



















































































































































































Текст
всесоюзный ордена трудового красного знамени
НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ (И Р Е А)
МЕТОДЫ ПОЛУЧЕНИЯ ХИМИЧЕСКИХ РЕАКТИВОВ И ПРЕПАРАТОВ
Выпуск 26
МОСКВА- 1974
СОДЕРЖАЛ ИЕ
Редакционная коллегия
Е. А. Божевольнов, А. В. Бромберг, В. Г. Брудзь,
В. М. Дзиомко, Р. П. Ластовский (гл. редактор), А. М. Лукин, Ю. П. Решетников, Б. Д. Степин,
В. Я. Темкина
Редактор С. С. Кузьмина
Корректоры: И. Р. Казарина и Л. А. Климанова Техн. ред. Е. Н. Данилова
Сдано в набор 18/Х—73 г. Подписано к печати 22/11-74 Л—52349J
Формат бумаги 60X90’/i6 Объем 22 печ. л. Уч.-изд. л. 14,5] Зак. 1164 Тираж 1000 экз. Цена 1 р. 36 коп.
Типография Военной Краснознаменной академии химической защиты •имени МАРШАЛА СОВЕТСКОГО СОЮЗА С. К. ТИМОШЕНКО
Алифатические азосоединения. В. С. Стопский, 3. И. Сергеева, Б. В. Иоффе..................................................... 9
Алкилтиоэтилакрилаты и алкилтиоэтилметакрилаты. М. А. Коршунов, Р. Г. Кузовлева, И. В. Фураева.............................12
Амиды и метиламиды у-оксикарбоновых кислот. В. А. Седавкина 15
5-Амил-2-пирролидои. В. А. Седавкина...............................19
9-Аминоакридин. И. И. Дыханов. Т. В. Перова, Р. Ф. Виденина, И. В. Покотыло, В. И. Базакин...................................23
о Амино Р,Р-ди(З иидолил)этилбензол. А. О. Гинзбург, С. В. Лылык, А. К. Шейнкман..................................................27
2-(2-Амино-5-нитробензамидо)бензойная кислота. И. С. Маркович, Н. А. Филягина, В, М. Дзиомко...................................29
8-Аминохинальдин. В. М. Дзиомко, И. А. Красавин, Б. В. Парусников, Ю. П. Радин................................................31
а-[(2-Аминоциклопеитил)метил]фуран. | А. А. Пономарев, I А. П. Кривенько .........................................................34
N-Арилпиридиниевые соли. Л. Г. Гах, А. К- Шейнкман, С. И. Баранов ............................................................36
1-Ацетиламино-2-ацетокси-3,5-диметилбеизол. В. М. Островская. М. М. Краснова, Л. В. Ломакина..................................40
2-Ацетиламинофеноксиуксусная кислота, В. М. Островская, М. М. Краснова, О. Т. Лушина ...................................42
2-Ацетиламинофенол. В. М. Островская, М. М. Краснова .... 44
К-Ацетил-N фурфуриламиноэтанол и 3- (N-Ацетил 5-метил-2-пирроли-дил) пропанол. |Л. А. Пономарев, | И. А. Маркушина, Т. И. Губина, М. В. Норицина............................................46
4-н-Ацилоксибензальдегиды. 3. С. Сиденко, В. А. Иншакова, Г. С. Чижова....................................................49
S-Бензилнзотиомочевипа гидрохлорид. Е. А. Бугай....................51
Бензиловый эфир бензойной кислоты. А. Л. Лифиц, Е. П. Агеев. С. И. Орел, И. А. Бару..........................................53
8-Бензоил амино-1-нафтол-3,6-дисульфокислота, двунатриевая соль.
Н. Н. Дыханов, Р. Ф. Виденина, И. В. Покотыло, Т. В. Перова, В. И. Базакин, П. Д. Якухный....................................55
3
N-Бензоил-о-толундин. Ю. Г. Бондарь...................................°8
Бис(п-нитрофенплгидразон) а,р-дикетобутиролактона. И. В. Хвостов 60 л-Бис(л-феноксифенокси) бензол. Н. Ю. Аронская, Е. И. Маховер,
Р. М. Гелыитейн, В. Д. Безуглый...................................63
7-Бром-1 -ацетил-5-феиил-1,2-дигидро-ЗН-1,4-бенздиазепип-2-он. 3. И.
Жилина, А. В. Богатский, С. .4. Андронати, Т. К. Чумаченко 66
4-Бром-3-метил-5-(5-бром-2-фурил) пиразол. | А. А. Пономарев, |
Л. В. Черкесова, А. А. Морщакова.................................68 •
а-Бромтетроновая кислота. И. В. Хвостов, О. А. Логинова ... 70
N-Бутилхинальдиний иодид. И. С. Маркович, Л. И. Блохина.
В. М. Дзиомко.......................................................
2,4-Гексадиеналь. О. С. Степанова, А, И. Галатина ..... 74
а-Гексилкротоновая кислота. Г. Ф. Танцюра, А. В. Богатский . 76
4-и-Гексоксибензилхлорид. 3. С. Сиденко, В. А. Иншакова, Г. С. Чижова ...........................................................79
1,4-Днацетилбензол. Е. И. Маховер..................................81
Диацетоловый эфир метионовой кислоты. А. Л. Фридман, Г. С. Исмагилова .......................................................83
Дибензоилметан. Н. Ю. Аронская, Р. М. Гельштейн, Е. А4. Иофиг 85
1,4-Дибром-2-метокси- и 1,4-дибром-2-этоксипентаны. А. В. Богатский, Г. Л. Камалов, Н. Г. Лукьяненко, В. И. Шарыгин .... 87
3,4-Дибутоксианилин. В. М. Дзиомко, А. В. Иващенко .... 90
4-N.N-(Дикарбоксиметил) аминометил-З-окси-2-нафтойная кислота.
Л. М. Тимакова, В. Я. Темкина, Г. Ф. Ярошенко, Н. Е. Хавченко 93 1,Г-Диметил-3,3'-Дн(тетрагидрофурил-2)дипропилоксид. М. Д. Липа нова...............................................................95)
Диметиловый эфир диэтиленгликоля. А. Л. Лифиц, А. А. Вейцмач 97’ цис- и траис-2,3-Диметнлоксетаны. Г. 4. Филип, С. А. Петраш, Г. В. Пьянкова.................................................99*
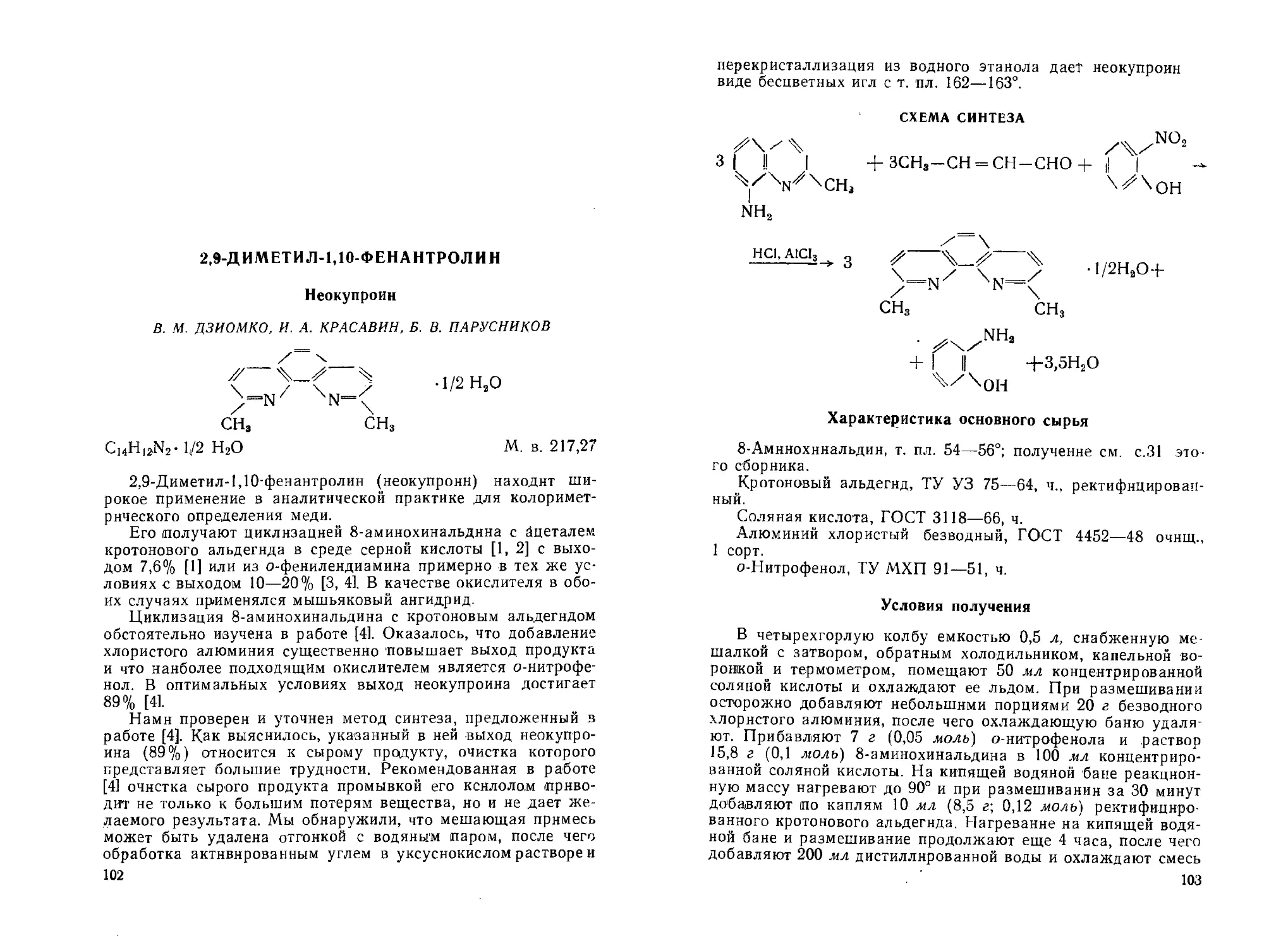
2,9-Диметил-1,10-фенантролин. В. М. Дзиомко, И. А. Красавин, Б. В. Парусников..................................................
М,Г4/-Диметилэтилеидиамино-Г4,Г'Г-бис (метиленфосфоновая
и Ь(.№'-ди(2-оксиэтил)этнлендиамино-М,Ь1/-бис(метилепфосфоио-вая кислоты). В. П. Мархаева, Ю. М. Поликарпов, М. В, Ру домино, Т. Я. Медведь...........................................
2,7-Диметокси-1,6-диоксаспнро[4,4]ионан. И. А. Маркушина, И. В. Шу-ляковская ........................................................
Дииитрил 1,10-фенаитролин-2,9-дикарбоновой кислоты. В. М. Дзиомко, Б. В. Парусников............................................
Диоксанат бис(трииитрометил) ртути. А. Л. Фридман, Т. Н. Ившина 2,6-Диоксибензилиминодиуксусная кислота. В. Я. Темкина, Н. В. Ци-рульникова, Л. Д. Меликсетян, Р. П. Ластовский . . . .
2,3-Диоксибензилиминодиуксусная кислота, мононатриевая соль.
В. Я. Темкина, И. В. Цирульникова, С. 4. Тарасова, Р. П. Ластовский ......................................................
М,1Ч'-Ди(2-окси-5-метилбензил)этилендиамино-М,Мл-диуксусная кислота. И. В. Цирульникова. В. Я. Темкина, Р. П. Ластовский 4,4/-Ди-н-пентоксназоксибензол. 3. С. Сиденко, В. А. Пятакова, Г. 'С. Чижова.....................................................
2,6-Дихлор-4-нитроаиилии. Р. Ф. Виденина, Н. Н. Дыханов .
2,6-Дихлор-1,4-феиилендиамнн. Р. Ф. Виденина. Н. И. Дыханов
102
10^ 10»
"I
116,
11<
12С|
1251 12?
126|
Диэтилентриамино-\>М,\!',Т'1/,\"- пента (метиленфосфоновая) кислот а.
М. В. Рудомино, Е. К. Колова, А. М. Орлов, Л. И. Мизрах, Т. Я. Медведь.................................................128
Диэтилфурфурилиденмалонат и этилфурфурилиденацетоацетат. Л. Г. Богатская, Г. Л. Камалов...................................130
D,L-Изолейцин. Е. П. Крысин, И. И. Громова, Л. Г. Андронова 132
Изомерные метил- и метоксибензолсульфохлориды. И. Н. Дыханов, А. Б. Джиджелава, Т. С. Рыжкова..................................134
Изо- (4,7,8,9-тетрагидро) бензо- 1,3-диметокситетрагидрофуран. | А. А.
Пономарев, | И. А. Маркушина, Г. Е. Мариничева .... 137
,и-Иоданилин. Г. А. Креймер, Е. А. Бугай..........................139
Кальцийацетоуксусный эфир. А. Е. Кожухова, М. В. Гренадерова, Т. К- Чумаченко, Л. Г. Деркач....................................141
5-(2-Карбоксиэтил)-1,2-дигидропирролизин. | А. А. Пономарев, |
Л. Н. Астахова, В. И. Волколупов . . i . 143
Комплекс серебряной соли тринитрометана с диоксаном. А. Л. Фридман, Т. Н. Ившина................................................145
Конифериловый альдегид. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева................................147
Конифериловый спирт. А. В. Богатский, 3. Д. Богатская, Л. В. Басалаева, Н. С. Уланова-Обезюк....................................150
Метакрилоилхлорид. И. Н. Дыханов, А. Б. Джиджелава .... 153
Метилбензилкетон. Г. Ф. Танцюра, Л. Я. Глинская, И. Ф. Герасименко ............................................................ J55
2-Метилбутаналь. Е. П Крысин, И. Н. Громова, Э. Д. Глинка, А. Г. Левина, Л. Г. Андронова....................................157
1-(3-Метил-1,2-дигидропнрролизил-5) -1-бутен-З-он. | ~А. А Пономарев, I Л. Н. Астахова........................................... 159
.Летилизопропилкетон. А. В. Богатский, Г. Л. Камалов .... 161
Метиловый эфир а-метил-р-меркаптопропиоиовон кислоты. А. В. Бо гатский, А. М. Турянская, А. И. Грень............................163
Метиловый эфир р-(5-метил-2-пирролидил) пропионовой кислоты.
| А. А. Пономарев, | М. В. Норицина, А. П. Кривенько . . 165
Метиловый эфир феруловой кислоты. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева, Н. С. Уланова-Обезюк ............................................................ 168
₽-(5-Метил-2-пнрролидил)пропионогидразид. | А. А. Пономарев, | М. В. Норицина...........................". ; ; . 171
I-Метил-5-пропил-2-пнрролидон. В. А. Седавкина....................173
<»-[!-Метил-3-(а-тетрагидрофурил)пропил]тетрагидрофуроат. М. Д.
Липанова......................................................176
Метилфеноксималоновый эфир. О. С. Степанова, А. С. Яворский, М. И. Побережная, Л. Б. Хагундокова..............................179
5
4
3-[2- (5-Метнлфурфурнлоксн)этокси]пропнламнн. j А. А. Пономарев, | Н. П. Масленникова, Г. В. Беспалова....................
3-^-Метилциклопентано(в)-2-пирролидил)пропилбензоат. | А. А. Пономарев,\ А. П. Кривенько, М. В. Но рицина.......................
2-Метоксн-М-ацетил-1,6-днокса-9-азаспнро[4,5]декан. | А. А. Пономарев, | И. А. Маркушина, Т. И. Губина.............................
5[2-(Метоксикарбоннл)-этнл]-1,2-днгидропирролизни. |/1. А, Понома-рев, | Л. Н. Астахова, В. И. Волколупов
(о-Метокснфеннл)оксиметилндендифосфоновая кислота, натриевая и свинцовая соли. И. Д. Колпакова, Л. В. Криницкая .
2-Метоксн-М-этил-1,6-диокса-9-азаспиро|'4,5]декан. | А. А. Пономарев, | И. А. Маркушина, Т. И. Губина..........................
Мопоалкнлгидразины. В. С. Стопский, 3. И. Сергеева, Б. В. Иоффе
2-М-Морфолил-2-оксо-5-метил-5-метоксиметил-1,3,2-дНоксафосфори-иан. А. В. Богатский, А. А. Колесник, Т. Д. Бутова ....
2-Нафтол-3,6-днсульфокнслота, двунатриевая соль. Н. П. Дыханов, Р. Ф. Виденина, И. В. Покотыло, Т. В. Перова, В. И. Базакин, П. Д. Якухный.................................................
2-Нитро-4,5-дибутокснанилин. В. М. Дзиомко, А. В. Иващенко .
5-(5-Питротенилиден) роданин. | А. А. Пономарев, | 3. В. Тиль . .
5-Нитрофенол (2-азо-Г) -2'- (0-пропноннлгидразино) нафталин и 5-нит-рофеиол(2-азо-Г)-2'-(fJ-бутирнлгидразнно)нафталин. К. А. Дуна веская, К- Е. Калинина, В. М. Дзиомко.........................
8-Нитрохинальднн. В. М. Дзиомко, И. А. Красавин, Б. В. Парусников, К). П. Радин.............................................
у-Оксикарбоновые кислоты, натриевые соли. В. А. Седавкина, А. Г. Басина . -.................................................
182
184
187
190
192
196
198
201
203
206
209
211
215
218
у-Оксипимелнновая кислота, динатрневая соль. В. А. Седавкина, А. Г. Басина..................................................... 222
М-(2-Оксиэтил)диэтилентрнамин. Л. М. Тимакова, В. Я. Темкина, Г. Ф. Ярошенко, Р. П. Ластовский............................., 224
N- (2-Окснэтнл) днэтнлентриам''ино-М,М'’Кл’,М//-тетраукСусиая кислота.
Л. М. Тимакова, В. Я. Темкина, Г. Ф. Ярошенко, Р. П. Ластовский .............................................................226
(1-Окснэтнлиден)дифосфонат серебра. И. Д. Колпакова, Л. В. Криницкая ......................................................... 228
(1-Окснэтилиден)дифосфоновая кислота, солн с аминами. И. Д.-Кол-пакова, Л. В. Криницкая, Е. И. Миронова.......................230
(1-Оксиэтилиден)дифосфоновая кислота, трннатриевая соль. Б. И.
Бихман, Е. М. Уринович, Т. А. Богомолова, Н. М. Дятлова 232
Ы-(2-Окснэтил) сукцинимид и N-(3-оксипропил) сукцинимид. В. М. Ост-
ровская, И. А. Шуйская, Г. В. Фомина, Л. В. Ломакина . 234
N- (2-Оксиэтнл)фталимид и М-(3-оксипропил)фталимнд. В. М. Островская, И. А. Шуйская, Т. В. Фомина, Л. В. Ломакина . . 237
у-Оксо-N- (5-гекснл-2-пирролидон) масляная кислота. В. А. Седавки на, Н. А. Морозова..............................................240
Пентафторазидобензол. А. В. Кашкин, Ю Л. Бахмутов, Н. Н. Чайникова .........................................................242
(2-Пнридилметил)имнноднуксусная кислота. Н. В. Цирульникова, В. Я. Темкина, Л. П. Разборская.................................244
р-(2-Пнрролидил) пропионовая кислота. | А. А. Пономарев, |
М. В. Норицина, А. П. Кривенько . . ~ . 246
З-Пнрролнзндон. I А. А. Пономарев, j М. В. Норицина, А. П. Кривенько .........................................................249
5-Пропил-1Ч-(Р-оксиэтил)-2-пирролидон. В. А. Седавкина, Н. А. Морозова .........................................................252
5-Пропнл-2-тнопирролидон и 1-метнл-5-пропнл-2-тиопирролидон.
В. А. Седавкина, Г. В. Беспалова...............................255
ц-транс-у-цис-Р-Стирнлакриловая кислота. Нгуен Ван Тонг, О. С.
Степанова, А. И. Галатина......................................258
.\,М,М',М'-Тетракнс(карбоксиметнл-5,5'-метиленднантраниловая кислота. Н. В. Цирульникова, Ю. Е. Дорошенко, Н. К, Харитонова, И. В. Хвостов...................................................262
Тетраэтил-1-окснэтилидендифосфонат. И. Д. Колпакова, Л. В. Криницкая, Е. И. Миронова.............................................265
Тлосемикарбазои 0-(5-хлор-2-фурил) акролеина. В. П. Керенцева, М. Д, Липанова..................................................268
Тримеллнтовая кислота. Г. А. Креймер..............................270
О,Ь-Триптофаи. Б. М. Котляревская. И. И. Губенко...................273
Трихлорангидрид тримеллнтовон кислоты. Я. А. Середницкий,
В. Н. Федына...................................................277
2,4,6-Трихлоранилин. Н. Н. Дыханов, Р. Ф. Виденина, Т. В. Перова, И. В. Покотыло, В. И. Базакин...................................280
Трнэтилфосфат. А. Л. Лифиц, Л. Я. Дистанова, А. А. Вейцман . 283
1,Ю-Феиантролин-2,9-днальдегнд. В. М. Дзиомко, Б. В. Парусников 285 1,10-Фенантролин-2,9-диальдокснм. В. М. Дзиомко, Б. В. Парусников 287 1,Ю-Фенантролин-2,9-днкарбоновая кислота. В. М. Дзиомко, Б. В.
Парусников.................................................... 289
2-Фенил-2-пропантиол. М. А. Коршунов, В. Е. Мазаев .... 291
Феруловая кислота. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева, Н. С. Уланова-Обезюк...................293
3-(а-Фурил)бутанал и 3-(а, а'-фурил)дибутанал. И. В. Антипова, И. М. Скворцов................................................ 295
1-(а-Фурил)-4-нитро-3-бутанол. И. А. Маркушина, Т. И. Губина . . 297
2-Хлорметилеипнрнднн. Н. В. Цирульникова, В. Я. Темкина, Л. Я-Шиленко, В. И. Сомова...........................................299
₽-Хлорпропноновая кислота. Л. Я. Глинская. 3. Д. Богатская, Г. Ф, Танцюра................................................. 301
7
6
7-Хлор-5-фенил-3-бензилидеи-1,2-дигидро-ЗН-1,4-бенздиазеш1Н-2-он.
А. В. Богатский, Т. К. Чумаченко, 3. И. Жилина, С. А. Андро-
нати........................................................303
п-Этилацетофенои. Е. И. Маховер.................................305
а-Этил-р-гексоксиметнлпропиоиоваи кислота. Е. А. Яценко, А. И.
Дроздовская............................................... 307
Этилеидиамиидиацетаты меди, кобальта, никеля. Е. М. Уринович, Н. М. Дятлова................................................309
5-Этил-5-феиоксибарбитуровая и 5-этил-5-феиокситиобарбитуровая 1
кислоты. О. С. Степанова, А. С. Яворский, М. И. Побережная, В. Н. Ганевич................................................312
Этилфеноксималоноваи кислота. О. С. Степанова, А. С. Яворский, М. И. Побережная, В. Н. Ганевич..........................314
Предметный указатель ...........................................316
АЛИФАТИЧЕСКИЕ АЗОСОЕДИНЕНИЯ
В. С. СТОПСКИЙ, 3. И. СЕРГЕЕВА, Б. В. ИОФФЕ r-n=n-r;
где R=C2H6— ; С3Н7—
R'=C2H5— ; С,Н7 — ; изо-С3Н,—
Важнейшим методом синтеза алифатических азосоединений до недавнего времени являлось окисление 1,2-диалкилгид-разинов [1, 2]. Исходные гидразины получали восстановлением азинов, ацил- и моноалкилгидразонов. Нами был предложен способ получения алифатических азосоединений изомеризацией моноалкилгидразонов альдегидов или кетонов с выходом 40—70% путем перегонки последних над твердой ще лочью [3]. Указанный метод исключает стадии восстановления гидразона в 1,2-диалкилгидразин и последующего окисления в азосоединение.
Подбирая оптимальные условия для синтеза азосоединений, мы изменили методику, используя для изомеризации в азосоединение не дважды перегнанный моноалкилгидразон, а неочищенный продукт конденсации моноалкилгидразина с альдегидом или кетоном; перегонка моноалкилгидразона проводится со щелочным раствором этиленгликоля, что позволяет значительно сократить продолжительность синтеза.
СХЕМА СИНТЕЗА
/R' r-nh~nh2 + о=с( — \ — П2О
R'
-> R—NH-N=C^ -> R-N = N-CH<’
9
где R=C2Hg— ; С3Н7 — ; «зо-С3Н7 —
R'—Н ; СН3-
R"=CH,- ; ^Не-
характеристика основного сырья
н-Пропилгидразин, т. кип. 121 —123°; см. с. 199 этого сборника.
Пропионовый альдегид, ТУ МХП 2682—51, ч.
Ацетон, ГОСТ 2603—63, ч. д. а.
ЭГиленгликоль, ГОСТ 10164—62, ч. д. а.
Этиловый эфир, ГОСТ 6265—52, ч.
Кали едкое гранулированное, 4203—65, ч. д. а.
Калий углекислый порошкообразный, 4221—65, ч. д. а.
Условия получения
Синтез 1,Г-азопропана. В колбу емкостью 250 мл, снабженную термометром, капельной воронкой, мешалкой и обратным холодильником, помещают 38,0 г (0,5 моль) н-пропилгидра-зина и 50 мл эфира и за 1 час при перемешивании прибавляют 29,0 г (0,5 моль) пропионового альдегида. Температуру реакционной смеси с помощью ледяной бани поддерживают в интервале 0—5° (см. примечание 1). Затем к смеси добавляют 4—-5 г едкого кали и отделяют отслоившийся водный слои. Эфирный раствор сушат в течение суток гранулированным едким кали. Эфир отгоняют, а к остатку (56 г, п?£ = 1,447— 1,449) прибавляют 40—50 мл 10—15%-ного раствора едкого кали в этиленгликоле и образующуюся смесь перегоняют на ректификационной колонке эффективностью 15 т. т. За 2 часа отгоняют 40,4 г азопропана с т. кип. НО—113°. Полученную фракцию для удаления следов воды встряхивают с порошкообразным поташом (~ 1 е), а затем перегоняют па ректификационной колонке.
Выход 1,1'-азопропана равен 34,2 (60%), т. кип. 113,0— 113,57761 мм, 1,4065, d4M = 0,7729.
По литературным данным [3], т. кип. 112,2—112,77760 мм, = 1,4065, tZ420 = 0,7730.
В условиях, аналогичных синтезу 1,Г-азопропана, были получены:
Азоэтан из этилгидразина и ацетальдегида, выход 67%, т. кип. 58,8—58,97760 мм, п™ = 1,3852, d420 = 0,7488. По литературным данным [3], т. кип. 58°, п^ = 1,3852.
1-(Этилазо)пропан из пропилгидразина и ацетальдегида, выход 58%, т. кип. 87,5—87,77765 мм, п™ = 1,3969, </420 = 10
= 0,7631. По литературным данным [3], т. кип. 86,4—86,9°/ 745 мм, п® = 1,3969, d?° == 0,7627.
2-(Этилазо)пропан из изопропилгидразина и ацетальдегида, выход 56%, т. кип. 75,5—76,17760 мм, п™ = 1,3887; ^420 = 0,7469. По литературным данным [3], т. кип. 75,57765 мм, п% = 1,3887, dt20 = 0,7468.
Получение 2-(пропилазо)пропана. В прибор, описанный в предыдущем опыте, помещают 38,0 г (0,5 моль) н-пропилгид-разина и при перемешивании за 1 час прибавляют по каплям 29,0 г (0,5 моль) ацетона с такой скоростью, чтобы температура реакционной смеси не поднималась выше 50°. Водный слой, отделяющийся после добавления едкого кали (~3 г), отбрасывают. Полученный гидразон (55,5 г, = 1,443— 1,447) сушат в течение суток гранулированным едким кали, а затем перегоняют со щелочным раствором этиленгликоля в условиях описанного выше опыта. За 8 часов (см. примечание 2) отгоняют 43,0 г вещества, собирая фракцию с т. кип. 95—102°, затем сушат поташом и перегоняют на ректификационной колонке.
Выход 2-(пропилазо)пропана равен 30,8 г (54%), т. кип. 100,5—100,87747 мм, п2° = 1,3987, d420 = 0,7596.
По литературным данным [3], т. кип. 101,8—102,37773 мм, п2° = 1,3987, dt20 = 0,7596.
Примечания:
1. Проведение конденсации моноалкилгндразина с альдегидом при комнатной или более высокой температуре, а также без растворителя приводит к уменьшению выхода гидразона.
2. Изомеризация моноалкилгидразонов кетоиов протекает в несколько раз дольше, чем у моноалкилгидразонов альдегидов.
ЛИТЕРАТУРА
1. Е. Muller. Allphatische Azo-und Azoxyverbindungen (Houben-Weil). Methoden der organischen chemie, 4 Aufl., X, 2, 757 (1967).
2. 4. Дж. Овербе prep, Ж. П. Ансел м, Дж. Г. Ломбардино Органические соединения со связями азот—азот. Л„ «Химия», 1970, с. 41.
3. Б. В. Иоффе, 3. И. Сергеева, В. С. Стопский. Авт. свид., № 174188; Бюлл. изобр., 17 (1965); Докл. АН СССР, 167, 831 (1966).
АЛКИЛТИОЭТИЛАКРИЛАТЫ И АЛКИЛТИОЭТИЛМЕТАКРИЛАТЫ
М. А. КОРШУНОВ, Р. Г. КУЗОВЛЕВА, И. В. ФУРАЕВА
CH2=CH-COOCH2CH2SCH3
CeH10OaS М. в. 146,22
CH2=C~COOCH2CH2SC2H5
сн8
C8H14OaS М. в. 174,28
СН2=С—COOCH2CH2SCeH13
сн3
C12H22O2S М. в. 230,37
Алкилтиоэтил акрилаты и алкилтиоэтилметакрилаты являются мономерами, содержащими в своем составе сульфидную серу. Введение серы позволяет получать полимеры с новыми свойствами: увеличивает их термостойкость, снижает температуру стеклования, приводит к меньшей набухаемости в органических средах [1].
Известен способ получения алкилтиоэтйлакрилатов, состоящий во взаимодействии хлорангидрида акриловой кислоты с алкилтиоэтанолами в присутствии триэтиламина в бензоле [2]. Недостатком этого способа является применение большого количества бензола и малодоступного хлорангидрида акриловой кислоты.
Нами разработан [3] новый способ получения алкилтио этилакрилатов и алкилтиоэтилметакрилатов, основанный нг переэтерификации метилакрилата или метилметакрилата алкилтиоэтанолами в присутствии алкоголятов натрия и титана в качестве катализаторов и фентиазина в качестве ингибитора полимеризации. Выход продуктов составляет 80—90%.
12
СХЕМА СИНТЕЗА
СН2=СНСООСН3 + CH3SCH2CH,OH
CH2=CHCOOCH2CHaSCH8 + снаон сн2=с-СООСНз + rsch2ch2oh
I
CHS
ch,=c-cooch2ch2sr + сн3он,
СНз
1де R==C2Hs— ; трет
Характеристика основного сырья
Метилакрилат, МРТУ 6—01—345—69, перегнанный.
Метилметакрилат, ТУ 2274—53, ч., перегнанный.
Метилтиоэтанол, т. кип. 81°/18 мм, = 1,4929; получение см. [2].
Этилтиоэтанол, т. кип. 58—59°/6 мм, n2g= 1,4860; получение см. [2].
грег-Гексилтиоэтанол, т. кип. 113°/1 мм, п™ = 1,4795 (см. примечание 1).
Тетрабутилат титана, МРТУ 6—09—2866—66, ч.
Метилат натрия, готовят растворением 1 а натрия (ГОСТ 3273—63) в 10 мл сухого метанола (ГОСТ 2222—65).
Фентиазин, ТУ 3288—52, ч., перегнанный.
Условия получения
Синтез метилтиоэтилакрилата. В трехгорлую колбу емкостью 150 мл, снабженную термометром, пробкой из мягкой резины для ввода катализатора и ректификационной колонкой (см. примечание 2), помещают 23 г (0,25 моль) метилтиоэтанола, 43 г (0,5 моль) метилакрилата и 0,2 г фентиазина. От смеси отгоняют около 3 мл метилакрилата для удаления возможных примесей влаги, затем вводят с помощью шприца через пробку из мягкой резины 3 мл тетрабутилата титана. Смесь кипятят, отгоняя образующийся в реакции метанол в смеси с метилакрилатом при температуре паров 62—63°. После прекращения образования метанола отгоняют при пониженном давлении избыточный метилакрилат, а затем остаток, собирая фракцию с т. кип. 62°/3 мм.
13
Выход метилтиоэтилакрилата равен 31,9 г (87,6%), dt2n = = 1,0584, п™ = 1,4812.
Найдено, %: С—49,49; Н—6,82; S—21,63; MR D = 39,24. C6H10O2S. Вычислено, %: С—49,38; Н—6,85; S—21,91; MR D = 39,09.
По литературным данным [2], т. кип. 6074 мм, = = 1,4790.
По аналогичной методике получают этилтиоэтнлметакрилат из 26,5 г (0,25 моль) этилтиоэтанола, 50 г (0,5 моль) метилметакрилата и 0,2 г фентиазина, вводя с помощью шприцг 0,4 мл метилата натрия, а затем через каждые 15 минут еще по 0,15 мл (всего 1 мл катализатора). Одновременно отбирают образующийся в реакции метанол 'В смеси с метилметакрилатом при температуре паров 64—66°.
Выход этилтиоэтилметакрилата равен 38 г (87,5%), т. кип. 73—7471,5 мм, tZ420 = 1,0166, /$= 1, 4777.
Найдено, %: S—18,50; MRD = 48,33.
CsHuOaS. Вычислено, %: S—18,38; MRD = 48,42.
По литературным данным [4], т. кип. 94—99711,5—14,5 мм.
По аналогичной методике из 40,76 г (0,25 моль) трет-гек-силтиоэтанола, 50 г (0,5 моль) метилметакрилата, 0,2 г фентиазина и 3,5 мл тетрабутилата титана получают 46,8 г (80,7%) трет-гексилтиоэтилметакрилата с т. кип. 120—121°/ 1 мм, = 0,9676, /г2,0 = 1,4765.
Найдено, %: С—62,95; Н—9,76; S—13,93; MR D= 67,07. Ci2H22O2S. Вычислено, %: С—62,61; Н—9,57; S—13,91; MR D = 66,80.
Примечания:
1. трет-Гексилтиоэтанол получен из трет-гексилмеркаптана [5] (т. кип. 119—122°, по= 1,4420) и зтилеихлоргидрина (МРТУ 6—09—4878—67) в присутствии водной щелочи (натр едкий, ГОСТ 4328—66, ч.).
2. Колонка представляет собой трубку из молибденового стекла (300X15 мм), заполненную насадкой, из многовнтковых спиралей размером 3X3 мм из нихромовой проволоки диаметром 0,2—0,3 мм. Колонка снабжена регулируемым электрообогревом и рубашкой для теплоизоляции. К головке колонки присоединен приемник для сбора смеси метанола с метилакрилатом.
ЛИТЕРАТУРА
1. Разводовский, Гусев. Хим. и технол. полимеров, 8, 43 (1965).
2. R. М. Me. Curdy, J. Н. Prager. J. Polymer. Set., А. 2, 1183
3. М. А. Коршунов, Р. Г. Кузовлева, И. В. Ф у р а е в а. Авг свид., 232248; Открытия, изобретения, промобразцы и товарные знаки, № 1 (1969).
4. S. Hashimoto, Т. Furukawa. J. Chem. High. Polym., 27, 110—115 (1970).
5. M. А. Коршунов, В. А. Бухарева, И. В. Ф у р а е в а, Л. В. Хохлова. Химия сераорганических соединений, содержащихся в нефтях и нефтепродуктах, VIII, 5 (1968).
14
АМИДЫ И МЕТИЛАМИДЫ у-ОКСИКАРБОНОВЫХ КИСЛОТ
В. А. СЕДАВКИНА
r-ch-ch2-ch2-
он
//°
R'-CH—СН2-СН2-С"
| 'NHCH;
ОН
где R=C,H,-; С4На-; С6НП-; изо-С8Нч-; С6Н18-R'=C3H, —; С4Н9—; С8НП-
Амиды и N-замещенные амиды у-оксикарбоновых кислот находят разнообразное применение. Некоторые из них обнаруживают высокую физиологическую активность {1], другие применяются в качестве загустителей для стабилизации композиций на основе высыхающих масел [2], коррозионных ингибиторов для жидких углеводородов при их контакте со сталью [3].
В литературе описаны различные методы синтеза амидов и N-алкиламидов у-оксикарбоновых кислот из аммиака или аминов и лактонов, из у-аминокарбоновых кислот, из амидов уталоидкарбоновых кислот, из амидов у-кетокарбоновых кислот [4]. '
Нами разработан метод получения амидов и метиламидов у-оксикарбоновых кислот гидрированием этиловых эфиров у-кетокарбоновых кислот с последующим насыщением растворов эфиров у-оксикарбоновых кислот аммиаком или метиламином. Выходы амидов достигают 90%. Все описанные амиды получены нами впервые.
15
СХЕМА СИНТЕЗА
Н,
NiR
II 0С2н6
о
^0 г-и сп „гы ____о'/
R'NH2
I XOC2H6
OH
уэ
-* R-CH-CH2-CHa-C< | 4NHR
OH
где R=C8H7—; C4Hg-~; С5Нц —; изо-С5Ни—; C6H13—
R'=H; СНз-
Характеристика основного сырья
Этиловые эфиры у-кетогептановой, у-кетооктановой, у-кето-нонановой, у-кетоизононановой, у-кетодекановой кислот; получение см. [5—7].
Аммиак газообразный.
Водород электролитический.
Метиламин солянокислый, ТУ ОРУ 3—58, ч.
Метиловый спирт, ГОСТ 6995—54.
Этиловый спирт, ГОСТ 5962—51.
Бензол, ГОСТ 5955—51, ч. д. а.
Никель Ренея, получен выщелачиванием сплава никеля и алюминия [81.
Условия получения
Получение амида у-оксигептановой кислоты. В стальной вращающийся автоклав емкостью 150 мл помещают 11,5 г (0,067 моль) этилового эфира у-кетогептановой кислоты в 30 мл этилового спирта и 2 г никеля Ренея. Гидрирование проводят при 100° и начальном давлении водорода 120 атм. После поглощения 1,5 л водорода автоклав охлаждают, катализатор отфильтровывают. Катализат переносят в стеклянную колбу емкостью 100 мл, взвешивают и насыщают аммиаком при 0° до увеличения веса на 4,5 г. Снова переносят катализат в тот же автоклав и нагревают при 120° в течение 7 часов. Охлаждают, отгоняют растворитель. Вещество перекристаллизовывают из бензола.
16
Выход амида у-оксигептаповой кислоты равен 8,2 г (89,3%), т. пл. 66—67°.
Найдено, %: С—58,24; 58,37; Н—10,36; 10,53; N—9,70; 9,89. C7H15NO2. Вычислено, %: С—58,54; Н—10,43; N—9,66.
По аналогичной методике из этиловых эфиров соответствующих у-кетокарбоновых кислот получены следующие амиды:
Амид у-оксиоктановой кислоты с выходом 82%, т. пл. 87— 88° (из бензола).
Найдено, %: С—59,84; 59,97; Н—10,75; 10,63; N- 8,79; 8,90. C3H17NO2. Вычислено, %: С—60,17; Н—10,70; N—8,80.
Амид у-оксинонановой кислоты с выходом 85%, т. пл. 85— 86° (из бензола).
Найдено, %: С—62,27; 62,33; Н—11,00; 10,80; N—8,08; 8,23. CgHigNOa. Вычислено, %: С — 62,42; Н—10,90; N — 8,09.
Амид у-оксиизононановой кислоты с выходом 80%, т. пл. 77—78° (из бензола).
Найдено, %: С—62,37; 62,28; Н—10,86; 10,90; N—8,22; 8,31. CgHigNOs. Вычислено, %: С—61,42; Н—10,90; N—8,09.
Амид у-оксидекановой кислоты с выходом 89%, т. пл. 95— 96° (из бензола).
Найдено, %: С—64,04; 64,35; Н—11,56; 11,13; N—7,25; 7,39. C10H21NO2. Вычислено, %: С—64,22; Н—11,32; N—7,49.
Получение метиламида у-оксигептановой кислоты. Во вращающийся автоклав емкостью 150 мл помещают раствор 7 г (0,04 моль) этилового эфира у-кетогептановой кислоты в 25 мл этилового спирта, добавляют 1 г никеля Ренея, подают водород под давлением 100 атм и ведут гидрирование при 100° до поглощения 900 мл водорода. Затем катализатор удаляют фильтрованием или центрифугированием. Раствор этилового эфира у-оксигептановой кислоты переносят в склянку Дрекселя и насыщают метиламином до привеса 4 г (см. примечание). Реакционную смесь снова помещают в автоклав и нагревают при постоянном перемешивании при 120° в течение 8 часов. Отгоняют растворитель и избыток метиламина, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 102—103°/3 мм, п™ = 1,4631, tZ420 = 1,0002.
Выход метиламида у-оксигептановой кислоты равен 5 г (79%).
Найдено, %: С—60,27; 60,15; Н—11,02; 11,04; N—8,98. C8Hi7NO2. Вычислнео, %; С — 60,43; Н — 10,78; N — 8,81.
По аналогичной методике из этиловых эфиров у-кетокарбоновых кислот получают следующие метиламиды;
Метиламид у-оксиоктановой кислоты, выход 83,8%, т. пл 90—91° (из бензола).
Найдено, %; С—62,34; Н—11,16; N—7,96. Ч>Н19]\Ю2. Вычислено, %; С—62,42; Н—10,90; N—8,09.
Зак. 1164 17
Метиламид у-оксинонановой кислоты, выход 82%, т. пл. 30—31° (из бензола).
Найдено, %; С—63,99; 64,08; Н—11,34; 11,22; N—7,76; 7,70.
CioH2iN02. Вычислено, %: С — 64,22; Н—11,32; N — 7,49.
Примечание.
Метиламин получают прибавлением по каплям 50%-ного раствора солянокислого метиламина к твердому едкому натру.
ЛИТЕРАТУРА
1 Пат. США 2922740; ZM., 3791 (1961).
2 Пат. США 28772332; РЖХим., 83224 П (1960).
3. Пат. США 2898301; С. А., Р18844а (1959).
4. Герм. пат. 1139483; С. А., Р78665 (1963).
5 В Г. Бухаров, Т. Е. Позднякова. Изв. АН СССР, сер. хим., 6, 1108 (I960).
6. А. А. Пономарев, В. А. С е д а в к и н а. Ж. общ. химии, 31, 984 (1961).
7. А. А. Пономарев, В. А. С е д а в к и н а. Ж- общ. химии, 32, 2540 (1962).
8. Синтезы органических препаратов, сб. 3. М„ ИЛ., 1952, с. 338.
5-АМИЛ-2-ПИРРОЛИДОН
В. А. СЕДАВКИНА
НщС.-ч ,==О
\N/
I Н
C,H17NO М. в. 155,23
Для получения 2-пирролидонов наиболе чаесто используется реакция у-лактонов с аммиаком [1, 21, электролитическое восстановление сукцинимида [3] и восстановление эфиров у-нитрокарбоновых кислот до соответствующих аминокислот с последующей циклизацией [41.
Нами разработан метод синтеза неизвестных ранее 5-ал-кил-2-пирролидонов, заключающийся в восстановительном аминировании этиловых эфиров у-кетокарбоновых кислот в присутствии никеля Ренея. Исходные эфиры легко получаются из фурилалкилкарбинолов (5—8].
СХЕМА СИНТЕЗА
С,НиСОСН8СНгСООСаН, Нис.-Ч
NIR \ N'
Н
Характеристика основного сырья
Этиловый эфир у-кетононановой кислоты, т. кип. 117— 118°/4 мм, п*> = 1,4354, d^Q = 0,9561; получение см. [51
2*
19
Этиловый спирт безводный, ГОС! 5962—51.
Аммиак из баллона.
Никель Ренея, получен выщелачиванием сплава никеля с, алюминием [91.
Водород электролитический.
Условия получения
Во вращающийся автоклав емкостью 150 мл помещают^ 16 г (0,08 моль) этилового эфира у-кетононановой кислоты,: 50 мл этилового спирта, насыщенного аммиаком при 0°, и 1 а никеля Ренея. Подают водород под давлением 90 атм и про-, водят реакцию при непрерывном перемешивании реакционной смеси при 120° до прекращения поглощения водорода (3 часа). По окончании реакции гидрогенизат фильтруют или центрифугируют, этиловый спирт и аммиак отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 126—12871 мм.
Выход 5-амил-2-пирролидона равен 8,46 г (68,4%). Веще-' ство хорошо растворяется в метиловом и этиловом спиртах, воде, нерастворимо в эфире, т. пл. 29—30° (см. примечание)
Найдено, %: С —69,42; 69,54; Н — 10,99; 10,96; N — 704. C9H17NO. Вычислено, %: С—69,67; Н—10,96; N—9,03.
Кипячением 2,3 г 5-амил-2-пирролидона с 5 а уксусного) ангидрида в течение 1,5 часа и последующей перегонкой при пониженном давлении получают 2,86 г (94%) N-ацетил-5 -амил-2-пирролидона, т. кип. 111—11272 мм, п2° = 1,4687.
Найдено, %; С—66,94; Н—10,02; N—6,93. C11H19NO2. Вычислено, %; С—67,06; Н—9,72; N—7,11.
Примечание.
Аналогичным методом из соответствующих этиловых эфиров у-кето1 •карбоновых кислот получены следующие 5-алкил-2-пирролндоны и их ацетильные производные: -•/'
5-Этил-2-пирролидои, выход 62,1%, т. кип. 127—130°/7 мм, nD =а = 1,4751; по литреатуриым данным [10], т. кип. 130°/8 мм, п™ “ 1,4800.
М-Ацетил-5-этил-2-пирролидон, выход 80,7%, т. кип. 103—10577 и.«, п2° = 1,4733.
Найдено, %: С—61,93; Н—9,00; N—8,90.
CsHiaNOs. Вычислено, %: С—61,99; Н—8,45;/№—9,04. •
5-Пропил.2-пирролидон, выход 62,2%, т. кип. 105—10671 мм, т. пл. 35—36°.
Найдено, %: С—65,90; 65,88; Н—9,99; 10,16; N—11,01. C7H13NO. Вычислено, %: С—66,06; Н—10,23; N—11,02.
М-Ацетил-5-пропил-2-пирролидон, выход 83,'4%, т.'кип. 108—10977 мм = 1,4704.
Найдено, %: С—63,84; Н—9,02; N—8,26. C9H115NO2. Вычислено, %: С—63,96; Н—8,95; N—8,29.
5-Бутил-2-пирролидон, выход 86,2%, т. кип. 118—12071 мм, т пл. 38’. Найдено, %: С—68,07; Н—10,65; N—10,01.
CjHisNO. Вычислено, %; С—68,08; Н—10,63; N—9,93.
Ы-Ацетил-5-бутил-2-пирролидои, выход 86,2%, т. кип. 108—1097'6 мм, = 1,4690.
Найдено, %: С—65,21; Н—9,42; N—8,08.
C10H17NO2. Вычислено, %: С—65,63; Н—9.36; N—7,70.
5-Изобутил-2-пирролидон, выход 69,1%, т. кип. 119—12071 мм, т. пл. 46-47°.
Найдено, %: С—67,93; Н—10,37; N—10,18.
CsHisNO. Вычислено, %: С—68,08; Н—10,63; N—9,93.
Ы-Ацетил-5-изобутил-2-пирролидон, выход 82%, т. пл. 107—108/6 мм. л™ = 1,4676.
Найдено, %: С—65,49; Н—9,78; N—7,95.
C10H17NO2. Вычислено, %: С—65,63; Н—9,36; N—7,70.
5-Изоамил-2-пирролидон, выход 60,3%, т, кип. 148—15075 мм, =
= 1,4778, = 0,9620.
Найдено, %: С—69,96; Н—11,09; N—9,09.
C9II17NO. Вычислено, %: С—69,66; Н—10,96; N—9,08.
М-Ацетил-5-изоамил-2-пирролидон, выход 82%, т. кип. 113—11576 мм, по ...^ 1-4680-
Найдено, %: С—66,94; Н—9,86; N—7,16.
C11H19NO2. Вычислено, %: С—67,06; Н—9,72; N—7,11.
5-Гексил-2-пирролидон, выход 59,5%, т. кип. 164—165°/7 мм, т. пл. 42’.
Найдено. %: С—71,00; Н—11,35; N—8,22.
C10H19NO. Вычислено, %: С—71,07; Н—11,24; N—8,28.
Ы-Ацетил-5-гексил-2-пирролидон, выход 85,5%, т. кип. 112—11377 мм, По = 1,4631.
Найдено, %: С—68,15; Н—10,34; N—6,45.
C1SH21NO2. Вычислено, %: С—68,21; Н—10,18; N—6,64.
5-Гептил-2-пирролидон, выход 64,4%, т. кип. 154—157°/2 мм, т. пл. 447 Найдено, %: С—72,18; Н—11,37; N—7,48.
C11H21NO. Вычислено, %: С—72,13; Н—11,47; N—7,67.
М-Ацетил-5-гептил-2-пирролидон, выход 86%, т. кип. 124—12677 мм, = 1,4628.
Найдено, %: С—69,55; Н—10,37; N—6,48.
C13H23NO2. Вычислено, %: С—69,28; Н—10,25; N—6,21,
5-Октил-2-пирролидон, выход 63%, т. кип. 169—160°, т. пл. 48°. Найдено, %: С—72,95; Н—11,74; N—7,11,
Ci2H23NO. Вычислено, %: С—73,03; Н—11,75; N—7,09.
М-Ацетил-5-октил-2-пирролидон, выход 86,6%, т. кип. 140—14277 мм, и о ~ 1,4622.
Найдено, %: С—70,10; Н—10,58; N—6,02.
L14H25NO2. Вычислено, %: С—70,29; Н—10,53; N—5,84.
5-Нонил-2-пирролидон, выход 67,4%, т. кип. 159—1617 т. пл. 51°.
Найдено, %: С—73,82; Н—6,73; N—7,30.
'-isHjsNO. Вычислено, %: С—74,0; Н—11,94; N—6,64.
М-Ацетил-5-нонил-2-пирролидон, выход 87,1%, т. кип. 170—17177 м.ч nD = 1,4602.
Р Найдено, %: С—70,96; Н—10,45; N—5,27.
'“15H27NO2. Вычислено, %: С—71,21; Н—10,76; N—5,54.
21
20
ЛИТЕРАТУРА
1. Ю. К. Юрьев. Уч. зап. МГУ, 132, 224 (1950).
2. Е. Spath, S. Z i n t h e г. Вег., 69B, 2727 (1936).
3. J. Tafel, O. Wassmuth. Ber., 40, 2831 (1907).
4. S. Colog, J. Pouchol. Bull. Soc. France, 3, 598 (1962).
5. А. А. Пономарев, В. А. Седавкина. Ж. общ. химии, 31, 984 (1961).
6. А. А. По и о м а р е в, В. А. Седавкина. Ж. общ. химии, 32, 2540 (1962).
7. В. Г. Б у х а р о в, Т. Е. Поздняков. Изв. АН СССР, сер. хим., 6, 1108 (1960).
8. А. А. Пономарев, В. А. Седавкина. Методы получения хи-' мических реактивов и препаратов, вып. 17. М., ИРЕА, 1968, с. 59.
9. Синтезы органических препаратов, сб. 3. М., ИЛ., 1952, с. 338.
10. Е. Spath. Вег. 66, 598 (1933).
9-АМИНОАКРИДИН
Я. Н. ДЫХАНОВ, Т. В. ПЕРОВА, Р. Ф. ВИДЕНИНА, И. В. ПОКОТЫЛО, В. И. БАЗАКИН
nh2
I II I I
C13H10N2
М. в. 194,24
Наиболее приемлемый для промышленного использования способ получения 9-аминоакридина заключается в циклизации N-фенилантраниловой кислоты избытком хлорокиси фосфора и последующей обработке образующегося 9-хлоракридина карбонатом аммония в расплаве фенола II]. В работе [2] этот способ усовершенствован путем замены фильтрации клейкого 9-хлоракридина экстракцией хлороформом. Однако продукт, получаемый по методике [2], не всегда удовлетворяет по своему качеству требованиям технических условий.
Нами установлено, что при выделении 9-хлоракридина из реакционной массы после циклизации феннлантраниловой кислоты использование концентрированного раствора аммиака (для связывания избыточной хлорокиси фосфора) нежелательно, поскольку в сильно щелочной среде 9-хлоракридин легко гидролизуется до 9-акридона. Эффективнее применять 10%-ный раствор аммиака, как это рекомендовано при синтезе замещенных 9-хлоракридинов [3], а для отделения 9-акридона, образующегося в небольших количествах в этих условиях, перед отгонкой экстрагента из хлороформного или дихлорэтанового экстракта последний необходимо фильтровать от взвеси акридона.
23
Схема синтеза
Характеристика основного сырья
Фенилантраниловая кислота, МРТУ 6—09—1648—64, ч. Хлорокись фосфора, МРТУ 6—09—337—63, ч.
Аммоний углекислый, ГОСТ 3770—64, ч.
1,2-Дихлорэтан, ГОСТ 5840—51, ч.
Ацетон, ГОСТ 2603—63, ч. д. а.
Условия получения
Синтез 9-хлоракридина. В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, термометром и капельной воронкой, помещают 50 г (0,235 моль) фенилантраниловой кислоты и приливают по каплям 160 мл (268 г; 1,75 моль) хлорокиси фосфора. Воронку заменяют обратным холодильником и содержимое колбы при перемешивании медленно нагревают до 65—70°. Начинается энергичная реакция, и колбу сразу же на короткое время погружают в стакан с холодной водой.
Через 15—20 минут, когда интенсивность кипения несколько уменьшится, температуру реакционной массы повышают до 108—110° и поддерживают в этих пределах 2 часа. После этого обратный холодильник заменяют на нисходящий и отгоняют избыток хлорокиси фосфора. Подвижный темный остаток охлаждают до 60—70° и немедленно (см. примечание 1) разлагают, выливая его небольшими порциями в энергично перемешиваемую смесь 500 г льда, 200 мл дихлорэтана и 300 мл 10%-ного раствора аммиака. Реакционную колбу ополаскивают дихлорэтаном дважды по 50 мл, который присоединяют к основной массе. В процессе разложения реакционной массы строго следят за щелочностью среды (pH 8—8,5), проверяя ее через каждые 2—3 минуты по универсальной индикаторной 24
бумажке; в противном случае перед добавлением очередной порции реакционной массы к уже разложенной ее части приливают еще 40—50 мл раствора аммиака.
Реакционную массу перемешивают 20 минут, помещают в делительную воронку и отделяют дихлорэтановый слой (нижний) от водного. Последний экстрагируют в той же воронке дважды по 50 мл дихлорэтана, присоединяют экстракт к основному органическому слою и фильтруют от взвеси акридо-на. Дихлорэтаиовый раствор сушат над 10 г хлористого кальция, вновь фильтруют и отгоняют экстрагент (275—280 мл), который используют в следующем опыте. Остаток охлаждают, выделившийся осадок 9-хлоракридииа отфильтровывают и сушат до постоянного веса либо на воздухе, либо при 70° в течение 20—25 минут (см. примечание 2).
Выход 9-хлоракридина равен 40 г (80%), т. пл. 118—119°, что соответствует литературным данным [11.
Получение 9-аминоакридина. В закрепленный на штативе стакан емкостью 1 л, снабженный мешалкой и термометром, помещают 40 г (0,187 моль) 9-хлоракридина и 200 г безводного фенола. Содержимое стакана нагревают, перемешивая 15— 20 минут при 70°, и к полученному расплаву постепенно прибавляют 30 г углекислого аммония (см. примечание 3) с такой скоростью, с какой позволяет обильное выделение углекислого газа. Затем температуру смеси быстро поднимают до 120° и нагревают при этой температуре 45 минут, не прекращая перемешивания.
Реакционную массу охлаждают до 30° и выливают в 500 мл ацетона, помещенного в охлаждаемый льдом стакан. Через 30—40 минут выпавший осадок гидрохлорида 9-амино-акридина отфильтровывают и промывают на фильтре еще 200 мл ацетона, который присоединяют к основному фильтрату. Из фильтрата отгоняют ацетон (650—700 мл), используя его в следующем опыте.
Осадок сырого гидрохлорида 9-амнноакридина трижды кипятят с водой (600, 300 и 100 мл). К последней порции воды добавляют 2 мл концентрированной соляной кислоты. Горячие водные вытяжки фильтруют и фильтраты соединяют вместе. Если выпадает осадок, то’его переводят в раствор нагреванием. К горячему раствору гидрохлорида 9-аминоакридина при перемешивании добавляют раствор 48 г едкого натра в 250 мл воды. Образовавшуюся суспензию охлаждают до 10— 15° и фильтруют с отсасыванием. Осадок на фильтре промывают холодной дистиллированной водой до отсутствия ионов хлора (проба с азотнокислым серебром), тщательно отжима ют и сушат при 100—105°.
Выход 9-аминоакридина равен 33 г (75% в расчете на 9-хлоракридин), т. пл. 232—234°, что совпадает с литературными данными 12]. Вещество по своим качествам соответству-
25
ёт требованиям МРТУ 6—09—4562—67 к препарату квалификации «ч».
Примечания:
1. 9-Хлоракридин легко гидролизуется как в кислом, так и в нейтральном растворах, поэтому после удаления избытка хлорокиси фосфора и до обработки аммиаком его нельзя долго хранить на открытом воздухе.
2. Если сушить 9-амииоакридии при более высокой температуре, то происходит потеря вещества вследствие возгонки.
3. Можно брать углекислый аммоний с более низким содержанием аммиака, чем предусмотрено ГОСТ 3770—64, при сохранении его общего количества.
ЛИТЕРАТУРА
1, О. Magidson. Вег., 66, 866 (1933).
2. Синтезы органических препаратов, сб. 1. М., ИЛ., 1952, с. 36.
3. Н. Н. Дыханов. Авт. свид. 126497; Бюлл. изобр., № 5 (1960).
о-АМИНО-р,р-ДИ(3-ИНДОЛИЛ)ЭТИЛБЕНЗОЛ
А. О. ГИНЗБУРГ, С. В. ЛЫЛЫК, А. К. ШЕИНКМАН
C24H21N3
М. в. 353,47
о-Амиио-р.р-ди(З-индолил) этилбензол (трииндол) получают при полимеризации индола в присутствии ортофосфорной кислоты [1, 2].
Способ, предлагаемый нами, основан на взаимодействии индола с водным раствором минеральной кислоты в присутствии азотистых оснований и отличается мягкими условиями проведения процесса, количественным выходом продукта, ие загрязненного смолистыми примесями.
СХЕМА СИНТЕЗА
27
Характеристика основного сырья
Иидол, ТУРУ—1064—54, ч.
Хинолин, ТУ МХП 93—47, свежеперегнанный.
Серная кислота, ГОСТ 4204—66, ч.
Условия получения
В колбу Эрлеимейера емкостью 100 мл с притертой пробкой помещают 11,7 г (0,1 моль) индола, растворенного в 12,9 г (0,1 моль) хинолина (см. примечание), й 27,5 Мл 20 % -ной серной кислоты. Смесь встряхивают 6 часов при комнатной температуре. Выпавший желтый осадок отфильтровывают и кипятят с 3—4-кратным избытком 25%-ного раствора аммиака в течение 0,5 часа. После охлаждения белый кристаллический осадок отфильтровывают и дважды перекристаллизовывают из 100 мл этанола.
Выход о-амино-£,0-ди(3-индолил)этилбензола равен 11,4 г (97—98%), т. пл. 168—170°; =0,54 (на окисн алюминия
в системе растворителей метанол : гексан : бензол : хлороформ 1 : 1 : 6 : 30.
По литературным данным [3], т. пл. 171°.
Найдено, %: С—81,45; 80,96; Н—6,29; 6,20; N—11,90; 12,05. C24H2iN3. Вычислено, %: С—81,55; Н—6,55; N—11,8.
Примечание.
В присутствии других азотистых оснований, например, пиридина, в аналогичных условиях время реакции увеличивается до 24—30 часов, а выход продукта падает (74%).
ЛИТЕРАТУРА
1. К. Keller. Вег., 46, 726 (1913).
2. G. Smith. Chem. and Chetn. Ind. 1451 (1954).
3. W. E. Noland, C. F. Hjmmer. I. Organ. Chem,, 25,9, 1525 (I960).
2-(2-АМИНО-5-НИТРОБЕНЗАМИДО) БЕНЗОЙНАЯ КИСЛОТА
И. С. МАРКОВИЧ, И. А. ФИЛЯГИНА, В. М. ДЗИОМКО
.соон
NH,
no2
СнНцМзОб М. в. 301,25
2-(2-Амиио-5-нитробеизамидо) бензойная кислота в литературе не описана. Нами это соединение получено сплавлением антраниловой кислоты с 5-нитроизатовым ангидридом.
СХЕМА СИНТЕЗА
О
соон Z\^yNO’
I II + I
^/4NH3 0Z
I
H
^\ZC°OH NH, ---* I II I
^/XNHCO—
NO2 29
Характеристика основного сырья
Антраниловая кислота, ВТУ МХП 2791—51, ч. 5-Нитроизатовый ангидрид, получение см. [1].
Условия получения
Смесь 0,7 г (0,005 моль) антраниловой кислоты и 1,04 г (0,005 моль) 5-ннтроизатового ангидрида постепенно нагревают до 120° и выдерживают при этой температуре до видимого прекращения выделения углекислого газа (~15—20 минут). Полученную порошкообразную массу дважды перекристаллизовывают из 100 мл смеси (1:1) диметилформамид— вода с углем и промывают 40 мл спирта.
Выход 2- (2-амино-5-нитробензамидо) бензойной кислоты равен 0,4 а (26,5%), т. пл. 284 — 285°;Хм,Кс = 274—278 нм, 340— 350 нм; 6276 = 94200, 6345 = 162000.
Найдено, %: С—56,01; Н—3;78; N—13,32. CuHuNjOb. Вычислено, %: С—55,81; Н—3,68; N—13,94.
ЛИТЕРАТУРА
1. Н. Koble. J. pract. Chem., 30, 477 (1884).
8-АМИНОХИНАЛЬДИН
В. М. ДЗИОМКО, И. А. КРАСАВИН, Б. В. ПАРУСНИКОВ.
Ю. П. РАДИН
сн8
NH,
C10H10N2
М. в. 158,21
............ г,.„. _______ ____J____ /восстановлением 8-нитрохинальдина хлористым оловом в соляной кислоте [1, 2], железом в разбавленной уксусной кислоте [3] нли каталитическим гидрированием на никеле Ренея [4]; при восстановлении железом выход достигает 89% [3], гидрирование дает количественный выход [4].
8-Аминохинальдин был получен также с количественным рыходом по реакции Бухерера путем нагревания оксисоедине-нйя с аммиаком при 200° в течение 10—15 часов в присутствии бисульфита натрия [5].
Другие способы получения 8-аминохинальдина, такие, как каталитическое восстановление 4-хлор-8-нитрохинальдин-1 -оксида [6] или восстановление 7-хлор-8-нитрохинальдина красным фосфором н иодистоводородной кислотой [7], едва ли имеют практическое значение из-за малой доступности исходных веществ.
Нами проверены два способа восстановления 8-нитрохи-нальдина: гидразин-гидратом в присутствии палладированно-го| угля в спиртовой среде и железом в разбавленной уксусной кислоте. В первом случае был получен высокий выход амина (до 95%), однако при хранении продукт быстро темнел. Восстановление железом дало, хотя и с более низким выходом, вполне устойчивый продукт хорошего качества.
31
no2
СХЕМА СИНТЕЗА
NHS
Характеристика основного сырья
8-Нитрохинальдин, т. пл. 137—139°; получение см. с. 215 этого сборника.
Железо карбонильное, ос. ч., марка А-2.
Уксусная кислота, ГОСТ 61—51, ч. д. а.
Натр едкий, ГОСТ 4328—66, ч.
Условия получения
В круглодонную колбу емкостью 2 л, заранее приспособленную для перегонки с паром, помещают 15,1 г (0,08 моль) 8-нитрохинальдина, 80 мл уксусной кислоты и 80 мл воды. При размешивании и нагревании смеси на кипящей водяной бане небольшими порциями к ней за 20—30 минут добавляют чер^з открытое горло колбы 15,2 г (0,27 г-а) порошкообразного карбонильного железа. Нагревание и размешивание продолжают еще 1 час. Слегка охладив содержимое колбы, подщелачивают его добавлением раствора 60 г едкого натра в 240 мл воды до pH 9—10, после чего перегоняют В-аминохи-нальдин с водяным паром. Перегонку продолжают до получе Ния совершенно прозрачного дистиллата (~3—3,5 л).
Дистиллат с осадком выдерживают 3—4 часа во льду, затем продукт отфильтровывают и сушат в эксикаторе над едким натром.
Выход 8-аминохинальдйна равен 8,5—9,2 г (67—72,3%), т. пл. 54—56°.
Фильтрат снова подвергают перегонке с паром и, получают еще 1—1,5 г амина. Общий выход 8-аминохинальдина равен 9,7—10,4 г (76,4—82%).
По литературным данным, т. пл. 8-аминохинальдина 50° [5]; 55—56° £7]; 56° [1, 3]; 57—58° [4, 6]. '
ЛИТЕРАТУРА
1.0. Doe bne г, W. Miller. Вег., 17, 1698 (1884).
2. М. А. Альперович, И. К- У ш е и к о. Ж. общ. химии, 29, 3384 (1959).
32
3. К. Madeja. J. prakt. Chem., 17, 97 (1962).
4. R. Roth, H. Erlenmeyer. Helv. chim. acta, 37, 1064 (1954).
5. Герм. пат. 666790 (1938); Chem., Zbl., 1939, 1, 533.
6. E. Ochiai, K. Sa take. J. Pharmac. Soc, Japan, 71, 1078
(1951); C. A., 46, 50451 (1952).
7. A. K. Sen. U. P. Basu. J. Indian., Chem. Soc.. 34, 833 (1954);
C. A., 52, 91221 (1958).
3 Зак, 1164
а-[(2- АМИНОЦИКЛОПЕНТИЛ )МЕТИЛ]ФУРАН
| А. А. ПОНОМАРЕВ, | А. П. КРИВЕНЬКО
CioHisNO
М. в. 165,10-
Ранее неизвестный а-[(2-аминоциклопентил)метил]фуран получен нами посредством восстановительного аминирования р-фурфурилиден-а-циклопентанона. Для выделения вещества использован метод, заключающийся в предварительной обработке реакционной смеси разбавленной минеральной кислотой; амин при этом превращается в соответствующую соль, которая легко отделяется. После разложения е'е щелочью амин подвергают вакуумной перегонке. Метод применен для получения других фурфурилзамещенных алициклических аминов [1, 2].
СХЕМА СИНТЕЗА
Н„ NH, NiR
Характеристика основного сырья
р-Фурфурилиден-а-циклопентанон, т. кип. 154°/15 мм; получение см. [3].
Водород, ГОСТ 3022—45.
Аммиак газообразный.
Метиловый спирт, ГОСТ 6995—54, х. ч
34
Соляная кислота, ГОСТ 3118—66, х. ч., 10%-ная.
Натр едкий, ГОСТ 2263—59, техн.
Никель скелетный, приготовление см. [4].
Условия получения
Во вращающийся автоклав емкостью 610 мл загружают 60 г (0,36 моль) свежеперегнанного р-фурфурилиден-а-цикло-пентанона, 140 мл метилового спирта, содержащего 30 г (1,8 моль) аммиака (см. примечание 1), 6 г скелетного никелевого катализатора. Начальное давление водорода 100 атм, температура 100°. Реакция заканчивается через 7 часов после поглощения рассчитанного количества водорода (16,6 л). Гид-рогенизат фильтруют от катализатора, спирт и избыток аммиака отгоняют при пониженном давлении. Колбу с остатком погружают в баню с холодной водой и при перемешивании к реакционной массе прибавляют 10%-ный раствор соляной кислоты до кислой реакции по конго. Смесь экстрагируют эфиром дважды порциями по 20 мл (см. примечание 2). Затем к водному раствору соли амина при охлаждении добавляют 16 г твердого едкого натра. Амин отделяют, водный раствор экстрагируют эфиром. Эфирные вытяжки соединяют с амином н сушат гранулированным едким кали. Эфир отгоняют, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 115°/14 мм.
Выход а-{(2-аминоциклопентил) метил]фурана равен 36 г (58%). Вещество представляет собой бесцветную жидкость с характерным аминным запахом, п™ = 1,5010, <А20 =' 1,0025.
Найдено, %: С—72,94; 72,92; Н—9,24; 9,12; N—8,75; 8,67; MR D = 48,60.
CiqHuNO. Вычислено, %: С—72,72; Н—9,04; N—8,48; MRD = 48,11.
Примечания:
1. Метиловый спирт насыщают аммиаком из баллона при 0°.
2. Из эфирного раствора отгоняют растворитель и при перегонке при пониженном давлении выделяют а[(2-аминоциклопентил)метил]фуран с выходом 13%, т. кип. 126—128715 мм, п™ = 1,5050, = 1,0780.
ЛИТЕРАТУРА
1. А. А. П о н о м а р е в, А. П. К р и в е н ь к о, М. В. Норицина. Химия гетероцикл, соед., 5, 580 (1967).
2. А. А. Пономарев, А. П. Кривенько, М. В. Но р и ц и н а. Ж общ. химии, 33, 1778 (1963).
3 Н. W. Walton. J. organ. Chem., 22. 1161 (1957).
4. Синтезы органических препаратов, сб. 3. М., ИЛ., 1952, с. 338.
3*
N-АРИЛПИРИДИНИЕВЫЕ СОЛИ
Л. Г. ГАХ, А. К. ШЕЙНКМАН, С. Н. БАРАНОВ
где Ar = C,Ht-; CH»CeH4—; Ci0H7-; NCeHe-; Х=С1;
Br; 1.
В литературе описан метод получения фенилпиридиний хлорида нагреванием 2,4-динитрофенилпиридиний хлорида с анилином в абсолютном спирте [1].
Нами проверен этот метод и использован для получения ряда новых арилпиридиниевых солей.
СХЕМА СИНТЕЗА
no2
ArNK„HX
Аг-N+ + O,N—^-NH,
Характеристика основного сырья
2,4-Динитрофеиилпиридиний хлорид, т. пл. 200°, см. приме-
43. НИС 1
Анилин, ГОСТ 5819—70, ч.
36
n-Толуидин, ТУ 6—09—66—70, ч.
а-Нафтиламин, ГОСТ 8827—58, ч.
З-Аминохинолин, ч.
Этиловый спирт, ГОСТ 8314—57.
Иодистоводородная кислота, ГОСТ 4200—48, ч.
Ртуть двухлористая, ГОСТ 4519—48, ч.
Условия получения
В круглодонной колбе емкостью 1 л растворяют при нагревании 0,1 моля 2,4-диннтрофенилпиридиний хлорида в 250— 300 мл абсолютного этилового спирта, затем добавляют пор циями при периодическом взбалтывании содержимого колбы 0,3 моля соответствующего ароматического амина (см. примечание 2). Смесь кипятят с обратным холодильником 20 часов, пока выпавший красный осадок не перейдет снова в раствор. Отгоняют наполовину спирт, а остаток разбавляют водой (0,8—11,0 1Л|) так, чтобы выпал в осадок весь динитроанилин. Смесь фильтруют, фильтрат взбалтывают с 35 г активированного угля в течение суток. Уголь отделяют, а фильтрат упаривают на водяной бане до 1/3 первоначального объема.
К полученному раствору добавляют 0,15 моля концентрированной галоидоводородной кислоты и осаждают пятикратным количеством насыщенного раствора хлорида ртути (проба на полноту осаждения). Через сутки осадок отфильтровы-ют, перекристаллизовывают из горячей воды, избегая больших разбавлений. Ртутную соль разлагают в горячем растворе сероводородом, фильтрат упаривают до получения густой массы. Осадок перекристаллизовывают из абсолютного спирта (см. таблицу 1, 2).
Примечания:
1. 2,4-Динитрофенилпиридиний хлорид получают нагреванием на водяной бане смеси эквимолярных количеств 2,4-динитрохлорбеизола и пиридина [1].
2. Кристаллический ароматический амин прибавляют в виде раствора его в минимальном количестве абсолютного этилового спирта.
ЛИТЕРАТУРА
1. Th. Zin eke, G. H e n s e r, W. Мб 11 er. Ann., 333. 296(1904).
2. Словарь органических соединений, т. Ill, М., Инлитиздат, 1949, с. 553.
3. В. А. Измаильский. ЖРФХО, S0, 190 (1918).
Таблица I
Условия получения и выход N-арилпиридиииевых солей
№ п/п N - Арилпиридиниевая соль Загрузка исходных веществ Выход, %
2,4-диннтрофеннл-пирндиннй хлорид, моль ариламин, моль НХ, г
1 1-Фенилпиридиний хлорид 0,1 Анилин, 0,3 НС1, 36%-ная; 78,2 60
2 1-Фенилпиридиний бромид 0,1 Анилин, 0,3 НВг, 70%-ная; 40,0 50
3 1-Фенилпиридиний иодид 0,1 Анилин, 0,3 HI, 57%-ная; 48,6 45
4 1 - (n-Толил) пиридиний иодид 0,09 л-Толуиднн 0,23 HI, 57%-ная; 44,0 45
5 1-(а-Нафтил) пиридиний иодид 0,07 а-Нафтиламни 0,075 HI, 57%-ная; 20,0 42
6 1 - (З-Хинолил) пиридиний иодид 0,019 3-Аминохинол ин 0,036 HI, 57%-ная; 10,0 40
Таблица 2
Константы и данные анализа N-арилпиридиниевых солей
Соединение Брутто-формула Мол. вес T. пл., °C Найдено, % Вычислено, %
экспериментальная литературная С Н N X С Н N X
1 CnH,0ClN 191,66 105106 105-106[1] — — —
2 C„H10BiN 236,12 156-157 155[2] __ — —
3 CnHioIN 283,12 209-210 209-210 [3] — — —
4 297,13 160-161 — 48,21 4,10 5,13 42,50 48,50 4,07 4,71 42,70
5 C1SH12IN 333,17 182-183 — 54,01 3,81 4,60 38,38 54,01 3,63 4,29 3,08
6 CuHjjINa 334,15 221-221,5 — 50,60 3,24 8,08 37,44 50,32 3,32 8,38 37,97
1-АЦЕТИЛ АМИ НО-2-АЦЕТОКСИ-3,5-ДИМЕТИЛБЕНЗОЛ
В. М. ОСТРОВСКАЯ, М. М. КРАСНОВА, Л. В. ЛОМАКИНА
H.CXX4/NH<° II I п •- Н,О
СН, сн‘
С1гН16МО3. — нао
2
М. в. 221,26
1-Ацетиламино-2-ацетокси-3,5-диметилбензол не описан в литературе и получен нами впервые с 100%-ным выходом ацетилированием 2-амино-4,6-диметилфенола уксусным ангидридом в присутствии избытка безводного ацетата натрия. В отсутствие ацетата натрия реакция не идет.
| ОН
СН,
СХЕМА СИНТЕЗА
Н.С NHC< \^\/ чсн, (СНаСОО),О . I |1 CH.COONa 'V'X
I ocf
СН, хсн,
Характеристика основного сырья
2-Амино-4,6-диметилфеиол, т. пл. 132°; получение см. [1].
Уксусный ангидрид, ГОСТ 5815—52, ч. д. а.
Натрий уксуснокислый плавленый, ТУ 6—09—246—70, ч.
40
Условия получения
В стакан к 40 мл уксусного ангидрида прибавляют при помешивании небольшими порциями 8 г (0,058 моль) 2-ами-но-4,6-диметилфенола, затем добавляют 4 г безводного ацетата; нат^ри^ и нагревают смесь 2—10 минут до растворения ацетата натрия. Реакционный раствор выливают в 400 мл воды, при этом выпадает белый волокнистый объемистый осадок. Осадок отфильтровывают (после промывки водой и сушки вес осадка 10,4 г) и сырым перекристаллизовывают из воды.
