены усовершенствования, которые значительно улуч-
шили технологические и экономические показатели метода. Кроме
того, усовершенствованы конструкции аппаратов, применены но-
вые конструкционные материалы, что позволило значительно удли-
нить срок их работы 209.
Предложено много способов, основанных на избирательной аб-
сорбции одного из компонентов — аммиака или СО2. Например,
избирательная абсорбция аммиака из газов дистилляции раство-
ром нитрата карбамида (способ «Инвента») 188’ 189’ 210-212_ Непогло-
щенную двуокись углерода или выбрасывают в атмосферу или по-
вторно используют в цикле 213-217 ДруГИМ примером является изби-
рательная абсорбция из газов дистилляции двуокиси углерода
(способ «Хемико») 199-200’ 218-221. По этому способу синтез прово-
дится при 170 ат и 175—185° в присутствии большого избытка ам->
миака (NH3: СО2 = 6 : 1), что позволяет повысить степень превра-
щения карбамата аммония в карбамид до 76%. Поглощение СО2
из газов дистилляции производится с помощью моноэтанол-
амина 222_
В настоящее время для этого способа предложен и несколько
иной технологический режим. Синтез проводится под давлением
280 ат и при температуре 205—232°, отношение NH3 : СО2 =
= 4—4,5: 1. Выход карбамида при этом составляет 80—85%. Для
большей устойчивости колонна синтеза футеруется цирконием.
Избыток тепла используется для получения пара. Процесс дистил-
ляции ведется в две ступени. Поглощение СО2 из газов дистилля-
ции производится селективно моноэтаноламином В * 10 *°.
Разработан и смешанный способ возврата газов в колонну син-
теза 12°. 223,224 — 750^ непрореагировавших NH3 и СО2 возвращают
9чо/ИКЛ в Виде водного РаствоРа аммонийных солей, а остальные
б/о с помощью избирательной абсорбции. При избирательной
сорбции СО2 водным раствором моноэтаноламина 225-229 процесс
°жет быть осуществлен с частичным или полным возвратом
1296
Гл. XXXIV. Карбамид
в цикл газов дистилляции. Разделение газов с полным возвратом
их в процесс может быть также осуществлено с применением двух
поглотителей — водного раствора моноэтаноламина для абсорбции
СО2 и раствора моноаммонийфосфата для поглощения аммиака 230.
Последний предложено также поглощать водным раствором поли-
оксисоединений (например, пентаэритрита или полигликоля) и бор-
ной кислоты231.
Способы, по которым возврат в цикл NH3 и СО2 осуществляется
в виде водного раствора аммонийных солей (жидкостный рецикл),
являются наиболее перспективными вследствие своей экономично-
сти. В последнее время многие фирмы ведут работу по усовершен-
ствованию этих способов 232-238.
По способу «Монтекатини» 239> 240 синтез карбамида проводят
в колонне футерованной нержавеющей сталью при давлении 190 ат,
температуре 175—180° и молярном соотношении NH3: СО2 = 5:1.
Для регулирования температурного режима колонна снабжена
охлаждающим кожухом. Синтез проводится в тщательно контро-
лируемых условиях и выход карбамида достигает 68%. Дистил-
ляция плава производится в две ступени под давлением 20 ат и
близком к атмосферному. Непрореагировавшие аммиак и двуокись
углерода полностью возвращаются в цикл в виде раствора аммо-
нийных солей. Упарку раствора карбамида производят под ва-
куумом до содержания влаги 0,6%. Готовый продукт получается
в виде гранул высокой чистоты и с малым содержанием биурета.
Полученный продукт годен для технических целей и для скармли-
вания животным.
По способу «Тоё-Коацу» (Toyo Koatsu, Япония)241-246 взаимо-
действие NH3 и СО2 проводится при несколько иных технологиче-
ских параметрах: давлении 220—230 ат, температуре 180—190° и
молярном отношении NH3 : СО2 = 3,5—4,5:1. При этом степень
конверсии СО2 в карбамид составляет ~58%. Плав подвергается
двухступенчатой дистилляции при 17 и 2—3 ат. В отличие от спо-
соба «Монтекатини» в данном способе часть аммиака, выделив-
шаяся в первой ступени дистилляции в виде чистой фракции жид-
кого аммиака, возвращается в цикл. Другая часть аммиака и СО2
возвращается в виде раствора аммонийных солей. Готовый про-
дукт выпускается как в мелкокристаллическом виде, так и в грану-
лированном 247-251. По способу «Стамикарбон» 2S2~257 рецикл непро-
реагировавшего NH3 и СО2 осуществляется аналогично способу
Тоё-Коацу. Синтез проводится при 175—190°, давлении 200 ат и
молярном отношении NH3 : СО2 = 4,5 : 1. Степень конверсии СО2
в карбамид составляет 62%. Дистилляцию плава ведут в две сту-
пени при давлениях соответственно 18 и 3 ат. Перед грануляцией
раствор упаривается до концентрации 99,8% CO(NH2)2 в двухсту-
пенчатой выпарной установке. Процесс выпарки идет под разре-
жением (в I ступени остаточное давление 300, во II ступени
Способы производства карбамида из NH3 и С02
1297
15 мм рт. ст-)- Технический карбамид получается непосредственно
из 70—74% раствора, предварительно обработанного, с целью
улучшения его качества, активированным углем 258’259.
В настоящее время фирма «Стамикарбон» ввела ряд усовер-
шенствований в свой способ, что позволило снизить расходные
коэффициенты и стоимость готового продукта. По новому способу
синтез карбамида проводят при давлении 130 ат, температуре 180°
и соотношении NH3 : СО2 = 4:1 (т. е. меньше, чем в старом спо-
собе). Плав из колонны синтеза под тем же давлением направ-
ляется в отдувочную колонну для разложения карбамата аммония.
Избыточный аммиак и продукты разложения карбамата аммония,
газообразные NH3 и СО2, отдуваются из плава с помощью свежей
двуокиси углерода, подаваемой в колонну компрессором, и возвра-
щаются в колонну синтеза. Далее следует обычная двухступенча-
тая дистилляция под давлением. Излишек трепла, образующийся
при синтезе, используется для производства пара 260-262.
Энергетические затраты при полной рециркуляции на 1 т гра-
нулированного карбамида составляют:
Пар, т ................
Вода, м3 (25°).........
Электроэнергия, кет ч
Отход пара (3,5 ат), т
Старый способ
(12 ат)
1,5
100
150
Новый способ
(25 ат)
0,95
55
120
0,25
Итальянская фирма SNAM (Societa Nationale Metanodotti),
в отличие от фирмы «Стамикарбон», для продувки плава приме-
нила газообразный аммиак: продувка ведется в одну ступень под
высоким давлением. Степень разложения карбамата составляет
98% 263-267. фирмой Тоё-Коацу предлагается комбинированный про-
цесс производства аммиака и карбамида, снижающий на 6—7%
эксплуатационные расходы и на 5—10% капиталовложения. Сущ-
ность способа заключается в переработке в карбамид двуокиси
углерода из газа, полученного конверсией метана и СО 268-27°.
Фирма «Инвента» предлагает способ с одноступенчатым разло-
жением карбамата аммония, проводимым при 130° и давлении
4 ат. При этом рециркуляция NH3 и СО2 достигает 99% 234.
Производство карбамида по схеме с жидкостным рециклом 271-274
На рис. 384 показана схема синтеза карбамида с дистилляцией
плава в две ступени с жидкостным рециклом. Газообразная дву-
окись углерода, предварительно очищенная от сероводорода и
органической серы, .освобожденная от механических загрязнений
и в которую добавлен кислород 275-279, сжимается в четырехступен-
а>ом компрессоре 280 от 1 до 200 ат и при 35—40° направляется
| смеситель 4. Жидкий аммиак в смеситель 4 подается плунжер-
пи насосом 3 также под давлением 200 ат. Туда же плунжерным
s
о
s
Способы производства карбамида из NH3 и С02
1299
насосом 14 под давлением 200 ат подаются в виде раствора аммо-
нийных солей не превращенные в карбамид NH3 и СО2. В смесите-
ле происходит перемешивание всех трех компонентов с одновре-
менным образованием карбамата аммония. Температура в смесителе
175°, давление 200 ат. Из смесителя реакционная смесь поступает
в колонну синтеза 5 281, в которой при давлении 200 ат и темпера-
туре 185° происходит образование карбамида 282’283. При времени
пребывания реакционной смеси в колонне синтеза 40—45 мин и
молярном отношении NH3 : СО2 : Н2О — 4,5 : 1 : 0,5 степень конвер-
сии СО2 в карбамид составляет около 62% 284. Образовавшийся в
колонне синтеза плав, содержащий 30—31% карбамида, 21—22%
карбамата аммония, 33—34% избыточного аммиака и 16—17%
воды, направляется на двухступенчатую дистилляцию. Агрегат ди-
стилляции для первой и второй ступеней состоит из трех аппара-
тов: ректификационной колонны 7, 10, подогревателя 8, 11 и сепа-
ратора 9, 12 285.
Плав карбамида, выходящий из верхней части колонны 5, дрос-
селируется от 200 до 18—20 ат и поступает в ректификационную
колонну агрегата дистилляции первой ступени 7 286. В ректифика-
ционной колонне происходит выделение в газовую фазу избыточ-
ного аммиака. Плав из колонны 7 поступает в подогреватель 8,
в котором его температура повышается до 163—165°. При этом из
него почти полностью выделяется избыточный аммиак и разла-
гается большая часть карбамата аммония. Образовавшаяся паро-
жидкостная смесь из подогревателя 8 направляется в сепаратор 9
для разделения. Газовая фаза из сепаратора возвращается в рек-
тификационную колонну 7, а жидкая фаза дросселируется до дав-
ления 3 ат и направляется на дистилляцию второй ступени.
Газовая фаза из ректификационной колонны 7, содержащая
75—76% аммиака, 21—22% двуокиси углерода и около 3% воды,
направляется в промывную колонну 6. Здесь происходит поглоще-
ние водой NH3 и СО2 с образованием аммонийных солей и очистка
газообразного NH3 от СО2. Температура в колонне поддержи-
вается около 100° в нижней части и 45—50° в верхней части. Для
регулирования температуры в нижнюю часть колонны подается
вар, а верхняя часть орошается жидким аммиаком. Газообразная
Фаза из промывной колонны, состоящая из чистого 121 аммиака
с нримесью небольшого количества инертных газов, направляется
в конденсатор первой ступени, где аммиак конденсируется и воз-
вращается через танк 2 в цикл.
ТКидкая фаза, представляющая собой концентрированный рас-
™°Р аммонийных солей, с температурой 100° поступает в плун-
ТееРннасос 14, которым под давлением 20Q ат подается в смеси-
содЬ 4' Раствор, поступающий на дистилляцию второй ступени,
Изб ЖИТ 60—61% карбамида, 4—5% карбамата аммония, 6—7%
ыточного аммиака и 29—30% воды. Дистилляция во второй
1300
Гл. XXXIV. Карбамид
ступени протекает так же, как и в первой, т. е. вначале раствор про-
ходит через ректификационную колонну 10, затем подогреватель 11,
в котором подогревается до 143—145°, и поступает в сепаратор 12.
где происходит разделение газообразной и жидкой фаз. Во второй
ступени дистилляции происходит окончательное разложение кар-
бамата аммония и отгонка аммиака и двуокиси углерода из рас-
твора. Раствор карбамида с концентрацией 70—72% из сепара-
тора 12 дросселируется и поступает в вакуум-испаритель 15, в ко-
тором при остаточном давлении 300 мм рт. ст. происходит его
концентрирование до 74—76% за счет самоиспарения. Далее этот
раствор через сборник 16 и маслоотделитель./? направляется на
переработку в готовый продукт. По схеме с замкнутым жидкостным
рециклом газовая фаза из ректификационной колонны 10, содержа-
щая 55—56% аммиака, 24—25% двуокиси углерода и 20—21%
водяных паров, направляется в конденсатор второй ступени 21 для
конденсации водяных паров. Образовавшийся в конденсаторе 21
слабый раствор аммонийных солей через сборник 22 насосом 23
подается в промывную колонну 6. Газовая фаза из конденса-
тора 21 и другие отходящие газы, содержащие NH3 и СО2, направ-
ляются в абсорбер 24, в котором NH3 и СО2 при 40° поглощаются
раствором аммонийных солей, циркулирующим через холодиль-
ник 27. Инертные газы из абсорбера выбрасываются в атмосферу.
Образовавшийся в абсорбере 24 раствор аммонийных солей подо-
гревается в теплообменнике 30 до 90—95° и подается в десор-
бер 26, в котором при 3 ат и 130—135° с помощью острого пара
происходит полное разложение аммонийных солей на NH3 и СО2.
Газообразные NH3 и СО2 вместе с водяными парами направляются
в конденсатор второй ступени 21, а оставшаяся вода удаляется
в .канализацию 287-289.
Переработка растворов карбамида в готовый продукт
Схема переработки растворов карбамида в готовый продукт
изображена на рис. 385. Раствор карбамида с дистилляции второй
ступени с концентрацией 74—76% поступает в сборник 1, из кото-
рого насосом 2 через напорный бак 3 подается в выпарной аппа-
рат 4.
Выпаривание растворов карбамида следует вести при условиях,
исключающих образование биурета 194’290-301. Обычно выпаривание
раствора карбамида ведут в выпарных аппаратах пленочного ти-
па под разрежением 550—600 мм рт. ст. зо2-зо4 Разрежение в выпар-
ном аппарате поддерживается конденсатором 5 и вакуум-насосом.
Для получения гранулированного продукта упаривание ведут до
концентрации плава 98—99% и выше. Плав из выпарки поступает
jb сборник 6, из которого насосом 7 через напорный бак 8 подается
в разбрызгиватель 9. Плав с помощью разбрызгивателя равно-
Способы производства карбамида из NH3 и СОг
1301
мерно распределяется по сечению грануляционной башни 10. Капли
плава, падая вниз, охлаждаются встречным потоком холодного
воздуха и затвердевают в гранулы 305-313 для получения продукта
с размером гранул 1—3 мм он подвергается сортировке на двух-
ситном грохоте 14\ частицы размером меньше 1 мм и больше 3 мм
собираются в баке 15, растворяются в воде и возвращаются на вы-
парку.
Гранулирование карбамида осуществляют также на наклонном
вращающемся грануляторе тарельчатого типа (фирма TVA). Мел-
Рис. 385. Схема получения гранулированного карбамида:
J — сбориик раствора карбамида; 2 — центробежный насос; 5 —напорный бак; 4 — выпарной
аппарат; 5 —конденсатор; £ —сборник для плава карбамида; 7 — центробежный насос для
плава; 8 — напорный бак для плава; 9 — разбрызгиватель плава; Ю — грануляционная башня;
Н — центробежный вентилятор; 12 — ленточный конвейер; 13 — элеватор; 14 — грохот; 15— бак
для растворения пыли и крупных гранул; 16 — ленточный конвейер.
кие гранулы ретура, находящиеся на тарелке, опрыскивают
99,5—99,8 %-ным плавом карбамида. Полученные гранулы охлаж-
дают и рассеивают 71’312.
Для получения карбамида в кристаллическом виде раствор упа-
ривают до концентрации 92—94%. Кристаллический карбамид по-
лучают кристаллизацией упаренного раствора в шнеках-кристалли-
заторах или в вакуум-кристаллизаторах при охлаждении раствора
до 40—45°. Отделение кристаллов от маточного раствора ведут на
Центрифугах с одновременной подсушкой воздухом. Так как кри-
сталлический карбамид главным образом используется для техни-
ческих целей, то его раствор предварительно подвергают очистке
активированным углем 197 •314-322.
Для получения карбамида с хорошими физическими свойствами
(неслеживающийся продукт) его сушат 323> 324, охлаждают 324>325 и
Црипудривают 326> 327.
1302
Гл. XXXIV. Карбамид
Карбамид, предназначенный для получения жидких удобрений,(
можно кристаллизовать на ленточных транспортерах, охлаждае-i
мых снизу водой 319. I
Проводились исследования использования газов дистилляции1
первой ступени с температурой 100—125° для сушки и грануляции
карбамида в псевдоожиженном слое. Получены положительные
результаты 328>329.
Запатентован 330 способ получения плава карбамида с содер-|
жанием воды 0,25—0,75% противоточным выпариванием в атмо-1
сфере инертного газа или воздуха, нагретого до 132—150°. В этом!
случае выпаривание ведут в две или три ступени. j
ЛИТЕРАТУРА I
1. А. Т. Зотов, Мочевина, Госхимиздат, 1963.— 2. Справочник азотчика,]
т. 2, Изд, «Химия», 1969, стр. 203.—3. Хр. Ал аминов, А. Вълева, С. Ге-1
о р г и е в а, Химия и Индустрия (болт.), 32, № 3, 71 (1960).—4. J. Agr. Food,?
Chem., 7, № 11, 762 (1959); 11, № 1, 39 (1963), —5. Nitrogen, Jan., № 15, 37.1
(1962). — 6. С. Казарновский, H. Малкина, ЖПХ, 3, № 3, 452 (1958).—
7. M. Михайлов, Хр. Аламинов, Хим. пром., № 11, 63 (1961). — 8. Е. L a s-
s a k, J. Proc. Roy. Soc., 100, № 3—4, 179, 1966 (1967). — 9. М. Frejaques,
Chim. et ind., 60, № 1, 22—35 (1948). —10. Б. Розиан, Л. Бородкина,
ЖПХ, 32, № 11, 2585 (1959).
11. S. Wicar, Brit. Chem. Eng., 8, № 12,. 818 (1963). —/2. В. П. Б л и н-
д и н, А. Я- Малахова, С. М. Асланов, ЖПХ, 30, № 4, 646 (1957).—
13. К. С у л а й м а н к у л о в, Труды молодых научных работников АН КиргССР,
Фрунзе, 1958, стр. 81. — 14. Авт. свид. 199851, 1967. —15. С. И. Вольфкович
и др.. Технология азотных удобрений, ОНТИ, 1935. — 16. А. И. Чеховских,
сб. «Исследования по химии и технологии удобрений, пестицидов, солей», Йзд
«Наука», 1966, стр. 112. — 17. Е. J а п е с k е, Z. Е 1 е к t г о с h е т., 35, 716
(1929). — 18. Е. Janecke, ibid., 36, 645 (1930); 38, 9 (1932). — 19. Г. Ш. Асла-
нян, Труды НИУИФ, в. 136, 1937, стр. 165. — 20. В. П. Бельский, Там же,
стр. 174.
21. J. Gasser, Chem. Ind., № 48, 2086 (1962). — 22. J. Y о p e z - F r e i g e г e,
Agriculture, № 367, 672 (1962).— 23. V. M о r a n i, Chim. et Ind., 83, Ns 2, 300
(I960), — 24. W. Temple man, J. Agr. Sci., 57, № 2, 237 (1961). — 25. A. Low,
F. Piper, ibid., 57, Ns 2, 249 (1961). — 26. G. Mees, T. Tomlinson, ibid., 62,
Ns 2, 199 (1964). — 27. Польск. пат. 46094, 1962, — 28. Швейц, пат. 363974, 1962. —
29. Б. Д. Мельник, Хим. пром., N? 5, 321 (1964). — 30. Пат. США 3103536,j
1963. I
31. Пат. США 3123637, 1964. — 32. В. Клевке, Н. Поляков, Л. А р с е-1
н ь е в а, Технология азотных удобрений, Госхимиздат, 1956.—33. J. Agr. Food’
Chem., 6, Ns 2, 87 (1958).—34. H. Tucker, Agr. Chem., 14, 50, 124 (1959). —
35. M. H. Набиев, H. Крылова и др., Узб. хим. ж., Ns 4, 8 (1962).—
36. Я. Шенкин, В. А. Клевке, Б. Людковская, ДАН СССР, 149, № 3,
656 (1953).—37. Информ, бюлл. о зарубеж. хим. пром. (НИИТЭХИМ), Ns.9,
8 (1960). — 38. G. W. W i n s о г, М. J. Long, J. Sci. Agr., 9, Ns 4, 185 (1958). —
39. А. Д. Фридман, В. А. Клевке, ЖПХ, 34, № 10, 2206 (1961).—
40. S. Datta, M. Idnani et al., Indian J. Appl. Chem., 25, Ns 1 (1962).
41 Nitrogen, № Г 43 (1960). — 42. Ibid., Ns 14, 42 (1961)..—43. Agr. Chem.,
18, Ns 1, 19 (1963). — 44. Nitrogen, № 17, 30 (1962). -^45. Chem. Ind., 13, Ns 2,
A93 (1961). — 46. Англ. пат. 869362, 1961. — 47. S. Datta, M. Idnani V. I s w a-
Литература
1303
rau, Indian J. Appl. Chem., 25, № 1, 42 (1962). — 48. Авт. свид. 160189, 1964. —
49. Япон. пат. 5372, 1962. — 50. Ф. В. Тур г ин, В, Соколова, Б, Блюм, Хи-
мия в сельск. хоз., № 2, 6 (1964).
51. А. Д. Фридман, В. А. Клевке и др„ ЖПХ, 38, № 5, 1091 (1965).—
52. Б. Блюм, Труды Н,-и. ин-та по удобрениям и инсектофунгицидам, в. 208,
1915, стр. 145. — 53. Пат. США 3235370, 1966. — 54. Пат. ЧССР 117168, 1966.—
55. Пат. США 3252786, 1966. — 56. С. Е. R е d em an n a. oth., Ind. Eng. Chem.,
50, № 4 (1958). — 57. А. В о s t i с с о, А. В о n о m i, Progr. Agr., 9, № 9, 1047
(1960),—58. Feedstuffs, July, 1960. — 59. H. Wet ter au, Сельск. хоз. за рубе-
жом. Животноводство, № 11, 14 (1960), — 60. Пат. США 3232740, 1966.
61. R. Moore, R. Parks, Chem. Eng. News, 37, № 47, 84 (1959).—
62. Юки Нумата, Юриа Дзюси, Никаи коче симбужа Токио, 1961; Но-
вые книги за рубежом, Сер. Б., № 9, 124 (1962), — 63. Н. S с h 1 i е f, Е. Leib-
nitz, Chem. Techn., 10, № 6, 345 (1958). — 64. J. Teubel, R. Schmiedel,
ibid., 12, № 8, 473 (I960). — 65. Ind. Eng. Chem., 53, № 12, 20A (1961).—
66. Chem. Process (USA), 27, № 16, 115 (1964). — 67. Res. a. Ind., 12, № 2, 87
(1967). — 68. Chem. Eng. News, 36, Ns 15, 72 (1958), — 69. T. Унанянц, Удобр.
и урожай, № 9, 53 (1959); Хим. пром., № 3, 188 (1957). — 70. Т, О t s u k a,
Chem. Ind. (Japan), 10, № 1, 24 (1959).
71. J. S z a f n i c k i, Chernik, 15, № 1, 4 (1962).—72. Chem. Eng., 2, № 5,
421 (1957). — 73. Chem. Age., 33, № 2123, 495 (i960). — 74, H. А. Симулин,
Хим. пром., № 7, 1 (1961). — 75. J. King, Chem. Eng., 66, № 20, 58 (1959).—
76. Информ, бюлл. о зарубеж. хим. пром. (НИИТЭХИМ), № 9, 8 (I960).—
77. Nitrogen, № 23, 1 (1963). — 78. М. D е u t с h, Chim. et ind., 80, № 3 bis,
12 (1958). — 79. J. Agr. Food. Chem., 6, № 2, 87 (1958). — 80. Chem. Eng. News,
36, № 7, 25 (1958).
81. Ibid., 36, № 50, 26 (1958). — 82. M. D e и t c h, Ind. chim. Beige, 24, № 11,
1342 (1959). — 83. Chem. Eng. News, 36, № 15, 72 (1958). — 84. Nitrogen,
№ 4, 40 (1959). —85. Ibid., № 9, 27 (1961). — 86. Chem. Ind., 13, Ns 4, 223
(1961).—87. Nitrogen, Ns 17, 43 (1962).—88. Agricultura (исп.), № 367, 672
(1962), — 89. G. Genin, Ind. chim., 51, № 565, 277 (1964), — 90. Г. Корень-
ков, H. Ромашова, Хим. пром., № 2, 62 (1965).
91. Хосино Тапэо, Пром., наука и техн. Японии, 3, Ns 8, 6 (1964),—
92. Chem. Eng. News, 43, № 35, 32 (1965). — 93. Cook Lucien H., Hydrocar-
bon Process a. Petrol. Ref., 45, Ns 2, 129 (1966). — 94. Inform, chim., Ns 40, 54, 57
(1966). — 95. H. C h о t i n e r, Oleagineux, 21, № 12, 753 (1966). — 96. Nitrogen,
№ 44, 23 (1966). — 97. Chem. Eng., 73, Ns 25, 76 (1966).— 98. Chem, Ind. (Ja-
pan), 16, № 1, 48 (1965). — 99. Итакава Хиромаса, Karaky К о z ё,
Chem. Ind. (Japan), 17, Ns 1, 18 (1966). —100. E. Gucci one, Chem. Factory,
10, Ns 11, 32 (1966).
101. I. Brunborg, Norsk hydro, 27, Ns 1, 26 (1967). — 102. A. G. Soo-
mar, Farm. J., 8, № 8, 27 (1967). — 103. A. Manciulescu, Revista de chim.,
12, № 8, 478 (1961). — 104. Chem. Eng. News, 38, № 51, 51 (I960). — 105. Nitro-
gen, № 9, 29 (1961). — 106. R. Franz, F. A p p 1 e g a t h, J, Org. Chem., 26,
№ 9, 3304 (1961). — 107. Пат. США 2857431, 1958. — 108. Пат. США 2966516,
1960. — 109, Франц, пат. 1366508, 1964. — 110. А. И о в и, Н, С. Торочешни-
ков, М. А. Людковская, В. А. Клевке, Хим. пром., № 11, 20 (1965).
111. Пат. ФРГ 1189981, 1965.— 112. Швед. пат. 213979, 1967, — 113. К- Ива-
нов, Г. Баланчивадзе, Труды Груз, политехи, ин-та, Ns 6, 1957, стр. 104.—
44. Н, Додонова, Вести. ЛГУ. Сер. физ. и хим., в. 3, № 16, 144 (1962).—
45. Япон. пат. 27055, 1965. — 116. А. В a s а г о f f, J. prakt. Chem., 1, 283
(1870). — 117. F. F i c h t e r, B. Becker, Ber., 44, 3473 (1911). — 118. С. M a t i g-
n о п, M. Frejacques, Compt. rend., 171, 1003 (19OQ); Bull. Soc. chim. France,
31, 307, 374 (1922); Ann. chim., 17, 257 (1922), — 119. Б. А. Болотов,
17 M. E. Позин
1304
Гл. XXXIV. Карбамид
А. Н. Попова, ЖХП, № 9, 32 (1934). —120. R. О. D a v i s, С. A. Black, Ind.
Eng. Chem., № 11, 1280 (1931).
121. H. А. Гольдберг, M. А. Л ю д к о в с к а я, X. Ф. Фридман,
В. И. Заграничный, Хим. наука и пром., № 6, 669 (1956). — 122. Е. В г 1-
п е г, J. chim. phys., 4, 266 (1906). — 123. Н. А. Гольдберг, А. Альтшулер,
Хим. пром., № 9, 14 (1962). —124. Г. Ефремова, Р. Королева, Г. Ле-
онтьева, Труды ГИАП. Химия и технология азотной промышленности, в. 12,
1961, стр. 5. — 125. Е. Otsuka, J. Chem. Soc. Japan. Ind. Chem. Sect., 63, № 8,
1344 (1960). —126. M. Frejacques, Chim. et ind., 60, № 1, 22 (1948).—
127. B. Neumann, A. Sonntag, Z. Elektrochem., 37, № 11, 805 (1931).—
128. Б. А. Болотов, A. H. Попова, ЖХП, № 9, 32 (1934). — 129. Б. А. Бо-
лотов, A. H. Попова, Ю. К- Соколова, Там же, № 9, 631 (1937).—
130. Г. А. Я к о в к и н, ЖПХ, 1, 70 (1928).
131. Е. Otsuka, J. Chem. Soc. Japan. Ind. Chem. Sect., 63, № 7, 1208
'(I960). — 132. Б. А. Болотов, A. H. Попова, ЖХП, № 14, 630 (1937).—
j33. Б. А. Болотов, В. P. Леман и др., Там же, 707. — 134. С. К a w a s u-
тп i, Bull. Chem. Soc. Japan, 27, 254 (1954). — 135. F. Fichter, H. Steiger,
5» Stanisch, Verhandll. Naturforschr. Ges. Basel, 28, № 11, 66 (1918).—
136. H. J. К r a s e, V. L. Gaddy, J. Am. Chem. Soc., 52, № 70, 3088 (1930).—
137. Kh. Ala mi no v, Khim. i. Ind. (Sofia), 29, № 5, 26 (1957). —138. И. Бе-
лоусов, авт. свид. 109714, 1956. — 139. И. Белоусов, авт. свид. 116575,
1957. — 140. Canad. Chem. Proc., 42, № 8, 51 (1958).
141. Н. А. Гольдберг, Жури. ВХО, 6, № 1, 49 (1961). — 142. Е. В 1 а-
s 1 a k, Przem. Chem., 39, № 3, 133 (1960). — 143. С. Cronan, Chem. Eng., 66,
№ 2, 44 (1959). — 144. Nitrogen, № 2, 1 (1959). — 145. J. Brunborg, Tidsskr.
kjemi, bergveseu of metallurg., 23, № 3, 58 (1963). — 146. Япон. пат. 3663, 1960.—
147. Япон. пат. 1473, 1958. — 148. Пат. США 3053891, 1959. — 149. Scaglione
Placido, Chim. е ind. 45, № 9, 1069 (1963). — 150. Швейц, пат. 369443, 1963.
151. Швейц, пат. 370762, 1963. — 152. Пат. ФРГ 1135889, 1963. — 153. Пат.
ЧССР 104589, 1962. — 154. Пат. США 3091637, 1963. —155. Франц, пат. 1318292,
1963. — 156. Е. G и с с i о n е, Chem. Eng., 72, № 2, 126 (1965). — 157. Nitrogen,
№ 33, 31 (1965). —/58. Франц, пат. 1356508, 1964. — 159. Бельг, пат. 620227,
1963. — 160. Англ. пат. 974411, 1964.
161. Англ. пат. 974412, 1964. — 162. Пат. США 3147304, 1964. — 163. Швейц,
пат. 387015, 1965. — 164. Пат. США 3155722, 1964. — 165. Пат. США 3155723,
1964.-/55. Пат. США 3172911, 1965. -/57. R. М. Reed, J. С. Reynolds,
Chem. Eng. Progr., 61, № 1, 62 (1965).— 168. Франц, пат. 1423412, 1965.—
169. Oil a. Gas. J., 64, № 33, 48 (1966). — 170. Бэссацу качаку кочё, 10, № 5,
.200 (1966) (Япон.).
171. Belli a Francesco, Chim. е ind., 48, № 11, Ц80 (1966).—
.172. Япон. пат. 22897, 1965. — 173. Y. Mayor, Chem. Rundschau, 20, № 4 54
(1967). — 174. Пат. ГДР 44383, 1966. —/75. Пат. США 3232985, 1966. —/75. Пат.
США 3255246, 1966. — /77. Б. П. Мельников, И. А. Кудрявцева, Про-
изводство мочевины, Изд. «Химия», 1965. — 178. В. В. Лебедев, Методиче-
ские указания и расчеты по технико-экономическим основам рациональной тех-
нологии производства мочевины, изд. Дзержинского филиала ГИАП, Дзержинск,
1963. — 179. S. Kawasumi, Bull. Chem. Soc. Japan, 26, 218 (1953).—
180. J. К i t a w a k i, S. Hori, J. S h i m о d a, Bull. Gov. Chem. Ind. Res. Inst.
Tokyo, 32, № 6, 10 (1937).
181. M. Tokuoka, J. Agr. Chem. Japan, № 10, 1933 (1934); № 11, 107, 174
(1935). — 182. N. W. Krase, V. L. Gaddy, Ind. Eng. Chem., 14, 611 (1922).—
183. H. J. Krase, V. L. Gaddy, ibid., 25, 1092 (1933). — 184. S. Kawasumi,
Bull Chem. Soc. Japan, 25, 227 (1952). — 185- R. К i j a m a, H. К i п о s h i t a,
Rev. Phys. Chem. Japan, 21, 11 (1951). — 186. Petrol. Ref., 36, Ko 11, 284 (1957).—»
Литература
1303
187. G. Chenoweth, Chem. Eng. Progr., 54, № 4, 55 (1958). —188. Англ. пат.
800444, 1958. —189. Ind. chim. Beige, 22, № 1, 69 (1957)'. —190. Бельг, пат.
472813, 1947. .
191. Франц, пат. 947448, 1949. — 192. V. Auerbach, Ind. chim. Beige, 19,
№ 4, 397—401 (1954). —193. W. H. T о n n, Chem. Eng., 62, № 10, 186—190
(1955). —194. L. H. Cook, Chem. Eng. Progr., 50, № 7, 327 (1954). — 195. R о o-
s eb oom, Chem. Eng., 58, № 3, 111—114 (1951). — 196. Chem. Eng. News, 43,
№ 36, 120 (1965). — 197. Япон. пат. 16221, 1961, — 198. Франц, пат. 826280,
1938. — 199. Р е с h i п е у, англ. пат. 753671, 1956. — 200. Chem Week., 22/11, 82,
№ 8, 40 (1958).
201. Chem. Eng. News, 37, № 13, 38 (1959). — 202. A. Smith, Gas Age,
July, 28, 32—35, 51 (1955). — 203. J. R e m о n d, Rev. Prod, chim., № 11—12,
197—200 (1952). — 204. Nitrogen, № 2, 34 (1959). — 205. Chem. Eng. News, 37,
№ 13, 38 (1959).— 206. Oil a. Gas J., 57, № 36, 158 (I960). — 207. G. Weyer-
muller, W. Smiley, Chem. Process (USA), 23, № 5, 122 (1960). — 208. Nitro-
gen, № 14, 29 (1961). — 209. Пат. США 3072721, 1963. — 210. J. R e m о n d, Rev.
Prod, chim., Jan., 1—6 (1956).
211. Petrol. Ref. 35, № 1, 200—202 (1956). — 212. W. F. Bland, Petrol.
Process, 7, № 10, 1457 (1952). — 213. Пат. США 2634826, 1953. — 214. Япон. пат.
5177, 1953. — 215. Япон. пат. 1793, 1950.—216. С. Cronan, Chem. Eng., 66,
№ 2, 78 (1959). — 217. Nitrogen, № 14, 14 (1961). — 218. Chem. Eng., 66, № 2,
48 (1959). — 219. Chem. Week., 87, № 20, 69 (I960). — 220. Chem. Age, 84,
№ 2162, 1025 (1960).
221. Chem. Week, 69 (1960). — 222. Англ. пат. 455865, 1936. — 223. Англ. пат.
756934, 1953. — 224. Chem. Eng., 65, № 7, 53 (1958).—225. M. Людковская,
А. Фридман, В. Клевке, авт. свид. 109674, 1957. — 226. М Людков-
ская, А. Фридман, Л. Савельева, Хим. пром., № 7, 35 (1958).—
227. М. А. Людковская, А. Д. Фридман, Л. Савельева, Труды ГИАП,
в. 10, 1959, стр. 248. — 228. А. Д. Фридман, М. А. Людковская, Труды
ГИАП. Химия и технология азотных удобрений, в. 12, 1961, стр. 255. —
229. М. А. Людковская, Т. Ухорская, Там же, стр. 283. — 230. М. Е. П о-
з и н, Б. А. Копылев, Л. Я. Т ерещенко, И. И. Орехов, авт. свид. 118094,
1958.
231. Г. В и л е с о в, В. Козин, П. Соколов, авт. свид. 122149, 1958.—
232. J. Morrison, Oil a. Gas Ind., 7, № 7, 39, 42, 44, 46 (1967).—
233. Л. Н. Альтшулер, В. И. Кучерявый, Хим. пром., № 1, 45 (1968).—
234. Швейц, пат. 387015, I960, —235. Пат. США 3270051, 1966,—235. Пат. США
3232982, 1966.—237. Пат. США 3232983, 1966.— 238. Chem. Process, 13, № 9,
22 (1967) (англ.). — 239. Nitrogen, № 1, 19 (1959). — 240. Пат. США 2777877,
1957.
241. Англ. пат. 885691, 1961. — 242. Япон. пат. 1072, 1959. — 243. Япон. пат.
2211, 1957. — 244. Hoshino Taneo, Japan Chem. Quart, 1, № 1, 20 (1965).—
245. Nitrogen, № 43, 26 (1966). — 246. Japan Chem. Quart., 2, № 4, 28 (1966). —
247. Chem. Eng., 66, № 25, 86 (1959). — 248. Nitrogen, № 4, 27 (1959).—
249. A. Kume, Petrol. Ref., 39, № 3, 200 (I960). — 250. Rev. prod. Chim., № 1030,
51 (1961).
251. J. Reynolds, C. Trimark e, Petrol. Ref., 41, № 12, 109 (1962).—
252. Англ. пат. 853220, 1959. — 253. Nitrogen, № 20, 7 (1962). — 254. G. Weyer-
niuller. W. Schellentrager, L. Marks, Chem. Process (USA), 25, № 16,.
19 (1962). — 255. Brit. Chem. Eng., 11, № 3, Suppl. (1966). — 256. Nitrogen, № 48,
31 (1967). — 257. S. K. Mitra, S. K. Mukherjee, Chem. Age (India), 18,
№ 11, 796 (1967). —258. Nitrogen, № 2, 25 (1959). — 259. O. N e u w i r t h,
°- Schulze-Bentrop, Erdol u. Kohle, 14, № 6, 456 (1961). — 260. Nitrogen,
№ 38, 29, 34 (1965).
17»
1306
Гл. XXXIV. Карбамид
261. Brit. Chem. Eng., 11, № 3, 1966. — 262. Chem. Process, 12, № 11, 4
(1966). — 263. Nitrogen, № 41, 34 (1966). — 264. Chim. e. ind., 49, № 6, 642
(1967). — 265. Coppa-Zuccari Giovanni, Europe a. Oil, 6, № 10—11,
24 (1967). — 266. Chem. Age (India), 18, № 11, 843 (1967). — 267. Chem. Process,
14, № 1, 10 (1968). — 268. Chem. a. Eng. News, 44, № 32, 24 (1966).—
269. P. A. Gaillard, P. G. Renaudin, Chim. et ind., 97, № 1, 37 (1967).—
270. Одзаки Кодзо, Chem. Factory, 10, Ns 11, 32 (1966),
271. В. Гаганов, сб. «Химия и технология азотных удобрений и продук-
тов органического синтеза», ОНТИ, 1963, стр. 19.— 272. Б. П. Мельников,
Бюлл. по обмену опытом в азотн. пром., Ns 8, 5 (1958). — 273. Nitrogen, № 20,
7 (1962). — 274. G. Weyermuller, W. Schellentrager, L. Marks, Chem.
Process (USA), 25, Ns 16, 19 (1962). — 275. А. Еремин, Г. Киркин и др.,
сб. «Коррозия и защита сталей», Машгиз, 1959, стр. 2221 — 276. Khr. Alami-
nov, Khim. i. Ind. (Sofia), 31, № 5, 135 (1959), —277. Corrosion, 16, № 7, 81
(I960).— 278. Chem. Eng., 65, Ns 16, 73 (1958).— 279. I. W i 1 e n i t z, Petrol.
Ref., 38, Ns 10, 98 (1959). — 280. R. Poricha, D. Rao, Technology (India), 3,
№ 2, 96 (1966).
281. Nitrogen, Ns 47, 34 (1967). —282. Швейц, пат. 337822, 1959, —283. Пат.
США 3041152, 1962, — 284. Пат. ФРГ 1097974, 1961, —285. Япон. пат. 7620,
1963. —286. Швейц, пат. 340503, 1959,—287. Пат. США 2913493, 1959,—288. Пат.
ФРГ 1081447, 1960. — 289. A. Manciulescu, Rev- chim., 15, № 12, 733
(1964). — 290. Пат. США 3232984, 1966.
291. В. И. Заграничный, Н. А. Гольдберг, Хим. пром., Ns 3, 14
(1962). — 292. Nitrogen, Ns 2, 38 (1959). — 293. Англ. пат. 885692, 1961.—
294. Австр. пат. 220154, 1962. —295. Пат. США 3025571, 1962. — 296. Япон. пат.
5323, 1961. — 297. К- Nowak, J. Wilczynska, S. N i k i е 1, Chernik, 16,
Ns 7—8, 189 (1963). —298. Швейц, пат. 376092, 1964.— 299. Австр. пат. 232000,
1964. —300. Пат. США 3151156, 1964.
301. Швейц, пат. 383352, 1964. — 302. Пат. США 2916516, 1959. — 303. Пат.
США 3147174, 1964. — 304. Пат. США 3251879, 1966. —305. Пат. США 2815376,
1958. —306. Пат. США 2933526, 2933527, 1960. — 307. Пат. США 2979421, 1961.—
308. Пат. США 3059280, 1962. — 309. Англ. пат. 932997, 1963. — 310. В. А. К н-
сельников, В, Я- Д е м ш и н, С. Г. Широков, Изв. вузов. Хим. и хим.
технол., 8, Ns 3, 504 (1965).
- 311. Авт. свид. 172759, 1965. — 312. Nitrogen, Ns 44, 31 (1966). — 313. Япон.
пат. 18803, 1965. — 314. Н. Schoch, J. Van den Bogaerde, Ind. Eng.
Chem., 53, Ns 2, 155 (1961). —3/5. Пат. США 2826612, 1958. — 316. Пат. ФРГ
1013644, 1957. — 317. Н. А. Шахова, А. И. Рычков, Хим. пром,, Ns 11, 856
(1963). —3/8. Пат. США 3124612, 1964. —319. Chem. Process (USA), 29, Ns 11,
55, 58 (1966).— 320. Nitrogen, Ns 47, 15 (1967).
32/. A. E. Bennett, J. Scarlett, J. B. S c u f f h a m, Chem. Process.
Eng., 48, № 1, 43 (1967).—322. Франц, пат. 1480235, 1967. —323. V. V a n e с e k,
M. Markvart, E. Lederer, Chem. Prum., 10/35, Ns 10, 521 (1960).—
324. E. А. Казакова, H. Мещеряков, Л. Музыченко, Хим. пром., Ns 5,
24 (1962). — 325. H. Schruber, Brit. Chem, Eng., 7, Ns 8, 587 (1962).—
326. Пат. ФРГ 1203244, 1966. —327. Япон. пат. 25320, 1964.— 328. В. Н. К н-
сельников, Я- И. Крейндель, В. Я- Демшин, С. Г. Широков, Изв.
вузов. Хим. и хим. технол., 10, Ns 3, 354 (1967).—329. В. И. Кисельников,
В. Я- Демшин, С. Г. Широков, Там же, Ns 10, 1172 (1967). — 330. Пат.
США 3223145, 1965.
Глава XXXV
КОМПЛЕКСНЫЕ ТВЕРДЫЕ СЛОЖНЫЕ УДОБРЕНИЯ
Комплексные удобрения содержат два или три основных пита-
тельных элемента — NP или NPK. Иногда в них вводят микроэле-
менты 1-4. Содержание питательных веществ в комплексных удоб-
рениях составляет обычно 30—60%, иногда больше5-7.
В мировом производстве удобрений доля комплексных удобре-
ний увеличилась с 24% в 1938 г, до 47,4% в 1966 г.8-13. Комплекс-
ные удобрения выпускаются в твердом и в жидком виде. Твердые
комплексные удобрения подразделяются (см. гл. I) на сложные,,
смешанные и сложно-смешанные. Множество промышленных спо-
собов производства твердых сложных удобрений, используемых в
настоящее время, можно разделить на две основные группы.
1) Способы, основанные на азотнокислотном разложении при-
родных фосфатов с последующей переработкой полученных раство-
ров и суспензий в готовый продукт — сложные удобрения типа нит-
рофоса и нитрофоски,
2) Способы, базирующиеся на использовании фосфорной кис-
лоты, дающие удобрения типа нитроаммофоски и диаммонитро-
фоски (или аммофоса и диаммофоса).
До недавнего времени основными видами сложных удобрений
являлись продукты азотнокислотной переработки фосфатов14.
В связи со стремлением увеличить концентрацию питательных ве-
ществ в удобрениях и обеспечить содержание в них Р2О5 пол-
ностью в водорастворимой форме15 стали развиваться способы,
основанные на использовании фосфорной кислоты с переработкой
ее в удобрения, содержащие фосфаты аммония.
В 1953/54 гг. на долю нитрофосфатов приходилось 65,5% от
общего выпуска комплексных удобрений в капиталистических стра-
нах и 34,5% на долю фосфатов аммония, а в 1965/66 гг. на долю
нитрофосфатов приходилось только 41,3jt, а на долю фосфатов
аммония 58,7%.
1308
Гл. XXXV. Комплексные твердые сложные удобрения
В таких странах, как ФРГ, Франция, Голландия, Бельгия i
Норвегия, основным направлением в производстве сложных удоб
рений остается азотнокислотное разложение фосфатов. В США
Канаде, Японии и Англии развитие производства сложных удобре
ний идет по линии использования фосфорной кислоты 16“26. В по
следнее время, по экономическим соображениям, в США снова
заинтересовались азотнокислотным разложением фосфатов и теми
Способами, которые с большой эффективностью используются j
Европе27. Д
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ РАЗЛОЖЕНИЯ ФОСФАТОВ
АЗОТНОЙ КИСЛОТОЙ
Впервые еще в 1908 г. Д. Н. Прянишников высказал соображе
ния о целесообразности азотнокислотного разложения фосфатов
Однако в то время, в условиях отсутствия производства дешевой
азотной кислоты, этот метод переработки фосфатов не мог быть
реализован. Возможность получения азотных и фосфорных удобре
ний путем азотнокислотного разложения фосфатов приобрела ре
альную основу лишь в 20-е годы, когда стало быстро развиваться
производство азотной кислоты из синтетического аммиака.
Азотнокислотная переработка фосфатов заключается в разло
жении фосфатов азотной кислотой и последующей переработке
образующегося раствора (вытяжки), содержащего нитрат кальция
и свободную фосфорную кислоту. В зависимости от метода пере
работки, вытяжки можно получать как односторонние азотные и
фосфорные удобрения, так и сложные, двойные или тройные удоб
рения с самым широким диапазоном соотношения питательных
веществ 28-32.
Разложение фосфатов азотной кислотой является сложным
процессом, протекающим согласно следующему основному уравне
нйю 33-35:
Ca5(PO4)3F + IOHNO3 = ЗН3РО4 + 5Ca(NO3)2 + HF
Содержащиеся в фосфатах примеси — карбонаты кальция и
магния, окислы железа, алюминия и редких земель и фторид каль
ция также взаимодействуют с азотной кислотой с образованием
нитратов:
(Mg, Са) СО3 + 2HNO3= (Mg, Ca)(NO3)2 + СО2 + Н2О
R2O3 + 6HNO3 = 2R(NO3)3 + ЗН2О
CaF2 + 2HNO3 = Ca(NO3)2 + 2HF
Окислы железа и алюминия разлагаются также выделяющейся
фосфорной кислотой с образованием не растворимых в воде фос-
фатов, что приводит к потере Р2О5 зв:
Fe2O3 +. 2Н3РО4 = 2FePO4 + ЗН2О
А1,О3 + 2Н3РО4 = 2А1РО4 + ЗН,О-
Основы разложения фосфатов азотной кислотой
1309
Поэтому фосфаты, содержащие больше 12% Fe2O3 (по отноше-
нию к Р2О5), считаются пригодными к химической переработке
только после предварительного обогащения.
Присутствие в фосфатах AI2O3 не так вредно, как ре20з, однако
также нежелательно, так как это приводит к загрязнению рас-
твора фосфорной кислоты (вытяжки). Выделяющийся фтористый
водород взаимодействует с кремневой кислотой, всегда сопутствую-
щей фосфатам, и обычно остается в растворе в виде кремнефтори-
стоводородной кислоты. Присутствующие в небольших количествах
в апатитовом концентрате нефелин и эгерин также разлагаются
азотной кислотой:
KAIS1O4 • 4NaAlSiO4 • nSiO2 + 20HNO3 =
= KNO3 + 4NaNO3 + 5Al(NO3)3 + nSiO2 + 10H2O
Na2O . Fe2O3 • 4SiO2 + 8HNO3 = 2NaNO3 + 2Fe(NO3)3 + 4SiO2 + 4H2O
Для полного разложения природных фосфатов процесс необхо-
димо вести в присутствии стехиометрической нормы азотной кис-
лоты, взятой из расчета на СаО в апатите или на СаО и MgO в
фосфоритах 37.
Если количество азотной кислоты меньше стехиометрического и
соответствует реакциям
2Ca5(PO4)3F + 14HNO3 = ЗСа(Н2РО4)2 + 7Ca(NO3)2 + 2HF
Ca5(PO4)3F + 4HNO3 = 3CaHPO4 + 2Ca(NO3)2 + HF
то первая из этих реакций будет проходить только при концентра-
ции исходной азотной кислоты не ниже 60%. Вторая реакция в
обычных условиях практически не идет.
Скорость гетерогенного процесса разложения природных фос-
фатов азотной кислотой в значительной степени определяется ве-
личиной поверхности соприкосновения реагентов. Практически сте-
пень измельчения фосфатов устанавливают, учитывая не только
продолжительность их разложения, но и расходы, связанные с их
измельчением 38>39.
Существенное влияние на скорость разложения фосфатов ока-
зывают консистенция пульпы и концентрации реагирующих компо-
нентов. Оптимальная концентрация раствора характеризуется его
плотностью, которая не должна превышать 1,55 г!смъ. Чтобы избе-
жать изменения установившихся концентраций, осуществляют хо-
рошее перемешивание фосфата и кислоты. Перемешивание обычно
производят механическим способом. Применение для этого сжатого
в°здуха связано с усилением вспенивания раствора. Образование
пены происходит вследствие выделения газообразных продуктов
Реакции — двуокиси углерода, фтористого водорода, паров воды
и окислов азота, получающихся при частичном разложении азотной
ислоты органическими примесями, которые содержатся в природных
1310
Гл. XXXV. Комплексные твердые сложные удобрения
Количество HNO3
6 % к стехиометрическому
Рнс. 386. Влияние количества
HNO3 на степень извлечения
Р2О5 из фосфата.
Концентрация HNOj,%
Рис. 387. Зависимость степени
извлечения Р2О5 из апатито-
вого концентрата от концен-
трации азотной кислоты.
Продолжительность процесса: за-
грузка 30 мин и перемешивание
1 ч 30 мин; температура 40—50°.
Числа на кривых — количество кис-
лоты в % от стехиометрического-
фосфатах. Перемешивание же раствора путем циркуляции с по-
мощью центробежного насоса сопряжено с трудностями, возникаю-
щими при эксплуатации насосов
в условиях сильно агрессивной сре-
ды (раствор азотной и фосфорной
кислот) и из-за наличия твердой
фазы (шлама).
Скорость разложения трикаль-
цийфосфата и степень извлечения
Р2О5 в раствор зависят от количе-
ства применяемой азотной кисло-
ты 40-44.
На рис. 386 показана степень из-
влечения P20s из кролевецкого фос-
форита при обработке его разными
количествами азотной кислоты в те-
чение 30 мин (15 мин приливание и
15 мин смешение) с изменением при
этом температуры от 17—20° до
40—52°«
При стехиометрическом количе-
стве азотной кислоты, по мере про-
текания реакции разложения, ско-
рость ее постепенно замедляется вследствие накопления в растворе
солей 45 и уменьшения кислотности рас-
твора. Для обеспечения постоянной
скорости разложения процесс обычно
проводят при 2—5 %-ном избытке азот-
ной кислоты против стехиометрическо-
го количества. В некоторых случаях
избыток азотной кислоты увеличивают
до 20—50% —это необходимо для обе-
спечения последующих операций пере-
работки полученного раствора в удо-
брение.
Обычно разложение природных фос-
фатов ведут при температуре 45—50°,
которая является оптимальной 41. При
уменьшении температуры (ниже 45°)
разложение замедляется. С повыше-
нием температуры уменьшается вяз-
кость раствора, улучшаются условия
диффузии вещества и скорость разло-
жения увеличивается 46. Однако выше
50° резко усиливается коррозия аппаратуры. Требуемая темпера-
тура (45—50°) поддерживается главным образом за счет теплового
Основы разложения фосфатов азотной кислотой
1311
эффекта реакции; температуру азотной кислоты, которая должна
составлять 30°, регулируют подогревом или охлаждением кислоты
в теплообменнике.
Степень извлечения P2Os в раствор мало зависит от концентра-
ции кислоты (рис. 387) 47.
ТАБЛИЦА 93
Растворы, получаемые при разложении
прир^цных фосфатов азотной кислотой
Компоненты раствора Состав раствора, %
при обработке апатита при обработке фосфоритов Каратау
HNO3 5,92 5,71
Н3РО4 13,52 9,53
Ca(NO3)2 37,78 32,10
Fe(NO3)3 0,45 0,89
Ce(NO3)3 0,47 —
Mg(NO3)2 ....... — 3,16
A1(NO3)3 — 0,56
H2SiF6 0,92 0,88
H2O 40,94 47,14
В табл. 93 приведен примерный состав растворов, получаемых
в результате разложения фосфатов азотной кислотой при 20%-ном
ее избытке против стехиометрического количества (из расчета на
СаО) и при коэффициенте разложения для Р2О5, CaO, MgO и
R2O3 — 0,98, для фтора — 0,95 и для Ре20з — 0,70.
Выделение фтора и редкоземельных элементов
из азотиокислотной вытяжки
Выделение фтора
При обработке природных фосфатов азотной кислотой большая
часть фтора, содержащегося в фосфатах, переходит в раствор,
называемый азотнокислотной вытяжкой, в виде кремнефтористо-
водородной кислоты 47-49. Распределение фтора по фазам при раз-
ложении апатита и фосфоритов Каратау в зависимости от темпера-
тУры приведено в табл. 94 s0’51.
Опыты проводились с 55 %-ной азотной кислотой, взятой
в количестве: для апатита 10% и для фосфорита 5% избытка от
стехиометрической нормы. Время разложения 30 мин. Присутствие
пРимесей фтористых соединений затрудняет переработку рас-
пора 47, 48,52,53, понижает концентрацию Р2О5 в конечных продуктах
на 3—4<у0, затрудняет процессы фильтрации и кристаллизации
1312
Гл. XXXV. Комплексные твердые сложные удобрения
ТАБЛИЦА
Распределение фтора (в %) по фазам при азотнокислотиом разложении
фосфатов
Темпера- тура, °C Фосфориты Каратау Апатит
твердая фаза жидкая фаза газообразная фаза твердая фаза жидкая фаза газообразная фаза ;
30 . 3,03 г 76,62 20,35
50 3,25 58,33 38,42 1,03 77,22 21,70
60 3,17 47,50 49,33 0,34 78,78 20,88 >
70 3,00 40,83 56,17 — — —
80 2,83 41,70 55,47 0,43 85,28 14,29 J
90 2,5 32,5 65,00 — — — <
и понижает устойчивость нержавеющей стали. Поэтому'
растворов
а также в связи с увеличением производства фтористых соедине
ний выделение фтора из азотнокислотной вытяжки целесообразно,
хотя и не является обязательным при переработке полученных
растворов на удобрения.
Фтор из раствора выделяют с помощью добавляемой к нему на
триевой соли 54~56. При введении натриевой соли в количестве 300%
от стехиометрического 80—85% фтора, содержащегося в растворе,
осаждается в виде кремнефторида натрия. Для осаждения рекомен
дуют применять карбонат или нитрат натрия. Осаждение нитратом
натрия протекает более спокойно; при употреблении карбоната
натрия образуется пена вследствие выделения СО2. Пользоваться
хлористым натрием не следует, так как ион хлора усиливает кор
розию аппаратуры из хромоникелевой стали; кроме того, часть
хлора при дальнейшей переработке фосфорнокислого раствора
перейдет в удобрение и ухудшит его качество. Раствор NaNO3 мо
жет иметь концентрацию 17—35%. Целесообразно использовать
раствор, получаемый при поглощении окислов азота из отходящих
газов в производстве азотной кислоты. Расход NaNO3 при введе
нии 300% избытка сверх стехиометрического количества состав
ляет 120—140 кг на 1 т апатита или фосфорита.
При использовании карбоната натрия необходимо применять
раствор, содержащий не больше 15% Na2CO3, для предотвраще
ния осаждения монокальцийфосфата; избыточная кислотность
обрабатываемого раствора не должна быть ниже ~4%. Расход
карбоната натрия с учетом 300%-ного избытка сверх стехиометри-
ческой нормы составляет 85 кг на 1 т апатита. Для получения азот-
нокислотнрй вытяжки с избыточной кислотностью не менее 4% раз
ложение фосфатов следует проводить при 10—20%-ном избытке
азотной кислоты сверх стехиометрического количества, рассчитан
ного на СаО.
Основы разложения фосфатов азотной кислотой
1313
Выделившийся кристаллический осадок Na2SiF6 отделяют от
раствора отстаиванием, затем фильтрацией. Скорость осаждения
кремнефторида натрия составляет 0,15 м/ч. Отделение осадка
Na2SiFe от вытяжки, из которой предварительно был выделен шлам,
не следует производить на отстойных центрифугах вследствие зна-
чительного уноса твердой фазы (иногда унос твердой фазы дости-
гает 80% при содержании жидкой фазы в осадке до 80%). Отстой-
ная центрифуга может быть использована для отделения кремне-
фторида натрия, полученного из вытяжки, содержащей шлам.
В этом случае степень извлечения твердого остатка составит 90%,
но в оставшейся твердой фазе (~10%), находящейся в жидкости,
будет преобладать NasSiFg.
При азотнокислотной переработке 1 т апатита получается 63 кг
Na2SiF6 с влажностью 30%; содержание Na2SiF6 в сухом веществе
составляет 87%.
Выделение редкоземельных элементов
В апатитовом концентрате содержится 0,9—0,95% редкоземель-
ных элементов цериевой группы (церий, лантан и др.). Выделение
редкоземельных элементов из раствора, полученного в результате
разложения апатитового концентрата азотной кислотой, основано
на малой растворимости фосфатов редкоземельных элементов
в слабокислых растворах (pH = 2—2,5). С повышением темпера-
туры при одной и той же степени нейтрализации раствора раство-
римость их понижается42. Так, при 35—40° и степени нейтрализа-
ции 40% содержание окислов редкоземельных элементов в рас-
творе составляет 0,12—0,13%, при той же степени нейтрализации
и при 60—80° содержание R2O3 не превышает 0,07—0,09%. Следо-
вательно, при более высоких температурах (60—80°) осаждение
фосфатов редкоземельных элементов начинается из более кислых
растворов.
Для осаждения 80—90% редкоземельных элементов от общего
количества их в апатитовом концентрате необходимы следующие
оптимальные условия 57’58: концентрация раствора — 35% Ca(NO3)2
и 9,5% Р2О5; температура ~60°; нейтрализация всей свободной
HNO3 и около 50% первого иона водорода фосфорной кислоты;
время осаждения около 2 ч. При этом из 1 т апатита получится
10,7 кг фосфатов редкоземельных элементов. Вместе с ними из рас-
твора осаждаются некоторые другие примеси, поэтому в получен-
ном сухом осадке содержится ~65% фосфатов редкоземельных
элементов.
Процесс осуществляют следующим образом. Азотнокислотную
вытяжку после выделения из нее шлама и фтористых соедине-
ний 59~бо нейтрализуют газообразным аммиаком или известняком
(суспензией, содержащей ~30% СаСОз).
1314
Гл. XXXV. Комплексные твердые сложные удобрения
Нейтрализацию проводят до величины pH не более 2—2,5. При
этом в осадок выделяются белые кристаллы фосфатов редкоземель-
ных элементов, например:
2Ce(NO3)3 + 2Н3РО4 + ЗСаСО3 = 2СеРО4 + 3Ca(NO3)2 + ЗСО2 + ЗН2О
Процесс проводят непрерывно в нескольких последовательно
соединенных реакторах с паровыми змеевиками при перемешива-
нии. Отделение осадка от раствора производят вначале деканта-
цией, затем на барабанных вакуум-фильтрах. Осадок с фильтров
содержит 15—25% влаги. Для получения чистых фосфатов редко-
земельных элементов необходимо произвести дополнительную их
очистку. Предложено выделять редкоземельные элементы из
азотнокислотной вытяжки хибинского апатита с помощью ионо-
обменных смол67.
ПЕРЕРАБОТКА РАСТВОРА, ПОЛУЧЕННОГО АЗОТНОКИСЛОТНЫМ 1
РАЗЛОЖЕНИЕМ ФОСФАТОВ I
Раствор, полученный азотнокислотным разложением, фосфатов,
содержит в основном фосфорную кислоту и нитрат кальция. Воз-
можным способом его переработки является выделение нитрата
кальция и последующее использование фосфорной кислоты для
получения фосфатов — дикальцийфосфата, монокальцийфосфата;
нитрат же кальция может быть выпущен в качестве продукта или
переработан, например, в аммиачную селитру. Однако такой путь
переработки азотнокислотной вытяжки не является лучшим в тех-
нико-экономическом отношении, так как отличается сложностью
технологических схем, необходимостью значительных капитальных
затрат и большими эксплуатационными расходами. Кроме того,
эффективность получаемых при этом простых удобрений ниже, чем
комплексных, а по своим физическим свойствам некоторые из них
(например, нитрат кальция) недостаточно доброкачественны. По-
этому распространение получила непосредственная переработка
вытяжки в комплексные азотно-фосфорные или азотно-фосфорно-
калийные удобрения. Методы этой переработки основаны на фазо-
вых равновесиях в системах СаО—Р2О5—N2O5—Н2О и СаО—NH3—
—Р2О5—N2O5—Н2О.
Физико-химические основы переработки
азотнокислотной вытяжки 40143150158’68-73
На рис. 388 и 389 показаны проекции полей насыщения на
координатные плоскости СаО—Н2О—Р2О5 и N2O5—Н2О—P2Os
в системе СаО—Р2О5—N2O5—Н2О при 25 и 50°. В табл. 95 приве-
дены составы жидких и твердых фаз в узловых точках изотерм
25 и 50°.
1
СаО.%
,°/о
Рис. 388. Система СаО—Р2О6—N2O5—Н2О при 25*.
Рнс. 389. Система СаО-[-Р2О5—N2O5—Н2О при 50*.
1316
Гл. XXXV. Комплексные твердые сложные удобрения
Узловые точки изотерм 25 и 50° в системе
CaO-P2Os—N2Os—Н2О
ТАБЛИЦА 93
Обо- значе- ние точки Состав раствора Твердые фазы
вес.®/о молей на 1000 моль Н2О
СаО Р2О5 N2O5 СаО Р2О5 NjOs
Изотерма 25° (рис. 388)
А 5,81 24,20 — 26,7 43,8 — Са(Н2РО4)2 • Н2О+СаНРО4
N 19,73 1,18 37,26 151,7 3,6 148,5 Са(Н2РО4)2 • Н2О+ +СаНРО4+ + Ca(NO3)2 • 4Н2О
М 19,79 Следы 37,74 149,8 Следы 149,8 CaHPO4+Ca(NO3)2 • 4Н2О+ + Са3(РО4)2 • Н2О
Р 14,24 18,21 30,36 . 122,9 62,9 136,0 Са(Н2РО4)2 • Н2О~Ь + Ca(NO3)2 • 4Н2О+ + Ca(NO3)2 • ЗН2О
В 11,03 — 51,89 95,4 — 233,1 Ca(NO3)2 4Н2О + + Ca(NO3)2 • ЗН2О
С 9,54 — 56,28 89,5 —• 274,3 Ca(NO3)2 • ЗН2О+ + Ca(NO3)2 • 2Н2О
D 2,79 — 62,84 26,1 — 304,6 Ca(NO3)2 • 2Н2О + Са (ЫО3)2
Изотерма 50° (рис. 389)
А 5,88 28,85 — 28,9 56,0 1 - Са(Н2РО4)2 • Н2О+СаНРО4
N 23,41 4,80 45,81 289,9 23,4 292,3 Са(Н2РО4)2 • Н2О + +СаНРО4 + +Ca(NO3)2 • 2Н2О
М 26,63 0,1 51,27 389,0 0,6 389,0 CaHPO4 + Ca(NO3)2 -2Н2О+ + Са3(РО4)2 • Н2О
Р 20,79 11,12 43,28 269,3 56,4 290,6 Са(Н2РО4)2 -Н2О+ + Ca(NO3)2 • 2Н3О+ + Ca(NO3)2
В 25,95 51,23 265,2 — 374,3 Ca(NO3)2 • 2H2O + Ca(NO3)2
t 8,2 20,6 8,3 СаНРО4+Са(Н2РО4)2 • Н2О
4 10,8 27,0 30,8 Са(Н2РО4)2 • H2O+Ca(NO3)2
Переработка азотнокислотной вытяжки
1317
В кислой среде фосфаты оказывают высаливающее действие
на нитрат кальция, растворимость кристаллогидратов которого
значительно уменьшается в присутствии фосфорной и азотной кис-
лот. В свою очередь, растворимость фосфатов уменьшается под
влиянием высаливающего действия нитратов.
В рассматриваемой системе изотермы характеризуются полями
насыщения моно- и дикальцийфосфатом, а также водным и безвод-
ным нитратом кальция. Эти твердые фазы образуются не только
в результате высаливания одних компонентов другими, но и в ре-
зультате различных химических реакций — разложения фосфатов
водой, взаимодействия между нитратом кальция и фосфорной кис-
лотой, а также между фосфатами и азотной кислотой.
Область образования стабильных насыщенных растворов ни-
трата кальция и фосфорной кислоты на изотерме 50° отвечает
линии MN, разделяющей поля дикальцийфосфата и дигидрата
нитрата кальция. Точка М соответствует растворимости дигидрата
нитрата кальция в воде; введение в систему Р2О5 приводит к умень-
шению растворимости нитрата кальция. В точке Л' раствор стано-
вится насыщенным также монокальцийфосфатом и содержит 4,8%
РгО5. Эта концентрация соответствует, если не учитывать содержа-
ния воды в монокальцийфосфате, 4,8 : 0,156 = 30,7% Р2О5 в исход-
ной кислоте [где 0,156 — содержание Р2О5 и воды в системе в долях
единицы, за вычетом Са (NO3)2-2H2O]. При учете воды, содержа-
щейся в моно- и дикальцийфосфате, концентрация исходной фос-
форной кислоты, соответствующая раствору в точке N, равна
29,2 % Р2О5. Отсюда следует, что в пределах концентраций от 0 до
29,2% Р2О5 при 50° образуется раствор фосфорной кислоты, насы-
щенный дигидратом нитрата кальция. Соответственно при 25°
в пределах концентрации от 0 до 36,1% Р2О5 образуется раствор
фосфорной кислоты, насыщенный четырехводным нитратом
кальция.
Азотнокислотная вытяжка, получающаяся при разложении апа-
титового концентрата 20%-ным избытком 47%-ной азотной кис-
лоты, содержит 29,8% N2O5, 12,4% СаО и 9,85% Р2О5. При таком
составе вытяжка может быть насыщена лишь двумя твердыми фа-
зами — моно- и дикальцийфосфатом. Но этому составу соответ-
ствует исходная концентрация Р2О5 20,7%, при которой раствор
фосфорной кислоты насыщен дигидратом нитрата кальция. Это
предопределяет образование монокальцийфосфата по реакции:
Ca(NO3)2 + 2Н3РО4 = Са(Н2РО4)2 + 2HNO3
или дикальцийфосфата по реакциям:
Са(Н2РО4)2 • Н2О + Ca(NO3)2 = 2СаНРО4 + 2HNO3 + Н2О
Са (Н2РО4)2 • Н2О 4- aq — СаНРО4 + Н3РО4 4- aq
1318
Гл. XXXV. Комплексные твердые сложные удобрения
»
Пользуясь данными диаграммы растворимости в системе
СаО—Р2О5—N2O5—Н2О, можно установить те пределы концен
траций азотной и фосфорной кислот, в которых протекают реакции
между ди- и монокальцийфосфатом и азотной кислотой, а также
между нитратом кальция и фосфорной кислотой. Прямая на проек
ции СаО — 0 — Р2О5, проведенная из точки 0 под углом наклона
отвечающим стехиометрическому отношению СаО к Р2О5 в
Са(Н2РО4)2, дает геометрическое место точек, в которых составы
растворов могут быть выражены через монокальцийфосфат и азот
ную кислоту. Отрезок этой прямой t — q, проходящий через поле
монокальцийфосфата, отвечает насыщенным растворам монокаль
цийфосфата в азотной кислоте. При этом граничная точка t соот
ветствует при 50° водному раствору азотной кислоты, насыщен-
ному моно- и дикальцийфосфатом, а граничная точка q — раствору
насыщенному монокальцийфосфатом и безводным нитратом каль
ция. Составы растворов в точке t: 8,2% СаО, 20,6% Р2О5 и 8,3%
N2O5; в точке q~. 10,8% СаО, 27,0% Р2О5 и 30,8% N2OS. Для образо
вания раствора, соответствующего по своему составу точке t, не
обходимо взять 8,3-1,165 : (1 — 0,3665) = 15,3% раствор азотной
кислоты (где 1,165 — переводной коэффициент от N2O5 к 2HNO3
а 0,3665 — доля Са(Н2РО4)2-Н2О в растворе). Концентрация исход
ного раствора азотной кислоты, необходимая для образования рас
твора состава q, равна 70,8% HNO3.
Таким образом, азотная кислота концентрацией от 15,3 до
70,8% HNO3 растворяет при 50° монокальцийфосфат без разложе
ния с образованием его насыщенных растворов. При использова
нии кислоты с концентрацией меньше 15,3% HNO3 монокальций
фосфат разлагается с выделением дикальцийфосфата (аналогично
разложению водой). Кислота концентрацией больше 70,8% HNO3
разлагает монокальцийфосфат с выделением в твердую фазу нит
рата кальция:
Са(Н2РО4)2 + 2HNO3 = Ca(NO3)2 + 2Н3РО4
При другой температуре пределы концентраций азотной кис
лоты, при которых существуют насыщенные растворы монокаль
цийфосфата в азотной кислоте, несколько изменяются и составляют
например, для 25° 10,5—69,8% HNO3. При этом нижний предел
концентраций возрастает с повышением температуры, а верхний —
почти не меняется.
Линии 0g на диаграммах представляют собой составы рас
творов, выраженные через дикальцийфосфат и азотную кислоту
Нижний предел концентрации азотной кислоты, при которой ди
кальцийфосфат растворяется в ней без образования новых фаз
можно принять весьма малым. Верхний предел (точка g) лежит
на кривой, разделяющей поля ди- и монокальцийфосфата. При 50°
раствор в точке g имеет состав 11,4% СаО, 14,5% Р2О5, 18,3% N2Os
Переработка азотнокислотной вытяжки
1319
Соответственно максимальная концентрация азотной кислоты, об-
разующей насыщенные растворы дикальцийфосфата, равна 20%
HNO3. При концентрации HNO3 более 20% в растворе происходит
разложение дикальцийфосфата с образованием в твердой фазе
монокальцийфосфата и в растворе нитрата кальция:
2СаНРО4 4- 2HNO3 4- aq = Са(Н2РО4)2 • Н2О 4- Са (NO3)2 4- aq
Весовое отношение СаО: Р2О5 в преципитате, определяющее
наклон линии 0g, составляет 0,79. Значение этого отноше-
ния в азотнокислотной вытяжке равно 1,31 [из соотношения
5Ca(NO3)2: ЗН3РО4, стр. 1308]. Поэтому составы растворов, соот-
ветствующие азотнокислотной вытяжке, располагаются в поле
СаНРО4 и могут рассматриваться как растворы нитрата кальция
в насыщенном растворе дикальцийфосфата и азотной кислоты.
Выделение СаНРО4 из раствора может быть осуществлено добав-
лением к нему СаО. При этом вначале может выделяться
Са(Н2РО4)2-Н2О (например, если раствор содержит ~20% СаО и
~ 15% Р2О5); затем, по достижении минимальной концентрации
азотной кислоты, равной 15,3% HNO3 при 50°, Са(Н2РО4)2-Н2О
перейдет в СаНРО4:
Са(Н2РО4)2 • Н2О 4- aq = СаНРО4 4- Н3РО4 4- aq
Выделившаяся в этих условиях фосфорная кислота также ней-
трализуется до дикальцийфосфата.
Количество окиси кальция (в виде известкового молока или
известняковой суспензии), добавляемое к вытяжке, определяют по
уравнениям:
5Ca(NO3)2 4-ЗН3РО4 4-ЗСаО = 5Ca(NO3)2 + ЗСаНРО4(тв.) 4-ЗН2О
ИЛИ
ЗСаНРО4(р-р) + 2Ca(NO3)2 + 6HNO3 4- ЗСаО =
= ЗСаНРО4(тв.) 4- 5Ca(NO3)2 4- ЗН2О
Азотнокислотная вытяжка, полученная разложением апатита
47%-ной азотной кислотой и содержащая 14,2% СаО, 10,7% Р2О5
и 27,4% N2O5, в процессе нейтрализации при 50° образует раствор,
не насыщенный монокальцийфосфатом. Поэтому практически моно-
кальцийфосфат не выделяется в качестве промежуточного про-
дукта (его выделение привело бы к образованию густых пульп, за-
трудняющих ведение процесса). Раствор становится насыщенным
Дикальцийфосфатом, когда степень нейтрализации первого иона
водорода фосфорной кислоты достигает 85%. При дальнейшей ней-
трализации в осадок будет выделяться дикальцийфосфат. На прак-
тике преципитируют вытяжки, разбавленные до 8—8,5% Р20з,
чтобы уменьшить пересыщение раствора дикальцийфосфатом и по-
лучить более крупные кристаллы преципитата.
1320
Гл. XXXV. Комплексные твердые сложные удобрения
Для разложения фосфата норму кислоты берут из расчета
2 моль HNO3 на 1 моль СаО в фосфате. В обычных условиях
уменьшение этой нормы приводит к уменьшению степени извле-
0,2 0,4 0,6 0,1
Са3(МОэ)в Са/РО,),
Рис. 390. Водная диаграмма
системы
СаО—Р2О5—N2O5—Н2О при 5°;
m — число молей воды иа 1 моль
сухих солей.
чения Р2О5 и СаО в раствор, состоя-
щий из нитрата кальция и фосфор-
ной кислоты. Как было указано вы-
ше, получающиеся растворы в этом
случае не насыщены монокальций-
фосфатом. Но при разложении фос-
фата с получением в растворе нитра-
та кальция и монокальцийфосфата
норма азотной кислоты снижается
на 30% в соответствии с реакцией::
Ca5(PO4)3F + 7HNO3 = \
= 1,5Са(Н2РО4)2 + 3,5Ca(NO3)2 +
При разложении апатитового кон-
центрата 45—50%-ной азотной кис-
лотой при 50° в этом случае должен
был бы образоваться раствор, со-
держащий 18—20% СаО, 14—16%
Р2О5 и 25—27% N2O5, насыщенный
Ht(NO3)e Щ(РОЩ
CajlNOjJs СаЦРоЖ
Природный фосфат
СаХР04)2%СаО
Рис. 391. Схема циклического про-
цесса получения преципитата и
нитрата кальция.
монокальцийфосфатом. Этот процесс можно осуществить с разбав-
ленной кислотой при 100—110° или применяя азотную кислоту кон-
центрацией выше 60%. На этом основано получение так называе-
мого нитросуперфосфата,
Переработка азотнокислотной вытяжки
1321
Используя изменение растворимости в системе СаО—P20s—
^_N2Os—НгО при разных температурах, возможно разделить ком-
поненты азотнокислотной вытяжки путем кристаллизации из нее
кристаллогидрата нитрата кальция. Как видно из рис. 390, рас-
творы, полученные разложением апатита 55, 52, 50 и 47%-ной азот-
ной кислотой (точки щ, а2, а3 и а4), находятся при 5° в поле четы-
рехводного нитрата кальция Са(ЫОз)2-4Н2О. Теоретический выход
последнего при охлаждении до +5° азотнокислотной вытяжки, по-
лученной с применением 30%-кого избытка (от стехиометрического
количества) 55%-ной азотной кислоты, составляет 71,5%, а с
47%-ной азотной кислотой — около 30%. Но он возрастает до 49%,
если избыток 47%-ной азотной кислоты, взятой на разложение
апатита, составляет 10% от стехиометрического количества. Для
дальнейшего повышения выхода Са (ЫОз)2-4Н2О требуется охла-
дить раствор до —5, —15°.
Разложение фосфата азотной кислотой с получением моно-'
кальцийфосфата и нитрата кальция может быть осуществлено
в циклическом процессе, где фосфат растворяется при 100° и четы-
рехводный нитрат кальция выделяется из раствора при его охлаж-
дении до 5°. Предварительно, до охлаждения раствора, в него вво-
дят азотную кислоту концентрацией выше 60% HNO3. После отде-
ления выделившегося четырехводного нитрата кальция к раствору
добавляют фосфат и нагревают до 100°. При этом выделяется
моиокальцийфосфат. К полученному после его отделения филь-
трату добавляют холодную азотную кислоту и, охлаждая раствор,
выделяют из него Ca(NO3)2-4H2O. Цикл, таким образом, возобнов-
ляется.
Циклическое получение дикальцийфосфага и нитрата кальция
возможно осуществить следующим образом (рис. 391). К азотно-
кислому раствору S, полученному после кристаллизации четырех-
водного нитрата кальция, добавляют природный фосфат, и система
переходит в точку Т. В результате разложения фосфата выделяется
в осадок дикальцийфосфат, и при охлаждении состав раствора со-
ответствует точке Q. После отделения дикальцийфосфата к рас-
твору добавляют азотную кислоту (точка /?), вследствие чего про-
исходит кристаллизация Ca(NO3)2-4H2O и система возвращается
в точку S. После этого цикл возобновляется.
Способы переработки азотнокислотиой вытяжки
Способы переработки азотнокислотной вытяжки по характеру
получаемых продуктов — односторонних или сложных удобре-
ний — можно подразделить на способы раздельного получения фос-
фатов (дикальцийфосфата, монокальцийфосфата) и нитратов
.(кальциевой или аммиачной селитры) 47-и способы получения
сложных удобрений 48,
1322
Г л. XXXV. Комплексные твердые сложные удобрения
Раздельное получение односторонних удобрений основано на
нейтрализации фосфорной кислоты в азотнокислотной вытяжке из-
вестняком или известковым молоком. При этом образуется осадок
дикальцийфосфата (преципитата), который отделяют от раствора
фильтрацией и высушивают. Остающийся раствор нитрата каль-
ция выпаривают и кристаллизуют. Более рационально конвертиро-
вать нитрат кальция в нитрат аммония с помощью карбоната ам-
мония 74’78. Возможно также раздельное получение монокальций-
фосфата и нитрата кальция (стр. 1321).
Получение из вытяжки односторонних удобрений отличается
сложностью и требует больших эксплуатационных и капитальных
затрат. Это обусловлено необходимостью обжига известняка, при-
готовления известкового молока, установки оборудования для тон-
кого измельчения известняка, осуществления разделения азотных
и фосфорных солей фильтрацией и выпариванием растворов с по-
следующей их кристаллизацией. Поэтому способы раздельного
получения односторонних фосфорных и азотных удобрений из азот-
нокислотной вытяжки не нашли широкого применения.
Основным отличием непосредственного производства сложных
удобрений из азотнокислотной вытяжки в большинстве случаев яв-
ляется нейтрализация ее и обезвоживание полученной пульпы без
разделения на отдельные компоненты. Отсутствие фильтрации зна-
чительно упрощает технологический процесс.
В большинстве применяемых в настоящее время способов вы-
тяжку нейтрализуют аммиаком. Получаемые таким путем сложные
удобрения содержат два питательных элемента — азот и фосфор;
их называют нитрофосами. Чаще перед гранулированием нейтра-
лизованной пульпы к ней добавляют соль калия (KCl, K2SO4) л
получают тройное удобрение — нитрофоску, — содержащее азот!
фосфор и калий. I
Состав нитрофосок различен, он зависит от способов их получе-
ния. Обычно нитрофоска содержит дикальцийфосфат, фосфаты ам-
мония, нитрат аммония и соли калия. Фосфор в нитрофоске нахо-
дится в цитратнорастворимой (СаНРО/,) и в водорастворимой
(NH4H2PO4) форме — последняя предпочтительнее. Поэтому стре-
мятся использовать способы производства нитрофосок, дающие по-
вышенное содержание в них водорастворимых соединений фосфора.
Нитрофоски выпускают с разным соотношением питательных
веществ N : Р2О5: К2О, например, 1:1:1; 1 : 1 : 1,5; 1:0,7: 1;|
1 : 1,5 : 1,5; 1:2:1. Это придает удобрению универсальность — оно]
может применяться на разных почвах и для разных культур. 1
В природных фосфатах отношение СаО : Р2О5 составляет: в апа-|
титвоом концентрате 1,32, в фосфоритах Каратау 1,62. С такими!
же соотношениями этих компонентов получаются и азотнокислот-1
ные вытяжки из этого сырья. В дикальцийфосфате СаО:Р2О5=1
=0,79. Поэтому при нейтрализации вытяжки аммиаком,.после того]
Переработка азотнокислотной вытяжки
1323
как вся фосфорная кислота израсходуется на образование дикаль-
цийфосфата, в растворе останется избыточный кальций в виде
Са(МОз)а. При высушивании пульпы полученный продукт будет
содержать нитрат кальция, что нежелательно ввиду его гигроско-
пичности. Этого можно избежать, если удалить из раствора избы-
точный кальций, достигнув соотношения СаО : Р2О5 = 0,79. В этом
случае весь фосфор будет находиться в продукте в цитратнораство-
римой форме (СаНРО4). Для получения же части фосфора в водо-
растворимой форме необходимо еще больше уменьшить соотно-
шение СаО : Р2О3 в реакционной массе, чтобы часть свободной
фосфорной кислоты превратилась при нейтрализации аммиаком
в фосфат аммония.
Применяют следующие способы уменьшения соотношения
СаО : Р2О5 в перерабатываемой системе 75’79'94: 1 ) вымораживание
нитрата кальция из вытяжки; 2) введение в систему фосфорной
кислоты (экстракционной или термической); 3) осаждение избыт-
ка кальция в виде CaSO4 серной кислотой или сульфатами аммо-
ния, натрия или калия; 4) осаждение избытка кальция в виде
СаСОз двуокисью углерода и аммиаком. Исследовалось также вы-
деление избытка кальция методом ионного обменаss.
Вымораживание нитрата кальция из вытяжки осуществляют на
заводах в Голландии, Франции, Норвегии 80> 8i> 96'*03.
При обработке фосфатов смесью азотной и серной кислот избы-
ток кальция связывается серной кислотой в сульфат, которьш
остается в удобрении в качестве балласта. Азотная кислота приме-
няется с концентрацией 42—55%, а серная 92—93%. Продукты
разложения обрабатывают газообразным аммиаком, в результате
чего получается пульпа, содержащая в растворе аммиачную се-
литру, а в осадке — дикальцпйфосфат и гипс. Этим методом про-
изводят удобрения во Франции, Англии, в США и в ФРГ 75’81’104’ *08.
При обработке фосфатов смесью кислот с содержанием 30% воды
образуется масса, которая в течение 10—30 мин затвердевает и
становится внешне похожей на суперфосфат. После двухдневного
хранения доля усвояемой Р2О5 достигает 98%. Затем продукт об-
рабатывают газообразным аммиаком; полученное удобрение (ни-
тросуперфосфат) содержит 6% N и 18% усвояемой Р2О5 82.
По карбонатному способу азотнокислотную вытяжку обрабаты-
вают вначале газообразным аммиаком (аммонизация), затем ам-
миаком и двуокисью углерода (аммонизация и карбонизация) и в
конце только двуокисью углерода 80’85’106’107. Недостатком этого
способа, более дешевого, чем другие, является невозможность
варьировать соотношение питательных веществ в готовом продук-
те; отношение N : P2Os может быть только 1 : 0,7—0,8 (стр. 1330).
Кроме того, Р2О5 содержится в удобрении не в водорастворимой,
а только в цитратнорастворимой форме, в виде преципитата. Все
это ограничивает возможность применения такого удобрения86.
1324
Гл. XXXV. Комплексные твердые сложные удобрения
Разлагая фосфаты смесью азотной и фосфорной кислот, можно
получить вытяжку с любым отношением СаО : Р2О5. По этому спо-
собу осуществляется производство удобрений в США, во Франции,
в ГДР и в Бельгии 96>97.
В нитрофосках, содержащих балласт — сульфат или карбонат
кальция — концентрация питательных веществ (N + P2O5+K2O)
составляет 35—36%, в нитрофосках же, не содержащих балласта,
получаемых с вымораживанием части нитрата кальция или с вве-
дением в процесс фосфорной кислоты, концентрация питательных
веществ достигает 45—50%.
Кроме упомянутых выше способов, нашедших применение в
'промышленности для производства различных видов удобрений,
представляют интерес следующие.
На швейцарском заводе Лонца79’108~из производят твердое азот-
иофосфорное удобрение типа суперфосфата по следующей схеме.
Фосфат смешивают с азотной кислотой концентрацией не ниже
70—80% без разделения продуктов реакции:
Са3(РО4)2 + 4HNO, = Са(Н2РО4)2 + 2Ca(NO3)2
После разложения в пульпу добавляют часть продукта, смесь
затвердевает, ее дробят и упаковывают в тару. По этой схеме по-
лучают двойное удобрение, содержащее 7,5% N (нитратного) и
14% Р2О5. Этим путем можно получать и тройное удобрение со-
става: 6% N, 12% Р2О5 и 8% К2О78.114.
Разработан 32 способ производства аналогичного продукта пу-
тем выпаривания азотнокислотной вытяжки с частичной регенера-
цией азотной кислоты (стр. 1346). Полученный продукт — нитро-
фос — представляет собой смесь кристаллов одноводного моно-
кальцийфосфата (~35%) и двухводного нитрата кальция (~65%)
с соотношением N : Р2О5, равным 1 : 2, и является водораствори-
мым удобрением.
В связи со все увеличивающимся количеством перерабатывае-
мого апатитового концентрата в удобрения, а нефелина в окись
-алюминия, представляет интерес проблема комплексной азотнокис-
лотной переработки апатито-нефелиновой руды без ее разделения и
обогащения.
Апатитовый и нефелиновый концентраты получают в результате
сложного трудоемкого процесса обогащения апатито-нефелиновой
руды, требующего значительных энергетических затрат, капиталь-
ных вложений и рабочей силы. Непосредственная азотнокислотная
переработка руды исключает эти процессы и позволяет комплексно
переработать сырье на удобрения, окись алюминия, технические
фосфаты и использовать фтор и другие ценные компоненты руды.
При разложении апатито-нефелиновой руды 40%-ной азотной кис-
лотой в раствор извлекается более 90% Р2О5 и А120з от количества
Переработка азотнокислотной вытяжки в сложные удобрения
1325
их в руде. При 50—60° процесс завершается в течение 2 ч. Затем
осуществляется ступенчатая нейтрализация полученной вытяжки,
например, аммиаком Н5.
ПЕРЕРАБОТКА АЗОТНОКИСЛОТНОЙ ВЫТЯЖКИ В СЛОЖНЫЕ
УДОБРЕНИЯ
На рис. 392 показана схема производства сложного удобрения
азотнокислотным разложением фосфатов. Разложение производят
азотной кислотой или смесью ее с серной или фосфорной кисло-
тами или другими реагентами. Процесс осуществляется непрерыв-
но в четырех реакторах 6. В первый реактор подается все количество
фосфата и вся норма азотной кислоты. Образовавшаяся пульпа
проходит последовательно все четыре реактора. В третий и чет-
вертый реакторы дозируется серная или фосфорная кислота в ко-
личестве примерно 60% от общей нормы (остальные 40% кислоты
подаются в аммонизаторы). Разложение фосфата идет при интен-
сивном перемешивании в течение 1 ч при температуре 45—60°, ко-
торая регулируется с помощью горячей воды, поступающей в ру-
башки реакторов. Степень разложения фосфатов достигает 98%.
Пульпа из четвертого реактора перетекает в реакторы аммониза-
ции 7.
Реактор (рис. 393) состоит из корпуса 1 U-образной формы,,
внутри которого помещена перегородка 8, разделяющая аппарат
как бы на две трубы, соединенных внизу калачом. В каждой трубе
имеется мешалка пропеллерного типа, делающая около 200 об/мин.
Снаружи реактор имеет водяную рубашку 3. Ввод газообразного
аммиака осуществляется по двум трубам в нижнюю часть реак-
тора. При вращении мешалок осуществляется циркуляция рас-
твора. Ввод и вывод раствора из реактора производится по тан-
генциально расположенным штуцерам. Реактор работает под ат-
мосферным давлением. Реакторы для разложения фосфатов имеют
аналогичную конструкцию, с той лишь разницей, что на вал наса-
живается одна лопасть (вместо двух) и электродвигатель устана-
вливается меньшей мощности.
