





























































































































Текст
ВСЕСОЮЗНЫЙ ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
«УТВЕРЖДАЮ» Зам. директора института
Р. П. Ластовский
МЕТОДЫ ПОЛУЧЕНИЯ ХИМИЧЕСКИХ РЕАКТИВОВ И ПРЕПАРАТОВ
Методические указания
Выпуск 25
МОСКВА—1973
Редакционная коллегия
Е. А. Божевольнов, А. В. Бромберг, В. Г. Брудзь, В. М. Дзиомко, Р. П. Ластовский (гл. редактор), А. М. Лукин, Ю. П. Решетников, Б. Д. Степин, В. Я- Темкина
Редактор С. С. Кузьмина
Техн, редактор А. И. Пирожкова
Корректоры Н. Р. Казарина и Л. А. Климанова
Сдано в набор 5/IV-1972 г. Подписано к печати 12/III-1973 г.
Формат бумаги бОхЭО1/^ Объем 16 печ. л. Уч-изд. л. 11,5
Л-96070 Зак. 1192, Тираж 1100 экз. Цена 92 коп.
Типография № 5 Управления издательств полиграфии и книжной торговли Мосгорнсполком.
Москва, Таганскя ул., 58
СОДЕРЖАНИЕ
З-Аллил-5-нитросалициловый альдегид. Д. А. Драпкина, Н. И. Дорошина . ?
д-Амипоацетофецон. В. В. Салий ............................... 9
а -Аминобензилидендифосфоновая кислота. Т. М. Балашова. И. Д.
Колпакова ...................................................Ч
1-Аминогексилидендифосфоновая кислота, Т. М. Балашова, И. Д.
Колпакова . . ........................................13
Аминофлуоресцеин 1. Г. И. Михайлов, Н. М. Рыбкина, А. А. Поло-женцева ......... 15
1-Аминоэтилидендифосфоновая кислота. Т. М. Балашова, И. Д. Колпакова .....................................................18
ж-Ацетобензолсульфонилфторид. К. М. Ройзен, Т. Н. Коровина, С. Б Макарова...............................................20
4-Бензилдифенил. И. А. Барц, Е. М. Иофис, Р. М. Гельштейн . . 22
N -Бензилфталимид. Ю. Г. Бондарь, Е. А. Чернини . . .25
Бензоил ацетонитрил и 4-хлорбензоилацетонитрил. Д. А. Драпкина,
В. Г. Брудзь, Н. И. Дорошина, Н. И. Соколова . . . .27
5-Бром-о-ванилин. Д. Л. Драпкина, Т. В. Скандалова, Н. А. Зу-
дилина .............................................. ..... 30
5-Бром-З-индолнлфосфат бария, В. М. Островская, И. А. Шуйская, Л. В. Ломакина . . . . ...................32
5-Бром-З-индолилфосфат натрия. В. М. Островская, И. А. Шуйская,
Л. В. Ломакина..............................................36
Бромкрезоловый зеленый. А. П. Болдырева, Г. И. Михайлов . . 39
Бромкрезоловый зеленый водорастворимый. А. П. Болдырева . . 42 Бромкрезоловый пурпуровый. Н. В. Факеева, А. П. Болдырева . 45 Бромкрезоловый пурпуровый водорастворимый. А. П. Болдырева . 48 5-Бром-4-оксиизофталевый альдегид. Д. А. Драпкина, Н. И. Доро-
шина ..................................................... 51
(З-Бромпропил) бензол. В. М. Островская, Н. Е. Каширина, Т. В.
Фомина, И. В. Хвостов ......... 53 трет-Бутилкарбазат. Е. А. Макарова, Г. Н. Кошелева . .56
4-втор-Бутнл-2-(а-метнлбензнл)фенол. В. М. Дзиомко, И. С. Мар-
кович, Н. В. Круглова.......................................59
Бутиловый эфир дихлоруксусной кислоты. И. Г. Хаскин, А. Л. Стол-
пер, Л. С. Лютая, Е. Р. Зарихта, В. С. Михайлов. Е. П. Бабин 61
Бутиловые эфиры метионина. И. И. Губенко, В. М. Котляревская . 63 трег-Бутиловые эфиры -D и La -оксиизовалериановых кислот.
Е. А. Макарова, Г. Н. Кошелева ... .... 66
Винил (дифенил) фосфииоксид. Г. Ф. Ярошенко, Л. М. Тимакова,
Н Е. Хавченко...............................................70
Гексаметилендиамино-N.N, N/ М'-тетракис(метилфосфоновая кислота)
В. В. Сидоренко, Н. В. Лапшина, Т. П. Коноплева . . . 73
Гексаметилфосфортриамид. А. Л. Фридман, В. Д. Сурков, Ф. М.
Мухаметшин..................................................75
3
Гексахлорацетон. И. Г. Хаскин, Л. О. Матвеев ..... 4 4’-Диалкоксиазоксибспзолы. Н. Г. Чернова, Г. И. Веселовская,
В. В. Салий, Е. И. Тарасюк, К. И. Мазыкина
1,6-Диаминогексан-1.1,6,6-тетрафосфоновая кислота. И. Д. Колпакова, Т. М. Балашова.............................................
Дибензил (дибензил амипометил)фосфиноксид. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я- Медведь
Дибензил (дибензиламинометил) фосфиноксид- N-оксид. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я, Медведь .
Ди-и-бутиловый и диаллиловый эфиры итаконовой кислоты. Е. И.
Тарасюк, Е. Ф. Цебриенко..................................
Дигексиловый эфир малеиновой кислоты. Н. Г. Чернова, Г. И. Веселовская, М. В. Мосенцева...................................
N, N-Днметил-л-нйтроанилин. Е. А. Бугай......................
4,4’- Диоксистильбеи-3,3’5,5’- тетракис(метилпмииодиуксусная кислота), тетранатриевая соль. Л. М. Тимакова, Г. Ф. Ярошенко, В. Я- Темкина, Р. П. Ластовский . . . . , .
Дифснил(диоктиламиноэтил)фосфиноксид. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я- Медведь
Дифенил(диэтиламинометил)фосфиноксид. Ю. М. Поликарпов, Г. Ф.
Ярошенко, Л. М. Тимакова, Т. Я- Медведь...................
N, N' Дифснил-л-фснилсндиамии. Б. И. Киссин, Е. Н. Куракин Дифенилхлорфосфип. Г. Ф. Ярошенко, Л. М. Тимакова, И. Е. Хав-яенко ..... .........................
Дихлорангидрид (3, {i, (3 -трнс(хлорметил) этилфосфористой кислоты.
Л. А. Власова, Е. В. Кузнецов .....................
н-Додециловый эфир галловой кислоты. Р. М. Гелыитейн, Е. И. Ма-ховер . . . .............................
п- и .и-Ксилилеидиамино-N, М'-бис(нзопропилфосфоиовые кислоты).
А4. В. Рудомино, Е. К. Колова, Т. Я- Медведь .... о -Метакриламидобеизойная кислота. С. И. Котенко, А. Г. Фадеиче-ва, И. С. Трохименко ......... DL - N -Метплвалип. Г. Н. Кошелева, Э. А. Башкир, В. И. Калмыков .........................................................
'ь-Монобутиловый эфир итаконовой кислоты. Р. А. Мелнгалве, А. Г.
Кайнова, С. Б. Макарова, Е. В. Егоров.....................
2- и 4-Нитро(3-бромпропил) бензолы. В. М. Островская, Н. Е. Каширина ........................................................
5-11итро-4,6-диметоксисалициловый альдегид. Д. А. Драпкина, Г. С.
Панова .......
Нитроксаминазо. Ю. М. Дедков . .....................
4-Нитро-2-метоксибензальдегид. Л. С. Зерюкина, Б. М. Болотин 5-Питро- Р -резорциловый альдегид. Д. А. Драпкина, И. М. Ролдугина, Н. И. Дорошина.........................................
З-Нитро-4,5,6 тримегоксифталевый ангидрид. Н. М. Рыбкина, М. Е.
Тихомирова. В. Н. Глушко..................................
7-Питро-4,5,6-триметоксифталид. Н. М. Рыбкина, М. Е. Тихомирова
В. Н. Глушко..............................................
Нитрофлуоресцеиндиацетат I. Н. М. Рыбкина, А. А. Положенцева, М. Е. Тихомирова...........................................\
2- и 4-Нитро(3-хлорпропил) бензолы. В. М. Островская, Н. Е. Каширина, Т. В. Фомина.....................................
н-Нониловый эфир п-питробензойной кислоты. Е. И. Маховер а-Оксибензилндендифосфоиовая кислота, натриевая и свинцовая соли. Р. П. Ластовский, И. Д. Колпакова, Л. В. Криницкая, Е. И. Миронова . .
4,4’-Оксибис(тетрахлорбензойная кислота). Е. П Бабин, Я П Ска-винский, И. А. Андрухов, А. А. Рыжков, А. С. Свириденко - .
77
80
84
86
88
90
92
94
96
99
101
103
166
109
111
ИЗ
116
118
121
123
125
127
131
133
135
138
140
143
145
147
151
Оксшидрохипоиаты свинца и кобальта. М. II. Русина, И. Я. Соло-мянникова, И. М. Дятлова . . . • 153
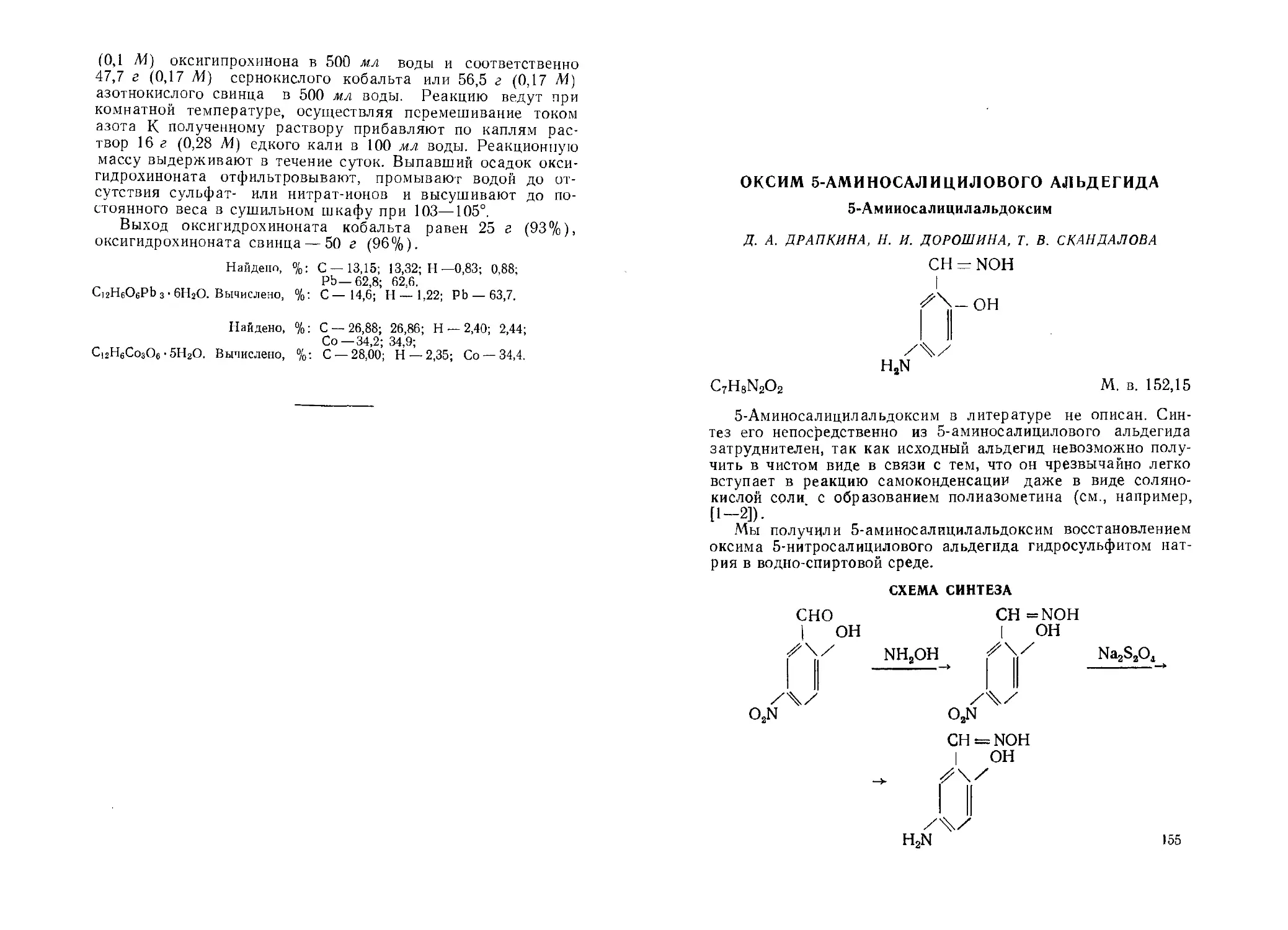
Оксим 5-аминосалицилового альдегида. Д. А. Драпкина, Н. И. Дорошина, Т. В. Скандилова ....................................155
9-Окси-5-метилфенил-1,3-бис (метиленимино) тетрауксусиая кислота.
В. Я. Темкина, И. В. Цирулъникова, И. П. Фадеева, Р. П. Л астанский . . • • • „ 158
8-Оксихинолин. Г. М. Бабчук, И. М. Храпова, Г. Д. Гандлевский . 160 (1-Оксиэтилиден) дифосфоиовая кислота, комплекс с медью. Л. В.
Криницкая, М. Н. Русина..........................Р ^2
(1-Оксиэтилиден)дифосфоновая кислота, монокалиевая соль. Б. И.
Бахман, Е. М. Уринович, Т. А. Богомолова, И. М. Дятлова . 164
N -(2-Оксиэтил)этилендиамино- X N'. N'-трис (метилфосфововая кислота). М. В. Рудомино, Е. К. Колова, Т. Я- Медведь . . 166
4-(н-н-Октилбензилокси) бутанол. В. А. Иншакова, 3. С. Сиденко,
Г. С. Чижова, Ю. С. Рябокобылко............................168
н-н-Октилбензилхлорид. 3. С. Сиденко, В. А. Иншакова, Г. С. Чижо-
ва, Ю. С. Рябокобылко......................................170
Октилиминобис[2-этил (дифенил) фосфиноксид]. Ю. М. Поликарпов,
Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я. Медведь .... 172
1-,2,3-Пирогаллол. А. Л. Лифиц, Е. П. Агеев....................1М
л-Стиролсульфонилфторид. К. М. Ройзен, Т. И. Коровина, С. Б. Ма-
карова ....................................................176
Те1ралкиловые эфиры этилендиампно- N, N' -бис(алкилфосфоновых кислот). М. В. Рудомино, И. В. Чурилина, Т. Я. Медведь 179
Тстраоктиловый эфир этилендиамино- N, N'-бис(бензилфосфоновоГ) кислоты). М. В. Рудомино, Е. К Колова, Т. Я- Медведь . . 183
Тиокарбамоилнитроеиний тетразолий. Л. А. Лабутина, В. М. Ост ровская, Л. В. Ломакина, Г. И. Михайлов....................185
Тиокарбогидразпд. В. М. Островская, Л. В. Ломакина . . 190
Тиомочевина. М. Ф. Кондрашова, Е. Я. Яровенко . . . 192
4,5,6-Триметоксифталид. Н. М. Рыбкина, М. Е. Тихомирова . .194
2,3,6- Триоксибензилиминодиуксусная кислота. Р. П. Ластовский
В. Я. Темкина, И. В. Цирульникова, Л. Я- Шиленко . . . 196
Трис( 3-цианэтил)фосфин. М. В. Рудомино, И. В. Чурилина, Т. Я-
Медведь ............ 199
Феноловый красный. А. П. Болдырева, А. У. Кагановская, Г. И. Михайлов . .................................... . . .201
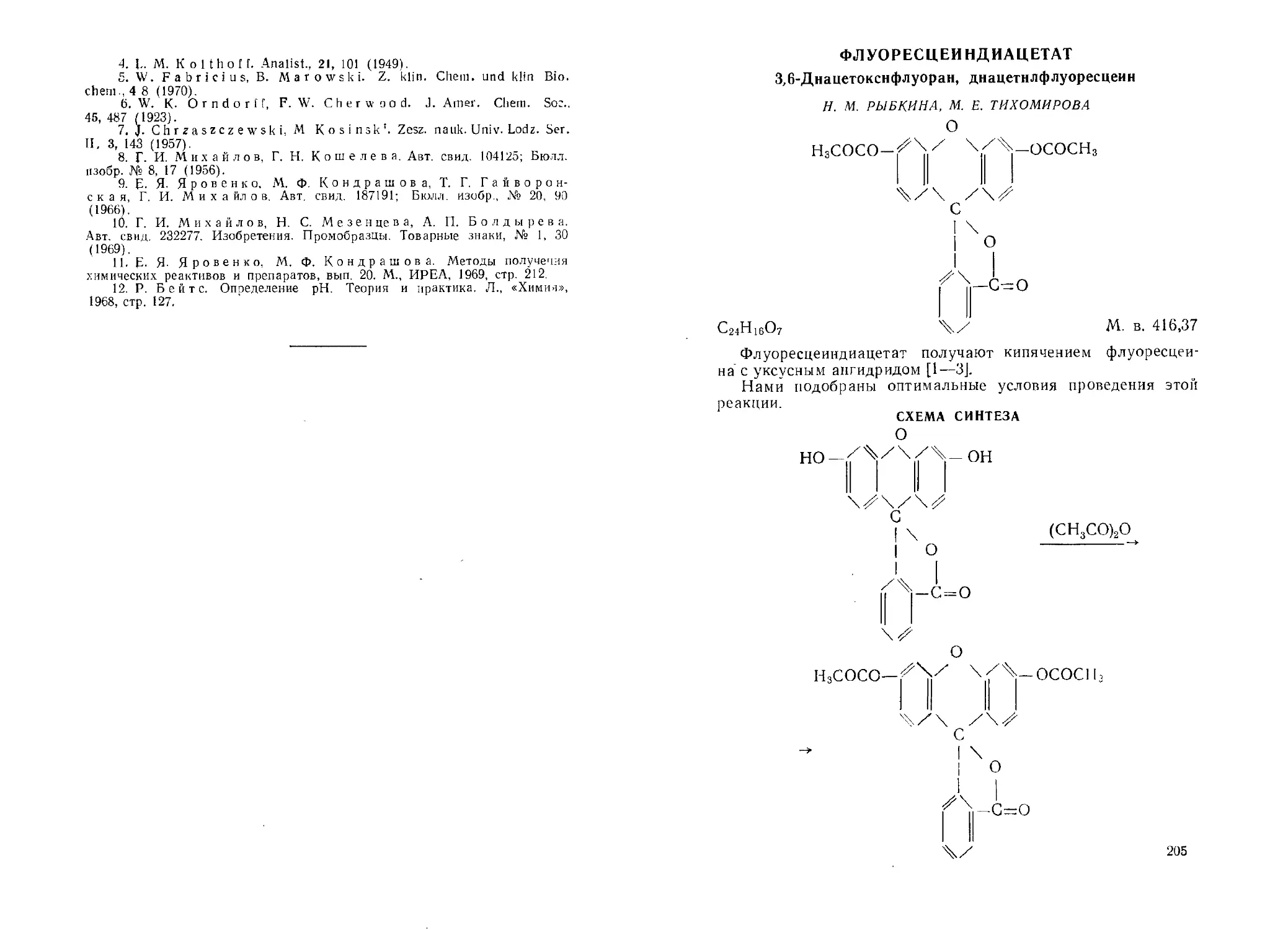
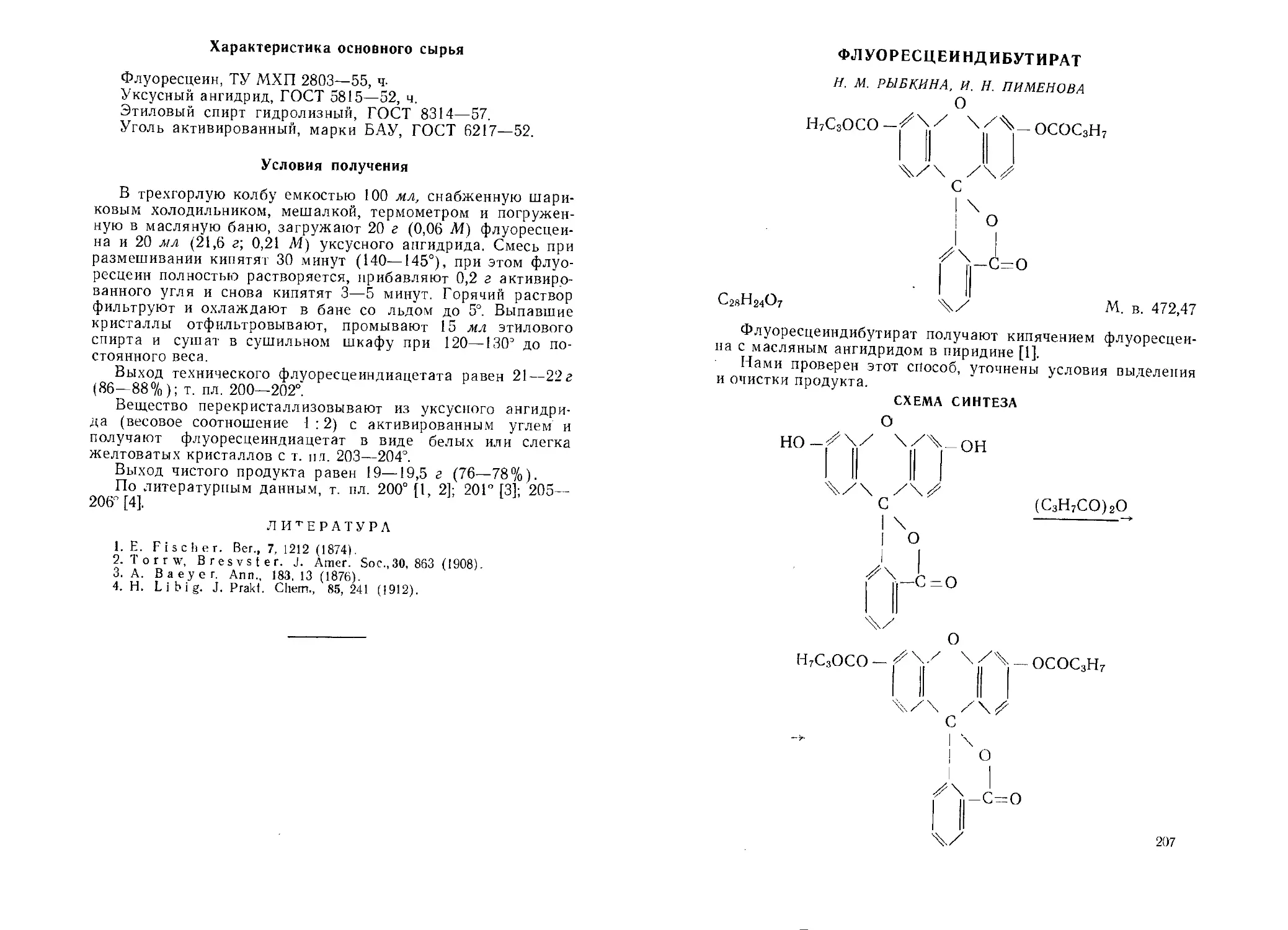
Флуоресцеиндиацетат. И. М. Рыбкина. Af. Е. Тихомирова . . . 205 Флуоресцеиндибутират. И. М. Рыбкина. И. И. Пименова . . . 207 а -Фруктоза.Н. В. Плеханова, М. И. Черняховская, Е. П. Крысин,
Г. П. Федорченко, И. Н. Голяева, В. И. Иванов . . . 209
8-Хинолинсульфокислота. Г. М. Бабчук, И. М. Храпова, Г. Д. Гандлевский ...................................................212
3 Хлор-5 питросалнциловый альдегид. Д. А. Драпкина, В. Г. Брудзь, И. И. Соколова.............................................214
(З-Хлорпропил) бензол. В. М. Островская, Т. В. Фомина . . 216
З-Хлорсалициловый альдегид. Д. А. Драпкина, В. Г. Брудзь, И. И.
Дорошина, И. И. Соколова .... .... 218
4-Циансалициловый альдегид. Р. У. Сафина, Б. М. Болотин . . 221
1,2,3-Циклогексантрион-1,3-диоксим. Г. С. Петрова, А. М. Лукин,
Е. И. Должникова, И. В. Титова, И. В. Хвостов .... 224 Циклогексантрион-1, 2, 3-триоксим. Г. С. Петрова, А. М. Лукин,
Е, Н. Должникова...........................................226
Этиловые эфиры п- и лг-ксили.теиднамино-N, М'бис(изопропилфосфо-новых кислот). М. В. Рудомино, Е. К- Колова, Т. Я- Медведь . 228
Предметный указатель . .................................231
5
З-АЛЛИЛ-5 НИТРОСАЛИЦИЛОВЫЙ АЛЬДЕГИД
Д. А. ДРАПДИНА, Н. И. ДОРОШИНА
сно
он
- СН2СН = сн2
o2n
c10h9no4
М. в. 207,18
З-Аллил-5-нитросалициловый альдегид был получен нами ранее [1] с выходом 31,4—34,8% нитрованием 3-аллилсалици-лового альдегида при комнатной температуре. Изменением температурного режима нам удалось повысить выход продукта на 10—15%.
СХЕМА СИНТЕЗА
СНО
Лг
Kz\
СН2СН = сн2
сно | он
HNO3
СНзСООН I II /ч/\ o2n сн2сн = сн2
Характеристика основного сырья
З-Аллилсалициловый альдегид, т- кип. 125—130713 мм;
<^0 = 1,5635— 1,5650; получение см [2].
Азотная кислота, ГОСТ 701—68, техн., пл. 1,51.
Уксусная кислота, ГОСТ 61—69, х. ч.
Петролейный эфир, ГОСТ 11992—62, т. кип. 40—60°.
Этиловый спирт ректификованный, ГОСТ 5962—67.
7
Условия получения
В круглодонную колбу емкостью 200 мл, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, помещают 16,2 г (0,1 Л1) 3-аллилсалицилового альдегида и 80 мл ледяной уксусной кислоты. Смесь нагревают до 40—50° и прибавляют за 40—50 минут раствор 5,1 мл (0,12 М) азотной кислоты в 25 мл уксусной кислоты, поддерживая температуру 50—55°. После 30-минутной выдержки при этой температуре реакционную смесь выливают в 400 г воды со льдом. На следующий день выпавший маслянистый осадок отфильтровывают, промывают 2—3 раза водой порциями по 10—15 мл, 3 раза этанолом порциями по 2—3 мл и сушат при комнатной температуре. Осадок помещают в экстрактор Сокслета и экстрагируют альдегид кипящим петролейным эфиром (200 мл); по охлаждении осадок отфильтровывают.
Выход З-аллил-5-нитросалицилового альдегида равен 9— 10 г (43—48%); т. пл. 81—82,5°. Дополнительная промывка несколькими миллилитрами спирта позволяет повысить температуру плавления продукта до 82—82,5° [1].
ЛИТЕРАТУРА
1. Д. А. Др а пк ина, В. Г. Брудзь, В. А. И и ш а к о в а, Ю. С. Рябо к о бы л ко. Ж. орган, химии, 5, 287 (1969).
2. Пат. США 2667442 (1954); С. А., 49, 1818 (1955).
tt-АМИНОАЦЕТОФЕНОН
В. В. САЛИЙ
С81 IgNO
М. в. 135,17
Описаны способы получения аминобензофенонов восстановлением металлами [1], гидразином [2], электрохимическим {3, 4] и другими методами восстановления [5—7].
Предлагаемый метод синтеза, основанный на восстановлении и-нитроацетофенона чугунными стружками, обеспечивает более легкое выделение n-аминоацетофенона из реакционной массы.
СХЕМА СИНТЕЗА
СОСПз
СОСНз
Fe
НС1
Nil;
9
Харакгеристика основного сырья
n-Нитроацетофенон, МРТУ 6—09—3371—67, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Стружки чугунньц, ГОСТ 1412—54.
Натр едкий, ГОСТ 4328—66, ч.
Этиловый спирт Щдролизный, ТУ 3—66—65, техн.
Условия получения
В трехгорлую колбу емкостью 0,7 л, снабженную термометром и мешалкой, пропущенной через обратный холодильник, загружают 60 г чугунных стружек, 30 мл воды, 12 мл концентрированной соляной кислоты. Смесь при перемешивании нагревают 10 мицуд на кипящей водяной бане и, продолжая нагревание и энергичное перемешивание, прибавляют небольшими порциями за 2 часа 49,5 г я-нитроацетофенона. Реакционную смесь выдерживают в тех же условиях еще 2,5 часа, после чего охлаждают до 60°. К смеси приливают 300 мл этилового спирта, энергично перемешивают 0,5 часа, отстаивают 0,5 часа и декантируют раствор на фильтр (воронка Бюхнера), снова приливают 200 мл спирта и повторяют ту же операцию. Осадок на фильтре промывают 100—150 мл этилового спирта до тех пор, пока фильтрат не станет бесцветным.
Полученный фильтрат нейтрализуют 10%-ным раствором щелочи до pH 7—8 (ц0 универсальной индикаторной бумаге) и фильтруют с отсасыванием через двойной бумажный фильтр. Из фильтрата отгоняет этиловый спирт (па водяной бане), оставляя в колбе око,ю 50 мл раствора. Остаток выливают в стакан и охлаждают до 15—20° при перемешивании./Выпав-шие кристаллы отживают, промывают на фильтре дистиллированной водой до нейтральной реакции промывнЦх вод и сушат в вытяжном шкафу. \
Выход n-аминоацетофенона равен 28 г (68,7%); т. пл. 106°.
По литературным данным [8], т. пл. 106°.
ЛИТЕРАТУРА
1. Drewsen. Ann.', 212, 152 (1882).
2. Общий практикум ц0 органической химии. Под ред. А. Н. Коста. М., «Мир», 1965, стр. 5]2. ’
3. W. Lob. Вег., 29,1894 (1895).
4. W. Lob. Z. Elektrochem., 2,46 (1896).
5. Geigy, Koenigs. Вег., 18, 2400 (1885).
6. Tatschaloff. J. prakt. Chem., [2|, 65, 308 (1902).
7. Gabriel,, Stelzner. Ber., 29, 1300 (1896).
8. Справочник химика, т. 2. M., «Химия», 1965, стр. 406.
10.
«-аминобензилидендифосфоновая кислота
Т. М. БАЛАШОВА, И. Д. КОЛПАКОВА
С6Н5 Р(О)(ОН)2
СХ
NH2 Р(О)(ОН)2
CzHnNOePz М. в. 267,04
а-Аминобензилидендифосфоновую кислоту получают взаимодействием треххлористого фосфора, бензонитрила и фосфористой кислоты [1, 2].
Нами проверены условия синтеза этой кислоты и показана возможность замены трехбромистого фосфора на более доступный и дешевый — треххлористый фосфор.
СХЕМА СИНТЕЗА
CCU
PC 13 + ЗН2О-► НзРОз + ЗНС1
С6Н5 Р(О)(ОН)2
Н2О \ /
C6H5CN + РС1з + Н3РО3—с
NH2 Р(О)(ОН)2
Характеристика основного сырья
Фосфор треххлористый, ГОСТ 91—41, ч.
Бензонитрил, МРТУ 6—09—7668—64, ч.
Четыреххлористый углерод, ГОСТ 5827—51, ч. д. а.
Ацетон, ГОСТ 2603—63, ч.
Условия получения
Получение фосфористой кислоты (см. примечание !) В трехгорлую колбу емкостью 250 мл,- помещенную в баню со
11
льдом и снабженную мешалкой, капельной воронкой и обратным холодильником с хлоркальциевой трубкой, загружают 80 мл четыреххлористого углерода и 21,3 г (0,15 (И) треххлористого фосфора. К смеси при перемешивании за 30 минут прибавляют по каплям 9 мл (0,2 Л1) дистиллированной воды и перемешивают еще 2 часа при комнатной температуре. Затем на силиконовой бане (50—60°) при пониженном давлении (8—10 мм) отгоняют четыреххлористый углерод. Остаток при охлаждении закристаллизовывастся в виде белых игл.
Выход фосфористой кислоты равен 13,5 г (количественный).
Получение а-аминобензилидендифосфоновой кислоты- К 13,5 г (0,15 М) фосфористой кислоты добавляют 5 г (0,05 М) бензонитрила и 13,9 г (0,1 М) треххлористого фосфора. Смесь перемешивают 3 часа при комнатной температуре и 8 часов при 55—60° (см. примечание 2), затем охлаждают ледяной баней до 0'\ прибавляют по каплям 30 мл ледяной воды и перемешивают до полного растворения.
Полученный слегка мутноватый раствор фильтруют с отсасыванием через двойной бумажный фильтр, фильтрат отделяют в делительной воронке от непрореагировавшего бензонитрила (-vl мл). К водному слою прибавляют равный объем ацетона (80 мл) и оставляют кристаллизоваться. Выпавший осадок отфильтровывают с отсасыванием, промывают па фильтре ацетоном 2 раза порциями по 15 мл, сушат сначала на воздухе, а затем в сушильном шкафу при 105 110 до постоянного веса.
Выход ц-аминобензилидендифосфоновои кислоты равен 4 г (42% в расчете на бензонитрил, вступивший в реакцию); т. пл. 223—225° (разлож.).
По литературным данным [1], т. пл. 225° (разлож.).° а-АминобензилидендифосфЬдовая кислота — белый порошок, растворим в холодной, лучше — в горячей воде, нерастворим в органических растворителях.
Найдено, %: С-30,9; 31,1; Н-4,3; 4,2; N-5,3; 5,2; Р—23,2; 22,9.
C7HuNOeP3 Вычислено, %: С-31,4; Н-4,1; N-5,3; Р-23.2.
Примечания.
1. Ввиду большой гигроскопичности фосфористой кислоты, последнюю необходимо Получить непосредственно перед синтезом а -аминобензили-дендифосфоновон кислоты, так как даже небольшое количество влаги в фосфористой кислоте значительно снижает выход продукта.
2. Повышение температуры приводит к осмолению реакционном масс >.
ЛИТЕРАТУРА
1. Пат. ФРГ 1002355 (1957); С. А., 53, 21814 1 (1959ф
2. Н. М Дятлова, В. В. Медынцев и др. Ж- общ. химии, 32, 2, 329 (1969).
12
1-АМИНОГЕКСИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА
Т. М. БАЛАШОВА, И. Д. КОЛПАКОВА
С5Нц Р(О)(ОН)2
4 с/
NH2Z Р(О)(ОН)г
C6H17NOeP2
М. в. 261.15
1-Аминогексилидендифосфоновую кислоту получают взаимодействием капронитрила, трехбромистого фосфора и фосфористой кислоты [1].
Нами проверены и уточнены условия синтеза и выделения этого соединения.
СХЕМА СИНТЕЗА
С5НП Р(О)(ОН)2 н2о \ /
C5HnCN + РВг3 + Н3РО3-> с
NH2 Р (ОКОН) 2
Характеристика основного сырья
п-Капронитрил, МРТУ 6—09—3814—67, ч.
Фосфор трехбромистып, МРТУ 6—09—338—63, ч.
Фосфористая кислота, ТУ МХП 518—41, ч.
Ацетон, ГОСТ 2603—63, ч.
Способ получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником с хлоркальциевой трубкой и термометром, помещают 10 а (0,1 (И) н-капропитрила, 54 г
13
(0,2 Л4) свежеперегнанного трехбромистого фосфора и 25,4 г (0,31 М) фосфористой кислоты. Смесь перемешивают 3 часа при комнатной температуре, затем еще 5 часов при 60—70° (температура бани) и оставляют на ночь. Загустевшую реакционную массу охлаждают (ледяной баней) до 0° и прибавляют к ней 50 мл ледяной воды; перемешивание продолжают до тех пор, пока масса полностью не растворится. Полученный раствор фильтруют с отсасываннем через подушку активированного угля. К бесцветному прозрачному фильтрату добавляют 100 мл ацетона, осадок отфильтровывают и промывают на фильтре 20 мл ацетона, сушат сначала на воздухе, затем в сушильном шкафу при НО—115°.
Выход 1-аминогексилидендифосфоновой кислоты равен 13 г (50%); т, пл. 226—228° (разлож.)-
По литературным данным [1], т. пл. 228° (разлож.).
1-Аминогексилидендифосфоновая кислота представляет собой блестящие кристаллы в виде пластиночек, хорошо растворимых в воде и нерастворимых в органических растворителях.
Найдено, %: С—27,8; 27,7; Н—6,7; 6,8; N—6,4; 6,5;
Р- 23 1 • 23 1
C5Hj7NO0P2 Вычислено, %; С—27,5; Н—6,5; N--6,4; Р—23,7.
Л ИТЕРАТУРА
1, Пат. ФРГ 1002355 (1954); С. А., 53, 21814 Г (1959).
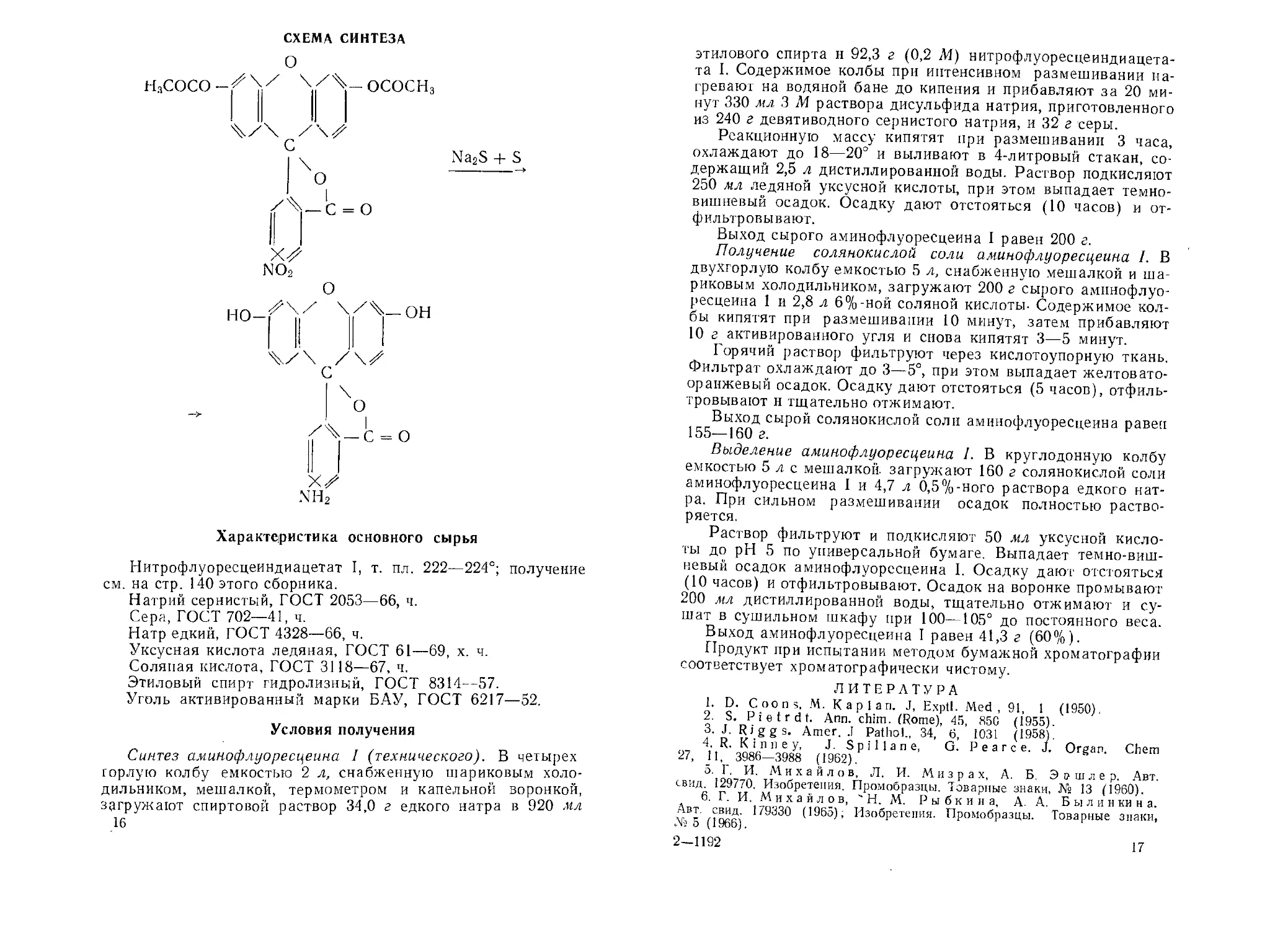
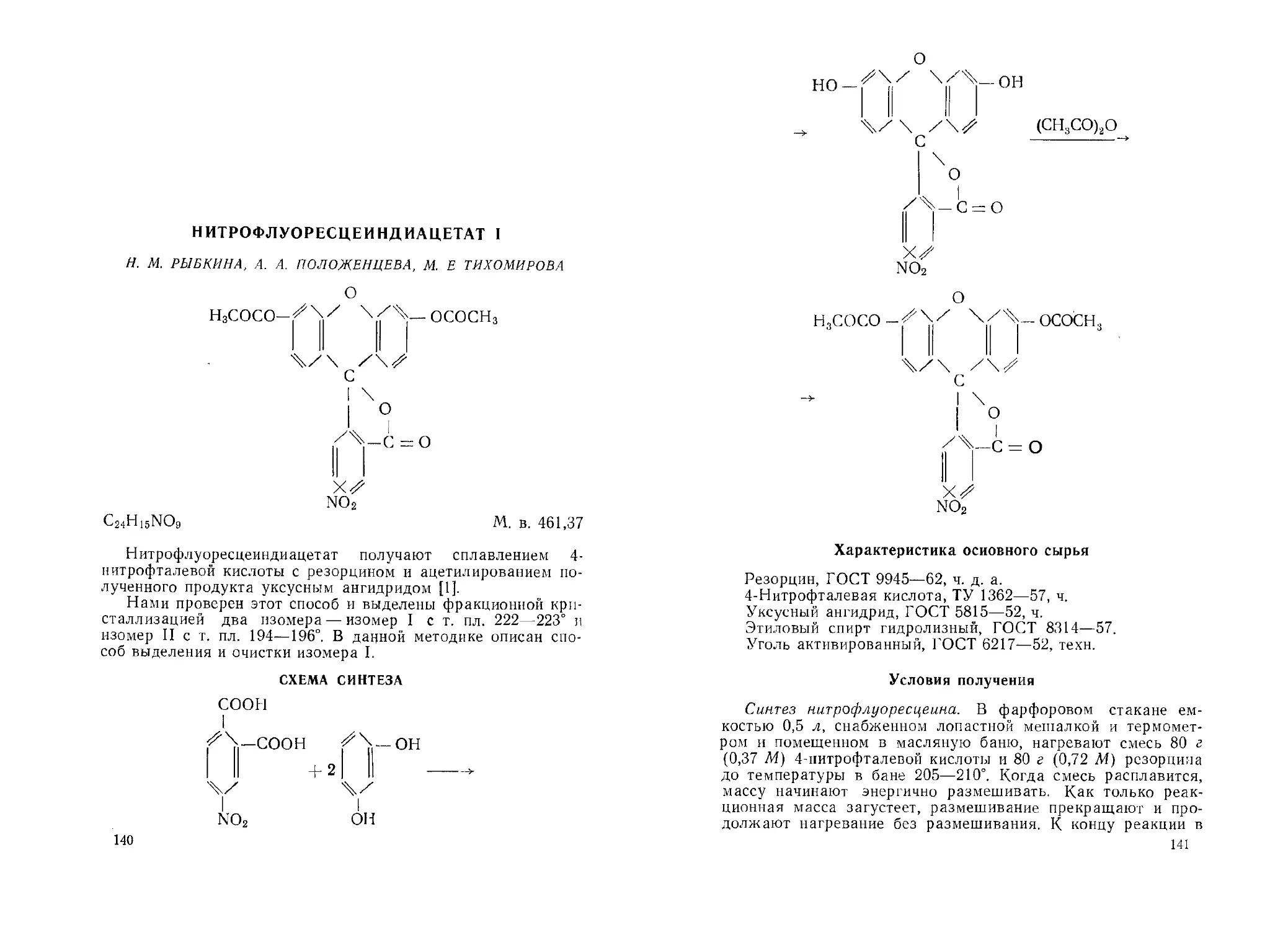
АМИНОФЛУОРЕСЦЕИН 1
Г. И. МИХАИЛОВ, Н. М. РЫБКИНА, А. А. ПОЛОЖЕНЦЕВА
C20H13NO5
М. в. 347,33
Амнпофлуоресцеин I является исходным продуктом для синтеза флуоресцентных метчиков белка.
В литературе описаны методы получения аминофлуоресцеина каталитическим гидрированием ннтрофлуоресцеина I на никеле Ренея {1, 2], на угольно-палладиевом катализаторе 13], восстановлением нитрофлуоресцеина сернистым натрием и Iидросульфидом {4] или гидразин-гидратом в присутствии никеля Ренея [5].
Нами предложен метод восстановления нитрофлуоресцеип-диацетата I раствором дисульфида натрия, найдены оптимальные условия проведения синтеза [6].
15
СХЕМА СИНТЕЗА
Ый2$ + S
Характеристика основного сырья
Нитрофлуоресцеиндиацетат I, т. пл. 222—224°; получение см. на стр. 140 этого сборника.
Натрий сернистый, ГОСТ 2053—66, ч.
Сера, ГОСТ 702—41, ч.
Натр едкий, ГОСТ 4328—66, ч.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Уголь активированный марки БАУ, ГОСТ 6217—52.
Условия получения
Синтез аминофлуоресцеина I (технического). В четырех гордую колбу емкостью 2 л, снабженную шариковым холодильником, мешалкой, термометром и капельной воронкой, загружают спиртовой раствор 34,0 г едкого натра в 920 мл
16
этилового спирта н 92,3 г (0,2 Л1) нитрофлуоресцеиндиацетата I. Содержимое колбы при интенсивном размешивании нагревают на водяной бане до кипения и прибавляют за 20 минут 330 мл 3 М раствора дисульфида натрия, приготовленного из 240 г девятиводного сернистого натрия, и 32 г серы.
Реакционную массу кипятят при размешивании 3 часа, охлаждают до 18—20° и выливают в 4-литровый стакан, содержащий 2,5 л дистиллированной воды. Раствор подкисляют 250 мл ледяной уксусной кислоты, при этом выпадает темновишневый осадок. Осадку дают отстояться (10 часов) и отфильтровывают.
Выход сырого аминофлуоресцеина I равен 200 г.
Получение солянокислой соли аминофлуоресцеина I. В двухгорлую колбу емкостью 5 л, снабженную мешалкой и шариковым холодильником, загружают 200 г сырого аминофлуоресцеина 1 и 2,8 л 6%-ной соляной кислоты- Содержимое колбы кипятят при размешивании 10 минут, затем прибавляют 10 г активированного угля и снова кипятят 3—5 минут.
Горячий раствор фильтруют через кислотоупорную ткань. Фильтрат охлаждают до 3—5°, при этом выпадает желтовато-оранжевый осадок. Осадку дают отстояться (5 часов), отфильтровывают н тщательно отжимают.
Выход сырой солянокислой соли аминофлуоресцеина равен 155—160 г.
Выделение аминофлуоресцеина 1. В круглодонную колбу емкостью 5 л с мешалкой загружают 160 г солянокислой соли аминофлуоресцеина I и 4,7 л 0,5%-ного раствора едкого натра. При сильном размешивании осадок полностью растворяется.
Раствор фильтруют и подкисляют 50 мл уксусной кислоты до pH 5 по универсальной бумаге. Выпадает темно-вишневый осадок аминофлуорссцеина I. Осадку дают отстояться (10 часов) и отфильтровывают. Осадок на воронке промывают 200 мл дистиллированной воды, тщательно отжимают и сушат в сушильном шкафу при 100—105° до постоянного веса.
Выход аминофлуоресцеина I равен 41,3 г (60%).
Продукт при испытании методом бумажной хроматографии соответствует хроматографически чистому.
ЛИТЕРАТУРА
1. D. С оо n s, М. К а р 1 а п. J, Exptl. Med , 91, 1 (1950).
2. S. Pietrdt. Ann. chim. (Rome), 45, 850 (1955).
3. J. Riggs. Amer. .1 Pathol., 34, 6, 1031 (1958).
4. R. Kinney, J. Spillane, G. Pearce. J. Organ. Chem 27, 11, 3986—3988 (1962).
5. Г, И. Михайлов, Л. И. Мизрах, А. Б. Э о ш л e p. Авт. свид. 129770. Изобретения. Промобразцы. Товарные знаки, № 13 (1960).
6. Г. И. Михайлов, 'Н. М. Рыбкина, А. А. Былинкина. Авт. свид. 179330 (1965); Изобретения. Промобразцы. Товарные знаки, № 5 (1966).
2—1192 17
1-АМИНОЭТИ Л ИД ЕНД И ФОСФОНОВАЯ КИСЛОТА
Т. М. БАЛАШОВА, И. Д. КОЛПАКОВА
СНз Р(О)(ОН)2
nh/ \(О)(ОН)2
C2H9NO6P2
М. в. 205,04
1-Аминоэтилидендифосфоновую кислоту получают взаимодействием трехбромистого фосфора, ацетонитрила и ледяной уксусной кислоты [1].
Нами проверены и уточнены условия синтеза и выделения зтого соединения.
СХЕМА СИНТЕЗА
СНз Р(О)(ОН)2 н2о \ / CH3CN + РВгз + СНзСООН-----_ С
NH2Z Р(О)(ОН)2
Характеристика основного сырья
Фосфор трехбромистый, МРТУ 6—09—338—63, ч.
Ацетонитрил, МРТУ 6—09—2508—65, ч.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч.
Способ получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником с хлоркальциевой трубкой и капельной воронкой, помещают 53 г (0,196 М) трехбромистого
18
фосфора и при охлаждении прибавляют по каплям 5 .? (0,12 Л4) ацетонитрила. Перемешивают при комнатной температуре 20 часов, затем прибавляют 32,5 г (0,54М) ледяной уксусной кислоты и перемешивают еще 20 часов при комнатной температуре. Реакционную массу охлаждают до 0° и осторожно прибавляют к ней 50 мл ледяной воды, при этом выделяется бромистый водород.
Полученный желтоватый раствор обесцвечивают активированным углем и фильтруют, фильтрат упаривают иа водяной бане до появления осадка и оставляют на ночь для более полного выделения продукта. На следующий день осадок отфильтровывают с отсасыванием и промывают на фильтре ацетоном дважды порциями по 20 мл-
Выход 1-аминоэтилидендифосфоновой кислоты равен 5,5 г (22%), т. пл. 275—277°.
Для очистки продукт перекристаллизовывают из водного (1:1) ацетона, отфильтровывают, промывают на фильтре 20 мл ацетона, сушат сначала на воздухе, а затем в сушильном шкафу при ПО—115°.
Выход перекристаллизованной 1-аминоэтилидендифосфоно-вой кислоты равен 5 г (20%); т. пл. 276—277°.
По литературным данным [1], т. пл. 277°.
Вещество представляет собой белый кристаллический порошок, хорошо растворим в воде, хуже — в спирте, не растворяется в органических растворителях.
Найдено, %: С—12,3; 12,2; Н—4,6; 4,8; N—6,8; 6,9;
Р—30,6; 30,4
C2H9NOtP2. Вычислено, %; С—11,8; Н—4,4; N—6,8; Р—30,09.
ЛИТЕРАТУРА
1. Пат. ФРГ 1002355 (1954); С. А., 53, 21814 1 (1959).
24
М -Ацетобензолсульфонилфторид
А. М. РОЙЗЕН, т. Н. КОРОВИНА, С. Б. МАКАРОВА
СОСНз
- so2f
CsHtFOsS
М. в. 202,20
-« Ацетобензолсульфонилфторид служит исходным сырьем для получения ж-стиролсульфокислоты, применяемой в синтезе ионообменных смол. , „
В литературе описан многостадийный синтез ж-ацетооен-Золсульфонилфторида из ж-хлорсульфонилбензойной кислоты
Нами разработан более простой.метод синтеза этого^оеди-Дения из ж-аминоацетофенона. иР°” у методу [2], причем и-ацетобензолсульфопилхлорид получе сто выход увеличен нами вдвое.
СХЕМА СИНТЕЗА
20
Характеристика основного сырья
ш-Аминоацетофенон, ТУ КРЗ 185—15, ч.
Натрий азотистокислый, ГОСТ 4197- 66, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Диоксан, ГОСТ 10455—63, ч.
Сернистый ангидрид.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч.
Медь однохлористая, ГОСТ 4164—61.
Условия получения
Синтез м-сщетобензолсульфонилхлорида. В колбе емкостью 0,5 л, снабженной мешалкой и термометром, растворяют при 60° 27 г (0,2 ЛГ) .u-амминоацетофенона в смеси 140мл концентрированной соляной кислоты и 20 мл воды. Раствор охлаждают до 0 —5° и через капельную воронку медленно приливают охлажденный раствор 14 г (0,2 М) нитрита натрия в 50 мл воды, следя за тем, чтобы температура реакции не превышала 5°. Реакционную массу выдерживают 15 минут при этой температуре, затем температуру доводят до комнатной.
В колбу емкостью 1 л, снабженную мешалкой и трубкой для отвода газа, помещают 250 мл ледяной уксусной кислоты и 3 г однохлористой меди. Смесь насыщают сернистым ангидридом в течение 1,5 часов до проскока газа и осторожно при комнатной температуре за 0,5 часа приливают через капельную воронку полученный раствор соли диазония. Реакционную массу выдерживают при 25° до прекращения выделения пузырьков азота и выливают на лед- Выделившийся осадок отфильтровывают и промывают ледяной водой до нейтральной реакции.