Выход 1-ацетиламино-2-ацетокси-3,5-диметилбензола равен 9,7 г (93%), т. пл. 158,5—160° (см. примечание).
Найдено, %: С—62,36; 62,12; Н—7,40; 7,42;
N—6,07; 5,98.
Ci2H15NO3 -у- Н2О. Вычислено, %: С—62,60; Н—6,96; N—6,09.
Примечание.
После сушки при 100° в вакууме над твердым едким кали.
Найдено, %: С—65,75; 65,60; Н—7,10; 7,11; N—6,53; 6,36.
Ci2Hi6NO3. Вычислено, %: С—65,14; Н—6,83; N—6,33.
ЛИТЕРАТУРА
1. В. М. Дзиомко, И. С. Маркович. Методы получения химических реактивов и препаратов, вып. 2. М., ИРЕА, 1961, с. 28.
2-АЦЕТИЛАМИНОФЕНОКСИУКСУСНАЯ КИСЛОТА
в. м. островская, м.'м. Краснова, о. т. лушина
.О
NHC<
I СН3
ОСН2СООН.
(X
C10HnNO4
М. в. 209,20
2-Ацетиламинофеноксиуксусную кислоту используют в синтезе 2-ам»нофеноксиуксусной кислоты. В литературе описан метод ее получения конденсацией 2-ацетиламинофенола с монохлоруксусной кислотой кипячением в водном растворе едкого натра [1].
Нами проверен и уточнен этот метод. Получаемая 2-аце-тиламинофеноксиуксусная кислота свободна от прнмеси 2-ацетиламинофенола, в то время как продукт реакции, проведенной нами по литературным данным, имел значительную примесь исходного вещества (данные по температурам плавления проб смешения, данные элементного анализа, а также наличие свободного фенола, обнаруживаемого пробной реакцией азосочетания с диазотированным п-нитроанилином).
СХЕМА СИНТЕЗА
NHC\ | хсн3
Ггон
4Z
С1СН2СООН NaOH; НС1
NHC^° | 4 CH,
^\-осн/:оон
42
Характеристика основного сырья
2-Ацетиламинофенол, т. пл. 206°; получение см. с. 44 этого сборника.
Натр едкий, ГОСТ 4328—66, ч. д. а.
Монохлор уксусная кислота, ГОСТ 5836—68, ч.
Условия получения
В стакан емкостью 2 л, снабженный мешалкой и обогревом, помещают 95 г (1 моль) монохлоруксусной кислоты, 700 мл воды, затем к полученному раствору приливают 132 мл (180 г, 1,5 моль) 33%-ного раствора едкого натра и добавляют 151 г (1 моль) 2-ацетиламинофенола.
К реакционной смеси, представляющей собой суспензию, добавляют еще 44 мл (0,5 моль) 33%-ного раствора едкого натра в 100 мл воды. Реакционная смесь становится прозрачной, pH среды 10. Смесь кипятят в течение двух часов, добав ляют раствор, состоящий из 47 г монохлоруксусной кислоты, 44 лфг 33%-ного раствора едкого натра и 350 мл воды, и кипятят еще один час, затем добавляют еще 44 мл 33%-ного раствора едкого натра и кипятят один час. К охлажденному реакционному раствору приливают 170 мл концентрированной соляной кислоты и 400 г толченого льда. Выпавший осадок отфильтровывают, промывают 600 мл охлажденной до 0° во дой и сушат.
Выход. 2-ацетиламинофеноксиуксусной кислоты равен 191 г (91%), т. пл. 143°
Продукт перекристаллизовывают из 600 мл кипящей воды с активированным углем. Выпавшие кристаллы промывают на фильтре 100 мл холодной воды и сушат.
Выход 2-ацетиламинофеноксиуксусной кислоты равен 174 г (83%), т. пл. 148—149°; содержание основного вещества 98%.
ЛИТЕРАТУРА
1. W. A. Jacobs, М. Н е i d е 1 b е г g е г. J. Amer. Chem. Soc«, 39 10, 2188 (1917).
2-АЦЕТИЛАМИНОФЕНОЛ
В. М. ОСТРОВСКАЯ, М. М. КРАСНОВА
ОН
1
II J ЧСН3
C8H9NOa
М. в. 151,17
2-Ацетиламииофенол получают ацетилированием 2-амино-феиола в 4%-ном растворе щелочи 20%-ным избытком уксусного ангидрида с последующей нейтрализацией соляной кислотой [1, 2J; нагреванием 2-аминофенола в уксусном ангидриде [3]; кипячением солянокислого 2-аминофенола с ледяной уксусной кислотой, ацетатом натрия и эквимолекулярным количеством уксусного ангидрида [4].
Нами предлагается способ получения 2-ацетиламинофено-ла ацетилированием 2-аминофеиола 20%-ным избытком уксусного ангидрида в среде этилацетата. Преимущества этого способа— возможность проведения реакции при комнатной температуре, малая продолжительность процесса, достижение высокого выхода продукта (96%).
СХЕМА СИНТЕЗА
NH,
ГУ0Н
(CH^OOJjO сн,соос,н5
/О
NHCf
I ХСН, /Ч—он
II I + сн,соон
44
Характеристика основного сырья
2-Аминофенол, МРТУ 6—09—6034—69, ч.
Уксусный ангидрид, ГОСТ 5815—69, ч. д. а.
Этиловый эфир уксусной кислоты, МРТУ 6—09—6515—70, х. ч.
Условия получения
В колбе емкостью 2 л, снабженной мешалкой, термометром, обратным холодильником и капельной воронкой, размешивают 218 г (2 моль) 2-аминофенола в 1200 мл этилацетата. К суспензии прибавляют по каплям 246 г (244 мл; 2,4 моль) уксусного ангидрида. Реакционную смесь размешивают при комнатной температуре 4 часа, затем нагревают 1 час при 50—60°. После охлаждения массы образовавшийся осадок отфильтровывают на воронке Бюхнера.
Выход 2-ацетнламннофенола равен 292 г (96%), т. пл. 205°.
По литературным данным, т. пл. 20Г [1, 2]; 202° [5]; 202— 203° (6]; 202,5—203,5° [7]; 209° [8].
ЛИТЕРАТУРА
1. A. L u m 1 ё г е, L. L u m i ё г е, Н. В а г b i е г. Bull. Soc. chim. [31, 33, 784 (1905).
2. L. C. Raiford, С. E. Greider. J. Amer. Chem. Soc., 46, 432 (1924).
3. Th. Z i n c k e, A. H e 1 e b r a n d. Ann., 226, 69 (1884).
4. R. Me idol a, G. H. Wool cot t, E. Wray. J. Chem. Soc., 69, 1323 (1947).
5. F. Bell, J. Kenyon. J. Chem. Soc., 1926, 1893,
6. B. Pawlewski. Ber. 35, 112 (1902).
7. S. N i e m e n t о w s ki. Ber., 30, 3069 (1897).
8. E. Bamberger. Ber., 36 , 2050 (1903).
\-Ли.ЕТИЛ-!\-ФУРФУРИЛАМИ НОЭТАНОЛ И 3-(.\-АЦЕТИЛ-5-МЕТИ Л-2-ПИРР0Л ИД ИЛ) ПРОПАНОЛ
А. А. ПОНОМАРЕВ, И. А. МАРКУШИНА, Т. И. ГУБИНА,
М. В. НОРИЦИНА
c,h„no8
CioHigNOa
\ J'-CH2NCH2CH2OH
4 (Г I
COCH.
H,C—l '-СН,СН,СН2ОН
I COCH,
M. B. 183,20
M. B. 185,26
Известно, что при ацетилировании аминоспиртов, содержащих первичную или вторичную аминогруппу, уксусным ангидридом, хлористым ацетилом или кетеном получают в основном N, О-диацетильные производные [1, 2].
Нами разработан метод избирательного ацетилирования N-фурфуриламиноэтанолов и N-пирролидилалканолов, содержащих вторичную аминогруппу [3]. Реакцию проводят в водном растворе при пропускании кетена до определенного привеса и получают с хорошим выходом N-ацетильные производные, строение которых подтверждено данными элементного анализа и ИК-спектрами, Валентному колебанию связи С=О в третичной амидной группе соответствует частота 1640 см~’; наличие гидроксильной группы подтверждается широкой полосой при 3400 слг'; полоса поглощения при 1750 см~', характерная для сложноэфирной группы, отсутствует.
46
СХЕМА СИНТЕЗА
И JL-CHjNHCHjCH.iOH Сн-^Р--^ тг
-> I J—CHjNCHjCHsOH XOZ |
COCH,
H»c-l J-CH,CH2CH,OH CHaC0 >
I H
H8C-I J-CH,CH,CHtOH NZ
I COCH,
Характеристика основного сырья
N-Фурфуриламиноэтанол, т. кип. 99—100°/2 мм, nfi — = 1,5026; получение см. [4].
3-(5-Метил-2-пирролидил) пропанол, т. кип. 104—10577 мм, п^= 1,4720; получение см. [5].
Условия получения
Синтез П-ацетил-П-фурфуриламиноэтанола. В раствор Юг (0,07 моль) N-фурфуриламиноэтанола в 4,3 г воды, помещенный в дрексель, пропускают кетен в течение 2—3 часов до привеса 4,6 г (см. примечание 1). Образовавшуюся в процессе реакции уксусную кислоту и воду отгоняют прн пониженном давлении, а остаток без предварительной сушки подвергают вакуумной перегонке.
Выход №-ацетил-Ы-фурфуриламиноэтанола равен 9,5 г (73%), т. кип. 139—14074 мм, п*> =1,5135, г/420 = 1.1730 (см. примечание 2).
Найдено, %: С—58,95, 59,23; Н—7,53, 7,57; N—7,53, 7,98;
MRd= 47,19.
C9H13NO3. Вычислено, %: С—59,30, Н—7,12, N—7,61; MR D = 47,17.
Получение 3-(М-ацетил-5-метил-2-пирролидил) пропанола. В раствор 7,2 г (0,046 моль) 3-(5-метил-2-пирролидил) пропанола в 8 мл воды, помещенный в Дрексель, в течение 2—3 часов пропускают кетен до привеса 4,2 г. Дрексель с ре-
47
акциоинои смесью при пропускании кетена энергично встряхивают и охлаждают ледяной водой. Воду и образовавшуюся уксусную кислоту отгоняют при пониженном давлении, остаток растворяют в эфире и сушат над прокаленным сернокислым магнием. Эфир отгоняют на водяной бане, остаток перегоняют в вакууме.
Выход 3-(№ацетил-5-метил-2-пирролидил)пропанола равен 7 г (75%), т. кип. 169—171°/3 мм, = 1,4880, rf420 = = 1,0278.
Найдено, %: С—64,67; 64,45; Н—10,01, 9,89; N—7,51; 7,48; MRd = 51,93.
CioHi9N02. Вычислено, %: С—64,95; Н—10,35; N—7,56; MRD = 51,77.
Примечания:
1. Необходимый для реакции ацетилирования кетен получают общеизвестными методами [6J.
2. Аналогично получают Лт-ацетнл-(5-метил-2-фурфурил)амнноэтанол из М-(5-метил-2-фурфури л) аминоэтанола [4], выход 89%, т. кип. 155— 15773 мм, nW = 1,5040, d?’ = 1,1290.
Найдено, %; С—60,99, 60,67; Н—7,77, 8,16; N—7,10, 7,02; MRD~ 51,97.
CioIIuNOa. Вычислено, %: С—60,90; Н—7,67; N—7,10; MRD =51(80.
ЛИТЕРАТУРА
1. А. А. Пономарев, М. В. Норицина, А. П. Кривенько. Докл. АН СССР, 156, 102 (1964).
2. А. А. Пономарев, Ю. Б. Исаев. Ж. общ. химии, 20, 1079 (1950).
3. А. А. Пономарев, И. А. Маркушина, М. В. Норнцнна. Авт. свнд. 183216; Изобретения. Промобразцы, Товарные знаки, № 13
4. А. А. П о н о м а р ев, Э. А. 3 а д у м и н а, И. А. Маркушина, И. М. Скворцов. Химия гетероцикл, соед. 4, 601 (1968).
5. А. А. Пономарев, М. В. Норнцнна, А. П. Кривенько. Химия гетероцикл, соед., 6, 923 (1966).
6. Органические реакции, т. 3. М., ИЛ, 1951, с. НО.
4-н-АЦИЛОКСИБЕНЗАЛЬДЕГИДЫ
3. С. СИДЕНКО, В. А. ИНШАКОВА. Г. С. ЧИЖОВА
онс-^ ^-ос^° ,
где R = н-СэН7—; h-C6Hi3—; h-C7Hib—
4-н-Бутирил-, 4-н-гептаноил- и 4-н-октаноилоксибензальде-гиды в литературе не описаны. Указанные альдегиды представляют интерес в качестве исходных соединений для получения жидко-кристаллических веществ.
4-н-Ацилоксибензальдегиды получены нами взаимодействием 4-оксибензальдегида с соответствующими хлористыми ацилами.
СХЕМА СИНТЕЗА
RCOC1
СИ О
Характеристика основного сырья
4-Оксибензальдегид, ТУ TCP 374р—61, ч.
н-Бутирил хлористый, т. кип. 99—102°; получение см. [1].
н-Гептаноил хлористый, т. кип. 175°; получение см. [1].
н-Октаноил хлористый, т. кип. 195,5°; получение см. [1].
Пиридин, ГОСТ 2747—44, ч.
Серная кислота, ГОСТ 4204—66, ч.
Хлористый кальций плавленый, ГОСТ 4460—66, ч.
4 Зак. 1164 49
Условия получения
В трехгорлой колбе емкостью 150 мл, снабженной мешалкой, капельной воронкой и обратным холодильником, смеши-зают 0,02 моля 4-оксибензальдегида, 20 мл сухого пиридина и при размешивании и охлаждении (4-2°) добавляют 0,02 моля соответствующего хлористого ацила. Йри этом сразу выпадает желтый осадок. Реакционную смесь перемешивают 5 часов при комнатной температуре, а затем выливают по каплям в охлажденную льдом разбавленную (1:6) серную кислоту. Коричневое масло отделяют, высушивают над хлористым кальцием и фракционируют при пониженном давлении.
Полученные соединения представляют собой подвижные светло-желтые масла с довольно резким запахом.
4-н-Ацилоксибензальдегиды охарактеризованы температурой кипения, показателем преломления и данными элементного анализа (таблица).
Константы н данные анализа 4-и-апилоксибензальдегндов
Ацилоксн-бензальдегиды Выход, % Т. кип., °C 1мм „20 nD Найдено, % Брутто - формула Вычисле-
НО, X
С Н с - Н
4-н-Бутирил-оксибензальдегид 40 129/2 1,5240 68,92 69,08 6,64 6,51 СцНиОз 68,75 6,25
4-и-гептаионл-оксибензаль-дегнд 53 162/2 1,5077 71,86 72,09 7,75 7,61 СцНцОз 71,80 7,69
4-н-Октаноил-окси-бензаль-дегид 51 158-160°/! 1,5038 72,52 72,48 8,12 8,09 CisHa0'?3 72,78 8,04
ЛИТЕРАТУРА
1. Синтезы органических препаратов, сб. 1. М., Инлнтиздат, 1949, с. 471.
S-БЕНЗИЛ ИЗОТИОМОЧЕВИНА ГИДРОХЛОРИД
£. А. БУГАИ
f\ NH,
-НС1
NH
CgHnCINzS M. в. 202,71
По литературным данным [1], S-бензилизотиомочевину получают взаимодействием тиомочевины с хлористым бензилом в спиртовой среде с выделением продукта вакуумупаркой.
Нами предложен метод получения солянокислой соли S-бензилизотиомочевины в водной среде без дополнительной очистки.
СХЕМА СИНТЕЗА
I II НС1
JJ-CHjGI + H.NCSNH,---------
NH,
” ^J-сн,—s-c^ hci.
NH
Характеристика основного сырья
Тиомочевина, ГОСТ 6344—52, ч.
Бензнл хлористый, МРТУ 6—02—538—69, техн.
Соляная кислота, ГОСТ 3118—67.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром и обратным холодильником, загружают 400 мл 4* 51
дистиллированной воды, 106 г (1,4 моль) тиомочевииы и 180 г (1,43 моль) хлористого бензила. Массу при перемешивании медленно нагревают до 65° и выдерживают 45 минут при 65— 70°. После растворения тиомочевины . (допускается слабая муть) в колбу добавляют 10 г активированного угля и продолжают размешивание еще 10—15 минут. Затем массу фильтруют от угля, фильтрат подкисляют 100 мл 35%-ной соляной кислоты (проба на полноту осаждения) и охлаждают до 5°. Выделившийся осадок отфильтровывают.
Выход S-бензилизотиомочевины гидрохлорида равен 240 г (85%), т. пл. 174—175°.
По литературным данным [2], т. пл. 174°; 176°.
ЛИТЕРАТУРА
1. R. Riemschneider, Н. Р е h 1 m a u п. Z. Naturforsch. b., 20 (6), 540 (1965).
2. Справочник химика, т. II. М.—Л., ГХИ, 1963, с. 332.
БЕНЗИЛОВЫЙ ЭФИР БЕНЗОЙНОЙ КИСЛОТЫ
Бензилбеизоат
А. Л. ЛИФИЦ, Е. П. АГЕЕВ, С. И. ОРЕЛ, И. А. БАРЦ
С14Н12О2
^_^-СООСН2-^
М/ в. 212,25
Бензиловый эфир бензойной кислоты применяется в фармацевтической промышленности и органическом синтезе.
Из описанных в литературе методов синтеза бензилбензоата наибольшую практическую ценность представляет получение его нз бензальдегида и бензилового спирта в присутствии метилара натрия [1] и из бензойнокислого натрия и хлористого бензила [2].
Нами разработан метод получения бензилбензоата из хлористого бензила и бензойной кислоты в присутствии кальцинированной соды и катализатора—диметилформамида в среде бензилбеизоата; метод позволяет получать бензилбеизоат высокой степени чистоты с выходом около 90%
СХЕМА СИНТЕЗА
\_/-СООН + \_/-СН,С1
* 0-соосн’-0
Характеристика основного сырья
Бензойная кислота, ГОСТ 6413—67, техн. Бензил хлористый, МРТУ 6—02—538—69, техн. Диметилформамид, ВТУ ЕУ 111—57, техн.
Сода кальцинированная, ГОСТ 5100—64, техн.
53
Условия получения
В трехгорлую колбу емкостью 3 л, снабженную мешалкой, термометром и. обратным холодильником, помещают 420 г (3,45 моль) бензойной кислоты, 280 г (2,65 моль) кальцини* рованной соды, 32 г (0,44 моль) диметилформамида, 460 г (3,65 моль) хлористого беизила и 100 мл бензилбензоата (см. примечание). Реакционную смесь нагревают на глицериновой бане до 120—130° и выдерживают при этой температуре 10— 12 часов. После охлаждения до 60—70° приливают 600— 700 мл воды и размешивают при нагревании (температура массы 80—90°) до полного растворения осадка.
После отстаивания верхний слой бензилбензоата отделяют и перегоняют при пониженном давлении, собирая фракцию при 160—165°/5 мм.
Выход бензилбензоата равен 650 г (89%), п™ = 1,5690; № =' 1,117, т. заст. 17—18°; содержание основного вещества 99%.
По литературным данным, т. пл. 21°, d418 —1,114;
= 1,5681 [3]; т. заст. 17° (4].
Примечание:
В качестве разбавителя используют первую фракцию после вакуум-пе-регоики бензилбензоата с т. заст. не ниже 5 , для первого синтеза бензнл-беизоата используют вместо первой фракции товарный продукт, причем выход эфира соответственно увеличивается.
ЛИТЕРАТУРА
1. Польск. пат. 38400 (1956).
2. Р. Scelba. Bull. chim. farm., 62, 33 (1923).
3. Словарь органических соединении, т. 1. М., Инлнтиздат, 1949, с. 240.
4. Laboratory Chemicals Catalogue, 1969, BDH England, s. 38.
8-БЕН30ИЛАМИН0-1-НАФТ0Л-3,6-ДИСУЛЬФОКИСЛОТА, ДВУНАТРИЕВАЯ СОЛЬ
N-Бензоил-Аш-кислота, двуиатриевая соль
Н. Н. ДЫХАНОВ, Р. Ф. ВИДЕНИНА, И. В. ПОКОТЫЛО, Т. В. ПЕРОВА, В. И. БАЗАКИН, П. Д. ЯКУХНЫИ
НО NH-CO ^_^
АЛ
NaO.S-J ч - SOsNa • 2Н,0
C17HHNNa2O8S2-2H2O М. в. 503,42
8-Амино-1 -нафтол-3,6-дисульфокислота (Аш-кислота), получаемая в промышленности гидролизом 1,8-диаминонафта-лин-3,6-дисульфокислоты [1] илн сплавлением 1-нафтиламин-3,6,8-трисульфокислоты со щелочами [2, 3]. всегда содержит примесь других изомерных аминонафтолсульфокислот, а также 1,8-диоксинафталин-3,6-дисульфокислоты (хромотроповой кислоты), образующейся при дальнейшем гидролизе Аш-кис-лоты как в кислой, так и в щелочной средах [4]. Бензоилиро вание технической Аш-кислоты бензоилхлоридом в водном растворе соды приводит сначала к образованию двунатриевой соли О, N-дибензоил-Аш-кислоты, которая при нагревании превращается в двунатриевую соль N'-бензоил-Аш-кислоты [5]. При добавлении к гидролизату хлорида натрия вместе с двунатриевой солью N-бензоил-Аш-кислоты частично высаливаются и примеси, содержавшиеся в Аш-кислоте. Повторное переоса-ждение из водного раствора хлоридом натрия и последующая однократная перекристаллизация из воды не устраняют полностью примеси, а при многократной перекристаллизации резко снижается выход продукта.
55
С более высокими выходом и содержанием основного Вещества двунатриевая соль N-бензоил-Аш-кислоты получается при очистке технического продукта по методу, предложенному одним из нас ранее для очистки натриевых солей других ароматических сульфокислот [6, 7]: экстракцией солей спиртами с последующим осветлением экстракта и отгонкой экстрагента.
Характеристика основного сырья
Двунатриевая соль N-бензоил-Аш-кислоты, техн., влажная паста, содержащая ~60% смеси сухих веществ.
Метиловый спирт синтетический, ГОСТ 6995—67, ч. Изоамиловый спирт, ГОСТ 5830—51, ч.
Условия очистки
Переосаждение технической двунатриевой соли N-бензоил-Аш-кислоты из водного раствора хлористым натрием проводят по методике переосаждения Р-соли, описанной иа стр. 204 этого сборника. Выход сухой переосаждеиной двунатриевой соли N-бензоил-Аш-кислоты составляет около 65% от веса смеси сухих веществ, содержавшихся в технической соли; для последующей очистки высушивание переосажденного продукта не обязательно.
В трехгорлую колбу емкостью 1 л, снабженную мешалкой с каучуковым абтюратором, водоуловителем (насадка типа Дина и Старка с нижним краном для удаления воды) и запасным отверстием, помещают 65—70 г влажной паеты пере-осажденной двунатриевой соли N-бензоил-Аш-кислоты (содержание сухого вещества ~50 г), 150 мл изоамилового спирта и 0,5 г гидросульфита натрия. Содержимое колбы нагревают на масляной бане до кипения (температура в бане 135—140°) и энергично перемешивают до прекращения накопления воды в водоуловителе (~1 час).
Образовавшуюся суспензию охлаждают до 30—40°, приливают к ней 450 мл метанола, кипятят 15 минут с 5 г активированного угля и фильтруют в горячем состоянии. Фильтрат (550—570 мл) помещают в широкогорлую колбу емкостью 0,5 л, снабженную мешалкой с каучуковым абтюратором и насадкой с нисходящим холодильником, и отгоняют 275—280 мл метанола (см. примечание 1). Остаток охлаждают до комнатной температуры, выпавшие кристаллы отфильтровывают (см. примечание 2), промывают на фильтре дважды по 10 мл безводного изопропилового спирта и сушат при пониженном давлении при 60—70°.
Выход дигидрата двунатриевой соли N-бензоил-Аш-кисло-ты равен 38—40 а (76—80%); вещество отвечает требованиям МРТУ 6—09—2400—65.
56
Примечаний:
1. Метанол отгоняют в таком количестве, чтобы в реакционной смеси его оставалось 150 мл (соотношение с нзоамиловым спиртом 1:1). В этих условиях получается легко фильтрующийся осадок дигидрата двуйатриевой соли N-бензоил-Аш-кислоты.
2. Из фильтрата фракционированием его при обычном давлении регенерируют исходные спирты и используют их в следующем опыте.
ЛИТЕРАТУРА
1. Герм. пат. 67062 (1890); Frdl., 3, 466.
2. Герм. пат. 69722 (1890); Frdl., 3, 468.
3. Takanashi, Snimada, J. Soc. Chem. Ind. Japan, 44, 697 (1941)
4. H. Доналдсон. Химия и технология соединений нафталинового ряда. М., ГНТИХимлит, 1963, с. 432.
5. BIOS. Documents FDX. 673, Frame 195.
6. H. H. Дыханов, Л. М. Егупова. Методы получения химических реактивов и препаратов, вып. 9. М., ИРЕА, 1964, с. 65.
7. Н. Н. Дыханов, Г. Н. Никитенко. Авт. свид. 162855; Бюлл. изобр. № 11 (1964).
N БЕНЗОИЛ-о-ТОЛУИДИН
N-(o -Толил)бензамид
К). Г. БОНДАРЬ
c14h13no
М. в. 211,26
N-Бензоил-о-толуиднн получают взаимодействием о-толуи-днца и бензоилхлорида в среде органического растворителя в 'присутствии щелочного агента [1—3].
Нами проверено влияние природы нейтрализующего агента и температуры реакции на скорость образования и выход продукта. Показано, что лучшие результаты могут быть получены при применении кальцинированной соды и проведении реакции при 100—110°. Использование толуола в качестве растворителя позволило исключить стадию выделения технического продукта.
СХЕМА СИНТЕЗА
NaaCOt
Характеристика основного сырья
о-Толуидин, ТУ МХП 1398—46, техн.
Бензоилхлорид, ТУ ГСНХ 10—76—60, техн.
58
Сода кальцинированная, ГОСТ 5100—64.
Толуол каменноугольный, ГОСТ 9880—61.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешалкой, капельной воронкой н обратным холодильником, помещают 56 г (0,52 моль) о-толундина, 65,5 г (0,62 моль) кальцинированной соды, 900 мл толуола и при перемешивании за 30 минут добавляют по каплям 84 г (0,595 моль) бензоилхлорида. Реакционную смесь нагревают до кнпеиия и кипятят 1 час, затем добавляют активированный уголь и фильтруют. Фильтрат охлаждают до 10°, осадок отфильтровывают.
Выход N-бензоил-о-толуиднна равен 94 г (84,5%), т. пл. 144°.
По литературным данным [4J, т. пл. 145—146°.
ЛИТЕРАТУРА
1. К. Auswers. Вег., 52, 1335 (1919).
2. К. В ей г а н д-Х и л ь г ет а г. Методы эксперимента в органической химии. М., Химия, 1968, с. 431.
3. Н. Н. Ворожцов. Основы синтеза промежуточных продуктов и красителей. М., Госхимиздат, 1955, с. 530.
4. Словарь органических соединений, т. Ш. М., Иилитиздат, 1949. с. 779.
БИС( п НИТРОФЕНИЛ ГИДРАЗОН) а, р-ДИКЕТОБУТИРОЛАКТОНА
3,4-Бис( л-нитрофенилгндразоио)-2-оксотетрагидрофуран
И. в. хвостов
O2N—f V-NH-N=C—C=N-NH-^ 5-NO2 , । \=/
H9C co xoz
C^HisNeOg
M. в. 384, 31
Бис(л-нитрофенилгидразон) а,р-дикетобутиролактона в литературе не описан. Вещество синтезировано взаимодействием а-бром-0-кетобутиролактона (а-бромтетроновая кислота) с л-нитрофенилдиазонием в присутствии карбоната калия с выделением промежуточного продукта — мононитрофенилгид-разона а, p-дикетобутиролактона, кратко описанного в работе [1]; последний превращен в конечный продукт действием л-иитрофенилгидразина в уксуснокислой среде.
СХЕМА СИНТЕЗА
ОС-СНВг
I I
Н2С со 'о/
O2N-^ ^-n2+oh
ос—C=N-NH-^ S-NO, -» I I
H,C CO
60
_ 0,N-^ NH—N—С—C = N—NH—/-N02
— I I
H2C co о
Характеристика основного сырья
а-Бром-р-кетобутиролактон, т. пл. ~ 180° (разл.); см. примечание.
n-Нитроанилин, ТУ 6—09—258—70, техн.
Соляная кислота, ГОСТ 3118—67, х. ч.
Натрий азотистокислый, ГОСТ 4197—66.
Калий углекислый, ГОСТ 4221—65.
n-Нитрофенилгидразин, МРТУ 6—09—5827—69.
Условия получения
Синтез п-нитрофенилгидразона а, $-дикетобутиролактона. В фарфоровом стакане растворяют 1 г (~0,055 моль) а-бром--p-кетобутиролактона в растворе 1 г углекислого калия в 20 мл воды. При этом наблюдается вспенивание раствора. К полученному еноляту калия при охлаждении льдом и постоянном размешивании медленно по каплям приливают холодный раствор п-нитрофенилдиазония, приготовленного из 0,7 г (~0,05 моль) п-нитроанилина, 10 мл 30%-ной соляной кислоты и 0,4 г (~0,05 моль) нитрита натрия. По мере прибавления диазораствора к калиевой соли бромлактона наблюдается выделение оранжевого осадка. Реакционный раствор размешивают еще 45 минут в ледяной баие, а затем оставляют на 30 минут при той же температуре. Продукт оранжевато-жел-того цвета отфильтровывают на стеклянном фильтре, промывают холодной водой и сушат в эксикаторе.
Выход п-нитрофенилгидразона а, р-дикетобутиролактоиа равен 0,8 г (58%), т. пл. 195—198°.
После двукратной перекристаллизации из ледяной уксусной кислоты получают продукт в виде золотистых пластинок с т. пл. 214°.
По литературным данным [1], т. пл. 214°.
Найдено, %: N—16,82; 16,9.
CioHtNsOs. Вычислено, %: N—16,86.
Получение бис(п-нитрофенилгидразона) а, ^-дикетобути-ролактона. Растворяют 0,15 г (0,0005 моль) п-нитрофенилгид-разоиа а, p-дикетобутиролактона при нагревании до 110° в небольшом количестве (~15 мл) ледяной уксусной кислоты и к полученному раствору при нагревании на водяной бане добав
61
ляют раствор 0,1 г (0,00066 моль) п-нитрофенилгидразнна в 20 мл 10%-ной уксусной кислоты. Очень скоро начинает выпадать ярко-красный мелкокристаллический осадок. Продукт отфильтровывают на стеклянном фильтре и промывают разбавленной уксусной кислотой.
Выход бис(п-нитрофенилгйдразона) аф-дикетобутиро лактона равен 0,2 г (~90%); вещество плохо растворимо в спирте, уксусной кислоте, этилацетате. После перекристаллизации из нитробензола вещество плавится при 298° с разложением (быстрое нагревание).
Найдено, %: N—21,73.
CisHizNeOe. Вычислено, %: N—21,86.
Пр имечание.
а-Бром-Р-кетобуТироЛактон был получен в соответствии с литератур ными данными [2] бромированием ацетоуксусного эфира в среде днэтило-вого эфира с последующей циклизацией а, у-дибромацетоуксусного эфира при нагревании в вакууме водоструйного насоса.
ЛИТЕРАТУРА
1. В. В. Ф е о ф й л а к т о в. Изв. АН СССР, ОХН, 4—5, 521 (1941).
2. L. Wolff, S. Schwabe. Ann.. 291, 231 (1896).
_и-БИС(.и-ФЕН0КСИФЕН0КСИ)БЕН30Л
Н. Ю. А РОНСКАЯ, Е. И. МАЛОВЕР, Р. М. ГЕЛЬ ШТЕЙН, В. Д. БЕЗУГЛЫЙ
С30Н22О4 М. в. 446,50
л-Бис(ж-феноксифенокси) бензол применяется в качестве рабочей жидкости для получения сверхвысокого вакуума [1], масляной основы, высокотемпературных радиационностойких смазок [2], в хроматографии.
По литературным данным, л«-бис(л«-феноксифенокси) бензол получают по реакции Ульмана конденсацией л-фенокси-фенола с л-дибромбензолом; л-феноксифеиол получают одним из известных способов: из л-феиоксиметоксибензола, зг-феноксибромбензола или из л<-феиоксиаиилина [3].
Нами усовершенствован способ получения лг-феноксифено-ла из резорцина .и бромбензола в среде N-диметилформамида [4, 5] н способ получения и очистки ж-бис(л«-феноксифенокси)-беизола [6], что позволило упростить процесс и получить продукт с высокой термоокислительной стабильностью.
СХЕМА СИНТЕЗА
НО__________ОМ Rr—\ ПМ<НА
—О—О—о___О—
I II I II I I I II I 5
Ч/ Ч/ Ч/ ч/
63
Характеристика- основного сырья
Резорцин, ГОСТ 9970—62, техн.
Бромбензол, МРТУ 6—09—5181—68, ч. л-Дибромбензол, МРТУ 6—09—6023—69,- ч. N-Диметилформамид, ГОСТ 5.703—70, ч.
Кали едкое, ГОСТ 4203—65, ч. д. а.
Бензол, ГОСТ 5955—68, ч. д. а.
Алюминий окись д/хроматографии, МРТУ 6—09—5296— —68, ч.
Условия получения
Синтез м-феноксифенола. В четырехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром, капельной воронкой, трубкой для подачи азота и елочной колонкой высотой 20 см с нисходящим холодильником, помещают 400 г (3,64 моль) резорцина, 572 г (3,64 моль) бромбензола, 480 мл N-диметилформамида и 20 а окиси меди. Реакционную смесь нагревают при перемешивании на глицериновой бане в токе азота др 140—145° и из капельной воронки постепенно добавляют раствор 190 г (2,88 моль) едкого кали в 100 мл воды, при этом отгоняется диметилформамид и бромбензол с водой, которые периодически возвращаются в реакционную колбу. Затем смесь нагревают до 150—155°, выдерживают при этой температуре 4 часа, охлаждают до 30° и выливают в колбу емкостью 3 л, 'содержащую 1,5 л воды. Выделившееся масло отделяют и дважды экстрагируют 15%-ным раствором едкого натра порциями по 350 мл (см. примечание).
Полученный щелочной раствор л-феноксифенола подкисляют 300 мл соляной кислоты до pH 2 и выделившийся масляный слой дважды экстрагируют бензолом порциями по 200 мл. Бензольный экстракт промывают водой 2—3 раза порциями по 500 мл до нейтральной реакции. Растворитель отгоняют, а остаток ректифицируют при пониженном давлении, отбирая фракцию с т. кип. 161—16275 мм.
Выход л-феноксифенола равен 136 г (20%), /1д° = 1,6005, di20 — 1,172; содержание основного вещества (метод ГЖХ) 99%.
По литературным данным [3], Яд = 1,6005.
Получение м-бис(м-феноксифенокси)бензола. В четырехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром, капельной воронкой и елочной колонкой высотой 20 см с нисходящим холодильником, помещают 300 г (1,61 моль) л/-феноксифенола, 185 г (0,784 моль) л-дибромбензола и 4 г меди в порошке. Смесь напревают на силиконовой бане до 170—180° и при этой температуре прибавляют из капельной воронки раствор 103 г (1,56 моль) едкого кали в 75 мл воды, 64
возвращая периодически в реакционную колбу отгоняющийся с водой л-дибромбензол. Затем смесь выдерживают 3 часа при 190—200° и 2 часа при 220—230°, охлаждают до 30—40°, добавляют при размешивании 1 л бензола и 300 мл 10%-ного раствора едкого натра. Бензольный раствор отделяют, промывают 500 мл 10%-ного раствора едкого натра, водой 2—3 раза порциями по 500 мл до нейтральной реакции и сушат хлористым кальцием. Отфильтрованный раствор эфира в бензоле для осветления пропускают при пониженном давлении (300— 350 мм) через стеклянную колонку (Л = 800 мм, Q = 60— 70 мм), заполненную на 2/3 окисью алюминия (~400 г). Растворитель сначала отгоняют при атмосферном давлении, затем при пониженном (~10 мм), а остаток фракционируют в вакууме, собирая фракцию с т. кип. 270—275°/0,1—0,05 мм.
Выход ж-бис (ж-феноксифенокси) бензола равен 250 г (70,0%), п% = 1,6300; л5о= 116 сст.
По литературным данным [3], = 1,6322.
Примечание.
После отделения л-феноксифенола остается бромбензол (270 г), который перегоняют и используют в последующих синтезах.
ЛИТЕРАТУРА
1. М. Л. Алашкевич, В. И. Мирнманова. Приборы и техника эксперимента, |1, 6, 157 (1966).
2. А. И. Д и и ц ес, Г. В. К л очко, Ю. И. Турский. Нефтехимия, 9, № 3, 450 (1969).
3. К. J. Sax, W. S. Saary, С. Z. М a h о п е у, J. М. О о г d о п. J. Organ. Chem., 25, 1590 (1960).
4. Пат. США 3294846; С. А., 66, 65281 (1967).
5. Н. Ю. Аронская, Р. М. Г е л ь ш т е й н, В. Д. Безуглый. Ав г. свид. 281481; Открытия. Изобретения. Промышленные образцы. Товарные знаки, № 29, 41 (1970).
4. Пат. США 3083234, С. А., 59. 8657с (1963).
5 Зак. ШИ
7-БР0М-1-АЦЕТИЛ 5-ФЕНИЛ-1,2-ДИГИДРО-ЗН-1,4-БЕНЗДИАЗЕПИН-2-ОН
3. Я. ЖИЛИНА. А. В. БОГАТСКИЙ, С. А. АНП.РОНАТИ. Т. к. ЧУ МАЧ ЕН КО
Н8ССО
I II сн /%/\c=Nz
Br I
С6н5
CnHiaBrNaOa
М. в. 356,17
1,2-Дигидро-ЗН-1,4-бенздиазепин-2-оны известны как психофармакологические препараты, применяющиеся в качестве транквилизаторов, седативных и гипнотических средств (1„ 2].
Ранее нами впервые была показана возможность получения 1-ацетильных производных указанного ряда соединений взаимодействием натриевых солей 1,2-дигидро-ЗН-1,4-бенз-диазепин-2-онов с хлористым ацетилом [3].
Неописанный ранее 7-бром-1-ацетил-5-фенил-1,2-дигидро-ЗН-1,4-бенздиазепин-2-он синтезирован действием хлористого ацетила на натриевую соль 7-бром-5-фенил-1.2-дигидро-ЗН-1,4-бенздиазепин-2-она.
СХЕМА СИНТЕЗА
/Ч/\
Вг
NH-CZ
сн2
C^N-/
С.Н,
CH3ONa , С1СОСН,
Н8ССО _
I Z° ^X/N-Cx I II сн3 Br/X/\C=N/
c,Hf
66
Характеристика основного сырья
7-Бром-5-фенил-1,2-дигидро-ЗН-1,4-бенздиазепин-2-он, т. пл. 220—221°, получение см. [3, 4].
Натрий металлический, ТУ 6—09—356—70, ч.
Бензол, ГОСТ 5955—68, х. ч.
Ацетил хлористый, ГОСТ 5829—51, ч.
Этиловый спирт, ГОСТ 8314—57.
Условия получения
В колбу емкостью 250 мл, снабженную мешалкой и обратным холодильником, вносят раствор 2,5 г (0,008 моль) 7-бром-5-феиил-1,2-дигидро-ЗН-1,4-бенздиазепин-2-она в 150 мл абсолютного бензола, нагревают на водяной бане до кипения и к кипящему раствору при помешивании прибавляют 0,81 г (0,015 моль) метилата натрия. Реакционную смесь кипятят до полного растворения алкоголята, после чего отгоняют 50 мл бензола и к раствору по каплям прибавляют 0,78 г (0,01 моль) хлористого ацетила. Раствор кипятят при перемешивании 40 минут, охлаждают до комнатной температуры, трижды промывают водой порциями по 50 мл, сушат прокаленным сульфатом натрия. Бензол полностью отгоняют при пониженном давлении, а остаток кристаллизуют в 50 мл этанола.
Выход 7-бром-1-ацетил-5-фенил-1,2-дигидро-ЗН-1,4-бензди-азепин-2-она равен 1,2 г (54%), т. пл. 184—185°.
Найдено, %: С—57,5; Н—3,4; N—7,9. CirHijBrNaCh. Вычислено, %: С—57,3; Н—3,4; N—7,9.
ЛИТЕРАТУРА
1. А. В. Богатский, С. А. Ан др он ат и. Успехи химии, 39, 2217 (1970).
2. Ю. И. Вихляев, Т. А. Клыгуль. Ки. «Итоги науки. Фармакология. Химиотерапевтические средства. Токсикология». М„ ВИНИТИ, 1968, с. 38.
3. С. А. А и д р о н а т и, А. В. Богатский, Ю. И. Вихляев, 3. И. Жилина, Б. М. Кац, Т. А. Клыгуль, В. Н. Худякова, Т. К. Чумаченко, А. А. Э и и а и. Ж- общ. химии, 40, 1881 (1970).
4. L. Н. Sternbach. R. 1. Fryer. W. Metleslcs, E. Reeder, Q. S a c h, Q. Saucy, A. S t e m p e 1. J. Organ. Chem., 27, 3788 (1962),
4-БРОМ-3-МЕТИЛ-5-(-БРОМ-2-ФУРИ Л) ПИРАЗОЛ
| А. А. ПОНОМАРЕВ, [ JL В. ЧЕРКЕСОВА, А. А. МОРЩАКОВА
Вг
I н
CgHeB^NzO М. в. 306,96
4-Бром-3-метил-5- (5-бром-2-фурил)пиразол получен нами впервые прямым бромированием 5-(2-фурил)-3-метйлпиразо-ла диоксандибромидом. Монобромзамещенное соединение этим методом получить не удалось.
СХЕМА СИНТЕЗА
1 U Н в JI J - сг 68 -сн3 СД^Оз'ВГэ Вг 4 j-CH3 N Н
Характеристика основного сырья
Бром, ГОСТ 4109—48, х. ч.
Диоксан, ГОСТ 10455—63, ч.
5-(2-Фурил)-3-метилпиразол, т. пл. 89—90° (из петролейно-го эфира); получение см. [1, 2].
Условия получения
В двугорлую колбу емкостью 100 мл, снабженную мешалкой и капельной воронкой, помещают 1 г (0,007 моль) фурил-метилпиразола в 8 мл эфира, постепенно при перемешивании прибавляют 3,4 г (0,014 моль) диоксандибромида, растворенного в 15 мл эфира (см. примечание 1). Вскоре выпадает желтый осадок, который постепенно темнеет. Осадок отфильтровывают, хорошо промывают эфиром и сушат при комнатной температуре.
Выход сырого 4-бром-3-метил-5-(5-бром-2-фурил) пиразола равен 1,8 г (87%).
После перекристаллизации из 50%-ного водного этанола получают иглообразные кристаллы желтого цвета с т. пл. 128—130°. Вещество хорошо растворимо в бензоле, диоксане, ацетоне, дихлорэтане и плохо растворимо в воде.
Найдено, %: С- 31,08; 31,47; Н—1,96; 2, 11; N—9,43; 9,53. CsH6Br2N2O. Вычислено, %: С—31,40; Н—1,98; N—9,15.
N'-Ацетильное производное имеет т. пл. 101—103° (из 50%-ного этанола), выход 90% (см. примечание 2).
Примечания:
1. Диоксандибромид готовят следующим образом [3, 4]: в фарфоровый стакан емкостью 1 л наливают 50 г свежеперегнаниого диоксана и быстро, при перемешивании и охлаждении ледяной водой, прибавляют 99 г (31,7 мл) брома. Полученный горячий раствор выливают при перемешивании в 200 лсл воды со льдом, причем образуется оранжевый кристаллический осадок. Осадок отсасывают и тщательно отжимают иа вороике Бюхнера, а затем досуха фильтровальной бумагой, выход 91%.
2. N-Ацетильное производное получают действием уксусного ангидрида на 4-бром-3-метил-5- (5-бром-2-фурил) пиразол.
ЛИТЕРАТУРА
1. И. И. Г р а н д б е р г, А. И. Кост, Д. В. С и б и р я к о в а. Ж. общ. химии, 30, 2920 (1960).
2. А. А. Пономарев, Л. В. Черкесова. Ж. общ. химии, 33, 3946 (1963).
3. Л. А. Яновская, А. П. Терентьев, А. И. Б е л е н ь к ий. Ж. общ. химии, 22, 1594 (1952).
4. А. П. Терентьев, А. И. Б е л е н ь к ий, Л. А. Яновская. Ж. общ. химии, 24, 1265 (1954).
а-БРОМТЕТРОНОВАЯ КИСЛОТА
а-Бром-р-кетобутиролактон;
3-бром-2,4-диоксотетрагидрофуран
И. В. ХВОСТОВ, О. А. ЛОГИНОВА
ОС=СВг
С4Н3ВГО3
ОС----СНВг
I I
Н2С СО
\о/
I I + н+
Н2С СО хох
М. в. 178,97
а-Бромтетроновая кислота применяется в фотографических процессах [1]; при синтезе жирно-ароматических азосоединений [2], антимикробных препаратов [31
а-Бромтетроновую кислоту получали нагреванием а, у-ди-бромацетоуксусного эфира при пониженном давлении: 15 мм [4J или 30—40 мм с выходом ~35% [5].
Нами внесены некоторые изменения в метод [41: реакция проведена при более низкой температуре и более высоком вакууме, что снизило количество побочных смолистых и лакри-могенных веществ.
СХЕМА СИНТЕЗА
О
Вг
СНз-СО—СН2-С—ОС2Н5 —
о
-> СН2—СО—СН—С—ОС2НВ ОС-СНВг
II II
Вг Вг Н2С СО
70
о
Характеристика основного сырья
Ацетоуксусный эфир, ГОСТ 9799—61.
Бром, ГОСТ 4109—64.
Условия получения
Синтез а.у-дибромацетоуксусного эфира. В колбу емкостью 1 л, снабженную мешалкой, помещают 150 г (140 мл; 1,15 моль) свежеперегнанного ацетоуксусного эфира и 140 мл диэтнлового эфира и при перемешивании по каплям добавляют 369 г (118 мл; 2,3 моль) брома, при этом наблюдается интенсивное выделение НВг. Реакционную массу оставляют на ночь. Окрашенную в красноватый цвет, сильно дымящую реакционную массу промывают 5—6 раз небольшим количествам холодной воды и сушат прокаленным хлористым кальцием. Диэтиловый эфир отгоняют.
Выход сырого а, удибромацетоуксусного эфира в виде желтого масла равен 304 г (~91%).
Получение а-бромтетроновой кислоты. В колбу Кляйзена емкостью 250 мл, снабженную капилляром, помещают 76 г (~0,26 моль) неочищенного а, у-дибромацетоуксуспого эфира. Колба через склянку Дрекселя соединена с водоструйным насосом. Содержимое колбы осторожно нагревают на масляной бане (температура банн НО—120°) при пониженном давлении 10—15 мм в течение 3 часов. Уже через 1,5—2 часа на чинают выпадать кристаллы бромлактона. Реакционную массу охлаждают до комнатной температуры и оставляют на ночь. Массу отфильтровывают на воронке со стеклянным фильтром, тщательно отжимают от маточного раствора, промывают небольшим количеством (5—10 мл) эфира.
Выход сырого бромлактона равен 15 г (32,3%).
После перекристаллизации из горячей воды получают 12 г а-бромтетроновой кислоты с т. пл. 181 —183° (разл.).
По литературным данным [41, т. пл. 183° (разл.).
Вещество показывает характерную для енолов реакцию с хлорным железом, окрашивает подобно сильным кислотам бумагу конго в синий цвет и выделяют йод из йодистого калия.
ЛИТЕРАТУРА
1. Франц, пат. 1374100 (1965); Chem. Zbl, 32, 3312 (1967).
2. В. В. Феофилактов. Изв. АН СССР, о. х. н., 526 (1941).
3. W. Kumler, J. Amer. Chem, Soc., 62, 2560 (1940).
4. Z. L. Wolff, K, Schwabe. Ann., 291,231 (1896).
5. W. Kumler. J. Amer. Chem. Soc., 60, 859 (1938).
NБУТИЛХИНАЛЬДИНИЙ ИОДИД
И. С. МАРКОВИЧ, Л. И. БЛОХИНА, В. М. ДЗИОМКО
CuHielN
М. в. 327,209
N-Бутилхинальдиний иодид в литературе не описан. Нами это соединение получено взаимодействием хинальдина с йодистым бутилом.
СХЕМА СИНТЕЗА
кААсн.
С4Н91
Характеристика основного сырья
Хинальдин, МРТУ 6—09—6234—69, ч.
Бутил иодистый, МРТУ 6—09—6512—70, ч.
Бутиловый спцрт, ГОСТ 6006—51, ч.
Условия получения
В круглодонную колбу, снабженную мешалкой и обратным холодильником, помещают 30,3 г (0,21 моль) хинальдина и 44,0 г (0,24 моль) йодистого бутила. Реакционную массу нагревают 24 часа при 125°с размешиванием.
72
Ёыпавший осадок отфильтровывают и перекристаллизовывают из бутилового спирта. Выход продукта равен 42 г (62,7%), т. пл. 162°.
После трехкратной перекристаллизации из бутилового спирта получают 32,6 г (50%) N-бутилхинальдиний иодида в виде желтых кристаллов с т. пл. 163—164°.
Найдено, %: С—51,10; 50,83; Н—5,52; 5,42; N—4,11; 4,11.
CuHisIN. Вычислено, %: С—51,4; Н—5,54; N—4,28.
'-миге = 302 нм, 390 нм, sH4Kc = 13750, 640 (соответственно).
2,4-ГЕКСАДИЕНАЛЬ
О. С. СТЕПАНОВА. А. И. САЛАТИНА
CgHgO
СНз—СН='СН—СН=СН—СНО
М. в. 96,06
2,4-Гексадиеналь, или сорбиновый альдегид, образуется при дегидрировании н-капронового альдегида в водной суспензии коллоидным палладием [II; как побочный продукт при конденсации ацетальдегида в присутствии ацетата натрия [2]; при восстановлении гексанола в атмосфере СО2 в присутствии ацетата никеля [31; при конденсации 1-пипер идин-1,3-бутадие-на с ацетальдегидом и последующей обработке продуктов реакции смесью уксусной кислоты с ацетангидридом [4].
Нами 2,4-гексадиеналь получен по видоизмененной методике Куна и Гоффера [51 конденсацией кротонового Альдегида с ацетальдегидом.
СХЕМА СИНТЕЗА
СН3СН=СН-СНО + СНз-СНО ->
-> CHS-CH=CH-CH=CH-CHO + Н2О
Характеристика основного сырья
Уксусный альдегид, МРТУ 6—09—5708—68, ч.
Кротоновый альдегид, ТУ 8П—244—69, ч.
Пиперидин, МРТУ 6—09—6355—69, ч., свежеперегнанный.
Натрий сульфат, безводный, ГОСТ 4166—66, ч.
Диэтиловый эфир, ГОСТ 6265—52, ч.
Условия получения
В трехгорлую колбу с трехрогим форштосом (либо в пя-тигорлую колбу) емкостью 1 л, снабженную мешалкой, обрат-74
ным холодильником (шариковым), капельной воронкой, термометром и газоподводной трубкой, помещают 140 г (2 моль) кротонового альдегида и 176 г (4 моль) ацетальдегида. Из системы вытесняют воздух током азота. Из воронки приливают 3 мл свежеперегна иного пиперидина и смесь оставляют стоять 1—2 часа. Затем реакционную смесь нагревают на водяной бане в течение 8 часов. При 40° жидкость начинает бурно кипеть, при 60—65° заметно выделение воды. К концу реакции количество реакционной воды составляет 30 мл. По достижении температуры 75—80° к реакционной смеси добавляют еще 5 мл пиперидина; кипение постепенно прекращается. После окончания реакции и охлаждения водный слой отделяют, экстрагируют трижды по 10 мл эфиром. Эфирную вытяжку соединяют с основной массой и сушат безводным сульфатом натрия. Эфир отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 54—57°/9 мм.
Выход 2,4-гексадиеналя равен 86,4 г (45%); <Л20 = 0,9591; п% = 1.5341.
Найдено, %: С—75,00, Н—8,60. С6Н8О. Вычислено, %: С—74,50; Н—8,30.
Данные ИК- и ЯМР-спектров показали, что полученный 2,4-гексадиеналь представляет собой смесь двух изомеров. В ИК-спектрах имеются полосы поглощения внеплоскостных деформационных колебаний СН-групп, характерных как для цис-замещения (650—750 см-1), так и для траяс-замещения (960—1000 см-1) при этиленовых связях. В области валентных С—С колебаний присутствуют асимметричные и симметричные полосы поглощения, характерные как для цис-(1560 и 1660 см-1), так и для транс- (1630—1700 см~‘) замещения.
Наличие двух изомеров вытекает также из ряда особенностей спектра ПМР сорбинового альдегида. Каждой неэквивалентной группе протонов СН3—, —СН=СН—СН=СН— и —-СНО отвечают две резонансные линии с различными значениями химических сдвигов: для метильной группы при ненасыщенном углеродном атоме —два дублета со средними химическими сдвигами 6 = 0,85 и 1,85 м. д., для олефиновых протонов — широкая мультиплетная линия в интервале 5,00— 7,16 м. д., для альдегидной группы — две острые линии с химическим сдвигом 9,35 и 9,45 м. д.
ЛИТЕРАТУРА
1. F. Baumgarten, О. Glatzel. Вег.. 859, 2661 (1948).
2, R. Zeisel, J. Neu wirth, Ann., 433, 126 (1926).
3. F. Hoaglin, J. Hirsch. J. Amer. Chem. Soc., 71, 3472 (1949), 4. H. Langenbeck, К. О о d d e. X. Atti de Congresse Internationale di Chimica. R. 1938, Br. 3, C. A, 34, (1940).»
5. R. Kuhn, M. Hoffer. Ber., 63,2168 (1930).
75
«-ГЕКСИЛКРОТОНОВАЯ КИСЛОТА
Г. Ф. ТАНЦЮРА, А. В. БОГАТСКИЙ
сна-сн=с-соон
СеН18
CioHreOs
170,25
а-Алкилкротоновые кислоты и их производные входят в состав природных соединений, используются в диеновом синтезе, при получении полимерных материалов. Синтезы а-алкилкротоновых кислот разработаны мало, что ограничивает их использование для практических целей. Некоторые из известных в настоящее время методов синтеза а-алкилкротоновых кислот позволяют получать их с незначительный выходом (10—12%) [1—3], другие, давая больший выход продукта, являются многостадийными [4—5].
Кислотное [6] и эфирное [7] расщепление алкил-а-алкокси-этилацетоуксусных эфиров позволяет предложить удобный метод получения а-алкилкротоновых кислот. Метод включает четыре стадии, суммарный выход а-алкилкротоновых кислот, считая на ацетоуксусный эфир, в среднем равен 20 %. а-Гек-силкротоновая кислота получена нами впервые.
СХЕМА СИНТЕЗА
II с-сн3
хс—ОС3Н6
С6Н5Вг
C2H5ONa
76
о II
OR CeH,3 С-СН3
Na; СНз-СН—С1 C2H5ONa ,
CHS-HCZ ХС-ОС2Н6
I II ОСНз о
СН3- СН=С-СООС2Н6 кон т сн3-сн=с-соон
свн13 с6н13
Характеристика основного сырья
Ацетоуксусный эфир, ГОСТ 9799—61, ч.
Этиловый спирт гидролизный, ТУ 19П—39—69.
Натрий металлический, ТУ 6—09—356—70, ч.
Бромистый гексил, ТУ 6—09—503—70.
Серная кислота, ГОСТ 4204—66—2, концентрированная.
Метанол, ГОСТ 6995—67, ч.
Углекислый натрий, ГОСТ 4201—66, х. ч.
Кали едкое, ГОСТ 4203—65, х. ч.
Условия получения
Синтез этилового эфира а-гексилкротоновой кислоты. В двугорлой колбе, снабженной обратным холодильником, к 45 мл этилового спирта добавляют 0,9 г металлического натрия. После полного растворения натрия по каплям добавляют 81,6 г (0,3 моль) гексил-а-метоксиэтилацетоуксусного эфира. Смесь кипятят с одновременной отгонкой на колонке Вигре высотой 60 см образующегося уксусноэтилового эфира. После полной отгонки уксусноэтилового эфира нагревание прекращают. К смеси добавляют 300 мл 5%-ной серной кислоты, выделившийся эфирный слой отделяют, а водный трехкратно экстрагируют эфиром по 20 мл. Соединенные эфирные вытяжки промывают 30 мл 10%-ного раствора бикарбоната натрия, затем 30 мл воды и высушивают прокаленным сульфатом натрия. Эфир отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 92°/3 мм.
Выход этилового эфира а-гексилкротоновой кислоты равен 21 г (35%); = 0,8812; = 1,4335.
Найдено, %: С—72,35; Н—11,34; MRD = 58,67 С12Н22О2. Вычислено, %: С—72,28; Н—11,81; MR D = 58,80.
Получение а-гексилкротоновой кислоты. В двухгорлой колбе с капельной воронкой и обратным холодильником к
77
приготовленному 10%-ному раствору едкого кали в 30%-ном спиртовом растворе по каплям добавляют 19,8 г (0,1 моль) этилового эфира а-гексилкротоновой кислоты. Смесь нагревают* на масленой бане 4 часа при 120°, после чего отгоняют спирт. После охлаждения реакционную смесь дважды экстрагируют эфиром по 20 мл. К остатку прибавляют 10%-ный раствор серной кислоты до кислой реакции по конго. Выделившуюся кислоту экстрагируют эфиром 3—4 раза по 25 мл. Эфирные вытяжки высушивают безводным сульфатом натрия. Эфир отгоняют, а остаток фракционируют при пониженном давлении, собирая фракцию с т. кип. 126—127°/3 мм.
Выход а-гексилкротоновой кислоты равен 10,2 г (60%); d4m = 0,9186, «“= 1,4530.
Найдено, %: С—70,56; Н—10,56; MRD = 50,05.
CioHisOj. Вычислено, %: С—70,49; Н—10,06; MRD = 49,48.
ЛИТЕРАТУРА
1. 1. Wislicenus, R. Р и с к е г t. Ann., 250, 234 (1880).
2. A. Michael, J. Ross. J. Amer. Chem. Soc., 55, 3684 (1933).
3. А. К. Плисов, А. В. ,Б о г а т с к и й. Ж. общ. химии, 27, 360 (1957).
4. А. В. Богатский, Н. А. Г о р я ч у к, Г. И. П а р н а к. Ж. общ. химии, 32, 1498 (1962).
. 5. А. В. Богатский, Г. Л. Камалов, Н. Л. Вострова. Ж. орган, химии, 75, 2147 (1969).
6. А. В. Б о г а т с к и й, Ю. Ю. С а м и т о в, Г. Ф. Танцюра, С. Г Соболева. Ж. орган, химии, 1, 1987 (1965).
7. А. В. Богатский, Ю. Ю. Самитов, Г. Ф. ТаИцюра, С. А. . Петр а ш. Ж. орган, химии, 3, 1376 (1967).
4-н-ГЕКСОКСИБЕНЗИЛХЛОРИД
3. С. СИДЕНКО, В. А. ИНШАКОВА, Г. С. ЧИЖОВА
освн18 I
СН2С1
С1зН19С1О
М. в. 226,735
4-нТексоксибензилхлорид в литературе не описан. Нами это соединение получено хлорметилированием н-гексоксибен-зола.
СХЕМА СИНТЕЗА
ос.н„
(СНаО)3, НС1 ч/
ОСвН18
I
ч/
I
СН,С1
Характеристика основного сырья
Гексоксибензол, т. кип. 109—110°/5 мм, п® = 1,4910; получение см. [1].
Бензол, ГОСТ 5955—51, ч. д. а.
Триоксан, ВТУ МХП 2711—55, ч.
Натрий сернокислый безводный, ГОСТ 4166—48, ч.
79
Условия получения
В четырехгорлую колбу загружают 54 г (0,3 моль) гексоксибензола, 95 мл сухого-бензола и 37,8 г (0,42 моль) триоксана, охлаждают смесь до 0—2° и пропускают ток сухого хлористого водорода в течение 4,5 часа. Затем реакционную смесь перемешивают при комнатной температуре 3 часа, отделяют бензольный слой, промывают 3—4 раза ледяной водой и сушат над сернокислым натрием. Бензол отгоняют, а остаток перегоняют в вакууме, собирая фракцию с т. кип. 162— 16377 мм.
Выход 4-н-гексоксибензилхлорида равен 35 г (51% в расчете на гексоксибензол), л2д = 1,5130.
Найдено, %: С—68,22; 68,47; Н—8,42; 8,14; С1—15,26; 15,46. C13H19CIO. Вычислено, %: С—68,87; Н—8,38; С1—15,67.
ЛИТЕРАТУРА
1. A. I. Vogel. J. Chem. Soc., 1948, 616.
1,4-ДИАЦЕТИЛ БЕНЗОЛ
Е И. МАХОВЕР
СюНщОг
СНзОС-^ _^-СОСН,
М. в. 162,19
В литературе описан метод получения 1,4-диацетилбензола окислением 4-этилацетофенона перманганатом калия в нейтральной среде [1, 2], однайо имеющиеся прописи разнятся условиями проведения синтеза и выходом продукта.
Нами проверены эти способы получения и установлены оптимальные соотношения компонентов и порядок их загрузки, температурные условия и метод очистки 1,4-диацетилбензола.
СХЕМА СИНТЕЗА
СН,СН2-^ _^-СОСН8 СН8ОС-^_^-СОСН,
Характеристика основного сырья
4-Этилацетофенон, ТУ TCP 647 р—62, ч.
Магний окись, ГОСТ 4526—67, ч. д. а.
Азотная кислота, ГОСТ 4461—67, ч.
Калий марганцовокислый, ГОСТ 5777—71, техн.
Изопропиловый спирт, ГОСТ 9805—69, техн.
Бензол, ГОСТ 5955—68, ч. д. а.
Условия получения
В четырехгорлую колбу емкостью 3 л, снабженную мешалкой, термометром и обратным холодильником и помещенную в глицериновую баню, загружают 1,5 л воды и 38 а (0,95 моль) окиси магния, приливают ~ 150 мл 60%-ной азотной 6 Зак. 1164 81
кислоты до достижения pH 7 и затем 90 г (0,6 моль) этилацетофенона. Содержимое колбы нагревают до 65° и за 2,5—3 часа прибавляют 130 г (0,82 моль) перманганата калия, добавляя каждую следующую порцию (8—10 г) после обесцвечивания предыдущей. Температура реакционной смеси при этом повышается до 70—80°.
К реакционной массе приливают 0,7 л бензола, кипятят 1 час, фильтруют, разделяют слои и из бензольного слоя полностью отгоняют бензол сначала при атмосферном, затем при пониженном давлении (200—300 мм). Остаток охлаждают до 3—5° и выделившийся осадок 1,4-диацетилбензола отфильтровывают на воронке Бюхнера. Фильтрат (неокисленный 4-этил-ацетофенон ~30 г) используют в следующих синтезах.
После перекристаллизации 40 г технического продукта из 200 мл изопропилового спирта с применением активированного угля получают 30 г 1,4-диацетилбензола (46% в расчете на вступивший в реакцию 4-этилацетофенон), т. пл. 113—114°.
По литературным данным [1], т. пл. 113—114°.
ЛИТЕРАТУРА
1. А. М. Сладков, С. В. Вит. Ж. общ. химии, 26, ИЗО (1956).
2. К. В е й г а и д-Х и л ь г е т а г. Методы эксперимента в органической химии. М., «Химия», 1968, с. 296.
ДИАЦЕТОЛОВЫЙ ЭФИР МЕТИОНОВОЙ кислоты
А. Л. ФРИДМАН, Г. С. ИСМАГИЛОВА
H3CCCH,OSO2-СН2-O2SOCH,CCH3
C7H12O8S2 М. в. 222,19
Эфиры сульфокислот применяются в качестве алкилирующих агентов и исходных соединений в различных синтезах. Однако производные метионовой кислоты являются труднодоступными и малоизученными соединениями. Нами разработан удобный метод синтеза не описанного ранее диацетолового эфира метионовой кислоты [1].
I СХЕМА СИНТЕЗА
CH2COCHN2+CH2(SO2OH)a—>CH2(SO2OCHiCOCH3)2
Характеристика основного сырья
Диазоацетон, т. кип. 46—47°/13 мм, d420 = 1,0864, п%= = 1,4608; получение см. [21.
Метионовая кислота, МРТУ 6—09—5018—68, техн.
Уксусный ангидрид, ГОСТ 5815—52, ч. д. а.
Бензол, ГОСТ 5955—51, ч.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, термометром и капельной воронкой, помещают 10 г (0,119 моль) диазоацетона в 100 мл уксусного ангидрида. К раствору при перемешивании и температуре 15—20° добавляют порциями 10,5 г (0,06 моль) метионовой кислоты до прекращения выделения азота. Реакционную смесь выдерживают 6* 8$
1 час при комнатной температуре, затем уксусный адгндрид гидролизуют 250 мл дистиллированной воды. Выпавший осадок отжимают на воронке Бюхнера, трижды промывают холодной водой порциями по 50 мл, сушат на воздухе и перекристаллизовывают из бензола.
Выход ди ацетолового эфира метионовой кислоты равен 12 г (70%). Вещество представляет собой белые иглы с т. пл. 108—108,5°.
Найдено, %: С—29,27; 29,06; Н—4,21; 4,16; S—22,30; 22,22. C7H12O8S2. Вычислено, %: С—29,17; Н—4,17; S—22,22.
ЛИТЕРАТУРА - t
1. А. Л. Фридман, Г. С. Исмагилова. Авт. свид. 235019; Открытия. Изобретения. Промобразцы. Товарные знаки, № 5 (1969).
2. Общий практикум по органической химии. М., «Мир», 1965, с. 535.
ДИБЕНЗОИЛМЕТАН
Н. Ю. АРОН СКАЯ, Р. М. ГЕЛЬШТЕИН, Е. М. ИОФИС
C1SH12O2
М. в. 224,26
В литературе описано получение днбеизоилметана действи-ем спиртового раствора метилата или этилата натрия иа дибромид бензальацетофенона [1—31. Выход продукта после выделения и перекристаллизации из метанола составляет 52% [1].
Нами использован в качестве щелочного агента бутилат натрия, изменен температурный режим синтеза и способ выделения продукта, что позволило упростить методику и исключить применение метанола.
СХЕМА СИНТЕЗА
f \ —снвг -вгне-с \
C4HaONa
Н2О
НС1
85
Характеристика основного сырья
Бензальацетофенона дибромид, МРТУ 6—09—1251—64, ч.
Бутилат натрия, 16%-иый раствор, получен азеотропной отгонкой воды из раствора едкого натра в бутаноле [41.
Соляная кислота, ГОСТ 3118—67, ч.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешал кой, термометром и холодильником и помещенную в водяную баню, загружают 1290 г (2,15 моль) 16%-ного раствора бутилата натрия и при размешивании прибавляют 394 г (1,07 моль) дибромида бензальацетофенона с такой скоростью, чтобы температура не превышала 25—30°. Затем реакционную смесь нагревают до 60°, выдерживают 2-часа при этой температуре, после чего добавляют 45 мл соляной кислоты до pH 2 и 335 мл воды и продолжают выдержку при 60° еще 1 час. По охлаждении до 25—30° реакционная масса расслаивается, верхний (бутанольный) слой отделяют и охлаждают до 0—5°. Выпавший осадок отжимают на воронке Бюхнера, промывают на фильтре 100 мл воды и 150 мл водного (1:1) изопропилового спирта и сушат на воздухе.
Выход дибензоилметана равен 135 г (56,5%), т. пл. 76,3— 77,3°.
По литературным данным [51, т. пл. 78°.
ЛИТЕРАТУРА
1. Синтезы органических препаратов, сб. 1. М., Инлитиздат, 1949, с. 186.
2. Pond, Moxwell, Norman. J. Amer. Chem. Soc., 21, 964 (1899).
3. S 1 u i t e r. Recuell trav. chim., 24, 368 (1905).
4. H. H. Дыханов, В. T. Скрипкина. Методы получения химических реактивов и препаратов, вып. 9, М., ИРЕА, 1964, с. 29.
5. Словарь органических соединений, т. 1. М., Йилитиздат, 1949, с. 665.
1,4-ДИБРОМ 2-МЕТОКСИ-И 1.4-ДИБРОМ-2-ЭТОКСИПЕНТАНЫ
А. В. БОГАТСКИЙ, Г. Л. КАМАЛОВ, Н. Г. ЛУКЬЯНЕНКО,
В. Н. ШАРЫГИН
СН2ВгСН (ОСНз) СНаСНВгСН3
СНгВгСН (ОС2Н5) СН2СНВгСН3
C6H,2Br2O. М. в. 259,98 С7НиВг2О. М. в. 274,00
Одним из наиболее распространенных способов получения Р.у'-дигалоидэфиров является реакция взаимодействия этиленовых углеводородов с соответствующими а,6-дигалоидэфира-ми в присутствии кислотных катализаторов [11.
Нами разработан способ синтеза 1,4-дибром-2-метокси- и 1,4-диб)ром-2-этоксипентанов из а,р-дибромэтилметилового ' и а,р-дибромэтилэтилового эфиров и пропилена в присутствии безводного (плавленого) хлористого цинка в качестве катализатора. Установлено, что количество катализатора существенно влияет на выход целевого продукта. Наилучшими условиями синтеза являются эфирная среда н 3%-ное содержание хлористого цинка.
СХЕМА СИНТЕЗА
CH2BrCHBrOR 4- СН2=СНСН3 —►
CH2BrCH(OR)CH2CHBrCH3, где R=CHS—; С3Н,—.
Характеристика основного сырья
а,р-Дибромэтилметиловый эфир, т. кип. 68—70716 мм; d4№ — 1,9698; = 1,5300 (см. примечание 1).
.87
а,0-ДиброМэтилэтиловый эфир, т. кип. 78—80°/15 мм; <Л20 = 1,7350; «д = 1,5048 (см. примечание 2).
Пропилен (см. примечание 3).
Цинк хлористый, ГОСТ 4529—69, ч„ (см. примечание 4).
Условия получения
В четырехгорлую колбу емкостью 250 мл, снабженную обратным холодильником, термометром, мешалкой и газоподводящей трубкой, помещают 1 моль соответствующего а, ₽-ди-бромэтилалкилового эфира, 50 мл абсолютного диэтилового эфира и хлористый цинк в количестве 3% от веса а, р-дибром-этилалкилового эфира. Через реакционную смесь барботйруют пропилен в течение 12 часов (см. примечание 5). Температуру поддерживают в пределах 18—20°. По окончании реакционную смесь последовательно промывают водой, 5%-ным раствором щелочи и вновь водой до нейтральной реакции. Отделяют нижний органический слой, сушат прокаленным хлористым кальцием и перегоняют при пониженном давлении в токе азота.
Выход 1,4-дибром-2-метоксипентана равен 117 г (45%); т. кйп. 71—7372 мм; d420 = 1,6163; я» = 1,4992.
Найдено, %: Вг—61,48.
CeHi2Br2O. Вычислено, %: Вг—61,51.
Выход 1,4-дибром-2-этоксипентана равен 145 г (53%); т. кип. 85—8774 мм; d420 = 1,5328; п™ = 1,4915.
Найдено, %: Вг—58,27.
СтНцВггО. Вычислено, %: Вг—58,39.
Методом ГЖХ (см. примечание 6) показано, что полученные продукты представляют собой смесь диастереомеров. Содержание высококипящего эритро-изомера составляет 74% в 1,4-дибром-2-метоксипентане и 77% в 1,4-дибром-2-этокси-пеитане. Эти изомеры выделены точной ректификацией (см. примечание 7):
Эритро-1,4-дибром-2-метоксипентан, т. кип. 61,571 мм; d420 = 1,6260; п% = 1,4971.
Найдено, %: Вг—61,40. C6Hi2Br2O. Вычислено, %: Вг—61,51.
Эритро-1,4-дибром~2-этоксипентан, т. кип. 7Г/1 мм; d420 = = 1,5493; Яд = 1,4945.
Найдено, %: Вг—58,37. С?Н14Вг2О. Вычислено, %: Вг—58,39.
Примечания:
1. а.р Дибромэтилметиловый эфир получен бромированием а-хлорэтил-метилового эфира [2]. Установлено, что иаилучший выход продукта наблюдается в случае прибавления а-хлорэтилметилового эфира к раствору брома в четыреххлористом углероде.
88
2. а.р Дибромэтилэтиловый эфир получен бромированием винилэтнло-вого эфира. Ввиду того, что реакция проходит гладко и с большим выходом, были сделаны попытки использовать иеперегнаниый продукт. Одиако выход при этом целевого продукта уменьшился на 10—15%.
3. Пропилен получен дегидратацией пропилового спирта на активированной окиси алюминия при 350—370° и очищен по методике [3].
4. Продажный безводный хлористый цинк плавили в фарфоровой чашке до стеклообразного состояния, помещали в эксикатор и употребляли по мере надобности. Перед синтезом катализатор быстро измельчали и взвешивали в бюксе с притертой пробкой.
5. Более длительное пропускание пропилена не приводит к увеличению выхода продукта.
6. Хроматографический анализ проводили на хроматографе ЛХМ-8М при 127°. Газ-носитель — гелий, длина колонки 2 м, 5% СКТФТ-50 на хромосорбе «G».
7. Изомеры разделили иа ректификациононй колонке полной конденсации со стеклянной насадкой высотой 190 см и числом теоретических тарелок 60; флегмовое число 114.
ЛИТЕРАТУРА
1. Ю. В. П око иов а. Галоидэфиры. М.-Л., «Химия», 1966, с. 275.
2. М. Ф. Ш о с т а к о в с к ий. Простые виниловые эфиры. М., Изд-во АН СССР, 1952, с. 98.
3. Ф. М. Р а п о п о р т, А. А. И л ь и и с к а я. Лабораторные методы получения чистых газов. М., Госхимиздат, 1963, с. 339.
3,4-ДИБУТОКСИАН ИЛИН
В. М. ДЗИОМКО, А. В. ИВАЩЕНКО
Ч/Чос4н9
C14H23NO2 М. в. 237,34
3,4-Дибутоксиаиилин находит применение в синтезе дисперсных красителей (аналогов целлитона желтого С) [1].
В литературе описано получение этого соединения восстановлением 3,4-дибутоксинитробензола [2, 3], который получают нитрованием дибутилового эфира пирокатехина [1, 3, 4]. Последний получают нз пирокатехина и бромистого бутила или бутилового эфира п-толуолсульфокислоты [1, 2, 4].
Нами уточнены условия синтеза 3,4-ди1бутоксианилина, 3,4-дибутоксинитробензола и дибутилового эфира пирокатехина и повышен выход на всех стадиях синтеза.
СХЕМА СИНТЕЗА
CtH9I ? кон
^\/осл
I II
HNO3
I I \OC4H9
Na2S
H*NW\/0C4H’
Характеристика основного сырья
Пирокатехин, МРТУ 6—09—5771—69, ч.
Кали едкий, ГОСТ 4203—65, ч.
90
Бутнл иодистый, МРТУ 6—09—6512—70, ч.
Азотная кислота, ГОСТ 4461—67, ч., пл. 1,42.
Уксусная кислота, ГОСТ 61—69, х. ч.
Натрий сернистый, ГОСТ 2053—66, ч.
Условия получения
Синтез дибутилового эфира пирокатехина. В трехгорлую колбу емкостью 2 л, снабженную мешалкой, обратным холодильником и капельной воронкой на шлифах, помещают ПО г (1,0 моль) пирокатехина и 368 г (2,0 моль) йодистого бутила. Колбу нагревают на водяной бане до 90° (температура бани) и прибавляют за 2 часа при хорошем размешивании раствор 112 г (2,0 моль) едкого кали в 560 мл этанола. Реакционную смесь кипятят 1 час, после чего добавляют 1 л воды и охлаждают до комнатной температуры. Отделяют органический слой, а из водного слоя экстрагируют эфиром трижды по 150 мл дополнительное количество вещества. Органический слой, соединенный с эфирными вытяжками, сушат безводным сульфатом натрия, отгоняют эфир и перегоняют при пониженном давлении.
