/















































































































































































































































































































Текст
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ НЕФТЕХИМИЧЕСКОГО СИНТЕЗА
им. А. В. ТОПЧИЕВА
ТЕОРЕТИЧЕСКАЯ
И ПРИКЛАДНАЯ
ПЛАЗМОХИМИЯ
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА
19 7 5
УДК 633.9.01
Теоретическая и прикладная плазмохимия. Полак Л. С.,
Овсянников А. А., СловецкийД. И., ВурзельФ. Б. М.у
«Наука», 1975, стр. 1—304.
Книга представляет собой обобщение результатов теоретических
и экспериментальных исследований в области плазмохимии. В ней изла-
гаются основы неравновесной химической кинетики, механизмы плазмо-
химических реакций, способы генерации и диагностики низкотемпера-
турной плазмы, химические реакции в плазменных струях и турбулент-
ных потоках, способы моделирования плазмохимических реакторов, а
также многочисленные прикладные плазмохимические процессы.
Книга представляет интерес для широкого круга специалистов — пре-
подавателей вузов, а также аспирантов и студентов, специализирую-
щихся в области плазмохин, низкотемпературной плазмы и физической
химии.
Таблиц 44. Иллюстраций 107. Библиогр. 1072 назв.
Ответственный редактор
доктор физико-математических наук
Л. С. ПОЛАК
Авторы:
Л. С. ПОЛАК, А. А. ОВСЯННИКОВ,
Д. И. СЛОВЕЦКИЙ, Ф. Б. ВУРЗЕЛЬ
20503-475
055(02)-75 115'75
(Й Издательство «Наука», 1975 г.
ПРЕДИСЛОВИЕ
Авторы этой книги исходили из того, что «подлинный ученый
скорее стремится установить факты, нежели искать подтвержде-
ния своему пониманию истины, ибо истина существует незави-
симо от того, будет она открыта или нет. И мудрый ученый со-
знает, что истина иногда может в глазах, созерцающих ее, ря-
диться в разные одеяния» х.
В течение последних 30—35 лет спорадически появлялись от-
дельные работы по важным вопросам плазмохимии, но оформи-
лась она в самостоятельную науку только за последнее десяти-
летие. Возникли теоретическая плазмохимия — новый раздел
физической химии и плазмохимическая технология — новая об-
ласть промышленной химической технологии.
Пришла пора подвести первые, конечно предварительные,
итоги бурно развивающейся науки с тем, чтобы в какой-то сте-
пени способствовать ее дальнейшему прогрессу.
Авторы будут считать свою задачу выполненной, если эта
книга послужит некоторым «катализатором» дальнейшего разви-
тия плазмохимии.
Первая и четвертая главы нанисаны А. А. Овсянниковым,
вторая — Л. С. Полаком, третья — Д. И. Словецким, пятая
глава — Ф. Б. Вурзелем.
За большую помощь в технической подготовке рукописи ав-
торы благодарят Н. М. Рытову.
Л. Полак
1 М. Кальвин. Химическая эволюция. М., «Мир», 1971, стр. 229.
1
ГЕНЕРАЦИЯ И ДИАГНОСТИКА
НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЫ
Организация любого промышленного химического процесса имеет своей
целью экономически целесообразное производство требуемого продукта из
исходного сырья. На рис. 1.1 представлена схема типичного химико-техно-
логического процесса [1]. Исходное сырье подвергается ряду физических
операций, доводится до состояния, в котором оно может испытывать хими-
ческие превращения, и пропускается через химический реактор. На выходе
_ Энергия
Рис. 1.1. Схема типичного химико-технологического процесса
химического реактора реакционная смесь подвергается дальнейшей обработ-
ке, задачей которой является сохранение, выделение и очистка целевого
продукта и т. п. После выделения целевого продукта часть непрореагировав-
шего сырья может вновь подаваться на вход схемы (рецикл) для повторной
обработки.
Схема плазмохимического процесса в принципе не отличается от приве-
денной. Однако в ряде случаев указанные на рис. 1.1 стадии процесса (за иск-
лючением, быть может, стадий разделения и очистки) могут совпадать и в
пространстве, и во времени. Обусловлено это основной особенностью плаз-
мохимических процессов, заключающейся в том, что по меньшей мере один
из компонентов реакционной смеси находится в состоянии плазмы х. Таким
образом, технологическая схема любого плазмохимического процесса дол-
жна включать в себя устройство для преобразования вещества в состояние
плазмы — генератор плазмы.
