

























































Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СОВЕТА МИНИСТРОВ СССР
ПО ХИМИИ
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ
ИНСТИТУТ ХИМИЧЕСКИХ РЕАКТИВОВ
ХИМИЧЕСКИЕ РЕАКТИВЫ
И ПРЕПАРАТЫ
Выпуск I
(Методы получения)
Группа научно-технической информации
MOCKBA-I960
Редакционная комиссия: Г. А. Певцов (председатель),
Ю. Л. Вийнштейн, А. И. Вольфсоп, Н. Е. Кожевникова,
Е. Я- Яровенко.
Составитель Р. М. Левина
Редактор Л. М. Воробьев
Корректоры В. И. Григорьева и В. Ф. Победимская
Технический редактор В. Н. Лутаенко
Я 88160 Подп. к печ. 28. 2. 1961 г.
Объем 3,38 п. л. Тир. 1000 экз. Зак. 991. Цена 50 коп.
Типолитография № В-15
СОДЕРЖАНИЕ
Стр.
Иминодиуксусная кислота. Р. П. Ластовский,
В. Я. Темкина 9
Гексаметилендиаминтетрауксусная кислота.
Р. П. Ластовский, В. Я. Темкина 12
2, 4, б-Триамин-1, 3, 5-триазин-Я N, N', N',
N", N" -гексауксусная кислота. Р. П. Ластов-
Ластовский, И. Д. Колпакова 15
2-Бромбензолфосфоновая кислота. А. М, Лукин,
И. Д. Калинина 18
2-Окси-5-хлорбензолфосфоновая кислота.
А. М. Лукин, И. Д. Калинина, Г. Б. Завара-
хина . .' 24
1-Антрахинонфосфоновая кислота и её натрие-
натриевая и аммонийная соли. А. М. Лукин,
Г. С. Петрова 29
2-Оксибензолфосфоновая кислота. А. М. Лукин,
И. Д, Калинина . . . 34
2-Оксибензоларсоновая кислота. А, М. Лукин,
И. Д. Калинина, Г. С. Петрова 38
4-Нитроанилин-2-арсоновая кислота. А. М. Лу-
Лукин, Г. С. Петрова . 41
4-Нитроаиилин-3-арсоновая кислота, А. М. Лу-
Лукин, Г. С. Петрова • . . 46
4-Нитроанилин-З-сульфокислота. А. М. Лукин,
Г. С. Петрова • 53
5-Нитроантраниловая кислота. А, М. Лукин,
Г. С. Петрова 57
Стр.
Хромотроповая кислота. Л. М, Лукин,
Г. Б. Заварихина 61
Фенилборная кислота. В. Г. Брудзь,
Д. А. Драпкина, И. С. Маркович 65
н-Бутиловын эфир борной кислоты.
В. Г. Брудзь, И. С. Маркович L 68
Монохлоруксусная кислота. Б. Г. Ясницкий,
A. П. Зайцев 71
2-Нитро-1-фталоиламинобензол. В. М. Дзиом-
ко, К. А. Дунаевская 78
2-Амино-1-фталоиламинобензол. В. М. Дзиомко,
К. А. Дунаевская 81
N-Окись пиридина. И. А. Красавин 83
N-Окись 4-нитропиридина. И. А. Красавин 89
8-Меркаптометилхинолин, его соли и соедине-
соединения с ионами металлов. Ю. А. Банковский,
3. В. Мисуловина, А. Ф. Иевиньш, М. Р. Бука 93
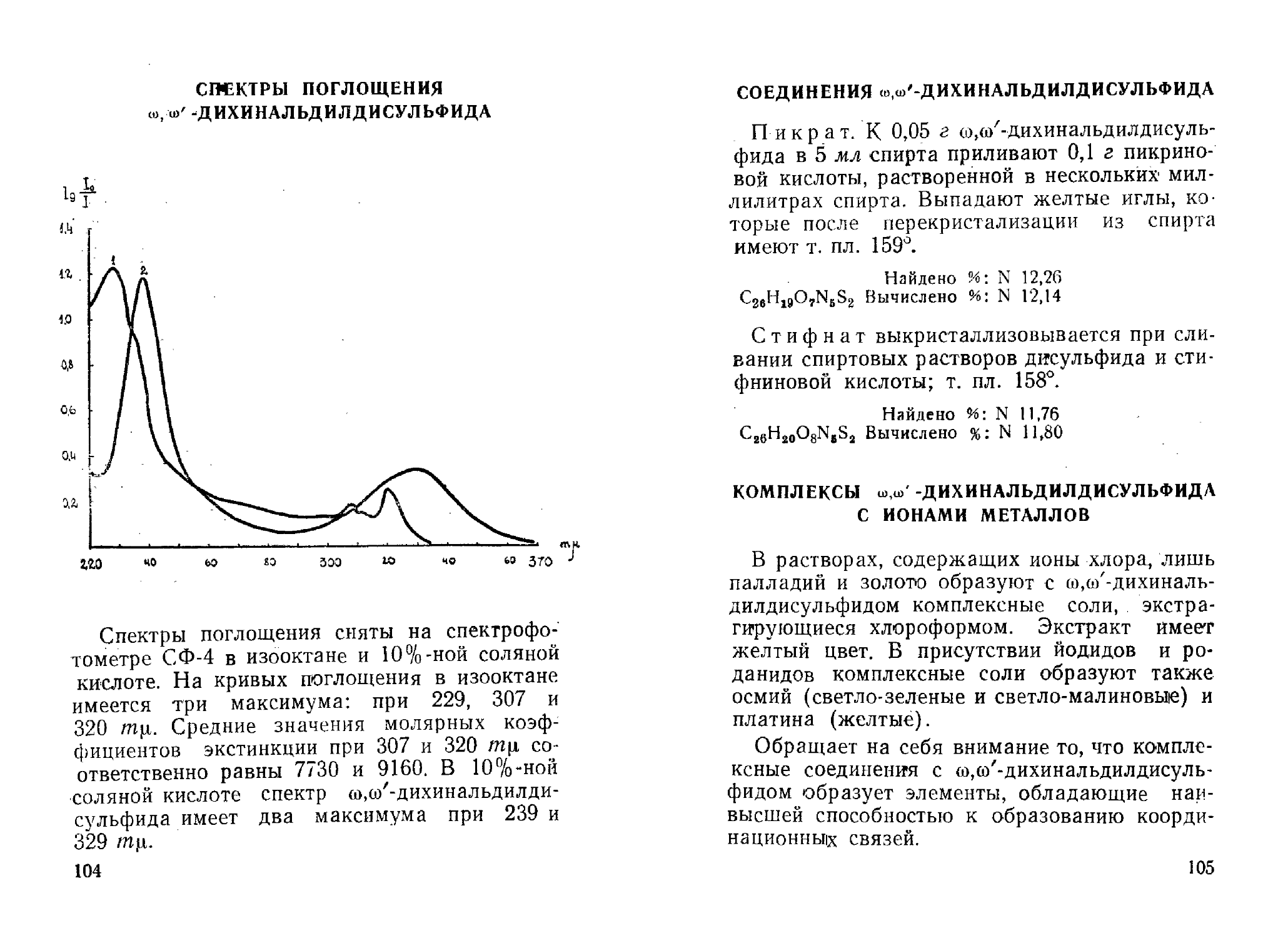
ш,со'-Дихинальдилдисульфид и его соединения
с кислотами и ионами металлов. Ю. А. Бан-
Банковский, Л. А. Федотова, А. Ф. Иевиньш 100
Удаление перекисей из диоксана с помощью
ионообменных смол. И. В. Хвостов,
B. В. Крохе, А. И. Вольфсон 107
Семилетним планом развития народного хо-
хозяйства перед промышленностью химических
реактивов поставлена задача обеспечить про-
производство и исследовательские работы во всех
областях науки и техники химическими реак-
реактивами и препаратами. Для выполнения этой
задачи промышленность химических реакти-
реактивов должна к концу семилетки производить
12—15 тысяч наименований различных соеди-
соединений.
Осуществление этой задачи невозможно без
наличия обширного фонда методических ма-
материалов по синтезу и анализу органических
и неорганических реактивов разных клас-
классов и групп. В различных научно-исследова-
научно-исследовательских учреждениях, лабораториях пред-
предприятий и т. д. имеется большое количество
материалов, представляющих большую цен-
ценность для предприятий химических реактивов.
В связи с этим Всесоюзный научно-иссле-
научно-исследовательский институт химических реактивов
приступил к подбору и систематизации имею-
имеющихся в различных организациях методов
получения и анализа различных соединений.
С целью информирования об этих методах
5
заводов химических реактивов и смежные от
раслей промышленности, химиков-синтетиков
и аналитиков научно-исследовательских инсти-
институтов и вузов, ВНИИ химических реактивок
начинает систематическое издание тематиче-
тематических сборников по синтезу и анализу «Хими-
«Химические реактивы и препараты» в сериях «Ме-
«Методы получения» и «Методы анализа».
Настоящим выпуском начинается издание
сборников по синтезу реактивов и препаратов.
В сборниках «Методы получения» предпо-
предполагается публиковать следующие материалы:
а) Отдельные методы получения химических
реактивов, растворителей, экстрагентов, ад-
адсорбентов и др.
б) Описанные в литературе и эксперимен-
экспериментально проверенные в лабораториях инсти-
институтов, заводов и других организаций методы'
получения с указанием наиболее ответствен-
ответственных1 моментов процессов и аппаратурного их
оформления.
в) Типовые методы получения групп соеди-
соединений.
г) Методы получения «узловых» соединений
для органического или неорганического син-
синтеза.
д) Оригинальные методы синтеза и т. п.
Первый выпуск серии посвящен методам
синтеза органических кислот, их производных и
некоторых других соединений.
Институт обращается с просьбой ко всем
организациям и отдельным лицам, работаю-
работающим в области синтеза и анализа химических
6
веществ, направлять для опубликования в
сборниках свои материалы. Все материалы, по-
пожелания, замечания и отзывы просьба присы-
присылать по адресу: Москва, Е-61, Богородский
вал, д. 3, ВНИИ химических реактивов.
Директор Института В. Г. Брудзь
ИМИНОДИУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИЙ, В. Я. ТЕМКИНА
(ВНИИ химических реактивов)
СН2СООН
ХСН9СООН
Иминодиуксусная кислота является узло-
узловым соединением при синтезе целого рядя
комплексометрических индикаторов: фталеин-
комплексона, тимолфталексоиа, ксиленолового
оранжевого, флкюресцеинкомплексона и дру-
других. !
В литературе описан ряд методов получе-
получения иминодиуксусной кислоты: из аммиака и
монохлоруксусной кислоты [1—4]; с примене-
применением синильной кислоты и ее солей [5—8];
расщеплением нитрилтриуксусной кислоты
под действием концентрированной соляной
кислоты [9—10]; взаимодействием гидразин-
диуксусной кислоты с нитритом натрия [11].
9
Нами разработан метод получения имиио-
диуксусной кислоты п>тем взаимодействия
нитрилтриуксусной и серной кислот. В резуль-
результате исследования влияния температуры, про-
продолжительности реакции, концентрации сер-
серной кислотьи, соотношения компонентов и спо-
способа выделения продукта на выход и каче-
качество иминодиуксусной кислоты, найдены опти-
оптимальные условия, обеспечивающие выход кис-
кислоты 75% теории.
30 г нитрилтриуксусной кислоты нагревают
с 50 мл 70%-ной серной кислоты при 140° в
течение 10 часов. Охлажденный до ~ 20° рас-
раствор выливают в 300 мл воды и обрабатыва-
обрабатывают при 80° активированным углем. Фильтрат
нагревают до 90° и прибавляют порошок гид-
гидрата окиси бария до полноты осаждения суль-
фат-ионов. После двухчасового нагревания
осадок сульфата бария отфильтровывают,
фильтрат упаривают до объема 40 мл и добав-
добавляют к нему 75 мл этилового спирта. Вьшак-
шую иминодиуксусную кислоту отфильтровы-
отфильтровывают и промывают спиртом. Выход кислоты
15,8 г, что составляет 75% теоретического.
Качество препарата регламентируется сле-
следующими техническими требованиями по ТУ
.190—58.
1. Внешний вид—порошок белого цвета.
2. Содержание основного вещества—не ме-
менее 97%.
3. Содержание нерастворимых в воде ве-
веществ—не более 0,1%.
10
4. Содержание свободной серной кислоты—
не более 1 %.
5. Остаток после прокаливания (в виде
сульфатов)—не более 0,5%-
Содержание основного вещества определя-
определяется титрованием навески препарата щелочью
i индикатором бромкрезоловым пурпуровым
(интервал рН перехода 5,2—6,8). Выбор ин-
индикатора сделан в результате проведения по-
тенциометрического титрования на потенцио-
потенциометре ЛП-5 со стеклянным и каломельным
электродами.
Литература
1. W. Heintz, Liebigs Ann. Chem. 124, 302 A862).
2. W. Heintz, Liebigs Ann. Chem. 122, 257A862).
3. W. Heintz, Liebigs Ann. Chem. 145, 49 A868).
4. Пат. США 2239617 A941).
5. H.'Franzen, J. Pharmacy and Pharmacology86,
142 A912).
6. K. I'olstorff, H. Meyer., Ber. 45, 1906 A912).
7. I. V. Dubsky. Ber. 54, 2659 A921).
8. Франц. пат. 746641 A933).
9. W. He'intz, Liebigs Ann. Chem." 149, 88 A869).
10. Q. Schwarzenbach, E. К a in p i t s с h., Helv.
Chlm. Acta 28, 1135 A945).
11. Y. Bailey, W. R e a d, J. Amer. Soc. 36,1759
A914).
ГЕКСАМЕТИЛЕНДИАМИНТЕТРА-
УКСУСНАЯ КИСЛОТА
р. п. л act овеют, в. я. т емкий а
(ВНИИ химических реактивов)
НООСН2С / СН2СООН
^N- (CH,),-N^
ноосн2с/ хсн2соон
Гексаметилендиаминтетрауксусная кислота
является комплексоном, предложенным ИРЕА
в 1954 роду [1]. В настоящее время ком-
плексон применяется для полярографического
определения примесей и разделения редкозе-
редкоземельных элементов [2].
