




























































































Текст
всесоюзный научно-исследовательский институт
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ
ХИМИЧЕСКИХ ВЕШЕСТВ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 15
МОСК ЗА - 1 96'/
Редакционная коллегия
Проф. Р. П. Ластовский (гл. редактор),
инженер-технолог А. М. Поспелов (зам. гл. редактора),
канд. техн, наук В. Г. Брудзь, проф, А. В. Бромберг,
канд. хим. наук Е. А. Божевольнов, проф, В. Л\. Дзиомко,
канд. хим. наук Г. А. Певцов
СОДЕРЖАНИЕ
Алкилдихлорфосфины. В. Г. Груздев, С. 3. Ивин, К. В. Кара-
ванов ........................... 7
З-Аминофталевая кислота, гидрохлорид. Р. И. Ластовский,
В. Я. Темкина, И. А. Егорушкина .............. 10
2-Антрилгидразин.’ Б. М. Болотин, Д. А. Драпкина,
И. И. Чернова, В. Г. Брудзь............................... 13
2-Антоол. И. И. Чернова, Д. А Драпкина, Б. М. Болотин,
В. Г. Брудзь.............................................. 15
Ацилхолини тиохолинйодилы. С. 3. Ивин................. 17
N Беизилэтилеидиамин-N, N', N'-триуксусная кислота.
Р. П. Ластовский, И. Д. Колпакова, Л. В. Криницкая,
Т. И. Иванова ............................................ 22
а, а'-Бис-(4-натрий-тегразолилазо-5)-этилацетат. И. С. Фру мина,
Г. С. Петрова, Н. И. Горюнова............................. 26
4 Бромбензальдегид. В. Г. Брудзь. Д. А. Драпкина,
Р. У. Судиярова ....................... 29
З-Бром-5-нитросалициловый альдегид. Д. А. Драпкина,
В. Г. Брудзь, В А. Иншакова, И. И. Плитина............... 32
2-Винилпиридин. Б. М. Куинджи, М. А. Зепалова, А. К. Валь-
кова, Л. Д. Глузман, Р. М. Цин, А. А. Рокк................ 34
Галлат кобальта. В. П. Чернецкий, Э А. Пономарева .... 37
Гидразид малеиновой кислоты. Р. М. Гелыитейн, Ф. В. Алек-
сандрова ........................... 40
Гидразобензол. И. Е. Задорожная....................... 43
б-Диазо-5-кето-Б-норлейцин. Г. И Кошелева, И. Налецкая 46
5,5-Диалкил-4-метил-4-окси-3-циантетрагидрофураны. Т. А. Фа-
ворская, Г. М. Толстопятов ......... .......... 50
Дибензиловый эфир. Л. Я. Дистанова ............ 56
1,4-Диметил-2-изопропилбензол и 1.4-димстил-2,5-диизопропил-
бензол. И. Г. Гах, Е. П. Бабин, Л. Г. Гах ........... 58
N, N-Диметил-о-нитроанилин. Р. ГТ. Ластовский, В. Я. Темки-
на, Л. М. Самылова........................................ 61
5,7-Диметил-8-оксихииальдоксим. В. М. Дзиомко, И. А. Краса-
вин, Т. Н. Егорова ....................... 63
3,3'-Диметил спиро-[бензоксазолин-2,2f-(2 ’ Н-1 '• бензопираны)}.
Д. А. Драпкина, В. Г. Брудзь, В. А. Иншакова, Н. И. Бадайкова 65
N, N-Диметнл-о-феиилендиамин. Р. П. Ластовский, В Я. Тем-
кина, Л. М. Самылова .................. ... 69
2,2'-Диокси-1,1'-нафтальдазин. Р. М. Гелыитейн. И. Е. Задо-
рожная ................................................... 71
2.8-Диоксихинолин. И. А. ‘ Красавин, В. М. Дзиомко,
Ю. П. Радин ................ ............ 73
N, N'-Дифеинлформамидинсульфиновая кислота. дигидрат.
Р, Л. Глобус, Р. П. Ластовский, Е. Я. Яровенко, С. П. Медведева 75
3
8,8'-Дихинолил. В. М. Дзиомко, 3. С. Сиденко, Г. С. Чижова 77
Дихлорангидрид трихлорметилтиофосфи новой кислоты.
Л. Е. Дмитриева, С. 3. Ивин, К. В. Караванов . . .......... 79
4,4'-Дихлордибутиловый эфир. Б. А. Розенберг, О. М. Чехута 81
2,2-Дихлорпропаи. С. А. Кочеткова, А. В. Афанасьева .... 83
N-Замещенные имиды малеиновой кислоты. В. С. Иванов,
В. К. Смирнова, Т. И. Сидорова, И. И. Мигунова, А. М. Абрамова,
Е. Г. Башина, Т. Р, Сеппен................................ 83
Комплексные соединения алкилтрихлорнолфосфинов и дналкил-
дихлорйодфосфинов и хлористого алюминия. В. Г. Груздев,
С. 3. Ивин, К. В. Караванов................................ 90
6-Метил-2-вииилпиридин. Б. М. Куинджи, М. А. Зепалова,
Л. Д, Глузман, А. К. Валькова, Р. М. Цин, И. В. Зайцева,
А. А. Рокк................................................. 93
Метил йодистый. И. Е. Задорожная........................ 96
N-Монометилолформамидинсульфиновая кислота. Р. Л. Глобус,
Р. И. Ластовский, Е. Я Яровенко, С. П. Медведева........... 98
1-М-Морфолнл-пропанол-2 и 1-М-пипериди.т-пропанол-2.
А. М. Самуилов, Г. Ф. Дрегваль........................... 100
Натриевая соль 2-антраценсульфокислоты. В. Г. Брудзь,
Д. А. Драпкина, Б. М Болотин, И. И. Чернова............... 102
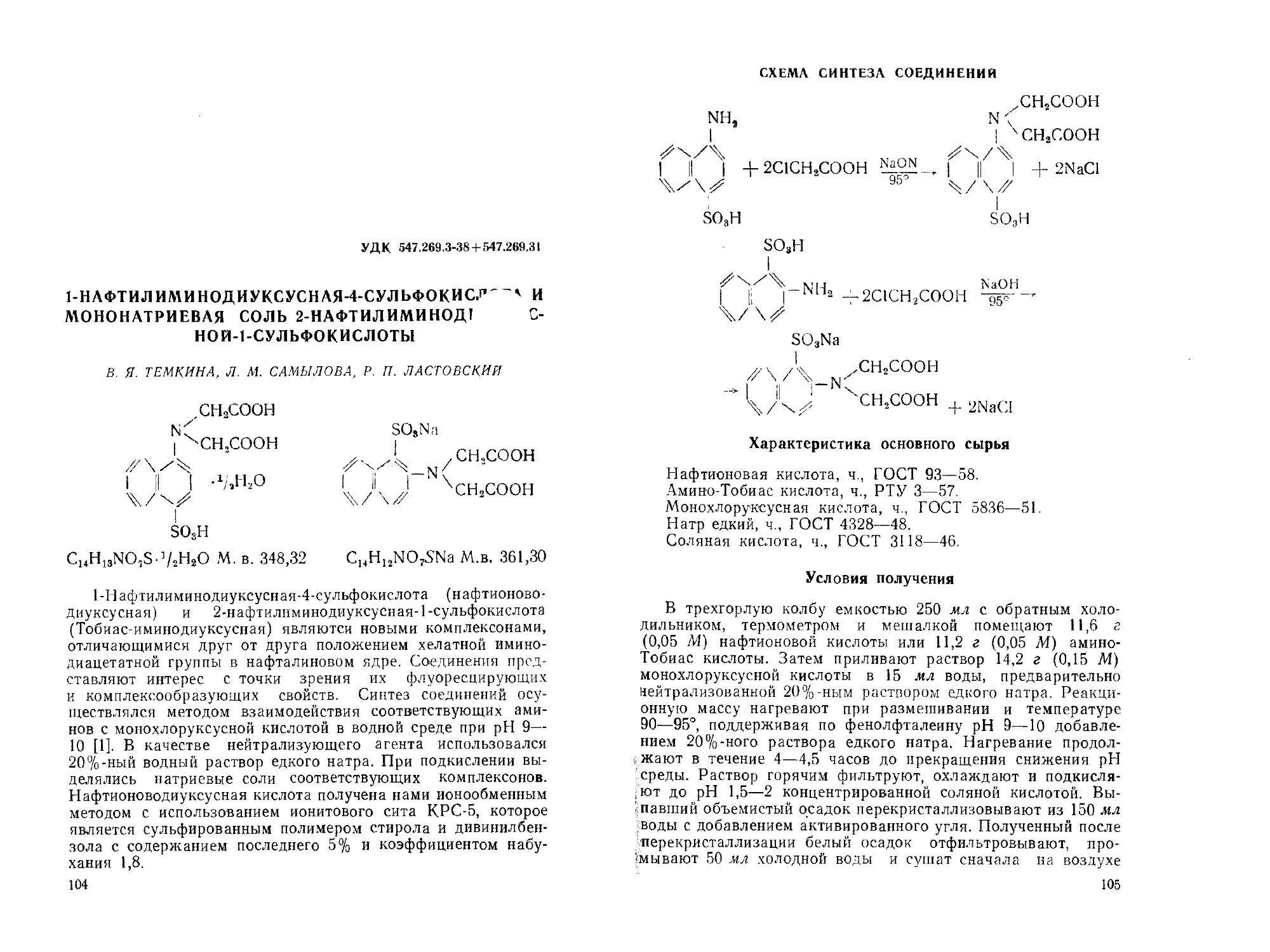
1-Нафтилиминодиуксусная-4-сульфокислота и мононатриевая
соль 2-нафтилиминодиуксуснон-1-сульфокислоть!. Р. П. Ластовс-
кий, В. Я- Темкина, Л. М. Самылова . ................. 104
5-Нитро-2-нафтиламин. В. М. Дзиомко, И. С. Маркович,
И. В. Круглова............................................ 107
5-Нитро-2-окси-3-метоксибензальдегид. В. Г. Брудзь,
Д. А. Драпкина, В. А. Иншакова, И. П. Платина............. 109
3- и 5-Нитросалиииловые альдегиды. В. Г. Брудзь, Д. А. Драп-
кина, В. А. Иншакова, И. И. Дорошина ............ 111
6-Н итрохинальдоксим. В. М. Дзиомко, 3. С, Сиденко.
Г. С. Чижова............................................. 114
б-Нитрохииолин-2-альдегид. В. М. Дзиомко, 3. С. Сиденко,
Г. С. Чижова.............................................. 116
1-(8-Окси-2-хинолил)-3,5-диметилпиразол. И. А. Красавин, В. М.
Дзиомко, И. И. Мирошкина, Ю. П. Радин ............ 118
1-(2-Пиридилазо)-2-нафтол. Г. С. Петрова, В. А. Платонова 120
Тетрабензоилацетоноевропиат пиперидиния. Р. П. Ластовский,
В. Я. Темкина, Н. В. Цирульникова........................ 123
Тетрадибензоилметаноевроииат пиперидиния. Р. П.Ластовский,
В. Я. Темкина, Н. В. Цирульникова........................ 125
1, 2, 4, 5-Тетраизопропилбензол. В. П. Марштупа,
Е. И. Бабин, Л. И. Марышкина, И. С. Морозова ........ 127
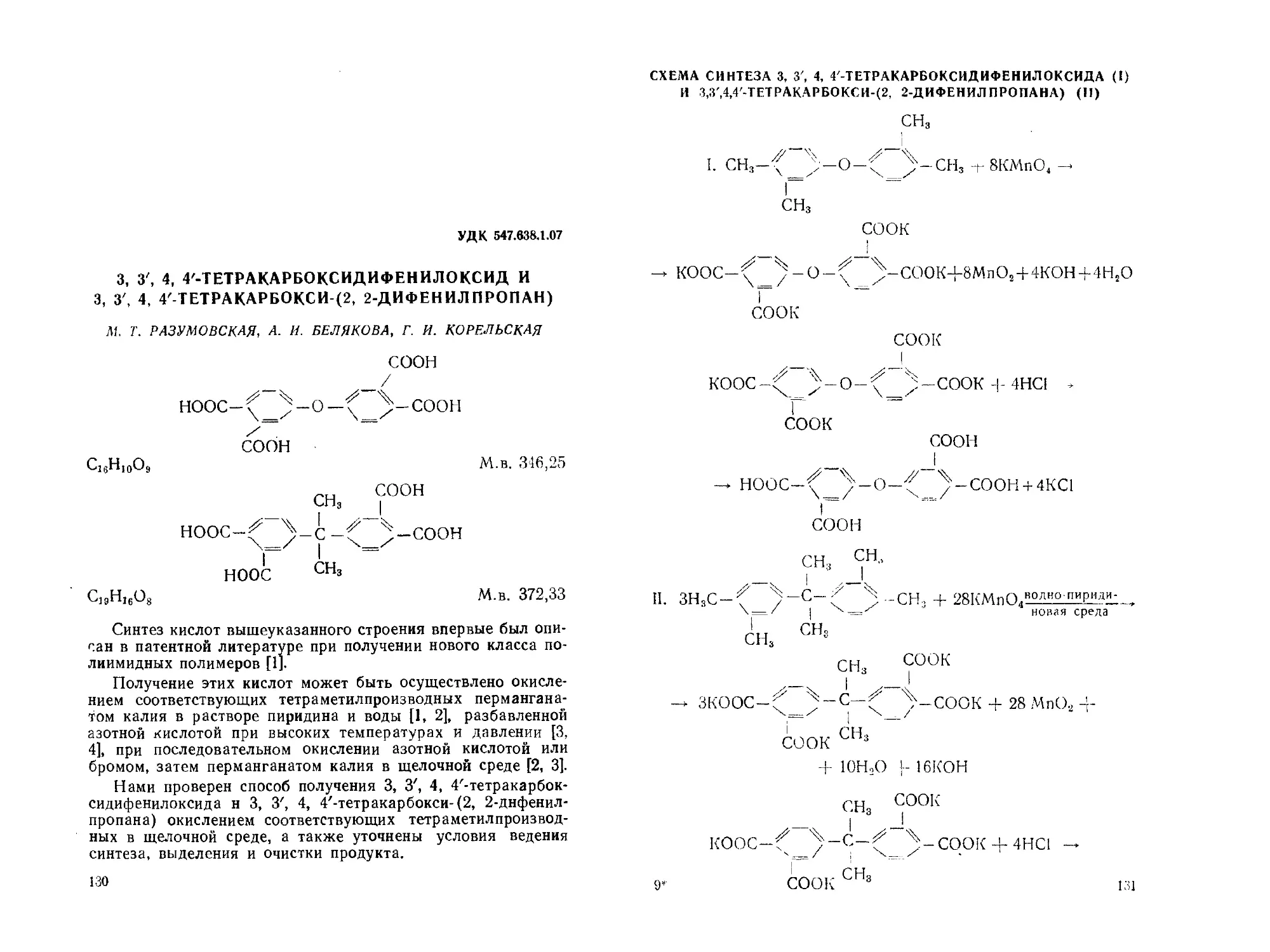
3, 3', 4, 4'-Тетракарбоксидифенилоксид и 3, 3', 4, 4'-тетракар-
бокси-(2,2дифенилпропан). М. Т. Разумовская, А. И. Белякова,
Г. И. Карельская......................................... 130
3, 3', 4, 4'-Тетраметилдифенилдиметилметан. С. А. Кочеткова,
А. В. Афанасьева................................... .... 136
р, р"-Триаминотриэтиламин солянокислый. Р. П Ластов-
ский, И. В. Хвостов, И. Д. Колпакова, Е. А. Злотина,
В. Н. Шкадова ........................ 138
Тридибензоилметанаммин-европий. Р. П. Ластовский,
В. Я- Темкина, Н. В. Цирульникова........................ 142
1, 3, З-Триметилспиро-[ (2'Н-1'-бсизопиран)-2, 2' - нндолины].
В, Г, Брудзь, Д. А. Драпкина, В. А. Иншакова, Н. И. Дорошина,
Н. И. Байдакова ....................... . 144
Трихлорметилдихлорфосфин. Л. Е. Дмитриева, К. В. Карава-
нов, С. 3. Ивин . . .................. . .............. 148
Уреид ацетоуксусной кислоты. В. Г. Кондратьева,
С. И. Завьялов............................................ 150
4
1-Фенил-3,3-диметил-2-метилениндолии. В. А. Иншакова,
Д. А. Драпкина, В. Г. Брудзь, И. ГТ. Платина................. 151
1-Фенил-3,.1-диметил спи ро-[(2'Н-1 '-бензопиран) -2,2'-ин долины].
Д. А. Драпкина, В. Г. Брудзь, В. А. Иншакова, И. П. Платина 154
Фениловый эфир антраниловой кислоты. В. М. Дзиомко,
И. С. Маркович, И. В. Круглова............................... 158
N-Фенилформамидинсульфиновая кислота. Р. Л. Глобус,
Р. И. Ластовский, Е. Я. Яровенко, А. П. Болдырева............ 160
л-фторбензальдегид. Д. А. Драпкина, И. И. Чернова,
В. Г. Брудзь................................................. 162
n-Фторэтилбензол. А. А. Артамонов, Р. Д, Бондарчук,
М. И. Котеленец.............................................. 165
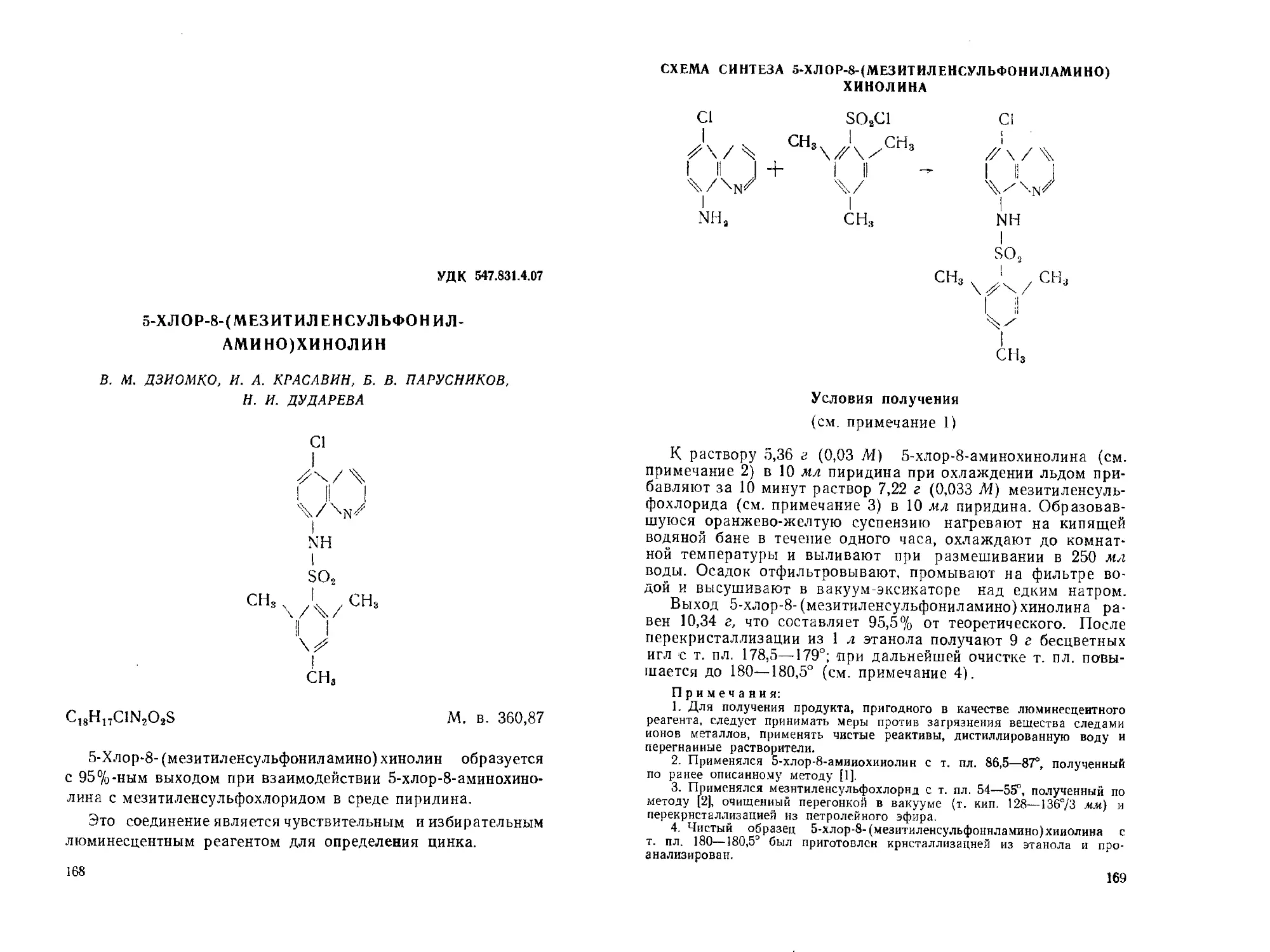
5-Хлор-8-(мезитиленсульфониламино)-хинолин. В. М. Дзиомко,
И. А. Красавин, Б. В. Парусников, И. И. Дударева............. 168
5-Хлор-З-нитросалициловый альдегид. Д. А. Драпкина,
В. Г. Брудзь, В. А. Иншакова, И. II. Платина................ 171
2-Хлор-4 (5)-фенилнмидазол. Г. И. Тюренкова. И. П. Беднягина 173
л-Хлорэтилбензол. А. А. Артамонов, Р. Д. Бондарчук .... 176
Щелочные соли галловой кислоты. В. П. Чернецкий,
Э. А. Пономарева, Т. Д. Белышева, Т. Г. Гладченко, Т. Г. Яровая 179
2-Эгилбензоксазол. Д. А. Драпкина, В. Г. Брудзь, В. А. Ин-
шакова, И. И. Бадайкова...................................... 184
Эфиры галловой кислоты. В. П. Чернецкий, Т. Д. Белышева,
В. Г. Волощук. Э. А. Пономарева ............... 186
Алфавитный указатель соединений, описанных в настоящем
сборнике,................................................ 191
УДК 661.718Л.07
АЛКИЛДИХЛОРФОСФИНЫ
В. Г. ГРУЗДЕВ, С. 3. ИВИН, к. в. КАРАВАНОВ
RPC12
Алкилдихлорфосфины являются исходными полупродукта-
ми в синтезе различных фосфорорганических соединений. В
литературе описано несколько способов их получения. Одним
из первых описан метод, основанный на реакции диалкил-
ртути с треххлористым фосфором [1]. Алкилдихлорфосфины
также могут быть получены алкилированием треххлористого
фосфора с помощью тетраэтилсвинца [2], кадмийорганических
соединений [3], прямым алкилированием красного фосфора
галоидалкилами [4], взаимодействием треххлористого фосфо-
ра с углеводородами [5J, восстановлением комплексных сое-
динений алкилтетрахлорфосфинов с хлористым алюминием:
а) порошкообразной сурьмой или цинком в диэтилфтала- '
те [6];
б) фенилдихлорфосфином в присутствии хлорокиси фос-
фора [7];
в) алюминием, фосфором или натрием в присутствии хло-
ристого калия [8], а также восстановлением алкилдихлор-
фосфиноксидов металлами и гидридами металлов [9, 10].
Нами разработан другой способ получения алкилдихлор-
фосфинов, заключающийся в термическом разложении комп-
лексных соединений алкилтрихлорйодфосфинов и хлористого
алюминия в присутствии свежепрокаленного хлористого ка-
лия или диэтилфталата (дибутилфталата) и их последующем
восстановлении металлами.
Выход алкилдихлорфосфинов ио данному методу состав-
ляет 60% и выше [11].
•7
ЭТИЛДИХЛОРФОСФИН
С3Н5РС13 М. в. 130,94
СХЕМЫ СИНТЕЗА ЭТИЛДИХЛОРФОСФИНА
C3H8PC13J • А1С1з + КС1 С3НВРС13 +J3 + КСЬ AICI3
(метод А)
CsH5PCl3.J-AlCl3+Me 4-Дэф C3HSPC134-J3 + Дэф-А1С13
(метод Б)
Характеристика основного сырья
Комплексное соединение этилтрихлорйодфосфина с хло-
ристым алюминием получают по методике, описанной в на-
стоящем сборнике (см. статью «Комплексные соединения ал-
килтрихлорйодфосфинов с хлористым алюминием»).
Магний в виде мелкой стружки.
Цинк в виде пыли.
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Диэтилфталат, чистый, обычный.
Условия получения
Метод А. В колбу Вюрца емкостью 0,5 л с прямым холо-
дильником и термометром помещают 128 г (0,3 М) комплекс-
ного соединения этилтрихлорйодфосфина с хлористым алю-
минием (см. применение 1), 22,3 г (0,3 М) хлористого калия.
Реакционную смесь тщательно перемешивают, а затем мед-
ленно нагревают до температуры 140° с отгоном жидких про-
дуктов реакции.
Вначале возгоняется йод, а затем отгоняются жидкие про-
дукты. Для освобождения от примеси йода полученный этил-
дихлорфосфин дважды перегоняют под вакуумом 1 мм без
нагревания перегонной колбы (т. е. при комнатной темпера-
туре).
Выход этилдихлорфосфина равен 17,6 г, что составляет
45% от теоретического; т. кип. 113—114°/752 мм; dA°—1,2592;
«о— 1,4930 (см. примечание 3).
По аналогичной методике могут быть получены и другие
алкилдихлорфосфины.
Метод Б. В колбу Кляйзена емкостью 0,5 л с прямым хо-
лодильником и термометром помещают 128 г (0,3 М) комп-
лексного соединения этилтрихлорйодфосфина с хлористым
алюминием и к нему, при охлаждении реакционной колбы
холодной водой, прикапывают 88,8 г (0,4 М) диэтилфталата.
8
При комнатной температуре к реакционной массе присыпают
4,8 г (0,2 Л4) магниевой стружки. Реакционную смесь нагре-
вают и при температуре НО—120° отгоняют образовавшийся
продукт.
Выход этилдихлорфосфина равен 23,5 г, что составляет
60% от теоретического; т. кип. 113—114°/752 мм; d420—1,2592;
tin - 1,493.
По аналогичной методике могут быть получены и другие
алкилдихлорфосфины.
Примечания:
1. Комплексное соединение этилтрихлорйодфосфина с хлористым алю-
минием гигроскопично, поэтому следует избегать излишнего соприкосно-
вения его с воздухом.
2. Этилдихлорфосфин окисляется на воздухе, в связи с чем его по-
лучение и перегонку желательно вести в токе азота или какого-либо дру-
гого инертного газа.
ЛИТЕРАТУРА
1. Fr. Gulch a rd, Вег., 32, 1572 (1890).
2. М. S. Kharascli. J. Organ Chem., 14. 429 (1949).
3. R. В. Fox. J. Amer. Chem. Soc., 72, 4147 (1950).
4. Л. 3. С о б о p о в с к и й, Б. М. Г л а д ш т е й и. Авт. свид. 130513;
Бюлл. изобр., № 15 (1960).
5. J, Р 1 a n f е 11 i, Q г 1 f m. J. Amer. Chem. Soc., 84, 5, 851 (1962).
6. J. L. Ferron, B. J. Perry. Nature (Engl.), 188,4746,22/(1960).
7. Паршалл. J. Inorg. and Nucl. Chem., 12, 3—4, 372 (1960).
8. И. П. К о м к о в, К. В. К a p а в а и о в, С. 3. И в и н Ж. общ. хи-
мии, 28, II, 2963 (1958); Методы получения химических реактивов н пре-
паратов, вып. 12, М., ИРЕА, 1965, стр. 5.
9. И. П. Комков, В. Г. Груздев, С. 3. Ивин. К. В. Карава-
нов. Проблемы органического синтеза, 1965, стр. 308; Методы получения
химических реактивов и препаратов, вып. 12, М„ ИРЕА, 1965, стр. 101;
Авт. свид. 160184; Бюлл. изобр., № 3, 20 (1964).
10. В. Г. Груздев, С. 3. Ивин, К. В. Караванов. Авт. свид.
159527; Бюлл. изобр., № 1, 14 (1964).
11. В. Г. Груздев, С. 3. Ивии, К. В. Караванов, И. С. М а-
зель, В. В. Тарасов. Ж. общ. химии, 36 (1966).
УДК 547.584.07
З-АМИНОФТАЛЕВАЯ КИСЛОТА. ГИДРОХЛОРИД
В. я. ТЕМКИНА, Н. А. ЕГОРУШКИНА, Р. П. ЛАСТОВСКИЙ
GOOH
^>|-СООН
C8H8C1NO4 М. в. 217,61
З-Аминофталевая кислота является промежуточным про-
дуктом в синтезе многих органических веществ. В литературе
известно несколько методов получения 3-аминофталевой кис-
лоты путем восстановления 3-нитрофталевой кислоты хлори-
дом олова в среде соляной кислоты [1, 2], цинковой пылью в
уксусной кислоте [3], сульфитом натрия [4], сульфатом желе-
за и баритовой водой в аммиачном растворе [5].
Нами проверен и уточнен метод получения 3-аминофтале-
вой кислоты посредством восстановления 3-нитрофталевой
кислоты хлоридом олова в среде соляной кислоты. Выход по-
лученного препарата составляет 78% от теоретического.
Исходная 3-нитрофталевая кислота получена нитрованием
фталевого ангидрида [6].
СХЕМА СИНТЕЗА 3-АМИНОФТАЛЕВОЙ КИСЛОТЫ
СО-О
HNO3.H2SQt
СООН
I
_ I^-COOH
СООН
Z\-COOH
NOa
10
СООН
СООН SnCla-2H2O, НС1
V'~no2
СООН
I
f^l-COOH
'^J'-NHs-HCl
Характеристика основного сырья
Фталевый ангидрид, ч.д.а., ГОСТ 5969—51.
Азотная кислота, х. ч., дымящая, уд. в. 1,51, ГОСТ
4461—48.
Азотная кислота, х. ч., концентрированная, уд. в. 1,42,
ГОСТ 4461—48.
Серная кислота, техническая, уд. в. 1,84, ГОСТ 4204—48.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Олово двухлористое, ч., ГОСТ 36—40.
Условия получения
Синтез 3-нитрофталевой кислоты. В трехгорлую колбу,
снабженную мешалкой, термометром, капельной воронкой,
холодильником с газоотводной трубкой, загружают 130 мл
(2,4 Л4) технической серной кислоты и 100 г (0,68 М) фтале-
вого ангидрида; смесь нагревают до 80° и при энергичном
размешивании прикапывают из капельной воронки 42 мл
(1 М) дымящей азотной кислоты, а затем 180 мл (4 М) кон-
центрированной азотной кислоты с такой скоростью, чтобы
температура реакционной массы не превышала 100—110°.
После двухчасовой выдержки при этой температуре и 12-ча-
совой при комнатной температуре реакционную массу выли-
вают в 300 мл воды. Выпавшую при охлаждении до 0° смесь
3- и 4-нитрофталевых кислот отфильтровывают и промывают
водой для удаления последней. Осадок 3-нитрофталевой кис-
лоты перекристаллизовывают из воды и сушат на воздухе.
Получают 34 г кислоты, что составляет 23,4%, считая на фта-
левый ангидрид; т. пл. 215—218".
По литературным данным, т. пл. вещества 215—218°, вы-
ход 24,2% [6].
Получение 3-аминофталевой кислоты. В трехгорлую кол-
бу, снабженную мешалкой, термометром и капельной ворон-
кой, загружают раствор 120 г (0,53 М) двухлористого оло-
ва в 300 мл (9,7 М) концентрированной соляной кислоты и
при непрерывном размешивании и температуре не выше 22°
вносят небольшими порциями 30 г (0,14 Л-1) 3-нитрофталевой
кислоты, полученной в первой стадии. Смесь перемешивают
при температуре 20—22° до полного растворения осадка.
11
После 12-часовой выдержки при комнатной температуре вы-
павший осадок гидрохлорида 3-аминофталевой кислоты от-
фильтровывают, промывают 50 мл концентрированной соля-
ной кислоты. Продукт перекристаллизовывают из концентри-
рованной соляной кислоты и сушат в вакуум-эксикаторе над
серной кислотой и щелочью.
Получают 24 г гидрохлорида 3-аминофталевой кислоты,
что составляет 78% от теоретического, считая на 3-нитрофта-
левую кислоту. Температура плавления гидрохлорида 225°.
По литературным данным, т. пл. гидрохлорида 225° [1].
Найдено, %: N — 6,54.
CgH8ClNO4. Вычислено, %: N—6,45.
ЛИТЕРАТУРА
1. J. R. Scott, J. В. Cohen. J. Chem. Soc.. 119, 666 (1921).
2. J. Amer. Chem. Soc., 42, 1873 (1920).
3. A. Bernthsen, A. Semper. Ber., 19, 166(1886).
4. P. Onnertz, Ber., 34, 3746 (1901).
5. H. Kaufmann, A. Belsswenger. Ber., 36, 2495 (1903).
6. Синтез органических препаратов, I, 1949, стр. 316.
Поступила в декабре 1965 г.
ИРЕА
УДК 547.556.8.07
2-АН ТР И Л ГИД РАЗ ИН
Б. М. БОЛОТИН, Д. А. ДР АННИНА, Н. И. ЧЕРНОВА, В. Г. БРУДЗЬ
I 1! I !
VVV
CUH12N2 М. в. 208,26
2-Антрилгидразин в литературе не описан. Единственным -
упоминанием о нем является работа [1], в которой автор пы-
тался получить 2-антрилгидразин из 2-антриламина диазоти-
рованием последнего и восстановлением диазониевой соли
двухлористым оловом. Однако в чистом состоянии продукт
выделить не удалось. Предлагаемый нами метод синтеза
2-антрилгидразина позволил получить продукт в чистом виде
и охарактеризовать его.
Метод заключается в нагревании 2-антрола с избытком
гидразин-гидрата при перемешивании в течение 17 часов при
атмосферном давлении.
СХЕМА СИНТЕЗА 2-АНТРИЛГИДРАЗИНА
] й | |-ОН 4-NH»—NHa
Ч/WW
—’ I || I I 2 “Г п2О
VW
Характеристика основного сырья
2-Антрол, ч., т. пл. 197—198° (см. статью «2-Антрол» в на-
стоящем сборнике, стр. 15).
Гидразин-гидрат, ч., ГОСТ 5832—51.
13
Условия получения
В круглодонную колбу емкостью 0,5 л, снабженную ме-
ханической мешалкой с затвором и обратным холодильни-
ком, загружают 12 г хорошо измельченного 2-антрола и
155 мл гидразин-гидрата. При постоянном перемешивании
смесь нагревают на песчаной бане до кипения и кипятят в
течение 17 часов (см. примечание), после чего смесь охлаж-
дают, выливают 400 мл ледяной воды и отфильтровывают.
Осадок промывают 100 мл холодной воды, обрабатывают
250 мл' 10 % -ного раствора едкого кали, отфильтровывают и
промывают 200 мл воды. Затем его переносят в стакан и
размешивают в 250 мл этилового спирта, снова отфильтровы-
вают, хорошо отжимают, промывают на фильтре 50 мл эти-
лового эфира и сушат при комнатной температуре. Получен-
ный 2-антрилгидразин флуоресцирует ярко-зеленым цветом.
Выход равен 10,7 г, что составляет 83,1% от теоретическо-
го в расчете на 2-антрол. После перекристаллизации из эти-
лового спирта с активированным углем 2-антрилгидразин
имеет желтоватый цвет и плавится при 198 -199э. Реакцию
можно вести с перерывами.
Найдено, %: С-81,02, 81,03; Н-5,73; 5,59.
CltH!3N2. Вычислено; %-. С —80,76; Н- 5,81.
Примечание.
При кипячении в течение 17 часов выход продукта составляет 83,1 %;
II часов — 72,6%;'6 часов — 70,6%.
ЛИТЕРАТУРА
1. О. F i н s t е г 1 е. Ital. Farmaco, Ed. sci. 10, 432 (1955); С. A., 50,
4115a.
Поступила в ноябре 1965 г.
ПРЕЛ
УДК 547.672.5.07
2-АНТРОЛ
Н. И. ЧЕРНОВА, Д. А. ДРАПКИНА, Б. М. БОЛОТИН. В. Г. БРУДЗЬ
\ / 4 /
СиН10О М. в. 194,23
2-Антрол может быть получен щелочным плавлением на-
триевой соли 2-антраценсульфокислоты [1]. Нами проверен и
уточнен этот метод.
СХЕМА СИНТЕЗА 2-АНТРОЛА
^"Х}/Y^-SOsNa ц_кон 310—320°
\ / X / Ч... он
-> I li | I + KNaSO3
Характеристика основного сырья
Натриевая соль 2-антраценсульфокислоты (см. статью
«Натриевая соль 2-антраценсульфокислоты» в настоящем
сборнике, стр. 102).
Кали едкое, ч„ ГОСТ 4203—48.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
В никелевый котелок (см. примечание 1) емкостью 750 мл
загружают 375 г едкого кали и прибавляют 3—5 капель во-
ды. Смесь при постоянном перемешивании никелевым шпа-
телем нагревают на бане со сплавом Вуда до 280° (едкое ка-
15
ли полностью расплавляется) и прибавляют 50 г тщательно
растертой сухой натриевой соли 2-аитраценсульфокислоты.
Температуру бани поднимают за 20- 25 минут до 320—330° и
при этой температуре, не прекращая перемешивания, выдер-
живают смесь до тех пор, пока она не окрашивается после-
довательно в оранжевый, бурый, коричневый цвет. При этом
часть натриевой соли 2-антраценсульфокислоты переходит в
антрацен, который возгоняется в виде серебристых кристал-
лов. Как только смесь окрасилась в коричневый цвет, через
каждые 2 минуты начинают брать пробы на полноту реакции
(см. примечание 2).
При получении положительной пробы сосуд вынимают из
бани (см. примечание 3). Когда масса наполовину застынет,
ее переносят небольшими порциями в помещенный в ледя-
ную баню двухлитровый стакан с 750 мл соляной кислоты и
1 кг льда (см. примечание 4).
Через 4—5 часов осадок отфильтровывают, переносят в
стакан емкостью 2л и кипятят с 0.75—1 л дистиллированной
воды в течение 15 минут. Затем осадок отфильтровывают,
промывают горячей водой и сушат при комнатной темпера-
туре. Сухой осадок имеет серо-зеленый цвет.
Выход 2-антрола равен 29,5 г, что составляет 85%, считая
на натриевую соль 2-антраценсульфокислоты. После перекри-
сталлизации продукта из 300 мл этилового спирта с активи-
рованным углем получают 25 г (72% от теории) антрола в
виде серовато-желтых серебристых кристаллов. Температура
плавления 197—198°.
Найдено, %: С-85,90; 86,18; Н—5,31; 5,36.
С]4Н]0О. Вычислено, %: С—86,61; Н—5,15.
По литературным данным, т. пл. 200° с разл. [1].
Примечания:
1. В работе [1] указывается, что сплавление производилось в сереб-
ряном сосуде. Нами испробован для этой цели стальной котел, однако
удовлетворительных результатов эта замена не дала. При сплавлении
натриевой соли 2-антраценсульфокислоты со щелочью в никелевом котле
был достигнут высокий выход антрола вполне удовлетворительного ка-
чества.
2. Проба на полноту реакции производится следующим образом. В
пробирку с соляной кислотой и льдом. (1: 1) вносят на палочке немного
раы.лава. Выпадает осадок светло-зеленого цвета, который в случае за-
кончившейся реакции полностью растворяется в эфире, окрашивая его в
желтый цвет.
3. В работе [1] смесь после удовлетворительной пробы на раствори-
мость в эфире выдерживалась еще 30 минут при температуре 300—310°.
Такая дополнительная выдержка в наших условиях приводит к осмоле-
нию 2-антрола и резкому снижению выхода.
4. Осторожно, сильное разбрызгивание! Обязательно надевать защит-
ные очки.
ЛИТЕРАТУРА
1. К. L a g о d z i n s k 1, Liebigs Ann. Chem., 342, 67 (1905).
Поступила в ноябре 1965 г.
16
ИРЕА
УДК 546.151.07
АЦИЛХОЛИН И ТИОХОЛИНЙОДИДЫ
С. 3. ИВИН
RCOXCH2CH2N(CH3)3 Х=О или S
J
В литературе описано несколько способов получения ука-
занных соединений. Так, ацетилтиохолинйодид по Реншоу [1]
получается из диметиламиноэтилмеркаптана и ацетилхлори-
да с последующим отщеплением хлористого водорода от об-
разующегося 2-ацетилтиоэтилдиметиламмоний хлорида из-
бытком гидроокиси серебра. Затем к выделенному 2-(ацетил-
тио) этилдиметиламину-прибавляется йодистый метил. Этот
способ трудно воспроизводится.
По указанному способу ацилхолии- и тиохолингалогениды
получали и другие авторы [2, 3, 4].
Нами найден более удобный способ получения ацилхолин-
и тиохолиннодидов, основанный на присоединении 2-йодэти-
ловых или тиоэтиловых эфиров карбоновых кислот к триме-
тиламину [5, 6].
АЦЕТИЛТИОХОЛИНЙОДИД
CHsCOSCH2CH2N(CH.,)s
J
C,H16NOSJ М. в. 289,17
СХЕМА СИНТЕЗА АЦЕТИЛТИОХОЛИНЙОДИДА
СН,-СН2 + KSCN СН2-СН2 + KOCN
xoz xsz
СН2-СН2 + CH3COJ -> JCH2CH2SCOCH3
CH3COSCH2CH3J + N(CH3)j + CH3COSCH3CH3N(CHs)3
2 Зак. 25 J 17
Характеристика основного сырья
Ацетилйодид, ч., ВТУ МХП 3842—53.
Этиленсульфид, т. кип. 54,9°/760 мм, получен из окиси
этилена и роданистого калия.
Триметиламин, обычно выделяемый из хлоргидрата перед
употреблением.
Условия получения
Синтез этиленсульфида. В круглодонную колбу емкостью
250 мл помещают 45 г продажного роданистого калия и
45 мл воды. В охлажденный (хлористый натрий + лед) до
минус 12—16° раствор прибавляют 36 г охлажденной окиси
этилена. Реакционную массу взбалтывают, колбу закрывают
ватой и оставляют на ночь в охлаждающем растворе (см.
примечание 1).
По мере естественного нагревания охлаждающего раство-
ра и реакционной массы до комнатной температуры образу-
ющийся этиленсульфид всплывает на поверхность реакцион-
ной массы. Верхний слой этиленсульфида с примесью воды и
окиси этилена декантируется, сушится над хлористым каль-
цием и перегоняется. Собирается фракция в пределах 53—56°.
Выход этиленсульфида составляет 40—45% от теоретичес-
кого (см. примечание 2).
Получение 2-йодтиоэтилового эфира уксусной кислоты
[5, 7]. В круглодонную колбу с обратным холодильником и ка-
пельной воронкой помещают 16 г (0,1 М) ацетилйодида и
25 мл абсолютного четыреххлористого углерода или бензола.
К полученному раствору постепенно из капельной воронки
прибавляют 6 г (0,1 М) этиленсульфида. Колба энергично
встряхивается (реакцию можно проводить с механическим
перемешиванием). По окончании прибавления реакционная
смесь нагревается в течение 30 минут при температуре ба-
ни 50—60°. Образовавшийся 2-йодтиоэтиловый эфир уксусной
кислоты перегоняется в вакууме; т. кип. 97°/10 мм; rf420—
1.8195; л2^ — 1,5190 .
Выход равен 17 г, что составляет 77% от теоретического.
Получение ацетилтиохолинйодида. В широкогорлую про-
бирку или одногорлую толстостенную круглодонную, колбу с
притертой пробкой (см. примечание 3) емкостью 150—200 мл
помещают 17 г (0,075 М) 2-йодтноэтилового эфира уксусной
1S
кислоты, полученного по методу, описанному выше, и 35 .ил
абсолютного эфира.