1 При этом плазма может являться как одним из реагентов в рассматриваемой хими-
ческой реакции, так и эффективным энергоносителем.
5
ГЕНЕРАТОРЫ НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЫ
Выбор генератора плазмы для осуществления данного плазмохимиче-
ского процесса определяется термодинамическими и кинетическими особен-
ностями этого процесса. Ввиду большого разнообразия возможных плазмо-
химических процессов для их осуществления требуются генераторы плазмы
самых разнообразных типов. С точки зрения организации промышленных
многотоннажных плазмохимических процессов наиболее перспективными
считаются в настоящее время электродуговые плазмотроны постоянного и
переменного тока (промышленной частоты). Достигнутый уровень мощности
таких плазмотронов составляет в настоящее время — 10 МВт при ресурсе
работы — 200—300 ч и КПД — 0,8. Конструкция этих плазмотронов допус-
кает работу нескольких плазмотронов на общий канал плазмохимического
реактора с соответствующим увеличением вкладываемой в плазму мощности.
В том случае, когда недопустимо загрязнение плазмы материалами эрозии
электродов, имеющее место в электродных дуговых плазмотронах, весьма
перспективными генераторами плазмы могут служить безэлектродные ВЧ- и
СВЧ-плазмотроны. Хотя достигнутый уровень мощности таких плазмо-
тронов сравнительно невелик (ВЧ-плазмотроны — не более 0,5 МВт,
СВЧ — не более 0,05 МВт) и КПД не превышает в лучшем случае 60%,
они успешно конкурируют с дуговыми плазмотронами в тех производствах,
где важнейшим фактором является стерильность генерируемой плазмы.
Кроме того, следует отметить, что ресурс работы таких плазмотронов в прин-
ципе всегда превышает ресурс работы дуговых плазмотронов (по крайней
мере в настоящее время).
Устройства, основанные на применении тлеющего, коронного и других
видов электрического разряда, менее широко применяются в промышленной
плазмохимии, чем дуговые и ВЧ-плазмотроны. Их использование тем не ме-
нее часто позволяет проводить химические реакции в неравновесной плаз-
ме, когда, например, температура тяжелых частиц находится на уровне
— 300" К, а электроны имеют среднюю энергию порядка нескольких элект-
рон-вольт. Условия проведения химических реакций в такой плазме еще слабо
изучены, но накопленные к настоящему времени результаты лабораторных
исследований позволяют надеяться, что именно использование неравно-
весной плазмы обеспечит возможность проведения сложных плазмохимиче-
ских синтезов.
Генераторы плазмы, основанные на применении рентгеновского, у- или
лазерного оптического излучения, а также ударных труб, используются
в настоящее время лишь в практике лабораторных плазмохимических ис-
следований. Получаемые с их помощью результаты имеют большое значение
для развития плазмохимии, позволяя рассчитывать коэффициенты скорости
и определять механизмы плазмохимических реакций.
В последнее время значительный интерес проявляется к способу генера-
ции низкотемпературной плазмы, основанному на адиабатическом сжатии
газов. Результаты, получаемые на установках одноимпульсного адиабати-
ческого сжатия, позволяют, как и в случае ударных труб, определять коэф-
фициенты скорости и энергии активации плазмохимических реакций.
Создание установок адиабатического сжатия непрерывного действия даст
возможность проводить многие плазмохимические процессы с высокой селек-
тивностью и большой производительностью.
1. Дуговые электродные плазмотроны
Надежно работающие дуговые плазмотроны появились после того, как
были разработаны способы стабилизации в пространстве столба электриче-
ской дуги и теплозащиты стенок разрядных камер. По-видимому, одним из
первых плазмотронов следует назвать устройство, предложенное Петерсом
[2]. Обзор ранних работ по дуговым генераторам плазмы можно найти в пе-
6
реводных сборниках [3, 4]. К настоящему времени разработка в СССР ду-
говых плазмотронов переменного и постоянного тока сильно продвинулась
вперед [5, 6].
Организация плазмохимического процесса на дуговых плазмотронах.
Требования, предъявляемые промышленностью к дуговым плазмотронам,
сводятся в основном к следующему: 1) высокий уровень мощности (несколько
МВт)\ 2) длительный ресурс работы (несколько сотен часов); 3) экономич-
ность, т. е. высокий электрический и тепловой КПД; 4) возможность нагрева
любых газов и газовых смесей до температуры, необходимой для осуществле-
ния данного технологического процесса. При организации конкретного плаз-
мохимического процесса к этим общим требованиям может добавляться ряд
частных (например, к материалу электродов, габаритам и т. п.).