Реакторы, применяемые для разложения апатита и для аммони-
зации пульпы, выполняются из нержавеющей хромоникелевой ста-
ли. При применении серной кислоты или сульфата аммония реак-
торы должны выполняться из нержавеющей стали, содержащей
2—3% молибдена. Производительность описанного реактора при
Диаметре каждой трубы 800—900 мм и высоте трубы ~ 2,5 м со-
ставляет 4—5 т азота в сутки 116'117.
Для аммонизации устанавливается от 10 до 15 последовательно
работающих реакторов. В первом аммонизаторе производят доме-
шивание пульпы, и в него реагентов не вводят. В последующие ам-
монизаторы подают оставшееся количество серной или фосфорной
Кислоты и газообразный аммиак.
Переработка азотнокислотной вытяжки в сложные удобрения
1327
При получении нитрофоски по карбонатной схеме в аммониза-
торы, вместо серной или фосфорной кислоты, вводят газообразную
двуокись углерода.
По i-i
Рис. 393. Реактор:
/ — корпус; 2 — крышка; 3 — охлаждающая рубашка; 4 — вал; 5 —ло-
пасть мешалки; 6 — электродвигатель; 7 —редуктор; 8 — перегородка.
Распределение вводимых реагентов по аммонизаторам произво-
дится в соответствии с заданным режимом по значению pH. Тем-
пературу в аммонизаторах поддерживают, в пределах 60—100°
1328
Гл. XXXV. Комплексные твердые сложные удобрения
с помощью охлаждающей воды, поступающей в рубашки. Повышение
температуры выше указанного предела не рекомендуется, так как
это связано с увеличением потерь аммиака и выделением в газовую
фазу фтористых соединений и окислов азота117. В аммонизаторах,
за счет выделившегося реакционного тепла испаряется 15—20% воды.
Газы из реакторов 6 перед выбросом в атмосферу промывают
водой, а газы из реакторов 7 — азотной кислотой. Совместное ула-
вливание газов производить не рекомендуется во избежание обра-
зования нитрита аммония 119’ 12°. В последний реактор-аммонизатор
вводят третий питательный элемент калий, обычно в виде КС1. При
этом протекает обменная реакция:
КС1 + NH4NO3 = KNO3 + NH4C1
Количество прореагировавшего по этой реакции КО зависит от
-продолжительности смешения. Обычно степень конверсии КС1 ко-
леблется в пределах 70—90%. После смешения с КС1 в пульпе
«остается 15—20% воды. Пульпа подвергается гранулированию и
сушке. Для снижения влажности и улучшения условий грануляции
в пульпу добавляют ретур (мелкую фракцию готового продукта).
В зависимости от метода грануляции и сушки количество добав-
ляемого ретура различно. Еще недавно процесс грануляции осуще-
ствляли в грануляторе шнекового типа 121 и сушку в барабанной
сушилке. Применение этих аппаратов требовало большого количе-
ства ретура (5—6-кратного по отношению к готовому продукту)122,
что было связано с необходимостью установки мощных транспорт-
ных механизмов, с повышенным расходом электроэнергии и боль-
шим пылевыделением.
В современных производственных схемах процессы грануляции
и сушки проводят в одном аппарате — сферодайзере /5 32-123~127.
Совмещение этих процессов сокращает количество ретура до 1 —
,2-кратного. При этом также упрощается внутрицеховой транспорт
и уменьшается пылевыделение. Сферодайзер представляет собой
барабан, например, с диаметром 4—5 м и длиной 12—30 м, внутри
которого имеется специальная насадка. Она обеспечивает пересы-
пание материала и создание равномерной завесы из него по всему
«сечению и всей длине барабана, что улучшает контакт материала
,с дымовыми газами и интенсифицирует процесс. Пульпа подается
в сферодайзер форсунками в распыленном состоянии с помощью
сжатого воздуха давлением 7—8 ат непосредственно на завесу из
падающих мелких частиц. Распыленная пульпа обвалакивает их,
И на их поверхности происходит кристаллизация солей из пульпы;
при этом образуются гранулы. Сушка гранул осуществляется ды-
мовыми газами, получаемыми при сжигании газообразного топ-
лива в топке 16. Температура газов на входе в сферодайзер 220—
•250°, на выходе 70—90°. Влагосъем составляет 18—20 кг/(м3-ч).
Конечная влажность продукта 0,5—1%.
Переработка азотнокислотной вытяжки в сложные удобрения
132»
Высушенные гранулы элеватором 17 подаются на 2-ситный гро-
хот 18 для рассева на три фракции. Самая мелкая фракция с раз-
мером частиц меньше 2 мм возвращается в сферодайзер в каче-
стве ретура. Крупная фракция с размером частиц более 4 мм по-
ступает в дробилку 19 и после измельчения также направляется в
ретур. Фракция с размером гранул 2—4 мм является продуктом,
который вначале охлаждается воздухом в барабане 20 до 35—45°,
затем поступает в аппарат-кондиционер 22, где он омасливается и*
опудривается, а затем направляется в склад.
Карбонатный способ
Этот способ заключается в нейтрализации азотнокислотной вы-
тяжки вначале аммиаком, а затем аммиаком и двуокисью угле-
рода ,28' 129:
5Ca(NO3\ + ЗН3РО4 + 3NH3 = 1,5Са(Н2РО4)2 + 3NH4NO3 + 3,5Ca(NO3)2
1,5Са(Н2РО4)2 + 3,5Ca(NO3)2 + 3NH3 = ЗСаНРО4 + 3NH4NO3 + 2Ca(NO3)2
2Ca(NO3)2 + 4NH3 + 2CO2 + 2H2O = 2CaCO3 + 4NH4NO3
5Ca(NO3)2 + 3H3PO4 + 10NH3 + 2CO2 + 2H2O = 3CaHPO4 + 10\H4NO3 + 2CaCO3
Разложение апатитового концентрата производят только азот-:
ной кислотой. В четвертый реактор вводят стабилизирующую до-
бавку 130 с целью предотвращения ретроградации Р2О5, при нейтра-
лизации вытяжки до pH = 8,5—9, по реакции:
2СаНРО4 + Ca(NO3)2 + 2NH3 = Са3(РО4)2 + 2NH4NO3
Без стабилизатора содержание доли усвояемой формы Р?СК рез-
ко падает в интервале pH=4—5 и при рН = 8 снижается с 90 до
32—35%. В качестве стабилизирующей добавки применяют соеди-
нения магния (из расчета 9 кг магния на 100 кг Р2О5) 130 — доло-
мит, каустический магнезит, каинит или сульфат магния. При пе-
реработке фосфоритов Капятау, содержащих магний, вводить,
стабилизатор не требуется. Образовавшийся раствор подвергают
нейтрализации. В первый реактор-нейтрализатор подают только
аммиак, во. все последующие, кроме последнего, аммиак и двуокись
Углерода. В последний реактор подают только двуокись углерода.
Основным условием нормальной работы реакторов-нейтрализа-
торов является равномерное поступление аммиака и двуокиси угле-
рода в строго выдержанном соотношении и при соблюдении опре-
деленной величины pH по реакторам, которая должна иметь при-
близительно следующие значения:
реакторэв-нейтрализа- 1 2 3 4 5 6 7 8
торов (по ходу раствора)
Значение pH........... 1-2 2,5-4,5 5-6 7,5 7.5 7,5-8 7,5-8 7,5-®
1330
Гл. XXXV. Комплексные твердые сложные удобрения
ТАБЛИЦА 96
Составы удобрений (в %),
получаемых по карбонатной
схеме
i Нитро- фоска Нитро- фос
СаНРО4 17,52 15,4
MgHPO4 3,08 3,83
NH4NO3 28,15 52,91
СаСО3 12,19 17,98
KNO3 21,13 —
NH4CI Нерастворимые при- 11,15 —
меси 3,40 9,88
КС1 1,78
Влага . 1,00 1,00
По карбонатной
В первой стадии аммонизации происходит образование моно-!
кальцийфосфата, который выделяется в твердую фазу. Это привоЛ
дит к загустеванию пульпы, преимущественно в интервале значеЯ
ний pH от 2 до 2,7. Чем большД
величина pH раствора, тем обильЯ
нее выделяется монокальцийфос-И
фат в осадок и тем гуще стано-"
вится пульпа. При значительных
pH пульпа загустевает настолько
сильно, что прекращается работа
мешалки и наступает полное рас-
стройство технологического режи-
ма. Приведенное выше распреде-
ление значений pH по нейтрали-
заторам обеспечивает получение
подвижной пульпы при условии
равномерной подачи реагентов
и интенсивного перемешивания, j
Температуру в нейтрализато-1
рах поддерживают в пределах!
60—80°. I
схеме может быть получено двойное (нитро-1
фос) или тройное (нитрофоска) сложное удобрение (табл. 96—98).
ТАБЛИЦА 97
Количество и соотношение питательных веществ
в сложных удобрениях, полученных по карбонатной
схеме
Нитрофоска Нитрофос
Общее содержание питательных
веществ, % ................
в том числе N . ..........
Р2О5 .........
к2о...........
Соотношение питательных ве-
ществ N : Р2О6 : К2О
(по весу) ................
Форма Р2О5 ...................
37,74
15,98
10,88
10,88
28,5
18,2
10,3
1,76:1
1,47:1:1 ______
Цитратнораст-
воримая
Приведенные данные по составу удобрений и расходные коэф-Я
фициенты (табл. 98) получены из расчета, в котором принята кон-"
центрация исходной азотной кислоты 47% и фосфатное сырье: апа-
титовый концентрат для нитрофоски и фосфориты Каратау для
нитрофоса. -
Переработка азотнокислотной вытяжки в сложные удобрения 1331
ТАБЛИЦА 98
Расходные коэффициенты иа производство
сложных удобрений по карбонатной схеме
Расход на 1 т нитрофоски Расход на 1 т нитрофоса
Фосфаты, т 0,285 0,393
Азотная кислота (100%), т . . . 0,366 0,43
Аммиак (100%), кг 97,0 111,5
Углекислый газ (100%), кг . . . 73,2 103
или .и3 37,2 52,5
Хлористый калий (95%), кг . . . 183,5 —
Магнезит (83% MgO), кг ... . 12,25 —
Условное топливо, кг . 64,2 60
Электроэнергия, кет • ч . ... . 96 67
Вода, м3 6 6
Сульфатные способы
Отличие этих способов от карбонатного состоит в том, что с по-
мощью сульфат-иона можно связать большее количество кальция,
так что оставшегося кальция не хватит для образования дикаль-
цийфосфата, и часть РгО5 останется в водорастворимой форме.
С карбонатом кальция эта форма несовместима из-за малого зна-
чения pH.
При введении в реакционную массу серной кислоты процесс мо-
жет быть представлен следующими суммарными реакциями:
2Ca6(PO4)3F + I2HNO3 + 4H2SO4 = 6Н3РО4 + 6Ca(NO3)2 + 4CaSO4 + 2HF
6Н3РО4 + 6Ca(NO3)2 + 4CaSO4 + 2HF + 13NH3 =
= 12NH4NO3 + 5CaHPO4 + NH4H2PO4 + 4CaSO4 + CaF2
2CaT(PO4)3F + 12HNO3 + 4H2SO4 + 13NH3 =
= 12NH4NO3 + NH4H2PO4 + 5CaHPO4 + 4CaSO4 + CaF2
В этом случае фосфат разлагается смесью азотной и серной
кислот или сначала азотной кислотой с последующей добавкой сер-
ной кислоты.
При одновременной подаче кислот для разложения апатита на
96% требуется 2 ч, а при последовательной — на 98% требуется
1 и. Полученную пульпу аммонизируют, смешивают, с хлористым
калием и с ретуром, гранулируют, сушат и охлаждают. Для полу-
чения готового продукта однородного гранулометрического состава
его рассеивают и дробят 131~133.
Большое значение имеет консистенция пульпы. Благодаря обиль-
ному осаждению CaSO4 в виде дигидрата, связывается часть воды,
что приводит к образованию малоподвижной пульпы, и процесс
1332
Гл. XXXV. Комплексные твердые сложные удобрения
затрудняется. Для того чтобы пульпа была разжиженной и подвиж-
ной, необходимо добавлять воду или к кислотам, поступающим на
разложение, или в стадии аммонизации. При содержании воды
53—40% образуется полугидрат сульфата кальция и пульпа обла-
дает достаточной подвижностью. Однако при дальнейшем ведении
процесса полугидрат очень быстро превращается в дигидрат, что
приводит к значительному увеличению вязкости пульпы9.
В процессе аммонизации пульпы распределение аммиака и сер-
ной кислоты по реакторам производят с расчетом, чтобы на вы-
ходе из последнего реактора величина pH пульпы не превышала
3,2 во избежание ретроградации Р2О5.
Азотно-сернокислотный способ позволяет получать удобрения с
различным соотношением питательных веществ 134~139.
В табл. 99 и 100 приводится состав продуктов (нитрофоски и
нитрофоса), получаемых этим способом в зависимости от соотно-
шения питательных веществ и содержания водной формы Р2О5. Это
расчетные данные для концентрации исходных кислот: 47% HNOj
И 92,5% H2SO4; фосфатное сырье: апатитовый концентрат для ни-
трофоски и фосфориты Каратау для нитрофоса.
таблица 99-
Содержание питательных веществ в сложных удобрениях в зависимости
от соотношения питательных веществ и содержания водорастворимой
формы Р2О5
Нитрофос (из фосфоритов Каратау) Нитрофоска (из апатита)
N : Р2О5 : К2О 1:1:0 1: 1,5:0 1:2:0 1:1:1 1: 1Л: 1,5 1:2:1
Содержание водо- растворимой формы Р2О5, % Содержание пи- 50 50 70 50 50 70
тательных ве- ществ, % . . . 22,9 21,40 20,20 35,4 35,6 31,62
На производство 1 т нитрофоски (из апатитового концентрата)
с весовым отношением N : Р2О5: КгО, равным 1:1:1, содержащей
50% Р2О5 от общего количества в водорастворимой форме, тре-
буется 0,309 т апатитового концентрата, 0,223 т HNO3 (100%)»
0,192 т H2SO4 (100%), 86,6 кг аммиака (100%), 0,1985 т хлори-
стого калия (95% КС1), 91 квт-ч электроэнергии, 60 кг условного
топлива, 10 м3 воды. На производство 1 т нитрофоса (из фосфо-
ритов Каратау) с весовым отношением N : Р2О5, равным 1:1, со-
держащего 50% Р2О5 в водорастворимой форме, требуется 0,44 т
фосфорита, 0,228 т HNO3 (100%), 0,278 т H2SO4 (100%), 78 кг
аммиака (100%), 60 квт-ч электроэнергии, 60 кг условного топ-
лива, 10 м3 воды.
Переработка азотнокислотной вытяжки в сложные удобрения
1333
ТАБЛИЦА 10Q
Солевой состав (в %) нитрофоса из фосфорита
Каратау (при N : Р2О5 = 1 : 1) и нитрофоски
из апатитового концентрата
(при N : Р2О6: К2О =1:1:1)
Нитрофос Нитрофоска
СаНРО4 6,90 11,3
MgHPo; 4,20
(NH4)2HPO4 10,60 10,93
CaSO4 • 0,5Н2О 40,62 28,22
NH4NO3 25,80 9,08
KNO3 — 22,70
NH4C1 —- 12,10
KC1 . . — 1,86
Нерастворимые примеси . . 10,88 2,81
Вода 1,00 1,00
В СССР производство нитрофоски из апатитового концентрата
по азотно-сернокислотной схеме осуществляется в 15 реакторах, из
которых первые четыре используются для разложения фосфата,
остальные для аммонизации. Грануляция и сушка продукта произ-
водится в сферодайзере. Требования к качеству нитрофоски при-
ведены в табл. 101.
ТАБЛИЦА 101
Требования к качеству нитрофоски (ГОСТ 11365—65)
(Содержание компонентов в вес. %)
Марка А Марка Б Марка В
Азота .(N) , 16,0-17,0 12,5 —13,5 11,0-12,0
Усвояемого Р2О5 Водорастворимого Р2О5 (% от уснояе- 16,0-17,0 8,5-9,5 10,0-11,0
мого), не менее . . . . 55 55 55
К2О 16,0-17,0 12,5-13,5 11,0-12,0
Влаги, не более . . . Гранулометрический состав (%): 2,0 2,0 2,0
фракция 2—4 мм, не менее .... 93 80 80
» 4 — 6 мм, не более .... 2,0 10,0 10,0
» меньше 2 мм, не более . . 5,0 10,0 10,0
Вм сто серной кислоты для осаждения кальция используют так-
же сульфат или бисульфат аммония6,140. (Возможно применение
сУльфатов и с другими катионами, например, К, Na.) Предпочти-
тельнее введение сульфата аммония в твердом виде, так как
ПРИ использовании его в виде раствора в систему вводится
1334
Гл. XXXV. Комплексные твердые сложные удобрения
дополнительное количество воды, которое значительно увеличивает
ретур (до 12—15-кратного). Количество добавляемого сульфата ам-
мония определяют, исходя из заданного содержания водораствори-
мой формы Р2О5 в готовом продукте. С сульфатом аммония вводится
часть аммиака (около 50% от требуемого количества)Остальной
аммиак вводится в виде газообразного. Аммонизацию ведут до
pH = 3,2—3,3 при 70—80°. Следует избегать значений pH в интер-
вале 2,1—2,7, при которых пульпа загустевает. Фирма TVA («Теп-
nessy Valley Authority», США) получает этим способом нитрофос-
фат с отношением N : Р2О5 = 2:1, содержащий 90% Р2О5 в водо-
растворимой форме.
Голландская фирма «Стамикарбон» осуществляет процесс с от-
делением гипса перед нейтрализацией азотнокислотной вытяжки
аммиаком и с конверсией этого гипса в сульфат аммония (стр. 1250),
который возвращают в производство 141- 142. Этот циркуляционный
метод позволяет обойтись без серной кислоты при разложении фос-
фата и без вводимых извне растворимых сульфатов. Здесь также
получается продукт, в котором отношение N : Р2О5= 1,67 : 1, т. е.
больше, чем желательное. Можно получить продукт с отношением
N : Р2О5 = 1 : 1, уменьшая количество (NH4)2SO4, вводимого на ста-
дии разложения фосфата, но при этом уменьшается доля водорас-
творимого Р2О5.
Фирма «Стамикарбон» при переработке марокканского фосфо-
рита использует сульфат аммония, являющийся отходом производ-
ства капролактама; получаемый нитрофосфат имеет состав (N—
—Р2О5—К2О) от 22,5—16—0 и до 26,5—14—О143. О разложении
апатитового концентрата бисульфатом аммония см. 144“146.
Рациональнее осуществлять производство сульфатной нитрофо-
ски с уменьшенным на 15—40% расходом азотной кислоты 32-147,
что помимо экономии кислоты и аммиака позволяет получать про-
дукт с более благоприятным отношением N : Р2О5— от 1,3:1 до
0,9 : 1. С этой целью сульфат или бисульфат аммония вводят в ре-
акционную массу в процессе разложения апатита азотной кисло-
той, а не после завершения этого процесса. (Вместо сульфата или
бисульфата можно вводить и серную кислоту нейтрализуемую за-
тем аммиаком.) В этом случае, образующийся при разложении
фосфата азотной кислотой нитрат кальция сразу же взаимодей-
ствует с сульфат-ионом. Кальций с самого начала процесса выво-
дится из сферы реакции в виде осадка сульфата кальция, и хими-
ческая активность жидкой фазы все время остается высокой. После
того как азотная кислота израсходуется, дальнейшее разложе-
ние фосфата идет за счет ненасыщенной соединениями кальция
фосфорной кислоты. Находящийся в растворе нитрат аммония (об-
разовавшийся в результате взаимодействия сульфата аммония с
нитратом кальция) не тормозит, в отличие от нитрата кальция, раз-
ложение апатита слабой фосфорной кислотой. В соответствии.с
Переработка азотнокислотной вытяжки в сложные удобрения
1335
этим процесс разложения апатита может быть представлен, напри-
мер, при применении азотной кислоты в количестве 90% от стехио-
метрического, следующим уравнением:
Ca5(PO4)3F + 9HNO3 + 3(NH4)2SO4 = 3CaSO4 + 6NH4NO3 +
+ 2H3POi + l,5Ca(NO3)2 +0,5Ca(H2PO4)2 + HF
При последующей нейтрализации пульпы аммиаком нейтрали-
зуются свободная фосфорная кислота и монокальцийфосфат, но
расход аммиака на нейтрализацию в этом случае соответственно
сокращается.
Степень разложения апатита, характеризуемая отношением в
пульпе Р2О5 усв : Р2О5 общ, при 90%-ном расходе азотной кислоты
от стехиометрического количества достигает 97%, т. е. остается
такой же, как и при применении 105—110%-ного количества азот-
ной кислоты. Это происходит за счет использования в процессе раз-
ложения химической энергии фосфорной кислоты. В то же время
снижение нормы азотной кислоты до 90% от стехиометрической
приводит к уменьшению расхода азотной кислоты и аммиака на
15—20%. После нейтрализации пульпы аммиаком до рН = 2,8—3,
добавки хлористого калия и сушки продукт содержит: 12,25% N,
9,87% Р205 общ, 9,57% Р2О5усв, 5,28% Р2О5 водораств, 14% К2О. От-
ношение N : Р2О5 = 1,25 : 1.
При применении азотной кислоты в количестве 80% от стехио-
метрического степень разложения апатита в пульпе составляет
~92%, а продукт содержит: 12,13% N, 10,5% Р2О5 ОбЩ, 9,24%
Р2Обусв, 5,о% Р2О3 волораств, 14,3% К2о. Отношение N : Р2О5= 1,15:1.
При уменьшении количества азотной кислоты уменьшается ко-
личество воды в пульпе, что позволяет уменьшить количество ре-
тура и облегчить гранулирование и сушку продукта.
Еще более рационален режим с применением всего 60% азот-
ной кислоты от стехиометрического количества при разложении
апатита смесью азотной и серной кислот. При этом серная кис-
лота используется не только для осаждения кальция, но и для раз-
ложения апатита. Общее количество кислот составляет около 90%
от стехиометрического. Степень разложения апатита в аммонизи-
рованной пульпе достигает 96,5%. Расход азотной кислоты в этом
случае сокращается на 40—42%, а расход аммиака — на 25—30%.
Готовый продукт содержит 10,6% N, 11,8% Р2О5 Общ, 10,4% Р2О5уСВ,
5,5% Р2О5 водораств, 13,6% К2о. Отношение N : Р2О5 = 0,9: 1.
При разложении апатита смесью азотной и серной кислот в
присутствии некоторого количества сульфата аммония или при од-
новременной частичной нейтрализации серной кислоты аммиаком
Из реакционной массы не выделяются окислы азота и в пульпе не
образуются нитриты. Если же апатит разлагать сначала одной
азотной кислотой, а затем добавлять серную, то не удается
18 М. Е. Позии
1336
Гл. XXXV. Комплексные твердые сложные удобрения
избежать образования небольшого количества нитритов и выделе|
ния окислов азота в газовую фазу.
Независимо от количества применяемой азотной кислоты мож-
но получать продукт, содержащий большую часть фосфора в виде
водорастворимых солей. Это достигается менее глубокой нейтрали-
зацией пульпы аммиаком — до pH = 1,8—2. вместо 2,8—3. Готовый
продукт в этом случае содержит больше 80% фосфора в водорас-
творимой форме. При этом на стадии грануляции и сушки в газо-
вую фазу выделяется большее количество фтористых соединений,
так как перерабатывается более кислая пульпа.
Вместо сульфата аммония может быть применен сульфат ка-
лия. В этом случае получается бесхлорная нитрофоска, которая мо-
жет быть использована с большой эффективностью для ряда сель-
скохозяйственных культур, чувствительных к хлору (виноград, та-
бак, цитрусовые и др.) 9> 148'153.
Определены оптимальные условия 154-156 разложения фосфори-
тов Каратау неполной нормой азотной кислоты в смеси с сульфа-
том калия. Изучена возможность конверсии Са(МОз)2, полученного
в условиях азотнокислотной переработки фосфатов газообразными
SO2 и NH3 в NH4NO3 и CaSO4 157' 158.
Фосфорнокислотный способ
Производство сложных удобрений по этому способу осуще-
ствляется по схемам, аналогичным рассмотренным выше. В раствор,
полученный после разложения фосфатов азотной кислотой, добав-
ляют фосфорную кислоту (термическую или экстракционную) или
производят разложение смесью этих кислот, причем процесс уско-
ряется — его продолжительность сокращается до 1 ч (при 35—45°).
Изменяя количество вводимой фосфорной кислоты, можно уста-
навливать любое соотношение СаО : Р2О5 в реакционной массе159.
При дальнейшей нейтрализации раствора аммиаком образуют-
ся дикальцийфосфат, фосфат и нитрат аммония, соотношение ме-
жду которыми зависит от относительного количества введенной в
процесс фосфорной кислоты, например:
l,5Ca(NO3)2 +ЗН3РО4 + 4,5NH3= 1,5СаНРО4 + 1,5NH4H2PO4 + 3NH4NO3
Водорастворимая форма Р2О5 получается за счет образования
фосфата аммония. Для выравнивания соотношения N : Р2О5 вводят
при разложении фосфатов избыток азотной кислоты, который с ам-
миаком образует нитрат аммония.
Обработка азотно-фосфорнокислотной вытяжки аммиаком про-
изводится последовательно в четырех нейтрализаторах. Распреде-
ление аммиака по нейтрализаторам следующее:
Нейтрализатор............................. 1 2
Количество вводимого аммиака, % . . . . 57 26
pH........................................ 1 1,8
3
13
2,4
4
4
3,7-4
Переработка азотнокислотной вытяжки в сложные удобрения 1337
При введении избытка аммиака ухудшается качество продукта
(снижается содержание усвояемой Р2О5). При правильной дози-
ровке продукт содержит до 98% Р2О5 в усвояемой форме. Темпе-
ратуру в процессе аммонизации поддерживают около 90—100°.
Пульпа, вытекающая из последнего реактора, содержит 28—32%
влаги. Ее подвергают сушке и грануляции. При осуществлении
этих операций в сферодайзере количество ретура уменьшается до
0,3—0,5 вес. ч. на 1 вес. ч. готового продукта. Такое количество
ретура необходимо и при применении для сушки пульпы распыли-
тельных сушилок с последующей грануляцией и досушкой гранул
в барабанных сушилках или при использовании комбинированных
аппаратов, в которых совмещены распылительная сушка и сушка
в кипящем слое.
По этому способу может быть получено двойное или тройное
удобрение с любым соотношением питательных веществ и часть
Р2О5 в удобрении находится в водорастворимой форме. При полу-
чении из апатитового концентрата, 55%-ной азотной и 32%-ной
фосфорной кислот нитрофоски с отношением N : Р2О5: К2О =
= 1:1:1; она содержит: 15,8% СаНРО4, 17,12% NH4NO3, 13%
NH4H2PO4, 31,89% KNO3, 16,78% NH4C1, 2,62% КС1, 1,46% нерас-
творимых примесей и 1 % воды; содержание питательных веществ:
16,5% N, 16,5% Р2О5, 16,5% К2О— всего 49,5%.
Для получения 1 т продукта указанного состава необходимо
израсходовать: 0,13 т апатитового концентрата, 0,34 т азотной
кислоты (100%), 0,16 т фосфорной кислоты (100%), 0,11 т ам-
миака (100%), 0,28 т хлористого калия (95%), 0,66 т пара, 50 м3
воды, 70 квт-ч электроэнергии и 25 кг условного топлива.
Универсальная установка фирмы РЕС
Универсальная промышленная установка французского кон-
церна Potasse et engrais chimique (PEC) предназначена для про-
изводства сложных удобрений любого состава и с различным соот-
ношением питательных веществ. Установка оборудована 20-ю реакто-
рами U-образной формы, сферодайзером, размольной и дробильной
аппаратурой, охлаждающим барабаном и кондиционером. Раз-
ложение фосфатов идет в первых четырех реакторах. Фосфат по-
дается в первые два реактора. В эти же оба реактора подается и
азотная кислота. В третий и четвертый реакторы добавляется ста-
билизирующая добавка — каустический магнезит. Температура
в процессе разложения около 80°. Степень разложения фосфатов
98—99%. Серная кислота или сульфат аммония подается в пятый
и шестой реакторы. Фосфорная кислота подается с третьего по пят-
надцатый включительно, кроме пятого и шестого. Хлористый калий
подается в реакторы с семнадцатого по двадцатый и аммиак — во
все реакторы, начиная с пятого. Аммонизация проводится при 105°.
Значение pH по реакторам регулируется в зависимости от осуще-
18'
1338
Гл. XXXV. Комплексные твердые сложные удобрения
ствляемого варианта в пределах от 1 до 5,2. Грануляция и сушка
в сферодайзере идет с помощью дымовых газов с температурой: на
входе 220° и на выходе 100°.
Готовый продукт (зерна 2—4 мм) содержит 50% РаО5 в водо-
растворимой форме при соотношениях N : Р2О5: К2О, равных
11,2:11,2:11,2; 14:8:14; 18:10:18; 16:16:16. Мощность уста-
новки меняется в зависимости от выпускаемой продукции. Приво-
дятся следующие энергетические расходы: электроэнергия от 44
до 60 квт-ч/т-, сжатый воздух (7—8 ат) на распыление пульпы
50 мъ!т (при нормальных условиях); природный газ (8500 ккал)м3)
от 50 до 77 Л13/т. Этот метод многократно исследовался и видо-
изменялся !61—173 I
Нитроаммофос и нитроаммофоска 1
При получении комплексных удобрений — нитрофосов и нит-
рофосок — азотнокислотным разложением фосфатов азотная кис-
лота является источником не только азота (наряду с аммиаком),
но и химической энергии, используемой для извлечения из природ-
ного фосфата фосфорной кислоты, превращаемой затем в фосфат-
ные компоненты сложного удобрения. Такое комбинированное
использование свойств азотной кислоты экономически весьма вы-
годно. Однако существенным недостатком этого способа является
необходимость перерабатывать азотнокислотную вытяжку, содер-
жащую наряду с фосфорной кислотой большое количество нитрата
кальция. Это вынуждает либо значительно усложнить производство
для удаления избытка кальция из системы, либо выпускать удоб-
рения с пониженным содержанием питательных веществ из-за при-
сутствия большого количества балласта (карбоната или сульфата
кальция). Кроме того, присутствие в вытяжке кальция не позволяет
получить, по крайней мере простыми путями, удобрение, в котором
фосфор был бы полностью в водорастворимой форме.
Всего этого можно избежать, если получать комплексные удоб-
рения, нейтрализуя аммиаком смесь азотной и фосфорной кислот,
с последующим гранулированием образующейся пульпы после до-
бавки к ней солей калия или без них. Получаемые таким путем
комплексные удобрения называют нитроаммофосами (N + Р) и
иитроаммофосками (N + Р + К).
Нитроаммофосы состоят из продуктов нейтрализации азотной
и фосфорной кислот аммиаком, т. е. из нитрата и фосфатов аммо-
ния, а нитроаммофоски, кроме того, и из продуктов взаимодействия
с этими солями добавленных к ним солей калия (КС1 или KaSCX).
Все эти соединения хорошо растворимы в воде, поэтому нитро-
аммофосы и нитроаммофоски являются водорастворимыми удобре-
ниями, и вся Р2О5 содержится в них также в водорастворимой
форме. Это значительное преимущество таких удобрений. Кроме
того, они совсем не содержат балласта и являются высококонцен-
Переработка азотнокислотной вытяжки в сложные удобрения
1339
трированными безбалластными удобрениями. Содержание пита-
тельных веществ в них может превышать 55%.
ЛТП совместно с Новомосковским химкомбинатом разработал
способ получения концентрированных сложных удобрений, содер-
жащих 45—50% питательных веществ, без использования фосфор-
ной кислоты извне. Сущность способа заключается в азотносерно-
кислотном разложении апатита или фосфоритов в присутствии обо-
ротного раствора; при этом в твердую фазу выделяются крупные,
хорошо фильтрующие и легко отделяемые от жидкой фазы кри-
сталлы полугидрата сульфата кальция. Полученный раствор, пред-
ставляющий собой концентрированную смесь азотной и фосфорной
кислот, перерабатывается в нитроаммофоску путем нейтрализации
аммиаком, смешения с КС1 и последующей сушки продукта.
Этим способом можно получать смесь 47—50 %-ной азотной
и 50—65%-ной по Р2О5 фосфорной кислот с молярным отноше-
нием HNO3 : Н3РО4 в растворе, равном от 0,5 : 1 до 3:1. Из этих
растворов могут быть получены концентрированные сложные удоб-
рения с отношением N : Р2О5, равным от 0,25 : 1 до 2 : 1.
При переработке этим способом апатитового концентрата раз-
мер выделяющихся кристаллов CaSO4-0,5H2O достигает 150 мк,
а при переработке каратаускихи кингисеппских флотоконцентратов
120 мк. Съем отмытого осадка с фильтра в пересчете на сухой
достигает в первом случае 5, во втором — 2 т/(м2-ч). Съем влажного
осадка 10—20 т/(м2-ч). После двухкратной промывки горячей водой
осадок содержит меньше 0,2% HNO3 и меньше 0,3% общего РгО5.
Получение концентрированной смеси азотной и фосфорной
кислот этим методом возможно и из низкокачественных (недообо-
гащенных или плохо обогащаемых) фосфоритов, например, содер-
жащих 20—15% Р2О5 и меньше. (Однако переработка некоторых
видов фосфоритов, например, Верхнекамского и Егорьевского
месторождений сопровождается выделением, окислов азота).
В связи с разработкой этого способа была изучена раствори-
мость сульфата кальция в кислотах 160. В азотной кислоте в пре-
делах 5—60% HNO3 при 20—80° наблюдается максимум раствори-
мости при концентрации —20% HNO3. В растворе, содержащем
30% HNO3 + Н3РО4, с увеличением относительной доли HNO3 рас-
творимость сульфата кальция монотонно возрастает, а для общей
концентрации кислот 50% характерен максимум растворимости
приблизительно при равном весовом содержании HNO3 и Н3РО4.
Получение нитрофоски с вымораживанием части
нитрата кальция
При охлаждении раствора, полученного при разложении фос-
фатов азотной кислотой, кристаллизуется четырехводный нитрат
кальция. При избытке азотной кислоты нитрат кальция выделяется
1340
Гл. XXXV. Комплексные твердые сложные удобрения
в виде двойной соли четырехводного кристаллогидрата с азотной
кислотой, отвечающей формуле 3Ca(NO3)2- 12H2O-HNO3. Наиболь-
шее влияние на степень осаждения нитрата кальция оказывает
концентрация исходной азотной кислоты (рис. 394). Чем она
больше, тем больше выделяется нитрата кальция. Следовательно,
используя для разложения фосфата азотную кислоту более высо-
кой концентрации, можно выделить из раствора то же количество
Температура, ’С Температура,°C
Рис. 394. Степень выделения
СаО в виде нитрата кальция
в зависимости от концентра-
ции исходной азотной кислоты
и температуры охлаждения
азотнокислотной вытяжки.
Рис. 395. Влияние нормы азотной кислоты
на степень выделения СаО в виде
Ca(NO3)2 • 4Н2О.
Концентрация кислоты: у— 52%; 2 — 50%;
3 — 47% HNO3. Норма от стехиометрического ко-
личества: а — 1,3, б — 1,5.
Концентрация азотной кислоты:
1 - 55°/0; 2 - 52°/о: 3 - 500/о;
4 - 470/0 HNOg.
иия СаО в виде нитрата кальция показано на рис. 395. Увеличение
нормы азотной кислоты сверх стехиометрической снижает степень
выделения СаО, в основном за счет разбавления раствора. (О выде-
лении СаО и MgO из растворов, полученных при азотнокислотном
разложении фосфоритов Каратау, см. 43’75’176). На рис. 396 при-
водятся данные о степени выделения СаО в виде твердого нитрата
кальция Ca(NO3)2 • 4Н2О (в % от исходного) в зависимости от
концентрации исходной азотной кислоты и температуры охлажден-
ного раствора (справа) и о содержании водорастворимой Р2О5 в
продукте в виде фосфата аммония при соответствующем количе-
стве выделенной СаО (слева). На рис. 397 приведено отношение
СаО к Р2О5 в растворе, получаемое в зависимости от концентра-
ции исходной кислоты и температуры охлаждения 1Т5-Л77_
Производство нитрофоски с вымораживанием избытка нитрата
кальция осуществляют следующим образом (рис. 398) 178-180. Рас-
Переработка азотнокислотной вытяжки в сложные удобрения 1341
твор, полученный при разложении фосфатов азотной кислотой и от-
деленный от нерастворившегося остатка (шлама), поступает
в сборник 1, из которого насосом 2 перекачивается в систему кри-
сталлизаторов 5. Предварительно раствор охлаждается водой с 50
до 25—30° в холодильниках 3 и 4. В кристаллизаторах 5 при охла-
ждении до —5, —10° рассолом, циркулирующим в змеевиках, из
раствора вымораживается нитрат кальция. (Предложен метод
охлаждения путем непосредственного соприкосновения раствора
Рнс. 396. Зависимость степени выделения СаО н со-
держания водорастворимой Р2О5 в продукте от кон-
центрации азотной кислоты и температуры ра-
створа.
с инертным хладагентом — бензином. Процесс осуществляется сле-
дующим образом. Бензин, предварительно охлажденный жидким
аммиаком, распыляется в растворе в виде мелких капель. Капли,
Поднимаясь вверх, охлаждают раствор и своим потоком поддержи-
вают во взвешенном состоянии образовавшиеся мелкие кристаллы.
По мере роста крупные кристаллы падают на дно кристаллизатора,
а бензин собирается в верхней части кристаллизатора, откуда
Вновь возвращается в цикл 181.)
Очень важно в процессе кристаллизации получить крупные кри-
сталлы нитрата кальция, которые затем более полно могут быть
1342
Гл. XXXV. Комплексные твердые сложные удобрения
Г вмпература, °C
Рис. 397. Отношение СаО; Р2О5
в растворе в зависимости от кон-
центрации исходной азотной
кислоты и температуры охлажде-
ния.
Концентрация азотной кислоты: I — 55%;
2 — 52%; 3 — 50%; 4~470/0 HNO3.
отделены от раствора. Поэтому охлаждение раствора производят
медленно, с постепенным понижением температуры до заданной.
Зависимость величины получаемых кристаллов Са (NO3)2-4H2O от
скорости охлаждения показана в табл. 102. Из приведенных дан-
ных видно, что наиболее крупные, хорошо фильтрующие кристаллы
Са (NO3)2 • 4Н2О получаются при медленном понижении темпера-
туры и общем времени кристаллизации 5—6 ч. Это время кристал-
лизации должно быть выдержано независимо от температуры кри-
сталлизации. При быстром вымора-
живании образующиеся мелкие кри-
сталлы нитрата кальция при филь-
трации проходят в маточный рас-
твор и увеличивают в нем отноше-
ние СаО : Р2О5, что способствует
ухудшению качества продукта.
Добавкой к раствору концентри-
рованной фосфорной кислоты, выса-
ливающей Са (NO3)2 • 4Н2О, можно
сократить вымораживание до Ц и
при этом охлаждать раствор лишь
до +3, +9° (в зависимости от коли-
чества кислоты)182.
В процессе охлаждения раство-
ра происходит налипание выделив-
шихся кристаллов на охлаждаю-
щую поверхность, которую необхо-
димо очищать. После окончания
кристаллизации и опорожнения кри-
вместо рассола подают пар или горя-
поверхности змеевиков от налипших
сталлизаторов, в змеевики
чую воду для оттаивания
кристаллов.
Смесь маточного раствора с кристаллами направляют для раз-
деления на автоматическую центрифугу 6 непрерывного действия.
Кристаллы промывают азотной кислотой, предварительно охла-
жденной до —10° в холодильнике 7. Промывную кислоту возвра-
щают в реакторы на разложение фосфатов.
Выделенный четырехводный кислый кристаллический нитрат
кальция обладает плохими физическими свойствами и содержит
около 10—11 % азота. Для получения готового продукта с лучшими
физическими свойствами и с более высоким содержанием азота
(15,5%) его растворяют в воде, нейтрализуют аммиаком до
pH = 9 и направляют на переработку (стр. 1215).
О новейших усовершенствованиях в переработке Ca(NO3)2-4H2O
см. 27’183'184. В частности, разработан способ, в котором смесь
Ca(NO3)2 и K2SO4 конвертируют при 300° в KNO3 и CaSO4; при
добавке карбамида или аммофоса получают удобрения, например,
Переработка азотнокислотной вытяжки в сложные удобрения
1343
типа 20—0—20 или 12—24—14 и др. 185. Осуществляют также кон^-
версию расплавленного в собственной кристаллизационной воде
нитрата кальция газообразными СОг и NH3; карбонат кальция от-
Рассол
Селитры на
переработку
ёготовый
лота на раз- реакторы амселитры
ложение срос- разложения
сватов фосфатов
продукт
Рис. 398. Схема производства нитрофоски способом вымораживания:
/-сборник раствора; 2, 9, 11, 13, 17 и 23 — центробежные насосы; 3 и 4 —водяные холодиль-
ники; 5 — кристаллизатор; 6 — центрифуга; 7 —рассольный холодильник для HNO3; 8 — сборник
нитрата кальция; 10 — сборник HNO3; /2 —сборник маточного раствора; 14 — запорный бак;
15 — нейтрализатор 1 ступени; /5—подогреватель; 13 — испаритель; 19 — барометрический кон-
денсатор; 20— ловушка; 21 — вакуум-насос; 22 —барометрический ящик; 23 — нейтрализаторы
И ступени; 24 — шнек для KCI; 25 — смесительный шнек; 26— шиек-гранулятор; 27 — скруббер;
29 — холодильник; 30 — вентилятор.
фильтровывают, а раствор NH4NO3 направляют в основной произ-
водственный поток 27.
Маточный раствор из сборника 12 подают на нейтрализацию.
Часть маточного раствора возвращают в цикл в реакторы разложе-
ния фосфатов. Возврат холодного маточного раствора в реакторы
ТАБЛИЦА 102
Зависимость величины кристаллов Ca(NO3)2 • 4Н2О от скорости охлаждения
(при концентрации исходной азотной кислоты 47—49% HNO3)
Снижение темпера- туры, град!ч Температура выморажива- ния, °C Величина кристаллов, мк Общая продолжи- тельность кристаллизации Примечание
15 -7 175 X 175 2 ч 50 мин
10 -10 350 X 520 3 » 25 » Имеетси большое количе- ство кристаллов 175 X 175 мк
5-6 — 12 875 X 875 5—6 ч Имеется некоторое коли- чество конгломератов
1344
Гл. XXXV. Комплексные твердые сложные удобрения
разложения производят в целях снижения температуры реакцион-
ной массы для уменьшения коррозии аппаратов, которая про-
исходит вследствие использования в этом процессе большого из-
бытка азотной кислоты (30—50%) против стехиометрической нор-
мы из расчета на СаО. Кроме того, фосфорная кислота действует
высаливающе на нитрат кальция.
Приводимые ниже данные характеризуют изменение выхода)
нитрата, возвращаемого в рабочий цикл:
Количество возвращаемого раствора (%Р2О5от i
количества Р2О5 в апатите).............О 50 75 100
Выход Ca(NO3)2, % .....................' 63 67 7S 76
Процесс нейтрализации маточного раствора газообразным ам-;
миаком проводится в две ступени с промежуточным выпариванием;
раствора. В первой ступени нейтрализацию ведут до pH = 3,5—3,8,'
а во второй — до pH = 6—6,8. Маточный раствор после выделения
из него части Ca(NO3)2 в основном содержит Н3РО4, Ca(NO3)2 и
HNO3. При нейтрализации этого раствора аммиаком до pH =
= 3,5—3,8 выделяется осадок, состоящий из дикальцийфосфата
с примесью небольших количеств трикальцийфосфата. Кроме того,
в осадке содержатся небольшие количества фторида кальция,
кремневой кислоты, фосфатов алюминия, железа и редкоземельных
элементов. В растворе находятся аммиачная селитра и моноаммо-
нийфосфат. В процессе выпаривания пульпы в осадок выделяется
также часть моноаммонийфосфата 186.
Первую ступень нейтрализации осуществляют непрерывным
способом последовательно в нескольких реакторах 15. Для этой
цели могут применяться реакторы различной конструкции, обеспе-
чивающие интенсивное перемешивание и хорошее распределение
аммиака в жидкой фазе. Процесс нейтрализации протекает с вы-
делением тепла. Температуру поддерживают на уровне 110°. Выпа-
ривание раствора ведут под разрежением 500—600 мм рт. ст. др
конечного содержания воды ~14%. Выпариваемый раствор цирку-
лирует между подогревателем 16 и вакуум-испарителем 18. После
выпарки раствор направляют в нейтрализаторы второй ступени 23
и затем в смесительный шнек 25, где в пульпу добавляют хлори-
стый калий. _ ,
При нейтрализации во второй ступени часть моноаммонийфос!
фата превращается в диаммонийфосфат. Приблизительное балан!
совое уравнение нейтрализации: I
ЗН3РО4 + l,5Ca(NO3)2 + 0,5HNO3 + 4,25NH3 - 3,5NH4NO3 + 1
+ 1,5СаНРО4 + 0,25NH4H2PO4 + 0,25(NH4)2HPO4 1
При добавке в пульпу КС1 и последующей ее тепловой обра4
ботке идет обменное разложение: 1
KC1 + NH4NOs = KNOs+NH4C1
Переработка азотнокислотной вытяжки в сложные удобрения
1345
Образующиеся нитрат калия и хлорид аммония также являются
компонентами получаемой нитрофоски.
Из смесительного шнека пульпа поступает в грануляционный
шнек 26, где ее смешивают с ретуром и затем сушат, охлаждают
и подвергают рассеву и дроблению для получения продукта необ-
ходимого гранулометрического состава с размером гранул
3—4 мм 187. Во избежание налипания материала на стенку су-
шилки, что сопровождается перегревом и разложением содержаще-
гося в удобрении нитрата аммония, количество влаги в гранулах,
направляемых на сушку, не должно превышать 4%. Для этого тре-
буется большое количество ретура, во много раз превышающее
количество готового удобрения.
В табл. 103 и 104 приводится состав готовых продуктов (ни-
ТАБЛИЦА 103
Содержание питательных веществ в зависимости от их соотношения
и доли Р2О5 в водорастворимой форме
Нитрофос (из фосфо* ритов Каратау) Нитрофоска (из апатита)
N : Р2О5: К2О 1:1 1:1,5 1:1:1 1:13:1,5
Доля Р 2О5 в водорастворимой форме, % Содержание питательных веществ, % 20 20 50 20
40,8 42,4 48 49,2
ТАБЛИЦА 104
Расходные коэффициенты на
1 т сложного удобрения
Нитрофос Нитрофоска
N : Р2О5 : К2О
1:1:0 1 ; 1,5 : О' 1:1:1 1 : 1,5 : 1,5
Апатитовый концентрат, т 0,41 0,48
Фосфориты Каратау, т 0,78 0,97 — —
Азотная кислота (100 %), г 1,12 1,23 0,73 0,62
Аммиак (100%), кг 148,5 144,8 113,0 95,5
Хлористый калий (95%), т — — •0,27 0,30
Холод, Мкал 250 312 100 155
Условное топливо, Кг 30 37,5 25 23
Пар, т 0,48 0,6 0,40 0,35
Вода, .и3 30 37,5 30 28
Электроэнергия,, кет ч 63 78,5 68 63
О т х оды
Кальциевая селитра в пересчете на го- 0,85 1,06 0,60 0,46
товый продукт [79,5% Ca(NO3)2], т Жаровой конденсат, т 0,34 0,48 0,32 0,28
134S
Гл. XXXV. Комплексные твердые сложные удобрения
трофоски и нитрофоса), полученных по способу с вымораживанием!
и расходные коэффициенты в зависимости от соотношения пита-|
тельных веществ и содержания водорастворимой формы Р2О5. я
Нитрофоска, полученная из апатитового концентрата при весоЛ
вом соотношении в ней N : Р2О5: КгО, равном 1:1:1, содержит"
15,3% СаНРО4, 13% NH4H2PO4, 16,3% NH4NO3, 30,9% KNOS, 2,5%
KC1, 4,5% нерастворимых примесей и 1% воды.
Получение водорастворимой нитрофоски с частичной
регенерацией азотной кислоты®
Этот способ основан на осуществлении реакции между компо-
нентами азотнокислотной вытяжки:
10Ca(NO8)2 + 6Н3РО4 = ЗСа(Н2РО4)2 + 7Ca(NO8)2 + 6HNO3
Реакция протекает при выпаривании вытяжки под атмосферным
давлением при НО—130° или под разрежением 500—600 мм рт. ст.
при 70—95°. При этом удаляется 75—85% воды от общего ее коли-
чества и около 30% от первоначально затраченной азотной кис-
лоты. В зависимости от интенсивности выпаривания вытяжки про-
цесс заканчивается в течение 20—50 мин в аппаратах с поверх-
ностным нагревом или в течение долей минуты в аппарате типа
распылительной сушилки.
Выпаривание вытяжки завершается на конечной стадии пере-
ходом реакционной массы при температуре реакции в сметанооб-
разное состояние. При охлаждении масса схватывается, а затем
затвердевает. Полученный полупродукт — нитрофос содержит Р2О5
в водорастворимой форме при отношении N : Р2О5, равном 1 : 2;
он представляет собой смесь кристаллов одноводного монокаль-
цийфосфата (~35%) и двухводного нитрата кальция (~65%) и
небольшого количества свободной кислоты или дикальцийфосфата
(в зависимости от условий осуществления процесса). Этот полу-
продукт отличается повышенной гигроскопичностью. Однако гигро-
скопические его свойства несколько улучшаются при добавке
к нему калийной соли и значительно улучшаются в присутствии
нейтрализующих веществ.
При добавке 5—10% фосфоритной муки к нитрофоске, содер-
жащей нитрат кальция, гигроскопичность ее не выше, а слеживае-
мость продукта в несколько раз меньше, чем у других видов нитро-
фоски (карбонатной или сернокислотной).
Процесс может быть осуществлен в аппаратах с паровыми ру-
башками, аналогичных по своему устройству аппаратам, приме-
няемым для получения нитрофоски по другим схемам или в рас-
пылительных сушилках.
Производство сложных удобрений на базе НзРО4
1347
ПРОИЗВОДСТВО СЛОЖНЫХ УДОБРЕНИЙ НА БАЗЕ ФОСФОРНОЙ
КИСЛОТЫ
На базе фосфорной кислоты существует несколько схем произ-
водства сложных удобрений типа нитроаммофоски и диаммонитро-
фоски 188. Эти удобрения характеризуются высоким содержанием
питательных веществ (50% и выше) и хорошими физическими свой-
ствами. Р2О5 в них находится полностью в водорастворимой форме.
Производственные схемы отличаются между собой методом ней-
трализации фосфорной кислоты и конструктивным оформлением
основных аппаратов.
Некоторые свойства водных систем, содержащих
фосфаты, нитраты аммония, карбамид и др.9
В процессе производства сложных удобрений фосфорная кис-
лота нейтрализуется до моноаммонийфосфата и диаммонийфос-
фата:
Н3РО4 + NHa = NH4H2PO4
Н3РО4 + 2NH3 = (NH4)2HPO4
Для этих целей может применяться как термическая, так и
экстракционная фосфорная кислота. Обычно применяется более
дешевая экстракционная кислота.
В сложных удобрениях могут быть лю-
бые соотношения между моно- и ди-
аммонийфосфатом.
Из диаграммы совместной раство-
римости NH4H2PO4 И (NH4)2HPO4 в
воде (рис. 399) видно, что раствори-
мость моноаммонийфосфата увеличи-
вается с повышением концентрации
диаммонийфосфата. Максимальная
растворимость моноаммонийфосфата
будет при молярно.м отношении
Н3РО4 : NH3 = 1,5. Во избежание по-
терь аммиака на практике нейтрализа-
цию ведут до отношения H3PO4:NH3 =
= 1,3—1,35 9.
Отношение N : Р2О5 в фосфатах
аммония составляет: для моноаммо-
(NH4)2HPO4, г/ЮОгНгО
Рис. 399. Диаграмма раство-
римости в системе
NH4H2PO4—(NH4)2 НРО4—Н2О
при 25°.
нийфосфата 1:5 и для диаммонийфосфата 1 :2,5. Для вырав-
нивания соотношения N : Р2О5 необходимо вводить в удобрения
азот. Обычно недостаток азота покрывают за счет добавки нитрата
аммония, карбамида или сульфата аммония.
1348
Гл. XXXV. Комплексные твердые сложные удобрения
Из диаграмм растворимости 189 при 25° для систем NH4H2PO4—
— (NH4)2SO4—Н2О (рис. 400) и (NH4)2HPO4— (NH4)2SO4—Н2О
(рис. 401) видно, что при увеличе-
нии содержания (NH4)2SO4 в рас-
творе, растворимость фосфатов ам-
мония уменьшается. При увеличении
Рис. 400.
в системе
Диаграмма растворимости
NH4H2PO4— (NH4)2SO4—н2о
при 25°.
Рис. 401. Диаграмма раствори-
мости в системе
(NH4)2HPO4— (NH4)2SO4—Н2Опри 25°.
концентрации фосфатов аммония растворимость (NH4)2SO4 из-
меняется незначительно. Нитрат аммония оказывает сильное вы-
Рис. 402. Диаграмма раство-
римости в системе
CO(NH2)2—NH4H2PO4— Н2О
при 50° (кривая MBL — по дан-
ным после 3-суточного пере-
саливающее действие на моноаммо-
нийфосфат 71’ 190-194.
В последнее время для выравнива-
ния соотношения N : Р2О5 в удобре-
ниях стали широко применять добав-
ку карбамида. Система NH4H2PO4—
—CO(NH2)2—Н2О изучена в интерва-
ле температур от 15,3 до 50° 195-198.
Изотерма этой системы при 50° пред-
ставлена на рис. 402.
В присутствии моноаммонийфосфа-
та карбамид гидролизуется:
CO(NH2)2 + Н2О = 2NH3 + СО2
С течением времени содержание
карбамида в растворе уменьшается,
но уменьшается и скорость его разло-
жения, что объясняется появлением в
растворе диаммонийфосфата, который
образуется при взаимодействии моно-
аммонийфосфата с выделяющимся при
разложении карбамида аммиаком. В присутствии диаммоний-
мешивания раствора; кри-
вая MB'L — по данным после 8-
суточного перемешивания рас-
твора).
фосфата разложение карбамида не происходит 199.
Производство сложных удобрений на базе Н3РО4
1349
Система CO(NH2)2—(NH4)2HPO4—Н2О изучена в интервале
температур от —16 до +30° 198’200. На рис. 403 представлена изо-
терма растворимости в четырехкомпонентной системе CO(NH2)2—
_NH4NO3—(NH4)2HPO4—
_Н2О при 50°; разложе-
ния карбамида не наблю-
дается ни в одной обла-
сти 201.
При производстве удо-
брений, содержащих три
основных питательных
элемента (NPK), в систе-
му вводят калий чаще
всего в виде хлористого
калия. Реакция
nh4no3 + kci
обычно не проходит до
конца и степень конвер-
сии КС! зависит от ре-
жима обработки материа-
ла. Повышение темпера-
туры и продолжительно-
сти взаимодействия ком-
понентов при хорошем
перемешивании увеличи-
вают степень конверсии
КС1 202, 203
Способы производства
Процессы получения
сложных удобрений из
фосфорной кислоты мож-
но разделить на три типа.
1) Процессы с двух-
ступенчатой аммониза-
Цией, в которых вначале аммонизируется фосфорная кислота до мо-
ноаммонийфосфата. Затем полученный раствор для выравнивания
отношения N : Р20з смешивается с плавом нитрата аммония или
карбамида 72’87- 204 и аммонизируется газообразным аммиаком до
Диаммонийфосфата. Доаммонизация осуществляется в аммониза-
торе-грануляторе, в который одновременно подаются хлористый ка-
лий и ретур. Доаммонизация проходит на поверхности гранул. Про-
цесс требует большого количества ретура (3,5—10-кратного и даже
1350
Гл. XXXV. Комплексные твердые сложные удобрения
1ВГ
более). Благодаря этому доаммонизация осуществляется в тонком
слое на поверхности гранул. Кроме того, большое количество ре-
тура позволяет поддерживать влажность материала на входе в су-
шильный барабан около 4%, что способствует получению прочных
гранул сферической формы 205~213.
2) Малоретурные процессы, в которых получение пульпы не-
обходимого состава осуществляется в реакторах, а грануляция и
сушка совмещаются в одном аппарате типа сферодайзера или
в сушилке типа РКСГ (с распылением и кипящим слоем материа-
ла сушилка — гранулятор). В этих аппаратах пульпа распыляется
форсунками на завесу падающих гранул или просто в объем, в ко-
тором происходит высушивание капель и формирование гранул.
Отношение количества ретура к количеству продукта для этой схе-
мы составляет от 0,3 до 1.
3) Расплавные процессы — в которых нейтрализуется смесь
кислот с использованием теплоты нейтрализации. Затем получен-
ный раствор выпаривается до состояния плава под вакуумом в вы-
парных аппаратах, смешивается с хлористым калием и гранули-
руется в башне или в барабанных грануляторах (в этом случае
в гранулятор поступают ретур и хлористый калий). По такой схеме
получаются очень прочные стекловидные гранулы.
Схемы с двухступенчатой аммонизацией
На рис. 404 представлена схема производства диаммонитро-
фоски (типа TVA). Фосфорная кислота концентрацией 40—42,5%
Р2О5 из сборника 1 насосом 2 подается в напорный бак 3, из кото-
рого она непрерывно поступает в реактор 8. Предварительно фос-
форная кислота проходит через промывные скрубберы 4; в них
осуществляется очистка газов, выходящих из аммонизатора-грану-
лятораЛииз сушильного барабана 16. В реактор 8, одновременно
с фосфорной кислотой, подается газообразный аммиак под давле-
нием 1,5—2 ат и при непрерывном перемешивании происходит ней-
трализация фосфорной кислоты. Количество вводимого в реактор ам-
миака обеспечивает получение в пульпе соотношения NH3: НзРО4 =
^=1,40: 1 (pH пульпы 5,6—5,7). При таком соотношении образуется
около 60% моноаммонийфосфата и 40% диаммонийфосфата:
Н3РО4 + 1,4NH3 = 0,6NH4H2PO4 + 0,4(NH4)2HPO4
За счет реакционного тепла температура в реакторе повышается
и поддерживается в пределах 115—120°. При этом около 20—30%|
воды, внесенной с кислотой, испаряется. На выходе из реактора
пульпа содержит ~25% воды. При такой влажности и таком соот-1
ношении фосфатов аммония пульпа достаточно подвижна и легка
распределяется в аммонизаторе-грануляторе 11, куда она подается
из реактора 8 через смеситель 10. В этот смеситель одновременна
ПмЛ nh4no?
1352
Г л. XXXV. Комплексные твердые сложные удобрения
подается: из напорного бака 9 плав аммиачной селитры концен-
трацией 96—98%, газообразный аммиак, хлористый калий ид
бункера 14 и ретур из бункера 13. В аммонизаторе-грануляторй
происходит донейтрализация моноаммонийфосфата до диаммоний!
фосфата и окончательное смешение всех компонентов с одновре!
менной грануляцией продукта. Балансовая реакция |
NH4H2PO4 + (NH4)2HPO4 + NH3 = (NH4)2HPO4
соответствует соотношению NH3: Н3РО4 = 2:1. Одновременно про-
исходит частичная конверсия КС1 с NH4NO3 в KNO3 и NH4C1.
Ретур подается из расчета снижения содержания влаги в гра-
нулированном продукте на выходе из аммонизатора-гранулятора
до 4—5%. Кратность регура при этом составляет 3—3,5- При сни-
жении влажности гранул до 1,3% кратность ретура возрастает до
7—10. Температуру в грануляторе, во избежание разложения про-
дукта, поддерживают не выше 70—75°.
В аммонизаторе-грануляторе происходит частичное испарение
влаги за счет выделяющегося тепла нейтрализации. Пары воды
и непрореагировавший аммиак удаляются вместе с воздухом, про-
сасываемым через гранулятор для удаления избыточного тепла и
направляются для очистки в промывной скруббер 4. Гранулы ди-
аммонитрофоски из гранулятора поступают в сушильньш барабан
16, где они сушатся до конечной влажности 1 % и меньше. Сушка
осуществляется топочными газами с температурой на входе 160—
180° и на выходе около 110°. Продукт выходит из сушилки с тем-
пературой 90° и элеватором 17 подается на двухситный грохот 18,
на котором он рассеивается на три фракции: ретур (размер частиц
меньше 2 мм), фракцию готового продукта с размером гранул
2—4 мм и на крупную фракцию с размером частиц более 4 мм,
которая поступает в дробилку 19, где она измельчается и вновь
элеватором 17 подается на рассев.
Фракция готового продукта через бункер 20 поступает в бара-
бан-кондиционер 21, где она подвергается омасливанию и при-
пудриванию и направляется в склад и на упаковку. В качестве
омасливающего вещества применяется парафинистый мазут, кото-
рый подается насосом-дозатором 22 и распиливается в первой
зоне кондиционера 21 форсункой. В качестве опудривающего мате-
риала применяются каолин, кизельгур, гипс и другие вещества,
подаваемые во вторую зону кондиционера.
Диаммонитрофоска, полученная по такой схеме, обладает хо-
рошими физическими свойствами, не слеживается длительное время
и малогигроскопична 214. Содержание и состав питательных веществ
в диаммонитрофоске N : Р2О5: КгО = 17:17: 17. Соотношение пи-
тательных веществ может меняться в зависимости от требований
потребителя.
Производство сложных удобрений на базе Н3РО4
1353
французская фирма «Спейшим», которая разработала проект
установки по приведенной на рис. 404 схеме (метод Пешине — Сен-
Гобен)210,215,216, предлагает для улучшения физических свойств го-
тового продукта сушку производить до конечной влажности 0,3%.
При такой низкой влажности готового продукта сушильный бара-
бан работает с очень малым влагосъемом — порядка 3,5 ка/(,и3-ч).
После сушки, перед кондиционированием, производится охла-
ждение продукта в кипящем слое воздухом, охлажденным
до +1и-
Английская фирма «Propane — Spenser Ltd» ввела в описан-
ную выше схему некоторые усовершенствования с целью улучше-
ния физических свойств готового продукта и технологических пока-
зателей. Исходным сырьем являются термическая фосфорная
кислота (54% Р2О5), аммиак, плав нитрата аммония (35% N) и хло-
ристый калий (60% К2О). В фосфорную кислоту перед нейтрализа-
цией вводится фосфоритная мука из расчета ~ 17 кг на 1 т про-
дукта для улучшения грануляции. Для этой же цели грануляция
в аммонизаторе-грануляторе производится в присутствии водяного
пара. Сушка проводится в две ступени последовательно в двух
сушильных барабанах. В первом гранулы высушиваются с 4 до
1,5—2% влажности. Часть гранул после первого сушильного бара-
бана возвращается в качестве ретура в аммонизатор-гранулятор,
а товарная часть гранул поступает во второй сушильный барабан,
где досушивается до конечной влажности 0,5%. Затем продукт
охлаждается в сушилке с кипящим слоем, где температура его по-
нижается на 10°. После этого продукт рассеивается. Фракция гото-
вого продукта перед кондиционированием вторично охлаждается
в сушилке барабанного типа.
Аналогичные схемы разработаны французской фирмой «Эр-
Жей» для получения удобрения состава 17—17—17 и итальянской
фирмой «Монтекатини» для получения удобрения состава
18: 18 : 18.
Японская фирма «Sogo Boeki Kaisha» предлагает производить
сложные удобрения по следующей схеме 207,217. Смесь фосфорной
(54,5% Р2О5) и азотной (48% HNO3) кислот аммонизируется по-
следовательно в двух форнейтрализаторах. Полученный раствор
солей поступает в смеситель шнекового типа, куда одновременно
Подаются хлористый калий и ретур. Затем масса перетекает в ам-
Монизатор-гранулятор, где она доаммонизируется. Полученные
гранулы с влажностью 4—5% высушиваются в сушилке топочными
газами с температурой 250° до конечной влажности 1%; продукт
рассеивается, охлаждается и кондиционируется. По данным фир-
мы, по этой схеме можно получить сложное удобрение состава
1:1,63:0,89 с общим содержанием питательных веществ более
60% и состава 1:1:1 с содержанием питательных веществ более
70% 126, 127.
1354
Гл. XXXV. Комплексные твердые сложные удобрения
Малоретурные схемы
Английская фирма «Фрейзер» 204-217’ 218'222 разработала метод
получения комплексного удобрения типа диаммонитрофоски со-
става 18: 18: 18, который отличается от других методов тем, что
процессы аммонизации, смешения компонентов, гранулирования и
сушки проводятся в одном аппарате — грануляторе барабанного
типа. Сушка продукта осуществляется только за счет почти полного
использования реакционного тепла. Процесс проводится по сле-
дующей схеме (рис. 405). Азотная кислота концентрации 67—69%
Рис. 405. Малоргтурная схема производства сложного удобрения:
/ — наборный бак для HNOa; 2 -напорный бак для Н3РО4; 5 - гранулят рр-аммонизатор»
4 — элеватор; 5 — бункер для КС1; 6 — охлаждающий барабан; 7 — элеватор; 8 —гроХоМ
9 — дробилка; 10 — шнек; 11 — ленточный конвейер.
из напорного бака 1 и фосфорная кислота концентрации 42—49%
Р2О5 из напорного бака 2 непрерывно поступают в гранулятор 3,
в который под давлением 1,5—2 ат также непрерывно подается
газообразный аммиак116. Гранулятор, в котором проходят все ста-
дии образования готового продукта, представляет собой горизон-
тальный барабан с концентрически установленным внутри вторым
барабаном меньшего диаметра. Фосфорная кислота и аммиак (из
расчета образования диаммонийфосфата) подаются во внутренний
барабан на слой гранул, поступающих из внешнего барабана. Во
внутренний барабан подается также и хлористый калий из бун-
кера 5. Во внешний барабан подается азотная кислота и аммиак ИЗ
расчета образования аммиачной селитры. Они поступают на слой
гранул, которые переходят во внешний барабан из внутреннего.
Циркуляция гранул между внешним и внутренним барабаном осу-
ществляется непрерывно. Гранулы при вращении перемещаются И®
Производство сложных удобрений на базе Н3РО4
1355-
длине внутреннего барабана и, дойдя до его конца, через край по-
купают во внешний барабан, в котором движутся в противополож-
ном направлении и, дойдя до конца, поступают в ковши и с их
помощью через течку направляются во внутренний барабан.
Хлористый калий и ретур подаются шнеком 10 в конец внутреннего-
барабана, противоположный подаче фосфорной кислоты.
Барабан вращается со скоростью ~14 об/мин. Отношение ко-
личества внутреннего ретура к готовому продукту колеблется
в пределах от 1 : 30 до 1 : 60 в зависимости от влажности выпускае-
мого готового продукта, которая составляет 0,3—0,5%. Количество-
Рис. 406. Схема производства сложного удобрения с аппа-
ратом РКСГ:
/ — сушилка РКСГ; 5—аппарат для охлаждения в кипящем слое;
3 — элеватор; 4 — грохот; 5 — дробилка; 6— барабан-КондициоНер;
7 — шиек; 8 — циклон; 9 — скруббер.
внешнего ретура очень невелико и составляет всего лишь 0,4 т на
1 т готового продукта. Процесс грануляции проводится при влаж-
ности массы менее 1%, что способствует образованию мелких гра-
нул; при этом выход товарной фракции достигает 70%. Темпера-
тура в грануляторе поддерживается в пределах 65—95°. Испарение
воды, вносимой с кислотами указанной концентрации, осуще-
ствляется только за счет тепла нейтрализации. Избыток тепла из
гранулятора удаляется воздухом, поступающим из охлаждающего-
барабана 6 и частично засасываемым через неплотности, имею-
щиеся в системе. Воздух проходит между барабанами и далее на-
правляется на очистку.
Готовый продукт из гранулятора с помощью ленточного кон-
вейера 11 и элеватора 4 направляется в охлаждающий барабан б,
где он охлаждается атмосферным воздухом и затем элеватором 7
подается для рассева на три фракции на двухситный грохот 8.
Мелкая фракция и крупная, измельченная в дробилке 9, возвра-
щаются в качестве ретура в гранулятор, а товарная фракция под-
вергается кондиционированию и направляется в склад.
1356
Гл. XXXV. Комплексные твердые сложные удобрения
К малоретурным схемам можно отнести также схемы, в кото-
рых грануляция и сушка осуществляются путем распыления или
в кипящем слое. В НИУИФ разработана конструкция аппарата
РСКГ — сушилки — гранулятора с распылением и кипящим слоем
материала. Работает этот аппарат по следующей схеме (рис. 406).
Готовая пульпа с влажностью около 50% распыляется в верхней
части аппарата в потоке дымовых газов с температурой 600—800°.
В факеле распыла происходит интенсивная сушка и образование
гранул. Гранулы с влажностью 15—18% падают вниз сушилки и
попадают в зону кипящего слоя. Здесь они подсушиваются посту-
пающим под решетку горячим воздухом до конечной влажности
~1%. Воздух предварительно нагревается до 160° за счет смеше-
ния с дымовыми газами. Температура в кипящем слое около 100°.
Дымовые газы из аппарата РКСГ проходят очистку в циклоне и
в скруббере, после чего выбрасываются в атмосферу. Этот аппарат
был испытан на опытной установке 223-226. О грануляции и сушке
в кипящем слое см. также226-228.
Получение комплексного удобрения
по безретурной схеме из расплава.
Получение комплексного удобрения по безретурной схеме из
расплава по методу фирмы «Стамикарбон» осуществляется следую-
щим образом (рис. 407). Фосфорная кислота (54,3% Р2О5) из сбор-
ника 1 насосом 2 подается в напорный бак 3. Азотная кислота
(47% HNO3) из сборника 4 насосом 5 перекачивается в напорный
бак 6. Из напорных баков 3 и 6 фосфорная и азотная кислоты по-
ступают в смеситель 7, где с помощью мешалки хорошо перемеши-
ваются. В смеситель 7 предусматривается подача конденсата в слу-
чае необходимости понижения концентрации смеси кислот. Смесь
кислот из смесителя 7 перетекает в сборник 8, из которого насо-
сом 9 подается в напорный бак 10 и далее в нейтрализатор 11.
Газообразный аммиак подается в нейтрализатор по пяти тру-
бам, погруженным в раствор. Нейтрализацию ведут до величины
pH = 2,8—3,2, при которой в растворе будут находиться моноаммо-
нийфосфат и нитрат аммония. За счет тепла реакции температура
в нейтрализаторе поддерживается около 120°. При этом часть воды
испаряется и концентрация раствора повышается до 76%. Раствор
солей из нейтрализатора перетекает в сборник 12, из которого
поступает на выпарку. Выпарку ведут при 170° и остаточном да-
влении 0,3 ат. Конечная концентрация раствора 98%. Греющим
агентом является насыщенный пар 13—15 ат. Выпарной аппарат
однокорпусный с выносной греющей камерой и с естественной цир-
куляцией. При указанных условиях соли находятся в расплавлен-
ном состоянии и твердая фаза не выделяется.
Плав из вып-арного аппарата стекает в сборник 14, в который
подается пылевидная фракция готового продукта. Из сборника
Производство сложных удобрений на базе Н3РО4
1357
насосом 15 плав перекачивается в напорный бак 16, установленный
на грануляционной башне. Из напорного бака плав переливается
в смеситель 17, где с помощью мешалки интенсивно перемеши-
вается с поступающим сюда КС1. Для предотвращения конверсии
КС1 с NH4NO3 и образования NH4C1, что значительно увеличивает
вязкость раствора и вызывает загустевание пульпы, емкость смеси-
1еля должна быть небольшой, чтобы время пребывания массы
в нем не превышало 40—50 сек.. Хлористый калий, поступающий на
смешение, должен быть предварительно высушен, тонко размолот
Рис. 407. Схема производства сложного удобрения из расплава:
/-сборник Н3РО4; 2 -насос Н3РО4; 3 — напорный бак Н3РО4; 4 —сборник HNO3;
5 —насос HNO3; 6 —напорный бак HNO3; 7 — смеситель; 8 — сборник смеси кислот;
9— насос для смеси кислот; /0 —напорный бак; 11 — нейтрализатор; 12 — сборник;
13 — выпарЦой аппарат с выносной греющей камерой; 14 — сборник для плава; 15— на-
сос для плйва; 16 — напорный бак для плава; 17 — смеситель для КС1; /8 — гранулятор;
19 — грануляционная башня; 20 — вентилятор; 21—скребок; 22—ленточный конвейер;
23 — охлаждающий барабан; 24 — элеватор; 25 — грохот; 26 — дробянка; 27 —циклон;
28 — вентилятор; 2.9 — барабан-кондиционер; 30 — бункер для припудривающей добавки;
31 — конденсатор; 32 — вакуум-насос; 33 — скруббер; 34 — насос.
и подогрет до 165—170°. Температура в смесителе поддерживается
равной 165—170°. Из смесителя масса самотеком поступает в гра-
нулятор 18 (~300 об/мин), с помощью которого разбрызгивается
в грануляционной башне 19. Гранулы с температурой ~90° падают
на дно башни и скребком 21 сбрасываются на ленточный кон-
вейер 22. Гранулы получаются очень прочными, благодаря чему
при дальнейшей их обработке и транспортировке образуется очень
мало пыли.
Далее готовый продукт охлаждается воздухом в барабане 23
и элеватором 24 подается на рассев на двухситный грохот 25. Са-
мая мелкая фракция из грохота, пыль из циклона 27 и измельчен-
ная крупная фракция в дробилке 26 направляются на смешение
с плавом в сборник 14. Товарная фракция с размером гранул
*>5—3,5 мм поступает в барабан-кондиционер 29, где опудривается
кизельгуром (или инфузорной землей, диатомовой землей) и
1358
Гл. XXXV. Комплексные твердые сложные удобрения
направляется в склад. Количество припудривающей добавки
~3,5% от веса продукта.
Получаемый по такой схеме продукт состава 17: 17: 17 со-
держит примерно: 40% NH4NO3; 30% NH4H2PO4; 27,5% КС1 и 1%
воды. По этой же схеме можно получать продукты 1 : 0,8 : 1 и
1 : 1 : 1,5 с общим содержанием питательных веществ около 50% 229_
По однотипной схеме, но с иным конструктивным оформлением
разработан процесс английской фирмой «Вайкон».
Плав, полученный аналогичным способом, предложено грану-
лировать на валке, погруженном на */3 диаметра в минеральное
масло с температурой —10°. На этом валке'образуются гранулы
сферической формы необходимого размера, которые, опускаясь на
дно масляной ванны, охлаждаются. После отделения от масла на
них остается защитная масляная пленка 230.
ЛИТЕРАТУРА
1. J. J. Мо г t v е d t, Agr. Chem., 20, № 6, 39, 128 (1965). — 2. V. Andri-
ska, Technika (Magyar), 10. № 12, 6 (1966).—3. A. J. Payne, Chem. Progress
Eng., 47, № 1, 5 (1966). — 4. Phos. a. Pot., Ns 21, 18 (1966). — 5. T. П. Уна-
н я н ц. Агрохимия, № 2, 146 (1964). — 6. А. В. Петербургский, А. В. Пост-
ников, Комплексные минеральные удобрения, Изд. «Московский рабочий»,
1966. — 7. Справочник по минеральным удобрениям, Сельхозгиз, 1960.—
8. Т. П. У н а н я н ц, Агрохимия, № 5, 146 (1966). — 9. Ф. Г. Марголис,
Т. П. У н а н я н ц, Производство комплексных удобрений, Изд. «Химия», 1968. —
10. Przem. Chem., 46, № 1, 38; № 2, 65 (1967).
11. Н. В a n t h i е n, Allgem. u. prakt. Chem., 18, № 4, 104 (1967).—12. ОГ
Tadeusz, Chernik, 18, № 2, 50 (1965). —13. J. Dankiewiez, Przem. Chem.,
44, Ns 10, 536 (1965). — 14. W. Janiczek, J. F a z a n, Chernik (польск.), 19,
№ 6, 184 (1966). — 15. Чехосл. пат. 109102, 1963. — 16. T. П. У н а н я н ц, Химия
в сельском хозяйстве, № 10, 62 (1966). — 17. Н. С h е р о г d, I. Mahan, Chem.
Eng. News, № 36, 82 (1966). —18. Comm Fertilizer, 112, № 4, 21 (1966).—
19. Statistisches Jahrbuch fiir die Bunderespublik Deutschland 1966, Berlin, 1967. —
20. Annuaire de Statistique industrielle 1966, Paris, 1967.
21. Farm Chemicals Handbook 1966, Ohia (USA), 1967. — 22. L’Engrais, 80,
№ 192, 8 (1966). — 23. World Fertilizer Atlas 1964, London, 1965. — 24. H. Ko-
table, Asahi Evening News, Ns 1, 60 (1965). — 25. Fertilizer Statistics India
1965—1966, Delli, 1967. — 26. T. H i g n e t, Farm. Chemie., 128, № 8, 36 (1965).—
27. R. Remirez, Chem. Eng., 74, № 22, 86, 90, 134 (1967).—28. С. И. Вольф-
кович, Ф. Г. Марголис, Н. Н. Поляков, Хим. пром., Ns 1, 34 (I960).—
.29. Ф. Г. Марголис, Т. В Глазова, А. И. Свердлова, Журн. ВХО,
7, Ns 5, 507 (1962). — 30. В. А. Клевке, Н. Н. Поляков, Л. 3. Арсенье-
ва, Технология азотных удобрений, Госхимиздат, 1963.
31. М. Н. Набиев, М. А. Касымова, сб. «Минеральные и органо-мине-
ральные удобрения, структурообразователи почв и гербициды», Ташкент, Изд.
«ФАН», 1967, стр. 17. — 32. М. Е. П о з и н, Б. А. К о п ы л е в, Новые методы
получения минеральных удобрений, Госхимиздат, 1962.— 33. Ле Ван 3 и,
Ф. М. Мирзаев, М. Н. Набиев, Узб. хим. ж., № 6, 7 (1967).— 34. К. Но-
ров, У. Ибрагимова, М. Н. Набиев, сб. «Минеральные и органо-мине-
ральные удобрения, структурообразователи почв н гербициды», Ташкент, Изд-
«ФАН», 1967, стр. 31. —35. Швейц, пат. 419201, 1967.— 36. Р. А. А близ ин а,
ДАН УзССР, Ns 4, 36 (1966). — 37. Л е Ван 3 и, Ф. М. М и р з а е в, М Н. Н а-
б и е в, сб. «Минеральные и органо-минеральные удобрения, структурообразова-
Литература
1359
Тели почв и гербициды», Ташкент, Изд. «ФАН», 1967, стр. 66.—38. В. М. Бег-
лов Ф. Ф. Г а н и х а н о в а, М. Н. Набиев, Узб. хим. ж., № 5, 3 (1967). —
да. Пат. ГДР 50043, 1966. —40. th. Р1 u s i е, L’ind. chim., № 374—376 (1948).
41. А. И. Логинова, Процесс разложения фосфатов азотной кислотой
, получением концентрированных удобрений, Автореф. канд. дисс., 1940. —
42. Г. И. Г о р ш т е й и, Т. И. Хахарина, ЖХП, № 1, 39 (1934).—
43 М. Н. Набиев, Азотнокислотная переработка фосфатов, Изд. АН УзбССР,
1957. — 44. В. Антонова, В. Тихонов, А. Беркутов, Изв. АН КазССР.
Сер. хим., № 101, 4, 76 (1951).—45. М. А. Ч е п е л е в е ц к и й, С. С. Рубино-
ва, Труды НИУИФ, в. 137, 1937, стр. 66.-46. С. Вольфковнч, А. Логи-
нова сб. «Памяти акад. Д. Н. Прянишникова», Изд. АН СССР, 1950,
стр. 253.-47. А. М. Поляк, ЖХП, № 11—12, 807 (1937). —48. С. И. Вольф-
кович, А. П. Белопольский, А. И. Логинова, Изв. АН СССР. ОХН,
К» 5, 705 (1940).—49. Пат. США 3205062, 1965. —50. М. Н. Набнев и др.,
Изв. АН УзССР, № 3, 25 (1954).
51 Б. И. Поляков, М. Н. Набиев, ДАН УзССР, № 1, 32 (1968).-
52. М. Л. Ч е п е л е в е ц к и й, У. Больц, ЖПХ, 10, № 7, 1183 (1937).—
53. М. Н. Набиев, Ю. И. Шакирова, ДАН УзССР, № 8, 25 (1954).—
54 В. Я- Аносов, С. К- Чирков, ЖПХ, 6, Ns 2, 224 (1933).—55. J. В г о-
s h ее г, F Lenfesty, J. Agr. Food Chem., 6, № 11, 827 (1958).—
56. С. И. Вольфковнч, А. И. Логинова, А. М. Поляк. Изв. АН СССР.
Сер. хим., Ns 101 (1938). —57. А. И. Логинова, ЖПХ, 15, № 1, 28 (1938). —
58. С. И. Вольфковнч, А. И. Логинова, ДАН СССР, 25, Ns 2, 124
(1939); Там же, 16, Ns 12 (1935). — 59. В. М. Беглов, Узб. хим. ж., № 4, 71
(1967).— 60. Б. М. Беглов, М Н. Набиев, М. И. С а н ж а р о в. Там же,
№ 6, 55 (1966).
61 К. Норов, У. Ибрагимова, М. Н. Набиев, сб «Минеральные
и органо-минеральные удобрения, структурообразователи почв и гербициды»,
Ташкент, Изд. «ФАН», 1967, стр. 26. — 62. В. М. Беглов, М Н. Набиев,
М. И. С а н ж а р о в, Узб. хим ж., № 6, 55 (1966). — 63. В. М Беглов. Там
же, № 2, 64 (1967). — 64. В. М. Беглов, А. И. Ильясов, сб. «Физико-хими-
ческие и технологические исследования минерального сырья», Ташкент, Изд.
«Наука», 1965, стр. 42. — 65. В. М. Беглов, М. Н. Набиев. Узб хим. ж.,
№ 6, 5 (1965). — 66. В. М. Беглов, Там же, № 2, 70 (1966). — 67. В. Bor-
kowski, К. Bayczyk, A. Borkowska, Zesz. pank. Uniw. Poznaniu. Mat,
fiz., Chim, Ns 65, 207 (1967).—68. А. П. Белопольский и др., ЖПХ, 10,
№ 3, 403 (1937); Ns 9, 1523 (1937). —69. R. Flatt e. al., Helv. chim. acta, 37,
№ 2, 607 (1954). — 70. R. Flatt e. al., ibid., № 7, 2363 (1954).
71. R. Flatt e. at, ibid., 39, Ns 2, 483 (1956). — 72. Я. С. Ш e н к и н,
В. А. Клевке, М. А. Людковская, ДАН СССР, 149, Ns 3, 656 (1963).—
73 M E П о з и н, Технология минеральных удобрений, Изд. «Химия», 1965. —
74. ф. г Марголис, А. М. Дубовицкий, Т. В. Глазова. ЖХП. Ns 1,
26 (1956); Ns 4, 27 (1957). —75. Chem. Age, 64, № 1658, 588 (1951).—
76. \v Mazgaj, J- Kwiecien, Przem. Chem., 11, № 8, 449 (1955)'.—
77. A. M. Дубовицкий, А. И. Ш e p e ш e в с к и й, Технология минеральных
Удобрений, Госхимиздат, 1941, стр. 218. —78. С. И. Вольфковнч, А. И. Л о-
гинова Усп. хим., 18, Ns 4, 453 (1949).—79. Г. И. Г о р ш т е й н, Т. И. Ха-
зарина ЖХП, № 10, 1057 (1935). — 80. А. М. Дубовицкий, Ф. Г. М а р-
г о л и с, ЖПХ. 29, № 3, 321 (1956).
, 81. L’ind. chim., 41, Ns 439, 42 (1954); 41. Ns 440, 76 (1954).—82. D. Knight,
' Anderson, M. S t r i p 1 i n, J. Agr. Food Chem., 1, № 2, 162 (1953).—
«3. F. Nielson, L. L a t e s, ibid., 1, № 10, 672 (1953).—84. Chem. Age 64,
№ 1655, 48 (1951).—85. L’ind. chim., 40, № 437, 367 (1953). —86. Д. Г. Гра-
’ев, Удобр, и урожай, Ns 2, 11 (1959).—87. М. Н. Набиев, Изв. АН УзССР,
5, 43 (1954).- 88. Chem. Eng., 63, Ns 10, 358 (1956).— 89. А. М. Дубо-
1360
Гл. XXXV. Комплексные твердые сложные удобрения
вицкий, Ф. Г. Марголис, сб. «Промышленность минеральных удобрений,
Госхимиздат, 1958, стр. 256. — 90. Л. 3. Арсеньева, Там же, стр. 270.
91. Б. Балла, Там же, стр. 274. — 92. Г. Ш три бель, Там же, стр. 284.
93. В. М а з г а й, Там же, стр. 288. — 94. Т. С. С т о б е ц к и й, Там же, стр. 292.
95. К- М. С а л д а д з е, Н. У. Р и з а е в, Н. А. Габриелян, М. Е. Позин
Б. А. Копылев, С. А. Сигов, Ф. М. Мирзаев, К- В. М е р е н к о в, сб
«Химия и технология минеральных удобрений», Ташкент, Изд. «ФАН», 196б"
стр. 182.—96. С h а п d г о п, L’Ind. chim., 39, № 414, 18 (1952). — 97. В. Ва11а’
G. Koranyi, Comm. XX Congres Inform, de Chimie Industrielle, Athenes, 17-J
24, Sept., 1957.—98. А. Б. Бектуров, В. И. Литвиненко, В. В. Тихо-
нов, В. К. Э с и к, В. Н. С о п и л и д и, сб. «Производительные силы Южного
Казахстана», т. 3, Алма-Ата, Изд. «Наука», 1966, - стр. 13. — 99. Авт. свид.
182743, 1966. —100. С. И. Вольфкович, Ф. Г. Марголис, Н. Н. Поля-
ков, Хим. пром., № 1, 34 (1960).
101. Ф. Г. Марголис, Т. В. Глазова, А. И. Свердлова, Жури. ВХО
7, № 5, 507 (1962).— 102. И. М. Каганский, 3. К. Федосеева, Г. С. Мух-
ля, сб. «Новые способы производства минеральных удобрений», Госхимиздат,
1962, стр. 17. — 103. Я. С. Ш е н к и н, В. А. Клевке, М. А. Людковская
ДАН СССР, 149, № 3, 656 (1963). —104. A. Constant, L’Ind. chim., 38, №411
280 (1951). — 105. G. Davies, Nature, 167, № 4255, 793 (1951). —106. Chem’
Eng., 60, № 8. 115 (1953). — 107. Ibid., 65, № 22, 60 (1958). — 108. Y. Martin
L’Ind. chim., 38, № 411, 278 (1951). — 109. J. T u r e n t i n e, Chem. Eng. News,
29, № 34, 3454 (1951). — 110. Chem. Eng., 63, № 1, 103 (1956).
111. I. Stinson, M. S triplin, N. Brown a. oth., J. Agr. Food Chem.,
4, № 3 (1956). —112. Agr. Chem., 11, 83 (1956). —113. Chem. Age, 64, № 1658,
585 (1951). — 114. Л. И Логинова, ЖХП. № 7, 10 (1950).— 115. M. Е. По-
зин, Э. Сабо, Труды ЛТИ им. Ленсовета, в. 36, 1956, стр. 93. — 116. Пат.
США 3248209, 1966, — 117. Пат. США 3248174, 1966, —118. Y. Lakota, Chem.
priimysl, 12, № 3, 113 (1962). —119. Франц, пат. 1408302, 1965.— 120. Англ. пат.
1019318, 1966.