ClCH2COOH+NaOH >ClCH2COONa+HaO
H2N-(CH2NNH2~f4ClCH2COONa+4NaOH—>
NaOOOyy "CH2COONa
+4NaCl+4H2O
12
NaOOCH2C
V
x
CHaGOONa
CH,COONa
4HC1-
NaOOCHjC"
HOOCH2C4
-> )N~(CH2N-N(- " +4NaCl
НООСН3(/ XCH2COOH
К раствору 152 г монохлоруксусной кисло-
кислоты, предварительно нейтрализованной едким
натром, прибавляют 53 мл 50%-ного раствора
едкого натра и 32,5 г гексаметилендиамина,
растворенного в 50 мл воды. Реакционную
массу нагревают при температуре 50° в тече-
течение 6 часов. Охлажденный раствор подкисля-
подкисляют концентрированной соляной кислотой до
рН 3, обрабатывают при нагревании активи-
активированным углем и упаривают на водяной ба-
бане до густой кашеобразной массы. При этом
выделяется комплексов в смеси с большим
количеством хлористого натрия. Последний
удаляют многократным промыванием осадка
ледяной водой.
Выход 58 г, что составляет 62% теории.
Качество препарата регламентируется сле-
следующими техническими требованиями по ТУ
1373—57.
1. Внешний вид—белый кристаллический
порошок.
2. Содержание основного вещества—не ме-
менее 99%.
3. Остаток после прокаливания (в виде
сульфатов)—не более 1%.
13
Литература
1. Р. П. Ластовский, Ю. И. Вайнштейн,
Н. М. Дятлова, В. Я. Т ё м к и н а, И. Д. К о л п а-
к о в а, Ж. аналит. химии, 10, 28 A955) t
2. В. Я. Т ё м к и н а, Р. П. Л а с т о в с к и и,
Н. М. Дятлова, «Сборник научно-технической ин-
информации ИРЕА», 1959, 3—4, стр. 5.
2, 4, 6-ТРИАМИН-1, 3, 5-
-ТРИАЗИН-N, N, N", N', N", N"-
-ГЕКСАУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИЙ, И. Д. КОЛ ПАКОВ А
(ВНИИ химических реактивов)
,СНаСООН
^снр оон
N N • 2НО
НООСНСЧ I II /XiCOOH ^
* N-C C-NJ '
ноосцс V чсн,соон
Новый комплексон, предложенный ИРЕА в
1958 г., представляет интерес для полярогра-
полярографического определения примесей катионов.
Ci
N ^ CHCOOH
f II *3HN
Я Я чснсоон
Cl N Cl
^снроон
V4CHpOOH
N N
С
C-< ^
V снроон
^CHpOOH
46 г иминодиуксусной кислоты нейтрализу-
нейтрализуют 20%-ным раствором едкого натра по уни-
универсальной бумажке. Отбирают третью часть
объема нейтрального раствора кислоты и при-
прибавляют к суспензии 18,4 г хлористого циану-
ра в 30 мл воды, охлажденной до 0°. Тем-
Температура в колбе в момент прибавления кис-
кислоты не должна превышать 10°. При этой тем-
температуре и рН 6—7 реакционную массу вы-
выдерживают 3 часа. Затем к раствору прили-
приливают остальную нейтрализованную иминоди-
уксусную кислоту, температуру реакционной
массы повышают до 45—50° и выдерживают
раствор в течение 1 часа. После этого темпе-
температуру в колбе доводят до 95—100° и, под-
поддерживая рН раствора 6—7, ведут реакцию
еще в течение 3 часов. Раствор фильтруют
горячим. Фильтрат охлаждают и подкисляют
10%-ным раствором соляной кислоты до рН 2,
При охлаждении выпадает кристаллический
осадок комплексона. Осадок отфильтровыва-
отфильтровывают и на воронке отмывают ледяной дистилли-
16
рованной водой от хлоридов. Очистку осуще-
осуществляют многократной перекристаллизацией
из горячей воды.
Вьиход кислоты 2,5 г.
2-БРОМБЕНЗОЛФОСФОНОВАЯ КИСЛОТА
А. М. ЛУКИН, Я. Д. КАЛИНИНА
(ВНИИ химических реактивов)
В основу синтеза 2-бромбензолфосфоновой
кислоты положены условия ее получения,
описанные Доком и Фридманом [1, 2]. При их
изучении была установлена целесообразность
введения нижеследующих изменений.
1. С целью уменьшения образования ди-
арилфосфоновых кислот, получающихся в ус-
условиях Дока и Фридмана, оказалось целесо-
целесообразным проводить реакцию при постоянном
наличии в реакционной массе избытка трех-
хлористого фосфора. Для этого, во-первых,
был изменен порядок загрузки (борфторид ди-
азония из о-броманилина вносят в смесь трех-
хлористого фосфора с этилацетатом); во-вто-
во-вторых, было увеличено количество треххлори-
18
стого фосфора примерно на 20%.
2. Для осаждения меди при выделении кис-
кислоты был использован сереводород [3], а не ед-
едкий натр [1], и соответственно изменены конеч-
конечные условия выделения.
о-Бромбензолфосфоновая кислота предста-
представляет интерес как исходный продукт для син-
синтеза реактивов и других фосфорорганических
соединений.
СИНТЕЗ 2-БРОМБЕНЗОЛФОСФОИОВОИ
КИСЛОТЫ*)
Характеристика основного сырья
1) о-Броманилин с т. пл. 31—32° (перегнан-
(перегнанный в вакууме).
*) См. примечание 1; в схеме уравнений в квадрат-
квадратные скобки заключен предполагаемый промежуточный
продукт; 'вторая и третья реакции протекают после-
последовательно в одном приборе.
19
2) Борфтористоводородная кислота уд. в.
1,24 (получение см, [4]).
3) «Абсолютный» этилацетат (см. приме-
примечание 2).
4) Фосфор треххлористый чистый.
Получение борфторида диазония о-бромани-
лина. К раствору 20,6 г свежеперегнанного
о-броманилина в 63 мл борфтористоводород-
ной кислоты добавляют при перемешивании и
при температуре от —3° до + 5°С (см. приме-
примечание 3) в течение 1 часа 9,1 г нитрита нат-
натрия. По мере добавления нитрита натрия на-
начинает выпадать белый осадок, который че-
через 30—40 минут отфильтровывают, промыва-
промывают дважды водой (по 30 мл) и дважды эфи-
эфиром (по 40 мл) и1 сушат на воздухе.
Получают 31 г мелкокристаллического осад-
осадка. Выход его составляет 95% теории, считая
па о-броманилин.
Получение о-бромбензофосфоновой кисло-
кислоты, К смеси 145 мл «абсолютного» этилацета-
та (см. примечание 2) и 10,5 мл треххлори-
стого фосфора при температуре 55° небольши-
небольшими порциями добавляют смесь 31 г борфтори
да диазония о-броманилина и 2,3 г бромистой
меди. По мере добавления борфторида диазо-
диазония температура поддерживается в пределах
50—60° до полного прекращения выделения
азота в течение 3—5 часов (см. примечание
4 и 5).
Реакционную массу охлаждают и при тем-
температуре 20—30° добавляют 30 мл воды. За-
Затем этилацетат отгоняют с паром (при этом
20
одновременно увлекаются некоторые побочные
продукты реакции), отфильтровывают выде-
выделившуюся ди-арилфосфоновую кислоту D,5 г),
далее в фильтрате сероводородом осаждают
медь, отфильтровывают сульфид меди, а маточ-
маточник кипятят с углем и отфильтровывают. Осве-
Осветленный раствор упаривают почти досуха,
затем охлаждают, выпавший осадок от-
отфильтровывают, промывают 50 мл водьи и
сушат при 100°. Получают 14,7 г технической
кислоты F2,5% теории, считая на борфторид
диазоиия о-броманилина).
Полученный осадок дважды перекристалли-
зовывают из разбавленной соляной кислоты
A:1). Получают 10,1 г D2% теории). Веще-
Вещество имеет т. пл. 199—201,5°, что соответст-
соответствует литературным данным [1].
Анализ на натрий [5] показывает его отсут-
отсутствие.
Найдено %: С 30,95; 30,74; Н 2,61; 2,51;
Р 13,00; 12,95.
CsH(,O3PBr. Вычислено %: С 30,41; II 2,55, Р 13,07.
Примечания:
1. В литературе отсутствуют какие-либо данные по
химизму реакции Дока и Фридмана [1]. Поэтому пред-
представленная схема образования 2-бромбензолфосфоновой
кислоты (как и других фосфоновых кислот, получаемых
по этому же методу) является весьма условной: она в
значительной степени основана на общепринятой схеме
течения аналогичной реакции образования арсоновых
кислот по Шеллеру. По нашим данным, реакция Дока
и Фридмана всегда сопровождается образованием ряда
побочных продуктов. Несмотря на все это, уравнения
могут быть полезны для стехиометрических расчетов.
21
2. Качество этилацетата и его чистота имеют боль-
большое влияние на конечный результат синтеза: наличие
даже небольших количеств этилового спирта и других
возможных примесей может привести к полному иска-
искажению результатов. Поэтому продукт, даже имею-
имеющий квалификацию «абсолютный», должен быть до-
дополнительно тщательно обезвожен и перегнан,
3. Сильное переохлаждение реакционной массы мо
жет привести к выпадению осадка борфторида амина
вместо борфторида диазония. Контролем нормального
хода процесса может служить наличие у выпавшего
осадка способности образовывать с Р-солью в содовой
среде азокраситель. Целесообразно также следить за
реакцией по форме кристаллов выпадающего осадка.
4. Начало реакции, которое можно наблюдать по
выделению азота, иногда возникает самопроизвольно
при 30—40°, иногда же лишь при подогреве до 50—60е.
В последнем случае реакция может идти чрезвычайно
бурно и требует немедленного прекращения нагрева
или даже охлаждения. Вообще целесообразно добав-
добавлять очередную порцию борфторида диазония лишь
после почти полного прекращения выделения азота ог
предыдущей порции. Помимо выделения азота, реак-
реакция сопровеждается выделением других побочных га-
газообразных продуктов кислотного характера, которые
следует улавливать или специально отводить в вытяж-
вытяжной канал.
5. Реакцию можно проводить в трехгорлой колбе,
снабженной термометром, мешалкой с затвором, отвер-
отверстием для загрузки (с пробкой) и отводной трубкой,
соединенной со склянкой Тищенко, содержащей этил-
ацетат (для контроля за ходом выделения азота). Все
соединительные части аппаратуры желательно иметь
на шлифах.
Литература
1. G. О. Do a k, L. D. Free dman.
Chem. Soc. 74, 753 A952).
2. G. О. D о а к, L. D. Freed m a n,
Chem. Soc. 75, 685 A953).
22
J. Anicr.
J, Aincr.
3. V L. Bell, G. M. Kosolapoff. J. Ашег.
Chem. Soc. 75, 4901 A953).
4. «Органические реакции», Сб. 5, стр. 164, ИЛ
1951 г.
5. А. М. Лукин, Г. Б. Завари хин а. Н. С. С и-
м о и о в а. «Вещества высокой чистоты и реактивы»,
Госхимиздат, 1959 г. № 23, стр. 106.
2-ОКСИ-5-ХЛОРБЕНЗОЛФОСФОНОВАЯ
КИСЛОТА
А. М. ЛУКИН, И. Д. КАЛИНИНА,
Г. Б. ЗАВАРИХИНА
(ВНИИ химических реактивов)
CI /vPQH,
ч/он
2-Окси-5-хлорбензолфосфоновая кислота в
литературе не описана. Синтез ее был осуще-
осуществлен на основе диазореакции Дока и Фрид-
Фридмана [1]. Целесообразно отметить, что по дан-
данным литературы, до нас осуществить эту ре-
реакцию в ряду о-аминофенола не удавалось и
настоящий пример синтеза является первым
случаем образования фосфоновой группы на
основе диазореакции в ряду замещенных
о-аминофенола [2].
При разработке условий образования выше-
вышеназванной кислоты были приняты во внима-
24
ние соображения, указанные в введении при
синтезе о-бромбензолфосфоновой кислоты (см.
стр. 18).
2-Окси-5-хлорбензолфосфоновая кислота
представляет интерес как осадитель ряда ка-
катионов, а также как полупродукт для синтеза
реактивов и других фосфорорганических со-
соединений.
СИНТЕЗ 2-ОКСИ-5-ХЛОРБЕНЗОЛФОСФОНОВОИ
КИСЛОТЫ*)
Характеристика основного сырья
1) 2-Амино-4-хлорфенол—технический про-
продукт, дважды перекристаллизованный из воды
(с углем), т. пл. 136,5—137,5°.
*) См. примечание 1 и указание ксхеме образования
2-бромбензолфосфоновой кислоты (см. стр. 18 и 21).
25
2) Борфтористоводородная кислота, уд. в.
1,24. Получение см. [3].
3) «Абсолютный» этилацетат.
4) Треххл-аристый фосфор, чистый.
Получение борфторида диазония 2-амино-
4-хлорфенола. К раствору 48 г 2-амино-4-
хлорфенола в 154 мл борфтористоводородной
кислоты при перемешивании и температуре от
—5° до +5° в течение 1 часа добавляют 28 г
нитрита натрия. Выпавший осадок через 30—
40 минут отфильтровывают, промывают 2 ра-
раза водой, 3 раза эфиром и сушат на воздухе.
Выход 49 г.
Получение 2-окси-5-хлорбензолфосфоновой
кислоты. К смеси 250 мл «абсолютного» этил-
ацетата с 23 мл треххлорисюго фосфора до-
добавляют порциями смесь 48,6 г борфторида
диазония с 4 г бромистой меди. По мере до-
добавления борфторида диазония температура
поднимается до 45—50° и начинается выделе-
выделение азота (см. примечание 1). По окончании
загрузки диазония реакционную массу выдер-
выдерживают при той же температуре до полного
прекращения выделения азота {3—5 часов).
После этого отфильтровывают выпавший оса-
осадок и к фильтрату добавляют по каплям при
20—30° 60 мл дистиллированной воды; далее
этилацетат отгоняют с паром, реакционную
массу охлаждают и сероводородом осаждаюi
медь. Выпавший сульфид меди отфильтровь*
Еают, а кислый фильтрат нагревают до пол-
полного удаления запаха сероводорода. Получен-
Полученный раствор усредняют сначала насыщенным
26
раствором едкого кали (до рН 2), а затем
кристаллическим бикарбонатом калия, конт-
контролируя процесс нейтрализации по прекраще-
прекращению появления синего пятна от капли раство-
раствора на бумажке конго. Далее массу упарива-
упаривают на водяной бане, при этом сначала выпада-
выпадают неорганические соли, которые отфильтровы-
отфильтровывают, а при упаривании до объема^ 50 мл вы-
выпадает кристаллический осадок соли синтези-
синтезированной кислоты (см. примечание 2). Полу-
Полученную соль кристаллизуют из воды и спирта.