Выход ш-ацетобензолсульфонилхлорида равен 34 г (80%); т пл. 41—42°, что соответствует литературным данным [2].
Получение м-ацетобензолсулъфонилфторида. В колбу емкостью 100 мл, снабженную мешалкой и термометром, помещают 10,5 г (0,05 АГ) ш-ацетобензолсульфонилхлорида, 30 мл диоксана и раствор 45 г (0,075 М) фтористого калия в 50 мл воды- Реакционную массу выдерживают 3 часа при 45° и выливают на лед. Выделившийся осадок отфильтровывают.
Выход лг-ацетобензолсульфонилфторида равен 8,2 г (85%). Вещество получают в виде белого кристаллического порошка с т. пл'. 90—92°, что соответствует литературным данным [1].
ЛИТЕРАТУРА
1. A. D. Cat, R. V. Poucke. J. Organ. Chem., 30, 1498 (1965).
2. C. Troltsch. J. pract. Chem., 22, 196 (1963).
21
CigHie
4-БЕНЗИЛДИФЕНИЛ
И. А. БАРЦ, Е. М. ИОФИС, Р. М. ГЕЛЬШТЕЙН
М. в. 244,32
4-Бензилдифенил применяется в газо жидкостной хроматографии в качестве неподвижной фазы [1, 2].
В литературе описан метод получения бензилдифенила конденсацией хлористого бензила с дифенилом [3, 4]. При этом образуется смесь изомеров, из которой 4-бензилдифенил в результате многократной перекристаллизации получают с выходом менее 10%.
Нами проверена и уточнена методика [4] получения 4-бен-зилдифенила восстановлением беизоилдифенила йодистоводородной кислотой в присутствии фосфора и разработан полярографический метод определения конца восстановления. 4-Бен-зоилдифенил получен по реакции Фриделя-Крафтса из хлористого бензоила и дифенила в среде дихлорэтана.
СХЕМА СИНТЕЗА
Характеристика основного сырья
Бензоил хлористый, ТУ ГСНХ 1079—60, техн. Дифенил, ГОСТ 4254—48, техн.
22
Алюминий хлористый, ГОСТ 4452—66, техн.
Дихлорэтан, ГОСТ 1942—63, техн.
йодистоводородная кислота, ГОСТ 4200—67, ч. д. а.
Изопропиловый спирт, ГОСТ 9805—61, техн.
Соляная кислота, ГОСТ 3118—67, ч.
Фосфор красный, ГОСТ 8655—57.
Толуол, ГОСТ 1930—56, ч-
Условия получения
Синтез бензоилдифенила. В трехгорлую колбу емкостью 3 л, снабженную мешалкой, обратным холодильником и термометром, помещают раствор 390 г (2,52 М) дифенила в 1,5 кг дихлорэтана. При размешивании приливают 354 г (2,52 М) хлористого бензоила и порциями за 1,5 часа при 20—25° прибавляют 402 г (3 А-1) хлористого алюминия. Реакционную массу нагревают на глицериновой бане до 50- 60° и после 30-минутной выдержки выливают в 10-литровый фарфоровый котел, содержащий 5 л 3%-ной соляной кислоты, охлажденной до 2—5°, и размешивают 2 часа. Органический слой отделяют от водного и отгоняют дихлорэтан. Остаток перегоняют в вакууме, собирая фракцию с т. кип. 234—235°/2 мм.
Выход бензоил дифенил а равен 520 г (80%); т. пл. 101 — 102°.
По литературным данным [5], т. пл. 102°.
Получение бензилдифенила. В трехгорлой колбе емкостью 3 л, снабженной мешалкой, обратным холодильником и термометром, кипятят при размешивании смесь 520 г (2 Af) бензоилдифенила, 500 г (1,75 М) йодистоводородной кислоты и 125 г (4 г-ат.) фосфора, причем температура кипения повышается от 120° до 135°. Смесь выдерживают при 135° до тех пор, пока концентрация бензоилдифенила в реакционной массе будет меньше 1% (см- примечание). Общая продолжительность кипячения около 40 часов.
Горячую (90—100°) реакционную массу дважды экстрагируют толуолом порциями по 550 мл. Толуольный экстракт при нагревании (90—100°) размешивают с 10 г активированного угля, фильтруют и из фильтрата отгоняют 700 мл толуола. Из остатка после охлаждения до 15° выпадает кристаллический осадок, который отфильтровывают, промывают 50 мл изопропилового спирта и сушат при 40—50°.
Выход бензилдифенила равен 385 г (78%); т. пл. 85—86°.
По литературным данным (6], т. пл. вещества 85°.
Пр и м е ч а и и е. Бепзоилдифенил в реакционной массе определяли полярографически в ячейке с донной ртутью с начальным напряжением 0,8 и на фоне 0,1 н. метанольного раствора гидрата окиси лития. Содержание бенгоилдифенила определяли по калибровочному графику.
23
ЛИТЕРАТУРА
1. Руководство по газовой хроматографии. Перевод с нем. под редакцией А. А. Жуховицкого. М., «Мир», 1969.
2 ['. Мак-Нейр, Э. Бонелли. Введение в газовую хроматографию. М., «Мир», 1970.
3. D. Costin N’enizesc'J. Am., 491,211 (1931).
4. М. А. Че ль цо в а, А. Д. Петров и др. Изв, АН СССР, сер. хим., 1, 129 (1965).
5. Справочник органических соединений, т. 3. М., Инлитиздат, 1949, cip. 384.
6. Справочник химика, т. 2, Л., «Химия», 1964, стр. 654.
N-БЕНЗИЛФТАЛ ИМИД
Ю. Г. БОНДАРЬ, Е. А. ЧЕРНИНА
СН2-
C15HuNO2
М. в. 237,26
N-Бензилфталимид получают взаимодействием фталимида калия и хлористого бензила [1], фталимида, хлористого бензила и поташа (2, 3], фталевого ангидрида и бензиламина в ледяной уксусной кислоте [4].
Нами проверены перечисленные методы и показано, что максимальный выход и лучшее качество продукта получается при проведении синтеза по последнему методу. Изменение соотношения компонентов, условий синтеза и выделения продукта позволили упростить процесс и увеличить выход N-бензил-фталимида.
СХЕМА СИНТЕЗА
СО
+ nh2ch2 -
25
Характеристика основного сырья
Фталевый ангидрид, ГОСТ 7119—54, 1 с., перегнанный. Бензиламин, МРТУ 6—09—5361—68, ч-
Уксусная кислота, ГОСТ 7071—54, техн.
Условия получения
В колбу емкостью 0,5 л с обратным холодильником помещают 430 г уксусной кислоты, 25 г (0,235 М) бензиламина, 50 г (0,335 Л4) фталевого ангидрида и кипятят 2 часа на глицериновой бане. В кипящую реакционную массу вносят активированный уголь, фильтруют и выливают фильтрат в стакан, содержащий 200 мл воды. После охлаждения до 20° осадок отжимают, промывают 150 мл воды и сушат при 40—50°.
Выход N-бензилфталимида равен 40 г.
Из уксуснокислого маточного раствора после разбавления равным по объему количеством воды получают осадок, который после перекристаллизации из уксусной кислоты даег дополнительно 5 г продукта-
Общий выход N-бензилфталимида равен 45 г (83%); т. пл. 115--1160; содержание основного вещества (по азоту) 99%.
По литературным данным [1], т. пл. вещества 115—116°.
ЛИТЕРАТУРА
1. S. Labrie. Вег., 20, 2227 (1887).
2. Синтезы органических препаратов, т. 2. М., Инлитиздат, 1952, стр. 84.
3. R. Manske. J. Cheni. Soc., 1926,2348.
4. Y. V a n a g s. Acia Univ. Latviensis Kim. Fakultat Ser. 4, 8, 405 (1939); C. A., 34, 1983 (1940).
БЕНЗОИЛАЦЕТОНИТРИЛ И 4-ХЛОРБЕНЗОИЛАЦЕТОНИТРИЛ
<й-Цианацетофенон, фенацилцианид
Д. А. ДРАПКИНА, В. Г. БРУДЗЬ, Н. И. ДОРОШИНА, Н. И. СОКОЛОВА
COCH2CN
/К
coch2cn
I Z4
C9H7NO М.В. 145,16
\Z
I.
Cl
CsHeCINO M. B. 179,60
Известный метод получения ароил ацетонитрилов [1, 2] основан на ацилировании циануксусного эфира хлорангидридами ароматических кислот с последующим щелочным гидролизом образовавшихся ароилциануксусных эфиров. Авторы на примере синтеза 4-нитропроизвюдного показали, что ацилирование можно проводить в присутствии едкого натра, если к циануксусному эфиру прибавлять одновременно растворы щелочи и хлорангидрида, поддерживая определенный pH среды.
Нами этот метод распространен на некоторые другие аро-илэфиры [3], упрощен процесс ацилирования, способ выделения и очистки конечных продуктов—ароилацетопитрилов.
СХЕМА СИНТЕЗА
Na ОН
С6Н5СОС1 + CH2(CN)COOC2H6
НС1
NaOH
-> C6H5COCH(CN)COOC2H3 —F7T"* C6H5COCH2CN
CU2
27
Характеристика основного сырья
Этиловый эфир циануксусной кислоты, ч.
Бензоил хлористый, ТУ МХП 92—51, техн. 4-Хлорбензоилхлорид, МРТУ 6—09—1036—64, ч. Ацетон, ГОСТ 2603—63, ч.
Натр едкий, ГОСТ 4328—66, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Углекислота пищевая, ГОСТ 8050—64 (из баллона).
Условия получения
Синтез этилового эфира бензоилциануксусной кислоты. В фарфоровый стакан емкостью 1 л- снабженный якорной мешалкой, термометром и капельной воронкой, помещают 22,4 г (0,16 Л4) бензоилхлорида, 19,9 г (0,176 А1) этилового эфира циануксусной кислоты и 150 мл ацетона. К полученному раствору при интенсивном размешивании и наружном охлаждении льдом с солью прибавляют из капельной воронки 40% -ный раствор едкого натра до достижения устойчивого pH 8—9. Щелочь прибавляют с такой скоростью, чтобы температура реакционной смеси не превышала 20° (15—20 минут). Затем удаляют баню, массу размешивают еще 2 часа, разбавляют 300 мл ледяной воды и осаждают эфир подкислением 10%-ной соляной кислотой по «конго». Осадок отфильтровывают с отсасыванием и промывают водой до нейтральной реакции.
Вьщод этилового эфира бензоилциануксусной кислоты равен 32 г (94—95% в расчете на хлорангидрид кислоты).
Получение бензоилацетонитрила. В трехгорлую колбу емкостью 1,5 л, снабженную мешалкой, термометром и воздушным холодильником, помещают 32 г этилового эфира бензо-илциаруксусной кислоты и 640 мл 10%-ного раствора едкого натра. Смесь размешивают 6 часов при 45—50°, затем оставляют на 15—20 часов при комнатной температуре. К раствору добавляют при размешивании 400 мл 10%-ной соляной кислоты и 1,5—2 г активированного угля (см. примечание 1), размешивают 30 минут и фильтруют; фильтрат частично нейтрализуют при размешивании 10%-ной соляной кислотой (pH 8,5—9), после чего насыщают углекислотой до прекращения выпадения осадка бензоилацетонитрила (см. примечание 2). Осадок отфильтровывают, тщательно отжимают и промывают 300 мл воды.
Выход бензоилацетонитрила равен 10,3—11,3 г (44—48% в расчете на бензоилхлорид); т. пл. 79,5—80,5°.
По литературным данным [1], т. пл. 79—80°.
Получение 4-хлорбензоилацетонитрила. По аналогичной методике из 28,0 г (0,16 М) м-хлорбензоилхлорида получают 28
32 г этилового эфира 4-хлорбензоилциануксусной кислоты. Для гидролиза эфира применяют 950 мл раствора едкого натра.
Выход 4-хлорбензоилацетонитрила равен 12,5—14,5 г (43— 50%); т. пл. 129—130°.
По литературным данным [1—4], т. пл. 129—130°-
Примечания.
1. Если раствор слабо окрашен, обработку углем исключают.
2. В этих условиях целиком осаждаются исключительно ароилацетонитрилы, все остальные примеси, как-то: исходный ароилциануксусный эфир, соответствующая кислота и другие остаются в растворе.
ЛИТЕРАТУРА
1. Ф. И- Степанов, Н. С. Вульфсон. Органические полупродукты и красители, вып. 1. М., НИОПиК, 1959, стр. 222.
2. Н. С. Вульфсон. Там же, вып. 2. 1961, стр. 123.
3. В. Г. Брудзь, Д. А. Драпкина, Н. И. Б а д а й к о в, Н. И. Дорошина. Авт. свид. 213831; Изобретения. Промобразцы. Товарные знаки, № 11, (1968).
4. J. Rabcewicz —Zubkowski, II. Kaflinska. Roczniki Chem., 10, 555 (1930).
5-БРОМ- oi-ВАНИЛИН
5-Бром-З-метоксисалициловый альдегид; 5-бром-2-окси-3-метоксибензальдегид
Д. А. ДРАПКИНА, Т. В. СКАНДАЛОВА, И. А. ЗУДИЛИНА
СвНтВгОз
М. в. 231,05
В литературе имеются противоречивые данные о возможности синтеза 5-бром-о-ванилина: бромированием о-ванилина бромом в ледяной уксусной кислоте альдегид был получен с выходом 73—80% [1—3j; однако, по данным работы [4], этим методом не удалось получить удовлетворительного результата; продукт был синтезирован с выходом 51 % при использовании сероуглерода в качестве реакционной среды.
Бромированием о-ванилина в среде ледяной уксусной кислоты в присутствии безводного уксуснокислого натрия мы получили 5-бром-о-ванилин с выходом 67—75%'.
СХЕМА СИНТЕЗА
СНО
П ~он
-ОСН3
Вг2
СНзСООН
СНО
(I 'ои AJ -осн-
Вг
30
Характеристика основного сырья
о-Вапилин, т. пл. 42—44°; получение см. [5].
Бром, ГОСТ 4109—64, ч.
Натрий уксуснокислый плавленый, МРТУ 6—09—2121 — -65, ч.
Уксусная кислота, ГОСТ 61—69, ч.
Этиловый спирт ректификованный, ГОСТ 5962—67.
Условия получения
В четырехгорлую колбу, снабженную мешалкой, капельной воронкой, воздушным холодильником и термометром, помещают 7,6 г (0,05 Л4) о-ванилина, 70 мл ледяной уксусной кислоты и 10 г плавленого ацетата натрия. Смесь размешивают, охлаждают до 10° и прибавляют за 30 минут раствор 2,6 мл [8,16 г (0,051 Л4)] брома в 15 мл уксусной кислоты, поддерживая температуру 10—15°. Через 10 минут реакционную смесь выливают в 200 мл воды. Через 4—5 часов выпавший осадок отфильтровывают, промывают водой до отрицательной реакции на бром (проба с азотнокислым серебром; небольшим помутнением можно пренебречь) и сушат при 50—60°.
Выход сырого 5-бром-о-ванилина равен 11,0—11,3 г (почти количественный); т. пл. 126—128°. Альдегид очищают кристаллизацией из 90—100 мл этанола.
Выход чистого продукта в виде светло-желтых или сероватых иголочек равен 8,0—8,6 г (67—75%); т. пл. 129—129,5°.
По литературным данным |[4]> т. пл. 128—129°.
ЛИТЕРАТУРА
1. Е. Rupp, К. Line k. Arch. Phartnazje, 253,37 (1915).
2. W. Davies J. Chem. Soc., 1923, 1575.
3. H. Fiedler. Arch. Pharmazie, 297, 226 (1964).
4. R. C. Fuson, R. G a e r t n e r, D. H. Chadwick. J. Organ. Chein., 13, 489 (1948).
5. A. П. Терентьев, E. Г. P у x а д з e, Г. П. Талызенкова, Г. В. Панова. Авт. евнд., 164589; Изобретения. Промобразцы. Товарные знак;!, № 16, 9 (1966); РЖХим., 2Н152 (1964).
5-БРОМ-З-ИНДОЛИЛФОСФАТ БАРИЯ
5-Броминдоксилфосфат бария
В. М. ОСТРОВСКАЯ, И. А. ШУЙСКАЯ, Л. В. ЛОМАКИНА
О
ОР
I \
I О
1 I
О—Ва-2Н2О
C8H5BaBrNO4P • 2Н2О
М. в. 463,39
5-Бром-З-индолилфосфат бария может быть использован в гистохимии в качестве субстрата для определения фосфатаз биологических тканей f 1J. Синтез этого соединения в литературе не описан. 5-Бром-З-индолилфосфат бария синтезирован нами фосфорилированием 5-бром-Ы-ацетилиндоксила хлорокисью фосфора в пиридине по методу [2], гидролизом продукта фосфорилирования в водном, а затем в щелочном растворе и действием на 5-броминдоксилфосфат натрия, образующегося в реакционном растворе, гидроокисью бария. Синтез 5-бром-N-ацетилиндоксила осуществляли, по литературным данным [3—5], гидролизом 5-бром-О, N-диацетилиндоксила.
СХЕМА СИНТЕЗА
Вг — г.-ОСОСНз Вг —Х\ г.-ОН
UU 2^. ии I н20 | >
СОСНз СОСНз
32
о
Br I II—Гор-С1 —° I II I С1 4/\NZ
I СОСНз
о
Br — I ii—и ~ ор~он I ll । \)Н
4/\NZ
СОСНз
NaOH,H2O
Ва(ОН)2
• 2Н2О
Характеристика основного сырья
5-Бром-О,М-диацетилиндоксил, т. пл. 123—124°, получение см. [6].
Натрий сернистокислый безводный, ГОСТ 195—66, ч.
Бария гидрат окиси, ГОСТ 4107—65, ч.
Пиридин, ГОСТ 2747—67, ч.
Уголь активированный марки БАУ, ГОСТ 6217—52, ч.
Фосфора хлорокись, МРТУ—6—09—337—63, ч. перегнанная.
Этиловый спирт ректификованный, ГОСТ 5962—67.
Бензол, ГОСТ 5955—51, ч. д. а-
Натр едкий, ГОСТ 4328—66, ч. д. а.
3—1192 33
Условия получения
Синтез 5-бром-П-ацетилиндоксила. В трехгорлой колбе емкостью 2 л, снабженной обратным холодильником и мешалкой, растворяют 47,5 г (0,34 А1) безводного сернистокислого натрия в 1320 мл воды, нагревают раствор до 70° и добавляют 99,7 г (0,34 М) 5-бром-О,М-диацетилиидоксила. Смесь размешивают 2,5 часа при 70°, затем охлаждают льдом, отфильтровывают осадок и сушат при 70°.
Выход 5-бром-Й-ацетилиндоксила равен 77,1 г (88,7%).
Вещество перекристаллизовывают дважды из бензола (710 мл и 630 мл) с активированным углем.
Выход 5-бром-М-ацетилиндоксила равен 38,1 г (44,6%). Вещество представляет собой кристаллы светло-розового цвета с т. пл. 182,5- 183,5°.
По литературным данным, т- пл. 184—185° [3], 188° [4], 187— 188° [5].
Получение 5-бром-З-индолилфосфата бария. В четырехгор-лой колбе емкостью 1 л, снабженной мешалкой с ртутным затвором, трубками для ввода и вывода газа и капельной воронкой, размешивают суспензию 38,1г (0,15Af) 5-6poM-N ацетил-ипдоксила в 425 мл сухого пиридина, охлаждают до минус 20° смесью сухой углекислоты в ацетоне, добавляют в токе сухого азота 18,3 мл (30,7 г; 0,20М) хлорокиси фосфора. Реакционную массу размешивают 20 часов при 0°.
Из раствора отгоняют растворитель, к остатку добавляют 430 г мелкоколотого льда и размешивают содержимое колбы при внешнем охлаждении льдом. К водной смеси прибавляют 10%-ный раствор едкого натра до pH 10 и кипятят 45 минут, затем добавляют активированный уголь (2 г), фильтруют и приливают полученный фильтрат к раствору 70 г (0,22 Л4) гидрата окиси бария в 2300 мл воды. Выпавший первичный осадок (24 г) отфильтровывают (он содержит 1% азота). Основной продукт осаждают добавлением к фильтрату 1750 мл этилового спирта. Смесь оставляют на ночь в холодильнике. Осадок отфильтровывают, промывают 40 мл этилового спирта и сушат в вакуум-эксикаторе над твердым едким кали.
Получают 44 г 5-бром-З-индолилфосфата бария.
Дополнительное количество продукта (2 г) выделяют, экстрагируя кипящей водой (2400 мл) первичный осадок и добавляя к экстракту 1200 мл регенерированного этилового спирта (см. примечание 1).
Общий выход 5-бром-З-индолилфосфата бария равен 46 г (66%). Вещество представляет собой блестящие кристаллы белого цвета с сероватым оттенком, не плавится, при нагревании постепенно темнеет (см. примечание 2).
Найдено, ' %: N—3,13, 2,95; Вг—17,41, 17,62. C8H5Ba,BrNO4P-2НгО. Вычислено, %: N—3,02; Вг—17,24.
34
Присутствие воды в молекуле 5-бром-З-индолилфосфата бария подтверждено ИК-спектром.
Примечания.
I. Из маточного раствора, полученного после фильтрования основного продукта, отгоняют этиловый спирт, добавляют в него 20 мл концентрированной серной кислоты и вновь перегоняют.
2. Реактив при стоянии в склянке с притертой пробкой сильно темнеет. Для очистки 5 г продукта в 1500 мл кипящей дистиллированной воды (при этом появляется темно-синий хлопьевидный осадок) добавляют 0,1г активированного угля и кипятят 5 минут. Осадок отфильтровывают, к фильтрату добавляют 750 мл этилового спирта и оставляют в холодильнике на ночь. Выпавшие кристаллы отфильтровывают. Выход 4 г (80%).
ЛИТЕРАТУРА
1. Э. Пирс. Гистохимия теоретическая и прикладная. М., Инлитиздат, 1962, стр. 368.
2. Т.Р. Hortwita, J. Chua е tai. J. Med. Chem., 9., 3.447 (1966).
3. H. C. F. S u, к. S. Tsou. J. Amer. Chem. Soc., 82, 1187 (I960).
4. S. J. Holt, A. E. Kellie et al. J. Chem. Soc., 1958, 1217.
5. S. J. Holt, V. Petrow. J. Chem. Soc., 1947,607.
6. В. M. Островская, И. А. Г op к ер. Методы получения химических реактивов и препаратов, вып. 18. М., ИРЕА, 1969, стр. 44.
3*
5-БРОМ-З-ИНДОЛИЛФОСФАТ НАТРИЯ
5-Броминдоксилфосфат натрия
В. М. ОСТРОВСКАЯ, И. А. ШУМСКАЯ, Л. В. ЛОМАКИНА
О
OP—ONa ^ONa
C8H5BrNNa2O4P М. в. 336,01
5-Бром-З-индолилфосфат натрия можно использовать в гистохимии в качестве субстрата для определения фосфатаз биологических тканей [1]. Синтез этого соединения в литературе не описан.
Нами разработан способ получения 5-бром-З-индолилфос-фата натрия обменной реакцией между бариевой солью 5-бром-3-инфолилфосфата и натриевой формой катионообменной смолы типа КРС или Дауэкс. Продукт очищают пере-осаждением из метанола этилацетатом иЛи диэтиловым эфиром.
Попытка выделить 5-бром-З-индолилфосфат натрия упариванием реакционного раствора, полученного при фосфорилировании N-ацетилиндоксила (см. ст. «5-Бром-З-индолилфосфаг бария») с последующим переосаждением из воды соляной кислотой или из этилового спирта диэтиловым эфиром не привела к получению чистого продукта.
36
СХЕМА СИНТЕЗА
О
ОР-О
I
О —Ва
NaOSO2- R
где R — остаток катиопообменной смолы.
Характеристика основного сырья
5-Бром-З-индолилфосфат бария, получение см. на стр. 32 этого сборника.
Смола КРС-8п (или Дауэкс-50).
Условия получения
К 8г катионообменной смолы КРС-8п приливают 800 мл 5%-ного раствора едкого натра, через 2 суток смолу отфильтровывают, промывают на фильтре дистиллированной водой до нейтральной реакции промывных вод и смешивают с суспензией из 2,4 г 5-бром-З-индолилфосфата бария в 120 мл дистиллированной воды. Смесь встряхивают в конической колбе на механической мешалке 5 часов.
Смолу отфильтровывают (фильтрат № 1) и снова встряхивают 2 часа со свежей порцией 120 мл дистиллированной воды. Смолу отфильтровывают (фильтрат № 2), из объединенных фильтратов отгоняют при пониженном давлении (10— 15 мм) воду. Остаток сушат в вакуум-эксикаторе, растворяют в кипящем метаноле (с активированным углем) и высаживают из метанола этилацетатом. Выпавший после стояния раствора в холодильнике осадок отфильтровывают и сушат в вакуум-эксикаторе
37
Выход 5-бром-З-индолилфосфата натрия равен 1,62 г (93%).
Вещество представляет собой голубовато-белый мелкокристаллический порошок, хорошо растворим в воде, не плавится.
ЛИТЕРАТУРА
1. Э. Пирс. Гистохимия теоретическая и прикладная. М., Инлит-пздат, 1962, стр. 308.
БРОМКРЕЗОЛОВЫЙ ЗЕЛЕНЫЙ
Т етрабром- м -крезолсу льфофталеин
А. П. БОЛДЫРЕВА, Г. И. МИХАЙЛОВ
М. в. 698,04
Бромкрезоловый зеленый, являющийся кислотно-основным индикатором, применяется для определения концентрации водородных ионов, для получения водорастворимого и смешанных индикаторов, для приготовления индикаторной бумаги, в бактериологии, в качестве адсорбционного индикатора при определении тиоцианата [1, 2].
Бромкрезоловый зеленый получают бромированием .«-крезолового пурпурового бромом в среде ледяной уксусной кислоты [3, 4] или гипобромитом натрия в водной среде [2]-
Нами предложен способ получения высококачественного бромкрезолового зеленого путем бромирования м-крезолового пурпурового бромом в этиловом спирте с одновременным получением в качестве побочного продукта бромистого этила.
39
СХЕМА СИНТЕЗА
Вг2
CSH5OH*
Характеристика основного сырья
л-Крезоловый пурпуровый, МРТУ 6—09—2458—65, ч. д. а.
Бром, ГОСТ 4109—64, ч.
Этиловый спирт гидролизный высшей очистки, ГОСТ 8314—57.
Уксусная кислота ледяная, ГОСТ 61—69, ч.
н-Бутиловый спирт, ГОСТ 6006—51, ч.
Условия получения
В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой с затвором, термометром, капельной воронкой и U-об-разной трубкой (см. примечание 1), другой конец которой погружен в приемник, охлаждений ледяной водой (для поглощения бромистого этила), загружают 40 г (0,1 М) тонкоиз-мельченного л-крезолового пурпурового (см. примечание 2) и
40
160 мл этилового спирт. Смесь нагревают при размешивании до 60° и в этих условиях выдерживают 30 минут, после чего за 1,5—2 часа прибавляют по каплям 128 г (40 мл; 0,8 Л4) брома, постепенно повышая температуру до 70—75° (не доводя до кипения спирта).
Реакционную массу выдерживают 2 часа при 70—75° (см. примечание 3), охлаждают до комнатной температуры и оставляют на 16—20 часов- Выпавший осадок бромкрезолового зеленого отфильтровывают, а из фильтрата отгоняют 50— 75 мл спирта. При охлаждении из маточного раствора выпадает дополнительное количество осадка, который объединяют с первой порцией, промывают 30—40 мл ледяной уксусной кислоты, тщательно отжимают и сушат при 100—105° до постоянного веса.
Выход бромкрезолового зеленого равен 48—50а (70—72%). Полученный индикатор является высококачественным элект-рофоретически однородным препаратом с содержанием основного вещества 95—97%, с максимальным поглощением в кислой области (pH 1) при 445 ±5 нм, в щелочной области (pH 11) при 615±5 нм.
По литературным данным [5], бромкрезоловый зеленый изменяет окраску от желтой к синей в интервале pH 3,8—5,4; максимальное поглощение в щелочной области (pH 11) при 617 нм.
Примечания.
1. Для улавливания бромистого этила более удобна U-образная трубка, погруженная в лед, так как водяного охлаждения с помощью холодильника Либиха недостаточно для полной конденсации паров бромистого этила.
2. лг-Крезоловый пурпуровый предварительно кипятят в н-бутиловом спирте для повышения в нем содержания основного вещества до 93— 95%.
3. Отогнанный бромистый этил отделяют от водного слоя, сушат над хлористым кальцием и отгоняют при 38—39°. Получают 13 мл (20 а) бромистого этила; п® = 1.4220, d — 1,4533. По литературным данным [6], По =1,4248; «1= 1,4555
ЛИТЕРАТУРА
1. И. М. Ко ль т го ф. Применение цветных индикаторов. Л., «Химия», 1926.
2. Г. Н. Кошелева. Методы получения химических реактивов и препаратов, вып. 3. М., ИРЕА, 1961, стр. 52.
3. В. Cohen Publ. Haith. Repts. 41, 3061 (1926).
4. W. R. Orndorff A. C, Purdy. J. Amer. Chcm., 48, 2218 (1926).
5. P. Бейтс. Определение pH. Теория и практика. Л., «Химия», 1968, стр. 127.
6. Краткий справочник химика. М., Госхимиздат, 1963.
41
БРОМКРЕЗОЛОВЫЙ ЗЕЛЕНЫЙ ВОДОРАСТВОРИМЫЙ
Тетрабром-м -крезолсульфофталеин, моноаммонийная соль
А. П. БОЛДЫРЕВА
Вг Вг
\/Ч А/
/\Z\Z\Z\ Вг | С | Вг сн3 I сн3
SO3NH4
\s
C21H17Br4NO5S
М. в. 715,10
Бромкрезоловый зеленый водорастворимый, являющийся кислотно-основным индикатором, применяется при колориметрическом определении концентрации водородных ионов [1].
Общий метод получения водорастворимых сульфофталеиновых индикаторов основан на взаимодействии спирторастворимых сульфофталеинов с водным или газообразным аммиаком [2].
Нами разработана методика получения высококачественного бромкрезолового зеленого водорастворимого действием газообразного или водного аммиака на бромкрезоловый зеленый спирторастворимый.
42
СХЕМА СИНТЕЗА
Вг Вг
ОН I | он
\/ч /ч/
II I II I /\^\/\^\
Вг | С | Вг NH3
СН3 \ СН3 ----
О
A-SO,
\z
Вг Вг
НО | |О
\/Ч
Вг I С | Вг
СН3 | CHS
|Z^j-SO3NH4
Характеристика основного сырья
Бромкрезоловый зеленый спирторастворимый, МРТУ 6— 09—2477—65, ч. д. а.
Аммиак водный, ГОСТ 3760—64, ч. д. а.
Условия получения
Взаимодействие с водным аммиаком. В фарфоровую чашку емкостью 0,5 л, помещенную на водяную баню, загружают 400 мл дистиллированной воды, 13,5 мл (0,18 М) 25%'-ного раствора аммиака и при нагревании до 50—60’ и перемешивании стеклянной палочкой осторожно прибавляют 40 г (0,055 Л4) растертого бромкрезолового зеленого спирторастворимого (см. примечание). При дальнейшем нагревании до 80—90° и перемешивании смесь растворяется. Раствор упаривают досуха, остаток извлекают из чашки, измельчают и сушат при 100—105° до постоянного веса.
43
Выход бромкрезолового зеленого водорастворимого равен 40—41 г (98—99%), содержание основного вещества 94—96%.
Взаимодействие с газообразным аммиаком. Помещают 40 г (0,055 Л1) тщательно растертого бромкрезолового зеленого спиргорастворимого в чашке Петри в атмосферу газообразного аммиака (можно использовать обычный эксикатор с плотно притертой крышкой, на дно которого помещают чашку Петри с 25%-ным водным аммиаком). Дают выдержку 2— 3 часа при комнатной температуре, после чего продукт сушат при 100—105е до постоянного веса.
Выход бромкрезолового зеленого водорастворимого равен 40—41 г (98—99%), содержание основного вещества 96—98%.
Образцы бромкрезолового зеленого водорастворимого, полученные обоими способами, являются электрофоретически однородными препаратами с максимальным поглощением в кислой среде (pH 1) при 445+5 нм и в щелочной среде (pH 11) при 615+5 нм. Интервал pH перхода окраски от желтой к синей 3,2—5,4.
По литературным данным [3], бромкрезоловый зеленый водорастворимый изменяет окраску от желтой к синей в интервале pH 3,2—5,4; максимальное поглощение в щелочной среде (pH 11) при 617 нм.
Примечание.
Исходный бромкрезоловый зеленый спирторастворимый должен иметь содержание основного вещества не менее 95%, что достигается кипячением реактива в н-бутиловом спирте.
ЛИТЕРАТУРА
I. Г. Н. Кошелева. Методы получения химических реактивов и препаратов, вып. 3. М., ИРЕА, 1961, стр. 52.
2. Р. П. Л а сто вс к ий, Г. И. Михайлов. Заводск. лаборатория, 19, 370 (1953).
3. Р. Бейтс. Определение pH. Теория и практика. Л., «Химия», 1968, стр. 127.
БРОМКРЕЗОЛОВЫЙ ПУРПУРОВЫЙ
Дибром-о-крезол сульфофталеин
Н. В. ФАКЕЕВА, А. П. БОЛДЫРЕВА
C2iII16Br2O5S
М. в. 540,74
Бромкрезоловый пурпуровый является кислотно-основным индикатором и применяется при колориметрическом определении концентрации водородных ионов, как адсорбционный индикатор, а в смеси с бромтимоловым синим — как витальный краситель,-для получения водорастворимого индикатора [1,2].
Бромкрезоловый пурпуровый получают бромированием крезолового красного бромом в уксусной кислоте [3—5].
Нами предложен способ получения высококачественного бромкрезолового пурпурового путем бромирования крезолового красного в уксуснокислой среде с одновременным улавливанием бромистого водорода-
45
СХЕМА СИНТЕЗА
Характеристика основного сырья
Крезоловый красный, ГОСТ 5849—51, ч. д. а-
Бром, ГОСТ 4109—64, ч.
Уксусная кислота ледяная, ГОСТ 61—69, ч.
Условия получения
В четырехгорлую колбу емкостью 1 л, снабженную мешалкой с затвором, термометром, капельной воронкой и U-образ-ной трубкой, другой конец которой опущен в приемник с ледяной водой (для поглощения паров бромистоводородной кислоты—см. примечание 1), загружают 106 г (0,26 .М) измельченного крезолового красного (см. примечание 2) и 500 мл ледяной уксусной кислоты, нагревают пра размешивании до 80° и в этих условиях выдерживают 30 минут. Затем охлаждают смесь до 60—65° и за 1,5—2 часа осторожно прибавляют 46
по каплям раствор 51 мл (158,5 г; 0,98 М) брома в 50 мл ледяной уксусной кислоты- постепенно повышая температуру до 75—80°.
Реакционную массу выдерживают в этих условиях 2 часа, охлаждают до комнатной температуры и оставляют на 16— 20 часов. Выпавший осадок отфильтровывают, промывают 90 мл ледяной уксусной кислоты, тщательно отжимают (см. примечание 3) и сушат при 60—70° до постоянного веса.
Выход бромкрезолового пурпурового равен 115—120 г (82—86%); содержание основного вещества 96—98%'. Полученный индикатор является электрофоретически однородным препаратом с максимальным поглощением в кислой среде (pH 1) при 435 + 5 ил и в щелочной среде (pH 11) при 590± 5 нм; интервал pH перехода окраски от желтой к пурпуровой 5,2- 6,8-
По литературным данным [6], бромкрезоловый пурпуровый имеет максимальное поглощение в щелочной среде (pH 11) при 591 нм; интервал pH перехода окраски от желтой к пурпуровой 5,2—6,8.
Примечания.
1. В укрупненных масштабах раствор бромистоводородной кислоты собирают от нескольких загрузок, отгоняют фракцию с т. кип. 122—125° и nt лучают 40 — 45%-ную бромистоводородную кислоту.
2. Крезоловый красный должен иметь содержание основного вещества не менее 90%; что достигается кипячением исходного реактива в н-бути-ловом спирте.
3. Из фильтрата отгоняют уксусную кислоту при 117—119° и используют ее при повторных загрузках.
ЛИТЕРАТУРА
1. В. Cohen. J. Amer. Chem. Soc., 44, 1851 (1922).
2. H. Petow, Е. Wittkower. Z. Exp. Med., 64, 747 (1929).
3. I. K. S о h о n. J. Amer. Chem. Soc., 20, 263 (1898).
4. X. Г. Бриттон. Водородные ионы. Л., ОНТИ, 1936.
5. П. Рабинович. Реактивы и препараты лабораторного назначения. М.-Л. ГОНТИ НКТП СССР, 1938, стр. 48.
6. Р. Бейтс. Определение pH. Теория и практика. Л., «Химия», 1968, стр. 127.
БРОМКРЕЗОЛОВЫЙ ПУРПУРОВЫЙ ВОДОРАСТВОРИМЫЙ
Дибром-о-крезолсульфофталеин, моноаммонийная соль
А. П. БОЛДЫРЕВА
Вг Вг
НО | | О
\/%
/\А/\/\
Н3С с сн3
/Л— so3nh4
C2iHI9Br2NO5S
М. в. 557,29
Бромкрезоловый пурпуровый водорастворимый является кислотно-основным индикатором и применятся при колориметрическом определении концентрации водородных ионов [1].
Известен общий метод получения водорастворимых сульфофталеиновых индикаторов .взаимодействием исходных снирторастворимых сульфофталеинов с аммиаком (водным или газообразным [1]).
Нами разработана методика получения высококачественного бромкрезолового пурпурового водорастворимого действием газообразного аммиака на бромкрезоловый пурпуровый спирторастворимый-
48
СХЕМА СИНТЕЗА
Характеристика основного сырья
Бромкрезоловый пурпуровый спирторастворимый, МРТУ 6—09—1573—64, ч. д. а.
Аммиак водный, ГОСТ 3760—64, ч. д. а. (или газообразный аммиак из баллона).
Условия получения
Помещают 40 г (0,07 М) тщательно растертого бромкрезолового пурпурового спирторастворимого в чашке Петри в атмосферу газообразного аммиака (можно использовать обычный эксикатор с плотно притертой крышкой, на дно которого помещают чашку Петри с 25%'-ным водным аммиаком). Дают выдержку 2—3 часа при комнатной температуре, после чего продукт сушат при 100° до постоянного веса.
4-1192 ’ 49
Выход бромкрезолового пурпурового водорастворимого равен 40—41 г (96—98%); содержание основного вещества 95—96%. Полученный образец является электрофоретически однородным препаратом с максимальным поглощением в кислой среде (pH 1) при 435 ± 5 нм и в щелочной среде. (pH 11) при 590 ± 5 нм. Интервал pH перехода окраски от желтой к пурпуровой 5,2—6,8-
По литературным данным (2,3], бромкрезоловый зеленый водорастворимый имеет максимальное поглощение в щелочной среде (pH 11) при 591 нм [2]; интервал pH перехода окраски от желтой к пурпуровой 5,2—6,8.
Примечание. Исходный бромкрезоловый пурпуровый ео.ртораст-ворнмый должен иметь содержание основного вещества не менее 95%, что достигается кипячением реактива в н-бутпловом спирте.
ЛИТЕРАТУРА
I. И. М. Кольтгоф. Применение цветных индикаторов. Л., «Химия», 1926.
2. Р. П. Л а с т о в ск и й, Г. И. Михайлов. Заводск. лаборатория, 19, 370 (1953).
•3. Р. Бейтс. Определение pH. Теория и практика. Л, «Химия», 1968, стр. 127.
5-БРОМ-4-ОКСИИЗОФТАЛЕВЫЙ АЛЬДЕГИД
З-Бром-5-формилсалициловый альдегид
Д. А. ДРАПКИНА, II. И. ДОРОШИНА
СНО
I
{ он
OHC-l^J — Вг
CgHgBfOg
М.в. 229,07
5-Бром-4-оксиизофталевый альдегид в литературе не описан; мы получили его бромированием 4-оксиизофталевого альдегида бромом в уксусной кислоте.
он с
СХЕМА СИНТЕЗА
СНО
Характеристика основного сырья
4-Оксиизофталевый альдегид, т. пл. 109—110°; получение см. [1].
Бром, ГОСТ 4109 -64, ч.
Уксусная кислота, ГОСТ 61—69, ч.
Этиловый спирт ректификованный, ГОСТ 5962—67.
4*
51
Условия получения
В круглодонную колбу емкостью 250 мл, снабженную мешалкой, капельной воронкой, термометром и обратным холодильником, помещают 15,0 г (0,1 М) 4-оксиизофгалевого альдегида и 35 мл уксусной кислоты. К полученной суспензии предварительно нагретой до 45—50° (водяная баня), прибавляют при интенсивном размешивании за 2 часа раствор 5,1 мл (16,0 г; 0.1 Л1) брома в 175 мл уксусной кислоты, поддерживая температуру 45—50°. После добавления части брома (за первые 15—20 минут) 4-октиизофталевый альдегид полностью растворяется. По окончании введения брома смесь размешивают еще 1 час при 45—50° и затем выливают на 250 г толченого льда; выпавший осадок отфильтровывают и промывают дважды водой порциями по 20 мл.
Выход 5-бром-4-оксиизофталевого альдегида равен 20,5— 21,5а (90—92%); т. пл. 133- 135°.
После кристаллизации из 260 мл спирта получают 14,0— 15,0 а (61—65%) чистого продукта в виде кристаллов белого цвета с т. пл. 140,5—141,5°. Повторная кристаллизация не изменяет температуру плавления.
Разбавлением спиртового маточника от кристаллизации равным объемом воды можно выделить около 4 г альдегида с т. пл. 136—138°, перекристаллизация которого из спирта дает дополнительно 1,6—1,8 г чистого 5-бром-4-оксиизофтале-вого альдегида.
Суммарный выход продукта равен 16—16,5 г (70—72%).
Найдено, %: С—-42,33: 42,44; Н—2,24; 2,14; N—34,73;
35,03;
СгН5ВгО3. Вычислено, %; С—41,97; Н—2,19; N—34,91.
Диоксим, т. пл. 2Ю—211° (50%-ный спирт).
Найдено, %: С—37,29; Н—2,89; N—10,90; Вг-30,54. C8H7BrNaO3. Вычислено, %: С—37,08; Н—2,73; N—10,81; Вг—30,84.
ЛИТЕРАТУРА
1. S. J. А п в у а 1, Р. J. М о г г i s et al. J. Chem. Soc., 1950, 2141.
(З-БРОМПРОПИЛ)БЕНЗОЛ
В. М. ОСТРОВСКАЯ, И. Е. КАШИРИНА, Т. В. ФОМИНА, И. В. ХВОСТОВ
У — СН2СН2СН2Вг
СдНцВг М. В. 199,09
(З-Бромпропил)бензол получают действием на З-феиил-1-пропанол трехбромистого фосфора [1, 2], пятибромистого фосфора [3], бромистоводородной кислоты [4—7] иногда в присутствии серной кислоты [8—10]; действием на фенил (3-фенил-пропиловый)эфир насыщенного раствора бромистого водорода в ледяной уксусной кислоте {11, 12]; алкилированием бензола 1-хлор-З-бромпропаном [13—15]; гидробромированием алЛилбензола бромистым водородом в присутствии перекиси ацетила или бензоила [16, 17].
Нами с хорошей воспроизводимостью и высоким выходом получен (3-бромпропил)бензол гидробромированием аллил-бензола в присутствии перекиси бензоила в бензоле. Бромистый водород мы получали бромированием бензола, в отличии от авторов {16—17], которые получали бромистый водород из красного фосфора и брома.
СХЕМА СИНТЕЗА
СН2СН = сн2 I
СН2СН2СН2Вг
НВг __—. —>
ч/
ч/
Характеристика основного сырья
Бензол, ГОСТ 5955—68, ч. д. а.
Бром, ГОСТ 4109—64, х. ч.
Аллилбензол, ТУ 16 п 518—69, ч., свежеперегнанный.
53
Перекись бензоила, техн., переосаждена из хлороформа метанолом и высушена в вакуум-эксикаторе.
Хлороформ, ГОСТ 3160—51, ч. д. а.
Ментол, ГОСТ 6995—67, х- ч.
Условия получения
Все приборы соединяются пружинками или резиновыми колечками (см. рис.), так как в системе во время реакции создается избыточное давление.
В круглодонную колбу 2 емкостью 0,5 л загружают 250 мл (2,8М) бензола, 4,5 г чугунной стружки. В реакционный сосуд 8 емкостью 120 мл со стеклянным пористым фильтром № 2 на дне (ставятся 2 сосуда, если требуется получить удвоенное количество продукта) наливают 65 мл (59 г; 0,5 Л1)
Схема установки:
1 — капельная воронка для брома; 2 — колба; 3 — обратный шариковый холодильник с верхним боковым отводом для газа; 4 — баня глицериновая с закрытой плиткой и терморегулятором; 5 — U-образная трубка, наполненная кусками нафталина; 6 — колонка с кусками плавленого бромистого кальция; 7 — трехходовой кран; 8 — реакционный сосуд; 9— склянка Тищенко для сухих веществ с плавленым хлористым кальцием;
10— склянка Тищенко с водой
аллибензола, охлаждают льдом и пропускают сухой бромистый водород, который получают при 60—70° добавлением в бензол 75 мл брома порциями из капельной воронки 3 со шлифом № 14 и длинной трубкой (80—90 см), пропущенной через обратный холодильник и достигающей дна колбы. Одновременно добавляют порциями суспензию 4 г перекиси бензоила в 10 мл бензола (см. примечание 1). Образование бромистого водорода начинается через 10—15 минут. Бромистый водород пропускают 3 часа, затем продувают 1 час воздух.
54
Массу экстрагируют 60 мл бензола, промывают водой, 5%'-ным раствором едкого натра до pH 7, снова водой (100 мл), 10%-ным раствором гидросульфита натрия (100мл) и сушат над хлористым кальцием с добавкой гидросульфита натрия- Отгоняют бензол при 80°, остаток перегоняют в вакууме, собирая фракцию с т. кип. 95—9774 мм.
Выход (3-бромпропил)бензола равен 80 г (81%); Пд — = 1,5469.
Повторная перегонка в вакууме дает 69 г (70%) вещества с т. кип. 95—9674 мм; п™ = 1,5463; п% = 1,5460-
По литературным данным, т. кип. 7672 мм [9]; 106— 10876 мм [13—15]; 107—11078—9 лъи [7]; 108—109°/10 мм [6]; 109°—1117Юмм [10]; 109711 мм [1]; 109712 мм [8]; 140712 мм [11]; -115/14 мм [12]; 111716 мм [3]; 113—115716 мм [16]; 1'14— 115716 мм [11]; 118—1197'17 мм [1,4]; 128—129729 мм [1];
=1,1720 [13—15]; лЬ° = 1,5436(9); = 1,5445(8], Лд =
= 1,5459 [17].
Примечание. Сразу всю перекись добавлять нельзя, так как произойдет разогрев реакционной массы.
ЛИТЕРАТУРА
1. D. Perlman, D. Davidson, М. Т. Boger t. J. Organ. Chem, I, 300 (1936).
2. H. Rupe, J. Burgin. Ber., 43, 172 (1910).
3. P. W, Cl utter buck, J. B. Cohen. J. Chem. Soc., 1923, 2509.
4. J. Braun. Ber., 43, 2842 (1910).
5, V. Grignard, E. Belief, C. Courtot. Ann. chim., [9], 12, 366 (1919).
6. J. W e i ns t о c k. J. Organ. Chem., 21, 540 (1956).
7. G. 0. Asp i nail, W. Baker. J. Chem. Soc., 1950, 743.
8. H. H. Bop ожцов, В. А. Коптюг. Ж- общ. химии, 28, 372 (1958).
9. J. Hase, H. Oura. Pharm Bull (Japan), 2,368 (1854);
C. A., 50, 11977b (1956).
10. R. Huisgen, W. Rapp et al. Ann., 586, 1, 59 (1954).
- 11. V. Grignard. Compt. rend., 138, 1049 (1904); Beilst., 5, 391 (1922).
12. H. teuchs, P. Sander. Ber., 58, 2201 (1925).
13. Л. В. Бугрова, И. П. Цукерваник. Ж. общ. химии, 32, 3575 (1962).
14. И. П.. Ц у к е р в а н и к, Л. В. Бугрова. Ж- общ. химии, 31. 2143 (196'1).
15. И. П Цукерваник, К. Яцимирскнй. Ж. общ. химии, 10 1076 (1940).
16. R. Que let, R. Durand-Dran, P. P i n e a u. Com pt. rend 244, 1218 (1957).
17. R. Durand-Dran. Ann. chim., [13), 4, 45—51 (1959).
55
трет- БУТИЛКАРБАЗАТ
Е. А. МАКАРОВА, Г. Н. КОШЕЛЕВА сн3
СН3 — С — О — СО — NH — NH2
СНз
C5HI2N2O2 М. В. 132,16
трет-Бутилкарбазат является одним из. важнейших препаратов, применяющихся при синтезе пептидов.
Описаны способы получения этого соединения из трет-бутилата натрия черз трет-бутил-З-метилтиокарбонат [1] и через метил-трег-бутилкарбонат [2]. В первом случае синтез связан с выделением метилмеркапт ана, поэтому мы предпочли второй способ, который проверили и уточнили- При этом в первой стадии часть избыточного трет-бутанола заменили на керосин, а во второй стадии серный эфир заменили на хлористый метилен, что удешевляет процесс, а также позволяет работать более безопасно.
СХЕМА СИНТЕЗА
CICOOCHs
(СНз)зС — ОН + Na-> (СНз)зС —ONa------
H2N — NH2
— (СНз)зС —О—СООСНз------------
— (СНз) 3С — О — CONH — NH2
Характеристика основного сырья
трет-Бутиловый спирт, ВТУ ХСНХ 355—60, ч.
Натрий металлический, ТУ МХП 1664—50, ч.
56
Метиловый эфир хлоругольной кислоты, ВТУ РУ 2083— 54, ч.
Гидразин-гидрат, ГОСТ 5832—65, ч.
Условия получения
Получение трет-бутилметилкарбоната. В четырсхгорлую колбу емкостью 3 л, снабженную воздушным холодильником с хлоркальциевой трубкой и тремя пробками, загружают 500 мл (5,4 М) трет-бутилового спирта и 600 мл керосина (см. примечание 1) и нагревают на водяной бане до 45°. Затем удаляют воду из бани и к реакционной смеси прибавляют 46 г (2 Л4) мелконарсзанного металлического натрия и оставляют стоять двое суток. За это время весь металлический натрий растворяется- К густой массе добавляют 800 мл керосина, в колбу вставляют мешалку с затвором, термометр, хлоркаль-цпевую трубку и капельную воронку, охлаждают реакционную смесь до 0° и при хорошем размешивании медленно, за 2 часа добавляют по каплям смесь 150 мл (2 Л4) метилового эфира хлоругольной кислоты и 100 мл керосина, поддерживая температуру в пределах 5—0°. После этого охладительную баню убирают и реакционную смесь перемешивают при комнатной температуре еще 4 часа.
На следующий день к реакционной массе добавляют 400 .ид воды, причем осадок растворяется. Верхний органический слой отделяют, высушивают сульфатом магния и отгоняют от него в циркуляционном испарителе смесь спирта и трет-бутилметилкарбоната при пониженном давлении (20 мм) и температуре термостата не выше 80° (см. примечание 2). Затем дистиллат разгоняют при нормальном давлении с дефлегматором, собирая фракции: до 90°, 90—120°, 120—135° и 135—140°. Фракции 90—120° и 135—140° объединяют и разгоняют повторно.
Выход фракции, содержащей трет-бутилметилкарбонат, равен 200—205 г (76—78%), т. кип. 120—135°; По = 1,3920— - 1,3980-
Получение трет-бутилкарбазата. В трехгорлую колбу емкостью 3 л, снабженную мешалкой, обратным холодильником и термометром, загружают 1 кг (7,6 Л4) трет-бутилметилкарбоната и 1,15 кг (23 Л4) гидразин-гидрата. Смесь кипятят па водяной бане 28 часов. Затем к реакционной массе добавляют 1 л воды и продукт экстрагируют хлористым метиленом 4 раза порциями по 400 мл. Экстракт высушивают сульфатом магния и упаривают при 90’—100°/20—40 мм. Остаток в колбе закристаллизовывается.
Выход трет-бутилкарбазата равен 460—530 г (46—53%); температура застывания 36°.
По литературным данным [2], после перегонки продут имеет т. пл. 38—40°
57
Примечания.
I. Керосин предварительно перегоняли. Для реакции во всех случаях применяли фракцию с т. кип. 80—100720 мм.
2. Остаток керосина можно употреблять для следующих загрузок 2—3 раза без дополнительной перегонки.