121. W. R. Horn, N. D. F о u z е г, Chem. Eng. Progr., 62, № 7, 109 (1966).—
122. D. G. Rands, Agr. Chem, 18, № 9, 35 (1963). —123. Пат. США 3239314,
1966. — 124. Франц, пат. 1435834, 1966. —/25. Англ. пат. 1039177, 1966,-
126. Япон. пат. 3193, 1965. —/27. Япон. пат. 10090, 1966, —/28. Ив. Кършев,
Д. Иванов, Р. Богданова, Годишнак хим.-технол. ин-та, 10, № 1, 101
(1964). —/29. Хуасюэ гунтье, № 2, 28 (1966). — 130. Ф. Г. Марголис,;
Т. В. Глазова, О. Н. Симонова, Хим. пром, № 2, И (1961). (
131. М. Н. Набиев, В. К. Дубовая, М. С. Нестерова, Химия и
технология минеральных удобрений, Ташкент, Изд. «ФАН», 1966, стр. 169.—
132. М. Н. Набиев, К- М. Камалов, В. К. Дубовая М. С. Нестеро-
ва, В. П. Иванов, Узб. хим. ж., № 2, 11 (1966). —/33. Англ. пат. 873578,
1961. — 134. М. Striplin, D. Meknight, Т. Hignett Ind. Eng. Chem, 44,
№ 1, 236 (1952). —/35. L’Ind. chim, 42, № 461, 390 (1955). —/36. Agr. Chem,
8, № 9, 58 (1953). — 137. T. H i g h e 11, M. Siegel, D. Knight, a. oth,
J. Agr. Food Chem, 6, № 11, 822 (1958). —/38. Farm. Chem, 116, № 2, 31, 33,
35, 37 (1953). —139. Tah-Ho-Huang, Agr. Chem, 10, № 11, 45, 109, 111, 1Ц
(1955).— 140. G. Kineses, J. Solymosi, M. Mehyhart, Nehezvegyipan
kutato int kozl, 3, № 1—2, 43 (1966).
141. Brit. Chem. Eng, 13, № 5, 1205 (1968).—142. P. Ell wood, Chem.
Eng, 75, № 5, 124 (1968). — 143. Phos. a. Pot, № 26, 20, 37 (1966).—
144. С. С Караханян, Г. О. Григорян, Изв. АН АрмССР. Сер. хим, 1°,
№ 5, 516, 521 (1965). — 145. С. С. К а р а х а и я н, Г. О. Григорян, Р. Л. Ми-
ру л я н, Там же, № 6, 615 (1965). —146. Пат. США 3172751, 1965.—
147. М. Е. Позин, Б. А. Копылев, И. Я. А з б е л ь, Л. Ф. Никитина,
Литература
1361
к А. Дмитриевский, ЖПХ, 37, № 10, 1 (1964). —148. Авт. свид. 205029.
.jgg—149. М Н. Набиев, А. В. Закржевская, У. И. Ибрагимова,
Э С. Му кимова, А. М. Амирова, сб. «Химия и технология минеральных
удобрений», Ташкент, Изд. «ФАН», 1966, стр. 154. —150. Авт. свид. 181666,
1966
151. М. Н. Набиев, А. Ф. Глаголева, А.' В. Закржевская.
3 С Мукимова, Узб. хим. ж., № 2, 5 (1965). —152. М. М о м и н о в,
ф М. Мирзаев, М. Н. Набиев, сб. «Химия и технология минеральных
удобрений», Ташкент, Изд. «ФАН», 1966, стр. 341. — 153. С. А. Сигов,
ф М. Мирзаев, Н. А. Чередниченко, Там же, стр. 217. — 154. М. Мо-
ли но в. Ф. М. Мирзаев, М. Н. Набиев, сб. «Минеральные и органо-мине-
ральные удобрения, структурообразователи почв и гербициды», Ташкент, Изд.
«фАН», 1967, сгр. 58. — 155. Ле Ван 3 и, Ф. М. Мирзаев, М. Н. Набиев,
ПАН УзССР, № 12, 26 (1967). —156. М. М о м и н о в, Ф. М. Мирзаев,
М Н. Н а б и е в, Узб. хим. ж., № 1,6 (1968). — 157. f\. Камалова, В. К. Ду-
бовая, М. Н. Набиев, сб. «Минеральные и органо-минеральные удобрения,
структурообразователи почв и гербициды», Ташкент, Изд. «ФАН», 1967, стр. 39.—
158. Mi. Н. Набиев, К- М. Камалов, В. К. Дубовая, Узб. хим. ж., № 1,
3 (1967). — 159. Б. А. Б е р е м ж а н о в, Г. Д. 3 о р о в, сб. «Химия и химическая
технология», т. 5, изд. Мин-ва высш, и средн, спец, образован. КазССР, 1966,
стр 130. — 160. Е. Л. В и л ь н и ц, Б. А. Дмитриевский, Б, А. Копылев,
М. Е. П о з и н, ЖПХ, 41, № 9, 1883 (1968).
161. Н, А. Габриелян, Н. У. Р и з а е в, Ф. М. Мирзаев, К. В, Ме-
ре н к о в, сб. «Минеральные н органо-минеральные удобрения, структурообразо-
ватели почв и гербициды», Ташкент, Изд. «ФАН», 1967, стр. 20.—162. Франц,
пат. 1448797, 1966. — 163. Пат. ФРГ 1047802, 1966. — 164. Пат. ФРГ 1047803.
1966.— /65. Авт. свид. 179468, 1966. — 166. Англ. пат. 1041823, 1966.—
167. Я- Б. Б л ю м б е р г, М. М. Ч е р н о б а е в а, А. И. Свердлова, сб. «Ис-
следование по химии и технологии удобрений, пестицидов, солей», Изд. «Наука»,
1966, стр. 136. — 168. Швейц, пат. 402901, 1966.— 169 Англ. пат. 1008402,
1965. — 170. Пат. США 3195980, 1965.
171. Пат. США 3005697. 1961, —/72. Пат. США 2985527, 1961.—/73. Франц,
пат. 74063, 1960. —174. М. Е. Позин, Б. А. Копылов, Ван Ли Шен,
С. И. Каганович, ЖПХ, 34, № 5, 994 (1961). —/75. М. Н. Набиев, О. Ша-
киров, Изв. АН УзССР. Сер. хим., № 4, 19 (1957). — 176. Б. Балла,
Д. К и н ч е т, Сообщение Исслед. ин-та осн. хим. пром. Вес. прем. (Венгрия),
т. I, в 1—2 1958 стр. 3. —/77. С. И. Вольфкович, А. И. Логинова,
ДАН СССР, 3, № 8, 729 (1946). — 178. М. К о 1 а с, N. Selkova, J. N у v 11,
Fr. М i s е k, Chem. priimysl., 16, № 9, 525 (1966). —179. F. W. Brandt,
V-H. Nees Phos. a. Pot., № 21, 22 (1966). —180. Бэссацу начаку кочё (япон.),
Ю. № 5, 204 ’(1966).
181. Я. Нывлт, Ф. М и ч е к, К. Гасс, ЖПХ, 35, № 7, 1424 (1962).—
182. М. Е. Позин, Б. А. Копылев, Б. А. Дмитриевский, Е. Б. Руда-
кова, Тезисы докладов научно-технической конференции ЛТИ им. Ленсовета,
Изд. «Химия» 1965, стр. 155. —183. Phos. a. Pot., № 30, 21 (1967); Brit. Chem.
Eng-, 12, № 1 i, Suppl. (1967). — 184. Chem. Age (India), 18, № 11, 839 (1967). —
185. 1. Moldovan, V. Pre da, Rev. chim. (рум.), 19, № 8, 448 (1968).—
Ж Франц, пат. 1443309, 1966.— /87. В. А. Клевке и др., Технология азот-
ййх удобрений, Госхимиздат, 1956. — 188. Пат. ГДР 49414, 1966. — 189. Ю.С.Ми-
Чвнко, М. Л. Чепелевецкий, Труды НИУИФ, в. 208, изд. НИУИФ, 1965,
16.— 190. А. Г. Б е р г м а и, П. Ф. Бочкарев, Изв. АН СССР, Сер. хим.,
* 1, 237 (1938).
. 191. R. Flatt, J. Brunisholz, Р. Rod, Helv. chim. acta, 38, № 3, 753
(1955). — 192. А. Г. Бергман, В. П. Радищев, И. Н. Никонова,
Н. Свешникова, Э. Б. Ш т е р н и н а, М. А. Я ц у к, ИСФХА АН СССР.
1362
Гл. XXXV. Комплексные твердые сложные удобрения
15, 157 (1947). —193. V. Schrodt, J. Chem. Eng. Data, 7, № 4, 476 (1962).-,
194. Ф. Г. Марголис, Т. В. Глазова, сб. «Исследования по химии и
технологии удобрений, пестицидов и солей», Изд. «Наука», 1966, стр. 82. __
195. В. П. Блидин, ЖОХ, 11, № И, 887 (1941). — 196. В. А. Полосин
А. Г. Тр ещов, ЖФХ, 27, № 1, 57 (1953). — 197. В. А. Полосин, А. Г. Тре;
щов, Изв. ТСХА, № 2, 203 (1953). — 198. Г. Л. Т у д о р о в с к а я, Ф. Г. Мар.
голис, Усп. хим., 34, № 12, 2124 (1965). — 199. Г. Л. Тудоровская
Ф. Г. Марголис, Труды НИУИФ, в. 208, изд. НИУИФ, 1965, стр. 7.J.
200. А. Г. Т р е щ о в, Автореф. канд. дисс.. Тимирязевская сельскохозяйственная
академия, 1953.
201. Г. Л. Тудоровская, Ф. Г. Марголис, Хим. пром, № 9, 678
(1966).— 202. А. Г. Бергман, Изв. АН СССР. Сер. хим., № 1, 203 (1938). —
203. А. И. Соклаков, Н. А. Василенко и др,-сб. «Исследования по хи-
мии и технологии удобрений, пестицидов и солей», Изд. «Наука», 1966,
стр. 91,— 204. Comm. Fertilizer, 110, № 4, 48 (1965). —205. Brit. Chem. Eng’
11, № 3 (1966).—206. Англ. пат. 1039973, 1966.—207. Бэссацу качаку кочё
(япон.), 10, № 5, 212 (1966).— 208. Пат. США 3241946, 1966. — 209. В. Вагга-
м а н, Фосфорная кислота, фосфаты и фосфорные удобрения, Госхимиздат,
1957. — 210. R. Jound, G. Hicks, G. Davis, J. Agr. Food Chem, 10, № 6
442 (1962).
211. A. Payne, Chem. Proc. Eng, 47, № 1, 5 (1966). — 212. M. Giorgini,
W. Weber, Technical Manufacturers Assocition, Stockholm, 1959.—213. Chem.
Eng. News, № 5, 10 (1965). — 214. E. П. Смирнова, С. T. Рындина,
T. И. Маркина, T. Ф. Абашкина, Хим. пром, № 1, 52 (1967).—
215. И. И. Орехов, Там же, № 1, 68 (1966). — 216. J. Agr. Food Chem, 10,
№ 6, 442 (1962). — 217. Бэссацу качаку кочё (япон.), № 5, 210 (1966).—
218. Англ. пат. 964354, 1964. — 21'9. Cheirn Z. Blatt, № 10, 2185, 2188 (1965).—
220. Chem. Proc. Europe. Suppl. to Brit. Chem. Eng, March, 47 (1966).
221. Brit. Chem. Eng, № 3, 73 (1966).—222. Nitrogen, № 42, 26 (1966).-
223. M. В. Лыков, Б. И. Леончик, Распылительные сушилки. Основы тео-
рии и расчета, Изд. «Машиностроение», 1966.—224. Авт. свид. 183190, 1966.—
225. Пат. ЧССР 110960, 1964, —226. М. И. Ниязов, В. А. Линкевич,
О. Б. Ерофеева, Ф. М. Мирзаев, М Н. Набиев, сб. «Минеральные и
органо-минеральные удобрения, структурообразователи почв и гербициды», Таш-
кент, Изд. «ФАН», 1967, стр. 90.—227. В. С. Вызго, А. И. Павлова,
В. П. Линкевич, О. Б. Ерофеева, М. И. Ниязов, М. Н. Набиев,
Там же, стр. 95. — 228. О. Б. Ерофеева, В. А. Линкевич, М. И. Ниязов,
Изв. вузов, Хим. и хим. технол., 11, № 2, 176 (1968). — 229. Авт. свид. 197631,
1967. — 230. Авт. свид. 197633, 1967.
Глава XXXVI
КОМПЛЕКСНЫЕ СМЕШАННЫЕ И СЛОЖНО-СМЕШАННЫЕ
УДОБРЕНИЯ
ОБЩИЕ СВЕДЕНИЯ
Смешанными называют комплексные удобрения, получаемые
смешением готовых удобрений. Процесс приготовления смешанных
удобрений называют тукосмешением.
По своим агрохимическим качествам смешанные удобрения
практически не отличаются от сложных 1-6. Преимуществом их про-
изводства является возможность выпуска очень широкого ассорти-
мента удобрений с любыми соотношениями питательных элементов,
удовлетворяющими разнообразным требованиям сельского хозяй-
ства. Преимущество же сложных удобрений заключается в возмож-
ности избежать ряда производственных процессов при их получе-
нии, используя полупродукты (например, плав нитрата аммония,
газообразный аммиак) вместо готовых продуктов, необходимых
для изготовления тукосмесей.
Применение смешанных удобрений очень распространено. В об-
щем балансе производства минеральных удобрений в капиталисти-
ческих странах7 смешанные удобрения составляют больше одной
трети. В отдельных странах (Англия, США) выпуск тукосмесей
составляет 60—70% от обшего производства удобрений.
Ассортимент смешанных удобрений весьма разнообразен.
В странах Западной Европы он исчисляется десятками марок
(сортов), а в США превышает 3000, однако наиболее распростра-
ненных марок тоже только десятки8. Тукосмеси готовят с различ-
ными соотношениями питательных веществ, причем каждый пита-
тельный элемент может входить в состав смеси в виде разных
Компонентов s~13. Например, азот в виде нитрата аммония, суль-
фата аммония, карбамида, фосфатов аммония; фосфор в виде су-
перфосфатов, фосфоритной муки; калий в виде хлорида и суль-
фата и проч. В зависимости от вида смешиваемых удобрений
1364 Г л. XXXVI. Смешанные и сложно-смешанные удобрения
общее содержание питательных веществ в тукосмеси может изме-
няться в широких пределах — от 25—30% при использовании про.
стого суперфосфата, сульфата аммония или аммиачной селитры
до 40% и больше в концентрированных смешанных удобрениях, по-
лучаемых смешением двойного суперфосфата, аммофоса, карба-
мида и других концентрированных удобрений |4“18.
В важнейших растениеводческих районах минеральные удобре.
ния применяются преимущественно в смешанном виде 19'21. В сов-
хозах Таджикской, Казахской и Узбекской республик на смешение
идет 55—70% всех расходуемых удобрений. В свекловичных сов-
хозах Украинской ССР и ЦЧО РСФСР почти все минеральные
удобрения применяются в смешанном виде.
Практика применения удобрений показала достаточность срав-
нительно небольшого числа вариантов соотношения питательных
элементов в смешанных удобрениях. Например, под хлопчатник
рекомендуются смеси с соотношением N : Р2О5: КгО, равным
1 : 2 : 0; 2 : 1 : 0; или 1:1:0; под зерновые культуры 1:2:2; 1:2:1
или 1:1:1; под сахарную свеклу 1:2:1; 1 : 1,5 : 1,5 и т. д.
Помимо основных питательных элементов (N + Р + К) сме-
шанные удобрения могут содержать микроэлементы, инсектофун-
гициды, гербициды, стимулирующие ростовые вещества и др. Для
нейтрализации избыточной кислотности и улучшения физических
свойств при изготовлении тукосмесей, в них часто вводят добав-
ки — наполнители, такие как костяная или фосфоритная мука, мел;
известняк, доломит и др. 2.
Смешанные удобрения производятся трех типов:
1) порошкообразные смешанные удобрения (тукосмесь) —про-
дукт механического смешения порошкообразных простых удоб-
рений;
2) гранулированные смешанные удобрения, получаемые меха-
ническим смешением простых гранулированных удобрений (в США
они известны под названием «балк-блендинг»);
3) сложно-смешанные гранулированные удобрения, получаемые
смешением порошкообразных простых удобрений с введением
в процесс жидких реагентов (раствора или плава нитрата аммо-
ния, аммиакатов, азотной, серной или фосфорной кислот, а также
газообразного аммиака). Для получения хороших тукосмесей
исходные компоненты должны быть сухими и рассыпчатыми, в про-
тивном случае их требуется сушить и измельчать или подвергать
этим операциям готовую смесь. Кроме того, желательно, чтобы
смешиваемые материалы не сильно различались по крупности и
плотности зерен. Материалы, не удовлетворяющие этим требова-
ниям, труднее превратить в однородную смесь. Кроме того, смеси,
состоящие из зерен разных размеров и разной плотности, подвер-
жены сегрегации, т. е расслаиваются при перевозке, встряхивании
и т. п. Поэтому предпочитают выпускать не смеси порошкообраз-
Синергизм и антагонизм удобрений
1365
вых или гранулированных продуктов, а гранулированные смеси,
т. е. гранулировать материал после или в процессе смешения ком-
понентов.
Ведутся исследования для получения нерасслаивающихся гра-
нулированных смесей7’16’17-22,23. g США в 1966 г. 88% смешанных
удобрений было выпущено в гранулированном виде и только 12%
в порошкообразном 6>8-24-30.
Раньше осуществляли главным образом гранулирование гото-
вых порошкообразных смесей, смачивая их водой, окатывая и вы-
сушивая гранулы. В последнее время все шире совмещают смеше-
ние удобрений с их дополнительной химической обработкой — ам-
монизацией газообразным аммиаком и жидкими аммиакатами,
введением в смеси кислот и нейтрализующих их материалов, раство-
ров и плавов взамен воды в процессе гранулирования и др. В ре-
зультате этого при смешении компонентов и гранулировании про-
текают химические реакции, а гранулы продукта получаются более
однородными и прочными. За счет тепла химических реакций про-
исходит высушивание гранул. Такие смешанные удобрения по су-
ществу мало отличаются от сложных; поэтому их и называют слож-
но-смешанными.
СИНЕРГИЗМ И АНТАГОНИЗМ УДОБРЕНИЙ
При получении смешанных комплексных удобрений следует учи-
тывать, что некоторые исходные соли и другие продукты нельзя
смешивать друг с другом, так как могут идти нежелательные хими-
ческие процессы, в результате которых будут теряться питательные
вещества (улетучиваться или ретроградировать в неусвояемую
форму) и ухудшаться физические свойства удобрения. Эти явле-
ния обусловливают так называемый антагонизм удобрений. В про-
тивоположность этому синергизмом удобрений можно назвать их
способность эффективного совместного действия без возникнове-
ния вредных побочных процессов, обусловливающую возможность
их смешения. Например, при смешении суперфосфата с аммиачной
селитрой в результате реакций
NH4NO3 + Н3РО4 = NH4H2PO4 + HNO3
2NH4NO3 + Са(Н2РО4)2 = 2NH4H2PO4 +Са (NO3)2
теряется некоторая часть азота (в виде паров HNO3 или окислов
азота) и физические свойства смеси оказываются худшими, чем
исходных компонентов, из-за появления более гигроскопичного со-
единения — нитрата кальция. При введении в эту смесь нейтрали-
зующих добавок или при аммонизации ее аммиаком образование
азотной кислоты предотвращается, поэтому исключается возмож-
ность потери азота, а благодаря превращению части монокаль-
Нийфосфата в дикальцийфосфат, связывающий некоторую долю
1366
Г л. XXXVI. Смешанные и сложно-смешанные удобрения
гигроскопической воды в кристаллизационную, улучшаются физи-
ческие свойства удобрения. При этом, однако, уменьшается содер.
жание водорастворимой Р2О5 за счет увеличения цитратнораство-
римой.
Для уменьшения ретроградации Р2О5 предложено31 добавлять
к смешанным удобрениям, содержащим суперфосфат, перед аммо-
низацией небольшие количества растворимых солей магния и же-
Рис. 408. Диаграмма смешения
удобрений:
/ — нитрат кальция: 2 —нитрат натрия;
3 -- сульфат-нитра г аммония; 4 — ка-
лийно-аммиачиая селитра; 5 — сульфат
аммония; 6 — хлористый аммоний;
7 —карбамид; 8 — цианамид кальция;
преципитат; 10 — суперфосфат;
11 — термофосфаты, томасшлак;
12 — сульфат калия и сульфат калия-
магния; 13 — калийные соли (50% КгО);
14 — калийные соли (20—40% КгО):
i: — известняк.
леза. При этом аммонизацию мож-
но вести до pH = 7, вместо 4,5, без
опасения ретроградации Р2О5, с по-
лучением продукта, содержащего
5% азота.
В некоторых случаях при смеше-
нии получаются удобрения, обла-
дающие лучшими физическими
свойствами, чем исходные компонен-
ты. Например, при смешении супер-
фосфата с сульфатом аммония в ре-
зультате реакции
(NH4)2SO4 + Са(Н2РО4)2 • Н2О + Н2О =
= 2NH4H2PO4 + CaSO4 • 2Н2О
слеживающиеся при хранении
смесь подсыхает и затвердевает
вследствие образования гипса. Она
обладает малой гигроскопичностью,
но для устранения слеживаемости
ее приходится измельчать после дли-
тельного хранения, когда приведен-
ная реакция завершится.
Удобрительные смеси с хороши-
ми физическими свойствами, с не-
большой гигроскопичностью и не
получаются, например, при смеше-
нии фосфатов аммония и хлорида калия с суперфосфатом и суль-
фатом аммония; при смешении их с нитратом аммония или карба-.
мидом получается удобрение, рассеваемость которого при хране-
нии во влажном воздухе ухудшается.
Для решения вопроса о возможности смешения тех или иных
удобрительных солей руководствуются различными диаграммами
смешения удобрений, составленными на основании теоретических
соображений и экспериментальных данных. Однако антагонизм
удобрений изучен еще недостаточно, и поэтому нередко разные диа-
граммы смешения дают противоречивые сведения. На рис. 408 при-
ведена одна из таких диаграмм. Горизонтальные и вертикальные
ряды на этой диаграмме обозначены числами, соответствующими
разным удобрениям. Возможность смешения характеризуется клет-
II роизводство смешанных удобрений
1367
ками, находящимися в месте пересечения рядов: белые клетки ука-
зывают на возможность, черные — на недопустимость смешения.
Заштрихованные клетки соответствуют смесям, в которых нежела-
тельные процессы идут сравнительно медленно. Буквой г обозна-
чены смеси, из которых могут выделяться газы, а буквой р — сме-
си, в которых возможна ретроградация Р2О5.
Из этой диаграммы следует, что, например, все калийные соли
можно смешивать с суперфосфатом, сульфатом аммония, нитра-
тами аммония и натрия и др.; сульфат аммония можно смешивать
с суперфосфатом, преципитатом, нитратом аммония и др. Возмож-
ности смешения удобрений значительно расширяются при добавке
к ним нейтрализующих материалов, что в приведенной диаграмме
не учитывается.
ПРОИЗВОДСТВО СМЕШАННЫХ УДОБРЕНИЙ
Смешанные удобрения изготовляют на химических предприя-
тиях и на специальных тукосмесительных станциях, расположен-
ных в районах потребления удобрений. Смешение удобрений при-
митивными методами производят и сами потребители.
Смесительные установки небольшой мощности работают по пе-
риодической схеме 32’33. Исходные компоненты просеивают и круп-
ные фракции измельчают. Отвешенные в соответствии с рецептурой
количества компонентов загружают в смеситель, перемешивают в
течение нескольких минут и выгружают готовый продукт. В каче-
стве смесителей используют вращающиеся барабаны, шнековые,
тарельчатые и другие аппараты. Емкость смесителей дости-
гает 6 т 34.
Заводское изготовление смешанных и сложно-смешанных удоб-
рений производят по непрерывным схемам 3S~38. Для возможности
непрерывной дозировки компонентов их просеивают через сита с
определенным размером отверстий, крупные фракции измельчают
и возвращают на рассев. Загрузка смесителя исходными материа-
лами и выгрузка из него готовой смеси происходят непрерывно.
При изготовлении гранулированных смешанных и сложносме-
шанных удобрений с добавкой к твердым смесям воды, растворов
солей, кислот, аммиакатов или с аммонизацией их газообразным
аммиаком, процессы смешения, аммонизации, грануляции совме-
щаются в одном непрерывно действующем аппарате — аммониза-
торе-грануляторе б’39’40. Продолжительность пребывания материа-
ла в аппарате ~10 мин. Вследствие выделения тепла реакций тем-
пература в нем повышается до 70—80° и происходит испарение
влаги, однако в большинстве случаев требуется дополнительное вы-
сушивание гранулированного смешанного удобрения топочными га-
зами в прямоточной сушилке до влажности меньше 3%. Высушен-
ный продукт охлаждается воздухом и рассеивается. Крупная фракция
19 M. Е. Поэин
1368
Г л. XXXVI. Смешанные и сложно-смешанные удобрения
дробится и возвращается на рассев, средняя выпускается в ка-
честве продукта, а мелкая возвращается в виде ретура в смеситель.
Количество ретура зависит от рецептуры и характера исходных ма-
териалов; обычно оно находится в пределах 0,1—1 вес. ч. на 1 вес. ч.
готового удобрения. Количеством ретура, а также подачей в слу-
чае необходимости в аммонизатор-гранулятор охлаждающего воз-
духа или греющего пара, можно регулировать температуру смеши-
ваемой массы и соотношение в ней твердых и жидкой фаз 41~43.
При получении гранулированных смешанных удобрений с высо-
ким содержанием азота некоторое его количество теряется при ам-
монизации, сушке и других операциях. Потери снижаются при пре-
дотвращении образования крупных гранул и регулировании темпе-
ратуры введением ретура или другими средствами 44. Получение гра-
нулированных смешанных удобрений облегчается, если часть исход-
ных твердых материалов имеет размер, приблизительно равный за-
данному размеру гранул. При этом увеличивается прочность гранул
и уменьшается расход пара, воды или кислоты при гранулировании45.
В качестве аммонизаторов-грануляторов используют вращаю-
щиеся барабанные, горизонтальные шнековые, наклонные тарель-
чатые46’47 и другие аппараты. Наклонный тарельчатый гранулятор
представляет собой плоский цилиндр диаметром, например, 3,6—
4,5 м, высотой 0,36 м, расположенный под углом 30—50° к гори-
зонту, вращающийся со скоростью 14 об/мин. Внутри имеется не-
подвижный скребок, расположенный вблизи от поверхности та-
релки—днища цилиндра. При работе гранулятора происходит
классификация материала — крупные гранулы перемещаются к
нижней части тарелки и выводятся, а мелкие остаются в аппарате,
пока не достигают требуемого размера. В таком грануляторе осу-
ществляют и аммонизацию удобрений.
Для усовершенствования работы тарельчатого гранулятора
предложено над ним установить конический или полусферический
зонт, смесь сухих материалов подавать на вершину зонта, где
устанавливается двойное сопло для подачи газообразного аммиа-
ка и кислоты. Смесь падает на днище тарельчатого гранулятора в
виде завесы, непрерывно смачиваемой кислотой и обрабатываемой
аммиаком 48’49.
Предложено также получать гранулированные удобрения мето-
дом прессования50'51.
На рис. 409 изображена схема установки для производства
сложно-смешанных удобрений. Установка является универсальной.
На ней может быть приготовлено сложно-смешанное гранулирован-
ное удобрение, составленное из любого сочетания компонентов с
любым соотношением питательных веществ7. Процесс смешения н
взаимодействия компонентов проводится в аммонизаторе-грануля-
торе 13. В зависимости от за_щнного состава удобрения в аммони-
затор-гранулятор подаются: из бункера 17, через ленточный весе*
Вова NH4N0, Н.РО* H»SO4 Аммиакаты
19:
1370
Гл. XXXVI. Смешанные и сложно-смешанные удобрения
вой дозатор 16, ленточным конвейером 15 сухие калийные, азотные
фосфорные удобрения и ретур; из напорных баков 1 через доза-
торы 2 дозируются: плав аммиачной селитры, фосфорная и серная
кислоты и аммиакаты. Газообразный аммиак поступает из испари-
теля аммиака 14. Для лучшего гранулообразования предусматри-
вается подача воды, которая распыляется с помощью сжатого воз-
духа. В аммонизаторе-грануляторе и за счет реакционного тепла
испаряется 30—35% введенной воды. Полученные гранулы элева-
тором 9 подаются в бункер 3, из которого поступают в сушильный
барабан 4, где они высушиваются до конечной влажности 1% (на-
чальная влажность около 5%). Сушка осуществляется топочными
газами с температурой около 200°. Температура гранул на выходе
из барабана 70—80°. Сухой продукт классифицируется на грохоте И
и измельчается на дробилке 10. Мелкая фракция возвращается в
качестве ретура в аммонизатор-гранулятор, а фракция готового
продукта с размером гранул 2—4 мм или 1—3 мм охлаждается
до 30—40° в охладителе с кипящим слоем 12 и затем поступает в
барабан-кондиционер 5, где подвергается омасливанию и опудри-
ванию. Продукт хранится в упакованном виде.
В СССР сконструирован и испытан полуавтомат для расфасов-
ки минеральных удобрений с производительностью 600 мешков в
1 ч по 30—50 кг при точности дозировки ±1%. О машинах и при-
способлениях для дробления слеживающихся минеральных удобре-
ний см. 52> 53.
РАСЧЕТ СОСТАВА ТУКОСМЕСЕЙ
Для расчета смешения удобрений пользуются треугольной диа-
граммой. Вершины равностороннего треугольника (рис. 410) соот-
ветствуют простым удобрениям, причем содержание в них пита-
тельных веществ — Р2О5, N и КгО — принимается за 100%. Фи-
гуративные точки, лежащие на сторонах треугольника, соответ-
ствуют составу двойных, а точки внутри треугольника — составу
тройных удобрений в пересчете на питательные вещества.
Например, требуется решить вопрос, в каком соотношении сле-
дует смешать аммонизированный суперфосфат, содержащий 14%
Р2О5 и 2,5% N, сульфат аммония, содержащий 21% N и калийную
соль (сильвинит, обогащенный хлористым калием), содержащую
условно 42% К2О, чтобы получить смешанное удобрение с соотно-
шением N : Р2О5: К2О, равным 2:2: 1.
Для этого наносят на диаграмму точку а, соответствующую со-
ставу аммонизированного суперфосфата (отношение отрезка N— в
к Р2О5 — а равно отношению содержания Р2О5 к N в аммонизиро-
ванном суперфосфате, т. е. 14:2,5). Затем наносят на диаграмму
точку Ь, соответствующую составу смешанного удобрения, и про-
водят через вершину треугольника, отвечающую 100% КгО, и точ-
ку b прямую линию до пересечения со стороной N — Р2О5 в точ-
Расчет состава тукосмесей
1371
too’/.
р,о.
too%
N
Рис. 410. Графический
смешения удобрений.
(100% N) : аммони-
супер фосфат [100%
1: 4‘7 ' "
Разделив
на процентное
питательных
исходных
too %
KtO
расчет
4-10
7+ 10 ‘ 7+10 “
последние
содержание
веществ в
удобрений,
удобрений
получим весовое отношение
1 . 1,65 . 2,35
в смеси: 42 . 21 • 14 + 2>5 -
ке с. Отношение длины отрезка ас к с — N дает отношение количе-
ства сульфата аммония в пересчете на 100% N к количеству ам-
монизированного суперфосфата в пересчете на 100% (P2O5 + N).
Измерив отрезки, устанавливаем, что это отношение равно 7 : 10.
Отношение длины отрезка Ьс к длине отрезка b — КгО дает отно-
шение количества калийной соли в пересчете на 100% КгО к сумме
сульфата аммония и аммонизиро-
ванного суперфосфата в пересчете
на 100% (N + P2O5). Это отношение
. равно 1:4. Из этих отношений сле-
дует, что для получения смешан-
ного удобрения заданного состава
необходимо, чтобы исходные удо-
брения были взяты в соотношении
калийная соль (100% КгО) : суль-
фат аммония
зированный
(РгО5 + N)] =
1 : 1,65 : 2,35.
числа
полезных
каждом из
натуральных исходных
= 0,024:0,079:0,145.
Приняв вес смеси за 100%, устанавливаем, что смешанное удо-
брение с заданным соотношением питательных веществ должно
состоять из 9,7% калийной соли, 31,9% сульфата аммония и 58,4%
аммонизированного суперфосфата.
Та же задача может быть решена аналитическим путем54. Обо-
значим: требуемое соотношение питательных веществ в смешанном
удобрении N : Р2О5: КгО=Л : В : С, процентное содержание этих
веществ в смешанном удобрении соответственно а, Ь, с, а в трех
исходных удобрениях 1) щ, blt clt 2) а2, b2, с2 и 3) а3, Ь3, с3. Иско-
мое процентное содержание исходных удобрений в смеси обозна-
чим соответственно х, у и z. Для вычисления шести неизвестных
(а, Ь, с, х, у и г) составляем шесть уравнений:
х , у , z
fl = aiT00+fl2T00 + “3 Т(Д:
6 = 61 Too + &2Тоо + &3Тоо; У=Т; 7 = -с : * + y+*-*w
В примере, рассмотренном выше, заданы следующие величины:
ci = 0
с2 = 0
с3 = 42
с = с‘ 100 +Са 100 +Сз 100 ’
- —• °.-А. х + у+г=100
Л=2
В = 2
С=1
bt = 14
Ь2=0
Ь3 = 0
01 = 2,5
02 = 21
о3 = О
1372
Гл. XXXVI. Смешанные и сложно-смешанные удобрения
Подставляя эти величины в уравнения и решая систему уравне-
ний, получим:
а = 8,16 6 = 8,16 с = 4,08
х = 58,32 у = 31,96 z = 9,72
т. е. тот же результат, что и с помощью треугольной диаграммы.
Если задано не соотношение, а содержание питательных эле-
ментов в смеси, то расчет ведут следующим путем55. Обозначим
содержание N, Р2О5 и К2О в 1 т каждого из исходных простых удо-
брений соответственно через 11, р и к %. Тогда 1 т смешанного удо-
брения будет содержать следующие количества простых компонен-
тов
N 4. Р + К 4-Г 1 ,
---1 1—— + С = I т
п р k
здесь N, Р и К — заданное содержание питательных веществ (N,
Р2О5, КгО) в смеси, а С — вес добавки для доведения смеси до еди-
ницы измерения. Например, для получения смешанного удобрения
N—Р2О5—КгО = 5—10—10 из сульфата аммония (п = 20,5); про-
стого суперфосфата (р = 20) и хлористого калия (£ = 60):
NPK 5 10 . 10 ПАЮ
---1--+ "т* с + + "гтг “ 0,912 т
п р k 20,5 20 60
Добавка, состоящая из инертного материала (доломита, гипса,
фосфогипса, кизельгура, извести, глины и т. п.):
С= 1 -0,912 = 0,082 т
Удобрительную смесь марки 10—10—10 из тех же компонентов
10 । 10 , 10 . . ..
приготовить невозможно, так как^у-ф —%--^0 = 1,155 > 1. Макси-
мальная концентрация питательных веществ в удобрении с соот-
ношением N : Р2О5: КгО = 1 : 1 : 1 будет в этом случае равняться
-Лее —гпк' —гцк" = 8,66 — 8,66 — 8,66. Для получения удобрения
1,100 1,1ОО 1,100 J
марки 10—10—10 было бы необходимо или использовать обогащен-
ный суперфосфат вместо простого, или заменить 20,5%-ный суль-
фат аммония 34,5%-ным нитратом аммония. В последнем случае
10 , 10 . 10 п
получается: -^ + 20 +%0 = °’957-
В случае Использования для смешения двойных удобрений рас-
чет ведут тем же способом, но в этом случае необходимо уравно-
весить отношение элементов в двойном удобрении для получения
необходимой общей формулы, выбрав для добавки соответствую-
щее простое удобрение. Например, нужно приготовить смесь 10—
—20—20 на основе моноаммонийфосфата 11—48—0, 60 %-ной ка-
лийной соли и третьего продукта, который еще необходимо вы-
брать. Отношение N : Р2О5 в моноаммонийфосфате 11:48 = 0,23
меньше требуемого для смеси отношения 10:20 = 0,5. Следова-
тельно, нужно рассматривать моноаммонийфосфат как источник
Расчет состава тукосмесей
1373
р20а и пополнить смесь соединением, которое служило бы источ-
ником азота. Имеем:
Р 20
Вес моноаммонийфосфата . . — = = 0,418 т
р 48
» калийной соли.................. — = — 0,334 »
й оО
Остаточный вес............ 1 — 0,418 — 0,334 = 0,248 »
Всего: 1,000 т
Но с моноаммонийфосфатом вносится уже 11-0,418=4,6% азо-
та. Дополнительно нужно внести 10—4,6=5,4 % азота. Минималь-
ное содержание азота в до-
бавке должно быть 5,4 :
: 0,248 = 21,8%, что больше,
чем в сульфате аммония;
значит для добавки следует
использовать нитрат аммо-
ния или карбамид.
Для определения количе-
ства исходных компонентов
смешанного удобрения мо-
жно воспользоваться номо-
граммой (рис. 411) 7’32. Для
этого через точку содержа-
ния питательного вещества в
заданной тукосмеси и точку
содержания его в исходном
компоненте (наклонная ли-
ния) проводят прямую до
пересечения с левой шкалой
количеств исходного веще-
ства в получаемой смеси.
Пусть, например, требуется
приготовить смесь состава
14—14—14 из аммиачной се-
литры (34,8% N), двойного
суперфосфата (46% Р2О5) и
хлористого калия (60%
КгО). Проводя прямую
(пунктир) из точки 14 на
правой шкале через точку
’14,8 на наклонной линии до
Рис.,411. Номограмма для определения ко-
личества компонентов в смешанных удо-
брениях.
левой шкалы, получаем здесь точку 40, т. е. на 100 вес. ч. смеси
следует взять 40 вес. ч. аммиачной селитры. Таким же образом на-
ходим расход двойного суперфосфата — 30 вес. ч. и хлористого
1374
Гл. XXXVI. Смешанные и сложно-смешанные удобрения
калия—23 вес. ч. Суммарное количество исходных простых удо-
брений равно 40 + 30 + 23 = 93%; остальные 7% составляют конди-
ционирующие добавки и нейтрализующие вещества.
ЛИТЕРАТУРА
1. Д. Г. Грачев, Хим. пром., № 5, 257 (1958).—2. Д. Г. Грачев, Сме-
шанные удобрения, Сельхозгиз, 1949.—3. Э. В. Брицке, Труды НИУ, в. 73,
стр. 35.—4. Л. Н. Прянишников, Агрохимия, Сельхозгиз, 1940.—
5. А. И. Дубовицкий, Ф. Г. Марголис, Научно-техн, информ, бюлл.,
№ 4, 1957, стр. 63. — 6. Т. П. У и а н я н ц, Там же, стр. 89. — 7. Ф. Г. Марго-
лис, Т. П. У и а н я н ц. Производство комплексных удобрений, Изд. «Химия»,
1968.—8. Farm Chemicals Handbook, 1966, Ohia (USA), 1967. — 9. T. R. Vis-
wanathan, Indian. Chem. Eng., 8, № 4, 116 (1966). —10. Phos. a. Pot., № 23,
22 (1966).
И. Франц, пат. 1466484, 1966. —12. Япон. пат. 7105, 1965. —13. Англ. пат.
1026937, 1966. —14. Хим. пром, за рубежом (НИИТЭХИМ), № 7, 55 (1964).—
15. Comm. Fertilizer, 110, № 4, 48 (1965). —16. Р. Travis, Т. Н i g n е t, Balk
Blending of Fertilizers, London, 1965. —17. G. Hoffmeister, S. Watkins,
J. Silver berg, J. Agr. Food Chem., 12, № 1, 64 (1964). — 18. J. Hardesty,
G. Dickey, В. 01 i k e, Farm. Chemie., 124, № 2, 32 (1961).'—19. A. M. Ду-
бовицкий, Ф. Г. Марголис, ЖПХ, 29, № 3, 321 (1956). — 20. Н. Д. Смир-
нов, ЖХП, № 1, 19 (1940).
21. О. Пудовкина, М. Канаш, Л. Арутюнова, Хлопководство, № 6,
9 (1951). —22. Япон. пат. 8893, 1967.— 23. Англ. пат. 1026084, 1966.—
24. I. О. Hardesty, Chem. Eng. Progr., 51, № 6, 291 (1955).— 25. A. V. Slack,
Agr. Food Chem., № 7, 568 (1955). — 26. Changes in Fertilizer Distribution and
Markating, Knoxville (TVA), 1965.—27. Производство и применение минераль-
ных удобрений, пер. с англ, под ред. А. В. Петербургского, Изд. «Колос»,
1965. — 28. Conference on the Development of Fertilizer Industry in Acta and
the Far Fast, Bombey, 1963. — 29. Г. В. Мельников, H. H. Ромашева,
Хим. пром, за рубежом (НИИТЭХИМ), № 6, 27 (1967).—30. J. F a z а и,
W. J a n i с z е k, Chernik (польск.), 19, № 1, 1 (1966).
31. L. S. Andress, Chim. et ind., 73, № 3, 531 (1955). — 32. Научно-техн,
информ, бюлл. НИУИФ, № 4 , 74 (1957). — 33. А. Д. Хоменко, О. Н. Сос-
новая, М. Н. Малеев, Хим. пром. Украины, № 4 (26), 23 (1966). —
34. R. Walsh, Farm Chemie., 127, № 10, 74 (1964). — 35. Chem. Eng., 61, № 7,
136 (1954).— 36. J. Agr. Food Chem., № 4, 318 (1956).—37. Д. Г. Грачев.
Л Б. Гриншпан, H. В. Бабенко, Жури. ВХО, 7, № 5, 513 (1965).—
38. R. Church; Farm. Chemie., 126, № 12, 52 (1963). — 39. F. Nilsson, Agr.
Chem., 16, № 5, 45 (1961). —40. G. Retting, Brit. Chem. Eng., 10, № 9, 608 (1965).
41. H. К о t a b 1 e, Asahi Evening News, № 1, 60 (1965). — 42. Kremagai,
Rapp, Hardesty, J. Agr. Food Chem., № 2, 25 (1954).— 43. Ch. Waters,
W. Arnold, W. Payne, ibid., 3, № 3, 218 (1955).— 44. T. P. H i g n e 11,
Farm. Chemie., 121, № 6, 41, 44, 46, 48 (1958). —45. H. B. Phillips a. oth.,
J. Agr. Food Chem., 6, № 6, 449 (1958).—46. Англ. пат. 1057459, 1967.—
47. R. D. J о u n g, J. W. M с C a m y, Canad. J. Chem. Eng., 45, № 1, 50 (1967) —
48. Пат. ФРГ 1140210, 1966. —49. Пат. ФРГ 1146511, 1967. — 50. Farm. Chemie.,
128, № 5, 48 (1965).
51. Авт. свид. 193538, 1967.—52. Н. В. Обыденкнн, Машины и приспо-
собления для дробления слежавшихся минеральных удобрений, изд. ВНИИП-
сельхозхим, Рязань, 1968. — 53. Временная инструкция по технике безопасности
прн работе на погрузочно-разгрузочных машинах в складах минеральных удоб-
рений, изд. ВНИИПсельхозхим, Рязань, 1967. — 54. J. Н. Payne, R. Т. W е fa-
te г, J. Agr. Food Chem., 8, № 3, 164 (I960).—55. Chim. et ind., 93, № 4, 391!
№ 5, 517 (1965).— 42. E. Mapstone, Chem. Eng., 72, № 22, 176 (1965).
Глава XXXVII
ЖИДКИЕ УДОБРЕНИЯ
ОБЩИЕ СВЕДЕНИЯ
Жидкие удобрения (ЖУ)1-8 могут быть простыми и комплекс-
ными. Простые жидкие удобрения представляют собой растворы
аммиачной селитры, карбамида, кальциевой селитры и их смесей
в жидком аммиаке или в концентрированной аммиачной воде —
так называемые аммиакаты 10’ ". В качестве жидких удобрений осо-
бенно широко применяют также жидкий аммиак и аммиачную
воду (20—22% NH3) 12- 13.
Жидкие комплексные удобрения (ЖКУ) приготавливаются
обычно на основе экстракционной или термической фосфорной кис-
лоты. В последнее время для этой цели стали использовать супер-
фосфорную и полифосфорные кислоты, что дает возможность полу-
чать более концентрированные жидкие удобрения. Производят
также суспендированные жидкие удобрения 14~18.
Еще в 1932—1935 гг. было показано, что аммиак и аммиакаты
так же усваиваются растениями и дают такой же эффект, как и
обычные, твердые азотные удобрения19-20. Производство же их
проще и дешевле, чем производство твердых удобрений. При рас-
творении в аммиаке нитрата аммония и нитрата кальция или их
смесей давление паров аммиака значительно снижается, и при
определенной концентрации солей для обычной температуры оно
равно атмосферному 21~23.
Аммиакаты на основе нитрата аммония и карбамида применяют
и для аммонизации суперфосфата и тукосмесей 24’25.
Особенно широкое распространение жидкие удобрения полу-
чили в США: в 1966 г. было произведено 1248 тыс. т жидких ком-
плексных удобрений (в натуре) 9’26'29, в 1969 г. общее производ-
ство жидких удобрений около 4 млн. т 30. Жидкие азотные и ком-
плексные удобрения применяются в большинстве стран Европы31,
а также в СССР32.
1376
Гл. XXXVII. Жидкие удобрения
Применение жидких удобрений обеспечивает- возможность пол-
ной механизации работ по погрузке, выгрузке и внесению удобре-
ний. Трудовые затраты их по внесению при условии допосевного
внесения фосфорных и калийных удобрений, сокращаются в 2—
3 раза по сравнению с затратами на внесение твердых удобрений.
Достигается более равномерное распределение удобрений в поч-
gg 33-36
Некоторые виды жидких удобрений могут применяться для под-
кормки растений с самолетов и автомобилей.
Непосредственное использование аммиака 37> 38 и аммиачной во-
ды в качестве жидких азотных удобрений обеспечивает возмож-
ность ускоренного строительства азотных заводов по «короткой»
схеме, без цехов переработки 'аммиака в азотную кислоту и ам-
миачную селитру. При этом сокращается объем строительных ра-
бот по энергетическому, транспортному и складскому хозяйству,
вспомогательным службам и жилью.
Значительно сокращаются также эксплуатационные затраты и,
следовательно, удешевляются удобрения, так как себестоимость
единицы азота в аммиаке приблизительно на 35% ниже, чем в са-
мом дешевом твердом азотном удобрении — аммиачной селитре.
Отпадает необходимость в бумажных мешках (или другой таре),
расход которых на упаковку продукции одного завода составляет
много миллионов штук. Кроме того, жидкие удобрения не обла-
дают такими отрицательными свойствами твердых удобрений, как
слеживаемость, сегрегация и т. п.39'41.
Наряду с преимуществами, в производстве и применении жид-
ких удобрений имеются известные трудности и недостатки. Стои-
мость Р2О5 в крепкой фосфорной кислоте, требующейся для произ-
водства концентрированных ЖКУ, выше, чем в более слабой кис-
лоте, пригодной для получения твердых удобрений.
В жидких удобрениях водных — растворах солей — в случае
большой их концентрации происходит высаливание, кристаллиза-
ция солей при понижении температуры42. Применение же менее
концентрированных растворов приводит к необходимости перево-
зить большие количества растворителя — воды. Поэтому стремят-
ся найти такие композиции жидких удобрений, которые имели бы
высокие концентрации и низкие температуры кристаллизации (за-
мерзания). Для хранения жидких удобрений требуется сооружение
резервуаров большой емкости, так как они используются лишь в
течение относительно короткого времени.
Жидкий аммиак экономичнее хранить при низком давлении и
искусственном охлаждении; для этой цели применяют сферические
емкости вместимостью 2—14 тыс. т 43. Применение жидких удобре-
ний требует капиталовложений на организацию распределительных
пунктов вблизи районов потребления, создания специального обо-
рудования для внесения удобрений в почву44, а также парка ци-
Физико-химические свойства
1377
стерн (железнодорожных и автомобильных) для их перевоз-
кИ 45-47; применение безводного аммиака в качестве удобрения тре-
бует специального оборудования, рассчитанного на повышенное
давление. Возможна транспортировка жидких удобрений по трубо-
проводам 48-49, а жидкого аммиака в контейнерах50'51.
Существенным недостатком некоторых жидких удобрений яв-
ляется их корродирующее действие4. В особенности это относится
к растворам аммиакатов нитрата аммония, обладающим повышен-
ными коррозионными свойствами по отношению к черным метал-
лам. Это затрудняет производство, хранение, транспортировку и
внесение удобрений в почву, так как связано с применением доро-
гостоящих материалов (нержавеющей стали, алюминия и др.).
Аммиакаты, аммиачную воду и особенно безводный аммиак во
избежание потерь азота требуется вносить в почву на глубину не
менее 12—15 см. Поверхностное внесение жидких удобрений типа
аммиакатов недопустимо, так как при этом будут большие потери
аммиака. Кроме того, возможны ожоги листьев и стеблей растений
при попадании на них аммиака и аммиакатов 52-53. Поэтому для
внесения в почву жидких удобрений применяют машины.
Оценка технико-экономических факторов, характеризующих
производство и применение твердых и жидких удобрений, показы-
вает, что в итоге преимущества остаются за жидкими удобре-
ниями 54-59.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Жидкий аммиак содержит .82,3% N и является самым концен-
трированным азотным удобрением. Под атмосферным давлением
аммиак кипит при —33,35° и замерзает при —77,7°. Абсолютное
давление пара над жидким аммиаком:
Температура, °C.....................—30 —10 0 10 20 40 50
Давление, ат ..................... 1,18 2,87 4,24 6,08 8,46 15,34 276
Жидкий аммиак хранят и транспортируют в стальных цистер-
нах, рассчитанных на давление 20—30 ат. Плотность газообразного
аммиака при нормальных условиях 0,77 кг/мъ. Согласно ГОСТ
6221 70, жидкий аммиак высшего, Г- и 2-го сортов должен содер-
жать, соответственно, не менее 99,95, 99,9, 99,6% NH3 и не более
0,05, 0,1, 0,4% влаги и 2,0, 8,0, 20,0 мг/л масла; железа в высшем и
1-м сортах должно быть не более 2,0 мг/л. По ГОСТ 5.1185—72 в
аттестованном продукте должно быть не менее 99,96%' NH3 и не
более 0,04% влаги, 2 мг/л масла и 1,0 мг/л железа.
в Аммиак очень хорошо растворяется в воде: 1 объем воды при
6 и при парциальном давлении NH3, равном атмосферному, рас-
творяет 1176 объемов аммиака (приведенных к нормальным усло-
виям), а при 20° — 702 объема. Пол. атмосферным давлением мак-
симальная концентрация NH3 в воде при 0° равна 46,7%, при 50°—
1378
Гл. XXXVII. Жидкие удобрения
18,6%. Зависимость между растворимостью аммиака в воде, давле-
нием его над раствором и температурой показана на рис. 412.
Аммиачную воду (аммиак водный технический, ГОСТ 9—67)
производят двух марок: А — для удобрения сельскохозяйственных
культур и Б — для промышленности. Каждая марка имеет 1 и 2
сорт. Для 1 сорта установлено содержание не менее 25%, для
2 сорта — 22% NH3. Содержание нелетучего остатка установлено
только для марки Б (не более
В продукте марки А допускает-
ся содержание не более 8 г/л
СО2 и 0,01 г/л меди.
Давление NH3, am NH3 20 зо 40 so 60 70 во во що
Рис. 412. Изотермы раствори- Рис. 413. Диаграмма растворимости в си-
мости аммиака в воде. стеме NH4NO3—NH3—Н2О.
Вследствие относительно небольшого давления пара над ам-
миачной водой ее хранят и транспортируют в стальных цистернах
или закрытых резервуарах, рассчитанных на давление 1,5—2 ат.
Твердые аммиакаты представляют собой комплексные кристал-
лические соединения, образующиеся при взаимодействии некото-
рых твердых солей (или их водных растворов) с жидким или газо-
образным аммиаком. Состав аммиакатов, полученных на основе
аммиачной селитры, соответствует формуле NH4NO3-nNH3-mH2O;
аммиакаты на основе кальциевой и аммиачной селитры имеют сле-
дующий состав: CalNOsC-xNIKNOs'/iNHs-^HsO. Твердый нитрат
аммония при температуре от —15 до +25° энергично поглощает
газообразный аммиак и переходит в жидкое состояние — обра-
зуется так называемая жидкость Дайверса. При —10° ее состав от-
вечает формуле NH4NO3-2NH3. С повышением температуры проис-,
ходит выделение аммиака и при 28° образуется твердая соль]
NH4NO3-NH3, которая легко теряет аммиак. Жидкие аммиакаты—1
светлые жидкости (допускается желтоватая окраска), плотность]
их зависит от состава и колеблется в пределах 0,9—1,25 г/см?. Да4
Физико-химические свойства
1379
вление пара над аммиакатами значительно ниже, чем над жидким
аммиаком.
Растворимость некоторых азотнокислых солей в воде, в аммиач-
ной воде и в жидком аммиаке приведена в табл. 105.
ТАБЛИЦА 105
Растворимость азотнокислых солей в разных растворителях
Соль Температура, °C Растворимость соли в г на 1000 г
воды 75%-ной ам- миачной ВОДЫ 85 %-ной ам- миачной воды 100%-ного жидкого аммиака
NaNO3 0 733 736,9 828,7 1274
25 927 — — 986,7
Ca(NO3)2 0 702,9 759,3 719,3 821,7
25 956,6 — — 803,5
KNO3 0 - 132,5 135,3 108,1 105,2
25 382,5 — — 103,4
NH*NO3 25 2090 — — 3587
Растворимости аммиачной селитрьци карбамида в аммиаке и
аммиачной воде показаны на рис. 413 и 41460-62. Растворимость
в системе CO(NH2)2—
—NH4NO3—NH3—Н2О
при 0 и 30° представлена
на рис. 41563. В этой си-
NH3 п л
Стеме ПРН NH3 + H2O =0’4
в твердой фазе образует-
ся комплекс CO(NH2)2-
•O,11NH3, а при отноше-
NH3 п к
НИИ nh?+h2o =0’5 ком‘
плекс имеет состав
CO(NH2)2-0,25NH3.
Температура высали-
вания аммиакатов пони-
жается с увеличением со-
держания в них аммиака
CO(NHX)X
Рис. 414. Диаграмма растворимости в системе
CO(NH2)2-NH3-H2O.
и уменьшением содержа-
ния воды. Аммиакат со-
става 55,14% NH4NO3,
25,69% NH3 и 18,97% Н2О
имеет температуру высаливания —27,5°. Введение в аммиакат
CO(NH2)2 или Ca(NO3)2 также приводит к понижению темпера-
туры высаливания 64-67.
1380
Гл. XXXVII. Жидкие удобрения
На рис. 416 и 417 показано изменение плотности аммиакатов
nh4no3, %
Рис. 415. Растворимость в системе
CO(NH2)2— NH4NO3—NH3— Н2О при 0 и 30°.
„ NH3
Числа на кривых — отношения .
NHg+HgU
NH3 + CO2 над растворами понижается,
зации повышаются ®8.
аммиака, а на рис. 418 и
419 — зависимость обще-
го давления пара над ам-
миакатами от содержания
соли, аммиака и воды в
растворе.
Производство аммиа-
катов, содержащих поми-
мо карбамида карбонат
аммония, позволяет с на-
ибольшей простотой ути-
лизировать отходящие га-
зы цеха карбамида.
О равновесиях в системе
(NH4)2CO3—CO(NH2)2—
—NH4NO3—Н2О см.®8.
При добавлении к таким
аммиакатам микроэлемен-
тов (соединений В, Со,
Mo, Zn, Си) в количестве
до 4% давление паров
а температуры кристалли-
Рис. 416. Плотность в системе
NH4NO3—NH3—Н2О при 15°.
Рис. 417. Плотность в системе |
CO(NH2)2—NH3—Н2О при 15°. п
Данные о некоторых свойствах аммиакатов, применяемых в
США, приведены в табл. 106. В табл. 107 и 108 приведены составь!
Физико-химические свойства
1381
аммиакатов, имеющих достаточно низкую температуру выделения
твердой фазы. Жидкие комплексные удобрения производятся дву-
мя способами: 1) нейтрализацией фосфорной кислоты аммиаком
с последующим смешением полученного раствора с КС1 или К2СО3,
Рис. 418. Общее давление пара
иад системой
NH4NO3—NH3—Н2О при 20°.
Рис. 419. Общее давление пара
над системой
CO(NH2)2—NH3—Н2О при 20°.
или другими солями, вводимыми для получения заданного соотно-
шения питательных веществ (горячий способ); 2) смешением пред-
варительно полученных растворов фосфатов или полифосфатов ам-
мония с другими солями — твердыми или растворенными (холод-
ный способ).
ТАБЛИЦА 106
Жидкие азотные удобрения, применяемые в США
Состав, вес. % Содержание азота, вес. % Абсолютное давление паров при 40°, ат Температура выделения твердой фазы, °C
nh3cbo6 NH4NO3 CO(NH2)2 Н2О
На основе нитрата аммония
26,3 55,5 18,2 41,0 2,12 -31,7
22,2 65,0 — 12,8 41,0 1,7 —6,1
16,6 66,8 16,6 1 37,0 1,07 +8,9
57,3 — 42,7 20,0 1,0 + 5,56
— 45,7 — 54,3 16,0 1,0 -11,7
На основе нитрата ам м о н и я и карбамид а
19,0 58,0 11,0 12,0 41,0 1,7 -13,9
44,3 35,4 20,3 32,0 1,0 7
— 39,5 30.5 30,0 28,0 1,0 -17,2
1382 Гл. XXXVII. Жидкие удобрения
ТАБЛИЦА 107
Составы аммиакатов иа основе нитрата аммония и карбамида
Состав, вес. % Содержание азота, вес. К Температура выделения твердой фазы °C
^Нзсвоб NH4NO3 CO(NH2)j Н2О
23-26 53-56 24-18 37,5-41 -25
20,1 45,1 15,2 19,6 39,4 -32,1
22,6 40,1 15,1 22,2 39,7 -36,2
20,1 40,3 20,0 19.6 • 40,0 -29,5
20,1 50,0 10,1 19,9 38,7 -28,3
ТАБЛИЦА 108
Составы аммиакатов с карбамидом и карбонатом аммония
Состав, вес. % Содержание азота, вес. % Температура выделения твердой фазы, °C
CO(NH2)2 NH3 со2 н2о
22,75 10,22 13,12 53,91 19,06 -15,2
24,04 9,18 10,74 56,04 18,77 -15,4
19.9 11,9 11,38 57,12 18,84 -24,5
Большое значение имеет взаимная растворимость компонентов
при различных условиях, так как одним из основных требований,
предъявляемых к жидким комплексным удобрениям, является —
низкая температура кристаллизации солей во избежание затрудне-
ний при их использовании70’71.
В системе NH4H2PO4—NH4NO3—КС1—Н2О 72 в диапазоне весо-
вых отношений NH4H2PO4: NH4NO3 от 0,5 до 2,66 и общей концен-
трации солей в насыщенном растворе 25, 28, 30, 32 и 34% увеличе-
ние содержания КС! в смеси солей до 25—35% приводит к пони-
жению температуры начала кристаллизации от 25—35 до 2—10°.
Дальнейшее увеличение содержания КС1 в смеси приводит к повы-
шению этой температуры. Стабильный насыщенный раствор, в ко-
тором отношение КО : NH4H2PO4: NH4NO3 = 35 : 32,5 : 32,5, при
24,5° имеет общую концентрацию солей 34 %, Наиболее низкая тем-
пература начала кристаллизации, равная 1,5°, наблюдается при от-
ношении КС! : NH4H2PO4: NH4NO3<=46,5 : 26,75 : 26,75 и общей кон-
центрации их в насыщенном растворе 28%. С повышением темпе-
ратуры содержание питательных веществ в растворе увеличивается,
а с уменьшением отношения N : Р2О5, т. е. при увеличении в
смеси доли Р2О5, не изменяется. Так, например, содержание пита-
Физико-химические свойства
1383
тельных веществ в растворе при отношении N : Р2О5: К2О=1 : 1 : 1
составляет около 17% при 15° и 19% при 25°, а для отношения
N:P2O5:K2O = 1 :2: 1 при 15° — 16,5%, при 25° — 18,9%.
В системе NH4H2PO4—NH4NO3-CO (NH2)2—Н2О 72'73 в интер-
вале температур от —7 до 50° и весовых отношений NH4H2PO4:
: NH4NO3 от 0,5 до 4 при общей концентрации солей в насыщенном
растворе 30, 35, 37 и 40% повышение содержания карбамида в
смеси сухих солей от 5 до 65% приводит к увеличению концентра-
ции насыщенного раствора. В указанном диапазоне концентраций
и температур наиболее низкая температура выделения твердой фа-
зы (от 0 до —7°) наблюдается при общем содержании в системе
30—35% солей и 50% карбамида в сухой массе солей. Понижение
температуры кристаллизации растворов от 30 до 10° при повышении
содержания в них солей может быть достигнуто либо при одном и
том же отношении NH4H2PO4: NH4NO3 за счет увеличения содер-
жания карбамида в общей массе солей, либо при одном и том же
содержании карбамида за счет изменения отношения NH4H2PO4:
: NH4NO3 от 1 до 4 и более.
О совместной растворимости компонентов жидких комплексных
удобрений, приготовленных на основе термической фосфорной кис-
лоты см.74-77. На рис. 420 представлены изотермы многокомпонент-
ных систем, позволяющих определить возможную концентрацию
питательных веществ в растворе в зависимости от заданного со-
отношения N : Р2О5: К2О, а также какой солью насыщен раствор и
какая твердая фаза будет из него выделяться при охлаждении
ниже 0°. Например, при соотношении N : Р2О5 : К2О=1 .-1:1, мож-
но приготовить жидкое комплексное удобрение из карбамида, фос-
форной кислоты, аммиака, хлористого калия и воды с общим со-
держанием питательных веществ 28% (рис. 420, а); при замене
карбамида нитратом аммония — 17% (рис. 420,6); при одновре-
менном применении карбамида и нитрата аммония — 22%
(рис. 420, в).
На рис. 421 представлена зависимость температуры кристалли-
зации жидких комплексных удобрений от концентрации питатель-
ных веществ 74.
На основе полифосфорных кислот 78-81 при их аммонизации га-
зообразным аммиаком с добавлением воды или аммиачной во-
дой получаются жидкие комплексные удобрения с соотношением
,М : Р2О5 = 1:3. Содержание питательных веществ в них при кон-
центрации исходной полифосфорной кислоты 74—83% Р2О3 соста-
вляет 42—48%, что на много превышает концентрацию питатель-
ных веществ в жидких комплексных удобрениях, полученных на
Основе термической или экстракционной фосфорной кислоты. Со-
отношение питательных веществ в удобрении может быть вырав-
нено растворением в них карбамида или добавлением смеси рас-
творов карбамида и нитрата аммония.
I
Физико-химическиесвойства
1385
Рис. 420. Изотермы многокомпонентных систем при 0° (за-
штрихована область кислых растворов):
а — система карбамид — аммиак — фосфорная кислота—хлористый калий-
вода (NH3 : РО4 — 1,6); б —система нитрат аммония — аммиак — фосфор-
ная кислота — хлористый калий —вода; в —система карбамид — нитрат
аммония — фосфорная кислота — хлористый калий — вода.
На диаграмме (рис. 422) показана растворимость фосфатов и
полифосфатов аммония80-82, полученных на основе ортофосфорной
кислоты (54% Р2О5) и полифосфорных кислот (76 и 79% Р2О5).
С повышением концентрации исходной фосфорной кислоты увели-
чивается концентрация питательных веществ в жидких удобрениях,
соответствующая определенной степени аммонизации кислот.
ТАБЛИЦА 109
Составы двойных жидких комплексных удобрений
Соотношение питательных веществ Марки
из ортофосфорной кислоты (52-54% Р2О5) из полнфосфорной кислоты (78-79% Р2О5)
4:1:0 16- 4-0 24- 6-0
3: 1: 0 18- 6-0 24- 8-0
2:1:0 16- 8-0 22-11-0
1:1:0 13-13-0 19-19-0
1:2:0 9-18-0 15-30-0
1:3:0 8-24-0 12-36-0
1386
Гл. XXXVII. Жидкие удобрения
В табл. 109 приведены составы двойных жидких комплексных
удобрений, которые могут быть получены на основе ортофосфорной
и полифосфорной кислот 80’83. В табл. НО приведены составы трой
ных жидких комплексных удобрений с соотношением N: Р2О5
: К2О = 1 : 1 : 1 и 1 : 2: 1, с температурой насыщения около 0°, кото
рые могут быть получены на основе жидких комплексных удобре
38
36
34
32
30
22
20
’8
6
и
5
5
Е
о
Е
5
е
5
36
29
26
24
22
20
19
16
-12-8-4 0 4
Температура, °C
16
8 Ю
за
*
Рис. 422. Влияние концентра-
ции Р2О5 и весового отноше-
ния N : Р2О5 на растворимость
солей в аммонизированных ра-
створах фосфорных кислот при
0°.
Твердые фазы-. I — NH4H2PO4;
11 - (NH4)2HPO4;
111 - (МН4)зНР207-Н20; ...
/Й--(NHjsPaOio • Н2О;
V-(NH4)2HPO4 2Н2О;
K/-(NH4)4P2O7 • H2O.
Рис. 421. Зависимость темпе-
ратуры кристаллизации жидких
комплексных удобрений от кон-
центрации питательных веществ
в системе карбамид — фосфаты
аммония — фосфорная кисло-
та — хлористый калий — вода
(в скобках — молярные отноше-
ния NH3: Н3РО4 в кристалли-
зующихся солях).
ний состава 11—33—0 и 8—24—0, приготовленных путем аммони
зации ортофосфорной кислоты 74.
При аммонизации экстракционной фосфорной кислоты выде
ляются осадки фосфатов железа и алюминия. При добавлении 1
раствору К.С1 последний взаимодействует с содержащейся в экс
тракционной кислоте кремнефтористоводородной кислотой с обра
зованием кремнефторида калия, который тоже выпадает в осадок
H2SiFe + 2КС1 + 2NH3 = K2SiFe + 2NH4C1
Разработан оптимальный режим аммонизации фосфорной кис-
лоты и фильтрации осадка74. Однако при использовании экстрак-
Производство жидких удобрений
1387
ТАБЛИЦА 11»
Составы тройных жидких комплексных удобрений
Добавляемые азотные компоненты Жидкие комплексные удобрения на основе марки 11 : 33 : 0; N : Р2О5 : КгО Жидкие комплексные удобрения на основе марки 8 : 24 ; С; N : Р2О5 : КгО
1:1:1 1:2:1 1:1:1 1:2:1
Раствор карбамида и нит- рата аммония (28% N) Нитрат аммония Карбамид 8-8-8 6-6-6 10-10-10* 8-16-8 7-14-7 8-16-8 7-7-7 5-5-5 9-9-9 7-14-7 5-10-5 7-14-7
* Температура насыщения 6—7®.
ционной фосфорной кислоты, можно получить жидкие комплекс-
ные удобрения лишь с низкой концентрацией питательных веществ,
около 24%, а для отделения осадка требуется фильтровальное обо-
рудование, что снижает экономичность процесса 83-88.
ПРОИЗВОДСТВО ЖИДКИХ УДОБРЕНИЙ
Схемы производства аммиачной воды и аммиакатов
на основе аммиачной селитры
Схема приготовления водного аммиака (аммиачной воды) пред-
ставлена на рис. 423 89-91. Сырьем является газообразный аммиак,,
подаваемый под избыточным давлением 2 ат из цеха синтеза ам-
миака или из хранилищ жидкого аммиака в колонну 3 тарельча-
того типа с колпачками. Используется также газообразный аммиак,
выделяющийся при заполнении цистерн жидким аммиаком. Ниж-
няя часть колонны 3 представляет собой трубчатый теплооб-
менник, предназначенный для отвода значительной части тепла
растворения аммиака. По трубкам теплообменника проходит охла-
ждающая вода, в межтрубном пространстве циркулирует водный
аммиак, через слой которого барботирует газообразный аммиак. Во-
избежание забивки колонны солями жесткости поглощение аммиа-
ка производится химически очищенной водой. Остаток непоглощен-
його аммиака поступает в верхнюю часть колонны, где газ прохо-
дит через колпачковые тарелки. На тарелках расположены охла-
ждающие змеевики, в которых циркулирует вода. Продукционный
водный аммиак (концентрация 25% NH3) перекачивают из ко-
лонны насосами 5 в хранилище 6.
Газообразный аммиак, выделяющийся из хранилища при дли-
тельном хранении в нем водного аммиака вследствие воздействия
тепла окружающего воздуха и солнечной радиации, направляют на
1388
Гл. XXXVII. Жидкие удобрения
поглощение в колонну 3. Из хранилища водный аммиак насосом 1
перекачивают в железнодорожные и автомобильные цистерны
Рис. 423. Схема получения водного аммиака:
] — сборник химически очищенной воды; 2, 5 н 7 — центробежные насосы;
3 —колонна для приготовления водного аммиака; 4 — сборник водного ам*
миака; £ —хранилище водного аммиака; 8—клапан с огнепреградителем;
9 — предохранительный клапан.
Рис. 424. Схема производства аммиакатов на основе
аммначнон селитры:
/ — нейтрализационный аппарат ИТН; 2 —труба с водяной ру
башкой; 3 — шнек-кристаллизатор; / — реактор; 5 — циркуля-
ционный насос; £ —холодильник; 7—улавливатель аммиака—
гндрозатвор.
в которых аммиачная вода перевозится потребителям. Заполнение
цистерн производится при помощи гибких шлангов.
На рис. 424 приведена схема производства аммиакатов, содер*|
жащих NH4NO3 и NH3. _ I
Производство жидких удобрений
1389
Раствор нитрата аммония с концентрацией 80—83% NH4NO3 и
температурой 135° из нейтрализационного аппарата 1 (в случае ис-
пользования для его получения 57%-ной азотной кислоты), или по-
сле предварительной выпарки (в случае использования 47%-ной
кислоты), поступает в шнек-кристаллизатор 3, предварительно ох-
лаждаясь в трубе с водяной рубашкой 2. В шнеке раствор NH4NO3
охлаждается водой до 40—30°. Образующуюся смесь кристаллов и
маточного раствора направляют в реактор 4. Дополнительное ко-
личество воды, необходимое для получения раствора аммиаката
Рис. 425. Схема производства аммиакатов на
основе аммиачной и кальциевой селитры:
1 — реактор; 2 и 7— центробежные насосы; 3 — холодильник;
4 —транспортер иа случай подачи Са(МОз)2 в виде соли;
5 — гидрозатвор; 6 — хранилище.
заданного состава, вводят в шнек-кристаллизатор или непосред-
ственно в реактор перед началом процесса. Газообразный аммиак
из общего коллектора передается в реактор через кольцевой рас-
пределитель, расположенный в нижней части аппарата. Это обес-
печивает не только равномерное распределение аммиака, но и пе-
ремешивание массы, благодаря чему исключается необходимость
применения мешалок. Для отвода тепла, выделяющегося в резуль-
тате взаимодействия газообразного аммиака с раствором, послед-
ний охлаждается водой при непрерывной циркуляции через двуххо-
довой трубчатый холодильник 6. В случае использования для при-
готовления аммиакатов жидкого аммиака специальное охлаждение
Не требуется. Отходящий из реактора непрореагировавший аммиак
Направляют в улавливатель 7, выполняющий одновременно роль
гидрозатвора.
В описанной схеме принята такая последовательность ведения
процесса: в реактор вводят определенное количество воды для обе-
спечения получения раствора аммиаката заданного состава, затем
подают газообразный аммиак и включают циркуляционный насос.
После получения 15—20 %-ной аммиачной воды из шнека-кристал-
1390
Гл. XXXVII. Жидкие удобрения
лизатора в реактор передают .раствор нитрата аммония. Предва.
рительное получение аммиачной воды создает наиболее благо-
приятные условия работы, так как препятствует возможной
кристаллизации соли. Когда состав циркулирующего раствора ам-
миаката достигает заданного, раствор передают центробежным на-
сосом в цистерны для отправки потребителям или в хранилище
готовой продукции.
Получение аммиаката, содержащего, кроме нитрата аммония,
нитрат кальция, осуществляют по схеме, изображенной на рис. 425.
В реактор 1 одновременно с раствором нитрата аммония (концен-
трация раствора нитрата аммония 80% при 80°) загружают рас-
считанное количество нитрата кальция в виде концентрированного
раствора или кристаллической соли. В остальном процесс ведут
в такой же последовательности, как в первом варианте 92~105.
Производство жидких комплексных удобрений в7> 89’105-119
На рис. 426 представлена (разработанная НИУИФ73) схема
производства жидких комплексных удобрений, реализованная в.
СССР. Фосфорная кислота разгружается из железнодорожных ци-
стерн при помощи вакуум-насоса 1 и сифонного устройства 4. Из
хранилища 5 она перекачивается в напорный бак 6, из которого че-
рез ротаметр 7 поступает в нейтрализатор 9. Одновременно из на-
порных баков 6 в нейтрализатор поступает аммиачная вода и хи-
мически очищенная вода.
Фосфорная кислота нейтрализуется аммиачной водой до рН =
= 6,5—7,5 (соотношение NH3 : Н3РО4 = 1,8—1,9, содержание Р2О3
в растворе 12,5—13%). Вместо аммиачной воды нейтрализацию
можно проводить аммиаком. Тепло, выделяющееся в процессе ней-
трализации, отводится холодной водой, циркулирующей в змеевике.
Процесс нейтрализации проводится при атмосферном давлении, при
50—60° и при непрерывном перемешивании. Раствор из нейтрали-
затора 9 непрерывно перетекает в реактор 10, в который дозируют-
ся в твердом виде карбамид и хлористый калий Растворение этих
компонентов протекает с поглощением тепла. Для поддержания
в реакторе 10 температуры ~50° в змеевики подается пар. Время,
необходимое для растворения твердых компонентов, составляет
20—30 мин. Из реактора 10 раствор непрерывно перетекает в сле-
дующий реактор 11, где происходит дополнительное перемешива-
ние раствора и охлаждение водой, циркулирующей в змеевике. Tod
товое жидкое удобрение перекачивается в хранилище 12. I
Для получения 1 т жидкого комплексного удобрения 1:1:1 <
•общим содержанием питательных веществ 27% требуется израс!
ходовать: термической фосфорной кислоты (53% Р2О5) 169,8
аммиачной воды (20,5% N) 155,1 кг, карбамида (46% N) 126,3
хлористого калия (60% КзО) 150,0 кг, воды 398,8 кг. I
Химически очищенная
вода
1392
Гл. XXXVII. Жидкие удобрения
Жидкие комплексные удобрения можно изготовить н без при-
менения аммиака, из диаммонийфосфата, карбамида и хлористого
калия (холодный способ). Эти вещества легко растворяются в во.
де, образуя почти нейтральный раствор (pH = 7,8) 12°.
Разработаны способы получения жидких комплексных удобре-
ний методом азотнокислотного разложения природных фосфатов
с последующей аммонизацией раствора 121-125.
Возможно использование различных отходов для производства
жидких комплексных удобрений. Так, используются сточные воды
пивоваренных заводов, образованные при производстве солода, пи-
ва или дрожжей, в которые добавляют 20—25% газообразного NH3
и получают жидкое удобрение, содержащее (в %): N — 20,5;
РгО5 — 0,0032; К2О — 0,05; органических веществ — 20125.
Предложено использовать отходы содовой промышленности127
И отбросные сульфитные щелока 128.
Суспендированные жидкие комплексные удобрения
Суспендированные жидкие комплексные удобрения характери-
зуются присутствием твердой фазы. Для предупреждения роста
кристаллов и выделения их в осадок при хранении в такие удобре-
ния вводят стабилизирующие добавки, увеличивающие вязкость
растворов, препятствующие росту кристаллов и уменьшающие ско-
рость их осаждения. В качестве стабилизирующих добавок реко-
мендуют применять аттапульгитовую глину, бентонитовую гли-
ну73’ 131>132, аэросил-175, нефелиновый шлам 133 и др. Для приготов-
ления суспендированных жидких удобрений используются те же
компоненты, что и для обычных жидких удобрений (экстракцион-
ная фосфорная кислота, полифосфорные кислоты, аммиак, карба-
мид, нитрат аммония, хлористый калий и др.). Имеются также ука-
зания на возможность приготовления устойчивых суспендирован-
ных удобрений без применения стабилизирующих добавок при
условии соблюдения определенного режима их приготовления130.
В настоящее время за рубежом производят суспендированные
удобрения на небольших промышленных установках как по «хо-
лодному», так и по «горячему» способам; выпускают различные
марки этих удобрений с общим содержанием питательных веществ
36—45%, что на много превышает содержание их в обычных жид-
ких удобрениях 134~137.
Разработан способ приготовления удобрений в виде тиксотроп-
ной пульпы 138.
ЛИТЕРАТУРА
1. Д. Г. Иванов, Ив. Н. Грънчаров, Химия и индустрия (бълг.), 39,
№ 1, 26 (1967). — 2. W. С. Scott, J. A. Wilbanks, Chem. Eng. Progr., 63,
№ 10, 58 (1967). — 3. Г. И. В илесов, С. Н. Г а н з, Р. И. Брагинская,
сб. «Химическая технология», в. 7, 1967, стр. 5. — 4. Пат. США 3197301, 1967,—
Литература
1393
, A Gusztav Andras, Mag yar kem. lapja. (венгр.), 19, № 10—11, 560
7io64).—6. Nitrogen, № 43, 13 (1966). —7. Ibid., № 44, 15, 36 (1967).—
я Ibid., № 45, 34 (1967).—9. A. Milosevic, Technics, 20, № 10; Hem. ind.
(сербо-хорв.), 19, № 10, 265 (1965).—10. С. И. Вольфкович и др., Техно-
логия азотных удобрений, ОНТИ, 1935.
11. А. И. Ш а т е н ш т е й н, Сжиженные газы как растворители, ч. 1, Гос-
химиздат, 1935; ч. II, Оборонгиз, 1939. — 12. W. Andrevs, Am. Fertilizer,
<п7 № 12, 9, 28; № 13, 11, 24, 26, 28 (1947).— 13. F. Н. Leavitt, ibid., НО,
№ 2, 7 (1949). —14. S. Wiliams, J. Newson, Farm. Chem., 122, № 12, 60
(1959). —15. D. W a 1 s t a d, J. Agr. Food Chem., 9, № 5, 348 (1961).—
16. A. Slack, M. Nason, ibid., 9, № 5, 343 (1961). —17. F. Achorn, Comm.
Fertilizer, 110, № 2, 23 (1965). —18 Ibid., № 1 (1965). —19. К. Зейденберг,
И. Ниселевич, Химстрой, № 6, 1635 (1932).—20. К. Зейденберг, сб.
«Советский азот», 1935, стр. 50.
21. К Зейденберг, А. Ильинская, ЖХП, № 7, 683 (1935).—
22. I. Her de gen, F. Nadolsky, Przem. Chem., 8, № 8, 455 (1955).—
23. С. H. Г а н з, А. В. Карчевский, P. И. Брагинская, Изв. вузов.
Хим. и хим. технол., 7, № 4, 619 (1964).—24. К. Зейденберг, сб. НИУ
«Минеральные удобрения», в. 113, 1933, стр. 74.—25. В. Ф. Гогин, Н. Редь-
кин, Д. Стеденко, Ценное дешевое удобрение, Изд. «Донбасс», 1967.—
26. Nitrogen, № 40, 1 (1966). — 27. Changes in Fertilizer, Distribution and Mar-
kating, Knoxville (TVA), 1965. — 28. Comm. Fertilizer, 114, № 4, 27 (1967).—
29. Nitrogen, № 49, 25 (1967).— 30. Chem. Eng., 76, № 10, 56, 58 (1969).
31. В. Д. Ванников, Растениеводство, № 2, 23 (1965). — 32. И. И. Б у р-
лаченко и др., Хим. пром., № 8, 492 (1958).—33. S. Aldrich, Agr. Chem.,
14, № 5, 41, 104 (1959). — 34. fl. И. Кильман, В. А. Клевке, Д. Ю. Гам-
бург, ЖХП, № 3, 135 (1957). — 35. Д. Коренков, Удобр. и урожай, № 4,
55 (1966). — 36. Ф. В. Турчин, Там же, № 2, 20 (1956). — 37. S. L. Jansson,
Vaxtnarings-nytt (шведок.), 22, № 4, 4/1 (1966). — 38. В. К о с а н, F. К о f е е п-
s к у, J. Neuberg, Chem. priimysl, 15, № 5, 260 (1965).—39. Т. Stobiecki,
Przem. Chem., № 2, 66 (1952).—40. R. Za n g g u t h a. oth., J. Agr. Food Chem.,
3, № 8, 650 (1955).
41. K. D. Jacob, W. Scholl, Comm. Fertilizer, 91, № ЗА, 94, 99, 102
(1955). — 42. S. Aldrich, Agr. Chem., 14, № 5, 41, 104 (1959).—43. Nitrogen,
№ 48, 35 (1967).—44. N. G a r t i g, Wiss-techn. Fortschr. Feldwirtsch., 6, № 2,
51 (1965). — 45. И. А б а з о в а, Св. Дудеков а, Д. Добров, Хим. и индуст-
рия (Бълг.), 38, № 8, 345 (1966). — 46. W. Н е у d е, Е. A. L i n d о w, S. R i-
ckelt, Chem. Techn., 17, № 9, 554 (1965).-—47. F. L. A p p 1 e g a t e, Chem.
Eng. Progr., 61, № 1, 66 (1965).— 48. Oil a. gas J., 65, № 52, 109 (1967).—
49. Ibid., 62, № 52, 163 (1964). — 50. Comm. Vehicles, 42, № 1, 46 (1968).
51. Nitrogen, № 30, 30 (1964). — 52. В. А. Клевке, fl. И. Кильман,
ЖПХ, 32, № 8, 1649 (1959).—53. Fertilizer Feeding Stuffs, 39, 18,
643 (1953). — 54. R. Luckhardt, Agr. Chem, 8, № 9, 45 (1953).—
55. D. W. Henderson, W. C. Bianchi, L. D. D о n e e n, Agr. Eng., 36,
№ 6 (1955). — 56. N. D. Abell, Agr. Chem., 22, № 11, 24 (1967). —57. В. И. Ми-
шари н a, Труды Уральского н.-и. ин-та с.-х., т. 5, 1964, стр. 135.—58. G. S е г-
v i s s, Grops a. Soils, 1, № 7, 91 (1949). — 59. F. W. Parker, Am. Fertilizer,
№ 13, 26 (1936).—60. E. A. W о r t h i n d t о n, R. C. D a t i n, D. P. Schutz,
Ind. Eng. Chem., 44, № 4, 910 (1952).
61. I. E. Schults, G. V. Elmorte, ibid., 38, № 3, 296 (1946).—
52. А. Г. Бергман, К. К. Мороз, ЖПХ, 39, № 12 (1966); ЖНХ, 12, № 2
(1967).— 63. В. Клевке, А. Кантор, ЖХП, № 6, 507 (1959).—
°4- В. А. Клевке, Я. И. Кильман, А. С. Кантор, Триады ГИАП, т. 9.
1959, стр. 183. —65. Н. Tucker, Agr. Chem., 14, Ns 4, 124 (1959).—
1394
Гл. XXXVII. Жидкие удобрения
66. J. Reynolds, ibid., 14, № 4, 47, 50 (1959).—67. W. Cryl, M. Hue]]
Przem. Chem., 46, № 1, 34 (1967). — 68. Д. С. Лесных, E. Ш. Муси'ня’
ЖПХ, 39, № 5, 1177 (1966). -69. С. H. Г а и з, P. И. Брагинская, Г. Б. oi
л о в с к а я, Изв. вузов. Хим. и хим. технол., 10, № 1, 116 (1967). — 70. Пат
США 3290140, 1966.
71. Франц, пат. 1469089, 1967. — 72. М. Е. П о з и и, Б. А. Копы лев
Н. к. Шиллинг, ЖПХ, 37, № 11, 2341 (1964); 38, № 1, 22 (1965).Д
73. Г. Л. Тудоровская, Ф. Г. Марголина, Труды НИУИФ, в. 208, 1965
стр. 7. — 74. Ф. Г. Марголис, Т. П. У н а н я н ц, Производство комплексных
удобрений, Изд. «Химия», 1968. — 75. A. Slack, J. Hatfield, Н. Shaffer
J. Duskell, Agr. Food Chem., 7, № 6, 404 (1959). — 76. Л. Г. Бергман’
А. Д. Дзуев, Уч. зап. Кабардино-Балкарск. ун-т. Сер., с.-х. и хим.-биол., в. 29
40 (1966). — 77. А. Г. Бергман, А. Д. Дзуев, 3.’М. Карманова, Там
же, 45 (1966). — 78. V. S a u с h е 11 i, J. Agr. Chem., 18, № 6, 54 (1963).—
79. Ван Везер, Фосфор и его соединения, ИЛ, 1962. — 80. A. Slack, Farm
Chemie., 128, № 4, 25 (1965).
81. W. Scott, J. W i 1 b u n k s, Agr. Chem., 16, № 5, 16 (1961).—
82. A. V. Slack, J. M. Potts, H. B. Shaffer, J. Agr. Food Chem., 13, № 2
165 (1965). —83. Япон. пат. 7102, 1965, — 84. Англ. пат. 1076901, 1967, —85. Пат’
США 3264087, 1966. — 86. Д. Г. Иванов, Ив. Н. Грънчаров, Хим. и инду-
стрия (Бълг.), 38, № 10, 454 (1966). —87. Пат. США 3109728, 1963.— 88 Япон.
пат. 22365, 1963. —89. Пат. США 3008801, 1961, — 90 Пат. США 3295927, 1967.
91. Пат. ЧССР, 112941, 1964. — 92. П. А. Баранов, Д. А. Кореньков,
И. В. Павловский, Жидкие азотные удобрения, Сельхозгиз, 1961,—
93. V. Sauchelli, The Chemistry and Technology of Fertilizers, London, 1960,
p. 513. — 94. Ф. В. Янишевский, В. А. Клевке, Журн. ВХО, 7, № 5, 534
(1962). — 95. В. А. Клевке, А. С. Кантор, Труды ГИАП, т. 14, 1963,
стр. 19. — 96. Е. Т. Саранча, А. М. Абросимова, Л. В. Андреева,
Хим. пром., 5, 383 (1965). — 97. Д. С. Лесных, Е. М. Мусина, ЖПХ, 39,
№ 5, 1177 (1966). — 98. X. Г. Бергман К. К. Мороз, Там же, № 12, 2825
(1966). — 99. W. Sadowski, Т. W г о п k е, Przem. Chem., 46, № 1, 18 (1967). —
100. S 1 а с к a, oth., J. Agr. Food Chem., 7, № 6, 404 (1959).
101. Пат. США 3144321, 1964. — 102. Франц, пат. 1410331, 1965. — 103. Япон.
пат. 22364, 1963. — 104. Пат. США 3117858, 1964. — 105. A. Slack, Farm.
Chemie., 128, № 3, 23 (1965). — 106. Пат. США 2969280, 1961. — 107. Пат.
США 3011875, 1961, — 108. Авт. свид. 182181, 1966. — 109. Япон. пат. 25206,
1965. —ПО. Пат. США 3234004, 1966.
111. Франц, пат. 1451568, 1966. — 112. Англ. пат. 969362 1964. — 113. Пат.
США 3183073, 1965. — 114. Англ. пат. 997932, 1965. — 115. Пат. США 3179509,
1965. — 116. Пат. США 3130033, 1964. — 117. Пат. США 3130034, 1964.-
118. S. В е п а г i, М. Man, Ch. V. Papa, V. S t а п a, Rev. Chim. (румын.). 16,
№ 1, 10 (1965). — 119. Япон. пат. 22363, 1963 — 120. J. Silverberg, G. Hof-
f m e i s t e г, Comm. Fertilizer, 97, № 2, 28 (1957).
121. M. H. Набиев, M. А. К а с ы м о в а, сб. «Минеральные и органомине-
ральные удобрения, структурообразователи почв и гербициды», Ташкент, Изд-
«ФАН», 1967, стр. 47. — 122. Япон. пат. 8093, 1965. —/23. Зи Ле В а н,
Ф. М. Мирзаев, М. Н. Набиев, Узб. хим. ж., № 5, 11 (1967). — 124. О. Б ер-
дим у р а т о в, сб. «Физико-химические и технологические исследования мине-
рального сырья», Изд. «Наука», Ташкент, 1965, стр. 48. — 125. М. Н. Набиев,
У. И. Ибрагимова, А. И. Ильясов и др., Жидкие сложные удобрения на
основе азотнокислотной переработки фосфатов, Изд. «Наука», Ташкент, 1965. —
126. Польск. пат. 48777, 1965. — 127. Е. А. Ш у м л я н с к а я, сб. «Приемы повы-
шения урожайности с.-х. культур», Киев, Изд. «Урожай», 1967, стр. 303. —
128. Польск. пат. 48777, 1965. — 129. Nitrogen, № 50, 31, 44 (1967). —130. J. Sil'
Литература 1395
verb erg, H. K. Walters, Comm. Fertilizer a. Plant Food Ind., 108, № 4, 26,
66 (1964).
131. П. А. Замягиченский, Глины СССР, в. 1,’ Изд. АН СССР, 1935.—
132. Ф. Д- Овчаренко, Гидрофильность глин и глинистых материалов, Изд.