Вьиход 5,5 г A2,5% теории).
Найдено %: Р 13,49; 13,49; С1 15,И; 15,63; К 8, 52
С„Н5О4РС1К. СеН.О^ РС1.
Вычислено %: Р 13,61; С1 15,58; К 8,5$
Примечания:
1. При проведении синтеза необходимо учесть все.
указания, сделанные в примечаниях к синтезу о-бром-
оензолфосфиновой кислоты (см. стр. 21-22).
2. Наиболее сложным моментом данного синтеза
является выделение калиевой соли. Обычно при ука-
указанных в прописи условиях выпадает эквимолекуляр-
эквимолекулярная смесь моиокалиевой соли со свободной кислотой.
При недостаточно точном соблюдении объема при
упаривании возможно выделение трудноразделимой
смеси неорганических калиевых солей с солями фосфо-
новой кислоты. Наилучшим контролем этой стадии яв-
является наблюдение под микроскопом за формой вы-
выпадающих кристаллов в пробе упариваемой массы: мч-
неральные соли выпадают в форме кубов, а соль
2-окси-5-хлорфосфоновой кислоты—продолговатых пла-
пластинок.
27
Литература
1. G. О. DoaK, L. D. Free dm an, J. Amer.
Cliem Soc. 73, 6658 A951).
2. A. M. Л у к и н, И. Д. К а л и н и н а, Ж- общ. хи-
химии, 30, 1597 (I960).
3. «Органические реакции». Сб. 5, стр. 164, ИЛ,
1951.
1 АНТРАХИНОНФОСФОНОВАЯ КИСЛОТА
И ЕЕ НАТРИЕВАЯ И АММОНИЙНАЯ
СОЛИ
А. М. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реиктивов)
1-Антрахшюнфосфоновая кислота впервые
описана нами [1]. В основе ее синтеза лежит
диазо-реакция Дока и Фридмана [2]. В отли-
отличие от общих условий проведения этой реак-
реакции, указанных ее авторами, в нашем случае
выход антрахинонфосфоновой кислоты дости-
достигает наибольшей величины при соотношении
треххлористого фосфора к борфториду диазо-
ния 2: 1 вместо 1:1. Условия выделения син-
синтезированного соединения также сильно от-
отличаются от типовых условий выделения, ус-
установленных Доком и Фридманом для соеди-
соединений бензольного ряда. 1-Антрахинонфосфо-
новая кислота представляет интерес как оса-
дитель ряда катионов, в частности, тория,
29
СИНТЕЗ I-АНТРАХИНОНФОСФОНОВОЙ
КИСЛОТЫ*)
о
о
9
II
о
О NBP
И I 2 ¦¦
6
осо
о
II
о
ЗНр ~
О РОД
Характеристика основного сырья
1) 1-Аминоантрахинон, чистый, не ниже
чем 95%-ный.
) См. примеч. 1 и 5 к описанию синтеза 2-бром-
бензолфосфоновой кислоты. В схеме уравнений в скоб-
скобки заключен предполагаемый промежуточный продукт
реакции.
30
2) Борфтористоводородная кислота, уд. в.
1,24 (приготовление, ее см. [3]).
3) «Абсолютный» этил ацетат.*)
4) Треххлористый фосфор, чистый.
Получение борфторида диазония из а-ами-
ноантрахинона. 11,8 г а-аминоантрахинона пе-
переосаждают из концентрированной серной ки-
кислоты и к полученной пасте добавляют 100 мл
40%-ной борфтористоводородной кислоты, пос-
после чего образовавшуюся суспензию диазотиру-
ют при 35—40° 40%-ньим водным раство-
раствором нитрита натрия G г). По окончании
диазотирования реакционную массу охлаж-
охлаждают до нуля, отфильтровывают выпав-
выпавший борфторид диазония, дважды промы-
промывают по 50 мл эфира и высушивают на воз-
воздухе. Получают 15,5 г светло-коричневого
кристаллического порошка. Вещество содер-
содержит непродиазотированный а-аминоантрахинон
в количестве 9°/о- Следовательно, выход диазо-
диазония составляет 87,5% теоретического.
Результаты анализа борфторида диазония,
дважды перекристаллизованного из кипящей
воды.
Найдено %: N 8,54, 8,58
CuHjOJ^BF^ Вычислено %: N 8,70.
Получение 1-антрахинонфосфоновой кисло-
кислоты. В смесь 80 мл «абсолютного» этил ацета-
ацетата и 8,4 мл @,096 г-моля) треххлористого фо-
фосфора при комнатной температуре небольши-
*) См. примечание 2 к описанию синтеза 2-бром-
бензолфосфоновой кислоты (стр. 22).
31
ми порциями загружают смесь 15,5 г @,041
г-моля) борфторида диазония из а-аминоантра-
хинона и 1 г однобромистой меди. По мерс
внесения диазония обычно начинается выделе-
выделение газа, в противном случае температуру ре-
реакционной маесьп постепенно повышают до
60°*). При этом повышении температуры вы-
выделение газа происходит уже всегда с одно-
одновременным изменением окраски раствора из
светло-желтого в темно-коричневый. Далее
реакционный раствор размешивают при
60—65° в течение 2 часов. На другой
день к реакционной массе при размешива-
размешивании и температуре 20—30° прибавляют по
каплям 12 мл воды, размешивают 1 час,
а затем из полученной смеси этилацетат
отгоняют с паром. Образовавшийся осадок
отфильтровывают, промывают 50 мл воды,
переносят в 400 мл 5%-ного раствора ед-
едкого натра, прибавляют 7 г активированно-
активированного угля, массу нагревают до кипения, после
чего полученный горячий раствор вновь от-
отфильтровывают, а фильтрат подкисляют кон-
концентрированной азотной кислотой. При этом
выпадает свободная 1-антрахинонфосфоновая
кислота, которую по охлаждении реакцион-
реакционной массы отфильтровывают, промывают
100 мл дистиллированной воды и высушивают
при 100°. Получают 7,6 г желтого порошка,
что составляет 60%, считая на борфторид ди-
диазония.
*) См. примечание 4 к описанию синтеза 2-бром-
бензолфосфоиовоп кислоты (стр. 22).
32
При кристаллизации из разбавленной A:1)
азотной кислоты с углем получают светло-
желтый мелкокристаллический порошок, поч-
почти нерастворимый в воде, но растворимый в
щелочных растворах, приобретающих при
этом желтую окраску. В большинстве органи-
органических растворителей кислота трудно раство-
растворима, плавится при температуре выше 300°,
Найдено %: Р 10,85; 11,06.
Ci4H9O5P. Вычислено %: Р 10,75.
Синтез натриевой и аммониевой солей аи-
трахинон-1-фосфоновой кислоты. Эти соли по-
получают растворением кислоты при нагревании
до кипения или в 1 н. растворе едкого натра
(соотношение 1 г :50 мл) или в 20%-ном рас-
растворе аммиака (соотношение 1 г: 50 мл). Го-
Горячие растворы фильтруют а фильтраты ох-
охлаждают. Выпавшие при этом соли отфиль-
отфильтровывают, промывают один раз водой и высу-
высушивают при 100°. Обе соли получают в виде
светло-желтьих порошков, хорошо раствори-
растворимых1 в воде.
Найдено %: Р 9,92; 9,62.
CMH8O5PNa. Вычислено %: Р 9,98.
Найдено %: Na 4,88, 4,76
Ci4HuO5NP. Вычислено 96: N 4,59.
Литература
1. А1. М. Л у к и н^ Г. С. Петрова. Ж. общ. хим.
27, 2171 A957).
2. G. О. D о a k, L. D. F r e e d m a n, J. Amer.
Chem. Soc, 73, 5658 A951).
3. «Органические реакции». Сб. 5, стр. 164, ИЛ,
1951.
33
2-ОКСИБЕНЗОЛФОСФОНОВАЯ КИСЛОТА
А. М. ЛУКИН, И. Д. КАЛИНИНА
(ВНИИ химических реактивов)
он
По литературным данным было несколько
попыток получить о-оксибензолфосфоновую
кислоту различными путями [1—3], из них
лишь один метод, а именно дебензилирование
с-бензилоксибензолфосфоновой кислоты, дал
положительный результат [3]. Исходная для
этого синтеза кислота в свою очередь можег
быть получена двумя путями: из о-бензилок-
сианилина и из о-бромбензолфосфоновой ки-
кислоты. В последнем случае выход достигает
10% [3]. Таким образом, в основе описанного
метода синтеза о-оксибензолфосфоновой кис-
кислоты по существу лежит обходный путь вве-
введения оксигруппы, а сам способ является от-
34
носительно сложным. В отличие от этого на-
нами найден простой метод ее получения непо-
непосредственно из 2-бромбензолфосфоновой кис-
кислоты с выходом 41% [4]; целесообразно отме-
отметить, что тот же путь в руках авторов [3] не
привел к положительным результатам.
2-Окагбензолфосфоновая кислота представ-
представляет интерес сама по себе как осадитель ряда
катионов, а также как исходный продукт для
синтеза более сложных аналитических р'еак-
тивов и других фосфорорганических соеди-
соединений.
СИНТЕЗ 2-ОКСИБЕНЗОЛФОСФОНОВОИ КИСЛОТЫ
Характеристика основного сырья
2-Бромбензолфосфоновая кислота с т. пл.
199—201,5 (условия ее синтеза см. стр. 19).
Закись меди—свежеприготовленная 50%-ная
паста (условия ее получения см. примечание).
Условия получения
К раствору 23,7 г о-бромбензолфосфоновой
кислоты в 400 мл раствора аммиака (уд, в.
0,9) добавляют в три приема (с интервалом
1 час) 36 г свежеприготовленной пасты заки-
закиси меди. Реакционную массу размешивают
35
при 70—80° в течение 18 часов. Для поддер-
поддержания концентрации аммиака через массу
каждые 2 часа пропускают газообразный ам-
аммиак (в течение 1 часа). Затем реакционную
массу охлаждают, подкисляют концентриро-
концентрированной соляной кислотой до рН 3—4, при
этом выпадает зеленоватый осадок, который
отфильтровывают, растворяют в разбавлен-
разбавленной соляной кислоте A : 2), осаждают серово-
сероводородом медь, выпавший сульфид меди от-
отфильтровывают. Маточный раствор кипятят с
углем, отфильтровывают уголь, фильтрат
охлаждают и нейтрализуют содой до прекра-
прекращения появления синего пятна от капли рас-
раствора на бумажке конго. Выпавший белый
кристаллический осадок промывают водой
до удаления хлор-ионов и сушат при 120°.
Выход кислоты 7,1 г D1% теории).
Анализ на натрий показывает его отсутст-
отсутствие [5].
Найдено %: С 41,48; 41,31; Н 4,56; 4,50; Р 17,61; 17,67
С6Н7О4Р
Вычислено %: С 41,39; Н 4,05; Р 17,79.
Примечание.
Получение закиси меди [6J: 50 г медного купороса
и 15 г глюкозы растворяют в 100 мл воды. Раствор до-
доводят до кипения и при перемешивании приливают хо-
холодный раствор 20 г едкого натра в 60 мл воды. Мгно-
Мгновенно начинает выпадать осадок гидроокиси, при на-
нагревании переходящий в оранжевую закись меди. Не-
Немедленно по прекращении перехода окраски реакци-
реакционную массу мгновенно охлаждают и нейтрализуют
уксусной кислотой до рН 8—9. Выпавший осадок от-
отфильтровывают и в виде пасты сразу же используют
36
для синтеза, так как на воздухе осадок сравнительно
легко окисляется.
Получают 36 г пасты, содержащей 50% воды.
Литература
1. J.Kennedy, E. S. Lane, J. L. Willans,
J. Chem. Soc. 4670 A956).
2. G. M. Kosolapoff „Organophosphorus compo-
compounds", J. Wiley N Y, 1950.
3. L. D. F r e e d m a n, G. O. D о a k, J. Org. Chem.
25, 140 A960).
4. A. M. Л у к и н, И. Д. К а л и н и н а. Ж. общ. хим.
30, 1597 A960$.
5. А. М. Л у к и н, Г. Б. 3 а в а р и х и н а, Н. С. Си-
Симонова. «Вещества высокой чистоты и реактивы»,
вып. 23, стр. 106, Госхимиздат, 1959.
6. С. Weygond; „Organic preparation", стр., 296.
2-ОКСИБЕНЗОЛАРСОНОВАЯ КИСЛОТА
А. М. ЛУКИН, И, Д. КАЛИНИНА, Г. С. ПЕТРОВА
(ВНИИ химических реактивов)
у
\/ ОН
Проверена и подтверждена литературная
методика получения о-оксибензодарсоновой
кислоты [1], представляющей интерес как ре-
актив-осадитель ряда катионов, а также как
исходный продукт для синтеза других реакти-
реактивов и мышьяксодержащих органических со-
соединений.
СИНТЕЗ СОЕДИНЕНИЯ
*
__/VAsO3H
Характеристика основного сырья
cr-Аминобензоларсоновая кислота, ч., ТУ РУ
722—52.
Условия получения
11 г о-аминобензоларсоновой кислоты рас-
растворяют при нагревании в 150 мл воды и 18 мл
концентрированной соляной кислоты; раствор
охлаждают и при температуре от 0 до 7° до-
добавляют раствор нитрита натрия C,8 г в
30 мл воды).
Полученную смесь нагревают при 50—60°
до полного прекращения выделения азота,
затем добавляют активированный уголь и на-
нагревают на кипящей водяной бане в течение
30 мин. Уголь отфильтровывают, а фильтрат
упаривают почти досуха.
Выпавший при охлаждении осадок отфиль-
отфильтровывают, промывают водой и сушат при
39
gO—90°. Получают 10 г технической кислоты,
которую перекристаллизовывают из воды (с
углем).
Выход 7 г белого кристаллического вещест-
вещества F3,5% теории).
Анализ на натрий показывает его отсут-
отсутствие.
Найдено %: As 33,97; 34,03.
C6H7O4As. Вычислено %: As 34,36.
Литература
1. N. A. Jacobs. J. Amer. Chem. Soc, 41, 1444
A919).
4-НИТРОАНИЛИН-2-АРСОНОВАЯ
КИСЛОТА
A. M. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов)
NH8
NQ
По литературным данным 4-нитроанилин-2-
арсоновую кислоту (можно получить либо арсе-
нированием «-нитроанилина [1], либо нитро-
нитрованием ацетильного производного о-амино-
бензоларооновой кислоты [2, 3].
