





























































Текст
всесоюзный ордена трудового красного знамени
НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
«УТВЕРЖДАЮ» Зам. директора института
Р. Ластовский
МЕТОДЫ. ПОЛУЧЕНИЯ ХИМИЧЕСКИХ РЕАКТИВОВ И ПРЕПАРАТОВ
Методические указания
Выпуск 24
МОСКВА—1972
Редакционная коллегия
Е. А. Божевольнов, А. В. Бромберг, В. Г. Брудзь, В. М. Дзиомко, Р. П. Ластовский (гл. редактор), Л. М. Лукин, Ю. П. Решетников, Б. Д. Степин, В. Я. Темкина
Редактор С. С. Кузьмина Техн, редактор А. И. Пирожкова Корректор Н. Р. Казарина
Сдано в набор 15/5-71 г. . . Подписано к печати 24/XII-71 г.
Формат бумаги 60 X 90716. Объем 7,25 печ. л. Уч.-изд. л. 5,2
Л-46029 Заказ 1029 Тираж 1100 экз. Цена 42 коп.
Московский нздатсльскошолиграфический техникум имени русского первопечатника Ивана Федорова. Дмитровский, 9
СОДЕРЖАНИЕ
Барий, кальций, магний, стронций метатитановодислые. С. А. Кутолин, А. И. Вулих, А. Е. Шаммасова...................5
Барий, кальций, стронций метациркониерокцелые. А. Е. Шаммасова, А. И. Вулих, С. А. Кутолин....................... 9
Барий и кальций метагафниевокислые. А. Е. Шаммасова, С. А. Кутолин, А. И. Вулих...................................11
Ванадий двуокись. Г. Н. Ходалевич, Е. Т. Лабыкина, Л. П. Гевлич, В. И. Берзин ...........................................13
Ванадий пятиокись особой чистоты. Б. И. Короткевич, А. И. Вулих, У. А. Саидахмедов, Д. А. Пахомов, Л. П. Жердиенко, А. А. Аловяйников ......................................... 14'
Ванадил бромистый. Г. Н. Ходалевич, Е. Т. Лабыкина, Л. П. Гевлич, В. И. Берзин . . '.............................17
Вольфрамовая кислота и вольфрамовый ангидрид. У. А. Саидахмедов, А. И. Вулих, Н. У. Ризаев...........................19
Галлий азотистый. С. А. Кутолин, А. И. Вулих, А. Е. Шаммасова . ............. 22
Галлий и индий сернокислые безводные. А. А. Яровой, В. Д. За-медянская . . .....................................25
Галлий и иидий фосфорнокислые. А. А. Яровой, Л, М. Петрова, Г. Е. Ревзин...............................' ... 27
Галлий (III) фтористый трехводный. Г. Е. Ревзин, Л. М. Петрова 29
Германий (IV) хлористый. Г. Е. Ревзин, Л. М. Петрова, А. А. Яровой . . ...................................31
Иидий (III) сернистый. Г. Е. Ревзин, А. А. Яровой . . 33
Индий сернокислый кислый семиводпый. А. А. Яровой . . 35
Индий углекислый основной. Г. Е. Ревзин..............37
Индий уксуснокислый основной. Г. Е. Ревзин .... 39
Кадмий сернокислый, олово (II) сернокислое и хлористое
Д. А. Пахомов, Т. М. Сосипатров, С. А. Крестовникова ... 41
Калий, рубидий и цезий алкоголяты. Г. Е. Ревзин ... 46
Калий-сурьма фтористая. Г. Е. Ревзин . . . . . 49
Литий бензойнокислый. Е. М. Шипилова, Т. В. Ревзина, И. А. Шишенкова . • .........................................51
Литий двухромовокислый двуводный. Е. М. Шипилова . . 53
Литий метафосфорнокислый. М. Ф. Фокина, Е. М. Шипилова . 55
Литий молибденовокислый. Е. М. Шипилова .... 57
Литий селенистокислый кислый. Е. М. Шипилова, Т. В. Ревзина 59
Литий щавелевокислый. Е. М. Шипилова, Е. С. Скрипченко . 90
Металлы III, IV и VIII групп йодистые безводные. А. А. Яровой, Л. М. Петрова, Г. Е. Ревзин.............................62
Молибденовая кислота и молибденовый ангидрид. У. А. Саидахмедов, А. И. Вулих, И. У. Ризаев ..........................67
Никель лауриновокислый. В. И. Веремей, Г. П. Медведецкая, А. В,. Страшненко, Р. К- Страшненко........................ 70
Олово сернокислое. Б. И. Короткевич, А. Й. By лих . . . 71
Рубидий азотнокислый кислый. С. М. Архипов; А. О. Лесовая, Н. И. Кашина.......................................................74
Рубидий и цезий гексафторалюминаты. С. М. Архипов, Т. В. Ревзина, Н. И. Кашина.................................................76
Рубидий и цезий фтористые кислые. С. М. Архипов, Н. И. Кашина, Т. В. Ревзина . . . ..........................78
Свинец лимоннокислый трехзамещенный.Н. Г. Чернова, Г. И. Веселовская, М. В. Мосенцева.........................................80
Свинец метатитановокислый, метациркониевокнслый, метапио-биевокнслый и метатанталовокислый. А. Е. Шаммасова, С, Л. Ку-талин, А. И. В у лих...............................................82
Сероводород, сернистый и углекислый газы. А. И. Вулих, А. А. Аловяйников, Г. Л. Никандров, М. К- Загорская , . . . 85
Таллий азотнокислый и сернокислый. У .Р. Кучкаров, У. Л. Саид-ахмедов, А. И. Вулих, Н. У. Ризаев .......... 88
Теллур двуокись. Г. Е. Ревзин..................................91
Теллуровая кислота. Г. Е. Ревзин А, И. Вулих, Л. П. Жердиенко 93
Цезий азотнокислый. А. И. Вулих, Л. Г. Сидорова, А. О. Лесовая 95
Цезий азотнокислый кислый, С. М. Архипов, А. О. Лесовая, Н. И. Кашина.......................................................98
Цезий муравьинокислый. Е. М. Шипилова, п Л. IT. Жердиенко, Б. Н. Пустовалов . . . . . ’. . . . . 100
Церий (1У)-аммоний сернокислый. С. М. Архипов, Л. М. ОТсолиг 102
Алфавитный перечень...........................................104
БАРИЙ, КАЛЬЦИЙ, МАГНИЙ, СТРОНЦИЙ МЕТАТИТАНОВОКИСЛЫЕ
С. А. КУТОЛИН, А. И. ВУЛИХ, А. Е. ШАММАСОВА
ВаТЮз М.в. 233,23
СаТЮз М.в. 135,97
MgTiO3 М.в. 120,20
SrT103 М.в. 183,51
Применение метатитановокислых солей бария, кальция, магния и стронция описано в работах [1—5].
Известны методы синтеза этих соединений, основанные на термической разложении предварительно приготовленных двойных солей органических кислот [6], на взаимодействии гидроокисей щёлочноземельных металлов с двуокисью титана в автоклаве [7], на 'спекании углекислых или сернокислых солей щелочноземельных металлов с двуокисью титана при 1300—1500° [2, 8, 9]. Однако все эти методы сложны в аппаратурном оформлении и длительны.
Метод [10] совместного осаждения углекислого бария и гидроокиси титана из растворов хлористого бария и хлористого титана (IV) с последующим прокаливанием осадка при 1100° позволяет, по-видимому, получать наиболее чистый метатитановокислый барий, ир метод слишком сложен для обычного препаративного синтеза.
Нами разработана методика количественного синтеза ме-татитановокислых солей щелочноземельных металлов, основанная на спекании шихты, содержащей углекислую соль щелочноземельного металла и двуокись титана, в токе аммиака при 750—900° и нагревании продукта реакции на воздухе при 650—800°. На первой стадии синтеза образуется частично восстановленная титановокислая соль щелочноземельного металла, на второй стадии происходит окисление ее кислородом воздуха и образование соединения стехиометрического состава МеТЮз (11, 12].
СХЕМА СИНТЕЗА
МеСО8 + TiO2 + t'NH8 MeTiO^-f- 4-N3-f-xH,O t -f-CO21, «5 и
МеТЮз-.Ч- ~O3->MeTiO8,
где Me = Ba, Ca, Mg, Sr; x — 0,2 —0,5.
Характеристика основного сырья
Барий углекислый, ГОСТ 4158—65, ч.
Кальций углекислый, ГОСТ 4530—66, ч. д. а.
Магний углекислый основной, ГОСТ 6419 -68, ч. д. а.
Стронций углекислый, МРТУ 6-09-4061-67, ч. д. а.
Титан двуокись, МРТУ 6-09-1211-64, ч. д. а.
Условия получения
Синтез проводят в электрической трубчатой печи с фарфоровой или кварцевой трубой, снабженной термопарой и терморегулятором, обеспечивающим регулирование температуры с точностью ±20°. Установка для синтеза представлена
на рисунке.
/ — баллон с аммиаком; 2 — редуктор; 3 — реометр; 4— л од Ска с шихтой; 5 — печь; 6 — пробка из шамотного кирпича; 7 —термопара; 8 — промежуточный сосуд; 9 — поглотительный сосуд с водой; 10—поглотительный сосуд с серной кислотой (1:1)
Шихту, составленную из стехиометрических количеств реагентов, измельчают в фарфоровой ступке или мельнице -до крупности 200 меш и загружают в фарфоровую лодочку, которую помещают в печь. К печи присоединяют источник аммиака и в течение 5—10 минут пропускают газ до полного вытеснения воздуха из системы. Затем, продолжая пропускание аммиака, повышают
температуру в печи до заданного значения и выдерживают шихту в токе аммиака 3—5 часов. Линейная скорсть пропускания аммиака в печи 3—5 ci/tjceK. По окончании процесса прекращают подачу аммиака, продувают печь инертным газом (азот) и соединяют ее с атмосферой. Полученный продукт выдерживают при заданной температуре на .воздухе 1—2 часа, после чего охлаждают; образовавшийся спек выгружают и измельчают в фарфоровой ступке или мельнице.
6
Ниже приведены условия синтеза отдельных соединений и физико-химические свойства полученных образцов.
Барий метатитановокислый. Шихту из 89 г углекислого бария и 35,5 г двуокиси титана выдерживают 5 часов в токе аммиака при 850°, затем прокаливают 2 часа на воздухе при 700°.
Получают 100 г спека желто-зеленого цвета с содержанием основного вещества 98,5—99%. Выход продукта составляет 97%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5205-68.
Кальций метатитановокислый. Шихту из 77 г углекислого кальция и 61 г двуокиси титана выдерживают 4 часа в токе аммиака при 900°. Образовавшийся спек прокаливают 2 часа на воздухе при 700°, охлаждают и измельчают.
. Получают 100 г порошка кремового или желтого цвета с содержанием основного вещества 99—99,5%. Выход продукта составляет 96%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5206-68.
Магний метатитановокислый. Тщательно смешивают 86 г углекислого основного магния (40% MgO) с 69 г двуокиси титана. Шихту загружают в фарфоровую лодочку и выдерживают 3 часа в токе аммиака при 850°. Полученный спек прокаливают 2 часа на воздухе при 700°, охлаждают и измельчают.
Получают 100 г порошка кремового цвета с содержанием основного вещества 98—99%. Выход продукта составляет 97%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-157-70.
Стронций метатитановокислый. Шихту из 84 г углекислого стронция и 45,5 г двуокиси титана нагревают 4 часа в токе аммиака при 900°, затем прокаливают 2 часа на воздухе при 700°.
Получают 100 г спека светло-коричневого цвета с содержанием основного вещества 98—99%. Выход продукта составляет 96%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5997-68.
ЛИТЕРАТУРА
1. Ф. Иона, Д. Ширане. Сегнетоэлектрические кристаллы. М., «Мир», 1965, стр. 153, 344.
2. И. А. Глозман. Пьезокерамика. М., «Энергия», 1967.
3. Ю. И. Романьков. Краткая химическая энциклопедия, т. 5. М., «Химия», 1967, стр. 189.
7
4. С. А. К у то л ин, А. И. Вулих. Физико-химические свойства i области применения металлатных соединений. М., ГОСИНТИ, 1967.
5. Пат. США 2764490 (1956).
6. Пат. США 2785911 (1956).
7. Пат. Швейцарск. 287553 (1953).
8. П. П. Будников, А. М. Гинстлинг. Реакции в смесях твер дых веществ. М., Стройиздат, 1965, стр. 360.
9. Н. F. Ключников. Руководство по неорганическому синтезу. М. «Химий», 1965, стр. 297.
10. Т. Ф. Лим ар ь, А. И. Савоськина. Сб. «Исследования в области химии и технологии минеральных солей и окислов», М.-Л., «Наука», 1965, стр. 276. .
11. С. А. К у т о л и н, А. Е. Ш а м м а с о в а, А. И. Вулих. Англ. наг. 1171875 (1970); С. А., 72, 33824 m (1970).
12. С. А. Ку тол ин, А. Е. Шам масов а, А. И. Вулих. Франц, пат. 1577983 (1969); С. А., 72, 91827 а (1970).
БАРИЙ, КАЛЬЦИЙ, СТРОНЦИЙ МЕТАЦИРКОЙИЕВОКИСЛЫЕ
А. Е. ШАММАСОВА, А. И. ВУЛИХ, С. А. КУТОЛИН
BaZrO3 М.в. 276,56
CaZrOs М. в. 179,30
SrZrO3 М. в. 226,84
Метациркониевокислые соли щелочноземельных металлов находят применение в качестве диэлектриков, например для конденсаторов, и пьезоэлектриков [1—4]. Описано применение этих соединений в керамической промышленности в качестве материалов для огнеупорных сосудов и покрытий (т. пл. 2350—2600°) и как компонентов стекол с высоким показателем преломления fl, 5, 6].
Известен метод получения метациркониевокислых солей щелочноземельных металлов спеканием соответствующих углекислых солей с двуокисью циркония при 1200—1500° [7—9]. Предложен также гидротермальный метод синтеза этих соединений, основанный на взаимодействии гидроокиси щелочноземельного металла с двуокисью циркония в водной среде при высоких температуре и давлении [10].
Однако эти методы длительны и сложны в аппаратурном оформлении.
Нами разработана методика количественного синтеза ме-тациркониевых солей щелочноземельных металлов спеканием углекислых солей щелочноземельных металлов с двуокисью циркония в токе аммиака при 900° [11, 12].
СХЕМА СИНТЕЗА
МеСО, + ZrO, + Ч NH3->MeZrO3_, 4- ^N, + хНгО + СОг;
Me ZrO3_,. + f О, Me Zr O3,
где Me = Ba, Ca, Sr; x = 0,2—0,5.
Характеристика основного сырья
Барий углекислый, ГОСТ 4158—65, ч.
Калий углекислый, ГОСТ 4530—66, ч. д. а.
9
Стронций углекислый, МРТУ 6-09-4061-67, ч. д. а.
Цирконий двуокись, МРТУ 6-09-965-63, ч.
Условия получения
Установка для проведения синтеза и общий порядок‘ведения процесса описаны на стр. 6 этого сборника.
Барий метациркониевокислый. Смешивают 74 г углекислого бария с 46 г двуокиси циркония. Шихту спекают 7 часов в токе аммиака при 900°, затем прокаливают 2 часа на воздухе при 800°. Образовавшийся спек измельчают. Получают 100 г порошка белого цвета с содержанием основного вещества 99%. Выход продукта составляет 97%,.
Метод позволяет получать продукт квалификации «ч.» в соответствии с ТУ 0-5-4-66.
Кальций метациркониевокислый. Шихту из 57 г углекислого кальция и 71 г двуокиси циркония выдерживают 5 часов при 900° в токе аммиака. Образовавшийся спек прокаливают 2 часа на воздухе при 700°, охлаждают и измельчают.
Получают 100 г порошка белого цвета с содержанием основного вещества 98—99%. Выход продукта составляет 97%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями ТУ 05-3-66.
Стронций метациркониевокислый. Тщательно смешивают 67 г углекислого стронция с 56 г двуокиси циркония. Шихту спекают 5 часов в токе аммиака при 950°. Полученный спек прокливают 2 часа па воздухе при 700°, охлаждают и измельчают.
Получают 100 г порошка белого цвета с содержанием" основного вещества 98—99%. Выход продукта составляет 97%.
Метод позволяет получить продукт квалификации «ч.» в соответствии с ТУ 0-5-5-66.
Л.ИТЕРАТУРЛ
1. П. П. Будников, А. М. Гинстлипг. Реакция в смесях твер дых веществ. М., Ст.ройиздат, 1965, стр. 367.
2. С. А. Кутолин, А. И. Вулих. Физико-химические свойства л области применения металлатных соединений. М., ГОСИНТИ, 1967.
3. Пат. США 2256750 (1961) и 3103442 (1963).
4. J. Koenig, В. Jaffe. J. Amer. Ceram. Soc., 47, 87 (1964).
5. M. А. Ко ле нк о в а, А. И. Лайнер. Изв. ВУЗ, Цветная металлургия, 4, 102 (1961).
6. Франц, пат. 1278632 (1962).
7. Э. К. К е л е р, Н. А. Година. Огнеупоры, 9, 416 (1953).
8. М. R. Nadler Е. S. Fitzsimmons. J. Amer. Ceram. Soc., 38, 215 (1955).
9. Э. К Келер, Н. Б. Карпенко. Ж. неоргаи. химии, 5,66'8 (1960).
10 A. N. Christensen, S. Е. Rasmussen. Acta chem. scand., 17, 845. (1963).
11. Англ. пат. 117.1875 (1970): С. А., 72, 33824 ш (1970).
12. франц, пат. 1577983 (1969); С. А., 72, 91827 а (1970).
10
БАРИЙ И КАЛЬЦИЙ МЕТАГАФНИЕВОКИСЛЫЕ
А. Е. ШАММАСОВА, С. А. КУТОЛИН, А. И. ВУЛИХ
СаНЮ3 М.в. 266,56
ВаНЮз М.в. 363,82
Метагафниевокислые соли щелочноземельных металлов имеют практическое значение для радиокерамической и огнеупорной промышленности [1], что определяется прежде всего их высокими температурами плавления (2500—2900°). Ведутся работы по изучению ферроэлектрических свойств твердых растворов Ba(Ti, Hf)O3 [2—4], (Pb, Ba)HfO3 [5], (Ca, Pb)HfO3 [6].
Описаны методы синтеза метагафниевокислого бария и кальция спеканием углекислых солей этих элементов с двуокисью гафния с многочасовой выдержкой на двух-трех температурных уровнях; заключительная стадия спекания проводится при 1400—1500° {3, 4, 7, 8].
Нами разработан метод низкотемпературного синтеза этих соединений спеканием углекислого бария или кальция с двуокисью гафния в токе аммиака и последующим прокаливанием продуктов реакции па воздухе [9, 10].
СХЕМА СИНТЕЗА
МеСО3 -ф НЮ2 + ^NH^MeHfO^ ф -^N2 ф- хН3О ф- СО,;
МеНЮ3^ф--^О3-->МеНЮа ,
где Me = Ва, Са; х = 0,2—0,5
Характеристика основного сырья
Барий углекислый, ГОСТ 4158—65, ч. д. а.
Гафний двуокись, ГФО-2, РЭ ТУ 880-62.
Кальций углекислый, ГОСТ 4530—66, ч. д. а.
11
Условия получения
Общие условия синтеза и описание установки для проведения процесса в токе аммиака приведено на стр. 6 этого сборника.
Кальций метагафниевокислый. Тщательно перемешивают 39 г углекислого кальция с 83 г двуокиси гафния. Смесь загружают в фарфоровую лодочку, помещают в печь и выдерживают 4 часа в токе аммиака при 900°, затем прокаливают на- воздухе 1 час при 700°. Спек охлаждают и измельчают.
Получают 100 г метагафниевокислого кальция с содержанием основного вещества 98—99%. Выход продукта составляет 98%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5211-68.
Барий метагафниевокислый. Смесь 53 г углекислого бария с 59 г двуокиси гафния спекают 4 часа в токе аммиака при 900°, затем прокаливают на воздухе 1 час при 700°. Спек охлаждают и измельчают.
Получают 100 г метагафниевокислого бария с содержанием основного вещества 98.5—99,0%. Выход продукта составляет 98%.
- Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 0-09-5209-68.
ЛИТЕРАТУРА
1. Н. А. Погодина, Э. К. Келер. Ж. неорган. химии, 4,884 (1959).
2. М. Л. Ш о лох о вич, А. Л. Ход а ков, Т. Н. Лезгинцева. Со. «Кристаллизация и фазовые переходы». Минск, Изд-во АН БССР, 1962, сир. 425.
3. М. Л. Ш о л о х о в и ч, А. Л. Ход ано.в, Т. Н. Лезгинцева. Сб. «Сегнетоэлектрики». Ростов-на-Дону, Изд-во РГУ, 1961, стр. 12.
4. Р. W i 11 i a m, V. J. Т е п п а г у. J. Amer. Ceram. Soc., 48, 413 (1965).
5. D. L. Wilcox, R. L. Cook. J. Amer. Ceram. Soc., 46, 343 (1963).
6. H. H. Крайдик. Ж. техн, физики, 28, 536 (1958).
7. Е. Г. Фесенко, О. И. Прокопало. Кристаллография, 6, 469 (1961).
8. В. Н. С т р ек а л о в с ки й, Г. В. Буров и др. Тр. ин-та электрохимии, УФ АН СССР, 5, 163 (1964).
9. Франц, пат. 1577983 (1.969); С. А., 72, 91827 а (1970).
10. Англ. пат. 1171875 (1970); С. А., 72, 33824 m (1970).
ВАНАДИЙ ДВУдКИСЬ
Г. Н. ХОДАЛЕВИЧ, Е. Т. ЛАБЫКИНА, Л. П. ГЕВЛИЧ, В. И. БЕРЗИН
V02 М. в. 82,94
Описаны способы получения двуокиси ванадия окислением окиси ванадия кислородом воздуха [1], нагреванием смеси окиси с пятиокнсью ванадия [2], разложением щавелевокислого ванадила [2]; однако все эти способы весьма длительны.
; Нами разработан метод получения двуокиси ванадия восстановлением пятиокнсн ванадия щавелевой кислотой.
СХЕМА СИНТЕЗА
V2O5+Н2С2О4 —> 2VOa + 2СО2 + Н2О
Характеристика основного сырья
Ванадий пятиокись, МРТУ 6-09-6594-70, ч. д. а.
Щавелевая кислота, ГОСТ 5873—68, ч.
Условия получения
В кварцевую трубку диаметром 20—25 мм, загружают хорошо растертую смесь 9,1 г пятнокиси ванадия с 18,9 г щавелевой кислоты (Н2С2О4 2Н2О), которую перед смешением предварительно подсушивают в эксикаторе.
Трубку помещают в электрическую трубчатую печь и непрерывно пропускают ток углекислого паза до выгрузки продукта. Температуру печи поднимают за 20—25 минут до 500° и выдерживают реакционную массу 1 час, периодически поворачивая трубку.
Полученный порошок черного цвета вновь смешивают с 12,6 г щавелевой кислоты (подсушенной) и повторяют эту операцию вторично.
Конечный продукт представляет собой порошок черно-синего цвета.
Получают 7,97 г двуокиси ванадия. Выход составляет 96%.
Во избежание окисления двуокиси ванадия кислородом воздуха его хранят в запаянных ампулах.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями ТУ У-247-51.
ЛИТЕРАТУРА
1. О. A. Cook. J. Amer:, Chem. Soc., 69, 33 (1947).
2. Руководство по препаративной и неорганической химии, под редакцией Г. 'Брауэра. М„ Инлитиздат, 1956, стр. 58-1.
13
ВАНАДИЙ ПЯТИОКИСЬ ОСОБОЙ ЧИСТОТЫ
Б. И. КОРОТКЕВИЧ, А. И. ВУЛИХ,' У. Л. САИДАХМЕДОВ,
Д. А. ПАХОМОВ, Л. П. ЖЕРДИЕНКО, А. А. АЛОВЯЙНИКОВ
V2O6 М.в. 181,88
Пятиокись ванадия устойчива в сухом воздухе; плотность— 3,357 г/см3 [1—3]; температура плавления 685°; температура кипения 1750° [4].
Нами разработан ионообменный способ получения пяти-окиси ванадия ос. ч. из технического сырья.
СХЕМА СИНТЕЗА
Реакции на катионите
RH + Me(VO3, А) -> RMe + H(VO3, А).
Реакции на анионите
R'VO3 + H(VO3, A) ->HVO3 + RA, где R и R7 — радикал функциональной группы соответственно катионита и анионита;
Me — катионы натрия и металлов-примесей;
А — анионы сильных кислот зримее ей.
Регенерация ионитов
RMe + HCl -л RH+MeCl,
R'A + NiaOH -> R'OH + NaA,
RZOH + HVO3 ->R'VO3 + H2O.
Характеристика основного сырья
Ванадий пятиокись, ЦМТУ 4637—54, техн.
Натр едкий, ГОСТ 4328—66, техн.
Соляная кислота, ГОСТ 3118—67, х. ч.
Катионит КУ-2, ГОСТ 13505—68.
Анионит АВ-17, ГОСТ 13504—68.
14
Условия получения
При нагревании и перемешивании 1 кг тонко измельченной.пятиокиси ванадия выщелачивают 0,6—0,65 кг 10—12%-ного раствора едкого натра [5]. Раствор ванадиевокислого натрия отфильтровывают от нерастворимого остатка и разбавляют водой до концентрации 0,3—0,4 г-экв!л V2O5. Разбавленный раствор содержит 0,40—0,42 г-экв]л катионов щелочных (преимущественно натрия) и щелочноземельных металлов, 0,08 г-экв!л анионов сильных кислот-примесей (SO42-, СР и др.), 0,002 г-экв/л катионов тяжелых металлов-примесей (N1, Со, Мп, Pb, Cd и др) и ~ 0,001 г-экв/л катионов железа.