Прообразом современных плазмохимических процессов, использующих
электродуговые генераторы плазмы, может служить процесс электрокре-
кинга природного газа. На рис. 1.2 представлен один из вариантов схемы
электродугового плазмотрона-реактора для процесса электрокрекинга
природного газа [7]. Дуга горит между стальными водоохлаждаемыми элект-
родами 1 и 2 в вихревом потоке природного газа, вдуваемого через кольцо
закрутки 3. Данное устройство позволило достигнуть довольно высоких
уровней мощности (~ 1—2 МВт) при КПД — 75—80%. Однако одно из
основных требований к таким установкам не выполнялось — ресурс плаз-
мотрона-реактора составлял всего лишь несколько часов, так как каналы
его электродов забивались сажей. Аналогичный результат был получен при
недавних полупромышленных испытаниях плазмотрона с двухсторонним
истечением конструкции ИТМО АН БССР [8] мощностью — 1 МВт. Этот
плазмотрон, позволивший получить вполне удовлетворительные результаты
в плазмохимическом процессе фиксации атмосферного азота, оказался непри-
годным для процесса получения ацетилена из природного газа. Как и в слу-
чае электрокрекинга, ресурс работы этого плазмотрона не превышал 10 ч
вследствие забивки сажей электродных каналов. Все это — результат стрем-
ления удовлетворить двум противоречивым требованиям, а именно: для
поддержания горения дуги в совмещенном плазмотроне-реакторе необходи-
мо нагреть рабочий газ до температуры JjlO4 ° К, метан при такой температуре
за времена ~10“7 сек диссоциирует на элементы — углерод и водород; в то
же время этот же рабочий газ является и перерабатываемым сырьем в плазмо-
химическом процесса получения ацетилена и по тре-
бованиям этого процесса не должен нагреваться
более 2000° К.
Рассматриваемое выше позволяет сделать по мень-
шей мере два основных вывода. Во-первых, при орга-
низации технологии плазмохимических процессов
целесообразно стремиться к максимальной степени
«развязки» отдельных узлов технологической схемы,
что позволит оптимизировать режим работы каждого
узла, исходя из требований всего процесса в целом.
Во-вторых, при организации конкретного плазмо-
химического процесса необходимо выбирать генера-
тор плазмы, исходя из требований, формулируемых
на основании результатов термодинамических и ки-
нетических исследований этого процесса. В частно-
сти, например, рассматривая тот же процесс полу-
чения ацетилена из метана, следует отдать предпоч-
тение процессу, осуществляемому в плазменной струе
[91. Плазменные струи можно получать различными
Рис. 1.2. Схема реактора электрокрекинга метана [7]
I, 2 — электроды; 3 — вихревая камера; 4 — измеритель давления
7
способами, но, исход! из макрокинетики указанного [процесса, наиболее
перспективными предзтавляются турбулентные плазменные струи, генери-
руемые дуговыми плазмотронами. Для этого процесса важно обеспечить мак-
симально возможную равномерность распределения температуры в плазмен-
ной струе, поступаю цей в реактор. Как известно, отдельная плазменная
струя дугового плазмотрона характеризуется распределением температуры,
близкими таковому в электрической дуге, однако использование недавно раз-
работанных многодуговых подогрэвателей [6] позволяет выполнить и это
технологическое требование. Действительно, температура газа на выходе
таких подогревателей практически мало изменяется по радиусу потока, рез-
ко снижаясь лишь в очень узкой пристеночной области течения (рис. 1.3).
Рис. 1.3. Радиальные распределения темпе-
ратуры газа на выходе камеры смешения
(длина камеры L = 2Z>, G2 = 30 г!сек)
7 — T2/Ti = 9,3, Gt = 60 г/сек; 2 — 11,3, 60;
3 — 10,5, 3
Рис. 1.4. Устройство камеры смешения многодугового подогревателя (а) и схема сме-
шения струй (б)
1 — фазные плазмотроны; 2 — камера смешения; 3 — ввод холодного газа
I
Устройство камеры смешения многодугового подогревателя и схема смеше-
ния струй в ней показаны на рис. 1.4. Оба рисунка заимствованы из моногра-
фии [6], выход которой в свет существенно облегчил написание данного раз-
дела; работу [6] можно -рекомендовать для изучения специалистам, рабо-
тающим в области практического использования дуговых плазмотронов.
Схемы конструкций дуговых плазмотронов. В настоящее время сущест-
вует довольно большое разнообразие конструкций дуговых плазмотронов,
предназначенных для использования как в научных исследованиях, так и
в промышленности.
Плазмотроны с дугой, стабилизированной стенками, используются в
основном для исследования свойств газов при высоких температурах и в
качестве генераторов мощного оптического излучения. Работают они при
очень малых расходах газа, а иногда приток газа полностью отсутствует.