ЛИТЕРАТУРА
1. L. A. Carpinn. J. Amer. Chem. Soc... 82, 2725 (1960).
2. M. Muraki. T. Mizoguchi. Chem. Pharm. Bull,, 18, 217 (1970).
4 -втор -БУТИЛ-2-(а-МЕТИЛБЕНЗИЛ)ФЕНОЛ
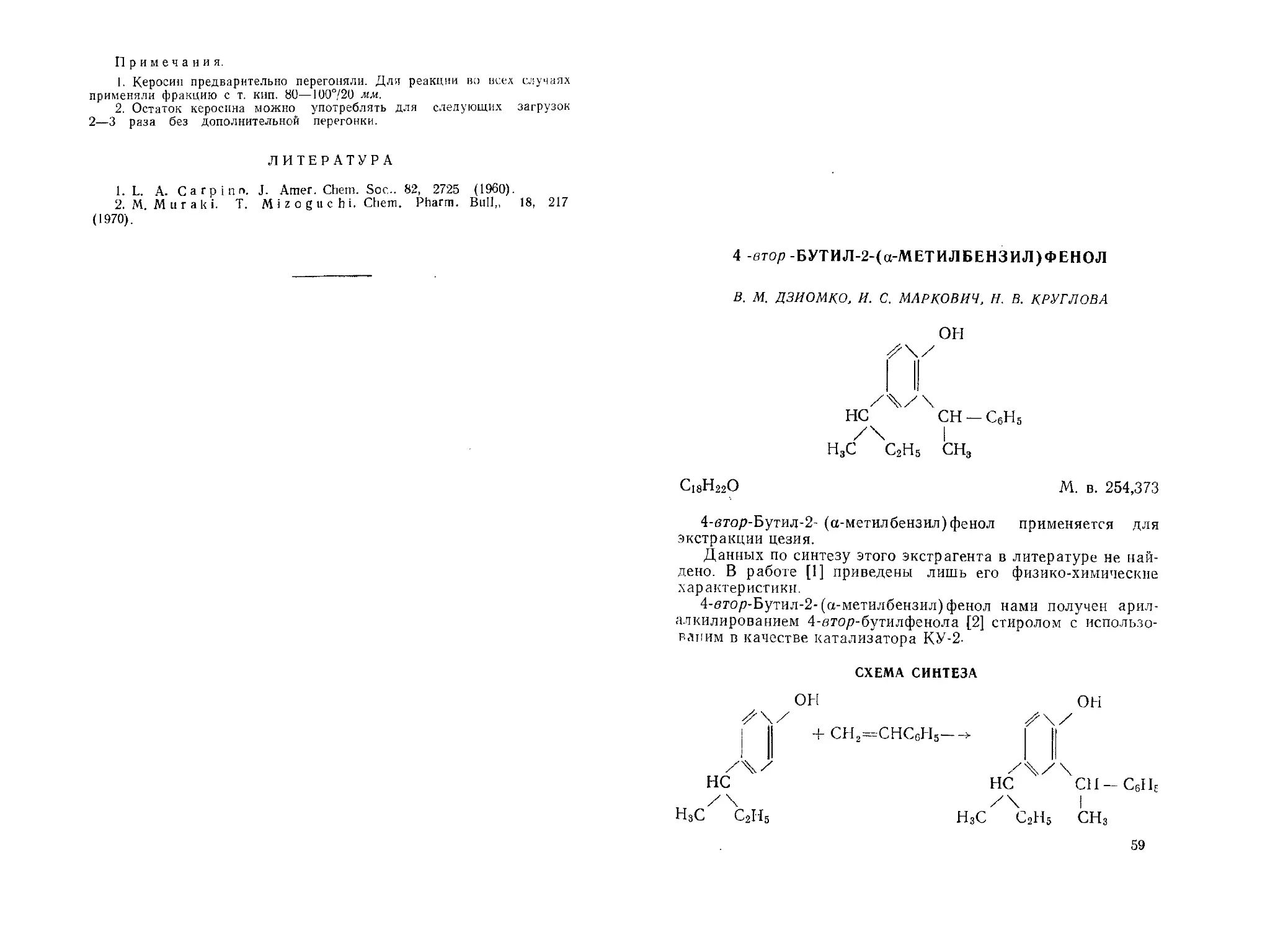
В. М. ДЗИОМКО, И. С. МАРКОВИЧ, И. В. КРУГЛОВА
он
CH - С6Н5
СН3
С18Н22О
М. в. 254,373
4-втор-Бутид-2- (а-метилбензил) фенол применяется для экстракции цезия.
Данных по синтезу этого экстрагента в литературе не найдено. В работе [1] приведены лишь его физико-химические характеристики.
4-втор-Бутил-2-(а-метилбензил)фенол нами получен арил-алкилированием 4-етор-бутилфенола [2] стиролом с использовании в качестве катализатора КУ-2-
СХЕМА СИНТЕЗА
ОН он
^\/ S\/
| I + CH2=CHCgH5—> I ||
/4/ /ч/\
НС НС СН-С6НЕ
НзС С2Н5 Н3С С2Н6 СН3
59
Характеристика основного сырья
4-втор-Бутилфенол, т. пл. 58—59°, получение см. [2]. Стирол, МРТУ 6—09—4055—67 (свежеперегнанный).
КУ-2, ВТУ М—661 55, в Н*-форме.
Бензол, ГОСТ 5955—68, ч. д. а.
Условия получения
В трехгорлую колбу емкостью 50 мл, снабженную мешалкой; термометром и обратным холодильником, загружают 10,5 г (0,07 М) 4-втор-бутилфенола, 7,3 г (0,07 А1) свежепере-гнанного стирола, 1,1 г КУ-2. Смесь нагревают до 165—170° и размешивают при этой температуре 6 часов, затем охлаждают до комнатной температуры и добавляют 30 мл бензола. Раствор отфильтровывают от катализатора и разгоняют в вакууме, собирая две фракции: при 100—115°/2 мм 4,0 г 4-втор-бутилфенола и при 177—180°3/3 мм 7,35 г 4-вго/?-бутил-2-(«-метилбензил)фенола.
Выход продукта составляет 66,8% в расчете на прореагировавший 4-втор-бутилфенол; По =1,5590; 1,5560; >-м'акс = = 279 нм; 284 нм; г279 = 2740; a2s4 = 2540.
По литературным данным [1], т. кип. 186°/6 мм; 207°/15 мм;
= 1,5570; = 279 нм; 284,5 нм ; е27з = 2618; e2S4, = 2474.
Найдено, %: С—-85,11; 85,13; Н—-8,91; 8,97.
С18Н22О. Вычислено, %: С—84,99; Н—8,78.
ЛИТЕРАТУРА
1. В. L. Egan, W. D. Arnold. Analyt. Chem., 38 (7), 950 (1966).
2. И. С. Маркович, Н. В. Круглова, В. М. Д з и о м к о. Методы получения химических реактивов и препаратов, выл. 20. М., ИРЕА, 1969, стр. 48.
БУТИЛОВЫЙ ЭФИР ДИХЛОРУКСУСНОЙ кислоты
Бутилдихлорацетат
И. Г. ХАСКИН, Л. л. СТОЛПЕР, Л. С. ЛЮТАЯ, Е. Р. ЗАРИХТА, В. С. МИХАИЛОВ, Е. П. БАБИН
СНС12СООС4Н9
СвНюС^Ог
М. в. 184,99
Бутиловый эфир днхлоруксусной кислоты является гербицидом [1].
Эфиры дихлоруксусной кислоты получают из хлораля и соответствующих спиртов в присутствии оснований и ацетонциангидрина, как катализатора процесса [2, 3]-
Нами этот метод применен для получения бутилового эфира дихлоруксусной кислоты, причем в качестве основания использован триэтиламип-
СХЕМА СИНТЕЗА
О
CN-
CCb--с -Ьн-СШоОН хг/^ ц -> СНС12СООС4Н9
\ М(С2Г15)з
н
Характеристика основного сырья
Хлораль, ТУ—6 УССР 90—68, техн., перегнанный.
Бутиловый спирт, ГОСТ 6006—51, ч.
Триэтиламип, МРТУ 6—09—2437—65, ч.
Ацетонциангидрин, ГОСТ 13198—67, техн.
Четыреххлористый углерод, ГОСТ 5827—68, ч.
61
Условия получения
В четырехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром, обратным холодильником и капельной воронкой, загружают 200 мл четыреххлористого углерода, 80,4 г (1,08 А1) бутилового спирта, 100,4 г (0,99 А1) триэтиламина и 10 мл ацетонциангидрина. К этой смеси из капельной воронки за 1 час при перемешивании прибавляют раствор 148,2 г (143,0 г 100%-ного; 0,97 М) хлораля в 125 мл четыреххлористого углерода, нагревают на глицериновой бане до 77° н кипятят 2 часа при перемешивании.
Полученную суспензию охлаждают до комнатной температуры и фильтруют с отсасыванием. Отжатый осадок солянокислого триэтиламина промывают па фильтре 50 мл четыреххлористого углерода. Объединенный фильтрат помещают в колбу Вюрца и отгоняют четыреххлористый углерод (см. примечание). Остаток в колбе перегоняют в вакууме, собирая фракцию с т. кип. 38—4975 мм; выход сырого эфира равен 124 г (69,1% в расчете на хлораль).
Повторная перегонка дает 117 г (65,2%) бутилдихлорацетата с г. кип. 48—5074 мм; di20= 1,1839; По = 1,4459-
По литературным данным [4], т. кип. вещества 192°, d^n~' 1,1848, nt =1,4425.
Найдено, %: CI—38,07 ; 38,16.
С6Н10С12О2. Вычислено, %: CI—38,32
Примечание. После регенерации четыреххлористын углерод используют в качестве растворителя для следующих операций получения бутилового эфира.
ЛИТЕРАТУРА
1. Д. Ф. Ширанков, К. А. Абрамова, А. Л. Столпер, И. Г. Ха ск нн. Авт. свид. 268798; Изобретения. Промобразцы. Товарные знаки, № 14 (1970).
2. В. А. М н х а л е в, М. И. Галченко, А. М. Желоховцева. Авт. евнд. 93037; Бюлл. изобр., № 12 (1951).
3, И. Г. Хаскин, Ю. А. Фиал ков, О. Д. Л нт в и «ч у к. Авт. свид. 103147; Бюлл. изобр., № 5 (1956).
4. A. J. V о g е 1 J. Chem. Soc., 1948, 1849.
БУТИЛОВЫЕ ЭФИРЫ МЕТИОНИНА
И. И. ГУБЕНКО, В. М. КОТЛЯРЕВСКАЯ
ROOC — СН — (СН2)2 - SCH3)
I
NH2
где R = н-CiHg —; i - С4Н9 —
Эфиры метионина применяются для разделения рацемической смеси на оптически активные изомеры.
В литературе (1] описано получение метилового и изопропилового эфиров метионина действием соответствующего безводного спирта и хлористого тионила (или хлористого водорода) на метионин.
По этому методу нами получены впервые бутиловый и изобутиловый эфиры метионина.
СХЕМА СИНТЕЗА
НООС —CH— (СН2)2—SCH3-I
nh2
ROOC - СИ — (СН2)2—SCH3
nh2- hci
SOC12
ROH*
NH4OH - —>
ROOC — CH — (CH2) 2 — SCH3
nh2
63
Характеристика основного сырья
Метионин, фармакопея IX.
Бутиловый спирт, ГОСТ 6006—51, ч. д. а.
Изобутиловый спирт, ГОСТ 6016—51, ч. д. а.
Хлористый тионил, МРТУ 09—2602—65, ч.
Аммиак водный, 25%-ный, ГОСТ 3760—64, ч. д. а.
Магний сернокислый, ГОСТ 4523—67, ч- д. а.
Хлороформ, ГОСТ 3160—51, ч. д- а.
Условия получения
Получение н-бутилового эфира метионина. В четырехгор-горлой колбе емкостью 1 л, снабженной капельной воронкой, термометром и обратным холодильником с трубкой, заполненной сульфатом магния, охлаждают до+5° 400 .ил (4,35М) н-бутилового спирта и прибавляют 50 мл (0,68 М) хлористого тионила с такой скоростью, чтобы температура реакционной массы не превышала +10°, а затем—50 г (0,335 М) метионина.
Реакционную смесь кипятят 6 часов на масляной бане при 150°. Отгоняют при пониженном давлении (40 мм) н-бутило-вый спирт. Остаток в виде темной вязкой массы растворяют в 100 мл теплой (55—60°) воды, раствор охлаждают до 20° и нейтрализуют 25%-ным раствором аммиака (-—15 мл) до pH 8,5. Отделяют в делительной воронке масляный слой, а водный экстрагируют хлороформом три раза порциями по 50 мл. Масляный слой и хлороформные вытяжки объединяют, сушат сульфатом магния в течение 6 часов, фильтруют и отгоняют хлороформ при пониженном давлении (100—120 мм), а затем— эфир, собирая фракцию с т. кип. 133—134°/2 мм.
Выход н-бутилового эфира равен 47,5 г (69,3% в расчете на метионин).
Найдено, %: С—53,07; Н—9,63; N —6,67. C9HlflNO2S. Вычислено, %: С—53,00; Н—9,58; N—6,85.
Получение изобутилового эфира метионина. В том же приборе (см. выше) охлаждают до +5° 400 мл (4,35А1) изобутилового спирта и прибавляют 49,0 мл (0,67 Л1) хлористого тионила с такой скоростью, чтобы температура реакционной массы не превышала +10°, а затем 49 г (0,33 Л1) метионина.
Реакционную смесь кипятят 7 часов на масляной бане при 140°.
Отгоняют при пониженном давлении (40 мм) изобутиловый спирт. Остаток в виде темной вязкой массы растворяют в 120 мл теплой (55—60°) воды, раствор охлаждают до 20° и нейтрализуют 25%-ным раствором аммиака (^45 мл) до pH 8,5. Масляный слой отделяют, а водный экстрагируют хлороформом три раза порциями по 85 мл-
61
Масляный слой и хлороформные вытяжки объединяют, сушат сернокислым магнием в течение 6 часов, фильтруют и отгоняют хлороформ при пониженном давлении (100—120 мм), а затем—эфир, собирая фракцию с т. кип- 128—13Г/4 мм.
Выход изобутилового эфира равен 56,32 г (84,5% в расчете на метионин).
найдено, %: С—52,9; И—9,65; N —6,72 C9H19NOaS. Вычислено, %: С—53,0; Н—9,58; N—6,85.
ЛИТЕРАТУРА
1.М. Brenner, V. Kocher. Helv. chim. acta, 32, 333 (1949).
5-1192
грет-БУТИЛОВЫЕ ЭФИРЫ D и L-a-ОКСИИЗОВАЛ Е-РИАНОВЫХ КИСЛОТ
Е. А. МАКАРОВА, Г. II. КОШЕЛЕВА
CgllisOa
СООС(СН3)3 I
НС — он
СН(СНз)2
СООС(СН3)3
НО —сн
СН(СНз)2
М. в. 174,23
трет-Бутиловые эфиры D- и L-a-оксиизовалериановых кислот находят применение в синтезе депсипептидов.
По литературным данным, эти эфиры получают из соответствующих D- и L-a-оксиизовалериановых кислот (1], которые, в свою очередь, получают из рацемической а-оксиизовалериа-новой кислоты [2].
По предлагаемому методу эфиры получают непосредственно из солей a-оксиизовалериановых кислот, служащих для деления кислот на оптические антиподы. Этот метод получения увеличивает выход продуктов и сокращает путь синтеза.
СХЕМА СИНТЕЗА
Для D-изомера
СН2ОН
I
НС - NH2-HOOC
| I СН3СОС1
НО —СН НС —ОН -----------*
I I
/%. СН(СН,)г
I NO.,
66
сн2он
I
ПС NH2-HC1
НО —СН
соон
I
+ НС — ОСОСНз
СН(СН3)2
no2 соон
НС —ОСОСНз +
СНз с=сн2 СНз
СООС(СНз)з СООС(СН3)3
-> ПС ОСОСНз NaOH, НС —ОН
I I
СН-(СНз)2 СН-(СН3)2
Схема синтеза L-изомера из L-a-оксиизовалериаповокис-лого D-треоамина аналогична.
Характеристика основного сырья
D-a-Оксиизовалериановокислый L-треоамин, т. пл. 159— 16Г, [к]о +28° (С 1, спирт); получение см. [2].
L-a-Оксиизовалериановокислый D-треоамин, г. ил. 160— 162°, [а]д +28°(С I, спирт); получение см. [2].
Ацетил хлористый, ГОСТ 5829—51, ч.
Изобутилен, техн.
Условия получения
Получение О-ацетил-ХУ-а-оксиизовалериановой кислоты. В четырехгорлую колбу емкостью 5 л, снабженную мешалкой, капельной воронкой, термометром и обратным холодильником с хлоркальциевой трубкой, загружают 1 кг (3,4 М) D-a-окси-изовалериановокислого L-треоамина и 2,5 л хлористого метилена, доводят температуру массы до 22—23° и по каплям добавляют к ней 256 мл (281 г, 3,8 Л4) хлористого ацетила. При этом температура самопроизвольно повышается до 24—28°. 5* 67
Размешивание при этой температуре продолжают еще 5—6 часов и оставляют массу до следующего дня.
Осадок хлоргидрата L-треоамина отфильтровывают, тщательно промывают хлористым метиленом (около 1 л) и хорошо отжимают (см. примечание).
Получение трет-бутилового эфира О-ацетил-Г)-а.-оксиизова-лериановой кислоты. Раствор О-ацегил-О-а-оксиизовалериано-вой кислоты в хлористом метилене (вместе с промывным раствором) переносят в колбу Бунзена емкостью 5л. В горло колбы вставляют резиновую пробку с трубкой, доходящей до дна (для ввода изобутилена), а к отводу присоединяют стеклянный кран. В колбу добавляют 25 мл. концентрированной серной кислоты, охлаждают смесью льда и соли и конденсируют в ней изобутилен до увеличения объема смеси на 2 л. Затем колбу Бунзена герметически закрывают и оставляют стоять при комнатной температуре 3 суток.
Осторожно открывают кран и постепенно спускают давление. Реакционную смесь выливают в банку емкостью 5 л с мешалкой, добавляют туда 1 л насыщенного раствора бикарбоната натрия (вспенивание!) и размешивают до тех пор, пока не испарится избыточный изобутилен: pH реакционной массы должен равняться 8—9 (по универсальной индикаторной бумаге).
Органический слой отделяют, высушивают сульфатом магния и отфильтровывают. Хлористый метилен упаривают в вакууме, а остаток перегоняют в вакууме, собирая фракцию с т. кип- 90—94°/10 мм.
Выход трет-бутилового эфира О-ацетил-О-а-оксиизовале-риановой кислоты равен 500—550 г (76—84%); По =1,4165;
+ 42,4° (С I, спирт).
Получение трет-бутилового эфира Т)-а-оксиизовалериано-вой кислоты. В круглодонной колбе емкостью 4 л растворяют 550 г трет-бутилового эфира О-ацетил-О-а-оксиизовалериано-вой кислоты в 450 мл метанола и 225 мл воды и к раствору медленно добавляют при размешивании и температуре 0—5° 1,3 л 2 н. раствора едкого натра. Реакционную массу размешивают 1 час при 5—10° и 4 часа при комнатной температуре, после чего экстрагируют эфиром 4 раза порциями по 500 мл. Эфирный экстракт промывают холодной водой дважды порциями по 500 мл и сушат сульфатом магния. Эфир отгоняют при пониженном давлении. Остаток закристализовывается при охлаждении.
Выход трет-бутилового эфира D-a-оксиизовалериановой кислоты равен 400—420 г (76—79%' в расчете на исходную соль); т. застывания 26—28°.
68
Продукт перекристаллизовывают из петролейного эфира при охлаждении до минус 20°. Получают кристаллическое вещество с т. пл. 30—31°.
Получение трет-бутилового эфира к-а-оксиизовалериано-вой кислоты. Из 1 кг L-a-оксиизовалериановокислого D-трео-амина аналогичным образом получают 400 г (75%) трет-бутилового эфира L-a-окслизовалериановой кислоты с т. застывания 26—28°
Примечание. Хлоргидрат L -треоамииа растворяют в 2,5 л воды, подщелачивают 10%-ным раствором едкого натра до pH 9—10 и получают основание с т. пл, 158—160°, которое употребляют для следующих загрузок. В случае получения вещества с более низкой температурой плавления его кристаллизуют из 25 кратного количества воды.
ЛИТЕРАТУРА
I. Р. Platt пег, К. Vogler et а]. Helv. chim. acta., 46, 927 (1963).
2. Л. А. Щукина, P. Г. Вдовина и др. Изв. АН СССР, сер. хим., 2, 310 (1962).
ВИНИЛ(ДИФЕНИЛ)ФОСФИНОКСИД
Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА, Н. Е. ХАВЧЕНКО
р - сн = сн2
С|4Н)3ОР
М. В. 228,23
Винил (дифенил) фосфиноксид можно получить из фенилдихлорфосфина и винилмагнийбромида с последующим мягким окислением полученного винил (дифенил) фосфина [1], реакцией дифенилхлорфосфина с окисью этилена [2] или взаимодействием этилового эфира дифенилфосфинистой кислоты с 1-2-дибромэтаном и последующим дегидробромированием образовавшейся окиси 2-бромэтилдифенилфосфина триэтилами-ном [3].
Нами проверен и уточнен последний метод получения винил (дифенил) фосфиноксида.
СХЕМА- СИНТЕЗА
(С6Н5)2РС1 + С2Н5ОН + (С2Нз)зЫ —>
(С6Н5)2РОС2Н5 Сг214—L
- (С6Н5)2Р(О)СН2СН2Вг
(C2H5)3N>
(СбН5) 2Р (О) СН == СН2 + (С2Н5) 3N • НВг
Характеристика основного сырья
Дифенилхлорфосфин, т. кип. 119—121°/1 мм; получение см. стр. 106 этого сборника.
Этиловый спирт абсолютный, ГОСТ 9674—61, ч.
70
Триэтиламин, МРТУ 6—09—2437—65, ч.
Дибромэтан, ГОСТ 2569—55, ч. д. а.
Натрий сернокислый безводный, ГОСТ 4166—66, ч Кчмол, МРТУ 6—09—2402—65, ч.
Гептан, ГОСТ 4375—48, ч.
Дпэтиловый эфир, ГОСТ 6265—52, ч.
Толуол, ГОСТ 5789—51, ч-
Азот (из баллона), ТУ МХП 4280—54-
Условия получения
Синтез этилового эфира дифенилфосфинистой кислоты. В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой, термометром, трубкой для ввода азота, капельной воронкой и обратным холодильником с хлоркальциевой трубкой, помещают 55 г (0,25 М) дифенилхлорфосфина в 250 мл сухого диэтилового эфира и 28,3 г (0,28 М) триэтиламина в 46 мл сухого эфира. Смесь охлаждают в ледяной бане до 5—0° и прибавляют по каплям 12,4 г (0,27 2И) абсолютного этилового спирта в 40 мл сухого эфира, поддерживая температуру не выше 5°. Затем охлаждающую баню удаляют, температуру массы доводят при перемешивании до 18—20° и оставляют на ночь. Выпавший осадок гидрохлорида триэтиламина отфильтровывают в токе азота, хорошо отжимают и промывают сухим эфиром 3—4 раза порциями по 75 мл. Фильтрат и промывную жидкость соединяют и в токе азоте под вакуумом отгоняют сначала диэтиловый эфир, затем этиловый эфир дифенилфосфинистой кислоты, собирая фракцию с т. кип. 120—12873—4 мм.
Выход эфира равен 42 г (73%); Яр =1,5880—1,5890, что совпадает с литературными данными [3].
Получение винил(дифенил)фосфиноксида. В четырехгор-лой колбе емкостью 1 л, снабженной мешалкой, обратным холодильником, термометром и капельной воронкой, нагревают до кипения (132°) 676 г (7,4 М) перегнанного дибром-этана и прибавляют по каплям за 30 минут 42 г (0,182 АП этилового эфира дифенилфосфинистой кислоты. Затем обратный холодильник заменяют на нисходящий и кипятят реакционную смесь два часа при атмосферном давлении, отгоняя ~10 мл бромистого этила (85—90%' от теоретического количества). Затем при остаточном давлении 10 мм отгоняют практически полностью 1,2-дибромэтан, добавляя в конце отгонки 45 мл кумола. Остаток, содержащий 2-бромэтил (дифенил) фосфиноксид и, возможно, некоторое количество винил-(дифенил) фосфиноксида, переносят в четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, и добавляют
71
200 мл толуола. К смеси по каплям прибавляют 20,2 г (0,2 Л4) триэтиламина, нагревают до кипения на масляной бане (при этом выпадает осадок гидробромида триэтиламина), добавляют еще 2,5 г триэтиламина и кипятят массу при перемешивании 1 час. Потом обратный холодильник заменяют на нисходящий и отгоняют избыток триэтиламина.
Реакционную смесь переносят в делительную воронку емкостью 0,5 л, колбу споласкивают 60 мл воды, воду присоединяют к основной массе. При энергичном встряхивании смеси осадок гидробромида триэтиламина растворяется в воде-Толуольный слой отделяют и отфильтровывают от объемистого белого осадка, который выпадает при стоянии- Толуольный фильтрат сушат над безводным сульфатом натрия, упаривают при пониженном давлении до объема 80 мл и разбавляют 50 мл гептана. Выпавший осадок отфильтровывают и после перекристаллизации из смеси (15:1) гептан-толуол получают 14,2 г винил (дифенил) фосфиноксида с т. пл. 115—117°.
Фильтрат упаривают в вакууме почти досуха и полученный осадок дважды перекристаллизовывают из смеси (15:1) гептана-толуол. Получают еще 24 г вещества с т. пл. 115—117°.
Осадок, полученный из толуола после разделения слоев, экстрагируют 100 мл кипящей смеси (2,5: 1) гептан — толуол. После охлаждения экстракта выпадает белый кристаллический осадок, который nepeKpncTaflflnaoBbiBatoT из смеси (15 :1) гептан—толуол. Получают 5,8 г винил (дифенил) фосфиноксида, т. пл. 115—117°.
Общий выход винил (дифенил) фосфиноксида равен 25,1 г (60%' в расчете на этиловый эфир дифенилфосфинистой кислоты); т. пл. 115—117°.
По литературным данным [3], т. пл. 115—117’-
ЛИТЕРАТУРА
1. М. И. Кабачник, Чжаи-Жун-’Юй, Е. И. Цветков. Докл. АН СССР, 135, 3, 603 (1960).
2. М. И. Кабачник, Т. Я. Медведь и др. Изв. АН СССР, сер. хим., 11, 2029 (1961).
3. R R a b i п о w i t z, J. P e 1 1 о n. J. Organ. Chem., 26, 4623 (1961).
ГЕКСАМЕТИЛЕНДИАМИНО-N, N, N', N ТЕТРАКИС-(МЕТИЛФОСФОНОВАЯ КИСЛОТА)
В. В. СИДОРЕНКО, Н. В. ЛАПШИНА, Т. П. КОНОПЛЕВА (Н2О3РСН2) 2N (СН2) eN (СН2РО3Н2) 2
CI0H28N2O12P4 М. в. 492,22
Синтез нового комплексона—гексаметилендиамипо-N, N, N', Х'-тетракис(метилфосфоновой кислоты) осуществлен нами впервые конденсацией гексаметилендиамина с формалином и фосфористой кислотой в кислой среде по методу (1].
СХЕМА СИНТЕЗА
II2N(CH2)6NH2 + 4Н3РО4 + 4СН2О—
-> (Н2О3РСН2) 2N (CH2)sN (СН2РО3Н2) 2
Характеристика основного сырья
Соляная кислота, ГОСТ 3118—67, ч., пл. 1,19.
Фосфористая кислота, ТУ МХП 518—41, ч.
Формалин, ВТУ МХП 2711—51, 35°/о1ный раствор.
Гексаметилендиамин, МРТУ 6—09—4720—67.
Условия получения
В трехгорлой колбе емкостью 0,5 л, снабженной мешалкой, обратным холодильником и капельной воронкой, к раствору 32,8 г (0,4 А1) фосфористой кислоты в 20 мл дистиллированной воды прибавляют при перемешивании по каплям 11,6 г (0,1 А1) гексаметилендиамина, затем 40 мл концентрированной соляной кислоты. Смесь нагревают на масляной бане до
73
100—110°, после чего за 1 час прибавляют по каплям 69 г (0,4 Л1) 35% -кого раствора формалина. Перемешивание продолжают при 100° еще 1 час, затем смесь охлаждают до комнатной температуры, продолжая перемешивание до выпадения осадка. Осадок отфильтровывают с отсасыванием, промывают на фильтре спиртом 3 раза порциями по 50 мл и высушивают.
Получают 34,8 г сырой гексаметилендиамино-N, N, N',N'-тетракис(метилфосфоновой кислоты).
Для очистки кислоту растворяют в 100 мл кипящей воды, фильтруют на воронке для горячего фильтрования, фильтрат кипятят 15 минут с активированным углем и фильтруют с отсасыванием. Раствор упаривают при пониженном давлении (15—20 мм) до объема 40мл; выпавший осадок отфильтровывают на стеклянном пористом фильтре (№ 3), промывают на фильтре спиртом 3 раза порциями по 50 мл и сушат над пяти-окисыо фосфора в вакуум-эксикаторе.
Выход чистой гексаметилендиамино-N, N, N', N'-тетракпс-(метил фосфоновой кислоты) равен 26,5 г (53,5%); т- пл- 224— 226°.
Найдено, %: С—24,6; Н—6,2; N—5,6, Р—25,0. Ci0H,8N8O12P4. Вычислено, %: С—24,4; Н—5,7; N—5,7; Р—25,0.
ЛИТЕРАТУРА
1. R. М а е d г 1 tzer, R. R. Irani. J. Organ. Chem., 31, 5, 1605 (1967).
ГЕКСАМЕТИЛ ФОСФОРТРИАМИД
А. Л. ФРИДМАН, В. Д. СУРКОВ, Ф. М. МУХАМЕТШИН
1CH3)2N
(CH3)2N-P = 0
(СН3) 2n
CeHiaN3OP М. в. 179,20
Гексаметилфосфортриамид принадлежит к числу высокополярных апротонных растворителей [1, 2]. Способ его получения основан на взаимодействии газообразного [1] или жидкого [3] диметиламина с хлорокисью фосфора в среде диизопропилового или дибутилового эфиров. При использовании газообразного диметиламина трубка, подводящая диметиламин, часто забивается побочным продуктом — гидрохлоридом диметиламина. Использование жидкого диметиламина усложняет процесс.
Нами разработана методика синтеза гексаметилфосфор-триамида в среде хлороформа. При этом значительная часть гидрохлорида диметиламина растворяется в хлороформе, а не-растворившаяся часть, благодаря большой плотности растворителя, вплывает на поверхность раствора и не мешает проведению процесса.
СХЕМА СИНТЕЗА
POCi3 + 6HN(CH3)2 -> [(CH3)2N]3PO + 3HC1-HN(CH3)2
Характеристика основного сырья
Диметиламин, газ, ГОСТ 9967—62, техн.
Фосфора хлорокись, СТУ 53—244—62, ч., перегнанная.
Хлороформ, ГОСТ 3160—51, ч.
Натр едкий, ГОСТ 4328—66, ч.
Натрий углекислый безводный, ГОСТ 83—63, ч.
75
Способ получения
В трехгорлую колбу емкостью 2 л, снабженную мешалкой с гидравлическим затвором и трубками для подачи и отвода газа, помещают 700 мл хлороформа и 267 г (1,74 М) хлорокиси фосфора. При перемешивании в колбу барботируют 520 г (11,5 Л4) диметиламина (см. примечание). Скорость подачи диметиламина 80—100 г/час. Температуру реакционной смеси поддерживают равной 20—25°, охлаждая колбу проточной водой- Затем смесь нагревают до 40°, выдерживают при этой температуре 1 час, охлаждают до 15° и отфильтровывают осадок гидрохлорида диметиламина, промывая его на фильтре 50 мл хлороформа.
Фильтрат небольшими порциями приливают к 800 мл 15%-ного водного раствора едкого натра. После перемешивания и отстаивания органический слой отделяют. Водный слой экстрагируют 2 раза хлороформом по 100 мл. Экстракт объединяют с органическим слоем и сушат безводным карбонатом натрия. После отгонки хлороформа на водяной бане остаток в колбе перегоняют в вакууме, собирая фракцию с т. кип. 9976 мм; п% =1,4540; df =1,026.
Выход гексаметилфосфортриамида равен 208 г (71%).
По литературным данным [2], т. кип. 9976 мм; п2о = = 1,4582°; dl® = 1,0253.
Примечание. При отсутствии технического диметиламина его получают разложением гидрохлорида диметиламина едким натром.
ЛИТЕРАТУРА
1. L. Robert. Chlm. et Ind ., 97, 337 (1967).
2. H. Normant, Angew. Chem., 79, 1029 (1967).
3. И. А. Ф а в о p ск а я, 3. А. Шевченко н др. Ж- орган, химии, 5, 201 (1969).
ГЕКСАХЛОРАЦЕТОН
И. Г. ХАСКИН, Л. О. МАТВЕЕВ
СС1з —СО —СС1з
С3С16О М. В. 264,78
Гексахлорацетон является активатором анионной полимеризации капролактама [1] и полупродуктом при получении трихлоруксусной кислоты [2], ее производных и ряда других соединений; применяется также в качестве гербицида в сельском хозяйстве [3]. ,
В литературе [4] описано получение гексахлорацетона хлорированием ацетона или изопропилового спирта при 25—95° с последующим дехлорированием продуктов при 150—160°. Последняя стадия ведется в вертикальных реакторах, заполненных активированным углем.
Предложенная нами методика [5] позволяет значительно снизить температуру хлорирования на этой стадии, исключить применение активированного угля и получить высокий выход продукта при проведении процесса в обычных реакторах для жидкофазного хлорирования, применяя в качестве катализаторов третичные амины или их гидрохлориды.
СХЕМА СИНТЕЗА
сн= х
) СНОП -----»СС13 - СО - СС13
СНз
Характеристика основного сырья
Изопропиловый спирт, ГОСТ 9805—69, техн., I с.
Хлор жидкий, ГОСТ 6718—68.
Натрий двууглекислый, ГОСТ 4201—68, ч.
Пиридин, ГОСТ 2747—67, ч.
77
Условия получения
В стеклянном вертикальном хлораторе диаметром 40 мм и высотой 400 мм, снабженном барботером, мешалкой, обратным холодильником, гильзой для термометра и рубашкой для обогрева и охлаждения, охлаждают до 10° 200 мл 80%-ного изопропилового спирта и при энергичном размешивании пропускают хлор со скоростью 24 г/час в течение 25 часов, следя за тем, чтобы температура в хлораторе не превышала 30°.
Затем температуру повышают до 35—40° и пропускают хлор при размешивании 8 часов со скоростью 16 г/час, после чего температуру постепенно увеличивают до 90—95°, пропуская хлор с той же скоростью в течение 17 часов, пока плотность реакционной массы не достигнет 1,6—1,62.
От хлорированного продукта, состоящего, в основном, из пентахлорацетона, отделяют верхний слой соляной кислоты и добавляют к нему 6 мл пиридина. Смесь вновь помещают в хлоратор и пропускают хлор еще 2,5 часа при 115° со скоростью 24 г/час до достижения плотности реакционной массы 1,720—1,735 (см. примечание). Сырой продукт отделяют от верхнего слоя пиридингидрохлорида и промывают последовательно при хорошем перемешивании 650 мл воды, 319 мл 3%-ного раствора бикарбоната натрия и водой два раза порциями по 650 мл. Массу сушат 10—12 часов безводным сернокислым натрием (33 г), фильтруют и перегоняют в вакууме.
Выход высушенного технического гексахлорацетона равен 695 г (95%’).
Разгонку технического продукта проводят на колонке, содержащей 15 теоретических тарелок, при остаточном давлении 40 мм.
При разгонке получают: 3,11 г пентахлорацетона с т. кип. 93—94°, «о —1,4905; промежуточную фракцию (5,33г) с т. кип. 94—108°, «о = 1,5060; фракцию гексахлорацетона с т. кип. 108—109°, «о = 1,5095; d12 = 1,744.
Выход гексахлорацетона равен 591 г (81% в расчете на изопропиловый спирт).
Найдено, %: С1 —80,25.
С3С1вО. Вычислено, %: С1 —80,03
Примечание.
В процессе хлорирования (в основном, на последней стадии) 25—30% хлора не вступают в реакцию, проскакивая через хлоратор. Более полное использование хлора достигается при проведении процесса в двух хлораторах; избыточный хлор в смеси с хлористым водородом нз первого хлоратора поступает во второй.
78
литература
1. Пат. США 2764479 (1956); С. А., 58, 8366; 59, 12099 g, (1963).
2. Пат. США, 3154525 (1961).
3. Пат. США, 2695918 (1954).
4. Пат. США, 2635117 (1953); Т. Ambrtis, A. Velnicerin Rev. chim (Bucharest), 14, [9], 506 (1963); С. A., 60,6739 f (1964).
5. И. Г. X а с к и н, Л. О. Матвеев, М. И. Левинский, А. А. Д а х и о, Р. Г. Г а л и м о в а, А. М. К у р и ш к о, В. Т. Вдовиченко. Авт. свид. 176279; Изобретения. Промобразцы. Товарные знаки, № 22,
4,4’-ДИАЛ КОКСИАЗОКСИБЕНЗОЛ Ы
Н. Г. ЧЕРНОВА, Г. И. ВЕСЕЛОВСКАЯ, В. В. САЛИН, Е. И. ТАРАСЮК, К. И. МАЗЫ КИН А
RO — f ^-N = N-^ —OR,
О
где R = h-C3H7—; Н-С4Н9—; н-С5Нц—; н-СеН13—;
H-CyHjs—; н-СвН17—; H-C9H19—; Н-С10Н21—;
Способ получения 4,4’-диалкоксиазоксибензолов основан на восстановлении 1,4-алкоксинитробензолов гидразин-гидратом [1], в щелочной среде—солями мышьяковистой кислоты [2,3] или металлами [4—7]. (
Нами проверена последняя методика и уточнены условия проведения реакции.
СХЕМА СИНТЕЗА z RJ z- 'X Zn
O2N —_/-ОН —O2N —__________________/-OR —~*
- RO— { N—N—— OR
\=/ ф \ = Z
О
Характеристика основного сырья
Децил йодистый, ТУ 79—II—877—63.
п-Нитрофенол, ТУ МХП 1614—47, техн.
Ацетон, ГОСТ 2603—63, и.
Калий углекислый безводный, ГОСТ 4221—65, ч.
Натр едкий, ГОСТ 4328—66, ч.
Цинковая пыль, ГОСТ 12601—67, техн.
Соляная кислота. ГОСТ 3118—67, ч.
Этиловый спирт гидролизный в/о, ТУ 3—66—65, -гехн.
Условия получения
Синтез п-(к-децилокси)нитробензола. В колбу емкостью 0,5 л, снабженную мешалкой и холодильником, загружают 80
0-1192
Условия получения и константы п-(н-алкокси)нитробензолов
Указано процентное содержание основного вещества.
81
210 мл ацетона, 148 г (1,07 Л1) мелкорастертого безводного карбоната калия; 41,36 г (0,3 М) «-нитрофенола и 142,5 г (0,53 М) йодистого децила. Содержимое колбы при интенсивном размешивании нагревают до кипения и выдерживают 21 час при температуре бани 70—80°. Реакционную массу охлаждают до комнатной температуры и фильтруют. Осадок на фильтре промывают 300 мл ацетона, присоединяя его к основному фильтрату. Из фильтрата отгоняют ацетон. Остаток в колбе перегоняют при пониженном давлении, собирая фракцию с т. кип. 231—236°/10 мм.
Выход п-(н-децилокси)нитробензола равен 64,9 г (78,1% в расчете на п-нитрофенол).
Условия получения и константы алкоксинитробензолов представлены в табл. 1 (см. примечание).
Получение 4,4'-ди(п-децилокси)азоксибензола. В трехгор-лой колбе емкостью 250 мл, снабженной мешалкой и обратным холодильником, растворяют при нагревании до 80— 90° 16 г (0,4 М) едкого натра в 120 мл этилового спирта. Полученный раствор охлаждают до 30—40°, прибавляют 27,9 г (0,1 Л4) п- (н-децилокси) нитробензола, 13 г (0,2 М) цинковой пыли и нагревают смесь 1 час при 80—90°. Массу охлаждают до комнатной температуры, выливают в 1 л воды и подкисляют 90 мл раствора (1:1) соляной кислоты до pH 2—3. Выпавший осадок отфильтровывают с отсасыванием, промывают 100 мл охлажденного до 15° этилового спирта и сушат 3 часа в вытяжном шкафу. Получают 34 г 4,4’-ди (н-децилокси) азоксибензола.
Продукт перекристаллизовывают из 3 л горячего этилового спирта.
Выход чистого 4,4’-ди (н-децилокси) азоксибензола равен 15,2 г [59,6% в расчете на п-(н-децилокси)нитробензол].
Условия получения и константы диалкоксиазоксибензолов представлены в табл. 2.
Примечание. Выход и-алкоксинитробензолов увеличивается до 90%, если в синтезе использовать свежепсрегианные йодистые алкилы.
ЛИТЕРАТУРА
1. Б. И. Богословский. Ж- прикл. химии, 9, 725 (1936).
2. Герм. пат. 77563; Frd. 4, 42
3. R. G a u d г у, К. Е. Keirstead. Canadian Journal of Research, 27B, 890 (1949).
4 В. А. Измаильский, В. H. К о л п е н с к и й. Ж. прикл. химии, 1, 4, 31 (1925).
5. В. А. Измаильский, Г. И. Арбузов. Ж. прикл. химии, 1, 5—6, 35 (1925).
6. В. О. Лукашевич. Анилинокрасочная пром-сть, 4, 605 (1934).
7. Н. Н. Ворожцов. Основы синтеза промежуточных продуктов и красителей, изд. 4. М., Госхимиздат, 1955, стр. 248.
8. С. Weygand, J. pract. Chem., 155, 332—41 (1940).
6*
83
82
1,6-ДИАМИН0ГЕКСАН-1,1,6,6-ТЕТРАФОСФОНОВАЯ КИСЛОТА
И. Д. КОЛПАКОВА, Т. М. БАЛАШОВА
(ОН)2Р(О) (О)Р(ОН)2
NH2 — С — (СН2) 4 — с — nh2 I I
(ОН)2Р(О) (О)Р(ОН)2
Cel 12oN2012P4 М. в. 436,12
Аминополифосфоновые кислоты могут применяться как комплексообразующие соединения [1].
В литературе |[1—3] описано получение аминоалкилиденди-фосфоповых кислот взаимодействием 2 молей тригалогенида фосфора на 1 моль нитрила в присутствии кислородсодержащих кислот (Н3РО3, СНзСООН и др.) Аминоалкилидептетра-фосфоновые кислоты могут быть получены аналогичным методом, если использовать в качестве исходного компонента органический динитрил.
Нами получен комплексон, синтез которого в литературе не описан.
СХЕМА СИНТЕЗА
(ОН)2Р(О) (О)Р(ОН)2
РС13 I I
CN (СН2)<CN NH2 — С — (СН2)4 — С - NH2
НзРОз ।
(ОН)2Р(О) (О)Р(ОН)2
Характеристика основного сырья
Треххлористый фосфор, ГОСТ 91—41> ч.
Четыреххлористый углерод, ГОСТ 5827—68, ч. Адипонитрил, МРТУ 6—09—4934—68, ч.
Ацетон, ГОСТ 2603—63, ч.
84
Условия получения
В четырехгорлой колбе емкостью 250 мл, снабженной мешалкой, капельной воронкой и обратным холодильником с хлоркальциевой трубкой, 24,5 г (0,3 Л4) свежеприготовленной фосфористой кислоты (см. примечание 1) приливают 27,5 г (0,2 Л4) треххлористого фосфора и при перемешивании прибавляют по каплям 5,4 г (0,05 перегнанного адипонитрила. Реакционную массу перемешивают 3 часа при комнатной температуре (см. примечание 2) и затем еще 5—6 часов при 55— 65° (температура бани) (см. примечание 3).
Содержимое колбы с помощью ледяной бани охлаждают до 0° и гидролизуют 50 мл ледяной воды. На следующий день образовавшуюся густую сметанообразную массу фильтруют с отсасыванием и промывают осадок на фильтре —ВО мл ацетона. Осадок сушат 30 минут на воздухе и 8—10 часов в сушильном шкафу при 80°.
Выход 1>6-диамнногексан-1,1,6,6-тетрафосфоновой кислоты равен 7,5 г (30%); т. пл. 232—234°.
Найдено», %: С—16,74; 16,62; Н—4,95; N—5,99; 6,10; р________________28 19- 28,29.
QH20N3O13P4 Вычислено, %: С—16,51; Н—4,65; N — 6,43; Р—28,41
Примечания.
1. Перед Началом синтеза 1,6-диаминогексан-1,1,6,6-тстрафосфоновой кислоты в том же приборе получают фосфористую кислоту по способу [1]: к охлажденному раствору 42,6 г (0,3 М) треххлористого фосфора в 160 мл четыреххлористого углерода прибавляют при перемешивании за 30 минут 18 мл (1 Л4) дистиллированной воды, после чего массу перемешивают еще 2 часа при комнатной температуре и отгоняют растворитель при пониженном давлении. Выход фосфористой кислоты почти количественный.
2. Через 1—1,5 часа реакционная масса переходит в стеклообразное состояние н мешалка останавливается.
3. Во время нагревания реакционную массу изредка перемешивают, прокручивая мешалку вручную.
ЛИТЕРАТУРА
I. D. Voigt, F. Gal lais. Inorgan. Synt. Vol. IV, 55—58 (1953).
2. Пат. ФРГ 1002355 (1957); С. A., 53, 21814 f (1959).
3. Пат. США 3303139 (1968); РЖХим., 9Н137П(1968).
4. С. Д. Мехтиев. Нитрилы. Баку. Азерб. гос. изд-во, 1966.
ДИБЕНЗИЛ (ДИБЕНЗИЛАМИНОМЕТИЛ) ФОСФИНОКСИД
Ю. М. ПОЛИКАРПОВ, Г. Ф. ЯРОШЕНКО, Л. М ТИМАКОВА, Т. Я. МЕДВЕДЬ
C29H3aNOP М. в. 439,54
Дибензил (дибензиламииометил) фосфииокСид является новым экстрагентом—комплексообразователем из класса а-ами-нозамещенных фосфиноксидов. Для синтеза этого соединения испопользована конденсация типа Маиниха дибенЗиламина, формальдегида и дибензилхлорфосфина в водной среде [1]. Исходный дибензилхлорфосфин получен иекаталитической реакцией диспропорционирования бензилдихлорфосфина [2, 3].
СХЕМА СИНТЕЗА
СН2О; (C6II5CH2)2NH
СеН5СН2РС12 (С6Н5СН2)2РС1---------------------
-> (C6H5CHs)2P(O)CH2N(CH2C6H5)2
Характеристика основного сырья
Бензилдихлорфосфин, т. кип. 75—7871 мм; получение см. [2,3].
Дибензиламин, МРТУ 6—09—2136—65, ч.
Формалин, ГОСТ 1625—61, ч., 37%'-ный раствор формальдегида.
Натр едкий, ГОСТ 3118—67, ч.
Бензол, ГОСТ 5955—68, ч.д.а.
Гексан, МРТУ 6—09—2937—66, ч.
Азот (из баллона), Ту МХП 4280—54.
86
Условия получения
Синтез дибензилхлорфосфина. В колбу Кляйзена емкостью 0,5 л с дефлегматором и капилляром для азота помещают 77,2 г (0,2 М) свежеперегн энного бензилдихлорфосфина и нагревают в слабом токе азота в бане со сплавом Вуда. Нагревание ведут с такой скоростью, чтобы реакционная смесь равномерно кипела и пары бензилдихлорфосфина конденсировались в реакционной колбе. Температуру бани медленно (в течение 3—4 часов) поднимают с 210° до 240°, отгоняя образующийся в ходе реакции треххлористый фосфор.
Отгонку ведут до тех пор, пока нс будет собрано около 10 мл треххлористого фосфора (60%' от теоретического количества). Остаток выдерживают 15 минут в вакууме водоструйного насоса и затем фракционируют при остаточном давлении 1 мм. Сначала отгоняют непрореагировавший бензилдихлорфосфин (т. кип. 75—78°/1 мм), затем собирают фракцию дибензилхлорфосфина с т. кип. 145—149°/1 мм.
Выход продукта равен 32 г (65%).
После повторной перегонки дибензилхлорфосфин кристаллизуется в виде белых кристаллов, с т. пл. 81,5—82°, что совпадает с литературными данными [3].
Получение дибензил(дибензиламинометил)фосфиноксида. В трехгорлую колбу емкостью 100 мл, снабженную мешалкой (60 об!мин), обратным холодильником и термометром, помещают 12,4 г (0,05 М) дибензилхлорфосфина, 24,6 г (0,125 Л4) дибензиламина и 7,8 мл 35%'-ного раствора формальдегида. Реакционную смесь нагревают при энергичном перемешивании 6 часов на кипящей водяной бане, охлаждают, промывают 20—25 мл 10%'-ного раствора щелочи, затем переносят в делительную воронку емкостью 250 мл и экстрагируют бензолом 3—4 раза порциями по 30—40 мл.
Бензольный экстракт сушат, бензол отгоняют в вакууме водоструйного насоса. К остатку добавляют гексан (^50 мл), выделившийся осадок перекристаллизовывают из смеси (10 :1) гексан—бензол.
Выход дибензил (дибензиламинометил) фосфиноксида па-вен 5,9 г (27%); т. пЛ'. 135—137°.
Найдено %: Р—7,1; N—3,2 СзоНзоМОР. Вычислено % : Р—7,1; N—3,2
ЛИТЕРАТУРА
1. М. И. Кабачннк, Т. Я- Медведь, Ю. М. Поликарпов, Н. С. Ступин, Л. А. Федорова. Авт. свид, 201397; Изобретения. Промобразцы. Товарные знаки, № 18 (1967).
2. Ю. И. Б а р а н о в, С. П. В а р ш а в с к и й, М. И. К а б а ч н и к, Н. К. Близнюк. Авт. свид. 199876; Изобретения. Промобразцы. Товарные знаки, № 16 (1967).
3. Ю. И. Баранов, С. В. Гореленко. Ж. общ. химии, 39, 4, 836 (1969).
87
ДИБЕНЗИЛ (ДИБЕНЗИЛАМИНОМЕТИЛ)ФОСФИН-ОКСИД-Ы-ОКСИД
Ю. М. ПОЛИКАРПОВ, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА, Т. я. МЕДВЕДЬ
C29H30NO2P
М. в. 455,54
Дибензил (дибензиламинометил) фосфиноксид-N-оксид получен окислением дибензил (дибензил аминометил) фосфиноксида перекисью водорода в среде уксусного ангидрида.
СХЕМА СИНТЕЗА
Н2О2; (СН3СО)2О (СеН6СН8)2,Р (О) CH2N (СН2С6Н5) 2 ---------—
(СбН5СН2)2Р(О)СН2М(СН2СбН5)2
О
Характеристика основного сырья
Дибензил (дибензиламинометил)фосфиноксид, т. пл. 135— 137°; получение см. стр. 86 этого сборника.
Уксусный ангидрид, ГОСТ 5815—52, ч. д. а.
Пергидроль, ГОСТ 177—55, мед., ЗО°/о'-ный раствор перекиси водорода.
Натр едкий, ГОСТ 3118—67, ч.
Этиловый спирт абсолютный, ГОСТ 9674—61, ч.
88
Условия получения
В трехгорлую колбу емкостью 50 мл, снабженную обратным холодильником, термометром и мешалкой, помещают 3 мл (0,029 М) 30%'-ного раствора перекиси водорода и 3 мл (0,029/4) уксусного ангидрида. Смесь охлаждают до 5—10° и при перемешивании прибавляют 0,77 г (0,00175 М) дибензил-(дибензиламинометил) фосфиноксида. Реакционную массу нагревают до 70° и выдерживают при этой температуре и непрерывном перемешивании 2 часа. По окончании выдержки содержимое колбы охлаждают до комнатной температуры и добавляют 7 мл (9,8 г; 0,39 Af) 40%-ного раствора едкого натра. Выпавший осадок отделяют, промывают 25 мл воды и очищают переосаждением из 50 мл спирта водой.
Выход дибензил (дибензиламинометил) фосфиноксида-N-оксида равен 0,28 г (35%); т. пл. 92—95° (с разложением).
Найдено, И: С—76,5; Н—6,7; Р—6,8; N—3,1. C29H30NO2P. Вычислено, %: С—76,0; Н—6,6; Р—6,8; N—3,1 .
ДИ-н-БУТИЛОВЫЙ И ДИАЛЛИЛОВЫЙ ЭФИРЫ ИТАКОНОВОЙ кислоты
Е. И. ТАРАСЮК, Е. Ф. ЦЕБРИЕНКО
сн2=-с —соос4н.
СН2 —СООС4Н9
сн2 = с - соосн2 — сн - с н, I
сн2 — соосн2 — сн = сн2
С1зН22О4 М. в. 242,32 СцН]4О1
М. в. 210,23
В литературе описано получение эфиров итаконовой кислоты прямой этерификацией итаконовой кислоты соответствующим спиртом в присутствии серной кислоты и ионообменной смолы [1].
Нами установлено, что применение в качестве катализатора n-толуолсульфокислоты увеличивает выход продукта.
СХЕМА СИНТЕЗА
СН2 = С — СООН СН2 = С — COOR
I I
СН2 — СООН + 2ROH - СН2 — C.OOR + 2Н2О,
где R = н-С4Н9—; СН2 = СН— СН2--.
Характеристика основного сырья
Итаконовая кислота, МРТУ 6—09—749—63, ч. н-Бутиловый спирт, ГОСТ 6006—51, ч.
..Аллиловый спирт, ТУ МХП 1880—48, ч.
Бензол, ГОСТ 5955—68, ч. д. а.
n-Толуолсульфокислота, ТУ МХП ОРУ 73—56, техн.
90
Условия получения
Получение ди-н-бутилового эфира итаконовой кислоты. В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, термометром, водоотделителем с нижним спуском и обратным холодильником и помещенную в парафиновую баню, загружают 52 г (0,4 М) итаконовой кислоты, 88,9 г (1,2 М) н-бути-лового спирта, 40 мл бензола, 10 г п-толуолсульфокислоты. Смесь нагревают, энергично перемешивая 3 часа при 120°, до полного отделения воды (14,5 мл). Затем температуру массы поднимают до 130° и выдерживают 0,5 часа, отгоняя избыток п-бутилового спирта и бензола (около 30 мл) через нижний спуск водоотделителя. Содержимое колбы охлаждают до 30— 40° и выливают в раствор 6,6 г карбоната натрия в 300мл воды. Смесь хорошо взбалтывают, эфирный слой отделяют и промывают дистиллированной водой до нейтральной реакции промывных вод (pH 6) по универсальной индикаторной бумаге. Эфирный слой сушат над свежепрокаленным сернокислым натрием в течение I часа, фильтруют и перегоняют при пониженном давлении, собирая фракцию с т. кип. 172— 175°/25 мм.
Выход ди-н-бутилового эфира итаконовой кислоты равен 63,7 г (65,7%); содержание основного вещества 97,0%; кислотность 0,5%.