Раствор ванадиевокислого натрия пропускают последовательно через катионитовую и анионитовую колонки [6], первая из которых высотой 1 м, диаметром ГО см, заполнена Зкг катионита КУ-2 в Н-форме, а вторая высотой 80 см, диаметром 10 см, заполнена 1 кг анионита АВ-17 в У03-форме (иониты взвешивают в воздушно-сухом состоянии и перед пропусканием раствора смачивают водой).
Элюацию раствора (см. примечание 1) через колонки проводят сверху вниз со скоростью 1,5 л/мин. На выходе из ка-тионитовой колонки измеряют pH фильтрата, желательно с точностью до 0,01 (потенциометрически). Элюацию прекращают при увеличении pH фильтрата более 1,60. Фильтрат представляет собой чистый коллоидный раствор ванадиевой кислоты (pH 1,60) [7, 8]. Затем через колонки в той же последовательности пропускают 30 л дистиллированной воды (примечание 2). Промывные воды объединяют с основным раствором и приливают 25%-ного раствора аммиака до соотношения КНз:УОз=1 : 3. Раствор нагревают до 90—100° и выдерживают при этой температуре и перемешивании 1 —1,5 часа до образования золотисто-желтой пульпы поливанадиевокис-лого аммония и последующего обесцвечивания раствора, имеющего pH 1,7—2,0. Осадок отфильтровывают (см. примечание 3), высушивают при 300°, а затем .прокаливают, периодически перемешивая, при 600° —550° в течение 2—3 часов.
Получают 700—800 г (в зависимости от содержания У20б в исходном техническом продукте) тонкокристаллического порошка пятиокиси ванадия (см. примечание 4). Выход пятиокиси ванадия особой чистоты составляет 90%.
Метод позволяет получать продукт в соответствии с требованиями МРТУ 6-09-6594-70.
Примечания:
1. При элюации раствора следят, чтобы образующийся раствор ванадиевой кислоты не находился в колонках более 1 часа.
2. Регенерацию катионита проводят после каждого цикла его работы пропусканием 1 н. раствора соляной кислоты через колонку сверху вниз со скоростью 5—10 л'час до отсутствия натрия в фильтрате (кбнтроли-
15
pytoT фотометрией пламени). Расход соляной кислоты составляет 0,8 кг на 1 кг катионита. По окончании регенерации катионит промывают водой до отсутствия кислоты в фильтрате (по метилоранжу).
Анионит подвергают регенерации после проведения 3-х циклов получения ванадиевой кислоты, так как за один цикл емкость анионитовой колонки используется на 25—30%. Регенерируют анионит пропусканием 1 н. раствора едкого натра до отсутствия в фильтрате на выходе из колонки хлор- н сульфат-иоиов, после чего анионит отмывают водой от раствора щелочи. Расход едкого натра составляет 0,6 кг иа 1 кг анионита. По окончании регенерации анионит промывают водой до отсутствия иона натрия в фильтрате, что контролируют спектральным методом.
3. Маточный раствор, содержащий 0,2 г/л V2O5, отбрасывают.
4. Для получения продукта в гранулированном виде (—80% гранул раамером 1—3 мм), коллоидный раствор ванадиевой кислоты нагревают до 90—100° и выдерживают при этой температуре 0,5—4 час. При этом ванадиевая кислота коагулирует в хорошо фильтрующийся осадок, который промывают дистиллированной водой. Осадок кислоты сушат при 300°, а затем прокаливают 2 часа при 500—550° для упрочения гранул, образующихся при сушке. Маточные растворы, содержащие 2—1,5 г/л V2O6, объединяют с промывными водами и перерабатывают иа порошкообразную пятиокись ванадия по описанной методике. Выход гранулированного продукта составляет 80—85%, порошкообразного 5—10%.
ЛИТЕРАТУРА
1. Справочник химика, т. 2. М.-Л., «Химия», 1965, стр. 42.
2. А. Ю. Поляков. Основы металлургии ванадия. М„ Металлургпз-дат, 1959.
3. Г. А. М е е р с о н, А. Н. 3 е л и к м а н. Металлургия легких металлов. М., Металлургиздат, 1955, стр. 258—259.
4. Краткий справочник химика. М., Госхимиздат, 1963.
5. М. Н. Ми тюре в и др. Промышленность химических реактивов, вып. 3 (9), М., ИРЕА, 1965, стр. 43.
6. А. И. Вулих, В. Л. Богатырев и др. Методы получения химических реактивов и препаратов, вып/ 16. М., ИРЕА, 1967, стр. 5.
7. Р. J. Antikaine.п. Suomen kern. 33, В94 (1960).
8. В. N. Ghosh, S. Р. М a i 11 k, et all. J. Indian, Chem. Soc., 2, 137 (1913); 7, 509—514 (1963).
ВАНАДИЛ БРОМИСТЫЙ
Г. И. ХОДАЛЕВИЧ, Е. Т. ЛАБЫКИНА, Л. П. ГЕВЛИЧ, В. И. БЕРЗИН
VOBr2-3,5H2O М.в. 289,82
Способы получения кристаллического бромистого ванадила в литературе не описаны; имеются сообщения [1, 2] о существовании буро-черного бромистого ванадила в твердом состоянии, не отвечающего, очевидно, приведенному составу.
Нами синтезирован бромистый ванадил взаимодействием двуокиси ванадия с бромистоводородной кислотой (первый способ) и сернокислого ванадила с бромистым барием (второй способ).
СХЕМА СИНТЕЗА
2VO2+4HBr+5H2O -> 2VOBr2 • 3,5Н2О VOSO4+'BaBr2 + 3,5H2O -> VOBr2 • 3,5H2O + BaSO4
Характеристика основного сырья
Барий бромистый, ч.
Барий гидрат окиси, ГОСТ 4107—48, х. ч.
Бромнстоводородная кислота, ГОСТ 2062—43, ч. д. а.
Ванадил сернокислый, ЦМТУ 2112—49, ч.
Ванадий двуокись, получение см. на стр. 13 этого сборника.
Условия получения
Взаимодействие двуокиси ванадия с бромистоводородной кислотой. Растворяют 10 г двуокиси ванадия в 130 мл 22—25 %-ной бромнстоводородной кислоты при нагревании до 60—90° на водяной бане. Полученный синий раствор фильтруют через стеклянный фильтр № 4. Фильтрат упаривают в чашке па водяной бане до образования черно-синего вязкого раствора. Чашку с раствором помещают в эксикатор с твердым едким кали на 48 часов, а затем в эксикатор с фосфорным ангидридом (см. примечание).
2 Зак. 1029
17
Получают 33,2 г бромистого ванадила. Выход составляет 95%.
Взаимодействие сернокислого ванадила с бромистым барием. Растворяют 23,5 г сернокислого ванадила в 350 мл воды при 60—80°, раствор слегка подкисляют бромистоводород-пой кислотой и добавляют при перемешивании раствор-33,31 г бромистого бария (ВаВгг • 2НгО) в 50 мл воды. Раствор оставляют на сутки для отстаивания осадка сернокислого бария, а затем отфильтровывают через бумажный фильтр. Полученный фильтрат бромистого канадила обрабатывают как указано в первом методе.
Получают 26,0 г бромистого ванадила. Выход составляет 89%.
Бромистый ванадил — мелкокристаллическое вещество черно-зеленого цвета, очень гигроскопичен и легко окисляется на воздухе. Хранят его в запаянных ампулах.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями ТУ 2П-2-68.
Примечание.
Бромистый ванадил очень трудно кристаллизуется. Для ускорения кристаллизации, перед установкой раствора в эксикатор с фосфорным ангидридом в него добавляют затравку или создают ее трением стеклянной палочки о дно чашки.
ЛИТЕРАТУРА
1. Б. В. Некрасов. Основы общей химии, т. 1. М., «Химия», 1965.
2. Г. Реми. Курс неорганической химии, т. 2. М., «Мир», 1966.
ВОЛЬФРАМОВАЯ КИСЛОТА И ВОЛЬФРАМОВЫЙ АНГИДРИД
У. А. САИДАХМЕДОВ, А. И. ВУЛИХ, Н. У. РИЗАЕВ
H2WO4 М. в. .249,86
WO3 М.в. 231,84-
Применение вольфрамовой кислоты и вольфрамового ангидрида описано в работах [1—3].
Известны методы получения чистой вольфрамовой кислоты осаждением ее кислотами из растворов вольфрамовокислого натрия или аммония [2—7], а также разложением вольфрамовокислого кальция смесью соляной и азотной кислот [7]. Общим недостатком этих методов является трудность очистки вольфрамовой кислоты от примесей солей, образующихся при обменной реакции. Выход вольфрамовой кислоты не превышает 90% [3].
Описано также получение чистых золей вольфрамовой кислоты пропусканием растворов ее солей через катионит в Н-форме (8—10].
Вольфрамовый ангидрид обычно получают термическим разложением вольфрамовой кислоты или вольфрамовокислого аммония. В обоих случаях полностью обезвоженный продукт может быть получен при 300° [2].
Нами установлены оптимальные условия ионообменного синтеза вольфрамовой кислоты. Разработанный метод значительно проще ранее применявшихся и обеспечивает получение продукта высокой чистоты.
СХЕМА СИНТЕЗА
Na2WO4+i2RH 2RNa + H2WO4
H2WO4-> WO3 + H2O
где R — радикал функциональной группы катионита. Регенерация ионита
RNa + HCl -> RH + NaCI.
2*
19
Характеристика основного сырья
Натрий вольфрамовокислый, МРТУ 6-09-2158-65, ч. Катионит КУ-2, МРТУ 6-05-903-65, техн.
Соляная кислота, ГОСТ 3118—67, ч.
Условия получения
Вольфрамовая кислота. Описание подготовки катионита и ионообменной установки приведено в работе [11].
Растворяют 140 г вольфрамовокислого натрия в 700 мл дистиллированной воды (см. примечание 1). Полученный раствор отфильтровывают, если необходимо, и пропускают через колонку, содержащую 300 г катионита КУ-2 в Н-фор-ме (в расчете на сухой ионит), со скоростью 25—30 мл/мин. Вытекающий -из колонки раствор представляет собой золь, содержащий 130—140 г/л вольфрамовой кислоты. После пропускания раствора колонку промывают 500 мл воды (со скоростью 40—50 мл/мин), что практически обеспечивает вытеснение всей оставшейся в колонке кислоты. Фильтраты соединяют, получая около 1 л коллоидного раствора (~100 г/л H2WO4), и нагревают до 80—100°. При этом образуется гель вольфрамовой кислоты; коагуляция практически заканчивается за 15 минут. Суспензию охлаждают до 40—50° и фильтруют через стеклянный фильтр № 3 (см. примечание. 2). Осадок, содержащий 40—50% влаги, высушивают при 100—110° (см. примечание 3) в течение 2 часов, а затем растирают в ступке.
Получают 100—102 г вольфрамовой кислоты. Выход составляет 95%.
Метод позволяет получать продукт значительно чище, чем предусмотрено требованиями МРТУ 6-09-243-1 -65 к квалификации «ч.».
Вольфрамовый ангидрид. В фарфоровый тигель или кварцевую кювету загружают НО г вольфрамовой кислоты (получение см. выше) слоем 1—2 см.' Кювету помещают в муфельную печь, нагревают до 300—350° и выдерживают* при этой температуре 2—3 часа. Если необходимо получить плотный, непористый продукт, температуру поднимают до 750—800° и выдерживают еще 2 часа.
Получают 99—101 г вольфрамового ангидрида. Выход составляет 98%.
Метод позволяет получить продукт значительно чище, чем это предусмотрено требованиями ТУ 6-09-397-70 к квалификации «ч.».
Примечания:
1. Для получения вольфрамовой кислоты и вольфрамового ангидрида пр данной методике могут быть использованы любые растворимые вольфрамовокислые соли, в частности вольфрамовокислый аммонии или
20
калий, а также техническая вольфрамовая кислота 1 сорта ГОСТ 2197—43, из которой получают раствор вольфрамовокислого аммония обработкой ее 5%-ным раствором аммиака квалификации «ч.».
2. Фильтрат содержит 1 г!л вольфрамовой кислоты (в форме золя). При получении значительных количеств продукта и сравнительно чистом сырье целесообразно использовать маточник для приготовления исходного раствора вольфрамовокислой соли в последующих циклах процесса.
3. Указание, приведенное в работе [5] о том, что при 100° вольфрамовая кислота теряет половину конституционной воды ошибочно. Однако, если желательно сохранить высокую реакционную способность кислоты (например, для последующего растворения в аммиаке) следует проводить сушку при более низкой температуре (60—80°) или вообще не сушить.
4. Вольфрамовую кислоту и ангидрид следует хранить в плотно закрытых банках из темного стекла.
ЛИТЕРАТУРА
1. А. К. Пик а ев. Краткая химическая энциклопедия, т. 1. М., «Химия», 1961, стр- 655.
2. А. Н. 3 е л и,к м а н, О. Е. Крейн, Г. В. Самсонов. Металлургия редких металлов. М., «Металлургия», 1964, стр. 29, 51.
3. Е. П. Бо гомельская, О. Е. Крейн. Сб. «Основы металлургии», т. 4. М„ «Металлургия», 1967, стр. 88, 100. '
4. Н. 3. С и и г а л о вс к и й. Соли редких п цветных металлов. М., Госхимтехнздат, 1932, стр. 198.
5. Ю. В. Карякин, И. И. Ангелов. Чистые, химические реактивы. М„ Госхимиздат, 1955, стр. 247.
6. Н. Г. Ключников. Руководство по неорганическому синтезу. М., «Химия», 1965, стр. 253.
7. Руководство по препаративной неорганической химии, под редакцией Г. Брауэра. М., Инлитиздат, 1956, стр. 651.
8. J. W. Ryznar. Ind. Eng. Chem., 36, 821 (1944).
, 9. В. N. Ghosh, P. K. Pal. J. Indian Chem. Soc., 39, 795 (1962).
10. А. И. В у л и x, В. А. К а з ь м ин с к а я, Л. П. Жердиенко. Промышленность химических реактивов, вып. 1 (7). М., ИРЕА, 1965, стр.9.
11. А. И. By лих, В. Л. Богатырев и др. Методы получения химических реактивов н препаратов, вып. 16. М., ИРЕА, 1967, стр. 5.
ГАЛЛИЙ АЗОТИСТЫЙ
С. А. КУТОЛИН, А. И. ВУЛИХ, А. Е. ШАММАСОВА
GaN М.в. 83,73
Нитрид галлия представляет собой порошок серого или желтоватого цвета, с водой не взаимодействует, устойчив на воздухе до 700°; разлагается при нагревании с раствором щелочи; плотность 6,1 г!смъ. Нитрид галлия обладает полупроводниковыми, люминесцентными и каталитическими свойствами, в связи с чем его использование перспективно в ряде областей техники [1—3].
Описано получение нитрида галлия нагреванием металлического галлия в токе аммиака при 1100—1200° [4—6], а также действием аммиака на нелетучие бинарные соединения галлия ( окись, фосфид, арсенид) при 1000—1200° [7, 8]. Однако эти методы не эффективны для производственного получения нитрида галлия ввиду их длительности и потери продукта за счет испарения [9]. Описанный в работах [10, И] метод получения нитрида галлия азотированием смеси металлического галлия с углекислым аммонием в токе аммиака при 1100—1200°, а также термическим разложением комплексного фторида (NH4)3GaF6 ]12] при более низких температурах (900—1000°) не может быть использован Для производственного синтеза ввиду низкого выхода продукта и сложности приготовления исходного соединения.
Нами разработан новый метод синтеза, удобный для тех- ‘ нического осуществления [13].
СХЕМА СИНТЕЗА
2Ga-M8NH3 + 3CuCl2 -> 2GaN+ 3Cu+6NH4CI
Характеристика основного сырья
Аммиак жидкий, ГОСТ 6221—62, 1 сорт.
Аммоний хлористый, ГОСТ 3770—64, ч. д. а.
Галлий металлический, РЭТУ 100 —59. Гл-0 или Гл-1.
Медь хлорная, ГОСТ 4167—61, ч. д. а.
22
Условия получения
В фарфоровой чашке тщательно смешивают 85 г расплавленного галлия и 40—45 г хлористого аммония (предварительно перекристаллизованного и высушенного) до получения однородной сыпучей массы. Приготовление смеси ускоряется, если подогреть чашку до 60—80°. Полученную шихту
загружают в кварцевый тигель или кювету емкостью 200 мл.
В кварцевый сосуд г емкостью 300 мл помещают 320 г хлорной мели (СпС12 2Н2О) (см. примечание 1) и выдерживают в вакуум-су-шильном шкафу при 120—130° и остаточном давлении не выше 50 мм в течение 2—3 часов.
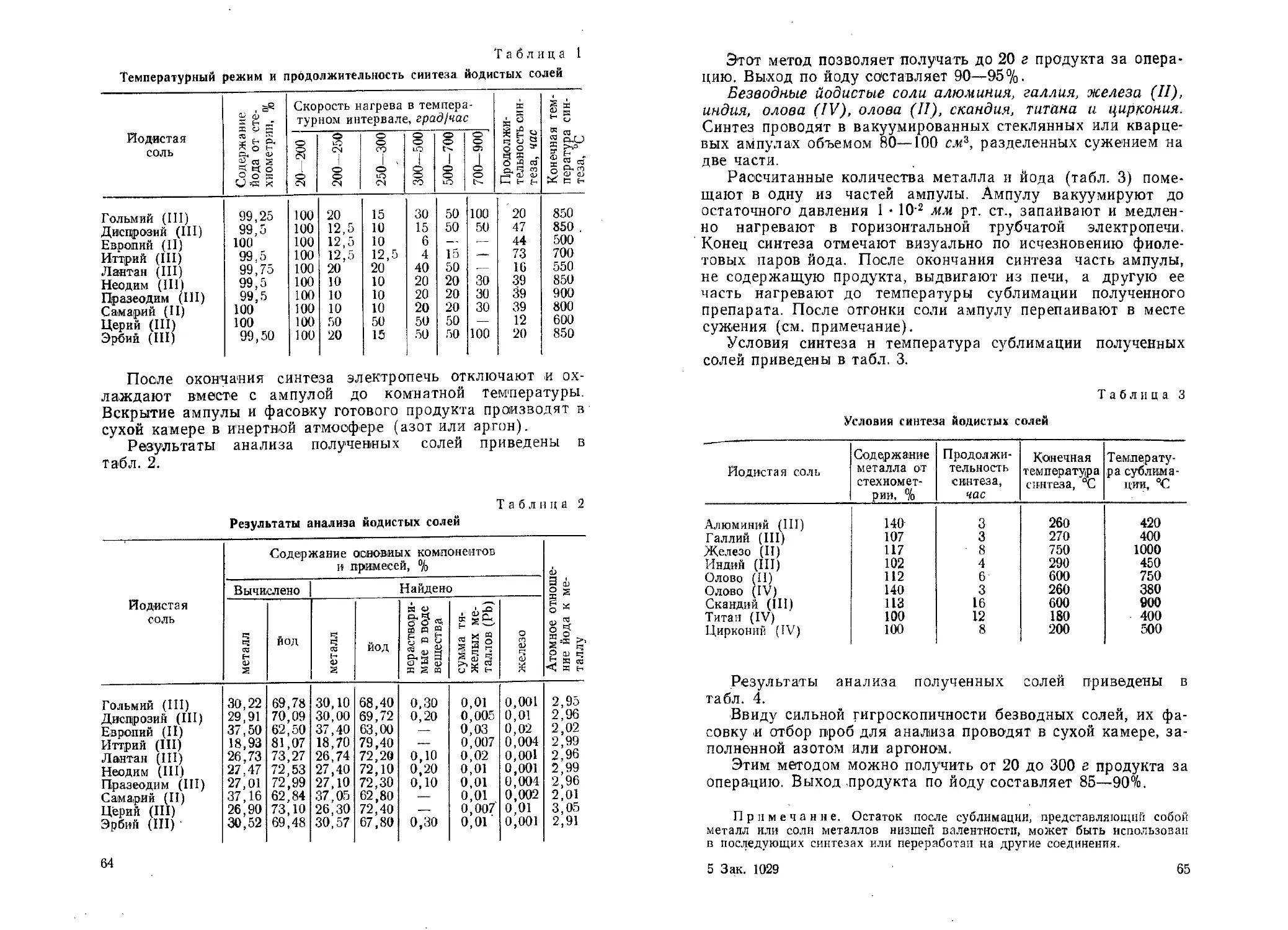
Установка для синтеза 1 — электрическая печь; 2 — реторта нержавеющей стали; 3 — кювета с шихтой; 4 — кювета с хлорной медью; 5 — термопара; 6 — конденсатор; 7 — редуктор; 8 — баллон с аммиаком
до 10 ати при 700°. Сосуды в реторте
Синтез проводят в горизонтальной реторте из нержавеющей стали (см. рисунок) с. рабочим объемом 5 л (см. примечание 2). Реторта рассчитана на давление размещают таким образом, чтобы смесь галлия и хлористого
из
аммония находилась в горячей зоне печи при 700°, а хлорная медь — в промежуточной при 200—500° (см. примечание 3).
Реторту в течение 3—5 минут продувают аммиаком для
вытеснения воздуха, затем повышают давление аммиака до 6—7 ати и температуру до 700°. При этих условиях процесс синтеза нитрида галлия заканчивается за 2—3 часа. Реторту охлаждают до 20—50°, вытесняют аммиак воздухом или азо-
том и продукт выгружают.
Получают 100 г нитрида галлия. Выход составляет 98%.
Отношение азота к галлию в продукте колеблется от 0,95 до 1,00. Метод позволяет получить продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-1806-64.
Примечания:
1. Если обезвоженная хлорная медь не используется немедленно, сс следует хранить ввиду высокой гигроскопичности в эксикаторе или вакуумном шкафу.
2. Оптимальные размеры реторты: внутренний диаметр 120 мм, длина 500 леи. Реторта герметично закрывается крышкой с конденсатором, термопарой и двумя патрубками для пропускания газа, к одному из которых' подключается баллон с аммиаком.
23
3. Хлорная медь вводится в систему для связывания свободного водорода, образующегося по реакции: 2Ga + 2NH3 -+ 2GaN + 3H2. При восстановлении хлорной меди водородом получается хлористый водород, который с аммиаком образует хлористый аммоний, удаляемый из газовой фазы за счет конденсации.
ЛИТЕРАТУРА
1. A. Rabenau. Compound Semiconductors, N-Y, m 1. 1962, стр. 171.
2. Б. Ф. Орм о нт. Ж. неорган. химии, 5, 255 (1960).
3. Пат. ФРГ 1077330 (1959).
4. W. Johnson, J. Parsons. М. Crew. J. Phys. Chem., 36, 2651 (1932).
5. Г. С. Жданов, Г. В. Лирмаи. Ж зксп. и теор. физики, 6, 1201 (1936),
6. R. J u z а, Н. Hahn. Z. Anorgan .und Allgem. Chem., 239, 282 (1938).
7. R. C. Schoonmaker, С. E. Burton. Inorg. Synt., 7, 16 (1963).
8. A. Addamiano. J. Electrochem. Soc., 108, 1072 (1961).
9. R. J. Sime, J. I,. Margrave. J. Phys. Chem. 60, 810 (1956).
10. Г. В. Самсонов, M. Д. Лютая. Ж. приют. химии, 35, 1680 (1962).
11. Г. В. Самсонов, М. Д. Лютая. Авт. свид. 156535; Бюлл. изобр. Xs 16 (1963). г
12. Н. Hahn, R. Juza. Z. anorgan. und allegem. Chem., 244, 111 (1940).
13. С. А. Кутолин, А. И. Вулих, A. E.‘Сергеева. Авт. свид.
18981.1; Изобретения. Промобразцы. Товарные знаки, № 1 (1967).
ГАЛЛИЙ И ИНДИЙ СЕРНОКИСЛЫЕ БЕЗВОДНЫЕ
А. А. ЯРОВОЙ. В. Д. ЗАМЕДЯНСКАЯ
In2(SO4)3 М.в. 517,83
Ga2(SO4)3 М.в. 427,63
Сернокислые галлий и индий представляют собой белые
кристаллические вещества, растворимые в воде и разбавлен,
ной серной кислоте. При нагревании на воздухе безводные соли разлагаются с образованием окисей галлия и индия. Температура разложения сернокислого галлия 520°, сернокислого индия 600° [1, 2].
Описан способ получения безводных сернокислых галлия и индия обезвоживанием их кристаллогидратов [2]. Однако сведения об условиях проведения процесса не приводятся.
Нами уточнены оптимальные условия растворения галлия и индия в серной кислоте .и дегидратации кристаллогидратов.
СХЕМА СИНТЕЗА
2Me + 3H2SO4 X Me2(SO4)3+3H2, где Me = In, Ga.
Характеристика основного сырья
Галлий металлический, ГОСТ 12797—67, Гл-1.
Индий металлический, ГОСТ 10297—62, Ин-1.
Пергидроль, ГОСТ 177—55, мед.
Серная кислота, ГОСТ 4204—66, х. ч.
Условия получения
Галлий сернокислый. В колбу емкостью 5 л помещают 500 г галлия, приливают 3,9 л 30%-ной серной кислоты. Растворение ведут 18—20 час. при нагревании на электроплитке, периодически добавляя небольшими порциями 1,34 л 30%-ной перекиси водорода. Полученный раствор упаривают в квар
25
цевых кюветах досуха на песчаной бане. Затем соль прокаливают 5—6 часов при 350—400° для удаления избытка серной кислоты.
Получают 1486 г сернокислого галлия. Выход составляет 97%.
Методика позволяет получать химически чистый продукт, отвечающий требованиям МРТУ 6-09-5285-68.
Индий сернокислый. В колбу емкостью 5 л помещают 500 г гранулированного индия и приливают небольшими порциями 2 л разбавленной (1:3) серной кислоты. Растворение металла в начальный момент проходит очень бурно, поэтому процесс проводят при комнатной температуре. В дальнейшем растворение продолжают при нагревании на электроплитке. Полученный раствор упаривают в кварцевых кюветах досуха на песчаной бане. Затем кюветы помещают в силитовую или муфельную печь и прокаливают 10 часов при 400°.
Получают 1095—1105 г сернокислого индия. Выход составляет 97—98%.
Методика позволяет получать химически чистый продукт, отвечающий требованиям МРТУ 6-09-5492-68.