*Н УССР, 1961. — 133. В. И. Гладушко, И. М. Астрели^, А. С. Плыгу-
нов А. Д. Хоменко, Хим. пром. Украины, № 4 (34), I (1967). — 134. Англ,
„ат ’973511, 1964; 984920, 1965. — 135. Пат. США, 3096170, 1963; 3155489, 1964;
3179496, 1965 ; 3206298, 1965. — 136. Япон. пат. 22366, 22368, 1963.—
137 F Р. А с h о г n, F. J. М у е г s, Agr. Chem., 24, № 3, 18,24 (1969). — 138. Пат.
США 3234005, 1966.
Глава XXXVIH
СОЛИ МЫШЬЯКА
Из солей мышьяка имеют наибольшее значение и изготовляют-
ся в больших количествах соли мышьяковистой и мышьяковой кис-
лот 1 — арсениты и арсенаты — главным образом кальциевые и на-
триевые. На основе этих солей производят также разнообразные
препараты, предназначенные для сельского хозяйства.
Исходным материалом для производства солей мышьяка слу-
жит мышьяковистый ангидрид, называемый белым мышьяком, по-
лучаемый путем окислительного обжига мышьяковых руд2.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Мышьяковистый ангидрид, или трехокись мышьяка, As2O3 су-
ществует в трех модификациях — аморфной, кубической (октаэдри-
ческой) и моноклинной. Точка превращения кубической и моно-
клинной модификаций (арсенолит^клаудетит) при давлении 1 ai
равна 3 240±30°. На рис. 427 показано изменение давления пара
As2O3 в зависимости от температуры. Ниже 315° давление пара не
является однозначной функцией температуры в связи с превра-
щением октаэдрической модификации As2O3 в моноклинную и об-
разованием твердых растворов. Аморфно-стекловидная модифика-
ция (плотность 3,74 г!см3) образуется при медленном охлаждении
паров As2O3 при 200°. Предполагают, что она является смесью обе-
их кристаллических форм. Аморфная трехокись мышьяка на воз-
духе постепенно становится фарфоровидной (непрозрачной) и пе-
реходит в октаэдрическую форму (плотность 3,64 г/см3). Послед-
няя образуется также при быстром охлаждении возгоняющегося
As2O3. При медленном нагревании до 200° аморфная форма пере-
ходит в моноклинную (плотность 4,0 г/см3). Она образуется также
при конденсации паров AS2O3 в интервале температур 275—315 •
Октаэдрические кристаллы плавятся при 275°; моноклинные — при-
Физико-химические свойства
1397
близительно при 315°; аморфно-стекловидная модификация устой-
чива выше -310°. Ниже 800° трехокись мышьяка существует в
виде димера As4O6, выше 1700° — только в виде As2O2.
Мышьяковистый ангидрид плохо растворим в воде: раствори-
мость при 0° равна 1,2 г, при 100° — 6 г в 100 г воды. Водные рас-
творы его имеют кислую реакцию, сладковаты, с металлическим
привкусом. Как амфотерный окисел As2O3 реагирует и со щелочами
и с кислотами. Однако кислотные
свойства As2O3 превалируют над Температура,°C
щелочными.
Образующаяся при растворении
As2O3 в воде и выделяемая из со-
единений мышьяковистая кислота
НзАзОз стабильна лишь в водных
растворах, в которых она диссоци-
ирует по схеме:
As3+ + 3OH“ As(OH)3 =
= H3AsO3 Н++ АзОг + Н2О
Константы диссоциации H3AsO3
при 20°: Я1=4-10'10, /\2 = 7-10~13,
К3 = 4-ИРМ Первая константа дис-
социации As(OH)3 равна 5-Ю-16.
Растворы мышьяковистого ан-
гидрида легко восстанавливаются 4
до элементарного мышьяка или до
арсина AsH3 (/Кип = —62,5°). Дав-
ление пара AsH3 (в см рт. ст.) мо-
жет быть определено по уравнению5:
lg Р = —1403,327-' — 9,43935 lg Т +
t/т- to4
Рис. 427. Давление пара As2O3:
АВС — давление пара над октаэдриче*
сними кристаллами (lg Р ~ — 6670Т~1 +
+ 13,728); BDE — давление пара над
жидким А$20з (lg Р = -2722Г'1 + 6,513);
FD — давление над моноклинными кри-
сталлами (Р — в мм. рт. ст.).
Дабление пара, мм pm.au.
+ 0,0080377 + 28,82835.
Мышьяковистая кислота легко окисляется в мышьяковую азот-
ной кислотой, галогенами и другими окислителями, но устойчива
по отношению к газообразному кислороду. Ее соли значительно
легче окисляются свободным кислородом, особенно в щелочной
среде и в присутствии катализаторов — солей меди, марганца,
иода6 и др. Раствор арсенита натрия с молярным отношением
As2O3:NaOH, равным 1:6, с концентрацией As2O3 — 14—17 г/л,
содержащей 5 г/л солей меди, при барботаже через него воздуха
(5—10 л/ч через 100 мл раствора), при 60° окисляется кислоро-
дом воздуха в течение 60 мин на 40—45%. Степень окисления 7
возрастает с повышением температуры до 70—75°, с увеличением
Молярного соотношения As2O3: NaOH и уменьшается с пониже-
нием концентрации As2O3. Реакция окисления As2O3 хромово-
кислым калием при 30°, pH = 8,3—10,6 и ионной силе раствора 1,75
1398
Гл. НИКУШ. Соли мышьяка
имеет первый порядок относительно концентраций каждого из обо-
их реагентов8. При рН>9 скорость реакции не зависит от концец-
трации Н+, а при рН<9 увеличивается с ростом концентрации Н+
При pH = 9,1 константа скорости реакции А=1,61-10“3 лЦг-мол.
сек).
Температура,°С
Рис. 428. Зависимость
степени окисления
As2O3 в As2O5 от
температуры (на воз-
духе в течение 1 ч).
С некоторыми галогенидами (КС1, KBr, KI) мышьяковистая
кислота образует в водных растворах комплексные соли перемен-
ного состава (MX-nAs2O3, где X — галоген), разлагающиеся при
кипячении. С NaCl и NH4C1 подобные ком-
плексы не образуются 9.
Сложное комплексное соединение
3Cu(AsO2)2 • Си(СН3СОО)г называется париж-
ской зеленью; оно не растворяется в воде, но
гидролизуется, выделяя свободную мышьяко-
вистую кислоту 10. При односуточном СТОЯНИИ
суспензии из 0,25 г парижской зелени и 100 мл
воды в раствор переходит ~ 0,005 г As2O3,
т. е. около 3—4% общего количества мышьяка.
Плотность технической парижской зелени ко-
леблется от 2,8 до 1,3 г)см3, а насыпная масса
от ~0,3 г!см3 у очень дисперсных образцов
до 1 г/см3 у образцов грубого помола.
Окисление трехокиси мышьяка в пятиокись
As2Os (мышьяковый ангидрид) можно произ-
водить нагреванием на воздухе до 600—700°
(рис. 428). Обрабатывая As2O3 50—60%-ной
азотной кислотой и затем выпаривая из реакционной массы воду,
не допуская перегрева выше 350°, можно получить продукт, содер-
жащий 99,5% и более As2O5. Разложение As2O5 с отщеплением О2
начинается выше 400° и.
Мышьяковая (ортомышьяковая) кислота H3AsO4 представляет
собой кристаллы ромбической системы, легко растворимые в воде.
Из водного раствора выделяется в гидратированной форме
2H3AsO4-H2O или As2O5-4H2O. При нагревании до 100° частично
обезвоживается, превращаясь в 3As2O5-5H2O, а выше 120° разла-
гается на As2O5 и Н2О. Полное разложение происходит при ~500°
без образования в качестве промежуточных продуктов пиро- и ме-
тамышьяковой кислот.
По своим кислотным свойствам мышьяковая кислота подобна
фосфорной. Константы диссоциации H3AsO4 при 20°: /<1=6-10"3,
А2 = 2-10-7, А3 = 3-10-12.
В системе As2O5—Na2O—Н2О при 20° образуются различные со-
единения 12. Кислота 4As2O5-5H2O существует только в растворе,
содержащем 70,5% As2O5. В равновесии с раствором, содержа-
щим 64—70,5% As2O5 (и 3% Na2O),существует3As2O5-Na2O-6H2O,
склонный к гидролизу. При дальнейшем понижении концентрации
Физико-химические свойства
1399
Д32О6 от 64 до 61% (при 3—8% Na2O) устойчивым является
2As2O5-Na2O-5H2O, также склонный к гидролизу. В области кон-
центраций 41—61% As2O5 (8—14,5% Na2O) образуется моноорто-
арсенат NaH2AsO4 • 7Н2О, отличающийся высокой растворимостью
(~200 г в 100 г Н2О). Динатрийарсенат Na2HAsO4-7H2O нахо-
дится в равновесии с растворами, содержащими 20—41% As2O5
(22,5—15% Na2O), и .характеризуется меньшей растворимостью
(85 а в 100 г Н2О), а тринатрийарсенат — в равновесии с раство-
рами 10,4—20% As2O5 (7,6—12,5% Na2O). В области концентраций
0,2—0,9% As2O5 (7—27% Na2O) существует двойная соль —псев-
д'оарсенат Na3AsO4-0,25NaOH-24H2O, кристаллографически иден-
тичная 12-водному тринатрийарсенату. (Псевдоарсенаты — про-
дукты замещения части кристаллизационной воды арсенатов мо-
лекулами NaOH). При добавлении Na2HAsO4 к раствору NaOH
образуются кристаллогидраты арсената: из раствора с 27—34%
Na2O выделяется Na3AsO4 • 8Н2О, при 34% Na2O—Na3AsO4-4H2O
(предположительно), в присутствии маточного раствора переходя-
щие постепенно в Na3AsO4-3H2O. Из растворов, содержащих боль-
ше 40,5% Na2O, кристаллизуется NaOH-H2O.
Мононатрийарсенат или дигидроарсенат натрия NaH2AsO4-Н2О
при нагревании обезвоживается, затем разлагается, подобно фос-
фату, по схеме:
NaH2AsO4 Na2H2As2O7 ——•> Na3H2As3O10 —(NaAsO3)x
Конечный продукт разложения — метаарсенат натрия плавится
при 615°13.
Взаимодействие арсената натрия с суспензией гидроокиси каль-
ция протекает в несколько стадий |4:
Na3AsO4 + Са(ОН)2 + 4Н2О = CaNaAsO4 • 4Н2О + 2NaOH
2CaNaAsO4 • 4Н2О + Са(ОН)2 + 2Н2О = Ca3(AsO4)2.10Н3О + 2NaOH
Ca3(AsO4)2 • 10Н2О + Са(ОН)2 = Ca3(AsO4)2 • Са(ОН)2.4Н2О + 5Н2О
В системе СаО—As2O5—Н2О при 17° устойчивыми являются сле-
дующие соединения:
При pH
» » от
» » »
» » »
» » »
до 2
2 » 6
4,6 » 6,8
6,8 » 7,8
7,8 » 9,6
более 9,6
СаН4( AsO4)2
Ca2H2(AsO4)2 • 2Н2О
Са2Н,( AsO4)2 • 4Н2О
5СаО 2As2O5 • ЮН2О
Ca3(AsO4), • хН2О
4СаО • As2O5 • 5Н2О
4СаО-As2O5-5H2O обезвоживается при нагревании до 200—
215°, а при 350° превращается в твердый раствор Са(ОН)2 в ди-
гидрате трикальцийарсенага ЗСаО-As2O5-2H2O, стойкий вплоть
До 500°.
Промышленные образцы арсената кальция, которым обычно
приписывают формулу тетракальцийарсената 4СаО’As2O5-5H2O
20 М. Е. Позии
1400
Гл. XXXVlll. Соли мышьяка
или Ca3(AsO4)2-Ca(OH)2-4H2O меняют состав, в зависимости от
производственного режима получения: молярное отношение СаО;
: As2O3 колеблется обычно в пределах 3,3—3,9.
При взаимодействии раствора гидроокиси кальция (не суспец-
зии) с ортомышьяковой кислотой выделяются прямоугольные приз-
мы 4СаО-As2O5-5H2O и ромбические пластинки ЗСаО • As2O5-ЮН20.
Последний метастабилен в присутствии избытка Са(ОН)2. Устой-
Содержание влаги в препаратах,
Рис. 429. Зависимость влажности арсената
кальция от относительной влажности воз-
духа:
/ — заводской продукт, содержащий 37,760/0 АзгОб,
1,20/0 водорастворимых AS2O5 + AS2O3, со средним
размером частиц 14 мк\ 2 — лабораторный препа-
рат, изготовленный нз чистых веществ н содержа-
щий 47,14°/о AS2O5, О,18О/о водорастворимых
AS2O5 + AS2O3, со средним размером частиц 18,4 мк\
3 — лабораторный препарат, изготовленный из тех-
нических веществ и содержащий 45,660/0 As20s>
О,86О/о водорастворимых AS2O5 + AS2O3 со средним
размером частиц 6,8 мк.
чивой фазой при 40—60°
являются твердые раство-
ры Са(ОН)2 в дигидрате
трикальцийарсената ЗСаО-
• As2Os • 2Н2О.
Водная суспензия 4СаО-
•As2O5-5H2O имеет значе-
ние pH = 9,6, а суспензия
3C^O-As2O5-10H2O — 9,1.
Эти соединения хорошо рас-
творяются (и потому обла-
дают высокой токсичностью)
при pH = 7,5—10. Водные
суспензии ЗСаО • As2O5 • 2Н2О
и твердых растворов
Са(ОН)2 в этой соли имеют
значения pH = 8,3—8,6 и
относительно мало раство-
римы (и менее токсичны)15.
По другим данным 16'17,
при 35° в системе СаО—
—As2O5—Н2О стабилен
Ca3(AsO4)2; при СаО:
: As2O5 > 3 осадок состоит
из Ca3(AsO4)2 и Са(ОН)2. При 62° установлено существование
Ca3(AsO4)2-2H2O и 4СаО-As2O5-xH2O, а в области 4>СаО:
: As2O5>3 — твердых растворов Са(ОН)2 в дигидрате трикальций-
арсената. При 90° стабильны Ca3(AsO4)2 и 3Ca3(AsO4)2-Ca(OH)2.
Арсенат кальция в водной суспензии гидролизуется:
CaHAsO4 + 2H2O Са(ОН)2 + H3AsO4
Избыток Са(ОН)2 предотвращает переход мышьяка в раствор.
Однако при длительном хранении или нахождении препарата на
воздухе содержащаяся в воздухе двуокись углерода карбонизует
известь, вследствие чего гидролиз интенсифицируется и появляется
свободная мышьяковая кислота. В отличие от лежалого продукта
свежий арсенат кальция, состоящий главным образом из основной
соли, выделяет водорастворимый мышьяк медленно и в небольших
количествах, чем объясняется его безопасность для растений.
Физико-химические свойства
1401
Склонность препарата к гидролизу, а также к слеживанию в зна-
чительной степени обусловливается гигроскопичностью арсената
кальция. На рис. 429 показано изменение содержания влаги в
препаратах арсената кальция в зависимости от относительной
влажности воздуха. Даже в районах с сухим климатом находя-
щийся на воздухе продукт поглощает больше влаги, чем допускает-,
ся ГОСТом (1% —см. стр. 1404). Поэтому его необходимо хранить
во влагонепроницаемой таре. Гигроскопичность арсената кальция
увеличивается при содержании в нем примесей NaOH или NaCl
(даже в количествах всего 0,5%). Однако распыляемость продукта
мало зависит от его влажности, если она не превышает 5% 18.
При смешении растворов динатрийарсената и сульфата магния,
кобальта, никеля, цинка, меди получаются медленно кристаллизую-
щиеся арсенаты этих металлов ,9~22.
Из арсенатов Fe, Си, Zn и Cd при одном и том же значении
pH наименее растворим арсенат железа. Растворимость гидрооки-
сей этих металлов больше растворимостей их арсенатов; этим мож-
но воспользоваться для выделения мышьяка из растворов при по-
мощи соответствующих гидроокисей. Из растворов, содержащих,
наряду с мышьяком, сульфаты цинка, кадмия и другие, можно се-
лективно выделить мышьяк, осаждая его при низких pH в форме
арсената железа 23. Ниже приведены произведения растворимости
некоторых арсенатов при 20° 24:
Мп3( AsO4)2
Pb3(AsO4)2
Co3(AsO4)2
Ni3( AsO4)2
A1AsO4 . .
(1,9±0,7) • 10 _
(4,1 ±3,6)-10 36
(7,6±4,2) • 10 9
(3,1 ±2,4)- 10_ ’
(l,6±0,7)- 10 16
CrAsO4............(7,8±1,5). 10Д;
FeAsO4............(5,7±4,2) • J0~2
BiAsO4............(4,4±2,9) • 10~‘°
Hg3AsO4 .... (l,9±0,6) • 10~37
При 20° произведения растворимости арсенитов25: цинка —
2,8-10~20, кобальта — 2,2-10~20, серебра — 1,1 • КГ17.
О растворимости в системах Na3AsO4—Na2SO4—НгО и
Cu3(AsO4)2— CuSO4—Н2О см.26.
При нагревании арсенатов большинство из них (трех- и четы-
рехзамещенные соли щелочноземельных металлов и ртути состав'
ляют исключение) в пределах 500—1000° разлагаются с образова'
Пием окисла или металла.
При взаимодействии As2O3 или сульфидов мышьяка с галогено-
водородами образуются галогениды мышьяка. Так, AsCh получает-
ся при пропускании сухого НС1 над нагретым до 180—200° As2O3:
As2O3 + 6НС1 = 2AsCl3 + ЗН2О
Треххлористый мышьяк (/пл=—18°, /Кип=130°) является рас-
творителем для FeCl3, SbCl3 и других соединений и может реаги-
ровать и как кислота (AsCl3 + Cl” = AsCl?) и как основание
20*
1402
Гл. XXXVIII. Соли мышьяка
(AsCh = AsClJ + СГ) . Фтористый мышьяк можно получить по
реакции
5As2O3 + 12HSO3F = 4AsF3 + 6H2SO4 + 3As2O(SO4)2
идущей с сильным экзотермическим эффектом 28. Сплавлением при
800° смеси KAsO3 и KF можно получить монофторарсенат калия
K2ASO3F. Монофторарсенаты Na, Ni, Со, Zn, Си, Cd и других ме-
таллов получаются обменным разложением их перхлоратов с
K2AsO3F. По растворимости и формам кристаллогидратов моно-
фторарсенаты имеют сходство с сульфатами 29’30 с которыми
образуют или двойные соли, например Zn'(SO4, AsO3F)-7Н2О,
(NH4)2Ni(SO4, AsO3F)-6H2O, или смешанные кристаллы, например
монофторарсенат и сульфат калия.
ПРИМЕНЕНИЕ МЫШЬЯКОВЫХ ПРЕПАРАТОВ И ТЕХНИЧЕСКИЕ
ТРЕБОВАНИЯ К НИМ
Соединения мышьяка сильно ядовиты (0,1 г As2O3 является
смертельной дозой для человека). Однако в малых количествах
некоторые из них применяют в качестве лекарственных препаратов.
Одним из сильнейших ядов является мышьяковистый водород.
Хлористый мышьяк и арсины (дик, адамсит, люизит и другие)
имеют военное значение как отравляющие и дымовые средства'31.
Белый мышьяк применяют в промышленности для осветления
стекла, консервирования мехов, изготовления пиротехнических
средств, для очистки газов от сероводорода (мышьяково-содовым
способом) и для других пелей. Его смеси с хроматами используют
для инициирования реакций полимеризации8. В основном же бе-
лый мышьяк перерабатывают на мышьяковые препараты, приме-
няющиеся в сельском хозяйстве в качестве ядов кишечного дей-
ствия для борьбы с вредителями растений — насекомыми и гры-
зунами, а также для борьбы с малярийным комаром 32-34 Арсенит
натрия применяют в качестве зооцида.
Некоторые мышьяксодержащие соединения используют для
борьбы с болезнями растений. Метиларсинсульфид является дей-
ствующйм началом препарата ризоктол, применяемого для про-
травливания семян хлопчатника. Метиларсин-бис (диметилдитио-
карбамат)— препарат урбацид — используют в борьбе с болез-
нями кофейного дерева. Препарат тузет (действующим началом
которого являются смесь урбацида, цирама — диметилдитиокарба-
мата цинка и тетраметилурамдисульфида) ускоряет созревание
плодов и применяется в борьбе с болезнями риса. Для борьбы с бо-
лезнями картофеля и риса в Японии применяют мышьяксодержа-
щие фунгициды общей формулы RnAs(Z)Xm, где R — алкил
(Ci—С4); X—SH, ОН, SNa; т + п = 3\ Z—О или S; например,
(C2Hs)2As(S)SH35. . .
П рименение мышьяковых препаратов
1403
В сельском хозяйстве мышьяковые препараты применяют в ви-
де тонких порошков или суспензий. Белый мышьяк и легко рас-
творимые в воде соединения мышьяка не применяют, так как они
ожигают растения.
В наибольшем количестве используются арсенаты кальция
(главным образом основные, обладающие наименьшей раствори-
мостью, а следовательно, и наименьшей фитоцидностью) и арсенат
свинца — джипсин, — выпускаемый преимущественно в виде
PbHAsO4 и обладающий еще меньшей фитоцидностью, чем основ-
ные арсенаты кальция. В меньшем количестве выпускают основ-
ные арсенаты свинца; они приближаются по составу к
Pb4(PbOH)(AsO4)3-3H2O.
За рубежом в качестве заменителя арсената свинца (для
борьбы с шелкопрядом и глистами овец) применяют кислый
арсенат олова SnHAsO4. В небольших количествах приме-
няют арсенат марганца, натрия, основной арсенат меди, цинк-
фторарсенат Zn3(AsO4)2 • ZnF2, цинкаммонийарсенат ZnNH4AsO4
и др.
Широкое использование органических инсектофунгицидов при-
вело к резкому сокращению потребления мышьяковых препаратов
в сельском хозяйстве. Это создало в ряде стран затруднения со
сбытом белого мышьяка, который получается в качестве побочного
продукта при переработке полиметаллических руд и использовался
в основном, как сырье для мышьяковых препаратов. В связи с
этим изыскиваются новые потребители мышьяксодержащих ве-
ществ.
За рубежом стали широко применять мышьяковые соли (в ча-
стности, арсенат натрия) для окорки древесины. Это облегчает
труд и позволяет производить заготовку древесины в любое время
года36. Их применяют также для антисептирования древесины37,
а, например, парижскую зелень и для окраски днищ судов с целью
предотвращения их обрастания.
Новой областью применения соединений мышьяка является
полупроводниковая техника, их используют в мазерах и лазерах
(арсенид галлия и др.); их стали применять и для дефолиации
растений в сельском хозяйстве.
Мышьяковистый ангидрид, согласно ГОСТ 1973—67 (табл. 111),
выпускают двух марок — рафинированный (порошок белого цве-
та) и технический (порошок серого цвета).
Арсенит кальция (ГОСТ 107—66) должен содержать не менее
62% Аз2О3, в том числе не более 0,5% свободного As2O3; содержа-
ние влаги не должно превышать 1,5%. Арсенит кальция, смешан-
ный с наполнителем — тальком или тонко размолотой фосфорит-
ной мукой, называют протарсом.
Арсенит натрия выпускают в виде водной пасты, содержащей
До 18% влаги и не меньше 52% АзгОз.
1404
Гл. XXXVIII, Соли мышьяка
таблица1ц
Качество мышьяковистого ангидрида
(Содержание компонентов в вес. %)
Рафинированный AS2O3 Технический As2O3 Я
чистый рядовой 1-й сорт 2-й сорт
As2O3, не более Остаток, не растворимый в водном растворе ам- 99,9 99,5 97,0 92,0
миака, не более .... 0,08 0,3 ’ 2 6
As2O3, не более Общая сера, не более . . Окрашивающие примеси, не более: — — 0,1 0,2
0,01 0,05 — —
Fe . 5- 10“3 0,02 — — idi.
Си, Мп, Ti (каждого) 5-10"? 0,01 — —
Сг 1 • 10 4 0,01 — —
N1 5-10“’ 0,01 — -ЯШ
Со 1•10“’ 0,01 — т
Влага, ие более 0.1 0,1 0,5 ко
Парижскую зелень — 3Cu(AsOgh• Си(СН3СОО)2 — выпускают
в виде порошка зеленого цвета, в котором, согласно ГОСТ 105—66,
должно быть: As2O3 не менее 53% (в том числе водорастворимого
Аз20з, а также растворимого в 90 %-ном этиловом спирте не более,
чем 3%), СиО не менее 28,5%, (СНзСО^О не менее 8%; влаж-
ность продукта не должна превышать 1%. Заменитель парижской
зелени — щелковская зелень содержит не менее 32% As2O3 (в том
числе не более 3,5% водорастворимого As2O3), не менее 17,5%
СиО и 5°/о (СН3СО)2О и не более 2% влаги.
Препарат арсмаль, применяемый для борьбы с личинками ма-
лярийного комара, получают, обрабатывая раствор мышьякови-
стой кислоты известью-пушонкой в смеси с инертным наполните-
лем — тальком, глиной или мелом и затем добавляя раствор мед-
ного купороса; отфильтрованную пасту, состоящую из арсенита
меди и наполнителя, высушивают и размалывают. Затем в препа-
рат вводят в качестве гидрофобной добавки 3% асидола или ней-
трального древесного креозотового масла для улучшения его нави-
гационных свойств, т. е. чтобы его зерна лучше удерживались на
поверхности воды. Препарат содержит: 9—11% As2O3, 7—10%
СиО и не больше 1,5% влаги.
Арсенат кальция (ГОСТ 156—66) должен содержать 38—42%
AS2O5, не более 1% As2O3, не более 0,6% суммы водорастворимых
AS2O5 и As2O3, не менее 2% щелочи (в пересчете на СаО) и не
более 1% влаги. Требования к дисперсности продукта такие же,
как и для других мышьяковых препаратов — 96% продукта должно
проходить через сито 6400 отв/см2, при этом максимальный размер
Сырье
1405
частиц не должен быть больше 85 мк, что обусловлено склонностью
продукта к флокуляции. Практически средний диаметр частиц
промышленных образцов арсената кальция 8—17 мк, а максималь-
ный ~ 40 мк38.
Арсенат кальция, разбавленный наполнителем — глиной или
каолином (в количестве 45%), с добавкой 3% асидола называют
меритолем.
Препарат купфермеритоль — комплексная соль арсенатов каль-
ция и меди, разбавленная образующимся в процессе получения
гипсом. Купфермеритоль получают, добавляя раствор медного ку-
пороса в смесь водных суспензий арсената кальция и извести-пу-
шонки. Образующуюся пульпу превращают на фильтре в пасту,
которую затем сушат, размалывают и гидрофобизируют асидо-
лом. Препарат содержит по 18—20% AS2O5 и СиО, не менее 1%
свободной окиси кальция, не более 0,5% As2O3 и 0,6% водораство-
римых As2O5 + As2O3, не более 1% влаги и 2—3% асидола.
За рубежом используют препарат, называемый лондонским
пурпуром, представляющий собой смесь арсенита и арсената каль-
ция. Он получается в виде отхода от производства фуксина и не-
которых других красителей и, помимо других примесей, содержит
краситель, придающий ему пурпурный цвет. Так как некоторые
образцы лондонского пурпура получались с большим содержанием
водорастворимого мышьяка, что приводило к ожогу листьев рас-
тений, то под этим названием стали выпускать и более токсичные
(за счет добавок белого мышьяка и арсената кальция) и более
стабильные и однородные по составу препараты. Состав некоторых
образцов лондонского пурпура: 26,8—28% общего мышьяка (As),
4,5—5,8% As2O3, 35,6—37,4% As2O5, 0,12—0,4% водорастворимого
мышьяка 39.
В связи с ядовитостью мышьяковых препаратов к их упаковке,
хранению и транспорту предъявляются особые требования. Тарой
для них служат барабаны из гофрированной или гладкой листо-
вой стали. После наполнения с помощью автоматической расфа-
совочной машины крышки барабанов завальцовывают или зали-
вают для уплотнения пеком. Затем барабаны вставляют в защит-
ные фанерные барабаны или деревянные бочки, а пространство
между их стенками засыпают древесными опилками.
СЫРЬЕ
Мышьяковые соединения весьма распространены в природе и
в небольших количествах содержатся во многих рудах, в морской
воде и в водах источников. В большинстве случаев мышьяк вхо-
дит в состав полиметаллических руд, содержащих цинк, свинец,
Никель, кобальт, медь, серебро, золото, олово, вольфрам и серу.
Количество мышьяка в таких рудах обычно меньше 1 % и его
1408
Гл. XXXVIII. Соли мышьяка
извлечение является попутным и зависит от условий извлечения
основных компонентов. Так, значительные количества белого
мышьяка получают в качестве побочного продукта из отходящих
газов ватержакетных медеплавильных печей путем улавливания
его в электрических фильтрах. Осаждающаяся пыль содержит
30—50% мышьяка; его очистку осуществляют возгонкой в отра-
жательных печах. Продукт, получаемый таким способом, называют
газовым мышьяком. Он составляет основную часть мирового про-
изводства белого мышьяка40, так как стоимость его значительно
ниже, чем продукта, получаемого специальным обжигом мышья-
ковых минералов.
Показана возможность выщелачивания мышьяка из железных
руд (с целью понижения содержания его в получаемой из руд
стали) горячими (80—100°) 5—6%-ными растворами NaOH и
КОН. Таким способом можно извлечь до 80% содержащегося
в руде мышьяка4'.
К мышьяковым рудам, имеющим самостоятельное значение, от-
носятся арсенопиритовые, скородитовые, леллингитовые, реаль-
гаро-аурипигментовые руды и вкрапленники в медистые и свин-
цово-цинковые руды. В них содержатся соединения мышьяка глав-
ным образом с серой и железом. К сернистым рудам мышьяка
относятся руды, содержащие реальгар As2S2 (оранжево-красный)
и аурипигмент As2Ss (желтый). Эти минералы имеют твердость
1,5—2 и плотность 3,4—3,6 г[см3. К железомышьяковым относятся
руды, содержащие мышьяковистый, мышьяковый колчедан и ско-
родит. Мышьяковистый колчедан (или лёллингит) FeAs2 (твер-
дость 5—5,5, плотность 7,1—7,4 г/сл3) встречается в плотных мас-
сах в виде примеси в серебряных, медных и других рудах цветных
металлов. Мышьяковый колчедан (или миспикель, арсенопирит)
FeAsS или FeS2 + FeAs2 образует натечные формы (твердость
5,6—6, плотность 5,9—6,2 г/с.и3) и содержит 46,1% As, 19,2% S
и 34,3% Fe; иногда 6—9% железа замещены кобальтом или нике-
лем. Мышьяковый колчедан — наиболее распространенный мышья-
ковый минерал, а его руды часто содержат золото и серебро в ко-
личествах, достаточных для промышленной разработки. Скородит
FeAsO4-2H2O встречается в виде кристаллических друз разного
цвета (твердость 3,5—4, плотность 3,1-—3,2 г/см3) 42 •43.
Мышьяковые руды месторождений Восточной Сибири содер-
жат: арсенопиритовые 9—10% As, скородитовые 10—12% As, по-
лиметаллические 6—9% As и золото-мышьяковые руды 4—15%
As44. В месторождениях реальгара и аурипигмента, имеющихся
на Кавказе и в Средней Азии, содержание мышьяка колеблется
в широких пределах — от нескольких процентов до 50% As в от-
дельных образцах породы 45-46. Бедные мышьяком руды после дроб-
ления и измельчения подвергают флотационному обогащению.
Концентрат содержит 18—22% мышьяка.
Получение белого мышьяка обжигом руд
1407
ПОЛУЧЕНИЕ БЕЛОГО МЫШЬЯКА ОБЖИГОМ
МЫШЬЯКОВЫХ РУД
При окислительном обжиге руд, содержащих сульфиды мышья-
ка, протекают следующие реакции:
2AS2S3 + 90з = 2AS2O3 + 6SO2
2AS2S2 -Ь 70 2 == 2AS2O3 -F 4SO2
2FeAsS + 5О2 = Fe2O3 + As2O3 + 2SO2
Образующаяся трехокись* мышьяка уходит из печей в виде
паров (см. рис. 427) вместе с другими газообразными продуктами
обжига, при охлаждении которых превращается в тонкодисперс-
ную пыль, выделяемую из газов в пылеуловителях. Побочной реак-
цией обжига является окисление трехокиси мышьяка до пятиокиси
AS2O3 + О2 *= AS2O5
давление пара которой очень мало, вследствие чего она переходит
в огарок.
Трехокись мышьяка, выгруженная из пылеуловителей, содер-
жит различное количество примесей. При достаточной чистоте ее
выпускают в виде готового продукта — белого мышьяка; если же
она загрязнена (серый или черный мышьяк), то ее очищают ра-
финированием путем повторного обжига при несколько меньшей
температуре, чем при обжиге руды.
Для обжига мышьяковых руд и концентратов2 применяют ме-
ханические печи разных конструкций — одноподовые с вращаю-
щимся подом или гребками и многоподовые. Иногда используют
отражательные и муфельные печи, дающие возможность получать
более чистый продукт, чем из механических печей. Преимуществом
муфельных печей является высокая концентрация паров AS2O3 в
газах, что резко уменьшает объем аппаратуры для конденсации
и улавливания AS2O3.
Степень извлечения As2O3 из руды зависит от размера зерен,
состава руды, температуры и длительности обжига. Оптимальный
размер частиц 3—5 мм. При обжиге слишком мелких частиц воз-
можно спекание материала, во избежание чего к руде добавляют
известняк (или другой наполнитель). Однако это приводит к уве-
личению содержания мышьяка в огарке и к снижению степени
извлечения As2Os вследствие образования арсенатов кальция и
сдвига равновесия реакции окисления As2O3 в сторону образова-
ния As2O5. Аналогично и при обжиге руд, содержащих значитель-
ные количества кальция, магния, свинца, трехокись мышьяка,
окисляясь в пятиокись, образует арсенаты этих металлов. Такие
руды обжигаются трудно, и для увеличения степени извлечения
мышьяка необходимо добавлять к ним уголь для восстановления
арсенатов и развития более высокой температуры. В присут-
ствии восстановителя (угля) обжигаются и скородитовые руды.
1408
Гл. XXXVIII. Соли мышьяка
содержащие арсенат железа FeAsO4 • 2Н2О, в . котором мышьяк
находится в виде AS2O5.
При обжиге смешанных руд, содержащих скородит и арсено-
пирит, помимо основных реакций обжига идет также восстанов-
ление As2O5 из скородита арсенопиритом
5As2O5 + 2FeAsS = Fe2O3 + 6As2O3 + 2SO2
что позволяет уменьшить содержание угля в шихте.
Сернистые руды подвергают окислительному обжигу при 500—
550°, арсенопиритные руды и концентраты обжигают при 600—
700°, восстановительный обжиг скородитовых- и смешанных руд
проводят при 800—900°. Огарок после обжига, содержащий цен-
ные металлы и 0,5—1,5% As2O3, направляют на дальнейшую пе-
реработку.
В обжиговых газах содержится 50—85 г/м3 As2O3. Газы вна-
чале очищаются от рудной пыли в сухих электрофильтрах, куда
они поступают с температурой 450—500° и откуда выходят с тем-
пературой ~350°. Дальнейшее охлаждение до 80—110°, при кото-
ром происходит кристаллизация твердого As2O3, осуществляют в
холодильниках и пылеуловителях разных конструкций — осади-
тельных камерах, полых башнях, рукавных фильтрах или электро-
фильтрах, в которых осаждается основная масса продукта в виде
серого мышьяка. Улавливание остатков As2O3 производят в ороси-
тельных башнях или в мокрых электрофильтрах, орошаемых во-
дой. Иногда из холодильников газ поступает непосредственно в
электрофильтры. Суспензию из электрофильтров отжимают на
нутч-фильтрах, и пасту высушивают47.
Получаемый технический белый мышьяк содержит 90—95%
As2O3. Для получения рафинированного продукта, содержащего
больше 97% As2O3, его подвергают возгонке при 500—600° и вто-
рично улавливают; печи и прочая аппаратура, применяемая для
рафинирования,-—такая же, как и для получения технического
продукта. Степень извлечения As2O3 при рафинировании ~85%.
Огарок после рафинирования, содержащий до 50% As2O3, приме-
шивают к исходному сырью, идущему на обжиг.
Стекловидный или аморфный мышьяковистый ангидрид полу-
чают путем медленной возгонки белого мышьяка при 500—600° и
последующей его конденсации. Стекловидный белый мышьяк мо-
жет быть получен также горячим прессованием (при 150°) рафи-
нированного белого мышьяка под давлением до 2500 ат.
АРСЕНИТ КАЛЬЦИЯ
Арсенит кальция получают растворением мышьяковистого
ангидрида в известковом молоке по реакциям
As2O3 4- Са(ОН)2 = Ca(AsO2)2 4- Н2О
As2O3 4-ЗСа(ОН)2 = Ca3(AsO3)2+ЗН2О
Арсенит кальция
1409
при этом арсенит кальция выделяется в осадок. Известь, приме-
няемая для получения арсенита кальция (и других мышьяковых
препаратов), должна легко гаситься водой и содержать не ме-
нее 92% активной окиси кальция (или 70% активной окиси каль-
ция в пушонке). Существуют два способа производства — мокрый
и полусухой.
Мокрый способ производства
В реактор с мешалкой и паровым барботером заливают извест-
ковое молоко, нагревают острым паром до 85—90° и вводят при
перемешивании мышьяковистый ангидрид (рис. 430). Количество
рат на гаше-
ние извести
Рис. 430. Схема получения арсенита кальция мокрым
способом:
1 — реактор; 2 —сборник для известкового молока; 3 — мерник
для известкового молока; 4 —бункер для белого мышьяка;
5 —весовой бункер-дозатор; 6 — рукавный фнльтр для улавли-
вания пыли; 7 — вакуум-насос; 8 — вакуум-фильтр; 9— вакуум-
сборник для фильтрата; /0 — барометрический конденсатор;
11 — шиековый питатель; 12 — сушилка; 13 — холодильный шнек;
/4 —бункер для арсенита; 15 — мокрый пылеуловитель;
16 — скруббер; 17 — вентилятор; 18 — мельничный бункер;
19— центробежная вакуум-мельница; 20— расфасовочный бун-
кер; 21 — шнек; 22 — расфасовочный аппарат; 23 — барабан
для готового продукта.
известкового молока берут с 10—15%-ным избытком (по СаО) от
теоретически необходимого. Белый мышьяк загружают в нагретое
молоко в течение 20—30 мин, затем берут пробу на полноту свя-
зывания мышьяка. Если в растворе больше 5 г/л As2O3, добав-
ляют рассчитанное количество известкового молока.
1410
Г л. XXXV III. Соли мышьяка
Полученная пульпа поступает на барабанный вакуум-фильтр.
Маточный раствор с фильтра направляют на приготовление из-
весткового молока и таким образом возвращают в процесс. От-
фильтрованную пасту — арсенит кальция с влажностью ~35%
высушивают в барабанной сушилке с наружным обогревом топоч-
ными газами. Водяные пары из сушильного барабана отсасы-
ваются вентилятором и подаются в пылеуловитель, орошаемый
водой, и в скруббер. Разрежение на выходе паров из сушилки
поддерживают не ниже 5 мм вод. ст. После сушки продукт с влаж-
ностью не более 1 % проходит через шнек, корпус которого охлаж-
дают снаружи водой, и поступает в нижний бункер, откуда пнев-
матическим способом подается в бункеры размольной установки.
Размолотый продукт направляют на расфасовку, которую осущест-
вляют с помощью герметизированных устройств, снабженных
пылеотводящими приспособлениями. На производство 1 т техниче-
ского арсенита кальция мокрым методом расходуют: 0,678 т бе-
лого мышьяка (100% As2O3), 0,335 т СаО (100%) в виде извест-
кового молока, 0,3 т условного топлива, 350 кет •ч электроэнергии
и 25 м3 воды.
Полусухой способ производства
По этому способу производят смешение извести-пушонки с бе-
лым мышьяком при ограниченном количестве воды. Исходные
сухие вещества загружают в вакуум-сушилку, представляющую
собой неподвижный горизонтальный цилиндр, вдоль оси которого
расположен вал мешалки с гребками; снаружи корпус сушилки
имеет паровую рубашку. Затем в сушилку заливают воду в коли-
честве, необходимом для разбавления пушонки в отношении 2:1,
и производят перемешивание массы в течение 1 ч без подачи пара
и при отключенном вакууме. Конец реакции контролируют по ана-
лизу пастообразной массы, которая не должна содержать больше
0,5% свободного As2O3. Затем для высушивания пасты включают
паровой обогрев и пускают вакуум-насос. Через каждые 0,5 ч
направление вращения вала мешалки автоматически изменяется,
причем изменяется и перемещение материала в сушилке — от се-
редины к краям или от краев к середине. Постепенно теряя воду,
паста через 3—4 ч загустевает и комкуется. Крупные комья при
дальнейшей сушке распадаются на более мелкие, чему способ-
ствует раздавливание их свободно лежащими в сушилке трубами,
передвигаемыми гребками. Сушку продолжают до содержания в
продукте 1—1,2% влаги. Постепенно на стенке корпуса сушилки
нарастает твердая корочка продукта, затрудняющая теплопередачу
и снижающая производительность аппарата. Для ее удаления су-
шилку 3—4 раза в месяц промывают водой. Выгружаемый из су-
шилки продукт поступает на размол и расфасовку. Схема произ-
водства этим способом изображена на рис. 431.
Арсенит кальция
1411
По другому варианту этого способа смешение исходных ве-
ществ— мышьяковистого ангидрида и извести-пушонки — произво-
дят в специальном реакторе в течение 10—15 мин. Затем в реактор
заливают горячую воду (60—70°) и перемешивают массу около 4 ч.
Полученную пасту высушивают в сушилке, обогреваемой топоч-
ными газами, имеющими на входе 700—850°, на выходе 130—160°,
Рис. 431. Схема получения арсенита кальция полусухим способом:
/ — приемник (разгрузочный аппарат) для белого мышьяка, подаваемого пнев-
матическим способом; 2 — расходный бункер для белого мышьяка; 3 — бункер
для нэвести-пушонки; 4 —весовой бункер; 5 —шнековый питатель; б —вакуум*
сушилка; 7 —бункер для продукта, выгружаемого нз сушилки; 8 — мельница;
Р —приемник (разгрузочный аппарат) для арсеиита кальция; /0—бункер для
готового продукта; 11 — расфасовочный аппарат; 12 — рукавный фильтр; 13 — мо-
крый фильтр-пылеуловитель; 14 — барометрический конденсатор.
Полусухой способ экономичнее, чем мокрый способ, вследствие
большей простоты производства, отсутствия операции фильтрации,
меньшего количества вредных отбросов и меньших потерь. На про-
изводство 1 т технического арсенита кальция полусухим методом
расходуют: 0,64—0,66 т белого мышьяка (100% AS2O3), 0,35—
0,365 т СаО (100%) в виде извести-пушонки, 0,35 т условного топ-
лива или 6 т пара, 260 кет • ч электроэнергии и 25 м3 воды.
Для того чтобы в атмосферу цеха не проникала вредная пыль,
все аппараты герметизированы и передача сухих порошкообразных
материалов (As2O3 и продукта) осуществляется с помощью ва-
куумного пневматического транспорта. Помимо того, все аппа-
раты, в которых передвигаются сухие, пылящие материалы, имеют
вентиляционные отводы. Уходящая по ним пыль улавливается
в мешочных фильтрах и возвращается в производство.
Н12 Гл. XXXVIII. Соли мышьяка
Для размола применяются вакуум-мельницы, в которых диск
дезинтегратора, устройство для пневматической классификации
материала и вентилятор, создающий воздушный поток, заключены
в один общий кожух.
АРСЕНИТ НАТРИЯ
Уравнение реакции получения арсенита натрия обычно пишут
следующим образом:
As2O3 4- 2Na2CO3 + Н2О м 2Na2HAsO3 4* 2СО2
Однако технический продукт содержит смесь разных солей ме-
та- и ортомышьяковистой кислот вследствие протекания реакций:
3Na2CO3 4- As2O3 = 2Na3AsO3 4- ЗСО2
Na2CO3 4~ As2O3 4- 2Н2О = 2NaH2AsO3 4- СО2
Na2CO3 4~ As2O3 « 2NaAsO2 4- СО2
Производство арсенита натрия заключается в варке мышьяко-
вистого ангидрида в растворе соды в реакторе, снабженном паро-
вым змеевиком. В нагретый до кипения раствор соды, содержащий
30—35% NasCOs, к которому добавлено небольшое количество
едкого натра (20—25% от веса Na2CO3), загружают отдельными
порциями в течение 45—60 мин мышьяковистый ангидрид, поддер-
живая температуру около 90—95°. Затем перемешивают массу
в течение нескольких часов при этой же температуре, тщательно
ее контролируя. Более низкая температура (ниже 80°) приводит
к прекращению растворения AS2O3, более высокая — к выбросам
массы из реактора вследствие интенсивного вспенивания, вызван-
ного выделением СО2. Конец реакции характеризуется исчезнове-
нием пены и началом спокойного кипения раствора. Раствор выпа-
ривают в том же реакторе в течение 16—20 ч до содержания в нем
не более 18% воды. Раствор при этом приобретает консистенцию
сиропа с большой вязкостью, что осложняет его переработку на
Сухой порошкообразный продукт. А так как арсенит натрия чаще
Всего применяют в виде растворов, для приготовления которых не
требуется сухой продукт, то его обычно выпускают в виде пасты,
содержащей до 18% влаги. Такая паста образуется при охлажде-
нии сиропообразного раствора в таре — барабанах из кровельного
железа, в которые его разливают после выпаривания. На произ-
водство 1 т технического арсенита натрия в виде пасты затрачи-
вается 0,528 т белого мышьяка (100% АэгОз), 0,237 т кальцини-
рованной соды (95% ЫагСОз), 0,05 т каустической соды (92%
NaOH), 12 мгкал пара, 32 квт-ч электроэнергии, 3,2 л3 воды.
’(Теоретически для образования 1 т метаарсенита натрия тре-
буется 0,525 т AS2O3 и 0,296 т 95%-ной кальцинированной соды.)
Парижская зелень '
1413
Пастообразный продукт, однако, имеет низкое качество. Он ха-
рактеризуется неоднородностью состава, что затрудняет его дози-
ровку при использовании. Помимо этого, затвердевший продукт
трудно удалить из барабанов, что сопряжено со значительными его
потерями. Поэтому более рационально получение порошкообраз-
ного арсенита натрия 47-49. Для этой цели густой раствор арсенита
натрия, выпаренный до содержания 20—25% воды, заливают в
стальные противни (длиной 1 м, шириной 0,2 м и высотой 0,1 м)
и высушивают в муфельной печи при 150—180°. Затем продукт
размалывают и расфасовывают.
Сухой кристаллический арсенит натрия (метаарсенит) можно
получить при взаимодействии белого мышьяка со смесью NaOH
И Na2COa в молярном отношении 2 : 1
2As2O3 + 2NaOH + Na2CO3 = 4NaAsO2 + CO2 + H2O
При смешении AS2O3 с раствором NaOH и ИагСОз (при общем
содержании воды 30—35%) при 60—70° образуется пульпа, на-
гревая которую до 85° получают студенистую массу черного цвета.
Затем ее высушивают при 160—200° и размалывают.
Сушку арсенита натрия без последующего размола, с получе-
нием порошкообразного или чешуйчатого продукта, содержащего
меньше 3% влаги, можно осуществить в вакуум-вальцовой су-
шилке, подавая в нее раствор с 33% воды49.
ПАРИЖСКАЯ ЗЕЛЕНЬ
Парижская или швейнфуртская зелень получается из мышья-
ковистого ангидрида, соды, медного купороса и уксусной кислоты.
•Схема производства изображена на рис. 432.
Вначале получают раствор метаарсенита натрия:
Аз20д "Ь Na2CO3 = 2NaAsO2 4- СО2
К этому раствору добавляют уксусную кислоту, которая, ней-
трализуя избыток соды, дает ацетат натрия:
Na2CO3 + 2СН3СООН = 2CH3COONa + СО2 + Н2О
Затем производят осаждение парижской зелени при прилива-
нии полученного раствора, содержащего метаарсенит и ацетат нат-
рия, к горячему (90—95°) раствору медного купороса (400 г/л):
6NaAsO2 +2CH3COONa +4CuSO4 = 3Cu(AsO2)2 • Cu(CH3COO)2 +4Na2SO4
Для полноты реакции и полного удаления из раствора ионов
меди избыток уксусной кислоты нейтрализуют содой, осадок от-
фильтровывают, тщательно промывают, полученную пасту, содер-
жащую ~35% влаги, сушат в вакуум-сушилке 10—12 ц да
1414
Гл. XXXVIII. Соли мышьяка
влажности 1% и измельчают50. Маточный раствор сульфата нат-
рия, содержащий до 0,03% As, не используется. Перед сбросом
в канализацию содержание мышьяка в нем снижают до санитарной
нормы (0,15 мг/л). С этой целью его обрабатывают раствором
железного купороса и известковым молоком. После отделения об-
разовавшихся осадков фильтрат, содержащий меньше 0,01%
мышьяка, разбавляют другими сточными водами, не загрязнен-
ными мышьяком, и лишь после этого отводят в канализацию. На
Рис. 432. Схема производства парижской зелени:
1 — весовой (разгрузочный) бункер для белого мышьяка; 2 —бункер для кальциниро-
ванной соды; 3 — бункер для медного купороса; 4 — растворитель соды; 5—центробеж-
ный насос для содового раствора; 6 — реактор метаарсенита натрия; 7 —ловушка;
8 — реактор; 9 —плунжерный насос; 10 — фильтрпресс; 11 — иутч-корыто; 12 — вакуум-
сушилка; 13 — пылеуловитель; 14— барометрический конденсатор; 15 — брызгоуловитель;
16 — барометрический приямок; 17 — ловушка; 18 — мокрый вакуум-иасос; 19 — продук-
ционный бункер; 20 — надмельничный бункер; 21 — мельница; 22— циклон; 23 — рукав-
ный фильтр; 24— эксгаустер; 25 —бункер для размолотого продукта; 26 — барабан для
готового продукта; 27—фильтры; 28 — мокрые вакуум-иасосы.
производство 1 т парижской зелени расходуют; 0,575 т белого
мышьяка (100% As2O3), 1 т медного купороса (100%
CuSO4-5H2O), 0,415 т кальцинированной соды (100%) и 0,127 т
уксусной кислоты (100%), 420 квт-ч электроэнергии, 8 т пара
и 55 м3 воды. Таким образом, на производство парижской зелени
расходуется большое количество дорогостоящего сырья.
Несколько более экономичным является получение парижской
зелени через углекислую медь так называемым полусухим спо-
собом51:
Cu(CH3COO)2 + 1,5СиСО3 • Си(ОН)2 + ЗАз2О3 =
= Си(СН3СОО)2 • 3Cu(AsO2)2 + 1,5Н2О + 1,5СО2
Взаимодействием раствора медного купороса (390—410 г/Л
CuSO4) с раствором соды (260 г/л Na2CO3) при 70° получают
Парижская зелень
1415
осадок основной углекислой меди. После отстаивания и удаления
раствора NazSCh осадок промывают в реакторе водой репульпа-
цИей. Затем пульпу, содержащую не более 0,2% NaoSCU спускают
в реактор, куда предварительно заливают 3/4 от общего необходи-
мого количества уксусной кислоты и загружают всю требуемую
навеску белого мышьяка с добавкой молотого каолина (50—100 кг
на 800 кг Аз20з). После смешения в реактор постепенно спускают
пульпу основной углекислой меди. После окончания выделения
двуокиси углерода и вспенивания реакционной массы к ней добав-
ляют остаток уксусной кислоты и подогревают массу вначале
до 70°. Затем подачей острого пара повышают температуру до 90°
и ведут перемешивание около 18 ч с постепенной передачей массы
на сушку после полного завершения процесса образования париж-
ской зелени (через 2,5—3 ч). Сушка продукта производится в две
стадии — до влажности 35—40% в гребковой двухвальной сушилке
воздухом, подогретым до 70—80°, и окончательная сушка до влаж-
ности не более 1,2% в вакуум-сушилке. При этом пастообразный
материал становится сыпучим. Высушенный продукт размалывают
и расфасовывают. На производство парижской зелени этим спо-
собом затрачивается: 0,56 т белого мышьяка (100% AS2O3), 0,982 т
медного купороса (100% CuSO4-5H2O), 0,447 т кальцинированной
соды (95% На2СОз), 0,116 т уксусной кислоты (100% СН3СООН),
5,6 мгкал пара, 282 кет -ч электроэнергии, 50 м3 воды. Этот способ
позволяет получать продукт I сорта из белого мышьяка, содержа-
щего всего 80—85% Аз20з-
Одно время с целью экономии соды вместо парижской зелени
производили щелковскую зелень, предложенную Щелковским хи-
мическом заводом в 1936 г. Она отличается от парижской тем, что
содержит примесь гипса; это понижает содержание AS2O3 (до
34—35%) и СиО (до 19—20%) и соответственно увеличивает рас-
ход продукта при применении.
Получение щелковской зелени аналогично получению париж-
ской зелени с той разницей, что взамен соды используют гашеную
известь. Процесс образования щелковской зелени состоит из сле-
дующих реакций:
As2O3 + Са(ОН)2 = Ca(AsO2)2 + Н2О
Са(ОН)2 + 2СН3СООН = Са(СН3СОО)2 + 2Н2О
3Ca(AsO2)2 + Са(СН3СОО)2 + 4CuSO4 = 3Cu(AsO2)2 Cu(CH3COO)2 + 4CaSO4
Так как в результате последней реакции в растворе не остается
примесей, то из производства исключается операция промывки
пастообразного осадка, что сильно сокращает общую продолжи-
тельность производственного процесса. На 1 т готового продукта
расходуют: 0,35 т белого мышьяка (100%), 0,148 т СаО в виде
пушонки, 0,6 т медного купороса (100% CuSO4-5H2O) и 0,075 т
Уксусной кислоты (100%)-
1416
Гл. XXXVI11. Соли мышьяка
МЫШЬЯКОВАЯ КИСЛОТА
Мышьяковая кислота является полупродуктом для производ.
ства различных мышьяковых препаратов. Сравнительно редко ее
применяют в качестве окислителя (например, при изготовлении
некоторых красителей) 47. Мышьяковую кислоту получают окисле-
нием белого мышьяка. Окисление может быть осуществлено азот-
ной кислотой52, смесью окислов азота и воздуха8’53"55, хлором*
хлорной известью, а также электрохимическим, термическим и дру-
гими способами.
Азотнокислотный способ '
Окисление белого мышьяка азотной кислотой
3As2O3 + 7Н2О + 4HNO3 = 6H3AsO4 + 4NO
является наиболее распространенным промышленным способом
получения мышьяковой кислоты, несмотря на то, что этот способ
осложняется необходимостью регенерации азотной кислоты ид
окислов азота, образующихся при ее восстановлении. Осуществле-
ние процесса несколько затрудняется сильным пенообразованием,.
возникающим под влиянием содержащихся в белом мышьяке при-
месей и вследствие интенсивного выделения газов в результате
реакции.
Окисление AS2O3 азотной кислотой осуществляют в реакторах
из кислотоупорного чугуна или из стали, футерованной кислото-
упорными плитками, или из нержавеющей стали 56. Для окисления:
3 т белого мышьяка в сутки требуется реактор с объемом 6—7 м3.|
Одновременно с приведенной выше основной реакцией происхо-
дит частичное восстановление азотной кислоты до ЫОг’.
2HNO3 + NO = 3NO2 + Н2О
Б результате процесс идет по уравнению: ]
As2O3 + 2Н2О + 2HNO3 = 2H3AsO4 4- NO + NO2
Этим уравнением выражается оптимальное соотношение между
Аз20з и HNO3. Оптимальной концентрацией кислоты является
30—32% HNO3. При этом получается мышьяковая кислота высо-
кой концентрации (50—58%). Однако скорость окисления 30—
32 %-ной азотной кислотой мала, и требуется применение катали-
заторов— НС1, HI. В реакцию вводят 2—3%-ный избыток AS2O3,
а по окончании окисления оставшийся осадок отделяют и вновь
возвращают на окисление. Для производства арсената кальция
требуется применять белый мышьяк 1 сорта, содержащий менее
5% примесей.
Выделяющиеся при окислении окислы азота поглощают водой,
и получаемую слабую 28—32%-ную азотную кислоту возвращают
на окисление белого мышьяка.
Мышьяковая кислота
1417
Производственный процесс заключается в следующем (см. ле-
вую часть схемы на рис. 434, стр. 1424). В реактор, снабженный
паровой рубашкой и мешалкой, заливают 30—32%-ную азотную
кислоту в количестве, равном ’/3 от необходимого для одной опе-
рации. Кислоту нагревают до 75—80° и производят постепенную
загрузку белого мышьяка. В качестве катализатора в реактор
вводят небольшое количество иода (0,5 г на 1 т AsoO5). Остальное
количество азотной кислоты подают постепенно в течение 3—4 ч.
К концу процесса реакционная масса занимает около половины
объема реактора. При реакции происходит сильное вспенивание,
для уменьшения которого рекомендуется вводить в аппарат суль-
фонафтеновые кислоты (контакт Петрова) из расчета 30—40 г
на 1 т арсената.
Через 5—6 ч, когда подача белого мышьяка оканчивается, тем-
пературу поднимают до 90—100° и перемешивание продолжают
еще 5—6 ч для полного восстановления азотной кислоты.
Полученная в реакторе мышьяковая кислота с концентрацией
до 700 а/л As2O5 отстаивается от твердых примесей (в том числе
от избыточного AS2O3) и окончательно отделяется от них филь-
трацией через нутч-фильтр.
Непрерывный способ окисления AS2O3 азотной кислотой в двух
каскадно расположенных реакторах с мешалками позволяет до-
стичь более высокого выхода мышьяковой кислоты, чем периоди-
ческий, обеспечивает спокойное ведение процесса, так как исклю-
чает резкое изменение скорости реакции и переброс реакционной
смеси (который иногда имеет место в первой стадии процесса при
осуществлении его периодическим способом) и, наконец, облегчает
регенерацию азотной кислоты из окислов азота вследствие по-
стоянства их концентрации.
Перспективный интерес может представить окисление белого
мышьяка азотной кислотой и кислородом под давлением. Чем
выше давление кислорода, тем быстрее идет окисление AS2O3 и
тем больше степень регенерации азотной кислоты. С наибольшей
скоростью процесс идет при 80° и при сильном перемешивании
реакционной массы.
Разработан способ комбинированного окисления соединений
трехвалентного мышьяка кислородом воздуха и азотной кислотой
на активированном угле; процесс идет с достаточной скоростью
при 20—30°54.
Возможно также окисление растворов мышьяковистых соеди-
нений кислородом воздуха (при 0—30°) с участием окислов азота
(~8% NO2 и ~1%NO) в присутствии катализатора иода (до
1%), вводимого в виде I2, HI, KI или IC1; рекомендуется осущест-
влять окисление противотоком в аппарате колонного типа53. При
Уменьшении pH раствора скорость окисления увеличивается; не-
обходимая кислотность раствора создается за счет образующейся
1418
Г л. XXXVIII. Соли мышьяка
при поглощении окислов азота азотной кислоты. Но повышений
кислотности раствора приводит к окислению I- в по реакций
2Г + 4Н+ + 2NO2 = 12 + 2Н2О + 2NO
и к последующему выдуванию из него иода, а потеря иода приво]
дит к замедлению, а затем к прекращению окисления арсенитов8
Другие способы получения мышьяковой кислоты I
При окислении водной суспензии белого мышьяка как газо-1
образным хлором, так и хлорной известью окислителем является
ион ОС1-: I
As2O3 + ЗН2О + 2ОСГ . 2H3AsO4 + 2СГ
При обработке суспензии газообразным хлором реакция про-
текает с достаточной быстротой, но наряду с мышьяковой кислотой
образуется соляная кислота. Для удаления НС1 смесь разбавлен-
ных кислот необходимо нагревать.
При окислении хлорной известью получается раствор мышья-
ковой кислоты с хлоридом кальция. Применяемая для окисления
хлорная известь должна содержать небольшой избыток Са(ОН)г
во избежание разложения ее с потерей хлора. Избыточная щелоч-
ность хлорной извести приводит к частичной нейтрализации мышья-
ковой кислоты с образованием труднорастворимого арсената каль-
ция. Окисление As2O3 в суспензии хлорной извести при 20—25°
достигает в лабораторных условиях 98%, если в хлорной извести
содержится не меньше 30% активного хлора, а суспензия не
очень концентрированная 51. Практически, однако, выход не боль-
ше 85%.
Получение мышьяковой кислоты окислением белого мышьяка
хлором и хлорной известью не имеет большого практического зна-
чения, так как расход окислителей велик, а продукт получается
загрязненным (хотя примесь хлорида кальция не препятствует
использованию мышьяковой кислоты для производства арсената
кальция). Кроме того, для разрушения остатков окислителя не-
обходимо затрачивать тиосульфат.
При электролизе мышьяковистой кислоты с применением пла-
тиновых электродов происходит электрохимическое окисление ее
с образованием мышьяковой кислоты. Анодная плотность тока
0,018 afcM2. В качестве катода, кроме платины, может быть при-
менен свинец.
Окисление трехокиси мышьяка до пятиокиси можно осуще-
ствлять прокаливанием в присутствии кислорода воздуха. При 700°
, за 1 ч окисляется до 97% As2O3 (рис. 428).
Арсенат кальция
1419
АРСЕНАТ КАЛЬЦИЯ
Существует несколько способов получения арсената кальция,
имеющих промышленное значение. Почти все они основаны на
окислении соединений трехвалентного мышьяка:
1) каталитическое окисление растворов арсенита кислородом
воздуха в щелочной среде;
2) осаждение арсената из мышьяковой кислоты, полученной
окислением белого мышьяка;
3) электрохимическое окисление арсенита;
4) окисление арсенита кальция воздухом при высоких темпе-
ратурах (термический способ).
Помимо перечисленных, существует способ получения арсената
кальция из окисленных мышьяковых руд, заключающийся в выще-
лачивании AS2O5 раствором едкого натра.
Наиболее экономичными, получившими широкое распростране-
ние в промышленности, является способ каталитического окисле-
ния растворов арсенита воздухом и азотнокислотный способ окис-
ления белого мышьяка с последующей переработкой мышьяковой
кислоты в арсенат.
Способ каталитического окисления растворов
арсенита воздухом
Каталитический способ производства арсената кальция заклю-
чается в окислении арсенита натрия в арсенат натрия кислородом
воздуха в присутствии катализатора (обычно медного купороса)
и в последующем осаждении арсената кальция обработкой рас-
твора известковым молоком. При этом вначале образуется двой-
ная соль CaNaAsO4 4Н2О, которая затем разлагается с образо-
ванием арсената кальция (см. стр. 1399). Схема производства этим
методом изображена на рис. 433.
В стальной реактор с мешалкой и барботером для острого пара
заливают маточные растворы и 40—45% раствор едкого натра в.
таком соотношении, чтобы концентрация NaOH была 75—85 г/л.
В жидкость, подогретую при работающей мешалке острым паром
до 80—90°, засыпают из весового бункера белый мышьяк в коли-
честве, отвечающем молярному отношению AS2O3 : NaOH от 1:6
до 1 : 6,5. После перемешивания в течение 30 мин при 85—95° в
результате реакции
As2O3 + 6NaOH = 2Na3AsO3 +ЗН2О
получается раствор арсенита натрия, содержащий не менее 50 г/л
As2O3; в этом растворе отношение NaOH : As2O3, определяемое па
отобранной на анализ пробе, должно находиться в пределах 1,21 —
1,31; в противном случае добавляют недостающий компонент и
Продолжают перемешивание массы. Общая продолжительность.
Арсенат кальция
1421
процесса образования арсенита натрия достигает 1,5—2 ч. В по-
лученный раствор загружают твердый или предварительно рас-
творенный в воде медный купорос (в количестве 2—3,5% от за-
груженного 100%-ного As2O3), который служит катализатором в.
процессе окисления арсенита. После растворения медного купороса
жидкость из реактора перекачивают в окислитель. При перера-
ботке белого мышьяка, содержащего значительные количества
примесей (сульфидов мышьяка и др.), замедляющих процесс окис-
ления, раствор арсенита натрия предварительно направляют на
отстаивание для отделения шлама, а затем в окислитель. В этом
случае раствор медного купороса добавляют к арсениту натрия
в окислителе.
Окислитель представляет собой стальной цилиндрический ре-
зервуар с коническим днищем, снабженный барботерами для воз-
духа и пара. В раствор, имеющий 65—85°, подают воздух в тече-
ние 1,5—4 ч. Отходящий воздух проходит через ловушку и про-
мывной скруббер, затем выбрасывается в атмосферу. Окисление
идет по реакции
2Na3AsO3 + O2 = 2Na3AsO4
которая считается- законченной, когда содержание AS2O3 в рас-
творе становится меньше 1 г/л.
Полученный раствор арсената натрия сливают в осадитель —
стальной цилиндрический резервуар с мешалкой и паровым змееви-
ком. Здесь, в подогретый до 85° раствор, при работающей
мешалке подают из мерника известковое молоко, содержащее
160 г/л CslO, в количестве, рассчитанном по содержанию As2Os
в растворе. Затем подогревают массу до 95—98°. Расчет осажде-
ния арсената кальция ведут по уравнению:
2Na3AsO4 + 4Са(ОН)2 = Са3( AsO4)2 • Са(ОН)2 + 6NaOH
Реакцию осаждения считают закончившейся, когда содержание
As2O5 в жидкой фазе ("в маточном растворе) становится меньше
1 г/л, а в твердой фазе суспензии содержится 38—42% As2Os
(в сухом веществе). В случае необходимости состав реакционной
массы корректируют, вводя недостающий реагент. Описанный ре-
жим не является оптимальным. Установлено16, что для получения
арсената кальция высокого качества и с максимальным выходом
осаждение необходимо вести при энергичном перемешивании при
температуре не ниже 90°, причем мышьяковый раствор следует
медленно прибавлять к известковому молоку. Молярное отноше-
ние СаО ; As2Os в исходных жидкостях должно быть 4, или по
весу 0,97. Практически берут 0,93—0,97 вес. ч. СаО на 1 вес. ч.
As2O5. Используемая известь должна быть высокого качества и
свежезагашенная.
1422
Гл: XXXVill. Соли мышьяка
По окончании осаждения прекращают перемешивание и подо-
трев и дают раствору, содержащему регенерированную щелочь
отстояться. Осветленный раствор с концентрацией 65—75 г/л
NaOH возвращают на приготовление арсенита натрия. Оставшуюся
в осадителе пульпу арсената кальция для отмывки от щелочи
взмучивают с водой и подвергают двукратной фильтрации на
•барабанных или ленточных вакуум-фильтрах непрерывного дей-
ствия. На первом фильтре осадок промывают водой; фильтрат,
разбавленный промывной водой, содержащий ~40 г/л NaOH,
используют для гашения извести. Осадок с первого фильтра ре-
пульпируется с водой в резервуаре с мешалкой и поступает на
вторичную фильтрацию. После этого пасту арсената кальция,
содержащую до 35% влаги, направляют на сушку до содержания
влаги не больше 1%.
Сушку производят в барабанной сушилке топочными газами,
которые по выходе из сушилки для улавливания уносимой ими
пыли проходят через батарейный циклон и мокрый фильтр или
через мокрый фильтр и скруббер, орошаемый водой.
Следует, однако, отметить, что более рациональна сушка
пасты не непосредственно дымовыми газами, а с помощью наруж-
ного обогрева. Это позволило бы избежать перегрева продукта
и снижения его токсичности. На некоторых заводах после пред-
варительного высушивания пасты до содержания 10—14% влаги
окончательную сушку производят в вакуум-сушилке с паровым
обогревом.
Выходящий из сушилки арсенат кальция охлаждают в шнеко-
вом холодильнике и направляют на размол в вакуум-мельницу,
а затем пневматическим способом подают в бункер готовой про-
дукции, из которого с помощью расфасовочного аппарата загру-
жают в тару.
Для размола арсената (и арсенита) кальция применяют
центробежную пневматическую мельницу вертикального типа
(ЦПВ) или горизонтального типа (ЦПГ). Мельница ЦПВ благо-
даря отсутствию редуктора проще по конструкции и расходует
в 2—3 раза меньше электроэнергии. Однако вследствие малого
числа оборотов размалывающего диска (1400 вместо 3000 об/мин
в мельнице ЦПГ) она дает помол низкого качества — в продукте
содержится больше крупных частиц, чем допускается стандартом.
Улучшение качества помола в мельнице ЦПВ может быть достиг-
нуто за счет равномерного ее питания, увеличения окружной ско-
рости и предотвращения попадания посторонних примесей сни-
жением их содержания в сырье (в извести и в белом мышьяке)59-
На производство 1 т арсената кальция описанным способом
расходуют: 0,35 т белого мышьяка (100% As2O3), 0,48 т извести
(100% СаО), 0,077 т едкого натра (92% NaOH), 0,014 т медного
купороса (100% CuSO4-5H2O), а также 2,5 т пара, 22,6 м3 воды-
Арсенат кальция
142J
430 квт-ч электроэнергии, 0,302 т условного топлива. Затраты
едкого натра объясняются потерями в производстве, которые могут
быть значительно уменьшены при рационализации и усовершен-
ствовании процесса 56.
Производство арсената кальция (как и других соединений
мышьяка) всегда сопровождается очисткой сточных вод, получае-
мых при промывке осадка на фильтрах, при улавливании пыли
из газов в мокрых пылеуловителях, вытекающих из мокрых ва-
куум-насосов или из барометрических и других конденсаторов,
образующихся при промывке аппаратов и полов и т. п. Все эти
ядовитые воды собираются в приямок с мешалкой и перекачи-
ваются центробежным насосом в отстойник-сгуститель. Осадок из
сгустителя возвращают в производство (в осадитель арсената
кальция), а осветленную воду обрабатывают известью для осаж-
дения из нее мышьяка в форме нерастворимых кальциевых солей.
Для этого воду отдельными порциями заливают в специальный
реактор с известью-пушонкой и после получасового перемешива-
ния отстоявшуюся воду сливают и заливают новую порцию. После
тридцати операций осадок, взмученный с водой, перекачивают в
отстойник-сгуститель и далее он возвращается в производство;
он состоит из арсената кальция (40—60%) и извести. Обработан-
ную известью осветленную воду вновь возвращают на производ-
ство, во все аппараты, потребляющие воду. Так как эта вода все
же загрязнена соединениями мышьяка, то стремятся получать воз-
можно меньшее количество сточной воды, чтобы можно было пол-
ностью возвратить ее в производство59. По нормам Государствен-
ной санитарной инспекции сточная вода должна содержать не
больше 0,15 мг!л As2O5.
Получение арсената кальция из мышьяковой кислоты
Способ производства арсената кальция из мышьяковой кислоты
нейтрализацией ее известью имеет преимущества перед каталити-
ческим и другими способами. Арсенат кальция, полученный этим
способом, обладает повышенной дисперсностью и пригоден для
авиационного опрыскивания концентрированными суспензиями.
Предварительное получение мышьяковой кислоты позволяет пере-
рабатывать ее не только на арсенат кальция, но и на другие
соли — арсенат свинца (джипсин), арсенат бария и проч. Арсенат
бария, являющийся хорошим зооцидом, получают взаимодействием
мышьяковой кислоты с хлоридом бария48.
Азотнокислотный способ
Наиболее совершенным способом получения арсената кальция
является азотнокислотный, использующий мышьяковую кислоту,
изготовленную путем окисления белого мышьяка азотной кислотой
(рис. 434).
1424
Гл. XXXVIII. Соли мышьяка
Способ основан на нейтрализации мышьяковой кислоты извест-
ковым молоком 32’60:
2H3AsO4 + 4Са(ОН)2 = Са3( AsO4)2 • Са(ОН)2 + 6Н2О
Реакцию проводят в осадителе — резервуаре с паровым змееви-
ком и мешалкой. В него заливают известковое молоко концентра-
цией 200 г/л, подогревают до 80—90° и подают при перемешива-
нии 10—12% раствор мышьяковой кислоты. Осаждение арсената
кальция заканчивается через 40—60 мин после подачи мышьяко-
вой кислоты. Затем пульпу арсената кальция направляют на ба-
Рис. 434. Схема производства мышьяковой кислоты и арсената кальция
азотнокислотным способом:
/ — бункер для белого мышьяка; 2—реактор для получения мышьяковой кислоты;
3 — холодильник; 4, 5 —отстойники; 6 — нутч-фильтр; 7 —сборник мышьяковой кислоты;
в—мерник азотной кислоты; 9 — окислительная башня; 10, // — абсорбционные башни;
12 — сборник азотной кислоты; 13 — сборник слабой азотной кислоты; 14 “Приемник
азотной кислоты; 15 — мерник для мышьяковой кислоты; 16 — мерник для известкового
молока; 17 — осадитель-реактор для получения арсената кальция; 18 — вакуум-фильтр;
19 — сборник фильтрата и промывных вод; 2о — сушилка; 2/ —бункер для арсената
кальция; 22 — мельница; 23 — расфасовочный бункер; 24 — камера расфасовки; 25 — ру-
кавный фильтр.
рабанный вакуум-фильтр. Фильтрат и промывную воду используют
для разбавления концентрированной мышьяковой кислоты и для
приготовления известкового молока. Для получения легкофильтрую-
щего осадка рекомендуют 60 вести осаждение при 50—60°. При этом
вместо тетракальцийарсената образуются кристаллы твердого рас-
твора гидроокиси кальция в дигидрате трикальцийарсената.
Паста арсената кальция снимается с фильтра с влажностью до
40% и поступает в вакуум-сушилку с паровым обогревом или в
барабанную сушилку, обогреваемую снаружи топочными газами.
Выходящие из сушилки пары проходят ловушку, где уносимая
ими ядовитая пыль улавливается при барботаже через воду. Вы-
гружаемый из сушилки продукт пневматическим способом транс-
портируется по трубе в бункер, из которого поступает на размол,
Арсенат кальция
1425
а затем в расфасовочный бункер для молотого продукта и из него
в тару. Пыль арсената, остающаяся в воздухе, выходящем из бун-
керов, улавливается в рукавных фильтрах. Воздух, отсасываемый
из этих фильтров ротационными вакуум-насосами, промывается
водой, которую периодически возвращают в цикл на разбавление
мышьяковой кислоты и приготовление известкового молока. На
1 т готового продукта расходуют: 0,37 т белого мышьяка, 0,07 т
49%-ной азотной кислоты, 0,5 т жженой извести, 0,02 кг иода,
70 м3 воды, 3 т пара, 540 квт-ч электроэнергии.
Помимо преимуществ, упомянутых выше, азотнокислотный спо-
соб производства арсената кальция имеет еще следующие преиму-
щества перед щелочным (каталитическим): возможность обойтись
без отстаивания суспензии арсената кальция перед фильтрова-
нием; последнее идет в 2—3 раза быстрее, фильтровальное полотно
разрушается значительно медленнее, а главное, фильтрат и про-
мывные воды полностью возвращаются в процесс.
Хлориый способ
При получении арсената кальция с применением в качестве
окислителя хлора подвергают хлорированию суспензию Аз20з в
воде, как и при получении мышьяковой кислоты (стр. 1418), н®
в этом случае смесь H3AsO4 и НС1 не разделяют.
Хлорированию подвергают суспензию AS2O3 в воде (185 кг
AS2O3 на 1 м3 воды). Процесс ведется в футерованных кислото-
упорной плиткой хлораторах с гуммированными мешалками и
протекает по уравнению:
As2O3 + 2С!2(г.) + 5Н2О(ж.) = 2H3AsO4 + 4НС1(р-р) +66,4 ккал
В получающуюся смесь разбавленных кислот в течение всего
процесса хлорирования, продолжающегося 3—4 ч, непрерывно
вводят известковое молоко. В хлораторах поддерживают 80—90°,
а дозировку известкового молока производят по pH реакционной
массы с таким расчетом, чтобы нейтрализовать только соляную
кислоту. По окончании реакции раствор содержит около 200 г/л
мышьяковой кислоты и хлорид кальция, образовавшийся при ней-
трализации НС1. Этот раствор отстаивается от шлама (нераство-
римые примеси, введенные с известковым молоком) и после подо-
грева до 80—90° вливается тонкой струей в известковое молоко
для осаждения арсената кальция. Осаждение заканчивается при
pH = 10. Такая последовательность осаждения позволяет получить
осадок хорошей структуры.
Осаждение мышьяковой кислоты может производиться и кар-
бонатом кальция, но реакция идет медленнее, чем с известью.
Осадок арсената кальция отделяют от маточного раствора на
барабанных вакуум-фильтрах. Отфильтрованная паста содержит
около 60% влаги и высушивается на вальцовых сушилках.
1426 Гл. XXXVIII. Соли мышьяка
Электрохимический способ Щ
-1
По этому способу вначале получают арсенит натрия раство-
рением белого мышьяка в 10% растворе NaOH. Щелочной рас-
твор арсенита натрия направляют на электролиз. В результате
электрохимического окисления на аноде протекает реакция
NaAsO2 + 2NaOH = Na3AsO4 + Н2
и образуется арсенат натрия. Выделяющийся на катоде водород
содержит мышьяковистый водород в количестве, равном 1 % от
веса перерабатываемого AS2O3. Степень окисления арсенита нат-
рия в арсенат достигает 92%.
Полученный раствор арсената натрия направляют в осадитель,
куда предварительно заливают известковое молоко. Образовав-
шийся осадок арсената кальция отделяют от жидкости на фильтре
и перерабатывают способами, рассмотренными выше. Маточный
щелочной раствор выпаривают до содержания 10% NaOH и воз-
вращают в процесс для получения арсенита натрия.
Термический способ
Э. В. Брицке был предложен способ получения арсената каль-
ция термическим путем, основанный на окислении паров мышья-
ковистого ангидрида кислородом воздуха при высокой температуре
в мышьяковый ангидрид и взаимодействии последнего с раскален-
ными кусками окиси кальция. Вначале процесс предполагалось
осуществить в шахтной печи при 700—900° с применением сухой
извести. Однако при этом возникли значительные трудности вслед-
ствие разности температур образования арсената кальция и воз-
гонки мышьяковистого ангидрида. Арсенат кальция образуется
при температуре более высокой, чем температура возгонки AsaOs-
В результате этого значительные количества возогнанного As^Os
не вступали в реакцию с окисью кальция. Эти затруднения были
впоследствии преодолены при использовании смоченной извести
в виде пасты. Разработанный вариант этого способа, так называе-
мый полусухой метод получения арсената кальция, заключается
в следующем61.
Предварительно смешением белого мышьяка, извести и воды
приготовляется полупродукт — паста, содержащая до 40% влаги,
которая после высушивания и прокаливания при 500—600° содер-
жит смесь арсенита кальция с избытком извести. При прокалива-
нии в окислительной атмосфере (в струе воздуха или топочных
газов, не содержащих восстановителей) образуется арсенат каль-
ция. Высушивание и окислительное прокаливание может прово-
диться раздельно в печах барабанного типа или в разных зонах
одной печи. В случае наружного обогрева барабана печи топоч-
Арсенат кальция
1427
дыми газами внутрь печи подается воздух, необходимый для окис-
ления. При внутреннем обогреве топливо должно сжигаться со
значительным избытком воздуха. В этом случае из печи выносится
больше продукта в виде пыли и требуются более громоздкие уста-
новки для ее улавливания. В случае объединения сушки и про-
калки в одной печи внутри нее необходимо устанавливать кольца
с шарами для размельчения продукта перед окислением. Темпе-
ратура окисления AS2O3 в As2O6 и образования арсената лежит
в пределах 600—650°. Получающийся продукт состоит из
4CaO-As2O5 и некоторого избытка окиси кальция. Избыток окиси
кальция полезен, так как понижает содержание в продукте водо-
растворимых соединений мышьяка.
Термический способ производства арсената кальция является
наиболее простым вследствие отсутствия громоздких операций
осаждения, фильтрации и промывки осадка. Однако этот способ
пока не используется в промышленности, так как дает продукт
с низкой токсичностью.
Получение арсената кальция из окисленных мышьяковых руд
Разработан способ получения арсената кальция не из белого
мышьяка, а непосредственно из окисленных скородитовых мышья-
ковых руд, содержащих 18—27% As, а также из бедных руд с со-
держанием 7—15% As62-34. Этот способ заключается в обработке
измельченной руды раствором едкого натра при 90—92°. При этом
As2O6 переходит в раствор в виде арсената натрия. Так, напри-
мер, если мышьяк содержится в руде в виде минерала скородита
(Fe2O3- As2O5-4H2O или FeAsO4 • 2Н2О), то протекает следующая
реакция:
FeAsO4 • 2Н2О +3NaOH = Na3AsO4 + Fe(OH)3 +2Н2О
Концентрация применяемого для выщелачивания руды рас-
твора едкого натра может колебаться от 5 до 40% в зависимости
от химического и минералогического состава руды. Бедные руды
требуют применения щелочи более высокой концентрации. Извле-
чение AS2O5 в раствор составляет 95—100%.
После выщелачивания и отстаивания пульпы осветленный рас-
твор арсената натрия перерабатывают на арсенат кальция осаж-
дением последнего известковым молоком. Известковое молоко с
концентрацией 20% СаО приготовляют из свежезагашенной изве-
сти и ведут осаждение при 90—95° путем приливания горячего
Раствора арсената натрия к горячему раствору известкового мо-
лока:
2Na3AsO4 + 4Са(ОН)2 = Ca3(AsO4)2 + Са(ОН)2 + 6NaOH
Осадок арсената кальция после отделения маточного раствора
Промывают водой до полного удаления NaOH, сушат при 120° и
1428
Гл. XXXVIII. Соли мышьяка
измельчают. При значительном содержании . водорастворимого
AsgOg в препарате к нему прибавляют Са(ОН)2.
Полученный продукт содержит 39—40% общего AsgOg, 0,15%
водорастворимого AS2O5, 0,15—0,2% свободной СаО и имеет объ-
емный вес 0,45 т/л3.
Вследствие сильной ядовитости соединений мышьяка при их
производстве должны особенно тщательно соблюдаться правила
те.хнйки безопасности и охраны труда. Необходимо, чтобы вся
аппаратура была герметизирована, а производственные помеще-
ния хорошо вентилировались. Так как отравление может происхо-
дить и через кожу, при впитывании в нее соединений мышьяка, то
работающие в цехах должны быть защищены специальными не-
проницаемыми комбинезонами, брезентовыми или резиновыми ру-
кавицами и сапогами и иметь респираторы. Предельно допустимая
концентрация AS2O5, AS2O3, AsH3 в воздухе рабочей зоны произ-
водственных помещений — 0,0003 мг/л.
ЛИТЕРАТУРА
1. М. Е. П о з и н, Технология минеральных удобрений н солей, Госхимиздат,
1957.—2. А. Н. Генварский, Производство мышьяка, ОНТИ, 1936,—
3. Н. Kirschning, К. Р ! i е t h, Z. anorg. Chem., 280, № 5—6, 346 (1955).—
4. А. К. Бабко, П. M. Марченко, Укр. хим. ж., 22, № 5, 665 (1956).—
5. R. Н. S h e r m a n, W. F. G i a q u e, J. Am. Chem. Soc., 77, № 8, 2154 (1955). —
6. Д. Я. Евдокимов, ЖПХ, 34, № 5, 1152 (1961). — 7. Д. Я- Евдокимов,
Укр. хим. ж., 17, № 2, 181 (1951). — 8. I. М. К о 11 h о f f, М. A. Finemann,
J. Phys. Chem., 60, № 10, 1383 (1956).—9. E. Tozefowitz e. al., Roczn. Chem.,
29, № 2—3, 243, 253 (1955); 30, № 1, 3 (1956). — 10. Г. П. Л учинений,
В. Ф. Чурилкииа, ЖПХ, 13, № 11, 1586 (1940).
11. Н. Guerin, Bull. Soc. chim. France, 1955, № 11—12, 1536; 1957, № 4,
545.— 12. H. Guerin, Compt. rend., 243, № 2, 156 (1956). — 13. E. Thilo,
I. Plaetschke, Z. anorg. Chem., 260, № 6, 297, 314 (1949). —14. С. M. Ш 0-
г а м, сб. «Исследования по прикладной химии», Изд. АН СССР, 1955, стр. 213—
15. С. М. Шотам, Н. И. Трушкина, ЖПХ, 22, № 1, 33 (1949). — 16. М. А. М о-
розова, Тезисы докладов НТК по инсектофунгицидам, изд. НИУИФ, 1944. —
17. М. Т. Серебренникова, ЖПХ, 13, № 11, 1539 (1940). —18. К. А. Тар,
Хим. пром., № 11, 11 (1946). — 19. R. Р u 1 о u, Compt. rend., 240, № 24, 2333
(1955).—20. М. Е. Купер м ан, В. И. Орлов, С. Н. Крутицкая,
Н. И. Трушкина, сб. «Исследования по прикладной химии», Изд. АН СССР,
1955, стр. 236; С. Н. Крутицкая, Труды НИУИФ, в. 167, 1960, стр. 156.
21. Tok ah a si, Sasaki, J. Chem. Soc. Japan. Ind. Chem. Sec., 56, № 11,
843 (1953). — 22. M. E. Куперман, А. П. Белопольск и й, сб. «Исследова-
ния по прикладной химии», Изд. АН СССР, 1955, стр. 225.—23. Л. М. Каба-
нова, В. Д. Пономарев, Труды Алтайского горнометаллургического н.-и.
ин-та, в. 3, 1956, стр. 136. — 24. В. Г. Ч у х л а н ц е в, ЖАХ, 11, № 5, 529
(1956).—25. В. Г. Чухланцев, ЖНХ, 2, № 5, 1190 (1957).— 26. И. Г. Дру-
жинин, А. А кб а ев, ЖПХ, 27, № 6, 1194 (1964). —27. J. L i n d q v i s t, Acta
chem. Scand., 9, № 1, 73, 79 (1955).—28. A. Engelbrecht, A. Aignesber-
ger, S. Hayen, Monatsh. Chem., 86, № 3, 470 (1955). — 29. N. K. Dutt,
A. K. Gupta, Current Sci., 24, № 6, 193 (1955); Z. anorg. Chem., 285, № 1—“
92 (1956).—30. G. Mitra, Sci. a. Culture, 19, № 4, 216 (1953).
Литература
1429
31. А.. Я. Берлин, Мышьяк на войне и в народном хозяйстве, ГНТИ,
1032. — 32. В. И. Орлов, П. В. Попов, М. А. Морозова, М. Г. Г а б р и е-
лова, Ядохимикаты (инсектисиды и фунгисиды), Госхимиздат, 1946,—
35. В. И. Орлов, М. А. Морозова, М. Н. Крутицкая, Журн. ВХО, 5, Ns 3,
968 (I960).—34. А. Л. Ефимов, И. А. К а з а с, Инсектициды и фунгициды,
Сельхозгиз, 1940.—35. С. Н. Иванова, Журн. ВХО, 9, № 5, 496 (1964).—
36. F. Cz abator, Е. Jahn, Paper Mill. News, 79, № 1, 14 (1956). — 37. В. И. О р-
л о в, М. Н. Крутицкая, А. С. Брук, Б. С. Иванова, Труды НИУИФ,
в. 167, 1960, стр. 201.—38. П. В. Попов, Тезисы НТК по инсектофунгисидам,
изд. НИУИФ, 1944. — 39. Д. Фрир, Химия инсектисидов и фунгисидов, ИЛ,
1948. — 40. Г. Л. Кореньков, Н. А. Устинова, Горнохимическое сырье
зарубежных стран, Изд. «Химия», 1965, стр. 137.
41. Н. А. Васютинский, Л. И. Васютинская, ЖПХ, 38, № 2, 251
(1965). — 42. Я. А. Марковский, Мышьяк, Госметаллургиздат, 1934.—
43. Л. Т. Белякова, 3. И. Мареева, Мышьяк. Требования промышленности
к качеству минерального сырья, в. 44, Госгеолтехиздат, 1961. — 44. И. М. Озе-
ров, Труды ЦНИГРИ, № 14, 1934. — 45. М. М. Константинов, Мышьяко-
вые руды СССР, Геолиздат, 1932. — 46. Л. А. Нечаев, Труды НИУ, в. 118,
1934. — 47. П. В. Дыбина, Технология минеральных солей, Госхимиздат, 1949,
гл. V. — 48. В. И. Орлов, К. А. Г а р, Хим. пром., № 9, 1 (1946).—
49. М. А. Морозова, Сборник НИУИФ, в. 167, 1960, стр. 110. — 50. П. В. М а н-
чев, Производство швейнфуртской зелени, Госхимиздат, 1933.