Выход дибутилового эфира пирокатехина равен 140—142 г (79—80%), т. кип. 156—158724 мм.
По литературным данным [4], т. кип. 156—157722 мм.
Получение 3,4-дибутоксинитробензола. В трехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром и капельной воронкой на шлифах, помещают 140 г (0,632 моль) дибутилового эфира пирокатехина и 490 мл уксусной кислоты. Колбу охлаждают в бане со льдом и прибавляют при интенсивном перемешивании из капельной воронки 245 мл азотной кислоты с такой скоростью, чтобы температура реакционной массы не превышала 10°. По окончании прибавления азотной кислоты массу перемешивают еще 30 минут и оставляют на ночь при комнатной температуре.
На следующий день к затвердевшей реакционной массе добавляют 1 л воды, смесь хорошо размешивают и фильтру-* ют. Осадок на фильтре промывают водой до нейтральной реакции промывных вод и сушат в вакуум-эксикаторе над твердым едким натром до постоянного веса.
Выход 3,4-дибутоксинитробензола равен 121 г (72%); по внешнему виду вещество представляет собой мелкие светло^ желтые иглы с т. пл. 55—56° (нз этанола).
По литературным данным, т. пл. 56° [3, 4]; 52—54° [2].
Получение 3,4-дибутоксианилина. В колбу емкостью 2 л, снабженную обратным холодильником, помещают 120 г (0,45 моль) 3,4-дибутоксинитробензола, 385 г (1,6 моль) девятиводного сульфида натрия, 240 мл воды и 720 мл этанола. Смесь нагревают на водяной бане до кипения, кипятят 3 часа.
91
Затем охлаждают и отделяют органический слой. Из водного слоя экстрагируют эфиром трижды 'По 100 мл дополнительное количество вещества. Органический слой, соединенный с эфирными вытяжками, сушат сульфатом натрия, отгоняют эфир, а остаток перегоняют при пониженном давлении.
Выход 3,4-дибутокснанилина равен 95—96 г (89—90%). По внешнему виду вещество представляет собой светло-жел-тр,е масло с т. кип. 193—195°/8 мм, быстро темнеющее при хранении.
По литературным данным, т. кип. 157—158°/1,5 мм [1J; 161 — 16273 мм [2].
ЛИТЕРАТУРА
1. N. Kuroki, A. Nishiura, К. Konishi. J. Chem. Soc. Japan. Jndustr. Chem. Soc.. 59, 1053 (1956).
2. F a r g h e r. J. Chem. Soc., 1920, 869.
3. S. К a w a i. Y. О k a w a, Y. Y a d a, H. H о s о i, T. Mur a koshi, J. Y a j i m a. Hlppon Kagaku Zasshi, 80, 551 (1959).
4. Brown, Robinson. J. Chem. Soc., 1917, 953.
4-N.N (ДИКАРБОКСИМЕТИЛ)АМИНОМЕТИЛ-З-ОКСИ-2-НАФТОЙНАЯ КИСЛОТА
Л. М. ТИМАКОВА, В. Я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО. Н Е. ХАВЧЕНКО
//~\
\_/ сн,соон
СН2СООН ноос он
C16H15NO7 М. в. 333,30
4-N.N- (Дикарбоксиметил) аминометил-З-окси-2-нафтойная кислота является аналогом известного реагента — 2-окси-З нафтойной кислоты, нашедшей широкое применение для флу-ориметрического определения ряда катионов [1—3]. Введение в молекулу реагента метилиминодиацетатной группировки, характерной для комплексонов, расширяет число катионов влияющих на флуоресценцию реагента, и значительно увеличивает чувствительность определений.
Синтез реагента осуществлен по реакции Манниха взаимодействием 2-окси-З-нафтойной кислоты с формальдегидом и иминодиуксусной кислотой [4, 5].
СХЕМА СИНТЕЗА
НООС ОН
+CHaO+NH^
сн,соон
сн,соон
93
/ CH,-N
ноос он
хсн2соон хсн3соон
Характеристика основного сырья
2-Окси-З-нафтойная кислота, МРТУ 6—09—2447—65, ч. Иминодиуксусная кислота, В ТУ МР УХП 190—58, ч. Формальдегид, ТУ МХП 2631—51, ч.
Уксусная кислота, ГОСТ 61—51.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, обратным холодильником и термометром, помещают 500 мл уксусной кислоты и при размешивании и температуре 45—50° добавляют 14.6 г (0,11 моль) иминодиуксусиой кислоты, нейтрализованной 30%-ным раствором едкого натра до pH 7—8, 12 г (0.13 моль) 35,8%-ного раствора формальдегида и 18,8 г (0,1 моль) 2-окси-З-нафтойной кислоты. Через 5—10 минут после растворения 2-окси-З-нафтойной кислоты начинает выпадать осадок комплексона, после чего температуру реакционной массы поднимают до 60—70° и выдерживают при этой температуре и размешивании 5 часов. Затем реакционную массу охлаждают, отфильтровывают желтоватый осадок, промывают его 500 мл этилового спирта и сушат в сушильном шкафу при 50°.
Выход 4-N,N-(дикарбоксиметил) аминометил-З-окси-2-иаф-тонной кислоты равен 28,3 г (90%).
Найдено, %: С— 57,3; 57,4; Н—4,8; 4,7; N—4,5; 4,4. CieHisNO?. Вычислено, %: С—57,7; Н—4,5; N—4,2.
ЛИТЕРАТУРА
1. А. И. Черкасов. Докл. АН СССР, 118, 309 (1958).
2. Т. С. Жигалки и а, А. И. Черкасов. Тр. Астрахаи. технолог, ин-та рыбн. пром-ти, 8, 25 (1962).
3. G. F. К irk bright, Т. S. West, С. Woodward. Analyt. Chem., 37, 137 (1965).
4. H. В. Ц и р ульн и к о в а, В. Я. Темкина, Г. Ф. Ярошенко, Р. П. Ластовскнй. Хим. пром-сть, 7, 502 (1971).
5. В. В u djejsji n s к у, Т. S. West. Analyt. chim. acta., 42, 3, 455 4968).
1,1/-ДИМЕТИЛ-3,3'-ДИ(ТЕТРАГИДРОФУРИЛ-2)-ДИПРОПИЛОКСИД
М. Д. ЛИПАНОВА
-СН3СН SCHOC НСН2СН,-'Чо J
СН3 СН8
С1бН30О3 М. в. 270,40
Простые эфиры тетрагидрофурановых спиртов могут найти применение в качестве пластификаторов высокополимеров и высококипящих растворителей [1, 2]. 1,Г-Диметил-3,3'-ди-(тетрагидрофурил-2)дипропилоксид получен нами впервые.
СХЕМА СИНТЕЗА
I J-CH2CHaCH(OH)CH, — хо
I J-CH2CHaCHOCHCHaCHa-l J + нао. | | хсг
СН» СН»
Характеристика основного сырья
1-(а-Тетра1гидрофурил)-3-бутанол, т. кип. 145758 мм; получение см. [3].
Дихлорэтан, ГОСТ 5840—51.
Условия получения
В круглодонную колбу емкостью 0,5 л, снабженную обратным холодильником с водоотделителем, помещают 72,1 г
95
(0,5 моль) тетрагидрофурилбутанола, 3 мл серной кислоты (пл. 1,83), 300 мл дихлорэтана и нагревают 7 часов на масляной бане при 120—130°. Затем к реакционной смеси прибавляют насыщенный раствор карбоната натрия и промывают водой до нейтральной реакции промывных вод по универсальной бумаге. Дихлорэтан и воду отгоняют при пониженном давлении, а остаток перегоняют, собирая фракцию, кипящую при 175—176°/9 мм.
Выход ^К-диметил-З.З'-ди (тетрагидрофурил-2) дипропил-оксида равен 36,6 г (58,6%). Вещество представляет собой бесцветную жидкость, с?420 = 0,960, «о = 1,4576 (см. примечание).
Найдено, %: С—71,05; 70,83; Н—10,92; 11,17; MRD = 76,78. С!6НзоОз. Вычислено, %: С—71,06; Н—11,18; MRn = 76,62.
Примечание.
По аналогичной методике получен впервые 3,3'-ди(тетрагидрофурнл-2)-дипропилоксид с выходом 69,5%, т. кип. 177—178°/9 мм, «/> = 1,4638, di20 = 0,9912.
Найдено, %: С—69,30; 69,07; Н—10,88; 10,88, MRD = 67,44. СиН260з. Вычислено, %: С—69,98; Н—10,81; MRD = 67,38.
ЛИТЕРАТУРА
1. А. А. Пономарев, А. А. Бенедиктова. Научный ежегодник Саратов, ун-та, 493 (1954).
2. А. А. Пономарев, 3. В. Тиль, М. Д. Л и п а и о в а. Вопросы использования пентозаисодержащего сырья. Рига, Изд-во АН СССР, 1958,
3. А. А. Пономарев. Синтезы и реакции фурановых веществ. Саратов. Изд-во СГУ, 1960, с. 112.
ДИМЕТИЛОВЫЙ ЭФИР ДИЭТИЛЕНГЛИКОЛЯ
Оксибис(2-метоксиэтан)
А. Л. ЛИФИЦ, А. А. ВЕЙИМАН
СНзОСНз— СН2ОСН2—СН2ОСН3
С6Н14О3 М. в. 134,18
Диметиловый эфир диэтиленгликоля получают нагреванием алкоголята натрия с р,р'-дихлордиэтиловым эфиром в течение 8—15 часов [11, из окиси этилена и метанола [2] или из р.р'-дихлордиэтилового эфира с водно-спиртовым раствором едкого натра [3, 4].
Нами предложен метод получения диметилового эфира диэтиленгликоля взаимодействием ^р'-дихлордиэтилового эфира со спиртовым раствором едкого кали; метод исключаег применение металлического натра и окиси этилена, менее продолжителен и позволяет получить продукт без примесей.
СХЕМА СИНТЕЗА
С1СН2-СНаОСН2-СН2С1 -+• СН3ОН
— СН3ОСН2-СН2ОСН2-СН2ОСН3
Характеристика основного сырья
р,р'-Дихлордиэтиловый эфир, ТУ МХП БУ 65—54, техн.
Метиловый спирт, ГОСТ 2222—65, техн.
Кали едкий, ГОСТ 4203—65, ч.
Кальций хлористый безводный, ГОСТ 450—58, техн.
7 Зак. 1164
97
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой с затвором, капельной воронкой и обратным холодильником, загружают 400 г (12,5 моль) метилового спирта и при перемешивании в один прием присыпают 360 г 85%-ного (5,5 моль) едкого кали. Содержимое колбы нагревают прй перемешивании до кипения и постепенно прибавляют 286 г (2 моль) р,р'-дихлордиэтилового эфира, следя за равномерностью кипения. Реакционную смесь кипятят еще 3 часа, затем охлаждают до комнатной температуры и отфильтровывают от хлористого калия. Осадок промывают 200 мл метанола. Из фильтрата отгоняют метанол, остаток помещают в делительную воронку, отделяют верхний слой, сушат хлористым кальцием и перегоняют при пониженном давлении, собирая фракцию с т. кип. 58—60°/15 мм.
Выход диметилового эфира диэтиленгликоля равен 95 а (35,5%), гР° = 1,4085, б/420 = 0,945 (см. примечание).
По литературным данным [5], = 1,4097; pi516 = 0,9514.
Примечание.
По аналогичной методике нами был получен диэтиловый эфир диэтиленгликоля с выходом 35%, Яд = 1,4120, dj® = 0,910. По литературным данным [5], р1515 = 0,9149.
ЛИТЕРАТУРА
1. L. Н. С г е t с h е г, W. Н. Р u 11 е n g ё г. J. Amer. Soc., 47, 163 (1925).
2. М. Ф. Леонов, И. А. Коршунов. Труды по химии и, хим. тех. нол., 3, 515 (1964).
3. Пат. ФРГ, 1129147; С. А. 57, 13613а (1962).
4. Пат. США 3020316; С. А., 57, 8438f (1962).
5. Словарь органических соединений, т. I. М., Инлитиздат. 1949, с. 828.
цис- И транс- 2,3-ДИМЕТИЛОКСЕТАНЫ
Г. А. ФИЛИП, С. А. ПЕТРАШ, Г. В. ПЬЯНКОВА
С5Н10О
М. в. 86,1.3
Оксетаны, являясь полупродуктами органического синтеза, находят применение в процессе полимеризации [1]. Однако методы синтеза оксетанов мало разработаны, а стереоизомеры 2,3- и 2,4-диалкилзамещенных оксетанов в работах, опубликованных до настоящего времени, не рассматриваются.
Нами впервые разработана методика разделения стереоизомерных оксетанов, а также усовершенствована известная методика получения оксетанов(2](использование сплава едких щелочей, проведение реакции в токе азота), что позволило увеличить выход продукта до 50%.
СХЕМА СИНТЕЗА
сн8
СН3—СН—СН—СН2С1
О Ас
СН3
> Н3С -< \ о
Характеристика основного сырья
Хлоргидринацетат 2-метил-1,3-бутандиола, т. кип. 60--62°/2 мм, di20 =<1,0438, Яд = 1,4340; получение см. [3].
Кали едкое, ГОСТ 4203—65, ч.
Натр едкий, ГОСТ 4328—66, ч. д. а.
7*
99
Условия получения
В круглодонную колбу емкостью 0,5 л, снабженную ме шалкой, капельной воронкой, термометром и нисходящим холодильником, соединенным с колонкой Вигре (20 см), помещают 200 г (5 моль) едкого натра и 168,3 г (3 моль) едкого кали. К расплавленной щелочи по каплям, со скоростью одной капли в секунду, прибавляют 164,6 г (1 моль) хлоргидринацетата 2-метил-1,3-бутандиола. Одновременно через систему пропускают ток азота, скорость которого определяют при выходе из ловушки (60 пузырьков в минуту). Для увеличения выхода целевого продукта систему продувают азотом еще 4 часа после окончания прибавления хлоргидринацетата 2-метил-1,3-бутандиола. Продукт циклизации собирают в ловушке, охлаждаемой смесью льда с солью, сушат сульфатом натрия и перегоняют, собирая фракцию с т. кип. 80—85°.
Выход 2,3-диметилоксетана равен 43 г (50%), d420 = = 0,8316; «“=1,4000.
Найдено, %: С—69,52; Н—11,74; Жд = 25,10.
С5НюО. Вычислено, %; С—69,73; Н—11,60; MRD = 25,27.
Разделение 2,3-диметилоксетанов на цис- и транс-изомеры. Разделяют 26 г смеси геометрических изомеров 2,3-диметилоксетанов (показано методами ТСХ и ГЖХ) 24 часа (круглосуточно) методом точной ректификации на колонке полной конденсации с числом теоретических тарелок 40 (стеклянная насадка). Режим колонки: сила тока на обогрев куба 102,5 а, на обогрев рубашки — 90 а. Число капель, скапывающих в минуту с нижнего счетчика — НО, с верхнего — 55. Пробы, емкостью 2 мл, отбирают с флегмовым числом 50. Хроматографирование фракций осуществляют на хроматографе ЛХМ-7А, длина колонки — 3 м, фаза 10% СКТФВ-803 на хромосорбе G, Т='85°, скорость газа-носителя (гелий) 21 мл!мин. Константы изомеров, выделенных с чистотой 99,9%, приведены в таблице.
Константы цис- н транс-2,3-диметилоксетанов
Изомер Т. кип., °C „20 nD Соотношение изомеров
Цис- Транс- 80 0,8469 1,4040 24,87 25,27 15
89 0,8203 1,3980 25,11 85
100
Строение указанных изомеров подтверждено Изучением ИК-спектров (980 см~х), спектров ЯМР (6, 10% СС1<, цис-4,25, транс- 4,65) и химическими методами [3].
ЛИТЕРАТУРА
1. Е. Goethals. Ind. chitn. beige, 30, 559 (1965).
2. C. R. Nolle r. Organ. Synth. 29, 92 (1949).
3. А. В. Богатский, Г. А. Филип, С. А. П e т p а ш, Л Се м e p д-ж и, Г. В. Пьянкова. Ж. орган, химии, 7, 577 (1971).
2.9-ДИМЕТИЛ-1.10-ФЕНАНТРОЛИН
Неокупроин
В. М. ДЗИОМКО, И. А. КРАСАВИН, Б. В. ПАРУСНИКОВ
•1/2 Н2О
М. в. 217,27
CI4H12N2- 1/2 Н2О
2,9-Диметил-1,10-фенантролин (неокупроин) находит широкое применение в аналитической практике для колориметрического определения меди.
Его получают циклизацией 8-аминохинальднна с ацеталем кротонового альдегида в среде серной кислоты [1, 2] с выходом 7,6% [1] или из о-фенилендиамина примерно в тех же условиях с выходом 10—20% [3, 4]. В качестве окислителя в обоих случаях применялся мышьяковый ангидрид.
Циклизация 8-аминохинальдина с кротоновым альдегидом обстоятельно изучена в работе [41. Оказалось, что добавление хлористого алюминия существенно повышает выход продукта и что наиболее подходящим окислителем является о-нитрофе-нол. В оптимальных условиях выход неокупроина достигает 89% [41.
Нами проверен и уточнен метод синтеза, предложенный в работе [4]. Как выяснилось, указанный в ней выход неокупроина (89%) относится к сырому продукту, очистка которого представляет большие трудности. Рекомендованная в работе [4] очистка сырого продукта промывкой его ксилолом приводит не только к большим потерям вещества, но и не дает желаемого результата. Мы обнаружили, что мешающая прнмесь может быть удалена отгонкой с водяным паром, после чего обработка активированным углем в уксуснокислом растворе и 102
перекристаллизация из водного этанола дает неокупроин виде бесцветных игл с т. пл. 162—163°.
СХЕМА СИНТЕЗА
z4/N0:
3 I II I + 3CHS-CH = CH-CHO + || |
nh2
HC1.A1CL о z----C Z-----%.
------3 Z Z .l/2HaO+
\=NZ X-N=Z
CH3 CH3
-f-3,5H2O
Характеристика основного сырья
8-Амннохннальдин, т. пл. 54—56°; получение см. с.31 этого сборника.
Кротоновый альдегид, ТУ УЗ 75—64, ч., ректифицированный.
Соляная кислота, ГОСТ 3118—66, ч.
Алюминий хлористый безводный, ГОСТ 4452—48 очнщ., 1 сорт.
о-Нитрофенол, ТУ МХП 91—51, ч.
Условия получения
В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой с затвором, обратным холодильником, капельной воронкой и термометром, помещают 50 мл концентрированной соляной кислоты и охлаждают ее льдом. При размешивании осторожно добавляют небольшими порциями 20 г безводного хлористого алюминия, после чего охлаждающую баню удаляют. Прибавляют 7 г (0,05 моль) о-нитрофенола и раствор 15,8 а (0,1 моль) 8-аминохинальдина в 100 мл концентрированной соляной кислоты. На кипящей водяной бане реакционную массу нагревают до 90° и при размешивании за 30 минут добавляют по каплям 10 мл (8,5 г; 0,12 моль) ректифицированного кротонового альдегида. Нагревание на кипящей водяной бане и размешивание продолжают еще 4 часа, после чего добавляют 200 мл дистиллированной воды и охлаждают смесь
103
До Комнатной температуры. Раствор декантируют в стакан емкостью 1 л, снабженный мешалкой, термометром и капельной воронкой, и охлаждают льдом. Из капельной воронки при размешивании добавляют при 8—10° 20%-ный раствор едкого натра до достижения pH 4. Раствор отделяют от выпавшей смолы, декантируя его в коническую колбу емкостью I л, кипятят 10—15 минут с 5 г активированного угля и фильтруют.
Фильтрат охлаждают до комнатной температуры и добавляют к нему при размешиваний 20%-ный раствор едкого натра до снльнощелочной реакции. Вьипавшнй осадок отсасывают, промывают водой и переносят в колбу для перегонки с паром. Отгоняют с паром летучие примеси до тех пор, пока дистиллат не станет совершенно прозрачным (объем днстнл-лата ~ 3 л).
К кубовому остатку прибавляют 50 мл уксусной кислоты, доводят объем водой до 1 л, кипятят 10—15 минут с 5 г активированного угля и фильтруют. После охлаждения до комнатной температуры раствор при размешивании подщелачивают водным аммиаком (~80 мл), суспензию перемешивают 2—3 часа, затем осадок отсасывают, промывают водой и отжимают.
Сырой продукт кристаллизуют из смеси 85 мл этанола и 145 мл воды с применением активированного угля (2 г).
Выход чистого гемигндрата 2,9-диметил-1,10-фенантроли-на в виде бесцветных игл равен 8—9,3 г (36,8—42,8%), т. пл. 161 — 163°.
По литературным данным, т. пл. 2,9-днметил-1,10-фенан-тролина равна 158—159° [3]; 159—160° [1]; 159,5° [2];. 161° [5]; 165° [4].
ЛИТЕРАТУРА
1. F. И. Case. J Amer. Chem. Soc. 70, 3994 (1948).
2. H. I r wing. D. H. Mellor. J. Chem. Soc., 1962, 2537.
3. E. J. O' Reilly, R. A. Plowman. Austral J. Chem., 13, 145 (1960); C. A., 54, 14251 b (1960).
4. К. M a d e J a, J. prakt. Chem. 17, 97 (1962).
5. E. С. M. Crigg, J. R. Hall, R. A. Plowman. Austral. J. Chem., 15, 425 (1962).
1\1,1\Т/-ДИМЕТИЛЭТИЛЕНДИАМИНО-М,М'-БИС-
(МЕТИЛЕНФОСФОНОВАЯ) и М,М'-ДИ(2-ОКСИЭТИЛ)-ЭТИЛЕНДИАМИНО-N, N' БИС
(МЕТИЛЕНФОСФОНОВАЯ КИСЛОТЫ)
В. П. МАРХАЕВА, Ю. М. ПОЛИКАРПОВ, М. В. РУДОМИНО,
Т. я. МЕДВЕДЬ
ноч .ОН
>р-HOZ II о -CH2-N-CH,CH2-N-CH,-P< 1 1 ' II хон R RO I R = CHS—
CcHigNaOePa M. в. 276,17 II R = OHCH2CH2-
CgHaaNsOgPa М. в. 336,23
М.ЬГ-Диметилэтнленднамино-К^М'-бис (метиленфосфоновая) и К,М'-дн(2-оксиэтнл)этилендиамнно-М, NT-бис (метнленфосфо-новая кислоты) являются новыми фосфорорганическими комплексонами, алкилированными по азоту аналогами этилендн-аминобис (метиленфосфоновой кислоты) [1]. Они отличаются от последней .повышенной водорастворимостью как самих кислот, так и комплексов с рядом ионов металлов, в частности, редкоземельных элементов.
Комплексоны получены нами конденсацией соответственно М,ЬТ-диметилэтилендиамина и М,ЬТ-ди(2-оксиэтил)этилеиди-амина с формалином и фосфористой кислотой по методу [2], а также конденсацией указанных диаминов с параформом и диэтилфосфитом с последующим омылением образовавшихся сложных эфиров до свободных кислот.
Продукты, полученные обоими методами, не давали депрессии температуры плавления смешанной пробы.
105
СХЕМА СИНТЕЗА
Способ I.
2Н3РО3 + 2СН2О + RNHCHaCHaNHR
-> (HO)sP-CH,NCH,CH8NCH2-P(OH)s
I! I I II
OR RO
Способ II.
2 (C2H6O)3PHO+2CH2O-bRNHCH2CH3NHR ——— — n jU
-* (C2H5O)2P-CH2NCH2CH2NCH2-Р(0С3НБ)21Г(Н-,0)
(HO),P-CH2NCH2CH8NCH,-P(OH),
II
OR RO
Характеристика основного сырья
Фосфористая кислота, МРТУ 6—09—6555—70, ч.
Формалин, 37-ный раствор, ГОСТ 1625—64.
Параформ, МРТУ 6—09—2128—65, ч.
Соляная кислота, ГОСТ 3118—66, ч.
Метиловый спирт синтетический, ГОСТ 6995—54.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Окись пропилена, т. кип. 36°.
Ы.Ы'-Диметилэтилендиамин, т. кип. 120°; получение см. [3].
Ы,Ы/-Ди(2-оксиэтил) этилендиамин, т. пл. 100—102°; получение см. [4].
Диэтилфосфит, ТУ TCP 1048р—63, т. кип. 68—70°/10 мм, п® = 1,4080.
Условия получения
Получение N ,П'-диметилэтилендиамино-\!,М'-бис( метиленфосфоновой кислоты) (способ I). В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, капельной воронкой и обратным холодильником, помещают 8,8 г (0,1 моль) ди-метилэтилендиамина, при охлаждении ледяной водой и перемешивании прибавляют 15 мл 2 н. соляной кислоты до достижения pH 2, затем раствор 19,6 г (0,24 моль) фосфористой кислоты в 25 мл воды. Температуру бани поднимают до 105— 106
110° и прибавляют по каплям за час 12 г 37%-ного раствора формалина (0,24 моль СН2О). Реакционную смесь выдерживают при этих условиях еще 3 часа. Затем избыток хлористого водорода удаляют при пониженном давлении, многократно добавляя воду (5 X 100 мл). Сиропообразную массу заливают 150 мл метилового спирта и выдерживают 1,5—2 суток до превращения ее в аморфный порошок, который затем растирают с дополнительным количеством спирта для удаления остатков хлористого водорода. Вещество сушат при пониженном давлении над пятиокисью фосфора при 100° до постоянного веса.
Выход М.М'-диметилэтилендиамино-М.М'-бис (метиленфосфоновой кислоты) равен 10,2 г (37%). Вещество представляет собой бесцветный мелкокристаллический или аморфный гигроскопичный порошок, т. пл. 256—257° (разл.).
Найдено, %: С—25,7; Н—6,1; N—10,0; Р 22,4. CeHi8N2OoP2. Вычислено, %: С—26,0; Н—6,5; N—10,1; Р—22,4.
П о лучение N,N'-du (2-оксиэтил ) этиле ндиамино-!\!,Х'-бис-(метиленфосфоновой кислоты). В аналогичных условиях из 12,2 г (0,1 моль) N.N'-ди(2-оксиэтил)этилендиамина, 19,6 а (0,24 моль) фосфористой кислоты и 12 г 37%-ного раствора формалина (0,24 моль СН2О) получают 15 г (45%) М,М'-ди(2-оксиэтил)этилендиамино-М,М'-бис (метиленфосфоновой кислоты). Вещество представляет собой бесцветный гигроскопичный порошок, т. пл. 80—82°.
Найдено, %: С—28,0; Н—8,5; N—7,9; Р—18,2. C6H22N2OeP2. Вычислено, %: С-28,5; Н—8,6; N—8,3; Р—18,5.
Получение этилового эфира \’,Х'-диметилэтилендиамино-. N^\г-бис(метиленфосфоновой кислоты) (способ II). В четырехгорлую колбу емкостью 50 мл, снабженную мешалкой, капельной воронкой, термометром и обратным холодильником, помещают 6,5 г (0,07 моль) М.М'-диметилэтилендиамина, прибавляют по каплям при перемешивании 19,8 г (0,15 моль) ди-этилфосфита, затем присыпают при охлаждении водой 4,6 а (0,15 моль) параформа, поддерживая температуру реакционной смеси не выше 90°. Реакционную массу перемешивают при температуре бани 105—110° в течение 2,5—3 часов. Избыток диэтилфосфита и формальдегида удаляют при пониженном давлении сначала при 20 мм, затем при 2—3 мм и температуре бани 80—90°. Полученный темно-коричневый сироп (24,5 г) очищают хроматографически на колонке: носитель А120з, элю-ант—бензол—этанол 6:1.
Выход этилового эфира Г^М'-диметилэтилендиамино-М.М'-бис(метиленфосфоновой кислоты) равен 16,2 г (60%), «Ь°= 1.4451, Rf = 0,7—0,8.
Найдено, %: С—43,0; Н—8,4; N—7,1; Р—16,0. С14Н34М2ОбР2. Вычислено, %: С—43,0; Н—8,7; N—7,2; Р—16,0.
107
Получение П,П'-диметилэтилендиамино-П,П'-бис(метилен-фосфоновой кислоты). В круглодонной колбе емкостью 50 мл, снабженной обратным холодильником, кипятят 6 часов 10,0 г (0,025 моль) этилового эфира кислоты с 20 мл концентрированной соляной кислоты, затем еще 0,5 часа с активированным углем- После охлаждения уголь отфильтровывают, от фильтрата отгоняют избыток хлористого водорода при пониженном давлении, многократно добавляя воду (5X100 мл). Остаток заливают 100 мл спирта и оставляют на ночь до превращения в порошкообразное вещество. Для удаления ионов хлора концентрированный водный раствор кислоты обрабатывают 50 мл окиси пропилена, верхний слой сливают, оставшееся сиропообразное вещество растирают со 150 мл спирта до превращения в бесцветный порошок. Последний отфильтровывают и сушат в вакууме над пятиокисью фосфора до постоянного веса.
Выход П,П/-диметилэтилендиамино-П,П'-бис( метиленфосфоновой кислоты) равен 5,8 г (83%), т. пл. 255—257°.
Найдено, %: N—9,7; Р—22,2.
CsH|SN2O6P2. Вычислено, %: N—10,1; Р—22,4.
Получение этилового эфира N,П'-ди(2-оксиэтил)этиленди-амино-N,N'-бис (метиленфосфоновой кислоты). Аналогичным способом из 3,34 г (0,024 моль) диэтилфосфита, 1,48 г (0,012 моль) М,М'-ди(2-оксиэтил)этилендиамииа и0,72 г (0,024моль) параформа получают 2,94 г (55%) этилового эфира N.N'-ди-(2-оксиэтил)этилендиамино-М,М'-бис(метиленфосфоновой кислоты), ft™ = 1,4390, У?, = 0,7—0,8.
Найдено, %: N—6,3; Р—14,3.
CisHaeNzOaPa- Вычислено, %: N—6,2; Р—14,0.
Получение N,П'-ди(2-оксиэтил)этилендиамино-П,N'-6uc* (метиленфосфоновой кислоты). Омылением 1,46 г (0,003 моль) полученного этилового эфира кислоты 5 мл концентрирован’ ной соляной кислоты аналогично описанному выше получают 0,89 г (80%) М,М,-ди(2-оксиэтил)этилендиамино-!Ч,П'/-бис(ме-тиленфосфоновой кислоты). Вещество представляет собой бесцветный гигроскопичный порошок, т. пл. 80—83°.
Найдено, %: С—28,0; Н—8,5; N—7,9; Р—18,5. OaHszNaOaPa- Вычислено, %: С—28,6; Н—8,5; N—8,3; Р—18,5.
ЛИТЕРАТУРА
1. Н. М. Д я т л о в а, М. И. К а б а ч н н к, Т. Я. М е д в е д ь, М. В. Р у-домино, Ю. Ф. Белугин. Докл. АН СССР, 161, 607 (1965).
2. К. М о е d г 11 ze г. R; R. Irani. J. Organ. Chem. 31, 1603 (1966).
3. I. R. Boon. J. Chem. Soc., 1947, 307.
4. Пат. ФРГ 635904 (1936); С. A., 31, 1113 (1938).
108
2,7 ДИМЕТОКСИ-1,6-ДИОКСАСПИРО[4,4]НОНАН
И. А. МАРКУШИНА, Н. В. ШУЛЯКОВСКАЯ
О
CH8O-f /~ОСНз
CgHisO^
М. в. 188,21
Ранее одним из нас был разработан метод электролитического интрамолекулярного алкоксилирования у-фурилалкано-лов [И. 2,7-Диметокси-1,6-диоксасриро[4,4]нонан получен нами впервые метоксилированием 1-(а-фурил)-З-пропаналя и последующим гидрированием образующегося 2,7-диметокси-1,6-ди-оксаспиро[4,4]нонена-3.
СХЕМА СИНТЕЗА
\ /~<Н2СН2СНО (NH<Br) —
х=ч ,ОСН3 ОН сн5о-< ।
ХСГ ХСН2СН2СН—осн3
-СН3ОН
СН3О-
Характеристика основного сырья
1-(а-Фурил)-3-пропаналь, т. кип. 81—82717 мм, = 1,4772; получение см. [21
109
Метанол, ГОСТ 6995—67, ч.
Аммоний бромистый, ГОСТ 4454—48, ч.
Водород, ГОСТ 3022—61.
Этиловый спирт 96%-ный, ГОСТ 6265—52.
Условия получения
Синтез 2,7-диметокси-1,6-диоксасгшро[4,4]нонена-3. В электролизер [3, 4] помещают раствор 63 г (0,5 моль) свежеперег-нанного 1-(а-фурил)-3-пропаналя и 10 г (0,1 моль) бромистого аммония в 200 мл метилового спирта. Смесь охлаждают до минус 20° (см. примечание 1). Электролиз проводят 7 часов при силе тока 4,0—3,5 а и напряжении 8—10 в. Затем раствор переносят в колбу и прибавляют метилат натрия (см. примечание 2). Метанол и аммиак отгоняют на водяной бане при пониженном давлении. Выпавший осадок бромистого натрия отфильтровывают на фильтре Шотта № 3 н промывают эфиром 3 раза порциями по 30 мл. Эфир отгоняют на водяной бане, остаток перегоняют, собирая фракцию с т. кип. 90—92°/ 2 мм.
Выход 2,7-диметокси-1,6-диоксаспиро[4,4]нопена-3 равен 66 г (74%), «д = 1,4570, = 1,1201 (см. примечание 3).
Найдено, %: С—58,18; 58,39; Н—7,56; 7,78.
CgHuOj. Вычислено, %: С—58,05; Н—7,58.
Получение 2,7-диметокси-1,6-диоксаспиро[4,4]нонана. В стальной вращающийся автоклав емкостью 250 мл помещают 8 г (0,04 моль) 2,7-диметокси-1,6-,диоксаспиро[4,41нонена-3, 90 мг абсолютного этилового спирта и 1 г никеля Ренея |5]. Гидрирование проводят прн комнатной температуре и начальном давлении водорода 100 атм и заканчивают по поглощении 0,53 л водорода. Катализат выгружают из автоклава, никель Ренея отфильтровывают и промывают на фильтре этиловым спиртом (2 раза порциями по 20 мл). Этанол отгоняют на водяной бане при пониженном давлении. Остаток перегоняют, собирая фракцию с т. кип. 92—93710 мм.
Выход 2,7-диметокси-1,6-диоксаспиро[4,41нонана равен 7,1 г (89%), «р = 1,4420, Л20 = 1,0816 (см. примечание 4).
Найдено, %: С - 57,60; 57,19; Н—8,94; 8,95.
CsHiaOj. Вычислено, °/о: С—57,43; Н—8,57.
Примечания;
1. В качестве охлаждающей смеси используют бутиловый или нзобу-тнловый спирт, в который добавляют твердую углекислоту до достижения и поддержания необходимой температуры.
2. Метилат натрия приготовляют из 2,4 г металлического натрия и 40 мл метанола. Прибавление метилата натрия необходимо для связывания свободного брома, образующегося в процессе электролиза.
ПО
3. Аналогичным путем получен впервые 2-метил-2,7-диметокси-1,6-ди-оксаспиро[4,4]нонен-3 из 1-(5-метил-2-фурил)-3.пропаналя [6] с выходом 71%, т. кип. 96—9879 мм, = 1,4530, rf?» = 1,0730.
Найдено, %: С—60,36; 60,38; Н—8,40, 8,38.
CioH16C>4. Вычислено, %; С—59,98; Н—8,07.
4. Аналогичным путем получен впервые 2-метил-2,7-диметокси- 1,6-ди-оксаспиро[4,4]нонаи с выходом 89%, т. кип. 88—8979 мм, лд = 1,4460, <№> = 1,0620.
Найдено, %: С—59,54; 59,39; Н—8,84; 8,63.
СюН|аО4. Вычислено, %; С—59,31, Н—8,97.
ЛИТЕРАТУРА
1. А. А. Пономарев И. А. Маркушина. Докл. АН СССР 126, 99 (1959).
2. А. А. Пономарев, 3. В. Тиль. Ж. общ. химии, 27, 1075 (1957).
3. А. А. Пономарев И. А. Маркушина. Ж. общ химии 30, 976 (1960).
4. А. А. Пономарев, И. А. Маркушина. Методы получения химических реактивов и препаратов, вып. 17. М„ ИРЕА, 1967, с. 99.
5. Синтезы органических препаратов, Сб. 3. М., ИЛ, 1952, с. 338.
6. Н. И. Ш у й к и н, А. Д. Петров. Изв. АН СССР, сер. хим., 9, 1682 (1964).
ДИНИТРИЛ 1,10 ФЕНАНТРОЛИН-2,9-ДИКАРБОНОВОЙ КИСЛОТЫ
В. М. ДЗИОМКО, Б. В. ПАРУСНИКОВ
c14h6n4
М. в. 230.23
Динитрнл 1,10-фенантролин-2,9-дикарбоновой кислоты в литературе не описан. Он получен нами с 87%-ным .выходом при нагревании раствора 1,10-фенантролин-2,9-диальдоксима в уксусном ангидриде. Дииитрил представляет собой желтоватое кристаллическое вещество с т. пл. 366°, нерастворимое в воде, растворяющееся при нагревании в пиридине, диметил-формамиде.
СХЕМА СИНТЕЗА
II
NOH
(СН3СО)2О
NOH
112
Характеристика основного сырья
1,10-Фенантролин-2,9-диальдоксим гидрат, т. разл. 257°; получение см. с. 287 этого ссбориика.
Уксусный ангидрид, ГОСТ 5815—69, ч. д. а., перегнанный
Условия получения
В круглодонной колбе емкостью 100 мл, снабженной мешалкой и обратным холодильником, кипятят 2 часа при размешивании смесь 4,26 г (0,015 моль) гидрата 1,10-фенантро лин-2,9-диальдоксим а и 35 мл уксусного ангидрида. Образуется прозрачный раствор, из которого затем выпадает желтый осадок дннитрнла. После охлаждения реакционной массы осадок отфильтровывают, промывают этанолом и высушивают в вакуум-эксикаторе над едким натром.
Выход динитрила 1,10-фенантролин-2,9-дикарбоновой кислоты равен 3,0 г (87%), т, пл. 364° (см. примечание).
Примечание:
Образец для анализа с т. пл. 366° был получен перекристаллизацией сначала из пиридина, затем из диметилформамида.
Найдено, %: С—72,83; 72,97; Н—2,88; 2,75; N—24,25; 24,50. Ci4H6N4. Вычислено, %: С—73,03; Н—2,63; N—24,34.
8 Зак. 1164
ДИОКСАНАТ БИС(ТРИ НИТРОМЕТИЛ )РТУТИ
А. Л. ФРИДМАН, Т. И. ИВШИНА
o(_^O-Hg|C(NOa)»J,
C6H8HgNeOH М. в. 588,75
Комплекс бис (тринитрометил) ртути с диоксаном может быть использован в реакциях тринитрометилмеркурирования. триннтрометилирования, присоединения бис(тринитроме-тил)ргути ло двойной связи, а также для синтеза гем-динит-роалкенов [1—5].
Диоксанат бис(триннтрометил)ртути синтезирован нами впервые. Диоксанат бис (тринитрометил) ртути в отличие от бис(тринитрометил)ртути устойчив и может храниться длительное время.
СХЕМА СИНТЕЗА
О\_/О + HgfC(NO,),b - о<_>O.Hg[C(NO2),J,
Характеристика основного сырья
Дноксан, ГОСТ 10455—63, ч.
Бис (тринитрометил) ртуть, бесцветное кристаллическое вещество, разлагается при температуре выше 230°, получение см. [1].
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, термометром и капельной воронкой, помещают 13,5 г (0,027 моль) бис (трннитрометил) ртути в 30 мл воды (см. примечание 1). К раствору при перемешивании н температуре 20—25° прибавляют по каплям 2,5 г (0,028 моль) дноксана. 114
Смесь перемешивают 15—20 минут. Выпавший белый крнстал-лический осадок отфильтровывают, промывают 20 мл воды и высушивают на воздухе. Комплекс трижды перекристаллизовывают из дихлорэтана.
Выход диоксаната бис(трннитрометил)ртути равен 8,5 г (85%),т. пл. 199° (разл.).Вещество хорошо растворимо в ацетоне, этаноле, эфире, хуже в дихлорэтане и хлоруглеводоро-дах (см. примечание 2).
Найдено, %: С—12,33; Н—1,40; N—14,37.
C6H»HgNsOM. Вычислено, %: С—12,23; Н—1,36; N—14,27.
Примечания:
1. Вне (трннитрометил) ртуть для синтеза комплекса получают непосредственно в виде водного раствора.
2. При обработке едким кали в метаноле комплекс расщепляется на эквимолекулярные количества диоксана, окиси ртути и калиевой соли три-нитрометана.
ЛИТЕРАТУРА
1. С. С. Новиков, Т. И. Годовиков а, В. А. Тартаковский. Изв. АН СССР, сер. хим., 3, 505 (1960).
2. С. С. Новиков, Т. И. Годовикова, В. А. Тартаковский. Изв. АН СССР, сер. хнм., 4, 669 (1960).
3. В. А. Тартаковский, С. С. Новиков, Т. И. Годовикова. Изв. АН СССР, сер. хим., 6, 1042 (1961).
4Ч А. Л. Фридман, В. П. И в ш и и, Т. Н. И в ш и и а, Н. Я. Волкова, В. А. Тартаковский, С. С. Новиков. Изв. АН СССР, сре. хим., 8, 1804 (1970).
5. А. Л. Фридман, В. П. И вшн н, Т. Н. И вши и а, В. А. Тарта-ковскнй, С. С. Новиков. Изв. АН СССР, сер. хим. 3, 729 (1970).
8*
2,6-ДИОКСИБЕНЗИЛ ИМИНОДИУКСУСНАЯ КИСЛОТА
В. я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА, Л. Д. МЕЛИКСЕТЯН, Р. П. ЛАСТОВСКИЙ
ОН
Jx/CHaN(CH2COOH),
I II Ч/Хон
CjiHuNOe М. в. 255,23
2,6-Диоксибеизилиминодиуксусная кислота является комплексообразующим реагентом, дающим с рядом катионов достаточно прочные комплексы.
Комплексон получен нами впервые, строение его доказано методом ЯМР [1].
СХЕМА СИНТЕЗА
ОН
I
СНаСООН
I || -i-cHjO+hn/ _>
Х./\он XCH2COONa
ОН
' z CHaN(CHaCOOH)a
- 1 II Xv/Xoh
Характеристика основного сырья
Резорцин, ГОСТ 9945—62, ч.
Формальдегид, 37%-иый раствор, ГОСТ 1625—61, техн.
116
Иминодиуксусная кислота, ВТУ МГ УХП 190—58, ч.
Натр едкий, ГОСТ 4328—66, ч.
Уксусная кислота ледяная, ГОСТ 61—51, х. ч.
Условия получения
В трехгорлую колбу емкостью 200 мл, снабженную мешалкой и капельной воронкой и помещенную в баню с обогревом и охлаждением, загружают 22 г (0,2 моль) резорцина и 100 мл ледяной уксусной кислоты. При размешивании и температуре 18—20° резорцин растворяется в ледяной уксусной кислоте. В полученный раствор приливают нейтрализованный раствор иминодиуксусной кислоты, состоящий из 26,6 г (0,2 моль) иминодиуксусной кислоты, 26 мл воды и 30%-ного раствора едкого натра, необходимого для нейтрализации кислоты до pH 7—8. Затем содержимое колбы охлаждают до 5— 7° и за 30 минут прибавляют 16,7 г (0,2 моль) 37%-ного раствора формальдегида, после чего температуру реакционной массы поднимают до 18—20° и размешивают 3 часа при этой температуре. Выпавший осадок отфильтровывают, промывают 300 мл воды до отсутствия запаха уксусной кислоты и сушат при 50°.
Выход 2,6-диоксибензилиминодиуксусной кислоты равен 30 г (—60%).
Найдено, %: С—51,5; 51,4; Н—5,2; 5,3; N—5,7; 5,6. СцН1зМО6. Вычислено, %; С—51,7; Н—5,1; N—5,5.
ЛИТЕРАТУРА
1. Н. В. Ци р у л ь н и к ов а, Н. А. Ко с т р о м и 11 а, Б. В. Жаданов, В. Я. Темкина. Ж- орган, химии, 7, 2, 327 (1971).
2,3-ДИОКСИБЕНЗИЛИМИНОДИУКСУСНАЯ КИСЛОТА, МОНОНАТРИЕВАЯ СОЛЬ
В. я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА, С. А. ТАРАСОВА, Р. П. ЛАСТОВСКИИ
ОН
А~он zCH,CO0H xCHsCOONa
CnHiaNNaO6
M. в. 277,21
2,3-Диоксибензнлимииоднуксусиая кислота (моионатриевая соль) является комплексообразующим реагентом и можег быть применена, в частности, для обработки активированного угля с целью использования последнего для сорбции из растворов ниобия и циркония.
Комплексон получен нами впервые; строение его доказано методом ЯМР [1].
СХЕМА СИНТЕЗА
ОН
СН,О + HN
/СНаСООН
\CH»COONa
1СН3СООН
ОН
I
j^Y*0H ^сн2соон
L CH.N<;
XCH»COONa
118
Характеристика основного сырья
Пирокатехин, ГОСТ 1872—62, ч. д. а.
Формальдегид, 37%-ный раствор, ГОСТ 1625—61, техн. Иминодиуксусная кислота, ВТУ МГ УХП 190—58, ч.
Натр едкий, ГОСТ 4328—66, ч.
Уксусная кислота ледяная. ГОСТ 61—51, х. ч.
Условия получения
В четырехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой и помещенную в баню с обогревом и охлаждением, загружают 100 мл ледяной уксусной кислоты и 11 г (0,1 моль) пирокатехина. К полученному раствору приливают при размешивании нейтрализованный водный раствор иминодиуксусной кислоты, состоящий из 13,3 г (0,1 моль) иминодиуксусной кислоты, 13 мл воды и 30%-ного раствора едкого натра в количестве, необходимом для нейтрализации кислоты до pH 7—8. Затем содержимое колбы охлаждают до температуры 5—10е и при этой температуре за 15—20 минут прибавляют по каплям 8,1 г (0,1 моль) 37%-ного раствора формальдегида,после чего реакционную массу нагревают до 50°, размешивают при этой температуре 4 часа, охлаждают и при сильном разме шивании выливают по каплям в стакан, содержащий 1,4 л ацетона. Выпавший хлопьевидный осадок через 1 —1,5 часа отфильтровывают, промывают 50 мл ацетона и сушат в ваку-ум-эксикаторе.
Выход мононатриевой соли 2,3-диоксибензилиминодиук-сусной кислоты равен 26,2 г (94,5%).
Найдено, %: N—4,8; 4,9; Na—18,7; 18—9. C))H)2NNaOe. Вычислено, %: N—5,0; Na—16,1.
ЛИТЕРАТУРА
1. Н. В. Цирульников а, Н. А. Костромина, Б. В. Жаданов, В. Я. Т е м к и и а. Ж- орган, химии, 7, 2, 327 (1971).
N,N' ДИ(2-ОКСИ-5-МЕТИЛБЕНЗИЛ)-ЭТИЛЕНДИАМИНО-Ч^-ДИУКСУСНАЯ КИСЛОТА
Н. В. ЦИРУЛЬНИКОВА, В. Я. ТЕМКИНА, Р. П. ЛАСТОВСКИИ
ОН ОН
|^X|-CH2-N-CH2—CH1-N-H,C-^2 * 4j| Ч/ | W
СН, СН,СООН СН.СООН СН,
C22H28N2O6 М. в. 416,48
N.N'-Ди (2-окси-5-метил бензил) этил ендиамино-ЧМ'-диук-сусная кислота является хелатореагентом, образующим с рядом катионов высокопрочные комплексы, в частности, с железом эта кислота образует комплекс практически уникальной прочности (IgK ~ 40).
Синтез комплексона осуществлен впервые по реакции Ман-ниха взаимодействием n-крезола, формальдегида и этиленди-амин-ЧЧ'-диуксусной кислоты в щелочной среде с последующим выделением продукта при подкислении реакционного раствора. Вещество представляет собой порошок желтоватого цвета, растворимый в спирте, щелочах, практически нерастворимый в воде.
СХЕМА СИНТЕЗА
ОН
2 + 2СНаО -f- HN-CH,-CH2-NH
X/ I I
I CHsCOONa CHgCOONa
CH,
120
он он
^\rCH,-N-CH.-CH.-N-CH,-[^ + NaCl + Н.0
СН, СН2СООН СН2СООН СНз
Характеристика основного сырья
я-Крезол, МРТУ 609—2769—66, ч.
Формальдегид, 37%-ный раствор, ГОСТ 1625—61, техн.
Этилендиамин-Г<,М7-диуксусная кислота, получение см. [1].
Натр едкий, ГОСТ 4328—66, ч.
Соляная кислота, ГОСТ 3118—66, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником и термометром и помещенную в баню с обогревом и охлаждением, загружают 6,5 -л (0,06 моль) n-крезола в 9 мл дистиллированной воды и 6 мл 30%-ного раствора едкого натра. К полученному раствору прибавляют 5,88 г (0,033 моль) этилендиамин-1Ч,ЬГ-диуксус ной кислоты, предварительно нейтрализованной 30%-ным раствором едкого натра до pH 9. Полученный раствор охлаждают до 10° и за 20 минут прибавляют по каплям 4,85 г (0,06 моль) 37%-ного раствора формальдегида. Реакционную массу размешивают 1 час при 18—20°, н 3 часа при 70°, охлаждают до 10—12° и подкисляют концентрированной соляной кислотой до достижения pH 2. Выпавший при этом маслянистый продукт отделяют, растворяют в 30 мл этилового спирта, фильтруют от неорганических солей и упаривают раствор при пониженном давлении досуха. Растворение вещества в этиловом спирте н отгонку растворителя повторяют 2—3 раза.
Выход М,М'-ди (2-окси-5-метилбензнл)этилендиамнно-М,М'-днуксуснон кислоты равен 5,4 г (39,9%).
Найдено, %: С—63,0; 63,1; Н—6,5; 6,6; N—7,0; 6,8. C22H2aN2O6. Вычислено, %; С—63,4, Н—6,8, N—6,7.
ЛИТЕРАТУРА
1. Н. В. Цирульникова, В. Я. Темкина, Г. А. Громова, Р. П. Ластовскнй. Методы получения химических реактивов и препаратов, вып. 18. М., «Химия», 1969, с 223.
4,4'-ДИ-н-ПЕНТОКСИАЗОКСИБЕНЗОЛ
3. С. СИДЕНКО, В. А. ИНШАКОВА, Г. С. ЧИЖОВА
^\—N=N—
HnCsO-J J ! I LoCeHn
C22H30N2O3 М. в. 370,47
4,4'-Ди-н-пентоксиазоксибензол может быть применен в качестве жидкого кристалла. Для его получения рекомендуется электролитическое восстановление 4-пентоксинитробензо-ла [1—4].
Предлагаемый метод позволяет получать 4,4'-ди-н-пенто-ксиазоксибензол восстановлением 4-пентоксинитробензола этилатом натрия в присутствии гидразин-гидрата.
СХЕМА СИНТЕЗА
no2
2 Йг i^Vn=n“iZXii
X/ Н’° HnC.O-^J £ ^/И-ОСзНн оеанп
Характеристика основного сырья
4-Пентоксинитробензол, т. пл. 73,5—74°; получение см. [2J.
Гидразин-гидрат, ГОСТ 5832—51, ч.
Натрий металлический плавленый, ТУ МХП 1664—50, ч.
Этиловый спирт синтетический, ГОСТ 9674—61.
Условия получения
В колбу емкостью 250 мл, снабженную обратным холодильником, загружают 130 мл этилового спирта и вносят не-122
большими порциями 10 г мелконарезаннбго металлически натрия. В горячий раствор этилата натрия вносят частями 12,54 г (0,06 моль) растертого в порошок 4-пентоксинитро-бензола, который при размешивании легко растворяется. К реакционному раствору быстро приливают 7,5 г гидразин-гид-рата и при перемешивании нагревают 1 час на кипящей водяной бане. Через 30 минут реакционную смесь выливают в 500 мл холодной воды, при этом выпадает желтый осадок, который отфильтровывают, промывают несколько раз холодной водой, сушат и кристаллизуют 2 раза из смеси (4: 1) спирт — циклогексан. Первую кристаллизацию проводят с добавлением активированного угля.
Выход 4,4'-ди-н-пентоксиазоксибензола равен 6,2 г (28%), т. пл. 73—74°.
По литературным данным [4], т. пл. 75,5°.
Найдено, %: С—71,04; 70,91; Н—7,85; 7,84; N—7,51; 7,68. СиНзоЫаОз. Вычислено, %: С—71,31; Н—8,16; N—7,56.
ЛИТЕРАТУРА
1. К. E lbs. Z. Elektrochem. angew. phys. Chem,, 7, 147 (1900).
2. C. Weygand, R. Gabler. J. prakt. Chem., 155.832 (1940).
3. C. Weygand, R. Gabler. Ber., 71 B, 2399 (1938).
4. H. Arnold. Z. phys. Chem., 226, 146 (1964).
2.6-ДИХЛОР-4-НИТРОАНИЛ ИН
Р. Ф. ВИДЕНИНА, Н. Н. ДЫХАНОВ
C5H4CI2N2O2
nh2
С1_('Уе1
no2
М. в. 207,0
Синтез 2,6-дихлор-4-нитроанилина с выходом около 70% был впервые осуществлен пропусканием газообразного хлора через раствор п-нитроанилина в разбавленной соляной кислоте (1]. Позднее это вещество было получено с близким к количественному выходом действием хлорноватистой кислоты на раствор п-ннтроанилнна в эфире [2].
Нами 2,6-дихлор-4-ннтроаннлин получен способом, аналогичным описанному для синтеза 3,5-дихлорсульфаниламнда [3] — действием перекиси водорода на раствор я-ннтроаиилина в разбавленной соляной кислоте. Этот способ удобен в технологическом оформлении н дает выход продукта близкий к количественному.
СХЕМА СИНТЕЗА 4НС1 + 2Н,О2 -> 2C1S + 4Н,О, NH2 NH,
NO2 NO,
124
Характеристика основного сырья
я-Ннтроанилин, ТУ 6—09—258—70, ч.
Пергидроль, ГОСТ 10929—64, х. ч.
Соляная кислота, ГОСТ 3118—67, ч.
Условия получения
В колбу емкостью 0,5 л, снабженную мешалкой и термометром, помещают 13,8 г (0,1 моль) я-нитроанилнна, 150 мл воды и 150 мл концентрированной соляной кислоты. Содержимое колбы нагревают до 50—55°, перемешивают до растворения осадка и приливают 22,5 мл 30%-ной перекиси водорода. Немедленно начинается экзотермическая реакция, в результате которой температура в массе самопроизвольно повышается до 74— 76° и удерживается в этих пределах 17—20 минут (см. примечание 1). За это время из реакционного раствора выпадает обильный осадок и масса сильно загустевает.
Как только температура в массе начнет понижаться, массу нагревают до 80—85° и фильтруют в горячем состоянии (см. примечание 2). Осадок на фильтре промывают 75— 100 мл горячей (80—85°) 15—20%-ной соляной кислоты, затем водой до нейтральной реакции и сушат при 85—90° (см. примечание 3).
Выход технического 2,6-дихлор-4-ннтроанилнна равен 19,5—19,8 г (95—96%), т. пл. 188—192°. После перекристаллизации из этанола (1:7) т. пл. 194—195° (см. примечание 4). что совпадает с литературными данными (2].
Примечания:
1. Дальнейшее повышение температуры реакции нежелательно, так как качество и выход продукта ухудшаются. В случае надобности реакционную колбу на короткое время погружают в баню с холодной водой.
2. Для более легкого извлечения продукта к реакционной массе следует добавить 150—200 мл горячей (80—85°) 15—20%-нон соляной кислоты.
3. Если сушить продукт при более высокой температуре, то происходит его частичная возгонка, и выход снижается.
4. Потери вещества при перекристаллизации не превышают 5% от веса технического продукта. Исходный растворитель регенерируется перегонкой н используется в аналогичном опыте.
ЛИТЕРАТУРА
1, О. N, Wi t1. Вег., 8, 145 (1875).
2. S. Goldschmidt, L. St r 6 m e n g e r. Ber., 55, 2457 (1922).
3. Синтезы органических препаратов, сб. 3. М., ИЛ, 1951, с. 351.
2.6-ДИХЛОР-1,4-ФЕН ИЛ ЕНДИАМИН
Р. Ф. ВИДЕНИНА. Н. Н. ДЫХАНОВ
C6H6C12N2
nh2 с\ A/ci I! I
I
nh2
M. B. 177,03
Известны два способа восстановления 2,6-дихлор-4-нитро-анилина в соответствующий диамин: цинком в соляной кислоте [1] и железом в серной кислоте [2]. В обоих случаях* процесс осуществляется в гетерогенной среде, выход 2,6-дихлор-1,4-фенилендиамина не превышает 60%, а извлечение его из цинкового или железного шлама представляет значительные трудности.
Нами 2,6-дахлор-1,4-фенилендиамин получен нагреванием 2,6-дихлор-4-ннтроанилина с избытком двухлористого олова в смеси концетрироваиной соляной кислоты и метилового спирта. Восстановление протекает в гомогенной среде, продукт легко выделяется из реакционного раствора в виде мало растворимого гидрохлорида с выходом 75%.
СХЕМА СИНТЕЗА
ЫН,
Ci\ A/ci
II I
W
NO,
ын2
nh2
126
Характеристика основного сырья
2,6-Дихлор-4-нитроанилин, т. пл, 194—195°; получение см. с. 124 этого сборника.
Олово двухлористое, ГОСТ 36—68, ч.
Метиловый спирт синтетический, ГОСТ 6995—67, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Условия получения
В колбу емкостью 0,5 л, снабженную мешалкой, термометром и обратным холодильником, помещают 20,7 г (0,1 моль) 2,6-днхлор-4-нитроаннлина, 90,5 г (0,4 моль) дитндрата дву-хлорнстого олова, 150 мл метилового спирта и 150 мл концентрированной соляной кислоты. Содержимое колбы размешивают до образования однородной суспензии, нагревают 3 часа при слабом кипении (75—80°) и полученный раствор оставляют охлаждаться во льду до 3—5°. Выпавший осадок отфильтровывают (см. примечание), растворяют в 500 мл воды и прибавляют 50 мл 20%-ного раствора едкого натра. Выпадает белый рыхлый осадок основания 2,6-днхлор-1,4-фенилен-днамнна, который отфильтровывают, промывают водой до нейтральной реакции и сушат при 75—80°.
Выход технического 2,6-дихлор-1,4-фенилендиамина равен 13,5—14 г (76—80%), т. пл. 120—123°. После перекристаллизации из 50%-ного метанола (потери при кристаллизации составляют около 10% от веса) т. пл. 123—123,5°, что соответствует литературным данным [2].
Примечание.
При добавлении к солянокислому маточному раствору избытка раствора едкого натра выпадает обильный белый осадок стаината натрия, который растворяют при разбавлении водой; нерастворившимся остается лишь небольшое количество 2,6-дихлор-1,4-фенилендиамина (не более 0,5 г). Дополнительное получение такого количества диамина не оправдывается слишком большим расходом щелочи.
ЛИТЕРАТУРА
1. О. N. Wi t L Вег. 8, 145 (1875).
2. R. Morgan, S. Cleage. J, Chem. Soc., 113, 594 (191Я1
Д И Э Т И Л Е Н Т Р И AM И Н О-N, N,N',N', N"- П Е Н ТА (МЕТИЛЕНФОСФОНОВАЯ КИСЛОТА)
М. В. РУДОМИНО, Е. к. К.ОЛОВА, А. М. ОРЛОВ, Л. И. МИЗРАХ, Г. Я. МЕДВЕДЬ
[(HO^PCHsbNCEUCHj-N—СН2СН,М[СН,Р(ОН),],
II I II
О СН4Р(О)(ОН), о
C9H28N3O15PS М. в, 574,01
Диэтил ентриамнно-№М1М/,М/,М"-пента (метиленфосфоновая кислота) (ДТПФ) является высокоэффективным металлоемким комплексообразующим соединением, способным связывать ионы широкого ряда металлов в прочные комплексы [1, 2]
Ранее [3] нами был предложен метод синтеза этой кислоты конденсацией диэтилентриамнна с формалином н фосфористой кислотой, одиако последняя является дефицитным н дорогостоящим продуктом. С целью замены фосфористой кислоты мы получили ДТПФ конденсацией днэтилентриамина с формалином и треххлористым фосфором аналогично методу, предложенному для синтеза аминоалкилфосфоновых кислот [4]. Очистка ДТПФ производилась хроматографически на катионите КУ-2.
СХЕМА СИНТЕЗА
Н,МСН4СН2ЫНСН2СН4КН4 + 5 СН2О + 5 РС13 + 10Н,О — - [(НО)2Р(о)СН,] tNCH1CH1NCHsCHsN [ CHSP(O)(OH) J, +15НС1
СН2Р(О)(ОН)5
Характеристика осиовиого сырья
Диэтилентриамин, МРТУ 6—09—3208—66, ч.
Формалин, 37%-нын, ГОСТ 1625—64.
128
Фосфор треххлористый, ГОСТ 91 —41, ч.
Метиловый спирт синтетический, ГОСТ 6995—54. Катионит КУ-2, ГОСТ 5695—53.
Условия получеиия
В четырехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром, капельной воронкой и обратным холодильником, помещают 10,3 г (0,1 моль) диэтилентриамина, 31,4 мл воды, 35,0 г 37%-ного формалина (0,55 моль СН2О), перемешивают до полного растворения и прибавляют по каплям при энергичном перемешивании 68,75 г (0,5 моль) трех-хлорнстого фосфора, поддерживая охлаждением водой температуру реакционной смеси до 30°. Затем реакционную массу нагревают до 105° и выдерживают при этой температуре 1,5 часа, последний час через систему продувают азот, затем понижают давление (15—20 мм) и отгоняют выделившийся хлористый водород. К остатку прибавляют 200 мл метилового спирта, выпавший осадок отделяют декантацией и растворяют в 350 мл воды. Раствор пропускают через колонку с катионитом КУ-2 (Н+-форма, 40X400 мм), смывая водой фракцию с pH 1—2. Элюат упаривают при пониженном давлении до объема 100 мл, полученный раствор прибавляют по каплям при перемешивании к 1 л метилового спирта. Выпавший осадок отфильтровывают и сушат в вакуум-эксикаторе над пяти-окисью фосфора.
Выход диэтилентриамино-М',М,ЬГ,М/,М"-пента (метиленфосфоновой кислоты) равен 37,5 г (66%).
Найдено, %: С—18,70; 18,38; Н—4,95; 5,04; N-7,17; 7,31. CgHaaNsOisPs. Вычисление, %: С—18,85; Н—4,89; N—7,33.
ЛИТЕРАТУРА
1( М. И. Кабачник, Н. М. Дит лов а, Т. Я. Медведь, Ю. Ф. Белугин, В. В. Сидоренко. Докл. АН СССР, 175, 351 (1967).
2. Л. И. Тихонова. Ж. неорган. химии, 13, 2687 (1968).
3. А. М. Орлов, Л. И. Миер ах, В. Г. Яковлев, Л. Б. Шагалов, М. И. К а б а ч и ик, Т. Я. Медведь, М. В. Р у д о м и и о. Авт. свид. 358320; Открытия. Изобретения. Промобразцы. Товарные знаки № 34
(1972).
4. Англ. пат. 1142294 (1969); РЖХнм., Зи144П (1970).
9 Зак. 1164
ДИЭТИЛФУРФУРИЛИДЕНМАЛОНАТ И ЭТИЛФУРФУРИЛИДЕНАЦЕТОАЦЕТАТ
Л. Г. БОГАТСКАЯ, Г. Л. КАМАЛОВ
II II II II „ /СОСНз
" "-сн=с(соос2н6)Л J-ch=c;
О О ХСООС2Н5
С12Н14О5 М. в. 238,23 CnH11S04 М. в. 208,20
Днэтнлфурфурилиденмалонат н этилфурфурилнденацето-ацетат получают по реакции Кновенагеля [1] взаимодействием фурфурола сответственно с малоновым и ацетоуксусным эфирами в присутствии каталитических количеств пиперидина. Эти вещества могут быть использованы для получения производных цис- и транс-фурнлакрнловых кислот и синтеза азотсодержащих гетероциклов.
Нами уточнена методика [1], показано, что в качестве катализаторов могут быть с успехом использованы аниониты, продукты реакции легко очищаются вакуумчперегонкой до кристаллического состояния. Методами ГЖХ и ЯМР показано, что этнлфурфурнлнденацетоацетат представляет собой смесь стереоизомеров (цис-транс) в процентном соотношении 60:40.
СХЕМА СИНТЕЗА
о
Н3С(СООС»Н5)2
J'—CH=C(COOGjH6)s О
Н3ССОСН,СООСгН8
СОСНз
—СН=С:
СООС8Н5
130
Характеристика основного сырья
Фурфурол, ГОСТ 10930—64, ч., свежеперегнанный.
Малоновый эфир, МРТУ 6—09—6046—69, ч., свежеперегнанный.
Ацетоуксусный эфир, ГОСТ 9799—61, ч., свежеперегнанный.
Анионит АВ-17П, ГОСТ 13504—68. перевод анионита в ОН-форму производили по методике [2].
Условия получения
В круглодонную колбу емкостью 250 мл, снабженную мешалкой и хлоркальцневой трубкой, помещают 160,2 г (1 моль) малонового либо 130,2 г (1 моль) ацетоуксусного эфира, 96,1 г (1 моль) фурфурола, 100 мл абсолютного спирта н 1 г анионита АВ-17П. Реакционную смесь перемешивают прн комнатной температуре 3—4 дня, катализатор отфильтровывают и промывают 30—50 мл абсолютного спирта. От фильтрата в токе сухого азота отгоняют спирт, а остаток, также в атмосфере сухого азота, перегоняют прн пониженном давлении.
Диэтнлфурфурилнденмалонат. Выход равен 178,5 г (75%), т. пл. 34—35° (см. примечание 1), т. кип. 163—16673 мм, d420 = = 1,1495; = 1,5363.
Найдено, %: С—60,54; Н—6,11. С12Н14О5. Вычислено, %: С—60,50; Н—6,00.
Этнлфурфурнлнденацетоацетат. Выход равен 135—140 г (65—68%), т. пл. 38—42° (см. примечание 2), т. кип. 152— 15472 мм, rfS050 = 1,4321; л™= 1,5458.
Найдено, %: С—63,68; Н—5,65. С11Н12О4. Вычислено, %: С—63,50; Н—5,77.
Примечания:
1. Вещество кристаллизуется при стоянии в течении недели в темноте либо при длительном трении стеклянной палочкой о дно колбы, что делает возможным определение его констант при 20°.
2. Широкий интервал температуры плавления обусловлен тем, что в результате реакции образуется смесь цис- и транс-нзомеров этилфурфу-рил идеи ацето ацет ат а.
ЛИТЕРАТУРА
1. Е. К п о е v е n a-g-e 1. Вег., 31, 735 , 2595 (1898).
2. К. И. С а л д а д з е, А. Б. Пешков, В. С. Титов. Высокомолекулярные ионообменные смолы. М., Госхимиздат, 1960, с. 85.
9
D.L-ИЗОЛЕЙЦИН
Е. П. КРЫСИН, И. И. ГРОМОВА, Л. Г. АНДРОНОВА
C6H13NOa
С2Н.Ч .СООН
Ун-сн<
CH3Z xnh2
М. в. 131,17
В основу метода получения D.L-изолейцина положен гидантоиновый синтез по способу Бухерера (1]и Штреккера [2], состоящий в превращении альдегида в циангидрин, дающий при обработке карбонатом аммония замещенный гидантоин. При гидролизе последнего едкими щелочами образуется аминокислота. Этот метод может быть положен в основу промышленного способа получения D.L-изолейцина, так как он технологически удобен и дает высокий выход аминокислоты.
СХЕМА СИНТЕЗА
С,Н*^СН-СНО -/кн^со—►С,Н,')СН-НС— СН/ (NH4),CO3 CHs/ J !
HN NH
II
О
с8н6
- >сн-сн-соон chZ i
NHj
Характеристика основного сырья
2-Метилбутаналь, т. кип. 91—92°; получение см. с. 157 этого сборника.
132
Натрий бисульфит, ГММХП 1944—49, ч.
Калий цианистый, МРТУ 6-09-3799—67, ч.
Аммоний углекислый, ГОСТ 3770—64, ч.
Барий гидрат окиси, ГОСТ 41—07—65, ч.
Изопропиловый спирт, ТУ 6—09—402—70, ч.
Условия получения
Синтез втор-бутилгидантоина. В четырехгорлую колбу емкостью 1 л, снабженную мешалкой, капельной воронкой, тер мометром и обратным холодильником, вносят 356 мл (1,4 моль) 99%-ного раствора бисульфита натрия и при энергичном перемешивании медленно добавляют 100 мл (1 моль) 2-метилбутаналя. Продолжая перемешивание, через 15— 20 минут небольшими порциями прибавляют 90,5 г (1,4 моль) цианистого калия. Образующийся слой отделяют, а маточный раствор экстрагируют 300 мл изопропилового спирта. Экстракты объединяют, переносят в колбу с обратным холодильником, добавляют 315 г углекислого аммония и кипятят на водяной бане 4—5 часов. Выпавший .при охлаждении осадок отфильтровывают.
Выход втор-бутилгидантоина равен 162 г (75% в расчете на 2-метилбутаналь), т. пл. 120—122°.
Получение Г),'С-изолейцина. В круглодонную колбу емкостью 1 л с обратным холодильником загружают 50 г (0,3 моль) втор-бутилгидантоина, 280 г (0,9 моль) гидрата окиси бария, 4Q0 мл воды. Смесь кипятят на электрической плитке 25 часов. При вспенивании добавляют высококипящий спирт (20 мл). Для полного окончания реакции ионы бария осаждают добавлением 250 г углекислого аммония и осадок отфильтровывают. При наличии ионов бария в фильтрате добавляют еще 50 г углекислого аммония. Фильтрат упаривают до объема 100 мл, охлаждают и выпавшие кристаллы отфильтровывают.
Выход DL-изолейцина равен 37,5 г (68% в расчете на вгор-бутилгидантоин); содержание основного вещества 99,2%.
Найдено, %: С—54,75 Н—9,89; N—10,67. C6Hi3NO2. Вычислено, %: С—54,90; Н—9,98; N—10,67.
ЛИТЕРАТУРА
1. Н. Т. Ви с here г, W. Steiner. J. pract. Chem., 140, 291 (1934). 2. A. Strecker. Ann, 75, 27 (1850).
ИЗОМЕРНЫЕ МЕТИЛ-
И МЕТОКСИБЕНЗОЛСУЛЬФОХЛОРИДЫ
Н. Н. ДЫХАНОВ, А. Б. ДЖИДЖЕЛАВА, Т. С. РЫЖКОВА
С7Н7СЮ2
SO2C1 (}сн3 М. в. 190,65
SOaCl
I /'Ч II +ОСН,
C7H7C1O3
М. в. 206,64
Общий метод синтеза орто-, мета- и лара-замещенных бензолсульфохлоридов заключается в действии избытка жидкой двуокиси серы или ее раствора в ледяной уксусной кислоте на соответственно замещенные хлориды феннлдиазониев в присутствии двухлорнстой меди [1]. Однако хлориды фенил-диазониев с электронодонорными заместителями весьма термолабильны, поэтому выходы сульфохлоридов из арнламинов с такими заместителями не превышают 30%.
Известно, что сульфаты арнлдиазониев более устойчивы к нагреванию, чем их хлориды [2]. Исходя из этого, для полу чения изомерных метил- и метоксибензолсульфохлоридов мы использовали сульфаты соответствующих метил- н метокси-фенилдиазониев действием на них избытка уксуснокислого раствора двуокиси серы в присутствии двухлористой меди и коя« центрированной соляной кислоты [3]. Катализатором этого процесса является не двухлористая медь, а образующаяся при ее восстановлении двуокисью серы однохлористая медь.
Выходы изомерных метнлбензолсульфохлоридов по этому способу достигают 80%, метоксибензолсульфохлоридов—40%.
СХЕМА СИНТЕЗА
NaNO, H2SO4
so?-
134
/ ч II +R Z\S SO2C1
где R = CHS— ; CH3O—.
Характеристика основного сырья
о-Толундин, МРТУ 6—09—2898—66, ч.
л-Толуиднн, ТУ 6—09—194—70, ч.
я-Толундин, ТУ 6—09—66—70, ч.
о-Анизидин, МРТУ 6—09—5888—69, ч. .'и-Анизндин, МРТУ 6—09—5770—69, ч. п-Анизидин, МРТУ 6—09—4817—67, ч. Уксусная кислота ледяная, ГОСТ 61—69, ч. Натрий азотистокислый, ГОСТ 4197—66, ч. Соляная кислота, ГОСТ 3118—67, ч.
Медь двухлористая, ГОСТ 4167—61, ч.
Бензол, ГОСТ 5955—68, ч. д. а.
Условия получения
В стакан емкостью 0,5 л, снабженный мешалкой, термометром и капельной воронкой, конец которой на 9—10 см не доходит до дна стакана, помещают 16,05 г (0,15 моль) толуидина или 18,45 г (0,15 моль) анизидина и раствор 12 мл концентрированной серной кислоты в 200 мл воды. Суспензию нагревают до полного растворения осадка (см. примечание 1). охлаждают до 0—20° и диазотируют раствором 10,35 г нитрита натрия в 35 мл воды, вводя его под поверхность жидкости. По окончании диазотирования кристаллы сульфата толуидина или анизидина должны полностью раствориться, а образовавшийся светло-желтый раствор сульфата диазония должен иметь кислую реакцию на конго (рН~2) и в течение 10 минуг давать положительную реакцию на свободную азотистую кислоту (проба с иод-крахмальной бумажкой). В противном случае приливают еще ~2—5 мл раствора серной кислоты или соответственно нитрита натрия.
В колбу емкостью 1 л, снабженную быстроходной мешалкой (120—150 об] мин). термометром и капельной воронкой, помещают 100 мл насыщенного уксуснокислого раствора двуокиси серы (см. примечание 2), 30 мл концентрированной соляной кислоты, 150—200 мл бензола и 6,5 г двухлористой меди. Смесь перемешивают до образования тонкой эмульсии, приливают к ней полученный раствор сульфата диазония, нагревают до 30—45° и выдерживают при этой температуре до полного прекращения выделения пузырьков азота, на что требуется от 10 минут до 4 часов (см. таблицу).
135
Реакционную массу выливают в тройной объем водопроводной воды, органический слой отделяют, промывают его водой (до нейтральной реакции в промывных водах), сушат 3 часа над 15 г хлорида кальция и фильтруют. Фильтрат помещают в .перегонную колбу и отгоняют бензол (его используют в очередном опыте), а остаток перегоняют при пониженном давлении.
Выходы и константы метил- н метоксибензолсульфохлори-дов приведены в таблице.
Условия разложения сульфатов диазоиия, выходы и константы сульфохлор ндов
Исходный амин Условия разложения сульфатов диазоиия Выход и константы сульфохлоридов
выход, % т. кип., °C !м м т. пл., °C
экспе-рнм. данные ЛИТ. данные [4]
темперу* время, мин
о-Толуидин 30—40 90 65,5 125-126/10 масло 10
тМ-ТолуиДНИ 30—40 10 74,7 145-146/22 » П,7
п-Толуидин 30-40 35 80,2 145- 146/15 70-71 69
о-Аиизидин 40-45 240 39,5 128—129/0,3 55-56 55-56
.м-Анизидии 40-45 15 40,4 158-159/20 масло масло
п-Анизндии 40—45 60 37,7 119-120/0,4 42- 43, 42-43
Примечания:
1. Переведение осадка сульфата ариламина в раствор при нагревании необходимо для получения его в тоикодисперсном состоянии прн последующем быстром охлаждении льдом.