Получение диаллилового эфира итаконовой кислоты. В круглодоппую колбу емкостью 0,75 л., снабженную мешалкой, водоотделителем, обратным холодильником и помещенную в [лицериновую баню, загружают 116,2 г (2 М) аллилового спирта, 32,5 г (0,25 М) итаконовой кислоты, 6 г п-толуолсуль-фокислоты, 200 мл бензола. Смесь нагревают, перемешивая 9—10 часов при температуре бани 108—110°, до полного отделения воды (около 14 мл). Затем через водоотделитель отгоняют бензол и избыток аллилового спирта (около 135 мл), повышая температуру в бане до 120°. Массу в колбе охлаждают до 25—30° и за 0,5 часа при перемешивании добавляют 10 г свежепрокаленной кальцинированной соды. Реакционный раствор фильтруют и перегоняют при пониженном давлении, собирая фракцию, кипящую при 141 —142°/25 мм.
Выход диаллилового эфира итаконовой кислоты равен 34 г (64,8%); содержание основного вещества 97,0%; кислотность 0,5%;
ЛИТЕРАТУРА
1. Пат. США 3056829 (1961).
91
ДИГЕКСИЛОВЫЙ ЭФИР МАЛЕИНОВОЙ КИСЛОТЫ
Дигексилмалеинат
Н. Г. ЧЕРНОВА, Г. И. ВЕСЕЛОВСКАЯ, М. В. МОСЕНЦЕВА
СН —СООСеН13
II
сн —соос6н13
С1вН28О4 М. в. 284,30
Общий способ получения эфиров'малеиновой кислоты заключается в действии малеинового ангидрида на соответствующие спирты. Катализаторами реакции этерификации обычно служат серная кислота и п-толуолсульфокислота [1, 2]. Однако полученный по этому методу дигексиловый эфир малеиновой кислоты содержит кислоту и другие примеси.
Нами разработана более простая методика получения дигексилового эфира малеиновой кислоты с применением ионообменной смолы КУ-2. При этом продукт получается чище, стабильного качества, с высоким выходом.
СХЕМА СИНТЕЗА
О
II
СН-С СН — СООС6Н13
II \ КУ-2 II
II рО + 2С6Н13ОН ------------ -||
СН-С СН —СООС6Н13
II
о
Характеристика основного сырья
Малеиновый ангидрид, ГОСТ 5854—68, ч.
Гексиловый спирт, МРТУ 6—09—5532—68, ч., пл. 0,8186.
Бензол, ГОСТ 5955—68, ч.д.а.
КУ-2, Н+-форма, МРТУ 6—09—903—63.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную трехрогим форштоссом, мешалкой, пропущенной через обратный холодильник, термометром, водоотделителем с обратным холодильником и помещенную в парафиновую баню, загружают 24,51 г (0,25 ЛТ) малеинового ангидрида. 76,6 г (0,75 М) гексилового спирта, 2,45 г смолы КУ-2, 150 мл бензола и нагревают 9— 10 часов при хорошем перемешивании до прекращения отделения воды (6 мл), при этоМ’реакционная масса имеет pH 7 (по универсальной индикаторной бумаге).
Из массы за 1,5 часа отгоняют бензол и избыточный спирт (всего 130 мл), повышая температуру массы до 120°. Охлажденную реакционную массу отделяют декантацией от катионита и перегоняют при пониженном давлении (см. примечание), собирая фракцию с т. кип. 198—200°/30 мм.
Выход дигексилового эфира малеиновой кислоты равен 57—59 г (80—83%' в расчете на малеиновый ангидрид); содержание основного вещества 98,5%, кислотность (в пересчете на малеиновую кислоту) 0,1 %1
По литературным данным [1, 2], т. кип. 127°/7 мм, 116°/1 мм.
Примечание. Предгон, собранный от нескольких синтезов, подвергают вакуум-перегонке и получают дополнительно к основному выходу 3—5% продукта в расчете на один синтез.
ЛИТЕРАТУРА
1. V. Sot tan et al. J. Amer. Soc., 75, 4101 (1953).
2. G. Kloban., S. Hudecek. Collect, 25,2307 (1960).
N, Х ДИМЕТИЛ к-НИТРОЛНИЛ ИН
Е. А. БУГАИ
C8H10N?O2
М. в. 116,18
М,М-Диметил-л«-нитроанилин получают нитрованием диме-тиланилина нитрующей смесью (1, 2j или метилированием .и-нитроанилина при высоких температурах {3]. Метилирование .«-питроанилина диметилсульфатом при высоких температурах приводит к образованию смеси моно- и диметиланилинов и частичному осмолению продуктов реакции.
Предложенная нами методика метилирования лг-нитроани-лина в растворителе (изопропиловый спирт) в присутствии щелочного агента (бикарбонат натрия) при температуре кипения спирта исключает образование N-метил-лг-нитроанилина, значительно упрощает очистку технического продукта (исключена стадия ацетилирования) и позволяет получить N.N-ди-метил-ж-нитроанилин высокой степени чистоты с выходом 58— 60%'.
СХЕМА СИНТЕЗА
no2
(CH3)2SO4 -----------—>
-nh2
NO.,
I
Mi_N(CH3)2
94
Характеристика основного сырья
л-Нитроанилин, ТУ МХП 153—40, техн.; содержание основного вещества 99,7 %'.
Диметилсульфат, ТУ МХП 3180—52, техн.
Натрий двууглекислый, ГОСТ 2156—68, техн-
Изопропиловый спирт, ГОСТ 9805—61, техн.
Условия получения
В четырехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром, обратным шариковым холодильником и капельной воронкой, загружают 1 л изопропилового спирта, 276 г (2М) лг-нитроанилина, 504 г (6М) бикарбоната натрия. Смесь при перемешивании нагревают на водяной бане до кипения и прибавляют за 1 —1,5 часа 760 г (6 М) диметилсульфата с такой скоростью, чтобы поддерживать слабое кипение (температура смеси 75—80°).
После охлаждения реакционной массы до 50—55° ее выливают в стакан, содержащий 3 л охлажденной воды. Выделившийся осадок отфильтровывают с отсасыванием, промывают 2—3 раза водой порциями по 1 л и сушат на воздухе.
Выход N.N-диметил-лг-нитроанилина равен 237 г (71,5%).
Технический продукт перекристаллизовывают из изопропилового спирта (на 1 г продукта 3—4 мл спирта).
Выход очищенного продукта равен 180—190 г (55—60%); т. пл. 58.5—59°; содержание основного вещества 99%'.
По литературным данным (1], т. пл. М,М-диметил-ж-нитро-анилина 59—60°.
ЛИТЕРАТУРА
1. Синтезы оргавическвх препаратов, т. 4. М., Инлитиздаг. 195.3, стр. 374.
2. Е. N о е 1 t i n g, E. Fo u r n 1 a u x. Ber,, 30, 2930 (1897).
3. F. U liman п, P. Wenner.' Ber.,33, 2476 (1900).
4,4-ДИОКСИСТИЛЬБЕН-3,3’,5,5 -ТЕТРАКИС-(МЕТИЛИМИНОДИУКСУСНАЯ КИСЛОТА), ТЕТРАНАТРИЕВАЯ СОЛЬ
Л. М. ТИМАКОВА, Г. Ф. ЯРОШЕНКО, В. Я. ТЕМКИНА,
Р. П. ЛАСТОВСКИИ
7 СН2СООН ch2n<
z ' CH2COONa
нс—€ У~~он
CH2COONa
CH2N(
4 СН2СООН
СН2СООН
CH,N\
__/ xCH2COONa
\_Z~OH
= \ ,CH2COONa
CH2N <
x CH2COOH
Сз4НзбМ4Ыа4О jg
M. в. 880,64
Тетрапатриевая соль 4,4'-диоксистильбен-3, 3', 5, 5'-тетра-кис(метилиминодиуксусной кислоты) является новым флуоресцирующим комплексоном, аналогом синтезированного нами ранее оксистильбенкомплексона [1]. Комплексон образует прочные комплексы с рядом катионов, что в некоторых случаях сопровождается изменением флуоресценции, и предложен нами в качестве первого металлфлуоресцентного индикатора для комплексонометрического определения скандия [2].
96
Комплексон получен взаимодействием 4,4’-диоксистильбена [3] с формальдегидом и иминодиуксусной кислотой по реакции Манниха [4].
СХЕМА СИНТЕЗА
SV /у—HN(CH2COOH)2
сн = сн он— н2со
СН2СООН
CH2N\
CH2COONa
нс-f Ч он
CH2COONa
CH2N^
сн2соон
СН2СООН
НС— / 'S—ОН
CH2COONa
CH2COONa
СН2СООН
Характеристика основного сырья
Иминодиуксусная кислота, ВТУ МГУХП 190—58, к.
4,4'-Диоксистильбен, получение см. [4].
Формальдегид, ТУ МХП 2631—51, ч.
Уксусная кислота, ГОСТ 61—69, ч.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником и капельной воронкой, помешают 6,4 г (0,03 М) иминодиуксусной кислоты, нейтрализованной 30%-ным раствором едкого натра до pH 7—8, и добавляют суспензию 2,1 г (0,01 М) 4,4'-диоксистильбена в 120 мл ледяной уксусной кислоты. К реакционной смеси, охлажденной до 5—7° банен со льдом, добавляют по каплям при перемешивании 9,2 г (0,12Л4) 37%-ного раствора формальдегида и оставляют при комнатной температуре на ночь. На другой день реакционную смесь нагревают 4 часа при 55—60° при 7—1192 97
размешивании и затем в течение часа поднимают температуру до 80°, при этом вся масса постепенно растворяется. Смесь выдерживают при 80° в течение 3—4 часов, по охлаждении переносят в стакан и при перемешивании добавляют 200 мл этилового спирта. Выпавший белый осадок фильтруют, промывают 50 мл этилового спирта и сушат в сушильном шкафу при 50°.
Выход комплексона равен 4,6 г (52%).
Комплексон хорошо растворяется в воде, не растворяется г органических растворителях.
Найдено, %: С—46,23; 46,15; Н—4,31; 4,25; N—6,41; 6,23 Вычислено, %: С—46,37; Н—4,12; N—6,36.
ЛИТЕРАТУРА
1. В. Я. Темкина, Г. Ф. Ярошенко, Л. М. Тимакова, Р. П. Л а ст о в ск ий. Методы получения химических реактивов и препаратов, вып. 18. М„ ИРЕА, 1969, стр. 86.
2. Р. П. Л а с т о в с к и й, В. Я. Т е м к и н а, Г. Ф. ЯР0ШенК0, Л. М. Тимакова. Авт. свид. 297920; Открытия. Изобретения. Промобразцы. Товарные знаки, № 10 (1971).
3. В. Я. Т е м.к и н а, Г. Ф. Ярошенко, Л. М. Тимакова, Р. П. Ластовский. А1етоды получения химических реактивов п пре пиратов, вып. 18. М., ИРЕА, 1969, стр. 89.
4. G. Schwarzenbach, G. Anderegg, R; Sall man. Helv. chim a-ta, 35, 1785 (1952).
ДИФЕНИЛ (ДИОКТИЛАМИНОЭТИЛ) фосфиноксид
Ю. М. ПОЛИКАРПОВ, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА, Т. Я. МЕДВЕДЬ
К
= Р-—СН2 —CH2—N(C8H17)2
/=\/II
о
C30H48NOP М. в. 469,69
Дифенил (диоктиламиноэтил) фосфиноксид является новым экстрагентом из класса р-аминозамещенных фосфиноксидов. Для получения этого соединения использован метод нуклеофильного присоединения диоктиламина к винил (дифенил) фос-финоксиду [1].
СХЕМА СИНТЕЗА
(С6Н5)2Р(О)СН=СН2 + (C8H17)2NH —
-> (C6H5)2P(O)CH2CH2N(C8II17)2
Характеристика основного сырья
Винил (дифенил) фосфиноксид, т. пл. 254—256°; получение см. стр. 70 этого сборника.
Диоктиламин, т. кип. 142—14773 мм.
Условия получения
Смесь 10 г (0,044 /И) винил (дифенил) фосфиноксида и 10,6 е (0,044 Л-1) диоктиламина нагревают 10 часов в запаянной ампуле при температуре бани 200—220°. Содержимое ампулы растворяют в 80 мл гексана, отфильтровывают непро-7* 99
реагировавший винил (дифенил) фосфиноксид. Из фильтрата отгоняют гексан, остаток выдерживают 30 минут в вакууме (1 мм) при 200°, отгоняя не вступивший в реакцию диокти-ламин. Остаток при охлаждении закристаллизовывается.
Выход дифенил (диоктиламиноэтил) фосфиноксида равен 12,4 г (60%); т. пл. 30—35°.
Найдено, %: С—76,3; Н—10,4;N—2,9; Р—6,3.
C,0H,SNOP. Вычислено, %: С—76,5; Н—10,3; N—3,0; Р—6,6.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я- Медведь и др. Изв. АН СССР, сер хим., 9, 1584 (1962).
ДИФЕН ИЛ (ДИЭТИЛ АМИНОМЕТИЛ) ФОСФИНОКСИД
Ю. М. ПОЛИКАРПОВ, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА,
Т. я. МЕДВЕДЬ
C17H22NOP
М. в. 287,34
Дифенил (диэтиламинометил) фосфиноксид является новым экстрагентом из класса а-аминозамещенных фосфинокси-дов. Для синтеза этого соединения использована конденсация типа Манниха диэтиламина, формальдегида и дифенилхлор-фосфина в водной среде [1].
СХЕМА СИНТЕЗА
(С6Н5)2РС1 + (C2H5)2NH + СН2О ->
-> (С6Н5) 2Р (О) CH2N (С2Н5) 2 + НС1
Характеристика основного сырья
Дифенилхлорфосфин, т. кип. 119—121°/1 мм; получение см. стр. 87 этого сборника.
Диэтиламин, СТУ 12—10—105—61, ч.
Формалин, ГОСТ 1625—62, ч„ 37%-ный раствор формальдегида.
Диэтиловый эфир, ГОСТ 6265—52, ч.
Гептан, ГОСТ 4375—48, ч.
101
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой,. капельной воронкой и обратным холодильником, помещают 154 мл (2,0 Л4) 35%-ного раствора формальдегида и 54,6 г (0,75 Л1) свежеперегнанного диэтиламина. Постепенно за 30 минут при непрерывном перемешивании из капельной воронки прибавляют 55 г (0,25 М) дифенилхлорфосфина. Реакционную смесь выдерживают 1 час при непрерывном перемешивании на кипящей водяной бане, переносят в делительную воронку емкостью 1 л и экстрагируют эфиром 3—4 раза порциями по 200 мл. Эфирный экстракт промывают водой, эфир отгоняют, остаток экстрагируют кипящим гептаном 6— 7 раз порциями по 100 мл. Выпадает белый кристаллический осадок, который перекристаллизовывают из 200 мл гептана.
Выход, дифенил (диэтиламинометил) фосфиноксида равен 28 г (39%' в расчете на дифенилхлорфосфин); т. пл. 92—93,5°.
Найдено, %: Р—11,6; N—5,2
C17H22NOP. Вычислено, %: Р—11,6; N—5,3.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я. Медведь, Ю. М. Поликарпов, Н. С. Ступин, Л. А. Федорова. Авт. свид. 201397; Изобретения. Промобразцы. Товарные знаки, № 18 (1967).
М,М’-Д ИФЕН ИЛ-га-ФЕН ИЛ ЕНД НАМИ Н
Б. И. КИССИН, Е. Н. КУРАКИН
CigHieNj
М. в. 260,34
М,1\т'-Дифенил-н-фенилендиамин применяется как антиоксидант в полимерных материалах.
Описаны способы получения этого соединения, основанные на взаимодействии анилина с гидрохиноном [1—9] или п-ами-нофенолом [10—11] с применением катализаторов.
Нами разработана методика получения Н,Ы'-дифенил-л-фенилендиамина взаимодействием гидрохинона с анилином с азеотропной отгонкой реакционной воды.
СХЕМА СИНТЕЗА
НО —V-ОН 4-2^ NH2 ——2-
-> Ч __ NH — - NH - Ч
Характеристика основного сырья
Гидрохинон, ГОСТ 2549—60, ч.
Анилин, ГОСТ 313—69, техн.
Хлорбензол, ГОСТ 646—60, техн. Дихлорбензол, ТУ 305—41, техн. Цинк хлористый, ГОСТ 4529—69, ч. Соляная кислота, ГОСТ 3118—67, ч. Уголь активированный, ГОСТ 4453—48.
103
Условия получения
В чугунный котелок емкостью 1 л, снабженный якорной мешалкой, термометром и обратным холодильником с водоотделителем, емкостью 20 м.л, помещают 121,1 г (1,3 Л1) анилина, 20 мл дихлорбеизола, 60,6 г (0,55 М) гидрохинона и 55,9 г (0,41 Л1) тонкоизмельченного хлористого цинка.
Смесь кипятят при постоянном размешивании на масляной бане. Образующаяся в ходе реакции вода отслаивается в разделителе и по мере накопления выводится через боковой слив, а дихлорбензол возвращается в реакционную массу. Через 2—2,5 часа температура реакционной массы поднимается до 230°. При этой температуре через обратный холодильник добавляют еще 84,2 г (0,9 ЛГ) анилина и вновь кипятят смесь столько, чтобы общая продолжительность кипячения составила 10 часов. После этого массу охлаждают до 180—190' и вносят за 5—10 минут еще 300 г (3,2 А1) анилина. Размешивание при 190° продолжают 1 час, затем охлаждают смесь до 20° и размешивают еще 2 часа. Осадок отфильтровывают с отсасыванием через двойной бумажный фильтр.
В стеклянный толстостенный или фарфоровый стакан емкостью 2 л, снабженный рамной мешалкой и термометром, вносят 800 мл воды, 205—210 Мл 30%-ной соляной кислоты и отжатую пасту дифенил-и.-фенилендиамина. Суспензию нагревают до 70° и размешивают при этой температуре 2 часа, следя по бумаге «конго» за наличием кислой среды в реакционной массе. Не давая остыть, суспензию фильтруют с отсасыванием и осадок на фильтре промывают теплой (50—60°) водой до нейтральной реакции промывных вод (pH 6—7) и сушат при 100°.
Выход Ы,М'-дифенил-я-фенилендиамина равен 98,8 г (69% в расчете на гидрохинон) , т. пл. 142—144°.
Для получения чистого продукта в трехгорлую колбу емкостью 1 л, снабженную лепестковой мешалкой, обратным холодильником и водоотделителем, помещают 250 мл хлорбензола и при размешивании постепенно вносят полученную водную пасту или сухой продукт. Прибавляют 18 г активированного угля и нагревают массу до кипения хлорбензола. После отгонки воды (в случае применения влажной пасты) размешивание при нагревании продолжают 1 час и быстро фильтруют горячий раствор. Уголь на фильтре промывают 50 мл горячего (100°) хлорбензола. Объединенный хлорбензольный раствор охлаждают и через 8 часов фильтруют. Осадок на фильтре промывают 45 мл хлорбензола и сушат при пониженном давлении (600—650 мм) при 60°.
Выход дифенил-п-фенилендиамина равен 79 г (55% в расчете на гидрохинон), т. пл. 150—152°.
По литературным данным [12], т. пл. 152°.
104
ЛИТЕРАТУРА
1. A. Calm. Вег., 16, 2786 (1883).
2. Пат. США 2238320 (1941); С. А., 35, 4778 (1941)
3. Пат. США 2503712 (1950); С. А., 44, 6434 (1950).
4. Франц, пат. 1179775 (1959); РЖХим., 19Л160 (1961).
5. Англ. пат. 864763 (1961); С. А., 55, 25859 (1961).
6. Б. И. К и с с и н, О. И. Сыпяк. Хим. пром-сть, 4, 26 (1961).
7. Б. И. К и с с и п, Е. Н. Куракин. Хим. пром-сть, 2, 24 (1965).
8. Б. И. К и с с и н, Е. Н. К у р а к и н. Материалы Всесоюзной науч-
но-технической конференции по синтезу химикатов для полимеров. 1966 г. Тамбов, «Тамбовская правда», 1969, стр. 263
9. Пат. США 3305584 (1967); С. А., 67, 21605 (1967).
10. Англ. пат. 481465 (1938); С. А., 32, 6259 (1938).
11. Пат. ФРГ 883751 (1953); РЖХим., 30300П (1956).
12. Е. Kirk. D. О th me г. Encyclopedia of chemical technology, V 5, 1950, P. 26.
ДИФЕНИЛХЛОРФОСФИН
Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА, И. Е. ХАВЧЕНКО
С12Н10С1Р
М. в. 220,64
Дифенилхлорфосфин получают термическим диспропорционированием фенилдихлорфосфина (1—3]. В отсутствие катализатора реакцию проводят под давлением при 300°, причем достигаются невысокие выходы продукта (около 35%) [4].
Нами использован метод получения дифенилхлорфосфина диспропорционированием фенилдихлорфосфина при нагревании в токе азота выше 200° в присутствии каталитического количества безводного хлористого алюминия (5]. Исходный фенилдихлорфосфин получен по реакции Фриделя-Крафтса взаимодействием треххлористого фосфора и бензола в присутствии хлористого алюминия и триэтиламина [6, 7],
СХЕМА СИНТЕЗА
А1С1з, (C2H5)3N А1С1з
С6Н6+РС13------------------* С6Н5РС12 ------
(С6Н5)2РС1 + РС1з
Характеристика основного сырья
Бензол, ГОСТ 5955—68, ч. д. а.
Фосфор треххлористый, ГОСТ 91—41, ч.
Алюминий хлористый безводный, ВТУ МХП 3500—52, ч, Триэтиламип, МРТУ 6— 09—2437—65, ч.
Азот (из баллона), ТУ МХП 4280—54.
106
Условия получения
Синтез фенилдихлорфосфина. В четырехгорлую колбу емкостью 1,5 л, снабженную мешалкой, обратным холодильником с хлоркальциевой трубкой, капельной воронкой и термометром, загружают 1 кг (7,3 М) треххлористого фосфора, 340 г (2,5 М) безводного хлористого алюминия и 195 г (2,5 Л1) сухого бензола. Смесь кипятят (80°) при перемешивании 3 часа, охлаждают и, не прекращая перемешивания, добавляют из капельной воронки 260 г (2,56 М) свежеиерегнаи-ного триэтиламина при температуре не выше 40°. На следующий день из реакционной массы отгоняют в токе азота избыток треххлористого фосфора, остаток перегоняют в вакууме, собирая фракцию с т. кип. 58—60°/1 мм.
Получают 281 г (63%) фенилдихлорфосфина.
Фенилдихлорфосфин представляет собой бесцветную, дымящую на воздухе жидкость ст. кип. 58—60°/1 мм; г с]) =1,5979; сг*20—1,3185, что совпадает с- литературными данными [6].
Получение дифенилхлорфосфина. В колбу Кляйзепа емкостью 1,5 л помещают 281 г (1,52 А1) фенилдихлорфосфина и 11,2 г безводного хлористого алюминия. Смесь нагревают на бане со сплавом Вуда при атмосферном давлении в слабом токе азота, при этом отгоняется образующийся в ходе реакции треххлористый фосфор. Нагревание ведут с такой скоростью, чтобы реакционная смесь равномерно кипела и .пары фенилдихлорфосфина конденсировались в реакционной колбе (при атмосферном давлении фенилдихлорфосфин кипит при 224,6°). Температуру бани поднимают за 2—2,5 часа от 220 до 300°. После того, как отгонят ^70 мл треххлористого фосфора (75% от теоретического количества), остаток выдерживают 10 минут в вакууме водоструйного насоса, а затем фракционируют при остаточном давлении 1 мм.
Сначала отгоняют непрореагировавший фенилдихлорфос-фип (т. кип. 58—6071 мм), затем собирают фракцию дифс-нилхлорфосфина с т. кип. 119—12171 мм.
Выход дифенилхлорфосфина равен 125 г (75%! в расчете на фенилдихлорфосфин); «о =1,6360; d20i = 1,1935; т. кип. 119—12171 мм, что совпадает с литературными данными [5, 7].
Дифенилхлорфосфин представляет собой бесцветную маслянистую жидкость с неприятным запахом, бурно реагирует с водой.
ЛИТЕРАТУРА
1. С. Dorken Вег., 21, 1505 (1888).
2. А. Е. Арбузов. ЖРФХО, 42, 395, 549 (1910).
107
3. В. А. Арбузов. Н. П. Гречкин. Ж. общ. химии, 20, 167 (1950).
4. A. Broglie. Вег,, 10,628 (1877).
5. М. И. К а б а ч н и к, Т. Я. Медведь и др. Изв, АН СССР, сер. хим., 11,2029 (1961).
6. О. М. К о s о 1 а р of 1. Organophosphorus Compounds. New-York, London. 42 (1950).
7. C. S t u e b e, W. M. le Suer, G. R. N о r m a n n. J. Amer. Chem , Soc., 77, 3526 (1955).
ДИХЛОРАНГИДРИД 0,₽,₽-ТРИС(ХЛОРМЕТИЛ)-ЭТИЛФОСФОРИСТОЙ кислоты
Л. А. ВЛАСОВА, Е. В. КУЗНЕЦОВ
(С1СН2)3С —СН2ОРС12
С5Н8С15ОР М. в. 292,24
Дихлорангидрид р, 0,0-трис (хлорметил) этилфосфористой кислоты может найти применение при синтезах фосфорорганических соединений.
Нами дихлорангидрид рдр-трис(хлорметил)этилфосфори-стой кислоты получен впервые [1] взаимодействием треххлористого фосфора с трихлоргидрином пентаэритрита или с 3,3-бис (хлорметил)оксациклобутаном при нагревании.
СХЕМА СИНТЕЗА
Из трихлоргидрина пентаэритрита
РС13 (С1СН2)3С- СН2ОН-------(С1СН2)3С - СН2ОРС12,
из 3,3-бис (хлорметил)оксациклобутана
СН2С1
I РС13
С1СН2 —С —СН2 -------(С1СН,)3С —СН2ОРС12
I
СН2-О
Характеристика основного сырья
Фосфор треххлористый, ГОСТ 91—41, ч.
Трихлоргидрин пентаэритрита, т. ил. 64°; получение см. [2].
3,3-Бис(хлорметил)оксациклобутан, т. кип. 80/10 мм; п® — = 1,4858; df =1,2996: получение см. [3].
109
Условия получения
В четырехгорлой колбе емкостью 0,5 л, снабженной мешалкой, капельной воронкой, термометром и обратным холодильником, нагревают до полного расплавления 191,5 г (1 Л1) трихлоргидрина пентаэритрита и при интенсивном перемешивании прибавляют по каплям 137 г (1 М) треххлористого фосфора. Реакционную смесь нагревают до 80° и выдерживают при этой температуре 4 часа (см. примечание). Из реакционной массы удаляют в токе сухой углекислоты на водоструйном насосе следы треххлористого фосфора, а остаток в колбе перегоняют в вакууме, собирая фракцию с т. кип. 120°/1,5 мм.
Выход дихлорангидрида 0, 0, 0-трис (хлорметил) этилфосфо-ристой кислоты равен 234,5 г (80%); n^° = l,5320; d^0 — 1,5298.
Найдено, %: Р—10,14; 10,21; Мцо 59,08.
CjHsCIsOP. Вычислено, % Р—10,59; MRo 59,40.
Получение дихлорангидрида 0. 0, 0-трис (хлорметил)этил-фосфористой кислоты из 3,3-бис(хлорметил)оксациклобутана проводят аналогичным образом: к 155 г (1 М) 3,3-бис(хлор-метил)оксациклобутана прибавляют по каплям 137 г (1 М) треххлористого фосфора и нагревают 10 часов при 80°. Выход продукта равен 219 г (75%').
Дихлорангидрид 0, 0, p-трис(хлорметил) этилфосфористой кислоты — прозрачная бесцветная жидкость с резким запахом, хорошо растворяется в органических растворителях, легко гидролизуется водой.
Примечание. Повышение температуры и более длительное нагревание реакционной массы может привести к побочным реакциям и образованию смеси продуктов.
ЛИТЕРАТУРА
1 Е В. Кузнецов, Л. А. Власова. Ж. общ. химии, 39, 698 (1969).
2. A. Mooradian, I. G 1 о k. J. Amer.. Chem. Soe., 67, 942 (1945).
3. Мономер для пентапласта. М.-Л., «Химия», 1965.
н-ДОДЕЦИЛОВЫЙ ЭФИР ГАЛЛОВОЙ КИСЛОТЫ
н-Додецилгаллат, лаурилгаллат
Р. М ГЕЛЬШТЕЙН, Е. И. МАХОВЕР
СООС12Н25
НО-1 J-- он он
С19Н30О5
М. в. 338,44
Додециловый эфир галловой кислоты, как и другие эфиры высших спиртов галловой кислоты, применяется в качестве антиокислителя жиров.
Эфиры галловой кислоты высших спиртов могут быть получены прямой этерификацией галловой кислоты избытком спирта в присутствии катализатора (серная кислота, п-толу-олсульфокислота) и растворителя (бензол, толуол, ксилол) с отгонкой образующейся воды в виде азеотропной смеси [1, 2] или взаимодействием галлоидхлорида со спиртом [1].
Нами усовершенствована методика синтеза и очистки до-децилгаллата: этерификация галловой кислоты додециловым спиртом проводится без растворителя при пониженном давлении в присутствии /1-толуолсульфокислоты. Технический продукт очищают перекристаллизацией из толуола.
СХЕМА СИНТЕЗА
соон
С12Н25ОН
СООС12Н25
I /\ но-J J-oh
I
ОН ш
Характеристика основного сырья
Галловая кислота, ТУ МХП 2726—55, ч.
н-Додециловый спирт, МРТУ 6—09—3735—67, ч. n-Толуолсульфокислота, ТУ МХП ОРУ 73—56, ч.
Толуол, ГОСТ 5789—51, ч. д. а.
Уголь активированный, ГОСТ 4453—48.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную термометром, мешалкой с затвором из резиновой трубки и елочной колонкой, помещают 170 г (1 М) безводной галловой кислоты (или 188 г одноводной), 400 г (2,15 М) н-додецилового спирта и 15 г (0,09 М) n-толуол сульфокислоты.
Отгонку воды ведут при пониженном давлении (25—40мм), постепенно за 1 час поднимая температуру реакционной массы от 80° до 140—155°, причем галловая кислота полностью растворяется. После 30-минутной выдержки при 150—155° реакционную массу выливают в 400 мл толуола, охлажденного до 5°. После охлаждения смеси до 7—10° осадок отфильтровывают с отсасыванием и сушат на воздухе.
Выход н-додецилового эфира галловой кислоты равен 340—345 г (102%, продукт, содержит додециловый спирт).
После перекристаллизации из 1380 мл толуола с применением активированного угля получают 270 г н-додецилгал-лата (79,5%); т. пл- 96—97°, содержание основного вещества 99% (определено методом осаждения в виде висмутовой соли); реакция — нейтральная, хлориды и сульфаты отсутствуют.
По литературным данным [1], т. пл 95—96°; 96—97°.
ЛИТЕРАТУРА
1. G. I. М. Van d е г Kern, 1. Н. Verbee k, I. С. F. Cleton. Recueil des Travaux chiiriques des Pavs — Bas, 3, 277 (1951).
2. Пат. США 2623897 (1952); С. A., 45, 7596 (1951).
п- и щ-КСИЛ ИЛЕНДИАМИНО М, \ БИС(ИЗОПРОПИЛ-ФОСФОНОВЫЕ КИСЛОТЫ)
М. В. РУДОМИИО, Е. К. КОЛОВА, Т. Я. МЕДВЕДЬ
(НО)2Р — С — NHCH2 — { —CH2NH — с — Р (ОН)2
II /\ \=/ /\ п
ОСН3 СНз _ СН3СН3О
(НО)2Р —с —nhch2—
II /\ (
О СНз СНз CH2NH —С —Р(ОН)2
II
СН3 СН3О
Ci4H26N2OsP2 М. в. 380,32
п- и лг-Ксилилендиамино-N, N'-бис (изопропилфосфоновые кислоты) являются новыми фосфорорганическими комплексонами с повышенной жесткостью цепи, соединяющей имино-диалкилфосфоновые группировки.
Обе кислоты получены нами впервые омылением их этиловых эфиров концентрированной соляной кислотой. Образующиеся при этом промежуточные хлоргидраты кислот превращались в свободные кислоты действием окиси пропиле-11а СХЕМА СИНТЕЗА
(C2H5O)2P-C-NHCH2
I! /\
О СНз СНз
= CH2NH - С - P(OC2HJ2 /\ I!
сн3сн3о
н3с СН3О
\/ II
—CH2--NH-C — Р(ОН)2
Г и
НС)
н2о"
СН2 — NH — С —
H3CZ сн3
Р(ОН)2
II
о
пНС1 ->
-НС1
1192
(НО)2Р — С — NHCFL-II /\ О СНз сн3
CH2NH — с —Р(ОН)2 /\ II СН3СНВО
из
Характеристика основного сырья
Этиловый эфир n-ксилилендиаминобис (изопропилфосфоновой кислоты), т. пл. 64—65°, получение см- стр. 228 этого сборника.
Этиловый эфир л-ксилилендиаминобис (изопропилфосфоновой кислоты), г. пл. 41—42°, получение см. стр. 228 этого сборника.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт, гидролизный, ГОСТ 8314—57.
Окись пропилена, т. кип. 36°.
Условия получения
Синтез дихлоргидрата п-ксилилендиаминобис(изопропил-фосфоновой кислоты). Раствор 20,4г (0,04144) этилового эфира п-ксилилендиаминобис (изопропилфосфоновой кислоты) в 40 мл концентрированной соляной кислоты кипятят с обратным холодильником 6 часов. Затем добавляют активированный уголь и кипятят еще 0,5 часа, уголь отфильтровывают, от фильтрата отгоняют избыток соляной кислоты при пониженном давлении, многократно добавляя воду (5x100 мл). Остаток растирают с этиловым спиртом до превращения в порошкообразное вещество.
Выход дихлоргидрата n-ксилилендиаминобис(изопропилфосфоновой кислоты) равен 13,9 г (75%), т. пл. 198—200°.
Найдено, %: О —12,7; 12,9.
СиНгбИгОвРг 2HCI. Вычислено, %: С1 —12,6.
Получение п- ксилилендиаминобис(изопропилфосфоновой кислоты). В четырехгорлой колбе, снабженной мешалкой, капельной воронкой, термометром и холодильником, к раствору 13 г (0-02941) дихлоргидрата в 85 мл 50%-ного этилового спирта прибавляют по каплям при перемешивании раствор^3,2 мл окиси пропилена в 7 мл спирта, поддерживая охлаждением водой температуру реакционной смеси 20±2°. Осадок отфильтровывают, растворяют в 150 мл воды, нагревают до кипения, фильтруют через воронку Шотта № 3, затем осторожно прибавляют ацетон до начала кристаллизации. После 12-часового стояния отфильтровывают бесцветное кристаллическое вещество и сушат на воздухе при 35°.
Выход н-ксилилендиаминобис(изопропилфосфоновой кислоты) равен 4,6 г (38%); т. пл. 215—218°.
Найдено, %: С—40,0; 39,8; Н—7,5; 7,4; N — 7,0; 7,1; Р—13,9; 14.0; Н2О — 8,4.
С14Н2ьМ20вРо-2Нг0. Вычислено, %: С — 40,5; Н — 7,2; N—6.7; Р — 14,9; Н2О — 8,7.
Получение монохлоргидрата м-ксилилендиаминобис(изо-пропилфосфоновой кислоты). Раствор 7,8г (0,016 44) этилово
114
го эфира лг-кислоты в 18 мл концентрированной соляной кислоты кипятят 7 часов, затем еще 15 .минут с активированным углем, последний отфильтровывают, фильтрат упаривают при пониженном давлении, многократно добавляя воду (5x50 мл). Остаток растворяют в 15 мл этилового спирта. Спустя двое суток выпавший кристаллический осадок отфильтровывают и сушат в вакуум-эксикаторе над пятиокисью фосфора.
Выход монохлоргидрата л-ксилилендиаминобисизопропил-фоофоновой кислоты равен 4,4 г (66%), т. пл. 185—193° (раз-лож.)
Найдено, %: С — 39,6; 39,9; Н — 6,4; 6,6; N— 7,2; 7,1;
Р — 14,6; 14,3; С1 -8,0; 7,8
CI4H20N2O6P2.HCl. Вычислено, %; С —40,3; Н — 6,5; N —6,7; Р — 14,8;
С1 — 8,5.
Получение м-ксилилендиаминобис(изопропилфосфоновой кислоты). К раствору 2,2 г (0,0053М) монохлоргидрата ж-кис-лоты в 15 мл 50%-ного этилового спирта прибавляют при перемешивании по каплям раствор 0,6 мл окиси пропилена в 10 мл спирта, поддерживая охлаждением водой температуру 20±2°. Осадок отфильтровывают, растворяют в 25 мл горячей воды и осторожно прибавляют ио каплям — 12 мл ацетона до начала кристаллизации. Спустя 2 дня отфильтровывают бесцветный кристаллический осадок и сушат на воздухе при 35°.
Выход м-ксилилендиаминобис (изопропилфосфоновой кислоты) равен 0,9 г (41%), т. пл. 212—215°.
Найдено, %: С — 39,9; 39.7; Н —7,2; 7,2; N-7,0-7,1; Р—14,4; 14,7, Н2О —8,3.
C14H26N2O6P2.2H2O. Вычислено, %: С — 40,5; Н — 7,2; N—6,7;
Р — 14,9; Н2О — 8,7.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я Медведь и др. Изв. АН СССР, сер .хим. 1501 (1967).
8*
о -МЕТАКРИЛАМИДОБЕНЗОЙНАЯ КИСЛОТА
С . И. КОТЕНКО, А. Г. ФАДЕИЧЕВА, И. С. ТРОХИМЕНКО
сн3
CH2 = C-C-NH—€
II
0 соон
CnHnNOs
М. в. 205,02
о-Метакриламидобензойная кислота применяется для синтеза ряда водорастворимых высокомолекулярных соединений, которые могут быть использованы как реактивы на тяжелые металлы.
В литературе описан способ получения о-метакриламидо-бензойной кислоты, основанный на ацилировании антраниловой кислоты хлор ангидридом метакриловой кислоты [1].
При проверке этой методики мы, используя в качестве реакционной среды хлороформ, а в качестве акцептора хлористого водорода триэтиламин, значительно увеличили выход продукта.
СХЕМА СИНТЕЗА
сн3 сн.(
сн2 - - С - С - С1 - II О H2N — сна= С - CNH соон ° соон
Характеристика основного сырья
Антраниловая кислота, ТУ 79П 686—61, ч.
Хлорангидрид метакриловой кислоты, перегнанный, т. кип.
97—99’, получение см. [2].
116
Триэтиламин, ГОСТ 9966—66, ч., перегнанный.
Хлороформ, ГОСТ 3160—51, ч., перегнанный над хлористым кальцием.
Соляная кислота, ГОСТ 3118—67, ч., пл. 1,18.
Этиловый спирт ректификованный, ГОСТ 5962—67.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную капельной воронкой, мешалкой и обратным холодильником, помещают 13,7 г (0,1 М) антраниловой кислоты, 20,2 г (0,2 М) триэтиламина и 50 мл хлороформа. Смесь охлаждают водой со льдом до 5—0° и по каплям, за 1,5—2 часа прибавляют 10,4 г (0,1 М) хлорангидрида метакриловой кислоты в 100-ил хлороформа. Массу перемешивают еще 1 час, прибавляют 2— 3 мл триэтиламина и оставляют на ночь.
К содержимому колбы прибавляют 150 мл воды, интенсивно перемешивают и избыточный триэтиламин нейтрализуют 10%-ной соляной кислотой (около 50 мл), доводя pH водного слоя до 2—3. Хлороформный слой отделяют и упаривают досуха (см. примечание). Остаток перекристаллизовывают из 400 мл водного (1 :3) этилового спирта.
Выход о-метакриламидобензойной кислоты равен 16 г (77%); т. пл. 170°.
По литературным данным [1], т. пл. 169—170°.
Найдено, %' N — 6,45; 6,51.
СнНпNO2. Вычислено , %: N — 6,83.
Примечание. Из водного слоя триэтиламин может быть регенерирован упариванием, подщелачиванием и отгонкой.
ЛИТЕРАТУРА
I. Г М. Четы рк нн а, Т. А. Соколова, М. М. К ото и. Высокомолекулярные соединения, 1, 248 (1959).
2. М. М. Котой, Т. А. Соколова, Г. М. Четы р к и н а. Ж. обит .химии, 27, 185 (1957).
D,L-N МЕТИЛВАЛИН
Г. Н. КОШЕЛЕВА, Э. А. БАШКИР, В. И. КАЛМЫКОВ
СНз
;сн —сн —соон СНз
NHCH3
c6h13no2
М. в. 131,18
D, L-N-Метилвалин применяется в синтезах пептидов и депсипептидов. В литературе описан метод получения метил-валина из рацемической а-бромизовалериановой кислоты и метиламина в водном растворе [1] или в избытке жидкого метиламина [2]. В данной работе использован метод с жидким метиламином {2], а для выделения продукта применен метод ионообменной колоночной хроматографии, что позволило устранить на стадии очистки абсолютный спирт и получить конечный продукт без примеси брома.
СХЕМА СИНТЕЗА
ч ch3nh2
/СН —сн —соон ----------
I
Вг
СНз
> ^сн —сн —соон
СНз NHCHs
Характеристика основного сырья
а-Бромизовалериановая кислота, ВТУ РУ 638—52, ч.
Метиламин,хлоргидрат, ТУ ОРУ 3—58, ч.
Аммиак водный 25%'-ный, ГОСТ 3760—64, ч.д.а.
Ионообменная смола КУ-2, ВТУ ГХПК М—661—55.
118
Условия получения
Получение жидкого метиламина. Помещают 200 г (5 Л1) едкого натра в литровую вращающуюся колбу 1 (см. рисунок) роторного испарителя 2, соединенного с обратным холодильником 4. Колбу с едким натром устанавливают на кипящей водяной бане 11, включают вращение и во вращающуюся колбу добавляют к щелочи из капельной воронки 5 раствор 250г
Схема прибора для получения жидкого метиламина.
1 — колба с NaOH; 2 — ротор; 3 — приемник для воды; 4— обратный холодильник; 5— капельная воронка с раствором гидрохлорида метиламина; 6— колонка с сухим КОН; 7 — ловушка; 8—хлоркальциевая трубка; 9 — приемник для жидкого метиламина; 10 — ацетоновая баня; // — водяная баня
(3,7 Л1) хлоргидрата метиламина в 0,5 л воды. Выделяющийся газообразный метиламин пропускают через обратный холодильник 4 и осушительную колонку 6 (высота 0,8 .и), наполненную гранулированным едким кали (0,8 кг). Затем его конденсируют в ловушке 7, охлаждаемой смесью сухого льда и ацетона, н собирают в приемник 9, охлаждаемый до минус 15—20°.
Выход жидкого метиламина равен 100 г (83%).
Получение D, L-N-метилвалина. В предварительно охлажденный до минус 30—40° автоклав загружают 275 г (1,5 М) а-бромизовалериановой кислоты и 420 г (13,3 А1) жидкого
119
метиламина. Автоклав закрывают, реакционную смесь перемешивают 48 часов при комнатной температуре, затем ее выгружают, упаривают досуха на водяной бане, сушат при 100° в течение ночи. Получают 325 г сухого остатка.
Сухой остаток растворяют в 1200 мл дистиллированной воды и раствор пропускают через колонку высотой 1,4 м, диаметром 80 мм, наполненную 3 л водной суспензии смолы КУ-2 (см. примечание 1). Затем промывают смолу 60 л дистиллированной воды до получения нейтрального элюата, после чего вымывают метилвалин 50 л 1 %'-ного водного раствора аммиака (см. примечание 2). Элюат собирают фракциями объемом по 3 л. В них определяют содержание метилвалина реакцией с нингидрином (см. примечание 3). Фракции, содержащие метилвалин (40—45 л), упаривают при пониженном давлении (15 мм) до объема 6 л. Раствор нагревают с углем, фильтруют через складчатый фильтр ц упаривают досуха при пониженном давлении (10—20 мм) на роторном испарителе.
Выход метилвалина равен 130 г (80—81%'); т. пл. 300° (возг.). По данным элементарного анализа, продукт не содержит брома.
По литературным данным {2], т. пл. вещества 300° (возг.).
Примечания.
1. Смолу КУ-2 до начала работы и для регенерации промывают 60 л 5%-ной соляной кислоты, а затем 60 л дистиллированной воды.
2. Вместо раствора аммиака можно применять 5%-ный раствор пиридина.
3. На фильтровальную бумагу наносят несколько капель элюата, высушивают, опрыскивают 1%-ным спиртовым раствором нингидрина и сушат 30 минут в шкафу при 100°. При положительной реакции появляется синее окрашивание пятна.
ЛИТЕРАТУРА
1. R. Hindetling, В. Prijs, Н. Erlenmejer. Helv. chim. acta, 38. 1415 (1955),
2. Ю. А. О в ч и и и и к о в, В. Т. Иванов, А. А. К и р ю ш к и н. Изв. АН СССР, сер. хим., И, 2046 (1962).
Р МОНОБУТИЛОВЫЙ ЭФИР ИТАКОНОВОЙ кислоты р-Монобутилитаконат
Р. А. МЕЛНГАЛВЕ, А, Г. КАИНОВА, С. Б. МАКАРОВА, Е. В. ЕГОРОВ
СН2 = С — СН2СООС4Но
I
СООН
С9Н14О1
М. в. 186,20
Монобутилитаконат является исходным мономером для синтеза полиитаконовой кислоты, различных сополимеров, ионообменных смол.
Обычно монобутилитаконат получают этерификацией итаконовой кислоты бутиловым спиртом. Однако синтезированный продукт содержит около 20% диэфира, причем монобутилитаконат состоит из смеси а- и р-моноэфиров [1]. Описан способ получения р-монометилитаконата, основанный на взаимодействии ангидрида итаконовой кислоты с метиловым спиртом; продукт содержит до 10%' диэфира [2].
Нами уточнены условия синтеза р-монобутилитаконата по методу [2] и предложен способ его очистки экстракцией диэтиловым эфиром.
СХЕМА СИНТЕЗА
СН2- с=о
СН2 = С +С4Н9ОН -> СН2 = С — СН2СООС4НЭ
ХС - О СООН
II
о
121
Характеристика основного сырья
Ангидрид итаконовой кислоты, ТУ TCP—927 р—62, ч. н-Бутиловый спирт, ГОСТ 6006—51, ч.
Этиловый эфир, ГОСТ 6265—52, ч.д.а.
Условия получения
В двухгорлую колбу емкостью 1 л, снабженную мешалкой, обратным холодильником и помещенную в глицериновую баню, загружают 390 г (3,5 М) итаконового ангидрида и 365 мл (4 ЛГ) н-бутилового спирта. Реакционную смесь интенсивно перемешивают при 115—120° в течение 8 часов. После охлаждения до комнатной температуры реакционную массу выливают в стакан и нейтрализуют 750 мл 55—60%-ного раствора углекислого калия (или углекислого натрия). Затем водный раствор экстрагируют этиловым эфиром дважды порциями по 250 мл. Водный слой отделяют, подкисляют концентрированной соляной кислотой до pH 4—5 и экстрагируют мо-нобутилитаконат эфиром три раза порциями по 250мл. Эфирные вытяжки собирают, эфир отгоняют. Остаток в перегонной колбе при охлаждении закристаллизовывается.
Выход р-монобутилитаконата равен ПО г (58—62%); т. пл. 32—33°; содержание основного вещества 99,0—99,5%.
По литературным данным [3], т. пл. 32—35°.
Для оценки чистоты р-монобутилитаконата использован метод тонкослойной восходящей хроматографии на окиси алюминия. Система растворителей: 10 мл хлороформа +5 мл бензола. Проявление йодом. Обнаружено одно пятно с 7?z — = 0,10.
По данным [1], для монобутилитаконата Rf =0,11; для бутилитаконата /?у=0,82.
Примечание.
Отделение дибутилитаконата от основного продукта основано па том, что диэфир растворим в этиловом эфире, а калиевая соль монобутилита-чоната в эфире не растворяется, по растворяется в воде.
ЛИТЕРАТУРА
1. М. А. Аскаров, Л. И. Семенова, Узб. хим. ж„ 6, 42 (1967).
2. В. R. Baker. J. Organ. Chem., 17, 116 (1952).
3. Пат. США 3484478 (1969).
2- и 4-НИТРО(3-БРОМПРОПИЛ)БЕНЗОЛЫ
В. М. ОСТРОВСКАЯ, Н. Е. КАШИРИНА
СН2СН2СН2Вг
СН2СН2СН2Вг
I
//\
I II
ч/
I
no2
СдНюВгЫОг
М. в. 244,09
2-Нитро (3-бромпропил) бензол и 4-нитро(3-бромпропил)-бензол образуются при нитровании (3-бромпропил) бензола азотной кислотой [1, 2]. Получены орто-изомер с njj = 1,5730 и пара-изомер с «д =1,5780, «д =1,5792 l[lj. По данным [2], при нитровании (3-бромпропил)бензола дымящей азотной кислотой образуется пара-изомер с выходом 82 %, по = = 1,5760.
Нами эти данные проверены и показано, что фракцию с показателем преломления пд = 1,5760 следует считать параизомером с примесью орто-изомера (установлено при искусственном смещении изомеров), а не чистым пара-изомером. Предложена методика нитрования (3-бромпропил) бензола азотной кислотой, пл. 1,475.
СХЕМА СИНТЕЗА
С3Н6Вг С3Н6Вг СзНеВг
' J 1
HNO3 n°2 +
2 I II -------I II I II
4/ 4/
I
no2
123
Характеристика основного сырья
(З-Бромпропил) бензол, т. кип. 95—9674 мм; получение см. на стр. 53 этого сборника.
Азотная кислота дымящая, техн., пл. 1,52.
Азотная кислота, ГОСТ 4461—67, ч.д.а.
Диэтиловый эфир, ГОСТ 6265—52, ч.
Кальций хлористый плавленый, ГОСТ 4460—66.
Условия получения
В трехгорлую колбу, снабженную мешалкой, капельной воронкой и термометром, помещают 313 мл (6,2 Л4) азотной кислоты, пл. 1,475 (готовят разбавлением дымящей азотной кислоты азотной кислотой, пл. 1,42), охлаждают до минус 19° смесью ацетона с твердой углекислотой и добавляют по каплям при размешивании 63 г (0,31 М) (3-бромпропил)бензола, поддерживая температуру минус 15°. Раствор размешивают 2 часа (в первый час температура поднимается до минус 8° во второй час—до 0°) и вливают в 1,1 л воды.. Отделяют выделившееся масло,экстрагируют его 500 мл диэтилового эфира. Водный слой промывают 50 мл эфира. Эфирные экстракты промывают водой 2 раза порциями по 200 мл, 10%-ным раствором бикарбоната натрия до pH 7, отделяют, сушат над плавленым хлористым кальцием. Эфир отгоняют, остаток медленно перегоняют в вакууме с дефлегматором, собирая фракцию орто-изомера с т. кип. 153—15772 мм, Лд = 1,5736, Лд = 1,5740; фракцию, представляющую смесь орто- и пара-изомеров (32 г) с т. кип. 158—165°/2 мм, fi'o = 1,5758, Лд =1,5763 и фракцию пара-изомера (26 г) с т. кип. 166—17072 мм, Лд = 1,5794, =1,5800. После раз-
гонки с дефлегматором средней фракции (обычно от нескольких опытов) получают еще 10 г орто-изомера с /г2д = 1,5740 и 10 г пара-изомера с =1,5790.
Выход орто-изомера равен 23 г (30%), пара-изомера—36 г (47%). Общий выход нитропродукров равен 71 г (92%).
По литературным данным, орто-изомер имеет т. кип. 11470,75 мм, 138—144°/2 мм, По= 1,573; пара-изомер имеет т. кип. 130—13670,4 мм, 138—14070,75 мм, 156—160°/2 мм, nf = 1,5780; «2д = 1,5792 [1]; т. кип. 152—156°/2 мм, ria -= 1,576 [2].
Л ИТЕРАТУРА
1. М. Davis, J. J. Roberts, W. С. J. R о s s. J. Chem. Soc.’ 1955, 890; C. A., 50, 1653 i (1956).
2. M. T. L ef f 1 e r., E. H. Volwiler. J. Amer. Chem. Soc. 60,896 (1938).
3. J. Braun, H. Deutsch. Ber., 45,2509 (1912).
124
5-НИТРО-4.6-ДИМЕТОКСИСАЛИЦИЛОВЫЙ АЛЬДЕГИД
Д. А. ДРАПКИНА, Г. С. ПАНОВА
сно
I
НзСО он
х|
ОСНз
c9h9no6
М. в. 227,18
5-Нитро-4,6-диметоксисалициловый альдегид описан лишь в одной работе |[1], где он получен нитрованием 4,6-димето-ксисалицилового альдегида концентрированной азотной кислотой в среде ледяной уксусной кислоты при температуре около 38°.Длительность реакции и выход продукта не указаны.
Мы проверили и уточнили этот метод; кроме того, на стадии очистки этанол заменили бензолом, что позволило сократить потери альдегида при кристаллизации и улучшить его качество.
СХЕМА СИНТЕЗА
СНО
I
НзСО -f\ — он
HNO3
СН3СООН
I
ОСНз
сно
НзСО -^\-он o2n -М
ОСНз
125
Характеристика основного сырья
4,6-Диметоксисалициловый альдегид, т. пл. 68—71°; получение см. [1].
Азотная кислота, ГОСТ 701—68, техн., пл. 1,51 —1,52.
Уксусная кислота, ГОСТ 61—69, х. ч.
Бензол, ГОСТ 5955—68, ч. д. а.
Условия получения
В колбу емкостью 100 мл, снабженную мешалкой, термо-метром, капельной воронкой и воздушным холодильником, помещают 3,27 г (0,018М) 4,6-диметоксисалицилового альдегида и 60 мл ледяной уксусной кислоты. К полученному раствору за 5—10 минут приливают из капельной воронки раствор 3,1 мл (0,074 М) азотной кислоты в 10 мл ледяной уксусной кислоты. Затем смесь, размешивая, нагревают на водяной бане при 40—50° в течение 1 часа, охлаждают до комнатной температуры и выливают на 200 г толченого льда. Выпавший осадок отфильтровывают, промывают водой до нейтральной реакции.