ЛИТЕРАТУРА
. 1. Справочник химика, т. 2. М., «Химия», 1964.
2. Химия редких и рассеянных элементов, под редакцией К. А. Большакова, т. 1. М„ «Высшая школа», 1965, стр. 82, 92.
ГАЛЛИЙ И ИНДИЙ ФОСФОРНОКИСЛЫЕ
А. А. ЯРОВОЙ, Л. М. ПЕТРОВА, Г. Е. РЕВЗИН
1пРО4 М. в. 164,69
GaPO4 М.в. 209,79
Фосфорнокислые галлий и индий получают осаждением из слабокислых растворов соответствующих солей растворами фосфорнокислых солей щелочных металлов или аммония [1—6]. Однако, этот метод обладает рядом недостатков, из которых следует отметить трудность получения продуктов стехиометрического состава.
Нами предложен способ получения фосфорнокислого галлия и индия, который позволяет получать продукты стехиометрического состава. Разработанный метод прост и экономичен.
СХЕМА СИНТЕЗА
Me+3HNO3+iHsPO4 --> MePO4+3NO2 + 3H2O, где Me = In, Ga.
Характеристика основного сырья
Азотная кислота, ГОСТ 4461—67, ч.
Галлий металлический, Р,Э ТУ 851—61, Гл-1.
Индий металлический, ГОСТ 10297—62, Ин-2.
Фосфорная кислота, ГОСТ 6552—58, ч.
Способ получения
Индий фосфорнокислый. Растворяют 5 г индия в концентрированной азотной кислоте, после чего добавляют 4,84 г 88%-ной фосфорной кислоты. Полученный раствор упа
27
ривают до сиропообразного состояния и прокаливают при 800° в течение 3 часов.
Получают 8,85 г фосфорнокислого индия. Выход составляет 97%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с тр’ебованиями МРТУ 6-09-5627-68.
Галлий фосфорнокислый. Растворяют 5 г галлия в концентрированной азотной кислоте, после чего добавляют 7,98 г 88%-ной фосфорной кислоты. Полученный раствор упаривают до сиропообразного состояния и прокаливают при 300° в течение 3 часов.
Получают 11,44 г фосфорнокислого галлия. Выход составляет 97%.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5859-69.
ЛИТЕРАТУРА
1. Б. В. Некрасов. Курс общей химии. М., Госхимиздат, 1963.
2. Г. Реми. Курс неорганической химии, т. 1. М., Иилитиздат, 1963, стр. 686.
3. Т. В. Черкашина. Сборник научных ’-трудов Гиредмела, т. 2. М., Металлургиздат, 1959, стр. 202.
4. И. В. Тан а наев, Н. Н. Чудинова, Ж. неорган. химии, 9, 2, 244 (1964).
5. Э. Н. Д е й ч м а н, И. В. Т а н а н а е в и др. Ж. неорган. химии, 13, 1, 47 (1968).
6. Англ. пат. 962182 (1964); С. А„ 61, Р9197в (1964).
ГАЛЛИЙ (Ш) ФТОРИСТЫЙ ТРЕХВОДНЫЙ
Г. Е. РЕВЗИН, Л. М. ПЕТРОВА
GaF3-3H2O М.в. 180,74
Физико-химические свойства фтористого галлия (III) трех-одного описаны в работах [1—5].
Фтористый талий (III) трехводный получают действием плавиковой кислоты на окись или гидроокись галлия [6, 7] или растворением галлия в плавиковой кислоте, осуществляемым в платиновой посуде [3]. Последний способ более удобен, поскольку он не связан с необходимостью предварительного получения окиси, но в работе [3] отсутствует подробное описание синтеза. Однако, как было нами обнаружено, в присутствии галлия плавиковая кислота частично растворяет пдатину и, вследствие сравнительно низкой скорости взаимодействия галлия с плавиковой кислотой, платиновая чашка выходит из строя после проведения нескольких циклов получения фтористого галлия^
Нами предложен способ получения этого соединения растворением галлия во фтористоводородной кислоте с добавлением перекиси водорода. Синтез осуществляют в сосудах из фторопласта-4 (политетрафторэтилена). Этот способ позволяет значительно сократить продолжительность процесса, повысить чистоту полученного продукта и исключить употребление платиновой аппаратуры.
СХЕМА СИНТЕЗА
Н2О2
G.a + 3HF + ЗН2О GaF3 • ЗН2О + 1,5 • Н2
Характеристика основного сырья
Галлий металлический, ГОСТ 12797—67, Гл-1.
Перекись водорода, ГОСТ 177—55, мед.
Фтористоводородная кислота, ГОСТ 10484—63, х. ч.
29
Условия получения
Во фторопластовую кювету емкостью 0,5 л помещают 20 г галлия н приливают 150 мл 40%-ной фтористоводородной кислоты. Реакционную массу нагревают до 50—60° н прн перемешивании фтороплакчтовоц мешалкой за 1—1,5 часа добавляют небольшими порюиями то мл 30%-ной перекиси водорода. Реакционную сме^ь подопревают (80—90°) при помощи инфракрасной лампы или на песчаной бане до полного растворения галлия. Объе<м раствора поддерживают постоянным, периодически добавляя дистиллированную воду.
Полученный раствор фтористого галлия (III) упаривают (110—120°) под инфракр^сной
лампой или на песчаной бане до образования влажной соли, которую затем сушат в сушильном шкафу при 50—(§0°
Получают 48 49,7 г прюдуКта. Выход составляет 93—96%.
Соль фасуют в поли^тиленовые мешочкн или склянки, парафинированные изнутри
Методика позволяет п олучать галлий трехфтористый, отвечающий требованиям М(руу 6-09-5625-68.
ЛИТЕРАТУРА
1. И. Г. Рысс. Химия фт^ра
и его неорганических соединений. М., Госхимиздат, 1956, стр. 544.
2. Справочник химика, М.-Л госхимиздат. 1964.
°' И' п Г.а Н а ев»’тт Б а усов а. Сб, «Химия редких элементов» вып .2. М„ Изд-во АН CQcp, 1955, стр. 23.
„ *“В. Т а н а и а е в, Т. 1g. Воротили на. Ж. неорган. химии, 14,
<э77 (19о9).
5. Химия редких и рассеятНЬ1х элементов, под редакцией К. А. Большакова т.1. М «Высшая школзд», 19б5> стр. 84.
°- у- Johns°n> J. В; ‘Parsons. J. Phys. Chem., 34, 1214 (1930).
rio>>n\ Klemm, H. Kill a n 2. anorgan. und allgem. Chem., 241,
•W (19o9).
ГЕРМАНИЙ (IV) ХЛОРИСТЫЙ
Г. Е. РЕВЗИН, Л. М. ПЕТРОВА, А. А. ЯРОВОЙ
GeCh М. в. 214,40
Физико-химические свойства хлористого германия (IV) описаны в работах [1—3].
Средн многочисленных способов получения хлористого германия (IV) наиболее удобен способ, заключающийся во взаимодействии стеклообразной двуокиси германия с концентрированной соляной кислотой [4, 5]. Недостатком метода является необходимость использования 10-кратного избытка концентрированной соляной кислоты [5] для предотвращения потерь двуокиси германия, растворимость которой повышается о понижением концентрации соляной кислоты [2, 3, 6—8].
Нами усовершенствован этот метод, что позволило увеличить производительность применяемой аппаратуры, а также несколько увеличить выход продукта [9].
СХЕМА СИНТЕЗА
GeO2+4HCl -> ОеС14+2НгО
Характеристика основного сырья
Германий двуокись, ЦМТУ 05-17-67, ос. ч
Соляная кислота, ГОСТ 3118—67, ч. а а.
Условия получения
В стеклянной колбе емкостью 1 л к 100 г стеклообразной двуокиси германия приливают 630 мл (750 г) соляной кислоты и в течение 6 часов насыщают водный раствор хлористом водородом, подаваемым через барботер со скоростью 180— 200 мл/мин. По окончании синтеза, определяемом визуально по образованию тяжелого слоя жидкого хлористого германия (IV), не содержащего твердых частиц непрореагиров-ав-
31
Шей двуокиси германия, его отделяют на делительной воронке от соляной кислоты и перегоняют, собирая фракцию, кипящую при 83—85°.
Получают 199 г продукта. Выход составляет 97%.
Метод позволяет получать препарат квалификации «ч.» в соответствии с требованиями МРТУ 6-09-3633-67.
ЛИТЕРАТУРА
1. Справочник химика, т. 2. М.-Л., «Химия», 1964.
2. Химия редких и рассеянных элементов, под редакцией К. А. Большакова, т. 1, М., «Высшая школа», 1965, стр. 171.
3. И. В. Тананаев, М. Я- Шпирт. Химия германия. М., «Химия», 1967, стр. 60.
4. О. Н. Бреусов, Л. М. Петрова. Методы получения химических реактивов и препаратов, вып. 16. М„ ИрЕА, 1967, стр. 141.
5. Л. М. Петрова, О. Н. Бреусов и др. Авт. спид. 163166; Билл, изобр. № 12 (1964).
6. (В. В. Удовенко, Ю. Я. Фиалков. Ж. неоргаи. химии, 2, 434 (1957).
7. О. В г а и е г, Н. М й 11 с f. Z. anorgan. und allqem. Chem., 287, 71 (1956).
8. D. Everest, J. Harrison. J. Chem. Soc., 1957, 1820, 4319.
9. Г. E, Ревзин, A. M. Петрова, А. А. Яровой. Авт. свид. 265093. Изобретения. Промобразцы. Товарные знаки, 10 (1970).
ИНДИИ (III) СЕРНИСТЫЙ
Г. Е. РЕВЗИН, А. А. ЯРОВОЙ
In2S3-0,5H2O М. в. 334,84
Физико-химическиё свойства сернистого индия (III) описаны в работах [1—5].
Известны следующие методы получения сернистого индия (III) [1,3—6]: сплавление в эвакуированной запаянной ампуле эквивалентных количеств индия и серы; осаждение сероводородом из нейтрального или уксуснокислого раствора соединений индия (III); осаждение сероводородом индия из растворов, содержащих малую концентрацию минеральной сильной кислоты; сульфидирование окиси индия сероводородом при 500—700°.
Синтез из элементов осложняется тем, что после образования на поверхности индия слоя тугоплавкой сернистой соли скорость взаимодействия компонентов резко падает, а повышение температуры вызывает увеличение давления паров серы и разрушение ампулы. Поэтому, несмотря на кажущуюся простоту, этот метод длителен н трудоемок. Осаждение сернистого индия (III) сероводородом из нейтрального и уксуснокислого раствора по нашим данным сопровождается соосаж-дением оксисульфидов индия переменного состава.
Как было установлено нами, наилучшие результаты дает метод осаждения сернистого индия (III) сероводородом из раствора сернокислого индия при 60—90° и pH 2,0—2,5.
СХЕМА СИНТЕЗА
In2 (SO4) з + ЗНгЗ —> ItisSs+SHzSOi
Характеристика основного сырья
Аммиак водный, ГОСТ 3760—64, ч.д. а., 25%-ный.
Железо сернистое, для аналитических целей, МРТУ 6-09-4531-67.
Индий сернокислый безводный, МРТУ 6-09-5492-68, ч.
Соляная кислота, ГОСТ 1382—42, техн.
3 Зак. 1029 33
Условия получения
Растворяют при нагревании 100 г сернокислого индия безводного в 500—600 мл дистиллированной воды. Полученный раствор (рН< 1,0) при энергичном перемешивании нейтрализуют аммиаком до pH 2,0—2,5. Если раствор мутный, его фильтруют и разбавляют дистиллированной водой до объема 870—900 мл, что соответствует концентрации индия ~50 г/л. В раствор сернокислого индия, подогретый до 60—90°, пропускают сероводород до достижения полноты осаждения индия. Температуру раствора во время осаждения поддерживают в пределах 60—90° с помощью водяной бани. Полнота осаждения проверяется качественно пропусканием сероводорода в прозрачный фильтрат. Если при этом не образуется желтый осадок, процесс считается законченным. Раствор фильтруют, осадок промывают на фильтре горячей дистиллированной водой до отсутствия сульфат-иона в промывной воде (реакция с хлористым барием в присутствии азотной кислоты). Отмытый осадок сушат до постоянного веса в вакуум-сушильном шкафу при 95—105° и остаточном давлении 10—20 мм рт. ст.
В фильтрате и промывных водах проверяют наличие индия нейтрализацией углекислым кислым натрием до pH 8. При наличии в растворе индия он осаждается в виде, углекислого основного индия [1п(ОН)СОз], который отфильтровывают, промывают 2—3 раза водой декантацией, отжимают на воронке Бюхнера и используют для получения сернокислого индия безводного.
Получают 55 г. продукта. Выход составляет 87%.
Метод позволяет получать чистый продукт, соответствующий требованиям МРТУ 6-09-3065-'66.
ЛИТЕРАТУРА
1. С. В. Б л еш,и некий, В. Ф. Абрамова. Химия индия. Фрунзе, Изд-во АН Киргиз. ССР, 1958, стр. 79—81.
2. Справочник химика, т. 2. М.-Л., Госхимиздат, 1964.
3. Руководство по препаративной неорганической химии, под редакцией Г. Брауера. М„ Инлитиздат, 1956, стр. 407.
4. Химия редких и рассеянных элементов, под редакцией К- А. Большакова, т. 1. М., «Высшая школа», 1965, стр. 96—97.
5. Е. С. Вачнадзе, Е. М. Нанобашвилн. Сообщения АН Груз. ССР, 21, 531 (1958).
6. Е. С. Вачнадзе. Сообщения АН Груз. ССР, 33 (2), 331 (1964).
ИНДИЙ СЕРНОКИСЛЫЙ КИСЛЫЙ СЕМИВОДНЫЙ
А. А. ЯРОВОЙ
In2(SO4)3 • II2SO4 • 7Н2О М.в. 742,01
Физико-химические свойства сернокислого кислого' индия описаны в работах [I, 2].
Типовой метод получения сернокислого кислого индия семиводного заключается в растворении индия в серной кислоте с последующим высаливанием продукта серной кислотой.
Нами установлено, что концентрация серной кислоты в маточном растворе должна быть около 52%, а оптимальная плотность маточного расвора 1,415—1,417.
СХЕМА СИНТЕЗА
2In+4H2SO4+7H2O In2(SO4)3.H2SO4-7H2O + 3H2
Характеристика основного сырья
Индий металлический, ГОСТ 10297—62, Ин-1.
Серная кислота, ГОСТ 4204—66, ч.
Уксусная кислота, ГОСТ 61—69, ч.д. а.
Условия получения
В колбу емкостью 1 л с обратным холодильником помещают 570 г 40%-пой серной кислоты (пл. 1,329) и постепенно добавляют 100 г гранулированного индия. Растворение проводят при осторожном нагревании на электрической плитке до 100—150°. Полученный раствор сливают в фарфоровую чашу и добавляют 292 г 90%-ной серной кислоты (пл. 1,814). Раствор охлаждают при постоянном перемешивании. Выпавшей соли дают отстояться, измеряют плотность раствора над осадком и при необходимости доводят плотность до величины 1,415—1,417, добавляя дистиллированную воду или концентрированную серную кислоту. Осадок выдерживают под раствором 5—6 часов, периодически перемешивая. Соль отфильтро
3*
35
вывают на воронке Бюхнера через перхлорвинилоВую ткань, промывают 500 мл концентрированной уксусной кислоты и сушат в вакуум-сушильном шкафу при 80—85° до полного удаления уксусной кислоты.
Получают 314 г продукта с содержанием основного вещества 98,5—99,5%. Выход составляет 97%.
Метод позволяет получать реактив квалификации «ч.» в соответствии с требованиями МРТУ 6-09-3633-68.
литература
1. Справочник химика, т. 2. М., «Химия», 1964.
2. Э. Н. Д е й ч м а и, Г. В. Р о д и ч е в а, Ж. А. Б р и ц ы н а. Ж. неор-ган. химии, 7, 877 (1962).
ИНДИЙ УГЛЕКИСЛЫЙ ОСНОВНОЙ
Г. Е. РЕВЗИН
1п(ОН)СО3 М.в. 191,84
Описан метод синтеза углекислого основного индия, заключающийся во взаимодействии растворов солей индия с углекислыми солями щелочных металлов [1, 2]. При этом образуется студенистый белый осадок, являющийся, по мнению авторов, смесью основного углекислого индия с гидроокисью индия.
Нами разработан метод синтеза углекислого основного индия стехиометрического состава, заключающийся во взаимодействии раствора хлористого индия с раствором углекислого кислого натрия. Получаемый продукт хорошо фильтруется и промывается и может быть использован в качестве исходного сырья для синтеза различных солей индия вместо гидроокиси индия, синтез и выделение которой в чистом состоянии связаны со значительными трудностями.
СХЕМА СИНТЕЗА
1П4-ЗНС1 -4- 1пС13 + 1,5Н2
InCl3 + 3NaHCO3 -> In(OH)GO3+3NaCl+H2O + 2CO2
Характеристика основного сырья
Индий металлический, ГОСТ 10297—<62, Ин-1.
Натрий углекислый кислый, ГОСТ 4201—66, х. ч.
Соляная кислота, ГОСТ 3118—67, х.ч.
Способ получения
В термостойкую колбу помещают 10 г гранулированного индия, приливают 35 мл соляной кислоты и растворяют индий при нагревании до 60—80°. Раствор хлористого индия разбавляют дистиллированной водой до получения раствора плотности 1,036, что соответствует концентрации индия в растворе ~25 г/л.
К раствору, помещенному в фарфоровый стакан, при интенсивном перемешивании стеклянной пропеллерной мешалкой (250—300 об/мин) приливают 450 мл 5%-ного
37
Ра£твора углекислого кислого натрия до pH 6. Полноту осаж-Де^ия углекислого основного индия проверяют добавлением Ра'Створа углекислого кислого натрия к небольшому количеству отфильтрованного раствора.
Осадок оставляют под раствором на сутки для старения, поСЛе чего его промывают дистиллированной водой ог хлор-ио'на декантацией 4—5 раз порциями по 500 мл, переносят на воронку Бюхнера и заканчивают промывку, периодически контролируя содержание хлор-иона в продукте (качественная реакция с азотнокислым серебром в азотнокислом рас-тв оре).
Промытый продукт отжимают на фильтре и сушат в сушильном шкафу при температуре не выше 50° в течение 5—8 часов, периодически перемешивая.
Получают 16,2—16,5 г углекислого основного индия. В^лход составляет 97—99%.
Метод позволяет получать продукт квалификации «ч.» в ^ответствии с требованиями МРТУ 6-09-5626-68.
ЛИТЕРАТУРА
1. И. П. Алвмарвн, Е. П. Цинцевнч, В. П. Бурлак. Заводск. ^Лаборатория, 25, 11, 1287 (1959).
2. Химия редких и рассеянных элементов под редакцией К- А. Боль-ш акова, т. 1. М., «Высшая школа», 1965, стр. 93.
ИНДИЙ УКСУСНОКИСЛЫЙ ОСНОВНОЙ
Г. Е. РЕВЗИН
In (ОН) (СНзСОО) 2 М. в. 249,92
Сведений о методах синтеза уксуснокислого основного индия и его свойствах в литературе не имеется. Известно лишь, что индиевые соли органических кислот обнаруживают комплексный характер; кроме того, имеются указания, что индий склонен к образованию с органическими монокарбоновыми кислотами основных солей состава In (ОН) (RCOO) 2 [1, 2].
Нами разработан метод синтеза основного уксуснокислого индия, заключающийся во взаимодействии углекислого основного индия с уксусным ангидридом в среде уксусной кислоты.
СХЕМА СИНТЕЗА
СНзСООН 1п(ОН)СОз+ (СН3СО)2О —--------->1п.(ОН) (СН3СОО)2 + СО2
Характеристика основного сырья
Индий углекислый основной, МРТУ 6-09-5626-68, ч.
Уксусный ангидрид, ГОСТ 581'5—69, ч.
Уксусная кислота, ГОСТ 61—69, х. ч.
Условия получения
Навеску 100 а тонкоизмельченного углекислого основного индия помещают в фарфоровую чашку и заливают смесью 150 мл ледяной уксусной кислоты и 75 мл уксусного ангидрида (см. примечание). Реакционную массу помещают на песчаную или масляную баню и упаривают, периодически перемешивая, досуха при 90—100°, а затем сушат 2—3 часа в сушильном шкафу при 80—100°.
Получают 125 г уксуснокислого основного индия с содержанием основного вещества 96%. Выход составляет 94%.
39
Метод позволяет получать продукт, удовлетворяющий требованиям МРТУ 6-09-5720-68.
Примечание.
Введение уксусного ангидрида предотвращает гидролиз продукта водой, выделяющейся при нейтрализации углекислого основного индия уксусной кислотой.
ЛИТЕРАТУРА
• I. И. П. Ал и мар ин, Е. П. Цинцевич, Т. Н. Леонова. Вест. МГУ, сер. хим., 6, 33 (1960).
2. Химия редких и рассеянных элементов, по*д редакцией К. А. Большакова, т. 1. М., «Высшая школа», 1965, стр. 93.
КАДМИЙ СЕРНОКИСЛЫЙ, ОЛОВО (II) СЕРНОКИСЛОЕ И ХЛОРИСТОЕ
Д. А. ПАХОМОВ, Т. М. СОСИПАТРОВ, С. А. КРЕСТОВНИКОВА
3CdSO4«8H2O М.в. 769,51
SnSO4 М.в. 214,75
SnCl2-2H2O М.в. 225,63
Получение чистых концентрированных растворов солей имеет практическое значение в промышленности и, в частности, в гальваностегии [1]. Применение для этих целей метода анодного растворения металлов в электролизерах с керамическими или тканевыми мембранами недостаточно эффективно.
Нами разработан метод синтеза сернокислого и хлористого олова и сернокислого кадмия анодным растворением металлов с применением в качестве диафрагмы анионитовых мембран марки МА-40-2с. Выделение солей из растворов (анолитов) осуществляли кристаллизацией. Наиболее эффективно применение описываемой методики для тоннажных производств.
Характеристика основного сырья
Кадмий, ГОСТ 1467—67, Кд-1 или Кд-0.
Олово, ГОСТ 860—60, 02 или 01.
Серная кислота, ГОСТ 4204—66, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Описание электролизёра
Электролизёр (см. рисунок) представляет собой ванну ящичного типа, в Которой на продольные борта подвешены подлежащие растворению аноды и диафрагмы с катодами. Диафрагмы изготовлены в виде прямоугольных плоских коробок, в которых верхняя стенка отсутствует, а две вертикальных (широких) заменены анион итовым мембранами (со стороны анодов), остальные стенки сделаны из винипласта. Крепление каждой мембраны к коробке осуществлено герметично с помощью прижимных планок и винтов. При этом по
41
обе стороны мембраны, т. е. со стороны планки и со стороны коробки, помещены резиновые прокладки такой же формы, как и планки. Катоды погружены в коробки на одинаковом расстоянии от мембран. На одном из продольных бортов ванны смонтирована анодная, а на другом—катодная шины, подключенные к источнику постоянного тока. Аноды и катоды подвешены на борта ванны так. что имеют контакт только с соответствуюш₽й "'иной.
Электролизёр (вид сверху)
1— корпус электролизера (Н-700 мм); 2 — растворимые аноды; 3 — корпус диафрагмы; 4 — полые катоды; 5—анионитовые мембраны (340X450 мм);
6— болты; 7—анодная шина; 8 — катодная шина;
9 — резиновые изоляторы; 10 — коллектор кислоты;
11 — штуцер; 12— коллектор охлаждающей воды;
13 — футеровка
В ванну заливают анолит, в диафрагмы — католит. Катоды изготовлены из нержавеющей стали (или из материала анода) полыми или с каналами для циркуляции охлаждающей воды. Корпус ванны изготовлен из стали-3 и футерован изнутри полиэтиленом или винипластом. Снаружи ванна имеет охлаждающую рубашку.
Для постоянного восполнения убыли кислоты в электролитах на продольных стенках ванны смонтированы два коллектора, один из которых обеспечивает подачу кислоты .в диафрагмы, а второй — в ванну. Коллекторы служат также и для слива электролита из диафрагм с помощью вакуума. Ванна имеет штуцер для слива анолита. При осуществлении непрерывного процесса электролиза штуцер монтируют на верхнем уровне анолита, при периодическом процессе — в дне ил-и в нижней части стенки ванны.
42
Условия получения
Кадмий сернокислый. Растворение кадмиевых анодов ведут в проточном анолите при .плотности тока до 10 а/дм2', мембранная плотность тока 7,5—10 а/дм2. Состав электролитов постоянный: анолит — 400—500 а/л сернокислого кадмия и 30—50 г/л H2SO4; католит—7%-ный раствор серной кислоты. Убыль кислоты в католите восполняют концентрированной, а в анолите — 50%-ной серной кислотой. Анолит в процессе электролиза перемешивают сжатым воздухом. Практические числа переноса сульфат-ионов — 50—60%. Кадмий осаждается на катоде в виде губки, легко всплывающей на поверхность электролита.
Для выделения соли анолит нейтрализуют до pH 5 окисленной кадмиевой губкой, фильтруют и упаривают при интенсивном перемешивании до пастообразного состояния, а затем кристаллизуют при медленном охлаждении и интенсивном перемешивании. Выпавшие кристаллы сернокислого кадмия отделяют от маточного раствора центрифугированием.
Выход продукта составляет 98%, выход по току >99%; проскок кадмия через мембрану 2,0—2,5%. Расход электроэнергии на растворение 1 кг кадмия — 2,87 кет-час [2].
Метод позволяет получать продукт квалификации «ч.» в соответствии с ГОСТ 4456—56.
Олово сернокислое. [3, 4]. В диафрагмы заливают 10— 12%-ный, и в ванну — 5%-ный растворы серной кислоты. Через электролизёр пропускают постоянный ток, и аноды растворяют до накопления в анолите (в ваине) 250—260 г/л сернокислого олова, а его кислотность к концу процесса снижают до 15—25 г/л серной кислоты. Концентрацию кислоты в диафрагмах и температуру электролитов (30—40°) поддерживают постоянной. Электролиз ведут при анодной плотности тока 4 а/дм2 в начале процесса и 1,5—1,35 а/дм2 в конце процесса.