2. Насыщенный раствор двуокиси серы в ледяной уксусной кислоте готовят пропусканием газа в жидкость при охлаждении до 5—10’ до привеса 50%. Полученный 30—33%-ный уксуснокислый раствор двуокиси серы хранят в силяике с притертой пробкой при температуре не выше 20°.
ЛИТЕРАТУРА
1. Г. Меервейн, Г. Дитмар, Г. Гельиер, К. Хафнер, Ф. Менш, О. Штейифорт. Химия и хим. технол. 6, 123 (1956).
2. С. Ф. Филиппычев. Химия и технология азокрасителей, т. I, М., Госхимнздат, 1938, с. 86.
3. Н. Н. Д ы х а и о в, А. Б. Д ж и д ж е л а в а, Т. С. Рыжкова. Авт. свид., 205013; Изобретения. Промобразцы. Товарные знаки, № 23 (1967).
4. Ч. Сью тер. Химия органических соединений серы, вып. 2. М., ИЛ, 1951, с. 286, 297.
И30(4,7,8,9-ТЕТРАГИДР0)БЕН30-1,3-ДИМЕТОКСИТЕТРАГИДРОФУРАН
А. А. ПОНОМАРЕВ, И. А. МАРКУШИНА, Г. Е. МАРИНИЧЕВА
СюН1бОз
М. в. 184, 23
При синтезе А4-тетрагндрофтальальдегида [1] в качестве побочного 'продукта был выделен изо (4,7,8,9-тетрагидро)бен-зо-1,3-диметокситетрагидрофуран. Нами это соединение синтезировано по реакции Дильса-Альдера взаимодействием 1,3-бу-тадиена с 2,5-диметокси-2,5-дигидрофураном [2].
СХЕМА СИНТЕЗА
Характеристика основного сырья
2,5-Диметокси-2,5-дигидрофуран, т. кип. 158—160°, Лд = = 1,4352; получение см. ГЗ].
1,3-Бутадиен, т. кип. —4,5°/760 мм, di20 = 0,6206; получение см. [4].
Гидрохинон (фото), ГОСТ 2549—60.
137
Условия получения
В стальной вращающийся автоклав емкостью 150 мл, охлажденный смесью твердой углекислоты с бутиловым или изобутиловым спиртом, помещают 39 г (0,3 моль) 2,5-диметокси-2,5-дигидрофурана, 32,4 г (0,6 моль) предварительно сжиженного 1,3-бутадиена (см. примечание 1), 0,5 г гидрохинона (см. примечание 2) и нагревают 13—15 часов при 200°. Затем реакционную смесь переносят в колбу Кляйзена с елочным дефлегматором высотой 3—4 см и перегоняют при пониженном давлении, собирая фракцию с т. кип. 76—80°УЗ мм.
Выход изо (4,7,8,9-тетрагидро) бензо-1,3-диметокситетрагид-рофурана равен 15 г (27%), = 1,4769, d420 = 1,066.
Найдено, %: С—65,09; Н—9,15.
СюНшОз. Вычислено, %: С—65,19; Н—8,75.
По литературным данным [1], т. кип. 64—65°/1,5 мм, п2} =i 1,4775.
Примечания;
1. Газообразный 1,3-бутадиен из баллона пропускают через хлоркальциевую трубку в ловушку, помещенную в смесь твердой углекислоты с бутиловым или изобутиловым спиртом.
2. Гидрохинон используют в качестве ингибитора побочных процессов— димеризации и полимеризации 1,3-бутадиена.
ЛИТЕРАТУРА
1. D. Z. Н u f f or d, D. S. T a r b e 11, T. R. Koszalka. J. Amer Chem. Soc., 74, 3014 (1952).
2. А. А. Пономарев, И. А. Маркушина, Г. E. Мариничева. Авт. свид. 253806; Изобретения. Промобразцы. Товарные знаки, № 31 (1969).
3. А. А. Пономарев. Синтезы и реакции фурановых веществ. Саратов, Изд-во СГУ, 1960. с. 176.
4. М. И. Дементьева. Анализ углеводородных газов. Л.—М., Гос-топтехиздат, 1959, с. 334.
л-ЙОДАНИЛИН
Г. А. КРЕЙМЕР, Е. А. БУГАЙ
C6H6IN'
М. в. 219,03
.w-Иоданилин получают восстановлением м-иодннтробензо-ла хлористым оловом [I] или гидразином в присутствии никеля Ренея [2]. В лабораторной практике нашел применение первый метод, требующий, однако, использования больших количеств дорогостоящего хлористого олова.
Нами разработана сравнительно простая методика восстановления м-иоднитробензола железным порошком в солянокислой среде.
СХЕМА СИНТЕЗА
NHj-HCI
no2
CH3COONa
Характеристика основного сырья
м-Иоднитробензол, МРТУ 6—09—5934—69, ч.
Железный порошок марки ПЖКН-2. Соляная кислота, ГОСТ 3118—67, ч. Натрий уксуснокислый, ГОСТ 199—68, ч. Хлористый метилен, ГОСТ 9968—62, техн.
139
Условия получения
В четырехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром, обратным холодильником и воронкой для загрузки сыпучих веществ, помещают 100 г (0,4 моль) лгиод-нитробензола, 360 мл воды и 280 мл соляной кислоты. Смесь нагревают до 70° н постепенно за 1,5 часа при интенсивном размешивании прибавляют 80 г (1,43 моль, 20%-ный избыток) железного порошка с такой скоростью, чтобы температура не превышала 78—82° (см. примечание). Далее нагревают смесь до 95° и выдерживают 30 минут при этой температуре, следя за тем, чтобы pH среды был равен 1; в случае необходимости добавляют немного соляной кислоты.
Реакционную массу охлаждают до 60° и нейтрализуют до pH 3,5, прибавляя горячий 40%-ный раствор уксуснокислого натрия, подкисленный уксусной кислотой до pH 6 — 7; всего расходуется около 280 г раствора. Затем массу охлаждают до 25° и выделившийся амин экстрагируют двумя порциями хлористого метилена по 50 мл. Объединенные экстракты сушат 5 г поташа, раствор фильтруют и осадок промываю/ 20 мл хлористого метилена.
Из раствора отгоняют хлористый метилен вначале при атмосферном, затем при пониженном давлении. Остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 132—13576—7 мм.
Выход л-иоданилина равен 61,5 г (70%), т. заст. 24°; содержание основного вещества (определено неводным титрованием хлорной кислотой) 99—100%.
По литературным данным [2], т. пл. 22—24°.
Примечание.
При более высокой температуре возможно образование значительных количеств анилина.
ЛИТЕРАТУРА
1. А. В a eye г. Вег., 38, 2761 (1905),
2. В. Е. Leggetter, R. К. Brown. Canad. J. Chem., 38, 2363 (1960); С. A., 55, 9312h (1961).
КАЛЬЦИЙАЦЕТОУКСУСНЫЙ ЭФИР
А. Е. КОЖУХОВА, М. В. ГРЕНАДЕРОВА, Т. К. ЧУМАЧЕНКО, Л. Г. ДЕРКАЧ
сн,-с=сн-с-ос,н5
I II
о о
ЧСа/2/
CiaHjgCaOe М, в. 298,36
Кальцийацетоуксусный эфир представляет интерес как исходное соединение для синтеза алкильных и ацильных производных ацетоуксусного эфира.
Известный способ получения кальцнйацетоуксусного эфира [1] взаимодействием ацетоуксусного эфира с цианистым кальцием мало удобен, так как продукт получается загрязненным исходным веществом и требует тщательной очистки.
Нами предложен простой и удобный метод получения кальцнйацетоуксусного эфира по аналогии с описанными методами получения енолятов щелочных металлов [2]. Метод применим для получения и других кальциевых производных (З-дикарбонильных соединений.
СХЕМА СИНТЕЗА
CH3COCH,COOCSH6 + (С2Н,О)»Са —
- СН,-С=СН-С-ОС2Н5
I II
о о
ХСа/2/
Характеристика основного сырья
Этиловый спирт ректификационный, ГОСТ 8314—57.
Кальций металлический (стружка).
Ацетоуксусный эфир, ГОСТ 9799—61, ч.
141
Условия получения
В круглодонную трехгорлую колбу емкостью 1л, снабженную мешалкой, обратным холодильником и капельной воронкой, помещают 40,0 г (1 г-атом) кальциевых стружек (см. примечание 1) и 500 мл абсолютного этилового спирта. Содержимое колбы нагревают на водяной бане до начала реакции (см. примечание 2), охлаждают до комнатной температуры и при перемешивании по каплям добавляют 269,0 г (2 моль) свежеперегнанного ацетоуксусного эфира. Выпавший при этом осадок отсасывают на воронке Бюхнера, промывают несколько раз абсолютным эфиром или спиртом и сушат в вакуум-эксикаторе.
Выход кальцийацетоуксусного эфира равен 271 г (91%); т. разл. выше 200°.
Найдено, %: С—47,90; Н—6,50; Са—13,35. CiaHjsCaOs- Вычислено, %: С—48,32; Н—6,04; Са—13,40.
Хелатная структура подтверждена изучением ИК-спектров. В ИК-спектре, снятом для чистого вещества в вазелиновом масле, имеется интенсивная полоса 1528 см 1 валентных колебаний С=С связи; полоса 1640 см-1 относится к валентным колебаниям карбонила. Обе полосы смещены в область меньших волновых чисел, что, очевидно, является 'следствием хе-лации и возникающего при этом сопряжения [3]. В спектре отсутствуют полосы свободной карбонильной группы (1740 и 1725 см~1).
Примечания:
1. Очищенные болванки металлического кальция измельчают на токарном станке, стружку промывают абсолютным эфиром.
2. Об окончании реакции судят по исчезновению металлического кальция и прекращению выделения пузырьков водорода.
ЛИТЕРАТУРА
1. Н. Franzen, W. Ryser. I. prakt. Chem., 88, 293 (1913).
2. Препаративная органическая химия, под ред. Н. С. Вульфсона. М.-Л., «Химия», 1964, с. 625.
3. S. W. I о г d а п о v, С h г. Ivan off, М. Arnaudo v, Р. Marko w. Chem., Ber, 99, 1518 (1966).
5-(2-КАРБОКСИЭТИЛ)-1,2-ДИГИДРОПИРРОЛИЗИН
| А. А. ПОНОМАРЕВ, ] Л. Н. АСТАХОВА, В. Н. ВОЛКОЛУПОВ
ноос- сн2-сн2
C10H13NO2
М. в. 179,21
В литературе 5-(2-карбоксиэтил)-1,2-дигидропирролизин не описан. Он синтезирован нами из 1,2-дигидро1Пирролизина и акриловой кислоты по методу 11], применявшемуся для получения аналогичных производных пиррольного ряда. В качестве катализатора использован фтористый бор.
СХЕМА СИНТЕЗА
VN\z
+ СН2=СН-СООН
НООС-СН2-СН2
Характеристика основного сырья
1,2-Дигидропирролизин, т. кип. 69—70°/15 мм, ti% =
= 1,5308, di10 = 1,0056; ^получение <см [2, 3].
Акриловая кислота, ТУ ГКХ РУ 1730—-62, ч.
Эфират фтористого бора, т. кип. 124—126°, cU15 — 1,135, получение см. [4].
143
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, капельной воронкой и обратным холодильником, помещают 3 г (0,028 моль) 1,2-дигидропирролизина, 2,6 г (0,036 моль) акриловой кислоты и при перемешивании добавляют по каплям за 10 минут 1,5 мл эфирата фтористого бора. Содержимое колбы сильно разогревается и при постепенном охлаждении затвердевает. Через 30 минут добавляют 50 мл 2н. раствора карбоната натрия. Водный раствор экстрагируют эфиром 3 раза порциями по 8 мл (эфирные вытяжки отбрасывают) и подкисляют 10%-ной соляной кислотой до кислой реакции по конго. Выделившийся маслянистый слой экстрагируют эфиром 5 раз порциями по 8 мл, эфирные вытяжки сушат сульфатом магния. Остаток, полученный после удаления эфира, быстро кристаллизуется.
Выход 5-(2-карбоксиэтил)-1,2-дигидропирролизина равен 4,2 г (84%) (см. примечание). Для очистки вещество перекристаллизовывают из гексана и получают 3 г (60%) почти бесцветных -кристаллов с т. пл. 94—96°; вещество хорошо растворимо в спирте, эфире, ацетоне, бензоле.
Найдено, %: С—67,11; 67,04; Н—7,39; 7,48; N—7,74; 7,82. Ci0Hi3NO2. Вычислено, %: С — 67,02; Н —7,31; N —7,83.
Примечание.
Аналогичным путем впервые синтезирован 3-метил-5-(2-карбоксиэтил)-1,2-дигидропирролизин из 3-метил-1,2-дигидропирролизииа (2, 3], выход 60%; т. пл. 89—91° (из гексаиа).
Найдено, %: С—68,86; 68,57; Н—8,24; 8,27; N—6,99; 7,05. CiiH15NO2. Вычислено, %; С—68,37; Н—7,82; N—7,24.
ЛИТЕРАТУРА
1. А. Т г е 1 b s, К. Н. Michl. Ann., 589, 163 (1954).
2. А. А. Пономарев, И. М. Скворцов. Ж. общ. химии, 97, 32 (1962).
3. А. А. Пономарев, И. М. Скворцов. Методы получения химических реактивов и препаратов, вып. 17. М., ИРЕА, 1968, с. 68.
4. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы. М., Госхимиздат, 1955, с. 97.
КОМПЛЕКС СЕРЕБРЯНОЙ СОЛИ ТРИНИТРОМЕТАНА С ДИОКСАНОМ
А. Л. ФРИДМАН, Т. Н. ИВШИНА
О O-AgC(NOs)3
C5H8AgN3O8
М. в. 346,00
Комплекс серебряной соли тринитрометана может быть использован для введения тринитрометильной группы в органические соединения и в других реакциях (1—6]. Комплекс устойчивее при хранении, чем серебряная соль тринитрометана, хорошо растворим во многих органических соединениях и облегчает точность дозировки серебряной соли тринитрометана. Соединение синтезировано нами впервые [7].
СХЕМА СИНТЕЗА
О О + AgC(NOs), -^2-- О O.AgC(NOs),
Характеристика основного сырья
Диоксан, ГОСТ 10455—63, ч.
Серебряная соль тринитрометана, т. пл. 110° (разл.); получение см. [5].
Условия получения
В стакан емкостью 100 мл, содержащий свежеприготовленный раствор 6 г (0,023 моль) серебряной соли тринитрометана в 15 мл воды, прибавляют по каплям 2,4 г (0,027 моль) диоксана при 20—25°, при этом образуется желтый кристаллический осадок. Реакционную смесь выдерживают 2 часа в бане
10 Зак. 1164
И5
с ледяной водой, осадок отжимают на вакуум-фильтре, промывают 3 мл холодной воды и сушат на воздухе в темном месте.
Выход комплекса серебряной соли тринитрометана с диоксаном равен 5 г (53%), т. пл. 86— 89° (разл.) (см. примечание). Комплекс представляет собой желтое кристаллическое вещество, растворимое в воде, этаноле, диметилформамиде, ацетоне.
Найдено, %: С—17,55; Н—2,31; N—12,21.
CsHsAgNsOg. Вычислено, %: С—17,34; Н—2,30; N—12,30.
Примечание.
При попытках перекристаллизации комплекс легко разлагается. При хранении т. пл. комплекса изменяется.
ЛЙТЕРАТУРА
1. С. А. Шевелев, В. И. Е р а ш к о, А. А. Ф а й н з и л ь б е р г. Изв. АН СССР, сер. хим., 9, 2113 (1968).
2. В. И. Ерашко, С. А. Шевелев, А. А. Ф а й н з и л ь б е р г. Изв АН СССР, сер. хим., 9, 2117 (1968).
3. С. А. Шевелев, В. И. Ерашко, Б. Г. Сайков, А. А. Ф айн-зил ь б е р г. Изв. АН СССР, сер. хим., 6, 1630 (1967).
4. С. А. Ш е в е л е в, В. И. Ерашко, Б. Г. Сайков, А. А. Ф а й н-зильберг. Изв. АН СССР, сер. хим., 2, 382 (1968).
5. В. И. С л овец кий, А. И. Иванов, С. А. Шевелев, А. А. Файизильберг, С. С. Новиков.. Ж- орган, химии, 8, 1445 (1966).
6. G. Hammond, W. Emmons, С. Parker, В. Graybill, М. Hawthorne. Tetrahedron, приложение 1, р. 177 (1963).
7. А. Л. Фридман, Т. Н. Ившина, В. П. И в ш и н, В. А. Т а р-таковский, С. С. Новиков. Изв. АН СССР, сер. хим., 10, 2336 (1969).
КОНИФЕРИЛОВЫЙ АЛЬДЕГИД
4-Окси-З-метоксикоричный альдегид
А. В. БОГАТСКИЙ, 3. Д. БОГАТСКАЯ, Л. М. НИКИФОРОВА, Л. В. БАСАЛАЕВА
сн=сн-сно
I хч х^чосн3 он
С10Н10Оз М. в. 178,19
Конифериловый альдегид является одним из важных органических веществ, входящих в состав природных соединений (лигнин). В литературе описаны различные методы синтеза альдегида [1—4]. Однако все они сложны, многостадийны, а выход продукта невелик.
Ранее нами был предложен [5J способ синтеза кониферило-вого альдегида восстановлением метилового эфира феруловой кислоты алюмогндридом лития в среде тетрагидрофурана. Продолжая работу в этом направлении, мы усовершенствовали методику синтеза кониферилового альдегида и применили абсолютный диэтиловый эфир в качестве растворителя вместо тетрагидрофурана, проводя реакцию при минут 80°.
СХЕМА СИНТЕЗА
СН=СН—СООСНз сн=сн—сно
Л L|A1H* :
Y4OCHS YXOCH,
он он
147
10*
Характеристика основного сырья
Метиловый эфир феруловой кислоты, т. кип. 183°/3 мм; т. пл. 59°, получение см. с. 168 этого сборника.
Алюмогидрид лития в порошке, РЭТУ 1067—63, ч. Диэтиловый эфир абсолютный, ГОСТ 6265—52, ч.
Условия получения
В четырехгорлой колбе емкостью 1 л, снабженной мешалкой, капельной воронкой, обратным холодильником с хлоркальциевой трубкой и термометром, помещают раствор 3 г алюмогидрида лития в 300 мл абсолютного диэтилового эфира. Раствор охлаждают до минус 80°. Для создания устойчивой низкой температуры; колбу устанавливают в глубокую баню, наполненную раздробленным сухим льдом и вместе с баней погружают в полиэтиленовый мешок, содержащий сухой лед. Сверху полиэтиленовый мешок плотно укутывают. Время от времени сухой лед смачивают ацетоном. Для проведения всего опыта требуется около 5 кг сухого льда. Заданную температуру (—80°) поддерживают до конца восстановления.
К охлажденному до минус 80° раствору алюмогидрида лития добавляют по каплям заМ часа, при перемешивании, охлажденный до 0° раствор 8 г (0,036 моль) метилового эфира феруловой кислоты в 200 мл абсолютного диэтилового эфира. По окончании прибавления реакционную смесь перемешивают еще 20 минут. После этого осторожно, по каплям, прибавляют сначала 100 мл дистиллированной воды, охлажденнной до 0° затем 50 мл уксусной кислоты, растворенной в 100 мл дистиллированной воды, при этом на дне колбы образуется осадок бледно-желтого цвета. Содержимое колбы переносят в перегонную колбу и отгоняют растворитель на закрытой водяной бане при 34°, причем, раствор из бледно-желтого превращается в мутный с белым коллоидным осадком на дне.
К содержимому колбы осторожно добавляют мелкие кусочки сухого льда до тех пор, пока он ие перестанет растворяться (реакция идет бурно!). Реакционный раствор желтого, иногда желто-зеленого цвета экстрагируют бензолом (5—6 раз). Бензольные вытяжки промывают несколько раз (2—3 раза) раствором бикарбоната натрия, затем водой и сушат порошкообразным прокаленным хлористым кальцием 10— 12 часов. Отфильтрованные бензольные вытяжки переносят в колбу для перегонки в вакууме, бензол отгоняют при пониженном давлении при 32—34°/100 мм. Остаток переносят в кристаллизатор и помещают в вакуум-эксикатор, наполненный парафиновой стружкой. После удаления бензола в кристаллизаторе остается вязкая масса желтого цвета, из которой на 148
следующий день выпадают крупные кристаллы Желтоватого цвета. Кристаллы отсасывают на воронке Бюхнера (или фильтре Шотта) и перекристаллизовывают из сухого бензола. Избыток бензола удаляют в вакуум-эксикаторе, наполненном парафиновой стружкой, в течение суток.
Выход кониферилового альдегида равен 5,9 г (87%). Вещество получают в виде крупных кристаллов бледно-желтого цвета с т. пл. 81—82°.
По литературным данным [5], т. пл. 82,5°.
Найдено, %: С—67,27; Н—5,52.
СюНюОз Вычислено, %: С—67,41; Н—5,61.
Конифериловый альдегид растворяется в воде, спирте, глицерине, в эфире, бензоле, сероуглероде, хлороформе. Вещество обесцвечивает бромную воду, что указывает на наличие двойной связи.
Характерен ИК-спектр кониферилового альдегида, снятый на приборе ИКС-14-А при толщине слоя 0,049 (скорость лепты 3, скорость сканирования 5, усиление 1). В качестве растворителя использован хлороформ. Спектр снимали на призмах NaCl и LiF. Спектр указывает на наличие в веществе альдегидной группы (v=1685 ел-1), кратной связи (v=1623 ел-1), ароматического кольца (v — 1589, 1500, 1421 см *), метоксильной группы (v = 2830 с.и1), а также (фенольной) гидроксильной группы (v = 3570 с.и-1). УФ-спектр кониферилового альдегида (снят на приборе СФ-4-А, при толщине слоя 0,058, 0,58 М раствор альдегида в этиловом спирте) указывает на максимум поглощения при Хиакс = 326 нм с интенсивностью Ige = 2,93.
ЛИТЕРАТУРА
1. Н. Pauly, К. Was chen. Ber., 56, 603 (1923).
2. Н. Pauly, К. Fenerstein. Ber., 62,297 (1929).
3. P. К lass on. Ben, 63, 912, (1930).
4. K. Freudenberg, R. Dillenberg. Ben, 84, 67 (1951).
5. А. В. Богатский, 3. Д. Богатская, T. E. Пипикова. Укр. хим. ж., 12, 12 (1966).
КОНИФЕРИЛОВЫЙ СПИРТ
3-(4-Окси-3-метоксифеиил)-2-пропеиол
А. В. БОГАТСКИЙ, 3. Д. БОГАТСКАЯ, Л. В. БАСАЛАЕВА И. С. УЛАНОВА-ОБЕЗЮК
СН = СН—СН2ОН I
^J-OCH»
он
C10H12O3
М. в. 180,01
Конифернловый спирт и его производные являются составной частью лнгннна [1—4].
В литературе описан способ получения кониферилового спирта восстановлением метилового эфира феруловой кислоты алюмогидридом лития [1].
Нами проверена методика (1], уточнена и несколько изменена. Предложено в качестве объекта восстановления брать феруловую кислоту.
СХЕМА СИНТЕЗА
СН=СН—COOR СН=СН-СН»ОН
LiAlH4_
\^-ОСН, '^'-ОСН,
он он
где R=H— или СН»— 150
Характеристика основного сырья
Метиловый эфир феруловой кислоты, т. пл. 59—60°, т. кип. 183°/3 мм, получение см. с. 168 этого сборника.
Феруловая кислота, т. пл. 173°, получение см. с. 293 этого сборника.
Алюмогндрнд лнтня в порошке фабричный, РЭТУ 1067— 63, ч.
Диэтиловый эфир (для наркоза) абсолютный, ГОСТ 6265—52, ч.
Условия получения
Синтез кониферилового спирта из метилового эфира феруловой кислоты. В четырехгорлую колбу емкостью 2 л, снабженную обратным холодильником, мешалкой, трубкой для ввода азота (над жидкостью), капельной воронкой н термометром, помещают 3 г алюмогидрида лития в 250 мл абсолютного диэтилового эфира. Колбу помещают в сухую баню и реакционную смесь нагревают 1 час при слабом кипении эфира и непрерывном перемешивании. Смесь охлаждают и колбу заполняют азотом. Содержимое колбы охлаждают смесью льда и соли до минус 19°. После этого по каплям прибавляют охлажденный до 0° раствор 7,5 г (0,036 моль) метилового эфира феруловой кислоты в 250 мл абсолютного днэтнлового эфира, следя за тем, чтобы температура в колбе не превышала минус 15°. При этом образуется серый осадок.
Реакционную смесь перемешивают еще 10 часов в атмосфере азота при температуре не выше минус 5° и оставляют на ночь. При повышении температуры до комнатной осадок окрашивается в белый цвет. На следующий день разлагают избыток алюмогидрида лития, прибавляя по каплям 100 мл влажного диэтилового эфира, а затем 250 мл дистиллированной воды, охлажденной до 0°, следя за тем чтобы температура реакционной смеси не превышала +5°. При этом осадок окра шнвается в желтый цвет. Водный слой отделяют с помощью делительной воронки, экстрагируют 250 мл диэтилового эфира и переносят снова в колбу. В колбу добавляют 300 г твердой углекислоты (сухой лед) для выделения кониферилового спирта. О полноте выделения судят по изменению цвета из желтого в серый.
Водный слой экстрагируют 10 раз порциями по 100 мл диэтилового эфира, энергично взбалтывая в делительной воронке (см. примечание 1). Объединенные эфирные вытяжки сушат прокаленным сульфатом натрия (порошок), затем Отгоняют на водяной бане днэтнловый эфир до объема 400 мл. Остаток дополнительно сушат сульфатам кальция7 (обезвоженного при 240°) и упаривают при пониженном давлении до
151
объема 150 мл. К остатку прибавляют 50 мл абсолютного Дй-этилового эфира и снова упаривают до объема 150 мл. После пяти-шестйкратного повторения Этой оперции раствор становится достаточно сухим и окончательно упаривается до появления маслообразного остатка. Маслообразный остаток растворяют при слабом нагревании в сухом метиленхлориде (см. примечание 2) и добавляют 10 мл петролейного эфира (фракция 30—45°). При этом раствор становится молочного цвета. После начала кристаллизации продукт помещают в хорошо закрытой колбочке на 12 часов в холодильник. Выпавшие кристаллы отсасывают, промывают смесью (1:1) метиленхлори-да — петролейного эфира и хорошо сушат в вакуум-эксикато-ре над пятиокисыо фосфора.
Выход кониферилового спирта равен 4,5 г (67%). Вещество получают в виде белых кристаллов с т. пл. 71—72°.
По литературным данным [2, 5], т. пл. 73°.
Коиифериловый спирт растворяется в диэтиловом эфире, спирте, хлороформе, четыреххлористом углероде, горячей воде.
Найдено, %: С-66,51; Н—6,37.
С10Н12О3. Вычислено, %: С—66,7; Н—6,67.
Получение кониферилового спирта из феруловой кислоты проводят в приборе, описанном выше. В колбу помещают 3,8 г (0,1 мюль ) алюмогидрнда лития в 150 мл абсолютного диэтилового эфира, аналогично описанному выше подготавливают восстановитель, колбу заполняют азотом, охлаждают смесью льда и соли до минус 10° и приливают по каплям за 3,5 часа раствор 10 г (0,06 моль) феруловой кйслоты в 1600 мл абсолютного диэтилового эфира (см. примечание 3). Реакционный раствор перемешивают 30 часов при комнатной температуре. Дальнейшее .проведение реакции и выделение конечного продукта проводят так же, как и в первом варианте.
Выход кониферилового спирта равен 3,1 г (36%). Вещество получают в виде кристаллов желтого цвета с т. пл. 72°.
Примечания:
1. Отделение эфира от суспензии А1(ОН)3 можно ускорить прибавлением после встряхивания 10—15 мл свежего диэтилового эфира.
2. При отсутствии метиленхлорида можно использовать хлороформ, предварительно хорошо очищенный.
3. необходимо брать большое количество абсолютного диэтилового эфира, так как феруловая кислота плохо растворима в диэтиловом эфире.
ЛИТЕРАТУРА
1. К. Freudenberg, Hubner. Вег., 85, 1181 (1952),
2. F. Tiernan, Haar man. Вег., 7, 611 (1874).
3. F. Tie man, B«r., 8; 1127 (1875).
4. A. Wacek, H. Kessering. Monatch., 2, 93 (1962).
152
МЕТАКРИЛОИЛХЛОРИД
Н. Н. ДЫХАНОВ, А. Б. ДЖИДЖЕЛАВА
CH2=C—СОС1
I сн,
С4Н5СЮ М. в. 104,54
Хлорангидрид метакриловой кислоты (метакрилоилхло-рид) применяется как непредельный ацилирующий агент, например, в синтезе эфиров целлюлозы [1]. Его получают либо нагреванием сухого метакрилата натрия с хлорокисью фосфора [2], либо действием треххлористого фосфора на метакриловую кислоту в присутствии однохлористой меди [1]. Выход метакрилоилхлорида по обоим способам составляет 64—68%.
Нами установлено, что примерно с таким же выходом метакрил оилхлор ид может быть получен подобно хлорангидри-дам других карбоновых кислот [3] нагреванием метакриловой кислоты с бензоилхлоридом при непрерывном удалении метакрилоилхлорида из сферы реакции.
СХЕМА СИНТЕЗА
СН2=С—СО -1- С6Н|СОС1 — CH2=C—СОС1 + с6несоон
I I I
СН, он сн,
Характеристика основного сырья
Метакриловая кислота, МРТУ 6—09—1768—64, ч.
Бензоил хлористый, МРТУ 6—09—6510—70, ч.
Условия получения
В колбу емкостью 0,5 л, снабженную елочным дефлегматорам высотой 35 см, отводная трубка которого соединена с
153
нисходящим холодильником, помещают 86,1 г (1,0 моль) метакриловой кислоты и 281,5 г (2,0 моль) хлористого бензоила. Смесь нагревают на масляной бане до слабого кипения и отгоняют образующийся метакрилоилхлорид до достижения в парах температуры 100°.
Сырой метакрилоилхлорид перегоняют из колбы с тем же дефлегматором, собирая фракцию с т. кип. 95—97°.
По литературным данным 11], т. кип. 95—97°.
Выход метакрилоилхлорида равен 70—73 г (68—70%); продукт по своим качествам отвечает требованиям МРТУ 6—09—6272—69.
ЛИТЕРАТУРА
1. А. А. Берлин, Т. А. Макарова. Ж- общ. химии, 21, 1267 (1951).
2. W. N. Haworth, Н. G г е g or i, L. F. Wiggins. J. Chem. Soc., 1948, 488.
3. P. К у г 1 d e s. J. Amer. Chem. Soc., 59, 208 (1937).
МЕТИЛБЕНЗИЛКЕТОН
Г. Ф. ТАНЦЮРА, Л. Я. ГЛИНСКАЯ, И. Ф. ГЕРАСИМЕНКО
свн,-сн2-со-снш
С9НюО , М. в. 134,17
Метнлбензилкетон получают различными методами, однако одни из них [1] дают незначительные выходы, другие отличаются трудоемкостью процесса и большим расходом исходного сырья [2, 3]. Нами предложен каталитический способ получения метилбензилкетона нз фенилуксусной и уксусной кислот в присутствии дешевого и доступного катализатора окиси алюминия.
СХЕМА СИНТЕЗА
С«Н5СН2СООН + СНзСООН свн6сн2сосн3
Характеристика основного сырья
Фенилуксусная кислота, ТУ МХП 2755—51, ч.
Уксусная кислота, ГОСТ 61—51, х. ч.> 98%-ная.
Окись алюминия черенковая, А—56, ВТУ 4618—56, измельчена и просеяна.
Условия получения
Реакцию проводят в кварцевой трубке длиной 85 см, 0 18 мм, соединенной через тройник с делительной воронкой и источником углекислого газа и помещенной в трубчатую печь с температурой 400—450°. Среднюю часть реактора (по длине 50 см) заполняют окисью алюминия. Реакцию проводят в атмосфере углекислого газа; скорость подачи—1 пузырек в секунду. В реактор приливают со скоростью 50—60 мл!час
155
раствор 136 г (1 моль) фенилуксусной кислоты в 120 мл ледяной уксусной кислоты, затем добавляют 10 мл ледяной уксусной кислоты (для вытеснения нз трубки продукта реакции).
Катализат собирают в охлаждаемый приемник с обратным холодильником. По окончании реакции катализат выливают в смесь 300 г льда и 100 г воды, затем нетрализуют по лакмусу 50%-ным раствором едкого натра. Водный слой отделяют и экстрагируют 2—3 раза бензолом порциями по 60 мл. Бензольные вытяжки соединяют с маслянистым слоем. Растворитель отгоняют, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 113—115°/20 мм.
Выход метилбензилкетоиа равен 80—90 г (60—67%) d420-= = 1,0239, п™= 1,5161.
По литературным данным [2], т. кип. 210—215°, d^— 1,003, п% = 1,5168.
ЛИТЕРАТУРА
1. Р. Mason, J. Terry. J. Amer. Chem. Soc., 62, 1622 (1940).
2. О. Ю. Мегидсон, Г. А. Г аркуш а. Ж. общ. химии, 11, 339 (1941).
3. Синтезы органических препаратов, сб. 2. М., ИЛ, 1950, с. 313.
2 МЕТИЛБУТАНАЛЬ
Е. П. КРЫСИН, И. И. ГРОМОВА, Э. Д. ГЛИНДА, А. Г. ЛЕВИНА, Л. Г. АНДРОНОВА
CsHi0O
CjHj-CH-CHO
I
сн3
М. в. 86,05
Для получения 2-метилбутаналя применяют один из методов окисления соответствующего бутилкарбинола [1], однако эти методы технологически неудобны и требуют труднодоступного сырья.
В данном синтезе был использован метод восстановления этилакроленна [2], полученного конденсацией формальдегида и н-масляного альдегида. Гидрирование этнлакролеина проводили в мягких условиях на катализаторе Pd-чернь.
СХЕМА СИНТЕЗА
СН.Ч
С3Н,СНО + )NH HC1 + Н»СО -CH8Z
C2Hb. ,N(CHs)3-HCr ;С(
CH3Z хсно
-+ С2Н3-СН-СНО I сн8
С2Н5-С-СНО II сн2
H2/Pd
Характеристика основного сырья
Диметнламин хлоргидрат, МРТУ 6—09—821—63, ч. н-Масляный альдегид, ТУ МХП 1523—51, ч.
Формалин, ГОСТ 1625—61, ч.
157
Условия получения
Синтез этилакролеина. В колбу емкостью 5 л, снабженную обратным холодильником, мешалкой и термометром, загружают 405 г (5 моль) хлоргидрата диметиламина, 400 мл (4,Ьмоль) н-масляного альдегида и 345 мл (5 моль) 40%-ного раствора формалина. Смесь перемешивают 6 часов при 50—60°, затем заменяют обратный холодильник иа прямой и отгоняют с водяным паром образовавшийся этилакролеин. Органический слой отделяют, сушат над сульфатом натрия и перегоняют на колонке с насадкой (кольца Рашига, 20—25 т. т.), отбирал фракцию с т. кип. 90—92°.
Выход этилакролеина равен 262 г (67% в расчете на н-масляный альдегид), п2$ — 1,4205.
По литературным данным [2], т. кип. 91—92°; = 1,4205.
Получение 2-метилбутаналя. В трехгорлую колбу емкостью 1 л, снабженную газоподводными трубками и мешалкой с ртутным затвором, вносят 150 г этилакролеина и быстро загружают катализатор (см. примечание). При комнатной температуре и интенсивном перемешивании пропускают водород. Скорость поглощения наблюдают по разнице прохождения водорода через контрольные склянки до и после реакционной колбы. Через 10—15 часов реакционную массу декантируют и перегоняют на той же колонке, собирая фракцию е т. кип. 90—92°.
Выход 2-метилбутаналя равен 107 г (72%), = 1,3940.
По литературным данным 13], т. кип. 91—92°; — 1,3896.
Примечание'.
Для приготовления катализатора 7 г хлористого палладия растворяют в 100 мл воды, слегка подкисленной соляной кислотой. Полученный раствор смешивают с 50 мл 33%-ного раствора формалина и охлаждают до КГ. При перемешивании за 10 минут добавляют 100 г 50%-иого раствора едкого кали и смесь нагревают 30 минут при 50°. После декантации воды осадок промывают дистиллированной водой до полного исчезновения ионов хлора (реакция с нитратом серебра).
ЛИТЕРАТУРА
1. L. Lardicci, R. Rossi. Atti Soc. Tosc. Sci natur, 1961, A68, 23-31.
2. C. S. Marvel, R; L. Myers, J. H. S a u n d e rs. J. Amer. Chem. Soc., 70, 1697 (1948).
3. Франц, пат. 1256630 (1961)
1-(3-МЕТИЛ-1,2-ДИГИДРОПИРРОЛИЗИЛ-5)-1--БУТЕН-З-ОН
| А. А. ПОНОМАРЕВ, | Л. Н. АСТАХОВА
wNxz
I I сн3—со—сн=сн сн,
C12H15NO м. в. 189,25
1-(3-Метил-1,2-днгидропирролизил-5)-1-бутен-3-он в литературе не описан. Он получен нами при взаимодействии 3-метил-5-формил-1,2-дигидропирролизина с ацетоном в водно-спиртовой среде в присутствии каталитических количеств щелочи.
СХЕМА СИНТЕЗА
I + CH3-CO-CH, — 4ZN\Z
I !
сно сн3
СН,-СО-СН=СН СНз
Характеристика основного сырья
3-Метил-5-формил-1,2-дигидропирролизин, т. кип. 96—97°/4 мм, п%= 1,5712, d^n= 1,0845; получение см. [1—3].
159
Ацетон, ГОСТ 2603—63, ч.
Этиловый спирт, ГОСТ 9674—61, ч.
Условия получения
В круглодонную колбу емкостью 50 мл, снабженную обратным холодильником, помещают 4,5 г (0,03 моль) З-метил-5-формил-1,2-дигидропирролизина, 5,2 г (0,09 моль) ацетона, 25 Цл этанола и добавляют 3 мл 10%-ного раствора едкого натра. Полученную смесь нагревают 10 минут на кипящей водяной бане и оставляют при комнатной температуре на 40— 48 часов (см. примечание). Затем раствор выливают в 50 мл воды, выделившееся масло экстрагируют эфиром 3 раза порциями по 10 мл. Объединенные эфирные вытяжки сушат сульфатом магния. Эфир удаляют, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 139—142°/2 мм.
Выход 1-(3-метил-1,2-дигидропирролизил-5)-1-бутен-3-она равен 3,4 г (59,6%). По внешнему виду вещество представляет собой очень вязкую жидкость желтого цвета.
Найдено, %: С—76,13; 75,82; Н—8,08; 7,99; N—7,51; 7,35. C12Hj5NO. Вычислено, %: С—76,15; Н—7,98; N—7,40.
Семикарбазон 1 - (3-метил-1,2-дигидропирролизил-5) -1 -бутен-1-оиа получен с выходом 89% в виде кристаллов желтоватого цвета с т. пл. 193—196° (разл. из этанола).
Найдено, %: N—22,56; 22,40.
CisHisN4O. Вычислено, %: N—22,75.
Примечание.
Сокращение времени реакции до 20 часов приводит к уменьшению выхода продукта на 10%. Без предварительного нагревания реакционной смеси выход продукта понижается до 35%.
ЛИТЕРАТУРА
1. Л. Н. Астахова, И. М. Скворцов, А. А. Пономарев. Ж. общ. химии, 34, 2410 (1964).
2. А. А. Пономарев, И. М. Скворцов, Л. Н. Астахова. Докл. АН СССР, 155, 861 (1964).
3. А. А. Пономарев, Л. Н. Астахова. Методы получения химических реактивов и препаратов, вып. 17. М., ИРЕА, 1968, с. 94.
МЕТИЛИЗОПРОПИЛКЕТОН
А. В. БОГАТСКИЙ, Г. Л. КАМАЛОВ
/СН3 СН3-С-СН<
II ХСН3 о
С5Н10О
М. в. 87,06
Распространенными способами получения метилалкилкето-нов являются способы кетониопо расщепления соответствующих алкилзамещениых ацетоуксусных эфиров разбавленными растворами щелочей [1] и окисления вторичных либо третичных спиртов [2]. Простым и удобным способом получения метил алкилкетонов представляется реакция парофазного гидролиза замещенных ацетоуксусных эфиров на окиси алюминия [3].
Нами предложен способ каталитического синтеза метил-изопропилкетона парофазным гидролизом диметил ацетоуксусного эфира на окиси алюминия ,при 320°.
СХЕМА СИНТЕЗА
С ЧС/С00С^ А1»°з
CHaZ хсосн3 н,°
сн3.
)СН-С-СН3
СН,/ II
о
Характеристика основного сырья
Диметилацетоуксусный эфир, т. кип. 74—75°/Ю мм; = = 0,9709; rip = 1,4179 (см. примечание 1).
Окись алюминия черенковая, не содержащая фтора, А-56, ВТУ 4618—65 (см. примечание 2).
11 Зак. 1164
161
Условия получения
Реактор для проведения парофазного гидролиза диметил-ацетоуксусного эфира представляет собой заполненную окисью алюминия кварцевую трубку 0 25 мм, помещенную в вертикальную трубчатую электропечь (см. примечание 3). Длина слоя катализатора 7 см, вес 12—15 г, насыпной объем 20—23 мл. Перед каждым опытом катализатор активируют током сухого воздуха при 600° в течение 2—3 часов, после чего воздух вытесняют азотом, в атмосфере которого проводит-1 ся реакция.
Диметилацетоуксусный эфир и воду в объемном соотношении 1:1 подают в реактор ,шприцевым дозатором со скоростью 10—11 мл!час при температуре 320° (см. примечание 4). Катализат собирают ? охлаждаемом льдом приемнике, снабженном обратным холодильником с газоотводной трубкой. Полученный катализат выливают в делительную воронку, насыщают водный слой хлористым натрием, после чего органический слой отделяют, сушат безводным сульфатом натрия и перегоняют.
Выход метилизопропилкетона 35% в расчете на вступивший в реакцию диметилацетоуксусный эфир (см. примечание 5), т. кип. 93—96°; d420 = 0,8056; п?" = 1,3889; т. пл. 2,4-ди-нитрофенилгидразона 116°, что соответствует литературным данным [4].
Примечания:
1. Диметилацетоуксусиый эфир получен действием йодистого метила на динатрийацетоуксусный эфир в среде абсолютного этанола по методике
2. Продажную окись алюминия измельчают и просеивают (фракция 2,5—3 лик).
* 3. Катализатор помещают в ту часть реактора, в которой ие имеется градиента теплового поля (предварительно найденное плато печи).
4. Температуру фиксируют предварительно откалиброванной платина-платииородиевой термопарой, э. д. с. которой измеряют потенциометром постоянного тока типа ПП-63. Точность регулирования температуры ±5°.
5. Промывка фракций, выкипающих до 90й, насыщенным раствором хлористого кальция с последующей сушкой их безводным сульфатом натрия и перегонкой, позволяет получать метнлнзопропилкетои с выходом до 50—60% в расчете на исходный днметилацетоуксусиый эфир.
ЛИТЕРАТУРА
1. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949, с. 274.
2. Синтезы органических препаратов, сб. 2. М., ИЛ, 1949, с. 324.
3. Г. Л. Камалов. Синтез й каталитические превращения некоторых эфиров и ацеталей. Автореферат канд. диссертации, Одесса, 1969.
4. Словарь органических соединений,. т. 2. М., ИЛ., 1949, с. 756
5. Н. Meyer, Monatsh., 27, 1087 (1906).
МЕТИЛОВЫЙ ЭФИР а-МЕТИЛ-0-МЕРКАПТОПРОПИОНОВОЙ КИСЛОТЫ
А. В. БОГАТСКИЯ, А. М. ТУРЯНСКАЯ, А. И. ГРЕНЬ
HS -СНз-СН-СООСН., I сн8
C5H10O2S М. .в. 134,19
Эфиры а-алкил-р-меркаптокислот мало доступные вещества, а между тем они являются более удобными исходными веществами в органическом синтезе, чем их кислоты, которые получают по методике [1]. Известный способ получения метилового эфира а-метил-р-меркаптопропионовой кислоты, основанный на взаимодействии метилового эфира а-метил-р-бромпропионовой кислоты с гидросульфидом калия {2], сопровождается побочными реакциями и выход продукта незначителен.
Предлагаемый способ синтеза метилового эфира а-метил-р-меркаптопропионовой кислоты отличается тем, что вместо гидросульфида калия применяют тиомочевину. Это позволяет получить продукт с хорошим выходом (60%) и практически полностью исключить побочные реакции.
СХЕМА СИНТЕЗА
ВгСН2-СН-СООСН8 (N”2h^S - HS-CHs-CH-COOCH8 сн3 сн8
Характеристика основного сырья
а-Метил-р-бромпропионовая кислота, метиловый эфир, т. кип. 73°/18 мм, = 1,4531, </420 = 1,4 1 81; получение
см. [3].
11* 163
Тиомочевина, ГОСТ 6344—52, х. ч.
Натр едкий, ГОСТ 4328—66, ч. д. а.
Этиловый спирт гидролизный, ТУ 19П—39—69.
Условия получения
В круглодонную колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником, капельной воронкой и трубкой для ввода газа, помещают 38 г (0,5 лголь) тиомочевины и 25 лй 95%-ного этилового спирта. Смесь нагревают 1 час, а затем к смеси постепенно при перемешивании прибавляют 90,5 г (0,5 моль) метилового эфира а-метил-р-бром-пропионовой кислоты. Полученный раствор кипятят с o6pai-ным холодильником в атмосфере азота 6 часов. К охлажденной смеси добавляют 20 г (0,5 моль) едкого натра в 60 мл этанола. Реакционную смесь перемешивают 1 час, а затем оставляют при комнатной температуре на ночь. Раствор подкисляют пропусканием углекислого газа до pH 5—6, спирт отгоняют, а остаток фракционируют, собирая фракцию, кипящую в пределах 75—79°/5 мм.
Выход метилового эфира а-метил-р-меркаптопропионовой кислоты равен 40 г (60%), п® = 1,4555, еЛ20 = 1,0470.
Найдено, %: S—23,00. C5H10O2S. Вычислено, %: S—23,88.
ЛИТЕРАТУРА
1. Общий практикум по органической химии', под ред. А. Н, Коста, М, 1965, с. 191.
2. В. Е. П е т р у н ь к и н, Н. М. Лысенко. Ж. общ. химии, 29, 309 (1959).
3. Синтезы органических препаратов, сб. 3, М., ИЛ, 1952, с. 308.
МЕТИЛОВЫЙ ЭФИР 0-(5-МЕТИЛ-2-ПИРРОЛИДИЛ)-пропионовой кислоты
| А. А. ПОНОМАРЕВ,\ М. В. НОРИЦИНА, А. П. КРИВЕНЬКО
СН3Х 4 NZ ЧСН2СН2СООСН3
н
C9H17NO2 М. в. 171,23
Метиловые эфиры пирролидилпропионовых кислот представляют интерес как потенциально физиологически активные вещества [1, 2].
Метиловый эфир р-(5-метил-2-, пирролидил) пропионовой кислоты получен нами впервые этерификацией р-(5-метил-2-лирролидил) пропионовой кислоты. Предлагаемым методом можно |Получать метиловые эфиры замещенных р-(2-пирроли-дил)пропионовых кислот, содержащих .в пирролидиновом цикле в положении 1 или 5 алкильный заместитель. В противном случае вместо ожидаемых эфиров получаются 3-пирролизидо-ны.
СХЕМА СИНТЕЗА
I---1 СН3ОН
I I НС1
ch/xn/xch2-ch2cooh
I н
СН,7 4 Nz 4 СН,СН,СООСНз н
165
Характеристика основного сырья
р-(5-Метнл-2-пнрролидил)лропиоиовая кислота, т. пл. 160°, получение см. с. 247 данного сборника.
Метиловый абсолютный спирт, ГОСТ 6995—54, х. ч., приготовление см. [3].
Этилацетат, СТ ГОХП 27—1839, ч.
Условия получения
Колбу емкостью 150 мл, снабженную обратным холодильником и содержащую 5 г (0,032 моль) р-(5-метал-2-пирроли-днл) пропионовой кислоты и 100 мл 3 н. раствора хлористого водорода в абсолютном метиловом спирте, нагревают 3,5 часа на водяной бане. Затем метиловый спирт отгоняют прн пониженном давлении, остаток растворяют в 5 мл воды, добавляют 50 мл этилацетата и быстро вносят 5 г (0,03 моль) безводного карбоната калия, причем метиловый эфир р-(5-метил-2-пирролидил)пропионовой кислоты переходит в 'слой этнлаце-тата. Этот слой отделяют, а остаток экстрагируют растворителем дважды порциями по 5 мл. Экстракты соединяют и сушат прокаленным сульфатом магния. Растворитель отгоняют, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 92—92,5°/8 мм.
Выход метилового эфира р-(5-метнл-2-пирролидил)пропн-оновой кислоты равен 2,8 г (52%), п™ = 1,4570, d420 — = 0,9889 (см. примечание).
Найдено, %: С—62,94; 62,97; Н—9,73; 9,95; N—7,60; 7,75; MRd =47,16.
C9H17NO2. Вычислено, %: С—63,10; Н—10,02; N—8,19; M/?D = 46,81.
Примечание.
Аналогичным путем получены впервые:
Метиловый эфир |3-(5-изобутил-2-пирролидил) пропионовой кислоты из 0-(5-изобутил-2-пирролидил)пропионовой кислоты, выход 60%, т. кип. 10273 мм, 1,4598, d?’ = 0,9642.
Найдено, %: С—68,07; 68,01; Н—10,83; 10,86; N—6,04; 6,52;
MRD = 60,57.
CijH2sNO2. Вычислено, %: С—67,57; Н—10,86; N—6,56; MR D = 60,67.
Метиловый эфир 0-(1-метил-2-пирролидил) пропионовой кислоты из р-(1-метил-2-пнрролидил) пропионовой кислоты, выход 36%, т. кип. 90— 91710 мм, = 1,4510, d420 = о,9819.
Найдено, %: С—62,70; 62,83; Н—10,03; 10,10; N—7,94; 7,90; MRd = 46,84.
C9Ht7NO3. Вычислено, %: С—63,10; Н—10,02; N—8,19; MRD = 47,16.
166
Метиловый эфир р-[циклопентано(в)-2-пирролидил]пропионовой кислоты из |3-[циклопентано(в)-2-пирролидил]пропионовой кислоты, выход 49%, т. кип. 101—10272 мм, г?£= 1,4800, <№= 1,0390.
Найдено, %: С—66,95; 66,53; Н—10,00; 9,71; N—7,01; 6,88;
MRD = 53,63.
C11H19NO2. Вычислено, %: С—66,97; Н—9,71; N—7,10; MRD = 53,85.
Метиловый эфир 0-(2-октагидроиндолил) пропионовой кислоты из Р(2-октагидроиидолил)пропионовой кислоты, выход 30%, т. кип. 11572 мм. п™ = 1,4852, d420 = 1,0370.
Найдено, %: С—68,49; 68,12; Н—10,14; 10,00; N—6,51; 6,45;
MRd = 58,48.
C12H2iNO2. Вычислено, %: С—68,21; Н—10,01; N—6,63; MRD = 58,37.
ЛИТЕРАТУРА
1. В. Matcovics, S. Fol de a k, S. Pozszasz. Acta Univ. Szeged iensis, 605 (1961).
2. Англ. пат. 702269 (1954).
3. ГО. К. Юрьев. Практические работы по органической химии, вып. I и II. М., Изд-во МГУ, 1961, с. 57.
МЕТИЛОВЫЙ ЭФИР ФЕРУЛОВОЙ кислоты
А. В. БОГАТСКИЙ, 3. Д. БОГАТСКАЯ, Л. М. НИКИФОРОВА, Л. В. БАСАЛАЕВА, Н. С. УЛАНОВА-ОБЕЗЮК
CH=CH-COOCHS
'Q-OCH,
он
СнН^С^
М. в. 208,22
Метиловый эфир феруловой кислоты был получен нами впервые двумя способами: этерификацией феруловой кислоты в присутствии концентрированной серной кислоты с выходом 25—38% или в присутствии катализатора — ионообменной смолы КУ-2 в Н-форме с выходом 54—60%.
СХЕМА СИНТЕЗА
СН=СН-СООН
А ^J-ОСНз
I он
сн=сн-соосн8
/ч
8ч/!-ОСН3
I он
Характеристика основного сырья
Феруловая кислота, т. пл. 173°, получение см. с. 293 этого сборника.
Метиловый спирт, ГОСТ 6995—54, ч., абсолютирован по методике [1].
168
Серная кислота, ГОСТ 4204—58, техн., пл. 1,84.
Ионообменная смола КУ-2, фабричная, ГОСТ 13505—68, обработана по методике [2] и переведена в Н-форму.
Условия получения
Вариант I. В круглодонной трехгорлой колбе емкостью 150 мл, снабженной мешалкой, капельной воронкой и обратным холодильником с хлоркальциевой трубкой, к 40 мл абсолютного метилового спирта прибавляют 3 мл концентрированной серной кислоты. Содержимое колбы взбалтывают и охлаждают,; после чего прибавляют 10 г (0,05 моль) феруловой кислоты и смесь нагревают 4 часа на масляной бане при слабом кипении реакционной смеси (температура бани 100— 110°). Затем отгоняют избыток спирта, а остаток после охлаждения выливают в 125 мл дистиллированной воды. В мутной молочно-белой смеси наблюдается расслоение, в результате чего выделяется маслянистый слой кофейного цвета, который отделяют. Водный слой экстрагируют диэтиловым эфиром 4 раз(а порциями по 30 мл. Эфирные вытяжки соединяют с маслянистым слоем, промывают от спирта дистиллированной водой, затем нейтрализуют 2,5%-ным раствором карбоната натрия и снова промывают водой. Эфирный слой сушат порошкообразным хлористым кальцием в течение 12 часов. Растворитель отгоняют, а остаток перегоняют при пониженном давлении на металлической бане в токе азота, собирая фрак цию с т. кип. 183°/3 мм (температура бани 240—250°).
Выход метилового эфира феруловой кислоты равен 2,7 г — 4,1 г (25—38%), т. пл. 59—60°.
Вариант 2. В прибор, описанный выше, помещают 10 г (0,05 моль) феруловой кислоты, 100 мл метилового спирта и 10 г ионообменной смолы КУ-2 и нагревают 8 часов на масляной бане прн 'слабом кипении реакционной смеси и непрерывном перемешивании. Реакционную смесь декантацией освобождают от смолы, смолу промывают абсолютным метиловым спиртом 3 раза порциями по 10 мл и присоединяют к общей массе. Избыток спирта отгоняют, а остаток перегоняют при пониженном давлении на металлической бане в токе азота, собирая фракцию с т. кип. 183°/3 мм (температура бани 230— 250°).
Выход метилового эфира феруловой кислоты равен 5,6— 5,8 г (52—55%), т. пл. 59—60°.
Вещество хорошо растворяется в хлороформе, абсолютном диэтиловом эфире, тетрагидрофуране, этиловом спирте, четыреххлористом углероде, нерастворимо в воде.
Найдено, %: С—63,19; Н—6,00. СцН12О<. Вычислено, %: С—63,40; Н—5,77.
169
ЙК-спёктр снят нй спектрометре ИКС-14-А при толщине слоя 0,245 см, скорость сканирования 4,7. Спектры снимались в вазелиновом масле и четыреххлористом углероде, призмы NaCl н LiF.
ИК-спектр, снятый в вазелиновом масле, указывает на наличие в продукте реакции кратной связи (v = 1623 слг1), гидроксильной группы (фенольной) (v ='3570 сж-1), ароматического кольца (v = 1589, 1500, 1420 сж-1), эфирной группировки (т = 1485 см~х) и метоксильной группы (v ='2830 см~1). Данные спектрограммы полностью подтверждают, что полученный продукт является эфиром феруловой кислоты.
УФ-спектр 0,56 М раствора метилового эфира феруловой кислоты в этиловом спирте снят на приборе СФ-4-А при толщине слоя 0,505 см. Метиловый эфир феруловой кислоты имеет максимум поглощения при кмакс = 310 нм и Ige —3,15.
ЛИТЕРАТУРА
1. Ю. К. Юрьев. Практические работы по органической химии, вып. 2. М„ Изд-во МГУ, 1957, с. 63.
2. К. М. С а л д а д з е, А. Б. Пашков, В. С. Титов. Ионообменные высокомолекулярные соединения. М., Госхимиздат, 1960, с. 84, 107.
р-(5-МЕТИЛ-2-ПИРРОЛИДИЛ)ПРОПИОНОГИДРАЗИД
| А. А. ПОНОМАРЕВ. | М. В. НОРИЦИНА
сн2-сн,- с
н
\nhnh,
C8H17N3O
М. в. 171,22
Гидразиды р-(2-пирролидил) пропионовых кислот в литературе не описаны. Нами они получены с хорошими выходами при взаимодействии соответствующих метиловых эфиров Р-(2-пирролидил) пропионовых кислот с гидразином. Аналогично гидразидам пиперидилкарбоновых кислот они обладают ярко выраженной антитуберкулезной активностью [1].
СХЕМА СИНТЕЗА
I l-CH2CH, COOCHs
ciCf н
NHa—NH,
-СН,-СН2С^
о
NHNH,
171
Характеристика основного сырья
Метиловый эфир 0- (5-метил-2-пирролидил) пропионовой кислоты, т. кип. 92—92,5°/8 мм, — 1,4570; получение см. с. 165 этого сборника.
Гидразин-гидрат, ГОСТ 5832—65.
Метанол, ГОСТ 6995—54, х. ч.
Петролейный эфир, ТУ МХП 279—59, ч.
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, вносят 3 г (0,017 моль) метилового эфира 0-(5-метил-2-пирролидил)пропионовой кислоты, 3 г(0,07 моль) 85%-ного раствора гидразин-гидрата, 60 мл метанола. Реакционную смесь перемешивают 4 часа при комнатной температуре, 5 часов при 50—60° и оставляют на ночь. Затем метанол, избыток гидразин-гидрата и воду отгоняют при пониженном давлении. К остатку добавляют 30 мл бензола и отгоняют в виде азеотропа оставшуюся в нем воду, при этом выпадают бесцветные кристаллы. Вещество перекристаллизовывают из петролейно-го эфира.
Выход 0-(5-метил-2-.пирролидил)пропионогидразида равен 2,6 г (87%), т. пл. 69—70° (см. примечание).
Найдено, %: С—55,97; 55,90; Н—10,15; 10,10; N—24,30; 24,45. CeHpNsO. Вычислено, %: С—56,10; Н—10,02; N—24,58.
Примечание.
Аналогичным путем получены впервые:
₽-(1-Метил-2-пирролидил)пропиоиогндразид из метилового эфира 0-(1-метил-2-пирролидил) пропионовой кислоты, выход 81%, т. пл. 68—69°.
Найдено, %: С—55,74; 55,84; Н—9,70; 10,07; N—24,90; 24,25. CsHnNsO. Вычислено, %: С—56,10; Н—10,02; N—24,58.
Р-(5-Изобутил-2-пнрролидил)пропиоиогидразид из метилового эфира (3-(5-изобутил-2 пнрролндил)пропионовой кислоты, выход 80%, т. пл. 36°.
Найдено, %: С—61,50; 61,37; Н—10,52; 10,91; N—19,33; 19,50. CnH23N3O. Вычислено, %: С—61,88; Н—10,86; N—19,73.
ЛИТЕРАТУРА
1. Н. L. Jale, К. Lessee. J. Amer, Chem. Soc., 75, 1933 (1953).
1-МЕТИЛ-5-ПРОПИЛ-2-ПИРРОЛИДОН
В. А. СЕДАВКИНА
Н7Сз-' 1=0
XNZ
СН3
C8H15NO М. в. 147,20
N-Метилзамещенные пирролидоны получают метилированием пирролидонов последовательной обработкой их металлическим натрием и йодистым метилом. Таким образом был получен 1-метил-2-пирролидон [1, 2]. Описаны также способы получения М-метил-2-пирролидона и 1,5-диметил-2-пирролидона из бутирон и валеролактонов [3—5], а также из янтарной и левулиновой кислот [6, 7].
В настоящей работе предлагается метод синтеза N-метил--5-алкил-2-пирролидонов, заключающийся в гидрировании смеси этиловых эфиров у-кетокарбоновых кислот с метиламином над скелетным никелевым катализатором. Получена группа неописанных ранее 1-метил-5-алкил-2-пирролидонов.
СХЕМА СИНТЕЗА
С,Н,-С-СН,-СН,-C(f 4- CH.NH,
I! ХОС,НЬ
О
Н, NiR
I СН,
173
Характеристика основного сырья
Этиловый эфир у-кетогептановой кислоты, т. кип. 94— 96°/2 мм; = 1,4338; получение см. [8].
Метиловый спирт, ГОСТ 6995—54.
Натр едкий, ГОСТ 4328—66, ч.
Метиламин солянокислый, ТУ ОРУ 3—58, ч.
Никель Ренея получен выщелачиванием сплава никеля с алюминием [9].
Водород электролитический.
Условия получения
В стальной вращающийся автоклав емкостью 150 мл помещают 17,2 г (0,1 моль) этилового эфира у-кетогепта,новой кислоты, 60 мл метилового спирта, содержащего 12 г метиламина (см. примечание 1) и 1,7 г никеля Ренея. Подают водород под давлением 100 атм и ведут гидрирование при постоянном перемешивании и нагревании (120—130°) до прекращения поглощения водорода. Затем катализатор удаляют фильтрованием или центрифугированием, избыток метиламина и растворитель отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 75—7677 мм.
Выход 1-метил-5-пропил-2-пирролидона равен 8,6 г (61,7%), л® =' 1,4706, d4№ = 0,9705 (см. примечание).
Найдено, %: С—68,18; 68,26; Н—10,50; 10,64; N—10,01; 10,64. C8H15NO. Вычислено, %: С—68,08; Н—10,63; N—9,93.
Примечания:
1. Для получения раствора метиламина в метиловом спирте поступают следующим образом. В кругло дойную колбу помещают твердый едкий натр и из капельной воронки прикапывают 50%-ный раствор солянокислого метиламина. Ток метиламина поступает во взвешенную склянку Дрекселя с метиловым спиртом. Количество поглотившегося метиламина определяется по привесу.
2. Аналогично получены:
1-Метил-5-бутил-2-пирролидои из этилового эфира у-кетооктановой кислоты, выход 60%, т. кип. 92—9471 мм, = 1,4695, dtw = 0,9558.
Найдено, %: С—69,41; 69,58; Н—10,70; 10,74; N—9,31; 9,33. CgHizNO. Вычислено, %: С—69,67; Н—10,96; N—9,03.
1-Метил-5-амил-2-пирролидон из этилового эфира у-кетононановой кислоты, выход 67%, т. кип. 100—1020/1 мм, = 1,4680, dt20 = 0,9462.
Найдено, %: С—71,05; 71,17; Н—11,06; 11,22; N—8,19; 8,14. C10H19NO. Вычислено, %: С—71,07; Н—11,24; N—8,28.
1-Метил-5-изоамил-2-пирролидои из этилового эфира у-кетоизононано-вой кислоты, выход 64,7%, т. кий. 97—9973 мм, = 1,4690, d,20 = 0,9490.
Найдено, %: С—71,08; 71,16; Н—11,36; 11,45; N—8,35; 8,48. CioHigNO. Вычислено, %: С—71,07; Н—11,24; N—8,28.
174
1-Метил-5-гексил-2-пирролидон из этилового эфира у-кетодекановой кислоты, выход 65,4%, т. кип. 150—15376 мм, = 1,4664, d^ = 0,9321. Найдено, %: С—72,22; 72,35; Н—11,40; 11,52; N—7,93; 7,85. ChHjiNO. Вычислено, %: С—72,13; Н—11,47; N—7,67.
1Метил-5-гептил-2-пирролидон из этилового эфира у-кетоуидекановой кислоты, выход 68,3%, т. кип. 153—15472 мм, — 1,4650, d^0 = 0,9236.
Найдено, %: С—71,98; 71,91; Н—11,70; 11,65; N—7,26, 7,29. Ci2H23NO. Вычислено, %: С—73,03; Н—11,75; N—7,09.
1-Метил-5октил-2пирролидон из этилового эфира у-кетододекановой кислоты с выходом 63%, т. кип. 160—16274 мм, п2$ = 1,4642, dtw = = 0,9628.
Найдено, %; С—74,01; 74,02; Н—12,08; 12,03; N—6,82; 6,89. CisHsNO. Вычислено, %: С—73,92; Н—11,85; N—6,63.
ЛИТЕРАТУРА
1. Е. Spath, Н. Bretsch eider. Вег., 62, 330 (1929).
2, С. С. Luman. J. Amer., Chem. Soc. 55, 297 (1933).
3. R. E. Kick, D. F. Othiuer. Encucl, Chem. Techn., 10, 759 (1954).
4. W. R e p p e. Polyvinilpyrrolldon. Weinheim, 1954, c. 354.
5. ГО. К. Юрьев. Уч. зап. МГУ, 132, 224 (1950).
6. В. И. Романовский, А. И. Соколова. Хим. пром-сть, 7, 491 (1963).
7. Герм. пат. 6092244 (1935); С. А., 29, 3116 (1935).
8. В. Г. Бухаров, Т. Е. Позднякова. Изв. АН СССР, сер. хим., 6, 1108 (1960).
9. Синтезы органических препаратов, сб. 3. М., ИЛ, 1952, с. 338.
а-/1-МЕТИЛ-3(ц-ТЕТРАГИДР0ФУРИЛ)ПР0ПИЛ/ ТЕТРАГИДРОФУРОАТ
М. Д. ЛИПАНОВА
L J-coochcHjCH,-! )
W I XOZ
сн3
С,3Н22О4 М. в. 242,30
Сложные эфиры тетрагидрофурановой кислоты и тетрагидрофурановых спиртов получены нами каталитическим гидрированием соответствующих фуроатов в присутствии никеля Ренея. Фуроаты синтезированы при нагревании фуроилхлори-да с тетрагидрофурановыми спиртами. При попытке этерифи-цировать пирослизевую кислоту тетрагидрофурановыми спиртами в присутствии серной кислоты образуется неразделимая перегонкой смесь фуроата и простого эфира тетрагидрофуранового спирта [1].
СХЕМА СИНТЕЗА
---II II-II X /1-СН,СН3СН(ОН)СН3
J-COOH Il J-COC1 -2----------------
0х ‘ 0х
!7~LcOOCHCH2CH ' J NTT,-» хОХ ] xoz ™ к
СН,
J—СООСНСН2СН2-'Ч 0х | ХО
СН,
176
Характеристика основного сырья
Пирослизевая кислота, т. пл. 133—134°, получение см. [2].
З-(а-Тетрагидрофурил) бутанол, т. кип. 145°/58 мм, п2" = = 1,4550; получение см. [3].
Хлористый тионил, МРТУ 6—09—2602—65.
Никель Ренея, получение см. 14].
Бензол, ГОСТ 5955—51.
Диоксан, ГОСТ 10455—63.
Условия получения
Синтез фуроилхлорида. В колбу Кляйзена ем,костью 100 мл, снабженную обратным холодильником с хлоркальциевой трубкой, вносят 22,4 г (0,2 моль) возогнанной пирослизе-вой кислоты и двумя равными порциями добавляют 41,3 г (3,5 моль) хлористого тионила. При этом происходит разогревание с обильным выделением хлористого водорода. Через 0,5 часа бурная реакция прекращается и реакционную смесь нагревают 1 —1,5 часа при 110—120°. После этого обратный холодильник заменяют на нисходящий и отгоняют хлористый тионил. Остаток перегоняют при атмосферном давлении, собирая фракцию с т. кип. 173—175°, или при пониженном давлении, собирая фракцию с т. кип. 89—90°/32 мм.
Выход фуроилхлорида равен 24,5 г (94,23%).
По литературным данным [5], т. кип. 173—175°.
Получение а-[1-метил-3-(а-тетрагидрофу рил)пропил]фуроа-та. В круглодонную колбу емкостью 200 мл, снабженную обратным холодильником, помещают 26,1 г (0,2 моль) фуроилхлорида, 28,8 г (0,2 моль) тетрагидрофурилбутанола, 100 мл сухого бензола и нагревают 5 часов на масляной бане при 120°. Содержимое колбы нейтрализуют насыщенным раствором карбоната натрия (60—65 мл), промывают водой и отго няют при пониженном давлении сначала бензол, а потом остаток, собирая фракцию, кипящую при 170—171°/9 мм.
Выход а-[1-метил-3-(а-тетрагидрофурил)пропил]фуроата равен 40,3 г (84,2%). Вещество представляет собой светло-желтое масло, = 1,4910, е/420 = 1,1026.
Найдено, %: С—65,67; 65,80; Н—7,37; 7,75; MRD = 62,28. CisHisCC Вычислено, %: С—65,53; И—7,61; MR D =61,84.
Получение а-[/-метил-3- (а-тетрагидрофурил')пропил}тетра-гидрофуроата. В автоклав емкостью 0,5 л загружают 24 г (0,1 моль) а-(1 -метил-3-(а-тетрагидрофурил)пропил]фуроата, 50 мл диоксана, 3 г никеля Ренея. Гидрирование ведут 3,5 часа при 140° и давлении водорода 50 атм. После поглощения рассчитанного количества водорода (5 л) автоклав разгружают, отфильтровывают катализатор и отгоняют при понижен-12 Зак. 1164 177
ном давлении сначала диоксан, а затем остаток, собирая фракцию, кипящую при 170—171°/9 мм.
Выход а-П -метил-3- (а-тетрагидрофурил) пропил ]тетрагид-рофуроата равен 21,75 г (90%) (см. примечание). Вещество представляет собой бесцветную подвижную жидкость, п2£ = = 1,4632, <Z42o= 1)0619
Найдено, %: С—64,42; 64,70; Н—9,00; 8,88; Л4/? D = 62,87. С1зН22О4. Вычислено, %: С—64,43; Н—9,15; MR р = 62,77.
Примечание.
По аналогичной методике впервые получен а-[1-метил-2-(а-тетрагид-рофурил)этил]тетрагидрофуроат с выходом 88,7%, т. кип. 173—175°/9 лъи, = 1,4704, dtw = 1,0955.
Найдено, %: С—63,00; 63,22; Н—8,65; 8,51; MRD =58,16. С12Н20О4. Вычислено, %: С—63,13; Н—8,83; MRD = 58,15.
ЛИТЕРАТУРА
1. А. А. Пономарев, М. Д. Липаиова, С. Г. Цыпкина. Научный ежегодник Сарат. уи-та, 492 (1954).
2. 3. В. Назарова, В. Н. Новиков. Методы получения химических реактивов и препаратов, вып. 17. М., ИРЕА, 1968, с. 160.
3. А. А. Пономарев. Синтезы и реакции фурановых веществ. Саратов., изд-во СГУ, 112 (1960).
4. Lies — Borart. Ann., 327 (1856).
5. A. A. Pavllc, Н. Adkins. J. Amer. Chem. Soc., 68, 1471 (1946).
МЕТИЛФЕНОКСИМАЛОНОВЫЙ ЭФИР
О. С. СТЕПАНОВА, А. С. ЯВОРСКИЙ, М. И. ПОБЕРЕЖНАЯ.
Л. Б. ХАГУНДОКОВА
СН»Ч ^.COOC^s с
СвН8О/ ХСООС2Н5
С14Н18О5
М. в. 266,30
Замещенные малоновые эфиры применяются для синтеза различных классов соединений: производных барбитуровой и тиобарбитуровой кислот [1], 1,3-диолов [2], замещенных дикарбоновых кислот и т. д.
Нами впервые получен метилфеноксималоновый эфир взаимодействием метилброммалонового эфира с фенолятом натрия.
СХЕМА СИНТЕЗА
СНэ^ /СООС,Н5
С + NaOCeH6
Вгх \соос2н5
СН„\ ^COOCjHj
С + NaBr
СВН6ОХ ХСООС3Н5
г/9
Характеристика основного сырья
Бромметилмалоновый эфир, т. кип. 112—114°/12 мм; d4m — = 1,3500; /zj° = 1,4500; получение см. [3].
Фенол, ГОСТ 6417—52, ч. д. а.
Этиловый спирт, ТУ 19П—39—69, абсолютирован по методике [4].
Натрий металлический, ТУ 1664—50. ч.
Условия получения
В трехгорлой колбе емкостью 1 л, снабженной мешалкой, капельной воронкой и обратным холодильником, растворяют порциями в 500 мл абсолютного этилового спирта 23 г (1 г-а) металлического натрия. Из капельной воронки при перемешивании сначала прибавляют 94 г (1 моль) фенола, растворенного в 20 мл абсолютного спирта, затем по каплям 256 г (1,01 моль) бромметилмалонового эфира. Реакционную смесь нагревают 8—12 часов, перемешивая, при ПО—130° до достижения нейтральной реакции среды по универсальному индикатору. Отгоняют спирт, растворяют выделившийся осадок соли в 300 мл воды, подкисляют двумя—тремя каплями концентрированной серной кислоты. Отделяют водный слой и экстрагируют эфиром дважды порциями по 150 мл; соединенные эфирные вытяжки и органический слой сушат прокаленным сульфатом натрия, отгоняют эфир, а остаток перегоняют при пониженном давлении, собирая фракцию 142—144°/6 мм.
Выход метилфеноксималонового эфира равен 196,8 г (73,9%); У420 = 1,1236; п*> = 1,4870.
Найдено, %: С—63,44; Н—6,91.
СцН18О5. Вычислено, %: С—63,14; Н—6,82.
Строение метилфеноксималонового эфира подтверждено ИК-спектрами. В спектре имеется характерный набор полос поглощения для монозамещенног.о бензольного кольца: деформационные колебания для =С—Н связи при 689 см~\ плоскостные колебания С = С скелета при 1493 см~'; валентные колебания С = С-связи при 1594 см1 и валентные колебания С—Н-связей бензольного кольца при 3046 и 3070 см~'. Кроме того имеется набор полос, характерных для замещенных малоновых эфиров: колебания С—О-эфирной связи карб-этоксила при 1031, 1070, 1113, 1139 и 1180 см~', и валентные колебания С = О связи (слабо расщепленный дублет) прч 1735 см~'. В спектре присутствует полоса поглощения при 1275 см~'. характерная для арилалифатических эфиров, что подтверждает структуру метилфеноксималонового эфира [5]. 180
ЛИТЕРАТУРА
1. Р. Я. Левина, Ф. К. Величко. Успехи химии, 29, 929 (1960).
2. А. В. Богатский, О. С. Степанова и др. Укр. хим. ж., 30, 1316 (1964).
3. М. Conrad, С. Bruckner, Вег., 24 , 2933 (1891).
4. Ю. К. Юрьев. Практические работы по органической химии. М, Изд-во МГУ, 1964, с, 52.
5. Л. Беллами. Инфракрасные спектры сложных молекул. М., ИЛ., 1963, с. 24, 30, 69.
3-[2-(5-МЕТИЛФУРФУРИЛОКСИ)ЭТОКСИ]-ПРОПИЛАМИН
| А. А. ПОНОМАРЕВ, | Н. П. МАСЛЕННИКОВА, Г. В. БЕСПАЛОВА
HsC-l J—СН2—О—СН,—СН2—О—СН2—СН2—СН2—NHa
CnH^NOa М. в. 213,15
3-[2- (5-Метилфурфурилокси) этокси]пропиламин впервые получен нами цианэтилированием 2-(5-метилфурфурилокси) -этанола с последующим гидрированием образующегося оксинитрила.
СХЕМА СИНТЕЗА
Н8сД j-CH,OCHaCH,OH 9H>=rCH~CN-.
- H8C-l J-CH2OCH,CH2OCH2CH2CN ►
\ф/ IN
-* Н.С-Г 1-CH,OCH,CH,OCH,CH2CH2NH2
Характеристика основного сырья
2-(5-Метилфурфурилокси) этанол, т. кип. 120—123710 мм, Лд = 1,4842; получение см. [1].
Акрилонитрил, ТУ РУ 797—53, ч., т. кип. 77,3°.
Никель Ренея, получение см. [2].
Водород электролитический.
Аммиак жидкий в баллонах.