Выход 5-нитро-4,6-диметоксисалицилового альдегида равен 2,2—2,4 г (52—59%); т. пл. 185—192°; вещество окраше-но'почти в черный цвет.
После кристаллизации из бензола (70 мл на 1 г вещества) в присутствии активированного угля получают 1,3—1,6 г (32—40%) чистого продукта в виде кристаллов, слегка окрашенных в песочный цвет с т. пл. 193—194°; после повторной кристаллизации из бензола т. пл. 195—196°.
По литературным данным (1J, т. пл. 192—193°.
ЛИТЕРАТУРА
1. Англ. пат. 1029230; С. А., 65, 2235 h (1966).
НИТРОКСАМИНАЗО
Пал ладион; 2'-сульфо-4'-нитрофенол-<6-азо-3>-7--4-аминонафталин-2,7-дисульфокислота
Ю. М. ДЕДКОВ
SO3H
C^HisNiO^Ss
М. в. 548,47
Нитроксаминазо(палладион) предложен в качестве специфического реагента для определения палладия [1, 2]. Его получают азосочетанием диазотированной 2-амино-4-нитрофе-нол-6-сульфокислоты с кислотой Фрейнда в кислой среде.
СХЕМА СИНТЕЗА
127
Характеристика основного сырья
2-Амино-4-нитрофенол-6-сульфокислота, техн.
1-Нафтиламин-3,6-дисульфокислота (кислота Фрейнда); МРТУ 6—09—3933—67, ч.
Натрий азотистокислый, ГОСТ 4197—66, ч.
Условия получения
Очистка 2-амино-4-нитрофенол-6-сульфокислоты. Растворяют 500 г технической кислоты в 0,5 л воды, добавляя необходимое количество углекислого натрия для достижения рг!^4, и раствор фильтруют с отсасыванием через стеклянный фильтр № 2. В фильтрат вносят 70—80 г хлористого натрия, нагревают на водяной бане до 60° и подкисляют разбавленной (1:1) соляной кислотой до pH 2. О достижении необходимой кислотности судят по отчетливому изменению окраски раствора от густой коричнево-красной (рН>4) до светло-желтой (рН<:2). Фильтрат охлаждают до 5е и выделившуюся смесь 2-амино-4-нитрофенол-6-сульфокислоты и ее натриевой соли отфильтровывают с отсасыванием, промывают на фильтре 50 мл этанола и суспендируют в 250 мл воды. Смесь нагревают до 60°, к суспензии приливают 50 мл соляной кислоты (пл. 1,19), охлаждают до 5°. Осадок отфильтровывают с отсасыванием, промывают на фильтре 50 мл. этанола, затем. 50 мл смеси (1 : 1) бензол—этанол и сушат в вакууме водоструйного насоса при 105°. Содержание основного вещества—85% в пересчете на кислоту—находят, титруя точную навеску продукта раствором азотистокислого натрия в кислой среде.
Очистка кислоты Фрейнда. Растворяют при нагревании 500 г кислоты в минимальном количестве воды, раствор нейтрализуют углекислым натрием до pH 5—6, фильтруют через плотный бумажный фильтр, добавляют сухой хлористый натрий из расчета 10 г на каждые 100 мл раствора, подкисляют соляной кислотой (пл. 1,19) до pH 2—3, после чего раствор охлаждают и выделившуюся смесь кислоты Фрейнда и ее натриевых солей отфильтровывают с отсасыванием, промывают 50 мл этанола и сушат в вакууме водоструйного насоса при 60°. Содержание основного вещества — 90%' в пересчете на кислоту—находят, титруя навеску раствором азотнокислого натрия.
Получение нитроксаминазо. Растворяют 29 г (0,10 АГ) 80%-ной 2-амино-4-нитрофенол-6-сульфокислоты в 150 мл 30%-ного водного пиридина, добавляя в случае неполного растворения углекислый натрий, и приливают соляную кислоту (пл. 1,19) до pH 2—3 по универсальной индикаторной бумажке. Суспензию тщательно перемешивают и охлаж-128
дают до 10° и, продолжая перемешивание и охлаждение, к ней приливают за 30 минут раствор 6,9 г (0,10 Л1) азотистокислого натрия в 70 мл воды,следя за тем, чтобы реакция среды все время оставалась отчетливо кислой. Через 20 минут пробой с йодкрахмальной бумагой убеждаются в наличии избытка нитрит-ионов. Еще через 30 минут избыток азотистой кислоты снимают, внося необходимое количество исходного амина (проба с йодкрахмальной бумагой).
Растворяют 60 г (0,12 М) 66%-ной кислоты Фрейнда в 100 мл 30%-ного водного пиридина и добавлением соляной кислоты (пл. 1,19) устанавливают pH раствора в пределах 3—4 (универсальная индикаторная бумага). Полученный раствор нагревают на водяной бане до 45“ и при постояннном интенсивном перемешивании за 1 час порциями по 10 мл вносят суспензию диазосоли 2-амино-4-нитрофенол-6-сульфо-кислоты. Реакция среды в ходе всего процесса должна оставаться отчетливо кислой, причем, кислотность раствора постепенно повышается до pH 1—2.
Реакционную массу перемешивают еще 1 час, в случае необходимости подкислением соляной кислотой (пл. 1,19) устанавливают pH смеси-хЛ по универсальной индикаторной бумаге и выдерживают 2 часа при 60°. Затем в смесь вносят хлористый натрий из расчета 20 г на каждые 100 мл смеси, охлаждают и оставляют на ночь.
На следующий день смесь натриевых солей нитроксами-назо отфильтровывают с отсасыванием, суспендируют в 300 мл воды, добавляют 1 г солянокислого гидроксиламина, затем 56%-ный раствор едкого кали (10—15 мл) до полного растворения осадка. Раствор фильтруют, водой доводят объем до 400—500 мл и вносят 100 г хлористого натрия, после чего нагревают на водяной бане до образования раствора. К нему приливают 30 мл соляной кислоты (пл. 1,19), смеси дают остыть до комнатной температуры и через 2 часа отфильтровывают осадок.
Полученный реагент проверяют на наличие продуктов разложения диазосоли, для чего хроматографируют на бумаге с применением в качестве элюанта 5%-ного раствора уротропина. При наличии на хроматограмме нескольких окрашенных зон переосаждение повторяют до получения однородного вещества. Реагент сушат в вакууме водоструйного насоса, постепенно повышая температуру с 60° до 105°. Полученный таким образом препарат пригоден для аналитических целей. Содержание основного вещества определяют спектрофотометрически: в 0,5 н. НС1 при 482 нм молярный коэффициент погашения нитроксаминазо равен 18,5-103.
Выход нитроксаминазо равен 45 г (80% в расчете на кислоту); содержание основного .вещества не ниже 70%. 9-1192 129
Для получения продукта в Н-форме 500 мл 0,1%-ного раствора пропускают со скоростью 2 капли в минуту через колонку, заполненную 100 г катионита КУ'2 в Н-форме, и полученный деминерализованный раствор упаривают в вакууме водоструйного насоса при 20°. Остаток сушат при 105°.
Найдено, %: N — 9,96; 10,05; 10,15.
CjeHj^NjO^Sg. Вычислено, %: N—10,21.
Л ИТЕРАТУРА
1. Ю. М. Дедков, А. Н. Ермаков, А. В Котов. Докл. АН СССР. 185, 3591 (1969).
2. Ю. М. Д е д к о в, А. Н. Ермаков. Авт. свид. 261767 (1968); Изобретения. Промобразцы. Товарные знаки, № 5 (1970).
4-НИТРО-2-МЕТОКСИБЕНЗАЛЬДЕГИД
Л. С. ЗЕРЮКИНА, Б. М. БОЛОТИН
СНО ^\-ОСНз
no2
C8H7NO4
М.в.181,15
По литературным данным, 4-нитро-2-метоксибензальде-гид был получен окислением 4-нитро-2-метокситолуола хромовым ангидридом fl], причем, выход продукта не указан.
Мы получили это соединение метилированием 4-нитроса-лицилового альдегида диметилсульф атом.
СНО
/ он
II I (CH3)2so^
no2
СНО
^\-ОСН3
Ч/
NO2
Характеристика основного сырья
4-Нитросалициловый альдегид, т. пл. 135—136°, получение см. [2].
Диметилсульф ат, МРТУ 6—09—3688—67, ч.
9*
131
Условия получения
ВНИМАНИЕ! Синтез необходимо проводить в хорошо работающем вытяжном шкафу, чтобы избежать воздействия ядовитых паров диметилсульфата.
В колбе емкостью 100 мл, снабженной мешалкой и термометром, растворяют при 80° 3,34 г (0,02 Л4) 4-нитроса-лицилового альдегида в 10 мл (0,02 М) 10%-ного водного раствора едкого кали. К раствору прибавляют при той же температуре за 30 минут 1,8 мл (0,02 М) диметилсульфата. Реакционный раствор перемешивают до исчезновения окраски, добавляют еще 5 мл (0,01 Л4| 10%-ного раствора едкого кали и 0,9 мл (0,01 М) диметилсульфата, опять перемешивают до исчезновения окраски и вновь прибавляют 3 мл щелочи и 0,5 мл диметилсульфата. После 15-минутной выдержки добавляют еще 2 мл щелочи и 0,4 мл диметилсульфата, массе дают остыть и экстрагируют в 2 приема 100 мл эфира. Эфирный экстракт промывают 20 мл 2 н. раствора едкого кали, высушивают над хлористым кальцием. Растворитель отгоняют при пониженном давлении, а остаток промывают 10 мл спирта.
Выход 4-нитро-2-метоксибензальдегида равен 1,3 г (40%); т. пл. 120—121°.
По литературным данным |[1], т. пл. 120—122°.
Л ИТЕРАТУРА
1. W. Berends, 4. М. W a i s v i s z, А. К. I. Mulder. Rec. trew. chim., 74, 1323 (1955); РЖХим., Il, 32448 (1956).
2. E. M. Bavin, R. J. Rees et al., J. РПггт, and Pharmazol., 2,764 (1950).
5-НИТРО р-РЕЗОРЦИЛОВЫЙ АЛЬДЕГИД
4-Окси-5-нитросалициловый альдегид;
2,4-диокси-4-нитробензальдегид
Д. А. ДРАПКИНА, Н. М. РОЛДУГИНА, Н. И. ДОРОШИНА
СНО
OsN-IJ
ОН
c7h5no5
М. в. 183,12
5-Нитро-р-резорциловый альдегид был получен [1] нитрованием p-резорцилового альдегида дымящей азотной кислотой в среде ледяной уксусной кислоты без указания выхода.
При воспроизведении данного метода нами получен продукт с выходом 16—25%, отвечающий по температуре плавления и внешнему виду литературным данным [1], однако, реакция нитрования протекала очень бурно, иногда с выбросом реакционной массы.
Мы применили для нитрования p-резорцилового альдегида разбавленную азотную кислоту и уточнили условия нитрования, в результате чего удалось стабилизировать выход прокачество и добиться спокойного проте-
СХЕМА СИНТЕЗА
СНО
I
HNO3 МГ°Н сн8соон* o2n-MI
I
он
дукта, повысить е кания реакции.
СНО
Ы1 ~~он
X/
ОН
133
Характеристика основного сырья
2,4-Диоксибензальдегид, МРТУ 6—09—3707—67, ч. Азотная кислота, ГОСТ 4461—67, ч., пл. 1,42.
Уксусная кислота, ГОСТ 61—69, ч. д. а.
Этиловый спирт, ректификованный, ГОСТ 5962—67.
Условия получения
В колбу емкостью 100 мл, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, помещают 4,14 г (0,03 Л4) 2,4-диоксибензальдегида и 15 мл ледяной уксусной кислоты. К суспензии при размешивании прибавляют за 0,5 часа 3,70 мл (0,06 А4) азотной кислоты в 15 мл ледяной уксусной кислоты, поддерживая температуру (в случае необходимости слабым охлаждением) в колбе не выше 45°. Тепло при этом выделяется за счет реакции нитрования. Смесь выдерживают 0,5 часа при 30—40°, разбавляют двойным количеством (по объему) воды и 3—4. часа охлаждают льдом, после чего отфильтровывают выпавший коричневато-желтый осадок и промывают его водой до нейтральной реакции промывных вод.
Выход технического продукта 1,80—1,85 г (33—35%); т. пл. 146—148°.
Продукт очищают однократной кристаллизацией из 130-ю 50%-ного этанола.
Выход чистого продукта в виде золотисто-желтых игл равен 1,40—1,50 г (26—28%); т. пл. 150—150,5°.
По литературным данным, вещество получали в виде желто-коричневых иризм (из бензола) с т. пл. 148—149° [1] или в виде золотисто-желтых игл (из 50%-ного этанола) с т. пл. 146—147° [2].
ЛИТЕРАТУРА
1. L. Qattermann. Lieb. Ann., 357, 313 (1907).
2. R. S. Desai, K. S. Radha, R. C. Shan. Proc. Indian, acad. Sci., 23A, 338 (1946).
З-НИТРО-4,5,6-ТРИМЕТОКСИФТАЛЕВЫИ АНГИДРИД
Н. М. РЫБКИНА, М. Е. ТИХОМИРОВА, В. Н. ГЛУШКО
О
ОСН3||
Н3СО | С \/\z \
I II 0
Z4Z\ Z
Н3СО | с
NO2 II о
CnH9NO8 М. В. 283,20
З-Нитро-4,5,6-триметоксифталевый ангидрид получают сублимацией З-нитро-4,5, 6-триметоксифталевой кислоты при 175° (1]. Проверка метода [lj показала, что ангидрид получается с выходом 20—25%.
Нами предложен более простой способ получения 3-нитро-4, 5, 6-триметоксифталевого ангидрида, основанный на кипячении З-нитро-4,5, 6-триметоксифталевой кислоты с уксусным ангидридом. Исходную З-нитро-4,5, 6-триметоксифталевую кислоту получают окислением 7-нитро-4,5,6-триметоксифта-лида марганцовокислым калием в щелочной среде [1]. Нами уточнены условия окисления.
СХЕМА СИНТЕЗА
ОСН3 Н3СО | СН2 \/\Z \ 1 II о КМп0* Z4Z\ z кон" НзСО | \z NOa С II ОСНз Н3СО | С(Ч к \Z\z HCI Z4Z\ Н3СО 1 COOK NO2
135
ОСНз ОСНз
Н3СО | СООН НзСО I со
\^\/ w\/ \
| ||х (СН3СО)гО | || 0
/Ч/\ /%./\ /
Н3СО | СООН НзСО ! СО
no2 no2
Характеристика основного сырья
7-Нитро-4,5,6-триметоксифталид, т. пл. 115—116°, получение см. стр. 138 этого сборника.
Калий марганцовокислый, ГОСТ 4527—65, ч. д. а.
Кали едкое, ГОСТ 4203—65, ч.
Ацетон, ГОСТ 2603—63, ч.
Уксусный ангидрид, ГОСТ 5815—52, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Уголь активированный, ГОСТ 6217—52, техн.
Условия получения
Синтез З-нитро-4, 5, 6-триметоксифталевой кислоты. В трех-горлую колбу емкостью 0,5 л, снабженную мешалкой, термометром и капельной воронкой, загружают 10 г (0,037 Л4) 7-нитро-4,5,6-триметоксифталида и 69 мл 10%-пого раствора едкого кали. Смесь нагревают на водяной бане до 40—45° и при размешивании за 9 часов порциями по 1—2 мл прибавляют раствор 13,3 г (0,084 Л4) марганцовокислого калия в 430 мл дистиллированной воды. Очередную порцию раствора марганцовокислого калия прибавляют только после перехода окраски реакционного раствора от темно-малиновой к бурой. Затем реакционную массу кипятят 2 часа, охлаждают до 18— 20° и отфильтровывают от двуокиси марганца. Фильтрат подкисляют 20 мл соляной кислоты до pH 1 по универсальной индикаторной бумаге и выпаривают досуха. Остаток экстрагируют ацетоном пять раз порциями по 100 мл. Отгоняют ацетон. Сухой остаток перекристаллизовывают из дистиллированной воды (1:4).
Выход чистой З-нитро-4, 5,6-триметоксифталевой кислоты равен 8,3 г (70,5%); т. пл. 162—163°.
По литературным данным (1], т. пл. 177,5—178°.
Найдено, %: С — 43,88; 43,76; Н —3,61; 3,81; N —4,75; 4,52. CuHnNOg. Вычислено, %: С —43,88; Н — 3,67; N — 4,37.
Получение З-нитро-4, 5, 6-триметоксифталевого ангидрида. В круглодонную колбу емкостью 100 мл с шариковым холодильником загружают 10 г (0,03 М) З-нитро-4,5,6-триметоксифталевой кислоты и 60 мл (64,8 г; 0,63 М) уксусного ангидрида. Реакционную смесь кипятят 10 минут, затем до-136
бавляют 0,3 г активированного угля и снова кипятят 3—5 минут. Горячий раствор фильтруют и охлаждают до 15—18°.
Выпавшие кристаллы отфильтровывают, промывают 10 мл этилового спирта и сушат до постоянного веса.
Выход З-нитро-4,5,6-триметоксифталевого ангидрида равен 6,1 г (68.5%); т. пл. 90—91°.
По литературным данным [1], т. пл. 90—91°.
ЛИТЕРАТУРА
1. Е. White, М. Burs еу. J. Organ. Chem., 31,6, 1912 1917 (1966).
7-НИТРО-4Д6-ТРИМЕТОКСИФТАЛ ИД
Н. М. РЫБКИНА, М. Е. ТИХОМИРОВА, В. Н. ГЛУШКО
Н3СО | С
N02 I! О
CuHnNCh
М. в. 269,22
Известные методы получения 7-нитро-4,5,6-триметокси-фталида основаны на нитровании 4,5,6-триметоксифталида концентрированной азотной кислотой [1] или азотной кислотой (пл. 1,49—1,51) в среде уксусной кислоты при 30—40° в течение нескольких дней [2].
Наши попытки воспроизвести синтез по данным этих работ ие имели успеха: 4, 5,6-триметоксифталид в условиях [1] полностью окисляется, а в условиях (2] был получен продукт с т. пл. 134—135°, что соответствовало т. пл. исходного 4,5,6-триметоксифталида.
Нами проведено нитрование 4,5,6-триметоксифталида 80 %-ной азотиой кислотой.
СХЕМА СИНТЕЗА
ОСН3
HNO3 ____—>
Н3СО с
II
Н3СО | с
no2 II о
о
138
Характеристика основного сырья
4,5,6-Триметоксифталид, т. пл. 135—136°, получение см. стр. 194 этого сборника.
Азотная кислота, ГОСТ 4461—67, ч.
Азотная кислота, ГОСТ 701—68, сорт 1, концентрированная.
Метиловый спирт, ГОСТ 6995—67.
Условия получения
В трехгорлую колбу емкостью 250 мл, погруженную в баню с охлаждающей смесью и снабженную мешалкой, термометром и загрузочной воронкой, помещают нитрующую смесь, приготовленную из 100 мл 98%-пой азотной кислоты и 50 мл 68 %-ной азотной кислоты, и постепенно прибавляют 20 г (0,08 Л4) 4,5,6-триметоксифталида, поддерживая температуру реакционной массы не выше 0°. Массу выдерживают 10 минут при 0° и выливают на 1 кг измельченного льда. Выпавший осадок отфильтровывают, промывают водой до нейтральной реакции промывных вод и сушат в сушильном шкафу при 100°.
Выход технического 7-нитро-4,5,6-триметоксифталида равен 7,8 г (32%); т. пл. ПО—112°. Продукт перекристаллизовывают из метанола в весовом соотношении (1 :10).
Выход 7-нитро-4,5,6-триметоксифталида в виде белого кристаллического порошка с т. пл. 115—116° равен 7,0 г (29,3%).
По литературным данным, т. пл. 116—116,5° [1], 118— 119° [2].
ЛИТЕРАТУРА
1. F. W е у g a n d, Н. W е b е г, Е, Maekawa. Вег. 90, 1879— 1895 (1957).
2. Англ. пат. 873935 (1959).
НИТРОФЛУОРЕСЦЕИНДИАЦЕТАТ I
Н. М. РЫБКИНА, А. А. ПОЛОЖЕНЦЕВА, М. Е ТИХОМИРОВА
О
НзСОСО-.^^/ \fV-ОСОСНз
C24H15NO9 М. в. 461,37
Нитрофлуоресцеиндиацетат получают сплавлением 4-нитрофталевой кислоты с резорцином и ацетилированием полученного продукта уксусным ангидридом [1].
Нами проверен этот способ и выделены фракционной кристаллизацией два изомера — изомер I с т. пл. 222—223° и изомер II с т. пл. 194—196°. В данной методике описан способ выделения и очистки изомера I.
СХЕМА СИНТЕЗА
соон
соон он
I II + 21 || --->
ч/ ч/
I I
no2 он
140
о
НО —'X|j,Z^j-'0H
Ч/\ /\<Х (СН3СО)2О с ------•
11 V=o
хх
no2
Характеристика основного сырья
Резорцин, ГОСТ 9945—62, ч. д. а.
4-Нитрофталевая кислота, ТУ 1362—57, ч.
Уксусный ангидрид, ГОСТ 5815—52, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Уголь активированный, ГОСТ 6217—52, техн.
Условия получения
Синтез нитрофлуоресцеина. В фарфоровом стакане емкостью 0,5 л, снабженном лопастной мешалкой и термометром и помещенном в масляную баню, нагревают смесь 80 г (0,37 А4) 4-питрофталевой кислоты и 80 г (0,72 М) резорцина до температуры в бане 205—210°. Когда смесь расплавится, массу начинают энергично размешивать. Как только реакционная масса загустеет, размешивание прекращают и продолжают нагревание без размешивания. К концу реакции в 141
стакане образуется твердый пористый плав. Общая продолжительность нагревания 6 часов.
Плав (140—145 г) тщательно растирают в фарфоровой ступке, просеивают через сито и кипятят 1 час с 1300 мл 2%-ной соляной кислоты. Колбу с реакционной массой охлаждают до 18—20°, осадок отфильтровывают на фарфоровой воронке через кислотоупорную ткань, промывают 300 мл горячей (70°) 2%-ной соляной кислотой, затем водой до отсутствия резорцина (проба с формалином). Полученную коричневую пасту сушат в сушильном шкафу при 105—110° до постоянного веса.
Выход сухого препарата равен 123—130 г (33—35%). Препарат представляет собой смесь изомеров нитрофлуоресцеина.
Получение нитрофлуоресцеиндиацетата I. В круглодонную колбу емкостью 1 л, снабженную шариковым холодильником и мешалкой, загружают 125 г технического питрофлуо-ресцеина (смесь изомеров) и 500 мл уксусного ангидрида. Содержимое колбы кипятят 2 часа при размешивании, затем добавляют 5 г активированного угля и снова кипятят 5 минут. Горячий раствор фильтруют и оставляют стоять на 10— 12 часов. Выпавшие желтовато-белые кристаллы отфильтровывают, хорошо отжимают, промывают на фильтре 25 мл уксусного ангидрида, а затем 25 мл этилового спирта и сушат в сушильном шкафу при 100° до постоянного веса.
Выход нитрофлуоресцеиндиацетата I равен 45—47 г (27— 29%); т. пл. 217—219°.
Технический продукт перекристаллизовывают из уксусного ангидрида в весовом соотношении 1 :2 и получают нитро-флуоресцеиндчацетат I в виде почти бесцветного кристаллического порошка с т. пл. 222—224°.
Выход чистого продукта равен 35,5—36,5 г (22—23%).
По литературным данным (1], т. пл. 222—223°.
ЛИТЕРАТУРА
1. A. Coons, М. Kaplan. J. Expt!. Med., 91, I (1950).
2- и 4-НИТРО(3-ХЛОРПРОПИЛ)БЕНЗОЛЫ
В. М. ОСТРОВСКАЯ, Н. Е. КАШИРИНА, Т. В. ФОМИНА
СН2СН2СН2С1
гг°2
СН2СН2СН2С1
C9H10C1NO2
no2
М. в. 199,64
Изомеры 4-нитро(3-хлОрпропил) бензол и 2-нитро(3-хлор-пропил)бензол образуются при нитровании (3-хлорпропил) бензола азотной кислотой (пл. 1,5) при минус 10°[1] или минус 15° (2]; азотной кислотой (пл. 1,47) при минус 15° |3].
В работах [1, 3] данных об образовании .орто-изомера не представлено, в работе [2] указано, что образуется 40% ортоизомера с т. кип. 103—10770,2 мм и 32% пара-изомера с т. кип. 112—11370,2 мм. Хотя условия нитрования (3-хлорпропил) бензола в методиках [2,3] практически сходны, по данным [3], образуется 84,5% пара-изомера с т. кип. 13870,25 мм.
При проверке нами методики [3] получен не пара-изомер, а смесь пара- и орто-изомеров.
СХЕМА СИНТЕЗА
СН2СН2СН2С1
СН2СН2СН2С1
HNO3
no2
143
Характеристика основного сырья
(З-Хлорпропил) бензол; получение см. стр. 216 этого сборника.
Азотная кислота дымящая, техн., пл. 1,52.
Азотная кислота, ГОСТ 4461—67, ч. д. а.
Диэтиловый эфир, ГОСТ 6265—52, ч.
Условия получения
В трехгорлую колбу емкостью 150 мл, снабженную мешалкой, термометром и капельной воронкой, помещают 50 мд азотной кислоты, пл. 1,475 (готовят разбавлением дымящей азотной кислоты азотной кислотой пл. 1,42), охлаждают до минус 15° и добавляют по каплям за 40 минут при размешивании 10,8 г (0,07 А!) (3-хлорпропил) бензола. Скорость добавления регулируют так, чтобы не повышалась температура.
Смесь размешивают 2 часа, раствор вливают в стакан со льдом (20 г), выделившееся масло экстрагируют 250 мл диэтилового эфира, экстракт промывают 100 мл воды, сушат над хлористым кальцием. Массу перегоняют в вакууме с дефлегматором, собирая фракцию орто-изомера (3,0 г; 21%) с т. кип. 134—13770,3 мм; /1^’= 1,5555; фракцию (2,4 г; 18%) с т. кип. 138—14070,3 мм, rifi — 1,5565 (см. примечание) и фракцию пара-изомера (4,8г; 34%) с т. кип. 140—14270,3мм, = 1,5579.
Общий выход изомеров 10,2 г (73 %)
Примечание. Среднюю фракцию, представляющую смесь орто- и пара-изомеров, собирают от нескольких параллельных опытов и разгоняют с дефлегматором.
ЛИТЕРАТУРА
1.1. Braun, Н. Deutsch. Вег., 45,2509 (1912).
2. A. Gliceto, A. Fava, A. Simeone. Gszz. chim. ital., 90, 600 (I960).
I. Buchi, I. Enezian et al Helv. chim. acta., 43, 1971 (1960)
н-НОНИЛОВЫЙ ЭФИР м-НИТРОБЕНЗОЙНОЙ кислоты
н- Нонил-п-нитробензоат
Е. И. МАХОВЕР
C16H23NO4
М. в. 293,36
н-Нониловый эфир n-нитробензойной кислоты применяется в качестве неподвижной фазы в газо-жидкостной хроматографии.
В литературе описаны методы получения высших эфиров нитробензойных кислот как из л-нитробензоилхлорида {1, 2J, так и непосредственно этерификацией нитробензойных кислот 2—3-кратным избытком высшего спирта в присутствии л-толуолсульфохлорида [3]. Для очистки эфиров рекомендуется перегонка в вакууме.
Нами показано, что проведение синтеза с эквимолярным соотношением компонентов и использование л-толуолсульфо-кислоты позволяет получить н-нонил-л-нитробензоат с высоким выходом и достаточно чистый без вакуум-перегонки.
СХЕМА СИНТЕЗА
СООН
I
|^Ч|| + С9Н19ОН снзС6Н4ЗО3Н
ZZ
no2
10-1192
СООС9Н19
4Z I
no2
145
Характеристика основного сырья
п-Нитробензойпая кислота, МРТУ 6—14—199—67, техн. н-Нониловый спирт, МРТУ 6—09—2878—66, ч.
n-Толуолсульфокислота, ТУ МХП ОРУ 73—56, ч.
Толуол, ГОСТ 4803—67, техн.
Уголь активированный, ГОСТ 4453—48, техн.
Алюминия окись, ТУ МХП 2962—54, д/хроматографии.
Кальций хлористый безводный, ГОСТ 450—58.
Сода кальцинированная, ГОСТ 5100—64.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром и водоотделителем с обратным холодильником, помещают 0,42 кг (2,5 М) n-нитробензойной кислоты, 0,36 кг (2,5 Л4) н-нонилового спирта, 30 г (0,175 М) п-толуолсульфо-кислоты и 800 мл толуола.
Смесь кипятят около двух часов при 115—125° до прекращения отделения воды (45 мл). После охлаждения до 30—40° реакционную массу нейтрализуют 500 мл 10%-ного раствора карбоната натрия до pH 9, затем промывают водой 3 раза порциями по 500 мл до pH 7 и сушат хлористым кальцием.
Отфильтрованный раствор эфира в толуоле для осветления пропускают при пониженном давлении (300—350 мм) через стеклянную колонку (60—70X800 мм), заполненную на 2/3 окисью алюминия (около 400 г). К осветленному раствору добавляют 30 г окиси алюминия и 30 г угля и отгоняют толуол сначала при атмосферном давлении, затем при пониженном давлении (^10 мм) вместе со следами нонилового спирта (температура бани до 170°).
После охлаждения до 100° эфир фильтруют от окиси алюминия и угля.
Выход н-нонилового эфира и-нитробензойной кислоты равен 550 г (74,5%); d™ = 1,065; я^ = 1,5072; кислотность 0,06% в пересчете на л-нитробензойную кислоту; содержание основного вещества 99,7% (определено омылением водноспиртовой щелочью).
По литературным данным [1], d2)) = 1,0719: «^= 1,5074.
ЛИТЕРАТУРА
1. Маг v u г D. Armstrong, J. Е. Copenhave,. J. Amer. Chem. Soc., 65, 2252 (1943).
2. I I a г о 1 d G. Rule, Annie H. Numhecs. J. Chem. Soc., 1926, 2121.
3. В. А. Аверин, В. P. Шилов, Э. И. Дорошко, И. Н. Дых а-н 6 в. Авт. свид. 166323; Бюлл изобр., № 22, 18 (1964).
146
а ОКСИБЕНЗИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА, НАТРИЕВАЯ И СВИНЦОВАЯ СОЛИ
Р. П. ЛАСТОВСКИИ, И. Д. КОЛПАКОВА, л. в. КРИНИЦКАЯ, Е. И. МИРОНОВА
о
О — р/
__ I он сон | О’
О-р/
О
3—
Na3+
C7H7NQ3O7P2
М. в. 334,04
О “ 4-о = р/ ___ I О
^-С-ОН рь?+-н2о ” |/°
о _
C7HeO7P2Pb2 • H2O М. в. 696,46
а-Оксибензилидендифосфоновая кислота (ОБДФК) является комплексообразующим реагентом. Подобно описанной ранее а-оксиэтилидендифосфоновой кислоте [1], ОБДФК и ее соли применяются для устранения жесткости воды [2—4], стабилизации перекисных соединений и поверхностно-актив-10* 147
пых веществ [3—6], удаления накипи в теплообменниках [4], как составляющая часть детергентов [4], для травления алюминия и его сплавов |[7], в парфюмерной промышленности [8, 9], а также в других областях народного хозяйства. Наиболее доступным способом получения ОБДФК является ацилирование фосфористой кислоты хлористым бензоилом [5, 7, 10- 12] или взаимодействие треххлористого фосфора с бензойной кислотой [3, 7, 11—13].
Нами проверен и уточнен последний способ. Получены натриевая и свинцовая соли кислоты.
СХЕМА СИНТЕЗА
РО(ОН)2
2РС1з + 3CGH5COOH + ЗН2О CGH5C- он + Хро;он), + 2С6Н5СОС1 + 4HCI,
О = Р(ОН)2
I с6н5с-он
I
О = Р(ОН)2
.NaOH
О
О РГ
р ОН
(f с— он
L0
О=Р<
4 О
3—
Na3+,
4 —
рь22+-н о
Характеристика основного сырья
Бензойная кислота, ГОСТ 10521—63, ч.
Фосфор треххлористый, ГОСТ 91—41, ч.
Натр едкий, ГОСТ 4328—66, ч.
Свинец азотнокислый, ГОСТ 4236—67, ч
148
Условия получения
Получение натриевой соли ОБДФК- В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, загружают смесь 91,5 г (0,75 М) бензойной кислоты и 13,5 мл (0,75 Л4) воды и постепенно при перемешивании прибавляют из капельной воронки 69 г (0,5 Л4) треххлористого фосфора с такой скоростью, чтобы температура в колбе не превышала 30—35°. Реакционную массу постепенно за 1 час нагревают до 120° и выдерживают 1 час при 120°, 5 часов при 130°, охлаждают до 90°, добавляют 500 мл воды и нагревают до полного растворения. При охлаждении реакционного раствора выпадает осадок бензойной кислоты, его отфильтровывают. Из фильтрата извлекают органические примеси горячим бензолом 2 раза порциями по 180 мл, нагревают с 5 г активированного угля. Уголь отфильтровывают, а раствор упаривают до получения густого сиропа с — 1,49—1,51. Раствор нейтрализуют 25%-ным раствором едкого натра до pH 7 по универсальной индикаторной бумажке, растирают со смесью (1:1) абсолютного спирта и ацетона до затвердевания массы, отфильтровывают и сушат при 80''.
Выход тринатриевой соли ОБДФК равен 38,3 г (51%).
Найдено, %: С—24,51, 24,39; Н — 2,10, 2,01;
Р—18,81, 18,69 (см. примечание 1)
C7H7Na3O7P2. Вычислено, %: С—24,80; Н—2,10, Р—18,55.
Получение свинцовой соли ОБДФК. Раствор 3 г натриевой соли ОБДФК в 15 мл воды осторожно приливают к раствору 30 г азотнокислого свинца в 75 мл воды. Выпадает белый мелкокристаллический осадок свинцовой соли ОБДФК-Реакционную смесь нагревают до 50—60°, при этом осадок оседает на дно. Соль отфильтровывают, промывают 20 мл спирта и сушат при 80°.
Выход свинцовой соли ОБДФК равен 5,6 г (90% в расчете на натриевую соль ОБДФК).
Найдено, %: С—11,96, 11,69; Н—1,16, 1,66;
РЬ —58,7; 59,3 (см. примечание 2), С7Н5О7Р2РЬ2 • Н2О. Вычислено, %: С—12,08; Н — 1,15; РЬ—59,5.
Примечания.
1. Образец для анализа дважды переосаждали из воды смесью (I : 1) абсолютного спирта и ацетона.
2. Содержание свинца определяли комплексонометрически после минерализации навески в концентрированной азотной кислоте [14, 15].
ЛИТЕРАТУРА
1. Р. П. Л а с т о в с к и й, И. Д. Колпакова и др. Методы получения химических реактивов и препаратов, вьш. 22. М., ПРЕД, 1970 стр. 150.
2. Пат. ФРГ 1082235 (1960); С. А., 55, 12722 h (1961).
149
3. Пат. ФРГ 1148235 (1963); 1148551 (1963); С. Z., 6/7, 239 (1964).
4. Пат. ФРГ 1251759 (1967).
5. Пат. ФРГ 1107207 (1961); С. А., 56, 12717а (1962).
6. Бельг, пат. 619619 (1963).
7. Пат. ФРГ 1206264 (1966); РЖХим., 13К76П (1967).
8. Пат. США 3202579 (1965).
9. Пат. США 3213129 (1965).
10. Бельг, пат. 591066 (1960).
11. Франц, пат. 1412865 (1965).
12. Пат. ФРГ 1194852 (1966).
13. Голланд, пат. 6604176 (1966); С. А., 66, 5260 (1967).
14. «Сульфарсазен». Инструкция по аналитическому применению. М., ИРЕА, 1961.
15. Г. С. Петрова, М. А. Ягодицы», А. М. Лукин. Заводск. лаборатория, 7, 776 (1970).
4,4 -0КСИБИС(ТЕТРАХЛ0РБЕН30ЙНАЯ КИСЛОТА)
Е. П. БАБИН, И. П. СКАВИНСКИЙ, Н. А. АНДРУХОВ.
А. А. РЫЖКОВ, А. С. СВИРИДЕНКО
О
Ч
I
НО-7
С1 С1 С1 С1
С1 С1 С1 С1
СцНгСиОб
М. в. 533,76
4,4'-Оксибис(тетрахлорбепзойная кислота) в литературе не описана. Она получена нами хлорированием 4,4'-оксиди-бензойной кислоты.
СХЕМА СИНТЕЗА
О
С1
С1
но
\ _/ - 0 -
CI CI
С1 С1
^_ч_с
С1=С1
Характеристика основного сырья
4,4'-Оксидибензойная кислота, т. пл. 337—338°; получение см. [1].
Хлор, ГОСТ 6718 68.
Серная кислота, ГОСТ 4208—66, 98%-ная.
Йод металлический, ГОСТ 4159—48.
151
Условия получения
В трехгорлый реактор цилиндрической формы высотой 250 мм, диаметром 20 мм, снабженный термометром, обратным холодильником и трубкой для подвода газа с пористой пластинкой, загружают 2,6 г (0,01 М) 4,4'-оксидибензойной кислоты, 80 г серной кислоты и 0,2 г металлического йода. Реакционную смесь нагревают на водяной бане до 90° и пропускают хлор со скоростью 30 г!час в течение 3,5 часа. Затем реакционную массу, охлажденную до комнатной температуры, выливают в смесь 50 мл воды с 100 г льда и охлаждают до 5—0е. Выпавший осадок желтого цвета отфильтровывают, промывают 300 мл холодной воды и сушат при 75—80°.
Выход 4,4'-оксибис (тетрахлорбензойной кислоты) равен 4,6 г (85,5%).
Полученное соединение представляет собой желтое кристаллическое вещество с т. пл. 268—270°, растворимо в спиртах, диоксане; нерастворимо в бензоле и четыреххлористом углероде.
Найдено, %: С —31,43; 31,40; С! — 53,20; 53,23. С14Н2С18О5. Вычислено, %: С—31.51; С1—53,14.
ЛИТЕРАТУРА
1. Е. Massarani, D. Nardi et. al. Farmaco (Paria), Ed. Sci , 18, 582 (1963).
ОКСИГИДРОХИНОНАТЫ СВИНЦА И КОБАЛЬТА
М. Н. РУСИНА, И. Я. СОЛОМЯННИКОВА, Н. М. ДЯТЛОВА
С03 (СеНзОз) 2 • 5Н2О РЬз(СвН30з) 2‘ 6Н2О
С12НбСо3Об • 5Н2О М. в. 513,05 С12Н60бРЬз • 6Н2О М. в. 975,62
Водонерастворимые комплексы свинца и кобальта с относительно высоким содержанием металла и кислорода необходимы для решения ряда задач новой техники. В связи с этим представляло интерес изучить возможность получения оксигидрохинонатов указанных металлов, в литературе не описанных.
СХЕМА СИНТЕЗА
ОН
I
/\-он кон
Ме(МОз)г + | ||
ч/ I он
где Me = Pb2+, Сог+.
Мез (СеНзОз) г • п Н2О,
Характеристика основного сырья
Оксигидрохинон, МРТУ 6—09—3982—67, ч.
Кобальт сернокислый, ГОСТ 4462—68, ч. д. а.
Свинец азотнокислый, ГОСТ 4236—67, х. ч.
Кали едкое, ГОСТ 4203—65, х. ч.
Условия получения
В четырехгорлую колбу емкостью 2 л, снабженную двумя делительными воронками и трубками для ввода и вывода газа, сливают одновременно за 15—20 минут растворы 12,6г
153
(0,1 М) оксигипрохинона в 500 мл воды и соответственно 47,7 г (0,17 Л4) сернокислого кобальта или 56,5 г (0,17 М) азотнокислого свинца в 500 мл воды. Реакцию ведут при комнатной температуре, осуществляя перемешивание током азота К полученному раствору прибавляют по каплям раствор 16 г (0,28 М) едкого кали в 100 мл воды. Реакционную массу выдерживают в течение суток. Выпавший осадок оксигидрохиноната отфильтровывают, промывают водой до отсутствия сульфат- или нитрат-ионов и высушивают до постоянного веса в сушильном шкафу при 103—105°.
Выход оксигидрохиноната кобальта равен 25 г (93%), оксигидрохиноната свинца — 50 г (96%).
Найдено, %: С —13,15; 13,32; Н—0,83; 0,88; РЬ—62,8; 62,6.
С12НбО6РЬз-6Н2О. Вычислено, %: С—14,6; Н —1,22; РЬ — 63,7.
Найдено, %: С — 26,88; 26,86; Н — 2,40; 2,44;
Со—34,2; 34,9;
С12Н6Со3О6 • 5Н2О. Вычислено, %: С —28,00; Н —2,35; Со — 34,4.
ОКСИМ 5-АМИНОСАЛИЦИЛОВОГО АЛЬДЕГИДА 5-Амииосалицилальдоксим
Д. А. ДРАПКИНА, Н. И. ДОРОШИНА, Т. В. СКАНДАЛОВА СН = NOH
C7H8N2O2 М. в. 152,15
5-Аминосалицилальдоксим в литературе не описан. Синтез его непосредственно из 5-аминосалицилового альдегида затруднителен, так как исходный альдегид невозможно получить в чистом виде в связи с тем, что он чрезвычайно легко вступает в реакцию самоконденсации даже в виде солянокислой соли с образованием полиазометина (см., например, [1-2])-
Мы получцли 5-аминосалицилальдоксим восстановлением оксима 5-нитросалицилового альдегида гидросульфитом натрия в водно-спиртовой среде.
СХЕМА СИНТЕЗА
O2N
CHO
I OH
NHsOH
СН =NOH | ОН
Na2S2O4
o2n
CH = NOH
I OH
h2n
155
Характеристика основного сырья
5-Нитросалициловый альдегид, МРТУ 6—09—4529—67, ч. Гидроксиламин гидрохлорид. ГОСТ 5456—65, ч. д. а.
Натрий гидросульфит, ГОСТ 246—67, техн.
Натрий углекислый безводный, ГОСТ 84—66, ч. Этиловый спирт ректификованный, ГОСТ 5962—67. Бензол, ГОСТ 5955—68, ч. д. а.
Эфир серный, ГОСТ 6265—68, мед.
Условия получения
Синтез 5-нитросалицилальдоксима. В двугорлую круглодонную колбу емкостью 250 мл, снабженную обратным холодильником и капельной воронкой, помещают 8,35 г (0,05 Л4) 5-нитросалицилового альдегида и 100 мл спирта. Смесь нагревают почти до кипения и к полученному раствору приливают струйкой, при встряхивании, предварительно нагретый до 60—70° раствор 3,82 г (0,055 Л4) солянокислого гидроксиламина в 15 мл дистиллированной воды. Почти тотчас выпадает осадок оксима. Реакционную смесь кипятят 5—10 минут, дают ей остыть до комнатной температуры и затем охлаждают 1—2 часа в бане со льдом. Осадок оксима отфильтровывают, дважды промывают спиртом, разбавленным (1 ' 1) водой (по объему) порциями по 5 мл.
Выход 5-нитросалицилальдоксима равен 8,4—8,5 г (92— 94%); т. пл. 219—220°.
Оксим кристаллизуют из 200 мл этилового спирта. Получают 6,4—6,5 г (70—71%) вещества с т. пл. 224—224,5°.
По литературным данным [3], т. пл. 225°.
Получение 5-аминосалицилальдоксима. Приготовляют раствор натриевой соли 5-нитросалицилальдоксима растворением при слабом нагревании 1,82 г (0,01 Л4) оксима в 60 мл спирта и вливают его в раствор 5,3 г (0,05 Л4) углекислого натрия в 250 мл воды. Раствор соли обладает интенсивнооранжевой окраской.
Затем в конической колбе емкостью 0,5 л растворяют 12 г гидросульфита натрия в 50 мл воды (см. примечание 1) и к этому раствору приливают порциями, по мере обесцвечивания, теплый (30—40°) раствор соли 5-нитросалицилальдокси-ма. Если окраска перестает изменяться, к смеси прибавляют еще некоторое количество сухого гидросульфата натрия, необходимое для обесцвечивания раствора, и снова продолжают добавлять раствор соли оксима. Общая длительность реакции 20—30 минут. По окончании восстановления полученный светло-желтый раствор охлаждают до комнатной температуры (см. примечание 2) и 5-аминосалицилальдоксим извлекают серным эфиром один раз 100 мл и четыре раза 156
порциями по 50 мл. Экстракт, не высушивая, концентрируют в вакууме водоструйного насоса, а остаток переносят в фарфоровую чашку. Осадок после полного испарения эфира отжимают на воронке и промывают 2—3 мл воды.
Выход 5-аминосалицилальдоксима равен 1,1 —1,3 г (72— 85%); т. пл. 165—167°.
После перекристаллизации из 370—400 мл бензола в присутствии активированного угля получают 0,6—0,7 г (39— 46%) чистого продукта в виде кристаллов песочного цвета с т. пл. 169—169,5°. Дополнительная кристаллизация температуру плавления не меняет.
Найдено, %: С — 55,43; 55,70; Н — 5,55; 5,40; N—18,40; 18,20. CyHsNsOs. Вычислено, %: С — 55,25; Н — 5,30; N — 18,42.
Примечания.
1. Гидросульфит натрия растворяют после приготовления раствора оксима и сразу же используют.
2. Обычно раствор после восстановления имеет pH 7,2—7,6, если pH выше, то перед экстракцией раствор осторожно нейтрализуют 20%-ной уксусной кислотой.
ЛИТЕРАТУРА
1. Н. Weil, М. Traun, S. Marcel. Ber., 55, 2664 (1922).
2. М. Tanaka. Bull. Chem., Soc. Japan, 37, 1210 (1964); 40, 1724 (1967); 41, 2807 (1968).
3. J. Mei senheitner, P. Zimmermann, (J. Ku mmer. Lieb. Ann., 446, 227 (1926).
2-ОКСИ-5-МЕТИЛФЕНИЛ-1,3-БИС(МЕТИЛ ЕНИМИНО)-ТЕТРАУКСУСНАЯ КИСЛОТА
В. я. ТЕМКИНА, И. В. ЦИРУЛЬНИКОВА, И. П. ФАДЕЕЕВА, Р. П. ЛАСТОВСКИИ
ОН
I
(НООСН2С) zNH2C — ^ j|“ CHaN (СН2С°ОН) 2
W ! СН3
C17H22N2O9 М. в. 398,38
2-Окси-5-метилфенил-1,3-бис (метиленимино) тетрауксус-ная кислота, являющаяся комплексообразующим реагентом с повышенной металлоемкостью, получена взаимодействием и-крезола с формальдегидом и иминодиуксусной кислотой [1]. Нами проверена и уточнена эта методика.
СХЕМА СИНТЕЗА
ОН
I СН2СООН
s\ /
| || + 2СН2О + 2HN\
Z/ СН2СООН
I
СН.з
NaOH
"на-
он । (НООСН2С) 2NH2C — — CH2N (СН2СООН) 2
Ч/ I СН.
158
Характеристика основного сырья
n-Крезол, МРТУ 6- 09- 2796—66, ч.
Формальдегид, ГОСТ 1625—61, техн., 37%-ный раствор. Иминодиуксусная кислота, ВТУ МГ УХП 190—58, ч.
Натр едкий, ГОСТ 4328—66, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57, техн.
Условия получения
В трехгорлой колбе емкостью 100 мл, снабженной мешалкой, капельной воронкой, обратным холодильником и помещенной в баню, к раствору 8,6 г (0,08 М) n-крезола в 12 мл воды и 8 мл 30%-ного раствора едкого натра постепенно приливают при размешивании нейтрализованный водный раствор 22,4 г (0,068 М) иминодиуксусной кислоты в 26 мл воды, затем 30%-ный раствор едкого натра до pH 7—8 по универсальной индикаторной бумаге. Содержимое колбы охлаждают до 10° и медленно за 20—30 минут по каплям прибавляют 13,6 г (0,168 М) 37%-пого раствора формальдегида.
Массу перемешивают при 20° один час, нагревают до 60—70°, выдерживают при этой температуре 2 часа, охлаждают до 7—10° и подкисляют при размешивании концентрированной соляной кислотой до pH 2. Раствор упаривают досуха в вакууме водоструйного насоса (15—20 мм) при 60— 70° (температура бани). Остаток кипятят с 500 мл этанола до тех пор, пока осадок в колбе не станет кристаллическим. Спиртовой раствор сливают, упаривают наполовину (на водоструйном насосе) и охлаждают. Выпавший осадок хлористого натрия отфильтровывают, а фильтрат упаривают досуха.
Выход 1-окси-4-метил фенил-2,6-бис (метиленимино) тетра-уксусной кислоты равен 28 г (87,9%).
Комплексон представляет собой кристаллическое вещество кремоватого цвета, гигроскопичен, хорошо растворим в спирте, щелочах, плохо — в воде.
Найдено, %: С —51,0; 51,1; Н — 5,7; 5,5; N — 7,2; 7,4, C17H22N2O9. Вычислено, %: С — 51,2; Н — 5,5; N — 7,0.
ЛИТЕРАТУРА
1. G. Schwarzetibaeh, G. A n d е г i g g, R. S а 11 m a n n. Helv. chim. acta., 35, 1785 (1952).
159
8-ОКСИХИНОЛИН
Г. М. БАБЧУК, И. М. ХРАПОВА, Г. Д. ГАНДЛЕВСКИЙ
он
C9H7NO
М. в. 145,16
8-Оксихинолин в настоящее время в промышленности получают из о-аминофенола с выходом^ 40 %.
Описаны способы получения 8-оксихинолина нагреванием 8-хлорхинолина с водным раствором щелочи [1], плавлением 8-хинолинсульфокислоты с твердой гидроокисью натрия (2— 4] или калия [5] при атмосферном давлении или с водным раствором щелочи в автоклаве [6, 7]. Однако 8-хлорхинолин малодоступен, а процесс плавления 8-хинолинсульфокислоты с твердыми гидроокисями сопровождается осмолением -продукта, который получается в виде твердого плава, трудно поддающегося дальнейшей обработке. В результате автоклавного процесса плавления 8-хиполинсульфокислоты с водным раствором щелочи получается жидкая, подвижная, легко эвакуируемая из автоклава масса, ч'О ликвидирует трудность в регулировании температуры и исключает опасность местных перегревов. ।
Нами исследован автоклавный метод щелочного плавления 8-хинолинсульфокислоты.
СХЕМА СИНТЕЗА
4/W
I N
SO3H
NaOH
НС1
z\/4
4Z\Z I N OH
160
Характеристика основного сырья
8-Хинолинсульфокислота, МРТУ 6—09—2456—65.
Натр едкий, ГОСТ 2263—59, техн.
Соляная кислота, ГОСТ 3118—67.
Условия получения
В автоклав из нержавеющей стали емкостью 1 л с мешалкой загружают раствор 216 г (5,41 М) едкого натра в 300 мл воды и 150 г (0,72 Л4) 8-хинолинсульфокислоты. Смесь при перемешивании нагревают до 270° и выдерживают при этой температуре 3,5 часа. Давление в автоклаве 30 атм.
Реакционную массу охлаждают до 100°, разбавляют 0,5 л воды, выгружают из автоклава и нейтрализуют 350 мл соляной кислоты до pH 7,2 (контроль по pH-метру). Из массы 8-оксихинолин отгоняют с паром, отфильтровывают и сушат прн 45°.
Выход 8-оксихинолипа равен 78,5 г (75,5%'); т. пл. 74,0— 74,5е; содержание основного вещества 99,5%.
Продукт удовлетворяет требованиям ГОСТ 5847—62, квалификации «ч. д. а.».
ЛИТЕРАТУРА
1. Н. Н. Ворожцов, С. Ф. Миценгендлер. Авт. свид. 38152; Билл. изобр. № 7—8 (1934).
2. A. Ynoye, М. Sugiura, G Tsuzuki. Bull. Chem. Soc. Japan, 27, 430 (1954).
3. Пат США 2489530 (1949).
4. Пат США 2999095 (1961).
5. Венгер, пат. 143478 (1957).
6. О. Ю. М а г и д с о н, М. В. Р у б ц о в. Химико-фармацевт. пром-сть, 1, 20 (1935).
7. Н. Н. Ворожцов, И. М. Коган. Вег., 63, 2354 (1930).
11-1192
(1-0КСИЭТИЛИДЕН)ДИФ0СФ0Н0ВАЯ КИСЛОТА, КОМПЛЕКС С МЕДЬЮ
Л. В. КРИВИЦКАЯ, М. В. РУСИНА
о—Р —о
Си НзС — С — ОН Си
\ I Z
о —'р — о
C2II4CU2O7P2
М. в. 329,07
Оксиэтилидендифосфоновая кислота (ОЭДФК) при взаимодействии с медью в водном растворе образует прочные протонированные и нормальные комплексы состава 1:1 и 2:1 [1].
Данных о синтезе твердых комплексов ОЭДФК с медью в литературе не приводится.
СХЕМА СИНТЕЗА
Н3С
Р(О) (ОН)
+ 2CuSO4 Na0H_
Р(О) (ОН)2
162
Характеристика основного сырья
Оксиэтилидендифосфоновая кислота, ТУ 14П 849—69, ч.
Медь сернокислая, ГОСТ 4165—68, ч.
Натр едкий, ГОСТ 4328—66, х. ч.
Условия получения
В стакане емкостью 1 л, снабженном мешалкой, растворяют 105 г (0,42 Л4) сернокислой меди в 400 мл воды и приливают раствор 43,82 г (0,21 М) ОЭДФК в 150 мл воды. К полученной смеси при тщательном перемешивании приливают раствор 29,9 г (0,78 Л4) едкого натра в 150 мл воды. Реакцию ведут при комнатной температуре. Реакционную массу выдерживают в течение 20—30 минут. Выпавший осадок бледно-голубого цвета отфильтровывают, промывают водой до отсутствия сульфат-ионов и сушат в сушильном шкафу при 105—110°.
Выход комплекса равен 60 г (90%).
Найдено, %: С — 6,59, 6,66; Н— 1,12, 1,18; Р—18,2;
18,6; Си-38,6, 38,0.