51. И. С. Розенкранц, Труды НИУИФ, в. 167, 1960, стр. 118.—
52. И. С. Розенкранц, Там же, стр 73. — 53. Д. Я. Евдокимов, ЖПХ, 33,
№ 7, 1664 (1960). — 54. Д. Я. Евдокимов, Там же, № 11, 2435 (I960).—
55. Д. Я. Евдокимов, Там же, 37, № 3, 465 (1964). — 56. И. С. Розен-
кранц, Тезисы докладов НТК по инсектофунгисидам, Изд. НИУИФ, 1944.—
57. С. А. Кац, А. И. Стрельцова, ЖХП, № 6, 435 (1937). — 58. И. С. Ро-
зенкранц, Хим. пром., № 1, 23 (1947). — 59. М. Г. Г а б р и ел о в а, М. А. Мо-
ро з о в а, Производство неорганических ядохимикатов, Изд. «Химия», 1964,
гл. 3. — 60. С. М. Ш о г а м, Труды НИУИФ, в. 167, 1960, стр. 43.
61. И. В. Голицын, Б. Б. Г и р ш г о р н, Л. С. Г а линкер,' Труды
НИУИФ, в. 123, 1935, стр. 7. — 62. В. И. Б рем пел ь, М. А. Морозова,
И. В. Голицын, Там же, стр. 53. — 63. И. В. Голицын, М. А. Моро-
зова, Там же, стр. 61.—64. И. В. Голицын, М. А. Морозова, Там же,
в. 135, 1939, стр. 5,
Глава XXXIX
СОЛИ КИСЛОРОДНЫХ кислот ХЛОРА
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Хлор образует ряд кислородных кислот — хлорноватистую
НСЮ, хлористую HClOj, хлорноватую НСЮ3 и хлорную НС1О4.
Эти кислоты и их соли — гипохлориты, хлориты, хлораты и пер-
хлораты — являются сильными окислителями.
Активность кислородных кислот хлора возрастает от хлорно-
ватистой к хлорной кислоте. Окислительная их активность увели-
чивается в обратном порядке — от хлорной к хлорноватистой, ко-
торая является наименее устойчивой.
Хлорноватистая кислота представляет собой продукт гидро-
лиза хлора:
С12 + Н2О нсю + н+ + сг
Константа гидролиза хлора в воде К — —1 имеет
следующие значенияY:
t, °C............... 10 20 25 35 40 50 60
К-IO4 ............. 2,58 4,06 '4,84 6,43 7,15 8,51 9,75
Равновесие устанавливается весьма быстро2’3.
В отсутствие хлора в растворе идет диссоциация хлорновати-
стой кислоты
НСЮ Н+ + С1О"
с образованием иона гипохлорита (СЮ-).
Константа диссоциации хлорноватистой кислоты4-6 при 10°
равна 3,23-10~8, при 30° — 6,61 • 10-8 и при 50° — 8,91 • 10-8. Хлор-
новатистая кислота вытесняется из ее солей — гипохлоритов даже
углекислым газом воздуха: СЮ" 4- СО2 4- Н2О = НСЮ + НСОз.
Физико-химические свойства
1431
3 равновесии с раствором хлорноватистой кислоты в парах7-8 на-
ходятся НСЮ, Н2О и С12О; последняя образуется по реакции:
2НС1О С12О + Н2О
Гипохлориты образуют в водном растворе ион СЮ", гидро-
лизующийся в щелочных растворах (pH = 8— 10):
С1О“ + Н2О нею + ОН"
Константа гидролиза гипохлорита4-6 равна 0,93-10"7 при 10°,
3,18-10"' при 30° и 6,14 • 10~7 при 50°.
В кислых растворах гипохлоритов (pH < 5) находятся хлор и
НСЮ, в слабощелочных (pH до 8,5) — гипохлорит и НСЮ9; в
сильнощелочных растворах (pH > 10) — только ионы СЮ-10.
Гипохлорит натрия выделяется из водного раствора при от-
гонке воды под вакуумом (при пониженной температуре) в виде
пятиводного кристаллогидрата NaC10-5H20, который легко пере-
ходит в одноводную соль NaClO-HsO. Последняя при нагревании
до 70° разлагается со взрывом.
Гипохлорит кальция получается при хлорировании известкового
молока в виде двуосновной Са(СЮ)2-2Са(ОН)2 или дветрети-
основной ЗСа(С1О)2 • 2Са(ОН)2 • 2Н2О или нейтральной Са(С1О)2-
•ЗН2О соли. Безводный гипохлорит кальция образуется при обез-
воживании трехводного кристаллогидрата сушкой при 60° в токе
сухого, не содержащего СО2 воздуха.
При хлорировании гидроокиси кальция (пушонки) образуется
хлорная известь, состав которой меняется в зависимости от усло-
вий получения. Она представляет собой сухой белый порошок с
запахом хлора. Наличие иногда желтоватого оттенка объясняется
содержанием примесей соединений железа. По своему химиче-
скому и фазовому составу хлорная известь является сложным
комплексом и, состоящим из соединений гипохлорита кальция
Са(С1О)2-2Са(ОН)2, ЗСа(С1О)2-2Са(ОН)2-2Н2О, Са(СЮ)2-ЗН2О,
хлоридов и оксихлоридов кальция12 СаС12-4Н2О, СаС12-2Н2О,
СаС12-ЗСа(ОН)2 • 12Н2О, СаС12• Са(ОН)2-Н2О, хлората кальция
и свободной гидроокиси кальция. При растворении хлорной изве-
сти в воде образуется раствор, идентичный раствору гипохлорита
кальция.
О качестве гипохлоритов и хлорной извести судят по содержа-
нию в них активного хлора и по нх стабильности, т. е. способности
сохранять активный хлор в течение длительного времени. Актив-
ным (иногда белящим или действующим) хлором называют то
количество хлора, которое выделяется при действии на продукт,
соляной кислотой, например:
4 Са(С1О)2+4НС1 = СаС12+ 2Н2О+2С12
21 М. Е. Позия
1432
Гл. XXXIX. Соли кислородных кислот хлора
Следовательно, одна молекула гипохлорита кальция эквива-
лентна двум молекулам активного хлора. При условном пересчете
на весовой процент оказывается, что чистый гипохлорит кальция
2 • 70 92
«содержит» ——-100 = 99,2% активного хлора; поэтому эту
соль называют «твердым хлором». В молекуле Са(СЮ)2 действи-
тельное содержание хлора в 2 раза меньше (49,6%). «Содержа-
ние активного хлора» является лишь условным выражением окис-
лительной способности продукта и не тождественно фактически
содержащемуся в нем количеству хлора.
Гипохлориты — мало стойкие соединения и при различных ус-
ловиях разлагаются с потерей активного хлора и с образованием
хлората (хлоратный распад), кислорода (кислородный распад)
или хлора (хлорный распад) по реакциям:
ЗС1О- = CIO3 + 2СГ
2С1О~= 2СГ + О2
СЮ” + СГ + Н2О = 2ОН" + С12
Скорость разложения мала в сильнокислой среде (pH ~ 1),
максимальна при pH ~ 6,7 и уменьшается в щелочной среде13.
В присутствии ионов бромата скорость разложения гипохлорита
возрастает14. Так как мерой окислительной способности является
потенциал, устанавливающийся в растворах на инертном элек-
троде (платине), то скорость реакций разложения гипохлорита в
зависимости от pH находится в соответствии 15 с различным ха-
рактером процессов анодного окисления и катодного восстановле-
ния в кислой и щелочной средах.
Ускорение разложения гипохлоритов (потеря ими активного
хлора) под влиянием двуокиси углерода объясняется вытеснением
ею свободной хлорноватистой кислоты, диссоциирующей с образо-
ванием иона СЮ', который затем распадается по одной из при-
веденных реакций. Разложение гипохлоритов ускоряется также в
присутствии аммиака и соединений аммония, что используют в про-
цессе отбелки 16-18 текстильных материалов (см. ниже). В водных
щелочных растворах гипохлориты относительно устойчивы. При
щелочности 2—3% растворы гипохлорита натрия могут храниться
в течение 10—15 суток. Повышается и их термическая стойкость19.
Гипохлорит натрия отличается высокой устойчивостью в водном
растворе тринатрийортоарсената вследствие образования комплек-
сной соли (ЫазАзО4 • 11Н2О)-NaClO • NaCl20.
Твердые соединения гипохлорита кальция в отсутствие приме-
-.сей и других веществ, ускоряющих процесс разложения (двуокиси
углерода и влаги), являются стабильными веществами21. При на-
гревании в течение 5 ч при 100° гипохлориты в среднем теряют
около 4% активного хлора. Техническая хлорная известь при тех
Физико-химические свойства 1J33
же условиях теряет около 40% содержащегося в ней активного
хлора, т. е. стабильность ее в 10 раз меньше.
Под каталитическим влиянием соединений некоторых металлов
(Ni, Со, Fe, Си, Мп) гипохлориты в присутствии влаги быстро
разлагаются 22'23 с выделением кислорода.
Смеси из предварительно подсушенных при 80—90° гипохлорита
кальция и окисей металлов в отсутствие влаги и без подогрева
сохраняются в течение нескольких лет. При нагревании этих сухих
смесей до 60—70° гипохлорит кальция разлагается с выделением
хлора и небольшого количества кислорода. Лучшими катализато-
рами для этой цели являются медно-кобальтовые и железо-кобаль-
товые 24.
Разложение чистого гипохлорита кальция в растворе является
реакцией второго порядка 25. Константа скорости реакции при 40°
равна 0,61 • Ю-3 и при 50° — 2,44-10'3 л/(г-мол-ч).
Разложение гипохлорита в растворе в присутствии каталитиче-
ски действующих гидроокисей кобальта, никеля, железа и меди
подавляется добавкой солей свинца, хрома, мышьяка и некоторых
других элементов. Гидроокись железа теряет свои каталитические
свойства в присутствии в растворе избытка твердой (нерастворен-
ной) гидроокиси кальция. Заслуживает внимания интенсивное
ингибиторное действие активных препаратов двуокиси кремния
на разложение растворов гипохлорита кальция, содержащих гид- 1
роокись железа 26.
Хлорная известь, содержащая 10% влаги, в диапазоне 60—130°
разлагается весьма интенсивно (почти полностью в течение 1 ч)
с выделением кислорода11. Высушенная хлорная известь до влаж-
ности 1,9% разлагается при 100° со скоростью, равной скорости
разложения влажной хлорной извести при 40°.
Разрушение хлорной извести при ее хранении протекает мед-
ленно. При нагревании разложение идет с выделением кислорода
и выше 150° сопровождается взрывом. Смеси хлорной извести с
органическими веществами воспламеняются при температурах
ниже 100°. Поэтому попадание в хлорную известь органических
примесей может привести к ее внезапному и быстрому разруше-
нию. Разложение начинается в точках местных перегревов, вызван-
ных окислительными процессами, и вследствие их экзотермичности
быстро распространяется по всей массе продукта.
Скорость каталитического распада хлорной извести под влия- .
нием примесей тяжелых металлов особенно увеличивается при по-
вышенной температуре (выше 40°) и в присутствии влаги27.
Разбавленные растворы хлорной извести могут сохраняться
без существенного разложения десятки суток при хранении их в
темноте в стеклянной посуде28.
Моноокись хлора С^О существует в виде раствора в четырех-
хлористом углероде при низких температурах (0° и ниже). Ее
21*
1434
Гл. XXXIX. Соли кислородных кислот хлора
готовят пропусканием хлора в суспензию желтой окиси ртути в ча-
тыреххлористом углероде. Изучены термодинамические характе-
ристики С1гО в растворе СС14 со щелочью и перекисью водорода2»
Двуокись хлора СЮо — газ от зеленовато-желтого до красно-
вато-желтого цвета (при температуре выше 11°) с плотностью
3,09 г/л (при 11°). При охлаждении ниже 11° двуокись хлора из
газообразного состояния конденсируется в жидкость, окрашенную
в красно-коричневый цвет. В твердом состоянии она образует оран-
жево-красные кристаллы, плавящиеся при —59°. Температура ки-
пения при 731 мм рт.ст. равна 9,9°. Двуокись хлора взрывается
при нагревании или в присутствии органических и других веществ,
способных окисляться. Она взрывоопасна в смесях с воздухом при
содержании СЮг выше 10%. Разбавление СЮг инертными газами
(Не, Ar, N2) снижает скорость ее детонации30.
Растворимость двуокиси хлора при 4° равна 2000 мл в 100 г
воды и прямо пропорциональна парциальному давлению газа над
раствором31-32. Значения 32-33 коэффициента К [в моль! (л • мм рт.
ст.)] в уравнении зависимости концентрации двуокиси хлора в рас-
творе с (в моль/л) от парциального ее давления Р (в мм рт. ст.)
с — КР при 0, 5, 10, 25 и 35°, соответственно равны: 70,6, 56,3, 46,2,
30,2 и 21,5. С повышением температуры растворимость двуокиси
хлора в воде резко уменьшается. Растворимость СЮг в других
растворителях (СС14, НгЗО4 и СН3СООН) также подчиняется За-
кону Генри34. В водных растворах на холоду двуокись хлора раз-
лагается крайне медленно, в горячей воде разлагается с образова-
нием HCIO3, С12 и Ог. Установлено существование кристалло-
гидрата СЮ2 • 6Н2О35.
Предполагают, что двуокись хлора является ангидридом36,
образующим с водой соответствующие кислоты H2CIO3 и
H2CI2O5, — весьма неустойчивые и восстанавливаемые металлами
до НСЮг- В отсутствие восстановителей скорость разложения этих
кислот выше скорости их образования. С перекисью водорода дву-
окись хлора реагирует, образуя хлористую кислоту37:
2СЮ2 + Н2О2 = 2НС1О2 + О2 Й
Двуокись хлора раздражает дыхательные пути и вызывает го!
ловную боль уже при разбавлении 45: 1 000 000. ।
Хлористая кислота 33-40 выделена и в свободном виде, но полу-
чается обычно в водных растворах. Константа диссоциации ее
равна 1,07 • 10-2 при 18°. Образование хлористой кислоты про-
исходит в значительных количествах лишь в сильнокислой среде
(pH <3). При этом в растворе наряду с хлористой кислотой нахо-
дится и двуокись хлора 41.
Хлориты — соли хлористой кислоты в твердом состоянии при
обычных условиях являются довольно устойчивыми соединениями.
Кислые водные растворы разлагаются тем быстрее, чем выше тем-
Физико-химические свойства .
• 1435
пература и меньше величина pH. Достаточно устойчивы щелочные
растворы42. Некоторые хлориты могут быть получены действием
свободной хлористой кислоты на нерастворимые карбонаты43. Хло-
рит натрия кристаллизуется из щелочного раствора в виде безвод-
ной соли NaClOz и тригидрата ХаСЮг-ЗНгО, переходящего в
безводную соль при 37,4°44. При нагревании до 175° разлагается
с выделением кислорода. Реакция идет с большой скоростью
вплоть до взрыва. В слабощелочных растворах, содержащих не бо-
лее 1 г-мол/л NaClOz, хлорит натрия не разлагается при кипяче-
нии. В более концентрированных растворах он разлагается по ре-
акциям 45< 46:
3NaClO2 = 2NaC103 + NaCl
NaC102 = NaCl + O2
Константы скорости этих реакций равны47 соответственно при
103°: 0,65- IO’6 и 1,2- 10-7; при 83°: 1,6- 10"7 и 0,2- 10'8.
Хлорноватая кислота в свободном виде может существовать
только в растворе. Она является сильной кислотой и энергичным
окислителем. Ее соли—хлораты — большей частью хорошо рас-
творимы в воде; в растворах не являются окислителями.
Хлорат калия или бертолетова соль КСЮз кристаллизуется в
безводной форме в виде прозрачных бесцветных кристаллов моно-
клинической системы плотностью 2,32 г/см3. Растворимость КСЮ3
в воде: при 0° —3,21%, при 104° (температура кипения)—37,6%.
При нагревании до 368,4° КС1О3 плавится, а затем начинает раз-
лагаться по реакциям:
2КС1О3 = 2КС1 + ЗО2 + 23,6 ккал
4КС1О3 = ЗКС1О4 + КС1 + 70,9 ккал
Образующиеся продукты (КС1 и KCIO4) ускоряют48 выделение
кислорода. При 610° образовавшийся перхлорат калия плавится
и разлагается:
КС1О4 = КС1 + 2О2 - 7,9 ккал
В присутствии катализаторов (МпОг и др.) хлорат калия раз-
лагается при более низких температурах с интенсивным выделе-
нием кислорода. Хлорат калия в кислой среде является сильным
окислителем. Смеси его с углем, серой и другими веществами
взрывают от удара. Хлорат калия (и другие хлораты) ядовит
(смертельная доза — 2—Зе КС1О3).
Хлорат натрия NaClO3 кристаллизуется в безводной форме,
сильно гигроскопичен, на воздухе расплывается. Насыщенный вод-
ный раствор содержит при —15° 41,9%, при 122° 74,1% NaClO3.
Температура плавления хлората натрия находится в пределах
248—264°. Отмечены случаи взрывов хлората натрия на складах
при хранении, а также воспламенения сухих частей растений, на
1436
Гл. XXXIX. Соли кислородных кислот хлора
которые попал хлорат натрия. В присутствии гигроскопических
веществ (СаС12, MgCl2 и др.) 4Э, а также полиборатов или мета-
боратов натрия взрыво- и огнеопасность хлората натрия сни-
жается. В системе NaC103—NaC102—Н2О 50 в диапазоне темпе-
ратур 15—45° кристаллизуются безводные NaC103 и NaC102, а
также NaC102-3H20.
Хлорат кальция Са(С10з)г кристаллизуется из водного рас-
твора в виде дигидрата51, плавящегося при 130°. Насыщенный вод-
ный раствор кипит при 182°. Безводный хлорат кальция разла-
гается при нагревании до 334°.
Гексагидрат хлората магния Mg(СЮ3)2 • 6Н2О представляет
собой ромбические кристаллы — длинные иглы или листочки. При
35° частично плавится и переходит в тетрагидрат. Растворимость
его в воде равна 53% при 0°, 56,5% при 18°, 60,23% при 29° и
63,65% при 35°. Он отличается высокой гигроскопичностью, не
взрывает и безопасен в пожарном отношении49.
Хлорная кислота52 образует два кристаллогидрата —
НС1О4 • 4Н2О и НСЮ4 • ЗН2О53 и является сильным электролитом54.
Коэффициент активности хлорной кислоты при 25° изменяется от
0,911 до 0,804 при изменении концентрации НСЮ4 от 0,01 до
0,1 М в 1 кг раствора55.
Перхлорат калия КСЮ4 образует ромбические кристаллы плот-
ностью 2,52 г/см3. При 0 в 100 мл воды растворяется 0,75 г, а при
100° — 21,8 г КСЮ4. Чистый перхлорат калия разлагается при
537—600° на КС1 и О2. В качестве промежуточного продукта об-
разуется КСЮз, который, расплавляясь, ускоряет разложение56.
Реакция ускоряется в присутствии КС1, KBr, KI57, Си, Fe, Со,
MgO и др.58.
Перхлорат магния образует кристаллогидраты с 2, 4 и 6 мо-
лекулами воды. Равновесное давление пара при 23° над
Mg(C104)2 • 6Н2О равно 20,9 мм рт. ст., над M.g(C104)2 • 4Н2О —
8,15 мм рт. ст., а над Mg(C104)2 • 2Н2О около 10~4—10~5 мм рт.ст.59.
При нагревании выше 400° Mg(C104)2 разлагается60.
Перхлорат аммония характеризуется наиболее высоким весо-
вым содержанием кислорода среди всех перхлоратов. В 100 а воды
при 0° растворяется 10,7 г, при 85° — 42,5 г NH4CIO4. Во взаимной
водной системе из перхлоратов и хлоридов .аммония и магния
наименее растворимой солью при 25° является NH4CIO461.
Кислородные соединения хлора высших степеней окисления —
пожаро- и взрывоопасны, особенно в присутствии примесей легко
окисляющихся, например органических веществ, от загрязнения
которыми их следует оберегать. Взрыв твердых сухих хлоратов и
перхлоратов может быть вызван ударом или сильным толчком, что
нужно учитывать при сушке, размоле и транспортировке этих ве-
ществ. Эти операции должны осуществляться в аппаратах, в ко-
торых исключена возможность ударов металлических частей.
Применение
1437
ПРИМЕНЕНИЕ
Соли низших кислородных кислот хлора являются хорошими
отбеливающими средствами вследствие их высокой окислительной
активности. Основным отбеливающим и окислительным хлорным
соединением является хлорная известь62. В настоящее время для
этих целей широко применяются также гипохлориты, хлориты и
двуокись хлора.
Наибольшие количества хлорной извести потребляют в текстиль-
ной и бумажной промышленности для отбелки тканей и целлюлозы
(хлорная известь часто называется белильной известью). Хлорную
известь применяют в качестве окислителя в некоторых химических
производствах (при получении хлороформа, хлорпикрина и других
продуктов), для дезинфекции питьевых и сточных вод, для дезин-
фекции овощехранилищ63 и в качестве хорошего дегазатора. Ее
используют также для очистки ацетилена и некоторых нефтепро-
дуктов.
Хлорную известь выпускают трех марок (табл. 112).
ТАБЛИЦА 112
Качество хлорной извести (ГОСТ 1692—58)
(Содержание компонентов в %)
Марки
А Б В
Активного хлора, не менее .... • . 35 35 32
Разница между содержанием общего и активного хлора, не более .... 2 2 4
Влаги, не более для длительного хранения .... 2 10 Не корми-
для текущего потребления .... 2 Не норм! ровано 1рэвано
Потери активного хлора в хлорной извести марки А должны
быть не больше 4% в течение 3 лет ее хранения со дня отгрузки
заводом.
Хлорную известь марок Б и В упаковывают в деревянные бочки
емкостью от 50 до 275 л, в фанероштампованные бочки или фа-
нерные барабаны емкостью 50 и 100 л, а также (для недлитель-
ного хранения) в сухие заливные деревянные бочки емкостью
от 50 до 250 л. Хлорную известь марки А, а также марки Б (для
Длительного хранения) упаковывают в стальные барабаны ем-
костью 100 л. Бочки или барабаны с хлорной известью герметично
закупоривают и хранят в сухом и прохладном помещении, защи-
щенном от действия прямых солнечных лучей. Вместо деревянных
бочек и барабанов применяют также полиэтиленовые мешки.
1438
Гл. XXXIX. Соли кислородных кислот хлора
Несмотря на эти предосторожности, хлорная известь при хра-
нении постепенно теряет активный хлор. При недостаточной гер-
метичности тары некоторые образцы продукта почти полностью
теряют активный хлор в течение одного года, а иногда значи-
тельно скорее. При 40—45° рядовая хлорная известь полностью
теряет активность в течение 2 месяцев.
Хлорная известь все более вытесняется другими более удоб-
ными в употреблении отбеливающими и окислительными вещест-
вами62— гипохлоритами, двуокисью хлора и др.
Гипохлорит натрия в виде водного раствора находит большое
распространение вследствие простоты изготовления его на месте
потребления. Он является полупродуктом 64 в производстве гидра-
зина, пластических масс, синтетических волокон и др. Предложен65
гипохлоритный способ переработки пылевидных отходов от заточ-
ки твердосплавного инструмента, основанный на окислении кар-
бида вольфрама в щелочных растворах NaClO и переходе воль-
фрама в раствор.
Согласно ГОСТ 11086—64, гипохлорит натрия должен быть
прозрачной зеленовато-желтой жидкостью без осадка и взвешен-
ных частиц, содержащей к моменту отгрузки потребителю не менее
185 г/л активного хлора и не более 0,07 г/л железа; содержание
NaOH должно быть в пределах 10—20 г/л. Раствор гипохлорита
натрия хранят и транспортируют в закрытых гуммированных или
защищенных винипластом цистернах и контейнерах при темпера-
туре не выше 25°.
Технический гипохлорит кальция, содержащий более 50% ак-
тивного хлора, транспортабельное, чем хлорная известь. С гипо-
хлоритом кальция перевозится менее 100% балласта (примеси и
тара), в то время как с хлорной известью 250—300%. Важным
преимуществом гипохлорита кальция, по сравнению с хлорной из-
вестью, является отсутствие значительного осадка при растворении
его в воде66 (при растворении хлорной извести образуется осадок
основных солей, в котором теряется иногда до 50% активного
хлора). Предложено67 использовать смесь 2 вес. ч. Са(ОС1)2 и
0,8 вес. ч. Na2SO4 в виде таблеток для обработки воды.
Гипохлорит кальция выпускают в виде дветретиосновной соли
ЗСа(С1О)2 • 2Са (ОН)2 • 2Н2О, обозначаемой ДТСГК, и реже в виде
двухосновного гипохлорита кальция Са (С1О)2 • 2Са(ОН)2, обозна-
чаемого ДСГК- ГОСТ 13392—67 предусматривает выпуск ДТСГК
I- и 2-го сорта. В них должно быть соответственно: активного
хлора не менее 55 и 50% и влаги не более 1 и 1,5%; содержание
общего хлора не должно превышать половинного содержания ак-
тивного хлора (%) плюс 6% для 1-го сорта, или плюс 7% для
2-го.
ДТСГК упаковывают в оцинкованные барабаны. Продукт необ-
ходимо хранить в сухом, неотапливаемом помещении.
Применение
1439
Двуокись хлора по своим окислительным свойствам занимает
промежуточное место между хлоратами и гипохлоритами. Основ-
ное ее преимущество как отбеливающего реагента заключается в
том, что она почти совершенно не действует разрушающим обра-
зом на клетчатку волокон. Поэтому ее широко используют как
лучшее отбеливающее средство для древесной (бумажной) массы
и целлюлозы, а также для стерилизации и дезодорации воды68 и
пищевых продуктов. Вследствие трудности хранения и перевозки,
СЮг обычно получают на месте потребления и используют в виде
10%-ной смеси с воздухом69.
Хлорит натрия широко применяется в текстильной промышлен-
ности для отбеливания тканей, пряжи, волокна. При этом дости-
гается высокое качество отбелки без уменьшения прочности во-
локна. Он используется также в качестве исходного материала для
получения небольших количеств двуокиси хлора.
Хлорат калия используют главным образом в спичечной про-
мышленности, в пиротехнике, в небольших количествах в фарма-
цевтической промышленности, а также во взрывной технике.
Состав технической бертолетовой соли должен соответствовать
данным табл. ИЗ.
ТАБЛИЦА 113
Состав технической бертолетовой соли (по ГОСТ 2713—70)
(содержание компонентов в %)
1-Й сорт 2-й сорт
Хлорат калия (в пересчете на сухое вещество), не ме- нее 99,85 99,6
Влага, не более 0,05 0,05
Не растворимые в воде вещества. не более 0,01 0,04
Хлориды (в пересчете на СаС12), не более 0,03 0,05
Сульфаты (в пересчете на CaSO4) не более 0,025 0,04
Броматы (в пересчете на КВгО3), не более 0,008 0,025
Щелочь (в пересчете на СаО), не более 0,01 0,02
Органические вещества, не более . 0,005 0,01
Тяжелые металлы (в пересчете на РЬ), не более .... 0,0005 0,005
Железо (Fe), не Солее 0,002 0,004
Хлорат натрия применяют в качестве гербицида и дефолианта
(в ограниченных количествах вследствие его гигроскопичности).
В основном его используют в качестве полупродукта для производ-
ства других хлоратов, перхлората калия, хлорной кислоты, дву-
окиси хлора и хлорита натрия. Некоторые (небольшие) количества
хлората натрия используют для беления целлюлозы. Описано при-
менение NaC103 для изготовления свечей, являющихся источником
кислорода на атомных подводных лодках70.
1440
Гл. XXXIX. Соли кислородных кислот хлора
Состав технического хлората натрия, кристаллического и рас-
твора (или пульпы), согласно ГОСТ 12257—66, должен соответ-
ствовать требованиям, приведенным в табл. 114.
таблица щ
Состав технического хлората натрия (ГОСТ 12257—66)
(Содержание компонентов в %)
Твердый
марка А марка Б
Влага, не более 0,05 3,0
Хлорат натрия (в пересчете иа сухое вещество), не менее 99,5 99,0
Хлорат натрия, в пределах — — 45-65
Хлориды (в пересчете иа NaCl), не бо- лее 0,3 п 0,7 0,7*
Сульфаты (SO4), не более 0,1 0,3 0,3*
Хроматы (СгО4), не более 0,04 0,1 ' 0,2*
Не растворимый в воде остаток, не бо- лее 0,01 0,05 0,05*
Железо, не более 0,015 0,015 0,015*
мешки из
в
* В пересчете на 100%-ный продукт.
Бертолетову соль и хлорат натрия упаковывают
полиэтиленовой или поливинилхлоридной пленки, вложенные
стальные оцинкованные или покрытые перхлорвиниловым лаком
барабаны, или в мешки из хлориновой ткани (также с вкладышем
из пленки).
Хлорат кальция является гербицидом общего действия и ши-
роко используется для уничтожения сорняков.
Хлорат магния также служит гербицидом и, кроме того, яв-
ляется дефолиантом, применяемым для предуборочного снятия
листьев хлопчатника 71>72, а в больших дозах может служить дес-
сикантом для предуборочного подсушивания хлопчатника и других
растений.
Хлорат магния (дефолиант), согласно ГОСТ 10483—66, дол-
жен содержать 60 ± 2% Mg(C103)2 • 6Н2О и не более 0,6% не рас-
творимого в воде остатка; температура начала его плавления
должна быть не ниже 44°. Его транспортируют в герметичных ба-
рабанах из черной кровельной стали или в бумажных битумиро-
ванных дублированных пятислойных мешках с вкладышем из
полиэтиленовой или поливинилхлоридной пленки.
Перхлораты применяют в производстве взрывчатых и пиротех-
нических материалов 52’73. Предложены74 смеси, содержащие
~60% КСЮ4, образующие гигроскопичный дым для регулирова-
ния атмосферных осадков.
Производство хлорной извести
1441
Среди перхлоратов особенное значение имеет перхлорат аммо-
ния, используемый для изготовления бездымных взрывчатых ве-
ществ75’76. Перхлораты тяжелых металлов и хлорную кислоту
используют в качестве электролитов в гальванопластике, при це-
ментации и др. В присутствии HCIO4 получают на электролитиче-
ски полированной меди плотные, блестящие осадки палладия77.
Указывают78 на возможность реэкстракции рения хлорной кисло-
той из органических растворителей.
ПРОИЗВОДСТВО ХЛОРНОЙ ИЗВЕСТИ
Хлорная известь получается в результате взаимодействия газо-
образного хлора с сухой гидроокисью кальция — гашеной известью
Са(ОН)2.
Получаемый при электролизе раствора NaCl хлор разбавляют
воздухом, и поступающий на хлорирование газ обычно содержит
40—50% С12. В газе не должно содержаться более 1,5% Н2 и более
0,4% СО2 (в расчете на 90—98%-ный газ по хлору). Содержание
влаги должно быть не более 0,06%.
Гашеную известь приготовляют из порошкообразной извести
с высоким содержанием кальция79.
Производство хлорной извести состоит из трех стадий: 1) об-
жига известняка, 2) получения пушонки (гашения извести) и
3) хлорирования пушонки.
Обжиг известняка
Известь получают обжигом при 950—1100° известняка или
мела. Известняк является более плотной породой, чем мел. Послед-
ний отличается значительной пористостью и повышенной влаж-
ностью 90.
Известняк, поступающий на обжиг, имеет размеры кусков
70—120 мм и должен содержать не менее 91% СаСО3, не более
1,7% MgCO3 и не более 0,2% R2O3. 1% примесей в известняке
дает почти 2% примесей в обожженной извести. Некоторые при-
родные высокосортные известняки содержат: 97—95% СаСО3,
1—1,5% MgCO3, 0,2—0,5% CaSCXi, 0,2—0,5% нерастворимых ве-
ществ, 0,8—1% влаги, 0,1—0,2% Fe<>03, 0,-2—0,4% А120з, 0,3—0,5%
SiO2, 0,2—0,4% других примесей81.
При обжиге известняка происходит диссоциация карбоната
кальция:
СаСОз = СаО + СО2 — 42,5 ккал
Необходимое тепло получают сжиганием кокса или других
видов топлива. Кокс должен содержать не более: 0,7% S, 12%
влаги, 15% золы, 1,7% летучих. Твердое топливо применяют в виде
кусков с размерами 40—60 мм.
1442
Гл. XXXIX. Соли кислородных кислот хлора
Диссоциация известняка идет с достаточной полнотой уже при
900°. (Давление СО2 в системе СаСОз^СаО + СО2 равно
760 мм рт. ст. при 897°.) Однако для ускорения обжига темпера-
туру в наиболее горячей зоне печи держат на уровне 1000—1100°.
При более низкой температуре часть известняка внутри крупных
кусков не успевает разложиться и образуется так называемый
«недожог» или «недопал», ухудшающий качество извести и увели-
чивающий количество отходов при ее гашении. Более высокая
температура приводит к оплавлению кусков извести вследствие
наличия в известняке и в золе угля примесей кремнезема, окислов
железа, алюминия, магния и других, образующих легкоплавкие
силикаты. Такая известь медленно гасится водой, и часть ее вместе
с «недопалом» отсеивается после гашения от дисперсной пушонки
в виде более крупных не успевших загаситься кусочков «пере-
жога» или «перепала».
Обжиг известняка осуществляют в шахтных пересыпных печах
или в печах с выносными топками81’82. Минимальное загрязнение
обожженной извести золой и несгоревшими частицами топлива до-
стигается в печах с выносными топками (газовые печи). Эти печи
имеют значительные преимущества перед пересыпными печами
также вследствие возможности применения более дешевых видов
топлива (по сравнению с коксом и антрацитом) 83-85.
Получаемая в результате обжига известняка жженая известь
имеет вид сероватых и желтоватых кусков. Плотность ее зави-
сит от температуры обжига, влияющей на степень уплотнения:
2,75 г!см3 при обжиге при 800°, 3,26—3,27 г/см2 — при 1000—1100°.
Объемный вес жженой извести, полученной из плотного известняка,
равен 1,45—1,70 г/см3, а из мела 0,7—1,1 а/сл3.
Получение пушонки (гашение извести)
Окись кальция при взаимодействии с водой гасится, образуя
пушонку — гидроокись кальция:
СаО + Н2О = Са(ОН)2 + 15,176 ккал
При этом происходит значительное увеличение объема — в
2—3 раза. Процесс гидратации извести ускоряется в 1,5—2 раза
при гашении слабым раствором (1—5%-ным) NaCl, CaCU или
других электролитов 86.
Скорость гашения извести зависит от ее состава и темпера-
туры обжига и может колебаться в пределах от нескольких минут
до нескольких часов. Сильно обожженная известь (при 1200° и вы-
ше) гидратируется медленно 87’88. Дисперсность пушонки также
уменьшается, если для получения ее применяется известь, обож-
женная при слишком высокой температуре.
Производство хлорной извести
1443
Обычно при гашении активной, быстро гасящейся извести тре-
буется отводить тепло реакции. При медленном гашении следует
проводить процесс без отвода тепла во избежание замедления
гидратации. Для получения тонкодисперсной пушонки необходим
предварительный подогрев воды, применяемой для гашения.
Аппарат для гашения извести представлен на рис. 4358‘. Ино-
гда аппарат снабжают рубашкой для поддержания требуемой тем-
пературы и просушки слоя гашеной извести, приставшей к стенкам
аппарата. Внутрь сетчатого барабана подают воду, нагретую до
85—95°. Известь гасится и рассыпается в мелкий порошок, просеи-
Рис. 435. Аппарат для гашения извести:
1 —барабан; 2 —вал с лопастями; 3 —загрузочная воронка; 4 — сетчатый барабан;
5 —труба для подвода воды; 6 — выгрузочное отверстие; 7 —труба для отвода водяного
пара.
вающийся при вращении сетчатого барабана через его стенки; она
попадает в наружный барабан, передвигается к его концу лопа-
стями вращающегося вала и выгружается. Воду дозируют так,
чтобы пушонка на выходе из аппарата содержала 0,5—2% влаги.
Гашение при недостатке воды протекает при более высокой тем-
пературе и приводит к усложнению процесса. Повышенное содер-
жание влаги в пушонке (больше 2%) затрудняет ее транспорти-
ровку. Предложено ускорять гашение путем разрыхления пушонки
продуванием через нее пара 89.
Для освобождения от кусочков «недопала» и «перепала» пу-
шонку пропускают через вращающееся сито—бурат. Это сито
представляет собой сетку с 64 отверстиями на 1 сл2, натянутую на
деревянную раму. Оно имеет форму восьмигранной усеченной пи-
рамиды, расположенной горизонтально и вращающейся вокруг
своей оси. Просеянная пушонка вылеживается в бункерах не ме-
нее 3 дней. За этот срок гашение полностью завершается, увели-
чивается дисперсность частиц, температура и влажность пушонкц
1444
Гл. XXXIX. Соли кислородных кислот хлора
уменьшаются, она приобретает однородный состав и может под-
вергнуться хлорированию.
После вылеживания в бункерах пушонка должна содержать
не менее 86% Са(ОН)2, не более 7% СаСОз, 0,6—1% свободной
влаги и менее 6% примесей (SiO2, R2O3, MgO и др.) при отсут-
ствии непогашенной окиси кальция. Обычно она содержит 90—92%
Са(ОН)2, 2—4% СаСОз, 0,5—0,9% влаги и до 5,5% других приме-
сей00. Ее плотность —2,08 г]см\ а объемный вес колеблется от 0,4
до I г!смъ (обычно 0,55—0,65).
Хлорирование пушонки
Теория процесса
Абсолютно сухие гидроокись кальция и хлор не вступают в
реакцию; хлор только адсорбируется известью. В производствен-
ных условиях хлорированию подвергают пушонку, содержащую
менее 1 % свободной влаги; применяющийся для этой цели раз-
бавленный воздухом хлор также содержит небольшое количество
влаги. Даже небольшая влажность исходных веществ обеспечивает
начало реакции гидролиза хлора с нейтрализацией образующихся
кислот известью. Затем гидролиз продолжается за счет воды, вы-
деляющейся при хлорировании из гидроокиси кальция, и все но-
вые количества извести вступают в процесс; образуется ряд соеди-
нений, из которых и состоит хлорная известь (стр. 1431).
В первый период хлорирования пушонки, по-видимому, обра-
зуется смесь двуосновного гипохлорита кальция и СаС12 • Са (ОН)2-
• Н2О по реакции:
5Са(ОН)2 + 2С12 = Са(СЮ)2 • 2Са(ОН)2 + СаС12 • Са(ОН)2 • Н2О + Н2О
Основной хлорид кальция значительно менее гигроскопичен,
чем хлорид кальция. Это обстоятельство облегчает удаление воды
с газом и способствует образованию сыпучего, не мажущего ма-
териала. Остающаяся гигроскопическая вода присутствует в виде
раствора, насыщенного Са(С1О)2, СаС12 и Са(ОН)2, находящегося'
в равновесии с двумя твердыми фазами — двуосновным гипохло-
ритом кальция и основным хлоридом кальция. Растворимость
Са(ОН)2 в присутствии Са(С1О)2 и СаС12 весьма невелика
(рис. 436) и, следовательно, содержание свободной извести в рас-
творе, образованном гигроскопической влагой, мало. При даль-
нейшем хлорировании двуосновной гипохлорит кальция разру-
шается и переходит в менее основную соль — ЗСа(С1О)2*
• 2Са(ОН)2-2Н2О — дветретиосновной гипохлорит кальция:
. 3[Са(СЮ)2 2Са(ОН)2] + 2Н2О = ЗСа(С1О)2 • 2Са(ОН)2 • 2Н2О + 4Са(ОН)2
- Освобождающаяся при этом гидроокись кальция переходит в
жидкую фазу и расходуется на взаимодействие с хлором:
2Са(ОН)2 + 2С12 = Са(С1О)2 + СаС12 + 2Н2О
Производство хлорной извести
1445
Но так как эта реакция протекает в растворе, насыщенном из-
вестью, гипохлоритом и хлоридом, то вновь образующиеся
Са(С1О)2 и СаС12 выделяются в твердую фазу в виде основных со-
лей— СаС12-Са(ОН)2-Н2О и ЗСа(С1О)2-2Са(ОН)2-2Н2О.
Переход двуосновного гипохлорита в дветретиосновной сопро-
вождается уменьшением количества жидкой фазы, так как вода
входит в состав твердого кристаллогидрата. На этой стадии хло-
рирования получается весьма сухой, рассыпчатый продукт.
Рис. 436. Изотерма растворимости в системе
Са(С1О)2—Са(ОН)2—СаС12—Н2О при 50°.
Дальнейшее хлорирование связано с разрушением дветрети-
основной соли и переходом ее в нейтральный гипохлорит кальция
Са(С1О)2-ЗН2О. После исчезновения дветретиосновной соли про-
дукт представляет собой, по-видимому, смесь нейтрального гипо-
хлорита и основного хлорида кальция. Наличие небольшого коли-
чества гигроскопической влаги приводит при продолжении хло-
рирования к частичному разрушению основного хлорида с
образованием тетрагидрата хлорида кальция, что увеличивает ги-
гроскопичность продукта. Однако после исчезновения жидкой фазы
поглощение хлора практически прекращается.
Получение хлорной извести
Хлорирование пушонки осуществляют в аппаратах непрерыв-
ного действия — механических полочных камерах Бакмана
(рис. 437). Это железобетонная башня, у которой сечение внутри —
круглое, а снаружи — восьмигранное. Размеры рабочей части ее:
высота 9,75 м, внутренний диаметр от 3,5 до 5,5 м. Для удобства
U4e>
Гл. XXXIX. Соли кислородных кислот хлора
обслуживания и экономии места камеры устанавливают блоками
по 4, 6, 8, 10, 12 и больше штук.
Камера разделена горизонтальными полками на восемь эта-
жей. По оси камеры проходит стальной вал с гребками, передви-
гающими известь по полкам. Пушонка поступает из расположен-
ного над камерой бункера через автоматический питатель на вто-
Рис. 437. Схема камер Бакмана для по-
лучения хлорной извести (разрез через две
смежные камеры):
—полки; $ —вал; 10 — гребки; ZZ — автоматиче-
ские питатели; 12 — трубы для ввода хлора;
13 — бункер для готовой продукции; 14 — бочка;
15 — труба для вывода газа.
рую (сверху) полку. Первая
полка служит для осажде-
ния пыли, уносимой газами.
На четных полках имеются
отверстия в центре, а на не-
четных — у периферии, че-
рез которые перегребаемая
известь проваливается с
полки на полку. Зубья греб-
ков перемешивают хлори-
руемую известь и медленно
передвигают ее — на четных
полках от периферии к цен-
тру, на нечетных — от цен-
тра к периферии. Над пер-
вой (пыльной) полкой греб-
ков не имеется. Над каждой
полкой в стенках камеры
имеются окна — отверстия,
закрытые деревянным щи-
том. Окна служат для
доступа внутрь камер с
целью их очистки, ремонта,
а также предохраняют ап-
парат от разрушения в слу-
чае аварийного повышения
давления газа, например от
взрыва, который может про-
изойти, если хлор содержит
значительное количество во-
дорода (это может случиться при нарушении режима работы цеха
электролиза, откуда поступает хлор). Продукт через отверстия
в восьмой полке попадает в бункер, откуда его выгружают в боч-
ки. Выгрузка производится при помощи механизма, устраняющего
пылеобразование.
Для защиты от разрушающего действия хлора и хлорной изве-
сти все внутренние части камер покрывают защитными пленками
(бетон и вал — кузбасским лаком, разведенным в полихлоридах,
скребки — гуммированы). Предложено также флюатировать по-
верхность бетона водным раствором кремнефтористого магния90-
П роизводство хлорной извести
1447
Хлор после осушки серной кислотой разбавляют воздухом до
концентрации 40—50% и подают по стальным гуммированным тру-
бам на седьмую и шестую полки. Он проходит камеру снизу вверх
навстречу извести. В холодное время года возможно работать с
более концентрированным газом89, содержащим до 60% С12. Ис-
пользованная для осушки хлора серная кислота содержит от 0,1 до
1,5% хлора. С целью регенерации кислоты хлор может быть уда-
лен из нее путем продувки воздухом91. Уходящий из камер сверху
выхлопной газ содержит не более 1,5 мг/л хлора. Его пропускают
для улавливания хлора через абсорбер — стальную гуммирован-
ную колонну, орошаемую известковым молоком, содержащим
80—НО г/л Са(ОН)2- При снижении концентрации Са(ОН)2
в молоке до 10—15 г/л его заменяют свежим. Очищенный газ, со-
держащий не более 0,2 мг;л хлора, проходит через брызгоотдели-
тель и выбрасывается в атмосферу. Предложено92 отходящие газы
использовать (вместо воздуха) для разбавления хлора.
Камера работает под небольшим разрежением: в верхней части
8—12 мм вод. ст., в нижней — 2—4 мм вод. ст. Недостаток разре-
жения приводит к смещению процесса хлорирования на нижние
полки и к газовыделению в помещение цеха. При избыточном раз-
режении увеличиваются потери хлора с выхлопным газом.
Хлорирование ведут при теоретически необходимом количестве
хлора. При избытке хлора увеличивается содержание в продукте
хлорида кальция. При недостатке хлора образуется недохлориро-
ванная известь. При хлорировании пушонки выделяется около
250 ккал на 1 кг поглощенного хлора. Для устранения перегрева
хлорной извести, который приводит к ее разложению с образова-
нием хлората кальция, необходимо, чтобы температура материала
не превышала 35—40°, а газа 45—50°. С этой целью хлор и раз-
бавляют воздухом, с которым удаляется часть реакционного тепла.
Помимо этого при монтаже аппарата внутрь шести нижних полок
закладывают плоские (спиральные) стальные змеевики. По этим
змеевикам циркулирует холодная вода или рассол, отводящий из
аппарата значительную часть тепла. На каждой полке уложено
220 м труб диаметром 25/з2 или 32/зв мм (поверхностью около
20 м2) в виде трех самостоятельных змеевиков с тремя вводами
и выводами. Однако стальные змеевики часто выходят из строя
из-за коррозии. Более стойкими являются змеевики, гуммирован-
ные с наружной поверхности. Они обеспечивают достаточный
теплообмен с полок и позволяют удлинить сроки межремонтного
пробега камер93. Слишком низкая температура в камерах приво-
дит вследствие замедления процесса к понижению качества хлор-
ной извести (рис. 438)94. Поэтому в зимнее время иногда пускают
в змеевики горячую воду.
Производительность камеры диаметром 5,2 м и высотой 9,7 м,
равная 8,5—9,5 т в сутки хлорной извести зависит от скорости враще-
1448
Гл. XXXIX. Соли кислородных кислот хлора
ния вала и может быть повышена на 30—50% при увеличении ско-
рости вращения вала от 1 оборота за 3,5 мин до 1 оборота за
2,1—2,2 мин95. Однако увеличение производительности камеры свя
зано с необходимостью улучшения отвода тепла реакции.
На производство 1 т хлорной извести (содержащей 35% актив-
ного хлора) расходуют: 0,45—0,46 т гашеной извести [100%
Са (ОН) 2], 0,37—0,38 т хлора (100%), 17,5—20 л3 воды, 19-L
20 кет ч электроэнергии.
>39
О го 40 60 во
Температура^
Рлс. 438. Зависимость
активности хлорной
извести от темпера-
туры хлорирования.
Для хлорирования пушонки можно применять аппараты и дру.
гих типов, например, наклонно расположенные- вращающиеся ба-
рабаны, в которых пушонка и хлор движутся
противотоком; аппараты, в которых пушонка
хлорируется во взвешенном состоянии 89’96-97.
Предложено98 также осуществление процесса
хлорирования пушонки с периодической про-
дувкой сухим воздухом — горячим для от-
гонки влаги или холодным для охлаждения
реакционной массы.
В странах с жарким климатом применяют
более стойкую при повышенных температурах
обезвоженную хлорную известь, содержащую
1—2% общей влаги при 34—35% активного
хлора59. Сушку хлорной извести можно осу-
ществить во взвешенном состоянии в потоке
воздуха, нагретого до 150—200°. При этом
в продукте можно снизить с И до 0,5% без
содержание влаги
уменьшения содержания активного хлора. Под вакуумом можно
высушивать продукт при 60—70°.
ГИПОХЛОРИТ НАТРИЯ
Чистый раствор NaClO можно получить нейтрализацией хлор-
новатистой кислоты щелочью или взаимодействием С1О2 в толуоле
с NaOH и Н2О2". Слабые растворы хлорноватистой кислоты с
содержанием 3—5% НСЮ могут быть получены 109 хлорированием
или суспензии желтой окиси ртути в воде при 0°, или водной сус-
пензии СаСОз при 0, —5°.
Технические растворы гипохлорита натрия всегда содержат так-
же хлорид натрия. Их получают: взаимодействием хлорной извести
с раствором соды или сульфата натрия, хлорированием раствора
NaOH или Na2CO3 и электролизом раствора поваренной соли.
При растворении хлорной извести в избытке воды составляю-
щие ее сложные соединения распадаются* растворимые соли —
Са(С1О)2 и СаС12 — переходят в раствор, а известь остается в
осадке.
В присутствии в растворе соды или сульфата натрия происхо-
дит обменное разложение с выделением в твердую фазу CaCOs
Гипохлорит натрия
1449
йЛи CaSO« и образованием в растворе NaClO и NaCl:
Ca(C10)s + Na2CO3 = 2NaC10 + CaCO3
Ca(C10)2 + Na2SO4 = 2NaC10 + CaSO4
Одновременно протекают реакции взаимодействия ЫагСО3 или
Na2SO4 с СаС1г. Процесс следует вести94 при температуре не
выше 30—35° во избежание хлоратного распада. Полученный после
отстаивания осадков раствор содержит NaClO и NaCl.
Представляет интерес получение раствора NaClO пропуска-
нием раствора Са(СЮ)г, содержащего около 5% активного хлора,
через слой катионита, регенерируемого водой, а затем 15% рас-
твором NaCl 101.
Хлорирование растворов NaOH или ИагСОз проводят или пу-
тем барботажа хлора через раствор щелочи в бетонных резервуа-
рах, или в башнях с насадкой, орошаемой циркулирующим щелоч-
ным раствором. Используют также абсорберы с мешалками и
охлаждающими змеевиками из полиэтилена 102. Для этой цели
предложены также аппараты в виде двух концентрических труб 103,
барботеры с тангенциальным вводом жидкости с целью придания
ей вращательного движения104, барботер и расположенный над
ним кожухотрубный теплообменник, смонтированные в одном кор-
пусе ‘°5, струйные инжекторы для смешения раствора с хлором
с завершением реакции в башне с насадкой 106.
Растворы едкого натра берут с концентрацией до 30% NaOH.
При хлорировании их выделяется значительное количество тепла:
2NaOH + С12 = NaClO + NaCl + Н2О + 25 ккал
Во избежание повышения температуры выше 35°, особенно в
конце процесса, абсорбцию хлора ведут медленно, чтобы щелок
успевал охладиться естественным путем, или применяют искусст-
венное его охлаждение.
С целью предупреждения повышения температуры рекомен-
дуют вести процесс при пониженном давлении 107, например, при
15—20 мм рт. ст., когда тепло реакции отводится за счет выпари-
вания воды.
Предотвращение разложения гипохлорита натрия достигается
также наличием в растворе свободной щелочи108. Процесс ведут
при избытке щелочи не менее 0,8 вес. ч. свободной NaOH на
1 вес. ч. NaClO. Возможно также применение добавки к щелочным
растворам гипохлорита (содержащим 80—90 г/л NaOH на 100—
НО г/л NaClO) бихромата калия109 в качестве стабилизатора
(0,01—0,1% от веса NaClO). При автоматическом регулировании
процесса на основе изменения во времени окислительно-восста-
новительного потенциала среды110, по-видимому, удается полу-
чать устойчивый 15—17% раствор NaClO при избытке NaOH всего
4 г/л 106.
1450
Гл. XXXIX. Соли кислородных кислот хлора
При получении гипохлорита натрия хлорированием растворов
соды процесс ведут по реакции:
2N а2СО3 + С12 + Н2О = NaClO 4- NaCl + 2NaHCO3
Дальнейшее хлорирование приводит к взаимодействию NaHCO3
с С12 и выделению СО2 и НСЮ. Двуокись углерода выделяет из
гипохлорита НСЮ, а свободная хлорноватистая кислота разлагает
гипохлорит (см. выше). При этом происходит почти полная потеря
активного хлора. Поэтому хлорирование раствора соды заканчи-
вают до начала выделения двуокиси углерода.
Электролитическое получение раствора гипохлорита натрия
осуществляют электролизом раствора поваренной соли в ваннах
без диафрагмы. При этом хлор, выделяющийся на аноде, реаги-
рует с едким натром, образующимся на катоде. Во избежание
образования хлората натрия вследствие окисления на аноде ионов
CIO по мере их накопления, электролиз ведут в условиях мини-
мального перенапряжения при выделении хлора и низкой кон-
центрации ионов СЮ~ в прианодном электролите. Для уменьше-
ния скорости разложения гипохлорита натрия процесс ведут при
20—25°, охлаждая циркулирующий раствор электролита. Электро-
дами служат платино-иридиевые сетки Можно также приме-
нять графитовые аноды и катоды. Электролиз проводят при плот-
ности тока до 1400 а1см2 и напряжении между электродами
3,7—4,2 в. В рассол добавляют хлорид кальция и ализариновое
или канифольное масло (~0,1%) для предотвращения катодного
восстановления. Выход по току по мере накопления активного
хлора до 10—12% г/л уменьшается от 95% в начале процесса
до 50—55%. При начальной концентрации раствора 100—120 г/л
NaCl и содержании в конечном растворе 15—20 г/л активного
хлора расход энергии составляет 5,5—6 кет•ч на 1 кг активного
хлора. При увеличении конечной концентрации активного хлора
расход энергии возрастает за счет снижения выходов по току.
Разработана 112 конструкция электролизера с засыпными элек-
тродами из зерен природного магнетита. В нижней и верхней ча-
стях слоя магнетита расположены токоподводящие электроды из
нержавеющей стали, служащие катодами и анодами. Для электро-
лиза применяют циркулирующие растворы, содержащие ~30 г/л
NaCl. Для этого можно использовать как искусственные растворы,
так и морскую воду и другие природные соляные рассолы.
ГИПОХЛОРИТ КАЛЬЦИЯ
Получение гипохлорита кальция основано на взаимодействии
известкового молока с хлором62'113'114.
Известковое молоко приготовляют гашением извести избытком
воды в аппаратах непрерывного действия — гасителях—вращаю-
Гипохлорит кальция
1451
щихся (8—12 об/мин) горизонтальных железных барабанах, рас-
положенных с небольшим уклоном. С одного конца барабана вво-
дят жженую известь и воду. Вытекающее из гасителя молоко очи-
щается от кусочков «недопала» и «перепала» и других примесей
отстаиванием, например в пескоочистителях непрерывного дей-
ствия. Молоко высокой концентрации, более вязкое и плохо отстаи-
вающееся от мелких частиц примесей, очищают фильтрацией во
вращающихся сетчатых барабанах или на вибрационных ситах115.
Очищенное известковое молоко поступает в резервуары-хра-
нилища, снабженные медленно вращающимися мешалками
(5—10 об/мин), предохраняющими суспензию от расслаивания.
В этих резервуарах молоко охлаждается с помощью водяных зме-
евиков от 50—60° до 15—20°. Очищенное и охлажденное молоко
пропускают через колонну, в которой улавливается непоглощен-
ный хлор из газа, уходящего из аппаратов для хлорирования. Из
колонн молоко поступает на хлорирование в абсорберы.
При хлорировании известкового молока протекает реакция:
2Са(ОН)2 + 2С12 = Са(СЮ)2 + СаС12 + 2Н2О + 52,6 ккал
Одновременно происходит образование основных солей и кри-
сталлогидратов гипохлорита кальция. Вследствие этого условия
осуществления производственного процесса сильно влияют на со-
став и качество продукта.
Существуют разные методы производства гипохлорита каль-
ция. Когда хлорированию подвергают известковое молоко концент-
рацией 50—100 г/л Са(ОН)2, то гипохлорит находится в растворе
и после удаления нерастворившейся части раствор выпаривают с
выделением кристаллов, представляющих собой нейтральный гипо-
хлорит кальция с небольшой примесью основной соли116. При вы-
паривании раствора и увеличении концентрации СаС12 раствори-
мость гипохлорита кальция резко уменьшается117. Кристаллы от-
деляют от маточного раствора и высушивают.
При хлорировании концентрированного известкового молока,
содержащего 450—600 г/л Са(ОН)2, образующийся гипохлорит
кальция выделяется в осадок. Вначале в твердой фазе находится
двуосновной гипохлорит кальция Са(С1О)2ф2Са(ОН)2. Отделением
осадка от маточного раствора можно получить продукт, содержа-
щий после сушки около 45% активного хлора (вместо теоретиче-
ских 49%), около 4% СаС12, 47% Са(ОН)2, 3% СаСОз и 1% вла-
ги. Этот продукт дает при растворении очень большое количество
известкового шлама, что затрудняет его использование.
Дальнейшим хлорированием суспензии двуосновного гипохло-
рита кальция, в зависимости от принятого режима процесса,
можно получить в осадке или дветретиосновной гипохлорит
ЗСа(С1О)2-2Са(ОН)2-2Н2О или нейтральный гипохлорит каль-
ция Са(С1О)2-ЗН2О. Дветретиосновной гипохлорит кальция легче
1452
Гл. XXXIX. Соли кислородных кислот хлора
отфильтровывается от маточного раствора, чем нейтральный, обра,
зующий по окончании хлорирования густую суспензию. Теоретиче-
ское содержание активного хлора в дветретиосновном гипохлорите
равно 70%. Практически можно получить продукт, содержащий
55—60% активного хлора и 18—20% Са(ОН)2.
Содержание активного хлора в чистом нейтральном гипохло-
рите кальция Са(СЮ)з • ЗН2О равно 71%. После его обезвожива-
ния сушкой при 60° в токе сухого не содержащего СО2 воздуха
можно получить безводный гипохлорит кальция, в котором теоре-
тически должно быть 99,2% активного хлора. При промышленном
производстве, вследствие частичного разложения при сушке и дру-
гих операциях и загрязнения примесями, продукт содержит
•65—80% активного хлора. Получаемые хлорированием суспензии
соединений гипохлорита кальция часто высушивают в распыли-
тельных сушилках до влажности 1% и ниже.
Хлорируют также известковое молоко с добавкой NaOH. При
этом в осадок выделяются кристаллы CaCl2-NaC10-NaCl-12Н2О,
•Осадок, отделенный от маточного раствора, можно обработать
суспензией, полученной хлорированием известкового молока118.
При соотношении СаС12 в суспензии гипохлорита кальция и NaOCl,
содержащемся в тройной соли, равном 1 :2, весь СаС12 переходит
при взаимодействии с NaOCl в Са (СЮ) 2. После отделения твер-
дой фазы от раствора и высушивания ее получают продукт — ги-
похлорит кальция, содержащий больше 70% активного хлора.
.Иногда для высаливания соединений гипохлорита в прохлориро-
ванную массу вводят раствор хлорида кальцияИ6. В несколько
измененном виде этот способ применяется для получения гипохло-
рита кальция из газов, отходящих из электролизеров производства
металлического магния119 и содержащих около 3,5% хлора, 1—2%
НС1 и примеси СО2. Часть охлажденного и очищенного от НО газа
направляют в абсорбционные колонны, орошаемые известковым
молоком, другую часть — в колонны, орошаемые раствором едкого
натра. Образующиеся растворы смешивают, в результате чего осу-
ществляется обменное разложение:
СаС12 +2NaClQ = Са(С1О)2 + 2NaCl
Готовый продукт Содержит около 40% активного хлора. Этот
:метод получения гипохлорита кальция может быть использован
на установках обезвреживания любых других хлорсодержащих
газов, на которых большей частью выпускают раствор хлорида
кальция.
Контроль процесса хлорирования осуществляют или химиче-
ским способом120, или измерением окислительно-восстановитель-
ного потенциала121’122. Последний метод позволяет осуществить
.автоматический, непрерывный и дистанционный контроль процесса
Двуокись хлора
I45S
в закрытом реакторе. При этом контролируется скорость реакции,,
режим подачи хлора, конец реакции, а также перемешивание ре-
акционной массы.
ДВУОКИСЬ ХЛОРА
Двуокись хлора можно получить или окислением хлора123-124
и гипохлоритов, или восстановлением хлоратов и перхлоратов.
Наиболее широкое распространение получили способы восстанов-
ления хлоратов в кислой среде3,>,25~127. Небольшие количества
С1О2 получают взаимодействием гипохлорита натрия и хлора.
Основное внимание при разработке и усовершенствовании спо-
собов производства С1О2 направлено к изысканию условий взрыво-
безопасное™ процессов и подбору достаточно стойких материалов
для аппаратуры. Пригодными для изготовления и защиты аппа-
ратуры являются пластикаты из группы поливинилхлоридов, а так-
же изделия из кислотоупорной керамики, цементные материалы,
стекло, кварц и фарфор. В некоторых случаях используется свинец
для изготовления трубопроводов, реакционных башен и других
аппаратов.
Сущность процесса восстановления кислых растворов хлората
заключается во взаимодействии образующихся при этом хлорно-
ватой и соляной кислот:
2НС10з + 2НС1 = 2С1О2 + С12 + 2Н2О
или
CIO3 + 2Н+ + е = С1О2 + Н2О
Для разложения хлората с образованием НСЮ3 применяют
концентрированный раствор кислоты (8—10 н. раствор H2SO4) 127.
Необходимая для реакции соляная кислота образуется в ре-
зультате взаимодействия некоторого количества НСЮз с восста-
новителем. В качестве восстановителя предложены различные ве-
щества: NO2, СО, MnSO4 и др. 128-132. В промышленных способах
производства двуокиси хлора применяют, главным образом, сер-
нистый газ и метанол 59- ,25'127’ ,32~135. При введении в реактор со-
ляной кислоты в качестве реагента применение специального вос-
становителя не требуется.
Аналогично соляной кислоте действуют и хлориды. На этом
основан способ получения двуокиси хлора восстановлением хло-
ратов хлоридами в присутствии серной кислоты31. Процесс состоит
из трех параллельно протекающих реакций:
СЮз + СГ + 2Н+ = СЮ2 + V3C12 + Н2О
5СЮз + СГ + 6Н+ = 6С1О2 + ЗН2О
СЮз + 5СГ + 6Н+ = ЗС12 + ЗН2О
1454
Гл. XXXIX. Соли кислородных кислот хлора
Превалирующей является первая реакция, идущая со значи-
тельно большей (в 100—1000 раз) скоростью, чем другие. При
восстановлении хлоратов хлоридами теоретически не расходуется
активный хлор, так как наряду с двуокисью хлора образуется
молекулярный хлор, который также обладает активными окисли-
тельными (отбеливающими) свойствами.
За рубежом большую часть двуокиси хлора получают восста-
новлением хлората сернистым газом, меньшую (~1/3)—метано-
лом и небольшое количество — соляной кислотой127. Продукт полу-
чается в виде разбавленного газа (с содержанием до 10% СЮ2)
или водного раствора.
Выход двуокиси хлора зависит от степени протекания побочной
реакции — дальнейшего восстановления до иона хлора:
С1О2 + 4Н+ + 5е = СГ + 2Н2О
Максимальный выход двуокиси хлора достигается при быстром
отводе ее из сферы реакции и предотвращении таким образом по-
бочной реакции 59’125. С этой целью предложено интенсифициро-
вать отвод продуктов реакции при помощи струи инертного газа
или воздуха 136’137.
Сернистокислотный способ
Способ основан на образовании НС1О3 при взаимодействии
NaClO3 с H2SO4
2NaC103 + H2SO4 = 2НС1О3 + Na2SO4
и НС1 при взаимодействии НС1О3 с SO2
НС1О3 + 2SO2 + 2Н2О = 2H2SO4 + НС1О
НСЮ + H2SO3 = НС1 + H2SO4
с дальнейшим восстановлением НС1О3 соляной кислотой.
При температуре до 20° и pH = 1—3,5 стадией, лимитирующей
скорость процесса, является реакция СЮГ с H2SO3138. Избыток
SO2 по сравнению с количеством, необходимым для образования
соляной кислоты, реагирует с двуокисью хлора с выделением соля-
ной и серной кислот.
На рис. 439 представлена схема получения двуокиси хлора по
сернистокислотному способу 69’125. В процессе используют раствор
хлората натрия, содержащий 45—47% NaClO3, и 75%-ную серную
кислоту. Соотношение ионов SO4- и СЮ3“ в реакционной смеси
рекомендуют139 поддерживать равным 1 : 2. Образующуюся дву-
окись хлора отделяют в промывной колонне от хлора и собирают
в виде водного раствора. Отмывку производят водой, в которой
растворимость СЮ2 превышает растворимость хлора в 10 раз и
мало зависит от концентрации хлора. Состав азеотропного рас-
твора в системе двуокись хлора — хлор в водной среде30 при Ю
Двуокись хлора
1455
90 мол. % С1О2 и 10 мол. % С1г- При 25 и 35° содержа-
и
ДО 20
NaOH
&
to
I
о
3
E
Рис. 439. Схема производства дву-
окиси хлора по сернистокислотному
способу:
/, 2 — дозировочные баки; 3, 4—реакторы;
5 — резервуар двуокиси хлора; 6 — промыв-
ная колонна; 7 — поглотитель хлора.
NaClOs
SOa + ЙозЗух
Рис. 440. Схема реактора непре-
рывного действия для производ-
ства двуокиси хлора по сернисто-
кислотному методу:
/ —выход газа на абсорбцию; 2 — скруб-
бер; 3 — основной реактор; 4 — подача
охлаждающей воды; 5 —вывод отрабо-
танного раствора; б — вспомогательный
реактор.
отвечает
Ние хлора в азеотропе соответственно увеличивается
30 мол. %. Соотношение С1О2 и NaC]Oj НгЗО4 нго
С12 в газе тем больше, чем мень-
ше концентрация применяемого
раствора NaClO3 14°. На этом ос-
нованы способы получения С1О2
с использованием разбавленных
растворов, содержащих около
50—60 г/л NaClO3141. Температу-'
ру реакции поддерживают около
75—90°. Отвод тепла реакции мо-
жет быть осуществлен за счет ис-
парения воды, уносимой инерт-
ным газом 142.
Для осуществления процесса
непрерывным путем применяют
аппарат, схематически изобра-
женный на рис. 440 127. Раствор хлората натрия поступает в основ-
ной реактор через скруббер, где взаимодействует с газом, не про-
реагировавшим в реакторе. Эф-
фективность процесса повышается
благодаря этому на 1—2%. Предло-
жено 143’144 для увеличения поверх-
ности контакта фаз добавлять суль-
фитные щелоки в качестве пенооб-
разователя. Процесс заканчивают
при снижении концентрации хло-
рата в реакторе до 10—20 г/л. Пе-
реработку хлората, содержащегося
в отходящей жидкости, производят
во вспомогательном реакторе. При
смешении продукта из обоих реак-
торов 145 можно получить газ, со-
держащий 10% С1О2. При введении
в реакционную массу NaCl в коли-
честве 5—10% от веса NaC103 вы-
ход С1О2 составляет 95—97% 14S-
Добавление хлората к циркулирую-
щей жидкости после вывода суль-
фата натрия позволяет поддержи-
вать постоянную концентрацию хло-
рата в растворе 147.
получения двуокиси хлора является
Сернистокислотный способ
весьма экономичным по капитальным затратам, а также простым
и удобным в эксплуатации. '
1456
Гл. XXXIX. Соли кислородных кислот хлора
Видоизменением этого способа является осуществление поо-
цесса без серной кислоты в присутствии 5—6% (от хлората нат-
рия) бихромата натрия как катализатора, с использованием непро.
реагировавшего хлората после вывода из цикла сульфата натрия.
Однако капитальные затраты в этом случае почти в 2 раза больше
и себестоимость 1 т активного хлора также выше, чем по методу
восстановления сернокислого раствора хлората сернистым газом59.
Помимо выпуска водного раствора С1О2, предложено получать
невзрывающийся гидрат 148-15°, содержащий около 25% С1О2, погло-
щением CIO2 насыщенным водным раствором при 5—15°, а также
жидкую смесь, состоящую из 2 вес. ч. хлора й 1 вес. ч. С1О2151.
Получить СЮ2 можно также, действуя сернистым газом на твердый
хлорат 162. Однако аппаратурное оформление процесса в этом слу-
чае значительно сложнее. Применение азотной 163- 154 или фосфор-
ной 155 кислоты, по-видимому, не имеет особых преимуществ по
сравнению с серной кислотой.
Представляет интерес разделение смеси С1О2 и С12 восстановле-
нием хлора в хлористый водород при пропускании газовой смеси
с добавкой SO2 через суспензию AS2O3166.
Метанольный способ
При обработке кислого раствора хлората метанолом соляная
кислота, необходимая для образования СЮ2 взаимодействием
Рис. 441. Схема реактора для получе-
ния двуокиси хлора восстановлением
хлората метанолом:
/ — выход газа к абсорбционным колоннам;
2 —подача охлаждающей воды; 3 — подача воз-
духа; 4 —отвод раствора на регенерацию;
5 —подача горячей воды.
НСЮз с НС1, так же как и
в сернистокислотном способе,
получается восстановлением
НСЮ3. Вначале образуется
хлорноватистая кислота, кото-
рая реагирует с метанолом с об-
разованием соляной кислоты:
НСЮ + СН3ОН = НС1 + НСНО + Н2О
НСЮ + НСНО = НС1 + нсоон
При этом восстановитель
применяют в жидком виде, и
весь процесс осуществляют в
жидкой фазе. Воздух, исполь-
зуемый для перемешивания ре-
агентов, способствует в то же
время десорбции двуокиси хло-
ра из жидкой фазы и служит
также для разбавления про-
дукта.
Реактор (рис. 441) состоит из двух стальных, внутри освинцо-
ванных аппаратов, снабженных рубашками. Жидкие реагенты —
раствор хлората натрия, метанол и серная кислота — подают снизу
Двуокись хлора
1457
0 первый — основной реактор. Процесс проводят при 60° и концен-
трации NaC103 100 г/л, H2SO4 400—500 г/л. Пройдя первый аппа-
рат, реакционная масса затем поступает во второй — вспомогатель-
ный реактор, в котором концентрацию NaClO3 поддерживают рав-
ной 10—20 г/л за счет непрерывного добавления метанола.
Образующиеся двуокись хлора и хлор в смеси с воздухом из
обоих аппаратов направляют в абсорбционные колонны, орошае-
мые водой, где двуокись хлора поглощается 127-167. По этому спо-
собу получают наиболее чистую двуокись хлора по стоимости не
намного выше, чем по сернистокислотному способу 59-158.
Солянокислотный способ
Преимуществом этого способа является отсутствие необходи-
мости в восстановителях. Продукт получается при непосредствен-
ном взаимодействии основных реагентов — хлората натрия и соля-
ной кислоты:
NaClO3 + 2НС1 = С1О2 + »/гС12 + NaCl + Н2О
Механизм процесса можно представить следующими реак-
циями 159:
НСЮз + НС1 = НС1О2 + НСЮ
НС1О2 + НСЮз = 2С1О2 + Н2О
НСЮ + НС1 = С12 + Н2О
Следы ионов марганца и серебра катализируют реакцию.
Образовавшийся раствор NaCl может быть использован для
электролитического получения хлората натрия. В результате в про-
цессе отсутствуют побочные солевые продукты и имеется возмож-
ность ускорить реакцию образования двуокиси хлора, не доводя ее
до конца. Отработанный раствор, содержащий непрореагировав-
ший хлорат натрия, циркулирует через электролитические ванны,
в которых происходит окисление хлорида в хлорат 69- 160'161.
Представляет интерес получение двуокиси хлора по этому спо-
собу с применением твердого хлората натрия и газообразного хло-
ристого водорода 162. Лучшие выходы получают в пределах темпе-
ратур от —10 до 0°. Процесс можно осуществить в трубах, загру-
женных хлоратом и погруженных в охлаждающую жидкость.
Предварительная добавка к хлорату пористого материала (пемзы,
обожженной глины или силикагеля) препятствует спеканию массы
и облегчает удаление NaCl после реакции 163.
Для получения двуокиси хлора взаимодействием раствора хло-
рата натрия с соляной кислотой требуются значительно большие
капитальные вложения в производство, чем по другим способам.
Однако в связи с рациональным использованием сырья двуокись
1458
Гл. XXXIX. Соли кислородных кислот хлора
хлора получается наиболее дешевой59. Она содержит до 50% (ц0
объему) хлора. Последний отделяют от двуокиси хлора и исполь-
зуют.
Хлоридный способ
Образование двуокиси хлора взаимодействием хлората и хло-
рида в сернокислотной среде можно представить следующими сум-
марными уравнениями:
Са(С1О3)2 + СаС12 + 2H2SO4 = 2С1О2 + С12 + 2CaSO4 + 2Н2О
ИЛИ
NaC103 + NaCl + H2SO4 = С1О2 + V2C12 + Na2SO4 + H2O
Исходные растворы хлората и хлорида могут быть получены
разными методами: хлорированием известкового молока, хлориро-
ванием содовых или содо-поташных растворов, являющихся отхо-
дами алюминиевого производства, или смешением растворов хло-
рата натрия и поваренной соли. Эффективность процесса в значи-
:ельной мере определяется молярным отношением хлората и
хлорида. При хлорировании известкового молока получаются рас-
творы, содержащие 150—200 г/л хлората кальция и 5 моль хлорида
на 1 моль хлората. Выход двуокиси хлора при восстановлении хло-
рата кальция в присутствии 93%-ной серной кислоты при 35—40°
в полузаводских условиях составил31 в среднем 70%, а степень
использования хлората достигала 98,5%- С учетом утилизации
хлора в виде гипохлорита фактически процесс получения двуокиси
хлора в этих условиях протекает без потерь.
Для производства 1 т двуокиси хлора по этому способу31 рас-
ходуется: 4,8 т негашеной извести (85% СаО), 3,6т хлора (100%)
и 4,4 т серной кислоты (93% H2SO4).
Растворы, полученные хлорированием содо-поташных смесей,
содержат наряду с хлоратом натрия некоторое количество хлората
кальция. Концентрация хлората натрия в образующихся растворах
не превышает 140 г/л при соотношении хлората натрия к хлориду!
равном 1 : 3. Восстановление хлората в этих растворах при опти-
мальном соотношении серной кислоты к хлорату, равном 3,26: 1>
позволяет использовать хлорат на 98,3% и достичь выхода дву-
окиси хлора, равного 80%.
Оптимальными условиями получения двуокиси хлора из рас-
твора хлората натрия и поваренной соли являются: концентрация
хлората в растворе — 350 г/л, концентрация серной кислоты — 93%
H2SO4, температура 35°, соотношение хлората к хлориду, равное
1 : 1,05, и соотношение серной кислоты к хлорату — 2,7. При этом
выход двуокиси хлора составляет 96%.
На 1 т двуокиси хлора расходуется: 1,75 т хлората натрия, 6,0 т
хлорида натрия и 4,35 г серной кислоты (93%).
Двуокись хлора
1459
Описано 164-167 производство двуокиси хлора хлоридным спосо-
бом с одновременным получением твердого сульфата натрия. Про-
цесс осуществляют в реакторе, соединенном с вакуум-испарителем
и кристаллизатором.
Получение двуокиси хлора из хлорита натрия
При взаимодействии хлорита натрия с хлором происходит обра-
зование хлористого натрия и выделяется двуокись хлора:
2NaC102 + С12 = 2NaCl + 2СЮ2
Этот способ ранее был основным для получения двуокиси
хлора; в настоящее время его применяют в ограниченных масшта-
бах. Более того, в связи с использованием сравнительно дешевых
методов получения С1О2 (восстановлением хлората натрия) дву-
окись хлора служит полупродуктом для производства хлорита
натрия. Однако при выработке небольших количеств двуокиси
хлора этот способ является простым и удобным. Процесс может
быть осуществлен сухим или мокрым путем 65. По сухому методу
в вертикальную башню загружают сухой технический хлорит нат-
рия. Снизу в башню подводят воздух, содержащий около 2% хлора,
а сверху отводят смесь воздуха с С1О2 (до 4%). Сухой способ
позволяет получить СЮ2 в «твердом» виде на носителе и избежать
коррозии, но он представляет собой значительную опасность вслед-
ствие возможности внезапного образования взрывчатой смеси при
быстром протекании реакции. Для предотвращения перегрева
в ходе реакции к хлориту прибавляют соли, содержащие значитель-
ное количество кристаллизационной воды, например Na2SO4- 10Н2О.
По мокрому методу хлор взаимодействует в башне с раствором
хлорита натрия. Через раствор (~0,3 кг NaC102 в 1 л воды)
пропускают воздух, содержащий ~3% С12. В выходящем газе со-
держится около 4% С1О2 и 1 % С12.
Двуокись хлора может быть получена из хлоритов и другими
методами ,5Э. При взаимодействии хлорита с сильными кислотами
протекает реакция:
5NaC102 + 4Н+ = 4СЮ2 + 2Н2О + NaCl + 4Na+
Помимо этой реакции протекает ряд побочных. Чтобы избежать
взрыва, хлорит вводят в реакционную среду в виде раствора. Вы-
ход С1О2 составляет от 60 до 80%. Действием персульфатов на
хлориты получают двуокись хлора в количестве 96% от теоретиче-
ского. При электролизе хлоритов получают С1О2 с выходом до 97%.
В некоторых случаях используют стабильные твердые смеси,
выделяющие двуокись хлора при растворении в воде.
Предложено 168 также получать С1О2 действием NClg в смеси
с разбавителем на сухой твердый хлорит натрия.
1460
Гл. XXXIX. Соли кислородных кислот хлора
ХЛОРИТ НАТРИЯ
Хлорит натрия получают 421169 обработкой двуокиси хлора вос-
становителями в щелочной среде
2С1О2 + 20 Н" = С1О~ + CIO3 + Н2О
или взаимодействием ее с перекисями 17°:
2С1О2 + Н2О2 + 2ОН- = 2С1О“ + 2Н2О + О2
Наибольшее распространение получили методы, использующие
восстановители5Э: углеродистые вещества, например, измельчен-
ный нефтяной кокс 171-173, цинковую пыль и другие металлические
порошки174, аммиак, диспергированную серу, низшие окислы и
гидроокиси Со, Мп, РЬ и других металлов с переменной валент-
ностью 175, а также другие вещества. При использовании газообраз-
ной С1О2, загрязненной хлором, ее можно очистить при помощи
известкового молока 17е. Полученный раствор хлорита и хлората
после отделения от твердой фазы фильтрацией выпаривают и кри-
сталлизуют. Благодаря малой растворимости NaC10244 в присут-
ствии NaC103 в твердую фазу выкристаллизовывается хлорит нат-
рия. В процессе выпаривания раствора происходит частичное раз-
ложение хлорита с образованием хлорида и хлората. Минимальные
потери хлорита натрия 47 наблюдаются при выпарке его растворов
под давлением около 100 мм рт. ст. (при температуре кипения
50—55°). При этих условиях степень разложения хлорита в тече-
ние 24-часового выпаривания не превышает 0,5% при содержании
1,5—2 г-мол/л NaC!O2 в растворе. Замедления разложения раство-
ров можно достигнуть добавлением .к ним пиро- или полифосфата,
а также Н2О2 177.
Очистку хлорита от NaCl и NaC103 можно осуществить обыч-
ной перекристаллизацией, или высаливанием его из раствора бен-
золом и сушкой 178. Очистку раствора от NaClO3 можно также осу-
ществить добавкой в реакционную массу водорастворимых соеди-
нений щелочноземельных металлов или добавкой едкого кали для
осаждения менее растворимого хлората калия l79-|f'2.
Выпускаемые за рубежом сорта хлорита натрия имеют разный
состав, например, 70—75% NaClO2, 10—11% NaCl, 10—12%
Na2CO3 59.
Представляет интерес предварительное получение хлорита ще-
лочноземельного металла и последующее обменное его разложение
с содой для получения чистого раствора хлорита натрия 183.
При использовании в качестве восстановителя цинковой пыли
также образуется вначале хлорит цинка, который затем вступает
в реакцию обменного разложения с едким натром 184. Полученный
после отделения осадка гидроокиси цинка раствор хлорита натрия
выпаривают и затем направляют на кристаллизацию. Этот способ
Хлораты натрия, калия, кальция и магния
1461
особенно удобен для получения хлоритов в лабораторных усло-
виях 174.
Из других предложенных восстановителей заслуживают внима-
ния Nal 185, Сг(ОН)з186 и мышьяковистая кислота173. Окисленные
продукты регенерируют и возвращают в процесс. При этом дости-
гаются высокие выходы как в процессе восстановления, так и в про-
цессе регенерации.
Хлорит натрия с высокими выходами (94—98%) можно полу-
чить при использовании в качестве восстановителя гемицеллю-
лоз 186, а также при взаимодействии двуокиси хлора с водным
раствором пербората натрия (NaBO3-4H2O) в щелочной среде187.
ХЛОРАТЫ НАТРИЯ, КАЛИЯ, КАЛЬЦИЯ И МАГНИЯ
Для получения хлоратов применяют электрохимический и хими-
ческий способы. Электрохимическим способом вначале получают
хлорат натрия, который затем перерабатывают химическим путем
в хлораты калия и др. Хлорат калия получают также электролизом
раствора КС1 188, но процесс осложняется из-за малой раствори-
мости КСЮ3. По химическому способу хлорированием известкового
молока вначале получают хлорат кальция, который подвергают
обменному разложению с хлоридом калия для образования хло-
рата калия 18Э.
Получение хлоратов электрохимическим методом предшество-
вало производству их химическими методами. Масштабы произ-
водства электролизом до первой мировой войны были больше, чем
химическим путем. Лишь в период между первой и второй миро-
выми войнами превалирующее значение имел химический способ
в связи с развитием хлорной промышленности. В настоящее время
основным методом производства хлората натрия является электро-
лиз раствора хлорида натрия 19°. За рубежом хлорат натрия произ-
водят химическим путем лишь на заводах, имеющих затруднения
в использовании хлора 59> 63.
Получение хлоратов натрия и калия электрохимическим методом
Электрохимическое получение хлоратов натрия и калия осно-
вано на анодном окислении хлорноватистой соли:
6С1О" + 6ОН- = 2CIO3 + 4С1- + 1V2O2 + ЗН2О
Теоретический выход хлората при электролизе нейтрального
раствора NaCl с платиновыми анодами составляет 66,67% 1П. Элек-
тролиз ускоряется в кислой среде при добавке НС1, а также при
повышении температуры вследствие ускорения химического окисле-
ния гипохлорита натрия. Добавка других кислот, например НВг,
не влияет на выход по токуй скорость реакции ,91. Теоретический
1462
Гл. XXXIX. Соли кислородных кислот хлора
выход хлората по току в кислом растворе может составить 100%
вследствие одновременного протекания наряду с разрядом ионов
СЮз химического окисления гипохлорита хлорноватистой кислотой
по реакции:
2НСЮ + СЮ” = СЮ3 + 2СГ + 2Н+
Но при большой кислотности может произойти выделение части
хлора в виде газа вследствие сдвига равновесия реакции гидролиза
хлора влево. Поэтому используют раствор с pH = 6,7, что соответ-
ствует соотношению хлората и свободной кислоты, равному 1 ; 2.
При этих условиях выхода по току хлората могут превысить
90%.
Предложено также для устранения изменения кислотности
в процессе электролиза предварительно насытить электролит хло-
ром 182. В раствор вводят 4—10 г/л хромата или бихромата натрия
для предотвращения восстановления хлорноватисто- и хлорновато-
кислой соли на катоде вследствие образования на нем пленки
основных соединений хрома. В присутствии NasCrC^ потери от вос-
становления снижаются до 1—3% вместо 70 % без добавки.
Электролиз раствора NaCl осуществляют в настоящее время
с применением графитированных анодов и стальных катодов вместо
платиновых; процесс ведут при 35—50°, при pH раствора около 6,7,
при объемной плотности тока 1,7—14 а/л, анодной плотности
300—1400 а/м2 и катодной плотности 250—540 а/м2. Выход по току
составляет в среднем 80—85%. Расход энергии на 1 т NaC103 со-
ставляет около 1500 квт-ч. Проведение электролиза при более
высокой температуре связано со значительным расходом графита.
Применение магнетитовых анодов вместо графитовых позволяет
повысить температуру до 70°5Э. Однако'магнетитовые аноды реже
применяются вследствие малой их электропроводности ш.
Имеются попытки еще больше увеличить плотности тока: объ-
емную до 64 а/л, анодную до 6000 а/м2 и катодную до 3100 а/м2193.
Для проведения процесса могут быть использованы электролизеры
с нагрузкой 15—18 тыс. п107.
Электролиз может быть осуществлен или с получением раствора
хлората низкой концентрации с последующей выпаркой и кристал-
лизацией, или в каскаде электролизеров с получением хлоратных
щелоков высокой концентрации ш’194 и кристаллизацией NaClOa
охлаждением.
Исходный раствор содержит 195: 270—280 г/л NaCl, 50—60 г/л
NaClO3, 5—6 г/л Na2Cr2O? и 0,5—0,6 г/л НС1. Его получают сме-
шением рассола поваренной соли и вторичного маточного раствора
после кристаллизации NaC103.
В выходящем слабом растворе, направляемом на выпаривание,
содержится 300—450 г/л NaClO3 и 150—180 г/л NaCl. Полученный
раствор необходимо освободить от непрореагировавшего гипохло-
Хлораты натрия, калия, кальция и магния
1463
рита для предотвращения коррозии. Это осуществляют нагрева-
нием раствора паром до 85—95° и последующим восстановлением
растворами муравьинокислой, сернистой соли и др. Обезвреженный
раствор отделяют от частиц графита в отстойнике и на песочном
фильтре, а затем выпаривают до плотности 1,5—1,6 г'см3. При вы-
паривании выделяется хлористый натрий, который после промывки
используют для приготовления исходного рассола.
Упаренный раствор содержит в среднем 900 г/л NaC103,
80—100 г/л NaCl и 17—18 г/л Na2Cr2O7. Его отделяют от NaCl,
нагревают до 100° и донасыщают хлоратом, выделенным из ма-
точных растворов. После донасыщения раствор плотностью
г-1,63 г[см3 и концентрацией около 1100 г/л NaClO3, охлаждают
в эмалированном чугунном кристаллизаторе до 30°. Выделившиеся
кристаллы хлората натрия отделяют от раствора центрифугирова-
нием, промывают водой от желтой пленки хромовокислой соли и
высушивают горячим воздухом.
Маточный раствор, полученный после кристаллизации основной
массы хлората, упаривают и выделенный после этого хлорат ис-
пользуется для донасыщения раствора, идущего на кристаллиза-
цию. Получаемый при этом вторичный маточный раствор направ-
ляют на смешение с соляным рассолом 188>196.
В некоторых случаях кристаллизацию NaClO3 из раствора
после электролиза осуществляют без предварительного его упари-
вания с направлением его непосредственно на охлаждение. В этом
случае получают при электролизе раствор, содержащий 550—
610 г/л NaClO3 и 100 г/л NaCl. После отстаивания частичек гра-
фита и дополнительной очистки на фильтре раствор подвергают
кристаллизации при охлаждении в аппаратах непрерывного дей-
ствия. Хлорат натрия отделяют от маточного раствора, высуши-
вают и измельчают. Маточный раствор, содержащий непрореагиро-
вавший NaCl, используют для растворения новых количеств соли.
Однако приход воды в процесс превышает ее расход на ~60 кг
на 1 т NaClO3. Поэтому во избежание разбавления растворов
рекомендуют 197 производить подпарку щелоков или сократить ввод
воды на отдельных стадиях производственного цикла. На производ-
ство 1 т NaClO3 по этому методу расходуют194: 5200—5500 квт-ч
электроэнергии, 4—8 кг электродов и холода около 200 тыс. ккал.
При работе с выпаркой при том же расходе электроэнергии вместо
холода расходуется 1,8—2,5 мгк.ал пара.
При производстве электрохимическим методом хлората калия 173
электролизу подвергают раствор, содержащий 250 г/л КС1, 50 г/л
КС1О3, 3 г/л К2СГ2О7, при pH ~ 5,5. Мощность электролизеров
равна 3000 а. Напряжение на ванне 3 в. Выходящий из ванны рас-
твор, содержащий 150—200 г/л КС1О3, после разложения гипохло-
рита направляют на кристаллизацию в бетонную колонну-холо-
дильник. Сверху в колонну распыливают раствор, а снизу подают
22 М. Е. Позин
1464
Гл. XX*XJX. Соли кислородных кислот хлора
вентилятором воздух при 15—20°. При этом происходит частичное
выпаривание раствора с одновременной кристаллизацией хлората
Вытекающую из нижней части колонны пульпу вначале сгущают
в отстойнике, а затем разделяют на центрифуге. Маточный раствор
возвращают в процесс после донасыщения его хлоридом калия.
Кристаллы хлората калия иногда растворяют и перекристаллизо-
вывают для получения высококачественного продукта.