Метиловый спирт, ГОСТ 6995—54, х. ч., т. кип. 65°. 182
Условия получения
Синтез 3-[2-(5-метилфурфурилокси)этокси]пропионитрила. В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром и обратным холодильником, помещают 25 г (0,16 моль) 2-(5-метилфурфурилокси)этанола, 100 мл толуола, 40 мл (0,05 моль) акрилонитрила и 15 мл 40%-ного раствора щелочи. Содержимое колбы энергично перемешивают 4 часа, следя за тем, чтобы температура не поднималась выше 40°j так как ,в течение первого часа возможно разогревание. Смесь 1оставляют на 12 часов при комнатной температуре, затем отделяют толуольный слой, отгоняют растворитель, остаток дерегоняют при пониженном давлении.
Выход 3-[2- (5-метилфурфурилокси) этокси]прбпионитрила равен 124,75 г (72%), т. кип. 126—13074 мм, п* = 1,4678; d420 = ,1057.
Получение 3-[2-(5-метилфурфурилокси)этокси]пропилами-на. Bq вращающийся автоклав емкостью 250 мл загружают 24 г (О,114 моль) 3-[2-(5-метилфурфурилокси)этокси]пропио-нитрила, 150 мл метилового спирта, насыщенного аммиаком при 0° & г никеля Ренея. Начальное давление водорода 115 атм. температура 95°. Гидрирование ведут до поглощения рассчитанного! количества водорода (2 л). Катализат фильтруют, спирт отгоняют, а остаток перегоняют при пониженном давлении.
Выход 3-[2-(5-метилфурфурилокси)этокси]пропиламина равен 7,4 г! Г56%); Т. кип. 119—12272 мм, п*> = 1,4823; сЦ20 = = 1,0512
I Найдено. %: С—62,23; 62,55; Н—8,93; 8,91; N—7,00; 7,07. CiiHwNO.J Вычислено, %: С—62,02; Н—8,99; N—7,58.
ЛИТЕРАТУРА
1. И./Ф. Бельский, С. Н. Харьков, Н. Н. Ш у й к и н. Изв. АН СССР, 1, 1966, с. 170.
2. Синтезы органических препаратов, сб. 3. М., ИЛ, 1952, с. 338.
3-[1М-МЕТИЛЦИКЛОПЕНТАНО(в)-2-ПИРРОЛИДИ1Л]-ПРОПИЛБЕНЗОАТ
|А А, ПОНОМАРЕВ,\ А. П. КРИВЕНЬКО, М. В. НОРИЦИНА
L A J-CH2-CH2-CH2-OOC— XN' х=/
СН8
C18H25NO2 М. zd7,18
В литературе описано получение эфиров спиртов йирроли-
динового ряда [1—3]. Нами синтезированы впервые сложные
эфиры 3-[М-метилциклопентано(в)-2-пирролидил]пррпанола, 3-(Ы-метил-2-пирролидил) пропанола и некоторых ароматических кислот с целью изучения физиологической активности полученных соединений.
СХЕМА СИНТЕЗА
J-(CH2)2-CH2OH + сюс—S ГГ \=/
I СНз
I
1-(СН2)з-ООС-’ J
N
I
СНз-НС1
NagCOg
184
'-(СН2)з-оос Nx
I
CHS
Характеристика основного сырья
3-[М-Метилциклопентано (в)2-пирролидил]пропанол, т.. кип. 125—128°/5 мм, п^= 1,4900; получение см. [4].
Бензоил хлористый, ГОСТ ТУ МХП 92—51.
Бензол абсолютный, приготовление см. [5].
Условия получения
В колбе емкостью 100 м.л смешивают раствор 4 г (0,02 моль) 3-[М-метилциклопентано(в)-2-пирролидил]пропанола в 20 м)л абсолютного бензола и раствор 2,8 г (0,02 моль) хлористого бензоила в 20 мл абсолютного бензола. При стоянии раствора п течение 2—3 часов при комнатной температуре выпадает белый кристаллический осадок. Кристаллы отсасывают на стеклянном пористом фильтре № 4, промывают абсолютным эфиром. После сушки в вакуум-эксикаторе получают 4 г (57%) гидрохлорида 3-[Ы-метилциклопентано(в)-2-пирро-лидил]пропилбензоата в виде бесцветных кристаллов с т. пл. 140—142°. Полученный гидрохлорид растворяют в 20 мл воды и к раствору приливают 15 мл насыщенного раствора карбоната натрия. Выделившееся масло отделяют, водный слой экстрагируют эфиром дважды порциями по 10 мл. Эфирные вытяжки соединяют с органическим слоем и сушат прокаленным карбонатом калия. Эфир отгоняют, остаток перегоняют прч пониженном давлении, собирая фракцию с т. кип. 180 — 182°/3 мм.
Выход 3-[К-метилциклопентано (в) -2-пирролндил)пропил •
бензоата равен 2,5 г (31%) (см. примечание). Вещество представляет собой жидкость с характерным запахом, = 1,5172, di№ = 1,0523.
Найдено, %: С—75,27; 75,20; Н—8,80; 8,73; N—4,49; 4,50; MRD = 82,85.
C,8H25NO2. Вычислено, %: С—75,23; Н—8,78; N—4,48; MRD = 82,95.
Примечание.
Аналогичным путем получен 3-[14-метилциклопентано(в)-2-пирролидил|-пропил-льметоксибензоат из 3-[-М-метилциклопентаио(в)-2-пирролидил]про-панола, выход 30%, т. кип. 184—185°/3 мм, = 1,5228, (U20 = 1,0826.
Найдено, %: С—71,77; 71,80; Н—8,45; 8,40; N—4,48; 4,42; MR D = 89,54.
Ci9H27NO2. Вычислено, %: С—71,90; Н—8,57; N—4,41; 447?D =89,19.
185
ЛИТЕРАТУРА
1. W. Kinnell, F. Perks. J. Pharmacy and Pharmacol., 12, 95 /(I960).
2. Белг. пат. 612947 (1963).
3. F. P. Doyle, M. D. Mehta, G. S. Sa ch, R. Ward. J. Chem, Soc., 1964, 578.
4. А. А. Пономарев, А. П. Кривенько, M. В. Норицина. Химия гетероцикл, соед., 6, 787 (1970).
5. Ю. К. Юрьев. Практические работы по органической химии, вып. I и П. М., изд-во МГУ, 1961, с. 47.
2-МЕТОКСИ-\АЦЕТИЛ-1,6-ДИОКСА-9АЗАСПИРО-[4,5]ДЕКАН
\ А. А, ПОНОМАРЕВ, | И. А. МАРКУШИНА, Т. И. ГУБИНА
СН8О
C10H17NO4
I СОСНз
М. в. 215,24
Единственным методом получения соединений ряда 1,6-дн-окса-9-азаспиро[4,5]декана является электролитическое интрамолекулярное алкоксилирование аминоспиртов фуранового ряда с последующим гидрированием образующихся 1,6-диок-са-9-азаспиро[4,5]деценов-3 {1, 2].
Ниже приводится несколько усовершенствованный нами препаративный способ получения 2-метокси-Н-ацетил-1,6-ди-окса-9-азаспиро!4,5]декана: после электролитического мето-ксилироваиия N-ацетилфурфуриламиноэтанола продукт не выделялся, а сразу подвергался гидрированию.
СХЕМА СИНТЕЗА
L J—CH2NCH2CHaOH ~— -Г
I а NH4Br
СОСНз
Характеристика основного сырья
N-Аце^лфурфуриламиноэтанол, т. кип. 139—140°/1 мм\ п*> = 1,5135, di20 = 1,173; см. с. 46 этого сборника.
Метанол, ГОСТ 6995—54, ч.
Аммоний бромистый, ГОСТ 4454—64, ч.
Водород, ГОСТ 3022—61.
Никель Ренея, приготовление см. [3].
Условия получения
В электролизер [4] помещают раствор 5 г (0,051 моль) бромистого аммония, 55 г (0,3 моль) свежеперегнанного N-ацетилфурфуриламиноэтанола в 220 мл метилового спирта и охлаждают до минус 20° (см. примечание 1). Электролиз проводят в течение 5 часов при силе тока 4,5—3 а и напряжении 12—15 в. Затем раствор переносят в колбу и прибавляют метилат натрия (см. примечание 2).
В стальной вращающийся автоклав помещают метанольный раствор 2-метокси-М-ацетил-1,6-диокса-9-азаспирс(4,5]де-цена-3 и 6 г катализатора никеля Ренея. Гидрирование проводят при комнатной температуре и начальном давлении водорода 95 атм и заканчивают по поглощении 2,9 л водорода. Катализат выгружают из автоклава, никель Ренея отфильтровывают и промывают на фильтре метанолом 2 раза порциями по 20 мл. Метанол отгоняют на водяной бане, остаток перегоняют при пониженном давлении (см. примечание 3), собирая фракцию с т. кип. 145—148°/2 мм.
Выход 2-метокси-Ы-ацетил-1,6-диокса-9-азаспиро!4,5]дека-на равен 40,6 г (63%), п™ = 1,4880, di20 = 1,1757 (см. примечание 4).
Найдено, %: С—56,08; 56,06; Н—7,86; 7,89; N—6,75; 6,81; = 52,74.
С,oHizNOt. Вычислено, %: С—55,80; Н—7,96; N—6,51; .W?n = 52,86.
Примечания:
1. В качестве охлаждающей смеси используют бутиловый или изобутиловый спирт, в который добавляют твердую углекислоту до достижения необходимой температуры в электролизере.
2. Метилат натрия приготовляют из 1,2 г металлического натрия и 20 мл метанола. Прибавление метилата натрия необходимо для связывания свободного брома, образующегося в процессе электролиза.
3. Перегонку проводят в колбе с дефлегматором в 1—2 наколки со стекловатой.
4. Аналогично получают 2-метокси-2-метнл-М-ацетил-1,6-диокса-9-аза-спнро[4,5]декан из М-ацетил(5-мстилфУрфурнл)аминоэтаиола, выход 70%, т. кнп. 137—13872 мм, 1,4865, = 1,1513.
Найдено, %: С—57,67; 57,38; Н—8,37; 8,36; N—6,21; 6,18; MRd =57,21.
ChHiuNOi. Вычислено, %: С—57,62; Н—8,35; N—6,11; .W?D = 57,48.
188
ЛИТЕРАТУРА
1. А, А. Пономарев, И. А. Маркушина, Л. И. Пивоварова, Т. И. Губина. Химия гетероцикл, соед. 2, 75 (1970).
2. А. А. Пономарев, И. А. Маркушина, Л. И. Пивоварова. Авт. свид., 191579; Изобретения. Промобразцы, Товарные знаки, № 4
3. Синтезы органических препаратов, сб. 3. М., ИЛ., 1952, с. 338.
4. А. А. Пономарев, И. А. Маркушина. Методы получения хи мнческнх реактивов и препаратов, вьш, 17. М., ИРЕА, 1967. с. 99.
5-[2-(МЕТОКСИКАРБОНИЛ)ЭТИЛ] 1,2-ДИГИДРОПИРРОЛИЗИН
I 4. А. ПОНОМАРЕВ, | Л. Н. АСТАХОВА, В. Н. ВОЛКОЛУПОВ
СН3ООС-СН2—СН,
C11H15NO2 м. в. 193,24
5-[2-(Метоксикарбонил) этил]-1.2-дигидропирролизин синтезирован нами впервые взаимодействием 1,2-дигидропирроли-зина с метиловым эфиром акриловой кислоты в присутствии фторида бора.
СХЕМА СИНТЕЗА
| | I + СН,=СН—СООСН3 -2^-^
k/N\/ ______________
LkJ
CHjOOC— сн3-сн2
Характеристика основного сырья
1,2- Дигидропирролизин, т. кип. 69—70°/15 мм, «^=1,5308, =' 1,0056; получение см. [2].
Метиловый эфир акриловой кислоты, ТУ TCP 320р—61, ч.
Эфират фтористого бора, т. кип. 124—126°, d^5 = 1,135; получение см. [3].
190
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, капельной воронкой и обратным холодильником, помещают 10,7 г (0,1 моль) 1,2-дигидропирролизина, 8,6 г (0,1 моль) метилового эфира акриловой кислоты и прн перемешивании и охлаждении прибавляют по каплям за 20 минут 6 мл эфирата фтористого бора. Через 30 минут к реакционной смеси добавляют 30 мл метанола и 150 мл воды. Выделившийся масляный слой экстрагируют эфиром 4 раза порциями по 15 мл, эфирные вытяжки сушат сульфатом магния. Эфир отгоняют, остаток перегоняют при пониженном давлении, собирая фракцию ст. кип. 118—119°/1 мм.
Выход 5-[2-(метоксикарбонил) этил-1,2-дигидропирролизи-на равен 12,4 г (64,2%), п*> = 1,5180; rf420 = 1,0975 (см. примечание) .
Найдено, %: С—68,06; 68,03; Н—7,98; 7,85; N—7,58; 7,51; MR D = 53,36.
C„Hi5NO2. Вычислено, %: С—68,37; Н—7,82; N—7,24; MRD = 53,25.
Примечание.
Аналогичным путем впервые синтезирован 3-метнл-5-[2-(метоксикарбо-иил)этил]-1,2-дигидропирролизин из 3-метил-1,2-дигидропирролизина [1, 2], выход 72%, т. кип. 122—12471 мм, = 1,5120, dtw = 1,0750.
Найдено, %: С—69,35; 69,20; Н—8,39; 8,23; N—6,89; 6,94. Cl2HirNO2. Вычислено, %: С—69,54; Н—8,26; N—6,75.
ЛИТЕРАТУРА
1. А. А. Пономарев, И. М. Скворцов. Ж. общ. химии, 97, 32 (1962).
2. А. А. Пономарев, И. М. Скворцов. Методы получения химических реактивов и препаратов, вып. 17. М., ИРЕА, 1968, с. 68.
3. Ю. В. Карякнн, И. И. Ангелов. Чистые химические реактивы. М., Госхимиздат, 1955, с. 97.
(о-МЕТОКСИФЕНИЛ) ОКСИМЕТИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА, НАТРИЕВАЯ И СВИНЦОВАЯ СОЛИ
(о-Метоксифенил )оксиметандифосфоновая кислота, натриевая и свинцовая соли
И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ
он
I
(НО)2ОР-С-РО(ОН)2
^рОСНз
СцН12О8Р2 М. в. 298,12
Являясь представителем а-оксиалкандифосфоновых кислот (о-метоксифенил) оксиметандифосфоновая кислота обладает высокой комплексообразующей способностью по отношению ко многим катионам в широком интервале pH [1, 2].
В литературе (о-метоксифенил) оксиметандифосфоновая кислота и ее соли не описаны. Кислота синтезирована нами в растворе взаимодействием о-метоксибензойной кислоты с треххлористым фосфором и водой по методу, описанному ранее для а-оксибензил-а.а-дифосфоновой кислоты [3—5], и выделена в виде натриевой и свинцовой солей.
СХЕМА СИНТЕЗА
соон
|^\-ОСНз
Ч/
РС13
Н2О
о оно
II I II (НО)аР-С-Р(ОН)2
,^-ОСНз + ч/
192
COCI
+А-°сн-Xz
+ HCl,
ООН,
' /POfOH), f \-Cr-OH
’ .11 \PO(OH)2
NaOH
Pb(CH3COO)3
OCH3
I /PCXONa), гД-ОИ C\pO(OHXONa)
г ОСН,
/PO(O),
P0(O)2
2-
Pb2+
Характеристика основного сырья
о-Мётоксибензоййая кислота, МРТУ 6—09—1444—64, ч.
Фосфор треххлрр истый, ГОСТ 91—41, ч.
Натр едкий, ГОСТ 4328—48, ч.
Свинец уксуснокислый, ГОСТ 1027—67, ч.
Условия получения
В чётырёхгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником, термометром й капельйой воронкой, загружают смесь 76,08 г (0,5 моль) о-метбксйбен • зойной кислоты и 9 г (0,5 моль) воды и постепенно прн перемешивании прибавляют из капельной воронки 41,25 г (0,3 моль) свежеперегнанного треххлористого фосфора с такой скоростью, чтобы температура в колбе не превышала 50’.
Температуру реакционной массы медленно за 2 часа поднимают до 120* (при этом выделяется хлористый водород, который поглощается водой) и выдерживают 3 часа при 120— 125°, Затем охлаждают до 90°, добавляют 200 мл воды и кипятят 1 час.
Выпавший при охлаждении осадок непрореагировавшей о-метоксибензойной кислоты отфильтровывают и промывают 200 мл воды.
13 Зак 1164 193
Фильтрат и промывные воды соединяют вместе, отгоняют с паром до отсутствия ионов хлора в дистиллате, упаривают до объема 50 мл и экстрагируют эфиром примесь о-мето-ксибензойной кислоты и ее хлорангидрида (5 раз по 25 мл). Нейтрализуют 25 мл раствора кислоты 42%-ным едким натром до pH 7 (по .универсальной индикаторной бумаге) ' и многократно прибавляют гидролизный спирт порциями по 50—100 мл до образования густого желтоватого масла, после чего те же операции проводят абсолютным спиртом до образования осадка, который отфильтровывают и сушат в эксикаторе над хлористым кальцием.
Выход тринатриевой соли (о-метоксифенил)оксиметандифосфоновой кислоты равен 18 г (64,8%).
Найдено, %: С—25,08; 25,15; Н—3,30; 3,40; Р—15,96. CnH9Na3O8P2• Н2О. Вычислено: %: С—25,05; Н—2,88; Р—16,2.
Приливают к 25 мл раствора кислоты раствор 56,9 з (0,15 моль) уксуснокислого свинца в 120 мл воды, отфильтровывают выпавший белый осадок, промывают его горячей водой (5 раз порциями по 100 мл) и сушат при 75°
Выход свинцовой соли (Ь-метоксифенил)оксйметаидифос-фоновой кислоты равен 33,0 г (60,6%).
Найдено,%: С—12,74; 12,98; Н—0,86; 1,15; Р 8,8; РЬ—56,7; 56,6 (см. примечание).
СцНвОвРгРЬг • Н2О. Вычислено, %: С—13,20, Н—1,37; Р—8,54; РЬ—57,0.
Прнмечанне.
Содержание свинца определяют комплексонометрически по следующей методике. В конической колбе растворяют навеску (~ 0,1 г) вещества в 15 мл смеси (1 :3) концентрированных азотной и соляной кислот при на греваннн и выпаривают содержимое колбы досуха. Эту операцию повторяют 3 раза. Остаток растворяют в 10 мл концентрированной азотной кислоты, нагревают до прекращения выделения бурых паров, добавляют ~ 50 м-л воды и нейтрализуют — 40%-ным раствором едкого натра до pH 1,5. Зате^ прибавляют точно 3 мл 0,-1 и, раствора динатриевой соли этилеидиаминтетрауксусной кислоты (НЭДТУ), 5 мл ацетатного буферного раствора с pH 5,5—6,0, 1 .мл раствора ксиленолового оранжевого н титруют 0,1 н. раствором НЭДТУ до перехода красной окраски в желтую.
Процентное содержание свинца (X) определяют по формуле
. (3 + И).0,001036.100-АГ
А ®® ,' .
8
где v —объем 0,1 н. раствора НЭДТУ, израсходованного на титрование, МЛ',
0.001036 — количество свинца, соответствующее 1 мл 0,1 н. раствопа НЭДТУ, г;
К — поправка к 0,1 и. раствору НЭДТУ;
g —навеска -препарата, г..
ЛИТЕРАТУРА
1. М. И. Кабачник, Р. П. Ластовекий, Т. Я. Медведь, В. В. Медынцев, И. Д. Колпакова, Н. М. Дятлова. Докл. АН СССР, 177, 582 (1967).
2. В. Blaser, К. Н. Worms, Н. G. Germacheid, К. Woll-mann, Z. anorgan. allgem. Chem., 381, 247 (1971),
3. P. П. Ластовскнй, И. Д. Колпакова, Л. В. Кривицкая, Е. И. Миронова. Методы получения химических реактивов и препаратов, вып. 25. М., ИРЕА, 1972, с. 147.
4. Бельг, пат. 619620 (1963); С. А., 59, 11566 (1963).
5. Фраиц. пат. 1412865 (1965); С. А., 64, 8237 (1966).
2-МЕТОКСИ-№ЭТИЛ-1,6-ДИОКСА-9-АЗАСПИРО-[4,5]ДЕКАН
1 А. А. ПОНОМАРЕВ. | И. А. МАРКУШИНА, Т. И. ГУБИНА
C10H19NO3
М, в. 201,25
2-Метокси-Г4-этил-1,6-диокса-9-азаспиро[4,5]декан получен нами впервые восстановлением 2-метокси-М-ацетил-1,6-диокса--9-азаспиро[4,5]декана алюмогидридом лития.
СХЕМА СИНТЕЗА
/°— \ ЫА1Н4
Хо/Х—n/
сосн8
сн,о-
Характеристика основного сырья
2-Метокси-М-ацетил-1,6-диокса-9-азаспиро{4,5]декан, т. кип. 145—148°/2 мм, п™ = 1,4880, d420 =' 1,1757; получение см. с. 187 этого сборника.
Алюмогидрид лития, РЭТУ 1067—63, ч.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным холодильником с хлоркальциевой трубкой и ка-196
пельной воронкой, помещают 3 г (0,08 моль) алюмогидрида лития в 100 мл абсолютного эфира (см. примечание 1). При перемешивании по каплям добавляют 7 г (0,04 моль) 2-мето-кси-Ы'-ацетил-1,6-диокса-9-азаспиро[4,51декана в 25 мл абсолютного бензола (см. примечание 1) и нагревают реакционную смесь 10 часов на водяной бане при 40—50°. Затем раствор с осадком непрореагировавшего алюмогидрида лития переносят в колбу Эрленмейера. При охлаждении колбы водой со льдом осторожно по каплям приливают • 20—40 мл воды для разложения избыточного алюмогидрида лития. Выпавший белый студнеобразный осадок отфильтровывают и промывают на фильтре эфиром 4 раза порциями по 50 мл. Эфирные вытяжки объединяют с основным эфирным раствором и сушат над безводным сульфатом магния. Эфир отгоняют на водяной бане, остаток перегоняют при пониженном давлении.
Выход 2-метокси-М-этил-1,6-диокса-9-азаспиро[4,5]декана равен 5,5 г (85%), т. кип. 97—100°/4 мм, п2° — 1,4630; d^ =-• = 1,051 (см. примечание 2).
Найдено, %: С—59,42; 59,18; Н—9,01; 9,69; N—6.79; 7,22; MR D = 52,76.
C,0Hi9NO3. Вычислено: %: С—59,68; Н—9,52; N—6,96; MR D = 52,85.
Примечания:
1. Абсолютный эфир и бензол приготовляют по методике [2].
2. Аналогичным путем получают 2-метокси-2-метил-М-этил-1,6-дпокса-9-азаспиро[4,5]декан, из 2-метокси-2-метил-1Ч-ацетил-1,б-диокса-9-азаспиро[4,51-декана fl] выход 80%, т. кип. 90—91“/4 ям, пв — 1,4639, dt20 = 1,036.
Найдено, %: С-61,42; 61,57; Н—9,90; 9,84; N—6,14; 6,44; MRB =57,13.
C11H22NO3. Вычислено, %: С—61,37; Н—9,83; N—6,50; MR D = 57,47.
ЛИТЕРАТУРА
1. А. А. Пономарев, И. А. Маркушина, Л. И. Пивоварова, Т. И. Губнна. Химия гетероцикл, соед., 2, 75 (1970).
2. Препаративная органическая химия. М.—Л., «Химия», 1964, с. 155.
МОНОАЛКИЛГИДРАЗИНЫ
В. С. СТОПСКИЙ, 3. И. СЕРГЕЕВА, Б. В. ИОФФЕ
CH.^CHs-NH-NHj
C2H6N'2 М. в. 60,10
СН,-СН2-СН2-NH-NH,
C3H8N2 М. в. 74,13
(CHS)2CH-NH-NH2
C3H8N2 м. в. 74,13
Моиоалкилгидразины получают с выходом 40—80% взаимодействием алкиламинов с хлорамином или гидроксиламин--0-сул.ьфокислотой [1, 3]. Однако наиболее доступными реагентами для синтеза моноалкилгидразинов являются галоген-алканы и гидразин-гидрат. В литературе описано получение этил-, н-пропил- и изопропилгидразинов взаимодействием гидразин-гидрата с бромистыми алкилами [4].
Нами предложен усовершенствованный вариант указанного метода: изменены температурные условия проведения реакции (что в случае этилгидразина, например, позволило увеличить выход с 22 до 66%), а также значительно сокращено количество гидразин-гидрата.
СХЕМА СИНТЕЗА
R—Вг 4-2NH2—NH, - R—NH-NH2 + NHaNH,-HBr
Характеристика основного сырья
Гидразин-гидрат, ГОСТ 5832—65, ч.
Этил бромистый, МРТУ 6—09—6467—69, ч.
Пропил бромистый, МРТУ 6—09—446—63, ч.
Изопропил бромистый, МРТУ 6—09—1696—64, ч.
Этиловый эфир, ГОСТ 6265—65, ч.
Кали едкое, ГОСТ 4203—65, ч. д. а.
198
Условия получения
Синтез этилгидразина. В колбу емкостью 1 л, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, помещают 500 мл (10,0 моль} гидразин-гйдра-та, нагревают до 35° и при энергичном перемешивании (см. примечание 1)' за 4—5 часов прибавляют 109 г (1,0 моль)' бромистого этила с такой скоростью, чтобы температура1 реакционной смеси не превышала 40°. Реакционную смесь нагревают еще 1 час при 50—55°. Затем экстрагируют этилгидра-зин эфиром в экстракторе непрерывного действия до установления постоянного объема экстрагируемого слоя (10—12 часов). Эфирный экстракт сушат в течение суток гранулированным едким кали; эфир отгоняют, а остаток перегоняют на ректификационной колонке эффективностью 15 т. т., собирая фракцию с т. кип. 99—101°.
Выход этилгидразина равен 36,6 г (66%). После повторной перегонки т. кип. 99—1017763 мм, Лд — 1,4308, d4® = = 0,8471 (см. примечание 2).
По литературным данным [4], т. кип. 1007774 мм, Ид = = 1,4276, d420 = 0,8461.
Получение п-пропилгидразина проводят по методике, описанной выше. Из 500 мл (10,0 .ноль) гидразин-гидрата и 246 г (2,0 моль) бромистого н-пропила получают 97 г (66%) н-про> пилгидразина с т. кип. 121 —123°. Реакцию проводят при 45— 50°, слегка нагревая реакционную смесь на водяной бане. По окончании добавления бромида нагревают смесь еще 1 час при 60—70°. После повторной перегонки получено вещество с т. кип. 121,9—122,27769 мм, ng = 1,4344, d420 = 0,8406.
По литертурным данным [4], т. кип. 119°, ng = 1,4343, di® = 0,8366.
Получение иэопропилгидразина. Из 400 мл (8,0 моль) гидразин-гидрата и 246 г (2,0 моль) бромистого изопропила получают 104,4 г (71%) иэопропилгидразина с т. кип. 105—108°. Реакцию проводят при 55—60°. По окончании добавления бромида нагревают реакционную смесь еще 1 час при 70°. Время экстракции 12—15 часов. После повторной перегонки получают 92 г (63%) вещества с т. кип. 108,6—109,0°/767 мм и т. пл. 10,9—11,2°, = 1,4292, di® = 0,8317.
По литературным данным [4}, т. кип. 106—107°, ng = = 1,4280, di® = 0,8361.
Примечания:
1. При уменьшении скорости перемешивания во время прибавления бромистого этила к гидразиитидрату выход этилгидразина может понизиться до 40%.
2. Полученный указанным методом этилгидразни содержит — 7—10% примеси 1,1-диэтилгидразииа, от которой не удается избавиться повторной перегонкой из-за близости температур кипения указанных соединений.
199
ЛИТЕРАТУРА
1. А. Н. Цост, Р. С. Сагитуллин. Успехи химии, 33, 361 (1964).
2. К. Be й г ан д-Х н л ь г ет а г. Методы эксперимента в органической хводи. М., «ХцйЙя». 1.868, с. 543.
3. Ч. Дж. ОверДергер, Ж-П. Анселы, Дж. Г. Л о.м б а р ди и о, Органические соединенна со связями азот —азот. Л., Химия, 1970, с. 1(1
4. А. Н. Кост, Р. С. Сагитуллин. Ж- общ. химии, 33, 867 (1863).
2-М-МО₽ФОЛИЛ-2тОКСО-5-МЕТИЛ-5-МЕТОКСИМЕТИЛ-1,3,2-Д И О КСАФОСФОР И НАН
А. В. БОГАТСКИЙ, А. А. КОЛЕСНИК. Г. Д. БУТОВА
CwHjoNOsP
СН3ОСН2
/С СНз
М. в. 295,35
2-Ы-Морфолил-2-оксо-5-метил-5-метоксиметил-1,3,2-диокса-фосфоринан получен нами впервые взаимодействием 2-хлор-5-метил-5-метоксиметил-1,3,2-диоксафосфоринана с морфолином.
Это соединение проявляет умеренную противоопухолевую активность.
СХЕМА СИНТЕЗА
снаосн,
СНз "сн*~°
СН2-ОЧ .О / х
+ 2HN о V Cl \_-х
СН3ОСНа
>с СНз
\) + HlZ
ХО • НС1
Характеристика основного сырья
2-Хлор-5-метил-5-метоксиметил-1,3,2-диоксафосфоринан, d420 = 1,284; п*> = 1,460, получение см. [1].
Морфолин, МРТУ 6—09—443—63. ч.
201
Условия получения
В трехгорлой колбе емкостью 0,5 л, снабженной мешалкой, обратным холодильником и капельной воронкой, к 15,0 г (0,16 моль) морфолина в 400 мл абсолютного эфира при перемешивании и охлаждении до минус 3° по каплям прибавляют раствор 18,0 г (0,08 моль) 2-хлор-2-оксо-5-метил-5-метоксимс-тил-1,3,2-диоксафосфоринана в 20 мл абсолютного эфира. При этом происходит медленное выделение солянокислого морфолина. Смесь нагревают на водяной бане 1—2 часа. Осадок отделяют, эфир отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 185°/1 мм.
Выход 2-М-морфолил-2-оксо-5-метил-5-метоксимегил-1,3,2-диоксафосфоринана равен 17 г (80%); rf420 = 1,2262, =
= 1,4820.
2-М-Морфолил-2-оксо-5-метил-5-метоксиметил-1,3,2-диокса-фосфоринан -представляет собой вязкое вещество, кристаллизующееся при стоянии.
Найдено, %: К—5,13; Р—11,93. C10H20NO5P. Вычислено, %: N—5,27; Р—11,65.
ЛИТЕРАТУРА
1. А. В. Богатский, А. А. Колесник, Ю. Ю. С а м и т о в, Т. Д. Бутова. Ж. общ. химии, 5, 1105 (1967).
2-НАФТОЛ-3,6-ДИСУЛЬФОКИСЛОТА, ДВУНАТРИЕВАЯ СОЛЬ
P-со л ь
Н. Н. ДЫХАНОВ, Р. Ф. ВИДЕНИНА, И. В. ПОКОТЫЛО, Т. В. ПЕРОВА, В. И. БАЛАКИН, П. Д. ЯКУХНЫИ
NaO3S-
CioHeN'aaOySa
М. в. 348,26
Сульфирование 2-нафтола при 100—150° приводит к образованию смеси 2-нафтол-6-сульфокислоты (кислота Шеффера), 2-нафтол-3,6-дисульфокислоты (Р-кислота) и 2-нафтол--6,8-дисульфокислоты (Г-кислота) . В зависимости от условий проведения процесса ту или другую нафтолсульфокислоту получают в преобладающем количестве [1—4], Наибольший выход Р-кислоты (84—85%) достигается при сульфировании 2-нафтола 10%-ным олеумом при 120—125° в течение 18 часов или при 130—135° в течение 12 часов; в этих условиях выход кислоты Шеффера составляет около 9% [5]. При выделении Р-кислогы из сульфомассы разбавлением ее водой и насыщением сухим хлористым натрием Все компоненты сульфомассы превращаются в натриевые соли, которые в виде примесей содержатся в технической Р-соли. Для очистки Р-соль переосаждают из водного раствора хлористым натрием, затем перекристаллизовывают из воды. Однако однократная очистка позволяет снизить суммарное содержание соли Шеффера и Г-соли лишь на 1,5—2%, а многократная очистка приводит к большим потерям продукта.
Основываясь на том, что Г-соль хорошо растворима в кипящем 80—90%-ном этаноле [5]. а соль Шеффера (как соль моносульфокислоты) хорошо растворяется в разбавленном 203
Сййртё, мы разработали способ очистки технической Р-солН путем экстракции из нее примесей 85%-ным этанолом с последующей перекристаллизацией остатка из 50%-ного спирта. После однократной очистки переосажденной Р-соли по этому способу содержание основного вещества в ней составляет не менее 95%.
Характеристика основного сырья
Р-соль, техн., влажная паста, содержащая не менее 60% смеси сухих веществ.
Кальций углекислый, ГОСТ 4530—66, ч.
Натрий хлористый, ГОСТ 4233—66> ч.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт, 96%-ный, ГОСТ 5962—69.
Условия очистки
Переосаждение. Растворяют 100 г влажной пасты техни ческой Р-соли в 250 мл воды, нагревают до 60—65° и прибавляют небольшими порциями 19—20 г карбоната кальция до достижения pH 8—8,5 по универсальному индикатору. Полученный темно-коричневый раствор нагревают до 92—95°, прибавляют 0,6 г гидросульфита натрия и 10 г активированного угля, перемешивают 30 минут и фильтруют в горячем состоянии. К фильтрату при энергичном перемешивании и температуре 92—95° осторожно приливают концентрированную соляную кислоту до слабокислой реакции по конго (~30 мл кислоты), затем в один прием присыпают 30 г хлорида натрия и, не прекращая перемешивания, охлаждают до 8—10°.
Выделившийся осадок отфильтровывают с отсасыванием, промывают на фильтре охлажденным до 8—10° 15%-ным раствором хлорида натрия до получения бесцветного фильтрата, тщательно отжимают и сушат в вакуум-сушильном шкафу при 70—80°. Получают Р-соль в виде белого или слегка сероватого порошка, содержащего 39—40 г сухого вещества.
Экстракция и перекристаллизация. В колбу емкостью 250 мл, снабженную мешалкой и обратным холодильником, помещают 40 г сухой Р-соли, прибавляют тройное (по весу) количество 85%-наго этанола, кипятят 15 минут и по возможности быстрее фильтруют в горячем состоянии. Осадок промывают на фильтре 60 мл кипящего 85%-ного этанола, присоединяя промывной спирт к основному фильтрату (см. примечание). Спиртовую экстракцию повторяют еще раз.
Сырой осадок Р-соли после экстракции (36—37 г) растворяют в 12-кратном (по весу) количестве горячего 50%-ного этанола, прибавляют 2 г активированного угля, кипятят 10 ми. нут и фильтруют в горячем состоянии. К горячему фильтра-204
ту прибавляют несколько кристалликов гидросульфита натрия и половинный объем 96%-кого этанола и оставляют охлаждаться во льду до 3—5°.
Выделившиеся бесцветные игольчатые кристаллы отфильтровывают, тщательно отжимают на фильтре, промывают 50 мл холодного 96%-ного этанола, еще раз отжимают и су шат.
Выход Р-соли равен 29—31 г (~50% от веса исходной технической соли в пересчете на смесь сухих веществ); содержание Г-соли — 2,5—3,0%, содержание соли Шеффера—1,8— 2,0%.
Примечание.
При охлаждении спиртового экстракта до 3—5° получают 4—5 г кристаллов чистой Г-соли. Из маточного раствора отгоняют 85—87%-ный этанол и используют его в следующем опыте.
ЛИТЕРАТУРА
1. Р. Griess. Вег. 13, 1956 (1880).
2. Герм. пат. 33229 и 33916 (1878); Frdl, 1, 377 и 378.
3. И. И. Воронцов. Ж. прикл. химии, 24, 332 (1951).
4. И. И. Воронцов, П. Н. Соколов. Аиил.-крас. пром-сть, 4, 17 (1934).
5. Н. Доналдсон. Химия и технология соединений нафталинового ряда. М., ГНТИХимлит, 1963, с. 349.
2-НИТРО-4.5-ДИБУТОКСИАНИЛИН
В. М. ДЗИОМКО. А. В. ИВАЩЕНКО
у>\^ОС4Н9
I II
OJNX'^/4OC4He
C14H22N2O4 М. в. ,288,34
2-Нитро-4,5-дибутоксианилин применяется в синтезе поли-гетероциклических систем [1].
В литературе описано получение этого соединения [1, 2] гидролизом 2-нитро-4,5-дибутоксиацетанилида, который получают нитрованием 3,4-дибутоксиацетанилида. Последний получают ацилированием 3,4-дибутоксианилина.
Нами разработаны новые условия ацилирования 3,4-дибутоксианилина, гидролиза 2-нитро-4,5-дибутоксиацетанилида и уточнены условия нитрования 3,4-дибутоксиацетанилида, благодаря чему выход конечного продукта повышен с 55% 100% в расчете на 3,4-дибутоксианилин.
СХЕМА СИНТЕЗА
H2N\^\^OC4Hg
j| (С ПзСО)3О
Х/ХОС4Нв
CH3COHN\ ,ОС4Н9
Y« hn°3--
206
CHaCOHN< ,ОС4Н9
YY +H.Q(H*)
H,N Wx/OC4H9
O2N/'^/'xOC4H9
Характеристика основного сырья
3,4-Дибутоксианилин, т. кип. 193—195°/8 мм; получение см с. 90 этого сборника.
Азотная кислота, ГОСТ 4461—67, ч.
Уксусный ангидрид, ГОСТ 5815—69, ч.
Условия получения
Синтез 2-нитро-4,5-дибутоксиацетанилида. В колбу емкостью I л, снабженную обратным холодильником, помещают 98,6 г (0,42 моль) свежеперег.нанного 3,4-дибутоксианилина, 200 мл бензола и 50 г (0,49 моль) уксусного ангидрида. Реакционную смесь нагревают на водяной бане до кипения и кипятят 1 час. Затем заменяют обратный холодильник на прямой и отгоняют бензол при пониженном давлении. Твердый остаток измельчают и растворяют в 630 мл уксусной кислоты.
Полученный раствор 3,4-дибутоксиацетанилида помещают в трехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром и капельной воронкой. Колбу охлаждают льдом и прибавляют при перемешивании из капельной воронки 295 мл азотной кислоты с такой скоростью, чтобы температура реакционной массы не превышала 15°. Перемешивание продолжают при комнатной температуре еще 2 часа, после чего прибавляют 1 л воды, хорошо размешивают смесь и фильтруют.
Осадок на фильтре промывают водой до нейтральной реакции промывных вод и сушат в вакуум-эксикаторе над твердым едким натром до постоянного веса.
Выход 2-нитро-4.5-дибутоксиацетанилида равен 120 г (100%). Вещество представляет собой мелкие иголочки лимонно-желтого цвета, т. пл. 83—84°.
По литературным данным, т. пл. 83—84° (!]; 85° [2].
Получение 2-нитро 4,5-дибутоксианилина. В двухгорлую колбу емкостью 1 л, снабженную обратным холодильником капельной воронкой, помещают 30 г (0,091 моль) 2-нитро-4,5-дибутоксиацетанилида, 40 мл соляной кислоты и 400 мл эта-207
нола. Реакционную смесь нагревают на водяной бане до кипения и кипятят 3 часа, после чего из капельной воронки по каплям прибавляют 50 мл аммиака. Реакционной массе дают охладиться до комнатной температуры, выпавшие при этом кристаллы отфильтровывают и сушат при 70—80° до постоянного веса.
Выход 2-нитро-4.5-дибутоксианилина равен 26 г (100%). По внешнему виду вещество представляет собой бледио-оран-жевые кристаллы с т. пл. 107—108°.
По литературным данным [1, 2], т. пл. 107—108°.
ЛИТЕРАТУРА
1. S. Kawai, Y. Okawa. Y. Yada, H. Hosoi, T. Murako-shi, J. Yajitna. Nippon Kagaku Zasshi, 80, 551 (1959).
2. F a r g h e r. J. Chem. Soc, 1920, 869.
5-(5-НИТРОТЕНИЛИДЕН)РОДАНИН
А. А. ПОНОМАРЕВ, | 3. В. ТИЛЬ
OC-NH
NOs-\ / — СН=С CS
CgH^NjOaSs
М. в. 272,32
Конденсация роданина с альдегидами может быть осуществлена в среде ледяной уксусной кислоты в присутствии уксусного ангидрида и ацетата натрия [1, 2]; в спиртовой среде в присутствии хлористого аммония [1, 2], уксусной кислоты или ацетата натрия [3]; в водной среде в присутствии ацетата натрия [4],
5-(5-Нитротенилиден) роданин ранее ие был описан и получен нами при конденсации 5-иитро-2-тиофенальдегида с роданином в кислой среде.
СХЕМА СИНТЕЗА
OC-NH
+ I I
HgC CS
Y
______ OC-NH no2-I^J-ch=c cs+H2o s Y
14 Зак- 1164
209
Характеристика основного сырья
5-Нитро-2-тиофенальдегид, т. кип. 118—120°/3 мм, т. >пл. 74° (см. примечание 1).
Роданин, т. пл. 168—170° (ом. примечание 2).
Уксусная кислота ледяная, ГОСТ 61—51.
Уксусный ангидрид, ГОСТ 5815—52, ч.
Натрий ацетат плавленый, ВТУ РУ 876—53, ч.
Условия получения
В круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, помещают 5 г (0,037 моль) роданина, 5,9 г (0,037 моль) 5-нитро-2-тиофенальдегида, 6 г ацетата натрия, 20 мл ледяной уксусной кислоты и 6 г уксусного ангидрида. Реакционную смесь нагревают 1 час на кипящей водяной бане, причем образуется осадок кирпичного цвета. Выпавшие кристаллы отфильтровывают, промывают водой до нейтральной реакции промывных вод, затем спиртом и эфиром.
Выход 5- (5-нитротенил идеи) роданина равен 9,1 г (91,9%), т. пл. 238—240°. Вещество хорошо растворяется в диметил-формамиде, диоксане, ацетоне, горячем спирте; нерастворимо в воде, дихлорэтане, эфире.
Примечания:
1. 5-Нитро-2-тиофенальдегид получают нитрованием в уксусном ангидриде 2-тиофенальдегнда с последующим гидролизом образующегося 5-нит-ро-2-тенилидендиацетата )5].
2. Роданин получают из монохлоруксусиой кислоты и роданистого аммония [6].
ЛИТЕРАТУРА
1. F. Allan, G. Allan. Canad. J. Chem. 36, 11, 1579 (1958); РЖхнм., 57165 (1959).
2. F. Allan, G. Allan. Recueil trav. chlm., 78, 5, 375 (1959); РЖХим., 1261 (1960).
3. R. Neu. Ber„ 90, 11, 2638 (1957); РЖХим., 32524 (1958).
4. А. А. Пономарев, П. С. Д и ч к о в а. Уч. Зап. Сарат. ун-та, 42, 45 (1955).
5. Р. F о win ari. J. Р. С h a n е. Bull. Soc. Chlm. France, 478 (1963); С. A., 159, 1569e (1963).
6. Л. M. Кульбер г. Синтезы органических реактивов для неорганического анализа. М.-Л., Госхимиздат, 1947.
5-НИТРОФЕНОЛ (2-АЗО-1')-2'-(Р-ПРОПИОНИЛ-ГИДРАЗИНО)НАФТАЛИН И 5-НИТРОФЕНОЛ (2-АЗО-Г)-2'-(р-БУТИРИЛ ГИДРАЗИНО)НАФТАЛИН
К. А. ДУНАЕВСКАЯ, К. Е. КАЛИНИНА, В. М. ДЗИОМКО
/x/N0’
I I
n^Y
NHNHCOCH8CH3
C19H17N5O4
C2oH19N504
M. в. 379,38
N I
II OH
N
f 4|Z NHNHCOCHgCH2CH8
M. в. 393.41
5-Нитрофен>ол (2-азо-И) -2'- (p-пропионилгидразино) нафталин и 5-нитрофенол(2-азо-1')-2'-(₽-бутирилгидразино)нафталин являются избирательными реагентами при определении цинка. Нами реактивы получены впервые.
М* 211
СХЕМА СИНТЕЗА
NH2—NH2 • Н20
nh-nh2
h2n
NaNO2 HCI ~+
NHNHCOR
^\/k/NHNHC0R
где R = C2H5—; C3H7—
Характеристика основного сырья
P-Нафтол, ГОСТ 5857—51, ч.
Гидразии-гидрат, ГОСТ 5832—51, ч.
Пропионовый ангидрид, МРТУ 6—09—1938—64, ч.
Масляный ангидрид, МРТУ 6—09—5541—68, ч.
Бензол, ГОСТ 8448—61, ч.
5-Нитро-2-аминофенол, СТУ 79—348—х—60, ч.
Натрий нитрит, ГОСТ 4197—48, х. ч.
Соляная кислота, ГОСТ 3118—46, пл. 1,18, ч. д. а.
Диметилформамид, СТУ 79—606—х—60, ч.
Этиловый спирт, ГОСТ 5963—51.
Серный эфир, ГОСТ 6265—52.
Условия получения
Синтез fi-нафтилгидразина. В двугорлую колбу емкостью 0,5 л, снабженную холодильником и трубкой для ввода тока 212
азота, загружают 100 г (0,694 моль) р-нафтола, 250 МЛ (5 моль) гидразин-гидрата. Смесь кипятят (температура в бане 140°) в токе азота 40—45 часов (см. примечание 1). Затем раствор выливают в 1,5 л 5%-ного раствора едкого кали. Осадок отфильтровывают, промывают ~300 мл 5%-ного раствора едкого кали и ~300 мл воды, быстро смешивают с 500 мл этилового спирта и фильтруют. Осадок на фильтре промывают ~200 мл серного эфира и сушат в эксикаторе, заполненном азотом, над хлористым кальцием; хранят в ампулах.
Выход p-нафтилгидразина равен 60—65 г (54,7—59,2%). По внешнему виду это мелкокристаллическое вещество кремового цвета; т. пл. 120—121°.
По литературным данным [1]- т. пл. 124—125°.
Получение пропионил-^-нафтилгидразина. В стеклянном стакане емкостью 100 мл смешивают 3,16 г (0,02 моль) p-нафтилгидразина с 20—30 мл бензола и при перемешивании прибавляют 1,56 мл (0,015 моль) пропионового ангидрида. По мере прибавления пропионового ангидрида происходит разогревание. Реакционную массу выдерживают 1 час и фильтруют; осадок на фильтре промывают ~ 10 мл бензола и перекристаллизовывают из разбавленного этилового спирта (40 мл спирта и 20 мл воды).
Выход пропионил-р-нафтилгидразина равен 1,4 г (34,4%): т. пл. 158—159°; вещество по внешнему виду представляет собой чешуйки белого цвета.
Найдено. %: С—72,69; 72,83; Н—6,70; 6,76; N—13,28; 13,30. C13H14N2O. Вычислено, %: С—72,89; Н—6,54; N—13,09.
Бутирил-^-нафтилгидразин синтезируют по аналогичной методике из 3,16 г (0,02 моль) p-нафтилгидразина в 20—30 мл бензола и 2,0 л|л< (0,012 моль) масляного ангидрида. После перекристаллизации из разбавленнного этилового спирта (100 мл спирта и 40 мл воды) получают 1,4 г (31%) бутирил-p-нафтилгидразина в виде чешуек белого цвета с т. пл. 144— 145°
Найдено, %: С—73,87; 74,09; Н—7,03; 7,08; N—12,12; 12,23. CuHioNsO. Вычислено, %: С—73,68; Н—7,01; N—12,23.
Получение 5-нитрофенол(2-азо-]')-2'-($-пропионилгидрази-но)нафталина. В стакане емкостью 100 мл смешивают 0,9 с (0,006 моль) 5-нитро-2-аминофенола с 16,5 мл воды и 3,6 мл соляной кислоты. При перемешивании прибавляют 0,45 •? (0,0065 моль) нитрита натрия в 3 мл воды и выдерживают массу при диазотировании 30 минут. Полученное диазосоединение сочетают с пропионил-р-нафтилгидразином. Для этого в стакан емкостью 0,5 л, снабженный, мешалкой, загружают 1,2 а пропионил-р-нафтилгидразина (0,006 моль) в 60 мл диметил-формамида и прибавляют диазосоединение. Массу выдерживают при перемешивании 24 часа. Выделившийся осадок от
213
фильтровывают, промывают водой и перекристаллизовывают из смеси 200 мл диметилформамида и 20 мл воды.
Выход 5-нитрофенол (2-азо-Г)-2'-(Р-пропиопилгидразино)-нафталина равен 1 г (45,4% в расчете на б-нитро^-аминофе-нол) по внешнему виду это мелкокристаллическое вещество красного цвета с т. пл. 201—203° (см. примечание 2).
Найдено, %: С—58,30; 58,16; Н—5,52; 5,87; N—18,35; 18,52.
CioHirNsO*- (CH3)2NCHO. Вычислено, %: С—58,40; Н—5,30; N—18,38.
5-Нитрофенол(2-азо - Г)-2'($-бутирилгидразино)нафталин, синтезируют по аналогичной методике из 1,3 г (0,006 моль) бутирил-р-нафтнлгидразина в 60 мл диметилформамида и полученного диазосоедннения. После перекристаллизации из смеси 100 мл диметилформамида и 20 мл воды получают 1,2 г (44,1 %) 5-нитрофенол (2-азо-1') ^'-(р-бутирилгидразино) нафталина в виде мелкокристаллического вещества красного цвета с т. пл. 196—197° (см. примечание 2).
Найдено, %: С—59,84; 59,77; Н—5,77, 5,76; N—17,93; 17,84.
C2oH19N504-(CH9)2NCHO. Вычислено, %: С—59,22; Н—5,57; N—18,02.
Примечания:
1. Азот последовательно проходит через пустую склянку, склянку, заполненную щелочным раствором пирогаллола, и пустую.
2. Методом газовой хроматографии найдено, что реагент содержит молекулу диметилформамида.
ЛИТЕРАТУРА
1. Е. Fischer. Ann., 232, 242 (1886).
8 НИТРОХИНАЛЬДИН
В. М. ДЗИОМКО, И. А. КРАСАВИН, Б. В. ПАРУСНИКОВ, Ю. П. РАДИН
I I I ^/XN^4CHS no2
С10Н8М2О2 М. в. 188,19
8-Нитрохинальдин получают циклизацией о-нитроанилина в условиях реакции Добнера-Миллера [1—3] или нитрованием хинальдина [4—6].
При циклизации о-нитроанилина в качестве второго компонента реакции применяют паральдегид [2, 3], кротоновый альдегид [3], винилбутиловый эфир [1]. Выход 8-нитрохинальдина составляет от 20% [2] до 44% [1]- при использовании в качестве окислителя мышьяковой кислоты — 62% [3].
Нитрование хинальдина осуществляют нагреванием предварительно приготовленного нитрата хинальдиния с серной кислотой [4], действием нитрующей смеси [6] или нитрата калия [5] на раствор хинальдина в серной кислоте.
При нитровании наряду с 8-нитрохинальднном (выход 33—35%) образуется 5-нитрохинальдин (55—59%) [4, 6]. Для разделения изомеров используют различие в их основности: при частичной нейтрализации кислых растворов в первую очередь выделяется 8-нитрохинальдин.
Предлагаемый нами способ нитрования, являющийся видоизменением описанного ранее метода [5], заключается в добавлении нитрата аммония к раствору бисульфата хинальди-иия в концентрированной серной кислоте. Поскольку бисуль фат легко поддается очистке путем кристаллизации, в качестве исходного сырья можно использовать наиболее доступный
215
И дешевый хинальДйн коКсохймйческоТо пройсхожденйя, Содержащий обычно много примесей. Для разделения смерй 5-и 8-нитрохинальдинов применена кристаллизация из этанола, при этом из раствора выпадает хуже растворимый 8-изойер с выходом ~33%.
СХЕМА СИНТЕЗА
+ H2SO4
СНа
I I! I HSO4-
H2SO4
NH4lto,
NO2
I
Z\/4
I II I -I II I + nh4hso4h H2O
\ снэ 4'CH3
NO,
Характеристика основного сырья
Хинальдин, ВТУ 2893—51, ч., перегнанный.
Хлороформ, ГОСТ 3160—51, ч. д. а.
Серная кислота, ГОСТ 4204—66, ч.
Этиловый спирт, ГОСТ 8314—57, гидролизный.
Аммоний азотнокислый, ГОСТ 3761—65, ч., высушенный.
Условия получения
Синтез бисульфата хинальдиния. В стакан емкостью 1 л, снабженный мешалкой, термометром и капельной воронкой, помещают 143 г (1 моль) свежеперегнанного хинальдина и 600 мл хлороформа. При охлаждении льдом и энергичном размешивании добавляют по каплям 105 г концентрированной серной кислоты при 15—20°. Размешивание продолжают еще 30 минут, затем осадок отсасывают, отжимают стеклянной пробкой, промывают на фильтре хлороформом и сушат при 60°.
Выход технического бисульфата хинальдиния равен 230— 240 г (95—99%), т. пл. 193—197°.
Для очистки техническую соль кристаллизуют из 1300 мл этилового спирта с применением 10 г активированного угля.
Выход очищенного бисульфата хинальдиния равен 170— 187 г (70,6—77,6%), т. пл. 208—212° (см. примечание).
216
Получение 8-нитрохинальдина. Ё трехгорлую колбу емкостью 0,5 л, снабженную мешалкой и термометром, помещают 120,6 г (0,5 моль) сухого перекристаллизованного бисульфата хинальдиния и 200 мл 95—98 %-ной серной кислоты. Суспензию размешивают до образования прозрачного раствора, охлаждают льдом и добавляют небольшими порциями 48 г (0,6 моль) сухого растертого азотнокислого аммония, поддерживая температуру не выше 40°. Перемешивание продолжают еще один час, затем выливают нитромассу на 750 г колотого льда, при размешивании и охлаждении льдом добавляют 650—700 мл водного аммиака до щелочной реакции, поддерживая температуру смеси не выше 20°. Суспензию размешивают при охлаждении еще 1—2 часа, после чего осадок отсасывают, промывают на фильтре водой и отжимают.
Влажный осадок размешивают с 400 мл хлороформа до растворения, водный слой отделяют, а органический промывают 5%-,ным раствором едкого натра 3 раза порциями по 100 мл и водой 4 раза порциями по 50 мл, подсушивают безводным сульфатом натрия, размешивают с 5 г активированного угля и фильтруют. При нагревании на водяной бане отгоняют хлороформ, остатки его удаляют в вакууме водоструйного насоса.
К полученной смеси 5- и 8-нитрохи.нальдинов прибавляют 600—650 мл этилового спирта, нагревают с обратным холодильником на водяной бане до полного растворения вещества, кипятят с 5 г активированного угля и горячий раствор фильтруют. При медленном охлаждении из фильтрата выпадают желтые кристаллы 8-нитрохинальдина. Через 5—10 часов их отсасывают, промывают этиловым спиртом и высушивают.
Выход 8-нитрохинальдина равен 29—31 г (31—33% в расчете на бисульфат хинальдиния), т. пл. 137—139°.
По литературным данным, т. пл. 129—131° [2]; 130° [5]; 137’ [3, 4, 6]; 139° [1].
Примечание.
Состав соли, определенный титрованием ее водного раствора 0,1 н. раствором едкого натра по фенолфталеину, соответствует формуле GoHsN • H2SO4.
ЛИТЕРАТУРА
1. Б. А. Порай-Кошиц, Л. С. Эфрос, В. Н. Вертки и а, В. В. Луцеико. Ж. общ. химии, 24, 895 (1954).
2. R. Roth, Н. Erlenmeyer. Helv. chlm. acta, 37, 1064 (1954).
3. К. Made) a. J. prakt. chem., 17, 97 (1962).
4. О. Doe bn er, W. Miller Ber., 17, 1698 (1884).
5. D. L. Ha mmick. J. Chem. Soc., 1926, 1302.
6. M. А. Альперович, И. К. У ш e н к о. Ж. общ. химии, 29, 3384 (1959).
217
Y-ОКСИКАРБОНОВЫЕ кислоты, натриевые соли
В. А. СЕДАВКИНА, А. Г. БАСИНА
CBHr-CH-CHa-CH11-COONa
I ОН
C,H13NaO3 М. в. 168,17
С4Н9—CH-CHa-CHj-COONa
I ОН
CBHi5NaO3 М. в. 182,20
СвНп- СН- СН,-CHg- COONa
ОН
С9Н17МаОз М. в. 196,25
В литературе описан метод получения натриевой соли у-оксимасляиой кислоты из у-бутиролактона [1].
Нами получены натриевые соли у-оксигептановой, у-оксиоктановой, у-оксинонановой кислот двумя методами. Первый метод [1] заключается в каталитическом гидрировании эфиров у-кетокарбоновых кислот в присутствии никеля Реиея .при 120° и давлении 100 атм. При этом в результате внутримолекулярной переэтерификации этиловых эфиров у-оксикарбоновых кислот получаются соответствующие у-лактоиы. Последние обработкой водно-спиртовым раствором натриевой щелочи превращаются в натриевые соли у-оксикарбоиовых кислот. Второй метод, предложенный нами, заключается в щелочном гидролизе этиловых эфиров у-кетокарбоиовых кислот и последующем гидрировании полученных таким образом натриевых солей у-кетокарбоновых кислот. Этот метод является 'более удобным, так как позволяет избежать выделения лактоиа и дает лучшие выходы солей. 218
СХЕМА СИНТЕЗА
Метод А
R—С—СН>—СН2—СООС2Н5 ;
II N1R
о
— R-CH-CH,-CHgCOOC2H5 -> он
---1 NaOH
1=0-----” R—СН2—СН2—CHt—COONa
4q/ он
Метод Б
R-C-CH2-CH2-COOCaH6
I! О
— R—С—CH2CH2COONa >R-CH-CH2CH2—COONa,
II N.R !
о он
где R = C3H7—; С4Н9—; С5НП—
Характеристика основного сырья
Этиловый эфир у-кетогептановой кислоты, т. кип. 94—967
2 мм, 1,4338; получение см. [2].
Этиловый эфир у-кетооктаиовой кислоты, т. кип. 98—100°/
3 мм, Дд = 1,4340; получение см. [2].
Этиловый эфир у-кетоионаиовой кислоты, т. кип. 107—1087
4 мм, = 1,4330; получение см. [3].
Натр едкий, ГОСТ 4203—65, ч.
Этиловый спирт, ГОСТ 5962—51.
Диоксан, ГОСТ ТУ 3112—52.
Никель Ренея, полученный выщелачиванием сплава никеля и алюминия [4].
Водород электролитический.
Условия получения
Синтез у-пропилбутиролактона. Метод А. Во вращающемся стальном автоклаве емкостью 150 мл гидрируют 10 г
219
(0,06 моль) этилового эфира у-кетогептановой кислоты, растворенного в 50 мл абсолютного этилового спирта, в присутствии 2 г никеля Ренея при 120° и давлении 115 атм до поглощения 1,344 л водорода. Затем катализатор отфильтровывают, растворитель отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 105—10778 мм,
Выход у-пропилбутиролактона равен 8 г (94%).
По литературным данным [5], т. кип. 111/°11 мм.
Получение натриевой соли у-оксигептановой кислоты. Метод А. В круглодонной колбе емкостью 150 мл с обратным холодильником, к 70 мл этилового спирта добавляют 1 мл воды и 1,5 г измельченного едкого натра и нагревают до полного растворения щелочи. Затем медленно при постоянном перемешивании прибавляют 5 г (0,037 моль) у-пропилбутиролак-тона и кипятят реакционную смесь 30 минут. Растворитель отгоняют, остаток через некоторое время закристаллизовыва-ется. Кристаллы тщательно промывают диоксаном.
Выход натриевой соли у-оксигептановой кислоты равен 5 г (89%).
Метод Б. В колбу емкостью 100 мл вносят 15 г (0,09 моль) этилового эфира у-кетогептановой кислоты, 3,3 г (0,082 моль) едкого натра, 50 мл дистиллированной воды и нагревают с обратным холодильником на водяной бане 5—6 часов до полного растворения эфира. Раствор переносят в автоклав, добавляют 3—4 г никеля Реиея и гидрируют при 120° и начальном давлении водорода 130 атм до поглощения 2,016 л водорода. Затем катализатор отфильтровывают, воду отгоняют при пониженном давлении. Закристаллизовавшийся остаток тщательно промывают диоксаном.
Выход натриевой соли у-оксигептановой кислоты равен 14 г (93,3%), т. пл. 135—136°.
Найдено, %: С—49,86; 49,79; Н—8,02; 8,06; Na—13,14. C7H13NaO3. Вычислено, %: С—50,00; Н—7,73; Na—13,69.
Получение натриевой соли у-оксиоктановой кислоты.
Метод А. По аналогичной методике из 15 г (0,079 моль) этилового эфира у-кетооктановой .кислоты получают 10 г (87,7%) у-бутилбутиролактона, т. кип. 112—114°/7 мм, п™ = — 1,4438. По литературным данным [6], т. кип. 122°/15 мм, п™ = 1,4430.
Из 5 г (0,035 моль) у-бутилбутиролактона и 1,5 г (0,037 моль) едкого натра получают 4,7 г (82,7%) натриевой соли у-оксиоктановой кислоты.
Метод Б. Из 15 г (0,079 моль) этилового эфира у-кетоокта-ново|й кислоты и 3,2 г (0,08 моль) едкого натра получают 220
14,3 г (94%) натриевой соли у-оксиоктановой кислоты, т. пл. 137—139°.
Найдено, %: С—52,55; 52,62; Н—8,60; 8,56; Na—12,40. CsHisNaOg. Вычислено, %; С—52,75; Н—8,24; Na—12,63.
Получение натриевой соли у-оксинонановой кислоты.
Метод А. По аналогичной методике из 10 г этилового эфира у-кетононановой кислоты получают 7,1 г (92,2%) у-амилбу тиролактона, т. кип. 134—134,5712 мм, n2g = 1,4468, rf420 — = 0,9662. По литературным данным [7], т. кип. 136713 мм, п2° = 1,4462, rf420 = 0,9672.
Из 5 г (0,032 моль) лйктоиа и 1,4 г (0,035 моль) едкого натра получают 4,9 г (80%) натриевой соли у-оксинонановой кислоты.
Метод Б. Из 15 г (0,079 моль) этилового эфира у-кетононановой кислоты и 3,1 г (0,079 моль) едкого натра получают 13,8 г (86%) натриевой соли у-оксинонановой кислоты, т. пл. 165—166°
Найдено, %: С—54,78; Н—8,63; Na—11,59. CgHnNaOs. Вычислено, %: С—55,10; Н—8,67; Na—11,73.
ЛИТЕРАТУРА
1. А. П. Арендорук, А. А. Серебрякова, А. П. Сколдинов. Мед. пром-сть СССР, 5, 6—8 (1963).
2. В. Г. Бухаров, Т. Е. Позднякова. Изв. АН СССР, сер. хим., 6, 1108 (1960).
3. А. А. Пономарев. Синтезы и реакции фурановых веществ. Саратов, Изд-во СГУ, 1960, с. 122.
4. Синтезы органических препаратов, сб. 3. М., ИЛ, 1952, с. 338.
5. Н. Rupe, М. Ron us, W. Lotz. Вег.. 35, 4272 (1903).
6. I. Colonge, P. Lasfargues. Bull. Soc. Chim. France, I, 177 (1962).
7. B. Rothstein. Bull. Soc. Chim. France, 2, 1936 (1935).
у-ОКСИПИМЕЛИНОВАЯ КИСЛОТА, ДВУНАТРИЕВАЯ СОЛЬ
В. А. СЕДАВКИНА, А. Г. БАСИНА
NaOOC—СНа—СН2—СН—СН,—СН2—COONa
ОН
C/HioNaaOs М. в. 240,15
Двунатриевая соль у-оксипимелиновой кислоты получена нами щелочным гидролизом диэтилового эфира у-кетопиме-линовой кислоты и последующим гидрированием полученной Динатриевой соли у-кетопимелиновой кислоты. Исходный диэтиловый эфир у-кетопимелиновой кислоты получен расщеплением фуранового цикла в этиловом эфире фурилакриловой кислоты по несколько измененной нами методике {1].
СХЕМА СИНТЕЗА
J-CH=CH-COOC,H5 -С1н£н—
Н5С,ООС-СН2-СН2-СО-СН2-СН,-СООС2Н6
—— NaOOC—СН2—СН,—СО—СН2—СН,COONa
-> NaOOC—СНа—СН2—СН—СН,—СН2—COONa
I
ОН
Характеристика основного сырья
Этиловый эфир фурилакриловой кислоты, т. кип. 233—235°; получение см. [2].
Натр едкий, ГОСТ 4203—65, ч.
Этиловый спирт абсолютированный, ГОСТ 9674—61.
Эфир серный, ГОСТ 6265—52.
222
Никель Ренея, получение см. [3].
Водород электролитический.
Калий углекислый, ГОСТ 4221—53, ч.
Условия получения
Синтез диэтилового эфира у-кетопимелиновой кислоты. В круглодонную колбу емкостью 250 мл, снабженную обратным холодильником с хлоркальциевой трубкой, помещают 40 г (0,22 моль) этилового эфира фурилакриловой кислоты и растворяют в 150 мл абсолютного этилового спирта, насыщенного сухим хлористым водородом до 15%-ной концентрации (26,4 г НС1). Реакционную смесь нагревают на кипящей водяной бане 3 часа. Затем заменяют обратный холодильник на нисходящий и отгоняют большую часть растворителя. К остатку приливают 150—200 мл насыщенного раствора карбоната калия и 50 мл серного эфира. Смесь встряхивают, эфирный слой отделяют, а водный экстрагируют эфиром 3—4 раза порциями по 30—40 мл. Объединенные эфирные вытяжки сушат безводным углекислым калием, эфир отгоняют, а остаток перегоняют при уменьшенном давлении, собирая фракцию с т. кип. 139—140°/5 мм, п2£ = 1,4462.
Выход диэтилового эфира у-кетопимелиновой кислоты равен 42,5 г (73%).
По литературным данным [1], т. кип. 182720 мм, п$ = = 1,4432.
Получение двунатриевой соли у-оксипимелиновой кислоты. В круглодонной колбе емкостью 150 мл к раствору 11 е едкого натра в 100 мл дистиллированной воды прибавляют 30 ? (0,14 моль) диэтилового эфира у-кетопимелиновой кислоты и нагревают до полного растворения эфира (4—5 часов). Раствор переносят во вращающийся автоклав емкостью 250 мл, добавляют 4 г никеля Ренея и ведут гидрирование при 100° и начальном давлении водорода 50 атм до поглощения 3,136 л водорода. Затем катализатор отфильтровывают, растворитель отгоняют, а остаток тщательно промывают этиловым спиртом порциями по 20—30 мл.
Выход двунатриевой соли у-оксипимелиновой кислоты равен 22 г (75,8%), т. пл. 226—228°.
Найдено, %: С—38,13; Н—4,88; Na—20,20. CjHioNaaOs. Вычислено, %: С—38,18; Н—4,54; Na—20,9.
ЛИТЕРАТУРА
1. М. В. Л и хот ер сто в, Е. Ф. Зеберг, Н. В. Корицкая. Ж. общ. химии, 20, 635 (1950).
2. А. А. Пономарев. Синтезы и реакции фурановых веществ. Саратов, изд-во СГУ, 1960, с. 122.
3. Синтезы органических препаратов, сб. 3. М., ИЛ, 1952, с. 338.
223
\-(2-ОКСИЭТИЛ)Д ИЭТИЛ ЕНТРИАМИН
Л. М. ТИМАКОВА. В. Я. ТЕМКИНА. Г. Ф. ЯРОШЕНКО, Р. П. ЛАСТОВСКИИ
NH2CH2CH2NHCH2CH2NHCH2CH2OH
C6H17N3O М. в, 147,22
М-(2-Оксиэтил)днэтилентриамин является исходным соединением для синтеза гидроксилсодержащих комплексонов — аналогов диэтилентриамннпентауксусной н днэтилентрнамин-пентафосфоновой кислот.
Обычно для введения оксиэтильной группы при получении аналогичных соединений используется взаимодействие аминов с окисью этилена [1, 2]. Для введения оксиэтильной группы нами предложен метод, в котором окись этилена заменена на более безопасный этиленхлоргидрин. Мт-(2-Оксиэтнл) диэтилентриамин получен взаимодействием диэтилентриамина с эти-ленхлоргидрином в водной среде.
СХЕМА СИНТЕЗА
NH2CH2CH2NHCH2CH2NH, + С1СН2СН2ОН ->
NH2CH2CH,NHCH2CH2NHCH2CH2OH + НС1
Характеристика основного сырья
Диэтилентрнамин, МРТУ 6—09—3208—66, ч.
Этиленхлоргидрин МРТУ 6—09—451—63, ч,
Натр едкий, ГОСТ 4203—65, ч,
Условия получения
В четырехгорлой колбе емкостью 0,5 л, снабженной мешалкой (100 об/мин), обратным холодильником, термометром и капельной воронкой, нагревают до кипения (107—110°) при 224
перемешивании раствор 103,2 а (1 моль) диэтилентриамина в 150 мл воды, после чего прибавляют по каплям из капельной воронкн 80,5 г (1 моль) этиленхлоргидрина с такой скоростью, чтобы реакционная масса равномерно кипела. Массу кипятят еще 1 час, затем понижают температуру до 60° и нейтрализуют выделившийся во время реакции хлористый водород добавлением в течение 2 часов 40 г (1 моль) едкого натра. После охлаждения реакционной массы отфильтровывают выпавший осадок хлорида натрия и отгоняют из фильтрата воду в вакууме водоструйного насоса (10 мм). Остаток фракционируют при пониженном давлении, отбирая фракцию с т. кип. 153—15571 мм.
Выход Ы-(?-оксиэтил)диэтилентриамина равен 55 г (37%). Вещество представляет собой маслянистую бесцветную жидкость, п2£— 1,4970.
Найдено, %: С—48,5; 48,7; Н—11,7; 11,5; N—28,7; 28,4. CeHjjNaO. Вычислено, %: С—48,9; Н—11,6; N—28,5.
ЛИТЕРАТУРА
1.1, К it sc hen, С. В. Pollard. J. Organ. Chem., 8 342 (1942).
2. N. Kazuo. Bull. Chem. Soc. Japan, 34, 5, 651 (1961).
15 Зак. 1164
\-(2-0КСИЭТИЛ )ДИЭТИЛ ЕНТРИАМИ НО-N.N.N'.N'-TETPAyKCyCHAH КИСЛОТА
Л. М. ТИМАКОВА, В. Я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Р. П. ЛАСТОВСКИИ
НОН2СН2С. _СН2СООН
)N-CH2CHs-N-CH,CH2NC
НООСН2СХ | хснасоон
СН2СООН
C14H25N3O9 М. в. 379,70
N - (2-Оксиэтил) диэтилентриамино-М,М/,М'",М//-тетрауксус-ная кислота является новым комплексообразователем—гидроксилсодержащим аналогом известного комплексона диэти-леитриаминпентауксусной кислоты, нашедшей широкое применение в аналитической практике для разделения р. з. э. и в других областях народного хозяйства (1, 2]. Синтез комплексона осуществлен взаимодействием N-(2-оксиэтил) диэтилеи-триамина с монохлоруксусной кислотой в воднощелочной среде.
СХЕМА СИНТЕЗА
N1OH
НОСН2СН2—NHCH2CH2N—CH2CH,NH2 ciCHaCOOH •*
НООСН2СЧ _СН2СООН
-» )N—CH2CH2-N-CH,CH2-N(
НОНаСНгС7 | ХСН2СООН
СН2
соон
Характеристика основного сырья
N-(2-Оксиэтил) диэтилентриамии, получение см. с. 224 этого сборника.
226
Моиохлоруксусная кислота, ГОСТ 5836—51, и.
Натр едкий, ГОСТ 203—65, ч., 20%-ный раствор.
Соляная кислота, ГОСТ 3118—67, ч.
Условия получения
В четырехгорлую колбу емкостью 0,5 л, снабженную мешалкой (100 об/мин), термометром, капельной воронкой н обратным холодильником, помещают раствор 14,7 г (0,1 моль) N1-(2-оксиэтил) диэтилеитриамина в 20 мл воды и при перемешивании приливают раствор 40,16 г (0,425 моль) монохлоруксусной кислоты в 50 мл воды, предварительно нейтрализованной раствором едкого натра до pH 8 по универсальной индикаторной бумаге. Реакционный раствор нагревают при размешивании 6 часов при 80—85°, затем повышают температуру до 90—95°, и размешивают 3—4 часа, поддерживая pH 9 (по фенолфталеину) добавлением раствора едкого натра. Реакцию ведут 9—10 часов до прекращения снижения pH среды. Из реакционного раствора отгоняют воду до 1/2 объема в вакууме водоструйного насоса (10—15 мм), отфильтровывают выпавший осадок хлорида натрия, фильтрат подкисляют концентрированной соляной кислотой до pH 1—1,5 (универсальная индикаторная бумага), после чего отгоняют воду до 1/3 объема. Отфильтровывают выпавший осадок, отгоняют воду до получения сиропообразного раствора, который по каплям приливают в 1 л метилового спирта. Получают 26 г комплексона с содержанием хлорида натрия 13%.
Для очистки 10%-иый раствор комплексона подвергают электродиализу в 4-х камерном электродиализаторе с ионообменными мембранами — анионообмеиной МА-40 и катионообменной МК-40, при i = 20 ма/см? и v = 5—30 в [3]. Процесс ведут до отсутствия качественной реакции на ион хлора. Из полученного обессоленного раствора комплексона отгоняют воду при пониженном давлении (10—15 мм).
Выход N- (2-оксиэтил) диэтилентриамино-И^г,И"И"-тетраук-сусной кислоты равен 18,5 г (49%) в расчете на исходный амии.
Найдено, %: С—44,2; 44,3; Н—6,6; 6,6; N—11,0; 10,9. СиНзвНзОд. Вычислено, %: С—44,3; Н—6,6; N—11,1.
ЛИТЕРАТУРА
1. Н. М. Дятлова, В. Я. Темкина, И. Д. Колпакова. Комплексоны. М., «Химия», 1970.
2. Г. Шварцеибах, Г. Флашка. Комплексонометрическое титрование. М., «Химия», 1970.
3. В. Я. Темкина, Л. М. Тимакова, Р. Т. Хлебородова, Г. Ф. Ярошенко, А. И. Рязанов. Химические реактивы и препараты, вып. 35. М„ ИРЕА, 1973, с. 50.
15*
227
(ЬОКСИЭТИЛИДЕН)ДИФОСФОНАТ СЕРЕБРА
1 -Оксиэтан-1,1 -дифосфонат серебра
И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ
г ° 1 О=Р— он -с 1 -TJ—O II о 1 Ag2
он сн» он _
CzHgAgzOyPa
М. в. 419,75
Твердый комплекс (1-оксиэтилвден)дифосфоновой кислоты с серебром (Ag — ОЭДФ) состава Ag:OЭДФ = 2:l был получен добавлением азотнокислого серебра непосредственно, к реакционной массе, полученной взаимодействием треххлорн: стого фосфора с уксусной кислотой без предварительного выделения ОЭДФ {1]. Комплекс ОЭДФ с серебром состава Ag: ОЭДФ = 3:1 описан в работе [2], однако, метод синтеза не приводится.
Нами получен комплекс Ag—ОЭДФ состава А^:ОЭДФ'=' — 2:1 взаимодействием ОЭДФ с азотнокислым серебром.
СХЕМА СИНТЕЗА
ОН ОН ОН
I I I
О=р------с----Р—О
I I I
ОН СНз ОН
AgNO3
О ОН О
I I I
О=Р—-~С----Р=о
2-
Ag»+
228
ОН СН» ОН
Характеристика основного сырья
(1-Оксиэтилиден) дифосфоновая кислота, ТУ П—849—69, ч.
Серебро азотнокислое, ГОСТ 1277—63, ч.
Условия получения
Растворяют 4,12 г (0,02 моль) ОЭДФ в 50 мл воды, добавляют 20%-ный раствор едкого кали до достижения pH 7 (по универсальной индикаторной бумаге) и приливают раствор 20,4 г (0,12 моль) азотнокислого серебра в 40 мл воды. Выпадает обильный белый осадок, который отфильтровывают, промывают ацетоном и сушат в вакуум-эксикаторе или в пистолете над пятиокисью фосфора.