Плотность тока уменьшают постепенно. Мембранная плотность тока 1,0—1,35 а/дм2. Уменьшение концентрации кислоты в электролитах восполняют добавлением концентрированной серной кислоты с помощью коллекторов. Практические числа переноса сульфат-ионов через мембрану составляют 53—44%. Полученный раствор (анолит) сливают из ванны и фильтруют от анодных шламов. Сернокислое олово высаливают серной кислотой (пл. 1,84) при температуре не выше 70° (3], выделяющиеся при этом кристаллы отделяют от маточного раствора центрифугированием, промывают этанолом и сушат при 90—100°. На 1 кг соли расходуют 1,5 кг серной кислоты я 0,17 кг этанола.
43
Шламы содержат примеси исходного олова; их накапливают, подвергают восстановительной плавке и рафинируют.
Выход продукта составляет 97%; выход по току 88—86%; проскок ионов олова к катоду — 5—6% (вместо 25—30% при использовании других мембран). Олово с катода направляют на переплавку в аноды.
Продукт соответствует квалификации «ч.» и «ч.д. а.» (ТУ МХП 2923—51).
Расход электроэнергии на растворение 1 кг олова при получении сернокислого олова 3,1—3,5 кет-час.
При осуществлении непрерывного процесса электролиза анодная плотность тока не должна превышать 2,0 а/дм2, а концентрация свободной кислоты в анолите—18—20 г!л H2SO4. Остальные условия процесса аналогичны описанным. Продукт соответствует квалификации «ч.», «ч.д. а.».
Для снятия пассивной пленки, образующейся в случае нарушения режима электролиза, может быть успешно использован способ, описанный в работе [5].
Олово хлористое. Растворение олова осуществляют в проточном анолите при плотности тока (анодной и мембранной) 9—10 а/дм2. Концентрация соляной кислоты в анолите составляет 20—40 г/л, в диафрагмах—150—175 г/л НС1. Электролиз ведут до получения раствора (анолита), содержащего 900—1100 г/л хлористого олова и 20—40 г/л соляной кислоты. Практические числа переноса хлор-ионов составляют 60%. Для компенсации убыли кислоты в электролитах используют концентрированную соляную кислоту [6]. Полученный раствор фильтруют, упаривают до содержания 1330 г/л хлористого олова и охлаждают до 15—20°. Выпавшие кристаллы хлористого олова отфильтровывают.
Выход соли по току близок к теоретическому: проскок олова к катоду 1—2%. Пассивирование анода не наблюдается.
Продукт отвечает по качеству квалификации «ч. д. а.», «ч.» (ГОСТ 36—40). Шламы перерабатывают так же, как и при получении сернокислого олова, а катодное олово (губку) растворяют в «сходном анолите.
Процесс электролитического растворения олова с постепенным накоплением соли в анолите до концентрации 1100 г/л хлористого олова и снижения содержания свободной соляной кислоты с 175 до 20 г/л протекает так же, как и описанный выше.
Расход электроэнергии на растворение 1 кг олова при получении хлористого олова — 2,37 кет-час.
44
ЛИТЕРАТУРА
1. Д. М. Чижиков. Кадмий, М„ «Наука», 1967.
2. Д. А. Пахомов, Т. М. Сосипатров, С. А. Крестов ник о-в а. Промышленность химических реактивов и особо чистых веществ, вып. 14 (20). М„ ИРЕА, 1969.
3. И. М. Селиванов, В. Ф. Гриценко, Р. И. Козлова. Авт свид. 141860 ;Бюлл. изобр., № 20 (1961).
4. Н. П. Гнусиа, И. М. Селиванов и др. Бюллетень ЦНИИОло-ва, № 3. Новосибирск, 1964.
5. Д. А. Пахомов. Авт. свид. 169260; Изобретения. Промобразцы Товарные знаки, № 6 (1965).
6. Д. А. Пахомов, Т. М. Сосипатров, С. А. Кр е с т о в н и ,к о-в а. Авт. свид. 202904; Изобретения. Промобразцы. Товарные знаки, № 20 (1967).
КАЛИЙ, РУБИДИЙ И ЦЕЗИЙ АЛКОГОЛЯТЫ
Г. Е. РЕВЗИН
СН3ОК М.в. 70,13
C3H7ORb М.в. 144,56
СН3ОСя М.в. 163,94
Физико-химические свойства и применение алкоголятов
щелочных металлов описаны в работах [1, 2].
Известны следующие методы синтеза алкоголятов щелочных металлов [2]: взаимодействие металлов со спиртами; взаимодействие металлов со спиртами в жидком аммиаке; разложение спиртами гидридов, металлорганических соединений, карбидов, нитридов, амидов и сульфидов калия, рубидия и .цезия; обменные реакции солей с алкоголятами; взаимодействие окислов или гидроокисей со спиртами; обменные реакции алкоголятов со спиртами, приводящие к синтезу новых алкоголятов; окисление алкильных производных металлов кислородом.
Однако перечисленные методы неприемлемы для получения чистых алкоголятов рубидия и цезия.
Разработанный нами метод получения алкоголятов калия, рубидия и цезия [3] заключается в обменном синтезе между соответствующими алкоголятами натрия, лития, магния, щелочноземельных металлов или алюминия и фтористыми солями калия, рубидия или цезия в среде абсолютного спирта. При этом образующиеся фтористые соли натрия или других указанных металлов количественно осаждаются из спиртового раствора, благодаря чрезвычайно малой их растворимости.
Для синтеза производных высших спиртов, растворимость в которых как фтористых солей, так и алкоголятов сравнительно низка, реакцию удобнее проводить в среде метанола. Образующийся на первой стадии метилат калия, рубидия или цезия в дальнейшем подвергается обменной реакции с высшим спиртом.
46
СХЕМА СИНТЕЗА
MeF + ROH + Na -» ROMe + NaF+ V2H2, где Me = К, Rb, Cs.
Характеристика основного сырья
Калии фтористый, ГОСТ 4522—65, ч.
Метиловый спирт синтетический, ГОСТ 6995—54, х. ч.
Натрий металлический плавленый, ТУ 6-09-356-70, ч.
Пропиловый спирт, МРТУ 6-09-6628-70, х.ч.
Рубидий фтористый, МРТУ 6-09-2969-66, ч.
Цезий фтористый, МРТУ 6-09-2545-65, ч.
Условия получения
Синтез проводят в трехгорлой колбе емкостью 250 мл, снабженной обратным холодильником и магнитной мешалкой и помещенной на глицериновую или масляную баню. Колбу предварительно высушивают в вакуум-шкафу при 300° в течение 0,5—1 часа, затем охлаждают в эксикаторе до комнатной температуры. Заполнение колбы аргоном (или азотом), очищенным от кислорода и влаги, производят после загрузки фторидов и спирта, пропуская газ через спирт с такой скоростью, чтобы внутри колбы создавалось небольшое избыточное давление инертного газа.
Калий и цезий метилаты. В колбу загружают 5,81 г фтористого калия (или 15,19 г фтористого цезия) и 100 мл обезвоженного метанола (см. примечание 1). Через колбу пропускают аргон и, создав с его помощью давление, быстро вносят 2,29 г натрия в виде проволоки диаметром около 2 мм. Смесь перемешивают 20 минут до прекращения выделения водорода и затем нагревают на бане до- кипения (для укрупнения осадка фтористого натрия).
После охлаждения раствор фильтруют, передавливая его током аргона через стеклянный пористый фильтр в колбу Вюрца.
Из реакционной смеси отгоняют метанол, а остаток высушивают в вакууме при 180° и остаточном давлении 15 мм рт. ст. в течение 0,5 час^( для метилата цезия — температура высушивания в вакууме 60°).
Получают 6,9 г метилата калия. Выход составляет 98,4% (в пересчете на натрий).
Найдено, % (см. примечание 2); К — 55,4; СН3О — 44,8;
Na —0.01; F —0,12.
СН3ОК. Вычислено, %: К — 55,75; СН3О — 44,25.
Получают 16 г метилата цезия. Выход составляет 98% (в пересчете н-а натрий).
47
Найдено, % (см. примечание 2): Cs — 80,7; СН3О— 20,1;
Na —0,01; F —0,20.
CH3OCs. Вычислено, %: Cs — 81,07; CH3O — 18,93.
Рубидий пропилат. В колбе растворяют 10,44 г фтористого рубидия в 150 мл обезвоженного н-пропилового спирта, затем вносят 2,29 г натриевой проволоки и проводят синтез как описано выше. Из реакционной смеси отгоняют н-пропиловый спирт, а остаток высушивают в вакууме при 150° и остаточном давлении 0,7 мм рт. ст. в течение 0,5 часа.
Получают 14 г н-пропилата рубидия. Выход составляет 97% (в пересчете на натрий).
Найдено, % (см примечание 2): Rb— 59,18; С3Н7О— 40,3;
Na —0,015; F —0,18.
C3H7ORb. Вычислено, %: Rb — 59,12; C3H7O— 40,88.
Примечания:
1. Метиловый и н-пропиловый спирты абсолютируют металлическим магнием и йодом по методике, описанной в работе [4], и перегоняют, предохраняя от влаги.
Фтористые калий, рубидий и цезий тщательно высушивают в вакууме при 200—2.50’, затем перекристаллизовывают из обезвоженного метанола и вновь высушивают при 300° в течение 2—3 часов.
2. Содержание щелочного металла в продуктах определено титрованием иавески, растворенной в воде; содержание алкоксильиой группы определено путем отгонки водно-спиртовой смеси после растворения навески в воде и определения спирта в дистилляте по удельному весу [5]. Примесь натрия определена пламенно-фотометрическим методом, ион-фтор определен колориметрическим методом с применением арсеназо и азотнокислого тория.
ЛИТЕРАТУРА
1. Н. Я- Турова, А. В. Новоселова. Успехи химии, 34, 3, 385 (1965).
2. G. Thomas. Ann. 12, 6, 367 (1951).
3. Г. Е. Ревзин. Ав,т. свид. 193477; Изобретения. Промобразцы. Товарные знаки, № 7 (1967).
4. Ю. К. Юр ь е в. Практические работы по органической химии, т. 2. М„ МГУ, 1957.
5. I. D. Mac Mahon, L. Е. Russel. Chem. Ind. 57, 462 (1945).
КАЛИЙ-СУРЬМА ФТОРИСТАЯ Калий пентафторантимонат (111)
Г. Е. РЕВЗИН
K2SbF6 М.в. 294,94
Физико-химические свойства пентафторантимоната (III) калия описаны в работах [1—3].
Калий пентафторантимонат получают взаимодействием водных растворов фтористых солей калия и сурьмы [1—3], Недостатком этого метода является возможность частичного гидролиза продукта в процессе упаривания раствора, а также термического разложения в результате местного перегрева. Кроме того, при упаривании досуха возможна сокристал-лизация продукта с иными солями, например, с KSb^Fis или KSbF< [1].
Нами разработан метод получения пентафторантим,оната калия взаимодействием исходных компонентов в водно-спиртовом растворе, причем получают чистый продукт с высоким выходом [4]. Аналогичный метод может быть использован также для получения комплексных соединений фтористого калия, рубидия или цезия с фтористыми солями многовалентных элементов.
СХЕМА СИНТЕЗА
, , (С2Н5ОН, Н2О) г
2KF + SbF3 K2SbFb
Характеристика основного сырья
Калий фтористый, ГОСТ 4522—65, ч.
Фтористоводородная кислота, ГОСТ 2567—54, техн.
Этиловый спирт, ГОСТ 5962—67, ректифицированный. Сурьма трехфтористая, МРТУ 6-09-2605-65, ч.
Условия получения
В сосуд из винипласта или органического стекла емкостью 1,5—2 л помещают 712 г фтористой сурь'мы (III) и растворяют в 400 мл горячей (50°) дистиллированной воды, подкис
4 Зак.' 1029 49
ленной 1—2 лл фтористоводородной кислоты для предотвращения гидролиза фтористой сурьмы (III). В другой емкости (колбе, стакане) растворяют при нагревании до 40—50° 753,3 г фтористого калия (KF-2H^O) в 400 мл 95%-ного этилового спирта. Оба раствора при необходимости фильтруют, фильтраты охлаждают до комнатной температуры и к спиртовому раствору фтористого калия приливают при интенсивном перемешивании водный раствор сурьмы (III) (см. примечание).
При сливании растворов образуется осадок пеитафторан-тимоната калия в виде белых чешуйчатых кристаллов с перламутровым блеском, причем реакционная смесь саморазо-гревается. Смесь охлаждают при перемешивании до комнатной температуры и отфильтровывают осадок на винипла-стовой воронке Бюхнера. Осадок промывают на фильтре 40—50 мл спирта, отжимают и сушат при 50—60° в вакуум-сушильном шкафу при остаточном давлении 20—30 мм рт. ст. в течение 1—2 час.
Получают 1,10—1,15 кг продукта. Выход составляет 94—98%.
Упариванием досуха маточного раствора в платиновой или фторопластовой посуде при 90—100° можно дополнительно выделить некоторое количество продукта или использовать маточник для растворения фтористой сурьмы (III) в следующей операции синтеза.
Методика позволяет получать продукт реактивной чистоты, удовлетворяющий требованиям МРТУ- 6-09-4826-67.
Примечание.
Избыток фтористой сурьмы (III) в растворе вызывает осаждение комплексов с большим содержанием сурьмы (KSbF4 или КЗЬДчз), поэтому для получения качественного продукта (KaSbFs) необходимо синтез проводить в условиях избытка в растворе фтористого калия, т. е. приливать водный раствор фтористой сурьмы (III) к спиртовому раствору фтористого калия.
ЛИТЕРАТУРА
1. И. Г. Рысс. Химия фтора и его неорганических соединений. М., Госхимиздат, 1956, стр. 270—27,1.
2. Gmelins. Hanrlbuch der anorganischen Chemie. Berlin, 8,34,1958, S. 792.
3. О. H. Бреусов, В. Г. Лаврентьева. Методы получения химических реактивов и препаратов, вып. 16. М., ИРЕА, 1967, стр. 160.
4. Г. Е. Ревзин. Авт. свид. 265089. Изобретения. Промобразцы. Товарные знаки, № 27 (1970).
ЛИТИЙ БЕНЗОИНОКИСЛЫЙ
£. М. ШИПИЛОВА, Т. В. РЕВЗИНА, И. А. ШИШЕНКОВА
C7HEO2Li М.в. 128,06
Бензойнокислый литий — белый мелкокристаллический порошок, растворимый в воде и спирте. Растворимость в г на 100 г растворителя составляет [1]: в воде 32,1 (28,5°); в 95%-ном спирте 4,5 (25°). Соль, по нашим данным, плавится при 365—370°, разлагается выше 420°.
Бензойнокислый литий применяется в фармакологии [2]. Нами уточнены условия получения этого соединения.
СХЕМА СИНТЕЗА
СбНбСООН + 1лОН —i^HgC^Li + HaO
Характеристика исходного сырья
Бензойная кислота, ГОСТ 10521—63, ч., х. ч. Литий гидроокись, МРТУ 6-09-4597-67, ч.
Условия получения
Растворяют 100 г гидроокиси лития в 300 мл горячей (80—100°) дистиллированной воды. К горячему раствору добавляют бензойную кислоту до pH 7 (по универсальному индикатору) и нагревают 1—2 часа при 80—90°. Раствор фильтруют от гидроокиси железа и механических примесей через тройной фильтр (бязь — бумага — бязь). К фильтрату добавляют бензойную кислоту до pH 6 и упаривают досуха (см. примечание 1). Остаток сушат в сушильном шкафу при НО—120° или на паровой бане до постоянного веса.
Получают 290 г бензойнокислого лития. Выход составляет 98% •
4* 51
Методика позволяет получить продукт реактивной чистоты, отвечающий требованиям МРТУ 6^09-6066-69 (см примечание 2).
Примечания:
1. Появляющееся розовое окрашивание раствора не влияет на качество получаемого продукта.
2. При получении продукта следует обращать внимание на качество сырья. В случае необходимости проводят очистку гидроокиси лития.
ЛИТЕРАТУРА
1. Lithium corporation of America Inc. 1967.
2. Б. И. Коган. Литий, области освоенного и возможного применения. М., ВИНИТИ, 1960, стр. 50.
ЛИТИЙ ДВУХРОМОВОКИСЛЫЙ ДВУВОДНЫЙ
Е. М. ШИПИЛОВА
Li2Cr2O7 • 2Н2О М. в. 265,90
Физико-химические свойства двухромовокислого лития двуводного описаны в работах [1, 2].
Двухромовокислый литий двуводный получают взаимодействием гидрата окиси лития с двухромовокислым аммонием [3] или гидрата окиси лития (углекислого лития) с хромовым ангидридом [4].
Нами уточнены условия получения этого соединения из гидроокиси лития и хромового ангидрида.
СХЕМА СИНТЕЗА 2LiOH+2CrO3 ->Li2Cr2O7+H2O
Характеристика основного сырья
Литий гидрат окиси, МРТУ 6-09-4597-67, ч.
Хромовый ангидрид, ГОСТ 3776—68, ч.
Условия получения
Растворяют 100 г гидрата окиси лития в 500 мл дистиллированной воды (80°). Добавляют к раствору хромовый ангидрид до pH 3—3,5 по универсальному индикатору. Полученный раствор упаривают на водяной бане примерно до половины объема для коагуляции гидрата окиси железа и окиси хрома (Ш). Затем доливают горячую дистиллированную воду (80°) до первоначального объема н фильтруют через 2 слоя бумажного фильтра на воронке Бюхнера. Прозрачный красно-оранжевый раствор двухромовокислого лития упаривают при периодическом перемешивании на водяной бане до образования кашицы (пл. 1,9), затем охлаждают на воздухе до комнатной температуры (периодически перемешивая). Кристаллы отделяют на центрифуге и сушат до постоянного веса на воздухе (см. примечание 1 и 2).
53
Получают 241 г двухромовокислого лития. Выход составляет 82%. При использовании маточных растворов выход продукта может быть увеличен до 96% (см. примечание 3).
Методика позволяет получить продукт реактивной чистоты, соответствующий требованиям МРТУ 6-09-4539-67.
Примечания:
1. Допустима сушка в сушильном шкафу при 30—35° в течение 1—1,5 часа. Увеличение времени или температуры сушки приводит к разложению продукта.
2. Полученную соль фасуют в банки, которые парафинируют.
3. Маточный раствор (если он ие изменил цвет) упаривают до пл. 1,6, охлаждают и сушат кристаллы как указано выше. Второй маточный раствор не используют.
ЛИТЕРАТУРА
1. Б. И. Котай. Литий. Области освоенного и возможного применения. М., ВИНТИ, 1960, стр. 67.
2. Справочник химика, 2-е изд., т. 2. М.-Л., Госхимиздат, 1963, стр. 113.
3. Руководство по препаративной неорганической химии, под редакцией Г. Брауера. М., ИЛ., 1956, стр. 451.
4. Gmelin-Kraus. Handbuch der anorganischen Chemie. Berlin, 8, 1962, S. 455.
ЛИТИЙ МЕТАФОСФОРНОКИСЛЫЙ
М. Ф. ФОКИНА, Е. М. ШИПИЛОВА
LiPO3 М.в. 85,91
Физико-химические свойства метафосфорнокислого лития приведены в работе [1]. Температура плавления 645—650°.
Описан способ получения этого соединения термической обработкой однозамещенных фосфорнокислых солей (2], а также способ его гранулирования [3].
Нами уточнены условия получения метафосфорнокислого лития термической обработкой однозамещенного фосфорнокислого лития.
СХЕМА СИНТЕЗА
LiOH + Н3РО4 —> LiHjPOi + HgO 500°-550°
LiH2PO4--------LiPO3+H20
Характеристика основного сырья
Литий гидроокись, МРТУ 6-09-4597-67, ч.
Ортофосфорная кислота, ГОСТ 6552—58, ч.
Условия получения
Растворяют 100 г гидроокиси лития в 300 мл горячей (80°) воды. К полученному раствору осторожно приливают 248 г ортофоофорной кислоты и нагревают 1—2 часа при 80°. Механические примеси и гидрат окиси железа отфильтровывают (ом. примечание 1).
Для очистки от примеси сульфат-ионов в горячий раствор добавляют порциями 3%-ный раствор гидроокиси бария; полнота очистки контролируется реакцией с хлористым барием.
Фильтрат упаривают на песчаной бане до образования пасты. Пасту пропускают через сито с редкими отверстиями диаметром 5 мм для получения гранулированного продукта. Гранулы помещают на противень, прокаливают 5—6 часов
55
trpg 500—550°, затем охлаждают до комнатной температуры и растирают.
Получают 170 г метафосфорнокислого лития. Выход составляет 89% (см. примечание 2).
Метод'позволяет получить продукт реактивной чистоты, отвечающий требованиям МРТУ 6-09-6161-69.
Примечания:
1. Для очистки раствора от примеси свинца через него пропускают в течение 10—15 минут сероводород; осадок отфильтровывают.
2. Низкйн выход обусловлен механическими потерями при растирании продукта.
ЛИТЕРАТУРА
1. Б. И. Коган. Литий. Области освоенного и возможного применения. М., ВИНИТИ, 1960, стр. 24.
2. Б. В. Некрасов. Курс общей химии. М., Госхимиздат, 1953, стр. 393.
3. Англ. пат. 1012410 (1964).
ЛИТИЙ МОЛИБДЕНОВОКИСЛЫЙ
Е. М. ШИПИЛОВА
М.в. 173,81
Физико-химические свойства молибденовокислого лития описаны в работах [1, 2].
Молибденовокислый литий может быть получен при нейтрализации до pH > 6,5 раствора гидроокиси лития молибденовым ангидридом [3] или спеканием гидроокиси лития (углекислого лития) с молибденовым ангидридом [4].
Нами уточнены условия получения этого соединения при взаимодействии гидроокиси лития и молибденового ангидрида в водном растворе.
СХЕМА СИНТЕЗА 2LiOH+MoO3 -> Li2MoO4+H2O
Характеристика основного сырья
Литий гидроокись, ГОСТ 8595—57, техн.
Молибденовый ангидрид, МРТУ 6-09-328-63, ч.
Условия получения
Растворяют 100 г гидроокиси лития в 1 л горячей (80°) дистиллированной воды и нейтрализуют молибденовым ангидридом до pH 7—7,5 по универсальной индикаторной бумаге. Полученный раствор нагревают на водяной бане 2 часа для коагуляции гидрата окиси железа и механических примесей н фильтруют. Фильтрат упаривают досуха под инфракрасной лампой, можно на паровой или песчаной бане (~>150°).
Получают 181 г молибденовокислого лития. Выход составляет 94%.
Метод позволяет получить продукт реактивной чистоты, отвечающий требованиям МРТУ 6-09-2261-65.
57
ЛИТЕРАТУРА
1. Справочник химика, изд. 2, т. 2., И.-Л., Госхнмнздат, 1963, стр. 109.
2. ASTM (12—763). Diffraction Data Cards and Alphabetical and Grouped Numerical Index of X — Ray Diffraction Data. Издание американского общества по испытанию материалов, Филадельфия, 1946—1963.
3. А. Н. Зеликман, О. Е. Крейн, Г. В. Самсонов. Металлургия редких металлов. М., Металлургия, 1964, стр. 98.
4. В. И. Спицыи, И. М. Кулешов. Ж. общ. химии, 21, 9, 552 (1951).
ЛИТИЙ СЕЛЕНИСТОКИСЛЫИ КИСЛЫЙ
Е. М. ШИПИЛОВА, Т. В. РЕВЗИНА
LiHSeOg М.в. 134,91
Литий селенистокислый кислый плавится при 300—340° разлагается при 380—480° до ПгЗеОз; плотность 2,84 а/си3.
Описаны методы получения этого соединения нейтрализацией селенистой кислоты углекислым литием или гидроокисью лития [1, 2].
Нами уточнены условия получения селенистокислого кислого лития с применением гидроокиси лития,
СХЕМА СИНТЕЗА
HsSeO3 + LiOH -> LiHSeO3 + Н2О
Характеристика основного сырья
Литий гидроокись, МРТУ 6-09-4597-67, ч.
Селенистая кислота, ГОС'!' 11081—64, ч.
Этиловый спирт, ТУ-3-66-65, гидролизный.
Условия получения
Растворяют 100 г селенистой кислоты в 300 мл горячей (60—70°) дистиллированной воды. В полученный раствор небольшими порциями добавляют 31,8 г гидроокиси лития до pH 4—4,5, после чего раствор нагревают 1—2 часа при 60—70°, затем фильтруют от механических примесей и гидроокиси железа. Фильтрат упаривают досуха, охлаждают до комнатной температуры. ОсаДок переносят на фильтр, промывают 45,5 мл спирта и сушат при 80° до постоянного веса.
Получают 90,0 г селенистокислого кислого лития. Выход составляет 95%•
Метод позволяет получить продукт реактивной чистоты, отвечающий требованиям МРТУ 6-09-6292-69.
ЛИТЕРАТУРА
1. Gmellin. Handbuch der anorganischen Chemie. Berlin, 1927, S. 218.
2. H. M. С e л и в а н о в a, T. А. С а з ы к и и а, Ж. иеоргаи. химии, 7 (2), 240 (1962).
59
ЛИТИЙ ЩАВЕЛЕВОКИСЛЫЙ
Е. М. ШИПИЛОВА, Е. С. СКРИПЧЕНКО
C2O4Li2 М.в. 101,90
Физико-химические свойства щавелевокислого лития и его применение описаны в работах {1—3]. .
Щавелевокислый литий получают нейтрализацией углекислого лития щавелевой кислотой [4].
Нами уточнен способ синтеза соединения.
СХЕМА СИНТЕЗА
2L1OH + H2C2O4 —> Ы2С2О4 + 2НгО
Характеристика основного сырья
Литий гидроокись, МРТУ 6-09-4597-67, ч.
Щавелевая кислота, ГОСТ 5873—68, ч. хч.