К охлажденному до минус 10—15° раствору небольшими
порциями прибавляют 5 г (0,08 М) охлажденного до минус
10° абсолютного триметиламина. После каждого прибавления
реактор закрывают и взбалтывают. После прибавления всего
количества триметиламина реактор закрывают пробкой,
пробку закрепляют к горловине колбы проволокой, оставля-
ют стоять на холоду 3—4 часа и затем выдерживают 2—4 дня
при комнатной температуре до прекращения увеличения
кристаллического осадка.
Выпавший осадок ацетилтиохолинйодида отфильтровыва-
ют, промывают абсолютным эфиром или кристаллизуют из
небольшого количества пропилового спирта. Образуются бле-
стящие белые кристаллы с т. пл. 204—205°. Выход равен
17,2 г, что составляет 80% от теоретического.
По описанной методике получаются и другие ацилтио-
холинйодиды и бромиды [6]:
CH3COSCH2CH2N(CHs)s, т. пл. 213°]6];
Вг
CH3CH2CH2COSCH2CH2N(CH3)3, т. пл. 140—143°(6],
Вг
АЦЕТИЛХОЛИНЙОДИД
CH3COOH2CH2N(CH3)s
C,HieNO2J J М. в. 273,11
СХЕМА СИНТЕЗА АЦЕТИЛХОЛИНЙОДИДА
СН2СН2 -т CHsCOJ CH3COOCH2CH2J
о
CHSCOOCH2CH2J + N(CH3)3 -> CHsCOOCH2CH2N(CH3)3
J
Характеристика основного сырья
Йодистый метил, ч., ТУ МХП 49—51.
Окись этилена, перегнанная, очищенная от окиси пропи-
лена,
2* 19
Триметиламин, выделенный из хлоргидрата триметилами-
на обычным методом и абсолютированный.
Получение 2-йодэтилацетата [8]. В круглодонную колбу с
обратным холодильником и трубкой (доходящей до дна) для
подвода газа помещают 16 г (0,1 М) ацетилйодида и 30 мл
абсолютного эфира или четыреххлористого углерода. Затем
при охлаждении колбы ледяной водой в реакционную массу
пропускают 4,5 г (0,1 Л4) окиси этилена. По окончании про-
пускания окиси этилена (10—15 минут) реакционную массу
нагревают 15—20 минут при 40—50°. Образовавшийся 2-йод-
этилацетат перегоняют в вакууме; т. кип. 184°/743 мм;
110°/60 мм.
Выход равен 16,1 г, что составляет 80% от теоретического.
Получение ацетилхолинйодида [8]. В круглодонную толсто-
стенную колбу емкостью 200—250 мл с притертой пробкой
(см. примечание 3) помещают 8 г (0,05 М) 2-йодэтилацетата.
К охлажденному до минус 15° раствору постепенно неболь-
шими порциями прибавляют 3,5 г (0,058 М) охлажденного
триметиламина. После каждого прибавления колба закрыва-
ется и встряхивается. По окончании прибавления колбу за-
крывают, пробку закрепляют латунной или алюминиевой
проволокой и реакционную массу оставляют на холоду в те-
чение 4—5 часов, а затем выдерживают 2—4 суток при ком-
натной температуре.
Выпавший осадок ацетилхолинйодида отфильтровывают
и промывают абсолютным эфиром или перекристаллизовы-
вают из абсолютного этанола; т. пл. 160—161°.
Выход равен 7,3 г, что составляет 64% от теоретического.
Другие ацилхолингалогениды получаются аналогично (6].
Получение промежуточных 2-галогенэтилацетатов из оки-
си этилена и бром (йод) ацилов описано в литературе [8]:
CH3COOCH2CHaN(CH3)3; т.пл. 143° [6};
Вг
CeH6COOCHaCH2N(CH3)3; т.пл. 218° (6].
Вг
Примечания:
1. Смешение раствора роданистого калия и окиси этилена обычно
осуществляют перед окончанием рабочего дии,
2. Этиленсульфид легко полимеризуется, нестоек при храпении. Поэ-
тому его следует получать только перед самым употреблением.
3. Колбу или другой реактор можно закрывать и резиновой трубкой.
Но при этом резина растворяется в триметиламине и реакционная масса
окрашивается. Однако на выход продукта и его качество это не влияет.
20
ЛИТЕРАТУРА
1. R. R. Renshaw, Р. F. Dreisbach, М. Z
J. Amer. Chem. Soc., 60, 1765 (1938).
2. в. H ansen. Acta chem. scand., 13, 159 (1979).
3. В н a n s e n. Acta chem. scand., 11, 537 (19э7).
4. J. к. Cline. C. A., 28. 4075 (1934),
5. С. 3. Ивин. Ж. общ. химии, 22, 267 (1952).
6. С. 3. И в и н. Ж. общ. химии, 28, 1332 (1958).
7. С. 3. Ивин. ж. общ. химии, 28, 177 (1958).
8. С. 3. И в и и. Ж. общ. химии. 28, 180 (1958).
Поступила в ноябре 1965 г.
УДК 547.292.07
N-БЕНЗИЛЭТИЛ ЕНДИАМИН-N, N', N'-ТРИУКСУСНАЯ
КИСЛОТА
N-Бензил-! ,2-диаминоэтан Ы,№,№-триуксусиая кислота
Р. И. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Т. И. ИВАНОВА
хсн2соон
XCH,COOH
ch3nch2ch,n
I I
//\ CH,СООН
M. в. 324,33
Бензилэтилендиаминтриуксусная кислота образует водо-
растворимые комплексы с медью и кальцием [1] и может ис-
пользоваться как комплексообразователь для этих металлов,
а также для ряда аналитических и технологических целей.
Железный комплекс этой кислоты может иайти применение
в качестве эффективного антихлорозного средства для нужд
сельского хозяйства.
Бруно, Чаберек и Мартелл [1] получили бензилэтиленди-
амиитриуксусную кислоту карбоксиметилированием бензил-
этилендиамина формальдегидом и цианистым натрием или
цианистым барием по методу [2]. Бензилэтилендиаминтрнук-
сусная кислота растворима в воде, а потому трудно отделяет-
ся от неорганических примесей. Авторы [1] выделяли кислоту
подкислением 30 %-ной серной кислотой, ступенчатым осаж-
дением спиртом и многократной перекристаллизацией из
90%-кого этилового спирта.
Нами синтезирована бензилэтилендиаминтриуксусная кис-
лота взаимодействием N-бензилэтилендиамина [3] с моно-
хлоруксусной кислотой в слабощелочной среде (pH 8,0—9,0).
22
Кислота получена в растворе и выделена в чистом виде дву-
мя способами:
1) подкислением соляной кислотой и многократной про-
мывкой комплексона 90%-ным метиловым спиртом;
2) пропусканием щелочного раствора комплексона через
катионит КУ-2.
В последнем случае наряду с бензилэтилендиаминтриук-
сусной кислотой получается ее моноаммонийная соль.
СХЕМА СИНТЕЗА N-БЕНЗИЛЭТИЛ ЕНДИАМИН-N,N', N' ТРИУКСУС-
НОИ КИСЛОТЫ
CH2NHCH2CH8NHa
CICH..COOH —NaOH
| ------=----------
CH8NCHsCH.N
I I
CH2COOH
XCH2COOH
\ch2cooh
Характеристика основного сырья
N-Бензилэтилендиамин, получают по методике [3].
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натр едкий, ч., ГОСТ 4328—48.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
В четырехгорлую колбу, снабженную механической ме-
шалкой, обратным холодильником, капельной воронкой и тер-
мометром, загружают 430 г (4,5 М) монохлоруксусной кис-
лоты, смачивают ее 60 мл дистиллированной воды и посте-
пенно, при охлаждении и перемешивании, нейтрализуют ед-
ким натром до pH 7 так, чтобы температура в колбе не под-
нималась выше 40°. На нейтрализацию идет 300—320 мл
42%-ного раствора или 310—330 мл 40%-ного раствора едко-
го натра. Затем из капельной воронки постепенно добавляют
136 г (1 Л1) бензилэтилендиамина с такой скоростью, чтобы
температура в колбе не превышала 40°. После окончания
прибавления амина температуру в колбе поднимают до 50—
60° и, не прекращая перемешивания, реакционную массу вы-
держивают при этой температуре, одновременно добавляя
порциями 40—42%-ный раствор едкого натра с таким расче-
том, чтобы pH смеси был равен 8—9 (по фенолфталеину и по
универсальной индикаторной бумажке). Выдержку продол-
жают до тех пор, пока при добавлении последней порции ще-
лочи pH не меняется в течение 30 минут. Всего выдержка
продолжается 8—10 часов. На нейтрализацию выделившейся
23
в ходе реакции соляной кислоты идет 185—200 мл 42%-кого
или 195—210 мл 40%-ного едкого натра. Реакционную массу
отфильтровывают от выпавшего осадка хлористого натрия.
Получают раствор с содержанием в нем комплексона 17—
18% (см. примечание).
Выделение N-бензилэтилендиамин- М,М',Ы'-триуксусной
кислоты.
1-й способ. Раствор комплексона подкисляют до pH 2—
2,5 10%-ной или концентрированной соляной кислотой. Вы-
павший после 1—2 суток стояния на холоду осадок отфиль-
тровывают, промывают небольшим количеством этилового
спирта, а затем 90%-ным метиловым спиртом до отсутствия
ионов хлора, отжимают и сушат при температуре 80°.
Выход вещества составляет 23% от технической бензил-
этиленднаминтриуксусной кислоты.
Найдено, %: N-780; 6,65; С—53,42; 53,69; И-6,33; 6,39.
C15H20N2O6. Н2О. Вычислено, %: N-8, 22; С-52,63; Н - 6,43.
2-й способ. Через колонку с катионитом КУ-2 в Н-фор-
ме пропускают щелочной раствор комплексона, полученный
указанным выше способом, втрое разбавленный дистиллиро-
ванной водой, и элюируют сначала водой, а затем 0,5%-ным
раствором аммиака. Собирают аммиачные элюаты (pH от
2 до 5), упаривают до сиропа, высаживают большим коли-
чеством ацетона, отфильтровывают и сушат в эксикаторе.
Из элюата с pH 3 выделяют бензилэтилендиаминтриуксус-
ную кислоту с т. пл. 129—132° (разл.).
Выход продукта составляет 13,5% от теоретического. По
литературным данным, т. пл. продукта 130—131° [1].
N-Бензилэтнлендиаминтриуксусная кислота представляет
собой порошок белого или слегка розоватого цвета, хорошо
растворимый в воде, хуже в спирте, нерастворимый в ацето-
не. На воздухе расплывается. Для удаления кристаллиза-
ционной воды продукт растворяют в абсолютном спирте с по-
следующей его отгонкой.
Найдено, %: N-8,55; 8,80; С-54,66; 54,98; Н— 6,65; 6,61.
C16H3oNsO6. Вычислено, %: N—8,64; С—55,55; Н—6,22.
Из элюата с pH 4—5 выделяют моноаммонийную соль
бензилэтилендиаминтриуксусной кислоты с т. пл. 150—155°.
Выход моноаммонийной соли составляет 17% от теорети-
ческого.
Продукт представляет собой кристаллический порошок
бледно-фиолетового цвета, растворимый в воде, нераствори-
мый в ацетоне.
Найдено, %; N-12.69; 12,89; С-53,02; 52,85; Н-7,30; 7,82.
C15HS3N3O6. Вычислено, %: N—12,30; С—52,80; Н—6,74.
Примечание.
Определение процентного содержания комплексона проводится титро-
ванием 0,05 М раствором сернокислого цинка в среде аммиачного бу-
фера (pH 9) в присутствии индикатора — эриохром черного Т.
24
Процентное содержание определяется по формуле:
х t>.М-0,05-Я-100
к-ЮОО
где х—процентное содержание комплексона в растворе;
М— молекулярный вес комплексона;
К — поправка титра 0,05 М раствора сернокислого цинка;
н — навеска, а;
v —количество мл 0,05 М раствора сернокислого цинка, пошедшее
на титрование.
ЛИТЕРАТУРА
1. A. J. В г u п о, S. Chaberek, А. Е. М а г t е 11. J. Amer. Chem.
Soc., 78, 2723 (1956).
2. R. Smith, J. L Bullock, F. C. Be r w о r t h, A. E. Martell.
J. Organ. Chem., 14, 355 (;949).
3. P. П. Ластовский, И. Д. Колпакова, Л. В. К р и н и ц к а я,
Т. И. Иванова. Методы получения химических реактивов и препаратов,
вып. 12, М„ ИРЕА, 1965, стр. 31.
Поступила в декабре 1965 г. ИРЕА
УДК 661.731.7.07
а, а'-БИС-(4-НАТРИЙТЕТРАЗОЛИЛАЗО-5)-
ЭТИЛАЦЕТАТ
Н. С. ФРУМИНА, Г. С. ПЕТРОВА. Н. Н. ГОРЮНОВА
N-N—Na Н
Na-N-N
\ I j
С—N—N—С—N—N—С
I I
N-n СООС2Н5 N-N
•ЗН2О
ЗН2О
М. в. 378,21
а, а'-Бис-(4-натрийтетразолилазо-5)-этилацетат, назван-
ный для краткости «тетра», впервые синтезирован и предло-
жен Джонасепом в качестве колориметрического реактива
на медь и никель [1, 2, 3]. Впоследствии Мустафиным, Горю-
новой и Фруминой было найдено, что он является ценным
комплек'сонометрическим индикатором на медь и может быть
применен для ее определения в бронзах и некоторых спла-
вах [4].
Реактив получают сочетанием 5-аминотетразола с ацето-
уксусным эфиром.
Нами уточнена методика его синтеза [I] и показано, что
применение для перекристаллизации продукта водного (1:1)
спирта обеспечивает сразу получение х. ч. соединения, в то
время как; автор, применявший для этой цели ректификат,
добился той же чистоты только после трехкратной перекри-
сталлизации.
СХ£МА СИНТЕЗА а, а'- БИС-(4-НАТРИЙТЕТРАЗОЛИЛАЗО-5)
ЭТИЛАЦЕТАТА
H,N - C-NH-NH2-HNOg-^^
NH
26
— H.N—C—NH—Na-NO3-C--" 0-^а HN —C—NH; + HNO3
II I II
NH NN
4-nZ
HN-C-NH2 hhT2-> HN-C-N=N-C1
I и на rn
NN NN
4N/ ^1/
2HN—C-N=N-Cl+CHsCO-CH2-COOC3Hs+5NaOH
I II
N N
4*‘N/
H
I
Na-N-C—N=N-C-N=N-C- N-Na-r
N N COOC2H6 N hl
4-2NaC14-CHsCOONa+4H,O
Характеристика основного сырья
Аминогуанидин нитрат, ч., ВТУ РУ 1027—54.
Ацетоуксусный эфир, ч., ТУ МХП 1887—48.
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
Синтез 5-аминотетразола [5}. Размешивают 68,5 г (0,5 М)
нитрата аминогуанидина в 100 мл 5 н. раствора азот-
ной кислоты и в смесь вносят 100 мл (0,5 М) 5 н. раствора
нитрита натрия. По мере прибавления при 40° раствора нит-
рита натрия образуется желтый раствор, к которому прибав-
ляют 140 г ацетата натрия (трехводного) и кипятят 5 минут.
По охлаждении раствора выпадают белые кристаллы.
Продукт отфильтровывают, отжимают, перекристаллизо-
вывают из воды и сушат при 80°. Получают43 г (88%,считая
на аминогуанидин нитрат) белого порошка 5-аминотетразола
с т. пл. 200—203°. Вещество содержит 2 молекулы воды
Получение а,а'-бис-(4-натрийтетразолилазо-5)-этилацетата.
Растворяют 9,7 г (0,1 М) 5-аминотетразола (двухводного)
в 40 мл 20%-ного водного раствора едкого натра, добавляют
раЬтвор 7,1 г (0,1 М) нитрита натрия в 25 мл воды и 60 г
толченого льда. Полученную массу, имеющую температуру
3—4°, выливают в смесь, состоящую из 28 мл концентриро-
ванной соляной кислоты, 120 мл воды и 200 г толченого льда.
Получают диазораствор с pH 2. После выдержки в течение
27
20 минут в колбу вносят раствор 50 г ацетата натрия в
100 мл воды, а затем 13 мл (0,1 М) ацетоуксусного эфира.
Раствор размешивают при 2—2,5° 2 часа, а затем вносят
100 г хлористого натрия. По мере растворения поваренной
соли выпадают оранжевые кристаллы реактива. Реакцион-
ную массу выдерживают в холодильнике 12 часов, а затем
фильтруют. Осадок промывают спиртом и сушат под ваку-
умом при 80° над пятиокисью фосфора.
Выход а,а'-бис-(4-натрийтетразолилазо-5)-этилацетата ра-
вен 9 г, что составляет 23%, считая на 5-аминотетразол.
Перекристаллизовывают 3 г реактива из 20 мл 50%-ного
спирта. Получают 1,8 г красно-оранжевого порошка.
Найдено, %: С—19,27: 19,07; II- 3,45; 3,37;
N —44,75; 44,20; Na—12,25;
11,91.
CdHeOaNpNaa-SHaO, Вычислено, 96: С-19,05; Н-3,20; N-44,49;
Na—12,15,
ЛИТЕРАТУРА
1. U. В. Jonassen, V. С. Ch a mbH п, V. Z. Wagner, R. А.
Hanry. Analyt. Chem., 30, 10 1660 (1958).
2. М. Frelgard, В. Jones. Analyst, 84, 1005, 716 (1959).
3. Р. S. Bowles, P. S. Nicks. Analyst, 86, 1024 (1961).
4. И. С. Мустафин, H. H. Горюнова, H. С. Фрумина. 3a-
водск. лаборатория, 7, 786 (1965).
5. Z. Vanina. Handbuch der praparativen Chemie, 2, 780 (1923).
Поступила в ноябре 1965 г,
ИРЕА
Саратовский гое.
универси тет
УДК 547.571.07
4-БРОМБЕНЗАЛЬДЕГИД
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, Р. У. СУДИЯРОВА
Вт~\~У~сн0
C,H5BrO "" М. в. 185,02
Наиболее доступными и отработанными методами полу-
чения 4-бромбензальдегида являются окисление 4-бромтолуо-
ла хромовым ангидридом [1] и бромирование 4-бромтолуола
до 4-бромбензилиденбромида с последующим омылением [2]
или бромирование до 4-бромбензилбромида и далее перевод
его в альдегид через уротропиновый комплекс по реакции
Соммле [3]. Недавно [4] был опубликован новый общий метод
синтеза альдегидов, заключающийся в непосредственной кон-
денсации соответствующих углеводородов с уротропином с
помощью полифосфорной кислоты. Выход 4-бромбензальде-
гида (из бромбензола) составлял 44%.
Нами 4-бромбензальдегид получен из 4-броманилина пу-
тем замены диазогруппы на формильную по способу, предло-
женному [5] для синтеза 2- и З-метил-4-бромбензальдегидов.
Этот способ представляет интерес, так как расширяет сырье-
вые источники 4-бромбензальдегида.
СХЕМА СИНТЕЗА 4-БРОМБЕНЗАЛЬДЕГИДА
Вг-\ /-NH2 N?N°2, Вг-^-NjCl ->
2Н3—N-OH[(CH3O)X, WH3OH] ; г Вг_^—X_CH==NOH I
CuSO4, Na,SO3. CH3COONa [ J
HCUbO^ Br_/—V-CHO
29
Характеристика основного сырья
4-Броманилин, ч., СТУ 79-565-Х—60.
Натрий азотистокислый, ч., ГОСТ 4197—48.
Кислота соляная, ч., ГОСТ 3118—46.
Пароформ, ч., ВТУ МХП 2711—51.
Гидроксиламин солянокислый, ч., ГОСТ 5456—51.
Медь сернокислая, ч., ГОСТ 4165—48.
Натрий сернистокислый, ч., ГОСТ 429—41.
Натрий уксуснокислый, ч., ГОСТ 199—52.
Натрий двууглекислый, ч., ГОСТ 4201—48.
Натрий сернистокислый кислый (раствор), ч., ТУ МХ-П
1944—49.
Условия получения
Получение 10%-ного раствора формальдоксима. В кругло-
донную колбу, снабженную обратным холодильником, загру-
жают 9 г ~ 0,3 М) параформа, 20,5 г ('-0,3 М) гидроксил-
амина солянокислого и 133 мл воды; смесь нагревают до по-
лучения прозрачного раствора, добавляют 40 г (—-0,3 М) аце-
тата натрия и слабо кипятят в течение 15 минут.
Диазотирование 4-6романалина. В фарфоровый стакан
емкостью 0,5 л, снабженный мешалкой, капельной воронкой
и термометром, загружают 34,4 г (0,2 М) 4-броманилина и
45 мл концентрированной соляной кислоты. Стакан погру-
жают в баню с охладительной смесью и при размешивании
прибавляют 40 мл воды и 75 г толченого льда, после чего
диазотируют раствором 14 г нитрита натрия в 20 мл воды в
течение 20—30 минут при температуре минус 5—0°. По окон-
чании диазотирования осторожно нейтрализуют (при охлаж-
дении) по бумажке конго раствором ацетата натрия (около
20 г в 20 мл воды).
Получение 4-бромбензальдегида. В круглодонную колбу
емкостью 1,5 л, снабженную мешалкой, термометром и ка-
пельной воронкой с длинной отводной трубкой, помещают ра-
створ формальдоксима и раствор, полученный из 4,9 г медно-
го купороса. 0,78 г сернистокислого натрия, 25 г уксуснокис-
лого натрия и 140 мл воды. Под поверхность этой смеси при
размешивании и температуре 5—15° вводят нейтрализован-
ный диазораствор за 20—30 минут. Затем размешивают в те-
чение 1 часа, подкисляют по конго соляной кислотой и добав-
ляют еще 180 мл концентрированной соляной кислоты. 'Уда-
ляют мешалку, термометр, капельную воронку и охлаждаю-
щую баню, колбу снабжают обратным холодильником и ре-
акционную смесь кипятят в течение 2 часов, после чего альде-
гид отгоняют с водяным паром. Дистиллят нейтрализуют би-
карбонатом натрия и альдегид извлекают эфиром (200 мл)
в 2—3 приема. Эфир отгоняют, остаток встряхивают с 75 мл
30
40%-ного раствора бисульфита натрия, нагревая время от
времени смесь до 60°. Осадок бисульфитного соединения про-
мывают несколько раз эфиром и разлагают 75 мл 20%-ной
серной кислоты при слабом нагревании (50—60°). Альдегид
извлекают эфиром (100 .м.-i) в 2 приема, эфирную вытяжку
сушат прокаленным сульфатом магния или натрия.
После отгонки эфира получают 11—13 г (30—34%) аль-
дегида (см. примечание), который кристаллизуют из 50—
60 мл водного этанола (ректификат разбавляют равным объ-
емом воды).
Выход альдегида с т. пл. 57,5—58° равен 7—8 г. Из ма-
точника дополнительно выделяют 1,5—2 г продукта с т. пл.
57—58°. Таким образом, общий выход составляет 8,5—9,5 г
(23—25% от теоретического выхода).
По литературным данным, т. пл. вещества 56—57° [6, 7];
55—57° [1—2]; 67° [4] (опечатка?).
Примечание.
Температуру плавления альдегида-сырца точно не удается определить
из-за примеси сульфата магния; начало его температуры плавления 57°.
При увеличении масштаба синтеза очистку целесообразнее осуществлять
вакуум-перегонкон.
ЛИТЕ Р АТ УРА
1. Сб. «Синтезы органических препаратов», т. 2, М., Госхимиздат,
1949, стр. 366.
2. Там же, стр. 105.
3. S. J. А п g у а 1. Р. J. М о г г i s, R. С R a s s а с k, J. A. W a s е г е г
J. Chem. Soc.. 1949. 2704.
4. D. A. Denton, Н. S u s с h i t z к у J. Chem. Soc.. 1963, 4741.
5. S. S. V e г п e к a r, S. D. Jabal, S. Rajagopal. Monatsheft,
93, 271, (1962).
6. M. M. К о т о н, E. П. Москвина, Ф. С. Ф л о p и н с к и й. Ж.
общ. химии, 21, 1843 (1951).
7. A SI s t 1, J. Bfirgmastcr, ;Ч. Fudini. J. Organ. Chem., 27, 279
(1962).
Поступила в ноябре 1965 г.
ПРЕЛ
УДК 547.576.07
3-БРОМ-5-НИТРОСАЛИЦИЛОВЫЙ АЛЬДЕГИД
3-Бром-5-нитро-2-оксибензальдегид
Д. А. ДРАПДИНА. В. Г. БРУДЗЬ, Г. А. ИНШАКОВА. И. П. ПЛИГИНА
сно
C7H4NO4Br М. в. 246,02
По литературным данным, З-бром-5-нитроса.тициловый
альдегид был получен [1] бромированием 5-нитросалицилово-
го альдегида в ледяной уксусной кислоте при 30—40° с выхо-
дом продукта, равным 70% от теоретического, но без указа-
ния его качества. Для очистки рекомендована разбавленная
уксусная кислота и этиловый спирт.
Нами проверен указанный способ и уточнены условия ре-
акции, выделения и очистки продукта.
СХЕМА СИНТЕЗА 3-БРОМ-5-НИТРОСАЛИЦИЛОВОГО АЛЬДЕГИДА
СНО
_ CHjCOOH ледяная
-Г tsr8 ---------------
СНО
Д/он
I II + НВг
O,N/'^/'4Br
32
Характеристика основного сырья
5-Нитросалициловый альдегид, ч., ТУ TCP 563р—61.
Бром, ч., ГОСТ 4109—48.
Уксусная кислота, ледяная, х. ч., ГОСТ 61—51.
Спирт этиловый технический, гидролизный, ГОСТ 8314—57.
Условия получения
В четырехгорлую колбу, снабженную механической ме-
шалкой, термометром, обратным воздушным холодильником
и капельной воронкой, загружают 33,4 г (0,2 Л4> 5-нитроса-
лицилового альдегида и 70 мл ледяной уксусной кислоты. К
полученной суспензии, нагретой на водяной бане до 60°, при-
бавляют раствор 32 г (0,2 М) брома в 350 мл ледяной уксус-
ной кислоты в течение 2 часов, поддерживая температуру ре-
акционной смеси 60—65°. Затем смесь размешивают еще один
час при той же температуре. К концу реакции осадок раст-
воряется.
По окончании выдержки реакционную смесь выливают в
500 г воды со льдом и оставляют стоять несколько часов для
полноты выделения осадка. Выпавший желтоватый осадок
отсасывают и промывают на фильтре водой. Продукт сушат
в шкафу при 70—80°.
Выход неочищенного продукта с т. пл. 139—142° равен
40—42 г, что составляет 81—85% от теоретического.
После перекристаллизации сначала из 1300 мл 50%-ной
уксусной кислоты, а затем из 400 мл гидролизного этилового
спирта получают 27,6—28 г альдегида или 56—56,9% от тео-
ретического выхода.
По внешнему виду это мелкие белые кристаллы с т. пл.
149—150°.
По литературным данным, т. пл. продукта 149—150° [1, 2].
ЛИТЕРАТУРА
1. R. О. С 1 i п t о п, S, С. Laskowski. J. Amer. Chem. Soc., 71,
3603 (1949).
2. E. Ber ma n, R. E. Fox, F. D. Thom so n. J. Amer. Chem. Soc.,
81, 5605 (1959).
Поступила в июле 1&6о г. ИРЕА
3 Зак. 25
УДК 547.821.4.07
2-ВИНИЛПИРИДИН
Б. М. КУИНДЖИ, М. А. ЗЕПАЛОВА, А. К. ВАЛЬКОВА,
Л. Д. ГЛУЗМАН, Р. М. ЦИН, А. А. РОКК
/V
li J
сн=сн3
C,H,N
М. в. 105,13
2-Винилпиридин получают из а-пиколина и формальдеги-
да с дальнейшей перегонкой образовавшегося 2-пиридилэта-
нола над едким кали или едким натром [1].
Описан также способ синтеза 2-винилпиридина из пири-
дил-0-бром-пропионовой кислоты в слабощелочном раство-
ре [2].
В следующих работах были изменены условия реакции, но
выход 2-пиридилэтанола был незначительным [3]. Описан
также метод получения его в автоклаве нагреванием а-пико-
лина с параформом [4]. Большое значение имеет молярное
соотношение а-пиколина и формальдегида [5].
2-Винилпиридин является исходным продуктом в произ-
водстве ионообменных смол, синтетического каучука, а также
лекарственных веществ.
Предлагаемый метод синтеза 2-винилпиридина позволяет
получать его с выходом 89% от теоретического.
СХЕМА СИНТЕЗА 2-ВИНИЛПИРИДИНА
/ Z ^1
XN’=/xCHj-PCH2O - xn//xch2ch2oh
/X
11 I . II I
WXcHiCtWH XN^\CH=-CH2
Характеристика основного сырья
«-Пиколин, ч., ВТУ РУ 771—52.
Формалин, техн., ГОСТ 1625—54.
Параформ, ч., ВТУ МХП 2711—51.
Натр едкий, ч., ГОСТ 4328—48.
Азот, из баллона, ТУ МХП 4280—54.
Условия получения
В автоклав емкостью 500 мл, снабженный мешалкой,
трубкой для подачи азота, карманом для термопары и элек-
трообогревом, загружают 150 г (1,613 М) а-пиколина и 41 г
27%-ного раствора формалина, содержащего 11 г (0,366 М)
формальдегида (см. примечание 1).
Автоклав продувают азотом, после чего доводят началь-
ное давление до 10 атмосфер, включают обогрев и мешалку.
Через 1 —1,5 часа температура достигает 165°, при которой
реакционную массу выдерживают 2 часа, давление при этом
повышается до 15—18 атмосфер.
После охлаждения автоклава давление спускают, содер-
жимое его выгружают и продукт разгоняют в вакууме, рав-
ном 5 мм.
Вначале, до 98°, отгоняется смесь в количестве 157,5 г,
состоящая из избыточного а-пиколина и воды (см. примеча-
ние 2), а затем в пределах от 98 до 130° получают 2-пири-
дилэтанол в количестве 28,2 г, который после определения в
нем гидроксильного числа подвергается дегидратации.
Для этого в колбу Клайзена емкостью 100 мл загружают
28,2 г (0,229 М) 2-пиридилэтаиола и 1,41 г (5% весовых от
2-пиридилэтанола) едкого натра, после чего эту смесь нагре-
вают в вакууме, равном 10 мм.
При 40°, по мере его образования, отгоняется сырой 2-ви-
нилпиридин в количестве 24,2 г, который затем вновь перего-
няется в том же вакууме. При этом вначале, до 45°, отгоня-
ется вода со следами 2-винилпиридина, а затем в пределах
от 45 до 53° получают 2-винилпиридин в количестве 22,6 г,
что составляет 89% от теоретического выхода: —1,5460.
Примечания:
1. Содержание формальдегида определяют титрованием технического
формалина, после чего последний разбавляется водой до удельного веса
при 20° 1,064 г, что соответствует 27%-ному раствору. Оптимальный вы-
ход 2-пиридилэтанола достигается применением 27%-ного раствора фор-
малина и соотношением я- пиколина и формальдегида 4,3 : 1.
3*
35
2. Избыточный о-пиколин используют в дальнейших операциях. Для
этого в отгоне до 98° при 5 мм титрованием определяют содержание
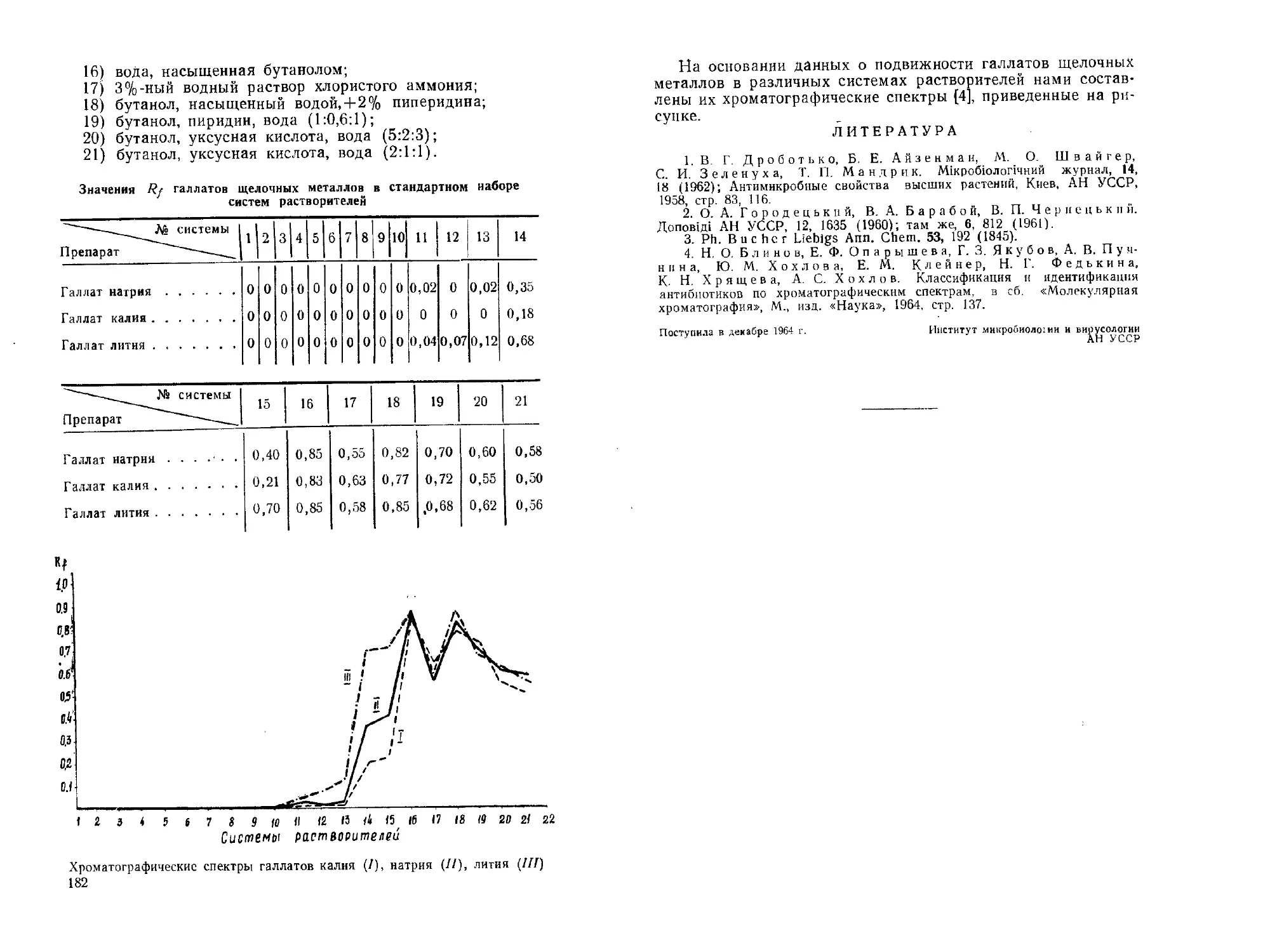
а- пиколина, затем к нему добавляют израсходованный на опыт «-пиколин
и параформ в таком количестве, чтобы сохранить их исходное соотноше-
ние 4,3 : 1.
ЛИТЕРАТУРА
1. A. Ladenburg. Liebigs Ann. Chcm., 39, 125 (1898).
2. A. Einhorn. Liebigs Ann. Chem., 265, 229 (1891).
3. S. M. T u 11 о c k, S. W. Me. El vein. J. Amer. Chem., Soc., 61,
962 (1939),
4. L. Kitchen, E. Hanson. J. Amer. Chem. Soc., 73, 1898 (1951).
5. Б. M. Куннджи, Л. Д. Глузман, M. A. 3 e п а л о в a,
А. К. Валькова, Р М. Ц и н, А. А. Рокк. Авт. свид. 135488; Бюлл.
изобр., № 3 (1961).
Поступила в 1965 г. МИИЯМ, УХИН
УДК 547.587.26.07
ГАЛЛАТ КОБАЛЬТА
В. П. ЧЕРНЕЦКИЙ, Э. А. ПОНОМАРЕВА
С14Н8О10Со2 • 4НаО М. в. 526,14
Галлат кобальта является производным галловой кисло-
ты, включающим микроэлемент кобальт, способный образо-
вывать в организме прочные внутрикомплексные связи с фер-
ментами, витаминами и другими жизненно важными соедине-
ниями.
В литературе описано несколько попыток получения гал-
лата кобальта. В зависимости от взятых исходных веществ
получаются соединения различного состава: Бухер [1] полу-
чил двухосновную соль из ацетата кобальта и галловой кис-
лоты—С7Н4О5СО 2Н2О, Глазивец [2] считает, что при реак-
ции между галловой кислотой и гидроокисью кобальта об-
разуется нечистая четырехосновная соль кобальта, и при-
писывает ей формулу ЗСцНзСозОю-СоО-ННаО.
При рассмотрении пространственных моделей Стюарта со-
лей галловой кислоты становится очевидным, что замещение
двухвалентным металлом любого радиуса двух атомов водо-
рода (в карбоксиле и одной из гидроксильных групп) одной
молекулы галловой кислоты, как это писалось до сих пор, не-
возможно. Поэтому галлату кобальта (и любой другой двух-
основной соли галловой кислоты) мы можем приписать толь-
ко приведенную выше структурную формулу.
37
Нами получен галлат кобальта при взаимодействии гал-
лата натрия с хлористым кобальтом. При этом было уста-
новлено, что образующаяся при реакции галловая кислота в
условиях опыта не реагирует с избытком хлорида кобальта.
Проводя реакцию в щелочной среде, можно превратить гал-
ловую кислоту в галлат натрия и таким образом ввести в ре-
акцию полностью. Однако из-за легкости окисления произ-
водных галловой кислоты в щелочной среде такой вариант
синтеза приводит к менее чистому препарату, а очистка его
ввиду практически полной нерастворимости чрезвычайно за-
труднена. Поэтому целесообразнее вести реакцию в соответ-
ствии с приведенным уравнением и регенерировать галловую
кислоту, выкристаллизовывающуюся из маточного раствора.
СХЕМА СИНТЕЗА ГАЛЛАТА КОБАЛЬТА
ONa
+ 2CoCls
ОН
ОН
о J 9П
ОН \сох
о „он
Ч.С/
/А
+ 2 fi H-4NaCl
НОХ^/ХОН
он
Характеристика основного сырья
Галлат натрия (получение см. на стр. 180).
Кобальт хлористый, ч„ ГОСТ 4525—48.
Ацетон, ч.д.а., ГОСТ 2603—63.
Условия получения
К теплому раствору 1,2 г (0,005 М) хлорида кобальта (см.
примечание 1) в 5 мл воды добавляют раствор 2,1 г (0,01 М)
галлата натрия в 20 мл воды (его лучше готовить в токе во-
дорода, см. примечание 2). Смесь прогревают на водяной ба-
не в течение 5 минут. Выпавший коричнево-фиолетовый оса-
38
док галлата кобальта отфильтровывают, промывают водой до
отрицательной реакции с хлорным железом и затем ацетоном.
Из маточного раствора после охлаждения выкристаллизовы-
вается галловая кислота (0,9 г) в виде длинных шелкови-
стых игл с температурой плавления 235°, не дающих депрес-
сии при сплавлении смешанной пробы с чистой галловой кис-
лотой.
Выход галлата кобальта равен 1,15 г, что составляет 87%
от теоретического. По внешнему виду вещество представляет
собой коричнево-фиолетовый порошок, не плавящийся до
500°, нерастворимый в воде и органических растворителях,
очень слабо (0,04 г в 100 г) растворим в феноле. С камфо-
рой не сплавляется.
Для анализа препарат высушивался в вакууме при 77° в
течение шести часов, кристаллизационная вода при этом не
удалялась.
Найдено, %: С—31, 77; 31, 65; 11-3,04; 3,13;
Со-22.37; 22,24.
(.^HjOnjCoj-lH-jO. Вычислено, %; С—31, 95; Н—3,06; Со—22,40.
Примечания:
1. Продажный хлорид кобальта содержит 6 молекул кристаллиза-
ционной воды.
2. Галлат натрия можно заменить приготовленным в токе водорода
раствором 1,7 г галловой кислоты и около 0,5 г карбоната натрия в горя-
чей воде (раствор должен иметь pH не выше 7).
ЛИТЕРАТУРА
1. Ph. Bucher. Liebigs Ann. Chem., 53, 192, (1845).
2. H. Hlasiwetz. Liebigs Ann. Chem., 142, 238 (1867).
Поступила в августе 1965 i.
Институт микробиологии
в вирусологии АН УССР
УДК 547.462.3.07
ГИДРАЗИД МАЛЕИНОВОЙ КИСЛОТЫ
1,2-Дигидропиридазиндион-3,6
Р. М. ГЕЛЬШТЕИН, Ф. В. АЛЕКСАНДРОВА
о
и
/с\
ПС NH
1! i
НС NH
c4h4n2o2
М..в. 112,08
Малеингидразид (1,2-дигидропиридазиндион-3,6) нашел
широкое применение в качестве вещества, препятствующего
росту растений [1].
Известны методы его получения взаимодействием малеи-
нового ангидрида и солен гидразин-гидрата в водной среде
[2, 3, 4, 5].
В производстве чаще применяют дешевую и легкодоступ-
ную сернокислую соль гидразина в виде суспенза в воде
(гидразин-сульфат плохо растворим в воде). Недостатком
метода является необходимость очистки полученного техни-
ческого продукта кристаллизацией из большего (1 :20) коли-
чества воды [3, 4].
Предложенная нами методика отличается тем, что с целью
исключения кристаллизации малеингидразида из воды про-
дукт получают путем прибавления малеинового ангидрида к
кипящему фильтрованному водному раствору гидразин-гид-
рата, гидразин-сульфата и сернокислого аммония.
40
СХЕМА СИНТЕЗА МАЛЕИНГИДРАЗИДА
НС-СО
|| /O + NH1-NH,-H2SO4 -
НС-СО
о
/С\
НС NH
—> II | + + H2SO4
НС NH
ЧСХ
о
Характеристика исходного сырья
/Малеиновый ангидрид, ч., ГОСТ 5854—51.
Гидразин-сульфат, техн., ГОСТ 3286—52.
Аммиачная вода, техн., ГОСТ 9—40.
Условия получения
В пятилитровую колбу, установленную в глицериновой ба-
не и снабженную мешалкой, обратным шариковым холодиль-
ником, термометром и загрузочным отверстием, помещают
нагретый до 50° и профильтрованный раствор 15 г (в пересче-
те на 100%-ный продукт) технического гидразин-сульфата,
500 мл 25 %-ной аммиачной воды и 2,4 л дистиллированной
воды.
Раствор кипятят 30 минут и затем при размешивании и
интенсивном кипении массы в течение 20—30 минут придают
600 г расплавленного (или чешуированного) чистого малеи-
нового ангидрида. После пятичасового кипения реакционную
массу охлаждают до 30°, выпавший осадок малеингидразида
отжимают, промывают дистиллированной водой до нейтраль-
ной реакции промывных вод и сушат при 60°.
Выход гидразида малеиновой кислоты квалификации
«чистый» (ТУ ХСНХ-2 09—59) равен 541 г, что соответствует
79% от теоретического, считая на малеиновый ангидрид.
ЛИТЕРА ТУ РА
1. Н. Н. Мельников, Ю. А. Бакса к о в. Химия гербецидов и ре-
гуляторов роста растений, М., Госхимиздат, 1962, стр. 613.
2. Амер, патент 2575954; С. А., 46, 6161 (1952).
41
3. Ю. А. Баскаков. Н. Н. Мельников. Ж общ. химии, 24,
1216 (1954).