Выход (1-оксиэтилиден)дифосфоната серебра равен 2,53 г (30,1%).
Найдено, %: С—5,40; Н—1,23; Р—15,20. C2HeAg2O7P2. Вычислено, %: С—5,72; Н—1,43; Р—14,75.
ЛИТЕРАТУРА-
1. Бельг, пат. 672168 (1966); С. А., 65, 12238 (1966).
2. F. Ко spare k. Monatch. Chem., 99, 2016 (1968).
(1-ОКСИЭТИЛИДЕН)ДИФОСФОНОВАЯ КИСЛОТА, СОЛИ С АМИНАМИ
И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ, Е. И. МИРОНОВА
О=Р(ОН)2
СН3—С-ОН • 2NHR1R,;
I О=Р(ОН)4
где Ri = R2=C2H5—
Ri = Н—; R2 = С6Н5-; о-СН3~; С6Н4-
Не описанные в литературе соли (1-оксиэтилидеи)дифос-фоиовой кислоты (ОЭДФ) с аминами получены нами взаимодействием соответствующего амииа со спиртовым раствором ОЭДФ. Соли растворимы в воде.
СХЕМА СИНТЕЗА
РО(ОН)2 РО(ОН)2
I I
СНа-С-ОН + 2NHR,R2 -> СН—С—ОН • 2NHRjR2
I I
PO(OH)S РО(ОН)2
Характеристика основного сырья
(1-Оксиэтилидеи) дифосфоновая кислота, ТУ П 849—69, ч.
Диэтиламии, ТУ 6—09—68—70, ч.
Анилин, ГОСТ 5819—70, ч.
о-Толуидии, МРТУ 6—09—2898—66, ч.
230
Условия получения
К раствору 20,6 г (0,1 моль) ОЭДФ в 120 мл этилового или изопропилового спирта прибавляют 1 моль соответствую щего амииа. При этом реакционная масса разогревается. По охлаждении выпадает осадок соли ОЭДФ с амином, который отфильтровывают, промывают 10 мл абсолютного этилового спирта и 25 мл ацетона и сушат при 80° или в вакуум-эксика-торе над пятиокисью фосфора. Очищают соль перекристаллизацией из водного спирта.
Выход солей, их температура плавления и элементный анализ приведены в таблице.
Выход, температура плавления и элементный анализ солей ОЭДФ с органическими основаниями
Амин Выход, % Растворитель при перекристаллизации Т. пл., °C Брутто- формула M. B. Найдено, % Вычислено, %
Днэтил-амнн 76,8 98,5% -ный изопропиловый спирт 157— 158 CgHgOfP t* • 2C4HnN 352,13 34,14; 33,29 9,27; 9,07; С—.34,15 H—8,52
7,32; 7,66 N—7,85
17,47. 17,51 Р-17,6
Анилин 71,4 50% -ный этиловый спирт 194— 195,5 (разл.) С2Н8О7Р,- •2C6H7N-H2O 410,30 41,48 41,58 5,59 5,86 С—41,00 Н—5,84
6,76 6,79 N—6,81
14,78 14,81 Р-15,10
о-Толуи- ДНН 70,0 50 %-ный этиловый спирт • 196-197 (разл.) CgHgO/jPg* 438,35 43,97 43,97 6,17 6,00 С—43,8 Н—6,40
6,24 6,06 N—6,40
14,57 14,52 Р-14,15
(1-0КСИЭТИЛИДЕН)ДИФ0СФ0Н0ВАЯ кислит А ТРИНАТРИЕВАЯ СОЛЬ
5. И. БИХМАН, Е. М. УРИНОВИЧ, Т. Д. БОГОМОЛОВА, Н. М. ДЯТЛОВА
?/0Na /P\QNa сн»-с 0Na
C2H5N'a3O7P2-4,5H2O М. в. 353,04
Щелочные соли (1-оксиэтилиден) дифосфоновой кислоты благодаря своей высокой комплексообразующей способности Находят применение в качестве моющих средств и детергентов [1], в медицине [2], для предотвращения окислитедв^р действия некоторых соединений [3]. Одним из наиболее интересных соединений этой кислоты является тринатриевая соль, водный раствор которой имеет почти нейтральную реакцию, что позволяет использовать это соединение для комплексообразования в средах, близких к природным.
Литературные данные о синтезе тринатриевой соли окси-этил ид ендифосфоновой кислоты малочисленны и противоречивы [1, 2, 4, 5].
Нами уточнены условия синтеза и состав тринатриевой С0-1 ли (1-оксиэтилиден)дифосфоновой кислоты.
СХЕМА СИНТЕЗА
О
ЪОН /р
СН,-С
NaOH
ОН
ОНГ\ „ II ХОН О
о
II / 0Na СНз-сГ X°Na | \ ХО№
II ЧОН О
232
Характеристика основного сырья
(1-Окси,этилиден)дифосфоновая кислота, ТУ 14 П 849—69, ч.
Натр едкий, ГОСТ 4328—66, ч.
Условия получения
В стакане емкостью 1 л, снабженном мешалкой и капельной воронкой, растворяют 309 г (1,5 моль) оксиэтилиденди-фосфоновой кислоты в 300 мл воды (показатель преломления раствора ri$ = 1,44—1,46). К полученному раствору при наружном охлаждении ледяной водой приливают по каплям раствор 180 г (4,5 моль) едкого натра в 270 мл воды. При достижении pH среды равного 7 раствор сразу же превращается в сплошную массу белоснежных кристаллов. Осадок растирают, переносят на фильтр и хорошо отжимают, после чего сушат сначала на воздухе, а затем в вакуум-эксикаторе при комнатной температуре до постоянной массы.
Выход тринатриевой соли (1-оксиэтилиден) дифосфоновой кислоты равен 410 г (77,3%); pH 5%-ного водного раствора соли равен 7,6.
Найдено, %: С—7,41; Н—4,44; Na—20,9; Н2О— 23,0 (ем. примечание).
СЛ15Ыа3О7Р2 • 4,5НзО. Вычислено, %: С—6,81; Н—3,97; Na—19,53; Н2О— —22,95.
Примечание.
Определение натрия методом пламенной фотометрии проведено О. В. Коньковой, определение кристаллизационной воды — на дериватографе системы Паулик—Паулик—Эрдей проведено Н. Ф. Шугал.
ЛИТЕРАТУРА
1. Пат. США 3366676 (1968).
2. М. D. F г а п с i s, R. G. G. R u s s е 1. Science, 165, 1264 (1969),
3. Франц, пат. 990660 (1965).
4. В. Blaser, К. Н. Worms, Н. G. Germscheid, К. Wol-Imann. Z. anorgan. und allgem. Chem., 381 247 (1971).
5. Пат. США 3484480 (1969); Пат. США 3579570 (1971).
М-(2-0КСИЭТИЛ)СУКЦИНИМИД И И-(З-ОКСИПРОПИЛ)-
сукцинимид
В. М. ОСТРОВСКАЯ, И. А. ШУЙСКАЯ, Т. В. ФОМИНА, Л. В. ЛОМАКИНА
Н2сх \
I n-ch2-ch2oh
н2сч /
<0
C6H9NO3 .4. в. 143,15
/<°
H2CZ \
I N-CH2-CH2-CH2OH
Н2СХ /
So
CzHnNOs м. в. 168,18
N-(2-Оксиэтил) сукцинимид и N-(3-оксипропил) сукцинимид являются промежуточными продуктами в синтезе органических реагентов, например, N-(алкоксиалкил) сукцинимидов, применяющихся в качестве сорбентов сернистого газа [1].
N-(2-Оксиэтил) сукцинимид получают взаимодействием моиоэтаноламина с янтарным ангидридом [2, 3] или янтарной кислотой [4, 5]; реакцией сукцинимида с этнленкарбоиатом [5]; нагреванием М-(р-диэгиламиноэтил) сукцинимида с моноэта-ноламином [6].
N-(3-Оксипропил)сукцинимид получают взаимодействием янтарного ангидрида с 3-аминопропаиолом [3].
234
Нами эти соединения Получены взаимодействием янтарной кислоты, соответственно, с моиоэгаиоламином и 3-амииопро-па.нолом.
СХЕМА СИНТЕЗА
соон со
сн2 сна \
I +HsN(CHs)nOH —< I N—(СН2)ЛОН + 2HSO,
сн2 сн5 /
хсоон хсо
где п — 2,3
Характеристика основного сырья
Янтарная кислота, ГОСТ 6341—52, ч.
Моноэтаноламин, МРТУ 6—09—3909—67, ч.
З-Аминопропанол МРТУ 6—09—3012—66, ч.
Условия получения
Синтез N- (2-оксиэтил) сукцинимида. В круглодонную колбу емкостью 0,5 л, снабженную термометром, загружают 118 а (1 моль) янтарной кислоты и постепенно добавляют 61 г (1 моль) безводного моноэтаноламина, при этом реакционная масса разогревается до 124°. Смесь нагревают на глицериновой бане при 150° в течение часа. После прекращения выделении паров воды и охлаждения вещество застывает в светло-желтую массу, которую перегоняют при пониженном давлении при 178°/5 мм и перекристаллизовывают из бензола.
Выход N-(2-оксиэтил) сукцинимида равен 136 г (95%); вещество представляет собой белый кристаллический порошок с т. пл. 58—60°.
По литературным данным, т. кип. 161 —16273 мм [5], 181 — 186°/6 мм [4], 21576 мм [6], т. пл. 54—58° (3], т. пл. 60° [6], т. пл. 60—61° [4].
М-(3-Оксш1ропил)сукцинимид получают аналогичным путем. В этом 'случае саморазогрев .происходит до 95°, нагревание реакционной массы проводят при 150—180° в течение двух часов. Продукт перегоняют при пониженном давлении 176— 178°/3 мм.
Выход N-(3-оксипропил)сукцинимида равен 80%, =
= 1,5042.
Найдено, %: С—53,06, 52,78; Н—7,16, 7,12; N—8,98, 8,91. СтНпЫОз. Вычислено, %: С—53,49; Н—7,05; N—8,91.
235
ЛИТЕРАТУРА
1. Пат. ФРГ 1273501 (1968).
2. А. Ф. Николаев, С. Н. Ушаков. Изв. АН СССР, 1, 1236 (1957).
3. Пат. США 3394144 (1968); С. А., 69, 78441р (1968).
4. О. Weitzel, F. S с h е i d е г, А. М. F г е t г d о г f, К. S е у п-sche, Н. Finger. Z. Phys. Chem., 334, 1, 25 (1963).
5. К. Jan age, S. Akioshi. J. Organ. Chem, 24, 122 (1959); C. A., 54, 5427a (1960).
6. K. Nakajima. Nippon Kagaku Zasshi, 81, 323 (1960); C. A, 56> 405a (1962).
М-(2-0КСИЭТИЛ)ФТАЛИМИД и М-(З-ОКСИПРОПИЛ)-ФТАЛИМИД
В. М. ОСТРОВСКАЯ, И. А. ШУЙСКАЯ, Л. В. ЛОМАКИНА
/Ч/с\
|| I n-ch»ch,oh
CjoHgNOs
/ч/с\
|| I N-CH,CH2CH2OH
ч
ChHuNOs
М. в. 191,19
М. в. 216,22
N- (2-Оксиэтил)фталимид и N-(3-оксипропил)фталимид являются промежуточными продуктами в синтезе органических соединений [1].
N-(2-Оксипропил) фталимид применяют в качестве неподвижной фазы в газо-жидкостной хроматографии при разделении терпенов (2].
В литературе описан способ получения N-(3-оксипропил) -фталимида взаимодействием фталевого ангидрида с 3-амино-пропанолом (2]. N1-(2-Оксиэтил) фталимид получают конденсацией фталевого ангидрида и моноэтанол амин а [3, 4]; нагреванием фталимида с этилеякарбонатом [5, 6]; нагреванием N-(p-диэтил аминоэтил) фталимида с моноэтаноламином [7J.
Нами проверены и уточнены условия получения этих соединений взаимодействием фталевого ангидрида соответственно с моноэтаноламином и 3-аминопропанолом.
237
СХЕМА СИНТЕЗА
II | О + H,N(CH2)„OH
где п — 2,3
Характеристика основного сырья
Фталевый ангидрид, ГОСТ 5869—67, ч.
Моноэтанол амин, МРТУ 6—09—3909—67, ч.
З-Аминопропанол, МРТУ 6—09—3012—66, ч.
Условия получения
Синтез №(2-оксиэтил)фталимида. В круглодонную колбу емкостью 0,5 л, снабженную термометром, вливают 60 мл (61 г, 1 моль) свежеперегнанного 'моноэтаноламина и добавляют 148 г (1 моль) фталевого ангидрида. Происходит вспенивание реакционной массы с разогревом до 174°. Сразу же колбу помещают в глицериновую баню, нагретую до 140° и выдерживают в течение часа при этой тем,пературе. Затем вещество перекристаллизовывают из 500 мл кипящей воды, сушат на воздухе, затем в сушильном шкафу.
Выход №-(2-оксиэтил)фталимида равен 181 г (95%), т. пл. 127—128°.
По литературным данным, т. пл. 127° [3]; 126—127° [4]; 128° [5], т. кип. 188°/6 мм [5].
Ы-(3-Оксипропил)фталимид получают аналогичным путем, Очищают переосаждением водой из этанола (2:5), сушат в вакуум-экоикаторе и перегоняют при пониженном давлении, собирая фракцию с т. кип. 200—203./5 мм:
Выход М-(З-оксипропил) фталимида составляет 85%, т. пл. 75,5—76,5°.
По литературным данным [2], т. пл. 75—78°, после трех перекристаллизаций из метанола т. пл. 78—8Г.
238
ЛИТЕРАТУРА
1. Пат. ФРГ 1273501 (1968).
2. R. L. Markus, J. G. O'Brien. J. Organ. Chem. 30, 6, 2063 (1965).
3. А. Ф. Николаев, С. H. Ушаков. Изв. АН СССР, 1, 1236 (1957).
4. Пат. США 3394144 (1968); С. А., 69 78441р (1968).
5. К. Yanage, S. A k i о s h i. J. Organ. Chem., 24; 122 (1959).
6. Яп. пат. 6862 (I960); С. A., 55, P7362d (1961).
7. К, Nakajima. Nippon Kagaku Zasshi, 31, 323 (1960); C. A. 56, 405a (1962).
у-ОКСО-М-(5-ГЕКСИЛ-2-ПИРРОЛ ИДОН) МАСЛЯНАЯ КИСЛОТА
В. А. СЕДАВКИНА, И. А. МОРОЗОВА
h18c(-’Xn>0
СОСН2СН2СООН
CI4H23NO4
М. в. 269,32
В литературе описано ацетилирование 5-алкил-2-пирроли-донов, осуществляемое кипячением последних с двукратным количеством уксусного ангидрида [1]. Нами разработан метод синтеза не описанной ранее у-оксо-Й-(5-гексил-2-,пирролидон) масляной кислоты, заключающийся в ацилировании 5-гексил-2-пирролидона янтарным ангидридом.
СХЕМА СИНТЕЗА
Н18С6-\ /-О
СОСН2СН2СООН
Характеристика основного сырья
5-Гексил-2-пнрролидон, т. кип. 165°/7 мм, т. пл. 42°; получение см. {1].
Этиловый спирт безводный, ГОСТ 9674—61.
Янтарный ангидрид, МРТУ 6—09—4248—67, т. пл. 119°.
240
Условия получения
В двугорлую колбу емкостью 50 мл, снабженную мешалкой и обратным холодильником, помещают 3 г (0,017 моль) 5-гексил-2-.пирролидона и 1,7 г (0,017 моль) янтарного ангидрида в 30 «л абсолютного этилового спирта. Реакционную смесь нагревают 3 часа на кипящей водяной бане, этиловый спирт отгоняют, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 145°/2 мм.
Выход у°ксо'№-(5-гексил-2-пирролидон) масляной кислоты равен 2,7 г (58%). Вещество представляет собой бесцветную вязкую жидкость с характерным запахом, хорошо растворяется в воде, этиловом и метиловом спиртах, диоксане, бензоле, дихлорэтане, = 1,4538, dt2a = 1,0121 (см. примечание).
Найдено, %: С—62,71; Н—8,37; N—5,60. C14H23NO4. Вычислено, %: С—62,51; Н—8,62; N—5,21.
Примечание.
Аналогичным путем из соответствующих 5-алкил-2-пирролидонов впервые получены:
у-Оксо-Ы-(5-пропил-2-пирролидон) масляная кислота, выход 54%, т. кип. 127—12872 мм, rty 1,4539, <АМ = 1,0666.
Найдено, %: С—58,50; Н—8,01; N—6,07. CnH17NO4. Вычислено, %: С—58,20; Н—7,55; N—6,17.
у-Оксо-Ы-(5-бутил-2-пирролидон) масляная кислота, выход 53,4%, т. кип. 122—12372 мм, rig = 1,4520, </420 = 1,0644.
Найдено, %: С—59,41; Н—8,21; N—6,02.
CisHigNOi. Вычислено, %: С—59,80; Н—7,95; N—5,81.
у-Оксо-Ы-(5-изобутил-2-пирролидои)масляная кислота, выход 53,7%, т. кип. 14272 мм, ng = 1,4526, <№ = 1,0639.
Найдено, %: С—59,70; Н—7,81; N—5,71.
CI2H19NO4. Вычислено, %: С—59,80; Н—7,95; N—5,81.
у-Оксо-И-(5-амил-2-пирролидон)масляная кислота, выход 57,7%, т. кип. 140—14272 мм, rig — 1,4528, d42“ = 1,0180.
Найдено, %: С—61,35; Н—8,00; N—5,52.
Ci3H21NO4. Вычислено, %: С—61,23; Н—8*30; N—5,49.
у-Оксо-И-(5-изоамил-2-пирролидои)масляиая кислота, выход 56%, т. йип. 15172 мм, ng = 1,4541, d4M = 1,0169.
Найдено, %: С—61,39; Н—8,15; N—5,71.
Ci3H2iNO4. Вычислено, %: С—61,23; Н—8,30; N—5,49.
у-Оксо-М-(5-нзогексил-2-пирролидон) масляная кислота, выход 57%, т. кип. 159—16072 мм, ng = 1,4549, d420 = 1,0119.
Найдено, %: С—62,63; Н—8,41; N—5,47. Ci4H23NO4. Вычислено: %: С—62,51; Н—8,62; N—5,21.
ЛИТЕРАТУРА
1. А. А. Пономарев, В. А. С е д а в к и н а. Химия гетероцикл, соед., 5, 809 (1969).
16 Зак. 1164
ПЕНТАФТОРАЗИДОБЕНЗОЛ
А. В. КАШКИН, Ю. Л. БАХМУТОВ, Н. Н. ЧАЙНИКОВА
N3
F—
—F
F
C6F5N3
М. в. 210,18
Пентафторазидобензол может служить исходным веществом для синтеза красителей и ряда других фторорганических продуктов 11, 2]. В литературе описано получение пентафтор-азидобензола нитрозированием гидразинопентафторбензола с выходом 30% [1].
Нами разработан способ синтеза этого соединения с выходом ~70% взаимодействием гексафторбензола с азидом натрия в среде диметилформамида.
СХЕМА СИНТЕЗА
F'\_/F
Е—^-F + NaNs — FX XF
F\ E
F-^_^-N3 + NaF. f/“\f
Характеристика основного сырья
Гексафтор бензол, ТУ TCP 2204—70, ч.
Азид натрий, синтезирован из гидравнн-гидрата, этнлнит-рита и метилата натрия; получение ем. [3, 4].
Диметилформамид, МРТУ 6—09—2068—65, ч.
242
Условия получения
В четырехгорлую колбу емкостью 200 мл, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, помещают раствор 7,75 г (0,12 моль) азида натрия в 70 мл диметилформамида. При перемешивании и температуре 40—45° прибавляют по каплям 22,2 г (0,12 моль) гексафторбензола. Содержимое колбы перемешивают 2 часа при 40—50°, охлаждают, выпавший осадок фторида натрия отфильтровывают. Из фильтрата продукт осаждают равным (по объему), количеством воды, отмывают от иона азида (проба с FeCl3) и перегоняют при пониженном давлении.
Выход пентрафторазидобензола равен 17,44 г (70%), т. кип. 52—547Ю мм, /$= 1,4703, dA20 = 1,6007.
По литературным данным [1], т. кип. 35°/5 мм.
ЛИТЕРАТУРА
1. Пат. США 3238230 (1966).
2. Англ. пат. 1029733 (1966).
3. Г. Б р а у е р. Руководство по препаративной неорганической химии. М., ИЛ„ 1956, с. 237.
4. В. В. Некрасов. Руководство к малому практикуму по органической химии. М., Госхимиздат, 1954, с. 85, 94.
16*
(2-ПИРИДИЛМЕТИЛ)ИМИН0ДИУКСУСНАЯ КИСЛОТА
Н. в. ЦИРУЛЬНИКОВА, В. Я. ТЕМКИНА, Л. П. РАЗБОРСКАЯ
I J-CH2N(CHgCOOH)2
C10Hi2N'2O4 М. в.224,22
(2-Пиридилметил)иминодиуксусная кислота является своеобразным комплексоном, содержащим в качестве дополнительного координационного партнера атом азота пиридинового цикла, что вносит определенную специфику в поведение этого соединения при взаимодействии с катионами. Известный в литературе метод получения этого соединения состоит в восстановлении пиридин-2-альдоксима до соответствующего амина и последующем взаимодействии последнего с монохлорускусной кислотой [1]. Однако получение исходного пиридин-2-альдокси-ма представляет собой чрезвычайно трудный многостадийный процесс.
Нами впервые предложено в качестве исходного реагента использовать доступный а-пиколин, который путем несложных превращений переводится через 2-хлорметиленпиридин в конечный продукт.
СХЕМА СИНТЕЗА
.сн,соон
+ HN< NaOH
'CH2COONa
//\
L JI-CHgN(CH2COONa)g >
CH2N(CH3COOH)a
244
Характеристика основного сырья
2-Хлорметиленпиридин, т. кип. 75—77°/12 мм; получение см. с. 299 этого сборника.
Иминодиуксусная кислота, ВТУ МГУХП 190—58, ч.
Натр едкий, ГОСТ 4328—66, ч.
Соляная кислота, ГОСТ 3118—66, ч.
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную термометром, обратным холодильником и мешалкой и помещенную в баню с обогревом, загружают 13 г (0,102 моль) 2-хлормети-ленпиридина и 20 мл воды. Полученный раствор нагревают при размешивании до 60° и приливают заранее приготовленный нейтрализованный 20%-ным раствором едкого натра до pH 7—8 раствор 14,2 г (0,107 моль) иминодиуксусной кислоты в 17 мл воды с такой скоростью, чтобы температура реакционного раствора не превышала 80°. При этой температуре и размешивании поддерживают pH раствора ранным 7 добавлением 20%-ного раствора едкого натра. Поскольку реакционный раствор интенсивно окрашен, значение pH определяют по индикаторной бумаге «Multi phan». После достижения леизме-няющегося в течение 1 часа значения pH реакционный раствор размешивают еще 2 часа при той же температуре, охлаждают до 7—10° (баня со льдом) и подкисляют при размешивании концентрированной соляной кислотой до pH 2. Выпавший осадок через 20 часов отфильтровывают, промывают на фильтре водным (1:5) этанолом от ионов хлора (проба с азотнокислым серебром) и сушат в вакуум-эксикаторе.
Выход (2-пиридилметил)иминодиуксусной кислоты равен 10 г (44,6%); т. пл. 178—179°. Вещество представляет собой порошок слегка кремого цвета, хорошо растворимый в щелочах, воде, нерастворимый в спирте, ацетоне,.
По литературным данным [1], т. пл. 174—175°.
ЛИТЕРАТУРА
1. Н. Irving. J.l.R.F. da Silva. J. Chem. Soc., 2, 945(1963).
0-(2-ПИРРОЛИДИЛ)ПРОПИОНОВАЯ кислота
| А. А. ПОНОМАРЕВ, ) М. В. НОРИЦИНА, А. П. КРИВЕНЬК
1 ’-сн2-снасоон
N
I н
C7H13NO2 М. в. 14Д1&
Известны способы получения 0- (2-пирролидил)пропиопо-вой кислоты многостадийными способами из пиррола илн'еЙ. производных [1—3]. Однако кислота в чистом виде не выделена и охарактеризован только ее этиловый эфир.
Нами предложен метод получения 0-(2-пирролидил)црр‘ пионовой кислоты и ее гомологов посредством окисления соответствующих пиролидиновых спиртов хромовым ангидридом;в 12 %-ном растворе серной кислоты [4]. Метод препаративйО прост, кислоты получаются с хорошими выходами.
СХЕМА СИНТЕЗА
)-СН,-сн2—снаон
I н
I j-CHa-CH,C^ хNZ ХОН
I
н
246
Характеристика основного сырья
3-(2-Пирролидил) пропанол, т. кип. 112—113°/15 мм, т. пл. 28—30°; получение см. [5].
Хромовый ангидрид, ГОСТ, 3776—47, ч.
Бария гидрат окиси, ГОСТ 4107—65, ч. д. а.
Этиловый спирт абсолютный, приготовлен по методу [6].
Условия получения
В колбу емкостью 0,5 л, содержащую раствор 5,2 о (0,04 моль) 3-(2-пирролидил)пропанола в 90 мл воды, при перемешивании приливают раствор 5,33 г (0,53 моль) хромового ангидрида в 150 мл 12%-ной серной кислоты. Реакционную смесь оставляют на 12 часов, затем приливают горячий раствор 95 г (0,3 моль) гидрата окиси бария в 180 мл воды и нагревают смесь на кипящей водяной бане 20 минут. Осадок отделяют на воронке Бюхнера, промывают 10 мл 2%-ного раствора гидрата окиси бария. К фильтрату прибавляют сухой лед до прекращения выделения осадка карбоната бария и снова фильтруют. Воду отгоняют досуха при пониженном давлении. Остаток растворяют в 100 мл абсолютного спирта, фильтруют, спирт отгоняют, а образовавшуюся стекловидную массу растирают с 50 мл эфира. При этом выпадают бесцветные кристаллы. Вещество перекристаллизовывают из смеси (1:3) спирт—ацетон.
Выход р-(2-,пирролидил)пропионовой кислоты равен 4,4 г (78%), т. пл. 156— 156,5° (разл.) (см. примечание).
Найдено, %: С—58,8; 58,41; Н—9,03; 8,89; N—9,91; 9,84. CzHisNOs. Вычислено, %: С—58,71; Н—9,15; N—9,78.
Примечание.
Аналогичным путем получены впервые;
Р-(5-Метил-2-пирролидил)пропионовая кислота из 3-(5-метил-2-пирро-лидил)пропанола, выход 79°/о, т. пл. 160° (разл.).
Найдено, %: С—60,90; 60,81; Н—9,87; 9,83; N—8,55, 8,67. CsHisNO2. Вычислено, %: С—61,20, Н—9,67; N—8,92.
Р (4 Этил-2-пирролидил)пропионовая кислота из 3-(4-этил-2-пирроли-дил)пропанола, выход 60%, т. пл. 157—157,5°.
Найдено, %; С—63,10; 62,95; Н—9,67; 10,09; N—8,30; 8,47. C9H17NO2. Вычислено, %: С—63,21; Н—10,02; N—8,19.
Р-(5-Изобутил-2-пирролидил) пропионовая кислота из 3-(5-изобутил-2-пирролидил)пропанола, выход 79%, т. пл. 159—160°.
Найдено, %; С—65,84; 66,32; Н -10,33; 10,77; N—6,98; 7,19. ChH2iNO2. Вычислено. %: С—66,29; Н—10,64; N—7,04.
р-[Цикпопеитано(в)-2-пирролидил]пропиоиовая кислота из 3-[цикло пентано(в)-2-пирролидил]пропанола, выход 85%, т. пл. 192—193° (разл.).
Найдено, %: С—65,24; 65,53; И—9,53; 9,55; N—7,66; 8,04. Ci0H17NO2. Вычислено, %: С—65,54; Н—9,35; N—7,64.
247
р-(2-Октагидроиидолил) пропионовая кислота из 3-(2-октагидроййДО-лил)пропанола, выход 83%, т. пл. 182—183° (разл.).
Найдено, %: С—66,54; 66,70; Н—9,39; 9,50; N—7,49- 740
CnHiaNOs. Вычислено, %; С—66,97; Н—9,71; N—7,10. ’
ЛИТЕРАТУРА
1. F.. GalinowsKy, A. Reichard. Вег., 77в, 138 (1944).
2. О. Clem о, N. Fletcher G, F u 11 о п, R. R а ре г. J. Chem Soc. 1950 1140.
3. Isamu Murakoschf. Yakudaku Zasshi 78, 594 (1958).
4. А. А. Пономарев, M. В. Норицина. Авт. свид. 193516; Изобретения. Промобразцы. Товарные знаки, № 7 (1967).
5 А. А. Пономарев, М. В. Норкцина, А. П. Крквенько. Химия гетероцикл, соед., 6, 923 (1966).
6. Ю. К. Юрьев. Практические работы по органической химии, выл. I и II. М., изд-во МГУ, 1961, с. 52.
З-ПИРРОЛИЗИДОН
| А. А. ПОНОМАРЕВ, | М. В. НОРИЦИНА, А. П. КРИВЕНЬКО
C7HnNO
М. в. 125,17
З-Пирроливидон и его гомологи являются физиологически активными веществами [1]. В литературе имеются лишь крат кие сведения о их простейшем представителе — 3-пирролизи-доне [2—4].
Нами предлагается метод синтеза 3-пирролизидона и его гомологов, ранее неописанных в литературе, посредством внутримолекулярного ацилирования соответствующих р-(2-пирро-лидил)пропионовых кислот.
СХЕМА СИНТЕЗА
I
Н
1-СН2-СН2СООН
Характеристика основного сырья
р-(2-Пирролидил)пропионовая кислота, т. пл. 155—156,5°; получение см. с. 246 этого сборника.
Условия получения
В перегонной колбе емкостью 10 мл нагревают до расплавления 5 г (0,035 моль) р-(2-пирролидил) пропионовой кис-
249
лоты, поддерживая температуру бани 180—185°. Затем осторожно понижают давление (до ~ 100 мм) и отгоняют образующуюся ,в результате дегидратации кислоты воду, после чего давление понижают до 10 мм и перегоняют остаток в колбе при температуре 113—119°. Дистиллат растворяют в эфире, сушат прокаленным сернокислым магнием, фильтруют и перегоняют при пониженном давлении, собирая фракцию с т. кип. 1157Ю мм.
Выход 3-пирролизидона равен 3,5 г (81%), я™ = 1,4980, (УД0 =: 1,0729 (см. примечание).
Найдено, %: С—66,69; 66.95; Н—9,00; 8,70; N—11,66; 11,69; MR D = 34,23
CjHnNO. Вычислено, %: С—67,16; Н—8,87; N—11,21; MR0 = 34,08.
По литературным данным [3], т. кип. 97—100710 мм.
Примечание.
Аналогичным путем получены впервые;
4-Метил 3-пирролизидон из р-(5-метил-2-пирролидил) пропионовой кислоты, выход 80%, т. кип. 117°/10 мм, = 1,4910, с1,'я = 1,0520.
Найдено, %: С—69,42; 69,26; Н—9,63; 9,36; N—10,32; 10,02; MR D = 38,37.
C8Hi3NO. Вычислено, %: С—69,12; Н—9,43; N—10,08; MRD =-38,79.
4-Изобутил-З-пирролизидон из р-(5-изобутил-2-пирролидил) пропионовой кислоты, выход 66%, т. кип. 139710 мм, — 1,4842, d420 = 0,9925.
Найдено, %: С—72,77; 72,55; Н—10,52; 10,49; N—8,04; 7,89: MR D = 52,30.
CiiHwNO. Вычислено, %: С—72,88; Н—10,52; N—7,74; MR D = 52,55.
5-Этил-З-пирролизидон из Р-(4-этил-2-пирролидил)пропионовой кислоты, выход 78%, т. кип. 133710 мм, Пр = 1,4880, d4w = 1,0249.
Найдено, %; С—70,09; 70,10; Н—9,94; 9,80; N—9,46; 9,28; MRd = 43,15.
C9H15NO. Вычислено, %: С—70,53; Н—9,88; N—9,13; MRd ’ = 43,31.
5-Изопропил-З-пирролизидон из, р- (4-изопропил-2-пирролидил) пропионовой кислоты, выход 75%, т. кип. 141710 мм, Пр = 1,4849; d4-a = 1,0070.
Найдено, %: С-71,84; 71,61; Н—12,01; 11,89; N—8,01; 7,96; MR D = 47,58.
C10H17NO. Вычислено, %: С—71,92; Н—12,07; N—8,39; MRD = 49,93.
Циклопеитано- (в) -3-пирролизидон из Р- циклопеитано(в)-2-пирролиднл]-пропиоиовой кислоты, выход 70%, т. кип. 126°/3 мм, — 1,5190, d420 = = 1,0980.
Найдено, %: С—72,40; 72,22; Н—9,26; 9,18; N—8,08; 8,32; MRd = 45,70.
C10Hi5N0. Вычислено, %: С—72,70; Н—9,15; N—8,47; M/?D = 45,73.
Циклогексаио- (в) -3-пирролизидон из р- (2-октагидроиндолил) -пропионовой кислоты, выход 50%, т. кип. 14173 мм. п™ — 1,5175, d4w = 1,08000.
Найдено, %: С—73,50; 73,60; Н—9,71; 9,62; N—7,82; 7,90; MRd =50,26.
C11H17NO. Вычислено, %: С—73,70; Н—9,56; N—7,81; МА>П = 50,36.
250
ЛИТЕРАТУРА
1. А. А. Пономарев, В. А. Седавкина, М. В. Норнцнна, И. В. Хромов-Борисов^ Е. В, Морева. II Всесоюзная конференция по химии пятичлеиных азотистых гетероциклов. Тезисы 12, Рига, изд:во РГУ, 1966.
2. Z. Oalinovsky, A. Reichard. Вег. 77b, “138 (1944).
3. Isamu Murakoshl. Yakudaku Zasshi, 78. o94 (1958); C. A.,52, 1840g (1958).
4. О. С e r v i n к a. Chem. Il sty, 52, 307 (1958),
5-ПРОПИЛ-М-(₽-ОКСИЭТИЛ)-2-ПИРРОЛИДОН
В. А. СЕДАВКИНА, Н. А. МОРОЗОВА
СН2- CHj-OH
C9H17NO2
М. в. 171,23
В литературе описано получение М-(р-оксиэтил)-2-пирро-лидона из 2-аминоэтанола и бутиролактона [1], из 2-пирролп-дона и окиси этилена [2].
Нами предложен метод получения ранее неизвестных 5-ал-кил-М-(р-оксиэтил)-2-пирролидонов, заключающийся вэтанол-аминировании этиловых эфиров у-кетокарбоновых кислот с использованием в качестве катализатора никеля Ренея.
Исходные этиловые эфиры у-кетокарбоновых кислот получены расщеплением цикла фурилалкилкарбинолов [3, 4].
СХЕМА СИНТЕЗА
С3Н7-С-СНа-СНа-С/ + HjNCHjCHjOH и чоеан6
о
I
СНа— сна-он
Характеристика основного сырья
Этиловый эфир у-кетогептановой кислоты, т. кип. 94-— 96°/2 мм, = 1,4338; получение см. (3].
252
Этиловый спирт безводный, ГОСТ 9674—61.
Этаноламин, т. кип. 172,2°, МРТУ 6—09—3909—67.
Никель Ренея получен выщелачиванием сплава никеля с алюминием [5].
Водород электролитический.
Условия получения
Во вращающийся автоклав емкостью 250 мл загружают 10 г (0,058 моль) этилового эфира у-кетогептановой кислоты в 20 мл абсолютного этилового спирта, 3,5 г (0,058 моль) эта-ноламина в 10 мл абсолютного этилового спирта и За никеля Ренея. Реакцию ведут при 100° и начальном давлении водорода 100—120 атм до поглощения рассчитанного количества водорода (1,51 л), на что требуется 10—12 часов. Катализатор отфильтровывают, отгоняют этиловый спирт, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 140— 142°/2 мм, = 1,4842; d420 =' 1,0445.
Выход 5-пропил Кт-(р-оксиэтил)-2-пирролидона равен 6,9 г (70,2%) (см. примечание).
Найдено, %: С—63,3; Н—10,3; N—8,5. C9H17NO2. Вычислено, %: С—63,2; Н—10,0; N—8,2
Примечание.
Аналогичным путем из этиловых эфиров соответствующих у-кетокарбоновых кислот получены следующие 5-алкил-Т4-(р-оксиэтил)-2-пирроли допы;
5-Бутнл-М-(Р-оксиэтил)-2-пирролидон, выход 70%, т. кип. 142— 14372 мм, и2? = 1,4860, d420 = 1,0271.
Найдено, % С—64,4; Н—10,3; N—7,6. Ci0H19NO2. Вычислено, %; С—64,8; Н—10,3; N—7,6.
5-Изобутил-М-(|3-оксиэтил)-2-пнрролидон, выход 70%, т. кип. 170— 17272 мм, «о = 1,4821, d4® = 1,0214.
Найдено, %: С—64,5; Н—10,1; N—7,7. Ci0H19NO2. Вычислено, %: С—64,8; Н—10,3; N—7,6.
5-Амил-Н-(Роксиэтил)-2-пирролидон, выход 70,5%, т. кип. 162— 16472 мм, «д = 1,4798, d4“ = 1,0088.
Найдено, %: С—66,4; Н—10,8; N—7,2. CiiH>iNO2. Вычислено, %: С—66,4; Н—10,6; N—7,0.
5-Изоамил-М-(р-оксиэтил)-2-пирролидои, выход 71%, т. кип. 192— 19472 мм, 1,4800, d42° = 1,0123.
Найдено, %: С—66,4; Н—10,9; N—6,9. ChH21NO2. Вычислено, %: С—66,4; Н—10,6; N—7,0.
5-Гексил-М-(Р-оксиэтил)-2-пирролидон, выход 71,5%, т. кип. 183— 18472 мм, tig = 1,4792, d42° = 1,0005.
Найдено, %; С—67,9; Н—10,9; N—6,7. Ci2H23NO2. Вычислено, %: С—67,6; Н—10,9; N—6,6.
253
5-Изогексил-М-(6оксиэтил)-2-пирролидон, выход 70%, т, кип. 196— 197°/2 мм, п™ = 1,4772, d42» = 0,9945.
Найдено, %: С—67,8; Н—10,8; N—6,3.
C12H23NO2. Вычислено, %: С—67,6; Н—10,9; N—6,6.
5-Гептил-Ы-(Р-оксиэтил)-2-пирролидон, выход 71%, т. кип. 195— 19672 мм, п* = 1,4709, d^> = 0,9698.
Найдено, %: С—69,4; Н—11,2; N—6,3.
C13H25NO2. Вычислено, %; С—68,8; Н—11,1; N—6,2.
5-Децил-М-(₽оксиэтил)-2-пирролидон, выход 72%, т. кип. 220-22272 мм. = 1,4640, d4™ = 0,8991.
Найдено, %: С—71,7; Н—11,5; N—5,4. Ci6H31NO2. Вычислено, %: С—71,4; Н—11,5; N—5,2.
ЛИТЕРАТУРА
1. В. Puetzer, L. Katz, L. И а г w i t z. J. Amer. Chem. Soc. 74, 4959 (1952).
2. M. Ф. Шостаковский, Ф. П. Сидельковская. Изв. АН СССР, о.х.н., 1, 111 (1958).
3. В. Г. Бухаров, Т. Е. Позднякова. Изв. АН СССР, сер. хим..
6, 1108 (1960).
4. А. А. П о и ом а р е в, В. А. С е д а в к и и а. Ж. общ. химии, 32, 2540 (1962).
5. Синтезы органических препаратов, сб. 3. М., ИЛ, 1952, с. 338.
5-ПРОПИЛ-2-ТИОП.ИРРОЛИДОН И 1-МЕТИЛ-5-ПР0ПИЛ-2-ТИОПИРРОЛИДОН
В. А. СЕДАВКИНА, Г. В. БЕСПАЛОВА
на-' J=s N сн3
C7H13N'S М. в. 143,25
C8Hi5NS М. в. 157,27
Известей метод синтеза М-аллил-2-тиопирролидона и Ы-винил-2-тиопирролидона взаимодействием соответствующих пирролидонов с пятисернистым фосфором [1].
Нами синтезирован ряд неизвестных ранее 5-алкил-2-тио* пирролидонов и 1-метил-5-алкил-2-тиопирролидонов по несколько измененной методике [1], что позволило увеличить выход продуктов до 80%.
СХЕМА СИНТЕЗА
НА-1 Ls +Раз5™^-> z=s>
N XNZ
I I
R R
где R-H; CH3-.
Характеристика основного сырья
5-Пропил-2-пирролидон, т. кнп. 105—106°/1 мм, т. пл. 36°; получение см. {2].
255
1-Метил-5-.пропил-2-пирролидон, т. кип. 75—76°/1 мм
= 1,4706, d^0 = 0,9705; получение см. [2].
Пятисернистый фосфор, ГОСТ 7200—54.
Пиридин, ГОСТ 2747—44.
л«-Ксилол, ТУ МХП 3600—53, ч.
Условия получения
Синтез 5-пропил-2-тиопирролидона. В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром и обратным холодильником, помещают раствор 3 г (0,021 .моль) 5-метил-2-пирролидона в смеси 50 мл сухого пиридина с 50 мл ксилола. При постоянном перемешивании к раствору добавляют 3 г (0,013 моль) пятисернистого фосфора и кипятят 1,5 часа при 115—120°, причем реакционная масса окрашивается в темно-красный цвет. По охлаждении смесь фильтруют, отгоняют растворители, а остаток перегоняют при пониженном давлении.
Выход 5-пролил-2-тиопирролидона равен 2,4 г (70%), т. кип. 163—164°/2 мм (см. примечание).
Найдено, %: С—58,6; Н—9,1; N—9,8; S—22,5. C7H13NS. Вычислено, %: С—58,8; Н—9,2; N—9,8; S—22,4.
Вещество представляет собой светло-желтую жидкость, кристаллизующуюся при охлаждении, хорошо растворяется в органических растворителях.
1-Метил-5-пропил-2-тиопирролидон. По аналогичной методике, при взаимодействии 5 г (0,031 моль) 1-метил-5-пропил-2-пирролидона с 5 г (0,035 моль) пятисернистого фосфора в смеси (1:1) пиридин-ксилол получают 4,5 г (80,3%) 1-метил -5-пропил-2-тиопи.рролидона, т. кип. 124—12571 мм, Пъ~ -- 1,5340 (см. примечание).
Найдено, %: С—61,4; Н—9,6; N—8,6; S—20,8. CeHisNS. Вычислено, %: С—61,2; Н—9,6; N—8,9; S—20,4.
Примечание.
По аналогичной методике получены следующие 5-алкил-2-тиопирроли-доны:
5-Бутил-2-тиопирролидон из 5-бутнл-2-пирролндона, выход 75%, т. кип. 185—187°/3 мм, т. пл. 62°.
Найдено, %: С—61,3; Н—9,5; N—8,8; S—20,3.
CeHisNS. Вычислено, %: С—61,2; Н—9,6; N—8,9; S—20,4.
5-Изобутил-2-тиопирролидои из 5-изобутил-2-пирролидона, выход 69%, т. кип. 178—17973 мм, т. пл. 64°.
Найдено, %: С—61,4; Н—9,6; N—8,8; S—20,8. CsH15NS. Вычислено, %: С—61,2; Н—9,6; N—8,9; S—20,4.
5-Амил-2-тиопирролидон из 5-амил-2-пирролидона, выход 76%, т. кил. 185—18673 мм, т. пл. 43°.
Найдено, %: С—63,4; Н—10,1; N—8,3; S—18,2. C9H17NS. Вычислено, %: С—63,2; Н—10,0; N—8,2; S—18,8.
256
5-Изоамил-2-тиопирролидон из 5-амил-2-пирролидона, выход 70%, л. кип. 172—17372 мм, Т- пл. 45°.
Найдено, %: С—63,4; Н—9,8; N—8,4; S—18,6.
CsHnNS. Вычислено, %: С—63,2; Н—10,0; N—8,2; S—18,8.
5-Гексил-2-тиопирролидои из 5-гексил-2-пирролидоиа, выход 62%. т. кип. 161—162°/1 мм, т. пл. 61°.
Найдено, %: С—64,4; Н—10,2; N—7,8; S—16,9.
CioHisNS. Вычислено, %: С—64,9; Н—10,4; N—7,6; S—17,3.
1 -Метил-5-бутил-2-тиопирролидон из 1 -метил-5-бутил-2-пирролидона,
выход 50%, т. кип. 134—13571 мм, Дц = 1,5310.
Найдено, %; С—63,1; Н—9,9; N—8,6; S—18,2.
C9H17NS. Вычислено, %: С—63,2; Н—10,0; N—8,2; S—18,8.
1 -Метил-5-изобутил-2-тиопирролидон из 1 -метил-5-изобутил-2-пирроли-
дона, выход 65%, т. кип. 135—13771 мм, Дц — 1,5355.
Найдено, %: С—63,4; Н—10,2; N—8,9; S—18,6.
CsHijNS. Вычислено, %: С—63,2; Н—10,0; N—8,2; S—18,8.
1-Метил-5-амил-2-тиопирролидои из 1 -метил-5-амил-2-пирролидона, выход 54%, т. кип. 126—12871 мм, п™ = 1,5380.
Найдено, %: С—64,5; Н—10,1; N—7,9; S—17,6.
C10H19NS. Вычислено, %: С—64,9; Н—10,4; N—7,6; S—17,3.
1-М.етил-5-изоамил-2-тиопирролидои из 1 -метил-5-изоамил-2-пнрролидо на выход 50%, т. кип. 133—13471 мм, Дц = 1,5350.
Найдено, %: С—64,5; Н—10,3; N—7,8; S—17,4.
CioHigNS. Вычислено, %: С—64,9; Н—10,4; N—7,6; S—17,3.
1 -Метил-5-гексил-2-тиопирролидон из 1 -метил-5-гексил-2-пирролидона, выход 66%, т. кип. 145—146°/1 мм, = 1,5410.
Найдено, %: С—66,9; Н—10,7; N—7,5; S—16,4.
ChH21NS. Вычислено, %; С—66,4; Н—10,6; N—7,0; S—16,1.
ЛИТЕРАТУРА
1. М. Ф. Ш о с т а к о в с к и й, Ф. Б. С и д е л ь к о в с к а я, А. А. Аветисян. Докл. АН СССР, сер. хим. 5, 153, 1089 (1963).
2. А. А. Пономарев, В. А. Седавкина. Химия гетероцикл, соед., 5, 809 (1969).
17 Зак. 1164
а-ятранс-у-цис-р-СТИРИЛАКРИЛОВАЯ КИСЛОТА
НГУЕН ВАН ТОНГ, О. С. СТЕПАНОВА, А. И. ГАЛАТИНА
Н\ /Н
н
соон
н
СпНюО2 М. в. 174.11
а-Гранс-у-^ас-р-стирилакриловая кислота является одним из 4-х возможных геометрических изомеров этой кислоты, представляющей интерес с точки зрения стереохимических особенностей и физиологической активности. Наиболее известным и распространенным изомером является а-транс-у-транс-форма. Способы ее получения известны [1, 2]. Некоторые из* менения в методике приводят к получению а-цис-у-транс-$-стирилакриловой кисЛоты [3]. Способы получения а-транс-у-цис-р-стирилакриловой кислоты в литературе фактически не описаны, за исключением одного многостадийного и малодоступного метода [4], причем автор не приводит доказательства строения этой кислоты.
Нами разработан новый способ получения а-транс-у-цис-fi-стирилакриловой кислоты путем селективного гидрирования метилового эфира 5-фенил-2-гранс-пентен-4-иновой кислоты, который образуется при дегидратации метилового эфира 3-окси-5-фенил-4-пентиновой кислоты. Последний получается по реакции Реформатского [5] взаимодействием фенилпропаргилового альдегида с метиловым эфиром бромуксусной кисло ты. Все промежуточные вещества получены впервые.
СХЕМА СИНТЕЗА
с4н5—с=с-сно + вг— сн3сооен,
Zn
258
CeHB-C=C-CH-CH2COOCHs -^Y— OH
> c6hs—c=c-ch=ch-coochs -ib—►
-* CeHs-CH=CH-CH=CH-COOCHs .
-> C6H,-CH^CH-CH=CH-COOH
Характеристика основного сырья
Катализатор Линдлара (дезактивированный палладий на карбонате кальция), получение см. [6].
Фенилпропаргиловый альдегид, получение см. [7, 8].
Метиловый эфир бромуксусной кислоты, получение см. [9]. Цинковая пыль.
Бензол, ГОСТ 5955—68, х. ч.
Хлорокись фосфора, МРТУ 6—09—337—63, ч.
Кали едкое, ГОСТ 4203—65, х. ч.
Уксусная кислота ледяная, ГОСТ 61—69, х. ч.
Пиридин, ГОСТ 2747—67, ч.
Серная кислота, ГОСТ 4204—66, ч.
Метиловый спирт, ГОСТ 6995—67, ч.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт, ректификат, ГОСТ 8314—57.
Условия получения
Синтез метилового эфира 3-окси-5-фенил-4-пентиновой кислоты. В круглодонную колбу емкостью 0,5 л помещают 8,1 г (0,214 г-атом) цинковой пыли, затем колбу нагревают на масляной бане до 100—110° и при перемешивании прибавляют пс| каплям смесь 13,0 г (0,1 моль) фенилпропаргилового альдегида и 18,36 г (0,12 моль) метилового эфира бромуксус-ной кислоты в 120 мл абсолютного бензола. Реакционную смесь перемешивают при 100—110° еще 20 минут. Содержимое колбы охлаждают и разлагают 12 мл уксусной кислоты в 150 мл воды. Отделяют верхний слой, а нижний экстрагируют трижды по 40 мл эфира. Эфир.ный раствор промывают 50 мл воды, 40 мл 10%-ного раствора карбоната натрия и снова 50 мл воды. Эфирный экстракт сушат сульфатом натрия и перегоняют при пониженном давлении, собирая фракцию с т. кип. 172—174°/8 мм.
Выход метилового эфира 3-оксн-5-фенил-4-пентиновой кислоты равен 12,2 г (60%); = 1,1394; «о = 1,5515.
Найдено, %: С—70,25; Н—6,1.
С^НцОз. Вычислено, %: С—70,58; Н—5,88.
17* 259
Получение метилового эфира 5-фенил-2-пентен-4-иновой кислоты. В колбу емкостью 250 мл, содержащую охлажденный льдом раствор 27,0 г (0,13 моль) метилового эфира 3-ок-си-5-фенил-4-пентиновой кислоты в 40 мл абсолютного пиридина, постепенно приливают охлажденный льдом раствор 74,0 г (0,488 моль) свежеперегнанной хлорокиси фосфора в 120 мл абсолютного пиридина. Реакционную смесь нагревают при 90—100°, перемешивая 3,5 часа. Охлажденную реакционную массу выливают в лед (1,5—2 кг) и подкисляют холодным 4 н. раствором серной кислоты до pH ~ 2. Смесь экстрагируют эфиром 5 раз порциями по 30 мл до прекращения окрашивания эфирных вытяжек. Промытый водой и раствором карбоната натрия эфирный раствор сушат сульфатом натрия и перегоняют при пониженном давлении, собирая фракцию с т. кип. 144—145°/8 мм.
Выход метилового эфира 5-фенил-2-пентен-4-иновой кислоты равен 16 г (66%), т. пл. 44—45°.
Найдено, %: С—77,7; Н—5,9.
С12Н10О2. Вычислено, %: С—77,41; Н—5,37.
Получение метилового эфира а-транс-у-цис-$-стирила'кри-ловой кислоты. В колбу емкостью 0,5 л помещают 9,3 г (0,05 моль) метилового эфира 5-фенил-2-пентен-4-иновой кислоты в 300 мл абсолютного метанола и прибавляют 2 г катализатора Линдлара. При непрерывном встряхивании реакционной смеси пропускают в течение 4—5 часов ток водорода. По окончании реакции (прекращается поглощение водорода) катализатор отфильтровывают, а метанол отгоняют. Остаток сушат сульфатом натрия и перегоняют при пониженном давлении, собирая фракцию с т. кип. ПО—11172 мм.
Выход метилового эфира а-гранс-у-цмс-р-стирилакриловой кислоты равен 8,46 г (90%); й?0 ='1,0611; Ло = 1,6010.
Найдено, %: С—76,3; Н—6,5.
С12Н12О2, Вычислено, %: С—76,6; Н—6,83.
Получение а-транс-у-цис-^-стирилакриловой кислоты. В колбу емкостью 250 мл помещают 12,9 г (0,068 моль) метилового эфира а-транс-у-цис-₽-стирилакриловой кислоты, раствор 5,7 г (0,102 лйодь) едкого кали в смеси 150 мл метанола и 2,5 мл воды. Смесь оставляют стоять в течение 4 суток при комнатной температуре. По мере стояния выпадают кристаллы калиевой соли а-транс-у-цис-р-стирилакриловой кислоты, которые затем переводят в саму кислоту подкислением водного раствора соли соляной кислотой( 1:3) до рН~ 1—2. Полученный продукт перекристаллизовывают из водно-спиртового раствора (1:1). Вещество кристаллизуется в виде белых игл с т. пл. 125—126°.
26ft
Выход а-транс-у-цис-р-стирилакриловой кислоты равен 4,2 г (36%).
Найдено, %: С—75,80; Н—5,40.
С11Н10О2. Вычислено, %: С—75,86; Н—5,74.
Строение полученной кислоты лодтверждено ИК- и ЯМР-спектрами. ИК-спектры: 996, 958 и 870 см~х. ЯМР-спектры; sJap = 16 гц, 3Jp7 = 12 гц, 3JsT = 12 гц; 3JTj = 12 гц.
ЛИТЕРАТУРА
1. О. Ddebner. Вег., 35. 2137 (1902),
2. A. Riedel. Ann., 364, 96 (1908).
3. С. Liebermann, Ber., 28, 1438 (1895).
4. Н Loh a us. Ann., 513, 219 (1934).
5. Ж- В e й г а н д-Х нльгетаг. Методы эксперимента в органической химии. М„ «Химия», 1969, с. 751.
6. Там же, с. 33.
7. Т. Zinck, D. Hagen. Ber., 17, 1814 (1884).
8. L. Claisen. Ber., 40, 3907 (1907).
9. R. Perkin. Ann., 108. 109 (1860).
\’.\?Х/,Х/-ТЕТРАКИС(КАРБОКСИМЕТИЛ)-5,5/ МЕТИЛЕНДИАНТРАНИЛОВАЯ КИСЛОТА
Н. В. ЦИРЮЛЬНИКОВА, Ю. Е. ДОРОШЕНКО, Н. К. ХАРИТОНОВА, И. В. ХВОСТОВ
НООССН,, )N. ..
НООССН/
НООС сн2
_СН2СООН N<
4 СН2СООН
СООН
O23H22N2O12 М. В. 518,44
Новый комплексон М,К,Мл,М'-тетракис(карбоксиметил)-5,5'-метилендиантраниловая кислота получена нами взаимодействием 5,5'-метилендиаитраниловой кислоты с хлоруксусной кислотой в присутствии 20%-ного раствора едкого натра. 5,5'-Метилендиантраниловая кислота синтезирована нами из антраниловой кислоты и формальдегида по методике [1].
СХЕМА СИНТЕЗА
H,NV4ho GH’-HN\/4
II I II I II I
ноос/ч^ X^4COOH HOOC/X^
HC1
/VNH2
CICH2COOH w NaOH
262
нооссн2 сн2соон
НООССН, II I II I кСНаСООН /\^\ /\^\ ноос сн2 соон
Характеристика основного сырья
Антраниловая кислота, ГОСТ 11121—65.
Формальдегид, 37%-ный раствор, ГОСТ 1625—61, техн.
Монохлоруксусная кислота, ГОСТ 5836—68.
Соляная кислота, ГОСТ 3118—67, концентрированная.
Условия получения
Синтез N,N'-метилендиантраниловой кислоты. В конической колбе емкостью 300 мл к суспензии 27,4 г (0,2 моль) антраниловой кислоты в 85 мл этилового спирта добавляют при перемешивании по каплям 8 мл 37%-него раствора формалина (0,1 моль СН2О). Смесь тщательно перемешивают 1 час и оставляют на ночь. Полученную густую массу разбавляют дистиллированной водой (~150—200 мл), фильтруют, осадок на фильтре промывают водой и сушат 5—6 часов при 50—60°.
Выход кислоты равен 27,8 г (97%), т. пл. 140°.
По литературным данным [1], т. пл. 154—156° (разл.).
Получение 5,5-метилендиантраниловой кислоты. В колбу емкостью 1 л, снабженную обратным холодильником, помещают 100 г (0,38 моль) Ы.М'-метилендиантраниловой кислоты, 200 мл соляной кислоты и 400 мл дистиллированной воды и нагревают смесь на кипящей водяной бане до растворения, после чего нагревание продолжают еще 5 часов. Затем к реакционной смеси добавляют 600 мл соляной кислоты и оставляют на ночь для кристаллизации. Выпавшие кристаллы хлор-гидрата 5,5'-метилендиантраниловой кислоты отфильтровывают иа фильтре Шотта, промывают смесью (2:1) соляной кислоты и воды и отсасывают.
Для получения чистого вещества к осадку прибавляют 200 мл соляной кислоты и 400 мл воды. Смесь нагревают на кипящей водяной бане до растворения, затем приливают 600 мл соляной кислоты и оставляют для кристаллизации. После двукратной перекристаллизации получают вещество серебристо-белого цвета. Очищенный хлоргидрат растворяют в 36%-ном растворе соляной кислоты, нейтрализуют 10%-ным раствором щелочи до pH 8—10 и постепенно прибавляют по каплям уксусную кислоту до достижения pH 5. Выпавший желтоватый осадок кислоты отфильтровывают, отмывают от
263
уксусной кислоты водой и сушат при пониженном давлении (15—20 мм).
Выход 5,5'-метилендиантраниловой кислоты равен 45 г (45%), т. пл. 233—234° (разл.).
По литературным данным [2], т. пл. 239°.
Получение N,N,N' ,М'-тетракис( карбоксиметил)-5,5'-мети-лендиантраниловой кислоты. В колбу емкостью 250 мл, снабженную мешалкой и обратным холодильником, загружают 10 г (0,035 моль) 5,5'-метнлендиантраниловой кислоты, 18 мл 20%-ного раствора едкого натра и 12 г (0,12 моль) монохдор-уксусной кислоты. Реакционную смесь нагревают до кипения иа водяной бане и кипятят, постепенно нейтрализуя выделяющийся хлористый водород добавлением 20%-ного раствора едкого иатра (по фенолфталеину). С момента установления pH 9—10 реакционный раствор кипятят еще 1 час, фильтруют, охлаждают и подкисляют 10%-иым раствором соляной кислоты до pH 1—2. Выпавший осадок через 15 часов отфильтровывают, отмывают от ионов хлора водным (—80%) спиртом и сушат при 50—60°.
Выход М,М,М',М'-тетракис(карбоксиметил) -5,5'метиленди-антраниловой кислоты равен 6,5 г (40%).
Найдено, %: С—52,3; 52,5; Н—4,8; 4,6; N—5,0; 4,8. C23H22N2O|2. Вычислено, %: С—52,8; Н—4,6; N—5,3.
ЛИТЕРАТУРА
1. О. Heller О. Fisselmann. Ann., 324, 118 (1902).
2. Н. Meh пег, J. prakt. Chem., 63. 255 (1901).
ТЕТРАЭТИЛ-1-ОКСИЭТИЛИДЕНДИФОСФОНАТ
Т етраэтил-1 -оксиэтаи-1,1 -дифосфонат, метилди(диэтилфосфои)карбииол
И. Л. КОЛПАКОВА, Л. В. КРИНИЦКАЯ, Е. И. МИРОНОВА
C2HeO CHsOCaHs
I I I
О=Р-С-Р=О
I I I
С2Н6О он осан,
С10Н24О7Р2 м. в. 318,24
Взаимодействием диэтилфосфористой кислоты с диэтиловым эфиром ацетофосфоновой кислоты при нагревании в присутствии диэтиламина [1, 2] или алкоголята натрия [3] было получено соединение, которое авторы этих работ считали тетраэтил-1-оксиэтилидеидифосфонатом. Это же соединение было получено также при взаимодействии этилфосфита натрия с хлористым ацетилом [4]. Позднее было показано, что под влиянием температуры и основных катализаторов происходит фоофонатнофосфатиая перегруппировка тетраалкил-1-окснал-килидендифосфонатов, т. е. синтезированное ранее [1—4] соединение представляет собой не тетраэтил-1-оксиэтилиденди-фосфонат, а изомерный ему диэтил-а-(диэтилфосфон-этил фосфат.
Тетраэтил-1-оксиэтилидеидифосфонат был получен реакцией диэтилфосфористой кислоты с диэтиловым эфиром ацетофосфоновой кислоты в мягких условиях в присутствии небольшого количества диэтиламина [5—7] при 40—50° с отгонкой диэтиламина и непрореагировавших веществ в вакууме при температуре, не превышающей 120° [5, 6] или той же реакцией при 80° с последующей кристализацией продукта при 0° [7].
265
В качестве побочного продукта тетраалкил-1-оксиэтили-денфосфонат образуется при реакции диэтил'фосфористой кислоты с винилацетатом [6], диэтиловото эфира ацетофосфоновой кислоты с этиловым спиртом в соотношении 1:1 в присутствии следовых количеств триэтиламина [8], диэтилового эфира ацетофоофоновой кислоты с диэтиламином в соотношении 1: 10 [8]. Однако и в этих случаях полученный продукт изомеризуется в диэтил-а-(диэтилфосфон) этилфосфат.
Эфиры (1-оксиэтилиден) дифоофоновой кислоты могут быть получены также азеотропной этерификацией кислоты спиртами [9] или переэтерификацией триалкилфосфитом [10].
Нами тетраэтил-1-оксиэтилидендифоофонат получен этерификацией (1-оксиэтилиден) дифосфоновой кислоты ортому-равьиным эфиром [11] при температуре, не превышающей 90’.
Тетраэтил-1-оксиэтилидендифосфонат может быть использован в качестве смазочного материала, пестицида и фармацевтического препарата [2, 4], в парфюмерной промышленности [12], в качестве добавок к детергентам [13].
СХЕМА СИНТЕЗА
РО(ОН)2 РО(ОС2Н6)2
/ I
СН3С-ОН 4-4(С2НВО)3СН -> СН3—С —ОН +
\ I
РО(ОНЬ РО(ОС2Нв)а
+ 4C2HsOH 4- 4НС(0)ОСаН6
Характеристика основного сырья
(1-Оксиэтилиден) дифосфоновая кислота, ТУ П 849—69, ч. Ортомуравьиный эфир, МРТУ 6—09—932—63, ч.
Условия получения
В трехгорлой колбе емкостью 1,0 л, снабженной мешалкой, обратным холодильником и термометром, нагревают до 50— 60° 82,4 г (0,4 моль) (1-оксиэтилиден) дифосфоновой кислоты и 356,8 г (2,4 моль) ортомуравьиного эфира и выдерживают при этой температуре при перемешивании 6 часов. Затем добавляют еще 119,0 г (0,8 моль) ортомуравьиного эфира, меняют обратный холодильник на нисходящий и, не выключая мешалку, отгоняют муравьиноэтиловый эфир и этиловый спирт. По мере отгонки температура реакционной массы постепенно повышается. Когда она достигает 90°, нагревание прекращают.
Отгоняют при пониженном давлении избыток ортомуравьиного эфира, следя за тем, чтобы температура реакционной 266
массы не поднималась выше 90°. В колбе остается вязкая жидкость желтоватого цвета (см. примечание).
Выход тетраэтил-1-оксиэтилидендифосфоната равен 127,2 г (90%), «“='1,4506, </4“ = 1,1815.
По литературным данным [5], «“ = 1,4510, d4“ = 1,1870.
Найдено, %: С—38,13; Н—8,02; Р—18,80.
CioHjiOtPs. Вычислено, %: С—37,7; Н—7,55; Р—19,50.
Примечание.
Прн перегонке при пониженном давлении (1—2 мм) тетраэтил-1-этнл ндендифосфоната происходит его перегруппировка в изомерный диэтил-а--диэтилфосфон) этилфосфат, сопровождающяся частичным разложением вещества.
ЛИТЕРАТУРА
1. R. L. Me. Connell, Н. W. Coove г. J. Amer. Chem. Soc., 78, 4450 (1956).
2. Швейц, пат. 316731 (1956); С. А., 52, 2066 (1958).
3. J. A. Cade J. Chem. Soc., 1959, 2272.
4. Швейц, пат. 321397 (1957); С. А., 52, 2066 (1958).
5. А. Н. Пудовик, И. В. Коновалова. Докл. АН СССР, 143, 875 (1962).
6. А. Н. П у до в и к, И. В. Ко н о в а л о в а. Ж. общ. химии, 32, 467 (1962).
7. С. J. Fitch, К. Moedritzer. J. Amer. Chem. Soc., 84, 1876 (1962).
8. А. П. П а ш и н к и н, T. X. Газизов, A. H. П у д о в н к. Ж. обш. химии, 40, 28 (1970).
9. Франц, пат. 1455979 (1966); С. А., 67, 3370 (1967).
10. D. A. Nicholson, Н. Vaughn, J. Organ. Chem., 36 , 3843 (1971).
11. D. A. N 1 с h о 1 s о n, W. A. C i 11 e у, О. T. Q u i m b y. J. Organ. Chem , 35, 3149 (1970).
12. Англ. пат. 990660 (1965); С. A., 63, 4092 (1965).
13. Франц, пат. 1506840 (1967); С. А., 70, 30328 (1969).
ТИОСЕМИКАРБАЗОН р-(5-ХЛОР-2-ФУРИЛ) АКРОЛЕИНА
Фуртизол
В. П. КЕРЕНЦЕВА, М. Д. Л И ПАНОВ А
О-С1Д J—СН—CHCH=N—NHCSNHj xoz
C8H8C1N3OS
M. в. 229,68
Фуртизон используется в качестве реактива для фотомет рического определения палладия и платины [1—3]. Реактив обладает высокой чувствительностью и избирательностью и по аналитическим свойствам ие уступает лучшим реактивам, предложенным ранее для определения указанных элементов.
СХЕМА СИНТЕЗА
С1Д Дсн=СН-СНО + h2nnhcsnh2 -> xoz
-> С1Д J-ch=chch=n-nhcsnh2 + нао хо
Характеристика основного сырья
Тиосемикарбазид, ТУ МХП 2490—51.
5-Хлорфурилакролеин, т. пл. 57—58°; получение см. [4].
Этиловый спирт, ГОСТ 5962—51.
Условия получения
В круглодонной колбе емкостью 100 мл, снабженной обратным холодильником, растворяют 0,91 г (0,01 моль) тиосе
268
микарбазида в 50 мл кипящей воды, охлаждают до комнатной температуры и добавляют 1,56 г (0,01 моль) 5-хлорфурил-акролеина, растворенного в 10 мл спирта. Реакционную смесь нагревают на кипящей водяной бане 10—15 минут. По охлаждении тиосемикарбазон кристаллизуется. Осадок отфильтровывают, промывают 25%-ным спиртом и сушат. Вещество перекристаллизовывают из 50%-ного спирта (25 мл).
Выход тиосемикарбазона Р-(5-хлор-2-фурил) акролеина равен 1,85 г (80%). Вещество представляет собой бесцветные кристаллы с т. пл. 152—153°.
Найдено, %: С—41,92; Н—3,52; N—18,52; CI—15,80. CsHeClN3OS. Вычислено, %: С—41,83; Н—3,51; N—18,30; С1—15,44.
ЛИТЕРАТУРА
1. В. П. Керенцева, И. С.Мустафин, М. Д. Липанова. Применение органических реактивов в анализе. Саратов, изд-во СГУ, 1968, с. 46.
2. В. П. Керенцева, И. С. Мустафин. Тезисы докладов межвузовского совещания по физико-химическим и аналитическим свойствам комплексных соединений редких и цветных металлов и их применению. Ростов н/Дону, 1964, с. 59.
ЗВ. П. Керенцева, И. С. Мустафин. Тезисы докладов II Всесоюзного совещания по применению органических реактивов в аналитической химии. Саратов, изд-во СГУ, 1966, с. 154.
4. А. А. П о и о м а р е в, М. Д. Л и п а н о в а. Ж. общ. химии, 32. 2535 (1962).
ТРИМЕЛАКТОВАЯ КИСЛОТА
Бензол-1,2,4-трикарбоновая кислота
Г. А. КРЕЙМЕР
^\-соон
HOOC-^Z'-COOH
CgHfiOe
М. в. 210,15
Тримеллитовую кислоту получают окислением псевдокумола и некоторых других 1,2,4-тризамещенных бензолов кислородом [1], азотной кислотой [2—4] или перманганатом калия [5, 6].
Нами разработана методика получения тримеллитовой кислоты окислением псевдокумола перманганатом калия в воднощелочной среде с выходом около 70%.
СХЕМА СИНТЕЗА
^\-сн8
КМпО4
н8с--Ч/(-сн,
-COOK
КООС-Ч/-СООК
НС1
^х-соон
НООС-^-СООН
270
Характеристика основного сырья
Псевдокумол, МРТУ 6—09—2596—65, ч.
Натр едкий, ГОСТ 4328—66, ч.
Перманганат калия, ГОСТ 5777—71, техн.
Соляная кислота, ГОСТ 3118—67, ч.
Этиловый спирт гидролизный, ГОСТ 8314—57.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешалкой, термометром и обратным холодильником, помещают 40 г (0,33 моль) псевдокумола, 1260 мл воды, 20 г едкого натра и 107 г перманганата калия. Реакционную смесь при интенсивном размешивании нагревают до кипения и кипятят 1 час. После охлаждения смеси до 80° прибавляют еще 107 г перманганата и вновь кипятят 1 час. Указанную операцию повторяют еще раз, после чего добавляют 36 г перманганата и кипятят 3 часа. Реакционная масса в конце кипячения должна содержать избыток перманганата (фиолетовая окраска раствора). Всего на окисление расходуется 357 г 95%-ного перманганата (2,14 моль; избыток 7%).
Массу охлаждают до 60°, избыток перманганата раскисляют 10—20 мл спирта, раствор фильтруют от двуокиси марганца, осадок на фильтре промывают тремя порциями воды по 200 мл. Фильтрат упаривают до объема 600 мл и нейтрализуют, медленно прибавляя 300 мл соляной кислоты (см. примечание 1). Раствор оставляют на 16 часов, причем в течение первого часа его несколько раз осторожно помешивают для того, чтобы выпадал более однородный осадок (см. примечание 2). Далее суспензию охлаждают льдом до 5—10°. Осадок фильтруют и промывают охлажденной до 10° дистиллированной водой четырьмя порциями по 50 мл. Сушат при 50—60°.
Выход трнмеллитовой кислоты равен 52 г (74,3%).
Кислота пригодна для препаративных целей без дополнительной очистки. В случае необходимости ее перекристаллизовывают из 4-кратното количества дистиллированной воды.
Выход трнмеллитовой кислоты после перекристаллизации равен 48,4 г (69%), т. пл. 228—230°; кислотное число: найдено— 798; вычислено — 801.
По литературным данным [3, 4, 6], т. пл. 228°.
Примечания:
1. Нейтрализация вначале сопровождается сильным вспениванием, по этому кислоту приливают медленно.
2. При быстром охлаждении выпадает мелкий, труднофильтрующий-ся осадок.
271
ЛИТЕРАТУРА
1. Пат. США 3155717 (1964); С. А. 62, 491а (1965).
2. Пат. США 3227751 (1966); С. А. 64, 8096е (1966).
3. Н. Я- Качу рн на, К. В. Прокофьев, В. Л. Казанский, А. Г. Трупанова. Нефтехимия, 5, 880 (1965).
4. Г. С. Миронов, В. Д. Шенн, М. И. Фарберов. Ж. прнкл. химии, 41, 868 (1968).
5. Пат. ГДР 10505 (1955); С. А., 53 199d (1959).
6. Н. М. М о р л я и, Ж- Л. Б а г р а т у н и, А. К. Гегамян. Методы получения химических реактивов и препаратов, вып. 23. М., ИРЕА, 1971, с. 133.
D,L-ТРИПТОФАН
Б. M. КОТЛЯРЕВСКАЯ, И. И. ГУБЕНКО
СН,-СН-СООН
nh2
CnHizN^Oa
М. в. 204,23
Триптофан применяется в биохимии, микробиологии, медицинской промышленности, сельском хозяйстве.
В литературе описаны синтезы D,L-триптофана циклизацией фенилгидразонов по Фишеру [1—4], из грамина [5—7], из индола [8—10].
Используя работы [8—10], мы разработали методику получения ВЛ-триптофана по схеме: индол—иидол-3-альдегид— индолальгидантоин — триптофан.
СХЕМА СИНТЕЗА
N(CH,)aCHO
РОС1, "*
н2с— со
I I
HN NH
^со/ диэтаноламин, абс. спирт
-> ----||~СН,=С----со
HN NH
I хсох
18 Зак. 1164
273
_ r ---------||-СН4-СН-СООН
'^Z'4N/ NH2
I H
Характеристика основного сырья
Индол, ВТУ РУ 1064—54, техн.
Диметилформамид, ГОСТ 5703—70, ч.
Хлорокись фосфора, МРТУ 6—09—337—63, ч. Диэтаноламин, СТУ 12—10—106—61, ч.
Сернистый аммоний, ГОСТ 3767—56, ч. д. а.
Соляная кислота, ГОСТ 3118—67, х. ч.
Натр едкий, ГОСТ 4328—66, ч.
Гидантоин, МРТУ 6—09—1873—64, ч.
Этиловый спирт, 99,8 %-ный РТУ РСФСР 622—57.
Этиловый спирт, 96%-ный, ТУ 3—66—65.
Условия получения
Синтез индолалъдегида. В четырехгорлую колбу емкостью 0,5 л, снабженную термометром, капельной воронкой, мешалкой и пришлифованным обратным холодильником с осушительной трубкой, заполненной безводным сернокислым кальцием, и погруженную в баню с ледяной водой, помещают 100 мл (1,315 моль) диметилформамида и по каплям за 1 час прибавляют 33 мл (0,358 моль) хлорокиси фосфора, при этом температура в колбе не должна превышать 20°. Затем к реакционной смеси при энергичном перемешивании и охлаждении по каплям добавляют раствор 36 г (0,307 моль) индола в 26 мл (0,342 моль) диметилформамида, следя за тем, чтобы температура реакционной массы не превышала 30°. Ледяную баню заменяют баней с теплой (30—35°) водой, доводят температуру в колбе до 35° и выдерживают реакционную смесь при 35° в течение 45 минут.
В стакан из термостойкого стекла емкостью 2 л загружают 400 г размельченного льда и выливают в лед реакционную массу. К полученному прозрачному раствору приливают порциями 200 мл раствора едкого натра (58 г в 309 мл дистиллированной воды) с такой скоростью, чтобы раствор оставался кислым, затем быстро добавляют оставшиеся 115 мл раствора едкого натра.
Реакционную массу нагревают на электрической плитке до кипения, кипятят 1 минуту и оставляют при комнатной температуре. Выпавшие кристаллы отфильтровывают, промывают, нй фильтре двумя порциями воды по 200 мл и сушат в сушильном шкафу при 100°. 274
Выход индолальдегида равен 39,4 г (89,6%), т. пл. 192— 193°.
По литературным данным [10], т. пл. 197—198°.
Получение индолальгидантоина. В двугорлую колбу емкостью 0,5 л, снабженную мешалкой и пришлифованным холодильником, загружают 25 г (0,172 моль) индолальдегида, 19 г (0,225 моль) гидантоина, 52 мл (0,448 моль) диэтаноламина и 75 мл абсолютного спирта. Смесь при энергичном перемешивании нагревают 2 часа на кипящей водяной бане. .При этом раствор постепенно густеет и образуется желтая тестообразная масса, которую отфильтровывают. Осадок загружают вновь в реакционную колбу, добавляют 100 мл дистиллированной воды и при энергичном перемешивании нагревают 15 минут иа кипящей водяной бане. Массу отфильтровывают, осадок промывают на фильтре 75 мл 96%-ного этилового спирта и сушат в сушильном шкафу при 100°.
Выход индолальгидантоина равен 34,1 г (87,0%), т. пл. 318—322°.