Условия получения
Растворяют 100 г гидроокиси лития в 300 мл горячей (80°) воды, добавляют щавелевую кислоту до pH 7—7,5 по универсальной индикаторной бумаге и нагревают 1—2 часа при 80°. Механические примеси и гидрат окиси железа отфильтровывают через слой фильтровальной бумаги и бязи. В горячий (80°) раствор пропускают сероводород в течение 10—15 минут для удаления свинца и отфильтровывают хлопьевидный осадок. Фильтрат проверяют на содержание суль-фат-иона качественной реакцией с хлористым барием. Суль-фат-ионы (еслиони присутствуют) осаждают 3%-ным раствором гидроокиси бария и отфильтровывают. Фильтрат подкисляют щавелевой кислотой до pH 5—6, упаривают при 80—90° до выпадения густой массы кристаллов и оставляют на 12 часов при комнатной температуре. Кристаллы отфильтровывают и сушат при 80—90° до постоянного веса.
60
Получают 100 г щавелевокислого лития. Выход составляет 88,6%.
Метод позволяет получить продукт реактивной чистоты, отвечающий требованиям МРТУ 6-09-5630-68.
ЛИТЕРАТУРА
1. Справочник химика, т. 2. М., Госхимиэдат, 1963, стр. 112.
2. В. В е a g 1 е у, R. W. Н. Small. Acta crvstallogr., 17 (6), 783 (1964).
3. В И. Коган. Литий. Области освоенного и возможного применения. М., ВИНИТИ, 1960, стр. 49.
4. Gm el in. Handbuch der anorganischen Chemie. Berlin, 1927, S 232.
МЕТАЛЛЫ HI, IV и VHI ГРУПП ЙОДИСТЫЕ БЕЗВОДНЫЕ
/1. А. ЯРОВОЙ, Л. М. ПЕТРОВА, Г. Е. РЕВЗИН
АП3 M. в. 407,69 EuI2 M. в. 405,77
Gal3 М.в. 450,43 Dyl, М.в. 543,21
Inls M. в. 495,53 Hol3 M. в. 545,64
Scl3 M. в. 425,67 Erl3 M. в. 547,97
YIs М.в. 469,62 Til4 М.в. 555,52
Lal3 М.в. 519,62 Zrl4 М.в. 598,84
Cel3 М.в. 520,83 Snl4 М.в. 626,31
Prl3 M. B. 521,62 Snl2 М.в. 372,50
Ndl3 М.в. 524,95 Fel2 M. в. 309,66
Sml2 М.в. 404,16
Безводные йодистые соли: A1I3, Gala, Inl3, Scl3, YI3, Lal3, Cel3, Prl3, Ndl3, Sml2, Eul2, Dyl3, Hol3, Erl3> Til4, Zrl4, Snl4, Snl2 и Fel2 представляют собой кристаллические гигроскопичные (за исключением Snl2) вещества, растворимые в воде и органических растворителях.
Для получения безводных йодистых солей используют следующие методы [1 —17]: синтез из элементов, дегидратацию кристаллогидратов при их термическом разложении в смеси с йодистым аммонием или в токе йодистого водорода и взаимодействие окислов металлов с йодистым аммонием, йодистым водородом или йодистым алюминием.
62
Нами разработан типовой метод получения безводных йодистых солей реактивной чистоты синтезом из элементов в вакуумированных ампулах, причем уточнены условия получения, соотношение исходных компонентов и определено их влияние на качество продукта и продолжительность процесса.
СХЕМА СИНТЕЗА
2 Ме+п12 дг2Ме1„
Характеристика основного сырья
Алюминий металлический, ГОСТ 6058—51, техн., ПА-1. Галлий металлический, ЦМ ТУ 4781—56, Гл-1.
Гольмий металлический, РЭТУ 1103—65, ГОМ-1. Диспрозий металлический, РЭТУ 1102—63, Ди-0. Европий металлический, РЭТУ 620—60, Ев-3. Железо порошкообразное, ГОСТ 9849—60, ПЖ-1. Индий металлический, ГОСТ 10297—62, Ин-2. Иттрий металлический, РЭТУ 1093—63, Им-3. Йод кристалический, ГОСТ 4159—64, ч. д. а. Лантан металлический, РЭТУ 1015—62, Ла-Э-1. Неодим металлический, РЭТУ 1097—63, Нм-1. Олово металлическое, ТУ МХП ОРУ 56—56, техн. Празеодим металлический, РЭТУ 1096—63, Пр-1. Самарий металлический, РЭТУ 1098—63, См-1. Скандий металлический, РЭТУ 629—60, СКМ-1. Титан металлический, МРТУ 1419—64, ТГ-100. Церий металлический, РЭТУ 1094—62, Се-Э-2. Цирконий металлический, МП ТУ 2545—60, техн. Эрбий металлический, РЭТУ 1104—63, Эрм-1.
Способ получения
Безводные йодистые соли иттрия и редкоземельных элементов. В кварцевую ампулу объемом 30—40 см3 помещают рассчитанные навески металла и йода (йод берется в количестве,. составляющем 99,25—99,75% от стехиометрически нербходимого). Для получения йодистых солей самария (II), европия (II) и церия (III) берут стехиометрические количества компонентов. Ампулу вакуумируют до остаточного давления 1 • 102 мм рт. ст., запаивают, помещают в горизонтальную трубчатую электропечь и медленно нагревают до исчезновения фиолетовых паров йода (табл. 1).
63
Таблица 1
Температурный режим и продолжительность синтеза йодистых солей
Йодистая соль Содержание йода от стехиометрии, % Скорость нагрева в температурном интервале, град/час Продолжительность синтеза, час Конечная температура синтеза, °C
20—200 О iC сч сч 250—300 300-500 500-700 700-900
Гольмий (III) 99,25 100 20 15 30 50 100 20 850
Диспрозий (III) 99,5 100 12,5 10 15 50 50 47 850 .
Европий (II) 100 100 12,5 10 6 — — 44 500
Иттрий (III) 99,5 100 12,5 12,5 4 15 73 700
Лантан (III) 99,75 100 20 20 40 50 — 16 550
Неодим (III) 99,5 100 10 10 20 20 30 39 850
Празеодим (III) 99,5 100 10 10 20 20 30 39 900
Самарий (II) 100 100 10 10 20 20 30 39 800
Церий (III) 100 100 50 50 50 50 — 12 600
Эрбий (III) 99,50 100 20 15 50 50 100 20 850
После окончания синтеза электропечь отключают и охлаждают вместе с ампулой до комнатной температуры. Вскрытие ампулы и фасовку готового продукта производят в сухой камере в инертной атмосфере (азот или аргон).
Результаты анализа полученных солей приведены в табл. 2.
Таблица 2
Результаты анализа йодистых солей
Йодистая соль Содержание основных компонентов и примесей, % Атомное отношение йода к металлу
Вычислено Найдено
металл ЙОД металл йод нерастворимые в воде । вещества сумма тя- 1 желых ме- ! таллов (РЬ) железо
Гольмий (III) 30,22 69,78 30,10 68,40 0,30 0,01 0,001 2,95
Диспрозий (III) 29,91 70,09 30,00 69,72 0,20 0,005 0,01 2,96
Европий (II) 37,50 62,50 37,40 63,00 — 0,03 0,02 2,02
Иттрий (III) 18,93 81,07 18,70 79,40 — 0,007 0,004 2,99
Лантан (III) 26,73 73,27 26,74 72,20 0,10 0,02 0,001 2,96
Неодим (III) 27,47 72,53 27,40 72,10 0,20 0,01 0,001 2,99
Празеодим (III) 27,01 72,99 27,10 72,30 0,10 0,01 0,004 2,96
Самарий (II) 37,16 62,84 37,05 62,80 — 0,01 0,002 2,01
Церий (III) 26,90 73,10 26,30 72,40 — 0,007 0,01 3,05
Эрбий (III) 30,52 69,48 30,57 67,80 0,30 0,01 0,001 2,91
64
Этот метод позволяет получать до 20 г продукта за операцию. Выход по йоду составляет 90—95%.
Безводные йодистые соли алюминия, галлия, железа (II), индия, олова (IV), олова (11), скандия, титана и циркония. Синтез проводят в вакуумированных стеклянных или кварцевых ампулах объемом 80—100 см3, разделенных сужением на две части.
Рассчитанные количества металла и йода (табл. 3) помещают в одну из частей ампулы. Ампулу вакуумируют до остаточного давления 1 • 102 мм рт. ст., запаивают и медленно нагревают в горизонтальной трубчатой электропечи. Конец синтеза отмечают визуально по исчезновению фиолетовых паров йода. После окончания синтеза часть ампулы, не содержащую продукта, выдвигают из печи, а другую ее часть нагревают до температуры сублимации полученного препарата. После отгонки соли ампулу перепаивают в месте сужения (см. примечание).
Условия синтеза н температура сублимации полученных солей приведены в табл. 3.
Таблица 3
Условия синтеза йодистых солей
йодистая соль Содержание металла от стехиометрии, % Продолжительность синтеза, час Конечная температура синтеза, °C Температура сублимации, °C
Алюминий (III) 140 3 260 420
Галлий (III) 107 3 270 400
Железо (II) 117 8 750 1000
Индий (III) 102 4 290 450
Олово (II) 112 6 600 750
Олово (IV) 140 3 260 380
Скандий (III) 113 16 600 800
Титан (IV) 100 12 180 400
Цирконий (IV) 100 8 200 500
Результаты анализа полученных солей приведены в табл. 4.
Ввиду сильной гигроскопичности безводных солей, их фасовку и отбор проб для анализа проводят в сухой камере, заполненной азотом или аргоном.
Этим методом можно получить от 20 до 300 г продукта за операцию. Выход продукта по йоду составляет 85—90%.
Примечание. Остаток после сублимации, представляющий собой металл или соли металлов низшей валентности, может быть использован в последующих синтезах или переработан на другие соединения.
5 Зак. 1029
65
Т а б л и ц а -г
Результаты анализа йодистых солей
Йодистая соль Содержание основных компонентов и примесей, % Атомное отношение йода к металлу
Вычислено | Найдено
металл ЙОД ме- талл ЙОД сумма тяжелых металлов (РЬ) железо
Алюминий (III) 6,62 93,38 6,63 92,81 0,002 0,0002 2,98
Галлий (III) 15,47 84,53 15,47 84,00 0,002 0,0003 2,98
Железо (II) 18,03 81,97 17,90 80,60 0,001 — 1,97
Ивдий (III) 23,17 76,83 23,16 75,95 0,002 0,001 2,95
Олово (11) 31,86 68,14 31,50 68,10 0,002 0,005 2,02
Олово (IV) 18,95 81,05 18,96 80,60 0,005 0,002 3,97
Скандий (III) 10,56 89,44 10,51 88,00 0,005 0,0005 2,97
Титан (IV) 8,62 91,38 8,69 91,25 0,003 0,001 3,96
Цирконий (IV) 15,23 84,77 15,20 83,80 0,002 0,001 3,94
ЛИТЕРАТУРА
1. М. D. Taylor, Chem. Revs. 62, 503 (1962).
2. М. D. Т а у 1 о г, С. Р. С а г t е г. J. Inorg. and Nucl. Chem., 24, 387 (1962).
3. G. J a n t s c h, H. G г и b i t s c h, F. H о f f m a n n, H. A1 b e r. Z. anorgan. und allgem. Chem., 185, 49 (1929).
4. M. Chaigneu. Bull. Soc. chim. de France, 886 (1957).
5. P. P. В h a t n a g a r, R. A. Sharma. J. Proc. Inst. Chem., 29, 97 (1957).
6. Инднйск. пат., ,54676 (1957); С. A., 51, P 13332 в.
7. А. А. Меньков, Л. H. Комиссарова. Ж., неорган. химии, 3, 766 (1964).
8. Руководство по препаративной неорганической химии. Под ред. Г. Брауера. М., Инлитиздат, 1956.
9. W. Blitz, A. Voigt. Z. anorgan. und allgem. Chem., 126, 50 (1923).
10. W. Klemm, W. T i 1 k, S. Mull'enhum. Z. anorgan. und allgem. Chem., 176, 15 (1928).
11. M. G. Gustavson. Ann. 172, 173 (1874).
12. W. Ne spit at. Phys. Chem., (B), 16, 164 (1932).
13. W. Fisher, O. luberm ann. Z. anorgan. und allgem. Chem., 227, 227 (1936).
14. F. Kutek. Collect. Czechosl. Chem. Communs., 31. 4, 1875 (1966).
15. Пат. США, 295334355 (1960); С. A. 55: P2039.
16. Ю. Б. К лете ник, О. А. Осипов. Изв. ВУЗов, Химия и хим. технология, 2, 679 (1959).
17. И. В. В и и а р о в, Е. И. Ковалева, Н. П. Хромова. Укр. хим. ж., 31, 10, 1107 (1965).
МОЛИБДЕНОВАЯ КИСЛОТА И МОЛИБДЕНОВЫЙ АНГИДРИД
У. А. САИДАХМЕДОВ, А. И. ВУЛИХ, Н. У. РИЗАЕВ
Н2Мо04 М.в. 161,96
МоОз М. в. 143,94
Физико-химические свойства и области применения молибденовой кислоты и молибденового анпидрида приведены в работах [1—7].
Описаны методы получения молибденовой кислоты осаждением ее из раствора молибденовокислого аммония азотной кислотой [6, 8, 9], а также гидролизом оксихлорида молибдена [10]. Недостатком этих методов является трудность очистки от примесей, кроме того, вследствие значительной растворимости молибденовой кислоты, велики ее потери с маточными растворами.
Молибденовый ангидрид получают термическим разложением парамолибденовокислого аммония или молибденовой кислоты. Ввиду обычно недостаточной чистоты исходных соединений продукт дополнительно очищают перегонкой при 800—900° [6, 9, 10].
В работе [11] отмечена возможность получения золя молибденовой кислоты при фильтрации раствора соли молибденовой кислоты через Н-катионит. При выпаривании коллоидного раствора, полученного этим способом, образуется стеклообразная молибденовая кислота [12]. Однако в этих работах не отражены конкретные условия получения золя и твердой кислоты.
Нами исследованы эти процессы и разработана методика получения молибденовой кислоты и ее ангидрида.
СХЕМА СИНТЕЗА
(NH4)бМо7024 • 4Н2О + 6RH -> 6RNH4 + 7Н2М0О4
Н2Мо04 -> МоО3 + Н2О
Регенерация ионита
RNH4+HCI -+rh + nh4ci,
где R — радикал функциональной группы катионита.
5*
67
Характеристика Основного сырья
Аммоний молибденовокислый, ГОСТ 3765—64, ч. (см. примечание).
Катионит КУ-2, МРТУ 6-09-903-65, техн.
Соляная кислота, ГОСТ 3118—46, ч.
Условия получения
Молибденовая кислота. Описание подготовки ионита и ионообменной установки приведено в работе [13].
Растворяют 115 г молибденовокислого аммония [(NH4)eMorO24 • 4Н2О] в 500 мл воды (~ 160 г/л МоО3) и пропускают раствор через колонку, содержащую 250 г катионита КУ-2 в Н-форме (в расчете на сухой ионит), со скоростью 20 мл/мин. Вытекающий .из колонки раствор содержит чистую молибденовую кислоту. Затем колонку промывают 400 мл воды со скоростью 30—40 мл/мин, что практически обеспечивает вытеснение всей оставшейся в колонке кислоты. Фйльтраты соединяют, получая 0,8 л коллоидного раствора с концентрацией молибденовой кислоты 120—130 г/л. Раствор выпаривают до объема 200—250 мл, причем молибденовая кислота выделяется в виде густой массы. Влажную массу (содержащую 60% влаги) охлаждают до 30—40°, переносят в стеклянный фильтр № 2, отделяют маточник и промывают 100 мл воды (см. примечание 2). Кислоту высушивают 3 часа при 90—100° (см. примечание 3).
Получают 99—101 г молибденовой кислоты. Выход составляет 94—96%.
Методика позволяет получить продукт квалификации ч. д. а. в соответствии с требованиями МРТУ 6-09-329-63.
Молибденовый ангидрид. Загружают в кварцевую кювету 115 г молибденовой кислоты и прокаливают 2 часа в печи при 350—400° (см. примечание 3).
Получают 99—401 г молибденового ангидрида. Выход составляет 97—99%.
Методика позволяет получить продукт квалификации ч. д. а. в соответствии с требованиями МРТУ 6-09-328-63.
Примечания:
1. Молибденовая кислота и молибденовый ангидрид квалификации «ч.» по данной методике могут быть получены также из технического молибденовокислого аммония 1 сорта (ГОСТ 2677—44), а также из молибденовокислого натрия или калия реактивной квалификации.
2. Фильтрат, содержащий 2 г/л молибденовой кислоты, может быть использован для растворения, исходной соли в следующей операции синтеза.
3. Молибденовую кислоту и ее ангидрид следует хранить в темных стеклянных банках с притертыми пробками.
68
ЛИТЕРАТУРА
1. Справочник по растворимости, т. 1. М,-Л., Госхимиздат, 1961, стр. 342.
2. А. Н. 3 е л и к м а и, О. Е. Крейн, Г. В. Самсонов. Металлургия редких металлов. М., Металлургия, 1964, стр. 97.
3. С. П. Воробьев, И. П. Давыдов. Ж. неорган. химии, 11, 2031 (1966).
4. О. А. С онг ин а. Редкие металлы. М., «Металлургия», 1964, стр. 51.
5. Л. В. Беляевская. Краткая химическая энциклопедия, т. 3. М.-Л., Госхимиздат, 1964, ст.р. 306.
6. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы. М., Госхимиздат, 1955, стр. 66, 262.
7. 3. Ф. Шахова. Ж. неорган. химии, 3, 1370 (1958); 6, 330 (1961); 7,1752.(1962).
8. Н. 3. С и н г а л о в с к и й. Соли редких и цветных металлов. М., Госхимтехиздат, 1932, стр. 189.
9. Руководство по препаративной неорганической химии, под редакцией Г. Брауэра. М., Инлитиздат, 1956, стр. 646.
10. Н. Г. Ключников. Руководство по неорганическому синтезу. М., «Химия», 1965, стр. 114, 251.
11. J. W. Ryznar. Ind. Eng. Chem., 36, 821 (1944).
12. E. Richardson. J. Inorg. and Nucl. Chem., 9, 267 (1959).
13. А. И. Вулих, В. Л. Богатырев и др. Методы получения химических реактивов и препаратов, выл. 16. М., ИРЕА, 1967, стр. 5.
НИКЕЛЬ ЛАУРИНОВОКИСЛЫЙ
В. Н. БЕРЕМЕН, Г. И. МЕДВЕДЕЦКАЯ, А. В. СТРАШНЕНКО, Р. К. СТРАШНЕНКО
(C^HaaOalaNi HjO . М.в. 475,36
Лауриновокислый никель получают взаимодействием уксуснокислого никеля с лауриновой кислотой в среде абсолютного этанола [1]. К недостаткам метода относятся трудность очистки соли и ее низкий выход (60%).
Нами разработан метод получения этого соединения взаимодействием сернокислого никеля с лауриновокислым калием в водной среде.
СХЕМА СИНТЕЗА
СН3 (СН2) юСООН + КОН -> СНз (СН2) юСООК + Н2О; 2СН3(СН2) 10COOK + NiSO4-> ,[СН3(СН2) 10CO2]2Ni + K2SO4
Характеристика основного сырья
Кали едкое, ГОСТ 4203—65, ч.
Лауриновая кислота, МРТУ 609-995-64, ч.
Никель сернокислый, ГОСТ 4465—61, ч. д. а.
Условия получения
Получение лауриновокислого никеля. В стакане емкостью 3 л, снабженном мешалкой, растворяют 34 г (0,6 М) едкого кали в 2,2 л воды. Раствор нагревают до кипения и при перемешивании загружают 120 г (0,6 М) лауриновой кислоты.
К полученному раствору лауриновокислого калия при перемешивании постепенно приливают раствор 196 г (0,7 М) сернокислого никеля (NiSO4-7H2O) в 250 мл воды, поддерживая температуру реакционной массы 85—90°. При этой температуре реакционный раствор выдерживают 30 минут, затем охлаждают до комнатной температуры. Выпавший осадок отсасывают на воронке Бюхнера, промывают водой до отрицательной реакции на сульфат-ионы и сушат при 40°.
Получают 280 г лауриновокислого никеля; выход количественный.
Метод позволяет получать продукт квалификации «ч.» в соответствии с требованиями ТУ 15ГТ501—69.
ЛИТЕРАТУРА
1. Whitmore Lanro. Ind. Eng. Chem., 22, 648 (1930).
70
ОЛОВО СЕРНОКИСЛОЕ
Б. И. КОРОТКЕВИЧ, А. И. ВУЛИХ
SnSO4 М.в. 214,75
Физико-химические свойства и области применения сернокислого олова приведены в работах [1—47].
Сернокислое олово получают растворением олова в серной кислоте, анодным растворением с различными мембранами, а также вытеснением меди из сернокислой меди оловом; однако продукт, полученный этими способами, содержит избыточную серную кислоту [1,4—7, 18—25].
Сернокислое слово может быть также получено осаждением гидрата закиси олова и последующим воздействием на него серной кислоты [5] или окислением олова кислородом и добавлением к нему серной кислоты [26]. Однако эти методы сложны и связаны с заметными потерями олова. Воздействием серной кислоты на хлористое олово (II) может быть также получено сернокислое олово, но оно содержит примесь двуокиси олова [27].
Нами разработан ионообменный способ получения сернокислого олова из хлористого олова или разбавленных растворов других солей олова. Способ основан на том, что катионит эффективно сорбирует олово из нейтральных или слабокислых растворов серной кислоты (до 2 н.), но в более концентрированных ее растворах (более 4н.) олово легко десорбируется вследствие перехода его в анионный комплекс.
СХЕМА СИНТЕЗА
2RII + SnCl2-> R2Sn+2HCl,
R2Sn + H2SO4 -> 2RH + SnSO4,
где R — радикал функциональной группы сильнокислотного катионита.
Характеристика основного сырья
Серная кислота, ГОСТ 4204—66, ч. д. а.
Соляная кислота, ГОСТ 3118—67, ч.д. а.
Олово хлористое, ГОСТ 36—68, ч., ч.д. а.
71
Условия получения
ВНИМАНИЕ! Все производные двухвалентного олова, в том числе сернокислое олово, ядовиты.
Растворяют 120 г хлористого олова (SnCI2-2H2O) в 1 л деминерализованной воды (см. примечание 2, 3) и полученный раствор пропускают через колонку, содержащую 300 г (в пересчете на сухую смолу) катионита КУ-2 в Н-форме крупностью 0,5—1 мм (см. примечание 4). Катионит промывают водой, пропуская ее в том же направлении, что и исходный раствор. По достижении нейтральной реакции фильтрата (pH 5—7 по универсальной индикаторной бумаге) кат.ионит выгружают на воронку Бюхнера и отжимают, а через колонку пропускают воду снизу вверх со скоростью 5 л!мин.
Отжатый катионит в Sn-форме помещают в высокий стеклянный стакан емкостью 3 л, приливают 1,5 л 50%-ной серной кислоты, перемешивают 15 минут и дают отстояться. По мере приливания кислоты и перемешивания образуются белые кристаллы продукта, оседающие на дно, а катионит всплывает. В стакане образуются три четко разграниченных слоя: осадок продукта на дне, слой чистого раствора кислоты и всплывший катионит. Катионит сливают вместе с частью кислоты на фильтр, отжимают под вакуумом и промывают на фильтре водой до нейтральной реакции фильтрата. Осадок продукта вместе с остальной кислотой переносят на фильтр, отжимают, промывают на фильтре 50 мл этилового спирта и высушивают 30—40 минут при 60—70° (см. примечание 4).
Получают 100 г сернокислого олова. Выход составляет 87% (см. примечание 5).
Метод позволяет получать продукт квалификации «ч.д. а.», в соответствии с требованиями МРТУ 6-09-1946-64.
Пр имечания:
1. Деминерализованную воду получают с помощью ионитов или дистилляцией.
2. Вместо раствора хлористого олова (II) могут быть использованы растворы любой соли олова, в том числе и сернокислое олово, получаемое анодным растворением. Концентрация кислоты в растворе соли ие должна Превышать 2 н.
3. Подготовка катионита (очистка и перевод в Н-фо<рму) должна проводиться согласно ГОСТ‘10896—64. Описание методики подготовки катионита и ионообменных операций можно иайти в общих пособиях по ионитам, например [28, 29], а также в статье [30].
4. Промывные растворы выводят из процесса. Маточные растворы собирают отдельно, избегая их разбавления, они содержат 8 г/л Sn и их используют в следующих операциях синтеза продукта. Выход по олову с учетом .использования маточника составляет не меиее 95%.
ЛИТЕРАТУРА
1. J. D. Donaldson, W. Moser. J. Chern. Soc. (London), 1960, 4000. 2. С. M a r i g n a c. Ann. Miner, 22, 1 (1857).
3. G. F. Reynolds. Analyst, 82, 46 (1957).
7o
4. H. G. D e n h a m, W. Е. К i n g. J. Chem.. Soc. (London), 1935, 1251
5. C. L. Man tell. Tin, 2-nd„ N-Y., 1949, pp. 175, 443.
6. В. И. Лайнер, H. T. Кудрявцев. Основы гальваностегии, ч. 1. М., Металлургиздат, 1953, стр. 601.
7. Encyclopedia of Chemical Technology, ed. by R. Kirk and O. Othmer, N-Y., vbl. 13, 1954.
8. А. Д. Деспиллер. Ж. физ. химии, 31, 736 (1957).
9. Пат. США 2862156 (1958).
10. Пат. США 2763555 (1956).
11. Пат. США 2917470 (1959).
12. Пат. США 2918357 (1959).
13. Б. С. К р а с и к о в, Б. И. Лаврентьев. Ж. прикл. химии, 30, 993 (1957).
14. W. Machu, М. G. Fouad. Werkstoffe und Korrosion, 9, 369 (1958).
15. Пат. ФРГ 1008056 (1957).
16. Яп. пат. 3060 (1959).
17. J. D. Donaldson, W. Moser, W. В. Simpson. Analyst, 86, 619 (1961).
18. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы. М., Госхимиздат, 1955, стр. 435, 438.
19. F. С. Mathers, Н. S. Rothrock. Ind. Eng. Chem., 23, 7, 83 (1931).
20. А. Я. Зытнер, В. Я. Бран дель. Тр. НИИМестпром, 1, 62 (1940); РЖХим., 9, 70 (1940).