4. Э. Э. Ду нк ель, С. А. Г ил л ер. Известия АН Латв. ССР, 2, 105
(1954).
5. Япон. пат. 7208/55; С. А., 51 18014с (1957).
Поступила в июле 1965 г. ЦЗЛ
УДК 547.537.07
ГИДРАЗОБЕНЗОЛ
И. Е. ЗАДОРОЖНАЯ
Н Н
CjsHjjNj
М. в. 184,24
Гидразобензол может быть получен восстановлением ни-
тробензола цинковой пылью в щелочной среде [1, 2, 3], амаль-
гамой натрия [4], чугунными стружками [5], электролитичес-
ким восстановлением [6, 7], а также восстановлением азобен-
зола активированной цинковой пылью в среде бензола [8] или
сероводородом [9].
Нами уточнена методика получения гидразобензола вос-
становлением азобензола цинковой пылью в среде изопро-
пилового спирта.
СХЕМА СИНТЕЗА ГИДРАЗОБЕНЗОЛА
^+Zn+2NaOH ->
^N-N^ 4 Na,ZnO2
H H
Характеристика основного сырья
Азобензол, ч., ТУ МХП 1862—48.
Цинковая пыль, ГОСТ 417—48.
Натр едкий, ч., ГОСТ 4328—48.
Спирт изопропиловый, техн., ГОСТ 9805—61.
43
Условия получения
В 10-литровую колбу с мешалкой, термометром н шари-
ковым обратным холодильником, установленную в бане с
паровым обогревом, загружают 2,5 л воды, 0,7 кг (17,5 Л1)
едкого натра, размешивают и к теплому раствору добавляют
1,84 л изопропилового спирта и 1 кг (5,42 Л4) азобензола.
Смесь нагревают до 50—60° при размешивании до полно-
го растворения продукта. Затем через каждые 45 минут пор-
циями по 70 г придают цинковую пыль. Первые 2—3 порции
загружают осторожно при 50—60°. Когда бурная реакция
прекратится, массу нагревают до 78—80° и держат эту тем-
пературу до конца внесения всей цинковой пыли. Всего при-
дают 510 г (7,8 М) 100%-ной цинковой пыли (применять
цинк с содержанием металла ниже 75% не рекомендуется).
После придачи всей цинковой пыли реакционную массу
кипятят при размешивании до полного обесцвечивания и за-
тем извлекают гидразобензол дважды по 7 л нагретым до
50—60° изопропиловым спиртом. После придачи спирта мас-
са нагревается до кипения и фильтруется (масса сильно ще-
лочная) .
Фильтрат охлаждают до 0—5° и выпавший гидразобензол
отфильтровывают, промывают разбавленным изопропиловым
спиртом (680 мл спирта и 1 л 330 мл воды), насыщенным
SO2, до нейтральной реакции. Полученный гидразобензол су-
шат на воздухе.
Выход технического продукта равен 800 г. Его кристалли-
зуют из изопропилового спирта в соотношении 1 :5,5 с углем,
охлаждают до 0—5°, фильтруют, промывают смесью дистил-
лированной воды и изопропилового спирта (1:2), насыщенно-
го SO2, и сушат на воздухе (под тягой).
Выход чистого гидразобензола равен 640 г, что составля-
ет 64%, считая на азобензол; т. пл. 127,1—127,5°.
По литепатурным данным, температура плавления веще-
ства 126°; 131° [10]; 126°—127° [И].
Продукт соответствует требованиям ТУ ГКХ РУ 1755—56.
ЛИТЕРАТУРА
1. Л. Гаттерман, Г. Виланд. Практические работы по органи-
ческой химии, М., Госхимиздат, 1948, стр. 223—224.
2. Препаративная органическая химия (Польск), М„ Госхимиздат,
.1959. стр. 515.
3. Герм. пат. 138496; Fridlander, VI, 1290.
4. Н. Н. Ворожцов. Основы синтеза промежуточных "продуктов и
красителей, М., Госхимиздат, 1955, стр. 247; П. А. Алексеев. Bulletin
de la Soclete chim. de France, (2) I, 324, 326 (1864).
5. С. Д. Топорков, H. И. Амиантов. Анилокрасочиая промыш-
ленность. I, 6, 3 (1931); H. И. Амиантов. Анилокрасочиая промышлен-
ность, 2, И, 24 (1932).
44
6. D a r m s t a d t е г. Chem. Zbl, 1907. II, 2002; Герм. пат. 189312.
7. Straub. Герм. пат. 79731.
8. В. О. Лукашевич. Ж. общ. химии, 6, 1064 (1936); Chem.Abstrs,
31, 1012 (1937).
9. К. Pa;sow. J. Pract. Chem., 84, 267 (1911).
10. Справочник химика, т. 11, М.—Л., Изд. «Химия», 1964, стр. 609.
11. Словарь органических соединений, т. II, стр. 203.
Поступила в июле 1965 г. !13.’'
УДК 547.466.6.07
б-ДИАЗО-5-KETO-L -НОРЛЕИЦИН
Дон
в-Диазо-5-OKco-L -норлейцин
Г. И. КОШЕЛЕВА, Г. Н. НАЛЕЦКАЯ
HOOC-CH-CH3-CH4-CO-CHN2
I
NHa
C6H9N8O8 М. в. 171,16
6-Диазо-5-кето- L-норлейцин (ДОН) применяется в каче-
стве ингибитора синтеза белка.
За основу синтеза ДОН'а была взята литературная про-
пись получения [I, 2, 3], но мы улучшили синтез, внеся ряд
изменений, а именно: ангидрид М-трифторацетил-Ь-глутами-
новой кислоты не выделялся, а сразу превращался в 5-Ди-
циклогексиламмониевую соль 1-этилового эфира N-трифтор-
аиетил-Ь-глутаминовой кислоты, были также изменены усло-
вия выделения этой соли и самого 6-диазо-5-кето-Ь-норлей-
цина.
Необходимый для синтеза трифторуксусный ангидрид по-
лучали перегонкой трифторуксусной кислоты над фосфорным
ангидридом [5].
СХЕМА СИНТЕЗА 6 ДИАЗО-5-КЕТО-L-НОРЛ ЕИЦИНА
2 CFSCOOH 4- Р2О, * CF3CO.
)О+2НРОЧ
CFSCO
46
НООС-СН-СНа-СООН 4- CFSCO,
| -4 CaH6OH 4-
NH., CF3COz
+ (CeH,i)aNH +
H5C2OOC-CH-CsH4-CH,-COOH-NH(CeHn),+
NHCOCFS
+ CF3COOH + H3O
HjCsOOC—CH—CH„—СН» -COOH-NH(CeHn)4 4- SOC12 -*
I
NHCOCF»
-> H6C2OOC-CH-CH2-CHt—coci + so2 +
NHCOCF3
+ (C6H„)3NH-HC1
H5C2OOC-CH—CH2-CH2-COC1 + CH3NS ->
I
NHCOCF3
-> HjC3OOC-CH-CH2-CH2-CO-CHN2 + HC1
NHCOCFg
H5C2OOC-CH-CH2-CH2 -CO—CHN2 4- NaOH
NHCOCF.
HOOC-CH—CH3-CH2-CO-CHNa 4- C3H6ONa
nh2
Характеристика основного сырья
Трифторуксусная кислота, т. кип. 71—72°.
Фосфорный ангидрид, ч.д.а., ВТУ 521—41.
L-Глутаминовая кислота, ч., ВТУМЗ 1605—52.
Хлористый тионил, ч., ВТУ МХП 3591—52.
Катионит КУ-2.
Дициклогексиламин [4], т. кип. 105—109°/7 мм, «д—1,489.
Нитрозометилмочевина [7], т. пл. 123—124°.
Диазометан [8], эфирный раствор.
47
Условия получения
Синтез трифторуксусного ангидрида. В двугорлую кругло-
донную колбу, соединенную с капельной воронкой и со зме-
евиковым холодильником, помещают 200 г (1,4 М) фосфор-
ного ангидрида, сразу же приливают 80 г (0,7 М) трифтор-
уксусной кислоты и смесь осторожно нагревают на водяной
или глицериновой бане до 100". В течение 1 —1,5 часа весь
трифторуксусный ангидрид отгоняется. Его еще раз перего-
няют с небольшим дефлегматором.
Выход трифторуксусного ангидрида равен 70 г, что со-
ставляет 90% от теоретического; т. кип. 39—40°.
По литературным данным, т. кип. вещества 39,5° [6].
Получение 5-дициклогексиламмониевой соли 1-этилового
эфира П-трифторацетил-Ь-глутаминовой кислоты. 17 г
(0,1 М) L-глутаминовой кислоты (высушенной в сушильном
шкафу при 120°) и 44 мл (0,20 (И) трифторуксусного ангид-
рида кипятят 30 минут с обратным холодильником до обра-
зования прозрачного раствора. Затем в вакууме при темпе-
ратуре бани около 50° отгоняют образовавшуюся трифтор-
уксусную кислоту и избыток трифторуксусного ангидрида.
Охлаждают и добавляют 102 мл абсолютного этилового спир-
та. Реакционной смеси дают стоять 1 час при комнатной тем-
пературе, кипятят 1 час и отгоняют в вакууме спирт. К ос-
татку добавляют 25 мл (1,4 Л1) дициклогексиламина, при
этом основная масса кристаллизуется в виде соли. Получен-
ную пастообразную массу растворяют в 50 мл бензола и
оставляют до следующего дня. Затем продукт отфильтровы-
вают, промывают петролейным эфиром, сушат на воздухе и
перекристаллизовывают из 400 мл воды с активированным
углем.
Выход 5-дициклогексиламмониевой соли 1-этилового эфи-
ра К’-трифторацетил-Ь-глутаминовой кислоты равен 29 г, что
составляет 65% от теоретического; т. пл. 191°.
По литературным данным, т. пл. вещества 189° [1].
Получение 5-хлорангидрада-1-этилового эфира N-трифтор-
ацетил-Ь-глутаминовой кислоты [5]. В колбу с обратным хо-
лодильником, на конце которого имеется хлоркальциевая
трубка, помещают 12,8 г (0,028 М) дициклогексиламмоние-
вой соли, 280 мл абсолютного бензола, 28 мл (0,33 М) хло-
ристого тионила и смесь кипятят 1,5 часа. Охлаждают до
0° и оставляют на 1 час при этой температуре. Отфильтро-
вывают образовавшийся дициклогексиламмонийхлорид.
Фильтрат упаривают досуха в вакууме водоструйного насоса.
Чтобы удалить весь хлористый тионил, остаток растворяют
дважды в 250 мл абсолютного бензола и каждый раз испаря-
ют бензол в вакууме. Получают кристаллический продукт
белого цвета.
48
Выход вещества равен 6,3 г, что составляет 80% от теоре-
тического.
Следующую стадию, реакцию диазотирования, следует
проводить сразу после получения хлорангидрида, так как
хлорангидрид неустойчив и уже на следующий день стано-
вится маслообразным.
Получение 1-этилового эфира &'-трифторацетил-6-диазо-5-
-кето-Ь-норлейцина. Растворяют 8 г (0,028 А4) 5-хлорангид-
рида 1-этилового эфира Ы-трифтораи.етил-Е-глутаминовой
кислоты в 100 мл эфира и вносят туда при температуре 5°
эфирный раствор диазометана (диазометан получают из 14 г
нитрозометилмочевины). По окончании выделения азота
оставляют стоять еще 1 час и отгоняют эфир в вакууме. По-
лучают зеленовато-желтое масло.
Выход вещества равен 6 г, что составляет 73% от теоре-
тического.
Получение 6-диазо-5-кето-Ь-норлейцина (ДОНа). К раст-
вору 6 г 1-этилового эфира М-трифторацетил-6-диазо-5-кето-
-L-норлейцина в 7 мл этилового спирта добавляют 60 мл 1 н.
едкого натра и встряхивают 30 минут при температуре в пре-
делах от 0 до 5°. Эфир, выпадающий при добавлении едкого
натра, снова переходит в раствор. Раствор подкисляют ка-
тионитом КУ-1 до pH 6 (по индикаторной бумаге «Рифан»),
на что уходит около 30 мл вещества. Раствор отделяют от
катионита, встряхивают с небольшим количеством активиро-
ванного угля и фильтруют. Фильтрат упаривают в вакууме
масляного насоса. К остатку добавляют ацетон до полного
осаждения 6-диазо-5-оксо-1_-норлейцина, который выпадает в
аморфном виде. Его очищают, растворяя в минимальном ко-
личестве воды и осаждают ацетоном (соотношение вода : аце-
тон = 1:3). Продукт отфильтровывают под вакуумом и су-
шат на воздухе.
Выход 6-диазо-5-оксо-Ь-норлейцина в виде аморфного
порошка светло-желтого цвета равен 1,65 г, что составляет
47% от теоретического.
Найдено, %: С—42,10; 42,01; Н—5,30; 5,45.
С6Н.,\'3О,. Вычислено, %: С—42,08; 11—5,30.
ЛИТЕРАТУРА
1. F. W е i g a n d, R. G е i g е г. Cheni. Вег., 90, 636 (1957).
2. F. W е 1 g а п d, М. R е i h е г. Chem. Вег., 88, 31 (1955).
3. F. W е i g a n d, R. G е i q е г. Chem. Вег., 90, 637 (1957).
4. А. Н. Кост. И. И. Г р а и д б е р г. Ж. обш. химии, 25, 1432 (1955).
5. F. Swarts. Bull, acad. ray. Belg., 8, 343, (1922).
6. F. Swarts. Bull. acad. ray. Belg., 8, 368 (1922).
7. B. Efstert. Angew, Chem., 54, 124 (1941),
8. C6. «Синтез органических препаратов», выл. 2, М„ ИЛ, 1949,
стр. 174.
Поступила в августе 1965 г-
4 Зак. 25
Институт химии
природных соединений
АН СССР
49
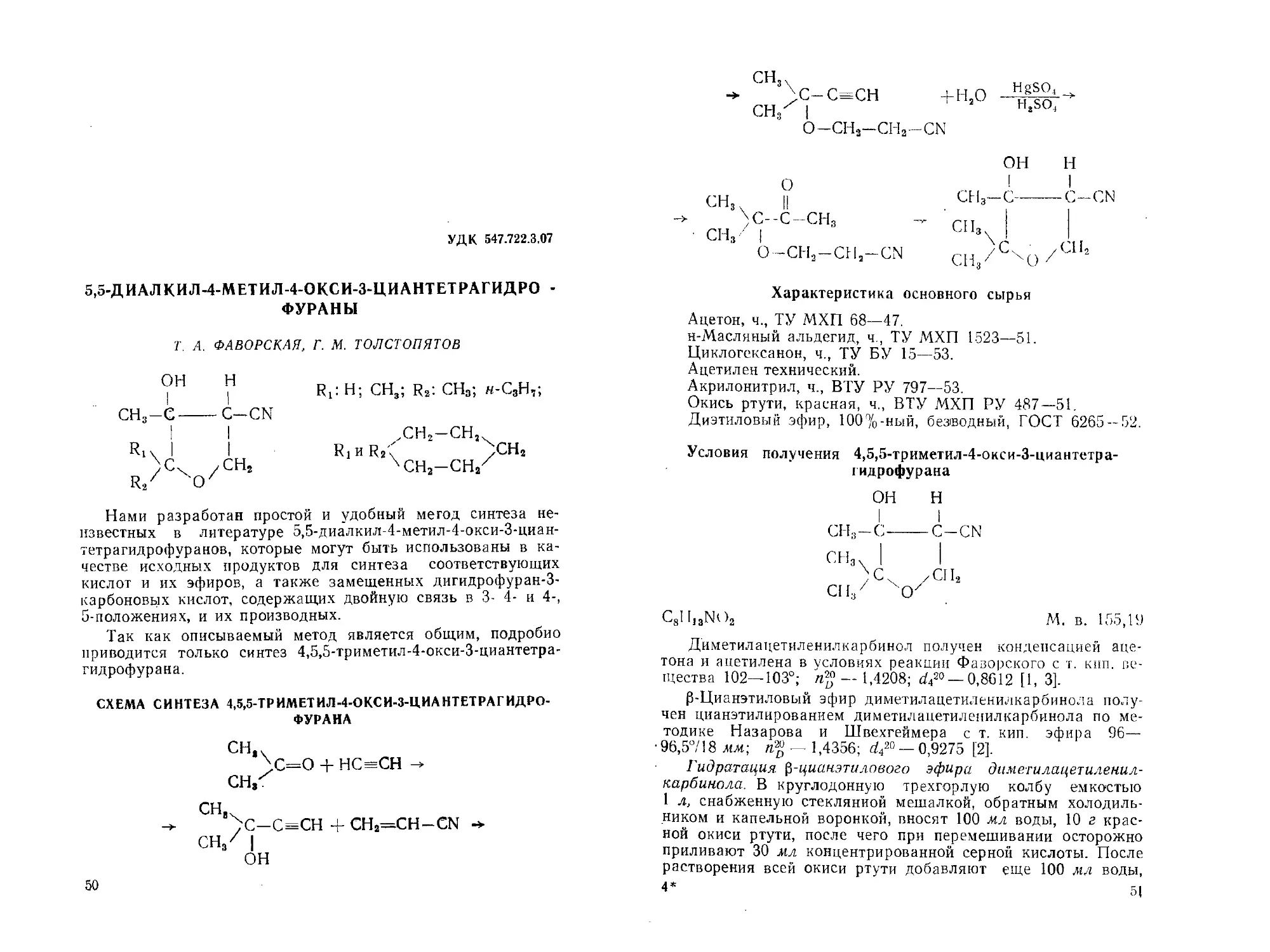
УДК 547.722.3.07
5,5-ДИАЛКИЛ-4-МЕТИЛ-4-ОКСИ-3-ЦИАНТЕТРАГИДРО -
ФУРАНЫ
Т. А. ФАВОРСКАЯ, Г. М. ТОЛСТОПЯТОВ
ОН н
I I
СН3-С------C-CN
I I
Ri4 I I
)сч ,сн2
R2Z 'О7
Rj! Н; СН3; R2*. CHgj H-CgH2j
ХН..-СН,.
R1 и Rg \ ,CH2
чсна-сн/
Нами разработан простой и удобный метод синтеза не-
известных в литературе 5,5-диалкил-4-метил-4-окси-3-циан-
тетрагидрофуранов, которые могут быть использованы в ка-
честве исходных продуктов для синтеза соответствующих
кислот и их эфиров, а также замещенных дигидрофуран-3-
карбоновых кислот, содержащих двойную связь в 3- 4- и 4-,
5-положениях, и их производных.
Так как описываемый метод является общим, подробно
приводится только синтез 4,5,5-триметил-4-окси-3-циантетра-
гидрофурана.
СХЕМА СИНТЕЗА 4,5,5-ТРИМЕТИЛ-4-ОКСИ-3-ЦИАНТЕТРАГИДРО-
ФУРАНА
сн,.
>с=о + нс=сн -+
СН3Х
СН,
-> ;С-С=СН + CH2=CH-CN
50
снзх
\С-С=СН +Нао
СН^ I
О—СН3—СН2—CN
Hgso4
HtSO4
о
сн3 II
)С--С-СН3
сн3 I
О -СН,—СНа—CN
он н
Характеристика основного сырья
Ацетон, ч., ТУ МХП 68—47.
н-Масляный альдегид, ч., ТУ МХП 1523—51.
Циклогексанон, ч., ТУ БУ 15—53.
Ацетилен технический.
Акрилонитрил, ч., ВТУ РУ 797—53.
Окись ртути, красная, ч., ВТУ МХП РУ 487—51.
Диэтиловый эфир, 100%-ный, безводный, ГОСТ 6265 — 52.
Условия получения 4,5,5-триметил-4-окси-3-циантетра-
гидрофурана
ОН н
I I
СНз-С-----C-CN
СНа. | I
;с. /Сн2
СН/ оу
C8II13NO2 М. в. 155,19
Д'иметилацетиленилкарбинол получен конденсацией аце-
тона и ацетилена в условиях реакции Фаворского с т. кип. ве-
щества 102—103°; /г® — 1,4208; — 0,8612 [1, 3].
р-Цианэтиловый эфир диметилацетиленилкарбинола полу-
чен цианэтилированием диметилацетиленилкарбинола по ме-
тодике Назарова и Швехгеймера с т. кип. эфира 96—
96,5718 лш; Д» — 1,4356; Д,20 —0,9275 [2].
Гидратация р-цианэтилевого эфира диметилацетиленил-
карбинола. В круглодонную трехгорлую колбу емкостью
1 л, снабженную стеклянной мешалкой, обратным холодиль-
ником и капельной воронкой, вносят 100 мл воды, 10 г крас-
ной окиси ртути, после чего при перемешивании осторожно
приливают 30 мл концентрированной серной кислоты. После
растворения всей окиси ртути добавляют еще 100 мл воды,
4* 51
охлаждают содержимое колбы до комнатной температуры и
приливают по каплям раствор 130 г (около 1 Л4) р-цианэти-
лового эфира диметилацетиленилкарбинола в 200 жл диэти-
лового эфира со скоростью, обеспечивающей равномерное сте-
кание эфира из обратного холодильника. Перемешивание
должно быть энергичным и равномерным во избежание бур-
ного вскипания эфира. После прибавления всего раствора
р-цианэтилового эфира диметилацетиленилкарбинола пере-
мешивание продолжают еще 4 -6 часов. Эфирный слой отде-
ляют, водный слой 6—8 раз экстрагируют эфиром. Эфирный
слой и вытяжки объединяют, промывают концентрированным
раствором соды и сушат над прокаленным сернокислым маг-
нием. После отгонки эфира остаток перегоняют в вакууме.
Выход р-цианэтилового эфира диметилацетилкарбинола
равен 100 г, что составляет 65% от теоретического; т. кип.
95—9672 мм; nf- 1,4361; z/2'7 — 1,0000.
Найдено, %: С-61,75; 61,84; Н—8,54; 8,48; N—9,07; 9,15;
MR/; —40,54.
C8H„NO2. Вычислено, %; С- 61,93; Н—8,38; N—9,02; MRD—40,62.
Циклизация fi-цианэтилового эфира диметилацетилкарби-
нола. В литровую колбу, снабженную мешалкой, капельной
воронкой и обратным холодильником с хлоркальциевой труб-
кой, вносят 750 мл безводного диэтилового эфира и 40 г су-
хого порошкообразного едкого кали. При сильном перемеши-
вании постепенно прибавляют 70 г (около 0,5 М) р-цианэти-
лового эфира диметилацетилкарбинола, при этом суспензия
светлеет и постепенно образуется рыхлый комплекс, оседаю-
щий на стенках и дне колбы в виде вязкой смолы. Переме-
шивание продолжают еще 2 часа, после чего смесь разлага-
ют 100 -150 мл воды до полного растворения щелочи, вхо-
дящей в комплекс. Водный слой многократно экстрагируют
эфиром, эфирный слой и вытяжки объединяют, нейтрализуют,
пропуская углекислый газ, и сушат над прокаленным серно-
кислым магнием (или сернокислым натрием).
После отгонки эфира из колбы с дефлегматором остается
иысококипящее, постепенно кристаллизующееся вещество, ко-
торое очищают перегонкой в вакууме или перекристаллиза-
цией из эфира.
Выход 4,5,5-триметил-4-окси-3-циантетрагидрофурана ра-
вен 61- 62 г, что составляет 78—80% от теоретического;
т. кип. 125—12676 мм; т. пл. 96,5—97,5° (см. примечание 1).
Для анализов 4,5,5-триметил-4-окси-3-циантетрагидрофу-
ран очищают многократной перекристаллизацией нз эфира.
Найдено, %: С 61,96, 61,88; Н—8,91; 8,65; N-9,06; 9,10.
С8Н13\’О5, Вычислено, %: С—61,93; Н-8,38; N-9,02.
Перекристаллизованный продукт представляет собой бе-
лое кристаллическое вещество, умеренно растворимое в эфи-
ре, хорошо в спирте и ацетоне.
52
Условия получения 4-метил-5-н-пропил-4-окси-3-
циантетрагидрофурана
ОН н
! !
СНз-С— С-СК
C9H15NO2 М. в. 169,22
н-Пропилацетиленилкарбинол получают конденсацией
н-масляного альдегида и ацетилена тю способу Фаворской [3];
т. кип. вещества 87—88°/Ю0 мм; — 1,4382; d204— 0,8710.
р-Цианэтиловый эфир н-пропилацетиленилкарбинола обра-
зуется путем цианэтилирования спирта; т. кип. 90—91°/6,5 мм;
— 1,4405; d2\~- 0,9243.
Гидратация $-цианэтилового эфира н-пропилацетиленил-
карбинола (75 г, 0,5 М) проводится в тех же условиях, что и
для р-цианэтилового эфира диметилацетиленилкарбинола.
Выход р-цианэтилового эфира н-пропилацетилкарбинола
равен 55—57 г, что составляет 65—68% от теоретического;
т. кип. 112—11373 мм; «о —1,4382; d2°4—0,9781.
Найдено, %: С—64,47; 64,68; Н-8,96; 9,04; N—8,30
8,28; MRp 45,38.
C„H15NO=. Вычислено, %: С-64,49: Н-8,88; N-8,28; MRD—45,23
Реакция циклизации $-цианэтилового эфира н-пропилаце-
тилкарбинола проводится аналогично циклизации р-цианэти-
лового эфира диметилацетилкарбинола. Верут 67 г (0,25 Л4)
Р-цианэтилового эфира н-пропилацетилкарбинола, 30 г едко-
го кали, 350 мл безводного эфира.
Выход 4-метил-5-н-пропил-4-окси-3-циантетрагидрофура-
на равен 42 г, что составляет 62% от теоретического; т. кип.
139—14076 мм (с разя.); п™ - 1,4776; d204 — 1,0505.
Найдено, %: С-64,65 , 64,90; Н-9,02; 9,14; N—8,32; 8,37;
MR^—44,74.
C9Hir>NO2. Вычислено, %: С—64,49; Н—8,88; N—8,28; MR0— 44,55.
Условия получения 4-метил-5,5-пентаметилен-4-окси-3-циап-
тетрагидрофурана
ОН Н
I I
СН3-С—C-CN
CnH17N'O2
М. в. 195,26
53
Ацетиленилциклогексанол получают конденсацией цикло-
гексанола и ацетилена в условиях реакции Фаворского [1, 3].
Т. кип. 74—76°/12 мм; т. пл. 22° (см. примечание 2).
Реакцию цианэтилирования проводят по вышеуказанной
методике [2]. Для синтеза р-цианэтилового эфира ацетиленил-
циклогексанола берут 124 г (1 А4) спирта, 350 мл диоксана,
53 г (1 М) акрилонитрила и в качестве катализатора 1,5 г
метилата натрия.
Выход продукта реакции равен 107 г, что составляет 64%
от теоретического; т. кип. 108—110/°3 мм; По— 1,4732;
(Р\ — 0,9884.
Найдено, %: С-7+,42; 74,39; Н-8,34; 8,63; N-8,27;
8,04; MR0 - 50,23.
Cj.H^NO, Вычислено, %: С-74,57; Н-8,51; N—7,91; MRo-50,26.
Реакция гидратации fi-цианэтилового эфира ацетиленил-
циклогексанола проводится в тех же условиях, что и для
Р-цианэтилового эфира диметилацетиленилкарбинола. Для
гидратации берут 5 г красной окиси ртути, 30 мл концентри-
рованной серной кислоты, 350 мл воды, 175 г (1 М) |3-циан-
этилового эфира ацетиленилциклогексанола.
Выход р-цианэтилового эфира ацетилциклогексанола ра-
вен 130 г, что составляет 69% от теоретического, т. кип. ве-
щества 128—13071 мм;пь — 1,4700; di20-- 1,0113.
Найдено, %; С-67,89; 67,74; Н-9,19; 9,09; N-6,95;
6,86; MR0 — 53,80.
CuHi7NO2. Вычислено, %: С-67,69; Н—8,72; N-7,18; MR0 —52,27.
Циклизация fi-цианэтилового эфира ацетилциклогексанола
проводится в условиях, аналогичных описанным выше. Бе-
рут 48 г (0,4 Л1) р-цианэтилового эфира, 60 г едкого кали,
600 мл безводного эфира. Продукт реакции перекристаллизо-
вывают из петролейного эфира.
Выход 4-метпл-5,5-пентаметил ен-4-окси-З-циантетрагидро-
фурана равен 39 г, что составляет 80% от теоретического;
т. пл. 103—104°.
Найдено, %: С-67,90; 67,80; Н-9,20; 9,07;
N-7,34; 7,20.
CnH17NO2 Вычислено, %; С -67,69; Н-8,72; N—7,18.
Продукт представляет собой белое кристаллическое веще-
ство, хорошо растворимое в органических растворителях, не-
растворимое в воде.
Примечания:
1. При увеличении количества диэтилового эфира н охлаждении ре-
акционной смеси до 0° (с разложением комплекса при комнатной темпе-
ратуре) выход вещества несколько увеличивается.
2. В положение 5-тетрагидрофуранового кольца могут быть введены
также и ароматические радикалы, однако пока не достигнуто достаточной
воспроизводимости в условиях реакции и выходах промежуточных и ко-
нечных продуктов.
54
ЛИТЕРАТУРА
1. А. Е. Фаворский, А. С. Онищенко. Ж. общ. химии, 11, 1111
(1941).
2. И. Н. Назаров, Г. А. Ш в е х г е й м е р. Ж. общ. химии, 24, 157
(1954); там же, 25, 504 (1955).
3. И. А. Фаворская, Э. М. А у в и н е и Ю. Г1. Арцы б а ш ев а.
Ж. общ. химии, 28, 1785 (1958).
Поступила в декабре 1965 г. ЛГУ
УДК 547.636.2,07
ДИБЕНЗИЛОВЫЙ ЭФИР
Л. я. ДИСТАНОВА
^\-СН2-О-СН2-Ч\
Ч/ ч/
С14Н14О М.в. 198,26
Дибензиловый эфир как хороший растворитель жиров и
масел находит широкое применение в органическом синтезе
и в промышленности.
В литературе описан ряд методов его получения [1—7].
С практической стороны наибольший интерес представля-
ет классический метод получения эфира из Спирта с приме-
нением серной кислоты как водоотнимающего средства.
Однако выход продукта по этому методу не стабилен
вследствие одновременно протекающих побочных реакций,
приводящих к его снижению.
Замена серной кислоты на более мягкий катализатор, на-
пример n-толуолсульфокислоту, себя не оправдывает, по-
скольку выход продукта при этом повышается лишь незначи-
тельно.
Нами разработана методика получения дибензилового
эфира с применением бензилата калия и бензила хлористого.
СХЕМА СИНТЕЗА ДИБЕНЗИЛОВОГО ЭФИРА
f \-СН2ОН С1СН,^ \ 1К0Н1
Ч/ ч./1
и сн, | (| +Hg0 + КС1
Ч/ Ч/
56
Характеристика основного сырья
Бензиловый спирт, ч., ГОСТ 8751—58.
Бензил хлористый, техн., ТУ МХП 588—49.
Кали едкое, марки А, ГОСТ 9285—59.
Кальций хлористый, те.хн., ГОСТ 450—58.
Условия получения
В литровую колбу с мешалкой, обратным холодильником,
термометром и капельной воронкой загружают 364 г (350 мл;
3.38 Л4) бензилового спирта и 266 г едкого кали. Смесь нагре-
вают при 120—130° в течение 2—3 часов до полного раство-
рения щелочи. Затем массу охлаждают до 80э и прибавляют
364 г (331 мл; 2, 88 М) хлористого бензила. После добавки
всего хлористого бензила смесь выдерживают при 120—130"
еще в течение 3—4 часов, охлаждают, отфильтровывают от
осадка, промывают, сушат.
Технический продукт очищают перегонкой в вакууме при
168—169°/20 мм.
Выход продукта, соответствующего ТУ МХП 3026—51,
равен 364 г, что составляет 54,5% от теоретического; т кип.
296,46; d — 1,040.
ЛИТЕРАТУРА
1. J. Leitner, В. Tara soft. Вег., 43. 943 (1910).
2. J. Meisenhelmer. Вег., 41, 1421 (1908).
3. L. S z р е г I, Т. W 1 е г u s z-K о w а 1 s к 1. Chem. Abstrs, 1, 909 (1918),
4. S. Cannizzaro. Ann. Chem., 92 113 (1864).
5. C. W. Lowe. Ann. Chem., 241, 374 (1887).
6. H. L im p r t c h t. Ann. Chem., 139.313 (1866).
7. R. Wegscheider. Mh. Chem., 21, 634 (1900).
Поступила в июле 1965 г.
ЦЗЛ
УДК 547.535.1.07
1,4-ДИМЕТИЛ-2-ИЗОПРОП И Л БЕНЗОЛ И 1,4-ДИМЕТИЛ-
-2,5-ДИИЗОПРОПИЛ БЕНЗОЛ
И. Г. ГАХ, Е. П. БАБИН, Л. Г. ГАХ
СИз /СН3
Агс\н
Y СН3
СН3
СН3
ОК
сн-
сн3
СИН16 М.в. 148,24
CUH2S М.в. 192,34
Алкилированием n-ксилола в присутствии катализаторов
Фриделя-Крафтса обычно получают трудноразделимую смесь
изомеров [1, 2].
Нами предлагается метод получения 1,4-диметил-2-изо-
лропилбензола и 1,4-диметил-2,5-диизопропилбензола посред-
ством алкилирования n-ксилола пропиленом в присутствии
катионообмеиной смолы КУ-2 как катализатора.
СХЕМА СИНТЕЗА ИЗОПРОПИЛКСИЛОЛОВ
сн3 CHS сн.
КУ-2 КУ-2
| || +С4Н.----Ч || + С3Н6----
Y у сп,
сн, сн3
Y сна
_сн3
CH^Y СНз
58 СН^ СН»
Характеристика основного сырья
rt-Ксилол, ЧМ ТУ 3601—53.
Катионит КУ-2, ГОСТ 5695—53, переведенный в Н-форму
и высушенный.
Пропилен, газообразный из баллона.
Условия получения
В колбу, снабженную механической мешалкой, обратным
Холодильником, термометром и барботером для пропускания
газа, помещают 106 г я-ксилола и 50 г катионита КУ-2. При
температуре 140° в колбу при сильном перемешивании подают
пропилен со скоростью около 3 л в час. Для получения моно-
изопропилксилола пропускают пропилен до тех пор, пока при-
вес реакционной массы будет равен 42 г. В этом случае алки-
лат состоит из 40% ксилола, 40% моноизопропилксилола и
20% диизопропилксилола. Если же поглощение пропилена
довести до 25 г (0,6 Л'1), то алкилат будет содержать 60%
ксилола, 30% моноизопропилксилола и 10% динзопропилкси-
лола. Для получения диизопропилксилола реакцию доводят
до прекращения поглощения пропилена реакционной массой
(55 г). В этом случае алкилат содержит 35% ксилола, 25%
моноизопропилксилола и 40% диизопропилксилола. Обработ-
ка реакционной массы после охлаждения заключается в отде-
лении катализатора (декантацией или фильтрованием) и
фракционной перегонке алкилата. При перегонке отбирают
следующие фракции:
1) 134—135°—ксилол; п$ — 1,4940;
2) 135—193°—промежуточная фракция:
3) 193—196° - 1,4-диметил-2-изопропилбензол; л® —
— 1,5010;
4) 196—233°—промежуточная фракция;
5) 233—236°—1,4-диметил-2,5-диизопропилбензол; «д—
- 1,5046.
Полученные 1,4-диметил-2-изопропилбензол и 1,4-диметил-
-2,5-диизопропилбензол не загрязнены другими изомерами.
1,4-Диметил-2-изопропилбеизол представляет собой бес-
цветную подвижную жидкость с температурой кипения
194—1957751 мм; n2j— 1,990; — 0,8712.
По литературным данным, т. кип. вещества 196,2° [1, 4);
nf — 1,4990 [1]; 1,4988 [4]; d^ — 0,8721 [1]; 0,8699 [4].
1,4-Диметил-2,5-диизопропилбензол представляет собой
кристаллическое вещество белого цвета с температурой
плавления 38—39° и температурой кипения 2367751 мм.
По литературным данным, т. пл. вещества 37,4° [2]; 37,2°
П, 3]; т. кип. 243,8° [1]; 244° [2].
59
ЛИТЕРАТУРА
1. Е. W. Kirkland, О. Р. Funderburck, F. Т, WadswortIt.
J. Organ. Chem., 23, 1G31 (1958).
2. А В. T о и ч и e в, P. H. В о л к о в С. В. За второ д и н ft. Доил.
АН СССР, 134, 844 (1960).
3. Е. С. Kooyman, A. Strang, Rec. trav. chirnog., 72, 329 (1953).
4. F, D. Rossini, K. S. Pitzer. R. 1.. A r u e t, R. M. Braun,
Q, C. Pimentel. Selected Values of Phisical and Thermodynamic Pro-
perties of Hydrocazhons and Related Compounds, Carnegie Press, Pittsburg,
1953,
Поступила в ноябре 1965 г. Донецкий филиал ИРЕА
УДК 547.551.5.07
М,М-ДИМЕТИЛ-о-НИТРОАНИЛИН
Р. П. ЛАСТОВСКИЙ, В. Я. ТЕМКИНА. Л. М. САМЫЛОВА
,сн3
N(
\снз
L А
X^/X'NO,
М.в. 166,17
c8h10n2o2
Из описанных б литературе способов получения N,N4m-
метилнитроанилинов практическое значение имеют 2 метода:
метилирование нитроанилина метилйодидом [1— 2] и взаимо-
действие нитрохлорбензола с диметиламином [3.] Второй ме-
тод, по данным автора, дает более высокий выход и хорошее
качество продукта.
Нами проверен второй метод получения и уточнены усло-
вия выделения о-изомера, заключающиеся в удалении раст-
ворителя и вакуум-нерегонке вещества.
СХЕМА СИНТЕЗА N,N -ДИМЕТИЛ- о-НИТРОАНИЛИНА
/СН8
/Ч ,N(
NH(CH3)2 ^\/ \сн3
--------> I II
4/4no2
Характеристика основного сырья
о-Нитрохлорбензол, ТУ МХП 2167—49.
Диметиламин солянокислый, ВТУ МХП 2885— 51.
Пиридин, ч., ГОСТ 2747—44.
61
Условия получения
Смесь 31,5 г (0,2 Л1) о-нитрохлорбензола, 300 мл пириди-
на и 50 г (0,6 М) двууглекислого натрия помещают в кругло-
донную колбу на 500 мл. К этой смеси прибавляют 27 г
(0,6 М) солянокислого диметиламина, растворенного в 10 мл
воды. Смесь кипятят с обратным холодильником в течение
10 часов. Затем горячий раствор отфильтровывают от неорга-
нической соли и осадок промывают 200 мл ацетона, который
присоединяют к фильтрату. Из полученного раствора отго-
няют растворитель и остаток перегоняют в вакууме при
20 мм, отбирая фракцию с температурой кипения 149—150°.
Получают 21 г М,М-диметил-о-нитроанилина, что составля-
ет 85% от теоретического выхода; n‘ff—1,6089.
По литературным данным, т. кип. продукта при 20 мл
149°; п™— 1,6080 [3].
ЛИТЕРАТУРА
1. Е. Bamberger, F. Tschirner, Вег., 32,1897, (1902).
2. Р. Friedlaender, М. 19,636, (1898).
3. Т. W. Campbell. J. Amer. Chem. Soc., 71,740(1949).
Поступила в декабре 1965 г.
ИРЕА
УДК 547.831.7.04
5,7-ДИМЕТИЛ-8-ОКСИХИНАЛЬДОКСИМ
Оксим 5,7-диметил-8-оксихинальдинового
альдегида
В. М. ДЗИОМКО, И. А. КРАСАВИН, Т. Н. ЕГОРОВА
СНз
AAnA
СН3 | 14 CH=NOH
ОН
C12H13N2O3
М.в. 216,24
В литературе 5,7-диметил-8-оксихинальдоксим не описан.
Он легко образуется из 5,7-диметил-8-оксихинальдинового
альдегида и гидроксиламина с почти количественным выхо-
дом и представляет собой желтые иглы (из этанола) с т. пл.
204—204,5°, нерастворимые в воде, но растворяющиеся во
многих органических растворителях.
5,7-Диметил-8-оксихинальдоксим является хелатообразую-
щим соединением. В щелочной среде он образует с солями
стронция комплекс, обладающий желтой люминесценцией в
ультрафиолетовых лучах и значительно менее растворимый,
чем кальциевый комплекс.
Получение 5,7-диметил-8-оксихинальдоксима
СН3
I II I
ch/Y4nZ4cho
+NH,OH
он
63
+на0
CH=NOH
В круглодонной колбе емкостью 250 мл с обратным холо-
дильником растворяют 2,0 г (0,01 М) 5,7-димети.ч-8-окси-
хннальдинового альдегида (см. примечание 1) в 35 мл чисто-
го этанола при нагревании на водяной бане.
Раствор 1,0 г (0,014 А4) солянокислого гидроксиламина
(ч. д. а.) в 4 мл дистиллированной воды точно нейтрализуют
30%-ным раствором едкого кали (х. ч.) и сразу прибавляют
к раствору альдегида. Смесь кипятят в течение 1 часа, затем
охлаждают и разбавляют образовавшуюся суспензию ди-
стиллированной водой (150 мл). Осадок отсасывают, промы-
вают водой н высушивают в вакуум-эксикаторе над едким
натром.
Выход оксима 2,0—2,1 г, что составляет 93,1—97,7% от
теоретического; т. пл. 200—201°. После перекристаллизации
из 25—30 мл этанола получают 1,6—1.7 г вещества с т. пл.
203—204°, которая при дальнейшей очистке повышается до
204- 204,5' (см. примечание 2).
Примечания:
1. Применялся 5,7-днметил-8-оксихинальдиновый альдегид с т. пл.
114,5—115°, полученный по описанному нами ранее методу и очищен-
ный посредством испарения его ацетоновою раствора, а затем перекрис-
таллизованный из циклогексана [1].
2. Чистый образец 5,7-диметил-8-оксихинальдоксима в виде желтых
игольчатых кристаллов с т. пл. 204—204,5“ был приготовлен перекристал-
лизацией из этанола и проанализирован.
Найдено, %: С -66,67; 66,5’; Н-5,92; 5.88; N-12,99; 13,12.
ClaH1;N,O2. Вычислено, %: С-66,65; Н-5,59; Н-12,96.
Л И Т Е Р А Т У Р А
1. И. А. Красавин, В. М. Дзиомко, Т. Н. Егорова. Методы
получения химических реактивов и препаратов, вып. 13, М., ИРЕА, 1965,
стр. 34.
Поступила в дакабре 1965 г. ИРЕА
УДК 547.642.07
3,3-ДИМЕТИЛСПИРО -[БЕНЗОКСАЗОЛИН-2,2' -<2'Н-1' -
БЕНЗОПИРАНЫ)]
Д. А. ДРАПКИНА. В. Г. БРУДЗЬ. В. А. ИНШАКОВА,
Н. И. БАДАИКОВА
сн3
I СНз
/
R"
Спиропираны представляют интерес в качестве фотохром-
ных веществ [1]. Идентичные прописи получения спиропиранов
этого ряда даны в английском и американском патентах [1]
без указания выходов и характеристик полученных продук-
тов. Нами проверен указанный метод и несколько уточнен
способ выделения спиранов. Применяя различные замещен-
ные салициловые альдегиды, мы получили ряд производных
3,3'-диметилспиро - [бензоксазолин-2,2'-(2/Н-1/-бензопирапа)].
Все полученные спиропираны и промежуточный продукт —
п-толуолсульфонат-2-этил-З-метилбензоксазолиния — оха-
рактеризованы температурой плавления и данными элемент-
ного анализа.
СХЕМА СИНТЕЗА 3,3'-ДИМЕТИЛСПИРО-
[БЕНЗОКСАЗОЛ ИН-2,2'-(2'Н-1'-БЕНЗОПИРАНОВ)]
СНз
\ II
z>\ /N (
I II Ч_СН.ГЦ. SO2OCH3
5 Зак. 25 О 65
сно
Л°н
R'/ \/\R„
пиридин, пиперидин
Характеристика основного сырья
2-Этилбензоксазол, получают по [1 j или [2].