Хотя первый метод одностадийный, а вто-
второй—трехстадийный, осуществление первого
связано с рядом существенных технологических
41
затруднений, а потому нами детально был
разработан второй путь, причем во все его
стадии был внесен ряд изменений. Так, реак-
реакцию ацетилирования оказалось целесообраз-
целесообразным осуществлять смесью уксусного ангидри-
ангидрида с уксусной кислотой вместо одного уксус-
уксусного ангидрида; при этом выход ацетилиро-
ванного продукта достигает 80—84% теории,
реакция идет спокойно, соединение выпадает
в кристаллическом состоянии (в условиях ли-
литературной прописи» происходит быстрое само-
разогревание реакционной массы и конечное
вещество получается в виде аморфного тягу-
тягучего продукта).
При нитровании о-ацетаминобензоларсоно-
вой кислоты было увеличено вдвое количество
азотной кислоты, что привело к повышению
выхода, причем одновременно была выявле-
выявлена возможность замены концентрированной
азотной кислоты кислотой уд. в. 1,35—1,36.
Стадию омыления 5-нитро-2-ацетаминобен-
золарсоновой кислоты удалось осуществить
непосредственно в реакционном растворе по-
после нитрования, без выделения ацетилирован-
ного соединения. Таким образом эта стадия
как самостоятельная отпала, а общая продол-
продолжительность синтеза значительно сократи-
сократилась.
4-Нитроанилин-2-арсоновая кислота явля-
является исходным продуктом для синтеза реакти-
реактивов—сульфарсазена и арсазена, а также пред-
представляет интерес для получения других мышь-
яксодержащих соединений.
42
СИНТЕЗ 4-НИТРОАНИЛИН-2-АРСОНОВОЙ
КИСЛОТЫ
/VNHCOCH3+
^
А
NHCQCR
Характеристика основного сырья
1) о-Аминофениларсоновая кислота, ч., ВТУ
РУ 722—52.
2) Ангидрид уксусный, ч., ГОСТ 5815—52.
3) Кислота уксусная 98%-ная, х. ч., ГОСТ
61—51.
4) Кислота азотная, уд. в. 1,36, ч.д.а.,
ГОСТ 4461—48.
Получение о-ацетаминобензоларсоновой ки-
кислоты. 22,6 г @,1 М) 96%-ной о-аминобензол-
арсоновой кислотьр загружают в круглодон-
ную колбу емкостью 100 мл, снабженную об-
обратным холодильником и мешалкой, и добав-
добавляют 5 мл уксусного ангидрида и 10 мл 98%-
ной уксусной кислоты. Содержимое колбы на-
нагревают на кипящей водяной бане при разме-
размешивании. Через 10—15 минут о-аминобензол-
43
арсоновая кислота растворяется и тут же вы-
выпадает ацетильное производное. Реакционную
массу охлаждают до комнатной температуры,
отфильтровывают, промывают 5 мл воды и
сушат на воздухе. Получают 20—22 г ацетиль-
ацетильного производного.
Получение 4-нитроанилин-2-арсоновой кис-
кислоты. 20 г о-ацетаминобензоларсоновой кис-
кислоты растворяют в 195 мл концентрированной
серной кислоты при размешивании, затем
охлаждают до 3—4° и прибавляют по каплям
смесь 12 мл азотной кислоты уд. в. 1,36 и
15 мл концентрированной серной кислоты.
Температура нитрования не должна превы-
превышать 10°. После прибавления смеси дают вы-
выдержку два часа и выливают реакцион-
реакционный раствор на 400 г льда. После растворения
льда реакционный раствор выдерживают 0,5
часа, затем при размешивании нагревают на
водяной бане в течение 0,5 часа до 70—80°;
при этом выпадает желтый кристаллический
осадок; далее реакционную массу выдержи-
выдерживают 1 час при 60—70°, охлаждают до ком-
комнатной температуры, осадок отфильтровыва-
отфильтровывают, промывают 15 мл воды и сушат при 60а.
Получают 17 г желтого кристаллического
порошка. Выход 65% теоретического, считая
на исходную о-аминобензоларсоновую кисло-
кислоту. После перекристаллизации из 50%-ного
спирта 4-нитроанилин-2-арсоновую кислоту
можно получить в химически чистом состоя-
состоянии.
44
Найдено %: As 28,3; 28,7
C6H7OBN,As Вычислено %: As 28,6
Л итератуpa
1. L. Benda, Ber 44, 3293 A911).
2 О. Winlersteiner. H. Lieb. Ber. 61,1130 A928).
¦i M A. P h i 111 p s, J. Chem. Soc. 1910 A930).
4-НИТРОАНИЛИН-З-АРСОНОВАЯ
КИСЛОТА
А. М. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов)
NHa
N02
По данным литературы 4-нитроанилин-З-
арсоновую кислоту можно получить нитрова-
нитрованием оксалильного производного ж-аминобен-
золарсоновой кислоты [1—2]. В свою очередь
последнюю получают либо из ж-нитроанилина
по реакции Барта [3], либо нитрованием бен-
золарсоновой кислоты [4] с последующим вос-
восстановлением нитросоединения до амитеа со-
солями железа [5, 6] или амальгамой натрия [7].
Из всех этих вариантов был вьвбран путь син-
46
теза из ж-нитроанилина ввиду большей досту-
доступности данного соединения по сравнению с
бензоларсоновой кислотой. В условия получе-
получения по данным литературы [1, 2, 3, 5, 6] мы
внесли лишь небольшие изменения и уточне-
уточнения и не исследовали их с целью возможного
повышения выхода. В основном полученные
результаты соответствуют литературным дан-
данным.
4-Нитроанилин-З-арсоновая кислота пред-
представляет интерес в качестве исходного соеди-
соединения для синтетических работ.
СИНТЕЗ 4-НИТРОАНИЛИН-З-АРСОНОВОЙ
КИСЛОТЫ*)
KBF4
*) В схеме показано образование оксаминовой ки-
кислоты в качестве промежуточного продукта, однако не
исключена возможность образования соответствующе-
соответствующего оксалиламина
47
АЮД
AsQH,
NHCOCOOH
нр
^NHCOCOOH
HNQ
NHCOCOOH
Hp
AsQH,
A.QH
NHCOCOOH
Характеристика основного сырья
1) ж-Нитроанилин, ч.д.а., ТУ МХП 360—41.
2) Кислота борфтористоводородная (ее
приготовление см. [8]).
3) Нитрит натрия, ч., ГОСТ 4197—48.
4) Ангидрид мышьяковистый, ч., Фармако-
Фармакопея, VIII, ст. 6.
5) Медь хлористая, ч., ГОСТ 4164—48.
48
6) Железо сернокислое закисное, ч., ГОСТ
4148—48.
7) Кислота щавелевая, ч., ГОСТ 5873—51.
8) Кислота азотная, уд. в. 1,4, ч.д.а.,
ГОСТ 4461—48.
Получение борфторида диазония м-нитро-
анилина. 34,5 г @,025 М) ж-нитроанилина
размешивают в 100 мл борфтористоводорной
кислоты. Суспензию диазотируют при 10—15°,
прибавляя в течение 1 часа 17,5 г нитрита на-
натрия. Выпавший борфторид диазония отфиль-
отфильтровывают, промывают 20 мл ледяной воды и
30 мл эфира. Получают 37—38 г розового по-
порошка.
Получение м-нитробензоларсоновой кисло-
кислоты. 23,6 г борфторида диазония размешивают
в 150 мл холодной воды, после чего получен-
полученную суспензию постепенно прибавляют в те-
течение 0,5 часа при 15—16° к заранее приго-
приготовленному раствору 20 г мышьяковистого
ангидрида, 16 г едкого натра и 3 г хлористой
меди в 300 мл воды (рН реакционной массы
<— 10) и выдерживают 10—15 часов при той
же температуре. Затем реакционную массу по-
подогревают до 60° и отфильтровывают от смо-
смолы. Фильтрат подкисляют концентрированной
соляной кислотой до рН 6—7, выпавший ко-
коричневого цвета осадок отфильтровывают, а к
фильтрату прибавляют 3 г активированного
угля и упаривают до объема оо 150 мл. Затем
уголь отфильтровывают, к фильтрату прибав-
прибавляют концентрированную соляную кислоту до
рН 2—3, при этом выпадает кристаллический
49
желтый осадок, который отфильтровывают и
сушат при 60°. Получают 15 г желтоватого
пюрошка. После перекристаллизации из воды
получают 7,5—9 г химически чистой ж-нитро-
бензоларсоновой кислоты.
Получение м-аминобензоларсоновой кисло-
кислоты. 9 г перекристаллизованной ж-нитробен-
золарсоновой кислоты растворяют в 20 мл
10%-ного раствора едкого натра. 71 г FeSO4 •
• 7Н2О растворяют в 200 мл воды при 40° в
литровой колбе. В полученный раствор соли
железа прибавляют 80 мл 25%-ного раствора
едкого натра (до рН 10—12). В образовав-
образовавшуюся голубую кашицу вливают сразу рас-
раствор нитрокислоты. Реакционная масса ста-
становится черной C0—40°). Колбу закрывают и
энергично встряхивают 7—8 минут, затем
массу отфильтровывают с отсасыванием через
бязевый мешок на воронке диаметром не ме-
менее 13 см. Фильтрование длится 4—5 часов.
Осадок на фильтре промывают 50 мл теплой
воды. Щелочной раствор амина подкисляют
концентрированной уксусной кислотой. В те-
течение 10 часов выпадает осадок, вес которого
после сушки на воздухе равен 3 г. Фильтрат
упаривают наполовину, отделяют выпавший
ацетат натрия и концентрированной соляной
кислотой доводят до рН 3—4. Это позволяет
получить еще 3 г амина. Полученные 6 г ами-
амина очищают растворением в 10%-ном раство-
растворе аммиака и выделением концентрированной
уксусной кислотой. Получают 5 г амина.
50
Получение м-оксалиламинобензоларсоновой
кислоты. 5 г л-аминобензоларсоновой кисло-
кислоты растирают с 8 г щавелевой кислоты. Смесь
помещают в трехгорлую колбу E0 мл), снаб-
снабженную обратным холодильником. Нагрева-
Нагревают смесь на масляной бане до 140°, масса при
этом плавится. После затвердевания массы ее
нагревают до 150° и выдерживают при этой
температуре 4—5 часов. Затем охлаждают, рас-
растворяют плав в 20 мл 2 н. раствора углекисло-
углекислого натрия. Продукт выделяют прибавлением
15 мл концентрированной соляной кислоты и
сушат на воздухе. Получают 5 г серого веще-
вещества.
Получение 4-оксалиламинонитробензол-2-
арсоновой кислоты. 5 г ж-оксалиламинобен-
золарсоновой кислоты вносят в 10 мл кон-
концентрированной серной кислоты при размеши-
размешивании. Полученный темный раствор охлажда-
охлаждают до 0° и прибавляют смесь из 1,5 мл азот-
азотной кислоты уд. в. 1,4 и 2 мл концентриро-
концентрированной серной кислоты. Выдерживают при
0—5° 2 часа. Раствор выливают на 70 г льда.
Выпадает желтый осадок, который отфиль-
отфильтровывают.
Получение 4-нитроанилин-З-арсоновой кис-
кислоты. Полученную пасту смешивают с 5 мл
соляной кислоты уд. в. 1,12 и кипятят в кол-
колбе с обратным холодильником в течение
0,5 часа; затем охлаждают, осадок отфиль-
отфильтровывают и сушат при 60°; вес его равен 2 г.
После двукратного растворения в 10%-ном
растворе аммиака и выделения концентриро-
51
ванной уксусной кислотой 4-нитроанилин-З-
арсоновую кислоту получают в химически чи-
чистом состоянии.
Найдено %: As 28,20; 28,41.
C6H7O8N2As Вычислено %\ As 28,6.
Литература
1. Гг. 11, 1035.
2. М. A. Phillips, J. Chera. Soc. 1910 A930).
3. Синтезы органических препаратов, сб. 4, стр. 379,
ИЛ, 1953.
4. А. М i с h a e I i s, H. L о е s n ег, Вег. 27, 265 A894).
5. W. A. Jakobs, M. Heide, Eberger, J. P.
Role. J. Amer. Chem. Soc. 40, 1580 A918).
6. Q. S. H a m i 11 о п, С G 1 y, J. Amer. Chem. Soc.
47, 435 A925).
7. A. Bertheim, L. В е n d a, Ber. 44 3299 A911).
8. «Органические реакции». Сб. 5, стр. 164, ИЛ,
1951.
4-НИТРОАНИЛИН-З СУЛЬФОКИСЛОТА
А. М. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов)
SQH
По литературным данным [1, 2] 4-нитроани-
лин-3-сульфокислоту получают нитрованием
ацетильного производного соответствующей
соли метаниловой кислоты; в основном даны
условия ацетилирования и нитрования; омы-
омыление происходит самопроизвольно при стоя-
стоянии массы после нитрования. Для получения
конечного продукта в химически чистом со-
состоянии авторы подвергают его несколько раз
кристаллизации из воды; выход не указан.
Проверка этих1 данных в лабораторном мае-
штабе подтвердила их. В условия получения
53
мы внесли лишь небольшие изменения и уточ-
уточнения, в основном, в стадии омыления и не
исследовали процесс с целью возможного по-
повышения выхода.
4-Нитроанилин-З-сульфокислота представ-
представляет интерес как исходное соединение для
синтетических работ.
СИНТЕЗ 4-НИТРОАНИЛИН-З-СУЛЬФОКИСЛОТЫ
NHCOCH,
NHCOCH, NHCOCH,
NO2
NHCOCH,
*Hp+HCl
Щ
NO8
Характеристика основного сырья
1) Кислота метаниловая, ч., ВТУ 410—51.
2) Ангидрид уксусный, ч., ГОСТ 5815—52.
3) Кислота азотная, уд. в. 1,4., ч.д.а., ГОСТ
4461—48.
Получение ацетильного производного мета-
ниловой кислоты. 5 г натриевой соли метани-
ловой кислоты (см. примечание), помещенной в
колбу с обратным холодильником, нагревают
2 часа с 20 мл уксусного ангидрида на кипя-
кипящей водяной бане. По охлаждении осадок от-
отфильтровывают и промьщают 5 мл воды. Су-
Сушат на воздухе. Получают 4 г ацетильного
производного в виде розоватого порошка.