По литературным данным [8, 9], т. пл. 320—322°.
Получение D.L-триптофана. В автоклав емкостью 1 л, рассчитанный на давление до 25 атм и снабженный мешалкой, электрообогревом, манометром и потенциометром для регистрации температуры, загружают 34 г (0,324 моль) индолальгидантоина, 486 мл (1,14 моль) 16%-ного раствора сернистого аммония.
Реакционную смесь нагревают 8 часов при 150—155°, давлении 18—20 атм и перемешивании. Реакционный раствор (желтого цвета) выгружают с помощью водоструйного вакуум-насоса и переносят в перегонную колбу. Раствор упаривают досуха нагреванием при пониженном давлении (80°/25 мм). К остатку, приливают 170 мл 96%-ного этилового спирта и образовавшуюся смесь фильтруют. Осадок, состоящий из смеси триптофана и серы, переносят с фильтра в стакан емкостью 0,5 4 добавляют 142 мл 1 н. соляной кислоты (для растворения триптофана); нерастворившуюся часть отфильтровывают.
В фильтрат добавляют 2,5 г угля и нагревают 30 минут при 60—70° для осветления раствора. Уголь отфильтровывают, а раствор триптофана нейтрализуют 130 мл 1 н. раствора едкого натра до достижения pH 5—6 (по универсальной индикаторной бумаге) и оставляют для кристаллизации на ночь в холодильнике.
Выпавшие кристаллы технического продукта отфильтровы вают, промывают 3 раза водой порциями по 50 мл и 50 мл холодного абсолютного спирта и сушат при 100°.
Выход технического D.L-триптофана равен 15 г (49,8%).
Для очистки 18 г технического продукта вносят в горячий (70°) раствор 4,5 г едкого натра в 165 мл воды, добавляют 1 г IS* 275
активированного угля и нагревают 10 минут при 70°. Раствор фильтруют, промывая фильтр 20 мл горячей дистиллированной воды. К охлажденному фильтрату добавляют 81 мл этилового спирта, 6,75 мл уксусной кислоты и оставляют для кристаллизации. Выпавший осадок отфильтровывают, промывают холодной водой и спиртом и сушат при 100°.
Выход триптофана равен 14,5 г (48,2% или 37,3% в расчете на индол), т. пл. 282—284°.
По литературным данным [8, 11], т. пл. 283—285° (из водного спирта).
ЛИТЕРАТУРА
1. А. А. М о е, D. I. Warner. J. Amer, Chem. Soc., 70, 2763 (1948).
2. P. F. A lb e r t s о n, S. Archer. J. Amer. Chem. !Soc., 66, 500 (1944).
3. C. Mannlch, W. Rock. Ber., 75, 809 (1942).
4. G. К a m a c h i a. Nippon Kagaku Zasshi, 86, 856 (1965).
5. H. R. Snyder, C. W. Smith, J. Amer. Chem. Soc., 66, 350 (1944).
6. D. J. Weisblatt, D. A. Lyttle. J. Amer. Chem. Soc., 71. 3079 (1949).
7. D. A. Lyttle, D. J. Weisblatt. J. Amer. Chem. Soc., 69, 2118 (1947).
8. W. 1. Boyd, Robson. Biochem. J. 29, 542, 546, 555, 2256 (1935)
9. Пат. США 2435399 (1948); С. A., 42, 4613d (1948).
10 C. W. Smith. J. Amer. Chem. Soc., 76, 3842 (1954).
11. Справочник химика, т. 2. M., ГХИ, 1963.
ТРИХЛОРАНГИДРИД ТРИМЕЛЛИТОВОЙ кислоты
я. А. СЕРЕДНЯЦКИЙ, В. Н. ФЕДЫНА
^\-СОС1
ClOC-^-COCl
C9H3CI3O3 м. в. 265,43
Трихлорангидрид тримеллитовой кислоты получают взаимодействием кислоты с пеитахлоридом фосфора [1]. Однако более удобным хлорирующим агентом, применяемым для синтеза хлорангидридов карбоновых кислот, является тионил-хлорид, активность которого в некоторых случаях повышают добавкой катализатора [2]. Из литературных данных известно, что тримеллитовая кислота и тримеллитовый ангидрид, реагируя с тионилхлоридом, образуют ангидридо-хлорангид-рид [1, 3].
Используя ранее разработанный метод получения хлоран-гидридов действием тионилхлорида на циклические ангидриды [4, 5], мы синтезировали в присутствии диметилформамида трихлорангидрид тримеллитовой кислоты.
СХЕМА СИНТЕЗА
ДМФА
SOC12 “*
/Ч-СОС!
CIOC-'L J-COC1
НС1 + so2
277
Характеристика основного сырья
Тримеллитовый ангидрид, ТУ 14П—994—69, ч.
Тримеллитовая кислота, ТУ 14П—974—69, ч. Тионилхлорид, МРТУ 6—09—2602—65, ч. Диметилформамид, ТУ 7П—11—69, х. ч.
Условия получения
Установка для синтеза состоит из стандартных деталей с нормальными шлифами: трехгорлой конической колбы емкостью 150 мл, обратного холодильника, капельной воронки и кармана с термометром. Перегонку трихлорангидрида ведут из реакционной колбы, используя обычную установку для пе< регонки при пониженном давлении. Применяют игольчатую колонку высотой 100 мм.
В трехгорлую колбу загружают 30 г (0,15 моль) тримелли-тового ангидрида и приливают из капельной воронки 37 мл (0,51 моль) тиоиилхлорида, к которому добавлено 0,55 г (2,5% по ангидриду) диметилформамида. Колбу с реакционной смесью помещают в терморегулируемую баню с температурой 130 + 5° и нагревают в течение 1 часа. Об окончании реакции судят по прекращении выделения газов (ом. примечание 1), а также по возрастанию температуры реакционной смеси до 110° (см. примечание 2, 3). Избыток тиоиилхлорида отгоняют при атмосферном давлении, а остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 142— 14372 мм.
Выход сырого трихлорангидрида равен 37,0 г (85%). После вторичной перегонки получают продукт с т. кип. 141 — 14272 мм, d420 = 1,516, «“= 1,592.
Найдено, %: С—40,92; Н—1,16; С1—39,71. С9Н3С13О3. Вычислено, %: С—40,74; Н—1,13; С1—40,06
По литературным данным [1], т. кип. 141—14272 мм, d420 — = 1,510, я® = 1,590.
Синтез трихлорангидрида из трнмеллитовой кислоты проводят в аналогичных условиях. Выход продукта составляет 80—85%.
Примечания:
1. За ходом реакции удобно следить, пропуская выделяющиеся газы в воду или раствор карбоната натрия.
2. Вследствие рециркуляции избытка тиоиилхлорида во время реакциг в колбе поддерживается температура 80—90°. После окончания реакции температура реакционной смеси резко повышается.
3. Нагревание реакционной смеси при температуре свыше 110° после прекращения выделения газов приводит к осмолеиию продукта.
278
Литература
1. Г. С. Миронов, В. Д. Шеин, М. И. Фарберов. Ж. прикл. химии, 41, 4, 868 (1968).
2. К. В е й г а н д-Х ильдетаг. Методы эксперимента в органической химии. М., «Химия», 1968, с. 231.
3. Пат. ГДР 31356 (1964).
4. Я. А. Середницкий, Е. М. Довбенчук, Я. Н. П и р И г. Сб. «Химия и хим. технол.». Изд-во Львовск. ун-та, 1969, с. 56.
5. Я- А. Середницкий, Е. М. Довбенчук, Д. К. Толопко, В. В. Шибанов. Вюник ЛШ, Х1м1я та технол. орг. речовин, 58, 21 (1971).
2,4,6-ТР ИХЛ ОРАН ИЛ И Н
Н. И. ДЫХАНОВ, Р. Ф. ВИДЕНИНА, Т. В. ПЕРОВА, И. В. ПОКОТЫЛО, В. И. БАЗАКИН
CkHiCIsN
М. в. 196.47
В отличие от бромирования анилина [1], его хлорирование (по наблюдениям авторов данной статьи) газообразным хлором даже в присутствии следов влаги сопровождается энергичным окислением и приводит, в основном, к образованию черного анилинового красителя; выход 2,4,6-трихлоранилина при этом не превышает 10%. Известны способы хлорирования анилина в неводной среде с помощью соединений, содержащих подвижный хлор, — треххлористого азота (2], М.ЬГ-дихлорэтилами-на (3], М,2,4-трихлорацетанилида [4] и хлористого сульфурила [5]. В первых трех случаях выход 2,4,6-трихлоранилина сравнительно невысок, а хлорирующие агенты 'мало доступны и взрывоопасны.
В основу предлагаемой методики положен метод [5], заключающийся в хлорировании анилина или его гидрохлорида избытком сульфурилхлорида в кипящем бензоле. Указанный в работе [5] выход 2,4,6-трихлоранилина (более 90%) относится к техническому продукту, но методика его очистки авторами не приводится. По нашим наблюдениям, выход чистого 2,4,6-трихлоранилина, полученного этим способом из гидрохлорида анилина примерно на одну треть выше, чем из анилина, и составляет около 60%.
280
СХЕМА СИНТЕЗА
NH2-HC1
+ SO2C12
nh2 I
и
I
Cl
Характеристика основного сырья
Аннлии гидрохлорид, ГОСТ 5822—69, ч. д. а.
Сульфурил хлористый, т. кип. 68—69°, получение см. [6]. Бензол, ГОСТ 5955—68, ч. д. а.
Метиловый спирт синтетический, ГОСТ 6995—67, ч.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, термометром и обратным холодильником, помещают 20 г (0,154 моль) гидрохлорида анилина, 200 мл сухого бензола и 40 мл (68 г; 0,535 моль) хлористого сульфурила. Образовавшуюся суспензию при перемешивании нагревают на водяной бане до начала выделения пузырьков газа (температура s массе ~40°), перемешивают 1 час, затем температуру в массе повышают до 70—80° (слабое кипение смеси) и выдерживают при этой температуре ~5 часов до полного прекращения выделения хлористого водорода (проба на конго). Об окончании реакции судят по исчезновению в реакционной массе кристаллов гидрохлорида анилина.
Заменяют обратный холодильник на нисходящий и при пониженном давлении (80—100 мм) досуха отгоняют смесь избыточного сульфурилхлорида и бензола (см. примечание). Сухой остаток суспендируют в 200 мл воды и фильтруют с отсасыванием. Осадок на фильтре промывают дистиллированной водой до отсутствия ионов хлора (проба с нитратом серебра) и сушат в вакуум-сушильном шкафу при 40—45°. Получают 27,5—28,0 г (90—92%) технического 2,4,6-трихлоранилина в виде темио-коричневого порошка с т. пл. 72—76°.
Технический продукт (27,5—28 г) растворяют в смеси 120 мл метилового спирта и 10 мл концентрированной соляной кислоты и при перемешивании осторожно добавляют 0,5— 0,6 г цинковой пыли. После 15-минутного кипячения раствор фильтруют, фильтрат охлаждают до 8—10°. Выделившиеся светло-сиреневые кристаллы отфильтровывают и, не высушивая, перекристаллизовывают из 100 мл метилового спирта с 1 г активированного угля. Получают 15—16 г (~5О°/о) 2,4,6-281
Трйхлорйнилина в виде бесцветных (или с легким розоватым оттенком) игл с т. пл. 77—78°.
Метанольные маточные растворы разбавляют в 5—6 раз водой, выделившийся осадок технического 2,4,6-трихлоранили-на отфильтровывают и вновь очищают так, как описано выше.
Суммарный выход чистого 2,4,6-трихлоранилина равен 18—19 г (~6О°/о в расчете на гидрохлорид анилина).
Примечание,
К отогнанному бензолу прибавляют примерно равный объем 5%-ного раствора едкого натра, отделяют бензольный слой, промывают водой до нейтральной реакции, сушат хлористым кальцием, перегоняют и используют в очередном опыте.
ЛИТЕРАТУРА
1. К. Silberstein. J. prakt. Chem., 27, 101 (1883).
2. W. Hentschel. Ber., 30, 1434 (1897).
3. A. Pierson, К. H e u m a n n. Ber., 16, 1047 (1883).
4. F. D. C h a 11 a w a y, R. С. T. Orton. J. Chem. Soc., 79 , 464 (1901).
5. W. Eller, L. Klemm. Ber., 55, 217 (1922).
6. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы. М., Госхимиздат, 1955, с. 508.
ТРИЭТИЛФОСФАТ
А. Л. ЛИФИЦ, Л. Я- ДИСТАНОВА, А. А. ВЕИЦМАН
с2н5о
С2Н6О-Р=О
с,н5о
М. в. 182,12
CeHisChP
Триалкилфосфаты применяются как растворители, пластификаторы, катализаторы, инсектициды или комплексообразующие агенты [1].
Общеизвестен метод получения триалкилфосфатов взаимодействием хлорокиси фосфора с соответствующим спиртом в присутствии пиридина [2, 3] или инертного растворителя [4].
Нами усовершенствован метод получения триэтилфосфата из хлорокиси фосфора и этилата натрия [5] и показана возможность получения продукта с высоким выходом без применения растворителя.
СХЕМА СИНТЕЗА
C2HeONa + РОС13 _-± (С2Н5О)3РО.
Характеристика основного сырья
15—16%-ный раствор этилата натрия в этаноле, техн. Хлорокись фосфора, СТУ 53—244—62, техн.
Условия получения
В четырехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром, обратным холодильником и капельной воронкой, помещают 680 г (1,58 моль) спиртового раствора эти
283
лата натрия и при перемешивании й температуре 20—22° медленно, за 1—1,5 часа, прибавляют по каплям 97,3 г (58,3 мл-, 0,63 моль) хлорокиси фосфора. По окончании прибавления хлорокиси фосфора среда должна быть нейтральной по фенолфталеину, в противном случае необходимо прибавить еще ие сколько капель хлорокиси фосфора. Реакционную массу перемешивают еще 2—3 часа и фильтруют. Из фильтрата отгоняют спирт, а остаток перегоняют при пониженном давлении, собирая фракцию, кипящую при 89—90°/5 мм.
Выход триэтилфосфата равен 87,5 г (76%), т. кил. 215,8°, содержание основного вещества 99,5%, с/420 = 1,0682, =
= 1,4052.
По литературным данным [6], т. кип. 216°, 98—98,5°/9 мм, d^=\№ti, = 1,40674.
ЛИТЕРАТУРА
1. Пат. ФРГ 1219013; С. А., 65, 6787а (1966).
2. I. Nolle г, R. Dutton. J. Amer. Chem. Soc., 55, 424 (1933).
3. Синтезы органических препаратов, сб. 2. М., Инлитиздат 1949, с. 138.
4. Пат. США 3079419; С. А., 59, 6257d (1963).
5. Н. L i m р г 1 с h t. Ann,, 134, 347 (1965).
6. Справочник химика, т. 2. Л.-М., Госхимиздат, 1963, с. 1146; т. 4, 1965, с. 826.
1,Ю-ФЕНАНТРОЛИН-2,9-ДИАЛЬДЕГИД
онс
В. М. ДЗИОМКО, Б. В. ПАРУСНИКОВ
сно
c14h8n2o2
М. в. 236,23
В литературе 1,10-фенантролин-2,9-диальдегид не описан. Его можно получить окислением 2,9-диметил-1,10-феиантроли-ка двуокисью селена в диоксане с выходом до 70%.
СХЕМА СИНТЕЗА
•1/2НгО SeO3 >
ОНС
СНО
Характеристика основного сырья
2,9-Диметил-1,10-фенантролин, гемигидрат, т. пл. 161—163°, юлучение см. с. 102 этого сборника.
Селенистый ангидрид, МРТУ 6—09—1553—64, ч., свеже-юзогнанный.
285
Диоксан, ГОСТ 10455—63, ч. д. а., перегнанный.
Пиридин, ГОСТ 2747—44, ч„ высушенный над едким натром и перегнанный.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, обратным холодильником и капельной воронкой, помещают 9,43 г (0,085 моль) ювежевозогнанного селенистого ангидрида, 300 мл свежеперегнанного диоксана и 6 мл дистиллированной воды. Прн нагревании на глицериновой бане (температура бани 125—135°) смесь доводят до кипения и, когда селенистый ангидрид растворится, добавляют за 4—5 минут раствор 8,69 г (0,04 моль) гемигидрата 2,9-диметил-1,10-фенантро-лина в 200 мл диоксана. Реакционную массу размешивают при кипении еще 30 минут, горячий раствор декантируют и охлаждают до 10—15°. Выпавший осадок отфильтровывают, растворяют при нагревании в смеси 40 мл 36—37%-ной соляной кислоты и 200 мл воды, раствор обрабатывают активированным углем и осторожно нейтрализуют 20%-ным раствором углекислого натрия. Осажденный продукт отфильтровывают, промывают водой и высушивают сначала в вакуум-эксикато ре над едким натром, затем в вакууме при 100° (см. примечание 1).
Выход 1,10-фенантролин-2,9-диальдегида равен 6,0—6,6 .? (64—70%)- Для очистки вещество перекристаллизовывают из пиридина (20 мл иа 1 г вещества) с применением активированного угля и получают около 4 г (42%) 1,10-фенантролин--2,9-диальдегида в виде розовых игл (см. примечания 2 и 3).
Примечания:
1. Переосажденное и высушенное в вакуум-эксикаторе вещество пред ставляет собой, вероятно, дигидрат 1,10-фенантролии-2,9-диальдегида. При последующем высушивании в вакууме при 100° потеря в весе составляет 13,6—14,0%; содержание воды, вычисленное дли формулы CnH8N2O2-2H2O, равно 13,23%.
2. Вещество не имеет четкой температуры плавления или разложения. При нагревании оно постепенно краснеет, затем чернеет и плавится около 245°.
3. Образец для анализа в виде желтоватых игл был получен путем пропускания раствора в хлороформе через колонку с окисью алюминия и последующей трехкратной перекристаллизацией из сухого пиридина.
Найдено, %: С—71,17; 71,44; Н—3,40; 3,73; N—11,48; 11,57. C,4H8N2O2. Вычислено, %: С—71,18; Н—3,41: N—11,86.
Мв-ФЕНАНТРОЛ И Н-2.9-ДИ АЛЬДОКСИМ
Диоксим 1,10-фенантролин-2,9-диальдегида
В. М. ДЗИОМКО, Б. В. ПАРУСНИКОВ
//--\____У----Ч .Hj0
\=N Z
НС СН
NOH NOH
Ci4H10N4Oa. Н2О
М. в. 284,28
В литературе 1,10-фенантролин-2,9-диальдоксим не описан. Он образуется с почти количественным выходом из 1,10-фе-нантролин-2,9-диальдегида и гидроксиламина в пиридиновом растворе.
Гидрат 1,10-фенаитролии-2,9-диальдоксима представляет собой мелкие бледно-кремовые иглы с температурой разложения 260°, растворимые в щелочах, очень плохо растворимые з органических растворителях, нерастворимые в воде.
СХЕМА СИНТЕЗА
V «I Г 1 . — , \
\ /-----\ NH8OH,HaO
онс сно
287
Характеристика основного сырья
1,10-Фена нтролин-2,9-диальдегид; получение см. с. 285 этого сборника.
Гидроксиламин солянокислый, ГОСТ 5456—65, ч.
Пиридин, ГОСТ 5747—44, ч., перегнанный.
Условия получения
В круглодонной колбе емкостью 100 мл растворяют 3,54 г (0,015 моль) 1,10-фенантролин-2,9-диальдегида в 40 мл кипя-щего пиридина, добавляют раствор 2,78 г (0,04 моль) солянокислого гидроксиламина в 20 мл воды. Реакционную массу кипятят 5 минут, охлаждают до комнатной температуры и выливают в 400 мл воды. Осадок отфильтровывают, промывают водой до исчезновения запаха пиридина и высушивают в ва-куум-эксикаторе над едким натром.
Выход моногидрата 1,10-фенантролин-2,9-диальдоксима равен 4,1—4,2 г (96—98,5%); т. разл. ~255°.
Для очистки вещество растворяют при нагревании в 250 мл 0,2 н. раствора едкого натра, обрабатывают углем и фильтруют. К горячему раствору приливают 40 мл 10%-ной уксусной кислоты, добавляют несколько кристалликов солянокислого гидроксил амина (см. примечание 1) и охлаждают. Светло-кремовый мелкокристаллический осадок отфильтровывают, промывают водой и высушивают в вакуум-эксикаторе над едким натром. Выход переосажденного гидрата диоксима равен 3,8 г (89%), т. разл. 257°, (см. примечание 2).
Примечания:
1. Если переосаждение проводить без добавки солянокислого гндро-ксиламииа, то переосаждеииый продукт имеет более низкую температуру разложения.
2. Образец для анализа с т. разл. 260° был приготовлен трехкратным переосаждеиием из щелочного раствора.
Найдено, %: С—59,50; 59,20; Н—4,20; 4,27; N—19,69;
19,92; НгО — 6,37% (сушка в вакууме при 100°).
C|4Hi0N4O2 • Н2О. Вычислено, %: С—59,15; Н—4,26; N—19,71; Н2О—6,34.
1.10-ФЕНАНТРОЛИН-2.Й-ДИКАРБОНОВАЯ КИСЛОТА
в: м. дзиомко, б. в. парусников
C14H8N2O4
М. в. 268,23
В литературе 1,10-фенаитролии-2,9-дикарбоновая кислота не описана. Она получена нами при щелочном гидролизе соответствующего динитрила.
Безводная кислота представляет собой желтоватое кристаллическое вещество, плавящееся с разложением при 216°, нерастворимое в воде и разбавленных минеральных кислотах, немного растворимое в горячем этаноле, хорошо растворимое в щелочах.
СХЕМА СИНТЕЗА
со,н СО8Н
Характеристика основного сырья
Динитрил 1,10-фенантролин-2,9-дикарбоновой кислоты,
т. пл. 364°; получение см. с. 112 этого сборника.
Натр едкий, ГОСТ 4328—66, х. ч.
Условия получения
Смесь 2,99 г (0,013 моль) днннтрила 1,10-фенантролнн-2,9-дикарбоновой кислоты и 100 мл 5 %-него раствора едкого нат-10 Зак. 1164 289
ра кипятят 2 часа с обратным холодильником, i ирячий раствор обрабатывают углем, фильтруют, разбавляют водой (200 мл) и подкисляют смесью 15 мл концентрированной соляной кислоты и 75 мл воды. Через 1—2 дня бесцветный осадок отфильтровывают (см. примечание 1), высушивают сначала в вакуум-эксикаторе над едким натром, затем в вакууме при 100°.
Выход 1,10-фенантролин-2,9-дикарбоновой кислоты равен 3,1 г (89%); т. пл. 214° (см. примечание 2).
Примечания:
1. При подкислении щелочного раствора образуется густая, плохо фильтрующаяся суспензия. При стоянии в водной среде аморфный осадок превращается в мелкие кристаллы, которые легко отфильтровать и промыть.
2. Образец для анализа с т. пл. 216° (разл.) получен переосаждени-ем из щелочного раствора и высушен в вакууме при 100°.
Найдено, %: С—62,58; 62,85; Н—3,12; 3,13; N—10,19; 10,50. Ci4H8N2O4. Вычислено, %: С—62,69; Н—3,01; N—10,44.
2-ФЕНИЛ-2-ПРОПАНТИОЛ
М. А. КОРШУНОВ, В. Е. МАЗАЕВ
SH
I
СНз-С-СНз
C9H12S
М. в. 152,26
2-Фенил-2-пропантиол в литературе не описан и получен нами впервые [1] присоединением газообразного сероводорода к а-метилстиролу в присутствии каталитических количеств хлористого алюминия.
СХЕМА СИНТЕЗА
SH
СНз-С=СН2 + H2S -^3
CH3-C-CH3
Характеристика основного сырья
а-Метилстирол, ГОСТ 12531—67, техн., перегнанный.
Толуол, ГОСТ 5789—69, ч. д. а., осушенный азеотропно.
Алюминий хлористый, ГОСТ 4452—66, техн.
Гидрохинон, марка А, ГОСТ 2549—60.
Натрий сернистый, ГОСТ 2053—66, ч,
19* 291
Серная кислота, ГОСТ 4204—66, ч.
Кальций хлористый, ГОСТ 4460—66, ч.
Фосфорный ангидрид, МРТУ 6—09—22—62, ч. д. а.
Условия получения
В четырехгорлую колбу емкостью 300 мл, снабженную двурогим форштосом, мешалкой, барботером сероводорода, термометром, капельной воронкой и обратным холодильником, помещают 65 мл толуола и 2,0 г (0,015 моль) хлористого алюминия. Содержимое колбы .насыщают сероводородом (см примечание) при минус 10° со скоростью 15 л/час и при пере мешивании по каплям за 1 час добавляют 59 г (0,5 моль) а-метилстирола, в котором растворено 0,5 г гидрохинона. Сероводород пропускают при тех же условиях еще 30 минут. Реакционную смесь промывают тремя порциями по 150 мл воды и перегоняют при пониженном давлении, собирая фракцию с т. кип. 48—50°/1 мм.
Выход 2-фенил-2-пропантиола равен 55,2 г (72,5%), п$= = 1,5500, di10 = 1,0074, молекулярный вес 152 (криоскопически в бензоле).
Найдено, %: С—69,94; Н—7,87; S—21,10. C9H12S. Вычислено, %: С—71,00; Н—7,94; S—21,06.
Вещество представляет собой подвижную бесцветную жИД‘ кость с неприятным запахом, растворяется в бензоле, толуоле, спирте, эфире.
Примечание.
Газообразный сероводород получают при постепенном смешении водных растворов сульфида натрия (насыщенный раствор) и серной кислоты (пл. 1,2—1,3 г/с.Ф) в генераторе сероводорода [2] или в приборе, состоящем из колбы Вюрца и капельной воронки. В последнем случае серную кислоту прибавляют по каплям к раствору сульфида натрия. Для осушки сероводород пропускают через четыре колонки, заполненные фарфоровыми кольцами, две колонки с прокаленным хлористым кальцием и колонку с фосфорным ангидридом. Фосфорный ангидрид желательно смещать с хорошо просушенной стеклянной ватой, это снижает сопротивление колонки и увеличивает срок службы осушителя. Скорость подачи сероводорода в реактор контролируется по реометру, заполненному керосином.
ЛИТЕРАТУРА
1. М. А. Коршунов, В. Е Мазаев. Авт. свид. 245088; Изобретения. Промобразцы. Товарные знаки, № 19 (1969).
2. М. А. К о р Ш у й о в, В. Е. Мазаев, Н. А. Преображенский. Заводск. лаборатория, 34, 3, 285 (1968).
ФЕРУЛОВАЯ КИСЛОТА
4-Окси-З^метоксикоричная кислота
А. В. БОГАТСКИЙ. 3. Д. БОГ АТСКАЯ. Л. М. НИКИФОРОВА, Л. В. БАСАЛАЕВА, Н. С. УЛАНОВА-ОБЕЗЮК
сн=сн-соон
/ч
г
он
С10Н10О4
М. в. 194,19
Феруловая кислота может быть получена различными способами (1—5]. Особый интерес, однако, представляет метод [5], основанный на реакции ванилина с малоновой кислотой. В качестве конденсирующего средства и растворителя применяют пиперидин и пиридин.
Нами проверена и уточнена методика [5] и предложены два новых варианта этой методики: в первом случае выход кислоты не ухудшается (70%), но в три раза сокращается время реакции; во втором — выход продукта уменьшается до 50— 45%, но его можно получить во много раз быстрее.
СХЕМА СИНТЕЗА
СНО
А ^J-ОСНз
ОН
СН2(СООН)2^ пиперидин пиридин
СН=СН-СООН
I /ч >х^!-осн8
I он 293
Характеристика Основного сырья
Ванилин, ГОСТ 16599—71, т. пл. 81—83°.
Малоновая кислота, МРТУ—6—09—397—63, ч., т. пл. 135°.
Пиридин, ГОСТ 2747—44; предварительно высушен над ед-ким кали [7] и перегнан, т. кип. 115°.
Пиперидин, ТУ 444—51.
Условия получения
В круглодонную колбу емкостью 1 л, снабженную змеевиковым обратным холодильником с хлоркальциевой трубкой, загружают 152 г (1 моль) ванилина, 230 г (2,2 моль) малоновой кислоты, 10 г (12 мл; 0,12 моль) пиперидина и 500 мл сухого пиридина. Реакционную смесь оставляют стоять неделю при комнатной температуре, во время реакции происходит выделение углекислого газа. Затем реакционную смесь нагревают 30 часов на водяной бане при 40—45°, охлаждают в ледяной бане и выливают при перемешивании в смесь 600 мл концентрированной соляной .кислоты и 1 кг мелкораздробленного льда.
Кислота сразу же начинает выпадать в осадок, образуя сплошную густую массу. Через час осадок отсасывают на воронке Бюхнера, промывают 100 мл 5%-ной соляной кислоты, затем ледяной водой дважды порциями по 100 мл и сушат между листами фильтровальной бумаги в темном месте в течение суток (выход сырого продукта почти количественный).
Для получения чистого продукта его перекристаллизовывают из 5 л кипящей воды (но не кипятят), стремясь избежать потемнения вещества. Для охлаждения сосуд оставляют при комнатной температуре в темноте на несколько часов, при этом нз раствора выпадают длинные иглы бледно-желтого цвета, которые отсасывают на воронке Бюхнера и сушат на -воздухе между листами фильтровальной бумаги (в темноте!), а затем в сушильном шкафу при 40—45°.
Выход феруловой кислоты равен 130 г (~70%), т. пл. 173°.
По литературным данным (5], т. пл. 173°.
М. в. вычислено 194, найдено по Расту 187.
По другому способу реакцию проводят в том же приборе с тем же соотношением исходных веществ, но реакционную массу не выдерживают при комнатной температуре, а ограничиваются 30-часовым нагреванием при 45—50°. Выход феруловой кислоты равен 77—97 г (40—50%).
ЛИТЕРАТУРА
1. Posner. J. prackt. Chem [2], 82. 434 (1910).
2. Т. Pacsu. L. Stieber. Ber., 82, 2977 (1929).
3. V о r s a t s. J. prackt. Chem., [2], 145, 265 (1936).
294
3-(а-ФУРИЛ)БУТАНАЛ И 3,(а,а'-ФУРИЛ)ДИБУТАНАЛ
И. В. АНТИПОВА, И. М. СКВОРЦОВ
I H-ch-qh2-cho ЧОХ I сня
:8HI0Oa М. в. 138,17
ОНС-Н,С-НС—II ll—CH-CHs-CHO
I ।
СН3 СНз
CiaHjeOg М.. в. 208,26
В основу методаполученняЗ-(а-фурнл)бутаналаизЗ-(а,а'-фурил)дибутанала положены данные по конденсации фурана с акролеином [1] и сильвана с кротоновым альдегидом [2]. В качестве катализаторов реакции были испытаны сернистая, уксусная, муравьиная и серная кислоты. Наилучшие результаты получены в опытах с 50%-ным раствором серной кислоты.
СХЕМА СИНТЕЗА
1|-сн=сн-c-f°
oz I чн сн.
II }-сн-сн2-ено+онс—нгс-нс-11 11-сн-снг-сно
I I I
СНз
295
Характеристика основного сырья
Фуран, ВТУ МХП, 2489—51, ч.
Кротоновый альдегид, т. кип. 102°, — 1*4362; получе-
ние см. [3].
Гидрохинон (фото), ГОСТ 2549—60.
Серная кислота, ГОСТ 4204—66, ч. д. а.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную мешалкой, обратным холодильником и капельной воронкой, помещают 42,6 г (0,6 моль) кротонового альдегида, 93,7 г (1,38 моль) фурана, 0,2 г гидрохинона и при перемешивании по каплям добавляют 7 мл 50 %-ной серной кислоты, после чего реакци онную смесь перемешивают еще 4 часа. Содержимое колбы нейтрализуют насыщенным раствором соды (pH 7), органический слой отделяют и сушат над прокаленным сульфатом магния. Из отфильтрованного раствора отгоняют избыток фурана, а остаток перегоняют при пониженном давлении, собирая фракцию кротонового альдегида, не вступившего в реакцию, с т. кип. 40—60750 мм (21 г), фракцию З-(а-фурил)бута-нала с т. кип. 78—82°/14 мм и фракцию диальдегида с т. кип. 136—14275 мм.
Выход 3-(«-фурил) бутанала равен 16,6 г (38% в расчете на прореагировавший кротоновый альдегид), ri$ = 1,4831, с/420 = 1,053.
Найдено, %: С—69,39; 69,57; Н—7,65; 7,23. СвНюОз. Вычислено, %: С—69,57; Н — 7,25.
Выход 3-(а,а/-фурил)дибутанала равен7,07г (13%), —
= 1,4990, <№<> = 1,077.
Найдено, %: С-68,92; 68,82; Н— 7,90; 7,84. С12НцО3. Вычислено, %: С—69,23; Н—7,69.
ЛИТЕРАТУРА
1. С. М. Шер л ин, А. Я. Берлин. Ж- общ. химии, 8, 7 (1938).
2. Н. И. Ш у й к и н, А. Д. П е т р о в, В. Г. Глуховцев, И. Ф. Бельский, Г. Е. Скобцева. Изв. АН СССР, о. х. и., 1682 (1964).
3. Препаративная органическая химия. М.-Л., «Химия», 1964, с. 857.
1-(а-ФУ₽ИЛ)-4-НИТРО-3-БУТАНОЛ
И. А. МАРКУШИНА, Т. И. ГУБИНА
"-СН,СН2СН(ОН)СН»МО2 о
С8НИШ4
М. в. 185,17
Нитроспирты фуранового ряда были получены реакцией конденсации фурфурола с алифатическими нитропарафинами [1], в качестве конденсирующего агента применялись карбонат и бикарбонат натрия. При использовании гидрата окиси натрия, как конденсирующего агента, в результате реакции конденсации 1-(а-фурил)-пропаналя и 1-(а-фурил)акролеина с нитрометаном были выделены лишь непредельные соединения [2].
Нами разработан метод получения б-нитроспиртов фуранового ряда из фурановых альдегидов и нитрометана в присутствии гидрата окиси натрия, благодаря чему значительно сокращено время реакции и увеличены выходы.
СХЕМА СИНТЕЗА
( J-CH2CH2CHO 4- CHsNOj -N-aQH ->
ХО
Г "!-CHaCH2CH-CH=NOO хох I он
Na+
~7-CH8CH*CH(OH)CH,NO2 0 х
297
Характеристика основного сырья
1-(а-Фурил)-2-пропаналь, т. кип. 80—82°/17 мм, = — 1,4776; получение см. [3].
Нитрометан, ГОСТ (ТУ) 129—59, ч., серия 3, т. кип. 101 — 102°
Этиловый спирт ректификационный, ГОСТ 5962—51.
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, термометром и капельной воронкой, помещают 13,6 г (0,11 моль) 1-(а-фурил)-2-пропаналя, 20,4 г (0,36 моль) нитрометана, 30 мл этилового спирта и охлаждают до 0°. При постоянном перемешивании прибавляют по каплям 11 мл 10 н. раствора едкого натра так, чтобы температура реакционной смеси не поднималась выше 10°, при этом образуется кашеобразный осадок желтого цвета. Перемешивание продолжают еще 30 минут при комнатной температуре, затем приливают ледяную воду (45—50 мл) до полного растворения осадка. Полученный раствор переносят в колбу Эрленмейера и подкисляют 10%-ным раствором уксусной кислоты до pH 6. Образовавшийся маслянистый слой отделяют иа делительной воронке. Водный слой 4 раза экстрагируют эфиром порциями по 50 мл. Маслянистый слой и эфирные вытяжки объединяют и сушат над прокаленным сернокислым магнием. Эфир отгоняют на водяной бане, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 131 —133°/1 мм.
Выход 1-(ц-фурил)-4-нитро-3-бутаиола равен 15,8 в (78%), = 1,5002, d420 = 1,2280 (см. примечание).
Найдено, %: С—52,09; Н—6,00; N—7,34. CeHnNO4. Вычислено, %: С—51,89; Н—5,99; N—7,56.
Примечание.
Аналогичным путем получены впервые:
1 - (5-Метил-2-фурил) -4-нитро-З-бутанол из 1 - (5-метил-2-фурил) -2-пропа паля, выход 73%, т. кип. 133—13872 мм, rip — 1,4990, dt2<> = 1,1807, Найдено, %: С—54,72; Н—6,54; N—7,04; = 49,54.
C9Hi3NO4. Вычислено, %: С—54,26; Н—6,58; N—7,03; MRD = 49,41.
1 -Метил(2-фурил)-4-нитро-З-бутанол из 1 - (2-фурил)-1 -метил-3-пропана-ля, выход 78%, т. кип. 130—13272 мм,п™ = 1,4970, <А2° = 1,1750.
Найдено, %; С—54,34; Н—6,79; N—6,90; MRD = 49,61. C9H13NO4. Вычислено, %: С—54,26; Н—6,58; N—7,03; WD= 49,41.
ЛИТЕРАТУРА
1. S. К ап а о. Pharm. Soc, Japan, 55, 1019—35 (1927).
2. О. Moldenh auer. Ann., 583, 46 (1953).
3. А. А. П о н о м a p e в, 3. В. Тиль. Ж. общ химии, 27, 1075 (1957).
298’
2-ХЛОРМЕТИЛЕНПИРИДИН
И. В. ЦИРЮЛЬНИКОВА, В. Я- ТЕМКИНА, Л. Я. ШИЛЕНКО.
В. И. СОМОВА
//\
N
CeHeCIN
М. в. 127,57
2-Хлорметиленпиридин является исходным полупродуктом в синтезе целой серии комплексонов, содержащих пиридиновый цикл.
Нами проверен и уточнен метод получения 2-хлорметилен-пиридииа, состоящий во взаимодействии N'-окси а-пиколина с хлор ангидридом трихлоруксусиой кислоты [1].
СХЕМА СИНТЕЗА
+Н.О, сн-с??н-> Hl ч/ N
I О
СС1,СОС1
СНС13 *
Характеристика основного сырья
а-Пиколии, МРТУ 6—09—2394—65, ч.
Перекись водорода, мед., ГОСТ 177—55.
299
Уксусная кислота ледяная, ГОСТ 61—51, х. ч.
Хлорангидрид трихлоруксусной кислоты, МРТУ 6—09— —773—63, ч.
Хлороформ, ГОСТ 3160—51, ч.
Условия получения
Синтез N-окиси а-пиколина [2]. В трехгорлую колбу емкостью 1 л, снабженную мешалкой, обратным холодильником и термометром и помещенную в баню с обогревом, загружают 46,6 г (0,5 моль) а-пиколина, 300 мл ледяной уксусной кислоты и 50 мл 30%-ного раствора перекиси водорода. Полученную смесь нагревают при размешивании 3 часа при 70—80°. затем добавляют-еще 35 мл 30%-ного раствора перекиси водорода и нагревают еще 9 часов в тех же условиях.
Реакционный раствор переносят в колбу Кляйзена и упаривают при пониженном давлении и температуре 35—40° до объема 100 мл; затем добавляют воду в количестве, равном отогнанному, и снова упаривают раствор до объема 100 мл. Остаток выливают в 250 мл хлороформа и встряхивают с водной суспензией карбоната натрия, состоящей из 50 мл воды и 30 г карбоната натрия, до тех пор, пока не прекратится выделение углекислого газа. Органический слой отделяют в делительной воронке, сушат над хлористым кальцием, затем помещают в колбу Кляйзена, отгоняют хлороформ при пониженном давлении и температуре 25—30°, а остаток перегоняют, собирая фракцию с т. кип. 123—124715 мм.
Выход N-окиси а-пиколина равен 27,3 г (50%).
Получение 2-хлорметиленпиридина. В двухгорлую колбу емкостью 0,5 л, снабженную обратным холодильником и помещенную в баню с обогревом, помещают 27,3 г (0,22 моль) све-жеперегнаннон N-окиси а-ииколина и 125 мл хлороформа. Раствор доводят до кипения, добавляют 55,2 г (0,33 моль) три-хлорацетилхлорида и кипятят 20 часов. Затем раствор охлаждают и вливают в него заранее приготовленный раствор 70 г бикарбоната натрия в 700 мл воды. Полученную смесь размешивают до тех пор, пока выделяется углекислый газ, и фильтруют. Органический слой отделяют в делительной воронке и перегоняют, собирая фракцию с т. кнп. 75—77712 мм.
Выход 2-хлорметилеипиридина равен 10,8 (30%); вещество представляет собой бесцветную жидкость, постепенно переходящую в темно-малиновый творожистый продукт.
По литературным данным [1], т. кип. 75—77°/\2 мм.
ЛИТЕРАТУРА
1. Т. Koenig, I. S. Nleczorek. J. Organ. Chem., 33, 4, 1530 (1968).
2. V. В о k e 1 h el d e, W. J. Linn. J. Amer. Chem. Soc., 78, 1286 (1954).
300
Р-ХЛОРПРОПИОНОВАЯ КИСЛОТА
Л. Я- ГЛИНСКАЯ, 3. Д. БОГАТСКАЯ, Г. Ф. ТАНЦЮРА
а—сн2—сн2—соон
C3H5CIO2 М. в. 108,52
Известно несколько способов получения р-хлорпропноно-вой кислоты (1—5], однако методики эти неудобны, так как исходные продукты либо ядовиты (акролеин) [1], либо сильные окислители (дымящие кислоты — азотная, соляная) (1, 2].
В литературе имеется краткое сообщение [3] о получении p-хлорпропионовой кислоты присоединением хлористоТо водорода к акриловой кислоте (методика синтеза не приводится).
Нами предложен метод синтеза p-хлорлропионовой кислоты из акриловой кислоты путем присоединения сухого хлористого водорода.
СХЕМА СИНТЕЗА
СН2=СН—СООН-Ь НС1 —> С1—сн2—сн2—соон
Характеристика основного сырья
Акриловая кислота, РУ 4730—62, ч.
Условия получения
В трехгорлой колбе емкостью 1 л, снабженной мешалкой, обратным холодильником и трубкой для подвода газа, охлаждают до 15° 800 г (11 моль) акриловой кислоты и за 4—5 часов при хорошем перемешивании пропускают сильный ток су хого хлористого водорода (ом. примечание) до начала кристаллизации реакционной массы и выделения избытка хлористого водорода через верхнее отверстие холодильника. Из реакционной массы отгоняют непрореагировавшую акриловую
301
кислоту, остаток перегоняют при пониженном давлении, собирая фракцию с т. кип. 8875 мм.
Выход р-хлорпро.пионовой кислоты равен 1080 г (90%), т. пл. 4 Г, т. кип..85°/5 мм, п™ = 1,4395; d450 — 1,2433.
Найдено, %: С1—23,00; MRQ = 23,00.
С3Н5СЮ2. Вычислено, %: С1—22,56; = 22,50.
По литературным данным [3], т. пл. 40—41°, т. кип. 203— 204° (с разл.).
ИК-спектры полученной кислоты идентичны описанным в литературе {7—9]. В спектрах, снятых в диоксане и в четыреххлористом углероде, имеются интенсивная полоса карбонильного поглощения при 1730—1700 см~г, широкая полоса при 1428—1430 см~', относящаяся к СНг-пруппе, находящейся по соседству с карбонильной группой; и полоса валентных колебаний С—С1 в интервале частот 750—700 см"'.
Примечание.
Хлористый водород получают и сушат по методике [6].
ЛИТЕРАТУРА
1. С. М о и г е и. R. С h а и х, Organ, synth,. 8, 54 (1928),
2. Н. Beckurts, R. Otto. Ber., 18, 226, (1885).
3. Ed. Linnemann Ann. 163, 96 (1872).
4, S, Powell, J. Amer. Chem, Soc. 46. 2879 (1924),
5. C. S m i t h R. Holm, J. Organ, Chem., 22. 746 (1957).
6. Ю. В. Карякин. Чистые химические реактивы. М., ГХИ, 1947, с. 276.
7. L. Flett. J. Chem. Soc., 1951, 962.
8. R. Shreve, A. H eether, A. Knight. Analyt; Chem., 22, 1498 (1950),
9. Л. Беллами. Инфракрасные спектры сложных молекул. М., И'1, 1963.
7-ХЛОР-5-ФЕНИЛ-3-БЕНЗИЛ ИДЕН-1,2-ДИГИДРО-ЗН-1.4-БЕНЗДИАЗЕПИН-2-ОН
А. В. БОГАТСКИЙ, т. к. ЧУМАЧЕНКО, 3. И. ЖИЛИНА,
С. А. АНДРОНАТИ
CasHigClNaO
^X^NH-cC
| II С=СН—С6Н6
Cl/'^/XC=N/
С6н6
М. в. 358,84
Препараты класса 1,4-бенздиазепинов находят широкое применение как успокаивающие, снотворные и миорелаксант-ные средства [1, 2]. С целью получения лекарственных препаратов с широким спектром действия нами осуществлена конденсация 1,2-дигидро-3-Н-1,4-бенздиазепин-2-она с бензальдегидом. Продукт конденсации выделяется в виде смеси изомеров (цис- и транс-), которые легко разделяются колоночной хроматографией.
СХЕМА СИНТЕЗА
^X/NH-C^
I II сн2+о-сн-с.н6
/4/\c===N/
V1 |
CH3COONa
(СНгСО)2О
са
303
о
^\/NH-c\
_ I II C=CH-CeHs
Cl/^/X4C=N/
C6H5
Характеристика основного сырья
7-Хлор-5-фенил-1,2-дигидро-3-Н-1,4-бенздиазепин-2-он, пл. 216—217°, получение см. (3].
Бензальдегид, ГОСТ 157—69, ч.
Уксусный ангидрид, ГОСТ 5815—69, ч.
Ацетат натрия, МРТУ 2121—65, ч.
Бензол, ГОСТ 5955—68, х. ч.
Метилен хлористый, МРТУ 6—09—5362—68, ч.
Окись алюминия, 2-й ст., МРТУ 6—09—5296—68.
Условия получения
В колбу емкостью 50 мл, снабженную мешалкой, обратным холодильником и термометром, помещают 1,9 г (0,007 моль) 7-хлор-5-фенил-1,2-дигидро-3-Н-1,4-бенздиазепин-2-она, 1,06 г (0,01 моль) бензальдегида, 0,62 г (0,0076 моль) ацетата натрия и 2 мл уксусного аннгидрида. Реакционную смесь нагревают на масляной бане 2 часа при перемешивании и слабом кипячении. Образовавшую смолообразную массу растворяют з хлористом метилене. Полученный раствор промывают 50 мл воды (3 раза) и 50 мл раствора бикарбоната натрия, сушат над прокаленным хлористым кальцием, растворитель отгоняют досуха. Остаток переносят в хроматографическую колонку, заполненную окисью алюминия 2-ой степени активности, и один из стереоизомеров элюируют бензолом, а второй—-смесью метиленхлорид—бензол (1 : 1).
Выход «fac-изомера с т. пл. 234—235° равен 1 г (40%).
Найдено, %: С—73,55; Н—4,45; N—7,90. C22Hi5C1N2O. Вычислено, %: С—73,40; Н—4,50; N—7,78.
Выход транс-изомера с т. пл. 223—225° равен 0,3 г (12%). Найдено, %: С—73,50; Н—4,50; N—7,75.
ЛИТЕРАТУРА
1, L. Sternbach. L, Randall, S, GastafsoH. Psychophar-macological Agents, N,-Y. Acad. Press., 1964, 137.
2. А. В. Богатский, Ю. И. Вихляев, С. А. А н д р о н а т и, Т. А. К л ы г у л ь, Т. К. Чумаченко, 3. И. Жилина. Хим. фярм. ж., 4, 5 (1970).
3. L. Н. Sternbach, R. 1. Fryer, W. Met lest es, F. Reeder, G. Sa ch, S. Sauscy, A. Stem pel. J. Organ. Chem., 27, 3788 (1962).
304
л-ЭТИЛАЦЕТОФЕНОН
1 -Этил-4-ацетилбензол
е: и. маховер
с2нб-^ ^-сосна
CioHijO
М. в. 148,20
По литературным данным, n-этилацетофенон может быль получен взаимодействием этилбензола с хлористым ацетилом в присутствии хлористого алюминия в растворителе [1] или без него [2|].
Нами предложен метод получения n-этйл ацетофенона в среде дихлорэтана с заменой хлористого ацетила уксусным ангидридом, что делает метод безопасным.
СХЕМА СИНТЕЗА
C2HS санб
I I
~-(СН,СО)2О
ч/ чх
I
сосна
Характеристика основного сырья
Этилбензол, ТУ ГКХ ОРУ 140—59, ч.
Уксусный ангидрид, ГОСТ 787—55, техн.
Алюминий хлористый безводный, ГОСТ 4452—66, техн.
Дихлорэтан, ГОСТ 1942—63, техи.
20 Зак. 1164
805
Условия получения
В четырехгорлую колбу емкостью 3 л, снабженную мешалкой, термометром, обратным холодильником и капельной воронкой и помещенную в баню для охлаждения, загружают 1,5 л (1,9 кг) дихлорэтана, предварительно высушенного хлористым кальцием и при перемешивании присыпают 635 г (4,8 моль) хлористого алюминия. Полученную суспензию охлаждают смесью льда и соли до 0° и за 30—40 минут прибавляют по каплям 230 г (2,26 моль) уксусного ангидрида при температуре не выше 5°. Затем охлаждают реакционнную смесь до минус 5—0° и за 1 час прибавляют по каплям 212 г (2 моль) этилбензола, поддерживая температуру минус 2—2Э, после чего охлаждение прекращают и продолжают размешивание до тех пор, пока температура реакционной массы достигнет 12—15°.
Содержимое колбы выливают при перемешивании в смесь 5 кг льда и 100 мл соляной кислоты. После отстаивания нижний слой отделяют, промывают 1 л воды, нейтрализуют 2 л 5%-ного раствора карбоната натрия, промывают 2 л воды до нейтральной реакции, сушат хлористым кальцием и фильтруют. Из фильтрата отгоняют дихлорэтан сначала при атмосферном, затем при пониженном давлении (~100 мм). Остаток фракционируют при пониженном давлении, отбирая фракцию с т. кип. 101 — 1047Ю мм.
Выход п-этилацетофеиоиа равен 240 г (81%), т. кип. 240J, <Л20 = 0,987, n2D°= 1,5290.
По литературным данным [1], т. кип. 236°, </420 = 0,991.
ЛИТЕРАТУРА
1. Н. Klages. Вег., 32, 1558 (1897).
2. Синтезы органических препаратов, сб. 4. М., ИЛ, 1953, с. 41.
а-ЭТИЛ-р-ГЕКСОКСИМЕТИЛ ПРОПИОНОВАЯ КИСЛОТА
Е. А. ЯЦЕНКО, А. И. ДРОЗДОВСКАЯ
С,Н13ОСН2СНСООН
I
Сгн5
СцНяА М. в. 202,17
а-Алкил-р-алкоксиметилпропионовые кислоты и их эфиры находят применение как пластификаторы в производстве целлюлозы, в качестве растворителей, смазок и полупродуктов [1].
а-Алкил-р-алкоксиметилпропионовые кислоты Чйолучают термическим расщеплением в атмосфере азота в установкедля пиролиза проточного типа. а-Этил-р-гексоксиметилпропионо-вая кислота получена нами впервые пиролизом а-этил-р-гексо-ксиметилмалоновой кислоты.
СХЕМА СИНТЕЗА
—СО,
С.Н13ОСНаС(СООН)2 ----------2----С6Н13ОСНаСНСООН
сан5 сан6
Характеристика основного сырья
а-Этил-р-гексоксиметилмалоновая кислота, т. ,пл. 53°, получение см. [2].
Азот, МРТУ 6—02—375—66, ос. ч. 1—3.
Натр едкий, ГОСТ 4328—66, ч.
Пирогаллол, ГОСТ 10451—63, ч.
Серная кислота, ГОСТ 4204—66, ч.
Условия получения
Пиролиз проводят в кварцевой трубке 0 12 мм. Реактор помещают в трубчатую печь, температуру печи (280—300°) 20* 307
регулируют лабораторным автотрансформатором марки РИО-250-2. Печь предварительно заполняют кварцевой насадкой (объем 15—16 мм, величина зерен 2—5 мм, 20—40 г). В редктор подают из капельной воронки 40 г (0,19 моль) расплавленной а-этил-р-гексоксиметилпропионовой кислоты со скоростью 0,35 г] мин. Пиролизат собирают в приемник, снабженный обратным холодильником для улавливания низкоки-пящих веществ. Обратный холодильник соединяют с газовыми часами для регулирования скорости подачи азота (см. примечание). Для улавливания углекислого газа отходящие газы проходят через склянку со щелочью. Скорость подачи азота 20 мл]мц.н..
Пиролизат перегоняют при пониженном давлении, собирая фракцию с т. кип. 154°/7 мм.
Выход а-этил-р-гексоксиметилпро.пионовой кислоты равен 24,2 г (74,6%).
Найдено, %: С-64,95;.Н—11,29.
С11Н22О3. Вычислено, %: С—65,30; Н—10,96.
Строение а-этил-р-гексоксиметилпропионовой кислоты подтверждено ЯМР- и ИК-спектрами. В ЯМР-спектрах имеется1 пик интенсивностью ЗН при б = 6,70 м. д. для CHg—О—СН-группы и раздвоенный широкий пик с б = 8,90 и 9,15 м. д. интенсивностью 18Н для остальных протонов. В ИК-слектрах — полоса поглощения для .карбонильной группы при 1690 см~' и широкая ’размытая полоса поглощения в области 3000— 3400 см~', характерная для ассоциированных гидроксильных групп.
Примечание.
К реакционной трубке присоединнют двурогий форштос, через который подают азот н реагент. Для очистки азота от примесей кислорода его предварительно пропускают через склянки Тищенко с пирогаллолом и серной кислотой, соединенные с прибором. Азот подается из газометра.
ЛИТЕРАТУРА
1. Пат. ФРГ 1067798 (1960); РЖХим, 14л80 (1961).
2. О. С. Степанова, Е. А. Яценко. Укр. хим. ж., 19, 612 (1963).
ЭТИЛЕНДИАМИНДИАЦЕТАТЫ МЕДИ, КОБАЛЬТА, НИКЕЛЯ
Е. М. УРИНОВИЧ, Н. М. ДЯТЛОВА
сн8—с=о
СН2—NtT о
| ;Ме • лН»О,
CH8-NH \
\н2—^С=О
где Me=Cu; Со; Ni
CgHioCuNjjCX • 4Н2С)
C6H10CoNaO4 • ЗН2О
C6H10N2NiO4- ЗНаО
М. в. 303,76
М. в. 281,13
М. в. 280,91
Этилендиамин-Ы.К'-диуксусная кислота является комплек-сообразователем, способным давать прочные комплексы с переходными металлами. В литературе описано образование ее комплексов с медью, кобальтом и никелем в водных растворах [1].
Нами синтез комплексов осуществлен взаимодействием ди-калиевой соли этилендиаминдиуксусной кислоты с эквивалентным количеством нитрата соответствующего металла. Комплексы впервые выделены в твердом состоянии.
СХЕМА СИНТЕЗА hoocch2nhch2ch2nhch3cooh — -Т koocch2nhchsch,nhch2cook ->
309
сн2—с=о
CH2-NH о
Me(NO3)2 | ' /
------------ ;Ме
СНЯ—NH Чо 'сн4-^с=±о
Характеристика основного сырья
Этилендиамин-Ы.ЬИ-диуксусная кислота, ТУ 5П-59—70, ч. Кали едкое, ГОСТ 4203—65, ч.
Медь азотнокислая, ГОСТ 4163—68, ч.
Кобальт азотнокислый, ГОСТ 4528—68, ч.
Никель азотнокислый, ГОСТ 4055—70, ч.
Условия получения
Синтез этилендиаминдиацетата меди. В трехгорлой колбе емкостью 0,5 л, снабженной мешалкой, обратным холодильником и капельной воронкой, растворяют при нагревании до 50—55° в 100 мл этилового спирта 24,1 г (0,1 моль) трехводной азотнокислой меди. К полученному раствору при тщательном размешивании приливают из капельной воронки раствор калиевой соли этилендиаминдиуксусной кислоты, приготовленный из 18,5 г (0,105 моль) этилендиаминдиуксусной кислоты и 11,5 г (0,205 моль) едкого кали в 120 мл 60%-ного водного спирта. Реакционную массу перемешивают еще 1 час при той же температуре, переносят в стакан и оставляют при комнатной температуре на ночь. Выделившийся густой темно-си* ний сироп постепенно закристаллизовывается. Кристаллы ярко-синего цвета отфильтровывают на воронке Бюхнера, промывают несколько раз небольшим количеством воды, охлажденной до 5—8°, и сушат сначала на воздухе, затем в вакуум-эк-сикаторе над хлористым кальцием.
Выход этилендиаминдиацетата меди равен 7,9 г (23%).
Найдено. %: С—23,57; Н—6,20; N—8,77; Си—19,31. CeHioNaCUCu • 4НзО. Вычислено, %: С—23,26; Н—5,85; N—9,05; Си—20,51.
Получение этилендиаминдиацетата кобальта. В стакане, снабженном мешалкой, растворяют при нагревании до 50—55° 11,6 а (0,04 моль) шестнводного азотнокислого кобальта в 20 мл воды. К полученному раствору приливают при перемешивании раствор дикалиевой соли этилендиаминдиуксусной кислоты, приготовленный из 7 а (0,04 моль) этилендиаминдиуксусной кислоты, и 4,4 а (0,08 моль) едкого кали в 20 мл воды. Реакционную массу нагревают при 50° еще 1 час, после 310
чего оставляют на ночь при комнатной температуре. Выпавшие кристаллы розовато-малинового цвета отфильтровывают на воронке Бюхнера, промывают несколько раз водой и сушат на воздухе.
Выход этилендиаминдиацетата кобальта равен 5,95 г (52%).
, Найдено, %: С—25,81; Н—5,46; N—9,45; Со—20,30.
CeHtoNACo • ЗН2О. Вычислено, %: С—25,09; Н—5,57; N—9,75; Со—20,52. Получение этилендиаминдиацетата никеля. По методике, описанной для этилендиаминдиацетата кобальта, из 29 г (0,1 моль) шестиводного азотнокислого никеля, 17,6 а (0,1 моль) этилендиаминдиуксусной кислоты и 11,2 г (0,2 моль) едкого; кали получают 15,5 а (54%) этилендиаминдиацетата никеля в виде голубых кристаллов.
Найдено, %: С—25,74; Н—5,43; N—9,72; Ni—19 72. C6Hi0Ns'O4Ni • ЗН2О. Вычислено, %: С—25,11; Н—5,56; N—9,78; Ni—20,46.
ЛИТЕРАТУРА
1. S. С h а b е г е с k, А. Е. Martell. J. Amer. Chem. Soc., 74, 6228 (1952).
5-ЭТИЛ-5-ФЕНОКСИБАРБИТУРОВАЯ И 5-ЭТИ.Й-5-ФЕНОКСИТИОБАРБИТУРОВАЯ КИСЛОТЫ
О. С. СТЕПАНОВА, А. С. ЯВОРСКИИ, М. И. ПОБЕРЕЖНАЯ,
В. Н. ГАНЕВИЧ
=0
Ci2H|2N2O4 М. в. 248,24
C12H12N2O3S М. ф 264,31
Замещенные барбитуровые и тиобарбитуровые Кислоты представляют интерес в качестве снотворных препаратов. Введение неразветвленных алкоксирадикалов в мрлекулу барбитуровой кислоты увеличивает ее физиологическую активность [I].
Нами синтезированы впервые 5-этил-5-феноксибарбитуро-вая и 5-этил-5-фенокситиобарбитуровая кислоты конденсацией этилфеноксималонового эфира с мочевиной или тиомочевиной в присутствии этилата натрия.
СХЕМА СИНТЕЗА
с.2н, .соос,н5 х + с6нвсг ХСООС2Н6
h2n4
)C=O(S) HaNz
X /==0(S)
CeH6OZ^|—NH/
312
Характеристика основного сырья
Этилфеноксималоновый эфир, т. кил. 150°/4 мм\ п® = = 1,4872.
Этиловый спирт, ТУ 19П—39—69, абсолютирован по методике (2].
Мочевина, ГОСТ 6691—67, ч.
Тиомочевина, ГОСТ 6344—52, ч.
Натрий металлический, ТУ 1664—50, ч.
Условия получения
В двугорлую колбу емкостью 250 мл, снабженную обратным холодильником с хлоркальциевой трубкой, к 100 мл абсолютного этилового спирта прибавляют порциями 6,9 г (0,3 г-а) металлического натрия. К полученному горячему раствору ал-коголята натрия прибавляют 12 г (0,2 моль) мочевины и 28 а (0,1 моль) этилфеноксималонового эфира. Реакционную смесь нагревают на масляной бане (108—110°) в общей сложности 24 часа, затем отгоняют этиловый спирт, твердый остаток растворяют в 100 мл горячей воды, и горячий раствор фильтруют. К холодному раствору при охлаждении ледяной водой приливают 10%-ный раствор соляной кислоты до кислой реакции по конго-рот. Выпавшие кристаллы отсасывают на воронке Бюхнера, промывают 100—150 мл воды и сушат в вакуум-эксикаторе. Очищают вещество трехкратной перекристаллиза цией из 70—100 мл этилового спирта.
Выход 5-этил-5-феноксибарбитуровой кислоты равен 14,8 г (60%), т. пл. 147—148°.
Найдено, %: N—11,11. C12H12N2O4. Вычислено, %: N—11,28.
По аналогичной методике получена с выходом 71 % 5-этил-5-фенокситиобарбитуровая кислота, т. пл. 132°.
Найдено, %: S—12,33. C12H12N2O3S. Вычислено, %: S—12,12.
Строение полученных барбитуровых кислот доказано ИК-спектрами. В ИК-спектрах имеются полосы поглощения, характерные для монозамещенного бензольного кольца, и полосы поглощения, характерные для барбитуровых кислот — для 5-этил-5-феноксибарбитуровой кислоты при 1700, 1738, 3100 и 3205 см~]. Аналогичные полосы поглощения в области 3180— 3400 сдг1 и 1680—1700 см~1 наблюдаются в ИК-cneicrpe 5-этил-5-фенокситиобарбитуровой кислоты [3].
ЛИТЕРАТУРА
1. Р. Я. Левина, Ф. К. Величко. Успехи химии, 29, 929 (1960).
2. Ю К. Юрьев. Практические работы по органической химии. М., Изд-во МГУ, 1961, с. 52.
3. А. Р. К а т р и ц к и й. Физические методы в химии гетероциклических соединений. М„ «Химия», 1966, с. 568.
313
ЭТИЛФЕНОКСИМАЛОНОВАЯ КИСЛОТА
О. С. СТЕПАНОВА, А. С. ЯВОРСКИЙ, М. И. ПОБЕРЕЖНАЯ,
В. Н. ГАНЕВИЧ
санБ соон
X
С2Н5О соон
С11Н12О5
М. в. 224,22
Алкилзамещенные феноксималоновые кислоты обладают физиологической активностью: проявляют гербицидное действие, а в малых количествах проявляют стимулирующую активность по отношению к растениям [1].
Этилфеноксималоновая кислота получена нами впервые омылением этилфеноксимало.нового эфира.
СХЕМА СИНТЕЗА
СаН5 СООС2Н5 С2Н5 соон
/е\ /с\ свнво соос2нБ С6НвО соон
Характеристика основного сырья
Этилфеноксималоновый эфир, т. кип. 150°/4 мм; di20 ~ = 1,1035; л» = 1,4872.
Кали едкое, ГОСТ 4203—65, ч. д. а.
Этиловый спирт, ТУ 19П—39—69.
Условия получения
В колбе емкостью 0,5 л, снабженной обратным холодильником и капельной воронкой, нагревают до кипения 250 мл 314
35%-ного водно-спиртового раствора едкого кали и прибавляют по каплям 70 г (0,25 моль) этилфеноксималонового эфира. Реакционную смесь кипятят 8 часов на масляной бане (температура бани ПО—115°). Затем прибавляют 50—70 мл воды до полного растворения образовавшейся калиевой соли и полностью отгоняют спирт. К остатку после охлаждения до комнатной температуры приливают 30—45 мл эфира, чтобы извлечь неомыленные продукты. Водный раствор калиевой соли фильтруют и продукты омыления выделяют разбавленной серной кислотой (1: 1), которую прибавляют до сильно кислой реакции (по конго-рот) при охлаждении льдом. Выделившееся масло экстрагируют трижды эфиром (по 100 мл за каждый прием), эфирные вытяжки дважды промывают водой (по 25 мл), а промывные воды экстрагируют 50 мл эфира. Соединенные эфирные вытяжки сушат сутки безводным сульфатом магния, раствор фильтруют от осушителя, отгоняют эфир, а остаток выливают в чашку и помещают в вакуум-эксикатор. После 70-часового стояния сиропообразная масса закристаллизовывается. Продукт очищают перекристаллизацией из 50—75 мл бензола.
Выход этилфеноксималоновой кислоты равен 43,7 г (78%); т. пл. 114°.
Найдено, %: С—58,81; Н—5,15. С11Н12О5. Вычислено, %: С—58,92; Н—5,35.
Строение этилфеноксималоновой кислоты подтверждено ИК-спектрами. В спектре имеются полосы поглощения, характерные для монозамещенного бензольного кольца при 645, 745, 1483, 1577, 3002 и 3042 еле-1, простой эфирной связи арил-али-фатических простых эфиров при 1286 см~' и для карбоксильных групп при 1682, 1719, 3587, 3613 и 3635 см~'.
ЛИТЕРАТУРА
1. О. С. Степанова, Ж. И. Безгудова. Укр. хим. ж., 33, 306 (1967).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ СОЕДИНЕНИЙ, ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
1,1'-Азопропан................................................ 10
Азоэтан........................................................ Ю
Амид у-оксигептановой кислоты...............................16
Амнд у-оксидекановф! кислоты................................17
Амид у-оксиизононановой кислоты ............................17
Амид у-оксннонановой кислоты................................17
Амид у-оксноктановой кислоты................................17
у-Амнлбутнролактон............................................221
5-Амил-Ы-(₽-2-оксиэтил)-2-пирролидон .........................253
5-Амнл-2-пирролидон............................................19
5-Амил-2-тиопнрролндон........................................256
9-Амнноакрнднн.................................................23
о-Амнно-р,р-ди(3-индолил)этнлбензол............................27
2-(2-Амнно-5-иитробензамидо) бензойная кислота.................29
8-Амннохииальднн.............................................. 31
а-[(2-Амнноцнклопеитил)метил]фуран.............................34
№Ацетил-5-амил-2-пирролидон....................................20
1-Ацетнламнио-2-ацетокси-3,5-диметнлбензол.....................40
2-Ацетнламинофенол............................................ 44
2-Ацетнламннофеноксиуксусная кислота...........................42
М-Ацетил-5-бутил-2-пирролидоп..................................21
Ы-Ацетил-5-гексил-2-пирролндон.................................21
Ы-Ацетил-5-гептил-2-пирролидои.................................21
К-Ацетнл-5-нзоамил-2-пирролидон................................21
М-Ацетил-5-изобутил-2-пирролидон...............................21
3-(Ы-Ацетнл-5-метил-2-пирролидил)пропанол......................46
Ы-Ацетнл-(5-метил-2-фурфурил)амииоэтанол.......................48
М-Ацетил-5-ноннл-2-пнрролндон..................................21
М-Ацетнл-5-октил-2-пирролндон..................................21
Ы-Ацетнл-5-пропил-2-пирролидон.................................20
Ы-Ацетил-Ы-фурфурнламиноэтаиол.................................46
Ы-Ацетил-5-этил-2-пирролндон...................................20
S-Беизилнзотиомочевииа гидрохлорид.............................51
316
8-Беизоиламино-1-нафтол-3,6-дисульфокислота, двунатрневая соль 55
N-Бензоил-Аш-кнслота, двунатриевая соль.....................55
Бензойная кислота, бензиловый эфир..........................53
N-Бензонл-о-толундин........................................... 5в
Бензол-1,2,4-трикарбоиовая кислота.........................270
Бнс(п-нитрофеннлгидразон) а,р-днкетобутиролактоиа ... 60
3,4-Бис (п-иитрофеинлгндразоио)-2-оксотетрагидрофуран . . .60
л-Бис(-и-феиоксифеиокси) бензол..................................65
7-Бром-1-ацетил-5-фенил-1,2-днгидро-ЗН-1,4-бенздиазепин-2-он . . 66
3-Бром-2,4-диоксотетрагидрофуран............................70
а-Бром-р-кетобутнролактон...................................... 70
4-Бром-3-метнл-5-(5-бром-2-фурил)-пнразол ...... 68
а-Бромтетроиовая кислота ....................................... 70
у-Бутилбутиролактон ........................................... 220
втор-Бутилгидантоин ........................................... 133
5-Бутнл-М-(р-окснэтил)-2-пнрролидои........................253
5-Бутил-2-пирролидои........................................20
5-Бутил-2-тиопирролидои , 256
N-Бутилхинальдииий иодид ....................................... 72
Бутнрнл-Р-нафтилгидразин , ............................ . 213
4-н-Бутирилокснбеизальдегид......................................50
2,4-Гексаднеиаль............................................... 74
а-Гексилкротоновая кислота ..................................... 76
«-Гексилкротоновая кислота, этиловый эфир........................77
5-Гексил-М-(Р-оксиэтил)-2-пирролидои............................253
5Тексил-2-пирролидои.............................................21
5-Гексил-2-тиопирролидои...................................... 257
трет-Гексилтиоэтилметакрилат.....................................14
4-н-Гексоксибензнлхлорид.........................................79
4-н-Гептаноилокснбензальдегид................................... 50
5-Гептил-М-(р-оксиэтил)-2-пирролндон............................253
5-Гептнл-2-пирролидон............................................21
5-Децнл-М-(р-оксиэтил)-2-пирролндон . . .....................253
1,4-Диацетилбеизол...............................................81
Дибензоилметаи...................................................85
а,у-Дибромацетоуксусиый эфир.....................................71
1,4-Дибром-2-метоксипеитаи.......................................87
1,4-Дибром-2-этоксипеитаи........................................87
3,4-Дибутоксианнлии..............................................90
3,4-Дибутоксииитробензол.........................................91
4-N.N-(Дикарбоксиметил) амииометил-З-окси-2-нафтойная кислота 93
1, И-Диметил-З,З'-ди (тетрагидрофурил-2) дипропилоксид . , . 95
транс-2,3-Диметилоксетан.........................................99
Ч“с-2,3-Днметилоксетаи...........................................99
2,9-Диметил-1,10-феиаитролии . 102
Ы,К'-Диметилэтилендиамино-М,Н'-бис(метилеифосфоновая кислота ............................................................105
317
М.М'-Днметнлэтиленднамннобис (метнленфосфоновая кислота) этиловый эфир.................................................107
2,7-Днметокси-1,6-диоксаспиро[4,4]ионан........................109
2,7-Диметоксн-1,6-дноксаспиро[4,4]нонен-3......................110
Динитрил 1,10-фенантролин-2,9-днкарбоновая кислота . .112
Диоксанат бис(триннтроммил)ртути................................Н4
2,6-Диоксибеизилиминодиуксусная кислота........................116
2,3-Диоксибензилимннодиуксусная кислота, моноиатрневая соль 118 МД'-Ди (2-окси-5-метилбеизил) этилеидиамино-М,5Г-диуксусиая кислота........................................................120
N.N'-Д и (2-оксиэтил) этилендиамино-М,М'-бис (метиленфосфоновая кислота)......................................................105
N.N'-Ди (2-оксиэтил) этилеидиаминобис(метиленфосфоиовая кислота),этиловый эфир.............................................108
3,3'-Ди-и-пентоксиазоксибензол.................................122
3,3'-Ди(тетрагидрофурнл-2)дипропилоксид..................... 96
2,6-Дихлор-4-питроанилнн.......................................124
2,6-Дихлор-1,4-феннлендиамин...................................126
Диэтиленгликоль, диметиловый эфир...............................97
Диэтиленгликоль, диэтиловый эфир................................99
Диэтилентриамино-М,М.1Ч',М',1Ч"-пента (метилфосфоновая кисло-
Днэтилфурфурилидеималонат......................................130
5-Изоамил-М-([$-оксиэтил)-2-пирролидон......................• 253
5-Изоамнл-2-пирролидон..........................................21
5-Изоамил-2-тиопирролидон . ... *....................257
5-Изобутил-1Ч-(р-оксиэтнл) -2-пнрролидон....................• 253
₽-(5-Изобутил-2-пирролиднл) пропионовая кислота .... 247
р-(5-Изобутил-2-пирролидил) пропионовая кислота, метиловый
эфир...................................................... 166
р-(5-Изобутил-2-пирролидил)пропионогидразид....................172
5-Изобутнл-2-пирролидои.........................................21
4-Изобутил-З-пирролизидон......................................250
5-Изобутил-2-тиопирролидон.....................................256
5-Изогексил-1Ч-(Р-оксиэтил)-2-пирролидон.......................253
D.L-Изолейцин..................................................132
Изопропилгидразин ............................................ 199
5-Изопропил-З-пирролизидон.....................................250
Изо(4,7,8,9-тетрагидро)беизо-1,3-димегокситетрагидрофуран . 137
Индолальгидаитоин............................................ 275
Индолальдегид..................................................274
.и-Иоданилин ..................................................139
Кальцийацетоуксусный эфир......................................141
5-(2-Карбоксиэтил)-1,2-дигидропирролизин.......................143
у-Кетопимелиновая кислота, диэтиловый эфир ..... 223
Конифериловый альдегид.........................................147
Конифериловый спирт............................................150
Метакрилоилхлорид ............................................ 153
318
Метиламид у-оксигептановой кислоты..............................17
Метиламид у-оксииоиановой кислоты...............................18
Метиламид у-оксиоктановой кислоты...............................17
1-Метил-5-амил-2-пирролндон....................................174
1-Метил-5-амил-2-тиопнрролндон.................................257
Метилбензилкетоп.............................................. 155
о-Метилбеизолсульфохлорид......................................134
м-Метилбензолсульфохлорид .....................................134
п-Метилбеизолсульфохлорид......................................134
2-Метилбутаналь............................................... 157
1-Метил-5-бутил-2-пирролидон...................................174
1-Метил-5-бутнл-2-тиопнрролидон................................257
1-Метнл-5-гексил-2-пирролидои..................................175
1-Метил-5-гептил-2-пирролидон..................................175
1-Метил-5-гексил-2-тиопирролидои...............................257
1-(3-Метил-1,2-дигидропирролизил-5)-1-бутен-3-он .... 159
2-Мети л-2,7-диметокси-1,6-диоксаспиро[4,4]иои ан..............111
2-Метил-2,7-днметокси-1,6-диоксаспиро[4,4]ноиеи-3 . . НО
Метилди (диэтилфосфон) карбинол....................................265
?},М'-Метиленднантраниловая кислота............................263
5,5'-Метилендиантраннловая кислота.............................263
1-Метил-5-изоамил-2-пирролидои.................................174
1-Метил-5-изоамил-2-тиопирролидон............................. 257
1-Метил-5-изобутил-2-тиопирролидон.............................257
Метнлизопропилкетон............................................161
3-Метил-5-(2-карбоксиэтил)-1,2-дигидропирролизии .... 144
а-Метил-р-меркаптопропионовая кислота, метиловый эфир 163
З-метил-5-(2-метоксикарбонил)этил-1,2-дигидропирролизии . . 191
1-Метил-5-октил-2-пирролидоп...................................175
р-(5-Метил-2-пирролидил) пропионовая кислота...................247
р-(,]-Метил-2-пирролидил)пропионовая кислота, метиловый эфир 166
Р(5-Метил-2-пнрролидил) пропионовая кислота .метиловый эфир 165
Р(1-Метил-2-пирролндил)пропионогидразид........................172
р (5-Метил-2-пирролидил)пропиоиогидразид.......................171
4-Метил-З-пирролизидон.........................................250
1-Метил-5-пропил-2-пирролидон..................................173
1-Метил-5-пропил-2-тиопирролидон...............................255
а-[1-Метил-3(а-тетрагидрофурил)пропил]фурат....................177
а-[ 1-Метил-3-(а-тетрагидрофурил)пропил]тетрагидроф урат . .176
а-[1-Метил-2-(а-тетрагндрофурил)этил]тетрагидрофурат ... 178
Метилтиоэтилакрилат.............................................13
Метилфеноксималоновый эфир.................................... 179
1-(5-Метил-2-фурил)-4-ннтро-3-бутанол..........................298
1-Метил (2-фурил)-4-иитро-3-бутанол............................298
3-[2-(5-Метнлфурфурилокси)этокси]пропиламин....................182
3-[2-(5-Метилфурфурнлоксн)этокси]пропионнтрил..................183
3-[М-Метилциклопентано(в)-2-пирролидил]пропилбензоат . . 184
319
3-[ N-Метилциклопентано (в) -2-пирролидил]пропил-л«-метоксибен-зоат..............................................................185
Метионовая кислота, днацетоловый эфир...........................83
2-Метокси-Г\1-ацетил-1,6-диокса-9-азаспнро[4,51дскьн .... 187
о-Метоксибензолсульфохлорид....................................134
л-Метоксибензолсульфохлорид....................................134
п-Метоксибепзолсульфохлорид.....................................134
5-[2-(Метоксикарбонил) этил]-1,2-дигидропирролизин .... 190
2-Метокси-2-метил-М-ацетил-1,6-диокса-9-азаспиро[4,5]декан . . 188
2-Метокси-2-метил-М-этил-1,6-диокса-9-азаспиро[4,5]декаи . , 197
(о-Метоксифенил )оксиметилидендифосфоиовая кислота, свинцо-
вая соль................................................192
(о-Метоксифенил)оксиметилидендифосфоновая кислота, натриевая соль......................................................192
2 Метокси-Ы-этил-1,6-диокса-9-азаспиро[4,5]декан..........196
2-М-Морфолил-2-оксо-5-метил-5-метоксиметил-1,3,2-диоксафосфо-рннан....................................................201
Р-Нафтилгидразин...............................................212
Ь(а-Нафтил) пиридиний иодид................................38
2-Нафтол-3,6-дисульфокислота,двунатриевая соль .... 203
Неокупроин................................................102
2-Нитро-4,5-дибутоксианилин....................................206
2-Нитро-4,5-днбутоксиацетанилид...........................207
5-(5-Нитротенилиден) роданин............................... - 209
п-Нитрофепилгндразон а,р-дикетобутиролактона...............61
5-Нитрофенол (2-азо Г) -2'-(0-бутирилтидразино) нафталин . 215
5-Нитрофенол (2-азо-1') -2'- (Р-пропионилгндразино) нафталин . 215
8-Нитрохинальдин........................................215
а-Ноннл-2-пирролидон............................................21
Оксибис(2-метоксиэтан) ....................................... 97
у-Оксигептановая кислота, натриевая соль...................... 220
4-Окси-З-метокснкорнЧная кислота..........................293
4-Окси-З-метоксикоричный альдегид..............................147
3-(4-Окси-3-метоксифенил)-2-пропенол......................150
у-Оксинонановая кислота, натриевая соль ..................... 221
у-Оксиоктановая кислота, натриевая соль......................220
у-Оксипимелиновая кислота, дниатриевая соль ................. 222
N-(3-Оксипропил) сукцинимид....................................234
N-(3-Оксипропил) фталимид ....................................237
3-Окси-5-феиил-4-пеитииовая кислота, метиловый эфир . . 259
N- (2-Оксиэтил) диэтилеитриамии ..............................224
N-(2-Оксиэтил)диэтилентриамнно-Г\,М',М",М"-тетрауксусная кислота ........................................................ 226
1-Оксиэтилиденднфосфонат серебра...........................228
Оксиэтилидеидифосфоновая кислота, соль с анилином . . 231
. Оксиэтилидеидифосфоновая кислота, соль с диэтиламином , 231
Оксиэтилидеидифосфоновая кислота, соль с о-толуиднном . 231
(1-Оксиэтилидеидифосфоновая кислота, тринатриевая соль . 232
Г4-(2-Оксиэтил)сукцинимид .....................................234
320
N-(2-Оксиэтил) фталимид >................................ . 237
у-Оксо-М-(5-амил-2-пирролидон) масляная кислота .... 241
у-Оксо-М-(5-бутнл-2-пирролндон) масляная кислота .... 241
у-Оксо-N-(5-гекснл-2-пирролидон)масляная кислота .... 240
у-Оксо-М-(5-изоамил-2-пирролидои) масляная кислота ... 241
у-Оксо-Ы-(5-изобутил-2-пирролидон) масляная кислота 241
у-Оксо-1У-(5-нзогексил-2-пирролидон)масляная кислота . . 241
у-Оксо-М-(5-пропил-2-пирролидон) масляная кислота .... 241
0-(2-Октагидроиндолил)пропионовая кислота.....................247
0- (2-ОктаГидронндолил)пропиОновая кислота, метиловый эфир . 167
Октаиоилоксибензальдегид.......................................50
5-Октил-2-пнрролндон...........................................21
Пеитафторазндобензол..........................................242
а-Пиколин, N-окнсь.......................................... 300
(2-Пиридилметил)имииодиуксусиая кислота.......................244
Пирокатехин, дибутнловый эфир..................................91
Р(2-Пнрролидил)пропиоиовая кислота .......................... 246
З-Пирролизидои................................................249
2-(Пропилазо)пропан............................................11
у-Пропилбутиролактон ........................................ 219
н-Пропилгидразин..............................................199
5-Пропил-М-(0-оксиэтил)-2-пирролнзидон ....... 252
5-Пропил-2-пирролидон..........................................20
5-Пропил-2-тиопирролидон......................................255
Пропионил-р-нафтилгидразин.................................. 213
Серебряная соль тринитрометана с диоксаиом, комплекс . 145
а-транс-у-цис-0-Стприлакрнловая кислота.......................258
а-транс-у-цис-р-Стнрилакриловая кислота, метиловый эфир . 260
N,N,N',N'-TeTpaKHC (карбоксиметил)-5,б'-метилендиаитраниловая кислота.......................................................262
Тетраэтил-1-оксиэтаи-1,1'-дифосфонат..........................265
Тетраэтил-1-оксиэтнлидендифосфоиат............................265
Тиосемнкарбазон 0-(5-хлор-2-фурил) акролеина..................268
Ы-(о-Толил) бензамид...........................................58
1-(п-Толил)пиридиний иодид................................... 36
Тримеллитовая кислота 270
Трнмеллитовая кислота, трихлорангидрид ...................... 277
D,L-Триптофан.................................................273
2,4,6-Трихлоранилин......................................... 280
Трнэтилфосфат.................................................283
1,10-Феиантролин-2,9-диальдегид...............................285
1,10-Фенаитролии-2,9-диальдоксим..............................287
1,10-Фенаитролин-2,9-дикарбоиовая кислота.....................289
5-Феиил-2-пеитеи-4-ииовая кислота, метиловый эфир .... 260
1-Феннлпиридиннй бромид........................................38
1-Фенилпирндиннй иодид.........................................38
1-Фенилпиридиний хлорид...................................... 38
2-Фенил-2-пропантиол..........................................291
21 Зак. 1164
321
м-Феноксифенол.............................................