.Метиловый эфир л-толуолсульфокислоты, ч., ВТУ РУ
G37- 52.
Ацетон, ч.д.а., ГОСТ 2603—51.
Пиридин, ч., ГОСТ 2747—44.
Пиперидин, ч„ ВТУ РУ 444-51.
5-Нитросалициловый альдегид, ч., ТУ TCP 563р—61.
З-Бром-5-нитросалициловый альдегид (получение см. в на-
стоящем сборнике).
5-Хлор-З-нитросалициловый альдегид (получение см. в на-
стоящем сборнике).
З-Метокси-5-нигросалициловый альдегид (получение см.
г. настоящем сборнике).
5-Бром-З-нитросалициловый альдегид (получение см. в
[3]).
Спирт этиловый, синтетический. 1ОСГ 9674—61.
Условия получения
Получение п-толуолсульфонат-2-этил-З-метилбензоксазолиния
СН3
А
II ^-СН2СН3
+
SOCeH4CH3
В трехгорлую колбу, снабженную механической мешал-
кой, обратным холодильником и капельной воронкой, загру-
жают 5 а (0,034 Л1) 2-этилбензоксазола и медленно при раз-
мешивании добавляют эквимолярное количество метилового
GG
эфира л-толуолсульфокислоты (6,4 г). Смесь нагревают на
глицериновой бане и выдерживают 2 часа при температуре
бани 150°. После примерно 40-минутного нагревания реакци-
онная масса затвердевает и перемешивание прекращают. По
окончании выдержки смесь охлаждают до комнатной темпе-
ратуры н растирают с 50 мл сухого ацетона, фильтруют, про-
мывают ацетоном до получения бесцветного фильтрата и су-
шат в вакуу.м-эксикаторе.
Получают 10—10,2 г вещества, что составляет 88,5—90,2%
от теоретического выхода; т. пл. 188,5—190°.
Найдено, %: S-9.4
CJ7HJ9NO4S. Вычислено, 0>'с: S—9,6
Получение 3,3'-диметилспиро-\бензоксазолин-2,2'-(2'Н-1'-
бензопиранов)]. В двугорлую колбу на 50 мл, снабженную
мешалкой и обратным холодильником, помещают 0,01 М со-
ответствующего салицилового альдегида и 17 мл пиридина в
качестве растворителя; при размешивании добавляют 0,01 Л-1
п-толуолсульфоната 2-этил-З-метилбензоксазолиния и 0,7 мл
пиперидина в качестве катализатора. Смесь нагревают на
кипящей водяной бане в течение 1 часа, после чего охлаж-
дают до комнатной температуры, выливают на 100 г толчено-
го льда и оставляют стоять в течение 18—20 часов. Получен-
ный осадок отфильтровывают, промывают 2—3 мл спирта и
кристаллизуют (1—2 раза) из спирта до постоянной темпе-
ратуры плавления. Данные по выходам, очистке и характе-
ристики полученных спиропиранов приведены в таблицах
1 и 2.
Таблица 1
Условия очистки и выходы 6R', 8'R" -3,3'-днметилспиро-
[бензоксазолин-2,2'-(2 Н-1'-бензопиранов)]
R"
№№ соедине- ний Заместители Кристаллизация Выход в % от теоретического
R' « количество спирта на 1 г спирана, мл число кристалли- заций сырца ЧИСТОГО
1 Вг NO, 90 1 20,5 1,0 .
2 NO, Вг 50 2 39,3 9,1
3 С1 NO. 40 1 15,7 11,6
4 NO, Н 30 2 27,7 21,9
О NO.. осн3 30 2 — 12,0
5 * С>7
Таблица 2
Характеристики 3,3'-димети леи иро-[бензокса зол ин-2,2'-(2'Н-1'-бе изо
пиранов)]
соедИ'
Темпера-
тура плав-
ления °C
Внешний
вид
спиранов
Брутто-
формула
Элементы Данные анализа
вычис- лено, % найдено, %
1 162,5-163,5 Желтые C17HI3BrN2O4 С 52,46 52,04; 52,44
кристаллы н 3,34 3,49; 3,57
N 7,20 7,03; 7,11
2 177-177,5 Желтые CnH^BrN.O, С 52,46 52,30; 52,47
блестящие н 3,34 3,50; 3,62
кристаллы N 7,20 7,07; 7,33
3 143-144,5 Зеленовато- CirH13CJN2O4 Вг 20,55 20,73; 20,53
желтые CI 10,30 10,18; 10,32
кристаллы N 8,12 8,10; 8,16
4 137-137,5 Желтые С17Н11\\О4 С 65,54 65,48; 65,13
кристаллы н 4,51 4,73; 4,83
N 9,03 9,48; —
5 204-205 Желтые C,SH16N?\3 С 63,53 63,80; 63,74
блестящие н 1,70 5,05; 5.02
кристаллы К 8,23 8,36; 8,06
Л И Т Е Р А Т У Р А
1. Брит. пат. 889186 (1962); Пат. США 3149120 (1964).
2. Д. А. Драпкина, В. Г. Брудзь, В. А. Иншакова, Н. И.
Ба дайкона. См. статью «2-Этилбензоксазол» в настоящем сборнике,
стр. 184.
3. R. О. С 11 n t о п, S. С. L a s к о w s k i. J. Amer. Chem. Soc., 71,3602
(1949).
Поступила в ноябре 1965 г. ИРЕА
УДК 547.553.1.07
N.N-ДИМЕТИЛ-о -ФЕНИЛЕНДИАМИН
Р. П. ЛАСТОВСКИЙ, В. я. ТЕМКИНА. Л. М. САМЫЛОВА
C8H12Na
^/XNH3
М. в. 136,19
По литературным данным, N.N'-диметил-о-фенилендиамин
получают восстановлением Ь1,Т^-диметил-2-нитроанилина цин-
ком в соляной кислоте [1] или в растворе хлористого аммо-
ния [2].
Нами проверен и уточнен последний метод получения.
СХЕМА СИНТЕЗА N.N -ДИМЕТИЛ- о -ФЕНИЛЕНДИАМИНА
Характеристика основного сырья
Ы,Ы-Диметил-2-нитроанилин (см. примечание).
Цинковая стружка.
Аммоний хлористый, ч., ГОСТ 3773—60.
Условия получения
В трехгорлую колбу емкостью 250 мл с обратным холо-
дильником, термометром и мешалкой помещают 8,3 г
(0,05 М 1\Г,М-диметил-2-нитроанилина, приливают 100 мл 5%-
69
него раствора хлористого аммония и нагревают до кипения.
В кипящий раствор в течение 1 часа постепенно добавляют
19,5 г (0,3 М) цинковой стружки. Смесь выдерживают 30
минут, отфильтровывают от неорганического осадка, кото-
рый промывают 50 мл воды, и промывные воды присоединя-
ют к фильтрату. Из полученного раствора отгоняют воду и
остаток перегоняют в вакууме при 25 мм, отбирая фракцию
с температурой кипения 100—101°.
Получают 6 г М',М-диметил-о-фенилендиамина, что состав-
ляет 87% от теоретического выхода.
По литературным данным, т. кип. продукта при 20 —
25 мм 99,5—101° [1—2].
Примечание.
Получение М,1\-диметил-2-иптроанилина изложено в соответствугощей
статье настоящего сборника.
ЛИТЕРАТУРА
1. 1. Pinnow, Вег., 32, 1668 (1902).
2. Е. Bamberger, F. Tschirner. Вег, 32, 1905 (1902).
Поступила в декабре U-63 г. ИРЕА
УДК 547.656.05
2,2-ДИОКСИ-1,1-НАФТАЛЬДАЗИН
Люмоген светло-желтый
С22Н igOyNg
Р. М. ГЕЛЫИТЕИН, И. Е. ЗАДОРОЖНАЯ
М.в. 340,38
Люмоген светло-желтый, относящийся к группе азомети-
новых красителей, люминесцирует желтым цветом под дейст-
вием ультрафиолетовых лучей.
Применяется для получения люминесцирующих красок.
Известен способ получения люмогена светло-желтого взаимо-
действием 2-окси-13-нафтальдегида и гидразин-гидрата в
спиртовой среде в присутствии кислоты [1].
Недостатком этого метода является применение дорогого
и опасного в обращении гидразин-гидрата. Кроме того, про-
ведение синтеза в присутствии кислоты затрудняет очистку
продукта.
Ниже приводится разработанный нами способ получения
люмогена светло-желтого взаимодействием 2-окси-1-нафталь-
дегида и гидразин-сульфата в водно-спиртовой среде.
СХЕМА СИНТЕЗА ЛЮМОГЕНА СВЕТЛО-ЖЕЛТОГО
он
2 | || | +H«N-NHa-H2SO4 + 2NH4OH
71
Н Н
I 1
С= N------N=C
I I
ИН нп
- , ц [-OHHO-I ц |+(Nh4),SO4+2H2O
Ч/W 4/W
Характеристика основного сырья
Гидразин-сульфат, ГОСТ 3186—52.
Аммиачная вода, ГОСТ 9—57.
2-Окси-1-нафтальдегид, «ч», МРТУ 6-09-670-63.
Спирт изопропиловый, ГОСТ 9805—61.
Условия получения
К суспензии 171 г (1,3 М) (в пересчете на 100%-ный гид-
разин-сульфат) в 540 мл воды, нагретой до 50°, придают при
размешивании 97 г (0,7 Л4) 25%-ной аммиачной воды. Полу-
ченный раствор фильтруют.
К нагретому до 50° раствору 450 а 2-окси-1-нафтальдегида
в 3,6 кг изопропилового спирта приливают при размешива-
нии горячий водный раствор гидразин-сульфата. После 45-
минутного кипячения реакционной массы краситель отфиль-
тровывают, отмывают от кислоты горячей дистиллированной
водой, промывают изопропиловым спиртом и сушат при 60—
70°
Выход люмогена светло-желтого равен 430 г, что состав-
ляет 97% от теоретического; продукт отвечает ТУ ГКХ
151—59.
ЛИТЕРАТУРА
I. И. М. Коган. Химия красителей, М., ГХИ, 1956, стр. 421.
Поступила в июле 1965 г, ЦЗЛ
УДК 547.831.7.07
2,8-ДИОКСИХИНОЛИН
8-Оксикарбостирил
И. А. КРАСАВИН, В. М. ДЗИОМКО, Ю. П. РАДИН
//\/\
I 11 I -1 I I
он ОН jij
CsH7NO2 М.в. 161,16
2,8-Диоксихинолин может быть получен путем нагревания
8-оксихинолина с расплавленной щелочью в серебряном
тигле до 380° [1] (выход 70%, т. пл. выше 260° с разложени-
ем) или щелочного плавления 8-оксихинолин-5-сульфокислоты
[2] (выход 24,6%, т. пл. 287—288°). Он образуется также при
гидролизе 8-ацетоксикарбостирила [3] (выход не указан,
т. пл. 250°) или при действии раствора щелочи на метосуль-
фат 8-окси- 1-метоксихинолиния [4] (выход 63—65%, т. пл.
288—289,5°).
Общим недостатком всех этих методов, в том числе и
предложенного недавно нами [4], является невысокая чи-
стота образующегося вещества, которое не всегда удается
освободить от загрязнений даже путем многократной пере-
кристаллизации (из диметилформамида).
Как нами установлено, наиболее чистый 2,8-диоксихино-
лин можно получить с выходом 79—84% при гидролизе
8-окси-2-метоксихинолина кипящей разбавленной соляной
кислотой. Продукт реакции без какой-либо дополнительной
очистки получается почти бесцветным и имеет т. пл. 290—
291°, т. е. более высокую, чем у образцов, приготовленных
известными ранее способами [1—4] и тщательно очищенных.
Строение продукта этой реакции подтверждено тем, что
его ИК-спектры тождественны спектрам 2,8-диоксихинолина,
73
полученного двумя другими методами [3, 4], а моноацетиль-
ные производные всех трех образцов не дали депрессии тем-
пературы плавления в пробах смешения.
Получение 2,8-диоксихинолина
Раствор 3,5 г (0,02 Л1) 8-окси-2-метоксихинолина (см. при-
мечание 1) в 100 мл 2%-ной соляной кислоты кипятят с об-
ратным холодильником в течение 6-8 часов (см. примеча-
ние 2). После охлаждения осадок отсасывают, тщательно
промывают водой и высушивают в вакуум-эксикаторе над
едким натром.
Выход 2,8-диоксихинолина равен 2,55—2,7 а, что состав-
ляет 79,2—83,9% от теоретического, т. ил. 290—291° (см.
примечания 3—5).
Примечания:
1. Применялся 8-окси-2-метоксихиног1Ин с. т. пл. 50—50,5”, полученный
по описанному ками недавно методу [5].
2. Продукт реакции нерастворим в разбавленных кислотах и начи-
нает выпадать в осадок из кипящего раствора примерно через 1 час пос-
ле начала нагревания.
3. При плавлении этого вещества образуется светлый расплав, тогда
как все образцы 2,8-диоксихииолииа, полученные другими методами [1 —
4], плавились с более или менее сильным почернением.
4. После перекристаллизации из диметилформамида (20 .ил) т. пл. не
изменилась.
5. Моноацетнльное производное (т. ил. 248,5—249°) было приготов-
лено кипячением образца с уксусным ангидридом. По литературным дан-
ным, т. 11.1. 244—247° (не испр.) [11; 247,5-248,5° [2]; 249—249,5° [6];
249,5-250° [4]; 250° [3].
ЛИТЕРАТУРА
J. J. Diamant. Monatsh. 16, 760 (1895).
2. Т. О h t a, Y. Mori, S, Takagl, Jr Vakugaku Zasshi. 78,697
(1958).
3. J. P. Phillips, E M. В а г r a I I, R. Breese. Trans. Kentucky
Acad. Sci., 17,135(1956); Chem. Abstrs, 51,11349a (195').
4. В. M. Дзиомко, И. А. Красавиц, IO. 11. Радин. Методы
получения химических реактивов и препаратов, вып. 13, М., ИРЕА, 1965,
стр. 44.
5. В. М. Дзиомко, И. А. Красавин, Ю. 11. Ради н. Методы по-
лучения химических реактивов и препаратов, вып. 14, М., ИРЕА, 1966,
стр. 86.
6. В. М. Дзиомко, И. А. Красавин, Ю. П. Радин, Н. И. М и-
рошкпна. Там же, стр. 88.
Поступила в декабре 1965 г.
ИРЕА
УДК 547.425.5.07
N, N'-ДИФЕН ИЛФОРМАМИДИНСУЛЬФИ НОВАЯ
КИСЛОТА, ДИГИДРАТ
Р. Л. ГЛОБУС. Р. П. ЛАСТОВСКИЙ, Е. Я. ЯРОВЕНДО
С. И. МЕДВЕДЕВА
c6h5n
>C-SOaH-2HaO
CeH5NH
CnH12N2O2S-2H,O М.в. 296,28
М,М'-Дифенилформамидинсульфиновая кислота [1] в лите-
ратуре не описана. Она, как и другие производные формами-
динсульфиновых кислот, может представить интерес как фи-
зиологически активное вещество.
СХЕМА СИНТЕЗА ЧКИ-ДИФЕНИЛФОРМАМИДИНСУЛЬФИНОВОЙ
КИСЛОТЫ
C6H6N CgH6N
X>-SH+2HaOa -> ^C-SO2H+2H,O
C6HBNH CeH5NH
Характеристика основного сырья
ЬГМ'-Дифенилтиомочевина, т. пл. 150—15Г.
Перекись водорода, медицинская, ГОСТ 177—55.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термометром
и капельной воронкой, загружают 5 г (0,02 А1) Ы,М'-дифенил-
тиомочевины и приливают 80 мл диоксана. Полученный раст-
вор охлаждают до 15° и добавляют 0,025 г молибденовокис-
лого натрия. При энергичном размешивании, при температу-
75
ре 15е по каплям приливают 6 мл (0,04 /И) 28,2%-ной пере-
киси водорода. По окончании прибавления перекиси водоро-
да температуру реакционной массы понижают до 5°, при этом
выпадает осадок, его отфильтровывают и сушат при темпе-
ратуре 60°.
Выход М,М'-дифенилформамидинсульфиновой кислоты ра-
вен 4 г, что составляет 61,6% от теоретического, считая на
М^'-дифенилтиомочевину.
Найдено, %: С-52,26; 52,60; Н -4,86; 5,08;
N—9,05; 9,13.
C13H12N3OaS.2HO. Вычислено, %: С-52,70; Н-5,40; N-9,46.
По внешнему виду вещество представляет собой белые
кристаллы с т. пл. 183—184°, не растворяется в воде и не-
полярных органических растворителях, плохо растворяется
в спирте.
ЛИТЕРАТУРА
1. Р. Л. Глобус, Р. П. Ластовский, Е. Я. Яровенко, С. И.
Медведева. Авт. свид. 178 803 (1965); Бюлл. нзобр., 4 (1966).
Поступила в ноябре 1963 г. ИРЕА
УДК 547.832.1.07
8,8-ДИХИНОЛИЛ
В. м. дзиомко, 3. С. СИДЕНКО, Г. С. ЧИЖОВА
C18Hi2N2 М.в. 256,29
По литературным данным, в.в'-дихинолил может быть по-
лучен скраупированием 2,2'-диаминодифенила в присутствии
мышьяковой кислоты [1], а также перегонкой 8,8'-дихинолил-
2,2-'дикарбоновой кислоты [2] или перегонкой 8,8'-дихинолил-
ртути с медным порошком [3].
8,8'-Дихинолил нами получен по способу [1] с тем измене-
нием, что выделение основания из реакционен смеси про-
ведено не трудоемким и малоэффективным методом фракцио-
нированного переосаждения, а путем экстрагирования бен-
золом.
СХЕМА СИНТЕЗА 8,8'-ДИХИНОЛ ИЛА
сн3он сно
I н I
СНОП -2^-> СН
| —2НаО ||
СН2ОН сн2
_р2
У СИ As2°5- >
H2N |Г -2Нз°
снг
77
Характеристика основного сырья
2,2'-Диаминодифенил, т. пл. 77—78°.
Глицерин, ч., ГОСТ 6259—52.
Мышьяковая кислота, 88—90%-ная.
Серная кислота, ч.д.а., ГОСТ 4204—48.
Аммиак водный, 25%-ный, ГОСТ 3760—47.
Бензол, ч.д.а., ГОСТ 5955—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Кали едкое, ч.д.а., ГОСТ 4203—48.
Условия получения
Смесь 12,5 г (—0,07 Л1) 2,2'-диаминодифенила, 20 г мышь-
яковой кислоты, 40 г глицерина, 40 г концентрированной сер-
ной кислоты осторожно нагревают в круглодонной колбе ем-
костью 0,25 л, снабженной обратным холодильником. Внача-
ле появляется сильное вспенивание, которое через 30 минут
уменьшается, после чего содержимое колбы кипятят еще 4—
5 часов. Затем добавляют 750 мл горячей воды и после ох-
лаждения фильтруют. Фильтрат подщелачивают аммиаком
до щелочной реакции по бриллиантовой желтой. Выпавший
при этом осадок экстрагируют горячим бензолом. Бензольные
вытяжки сушат едким кали, затем бензол отгоняют, а остаток
дважды перекристаллизовывают из спирта.
Выход равен 3,1 г, что составляет 19% от теоретического,
считая на 2,2'-диаминодифенил; т. пл. вещества 204—205°.
По литературным данным, т. пл. вещества 205—207° [1],
выход не указан.
ЛИТЕРАТУРА
I. S. Niementowski, М. Seifert. Вег., 38, 762 (1905).
2. Z Jakubowski, S. Niementowski. Ber., 42, 634 (1909).
3. Т. Ukai. J. Pharmac. Soc. Japan, 48, 172 (1928).
Поступила в декабре 1965 г.
ИРЕА
УДК 543.847
ДИХЛОРАНГИДРИД ТРИХЛОРМЕТИЛТИОФОСФИ-
НОВОЙ кислоты
Л. Е. ДМИ ГРНР.ВА, С. 3. ИВИН, К,. В. КАРАВАНОВ
СС13Р8СК
CPSCU М.в. 252.31
До последнего времени дихлор ангидрид трихлорметилтио-
фосфиновой кислоты в литературе не был описан.
Нами разработан способ его получения, который основан
на реакции комплексного соединения трихлормегилтетрахлор-
фосфина и хлористого алюминия с этиленсульфидом (метод
А) или этилмеркаптаном (метод Б).
СХЕМЫ СИНТЕЗА ДИХЛОРАНГИДРИДА ТРИХЛОРМЕТИЛТИО-
ФОСФИновой кислоты
СС1,РС14-А1С13Ц-СН2—CH2S —
CC13PSC12+ C1CH2-CH.2CI + А1С13-КС1 (метод А)
КС1
СС13РС14-А1С13 + C2HeSH----->
-> CClgPSClo 4- С2Н,С1 + КС1-А1С1, 4- НС1 (метод Б)
Характеристика основного сырья
Комплексное соединение трихлорметилтетрахлорфосфина
и хлористого алюминия получают из треххлористого фосфо-
ра, треххлористого алюминия и четыреххлористого углерода
по методике [1, 3].
Этиленсульфид, т. кип. 54,9°/760 мм, получают из рода-
нистого калия и окиси этилена [2].
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Этилмеркаптан, ч., обычный.
79
Условия получения
Метод А. В трехгорлую колбу с мешалкой, капельной во-
ронкой и обратным холодильником помещают тщательно пе-
ремешанную смесь 63 г (0,15 М) комплексного соединения
трихлорметилтетрахлорфосфина и хлористого алюминия и
11,2 а (0,15 М) свежепрокаленного хлористого калия.
К реакционной массе, охлажденной до минус 2—3°, прн
перемешивании по каплям прибавляют 14,7 г (0,15 А4) эти-
ленсульфида. После прибавления, не прекращая перемеши-
вания, температуру в колбе медленно повышают до комнат-
ной, а затем нагревают 30 минут при 80°. Затем обратный
холодильник заменяют на прямой н отгоняют образовавший-
ся дихлорангидрид трихлорметилтиофосфиновой кислоты.
После повторной перегонки выход вещества равен 15,1 а,
что составляет 40% от теоретического; т. кип. 95°/10 мм:
т. пл. 120°.
При разгонке получают также 4 г (25%) дихлорэтана с
т. кип. 847760 мм; z^20 — 1,2530; — 1,4432.
Метод Б. В трехгорлую колбу с мешалкой, обратным хо-
лодильником и капельной воронкой помещают смесь 63 г
(0,15 М) комплексного соединения трихлорметилтетрахлор-
фосфина и хлористого алюминия и 11,2 г (0,15 Л1) свеже-
прокаленного хлористого калия. При перемешивании по кап-
лям прибавляют 9,3 г (0,15 Л4) этилмеркаптана. Взаимодей-
ствие протекает при комнатной температуре. После прибав-
ления этилмеркаптана колбу при перемешивании реакцион-
ной массы нагревают 30 минут до 50°. Затем обратный холо-
дильник меняют на прямой и отгоняют образовавшийся про-
дукт в вакууме 10 мм, собирая его в охлажденной ловушке
и приемнике.
При повторной перегонке получают 27 г дихлорангидри-
да трихлорметилтиофосфиновой кислоты, что составляет 75%
от теоретического; т. кип. 957Ю мм; т. пл. 120°.
В охлажденной ловушке собрано также 6,1 г хлористого
этила с т. кип. 12°.
ЛИТЕРАТУРА
1. А. М. К i n п с г, Е. А. Р е г г е n. J. Chem. Soc., 3437 (1952).
2. И. В. Враз. Ж. общ. химии, 21, 688 (1951).
3. И. П. Комков, К. В. Караванов, С. 3. Ивин. Методы полу-
чения химических реактивов и препаратов, вып. 12, М„ ИРЕА, 1965,
стр. 73.
Поступила в ноябре 1965 г.
УДК 547.264'11.07
4,4'-ДИХЛОРДИБУТИЛОВЫЙ ЭФИР
Б. А. РОЗЕНБЕРГ, О. М. ЧЕХУТА
с1(сн2)4О(сн2)4а
С8НкОС1а. М.в. 198,92
4,4'-Дихлордибутиловый эфир представляет значительный
практический интерес как исходное сырье для синтеза раз-
личных бифункциональных соединений (динитрилов, дикис-
лот, диаминов, диолов и др.), широко применяемых в про-
мышленности органического синтеза и полимерных материа-
лов [1].
Описанные в литературе способы получения 4,4"-дихлор-
дибутилового эфира основаны на взаимодействии тетрагидро-
фурана с хлористым водородом или хлорангидридами неорга-
нических кислот в присутствии льюисовских [2, 3] или про-
тонных кислот [1]. Выход 4,4'-дихлордибутилового эфира при
использовании указанных способов не превышает 60—70%,
так как часть тетрагидрофурана расходуется на образование
мономерного дихлорида (1,4-дихлорбутана) или более высо-
комолекулярных, чем димер, дихлоридов.
Предлагаемый нами способ получения 4,4'-дихлордибути-
лового эфира путем взаимодействия тетрагидрофурана с хло-
ристым тионилом в растворе полярного и достаточно высоко-
кипящего растворителя и в присутствии ранее не применяв-
шейся для указанных целей пятихлористой сурьмы повышает
выход эфира до 90%.
СХЕМА СИНТЕЗА 4,4-ДИХЛОРДИБУТИЛОВОГО ЭФИРА
2 |___/ О + SOC12 С1(СН2)4О(СН2)4С1
Характеристика основного сырья
Тетрагидрофуран, технический, т. кип. 64—66° (см. при-
мечание 1).
Зак. 25 81
Хлористый тионил, технический.
Пятихлористая сурьма, ч., МПТУ 4428—54.
Нитрометан, ч., ТУ ГКХ ОРУ 129— -59 (см. примечание 1).
Условия получения
В кипящий раствор 15 мл нитрометана с 1,8 г (0,006 Л4)
пятихлористой сурьмы в трехгорлой колбе, снабженной тер-
мометром, обратным холодильником и капельной воронкой,
постепенно добавляют раствор 20,2 мл (0,25 М) тетрагидро-
фурана в 13 мл (0,165 М) хлористого тионила.
После двухчасового нагревания из реакционной смеси от-
гоняют непрореагировавший избыток хлористого тионила и
нитрометан, а остаток охлаждают до комнатной температуры.
За время реакции температура в парах постепенно под-
нимается с 68° до 97—100°. Достижение указанной темпера-
туры в парах реакционной смеси может служить признаком
окончания реакции.
Охлажденную реакционную смесь выливают в воду, про-
дукт реакции экстрагируют 100 мл эфира, эфирный раствор
промывают водным раствором бикарбоната натрия, а затем
водой до нейтральной реакции. После высушивания эфирно-
го раствора над свежепрокаленным хлористым кальцием и
отгонкой растворителя продукт реакции выделяют перегонкой
в вакууме (см. примечание 2).
Выход 4,4'-дихлордибутилового эфира с т. кип. 11075 мм
рацен 22 г, что составляет 88,7% в расчете на тетрагидро-
фуран; п'^—1,4570; с?420— 1,0760. По литературным дан-
ным, т. кип. вещества 126—128713 мм; — 1,4568; <№—
1,0810 [3].
Примечания:
1. Исходные продукты предварительно тщательно высушивают и пе-
регоняют; нитрометан — иад пятиокисью фосфора, тстрагидрофураи— над
прокаленным поташом или твердым едким кали, а затем иад проволокой
из металлического натрия.
2 Выделение 4,4/-дихлордибутилового эфира из реакционной смеси
можно проводить также перегонкой с паром.
ЛИТЕРАТУРА
1. К. Alexander, L. Schniepp. J. Amer. Chem. Soc., 70, 1839
(1918).
2. В. И. Л у т к о в a, H. И. Куценко, М. И i к и и а. Ж. общ. химии,
25, 2102 (1955).
3. В. И. Л у т к о в а, Н. И. Куценко. Ж. прикл. химии, 32, 1635
(1959). ;
Поступила в октябре 1964 г. Донецкий филиал ИРЕА
УДК 547.284.3.05
2,2-Д ИХЛОРПРОПЛН
Ацетонхлорид
С. А. КОЧЕТКОВА, А. В. АФАНАСЬЕВА
СН9-СС12-СН,
С9Н9С12 М.в. 112,98
2,2-Дихлорпропан применяется в органическом синтезе. По
литературным данным, 2,2-дихлорпроиан можно получить
взаимодействием ацетона с пятихлористым фосфором [1, 2, 3j.
Нами был проверен этот метод получения 2,2-дихлорпро-
пана.
СХЕМА СИНТЕЗА 2,2-ДИХЛОРПРОПАНА
СН3-СО-СНз+РС15 -* СН3~СС12-СН3 + РОС13
Характеристика основного сырья
Ацетон, ч.д.а., ГОСТ 2603—51.
Пятихлористый фосфор, ч.д.а., ГОСТ 386 -41.
Натрий сернокислый, ч., безводный. ГОСТ 4156—48.
Фосфорный ангидрид, х. ч., МРТУ-6-09-22—62.
Условия получения
В трехгорлую колбу емкостью 2 л с двурогим форштосом,
ртутным затвором, мешалкой, капельной воронкой, термомет-
ром, шариковым холодильником, соединенным с двумя склян-
ками Дрекселя, охлаждаемыми твердой углекислотой для
улавливания летучих продуктов, загружают 624 г (3 М) пя-
тихлористого фосфора. Колбу помещают в баню с охлаждаю-
щей смесью (лед и поваренная соль), охлаждают до 0° и в
течение часа осторожно, при непрерывном перемешивании,
прибавляют 436 мл (348 г, 6 М) ацетона, следя за тем, чтобы
6« S3
температура реакционной массы не превышала 15—20°. Пос-
ле прибавления ацетона реакционную массу выдерживают
при слабом кипении (температура бани 60—70°) в течение
3 часов, затем снова охлаждают до 0° и при постоянном пере-
мешивании очень осторожно вносят небольшими кусочками
I кг льда. Органический слой отделяют и промывают не-
сколько раз дистиллированной водой до нейтральной реак-
ции по универсальной индикаторной бумажке. Сырой про-
дукт (138,7 г) сушат вначале над безводным сернокислым
натрием, а затем над фосфорным ангидридом.
Выход 2,2-дихлорпропана равен 120 г, что составляет 35%
от теоретического, считая на фосфор пятихлористый.
Высушенный продукт очищают фракционной перегонкой
при атмосферном давлении, применяя для разгонки колбу с
елочным дефлегматором. Отбирают фпакцию с т. кип. 66—
69°.
Получают 66 г вещества, что составляет 19% от теорети-
ческого. Свежеперегнанный 2,2-дихлориропан представляет
собой бесцветную жидкость с — 1,4145.
По литературным данным, п™— 1,40932 [4]; 1,4093
[51
ЛИТЕРАТУРА
1. С. Fridel, A. Ladenburg. Liebigs Ann. Chem., 142, 315.
2. A L. Henne, M. W. Ren о 11, J. Amer. Chem. Soc., 59, 2435
(1937).
3. C. A., 52, 1253g (1958); Брит. пат. 753384 (1956).
4. Beilst, 1, 105.
5. Справочник химика, II, M„ Госхимиздат, 1961, стр. 1042, № 238.
Поступила в ноябре 1S65 г. ИРЕА
УДК 547.462.3.07
N-ЗАМЕЩЕННЫЕ ИМИДЫ МАЛЕИНОВОЙ КИСЛОТЫ
N-Мальимиды
В. С. ИВАНОВ, В. к. СМИРНОВА, Т. И. СИДОРОВА,
И. И. МИГУНОВА, А. М. АБРАМОВА, Е. Г. БАШИНА, Т. Р. СЕППЕН
//
сн-сх
II /N-R
СН-С
сн3
R • ~ СН3;
С1
_^~4_NOa; ; -СН3-^—
N-Замещенные имиды малеиновой кислоты, особенно
N-фенилмальимид и его производные, могут быть применены
в качестве мономеров [1], вулканизующих агентов (2[ эффек-
тивных сенсибилизаторов радиационной вулканизации поли-
диеновых эластомеров [3], антибактериальных средств [4],
фунгисидов [5], репеллентов грызунов [6], дефолиантов и про-
тиворостовых веществ [7].
Описанные в литературе синтезы N-фенилмальимида п
некоторых его производных основаны на взаимодействии ма-
леинового ангидрида с ароматическим амином, приводящем
к образованию соответствующей малеаминовой кислоты, ко-
торая при дегидратации циклизуется в имид [8—11]. Кроме
того, имеются указания на возможность получения имидов из
яблочной кислоты и соответствующих аминов [12], при сплав-
лении аминов и кислот [13] или ангидридов [14], при кипяче-
нии водных суспензий аминов и ангидридов [15].
85
Нами разработаны условия получения, обеспечивающие
наибольший выход и чистоту описанных ранее N-фенил-,
N-n-толил-, N-п-хлорфенил-, N-н-нитрофеиил-, и N-бензил-
мальимидов через соответствующую малеаминовую кислоту
путем дегидратации последней в присутствии уксусного ан-
гидрида и уксуснокислого натрия, а также впервые получены
указанным способом Г4-2,4-диметилфенил- и N-л-хлорфенил-
мальимиды.
Синтез проводится в две стадии. Установлено, что выход и
чистота образующихся имидов в большой степени зависят от
чистоты исходных вешеств и растворителей. В качестве ра-
створителя на первой стадии нами предложен хлороформ
вместо наиболее часто употребляемого эфира. Это способст-
вует безопасности проведения процесса при обеспечении не-
обходимого качества синтезируемых продуктов.
СХЕМА СИНТЕЗА N -ЗАМЕЩЕННЫХ МАЛЬИМИДОВ
1-я стадия:
СН-СХ CH-CONHR
;o+NH,-R - 'I
СН-С7 сн-соон
'^о
N-R-малеамииовая кислота
2-я стади я:
^0
CH-CONHR СН-С\
1 -Н;о II )N-R
СН-СООН СН-С
Характеристика основного сырья
/Малеиновый ангидрид, ч., ГОСТ 5854—51.
Анилин, ч., ГОСТ 5819—51.
n-Толуидин, ч.', ТУ ОРУ 47—56.
.м-Ксилидин, ч., ТУ МХП 197—51.
л-Хлоранилин, ч., СТУ 79—484—X—60.
з«-Хлоранилин, ч., СТУ 79—496—X—60.
я-Нитроанилин, техн., марка Б, ГОСТ 4398—59.
Бензиламин, ч., ГОСТ (ТУ) 3724—53.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Уксуснокислый натрий, ч.д.а., ГОСТ 199—52.
S6
Условия получения
1-я стадия. Получение малеаминовых кислот. В литро-
вую круглодонную колбу с мешалкой, обратным холодильни-
ком и капельной воронкой помещают 39,2 г (0,4 М) размель-
ченного малеинового ангидрида и 50 мл свежеперегнанного
сухого хлороформа (или абсолютного эфира). После раство-
рения малеинового ангидрида добавляют 0,4 М анилина
(37,2 г), п-толуидина (42,9 г), лг-ксилидина (48,5 г), га-нитро-
анилина (55,3 г), га-хлоранилина (47,1 г), л-хлоранилина
(47,1 г) ил.и бензиламина (42,9 г).
Содержимое колбы перемешивают около одного часа до
полного охлаждения. Образовавшуюся кислоту отфильтро-
вывают на воронке Бюхнера и сушат. Выходы кислот состав-
ляют 70—95% от теоретического.
Примечания:
1. Предварительно перегнанные анилин, Л1-кси.тпдин и .и-хлорапилип
разбавляют 50 мл хлороформа (абсолютного эфира) и добавляют к ра-
створу малеинового ангидрида в том же растворителе.
2. Бензиламин, растворенный в 50 мл хлороформа (или абсолютного
эфира), следует приливать очень быстро, чтобы предотвратить поглощение
углекислоты из воздуха.
3. При получении М-2,-4-диметилфепилмалеамиповой кислоты (из
л-ксилидина) требуется особая чистота исходных веществ. В качестве ра-
створителя следует использовать только абсолютный эфир. Кислота два-
жды перекристаллизовывается из диоксана.
2-я стадия. Получение N-мальимидов. В полулитровую
колбу Эрленмейера с мешалкой и обратным холодильником,
снабженным хлоркальциевой трубкой, помещают 160 мл све-
жеперегнанного уксусного ангидрида, 15,5 г безводного ук-
суснокислого натрия и. 0,4 М мальаниловой (76,5 г), N-n-то-
лил- (82,1 г), М-2,4-Диметилфенил- (87,7 г), N-n-нитрофенил-
(94,5 г), N-ra-хлорфенил- (86,3 г), М-льхлорфенил- (86,3 г)
или N-бензил- (82,1 г) малеаминовой кислоты. Нагреванием
на водяной бане (80—90°) при перемешивании добиваются
растворения кислоты. После того как раствор станет проз-
рачным, содержимое колбы нагревают еще полчаса при пе-
ремешивании. Нерастворившуюся часть отфильтровывают на
воронке горячего фильтрования. Фильтрат охлаждают в бане
с ледяной водой до начала кристаллизации и выливают в
стакан с холодной водой. Образовавшийся при охлаждении
и периодическом перемешивании осадок имида отфильтровы-
вают на воронке Бюхнера, многократно промывают холодной
водой, один раз петролейным эфиром (т. кип. 30—60°) и су-
шат.
Выход сырого продукта составляет 80—95% от теорети-
ческого. Сырой продукт перекристаллизовывают из сухого
бензола до получения вещества с постоянной температурой
Плавления.
87
Температура плавления v
малеаминовой кислоты, °C Характеристика имида
Примечания:
1. Выход сырого М-2,4-диметилфенилмальимида не превышал 30%.
2. N-n-нитрофенилмальимид перекристаллизовывали из диоксана.
3. Сушка кислот и имидов эффективно проходит под действием ин-
фракрасного излучения (лампа ЗС-1).
Температуры плавления малеаминовых кислот и соответ-
ствующих имидов, а также данные элементарного анализа
приведены в таблице.
ЛИТЕРАТУРА
1. В. С, Иванов, Т. А. Сухих, А. X. Б регер, В. Б. Осипов,
Б. А. Гольдин. Авт. свид. 166832; Бюлл. изобр., 23, 59 (1964).
2. Р. О. Tawney, W. J. Wenisch. S van d er Burg, D. J.
Pely ea. J. Appl. Polymer Sci., 8, 2281 (1964); Химия и техн, полимеров,
4, 108 (1965).
3. R. Roberts. Proceedings of the 1962 Tihany Symposium Radiation
Chemistry Akademial Kiado, Budapest, 1964, p. 79.
4. D. M a r r I a n и др. Biochem. J., 54, 65 (1953).
5. L. Ferenczy. Acta biol. Acad, scient, hung., 10, 77 (1959).
6. E. В e 11 a c k, J. B. D e wi 11 o. J. Agric. and Food Chem., 2,1176
(1954).
7. J. Van Overbeek, B. Biondean, V. Horne. Amer.
J. Bot., 42, 205 (1955).
8. Сб. «Синтезы органических препаратов», 12, M., изд. «Мир», 1964,
стр. 181.
9. М. X. Глузмаи. Ж. общ. химии, 28, 2987 (1958).
10. А. Е. К р е т о в, Н. Е. К у л ь ч и ц к а я. Ж. общ. химии, 26, 208
(1956).
11. И. Б. Крачков. Методы получения химических реактивов и пре-
паратов, вып. 8, М., ИРЕА, 1964, стр. 83.
12. R. Lukes, М. Per gal. Coll. Czech, Chem. Comm., 27, 1387
(1962).
13. A. Michael. Ber., 10, 577 (1887).
14. F. D. C h a 11 w а у, G. D. P a r k e s. J. Chem. Soc., 1923,123,663.
15. G. KOH er. Ber., 37, 1598 (1904).
16. Piutti. Gazz. chim ital., 261, 439 (1866).
17. United States Kubber Co. Англ. пат. 880947 (1960); С. A., 56,6144 в
(1962).
Поступила в июле 1965 г. ЛГУ
s
S
к
•е
о
о.
X
Г
е
X
Z
88
УДК 546.823:541.49
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ АЛКИЛТРИХЛОР-
ЙОДФОСФИНОВ И ДИАЛКИЛДИХЛОРЙОДФОСФИНОВ
И ХЛОРИСТОГО АЛЮМИНИЯ
В. Г. ГРУЗДЕВ, С. 3. ИВИН, к. В. КАРАВАНОВ
R v
RPCI,JA1C1, 4PCl2J-AlCla
R'x
Комплексные соединения алкилтрихлорйодфосфинов и ди-
алкилдихлорйодфосфинов и хлористого алюминия впервые
были получены нами [1J. Синтез их осуществлялся простым
смешением хлористого алюминия, треххлористого фосфора
(или алкилдих'лорфосфина в случае получения вторичных
комплексов) и йодистого алкила в одногорлой колбе с обрат-
ным холодильником, защищенным от влаги воздуха хлор-
кальциевой трубкой. Вместо одногорлой колбы для получения
комплексов можно использовать любую емкость, защищен-
ную от влаги воздуха. Дополнительного нагревания не тре-
буется, выход комплексов — количественный.
КОМПЛЕКСНОЕ СОЕДИНЕНИЕ МЕТИЛТРИХЛОРЙОД-
ФОСФИНА И ХЛОРИСТОГО АЛЮМИНИЯ
CH,PC13J-A1C18
CH3AlPCleJ М. в. 412,61
СХЕМА СИНТЕЗА КОМПЛЕКСА МЕТИЛТРИХЛОРЙОДФОСФИНА
И ХЛОРИСТОГО АЛЮМИНИЯ
CH3J + РС13 + А1С13-► CH3PCI3J • AlCIg
Характеристика основного сырья
йодистый метил, ч., ТУ МХП 49—51.
Алюминий хлористый, ос. ч., безводный, ВТУ МХП 3500—
52 или свежевозогнанный.
Фосфор треххлористый, ч., перегнанный, ГОСТ 91—41,
90
Условия получения
В круглодонную колбу емкостью 1 л с обратным холо-
дильником, защищенным хлоркальциевой трубкой, загружают
133,3 г (1 М) хлористого алюминия и 137,3 г (1 Л4) треххло-
ристого фосфора. К смеси приливают 141,9 г (1 Л4) йодисто-
го метила (см. примечание 1). При небольшом взбалтывании
реакционная смесь самопроизвольно разогревается до 80—
100°. Взбалтывание продолжают в течение 5—10 минут. Со-
держимое колбы при остывании закристаллизовывается в
однородную массу светло-желтого цвета, которую затем вы-
держивают 1 час под вакуумом 10 мм.
Выход комплексного соединения метилтрихлорйодфосфи-
на с хлористым алюминием равен 408,3 г, что составляет
99,0% от теоретического; т. пл. 94°.
По описанной методике можно получить комплексные сое-
динения алкилтрихлорйодфосфинов и хлористого алюминия с
различными алкильными радикалами при фосфоре, используя
для этого соответствующие йодистые алкилы.
Комплексное соединение CjHjPChJ-AlCb имеет т. пл. 137°;
CHs = CHCHaPCla-J • А1С13 — маслообразная коричневая жид-
кость.
КОМПЛЕКСНОЕ СОЕДИНЕНИЕ ДИМЕТИЛДИХЛОР-
ЙОДФОСФИНА С ХЛОРИСТЫМ АЛЮМИНИЕМ
(СН3)гРС1Д-А1С13
C2HeAlPCl5J
М. в. 392,19
СХЕМА СИНТЕЗА КОМПЛЕКСА ДИМЕТИЛДИХЛОРИОДФОСФИНА
С ХЛОРИСТЫМ АЛЮМИНИЕМ
сн,
CH8J + СН3РС12 + А1С13--* ^PC12J- А1С1з
СН3/
Характеристика основного сырья
йодистый метил, ТУ МХП 49—51.