С2Н4Си20тР2. Вычислено, %: С — 7,3; II—1,21; Р—18,9; Си —37,2.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я- Медведь, Р. П Ластовский и др. Докл. АН СССР, 177, 3,582 (1967).
И*
(1-0КСИЭТИЛИДЕН)ДИФ0СФ0Н0ВАЯ КИСЛОТА, МОНОКАЛИЕВАЯ СОЛЬ
Б. И. БИХМАН, Е. М. УРИНОВИЧ, Т. Л. БОГОМОЛОВА, И. М. ДЯТЛОВА
О
Р- он
Z ХОН
СНз—С
I \ он
ОН \ / р
|| \ о ок
С2Н7КО7Р2 • 2Н2О
2Н2О
М. в. 280,16
Оксиэтилидендифосфонаты щелочных металлов находят применение в качестве детергентов н моющих средств [1—4], для смягчения воды и стабилизации поверхностно-активных веществ и перекисных соединений 1[5], для предотвращения окислительного действия некоторых соединений [6], в составе калийных удобрений [7]. \
В литературе практически отсутствуют сведения относительно синтеза калиевых солей оксиэтилидендифосфоновой кислоты, лишь в ряде патентов имеются указания о трудности их выделения в твердом состоянии [4, 8, 9].
Нами разработаны условия синтеза и выделения монока-лиевой соли оксиэтилидендифосфоновой кислоты.
164
СХЕМА СИНТЕЗА
// ^ОН К0Н, СНз —С -2Н2О
он4 \ /0Н Р охок
Характеристика основного сырья
Оксиэтилидендифосфоновая кислота, ТУ 14П849—69, и. Кали едкое, ГОСТ 4203—65, ч.
Условия получения
В стакане емкостью 100 мл, снабженном мешалкой, термометром и капельной воронкой, растворяют 8,4 г (0,15 Л4) едкого кали в 13 мл дистиллированной воды. К полученному раствору, охлажденному льдом до комнатной температуры, медленно приливают из капельной воронки раствор 31 г (0,15 Л4) оксиэтилидендифосфоновой кислоты в 31 мл дистиллированной воды, следя за тем, чтобы температура в реакционной массе не превышала 40°, и продолжают перемешивание до начала образования осадка. Затем стакан с реакционной массой помещают в охлажденную смесь льда с поваренной солью и оставляют на холоду до полного выпадения осадка (^ 1 час). Выделившиеся при стоянии белые кристаллы отфильтровывают, промывают небольшим (^10—15 мл) количеством ледяной воды и сушат на воздухе до постоянного веса.
Выход моиокалиевой соли оксиэтилидендифосфоновой кислоты равен 33 г (84%).
Найдено, * %: С — 9,08; Н — 4,10; Н2О — 13,40. К — 13,70; С2Н7КО7Рг.2НгО. Вычислено, %: С — 8,57; К — 13,93; Н — 3,93; Н2О—12,84.
ЛИТЕРАТУРА
I. Пат. США 3366676 (1968); РМХим., 13Н149П (1969).
2. Пат. США 3366677 (1968); РЖХим, 16Н143П (1969).
3. Пат. США 3159581 (1964); С. A., 4225g (1965) .
4. Пат. США 3527795 (1970).
5. Пат. ФРГ 1148235 (1963); С. Z., 6/7, 239 (1964).
6. Пат. США 3202579 (1965).
7. Франц, пат. 1521412 (1968).
8. Франц, пат. 1460160 (1966).
9. Пат. США 3484480 (1969).
* Анализ соединений проводили сотрудники лаборатории стандартизации Л. Д. Меликситян и В. И. Сомова.
N (2-0КСИЭТИЛ)ЭТИЛЕНДИАМИНО-
N, N', Ы'-ТРИС(МЕТИЛФОСФОНОВАЯ КИСЛОТА)
М. В. РУДОМИНО, Е. к. КОЛОВА, Т. Я. МЕДВЕДЬ,
НОСН2—сн2 СН2Р(О) (ОН)2
4 nch2 — ch2n у Н2°
(НО)2(О)РН2С СН2Р(О) (ОН)2
c7h2In2o10p3 • -Н2о М. в. 395,20
N- (2-Октиэтил)этилендиамино-М,Ы',1\т/-трис( метилфосфоновая кислота) является новым фосфорорганическим комплексоном.
Методика получения этой кислоты основана на взаимодействии N-(2-оксиэтил) этилендиамина с формалином и фосфористой кислотой в сильнокислой среде. Во избежание получения примеси неполностью фосфорилированного амина были применены 20%-ные избытки формалина и фосфористой кислоты по отношению к амииной компоненте- Кислоту дополнительно очищали хроматографированием на катионите КУ-2 (ВТ-форма) с последующим переосаждением из метилового спирта.
СХЕМА СИНТЕЗА
НОСН2 — CH2NHCH2 — CH2NH2 + ЗСН2О + ЗН3РО3
HCI
(НО)2Р(О)СН2 СН2Р(О)(ОН)2
nch2—ch2n
НОСН2 — СН2 СНгР (О) (ОН) з
166
Характеристика основного сырья
N-(2-Оксиэтил) этилендиамин, ТУ 16П—339—68, ч. Формалин, ГОСТ 1625—64, 37%-ный раствор. Фосфористая кислота, ТУ НКХП 518—41, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57. Метиловый спирт синтетический, ГОСТ 6995—67. Катионит КУ-2, ГОСТ 5695—53.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, шариковым холодильником и капельной воронкой, помещают 10,4 г (0,1 М) N-(2-оксиэтил)этилендиамина и при перемешивании и охлаждении ледяной водой прибавляют по каплям 30 мл концентрированной соляной кислоты, затем раствор 29,6 г (0,36 М) фосфористой кислоты в 30 мл воды. Реакционную массу нагревают до кипения (температура бани 130°) и за 1 час прибавляют по каплям 40 г (0,36 М) 37% -ного раствора формалина. Нагревание при этой же температуре продолжают еще 1,5 часа, затем отгоняют воду и хлористый водород при пониженном давлении. Остаток растирают с 500 мл этилового спирта до превращения в порошкообразное вещество. Последнее отфильтровывают и сушат в эксикаторе над хлористым кальцием.
Вещество растворяют в 220 мл воды и хроматографируют на колонке (40X400 мм) с катионитом КУ-2 (Н+-форма). Элюированием водой собирают фракцию с pH 1—2. От элюата отгоняют досуха при пониженном давлении воду, остаток растворяют в 50 мл воды, раствор прибавляют по каплям при перемешивании к 600 мл метилового спирта. Выпавший осадок отфильтровывают и сушат в эксикаторе над хлористым кальцием.
Выход N- (2-оксиэт ил) этилендиамино-N. N', N'-трис (метилфосфоновой кислоты) раве,п 21,7 г (56,5%); т. пл. 165—170° (разлож.).
Найдено, %: С —21,2; 21,3; Н — 5,8; 5,6;
N —6,7; 6,5; Р — 22,9; 22,8;
1 Н2О—2,6.
C7H2i N2OioPs• Н2О. Вычислено, %; С — 21,3; Н — 5,6; N — 7,1;
Р—23,5; Н2О —2,2.
4 (г-п-ОКТИЛ БЕНЗИЛ ОКСИ) БУТА НОЛ
В. А. ИНШАКОВА, 3. С. СИДЕНКО, Г. С. ЧИЖОВА, Ю. С. РЯБОКОБЫЛКО
С8Н17— — сн2 — О— (СН2)4 —он
С19Н32О2
М. в. 292,57
В литературе 4- (n-н-октилбензилокси)бутанол не описан. Это соединение, как и другие моноалкилбензиловые эфиры алкандиолов (1], может применяться в качестве смазочного материала.
СХЕМА СИНТЕЗА
С8Н)7-^ —СН2С1 + НО—(СН2)4 —он
С8н17 -<( - СН2 - О - (СН2) 4 — он
Характеристика основного сырья
н-н-Октилбензилхлорид, т. кип. 143—146°/2 мм; rift — — 1,5062; получение см. стр. 170 этого сборника.
1,4-Бутандиол, ТУ 67—2—ВП, ч.
Натр едкий, ГОСТ 4328—66, х. ч.
Калий углекислый, ГОСТ 4221—65, ч.
Диэтиловый эфир, ГОСТ 6265—52.
Условия получения
В двугорлую колбу емкостью 100 мл, снабженную мешалкой и обратным холодильником, помещают 35,8 г (0,15 Af) н-н-октилбензилхлорида, 14,2 г (0.17АТ) 1,4-бутапдиола и
J68
26,4 г 50%-ного раствора едкого натра. При этом реакционная масса заметно затвердевает и ее постепенно нагревают. Когда масса станет достаточно подвижной, включают мешалку и нагревают массу 4 часа при температуре бани ПО— 120°. затем охлаждают до комнатной температуры и экстрагируют 150 мл эфира. Эфирную вытяжку промывают дистиллированной водой до нейтральной реакции и сушат поташом. Эфир отгоняют, а остаток перегоняют в вакууме, собирая фракции предгона, содержащего непрореагировавший н-н-октилбензилхлорид, с т. кип. до 20072 мм; =1,4920 (~3 г), и 4-(n-н-октилбензилокси)бутанола с т. кин. 200— 20572 мм; п™ = 1,4983.
Выход 4-(n-н-октилбензилокси)бутанола равен 18,2 г (41,5% в расчете на я-н-октилбензилхлорид).
Вещество представляет собой бесцветную маслянистую жидкость с резким запахом, растворимо в спирте и ацетоне.
Найдено, %: С — 78,38; 78,42; Н— 10,74; 11,13.
С19Н32О2. Вычислено, %; С —78,10; Н—10,96.
В ИК-спектре (таблетки КВг) обнаружены полоса поглощения валентного колебания О—Н-связи при 3400 с.и*1 (средняя, очень широкая) и интенсивный дублет валентных колебаний С — О-связи при 1068 и 1100 см~ ’.
ЛИТЕРАТУРА
1. Пат. ГДР 44784 (1966).
л-н-ОКТИЛБЕНЗИЛХЛОРИД
3. С. СИДЕНКО, В. А. ИНШАКОВА, Г. С. ЧИЖОВА, Ю. С. РЯБОКОБЫЛКО
С8Н17-^_^-СН2С1
C15H23CI
М. в. 238,50
n-н-Октилбензилхлорид получают хлорметилированием н-октилбензола формальдегидом (или параформом) и хлористым водородом в присутствии хлористого цинка (реакция Блана) [1—4] или хлорметилированием и-октилбензола формальдегидом и хлорсульфоновой кислотой в среде метанола [5].
Нами проверены и уточнены условия получения гг-н-октил-бензцлхлорида по методу Блана, исходный н-октилбензол получен по реакции Вюрца-Фиттига [6].
СХЕМА СИНТЕЗА
CsI 1)7 —
ZnCl2,HCl UH17—
СН2С1
Характеристика основного сырья
н-Октилбензол, т. кип. 109—110°/2 мм; п™ =1,4850, получение см. [6].
Параформ, ВТУ МХП 2711—55, ч.
Цинк хлористый безводный, ГОСТ 4529—48, ч.
Серная кислота, ГОСТ 4204—66, ч.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч.
Натрий двууглекислый, ГОСТ 4201—66, ч.
Калий углекислый, ГОСТ 4221—65, х. ч.
Натрий хлористый, ГОСТ 4233—66, ч.
170
Условия получения
В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником, термометром и трубкой для ввода хлористого водорода, помещают в указанном порядке 57 s (0,3 М) н-октилбензола, 72 г (1,2 Af) ледяной уксусной кислоты, 18 г (0,2 М) параформа и 32,4 г (0,24М) безводного хлористого цинка. Смесь нагревают до 50° и при этой температуре пропускают 2 часа быстрый ток сухого хлористого водорода. (
Реакционную смесь охлаждают до комнатной температуры, отделяют органический слой, промывают его 100 мл горячей (50—60°) воды, 2 раза 10%-ным раствором бикарбоната натрия порциями по 75 мл и снова 2 раза горячей водой порциями по 100 мл. Промытый органический слой сушат карбонатом калия и перегоняют в вакууме. При перегонке отбирают предгон (содержит непрореагировавший н-октилбензол) с т. кип. до 11672 мм, = 1,4828 (—25 г); промежуточную фракцию с т. кин. до 14172 мм; я®’ = 1,4792 ( ~3 г); и я-н-октилбензилхлорид, т. кип. 143—146°/2 мм, п™ = 1,5062.
Выход я-н-октилбензилхлорида равен 24,3 г (34% в расчете на н-октилбензол). Вещество представляет собой бесцветную маслянистую жидкость с резким запахом.
Найдено, %: С —75,80; 75,98; Н — 9,36, 9,40; С1—14,90; 14,70. С15Н23С1. Вычислено, %: С — 75,5; Н — 9,6; С1—14,9.
В ИК-спектре (таблетки КВг) обнаружены полосы поглощения валентного клебания С—С1 при 676 си-1 (интенсивная) и неплоских деформационных колебаний С — Н-связей пара-дизамещенного бензольного кольца при 828 см~1 (средняя).
ЛИТЕРАТУРА
I. Пат. США 2640079 (1953); С. А., 48, 7635 d (1954).
2. Англ. пат. 788881 (1958); С. А., 52, 11919b (1958).
3. Англ. пат. II18685 (1968); С. А., 69, 78396с (1968).
4. Тоги Kusano, Типе о Suzuki. Kogyo Kogaku. Zasshi, 65, 223 (1962); С. A., 58, 4446c (1963).
5. Пат. США 2630459 (1953); C.A., 48, 1435g (1954).
6. E. A. Schweinitz. Ber., 19,641 (1886).
ОКТИЛ ИМИН0БИС[2-ЭТИЛ (ДИФЕН ИЛ )-ФОСФИНОКСИД]
Ю. М. ПОЛИКАРПОВ, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА, Т. Я. МЕДВЕДЬ
~ Р — СН2 СН2— N — СН2СН2 — Р “
I II\/=\
\=/ О CsHj7 О %_____к?
C36H45NO2P2 М. в. 585,71
Октилиминоби<ф2-этил (дифенил) фосфиноксид] является новым экстрагентом—комплексообразователем из класса |3-аминозамещенных фосфиноксидов.
Для синтеза этого соединения использован метод нуклеофильного присоединения октиламина к винил (дифенил) фос-финоксиду [1].
СХЕМА СИНТЕЗА
(C8Hi7)NH2-I-IC1
2 (С6н5) Р (О) CH = СН2 + NH2 (СкН17) -----------
-> (С3Н5) 2Р (О) C2H4NC2H4 (О) Р (C6Hs) 2
I
С8Н17
Характеристика основного сырья
Винил (дифенил) фосфиноксид, т. пл. 115—117°; получение см. стр. 70 этого сборника.
Бензол, ГОСТ 5955—68, ч. д. а.
Монооктиламин. т. кип. 73—78°/3 мм; МРТУ 6—09— 3272—66.
172
Условия получения
Получение хлоргидрата монооктиламина. В пробирку с отводом помещают 1 г монооктиламина в 10 мл абсолютного серного эфира. Пробирку погружают в стакан со смесью льда и поваренной соли и охлаждают до минус 10°. При этой температуре через эфирный раствор амина пропускают ток сухого хлористого водорода, который получают из чистых хлористого натрия и серной кислоты. По мере пропускания хлористого водорода выпадает белый хлопьевидный осадок хлоргидрата монооктиламина, который отфильтровывают и сушат. Получают 0,7 г хлоргидрата монооктиламина с т. пл. 196—197°.
По литературным данным, т. пл. 198° [2].
Получение октилиминобис[2-этил(дифенил)фосфиноксида]. Смесь 4,6 г (0,02 М) винил (дифенил) фосфиноксида, 1,3 г (0,01 Л1) монооктиламина и 0,2 г хлоргидрата монооктиламина нагревают 8 часов в запаянной ампуле при 180°. Содержимое ампулы выливают горячим в бюкс. Застывшую массу перекристаллизовывают из 25 мл бензола.
Выход октилиминобис(2-этил (дифенил) фосфиноксида] равен 3,6 г (60%); т. пл. 153—154°.
Найдено, %: Р— 10,4; N — 2,2 C36H45NO2P2. Вычислено, %: Р—10,6; N—2,4.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я. Медведь и др. Изв. АН СССР, сер. хим., 9, 1584 (1962).
2. I. V- Braun, G. Blessing, F. Zobel. Ber., 56, 1988 (1923).
1,2,3-ПИРОГАЛЛОЛ
1,2,3-Триоксибензол
А. Л. ЛИФИЦ, Е П. АГЕЕВ
НО-Ml- он
он
СбНбОз
М. в. 126,11
Пирогаллол применяется в аналитической химии, в газовом анализе в качестве поглотителя кислорода, в фотографии как проявитель. |
Наиболее известные методы синтеза пирогаллола основаны на реакции декарбоксилирования галловой кислоты в различных условиях: декарбоксилирование галловой кислоты в жидкой фазе при 190—200° [1], нагревание водных растворов галловой кислоты при 200—240° под давлением [2—4]. Проверка первого метода показала его непригодность для получения продукта реактивной квалификации; осуществление второго метода в производственных условиях затруднено вследствие применения специальных автоклавов с масляным или электрообогревом. ,
Для получения пиррогаллола реактивной квалификации в производстве до последнего времени использовался метод декарбоксилирования сухой галловой кислоты при 200—240’ с последующей вакуум-перегонкой образовавшегося технического продукта (3]. Однако процесс сопровождается значительным осмолением продукта и выход его не превышает 49%'.
Предлагаемый метод декарбоксилирования водных растворов галловой кислоты при 150—160° под давлением до 12 атм, исключает осмоленис продукта и увеличивает выход до 75%'.
СХЕМА СИНТЕЗА
СООН
I
/\
I II Н2° I II
НО—I^JI-OH но-^-он
I I
он он
174
Характеристика основного сырья
Галловая кислота, ТУ—5 ГХФП, техн.
Условия получения
В автоклав из нержавеющей стали емкостью 50 л, снабженный отводной трубкой с вентилем, манометром на 20 атм, предохранительным клапаном, подводкой пара и воды в рубашку автоклава, загружают 30 кг галловой кислоты и 15 л водопроводной воды. Автоклав нагревают пропусканием пара в рубашку аппарата до давления 6—6,5 атм. Примерно через 1 —1,5 часа давление в автоклаве начинает медленно возрастать и через 3 часа достигает 10—12 атм. Температура реакционной массы при этом достигает 145—150°. По достижении указанного давления вентиль на отводной трубке слегка приоткрывают и, продолжая нагревание автоклава, выпускают в атмосферу образующуюся в процессе реакции углекислоту, следя за тем, чтобы давление в автоклаве оставалось не ниже 5 атм. Затем вентиль на отводной трубке закрывают и продолжают нагревание до достижения давления в автоклаве 12 атм. Реакция считается законченной, если давление в автоклаве при дальнейшем нагревании не повышается. Продолжительность процесса 16 часов.
Реакционный раствор, содержащий не более 2—3% галловой кислоты (см. примечание), упаривают в чугунной эмалированной чаше емкостью 50 л до образования (при охлаждении пробы) твердой серой массы. Массу загружают в колбу емкостью 5 Л и перегоняют при пониженном давлении, собирая фракцию с т. кип. 220720 мм.
Выход 1, 2, 3-пирогаллола квалификации «ч. д. а.» равен 15—16 кг (75—80%); т. пл. 132—134°.
По литературным данным (5, 6], т. пл. 132,5—133,5°.
Примечание. Для определения содержания галловой кислоты растворяют 2 г технического пирогаллола в 20 мл дистиллированной воды и титруют 0,1 п. раствором едкого натра в присутствии смешанного (1:1) индиктора — бромкрезолового пурпурного и бромтимолового синего; 1 мл 0,1 н. раствора NаОН соответствует 0,017 г галловой кислоты.
ЛИТЕРАТУРА
1. L. Bart, .1. S с h о d е г., Вег. 12, 1259 (1879).
2. Герм. пат. 333153, Frd., 13, 253.
3. Ullmann, Enzyklopadie der technischen Chemie, IX. 298 (1921).
4. Ю. Швицер. Производство химико-фармацевтических препаратов, М.-Л., изд-во ОНТИ НК.ТП СССР, 1934.
5. Справочник «Химические реактивы п препараты». Под редакцией В. И. Кузнецова. М.-Л., Госхимиздат, 1953.
6. Синтезы органических препаратов, т. Ill, М., Инлитиздат, 1949, стр. 558.
175
м-СТИРОЛСУЛЬФОНИЛФТОРИД
К. М. РОИЗЕН, Т. Н. КОРОВИНА, С. Б. МАКАРОВА
СН - сн2
c8h7fo2s
so2f
М. в. 186,20
ти-Стиролсульфонилфторид является исходным сырьем для получения линейных и трехмерных полимеров.
В литературе [1—2] описан синтез ж-стиролсульфопилфто-рида прямым гидрированием ж-ацетобензолсульфонилфтори-ла водородом при давлении 70 атм с последующей дегидратацией полученного карбинола при пониженном давлении (1 мм) и 350°.
Нами разработан более простой метод синтеза .м-стирол-сульфонилфторида по аналогии с работами [3, 4].
СХЕМА СИНТЕЗА
СН(ОН)СНз
I
Al (С3Н;) з
J —SO2F
сн = сн2
U-SO’F
H3PO3
176
Характеристика основного сырья
ж-Ацетобензолсульфонилфторид, т. пл. 90—92°; получение см. стр. 20 этого сборника.
Алюминия изопропилат, МРТУ 6—09—5253—68, 30%-ный раствор, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Ортофосфорная кислота, ГОСТ 6552—58, ч.
Условия получения
Синтез м~оксиэтилиденбензолсульфонилфторида. В круглодонную колбу емкостью 250 мл помещают 20,2 г (0,1 М) льацетобензолсульфонилфторида и 100 мл 30%-ного раствора изопропилата алюминия (0,1 А4). Колбу соединяют с коротким обратным холодильником, вернюю часть которого с помощью изогнутой трубки соединяют с небольшим нисходящим холодильником. Медленно отгоняют на водяной бане при 70—80° смесь ацетона с изопропиловым спиртом, при этом пары спирта частично конденсируются. Смесь в реакционной колбе кипятят так, чтобы перегонка шла со скоростью 7— 10 капель в минуту. Перегонку ведут 10—12 часов, до получения в дистилляте отрицательной реакции на ацетон (реакция с 2,4-динитрофенилгидразином).
Затем из смеси при пониженном давлении удаляют избыток изопропилового спирта, полученный осадок охлаждают и гидролизуют 210 мл холодной разбавленной (1:5) соляной кислотой.
Органический слой извлекают 250—300 мл серного эфира, эфир отгоняют.
Выход ж-оксиэтилиденбензолсу чьфонилфторида равен 20 г; п% = 1,5085.
По литературным данным |[2], rig = 1,5095.
Получение м-стиролсульфонилфторида. В колбу емкостью 250 мл, снабженную мешалкой и термометром, помещают смесь 20 г (0,1 /VI) ж-оксиэтилиденбензолсульфонил-фторида и 160 мл (273,4 г, 3,3 М) ортофосфорной кислоты. Смесь нагревают на глицериновой бане за 15 минут до 125° и выдерживают при этой температуре 3—4 минуты. Затем содержимое колбы выливают в 1 л воды.
Органический слой экстрагируют 250—300 мл серного эфира, экстракт сушат над прокаленным сульфатом натрия и перегоняют, собирая фракцию с т. кип. 111 — 112°/4 мм; ti?g = 1,5270.
Выход м-стиролсульфонилфторида равен 9,0 г (45%). Вещество представляет собой бесцветную жидкость, не смешивающуюся с водой.
По литературным данным [1], = 1,5270.
12-1192 177
ЛИТЕРАТУРА
1. R. Hart. Mskromolek. Chem.. 49,33 (1961).
2. Пат. ФРГ 1134831; Zbl, 21030(1963).
3. Г. С. Колесников. Синтез винильных производных ароматических п гетероциклических соединений. М„ Изд-во АН СССР, 1960, стр. 12(1
1. С. L. Arcus, R. Г.. Schaffer. J. Chem. Soc., 24. 28 (1958).
ТЕТРААЛКИЛОВЫЕ ЭФИРЫ ЭТИЛЕНДИАМИНО-N, N-БИС(АЛКИЛФОСФОНОВЫХ КИСЛОТ)
М. В. РУДОМИНО, И. В. ЧУРИЛИНА, Т. я. МЕДВЕДЬ
(RO) 2Р — С - - NHCH2 — С H2NH — С — Р (OR) 2,
II /\ /\ II
OR' R" R' R"O
где R == Н-С4Н9 —; Н-С5Нц—; H-CgHi7—; R' = CH3—
R" = H-CeHi3 —.
Нерастворимые в воде тетраалкиловые эфиры этилендна-мино-N, N'-бис(алкилфосфоновых кислот) могут быть использованы в качестве экстрагентов металлов. Синтез этих соединений мы осуществили впервые конденсацией этилендиамина с кетонами и диалкилфосфитами.
СХЕМА СИНТЕЗА
О
2(RO)2P + 2R' —С —R" + H2NCH2 — CH2NH2 -------щттГ
Ч И -2H2O
О о
-> (RO)2P —C —NHCH2 —CH2NH —С — P(OR)2.
II z\ Z\ II
OR' R" R' R" О
Характеристика основного сырья
Дибутилфосфит, т. кнп. 9373 мм; п™ =1,4240; получение см. [1].
Диамилфосфит, т. кип. 138—-13976 мм; =1,4300; получение см. [2].
12* 179
Таблица 1
Константы и данные анализа
тетраалкиловых эфиров этилендиамино-N, Н-'бис(алкилфосфоновых кислот)
(RO)2 Р - С - NHCH2CH2NH — С - P(OR).,
II /\ /\ II
О R' R" R' R"O
Название эфира и брутто-формула
Найдено, %
II N
Вычислено, %
Тетрабутиловый эфир этиленднаминобис (изопропилфосфоновой кислоты), C24H54N2O6P2 СН3 СНз 72 1,0097 1,4590 143,1 143,0 54,6 54,4 10,3 10,2
Тетрабутиловый эфир этиленднаминобис (изооктилфосфоновой кислоты), CS4H74N2O6P2 СНз CsHig 89 0,9772 1,4605 187,8 188,1 60,7 60,8 11,3 11,4
Тетраамиловый эфир этиленднаминобис (изопропилфосфоновой кислоты), C2SHC2N2O6P2 СНз СНз 86 0,9961 1,4560 159,7 160,4 56,8 56,7 10,9 11,0
Тетраоктиловый эфир этиленднаминобис(изопропилфосфоновой кислоты), C4oH86N206P2 СНз СНз 55 0,9464 1,4579 216,3 215,9 64,9 64,7 11,8 11,7
Р
С Н N Р
Таблица 2
Константы и данные анализа пикнатов тетнаалкиловых эфиров этилендиамино-N, N'-бис(алкилфосфоновых кислот) (RO)2P—с — NHCH2CH2NH - С — Р (OR)2.2C6H2( NO,)3OH
OR' R" R^ITO
R R' R" Т. пл. (из спирта), ’С Найдено, % Вычислено, %
С Н N Р Н2О С Н N Р Н2О
Н-С4Н9 СНз СНз 151,5—152 42,5 42,7 6,0 6,2 11,5 11,5 5,9 6,0 3,2 42,2 6,0 11,0 6,0 3,5
н-С4Н9 СН3 С6Н13 100—102 49,2 49,6 7,6 7,3 10,4 10,4 5,2 5,2 — 49,1 7,2 10,1 5,5 —
н-С5Нц СНз СНз 136—138 46/) 46,0 6,8 7,1 10,9 10,9 5Л — 46,1 6,6 10,8 5,9 —
Н-С8Н|7 СН3 СНз 124—126 51,7 51,9 7,8 7,9 9,3 9,5 4,7 4,7 51,5 7,7 9,3 5,1 —
Диоктилфосфит, т. кип, 175—185°/2 мм; п2£ = 1,4422; получение см. [1].
Этилендиамин, безводн., т. кип. 116,5°; п2^ =1,4540.
Ацетон, ГОСТ 2603—63, ч.
Метилгексил кетон, МРТУ 6—09—2681—65, ч.
Бензол, ГОСТ 5955—68, ч.
Калий углекислый, ГОСТ 4221—65, ч. д. а.
Натрий сернокислый, ГОСТ 4166—66, ч. д. а.
Окись алюминия, для хроматографии, ТУ МХП 2962—54.
Условия получения
В четырехгорлой колбе емкостью 250 мл, снабженной мешалкой, термометром, обратным холодильником и Ч-образной насадкой с двумя капельными воронками, нагревают до 60° 0,25 моля этиленднамина и прибавляют по каплям при перемешивании одновременно из двух воронок 0,5 моля диалкил-фосфита и 0,5 моля кетона, поддерживая температуру реакционной смеси около 60°. Через 15 минут реакционную смесь охлаждают и экстрагируют 500 мл бензола. Бензольный экстракт промывают 40 мл насыщенного раствора углекислого калия, затем 40 мл воды и сушат сернокислым натрием. Бензол удаляют при пониженном давлении. Остаток растворяют в 5-кратном объеме сухого бензола и хроматографируют на колонке с окисью алюминия.
Все полученные эфиры — светлые прозрачные маслообразные вещества; с пикриновой кислотой они образуют кристаллические пикраты. Перечень веществ, их константы и данные анализа представлены в табл. 1 и 2.
ЛИТЕРАТУРА
1. Б. А. Арбузов, Г. И. Виноградова. Изв. АП СССР, сер. хим., 617 (1947).
2. D. Rarnaswatini, Е. R. Kirch. J. Amer. Chem. Soc 75, 1763 (1963).
ТЕТРАОКТИЛОВЫЙ ЭФИР ЭТИЛ ЕНДИАМИНО-N, N'-БИС (БЕНЗИЛФОСФОНОВОЙ КИСЛОТЫ)
М. В. РУДОМИНО, Е. К. КОЛОВА, Т. Я. МЕДВЕДЬ
(Н17С6О)2 Р — сн — NHCH2 — CH2NH — сн — Р (ОС8Н17) 2
II I I II
о С6Н5 С6Н5 о
С48Н8бЬ12ОбР2 М. в. 849,13
Нерастворимый в воде тетраоктиловый эфир этилендиа-mhho-N>N'-6hc (бензилфосфоновой кислоты) может быть использован в качестве экстрагента металлов. Синтез этого соединения осуществлен нами впервые конденсацией диоктилфос-фита с М,'№-этилепбис(бензилиденимином). Последний получен взаимодействием этилендиамина с бензальдегидом в бензольном растворе {1] и очищен перекристаллизацией из эфира.
СХЕМА СИНТЕЗА
H2NCH2 — CH2NH2 + 2C6HSCHO —
(С8Н17О)2РНО
— С6Н5СН = NCH2CH2N = СНСбНг,---------------*
-> (С8Н17О) 2Р — CHNHCH2 — CH2NHCH — Р (ОС8Н17) 2
II I I II
о С6Н5 свн3 о
Характеристика основного сырья
Диоктилфосфит, т. кип. 175—18572 мм; п2^ = 1,4420; получение см. [2].
Этилендиамин безводный, т. кип. 116,5°; п2° = 1,4540.
Бензальдегид, ГОСТ 157—51, ч.
Бензол, ГОСТ 5955—68, ч.
183
Калий углекислый, ГОСТ 4221—65, ч. д. а.
Натрий сернокислый безводный, ГОСТ 4166—66, ч. д. а.
Этиловый эфир, ГОСТ 6265-—52, ч.
Пикриновая кислота, ВТУ 26—55.
Условия получения
Синтез П,П'-этиленбис(бензилиденимина). В круглодонной колбе емкостью 100 мл, снабженной насадкой Дина-Старка и обратным холодильником, кипятят раствор 6,0 г (0,1 М) этилендиамина и 21,2 г (0,2 Л4) бензальдегида в 50 мл бензола до отделения 3,4 мл воды (95% от теории). Затем бензол отгоняют при пониженном давлении; остаток перекристаллизовывают из эфира.
Выход М,М'-этилепбис(бензилиденимина) равен 17,3 г (73,5%); т. пл. 53—54°.
По литературным данным [1], г. пл. 53—54°.
Получение тетраоктилового эфира этилендиамино-N .N' -бис (бензилфосфоновой кислоты). В четырехгорлую колбу емкостью 100 мл, снабженную мешалкой, термометром, капельной воронкой и обратным холодильником, помещают раствор 4,0 г (0,017 М) М,М'-этиленбис(бензилиденимина) в 25 мл бензола и прибавляют при нагревании до 60° и перемешивании по каплям 11,1 г (0,034 М) диоктилфосфита. Массу выдерживают при этой температуре 30 минут, охлаждают, промывают 10 мл насыщенного раствора углекислого калия и 10 мл воды, сушат сернокислым натрием и отгоняют бензол при пониженном давлении.
Выход эфира равен 12,4г (82%); =1,4881; d^= 0,9948.
Вещество представляет собой светлую маслообразную жидкость, закристаллизовывающуюся при стоянии.
Найдено, %: С — 67,6; 67,9; Н — 10,4; 10,5; N—3,5; 3,8; Р —6,9; 7.0;MRD 246,0.
C4SH86N2OeP2. Вычислено, %: С — 67,9; Н—10,2; N — 3,3; Р — 7,3 MRD 245,7.
Вещество образует с пикриновой кислотой кристаллический пикрат, т. пл. 146—148° (из спирта).
Найдено, %: С — 55,0; 55,3; Н — 7,2;
7,3; N —9,0; 8,9; Р — 4,1; 4,0.
C4SHseN2O6P2-2CeH2(NO2)3OH. Вычислено, %: С —55,1; Н —7,1; N — 8,6; Р —4,7.
ЛИТЕРАТУРА
1. С. Mason. Вег., 20,270 (1887).
2. Ф. Л. Мак л я ев, Н. К- Близнюк, Г. И. Еремин. Ж- общ. химии, 30, 4053 (1960).
.184
ТИОКАРБАМОИЛ НИТРОСИНИЙ ТЕТРАЗОЛ Ий
2,2'-Ди-п-нитрофенил-5,5'-ди-га-тиокарбамоилфенил-3,3'-(3,3'-диметокси-4,4'-бифенилен)дитетразолий дихлорид
Л. А. ЛАБУТИНА, В. М. ОСТРОВСКАЯ, Л. В. ЛОМАКИНА, Г. И. МИХАИЛОВ
C42H32O2N12O6S2
М. в. 935,72
В литературе [1] описан метод получения тиокарбамоил -нитросипего тетразолия в четыре стадии: 1) конденсацией п-цианбензальдегида с га-нитрофенилгидразином с образованием га-нитрофенилгидразона га-цианбензальдегида; 2) сочетанием п-нитрофенилгидразона га-цианбензальдегида с диазотированным 3,3'-диметоксибензидином с образованием 2,2'-ди-п-нитрофенил-5,5'-ди-га-цианфенил-3,3'-(3,3'-диметокси -4,4'-бифенилен) диформазана; 3) окислением диформазана до ни-аннитросинего тетразолия — 2,2’-ди-п-питрофепил-5,5’-ди-п-ци-анфенил-3,3'- (3,3'-диметокси-4,4'-бифенилен) дитетразолия ди-185
хлорида; 4) превращение цианнитросипего тетразолия в тио-карбамиолнитросиний тетразолий под действием тиоацетамида.
Нами проверен этот синтез.
СХЕМА СИНТЕЗА
186
Характеристика основного сырья
Аммоний хлористый, ГОСТ 3773—60, ч.
Тиоацетамид, МРТУ 6—09—4095—67, ч.
Изоамилнитрит, ТУ № ОРУ 22—55, ч.
Диметилформамид, МРТУ 6—09—2068—65, ч.
Метанол, ГОСТ 6995—67, х. ч.
3,3’-Димет оксибензидин, МРТУ 6—09—2307—65, ч.
Диоксан, ГОСТ 10455—63, ч., высушен над твердым едким калием и перегнан.
Тетрагидрофуран, высушен над твердым едким калием и перегнан. /
Серная кислота, ГОСТ 4204—66, ч., концентрированная.
Нитрофенилгидразин, ТУ НКХП 387—41, ч.
Уксусная кислота, ГОСТ 61—69, ч. д. а.
4-Цианбензальдегнд, т. пл. 100—ЮГ, получение см. этот сборник, вып. 22.
187
Соляная кислота, ГОСТ 3118—67, ч.
Натрий азотистокислый, ГОСТ 4197—66, ч.
Кали едкое, ГОСТ 4203—65, ч. д. а-
Серный эфир. ГОСТ 6265—52, ч.
Бензол, ГОСТ 5955—68, ч. д. а.
Условия получения
Получение п-нитрофенилгидразона п-цианбензальдегида. В трехгорлую колбу емкостью 250 мл, снабженную шариковым холодильником, мешалкой и делительной воронкой, загружают 15,3 г (0,1 Л1) n-нитрофенилгидразина и 175 мл ледяной кусусной кислоты и при интенсивном размешивании прибавляют 13,1 г (0,1 Л4) п-цианбензальдегида. Выпавший желтый осадок отфильтровывают, промывают 25 мл ледяной уксусной кислоты и сушат в вакуум-эксикаторе.
Выход n-нитрофенилгидразона п-цианбепзальдегида равен 27,5 г (89,8%); т. пл. 208—210°.
После перекристаллизации из 500 мл ацетона получают 21.5 г (70,3%) продукта; т. пл. 216ф218о.
По литературным данным [1], г. пл. вещества 218°.
Получение 2,2’-ди-п-нитрофенил-5,5’-ди-п-цианфенил-313’~ (3,3'-диметокси-4,4'-бифенилен)диформазана. В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, капельной воронкой, термометром и помещенную в охлаждающую баню, загружают 10 г (0,032М) 3,3’-диметоксибензидина солянокислого и 48 мл дистиллированной воды. К суспензии прибавляют 12 мл концентрированной соляной кислоты и охлаждают до минус 5°.
Отдельно готовят раствор 4,8 г (0,07 М) азотистокислого натрия в 12 мл воды, охлаждают его до 0° и за 30 минут прибавляют к реакционной смеси, поддерживая температуру минус 5—0°. Конец диазотирования определяют по реакции пробы раствора с йодкрахмальной бумагой (диазотирование считают оконченным, если через 10 минут после добавления 1 капли диазораствора иодкрахмальная бумажка синеет). Полученный диазораствор, не вынимая из охлаждающей бани, быстро фильтруют от незначительного количества черного осадка и оставляют раствор в ледяной бане.
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром, капельной воронкой и помещенную в охлаждающую баню, загружают 17,2 г (0,064 Л1) п-нитрофенилгидразона п-цианбензальдегида и 140 мл тетрагидрофурана. Поддерживая температуру реакционной смеси минус 28°±30° охлаждением смесью сухой углекислоты в ацетоне, прибавляют при тщательном перемешивании диазораствор. Затем прибавляют струйкой раствор 8 г едкого кали в 15 мл воды, поддерживая температуру в колбе минус 25е в течение 188
1 часа. После этого температуру массы доводят до комнатной, добавляют 64 мл воды и 140 мл метанола и оставляют на ночь. Осадок отфильтровывают, промывают 1 л метанола и высушивают на воздухе.
Выход диформазана равен 20,1 г (38,5%).
Вещество загружают в экстрактор Сокслета и экстрагируют бензолом в течение 16 рабочих дней.
Выход продукта после экстракции равен 6,6 г (10,8%); т. пл. 300—304° (разлож.).
По литературным данным [1], т. пл. вещества 300°.
Получение цианнитросинего тетразолия. В трехгорлую колбу емкостью 250 мл, снабженную капельной воронкой, мешалкой, барботером и помещенную в охлаждающую баню, загружают 5,0 г (0,006 А4) диформазана, 80 мл диоксана и 80 мл тетрагидрофурана. К полученной суспензии по каплям прибавляют 6 мл (0,044 М) изоамилнитрита. Поддерживая температуру реакционной смеси 0°, за 2,5 часа пропускают хлористый водород, после чего добавляют еще 2 мл (0,015 М) изоамилнитрита. Реакционную массу размешивают при комнатной температуре 4 часа и оставляют на ночь. Выпавший осадок отфильтровывают.
Выход тетразолия равен 3,2 г (59,2%); т. пл. 198—199°.
Вещество растворяют в метаноле и осаждают эфиром (1:10).
Выход тетразолия равен 2,2 г (40,7%); т. пл. 208°т-209° (разлож).
По литературным данным [1],т. пл. вещества 208° (разлож.).
Получение тиокарбамоилнитросинего тетразолия. В трехгорлую колбу емкостью 250 мл, снабженную холодильником, .хлоркальциевой трубкой, мешалкой, барботером и помещенную в охлаждающую баню, загружают 1,4 г (0,0016 Af) цианнитросинего тетразолия и 90 мл диметилформамида. Поддерживая температуру реакции 0°, в течение 1 часа пропускают хлористый водород, при этом масса затвердевает.
Реакционную смесь нагревают на глицериновой бане (температура бани 100°), добавляют 2,2 г (0,03 Af) тиоацетамида и выдерживают 1 час при 100°. Затем температуру смеси доводят до комнатной и вливают в нее 250 мл дистиллированной воды. Выпавший осадок отфильтровывают.
Выход тетразолия равен 1,2 г (80%). Вещество растворяют в диметилформамиде с активированным углем, фильтруют и высаживают эфиром (1 :10).
Выход тетразолия равен 0,9 г (60%); т. пл. 192ф194° (раз-ложЛ.
По литературным данным |[1], г- 11л- вещества 192° (раз-ЛОЖ')' ЛИТЕРАТУРА
1. А. М. Seligman, Н. Ueno et al. J. Histochem. and Cytochem., 15,1 (1967).
189
ТИОКАРБОГИДРАЗИД
Тиоугольная кислота, дигидразид
В Л1. ОСТРОВСКАЯ, Л. В. ЛОМАКИНА
NHNH2
s = с
NHNH2
CHeN4S
М. в. 106,15
Тиокарбогидразид применяется в электронно-гистохимических исследованиях в качестве контрастирующего реагента, для улучшения осмиофильности продуктов реакции в гистохимических срезах [1].
Тиокарбогидразид получают действием тиофосгеиа на гидразин-гидрат (2,3], кипячением этилового эфира этилксан-тогеновой кислоты с гидразин-гидратом в этаноле [4].
Нами проверен первый способ получения тиокарбогидразида.
СХЕМА СИНТЕЗА
2H2N —NH2+S = CZ —> ^Cl
nhnh2
S =- CZ
\ nhnh2
Характеристика основного сырья
Гидразин-гидрат, ГОСТ 5832—65, ч. д- а.
Тиофосген, ТУ ОЭЗ 57—65, ч.
Диэтиловый эфир, ГОСТ 6265—52, ч.
190
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой с затвором, капельной воронкой и термометром, загружают 82 мл (1,69 М) гидразин-гидрата и 100 мл диэтилового эфира, охлаждают при размешивании до 5° и прибавляют по каплям смесь 12 г (0,1 М) тиофосгена с 20 мл диэтилового эфира. Массу размешивают несколько часов и оставляют на двое суток при комнатной температуре. Выпавший осадок отфильтровывают, промывают на фильтре небольшим количеством охлажденной до 0° воды и перекристаллизовывают из 115 мл дистиллированной-воды. Осадок сушат в вакуум-эксикаторе над фосфорным ангидридом.
Выход тиокарбогидразида равен 7,6 г (71%). Вещество представляет собой блестящие белые кристаллы с т. пл. 170° (разл.).
По литературным данным, т. пл. 168° (разл.) [2, 3], 170° (разл.) [4].
ЛИТЕРАТУРА
1. J. S. Hanker, Ch. Deb etal. Science, 152, 1631 (1966).
2. R. S to lie, P. E. Bowles. Ber., 41, 1099 (1908).
3. W. Autenrieth, H. Hefner. Ber., 58,2154 (1925).
4. P. Ch. Guha, S. Ch. De. J. Chem. Soc„ 1924, 1216.
ТИОМОЧЕВИНА
И. Ф КОНДРАШОВА, Е. Я. ЯРОВЕИКО
H?N
\С = S
H2N
ch4n2s
М. в. 76,12
Наиболее распространенным способом получения тиомочевины является способ взаимодействия цианамида кальция с сульфидом аммония [1—3]. Нами предложен метод получения тиомочевины из цианамида кальция и сульфида аммония в присутствии соляной кислоты в качестве катализатора, что увеличивает выход продукта (с 80—85% до 96—98%) и улучшает его качество {4].
СХЕМА СИНТЕЗА
н£0
CaCN2 + (NH4)2S -------(NH2)2CS + Ca(OH)2
Характеристика основного сырья
Цианамид кальция, ГОСТ 1780—56, техн.
Аммоний односернистый, раствор, ГОСТ 3767—56, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Аммоний углекислый, ГОСТ 9325—60, ч.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой и термометром, загружают 222 мл раствора тиомочевины (концентрация 115,7 г/л) (см. примечание 1), 200 г (1,4 М) 132
цианамида кальция (содержание азота 19,5%), 0,2 мл соляной кислоты и 462 мл раствора сульфида аммония (концентрация 205,7 г/л). При энергичном размешивании поднимают температуру реакционной массы за 0,5 часа до 80° и при этой температуре смесь выдерживают 1 час 40 минут. Выпавшую гидроокись кальция отфильтровывают и промывают 800 мл горячей воды до исчезновения тиомочевины в промывных водах (проба на азотновисмутовую бумажку). К фильтрату добавляют 10 г углекислого аммония и 1 г активированного угля и нагревают 10 минут при 55—60°. Осадок углекислого кальция отфильтровывают и промывают водой до исчезновения тиомочевины в промывных водах. Фильтрат упаривают при пониженном давлении 50—55/°60—65 мм (см. примечание 2). Осадок отфильтровывают и сушат при 80—85°.
Выход тиомочевины равен 101,6—103,9 г (96—98% в расчете на цианамид кальция); содержание основного вещества 98—99,5% (определено меркурометрическим титрованием).
Примечания.
1. Водный раствор тиомочевины берется для понижения растворимости продукта и уменьшения образования побочных продуктов.
2. Фильтрат (136 мл) содержит 189 г/л тиомочевины и может быть использован для следующего синтеза.
ЛИТЕРАТУРА
1. В. Г. Б р у д з ь, Р. Л. Глобус. Авт. евнд. 101314 (1956); Бюлл. изобр., 9, 11 (1955).
2. Англ. пат. 903864 (1962); С. А., 58, 4433 (1963).
3. Франц, пат. 655457 (1927); С. А. II, 487 (1927).
4. Е. Я. Яровенко, М. Ф. Кондрашова, С. П. Медведева. Авт. свид., 318574; Открытия, Изобретения. Промобразцы. Товарные знаки, № 32 (1971).
13—1192
4,5,6-ТРИМЕТОКСИФТАЛ ИД
И. М. РЫБКИНА, М. Е. ТИХОМИРОВА
ОСНз
НзСО I сн.
/ч/\ /
Н3СО С
II
О
СПН12О5 м. в. 224,21
Из литературных данных известно, что 4,5,6-триметокси-фталид получают кипячением диоксанового раствора 3,4,5-триметоксибензойной кислоты с формалином и соляной кислотой [1].
Нами проверен и уточнен этот метод.
СХЕМА СИНТЕЗА
ОСНз НзСО 1 JL-COOH НзСО осн3 НзСО I снг HCHO \ / \ / \ НС1 || z^/\ / НзСО с II О
194
Характеристика основного сырья
3,4,5-Триметоксибензойная кислота, т. пл. 167°—168°; получение см. [2].
Формалин, марки ФМ, ГОСТ 1625—61.
Диоксан, ГОСТ 10455—63, ч.
Соляная кислота, концентрированная, ГОСТ 3118—67, ч Хлороформ, ГОСТ 3160—51, х. ч.
Натр едкий, ГОСТ 4328—66, ч.
Уксусноэтиловый эфир, СТ ГОХГ 27—1839, ч.
Условия получения
В круглодонную колбу емкостью 2 л, снабженную шариковым холодильником, загружают 100 г (0,47 М) 3,4,5-три-метоксибензойной кислоты в 600 мл диоксана, 250 мл 37%-ного (3,3 М) раствора формалина и 450 мл соляной кислоты. Реакционную смесь кипятят 1 час на водяной бане, охлаждают до 18—20°, выливают в 2,5 л дистиллированной воды и оставляют стоять 12 часов. Осадок отфильтровывают и сушат в сушильном шкафу при 65—70° до постоянного веса.
Технический продукт растворяют в 300 мл хлороформа и хлороформный раствор промывают 1 н. раствором едкого натра 5 раз порциями по 100 мл. Хлороформ отгоняют. Остаток перекристаллизовывают из 300 мл уксусноэтилового эфира.
Выход 4,5,6-триметоксифталида равен 38 г (35,9%); т. пл. 135—136°.
По литературным данным [1], т. пл. 135—136°.
ЛИТЕРАТУРА
1. Е. W h i t е, М. В и г s е у. J. Organ. Chem., 31, б, 1912—1917 (1966).
2. Синтезы органических препаратов, вып. 1. М., Инлитиздат, 1953, стр. 406.
13*
2,3,6-ТРИОКСИБЕНЗИЛИМИНОДИУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИИ, В. я. ТЕМКИНА, Н, В. ЦИРУЛЬНИКОВЛ, Л. я. ШИЛЕНКО
он
- он
CH2N(CH2COOH)2
он
CuHi3NO7 М. в. 271,23
Синтез 2,3,6-триоксибензилиминодиуксусноЙ кислоты в литературе не описан. Вещество может найти применение в качестве хелатореагента.
2,3,6-Триоксибензилиминодиуксусная кислота получена взаимодействием оксигидрохипона с формальдегидом и ими-нодиуксусной кислотой в уксуснокислой среде. Строение соединения предложено на основании изучения спектров ЯМР [1].
СХЕМА СИНТЕЗА
ОСОСНз ОН
^\~ОСОСН3 НС1 ОН CH2O;HN(CH2COOH)2
II сдан I I СН..СООН
I I
ОСОСНз он
он
{ \ - он
' J—CH2N(CH2COOH)2
196 ОН
Характеристика основного сырья
Пирогаллол «А», ГОСТ 6408—52, ч. д. а.
Формальдегид, ГОСТ 1625—61, техн. 37%-ный раствор.
Иминодиуксусная кислота, ВТУ МГУХП 190—58, ч.
Соляная кислота, ГОСТ 3118—67, ч., пл. 1,19.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч. Натр едкий, ГОСТ 4328—66, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57, техн.
Условия получения
Синтез 1,2,4-триоксибензола. В колбу емкостью 100 мл, снабженную обратным холодильником и погруженную в баню с обогревом, помещают 16,5 г (0,065 Л4) пирогаллола «А», 33 мл этилового спирта, 2,5 мл концентрированной соляной кислоты и 1,6 г активированного угля. Содержимое колбы кипятят 1 час при 75—78°. Горячий раствор фильтруют; фильтрат переносят в колбу Кляйзена емкостью 100 мл и отгоняют при пониженном давлении (50—100 мм) избыток этилового спирта и образовавшийся в ходе реакции этилацетат (температура бани не выше 60°) до появления кристаллов на стенках колбы. Остаток выливают в фарфоровую чашку и оставляют кристаллизоваться на 20 часов. Выпавший осадок отфильтровывают и сушат в вакуум-эксикаторе.
Выход 1,2,4-триоксибензола равен 4,8 г (57,9% в расчете на пирогаллол «А»).
1,2,4-Триоксибензол представляет собой кристаллическое вещество серовато-голубого цвета; перекристаллизованный из воды продукт имеет т. пл. 140°.
По литературным данным [2], т. пл. 140°.
Получение 2,3,6-триоксибензилиминодиуксусной кислоты.
В трехгорлой крлбе емкостью 50 мл, снабженной мешалкой, капельной воронкой, обратным холодильником и помещенной в баню, растворяют при размешивании 2,5 г (0,02 М) 1,2,4-триоксибензола в 25 мл ледяной уксусной кислоты. Затем в колбу приливают водный раствор мононатриевой соли имиподиуксусной кислоты, при! отовленный следующим образом: 2,7 г (0,02 М) иминодиуксусной кислоты помещают в стакан, туда же приливают 3 мл дистиллированной воды и при размешивании медленно нейтрализуют полученную суспензию 30%-ным раствором едкого натра до pH 7—8 по универсальной индикаторной бумаге.
Содержимое колбы охлаждают в ледяной бане до 5—10° и, поддерживая эту температуру, при перемешивании за 20 минут прибавляют по каплям 1,5 г (0,02 М) 37%-ного раствора формальдегида. Затем температуру в бане поднимают до 20° и раствор размешивают при этой температуре 2 часа.
197
Выпавший осадок комплексона отфильтровывают, промывают на фильтре 100 мл дистиллированной воды и сушат при 55—60°.
Выход 2,3,6-триоксибензилиминодиуксусной кислоты равен 4,3 г (77,7% в расчете па оксигидрохинон).
Вещество представляет собой порошок слегка кремового цвета, плохо растворимый в воде, нерастворимый в органических растворителях.
Найдено, %: С — 49,0; 49,0; Н — 4,6; 4,8; N — 5,1; 5,3.
CnHi»NO-. Вычислено, %: С — 48,7; Н — 4,8; N—5,2.
ЛИТЕРАТУРА
1. Н. В. Ц и р у л ь н и к о в а, Н. А. Костромина и др. Ж. орган, химии, 7. 327 (1971).
2. Справочник химика, т. 2. М.-Л., Госхимиздат, 1963, стр. 616.
ТРИС(Р-ЦИАНЭТИЛ)ФОСФИН Фосфинидинтрис(3,3'3-пропионитрил)
М. В. РУДОМИНО, И. В. ЧУРИЛИНА, Т. я. МЕДВЕДЬ,
ncch2ch2- р—ch-ch2cn
ch2ch2cn
C9H12N3P М. в. 193,19
Трис ((3-цианэтил) фосфин применяется в качестве комплек-сообразователя в синтезе катализаторов для полимеризацион-ных процессов [1].
Трис (р-цианэтил) фосфин получают взаимодействием акрилонитрила с газообразным фосфином (1,2] или с трис-(оксиметил) фосфином [3,4]. Проверка показала, что последний метод, хотя и является более удобным в лабораторных условиях, дает низкий выход. Это связано с крайней неустойчивостью и склонностью к окислению исходного трис(окси-метил) фосфина, который получают обычно действием оснований на тетра (оксимегил)фосфонийхлорид [5, 6].