21. И. М. Селиванов, В. Ф. Грищенко, Р. И. Козлова. Авт. свид. 141860; Бюлл. изобр., № 20 (1961).
22. Н. П. Гиусин. Авт. свид. 157342; Бюлл. изобр., № 18 (1963).
23. Н. П. Г н у с и н, И. М. Селиванов, Т. М. С о 'с и п а т р о в. Уч.
зап. ЦНИИОлово, 3, 70 (1964).
24. С. А. Плетенев, Н. Н. Розов. Цветные металлы, 9 (1936).
25. А. Н. Новиков, Г. С. Прокопенко. Ж. прикл. химии, 10,
257 (1937).
26. Пат. США 2726929 (1955).
27. R. J. Durrant. J. Chem. Soc, (London), 107, 625 (1915).
28. К. M. С а л д а д з е, А. Б. Пашков, В. С. Титов. Ионообменные высокомолекулярные соединения. М., Госхимиздат, 1960.
29. О. Самуэльсон. Ионообменные разделения в аналитической химии. М.-Л., «Химия», 1966.
30. А. И. Вулих, В. Л. Богатырев и др. Методы получения химических реактивов и препаратов, вып. 16. М., ИРЕА, 1967, стр. 5.
РУБИДИИ АЗОТНОКИСЛЫЙ КИСЛЫЙ
С. Af. АРХИПОВ, А. О. ЛЕСОВ АН, Н. И. КАШИНА
RbNO3-2HNO3 M.s. 273,50
Физико-химические свойства азотнокислого кислого рубидия (RbNO3• 2HNOs) приведены в работах fl, 2].
На основании литературных данных [2] по растворимости в системе RbNO3—HNO3—Н2О и собственных исследований этой системы при температурах выше 35° нами разработана методика получения азотнокислого кислого рубидия.
СХЕМА СИНТЕЗА
RbNO3 + 2HNO3 -> RbNO3 • 2HNO3
Характеристика основного сырья
Азотная кислота, ГОСТ 701—68, сорт 1, концентрированная.
Рубидий азотнокислый, МРТУ 6-09-4698-67, ч.
Условия получения
В трехгорлую колбу, снабженную мешалкой с ртутным затвором, обратным холодильником, капельной воронкой и термометром, загружают 100 г азотнокислого рубидия и прибавляют по каплям за 15 минут при перемешивании 76 мл концентрированной азотной кислоты пл. 1,506, поддерживая температуру 35—38°. После полного растворения азотнокислого рубидия колбу помещают в ледяную баню. Кристаллизация соли, продолжающаяся 1 час., сопровождается выделением значительного количества тепла. Выпавшие кристаллы отфильтровывают через перхлорвиниловую ткань.
Получают 140 г азотнокислого кислого рубидия. Выход составляет 75%.
74
Маточный раствор обрабатывают углекислым рубидием до нейтральной реакции среды, упаривают досуха и полученный азотнокислый рубидий используют для получения азотнокислого кислого рубидия.
Общий выход продукта с учетом переработки маточных растворов составляет 95%. Содержание основного вещества 98—99%.
ЛИТЕРАТУРА
1. G. Mascerp а, I. Р о t i е г, А. Р о t i е г. Compt. rend. Acad. Seients, 249, 123 (1958) .
2. M. А. Якимов, В. К. Филиппов, Н. Г. Константинова. Ж. неорган. химии, 12, 2230 (1967).
РУБИДИИ И ЦЕЗИИ ГЕКСАФТОРАЛ ЮМИНАТЫ
С. М. АРХИПОВ, Т. В. РЕВЗИНА, Н. И. КАШИНА
Rb3AlF6 М.в. 397,38
Cs3AlF6 М.в. 539,69
Физико-химические свойства гексафторалюминатов рубидия и цезия приведены в работе [1].
Гексафторалюминаты рубидия и цезия получают сплавлением безводных фтористого алюминия с фтористыми солями рубидия и цезия в молярном соотношении 1 :3 [1, 2].
Нами уточнены условия получения гексафторалюминатов рубидия и цезия этим методом.
СХЕМА СИНТЕЗА
3MeF + AlF3 ->Me3AlF6, где M = Rb, Cs.
Характеристика основного сырья
Алюминий фтористый, МРТУ 6-09-125-63, ч.
Рубидий фтористый, МРТУ 6-09-2969-66, ч.
Цезий фтористый, МРТУ 6-09-5330-68, ч.
Условия получения
Рубидий гексафторалюминат. Смесь 50 г фтористого рубидия и 13,2 г фтористого алюминия пометают в платиновую чашку и высушивают в печи при 400—500е в течение 1,5—2 часов, а затем нагревают до 980—1000° и выдерживают при этой температуре 10—15 минут. Полученный сплав охлаждают и растирают.
.Получают 62 г гексафтор алюмината рубидия. Выход составляет 99,6%,
76
Цезий гексафторалюминат. Смесь 50 г фтористого цезий И 8,94 г фтористого алюминия помещают в платиновую чашку и высушивают в печи при 400—500°, а затем при 820—840° выдерживают 10—15 минут. Полученный плав охлаждают и растирают.
Получают 55,7 г гексафторалюмината цезия. Выход составляет 99%.
Метод позволяет получать продукты квалификации «ч.» согласно требованиям РЭТУ 1314—66 и РЭТУ 1313—66 для гексафторалюминатов рубидия и цезия, соответственно.
ЛИТЕРАТУРА
I. Holm. Jan Lietraw. Acta Chem Scand., 19 (1), 261 (1965)
2. E. П. Дергунов. Докл. АН СССР, 60, 1185 (1948).
рубидии и цезии фтористые кислые
С. М. АРХИПОВ, И. И. КАШИНА, Т. В. РЕВЗИНА
RbHF2 М.в. 124,47
CsHF2 М.в. 171,91
Физико-химические свойства и -применение фтористых кислых солей рубидия и дезия приведены в работах [1—3].
Фтористые кислые соли рубидия и цезия получают действием фтористого водорода на углекислую фтористую соль рубидия или цезия |[4], а также взаимодействием углекислых солей этих металлов с плавиковой кислотой [2].
Нами уточнены условия получения фтористых кислых солей рубидия и цезия в водных растворах.
СХЕМА СИНТЕЗА
Me2CO3 + 4HF ->2MeHF2 + H2O + CO2, где Me = Rb, Cs.
Характеристика основного сырья
Рубидий углекислый, МРТУ 6-09-29|67-66, ч.
Фтористоводородная кислота, ГОСТ 10484—63, ч Цезий углекислый, МРТУ 6-09-4552-67, ч.
Условия получения
Рубидий фтористый кислый. Растворяют 100 г углекислого рубидия в 96,03 г 35%-ной фтористоводородной кислоты при комнатной температуре. Соотношение исходных компонентов стехиометрическое. Полученный раствор фильтруют и упаривают досуха в (платиновых чашках под инфракрасными нагревателями или на песчаной бане при температуре не выше 100° во избежание разложения продукта. Окончательное высушивание соли проводят в вакуум-сушильном шкафу при 50-—60° в течение 5—6 часов.
Получают 103 г фтористого кислого рубидия. Выход составляет 98,5%.
78
Цезий фтористый кислый. Получают по аналогичной Методике. Для синтеза берут 100 г углекислого цезия и 68,8 г фтористоводородной кислоты. Упарку раствора проводят при 80°.
Получают 101 г фтористого кислого цезия. Выход составляет 97,5%.
Метод позволяет получать продукты квалификации «ч.» согласно требованиям ТУ 05-6-66 и ТУ 05-7-66 на фтористокислые соли рубидия и цезия, соответственно.
ЛИТЕРАТУРА
1. И. Г. Рысс. Химия фтора и его неорганических соединений. М., Госхимиздат, 1956, стр. 122.
2. R. Кг uh, К- Fuwa, Т. Ever. J. Amer. Chem. Soc., 78 (17), 4256 (1956).
3. Wartenbery, Bosse. Z. Eleklrochem, 28, 384 (1922).
4. В. С. Ятлов, И. Г. Рысс, П. И. Лапин. Сборник работ по химической технологии минеральных веществ. Труды УНИХИМ, т. 2, 1933.
СВИНЕЦ ЛИМОННОКИСЛЫЙ ТРЕХЗАМЕЩЕННЫЙ
Н. Г. ЧЕРНОВА, Г. И. ВЕСЕЛОВСКАЯ. М. В. МОСЕНЦЕВА
(СеН6О7)2РЬ3 • 2Н2О М.в. 1035,81
При применении заводского метода синтеза лимоннокислого свинца трехзамещенного квалификации «х. ч.» взаимодействием лимоннокислого натрия с азотнокислым свинцом приходится проводить многократную промывку продукта от нитратов в течение трех суток.
Нами разработан метод получения этой соли взаимодействием лимоннокислого натрия трехзамещенного с уксуснокислым свинцом. По этому методу на синтез продукта затрачивается только 4 часа.
СХЕМА СИНТЕЗА
3Pb(CH3COO)2 + 2C6HsO7Na3 (СбН5О7)2РЬ3 + 6СН3СООМа
Характеристика основного сырья
Натрий лимоннокислый трехзамещенный, ГОСТ 3161—57, ч.
Свинец уксуснокислый, ГОСТ 1027—67, ч.
Условия получения
ВНИМАНИЕ! Производство лимоннокислого свинца трехзамещенного относится к группе особо вредных производств, и поэтому при его получении необходимо строго соблюдать правила техники безопасности и промсаиитарии [1].
В конической колбе емкостью 1 л, растворяют 69,6 е лимоннокислого натрия трехзамещеиного в 250 мл дистиллированной воды при 80—90°.
Полученный горячий раствор фильтруют и приливают небольшой струйкой в нагретый до 80—90° и профильтрованный раствор 113,8 г уксуснокислого свинца в 250 мл воды. Реакционную смесь перемешивают 1 час при нагревании (80—90°).
80
Осадок лимоннокислого свинца трехзамещенного тщательно отжимают на воронке Бюхнера, промывают небольшими порциями 1,5—2 л горячей (80—90°) дистиллированной воды и сушат 48 часов на воздухе или 6—7 часов в сушильном шкафу при 100—110°.
Получают 93,3 г лимоннокислого свинца трехзамещенного. Выход составляет 90%.
Метод позволяет получать лимоннокислый свинец трехзамещенный квалификации «х. ч.» в соответствии с требованиями МРТУ 6-09-5607-68.
ЛИТЕРАТУРА
1, Вредные вещества в промышленности, под общей редакцией Н. В. Лазарева, т. 2. М., «Химия», 1965, стр. 450.
СВИНЕЦ МЕТАТИТАНОВОКИСЛЫЙ, МЕТАЦИРКОНИЕВОКИСЛЫЙ, МЕТАН ИОБИЕВОКИСЛЫЙ И МЕТАТАНТАЛОВОКИСЛЫЙ
А. Е. ШАММАСОВА, С. А. КУТОЛИН, 4. И. ВУЛИХ
РЬТЮз М.в. 303,09
PbZrO3 М.в. 346,41
PbNb2O6 М.в. 489,00
РЬТа2О6 М.в. 665,09
Физико-химические свойства названных соединений свинца и области их применения приведены в работах [1—9].
Известны методы синтеза метаниобиевокислого, метатан-таловокислого, мета'титановокислого и метациркониевокисло-го свинца, основанные на термическом разложении двойных солей органических кислот [10], на взаимодействии хлористого титана (IV) с раствором хлористого или азотнокислого свинца в присутствии перекиси водорода с последующим прокаливанием при 800—1000° [11], на спекании углекислого свинца или окиси свинца с окислами титана, циркония, ниобия и тантала в атмосфере окиси свинца при 1100—1450° [12, 13], а также работы по снижению температуры спекания при получении упомянутых соединений свинца [14, 15].
Нами разработана методика синтеза этих соединений спеканием углекислого основного свинца с двуокисями титана и циркония и пятиокисями ниобия и тантала при 850— 950°.
СХЕМА СИНТЕЗА
2РЬСО3 • РЬ (ОН) 2+ЗМеО2 ЗРЬМеО3 + 2СО2+ Н2О, где Me = Ti, Zr;
2РЬСО3 • РЬ (ОН)2 + ЗМе2О5 ЗРЬМе206 + 2СО2 + Н20.
где Me = Nb, Та.
Характеристика основного сырья
Ниобий пятиокись, РЭТУ 1296—66, ч.
Свинец углекислый основной, ГОСТ 11840—66, х. ч.
82
Тантал пятиокись, МРТУ 6-09-4074-67, ч.
Титан двуокись, МРТУ 6-09-1211-64, ч.д. а.
Цирконий двуокись, МРТУ 6-09-965-63, ч.
Способ получения
Синтез проводят в муфельной электрической печи, снабженной термопарой и терморегулятором, обеспечивающим регулирование температуры с точностью ±20°.
Стехиометрически составленную шихту измельчают в фарфоровой ступке или мельнице до крупности 200 меш, загружают в корундовый тигель и помещают в печь. Шихту выдерживают при заданных температуре и времени, -после чего тигель охлаждают, выгружают образовавшийся опек и измельчают его в фарфоровой ступке или мельнице.
Свинец метатитановокислый. Шихту из 89 г углекислого основного свинца (84% РЬО) с 27,5 г двуокиси титана выдерживают 3 часа при 850°.
Получают 100 г порошка зеленовато-желтоватого цвета, содержащего 98% РЬТЮз. Выход продукта составляет 97%.
Свинец метациркониевокислый. Шихту из 78 г углекислого основного свинца и 38 г двуокиси циркония выдерживают 4 часа при 850°.
Получают 100 г порошка кремового цвета, содержащего 99% PbZrO3- Выход продукта составляет 93%.
Свинец метаниобиевокислый. Шихту из 54 г углекислого основного свинца и 56 г пятиокиси ниобия напревают 4 часа при 850°.
Получают 100 г порошка желтого цвета, содержащего 98—99% PbNb2Oe. .Выход продукта составляет 97%.
Свинец метатанталовокислый. Шихту из 40 г свинца углекислого основного и 70 г пятиокиси тантала выдерживают 5 часов -при 950°.
Получают 100 г порошка желтоватого цвета, содержащего 98—99% PbTasOg. Выход продукта составляет 95%.
Метод позволяет получать метатитановокислый, метациркониевокислый, м,етаниобиевокислый и метатанталовокислый свинец в соответствии с требованиями МРТУ 6-09-5859-67, МРТУ 6-09-5860-69, МРТУ 6-09-5857-69 и МРТУ 6-09-5858-69, соответственно.
ЛИТЕРАТУРА
1. Я. Г. Горощенко. Химия ниобия и тантала. Киев, «Наукова думка», 1965.
2. Ф. Иона, Д. Ширане, Сегнетоэлектрические кристаллы. М, «Мир», 1965.
3. И, Л. Гл оз май. Пьезоксраиика. М., «Энергия», 1967.
6*
83
4. Пат. США 2525627 (1950); С. А., 45, 324 (1951).
5. Пат. США 2541833 (1951); С. А., 45, 4421 (1951).
6. Пат. США 3279947 (1966); С. А., 66, 41705 (1967).
7. W. Р. Mason, R. F. W i с к. Proc. Inst. Radio Engrs., 42, 1606 (1954).
8. Брит. пат. 843919 (1960); С. А., 55, 5904 (1961).
9. Пат. США 2892955 (1959); С. А., 53, 20744 (1959).
10. R. V. D a m 1 е. Phys, status, solidi, 22,4, 64 (1967).
11. В. В. Климов, В. А. 3 в о ник, Т. П. Евстратова, О. С. Д и д-ковская, В. Н. Козаченко. Авт. свид. 159809; Бюлл. изобр., № 2, 14 (1964).
12. Г. А. Смоленский, А. И. Аграновская. Докл. АН СССР, 92, 143 (1954).
13. Г. А. Смоленский. Докл. АН СССР. 85, 985 (1952).
14.0, С. Д идк о в ск а я, В. В. Климов, IO. Н. Венсвцев. Изв. АН СССР, сер. неорг. мат,, 4, ,1, 157 (1968),
15. О. С. Дидковская. Получение, исследование и разработка новых пьеэокерамических материалов. Автореферат кандидатской диссертации. М., 4968
СЕРОВОДОРОД, СЕРНИСТЫЙ И УГЛЕКИСЛЫЙ ГАЗЫ
А. И. ВУЛИХ, А. А. АЛОВЯИНИДОВ, Г. А. НИКАНДРОВ, М. К. ЗАГОРСКАЯ
СО2 М.в. 44,01
SO2 М. в. 64,06
H2S М.в. 34,08
Лабораторные методы получения двуокиси углерода, двуокиси серы и сероводорода основаны на действии водных растворов сильных неорганических кислот (обычно серной или соляной) на соответствующие соли щелочных или щелочноземельных металлов (1, 2]. Недостатком этих методов является то, что образующийся газ загрязняется парами или туманами применяемых кислот, а также мелкими каплями раствора, содержащими кислоту и образующуюся соль. Кроме того, скорость выделения газа изменяется во времени.
Разработанный нами метод, основанный на взаимодействии раствора соответствующей соли и катионита в Н-форме или анионита в соответствующей солевой форме и кислоты, позволяет получать газы с незначительным загрязнением их электролитом и упрощает регулирование процесса с целью достижения постоянного тока газа [3] (см. примечание 1).
СХЕМА СИНТЕЗА
RH + NaHCO3 -> RNa + H2O + CO2, где R — радикал функциональной группы катионита
2R/HSO3 + H2SO4 R'2SO4+2H20+2S02:
где R'—радикал функциональной группы анионита
2RH + ZnS H“R2Zn + H2S
Характеристика основного сырья
Аниоиообменная смола АВ-17, ГОСТ 13504—68, техн.
Катионообменная смола КУ-2, ГОСТ 13505—68, техн.
Натрий гидрат окиси, ГОСТ 4328—48, х.ч.
85
Натрий сернистокислый кислый (бисульфит), ГОСТ 902—41, технический.
Натрий углекислый кислый, ГОСТ 2156—52, техн.
Серная кислота, ГОСТ 4204—66, х. ч.
Соляная кислота, ГОСТ 3148—67, х. ч.
Цинк сернистый, МРТУ 6-09-1945-64, ч.
Условия получения
Подготовка катионита КУ-2. Катионит выдерживают в течение суток в насыщенном растворе хлористого натрия, затем промывают водой, осторожно декантируя при легком перемешивании, чтобы смыть инородные механические примеси и продукты деструкции смолы. Набухший катионит загружают в колонку и попеременно промывают водой, 1 М раствором едкого натра и 1 М соляной кислотой до отсутствия в фильтрате железа (качественная реакция с роданистым аммонием) и минимального содержания органических веществ, которые определяют по прозрачности раствора и окисляемо-сти (проба с марганцевокислым калием). Последнюю обработку проводят 1М соляной кислотой до полного перевода катионита в Н-форму, промывают водой до pH 6 (проба на отсутствие хлор-иона в фильтрате).
Подготовленный катионит высушивают при комнатной температуре и хранят в банках с притертыми пробками.
Подготовка анионита АВ-17. Набухший в воде анионит несколько раз промывают водой, осторожно декантируя каждый раз после перемешивания. Промытый анионит загружают в колонку и обрабатывают 10%-ным раствором соляной кислоты до отсутствия железа в фильтрате на выходе из колонки (качественная реакция с роданистым аммонием). Обработанный кислотой анионит отмывают от избытка кислоты пятью объемами дистиллированной воды (объем воды должен соответствовать объему взятой смолы). Затем анионит переводят в ОН-форму, для чего пропускают через него 5%-ный раствор едкого натра до выравнивания ее концентрации на входе и выходе колонки и промывают дистиллированной водой до отсутствия щелочной реакции по фенолфталеину.
Для приготовления HSOs-формы анионита гидроксильную форму его обрабатывают избытком раствора кислого сернистокислого натрия.
Готовый анионит отжимают на воронке Бюхнера и хранят в банках с притертыми пробками.
Углекислый газ. В круглодонной колбе емкостью 1 л с отводной трубкой перемешивают с помощью магнитной мешалки. 800 мл 0,5М раствора углекислого кислого натрия с 100г катионита К.У-2 в Н-форме (фракция 0,5—1 мм- емкость
86
4,8 мг-экв!г смолы). С учетом растворимости газа и разбавления за счет свободного объема колбы получают 9 л газа; 80—90% этого объема выделяется с постоянной скоростью ~2 л/мин (см. примечание 2).
Выход углекислого газа составляет 95%.
Сернистый газ. Перемешивают в колбе 200 г анионита АВ-17 в HSOs-форме (фракция 0,5—1 мм; емкость 3,8 мг-экв/г смолы) н 500 мл 2н. соляной кислоты.
Получают 15 л газа, выделяющегося с постоянной скоростью ~ 0,5 л]мин.
Выход сернистого газа составляет 90%.
. Сероводород. Перемешивают в колбе суспензию 30 г сернистого цинка (98,5% ZnS) и 100 г катионита КУ-2 в Н-фор-ме в 500 мл воды. С учетом растворимости газа и разбавления за счет свободного объема получают 5 л газа. В отсутствие кислоты скорость выделения сероводорода составляет 2 мл!мин. В присутствии 0,005 н. НС1 скорость выделения составляет 5 мл!мин; 0,01 н. НС1—10 мл)мин; 0,05 н. НС1 — 37 мл/мин; 0,1 н. НС1 — 65 мл/мин и сохраняется постоянной до выделения 85—90% газа.
Выход сероводорода составляет 95%.
Примечания:
1. Возможные приемы получения газов с помощью ионообменных смол не ограничиваются приведенными примерами; метод допускает широкое варьирование как ассортимента ионообменных смол в качестве источников ионов, так и веществ, обработка которых ионитами приводит к выделению газа.
2. С целью непрерывного получения больших количеств газов процесс предпочтительнее проводить .в аппаратах с саморегулированием, например, в аппарате Киппа,
ЛИТЕРАТУРА
1. Ф. М. Рапопорт, А А. Ильинская. Лабораторные методы получения чистых газов. М., Госхимиздат, 1963.
2. Г. Лукс. Экспериментальные методы в неорганической химии. М„ «Мир», 1965, стр. 350.
3. А. И. Вулих, Г. А. Никандров, А. А. Аловяйников, М. К. Загорская. Авт. свид, 270705: Изобретения. Промобразцы. Товарные знаки, № 17 (1970).
ТАЛЛИЙ АЗОТНОКИСЛЫЙ И СЕРНОКИСЛЫЙ
У. Р. КУЧКАРОВ, У. А. САИДАХМЕДОВ, А. И. ВУЛИХ, И. У. РИЗАЕВ
T1NO3 М.в. 266,37
TI2SO4 М.в. 504,80
Физико-химические свойства и области применения азотно-«ислого и сернокислого таллия приведены в работах [1—4]. Азотнокислый и сернокислый таллий ядовиты.
В разработанной нами методике описывается ионообменный синтез солей таллия с использованием слабокислотного карбоксильного катионита. Так как Н-форма такого катионита представляет собой слабую кислоту (рК ~5), ее соли легко разлагаются сильными кислотами. Это позволяет получать при десорбции катионов такими кислотами растворы солей, концентрация которых ограничивается лишь их растворимостью.
Этот метод может быть попользован не только для взаимопревращений солей таллия, но и при извлечении таллия из растворов, образующихся при получении или применении его солей.
СХЕМА СИНТЕЗА
Подготовка ионита
RH + NaOH RNa+H2O
Сорбция таллия
RNa+TlNO3(Tl2SO4) -> RT1 + NaNO3(Na2SO4)
Десорбция таллия
2RT1 + H2SO4 (HNO3) -> 2RH + T12SO4 (T1NO3),
где R — радикал карбоксильного катионита (КБ-4, КБ-4-П2 и др.).
Характеристика основного сырья
Азотная кислота, ГОСТ 4461—67, ч.
Катионит КБ-4 или КБ-4-П2, МРТУ 6-05-952-65.
Натр едкий, ГОСТ 4328—66, ч.
Серная кислота, ГОСТ 4204—66, ч.
Таллий азотнокислый, МРТУ 6-09-6068-69, ч.
Таллий сернокислый, МРТУ 6-09-449-70, ч.
88
Условия получения
Подготовка и регенерация катионита. В колонку диаметром 35—40 мм и длиной 60—80 см загружают 100 г катионита КБ-4 или КБ-4-П2 (в расчете на сухой ионит в Н-форме). Через слой ионита пропускают 1 л 5 %-ной серной (или соляной) кислоты, 200—300 мл воды, затем пропускают 2%-ный (0,5 н.) раствор едкого натра до выравнивания концентрации щелочи в исходном растворе ифильтрате и промывают500мл воды.
Растворы и воду пропускают со скоростью 30 мл!мин. После указанной обработки катионит содержит около 10 мг-экв/г Na+.
Регенерацию ионита проводят аналогично, исключая операцию обработки кислотой.
Таллий азотнокислый. Растворяют 300 г сернокислого таллия в 6 л дистиллированной воды и полученный раствор пропускают через колонку с ионитом со скоростью 50 мл/мин до уравнивания концентрации таллия в исходном растворе и фильтрате (контроль по плотности раствора). Затем промывают ионит, пропуская 1 л воды с той же скоростью. Качественное присутствие или отсутствие Т1+ в фильтрате устанавливается по реакции с йодистым калием. Ионит после сорбции таллия содержит около 200 г Т1+ (1 г-эке). Основная часть фильтрата (5—5,5 л) предствляет собой раствор сернокислого натрия, который удаляется; последняя часть фильтрата (-—'0,5 л) содержит сернокислый таллий в смеси с сернокислым натрием и в следующем цикле процесса пропускается через катионит в Na-форме перед пропусканием раствора сернокислого таллия. Промывной раствор содержит только сернокислый таллий и используется для растворения исходной соли в следующем цикле синтеза.
Десорбцию таллия проводят раствором азотной кислоты, нагретым до 60—80°. Получают 800—850 мл нейтрального фильтрата, содержащего 250 е/л TINO3 (80—85% от содержания Т1+ в ионите) и 700—800 мл кислого фильтрата, содержащего 50—60 г/л TINO3 и 40—45 г/л HNO3. Ионит промывают водой (300—500 мл), вымывая оставшуюся в слое ионита кислоту, затем регенерируют.