Метилдихлорфосфин, перегнанный (см. статью «Алкилди-
хлорфосфин» в настоящем сборнике).
Алюминий хлористый, безводный, ВТУ МХП 3500—52 или
свежевозогнанный.
Условия получения
В круглодонную колбу емкостью 1 л с обратным холо-
дильником, защищенным хлоркальциевой трубкой, загружают
133,3 г (1 М) хлористого алюминия и 116,9 г (1 /И) метил-
91
дихлорфосфина. К смеси приливают 141,9 г (1 Л1) йодистого
метила (см. примечание 1).
При слабом взбалтывании реакция начинается немедлен-
но, реакционная смесь самопроизвольно разогревается до
80—100°. Содержимое колбы по охлаждении до комнатной
температуры выдерживают 1 час в вакууме 10 мм, после чего
оно закристаллизовывается в однородную массу светло-ко-
ричневого цвета (см. примечание 2).
Выход комплексного соединения диметилдихлорйодфос-
фина с хлористым алюминием равен 388,9 г, что составляет
99,2% от теоретического; т. пл. 120°.
Примечания:
1. йодистый мстил можно приливать за один прием, однако обрат-
ный холодильник в данном случае во избежание потери йодистого метила
должен быть высотой не менее 600 мм и диаметр внутренней трубки не
менее 25 мм.
2. Комплексное соединение легко гидролизуется, в связи с чем кон-
такт его с воздухом необходимо ограничить.
Другие комплексные соединения диалкилдихлорйодфос-
финов и хлористого алюминия с одноименными или разно-
именными алкильными радикалами получают по описанной
методике с выходом 98—99%. Комплексное соединение
СН»,
xPC12J-A1C13
с2н5^
имеет т. пл. 91°.
ЛИТЕРАТУРА
1. В. Г. Груздев, С. 3. Ивин, К. В. Караванов, И. С. М а-
зель, В, В. Тарасов. /К. общ. химии, 36, (1966).
Поступила в ноябре 1965 г.
УДК 547.821.41.07
6-МЕТИЛ-2-ВИНИЛ ПИРИДИН
Б. М. КУИНДЖИ, М. А. ЗЕПАЛОВЛ, Л. Д. ГЛУЗМАН,
А. К. ВАЛЬКОВА, Р. М. ДИН, И. В. ЗАЙЦЕВА, А. А. РОКК
/Ч
HjC^N^CHxxCH,
CgH9N М. в. 119,16
6-Метил-2-винилпиридин получают из 2,6-лутидина и
формальдегида с последующей обработкой образующегося
6-метил-2-пиридилэтанола прг! нагревании едким кали или
натром [1, 2].
Поскольку 2,6-лутидин имеет две реакционноспособные
группы, то при реакции его с формальдегидом образуется
смесь продуктов метил-пиридилэтанола и пиридилдиэтанола
[3, 4, 5].
Однако во всех этих работах выход винильных соедине-
ний был незначительным, что подтверждается и более позд-
ними работами [6].
6-Метил-2-винилпиридин является исходным мономером в
производстве ионообменных смол, а 2,6-дивинилпиридин —
сшивающим агентом при их синтезе.
Предлагаемый метод получения 6-метил-2-винилпиридина
дает возможность получать его с выходом до 70% от теоре-
тического и, кроме того, дивинилпиридин с выходом до 5%
от 6-метил-2-винилпиридина по весу.
СХЕМА СИНТЕЗА 6-МЕТИЛ-2-ВИНИЛ ПИРИДИНА
/ ^1 /
H3C/XN^XCH3 + CH,O -> HsC/XN/’'XCH2CH.2OH
93
H 0 / ч
II | а- || I
А A NaOH А /\
H3C N CH,CH2OH Н3С Ч/чСН=СН,
и / Ч
н2с = нс/44 сп—сн,_>
Характеристика основного сырья
2,6-Лутидин, ч., ТУ TCP 180р—60.
Формалин, техн., ГОСТ 1625—54.
Параформ, ч., МХП 2711—51.
Натр едкий, ч., ГОСТ 4328—48.
Азот, из баллона, ТУ МХП 4280—54.
Условия получения
В1 автоклав емкостью 500 мл, снабженный мешалкой,
трубкой для подачи азота, карманом для термопары и элек-
трообогревом, загружают 250 г (2,34 М) 2,6-лутидина н
126,2 г 37%-ного формалина, содержащего 46,7 г (1,55 /И)
формальдегида (см. примечание 1).
Автоклав продувают азотом, после чего ловоцят началь-
ное давление до 10 атмосфер и включают обогрев и мешал-
ку.
Через 1 — 1,5 часа температура достигает 165°, при кото-
рой реакционную массу выдерживают 2 часа, давление при
этом в автоклаве повышается до 15—18 атмосфер.
После охлаждения автоклава давление спускают, содер-
жимое выгружают и в вакууме, равном 20 мм, отгоняют при
температуре до 80° избыток 2,6-лутидина с водой в количест-
ве 246 г (см. примечание 2), а затем в пределах ПО—170°
при 5 мм перегоняют смесь метилпиридилэтанола и пиридил-
диэтанола в количестве 103 г, которую затем подвергают де-
гидратации.
Для этого в колбу Клайзеиа емкостью 500 мл загружают
103 г (0,754 М) смесн пиридилэтанолов, 5,15 г едкого натра
(5% по весу) и нагревают в вакууме 20 мм. В пределах 65—
85° отгоняется, по мере образования, 88 г продукта дегидра-
тации с водой. После отделения воды смесь винильных про-
изводных разгоняется при 20 мм на две фракции:
1-я фракция в количестве 62 г, полученная в пределах от
66 до 73°, является 6-метил-2-винилпиридином; п™ — 1,5380;
2-я фракция в количестве 3,7 г выкипает в пределах от
85 до 95“ она представляет собой дивинилпиридин; tip —
1,5620.
94
Выход 6-метил-2-винилпиридина равен 69% от теорети-
ческого, а выход 2,6-дивинилпиридина составляет около 5%
от 6-метил-2-винилпиридина по весу.
Примечания:
I. Оптимальный выход 6-метил-2-пиридилэтанола и пиридилдиэтано-
ла достигается сохранением соотношения исходных компонентов 1,5:1.
2. Избыток 2,6-лутндииа используется в следующих опытах. Для это-
го в отгоне до 80° при 20 .ил1 титрованием определяется содержание
2,6-лутидпна, затем к Нему добавляют израсходованный на опыт 2,6-лу-
тпдин и параформ так, чтобы сохранить необходимое соотношение.
ЛИТЕРАТУРА
1. W. Kolnig, G. Нарре. Вег., 36, 2904 (19СЗ),
2. К. Loffler, F. Thiel. Ber. 42, 132 (1909)
3. Е. Prof ft. Chem. Teclni., 7, .'11 (1955).
4. E. Profit. Chem. Techn.. <8, 878 (1956).
5. I. Michalski, K. Studniarski. Roczn. chem., 9,29,1141
(1955).
6- E. Profit. Client. Techn., 5, 280 (1957).
Поступила в ноябре 1965 г.
пиипм
у хин
УДК М7.539.4.07
МЕТИЛ ЙОДИСТЫЙ
И. Е. ЗАДОРОЖНАЯ
Н
H-C-J
н
CH3J М. в. 141,93
Метил йодистый может быть получен из натрия йодистого
[1] или калия йодистого [2, 3] и диметилсульфата в водной
среде; действием метилового эфира толуолсульфокислоты на
водный раствор йодистого калия {3, 4}, взаимодействием ме-
тилового спирта с трехйодистым фосфором или смесью йода
и фосфора [5].
Синтезы метила йодистого с применением йода и фосфора
или диметилсульфата опасны, а метиловый эфир толуол-
сульфокислоты трудно доступен. В связи с этим нами разра-
ботана методика получения метила йодистого взаимодейст-
вием метилового эфира бензолсульфокислоты с йодистым
натрием в водной среде.
СХЕМА СИНТЕЗА ИОДИСТОГО МЕТИЛА
NaJ -ф CH3OSO2-^ -> CH3J +• NaSO3
Характеристика основного сырья
Натрий йодистый, ч., ГОСТ 8422—57.
Метиловый эфир бензолсульфокислоты, ч., МРТУ
6-09-99—62.
96
Условия получения
В двухлитровую колбу с мешалкой, термометром, капель-
ной воронкой н насадкой Вюрца, соединенной с холодильни-
ком Либиха, конец которого опущен в приемную колбу под
воду со льдом (приемная колба с обратным шариковым хо-
лодильником), загружают 1,35 кг натрия йодистого (в пере-
счете на 100%-ный продукт) и 1,35 л воды. Массу размеши-
вают до растворения кристаллов и по каплям прибавляют
1,55 кг (в пересчете на 100%-ный продукт) метилового эфи-
ра бензолсульфокислоты.
После введения примерно третьей части эфира массу осто-
рожно нагревают до 60—70°, после чего продолжают прида-
чу оставшегося эфира и затем при этой же температуре отго-
няют йодистый мстил. Когда выделение продукта прекра-
щается, массу нагревают до 100° до полной отгонки вещества.
Отделенный от воды продукт сушат хлористым кальцием.
Сухой метил йодистый перегоняют с елочной колонкой при
температуре 42°.
Выход метила йодистого равен 1,167 кг, что составляет
91,5% от теоретического, считая на натрий йодистый. Веще-
ство соответствует ТУ МХП 49—51.
Примечание.
В укрупненном производстве хлористый кальций после сушки раство-
ряют в воде, отделившийся метил йодистый объединяют с полученными
фракциями, сушат хлористым кальцием и перегоняют.
Вместо натрия йодистого чистого можно использовать технический
продукт или отходы производства монокристаллов.
Метиловый эфир бензолсульфокислоты можно брать технический.
ЛИТЕРАТУРА
1. М. Н. Б а ис о в а, Н. Н. Дых а ио в. Методы получения химичес-
ких реактивов и препаратов, вып. 10, М., ИРЕА, 1964, стр. 55.
2. R. F. W е i n 1 е n d, К. Sc 11 mid. Ber.. 38, 23.7 (1905); Герм,
пат. 17f.2O1’: Fridlander, 8. 17
3. Л. Гаттермаи, Г. Виланд. Практические работы по органи-
ческой химии, М.—Л., Госхимиздат, 1948, стр. 138, 282.
4. И. Губен. Методы органической химии, т. 3. М„ ОНТИ, Главная
редакция химической литературы, 1935, стр. 582.
5. К. Dumas, Peligot. Liebigs. Ann, Chem.. 15, 20 (1835),
P. C. Haywood. J. Chem. Soc., 121, 1911 (1922); J. W. Walker;
F. M. Z. J о h о n s о n. J. Chem. Soc., 87, 1592 (1905).
Поступила в мюле 1965 г. ЦЗЛ
7 Зак,2ё
УДК 547.542.07
N -МОНОМЕТИЛОЛФОРМАМИДИНСУЛЬФИНОВАЯ
КИСЛОТА
Р. Л. ГЛОБУС, Р. п. ластовский, е. я. яровенко,
С. II. МЕДВЕДЕВА
HN
^C-SO2H
ch2ohnh
C2HeN2O3S
М. в. 138,08
N-Монометилолформамидинсульфиновая кислот;! в лите-
ратуре не описана.
Она, как и другие производные формамидинсульфиновых
кислот, может представить интерес как физиологически ак-
тивное вещество.
СХЕМА СИНТЕЗА N -МОНОМЕТИЛОЛФОРМАМИДИНСУЛЬФИНОВОЙ
КИСЛОТЫ
HN
/С -- SH + 2НаО2
CHSOHNH
HN
/С - SO2HH-2H2O
CHjOHNH
Характеристика основного сырья
N-Монометилолтиомочевина, т. пл. 95—96°.
Перекись водорода, медицинская, ГОСТ 177—55.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термометром
и капельной воронкой, загружают 3 г (0,03 М) монометилол-
тиомочевины и приливают 6 мл этилового спирта. Получен-
ие
ную суспензию охлаждают до 3° и при этой температуре мед-
ленно приливают 6 мл (0,06 Л4) 31,2%-ной перекиси водоро-
да. По окончании добавления всей перекиси водорода реак-
ционную смесь выдерживают при 3° в течение 30 минут, за-
тем полученный продукт отфильтровывают и сушат при тем-
пературе 50°.
Получают 0,8 г N-монометилолформамидинсульфиновой
кислоты, что составляет 20% от теоретического выхода.
Найдено, %: С-17,93; 17.73; И—4,17; 4,35.
СгН,М2О85. Вычислено, И: С -17,39; Н-4,35.
По внешнему виду вещество представляет собой белые
кристаллы с т. пл. 97—98°, не растворяется в воде и непо-
лярных органических растворителях, в спирте растворяется.
Поступила в ноябре 1965 г. ИРЕА
УДК 547.822.3.07
l-N-МОРФОЛИЛ-ПРОПАНОЛ-г И 1-N -ПИПЕРИДИЛ -
ПРОПАНОЛ-2
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
сн,-сн2 ч
of )N-CH2-CH-CH3
ХСН2-СН2 / |
он
С,Н]5МО2 М. в. 145,20
C8H17NO
,СН2-СН2.
сн2< fN-CH2-CH-CH3
\сн2- -СН/ I
он
М. в. 143,22
В литературе имеется сообщение о синтезе l-N-морфолил-
пропанола-2 [1].
Нами проверена и уточнена эта методика. Аналогично по-
лучен 1-Ы-пиперидил-пропанол-2.
Полученные соединения могут быть использованы в каче-
стве поверхностно-активных веществ и исходных веществ для
органического синтеза.
СХЕМА СИНТЕЗА I-N-МОРФОЛИЛ-ПРОПАНОЛА 2 И
1-М-ПИПЕРИДИЛ-ПРОПАНОЛА-2
СН3— сн2,
СН3-СН-СН2 4 of \NH->
ХСН2-С.Н/
,сн2- сн2.
-> о( ;n-ch2-ch-ch3
ХСН2- СН/ I
он
100
.сн,- сн2.
сн9—сн-сна+сн/ ;nh->
\_/ хсн,-сн/
О 2 2
сн2- сн.2ч
-> сн/ . N-СН,—СН-СН3
ЧСН,-СН/ ' |
ОН
Характеристика основного сырья
Морфолин, ч., МРТУ 6-09-443—63.
Пиперидин, ч., ТУ РУ 444—51.
Окись пропилена, свежеперегнанная, т. кип. 35°.
Условия получения
В круглодонную колбу, снабженную обратным холодиль-
ником, помещают 85 г (1 М) пиперидина, 5 мл воды и при
перемешивании прибавляют 58 а (1 М) окиси пропилена.
Экзотермическая реакция начинается только через 30 минут,
после чего реакционную смесь охлаждают водой до 20° и
оставляют при этой температуре на 10—15 часов. Затем на-
гревают 30 минут на кипящей водяной бане и отгоняют лег-
кокипящие продукты. Остаток перегоняют в вакууме.
Выход 1 -М'-пиперидил-пропанола-2 равен 128 а, что сос-
тавляет 89% от теоретического: т. кип. 59—6Г/6 мм; d,;21’ —
0,9263; — 1,4623.
Найдено, %: N—9,81; MRO —42,51.
C8H17NO. Вычислено, %: N-9,78: MRO —42,41.
Аналогично получают и 1-М-морфолил-пропанол-2. Для ре-
акции берут 87 а (1 М) морфолина, 5 мл воды и 58 а (1 М)
окиси пропилена.
Выход 1-М-морфолил-пропанола-2 равен 123 а, что состав-
ляет 88% от теоретического; т. кип. 77—79°/5 мм; du20—1,019;
п2° 1,4648.
Найдено, %; С -57,62; Н 10,59; MRD -39,36.
C7H)8NO,. Вычислено, %: С-57,90; Н—10,41; М%-39,43.
По литературным данным, т. кип. вещества 97—98°/13 мм;
— 1,4621 [1].
ЛИТЕРАТУРА
I. М. С. Малиновский. Окиси олефинов и их производные, М.,
Госхимиздат, 1961, стр. 309.
Поступила в июле 196.' г. Донецкий филиал ПРЕД
101
УДК 547.672.3.07
НАТРИЕВАЯ СОЛЬ 2-АНТРАЦЕНСУЛЬФОКИСЛОТЫ
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, Б. М. БОЛОТИН, Н. И. ЧЕРНОВА
C14H9OaNaS
SO;1Na
М. в. 280,27
Натриевая соль 2-антраценсульфокислоты может быть по-
лучена восстановлением натриевой соли 2-антрахинонсульфо-
кислоты цинковой пылью в среде аммиака. В заводских усло-
виях эта реакция проводится в автоклаве под небольшим дав-
лением. В целях осуществления упрощенного лабораторного
метода получения нами проверена и уточнена литературная
пропись [1], позволяющая вести синтез без избыточного дав-
ления.
СХЕМА СИНТЕЗА НАТРИЕВОЙ СОЛИ
2-АНТРАЦЕНСУЛЬФОКИСЛОТЫ
н- 2Zn -ь 4NH4OH
^YY4/S°sNa
+ 2 (NH4)tZnO8-l- 2HSO
Характеристика основного сырья
Натриевая соль 2-антрахинонсульфокислоты (серебристая
соль), ч„ ТУ МХП 4075—53.
102
Цинковая пыль, 85%-ная.
Аммиак водный, ч.д.а., 25%-ный, ГОСТ 3760—47.
Уксусная кислота, ледяная, х. ч., ГОСТ 61—51,
Натрий хлористый, ч., ГОСТ 4233—48.
Условия получения
В трехгорлую колбу емкостью 4 л, снабженную механи-
ческой мешалкой с затвором, обратным холодильником и тер-
мометром, загружают 150 г (0,48 М в пересчете на 100%-ную)
натриевой соли 2-антрахинонсульфокислоты, 192 г цинковой
пыли,. 5,1 мл ледяной уксусной кислоты, 150 мл водного ам-
миака и 600 мл дистиллированной воды. Смесь размешивают
при комнатной температуре 10 минут, нагревают на водяной
бане до 90° в течение 1 часа, размешивают при этой темпе-
ратуре 2 часа (см. примечание 1) и охлаждают до комнатной
температуры. Масса становится светло-серой. Осадок (I) от-
фильтровывают и промывают в три приема 450 мл 10%-ного
раствора хлористого натрия. Фильтрат и промывные воды на-
гревают до 90°, подкисляют до pH 3 концентрированной соля-
ной кислотой (~ 100 мл) и охлаждают. Выпавший оса-
док (II) отфильтровывают, промывают в два приема 100 мл
10%-ного раствора хлористого натрия.
Промытый реакционный осадок (I) переносят в четырех-
литровую колбу, снабженную механической мешалкой, термо-
метром и капельной воронкой, вносят 1,8 л воды и 300 г хло-
ристого натрия. Смесь размешивают, нагревают до 90° и за
4—5 часов приливают 900 мл концентрированной соляной
кислоты. (При быстром приливании кислоты образуется мно-
го пены). После растворения цинка суспензию охлаждают до
20°, осадок (1а) отфильтровывают, промывают в три приема
300 мл 10%-ного раствора хлористого натрия. Осадки 1а и
II объединяют (см. примечание 2) и сушат в сушильном
шкафу при температуре 100°.
Всего получают 125 г натриевой соли 2-антраценсульфо-
кислоты, что составляет 92 % от теоретического выхода. По
внешнему виду полученная натриевая соль 2-антраценсульфо-
кислоты серовато-белого цвета, довольно интенсивно флуо-
ресцирующая синим, вполне пригодна для получения из нее
2-антрола.
Примечания:
1. Необходимо регулировать скорость перемешивания массы, так как
образуется обильная пена.
2. Осадок I а по своему качеству хуже осадка II вследствие неболь-
шого содержания в нем цинка, но для переработки на 2-антрол оба осад-
ка вполне пригодны.
Л ИТЕРАТУРА
1. С. Liebermann. Liebigs. Ann. Chem.. 212, 57 (1882).
Поступила в ноябре 1965 г. ИРЕА
103
УДК 547.269.3-38 + 547.269.31
1-НАФТИЛИМИН0ДИУКСУСНАЯ-4-СУЛЬФ0КИСЛ'и
МОНОНАТРИЕВАЯ СОЛЬ 2-НАФТИЛИМИН0Д1 С-
НОЙ-1-СУЛЬФОКИСЛОТЫ
В. Я. ТЕМКИНА, Л. М. САМЫЛОВА, Р. П. ЛАСТОВСКИЙ
,снасоон
I ХСН,СООН
I II I -1/,н2о
I
SO3H
C14H13NO,S]/2H2O М. в. 348,32
SOsNfi
/СН2СООН
VHsCOOH
C14H12NO7SNa М.в. 361,30
1-Нафтилиминодиуксусная-4-сульфокислота (нафтионово-
диуксусная) и 2-нафтилиминодиуксусная-1 -сульфокислота
(Тобиас-иминодиуксусная) являются новыми комплексонами,
отличающимися друг от друга положением хелатной имино-
диацетатной группы в нафталиновом ядре. Соединения пред-
ставляют интерес с точки зрения их флуоресцирующих
и комплексообразующих свойств. Синтез соединений осу-
ществлялся методом взаимодействия соответствующих ами-
нов с монохлоруксусной кислотой в водной среде при pH 9—
10 [1]. В качестве нейтрализующего агента использовался
20%-ный водный раствор едкого натра. При подкислении вы-
делялись натриевые соли соответствующих комплексонов.
Нафтионоводиуксусная кислота получена нами ионообменным
методом с использованием ионитового сита КРС-5, которое
является сульфированным полимером стирола и дивинилбен-
зола с содержанием последнего 5% и коэффициентом набу-
хания 1,8.
104
СХЕМА СИНТЕЗА СОЕДИНЕНИЙ
,СН2СООН
NH, N;
| | ХСН2СООН
| II I + 2С1СН2СООН AAA. I II | + 2NaCl
А/\А 95’ Ч/\/
SOaH SO3H
SO3H
\]|j NaOH
I I; |-M12 __2CICH,COOH —50- —
X/\/
SO3Na
A_^CH’C°OH
A/А XCH,COOH + 2NoCl
Характеристика основного сырья
Нафтионовая кислота, ч., ГОСТ 93—58.
Амино-Тобиас кислота, ч., РТУ 3—57.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натр едкий, ч., ГОСТ 4328—48.
Соляная кислота, ч., ГОСТ ЗН8—46.
Условия получения
В трехгорлую колбу емкостью 250 мл с обратным холо-
дильником, термометром и мешалкой помещают Н,6 г
(0,05 /И) нафтионовой кислоты или Н,2 г (0,05 М) амино-
Тобиас кислоты. Затем приливают раствор 14,2 г (0,15 М)
монохлоруксусной кислоты в 15 мл воды, предварительно
нейтрализованной 20%-ным раствором едкого натра. Реакци-
онную массу нагревают при размешивании и температуре
90—95°, поддерживая по фенолфталеину pH 9—10 добавле-
нием 20%-ного раствора едкого натра. Нагревание продол-
; жают в течение 4—4,5 часов до прекращения снижения pH
среды. Раствор горячим фильтруют, охлаждают и подкисля-
,ют до pH 1,5—2 концентрированной соляной кислотой. Вы-
спавший объемистый осадок перекристаллизовывают из 150 мл
своды с добавлением активированного угля. Полученный после
‘перекристаллизации белый осадок отфильтровывают, про-
мывают 50 мл. холодной воды и сушат сначала на воздухе
105
без доступа света, затем в сушильном шкафу при температу-
ре 90—95° до постоянного веса.
Получают 10 г мононатриевой соли нафтионоводиуксусной
кислоты (I), что составляет 54% от теоретического выхода,
или 9,5 г мононатриевой соли Тобиас-иминодиуксусной кис-
лоты (II), что составляет 51 % от теоретического.
Для соединения!*:
Найдено, %: N -4,27; 4,36; Na—5,81; 6,05.
C14H12NO7SNa. 1/2Н2О. Вычислено, %: N-3,76; Na-6,21.
Для соединения II*:
Найдено, %: N—3,89; 4,18; Na—6,58: 6,73.
C14H12NOTSNa. Вычислено, %: N—3,88; Na—6,38.
Оба соединения хорошо растворимы в горячей воде и ще-
лочи, плохо растворимы в холодной воде, метиловом и этило-
вом спиртах .нерастворимы в бензоле, эфире, ацетоне.
Выделение нафтионоводиуксусной кислоты проводят сле-
дующим образом. Через колонку емкостью 50 мл, диаметром
14 мм с катионитом КРС-5 пропускают раствор 500 мг комп-
лексона в 100 мл воды. Объем фильтрата до проскока комп-
лексона 13 мл. Собирают фракцию фильтрата со значением
pH 1—2, отгоняют воду в вакууме при температуре 50°, в
конце отгонки добавляют в несколько приемов 100 мл абсо-
лютного этанола и отгоняют. Выпавший осадок фильтруют,
промывают 25 мл абсолютного этанола и сушат в сушильном
шкафу при температуре 85°.
Получают 450 мг нафтионоводиуксусной кислоты (III).
Для соединения III**:
Найдено, %: С—18,31; 48,59; 14—4,61; 4,55;
N 1,12; 4,31.
C]tH13NOTS. 1/2Н..О. Вычислено, %: С-48,27; И 4,05; N-1.02.
ЛИТЕРАТУРА
1. J. К. Aiken. Chem. h-d., 17, 1331 (1956).
Поступила в декабре I9v5 г. ИРЕА
* Соединение I представляет собой порошок бело-розового цвета, а
соединение II — порошок светло-желтого цвета.
** Соединение III представляет собой порошок светло-бежевого цве-
та, хорошо растворимый в воде, хуже в метиловом и этиловом спиртах,
нерастворимый в бензоле, эфире, ацетоне.
УДК 547.554.07
5-НИТРО-2-НАФТИЛАМИН
В. М. ДЗИОМКО, И. С. МАРКОВИЧ, И. В. КРУГЛОВА
C)0HsN2O3
I
NO3
М. в. 188,18
По литературным данным, 5-нитро-2-нафтиламин получа-
ют частичным восстановлением 1, 6-динитронафталина [1, 2];
из 2-нафтиламина обработкой нитрозилсерной кислотой [3, 4];
из азотнокислой соли р-нафтиламина при действии серной
кислоты [3, 5].
Нами были проверены как наиболее доступные пути син-
теза из 2-нафтиламина и азотнокислой соли 2-нафтиламина.
При действии нитрозилсерной кислоты на 2-нафтиламин нам
не удалось получить чистый 5-нитро-2-нафтиламин. Послед-
ний был получен по методу [5].
СХЕМА ПОЛУЧЕНИЯ 5-НИТРО-2-НАФТИЛАМИНА
Характеристика исходного сырья
2-Нафтиламин, ч., ТУ МХП 1543—47.
Азотная кислота, ч., ГОСТ 4461—48.
Серная кислота, ч., ГОСТ 4204—48,
107
Условия получения
Синтез азотнокислой соли 2-нафтиламина. Тщательно раз-
мешивают 143 г (1 М) 2-нафтиламина с 90 мл азотной кисло-
ты (d—1,36, см. примечание 1). Полученную азотнокислую
соль 2-нафтиламина перекристаллизовывают из воды и тща-
тельно высушивают при 100° (см. примечание 2).
Получение 5-нитро-2-нафтиламина. К 300 мл концентриро-
ванной серной кислоты при температуре от минус 5° до 5°
прибавляют 50 г азотнокислого 2-нафтиламина. Прибавление
проводят по возможности быстро. После растворения азотно-
кислой соли темно-красную реакционную смесь выливают в
6—8-кратное количество гводы (~2 л) и нагревают до кипе-
ния. Горячий раствор отфильтровывают от смолы (см. при-
мечание 3) и нерастворимого остатка. При охлаждении ма-
точника выпадает сернокислая соль 5- и 8-нитро-2-нафтилами-
на, которую переводят в основание с помощью обработки
10%-ным раствором аммиака. Смесь аминов перекристалли-
зовывают из этанола, при этом 5-нитро-2-нафтиламин выпада-
ет в осадок, который еще раз кристаллизуют из этилового
спирта.
Выход 5-нитро-2-нафтиламина равен 8 г, что составляет
18% от теоретического; т. пл. 140°.
По литературным данным, т. пл. вещества 140° [5].
Примечания:
1. При применении более концентрированной азотной кислоты наблю-
дается осмоление.
2. При температуре, выше 100° происходит разложение продукта.
3. Количество смолы увеличивается с повышением температуры прн
прибавлении азотнокислого 2-нафтиламина.
ЛИТЕРАТУРА
1 Н V е s е 1 v, М. Dvorak. Bull. Soc. ehlm. de France,, [4], 33
319(1923).
2. H. Hodgson, II. S. Turner. J. Chem. Soc , 1943, 318.
3. P. Friedlander, St. Szymanski. Ber , 25, 2078 (1892).
4, H, H. Hodgson. H Davey. J. Chem. Soc, 1939, 348.
5. G. T. Morgan, S. C h a r a n. J. Soc., Chem. Ind. 41, 1т (1922).
Поступила в декабре 1965 г. ИРЕА
УДК 547.571.07
5-НИТРО-2-ОКСИ-3-МЕТОКСИБЕНЗАЛЬДЕГИД
5-Нитро-З-метоксисалициловый альдегид
5-Нитро-О-ванилин
Д. Г. БРУДЗЬ, Д. А. ДРАПКИНА, В. А. ИНШАКОВА. И. П. ПЛИТИНА
СНО
I
<Л-0Н
O2N-^ '•CH,
C8H7NOs M. в. 197,14
5-Нитро-о-ванилин был получен нитрованием о-ванилина
в среде уксусной кислоты [1, 2, 3]. По [1] о-ванилин растворя-
ют в ледяной уксусной кислоте и прибавляют раствор азот-
ной кислоты (а= 1,42) в ледяной уксусной кислоте при 10—
20°. Выход сырца равен 67%. По прописи [2—3] к раствору
о-винилина в ледяной уксусной кислоте прибавляют дымя-
щую азотную кислоту при температуре ниже 5° [3]. Выход
продукта составлял 64,2% [2] и 78,7 7о [3].
Нами проверены методы нитрования [1, 3]. Ни в одном
случае не удалось полностью воспроизвести указанные в про-
писях результаты. Взяв за основу способ [1], мы уточнили вре-
мя прибавления азотной кислоты, время выдержки и усло-
вия выделения продукта.
СХЕМА СИНТЕЗА 5-НИТРО 2-ОКСИ-З-МЕТОКСИБЕНЗАЛЬДЕГИДА
СНО СНО
I I
ОН СН3СООН //\_ОН
^J-OCH3+HNO3 -* Q^-I^J — OCHa +Нз°
109
Характеристика основного сырья
Азотная кислота, ч., ГОСТ 4461—48.
Уксусная кислота, ледяная, х. ч., ГОСТ 61—51.
о-Ванилин с т. пл. 42,5—43,5°, выделенный из отходов про-
изводства ванилина по методу [4].
Условия получения
В фарфоровый стакан, снабженный мешалкой, термомет-
ром и капельной воронкой, загружают 70 мл ледяной уксус-
ной кислоты и 15,2 а (0,1 М) о-ванидина. Смесь охлаждают
до 10—14° и при размешивании в течение 1 часа прикапыва-
ют раствор 5 мл азотной кислоты (d— 1,42) в 20 мл ледяной
уксусной кислоты. Температуру смеси поддерживают в пре-
делах от 10 до 17°. Затем, сняв охлаждение, дают выдержку
1 час и реакционную смесь выливают на 100 г толченого льда.
Через 1 час осадок отфильтровывают, промывают водой до
нейтральной реакции. Получают 10—10,2 г сырого 5-ннтро-
о-ванилииа, что составляет 50,8—51,8% от теоретического;
т. пл. 137—138°. Альдегид растворяют при нагревании в
140 мл спирта. От полученного раствора отгоняют примерно
половину спирта; из остатка выкристаллизовывается 5-нитро-
о-ванилин, который повторно растворяют в 60 мл кипящей
ледяной уксусной кислоты. К горячему раствору добавляют
равный объем кипящей воды. По охлаждении выкристалли-
зовывается чистый 5-нитро-о-ванилин.
Выход равен 7,4 г, что составляет 37,6% от теоретического;
т. пл. 139—140°.
По литературным данным, т. пл. вещества 140—14Г [1];
138,5—140° [2]; 142° {3]; 136,5—137,5° [5].
ЛИТЕРАТУРА
1. W. Davies. .1. Chem. Soc., 123, 1583 (1923).
2. М. Murakami. Liebigs Ann. Chem., 496, 122 (1932),
3. В. B. D e у, V. Л. К u 11 i. Proc. Nat. Inst. Sci. India, 6, 641 (1940),
4. И. H. Бра тус, В. Г. Воронин, К. А. Богданов. Труды
ВНИИСНДв, выпуск V, 111 (1961).
5. J. Koetschet. Р. Koetschet. Helv. chlm. acta, 13, 482
(1930).
Поступила в ноябре 1965 г.
ИРЕА
УДК 547.571.07
3- и 5-НИТРОСАЛИЦИЛОВЫЕ АЛЬДЕГИДЫ
3- и 5-Нитро-2-оксибензальдегиды
В. Г. БРУДЗЬ, Д. А. ДРАПДИНА, В. А. ИНШАКОВА,
Н. И. ДОРОШИНА
СНО сно
JXX°H J\/0H
Г ' г <
C,HsNO4 М. в. 167,05
По литературным данным, 3- и 5-нитросалициловые аль-
дегиды получают нитрованием салицилового альдегида ды-
мящей азотной кислотой в ледяной уксусной кислоте, разде-
лением изомеров через их натриевые соли [1] или соли бария
[2] с последующей очисткой обоих альдегидов путем перекри-
сталлизации из разбавленной уксусной кислоты [1].
Однако при проверке метода [1] оказалось, что не всегда
удается получить 3-нитросалициловый альдегид с требуемой
температурой плавления кристаллизацией только из разбав-
ленной уксусной кислоты. Чистый продукт был получен после
дополнительной кристаллизации из изопропилового спирта,
разбавленного водой в отношении 1:1 по объему.
СХЕМА СИНТЕЗА 3- И 5-НИТРОСАЛ ИЦИЛОВЫХ АЛЬДЕГИДОВ
СНО
+ HNO.,
СН3СООН
ш
Характеристика основного сырья
Салициловый альдегид, ч.д.а., ГОХП 27-18—65.
Уксусная кислота, ледяная, х. ч„ ГОСТ 61—51.
Азотная кислота, ч., ГОСТ 4461 — 48.
Изопропиловый спирт, оч., ТУ ОРУ 50—57.
Натр, едкий, ч., ГОСТ 4328—48.
Условия получения
В фарфоровый стакан емкостью 1,5 л, снабженный мешал-
кой, капельной воронкой и термометром, помещают 100 г
(0,82 Л1) салицилового альдегида и 500 г ледяной уксусной
кислоты. Смесь охлаждают до 5—8° и примерно за один час
при перемешивании прибавляют 150 г азотной кислоты уд.
веса 1,5, поддерживая температуру реакционной смеси не
выше 10—15°. По прибавлении всей азотной кислоты снимают
охлаждение и дают температуре смеси подняться до 40°, пос-
ле чего реакционную массу немедленно выливают на 2000 г
воды со льдом (см. примечание).
Полученный осадок смеси 3- и 5-нитросалициловых альде-
гидов отсасывают, промывают водой до нейтральной реакции
и растворяют при нагревании в 1200 мл воды, содержащей
38—40 г едкого натра. Раствор оставляют на 15—20 часов
для кристаллизации. На следующий день отсасывают красно-
оранжевые игольчатые кристаллы натриевой соли 5-нитроса-
лицилового альдегида, содержащей примесь соли 3-нитроса-
лицилового альдегида. Соль тщательно отжимают и, не про-
мывая, перекристаллизовывают из минимального количества
дистиллированной воды.
Выпавший осадок фильтруют на следующий день, раство-
ряют в воде при нагревании и высаживают подкислением кон-
центрированной соляной кислотой.
Полученный светло-желтый осадок 5-нитросалицилового
альдегида кристаллизуют из 10 %-ной уксусной кислоты (на
1 г альдегида берут 45 мл растворителя).
Получают 38—45 г 5-нитросалицилового альдегида, что со-
ставляет 27,7—33% от теоретического выхода; т. пл. 125—
126°.
По литературным данным, т. пл. продукта 126° [1], 125° [2].
Оба фильтрата от натриевой соли альдегида объединяют
и упаривают до 1/4 первоначального объема. Выпавший ио
охлаждении осадок представляет собой натриевую соль
3-нитросалицилового альдегида. Ее отсасывают и кристалли-
зуют из воды. Перекристаллизованную соль 3-нитросалици-
лового альдегида растворяют в горячей воде и подкисляют
концентрированной соляной кислотой. Полученный 3-нитро-
салициловый альдегид очищают кристаллизацией из 10%-ной
112
уксусной кислоты и повторно из изопропилового спирта, раз-
бавленного водой в отношении 1=1 по объему (на 1 г альде-
гида берут 10 мл растворителя).
Выход 3-нитросалицилового альдегида равен 20—23 г, что
составляет 14,6—16,8% от теоретического; т. пл. 108—109°.
По литературным данным, т. пл. вещества 109—110° [1];
107° [2].
Примечание.
Обычно подъем температуры длится около 30 минут. К концу вы-
держки температура поднимается быстро и, если замедлить, выливание
реакционной смеси на воду со льдом, может произойти вскипание с вы-
бросом массы.
ЛИТЕРАТУРА
1. W. Miller. Вег., 20, 1927 (1887).
2. С. Tag е. Вег...2О, 211)9 (1887).
Поступила в июле 1965 г. ИРЕА
8 Зак. 25
УДК 547.574.2.07
6-НИТРОХИНАЛЬДОКСИМ
Оксим 6-нитрохинолин-2-альдегида
В. М. ДЗИОМКО. 3. С. СИДЕНКО. Г. С. ЧИЖОВА
V\nAch=noh
CioH7N303 М. в. 217,18
6-Нитрохинальдоксим в литературе не описан.
СХЕМА СИНТЕЗА 6-НИТРОХИНАЛЬДОКСИМА
0,N-Q( )_сно + NH=0H rjcr
OaN-/V '
\\ /\ //-CH-NOH
44 7 Ж
Характеристика основного сырья
Гидроксиламин солянокислый, ч.д.а., ГОСТ 5456—51.
Пиридин, ч., ГОСТ 2747—44.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Условия получения
В колбе, снабженной обратным холодильником, растворя-
ют при нагревании 1,8 г (0,01 М) 6-нитрохинолии-2-альдегида
в смеси 100 мл спирта и 20 мл пиридина. Затем добавляют
раствор 1 г (0,015 М) солянокислого гидроксиламина в смеси
3 мл спирта и 2 мл воды. Реакционную смесь кипятят
1,5 часа.
114
После охлаждения добавляют содового раствора до ще-
лочной реакции. Оксим выделяется в виде желтого осадка,
его отфильтровывают и кристаллизуют из спирта.
Выход аналитически чистого продукта равен 0,6 г, что со-
ставляет 33% от теоретического, считая на 6-нитрохинолин-2-
альдегид, т. пл. 248—249°. Мелкие кристаллы желтого цвета,
хорошо растворимы в спирте, уксусной кислоте, пиридине,
плохо в хлороформе, ацетоне, нерастворимы в воде.
Найдено, %: С—55, 61; 55,49; II -3,28; 3,47; N —19,22: 19,51.
C]0H,N3O3. Вычислено. %: С 55,25; Н—3,24j N-19,35.
Поступила в декабре IS65 г. ИРЕА
8*
УДК 547.571.07
6-НИТРОХИНОЛ ИН-2- АЛЬДЕГИД
В. М. ДЗИОМКО, 3. С. СИДЕНКО, Г. С. ЧИЖОВА
O2N-/X/
VU-CH0
CI0H6N2O3
М. в. 202, 161
6-Нитрохинолин-2-альдегид может служить исходным
продуктом для- синтеза различных соединений и описан срав-
нительно недавно [1]. В литературе имеется лишь одно сооб-
щение о получении 6-нитрохинолин-2-альдегида окислением
6-нитрохинальдина двуокисью селена.
Нами проверен указанный метод получения, уточнены
условия синтеза, выделения и очистки продукта.
СХЕМА СИНТЕЗА 6-НИТРОХИНОЛ ИН-2-АЛЬДЕГИДА
SeO;
(),N
L 1—сно
Характеристика основного сырья
Двуокись селена, ч., ТУ ГКХ 1502—61.
Пиридин, ч., ГОСТ 2747—44.
Мета-ксилол, ч., ТУ МХП 3600—53.
Условия получения
В трехгорлой колбе емкостью 200 мл, снабженной меха-
нической мешалкой, обратным холодильником и термометром,
растворяют при 70° 18,8 г (0,1 М) 6-нитрохинальдина, синте-
зированного по способу [2], в НО г мета-ксилола. К получен-
ие
ному раствору добавляют постепенно в течение 30 минут при
эффективном перемешивании 15,6 г (0,13 М) свежеприготов-
ленной двуокиси селена (см. примечание 1). После окончания
прибавления двуокиси селена реакционную смесь нагревают
три часа при 120°. Затем реакционную смесь еще в горячем
состоянии отфильтровывают от селена. По охлаждении из
фильтрата выпадают желтые кристаллы 6-нитрохинолин-2-
альдегида, которые перекристаллизовывают из пиридина.
Выход 6,2 г, что составляет 37% от теории, считая на
6-нитрохинальдин; т. пл. 198—200° (см. примечание 2). По ли-
тературным данным, т. пл. вещества 202—204° [1], выход не
указан.
Найдено, %: С—58,92; 59,07; 11-2,97; 3,10; N —13,97; 14,08
CjoH6N203 Вычислено, %: С-59,40; H-2,99;N- 13,85.
Примечания:
1. Продажную двуокись селена перед употреблением в реакцию воз-
гоняют по [3].
2. При многократных перекристаллизациях из пиридина, л-ксилола
поднять т. пл. до указанной в литературе не удалось.
ЛИТЕРАТУРА
1. Брит, патент 874 980 (1958).
2. И. К. У шеи к о, Л. И. Ч о в н и к. Ж. общ. химии, 30, 2662 (1960).
3. И. А. К р а с а в и н, О. В. И в а н о в, В. М. Дзиомко. Методы
получения химических реактивов и препаратов, вып. 9, М., ИРЕА, 1964,
стр, 11.
Поступила в декабре 1965 г, ИРЕА
УДК 547.831.7.07
1-(8-ОКСИ-2-ХИНОЛИЛ)-3,5-ДИМЕТИЛ ПИРАЗОЛ
И. А. КРАСАВИН, В. AI. ДЗИОМКО, Н. И. МИРОШКИНА,
Ю. И. РАДИН
ChHisNsO
М.в. 239,28
В литературе 1-(8-окси-2-хинолил)-3,5-диметилпиразол
не описан. Он получен нами с 87%-ным выходом при нагре-
вании 8-окси-2-гидразинохинолина с 2,4-пентандионом в ки-
пящем этаноле.
Чистый 1 - (8-окси-2-хинолил) -3,5-диметилпиразол крис-
таллизуется из этанола в бесцветных иглах с т. пл. 134—
134,5°; вещество практически нерастворимо в воде, но хоро-
шо растворяется в большинстве органических растворителей
и в водных растворах щелочей и кислот, очень медленно пере-
гоняется с водяным паром.
1-(8-Окси-2-хинолил) -3,5-диметилпиразол является хела-
тообразующим соединением.
Схема синтеза 1-(8-оксн-2-хинолил)-3,5-диметнлпиразола
ОН
+CHSCOCH2COCH3 ->
NHNH3
118
ОН
+ ‘2Н2О
СНз
Условия получения
В круглодонной колбе емкостью 100 мл с обратным холо-
дильником растворяют 3,50 г (0,02 Л4) 8-окси-2-гидразинохи-
нолина (см. примечание 1) в 40 мл этанола при нагревании
на водяной бане и добавляют 2,0 г (0,02 М) свежеперегнан-
пого 2,4-пентандиона. Раствор кипятят в течение 2—2,5 часов
и оставляют па ночь. Перед выделением продукта жидкость
с выпавшим из нее осадком выдерживают в ледяной бане
1—2 часа, затем кристаллы отсасывают, промывают этано-
лом (5—7 мл) и высушивают.