Получение 4-нитроацетанилин-З-сульфокис-
лоты. 4 г ацетильного производного метани-
ловой кислоты растворяют в 10 мл концен-
концентрированной серной кислоты при 20°. Раствор
охлаждают до 0° и к нему по каплям при-
прибавляют смесь из 1,5 мл азотной кислоты уд.
в. 1,4 и 3 мл концентрированной серной кис-
кислоты. Нитрование проводят при 2—4°. Полу-
Полученный раствор выдерживают при размешива-
размешивании 2 часа, а затем выливают на 100 г льда.
Через несколько часов выпадает желтоватый
осадок.
Получение 4-нитроанилин-З-сульфокислоты.
Отфильтрованный осадок в виде пасты сме-
смешивают с 20 мл соляной кислоты уд. в. 1,12
и кипятят 0,5 часа. За это время желтоватый
«садок переходит в белый, который отфиль-
отфильтровывают, промывают 5 мл воды и сушат на
55
воздухе. Получают 3 г. Вещество один раз
перекристаллизовывают из 50%-ного спирта.
Найдено %: N 11,69; 11,59
jOsS.HjO. Вычислено %: N 11,85.
Примечание.
По данным A] метаниловую кислоту, как и другие"
сульфокислоты аминов, для ацетилирования необходи-
необходимо брать не в форме свободной кислоты, а в виде
соли, например, натриевой [1] или бариевой [2].
Литература
1. R. Nietzki, Th. BencKiser, Ber., 17, 707
A884).
2. E. Eger, Ber. 21, 2579 A888).
5-НИТРОАНТРАНИЛОВАЯ КИСЛОТА
A. M. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов)
QN'N/COOH
По литературным данным 5-нитроантранн-
ловую кислоту можно получить или нитрова-
нитрованием о-бромбензойной кислоты с последую-
последующим замещением брома на аминогруппу [1J
или нитрованием ацетильного производного
антраниловой кислоты с последующим омы-
омылением ацетильной группы [2, 3]. В основу по-
получения был взят последний путь ввиду боль-
большей доступности исходного вещества и боль-
.шей простоты условий его осуществления.
Проверка описанных приемов нитрования
показала, что в среде концентрированной сер-
серной кислоты [2] не удается получить положи-
положительных результатов, в то время как нитро-
57
вание в дымящей азотной кислоте [3] дает
продукт удовлетворительного качества. В ус-
условия получения по выбранному пути мы соч-
сочли целесообразным внести лишь небольшие
изменения и не исследовали процесс с целью
возможного повышения выхода.
5-Нитроантраниловая кислота представляет
интерес в качестве препарата для синтетиче
ских работ.
СИНТЕЗ 5-НИТРОАНТРАНИЛОВОЙ КИСЛОТЫ
/VNHCOCH
\/соон
нсосн.
Характеристика основного сырья
1) Кислота антраниловая, ч., ВТУ МХП
2791—51.
2) Ангидрид уксусный, ч., ГОСТ 5815—52.
58
3) Бензол, ч.д.а., ГОСТ 5955—51.
4) Кислота азотная, уд. в. 1,52.
Получение ацетантраниловой кислоты. В
колбе с обратным холодильником растворяют
34 г @,25 М) антраниловой кислоты в 250 мл
бензола при 50—60° и при этой температуре
в раствор прибавляют небольшими порциями
25 мл уксусного ангидрида, после чего через 5
минут наблюдается выпадение осадка. Реак-
Реакционную массу кипятят с обратным холодиль-
холодильником 2 часа. По охлаждении отсасывают вы-
выпавший розоватый кристаллический осадок.
После сушки при 60° получают 32,5 г ацетан-
ацетантраниловой кислоты.
Получение 5-нитроацетантраниловой кисло-
кислоты. В 40 мл концентрированной азотной кис-
кислоты уд. в. 1,52 прибавляют при размешива-
размешивании небольшими Лорциями 20 г ацетантрани-
ацетантраниловой кислоты. Температура при этом не дол-
должна превышать 4°. По окончании прибавле-
прибавления реакционную массу размешивают 2 часа
при 5—7° и затем вьпливают на 200 г льда.
Выпадает желтый осадок, его отфильтровы-
отфильтровывают, промывают 300 мл дистиллированной
воды и сушат на воздухе. Получают 14,5 г
светло-желтого порошка.
Получение 5-нитроантраниловой кислоты.
4 г 5-нитроацетантраниловой кислоты кипятят
1 час в колбе с обратным холодильником с
35 мл соляной кислоты уд. в. 1,12. Затем ре-
реакционную массу выливают в 100 мл горя-
горячей дистиллированной водьц. По охлаждении
отфильтровывают желтый осадок; после од-
59
нократной перекристаллизации из 50°/сгного
спирта и сушки при 70° получают 1,5 г свет-
светло-желтого вещества.
Найдено %: С 45,47; 45,62; Н 4,17; 4,04; N 15.68; 15,63
СНОГ$
4»
Вычислено %: С 46,17; Н 3,4; N 15,4.
Литература
1. М. RfaaHs, Lieb. Ann. 194. 99 A879).
2. Н. Rupe, Ber. 30, 1097 A897).
3. О. Ю. Магидсон и А. И. Травин. Ж- общ.
химии 7, 842 A937).
ХРОМОТРОПОВАЯ КИСЛОТА
А. М. ЛУКИН, Г. Б. ЗАВАРИХИНА
(ВНИИ химических реактивов)
но он
sqH
В основу получения хромотроповой кислоты
в свободном состоянии был положен распро-
распространенный метод выделения свободных суль-
фокислот путем обработки их бариевых солей
серной кислотой. Особенностью нижеприведен-
нижеприведенной методики является применение серной
кислоты на 2—3% меньше, чем это необходи-
необходимо для полного переведения взятого количе-
количества бариевой соли хромотроповой кислоты.
Этот прием обеспечивав легкость выделения
свободной хромотроповой кислоты сразу в
химически чистом состоянии.
61
Хромотроповая кислота в свободном состо-
состоянии имеет ограниченное применение (лишь
для некоторый специальных случаев в анали-
аналитической практике).
СИНТЕЗ ХРОМОТРОПОВОЙ КИСЛОТЫ
ноон
но он
2NaCI
но он
но он
+BaSo4
Характеристика основного сырья
1) Хромотроповая кислота, ч., ВТУ МХГТ
4045—53 (в виде динатриевой соли).
2) Хлористый барий, ч., ГОСТ 4108—48.
Получение бариевой соли хромотроповой
кислоты. 100 г динатриевой соли хромотропо-
хромотроповой кислоты растворяют в 500 мл воды, под-
подкисленной 5 мл концентрированной соляной
кислоты. Раствор нагревают до кипения и бы-
быстро загружают 64 г хлористого бария. (Ко-
62
личество хлористого бария берут с 5%-ным
избытком по сравнению с теоретически рас-
рассчитанным). По мере загрузки хлористого ба-
бария из реакционной массы начинает тут же
выпадать осадок бариевой соли хромотропо-
хромотроповой кислоты. После внесения всего количества
хлористого бария реакционную массу выдер-
выдерживают при размешивании и температуре ки-
кипения 5—10 минут, после чего охлаждают до
комнатной температуры, образовавшийся оса-
осадок отфильтровывают с отсасыванием, про-
промывают на фильтре два раза водой (по 100
мл), после чего сушат при 80—90°. Соль по-
получается в количестве 120 г*), что составляет
97,5% теоретического выхода. Полученная
соль натрия не содержит.
Найдено %: Ва 28,36.
CjoHgOsSaBa^t^O Вычислено %: Ва 27,98
Получение хромотроповой кислоты в сво-
свободном виде. 107 г бариевой соли хромотро-
хромотроповой кислоты вносят в 500 мл воды, после
образования однородной суспензии (при раз-
размешивании) к реакционной массе осторожно
добавляют 22,2 г 93,5%-ной серной кислоты
Смесь нагревают до кипения, выдерживают
30 мин. при этой температуре, после чего оса-
осадок отфильтровывают и дважды промывают
по 50 мл горячей воды. Основной фильтрат и
*) Для получения хромотроповой кислоты в свобод-
свободном виде нет необходимости сушить бариевую соль.
Можно применять последнюю и в виде пасты, но в
таком случае в ней необходимо определить процент-
процентное содержание соли.
63
промывные воды упаривают досуха на водя-
водяной бане. Получают 70 г свободной хромо-
троповой кислоты, что составляет 90% теоре-
теоретического выхода, считая на взятое количест-
количество бариевой соли, или 88%, считая на динат-
риевую соль.
Найдено %: S 17,83; 17,60.
•C10H8O8S,.2HsO. Вычислено %: S 18,10.
ФЕНИЛБОРНАЯ КИСЛОТА
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, И. С. МАРКОВИЧ
(ВНИИ химических реактивов)
По литературным данным фенилборную ки-
кислоту получают взаимодействием фенилмаг-
кийбромида с бутилборатом [1—3] или с трех-
фтористым бором [4], а также взаимодействи-
взаимодействием фениллития с бути-лборатом [5].
Проверен метод получения фенилборной ки-
кислоты по Бину и Джонсону [2] из фенилмагний-
бромида и н-бутилбората.
ЗН2О
C6H6MgBr+B(OC4H9K >С6Н5В(ОНJ+
-f-MgBr(OH)+3C4H9QH
Предлагаемая методика отличается от лите-
литературной следующим:
65
1. Реакцию проводят в обратном порядке,
прибавляя эфирный раствор н-бутилбората к
охлажденному раствору фенилмагнийброми-
да.
2. Выделение фенилборной кислоты осуще-
осуществляют путем отгонки бутилового спирта,
исключив подщелачивание едким кали.
В круглодонную колбу емкостью 500 млу
снабженную мешалкой с ртутным затвором,
обратным холодильником с хлоркальциевой
трубкой, капельной воронкой и термометром,
загружают 39,25 г @,25 М) бромбензола, 6 г
@,25 М) магниевых стружек и несколько кри-
кристаллов йода. К смеси при размешивании в
течение часа прибавляют 250 мл эфира, при
этом большая часть магния растворяется. За-
Затем реакционную смесь нагревают на водяной
бане в течение 40—50 мин. до полного раство-
растворения магния.
Получение фенилборной кислоты. Получен-
Полученный раствор фенилмагнийбромида охлажда-
охлаждают до —71 —73° (сухой лед и ацетон) и по-
постепенно, в течение 2 часов, прибавляют рас-
раствор 58 г н-бутилбората @,25 М) в 350 мл
эфира, поддерживая указанную температуру.
После окончания прибавления смесь разме-
размешивают при той же температуре в течение 1
часа и выливают в 150 мл 10%-ной серной ки-
кислоты (предварительно охлажденной льдом).
Эфирный слой отделяют; водный слой дваж-
дважды экстрагируют эфиром (порциями по
100 мл). Эфирные вытяжки соединяют и от-
отгоняют на водяной бане эфир. К оставшейся
66
смеси добавляют 100 мл воды и отгоняют бу-
бутиловый спирт в вакууме (при температуре
бани не более 35—40°), остаток перекристал-
лизовывают из воды. Получают 8 г фенилбор-
фенилборной кислоты в виде белых иголочек, имеющих
т. пл. 215°. Маточный раствор упаривают до
половины первоначального объема. По охла-
охлаждении получают дополнительно 4—5 г фе-
фенилборной кислоты того же качества.
Литература
1. Е. Kh о tin sky, M. Melamed, Ber., 42.3090
A909).
2. F. R, Bean, J. R. J о h n s о n, J. Amer, Chem.
Soc, 54, 4416 A932).
3. W. К б n i g, \V. S с h а г r n b e с k, J. prakt.
Chem.. 128, 153 A930).
4. E. Krause, R. N i t s с h e, Ber., 55, 1261 A922).
5. Б. М. Михайлов, П. М. А р о и о в и ч, Изв.
АН СССР, Отд. хим. наук 3, 322 A956).
н-БУТИЛОВЫЙ ЭФИР БОРНОЙ КИСЛОТЫ
(три-н-бутил борат)
В. Г. БРУДЗЬ, И. С. МАРКОВИЧ
(ВНИИ химических реактивов)
В(ОС4Н9K
Из описанный в литературе методов полу-
получения эфиров борной кислоты наиболее удоб-
удобным является метод этерификации борной ки-
кислоты спиртом с одновременной отгонкой об-
образующейся воды [1]. Способы получения бор-
борных эфиров взаимодействием спиртов с бор-
ноуксусным ангидридом [2. 3] хлористым бо-
бором [4], борилсульфатом [5], этерификацией в
присутствии соляной кислоты [6], а также из
борной кислоты и абсолютного спирта [7]
являются менее удобными.
Нами проверен первый из указанных выше
методов.
Н3ВО3 + ЗС4Н9ОН ^ B(OQH9K + ЗН2О
68
Реакцию проводят в круглодонной колбе
емкостью 1 л, снабженной лопастной мешал-
мешалкой, термометром и градуированной водоот-
борной пробиркой емкостью 100 мл. Послед-
Последняя снабжена обратным холодильником с
хлоркальциевой трубкой. В колбу загружают
61,8 г борной кислоты (х.ч.) и 222 г бутило-
бутилового спирта (ч.).
Смесь нагревают до 110—120° и при этой
температуре размешивают в течение 4,5 часов.
За это время в водоотборной пробирке накап-
накапливается 50—60 мл воды. Если воды отгоня-
отгоняется меньше, размешивание продолжают еще
в течение 1 часа и затем отгоняют невступив-
ший в реакцию бутиловый спирт G4—82 г,
который может быть использован для после-
последующего синтеза).
Оставшуюся после отгонки бутилового спир-
спирта массу перегоняют в вакууме и собирают
фракцию 104—105°/10 мм рт. ст., представля-
представляющую собой бесцветную жидкость с удель-
удельным весом
d" 0,8620 -0,8616 и d2?l5 0,8566
м 27,5
Выход 320—330 г, что составляет 92—93%
теории, считая на борную кислоту (по лите-
литературным данным 96,5%).
Содержание основного вещества 99—100%.
Метод определения основного вещества раз-
разработан в Институте химических реактивов
Новиковской Н. А. и Ротенберг И. Л.
69
Литература
1. В. К. Кусков, В. А. Жукова, Изв. АН СССР
733 A956)\
2. A. Pictet, A. Geleznoff, Ber 36, 2219 A903).
3. Н. G. Cook, J. D. Ilett, В. С. S a u n d е г s,
G. J. Stacey, J. Chem. Soc. 3125 A950).
4. D. R. Martin, J. Amer. Chem. Soc. 73, 2674 A951).
5. A. Pictet, G. Karl, Bill. Soc. Chim. 4, 3, 1122
A908).