8
a — стенки [из электроизолятора •
б — металлическая стенка; в —ме-
таллическая секционированная
1 — катод; 2 — анод; 3 — столб,
дуги; 4 — изолятор; 5 —межэлект-
родная металлическая вставка
Рис. 1.5. Схемы плазмотронов с дугой, стабилизированной стенками
Рис. 1.6. Схемы плазмотронов с дугой, стабилизированной вихревым потоком рабочего
тела, и самоустанавливающейся длиной
а — однокамерный; б — однокамерный с полым электродом; в — двухкамерный; г — однокамерный,
с двухсторонним истечением газа. 1 — электроды; 2 — столб дуги; 3 — канал шунтирующего про-
боя; 4 — катушки электромагнитов; <5 — вихревая камера; 6 — термокатод; 7 — зона перемещения
конца дуги по электроду
Рис. 1.7. Схемы плазмотронов с фиксированной длиной дуги
а — с секционированной межэлектродной вставкой; б — с газодинамической фиксацией
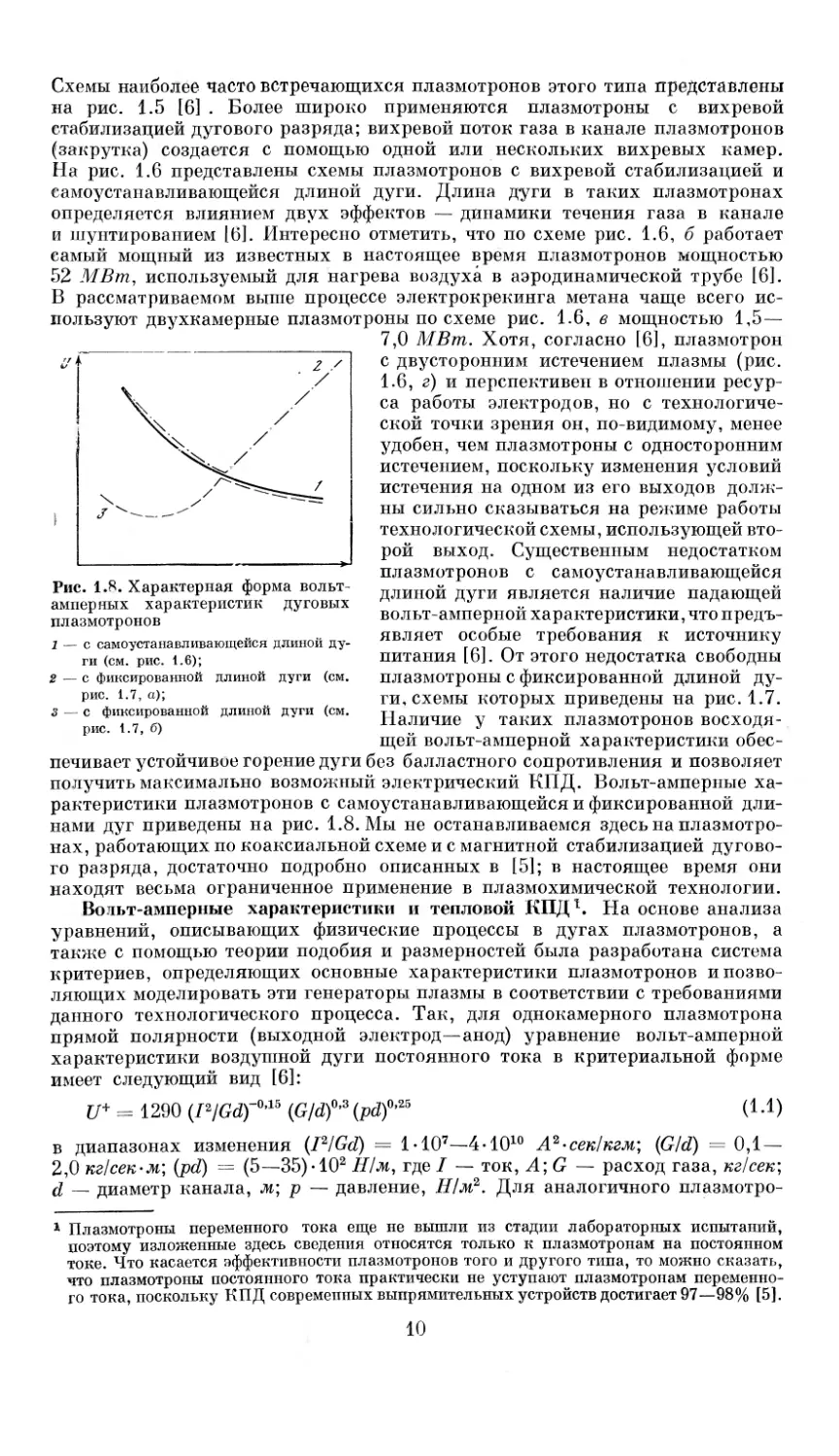
Рис. 1.8. Характерная форма вольт-
амперных характеристик дуговых
плазмотронов
1 — с самоустанавливающейся длиной ду-
ги (см. рис. 1.6);
2 — с фиксированной длиной дуги (см.
рис. 1.7, а);
3 — с фиксированной длиной дуги (см.
рис. 1.7, б)
Схемы наиболее часто встречающихся плазмотронов этого типа представлены
на рис. 1.5 [6] . Более широко применяются плазмотроны с вихревой
стабилизацией дугового разряда; вихревой поток газа в канале плазмотронов
(закрутка) создается с помощью одной или нескольких вихревых камер.