Выход сырого вещества равен 3,85—4,15 а, что составляет
80,5—86,8% от теоретического; т. пл. 133—134°.
После перекристаллизации из 50—55 мл этанола полу-
ч'ают 3,2—3,5 г бледно-желтых игл с т. пл. 133,5—134,5°
(см. примечания 2 и 3).
Примечания:
1. Применялся 8-окси-2-гидразинохинолин с т. пл. 178,5—179,5°, полу-
ченный по описанному нами ранее методу [I].
2. Совершенно бесцветное вещество можно получить посредством
перегонки с водяным паром, но такой способ пригоден для очистки
.тишь небольших количеств продукта (0,1 0,2 г), так как вещество пере-
гоняется крайне медленно.
3. Чистый образец 1-(8-окси-2-хинолил)-3,5-днметплпиразола в виде
бесцветных игл с т. пл. 134—134,5° был приготовлен перегонкой с. водя-
ным паром и перекристаллизацией из этанола и проанализирован.
Найдено, %: С-69.95; 69,93; Н-5,43; 5,45; N-17,29; 17,38
C]4H13N,O. Вычислено, %: С—70,27; Н—5,48; N —17,56,
ЛИТЕРАТУРА
I. И. А. К P а с а в и н, В. М. Дзиомко, Н. И. М и р о ш к и и а.
Методы получения химических реактивов и препаратов, вып. 14, М.,
ИРЕА, 1966, стр. 86.
Поступила и октябре 1966 г.
ИРЕА
УДК 547.655.4.07
1-(2-ПИРИДИЛАЗО)-2-НАФТОЛ
Г. С. ПЕТРОВА, В. А. ПЛАТОНОВА
C16HnN3O
М. в. 249,27
1-(2-Пиридилазо)-2-нафтол (ПАН) впервые был синтези-
рован Чичибабиным [1, 2]. В 1955 г. была установлена его
практическая ценность как реактива для аналитических опре-
делений. ПАН в настоящее время широко применяется как
комплексонометрический индикатор при определении меди,
цинка, кадмия, висмута, таллия и ряда других элементов
[3, 4].
Реактив получают сочетанием диазосоли 2-аминопиридина
с р-нафтолом в среде абсолютного спирта.
В основу прописи синтеза ПАНа, представленной ниже,
использована рецептура Чичибабина, в которую были вне-
сены некоторые видоизменения, а именно: после сочетания в
реакционную массу прибавлялась твердая углекислота взамен
пропускания углекислого газа. Использованный прием позво-
лил добиться не только нужного pH реакционной массы, но
благодаря охлаждению и наличию кристаллизационных цент-
ров обеспечил быстрое выделение реактива, не требующее
48-часовой выдержки. В этих условиях 1-(2-пиридилазо)-2-
нафтол получается в состоянии, близком к химически чистому,
без дополнительной перекристаллизации и удовлетворяет ана-
литическим требованиям *.
* Испытание аналитической пригодности реактива произведено
М 3. Парташниковой.
120
СХЕМА СИНТЕЗА 1-(2-ПИРИДИЛАЗО)-2-НАФТОЛА
J-NH2 + C6HnONO -Ь C3H6ONa. —
//\
I J-N=N—ONa + C6HnOH -f- C2H6OH
J-N=N-ONa
Характеристика основного сырья
2-Аминопиридин, ч., ВТУ МХП 3141—52.
Изоамилнитрит, ч., свежеперегнанный, ТУ ОРУ 22—55 [5].
Натрий металлический, ч.д.а., ТУ МХП 1664—50.
Спирт этиловый, абсолютный.
P-Нафтол, ч.д.а., 98,8%-ный, ГОСТ 5838—51.
Этиловый эфир, ч., ГОСТ 6265—52.
Условия получения
В круглодонную колбу емкостью 1 л загружают 300 мл
абсолютного этилового спирта и небольшими порциями до-
бавляют 20 г металлического натрия.
Для полного растворения массу нагревают на глицерино-
вой бане с обратным холодильником. К 'полученному этилату
натрия, охлажденному до комнатной температуры, прибавля-
ют 175 г (1,4 М) изоамилнитрита и 141 г а-аминопиридина
(1,5 М). Смесь перемешивают до полного растворения и ки-
пятят на песочной бане с обратным холодильником в течение
8 часов. По охлаждении выпадает в осадок диазосоль. К по-
лученной массе приливают трехкратное количество серного
эфира и оставляют на 12 часов. Диазосоль отфильтровывают,
промывают эфиром и сушат на воздухе до удаления запаха
эфира.
Получают 85 г светло-желтого порошка.
Сочетание с ^-нафтолом. В стакан емкостью 200 мл загру-
жают 29 г диазосоли и 130 мл абсолютного спирта, размеши-
121
вают 1 час, а затем прибавляют небольшими порциями 29,5 г
Р-нафтола (0,2 М). Размешивают 2 часа и дают суточную
выдержку. Затем в раствор при перемешивании загружают
небольшими порциями сухой лед до загустения массы. Вы-
павший осадок отфильтровывают, отжимают и переносят в
колбу, куда прибавляют 400 мл спирта. Кипятят с обратным
холодильником 15 минут, а затем фильтруют. По охлаждении
из фильтрата выпадают красные кристаллы азосоединения.
Кристаллы отфильтровывают, а фильтрат используют для по-
вторной операции экстрагирования.
Полученный раствор оставляют на 12 часов, выпавший
осадок отфильтровывают, отжимают и сушат до постоянного
веса при 60°.
Выход 1-(2-пиридилазо)-2-нафтола равен 15,2 г, что со-
ставляет 32% от теоретического, считая на р-нафтол.
По внешнему виду вещество представляет собой оранже-
вый порошок с т. пл. 137°.
По литературным данным, т. пл. продукта 137° [1, 2].
ЛИТЕРАТУРА
I. А. Е. Чичибабии, М. Д. Р я з а и ц е в. ЖРФХО, 47, 1571 (1915).
2. А. Е. Ч и ч и б а б и н, М. Д. Р я з а н ц е в. ЖРФХО, 50, 513 (1918).
3. А. И. Б у с е в, А. Г. Петренко. Заводск. лаборатория, 24, 12,
1449 (1958).
4. А. И. Б у сев, В. М. Иванов. Ж. аналит. химии, 19, 10, 1238
(1964).
5. А. И. Б у с е в, В. М. И в а н о в, Л. Л. Т а л и и о в а. Методы по-
лучения химических реактивов п препаратов, вып. 6, М., ИРЕА, 1962,
стр. 40.
Поступила в ноябре 1965 г.
ИРЕА
УДК 647.822.3.07
ТЕТРАБЕНЗОИЛ АЦЕТОНОЕВРОПИАТ ПИПЕРИДИНИЯ
Р. П. ЛАСТОВСКИИ, В. Я. ТЕМКИНА, Н. В. ЦИРЮЛЬНИКОВА
о- О
II
[Eu(C.Hs-C=CH-C-CHs)4]-C6H10NH8+
C45EuH48O8N М.в. 882,84
Комплексы р.з.э. с р-дикетонами, к которым относится и
тетрабензоилацетоноевропиат пиперидиния, находят примене-
ние в аналитической химии [1—3] и квантовой электронике
14]. По литературным данным, это соединение получают при
взаимодействии спиртовых растворов хлорида европия, бензо-
илацетона и пиперидина, взятых в стехиометрическом соотно-
шении [4, 5].
Нами был проверен описаний выше метод. Внесены уточ-
нения в отношении количества спирта, содержания в нем во-
ды и выхода готового продукта, а также оптимальной темпе-
ратуры реакции. Показано, что максимальный выход и луч-
шие люминесцентные свойства комплекса получаются при
использовании в качестве растворителя 90%-ного этилового
спирта в количестве 3 мл на 1 мМ бензоилацетона.
СХЕМА СИНТЕЗА ТЕТРАБЕНЗОИЛАЦЕТОНОЕВРОПИАТА
ПИПЕРИДИНИЯ
Eu(NO8)94-4CeHsCOCH2COCH5+4C6H10NH —
—> [Eu(C6HsCOCHCOCHs)4]C6H10NH2++ 3C6H10NH-HN(.)3
Характеристика основного сырья
Нитрат европия, ч., РЭТУ 711—61.
Бензоил ацетон, ч., ТУ ГКХ РУ 1756—62.
Пиперидин, ч., ТУ РУ 444—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
123
Условия получения
В колбу емкостью 50 мл с мешалкой и обратным холо-
дильником помещают 1,08 г (6,66 мМ) бензоилацетона, при-
ливают 20 мл 90%-иого этилового спирта и раствор нагрева-
ют до 50°.
К раствору бензоилацетона в спирте добавляют при раз-
мешивании заранее приготовленный и подогретый до темпе-
ратуры 50° раствор 0,4 г (1,18 лА1) нитрата европия в 1 мл
воды, К смеси растворов приливают 0,91 г (10,7 мМ) пипери-
дина. Раствор охлаждают, при этом выпадает желтый кри-
сталлический осадок, который отфильтровывают и сушат в
эксикаторе над СаС12.
Выход тетрабензоилацетоноевропиата пиперидиния равен
0,7 г, что составляет 68% от теоретического.
Найдено, %: Ей —17,22; 17,44; С-61,06; 60,80; Н-6,08; 6,05.
N—1,52; 1,57;
C^EuHisOsN. Вычислено, %: Ей- 17.22, С-61,28; Н—5,37: М-1,58.
ЛИТЕРАТУРА
1. L. Hol zbe cher. Chem. llsty, 47, 680(1953).
2. К. К a 1 i s t r a t о v, A, Plan, B. Osso ws t v. Analyt. chitn
acta, 22, 195(1960).
3. H. С. Полуэктов, Л. И. Кононенко, P. А. Витку н,
M. П. Никонова, Оптика и спектроскопия, 17, 73 (1964).
4. С. В г echer, Н. Samelson, A. Lempicki. J. Chem Phvs.,
42,1081(1965).
5. Н. Bauer, J. Blanc, D. L. Ross. J. Amer. Chem. Soc., 86,
5125(1964).
Поступила в декабре 19C5 г.
ИРЕА
УДК 547.822.3.07
ТЕТРАДИБЕНЗОИЛМЕТАНОЕВРОПИАТ
ПИПЕРИДИНИЯ
Р. п. ластовский, в. я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА
9
[Еи(С6Н6С—СН - С - С,Н5)4] -C6H6NH2+
Ce5EuH56O8N Mb. 1131,12
Комплексы р.з.э. с р-дикетонами, к числу которых принад-
лежит тетрадибензоилметаноевропиат пиперидиния, находят
применение в аналитической химии [1—3] и квантовой элек-
тронике [4].
По литературным данным, это соединение, содержащее
4 молекулы р-дикетона, получают при взаимодействии спир-
товых растворов хлорида европия, дибензоилметана и пипе-
ридина, взятых в стехиометрическом соотношении [4, 5].
Нами был проверен и уточнен указанный выше метод.
Оказалось, что наибольший выход вещества и лучшие люми-
несцентные свойства получаются при использовании в каче-
стве растворителя абсолютного этилового спирта в количе-
стве 24 мл на 1 мМ нитрата европия.
СХЕМА СИНТЕЗА ТЕТРАДИБЕНЗОИЛМЕТАНОЕВРОПИАТА
ПИПЕРИДИНИЯ
Eu(NO3j3 + 4CeH5COCH2COC6H5+4C6H1()NH—*
—> [Eu(C.H5COCHCOC6H5)4]-C5H]0NH2++3C6H]0NH-HNO3.
Характеристика основного сырья
Нитрат европия, ч., РЭТУ 711—61.
Дибензоилметан, ч., ТУ 6-09-430—63.
Пиперидин, ч„ ТУ РУ 444—51.
Спирт этиловый, абсолютный, ректифицированный. ГОСТ
5962—51.
125
Условия получения
В колбу емкостью 50 мл с мешалкой и обратным холо-
дильником помещают 0,5 г (1,47 мМ) нитрата европия. За-
тем приливают 10 мл этанола и при размешивании добавляют
заранее приготовленный раствор 1,5 г (6,69 мМ) дибензоил-
метана в 25 мл этанола. К смеси растворов приливают 0,86 г
(10,1 мМ) пиперидина. Выпадает осадок желтого цвета. Оса-
док отфильтровывают и сушат в эксикаторе над СаОг-
Выход тетрадибензоилметаноевропиата пиперидиния ра-
вен 1,3 г, что составляет 78% от теоретического.
Найдено, %: Еи-12,82: 13,07..
C65EuH6eOsN. Вычислено, %: Ен—13,44.
ЛИТЕРАТУРА
1. L. Holzbecher. Chem. llsty, 47,680 (1953).
2. К. Kallstratov, A. Plan, В. Ossowsky. Analyt. Chlm
acta, 22,195 (1960).
3. H. С. Полуэктов, Л. И. Кононенко, P. А. Витку»,
M. П. Никонова. Оптика и спектроскопия, 17, 73 (1964),
4. С. В г е с h е г, Н. S a m е 1 s о п, A. L е m р 1 с к 1. J. Chem. Phys.,
42,1081 (1965).
5. Н. Bauer, J. Blanc, D. L Ross. J. Amer. Chem. Soc., 86,
5125 (1964).
Поступила в декабре 1965 г.
ИРЕА
УДК 547.535.1.07
1, 2, 4, 5-ТЕТРАИЗОПРОПИЛБЕНЗОЛ
В. П. МАРШТУПА, Е. П. БАБИН, Л. И. МАРЫШКИНА,
И. С. МОРОЗОВА
CjgH3g
,сн3
- сн<
сн3
/СН3
- сн<
сн3
М.в. 246,43
В литературе известны методы получения 1, 2, 4, 5-тетра-
изопропилбензола, главным образом, алкилированием бензо-
ла изопропилхлоридом, пропиленом и другими алкилирующи-
ми агентами в присутствии катализаторов Фриделя-Крафтса-
Густавсона [1]. Методика же выделения 1, 2, 4, 5-тетраизо-
пропилбензола не описана и в связи с этим не уточнены вы-
ходы указанного алкилбензола.
В данной методике предложен способ получения чистого
I, 2, 4, 5-тетраизопропилбензола с выходом 75—80%.
СХЕМА СИНТЕЗА 1, 2, 4, 5 ТЕТРАИЗОПРОПИЛБЕНЗОЛА
сн3
, -СН
СдНе . « \ С3Н6
A1CI- СН3 А1С1з
Нзсх /СН3
нс+ 4-си
СНз
Н3с
СзН6
A1CI,
127
H3c CHS
сн
Н3С сн3 н.с сн3
си,
сн3
н3с
н3с
не-
,СН,
-сн<
сн3
Характеристика основного сырья
Бензол, х. ч., ГОСТ 5955—51, температура кипения 80,2°.
Алюминий хлористый, безводный, ГОСТ 4452—48.
Пропилен, 96—98%-ный.
Условия получения
В круглодонную трехгорлую колбу емкостью 5 л, снаб-
женную обратным холодильником, шнековой мешалкой, вра-
щающейся со скоростью 1,0—1,5 тысячи оборотов в минуту, и
барботером для подачи пропилена, загружают 1 кг высушен-
ного над хлористым кальцием бензола и засыпают 0,05 кг
безводного хлористого алюминия. Колбу помещают в водяную
баню, имеющую температуру 60° (±5°).
После достижения в реакционной среде заданной темпе-
ратуры подают с определенной скоростью высушенный над
серной кислотой пропилен, полученный дегидратацией изопро-
пилового спирта над природным алюмосиликатом. Скорость
подачи газа должна быть равна скорости его поглощения, что
определяется минимальным проскоком его через реакционную
колбу.
После подачи 2,3 кг (1230 л в пересчете на 100%-ный)
пропилена содержимое колбы промывают горячей (60—80°)
водой до нейтральной реакции промывных вод на ион хлора.
Из охлажденного до 15—20° алкилата отжимают на прессе
тетраизопропилбензол. Фильтрат после пресса перегоняют из
колбы Вюрца вплоть до появления в холодильнике кристал-
лов тетраизопропилбензола. Выделенный продукт кристалли-
зуют из этилового спирта.
128
Выход очищенного 1, 2, 4, 5-тетраизопропилбензола равен
2320 г, что составляет 78% от теоретического; т. пл. 117°;
т. кип. 2607775 мм.
1, 2, 4, 5-Тетраизопропилбензол растворим в диэтиловом
эфире, слабо растворим в бензоле, плохо растворим в этило-
вом спирте, нерастворим в воде.
ЛИТЕРАТУРА
1. Е. П. Бабин. Химическая промышленность, 6, 381 (1961).
1оступила в ноябре 1965 г. Донецкий филиал ИРЕА
9 Зак.
УДК 547.838.1.07
3, з; 4, 4'-ТЕТРАКАРБОКСИДИФЕНИЛОКСИД И
3, 3', 4, 4'-ТЕТРАКАРБОКСИ-(2, 2-ДИФЕНИЛПРОПАН)
Л1. Г. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И. КОРЕЛЬСКАЯ
СООН
НООС-^ /-О—€ ^-соон
СООН
С1вН10О9 М.в. 346,25
СООН
СН3 j
НООС-^ ^-С-^ ^-СООН
НООС СНз
С]8Н16О8 М.в. 372,33
Синтез кислот вышеуказанного строения впервые был опи-
сан в патентной литературе при получении нового класса по-
лиимидных полимеров [1].
Получение этих кислот может быть осуществлено окисле-
нием соответствующих тетраметилпроизводных пермангана-
том калия в растворе пиридина и воды [1, 2], разбавленной
азотной кислотой при высоких температурах и давлении [3,
4], при последовательном окислении азотной кислотой или
бромом, затем перманганатом калия в щелочной среде [2, 3].
Нами проверен способ получения 3, 3', 4, 4'-тетракарбок-
сидифенилоксида н 3, 3', 4, 4'-тетракарбокси-(2, 2-днфенил-
пропана) окислением соответствующих тетраметилпроизвод-
ных в щелочной среде, а также уточнены условия ведения
синтеза, выделения и очистки продукта.
130
СХЕМА СИНТЕЗА 3, У, 4, 4-ТЕТРАКАРБОКСИДИФЕНИЛОКСИДА (I)
И 3,3',4,4-ТЕТРАКАРБОКСИ-(2, 2-ДИФЕНИЛ ПРОПАНА) (II)
СН3
I. сн3-/:
о-
-СН3 • 8КМпО
СН3
COOK
коос-
-О-
:-COOK+8MnOs4-4KOH+4HsO
СООК
COOK
коос-
-о-
СООК Н- 4НС1
СООК
СООН
НООС-?
-O-'f ^-СООН + 4КС1
СООН
II. ЗНзС-^ /“С~Л /-СН, + 281<МпО/°лнопиРиДи-
\ = / | новая среда
сн, сн-
СН, COOK
__ I ______ I
-> ЗКООС-^ )"-С ^-СООК + 28 Мп0.2 Ч-
СН,
COOK 3
+ ЮНоО Ч 16КОН
сн3 С001<
__ I _!
КООС-^_^-С-^ ^-СООКЧ-4НС1 -
9* COOK СНз 1
СН3 ХСООН
-> Н00С-^~^— С-^-^-С00Н-НКС1
соон СНз
ПОЛУЧЕНИЕ 3,3' 4,4’-ТЕТРАКАРБОКСИДИФЕНИЛОКСИДА
Характеристика основного сырья
З.З'ДД'-Тстраметилдифенилоксид, ч. (см. примечание 1).
Пиридин, ч., ГОСТ 2747—44.
Кислота соляная, ч., ГОСТ 3118—46.
Калий марганцевокислый, техн., ГОСТ 5771—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Условия получения
В четырехгорлую круглодонную колбу емкостью 3 л,
снабженную механической мешалкой с вазелиновым затво-
ром, обратным холодильником и термометром, загружают
1000 мл воды, 200 мл пиридина и 36 г (0,16 М) 3, 3', 4, 4'-тет-
раметилдифенилоксида. Смесь интенсивно перемешивают,
нагревают до 85° и порциями по 25 г вводят марганцевокис-
лый калий в количестве 250 г (1,58 А!) в течение 4—5 часов
(см. примечание 2). После прибавления всего перманганата
реакционную массу охлаждают до 75° и отфильтровывают
на фарфоровой воронке через двойной бумажный фильтр.
Двуокись марганца промывают на фильтре сначала 50 мл
горячего пиридина, затем 4 раза горячей водой по 100 мл.
Основной фильтрат и промывные воды соединяют вместе,
переносят в ту же реакционную колбу, нагревают до 85°
и заканчивают окисление прибавлением 200 г (1,26 А4) пер-
манганата калия в течение 5 часов. После прибавления
всего количества перманганата калия реакционную массу
выдерживают в течение часа (см. примечание 3), затем горя-
чую реакционную смесь отфильтровывают на фарфоровой
воронке. Для полноты выделения продукта двуокись мар-
ганца промывают на фильтре горячей водой 3 раза по
100 мл. Основной фильтрат и промывные воды соединяют
вместе, переносят в перегонную колбу и упаривают на кипя-
щей водяной бане в вакууме от водоструйного насоса до
объема 500 мл. Полученный раствор переносят в двухлитро-
вый стакан, охлаждают ледяной водой до 5° и при перемеши-
вании постепенно прибавляют к нему около 100 мл концент-
рированной соляной кислоты до кислой реакции по универ-
сальной индикаторной бумаге (см. примечание 4). Кислый
132
раствор оставляют стоять в течение ночи при комнатной
температуре. Выпавший белый мелкокристаллический осадок
отсасывают на фарфоровой воронке и тщательно отжимают.
Сырой продукт в количестве 70—75 г перекристаллизовыва-
ют из 1400 мл 5%-ного раствора соляной кислоты, промыва-
ют охлажденной дистиллированной водой до отсутствия
ионов хлора в промывных водах и сушат при 80° до постоян-
ного веса. Из маточника после выделения путем упаривания
и перекристаллизации из воды получают дополнительно
около 4 г продукта.
Общий выход 3,3',4,4'-тетракарбоксидифенилоксида 30,6—
33.6 г, что составляет 55—60% от теоретического; т. пл.
227—229°, что соответствует литературным данным [2].
ПОЛУЧЕНИЕ 3,3', 4,4’-ТЕТРАКАРБОКСИ-
(2,2-ДИФЕНИЛПРОПА НА J *
Характеристика основного сырья
3,3',4,4'-Тетраметилдифенилдиметилметан, ч. (см. соответ-
ствующую статью в настоящем сборнике).
Пиридин, ч., ГОСТ 2747—44.
Калий марганцевокислый, ч.д.а., ГОСТ 4527—48.
Натр едкий, х.ч„ ГОСТ 4328—48,
О-Ксилол, ч„ ГОСТ 2949—51.
Кислота соляная, ч., ГОСТ 3118—42.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Ацетон, ч.д.а., ГОСТ 2603—63.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную механиче-
ской мешалкой с ртутным затвором, обратным холодильни-
ком и термометром, загружают 40 г (0,15 М) З.З'ЛД'-тетра-
метилдифеиилпропана, 800 мл пиридина и 400 мл воды. Смесь
нагревают при перемешивании до 96—98° и небольшими пор-
циями в течение 1,5—2 часов прибавляют 100 г (0,6 Л1) мар-
ганцевокислого калия, затем, не прекращая перемешивания,
реакционную массу выдерживают при указанной температуре
в течение 3 часов. После выдержки горячую смесь фильтруют
па фарфоровой воронке через двойной бумажный фильтр.
Двуокись марганца промывают на фильтре сначала 50 мл
горячего пиридина, а затем 4 раза горячей водой по 100 мл.
Промывные воды и основной фильтрат соединяют вместе, пе-
* В получении 3,3',4,4'-тетракарбокси-(2,2-дифеннлпропана) участво-
вали С. А. Кочеткова и А. В. Кочеткова.
133
реносят в перегонную колбу, упаривают под вакуумом от во-
доструйного насоса до объема 100 мл, переносят в ту же
реакционную колбу, прибавляют 1 л 7%-ного раствора едкого
натра, нагревают до 96—98° и заканчивают окисление прибав-
лением 150 г (1,0 Л1) перманганата калия, как описано выше.
После окисления реакционную массу выдерживают при 96—
98° в течение одного часа (см. примечание 3), затем фильт-
руют на фарфоровой воронке через двойной бумажный
фильтр. Двуокись марганца промывают на фильтре несколько
раз горячей водой. Основной фильтрат и промывные воды
соединяют вместе и упаривают до объема 600 мл. Упаренный
фильтрат переносят в двухлитровый стакан, охлаждают ледя-
ной водой до 10° и небольшими порциями при перемешива-
нии прибавляют к нему концентрированный раствор соляной
кислоты до pH 1 по универсальной индикаторной бумаге, на
что расходуется около 240 мл кислоты (см. примечание 4).
Кислый раствор оставляют на ночь при комнатной темпера-
туре. Выпавший мелкокристаллический осадок фильтруют на
фарфоровой воронке и тщательно отжимают. Сырой продукт
(90—95 г) обрабатывают 5%-ным раствором соляной кисло-
ты следующим образом: 90 г сырого продукта растворяют
при нагревании в 70 мл 5%-ного раствора соляной кислоты;
раствор доводят до кипения, кипятят в течение 10 минут и
фильтруют в горячем виде.
Фильтрат охлаждают до 10° и выдерживают при этой тем-
пературе около 2 часов. Выпавший осадок отфильтровывают
и сушат при 80° до постоянного веса. Выход 3,3',4,4'-тетракар-
боксидифепилпропана составляет 37—40 г, т. пл. 163—165°.
Очистка технического продукта. Переносят 40 г сухого
растертого в ступке продукта в круглодонную емкостью
0,75 л колбу, снабженную обратным холодильником и меха-
нической мешалкой, добавляют 450 мл ацетона и нагревают
при перемешивании на водяной бане с закрытым электрообо-
гревом до кипения. Кипящий раствор выдерживают 15 минут,
затем фильтруют через бумажный фильтр. Фильтрат упари-
вают на водяной бане под вакуумом от водоструйного насоса
до образования подвижного светло-желтого масла; затем при-
бавляют 100 мл о-ксилола, нагревают до 70° и перемешивают
в течение 30 минут. Выпавший осадок отфильтровывают, су-
шат при 80° до постоянного веса.
Выход чистого продукта равен 36—39 г, что составляет
61—64,3% от теоретического; т. пл. 184—185°, что соответст-
вует литературным данным [3].
Примечания:
1. Получение 3,.3/,4,4/-тетраметилдифенилоксида см. «Методы полу-
чения химических реактивов и препаратов», вып. 12, стр. 105.
2. Каждую последующую порцию перманганата калия добавляют пос-
ле раскисления предыдущей. Темно-малиновая окраска раствора от пер-
1.34
манганата калия должна перейти в бурую от образовавшейся двуокиси
марганца.
3. В случае иераскисления последней порцнн перманганата калия
после выдержки реакционную массу охлаждают до 60° и при перемеши-
вании прибавляют 30 мл этилового спирта.
4. Во избежании вспенивания с выбросом реакционной массы кисло-
ту прибавляют небольшими порциями н при перемешивании.
ЛИТЕРАТУРА
1. W. F. Gresham, М. A. Naylor. Пат. США 2731447; Р. ж.
21029 п. (1957).
2. С. S. М а г v е 1, J. Н. Rossweiler. J. Amer. Chem. Soc. 80,.
6600 (1958). Пат. США 2712543.
3 W. F. G г е s h a m, M. A. N а у 1 о r. C. A., 50, 5755 (1956).
4. Англ. пат. 662139; С. A., 46, 11242 (1952).
Поступила в ноябре 1965 i. HPFA
УДК 547.638.1.07
3, 3', 4, 4/-ТЕТРАМЕТИЛДИФЕНИЛДИМЕТИЛМЕТАН
3, 3', 4, 4'-Тетраметилдифенилпропан
С. А. КОЧЕТКОВА, А. В. АФАНАСЬЕВА
Н3С СН3 СНз
сн8
М. в. 252,40
3,3',4,4'-Тетраметилдифенилдиметилметан применяется для
синтеза 3, З'ЛЛ'-тетракарбоксидифенилдиметилметана. В па-
тентной литературе описаны методы получения З.З'ЛЛ'-тетра-
метилдифенилдиметилметана конденсацией 2,2-дихлорпропана
с о-ксилолом в присутствии безводного хлористого алюминия
на холоду [1, 2].
Нами проверены и подтверждены патентные данные полу-
чения 3,3',4,4'-тетраметилдифенилдиметилметана.
СХЕМА СИНТЕЗА 3,3',4,4' ТЕТРАМЕТИЛДИФЕНИЛДИМЕТИЛМЕТАНА
сн3 С1
Cl
НзС СН3 СИ»
— НаС-^~^-С-^~^-СНз+2НС1
\=/ |
СН3
136
Характеристика основного сырья
о-Ксилол, ч., ГОСТ 2949—51.
2,2-Дихлорпропан (см. статью «2,2-Дихлорпропан» в на-
стоящем сборнике, стр. 83).
Алюминий хлористый, субл., ГОСТ 3759—47.
Магний сернокислый, ч.д.а., безводный, ГОСТ 4523—48.
Условия получения
В трехгорлую колбу емкостью 1 л снабженную двурогим
форштосом, ртутным затвором, мешалкой, капельной ворон-
кой, термометром, шариковым холодильником, помещают 26 г
(0,2 Л1) хлористого алюминия, охлаждают до 0° и при непре-
рывном перемешивании прибавляют из капельной воронки
360 мл (318 г; ЗЛ4) предварительно охлажденного до 0°о-кси-
лола. Реакционную массу интенсивно перемешивают, охлаж-
дают до минус 4—5° и к ней в течение полутора часов при-
бавляют из той же капельной воронки смесь, состоящую из
113 г (1 М) 2,2-дихлорпропана и 120 мл (106 г; 1 М) о-кси-
лола. После прибавления смеси реакционную массу, не пре-
кращая перемешивания, выдерживают в течение 4 часов при
температуре минус 4—5°, затем выливают на лед (500 г).
Верхний органический слой отделяют, промывают последова-
тельно 3%-ным водным раствором едкого натра (3 раза по
50 мл) и дистиллированной водой до слабокислой реакции
по универсальной индикаторной бумажке, сушат над серно-
кислым магнием в течение 24 часов и отгоняют избыток
о-ксилола при 40—45°/20—30 мм. Остаток после отгонки о-кси-
лола переносят в химический стакан и охлаждают до 14—15°;
выпавшие кристаллы ЗД'ДД'-тетраметилдифенилдиметилме-
тана отфильтровывают и сушат до постоянного веса в ваку-
ум-эксикаторе над хлористым кальцием.
После перекристаллизации из метанола выход чистого
З.З'ЛЛ'-тетраметилдифенилдиметилметана равен 89 г, что со-
ставляет 35,3% от теоретического; т. пл. 52—53°.
Найдено, %: С—90,57; 90,30, Н—9,58; 9.48.
С19Н24. Вычислено, %: С—90,40; Н—9,57.
ЛИТЕРАТУРА
1. W. F. Gresham, М. A. Naylor. Пат. США 27Г2543; С. А.,
50, 5755 (1956).
2. W. F. Gresham, М. A. Naylor. Пат. США 2731447; Р. ж.,
1957, 21029 П.
УДК 547.416.07
р, 3 ,р"-триаминотриэтиламин солянокислый
Трен
Р. П. ЛАСТОВСКИЙ, И. В. ХВОСТОВ, И. Д. КОЛПАКОВА,
Е. А. ЗЛОТИИА, В. Н. Ш КАДО В А
N(CH2CH2NH2)3-3HC1
CeH21N4CI3 М. в. 255, 62
Триаминотриэтиламин образует стабильные комплексы с
рядом катионов (Cu2+, Zn2T, Со2+, Со8 + ), а также может
служить полупродуктом для синтеза других комплексообра-
зующих соединений [1]. Кристаллы солянокислой соли триа-
минотриэтиламина характеризуются значительным эффектом
Поккельса, оптической активностью, стабильны на воздухе
[2]. Известны и другие области применения трена [3].
Триаминотриэтиламин получают по методике Ристенпарта
[4] пропусканием тока сухого аммиака через расплавленный
бромэтилфталимид [5] при 140—150° в течение 5—8 часов.
При последующем кислом гидролизе выделенного трис-(фтал-
имидоэтил) амина получают хлоргидрат трена—белый кри-
сталлический порошок, разлагающийся выше 300°, Путем об-
работки хлоргидрата щелочью и экстракции спиртом выде-
ляют само основание, которое представляет собой бесцвет-
ную вязкую жидкость, растворимую в спирте и хлороформе
и нерастворимую в эфире, присоединяющую на воздухе
угольную кислоту. Продукт перегоняется при 263°/744 мм и
имеет d19—0,977.
Описан также способ получения трена по реакции взаимо-
действия трихлорэтиламина (или его солянокислой соли) со
фталимидом калия [6].
Нами проведен синтез трена по методике Ристенпарта,
проверены и усовершенствованы отдельные стадии этой мето-
дики, а также разработаны технологические условия про-
цесса.
138
СХЕМА СИНТЕЗА ТРИАМИНОТРИЭТИЛАМИНЛ СОЛЯНОКИСЛОГО
/СО--
BrCH2CH2Br+KN/
XC0-t
CO-AX
BrCH2CH3N\ | j| 4-KBr
4NH3+3BrCH2CH2N/
\со-
N | CH2CH2N /
/ /CO-f>,\ HC1
N CH,CHsn/ + 8H2O -Ad__>
\ XcoAA
AX ~cOOH
— N(CH2CHtNH2)8-3HCl+3| |l
^y-COOH
Характеристика основного сырья
Фталимид-калий, ч., МРТУ 6-09-2185—65.
Дибромэтан, ч.д.а., ГОСТ 2569—44.
Диметилформамид, ч., МСТУ 6-09-2068—65.
Углерод четыреххлористый, ч.д.а., ГОСТ 5827—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Уксусная кислота, ледяная, х.ч., ГОСТ 61—51.
Аммиак (из баллона), осушенный над твердым едким
кали.
Соляная кислота, ч.д.а., ГОСТ 3118—46.
Условия получения
Синтез бромэтилфталимида *. В трехгорлую колбу емко-
стью 1 л, снабженную мешалкой, обратным холодильником и
термометром, помещают 87 г (0,47 А1) фталимида калия,
45 мл (0,52 Л1) дибромэтана и 100 мл диметилформамида.
Смесь при перемешивании нагревают на водяной бане, дово-
Синтез проведен по методике [7].
139
дя температуру бани до 60—70°. При этой температуре на-
чинается экзотермическая реакция с резким скачком темпе-
ратуры в реакционной массе до 120°. После этого нагревание
прекращают, дают смеси охладиться до комнатной темпера-
туры, добавляют 500 мл четыреххлористого углерода и выли-
вают в 2000 мл воды. Водный слой отделяют от органического
и экстрагируют дважды порциями по 200 мл четыреххлори-
стого углерода. Общий органический слой промывают 400 мл
0,2 н. едкого натра и 400 мл воды.
После отгонки четыреххлористого углерода в вакууме по-
лучают 90 г бромэтилфталимида — белого кристаллического
вещества с т. пл. 76—80° (по литературным данным, 79—
81°).
Найдено, %: С-47,96; Н -3,28; N -5,61; Вг—31,5.
Ctl)Ht2NO2Br. Вычислено, %: С -47,3; Н— 3,14; N—5,51; Вг 31,4.
Получение трис-(фталимидэтил)амина. В трехгорлую кол-
бу емкостью 150 мл, снабженную мешалкой, трубкой для про-
пускания аммиака, обратным холодильником, соединенным с
поглотителем аммиака, и масляной баней для обогрева, по-
мещают 80 г (0,32 М) бромэтилфталимида и включают обог-
рев. После того как температура бани достигнет 150°, вклю-
чают перемешивание и подачу сухого аммиака. Расход амми-
ака контролируют по предварительно отградуированному рео-
метру. На полное аминирование расходуется 12 л (0,5 М) ам-
миака. Через 8 часов прекращают подачу аммиака и в горя-
чем состоянии полученный продукт трижды экстрагируют
97%-ным спиртом порциями по 50 мл. Осадок хорошо отжи-
мают и перекристаллизовывают из 150 мл кипящей ледяной
уксусной кислоты. При этом получают 45 г бромгидрата в
виде белого кристаллического вещества с т. пл. 232°.
Найдено, %: С 56,17; Н—4.67; Вг —12,93.
Ca0H87N4OeBi. Вычислено, %: С—58,32; В—4,08; Вг —12,94.
После обработки бромгидрата щелочью получают 39 г бе-
лого порошкообразного продукта с т. пл. 185—187° (по ли-
тературным данным, 187°).
Найдено, %: С—67,05; Н-4,65; N—10,44.
C»oH3eN4Oe. Вычислено, %: С—67,16; Н—4,51; N—10,44.
Получение $,$\$"-триаминотриэтиламина солянокислого.
В колбу емкостью 500 мл загружают 15 г трис-(фталимидо-
этил) амина и очень осторожно кипятят с обратным холодиль-
ником с 300 мл разбавленной (Г.1 по объему) соляной кисло-
ты в течение ~7 часов. По охлаждении прозрачного раствора
выпадает фталевая кислота; ее отделяют, а фильтрат кон-
центрируют до небольшого объема, при смешении со спир-
том выпадает трен-хлоргидрат—белый кристаллический по-
рошок, разлагающийся выше 300°.
140
Выход равен 5 г. Были получены пикрат трена с т. пл. 229°
(по литературным данным, т. пл. 228—230°) и трибензоильное
производное трена с т. пл. 146—147° (по литературным дан-
ным, т. пл. 146—148°).
Найдено, %: С—28,09; Н- 8,06; С1—41.67.
CeH2lN4Cle. Вычислено, %: С-28,2; Н-8,22 С1—41,68.
Общий выход триаминтриэтиламина солянокислого 12,8%,
считая на исходный фталимид-калия.
ЛИТЕРАТУРА
1. N. Barclay. J. Chem. Soc., 1958, 2540.
2. К. Burer. Applied Optics, 4, 5, 545 (1965).
3. M. Jonassen и лр. J. Phys. Chem., 63, 411 (1959); C. R e i 11 e у
и др. Hemist-Analyst, 46, 3, 59 (1957).
4. В. Ris ten part. Ber.. 29 ,2531 (1896).
5. S. Jabriel. Ber., 20, 27'7 (1887); Ber., 21, 566 (1888); Ber., 25, 2753
(1892).
6. Венгерский пат. 143607; Chem. Zbl., 1961, 14097.
7. J. Sheehan и др. J. Amer. Chem. Soc., 1950, 72, 2786.
Поступила в декабре 1965 г. ИРЕА
УДК 547.554.07
ТРИДИБЕНЗОИЛМЕТАНАММИН-ЕВРОПИЙ
Р. И. ЛАСТОВСКИЙ, В. я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА
2Eu-nh3
3
C«EuH38OeN
М. в. 838,74
Тридибензоилметанаммин-европий наряду с другими ком-
плексами р.з.э. с р-дикетонами находит применение в анали-
тической химии [1—3] и квантовой электронике [4].
По литературным данным, это соединение получают при
добавлении аммиака к водно-спиртовым растворам нитрата
европия и р-дикетона [5].
Нами был проверен и уточнен описанный метод. Оказа-
лось, что лучшие результаты получаются при использовании
в качестве растворителя абсолютного этилового спирта.
СХЕМА СИНТЕЗА ДИБЕНЗОИЛМЕТАНАТА ЕВРОПИЯ
Eu(NOs)3 + 3C6H6COCH2COC6H54 4NH3 —
Eu(C6H6COCHCOC6H6)»-NHs + 3NH4NO3
Характеристика основного сырья
Нитрат европия, РЭТУ № 711—61.
Дибензоил метан, ТУ 6-09-430—63.
Аммиак (из баллона).
Спирт этиловый, абсолютный, ректифицированный, ГОСТ
5962—51.
Условия получения
В колбу емкостью 250 мл с мешалкой и обратным холо-
дильником помещают 0,85 г (2,51 мМ) нитрата европия. За-
142
тем прйлив^ют 10 мл абсолютного спирта и при размешива-
нии добавляют заранее приготовленный спиртовой раствор
2 г (8,92 мМ) дибензоилметана в 150 мл абсолютного спирта.
К смеси растворов приливают 17,5 мл аммиачного спиртового
раствора, содержащего 0,2 г (12,6 мМ) аммиака. Выпавший
осадок желтого цвета отфильтровывают и сушат в эксикаторе
над СаС12.<
Выход тридибензоилметанаммин-европия равен 1,6 г, что
составляет 75% от теоретического; т. пл. 120—125°.
Найдено, %: С-64,15; 64,25; Н-4,43; 4,75; N—1,53; 1,31;
0 __21' 21 4
CisEuI ls6O6N. Вычислено, %: С—64,43; Н- 4,32: N—1,67; Eu*Os—20,98
ЛИТЕРАТУРА
1. L. Holz be с lie г. Chem. listy, 47, 680 (1953).
2. К. Kalislratov, А. Р f а п, В. Ossowskv. Analyt. chim.
acta, 22, 195 (1960).
3. H. С. Полуэктов. Л. И. Кононенко, P. А. Виткун,
M. II. Никонова. Оптика и спектроскопия, 17, 73 (1964).
4. С. В г е с li е г, Н. Sa me Ison, A. L с т р 1 с к 1. J. Chem. Phys.
42, 1081 (1965).
5. L. Sa с coni, R. Ercoli. Gazz. chim. ital., 79, 731 (1949).
Поступила в декабре 1965 г. ИРЕА
УДК 547.754.07
1, 3, 3-ТРИМЕТИЛСПИР0-|(2'Н-1'-БЕН30ПИРАН)-2, 2'-
ИНДОЛИНЫ]
В. Г. БРУДЗЬ, Д. А. ДРАНКИНА, В. А. ИНШАКОВА,
Н. И. ДОРОШИНА, И. И. БАДАИКОВА
сн, СН,
Спиропираны представляют интерес в качестве фотохром-
ных веществ [1—5].
Пропись получения спиропиранов этого ряда дана в рабо-
те [1].
Нами проверен этот метод получения, подобраны более
подходящие растворители для очистки 6'-бром-8'-нитро- и
в'-бром-б'-нитро-производных; получен и охарактеризован
8'-нитроспиропиран этого ряда, не описанный в упомянутой
работе.
СХЕМА СИНТЕЗА 1ДЗ-ТРИМЕТИЛСПИРО-[(2'Н-1'-БЕНЗОПИРАН)-
-2,2'-ИНДОЛИНОВ]
СН
СНО
;\/0H
R I R"
R’
спирт
144
СНз сн3
Характеристика основного сырья
Спирт этиловый, абсолютированный, ГОСТ 9674—61.
Спирт этиловый, синтетический, ГОСТ 9674—61.
Пиридин, ч., ГОСТ 2747—44.
Диметилформамид, ч., ВТУ РУ 1193—56.
5-Нитросалициловый альдегид, ч., ТУ TCP 563 р—61.
4-Нитросалициловый альдегид (получение см. в [6]).
З-Нитросалициловый альдегид (см. стр. 111 в настоящем
сборнике).
5-Бромсалициловый альдегид, ч., СТУ 79-443—60.
З-Бром-5-нитросалипиловый альдегид (см. стр. 32 в на-
стоящем сборнике).
5-Хлорсалипиловый альдегид, ч., ТУ 338—65.
5-Хлор-З-нитросалициловый альдегид (см. стр. 171 в на-
стоящем сборнике).
5-Нитро-о-ванилин (см. стр. 109 в настоящем сборнике).
1,3,3-Триметил-2-метилениндолин (получение см. [8]).