Иногда хлорат калия производят комбинированным методом
в две стадии. Вначале ведут электролиз раствора хлорида натрия,
содержащего также некоторое количество КСЮз (от оборотных
растворов). Затем проводят обменную реакцию NaC103 с хлоридом
калия 198. Предварительно щелок подвергают хлорированию. При
хлорировании образуется дополнительное количество ЫаСЮз за
счет неокислившегося при электролизе NaClO. При этом NaC103
получается взаимодействием гипохлорита и хлорноватистой кис-
лоты 200 (см. выше).
При электролизе смешанного раствора NaCl и КС1 конверсия
NaC103 при помощи КС1 осуществляется в меньшем объеме вслед-
ствие образования значительных количеств КСЮ3 электрохимиче-
ским путем. Исходный раствор содержит 70—100 г/л КС1О3 (от
оборотных растворов), 180—220 г/л NaCl, 100—130 г/л КС1,
5—6 г/л NaaCraO? и 0,6—0,7 г/л НС1. В результате электролиза по-
лучают раствор, содержащий 150—200 г/л КС1О3, 80—120 г/л
NaClO3, 60—70 г/л КО, 140—160 г/л NaCl. Его нагревают до 100°
.в аппарате с мешалкой, куда подают твердый хлорид калия. Кон-
вертированный раствор, содержащий 270—300 г/л КС1О3,
180—200 г/л NaCl и 100—130 г/л КС1, охлаждают до 35—40°
для кристаллизации КСЮ3. После отделения выделившихся кри-
сталлов маточный раствор возвращают на электролиз, доводя его
состав до первоначального.
На получение 1 т КСЮз электролизом смешанного раствора
расходуется 0,61—0,65 т КС1, 15—20 кг НС1, 1,5—2,0 кг К2СГ2О7
и около 6000 квт-ч электроэнергии.
Получение хлората калия известковым методом
Физико-химические основы
Обычно хлорированию подвергают известковое молоко с кон-
центрацией 130—140 г/л СаО. Реакция идет до полного исчезнове-
ния свободной извести по уравнению:
2Са(ОН)2 + 2С12 = Са(С1О)2 + СаС12 + 2Н2О + 52,6 ккал
При дальнейшем хлорировании образующаяся при гидролизе
хлора хлорноватистая кислота окисляет гипохлорит кальция в хло-
рат:
Са(С1О)а + 4НС1О = Са(С1О3)2 + 4НС1
Хлораты натрия, калия, кальция и магния
1465
Образующаяся соляная кислота также реагирует с гипохлори-
том, превращая его в хлорид кальция и выделяя малодиссоцииро-
ванные молекулы НСЮ:
2Са(СЮ)2 + 4НС1 = 2СаС12 + 4НС1О
Хлорноватистая кислота окисляет новое количество Са(С1О)2
и т. д. до тех пор, пока весь гипохлорит не перейдет в хлорат. Та-
ким образом, хлорноватистая кислота является лишь катализато-
ром — переносчиком кислорода.
Суммарная реакция второй стадии процесса выражается урав-
нением:
ЗСа(С1О)2 = Са(С1О3)2 +2СаС12 +36,7 ккал
Общим уравнением процесса образования хлората при хлори-
ровании известкового молока является:
6Са(ОН)2 + 6С12 = Са(СЮ3)2 + 5СаС12 + 6Н2О + 194,5 ккал
Из этого уравнения видно, что при хлорировании лишь 7в часть
щелочи превращается в хлорат, а 5/е. переходят в хлорид. Этим
объясняется невыгодность прямого получения хлората калия хло-
рированием едкого кали по реакции
6КОН + ЗС12 = КС1О3 + 5КС1 + ЗН2О
так как при этом ~83% ценной щелочи КОН, получаемой электро-
лизом КС1 с большой затратой энергии, вновь превращаются в КС1.
При хлорировании же известкового молока с последующим обмен-
ным разложением Са(СЮз)2 и КС1 расходуется значительно менее
ценная щелочь — Са(ОН)2.
Основным фактором, обусловливающим образование хлората из
гипохлорита с наибольшей скоростью, является нейтральность
среды. В отличие от прежних взглядов, согласно которым стреми-
лись создать кислую среду, последующими исследованиями устано-
влено 20)> 202, что максимальная скорость образования хлората на-
блюдается при pH раствора 7—7,4. Окислителем в этом процессе
может быть как НСЮ, так и СЮ-, и образование хлората можно
выразить следующими реакциями:
2НС1О + СЮ- = ClOg + 2Н+ + 2СГ -
2СЮ-+ НСЮ = CIO3 + Н++ 2СГ
Чем выше температура, тем быстрее идет окисление гипохло-
рита хлорноватистой кислотой. Однако повышение температуры
приводит не только к ускорению образования хлората, но вызывает
частичное разложение гипохлорита с потерей кислорода. Опти-
мальной температурой хлорирования считается 75—80е, при кото-
рой потеря кислорода невелика. Для хлорирования может приме-
няться хлор любой концентрации.
22*
1466
Гл. XXXIX. Соли кислородных кислот хлора
На рис. 442 изображена клинографическая проекция на гори-
вонтальную плоскость диаграммы растворимости в системе 2°з
Са(С1О8)2 + 2КС1 = 2КС1О3 + СаС12 при 0°. Поле кристаллизации
хлората калия занимает почти всю диаграмму, и его окаймляют
узкие поля остальных солей. Стабильной является пара КСЮ3 -р
+ СаС12, так как линия соприкосновения их полей пересекает сре-
динную линию АОС. Поля Са(С1О3)2-2Н2О и КС1 не соприка-
саются, и эти соли не могут находиться совместно в твердой фазе.
Рис. 442. Система Са(С1Оз)2 + 2КС1 =* 2КС1О3 + СаС12 при 0°.
Концентрации солей выражены в молях на 1000 моль воды.
Вертикальная плоскость АОС делит диаграмму на две части.
В левой — растворы состоят из КСЮ3, СаС12 и Са(С1О3)2, в пра-
вой— из КСЮ3, СаС12 и КС1. По линии АОС растворы состоят
только из СаС12 и КСЮ3. Получаемые при хлорировании известко-
>вого молока растворы содержат на 1 моль Са(С1О3)2 5 моль СаС12,
и их фигуративные точки располагаются на прямой OR в системе
СаС12—Са(СЮ3)2—Н2О. Для любой точки этой прямой, напри-
мер а, соотношение
Са(С1О,)2:СаС12~ = -|
При растворении КС1 состав раствора перемещается парал-
лельно оси ОБ и достигает линии АОС в точке с, когда количество
КС1 эквивалентно хлорату кальция. На практике обычно вводят
избыток КС1, и состав раствора перемещается в правую часть
диаграммы, например в точку р. После выделения КСЮ3 фигура-
тивная точка маточного раствора несколько поднимется по оси
СаС12 ввиду добавочно образующегося по реакции обменного раз-
ложения хлорида кальция. Таким образом, состав конечного рас-
твора изображается точкой Ь', находящейся в правой части диа-
граммы.
Хлораты натрия, калия, кальция и магния
1467
Способы и режимы производства
Для абсорбции хлора известковым молоком применяют аппа-
раты различных конструкций. Используют, например, стальные
цилиндрические аппараты, диаметром 2—2,5 м и высотой 4,5—5 м,
футерованные диабазовой плиткой или кислотоупорным кирпичом,
в которые заливают известковое молоко и подводят хлор через бар-
ботажную трубу 204. По внешнему сходству их называют башнями.
Внутри башни имеется стальной змеевик, по которому циркулирует
холодная вода для отвода тепла реакции. Сухой хлор подают в
башню по железной гуммированной трубе.
В хлорированном щелоке должно быть 76—80 г/л Са(СЮз)2,
230—240 г/л СаС12 и не более 3—4 г/л Са(СЮ)2. Выход хлората,
по поглощенному хлору составляет около 95%. Прохлорированный
щелок продувают воздухом (для удаления растворенного хлора).
Затем его подвергают «обезвреживанию», т. е. разрушают содер-
жащийся в нем гипохлорит восстановлением СЮ". Для этого к го-
рячему щелоку добавляют раствор аммиака, гипосульфита или
органические вещества — мелассу, муку, древесные опилки и т. п.
Обезвреживание производят в больших резервуарах (емкостью
15—20 л3), футерованных кислотоупорным материалом, снабжен-
ных мешалками и паровыми змеевиками для поддержания темпе-
ратуры щелока на уровне 60—70°. Хорошо прохлорированный ще-
лок до обезвреживания имеет ярко-розовый цвет, обусловленный
содержанием в нем примеси железа в виде феррата кальция
CaFeCU При обезвреживании феррат кальция разрушается, обра-
зуя нерастворимый Ре(ОН)з, и щелок приобретает грязно-желтый
цвет.
После обезвреживания щелок отделяют от шлама — нераство-
римых примесей, перешедших из извести [СаСОз, SiO2, Fe(OH)a
и др.], — отстаиванием или фильтрацией (при работе с концентри-
рованным щелоком, полученным из известкового молока высокой
концентрации). Шлам промывают для уменьшения потерь хлората,
и промывные воды используют при приготовлении известкового
молока. Очищенный от шлама щелок выпаривают. В тех случаях,
когда для хлорирования используют известковое молоко высокой
концентрации, хлорированный щелок получается достаточно кон-
центрированным и не требует выпарки. В этом случае для обмен-
ного разложения применяют твердый КС1, чтобы не разбавлять
раствор перед кристаллизацией. Однако при этом в связи с по-
вышенной вязкостью суспензии затрудняется отделение шлама
от прохлорированного щелока, а потери Са(СЮз)2 увеличива-
ются.
Когда процесс осуществляют по схеме с выпаркой щелоков
(после обменного разложения), то обычно для обменного разло-
жения применяют раствор КС1, так как при этом облегчается его
1468
Г л. XXXIX. Соли кислородных кислот хлора
дозировка и ускоряется реакция. Обменную реакцию ведут при по-
вышенной температуре; затем щелок выпаривают. Выпаренный'
щелок, содержащий 160—170 г/л КС1О3 450—460 г/л СаС12 на-5
правляют на кристаллизацию. /
На разных заводах применяются различные схемы кристалли-
зации. Например, щелок после обменного разложения охлаждают
в непрерывно действующих кристаллизаторах барабанного типа
с водяным или воздушным охлаждением до 15—20°; после отделе-
ния основной массы кристаллов КС1О3 производят дополнительное
вымораживание КС1О3 при охлаждении маточного раствора до
—1*0, —20°. Кристаллы бертолетовой соли отфуговывают и сушат.
Для получения более чистой соли часть отфугованной соли подвер-
гают перекристаллизации.
Из прохлорированного щелока перед реакцией обменного раз-
ложения можно выделить значительную часть СаС12. При охлажде-
нии щелока до 10° из него выделяются кристаллы СаС12-6Н2О, так
как Са(С1О3)2 обладает большой растворимостью. Другим вариан-
том является осаждение СаС12 (в частности, после его предвари-
тельной кристаллизации) добавлением к горячему раствору из-'
вес-ти. При охлаждении выделяется оксихлорид примерного состава
ЗСаО • СаС12 15Н2О. Этими способами можно перед обменным раз-
ложением получить из щелока до 95% СаС12.
По другой схеме обменное разложение между КС1 и Са(С1О3)2
производят до выпарки и из выпаренного щелока охлаждением до
30—35° выделяют до 80% КС1О3. Пульпу разделяют на центри-
фуге. Бертолетова соль с содержанием 75—80% КС1О3 и 5—6%
СаС12 идет на перекристаллизацию, а маточный раствор (~40 г/л
КС1О3 и 460—470 г/л СаС12) — на дополнительное охлаждение.
При охлаждении маточного раствора от 30 до —2, —3° в нем
остается около 5% КС1О3 (от первоначального его количества).
Выделившийся КС1О3 после отделения от раствора также подвер-
гают перекристаллизации. Оставшийся после вымораживания ще-
лок, содержащий 470—480 г/л СаС12 и не более 14 г/л КСЮ3,
является отходом производства, используемым для получения хло-
ристого бария или для выделения товарного хлористого кальция
(стр. 745).
Производство бертолетовой соли описанным способом является
периодическим, громоздким и многостадийным. По более совершен-
ной схеме 205 хлорирование известкового молока и обменную реак-
цию хлората кальция с хлоридом калия осуществляют в одном
аппарате. Хлорид калия подают в бак с известковым молоком
с 15—20%-н.ым избытком по отношению к СаО для обеспечения
более полного выделения хлората калия. Первая стадия про-
цесса — образование гипохлорита — не изменяется от присутствия
хлорида калия. Однако вторая стЭ1Ья—переход гипохлорита
в хлорат и хлорид в присутствии КС — характеризуется более
Хлораты натрия,, калия, кальция и магния
1469
устойчивым пенообразованием. Разрушение пены производят путем
разбрызгивания на ее поверхности избыточного маточного рас-
твора второй кристаллизации. Образовавшийся хлорированный
щелок направляют на выпарку и последующее охлаждение для
кристаллизации хлората калия и его переработки, аналогичной
описанной выше.
После отжима на центрифуге хлорат калия содержит 4—8%
влаги. Так как нагревание его до высоких температур может при-
вести к разложению и взрыву, сушку соли производят при низких
температурах в вакуум-сушилках. После высушивания хлорат ка-
лия просеивают или сепарируют на фракции воздушным сепара-
тором. Часть более крупной соли иногда подвергается размолу.
Перед размолом соль пропускают через магнитный сепаратор для
извлечения случайно попавших частиц железа, которые могут
вызвать взрыв соли при размоле. Размол производят в дезинтегра-
торе или в бегунах. Ввиду взрывоопасности операции сушки и раз-
мола организуются в отдельном здании, удаленном от других це-
хов завода.
На производство 1 т хлората калия по известковому, методу
расходуют: 2,15—2,17 т извести (100% СаО), 2,2—2,3 т хлора
(100%), 0,9—0,98 т хлористого калия (100% КС1), 32—34 кг ме-
лассы (концентрации 4,6%), 400 квт-ч электроэнергии, 600—610 м3
воды и 7—7,1 мгкал пара (4 атм)\ в качестве отхода получается
~3т СаС12 в виде раствора указанной выше концентрации.
Хлорат калия может быть получен и многими другими хими-
ческими способами, в основе которых лежат хлорирование суспен-
зий Mg(OH)2 206, СаСОз, ZnO и других соединений и обменное раз-
ложение полученных хлоратов с КС1 или другими солями калия
(K2SO4, К2СО3).
Хлорат натрия
Выделение СаС12 из хлоратного щелока, образовавшегося при
хлорировании известкового молока, лежит в основе получения хло-
рата натрия. Оставшийся маточный раствор после отделения хло-
рида кальция разбавляют и обрабатывают сульфатом натрия или
содой. После отделения осадка сульфата или карбоната кальция
получается щелок, содержащий NaClO3 и NaCl. Его направляют
на выпарку, при которой происходит кристаллизация NaCl. После
отделения NaCl концентрированный щелок охлаждают, и из него
кристаллизуется NaClO3.
Хлорат натрия можно также получить каустическим или содо-
вым способом, хлорируя раствор едкого натра, соды или более
дешевого бикарбоната натрия 207.
Полученные щелоки, содержащие NaClO3 и NaCl, выпаривают
и обрабатывают, как указано выше.
1470 Гл. XXXIX. Соли кислородных кислот хлора
Хлораты кальция и магния
Хлорат кальция можно получить из хлорированных щелоков
известкового молока упариванием или охлаждением с разделением
системы Са(СЮз)2—СаС12—Н2О, а также обменным разложением
хлората натрия с хлоридом кальция 208:
2NaC103 + СаС12 = Са(СЮ3)2 + 2NaCl
Образующийся при этом раствор хлорида натрия используют
для производства хлората натрия электролизом. В последнем слу-
чае единственным сырьем оказывается хлорид кальция, что позво-
ляет получать хлорат кальция из отбросного хлорида кальция со-
довых заводов.
Хлорат магния Mg(C103)2 образуется при хлорировании суспен-
зии гидроокиси 49 или окиси магния в воде. Из полученного хлорат-
хлоридного раствора хлорат выделяют дробной кристаллизацией
или экстракцией ацетоном. В продукте содержится до 10% MgCI2.
Получение хлората магния электролизом раствора MgCl2 в при-
сутствии бихромата затрудняется вследствие образования трудно-
растворимой в воде гидроокиси магния.
В небольших количествах хлорат магния получают 209 обменным
разложением хлората натрия с хлоридом магния или сульфатом
магния:
2NaC103 + MgCl2 = Mg(C103)2 + 2NaCl
2NaC103 + MgSO4 = Mg(C103)2 + Na2SO4
Лучшие выходы достигаются при осуществлении этих реакций
не в водной среде, а в ацетоне. Однако в этом случае требуется
возвращение ацетона, и процесс может быть экономически целесо-
образным лишь при обеспечении небольших потерь ацетона.
Простым способом получения хлоратмагниевого дефолианта
является приготовление смеси хлората и хлорида магния, являю-
щейся достаточно эффективной, несмотря на пониженное содержа-
ние хлората. Такая смесь с содержанием 55—60% Mg(CIO3)2-6H2O
может быть приготовлена сплавлением хлората натрия с шести-
водным хлоридом магния210. Реакцию проводят при 110—120°
в реакторе, снабженном мешалкой, куда подают расплавленный
шестиводный хлорид магния и хлорат натрия 209. Суспензия хло-
рида натрия в расплаве подается на барабаны-кристаллизаторы,
охлаждаемые водой. При отношении MgCI2-6H2O : NaCIO3 (в экви-
валентах), равном 1,3 4- 1,4, полученный дефолиант имеет темпера-
туру плавления не ниже 45°. На производство 1 т продукта, содер-
жащего 58% Mg(CIO3)2-6H2O, расходуют 0,44 т хлората натрия
и 0,56 т шестиводного хлорида магния.
Перхлораты натрия, калия и аммония
1471
ПЕРХЛОРАТЫ НАТРИЯ, КАЛИЯ И АММОНИЯ И ХЛОРНАЯ
КИСЛОТА501 59> 1111 211
Производство перхлоратов осуществляют почти исключительно
электрохимическим путем. Электролизу подвергают водный рас-
твор хлората натрия. Однако получающийся перхлорат натрия не
находит широкого применения вследствие его сильной гигроскопич-
ности и расплывания на воздухе. Обменным разложением его
с хлоридами или сульфатами калия или аммония получают пер-
хлорат калия или аммония.
Перхлорат калия может быть получен также при нагревании
хлората калия при 350—450°:
4КС10з = ЗКСЮ4+КС1
В результате выщелачивания застывшего плава водой (или обо-
ротными растворами) КО переходит в раствор, а труднораствори-
мый KCIO4 остается в осадке. Однако этот метод в промышлен-
ности не применяется.
Аналогичным образом — термическим разложением NaClOs при
400—600° можно получить и перхлорат натрия212. Предложено 213
осуществлять окисление хлората калия до перхлората двуокисью
свинца в сернокислой среде.
Перхлорат натрия
Электрохимическое окисление хлорат-иона на аноде протекает
с образованием хлорной и хлорноватой кислот:
2CIO3 + Н2О = НС1О4 + НС1О3 + 2е
Одновременно на катоде происходит разряд ионов водорода и
образование щелочи, взаимодействующей с НС1Оа и НС1О4:
2Н2О + 2е = Н2 + 2ОН"
НС1О4 + НСЮ3 + 2ОН" = сю; + сю; + 2Н2О
Таким образом, суммарный процесс может быть представлен
уравнением:
сю; + н2о - сю; + н2
Электролиз осуществляют с применением в качестве анодов
платиновых сеток, а в качестве катодов — перфорированных нике-
левых пластинок 214.
В качестве катодных материалов могут быть также использо-
ваны нержавеющая сталь и графит. Во избежание потерь тока
вследствие частичного разряда ионов ОН", особенно увеличиваю-
щихся в конце процесса (при малой концентрации ионов СЮ3),
поддерживают небольшую кислотность электролита в пределах
1472
Гл. XXXIX. Соли кислородных кислот хлора
0,1—0,15 г/л НС1195. При избытке кислоты может произойти хими-
ческое разложение хлората, а также разряд ионов хлора на
аноде.
При концентрации электролита 500—700 г/л ЫаСЮз, темпера-
туре 40—60°, анодной плотности тока 3000—7000 а/м2 и катодной —
1000—2000 а/м2, напряжении 6—10 в, начальный выход по току
составляет 95—98%. К концу процесса при концентрации ЫаСЮ4,
равной 900—1000 г/л, и ЫаСЮз ниже 50 г/л выход по току умень-
шается до 40—50%. В среднем он составляет около 85%, и расход
энергии на 1 т ЫаСЮз колеблется в пределах 3000—3500 квт-ч.
Некоторое снижение температуры или повышение ее до 60—70°
не влияет заметно на выход по току. Повышение температуры при-
водит к увеличению износа платинового анода, но к уменьшению
напряжения на электролизере215. Поэтому обычно работают при
40—60°. Считают 215, что оптимальной плотностью тока на аноде,
при которой достигается минимальная стоимость продукта, должна
быть 0,4—0,5 а/см2.
Когда электролиз осуществляют с применением платиновых
анодов и стальных катодов, в электролит добавляют 3—4 г/л
ЫааСггО? для предотвращения восстановления катодов. В этом слу-
чае наилучшие выходы по току достигаются при 30—40°. Тепло
реакции отводят искусственным охлаждением. После нейтрализа-
ции конечного раствора, содержащего 900—1000 г/л ЫаС1О4 и
50 г/л ЫаСЮз, и выпаривания его до концентрации 1300—1350 г/л
NaClO4 из него кристаллизуют твердый перхлорат натрия.
Представляет интерес осуществление процесса с получением
раствора, содержащего минимальное количество хлората, который
непосредственно может быть использован для получения перхло-
рата аммония211. Указывают215, что при электролизе начального
раствора, содержащего 600—700 г/л ЫаСЮз, в серии ванн с цир-
куляцией электролита концентрация в конечном растворе перхло-
рата достигает 1000 г/л, а хлората составляет 3—5 г/л.
В последнее время большое внимание уделяется сокращению \
количества платины в производстве перхлората, или замене ее дру-
гими материалами для изготовления анодов211. Возможно исполь-
зование тантала или титана в качестве токоподводящих металлов,
на которые наносится слой платины 50’ 215-217 Описано 216 использо-
вание в производстве перхлората натрия анодов из двуокиси
свинца, нанесенной электроосаждением на токоподводящую основу
из графита. Устранение механических повреждений покрытия и
придание ему однородности достигается обработкой эпоксидной
смолой, силиконовым каучуком или другими аналогичными мате-
риалами. В качестве токоподводящей основы для анодов из дву-
окиси свинца вместо графита можно также применять тантал 50>215.
При использовании анодов из двуокиси свинца, выход перхло-
рата натрия по току несколько ниже (на ~5%), чем при работе
Перхлораты натрия, калия и аммония
1473
с платиновыми анодами. Это наблюдается в особенности на послед-
них стадиях электролиза — при малых концентрациях хлората
в конечном растворе.
Получение перхлората с применением РЬО2 в качестве анода
ведут при анодной плотности тока 0,15 а/см2 и катодной —
0,07 а/см2 при напряжении 4,7—5,7 в. К раствору добавляют
0,5 г/л NaF и не вводят Na2Cr2O7. Добавка фторида позволяет
завершить процесс при конечной концентрации ЫаСЮз в растворе
такой же, какая достигается на платиновых анодах без резкого
уменьшения выхода по току, который составляет в среднем 90%.
Перхлорат калия
Перхлорат калия получают обменным разложением NaClO4 и
КС1. Раствор, содержащий 600 г/л NaClO4, после фильтрации от
механических примесей направляют в кристаллизатор, куда одно-
временно подают раствор хлорида калия. Массу охлаждают не
ниже 20—25° во избежание образования твердых растворов 219. Вы-
ход перхлората калия составляет 95%. Кристаллы отделяют на
центрифуге, высушивают до содержания 0,01—0,04% влаги и из-
мельчают в дезинтеграторе. В готовом продукте содержится до
99,5% КС1О4.
Перхлорат аммония
Перхлорат аммония производят обменным разложением пер-
хлората натрия и хлорида аммония. Исходными материалами слу-
жат перхлорат натрия, аммиак и соляная кислота 220’221.
Рис. 443. Схема производства перхлората аммония:
/ — реактор; 2 — бак; 3, 22 — кристаллизаторы; 4, 8, 2/ —питатели центрифуг; 5, 17,
20 —резервуары для маточных растворов; 6, 9, /9 — центрифуги; 7—мешалка для
пульпы; 10, /2 —скрубберы;//— вибрационная сушилка; /3 —бункер; 14 — вибрационное
сито; 15 — смеситель; 16 — растворитель пыли; /3 —запасной сборник для маточного
раствора.
На рис. 443 представлена схема производства перхлората ам-
мония. В реактор загружают 56% раствор перхлората натрия,
рециркулирующие растворы и промывные воды. Затем при
1474
Гл. XXXIX. Соли кислородных кислот хлора
медленном перемешивании подают в раствор 35%-ную соляную
кислоту и аммиак в количестве, меньшем, чем стехиометрическое
(pH = 5,5 при 70—80°). Из реактора раствор поступает в бак 2,
куда подают дополнительное количество аммиака, необходимое
для завершения реакции обменного разложения. Из полученного
раствора выкристаллизовывают NH4CIO4, который характеризуется
наименьшей растворимостью в образующейся системе. Его отде-
ляют центрифугированием, предварительно подсушивают до влаж-
ности 0,5% в центрифуге-сушилке и окончательно до влажности
0,02% в трехступенчатом вибрационном агрегате222. Из маточного
раствора выделяют кристаллизацией NaCl, который также отде-
ляют на центрифуге. Оставшийся маточный раствор используют
для приготовления исходной смеси. Для использования отхода —
кристаллического NaCl (например, для производства хлората
натрия) предложено 223 очищать его от примесей NH4CIO4 топоч-
ными газами, нагретыми до 500—800°.
Неслеживающийся перхлорат аммония можно получить путем
обрызгивания кристаллов NH4CIO4 водным раствором солей три-
сульфокислот аминоарилметилкарбинола 224.
Хлорная кислота
Наиболее распространенным способом производства хлорной
кислоты является солянокислотный, заключающийся в вытеснении
хлорной кислоты из перхлората натрия:
NaClO4 + НС1 = НСЮ4 + NaCl
Хлорид натрия почти полностью выделяется в осадок. Во избе-
жание взрыва применяемый перхлорат должен содержать не
больше 0,5% хлорита.
При взаимодействии раствора NaC104 с 35%-ной соляной кис-
лотой образуется раствор, содержащий около 32% НСЮ4, 8% НО,
6% NaCl и непрореагировавшего NaC104 и 54% Н2О, с плотностью
1,35 г!см3. Выделившийся в осадок NaCl отделяют от раствора
фильтрованием и возвращают на электролиз для получения хло-
рата и перхлората. Полученный раствор выпаривают в три стадии.
Вначале происходит доразложение NaC104 и отгоняется хлористый
водород, конденсирующийся в виде 35%-ной соляной кислоты. При
этом образуется раствор, содержащий 39,5% НСЮ4, ~0,8—0,9%
НС1, 7—7,5% NaCl, 52,5% Н2О, с плотностью 1,395 г/см?. Во вто-
рой стадии отгоняется в основном вода, а оставшийся NaCl при
повышенной температуре реагирует с НСЮ4. Раствор, выпаренный
до плотности 1,58 г!см\ имеет состав: 57% НСЮ4, ~ 0,05% НС1,
10,5% NaC104, 32,5% Н2О. Его подвергают дальнейшему выпари-
ванию при 200—270°, в процессе которого выкристаллизовывается
NaC104, возвращаемый в процесс, и образуется азеотропная смесь,
содержащая 72,4% НСЮ4 с температурой кипения 203°. После
Перхлораты натрия, калия и аммония
1475
очистки от примесей получается продукт, содержащий 70—72%
HCIO4. Безводную хлорную кислоту можно получить перегонкой
72%-ной хлорной кислоты 225 в присутствии 15—20%-ного олеума
при температуре ~50° под давлением 1 мм рт. ст.
Предложено 226 в качестве стабилизирующих агентов, препят-
ствующих разложению безводной НСЮ4, применять низшие ке-
тоны, алкилнитрилы, амиды предельных кислот и хлоропроизвод-
ные этих соединений. Наибольшей эффективностью обладает
1,3-дихлорацетон.
Перспективным является непосредственное получение хлорной
кислоты электролизом соляной кислоты, известное еще с конца
прошлого века. В настоящее время этот процесс осуществляется
электролизом водного раствора хлора в присутствии хлорной кис-
лоты 227. При этом на аноде протекает одновременно окисление
растворенного хлора, образование хлорноватой кислоты и окисле-
ние хлората в перхлорат; на катоде происходит выделение водо-
рода. Суммарный процесс может быть представлен уравнением:
С12 + 8Н2О = 2НС1О4 + 7Н2
Считают 227, что прямой электрохимический способ получения
хлорной кислоты может оказаться целесообразнее химического,
вследствие уменьшения расхода первоначальных материалов и ко-
личества производственных стадий, потребных для образования
конечного продукта. По этой же причине предпочтительнее окис-
лять хлор в растворе хлорной кислоты, чем окислять на аноде
соляную кислоту. В последнем случае имеется дополнительная
электрохимическая стадия окисления иона хлора и, кроме того,
раствор больше загрязнен примесями, в частности хлорноватой
кислотой, которая препятствует получению в дальнейшем перхлора-
тов. Образование хлорной кислоты при электролизе соляной кис-
лоты происходит при применении растворов с концентрацией 0,1 н.
НС1 при плотности тока на гладком платиновом аноде 4000 а/м2.
В этих условиях выход по току в расчете на хлорную кислоту со-
ставляет 40—45%. При большей или меньшей концентрации соля-
ной кислоты, а также при меньшей анодной плотности выход по
току резко уменьшается.
Для производства хлорной кислоты электролитическим окисле-
нием хлора используют 40%-ную хлорную кислоту, которую насы-
щают при —5° хлором, концентрация которого составляет 3 г/л.
Процесс осуществляется в электролизере фильтрпрессного типа, со-
стоящем из пяти катодов и четырех анодов. В качестве анодов при-
меняется платиновая фольга, укрепленная на танталовой рамке,
в качестве катода — серебряная пластина. Раствор подают в анод-
ное пространство, отделенное перегородками от катодного про-
странства. Отбор кислоты производится из катодного пространства.
Этим удается электрохимически осадить на катоде некоторое
1476
Гл. XXXIX. Соли кислородных кислот хлора
количество платины, переходящее в.раствор вследствие коррозии
Одновременно исключается возможность переноса анионов хлорной
кислоты и изменения вследствие этого состава электролита.
На катоде выделяется водород, содержащий некоторое количе-
ство хлора (5—7%). Его отводят из катодного пространства; из
анодного пространства отводят газообразные продукты окисления.
В них содержится 25—27% хлора.
Кислота из электролизера направляется в отгонную колонну,
обогреваемую паром, где она очищается от воды, хлористого водо-
рода, образовавшегося при гидролизе хлора и непрореагировав-
шего хлора. Получаемая кислота содержит 68—72% НС1О4.
ЛИТЕРАТУРА
1. А. А. Яков кин, ЖРФХО 32, 673 (1900). —2. Е. А. Ш и л о в, С. Н. Со-
лодущенко в, ЖФХ, 19, № 9, 405 (1945). — 3. Е. А. Шилов, С. Н. С о л о-
душенков, Там же, 21, № 10, 1159 (1947). —4. Б. П. Никольский,
И. Е. Флис, Труды ЛТИ, в. 1, 1949, стр. 61. —5. И. Е. Флис, К. П. Мищен-
ко, Н. В. Пахомова, ЖНХ, 3, № 8, 1772 (1958). — 6. Т. G о г s t о n, Pulp а.
Pap. Mag. Canada, 31, 374 (1931). — 7. J. Ourisson, M. Kastner, Bull. Soc.
chim. France, [5] 6, Ns 8—9, 1307 (1939). — 8. A. J. I b a r s, V. J. Virgin, An.
Real. Soc. espanola fis. у quim., 49, № 5, 341 (1953). — 9. И. E. Флис, ЖПХ,
31, № 8, 1194 (1958). —10. Б. П. Никольский, И. Е. Флис, ЖОХ, 22, № 8,
1298 (1952).
11. С. Е. М а к а р о в, Сборник статей Всесоюзн. заочи. политехи, нн-та, в. 9,
1955, стр. 52.-72. И. И. Вольнов, ИСФХА АН СССР, 21, 251 (1956).—
13. R. М. Chapin, J. Am. Chem. Soc., 56, 2211 (1934). —14. M. Lewin, M. Av-
raham!, Bull. Res. Coujncil Israel, 3, № 4, 445 (1954); J. Am. Chem. Soc., 77,
№ 17, 4491 (1955). —/5. И. E. Флис, И. M. Воробьев, ЖФХ, 37, 1865
(1963). —16. А. П. Закощиков, Р. А. Нежельская, Н. А. Пихунова,
ЖПХ, 10, № 1, 36 (1937). —/7. А. П. Закощиков, Н. А. Пихунова,
Там же, 46. —18. А. П. Закощиков, Р. А. Нежельская, Н. А. Пиху-
нова, Там же, № 8, 1380 (1937). — 19. Н а к о в, Лека пром., 5, № 1, 42
1956.— 20. Е. С. Braaka, oth., J. Am. Chem. Soc., 78, № 20, 5160 (1956).
21. M. E. Позин, ЖХП, № 11, 23 (1932).—22. G. H. А у r e s, M. H. В о о t h,
J. Am. Chem. Soc., 77, № 4, 825 (1955). — 23. Ф. M. Перельман, А. Я. 3 во-
ры x и н, ЖФХ, 29, № 6, 980 (1955). —24. В. М. Г а л л а к, ЖПХ, 24, Ns 8,
798 (1951). —25. А. Ю. П р о к о п ч и к, ЖФХ, 29, № 6, 1020 (1955).—
26. А. Ю. Прокопчик, И. В. Яницкий, Там же, 28, Ns 11, 1999 (1954);
Труды Каунасск. политехи, ин-та, т. 4, 1955, стр. И, —27. Н. G. Bijawat,
Р. К. Sard, J. Appl. Chem., 6, № 2, 60 (1956). — 28. М. Н. Клюка но в, Водо-
снабж. и сантехника, № 1, 43 (1940). — 29. К- П. Мищенко, И. Е. Флис,
В. А. К у ст од ин а, ЖПХ, 33, 267 (1960); 34, 125 (1961); 34, 306 (1961); 35,
1374 (1962). — 30. М. Ben Said, J. Н a j а 1, J. Combourien, Bull. Soc.
chim. France, 1965, Ns 10, 2908.
31. К. Д. Д о б p ы ш и н, Новые отбеливающие вещества, Изд. «Лесная про-
мышленность», 1968. — 32. К. Д. Д о б р ы ш и н, И. И. Б л о ш т е й н, И. Е. Ф л и с,
М. К. Быняева, ЖПХ, 37, Ns 3, 498 (1964). —33. К. Д. Добрынин,
И. Е. Ф л и с, С. К. Фи ш, Там же, Ns 11, 2382 (1964). — 34. W. М о s s с h е 1 е i п,
Chim. et ind., 97, № 1, 419 (1967). — 35. М. Bigorgne, Compt. rend., 236, №20,
1966 (1953).—36. M. Bigorgne, ibid., 240, № 3, 311 (1955). —37. R. C u r 11,
E. Mon tai di, Gazz. chim. ital., 83, № 9 748 (1953). — 38. И. E. Флис,
T. M. Васильева, ЖОХ, 26, № 5, 1272 (1956). — 39. G. F. Davidson, J.
Chem. Soc., 1954, 1649. — 40. G. R. Levi, R. Curti, Ricerca sci., 23, Ns 10,
1798 (1953).
Литература
1477
41. И. Е. Ф л и с, ЖПХ, 29, № 5, 633 (1956). —42. М. Козлов, Текст, пром.,
№ 4, 25 (1945). —43. R. С. Вег tog Но, Ann. Chim., 43, № 8—9, 564 (1953).—
44. G. L. Cunningham, Tong San О e y, J. Am. Chem. Soc., 77, № 3, 799
(1955). — 45. M. C. Taylor, J. F. White, G. P. Vincent, G. L. Cunning-
ham, Ind. Eng. Chem., 32, № 7, 899 (1940). — 46. J. F. White, M. C. Taylor,
G. P. Vincent, ibid., 34, № 7, 782 (1942). —47. Ш. С. Щеголь, ЖПХ, 31,
№ 5, 680 (1958). — 48. A. Glasner, L. Wei den f eld, J. Am. Chem. Soc., 74,
№ 10, 2464 (1952).—49. Ю. А. Баскаков, H. H. Мельников, Хим. пром.,
№ 5, 51 (1955). — 50. G. L. Cunningham, Tong San Oey, J. Am. Chem.
Soc., 77, № 17, 4498 (1955).
51. И. И. Вольнов, ИСФХА АН СССР, 24, 160 (1954). —52. И. Шу-
махер, Перхлораты. Свойства, производство и применение, Госхимиздат, 1963. —
53. А. Клочко, М. Ш. Курбанов, ИСФХА АН СССР, 24, 237 (1954).—
54. D. К. Bidinnsti, W. J. В iе гm апn, Canad. J. Chem., 34, № И, 1591
(1956). — 55. А. К. Covington, J. Е. Prue, J. Chem. Soc., 1957, 1567.—
56. L. L. Birciemchaw, T. R. Phillips, ibid., 1957, 703. — 57. A. Glasner,
L. Weidenfeld, J. Am. Chem. Soc., 74, № 10, 2467 (1952). — 58. A. E. S i ni-
che n, A. Glasner, Bull. Soc. chim. France, 1953, № 1, 127. — 59. L. E. Co-
peland, R. H. Bragg, J. Phys. Chem., 58, № 12, 1075 (1954). — 60. А. А. Зи-
новьев, Л. И. Чудинова, ЖНХ, 1, № 8, 1722 (1956).
61. И. Н. Ле Пешков, В. И. Лебошина, Уч. зап. Яросл. пед. ин-та,
в. 59, 1966, стр. 48. — 62. Г. А. Дмитриев, А. И. Качалов, А. Г. Симон,
Хим. наука и пром., 2, № 6, 743 (1957). — 63. В. И. Орлов, П. В. Попов,
М. А. Морозова, М. Г. Габриелов а, Ядохимикаты (инсектисиды и фун-
гисиды), Госхимиздат, 1946. — 64. Chem. Eng., 60, № 9, 117, 120 (1953).—
65. А. Н. 3 е л и к м а н, Ф. А. Г и м е л ь ф а р б, Изв. вузов. Цветная металлург.,
№ 2, 110 (1966). — 66. М. Е. Позин, ЖХП, № 10, 1023 (1935).—67. H.L. Rob-
son, пат. США 3234141, 1966.—68. С. R. Н а г 1 о с k, М. R. D о w И п, Water а.
Sewage Works, 100, № 2, 74 (1953). — 69. Chem. Trade J., 125, № 3263, 725;
№ 3264, 775 (1949). —70. P. R. Gustafson, пат. США 3207695, 1965.
71. Ю. Ракитин, Хлопководство, № 9, 36 (1955); Л. И. Королев,
В. А. В о й т е х о в а, Л. Д. Стонов, Труды НИУИФ, в. 167, 1960, стр. 208. —
72. W. Wallace, J. Electrochem. Soc., 99, № 11, 309 (1952). —73. И. Ф. Бли-
нов, Хлоратные и перхлоратные взрывчатые вещества, Оборонгиз, 1931.—
74. L. А. В u г n а г d t, W. G. Finnegan, пат. США 3274035, 1966. — 75. Chem.
Eng. News, 35, № 19, 88 (1957). — 76. J. Schumacher, D. K. Stern, Chem.
Eng. Progr., 53, № 9, 428 (1957). — 77. E. A. Hahn, T. R. Ingraham, Trans.
Metallurg, Soc. A1ME, 236, № 8, 1098 (1966). — 78. P. E. Church-Ward, пат.
США 3244475, 1966. — 79. M. П. Сытин, Производство извести, изд. Мии. пром,
стройматериалов РСФСР, 1947. — 80. П. Н. Григорьев, А. М. Кузнецов,
Известь. Ее производство и применение, Госстройиздат, 1944.
8/. В. М. Горчаков, Производство хлорной извести, ОНТИ, 1936.—
82. Г. В. Брусиловский, Производство извести, Госхимиздат, 1954.—-
83. А. С. К р а м м, Обжиг известняка, изд. Мин. пром, стройматериалов РСФСР,
1938.— 84. А. С. Крамм, Основные элементы теории и расчета шахтных полу-
газовых известково-обжигательных печей, БТИ, 1949. — 85. Б. К. Буткевич,
Производство извести, Гизместпром, 1944. — 86. М. Е. Позин, ЖХП, № 12, 43
(1934). — 87. М. Е. Позин, Там же, № 9, 926 (1935). — 88. М. Е. П о з и и, Зав.
лаб., № 11, 1041 (1934). — 89. К- Beckenbach, англ. пат. 702001, 1954.—
77. Н. А. Рапопорт, Производство хлорной извести, ОНТИ, 1939. —
90. Е. М. X а и и н, ЖХП, № 10, 40 (1939).
91. М. Е. Позин, Труды ГИПХ, в. 15, 1932, стр. 43. — 92- Н. Seidel,
W. Kuhn, пат. ГДР 11831, 1956. — 93. А. Ф. Присте некий, Г. А. Н о в о-
крещенов, В. Н. Копанев, А. Д. Горбачев, С. К. Кирохосян,
Д. Д. Савелов, Обмен опытом, изд. ин-та им. Л. Я- Карпова, № 1 (46), 1958,
стр. 7. — 94. М. Е. Позин, Белящие соединения хлора, Госхимиздат, 1933.—
1478
Г л. XXXIX. Соли кислородных кислот хлора
95. X. И. Сидякин, Хим. пром., № 5, 301 (1955). — 96. Б. А. Сасс-Тисов-
ский, 3. И. Л и ф а т о в а, Там же, № 2, 1 (1947). — 97. S. Ramaswamg
Altech., № 5, 31 (1955). — 98. О. Karsten, М. V о е s t е, пат. ФРГ 887037'
1953. — 99. Т. Jamakoshi, япон. пат. 2426, 1954. — 100. М. Е. Позин, ЖХГ1
№ 8, 810 (1935).
101. J. G. М a t а г е s е, J. К. Moorhead, англ. пат. 686968, 1953 —
102. Ind. Chem., № 314, 115 (1951); № 333, 451 (1952). —103. К. Peters, пат
ГДР 6400, 1954. — 104. R. L. С а г г, пат. США 2639981, 1953. — 105. R. F. Hen-
ri, франц, пат. 1349076, 1963. —106. Chem. Eng., 61, № 3, 128 (1954). — 107. Пат
ФРГ 941427 и 941970, 1956. — 108. R. J. Phadke, Current Sci., 26, № 1, 12
£1957). — 109. R. C. Brandon, пат. США 2662858, 1953, — ПО. F. С 1 е b о i з,
Р. Antoine, пат. США 3199949, 1965.
111. В. Г. Хомяков, В. П. М а ш о в е ц, Л. Л. Кузьмин, Технология
электрохимических производств, Госхимиздат, 1949. — 112. Н. А. Масленни-
ков, В. А. Казарян, сб. «Качество подготовки морской воды», № 2, 1967,
стр. 68. —113. М. Е. Позин, ЖХП, № И, 672 (1936).— 114. И. А. Эльмано-
вич, Труды ГИПХ, в. 20, 1934, стр. 59. —115. В. Ф. Чернов, Производство
кальцинированной соды, Госхимиздат, 1957. —116. Тао Жунь-чэин, Хуасюэ
шицзе, 9, № 9, 395 (1954). — 117. М. Е, Позин, Ц. А. Левина, ЖХП, № 14,
864 (1936). —118. В. Н. Nicalaisen, пат. США 3241912 и 3251647, 1966.—
119. J. М. Avery, R. F. Evans, Chem. Met. Eng., 52, № 4, 94 (1945).—
120. M. Г. Беркович, Зав. лаб., 22, № 4, 423 (1956).
121. И. Л. Ломакин, Хим. пром., № 4, 15 (1946). — 122. Е. Р. D i г п с а п,
Pulp. Paper Ind., 9, № 2, 12 (1956). — 123. G. Holst, Chem. Revs, 54, № 1,
169 (1954). — 124. Франц, пат. 1032565, 1953. — 125. H. T u d ё n, Svens pappers-
tidn, 57, № 16, 583 (1954). — 126. Kuexapa, J. Soc. Pressure Gas Ind., Japan,
20, № 7, 6 (1956). —/27. W. H. R a p s о n, Canad. J. Chem. Eng., 36, № 6, 262
(1958). — 128. H. C. Marks, R. R. Joiner, канад. пат. 513016, 1955.—
129. H. C. Marks, R. R. Joiner, канад. пат. 523827, 1956. — 130. Ito, япон.
пат. 1978, 1954.
131. W. H. Rapson, M. Wayman, швед. пат. 151521, 1955,—
132. W. Н. Rapson, Tappi, 37, № 4, 129 (1954). — 133. H. Beduneau, Rev.
prod, chim., 57, № 1201, 173 (1954). —/34. W. H. Rapson, Pulp a. Paper Mag.
Canada, 55, № 5, 92 (1954). — 135. M. Wayman, W. Rapson, канад. пат.
499846, 1954. —136. С. H. Evans, пат. ФРГ 884040, 1953. —137. Н. С. Mark s,
V. G. Sarli, пат. США 2710246, 1955. — 138. Е. Н. Gleason, G. Mino,
W. М. Thomas, J. Phys. Chem., 61, № 4, 447 (1957).— 139. I. Sevon,
E. V. S u n d m о п, пат. ФРГ 937470, 1955.—140. J. К e p i n s k i, Przem. Chem.,
10, № 8, 387 (1954); 11, № 3, 124 (1955).
141. Англ. пат. 688502, 1953. — /42. E. Wagner, пат. США 2653079, 1953,—
143. E. Wagner, пат. ГДР 6667, 1954. —144. E. Wagner, пат. ГДР 7751,
1954.— /45. Амадзу, Исихара, япон. пат. 6123, 1954. — 146. W. W. North-
graves, В. Н. N i с о 1 a i s е n, Т. Н. Dexter, D. J. J a s г k a, South. Pulp а.
Paper Manufact., 20, № 2, 70, 72, 74 (1957). —147. S. U. E к m а п, швед. пат.
138858, 1953. —148. H. V. Williamson, C. A. Hampel, пат. США 2683651,
1954. —149. H. V. Williamson, С. A. Hampel, канад. пат. 515449, 1955. —
150. J. С. Н е s s о п, пат. США 2704703, 1955.
151. W. A. S t о п е, пат. США 2678922, 1954. — 152. J. S е v о п, F. V. S е i п d-
m а п, пат. ФРГ 940893, 1956. — 153. F. Н. Dole, франц, пат. 1054039, 1954.—
154. Англ. пат. 687099, 1953.-— 155. М. L. Au do gn a Cd, канад пат. 512954,
1955. — 156. A. Schmidt, пат. ФРГ 942686, 1956,—/57. L. А. В eu dr у, Paper.
Mill News, 77, № 12, 83 (1954).— 158. Tappi Monograph., Ser., 10 (1953).—
159. W. Mosschelein, Chim. et ind., 97, № 1, 49 (1967). — 160 E Ke s t i n g.
Paper Mill News, 76, № 49, 96, 98, 104, 106 (1953).
Литература
1479
161. Е. Ke sting, пат. ФРГ 924689, 1955. — 162. Франц, пат. 1026630, 1953. —
163. W. Krings, Osterr. chem. Z., 56, № 19—20, 287 (1955). —164. Chem. Week,
100, № 7, 78 (1967). — 165. R. J. Read, Paper Trade J., 151, № 6, 44 (1967).—
166. Canad. Chem. Process, 57, № 3, 64 (1967). — 167. W. H. Rapson, англ,
пат. 1056790, 1967. — 168. G. M. Booth, канад. пат. 506640, 1954.—
169. А. С. Чернышев, В. В. Штуцер, Н. Г. Семенова, Усп. хим., 25,
№ 1, 91 (1956). — 170. R. Kirk, D. О t h m е г, Encyclopedia of Chemical Techno-
logy, v. Ill, N. Y„ 1949, p. 700.
171. K. Cederquist, B. Lunde п, швед. пат. 143965, 1954. —172. Англ,
пат. 687246, 1953. —/73. Франц, пат. 1025778, 1953, —/74. В. В. Штузер,
А С. Чернышев, Труды Московск. текстильн. ин-та, т. 13, 1954, стр. 112.—
175. G. Holst, Ind. Eng. Chem., 42, № 11, 2359 (1950). — 176. Пат. ФРГ 873993,
1953. — 177. R. N. Aston, швед. пат. 145633, 1954. —178. R. Weiner, Z. Elek-
trochem., 52, № 3, 234 (1948). —179. F. H. Dole, канад, пат. 515258, 1955,—
180. Швейц, пат. 290581, 1953.
181. S. Saito, япон. пат. 1668, 1953. —182. Франц, пат. 1027783, 1953.—
183. Англ. пат. 706667, 1959. —184. F. Erbe, пат. ФРГ 892890, 1953. —185. Т. Ito,
япон. пат. 1576, 1577, 1954. —186. Ямамото, Фукусима, Накамура, япон.
пат. 7576, 1954. — 187. S. М. Mehta, D. J. Mehta, Proc. Indian Acad. Sci., A44,
№ 6, 392 (1956). — 188. N. C. White, Trans. Electrochem. Soc., 92, 15 (1947).-
189. С. С. Ш p а й б м а н, Производство бертолетовой соли и других хлоратов,
ОНТИ, 1938. — 190. В. Г. Флейшман, Хим. пром., № 1, 9 (1959).
191. Д. П. М а н о е в, Труды ГИПХ, в. 3, 1925, стр. 27. — 192. W. К. Ear-
nest, Е. Н. Karr, швед. пат. 143029, 1953. — 193. N. Subramanian,
В. К. Rao, Н. V. U d и р а, В. В. Dey, J. Sci. Ind. Res., 15, 11, 665 (1956).—
194. Ullmans Encyklopedia der technischen Chemie, B. 5, Berlin, 1954.—
195. H. П. Федотьев, А. Ф. Алабышев, А. Л. Ротинян, П. M. Вяче-
славов, П. Б. Жив отин ский, А. А. Б ал ьн бек, Прикладная электрохи-
мия, Изд. «Химия», 1967, стр. 434. — 196. С. L. М а п t е 11, Industrial Electroche-
mistry, N. Y., 1950. — 197. В. А. Шляпников, С. С. Ш р а й б м а н, Хим. пром.,
№ 3, 66 (1967). — 198. В. Г. Флейшман, Там же, № 7, 27 (1957).—
199. J. D. Ans, Н. Е. Freund, Z. Elektrochem., 61, № 1, 10 (1957).— 200. A. N а 1-
1 е t, R. A. Paris, Bull. Soc. chim. France, 1956, № 3, 488.
201. И. E. Флис, Автореф. докт. дисс., ЛТИ им. Ленсовета, 1959. —
202. И. Е. Флис, М. К. Б ы н я е в а, И. И. Б л о ш т е й н, ЖПХ, 33, № 4, 779
(1960).—203. А. И. Заславский, Труды ГИПХ, в. 23, 1935, стр. 67.—
204. Н. С. Спирин, Калий, № 6, 20 (1936). — 205. Е. Н. Токарева, Обмен
опытом, изд. Ин-та им. Л. Я. Карпова, № 5 (50), 1958, стр. 7. — 206. И. С. М о р о-
зо в, Труды ГИПХ, в. 15, 1932, стр. 46. — 207. Е. П. К а ц у к, ЖХП, № 6, 586
(1935). — 208. Н. А. Эльманович, ЖХП, Ns 4, 69 (1934). — 209. Ю. М. Мар-
тынов, М. А. Матвеев, Л. М. Якименко, А. А. Фурман, Хим. пром.,
№ 7, 420 (1958). — 210. М. L. Somerville, франц, пат. 1102374, 1955.
211. М. Я. Ф и о ш и н, А. М. М е ж е р и ц к и й, Хим. пром., № 9, 36 (1967). —
212. J. С. Schumacher, пат. США 2733982, 1956. — 213. Е. О 11, Chem. Вег.,
86, Ns 9, 1065 (1953). — 214. К. Ф. Сагайдачный, М. И. Василенко,
А. О. Скирстимонский, Труды ГИПХ, в. 19, 1934, стр. 57. — 2/5. A. La-
ge п d г е, Chem. Ing. Techn., 34, 379 (1962). — 216. Chem. Eng., 72, 82 (1965).—
217. Канад, пат. 640131, 1962. — 218. Roczn. chem., 36, 685 (1962).—
219. А. С. Карнаухов, А. В. M о к и н, ЖНХ, 2, № 4, 910 (1957).—
220. N. A. Bell, J. Whetstone, англ. пат. 689283, 1953.
221. J. С. Schumacher, пат. США 2739873, 1956. — 222. Chem. Eng., 62,
№ 12, 334 (1955), —223. D. R. Stern, пат. США 3254947, 1966.— 224. Норв.
пат. 81257, 1953, —225. G. F. Smith, J. Am. Chem. Soc., 75, Ns 1, 184 (1953). —
226. R. D. Stewart, M. A. Prieto, S. E. Gordon, пат. США 3258309,
1966. — 227. W. Miiller, P. J 6 n e k, Chem. Ing. Techn., 35, 78 (1963).
Глава XL
ПРОДУКТЫ ВЫСОКОТЕМПЕРАТУРНОГО ХЛОРИРОВАНИЯ
РУД
ОБЩИЕ СВЕДЕНИЯ
Хлор является весьма активным реагентом. При высоких тем-
пературах он способен вытеснять серу из сульфидов, а в присут-
ствии восстановителей хлорировать окислы различных металлов и
вытеснять из сульфатов, фосфатов, силикатов кислородные соеди-
нения серы, фосфора, кремния с образованием соответствующих
хлоридов. Это используют в технологии благородных и цветных
металлов при рафинировке золота, алюминия, свинца и олова 1-3,
а также в металлургии титана и редких металлов — циркония, тан-
тала, ниобия и др.4-э. При хлорировании полиметаллических руд
образующиеся хлориды могут быть разделены на основе различия
в температурах испарения, а также методами экстракции 10.
Гидролиз хлоридов металлов является одним из методов полу-
чения металлических порошков. В частности, он применяется для
получения железного порошка из стальной стружки и. При гидро-
лизе хлористого бора образуется борная кислота 12. Хлориды яв-
ляются в некоторой степени растворителями металлов (РЬ в рас-
плавленном РЬС12, Cd в CdCl2 и др.) 13. Представляет интерес
хлорирование различных сульфатов с получением хлоридов и серни-
стого газа 14’ 1S, в частности с осуществлением процесса в среде рас-
плавленных хлоридов16. При хлорировании апатита в расплаве
СаС12 при 850—900° образуется РС1з и товарный хлорид кальция 17.
Заслуживает внимания хлорирование сульфидных руд хлором
в жидкой хлористой сере, вследствие низкой температуры процесса
(130—150°), высокой скорости его и отсутствия взаимодействия
с пустой породой. Процесс исследован применительно к извлече-
нию меди, свинца и цинка из бедных и богатых руд 18. .
Для интенсификации процессов хлорирования все больше ис-
пользуют аппараты со взвешенным слоем 19 обрабатываемого мате-
риала.
Четыреххлористый титан
1481
Из неорганических хлоропродуктов, образующихся в резуль-
тате горячего хлорирования руд, наибольшее значение в химиче-
ской промышленности имеют четыреххлористые титан и кремний,
треххлористый алюминий, хлорокись фосфора (стр. 1063), а также
безводный хлорид магния (стр. 278).
ЧЕТЫРЕХХЛОРИСТЫЙ ТИТАН
Физико-химические свойства20’21
Четыреххлористый титан TiCl4 при 20° — бесцветная жидкость
с плотностью 1,27 г[смъ, замерзающая при —23° и кипящая при
136°. При загрязнении низшими хлоридами, а также хлориым же-
лезом, оксихлоридом ванадия и другими он приобретает желтую
окраску. Во влажном воздухе его пар
гидролизуется с образованием густого
белого дыма:
TiCl4 + ЗН2О = H2TiO3 + 4НС1
Давление пара TiCh (в ммрт.ст.)
может быть вычислено по формуле
lg Р = 7,64433— 1947,6 7’-1 (рис. 444).
В присутствии водорода при 500—
800° четыреххлористый титан образует
фиолетовый треххлористый титан, а
при избытке водорода — двуххлори-
стый титан; при большом избытке во-
дорода и повышении температуры до
900—1000° восстановление идет до ме-
таллического титана.
Аналогично четыреххлористый ти-
Рис. 444. Давление пара над
жидким TiCl4.
тан восстанавливается и другими вос-
становителями, например, магнием, алюминием. В присутствии
кислорода при 600—1000° четыреххлористый титан окисляется
с образованием молекулярного хлора;
Т1С14 + О2 = TiO2 + 2С12
Прежние сведения о частичном образовании при этом оксихло-
рида
4TiCl4 + ЗО2 = 2Ti2O3Cl + 6С12
экспериментально не подтвердились22.
Четыреххлористый титан бурно реагирует с водой с выделением
большого количества тепла. Вначале образуются гидраты, а затем
начинается гидролиз с образованием метатитановой кислоты
Н2ТЮ3 и соляной кислоты. В избытке воды образуется пятиводный
четыреххлористый титан TiC14-5H2O, а при низких температурах
1482 Гл. XL. Продукты высокотемпературного хлорирования руд
и недостатке воды — двухводный TiCU-2H2O, а иногда и одновод.
ный кристаллогидрат — TiC14-H2O. При взаимодействии с парами
воды продуктами реакции до 450° являются окси- и гидрооксихло-
риды титана, при более высоких температурах (~750°)— двуокись
титана:
TiCl4 + 2Н2О = TiO2 + 4НС1
С повышением температуры в продуктах гидролиза умень-
шается содержание оксихлоридного титана и увеличивается содер-
жание двуокиси титана, В этой системе существует непрерывный
ряд соединений с плавным изменением в их составе атомного отно-
шения титана и хлора. Предложено состав основных хлоридов
четырехвалентного титана, образующихся при взаимодействии
с парами воды, выражать формулами23:
Т1Ов(ОН)2_2аС12 (где а = 0 ч- 1)
Ti(OH)xCI4-x • пН2О (где х : (4 — х) •= 1 ч- 3]
Основные хлориды титана — термически нестойкие соединения;
полученные при комнатной температуре они начинают разла-
гаться уже при 50—60° и полностью переходят в двуокись титана
выше 300°. На воздухе постепенно разлагаются под влиянием
влаги. Растворимость основных хлоридов в воде и 5%-ной серной
кислоте тем больше, чем ниже температура их образования.
В четыреххлористом титане они образуют структурированные
взвеси вследствие анизодиаметрической формы частиц и высокой
их дисперсности24.
Из аналогов четыреххлористого титана наибольшее значение
в металлургии титана сверхвысокой частоты имеют иодиды ти-
тана 25~28.
Применение
Четыреххлористый титан имеет большое значение как сырье
для производства металлического титана 29-33, находящего примене-
ние в качестве конструкционного материала, в частности в химиче-
ской промышленности и ядерной технике 34’35.
Получение чистого металла из четыреххлористого титана проще,
чем восстановление его из кислородных соединений из-за большого
сродства титана к кислороду36> 37. При содержании же больше
0,2% кислорода титан теряет способность к пластической деформа-
ции. Из четыреххлористого титана можно получать ковкий титан
с минимальным количеством примесей — магнийтермическнм (вос-
становление TiCU магнием), натрийтермическим (восстановление
TiCl4 натрием) и электрохимическим методами6'38-4?. Наиболее
освоенным и эксплуатируемым в промышленных условиях является
магнийтермическое восстановление T1CI4.
Четыреххлористый, титан
1483
В связи с расширяющимся производством титана и уменьше-
нием его стоимости перспективным является применение его в ка-
честве декоративного материала в строительстве41. Улучшение
внешнего вида его поверхности и придание ей разнообразных
оттенков возможно методами механической полировки, анодирова-
ния, термического травления, а также никелирования 42 и др.
Титанокарбидные сплавы характеризуются меньшей температу-
рой резания и меньшим коэффициентом трения, чем вольфрамокар-
бидный сплав. Поэтому они пригодны для обработки высоко-
прочных и жаростойких материалов. Основные достоинства
титанокарбидных сплавов заключаются в высокой твердости и
очень высоком сопротивлении абразивному износу43. Во многих
электролитических процессах применяют титановые аноды вместо
платины или других благородных металлов. Показана44 возмож-
ность электрохимического получения двуокиси марганца с исполь-
зованием титановых анодов.
В лакокрасочной промышленности четыреххлористый титан
используют для получения двуокиси титана45"49. При взаимодей-
ствии раствора TiCl4 с аммиаком и концентрированной серной
кислотой образуется двойная соль (NH^aSOrTiOSO^HaO, из ко-
торой можно получить легкую двуокись титана (с кажущейся плот-
ностью 0,25 г/сж3), пригодную для синтеза драгоценных камней50.
Четыреххлористый титан является интенсивным дымообразовате-
лем, используемым в военном деле51. Образование дыма происхо-
дит в результате гидратации и гидролиза TiCI4 водяными парами,
находящимися в воздухе, и значительно интенсифицируется в при-
сутствии аммиака.
Сырье6’36
Содержание титана в земной коре составляет 0,6 вес. %; по весу
он занимает четвертое место среди наиболее распространенных
конструкционных металлов (после алюминия, железа и магния).
Несмотря на распространенность, скопления титановых соедине-
ний встречаются редко. Наиболее распространенные титановые ми-
нералы — ильменит и титаномагнетит, рутил, перовскит и сфен, или
титанит. Ильменит FeTiO3—метатитанат железа — впервые был
найден на Урале в Ильменских горах. При его выветривании об-
разуются тонкодисперсные окислы титана и железа.
Электромагнитным разделением магнетита и ильменита полу-
чают ильменитовый концентрат (слабомагнитная фракция), же-
лезный концентрат (магнетитовый) и хвосты (немагнитная фрак-
ция). Двуокись титана встречается в магматических горных
породах в трех модификациях — рутил, анатаз и брукит, из кото-
рых наиболее распространенной является рутил. Встречаются
также концентрированные месторождения рутила осадочного типа
на морском побережье, образовавшиеся при распаде горных пород.
1484 Гл. XL. Продукты высокотемпературного хлорирования, руд
Рутил, освобожденный от пустой кремнистой породы47, является
наиболее концентрированным титановым сырьем. Он содержит от
91 до 99% TiO2 с небольшими примесями циркония, ниобия, ва-
надия, хрома, железа, кремния и алюминия.
В СССР имеются крупные месторождения перовскита — тита-
ната кальция CaTiO3.
Сфен, или титанит — титаносиликат кальция СаО • TiO2 • SiO2,
в котором часть СаО может замещаться на FeO и MgO. Источни-
ком титана могут также служить руды, содержащие комплексные
минералы — лопарит, пирохлор и другие титано-ниобиево-тантало-
вые минералы.
Ильменит в настоящее время является основным видом титано-
вого сырья.
В 1963 г. добыча ильменита в капиталистических странах
составила 52 больше 2 млн. г, а рутила ~0,2 млн. т.
В табл. 115 приведен примерный состав титановых руд8’53.
ТАБЛИЦА 115
Содержание главных компонентов в титановых рудах
(Содержание компонентов в %)
Компонент Ильменит с высоким содержанием TiO2 Ильменит с низким содержанием TiO2 Титано- магнетит Ильменитовый концентрат Перовскит Сфен
тю2 57,5-61 38,5-41,5 8-11 40-44 44-47 30-33
FeO 9-19 28-31 30-34 27-32 — 2-4
Fe2O3 20-26 18-21 46-50 17-23 4,5-6 —
'MgO 0,5-1,2 3,5-4,5 1,5-3 2-3 2,5 1
СаО 0,1-0,2 0,3-1 0,6-1,5 0,5-1,5 34,6—36,8 27-29
SiO2 0,5-2 2-9 1,5-4,5 1,5-2 5,7-7,8 27-29
А12О3 1-2 2-4 3-6 0,5-3,5 0,9 1,5-2,5
В связи с малым содержанием примесей в рутиловом концен-
трате получение TiCU из этого вида сырья проще, чем* при исполь-
зовании ильменита. Однако рутил — более дорогое и менее распро-
страненное сырье.
Непосредственное хлорирование ильменита затрудняется обра-
зованием большого количества хлорного железа. Поэтому ильменит
до хлорирования подвергают обезжелезиванию.
Способы обезжелезивания ильменитового концентрата
Наибольшее практическое значение имеют следующие способы
удаления основной массы железа из ильменитового концентрата:
1) прямое восстановление ильменита, 2) восстановительная плавка
и 3) карбидизация ильменита.
Четыреххлористый титан
1485
Прямое восстановление ильменита осуществляют углеродом,
водородом или газообразным топливом при 900—1200°:
FeTiO3 + С = Fe + TiO2 + СО
FeTiO3 + Н2 = Fe + TiO2 + Н2О
Образующееся металлическое железо отделяется от двуокиси ти-
тана электромагнитной сепарацией. После отмывки от остатков
железа слабокислым раствором хлорного железа двуокись титана
высушивают и направляют на хлорирование.
Предложено осуществлять восстановление ильменита в при-
сутствии небольших количеств шлакообразующих веществ 54. При
восстановлении железотитанового сырья коксом в присутствии
CaSO4 при 1100—1200° можно получить 55 концентрат с содержа-
нием 90—95% TiO2.
При восстановительной плавке происходит избирательное вос-
становление ильменита углеродом. Ее проводят в электродуговой
печи при 1400—1500°, причем образуются богатые титаном шлаки
и чугун с содержанием около 1% Си 0,5% S 56-58. Получаемые
титанистые шлаки содержат 65—90% TiO2, 0,5—3% FeO, 1—7%
SiO2, 0,5—6% А120з, 0,1—3,5% СаО, 5,5—20% MgO, 0,15-0,4%
V2O5, 0,02—0,2% Н2О. Плавку можно .вести и в присутствии флю-
сов (окиси кальция, окиси магния и др.) при пониженной темпера-
туре 6'59. Однако в этом случае затрудняется процесс последую-
щего хлорирования титанистых шлаков вследствие образования
легкоплавкой пленки СаС12, MgCl2 и др.
Заслуживает внимания восстановление титанистого сырья при
1200—1400° коксом в отсутствие флюсов с получением смеси низ-
ших окислов титана и небольшого количества карбида кальция 60.
Такая смесь хлорируется при более низкой температуре (200—
500°), чем шлаки, содержащие двуокись титана.
Представляет также интерес переработка ильменитового кон-
центрата и других титано-железистых материалов в сульфиды ти-
тана, которые хлорируются даже при 175°. Наиболее перспектив-
ным методом получения сульфидов титана является плавка титано-
вого сырья на штейн. Титановый штейн получается 61 при плавке
титанового сырья с коксом и пиритом при температуре около 1500°.
Полученный продукт содержит 37—45% Ti (в виде TiS, Ti2S3, TiS2,
TiO и др.), 10—20% Fe (в элементарном виде и в виде TiFeS2),
20—30% S и 1,5—3,5% С.
Карбидизация ильменита происходит при повышении темпера-
туры восстановительной плавки до 1800—2200° и увеличении содер-
жания углерода в шихте 6; выплавляется чугун и образуется кар-
бид титана:
FeTiO3 + 4С = TiC + Fe + ЗСО
Образующаяся при плавке в присутствии воздуха пористая
масса представляет собой твердый раствор карбида и нитрида
1486 Fa. XL. Продукты высокотемпературного хлорирования руд
титана Ti(C, N) или оксикарбонитрид Ti(C, N, О). После измель-
чения и отделения основной массы железа (чугуна) электромагнит-
ной сепарацией и обработкой разбавленной соляной кислотой, в ко-
торой карбид и карбонитрид титана не растворяются, полученный
полупродукт направляют на хлорирование.
Из других методов обезжелезивания ильменитового концентрата
заслуживает внимания растворение его в кислоте 58. Из сернокис-
лотного раствора выделяют гидролизом двуокись титана, которую
можно использовать для хлорирования. Обычно таким путем по-
лучают из ильменита (и рутила) титановые белила62. Возможна
также переработка ильменита соляной кислотой. Образующийся
при этом хлорид титана подвергают гидролизу 63.
Получение четыреххлористого титана из рутилового
концентрата и титанистых шлаков
При использовании в качестве сырья рутилового концентрата
или продуктов обезжелезивания ильменитового концентрата пря-
мым восстановлением или восстановительной плавкой хлорирова-
нию подвергают материал, содержащий двуокись титана. Не-
посредственное взаимодействие двуокиси титана с хлором идет по
реакции
TiO2 + 2С12 = TiCl4 + О2 — 45 ккал
без образования оксихлоридов титана64, но протекает с небольшой
скоростью даже при высоких температурах (800—1000°).
В отсутствие восстановителя хлорирование ильменита практи-
чески приводит к образованию FeCl3 и чистой TiO2 65. В присут-
ствии угля хлорирование TiO2 наблюдается уже при 400—450°,
а при 600—800° протекает со скоростью, достаточной для практи-
ческого его использования. При этом в зависимости от условий
осуществления процесса (главным образом, от температуры) об-
разуется окись углерода или двуокись углерода, или фосген66'71:
TiO2 + 2С12 4" 2С = TiCl4 4~ 2СО 4- 7,8 ккал
Т1О2 4" 2С12 4“ С == TiCU 4" СО2 4~ 49 ккал
ТЮ2 4- 4С12 + 2С == TiCl4 + 2СОС12 + 62 ккал
Образование низших хлоридов титана (ПС1з и TiCl2) является
термодинамически маловероятным 66.
Ниже 600° хлорирование идет с образованием двуокиси угле-,
рода, а выше 600° — преимущественно окиси углерода72. При
900—1000°, помимо окиси углерода, образуется некоторое коли-
чество фосгена. В диапазоне 600—800° скорость образования фос-
гена невелика. При хлорировании выше 1300° газообразные про-
дукты содержат73 практически только окись углерода (2/з объ-
ема) и четыреххлористый титан (’/3 объема).
Четыреххлористый титан
1487
Скорость хлорирования зависит от размеров частиц сырья,
скорости потока хлора и температуры. В присутствии катализатора
(около 0,1% двуокиси марганца) хлорирование ТЮг ускоряется
особенно при низких температурах. Степень превращения ТЮ2
в ИСЦ в лабораторных условиях при 400° достигает 70% (за 4 ч)
вместо 15% без катализатора. Так как хлорирование ведут обычно
в шахтных печах, то сырьевые материалы предварительно брике-
тируют,
Скорость хлорирования брикетов из двуокиси титана и 26%
нефтяного кокса зависит от величины брикетов 74. На рис. 445 по-
казано изменение степени хлори-
рования ТЮ2 в зависимости от
первоначального веса брикетов
при скорости хлора 20 см/лшн.
Для брикетов цилиндрической
формы глубина хлорирования х,
Рис: 445. Зависимость степени
хлорирования двуокиси титана
от веса брикетов при 700°.
Рис. 446. Влияние темпера-
туры на скорость хлорирова-
ния TiO2.
представляющая собой толщину слоя брикета в см, вступившего
в реакцию за время т, может быть вычислена по формуле
где р — плотность брикета, г/см3-,
Ро —. начальный вес брикета, г;
г] — степень хлорирования.
Величина х вычисляется как полуразность определяющих диа-
метров брикетов 'Л (До— D) до реакции (До) и оставшегося (Д)
к моменту т. Можно принять, что скорость хлорирования пропор-
циональна внешней поверхности брикета, f. е. ~^~ = kS (где S —
переменная величина внешней поверхности без учета пористости);
r dx 1
так как dp = pSdx, то — (где — удельная скорость хло-
рирования), и, следовательно, глубина хлорирования прямо про-
порциональна времени. С уменьшением размера брикетов
1488
Гл. XL. Продукты, высокотемпературного хлорирования руд
увеличивается общая реакционная поверхность и тем самым уско-
ряется процесс хлорирования. Но при очень малых размерах бри-
кетов возрастает гидравлическое сопротивление. На рис. 446 пока-
зано влияние температуры на скорость хлорирования двуокиси
титана при скорости потока хлора 16 см)мин. При 550° процесс при
малой линейной скорости хлора переходит из кинетической в диф-
фузионную область. Для кинетической области эффективная энер-
гия активации реакции равна 37,5 ккал!молъ.
Рис. 447. Зависимость
скорости хлорирования
Т1О2 От концентрации
хлора в газовой фазе.
Скорость хлорирования пропорциональна
линейной скорости хлора в степени 0,43
(измерено в пределах от'13 до 41 см/мин)
и возрастает пропорционально содержанию
хлора в газовой фазе при концентрациях
выше 30% (рис. 447).
Хлорирование ведут при 800—900°. По-
догрев массы до температуры реакции осу-
ществляется частично электрическим током
при помощи угольных электродов, частично
за счет выделяющегося тепла реакции. Га-
зы, пройдя пылеуловитель, поступают в
конденсационную систему, состоящую из
скруббера и трубчатых холодильников, где
происходит ожижение и отделение Т1С14.
Несконденсировавшиеся газы после допол-
нительной очистки выбрасываются в атмо-
сферу. Жидкий TiCl4 загрязнен твердыми, а также растворенными
хлоридами. После фильтрации и дистилляции его очищают от
соединений ванадия при помощи медного порошка и от четырех-
хлористого кремния— ректификацией (стр. 1493).
Примеси, содержащиеся в сырье, особенно при переработке ти-
танистых шлаков из ильменита, увеличивают расход хлора 75 и за-
трудняют ведение процесса. Наряду с TiCl4 образуются также ле-
тучие и нелетучие хлориды и оксихлориды. В табл. 116 приведены
температуры плавления и кипения некоторых хлоридов.
ТАБЛИЦА 116
Температуры плавления и кипения некоторых хлоридов и оксихлоридов
Т1С14 FeCl3 А1С1з VC13 VOCI3 SiCl4 СгО2С12
Температура плавления, °C -23 303 192,4 (под давле- нием) -26 ниже —77 -67 —96,5
Температура кипения, °C 136 319 182,7 (возгонка) 164 127 57 116,7
Четыреххлористый титан
1489
Летучие вещества — FeCU, SiCl4, ZrCl4, AICI3, VOCI3, СОСЬ,
SCh, SnCh, HC1 — удаляются из печи и конденсируются вместе
с T1CI4 частично в твердом, а частично в жидком состоянии.
Часть нелетучих и высококипящих хлоридов — кальция, магния,
марганца, алюминия, железа, нехлорированные соединения крем-
ния (в количестве около 50% от общего количества кремния)
остаются в печи и выпускаются в расплавленном виде.
Присутствие в шлаке свободного кремнезема затрудняет удале-
ние жидких хлоридов из печи76. Свободный кремнезем медленнее
хлорируется и хуже удаляется из печи, чем связанный. Наиболее
вредными примесями являются соединения алюминия. Они при-
водят к значительному увеличению расхода хлора и засорению
коммуникаций — хлористый алюминий конденсируется из газа в
твердом виде в газоходах, пылеуловителях и других аппаратах.
Содержание его в пыли, осадках фильтров, скрубберов и вентиля-
торов составляет 30—60%. Для уменьшения содержания алюминия
в сырье, используемом для хлорирования, производят отмывку гли-
ны из руды или выделяют из руды соединения алюминия другими
методами (например, флотацией). Аналогичным образом ведет
себя и хлорное железо. Растворимость его77 в TiCl4 равна 0,003%
при 40° и 0,043% при 110°.
Основное количество образующегося MgCl2 (около 80%) извле-
кается из печного газа при очистке его от пыли и частично (около
15%) выходит из печи с плавом. Остальное количество MgCl2 отде-
ляется с осадком при фильтрации жидкого четыреххлористого ти-
тана.
Для отделения хлоридов Fe2+, Мп2+ и Mg2+, плавящихся при
712—650°, предложено 78 охлаждать газы до 200—300° в верхней
части печи, орошаемой суспензией твердого инертного материала
(например, песка) в жидком четыреххлористом титане. Инертный
материал вместе с конденсировавшимися на его частицах хлори-
дами железа, марганца и алюминия выгружают из зоны охлажде-
ния, откуда также отводятся очищенные пары TiCl4.
Хлористый кальций в основном остается в печи и выходит вме-
сте со шлаком. Возможно, что расплавленный шлак абсорбирует
немного T1CI4, который отгоняется при продувке шлака возду-
хом 79.
Соединения ванадия переходят в технический хлорид титана и
отделяются лишь при очистке продукта.
Хлорирование титанистого сырья осуществляют в шахтных элек-
трических печах периодического действия, а также в хлораторах
непрерывного действия с применением в качестве рабочей среды
расплава хлоридов. Хлорирование титанистого сырья возможно
также осуществить в печи со взвешенным слоем 80-82.
Электрическая печь (рис. 448) состоит из стального кожуха, фу-
терованного изнутри плотным шамотным или динасовым кирпичом.
1490
Гл. XL. Продукты высокотемпературного хлорирования руд
В нижней части печи помещаются два ряда (верхний и нижний)
угольных электродов, по три электрода в каждом ряду. Про-
странство между рядами электродов заполняют насадкой из уголь-
ных цилиндров, служащей электросопротивлением, с помощью
которого в печи поддержи-
вается необходимая темпе-
ратура.
Брикеты шихты загружа-
ют в печь периодически, так
чтобы было заполнено про-
странство' над угольной на-
садкой. Хлор подают в печь
по фурмам, расположенным
под углом несколько выше
нижнего ряда электродов.
Газообразные продукты хло-
рирования вместе с непроре-
агировавшим (избыточным)
хлором выходят из верхней
части печи и направляются
в систему пылеочистки и для
• дальнейшей переработки.
Вместо брикетов можно за-
гружать в печь гранулы,
приготовленные83 с приме-
нением в качестве связки
бентонита и обожженные
при 1000—1100°.
Предложено получать
TiCU хлорированием порош-
кообразного сырья, содер-
жащего окислы титана, в
потоке хлора и окиси угле-
рода 84. Отмечается благо-
приятное действие примеси
СО к хлору при хлорирова-
нии в электрических печах85.
Заслуживает внимания по-
Рис. 448. Схема электрической шахтной
печи для хлорирования титанистого сырья;
I —кожух из листовой стали; 2 —футеровка;
5 —угольный электрод; 4 — загрузочное устройство;
5— труба для выхода газов; 6 — фурма для подачи
хлора.
стадийное осуществление процесса хлорирования в двух или не-
скольких аппаратах86. В первом аппарате проводят хлорирование
при пониженной температуре с образованием легко летучих хлори-
дов и хвостовых газов. После отделения хлоридов газы направляют
во второй аппарат.
В СССР помимо хлорирования брикетов из титановых шлаков
и кокса в шахтных электрических печах периодического действия,
четыреххлористый титан получают также хлорированием измель-
Четыреххлористый титан
1491
ченных титановых шлаков и кокса, взвешенных в расплаве хлори-
дов, в хлораторах непрерывного действия.
Этот способ является весьма перспективным вследствие воз-
можности осуществления процесса в высокоинтенсивных аппара-
тах непрерывного действия и уменьшения капитальных и экс-
плуатационных расходов. В качестве реакционной среды приме-
няют расплавленный карналлит 87, а также смесь хлоридов калия
и натрия или чистые хлориды, например, расплав NaCl88 для хло-
рирования смеси ТЮ2 и древесного угля при 900°. При содержании
в расплаве ~2% хлорного железа интенсифицируется массопере-
нос хлора к поверхности частиц двуокиси титана. Установлено89,
что количество хлора, транспортируемого растворенным хлорным
железом от поверхности пузырька к твердой хлорируемой поверх-
ности, примерно в 100 раз больше количества растворенного хлора,
транспортируемого через расплав. Аналогично действует также до-
бавка в расплав хлористого алюминия90. При температурах выше
750° скорость процесса хлорирования тормозится массопередачей
реагирующих веществ в расплаве, окружающем пузырек хлора и
твердые частицы ТЮ2 и кокса91. Процесс может быть осуществлен
в барботерах, снабженных механическими мешалками, аппаратах
газлифтного типа и других, в которых не происходит осаждения
твердых частиц суспензии.
Рекомендуют 92 хлорирование шлаков в расплавленных хлори-
дах проводить при пониженных температурах (650—750°) для уве-
личения накопления хлоридов железа в расплаве и уменьшения
количества возгонов твердых хлоридов, затрудняющих работу ап-
паратов конденсации. Максимальная скорость хлорирования дости-
гается при концентрации в суспензии 5—6% нефтяного кокса и
10—12% двуокиси титана (при средних размерах их частиц
~25 мк). При этом следует учитывать общую суммарную поверх-
ность твердых частиц в расплаве, которая не должна превышать
280 000 см^л расплава.
Состав рабочего расплава по количеству хлоридов калия и на-
трия необходимо регулировать так, чтобы при соответствующей
температуре вязкость его не превышала 3,5 сп.
Недостатком способа хлорирования в расплавах хлоридов яв-
ляются повышенные потери титана с возгонами, идущими в отвал.
Из них 30—40% уносится в виде шихты из хлоратора и 10—12%
приходится на долю окси- и гидрооксихлоридов титана, образую-
щихся при взаимодействии TiCl4 с кислородом и парами воды при
высокой температуре93. Уменьшения потерь можно достигнуть при
использовании шихты более крупного помола, полной герметиза-
ции конденсационной системы и тщательной осушке сырья. Пред-
ставляет также интерес дохлорирование возгонов на угольной на-
садке 94, разработанное применительно к получению хлоридов нио-
бия.
1492
Гл. XL. Продукты высокотемпературного хлорирования руд
Жидкий четыреххлористый титан, полученный при охлаждении
продуктов хлорирования, содержит много примесей в виде твердых
взвешенных частиц и растворимых веществ.
С целью предотвращения осаждения хлорного железа и других
примесей на теплопередающих поверхностях при охлаждении ре-
комендуют 95 образующуюся суспензию взвесей в четыреххлори-
стом титане пропускать через охлаждающие устройства со ско-
ростью 2—5 м]сек.
Из продуктов хлорирования титанистых шлаков в расплаве
NaCl или отработанного магниевого электролита выделяются при
их охлаждении в системе конденсации плавкие возгоны, затруд-
няющие отделение примесей. Установлено96, что такие возгоны по-
являются при уменьшении в расплаве концентрации TiOs ниже
0,8% и интенсивном испарении из расплава комплексных хлори-
дов (К, Na)FeCl4 и (К, Na)AlCU, которые конденсируются в си-
стеме охлаждения отходящих газов хлоратора.
Хлориды железа и алюминия отделяются из жидкого Т1С14 вме-
сте с другими твердыми примесями при отстаивании или при филь-
тровании конденсата через пористые керамические фильтры или
активированный уголь. Содержание железа в хлориде титана после
фильтрации составляет не более 0,02%, а другие нерастворимые
примеси практически отсутствуют 97.
Из отделенных примесей можно выделить FeCl3 путем расплав-
ления их при 350—450° под давлением, достаточным для предо-
твращения испарения хлорида98. После отстаивания расплава он
расслаивается на три части — верхнюю, обогащенную углеродисты-
ми частицами, нижнюю — частицами двуокиси титана и среднюю,
состоящую в основном из хлорного железа. Нижнюю и верхнюю
части выводят из зоны плавления и возвращают на хлорирование.
Среднюю часть отводят в качестве продукта или окисляют до РегОз
в присутствии кислорода.
При наличии в продукционных газах хлористого железа его мо-
жно выделить99 охлаждением до 500—550° в виде сухих стабиль-
ных кристаллов FeCh. Их отделяют от газового потока в циклон-
ном сепараторе. Из оставшейся газовой смеси конденсируют Т1С14.
Отфильтрованный от твердых примесей конденсат — технический
четыреххлористый титан имеет красновато-желтый цвет и содержит
около 97—99% TiCl4, 1,5—2,5% SiCl4, 0,02—0,002% Fe, 0,1—0,3%
Cl и 0,06—0,1% V. Хлор ограниченно растворим в Т1С14 10°, a S1C14
образует с Т1С14 непрерывный ряд жидких смесей (рис. 449).
Очистка технического продукта для получения TiCl4, пригодного
для производства ковкого титана, производится при помощи меди
с последующей дистилляцией образовавшихся продуктов. При вза-
имодействии с порошком меди хлор, растворенный в Т1С14, связы-
вается в хлорид ме'ди, а хлориды железа и ванадия, а также хлор-
окись ванадия восстанавливаются до низших хлоридов, имеющих
Четыреххлористый титан
1493
более высокую температуру кипения. (Температура кипения хлор-
окиси ванадия незначительно отличается от температуры кипения
четыреххлористого титана (127 и 136°), поэтому отделение VOC13
от TiCU физическими методами затруднительно.)
После обработки медью четыреххлористый титан для удаления
примесей подвергают двухступенчатой ректификации. Вначале от-
деляют наиболее низкокипящий компонент — четыреххлористый
кремний, а хлорид титана накапливается в кубовом остатке. При
повторной ректификации в дистилляте получается чистый четырех-
хлористый титан с содержанием
98,5—99% TiCl4, а в кубовом остат-
ке — примеси.
Предложено много способов об-
работки технического продукта, при-
готовления активной меди и очистки
TiCU от примесей. Так, обработку
конденсата можно осуществить про-
пусканием его в жидком или паро-
образном виде через колонну, за-
полненную медной стружкой, или
путем размешивания его с порош-
ком меди, полученным восстановле-
нием окиси меди 101’ 102.
Из образующейся пульпы отго-
няют TiCl4 и выделяют ванадий и
Рис. 449. Диаграмма температур
кипения в системе TiCl4—SiCl4.
медь разными методами. Например103, обработкой щелочью для
перевода в раствор ванадия и затем кислотой для извлечения ме-
ди. При пропускании паров TiCU через колонну с насадкой из из-
мельченной меди соединения ванадия полностью задерживаются на
меди 104’105. Для их извлечения и регенерации меди колонну мож-
но периодически промывать разбавленной кислотой 104. Для увели-
чения активности металлической меди рекомендуется 106 обрабаты-
вать ее летучими спиртами, альдегидами или кетонами с последую-
щим испарением этих веществ под вакуумом. Активированную
таким образом медь необходимо до употребления предохранить от
соприкосновения с воздухом.
Разрушение хлорокиси ванадия можно, по-видимому, осуще-
ствить и при добавке 0,15—0,3% иода или иодидов 107-108 или при
помощи сероводорода 10Э.
Предложены также методы очистки технического четыреххлори-
стого титана, основанные на адсорбции примесей твердыми погло-
тителями 6. В качестве адсорбентов можно применять ламповую или
газовую сажу, или древесный уголь. Четыреххлористый титан осо-
бо высокой чистоты, содержащий 4-10'®% примесей, можно полу-
чать при использовании трехслойного адсорбционного фильтра, со-
стоящего из угля, алюмогеля и силикагеля ио.
1494 Гл. XL. Продукты высокотемпературного хлорирования руд
При пропускании паров четыреххлористого титана через насад,
ку из углеродсодержащих реагентов одновременно с очисткой от
ванадия протекает также очистка TiCU от хлора и кислорода111.
Четыреххлористый титан высокой чистоты с содержанием
99,999 мол. % TiCh 112 может быть получен при перегонке очищен-
ного продукта под разрежением. Возможна также очистка дистил-
ляцией TiCl4 в присутствии небольшого количества минеральных
масел (0,1—0,3 вес.%) с применением отдувки инертным газом113.
Предложено очистку технического TiCl4, содержащего до 1 % VOC13
и 0,5—3% TiCl3 (в суспендированном виде), производить с приме-
нением вазелинового масла114 в количестве 0,2—0,3 вес. ч. на 1 вес.
ч. VOCI3 в исходном материале. Смесь нагревают при перемеши-
вании и выдерживают 5—10 мин при 120—150°. Затем из нее отго-
няют TiCl4. В получаемом продукте содержится меньше 0,01 вес. %
VOCI3. При отсутствии в техническом TiCh примеси Т1С13 послед-
ний добавляют вместе с вазелиновым маслом в количестве 1,5—
2 вес. ч. на 1 вес. ч. VOC13.
Получение четыреххлористого титана из других видов сырья
Высокая температура хлорирования рутилового концентрата и
титанистых шлаков, а также необходимость предварительной обра-
ботки ильменита являются причинами изыскания других методов
получения четыреххлористого титана с применением других видов
сырья. К ним относятся: хлорирование карбида титана при 300—
400°73, хлорирование низших окислов титана при 200—500°60, хло-
рирование сульфидов титана при 200—250°61 и непосредственное
хлорирование ильменитового концентрата115-118.