Кислый фильтрат используют в следующем цикле процесса (пропуская его при десорбции таллия в первую очередь), а промывной раствор — для приготовления 1 н. раствора азотной кислоты (пропускается при десорбции таллия ВО' вторую очередь).
Нейтральный фильтрат выпаривают до объема 150 мл и охлаждают при интенсивном перемешивании до 20°. Фильтруют маточный раствор (около 50 мл, концентрация T1NO3 90—1Q0 г/л). Кристаллы высушивают при 100°.
89
Получают 200 г (65%) азотнокислого таллия квалифика ции «ч.».
Маточник присоединяется к раствору, поступающему на выпаривание в следующем цикле. В следующих циклах, $ учетом возврата в процесс побочных фильтратов, получают) 260 г соли. Выход составляет ~95%. Во втором цикле сернокислого таллия берут не 300, а 250 г.
Метод позволяет получить продукт квалификации «ч.» в* соответствии с требованиями ТУ 6-09-6068-69.
Таллий сернокислый. Растворяют 300 г азотнокислого таллия в 2 л дистиллированной воды и полученный раствор пропускает через колонку с ионитом со скоростью 30 мл!мин до уравнивания концентрации соли в исходном растворе г фильтрате, затем вымывают оставшийся в колонке раство{ 1 л горячей (90°) воды. (Использование фильтратов здесь i далее аналогично указанному при синтезе азотнокислого таллия).
Десорбцию таллия проводят 0,5 н. раствором серной кис^ лоты; раствор кислоты напревают до 90—95° и пропускают через колонку непосредственно вслед за промывкой ионита горячей водой; для предотвращения кристаллизации соли в колонке температура фильтрата на выходе из колонки должна быть не ниже 60°. Скорость фильтрации 50 мл{мин. Полу-, чают 1,6—4,7 л нейтрального раствора, содержащего 120 г/л TI2SO4, и 1,5 л кислого фильтрата, содержащего 25 г/л TI2SO4 и 20 г/л H2SO4. Ионит промывают холодной водой (300—500 мл) и регенерируют.
Нейтральный фильтрат выпаривают до объема ~200 мл (при этом выделяется часть соли) и охлаждают при перемешивании до 20°. Отфильтровывают маточный раствор (около 100 мл, концентрация TI2SO4 50 г/л). Кристаллы высушивают при 100°.
Получают 195 г (70%) сернокислого таллия. Во втором цикле выход соли достигает 95%.
Метод позволяет получить продукт квалификации «ч.» в соответствий с требованиями ТУ 6-09-449-70.
ЛИТЕРАТУРА
1. Краткая химическая энциклопедия, т. 5. М., 1967, стр. 14, 17.
2. Справочник химика, т. 2. Л.-М., 1963, стр. 217, 218.
3. О. Самуэльсон. Ионообменные разделения в аналитической химии. М.-Л., «Химия», 1966, стр. 67.
4. А, Ю. Д ад аба ев, С. Hf С у ш е и к о, В. Д. Пономарев. Сб. «Теория и практика ионного обмена». Алма-Ата, 1963, стр. 137.
90
ТЕЛЛУР ДВУОКИСЬ
Г. Е. РЕВЗИН
ТеО2 М. в. 159,60
Физико-химические свойства двуокиси теллура приведены в работах |fl—5].
Обзор методов получения двуокиси теллура приведен в работе [6]. Там же подробно описан один из наиболее удобных методов получения чистой двуокиси теллура осаждением её азотной кислотой из раствора теллуристокислого натрия или калия. Однако этот метод сопряжен с выделением в атмосферу значительных количеств окислов азота, что препятствует его реализации в крупных масштабах.
Нами разработан новый метод окисления теллура, заключающийся в нагревании смеси теллура с азотнокислыми солями щелочных металлов на воздухе [7]. Преимуществом предложенного метода является попутная очистка теллура от селена не только в процессе осаждения двуокиси теллура из раствора, но и при окислении. Температура спекания теллура с азотнокислым калием или натрием составляет 400—430°; образующаяся при этом параллельно с основным продуктом двуокись селена возгоняется (температура сублимации 317—337° [1, 2]).
СХЕМА СИНТЕЗА
Te + 2KNO3—> ТеО2 SKNOsi ТеО2 + 2КОН-> К2ТеО3 + Н2О;
КэТеОз + 2HNO3 —TeO2-f-2KNO3 + H2O.
Характеристика основного сырья
Азотная кислота, ГОСТ 4461—67, ч.д. а.
Калий азотнокислый, ГОСТ 4217—65, ч.
Кали едкое, ГОСТ 4203—65, ч.
Теллур металлический, ГОСТ 9514—60, Т-1.
Условия получения
Смешивают в ступке 100 г порошкообразного теллура (см. примечание 1,2) с 200 г азотнокислого калия. Смесь помещают в фарфоровую чашку или кварцевую кювету и нагревают 1 час в муфельной печи при 400—430° (см. примечание 3). Спек охлаждают и выщелачивают при перемешивании нагретым. (80—90°) раствором 90 г едкого кали в 900 мл воды.
91
Раствор теллуристокислого калия отфильтровывают от неокисленного теллура (см. примечание 4). Фильтрат нагревают до ~100° и при постоянном перемешивании осаждают двуокись теллура азотной кислотой (пл. 1,18—1,20) порциями по 10—20 мл.
Осаждение считают законченным, когда pH раствора достигнет 3,5—4 и остается неизменным 15—20 минут при 100° и интенсивном перемешивании. Затем реакционную смесь охлаждают, осадок двуокиси теллура отфильтровывают (см. примечание 5), промывают на фильтре дистиллированной водой до отсутствия в фильтрате нитрдт-иона (проба с дифениламином) и сушат 2—3 часа в сушильном шкафу при 150—170°.
Получают 122 г двуокиси теллура. Выход составляет 97,5%.
Метод позволяет получать продукт квалификации «ч. д. а.» в соответствии с требованиями МРТУ 6-09-686-63.
Примечания:
1. Аналогичным образом двуокись теллура может быть получена при окислении теллура азотнокислым натрием, однако црн этом продолжительность процесса окисления должна быть увеличена до 2-х часов, а выход продукта составляет 90—95%.
2. Окислению может подвергаться не только порошкообразный, но и кристаллический теллур. Последний измельчают до крупности 0,1—0,2 м'м; количество вводимого в шихту окислителя увеличивается на 10—20%. а прямой выход двуокиси теллура снижается до 85—90%.
3. Температура в зоне окисления не должна превышать 430°, так как это вызовет плавление теллура (т. пл. 450°), что приведет к образованию двух жидких фаз: верхняя — расплав азотнокислого калия (натрйя) и нижняя—расплав теллура. При этом резко уменьшается поверхность .окисляемого металла, снижается скорость окисления и выход продукта, а также производительность процесса.
4. Вес остатка, представляющего собой неокмеленный теллур, а.также примеси железа и тяжелых металлов в виде окислов, гидроокисей или теллуритов составляет 2—2,5 г.
5. Маточный раствор, содержащий азотнокислый калий, упаривают досуха, высушивают н .вновь используют для окисления следующих порций теллура.
ЛИТЕРАТУРА
1. Справочник химика, т. 2. М.-Л., «Химия», 1964, стр. 220.
2. А. А. Кудрявцев. Химия и технология селена и теллура. М, «Высшая школа», 1961, стр. 85.
3. Руководство по препаративной неорганической химии, под редакцией Г. Брауэра. М., Иплитиздат, 1956, стр. 227.
4. А. Н. Крестовников, А. С. Шахов. Физико-химические и термодинамические свойства редких элементов. М., Металлургиздат, 1943, стр. 147.
5. I. I ssa, S. A w a d, I. Phys. Chem., 58, 948 (1954).
6. Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 16. М., ИРЕА, 1967, стр. 168.
7. Г. Е. Рев зии. Авт. свид. 225144. Изобретения. Промобразцы. Товарные знаки, № 27 (1968).
92
ТЕЛЛУРОВАЯ КИСЛОТА
Ортотеллуровая кислота
Г. Е. РЕВЗИН, А. И. ВУЛИХ, Л. П. ЖЕРДИЕНКО
Н2ТеО4-2Н2О М.в. 229.64
Физико-химические свойства теллуровой кислоты приведены в работах [1, 2].
Обзор свойств методов получения теллуровой кислоты приведен в работе [3], где описан ее синтез, основанный на окислении перекисью водорода гидратированной двуокиси теллура, свежеосажденной при pH 6 из раствора теллуристокислого натрия (калия) [4]. Однако применение этого метода ограничено необходимостью использования свежеосажденной гидратированной двуокиси теллура.
Нами разработан новый метод синтеза чистой теллуровой кислоты, основанный на окислении суспензии двуокиси теллура (полученной любым способом) раствором перекиси водорода (различных концентраций) в присутствии углекислого или углекислого кислого натрия или калия (5] с последующей обработкой раствора на сильнокислотном катионите, КУ-2 в Н-форме (6].
СХЕМА СИНТЕЗА
ГеОа + К2СО3 + Н2О2 —> К2ТеО4-)- Н2О + СО2 К2ТеО4 + 2Н2О +>2RH -> 2RK+Н2ТеО4 • 2Н2О,
где R — радикал функциональной группы катионита.
Характеристика основного сырья
Калий углекислый, ГОСТ 4221—65, ч.
Катионит КУ-2, МРТУ 6-05-903-65.
Перекись водорода, ГОСТ 177—55, мед.
Теллур двуокись, МРТУ 6-09-3108-66, ч.
93
Условия получения
К ЮО г (0,626 М) измельченной двуокиси теллура добавляют 88 г (0,63 М) безводного углекислого калия, 150 мл дистиллированной воды и 75 мл 30%-ной перекиси водорода (0,7 М). Смесь перемешивают 30 минут, при 105—110°. После прекращения выделения газообразных продуктов реакции (углекислоты и кислорода) и образования гомогенного раствора его охлаждают, фильтруют, разбавляют водой до объема 600 мл.
К раствору при перемешивании добавляют около 300 г воздушно-сухого катионита КУ-2 в Н«форме до установления pH меньше 3. При добавлении к раствору теллуроворислого калия первых порций катионита наблюдается образование осадка малорастворимых полителлуратов, который растворяется при дальнейшем добавлении смолы. Раствор фильтруют от катионита, промывают катионит дистиллированной водой 3 раза порциями по 130—150 мл и объединенный с промывной водой раствор пропускают через ионообменную колонку, содержащую около 50 г катионита КУ-2 в Н-форме, со скоростью 25—50 мл/мин. Колонку промывают 100 мл дистиллированной воды и упаривают раствор теллуровой кислоты до /появления кристаллической пленки.
По охлаждении до комнатной температуры к раствору добавляют двукратное по объему количество 90—95 %-наго этилового спирта. Через 30 мин. отфильтровывают кристаллы кислоты, промывают их на фильтре 50 мл спирта и сушат в вакуум-эксикаторе над фосфорным ангидридом.
Получают , 134 г теллуровой кислоты. Выход составляет 93%.
Метод позволяет получать продукт квалификации «ч.», в соответствии с требованиями МРТУ 6-09-1655-64.
ЛИТЕРАТУРА
1. А. Н. Крестовников, А. С. Шахов. Физико-химические и термодинамические свойства редких элементов. М., Металлургиздат, 1943, стр. 147.
2. А. А. Кудрявцев. Химия и технология селена и теллура. М, «Высшая школа», 1961, стр. 90.
3. Г. Е. Р е в з и н. Методы получения химических реактивов и препаратов, вып. 16. М., ИРЕА, 1967, стр. 172.
4. Г. Е. Рев зип, Авт. свид. 167834, Изобретения. Промобразцы. Товарные знаки, № 3 (1965).
. 5. Г. Е. Р е в з и и, А. И. Вулих, Л. П. Ж'-РДненко. Авт, свид. 202893, Изобретения. Промобразцы. Товарные знаки, № 20 (1967).
6. А. И. Вулих, В. Л. Богатырев и др. Методы получения химических реактивов и препаратов, вьш. 16, М., ИРЕА, 1967, стр. 5.
94
ЦЕЗИЙ АЗОТНОКИСЛЫЙ
А. И. ВУЛИХ, Л. Г. СИДОРОВА, А. О. ЛЕСОВАЯ
CsNO3 М.в. 194,91
Физико-химические свойства и применение азотнокислого цезия 'приведены в работах [1—37].
Азотнокислый цезий получают действием азотной кислоты на хлористый цезий [117, 23], нейтрализацией углекислого цезия азотной кислотой [38]. Однако в литературе отсутствует детальное описание синтеза продукта.
Нами отработана методика получения азотнокислого цезия нейтрализацией углекислого цезия азотной кислотой.
СХЕМА СИНТЕЗА
Cs2CO3+2HNO3 2CsNO3 +Н2О +COS
Характеристика основного сырья
Азотная кислота, ГОСТ 1461—48, ч.
Цезий углекислый, МРТУ 6-09-1721-64, ч.
Условия получения
Растворяют 100 г углекислого цезия в 300 мл деминерализованной дистиллированной воды, причем температура раствора повышается до 50° и растворение заканчивается за 5 минут.
Устанавливают pH раствора 7ч-7,5 (по индикаторной бумаге) добавлением при перемешивании азотной кислоты. Затем в горячий (70—80°) раствор азотнокислого цезия пропускают сероводород в течение 10—15 минут и выдерживают его при этой температуре около 1 часа, после чего осадок отфильтровывают и промывают на фильтре 50 мл теплой (40—50°) воды. Полученный раствор подкисляют азотной кислотой до pH 4,5ч-5,0 по индикаторной бумаге, напревают до 70—80° и повторно пропускают через него сероводород
95
(10—il5 минут), а осадок отфильтровывают и промыва)оТ. Очищенный от ионов трехвалентных и тяжелых металлов раствор выпаривают до начала выделения кристаллов (плотность горячего раствора 1,85—1,87) и охлаждают при постоянном перемешивании до 20—25°. Кристаллы отфильтровывают, промывают на фильтре 30—50 мл холодной воды, переносят в чашку, где распределяют по ее поверхности слоем толщиной ~1 см и высушивают при 110—120° в течение часа, периодически растирая.
Получают 95 г азотнокислого цезия квалификации «ч.д. а.». Выход составляет 80%.
Маточный раствор, содержащий 20% общего количества азотнокислого цезия, выпаривают до начала выделения кристаллов, после чего кристаллизуют в условиях, аналогичных описанным выше. После отделения кристаллов и их высушивания получают ~20 г азотнокислого цезия квалификации «ч.», т. е. дополнительно 15%.
Для получения продукта квалификации «ч. д. а.» кристаллы, полученные из маточного раствора, расворяют в воде (1 : 1), выпаривают раствор до начала кристаллизации, охлаждают до 20—25°, отфильтровывают кристаллы и высушивают их.
Общий выход продукта квалификации «ч.д. а.» составляет 92% .(110 г).
Для .получения азотнокислого цезия квалификации «х. ч.» аналогично перекристаллизовывают соль квалификации «ч. д. а.».
Выход соли квалификации «х.ч.» составляет 85% (100 г) (см. примечание).
Примечание: Если в качестве исходного сырья взяты углекислый цезий и азотная кислота квалификации «ч. д. а.», то получают за одну операцию азотнокислый цезий квалификации «х. ч.» с выходом 95% (115 г).
За одну операцию кристаллизации продукта (в выше указанных условиях) содержание калия уменьшается примерно в 5 раз, а рубидия — в 3 раза (при исходном содержании К>0,01 % и Rb>0,05%). Метод позволяет получить продукт квалификации «ч.» и «ч. д. а.» в соответствии с требованиями ТУ 6-09-437-70.
ЛИТЕРАТУРА
1. L. W а 1 d b а и е г, D. С. М е С a n n. J. Chem. Phys., 2, 616 (1934).
2. В. Gossner. Z. Krystallogr,, 38, 142 (|1904).
3. F. Wall er a nt. Bl. Soc, Mineral., 28, 326 ,(1905).
4. P. W. Bridgeman. Proc. Amer.’Acad. Sei., 51, 590 (1916).
5. C. D. West. Z. Krystallogr., 88, 98 (1934).
6. S. Gordon, C. Campbell. Analyt. Chem., 27, 1102 (1955); cm, перевод в Сб. «Рубидий», М., Иилитиздат, 1959, стр. 44.
7. N. A. Puschin, М. Radoicic. Z. anorgan. und allgern. Chem., 233; 41 (1937).
8. В. E. Плющев, И. В. Маркина, Л. П. Шкловер, Докл. АН СССР, 108, 645 (1956).
96
9. L. E r d e у, G. L ip t ay, S. Gal. T a 1 a n t a, 12, 257 (1965).
10. В. П. Б лидин. Изв. сектора физико-химического анализа, 23, 233 (4953).
11. F. М. Jaeger. Z. anorgan. und allgem. Chem., 101, 193 (1917).
12. П. И. Проценко, А. В. Проценко. Изв. ВУЗ, хим. и хим. технология, 8, 160 (1965).
13. М. G е n t п е г s z w е г. J. Chim. phys. 27, 9 (1930).
14. F. Z. Haigh. J, Amer. Chem. Soc„ 34, 1140 (1912).
15. E. Berkeley. Phil. Trans. Roy. Soc., 208 A, 213 (1904).
16. E. W. Washburn. J. Amer. Chem. Soc., 33, 1687 (1911).
17. В. M, Jones. J. Chem. Soc., 93, 1743 (1908).
18. Caesium Salts. Ed. irom British Drug Hauses. London, 1964, p. 23.
19. P. B. Davis, H. C. Jones. J. Amer. Chem. Soc., 37, 2639 (1915).
20. И. С. Пехташев, С. А. Вознесенский. Изв. ВУЗ, хим. и хим. технология, 2, 827 (1959L
21. Г. Мас шер па, Ж. Потье, А. Потье. Сб. «Цезий». М., ИЛ, 1963, стр. 56.
22. Справочник по плавкости солевых систем, т. 1. М.-Л., Изд-во АН СССР, 1961.
23. Н. Z. Wells. Amer. Chem. J., 26, 267 (1901).
24. P. M. Шкловская, И. M. Суханова. Промышленность химических реактивов, вып. 3, М., ИРЕА, 1963, стр. 61.
25. R. Е. Kirk, D. F. Othmer. Encyclopedia of Chemical Technology, 2-nd eg. Vol, 4. N-Y 1964, p. 863.
26. А. И. Вулих, А. О. Лесовая и др. Сб. «Редкие щелочные элементы», вып. 2. Новосибирск, «Наука», 1967, стр. 312, 320.
27. Р. М. Шкловская. Методы получения химических реактивов и препаратов, вып. 16. М., ИРЕА, 1967, стр. 94,
28. Е. L. Simons et al. Taianta, 13, 199 (1966).
29. A. H. Киргинцев, E. Г. Аввакумов, А. И. Вулих. Докл. АН СССР, 164, 1315 (1965); Ж. неорган. химии, 11, 2328 (1966).
30. R. Tri.edman et al. Progr. Astronaut. Aeronaut., 12, 379 (1962), C. A„ 62, 11620 d (1965).
31. N. W. Rosenberg, D. Golomb. Progr. Astronaut. Aeronaut., 12, 395 (1962); C. A., 63, 1338b (1965).
32. M. S. Jones et al. US At. Energy Comm., MDH-646 (1963); C. A., 61, 14019 (1964).
33. K. Baldwin, W. Balwanz. Trans. Nucl. Sci., 11, 2, 51 (1964);
C. A., 61, 4137 (1964).
34. Франц, пат. 1184312 (1959),
35. Брит. агат. 965894 (1964).
36. А. Я. Даицигер. Физика твердого тела, 7, 2279 (1965).
37. В. В. Гладкий, И. С. Ж ел у дев. Кристаллограф113, Ю, 1, 63 (1965).
38. F. М. Jaeger, В. Karma. Z. anorgan und allgem. Chem., 113, 51 (1920).
ЦЕЗИЙ АЗОТНОКИСЛЫЙ кислый
С. М. АРХИПОВ, А. О. ЛЕСОВАЯ, Н. И. КАШИНА
CsNO3-HNO3 М.в. 257,92
Физико-химические свойства азотнокислого кислого цезия описаны в работах [1-3].
Установлено [2, 3], что в системе CsNO3—HNO3—Н2О при различных температурах образуются соединения CsNO3- HNO3 и CsNO3-2HNO3,
На основании данных по растворимости в системе CsNO3—HNO3—Н2О нами (разработана методика получения азотнокислого кислого цезия.
СХЕМА СИНТЕЗА
CsNO3 + HNO3 -> CsNO3- HNO3
Характеристика основного сырья
Азотная кислота, ГОСТ 701—68, сорт 1, концентрированная. Цезий азотнокислый, МРТУ 6-09-5930-69, ч.
Условия получения
В трехгорлую колбу, снабженную мешалкой с ртутным затвором, обратным холодильником, капельной воронкой и термометром, загружают 100 г азотнокислого цезия, 18 мл дистиллированной воды и прибавляют по каплям за 10 минут при перемешивании 43 мл концентрированной азотной кислоты (пл. 1,506), поддерживая температуру в пределах 60—65°.
После полного растворения азотнокислого цезия колбу помещают в ледяную баню и охлаждают до 10°. Выпавшие кристаллы отфильтровывают на перхлорвиниловой ткани.
Получают 106 г азотнокислого кислого цезия. Прямой выход составляет 80% в пересчете на азотнокислый цезий. Содержание основного вещества в продукте 98—99%.
98
Маточный раствор от кристаллизации продукта обрабатывают углекислым цезием до нейтральной реакции, упаривают досуха и полученный азотнокислый цезий используют для получения азотнокислого кислого цезия.
Общий выход продукта с учетом переработки маточных растворов составляет 97%.
ЛИТЕРАТУРА
1. G. Mascherpa, A. Potier. Compt. rend. Acad. Scients, 249 (1), 123—125 (1959).
2. С. А. Щукарев, M. А Якимов, В. Я. Мишин. Ж. неорган. химии, 3, 7, 1661 (1958).
3. М. А. Якимов, В. Я. Мишин. Ж- неоргал. химии, 8,1,226 (1963.)
у*
ЦЕЗИЙ МУРАВЬИНОКИСЛЫЙ
Цезий формиат
Е. М. ШИПИЛОВА, Л. П. /КЕР ДИЕН К. 0, Б. И. ПУСТОВАЛОВ
CsHCO2 М.в. 177,92
Физико-химические свойства муравьинокислого цезия приведены в работах [1, 2]. Вещество гигроскопично, на воздухе расплывается; температура плавления 265° [2], разлагается при 420°.
Известен способ получения муравьинокислого цезия нейтрализацией муравьиной кислоты углекислым цезием [3].
Нами уточнены условия получения муравьинокислого цезия.
СХЕМА СИНТЕЗА
Cs2CO3 + 2HCOOH -н 2CsHCO2 + H2O + CO2
Характеристика основного сырья
Муравьиная кислота, ГОСТ 5848—60, ч., 85%.
Цезий углекислый, МРТУ 6-09-1721-64, ч.,
Условия получения
Растворяют 100 г углекислого цезия в 100 мл горячей (80°) воды и отфильтровывают от механических примесей. К фильтрату осторожно небольшими порциями при постоянном перемешивании добавляют муравьиную кислоту до pH 7—6,5 (см. примечание 1). Раствор упаривают досуха (150°), и сушат 15 часов в вакуум-сушильном шкафу при 50° до постоянного веса (см. примечание 2).
100
Получают 96 г муравьинокислого цезия с содержанием основного вещества 98,0%. Выход составляет 90%.
Метод позволяет получать продукт, отвечающий требованиям МРТУ 6-09-2005-64.
Примечания:
1. Если в растворе после нейтрализации при нагревании появляются хлопья, их отфильтровывают.
2. Высушенную соль расфасовывают в ампулы в герметичной сухой камере.
ЛИТЕРАТУРА
1. Справочник по растворимости солевых систем, т. 3. М., Госхимнз-дат, 1961, стр. 2157.
2. Справочник химика, т. 2, М., Госхимиздат, 1963, стр. 246.
3. Gmelin. Handbuch dcr anorganischen Chemie. Berlin, 1938, s. 240.
ЦЕРИЙ (1У)-АММ0НИЙ СЕРНОКИСЛЫЙ
С. М. ЛРХИПОВ, л. м. отсолиг
Ce(SO4)2-2(NH4)2SO4-2H2O М.в. 632,55
Физико-химические свойства церий (IV)-аммоний сернокислого приведены в работах [1, 2].
Церий (IV)-аммоний сернокислый с содержанием двуокиси церия в пределах 22—32% получали [1] смешением кислого раствора сернокислого церия (IV) с раствором сернокислого аммония с последующим упариванием и выделением кристаллического продукта. По этому методу получена смесь следующих солей: 2Ce(SO4)2 • 3(NH4)2SO4 ЗН2О; Ce(SO4)2-•2(NH4)2SO4-2H2O и Ce(SO4)2-3(NH4)2SO4-4H2O.
Нами уточнены условия получения церий (IV)-аммоний сернокислого. Содержание двуокиси церия в соли, полученной разработанным нами способом, колеблется в пределах от 26,40 до 27,75%, что соответствует формуле Ce(SO4)2-• 2(NH4)2SO4 • 2Н?О.
СХЕМА СИНТЕЗА
H2so4
Ce(SO4)2 + 2(NH4)2SO4+nH2O----->
-> Ce(SO4)2 -2(NH4)2SO4 • 2Н2О.
Характеристика основного сырья
Аммоний сернокислый, ГОСТ 3769—60, х. ч.
Серная кислота, ГОСТ 4204—66, ч.
Этиловый спирт, ГОСТ 5062—63.
Церий двуокись, РЭТУ 144—59, Цо-1.
Церий (IV) сернокислый, МРТУ 6-09-2436-65. ч.
Условия получения
.Раствор, содержащий 100 г сернокислого церия (см. примечание 1) и 60 мл серной кислоты, упаривают при 80—90° до начала выпадения кристаллов. Добавлением воды кристал-102
лы растворяют и к полученному раствору при постоянном перемешивании добавляют горячий (90°) насыщенный раствор сернокислого аммония, содержащий 81,80 г (NH4)2SO4. В момент смешения на дне чашки не должно быть кристаллов. До начала выделения кристаллов реакционную смесь нагревают при 80°, затем температуру снижают до 70° и упаривают раствор до выделения основной массы кристаллов. Затем раствор постепенно охлаждают до 20—25°, кристаллы отфильтровывают через перхлорвиниловую ткань (см. примечание 2), промывают спиртом до отсутствия свободной серной кислоты и сушат на воздухе.