На рис. 1.6 представлены схемы плазмотронов с вихревой стабилизацией и
самоустанавливающейся длиной дуги. Длина дуги в таких плазмотронах
определяется влиянием двух эффектов — динамики течения газа в канале
и шунтированием 1б]. Интересно отметить, что по схеме рис. 1.6, б работает
самый мощный из известных в настоящее время плазмотронов мощностью
52 МВт, используемый для нагрева воздуха в аэродинамической трубе [6].
В рассматриваемом выше процессе электрокрекинга метана чаще всего ис-
пользуют двухкамерные плазмотроны по схеме рис. 1.6, в мощностью 1,5 —
7,0 МВт. Хотя, согласно [6], плазмотрон
с двусторонним истечением плазмы (рис.
1.6, г) и перспективен в отношении ресур-
са работы электродов, но с технологиче-
ской точки зрения он, по-видимому, менее
удобен, чем плазмотроны с односторонним
истечением, поскольку изменения условий
истечения на одном из его выходов долж-
ны сильно сказываться на режиме работы
технологической схемы, использующей вто-
рой выход. Существенным недостатком
плазмотронов с самоустанавливающейся
длиной дуги является наличие падающей
вольт-амперной характеристики, что предъ-
являет особые требования к источнику
питания [6]. От этого недостатка свободны
плазмотроны с фиксированной длиной ду-
ги, схемы которых приведены на рис. 1.7.
Наличие у таких плазмотронов восходя-
щей вольт-амперной характеристики обес-
печивает устойчивое горение дуги без балластного сопротивления и позволяет
получить максимально возможный электрический КПД. Вольт-амперные ха-
рактеристики плазмотронов с самоустанавливающейся и фиксированной дли-
нами дуг приведены на рис. 1.8. Мы не останавливаемся здесь на плазмотро-
нах, работающих по коаксиальной схеме и с магнитной стабилизацией дугово-
го разряда, достаточно подробно описанных в [5]; в настоящее время они
находят весьма ограниченное применение в плазмохимической технологии.
Вольт-амперные характеристики и тепловой КПД1. На основе анализа
уравнений, описывающих физические процессы в дугах плазмотронов, а
также с помощью теории подобия и размерностей была разработана система
критериев, определяющих основные характеристики плазмотронов и позво-
ляющих моделировать эти генераторы плазмы в соответствии с требованиями
данного технологического процесса. Так, для однокамерного плазмотрона
прямой полярности (выходной электрод—анод) уравнение вольт-амперной
характеристики воздушной дуги постоянного тока в критериальной форме
имеет следующий вид [6]:
V+ = 1290 (/2/Gd)“0,15 (G/d)0’3 * * (pd)0’25 (1 Л)
в диапазонах изменения (F/Gd) = 1-107—4-1010 А2-сек/кгм; (Gid) = 0,1 —
2,0 кг! сек(pd) = (5—35)-102 Н1м, где I — ток, A; G — расход газа, кг/сек;
d — диаметр канала, м; р — давление, Н!м2. Для аналогичного плазмотро-
1 Плазмотроны переменного тока еще не вышли из стадии лабораторных испытаний,
поэтому изложенные здесь сведения относятся только к плазмотронам на постоянном
токе. Что касается эффективности плазмотронов того и другого типа, то можно сказать,
что плазмотроны постоянного тока практически не уступают плазмотронам переменно-
го тока, поскольку КПД современных выпрямительных устройств достигает 97—98% [5].
10
на обратной полярности (выходная электрод—катод).
Z7“ = 197O;(72/Gd)-0^ (G/d)Mi(pd)0’25. (1.1а)
Уравнения (1,1) и (1.1а) описывают экспериментальные данные с погреш-
ностью не более ± 8%. Вольт-амперная характеристика дуги в плазмотроне
двустороннего истечения вполне удовлетворительно описывается уравне^
нием
U = 3060 (72/Gd)~°’1^(®/d)0’12 (pd)0’25, (1.2)
а соответствующее уравнение для двухкамерного плазмотрона (погреш-
ность описания экспериментальных данных ±12%) имеет вид
[7+ = 1360 (72/Gd)-0,2 (G/d)0,25* (pd)0’35, (1.3)
оно справедливо в диапазоне изменения комплексов (P/Gd) = 1-106—
4-109 А^-сек/кем, ((G/d) = 5-10"2—26 кг/сек-м, pd~ 103—8-105 Н/м и опре-
деляющих параметров: I — 50—5000 A, G — 1 • 10~3 — 3,5 кг/сек, d =
~ (5—76) • 10-3jh, р = (1—1000) • 105 Н/м2. Следует отметить, что критериаль-
ные уравнения, полученные при работе на других газах, могут отличаться
от приведенных. Так, критериальное уравнение вольт-амперной характери-
стики дуги в ‘однокамерном плазмотроне с вихревой стабилизацией метаном
имеет вид [7] (ср. с формулой (1.1а))]
Ш/1 = 8,33 • 103 (72/Gd)~0,766 (G/d)"0’234 (pd)0’422 (1.4)
и для случая стабилизации водородом [10]
U = 3190 (G/7)0’4 (G/df'* (pdf'™ (1.4а)
в диапазонах изменения критериев (G/7) = 10“3—10“2 г/А»сек, (G/d) =*
= 0,04—0,25 г/сек -мм, (pd) = 10—30 ата-мм.