Предложено много способов непосредственного хлорирования
ильменита при условиях, обеспечивающих связывание железа в не-
летучие или мало летучие соединения путем добавки к шихте 3—
5% Na2CO3119 или обработки шихты, содержащей кальций и маг-
ний, серной либо фосфорной кислотой *20’ 121 или добавки к шихте
фосфата титана *22. Возможно также выделение Т1С14 из его смеси
с FeCl3 обработкой ее перегретыми до 150—300° парами четырех-
хлористого титана 123 в количестве, необходимом для полного испа-
рения Т1С14, содержащегося в смеси. Отогнанные пары хлорида ти-
тана затем конденсируют. Из газовой смеси с небольшим содержа-
нием TiCl4 (до 10%) последний может быть извлечен адсорбцией124
на активированном угле при 20°. Десорбция TiCl4 из угля произ-
водится при помощи горячего инертного газа.
Помимо сложности разделения хлоридов железа и титана, не-
посредственное хлорирование ильменита связано также с разреше-
нием вопроса использования значительных количеств образующе-
гося хлорного железа.
Четыреххлористый кремний
1495
ТРЕХХЛОРИСТЫЙ ТИТАН
Треххлористый титан образуется при восстановлении четырех-
хлористого титана водородом на горячей поверхности при 1000° 125.
В полученном продукте содержится 95—98% TiCls 126’127. Значи-
тельно быстрее (почти мгновенно) протекает реакция при пропу-
скании водорода, насыщенного парами TiCh, через расплавленную
смесь хлоридов NaCl и КС1 или MgCl2 и CaClg. Расплав, содержа-
щий низшие хлориды титана, легко затвердевает. Его сохраняют в
инертной атмосфере.
Треххлористый титан можно также получить взаимодействием
смеси 128 из двуокиси титана или ильменита и угля с газообразным
TiCU при 800°. Образующуюся смесь Т1С1з, СО и непрореагировав-
шего Т1С14 быстро охлаждают до температуры ниже 650°, и отде-
ляют твердый треххлористый титан. Охлаждение следует произво-
дить быстро, во избежание взаимодействия между непрореаги-
ровавшим газообразным четыреххлористым титаном и окисью
углерода 129.
Предложено 130 получать кристаллический фиолетовый треххло-
ристый титан восстановлением четырххлористого металлическим
алюминием в присутствии А1С13 или НС1, являющихся катализато-
рами реакции. Процесс проводят в обогреваемой шаровой мель-
нице, вращающейся со скоростью 75 об/мин, при 150—250° в тече-
ние 5—10 ч. Треххлористый титан получают также восстановле-
нием Т1С14 кремнием.
Взаимодействием паров TiCU с порошком металлического алю-
миния при 450—650° получают двуххлористый титан. Предложе-
но 131 осуществлять процесс в псевдоожиженном слое, создаваемом
смесью паров четыреххлористого титана с инертным газом.
Треххлористый титан можно очистить от примесей TiCh и те-
трагалогенидов других металлов 132 путем его промывки эфирами
алифатического или ароматического ряда при 20°. После удаления
адсорбированного растворителя сушкой при 40° и отгонки инерт-
ным газом получается продукт, содержащий 0,5% TiCl4 и обладаю-
щий высокой активностью в качестве составной части катализа-
тора синтеза полипропилена.
ЧЕТЫРЕХХЛОРИСТЫИ КРЕМНИИ
Физико-химические свойства
Четыреххлористый кремний — простейшее соединение кремния
с хлором. Существуют также многочисленные галогениды общей
формулы Si„X2n+2, в которых атомы кремния связаны друг с дру-
гом в виде цепей. При максимальной длине цепи, содержащей
25 атомов кремния, S125CI52 имеет вид бесцветной пластической
массы. Известны также смешанные галогениды SiClsBr, SiClsF
и др.
23 М. Е. Позин
1496 Гл. XL. Продукты высокотемпературного хлорирования руд
Рис. 450. Давление пара
над жидким SiCl4.
Четыреххлористый кремний или тетрахлормоносилан при обыч-
ных условиях представляет собой бесцветную жидкость плотностью
1,487 г/см3 (при 20°), кипящую при 57° и замерзающую при —67°.
Давление пара над жидким SiCl4 при 20° равно 195,86 мм рт. ст.
(рис. 450) и в зависимости от температуры может быть подсчитано
по формуле lgP = 7,644—1572,IT*1.
Во влажном воздухе четыреххлористый кремний дымит вслед-
ствие гидролиза и образования хлористого водорода. С газообраз-
ным аммиаком дает очень густой дым. С
водой S1CI4 бурно реагирует с выделением
большого количества тепла и образованием
бесцветного студенистого осадка ,33. С окис-
лами многих металлов образует соответ-
ствующие хлориды.
При взаимодействии с водородом (и дру-
гими восстановителями) SiCU образует три-
хлорсилан HS1CI3 и другие хлорзамещен-
ные силана, имеющие большое значение в
органическом синтезе.
Применение
Четыреххлористый кремний является ис-
ходным материалом при синтезе кремний-
органических соединений, используемых
для получения диэлектриков, лакокрасоч-
ных жаростойких покрытий, смазочных ма-
териалов, уплотнительных материалов, гидрофобизирующих
средств для защиты от влаги различных изделий и т. д.134. Среди
кремнийорганических соединений известны кремнийорганические
смолы, кремнийорганический каучук, широко применяемый для
получения теплостойкой резиновой изоляции проводов, теплостой-
ких прокладок и др. 135~138. Четыреххлористый кремний используют
в качестве средства для создания дымовых завес. Он служит
для получения аэросила — безводной высокодисперсной двуокиси
кремния, используемой в качестве наполнителя в производстве
термостойких резин на основе силиконового каучука. При ги-<
дролизе SiCh в пламени водорода при 750—1000° образуется139’140
весьма однородная двуокись кремния с размерами частиц от 10 до
40 ммк. В зависимости от режима гидролиза можно получать крем-
незем с удельной поверхностью от 50 до 450 лс2/г.
Наполнители типа аэросила отличаются от кремнезема, полу-
ченного методом осаждения, однородностью структуры, отсутствием
внутренних пор, низкой концентрацией поверхностных гидроксилов.
Это обеспечивает лучшее совмещение наполнителя с молекулами
органических полимеров.
Четыреххлористый кремний
1497
SiCU употребляют для получения чистого кремния-полупровод-
ника •— восстановлением его парами цинка при высокой темпера-
туре. Образование элементарного кремния возможно также путем
диспропорционирования 141>142 субхлорида, полученного восстанов-
лением SiCl4 водородом:
S iCl 4 + Н2 = SiC 12 + 2НС1
2SiCl2 Si + SiCl4
Согласно ГОСТ 8767—58, четыреххлористый кремний, получае-
мый хлорированием металлического кремния и ферросилиция, дол-
жен представлять собой прозрачную бесцветную или желтоватую
жидкость плотностью 1,48—1,50 г)см3 (20°). Максимально допусти-
мое содержание в нем железа равно 0,001%- Четыреххлористый
кремний должен иметь следующий фракционный состав, опреде-
ляемый при барометрическом давлении 760 мм рт. ст.‘. темпера-
тура начала перегонки не менее 55°, температура конца перегонки
не более 59°, остаток после перегонки не более 2,5%. Четыреххло-
ристый кремний транспортируют в стальных цистернах и в сталь-
ных бочках. Цистерны снабжены сифонами и защитными зонтами
от солнечных лучей, а бочки — пробками с колпаками. При транс-
портировании SiCU в цистернах допускается наличие в нем легкой
мути.
Получение четыреххлористого кремния
При производстве четыреххлористого титана хлорированием ти-
танистого сырья входящие в его состав соединения кремния пре-
вращаются почти на 50% в четыреххлористый кремний. Образую-
щиеся пары SiCl4 конденсируются вместе с TiCl4; после очистки
Т1С14 от других примесей отделение SiCl4 производят ректифика-
цией. Четыреххлористый кремний получается в виде дистиллята,
загрязненного небольшим количеством TiCl4. После дополнитель-
ной дистилляции получают чистый продукт.
Аналогичным образом получают SiCl4 как побочный продукт
при переработке хлорированием титано-ниобиево-танталового
сырья6. Из отходящих газов производства хлористого алюминия
SiCl4 получают абсорбцией керосином при —15° с последующей от-
гонкой *43.
К старым способам получения SiCl4 относятся хлорирование
кремния, обработка кремнезема треххлористым бором, а также
хлорирование ферросилиция, прокаливание карборунда в токе хло-
ра при 1000—1200°, обработка смеси кремнезема и угля полухлори-
стой серой или фосгеном 1441145. Получение четыреххлористого крем-
ния из элементарного кремния наиболее просто. Взаимодействие
кремния с хлором с образованием SiCl4 протекает ниже 1000°.
23*
1498
Гл. XL. Продукты высокотемпературного хлорирования руд
Выше 1000° отношение Cl: Si уменьшается146 и составляет при
1400° ~2,1 вследствие реакции:
SiCl4 + Si 2SiCl2
В присутствии катализатора из активированного металла (на-
пример, меди) синтез SiCh может быть осуществлен 147 при темпе-
ратуре ниже 150°. Это позволяет почти полностью исключить хло-
рирование примесей, содержащихся в кремнии. Вместо активиро-
ванной меди можно применить добавку к кремнию порошка
меди 148 с предварительной обработкой смеси водородом при 250°.
В присутствии катализаторов возможно не только снижение тем-
пературы, но и изменение направления хлорирования. Так, в при-
сутствии хлорида аммония или хлоридов щелочных149 или ще-
лочноземельных металлов150 взаимодействие кремния, а также
ферросилиция с хлором при 150—250° приводит к образованию
гексахлорида кремния Si2C16.
Карбид кремния (карборунд) реагирует с хлором 151 при 700— :
950°: j
SiC+ 2С12 = SiCl4 + С
Для очистки реактора от накапливающегося элементарного уг- :
лерода в зону реакции вместо хлора периодически вводят воздух, '
который частично окисляет углерод до окиси и двуокиси углерода
и частично механически уносит мелкодисперсную сажу.
Наибольшее количество четыреххлористого кремния получают
из ферросилиция, содержащего 70—90% Si. Он начинает реагиро-
вать с хлором ниже 300°. Оптимальная температура хлорирования
550—600°152. Выход SiCl4 составляет 90—95%. При пониженных
температурах увеличивается выход побочных продуктов — гекса-
хлордисилана и других хлоропроизводных, например Si2Cle, Si3Cl8.
При очень тонком измельчении ферросилиция (а также элемен-
тарного кремния) и быстром потоке хлора с отводом продуктов ре-
акции образование SiCK с выходом 96% протекает при обычной
температуре 163. Представляется перспективным освоение производ-
ства четыреххлористого кремния из более дешевого, чем ферроси-
лиций, кремнеземистого сырья, в частности диатомита, содержа-
щего небольшое количество Fe2Os, что облегчит очистку продукта
от FeCl3. Вследствие пористой структуры и активности находяще-
гося в нем кремнезема диатомит хлорируется 154 в присутствии угля
при пониженной температуре 730—750°. Выход SiCl4 составляет
45—50% при хлорировании брикетов из диатомита и угля, приго-
товленных с применением сульфитного щелока в качестве связую-
щего 154. При осуществлении процесса в среде расплавленных солей
(эквимолекулярной смеси хлоридов натрия и калия) создается хо-
роший контакт между измельченной шихтой из диатомита и угля
и хлором. В этих условиях степень перехода SiO2 из диатомита в
SiCU составляет при 750° 95—97% 155.
Четыреххлористый кремний
1499
Заслуживает внимания получение четыреххлористого кремния
хлорированием мелкозернистых силикатов или кварца в электриче-
ской печи кипящего слоя в присутствии избытка углерода 156. Рас-
ход электроэнергии на 1 г SiCU составляет 1100—1300 квт-ч.
Четыреххлористый кремний образуется в качестве побочного
продукта при хлорировании руд редких металлов 157 и др. При хло-
рировании циркона в присутствии кокса в псевдоожиженном слое
при 900—1400° процесс значительно интенсифицируется при добав-
ке 0,5—2% КС1 (по отношению к весу хлорируемого материала) ,58.
Степень использования газообразного хлора в присутствии добавки
КС1 увеличивается на 7—10% и достигает ~98%. Образующиеся
продукты хлорирования ZrCi4 и SiCl4 раздельно конденсируют и
очищают.
Представляет интерес выделение SiCl4 из газов хлорирования
каолина с применением в качестве абсорбента монохлорбензола
или парафинового масла 159. Поглощенный SiCl4 выделяется путем
дистилляции и последующей ректификацией получают продукт вы-
сокой чистоты.
Очистка сырого четыреххлористого кремния осуществляется в
основном ректификацией и мало чем отличается от очистки сырого
Т1С14, за исключением химической обработки для удаления соеди-
нений ванадия, которая в этом случае зачастую не нужна, особенно
при получении SiCl4 из элементарного кремния или диатомита. При-
месь PCls можно отделить добавкой А1С13; образующийся ком-
плекс А1С13• РС15 отделяют от SiCl4 дистилляцией 16°.
Вместо А1С13 рекомендуют 161 применять в качестве адсорбента
А12О3 с покрытием из PtCl4, образующей с РС13 хелатное соеди-
нение.
Очистка SiCl4 от следов хлорида бора может быть произведена
с помощью металлического алюминия 162. Процесс основан на вза-
имодействии при 200—260° хлорида бора с алюминием (в отличие
от S1C14) с образованием чистого бора и А1С13, который уходит с
парами. Последовательной конденсацией паро-газовой смеси полу-
чают чистые SiCl4 и А1С13.
Предложено производить очистку сырого четыреххлористого
кремния от примеси TiCl4 при помощи 95 %-ной серной кислоты или
смеси ее с хлорсульфоновой кислотой или фосфорной кислотой 163.
Эти кислоты образуют нелетучие соединения с TiCl4, хорошо рас-
творимые в кислоте и не растворимые в хлориде кремния. При об-
работке кислотой жидкого SiCl4 через некоторое время после тща-
тельного перемешивания жидкостей отделяется слой очищенного
хлорида кремния. Более предпочтительной является обработка га-
зообразного SiCl4, при которой одновременно отделяются и более
высококипящие силоксаны, конденсирующиеся и собирающиеся на
поверхности кислоты.
1500
Гл. XL. Продукты высокотемпературного хлорирования руд
БЕЗВОДНЫЙ ХЛОРИСТЫЙ АЛЮМИНИЙ
Физико-химические свойства
Хлористый алюминий А1С13 — белый кристаллический порошок
9 плотностью 2,47 г!смг возгоняется при 182,7°, под давлением
2,5 ат плавится при 192,4°.
Давление паров А1С13 равно 760,0 мм рт. ст. при 180,2° и
2277,5 мм рт. ст. при 213°.
ТАБЛИЦА 117
Давление паров хлоридов алюминия и железа
А1С13 [1пл= 192.4° (под давлением), <в03г = 182.7°] FeC13 = 303°. fK = 319°)
t, °C Р, мм рт. ст. t, °C P, мм рт. CT.
100 1 194 1
116,4 5 222,8 2
123,9 10 235,5 10
131,8 20 246 20
139,9 40 256,8 40
145,4 ' 60 363 60
152,0 100 272,5 100
161,8 200 285 200
171,6 400 298 400
180,2 760 319 760
В табл. 117 приведены значения давления паров А1С1з и FeCl3
при разных температурах, а в табл. 118 — состав и давление паров
в системе FeCl3—AICI3.
ТАБЛИЦА Н8
Состав и давление паров в системе FeCl3—А1С1з
Температура, °C Состав плава, вес, % Состав паров, вес. % Давление паров, мм рт. ст.
А1С1з FeClg А1С1з РеС1з А1С1з FeCl3 общее
150 75 25 97,1 2,9 54,0 1,3 55,3
183 75 25 97,2 2,8 370,7 8,8 379,5
Растворимость А1С13 в 100 г воды при 20° равна 46 г, в горячей
воде разлагается. Хорошо растворяется во многих органических
растворителях. Из водного раствора кристаллизуется АЮз-бНгО
с плотностью 2,4 г! см3. расплывающийся на воздухе. При нагрева-
нии отщепляет воду и НС1 с образованием А12О3.
В воде хлористый алюминий гидролизуется с образованием ос-
новных хлоридов алюминия. Предполагают 164, что они отвечают
Безводный хлористый алюминий
1501
общей формуле А1С13-пА1 (ОН)3. Вероятно, что при взаимодействии
А1С13 с водой образуются также комплексные кислоты
Нз[А1С13(ОН)з] и Нз[А1С12(ОН)4].
С газообразным аммиаком хлористый алюминий образует ам-
миакаты: A1C13-6NH3, частично разлагающийся при 180°, и А1С13-
• NH3, стойкий до 400°. Хлористый алюминий образует соединения
и со многими другими неорганическими и органическими вещества-
ми. С галогенидами одновалентных металлов хлористый алюминий
образует комплексные соединения типа М[А1С14]. Этим обусловли-
вается его каталитическая активность. В присутствии А1С13 повы-
шается давление пара других хлоридов. Из расплава NaCl—AICI3,
содержащего около 50 мол.% А1С1з, выше 550° отгоняются значи-
тельные количества NaCl, возможно вследствие образования лету-
чего соединения NaAICU 165. При прокаливании хлористого алюми-
ния в токе воздуха уже при 400° образуются окись алюминия и
хлор 166.
Помимо треххлористого алюминия известен монохлорид алюми-
ния А1С1, образующийся при взаимодействии металлического алю-
миния с газообразным хлористым водородом выше 1100° при давле-
нии 10 мм рт. ст. При 1020° получается продукт состава А1С1г,2з167.
Монохлорид алюминия образуется также при действии пара А1С13
на алюминий при высоких температурах I6S. Исследуется получе-
ние металлического алюминия высокой чистоты169 разложением
А1С1 при 700—800°.
Применение
Хлористый алюминий применяют, главным образом, в качестве
катализатора при крекинге нефтепродуктов, а также для ряда ор-
ганических синтезов 17°.
Он обладает также полимеризующими свойствами. Это имеет
большое значение для производства смазочных масел и моторного
топлива, синтетического каучука и других полимеров. Гидролизом
А1С13 в паровой фазе получают тонкодисперсную окись алюми-
ния 1П.
Технический безводный хлористый алюминий выпускают двух
сортов. Согласно ГОСТ 4452—66, продукт должен иметь белый или
слабо-желтый цвет и содержать в 1 и 2 сорте соответственно: не ме-
нее 99,0 и 98,5% А1С13 и не более 0,05 и 0,15% железа (в пересчете
на FeCl3) и 0,5 и 0,8% титана (в пересчете на Т1С14). Частицы хло-
ристого алюминия обоих сортов должны быть не больше 5 мм.
Получение безводного хлористого алюминия
Вследствие гидролиза водных растворов А1С13 и разложения его
при высоких температурах получение безводного А1С13 из раство-
ров или шестиводного хлорида алюминия весьма затруднено.
1502
Гл. XL. Продукты высокотемпературного хлорирования руд
Поэтому основным методом получения безводного А1С13 является!
хлорирование материалов, содержащих алюминий 172. |
Металлический алюминий — дорогой вид сырья, и он приме-я
няется для производства хлористого алюминия действием хлор а173 4
или сухого хлористого водорода лишь в ограниченных количествах,
главным образом в лабораторных условиях. Изучено 174 хлорирова-
ние алюминиевого порошка газообразным хлором в расплаве, со-
держащем FeCl3. Обычным же сырьем служат окись алюминия,
соединения, содержащие глинозем, бокситы и алюмосиликаты, на-
пример лейциты, каолин и глина. Чаще всего используют глинозем
и каолин и их смеси 1SS.
Получение безводного хлористого алюминия из материалов, со-4
держащих глинозем, основано на реакции хлорирования окисий
алюминия в присутствии углерода как восстановителя: ?
А12О3 + ЗС + ЗС12 = 2А1С13 + ЗСО
2А12О3 + ЗС + 6С12 = 4 А1С13 + ЗСО2
Тепла, выделяющегося по второй реакции, достаточно для обес-'
печения автотермичности процесса 175.
Окись алюминия в виде брикетов с коксом, приготовленных на
смоле, практически полностью хлорируется при 650—800° в течение
40—60 мин при полном использовании хлора. В полученном про-
дукте содержится до 98—99% А1С13 (остальное — непрореагиро-
вавший А12О3). В присутствии небольших количеств SiO2 хлориро-1
вание смеси А12О3 + С ускоряется 176. ?
Процесс можно осуществить взаимодействием хлора и окиси уг-“',
лерода с порошкообразной окисью алюминия в присутствии хлори-1
дов щелочного металла и алюминия177. Отношение хлоридов к";
А12О3 поддерживают равным 1:1. Хлорирование смеси окиси алю-
миния с углем в аппарате со взвешенным слоем 178 позволяет ис-4
ключить операцию брикетирования и осуществить процесс непре-
рывным путем. Для хлорирования можно использовать, помимо
хлора, фосген 17S’ 180. Для уменьшения уноса с газами тонкодис-
персной окиси алюминия рекомендуют 181> 182 применять глинозем в
виде гранул с размерами частиц 0,5—1 мм.
При хлорировании брикетов из боксита, каолина или глины,
помимо А1С13, образуются также другие хлориды вследствие вза-
имодействия с хлором примесей Ре20з, SiO2, TiO2 и др. Описано
производство хлористого алюминия из боксита с низким содержа-
нием Si и Fe 182. Боксит вначале прокаливают при 950—1000° во
вращающейся печи для удаления влаги. К прокаленному, измель-
ченному бокситу добавляют равное количество кокса, расплавлен-
ный асфальт или другое связующее и приготавливают брикеты,
которые подогревают в шахтной печи горячим газом до 800° для
удаления углеводородов и влаги, а затем хлорируют в течение
8—10 ч при 850°. Для получения продукта, содержащего 94—95 %
Безводный хлористый алюминий
1503
А1С13, следует применять боксит с высоким содержанием AI2O3
(55—60%) и низким содержанием SiOs (менее 5%) и Fe2O3 (ме-
нее 3%).
Газообразный продукт хлорирования улавливается в стальных
цилиндрических конденсаторах вертикального типа. Внутри имеют-
ся мешалки, сбрасывающие в бункеры оседающий на стенках гото-
вый продукт.
Большим недостатком этого способа является сложность очист-
ки полученного продукта от примесей других хлоридов. Предло-
женный способ осуществления процесса под разрежением 700—
750 мм рт. ст. при высокой температуре (1000—1510°) с целью
разложения образующихся примесей хлоридов 183 требует проверки
и представляется сложным в технологическом отношении.
При хлорировании каолина помимо А12О3 хлорируется также и
SiO2. Степень использования хлора на хлорирование А12О3 из као-
лина при 550—800° в среднем составляет 45—50% 184- Остальное
количество хлора расходуется на хлорирование примесей. Ниже
900° скорость хлорирования А12О3 в каолине больше скорости хло-
рирования SiO2185. В присутствии фосгена выход А1С13 возрастает
с повышением температуры до 1000° 186’187. По мере протекания
процесса во времени скорость хлорирования А12О3 при температу-
рах ниже 1000° уменьшается быстрее, чем скорость хлорирования
SiO2, вследствие чего непрерывно возрастает отношение прореаги-
ровавших SiO2 и А12О3 184’188. При 1000° и выше скорость хлориро-
вания SiO2 и А12О3 по времени снижается в одинаковой сте-
пени и отношение прохлорированных SiO2 и А12О3 остается по-
стоянным.
Влияние температуры на скорость хлорирования каолина и глин
и на степень использования хлора для образования А1С13 связано
с фазовыми превращениями, происходящими при нагревании као-
лина и образованием модификаций А12О3 и SiO2, обладающих раз-
личной реакционной способностью 188. При нагревании каолинита
он вначале переходит в метакаолинит или каолинитовый ангидрид
2SiO2-Al2O3, который при 970° превращается в силлиманит
SiO2 • А12О3 189-195 (стр. 639). Силлиманит представляет собой соеди-
нение с более упорядоченной кристаллической структурой по срав-
нению с каолинитовым ангидридом. Этим объясняется понижение
скорости хлорирования каолина в пределах 950—1000° 185> 187> 196.
При более высоких температурах скорость хлорирования вновь
возрастает и при 1200° можно получить хороший выход А1С13IS7.
При предварительной обработке прокаленного каолина соляной
кислотой условия хлорирования улучшаются. Степень использова-
ния хлора на образование А1С13 возрастает до 70—80%. Соответ-
ственно снижается доля от общего количества хлора, расходуемая
на образование четыреххлористого кремния175. Наиболее рацио-
нальным является хлорирование шихты из каолина и глинозема155.
1504
Гл. XL. Продукты высокотемпературного хлорирования руд
При подборе рационального состава шихты удается достичь мак-
симального использования хлора и А12О3 из каолина.
Технический хлористый алюминий содержит примеси SiCl4,
TiCl4, а также FeClj. Четыреххлористые кремний и титан легко
удаляются, так как они кипят при температурах, намного ниже
температуры возгонки хлористого алюминия. Основные затрудне-
ния по очистке хлористого алюминия связаны с удалением хлор-
ного железа. Большинство предложенных методов основано на вос-
становлении хлорного железа до металлического железа путем на-
гревания с другим металлом, имеющим большее сродство к хлору,
чем железо. Чаще всего для этой цели применяют возгонку сырого
продукта над алюминиевыми стружками в алюминиевом со-
суде 170’ 198.
Технический хлористый алюминий окрашен в желтый цвет
вследствие содержания в нем хлорного железа в количестве до 2—
3%. Наряду с хлорным железом он содержит окислы и оксихло-
риды железа и алюминия, образующиеся при частичном гидролизе
этих солей на воздухе. '
Химически чистый безводный хлористый алюминий можно по-
лучить:
1. Действием хлора или хлористого водорода на металлический
алюминий при 400—500° 199-201.
2. Восстановлением хлорного железа, содержащегося в техни-
ческом хлористом алюминии в железо при нагревании с алюминие-
выми стружками или в хлористое железо при нагревании с желез-
ными стружками в запаянных трубках при 200—250° 202. Получен-
ный продукт подвергают возгонке.
3. Нагреванием хлористого алюминия с алюминиевым порош-
ком в расплаве с 4—5% NaCl при нормальном давлении 203’204 с
последующей сублимацией очищенного хлористого алюминия. Од-
нако при атмосферном давлении разделение хлоридов железа и
алюминия затруднительно. Процесс значительно упрощается, если
технический хлористый алюминий сублимировать через нагретые
до 170° алюминиевые стружки в вакууме.
ЛИТЕРАТУРА
1. Г. Г. Уразов, И. С. Морозов, Труды ГИПХ, в. 20, 1934, стр. 7,—
2. Г. Г. Уразов, Л. Р. Эдельсои, Материалы по металлургии цветных ме-
таллов, Изд. Кубуч, 1932.—3. Д. М. Ч и ж и к о в, Б. С. Ф р е н ц, Хлорный ме-
тод переработки оловянных руд и концентратов, Изд. АН СССР, 1941. —
4. Г. Г. Уразов, И. С. Морозов, Н. П. Шманцарь, ЖПХ, 10, № 1, 6
(1937).—5. Я. Д. Фридман, О хлорных методах переработки вольфрамовых
руд, Изд. Кирг. АН СССР, 1947.—6. Г. А. Меерсон, А. Н. 3 е л и к м а н, Ме-
таллургия редких металлов, Госметаллургиздат, 1955. — 7. И. С. Морозов,
Физико-химические основы применения хлора в металлургии редких и цветных
металлов, Изд. «Наука», 1965. — 8. И. С. Морозов, ЖПХ, 33, № 8, 1185
(1960). — 9. В. В. Печковский, А. Л. Софронов, ЖПХ, 39, № 10, 2153
Литература
1505
(I960)—10. Ф. С. Куликов, сб. «Чистые металлы и проводники», Госметал-
лургиздат, 1959, стр. 283.
11. М. В. Ионин, Порошковая металлургия, Труды иаучио-технической
сессии, Госметаллургиздат, 1954. — 12. Э. И. Креч, Н. Е. 3 а х а р ч е и к о, ЖПХ,
13, № 6, 846 (1940). —13. Г. Г. Уразов, А. С. Карнаухов, ДАН СССР,
96, № 3, 535 (1954). —14. А. Я. Зворыкин, ЖПХ, 9, № 1, 1 (1936).—
15. Б. А. Ко пыл ев, Л. П. С в я т н а я, ЖПХ, 14, № 6, 783 (1941).—
16. В. В. Печковский, А. Л. Софронов, Изв. вузов. Хим. и хим. технол.,
19, № 2, 280 (1966). — 17. И. С. Морозов, Применение хлора в металлургии
редких и цветных металлов, Изд. «Металлургия», 1966, стр. 81, 220.—
18. Н. С. Фортунатов, 3. А. Фокина, Л. А. П а р а х н е в и ч, Ю. М. Т р е-
скунова, С. М. Рябоконь, Укр. хим. ж., 32, № 4, 406 (1966). — 19. С. S. R а о,
Chem. Age (India), 17, № 7, 545 (1966). — 20. Г. П. Л учинский, Химия ти-
тана, Госхимиздат, 1941..
21. Г. П. Лучииский, 'Четыреххлористый титан, Оборонгиз, 1939.—
22. Л. Н. Щ е г р о в, Труды Уральск, политехи, ин-та, в. 96, 1960.—
23. Л. Н. Щ е г р о в, Я- Е. Вильня некий, ДАН СССР, 140, № 3 (1961).—
24. Т. к. 3 а в а р и ц к а я, М. Н. П о д г а й с к а я, С. Т. П р о х о р о в, ЖПХ, 40,
№ 3, 662 (1967). —25. J. S. Fast, Rec. trav. chirm, 58, 174 (1939). — 26. W. В lu-
men t a 1, H. Smith, Ind. Eng. Chem., 42, № 2, 249 (1950). — 27. Z. anorg.
Chem., 241, 42 (1939). — 28. A. Herczog, L. Pidglou, Canad. J. Chem., 34,
№ 12, 1687 (1956). — 29. W. Kroll, Metaux Corrosion Industries, 26 (313), 329
(1951). — 30. Сб. «Титан и его сплавы», под ред. А. И. Евстюхина, ч. 1, ИЛ,
1953.
31. Сб. «Титан и его сплавы», Изд. АН СССР, 1958. — 32. Я. М. Липкес,
Титан и его применение в технике, Оборонгиз, 1945. — 33. W. Waggaman,
Е. Gee, Chem. Eng. News, 26, № 6, 377 (1948). — 34. A.. Stelmaszczyn,
Chernik, 19, № 6, 199 (1966). — 35. S t a g n i, Galvanotecnica, 17, № 6, 121
(1966). — 36. А. Д. M а к к в и л л э н, М. К. М а к к в и л л э н, Титан, Госметал-
лургиздат, 1958. — 37. Е. Junker, L. anorg. Chem., 228, 97 (1936). — 38. Н. S i m-
h a, H. Werner, Trans. Indian. Inst. Metals, № 8, 269 (1954—1955).—
39. O. Q. Leone, H. Knudsen, D. Couch, J. Metals, 19, № 3, 18 (1967).—
40. H. И. А н у ф p и e в а, ЖПХ, 40, № 6, 1288 (1967).
41. J. W. Cotton, C. S. Hayfield, Trans. Inst. Metal Finish, 45, № 2,
48 (1967). — 42. Я. И. Вальсюнене, Ю. А. П p о к о п ч и к, Труды АН ЛитССР,
Б, № 2, (45), 1966, стр. 61, — 43. Т. W. Judson, Cutt. Tool Eng., 19, № 3, 9
(1967). — 44. C. Drotschmam, Batteries, 20, № 11—12, 990 (1967).—
45. J. Barksdale, канад. пат. 506095, 1954. — 46. J. К г ch ma, H. Schau-
mann, канад. пат. 517816, 1955.—47. J. Barksdale, Titan, Ronald Press,
1949. — 48. W. F г e у, канад. пат. 510545, 1955. — 49. Кавасаки, япон. пат. 8178,
1959. — 50. К У н и т о м и, япон. пат. 7469, 1954.
51. Ю. И. Вейцер, Г. П. Л учи некий, Химия и физика маскирующих
дымов, Оборонгиз, 1938. — 52. J. W. Stamper, Bull. Bur. Mines, № 630, 971
(1965). — 53. W. E. О p p i e, J. Metals, 6, 807 (1954). — 54. Франц, пат. 1092910,
1955. — 55. Тахимото, J. Chem. Soc. Japan. Ind. Chem. Sec., 58, № 10, 732
(1955).— 56. W. Kroll, Metallkund, 45, № 2, 67 (1954). —57. Eng. Min. J., 152,
(3), 76 (1951). — 58. P. H. Brace, J. Soc. Electrochem., 94, № 4, 70 (1948).—
59. X а т т о p и, J. Chem. Soc. Japan. Ind. Chem. Sec., 58, № 4, 250 (1955).—
60. С. H. Gorski, J. Metals, 191, № 2, 131 (1951).
61. R. G. Knickerbocker, С. H. Gorski, H. Kenworthy, A. G. Star-
11 p er, J. Metals, 1, № 11, 785 (1949). — 62. E. В. Беленький, И. В. Рис.
кин, Химия и технология пигментов, Госхимиздат, 1949. — 63. Н. Burgholz,
Stahl u. Eisen, 69, 247 (1949). — 64. M. Farber, A. Darnell, J. Chem. Phys.,
23, № 8, 1460 (1955). — 65. Я. И. Ивашеицев, В. И. Максименко, Труды
1506
Гл. XL. Продукты высокотемпературного хлорирования руд
Сибирск. техиол. ин-та, № 38, 1966, стр. 94. — 66. В. А. Рябов, Г. Н. 3 в и а-
дадзе, О. В. Альтшулер, Д. М. Чижиков, Труды Института металлур-
гии им. А. А. Бойкова, в. 1, 1957. — 67. А. В. Памфилов, А. С. Худяков
Е. Г. Штандель, ЖОХ. 5, № 5, 605 (1935). — 68. А. В. Памфилов’
М. Г. Ши хер, Там же, 7, № 22, 2760 (1937). — 69. А. В. Памфилов’
Е. Г. Штандель, ЖПХ, 9, № 10, 1770 (1936). — 70. А. В. Памфилов’,
Е. Г. Ш т а и д е л ь, Там же, 1781.
71. А. В. Зверев, 3. С. Барсукова, сб. «Минеральное сырье», в. 13,
Изд. «Недра», 1966, стр. 42. — 72. И. Н. Г о д и е в, А. В. П а м ф и л о в, ЖОХ,
7, № 8, 1264 (1937). — 73. А. Б. Безукладников, ЖПХ, 34, № 1, 49
(1961). — 74. И. С. Морозов, С. Л. С т е ф а и ю к, ЖНХ, 3, № 10, 2366
(1958). — 75. Такимото, J. Chem. Soc. Japan. Ind. Chem'. Sec., 58, № 7, 496
(1955). — 76. Г. А. Меерсои, Хим. наука и пром., 1, № 5, 496 (1956).—
77. Ацума, Гото, J. Mining Inst. Japan, 71, № 809, 687 (1955).—
78. Т. G. Craig, аигл. пат. 1050527, 1966. — 79. И с и ц у к а, япон. пат. 1869,
1955.— 80. L. Rowe, W. Е. Oppie, J. Metals, 7, № 11, Sec. 1, 1189 (1955).
81. Е. И. М а ч к а с о в, Э. Н. Сулеймеиов, В. Д. Пономарев, Труды
Ин-та металлургии и обогащения АН КазССР, в. 18, 1966, стр. 58. — 82. А. К и-
1 i и g, S. Hartmann, пат. ФРГ 1186835, 1965.—83. W. Т. Shannon,
В. F. Kennedy, N. Z. J. Sci., 9, № 2, 303 (1966). — 84. Иид. пат. 47771, 1953. —
85. Та хим ото, X аттори, J. Chem. Soc. Japan. Ind. Chem. Sec., 58, № 4,
253 (1955). — 86. Франц, пат. 1075944, 1954. — 87. А. Б. Безукладников,
ЖПХ, 35, № 11, 2389 (1962). — 88. Савада, Фудзи и, япои. пат. 264, 1956.—
89. А. Б. Безукладников, Я- Е. Вильияиский, ЖПХ, 34, № 1, 49
(1961); Металлургия и химия титана. Труды Ин-та металлургии им. А. А. Бай-
кова, в. 5, Изд. АН СССР, 1961, стр. 135. — 90. А. Б. Безукладников, ЖПХ,
36, № 2. 451 (1963).
91. А. Б. Безукладников, Там же, 40, № 1, 31 (1967). — 92. А. Б. Бе-
зукладников, Автореф. канд. дисс., Уральск, политехи, ии-т, Березники,
1963. — 93. Л. Н. Щ е г р о в, Автореф. канд. дисс., Уральск, политехи, ии-т,
Свердловск, 1962. — 94. А. Н. К е т о в. И. М. Колесов, Изв. вузов. Цветная
металлургия, 3, 77 (1966). — 95. Т. Т. G п i е w е k, Е. О. К 1 е i п f е 1 d е г, пат.
США 3258064, 1966. — 96. А. Б. Безукладников, В. А. Безворитний,
Е. А. Ионов, Цветные металлы, № 7, 88 (1967). — 97. F. К. McTaggart,
J. Concil Sci. Ind. Res., 18, № 1, 5 (1945). — 98. E. W. N els on, M. C. Godwin,
T. J. Crossley, англ. пат. 1043790, 1966. — 99. E. L. Cairns, E. О. К 1 e i n-
I e 1 d e г, пат. США 3261664, 1966. — 100. W. К r i v e, D. Mason, J. Phys. Chem.,
60, № 3, 374 (1956).
101. С. к. S t о d d a r t, E. Pietz, U. S. Bureau of Mines. Rept Invest., 4153,
1947. — 102. J. Levi, D. Pickard, франц, пат. 1097736, 1955. — 103. В. И. Бо-
родин, В. П. Василенко, И. П. Сорокин, В. И. Тыква, авт. свид.
187306, 1966. — 104. Н. Н. Schakmann, канад. пат. 493166, 1953. — 105. И с и-
цука, япои. пат. 971, 1954. —106. J. Lewy, пат. США 2725350, 1955.—
107. Н. Espenschied, пат. ФРГ 931104, 1955. — 108. Англ. пат. 690470, 1953. —
109. Н. Н, Рубаи, Л. А. Карташева, Труды Ин-та металлургии и обога-
щения АН КазССР, в. 18, 1966, стр. 49.— ПО. А. Д. Ефремов, Я. Д. 3 е л ь-
веиский, И. П. Оглоблина, Хим. пром., № 10, 758 (1969).
111. Р. С. Исламов, И. М. Ч е п р а с о в, В. Д. Пономарев, Труды
Ин-та металлургии и обогащ. АН КазССР, в. 22, 1967, стр. 87. — 112. W С 1а-
b a u gh, R. Les li е, J. Res. Nat. Bur. Stand., 55, № 5, 261 (1955). — //3. F. F a li-
no e, J. D. Sturm, пат. США 3156630. 1964. — 114. C. L. Boyd, пат. США
3253885, 1966. — 115. H. С у г, F. Griffith, пат. США 2723903, 1955.—
116. W. Frey W. Weber, канад. пат. 517913, 1955. —117. W. Frey, пат. США
2675891, 1954.— //8. Австрал. пат. 151612, 1953. —119. Англ. пат. 725555, 1955.—
120. Австрал. пат. 156522, 1954.
Литература
1507
121. L. A a g а а г d, L. Rowe, канад. пат. 519655, 1955. — 122. L. A a g а -
а г d, S. Cole, канад. пат. 519654, 1955. —/25. J. К г china, канад. пат.
492989, 1953. —124. J. К г ch та, пат. США 2682930, 1954; каиад. пат. 517556,
1955. — 125. N. S i d h a n t a, К. S h e n о у, В. Dey, Bull. Centr. Electrochem.
Res. Inst., 1, № 4, 30, 1954. — 126. H. Sinha, Proc. Austral. Inst. Mining a.
Metallurgy, № 178, 7 (1956). —127. A. M. Владимиров, Б. M. Воловик,
А. А. Г аврилова, В. И. Каменецкий, В. А. Кроль, Хим. наука и пром.,
4, № 1, 132 (1959). — 128. R. Ruehrwein, пат. США 2720445, 1955.—
129. R. Ruehrwein, G. Skinner, пат. США 2270281, 1955. —130. L. La-
ci a k i, G. С о г s i, австр. пат. 250306, 1966.
131. Аигл. пат. 1043701, 1966. —132. О. Н. Пирогов, Н. М. Чирков,
Д. В. Иванюков, В. И. Цветкова, П. В. Крымов, В. Ф. Петрова,
авт. свид. 186997, 1966. — 133. А. П. Крешков, В. А. Борк, Труды МХТИ
им. Д. И. Менделеева, т. 14, 1951, стр. 12. —134. В. Г. Флейшман, Хим.
пром., № 1, 9, 1959. — 135. К. А. Андрианов, М. В. Соболевский, Вы-
сокомолекулярные кремиийоргаиические соедииеиия, Обороигиз, 1949. — 136. При-
менение силиконов в качестве смазок, под ред. Н. М. Долгова, Госхимиздат,
1949. —137. К- А. Андрианов, А. И. Петрашко, Кремиийоргаиические
полимеры в народном хозяйстве, Изд. АН СССР, 1959. —138. К- А. Андриа-
нов, А. А. Жданов, Хим. пром., № 11, 329 (1950). — 139. В. Я- Николина,
Там же, № 1, 48 (1962). —140. В. Я- Николина, Л. И. К и ы ш, Н. А. Де-
ринг, сб. «Работы по технологии производства наполнителей и адсорбентов
минерального происхождения», в. 15, Госхимиздат, 1963, стр. 64.
141. Н. Н. Мурач, Хим. наука и пром., 1, № 5, 492 (1956). — 142. В. Н. Ч е-
чеицев, Л. А. Фирсанова, В. Н. Зайцев, Л. В. Матвеенко, сб. Мо-
сковского ин-та стали и сплавов, в. 41, 1966, стр. 281. — 143. Г. М. Строигин,
Хим. наука и пром., 2, № 6, 785 (1957). —144. Б. Н. Долгов, Химия кремие-
оргаиических соединений, Госхимиздат, 1933. —145. К- А. Андрианов,
О. И. Грибанова, Кремиийоргаиические полимерные продукты для промыш-
ленности, ч. II, Госэиергоиздат, 1946; Кремиийоргаиические соединения, Госхим-
издат, 1955. —146. П. Ф. Антипин, В. В. Сергеев, ЖПХ, 27, № 7, 784, 1954.—
147. Фраиц. пат. 1040049, 1953. — 148. К. N i s h i k a m а, япон. пат. 1477, 1954.—
149. F. D. Stedman, каиад. пат. 496501, 1963.— 150. F. D. Stedman, англ,
пат. 712279, 1954.
151.1. Andersen, пат. США 2739041, 1956. —152. Л. И. Кузнецов-
Фетисов, А. И. Милютин, Труды Каз. х.-т. ии-та им. С. М. Кирова, в. 14,
1949, стр. 107. —153. A.. Koster, Angew. Chem., 69, 563 (1957). —154. Р. S р а-
cu, Р. Voichescu, A. Ovanesian, Studii si cercetari chim., 3, № 3—4, 195
(1955). — 155. В. Г. Флейшман, Хим. пром., № 7, 27, 1957. — 156. R. Р 2 t s с h,
пат. ФРГ 1187586, 1965. —/57. С. А. Амирова, Автореф. докт. дисс., ЛТИ
им. Ленсовета, 1965. —158. A. EL Callow, В. Harris, аигл. пат. 1031371,
1966.— 159. D. Curcaneanu, С. Todereanu, A. Teodor у, V. A n a s t а-
siu, Studii si cercetari chim. Acad. RSR, 14, № 9, 663 (1966). — 160. J. W. W h e-
1 a n, пат. США 2821460, 1958.
161. J. A. Baldrey, J. C. Weaver, аигл. пат. 1015604, 1966. —162. S. La-
n i n i, A. G a u m a n n, швейц, пат. 395949, 1966. — 163. H. Melzer, пат. ФРГ
948149, 1956. — 164. Т а и а б э, J. Pharm. Soc. Japan, 74, № 8, 866 (1954).—
165. Е. Dewing, J. Am. Chem. Soc., 77, № 9, 2639, 1955. —166. В. I m e 1 i k,
M. Mathieu, Compt. rend., Sci., 242, № 6, 773 (1956). —167. E. Gastinge r,
Angew. Chem., 67, № 3, 108 (1955). —168. С. А. Семеикович, Автореф. докт.
дисс., Ии-т полупроводников АН СССР, 1959. —169. Canad. Chem. Process, 51,
№ 2, 45 (1967). — 170. Ч. Томас, Безводный хлористый алюминий в органиче-
ской химии, ИЛ, 1949.
171. В. Л. Закутииский, И. Г. Бляхе р, Труды УНИХИМ, вып. 14,
1967, стр. 121. —172. О. С. Рольстон, Безводный Хлористый алюминий, Изв.
1508 Гл. XL. Продукты высокотемпературного хлорирования руд
Сов. нефт. пром., 1924. —173. L. R. Krishnamurthi, Chem. Age (India), 16,
№ 5, 422 (1965). —174. В. M. Коган, Б. Г. Рабовский, А. А. Фурман,
Хим. пром., № 5, 365 (1967). — 175. П. П. Федотьев, А. А. Чижик, Труды
ГИПХ, в. 20, 1934, стр. 76. —/76. Э. И. Креч, ЖОХ, 7, № 8, 1249 (1937).—
177. Франц, пат. 1026002, 1953. —178. Т. Adamski, Е. Buntner, Przem.
Chem., 10, № 12, 599 (1954). —179. Франц, пат. 1069243, 1954. — 180. Англ, пат
718773, 1954.
181. A. lohannsen, W. Pfannmiilier, W. Klinger, пат. ФРГ 948972
1956. — 182. S. McAfee, Ind. Eng. Chem., 21, № 7, 670 (1929). — 183. P. Weiss,
пат. ФРГ 933688, 1955. — 184. H. H. Воронин, И. С. Г а линкер, ЖПХ, 7,
№ 2—3, 143 (1930). —185. A. Kaczorowski, Przem. Chem., 20, 221 (1936).—
186. П. II. Будников, Z. anorg. Chem., 37, № 1, 100 (1924). — 187. И. Е. Ада-
ДУров, ЖПХ, 5, № 21—22, 1288 (1928). —188. Б. Г. Рабовский, ЖПХ, 33,
№ 3, 540, 1960. — 189. Э. К- Келлер, А. И. Леонов, Усп. хим., 22, № 3,
334 (1953). — 190. П. Т. Данильченко, Л. А Булыгин, Труды Крымсю
фил. АН СССР, т. 4, 1953, стр. 25.
191. Е. Н. Никитин, ДАН СССР, 105, № 5, 1014 (1955). —192. R. Roy,
D. М. Roy, Е. Е. F г а п с i s, J. Am. Ceram. Soc., 38, 198 (1955). — 193. J. W i e g-
m a n п, С. H. Horte, Z. anorg. Chem., 286, № 5—6, 268 (1956).—
194. A. M. Калинина, E. А. Порай-Кошиц, ДАН СССР, 114, № 2, 365
(1957). —195. A. M. Фишер, Б. Г. Рабовский, В. И. Финкельштейн,
Труды по химии и химической технологии, т. 1, Горький, 1958, стр. 102. —
196. Н. К- Аитоиевич, Труды Всес. и.-и. ии-та керамики (ВНИИК), т. 45,
1934, стр. 3. —197. И. А. Казарновский, авт. свид. 23970, 1931.—
198. L. J. Derham, англ. пат. 1035807, 1035808, 1966. —199. Ю. В. Карякин,
И. И. Ангелов, Чистые химические реактивы, Изд. «Химия», 1963, стр. 25.—
200. Н. Г. Ключников, Руководство по неорганическому синтезу, Изд. «Хи-
мия», 1965, стр. 184.
201. Руководство по препаративной неорганической химии, под ред.
Г. Брауера, ИЛ, 1956, стр. 384.—202. X. И. Крягова, ЖОХ, 9, № 19, 1755
(1939).—203. Н. И. Грацианский, Укр. хим. ж., 9, 432 (1934).—
204. Ю. К Делимарский, Е. Н. Скобец, Л. С. Береиблюм, ЖФХ,
22, 1108 (1948).
Глава XLI
ЦИАНИСТЫЕ СОЕДИНЕНИЯ
К цианистым соединениям относятся вещества, содержащие од-
новалентную группу циана CN и его производные В свободном
виде может быть выделен газообразный циан (CN)X, представляю-
щий собой в большинстве случаев дициан (CN)2, находящийся в
равновесии 2 при высоких температурах с CN. Дициан является од-
ним из наиболее реакционноспособных газов. Он легко реагирует
с металлами и окислами металлов (образуя цианиды и цианами-
ды), с галогенами и различными органическими веществами. Из '
неорганических соединений циана наибольшее техническое значе-
ние имеют: синильная кислота HCN и цианиды калия, натрия и
другие, цианамид CN-NH2 и кальцийцианамид, а также ферро- и
феррицианиды калия и железа.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Цианистоводородная или синильная кислота HCN — бесцветная
жидкость с запахом горького миндаля. Она кипит при 25,63°, за-
мерзает при —13,4°. Плотность жидкости при 18° 0,6967 г!см?. С
водой смешивается во всех отношениях. На рис. 451 показана кри-
вая давления пара чистой синильной кислоты, а на рис. 452 давле-
ние пара ее водных растворов при 18°. Пары HCN горят голубо-
ватым пламенем.
Синильная кислота и ее водные растворы устойчивы лишь в при-
сутствии стабилизаторов — небольших количеств свободных мине-
ральных кислот. Без стабилизаторов, и особенно в присутствии
следов щелочей, синильная кислота постепенно темнеет, что свя-
зано с образованием продуктов ее полимеризации. При кипячении
водных растворов (а также в щелочной среде) частично идет гид-
ролиз HCN с образованием формиата аммония3:
HCN + 2Н2О HCOONH4
1510
Гл. XLI. Цианистые соединения
Кислотные свойства синильной кислоты выражены весьма ела-.
бо, и она легко вытесняется из своих солей другими кислотами, в <
том числе угольной. Поэтому под действием атмосферного воздуха,!
содержащего двуокись углерода, из цианидов выделяются пары
HCN.
Цианистый натрий NaCN образует бесцветные кристаллы ку-
бической формы. Кристаллизуется с двумя и с одной молекулой
Температура, °C
Рис. 451. Зависимость
давления пара синиль-
ной кислоты от тем-
пературы.
воды, а выше 34,7° — в безводной форме.
Температура плавления безводной соли
563,7°. Насыщенный водный раствор содер-
жит при 0° 43,4%. при 34,7° 82,0% NaCN.
HCN в жидкости, мол. n/t
Рис. 452. Давление паров над водными
растворами синильной кислоты при 18°:
Р — общее давление паров, — давление циа-
нистого водорода, р
н2о
— давление водяного пара.
Цианистый калий KCN кристаллизуется в безводной форме, об-
разуя бесцветные октаэдрические кристаллы. Плавится при 634,5°.
Расплывается на воздухе.
Цианиды натрия и калия достаточно устойчивы при нагревании
вплоть до 800°. В смеси с хлоридами щелочноземельных металлов
в расплавленном состоянии они превращаются в цианамидные
соли 4:
2NaCN + ВаС12 = 2NaCl + Ba(CN)2
Ba(CN)2 = BaCN2 + C
BaCN2 + 2NaCl = Na2CN2 + BaCl2
Цианистый кальций Ca(CN)2 выделяется из водных растворов
в виде кристаллов кубической формы. Раствор соли и твердая соль
во влажном воздухе легко теряют синильную кислоту, переходя при
этом в основные соединения.
Синильная кислота (как жидкая, так и парообразная) и ее соли
чрезвычайно ядовиты. Для человека смертельной дозой является
0,05 г HCN.
Физико-химические свойства
1511
Цианамид кальция CaCNa— кальциевая соль цианамида H2CN2
представляет собой бесцветные кристаллы с плотностью 2,3 г]см\
Технический продукт имеет темно-серый цвет вследствие присут-
ствия в нем 10—15% углерода. Растворимость CaCN2 в 100 г воды
при 25° равна 2,5 г. В водном растворе CaCN2 гидролизуется:
2CaCN2 + 2Н2О = Ca(HCN2)2 + Са(ОН)2
С течением времени кисл-ая соль цианамида кальция разлагает-
ся с образованием карбамида CO(NH2)2, дицианамида (H2CN2)2 и
других продуктов. На воздухе под влиянием двуокиси углерода и
влаги вначале образуется соединение CaCN2-CO2-5H2O, которое
затем гидролизуется.
При взаимодействии CaCN2 с СО2 в воде образуется цианамид,
который при нагревании раствора до 80—90° полимеризуется в ди-
цианамид5. Чистый CaCN2 устойчив в атмосфере азота до 1200°,
плавится при 1340°. При нагревании диссоциирует в твердом со-
стоянии. При нагревании смеси CaCN2 с NaCl или К.С1 вначале
идет обменная реакция 6’7 с образованием цианамида щелочного
металла, который разлагается 8:
Na2CN2 - NaCN + Na + V2N2
Отравление цианамидами происходит при вдыхании пыли
вследствие образования дициана.
Цианамид кальция обладает сильно раздражающими свойства-
ми, вызывает воспалительные заболевания кожи. Он оказывает об-
щее действие на сосудисто-двигательный и дыхательный центры,
вызывает головные боли, желудочно-кишечные заболевания и
т. п.9’ ’°. Раздражающее действие цианамида кальция CaCN2 на
слизистые оболочки и кожу обусловлено содержанием в продукте
окиси кальция.
Цианат калия KNCO хорошо растворим в воде. При продол-
жительном стоянии водный раствор разлагается с образованием
двуокиси углерода и аммиака; в растворе NH4NO3 или NH4C1 в ам-
миаке превращается в карбамид:
KNCO + NH4NO3 = KNO3 + CO(NH2)2
Роданистоводородная кислота HNCS ниже 0° — бесцветная, бы-
стро желтеющая жидкость с острым запахом, напоминающим за-
пах уксусной кислоты. Сильно летуча, на воздухе легко разлагает-
ся на HCN и персульфоциановую кислоту. Разбавленные водные
растворы (3—5% HNCS) достаточно устойчивы. В более концен-
трированных растворах разлагается с образованием CS2 или H2S:
2SCNH + 2Н2О = СО2 + CS2 + 2NH,
SCNH + 2Н2О - СО2 + H2S + NH3
1512
Гл. XLI. Цианистые соединения
Роданистый аммоний NH4NCS выделяется из водного раствору
в виде белых кристаллов, плавящихся при 159°. При 0° в 100 г во]
ды растворяется 122,1 г, при 20°— 162,2 г роданида аммония. При
нагревании до 150—220° роданид аммония превращается в тиомо-
чевину. В системе NH4NCS—(NH2)2CS переходная точка лежит
при 142° и соответствует составу 40% NH4NCS и 60% (NH2)2CS.
Предполагают п, что процесс протекает с образованием промежу-
точного продукта HN=C(SH)NH2. В системе NH4NCS—
— (NH2)2CS—Н2О 12 помимо основных компонентов кристалли-
зуется двойная соль NH4NCS- (NH2)2CS.
Роданистый калий KNCS при нагревании на воздухе до 600°
разлагается с образованием цианистого калия, синильной кислоты
и сернистого газа.
Роданид железа отличается интенсивной кроваво-красной
окраской в водном растворе. Это служит характерной реакцией на
железо или родан 13.
Цианистые соли обладают большой способностью к комплексо-
образованию, причем ион CN~ входит во внутреннюю сферу ком-
плекса. Примером комплексных цианистых солей могут служить
цинковые цианистые комплексы 14 [Zn(CN)3]' и [Zn(CN)4]2-, обра-
зующиеся при растворении цинка в растворах цианидов, а также
ферроцианид натрия Na4Fe(CN)6 (железистосинеродистый натрий
или желтый синь-натр, или желтая кровяная соль) и феррицианид
натрия Na3Fe(CN)6 (железосинеродистый натрий или красный
синь-натр, или красная кровяная соль). Эти названия появились в
связи с тем, что: 1) ферроцианиды окрашены в желтый, а ферри-
цианиды в красный цвет; 2) впервые эти соли получали в промыш-
ленности прокаливанием сухой крови животных с щелочными кар-
бонатами и железными опилками; 3) ферроцианиды выделяют из
растворов солей трехвалентного железа нерастворимый осадок
Fe4[Fe(CN)6]3 ярко-синего цвета (берлинская лазурь), а ферри-
цианиды из растворов солей двухвалентного железа выделяют оса-
док Fe3[Fe(CN)6]2 также синего цвета (турнбуллева синь). Берлин-
ской лазури приписывают также формулу Fe[Fe (CN)6]15.
Железистосинеродистый натрий кристаллизуется из водных рас-
творов в виде декагидрата Na4Fe(CN)6- 10Н2О, а выше 81° — в без-
водной форме. Насыщенный раствор содержит при 25° 17,04%, при
80° — 38,0% Na4Fe(CN)e. Декагидрат на воздухе легко выветри-
вается (давление водяного пара над декагидратом при 25° равно
9 мм рт. ст., а при 60° — 99,5 мм рт. ст.).
Железистосинеродистый калий кристаллизуется с тремя моле-
кулами воды —• K4Fe(CN)6-3H2O. Растворимость его в воде при 25°
составляет 24,15%.
Железосинеродистый калий КзРе(СМ)6 кристаллизуется безвод-
ным. Растворимость в воде при 4° — 24,8%, при 104,4° — 45,2%.
Применение
1513
ПРИМЕНЕНИЕ
Способность цианидов образовывать комплексные соединения
широко используется для извлечения драгоценных металлов (зо-
лота, серебра) из руд. Ядовитые свойства синильной кислоты ис-
пользуются при применении цианидов в качестве фумигантов для
борьбы с паразитами в сельском хозяйстве (окуривание расте-
ний) и при санитарной обработке (окуривание пароходов, железно-
дорожных вагонов, казарм и пр.) 1б-18. Цианиды используют в галь-
ваностегии, в производстве пластических масс, искусственных смол,
лаков, красок, для цементации стальных изделий, в текстильной
промышленности в качестве протрав при крашении тканей (ком-
плексные соли) и пр. Указывают 19-20, что небольшие добавки ком-
плексных цианидов увеличивают растворимость хлоридов натрия
и калия.
Имеются патенты, предлагающие использовать продукты поли-
меризации синильной кислоты в качестве удобрений 21’22.
Синильная кислота имеет важное значение в качестве исходного
сырья для получения акрилонитрила СН2=СН—CN, являющегося
полупродуктом в производстве нитрильного каучука и синтетиче-
ских волокон 23-27. Получаемые из синильной кислоты акрилаты пе-
рерабатывают в органическое стекло.
Цианистый натрий и цианистый калий, а также цианплав (чер-
ный цианид), получаемый из цианамида кальция (стр. 1525), ис-
пользуют в машиностроении для закалки специальных сталей, для
извлечения золота из руд (цианирование руд), при обогащении по-
лиметаллических руд, а также в сельском хозяйстве для борьбы с
вредителями и в ряде’ других отраслей промышленности и народ-
ного хозяйства 28. Особенное значение они имеют для борьбы с сус-
ликами 29 и с вредителями цитрусовых растений 30.
Цианплав служит сырьем для получения цианидов натрия, ка-
лия, а также желтой кровяной соли. Последнюю применяют В тек-
стильной промышленности и в производстве красок.
Цианамид кальция используют в качестве азотного удобрения,
в особенности под технические культуры — хлопок, сахарную свек-
лу и др. Предложены различные смеси на основе цианамида каль-
ция с целью увеличения длительности его действия 31> 32 или при-
дания ему также инсектицидных и фунгицидных свойств 33. Циана-
мид кальция в смеси с кремнефторидом натрия применяют для
дефолиации хлопчатника перед механизированной уборкой хлоп-
ка 34-35. Он обладает также гербицидными свойствами и исполь-
зуется для уничтожения сорняков. Помимо указанных областей
применения, цианамид кальция служит исходным материалом в хи-
мической промышленности для получения цианплава и цианидов, а
также дицианамида, применяемого в производстве пластических
масс, искусственных смол и лаков.
1514
Гл. XLI. Цианистые соединения
Описаны каталитические свойства цианамида кальция, напри-
мер, при окислении раствора индигокармина перекисью водорода
при 35—40° и применение его в качестве активатора и носителя для
приготовления активных смешанных контактов36.
Роданид аммония и другие роданиды находят ограниченное
применение. Некоторое количество роданида аммония получают
при очистке коксового газа. Возможна переработка роданида в
тиомочевину 12, употребляемую в различных областях техники. Ро-
данид аммония используется для синтеза роданина37, производные
которого находят применение в неорганическом анализе38, фото-
графии, а также в органическом синтезе для получения амино-
кислот, нитрилов, аминов39.
Из роданистых солей можно получать полироданоалканы, об-
ладающие инсектофунгицидными и бактерицидными свойствами40.
В табл. 119 приведены качественные показатели, которым дол-
жны удовлетворять технические цианистый натрий и цианистый ка-
лий.
ТАБЛИЦА 11»
Качество технических цианистого натрия (ГОСТ 8464—69)
и цианистого калия (ГОСТ 8465—69)
(Содержание компонентов в %)
Цианистый натрий Цианистый калий
1-й сорт 2-й сорт 1-й сорт 2-й сорт
Основного вещества (NaCN или KCN), 93
не менее 90 87 95,
Углекислого натрия, не более .... 1,4 3,0 — —
Углекислого калия, не более — — 1.0 1,0
Хлористого натрия, не более 3,0 3.0 — —
Едкого натра, не более Едких щелочей (в пересчете на КОН), 1,0 2,0 1.0 1,0
не более — —
Сульфидов (в пересчете на S), не бо- лее Не растворимого в воде остатка. 0,2 0,2 0,003 0,003
не более —•
Влаги, не более 3,0 5,0 2.0 2,0
Цианид натрия и калия упаковывают в железные барабаны
весом до 100 кг или в железные банки весом до 10 кг. Барабаны и
банки помещают в фанерные барабаны с дощатыми днищами.
При работе с цианидами натрия и калия необходимо строго со-
блюдать правила безопасности обращения с высокотоксичными от-
равляющими веществами. Аппаратура должна быть абсолютно гер-
метизирована. При вскрытии аппаратуры и тары, а также при со-
держании в воздухе более 0,05—0,06 мг HCN на 1 л необходимо
пользоваться противогазом.
Применение
1515
Цианплав должен удовлетворять по своему качеству требова-
ниям ГОСТ 452—66 (табл. 120). Цианплав марки А — пластинча-
тый — применяется для промышленных целей. Цианплав марки
Б — молотый — применяется для борьбы с вредителями в сельском
хозяйстве, а также как дезинсекционное и противоэпидемическое
средство.
ТАБЛИЦА 12»
, Качество цианплава (черного цианида) по ГОСТ 452—66
(Содержание компонентов в %)
Марки
А Б
1-й сорт 2-й сорт 1-й сорт 2-й сорт
Цианидов в пересчете на NaCN, не ме- нее 47 42 45 42
Карбида кальция, не более 1,0 2,0 0,8 13
Серы, не более Углерода свободного, не более .... 0,4 0,7 Не нормируется
3,0 4,0 » »
Размеры частиц цианплава, применяемого в качестве противо-
эпидемического средства, должны быть от 0,3 до 3,0 мм. В сель-
ском хозяйстве цианплав применяют в порошкообразном виде с
размером частиц от 0,3 до 1,5 мм.
Цианамид кальция выпускают двух марок: грубомолотый (мар-
ка А), предназначаемый для переработки в другие химические про-
дукты, и тонкомолотый, умасленный минеральным маслом (марка
Б), используемый в качестве дефолианта41. Чистый CaCN2 содер-
жит 34,98% азота. В техническом продукте имеются примеси выде-
лившегося углерода и непрореагировавших СаС2 и СаО (табл. 121).
ТАБЛИЦА 121
Требования к качеству цианамида кальция по ГОСТ 1780—56
(Содержание компонентов в %)
Марки
А в
1-сорт 2-сорт
Цианамидного азота, не менее Карбида кальция, не более 20 18,5 19
1,0 2,0 0,2
Минерального масла, в пределах Остаток, не более — — 0.5-1Д
иа сетке № 0071 — — 5,0
» » № 06 0,5 03 —
Цианамид кальция марки Б, отгружаемый в хлопкосеющие райо-
ны Узбекской, Туркменской, Таджикской, Киргизской и Казахской
1516
Гл. XLI. Цианистые соединения
ССР, упаковывают в барабаны емкостью 50—100 л, при отгрузке
в другие районы продукты упаковывают в пятислойные бумажные |
битумированные мешки весом 35 кг. |
Требования к качеству технических железистосинеродистых нат- 3
рия и калия приведены в табл. 122. Их упаковывают в многослой- $
ные бумажные мешки с полиэтиленовым вкладышем или битуми- -
рованные с непропитанным внутренним слоем бумаги.
ТАБЛИЦА 122
Требования к качеству железистосинеродистых натрия
(ГОСТ 6817—54) и калия (ГОСТ 6816—75)
(Содержание компонентов в %)
Железистосинеро- дистый натрий Железистосинеро- дистый калий
1-й сорт . 2-й сорт 1-й сорт 2-й сорт
Na ;Fe(CN)6 • 10Н2О или K4Fe(CN)6 X X ЗН2О, не меиее 97 95 98 96
Хлориды (в пересчете на NaCN), не более . 1,0 2,0 1.1 1,3
Цианиды (в пересчете на NaCN), не более 0,01 0,02 0,001 0,001 |
Не растворимый в воде остаток . . . 0,02 . 0,04 0,01 0,01
Не растворимый в соляной кислоте остаток, не более Влага, ие более 0,01 0,03 —. —. Я
2,0 3,0 — —
СЫРЬЕ
В первый период своего развития (до 90-х годов прошлого ве- J
ка) производство цианидов базировалось почти исключительно на М
переработке животных и растительных отбросов. В дальнейшем все В
большее значение стали приобретать синтетические методы, осно- i
ванные на переработке аммиака, на фиксации атмосферного азота, I
а также методы, связанные с переработкой отходов коксогазового I
производства, паточной барды и др. В настоящее время большие I
количества цианидов производят путем переработки цианамида I
>• кальция, и превалирующее значение приобретает синтез синильной И
кислоты из природного газа (метана) и аммиака.
ЦИАНАМИД КАЛЬЦИЯ
Получение цианамида кальция основано на взаимодействии
карбида кддьция с азотом по обратимой реакции:
CaC2 + N2 CaCN2 + C + 70 ккал
Реакция азотирования карбида кальция начинается уже при
700—800° и протекает с большой скоростью при 1000—1100°42>43.
Цианамид кальция
1517
На рис. 453 представлена изобара реакции образования циана-
мида кальция при атмосферном давлении. С повышением темпера-
туры уменьшается содержание азота в твердой фазе (цианамиде
кальция) и возрастает равновесное давление азота в системе
CaCN2—N2, которое зависит также от степени азотирования кар-
бида кальция (рис. 454).
Цианамид кальция (с максимальным содержанием азота, рав-
ным 29%) вполне стабилен вплоть
ИЗО—1200° происходит уменьше-
ние содержания азота в твердой
фазе. Температура 1200° соответ-
Рис. 454. Давление азо-
та в системе CaCN2—N2
в зависимости от степени
азотирования карбида
кальция.
Рис. 453. Равновесие в системе
CaCN2—N2 при атмосферном давле-
нии.
ствует стабильному существованию цианамида кальция с содер-
жанием 21% азота. Обратимость реакции имеет место вплоть до
1400° при уменьшающемся содержании азота в твердой фазе. Выше
1400° цианамид кальция диссоциирует необратимо по реакции:
C + CaCN2 = Ca + 2C + N2
Высокопроцентный цианамид кальция можно получить при тем-
пературе не выше 1100°, когда разложение цианамида кальция еще
незначительно. Степень азотирования или содержание азота в по-
лученном продукте зависит также от чистоты и качества применяе-
мых азота и карбида кальция. Последний должен иметь литраж
290—300 л/кг. (Под литражом карбида кальция понимают коли-
чество литров ацетилена при 20° и 760 мм рт. ст., выделяемого 1 кг
карбида кальция при обработке его водой). Для азотирования при-
меняют азот, содержащий 99,8% азота и лишь 0,2% примесей
(кислорода, аргона и др.). Скорость реакции возрастает в присут-
ствии 10% СаС12 или 2—3% CaF2. В промышленных условиях
к карбиду кальция добавляют 2—3% плавикового шпата и процесс
ведут при 800—1150°.
1518
Гл. XLI. Цианистые соединения
Во избежание снижения выхода цианамида содержание в кар-
биде кальция примесей SiOz, глинозема, окиси магния, силикатов
и других примесей должно быть минимальным 44.
Получение цианамида кальция в электрических печах
Процесс азотирования карбида кальция — экзотермический
при 800—1000° протекает автотермично. Предварительный разогрев
шихты до температуры начала реакции осуществляют преимуще-
ственно с помощью электрического тока. Применяют также газо-
вый обогрев.
Рис. 455. Схема производства цианамида кальции:
1, 15 — дробилки; 2, 9, 16, 23 — элеваторы; 3, 8, 11, 18, 22—шнеки; 4, 7, 10, 19, 24—бун-
керы; 5, 20— питатели; 6, 21—трубчатые мельницы; 12 — загрузочный цилиндр; 13 — циана-
мидные печи; 14 — охладительные стаканы; 17 — шнековый транспортер; 25 — барабан
для укупорки; 26 — вагонетка.
Получение цианамида кальция осуществляют большей частью
в печах периодического действия вследствие необходимости дли-
тельной обработки материала. Из различных конструкций печей
непрерывного действия эксплуатируются преимущественно каналь-
ные печи при использовании шихты с добавкой хлористого каль-
ция. Возможно использование и вращающихся трубчатых печей 43.
Основным типом цианамидной печи периодического действия,
является ретортная печь 43'45, получившая свое название от способа
загрузки в нее шихты с помощью бумажной или картонной «ре-
Цианамид кальция •
151»
торты». В настоящее время все больше используют печи периодиче-
ского действия без реторт, и карбидную шихту загружают не-
посредственно в печь.
На рис. 455 показана схема производства цианамида кальция 4б.
Карбид кальция с размерами кусков до 100 мм вначале дробят, за-
тем измельчают в трубчатой мельнице
до тонкости, при которой 98,5% материа-
ла проходит через сито с 900 отв1см2. В
трубчатую мельницу подают также пла-
виковый шпат и около 1-5% цианамида
кальция. Введение в шихту возвратного
цианамида кальция позволяет предотвра-
тить или уменьшить спекание шихты и
снизить температуру процесса, так как
CaCN2 является катализатором реакции
азотирования. Измельченную шихту по-
дают через бункер к загрузочным ци-
линдрам, служащим для непосредствен-
ной загрузки шихты в цианамидные печи
и расположенным в печном отделении.
В цилиндр загружают автоматически
1200—1300 кг шихты и устанавливают
его при помощи мостового крана на
съемную воронку цианамидной печи. За-
грузку шихты в печь производят при от-
крывании затвора загрузочного цилинд-
ра с помощью воронки, снабженной виб-
ратором, служащим для уплотнения ших-
ты в печи.
Цианамидная печь с непосредствен-
ной загрузкой шихты представляет собой
стальной цилиндр, футерованный изнут-
ри шамотным кирпичом (рис. 456), вы-
сотой около 3 м, диаметром вверху
0,8 м и внизу 0,76 м. Сужение шахты
печи книзу облегчает выгрузку циан-
амидного блока. Печь закрывается
крышкой с песочным затвором. В ниж-
ней части имеется зольная коробка с
трубопроводом для подачи азота. Рас-
Рис. 456. Цианамидная печь:
/ — цилиндр (кожух); 2 — футе-
ровка (шамотный кирпич); 3 — чу-
гунный конус; 4 — зольная ко-
робка с контактной плитой;
5 — люк; 6 — трубопровод для
азота; 7 —электрод; 5 —каналы;
Р —песочный затвор; /0—крышка;
11 — штуцер.
пределение азота в шихте производится
по трем вертикальным каналам, образуемым при помощи трех
стальных труб, вставляемых в печь перед загрузкой шихты и
удаляемых после загрузки. Таким же образом устроен централь-
ный канал, в который вводят нагревательный электрод; с его по-
мощью осуществляют начальный подогрев карбида. На дно печи
1520
Гл. ХЦ. Цианистые соединения
укладывают чугунный конус, который сцепляется с металлической
штангой при извлечении цианамидного блока из печи с помощью
мостового крана. Выгруженный цианамид помещают в стальные
стаканы, где он охлаждается до 40—50° в течение 24—40 ч. Затем
его измельчают и основную массу измельченного продукта обраба-
тывают водой с целью разложения непрореагировавшего карбида
кальция и минеральным маслом для уменьшения пыления. При
переработке цианамида кальция в цианплав и другие химические
продукты его подвергают более грубому помолу (стр. 1524). В этом
случае не производят обработки его водой и маслом.
Процесс азотирования карбида проводят следующим образом.
Загрузку шихты в печь производят в течение 10—12 мин. Затем
закрывают печь, продувают ее азотом в течение 8—10 мин для
удаления воздуха и включают нагрев электрода. Нагревание
шихты до температуры интенсивного взаимодействия карбида
с азотом продолжается в течение 2—3 ч при непрерывной подаче
азота под избыточным давлением 5—10 мм вод. ст. После этого
прекращают нагрев, электрод удаляют из печи, закрывают элек-
тродный канал и увеличивают давление подаваемого азота до
20 мм вод. ст. (сверх атмосферного). Спустя 4 ч начинают удалять
из печи выделяющиеся газы (ацетилен и др.) через отвер-
стие в крышке, закрывающей электродный канал. Подачу азота
в печь продолжают, пока температура в печи не снизится до
600—700°.
После этого подачу азота прекращают и оставляют цианамид
в печи на 1—2 ч для остывания, и затем его выгружают. Весь про-
цесс с учетом времени загрузки и выгрузки длится 40—50 ч. Про-
изводительность описанной печи за 1 цикл составляет около 1,5 т
цианамида кальция. На производство 1 т цианамида кальция
(в пересчете на продукт, содержащий 18% азота) расходуют:
0,785 т карбида кальция, 630 м3 азота, 0,020 т плавикового шпата
(85% CaF2) и 90 квт-ч электроэнергии.
Производство цианамида кальция является взрывоопасным
вследствие возможности выделения ацетилена при соприкосновении
карбида кальция с влагой и образования взрывчатой смеси из аце-
тилена и воздуха. Поэтому все аппараты должны быть заполнены
азотом (вместо воздуха) и хорошо герметизированы, а производ-
ственные помещения должны вентилироваться для предотвращения
проникания вредной цианамидной пыли. В присутствии ничтожных
количеств спирта в организме (даже от лекарств, приготовленных
на спирте) вдыхание цианамидной пыли вызывает приступы уду-
шья. Рабочий персонал, обслуживающий производство цианамида
кальция, должен быть снабжен защитными средствами — респира-
тором, очками и сухой спецодеждой. Перед началом работы надо
смазать лицо и руки тонким слоем вазелина.
Цианамид кальция
1521
Получение гранулированного цианамида кальция
Помимо порошкообразного цианамида кальция выпускают гра-
нулированный в виде зерен размером 2—5 мм, который не пылит
и более удобен при пользовании. Перед грануляцией цианамид
кальция увлажняют с целью гашения содержащейся в цианамиде
окиси кальция и разложения остатков карбида кальция. Приме-
няют разные способы гранулирования цианамида кальция 45>47.
Наиболее простой — это грануляция в мешалках непрерывного дей-
ствия. Более совершенный гранулятор представляет собой два
вальца, из которых один сплошной, другой — полый с дырчатой
поверхностью. Массу вводят между цилиндрами — вальцами гра-
нулятора. При вращении полого цилиндра цианамид кальция про-
давливается через отверстия внутрь цилиндра и превращается при
этом в палочки, которые удаляются из внутренней части цилиндра
при его вращении, и ленточным транспортером передаются в су-
шильное отделение.
При увлажнении цианамида кальция происходит частичная по-
теря азота вследствие взаимодействия CaCN2 с водой:
CaCN2 + ЗН2О = 2NH3 + СаСОя
Поэтому более целесообразным, по-видимому, является связы-
вание свободной окиси кальция не водой, а двуокисью углерода 43,
а также использование для гидратации раствора Са (NO3) 2 48,49.
Для придания гранулам цианамида кальция большей механиче-
ской прочности предложено 50 их дополнительно увлажнять и вы-
сушивать нагретым воздухом при 80—90°. При этом 10—15% воды
химически связывается с цианамидом кальция, а остальное коли-
чество воды удаляется при сушке.
Другие методы получения цианамида кальция
Азотирование карбида кальция непрерывным путем возможно
в печах различной конструкции43: канальных (или туннельных)
печах с газовым или электрическим обогревом, вращающихся труб-
чатых печах, в шахтных печах, в вертикальной печи, выполненной
в виде усеченного конуса и др.51. Из них некоторое применение
нашли канальные и вращающиеся печи.
Основными причинами, затрудняющими проведение процесса
непрерывным путем, являются большая длительность реакции азо-
тирования, трудность регулирования температуры и спекание обра-
зующегося продукта.
Для ускорения процесса предлагают52 применять порошко-
образный карбид.
Описано53 производство цианамида кальция во вращающихся
печах производительностью ~20—22 тыс. т в год (55—60 т в
1522
Гл. XLI. Цианистые соединения
сутки) цианамида кальция с применением карбида кальция разме-
ром 0,075—2 мм с добавкой 1—2% СаС12. Печь общей длиной 12 м
и наружным диаметром 3 м установлена под наклоном 3° и вра-
щается со скоростью 5 об/мин. Азотирование начинается при 750°.
Отвод тепла реакции производится при помощи ребер, приварен-
ных на наружной поверхности средней части барабана, где темпе-
ратура поднимается до 1000°. Пуск печи осуществляется сжиганием
антрацита и после достижения необходимой температуры в даль-
нейшем реакция азотирования протекает автотермически.
Получение порошкообразного цианамида кальция (вместо спек-
шегося продукта) возможно 54’55 при вдувании тонкодисперсного
карбида (с размерами частии 0,09—0,058 мм) сжатым углеводо-
родным газом в башню — печь, куда одновременно поступает азот,
и для поддержания ~800° кислород в количестве, необходимом
только для сжигания углеводородов до окиси углерода и водорода
(но не до двуокиси углерода и Н2О). Азотирование карбида каль-
ция в печи со взвешенным слоем 56 возможно явится лучшей осно-
вой для создания совершенного непрерывного способа получения
цианамида кальция.
Представляют также интерес предложения по ускорению азоти-
рования в периодических печах. Так, установка в цилиндрических
печах трех электродов (вместо одного, центрального) позволяет
сократить время азотирования57 на ~20%. При загрузке цен-
тральной части печи высоколитражным карбидом (260—270 л/кг),
а периферийных частей — мелочью достигается лучшее использо-
вание тепла и более быстрое азотирование мелкого карбида за счет
избытка тепла, получаемого в начале процесса 53.
Некоторые преимущества в общем технологическом процессе
получения карбида и цианамида кальция может дать введение до-
бавки CaF2 в шихту, используемую для производства карбида59.
С другой стороны, имеются указания 60 на большую эффективность
в процессе получения цианамида кальция добавок NaF, A1F3 или
NasAlFe по сравнению с CaF2.
Рекомендуют 61> 62 производить цианамид кальция, содержащий
до 2,5% азотнокислого кальция, обработкой его распыленной
40—60%-ной азотной кислотой.
Представляют интерес бескарбидные способы получения циан-
амида кальция (с содержанием до 35% азота) взаимодействием
аммиака с карбонатом или окисью кальция 63-66 при 700—800
я присутствии в качестве катализатора CaF2 по реакции:
СаСОз + 2NH3 = CaCN2 + ЗН2О
Равномерная температура (~750°) в реакционном слое может
быть достигнута 64 применением гранулированного карбоната каль-
ция с постоянной величиной гранул.
Производство цианидов натрия и калия
1523
При использовании окиси кальция реакция протекает в при-
сутствии ^углеродсодержащих соединений (СОг, СН4, СО и др.):
СаО + СО2 + 2NH3 = CaCN2 + ЗН2О
СаО + СИ4 + 2NH3 = CaCN2 + Н2О + 4Н2
СаО + 2СО + 2NH3 = CaCN2 + СО2 + Н2О + 2Н2
Продукт, образующийся по последней реакции, содержит до
31% азота 67’68 (вместо 18—20% в продукте, полученном из кар-
бида кальция). Взаимодействием известняка с аммиаком в при-
сутствии метана или пропан-бутановой смеси при 1000—1150° были
получены в лабораторных условиях продукты, содержащие до
33,5% азота69. Примеси соединений Ре20з и Р2О5, содержащиеся
в известняке, снижают концентрацию азота в продукте вследствие
взаимодействия их с углеводородами. Примесь водяных паров в га-
зовой смеси уменьшает использование аммиака70.
Процесс можно осуществить в аппаратах со взвешенным сло-
ем 71, во вращающихся барабанах и в печах других конструкций
с применением известняка или извести с мелкими или крупными
размерами зерен или кусков.
Отходящие из реактора газы, содержащие HCN, NH3 и СО2,
можно регенерировать 72 и вновь использовать.
Чистый цианамид кальция, не содержащий графита и состоя-
щий на 97% из CaCN2, можно получить из карбамида и извести73.
Вначале получают при 300° промежуточный продукт — смесь ци-
анурата, цианата и окиси или карбоната кальция, который затем
после охлаждения и измельчения прокаливают при 700° в течение
14 ч. Предложено 74 получать белый цианамид кальция из Са(ОН)2,
СО и N2 в печи со взвешенным слоем, состоящей из нескольких
камер, разделенных подами и каналами. В нижнюю камеру подво-
дятся реакционные газы, и из нее отводится готовый продукт.
В верхнюю камеру подается гидроокись кальция, и из нее отво-
дятся отходящие газы. Вместо Са(ОН)2 можно также использовать
СаО75, предварительно обработанную при помощи НС1 в псевдо-
ожиженном слое при 500—950°.
В связи с развитием производства синильной кислоты из метана
и аммиака, перспективным является получение цианамида кальция
взаимодействием карбоната кальция с цианистым водородом при
850—900°. Выход цианамида кальция при этом достигает 98%, и
образующийся продукт не содержит полимеров цианистого водо-
рода 76.
ПРОИЗВОДСТВО ЦИАНИДОВ НАТРИЯ И КАЛИЯ
Существует много способов получения цианистых солей натрия,
калия и других металлов:
1) нейтрализация синильной кислоты или поглощение циани-
стого водорода растворами щелочей или карбонатов;
1524
Гл. XL1. Цианистые соединения
2) взаимодействие цианамида кальция с углем и карбонатом
натрия или хлоридом натрия (получение цианплава);
3) фиксация азота смесью соды с углем;
4) восстановление цианатов, полученных из NH3 и карбонатов;
5) сплавление амида натрия с углем;
6) сухая перегонка барды;
7) получение цианидов из коксового газа.
До недавнего времени основными методами получения циани-
стых солей были переработка цианамида кальция в цианплав и
сплавление амида натрия с углем 77. Полученные таким образом
цианиды использовались для производства синильной кислоты 78’79.
В настоящее время освоены в промышленности экономичные ме-
тоды прямого синтеза синильной кислоты из природного газа и ам-
миака (стр. 1532). По мере расширения производства синильной
кислоты, она может быть использована и для получения цианистых
солей.
Получение цианистых солей из синильной кислоты
Нейтрализация синильной кислоты растворами щелочей или
карбонатов является наиболее простым способом, позволяющим
получить чистые цианистые соли, например:
HCN + NaOH = NaCN + Н2О
Расход реагентов можно регулировать потенциометрическим ме-
тодом 80.
Концентрированные растворы чистых солей высушивают в ва-
куум-вальцовых сушилках или другими способами, или из них кри-
сталлизуют 81 цианид, а маточный раствор после отделения центри-
фугированием от кристаллов возвращают в процесс. При предвари-
тельной подсушке кристаллов на центрифуге инертным газом,
подогретым до 30—40°, получается соль, содержащая около 6%
влаги82. Окончательную сушку продукта можно производить при
ломощи газов, имеющих температуру ~90°. В высушенной соли
остаются примеси щелочи.
Для понижения содержания щелочи в сухом цианиде натрия
предложено 83 пропускать над ним в течение 5—7 мин струю сухого
воздуха при ПО—120°, содержащего 0,5—1 % СОг.
Представляет интерес получение цианида калия в результате
обменной реакции в расплаве84:
КС1 + NaCN KCN + NaCl
Получающийся этим методом цианид калия практически не со-
держит других анионов.
Производство цианидов натрия и калия
1525
Получение цианплава из цианамида кальция
При высоких температурах технический цианамид кальция
в присутствии угля превращается в цианид кальция:
CaCN2+C 7—Ca(CN)2 — 24,6 ккал
На этом основано промышленное получение цианидов сплавлением
цианамида кальция с углем и с поваренной солью или содой:
CaCN2 + C + 2NaCl = 2NaCN +СаС12
CaCN2 + С + Na2CO3 = 2NaCN + СаО + СО2
Реакция с содой идет при 800—850°. Реакция с поваренной
солью протекает при значительно более высокой температуре
(1400—1500°). Несмотря на это, в промышленности чаще приме-
няется поваренная соль, более дешевая, чем сода. Кроме того, реак-
ция с поваренной солью протекает намного быстрее, чем с содой.
Сплавление производят в электрических печах.
Образовавшийся цианплав выпускают из печи через летку при
~ 1500°. Так как плав нестоек и при медленном остывании в пре-
делах от 800 до 400° распадается обратно на цианамид и уголь,
то его необходимо охладить быстро. Охлаждение производят на
поверхности вращающегося барабана, внутрь которого подают
проточную воду. Застывший плав снимается с барабана ножом и
поступает на упаковку в стальные барабаны, которые быстро за-
вальцовывают во избежание потери продуктом синильной кислоты
под действием двуокиси углерода и влаги воздуха. Полученный
таким способом продукт называется цианплавом или черным ци-
анидом (черный цвет обусловлен присутствием угля). Цианплав
содержит цианиды в виде NaCN и Ca(CN)2 и примеси NaCl, СаС12>
С, СаС2 и др. На производство 1 т цианплава (45% NaCN) расхо-
дуют 0,72 т цианамида кальция, 0,3 т поваренной соли и 960—
1000 квт-ч электроэнергии.
Большую часть вырабатываемого цианплава используют не-
посредственно без дальнейшей его переработки на чистые циани-
стые соли, например для окуривания растений и для дезинсекции.
Цианплав смачивают водой, в результате чего он разлагается на
воздухе с выделением газообразной синильной кислоты:
Ca(CN)2 + СО2 + Н2О = СаСОз + 2HCN
Выделение цианистого водорода ускоряется при измельчении
цианплава до величины частиц менее 1 мм и подогреве смеси его
с водой 85.
Фиксация азота смесью соды с углем
При пропускании азота через нагретую смесь натрия и угля об-
разуется цианид натрия:
2Na + 2С + Ы2 = 2NaCN + 46 ккал
1526
Г л. XLI. Цианистые соединения
Эта реакция значительно ускоряется в присутствии металлов Я
(Fe, Ni, Мп). Ввиду того, что натрий является весьма дорогим ма- |
териалом, получение цианида натрия осуществляют фиксацией 1
азота смесью соды и угля при 950—1000° под давлением 15 ат |
в присутствии катализаторов86. При этом, по-видимому, сода вое- |
станавливается до натрия I
Na2CO3 + 2С = 2Na + ЗСО — 186 ккал .'1
а натрий реагирует с углем и азотом. В общем процесс идет по
суммарной реакции: |
Na2CO3 + 4С + N2 = 2NaCN + ЗСО - 140 ккал 1
Вместо соды могут применяться другие карбонаты (К2СО3, I
ВаСОз и пр.), причем получаются соответствующие цианиды. Пред- 1
ложено 87 осуществлять процесс в присутствии паров воды с целью 1
получения непосредственно цианистого водорода. Д
Восстановление цианатов, полученных из аммиака и карбонатов I
По этому способу за рубежом получали значительные количе- I
ства цианистых солей. Он заключается в обработке газообразным 1
аммиаком раскаленной смеси щелочного карбоната и угля. При Ч
этом вначале образуется цианат: 1
K2CO3 + NH3 = KCNO + KOH + H2O 1
Затем цианат восстанавливается углем в цианид: I
KCNO + С = KCN + СО J
Из полученного плава цианид выщелачивают водой, затем вы-
деляют выпаркой и кристаллизацией. 1
Возможно88 получение цианида из ВаСОз и NH4C1 в присут- I
ствии металлического калия при 600—700°. Реакция заканчивается I
в течение 1 ч. По-видимому, процесс протекает по следующей I
схеме: I
2ВаСО3 + 2NH4C1 = Ba(CNO)2 + ВаС12 + 4Н2О I
Ba(CNO)2 + 2К - 2KCN + ВаО + 1/2О2 I
Обработкой плава кислотой отгоняют HCN в раствор щелочи.