6. E. Хотинский, С. Пупко, Укр. хим. ж. 4,
13 A929).
7. «Синтезы органических препаратов», сб. 2,
стр. 133, ИЛ, 1949.
МОНОХЛОРУКСУСНАЯ КИСЛОТА
Б. Г. ЯСНИЦКИЙ, А. П. ЗАЙЦЕВ
(Лаборатория химической технологии Харьковского на-
научно-исследовательского химико-фармацевтического ин-
института)
Существующая в настоящее время техноло-
технология производства монохлоруксусной кислоты
путем хлорирования уксусной кислоты в при-
присутствии различных катализаторов позволяет
получать технический продукт с выходом ме-
менее 80% и содержанием монохлоруксусной
кислоты 98% и примесей дихлоруксусной ки-
кислоты @,1—0,2%) и катализаторов. Очистка
ее представляет значительные трудности вви-
ввиду близости свойств, в частности, точек кипе-
кипения моно- и дихлоруксусных кислот и летуче-
летучести катализаторов (FeCl3, SCb, PC13 и др).
Так, в результате ее разгонки и последующей
перекристаллизации из бензола получают ре-
реактивную монохлоруксусную кислоту с выхо-
выходом 50% от технической, содержание основ-
основного вещества 99%; остаток после прокалива-
71
ния 0,005%, Метод является дорогим (общий
выход не превышает 40%, считая на уксусную
кислоту) и не обеспечивает получения моно-
монохлоруксусной кислоты достаточной чистоты.
Нами разработан метод получения техни-
технической монохлоруксусной кислоты путем оки-
окисления хлорацетальдегида азотной кислотой
[1]. Это исключает возможность образования
ди- и трихлоруксусных кислот. Окисление про-
проходит количественно. В силу этого других ор-
органических примесей в реакционной массе
также не образуется.
Процесс окисления осуществляется непре-
непрерывно, для его инициирования добавляют не-
небольшое количество нитроолеума. Таким об-
образом реакционная масса, при использовании
коррозионностойкой аппаратуры (стекло,
эмаль), может содержать только те примеси,
которые внесены с азотной кислотой и раство-
раствором хлорацетальдегида.
Продукт окисления, содержащий монохлор-
уксусную и азотную кислоты, воду и раство-
растворенные окислы азота, подвергают разгонке
на ректификационной колонне под вакуумом
100 мм рт. ст., причем полностью отгоняются
окислы азота, азотная кислота и вода. Кубо-
Кубовый остаток после охлаждения представляет
собой кристаллическую монохлоруксусную ки-
кислоту «техническую» с практическим выходом
98,5—99,5%. затвердевавшую при 61° и со-
содержащую по анализу:
монохлоруксусной кислоты 99,0—99,5%
ди- и трихлоруксусных кислот отсутствие
72
хлоридов отсутствие
нитритов и нитратов отсутствие
остаток после прокаливания (при использо-
использовании хлорацетальдегида, дающего 0,005%,
и технической азотной кислоты—0,018%
остатка после прокаливания) составляет
0,03%.
С целью дальнейшей очистки производится
перегонка монохлоруксусной кислоты из ку-
кубового остатка при давлении 100 мм рт. ст. и
температуре 130—13Г. Отогнанная монохлор-
уксусная кислота представляет собой продукт,
имеющий температуру застывания 62,4°"и со-
содержащий по анализу:
монохлоруксусной кислоты (при хранении
в герметичной таре или над фосфорным
ангидридом) 100±0.1%
ди- и трихлоруксусных кислот отсутствие
хлоридов отсутствие
нитритов и нитратов отсутствие
остаток после прокаливания B0 г моно-
монохлоруксусной кислоты [2] на аналитиче-
аналитических весах с точностью до 0,0001 г) не об-
обнаруживается, т. е. составляет менее
0,0001%.
Выход очищенной монохлоруксусной кисло-
кислоты составляет 88—90%, считая на техниче-
техническую, или 87—89%, считая на исходный хлор-
ацетальдегид.
Таким образом, разработанный метод не
только обеспечивает более высокую чистоту
продукта, но и имеет существенное экономи-
ческое преимущество перед методом, применяе-
применяемым в настоящее время (себестоимость нового
метода в 5—6 раз меньше существующего).
Приводим описание работы одного из вари-
вариантов лабораторной установки для непрерыв-
непрерывно-периодического получения монохлоруксус-
ной кислоты по разработанному методу.
Схема, изображенная на рисунке, выполне-
выполнена из стекла и собрана на нормальных шли-
шлифах. ^
Из дозаторов 1 и 2 поплавкового типа [3]
с постоянной скоростью в окислитель 3 посту-
поступает 50—55%-ный раствор хлорацетальдегида
и 96%-ная азотная кислота. Соотношение ско-
скоростей поступления этих растворов обеспечи-
обеспечивает постоянное молярное соотношение ин-
гридиентов (хлорацетальдегид : азотная кис-
кислота : вода) 1:1; 5 : 4,2.
Окислитель представляет собой ^/-образный
сосуд, рабочая часть которого заполнена сте-
стеклянными кольцами Рашига и снабжена ру-
рубашкой для охлаждения. Температура в реак-
реакционной массе строго поддерживается в пре-
пределах 30—40°. При более низкой температу-
температуре процесс не успевает пройти в окислителе и
заканчивается в приемнике; более высокая
температура не допустима, так как при этом
происходит гидролиз хлорацетальдегида, что
ведет к понижению выхода и загрязнению
продукта. Реакционная масса самотеком по-
поступает в приемник 4. Выделяющиеся окислы
азота по трубопроводам а и Ь удаляются с
помощью водоструйного насоса.
74
^
«J
5
о.
о
ч
и
ИЗ
о
X
с
о
=;
X
о
я
с
S
и
к
ГС
ч
о
с
5
к
a
о
x
о.
о
н
са
с
о
Ю
X
Из приемника 4 реакционная масса с помо-
помощью вакуума периодически засасывается в
куб 5, снабженный ректификационной колон-
колонкой 6, холодильником 7 и приемниками 8 я 9.
Отгонка из реакционной :массы окислов азота,
азотной кислоты! и воды производится под
вакуумом 100 мм рт. ст. до температуры па-
паров 110°. Отгон, собираемый в приемник 8,
содержит примерно 30%-ную азотную кисло-
кислоту, свободную от монохлоруксусной кислоты;
в пределах температуры 110—125° отбирает-
отбирается фракция в приемник 9, содержащая оста-
остаток воды и 1—2% загруженной монохлорук-
монохлоруксусной кислоты.
Кубовой остаток представляет собой техни-
техническую монохлоруксусную кислоту и в рас-
расплавленном состоянии по трубке перекачива-
перекачивается с помощью вакуума в куб 10, снабжен-
снабженный ректификационный колонкой 11, воздуш-
воздушным холодильником 12 и приемником чистой
монохлоруксусной кислоты 13.
Из куба 10 при давлении 100 мм рт. ст. и
температуре 130—131° производится отгон мо-
монохлоруксусной кислоты. Собранная в прием-
приемнике 13 монохлоруксусная кислота является
продуктом квалификации «чистый». Выход ее
достигает 87—89% теории, считая на исход-
исходный хлорацетальдегид. При емкости окисли-
окислительного аппарата 175 мл производительность
его составляет 700 г/час реакционной массы
или 300 г/час монохлоруксусной кислоты, т. е. 2
тонны в год.
76
Экспериментальные исследования, послу-
послужившие основанием для выбора оптимальных
условий процессов окисления и ректификации,
опубликованы ранее [4, 5].
Литература
1 Б Г. Ясницкий и А. П. Зайцев. Авт. свид.
119 875 A959)
2. ГОСТ 5836—51.
3 Б. Г. Ясницкий и А. П. Зайцев. Заводск.
лаборатория, 25, 4, 499 A959).
4 Б Г Ясницкий и А. П. Зайцев. Химиче-
Химическая промышленность, 2, 1960. Орган Института тех-
технической информации. ГНТК Совета Министров YLLP
5. Б. Г. Ясницкий и А. П. Зайцев. Там же.
2-НИТРО 1-ФТАЛОИЛАМИНОБЕНЗОЛ
В. М. ДЗИ0МК0, К. А. ДУНАЕВСКАЯ
(ВНИИ химических реактивов)-
О
N -СО
NOB
Согласно литературным данным 2-нитро-1-
фталоиламинобензол можно получать сплавле-
сплавлением о-нитроанилина с фталевым ангидридом
[1], а также конденсацией последних в ледя-
ледяной уксусной кислоте [2, 3]. Нами найдено, что
лучшие результаты получаются при нагрева-
нагревании компонентов в присутствии небольшого
количества нитробензола [4].
К тщательно перемешанной смеси тонкоиз-
мельчениых 55 г @,4 М) о-нитроанилина и
118 г @,8 М) фталиевого ангидрида добавля-
добавляют 15 мл нитробензола (см. прим. 1). Смесь
78
нагревают при температуре бани 180—190° в
течение 20 часов (см. прим. 2), расплавлен-
расплавленную реакционную массу выливают при разме-
размешивании в 800 мл воды (см. прим. 3), про-
продукт отфильтровывают, промывают на филь-
фильтре водой A,5 л) и перекристаллизовывают
из ацетона A : 9).
Выход 80 г G3% теории). Температура
плавления 203—204° (по литературным дан-
данным 202—203° [5].
2-Нитро-1-фталоиламинобензол с т. пл.
201,5° может быть получен без перекристал-
перекристаллизации из ацетона. Продукт после промыв-
промывки водой на фильтре промывают ацетоном
A :3). При этом выход составляет 93% тео-
теории.
Примечания:
1. Синтез проводят в круглодонной колбе, снабжен-
снабженной трубкой @ 2,5 см, /35—40 см), помещенной в
масляную баню.
2. Нагревание можно проводить с перерывами.
3. Выливать расплавленную реакционную массу в
воду следует небольшими порциями, осторожно.
79
Литература
1. J. Maier, Lieb. Ann. 327, 17 A903).
2. G. Wanag, Ber. 75, 719 A942).
3. G. Wanag, A. Vernhergs, Ber. 75, 1558A942).
4. В. М. Дзиомко, Ki- А. Дунаевская. Ж.
обш. химии, 30, 628—632 (I960)
5. Н. R u р е, К. G. T h i e s s, Ber, 42, 4303 A909).
2-АМИНО-1-ФТАЛОИЛАМИНОБЕНЗОЛ
В. М. ДЗИОМКО, К. А. ДУНАЕВСКАЯ
(ВНИИ химических реактивов)
/vN-CO
Согласно литературным данным 2-амино-1-
фталоиламинобензол получают восстановле-
восстановлением 2-нитро-1-фталоиламинобензола желе-
железом в спиртово-водной среде [1], гидросуль-
гидросульфитом натрия в среде водцого ацетона [2] и
каталитическим восстановлением с катализа-
катализатором палладированным углем [3].
Нами найдено, что лучшие результаты по-
получаются при восстановлении железом в сре-
среде водного ацетона [4].
81
CHJCOOH
CHgCH,
О
К раствору 10 г @,04 M) 2-нитро-1-фтало-
иламинобензола в 250 мл ацетона добавляют
250 мл воды, 10 г @,17 М) железа в порош-
порошке и 2 мл уксусной кислоты (см. прим. 1).
Реакционную массу перемешивают 3 часа при
температуре 35—45°, после чего добавляют ак-
активированный уголь, смесь кипятят 10 мину г
и фильтруют, фильтрат выливают на лед
D00 г). Осадок отфильтровывают и перекри--
сталлизовывают из этилового спирта A :50).
Выход 6—7 г D0—45% теории). Т. пл. 187—-
190°; по литературным данным 180—189° [1].
Найдено %: N |1,89
Вычислено %: N 11,76
Примечание.
Синтез проводят в трехгорлой 'колбе, снабжен-
снабженном термометром, мешалкой и холодильником, поме-
помещенной в водяную баню
Литература
1. J. M aier, Lieb. Ami 327, 17 A903).
2. Н. Rupe, К. G. T h i e s s. Ber. 42, 4.303 A909).
3. E. I). Bergmann, M Beniov. .1. Org. Ch. 20,
1654 A955).
4 В. М. Дзиомко, К. А. Дунаевская Ж,
общ. химии 30, 628—632, 1960.
82
N-ОКИСЬ ПИРИДИНА
( Пиридин-М-оксид; пиридин-1 -оксид)
И. А. КРАСАВИН
(ВНИИ химических реактивос)
N-Окись пиридина является важным исход-
исходным продуктом при получении многих произ-
производных пиридина, замещенньих в 4, 3, 2 поло-
положениях. Имеется ряд обзоров по получению,
свойствам и применению N-окиси пиридина
[1-5].
N-Окись пиридина получают окислением пи-
пиридина при помощи надбензойной [6], моно-
надфталевой [7], 40%-ной надуксусной кислот
83
[8], перекиси водорода в присутствии уксусной
[9—12] или других карбоновых кислот [11].
Методика, предложенная в «Синтезах органи-
органических препаратов» [8], основана на примене-
применении .40%-ной надуксусной кислоты, дефицит-
дефицитной и опасной в обращении. При окислении
пиридина перекисью водорода в присутствии
уксусной кислоты выход N-окиси возрастает
при увеличении избытка кислоты [12]. Авторы
ряда работ [1, 12] получили 94—96%-ный вы-
выход, применяя 10 М уксусной кислоты на 1 М
пиридина.
Нами бьгло установлено, что окисление идет
успешно также и при молярном соотношении
кислоты и пиридина 2:1. Выход при этом не-
несколько снижается, но значительно уменьша-
уменьшаются реакционные объемы и расход уксусной
кислоты.
Образующийся после отгонки воды и ук-
уксусной кислоты неочищенный ацетат N-окиси
пиридина может быть переработан как в
N-окись пиридина, так и в ее соли, например.
в гидрохлорид. Для ряда целей (например,
для нитрования) можно применять N-окись
пиридина в виде гидрохлорида. В то же вре-
время выделение, очистка и хранение этой соли
по сравнению с основанием значительно про-
проще. Было установлено, что при получении
гидрохлорида можно с успехом отказаться от
упаривания реакционной массы в вакууме с
перемешиванием [8] и, тем самым, упростить
аппаратуру. Перекристалл изовывать гидрохло-
84
рид более удобно из этилового спирта, чем из
изопропилового [8].