Возможны и другие формы критериального описания вольт-амперных
характеристик дуговых плазмотронов [8, 11]. Характеристика интересного
с точки зрения практического использования плазмотрона с фиксированной
длиной дуги (плазмотрон с «уступом», см. рис. 1.7, 6) описывается следующим
критериальным уравнением (восходящая ветвь) [6]:
Z7+ = 4,55 (1 + 4,6 • IO"® Z/d2) (G/d2)0’22 (L2/d2)0’86 (М)0’23, (1.5)
выведенным с погрешностью около ±11% в следующем диапазоне измене-
ния определяющих комплексов и критериев:
(7/d2) - (0,8-4). 104 А/м, (L2/d2) = 5,6-14,5,
(G/d2) - 0,8-6,5 кг/сек-м, (pd2) = (2-40).103 Н/м.
Второй важной характеристикой генератора плазмы является тепловой
коэффициент полезного действия, определяемый как отношение количества
энергии, уносимое в единицу времени газом из плазмотрона, к мощности ду-
ги. Анализ зависимостей теплового КПД от различных факторов имеется
в [6]; из этой же работы взята и приводимая ниже критериальная формула
для теплового КПД ц двухкамерного плазмотрона, работающего на воздухе:
(1 — Т])/ я = 5,85 • 10~6 (Z2/Gd)~°’2e5 (G/d)0,265 (pd)0’3 (L/d)0'5 = if. (1.6)
Эта формула описывает экспериментальные данные с погрешностью
±10% (рис. 1.9) в диапазоне изменения 7 = 50—3600 A, G = 1 • 10“3—
2.2 кг/сек, d = 1-10-2 — 7,6-10~2 м (L — суммарная длина выходного и тор-
цевого электродов).
Как отмечают авторы [6], ею можно пользоваться при расчете плазмо-
тронов и других типов — однокамерных, с двусторонним истечением,
И
7
Рис. 1.9. Аппроксимация экспериментальных данных по тепловому КПД двухкамерного
плазмотрона постоянного и переменного тока (при работе на воздухе)
Кривая — расчет по формуле (1.6), точки — эксперимент (см. [6])
с гладким и ступенчатым электродами и т. д.— в следующем диапазоне из-
менения комплексов: (PIGd) = (5-106—5-109) А2-сек1к м, (fild) = 0,5 —
56 кг!сек-м, (pd) = 1 -103—8-105 Н/м, Lid = 5-40.
Следует отметить, что достижение высоких уровней энтальпии нагревае-
мого газа сопряжено со значительными потерями тепла и снижением КПД;
поэтому представляют интерес плазмотроны с распределенной подачей газа
по каналу и электродами из пористых материалов. Это позволяет получать
КПД — 0,75—0,8 при весьма высоких уровнях подогрева [6]. Выше упо-
миналось, что многодуговые подогреватели являются весьма перспективны-
Рис. 1.10. Аппроксимация эк-
спериментальных данных по
тепловому КПД камеры сме-
шения
Кривая — расчет по формуле (1.7),
точки — эксперимент [6]
ми генераторами плазмы с точки зрения плазмохимической технологии.
КПД камер смешения таких подогревателей описывается следующим при-
ближенным уравнением [6] (для воздуха):
(1 _ n)/n = 145 (Л/d)0’5 Re"0,75 = g, (1.7)
где L и d — соответственно длина и диаметр камеры, Re — число Рейнольд-
са, рассчитываемое по усредненному значению температуры Л3, статическо-
му давлению р3 (1 ата) и скорости газа г?3 в камере смешения (см. рис. 1.4).
Соответствие между результатами расчета по формуле (1.7) и результатами
эксперимента иллюстрируется рис. 1.10.
Параметры плазменных струй, генерируемых дуговыми плазмотронами.
Распределения температуры и скорости газа в плазменных струях на выходе
электродуговых плазмотронов мало отличаются от таковых в столбе дуги
19, 12, 13]. В качестве примера на рис. 1.11—1.12 приведены радиальные и
осевые распределения температуры и скорости-в свободной аргоновой струе,
истекающей из сопла плазмотрона постоянного тока, а на рис. 1.13 — рас-
пределение массового расхода в той же струе. Значительная неравномер-
ность- распределения температуры по сечению струи может вызывать опре-
12
Рис. 1.11. Радиальные распределения температуры (а) и скорости (б) в плазменной струе
аргона в сечениях, нормальных оси струи и удаленных на различные расстояния от среза
сопла электродугового плазмотрона
1 — срез сопла; 2 — 4 мм; з — 8;, 4 — 14. Диаметр сопла 3,5 мм, среднемассовая энтальпия 2-10-
дж/кг; ток дуги 50 А, напряжение 20 В, расход аргона 10 л/мин: соеднемассовая температура струи
на срезе сопла 3000° К
Рис. 1.12. Изменение осевых
аргона по мере удаления от
Параметры струи см. рис. 1.11
значений температуры (а) и
среза сопла плазмотрона
скорости (б) плазменной струи
Рис. 1.13. Распределение массового
расхода аргона в плазменной струе
на срезе сопла электродугового
плазмотрона постоянного тока
Параметры струи см. рис. 1-11
деленные затруднения при проведении в таких струях плазмохимических
процессов. Величина скорости нагретого газа на выходе дуговых плазмо-
тронов может варьироваться в очень широких пределах — от — 10 м!сек
до нескольких километров в секунду и зависит от расхода плазмообра-
зующего газа, диаметра сопла плазмотрона, мощности в дуге и других фак-
торов. Максимальная температура на оси струи может варьироваться в
пределах от 104 до 5-104°К; среднемассовая температура нагретого газа
достигает — 104 ° К при работе на одноатомных газах и (4—5) • 103 ° К при
работе на двухатомных газах (азот, водород).