Выход HCN составляет 93—95%. При использовании металличе-
ского натрия вместо калия выход HCN снижается до 50%.
Сплавление амида натрия с углем
При обработке расплавленного натрия сухим газообразным ам-
миаком при 350—500° в периодически или непрерывно действую-
щих чугунных ретортах образуется амид натрия:
2Na + 2NH3 = 2NaNH2 + Н2
Производство цианидов натрия и калия
1527
Расплавленный амид натрия обрабатывают в стальных котлах
древесным углем при 600—800°. Образующийся вначале цианамид
натрия
2NaNH2 + С - Na2CN2 + 2Н2
превращается затем в цианид
Na2CN2 + С - 2NaCN
Суммарной реакцией получения цианида натрия этим методом
является:
2Na + 2С + 2NH3 - 2NaCN + ЗН2
На некоторых заводах обе стадии процесса — получение амида
натрия и переработку его в цианид — осуществляют в одном аппа-
рате — в котле, заполненном углем, в который подают аммиак и
натрий. Этот способ дает возможность получать цианистый натрий
с большим выходом — 98—99%. Полученный расплавленный циа-
нистый натрий отделяют от угля фильтрованием. Продукт содер-
жит до 97% NaCN. По другому варианту цианистый натрий выще-
лачивают из остывшего плава водой, растворы очищают и перера-
батывают на твердый продукт.
Сухая перегонка барды
Отходы свеклосахарных производств — черная патока или ме-
ласса и винокуренных заводов — барда — содержат до 2% азота.
При сухой перегонке этих отходов в закрытых ретортах остается
бардяной кокс и выделяются газы, содержащие СО2, СО, Н2, СН4,
С2Н2, NH3, N2 и метиламины. Эти газы перегревают до 1000—1100°,
причем метиламины разлагаются с образованием цианистого водо-
рода:
3ch3nh2 - NH4CN + HCN + СН4+ЗН2
(CH3)3N= HCN + 2CH4
Возможно, что происходит и взаимодействие аммиака с углем
nh3 + c=hcn + h2
а также и ряд других реакций, ведущих к образованию HCN.
В результате этих реакций газ, выходящий из перегревателя,
содержит до 7% HCN и до 7% NH3. Для поглощения аммиака
охлажденный газ промывают серной кислотой, после чего циани-
стый водород поглощают растворами щелочей.
При сухой перегонке барды из общего количества содержаще-
гося в ней азота лишь 25% превращается в цианистые соединения,
примерно столько же — в аммиак; половина азота теряется в виде
элементарного азота. : .
Предложены различные способы выделения HCN из газовой
смеси, при которых не расходуется серная кислота;, нацример,
24 м, е. позин
1528
Гл. XLI. Цианистые соединения
промывкой водным раствором NH4OH при 50—60° 89’90 двуокись ,
углерода можно связать в (ЫН^зСОз89. В присутствии СО2 раство- >
римость HCN мала и основная его масса остается в газе90, из кото-
рого он поглощается щелочным раствором.
Из водных растворов HCN, содержащих также другую кислоту
и аммиачную ее соль, можно отогнать под вакуумом практически
весь цианистый водород и небольшое количество воды 91.
Получение цианидов из коксового газа
Коксовый газ содержит небольшие количества цианистых со-
единений, в которые переходит всего 1—2% азота, содержащегося
в коксуемом угле. Образование цианистого водорода происходит •
из содержащегося в газе аммиака главным образом по следующим
реакциям:
NH3 + CO = HCN + H2O
2NH3 + СгН2 = 2HCN + ЗН2
Некоторые количества HCN образуются и по другим реакциям,
в результате взаимодействия аммиака с углеродом, метаном, эти-
леном, а также в результате разложения имеющегося в газе три-
метиламина на HCN и СН4. При охлаждении газа образуется <
легколетучий цианистый аммоний, который, взаимодействуя с на-
ходящимся в газе сероводородом, дает роданистый аммоний:
2NH4CN + 2H2S + О2 = 2NH4NCS + 2Н2О
При водной промывке коксового газа в раствор переходят, на-
ряду с HCN, также NH3, H2S и СО2. Дистилляцией полученного
раствора можно выделить в виде дистиллята, отводимого из верх-
ней части колонны, до 98% от всего количества HCN в смеси с не- '
большим количеством аммиака92. Сероводород и двуокись угле-
рода выходят из колонны в газообразном виде. В кубе остается
до 99,5% NH3 в виде разбавленного раствора. -•
Представляет интерес выделение небольших количеств синиль-
ной кислоты из сточных вод производства сульфата аммония из
аммиака коксового газа. Они образуются при промывке, ведущейся
с целью обезвреживания газов, отходящих из сатураторов погло- '
щения аммиака серной кислотой и содержащих 100—3000 мг/л
синильной кислоты и сероводорода и незначительные количества
аммиака. Предложена 93 двухступенчатая очистка сточных вод, за-
ключающаяся в раздельной отдувке из них воздухом сероводорода
и синильной кислоты. Скорости диффузии HCN и H2S из жидкой
фазы в газовую почти одинаковы, но коэффициент растворимости
синильной кислоты значительно больше. Поэтому сероводород от-
дувается в ~ 100 раз быстрее и выделяется в первую очередь. От-'
дувочные газы первой ступени циркулируют в процессе и исполь-
Получение роданидов натрия и аммония
1529
зуются для репульпации суспензии в сатураторе сульфата аммо-
ния (рис. 457). Воздух после сатуратора вместе с остаточными
компонентами коксового газа и выделившимися парами поступает
в водяной холодильник, где охлаждается с 95 до 30°. Полученный
конденсат вместе с паро-газовой фазой направляется на промывку,
где достигается очистка раствора от сероводорода. Выделившийся
сероводород отводят из про-
мывной башни в систему
сероочистки или на сжига-
ние или для других целей.
Очищенный от сероводоро-
да раствор направляется во
вторую ступень, где допол-
нительным потоком воздуха
отдувается цианистоводо-
родная кислота, перераба-
тываемая далее в циани-
стый натрий.
При сероочистке газа
мышьяково-содовым спосо-
бом цианистые соединения
переходят в роданиды (см.
ниже). Из мокрых способов
очистки для улавливания
цианистых соединений наи-
большее значение имеет
Рнс. 457. Схема получения синильной
кнсдоты' и сероводорода из сточных вод
коксового производства:
/—сатуратор. 2 —барботер; 3 ~ холодильник;
4 — промыватель; 5 и 5 —отдувочные колонны.
промывка газа раствором железного купороса. Образующиеся при
этом нерастворимые ферроцианиды отделяются в виде так назы-
ваемого цианидного ила, являющегося сырьем для получения фер-
роцианидов. Ферроцианиды образуются также при сухой очистке га-
зов окислами железа и накапливаются в газоочистительной массе.
ПОЛУЧЕНИЕ РОДАНИДОВ НАТРИЯ И АММОНИЯ ИЗ ОТБРОСНЫХ
РАСТВОРОВ МЫШЬЯКОВО-СОДОВОЙ ОЧИСТКИ ГАЗОВ
В процессе очистки водяного и коксового газов от сероводорода
мышьяково-содовым способом цианистые соединения, содержа-
щиеся в газе, переходят в роданиды:
HCN + NaHS + V2O2 = NaNCS + Н2О
Основной побочной реакцией в этом процессе является окисле-
ние гидросульфида натрия и образование тиосульфата натрия
(стр. 466):
2NaHS + 2О2 = Na2S2O3 + Н2О
Для уменьшения количества накапливающихся солей в рабочем
растворе часть его выводят из цикла и заменяют свежим. Из
24*
1530
Г л. XL1. Цианистые соединения
выведенного раствора выделяют тиосульфат натрия. В маточных
растворах, остающихся после кристаллизации тиосульфата, содер-
жатся 150—200 г)л ЫагЗгОз и 300—350 г/л роданида натрия. Рас-
творы обрабатывают серной кислотой для разрушения тиосульфата
натрия:
3Na2S2O3 + H2SO4 = 3Na2SO4 + 4S + H2O
При этом происходит разложение тиосульфата натрия на 90%,
В то время как роданид разлагается лишь на 15—18% 94.
Разложение тиосульфата натрия производят при одновременном
выпаривании раствора95. К нейтрализованному маточному рас-
твору с тиосульфатной установки (или нейтрализованному для вы-
деления мышьяка рабочему раствору мышьяково-содовой очистки)
добавляют серную кислоту и нагревают его в выпарном аппарате
до кипения. После 30—45-минутного выдерживания для лучшего
разложения тиосульфата раствор выпаривают до содержания
520—580 г/л роданида натрия. При выпаривании раствора кристал-
лизуется сульфат натрия. При достижении концентрации NaNCS
в растворе более 500 г/л, Na2SO4 выделяется на 97—99%. Даль-
нейшим выпариванием повышают концентрацию NaNCS до 800 г/л,
после чего при охлаждении раствора до 25—30° из него кристалли-
зуется двухводный роданид натрия. Выход роданида натрия со-
ставляет 60—70%. Полученный продукт содержит 0,2—0,5% тио-
сульфата и 1—4% сульфата натрия96.
Для получения роданида аммония раствор роданида натрия
после первой выпарки, до отделения от сульфата натрия, обраба-
тывают сульфатом и хлоридом аммония. Затем раствор роданида
аммония отделяют от осадка и подвергают концентрированию и
кристаллизации.
Роданид аммония можно получить при промывке коксового газа
суспензией серы 97 при 30—35° или раствором сульфида аммония,
в котором предварительно растворено около 20% серы98.
Роданиды щелочных и щелочноземельных металлов можно
также получить действием разбавленного раствора HCN на рас-
творы сульфидов в смеси с серой или в смеси с высшими полисуль-
фидами99. Для образования тонкодисперсной эмульсии или сус-
пензии серы рекомендуется ввести в раствор ациклический спирт
(например, метилциклогексанол) 100 или натриевую соль алкилиро-
ванной нафталинсульфокислоты 101.
ПРОИЗВОДСТВО ЖЕЛЕЗИСТО- И ЖЕЛЕЗОСИНЕРОДИСТЫХ СОЛЕИ
Железистосинеродистые соли
Источником сырья для получения ферроцианидов являются от-
работанная газоочистительная масса и циановый ил (стр. 1529). Эти
массы обрабатывают вначале водой для извлечения растворимых
Производство железисто- и железосинеродистых солей
1531
соединений — роданида аммония, сульфата аммония и других,
а затем их смешивают с известковым молоком и нагревают до ки-
пения. При этом нерастворимые комплексные соединения железа
переходят в растворимый железистосинеродистый кальций:
Fe4[Fe(CN)6]3 + 6Са(ОН)2 = 3Ca2Fe(CN)6 + 4Fe(OH)3
Железистосинеродистый кальций перерабатывают на железисто-
синеродистый натрий или калий обменным разложением с пова-
ренной солью, содой, сульфатом натрия, хлоридом калия, поташом
и т. п.
Железистосинеродистый натрий получается также при не-
посредственной промывке коксовых газов раствором соды в при-
сутствии углекислого железа i02. В растворе образуется Na4Fe(CN)e,
который очищают кристаллизацией.
Промышленное значение имеет способ получения железисто-
синеродистых калия и натрия из цианплава. При обработке циан-
плава раствором железного купороса цианиды переходят в ферро-
цианиды:
4Ca(CN)2 + 4NaCN + 2FeSO4 = Ca2Fe(CN)6 + Na4Fe(CN)e +2CaSO4
После отделения от шлама, в котором содержатся гипс и нерас-
творимые примеси цианплава (уголь, Са(ОН)2 и пр.), раствор фер-
роцианидов перерабатывают на чистые соли методами обменного
разложения.
Предложено 103 получать ферроцианид калия обработкой циан-
плава хлористым железом и после обменной реакции с КС1
разложением двойной калий-кальциевой ферроцианидной соли рас-
твором едкого кали.
Способы получения ферроцианидов натрия и калия непосред-
ственно из цианистых натрия и калия и соединений железа в на-
стоящее время утратили свое значение и применяются лишь в не-
больших масштабах.
Получение железистосинеродистого калия из животных отбро-
сов, содержащих азот (сухая кровь, рога, копыта, волос, обрезки
кожи ит. п.), — в свое время единственный промышленный способ
получения цианистых соединений — теперь почти не применяется.
Этот способ заключается в сплавлении животных отбросов с пота-
шом и железными стружками в чугунных котлах с мешалками. Из
застывшего плава выщелачивают K4Fe(CN)6, который затем вы-
кристаллизовывают.
Железосинеродистые соли
Эти соли получают окислением железистосинеродистых солей
электрохимическим способом или с помощью окислителей, напри-
мер хлора. При электролизе растворов железистосинеродистых
1532
Гл. XLI. Цианистые соединения
солей ион Fe(CN)g" окисляется на аноде в ион Fe(CN)g", а на
катоде образуется щелочь и выделяется водород.
В опытном электролизере, разделенном керамической диафраг-
мой на анодную и катодную части, с применением пластинчатых
анодов из графита, покрытых эпоксидной смолой, и ртутного ка-
тода, получали продукт 104, содержащий в среднем 99,23%
K3Fe(CN)6, 0,35% K4Fe(CN)6 и 0,42% Fe(OH)3. При плотности
тока 3,5—4,3 а/см2, напряжении на ванне 4,5—5,0 в выход по току
составил 96,5—98,0%. Электролиз проводят при 50° с циркуляцией
маточного раствора, получаемого после выделения из анолита
охлаждением до 15° кристаллов КзРе(СЫ)6- К циркулирующему
раствору добавляют железистосинеродистый калий до первоначаль-
ного состава.
При окислении хлором реакция
2Na4Fe(CN)6 + С12 = 2Na3Fe(CN)e + 2NaCl
идет как в растворе, так и при действии газообразного хлора на
твердую соль. Полученные соли разделяют кристаллизацией. При
обработке хлором твердого ферроцианида иногда выпускают смесь
солей (феррицианида и хлорида), так как хлориды обычно не яв-
ляются вредной примесью.
ПРОИЗВОДСТВО СИНИЛЬНОЙ кислоты контактными
СПОСОБАМИ23
Производство синильной кислоты взаимодействием цианистого
натрия или цианплава с серной кислотой является громоздким и
неэкономичным. Получение синильной кислоты этими методами
связано со значительными затратами энергии (до 16 500 квт-ч на
1 т синильной кислоты) и серной кислоты. При этом образуются
большие количества неиспользуемых отходов — сернокислого нат-
рия или сернокислого кальция, сернистых соединений. Эти способы
не могут обеспечить все возрастающие потребности в синильной
кислоте для производства высокополимерных продуктов — произ-
водных акриловой и метакриловой кислот.
В течение последних десятилетий широко распространяются
различные способы прямого синтеза синильной кислоты 105-|1С, осно-
ванные на термическом или каталитическом взаимодействии угле-
водородов с азотом или аммиаком. Из них наибольшее значение
получили контактные способы образования синильной кислоты из
аммиака и углеводородов или продуктов их неполного окисления.
Способы синтеза синильной кислоты
Разработаны и частично практически осуществлены следующие
епособы синтеза синильной кислоты.
1. Взаимодействие аммиака с окисью углерода в присутствии
катализатора — окиси алюминия или смеси окислов СегОз и
Производство HCN контактными способами
1533
VjOs ш. Реакция
NH3 + СО = HCN + Н2О
протекает с небольшой скоростью. Выход синильной кислоты при
700° и 30-кратном избытке окиси углерода 105 не превышает 65%
(в расчете на аммиак). Для циркуляции газов после выделения
HCN и NH3 предложено 118 окислить содержащийся в них водород
при 300—450° в присутствии медного, кобальтового и других ката-
лизаторов.
2. Пирогенетическое разложение (электро- или термокрекинг)
углеводородов с азотом или аммиаком. Процесс сводится к взаимо-
действию продуктов разложения — углерода, водорода и азота
или углерода с аммиаком
2С + Н2 +N2 - 2HCN
или
C + NH3 = HCN + H2
Способ электрокрекинга связан с большим расходом электро-
энергии (50—70 квт-ч на 1 кг синильной кислоты) и затрудне-
ниями технического порядка вследствие отложения сажи. Термо-
крекинг смеси метана и аммиака при 1200—1400° идет с большой
скоростью и выходом HCN, превышающим 90% 106 и достигающим
даже 99,5% 11S- Однако аппаратурное оформление процесса сопря-
жено с большими трудностями, как и при электрокрекинге.
Имеются сведения 120 об осуществлении процесса синтеза HCN
из NH3 и легких углеводородов в псевдоожиженном слое с благо-
приятными технико-экономическими показателями.
Псевдоожижению подвергаются частицы кокса, движущиеся во-
круг обогревающих электродов, в печи специальной формы. Обра-
зование цианистоводородной кислоты протекает при 1350—1650°
и атмосферном давлении без катализатора по реакции (например,
в случае применения пропана):
С3Н8 + 3NH3 = 3HCN + 7Н2
Выход HCN достигает 85—90%. На 1 кг HCN расходуется
0,64 кг пропана, 0,74 кг аммиака и около 6 квт-ч электроэнергии.
В качестве побочного продукта получается 2,3—2,4- м3 98%-ного
водорода.
Описаны 121 результаты лабораторных опытов получения циа-
нистоводородной кислоты из угля и аммиака. Уголь, измельченный
на сите 0053, предварительно нагревали до 400—500° для предот-
вращения забивания реактора продуктами коксования. Выход HCN
при 1250° составил от 75 до 100% от теоретического в зависимости
от скорости подачи газа. Предварительная аммонизация угля
позволяет значительно ускорить процесс.
3. Синтез синильной кислоты из метана и аммиака в элек-
троразрядах высокого напряжения или высокой частоты. При
1534
Гл. XLL Цианистые соединения
напряжении 4500—5000 в под уменьшенным давлением и соотноше-
нии NH3: СН4, равном 1:1, выход синильной кислоты составляет
70% по аммиаку 107- 108. При избытке метана или аммиака можно
полностью превратить в синильную кислоту соответственно аммиак
или метан. Расход энергии составляет 25—30 квт-ч на 1 кг HCN.
При осуществлении процесса при атмосферном давлении в высоко-
частотном разряде с частотой 106—107 периодов в секунду расход
электроэнергии снижается и составляет при переработке смеси
метана и азота 23 квт-ч, а при переработке смеси керосиновых
погонов и азота— 17 квт-ч на 1 кг HCN 109. Изучено 122 образова-
ние HCN при облучении смесей аммиака с углеводородами в атом-
ном реакторе.
4. Каталитическое прямое окисление метана и аммиака. Сущ-
ность способа 110’111 заключается в частичном окислении кислоро-
дом смеси метана и аммиака в присутствии катализатора:
СН4 + NH3 + 1,5О2 = HCN + ЗН2О + 113,3 ккал
Взаимодействие метана и аммиака в присутствии воздуха яв-
ляется сильно экзотермической реакцией. Это позволяет осуще-
ствлять процесс автотермично. В качестве катализаторов могут
быть использованы металлы платиновой группы (Pt, Ir, Rh, Pd,
Os), а также окислы металлов A12O3, Fe2O3.
В связи с отсутствием энергетических затрат на проведение
реакции образования синильной кислоты этот метод оказался наи-
более экономичным, используемым в довольно крупных производ-
ственных масштабах (до 10 000 т и больше в год). Большое значе-
ние этого способа обусловливает целесообразность его непрерыв-
ного усовершенствования123. Наряду с этим проводятся
изыскания по осуществлению синтеза цианистого водорода из ме-
тана и аммиака без добавления воздуха. Описана полузаводская
установка производительностью 50 т HCN в месяц, на которой вы-
ход по метану составляет 91%, а по аммиаку 83% 115. При 1600°
в продуктах каталитического взаимодействия СН4, NH3 и О2 содер-
жится до 8,8% HCN ,24.
5. Формамидный способ является разновидностью способа по-
лучения синильной кислоты из окиси углерода и аммиака. Он осно-
ван на дегидратации формамида, который получается при взаимо-
действии окиси углерода и аммиака в щелочном растворе
метилового спирта.
Из новых направлений представляет интерес образование циа-
нистого водорода при очень высоких температурах (3000—4000°),
достигаемых при адиабатическом сжатии исходных газов до
5—10 тыс. ат или развиваемых в плазменных реакторах. Добавка
к смесям СН4 и N2 или С2Н2 и N2 значительных количеств Аг спо-
собствует повышению температуры, развиваемой при сжатии '06.
Максимальный выход HCN равен 1% от начального объема смеси,
Производство HCN контактными способами
1535
содержащей 10% СН4, 13% N2, 77% Аг при давлении 8500 ат и
4% для смеси 25% С2Н2, 32% N3, 43% Аг при давлении 7400 ат.
В настоящее время во многих странах широко развернулись
исследования получения синильной кислоты, а также циана (ди-
циана) в плазменных реакторах (в потоке низкотемпературной
плазмы). Высокие температуры и особые свойства плазмы позво-
ляют осуществить в ней синтез синильной кислоты как из элемен-
тов, так и из метана и азота или углерода и аммиака или метана
и аммиака 125-128.
Производство синильной кислоты контактным окислением
метана и аммиака23’129
Непосредственное образование цианистого водорода из метана
и аммиака основано на эндотермической реакции:
CH4 + NH3= HCN + 3H,
Скорость этой реакции резко повышается при одновременном
сжигании водорода, т. е. при неполном окислении метана и ам-
миака. При этом реакция становится экзотермической. Высокие
выходы с одновременным значительным повышением скорости
реакции достигаются при каталитическом осуществлении процесса
(например, на платиновом и других катализаторах).
Существуют различные представления о механизме образова-
ния цианистого водорода при неполном окислении метана и ам-
миака на катализаторе. Предполагают, что процесс протекает с об-
разованием промежуточного соединения — нитроксила HNO или
распадом метана и аммиака на атомы или радикалы ,3°.
Углеводородные радикалы, образующиеся из углеводородов .и
атомов О и Н, могут реагировать 131 с NH3 в зоне горения даже
при отсутствии атомов азота. Более вероятной является радикаль-
но-цепная теория 132’133. Согласно этой теории механизм процесса
складывается из следующих стадий: 1) адсорбции кислорода на
платиновом катализаторе с образованием весьма активных поверх-
ностных соединений, 2) взаимодействия молекул метана и аммиа-
ка с поверхностными соединениями на катализаторе с образова-
нием новых радикалов и бирадикалов (СН3. NH2, ОН, СН2, NH),
3) взаимодействия образующихся радикалов с метаном и аммиа-
ком, в результате чего получается метиламин CH3NH2 по реакциям:
CH2 + NH3 = CH3NH2
nh + ch4 = ch3nh2
4) окисления метиламина кислородом воздуха в синильную кис-
лоту:
CH3NH2 + О2 = HCN + 2Н2О + 77,6 ккал
1536
Гл. XL1. Цианистые соединения
Одновременно с образованием цианистого водорода идут реак-
ции разложения азотных радикалов на азот и водород и окисления
углеродных радикалов в окись углерода и водород. Полезно ис-
пользуется для образования цианистого водорода 60—63% ам-
миака и 56—57% метана. Остальное количество аммиака частично
разлагается (12—18%) на азот и водород, частично (22—28%)
остается непрореагировавшим. Для более полного использования
аммиака необходим небольшой избыток метана и некоторый не-
достаток кислорода по сравнению со стехиометрическим количе-
ством. Оптимальное соотношение газов (по объему) СН4 : NH3
равно 1,1 иО2: СН4 — 1,33—1,43.
Предложено 134 использовать смеси и с иными соотношениями,
например, 16,2% СН4, 10,8% NH3, 18,3% О2 и 54,7% N2. Избыток
кислорода ведет к повышению температуры катализатора и ча-
стичному сгоранию цианистого водорода, т. е. к снижению его вы-
хода.
Скорость процесса зависит от условий диффузии реагентов из
объема к поверхности катализатора и продуктов реакции в обрат-
ном направлении. При большой линейной скорости газа реагенты
не успевают продиффундировать к поверхности катализатора. При
малой линейной скорости продукты реакции, вследствие длитель-
ного их пребывания на катализаторе, разлагаются с отложением
углерода. Обычно выбирают линейную скорость газового потока
в пределах 0,8—1,4 м]сек (считая на холодный газ).
В качестве катализатора применяют сплав платины с ро-
дием или иридием в форме сетки, изготовленной из нитей тол-
щиной от 0,04 до 0,16 мм с количеством отверстий на 1 см2 от
3600 до 500. Свободный зазор между нитями равен от 0,127 до
0,287 мм.
Платиновый катализатор весьма чувствителен к действию раз-
личных примесей газообразных и твердых (пыли) веществ. Осо-
бенно вредным является углерод, образующийся при разложении
нестойких в условиях синтеза углеводородов. Катализатор отрав-
ляется необратимо под влиянием этилена, пропилена и высших
олефинов и особенно при наличии в газе ~0,1% ацетилена. При-
сутствие в газе до 0,1% сероводорода приводит к обратимому от-
равлению катализатора. В отсутствие сероводорода в газе катали-
затор, ранее отравленный сероводородом, быстро восстанавливает
свою активность. Содержание окиси углерода до 8—10% не оказы-
вает влияния на действие катализатора, а присутствие водорода
в некоторой степени благоприятно сказывается на работе катализа-
тора, предотвращая отложение углерода на его поверхности 133.
Резкое снижение активности катализатора происходит при попада-
нии на него железа, меди, свинца, а также при содержании в газе
ничтожных количеств (0,00001%) соединений фосфора и мышьяка.
Поэтому исходные реагенты — метан, аммиак и воздух — тша-
Производство HCN контактными способами
1537
тельно очищают от механических примесей, а также от веществ,
отравляющих катализатор.
Регенерацию отравленного катализатора предложено произво-
дить продувкой через него воздуха, а затем азото-водородной
смеси *35.
Источником метана служат метановые фракции нефтяного или
коксового газа, а также природный газ, содержащие 97% и больше
метана. Очищенные газы смешивают в указанном выше соотно-
Рис. 458. Схема производства синиль-
ной кислоты каталитическим окислением
метана и аммиака:
/ — скруббер; 2, 2', 2° — фильтры; 3, 3'9
3” — расходомеры; 4 —смеситель; 5 — контакт-
ный аппарат; 6 — котел утилизатор; / — погло-
тительная башня; 8, /5 — насосы; 9 — компрес-
сор’. 10 — тарельчатая колонна; п—дистилля-
ционная колонна; 12— конденсатор; 18 — мер-
ник; М—приемник.
шении и направляют в кон-
тактный аппарат (рис. 458),
аналогичный по своей кон-
струкции аппаратам, приме-
няемым для окисления аммиа-
ка в производстве азотной кис-
лоты. Аппарат в верхней части
представляет собой два кону-
са, сложенные основаниями. В
расширенной части помещены
3—6 сеток из платинового
сплава. Нижняя цилиндриче-
ская часть аппарата представ-
ляет собой котел-утилизатор,
служащий для использования
тепла реакции и охлаждения
контактных газов.
Вначале сетку аппарата
разогревают до 300—500°, за-
тем в аппарат вводят аммиач-
но-воздушную смесь (содержа-
щую 10% NH3), которую продолжают пропускать через аппарат в
течение 3—4 ч, пока устанавливается температура 800—900° и до-
стигается определенная .активность катализатора. После этого в
смеситель газов через специальный фильтр и расходомер начинают
подавать метан. В выходящих из контактного аппарата газах со-
держится (при работе на метановой фракции нефтяного газа, содер-
жащей 97—97,5% СН4) около 6% HCN, 1,5—1,7% NH3, 0,2% СО2,
4,3—4,5% СО, 0,5% СН4, 7,5% Н2, 0,1% О2, 56,7% азота, 23—23,5%
водяного пара. Степень превращения аммиака в HCN в зависимо-
сти от условий составляет 63—70%. После контактирования газы
с температурой 900—1000° проходят котел-утилизатор, где быстро
охлаждаются до 150° (т. е. до температуры несколько выше точки
росы с целью предотвращения гидролиза синильной кислоты).
Охлажденную газовую смесь направляют в башню, орошаемую
слабым раствором смеси серной кислоты и сульфата аммония
для поглощения непрореагировавшего аммиака и предотвраще-
ния образования полимеров цианистого водорода 128> гзг-гзэ.
1538 Гл. XLI. Цианистые соединения
Образовавшийся в башне раствор сульфата аммония перерабаты- J
вают в твердый продукт. Оставшуюся газовую смесь сжимают до У
7 ат и подают в тарельчатую колонну, где ее промывают слегка J
подкисленной водой. Цианистый водород, растворяясь, образует 1
синильную кислоту. Оставшийся газ, не содержащий аммиака и q
синильной кислоты, выбрасывают в атмосферу или сжигают.
В выбрасываемых в атмосферу газах допустимое санитарными J
нормами содержание HCN составляет 0,0003 мг)л. Очистка воз- -ч
душных выбросов от синильной кислоты является важной пробле- Я
мой и в процессах, осуществляемых с использованием синильной 1
кислоты. Например, в производстве акриловой кислоты промывка Ц
отбросных газов растворами едкого натра при 60° в насадочных Я
скрубберах не позволяет достичь санитарной нормы. Предложе- 1
но 140 для полной очистки газа, отходящего из щелочного абсор- Ч
бера, от синильной кислоты проводить дополнительную его про- Ч
мывку небольшим количеством чистого раствора щелочи низкой *1
концентрации (0,85—3,5 г/л), не содержащего цианида натрия. 1
Полученный водный раствор синильной кислоты подвергают ди- Ч
стилляции в колпачковой колонне с отгонкой жидкой синильной I
кислоты; кубовый остаток представляет собой слегка подкислен- I
ную воду, возвращаемую в колонну для улавливания цианистого J
водорода. К жидкой синильной кислоте, содержащей 98,5% HCN и 1
1,5% воды 141, а при дополнительной ректификации 142 до 99,5% -1
HCN, добавляют стабилизатор — фосфорную кислоту в количестве '1
0,1—0,2% (или другие кислоты). На производство 1 т HCN расхо- "I
дуют: 1,05—1,08 т метана и 1,05 т аммиака, из которых 0,3 т пре- .1
вращается в сульфат аммония. I
Извлечение аммиака из газовой смеси по описанной схеме яв- I
ляется малосовершенным, связанным с затратами серной кислоты /I
и необходимостью специальной обработки раствора для получения I
твердого сульфата аммония. Более совершенным способом разде- I
ления контактных газов является избирательная абсорбция из них I
аммиака водным раствором пентаэритритбората (или любого кис- I
лого эфира многоатомного спирта и борной кислоты). Образую- I
щаяся аммонийная соль при нагревании выше 100° разлагается I
с выделением аммиака и регенерацией поглотителя. Охлажденные ‘I
в котле-утилизаторе контактные газы направляют в насадочную I
колонну (рис. 459), орошаемую холодным насыщенным водным "1
раствором пентаэритритбората. При этом из газа переходят в рас- I
твор аммиак и цианистый водород. Непоглотившиеся газы сжигают I
в котельной. Вытекающий из поглотительной колонны слабокислый I
раствор после подогрева в теплообменнике направляют в первую . I
десорбционную колонну, где при 80° отгоняется цианистый водород I
попутно с небольшим количеством аммиака. Десорбцию цианистого 4|
водорода осуществляют под пониженным давлением (200—
250 мм рт. ст.). В верхнюю часть колонны подают свежий раствор »
Производство HCN контактными способами
1539
пентаэритритбората, который частично связывает аммиак, отгоняе-
мый в процессе десорбции цианистого водорода. Основное количе-
ство аммиака выделяют в регенерационной колонне. После охлаж-
дения и конденсации воды аммиак возвращают в процесс. Из
полученной аммиачной воды выделяют остаток аммиака в отгон-
ной колонне.
Недостатками описанного метода получения синильной кислоты
являются не полное использование аммиака, образование в каче-
стве побочного продукта окиси углерода и низкая концентрация
Рис. 459. Схема производства цианистого водорода
с разделением аммиака и синильной кислоты:
1 — фильтр; 2— смеситель; 3 — контактный аппарат; 4—котел-
утилизатор; 5 —поглотительная колонна; 6, 10, 14, —
Обменники; 7 —десорбционная колонна; S — скруббер; 9 — ва-
куум-насос; // — регенерационная колонна; 12 — холодильник;
13 — отгонная колонна.
HCN в образующейся газовой смеси, а также определенные потери
платины. Перспективным представляется некаталитическое получе-
ние цианистого водорода из метана и аммиака в псевдоожиженном
сЛОе 121, 127, из (см выше). Нагревание газов до 1400—1600° про-
изводится с помощью специальных графитовых электродов, поме-
щенных в кипящий слой из электропроводных частиц кокса. В этих
условиях концентрация HCN в газовых продуктах достигает
20—25% и основным побочным продуктом является водород. Про-
цесс протекает в кипящем слое с большой скоростью и характери-
зуется быстрым подъемом температуры в реакторе и легкостью
регулирования режима путем изменения скорости прохождения
газа, глубины погружения электродов в кипящий слой и подводи-
мой к электродам мощности. Эти реакторы отличаются длитель-
ностью работы при высоких температурах и могут работать как на
переменном, так и на постоянном токе при напряжении, не превы-
шающем 100 в.
1640
Гл. XLI. Цианистые соединения
Получение цианистого водорода из аммиака и окиси
углерода (формамидный способ)
Этот процесс осуществляют в три ступени 144. Вначале из мети-
лового спирта и окиси углерода получают метилформиат:
СН3ОН + СО = СНзООСН
Реакцию ведут периодическим способом в автоклаве, куда за-
гружают метиловый спирт и подают СО под давлением 10,5 ат.
Катализатором служит метилат натрия, для получения которого
к метиловому спирту добавляют 1 % металлического натрия. Реак-
ция начинается при 60° и проводится при 100°, для чего автоклав
охлаждают, так как реакция экзотермическая. Постепенно давле-
ние повышают до 210 ат. За 4 ч реакция проходит на 80%.
Метилформиат отгоняют вместе с 5—10% метилового спирта и
нагревают до 40°. Через смесь пропускают безводный аммиак под
давлением около 14 ат. Образуется формамид:
СНзООСН + NH3 = HCONH2 + СН3ОН
Эта реакция экзотермическая и при 3%-ном избытке аммиака
завершается за 20 мин на 98%. Основную массу регенерирован-
ного метилового спирта и непрореагировавшего аммиака отгоняют.
Формамид перегоняют в вакууме, после чего он содержит до 99,7%
основного вещества. Отогнанные метиловый спирт, аммиак и метил-
формиат вновь возвращают в процесс.
Полученный формамид подвергают дегидратации, пропуская
его в присутствии газообразного аммиака при 200—300° над акти-
вированным глиноземом. При этом образуется HCN:
HCONH2 = HCN + Н2О
Предложено145 разложение паров формамида в присутствии
NH3 проводить в обогреваемом контактном аппарате.
Суммирование реакций, протекающих в каждой стадии, дает
общее уравнение процесса:
NH3 + CO = HCN + Н2О
Получение синильной кислоты плазменным методом 125'127
Известно, что вещества в плазменном (высокотемпературном)
состоянии существенно отличаются от газов своими электриче-
скими и магнитными свойствами. При 20—50 тысячах градусов
в газообразном веществе практически отсутствуют нейтральные
атомы и оно состоит исключительно из ионов. Даже в условиях
низкотемпературной плазмы, образующейся при температурах в не-
сколько тысяч градусов, химические процессы протекают с огром-
ной скоростью. Это позволяет осуществить их высокоинтенсивным
Производство HCN контактными способами
1541
путем в компактных аппаратах малых размеров и достичь больших
выходов при эндотермических реакциях.
Низкотемпературная плазма получается в плазмотронах —
алектродуговых устройствах, в которых электрическая дуга под-
вергается тепловому или магнитному сжатию. На рис. 460 пока-
вано принципиальное устройство плазмотрона с магнитной стаби-
лизацией электрической дуги. Образующаяся дуга между катодом
и анодом под влиянием магнитного
образно по поверхности анода.
Это позволяет сосредоточить теп-
ловую энергию дуги в большем
пространстве и большем объеме
газа по сравнению с обычным го-
рением дуги. В результате значи-
тельное количество энергии дуги
идет на нагревание вводимого
агента. Тепловой коэффициент
полезного действия плазмотрона,
выраженный отношением энергии,
переданной газовому потоку, к
энергии электрической дуги, со-
поля перемещается вихре-
Газ
Рис. 460. Схема плазмотрона с маг-
иитновихревой стабилизацией дуги:
/ — катод; 2 — соленоид; 3 —анод; 4 — дуга.
составляет 60—70%, а в плазмотронах с предварительным подо-
гревом газа, используемого для охлаждения электродов, может
быть доведен до 95—98 %.
В качестве плазмообразующей среды используются аргон, азот,
водород, метан и др. Изолирующими материалами служат нитрид
бора, асбоцемент, фторопласт и др.
Образование цианистого водорода в плазменной среде основано
на взаимодействии между атомами (в нормальном либо возбуж-
денном состоянии), получаемыми при разложении плазменных и
реагирующих газов. При получении синильной кислоты из эле-
ментов
2С + N2 + Н2 = 2HCN — 60,2 ккал
процесс можно осуществить в азотной плазме, т. е. с одновремен-
ным использованием азота в качестве газа разряда. Источником
углерода служит расходуемый графитовый катод, а водород вво-
дится непосредственно в область разряда (в плазму). Вследствие
большого эндотермического эффекта реакции достаточно высокие
выходы HCN могут быть получены лишь при температурах выше
3000°. С понижением температуры равновесие реакции сдвигается
в сторону разложения HCN на исходные вещества. Поэтому
с целью сохранения возможно большего количества HCN в газе
газовая смесь подвергается закалке—резкому охлаждению.
На рис. 461 представлена схема электродного узла реактора, ис-
пользованного в опытах получения HCN в плазме 125. Электродный
1542
Гл. XLI. Цианистые соединения
узел реактора состоит из вольфрамового (~2% Th) стержне-
вого катода диаметром 3 мм и съемного медного анода, запрессо-
ванного в держатель из нержавеющей стали. Медный анод поме-
щают внутрь графитового цилиндра с целью более равномерного
распределения разряда по поверхности анода. При использовании
Рис. 461. Конструкция электродного узла плаз-
менной форсунки:
/ —труба для подачи газа разряда; 2 — ниппель; 3— верх-
няя пластинка катода; 4 — катодный узел; 5 — газораспре-
делительная тарелка; 6 — катод; 7—медная анодная на-
кладка; 8— анодный узел; 9— чехол анодного узла;
/0—брусок, снимающий катод; //— найлоновый изоля-
тор; 12— втулка для снятия катода.
в качестве исходного материала элементарного углерода вольфра-
мовый катод заменяют графитовым, который по мере испарения
продвигается вниз, к отверстию форсунки. Катод и анод интенсивно
охлаждаются водой и изолированы друг от друга корпусом из най-
лона, который служит также держателем катодного узла. Плазмо-
образующий газ (аргон или азот) поступает через распределитель-
ную тарелку с шестью отверстиями. Мощность реактора составляла
10—12 кет.
Реагирующие газы вводятся при помощи охлаждаемых водой
устройств через отверстия диаметром 0,75 мм, расположенные
в съемном аноде перпендикулярно или под углом 30° к разряду.
Литература
1543
Продукты реакции охлаждают и после отделения сажи они посту-
пают на переработку в синильную кислоту или цианистые соли.
Процесс может быть осуществлен с применением других исходных
Веществ, подаваемых в азотную плазму, например, по реакциям:
c + nh3 = hcn + h2
2СН4 + N2 = 2HCN +ЗН2
NH3 + CH4 = HCN + ЗН2
Во всех случаях при получении 1 г-мол цианистого водорода
расходуется около 2,7 кет • ч электроэнергии. Выходы HCN зави-
сят 126, помимо других условий, также от соотношения С : N : Н.
Выход синильной кислоты составляет около 50% при синтезе из
элементов, ~40% при синтезе из углерода и аммиака, 45—46% —
из метана и азота и выше 50% — из метана и аммиака.
Получение дициана в плазме основано на синтезе циана при
3500—7000° по реакции
• 2C + N2 = 2CN
и полимеризации его в дицнан при охлаждении продуктов реакции.
Процесс изучали 125 при температуре плазмы 4000—4500° и продол-
жительности контакта реагентов 5-10-6 сек с применением азота
в качестве плазмообразующего газа и углерода, поступавшего
в зону реакции за счет испарения графитового анода.
Теоретический выход дициана при атмосферном давлении и сте-
хиометрическом соотношении компонентов может составить 53,5%
при оптимальной температуре 4150° На 1 т связанного азота в ди-
циане расходуется (без учета рекуперации тепла) 13 600 квт-ч
энергии.
ЛИТЕРАТУРА
АГ. Келер, Технология цианистых соединений, Госхимиздат, 1934. —
2. R. R u the г, W. McLain, J. Chem. Phys., 24, № 1, 173 (1958).—
3. J. D. F. Marsh, M. J. Martin, J. Appl. Chem., 7, № 5, 205 (1957).—
4. В. А. Шушунов, A. M. Павлов, К. Ф. Федякова, ЖПХ, 29, № 11,
1637 (1956). — 5. Сим о, Икарам э, Sci. Repts Res. Insts Tohoku Univ., A5,
№ 2, 185 (1955).—6. В. А. Шушунов, A. M. Павлов, ДАН СССР, 89, №6,
1033 (1953). —7. В. А. Шушунов, А. М. Павлов, ЖПХ, 28, № 11, 1161
(1955). — 8. В. А. Шушунов, А. М. Павлов. Там же, № 9, 934 (1955).—
9. Вредные вещества в промышленности, под ред. Н. В. Лазарева, т. II, Госхим-
издат, 1963. —10. Правила и нормы техники безопасности и промышленной са-
нитарии для проектирования, строительства и эксплуатации производства циан-
амида кальция и цианплава, Госхимиздат, 1960.
И. Т. J а п a g i m о t о, Rev. Phys. Chem. Japan, 24, № 1, 1 (1954).—
12. А. В. Соколов, Труды Н.-и. ин-та хим. реактивов, в. 20, 1951, стр. 168. —
13. Ивасаки, Симодзима, J. Chem. Soc. Japan. Pure Chem. Sec., 76, № 7,
749 (1955). —/4. О. К. Щербаков, И. А. Каковский, Цветные металлы,
№ 6, 15 (1966). — 15. R. S. S a x e m а, А. К. Bhattacharya, J. Indian Chem.
Soc., 31, № 1, 53 (1954)— 16. Г. Петерс, Цианистые соединения и их приме-
нение в борьбе с вредителями и паразитами, ОНТИ, 1935. — 77. А. Л. Е ф и-
м о в, И. А. К а з а с, Инсектициды н фунгициды, Сельхозгнз, 1940. —
1644
Гл. XLI. Цианистые соединения
18. Г. И. Войнилович, Л. К. Ах pan, ЖПХ, 9, № 10, 1790 (1936).—
19. Н. Е. Gunning, пат. США 3215471, 1965. — 20. С. Т. R 01 a n d, Р. Н. R а 1-
ston, пат. США 3213017, 1965.
21. С. Кап ter, пат. ФРГ 911018, 1954. —22. Англ. пат. 733269, 1955.—
23. С. С. Бобков, Хим. наука и пром., 2, № 1, 34 (1957).—24. Chem. Eng
News, 23, № 20, 1840 (1945). — 25. Ed. S t a f f, Chem. Ind., 54, № 6, 835 (1944).—
26. H. Rein, Angew. Chem., 60, 159 (1948); 61, 241 (1949).— 27. M. S о u 1 a d,
Industrie Textile, 69, 673 (1952). — 28. Л. А. Кузнецов, Производство кар-
бида кальция, Госхимиздат, 1950. — 29. В. С. Чувахин, К. С. Муш ников а,
Б. Н. Пастухов, С. Д. Попов, Б. А. Герасимов, П. В. 3 а р и н г, По-
собие по борьбе с вредителями сельскохозяйственных культур, Сельхозгиз,
1945. — 30. В. И. Орлов, П. В. Попов, М. А. Морозова, М. Г. Г а б р и е-
л о в а. Ядохимикаты (инсектисиды и фунгсгсиды), Госхимиздат, 1946.
31. К. Milles, пат. ФРГ 923792, 1955. — 32. К. М i 11 е s, пат. ФРГ 925479,
1955. — 33. К. Milles, франц, пат. 1050539, 1954.—34. Ю. В. Ракитин,
К. Е. Овчаров, Л. Г. Брегет о в а, Физиология растений, 2, № 2, 177
(1955). — 35. Ю. А. Баскаков, Н. Н. Мельников, Хим. пром., № 7. 435
(1955). — 36. A. Krause, Roczn. chem., 29, № 2—3, 300 (1955). — 37. А. П. Г р и-
щ у к, С. Н. Баранов, ЖПХ, 32, № 11, 2601 (1959). — 38. В. И. Кузнецов,
ДАН СССР. Нов. сер., 50, 233 (1949). — 39. Адамс, Органические реакции,
ИЛ, 1948. — 40. М. А. Марова, М. Г. Воронков, Б. Н. Долгов, ЖПХ,
30, № 4. 650 (1957).
41. П. Е. К а з а р ь я н, Цианамид кальция и обращение с ним, изд. Мин. хлоп-
ководства СССР, Гл. Упр. сельскохоз. проп., 1951.—42. A. Cochet, Z. angew.
Chem., 44, № 21, 367 (1931).—43. Л. А. Кузнецов, М. Д. Шварц, Общая
технология карбида и цианамида кальция, ОНТИ, 1935.—44. G. W а 1 d е,
Н. Rock, Chem. Z. Chem. Apparat., 87, № 23, 839 (1963).—45. Л. А. Кузне-
цов, Производство карбида кальция, цианамида кальция и цианистого плава,
Госхимиздат, 1940. — 46. В. А. Клевке, Н. Н. Поляков, Л. 3. Арсеньева,
Технология азотных удобрений, Госхимиздат, 1963.—47. J. Cerny, Chem. Pru-
mysl., 5, № 3. 117 (1955).—48. Г. А о п о, япон. пат. 1714, 1953. — 49. Франц,
пат. 1112078, 1956. — 50. Франц, пат. 1028482, 1953.
51. Судзуки С ю с а к у, япон. пат. 1101, 1966. — 52. F. К а е s s, Т. Fi-
scher, Н. Kronacher, швед. пат. 200249, 1965. — 53. R. Sevin, J. four
felectr. et inds electrochim., 66, № 7, 229 (1961). — 54. Минэмура, Кудо, япон.
пат. 423, 1955.—55. J. Daniels, пат. США 2687945, 1954. — 56. Англ. пат.
688188, 1953. — 57. В. Ф. Постников, Т. И. Кунин, Труды Ивановского хи-
мико-технологического ин-та, вып. 3, 1940, стр. 117. — 58. Н. В. Пятницкий,
И. Е. Николаев, Г. М. С т р о н г и н. Предложения по эконом, электр. и тепл,
внергии, премированные на 9 Всес. конкурсе, Госметаллургиздат, 1955.—
59. F. К a ess, пат. ФРГ 934588, 1955. — 60. Нага и, Какандзава, Исида,
J. Chem. Soc. Japan, Ind. Chem. Sec., 56, № 4, 241; № 5, 321 (1953).
61. F. К a e s s, E. D о c h 1 e m a n n, пат. ФРГ 897842, 1953. — 62. F. К a ess,
E. Dochlemann, канад. пат. 502740, 1954. — 63. W. Stoll, австр. пат. 178632,
1954.—64. A. Dinelli, О. M e z z e 11 i, франц, пат. 1085940, 1955.—
65. E. R i n d t о г f, W. Ruschmann, J. Daniels, пат. ФРГ 909454, 1954.—
66. Франц, пат. 1065576, 1954. — 67. С. А. Сигов, 3. М. Лейкин, Р. И. Д а й-
ч и, Ш. А. Якубов, Рефераты докладов и сообщений IX Менделеевского съез-
да, № 7, 1965, стр. 81; сб. «Химия и технология минеральных удобрений», Таш-
кент, Изд. «ФАН», 1966, стр. 210. — 68. С. А. Сигов, 3. М. Л е й к и н,
Р. И. Д а й ч и, сб. «Химия и технология минеральных удобрений», Ташкент, Изд.
«ФАН», 1966, стр. 204. — 69. X. А. Григорян, Л. А. Д а с о я н, Пром. Арме-
нии, № 2 и 9 (1963). — 70. М. С. Е ч и к я н, Л. А. Д а с о я н, Арм. хим. ж., 19,
№ 6, 453 (1968); № 7, 547 (1966).
Литература
1545
71. С. А. Сигов, Узб. хим. ж., № 3, 48 (1967). — 72. Е. Becker, Е. Gla-
ser, Н. Kronacher, пат. ФРГ 1188058, 1965. — 73. J. Р. Р i с а г d, М. L. В 1 a i з,
пат. США 3173755, 1965. — 74. F. К а е s s, Н. Kronacher, Е. Becker-
Boos t, пат. ФРГ 937770, 1956. — 75. Н. Н б g е г, Н. Kronacher, пат. ФРГ
1206410, 1966. — 76. В. А. Шушунов, А. М. Павлов, ЖПХ, 28, № 1, 98
(1955). — 77. А. Е. Кретов, Кальцийцианамид и продукты его переработки,
ОНТИ, 1934.— 78. G. Н. В u chem ап, Chim. е. ind., 28, № 5, 1024 (1932).—
79. Р. J. С а г 1 е s 1 е, Ind. Eng. Chem., 25, № 9, 959 (1933). — 80. Е. Hartert,
Н. Schwarz, К. Hengst, пат. ФРГ 1211135, 1966.
81. Т. D. McMinn, пат. США 2708151, 1955. —82. Н. Kronacher,
Н. Н о е g е г, пат. ФРГ 935188, 1955. — 83. Н. С о р е 1 i п, пат. США 2710788,
1955, — 84. С. Н. Lemke, J. Electrochem. Soc., 101, № 4, 203 (1954).—
85. Н. И. Аксенов, Л. Л. Ш п е к т о р о в, Р. Я- К и п и я н и, Сельхозмашина,
№ 2, 12 (1954).—86. Н. А. Флейшер, Труды ГИПХ, в. 10, 1928, стр. 5.—
87. Франц, пат. 1095389, 1955. — 88. F. L. S i х m а, Н. Н е m d е г i k s, К. Helle,
U. Holldstein, R. Ling, Rec. trav. chim., 73, № 3, 161 (1954). — 89. Англ,
пат. 697505, 1953.—90. W. Wehrheim, E. Becker, H. Kronacher, пат.
ФРГ 912690, 1954.
91. L. H. С о ok, J. Heights, W. J. Rosenbloom, канад. пат. 492770,
1953. — 92. E. Asendorf, пат. ФРГ 953791, 1956. — 93. P. К a u n e r t, H. Do-
ring, Chem. Ingr. Techn., 34, № 12, 808 (1962). — 94. H. H. Поляков,
А. В. Гранжан, Хим. пром., № 7, 199 (1950). — 95. П. А. Хода к, И. Н. 3 а ft-
де л ьм ан, Б. В. Волков, Бюлл. по обмену опытом в азотной промышлен-
ности, № 1, 25 (1956). — 96. А. Е. Телепнева, Т. Д. Авербух, Труды
УНИХИМ, в. 8, 1959, стр. 122. — 97. Хасимото, Кири на я, Саэпи, япон.
пат. 4784, 1954. — 98. N. Russell, пат. США 2678871, 1954. — 99. О. Kubik,
чехосл. пат. 83654, 1955. —100. J. Schneider, С. Kozak, чехосл. пат. 83871,
1955.
101. Англ. пат. 739635, 1955. — 102. Н. A. G о 11 m а г, J. Н. М е г е - d i h,
пат. ФРГ 908134, 1954. — 103. И. Я. Кириченко, Р. П. Сидорова,
Е. Ф. Рутман, Т. У. Ищенко, авт. свид. 176574, 1965. — 104. A. Reg пег,
J. Balej, 1. Ronsar, Chem. promysl., 12, № 1, 8 (1962). — 105. G. В г е d i g,
Е. Е 1 б d, Е. D е m m е, Z. Elektrochem., 36, № 12, 1003 (1930). — 106. Р. W. S h e r-
w о о d, Petr. Eng., 12, 11 (1950). —107. F. Fischer, K. Peters, Chem. Zbl.,
2, 1107 (1927). — 108. K. Peters, K. Kiister, Brennst. Chem., 12, 122 (1931).—
109. Em. Briner, J. D e s b a i 11 e t s, M. W a r t h e i m, Helv. chim. acta, 21,
№ 4, 859 (1938). —110. L. A n d r u s s о w, Bull. Soc. chim. France, ser. 5, 18,
№ 1—2, 45 (1951).
111. L. Andrussow, Angew. Chem., 48, № 37, 593 (1935). — 112. Rev. prod,
chim., 57, № 1202, 222 (1954). —113. R. Wendlaut, англ. пат. 737995, 1955.—
114. Англ. пат. 722025, 1955. — 115. F. Е n d t е г, Chem. Ingr. Techn., 30, № 5,
805 (1958). —116. A. M. Маркевич, И. И. Тамм, Ю. Н. Рябинин, ДАН
СССР, 113, № 4, 856 (1957). — 117. Франц, пат. 1415207, 1965. — 118. Е. Becker-
Boost, Н. Kronacher, W. Lane, пат. ФРГ 1206409, 1966. — 119. Бзссацу
кагаку когё, 10, № 5, 228 (1966). — 120. Chem. Eng., 68, № 19, 72 (1961).
121. Chem. Process, 14, № 4, 12 (1968). — 122. Б. Г. Д з а н т и е в,
В. Н. Трубников, В. Н. Попов, Изв. АН БССР. Сер. физ.-техн, н., № 3,
8 (1966). — 123. W. R. Jenks, A. W. Andersen, пат. США 3215495, 1965. —
124. Ляо Ши-цзян ь, Цзяо Хуа-цзю-ань, У Чан-сянь, Ли Ю й -
лань. Ли Цун-ци, Сюй Лань-ии, Кэсюэ тунбао, Kexue tongbao, 17, № 3,
128 (1966). — 125. Н. W. L е и t п е г, Ind. Eng. Спет. Process. Disigne Develop-
ment, 1, 166 (1962); 2, 315 (1963). —126. С. H. Г а н з, В. Д. Пархоменко,
А. П. Мельник, Ю. И. Краснокутский, М. Н. Полторацкий, Плаз-
мы в промышленности, Киев, 1966. — 127. С. Н. Г а н з, А. П. Мельник,
В. Д. Пархоменко, Получение связанного азота в плазме, Киев, 1967.--
1546
Гл. XLI. Цианистые соединения
128. I. A. De Vynek, Silicates industr., 31, № 1, 21 (1966); 32, № 5, 181
(1967). —129. P. W. Sherwood, Petrol. Eng., 81, № 2, 22 (1959).—
180. И. E. А д а д у p о в, Укр. хим. ж., 11, кн. 3, 28/ (1936).
181. D. R. Safrang, R. R. Reeves, P. Harteck, J. Am. Chem. Soo.,
86, № 15, 316 (1964). — 132. H. И. Семенов, Усп. хим., 20, № 6, 673 (1951).—
133. H. Н. Семенов, Развитие цепной теории окисления (проблемы окисления
углеводородов), Изд. АН СССР, 1954. —134. С. Фудзисэ, М. Мацуда,
Т. Н у м а д а, япон. пат. 17661, 1964. — 135. П. А. Т е с н е р, И. С. Р а ф а л ь ке с,
ДАН СССР, 87, № 5, 821 (1952). —136. J. К. Pan Bingham, R. О. Roth,
пат. США 3244479, 1966. —137. U. Naffezzons, Chim. L’ind., 8, 460 (1952).—
188. N. Updergaff, Petrol. Ref., 32, 196 (1958). —189. Ibid., 38, № 11, 261
(1959). — 140. В. В. Ж и т и н е в а, В. И. Сидоров, Г. К. Митр юшки на,
Г. Н. Федорина, Хим. пром., № 1, 36 (1967).
141. Hydrocarbon Process a. Petrol. Ref., 40, № II, 253 (1961).—
142. U. Kuhlmann, Inform, chim., № 50, 29 (1967). —143. Н. S. Johnson,
A. H. Andersen, швейц, пат. 397614, 1966; 19914, 1965. — 144. H. Burrell,
Chem. Eng. News, 25, № 27, 1939 (1947). —145. E. Asendorf, F, Bittner,
швед. пат. 203021, 1966.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авициды 32
Азот 20, 38
Азурит 662
Акарициды 31
Аллофан 637
Алунит 142, 181, 183, 634, 640
Альгициды 33
Алюминий
бисульфат 643
гидроокись 645, 1126
монохлорид 1501
окись 388, 636, 638, 1324
сульфат 632 и сл.
сульфит 634, 651
сульфит-бисульфит 653
хлорид 387, 735, 1500 и сл.
фторид 1101, 1126, 1160
Алюмогель 601
Алюмокарбонат натрия 1132
Алюмосиликаты 637
Амберлит 249
Амид натрия 1526
Аммиакаты 566, 855, 1375 и сл., 1501
Аммонал 1185
Аммоннзатор-гранулятор 1349, 1368
Аммонит 64, 1185
Аммоний
бикарбонат 1259, 1263, 1284
бисульфат 623
бисульфит 508 и сл., 534
бифторид 358, 1100, 1162 и сл.
бихромат 570, 617
карбамат 1259, 1284 и сл.
карбонат 1203, 1205
нитрат 1178 и сл.
нитрит 1209 и сл.
пиросульфит 508
роданид 1529
сульфат 1236 и сл.
Аммоний
сульф ат-нитр ат 1206
сульфит 508, 520, 534, 1239
тиосульфат 510
фосфаты 1264 и сл.
фторид 1101, 1162 и сл.
хлорид 1253
хромат 569
Аммофос 1264 и сл.
Анатаз 1483
Ангидрит см. Кальций сульфат
Апатит 801 и сл.
Арканит 142
Арсенаты 1397 и сл.
Арсениты 1397 и сл.
Арсенопирит 1406, 1408
Арсмаль 1404
Астраханит 46 и сл.. 98 и сл.; 270
Атакомит 662
Аттрактанты 32
Аурипигмент 1406
Афициды 31
Ашарит 319 и сл.
Бакмана камеры 1445 и сл.
Бактерициды 32
Барит 424, 427 и сл.
Барий
алюминат 430
гидрат окиси 416, 456 и сл.
гидросульфид 296, 438
гипохлорит 443
дипероксигидрат 462
дисилицид 423
карбонат 420, 440, 543 и сл.
кремнефторид 420, 421
метасиликат 419, 459
нитрат 418, 451 и сл.
1548
Предметный указатель
Барий
окись 418, 460
ортосиликат 418, 430
перекись 417, 420, 460
полисульфиды 419
силикат 459
сульфат 419 и сл., 425 и сл.
сульфид 296, 419, 427 и сл.
сульфит 429
тетрафторборат 312
тиоферрит 430
титанат 419
феррит 418, 430
фторид 420, 421
хлорид 434 и сл.
цинкат 459 ••
Бекстремит 751
Белая сажа 1154, 1163 и сл.
Белый матт 676, 682
Беркеит 98, 171, 179
Берлинская лазурь 1512
Бертолетова соль 1435, 1439, 1461 и сл.
Бескова камера 831
Биксбиит 751
Бисульфитформальдегид 545
Биурет 1282
Бихроматы 564 и сл.
Бишофит 173, 262, 270, 273 и сл.
Бланфикс 421, 425
Боксит 637, 652, 1133
Бор 311 и сл.
ангидрид 311, 356
в микроудобрениях 318
карбид 311, 315
минералы 319
нитрид 311, 315
фосфат 314
фторбораты 357
фторид 311, 357
Бораты 313 и сл.
аммония 314
калия 313
кальция 313, 337
магния 314
марганца 532
меди 532
натрия 313
Борацит 319
Бордосская жидкость 664
Бориды 311, 315
Борил 315
Бородвойной суперфосфат 348
Борокальцит 319
Боронатрокальцит 319, 341
Боросульфатаммоний 346
Боросуперфосфат 347
Браунит 751
Браунмиллерит магниевый 586
Бром ИЗ, 206 и сл.
Броматы 208, 233
Бромиды S07, 231
Бромкарналлит 142
Бротфильда способ 831
Брукит 1483
Брусит 265
Брушит 1017
Бура 313 и сл., 337 и сл.
Вернадит 751
Витерит 418, 424
Вода бромная 211
буровая 209, 240, 242
морская 46, 109, 145, 272, 294
иадсмольная 1243
океанская 109
хлорная 215
Воды 751
Галит 47 и сл., 60 и сл.
Галитовые отходы 89
Галлуазит 637
Галмей 720
Гаусманит 751
Геденбергит 335
Гематит 637
Гербициды 33
Гиббсит 637
Гидроборацит 319
Гидровруб 54
Гидрогаусманит 751
Гидросульфиды 466 и сл., 499
Гидросульфит 508, 538 и сл.
Гипоманганат калия 755
Гипохлорит 1430, 1448, 1450
Гипс см. Кальций сульфат
Глазерит 98, 142, 145, 179
хромовый 569
Глауберит 51, 98, 104
Глауберова соль 102, 134
Глауконит 142, 637, 803
Глина 637
Глинозем 183, 635 и сл., 944, 1324
Гранат 335
Гранатка 67
Гроутит 751
Грэма-соль 1071
Данбурит 319
Датолит 314, 319, 335, 346
Дессиканты 33
Предметный указатель
154»
Дефолианты 33
Диаспор 637
Дигофат 785
Диккит 637
Дицианамид 1511
Доломит 266, 270, 277, 581
Дунит 305
Дуст 33
Железо
гидрат закиси 699, 708 и сл.
гидрат окиси 528, 639, 699, 708 и сл.
закись-окись 699, 711
окись 699, 710
оксисульфат 711
оксихлорид 698
сульфаты 694 и сл.
углекислое 711
фосфаты 886
хлориды 707
Железистосинеродистые соли 1512, 1530
Железосинеродистые соли 1512, 1531
Зооциды 32
Ильменит 637, 803, 1483
Ингибиторы 370, 518, 572
Индерит 314, 319
Инсектициды 31
Инсектофунгициды 32
Иньоит 313, 319
Иод 237 и сл.
Иодаты 239 и сл., 256 и сл.
Йодиды 238 и сл., 256 и сл.
Иодирование соли 82
Каинит 46, 142, 174, 270, 637
Каламин 720
Калий
арсенат 1402
бикарбонат 187, 607
бисульфат 374
бифторид 1100
бихромат 570, 606 и сл.
галогениды 138 и сл.
карбонат 186 и сл.
кремнефторид 1147, 1154
минералы 142
нитрат 1222 и сл.
сульфат 139, 173 и сл.
фторид 1101, 1154
хлорид 138 и сл.
хромат 568
Калиборит 142, 319
Калимагнезия 141
Калише 1217
Калушит 142
Кальтенбаха колонна 211
Кальцит см. Кальций карбонат
Кальций
алюминаты 576, 586 и сл.
алюмосиликат 586
алюмоферриты 586
арсенат 1399, 1419 и сл.
арсенит 1408 и сл.
бикарбонат 69, 72
бисульфит 511 и сл.
гидрат окиси 1442
гидрооксихлорид 746
гидросульфид 500
карбонат 581, 1329, 1441
кремнефторид 1007, 1104, 1147, 1158
нитрат 1210 и сл., 1339
окись 577, 1442
оксихлорид 740, 1431
оксихромит 582
ортосиликат 1039 и сл.
субхлорид 739
сульфат 832 и сл., 884 и сл., 895
и сл.
сульфид 498
сульфит 508, 521
хлорид 738 и сл.
хромат 569 и сл.
Каолин 637 и сл.
Каолинит 637 и сл.
Карбамид 1282 и сл., 1362 и сл., 1379
и сл., 1511
нитрат 1283
фосфат 1284
Карбоксиметилцеллюлоза 78
Карналлит 46, 142 и сл., 270
Квасцы
алюминиевые 632 и сл.
железные 658, 696, 698
хромовые 566, 613 и сл.
Квенстетит 696
Кернит 319, 342
Кианит 637 ।
Кизерит 76, 132, 160, 270
Кислота
азотная 451, 1186, 1308 и Сл.
арилборная 315
борная 312 и сл.
бромистоводородная 227 и сл
бромноватая 207
бромноватистая 207
гексафторфосфорная 1105
гидросернистая 506 и сл.
двухромовая 566
1550
Предметный указатель
Кислота
дитионовая 506 и сл.
иодистоводородная 251 и сл.
йодноватая 238
иодноватистая 238
кремневая 1160
кремнефтористоводородная 1103 и сл.
марганцовая 754
марганцовистая 754
метаборная 312, 357 *
метамаргаицовистая 752
метафосфиновая 1274
метафосфориая 832, 957
мышьяковая 1398, 1416 и сл.
мышьяковистая 1397
иитрозилсериая 1240
ортоборная 312 и сл.
ортомаргаицоватистая 752
ортомышьяковая 1398
ортофосфорная 881
пиросериистая 506 и сл.
пирофосфорная 881, 939
плавиковая 1097 и сл.
полиборная 313
полиметафосфорная 832
политионовые 506 и сл.
полифосфорные 881, 939
роданистоводородиая 1511
сериая 611, 640, 1239
сернистая 506 и сл.
синильная 1509 и сл.
соляная 363 и сл.
сульфоксиловая 538, 549
суперфосфорная 832, 882, 940
тетраборная 312
тетраполифосфорная 832
тетрафторборная 312
тиосерная 506 и сл., 547 и сл.
тиосернистая 506 и сл.
триполпфосфорная 831
тритионовая 467
угольная 603, 765
уксусная 1413
фосфорная 881 и сл., 938 и сл.
фторалюминиевая 1124
фторборная 1106
фторсульфоновая 358, 1100 и сл.
хлористая 1430
хлорная 1430
хлорноватая 1430
хлорноватистая 1430
хлорсульфоиовая 1499
хромистая 573
хромовая 566 и сл.
хромосерная 567
циановая 1282
циануровая 1282
Коагулянт 634, 640 и сл., 706
Колеманит 313, 319
Коквимсбит 696
Колчедан
мышьяковистый 1406
мышьяковый 1406
огарок 686, 705
Корма минеральные 34
прицепитат 1032 и сл.
трнкальцийфосфат 1042, 1043
, Кремнефторид
алюминия 1126, 1147, 1148
аммония 1147, 1162
бария 1104
калия 1167, 1154
кальция 1147
магния 1147
натрия 1108, 1142
цинка 1147
Кремний
карбид 1498
субхлорид 1497
хлорид 1495 и сл.
Криолит 1108, 1124, 1160
Криптомелан 752
Кубиершского колонна 210 и 'сл.
Купфермеритоль 1405
Курнакит 751
Курнаковит 319
Курскит 803
Купоросы 663 и сл., 694 и сл., 715 и сл.
Курроля соль 1071
Лангбейнит 142 и сл., 174, 270
Лейцит 142, 1502
Леонит 142, 270
Леллингит 1406
Лимациды 31
Лимонит 631, 804
Литопон 459, 724, 727
Лондонский пурпур 1405
Лопарит 1483
Лярвициды 31
Магнезит 265, 270
каустический 269
Магнезия 268, 281 и сл.
Магнетит 1483
Магний
аммоиийфосфат 20
бикарбонат 266, 282 и сл.
бисульфит 521
гидрат окиси 265, 283 и сл.
гидрооксихлорид 300
Предметный указатель
1551
Магний
гидроснликат 267
карбонат 265, 283 и сл.
минералы 270
нитрат 1308
окись 265 и сл., 281 и сл.
оксихлорид 263, 267
сили^офосфат 305 -
сульоат 267, 270, 301 и сл.
еульоид 303
сульфит 508
хлорид 263, 272 и сл.
хромат 582
Маддреля соль 1071
Мажеф 785
Малахит 662
Манганат 754, 779
Манганит 751
Манганозит 751
Манганокальцит 760
Марганец
арсенат 1403
карбонат 752, 762 и сл,
нитрат 722, 753
окислы 751 и сл.
руды 759 и сл.
сульфат 752 и сл.
фосфат 785 и сл.
хлорид 753, 767, 773
Марганцовый шпат 751
Марсы 708
Медь
гидраты окислов 661 и сл.
гранулирование 667, 670
карбонат 662, 1414
окислы 661 и сл.
оксихлорид 676
сульфат 663 и сл.
сульфит 685
хлорокись 664, 676, 690
хлорид 662
Мейергофферит 313, 319 •
Метаморфизация рассолов 48 и сл.
Метафосфаты 1055, 1070 и сл.
Метиламин 1527
Микроудобрения 21, 666, 718, 775
Микроэлементы 21 и сл., 666, 718
Мирабилит 46 и сл., 98 и сл.
Миспикель 1406
Моллюскоциды 31
Монетит 1017
Монтмориллонит 637
Мориц—Стандерт камера 858
Мочевина см. Карбамид
Муллит 639
Мышьяк
пятиокись 1407
Мышьяк
сульфиды 1406, 1407
трехокись 1397 и сл.
трикальцийарсенат 1399
фториды 1402
хлориды 1401
Нагельшмитит 1039
Накрит 637
Натрий
алюминат 586, ИЗО, 1160 и сл.
арсенат 1399, 1419 и сл.
арсенит 1403, 1412
бикарбонат 341, 523 и сл.
бисульфат 372 и сл., 611 и сл.
бисульфит 506 и сл.
гидросульфид 437, 466 и сл.
гипохлорит 1431, 1448
метасиликат 475
нитрат 1216 и сл.
политиоиаты 506
сульфат 98 и сл., 371 и сл.
сульфит 240, 471 и сл., 506 и сл.
тиосульфат 340 и сл.
хлорид 46 и сл., 60 и сл.
хроматы 568 и сл.
Немалит 265
Нематоциды 31
Нефелин 142, 183, 196, 640 и сл.
Никель
карбонат 731
окислы 731
сульфат 731, 732
хлорид 731
Нитриты 1216 и сл.
Нитрификация 1204
Нитроаммофос 1338
Нитроаммофоска 1338
Нитросуперфосфат 1323
Нитрофос 1302 и сл.
Нитрофоска 1302 и сл.
Новосадка 51
Норденгрена способ 831
Ныовель 304
Обессоливание воды 87
Овициды 31
Озера соляные 47 и сл.
Оливин 270
Опал 637
Пандермит 319
Паркера порошок 785
1552
Предметный указатель
Парижская зелень 1398, 1413
Патерноит 319
Пенные аппараты 221, 748, 813
Пенный режим 218, 221, 1140
Пербораты 313, 353
Периклаз 265
Перкарбонат 355
Перманганаты 754 и сл., 779 и сл.
Перовскит 1483
Перхлораты 1436, 1440, 1471
Пестициды 31
Пиниоит 319
Пирит 763
Пироксен 803
Пиролюзит 751 и сл.
Пиросульфиты 506, 511, 535 и сл.
Пирофиллит 637
Пирофосфаты 1083 и сл.
Пирохлор 1483
Пирохроит 751
Плавиковый шпат 357, 1107, 1109 и сл.
Плавленый фосфат 1038 и сл.
Поваренная соль 60 и сл.
иодированная 82
Полевой шпат 637, 804
Полиакриламид 156
Полианит 751
Полибораты 313
Полибромиды 244
Полигалит 142, 183, 270
Полиодиды 252
Полисульфиды 466 и сл.
Полифосфаты 1071 и сл.
Поташ 187 и сл.
Преципитат 1017 и сл. .
Прицеит 319
Протарс 1403
Протравители 32
Псиломелан 751
Пуссьера 720
Разорит см. Кернит
Рансьеит 752
Рамсделлит 751
Ратициды 32
Реальгар 1406
Ренанит 1040
Репелленты 32
Ретроградация Р2Оз 849
Реформкалий 179
Роговые обманки 637
Роданиды 1511 и сл.
Роденцитиды 32
Родонит 751
Родохрозит 751, 752, 760
Романешит 752
Роигалит 510 и сл., 545 и сл.
Рутил 637, 1483
Сакетта способ 831
Сакиит 270
Сассолин 319 см. Кислота борная
Селеносульфаты 549
Селитра
аммиачная 1178 и сл.
известково-аммиачная 1204 и сл.
калийная 1222 .
калийно-аммиачная 1208
кальциевая 1210 и сл.
натриевая 1216 и сл.
Сера 388, 501
гексафторид 1105
двуокись 379, 507 и сл.
Серицит 637
«Серная печень» 502
Сероводород 407 и сл.
Серпентин 270
Сесквикарбонат 1259, 1285
Сидерит 637
Сиенит 640
Силикат
кальция 586, 1131
натрия 579, ИЗО
магния 885
цинка 720
Силикокарнотит 104Ьи сл.
Силлиманит 639, 1503
Сильвин 46. 142, 149 и сл.
Сильвинит 142 и сл.
Сингенито-гипс 182
Системы безводные
HF—NH4F 1100
H2SO4—Na2SO4 374
H2SO4—K2SO4 374
B2O—SiO2—P2O5 314
NH4NCS— (NH4)2CS 1512
(NH2)2CO—NH2COONH4 1289
(NHahCO-NIFCOONH,—NH3 1288
Na2—S 469
Na2SO4—Na2S 472
Na2SO4—NiSO4 733
NaCl—Na3H(SO4)2 374
NaCl—Na2SO4 379
Na2CrO4—Na2CO3 578
Na2CrO4—CaCrO4 579
MgO+Cl2^MgCl2+V2O2 280
CaCN2+N2 1517
CaO—SiO2 945
CaO—SiO2—3CaO • P2O5 1044, 1045
CaO—P2O5—SiO2 1039
CaO • Сг20з—CaO А1аОз 576
Предметный указатель
1553
Системы безводные
CaF2—Na2CO3 ИЗО
CaF2—СаО ИЗО
СаСгО4—Na2CO3 579
Сг2О3—SiO2 566
ZnSO4—Na2SO4 716
ZnSO4—NaCl 716
ZnSO4—KC1 716
ZnSO4-ZnCl2 716
ZnCl2—NH3 717
TiCl4—SiCl4 1493
BaSO4—NaCl 419
BaCl2—CaCl2 418
ВаС12—KC1—NaCl 418
Br2—Cl2 212
Системы водные
P2OS-H2O 942
H2SO4—H3PO4—H2O 887 и сл.
H3BO3—MgSO4—H2O 332
H3BO3—MgSO4—H2SO4—H2O 330
H3BO3—KH2PO4—H2O 334
Н3ВО3—K2SO4—Н2О 334
НзВОз-NaF—Н2О 314 '
HF—Н2О 1098
HF—H2SO4—Н2О 1099
HF—SiF4—Н2О 1103
HF—A1F3—Н2О 1127
H2S—Na2CO3— NaHCOj—NaHS—НгО
468
H2S—K2CO3—КНСОз—KHS—HsO
468
HC1—H2O 365, 397
HC1—H2SO4—H2O 368
HC1—CaCl2—H2O 740
HC1—MgCl2—H2O 275
B2O3—H2O 356
B2O3—MgO—H2O 332
NH3—H3PO4—H2O 1264
NH3—H2O 1378
NH3—CO2—H2O 1261
NH4HF2—NH4CI— H2O 1100
nh4hf2—nh4f—h2o 1100
NH4H2PO4— (NH4)2HPO4—H2O 1347
NH4H2PO4— (NH4)2SO4—H2O 1348
NH4H2PO4— (NH2)2CO—H2O 1348
NH4H2PO4—NH4NO3—H2O 1265
NH4H2PO4—NaNO3—H2O 1265
(NH4) 2HPO4-NH4NO3— (NH2) 2CO—.
H2O 1349
NH4NCS-(NH4)2CS-H2O 1512
(NH2)2CO—NH3—H,0 1284 1379 и сл.
(NH2j2CO—NH4NO3—NH3—HqO 1380
(NH2)2CO-NH3—H3PO4-KC1-H2O
1385
NH4NO3—NH3—H2O 1378 и сл.
NH4NO3— (NH4)2SO4— H2O 1207
NH4NO3-KC1—HSO 1228
Системы водные '
, (NH4)2SO4—H2SO4—H2O 1241
' (NH4)2SO3—NH4HSO3—H2O 509
(NH4)2S63-NH4HSO3— (NH4)2SO4—
H2O 509
NH4C1—HC1—H2O 365
NH4C1—NaCl—H2O 1256
Na2B4O7—HBO3—Na2SO4—H2O 340
Na2B4O7—Na2SO4—H2O 339
NaNO3—NaNO2—H2O J216
Na2O—B2O3—H2O 340
Na2O—P2O5—H2O 1066
Na2O—CrO3—SO3—H2O 611
NaF—AIF3—H2O 1102
Na2SiF6—NaCl—H2O 1143
Na2S—Na2SO4—H2O 484
Na2S2O3—Na2SO3—H2O 553
Na2S2O3—Na2SO3—-Na2SO4—H2O 552
Na2SO3—HSO 508
Na2SO4— H2O 99
Na2SO4—H2SO4—H2O 372
Na2SO4—NaOH—H2O 101
Na2SO4—NaCl—H2O 100, 101
Na2SO4—NaCl—NaOH—H2O 101
Na2SO4—A12(SO4)3—H2O 632
NaCl—H2O 61
NaCl — NH4HSO3—H2O 533
NaCl—(NH4)2SO3—H2O 533
NaCl—MgSO4—H2O 71, 87, 110, 111,
272 273
NaCl—MgCl2—H2O 66, 88
NaCl—KC1—Na2SO4—K2SO4—
Na2CO3—K2CO3—H2O 194
NaCl—KC1—MgCl2—H2O 274
NaCl—K2Cr2O7—H2O 608
NaClO3—NaClO2— H2O 1436
Na2CrO4—Na2CO3—H2O 568
Na2CrO4—Na2SO4—H2O 569
Na2CrO4—Na2Cr2O7—H2O 604
Na2Cr2O7—Na2SO4—H2O 601
NaAl(SO4)2—KA1(SO4)2—H2O 656
NaBr—NaBrO3—H2O 225
NaBr—NaBrO3—NaHCO3—H2O 226
Nal— NaIO3—H2O 256
Nal—Na2CO3—NaHCO3—H2O 257
MgO—HC1—H2O 300
MgO—P2OS—H2O 851
MgO—CaO—P2O5—H2O 852
A1(OH)3—A12(SO4)3—H2O 633
P2o5—H2O 942
SO2—H2O 519
K2CO3-H2O 187
K2CO3—Na2SO4—H2O 198
K2CO3—Na2CO3—H2O 198
K2CO3—NaCl—H2O 193, 194
КгСОз—KC1—H2O 191
K2CO3—KC1—K2SO4—HSO 191
1554
Предметный указатель
Системы водные
KF—HF—Н2О 1100
КС1—NaNOs—Н2О 1228
KCI—NaCl—Н2О 147
KCI—MgSO4—Н2О 178
KCI—MgCl2—Н2О 162
К2СгО4—Na2SO4—Н2О 569
К2СгО4—Na2CrO4—Н2О 569
К2СгО4— K2SO4-H2O 568
К2Сг2О7—K2so4—Н2О 570
К2Сг2О?—СаСггО?—Н2О 570
К1-К2СО3-КНСОз-Н2О 257
KI—КОН—К2СО3—Н2О 257
К1—КОН—Н2СОз—Н2О 257
KI-Nal— Н2О 259
KI—КЮз—Н2О 256
Ca(NO3)2—Н2О 1210
Ca(NO3)2—HNO3—Н2О 1212
СаО—Р2О5—Н2О 842, 844, 852, 975,
980, 1018
СаО—Р2О5—H2SiF6—Н2О 1025
СаО—Р2О6—N2O5—Н2О 1315 и
сл.
СаО—Р2О5—CaSiFs—Н2О 974
СаО—As2O5—Н2О 1399
CaSO4—Н3РО4—Н2О 901
СаС12—Н2О 739
СаС12—NaCl—Н2О 66, 740
СаС12—MgCh—Н2О 740
СаС12—MgCl2—NaCl—Н2О 66
СаС12—MgCl2—КС1—Н2О 740
СаС12—Са(ОН)2—Н2О 739
Са(С1О3)2-КС1—Н2О 1466
Сг2О3—Н2О 564
СгОз—HF—Н2О 567
СгОз—H2SO4—Н2О 566
МпО—Р2О5—Н2О 786
FeSO4—Н2О 694
FeSO4—H2SO4—Н2О 695
FeBr2—Н2О 208
NiSO4—H2SO4—Н2О 732
NiSO4—CoSO4—Н2О 733
CuCl—HC1— H2O 365
CuCl—NH4C1—HC1—H2O 365
CuSO4—FeSO4—H2SO4—H2O 663
ZnSO4—H2SO4—H2O 716
ZnSO4—(NH4)2SO4—H2O 716
ZnSO4— (NH4)2SO4—Na2SO4—H2O
716
ZnSO4—Na2SO4—H2O 716
ZnSO4—CuSO4—FeSO4—H2SO4—H2O
716
ZnCl2—H2O 717
ZnCl2—HC1—H2O 365
Ba(HS)2—NaCl—H2O 437
ВаСОз—Na2SO4—H2O 418
BaCO3—Ca(NO3)2—H2O 453
Системы водные
Ba(NO3)2—Ca(NO3)a—H2O 418
BaSO4—K2CO3—H2O 418
BaCl2— HC1—H2O 436
BaCl2—NH4NO3—H2O 453
BaCl2—NaOH—H2O 457
Ba(OH)2—H2O 416
Ba (OH) 2— H2O2-H2O 417
Скородит 1408
Смитсонит 720
Совелит 304
Сольбар 421
Ссайбелеит 319
Старосадка 51
Стассфурит 319
Стедит 1041
Стенгеля способ 1199
Стимулятор роста 32
Сульфаны 544
Сульфгидрат см. Гндросулырнд
Сульоигран 492
Сульфид
аммония 467
барня 419, 427 и сл.
железа 435
кальция 498
натрия 466 и сл.
Сульфоаммофос 1266
Сульфоборит 319
Сульфоксилат натрия 538
Сульфоксилаты 513
Сульфоксилатформальдегид 513
Сульфурилфториды 1105
Суперфосфат
двойной 971 и сл.
обогащенный 971, 1010 н сл.
простой 828 и сл.
Сф’алерит 720
Сфен 803, 1483
Сферодайзер 1337
Тальк 270
Тахгидрит 144
Тенардит см. Сульфат натрия
Термофосфаты 1038 и сл.
Термощелочиые фосфаты 1038
сл.
Тетракальцийфосфат 1039
Тетратионат натрия 558
Тетрафторалюминат натрия 1132
Тетрахлормоносилан см. Кремний хло-
рид
Тинкал 319
Тинкалкернит 319
Тиомочевина 1512
Предметней указатель
1555
Тиомышьяковый натрий 556
Тиооксимышьяковый натрий 556
Тиосульфат
аммония 510, 559 и сл.
кальция 559
иатрия 508, 547 и .сл.
Тнофторид 1105
Тнтан
двуокись 1482, 1486
иодид 1482
карбид 1485, 1491
нитрид 1485
оксикарбоннтрид 1486
оксихлорид 1482
сульфид 1485, 1494
фосфат 1494
хлорид 1481 и сл.
Титановые белила 1486
руды 1483
Титаномагнетит 1483
Томасит 1041
Томасшлак 1041
Травильные растворы 701
Трепел 181, ИЗО
Тринатрийгидросульфат 373
Трихлорсилан 1496
Трона 145
Тукосмеси 1361 и сл.
Турмалин 319, 349, 637
Турнбуллева синь 1512
Туттона соль 305
Тяжелый шпат 424 и сл.
Удобрения минеральные
бактериальные 24
безбалластные 27
гранулированные 29
двойные 27
диаграмма смешения 1366
жидкие 1375 и сл.
концентрированные 27
косвенные 27
микроудобрения 23
органические 24
органо-минеральные 24
подкормки 24
полные 27
предпосевные 24 а
припосевные 24
простые 27
прямые 27
сложно-смешанные 27, 1363
сл.
сложные 27, 1307 и сл.
смешанные 27, 1363 и сл,
тройные 27
Удобрения минеральные
уравновешенные 28
условные единицы 38
физиологическая реакция 30
Улексит 319
Уртит 637
Феррат натрия 577, 580
Феррит натрия 577
Феррицианиды 1512, 1531 и сл.
Ферросилиций 1498
Феррохром 576, 615
Ферроцианиды 1512, 1530 и сл.
Флюорит 282, 1109
Фосфаль 1040
Фосфатирование 785
Фосфатный комплекс суперфосфата
842
Фосфаты
алюминия 833, 886, 1308 и сл.
аммония 1085, 1264
железа 833, 886
калия 1065
кальция 1089
карбамида 1284
конденсированные 1070
магния 850, 885, 1019
мировые запасы 808
натрия 1065 и сл., 1070
обесфторенные 1043 и сл.
природные 801 и сл.
состав и свойства 801 и сл.
Фосфор 938 и сл.
окись 938, 957
сульфид 1062
фосфиды 1064
хлориды 1063
хлорокись 1063
Фосфоритная мука 819 и сл.
Фосфоритование почв 819
Франколит 803
Фтор 1095, 1110
Фтора окись 1095, 1112
Фторборат калия 1106 .
Фториды 1093 и сл.
Фтористые газы 858, 1134 и сл.
Фтористый водород 834, 885, 1096
и сл.
Фторсульфинаты 1105
Фумиганты 33
Фунгициды 32, 664
Халцедон 637
Хальконтит 663
Халькопирит 685
1556
Предметный указатель
Хартзальц 144, 159
Хильгенштокит 1041
Хиолит 1102, 1127, 1165
Хлор 408 и сл., 1229, 1430 и сл., 1480
и сл.
активный 1431
двуокись 1434 и сл., 1453 и сл.
Хлораты 1461 и сл.
Хлорид
алюминия 1500 и сл.
аммония 1253 и сл.
бария 296, 417, 434 и сл.
железа 408, 435, 698, 707 и сл.
калия 138 и сл..
кальция 738 и сл.
магния 87, 113, 161, 272 и сл.
марганца 753, 777 и сл.
меди 365, 409 и сл.
мышьяка 1401
натрия 60 и сл.
олова 365
серы 410
титана 1481 и сл.
фосфора 1063
хрома 565, 621 и сл.
цинка 365, 728 и сл.
Хлористый
водород 363 и сл.
нитрозил 412. 1229
сульфурил 380, 387
тетраметиламмоний 406
Хлорит натрия 1435, 1460
Хлорная известь 1431, 1441 и сл.
Хром
аммиакаты 566
бихромат 569 и сл., 596, 606
гидрат окиси 565, 615
декахромат 567
карбиды 576
кислый сульфат 565
монохромат 568, 576
нитрат 565
окислы 564 и сл.
оксихлорид 567
оксифторид 567
сульфат 565, 6(3 и сл.
сульфито-сульфат 618
хлорид 565, 621 и сл.
Хромаль 601
Хроматы 568 и сл.
Хромиты 573 и сл.
Хроми-хромат 617
Хромовые
квасцы 566, 613 и сл.
руды 573
глазерит 569
Хромпик 564 и сл.
Цеолит 1221
Цианамид
кальция 1511, 1516 и сл.
натрия 1511, 1527
Цианаты 1511
Цианиды 1510 и сл., 1523 и сл.
Цианистый водород 1509. 1532 и сл.
Цианплав 1515, 1525
Цинк
аммиакаты 717
бисульфит 523
гидрат окиси 714 и сл.
карбонат 539
окись 714
сульфат 715, 721 и сл.
сульфид 460, 715
сульфит 545
хлорид 717, 728
Цинковый
каинит 716
купорос 714, 721 и сл.
шпат 720
Шевреля соль 680
Шенит 142, 174, 270
Шлаки металлургические 1040
Штаффелит 803
Швейнфуртская зелень 1413
Щелковская зелень 1415
Эгирии 803
Эвдиалит 803
Энгеля соль 190
Эпсомит 46, 270, 301 и сл.
МАКС ЕФИМОВИЧ ПОЗИН
ТЕХНОЛОГИЯ МИНЕРАЛЬНЫХ СОЛЕЙ
(удобрений, пестицидов, промышленных солей,
окислов и кислот)
Часть II
Редактор 3. И. Грива
Техн, редактор 3. Е. Маркова
Переплет художника А. П. Рыбакова
Корректоры: В. Б. Генгут, Л. А. Любович
М 09439, Подп. к печ. 15/IV 1974 г. Формат бумаги 60X90’/^.
Бумага тип. № 2. Усл. печ. л. 48,0. Уч.-изд. л. 53,31.
Тираж 5500 экз. Заказ 838. Изд. № 529. Цена 2 р. 90 к.
Издательство «Химия», Ленинградское отделение
191186, Ленинград, Д-186. Невский пр.. 28
Отпечатано с матриц ордена Трудового Красного Знамени
Ленинградской типографии № 2 им. Евгении Соколовой
в Ленинградской типографии № 6
Союзполиграфпрома при Государственном комитете Совета Министров СССР
по делам издательств, полиграфии в книжной торговли
196006, Ленинград, Московский пр., 91