Мы установили, что выход трис (р-цианэтил) фосфина существенно увеличивается, если трис (оксиметил) фосфин получать действием спиртовой щелочи на спиртовый раствор тетр а (оксиметил) фосфонийхлорида и далее без выделения его из раствора вводить в реакцию с акрилонитрилом.
СХЕМА СИНТЕЗА
С2Н.-,ОН CH.—CHCN
(НОСН2)4РС1 + NaOH а - (ОНСН2)3Р -------------------
-> (NCCH2CH2)3P + ЗСН2О
199
Характеристика основного сырья
Тетра (оксиметил) фосфонийхлорид, т. пл. 143—146°; получение см. [7].
Акриловой кислоты нитрил МРТУ 6—09—2487—65, ч.
Этиловый спирт абсолютный, ГОСТ 9674—61, ч.
Натр едкий, ГОСТ 4328- 66, ч.
Условия получения
В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой, капельной воронкой, обратным холодильником и трубкой для подвода инертного газа, помещают раствор 41,9 г (0,22 М) тетра (оксиметил) фосфонийхлорида в 160 мл абсолютного этилового спирта; колбу тщательно продувают сухим аргоном, затем при перемешивании прибавляют по каплям горячий (70°) раствор 8,8 г (0,22 М) едкого натра в 175 мл абсолютного этилового спирта. Реакционную смесь фильтруют в токе аргона в трехгорлой колбе. По окончании фильтрования колбу снабжают мешалкой, капельной воронкой и обратным холодильником и к ее содержимому прибавляют по каплям при перемешивании 35 г (0,66 Л1) нитрила акриловой кислоты, поддерживая температуру 40—45°.
Через 10—15 минут выпадает бесцветный кристаллический осадок. После 12-часовой выдержки при 20° его отфильтровывают и сушат в вакуум-эксикаторе над хлористым кальцием.
Выход трис(0-цианэтил)фосфина равен 34,2 г (80%); т. пл. 96—97°.
По литературным данным (3, 4], т. пл. 97°.
ЛИТЕРАТУРА
1. Пат. США 2822376 (1958); РЖХим., 1959, 83178.
2. М. М. Rahu t, J. Hechenbleickner et al. J. Amer. Chemi Soc., 81, 1103 (1959).
3. Пат. ФРГ 1082910 (1960); РЖХим., 1962, 5л112П.
4. Р. К. Ва л е т д и и о в, Е. В. Кузнецов, С. Л. Комиссарова. Ж. общ. химии, 39, 1744 (1969).
5. К. А. Петров, В. А. Паршина, М. В. Л у з а н о в а. Ж- общ. химии, 32, 554 (1962).
6. К J. С о s k г a n, J. С. V е г к a d е. Inorgan. Chem.,4, 1655 (1965).
7. W. A. Reeves, F. F. Flin, J. D Guthrie. J. Amer. Chem-Soo., 77, 3923 (1955).
ФЕНОЛОВЫЙ КРАСНЫЙ Фенол сульфофталенн
Л. П. БОЛДЫРЕВА, А. У. КАГАНОВСКАЯ, Г. И. МИХАИЛОВ
НО он
\/х /Ч/
W\ /V с
I Хо
S02 • Н2О
C19H14O5S • — Н2О М. в. 358,87
4
Феноловый красный является кислотно-щелочным индикатором с переходом окраски от желтой к красной при pH 6,8—8,4. Нами установлено, что чистый образец фенолового красного имеет второй переход окраски в кислой области ог розовой к желтой при pH 0,7—2,0. Реактив применяется при определении концентрации водородных ионов, для получения галоидзамещенных, водорастворимых и универсальных индикаторов [1—4], в медицине и вирусологии [5].
Феноловый красный получают конденсацией фенола с О-сульфобензойной кислотой, ее дихлорангидридом, ангидридом или моноамонийной солью. Синтез проводят в присутствии хлорокиси цинка {6], пятиокиси фосфора [7], смеси хлорокиси фосфора и хлористого цинка [8] или смеси хлорокиси фосфора и хлорной кислоты [9].
201
Нами предложен способ конденсации о-сульфобензойной кислоты с избытком фенола в присутствии смеси хлорокиси фосфора и хлорной кислоты [10]. Очистку фенолового красного путем переоеаждения из аммиака соляной кислотой [11] мы заменили промывкой горячен водой и экстракционной промывкой п-бутиловым спиртом, что позволило получить высокрчистый (96—98%-ный) электрофоретически однородный индикатор.
СХЕМА СИНТЕЗА
Характеристика основного сырья
о-Сульфобензойная кислота, МРТУ 6—09—811—63, ч.
Фенол, ГОСТ 6417—52, ч.
Хлорокись фосфора, МРТУ 6—09—337—63, ч.
Хлорная кислота, МРТУ 6—09—6604—70, ч.
н-Бутиловый спирт, ГОСТ 6006—51, ч.
Условия получения
В четырехгорлую колбу емкостью 2,5 л, снабженную мешалкой с затвором, обратным холодильником (см. примечание 1), капельной воронкой и термометром, загружают 103 г (0,5 М) безводной о-сульфобензойной кислоты (см. примечание 2), 15 мл 57%’-ной хлорной кислоты, 15 мл хлорокиси фосфора (см. примечание 3) и 150 мл предварительно расплавленного фенола (см. примечание 4). За 15—20 минут
202
при энергичном размешивании нагревают реакционную смесь до 120—130° и при этой температуре в течение 1 —1,5 часа прибавляют по каплям 45 мл хлорокиси фосфора, не допуская сильного вспенивания реакционной массы. Затем реакционную массу выдерживают в тех же условиях еще 30—40 минут до образования однородной кашеобразной консистенции, после чего прибавляют при размешивании 1 —1,5 л горячей воды и кипятят 1 час.
Горячий раствор отфильтровывают и осадок промывают 5—6 л горячей воды при размешивании (см. примечание 5), отжимают и кипятят 2—3 раза с 400 мл н-бутилового спирта по 40—50 минут, каждый раз применяя свежие порции спирта (см. примечание 6). Осадок сушат при 100—110° в течение 4—5 часов до постоянного веса.
Выход фенолового красного равен ПО—115 г (60—63% в расчете на о-сульфобензойную кислоту); содержание основного вещества 96—98%.
Индикатор обладает двумя переходами окраски: от желтой к красной при pH 6,8—8,4 и от розовой к желтой при pH 0,7—2,0; максимальное поглощение в кислой среде (pH 1) при 505 нм, в щелочной среде (pH И) при 558 нм.
По литературным данным [12], феноловый красный имеет интервал перехода окраски от желтой к красной при pH 6,8— 8,4; максимальное поглощение в щелочной среде (pH 11) при 558 нм.
Примечания.
1. Выделяющийся в процессе синтеза хлористый водород улавливают раствором едкого натра в склянке Тищенко, присоединенной к обратному холодильнику.
2. Для получения безводной о-сульфобензойпой кислоты 130 г кристаллической о-сулвфобензойной кислоты нагревают 3—4 часа в открытом фарфоровом стакане при размешивании и температуре 140—145°, после чего расплавленную кислоту выливают в фарфоровую чашку и хранят в эксикаторе иад безводным хлористым кальцием. Выход безводной о-сульфобензойной кислоты равен 103 г; т. пл. 139—141°.
3. Применяют свежеперегнанную хлорокись фосфора, кипящую при 106—108°.
4. Применение избытка фенола устраняет к концу синтеза загустевание реакционной массы и обеспечивает хорошее размещивание.
5. Фенол может быть регенерирован из промывных вод в виде азеотропа.
6. н-Бутиловын спирт после экстракционной промывки фенолового красного может быть регенерирован путем отгонки с выходом 80% и использован при последующих синтезах.
ЛИТЕРАТУРА
1. Л. М. Кульберг. Тр. Всес. конференции по аналит. химии. 2 323 (1943).
2. A. Meretoja. Suomen. Kemistileht, 18А, 121 (1945).
3. Г. И. Михайлов Заводск. лаборатория, 21, 156 (1955).
203
4. L. М. К о 1 th о f f. Analist., 21, 101 (1949).
5. W. Fabricios, В, Marowski. Z. klin. Chem. und klin Bio. chem., 4 8 (1970).
6. W. K- Orndorff, F. W. Ch er wood. J. Amer. Chem. So?.. 45,487 (1923).
7. J. Chrzaszczewski, M Kosinsk'. Zesz. nauk. Univ. Lodz. Ser. II, 3, 143 (1957).
8. Г. И. Михайлов, Г. H. Кошелева. Авт. свид. 104125; Бюлл. пзобр. № 8, 17 (1956).
9. В. Я- Я р о в е н к о. М. Ф. Кондрашова, Т. Г. Г а й в о р о н-с к а я, Г. И. Михайлов. Авт. свид. 187191; Бюлл. изобр., № 20, 90 (1966).
10. Г. И. Михайлов, Н. С. Мезенцева, А. П. Болдырева. Авт. свид. 232277. Изобретения. Промобразцы. Товарные знаки, № 1, 30 (1969).
11. Е. Я- Я P о в е н к о, М. Ф. Кондрашова. Методы получения химических реактивов и препаратов, вып. 20. М., ИРЕА, 1969, стр. 212.
12. Р. Бейтс. Определение pH. Теория и практика. Л., «Химия», 1968, стр. 127.
ФЛУОРЕСЦЕИНДИАЦЕТАТ
3,6-Днацетокснфлуоран, днацетнлфлуоресцеин
И. М. РЫБКИНА, М. Е. ТИХОМИРОВА О
Н3СОСО-|^\/ \/^|-ОСОСН3
Ч/\ Z\/
С
I \
I О
С24Н16О7 4Z М. в. 416,37
Флуоресцеиндиацетат получают кипячением флуоресцеина с уксусным ангидридом [1—3].
Нами подобраны оптимальные условия проведения этой реакции.
СХЕМА СИНТЕЗА
О
он
\s\s\s с
I \ (СН3СО)2О
I о -----
/ч 1 п
II г
о
НзСОСО—ОСОСНз
Ч/\ /W
с
I \
I О
\ Г1_0
I г
4z
205
Характеристика основного сырья
Флуоресцеин, ТУ МХП 2803—55, ч-
Уксусный ангидрид, ГОСТ 5815—52, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Уголь активированный, марки БАУ, ГОСТ 6217—52.
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную шариковым холодильником, мешалкой, термометром и погруженную в масляную баню, загружают 20 г (0,06 М) флуоресцеина и 20 мл (21,6 г; 0,21 М) уксусного ангидрида. Смесь при размешивании кипятят 30 минут (140—145°), при этом флуоресцеин полностью растворяется, прибавляют 0,2 г активированного угля и снова кипятят 3—5 минут. Горячий раствор фильтруют и охлаждают в бане со льдом до 5°. Выпавшие кристаллы отфильтровывают, промывают 15 мл этилового спирта и сушат в сушильном шкафу при 120—130' до постоянного веса.
Выход технического флуоресцеиндиацетата равен 21—22г (86—88%); т. пл. 200—202°
Вещество перекристаллизовывают из уксусного ангидрида (весовое соотношение 1:2) с активированным углем и получают флуоресцеиндиацетат в виде белых или слегка желтоватых кристаллов с т. пл. 203—204°.
Выход чистого продукта равен 19—19,5 г (76—78%).
По литературным данным, т. пл. 200° [1, 2]; 201° [3]; 205— 206” [4].
ЛИТЕРАТУРА
1. Е. Fischer. Вег., 7, 1212 (1874).
2. Torr w, В res vs ter. J. Amer' Soc., 30, 863 (1908).
3. A. Baeyer. Ann., 183, 13 (1876).
4. H. L i b i g. J. Prakt. Chem., 85, 241 (1912).
ФЛУОРЕСЦЕИНДИБУТИРАТ
И. М. РЫБКИНА, И. Н. ПИМЕНОВА О
ГЬСзОСО-^/ \/^-ОСОСзН7 ч/\ zw с
I \ | о ' 1 ^^-С=О C2SH24O7 'Чк/ М.. в. 472,47
Флуоресцеиндибутират получают кипячением флуоресцеина с масляным ангидридом в пиридине [1].
Нами проверен этот способ, уточнены условия выделения и очистки продукта.
СХЕМА СИНТЕЗА
О НО—\ZX_q ч/\ ZV с 1 \ 1 О \ Г1 _ Г) 1II ZZ О H7C3OCO-^\-z 4 / Ч CZ 1 '> 1 1 z\ L/ и (С3Н7СО)2О —> - ОСОС3Н7 z\z 4 0 1 —С=О 207
Характеристика основного сырья
Флуоресцеин, ТУ МХП 280355, ч.
Масляной кислоты ангидрид, ВТУ УХП 50—58, ч.
Пиридин, ГОСТ 2747—67, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Активированный уголь, ГОСТ 6217—52, те.хн.
Условия получения
В трехгорлую колбу емкостью 100 мл, погруженную в масляную баню и снабженную шариковым холодильником, мешалкой и термометром, загружают 10 г (0,03 Л1) флуоресцеина. 15 мл (14,9 г; 0,09 Л4) масляного ангидрида и 7 мл пиридина. Смесь при размешивании кипятят 1 час, охлаждают до 15—20° и при энергичном размешивании выливают в 200 мл ледяной воды. Флуоресцеиндибутират выпадает вначале в виде масла, которое по мере размешивания закристал-лизовывается. Коричневые маслянистые кристаллы отфильтровывают и промывают 20 мл этилового спирта.
Выход сырого флуоресцеиндибутирата равен 13—13,5 г, (92—95%); т. пл. 107—110°.
Вещество дважды перекристаллизовывают из этилового спирта (весовое соотношение 1:5) с активированным углем и получают белый с сероватым оттенком порошок с т. пл. 114—116°.
Выход чистого флуоресцеиндибутирата равен 10—10,3 г (70-72,5%).
По литературным данным [1], т. пл. 118—119°.
ЛИТЕРАТУРА
I. D. N. Kramer, О. G. Guilbault. Analyt Chem., 35,588 (1963).
208
а-ФРУ КТО ЗА
Левулеза
Н. В. ПЛЕХАНОВА, М. И. ЧЕРНЯХОВСКАЯ, Е. П. КРЫСИН, Г. П. ФЕДОРЧЕНКО, Н. Н. ГОЛЯЕВА, В. И. ИВАНОВ
CeHijOe
М. в. 180,16
а-Фруктоза используется в микробиологической практике. Источником получения ее служит инулин {1]. Нами предложено использовать в качестве сырья для получения инулина и а-фруктозы корни инули крупной высокой. Разработан метод получения этих соединений [2].
Характеристика основного сырья
Корни инули крупной высокой.
Кальций углекислый, ГОСТ 4530—66.
Свинца окись, ГОСТ 9199—68.
Свинец уксуснокислый, ГОСТ 1027—67-
Натрий сернистый, ГОСТ 2053—66.
Этиловый спирт гидролизный, ГОСТ 5962—67, в/о.
Серная кислота, ГОСТ 4204—66-
Условия получения
Получение инулина. В двугорлую колбу емкостью 2 л, снабженную мешалкой и термометром, помещают 150 г мел-коизмельченных корней инули (см. примечание 1), 1,5 л горячей (85°) воды, прибавляют 3 г углекислого кальция (pH 14-1192 209
6—6,5). Содержимое колбы нагревают при перемешивании до 85° и выдерживают в этих условиях 40 минут.
Смесь быстро фильтруют с отсасыванием через бязевый фильтр. Шрот промывают дистиллированной горячей (85°) водой два раза порциями по 50 мл. Промывные воды присоединяют к фильтрату. Полученным экстрактом (^1500 мл) быстро заливают новую порцию тоикоизмельчснных корней (150 г) и вновь проводят описанные выше операции. В результате обработки 600 г корней получают 1500 мл экстракта с pH 6,5. t :
К 1500 мл экстракта при нагревании до 50—55° и перемешивании прибавляют 6 г активированного угля, 6 г силикагеля и 17,5 мл раствора уксуснокислого свинца (см. примечание 2) и делают пробу на полноту осаждения (см. примечание 3). Реакционную массу выдерживают без перемешивания 30 минут, затем фильтруют с отсасыванием. Остаток на фильтре промывают 150 мл дистиллированной горячей (55°) воды.
К объединенному фильтрату прибавляют 1,5 г активированного угля и 1,5 г силикагеля и, осторожно, частями при перемешивании приливают 12 мл 40%-ного водного раствора сернистого натрия, после чего делают пробу на полноту осаждения (см. примечание 4).
Реакционную смесь перемешивают 15 минут, выпавший осадок отфильтровывают с отсасыванием через двойной бумажный фильтр и промывают 50 мл дистиллированной горячей (50—55°) воды.
Объединенный фильтрат (1500—1530 мл) упаривают при пониженном давлении (40—60 мм) на водяной бане вначале при 60° (температура бани), а к концу упарки при 55° до получения раствора с показателем рефракции 1,373—1,375. К раствору прибавляют 1,5 г активированного угля и 1,5 г силикагеля и фильтруют через воронку для горячего фильтрования.
К фильтрату добавляют двойной объем этилового спирта, размешивают и оставляют на 36—40 часов. Смесь фильтруют с отсасыванием, первоначально декантируя верхний прозрачный слой. Осадок промывают на фильтре спиртом дважды порциями по 50 мл и сушат при 25°.
Получают 120 г инулина.
Получение фруктозы. В трехгорлой колбе емкостью 1,5 л, снабженной мешалкой, холодильником и термометром и помещенной в водяную баню, нагревают до 85° 900 мл 0,5 %-ной серной кислоты и при медленном размешивании растворяют 100 г инулина.
Смесь нагревают 15 минут при 85° с 1 г активированного угля, охлаждают за 3-^5 минут до 55—50° и нейтрализуют
210
4,5 г углекислого кальция до pH 6,0—6,5 по универсальной индикаторной бумаге.
Осадок сернокислого кальция и угля отфильтровывают через двойной бумажный фильтр и промывают дистиллированной горячей (50°) водой дважды порциями по 30 мл. Объединенный фильтрат упаривают в вакууме водоструйного насоса (20—35 мм) до образования сиропа с показателем рефракции 1,498—1,499.
Полученный сироп растворяют при нагревании (72—74°) и постоянном перемешивании в 400 мл 96%-ного этилового спирта, добавляют 2 г активированного угля и продолжают перемешивание егце 5 минут. Раствор фильтруют. Осадок на фильтре промывают горячим (50°) 96%-ным этиловым спиртом дважды порциями по 25 мл. Промывные воды присоединяют к фильтрату, дают охладиться до 32—34° и прибавляют 0,1 г кристаллической а-фруктозы в качестве затравки. Раствор перемешивают и оставляют кристаллизовываться при 5° в течение 4—5 дней. Выпавшие кристаллы отфильтровывают п сушат при 30°.
Выход фруктозы равен 82 г (82% от инулина и 16,4% от веса корней инули крупной высокой).
Препарат, представляющий собой белый кристаллический порошок, хроматографически однороден эй"—92°.
Примечания.
1. Измельченные корни инули должны содержать не менее 13% инулина и не больше 11% влаги. Срок храпения корней 1 год.
2. Раствор основного уксуснокислого свинца приготавливают следующим образом: 165 г окиси свинца тщательно взмучивают в 165 мл воды и при перемешивании приливают горячий (почти кипящий) раствор 270 г уксуснокислого свинца в 900 мл воды. После 15 минут перемешивания раствор оставляют для отстаивания и используют в дальнейшем верхний прозрачный раствор.
3. К 5 мл отфильтрованного горячего (50°) экстракта прибавляют 2 капли раствора основного уксуснокислого свинца. В течение полминуты не должен выпадать осадок.
4. К 5 мл отфильтрованного раствора прибавляют 1 каплю 10%-ного раствора сернистого натрия, при этом испытуемый раствор должен остаться бесцветным.
ЛИТЕРАТУРА
1. И. П. Ром и п с кий. Фруктоза и инулин. Изд-во АН УССР, 1959.
2. Е. П. К р ы с и н, И. В. Плеханова, М. И. Черняховская, В И. Иванов. Авт. свид. 183146;. Изобретения. Промобразцы. Товарные знаки, № 12 (1966).
14*
8-ХИНОЛИНСУЛЬФОКИСЛОТА
Г. М. БАБЧУК, И. М. ХРАПОВА, Г. Д. ГАНДЛЕВСКИЙ
C9H7NO3S
М. в. 209,21
8-Хинолинсульфокислоту получают сульфированием хинолина 20%-ным олеумом {!], 50%-ным олеумом [2] или концентрированной серной кислотой с последующей обработкой 60— 65%’-ным олеумом {3].
Нами разработана методика получения 8-хинолипсульфо-кислоты сульфированием технического хинолина высокопроцентным олеумом.
СХЕМА СИНТЕЗА
олеум
SO3H
Характеристика основного сырья
Хинолин, ЧМ ТУ 3183—52, техн.
Олеум высокопроцентный, ТУ ГАПУ 189—53.
212
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, контактным термометром и обратным холодильником, загружают 72 мл (146 г) олеума и через капельную воронку при охлаждении и перемешивании медленно приливают 50мл (54,5 г) хинолина, поддерживая температуру реакционной массы в пределах 25—40°.
Массу нагревают на масляной бане до 140°, выдерживают 2 часа при этой температуре, охлаждают и выливают при перемешивании в 500 мл холодной воды. Содержание серной кислоты в маточном растворе должно быть 25—30%. Выпавший осадок отфильтровывают, отмывают до отсутствия суль-фат-ионов (проба на осаждение с хлористым барием) и сушат при 120°.
Выход 8-хинолинсульфокислоты равен 55,2 г (70%); содержание основного вещества 99%' (см. примечание).
Полученная 8-хинолинсульфокислота отвечает требованиям МРТУ 6—09—2456—65 на реактив квалификации «чистый».
Примечание. Исследования сульфирования хинолина проводили также в аппарате из нержавеющей стали; результаты этих опытов совпадают с описанными выше.
ЛИТЕРАТУРА
1. О. Ю. Магидсон, М. В. Рубцов. Химико-фармацевт. пром-сть, I, 20 (1935).
2. А. I п о у е, М. S u g i u г a, G. Т s и г и k i. Bull. Chem. Soc., Japan-27, 430 (1954).
3. Ю. А. Б а н к о в с к и Й, А. Ф. И е в и н ь ш, Э. А. Л у к ш а. Ж- общ. химии, 28, 8, 2273—76 (1958).
3-ХЛОР-5-НИТРОСАЛ ИЦИЛОВЫИ АЛЬДЕГИД
3-Хлор-5-нитро-2-оксибензальдегид
Д. А. ДРАПК.ИНА, В. Г. БРУДЗЬ, Н. И. СОКОЛОВА
сно
Ггон
/\/\
O2N Cl
c7h4cino4
М. в. 201,61
З-Хлор-5-нитросалициловый альдегид описан лишь в одной работе [1], в которой его получали нитрованием 3-хлорсали-цилового альдегида азотной кислотой в ледяной уксусной кислоте с выходом сырца 83%'..
Мы проверили этот метод, увеличив загрузку и уточнив некоторые детали синтеза.
СХЕМА СИНТЕЗА
СНО
,f\-OH СН3СООН
+ HNO3 --------
\ / ci
сно
I
/Ч QH
“Ь
o2n
Характеристика основного сырья
З-Хлорсалициловый альдегид, т. пл. 52—54°; получение см. стр. 218 этого сборника.
Азотиая кислота, ГОСТ 701—68, ч„ пл. 1,42.
Уксусная кислота, ГОСТ 61—69, х, ч.
Этиловый спирт ректификованный, ГОСТ 5962—67, ч.
214
Условия получения
В четырехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, помещают 15,7 (0,1 М) 3-хлорсалицилового альдегида и 80 мл ледяной уксусной кислоты. Смесь нагревают на водяной бане до 40—45° и за 30—40 минут прибавляют раствор 6,9 мл (0,11 Л4) азотной кислоты в 30 мл уксусной кислоты, поддерживая температуру 40—45°. Затем удаляют баню, выдерживают смесь 1 час при комнатной температуре (к концу выдержки выпадает обильный осадок), добавляют 100 мл воды. Через несколько часов осадок отфильтровывают и промывают водой до нейтральной реакции.
Получают 15,8—16,2 г альдегида (78—80%); т. пл. 124— 126°.
После перекристаллизации из разбавленного (3: 1 по объему) спирта выход З-хлор-5-нитросалицилового альдегида равен 11,5—12,6 г (57—62%); т. пл. 128—129°.
По литературным данным [1]> т. пл. 129°.
ЛИТЕРАТУРА
1. W. Davies, S. R u b е n s t е i n. J. Chem. Soc., 1923,2839.
(З-ХЛОРПРОПИЛ)БЕНЗОЛ
В. М. ОСТРОВСКАЯ, т. В. ФОМИНА
X — СН2СН2СН2С1
СяНцС1 м. в. 154,64
(З-Хлорпропил)бензол получают действием на 3-фенил-пропанол тионилхлорида в диэтиламине [1—2] или в толуоле [3], пятихлористого фосфора [3], концентрированной соляной кислоты [4—10], действием бензилмагнийхлорида на ди (2-хлорэтил)сульфат [11] или n-толуолсульфокислоты на 2-хлор-этиловый эфир [4—5].
Нами (3-хлорпропил) бензол получен взаимодействием 3-фенилпропанола с хлористым тионилом в смеси .хлороформа и пиридина.
СХЕМА СИНТЕЗА
SOC12
—СН2СН2СН2ОН ----------
-> СН2СН2СН2С1
Характеристика основного сырья
З-Фенилпропанол, т. кип. 129°/20 мм, =1,5260; получение см. [12].
Хлороформ, ГОСТ 3160—51, ч. д. а.
Пиридин, ГОСТ 2747—67, ч„ дополнительно перегнанный над металлическим натрием.
Тионил хлористый, МРТУ 6—09—2602—65, ч.
Диэтиловый эфир, ГОСТ 6265—52, ч.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой с затвором, обратным холодильником с хлоркальциевой 216
трубкой и капельной воронкой, помещают 21 г (0,15 Л1) 3-фе-нилпропанола, 190 мл хлороформа и 23 мл пиридина, охлаждают до 0° и прибавляют по каплям при размешивании 28 мл (47 г, 0,39 М) тионилхлорида. Смесь нагревают на глицериновой бане 2 часа при 90°, затем отгоняют хлороформ (т. кип. 6Г), пиридин (т. кип. 115°). Остаток экстрагируют 250 мл диэтилового эфира, промывают экстракт 100 мл воды, сушат над плавленым хлористым кальцием. Эфир отгоняют, остаток перегоняют в вакууме, собирая фракцию с т. кип. 72— 76°/5 мм.
Выход (3-хлорпропил)бензола равен 15 г (62%'); —
= 1,5205.
По литературным данным, т. кип. 89—9376 мм [13], 85— 8779 мм [4—5]; 219—220/705 мм [3].
Найдено, %: С1—22,85; 22,90.
С9НцС1. Вычислено, %: С1—22,93.
ЛИТЕРАТУРА
1. W. Н. Gray. J. Chem. Soc., 127, 1156 (1925).
2. К. Kindler. Ann., 452, 119 (1927).
3. L. Ber me jo, J. Herrera. IX Congr. internguim. pura aplicada, 4, 238 (1934); C. A., 30, 34 1 87, (1936).
4. H. Gilman, N. J. В e a b e r. .1. Amer. Chem. Soc., 45, 842 (1923). 5. L. Bert. Compt. rend., 186, 374 (1928); Beilst., 5, 2,305 (1943). 6. Errera. Gazz. Cihm. ital., 16,313 (1886); Beilst., 5,391 (1922).
7. J. В. С о n a n t, W. R. Kirner. J. Amer. Chein. Soc,
46, 242 (1924).
8. J. F. Norris, H. B. Taylor. J. Amer. Chem. Soc.,
46, 756 (1924); Beilst. 5, 2, 305 (1943).
9. J. Braun. Ber., 43,2842 (1910).
10. V. G r i g n a rd, E. В el I e t, С. С о u r t о t. Ann. chim., [9], 12, 366 (1919).
11. С. M. Suter, P. B. Evans. J. Amer. Chem. Soc., 60, 536 (1938); C A 32 333J8 (19381
12. P. W. Clutterbuck, J. B. Cohen. J. Chem. Soc., 123, 2509 (1923).
13. S. S. Rossander, C. S. Marvel. J. Amer. Chem. Soc., 50, 1495 (1928).
3-ХЛОРСАЛИЦИЛОВЫИ АЛЬДЕГИД
З-Хлор-2-оксибенз альдегид
Д. А ДРАПКИНА, В. Г. БРУДЗЬ, Н. И. ДОРОШИНА, Н. И. СОКОЛОВА
СНО
^\-ОН
С7Н5С1О2
М. в. 156,60
Известен способ синтеза 3-хлорсалицилового альдегида из о-хлорфенола по реакции Реймер-Тимана [1—3], но экспериментальные данные ни в одной из этих работ не сообщаются. По данным [1], выход альдегида, очищенного перегонкой с паром переводом через бисульфитпое соединение и кристаллизацией из метанола, составлял 10—15%'.
Мы получили 3-хлорсалициловый альдегид таким же способом.
СХЕМА СИНТЕЗА
if Y-OH
II I + СНС13 + NaOH
СНО
218
Характеристика основного сырья
о-Хлорфенол, МРТУ 6—09—6509—70, ч.
Хлороформ, ГОСТ 3160—51, ч.
Натр едкий, ГОСТ 4328—66, ч.
Серная кислота, ГОСТ 4204—66, ч.
Натрий бисульфит, ГОСТ 902—68, техн.
Метиловый спирт, ГОСТ 6995—67, ч.
Диэтиловый эфир, фарм. ф. IX.
Условия получения
В чстырехгорлой колбе емкостью 1 л, снабженной мешалкой, обратным холодильником, капельной воронкой и термометром, к раствору 160 г (4 М) едкого натра в 200 мл воды добавляют при размешивании 64,3 г (0,5 М) о-хлорфенола. Смесь нагревают на водяной бане до 70° и за 2 часа прибавляют 80 мл (1 М) хлороформа при слабом кипении массы. Реакционную массу кипятят еще 15 минут, охлаждают до комнатной температуры, разбавляют 300 мл воды и подкисляют 50%-ной серной кислотой (~50 мл) до pH 2—3. З-Хлор-салициловый альдегид и непрореагировавший о-хлорфепол отгоняют с водяным паром, собирая около 2 л дистиллата (см. примечание 1).
Выделившееся тяжелое масло отделяют от водного слоя, а последний трижды экстрагируют серным эфиром (200, 50 и 25 мл). .Масляный слой и эфирные вытяжки встряхивают 1 —1,5 часа на механической качалке с 150 мл 40%-ного раствора бисульфита натрия. Осадок бисульфитного соединения отфильтровывают с отсасыванием и тщательно промывают серным эфиром от непрореагировавшего о-хлорфенола (см. примечание 2). Бисульфитное соединение разлагают непродолжительным нагреванием при 60—70° с 80—100 мл 20%-ной серной кислоты. Выделяющийся в виде масла альдегид через несколько часов кристаллизуется; кристаллы на следующий день растирают, фильтруют, промывают небольшим количеством воды и сушат при комнатной температуре.
Выход 3-хлорсалицилового альдегида равен 11,6—13,6 г (14,8—16,2%); т. пл. 52—53°. Продукт такого качества вполне пригоден для синтеза З-хлор-5-нитросалицилового альдегида.
Перекристаллизацией из водного (1:1 по объему) метилового спирта можно получить 3-хлорсалициловый альдегид с т. пл. 53—54°; выход чистого продукта составляет 60—70% от веса технического продукта.
По литературным данным, т. пл. 56° (2], 55° [1], 54,5° [3], 53,7 — 54,5° [4].
219
Примечания.
1. Приемник необходимо снабдить обратным холодильником.
2. Качество продукта зависит от тщательности отмывки бисульфитно-го соединения от о-хлорфенола.
ЛИТЕРАТУРА
1. W. Davies, S. R u b ens t е i n. J. Chem. Soc., 1923,2839.
2. R. J. Smith, W. H. R e a d. Ann. Appl. Biol., 49, 233 (1961).
3. R. Clarke, R. A. Cowen et al. J. Chem. Soc., 1963,245.
4. C. Postmus, J. A. Kaye et al. J. Organ. Chem, 29,2693 (1964).
4-ЦИАНСАЛИЦИЛОВЫЙ АЛЬДЕГИД
2-Окси-4-цианбензальдегид
Р. У. САФИНА, Б. М. БОЛОТИН
C8H5NO2
М. в. 147,13
4-Циансалициловый альдегид в литературе не описан. Мы получили его окислением 2-окси-4-циантолуола хромовым ангидридом.
СХЕМА СИНТЕЗА
СНз СНз
I I
SOC12 HNO3
I l| ВМФ A* | ||
X/
I I
conh2 cn
СНз
I no2 Sn + HCI
I II [H]
4/
CN
CH3 CH3 CHO
I nh2 i oh I oh
NaNO2 CrO3
I И T^SOr | || Ac/Г I ||
4/ 4/ 4/
I I I
CN CN CN
Характеристика основного сырья
га-Толуиловой кислоты амид., ТУ 14П—524—67, ч.
Хлористый тионил, МРТУ 6—09—2602—65, ч.
221
Диметилформамид, МРТУ 6—09—2068—65, ч.
Азотная кислота, ГОСТ 701—68, техн.
Олово гранулированное, МРТУ 6—09—481—63, ч.
Натрий азотистокислый, ГОСТ 4197—66, ч.
Хромовый ангидрид, ГОСТ 3776—68, ч.
Уксусный ангидрид, ГОСТ 5815—69, ч.
Условия получения
Синтез 1,4-толунитрила. В трехгорлой колбе емкостью 0,5 л, снабженной термометром, мешалкой, капельной воронкой и охлаждающей баней, растворяют при перемешивании 57 г (0,42 М) n-толуамида в 200 мл диметилформамида. К полученному раствору при нагревании (30—40°) за 1 час прибавляют 72 мл (1 М) хлористого тионила. Реакционную массу выдерживают 30 минут при комнатной температуре и выливают при перемешивании в смесь 1 л воды и 0,5 кг льда. Выпавший осадок через 1 час отфильтровывают и добавляют к нему 50 мл бензола. Из бензольного экстракта отгоняют на водяной бане бензол, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 106—108720 мм.
Выход 1,4-толунитрила равен 35 г (71,4%).
По литературным данным [1], т. кип. 218°.
Получение З-нитро-1,4-толу нитрила. В стакан, снабженный мешалкой, термометром и капельной воронкой, помещают 32,6 мл серной кислоты (пл. 1,84) и 12,6 мл дымящей азотной кислоты (пл. 1,52) и при комнатной температуре (20—25°) прибавляют по каплям 15 г (0,13 М) и-толунитри-ла. Смесь перемешивают еще 30 минут, а затем выливают в 300 мл ледяной воды. Выпавший осадок отфильтровывают и перекристаллизовывают из воды.
Выход З-нитро-1,4-толунитрила равен 15 г (72%); т. пл. 106—107°. I
По литературным данным [2], т. пл. 107°.
Получение З-амино-1,4-толу нит рила. В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой и термометром, помещают 75мл соляной кислоты (пл. 1,18) и 37,5г гранулированного металлического олова и при температуре 23—30° прибавляют за 30 минут 15 г (0,092 М) З-нитро-1,4-толунитрила. Для поддержания требуемой температуры колбу охлаждают водой. Затем в реакционную смесь при той же температуре за 1 час в 5 приемов прибавляют еще 75 мл кислоты и 37,5 г олова. Содержимое колбы выливают в 300 мл воды и фильтруют.
Фильтрат охлаждают с помощью ледяной бани и подщелачивают 7%-ным раствором едкого натра до сильнощелоч-иой реакции (pH 10). 2-Амино-4-циантолуол экстрагируют 222
500 мл бензола. Бензольный экстракт высушивают над хлористым кальцием, растворитель отгоняют на водяной бане, а остаток перекристаллизовывают из 150 мл 30%-ного водного спирта.
Выход З-амино-1,4-толунитрила равен 7,1 г (58%); т. пл. 78—80°.
По литературным данным [2], т. пл. 81°.
Получение З-окси-1,4-толунитрила. В стакане, помещенном в охлаждающую баню и снабженном мешалкой и термометром, при температуре 5—10° к раствору 7 г (0,053 М) З-амино-1,4-толунитрила в 140 мл 10%-ной серной кислоты прибавляют по каплям раствор 5,25 г нитрита натрия в 20 мл воды. Полученный раствор диазосоли фильтруют и нагревают фильтрат на кипящей водяной бане до прекращения выделения азота. Образовавшееся желтое масло (5,4 г) за-кристаллизовывается при охлаждении.
После кристаллизации из 100 мл циклогексана получают 3,6 г (51%) З-окси-1,4-толунитрила в виде желтых кристаллов с т. пл. 98,5°.
По литературным данным (21, т. пл. 99°.
Получение 4-циансалицилового альдегида. В трехгорлой колбе емкостью 50 мл, снабженной мешалкой, термометром и капельной воронкой, к охлажденной до 0° смеси 10 мл уксусного ангидрида и 1,2 г (0,009 М) З-окси-1,4-толунитрила прибавляют 2 мл серной кислоты (пл. 1,84). К содержимому колбы при охлаждении смесью льда и соли до минус 4—0° за 30 минут добавляют охлажденный до 0° раствор 2,5 г (0,025 Л4) хромового ангидрида в 11,5 мл уксусного ангидрида. Реакционную массу оставляют на 2,5 часа в бане со льдом, после чего выливают в смесь 50 г льда и 100 мл воды. Полученный триацетат 4-циансалицилового альдегида извлекают в 2 приема 100 мл бензола, бензол отгоняют на водяной бане, а остаток растворяют в смеси 12,5 мл спирта, 12,5 мл воды и 1,2 мл серной кислоты (пл. 1,84). Смесь кипятят 30 минут в колбе с обратным холодильником и быстро фильтруют. Фильтрат охлаждают в ледяной бане. Выпавшие кристаллы отфильтровывают и перекристаллизовывают из 50 мл циклогексана.
Выход 4-циансалицилового альдегида равен 0,16 г (12%). Но внешнему виду вещество представляет собой чуть желтоватые кристаллы с желтой люминесценцией; т. пл. 122,5— 123°.
Найдено, %: С — 69,39; Н — 3,38; N —9,58. CeHsNOj. Вычислено, %: С — 69,30; Н — 3,42; N — 9.52.
ЛИТЕРАТУРА
1. Н. М. Рыбкина, Л. А. Лабутина и др. Химические реактивы и ирепаоаты, выл. 22. М.. ИРЕА, 1970, стр. 189.
2. W. Borsche, Е. В о с к е г. Вег., 36, 4359 (1903).
223
1,2,3-ЦИКЛОГЕКСАНТРИОН-1,3-ДИОКСИМ
2,6-Дннзонитрозоциклогексанон
Г. С. ПЕТРОВА, А. М. ЛУКИН, Е, Н. ДОЛЖНИКОВА, Н. В. ТИТОВА, И. В. ХВОСТОВ
О
HON =,/\-NOH
C6H8N2O3
М. в. 156,12
По данным литературы, 2,6-диизонитрозоциклогексанон получают в свободном виде или в виде мононатриевой соли обычными методами нитрозирования кетонов, содержащих активную метиленовую группу. Нитрозирующими средствами в большинстве случаев являются различные алкилнитриты [1—41 и нитрит натрия [5]. ;
2,6- Диизонитрозоциклогексанон получен нами по методу [5]. По данным этой работы выход мононатриевой соли 2,6-диизонитрозоциклогексанона после перекристаллизации из метанола составляет 95%!. Как показали наши исследования, продукт до перекристаллизации содержит значительные количества (до 50%) ацетата натрия; перекристаллизация из метанола способствует значительному снижению количества ацетата натрия, однако, полностью его не устраняет. Нами найдены условия получения мононатриевой соли 2,6-дииЗо-нитрозоциклогексанона, свободного от ацетата натрия, с выходом 70%.
СХЕМА СИНТЕЗА
О О
/ \ сн3соон H0N / \ N0Na NaNO2
224
Характеристика основного сырья
Циклогексанон, ТУ БУ 15—53, ч.
Натрий азотистокислый, ГОСТ 4197—66, ч.
Уксусная кислота, ГОСТ 61—69, х. ч.
Условия получения
Синтез мононатриевой соли 2,6-диизонитрозоциклогексано-на. В колбу емкостью 5 л, снабженную мешалкой, капельной воронкой и трубкой для отвода газа, загружают растворы 350 г (3,5 М) циклогексанона в 1 л этилового спирта и 980 г (14,0 М) нитрита натрия в 1,4 л воды. В течение трех дней при комнатной температуре (18—22°) прибавляют постепенно 840 мл ледяной уксусной кислоты. На третий день появляется обильный осадок. Реакционную массу выдерживают еще один день без размешивания. Затем прибавляют 700 мл 50%-ного водного этилового спирта, тщательно размешивают п фильтруют. Осадок на фильтре промывают 1,5 л 96%-ного этилового спирта, тщательно отжимают и сушат при комнатной температуре.
Выход мононатриевой соли 2,6-диизонитрозоциклогексано-ira равен 425 г (70% в расчете на циклогексанон).
Найдено, %: Na—12,52; 12,58.
CeH7N2NaO3. Вычислено, %: Na—12,91.
Получение 2,6-диизонитрозоциклогексанона. Размешивают 20 г мононатриевой соли 2,6-диизонитрозоциклогексанона в 250 мл теплого (35°) этилового спирта и прибавляют 10 мл концентрированной соляной кислоты до pH 1—2. Выпавший хлористый натрий отфильтровывают, а фильтрат охлаждают до минус 5°. Осадок отфильтровывают и сушат при пониженном давлении при комнатной температуре*.
Выход 2,6-диизонитрозоциклогексанона равен 12,5 г (72% в расчете на мононатриевую соль 2,6-диизонитрозоциклогексанона), т. пл. 200° (разлож.).
По литературным данным, т. пл. 200° [1], 233° [3].
Найдено, %: С —46,61; Н — 5,38; N—17,91.
C6H8N2O3. Вычислено, %: С —46,20; Н —5,20; N —17,90.
ЛИТЕРАТУРА
1. A. Ferris, Н. La t curette. N. S., 2, 999, 875, Appl Nov, pl (1957); РЖХим., 56; P, 1524C (1962).
2. M u r a k a m i M э s u о. V u k a m a V a s u li i 1 1 e. Met. Inst. Scient ar.d Industr. Re Osaka. Unit.., 16, 217 (1959).
3. N. Ctiopre, W. Cocker, B. Cross. J. Chem., Soc. 1960,588. 4. D. Batesky, N. Moon. J. Oryan. Chem., 24, 11, 1694 (1959). 5. A. F r e i b s, A. К u h n . Ber., 90, 1691 (1957).
* Строению этого соединения будет посвящено специальное сообщение.
15-1192 225
ЦИКЛОГЕКСАНТРИОН-1,2,3-ТРИОКСИМ
Г. С. ПЕТРОВА, А. М. ЛУКИН, Е. Н. ДОЛЖНИКОВА
NOH
II
HON = < \ = NOH
CeHgNgOg
М. в. 171,15
Данное соединение рекомендовано для спектрофотометрического определения кобальта 1[1] и никеля {2]. Получают его оксимированием мононатриевой соли 1,2,3-циклогексантрион-1,3-диоксима в среде метилового спирта (3] или в присутствии метилата натрия (4]. Нами выбран и проверен первый способ получения.
СХЕМА СИНТЕЗА
О NOH
II II
HON =i/\= NONa NH2OH-HC1^ HON=/\-NOH
Характеристика основного сырья
1,2,3-Циклогексантрион-1,3-диоксим, мононатриевая соль, см. стр. 224 этого сборника.
.Гидроксиламин солянокислый, ГОСТ 5456—65, ч.
Метиловый спирт, ГОСТ 6995—54, ч.
Условия получения
К раствору 20 г (0,5 М) мононатриевой соли 2,6-диизо-нитрозоциклогексанона в 240 мл метилового спирта добавляют 8 г (0,25 А4) солянокислого гидроксиламина и кипятят 2 часа в колбе с обратным холодильником на водяной бане. 226
Раствор фильтруют, упаривают до половины объема на водяной бане и охлаждают до 0°. Выпавший розоватый осадок отфильтровывают. Продукт дважды перекристаллизовывают из воды.
Выход циклогексантрион-1,2,3-триоксима равен 9 г (78%), т. пл. 176°.
По литературным данным [3], т. пл. 176—177°.
Найдено, %: С — 42,56; Н — 5,57; N — 24,90.
CeIleNsO3. Вычислено, %: С —42,48; Н — 5,33; N—24,65.
ЛИТЕРАТУРА
1. W. Frierson, N. Patterson. Analyt. Chem., 33, 8, 1096 (1961).
2. F r i e r s о n, N. Marable. Analyt, Chem., 34,2,210 (1962).
3. A. Freibs. A. Kuhn. Bet., 90,1691 (1937).
4. D. Batesky. J. Organ. Chem.’ 24, 11, 1697 (1959).
15*
ЭТИЛОВЫЕ ЭФИРЫ и- И л-КСИЛ ИЛЕНДИАМИНО-N,N'-BHC( ИЗОПРОПИЛ ФОСФОНОВЫХ КИСЛОТ)
М. В. РУДОМИНО, Е. К. КОЛОВА, Т. я. МЕДВЕДЬ
(C2HSO)2P — С — NHCH2 — CH2NH — С — P(OC2HS)2 II /\ Х=Х /\ II
О СНзСНз СНз СН3О
(С2Н5О)2Р — С — NHCH2
II /\ х=х
О СН3 СН3 CH2NH — С — Р (ОС2Н5) ,
/\ ||
СН3СН3О
С22Н42Н20бРг
М. в. 492,55
Этиловые эфиры п- и ж-ксилилендиамино-М,М'-бис (изопропилфосфоновых кислот являются исходными продуктами в синтезе фосфорорганических комплексонов с повышенной жесткостью цепи, соединяющей иминодиалкилфосфоновые группировки.
Оба соединения получены нами впервые конденсацией ди-этилфосфита и ацетона с п- и л-ксилилендиаминами в соответствии с общим методом (1]. Для дополнительной характеристики указанных эфиров получены их кристаллические пикраты.
СХЕМА СИНТЕЗА н
2(С2Н5О)2Р 4- 2CH3COCH34-H2NCHa —
Ч^ — CH2NH2
(С21[5О);!Р-С —nhch2-/ Ч
О СН^СНз CH2NH -С - Р (ОСгНб)2
/\ II
СНзСНзО
228
Характеристика основного сырья
л-Ксилилендиамии, т. кип. 245—248°, получение см. [2]. п-Кснлилендиамин, т. пл. 35°, получение см. [3].
Диэтилфосфит, ТУ 14П 763—68.
Ацетон, ГОСТ 2603—63, ч.
Бензол, ГОСТ 5955—68, ч-
Калий углекислый, ГОСТ 4221—65, ч.д.а.
Пикриновая кислота, ВТУ 26—55, ч.
Условия получения
Синтез этилового эфира п-ксилилендиаминобис(изопропил-фосфоновой кислоты). В четырехгорлой колбе, снабженной мешалкой, термометром, обратным холодильником и Ч-образной насадкой с двумя капельными воронками, к 13,6 г (0,1 М) расплавленного при 60° /т-ксилилендиамина прибавляют по каплям одновременно 27,6 е (0,2 А!) диэтилфосфита и 11,6 г (0,2 М) ацетона, поддерживая охлаждением водой температуру реакционной смеси 60*2°. По окончании прибавления реагентов массу выдерживают при указанной температуре 0,5 часа, охлаждают до комнатной температуры и экстрагируют 200 мл бензола. Бензольный экстракт промывают 8 мл насыщенного раствора углекислого калия и сушат сернокислым натрием. После удаления растворителя при пониженном давлении остаток кристаллизуется при стоянии. Вещество перекристаллизовывают из эфира.
Выход этилового эфира л-ксилилендиаминобис(изопропил фосфоновой кислоты) равен 38,6 е (85%); т. пл- 64—65° (в запаянном капилляре).
Найдено: %: С — 54,1; 53,6; Н — 8,7; 8,8; N — 5,5; 5.7; Р— 12,6; 12,4;
C^H.^N 2О5Р2. Вычислено, %: С — 53,6; Н — 8,6; N—5,7; Р—12,6.
Дипикрат этилового эфира п-ксилилендиаминобис(изопро-пнлфосфоновой кислоты), т. пл. 165—166° (разлож.) [из ледяной уксусной кислоты].
Найдено, %: С —42,1; 42,4; Н — 4,8; 5,0; N —11,3; Р—6,7; 6,4.
Сз4Н48Н802оР2. Вычислено, %: С — 42,9; Н —5,1; N—11,7; Р — 6,5.
Этиловый эфир дт-ксилилендиаминобис(изопропилфосфоновой кислоты), синтезирован по аналогичной методике, выход 91%', т. пл. 41—42° (в запаянном капилляре).
Найдено, %: С — 53,3; 53,6; Н — 8,7; 8,9; N —6.2; 6,2;
Р — 11,9; 12,0.
C22H42N2O6P2. Вычислено, %: С — 53,6; Н — 8,6; N — 5,7; Р—12,5.
229
Дипикрат этилового эфира ж-ксилилендиаминобис(изопро-пилфосфоновой кислоты), т. пл. 132—132,5° (из этилового спирта).
Найдено, %: С —43,1; 43,3; Н — 5,2; 5,1; N- 11,3 11,2;
Р —6,4; 6,3.
СзбН49Н8О20Р2. Вычислено, %: С — 42,9; Н —5,1; N— 11,7; Р —6.5.
литература
1. Т. Я. Медведь, М. В. Руд ом ин о и др. Изв. АН СССР, сер. хим., 353 (1967).
2. R. Brome. Вег., 21,2705 (1888).
3. F. Lustig. Вег., 28,2992 (1895).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ СОЕДИНЕНИЙ, ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
З-Аллил-5-нитросалициловый альдегид .............................7
,1-Аминоацетофенон...............................................9
а -Аминобензилидендифосфоновая кислота . . . . ..11
1-Аминогеки1лидендифосфоновая кислота...........................13
2-Амино-4-нитрофенол-6-сульфокислота...........................128
3-Амино- 1,4-толунитрил . . . ...........................222
5-Ампносалицилальдоксим........................................155
Аминофлуоресцеин I . . . ...........................15
1-Аминоэтилидендифосфоновая кислота ...... 18
О-Ацетил-О-я-окснизовалериановая кислота . . ... 67
л-Ацетобензолсульфонилфторид . 20
л-Ацетобензолсульфонилхлорид ....... 21
4-Бензилдифенил . . . ...........................22
N -Бензилфталимид...............................................25
Бензоилацетонитрил ........................................... .27
4-Бензоилдифенил . . . ................................23
5-Бром-N-ацетнлиндоксил . ...........................34
5-Бром-о-ванилин............................................ . 30
5-Бромнвдоксилфосфат бария .... .... 32
5-Броминдоксилфосфат натрия.....................................36
5-Бром-З-индолилфосфат бария....................................32
5-Бром-З-индолилфосфат натрия . . . . .... 36
Бромкрезоловый Зеленый .............................39
Бромкрезоловый зеленый водорастворимый ...... 42
Бромкрезоловый пурпуровый .............................45
Бромкрезоловый пурпуровый водорастворимый......................48
5-Бром-З-метоксисалициловый альдегид . . .... 30
5-Бром-2-окси-3-метоксибензальдегид . . ...................30
5-Бром-4-оксиизофталевый ангидрид . . . .... 51
(З-Ббромпропил)бензол . 5.3
З-Бром-5-форМилсалициловый альдегид . . ... 51
Бутилдихлорацетат ..............................................61
трет-Бутилкарбазат..............................................56
4-б7'ор-Бутил-2-(я-метнлбензил)фенол , . . . ... 59
трет-Бутилметилкарбонат . . . . . . .... 57
трет-Бутиловый эфир О-ацетил-Б-з-оксиизовалериановой кислоты . 68
Бутиловый эфир дихлоруксуснон кислоты...........................61
Бутиловый эфир метионина ......... 63 трет-Бутиловый эфир D-т-оксиизовалериановой кислоты . .66
трет-Бутиловый эфир L «-оксиизовалериановой кислоты ... 66
гс-(н-Бутокси) нитробензол . . ...........................81
Винил (дифенил) фосфиноксид . . . ..... 70
Гексаметилендиамнно- М,М,14',Ы'-тетракис(метилфосфоновая кислота) 73
Гексаметилфосфортрнамид . . . . . .... 75
Гексахлорацетон ............................................... 77
231
п- (н-Гексилокси) нитробензол . 81
п-(н-Гептилокси)нитробензол . , . .... 81
п-(н-Децилоксн)! тробензол.......................................81
Диаллиловый эфир итаконовой кислоты..............................90
1,6-Диаминогексан-1,1,6,6-тетрафосфоновая кислота ... 84
Диацстилфлуоресцеин................................ 205
Диацетоксифлуоран................................................205
Дибензил (дибензиламинометил)фосфиноксид . . ... 86
Дибензил (дибензиламинометил)фосфинокснд-М-оксид . . .88
Дибспзнлхлорфосфин................................................87
Дибром-о-крезолсульфофталеин .....................................45
Дибром-о-крезолсульфофталеин, моноаммонийная соль . . .48
Ди-н-бутиловый эфир итаконовой кислоты . . . ... 90
4,4'-Дибутоксиазоксибензол.......................................82
Дигексилмалеинат . . . . . ......................92
Дигексиловый эфир малеиновой кислоты ...... 92
4,4’-Дигексилоксиазоксибензол . . . . .... 82
4,4’-Дцгептилоксиазоксибензол . . ..... 82
2,6-Диизонитрозоциклогсксанон ........ 224
2,6-Диизонитрозоциклогексанон, моноиатриевая соль . . . 225
4,4’-Дидецплокспазоксибензол . . ......................82
N, N'-Диметил-л-нитроанилин ....... 94
2,2'-Ди-и-нитрофенил-5,5'-ди-и-тиокарбамоилфенил-3,3'-(3,3'-димето-кси-4,4’-бифеиилеи) дитетразолий хлористый..................185
2,2’- Ди-/г-иитрофеннл-5,5’- ди-п-нианфенил-3,3’- (3,3’-диметокси-4,4’-бнфепилен) диформазан.......................................188
4,4’-Динонилоксназоксибспзол .....................................82
2,4-Диокси-4-нитробензальдегид .................................133
4,4’-Диоксистильбен-3,3’,5,5’-тетракис(метнлиминодиуксуспая кислота).