2. Высокочастотные индукционные (ВЧИ) плазмотроны
ВЧИ-плазмотроны имеют определенные преимущества по сравнению с
электродуговыми генераторами плазмы. Их использование позволяет полу-
чать плазму, не загрязненную материалом эрозии электродов, обеспечить
длительный ресурс эксплуатации и работать практически на любом газе.
Несмотря на широкое распространение ВЧИ-плазмотронов в лабораторной
и исследовательской практике, до последнего времени считалось, что из-за
сравнительно низкого КПД и невысокого уровня мощности эти плазмотро-
ны не будут широко применяться в промышленности. Однако существуют
работы, в которых показано, что можно создать ВЧИ-нлазмотроны мощно-
стью до 1 МВт [14] и достигнуть КПД — 70% [15]. Более того, при работе
на агрессивных газах затраты на эксплуатацию ВЧИ-установки мощностью
1 МВт. с КПД 55% в несколько раз меньше, чем затраты на эксплуатацию
электродугового подогревателя того же уровня мощности [16].
Первые ВЧИ-плазмотроны достаточно большой мощности были разрабо-
таны Г. И. Бабатом [17]. Однако они не имели достаточно надежной тепло-
защиты стенок. Близкий к существующим в настоящее время ВЧИ-плазмо-
трон с газовой защитой стенок разрядной камеры был предложен Ридом [18].
После этого конструкция ВЧИ-плазмотронов изменилась мало, за исключе-
нием разработки металлических разрезных водоохлаждаемых разрядных ка-
мер [19, 20], использование которых может явиться основой для широкого
внедрения ВЧИ-плазмотронов в промышленность.
Обзоры результатов теоретических и экспериментальных исследований
ВЧИ-плазмотронов можно найти в [13, 21]. Там же приводятся описания ря-
да конструкций ВЧИ-плазмотронов, в том числе и с металлическими раз-
резными водоохлаждаемыми камерами.
Плазменные струи, генерируемые с помощью ВЧИ-плазмотронов, ис-
пользуются сравнительно редко. Чаще всего предпочитают проводить про-
цесс непосредственно в зоне самого ВЧ-разряда. О возникающих при этом
проблемах и некоторых путях их решения может дать представление рас-
смотрение довольно типичного технологического процесса получения ульт-
радисперсных порошков чистых металлов в плазме ВЧ-разряда [22]. Схема
ВЧИ-плазмотрона показана на рис. 1.14. Рабочие частоты этого плазмотро-
на — 1,7 и 3,3 Мгц. Он состоит из внутренней разрядной камеры (кварц) 2
и внешней (кварцевой) камеры 3. Индуктор 4 располагается в нижней части
камер. Показанная на рис. 14 форма плазмы обусловлена взаимодействием
разряда с потоками рабочего газа. Один из этих потоков — тангенциальный
периферийный поток аргона — формируется в верхней части разрядной
камеры и служит для стабилизации разряда в пространстве и тепловой за
щиты стенок камеры. Второй поток — рабочий, газ которого содержит пе-
рерабатываемый порошок,— направляется по водоохлаждаемой трубке 1
в центр разряда.
Авторы [22] были вынуждены выбирать оптимальные размеры разряд-
ной камеры, исходя из требований технологии процесса. Для этого необ-
ходимо было оценивать диаметр разряда. На основе литературных данных
[18, 23—27] и собственных оценок авторы [22] получили критериальное соот-
ношение между диаметром разряда, колебательной мощностью и частотой.
14
Рис. 1.14. Схема конструкции высокочастотного индукционного плазмотрона
1 — трубка для ввода порошка; 2 — внутренняя разрядная камера (кварц); 3 — внешняя камера
(кварц); 4 — индуктор; 5 — светящееся плазменное образование; 6 — устройство для закалки про-
дукта; 7 — подача аргона; 8 — подача охлаждающей воды
Оно представлено на рис. 1.15 и, очевидно, может быть полезным при
разработке и других технологических процессов в плазме ВЧ-разряда.
Следующей задачей, решением которой были вынуждены заниматься ав-
торы [22], являлось определение расходных характеристик плазмотрона.