HCI
Получение ацетата N-окиси пиридина. В
3-литровой колбе смешивают 474 г F М) пи-
пиридина с 720 г A2 М) 98—99%-ной уксусной
кислоты и добавляют 1320 мл 28%-ной пере-
перекиси водорода (см. прим. 1). Нагревают
15 часов на водяной бане при 70—80° (см.
прим. 2). К охлажденному раствору осторож-
осторожно добавляют 50 мл 20%-ного раствора фор-
формалина (см. прим. 3), отгоняют воду и ук-
уксусную кислоту на кипящей водяной бане в
вакууме водоструйного насоса B0—30 мм рт«
ст.) до полного прекращения отгонки. Добав-
Добавляют 500 мл воды и снова отгоняют в тех же
условиях (см. прим. 4). Остаток в колбе
G50—775 г) представляет собой технический
ацетат N-окиси пиридина с содержанием ос-
основного вещества 90—92% (см. прим. 5).
85
Получение N-окиси пиридина.
ВНИМАНИЕ! ПЕРЕД ПЕРЕГОНКОЙ НЕОБХОДИМО
УБЕДИТЬСЯ В ОТСУТСТВИИ ПЕРЕКИСЕИ, ЧТО
ДЕЛАЕТСЯ ПРОБОЙ С ЙОДИСТЫМ КАЛИЕМ
Для работы применяют прибор, пригодный
для перегонки твердьих веществ в вакууме
(см. прим. 6). Прибор снабжают приспособ-
приспособлением для улавливания уксусной кислоты,
присоединив отвод приемника к холодильнику
Либиха, другой конец которого присоединяют
к вакуум-насосу через ловушку, охлаждаемую
до —35° смесью сухого льда и ацетона.
В куб прибора помещают 250 г техническо-
технического ацетата N-окиси пиридина и несколько
комков стеклянной ваты. Нагревают куб на
масляной бане (см. прим. 7) и перегоняют
жидкость в пределах 100 — 10571 мм. При
этом N-окись пиридина собирается в прием-
приемник, а пары уксусной кислоты переходят в хо-
холодильник и удерживаются в ловушке. Выход
125—133 г F6—70% теоретического) т. ил.
04—65° (см. прим. 8).
Получение гидрохлорида N-окиси пиридина.
В трехгорлую колбу емкостью 1,5—2,0 л, сна-
снабженную мешалкой, прямой трубкой для вво-
ввода газа (см. прим. 9) и отводной трубкой,
ведущей к ловушке, помещают 380 г неочи-
неочищенного ацетата N-окиси пиридина и, при
размешивании и охлаждении, пропускают хло-
хлористый водород.
86
При достижении привеса 100—ПО г, когда
масса сильно загустеет, насыщение прерыва-
прерывают и белый осадок отфильтровывают с отса-
отсасыванием. Фильтрат упаривают в вакууме во-
водоструйного насоса на водяной бане и сно-
снова насыщают хлористым водородом, получая
дополнительно некоторое количество гидро-
гидрохлорида.
Для очистки продукты соединяют и раст-
растворяют примерно в двукратном по весу коли-
количестве этилового спирта и насыщенный рас-
раствор оставляют медленно охлаждаться. Вы-
Выпавшие крупные кристаллы отфильтровывают,
промывают холодным спиртом и сушат. Вы-
Выход 296г G5% теоретического), т. пл. 178—
180°, после повторной кристаллизации т. пл.
179,5—181°.
Примечания:
1. Применялась продажная стабилизированная пе-
перекись водорода.
2. Можно производить нагревание с перерывами и
течение 2—3 дней.
3. Раствор формалина применяется для разложений
нерекисных соединений, Добавлять его следует осто-
осторожно при размешивании.
4. Следует возможно более полно отогнать воду и
остатки уксусной кислоты.
5. Продукт не следует долго хранить, так как он
медленно разлагается при стоянии и в нем вновь об-
образуются перекненые соединения.
6. Пригоден прибор, описанный в «Синтезах орга-
органических препаратов» [13].
7. Температура в бане не выше 130—135°.
8. Продукт сильно гигроскопичен. Его следует хра-
хранить в запаянных ампулах в темноте.
87
9. В качестве вводной трубки удобно применять
стеклянный тройник, установленный на пробке в горле
колбы. К боковому отростку подводят газ, а через
прямую часть пропускают топкую стеклянную палоч-
палочку, укрепленную на верхнем конце тройника с помо-
помощью отрезка резиновой трубки. Перемещением палоч-
палочки можно прочищать трубку, предотвращая забивание
ее образующейся солью.
Литература
1. Е. Ochiai, J. Org. Chem., 18, 534 A953).
2. Z. Taiikowa, Wiadomosci Chem., 7, 169 A953).
3. F. E. Cislak, Ind. Eng. Chem. 47, 800 A955).
4. A. R. Katritzky, Quaterly Rewievs 10, 395 A956).
5. A. J. Cox, Manufact. Chem. 28, 463 A957).
6. J. M e i s e n h e i m e r, Ber. 59, 1848 A926).
7. B. Bobzanski, L. Kochanska, А. К. о wa-
wale wska, Ber. 71, 2385 A938).
8. «Синтезы органических препаратов», сб. 5, стр. 56,
56, ИЛ, 1954.
9. Е. Ochiai, M. I s h i k a w a, S, Zai Ren.
J. Pharra. Soc. Japan 65, 72 A945); С. А. 45, 8526d A951).
10. H. J. den Ilertog, W. P. Com be, Rec. trav.
chim. 70, 581 A951), С A. 46, 8654i A952).
11. E. Ochiai, M. Katada, E. Hayaslii,
J. Pharm. Soc. Japan. 67, 33 A947); С. А. 45, 954Ii A951).
12, M, А ли-Заде, Уч. зап. Ленинабадского Гос.
пед.'ин-та, вып. 3, 111 A95ti); РЖХим, 1957, 71654.
13. «Синтезы органических препаратов», сб. 3, стр.
116, ИЛ, 1952 г.
N-ОКИСЬ 4-НИТРОПИРИДИНА
D- Нитропири дин-М-оксид;
4-нитропиридин-1 -оксид)
И. А. КРАСАВИН
(ВНИИ химических реактивов)
N-Окись 4-нитропиридина является проме-
промежуточным продуктом при получении многих
производных пиридина, замещенных в положе-
положении 4 [1—5].
N-Окись 4-нитропириди'на получают нитро-
нитрованием N-окиси пиридина (или ее солей) в
концентрированной или дымящей серной кис-
кислоте при- 90—130° нитратом калия [1,6—9]
азотной кислотой [1] или дымящей азотной
89
кислотой [1, 10, 11]. Вьиход достигает 95% [11].
Для получения N-окиси 4-нитропиридина мы
применили вместо основания гидрохлорид
N-окиси пиридина, так как эта соль более
легко синтезируется и более удобна при хра-
хранении.
Получение N-окиси 4-нитропиридина. В кру-
глодонной колбе емкостью 2 л со шлифом к
148,9 г A,13 М) гидрохлорида N-окиси пири-
пиридина прибавляют по каплям при охлаждении
205 мл олеума {содержание SO3 7%), отводя
выделяющийся хлористый водород в ловушку.
После прекращение выделения газа осторож-
осторожно добавляют 135 мл 60%-ного олеума и
171,6 г A,7 М) тонкорастертого нитрата ка-
калия. К колбе присоединяют на шлифе обрат-
обратный холодильник, защищенный хлоркальцие-
вой трубкой. Колбу очень осторожно подогре-
подогревают на водяной бане до появления в ней оки-
окислов азота, что является признаком начала
реакции. Баню с горячей водой немедленно
удаляют и колбу охлаждают холодной водой,
чтобы умерить течение реакции (см. прим. 1).
Когда течение реакции замедлится, кол-
колбу вновь нагревают на кипящей водяной бане
и выдерживают в течение 4 часов. Затем мас-
массу охлаждают, выливают на лед и обрабагы-
90
вают твердым карбонатом натрия до нейтра-
нейтральной реакции. Осадок отсасывают, промы-
промывают ледяной водой и сушат. Выход неочи-
неочищенного продукта 95 г (см. прим. 2).
Перекристаллизацию осуществляют путем
растворения неочищенного продукта в 1250 мл
кипящего ацетона, фильтрования и упарива-
упаривания раствора до начала кристаллизации. При
охлаждении и стоянии выделяется 73 г N-оки-
N-окиси 4-нитропиридина, имеющего т. пл. 160—
161,5°.
Упариванием ацетонового маточника полу-
получают еще 9,5—10 г продукта, имеющего т. пл.
159,5—161°. Общий выход 82,5—83 г E2% те-
теоретического) (примечание 2).
Примечания;
1. Реакция проходит бурно и сопровождается обиль-
обильным выделением окислов азота.
2. Выход продукта, по-видимому, может быть по-
повышен, если экстрагировать хлороформом из фильтра-
фильтрата растворенную б нем N-окись 4-ннтропиридина.
Литература
1. Е. О с hi a i, J. Or#. Chein. 18, 534 A953).
2. Z. T alikowa, Wiadomosci Chcm. 7 169 A953).
3. V. Е. Cislak, Ind. Eng. Chem. 47, 800 A955).
4. A. R. К a t r i t z k y, Quaterly Rcwicvs 10, 395 A956).
5. A. j. Cox, Manufact. Chem. 28, 463 A957).
6. E. Ochiai, K. A r i in a, M. I s li i k a w a,
J. Pharm. Soc. Japan 63, 79 A943); C. A. 45, 515tf (H+51).
7. H. Ochiai, M. I s h i k a w ;i, К. А г i m a,
J. Pharm. Soc. Japan 64, 210A944); С A. 45, 51541 A951).
8. E. Ochiai, S. Zai-Ren, J. Phann. Soc. Japan
65, 73 A945); С. А. 45, 8526h A951).
91
9. Е. О с h i a i, Е. Н а у a s h i, М. К. a t a d a, J.
Pharm, Soc. Japan 67, 79 A947); С. А. 45, 9538а A951).
10. Н. J. den Her tog, J. Overhoff, Rec. trav. chim.
69, 468 A950); С. А. 44, 6418c A950).
П. Н. J. den Hertog, W. P. Combe, Rec.
trav. chim. 70, 581 A951); С. А. 46 86541 A952).
8-МЕРКАПТОМЕТИЛХИНОЛИН, ЕГО
СОЛИ И СОЕДИНЕНИЯ С ИОНАМИ
МЕТАЛЛОВ
/о. л. банковский, з. в. мисуловинл,
А, Ф. И ЕВИН МП, М. Р. БУКА
(Институт химии АН Латвийской ССР)
8-Меркаптометилхинолин в литературе не
описан. В связи с этим для его синтеза при-
принята обычно применяемая схема:
снл
CHBr CH2SH
Получение 8-бромметилхинолина. В литера-
литературе описано несколько способов получения
8-бромметилхинолина. При синтезе 8-дибром-
метилхинолина с небольшим выходом полу-
получается 8-бромметилхинол1-гн [1]. Бромирование
8-метилхинолииа N-бромсукцинимидом в сре-
9:?
де четыреххлористого углерода [2] в ультра-
ультрафиолетовых лучах, по нашим данным, даст
вьиход 8-бромметилхинолина 16—20% (в ра-
работе авторов этого метода выход не указан).
При более длительном проведении реакции
A2 часов) [3] выход 8-бромметилхинолина по-
повышается, по нашим данным, до 20—25%.
Наиболее подходящим оказался метод с при-
применением перекиси бензоила в качестве ката-
катализатора (выход 60%) [4]. В наших опытах
было установлено, что выход 8-бромметилхи-
нолина в большой степени зависит от качества
перекиси бензоила.
Смесь 23 г свежеперегнанного 8-метилхи-
нолина, 29 г N-бромсукцинимида и 1 г пере-
перекиси бензоила в 300 мл четыреххлористого
углерода нагревают на водяной бане с обрат-
нам холодильником в течение 4 часов. Реак-
Реакционная смесь в колбе постепенно темнеет и
к концу реакции становится винно-красной.
После охлаждения до 0° раствор фильтру-
фильтруют. Осадок на фильтре промывают неболь-
небольшим количеством холодного четыреххлори-
четыреххлористого углерода, который присоединяют к филь-
фильтрату. Четыреххлористый углерод отгоняют
па водяной бане, а из остатка в колбе 8-бром-
метилхинолин экстрагируют 80—100 мл пе-
тролейного эфира путем кипячения с обрат-
обратным холодильником. Горячий раствор 8-бром-
8-бромметилхинолина в петролейном эфире осто-
осторожно сливают и быстро охлаждают в ледя-
ледяной ванне. Выкристаллизовываются розовые
иглы, имеющие т. пл. 33—34°. Выход 22 г,
94
F2% теоретического). После перекристалли-
перекристаллизации из петролейного эфира или спирта, вы-
выпадают бесцветные иглы.
Получение 8-меркаптометилхинолина. К
спиртовому раствору гидросульфида калия
(полученному тщательным насыщением серо-
сероводородом 12 г едкого кали в 80 мл спирта)
прибавляют 6 г 8-бромметилхинолина. Уже
при комнатной температуре начинается реак-
реакция и выпадает осадок бромистого калия,
Смесь нагревают на водяной бане 15—20 мин.
После охлаждения реакционную смесь выли-
выливают в 200 мл дистиллированной воды. Эмуль-
Эмульсию 8-меркаптометилхинолина экстрагируют
100—150 мл эфира. После высушивания эфир-
эфирного экстракта в течение 12—16 часов безвод-
безводным сульфатом натрия эфир отгоняют. Полу-
Получают 4,5—4,6 г 8-меркаптометилхинолина (96%
теоретического по 8-бромметилхинолину или
60% по 8-метилхинолину). Полученный про-
продукт представляет собой светло-желтое масло
с неприятным запахом и т. кип. 168—170° при
15 мм рт. ст., которое разложилось при попыт-
попытке перегнать его при атмосферном давлении.
При хранении на воздухе в течение 1—2 ча-
часов окисляется в дисульфид. Перегнанный в
вакууме 8-меркаптометилхинолин сохраняется
в закрытом сосуде 3—4 недели с постепенным
образованием дисульфида.
Найдено %: С 67,70; Н 5,29; N 7,92; S 18,49
CI0H9NS Вычислено %: С 68,53; Н 5,13: N 7,99; S 18,2^
Получение 8,8'-метилхинолиндисульфида
может быть осуществлено окислением пере-
95
кисью водорода или кислородом воздуха ще-
щелочных растворов 8-меркаптометилхинолина.