тетранатриевая соль...........................................96
4,4'-Диоктилоксиазоксибензол . 82
4,4’-Днпентилоксиазоксибензол . ......................82
4,4’-Дипропоксиазоксибензол . . . ..................82
Дихлорангидрнд 3,₽,3-трис(хлорметил)этилфосфористой кислоты . 109
Дифенил (диоктиламиноэтил)фосфиноксид . . . ... 99
Дифенил (диэтиламинометил)фосфиноксид . . . . . .101
N. N'-Дифенил-и-фенилендиамин...................................103
Дифенилхлорфосфин .............................................106
н-Додецилгаллат . . ...................................111
п-Додецпловый эфир галловой кислоты . . . , . .111
Изобутиловый эфир метионина......................................63
Инулин...........................................................209
и-Ксилилендиамино- N, М'-бис(изопропилфосфоновая кислота) . 113
л-Ксилилендиамино-N, М'-бнс(изопропилфосфоновая кислота) . ИЗ
и-Ксилилендиамино-N. N'-бис (изопропилфосфоновая кислота), ди-
хлоргидрат..................................................114
лг-Ксилилендиамино- N N'-бис(изопропилфосфоновая кислота), мо-нохлоргидрат .................................................. 114
Лаурилгаллат....................................................111
Левулеза......................................................._ 209
о-Метакриламидобензойиая кислота................................116
Метиламин . . .........................................119
D, L-N -Метилвалин . . . . . . , . .118
₽ -Монобутилитаконат......................, .... 121
233
Р -ЛАоНобутиловый эфир итаконовой кислоты . . . . 121
Мопооктиламин, хлоргидрат.......................................173
2-Нитро(3-бромпропил) бензол . . . . . • . .123
4-Нитро(3-бромропил) бензол . ......................123
5-Нитро-4,6-диметоксисалициловый альдегид.......................125
Нитроксаминазо..................................................127
4-Нитро-2-метоксибензальдегид . . ......................131
5-Ннтро- р -резорциловый альдегид...............................133
5-Нитросалицилальдоксим . ............................156
7-Нитро-4,5,6-триметоксифталид..................................138
З-Нитро-4,5,6-тримебоксифталевая кислота........................136
З-Нитро-4,5,6-триметоксифталевая кислота ..... 135
3-Нитро-1,4-толунитрил 222
Нитрофлуоресцеин . 141
Нитрофлуоресцеиндиацетат I......................................140
2-Нитро(3-хлорпропил)бензол.....................................143
4-Нитро(3-хлорпропил) бензол . . ......................143
н-Нонил-п-нитробензоат . . . . ......................145
п-(н-Нонилокси) нитробензол.....................................,81
н-Нониловый эфир ц-нитробензойной кислоты.......................145
а-Оксибензилидендифосфоновая кислота, свинцовая соль . . . 147
а -Оксибензилидендифосфоновая кислота, тринатриевая соль . . 147
4.4’-Оксибис(тетрахлорбензонная кислота)........................151
Оксигидрохинонат кобальта .... 153
Оксигидрохинонат свинца . . 153
Оксим 5-аминосалицилового альдегида ...... 155 2-Окси-5-метилфенил-1,3-бис(метиленпмино)тетрауксусная кислота . 158 4-Окси-5-нитросалицИловый альдегид . . . . . .133
8-Оксихинолин...................................................160
З-Окси-1,4-толу нитрил . . ...........................223
2-Окси-4-цианбензальдегид.......................................221
ж-Оксиэтилиденбензолсульфонилфторид , . . . .177
(1-Оксиэтилиден) дифосфоновая кислота, монокалиевая соль . . 164
(1-Оксиэтилиден)дифосфоновая кислота, комплекс с медью .. , 162
N- (2-Оксиэтил)этилендиамино- N, N', ГЧ'-трис(метилфосфоповая кислота) .......................................................166
4-(n-н-Октилбензилокси) бутанол . ..................168
л-н-Октилбензилхлорид...........................................170
Октилиминобис[2-этил (дифенил) фосфиноксид] . . . . .172
/1-(н-Октилокси) нитробензол . , . . ..................81
Палладион.......................................................127
п-(н-Пентилокси)нитробензол .................................... 81
1.2,3-Пирогаллол ........ ... 174
п-(н-Пропилокси)нитробензол . . . . .... 81
лг-Стиролсульфонилфторид........................................176
2'-Сульфо-4-'-нитрофенол (6-азо-3)-4-аминопафталин-2,7-Д1|сульфокис-лота.........................................................127
Тетраамиловый эфир этилендиаминобиспзопропилфосфоновой кислоты ........................................................181
Тетрабром-лг-крезолсульфофталсин ................................39
Тетрабром-.и-крезолсулыфофталеин, моноаммонийная соль . 42
Тетраоктиловый эфир этилендиамино-N, Г4'-бис(бензилфосфоновой кислоты).....................................................183
Тетрабутиловый эфир этилендиаминобисизооктилфосфоновой кислоты . ................................181
233
Тетрабутиловый эфир этилендиаминобисизопропилфосфоновой кислоты . . . . . . . .181
Тетраоктиловый эфир этилендиаминобисизопропи.чфосфоновой кислоты .......................................................181
Тиокарбамоилнитросиний тетразолий...............................185
Тиокарбогидразид ........... 190
Тиомочевина.....................................................192
Тиоугольная кислота, дигидразид.................................190
1.4-Толунитрил .................................................222
4,5,6-Триметоксифталид..........................................194
2,3,6 Триоксибензилимиподиуксусная кислота ..... 196
1,2,3-Триоксибензол.............................................174
1,2,4-Триоксибензол.............................................197
Трис (g-цианэтил) фосфин . . ...........................199
Фснацилцианид .................................................. 27
Фенилдихлорфосфин...............................................107
Феноловый красный ... ................................201
Фенолсульфофталеин . ....................................201
Флуоресцеипдиацстат .......................................... 205
Флуореснеиидибутират.......................................... 207
Фосфинидинтрис (3,3',3"-пропионитрил) ...... 199
-Фруктоза.....................................................209
8-Хинолинсульфокислота . . . .......................212
4-Хлорбензоилацетонитрил.........................................27
3-Хлор-5-нитро-2-оксибензальдегИд . . . . . . .214
З-Хлор-5-иитросалициловый альдегид . . . . . . .214
З-Хлор-2-оксибензальдегид ..... 218
(З-Хлорпропил) бензол . . ...........................216
З-Хлорсалициловый альдегид .....................................218
о> -Циаиацетофепои ...............................................27
Цианнитросиний тетразолий ......... 189
4-Циансалициловый альдегид ........ 221
1,2,3-Циклогексантрион-1,3-ди оксим ....... 224
Циклогексантрион-1,2,3-триоксим.................................226
N,N' -Этиленбис(бензилиденимин) . . .... 184
Этиловый эфир беизоилциаиуксусной кислоты........................28
Этиловый эфир дифенилфосфинистой кислоты . . . . . 71
Этиловый эфир д-кснлилсидиамино-N , М'бис(изопропилфосфоиовой кислоты).....................................................228
Этиловый эфир л-ксилилендиамиио- Ы,М'бис(изопропилфосфоновой кислоты).....................................................228
УДК 547.576'313.3'117,5
3-АЛЛИЛ-5-НИТРОСАЛИЦИЛОВЫИ АЛЬДЕГИД. Д. А. Драп-кина, Н. И. Дорошина. Методы получения химических реактивов и препаратов, вып. 25. М. ИРЕА, 1971, стр. 7.
З-Аллил-5-питросалициловый альдегид, т. пл. 81—82,5°, получен с выходом 43—48% нитрованием 3-аллнлсалицилового альдегида азотной кислотой в уксуснокислой среде при 50—55°. Библ. 2 назв.
УДК 547.572'117.12
n-АМИНОАЦЕТОФЕНОН. В. В. Салий. Методы получения химических реактивов и препаратов, вып. 25, М., ИРЕА, 1972, стр. 9.
п-Аминоацетофеьон, т. пл. 106°, с выходом 68,7%получен восстановлением и-нитроацетофенона чугунными стружкми в соляно кислой среде. Библ. 8 назв.
УДК 547,554'118.5
а -АМИНОБЕНЗИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА. Т. М. Ба лашова, И. Д. Колпакова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 11.
а -Аминобензилидендифосфоновая кислота, т. пл. 223—225°, с выходом 42% получена взаимодействием треххлористого фосфора, бензонитрила и фосфористой кислоты с последующим гидролизом продукта реакции. Библ. 2 назв.
УДК 547.419.1'117.12
1-АМИНОГЕКС.ИЛ И ДЕН ДИФОСФОНОВАЯ КИСЛОТА. Т. М Балашова, И Д. Колпакова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 13
1-Аминогексилидендифосфоновая кислота, т. пл. 226—228° (раз-лож.), с выходом 50% получена взаимодействием трехбромистого фосфора, и-капронитрила и фосфористой кислоты с последующим гидролизом продукта реакции. Библ. 1 назв.
УДК 547.633.6'117.12
АМИНОФЛУОРЕСЦЕИН I. Г. И. Михайлов, Н. М. Рыбкина, А. А. Положенцева. Методы получения химических реактивов и Препаратов. вып. 25. М„ ИРЕА, 1972, стр. 15.
Аминофлуоресцеин I с выходом 60% получен восстановлением нитрофлуоресцеиндиацетата I раствором дисульфида натрия. Библ. 6 назв,
235
УДК 547.419.1
1-АМИПОЭТИЛИДЕНДИ ФОСФОНОВАЯ КИСЛОТА. Т. М. Балашова, И. Д. Колпакова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 18.
1-Аминоэтилидендифосфоиовая кислота, т. пл. 276—277°, с выходом 20% получена взаимодействием трехбромистого фосфора, ацетонитрила и ледяной уксусной кислоты с последующим гидролизом продукта реакции. Библ. 1 назв.
УДК 547.541.512'384
л-АЦЕТОБЕНЗОЛ СУЛЬФОНИЛ ФТОРИД. К. М. Ройзен, Т. Н. Коровина, С. Б. Макарова. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 20.
л-Ацетобензолсульфонилфторнд, т. пл. 90—92°, получен с выходом 85% из ацетобензолсульфонилхлорида, который получают из .к-амцноацетофенона. Библ. 2 назв.
УДК 547.622
4-БЕНЗИЛДИФЕНИЛ. И. А. Барц, Е. М. Иофис, Р. М Гельштейн. Методы получения химических реактивов и препаратов, вып. 25. М..
ИРЕА, 1972, стр. 22.
4-Бензилдифенил, т. пл. 85—86°, с выходом 78% получен восстановлением бензонлднфеиила йодистоводородной кислотой в присутствии фосфора. Разработан полярографический метод определения конца восстановления. 4-Бензонлднфенил получен по реакции Фриделя-Крафтса нз хлористого бензоила и дифенила в среде дихлорэтана. Библ. 6 назв.
УДК 547.584'533
N -БЕНЗИЛФТАЛИМИД. Ю. Г. Бондарь, Е. А. Чернина. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА 1972, стр. 25.
N-Бензилфталимид, т. пл. 115—116°, с выходом 83% получен взаимодействием бензиламина и фталевого ангидрида в среде ледяной уксусной кислоты. Бнбл. 4 назв.
236
УДК 547.589,4'052.1
БЕНЗОИЛАЦЕТОНИТРИЛ И 4-ХЛОРБЕНЗОИЛАЦЕТОНИТРИЛ. Д. А. Драпкина. В. Г. Брудзь, И. И. Дорошина, Н. И. Соколова. Методы получения химических реактивов и препаратов, вып. 25, М., ИРЕА, 1972, стр. 27.
Бензонлацетонитрил, т. пл. 79,5—80,5° и 4-хлорбензоилацетонит-рил, г. пл. 129—130°, получены с выходом 43—50% взаимодействием хлорангидридов соответствующих кислот с этиловым эфиром циануксусной кислоты и последующим щелочным гидролизом. Библ. 4 назв.
УДК 547.576'114
5-БРОМ-о-ВАНИЛИН. Д. А. Драпкина, Т. В. Скандалова, Н. А. Зу-дилина. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 30.
5-Бром-о-ванилнн, т. пл. 129—129,5°, с выходом 65—67% получен бромированием о-ваннлина бромом в уксусной кислоте в присутствии плавленого ацетата натрия. Библ. 5 назв.
УДК 547.755'118.5'114
5-БРОМ-З-ИНДОЛИЛФОСФАТ БАРИЯ. В. М. Островская, И. А. Шуйская, Л. В. Ломакина. Методы получения химических
реактивов н препаратов, вып. 25. М., ИРЕА, 1972, стр. 32.
5-Бром-З-нндолилфосфат бария с выходом 66% получен фосфорилированием 5-бром-Ь1-ацетилиндоксила хлорокисью фосфора в пиридине, гидролизом продукта фосфорилирования в водном и щелочном растворах и действием на 5-бромидоксилфосфат динагриевоп соли гидроокисью бария. Библ. 6 назв.
УДК 547.755'118.5'114
5-БРОМ-З-ИНДОЛИЛФОСФАТ НАТРИЯ. В. М. Островская, И. А. Шунская, Л. В. Ломакина. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 36.
5-Бром-З-индолилфосфат натрия получен с выходом 93% обменной реакцией между бариевой солью 5-броминдолилфосфата и натриевой формой катионо-обменной смолы. Библ, 1. назв.
237
УДК 547,633.6'563.13.022'114—43
БРОМКРЕЗОЛОВЫИ ЗЕЛЕНЫЙ. А. П. Болдырева, Г. И. Михайлов. Методы получения химических реактивов и препаратов, вып.
25. М„ ИРЕА, 1972, стр. 39.
Бромкрезоловый зеленый высокого качества получен с выходом 70—72% бромированием .и-крезолового пурпурового свободным бромом в среде этилового спирта с одновременным получением в качестве побочного продукта бромистого этила. Библ. 6 назв.
УДК 547.633.6'563.13.022'114—43
БРОМКРЕЗОЛОВЫЙ ЗЕЛЕНЫЙ ВОДОРАСТВОРИМЫЙ. А. П. Болдырева. Методы получения химических реактивов и препаратов, вып. 25. М.. ИРЕА, 1972, стр. 42.
Бромкрезоловый зеленый водорастворимый получен с' выходом 95—98% действием газообразного или водного аммиака па спирторастворимый бромкрезоловый зеленый. Библ. 3 назв.
УДК 547.633.6'563.13.021'114—43
БРОМКРЕЗОЛОВЫИ ПУРПУРОВЫЙ. Н. В. Факеева, А. П. Болдырева. Методы получения химических реактивов и препаратов, вып.
25. М, ИРЕА, 1972, стр. 45.
Бромкрезоловый пурпуровый высокого качества получен с выходом 96—98% бромированием крезолового красного бромом в уксуснокислой среде с одновременным получением бромистоводородной кислоты в качестве побочного продукта. Библ. 6 назв.
УДК 547.633.6'563.13.021'114—43
БРОМКРЕЗОЛОВЫИ ПУРПУРОВЫЙ ВОДОРАСТВОРИМЫЙ. А. П. Болдырева. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 48.
Бромкрезоловый пурпуровый водорастворимый высокого качества получен с выходом 96—98% взаимодействием спирторастворимого бромкрезолового пурпурового с газообразным аммиаком. Библ. 3 назв.
238
УДК 547.587.3'11-1
5-БРОМ-4-ОКСИИЗОФТАЛЕВЫИ АЛЬДЕГИД. Д. А. Драпкина, Н. И. Дорошина. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 51.
5-Бром-4-оксиизофталевый альдегид, т. пл. 140,5—141,5°; получен с выходом 70—72% бромированием 4-оксиизофталевого альдегида. Библ. 1 иазв.
УДК 547.539.312'223.3
(3-БРОМПРОПИЛ) БЕНЗОЛ. В. М. Островская, И. Е. Каширина, Т. В. Фомина, И. В. Хвостов. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 53.
(З-Бромпропил) бензол, т. кип. 95—97°/4 мм, получен с выходом 70% гидробромированием аллилбензола в присутствии перекиси бензоила в бензоле. Рис. 1. Библ. 17 назв.
УДК 547.497.1'264—125
грет-БУТИЛ КАРЕ АЗАТ. Е. А. Макарова, Г. Н. Кошелева. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 56.
трет-Бутилкарбазат, т. заст. 36°, с выходом 46—53% получен действием гидразин-гидрата „а трет-бутилметилкарбонат. Последний получен при взаимодействии трет-бутанола, металлического натрия и метилового эфира хлоругольной кислоты с выходом 76— 78%. Библ. 2 назв.
УДК 547.636.5'214—125
4-втор-БУТИЛ-2-(а-МЕТИЛБЕНЗИЛ)ФЕНОЛ. В. М. Дзиомко, И. С. Маркович, Н. В. Круглова. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 59.
4-<?гор-Бутил-2-(я-метилбензи.т)фенол, т. кип. 177—180°/3 мм, с выходом 66,8% получен арилалкилированием 4-втор-бутилфе;юла стиролом в присутствии катализатора КУ-2. Библ, 2 назв.
239
УДК 547.464.213'26'264
БУТИЛОВЫЙ ЭФИР ДИХЛ ОРУ КСУСНОИ КИСЛОТЫ. И. Г. Хаскин, А. Л. Столпер, Л. С. Лютая, Е. Р. Зарихта, В. С. Михай-юн, Е. П. Бабин. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 61.
Бутиловый эфир дихлоруксусной кислоты, т. кип. 48—50°/4 мм, с выходом 65,2% получен взаимодействием хлораля с бутиловым спиртом и триэтиламином в присутствии ацетонциангидрина в среде четыреххлористого углерода. Библ. 4 назв.
УДК 547.425.5
БУТИЛОВЫЕ ЭФИРЫ МЕТИОНИНА. И. И. Губенко, В. М. Кот-ляревская. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 63.
Бутиловый эфир метионина, т. кип. 133—134°/2 мм и изобутиловый эфир, т. кип. 128—13Г/4 мм, получены с выходом 69,3% и 84,5%, соответственно, этерификацией метионина н присутствии хлористого тиопила. Библ. 1 назв.
УДК 547.472.4—125’26’264—125
трет-БУТИЛ ОВЫЕ ЭФИРЫ D и L-a-ОКСИИЗОВАЛЕРИА-НОВЫХ КИСЛОТ. Е. А. Макарова, Г. Н. Кошелева. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 66.
трет-Бутиловые эфиры D и L-a-оксиизовалериановых кислот получены с выходом 70—78% из солей соответственно О-а-оксиизо-валериановокислого L-треоамина и L-x-оксиизовалериановокислого D -треоамина. Библ. 2 назв.
УДК 547.558.1—31
ВИНИЛ (ДИФЕНИЛ) ФОСФИНОКСИД. Г. Ф. Ярошенко, Л. М. Тимакова, Н. Е. Хавченко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 70.
Винил (дифенил) фосфиноксид, т. пл. 115—117°, с выходом 60% получен взаимодействием эфира дифенилфосфинистой кислоты с 1,2-дибромэтаном и последующим дегидробромировапием образовавшейся окиси 2-бромэтилдифеиилфосфина триэтиламином. Библ. 3 назв.
240
УДК 547.415.1’241
ГЕКСАМЕТИЛЕНДИАМИНО-N, N.N', N'-ТЕТРАКИС(МЕТИЛ-ФОСФОНОВАЯ КИСЛОТА). В. В. Сидоренко, Н. В. Лапшина, Т. П. Коноплева. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 73.
Гексаметилендиамино- N,N, N', М'-тетракис(метилфосфоновая кислота), т. пл. 224—226°, получена с выходом 53,5% взаимодействием гексаметилендиамина с фосфористой кислотой и формалином в кислой среде. Библ. 1 назв.
УДК 547.419.1
ГЕКСАМЕТИЛФОСФОРТРИАМИД. А. Л. Фридман, В. Д. Сурков, Ф. М. Мухаметшин. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 75.
Гексаметилфосфортриамид, т. кип. 99/6 мм, с выходом 71% по-’ лучен взаимодействием газообразопого диметиламина с хлорокисью; фосфора в среде хлороформа. Библ. 3 наяв.
УДК 547.445.5
ГЕКСАХЛОРАЦЕТОН. И. Г. Хаскин, Л. О. Матвеев. Методы получения химических реактивов н препаратов, вып. 25. М„ ИРЕА, 1972, стр. 77.
Гексахлорацетон, т. кип. 109740 мм, с выходом 81% получен хлорированием 80%-ного изопропилового спирта в течение 52 часов в интервале температур 30—115°. Библ. 5 назв.
УДК 547.556.2’263 + 268
4,4'-ДИАЛКОКСИАЗОКСИБЕНЗОЛЫ. Я. Г. Чернова, Г. И. Веселовская, В. В. Салий, Е. И. Тарасюк, К. И. Мазыкина. Методы получения химических реактивов и препаратов, вып 25. М., ИРЕА, 1972, стр. 80.
4,4’-Диалкокс’иазоксибензолы получены с выходом 58—75% действием цинковой пыли в щелочной среде на 1.4-алкоксинитробензолы. Табл. 2, библ. 8 назв.
16—1192
241
УДК 547.415.1’241
1,6-ДИАМИНОГЕКСАН-1,1,6,6-ТЕТРАФОСФОНОВАЯ КИСЛОТА. И. Д. Колпакова, Т. М. Балашова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1972, стр. 84.
1,6-Диамииогексан-1,1,6,6-тетрафосфоновая кислота, т. пл. 232— 234°, получена с выходом 30% взаимодействием адипонитрила, треххлористого фосфора и свежеприготовленной фосфористой кислоты с последующим гидролизом полупродукта реакции. Библ. 4 назв.
УДК 547.558-31
ДИБЕНЗИЛ (ДИБЕНЗИЛАМИНОМЕТИЛ) ФОСФИНОКСИД.
Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я. Медведь. Методы получения химических реактивов и препартов, вып.
25. М„ ИРЕА, 1972, стр. 86.
Дибензил (дибензиламинометил) фосфииоксид, т. пл. 135—137°, с выходом 27% получен взаимодействием дибензиламина, формальдегида и дибензилхлорфосфина в водной среде. Исходный дибензил-хлорфосфин получен некаталитической реакцией диспропорциониро вания бензилдихлорфосфина. Библ. 3 пазв.
УДК 547.558—31
ДИБЕНЗИЛ (ДИБЕНЗИЛАМИНОМЕТИЛ) ФОСФИНОКСИД-N-ОКСИД. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М Тимакова, Т. Я- Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 88.
Дибензил(дибензиламииометил)фосфииоксид-N-оксид, т. пл. 92— 95° (разлож.), получен с выходом 35% окислением дибензил (дибензиламинометил) фосфинокси да перекисью водорода в среде уксусного ангидрида.
УДК 547.462.3’26’264+547.462.3'26'361
ДИ-п-БУТИЛОВЫЙ И ДИАЛЛИЛОВЫЙ ЭФИРЫ ИТАКОНОВОЙ КИСЛОТЫ. Е. И. Тарасюк, Е. Ф. Цебриенко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 90.
Ди-н-бутиловый эфир итаконовой кислоты, т. кип. 172—175'725 .юч, с выходом 65,7% и диаллиловый эфир итаконовой кислоты, т. кип. 141 —142725 мм, с выходом 64,3% получены этерификацией итаконовой кислоты соответствующим спиртом в присутствии п-толуол-сульфокислоты. Библ. 1 назв.
242
УДК 547.462.3’26’266
ДИГЕКСИЛОВЫЙ ЭФИР МАЛЕИНОВОЙ КИСЛОТЫ. Я. Г. Чернова, Г. И. Веселовская, М. В. Мосенцева. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 92.
Дигексиловый эфир малеиновой кислоты, т. кип. 198—200730 мм, с выходом 83—85% получен действием малеинового ангидрида на гексиловый спирт в присутствии ионообменной смолы КУ-2. Библ. 2 назв.
УДК 547.551.54.022
N,N-ДИМЕТИЛ-л-НИТРОАНИЛИН. Е. А. Бугай. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 94.
N.N-Диметил-л-нитроанилин, т. пл. 58,5—59°, с выходом 55—60% получен метилированием ,и-нитроанилина диметилсульфатом в среде изопропилового спирта в присутствии бикарбоната натрия при 75—80°. Библ. 3 пазв.
УДК 547.633.3’466.22'292'211
4,4’-ДИОКСИСТИЛЬБЕН-3,3’,5,5’-ТЕТРАКСИС(ИМИНОМЕТИЛ-ДИУКСУСНАЯ КИСЛОТА), ТЕТРАНАТРИЕВАЯ СОЛЬ. Л. М.Ти-макова, Г. Ф. Ярошенко, В. Я- Темкина, Р. П. Ластовский. Методы получения химических реактивов н препаратов, вып. 25. М., ИРЕА, 1972, стр. 96.
Новый комплексон получен с выходом 52% по реакции Манни-ха взаимодействием 4,4’-диоксистильбена с иминодиуксусной кислотой и формальдегидом. Библ 4 назв.
УДК 547.558—31
ДИФЕНИЛ (ДИОКТИЛАМИНОЭТИЛ) ФОСФИНОКСИД. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 99.
Дифенил(диоктиламиноэтил)фосфиноксид, т. пл. 30—35°, с выходом 60% получен взаимодействием винил (дифенил) фосфиноксида с диоктиламином прн 180° в течение 10 часов. Библ. 1 назв.
16*
243
УДК 547.558—31
ДИФЕНИЛ (ДИЭТИЛАМИНОМЕТИЛ) ФОСФИНОКСИД. ю. м. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я- Медведь, Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 101.
Дифенил (диэтиламинометил) фосфиноксид, т. пл. 92—93,5°, с выходом 39% получен взаимодействием диэтиламина, формальдегида и дифенилхлорфосфина. Библ. 1 назв.
УДК 547.553.2
N, N -ДИФЕНИЛ-п-ФЕНИЛЕНДИАМИП. Б. И. Киссин, Е. Н. Куракин. Методы получения химических реактивов и препаратов, вып 25. М„ ИРЕА, 1972, стр. ЮЗ.
N, N'-Дифенил-л-фенилендиамин, т. пл. 150—152”, с выходом 55°/( (по гидрохинону) получен взаимодействием анилина с гидрохинонов в присутствии хлористого цинка с азеотропной отгонкой реакцион ной воды. Для образования азеотропа вводят дихлорбензол. Библ 12 назв.
УДК 547.468.1’113
ДИФЕНИЛ ХЛОРФОСФИН. Г. Ф. Ярошенко, Л. М. Тимакова, Н. Е. Хавченко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 106.
Дифенилхлорфосфин, т. кип. 119—12171 мм, получен с выходом 75% каталитическим диспропорционированием днфенилхлорфосфи-на, который синтезирован по реакции Фриделя-Крафтса взаимодействием треххлористого фосфора и бензола в присутствии хлористого алюминия и триэтиламина. Библ. 7 назв.
УДК 547.412.614’118.3’113
ДИХЛОРАНГИДРИД ₽,р,8-ТРИС(ХЛОРМЕТИЛ)ЭТИЛФОС-ФОРИС'ГОЙ КИСЛОТЫ. Л. А. Власова, Е. В. Кузнецов. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА. 1972, стр. 109.
Дихлорангидрид fi, -трис(хлорметил)этилфосфористой кислоты, т. кип. 12071,5 мм, получен взаимодействием треххлористого фосфора с трихлоргидрином пентаэритрита (выход 80%) или треххлорис-гого фосфора с 3,3-бис(хлорметил)оксациклобутаном (выход 75%) Библ. 3 назв.
244
УДК 547.587.26'26’268.13
н-ДОДЕЦИЛОВЫЙ ЭФИР ГАЛЛОВОЙ КИСЛОТЫ. Р. М Гель-штейн, Е. И. Маховер. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 111,
н-Додециловый эфир галловой кислоты, т. пл. 96—97°, с выходом 79,5% получен этерификацией галловой кислоты додециловым спиртом без растворителя при пониженном давлении в присутствии п-толуолсульфокислоты. Библ. 2 назв.
УДК 547.415.1’241.
п- и -и-КСИЛИЛЕНДИАМИНО-N, 1У'-БИС(ИЗОПРОПИЛФОС-ФОНОВЫЕ КИСЛОТЫ). М. В. Рудомино, Е. К Колова, Т. Я. Медведь. Методы получения химических реактивов н препаратов, вып.
25. М„ ИРЕА, 1972, стр. ИЗ.
Омылением тетраэтиловых эфиров п- и л-ксилилендиамипо-К^'-бис(изопропилфосфоиовых кислот) концентрированной соляной кислотой получены впервые хлоргидраты п- и л-ксилилендиамино-N, N'-бис (изопропилфосфоновых кислот) (I и II), которые последующей обработкой окисью пропилена превращены в свободные п- и л-кси-лилендиаминобис(изопропилфосфоновые кислоты) (III и IV), выделенные в виде дигидратов. Приведены выход (%), т. пл. (разлож.): 1—75: 198—200°; II—66; 185—193°; 111—38; 215-218° (из водного ацетона); IV—41; 212—215° (из водного ацетона). Библ. 1 назв.
УДК 547.551.44.
о-МЕТАКРИЛАМИДОБЕНЗОЙНАЯ КИСЛОТА. С. И. Котенко, А. Г. Фадеичева, И, С. Трохименко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 116.
о-Метакриламидобензойпая кислота, т. пл. 170°, с выходом 77% получена ацилированием антраниловой кислоты хлорангидридом метакриловой кислоты в среде хлороформа в присутствии триэтиламина. Библ. 2 назв.
УДК 547.466,25.
D, L-N-МЕТИЛ ВАЛИН. Г. Н. Кошелева, Э. А. Башкир, В. И. Калмыков. Методы получения химических реактивов и препаратов вып. 25. М„ ИРЕА, 1972, стр. 118.
D, L-N -Метилвалин, т. пл. 300° (возг.), с выходом 81% получен при взаимодействии а-бромизовалериановой кислоты и жидкого метиламина, который получают разложением хлоргидрата метиламина щелочью. Для очистки продукта применен метод ионообменной колоночной хроматографии. Рис. 1, библ. 2, назв.
245
УДК 547.462.3’26’264
Р-МОНОБУТИЛОВЫЙ ЭФИР ИТАКОНОВОЙ кислоты. Р. А. Мелнгалве, А. Г. Кайнова, С. Б. Макарова, Е. В. Егоров. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 121.
₽-Монобутиловый эфир итаконовой кислоты, т. пл. 32—35°, получен с выходом 58—62% взаимодействием ангидрида итаконовой кислоты с метиловым спиртом. Библ. 3 назв.
УДК 547.548’114
2- и 4-НИТРО(3-БРОМПРОПИЛ) БЕНЗОЛЫ. В. М. Островская, Н. Е. Каширина. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 123.
2-Нитро (3-бромпропил) бензол, т. кип. 153—15772 мм, и 4-нит-ро (3-бромпропил) бензол, т. кип. 166—17072 мм, с выходом 30% и 47%, соответственно, получены нитрованием (3-бромпропил)бензола азотной кислотой, пл. 1,475. Библ. 3 иазв.
УДК 547.576’117.5
5-НИТРО- 4,6- ДИМЕТОКСИСАЛИЦИЛОВЫЙ АЛЬДЕГИД. Д. А. Драпкина, Г. С. Панова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 125.
5-Нитро-4,6-диметоксисалнциловый альдегид, т. пл. 193—1947 получен с выходом 32—40% нитрованием 4,6-диме.токсисалицилового альдегида концентрированной азотной кислотой в среде ледяной уксусной кислоты. Библ. 1 иазв.
УДК 547.645.3
НИТРОКСАМИНАЗО. Ю. М. Дедков. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 127.
Азокраситель 2-су льфо-4-нитрофеиол (6-азо-2) - Г-иафтил амин-3,6-ди-сульфокислота предложен в качестве селективного реагента для фотометрического определения палладия. Реактив с выходом 80% получен азосочетанием дназосоли, приготовленной из 2-амино-4-нит-рофенол-6-сульфокислоты, с избытком кислоты Фрейнда в кислой среде в присутствии пиридина. Краситель очищают многократным переосаждеиием. Библ, 2 назв.
246
УДК 547.576’117.5
4-НИТРО-2-МЕТОКСЙБЕНЗАЛЬДЕГИД. Л. С. Зерюкина, Б. М. Болотин. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА. 1972, стр. 131.
4-Нитро-2-мстоксибснзальдсгид, т. пл. 120—121°, получен с выходом 40% метилированием 2-окси-4-нитробензальдегида диметилсульфатом. Библ. 2 назв.
УДК 547.565’117.5
5-НИТРО-З -РЕЗОРЦИЛОВЫИ АЛЬДЕГИД. Д. А. Драпкино Н. М. Ролдугина, Н. И. Дорошина. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 133.
5-Нитро-3 -резорциловый альдегид, т. пл. 150—150,5°, получен с выходом 27% нитрованием 2,4-диоксибензальдегида. Библ. 2 назв.
УДК 547.584’261’117.5
З-НИТРО-4,5,6-ТРИМЕТОКСИФТАЛЕВЫЙ АНГИДРИД. И. М. Рыбкина, М. Е. Тихомирова, В. Н. Глушко. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 135
З-Нитро-4,5,6-триметоксифталевый ангидрид, т. пл. 90—91°, с выходом 68,5% получен кипячением З-нитро-4,5,6-триметоксифталевой кислоты с уксусным ангидридом. Библ. 1 назв.
УДК 547.588.21'261'117.5
7-НИТРО-4,5,6-ТРИМЕТОКСИФТАЛИД. Н. М Рыбкина, М. Е. Тихомирова, В. Н. Глушко. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 138.
7-Нитро-4,5,6-триметоксифталид, т. пл. 115—116°, с выходом 29,3% получен нитрованием 4,5,6-триметоксифталида 80%-иой азотной кислотой. Библ. 2 назв.
УДК 547.633.6’117.5
НИТРОФЛУОРЕСЦЕИНДИАЦЕТАТ I. Н. М. Рыбкина, А. А. Положенцева, М. Е. Тихомирова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 140.
Нитрофлуоресцеиндиацетат I получен с выходом 33—35% сплавлением 4-нитрофталевой кислоты с резорцином, с последующим разделением изомеров. Библ. 1 назв.
247
УДК 547.548’113
2- и 4-НИТРО(3-ХЛОРПРОПИЛ) БЕНЗОЛЫ. В. М. Островская, Н. Е. Каширина, Т. В. Фомина. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 143.
2-Нитро(3-хлорпропил) бензол, т. кип. 134—13770,3 мм, с выходом 21% и 4-нитро(3-хлорпропил) бензол, т. кип. 140—142°/0,3 мм, с выходом 34% получены нитрованием (3-хлорпропил) бензола. Библ. 3 назв.
УДК 547.581.2'117.5'26’268.12
н-НОНИЛОВЫЙ ЭФИР п-НИТРОБЕНЗОЙНОЙ кислоты.
Е. И. Маховер. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 145.
н-Нонил-и-нитробеизоат, d?- =1,065, /г/Д =1,5072, с выходом 74,5% получен этерификацией нитробеизойиой кислоты нониловым спиртом (эквимолярное соотношение) в присутствии л-толуолсуль-фокислоты. Библ. 3 назв.
УДК 547.568.1’118.5
а-ОКСИБЕНЗИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА, НАТРИЕВАЯ И СВИНЦОВАЯ СОЛИ. Р. П. Ластовский, И. Д. Колпакова, Л. В. Кривицкая, Е. И. Миронова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 147.
а-Оксибензилидендифосфоновая кислота (ОБДФК) получена нагреванием треххлористого фосфора с бензойной кислотой. Нейтрализацией едким натром выделена тринатриевая соль ОБДФК с выходом 51% в расчете на треххлористый фосфор. Взаимодействием последней с азотнокислым свинцом получена свинцовая соль ОБДФК с выходом 90% в расчете на тринатриевую соль. Библ. 15. назв.
УДК 547. 583.1
4,4'-ОКСИБИС(ТЕТРАХЛОРБЕНЗОЙНАЯ КИСЛОТА). Е. П. Бабин, Я. П. Скавинский, Н. А. Андрухов, А. А. Рыжков, А. С. Свириденко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 151.
4,4’-Оксибис(тетрахлорбензойиая кислота), т. пл. 268—270°, с выходом 85,5% получена хлорированием 4,4'-оксидибензойноп кислоты в растворе 98%-ной серной кислоты в присутствии йода как катализатора. Библ, 1 назв.
248
УДК 547.565.3’181,7 + 547.565.3’173.2
ОКСИГИДРОХИНОНАТЫ СВИНЦА И КОБАЛЬТА. М. Н. Русина, И. Я- Соломянникова, Н. М. Дятлова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 153.
Оксигидрохинонаты кобальта и свинца получены взаимодействием оксигидрохинона с солью соответствующего металла в щелочной среде с выходом 96 и 93% соответственно.
УДК 547.576’547.2’117.12
ОКСИМ 5-АМИНОСАЛИЦИЛОВОГО АЛЬДЕГИДА. Д. А. Драпкина, Н. И. Дорошина, Т. В. Скандалова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 155.
Оксим 5-аминосалицилового альдегида, т. пл. 169—169,5°, получен с выходом 39—46% восстановлением оксима 5-нитросалицило-вого альдегида гидросульфитом натрия. Библ. 3 назв.
УДК 547.563.13’466.22’292’211
2-ОКСИ-5-МЕТИЛ ФЕНИЛ -1,3-БИС (МЕТИЛЕНИМИНО) ТЕТРД-УКСУСНАЯ КИСЛОТА. В. Я. Темкина, Н. В. Цирульникова, И. П. Фадеева, Р. П. Ластовский. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 158.
2-Окси-5-метилфенил- 1,3-бис (метилениминс) тетра уксусная кислота получена с выходом 90% по реакции Манника взаимодействием и-крезола с формал: дегидом и иминодиуксусной кислотой в водио-щелочиой среде. Библ. 1 назв.
УДК 547.831.7
8-ОКСИХИНОЛИН. Г. М. Бабчук, И. М. Храпова, Г. Д. Гандлевский. Методы получения химических реактивов и препаратов вып. 25. М„ ИРЕА, 1972, стр. 160.
8-Оксихинолин, т. пл. 74,0—74,5°, получен с выходом 75,5% щелочным плавлением 8-хннолинсульфокислоты в автоклаве под давлением. Продукт удовлетворяет требованиям ГОСТ 5847—62 квалификации «ч.д.а.». Библ. 7 назв.
249
УДК 547.262’241’156
(1-0КСИЭТИЛИДЕН)ДИФ0СФ0Н0ВАЯ КИСЛОТА, КОМПЛЕКС С МЕДЬЮ. Л. В. Криницкая, М. Н. Русина. Методы получения химических реактивов и препаратов, вып. 25. М..ИРЕА, 1972, стр. 162.
Комплекс меди с оксиэтилидендифосфоновой кислотой получен с выходом 90% путем взаимодействия оксиэтилидендифосфоновой кислоты с солью меди в щелочной среде. Библ. 1 назв.
УДК 547.262'241’132
(1-ОКСИЭТИЛИДЕН)ДИФОСФОНОВАЯ КИСЛОТА, МОНОКА-ЛИЕВАЯ СОЛЬ. Б. И. Бихман, Е. М. Уринович, Т. Л. Богомолова, А. М. Дятлова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 164.
Монокалиевая соль (1-оксиэтилиден) дифосфоновой кислоты получена с выходом 84% взаимодействием оксиэтилидендифосфоновой кислоты с едким кали. Библ. 9 иазв.
УДК 547.435.21’241
N- (2-ОКСИЭТИЛ)ЭТИЛЕНДИАМИНО- N,N' N'-ТРИС(МЕТИЛФОСФОНОВАЯ КИСЛОТА). М. В. Рудомино, Е. К. Колова, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 166.
N- (2-Оксиэтил) этиленди амино- N,N,’N'-трис (метилфосфоновая кислота), моногидрат, т. пл. 165—170°, с выходом 56,5% получена конденсацией N-(2-оксиэтил) этилендиамина с формалином и фосфористой кислотой.
УДК 547.562’264’217.2
4- (п-н-ОКТИЛБЕНЗИЛОКСИ)БУТАНОЛ. В. А. Иншакова, 3. С. Сиденко, Г. С. Чижова, Ю. С. Рябокобылко. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 168
4-(n-н-Октилбензилокси) бутанол, т. кип. 200—205°/2 мм, с выходом 41,5% получен взаимодействием п-н-октилбензилхлорида с 1,4-бутаидиолом в щелочной среде. Библ. 1 назв.
250
УДК 547.539.212'222.1’217.2
n-н-ОКТИЛБЕНЗИЛХЛОРИД. 3. С. Сиденко, В. А. Иншакова, Г. С. Чижова, Ю. С. Рябокобылко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 170.
п-н-Октилбензилхлорид, т. кип. 143—146°/2 мм, с выходом 34% получен взаимодействием н-октилбензола с параформом и хлористым водородом в присутствии хлористого циика. Библ. 6 назв.
УДК 547.419.1
ОКТИЛИМИНОБИС[2-ЭТИЛ (ДИФЕНИЛ) ФОСФИНОКСИД]. Ю. М. Поликарпов, Г. Ф. Ярошенко, Л. М. Тимакова, Т. Я- Медведь. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 172.
Октилиминобис[2-эт ил (дифенил) фосфиноксид], т. пл. 153—154°, с выходом 60% получен гзаимодействием винил (дифенил) фосфиноксида с монооктиламином в присутствии каталитических количеств хлоргидрата монооктиламина. Библ. 2 назв.
УДК 547.565.3
1,2,3-ПИРОГАЛЛОЛ А. Л. Лифиц, Е. П. Агеев. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 174.
Пирогаллол реактивного качества получен декарбоксилированием водных растворов галловой кислоты при 145—150° и давлении 10— 12 атм. Библ. 6 назв.
УДК 547.541.512
ж-СТИРОЛСУЛЬФОНИЛФТОРИД. К. вина, С. Б. Макарова. Методы получения препаратов, вып. 25. М., ИРЕА,
М. Рорзен, Т. II. Корс-химических реактивов и 1972, стр. 176.
ж-Стиролсульфонилфторид, т. кип. 111—11274 мм, г.ер =1,5270, получен с выходом 45% из ж-оксиэтилиденбензолсульфонилфторида, который получают восстановлением ж-ацетобепзолсульфонилфтори-да. Библ. 4 назв.
251
УДК 547.415.1’241’26’118
ТЕТРААЛКИЛОВЫЕ ЭФИРЫ ЭТИЛЕНДИАМИНО-N,N'-БИС-(АЛ КИЛ ФОСФОНОВЫХ КИСЛОТ). М. В. Рудомино, Н. В. Чурилина, Т. Я- Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 179.
Конденсацией этилендиамина с диалкилфосфитами и кетонами получены тетраалкиловые эфиры этилендиамино-N, N'-бис(алкилфосфоновых кислот) [( RO)2P(O)CR'R"N НСН2]2, для которых приведены R, R', R", брутто-формула, выход т. пл. дипикра-
та (из спирта):
н-С4Н9, СНз, СНз, C24HS4N2O6P2, 72, 1,4590, 1,0097, 151,5—152=; н-С4Н9, СНз, С6Н|з, C34H74N2O6P2, 89, 1,4605, 0,9772, 100—102°, н-СзНп, СНз, СНз C2BH62N2O6P2, 86, 1.4560, 0,9961, 136—138=;
н-СвН,7, СНз, СНз, C40HS6 N2O6P2, 55, 1,4579, 0,9461, 124—126° Табл. 2, библ. 2 назв.
УДК 547.554’26’118
ТЕТРАОКТИЛОВЫИ ЭФИР ЭТИЛЕНДИАМИНО-N.N'-БИС-(БЕНЗИЛФОСФОНОВОИ КИСЛОТЫ). М. В. Рудомино, Е. К. Конова, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 183.
Тетраоктиловый эфир этилендиамино- N,N'-бис (бензилфосфоновой кислоты) Лд11 — 1.4881,4/4° =0,9948 (закристаллизовывается при стоянии) получен с выходом 82% конденсацией N,N'-этиленбис(бензил-иденимнна) с диоктилфосфитом. Библ. 2 назв.
УДК 547,796.1
ТИОКАРБАМОИЛНИТРОСИНИИ ТЕТРАЗОЛИЙ. Л. А. Лабутина, В. М. Островская, Л. В. Ломакина, Г. И. Михайлов. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 185.
Тиокарбамоилнитросиний тетразолий, т. пл. 192—194°, с выходом 60% получен: 1) конденсацией и-циапбепзальдегида с и-нитрофенил-гидразином с образованием п-нитрофенилгидразона п-цианбензаль-дегида; 2) сочетанием полученного гидразона с диазотированным 3,3'-диметоксибензидином с образованием 2,2’-ди-п-питрофснн.т-5,5’-ди-п-цианфепил-3,3' - (3,3'-диметокси -4,4’- бифеяилен)диформазана; 3) окислением диформазана до цианнитросинего тетразолия; 4) превращением последнего под действием тиоацетамида в конечный продукт. Библ. 1 назв.
252
УДК 547.497.1
ТИОКЛРБОГИДРЛЗИД. В. М. Островская, Л. В. Ломакина. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 190.
Тиокарбогидразид, т. пл. 70°, получен с выходом 71% взаимодействием тиофосгена е гидразин-гидратом. Библ. 4 назв.
УДК 547.496.3
ТИОМОЧЕВИНА. М Ф. Кондрашова, Е. Я. Яровенко. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 192.
Тиомочевипа, содержание основного вещества 98—99,5%, с выходом 96—98% получена взаимодействием цианамида кальция и сульфида аммония в присутствии соляной кислоты. Библ. 4 назв.
УДК 547.588.21’261
4,5,6-ТРИМЕТОКСИФТАЛИД. Н. М. Рыбкина, М. Е. Тихомирова. Методы получения химических реактивов и препаратов, вып. 25 М., ИРЕА, 1972, стр. 194.
4,5,6-Триметоксифталид, т. пл. 135—136°, с выходом 35,9% получен кипячением диоксанового раствора 3,4.5-триметОксибензойпон кислоты с формалином и соляной кислотой. Библ. 2 назв.
УДК 547.565.3’466.22’292'211
2,3,6-ТРИОКСИБЕНЗИЛИМИНОДИУКСУСНАЯ КИСЛОТА. Р. П. Ластовский, В. Я. Темкина, Н. В. Цирульникова, Л. Я. Ши-ленко. Методы получения химических реактивов и препаратов, вып.
25. М., ИРЕА, 1972,’ стр. 196
2.3,6-Триоксибензилиминодиуксусная кислота получена впервые с выходом 77,7% по реакции Манниха взаимодействием оксигидрохинона с формальдегидом и иминодиуксуспоп кислотой в уксусной среде. Оксигидрохинон получен омылением пирогаллола «А» в еппр-тово-кислой среде. Библ. 2 назв.
253
УДК 547.468. Г052.1
ТРИС (З-ЦИАПЭТИЛ) ФОСФИН. М. В. Рудомино, Н. В. Чурилина, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 199.
Трис(?-цианэтил)фосфин, т. пл. 96—97°, с выходом 80% получен взаимодействием тетраоксиметилфосфонийхлорида со спиртовой щелочью и последующей обработкой реакционного раствора нитрилом акриловой кислоты. Библ. 7 назв.
УДК 547.633.6—43
ФЕНОЛОВЫЙ КРАСНЫЙ. А. П. Болдырева. А. У. Каганов-ская, Г. И. Михайлов. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 201.
Феноловый красный получен с выходом 59—63% конденсацией о-сульфобейзойной кислотой с избытком фенола в присутствии смеси хлорокиси фосфора и хлорной кислоты с последующей очисткой продукта путем промывки горячей водой и экстракционной промывки и-бутиловым спиртом. Полученный индикатор электрофорегиче-ски однороден, имеет 'максимальное поглощение в кислой области при 505±5 нм и щелочной области при 558±5 нм, обладает двумя переходами окраски: от розовой к желтой при pH 0,7—2,0 и от желтой к красной при pH 6,2—8,4. Библ. 12. назв.
УДК 547.633.6
ФЛУОРЕСЦЕИНДИАЦЕТАТ. Н. М. Рыбкина, М. Е. Тихомирова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 205.
Флуоресцеиндиацетат, т. пл. 203—204°, с выходом 76—78% получен кипячением флуоресцеина с уксусным ангидридом. Библ. 4 назв.
УДК 547.633.6
ФЛУОРЕСЦЕИНДИБУТИРАТ. Н. М. Рыбкина, И. Н. Пименова. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 207.
Флуоресцеиндибутират, т. пл. 114—116°, с выходом 70—72,5% получен кипячением флуоресцеина с масляным ангидридом в среде пиридина. Библ. 1 назв.
254
УДК 547.455.632
а -ФРУКТОЗА. Я. В. Плеханова, Л1. И. Черняховская, Е. П. Крысий, Г. П. Федорченко, И. И. Голяева, В. И. Иванов. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 209.
а-Фруктоза получена с выходом 16,4% из корней инули крупной высокой экстрацией инулина н последующим гидролизом его в 0,5%-ной серной кислоте. Библ. 2 назв.
УДК 547.833.4
8-ХИНОЛИНСУЛЬФОКИСЛОТА. Г. М. Бабчук, И. М. Храпова, Г. Д. Гандлевский. Методы получения химических реактивов и препаратов, вып. 25. М„ ИРЕА, 1972, стр. 212.
8-Хпиолиисульфокислота получена с выходом 70% сульфированием технического хинолина высокопроцентным олеумом. Полученный реактив отвечает требованиям МРТУ 6—09—2456—65 квалификации «чистый». Библ. 3 назв.
УДК 547.576’117.5’113
З-ХЛОР-5-НИТРОСАЛИЦИЛОВЫй АЛЬДЕГИД. Д. А. Драпкина, В. Г. Брудзь, Н. И. Соколова. Методы получения химических реактивов и препаратов, вып. 25, М„ ИРЕА, 1972, стр. 214.
З-Хлор-5-нитросалициловый альдегид, т. пл. 128—129°, с выходом 57—62% получен нитрованием 3-хлорсалнцилового альдегида в уксуснокислой среде. Библ. 1 назв.
УДК 547.539.212’223.3
(З-ХЛОРПРОПИЛ)БЕНЗОЛ. В. М. Островская, Т. В. Фомина. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 216.
(З-Хлорпропил) бензол, т. кип. 72—76,5°/5 получен с выходом
62% взаимодействием 3-фенилпропанола с хлористым тионилом в смеси хлороформа и пиридина. Библ. 13 назв.
УДК 547.576’113’022
3-ХЛОРСАЛИЦИЛОВЫЙ АЛЬДЕГИД. Д. А. Драпкина, В. Г. Брудзь, Н. И.Дорошина, Н. И. Соколова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 218.
З-Хлорсалициловый альдегид, т. пл. 53—54°, с выходом около 10% получен из о-хлорфенола по реакции Реймер-Тимана. Библ. 4 назв.
255
УК 547.576.491.
4-ЦИАНСАЛИЦИЛОВЫИ АЛЬДЕГИД. Р. У. Сафина, Б. М. Болотин. Методы получения химических реактивов и препаратов, вып.
25. М„ ИРЕА, 1972, стр. 221.
4-Циансалициловый альдегид, т. пл. 122—123°, получен окислением 2-окси-4-циантолуола хромовым ангидридом в уксусном ангидриде. Необходимый для реакции 2-окси-4-циаитолуол получен по следующей схеме: п-толуамид — п-толунитрил— 2-нитро-4-цианто-луол — 2-амино-4-циантолуол—2-окси-4-циантолуол. Библ. 2 назв.
УДК 547.594.3’288.4
1,2,3-ЦИКЛОГЕКСАНТРИОН-1,3,-ДИОКСИМ. Г. С. Петрова, А. М. Лукин, Е. Н. Должникова, Н. В. Титова, И. В. Хвостов. Методы получения химических реактивов и препаратов, вып. 25. М..
ИРЕА, 1972, стр. 224.
1,2,3-Циклогексантрион-1,3-диоксим, т. разлож. 200°, получен с выходом 72% нитрозированием циклогексанона нитритом натрия в среде ледяной уксусной кислоты с последующим гидролизом полученной соли. Библ, 5 назв.
УДК 547.592.12’288.4
ЦИКЛОГЕКСАНТРИОН-1,2,3-ТРИОКСИМ. Г. С. Петрова, А. М. Лукин, Е. Н. Должникова. Методы получения химических реактивов и препаратов, вып. 25, М„ ИРЕА, 1972, стр. 226
Циклогексаитрион-1,2,3-триоксим. т. пл. 176°, получен с выходом 78% окислением мононатриевой соли 1,2,3-циклогексантрион-1,3-диоксима в среде метилового спирта гидроксиламином солянокислым. Библ. 4. назв.
УДК 547.558.1’26’118
ЭТИЛОВЫЕ ЭФИРЫ п- п л-КСИЛИЛЕНДИАМИНО-N, N' БИС (ИЗОПРОПИЛ ФОСФОНОВЫХ КИСЛОТ). М. В. Рудомино, Е. К. Колова, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, стр. 228.
Конденсацией п- и л-ксилилендиаминов с ацетоном и диэтил-фосфином получены впервые тетраэтиловые эфиры п- и ж-ксилилен-диамино-М,М'-бпс(изопропилфосфоповых кислот), соответственно I и И, для которых приведены: выход (%), т. пл. (из эфира), т. пл. (разлож.) дипикрата: 1—85; 64—65°; 165—166° (из лед. уксусной кислоты); 11—91; 41—42°; 132—132,5° (нз спирта). Библ. 3 назв.
256
Замеченные опечатки
Стр. Строка НАПЕЧАТАНО ' СЛЕДУЕТ.ЧЙТЖЬ
17 7 снизу 27, 27,
60 14 » —180’3/3 —180°/3
127 2 сверху , <6-азо-3>-7- <6-азо-3>-
199 2 » (З.З’.З-.., (33’,3”-... , ,
243 16 снизу -тетраксис (иминомети л; -тетра кис (метилйминО-
Тип. № б УИМ. Зак. 1192а—1100