Условия получения
В двугорлую колбу на 50 мл, снабженную мешалкой и об-
ратным холодильником, загружают 0,01 Л4 соответствующего
замещенного салицилового альдегида и 25 мл абсолютиро-
ванного спирта; при размешивании добавляют 1,73 г (0,01 Л4)
свежеперегнанного 1,3,3-триметил-2-метилениндолина. Смесь
нагревают на водяной бане при температуре кипения спирта
в течение 3—4 часов. На следующий день фильтруют выпав-
ший осадок и промывают 3—5 мл спирта. Полученный спиро-,
пиран кристаллизуют 1—3 раза. Несколько иначе очищают
б'-бром- и 6'-хлор-8'-нитропроизводные. Для отделения нуж-
ного спиропирана от продукта дальнейшей конденсации
с 1, 3,3-триметил-2-метилен-индолином (см. [8]) осадок кипя-
тят 2—3 раза с небольшим количеством спирта (15—20 мл)
в течение 10 минут и фильтруют горячим от нерастворившей-
ся части. Фильтраты объединяют и концентрируют до не-
большого объема (~'/’з первоначального объема). Выпав-
ший осадок кристаллизуют 1—2 раза из спирта.
Данные по выходам, очистке и характеристике получен-
ных соединений приведены в таблицах 1 и 2.
10 Зак. 25 145
Таблица 1
Условия очистки и выходы 6'R, 7'R', 8'R" -1,3,3-триметилспиро-
[(2'Н-Г-бензопиран)-2,2'-нндолинов)
№ соеди- нения Заместители Кристаллизация Выход от теоретиче- ского, %
R R’ R" растворитель мл иа 1г спирана число кристал- лизаций1
сырца ЧИСТО- ГО
1 no2 Н Н спирт 50 1 90,0 80,7
2 н NO, Н спирт 40 1 40,4 28,0
3 н 11 no2 спирт 20 2 80,1 48,8
4 Вг Н н спирт 20 2 11,8 8,4
5 no2 н Вг 1) пиридин 4- вода (Ы) 2) диметилформ- амнд + вода (1:1) 40 30 }2 92,2 74,3
6 н С1 Н спирт 20 1 44,8 34,6
7 С1 н no2 спирт 30 1 70,0 33,4
8 NOa н осн3 1) спирт 65 2
2) ацетон + вода (2:1) 27 1 73,0 56,8
Таблица 2
Характеристика 6'R, 7'R', 8R"-1,Э,3-триметилспиро-(2'Н-1'-бензопн-
ран-2,2'-индолинов)
соеди- нил Температура плавления, °C Внешний вид спиропи- ранов Брутто- (формула Элементы Данные анализа
вы- чис- лено, И найдено, %
чистого полите- ратурн. данным
£ О)
1 177,5- — 178 176—177 И] желтый С н N 70,78 5,63 8,69 70,46; 69,85 5,85; 6,12 8,42; 8,73
2 113-114 107—108 1 желтый 1,1 I c19h,8n2o, С Н N 70,78 5,63 8,69 70,65; 70,87 5,72; 5,42 8,54; 8,37
3 147-148 — серый C[9H18N2Oe С н N 70,78 5,63 8,69 71,45; 71,22 5,89; 5,79 8,58; 8,52
146
Продолж. табл. 2
№ соеди- нения Температура плавления, °C Внешний вид спиропи- ранов Бру гто- формула I Элементы Данные анализа
вы- ЧИСт л ено. % найдено, %
чистого по лите- ратурн. данным
4 85,5-87 86-87 белый C,sH]sNOBr с 64,07 64,0; 63,87
[9] н 5,09 5,38; .5,31
N 3,93 3,4п; 3,75
Вг 22,16 22,28; 21,98
5 248 -249 255-256 коричне- CjgH^N^OgBr С 56,87 56,67; 56,33
с разло- Г] вый с н 4,3 4,94; 4,93
жением блеском N 6,97 6,26; 6,54
6 118—119 116-117 белый С! Hl8NOCl С 73,2 72,88; 73,30
и 5,8 5,83; 6,07
N 4,49 4,62; 4,42
7 134-135 134-135 ^грязно- Cl9H17N2O3Cl N 7,85 8,04; 7,98
[1] желтый
8 165—166 152,5— сине- C20H20N2O4 С 68,2 68,14; 68,61
-153,5 зеленый н 5,7 6,10; 6,03
II] N 7,9 8,45 8,06
ЛИТЕРАТУРА
1. Е. В е г m а п, К. Е. F о х, F. D. Thomso n. J. Anier. Chem. Soc.,
81, 5605 (1959).
2. Брит. пат. 973028 (1962).
3. Пат. США 3100778 (1963).
4. Пат. США 3116148 (1963).
5. Брит. пат. 969754 (1964).
6 J. R. Segesser, М. Calvin. J. Amer. Chem. Soc., 64, 825
(1942).
7. И. И. Левкоев, А, Я. Башкирова. Ж. прикл. химии, 35, 3,
688 (1962).
8. С. F. К о е 1 s с 11, W.R. Workmen. J. Amer. Chem. Soc., 74,
6288 (1952).
Поступила в декабре 1965 г. ИРЕА
10’
УДК 547.416.07
ТРИХЛОРМЕТИЛДИХЛОРФОСФИН
Л. Е. ДМИТРИЕВА, К. В. КАРАВАНОВ, С. 3. ИВИН
СС13РС1а
CPC1S М. В. 220,24
В литературе описано несколько способов получения три-
хлорметилдихлорфосфина.
Один из них основан на превращении метилдихлорфосфи-
на в трихлорметилтетрахлорфосфин, который затем восста-
навливается до трихлорметилдихлорфосфина с помощью ме-
тилдихлорфосфита [1].
Второй способ основан на восстановлении комплексного
соединения трихлорметилтетрахлорфосфина и хлористого
алюминия белым фосфором [2]. Трихлорметилдихлорфосфин
получают также взаимодействием белого фосфора с четырех-
хлористым углеродом в вакуумированной трубке при нагре-
вании [3].
Перечисленные методы являются трудоемкими, но глав-
ным недостатком их являются низкие выходы трихлорметил-
дихлорфосфина.
Нами изучен способ получения трихлорметилдихлорфос-
фина, основанный на взаимодействии комплексного соедине-
ния трихлорметилтетрахлорфосфина и хлористого алюминия
с фенилдихлорфосфином в присутствии хлорокиси фосфора.
По этому методу выход трихлорметилдихлорфосфина дости-
гает 60%.
СХЕМА СИНТЕЗА ТРИХЛОРМЕТИЛДИХЛОРФОСФИНА
СС13РС14 • AICk + СвН6РС12 + РОС13 ->
СС13РС12 ф C6H5PC14-A1C1s + РОС13
148
Характеристика основного сырья
Комплексное соединение трихлорметилтетрахлорфосфина с
хлористым алюминием (см. примечание 1), получают из трех-
хлористого фосфора, треххлористого алюминия и четыреххло-
ристого углерода по методике [4].
Хлорокись техническая, перегнанная (см. примечание 2).
Фенилдихлорфосфин (см. примечание 2), получают из бен-
зола, треххлористого фосфора и алюминия по методике [5].
Условия получения
В трехгорлую колбу с мешалкой, обратным холодильни-
ком и капельной воронкой помещают 42,5 г (0,1 М) комплек-
сного соединения трихлорметилтетрахлорфосфина и хлористо-
го алюминия, 15,3 г (0,1 А4) хлорокиси фосфора и при ком-
натной температуре прикапывают 18,9 г (0,1 М) фенилди-
хлорфосфина.
Реакционную массу нагревают в течение 1—1,5 часа до
60°. Затем продукты реакции отгоняют в вакууме 20 мм в
токе азота.
При повторной перегонке получают 12 г трихлорметилди-
хлорфосфина, что составляет 60% от теоретического выхода;
т. кип. 65—67°/20 мм; т. пл. 41° (см. примечание 3).
Примечания-.
1. Комплексное соединение трихлорметилтетрахлорфосфина и хлори-
стого алюминия гигроскопично, поэтому следует избегать излишнего сопри-
косновения его с воздухом.
2. Хлорокись фосфора и фенилдихлорфосфины — свежеразогнанные.
3. Трихлорметилдихлорфосфин — полукристаллический прозрачный
продукт, легко окисляется на воздухе.
ЛИТЕРАТУРА
1. L. Q u I п, Н. R о 1 s I о п. J. organ. Chem., 23, 1693 (19.58).
2. J. L. Van Winkle, S. В e 11, G. M о r r I s. Пат, СШ A 2875 224;
C. A., 53, 13054 (1958).
3. J. E. Nixon. J. Chem. Soc., 2471 (1964).
4. A. M. К i n n e r, E. A. Perren. J. Chem. Soc., 3437 (1952).
5. E. Л. Г e ф т e p. Фосфорорганические мономеры и полимеры, М.
АН СССР, 1960, стр. 98.
Поступила в ноябре 196.'» г.
УДК 547.495.8.07
УРЕИД АЦЕТОУКСУСНОЙ КИСЛОТЫ
Г. В. КОНДРАТЬЕВА, С. И. ЗАВЬЯЛОВ
H3NCONHCOCHaCOCH3
C6H8N2O3 М. в. 144,13
Уреид ацетоуксусной кислоты в литературе не описан.
Данное соединение получено нами путем конденсации моче-
вины и дикетена в толуоле в присутствии пиридина как ката-
лизатора.
СХЕМА СИНТЕЗА УРЕИДА АЦЕТОУКСУСНОЙ КИСЛОТЫ
H,NCONH2 + СН3 = С----СН2 -> H2NCONHCOCH2COCHa
I I
о—с=о
Характеристика основного сырья
Мочевина, ч., ГОСТ 6691—53.
Дикетен, ч.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешал-
кой, холодильником и термометром, помещают 7,9 г мочеви-
ны и 65 мл сухого толуола. К полученной суспензии при пе-
ремешивании добавляют 15 мл дикетена, а затем 2,5 мл су-
хого пиридина. В течение последующих 30 минут температура
смеси достигает 65°. После охлаждения до комнатной темпе-
ратуры выпавший осадок отделяют и промывают последова-
тельно толуолом, эфиром, водой и метанолом.
Выход уреида ацетоуксусной кислоты равен 14,3 г, что со-
ставляет от теоретического 74%; т. пл. 139—141°.
После перекристаллизации из водного спирта получают
9,5 г уреида с т. пл. 146—147°.
Найдено, %: С—41,79; Н—5,70; N 19,65.
C,H8N2O3. Вычислено, %: С—4'.67; Н—5,59; N — 19,43.
Поступила в июль 1965 г. ИОХ АН СССР
150
УДК 547.754.07
1-ФЕНИЛ-3.3-ДИМЕТИЛ-2-МЕТИЛ ЕНИНДОЛИН
В. А. ИНШАКОВА, Д. 4. ДРАПКИНА, В. Г. БРУДЗЬ,
И. П. ПЛИТИНА
СН3 СН3
C17H17N
М. в. 235,32
1 -Фенил-3,З-диметил-2-метилениндолин получается цикли-
зацией дифенилгидразона метилизопропилкетона. Бруннер в
качестве конденсирующего средства использовал двухлори-
стое олово [1] и йодистоводородную кислоту [2]. По прописи,
данной в патенте [3], циклизацию проводят в присутствии
плавленого хлористого цинка без предварительного выделе-
ния гидразона.
Нами проверен и несколько упрощен метод [3] и более пол-
но охарактеризован продукт.
СХЕМА СИНТЕЗА 1-ФЕНИЛ-3.3-ДИМЕТИЛ-2-МЕТИЛЕНИНДОЛ ИНА
J'—N—NH2-HC1 + СНз-С-СН
сн,
сн3
151
• НС1
Характеристика основного сырья
Дифенилгидразнн солянокислый асимметричный, ч., ТУ
МХП 2665—51.
Метилизопропилкетон [4].
Спирт этиловый, абсолютированный, ГОСТ 9674—61.
Кали едкое, ч„ ГОСТ 4203—48.
Цинк хлористый, безводный, ч., ГОСТ 4529—48.
Этиловый эфир, ч., ГОСТ 6265—52.
Условия получения
В колбе, снабженной мешалкой и обратным холодильни-
ком, запертым хлоркальциевой трубкой, нагревают на водя-
ной бане при температуре кипения спирта в течение 1,5 часа
28,7 г (0,13 М) N.N-дифенилгидразина солянокислого, 11,9 г
(0,138 М) метилизопропилкетона и НО мл абсолютного спир-
та, затем раствор охлаждают до комнатной температуры (см.
примечание). К смеси добавляют 120 г плавленого хлористого
цинка и снова кипятят 1,5 часа с перемешиванием. После это-
го реакционную смесь охлаждают, добавляют 40%-ный раст-
вор едкого кали до сильно щелочной реакции (200 мл) и
экстрагируют 5 раз эфиром (400 мл). Эфирный экстракт про-
мывают 2 раза водой по 75 мл и сушат над плавленым едким
натром. Эфир отгоняют, остаток перегоняют в вакууме, соби-
рая фракцию с т. кип. 150—155°/8 мм.
Выход Гфенил-3,3-диметил-2-метилениндолина равен
17,6 г, что составляет 57,8% от теоретического, считая на
N.N-дифенилгидразин солянокислый; п2° —1,6156. Вначале
152
вещество представляет собой почти бесцветное масло, при сто-
янии постепенно краснеет.
По литературным данным, т. кип. продукта 100—102°/
/0,01 мм [31; 208—208,5752 мм [1]; 183—185732 мм [2].
Примечание.
При охлаждении раствора выпадает небольшой осадок. В патенте
[3] рекомендуется удалить его фильтрованием; однако, по нашим данным,
с фильтрацией и без нее получаются одинаковые результаты.
ЛИТЕРАТУРА
I. К. Brunner. Monatsh., 21, 167 (1901).
2. К. Brunner. Ber., 31, 1948 (1898).
3. Брит. пат. 883803 (1961).
4. Синтез органических препаратов, т. 2, М., 1949, стр. 323.
Поступила в декабре 1965 г. ИРЕА
УДК 547.754.07
1-ФЕН ИЛ-3,3-ДИМЕТИЛ СПИРО-Цг'Н-Г-БЕНЗОПИ-
РАН)-2,2'-И НДОЛ ИНЫ]
Д А. ДРАПКИНА, В. Г. БРУДЗЬ, В. А. ИНШАКОВА, И. П. ПЛИТИНА
сн3 сн3
Спиропираны представляют интерес в качестве фотохром-
ных веществ [1]. Пропись получения спиропиранов этого ряда
дана в патенте [1]; однако соединения не охарактеризованы, за
исключением б'-метокси-б'-нитропроизводного, для которого
приведена температура плавления.
Нами проверен указанный метод и подобраны условия
очистки отдельных спиропиранов. Полученные 1-фенил-3,3-
-диметилспиро-[(2/Н-1,-бензопиран)-2,2,-индолины] охаракте-
ризованы температурой плавления и данными элементного
анализа.
СХЕМА СИНТЕЗА 1-ФЕНИЛ-3,3-ДИМЕТИЛСПИРО-[(2'Н-1-
-БЕНЗОПИРАН)-2,2'-ИНДОЛИНОВ]
R СНО
Х|^Х,ОН
Да
R' R"
спирт
154
сна сн3
Характеристика основного сырья
5-Нитросалициловый альдегид, ч., ТУ TCP 563 р—61.
Спирт этиловый, синтетический, ГОСТ 9674—61.
З-Метокси-6-нитросалициловый альдегид (получение см.
в [2]).
5-Нитро-о-ванилин (см. стр. 109 в настоящем сборнике).
5-Метокси-З-нитросалициловый альдегид (получение см.
в [3]).
З-Бром-5-нитросалициловый альдегид (см. стр. 32 в на-
стоящем сборнике).
5-Бром-З-нитросалициловый альдегид (получение см. в
[4])-
5-Хлор-З-нитросалициловый альдегид (см. стр. 171 в на-
стоящем сборнике).
З-Нитросалициловый альдегид (см. стр. 111 в настоящем
сборнике).
1-Фенил-3,3-диметил-2-метилениндолин (см. стр. 151 в на-
стоящем сборнике).
Условия получения
В двугорлую колбу на 50 мл, снабженную мешалкой и
обратным холодильником, помещают 2,35 г (0,01 М) свежепе-
регнанного 1-фенил-3,3-диметил-2-метилениндолина и 10 мл
спирта. При размешивании добавляют 0,01 А4 соответствую-
щего альдегида и 15 мл спирта. Реакционную смесь кипятят
при нагревании на водяной бане в течение 2 часов. Для более
полного выделения осадка смесь охлаждают в бане со льдом.
Затем продукт отфильтровывают, промывают 3—5 мл спирта
и кристаллизуют (2—3 раза) из спирта.
Несколько иначе очищают 6/-хлор-8/-нитропроизводное.
Кипятят 3 г спирана-сырца (т. пл. 157—159°) с 200 мл спирта
в течение 10 минут; полученный раствор фильтруют от нера-
створившейся части (побочный, более высокоплавкий про-
дукт). Из фильтрата кристаллизуется осадок (1,9 а), плавя-
щийся при 158—159°. Фильтрат от него концентрируют до
КЗ первоначального объема и по охлаждении получают
спиран с т. пл. 163—164°. Повторными кристаллизациями
155
фракции, плавящейся при 158—159°, разделяют ее на основ-
ной продукт и вцсокоплавкий. Все фракции спирана, плавя-
щиеся при 163—164°, кристаллизуют 1—2 раза обычным пу-
тем из спирта, после чего получают чистый 1-фенил-3,3-диме-
тил-6' -хлор-8' -нитроспиро-[(2'Н-Г-бензо'Пиран)-2,2' -индо-
лин].
Данные по выходам, очистке и характеристики получен-
ных спиропиранов приведены в табл. 1 и 2.
Таблица 1
Условия очистки и выходы 5'R, 6’R', 8'Р"-1-фенил-3,3-днметилспиро-
[(г'Н-Ц-бензопиран^г.г'-индолинов]
№ соеди- нения Заме стители Кристаллизация Выход от теорети- ческого, %
R R' R'' л/л спирта на 1 г спирана ЧИСЛО кристаллы- эацнй”1 сырца ЧИСТОГО
1 Н no2 осн3 175 20 2 74,3 48,0
2 Н ОСН3 NCK 70 2 78,5 64,0
3 NOo н ОСН3 375 2 77,8 57,8
4 Н no2 Вг 75 2 67.7 51,6
5 Н Вг no2 200 2 71 ,7 47.8
6 н С1 NO, 70 3* 76,1 39,0
7 н no2 н 130 3 64,3 31,5
8 н н NO, 75 2 71,0 47,9
* Указано число кристаллизаций основной фракции требуемого сии-
ропираиа.
Таблица 2
Характеристики 1-фенил-3,3-диметилспиро-[(2'Н-1'-бензопиран)-
-2,2'-индолннов]
№ сое- дине- ния Темпера- тура плавления, °C Внешний вид спиропнра- нов Брутто-формула Эле- мен- ты Данные анал иза
вычис- лено, % найдено, %
1 181,5-181,8 Зеленовато- CjsH22N2O4 с 72,46 71,96; 71,91
ПО лит. желтый н 5,35 5,41; 5,28
данным N 6,76 6,41; 6,70
179,5-181(1]
2 167—168 Ярко-жел- ^25^22^2^4 С 72,46 72,81; 72,23
тый н 5,35 5,55; 5,51
N 6,75 6,66; 6,87
156
Продолж. табл. 2
Л® сое- дине- ния Темпера- тура плавления, °C Внешний вид спиропнра- нов Брутто-формула Эле- мен- ты Данные анализа
вычис- лено, % найдено, %
3 225 - 226 Светло- желтый с н N 72,46 5,35 6,76 72,80, 72,40 5,55, 5,49 6,65, 6,45
4 165-166 Ярко-жел- тый С24Н1ЭВг1Ч2О8 С 11 N Вг 62,20 4,11 6,04 17,26 62,44; 62,47 4,27; 4,26 6,54; 6,52 16,94; 16,96
5 187,5- 188,5 Желтый C24H19BrN2O3 С Н N 62,20 161,97 ; 62,27 4,11 4,47; 4,37 6,04' 6,32; 6,04
6 167—168 Желтый С2.1Н19С11\20з С н С1 68.79 4,57 8,47 69,11; 69,01 4,57; 4,57 8,06; 8,63
7 148—149 Желтый с н N 74,98 5,24 7,29 75,02; 75,06 5,17; 5,16 7,43; 7,54
8 171-172 Желтые иголки C24H2CN2O3 С н N 74,98 5,24 7,29 74,66; 74,70 5,18; 5,19 7,30; 7,60
ЛИТЕРАТУРА
1. Брит. пат. 883803 (1961).
2. W. R е i d, Н. Schiller. Ber., 85, 216 (1952).
3. L. R u b i n s t е 1 n. J. Chem. Soc. 127, 2000 (1925).
4. R. O. Clinton, S. C. Laskowski. J. Amer. Chem. Soc., 71,
3602 (1949).
Поступила в декабре 1965 г.
ИРЕА
УДК 547.583.5.07
ФЕНИЛОВЫЙ ЭФИР АНТРАНИЛОВОЙ КИСЛОТЫ
В. М. ДЗИОМКО, И. С. МАРКОВИЧ, И. В. КРУГЛОВА
,соосвн6
I f
^/Xnh2
М. в. 229,23
СцНцЫОг
По литературным данным, фениловый эфир антраниловой
кислоты получают нагреванием изатового ангидрида с фено-
лом [1] или восстановлением фенилового эфира о-нитробен-
зойной кислоты оловом и соляной кислотой [2], на никеле
Рэнея [3], железом и уксусной кислотой [3].
Нами был проверен метод [2]. В процессе работы были
уточнены соотношения компонентов, температура проведения
реакции и способы выделения продукта.
СХЕМА СИНТЕЗА ФЕНИЛОВОГО ЭФИРА АНТРАНИЛОВОЙ
КИСЛОТЫ
,соосвн5
Sn; иС|._
1С3Н6ОН)
"'NO,
^х/соосвнБ
I II
Характеристика основного сырья
Олово, ч., ТУ МХП ОРУ 56—56.
Соляная кислота, ч., ГОСТ 3118—46.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Условия получения
В трехгорлую колбу, снабженную холодильником и ме-
шалкой, загружают 4,4 г (0,02 Л4) фенилового эфира о-нитро-
158
бензойной кислоты (см. примечание) и 20 мл этилового спир-
та. Раствор нагревают до 70° и осторожно добавляют к нему
20 мл концентрированной соляной кислоты. Поддерживая
температуру 85°, постепенно, небольшими кусочками, при по-
стоянном размешивании, вносят в реакционную смесь 7 е
(0,06 М) гранулированного олова. После окончания добав-
ления олова нагревание продолжают в течение 50 минут. По
окончании восстановления отгоняют спирт, к реакционной
смеси добавляют 30 40 мл бензола и нейтрализуют насыщен-
ным раствором соды до pH 8—9. Осадок отфильтровывают
и промывают бензолом. Бензольные вытяжки собирают и
упаривают при комнатной температуре.
Выпавшие кристаллы фенилового эфира антраниловой
кислоты перекристаллизовывают из этилового спирта. Выход
равен 1,5 г, что составляет 40% от теоретического; т. пл. 70°.
По литературным данным, т. пл. вещества 70° [1]; 72—73°
[3].
Примечание.
Фениловый эфир о-нитробензойной кислоты получен по методу [3].
ЛИТЕРАТУРА
1. G. Schmid t. J. Pract. Chem., [2]. 36, 377 (1877).
2. H. G. Rule, E. Brets ch er. J. Chem. Soc., 1927, 926,
3. R. A. H e а с о c k, D. H. Iley. J. Chem. Soc., 1954, 2482.
Поступила в декабре 1965 г. ИРЕД
УДК 547.425.5.07
N-ФЕНИЛФОРМАМИДИНСУЛЬФИНОВАЯ КИСЛОТА
Р. Л. ГЛОБУС. Р. П. ЛАСТОВСКИЙ, Е. Я. Я РОВ ЕН КО,
А. П. БОЛДЫРЕВА
HN
^C-SO2H
CeH6NH
C,H8N2O2S М.в. 184,15
N-Фенилформамидинсульфиновая кислота [1] в литературе
не описана.
Она, как и другие производные формамидинсульфиновых
кислот, может представить интерес как физиологически ак-
тивное вещество.
СХЕМА СИНТЕЗА N-ФЕНИЛФОРМАМИДИНСУЛЬФИНОВОЙ
КИСЛОТЫ
HN HN
- SH + 2Н2О2 - ^C-SO2H+2H,O
C,H6NH C6H6NH
Характеристика основного сырья
N-Фенилтиомочевина, т. пл. 150—152°.
Перекись водорода, медицинская, ГОСТ 177—55.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термометром
и капельной воронкой, загружают 2 г (0,01 М) N-фенилтио-
мочевийы, 0,005 г молибденовокислого натрия и приливают
10 мл воды.
160
Полученную реакционную смесь охлаждают до 0—5° и до-
бавляют по каплям 3 мл (0,03 М) 31,2%-ной перекиси водо-
рода, поддерживая ту же температуру. По окончании при-
бавления всей перекиси водорода реакционную массу при
размешивании выдерживают в течение 1 часа при темпера-
туре в пределах от 0 до 5°. По окончании выдержки получен-
ный белый осадок отфильтровывают и высушивают при тем-
пературе 60° .1
Выход N-фенилформамидинсульфиновой кислоты равен
1 г, что составляет 41,4% от теоретического, считая на N-фе-
нилтиомочевину.
Найдено, %: С-45; 45,99; Н-4,52; 4,58; N—15,10; 15,20.
C7H8N,O2S. Вычислено, %: С-45,60; Н—4,35; N- 15,20.
По внешнему виду вещество представляет собой белые
кристаллы с т. пл. 132—133°, не растворяется в воде и не-
полярных органических растворителях, в спирте растворимо.
ЛИТЕРАТУРА
1. Р. Л. Глобус, Р. П. Ластовский, Е. Я. Я ров ей к о.
С. П. Медведева. Авт. свид. 178803 (1965); Бюлл. изобр. № 4 (1966).
Поступила в ноябре 1S65 г. ИРЕА
11 Зак. 25
УДК 547.571.07
л-ФТОРБЕНЗАЛ ЬДЕГИД
Д. А. ДРАПКИНА, Н, И. ЧЕРНОВА, В. Г. БРУДЗЬ
С7Н5РО
F-^ ^-СНО
М.в. 124,11
п-Фторбензальдегид был получен из п-фтортолуола
хлорированием [1, 2] или бромированием [3] его до п-фтор-
бензальгалогенида с последующим омылением или до бензил-
галогенида с последующей обработкой азотнокислым свин-
цом [4]. Шиман [5] и другие [6] окисляли n-фтортолуол до аль-
дегида хромилхлоридом. п-Фторбензальдегид получали так-
же реакцией Мак-Фадиена и Стивенса из /г-фторэтилбензоата
[7] и из n-фторбензанилида обработкой хлористым тиони-
лом [8].
Мы синтезировали n-фторбензальдегид реакцией Соммле
в соответствии с прописью [9], дайной для 4-стильбензальде-
гида.
СХЕМА СИНТЕЗА п -ФТОРБЕНЗАЛЬДЕГИДЛ
- сна-со
СНз-f /-F+ I /NBr
' СН2—COZ
(Свн.,со)2ог
ч сн,-сс\
BrH,C-f f-F-r I ‘ ;NH;
сн2-сох
BrH2C-^_)--F + (CH,)6N4 -
(CH2)6N4.BrH2C-^ F Cfo-COOH.^
— F-^«-^-CHO
162
Характеристика основного сырья
п-Фтортолуол, ч., ТУ TCP 450 р—61.
N-Бромсукцинимид, ч., ВТУ РУ 1085—54.
Уротропин, ч., ф — IX, фарм.
Уксусная кислота, х. ч., ледяная, ГОСТ 61—51.
Натрий углекислый, ч., ГОСТ 199—52.
Хлороформ, х. ч., ГОСТ 3160—51.
Четыреххлористый углерод, ч., ГОСТ 5827—51.
Условия получения
В круглодонную колбу емкостью 0,5 л, снабженную об-
ратным холодильником и механической мешалкой, загружают
20,45 г (0,186 М) п-фтортолуола, 37,8 г (0,212 М) N-бромсук-
цинимида, 0,4 г очищенной перекиси бензоила и 430 мл че-
тыреххлористого углерода. Смесь кипятят на водяной бане в
течение 3 часов при постоянном перемешивании, затем горя-
чую суспензию отфильтровывают, к фильтрату добавляют
42,55 г уротропина в 280 мл хлороформа и кипятят при пере-
мешивании в течение двух часов, после чего смесь оставляют
на ночь. Выпавший белый осадок отфильтровывают, промы-
вают хлороформом, тщательно отжимают и переносят в дву-
горлую колбу на 3 л с обратным холодильником и мешалкой.
Добавив 445 мл ледяной уксусной кислоты, полученный рас-
твор кипятят в течение 3 часов; в горячий раствор приливают
800 мл дистиллированной воды и кипятят еще 2 часа. По
охлаждении раствор переносят в четырехлитровый фарфоро-
вый стакан, снабженный мешалкой, и нейтрализуют до
pH 6,5 800 г углекислого натрия. n-Фторбензальдегид экстра-
гируют в три приема 150 мл серного эфира. Эфирный экстракт
сушат над прокаленным сернокислым натрием, затем эфир
отгоняют, а остаток разгоняют в вакууме, отбирая фракцию
84—85723 мм.
Выход вещества равен 6,64 г, что составляет 29% от тео-
ретического; Пд — 1,5141.
По литературным данным, п™ —1,5210 [4]; п‘% —1,5183
[2]; 181,57763 [3]; 71—73718—20 [5]; 71—73/15 [2}; 96—
99°/15 ПО].
ЛИТЕРАТУРА
1. 1. J. Renkes. Zbl., 1919, 1, 8'20.
2. К е n I е i Fukui, Н i s а о Kitano и др. С. А., 54, 5519 (1960).
3. I. В. Shoe smith, С. Е. Sos son, R. Н. Slater. J. Chem.
Soc., 1926. 2760.
4. Чехослов. пат. 96506 (1960).
5. G. Schiemann. Z. phys. Chem., 156, 397 (1931).
6. Ch’eng Yeh Y ii a n, C h’u n Nien Chang, J. Fang Yeh.
C. A., 54, 12097 c (1960).
11* 163
7. E. L, Be n n e tt, C. N i 1 m a n n. J. Amer. Soc., 72, 1800 (1956).
8, F. F. EHenbersrer, R. О lei ter. Ber., 97, 2, 480 (1964).
9. Q. Drefahl, W. Hartrodt. J. pract. Chem.. 4, (276), ПЭ-
124.
10. G. Olah, A. Pa va lath, J. Kuhn. Acta chim. Acad. Sclent,
hung., 7, 85 (1955).
Поступила а декабре 1965 r.
ИРЕА
УДК 547.534.1.07
и-ФТОРЭТИЛБЕНЗОЛ
А. А. АРТАМОНОВ, Р. Д. БОНДАРЧУК, М. И. КОТЕЛЕНЕЦ
С2н5
CgHgF
М. в. 124,15
В литературе описан синтез n-фторэтилбензола из гс-фтор-
бромбензола и бромистого этила в присутствии металличе-
ского натрия с выходом около 10% [1]. n-Фторэтилбензол был
получен также из n-аминоэтилбензола диазотированием пос-
леднего и приливанием диазораствора к нагретой фтористо-
водородной кислоте. Выход вешества в этом случае не превы-
шал 24% [2].
Нами разработан препаративный метод получения п-фтор-
этилбензола из n-аминоэтилбензола с выходом 62%, основан-
ный на реакции Шимана [3].
СХЕМА СИНТЕЗА п- ФТОРЭТИЛБЕНЗОЛА
НЕ + Н3ВОз = HBF4 + ЗНгО
HBF4 + NaOH = NaBF4 + Н2О
СгН5
NHa
N=N
N=N
165
С2Н5
А-4'
N^N
Характеристика основного сырья
Плавиковая кислота, ч., ТУ МХП 289—47.
Борная кислота, ч., ст. ГОХП 27—1830.
Соляная кислота, х. ч., ГОСТ ЗП8—46.
Нитрит натрия, ч., ГОСТ 4197—48.
Натр едкий, х. ч., ГОСТ 4328—48.
л-Аминоэтилбензол (см. примечание).
Условия получения
Синтез борфтората натрия. В полиэтиленовом флаконе
емкостью 500 мл взвешивают 123,9 г (6,2 М) 42%-ной плави-
ковой кислоты и при постоянном взбалтывании и охлаждении
смесью льда и соли прибавляют к ней небольшими порциями
38,7 г (0,624 М) борной кислоты. После стояния при комнат-
ной температуре в течение ночи смесь нейтрализуют 5 н. рас-
твором едкого натра по метиловому оранжевому.
Получение борфторида п-этилбензолдиазония. В трехгор-
лую колбу емкостью 1 л, снабженную механической мешал-
кой, термометром и капельной воронкой, охлаждаемую льдом
и солью, вносят 79,8 мл (0,945 М) концентрированной соля-
ной кислоты и при механическом перемешивании добавляют
48 мл (0,384 М) п-аминоэтилбензола. Охлаждают реакцион-
ную смесь до минус 5° и прибавляют по каплям раствор нит-
рита натрия в 108 мл воды, следя за тем. чтобы температура
не повышалась выше 5°. По окончании диазотирования дизо-
раствор охлаждают до минус 10° и приливают к нему полу-
ченный ранее раствор борфтората натрия, охлажденный до0°.
Смесь перемешивают еще 30 минут и отфильтровывают вы-
павший борфторид п-этилбензолдиазония. Затем его промы-
вают на фильтре 75 мл ледяной воды и тщательно отжимают.
Полученную лепешку измельчают и сушат на фильтровальной
бумаге. Получают 67,35 г кремового борфторида и-этилбен-
золдиазония.
Выход вещества составляет 79,3%.
Получение п-фторэтилбензола. В колбу Вюрца емкостью
250 мл. конец отвода которой загнут под прямым углом вниз,
загружают измельченный в ступке и совершенно сухой бор-
166
фторид п-этилбензолдиазония. Через загнутый отросток от-
вода колбу Вюрца соединяют с круглодонной колбой на
100 мл, снабженной обратным шариковым холодильником.
Верхний конец холодильника соединяют с ловушкой, кото-
рую помещают в смесь льда и соли.
Нагревают дно колбы Вюрца в одном месте пламенем
спиртовки до начала разложения борфторида, которое про-
должается дальше самостоятельно. Когда самопроизвольный
процесс закончится, разложение заканчивают, нагревая кол-
бу на плитке до тех пор, пока перестанет выделяться фтори-
стый бор и в ней останется лишь небольшое количество тем-
но-окрашенного остатка.
n-Фторэтилбензол из круглодонной колбы и ловушки сое-
диняют вместе, встряхивают 4 раза с 10%-нь1м раствором
едкого натра, беря для каждой операции по 20 мл щелочи,
промывают водой, сушат над хлористым кальцием и перего-
няют.
Получают 29,58 г (77,92%) n-фторэтилбензола с т. кип.
141—141,5°; п$—1,4724; dt20—0,98153.
По литературным данным, т. кип. его 141°; d02Q—0,967;
d°0~0,994 [2].
Выход n-фторэтилбензола в расчете на п-аминоэтилбензол
составляет около 62%.
n-Фторэтилбензол может быть превращен каталитическим
дегидрированием в п-фторстирол — мономер в синтезе термо-
стойких полимеров [4].
Примечание.
n-Ампноэтилбензол получен нитрованием этилбензола, ректификацией
смеси п- и о-нитроэтилбензолов в вакууме и последующим восстановле-
нием индивидуального n-нитроэтилбензола [5]; т. кип. 95—96°/Ю мм;
П 20—1,5558; 96885.
ЛИТЕРАТУРА
1. G. S с h i е га a n, R. Р i 11 а г s к у. Вег., 64, 1340 (1931).
2. О. R. Qu г lie, Е. Е. Reid. .1. Amer. Chem. Soc., 47, 2357
(1925).
3. А. Роэ. Органические реакции, ИЛ, М., 5, 1951, стр. 155.
4. Пласт, массы, 5 (I960).
5. П. М. Кочергин. Ж. общ. химии, 27, 3204 (1957).
Поступила в октябре 1965 г.
Донецкий филиал ИРЕА
УДК 547.831.4.07
5-ХЛОР-8-(МЕЗИТИЛЕНСУЛЬФОНИЛ-
АМИНО)ХИНОЛИН
В. М. ДЗИОМКО, И. А. КРАСАВИН, Б. В. ПАРУСНИКОВ,
Н. И. ДУДАРЕВА
C18H17C1N2O2S
М. в. 360,87
5-Хлор-8- (мезитиленсульфониламино) хинолин образуется
с 95%-ным выходом при взаимодействии 5-хлор-8-аминохино-
лина с мезитиленсульфохлоридом в среде пиридина.
Это соединение является чувствительным и избирательным
люминесцентным реагентом для определения цинка.
168
СХЕМА СИНТЕЗА 5-ХЛОР-8-(МЕЗИТИЛЕНСУЛЬФОНИЛАМИНО)
ХИНОЛИНА
Условия получения
(см. примечание 1)
К раствору 5,36 г (0,03 М) 5-хлор-8-аминохинолина (см.
примечание 2) в 10 мл пиридина при охлаждении льдом при-
бавляют за 10 минут раствор 7,22 г (0,033 М) мезитиленсуль-
фохлорида (см. примечание 3) в 10 мл пиридина. Образовав-
шуюся оранжево-желтую суспензию нагревают на кипящей
водяной бане в течение одного часа, охлаждают до комнат-
ной температуры и выливают при размешивании в 250 мл
воды. Осадок отфильтровывают, промывают на фильтре во-
дой и высушивают в вакуум-эксикаторе над едким натром.
Выход 5-хлор-8-(мезитиленсульфониламино)хинолина ра-
вен 10,34 г, что составляет 95,5% от теоретического. После
перекристаллизации из 1 л этанола получают 9 г бесцветных
игл с т. пл. 178,5—179°; при дальнейшей очистке т. пл. повы-
шается до 180—180,5° (см. примечание 4).
Примечания:
1. Для получения продукта, пригодного в качестве люминесцентного
реагента, следует принимать меры против загрязнения вещества следами
ионов металлов, применять чистые реактивы, дистиллированную воду и
перегнанные растворители.
2. Применялся 5-хлор-8-амииохииолин с т. пл. 86,5—87°, полученный
по ранее описанному методу [1].
3. Применялся мезнтиленсульфохлорнд с т. пл. 54—55°, полученный по
методу [2], очищенный перегонкой в вакууме (т. кип. 128—136°/3 мм) и
перекристаллизацией из петролейного эфира.
4. Чистый образец 5-хлор-8-(мезитиленсульфоннламино)хииолина с
т. пл. 180—180,5° был приготовлен кристаллизацией из этанола и про-
анализирован.
169
Найдено, %: С—60,42; 60,40; Н 5,00; 4,99; CI-9,73;
9.62; N -8,05; 7,99.
C1fliInClN2O2S. Вычислено, %: С—69,91; Н-4,75; С1--9,82; N-7,76.
ЛИТЕРАТУРА
1. В, М. Дзиомко, И. А. Красавин. Методы получения хими-
ческих реактивов и препаратов, вып. 2, М., ИРЕА, 1961, стр. 98.
2. С. Н. Wang. S. Q. Cohen. J. Amer. Chem. Soc., 7) 1928
(1954).
Поступила в декабре 1963 г. ИРЕА
УДК 547.571.07
5-ХЛОР-З-НИТРОСАЛ ИЦИЛОВЫЙ АЛЬДЕГИД
5-Хлор-3-нитро-2-оксибензальдегид
Д. А. ДРАПКИНА, В. Г. БРУДЗЬ, В. А. ИНШАДОВА, И. П. ПЛИТИНА
СНО
I
Пгон
C1-!^J-NO2
C7H4NO4C1 х м. в. 201,56
В литературе описан один способ [1] синтеза
5-хлор-З-нитросалицилового альдегида путем нитрования
5-хлорсалицилового альдегида азотной кислотой (уд. веса
1,42) в ледяной уксусной кислоте при 60—70°. После двукрат-
ной кристаллизации из лигроина (т. кип. 80—100°) выход
продукта составлял 52,4% от теоретического.
При проверке предложенного метода нами уточнены усло-
вия реакции и выделения продукта. В результате был дос-
тигнут выход почти чистого продукта, равный 77—80% от
теоретического, с т. пл. 106—107° без дополнительной очистки.
Однократной кристаллизацией из петролейного эфира удалось
повысить температуру плавления еще на один градус.
СХЕМА СИНТЕЗА 5-ХЛОР-З-НИТРОСАЛИЦИЛОВОГО АЛЬДЕГИДА
СНО
Г| f¥0H + ™о.с-^^
С1~\/
сно
I
, он
С1 1 11 NO +сн3соон+н2о
a-^/-NO2 171
Характеристика основного сырья
5-Хлорсалициловый альдегид, ч., ТУ В338—65.
Азотная кислота, ч., ГОСТ 4461—48.
Уксусная кислота, ледяная, х. ч., ГОСТ 61—51.
Петролейный эфир, ч., МХТУ 279—59.
Условия получения
В колбу, снабженную мешалкой, термометром, обратным
холодильником и капельной воронкой, помещают 15,65 г
(0,1 М) 5-хлорсалицилового альдегида и 200 мл ледяной
уксусной кислоты. Смесь нагревают до 65° и к полученному
раствору при размешивании в течение одного часа прибавля-
ют 15,5 мл 73%-ной азотной кислоты (уд. вес 1,426, см. при-
мечание), поддерживая температуру в пределах 65—70°. За-
тем дают выдержку 10—15 минут при той же температуре и
реакционную смесь выливают на 200 г воды со льдом. Через
1—2 часа осадок отсасывают и промывают на воронке 2 раза
водой (по 100 мл).
Получают 15,6—17 г (77—80% от теоретического выхода)
технического продукта с т. пл. 106—107°.
После кристаллизации из 1200 мл петролейного эфира вы-
ход 5-хлор-З-нитросалицилового альдегида равен 13,4—13,7 г,
что составляет 60,4—60,5% от теоретического; т. пл. 107—
108°.
По литературным данным, т. пл. продукта 105—107° [1],
108—109° [2].
Примечание.
Молярное отношение альдегид:азотная кислота = 1:2,55.
В опытах с азотной кислотой уд. веса 1,38 и 1,40 и при молярном
отношении альдегид-.азотная кислота = 1:2,4 не удалось получить 5-хлор-
-3-нитросалициловый альдегид с требуемой температурой плавления.
ЛИТЕРАТУРА
1. А. В. Lovett, Е. Roberts. J. Chem. Soc., 1928, 1978.
2. М. Crawford, J. W. Rasburn. J. Chem. Soc., 1956, 2155.
Поступила в июле 1965 г.
ИРЕА
УДК 547.781.4.07
2-ХЛОР-4(5)-ФЕНИЛИМИДАЗОЛ
Г. Н ТЮРЕНКОВА, Н. П. БЕДНЯГИНА
C9H,C1Nj
М. в. 178,62
В литературе 2-хлор-4(5)-фенилимидазол не описан.
СХЕМА СИНТЕЗА 2-ХЛОР-4(5)-ФЕНИЛ ИМИДАЗОЛА
NH
/ч
С6Н5-С-СН -NH2 + HCNO - II V п
II II
0 H6C6/'nH
NH
X к
х ' / Ч.
|1 )С = О+РОС13- | ^с-CI
H6C6/XN'H Н5Св/\н
Характеристика основного сырья
Хлоргидрат о) -аминоацетофенона, получен из ш-бромаце-
тофенона по [1].
Цианат калия, полученный по [2].
Хлорокись фосфора, свежеперегнанная, т. кип. 105—107°
Натр едкий, ч., ГОСТ 4328—48, 30%-ный раствор.