Феруловая кислота .........................................
Феруловая кислота, метиловый эфир..........................
3-(а-Фурил)бутанал.........................................
3-(а-Фурнл)днбутанал...................................
1-(а-Фурнл)-4-нитро-3-бутаиол..............................
Фуронлхлорид ..............................................
Фуртизон...................................................
Хинальдиний бисульфат .....................................
1(3-Хинолил) пиридиний иодид...............................
9-Хлоракридин .............................................
2-Хлорметиленпиридии.......................................
p-Хлорпропиоиовая кислота .................................
7-Хлор-5-фенил-3-беизилиден-1,2-дигидро-ЗН-1,4-бенздиазепин--2-он...........................« ;......................
64
293 168
293 295 297 177
268 216 ¥ 24
299
301
303
Циклогексано(в)-3-пнрролизидон.................................250
р-[Циклопентаио(в)-2-пирролидил]пропионовая кислота . . . 247
Р-[Циклопентано(в)-2-пирролидил]пропионовая кислота , метиловый эфир....................................................166
Циклопентано(в)-3-пирролизидон.................................250
1-(Этилазо) пропан........................................... 10
2-(Этилазо) пропан.............................................11
Этилакролеин .................................................. 158
1 -Этил-4-ацетилбензол.........................................305
п-Этилацетофенон........................................... 305
а-Этил-р-гексоксиметилпропиоиовая кислота...................307
Этнлгндразии .............................................. 199
Этилендиаминдиацетат кобальта...............................309
Этилендиаминднацетат меди ..................................309
Этилендиаминдиацетат никеля.................................309
Р-(4-Этнл-2-пирролидил) пропионовая кислота ... . . 247
5-Этил-2-пнрролидон..........................................20
5-Этил-З-пирролизидои...................................... 250
Этилтиоэтилметакрилат ...................................... 14
5-Этил-5-феноксибарбитуровая кислота........................312
Этилфенокснмалоновая кислота................................314
5-Этил-5-фенокситиобарбитуровая кислота.................... 312
Этилфурфурилиденацетоацетат . 130
УДК 547.235.2'21.
АЛИФАТИЧЕСКИЕ АЗОСОЕДИНЕНИЯ. В. С. Стопский, 3. И. Сергеева, Б. В. Иоффе. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 9.
Перегонкой с раствором щелочи в этиленгликоле продуктов конденсации моиоалкилгидразинов с альдегидами или кетонами получены (при веществах указаны выход и т. кип.): азоэтаи, 67%, 58,8 — 58,97760 мм; 1,1'-азопропан, 60%, 113,0—113,57761 мм; 1-(этила-зо)пропан, 58%, 87,5—87,7°/765 мм; 2-(пропилазо)пропан, 56%, 75,5— 76,17760 мм. Бнбл. 5 назв.
УДК 547.391.126'122.07(088.8)
АЛКИЛТИОЭТИЛАКРИЛАТЫ и АЛКИЛТИОЭТИЛМЕТАКРИ-ЛАТЫ. М. А. Коршунов, Р. Г. Кузовлева, И. В. Фураева. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 12.
Переэтерификацией метилакрилата или метилметакрилата соответствующими алкилтиоэтанолами в присутствии алкоголятов натрия и титана как катализаторов н фентназина как ингибитора полимеризации получены (при веществах указаны т. кип. и выход): метил-тиоэтилакрнлат, 6273 мм, 87,6%; этилтиоэтнлметакрилат, 73— 7471 5 мм, 87,5%; грег-гексилтиоэтилметакрилат, 120—12171 мм, 80,7%. Библ. 5 назв.
УДК 547.473.fc
АМИДЫ и МЕТИЛАМИДЫ у-ОКСИКАРБОНОВЫХ КИСЛОТ В. А. Седавкина. Методы получения химических реактивов н препаратов, вып. 26, М., ИРЕА, 1974, с. 15.
Амиды и метиламиды у-оксикарбоновых кислот получены путем каталитического гидрирования этиловых эфиров у-кетокарбоновых кислот с последующим насыщением растворов эфиров у-оксикарбо-новых кислот аммиаком или метиламином и нагреванием при 120°: амид у-оксигептановой кислоты, т. пл. 66—67°, выход 89,3%; амид у-оксиоктановой кислоты, т. пл. 87—88°, выход 82%;
амид у-оксинонановой кислоты, т. пл. 85—86°, выход 85%;
амид у-оксиизоионановой кислоты, т. пл. 77—78°, выход 80%; амид у-оксндекановой кислоты, т. кип. 95—96°, выход 89%;
метиламид у-окснгептановой кислоты, т. кип. 102—103°/3 мм, По = 1,4631, </420 = 1,0002, выход 79%;
метиламнд у-оксиоктановой кислоты, т. пл. 90—91°, выход 83,8%; метиламид у-оксиноиановой кислоты, т. кип. 30—31°, выход 82%. Библ. 8 назв.
21*
323
УДК 547.466.3—318
5-АМИЛ-2-ПИРРОЛИДОН. В. А. Седавкина. Методы получения химических реактивов и препаратов, вып.26. М.,- ИРЕА, 1974, с. 19.
5-Амил-2-пирролидон, т. кип. 126—12871 мм; т. пл. 29—30°, по-| лучен с выходом 68,4 % восстановительным аминированием этилового эфира у-кетоионановой кислоты в присутствии никеля Ренея при 120° и давлении водорода 90 атм. Кипячением 5 амил-2-пнрролидона с избытком уксусного ангидрида получен Ы-ацетил-5-амил-2-пирроли-дон с выходом 94%, т. кип. 111—11272 мм, ~ 1,4687.
Из соответствующих этиловых эфиров у-кетокарбоновых кислбт получен ряд других 5-алкил-2-пирролидонов и их ацетильные производные. Библ. 10 назв.
УДК 547.835.3
9-АМИНОАКРИДИН. Я. Н. Дыханов, Т. В. Перова, Р. Ф. Виде-нина, И. В. Покотыло, В. И. Базакин. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 23.
9-Амнноакридии, т. пл. 232—234°, получен с выходом 75% взаимодействием N-фенилантраниловой кислоты с избытком хлорокиси фосфора при 108—110° последующей обработкой полученного 9-хлоракридина избытком углекислого аммония в расплаве фенола при 70° и очисткой технического амина осаждением едким натром из солянокислого раствора и перекристаллизацией из ацетона. Библ; 3 назв.
УДК 547.751
о-АМИНОР,₽-ДИ(3-ИНДОЛИЛ)ЭТИЛБЕНЗОЛ. А. О. Гинзбург, С. В. Лылык, А. К. Шейнкман. Методы получения химически^ реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 27.
о-Амино-р,6-ди(3-иидолил)этилбеизол, т. пл. 168—170°, получен с выходом 97% взаимодействием индола с 20%-ной серной кислотой в присутствии азотистых оснований при комнатной температура. Библ. 3 иазв.
УДК 547.583.5
2-(2-АМИНО-5-НИТРОБЕНЗАМИДО) БЕНЗОЙНАЯ КИСЛОТА. И. С. Маркович, Н. А. Филягина, В. М. Дзиомко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 29.
2-(2-Амино-5-иитробензамидо)беизойная кислота, т. пл. 284—285°. получена впервые с выходом 26,5% сплавлением антраниловой кислоты с 5-нитроизатовым ангидридом. Библ. 1 назв.
324
УДК 547.831.6
8-АМИНОХИНАЛЬДИН. В. М. Дзиомко, И. А. Красавин, Б. В. Парусников^ Ю. П. Радин. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЁА, 1974, с. 31.
8-Аминохинальдин, т. пл. 54—56°, получен с выходом 76—82% восстановлением 8-нйтрохпиальдина порошкообразным железом в разбавленной уксусной кислоте. Библ. 7 назв.
УДК 547. 747 + 542.951.1
а-[(2-АМИНОЦИКЛОПЕНТИЛ)МЕТИЛ]ФУРАН.|Л. А. Пономарев, I А. П. Кривенько. Методы получения химических препаратов, вып. 26. М, ИРЕА, 1974, с. 34.
а-[2- (Аминоциклопентил) метил]фуран, т. кип. 115°/14 мм, Пр =
= 1.5010, с выходом 58% получен впервые реакцией восстановитель ного аминирования р-фурфурилиден-а-циклопеитана. Библ. 4 назв.
УДК 547.821.3
N-АРИЛПИРИДИНИЕВЫЕ СОЛИ. Л. Г. Гах, А. К. Шейнкмач, С. Н. Баранов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 36.
Проверен метод получения N-фенилпиридиний хлорида нагреванием 2,4-динитрофенилпиридиний хлорида с анилином в абсолютном спирте. Указанным способом получены (при веществах указан выход н т. пл.); 1-фенилпиридиний хлорид, 60%, 105—106°; 1-фенилпириди-иий бромид, 50%, 156—157°; 1-фенилпиридииий иодид, 45%, 209 — 210°; 1-п-толилпиридиний иодид, 45%, 160—161°; 1-а-нафтилпириди-ний иодид, 42%, 182—183°; 1,3-хинолилпирпдиннй иодид, 40%, 221 — 221,5°. Табл. 2, бнбл. 3 назв.
УДК 547.552.2
1-АЦЕТИЛАМИНО-2-АЦЕТОКСИ-3,5-ДИМЕТИЛБЕНЗОЛ.
В. М. Островская, М. М. Краснова, Л. В. Ломакина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 40.
1-Ацетиламиио-2-ацетокси-3,5-диметилбензола, т. пл. 158.5—160°. получен с выходом 93% ацетилированием 2-амиио-4,6-диметилфенола в уксусном ангидриде в присутствии ацетата натрия. Библ. 1 назв.
325
УДК 547.551.4
2-АЦЕТИЛАМИНОФЕНОКСИУКСУСНАЯ КИСЛОТА. В. М. Островская, М. М. Краснова, О. Т. Лушина. Методы получения химических реактивов и препаратов, вып. 26. М. ИРЕА, 1974, с. 42.
2-Ацетиламииофеиоксиуксусиая кислота, т. пл. 149°, получена с выходом 78% взаимодействием 2 ацетиламииофеиола с монохлор-уксусной кислотой. Библ. 1 иазв.
УДК 547.564.4
2-АЦЕТИЛАМИНОФЕНОЛ. В. М Островская, М. М. Краснова. Методы получения химических реактивов и препаратов, вып. 26. М.. ИРЕА, 1974, с. 44.
2-Ацетиламииофеиол, т. пл. 205°, получен с выходом 96% ацетилированием 2-аминофенола уксусным ангидридом в этилацетате. Библ. 8 иазв.
УДК 547.435.07
И-АЦЕТИЛ-И-ФУРФУРИЛАМИНОЭТАНОЛ И 3-(N-АЦЕТИЛ. 5-МЕТИЛ-2-ПИРРОЛИДИЛ) ПРОПАНОЛ. |Д. А. Пономарев], И. А. Маркушина, Т. И. Губина, М. В. Норицина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 46.
N-Ацетил-М-фурфуриламииоэтаиол, т. кип. 439—140°/4 мм, с выходом 73% и 3-(Й-Ацетнл-5-метил-2-пирролидил) пропанол, т. кип. 169—17Г/3 мм, с выходом 75%, получены ацетилированием кетеиоМ водных растворов N-фурфуриламиноэтаиола и 1-(5-метил-2-пирроли-дил)-3-пропанола. Библ. 6 назв.
УДК 547.571
4-и-лЦИЛОКСИБЕНЗАЛЬДЕГИДЫ, 3. С. Сиденко, В. А. Ин шакова, Г. С. Чижова. Методы получения химических реактивов препаратов, вып. 26. М., ИРЕА, 1974, с. 49.
4-и-Бутирил-, 4-и-гептоил-, 4-н-каприлилбеизальдегиды получен с выходами 40, 53 и 51 % соответственно, взаимодействием 4-окс бензальдегида с хлористыми ацилами. Табл. 1, библ. 1.
326
УДК 547.496.3
S-БЕНЗИЛИЗОТИОМОЧЕВИНА ГИДРОХЛОРИД. Е. А. Буга-1, Методы получения химических реактивов и препаратов, вып. 26. М, ИРЕА, 1974, с. 51.
S-Бензилизотиомочевина гидрохлорид, т. пл. 174—175°, с выходом 85% получен при взаимодействии тиомочевины и хлористого бензила в водной среде. Библ. 4 иазв.
Удк 547.582.2
БЕНЗИЛОВЫЙ ЭФИР БЕНЗОЙНОЙ КИСЛОТЫ. А. Л. Ли-фиц, Е. П. Агеев, С. И. Орел, И. А. Барц. Методы получения химических реактивов, вып. 26. М., ИРЕА, 1974, с. 53.
Разработана методика получения бепзилбеизоата с выходом 89% из бензойной кислоты и хлористого бензила в присутствии кальцинированной соды и катализатора — диметилформамида в среде беч-знлбензоата. Библ. 4 иазв.
УДК 547.653.1
8-БЕНЗОИЛ АМИНО-1 -НАФТОЛ-3,6-ДИСУЛЬФОКИСЛ ОТА. ДВУНАТРИЕВАЯ СОЛЬ. Н. Н. Дыханов, Р. Ф. Виденина, И. В. Покотыло, Т. В. Перова, В. И. Базакин, П- Д- Якухный. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 55.
Двунатриевая соль 8-бензоиламино-1-нафтол-3,6-дисульфокнсло-ты выделена из технического продукта экстракцией смесью метилового и н-амилового спиртов с последующей очисткой экстракта активированным углем и крнсталлнзацией соли при охлаждении. Библ. 7 назв.
УДК 547.552.1
N-БЕНЗОИЛ-о-ТОЛУИДИН. Ю. Г. Бондарь. Методы получения химических реактивов и препаратов, вып. 26, М., ИРЕА, 1974, с. 58.
N-Бензоил-о-толуидин, f. пл. 144°, с выходом 84,5% получен взаимодействием о-толуидина с бензоилхлоридом в среде толуола в присутствии кальцинированной соды. Библ. 4 назв.
.327
УДК 547.473.2—314
БИС(н-НИТРОФЕНИЛ ГИДРАЗОН) а,₽-ДИКЕТОБУТИРОЛ-АКТОНА. И. В. Хвостов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 60.
Бис(п-иитрофеиилгидразои) а,р-дикетобутиролактоиа, т. пл. 298°, с выходом ~ 90% получен при действии n-иитрофеиилдиазония на а-бром-0-кетобутиролактои в среде углекислого калий через промежу точный продукт — п-иитрофеиилгидразон а,р-дикетобутиролактоиа с. последующим его превращением в конечный продукт в уксуснокислой среде. Библ. 2 назв.
УДК 547. 562
л-БИС(л-ФЕНОКСИФЕНОКСИ) БЕНЗОЛ. Н. Ю. Аронская, Е. И. Маховер, Р. М. Гелыитейн, В. Д. Безуглый. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 63.
.и-Бис(л-феиоксифеиокси) бензол, Яд = 1,6300, с выходом 70% получен из л-дибромбензола и лг-феиоксифеиола, синтезированного из резорцина и бромбеизола в диметилформ амиде. Библ. 6 назв.
УДК 547.89
7-БРОМ-1-АЦЕТИЛ-5-ФЕНИЛ-1,2-ДИГИДРО-ЗН-1,4;БЕНЗДИ-АЗЕПИН-2-ОН. 3. И. Жилина, А. В. Богатский, С. А. Андронати, Т. К. Чумаченко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 66.
7-Бром- 1-ацетил-5-феиил- 1,2-диги дро-ЗН-1,4-беиздиазепии-2-он, т. пл. 184—185°, с выходом 54% получен взаимодействием натриевой соли 7-бром-5-феиил-1,2-дигидро-ЗН-1,4-бенздиазеппн-2-она с хлористым ацетилом. Библ. 4 назв.
УДК 547.773.07
4-БРОМ-3-МЕТИЛ-5- (5-БРОМ-2-ФУ РИЛ) ПИРАЗОЛ.
|А. А. Пономарев, | Л. В. Черкесова, А. А. Морщакова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с.68.
4-Бром-3-метил-5-(5-бром-2-фурил) пиразол, т. пл. 128—130°, с выходом 87% получен прямым бромированием 5-(2-фурил)-3-метил-пиразола диоксандибромидом в эфире. Библ. 4 назв.
328
УДК 547.473.2-314
а-БРОМТЕТРОНОВАЯ КИСЛОТА. И. В. Хвостов, О. А. Логинова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 70.
а-БромтетроИовая кислота, т. пл. 181—183°, (разл.), получена с выходом бромированием ацетоуксусного эфира с последующей цик лизацией а.у-дибромацетоуксусиого эфира при нагревании 110—120° и пониженном давлении (10—15 мм). Библ. 5 иазв.
УДК 547.831.2
N-БУТИЛХИНАЛЬДИНИй ЙОДИД. И. С. Маркович, Л. И. Блохина, В. М. Дзиомко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 72.
N-Бутилхинальдииий йодид, т. пл. 162°, получен впервые с выходом 62,7% взаимодействием хииальдииа с йодистым бутилом.
УДК 547.38
2,4-ГЕКСАДИЕНАЛЬ. О. С. Степанова, А. И. Галатина. Методы получения химический реактивов и препаратов вып. 26. М. ИРЕА, 1974, с. 74.
2,4-Гексадиеналь, т. кип. 57°/9 мм, с выходом 45% получен коп денсацией кротонового альдегида с ацетальдегидом. Библ. 5 иазв.
УДК 547.725
а-ГЕКСИЛКРОТОНОВАЯ КИСЛОТА. Г. Ф. Танцюра, А. В. Богатский. Методы получения химических реактивов и препаратов, вып. 26, М., ИРЕА, 1974, с. 76.
а-Гексилкротоновая кислота, т. кип. 126—127°/3 мм, с выходом 60%, получена путем эфирного расщепления гексил-а-метоксиэти.:-ацетоуксусного эфира. Библ. 7 назв.
УДК 547.562.
4-и-ГЕКСОКСИБЕНЗИЛХЛОРИД. 3. С. Сиденко, В. А. Ишиа-кова, Г. С. Чижова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 79.
4-и-Гексоксибеизилхлорид, т. кип. 162—163°/7 мм, получен впервые с выходом 51% хлорметилироваиием и-гексоксибензола.
329
УДК 547.5723
1,4-ДИАЦЕТИЛБЕНЗОЛ. Е. И. Маховер. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 81.
1,4-Диацетилбеизол, т. пл. 113—114°, с выходом 46% получен окислением 4-этилацетофеиоиа пермаигаиатом калия в водной среде в присутствии азотнокислого магния. Библ. 2 назв.
УДК 547.425.5
ДИАЦЕТОЛОВЫЙ ЭФИР МЕТИОНОВОЙ КИСЛОТЫ. А. Л. Фридман, Г. С. Исмагилова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 83.
Диацетоловый эфир метионовой кислоты, т. пл. 108—108,5°, получен впервые с выходом 70% взаимодействием диазоацетоиа с метионовой кислотой в уксусном ангидриде. Библ. 2 иазв.
УДК 547.572.3
ДИБЕНЗОИЛМЕТАН. И. Ю. Аронская, Р. М. Гелыитейн, Е. М. Иофис. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 85.
Дибензоилметаи, т. пл. 76,3—77,3°, с выходом 56,5%, получен омылением дибромида бензальацетофеноиа 16%-ным раствором бутилата натрия. Библ. 3 назв.
УДК 547.223.271.272
1,4-ДИБрОМ-2-МЕТОКСИ- И 1.4-ДИБРОМ-2-ЭТОКСИПЕНТА-НЫ. А. В. Богатский, Г. Л. Камалов, Н. Г. Лукьяненко, В. И. Ша-рыгин. Методы получения химических реактивов и препаратов, выл. 26. М., ИРЕА, 1974, с. 87.
1,4-Дибром-2-метоксипентаи, т. кип. 71—73°/2 мм, с выходом 45% и 1,4-дибром-2-этоксипеитан, т. кип. 85—8774 мм, с выходом 53% получены взаимодействием соответствующих а,р-дибромэтил-алкиловых эфиров с пропиленом. Библ. 3 назв.
УДК 547.551
3,4-ДИБУТОКСИАНИЛИН. В. М. Дзиомко, А. В. Иващенко. Методы получения химических реактивов и препаратов, вып. 26. М, ИРЕА, 1974, с. 90.
3,4-Дибутоксиаиилин, т. кип. 193—f95°/24 мм. получен с выходом 89—90% восстановлением 3,4-дибутоксинитробеизола сульфидом натрия в водном этаноле. Библ. 4 назв.
330
УДК 547.657
4-М,М-(ДИКАРБОКСИМЕТИЛ)АМИНОМЕТИЛ-3-ОКСИ-2-НАФТОИНАЯ КИСЛОТА. Л. М. Тимакова, В. Я, Темкина, Г. Ф. Ярошенко, Н. Ё. Хавченко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 93.
4-N.N- (Дикарбоксиметил) аминометил-З-окси-2-иафтойная кислота получена по реакции Маиииха с выходом 90% взаимодействием 2-окси-З-иафтойиой кислоты с формальдегидом и имииодиуксусиой кислотой в уксуснокислой среде. Библ. 5 иазв.
УДК 547.723+542.951.1
1,1'-ДИМЕТИЛ-3,3'-ДИ(ТЕТРАГИДРОФУРИЛ-2) ДИПРОПИЛ ОКСИД.М Д, Липанова. Методы получения химических реактивоа и препаратов, вып. 26. М., ИРЕА, 1974, с. 95.
1>1'-Диметил-3,3'-ди(тетрагидрофурил-2)дипропилоксид, т. кип. 175—176°/9 мм, с выходом 58,6% получен нагреванием 1-(а-тетрагид-рофурил)-3-бутанола с серной кислотой в дихлорэтане. Библ. 4 назв.
УДК 547.421.5
ДИМЕТИЛОВЫЙ ЭФИР ДИЭТИЛЕНГЛИКОЛЯ. А. Л. Лифиц, А. А. Вейцман. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с.97.
Диметиловый эфир диэтилеигликоля, = 1,4085, с выходом 35,5% получен взаимодействием f.f'-дихлордиэтилового эфира со спиртовым раствором едкого кали. Библ. 5 иазв.
УДК 547.71
ЦИС- И ТРАЯС-2,3-ДИМЕТИЛОКСЕТАНЫ. Г. А. Филип, С. А, Петраш, Г. В. Пьянкова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 99.
Получена смесь стереоизомерных цис- и транс-2,3-диметилоксе-танов: т. кип. 80°, 0,8469, Лц = 1,4040 и т. кип. 89°, d?’ ••=
= 0,8203, п™ = 1,3980, соответственно, с выходом 50% взаимодействием хлоргидрииацетата 2-метил-1,3-бутандиола со Щелочами. Библ. 3 назв.
331
УДК 547 8363
2,9-ДИМЕТИЛЛ,Ю-ФЕНАНТРОЛИН. В. М Дзиомко, И. А. Красавин, Б. В. Парусников. Методы получения Химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 102.
2,9-Диметил-1,10-фенантролин, т. пл. 161—163°, получен с выходом 36,8—42,8% циклизацией 8-аминохинальдина с кротоновым альдегидом в среде соляной кислоты в присутствии хлористого алюминия. Библ. 5 иазв.
УДК 547.415.1'241'118
КМ'-ДИМЕТИЛЭТИЛЕНДИАМИНО-М,М'-БИС(МЕТИЛЕН-ФОСФОНОВАЯ И М,Ы'-ДИ(2-ОКСИЭТИЛ)ЭТИЛЕНДИАМИНО-N.N'-БИСМЕТИЛЕНФОСФОНОВАЯ КИСЛОТЫ. В. П. Мархаева, 10. М. Поликарпов, М. В. Рудомино, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 105.
М,М'-Диметилэтилендиамино-М,М'-бисметиленфосфоиовая и N.N'--ди(2-окснэтил)этнлеидиамино-М,М'-бисметиленфосфоновая кислоты получены конденсацией соответственно М,М'-диметилэтилендиамииа й М,М'-ди(2-оксиэтил)этилендиамнна с формалином н фосфористой кислотой, а также конденсацией указанных диаминов с параформом и днэтнлфосфнтом с последующим омылением образовавшихся сложных эфиров до свободных кислот. Библ. 4 назв.
УДК 547.642.07
2,7-ДИМЕТОКСИ-1,6-ДИОКСАСПИРО[4,4]НОНАН. И. А. Маркушина, Н. В. Шуляковская. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 109.
2,7-Диметокси-1,6-диоксаспиро[4,4]нонан, т. кип, 92—93°/10 мм, с выходом 89% получен впервые электролитическим интрамолекулярным метокснлированием 1-(а-фурил)-3-пропаналя с последующим гидрированием образующегося 2,7-днметокси- 1,6-дноксаспиро[4,4]но-нена-3 при комнатной температуре в присутствии никеля Ренея. Библ. 6 назв.
УДК 547 836.3
ДИНИТРИЛ 1,10-ФЕНАНТРОЛИН-2,9-ДИКАРБОНОВОИ КИСЛОТЫ. В. М. Дзиомко, Б. В. Парусников. Методы получения химических реактивов н препаратов, вып. 26, М., ИРЕА, 1974, с. 112.
Динитрнл 1,10-фенантролин-2,9-дикарбоновой кислоты, т, пл. 364°, получен с выходом 87% при взаимодействии 1,10-феиантро-лнн-2,9-днальдоксима с уксусным ангидридом.
332,
УДК 547.841.254.9
ДИОКСАНАТ БИС (ТРИНИТРОМЕТИЛ) РТУТИ. А. Л. Фридман, Т. Н. Ившина. Методы получения химических реактивов н препаратов, вып. 26. М., ИРЕА, . 1974, с. 114.
Диоксанат бис(тринитрометил) ртути, т. разл. 199°, с выходом 85% получен впервые взаимодействием бис (тринитрометил) ртути с диоксаном в воде. Библ. 5 назв.
УДК 547.565 2'466.22'292'211
2,6-ДИОКСИБЕНЗИЛИМИНОДИУКСУСНАЯ КИСЛОТА. В. Я. Темкина, Н. В. Цирульникова, Л. Д. Меликсетян, Р. П. Ластовский. Методы получения химических реактивов и препаратов, вып. 26. М , ИРЕА, 1974, с.116.
2,6-Дноксибензилиминоднуксусная кислота получена с выходом 60% по реакции Манниха взаимодействием резорцина, формальдегида и иминодиуксусной кислоты в среде ледяной уксусной кислоты. Библ. 1 назв.
УДК 547.565.2'466.22'292'211
2,3-ДИОКСИБЕНЗИЛИМИНОДИУКСУСНАЯ КИСЛОТА, МО НОНАТРИЕВАЯ СОЛЬ. В. Я. Темкина, Н. В. Цирульникова, С. 4 Тарасова, Р. П. Ластовский. Методы получения химических реактивов н препаратов, вып. 26. М., ИРЕА, 1974, с. 118.
Мононатриевая соль 2,3-диоксибензилиминодиуксусиой кислоты получена с выходом 94,5% по реакции Манниха взаимодействием пирокатехина, формальдегида и иминодиуксусной кислоты в уксуснокислой среде с последующим высаживанием полученного вещест ва из реакционного раствора ацетоном. Библ. 1 назв.
УДК 547.563'466.22'211
М,М'-ДИ(2-ОКСИ-5-МЕТИЛБЕНЗИЛ)ЭТИЛЕНДИАМИНО-N.N'-ДИУКСУСНАЯ КИСЛОТА. Н. В. Цирульникова, В. Я. Темкина, Р. П. Ластовский. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 120.
N.N'-Дн (2-окси-5-метнлбензил) -этилендиамнно-М,М'-диуксусиая кислота получена с выходом ~ 40% по реакции Манниха взаимодействием n-крезола с формальдегидом н этнлеиднамнн-1Ч,М'-диук-сусной кислотой в щелочной среде. Библ. 1 назв.
333
УДК 547.5562
З.З'-ДИ-Н-ПЕНТОКСИАЗОКСИБЕНЗОЛ. 3. С. Сиденко, В. А. Иншакова, Г. С. Чижова. Методы получения химических реактивов Л препаратов, вып. 26. М., ИРЕА, 1974, с. 122-
4,4'-Ди-н-пентоксиазоксибеизол, т. пл. 73—74°, получен с выходом 28% восстановлением 4-пеитоксниитробензола этилатом натрия в присутствии гидразин-гидрата. Библ. 4 назв.
УДК 547.551.54'51
2.6-ДИХЛОР-4-НИТРОАНИЛИН. Р. Ф. Виденина, Н. И. Дыханов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 124.
2,6-Дихлор-4-иитроанилин, т. пл. 194—195°, получен с 95%-ным выходом действием перекиси водорода на солянокислый раствор п-ннтроаиилина прн 50—55°. Библ. 3 назв.
УДК 547.533.1'539.221
2,6-ДИХЛОр-1,4-ФЕНИЛЕНДИАМИН. Р. Ф. Виденина, Н. Н. Дыханов. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 126.
2,6-Дихлор-1,4-феиилендиамин, 123—123,5°, получен с выходом 75% восстановлением 2,6-днхлор-4-иитроаиилииа двухлорнстым оловом в кипящей смеси концентрированной соляной кислоты и метанола. Библ. 2 иазв.
УДК 547.415.1'241
ДИЭТИЛ ЕНТРИАМИНО-N.N,N',N',N''-ПЕНТАМЕТИЛЕНФОС-ФОНОВАЯ КИСЛОТА. М. В. Ру домино, Е. К. Колова, А. М. Орлова, Л. И. Мизрах, Т. Я. Медведь. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 128.
Диэтнлеитриамино-М,М,М',М',М"-пентаметиленфосфоиовая кислота получена с выходом 66% конденсацией диэтилентриамнна с формалином треххлористым фосфором. Библ. 4 назв.
334
УДК 547.462.26.725.484.34
ДИЭТИЛФУРФУРИЛИДЕНМАЛОНАТ И ЭТИЛФУРФУРИЛИ-ДЕНАЦЕТОАЦЕТАТ. Л. Г. Богатская, Г. Л. Камалов. Методы получения химических реактивов и препаратов, вЫп. 26. М., ИРЕА, 1974, с. 130.
Диэтилфурфурилиденмалоиат, т. кип. 163—16673 мм, с выходом 75% и этилфурфурилиденацетоацетат, т. кип. 152— 15772 мм, с выходом 65—68%, получены взаимодействием фурфурола, соответственно, с малоновым и ацетоуксусным эфирами в присутствии анионо-обменной смолы АВ-17П. Библ. 2 назв.
УДК 547.466.26
Д.ЬИЗОЛЕЙЦИН. Е. П. Крысий, И. И. Громова, Л. Г. Андронова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 132.
О,Ь-Изолейцнн получен с выходом 68% гидролизом втор-бутил-гидантоина, синтезированного последовательной обработкой метнл-этил ацетальдегид а бисульфитом иатрня, цианистым калием н карбонатом аммония. Бнбл. 6 назв.
УДК 547.541.513
ИЗОМЕРНЫЕ МЕТИЛ- И МЕТОКСИБЕНЗОЛСУЛЬФОХЛОРИДЫ. Н. Н. Дыханов, А. Б. Джиджелава, Т. С. Рыжкова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 134.
Изомерные метилбензолсульфохлориды (выход 80%) и метокси-беизолсульфохлориды (выход 40%) получены действием избытка уксуснокислого раствора двуокиси серы на сульфаты соответственно замещенных феиилдназониев в присутствии двухлористой меди (катализатор) и соляной кислоты с одновременной экстракцией образующихся сульфохлоридов бензолом. Библ. 4 назв.
УДК 547.663:727 + 542.942,7:953.1
ИЗО (4,7,8,9-ТЕТрАГИД РО) БЕНЗО-1,3-ДИМЕТОКСИТЕТРА-ГИДРОФУРАН. | А. А. Пономарев, | И. А. Маркушина, Г. Е. Мариничева. Методы получения химических реактивов, вып. 26. М., ИРЕА, 1974, с. 137.
Изо (4,7,8,9-тетрагидро) бензо- 1,3-диметокснтетрагидрофураи, т. кип. 76—8073 мм, н% — 1,4769, с выходом 27% получен взаимодействием 1,3-бутадиеиа с 1,5-диметокси-2,5-дигидрофураиом в присутствии гидрохинона. Библ. 4 назв.
335
УДК 547.551.51
л-ИОДАНИЛИН. Г. А. Креймер, Е. А. Бугай. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 139.
.и-йодаиилин, т. заст. 24°, с выходом 70% получен восстановлением м-йоднитробензола железным' порошком в солянокислой среде. Библ. 2 назв.
УДК 547.254.1
КАЛЬЦИИАЦЕТОУКСУСНЫИ ЭФИР. А. Е. Кожухова, М. В. Гренадерова, Т. К. Чумаченко, Л. Г. Деркач. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 141.
Кальций ацетоуксусный эфир, т. разл. >200°, с выходом 91% получен взаимодействием ацетоуксусного эфира с этилатом кальция. Библ. 3 назв.
УДК 547.741.07
5-(2-КАРБОКСИЭТИЛ)-1,2-ДИГИДРОПИРРОЛИЗИН.
| А. А. Пономарев, | Л. Н. Астахова, В. Н. Волколупов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 143.
5-(2-Карбоксиэтил)-1,2-дигидропирролизин, т. пл. 94—96° (из гексаиа), с выходом 84% получен взаимодействием 1,2-дигидропир-ролизииа с акриловой кислотой в присутствии фтористого бора. Библ. 4 назв.
УДК 547.841.255.61
КОМПЛЕКС СЕРЕБРЯНОЙ СОЛИ ТРИНИТрОМЕТАНА С ДИОКСАНОМ. А. Л. Фридман, Т. И. Ившина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 145.
Комплекс серебряной соли тринитрометана с диоксаном, т. разл 86—89°, получен впервые с выходом 53% взаимодействием серебряной соли трипитрометана с диоксапом в воде. Бнбл. 7 назв.
УДК 547.415.1'562.1
КОНИФЕРИЛОВЫЙ АЛЬДЕГИД. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 147.
Конифериловый альдегид, т. пл. 81—82°, с выходом 87% получен восстановлением алюмогидридом лития метилового эфира феру ловой кислоты прн температуре минус 80?. Библ. 5 назв.
336
УДК 547.415.1'562.1
КОНИФЕРИЛОВЫЙ СПИРТ. А. В. Богатский, 3. Л. Богатская, Л. В. Басалаева, Н. С. Уланова-Обезюк. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974; с. 150.
Конифериловый спирт, т. пл. 71—72°, с выходом 67—68% получен восстановлением метилового эфира феруловой кислоты алюмо-гидридом лнтия при температуре минус 19°. Библ. 4 назв.
УДК 547.397'393
МЕТАКРИЛОИЛХЛОРИД. Н. И. Дыханов, А. Б. Джиджелава. Методы получения химических реактивов и препаратов, вып. 26. М, ИРЕА, 1974, с. 153.
Хлорангндрид метакриловой кислоты, т. кип. 95—97°, получен с выходом 70% нагреванием метакриловой кислоты с избытком хлористого бензоила при непрерывном удалении метакрилоилхлорида из сферы реакции. Библ. 3 назв.
УДК 547.284
МЕТИЛБЕНЗИЛКЕТОН. Г. Ф. Танцюра, Л. Я. Глинская, И. Ф. Герасименко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 155.
Метилбензилкетон, т. кип. 113—115°/20 мм, с выходом 60—67% получен взаимодействием фенилуксусной кислоты с ледяной уксусной кислотой при 400—45(г в атмосфере углекислого газа. Библ. 3 назв.
УДК 547.264
2-МЕТИЛБУТАНАЛЬ. Е. П. Крысин, И. Н. Громова, Э. Д. Глинка, А. Г. Левина, Л. Г. Андронова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 157.
2-Метилбутаиаль, т. кип. 91—92°, с выходом 72% получен восстановлением на Pd-черни этилакролеина, образуемого конденсацией формальдегида н н-масляного альдегида в присутствии хлоргид-рата диметиламииа. Библ. 4 назв.
УДК 547.741.07
1-(3-МЕТИЛ-1,2-ДИГИД РОПИРРОЛИЗИЛ-5)-1-БУТЕН-3-ОН. | А. А. Пономарев,] Л. И. Астахова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 159.
1-(3-Метил-1,2-дигидропирролизил-5)-1-бутен-3-он, т. кип. 139— 142°/2 лои, с выходом 59,6% получен конденсацией З-метил-5-формил--1,2-дигидропирролизина с ацетоном в водиоспиртовой среде в присутствии едкого натра. Библ. 3 назв.
22 Зак. П64 337
УДК 547.284
МЕТИЛИЗОПРОПИЛКЕТОН. А. В. Богатский, Г. Л. Камалов. Методы получения химических реактивов и препаратов, вып. 26. М.,. ИРЕА,1974, с. 161.
Метилизопропилкетрн, т. кип. 93—96°, с выходом 50—60% получен парофазным гидролизом диметилацетоуксусного эфира при 320е Библ. 5 иазв.
УДК 547.271.569.4
МЕТИЛОВЫЙ ЭФИР. а-МЕТИЛ-р-МЕРКАПТОПРОПИОНО-ВОй КИСЛОТЫ. А. В. Богатский, А. Н. Турянская, А. И. Грень. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 163.
Метиловый эфир а-метил-р-меркаптопропионовой кислоты, т. кит. 75—79°/5 мм, с выходом 60% получен взаимодействием метилового эфира а-метил-Р бромпропионовой кислоты с тиомочевиной. Библ. 3 назв.
УДК 547.747.271+542.951.3
МЕТИЛОВЫЙ ЭФИР р-(5-МЕТИЛ-2-ПИрРОЛИДИЛ) ПРОПИОНОВОЙ КИСЛОТЫ. | А. А. Пономарев, | М. В. Норицина, А. Н. Кривенько. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 165.
Метиловый эфир р-(5-метил-2-пирролидил) пропионовой кислоты, т. кип. 92—92,578 мм, Ир = 1,4570, с выходом 52% получен впервые этерификацией 0-(5-метил-2-пирролидил) пропионовой кислоты метиловым спиртом в присутствии хлористого водорода. Библ. 3 назв.
УДК 547.415.1'562.1
МЕТИЛОВЫЙ ЭФИР ФЕРУЛОВОЙ КИСЛОТЫ. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева, Н. С. Уланова-Обезюк. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 168.
Метиловый эфир феруловой кислоты, т. кип. 18373 мм, т. пл. 59—60°, с выходом 25—38% получен взаимодействием феруловой кислоты с метиловым спиртом в присутствии серной кислоты и с выходом 52—55% взаимодействием феруловой кислоты с метиловым спиртом в присутствии ионообменнной смолы КУ-2. Библ. 2 назв.
338
УДК 547.747.23S
Р- (5-МЕТИЛ-2-ПИРРОЛИД ИЛ) ПРОПИОНОГИДРАЗИД.
| А. А. Пономарев, | М. В, Норицина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 171.
р-(5-Метил-2-пирролидил)пропиоиогидразид, т. пл. 69—70°, с выходом 87% получен впервые при взаимодействии метилового эфира Р-(5-метил-2-пирролидил) пропионовой кислоты с гидразином в мета-иоле. Библ. 1 иазв.
УДК 547.466.3-318
1-МЕТИЛ-5-ПРОПИЛ-2-ПИРРОЛИДОН. В. А. Седавкина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 173.
1-Метил-5-пропил-2-пирролидои, т. кип. 75—7677 мм, ~ = 1,4706, d420 = 0,9705, получен с выходом 61,7% гидрированием смеси этилового эфира у-кетогептановой кислоты и метиламина над скелетным никелевым катализатором при 120—130° и давлении водорода 100 атм. Библ. .9 иазв.
УДК 547.725+542.951.4
а-[1-МЕТИЛ-3-(а-ТЕТРАГИДРОФУРИЛ)ПРОПИЛ]ТЕТРА-ГИДРОФУРОАТ. М. Д. Липанова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 176.
а-[1-Метнл-3- (а-тетрагидрофурил) пропил]тетрагидрофуроат, т. кип. 170—171°/9 мм, с выходом 90% получен каталитическим гидрированием в присутствии никеля Ренея. Библ. 5 назв.
УДК 542.951.3
МЕТИЛФЕНОКСИМАЛОНОВЫЙ ЭФИР. О. С. Степанова, А. С. Яворский, М. И. Побережная, Л. Б. Хагундокова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 179.
Метилфеиоксималоновый эфир, т. кип. 142—14476 мм, с выходом 73,9% синтезирован взаимодействием фенолята натрия с бром-метилмалоновым эфиром в абсолютном этаноле. Библ. 5 назв.
22*
339
УДК 547.722.6.07
3-[2-(5-МЕТИЛФУРФУРИЛОКСИ)ЭТОКСИ]ПРОПИЛАМИН. | А. А. Пономарев, | Н. П. Масленникова, Г. В. Беспалова. Мето-ды получения химических реактивов и препаратов, вып. 26. Мх ИРЕА, 1974, с. 182.
3-[2-(5-Метилфурфурилокси)этокси]пропиламии, т. кип. 119— 122°/2 мм, с выходом 56% получен цианэтилированием 2-(5-метил-фУРФурнлокси)этанола с последующим гидрированием образующегося оксипитрила. Библ. 2 назв.
УДК 547.435+66.095.11
3-[N-МЕТИЛ ЦИКЛОПЕИТАНО (в)-2-ПИРРОЛ ИД ИЛ]ПРО-ПИЛБЕНЗОАТ. | А. А. Пономарев, | А. П. Кривенько, М. В. Нори-цина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 184.
3-[Г4-Метилциклопентано(в) -2-пирролидил]пропилбензоат, т. кип. 180—18273 мм, Лр = 1,5172, с выходом 31% получен впервые ацилированием 3-[М-метилциклопентано(в) 2-пирролидил]пропанола хлористым бензоилом в абсолютном бензоле. Библ. 5 назв.
УДК 547.722.07
2-МЕТОКСИ-М-АЦЕТИЛ-1,6-ДИОКСА-9-АЗАСПИРО[4,5]ДЕ-КАН. | А. А. Пономарев, | И. А. Маркушина, Т. И. Губина. Методы получения химических реактивов и препаратов, вып. 26. М , ИРЕА, 1974, с. 187.
2-Метокси-Г4-ацетил-1,6-диокса-9-азаспиро[4,5]декан, т. кии. 136—13871 мм, с выходом 63% получен электролитическим метокси-лированнем N-ацетилфурфуриламиноэтанола с последующим гидрированием образующегося 2-метокси-Г4-ацетил-1,6-диокса-9-азаспиро--[4,5]децеиа-3. Библ. 4 назв.
УДК 547.741.07
5-[2- (МЕТОКСИКАРБОНИЛ) ЭТИЛ ] 1,2-ДИГИД РОПИРРОЛ И-ЗИН. | А, А. Пономарев,\ Л. И. Астахова, В. И. Волколупов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 190.
5-[2-(Метоксикарбоиил)этил]-1,2-дигидропирролизии, т. кип. 118— 11971 мм, л® = 1,5180, d?0 = 1,0975, с выходом 64,2% получен взаимодействием 1,2-дигндропирролизииа с метиловым эфиром акриловой кислоты в присутствии фтористого бора. Библ. 3 назв.
340
УДК 547.558.1
(о-МЕТОКСИФЕНИЛ)ОКСИМЕТИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА, НАТРИЕВАЯ И СВИНЦОВАЯ СОЛИ. И. Д. Колпакова, Л. В. Кравицкая. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 192.
(о-Метоксифеиил)оксиметилидеидифосфоиовая кислота синтезирована взаимодействием о-метоксибеизойиой кислоты с треххлористым фосфором и водой при 120— 125° с последующим гидролизом водой. Выделена в виде натриевой (с выходом 64,8%) и свинцовой (с выходом 60,6%) солей. Библ. 5 назв.
УДК 547.722.07
2-МЕТОКСИ-И-ЭТИЛ-1,6-ДИОКСА-9-АЗАСПИРО[4,5]ДЕКАН | А. А. Пономарев, | И. А. Маркушина, Т. И. Губина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, е. 196.
2-Метокси-Ы-этил-1,6-диокса-9-азаспиро[4,5]декан, т. кип. 97— 100°/4 мм, с выходом 85% получен восстановлением 2-метокси-И-ацетил-1,6-диОкса-9-азаспиро[4,5]декана алюмогидридом лития. Библ. 2 назв.
УДК 547.234.1'21
МОНОАЛКИЛГИДРАЗИНЫ. В. С. Стопский, 3. И. Сергеева. В. В. Иоффе. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 198.
При взаимодействии бромистых алкилов с гидразин-гидратом получены: этилгидразии, т. кип 99—101°/763 мм, выход 66%; про-пилгидразин, т. кип. 121,9—122,27769 мм, выход 66%; изопропилгид-разин, т. кип. 108,6—109,07767 мм, т. пл. 10,9—11,2°, выход 63%. Библ. 4 пазв.
УДК 54,7.879
2-И-МОРФОЛИЛ 2-ОКСО-5-МЕТИЛ-5-МЕТОКСИМЕТИЛ-1,3,2--ДИОКСАФОСФОРИНАН. А. В. Богатский, А. А. Колесник, Т. Д. Бутова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 201.
2-Г4-Морфолил-2-оксо-5-метил-5-метоксиметил-1,3,2-диоксафосфо-рннан, т. кип. 185—18871 мм, с выходом 80% получен взаимодействием 2-хлор-2-оксо-5-метил-5-метоксиметил-1,3,2-диоксафосфорина-на с морфолином. Библ, 1 иазв.
341
УДК 547.653.1
2-НАФТОЛ-3,6-ДИСУЛЬФОКИСЛОТА, ДВУНАТРИЕВАЯ СОЛЬ Н. Н. Дыханов, Р. Ф. Виденина, Т. В. Перова, И. В. Покаты-ло, В. И. Базакин, П. Д. Якухный. Методы получения химически» реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 203.
Чистая двунатриевая соль 2-иафтол-3,6-дисульфокислоты выделена из технического продукта (получаемого известным способом) перекристаллизацией из 50%-ного этанола после предварительной экстракции основного количества примесей 85%-ным этанолом. Библ 5 иазв.
УДК 547.551.54
2-НИТРО-4,5-ДИБУТОКСИАНИЛИН. В. М. Дзиомко, А. В Иващенко. Методы получения химических реактивов и препаратов вып. 26. М., ИРЕА, 1974, с. 206.
2-Нитро-4,5-дибутоксианнлин, т*. пл. 107—108°, получен с количественным выходом нитрованием 3,4-дибутоксиацетанилида с последующим гидролизом 2-интро-4,5-дибутоксиацетанилида. Библ. 2 иазв.
УДК 547.7324-542.953.2
5-(5-НИТРОТЕНИЛ ИДЕН) РОДАНИН. |А. А. Пономарев] 3. В Тиль. Методы получения химических реактивов и препаратов, вып. 26. М. ИРЕА, 1974, с. 209,
5-(5-Нитротеиилиден)родании, т. пл. 238—240°, с выходом 91,8% получен конденсацией 5-иитро-2-тиофеиальдегида с роданином в ледяной уксусной кислоте. Библ. 6 назв.
УДК 547.654.3
5-НИТРОФЕНОЛ (2-АЗО- 1') -2'- (^-ПРОПИОНИЛ ГИДРАЗИ-НО)НАФТАЛИН И 5-НИТРОФЕНОЛ (2-АЗО-1')-2'-(р-БУТИРИЛ-ГИДрАЗИНО) НАФТАЛИН. К. А. Дунаевская, К. Е. Калинина, В. М. Дзиомко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 211.
5-Нитрофенол (2-азо- Г) -2'- (р-пропиоиилгидразино) нафталин (1) и 5-иитрофеиол(2-азо-1/)-2/-(Р-бутирилгидразнно) нафталин (II) получены диазотированием 5-нитро-2-аминофеиола и сочетанием полученных диазосоединений, соответствеиио, с пропионил-6-иафтилгид-разииом и бутирил-р-яафтилгидразяном. Выход 1 45,4%; выход 11 44,1% в расчете на 5-нитро-2-аминофенол. Библ. 1 иазв.
342
УДК 547.831.1
8-НИТРОХИНАЛЬДИН. В. М. Дзиомко, И. А. Красавин, Б. В. Парусников, Ю. П. Радин. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 215.
8-Нитрохииальдин, т. пл. 137—139°, получен с выходом 31—33% иятроваиием хинальдина действием нитрата аммония на раствор бисульфата хинальдиния в серной кислоте. Библ. 6 иазв.
УДК 547.473.2
у-ОКСИКАРБОНОВЫЕ КИСЛОТЫ, НАТРИЕВЫЕ СОЛИ. В. А. Седавкина, А. Г. Басина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 218.
Натриевые соли у-оксикарбоновых кислот получены 2 методами: каталитическим гидрированием или щелочным гидролизом этиловых эфиров у-кетокарбоиовых кислот. Натриевая соль у-оксигептановой кислоты с выходами: 89% и 93,3%, т. пл. 135—136°; натриевая соль у-ойсиоитановои кислоты с выходами 82,7% и 94%, т. пл. 137—139°; натриевая соль у-оксиноиаиовой кислоты с выходами 80 и 86%, т. пл. 165—166°. Библ. 6 назв.
УДК 547.473.2
у-ОКСИПИМЕЛИНОВАЯ КИСЛОТА, ДИНАТРИЕВАЯ СОЛЬ. В. А. Седавкина, А. Г. Басина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 222.
Дииатриевая соль у-оксипимелиновой кислоты, т. пл. 226—228°, получена с выходом 75,8% гидрированием дннатриевой соли у-кето-пнмелнновой кислоты в присутствии никеля Ренея при 100° и давлении 50 а/гм. Библ. 3 назв.
УДК 547.415. 5
N-(2-ОКСИЭТИЛ) ДИЭТИЛЕНТРИАМИН. Л. М. Тимаков г, В. Я. Темкина, Г. Ф. Ярошенко, Р. П. Ластовский. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 224.
N-(2-Оксиэтил)днэтилентриамнн получен с выходом 37% взаимодействием диэтнлентриамииа с этиленхлоргидрином в водной среде. Библ. 2 назв.
343
УДК 547.415.5
М-(2-0КСИЭТИЛ)ДИЭТИЛЕНТРИАМИН0-М,М',М",И"-ТЕТРА-УКСУСНАЯ КИСЛОТА. Л. М. Тимакова, В. Я. Темкина, Г. Ф. Ярошенко, Р. П. Ластовский. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 226.
N-(2-Оксиэтил) диэтилеитрцамино-М,М',М",М"-тетрауксуснаи кислота получена с выходом 49% взаимодействием N- (2-оксиэтил)ди-этилеитриамнна с монохлоруксусиой кислотой в водио-щелочной среде) п0и 90°. Для отделения комплексона от хлористого натрия не пользован метод электродиализа. Библ. 3 назв.
УДК 547.419.11'571
(1-ОКСИЭТИЛ И ДЕН) ДИФОСФОНАТ СЕРЕБРА. И. Д. Колпакова, Л. В. Криницкая. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 228.
(1-Оксиэтилидеи)дифосфоиат серебра состава Me : L = 2 : 1, получен с выходом 30,1% взаимодействием окснэтилидендифосфоно-вой кислоты с азотнокислым серебром при pH 7. Библ. 2 иазв.
УДК 547.419.1
1-ОКСИЭТИЛИДЕНДИФОСФОНОВАЯ КИСЛОТА,, СОЛИ С АМИНАМИ. И. Д. Колпакова, Л. В. Криницкая, Е. И. Миронова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА,1974, с. 230.
Соли 1-оксиэтилидеидифосфоиовой кислоты с диэтиламином, анилином, о-толуидином СаНвОтРа • 2RiR2NH • Н2О получены реакцией кислоты с соответствующим амином в спиртовом растворе. Соли очищены перекристаллизацией из водного спирта.
Даны: Ri, Ra; выход (%); т. пл.: С2Н5, С2Нз 76,8, 157—158°; Н. С6Н5, 71,4, 194—195,5° (разл.); Н, о-СН3СвН4, 70, 196— 197° (разл ).
УДК 547.419.11'33
(ЬОКСИЭТИЛИДЕН)ДИФОСФОНОВАЯ КИСЛОТА, ТРИНАТ-РИЕВАЯ СОЛЬ. Б. И. Бихман, Е. М. Уринович, Т. А. Богомолова, Н. М. Дятлова. Методы получения химических реактивов и препаратов, вып. 2Q. М., ИРЕА, 1974, с. 232.
Тринатриевая соль (1-оксиэтнлиден) дифосфоновой кислоты получена с выходом 77,3% взаимодействием оксиэтилидендифосфоновой кислоты с едким натром. Библ. 4 назв.
344
УД К 547.461 4
N-(2-ОКСИЭТИЛ) СУКЦИНИМИД И N-(3-ОКСИПРОПИЛ)-СУКЦИНИМИД. В. М. Островская, И. А. Шуйская, Т. В. Фомина. Л. В. Ломакина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 234.
N-(2-Оксиэтил) сукцинимид, т. пл. 58—60°, с выходом 95% и N-(3-оксипропил) сукцинимид, т. кип. 176—17873 мм, с выходом получены взаимодействием янтарной кислоты с моиоэтаиоламином или 3-аминопропаиолом, соответственно. Библ. 7 назв.
УДК 547.584'054.211
М-(2-ОКСИЭТИЛ)ФТАЛИМИД И М-(3-ОКСИПРОПИЛ)ФТА-ЛИМИД. В. М. Островская. И. А. Шуйская, Т. В. Фомина, Л. В. Ломакина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 237.
N-(2-Оксиэтил)фталимид, т. пл. 127—128°, с выходом 95% и N-(2-оксйпропил) фталимид, т. пл. 75,5—79°, с выходом 85% получены взаимодействием фталевого ангидрида с моиоэтаиоламином и 3-аминопропаиолом, соответственно. Библ. 7 иазв.
УДК 547.466.3—318
у-ОКСО-И-(5-ГЕКСИЛ-2-ПИРРОЛИДОН)МАСЛЯНАЯ КИСЛОТА. В. А. Седавкина, Н. А. Морозова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 240.
у-Оксо-N-(5-гекснл-2-пирролидои) масляная кислота, т. кип. 14572 мм, с выходом 58% получена ацилированием 5-алкил-2-пирро-лидона янтарным ангидридом. Аналогично получены другие у-оксо--М-(5-алкил-2-пирролидоп)масляные кислоты. Библ. 1 назв.
УДК 547.539.546.33
ПЕНТАФТОРАЗИДОБЕНЗОЛ. А. В. Кашкин, Ю. Л. Бахмутов, Н. Н. Чайникова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 242.
Пеитафторазидобеизол, т. кнп. 52—54710 мм, =1,4703, получен с выходом 70% при взаимодействии гексафторбеизола с азидом натрия в среде диметилформамида. Библ. 4 назв.
345
УДК 547.821.466.22'292'211
(2-ПИРИДИЛМЕТИЛ)ИМИН0ДИУКСУСНАЯ КИСЛОТА. Н. В. Цирульникова, В. Я. Темкина, Л. П. Разборская. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 244.
(2-Пиридилметил)имииодиуксусная кислота т. пл. 178—179°, по лучена с выходом 44,6% взаимодействием 2-хлорметиленпиридииа с иминодиуксусной кислотой в водно-щелочной среде с последующим выделением ее путем подкисления реакционного раствора концентрированной соляной кислотой. Библ. 1 назв.
УДК 547.747 + 542.951.1
0-(2-ПИРРОЛИДИЛ)ПРОПИОНОВАЯ КИСЛОТА. |Д. А. Пономарев, | Л4. В. Норицина, А. П. Кривенько. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 246.
Р-(2-Пирролндил) пропионовая кислота, т. пл. 156—156,5°, с выходом 78% получена впервые окислением 3-(2-пнрролидил) пропанола хромовым ангидридом в 12%-ном растворе серной кислоты. Библ. 6 иазв.
УДК 547.318 + 542.953.2
З-ПИРРОЛИЗИДОН. \ А. А. Пономарев,\ М. В. Норицина, А. П. Кривенько. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 249.
З-Пирролизидои, т. кип. 1157Ю мм, Дц = 1,4980, получен с выходом 81% внутримолекулярным ацилированием р- (2-пирроли-днл) пропионовой кислоты. Библ. 4 иазв.
УДК 547.466.3—318.
5-ПРОПИЛ-М-(Р-ОКСИЭТИЛ)-2-ПИРРОЛИДОН. В. А. Седавкина, Н. А. Морозова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 252.
5-Пропил-М-(3-оксиэтил)-2-пнрролндои, т. кип. 140—142°/2 мм, с выходом 70,2% получен этаноламинированнем этилового эфира у-кетоиогептановой кислоты в абсолютном этиловом спирте с использованием никеля Ренея. Аналогично получен ряд других 5-алкил-1Ч-(Р-оксиэтил)-2-пирролидоиов. Библ. 5 назв.
346
УДК 547,466.3-316
5-ПРОПИЛ-2-ТИОПИРРОЛИДОН И 1-МЕТИЛ-5-ПРОПИЛ-2-ТИОПИРРОЛИДОН. В. А. Седавкина, Г. В. Беспалова. Методы получения 'химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 255.
5-Пропил-2-тиопирролидон, т. кип. 16472 мм, с выходом 70% и 1-метил-5-пропил-2-тиопирролидон, т. кип. 124—12571 мм, с выходом 80% получены тиолнрованнем 5-пропил-2-пирролидоиа и 1-ме-тил-5-пропнл-2-пирролидона, соответственно, пятисернистым фосфором в среде пиридин-ксилол. Аналогично получены ряд других 5-алкил-2-тиопирролидолов и 1-метил-5-алкнл-2-тнопирролидолов. Библ. 2 назв.
УДК 547.39
, а-транс-у-цмс-р-СТИРИЛАКРИЛОВАЯ КИСЛОТА. Нгуен Ван Тонг, О. С. Степанова, A. ff. Галатина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 258.
а-Транс-у-цис-р-стирнлакриловая кислота, т. пл. 125—126°, с выходом 36% получена путем селективного гидрирования метилового эфира 5-фенил-2-гранс-пентен-4-иновой кислоты, который образуется при дегидратации метилового эфира 3-оксн-5-феиил-4-пентииовон кислоты. Библ. 9 назв.
УДК 547.583.5'292
N,N,N',N'-TETPAKHC (КАРБОКСИМЕТИЛ) -5,5'-МЕТИЛЕНДИ-АНТРАНИЛОВАЯ КИСЛОТА. Н. В. Цирульникова, Ю. Е. Дорошенко, И. К. Харитонова, И. В. Хвостов. Методы получения химических реактивов и препаратов, вып. 26. М„ ИРЕА, 1974, с. 262.
N,N,N',N'-TeTpaKHc (карбоксиметил) -5,5'-метнлендиантраниловая кислота получена с выходом 40% из 5,5'-метилендиантраниловой кислоты и хлоруксусной в присутствии 20%-него NaOH. 5,5'-Мети-лендиантраниловая кислота получена из антраниловой кислоты и формальдегида (37%-ный раствор) через промежуточный продукт 51,Ь1'-метилендиантраинловую кислоту. Библ. 2 назв.
УДК 547.419.1
ТЕТРАЭТИЛ-1-ОКСИЭТИЛИДЕНДИФОСФОНАТ. И. Д. Колпакова, Л. В. Криницкая, Е. И. Миронова. Методы получения химических реактивов н препаратов, вып. 26. М_, ИРЕА, 1974, с. 265.
Тетраэтил-1-оксиэтилидендифосфоиат получен с выходом 90% этерификацией 1-оксиэтилиденднфосфоиовой кислоты ортомуравьи-иым эфиром при температуре, не превышающей 90°. Побочные продукты и избыток ортомуравьиного эфира отгоняют при пониженном давлении. При перегонке в вакууме тетраэтил-1-оксиэтнлиденднфое-фонат изомеризуется в диэтил-а-(диэтокснфосфонил) этилфосфат. Библ. 13 назв.
347
УДК 547.724
ТИОСЕМИКАРБАЗОН (5-(5-ХЛОР-2-ФУРИЛ) АКРОЛЕИНА. В. П. Керенцева, М. Д. Липанова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 268.
Тиосемикарбазои р-(5-хлор-2-фурил) акролеина, т. пл. 152—153°, с выходом 80% получен конденсацией тиосемикарбазида с р-(5-хлор--2-фурил) акролеином в водио-спиртовом растворе. Библ. 4 назв.
УДК 547.585
ТРИМЕЛЛИТОВАЯ КИСЛОТА. Г. А. Креймер. Методы получения химических реактивов и препаратов, вып. 26. М, ИРЕА, 1974, с. 270.
Тримеллитовая кислота, т. пл. 228—230°, с выходом 69% получена окислением псевдокумола перманганатом калия. Библ. 6 иазв.
УДК 547.7-57
D.L-ТРИПТОФАН. Б. М. Котляревская, И. И. Губенко. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 273.
Разработана методика синтеза В,Е-триптофана по схеме: пн-дол-иидолальдегид-иидолальгидантоин-триптофаи. Триптофан получки; с выходом 37,3% в расчете иа индол; т. пл. 282—284’. Библ. 11 .назв.
УДК 547.585'397
ТРИХЛОРАНГИДРИД ТРИМЕЛЛИТОВОЙ КИСЛОТЫ. Я. А. Середницкий, В. Н. Федына. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 277.
Трихлорангидрид тримеллитовой кислоты получен с выходом 85% взаимодействием ангидрида тримеллитовой кислоты с тионил-хлоридом в присутствии катализатора — диметилформамида. По настоящей методике трихлорангидрид тримеллитовой кислоты может быть получен из кислоты с выходом 80—85%. Библ. 5 иазв.
348
УДК 547. 551. 51
2,4,6-ТРИХЛОРАНИЛИН. Н. Н. Дыханов, Т. В. Перова, Р. Ф. Виденина, И. В. Пакотыло, В. И. Базакин. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 280.
2,4,6-Трихлораиилин, т. пл. 77—78°, получен с выходем 60% хлорированием бензольной суспензии гидрохлорида анилина сульфурил-хлоридом при 70—80°. Библ. 5 назв.
УДК 547.212'118.5
ТРИЭТИЛФОСФАТ. А. Л. Лифиц, Л. Я. Дистанова, А. А. Вейц-ман. Методы получения химических реактивов, вып. 26 М., ИРЕА, 1974, с. 283.
Усовершенствована методика получения триэтилфосфата из хлорокиси фосфора и этилата натрия без растворителя; выход 76%. Библ. 6 назв.
547.836.3'571
1,Ю-ФЕНАНТРОЛИН-2,9-ДИАЛЬДЕГИД. В. М. Дзиомко, Б. В. Парусников. Методы получения химических реактивов и препаратов, вып. 26. М, ИРЕА, 1974, с.285.
1,10-Фенаитролин-2,9-диальдегид получен с выходом 64—70% окислением 2,9-диметил-1,10-феиаитролипа двуокисью селена в диоксане.
УДК 547.836.3'574.2
1,Ю-ФЕНАНТРОЛИН-2,9-ДИАЛЬДОКСИМ. В. М. Дзиомко, Б. В. Парусников. Методы получения химических реактивов и препаратов, вып 26. М., ИРЕА, 1974 с. 287.
1,10-Фенантролии-2,9-диальдоксим, т. разл. 257°, получен с почти количественным выходом при взаимодействии 1,10-фенаитролин-2,9-диальдегида с солянокислым гидроксиламином в пиридиновом растворе.
УДК 547.836.3'54 —32
1.Ю-ФЕНАНТРОЛИН-2,9-ДИКАРБОНОВАЯ КИСЛОТА. В. М. Дзиомко, Б. В. Парусников. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 289.
1,10-Феиантролии-2,9-дикарбоиовая кислота, т. пл. 214°, получена с выходом 89% при щелочном гидролизе динитрила 1,10-феиан-тролин-2,9-дикарбоновой кислоты.
349
УДК 547.569.1.07(088.8)
2-ФЕНИЛ-2-ПРОПАНТИОЛ. М. А. Коршунов, В. Е. Мазаев. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 291,
2-Фенил-2-пропантнол, т. кип. 48—50°/1 лои, получен впервые с выходом 72,5% присоединением газообразного сероводорода к а-метилстнролу в присутствии каталитических количеств хлористого алюминия. Библ. 2 назв.
УДК 547.415.1'562.1
ФЕРУЛОВАЯ КИСЛОТА. А. В. Богатский, 3. Д. Богатская, Л. М. Никифорова, Л. В. Басалаева, Н. С. Уланова-Обезкж. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 293.
Феруловая кислота, т. пл. 173°, с выходом 70% получена взаимодействием ваннлнна с малоновой кислотой в присутствии пиперидина в сухом пиридине. Библ. 3 назв.
УДК 547.722.1'281
3- (а-ФУРИЛ )БУТАНАЛ И 3-(а,а'-(ФУРИЛ)ДИБУТАНАЛ. И. В. Антипова, И. М. Скворцов. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 295.
3-(а-Фурил)бутанал, т. кнп. 78—82°/14 мм, Яр = 1,4831, с выходом 38% получен конденсацией фурана с кротоновым альдегидом в присутствии серной кислоты. Вторым продуктом реакции является 3-(а,а'-фурил)дибутанал, т. кнп. 136—142°/5 мм, — 1,4990, выход 13%.
УДК 547.723.07
1-(а-ФУРИЛ)-4-НИТРО-3-БУТАНОЛ. И. А. Маркушина, Т. И. Губина. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 297.
1-(а-Фурил)-4-нитро-З-бутанол, т. кнп. 131—133^/1 мм, с выходом 78% получен конденсацией 1-(фурил)-2-пропаналя с нитрометаном в присутствии щелочного агента. Библ. 3 назв.
350
УДК 547.821
2-ХЛОРМЕТИЛЕНПИРИДИН. Н. В. Цирульникова, В. Я. Тен-кина, Л. Я. Шиленко, В. И. Сомова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 299.
Проверен и уточнен способ получения 2-хлорметнленпнридина, т. кип. 75—77°/12 мм, взаимодействием N-оксн а-пиколнна с три-хлорацетнлхлоридом в среде хлороформа; выход 30%. Библ. 2 назв.
УДК 547.725
3-ХЛОРПРОПИОНОВАЯ КИСЛОТА. Л. Я. Глинская, 3. Д. Богатская, Г. Ф. Танцюра. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 301.
p-Хлорпропноновая кислота, т. кип. 88°/5 леи, т. пл. 41°, с выходом 90% получена взаимодействием газообразного хлористого во дорода с акриловой кислотой. Библ. 9 назв.
УДК 547.89
7-ХЛОр-5-ФЕНИЛ-ЗБЕНЗИЛИДЕН-1,2-ДИГИДРО-ЗН-1,4-БЕНЗДИАЗЕПИН-2-ОН. А. В. Богатский, Т. К. Чумаченко, 3. И. Жилина, С. А. Андронати. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 303.
7-Хлор-5-феннл-3-бензилиден-1,2-днгндро-ЗН-1,4-бенздназепин-2-он получен в виде смеси цис, транс-изомеров взаимодействием 7-хлор-5-фенил-1,3-днгидро-2Н-1,4-бенздназепнн-2-она с бензальдегидом. Разделение изомеров легко достигается колоночной хроматографией. Библ. 3 назв.
УДК 547.572.1
и-ЭТИЛАЦЕТОФЕНОН. Е. И Маховер. Методы получения химических реактивов н препаратов, вып. 26. М., ИРЕА, 1974, с. 305.
п-Этиланетофенон, т. кип. 240°, с выходом 81% получен по реакции Фриделя-Крафтса взаимодействием этилбензола с уксусным ангидридом в присутствии хлористого алюминия в среде дихлорэтана. Библ. 2 назв.
УДК 547.725
а-ЭТИЛ-Р-ГЕКСОКСИМЕТИЛПРОПИОНОВАЯ КИСЛОТА. Е. А. Яценко, А. И. Дроздовская. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 307.
а-Этил-р-гексоксиметилпропионовая кислота, т. пл. 154°С/7 мм, с выходом 74,6% получена декарбоксилированием а-этил-р-гексокси-метилмалоновой кислоты. Библ. 2 назв.
351
УДК 547.415.1
ЭТИЛЕНДИАМИНДИАЦЕТАТЫ МЕДИ, КОБАЛЬТА НИКЕЛЯ. Е. М. Уринович, И. М. Дятлова. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 309: '
Этилендиамнндиацетаты меди, кобальта и никеля получены с выходом 23, 52, и 54%, соответственно, взаимодействием дикалиевой соли этилендиаминднуксусной кислоты с эквивалентным количеством нитрата соответствующего металла. Библ. 1 назв.
УДК 547.854.5
5-ЭТИЛ-5-ФЕНОКСИБАРБИТУРОВАЯ И 5-ЭТИЛ-5-ФЕНО-КСИТИОБАРБИТУРОВАЯ КИСЛОТЫ. О. С. Степанова, А. С. Яворский, М. И. Побережная, В. П. Ганевич. Методы получения химических реактивов и препаратов, вып. 26. М., ИРЕА, 1974, с. 312.
5-Этил-5-феноксибарбитуровая, т. пл. 147—148°, с выходом 60% и 5-этнл-5-фенокснтнобарбитуровая кислоты, т. пл. 132°, с выходом 71% получены конденсацией этнлфеноксималонового эфира с мочевиной (тиомочевиной) в абсолютном этиловом спирте в присутствии этилата натрия. Бнбл. 3 назв.
УДК 547.476.1
ЭТИЛФЕНОКСИМАЛОНОВАЯ КИСЛОТА. О. С. Степанова, А. С. Яворский, М. И. Побережная, В. Н. Ганевич. Методы получения химических реактивов н препаратов, вып. 26. М., ЙРЕА, 1974, с. 314.
Этилфеноксималоновая кислота, т. пл. 114°, с выходом 78% получена омылением этилфенокснмалонового эфира водно-спиртовым раствором едкого кали. Библ. 1 назв.