Получают 173 г церий-аммоний сернокислого [Ce(SO4)2-• 2(NH4)2SO4 • 2Н2О]. Выход составляет 90%.
Метод позволяет получить продукт квалификации «ч.» в соответствии с требованиями МРТУ 6-09-5637-68.
Примечания:
4. При отсутствии сернокислого церия (IV) в качестве исходного сырья используют двуокись церия после ее предварительной очистки от железа и тяжелых металлов. Очищенную двуокись церия (100 г) помещают в фарфоровую чашку, приливают 160 мл серной кислоты (пл. 1,83) и при периодическом перемешивании нагревают при 200—250°. Полученная масса должна полностью растворяться в 933 мл холодной дистиллированной воды. Отстоявшийся прозрачный раствор, содержащий сернокислый церий (IV) и свободную серную кислоту, может быть использован для получения церий (IV)-аммоний сернокислого.
2. Маточный раствор, содержащий 5—10 г/л двуокиси церия (IV) и 400-—500 г/л серной кислоты нейтрализуют водным раствором аммиака для выделения гидроокиси церия.
ЛИТЕРАТУРА
1. В. А. Головня, Л. А. Поспелова. Ж. неорган. химии, 6 (3), 638 (1961).
2. В. В. Серебренников. Химия редкоземельных элементов. Томск, ТГУ, 1959, стр. 324.
7*
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ, ОПИСАННЫХ В НАСТОЯЩЕМ ВЫПУСКЕ
Алюминий йодистый . . ..............................65
Барий метагафниевокислый . , ....... 11
Барий метатитановокислый......................................... 5
Барий метациркониевокислый...................................... 9
.Ванадий двуокись .............................................. 13
Ванадий пятиокцсь .............................................. 14
Ванадил бромистый .............................................. 17
Вольфрамовый ангидрид............................................19
Вольфрамовая кислота.............................................19
Галлий азотистый............................................... 22
Галлий (III) йодистый............................................65
Галлий сернокислый безводный.....................................25
Галлий фосфорнокислый............................................27
Галлий (III) фтористый, трехводный...............................29
Германий (IV) хлористый..........................................31
Гольмий (III) йодистый...........................................64
Диспрозий (III) йодистый................................' . . 64
Европий (II) йодистый............................................64
Индий (III) йодистый.............................................65
Иидий (III) сернистый............................................33
Индий сернокислый безводный.....................................2,5
Индий сернокислый кислый семпводный..............................35
Ивдий углекислый основной......................................37
Иидий уксуснокислый основной.....................................39
Индий фосфорнокислый.............................................27
Иттрий (III) йодистый .... 64
Железо (II) йодистое............................................ 65
Кадмий сернокислый...............................................41
Калий метилат....................................................46
Калий пентафтораитимонат....................................... 49
Калий-сурьма фтористая...........................................49
Кальций метагафниевокислый .... 11
Кальций метатитановокислый .... ..... 5
Кальций метациркониевокислый ............................... 9
104
Лантан (III) йодистый...........................................64
Литий бензойнокислый..........................................51
Литий двухромовокислый........................................53
Литий метафосфорнокислый......................................55
Литий молибденовокислый.......................................57
Литий селенистокислый кислый..................................59
Литий щавелевокислый..........................................60
Магний метатитановокислый....................................... 5
Молибденовый ангидрид...........................................67
Молибденовая кислота............................................67
Неодим (III) йодистый...........................................64
Никель лауриновокислый........................................ 70
Олово (II) йодистое.............................................65
Олово (IV) йодистое.............................................65
Олово сернокислое . . . 41,71
Олово хлористое ......................................' • • 41
Празеодим (III) йодистый....................................... 64
Рубидий азотнокислый кислый.....................................74
Рубидий гексафторалюминат.......................................76
Рубидий и пропилат..............................................46
Рубидий фтористый кислый........................................78
Самарий (II) йодистый...........................................64
Свинец лимоннокислый трехзамещенный.............................80
Свинец мета ниобиевокислый......................................82
Свинец метатаиталовокнслый......................................82
Свинец метатитановокислый.......................................82
Свинец метациркониевокислый.....................................82
Сероводород .................................................
Сернистый 1 аз...............................................
Скандий йодистый ............................................
Стронций метатитановокислый . . .......................
Стронций метациркониевокислый ......................... .
Таллий азотнокислый...............................................88
Таллин сернокислый ............................................. 88
Теллур двуокись ................................................. 91
Теллуровая кислота ............................................. 93
Титан (IV) йодистый...............................................65
Углекислый газ....................................................85
Цезий азотнокислый............................................. 95
Цезий азотнокислый кислый........................................98
Цезий гексафторалюминат ..........................................76
Цезий метилат....................................................46
Цезий муравьинокислый...........................................100
Цезий фтористый кислый............................................78
Церий (IV) -аммоний сернокислый.................................102
Церий (III) йодистый . 64
Цирконий (IV) йодистый .......................................... 65
Эрбий (III) йодистый..............................................64
105
УДК 546.824'442.07
Барий, кальций, магний, стронций метатитановокислые. С. А. Куто-лин, А. И. Вулих, А. Е. Шаммасова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 5.
Метатитановокислые барий, кальций, магний, стронций получены спеканием углекислых солей этих металлов с двуокисью титана в стехиометрических количествах в токе аммиака при 850—900°. Получали стехиометрические соединения состава МеТЮз. Содержание основного вещества 98—99%, Выход продуктов 97%. Библ. 12 назв., рис. 1.
УДК 546.831.4'442.07
Барий, кальций, стронций метациркоииевокислые. А. Е. Шаммасова, А. И. Вулих, С. А. Кутолин. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 9. .
Метациркоииевокислые барий, кальций, стронций получены с выходом 97% спеканием углекислых солей этих металлов с двуокисью циркония, взятых в стехиометрических количествах, в токе аммиака при 900—950°. Получили стехиометрические соединения состава MeZrO3 . Содержание основного вещества 98—99%. Библ. 12 назв.
УДК 546.832'442.07
Барий и кальций метагафниевокислые. А..Е. Шаммасова, С. А. Ку-толин, А. И. Вулих, Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 11.
Метагафниевокислые барий и кальций получены с выходом 98% спеканием их углекислых солей с двуокисью гафния в стехиометрических количествах в токе аммиака при 900°. Получали стехиометрические соединения состава МеНЮз. Содержание основного вещества 98—99%. Библ. 10 назв.
УДК 546.881.4 : 661.888.1
Ванадий двуокись. Г. Н. Ходалевич, Е. Т. Лабыкина, Л. П. Гевлич, В. И. Берзин. Методы получения химических реактивов и препаратов, вып. 24. М, ИРЕА, 1972, стр. 13.
Двуокись ванадия получена с выходом 96% восстановлением пятиокиси ванадия щавелевой кислотой в токе углекислого газа при 500°. Содержание основного вещества 83%. Библ. 2 назв.
106
УДК 546,881.5:661.888.1
Ванадий пятиокись особой чистоты. Б. И. Короткевич, А. И. Вулих, У. А. Саидахмедов, Д. А. Пахомов, Л. П. Жердиенко, А. А. Аловяйников. Методы получения химических реактивов и прс- паратов, вып. 24. М.., ИРЕА, 1972, стр. 14.
Пятиокись ванадия особой чистоты получена с выходом 90% ее растворерием в едком натре и пропусканием образующегося раствора метаванадата натрия через последовательно соединенные колонки с катионитом КУ-2 в Н-форме и анионитом АВ-17 в \гОз-форме с получением на выходе из колонки золя метаванадиевой кислоты. Приводятся условия синтеза и очистки HVO3 (концентрация исходного раствора — 0,4 г-экв/л V2Os; скорость элюации 0,2—0,4 см/сек и др.), а также условия переработки ее на порошкообразную и гранулированную пятиокись ванадия. Содержание основного вещества 97—98%. Библ. 8 назв.
УДК 546.881.4—ЭГ141
Ванадил бромистый. Г. Н. Ходалевич, Е. Т. Лабыкина, Л. П. Гевлич, В. И. Берзин. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 17.
Бромистый ванадил получен взаимодействием двуокиси ванадия с бромистоводородной кислотой (выход 95%) или сернокислого ванадила с бромистым барием (выход 89%). Полученный раствор бромистого ванадила упаривают, охлаждают и кристаллизуют в экси'-каторе. Хранят в ампулах. Содержание основного вещества 50%. Библ. 2 иазв.
УДК Мб.786—31/32
Вольфрамовая кислота и вольфрамовый ангидрид. У. А. Саидахмедов, А. И. Вулих. И. У. Ризаев Методы получения химических реактивов п препаратов, вып. 24. М., ИРЕА, 1972, стр. 19.
Вольфрамовая кислота получена с выходом 95% из вольфрамовокислого натрия ионообменным способом. Осадок высушивают при 100—110°. Содержание основного вещества — 99%.
Вольфрамовый ангидрид с выходом 98% получен прокаливанием вольфрамовой кислоты при 300—350°. Библ. 11 назв.
107
УДК 546.681'171.4
Галлий азотистый. С. А. Кутолин, А. И. Вулих. А. Е. Шаммасова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 22.
Нитрид галлия получен с выходом 98% азотированием смеси галлия и хлористого аммония. Синтез проводят в горизонтальной реторте из нержавеющей стали при 700° под давлением аммиака 6—7 ати. Молярное отношение азота к галлию в продукте колеблется в интервале от 0,95 до 1,00. Содержание основного вещества 97%. 1 рис., библ. 13 назв.
УДК 546.681.3’226 + 546,682.3'226
Галлий и индий сернокислые безводные. А. А. Яровой, В. Д. За-медянская. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 25.
Сернокислые галлий (I) и индий (II) получены растворением галлия в смеси 30%-ной серной кислоты и перекиси водорода и индия — в 30%-иой серной кислоте. Выделение I и 11 из раствора осуществляют упаркой и прокалкой солей при 350—400°. Выход I 97%; содержание основного вещества 99,5—99,9%. Выход II 97—98%; содержание основного вещества 99%. Библ. 2 назв.
УДК 546.681.3’185 + 546.682.3’185
Галлий и индий фосфорнокислые. А. А. Яровой, Л. М. Петрова, Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972,' стр. 27.
Фосфорнокислые галл.ий и индий получены с выходам 97% растворением металлов в азотной кислоте, добавлением стехиометрического количества фосфорной кислоты и выделением продукта упариванием раствора с последующей прокалкой. Содержание основного вещества в фосфорнокислом галлни 99,6% и в фосфорнокислом индии— 99,5%. Бнбл. 6 назв.
УДК 546.682.3'161
Галлий (III) фтористый трехводиый. Г. Е. Ревзин, Л. М. Петрова. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 29.
Галлий фтористый трехводный получен с выходом 93—96% рарт-ворением при нагревании галлия в смеси 40%-ной фтористоводородной кислоты и 30%-ной перекиси водорода с последующим выделе-' нием из раствора упариванием и сушкой. Содержание основного вещества 98,5%. Библ. 7 назв.
108
УДК 546.289.4’131.07
Германий (IV) хлористый. Г. Е. Ревзин, Л. М. Петрова, А. А. Яровой. Методы получения химических реактивов и препаратов, вып. 24.
М., ИРЕА, 1972, стр. 31.
Хлористый германий (IV) получен с выходом 97% обработкой стеклообразной двуокиси германия концентрированиой соляной кислотой при насыщении реакционной массы газообразным хлористым водородом. Содержание основного вещества 99,8%. Библ. 9 иазв.
УДК 546.683.3’221
Иидий (III) сернистый. Г. Е. Ревзин, А. А. Яровой. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 33.
Сернистый индий (III) получен с выходом 87% действием сероводорода на раствор сернокислого индия с концентрацией иидия 50 г/л при pH 2,0—2,5 и температуре 60—90°. Осадок после фильтрации и промывки от SO/'-иОна высушивают при 95—105° и остаточном давлении 10—20 мм pi. ст. Индий из маточного раствора осаждается в виде 1п(ОН)СОз. Содержание основного вещества 96%. Библ. 6 назв.
УДК 546.682.3’226
Иидий сернокислый кислый семиводный. А. А. Яровой. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 35.
Описан метод синтеза In2(SO4)3 • H2SO4 • 7Н2О с выходом 97% растворением индия в 40%-ной серной кислоте с последующим высаливанием серной кислотой до пл. маточного раствора 1,415—1,417. Содержание основного вещества 98,5—99,5%. Библ. 2 назв.
УДК 546.682.3’264
Иидий углекислый основной. Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 37.
Описан метод получения 1п(ОН)СОз (I) с выходом 97—98% взаимодействием хлористого индия с 5%-ным раствором углекислого кислого нат.рня. Осадок 1 выдерживают 24 часа под раствором, отмывают от хлор-иона дистиллированной водой декантацией, затем отжимают на фильтре и сушат 6—8 часов при 50°. Содержание основного вещества 95%. Библ. 2 иазв.
109
I
УДК 546.682.3’292
Иидий уксуснокислый, основной. Г. Е. Ревзин. Методы получения химических реактивов н препаратов, вып. 24. М., ИРЕА, 1972, стр. 39.
Описан метод получения уксуснокислого основного индия (I) с выходом 94% обработкой углекислого основного индия уксусным ангидридом в среде уксусной кислоты. Смесь упаривают досуха при 90—100° и сушат 2—3 часа при 80 -100°. Содержание основного вещества в продукте Квалификации «ч.» 95%, а «х. ч.» 97,5%. Библ. 2 назв.
УДК 546.812’226 + 546.812(088.8) +546.48’226
Кадмий сернокислый, олово (II) сернокислое и хлористое. Д. А. Пахомов, Т. М. Сосипатров, С. А. Крестовникова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 411.
Описан синтез сернокислого кадмия, сернокислого олова и хлористого олова анодным растворением металлов в соответствующих кислотах в электролизере с аиионитовыми мембранами марки МА-40-2с. Выход по току соответственно — 99, 100 и 86—88%. Режим электролиза при получении SnCl2.2Н2О и 3CdSO4 • 8Н2О— непрерывный, a SnSO< — периодический. Рнс. 1. Библ. 6 назв.
УДК 547.26' 132 + 547.26’ 135 + 547.26’ 136
Калий, рубидий и цезий алкоголяты. Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М., 1972, стр. 46.
Описан метод синтеза алкоголятов калия, рубидия н цезия с выходом 97—98,5% обменной реакцией их фтористых солей с алкого-лятами натрия в.среде абсолютного спирта. Содержание основного вещества 90%. Библ. 5 назв.
УДК 546.863’32’161.07
Калий-сурьма фтористая. Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 49.
Пентафтораитимонат калия получен с выходом 94—98%-взаимо-действнем водного раствора фтористой сурьмы (111) с водно-спиртовым раствором фтористого калия. Содержание основного вещества 99—101 %. Библ. 4 назв.
110
УДК 547.581.2’34
Литий беизойиокислый. Е. М. Шипилова, Т. В. Ревзина, И. А. Ши-шенкова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 51.
Бензойнокислый литий получен с выходом 98% нейтрализацией раствора гидроокиси лития бензойной кислотой до pH 7. Содержание основного вещества 99%. Библ. 2 назв.
УДК 546.766’34
Литий двухромовокислый двуводный. Е. М. Шипилова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 53.
Двухромовокислый литий двуводнын получен с выходом 96% взаимодействием раствора гидрата окиси лития с хромовым ангидридом. После фильтрации раствор упаривают отделяют кристаллы и сушат. Содержание основного вещества 98%. Библ. 4 назв.
УДК 546.185.325+546.34
Литий метафосфорнокислый. Af. Ф. Фокина, Е. М. Шипилова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 55.
Метафосфорнокислый литий получен с выходом 89% термической обработкой однозамещепиого фосфорнокислого лития, последний получают нейтрализацией гидрата окиси лития ортофосфорной кислотой. После очистки и фильтрации раствор упаривают до получения пасты, которую гранулируют, затем прокаливают при 500—550°. Содержание основного вещества 98,5—99%. Библ. 3 назв.
УДК 546.776’34
Литий молибденовокислый. Е. М. Шипилова. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 57.
Молибденовокислый литий получен с выходом 98% взаимодействием раствора гидроокиси лития с молибденовым ангидридом. После фильтрации раствор упаривают досуха. Содержание основного вещества 98%. Библ. 4 назв
111
УДК 546’484.34
Литий селеиистокислый кислый. Е. М Шипилова, Т. В. Ревзина. Методы получения химических реактивов и препаратов, вып. 24. М, ИРЕА, 1972, стр. 59.
Селеиистокиелый кислый литий получен с выходом 95% нейтрализацией гидроокисью лития раствора селенистой кислоты до pH 4—4,5. После фильтрации раствор упа.ривают досуха, промывают спиртом и сушат при 80°. Содержание основного вещества 99%. Библ. 3 назв.
УДК 547.461.2 + 546.34
Литий щавелевокислый. Е. М. Шипилова, Е. С. Скрипченко. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 60.
Щавелевокислый литий получен с выходом 88,6% нейтрализацией раствора гидроокиси лития щавелевой кислотой до pH 6—5. Раствор упаривают до появления большой массы кристаллов и сушат. Содержание основного вещества 99—99,5%. Библ. 4 пазв.
УДК 661.472
Металлы III, IV и VIII групп йодистые безводные. А. А. Яровой, Л. М. Петрова, Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 62.
Описан метод синтеза 19 безводных йодистых солей металлов III, IV и VIII групп, получаемых взаимодействием металла с йодом в вакуумировявных стеклянных или кварцевых ампулах.
Уточнены условия синтеза и сублимации продуктов. Приведены результаты анализов опытных образцов. Библ. 17 назв.
УДК 546.776—31/32
Молибденовая кислота и молибденовый ангидрид. У. А. Саидахмедов, А. И. Вулих, И. У. Ризаев. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 67.
Молибденовая кислота (1) получена с выходам 94—96% из мо- либденовакислого аммония ионообменным способом.
Молибденовый ангидрид получен с выходом 97—99% прокалива-‘ нием I 2 часа при 350—400°. Содержание основного вещества 99%. Библ. 13 назв.
112
УДК 546.742 + 547.295.72
Никель лаурииовокислый. В. Н. Веремей, Г. П. МедвеЬецкЛя, А. В. Страшненко, Р. К. Страшненко. «Метоны получении химических реактивов и препаратов», вып. 24. М., ИРЕА, 1972, стр. 70.
Описал синтез лауриновокислого никеля взаимодействием сернокислого никеля с лаурииовокислым калием в водной среде; выход количественный. Библ. 1 назв.
УДК 546.812’226.07
Олово сернокислое.
чения химических
Б. И. Короткевич, А. И. Вулих. Методы полу-реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 71.
Разработан метод получения сернокислого олова нз хлористого олова или разбавленных растворов других его солей, основанный иа эффективной сорбции олова катионитом из нейтральных или слабокислых растворов (до 2 н. H2SO4) и лепкой десорбируемости его в концентрированных растворах серной кислоты (более 4и.). Выход продукта квалификации «ч.д. а.» составляет 87%, при использовании маточного раствора — не менее 95%. Библ. 30 назв.
УДК 546.35475
Рубидий азотнокислый кислый. С. М. Архипов, А. О. Лесовая, Н. И. Кашина. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 74.
Азотнокислый кислый рубидий получен с выходом 95% смешением азотнокислого рубидия с концентрированной азотйой кислотой при 35—38° и последующей кристаллизацией полученного раствора. Маточный раствор солн нейтрализуют углекислым рубидием, а полученный азотнокислый рубидий вновь возвращается в процесс. Содержание основного вещества 98—99%. Библ. 2 назв.
УДК 546.35’623’161 +546.36’623’161
Рубидий и цезнй гексафторалюмииаты. С. М. Архипов, Т. В. Ревзина, II. И. Кашина. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 76.
Гексафторалюмииаты рубидия н цезия получены с выходом 99% сушкой и прокаливанием смеси фтористого алюминия с фтористыми солями рубидия и цезия, взятых в молярном соотношении 1 : 3. Прокаливание гексафторалюмината рубидия осуществляют при 980—1000°, а гексафторалюмината цезия при 820—840°. Содержание основного вещества 98%. Библ.. 2 назв.
нз
УДК 546.162+'546.35’36
Рубидий и цезий фтористые кислые. С. М. Архипов, Н. И. Кашина, Т. В. Ревзина. Методы получения химических реактивов и препаратов. вып. 24. М„ ИРЕА, 1972, стр. 78.
Фтористый кислый рубидий получен с выходом 98,5% растворением углекислого рубидия во фтористоводородной кислоте. Раствор фильтруют и упаривают досуха гари температуре не выше 100°. Остаток высушивают в вакуум-сушильном шкафу при 50—60'»
Фтористый кислый цезий получен по аналогичной методике с выходом 97,5%. Содержание основного вещества 96%. Библ. 4 назв.
УДК 546.815+ 547.477.1 '
Свинец лимоннокислый трехзамещенный. Н. Г. Чернова, Г. И. Веселовская, М. В. Мосенцева. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 80.
Лимоннокислый свинец трехзамещеппый, получен с выходом 81,5% взаимодействием уксуснокислого свинца с лимоннокислым натрием трехзамещенным. Содержание основного вещества 99%. Библ. 1 назв.
УДК 546.817’824’831,4’882’883
Свииец метатитановокислый, метациркониевокислый, метаниобиево-кислый н метатанталовокислый. А. Е. Шаммасова, С. А, Кутолин, А. И. Вулих. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 82.
Описан метод синтеза метатитановокислого, метаццрконпевокис-лого п метатанталовокислого свинца спеканием углекислого основного свинца с двуокисями титана и циркония и пятиокисями ниобия и тантала, взятых в стехиометрических количествах, в муфельной печи при 850—950° в течение 3—5 часов Выход продуктов составляет 97—98%. Содержание основного вещества 97%. Библ. 15 назв.
УДК 546.221.1 +546.224—31 + 546.264—31
Сероводород, сернистый и углекислый газы. Л. И. Вулих, А. А. Ало-вяйников, Г. А. Никандров, М. К- Загорская. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 85.
Описаны лабораторные методы получения сероводорода, сернистого и углекислого газов перемешиванием раствора соответствующей соли и катионита в Н-форме или анионита в соответствующей солевой форме и кислот. Приводятся данные по соотношениям реагентов, количествам выделяющихся газов и скоростям их выделения. Выход газа 90—95%. Библ. 3 назв
114
УДК 546.683.1’175 + 546.683.1’226
Таллий азотнокислый И сернокислый. У. Р. Кучкаров, У. А. Саидахмедов, А. И. Вулих, Н. У. Ризаев, Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 88.
Описаны ионообменные способы получения азотнокислого и сернокислого таллия на катионите КБ-4-П2. Полученные ионообменным методом растворы солей упаривают и кристаллизуют. Выход составляет ~ 95%. Содержание основного вещества 98—99%. Библ. 4 назв.
УДК 546.244—31.07
Теллур двуокись. Г. Е. Ревзин. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 91.
Описан метод синтеза двуокиси теллура (I) окислением теллура при сплавлении с азотнокислым калием (натрием). Молярное отношение МеМОз : Те=,2-^2,5; температура окисления 400—430°; время процесса— 1—2 часа. Спёк растворяется в 10%-ной едкой щелочи, р-р доводят до pH 3,5—4 азотной кислотой, причем осаждается I. Выход I при окислении теллура азотнокислым калием составляет 97,5%. Содержание основного вещества 99,6—99,9%. Библ. 7 назв.
УДК 546.246 = 325.04.07
Теллуровая кислота. Г. Е. Ревзин, А. И. Вулих, Л. П. Жердиенко. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 93.
Теллуровая кислота с выходом 93% получена окислением суспензии двуокиси теллура перекисью водорода в присутствии углекислого или углекислого кислого калия или натрия с последующим ионообменным превращением теллуровокислого калия на ионите КУ-2. Содержание основного вещества 99,7—99,9%. Библ. 6 назв.
УДК 546.36’175
Цезий азотнокислый. А. И. Вулих, Л. Г. Сидорова, А. О. Лесовая. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 95.
Азотнокислый цезий получен с выходом 98—99,5% нейтрализа-\цией углекислого цезия азотной кислотой. Полученный раствор очищают сероводородом, затем его упаривают, кристаллизуют и сушат. Содержание основного вещества 98—99,5%. Библ. 38 иазв.
115
УДК 546.36’175
Цезий азотнокислый кислый. С. М. Архипов, А. О. Лесовая, Н. И. Кашина. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 98.
Азотнокислый кислый цезий с выходом 97% пол уч ед—растворением при 60—65° азотнокислого цезия в азотной кислоте с последующим охлаждением раствора до 10°. Кристаллы продукта отфильтровывают, а маточник после нейтрализации углекислым цезием возвращают вновь в процесс. Содержание основного вещества 98—99%. Библ. 3 назв.
УДК 546.36 +547.291
Цезий муравьинокислый. Е. М. Шипилова, Л. П. Жердиенко, Б. Н. Пустовалов. Методы получения химических реактивов и препаратов, вып. 24. М„ ИРЕА, 1972, стр. 100.
Муравьинокислый цезий с выходом 90% получен нейтрализацией углекислого цезия до pH 7—6,5. Раствор упаривают досуха. Сушат в вакуум-сушнльном шкафу при 50° в течение ~ 15 часов. Содержание основного вещества 98%. Библ. 3 назв.
УДК 546,655.4’226 + 546.171.1’226
Церий (1У)-аммоний сернокислый. С, М. Архипов, Л. М. Отсолиг. Методы получения химических реактивов и препаратов, вып. 24. М., ИРЕА, 1972, стр. 102.
Церий-аммоинй сернокислый [Ce(SO4)2.2(NH4)2SO4 • 2НзО] получен с выходом 90% упариванием растворов сернокислого церия, сернокислого аммония н серной кислоты при 70° до выделения основной массы кристаллов с последующим охлаждением до 20—25°. Кристаллы отделяют от маточника, промывают спиртом и сушат. Содержание основного вещества 98—<100%. Библ. 2 назв.