При стоянии на воздухе в течение нескольких
часов спиртовых растворов 8-меркаптометил-
QQ СО
хинолина образуются длинные слегка желто-
желтоватые иглы с т. ил. 121°. Соединение нераст-
нерастворимо в воде и щелочах, растворимо в раз-
разбавленных кислотах, спиртах, бензоле, хлоро-
хлороформе и других органических растворителях.
Найдено %: N 8,06; S 18,32
C20HieN,S3 Вычислено %: N 8,04; S 18,40.
СОЛИ 8-МЕРКАПТОМЕТИЛХИНОЛИНА
Меркаптогруппа, обладающая в 8-меркапто-
метилхинолине слабо кислым характером, дает
возможность получить соли щелочных метал-
металлов. Нам не удалось выделить эти соли в до-
достаточно чистом состоянии, чтобы охаракте
ризовать их, так как они чрезвычайно быстро
окисляются в дисульфид. Устойчивые соли
8-меркаптометилхинолина можно получить
путем его взаимодействия с кислотами: такие
соли долго сохраняются, не подвергаясь изме-
изменениям.
06
Перхлорат 8-меркаптометилхинолина. 4,7 г
8-меркаптометилхинолина небольшими порци-
порциями растворяют в 8 мл 57%-ной хлорной кис-
кислоты, после чего вскоре появляются первые
кристаллы, а затем закристаллизовывается
вся масса, превращаясь в густую кашу. Пос-
После отсасывания на нутч-фильтре получают
7,2—7,3 г перхлората или 97—98% теоретиче-
теоретического. Перекристаллизация из 30 мл дистилли-
дистиллированной воды дает 6 г бесцветных или слег-
слегка желтоватых кристаллов, малораст&оримых
в холодной воде. Перхлорат взрывается при
нагревании; в сухом состоянии устойчив при
хранении (при хранении на воздухе в течение
года не замечено признаков окисления).
Найдено %: С 43,57; N 5,08; S 11,71
C10H9NS.HC104 Вычислено %: С 43,56; N 5,07; S 11,63
Получены также соли: бромистоводородная
CioHgNS * НВг, йодистоводородная CioHgNS •
• HJ, виннокислая CioHgNS • С4Н6Об и пикри-
новокислая C10H9NS • СбН2(Ж>2)зОН, состав
которых установлен элементарным,анализом.
Эти соли при хранении достаточно устойчивы,
кроме последней, которая постепенно разла-
разлагается.
ВЗАИМОДЕЙСТВИЕ 8-МЕРКАПТОМЕТИЛХИНОЛИ-
8-МЕРКАПТОМЕТИЛХИНОЛИНА С ИОНАМИ МЕТАЛЛОВ
В опытах применялся 1%-ный раствор пер-
перхлората 8-меркаптометилхинолина в ацетоне,
0,1 мл которого вводили в 5 мл соответству-
соответствующего буфера (рН 2,2; 6,8; 9,1). В получен-
97
ный таким образом раствор 8-меркаптометил-
8-меркаптометилхинолина приливали по каплям 2 мл 0,0005 М
раствора соли катиона. Образовавшийся оса-
осадок испытывали на растворимость в различ-
различных органических растворителях. Результаты
опытов представлены в нижеследующей таб-
таблице.
Таблица
Взаимодействие 8-меркаптометилхинолина
с ионами металлов
«ент
ч
1
Си
Pd
Hg
Pt
Os
Ag
Аи
Bi
рН 2,2
цвет
комплекс-
комплексной соли
2
буроватс-
жёлтый
оранжево-
жёлтый
бесцветный
бесцветный
фиолетово-
коричневый
бесцветный
бесцветный
светло-жёл-
светло-жёлтый
рН 6,8
цвет ком-
комплексной
соли
3
буровато-
жёлтый
оранжево-
жёлтый
бесцветный
бесцветный
фиолетово-
коричневый
бесцветный
бесцветный
светло-жёл-
светло-жёлтый
рН 9,5
цвет ком-
комплексной
соли
4
буровато-
жёлтый
оранжево-
жёлтый
бесцветный
бесцветный
фиолетово-
коричневый
бесцветный
бесцветный
светло-жёл-
светло-жёлтый
98
Продолж. табл.
1
Cd
Pb
In
Ni
Co
Fe
2
3
бесцветный
бесцветный
бесцветный
4
бесцветный
бесцветный
бесцветный
красно-ко-
красно-коричневый
оранжево-
коричневый
красно-корич-
красно-коричневый, быст-
быстро разрушаю-
разрушающийся
Литература
1. J. Howitz, P. N other, Ber. 39, 2709 A906).
2. Masaru Hasegawa, J. Pharm. Soc. Japan 71, 256
{1951).
3. Von Ng. Ph. Bun.—Hoi, Rec. tra/. chim. 73, 197
<1954).
4. B. Prijs, R. Gall, Helv. Chim. Acta 37, 90 A954).
(о, ш'-ДИХИНАЛЬДИЛДИСУЛЬФИД И
ЕГО СОЕДИНЕНИЯ С КИСЛОТАМИ И
ИОНАМИ МЕТАЛЛОВ
Ю. А. БАНКОВСКИЙ, Л. А. ФЕДОТОВА,
А. Ф. ИЕВИНЬШ
(Институт химии АН Латвийской ССР}
chs-s-s-chs
Необходимый для синтеза со,й/-дихинальдил-
дисульфида ш-монобромхинальдин мы пытались
получить бромированием хинальдина N-бром-
сукцинимидом. Этот метод дает хорошие ре-
результаты при бромировании 8-метилхинолина.
В случае хинальдина во всех вариантах по-
поставленных опытов имело место сильное ос-
моление реакционной смеси, из которой вьп-
делить ш-монобромхинальдин не удалось. По-
Поэтому со-монобромхинальдин получен восста-
100
иовлением и-трибромхинальдина в ацетоно-
ацетоновом растворе хлористым оловом по методу,
описанному Хеммиком [1]. По этой прописи
всегда получается хинальдин и незначитель-
незначительное количество со-монобромхинальдина. Ока-
Оказалось, что указываемое Хеммиком количест-
количество хлористого олова слишком велико и при-
приводит к исчерпывающему восстановлению
со-трибромхинальдина в хинальдин. Кроме то*
го, для нейтрализации реакционной смеси
нельзя применять рекомендуемый Хеммиком
гидрат окиси кальция, который приводит к
отщеплению брома и образованию хинальди-
хинальдина. Вместо гидрата окиси кальция применен
углекислый кальций.
Синтез о^со'-дихинальдилдисульфида состоит
из следующих стадий:
Получение (?>-трибромхинальдина. (о-Три*-
бромхинальдин является весьма важным про-
промежуточным продуктом в ряде синтезов, в
частности, в синтезе хинальдиновой кислоты.
Наиболее удовлетворительная пропись его по-
получения дана Хеммиком [I]. Мьв уточнили ус-
условия проведения отдельных стадий, следуя
которым можно получить более чистый про-
продукт, чем непосредственно по прописи Хем-
мика.
80 г безводного ацетата натрия нагревают
до растворения в 250 мл ледяной уксусной ки-
кислоты и приливают 25 г хинальдина. Смесь
охлаждают до 70° и при механическом пере-
перемешивании в течение 30 мин. прибавляют из
101
капельной воронки раствор 27,8 мл брома в
100 мл ледяной уксусной кислоты.
По окончании введения раствора колбу на-
нагревают 2 часа в водяной бане. В осадок вы-
выпадает бромистый натрий, а по охлаждении
смеси—(о-трибромхинальдин. Осадок отфиль-
отфильтровывают и промывают водой до полного
растворения бромистого натрия. Белые пла-
пластинки имеют т. пл. 126°, а после перекристал-
перекристаллизации из спирта 128° (по литературным
данным 128°).
При разбавлении фильтрата водой осажда-
осаждается еще некоторое количество менее чистого
(й-трибромхинальдина. Выход 35,5 г (92% те-
теории).
В большинстве случаев общий выход до-
достигает 80% теоретического.
На выход ш-трибромхинальдина очень си-
сильно влияет качество уксусной кислоты и аце-
ацетата натрия. Должна применяться только ле-
ледяная уксусная кислота, а ацетат натрия сле-
следует тщательно обезвоживать как описано в
сборнике «Препаративная органическая хи-
химия».
Получение ы-монобромхинальдина. К. 40 ?
охлажденному до —5° раствору w-трибром-
хинальдина в 300 мл ацетона при интенсивном
механическом перемешивании приливают из
капельной воронки в течение 1 часа раствор
60 г хлористого олова (SnCl2-2H2O) в 150 мг
ацетона. В течение всего периода приливания
раствора хлористого олова температуру смеси
поддерживают не выше —2°, так как повыше-
102
ние температуры даже до 0° значительно
снижает выход ш-монобромхинальдина. При
введении половины объема раствора двуххло-
ристого олова вькпадает светло-желтый оса-
осадок «-дибромхинальдина, который при введе-
введении остальной части раствора полностью рас^
творяется. Ацетон отгоняют, остаток в колбе
осторожно нейтрализуют избытком порошка
углекислого кальция, ш-монобромхинальдин
отгоняют с паром. Из дистиллата выкристал-
выкристаллизовываются белые иглы, имеющие т. пл. 50°
(по литературным данным 53°). Выход 11 г
или- 46,6% теоретического. Средний выкод
со-бромхинальдина равен 40%.
Получение <й,(х>'-дихинальдилдисульфида.
4 г едкого кали в 100 мл спирта тщательно на-
насыщают сероводородом. К полученному рас-
раствору приливают раствор 2 г о)-монобромхи-
нальдина в 20 мл спирта. Выпадает осадок
бромистого калия. Реакционную смесь нагре-
нагревают 15 мин. на водяной бане, охлаждают и
разбавляют водой до образования мути. Пр**
этом бромистый калий растворяется и из рас-
раствора выкристаллизовываются белые иглы
<й,<й'-дихинальдилдисульфида. Перекристалли--
зованные из гексана слегка буроватые листо-
листочки имеют т. пл. 96°. Выход 1,5 г или 97% те-
теоретического.
Найдено: %: N 7,96; S 18,63; С 68,44
Ca0Hl6N8S2 Вычислено %: N 8,03; S 18,42; С 69,02
Активный водород отсутствует.
103
СП€КТРЫ ПОГЛОЩЕНИЯ
со, и/ -ДИХИНАЛЬДИЛДИСУЛЬФИДА
W 370
.,
Спектры поглощения сняты на спектрофо-
спектрофотометре СФ-4 в изооктане и 10%-ной соляной
кислоте. На кривых поглощения в изооктане
имеется три максимума: при 229, 307 и
320 m\i. Средние значения молярных коэф-
коэффициентов экстинкции при 307 и 320 mji со-
ответственно равны 7730 и 9160. В 10%-ной
соляной кислоте спектр &>,о/-дихинальдилди-
сульфида имеет два максимума при 239 и
329
104
СОЕДИНЕНИЯ «в.ш'-ДИХИНАЛЬДИЛДИСУЛЬФИДА
П икр а т. К 0,05 г со>о/-дихинальдилдисуль-
фида в 5 мл спирта приливают 0,1 г пикрино-
пикриновой кислоты, растворенной в нескольких мил-
миллилитрах спирта. Выпадают желтые иглы, ко-
которые после перекристализации из спирта
имеют т. пл. 159°.
Найдено %: N 12,26
C2eHl9O7N5S2 Вычислено %\ N 12,14
Стифнат выкристаллизовывается при сли-
сливании спиртовых растворов дисульфида и сти-
фниновой кислоты; т. пл. 158°.
Найдено %: N 11,76
C26H2o08NeS2 Вычислено %: N 11,80
КОМПЛЕКСЫ io,io' -ДИХИНАЛЬДИЛДИСУЛЬФИДА
С ИОНАМИ МЕТАЛЛОВ
В растворах, содержащих ионы хлора, лишь
палладий и золото образуют с ю,о/-дихиналь-
дилдисульфидом комплексные соли, экстра-
экстрагирующиеся хлороформом. Экстракт имеет
желтый цвет. В присутствии йодидов и ро-
данидов комплексные соли образуют также
осмий (светло-зеленые и светло-малиновые) и
платина (желтые).
Обращает на себя внимание то, что компле-
комплексные соединения с &>,<й'-дихинальдилдисуль-
фидом образует элементы, обладающие наи-
наивысшей способностью к образованию коорди-
координационный связей.
105
Экспериментально установлено, что о),со'-ди-
хинальдилдисульфид не удается восстановить
в to-меркаптохинальдстн даже энергичными вос-
восстановителями. Но одновалентная медь вос-
восстанавливает о),о/-дихинальдилдисульфид в
ю-меркаптохинальдин с образованием внутри-
комплексной соли, растворяющейся в органи-
органических растворителях с бурой окраской.
К 2 мл раствора двухвалентной меди при-
приливают 0,5 мл 20%-ного раствора восстанови-
восстановителя (гидроксиламина, аскорбиновой кисло-
кислоты и др.). 1 мл 0,3%-ного раствора ю,со'-ди~
хинальдилдисульфида в 0,1%-ной соляной ки-
кислоте и затем 20%-ным раствором ацетата на-
натрия доводят рН до 4—5. Образовавшийся
ю-меркаптохинальдинат меди экстрагируют
2 мл хлороформа.
JlHtepaTypa
1. D. Z. Hammick, J. Chem. Soc. 1923, 2882; 1926,
1302.
2. Препаративная органическая химия. Госхимиздат
Москва, 1959 г.
УДАЛЕНИЕ ПЕРЕКИСЕИ ИЗ ДИОКСАНА
С ПОМОЩЬЮ ИОНООБМЕННЫХ СМОЛ
И. В. ХВОСТОВ, В. В. КРОХВ, А. И. ВОЛЬФСОН
(ВНИИ химических реактивов)
В литературе имеются указания на возмо-
возможность удаления перекисей из простык эфи-
ров. Так, например, пропускание окисленного
диэтилового эфира через ионообменную ко-
колонку, заполненную смолой «Дауэкс—1», ос-
освобождает его от перекисных соединений. До-
Добавка указанной смолы делает эфир устойчи-
устойчивым к окислению даже при хранении на сол-
солнечном свете.
В соответствии? с ТУ МХП 3111—52 и ВТУ
266—58 содержание перекисных соединений в
диоксане не должно превышать 0,0015% (в
пересчете на активный кислород). Удаление
перекисей из окислившегося диоксана осуще-
осуществляется следующим образом:
К 100 мл диоксана добавляют 2 г воздушно-
сухого анионита ЭДЭ-10П в ОН-форме и смесь
107