173
"Условия получения
Синтез 4(5)-фенилимидазолона-2. Растворяют 27,4 г
(0,16 М) хлоргидрата <и -аминоацетофенона при нагревании
в 100 мл этанола и в горячий раствор вносят суспензию 15 ’
(0,18 М) цианата калия в 25 мл воды. Смесь кипятят в колбе
с обратным холодильником на водяной бане в течение трех
часов, причем в процессе нагревания выпадает осадок 4(5)-
-фенилимндазолона-2. После охлаждения реакционной массы
осадок отфильтровывают, промывают водой н очищают пере-
осаждением из щелочного раствора соляной кислотой в при-
сутствии активированного угля.
Выход продукта с т. пл. выше 300" равен 11 г, что состав-
ляет 54%, считая на хлоргидрат ш-аминоацетофенона (см.
примечание 1). При перекристаллизации из диметилформами-
да получают бесцветные пластинки.
Получение 2-хлор-4(5)-фенилимидазола. Нагревают 4 г
(0,025 М) 4(5)-фенилнмидазолона-2 и 20 мл (0,15 Л4) свеже-
перегнанной хлорокиси фосфора (см. примечание 2) в запа-
янной ампуле при 170—180° в течение четырех часов. Избы-
ток хлорокиси фосфора отгоняют, а остаток разлагают ледя-
ной водой, причем в процессе разложения выпадает осадок.
К реакционной массе при охлаждении добавляют 30%-ный
раствор едкого натра для нейтрализации кислоты, образую-
щейся в процессе разложения хлорокиси фосфора, и раство-
рения осадка (pH 9—10), (см. примечание 3). Щелочной рас-
твор кипятят с активированным углем в течение 2—3 минут,
фильтруют и при нейтрализации фильтрата соляной кислотой
до pH 6—6,5 выпадает в виде бесцветных игл 2-хлор-4(5)-
-фенилнмидазол.
Выход равен 3,7 г, что составляет 77%, считая на 4(5)-фе-
нилимндазолон-2. После перекристаллизации из 50%-ного
водного спирта вещество имеет т. пл. 162—164°. Продукт рас-
творим в спирте, ацетоне, дноксане, хуже — в эфире, бензоле;
нерастворим в воде.
Найдено, %: С1—19,25; N —15,25.
C9H7C1N2. Вычислено, %: С1—19,73; N—15,59.
Примечания:
1. По литературным данным [3, 4], 4(5)-фенилимидазолон-2 имеет
т. пл. 325°.
2. Необходима свежеперегпаиная хлорокись фосфора, так как незна-
чительная примесЕ, соляной кислоты увеличивает выход побочных продук-
тов. Уменьшение расхода хлорокнси фосфора также влечет за собой сни-
жение выхода 2-хлор-4(5)-фенилимидазола.
3. Прн добавлении щелочи полного растворения осадка не наблюда-
ется. Остается смолистый остаток, представляющий собой, очевидно, по-
лимерные фосфорные эфиры, который отфильтровывают вместе с акти-
вированным углем.
174
ЛИТЕРАТУРА
I. П. М. К о ч е р г и н, М. Н. Щукина. Ж. общ. химии, 25, 2182
(1955).
2. И. Губен. Методы органической химии, т. IV, книга 2, М., Гос-
химиздат, 1949, стр. 1073.
3. Н. Rupe. Вег., 28, 251 (1895).
4. A. Lawson. J. Lhetn. Soc., 1957. 1443.
Поступила я ноябре 196“* г.
Уральский политехнический
институт
УДК 547.534.1.07
и-ХЛОРЭТИЛБЕНЗОЛ
А. А. АРТАМОНОВ, Р. Д. БОНДАРЧУК,
CSHS
W
1
Cl
С8Н,С1 М. в. 140,61
гс-Хлорэтилбензол является полупродуктом в получении
п-хлорстирола [1, 2]— мономера, который предложено приме-
нять для получения трудновоспламеняющихся полимеров с
высокой температурой размягчения [3].
При алкилировании хлорбензола галоидными этилами в
присутствии хлористого алюминия [4] и металлического алю-
миния [5] был получен хлорэтилбензол, который, как показа-
ли авторы цитированных выше работ, содержит в основном
и-изомер. Доказательством служила температура плавления
хлорбензойной кислоты, полученной при окислении хлорэтил-
бензолов. Проведенное нами алкилирование хлорбензола в
присутствии обоих катализаторов и анализ продуктов реак-
ции методом газо-жидкостной хроматографии показало, что
при этом получаются смеси изомеров, содержащие от 20 дс
40% о-хлорэтилбензола. Ввиду того что нам не удалось раз-
делить эти смеси ректификацией, 4-хлорэтилбензол был полу
чен нами из ц-аминоэтилбензола с выходом около 79% п(
реакции Зандмейера.
176
СХЕМА СИНТЕЗА п-ХЛОРЭТИЛБЕНЗОЛА
С2н5 I CSH5 сгн5
NaNO. 1 I? —ь ч/ НС1 ^\ci- Cu2ci» , НС1
nh2 NX Cl
Характеристика основного сырья
и-Аминоэтилбензол (см. примечание в статье «п-Фтор-
>тилбензол» в настоящем сборнике, стр. 165).
Соляная кислота, х. ч., ГОСТ 3118—46.
Нитрит натрия, х. ч., ГОСТ 4197—48.
Медь однохлористая, ч., 95%-ная.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную механи-
ческой мешалкой, термометром и капельной воронкой, загру-
жают 62 мл (0,5 М) д-аминоэтилбензола, 109 мл концентри-
рованной соляной кислоты и 240 мл воды. Охлаждают реак-
ционную смесь до 0—2° и добавляют к ней по каплям раствор
34,9 г (0,506 М) нитрита натрия в 80,5 мл воды, строго следя
за тем, чтобы температура не поднималась выше 5° и чтобы
конец капельной воронки постоянно находился в смеси.
Одновременно в другую трехгорлую круглодонную колбу
емкостью на 1 л, снабженную механической мешалкой и об-
ратным холодильником, загружают 20,84 г (0,1 М) однохло-
ристой меди и 218,15 мл (2,5 М) концентрированной соляной
кислоты. Смесь нагревают на водяной бане до 80° и прили-
вают к ней полученный ранее диазораствор. При последую-
щем нагревании и перемешивании реакционную смесь выдер-
живают еще 30 минут. Затем ее охлаждают и экстрагируют
эфиром. Экстракт промывают несколько раз разбавленным
раствором щелочи, водой, сушат над хлористым кальцием и
разгоняют, отогнав вначале на водяной бане эфир.
Выход я-хлорэтилбензола равен 55,5 г, что составляет
79% в расчете на и-аминоэтилбензол; т. кип. 184—184,5°;
«и» _ 1,5182; d-o—1,4393.
По литературным данным, т. кип. продукта 184,42°; п2£ —
1,5175; 6/2°—1,0455 [6].
12 Зак. 23 172
ЛИТЕРАТУРА
1. Канад, пат. 499681; 1954; РЖхим, 14, 44472 (1956).
2. А. А. Баландин, Т. М. М а р у к я и, Т. К. Лавровская
Р. Г. Сеймовая, Л. В. Грызлова. Изв. АН СССР, ОХН, 11, 2031
(1962).
3. Пат. ФРГ 1065409, 1960; РЖхим, 14Л153 (1961).
4. Schreiner. J. pract. Chem., 81, 557 (1910).
5. М. Б. Турова-Поляк, М. А. Маслова. Ж. общ. химии, 27,
897 (1957).
6. R. Dreisbach. Ind. Eng. Chem., 41, 2875 (1949).
Поступила в октябре 1965 г. Донецкий филиал ИРЕА
УДК 547.587.26-381.07
ЩЕЛОЧНЫЕ СОЛИ ГАЛЛОВОЙ КИСЛОТЫ
В. П. ЧЕРНЕЦКИЙ, Э. А. ПОНОМАРЕВА, Т. Д. БЕЛЫШЕВА,
Т. Г. ГЛАДЧЕНКО, Т. Г. ЯРОВАЯ
НО
НО-^ с^°
О-Ме
ОН
Me: Na, К, L1
Производные галловой кислоты обладают широким спект-
ром биологического действия — антимикробного, противови-
русного, антилучевого [1]. Физиологическая активность их, в
частности лечебное действие при острой лучевой болезни, воз-
растает с увеличением растворимости препаратов в воде [2].
Получение чистых галлатов щелочных металлов требует
соблюдения специальных условий, так как производные гал-
ловой кислоты в щелочной среде чрезвычайно легко окисля-
ются, из-за чего описанные в литературе галлаты натрия и
калия были получены [3] в недостаточно чистом состоянии.
Галлат лития в литературе не описан.
Нами разработан способ получения галлатов натрия, ка-
лия, лития действием метилатов этих металлов на спиртовый
раствор галловой кислоты в токе водорода. При получении
солей существенным моментом является обработка их ацето-
ном, так как в присутствии даже следов влаги или спирта эти
соли темнеют при выделении и дальнейшем хранении. После
кипячения с ацетоном галлаты щелочных металлов сохраня-
ются годами, не разлагаясь.
СХЕМА СИНТЕЗА ЩЕЛОЧНЫХ СОЛЕЙ ГАЛЛОВОЙ КИСЛОТЫ
ОН
НО-^ ^-СООН + СН3О Me -*
12* ОН 179
он
— HO-f СООМе + СН3ОН
ОН
Характеристика основного сырья
Галловая кислота, ч., ТУ МХП 2726—55.
Метиловый спирт, ч., ГОСТ 2222—54.
Натрий металлический, ч., ГОСТ 3273—55.
Калий металлический, ч., ТУ МХП 2010—55.
Литий металлический, ч.
Ацетон, ч. д. а., ГОСТ 2603—63.
Условия получения
Галлат натрия. К раствору 20,4 г (0,12 М) галловой кис-
лоты в 80 ж-i метанола при перемешивании и пропускании
водорода добавляют по частям в течение двух минут раствор
метилата натрия, приготовленный из 2,5 г (0,11 М) натрия и
50 мл метанола. Реакцию контролируют измерением pH, ко-
торый поддерживают в пределах 6—7. Раствор перемешивают
в токе водорода в течение 30 минут, после чего метанол упа-
ривают наполовину в вакууме в токе метана или азота (см.
примечание 1). Выкристаллизовавшийся галлат натрия пос-
ле охлаждения отфильтровывают в атмосфере метана илн
азота и промывают 200 мл ацетона. Выделившееся из маточ-
ного раствора при добавлении ацетона дополнительное коли-
чество галлата натрия отфильтровывают и промывают 300 мл
ацетона.
Всего получают 23 г белого мелкокристаллического по-
рошка, не плавящегося до 500° (выше 200° вещество посте-
пенно темнеет), хорошо растворимого в воде, спирте, не рас-
творимого в ацетоне. Выход количественный.
От примеси галловой кислоты галлат натрия отделяют ки-
пячением в ацетоне. После высушивания в вакууме при 77°
в течение четырех часов найдено, %: С—40,31; 40,39; Н—3,29;
3,31; Na—11,19; 11,26. C7H5O5Na • Н2О; м. в. 210,11. Вычисле-
но, %: С—40,03; Н—3,36; Na—10,95.
Галлат калия. К раствору 3,9 г (0,0235 Л!) галлоной кис-
лоты в 12 мл метанола при перемешивании и пропускании
водорода добавляют раствор метилата калия, полученный из
0,9 г (0,023 М) металлического калия и 25 мл метанола; pH
раствора во время реакции поддерживают в пределах 6—7.
Через 2 минуты выделяется осадок галлата калия. К реакци-
онной смеси добавляют 300 мл ацетона, осадок отфильтровы-
вают и промывают 200 мл ацетона.
180
После кипячения в ацетоне получа!от 4,65 г (95%) белого
мелкокристаллического порошка, не плавящегося до 500° (ве-
щество постепенно темнеет выше 200°), хорошо растворимого
в воде и спирте, не растворимого в ацетоне.
После высушивания в вакууме при 77° в течение четырех
часов найдено, %: С—37,41; 37,48; Н—2,95; 2,92; К—17,69;
17,72. С7Н5О5 Н2О; м. в. 226,23. Вычислено, %: С—37,16;
Н—3,12; К—17,28.
Галлат лития. К раствору 12 г (0,07 44) галловой кислоты
в 50 мл метанола при перемешивании в токе водорода добав-
ляют в течение одной минуты при pH 6—7 раствор метилата
лития, приготовленный из 0,45 г (0,065 М) металлического
лития и 30 мл метанола. Полученный раствор сильно охлаж-
дают сухим льдом, выделившийся осадок фильтруют в атмо-
сфере метана и промывают ацетоном. Маточный раствор упа-
ривают в вакууме в токе метана до 1/3 объема и обрабатыва-
ют 200 мл ацетона. Выделившийся дополнительно осадок гал-
лата лития хорошо промывают ацетоном (см. примечание 2).
Всего получают 12,45 г (99%) галлата лития в виде белых
призм, не плавящихся до 500° (выше 200° вещество постепен-
но темнеет), хорошо растворимых в воде и спирте, не раство-
римых в ацетоне.
После высушивания в вакууме при 77° в течение четырех
часов найдено, %: С—42,98; 43,30; Н—3,83; 3,92; Li—3,15;
3,28. C7H5O,3Li Н2О; м. в. 194,06. Вычислено, %. С—43,32;
Н—3,64; Li —3,58.
Примечания:
1. Если кристаллизация начинается до упаривания, дальнейшая об-
работка производится, как описано при получении галлата калия.
2. Охлаждение сухим льдом и упаривание в вакууме можно заме-
нить осаждением ацетоном, однако при этом расходуется около 3 л аце-
тона на 12 г препарата.
В таблице приведены значения R/ солей галловой кис-
лоты в следующем стандартном наборе систем растворителей:
1) бензол;
2) этилацетат;
3) бутилацетат;
4) петролейный эфир, насыщенный водой;
5) четыреххлористый углерод, насыщенный водой;
6) бензол, насыщенный водой;
7) хлороформ, насыщенный водой;
8) этилацетат, насыщенный водой;
9) бутилацетат, насыщенный водой;
10) ацетон;
11) бутанол, насыщенный водой;
12) изопропиловый спирт +10% воды;
13) ацетон + 10% воды;
14) метанол;
15) метанол + 10% воды;
181
16) вода, насыщенная бутанолом;
17) 3%-ный водный раствор хлористого аммония;
18) бутанол, насыщенный водой,+2% пиперидина;
19) бутанол, пиридин, вода (1:0,6:1);
20) бутанол, уксусная кислота, вода (5:2:3);
21) бутанол, уксусная кислота, вода (2:1:1).
Системь! растворителей
Хроматографические спектры галлатов калия (/), натрия (//), лития (III)
182
На основании данных о подвижности галлатов щелочных
металлов в различных системах растворителей нами состав-
лены их хроматографические спектры (4J, приведенные на ри-
сунке.
ЛИТЕРАТУРА
1. В. Г. Д р о б о т ь к о, Б. Е. А й з е н м а и, М. О. Швайгер,
С. И. Зелену ха, Т. П. Мавдрик. М1кроб1олог1чний журнал, 14,
18 (1962); Антимикробные свойства высших растений, Киев, АН УССР,
1958, стр. 83, 116.
2. О. А. Г о р о д е ц ь к и й, В. А. Б а р а б о й, В. П. Ч е р и е ц ь к и й.
Допов1д1 АН УССР, 12, 1635 (1960); там же, 6, 812 (1961).
3. Ph. Bucher Liebigs Ann. Chem. 53, 192 (1845).
4. H. О. Блинов, E. Ф. О п a p ы ш e в а, Г. 3. Якубов, А. В. Пу н-
н и н а, Ю. М. Хохлова, Е. М. Клейнер, Н. Г. Федькина.
К. Н. X р я ш, е в а, А. С. Хохлов. Классификация и идентификация
антибиотиков по хроматографическим спектрам, в сб. «Молекулярная
хроматография», М., изд. «Наука», 1964, стр. 137.
Поступила в декабре 1964 г.
Институт микробиологи и вирусологии
АН УССР
УДК 547.787.3.07
2-ЭТИЛБЕНЗОКСАЗОЛ
Д. 4 ДРАПКИНА, В. Г. БРУДЗЬ, В. Л. ИНШАКОВА,
Н. И. БАДАИКОВА
I i| >-chsch3
/ \ о /
C0H,,NO М.в. 147,18
2-Этилбензоксазол был получен взаимодействием о-амино-
фенола с пропионовым ангидридом — выход 68% [1] и 28%
[2]; пропиоиитрилом—выход 61,2% (31 и триэтилортопропио-
натом— выход 75% 14].
Нами для синтеза этого вещества был использован хлор-
ангидрид пропионовой кислоты.
СХЕМА СИНТЕЗА 2-ЭТИЛБЕНЗОКСАЗОЛ А
ОН
I
C.H»gH?-C.°C1 Ч
| II 3 I II /—CH4CHj
W Ч/\о/
Характеристика основного сырья
о-Аминофенол, ч., ТУ МХП 1979—49.
Хлорангидрид пропионовой кислоты, ч., ВТУ ХСНХ
178—53.
Условия получения
В круглодонную колбу, снабженную обратным холодиль-
ником, помещают 18,5 г (0,07 М) о-аминофенола и 25 г
(0,27 /И) хлорангидрида .пропионовой кислоты и нагревают
184
смесь На глицериновой бане до 120° (температура бани). Ре-
акционную массу выдерживают в этих условиях в течение
1,5 часа. Затем температуру бани повышают до 150° и, уда-
лив холодильник, дают выдержку при этой же температуре в
течение 5 часов. По окончании выдержки к колбе присоеди-
няют нисходящий холодильник и отгоняют фракцию, выки-
пающую до 160° (в парах). Эта фракция в основном представ-
ляет собой избыточный пропионилхлорид. Остаток охлажда-
ют до комнатной температуры, добавляют 50 мл 10%-ного
раствора углекислого натрия и экстрагируют этиловым эфи-
ром (3 раза по 70 мл). Эфнрный экстракт сушат над прока-
ленным углекислым калием, эфир отгоняют, а остаток пере-
гоняют в вакууме, отбирая фракцию при 96—97°/9 мм.
Выход 2-этилбензоксазола равен 10—11 г, что составляет
40—44% от теоретического; т. кип. 215—2177760 мм; ntg —
1,5419.
По литературным данным, т. кип. продукта 214—
2177760 мм Ш; 75—7672 мм [3]; 129723 мм 141.
Примечание.
Примерно такой же выход 2-этилбензоксазола получают при несколько
иной обработке: после отгонки фракций, выкипающей при 160°, темпера-
туру поднимают выше и перегоняют 2-этнлбензоксазол, основное количе-
ство которого переходит при 214—220°.
Его промывают 2 раза 10%-ным раствором соды (по 70 мл), сушат
и перегоняют в вакууме. Этот метод выделения менее удобен вследствие
кристаллизации в форштоссе холодильника солянокислой соли 2-этилбен-
зоксазола.
ЛИТЕРАТУРА
1. Брит. пат. 889186 (1962); Пат. США 3149120 (1964).
2. F. М. Hamer. J. Chem. Soc., 1956, 1480.
3. L. E. H alljes, E. C. Wagner. J. Organ. Chem., 9, 31(1944).
4. G. L. Lenkins, A. M. К n e v e 1, Ch. S. Davis. J. Or?an
Chem., 26, 274 (1961).
Поступила s ноябре 1965 r.
ИРЕА
УДК 547.587.26.07
ЭФИРЫ ГАЛЛОВОЙ кислоты
В. П. ЧЕРНЕЦКИЙ, Т. Д. БЕЛЫШЕВА, В. Г. ВОЛОЩУК.
Э. А. ПОНОМАРЕВА
НО
НО-/
р 4 OR
НО
R: СН3; СаН6; н-С3Н7; н-С4Н9; п-С6Нп; i-C6Hu;
н-С6Н13; н-С,Н15; н-С8Н17.
Эфиры галловой кислоты широко применяются в качестве
антиоксидантов в пищевой и парфюмерной промышленности.
Они обладают высокой активностью против бактерий [1] и
вирусов [2]. В последнее время установлено противоопухоле-
вое [3] и антилучевое [4] действие пропилгаллата и других
эфиров галловой кислоты.
В литературе описано получение эфиров галловой кисло-
ты из галловой кислоты и спирта в присутствии хлористого
водорода [5, 15], серной кислоты [6] и алкилсульфатов [7].
В качестве катализатора этерификации применяют также
ионообменную смолу типа Амберлит IR-120 [8].
При прямой этерификации галловой кислоты высшими
спиртами используют высококипящий полярный инертный ра-
створитель (например, анизол или нитробензол) и раствори-
тель, с которым вода удаляется азеотропно [9]. Разработан
также способ получения эфиров через хлорангидрид галловой
кислоты: галловая кислота реагирует с хлористым тионилом
при 120°, галлоилхлорид выделяется и затем вводится в реак-
цию с высшими спиртами [10, 12].
Мы установили, что этот процесс можно значительно
упростить: этерификация идет при добавлении хлористого
186
тионила к смеси спирта и галловой кислоты. Нами показано
также, что этерификацию н-гексиловым, н-гептиловым и
н-октиловым спиртами можно проводить классическим спосо-
бом — при насыщении хлористым водородом. Этот простой
способ дает удовлетворительные выходы.
Применение нами в качестве катализатора ионообменной
смолы давало хорошие результаты, когда использовалась
ионообменная смола № 1, сильнокислая, катионообменная,
для аналитических целей. В присутствии катионита КАУ-1
этерификация не идет.
СХЕМА СИНТЕЗА ЭФИРОВ ГАЛЛОВОЙ КИСЛОТЫ
НО
L /О
но—Ч-с^
он
но
но
1_ о
+ROH * НО—+Н2о
OR
но
Характеристика основного сырья
Галловая кислота, ч., ТУ МХП 2726—55.
Метиловый спирт, ч., ГОСТ 2222—54.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
н-Пропиловый спирт, очищенный, ст. ГОХП 1523—54.
н-Бутиловый спирт, ч., ГОСТ 6006—51.
н-Амиловый спирт, ч., СТУ 12-10-158—61.
Изоамиловый спирт, ч., ГОСТ 5830—51.
н-Гексиловый спирт, очищенный, СТУ 12-10-154—61.
н-Гептиловый спирт, ч., СТУ 12-10-157—61.
н-Октиловый спирт, ч., СТУ 12-10-156—61.
Тионил хлористый, ВТУ МХП 3591—52.
Серная кислота, х. ч., ГОСТ 4204—48.
Ионообменная смола № 1, сильнокислая, катионообмен-
ная, для аналитических целей.
Условия получения
а) Смесь галловой кислоты и спирта (1:4) насыщают при
минус 5—0" хлористым водородом и кипятят с обратным
холодильником несколько часов. Галловая кислота, как пра-
вило, растворяется через час после начала кипения. После
отгонки спирта в вакууме эфир промывают ледяной водой и
очищают кристаллизацией.
б) Смесь 34 г галловой кислоты, ионитного катализатора
(см. примечание 1) кипятят с обратным холодильником не-
сколько часов. Если спирт образует с водой азеотропную
187
смесь (например, изоамнловый), выделяющуюся при реакции
воду отбирают в водоуловителе (ловушка Дина — Старка; см.
примечание 2). Катализатор отфильтровывают, раствор обра-
батывают, как описано выше.
в) К смеси галловой кислоты и спирта (1:10) при переме-
шивании и охлаждении добавляют по каплям 3 мл хлористо-
го тионила (см. примечание 3). Смесь кипятят с обратным
холодильником в течение 2 часов. Дальнейшая обработка по
предыдущему.
г) Смесь 160 г галловой кислоты, 320 мл спирта и 16 мл
концентрированной серной кислоты кипятят с обратным холо-
дильником в течение 8 часов. Дальнейшая обработка по пре-
дыдущему.
д) Смесь 68 г галловой кислоты, 240 мл н-бутанола и 1 мл
концентрированной серной кислоты кипятят с отбором воды
в ловушке Дина — Старка до полного растворения галловой
кислоты (см. примечание 4). Дальнейшая обработка по пре-
дыдущему.
е) Смесь 100 г галловой кислоты, 270 мг этилового спирта
и 20 мл концентрированной серной кислоты кипятят с обрат-
ным холодильником в течение 4,5 часов. Серную кислоту ней-
трализуют 40 г карбоната бария. Избыток этилового спирта
отгоняют в вакууме после разбавления смеси небольшим ко-
личеством горячей воды. Оставшуюся массу обрабатывают
горячей водой, осадок солей удаляют центрифугированием
при 6 000 оборотов в минуту в течение 20 минут. Из ма-
точного раствора при охлаждении выделяется эфир в .виде
друз длинных игл. После перекристаллизации из воды с об-
работкой активированным углем получают кристаллогидрат
эфира с т. пл. 70°. Кристаллизационную воду удаляют кипя-
чением в бензоле с ловушкой Дина — Старка.
В таблице приведены данные о способах получения и кон-
станты полученных эфиров.
Примечания:
1. Ионитный катализатор предварительно обрабатывают разбавленной
серной кислотой с последующей тщательной отмывкой водой до исчез-
новения сульфат-иона в фильтрате ГН] и сушат в вакууме водоструйного
насоса при SO'' до постоянного веса.
2. Процесс прекращается, когда количество выделившейся воды соот-
ветствует уравнению реакции. Превышение этого количества свидетельст-
вует о нежелательном превращении избыточного спирта в эфир [17].
3. Хлористый тионпл берут свежеперегнаниый над льняным маслом.
4. Необходимо избегать перегрева. Бутилгаллат легко осмоляется.
5. Этилгаллат по свойствам отличается от остальных эфиров. Он не
получается в присутствии иоиитиого катализатора, легко гидролизуется
водой. При получении в присутствии серной кислоты перед упариванием
в вакууме необходима нейтрализация карбонатом бария. Полученный нами
препарат имел температуру плавления ниже, чем описанная в литературе,
но элементарный анализ подтверждает его аналитическую чистоту.
Найдено, %: С—54,78; 54,79; Н—5,03; 5,10
С9Н)0О5. Вычислено, %: С 54.55; Н—5,05.
188
6. Полученный па мил галлат плавится при температуре па 3° ниже,
чем описано в литературе.
Найдено, %: С-59,90; 59,88; Н-6,61; 6,71.
ClsH10O3. Вычислено, %: С -60,00; Н—6,67.
7. Интересно сопоставить температуры плавления полученных эфиров.
При увеличении числа углеродных атомов в спиртовом радикале наблю-
дается вначале резкое понижение температуры плавления. Начиная с
н-гексил-галлата, при переходе к высшим гомологам температура плавления
практически не меняется: н-гексил—94°; н-гептпл—96°; н-октил—96—
97°; и-ноиил — 96—97°; н-децил—94—95°; н-уидецнл- —96—97°; н-додецнл-
—96—97°; н-тетрадецил—97°; и-гексадецилгаллат —99° [12].
8. Две последние колонки таблицы содержат значения R/ эфиров в
бутаноле, насыщенном водой, и в этилацетате. Ry галловой кислоты в пер-
вой системе растворителей был равен 0,55, во второй — 0,78. Проявление
бумажных хроматограмм проводилось 1%-ным раствором хлорного же-
леза.
R Способ получения Время кипяче- ния (часы) Растворитель для кристал- лизации Выход, % Температура плавле- ния* ** ***", °C Rf****
в бутаноле, насыщен- ном водой в этелаце- татс
наши данные литера- турные данные
-сн3 б г 2 8 метанол - вода (2:3) 81,8 95,0 196 198 200 - 202 199-202 18] 0,55 0,66
(%!;.' а е 6 4,5 вода 64,7 66,4 152-152,5 146-147 141, 160 158 [9,13] 0,88 0,95
— н-С3Н7 б г 10 4 вода 56,6 51,0 150, 148-150 147-148 [14,6] 0,85 0,82
- н-С,П„ б д 5 1 вода 87,5 70,4 141 143 142 143-144 [14,6] 0,80 0,76
—и-С8Нп** а в 2 2 вода 86,0 69,2 124-125 124 127 [15] 0,82 0,82
—/-С5Нц б 4 вода 70,5 142-143** 145-146 [16] 0,85 0,86
н-СеН13 а 6 вода 51,2 93-95 93-94 [12] 0,68 0,65
— И-С,Н,5 а 6 вода, аце- тон- вода, метанол— вода, ССЦ 48,0 94-95 96 [121 0,72 0,68
— |Г-С8НП а 6 СС1О мета- нол—вода, ацетон — вода 67,8 96-97 96-97 [12] 0,81 0,75
* См. примечание 5.
** См. примечание 6.
*** См. примечание 7.
**** См. примечание 8.
189
Литература
1. В. D о n а 1 d, Johnstone, М. W. Т о о t е, W. I. Rogers,
t. Е. Little. Antibiotics a. Chemotherapy, 3, 203 (1953).
К. J. Н. Sluts. Chem. Courant, 50, 383 (1951).
2. С. М. Гершензон, Т. Г. Б р е з г у и о в а, В. П. Ч е р и е ц к н й.
Докл. АН СССР, 431, 442 (1960).
3. Н. М. Эмануэль, Т. Э. Липатова. Докл. АН СССР, 130,
221 (1960).
4. А. А. Городецкий, В. А. Б а р а б о й, В. И. Чернецкий.
Радиобиология, 781 (1961).
5. С. S с h б р f, L. Winterhalder. Liebigs Ann. Chem., 544.,
62 (1940).
6. В. H e p n e r, L. Zyto. Roczn. chem., 12, 100(1932).
7. H. V о п В r a m e r, M. L. Clemens. Англ. пат. 2615042; С.A., 47,
9362a (1953).
8. J. Haas, M.R. Z e n t n e r. J. Amer. Pharmac. Assoc. 43,
635 (1954).
9. W. С. H u 1 t, J. K. W e i I, G. C. N u 11 i n g. C. J. jC о w a n. J.
Amer. Chem. Soc., 69, 2003 (1947).
10. N. V. Chemische. Fabrick „Naarden". Голл. пат. 63319' С.A., 43,
6665rf (1949).
11. Препаративная органическая химия, ГИТИ, Химлит., 1959, стр. 852.
12. G. J. М. V a n d е г К е г k, J. Н. Verbee к, J. С. F. С 1 е t о и.
Rec. trav. chim., 70, 277 (1951).
13. С. Etti., Ber. Il, 1882 (1887); H. C. Bidle. J. Amer. Chem. Soc.,
35, 96 (1913).
14. J. Clarke, R. Robinson, I. C. Smith. Soc. 1927, 2647.
15. A. Russel, W. О. Те bions. J. Amer. Chem. Soc., 64, 2274
(1942).
16. A. M с. К e n z i e, H. A. M u lie r. J. Chem. Soc., 95, 547 (1909).
17. H. H. Д ы x а н о в, В. P. Шилов. Методы получения химических
реактивов и препаратов, вып. 10, М., ИРЕА, (1964), стр. 90.
Поступила в августе 1965 г. Институт микробиологии и
вирусологии АН УССР
АЛФАВИТНЫЙ УКАЗАТЕЛЬ СОЕДИНЕНИЙ, ОПИСАННЫХ
В НАСТОЯЩЕМ СБОРНИКЕ
стр.
Азотнокислая соль 2-иафтиламина................................... 108
Алкилдихлорфосфины.................................................. 7
н-Амиловый эфир галловой кислоты . . •.......................... 189
Аминотетразол..................•................................... 27
З-Амииофталевая кислота, гидрохлорид............................... 10
2-Аитрилгидразин................................................... 13
2-Антрол :......................................................... 15
6-Ацетилеиилииклогексаиол.......................................... 54
Ацетилтиохолинйодид ............................................... 17
Ацетилхолннйодид................................................... 19
N-Бензилмальимид ........................•......................... 88
Г^-Бензилэтилендиамнн-М.М'.К'-триуксусная кислота.................. 22
а,а-Бис-(4-натрийтетразолилазо-5)-этилацетат ...................... 26
Борфторнд м-этилбепзолдиазоиий.................................... 166
4-Бромбеизальдетнд................................................. 29
6'-Бром-8'-нитро-3,3'-диметилспиро-[бензоксазол-2,2'- (2'Н-1'-бензо-
пиран)]....................................................... 67
8'-Бром-6'-иитро-3,3'-диметилспиро- [бензоксазол-2,2'-(2’Н-Г-бензо-
пиран)] ................................•...................... 67
З-Бром-5-нитросалициловый альдегид................................. 32
6'-Бром-8'-иитро-1,3,3-триметилспиро-|(2'Н-Г-бвизопиран)-2,2г-иидо-
лни].......................................................... 146
8'-Бром-6'-нитро-1,3,3-триметилспиро- |(2'Н-1 'бвнзопираи)-2,2'-иидо-
лии]......................................................... 146
6'-Бром-8'-иитро- 1-феиил-3,3- диметилспиро- [(2'Н-Г-бензонираи)-
2,2'-индолин]................................................ 156
8,-Бром-6'-нитро-1-фенил-3,3-диметилспиро-[(2'Н-1'-беизопираи)-2,2'-
иидолин]...................................................... 156
6'-Бром-1,3,3-триметилспиро-[(2'Н-Г-беизопиран)-2,2'иидолин] . . . 146
Бромэтилфталимид ................................................ 139
и-Бутиловый эфир галловой кислоты...........•.................... 189
2-Винилпиридии..................................................... 34
Галлат калия .................................................... 180
Галлат кобальта.................................................. 37
191
Галлат лития .................................................... 180
Галлат натрия................................................... 180
н-Гексиловый эфир галловой кислоты............................... 189
н-Гептиловын эфир галловой кислоты............................... 189
Гидразид малеиновой кислоты...................................... 40
Гидразобензол.............................................. . 43
6-Диазо-5-кето1-норлейцин........................................ 46
5,5-Диалкил-4-метил-4-окси-3-циантетрагидрофураны................ 50
Дибензиловый эфир............................................... 50
Диметилацетиленилкарбинол........................................ 51
1,4-Диметил-2,5-диизопропилбепзол................ . . . . 58
1,4-Диметил-2-нзопропилбепзол....................•.............. 58
N.N-Диметил-о-нитроаиилии....................................... 61
5,7-Диметнл-8-оксихинальдоксим.................................. 63
3,3-Диметилспиро-[беизоксазолин-2,2'-(2'Н-1'-бензопираны)] .... 65
N.N-Днметил-о-фенилендиамин . .................................. 69
М-2,4-Димети11феиилмальимид . . . •..........................1 88
2,2'-Диокси-1,Г-нафтальдазин.................................... 71
2,8-Диоксихинолин............................................... 73
N-N'-Дифенилформамидинсульфиновая кислота, дигидрат........... 75
8,8-Дихинолин................................................... 77
Дихлорангидрид трихлорметилтиофосфиновой кислоты................ 79
4,4'-Дихлордибутиловый эфир..................................... 81
2,2- Дихлорпропан.............•.......................• . . . 83
5-Днциклогексиламмониевая соль 1-этилового эфира N-трифтор-
ацстил-Г-глутаминовой кислоты ................................ 48
N-Замещснные имиды малеиновой кислоты............................ 85
Изоамиловый эфир галловой кислоты.............................. 189
2-Йодтиоэтиловый эфир уксусной кислоты........................... 18
2-Йодэтилацетат ................................................. 20
Комплексное соединение метилтрихлорйодфосфина и хлористого
алюминия...................................................... 90
Комплексное соединение диметилдихлорфосфина и хлористого
алюминия ................................................... 91
Малеаминовые кислоты.........................................• 87
6-Метил-2-вииилпиридин.......................................... 93
Метил йодистый.................................................. 96
Метиловый эфир галловой кислоты............................... 189
4-Метил-5,5-пеитаметилен-4-окси-3-циантетрагндрофуран............ 53
4-Метнл-5-н-пропил-4-окси-3-циантетрагидрофуран ....... 53
8'-Метокси-6'-нитро-3,3'-диметилспиро- [бензоксазол-2,2'- (2' Н-1 '-бен-
зопиран)] .................................................... 67
8'-Метокси-6'-нитро-1,3,3-триметил спиро -[(2'Н -1'-беизопиран)-2-2'-
икдолин]..................................................... 146
6'-Метокси-8'-нитро-1-фенил-3,3-диметилспиро- [(2'Н-1'-бензол иран)-
2,2'-индолин]............................................... 156
8'-Метокси - 6' - иитро-1-фенил-3,3-диметилспиро- [(2'Н-1'-бензопи-
ран)-2,2'-индолин]......................•...................... 156
8'-Метокси-5'-нитро-1-феннл-3,3-диметил спиро-[(2'Н-Г-беизопиран)-
-2,2'-иидолин]............................................... 156
N-Монометилолформамидинсульфииовая кислота...................... 98
Моионатриевая соль 2-нафтилимииодиуксусной-1-сульфокислоты . 104
1-М-Морфолил-пропаиол-2.......................................... ИХ)
192
Натриевая соль 2-антраценсульфокислоты....................... 102
1-Нафтилимннодиуксусиая 4-сульфокнслота.............. .... 104
Нафтионоводиуксусная кислота ...................................... 166
6'-Нитро-3,3'-диметнлспиро-[бензоксазол-2,2'-(2'Н-1 ’-бензопиран)] . 67
5-Нитро-2-нафтиламин.............................................. 107
5-Нитро-2-оксн-3-метоксибеизальдегид............................. 109
З-Нитросалициловын альдегид.................................. 111
5-Нитросалициловый альдегид.................................. 111
6'-Нитро-1,3,3-трнметилспиро-[(2'Н-1'-беизопираи)-2,2'-индолин] . . 146
7'-Нитро-1,3,3-триметилспиро-[(2'Н-Г-бензопиран)-2,2'-нндолнн] . 146
8'-Нитро-1,3,3-триметилспиро- [(2'Н-1'-бензопиран)-2,2'-индолнн] . 146
6'-Нитро-1-фенил-3,3-димстилспиро-[(2'Н-Г-бензопиран) -2,2'-иидо-
лии].......................... . • . . •..................... 156
8'-Нитро-1-фенил-3,Здиметилспнро - [(2'Н-1'-бензопиран)-2,2'-ипдо-
лин] ......................................................... 156
N-n-Нитрофенилмальимид............................................ 88
З-Нитрофталевая кислота..........................................• 11
б-Нитрохинолин-2-альдегид......................................... 116
6-Нитрохинальдоксим............................................... 114
1-(8-Окси-2-хинолил)-3,5-диметилпиразол........................... 118
Октиловый эфир галловой кислоты................................... 189
1-ГЧ-Пиперидил-пропанол-2......................................... 100
1-(2-Пиридилазо)-2-нафтол........................................ 120
п-Пропилацетиленкарбинол........................................... 53
н-Пропп.ювый эфир галловой кислоты...................• . . . . 18!)
Тетрабензоилацетоноевропиат пиперидиния .............’..... 123
Тетрадибеизоилметаноевропиат пиперидиния . . •.................... 125
1,2,4,5-Тетраизоиропилбеизол...................................... 127
3,3'4,4'-Тетракарбоксидифенилоксид............................... 130
3,3',4,4'-Тетракарбокси-(2,2'-дифеиилпропан)...................... 130
3,3',4,4'-Тетраметнлдифенилднметнлметан.................. . . 136
N-n-Толилмальимид................................................... 88
n-Толуолсульфонат 2-этил-З-мстилбепзоксазолипия..................... 66
Р,Р',Р”-Триаминотриэтиламин солянокислый.......................... 138
Тридибензоилмеганаммип-европий . . .............................. 142
4,5,5-Триметил-4-окси-3-пиантетрагидрофуран........................ 50
1,3,3-Триметилспнро-[(2'Н-1'-бензопиран)-2,2'-нндолииы]........... 144
Трис-(фталимндэтнл)-амин.......................................... 146
Трифторуксусный ангидрид...................................• . . 48
Трихлорметилдихлорфосфин.......................................... 148
У рейд ацетоуксусной кислоты...................................... 150
1-Фепнл-3,3,-диметил-2-метиленинлолии ............................ 151
1-Феиил-3,3-диметилспиро-[(2'Н-1'-бензопнран)-2,2'-пндолииы] . . 154
4(5)-Феиилимидазон-2.............................................. 174
N-Фенилмальнмид..................................................... 88
Фениловый эфир антраниловой кислоты............................... 158
N-Феиплформамндинсульфиновая кислота............................... 160
П-Фторэтилбензол.................................................. 165
п-Фторбеизальдегид............................•.................... 162
5-Хлораигидрид 1-этилового эфира Г4-трифторацетил-1-глутамино-
вой кислоты . . . ............................................. 48
5-Хлор-8-(мезитиленсульфониламино)-хииолин.................... 168
5-Хлор-З-нитросалициловый эфир.......................• . . . . 171
6'-Хлор-8'-нитро-3,3'-диметилспиро-[бензоксазол-2,2'- (2'Н-1'-бензо-
пиран)! ...................................................... 67
13 Зак. 25 193
6'-Хлор-8'-ннтро-1,3,3-триметилспнро-[(2'Н-Г-беизопнран)- 2,2’ -;ин-
долин]......................................................... 146
б’-Хлор - 8' - нитро - 1- фенил-3,3-диметнлспиро-[(2'Н-Г-беизопираи)-
2,2'-нндолнн].................................................. 156
7'-Хлор-1,3,3-триметилспиро-[(2'Н-Г-бензопиран)-2,2'-индолин] . . 146
2-Хлор-4(5)-фенилимидазол...................................... 173
N-n- Хлорфеннлмальимид ......................................... 88
N-лг-Хлорфенилмальимнд . . .................................... 88
п-Хлорэтнлбензол............................................... 176
Шелочные соли галловой кислоты.................................... 179
2-Этилбензоксазол................................................. 184
Этнлдихлорфосфнн..........................•...................... 8
Этилеисульфид.............•..................................... 18
Этиловый спирт галловой кислоты .... ................... ..... 189
I-Этиловый эфир М-трифторацетил-6-диазо-5-кето-Е-норлейцина . . 49
Эфиры галловой кислоты......................................... 186
Редактор Б. г. Козлов
Техн, редактор А. И. Пирожкова
Корректоры: Л. Г. Черкасова, Л. А. Климанова
Сдано в набор ?0. 12.1965 г. Подо. к печати 24.1.1967 г.
Л-73056 Формат бум. 60x90 lJM Печ. л. 12,25 Уч.-изд. л. 6,9
Зак. 25 Тираж 1300 экэ. Цена 62 коп.___________
типография ВАХЗ
Замеченные опечатки
Стр, Строка Напечатано Следует читать
14 8 сверху 400 мл I в 400 мл
19 3 снизу Йодистый метил, ч., Ацетил йодистый, ч.,
ТУ МХП 49-51. ВТУ МХП 3842— 53.
20 3 » трубкой пробкой
24 16 сверху N-780 N—7,80
,СН8-СН2ч ,СН2-СН2.
50 6 , Ri и R2f ХСП2 R, и R2:f хСН2
хснг-сн/ XCH2-CH2Z
64 6 снизу Н—12,96 N—12,96
66 5 . SOCcH4CH3 SO3C6H5CH3
68 Табл. 2, гра- CnHj.BrNjO, С. CjyHigBrNgO^ С
фы 4, 5, 6, 7 н II
н N
Вг
71 12 сверху 2-окси-13-иафтальдегид 2-окси-1-иафтальдегил
74 13 снизу Vakugaku Yakugaku
74 4 „ стр. 86 стр. 88
74 2 стр. 88 стр. 18
79 14 сверху, СС12РС14-Д1С13 CCIjPCIj-А1С13
109 12 сверху о-винилина о-ванилина
водно-пириди-
новая среда
133 1 снизу А. В. Кочеткова А. В. Афанасьева
146 Табл. 1, гра- 33,4 43,4
фа 9
147 Табл. 2, гра- 116-117 116-117 [1]
фы 3 и 5 CjHi8NOC1 C19H18NOCI
СНг ^СНз СН3\ ^СНз
154 Формула 1 1 >сн* 1 1 хЧ = СНз
А 1 /X
U 1 II
170 3 сверху С-69,91 С—59,91
184 2 снизу (0,07 М) (0,17 М)
185 3 . 4. G. L. Lenklns 4. О. L. Jenkins