Это было связано с необходимостью выбора такого соотношения линейных
скоростей потоков газа по периферии и в центре трубки плазмотрона, кото-
рое обеспечивало бы оптимальную переработку исходного порошка. На
рис. 1.16 представлена зависимость мощности Рр в разряде от расхода газа
Gt в центральном потоке при различных расходах газа G2 в стабилизирующем
(защитном) тангенциальном потоке на периферии разряда. При переходе
с увеличением G± через точки максимума Рр разряд становится неустойчивым
15
и в отдельных случаях срывается. Такой срыв разряда практически проис-
ходил всегда при работе на частоте 1,7 Мгц. Было установлено, что в режиме
устойчивой работы можно получить одинаковую мощность в разряде, из-
меняя величину расхода газа в тангенциальном потоке и не меняя расхода
плазмообразующего газа в центре разряда. Это обстоятельство имеет боль-
шое значение для подбора оптимального режима переработки сырья.
Ввод порошка приводит к изменению мощности в разряде. Этот эффект
иллюстрируется рис 1.17. Видно, что относительно небольшая скорость по-
дачи порошка в плазму, составлявшая 30 г/ч (т. е. 0,05 кг на 1 кг газа), вы-
зывает заметное увеличение мощности. Обусловлен этот эффект, по мнению
мм кбт
/17 f, МГц
Рис. 1.15. Обобщение экспе-
риментальных данных по за-
висимости диаметра разряда
в ВЧИ-плазмотроне от частоты
и колебательной мощности
7 — [18]; 2 — [22]; 3 — [23];
4 — [24]; 5 — [25]; 6 — [26, 271
авторов [22], влиянием порошка не только на степень ионизации в плазме
разряда, но и на газодинамические характеристики потока.
Приведенный здесь пример показывает, что при организации конкрет-
ного технологического процесса непосредственно в плазме ВЧ-разряда тре-
буется сложная комплексная отработка технологии процесса совместно с
нахождением устойчивых оптимальных режимов работы генератора плазмы.
По-видимому, задачу можно облегчить, проводя процесс в струе плазмы,
генерируемой ВЧ-плазмотроном, параметры которой в настоящее время оп-
ределить сравнительно несложно. Кроме того, с точки зрения химической
технологии процесс, проходящий в системе с менее ярко выраженными гра-
диентами параметров (например, температуры), легче рассчитывать и оптими-
зировать; именно такую систему можно получить, вводя сырье в струю плазмы
на выходе генератора спустя некоторое время, необходимое для релак-
сации параметров смеси. Время этой релаксации практически в большин-
стве случаев удается снизить до такой величины, что степень конверсии
исходных веществ на нестационарном участке будет составлять небольшую
долю общей конверсии (см. стр. 213).
По-видимому, генераторы с вихревой стабилизацией ВЧ-разряда, подоб-
ные изображенному на рис. 1.14, не могут обеспечить получение плазменной
струи с достаточно высокими технико-экономическими показателями. Так
как основная масса газа проходит в холодной пристеночной области, уровень
достигаемых среднемассовых температур низок и экономические показатели
всего процесса ухудшаются. Необходимость в создании специально сформи-
рованного потока газа для защиты стенок и стабилизации разряда является
следствием использования для получения плазмы ВЧ-генераторов с нагру-
зочными характеристиками, аналогичными характеристикам генераторов,
применяемых для индукционного нагрева твердых проводников [26]. В от-
личие от последних между режимом работы генератора и параметрами разря-
да существует сильная связь. Следовательно, необходимо рассчитывать ВЧ-
генератор с учетом влияния на его работу нагрузки — плазмы. В работе
529] система уравнений, описывающих систему ВЧ-генератор — разряд,
была решена для каналовой модели индукционного разряда. В этой модели
учитывались излучения разряда, теплопроводность, наличие потока газа,
16
Рис. 1.16. Зависимость мощности, вкладываемой в ВЧИ-разряд, от расхода газа Gr
в осевом потоке при различных расходах газа G2 в периферийном тангенциальном потоке
Частота 4,3 МГц', диаметр внутренней разрядной камеры 22 мм; диаметр трубки для осевой подачи’
газа 6 мм; колебательная мощность 6 кВт. Расход G2 (в л/ч): 1 — 140; 2 — 160; 3 — 180; 4 — 220;.
5 — 280; 6 — 340
Рис. 1.17. Зависимости мощности, вкладываемой в разряд, от расхода газа в осевом по-
токе при наличии (1) и отсутствии (2) подачи порошка с расходом 30 г!ч
Частота 1,7 МГц, расход газа в периферийном потоке G2 — 500 л/ч
конечность длины индуктора и характеристики реального ВЧ-генератора..
Анализ результатов решения показывает, что, выбирая определенный диа-
метр разрядной камеры, можно в равномерном по сечению разрядной камеры
аксиальном потоке газа получить отделенный от стенок, устойчивый так.
называемый контрагированный индукционный разряд [29]. Для этого сле-
дует использовать такой ВЧ-генератор, для которого зависимость переда-
ваемой в разряд мощности от диаметра разряда имеет крутопадающий уча-
сток. Авторами [28—30] был не только выполнен расчет такого генератора, но
и изготовлен действующий макет с выходной мощностью 60 кВт и рабочей
частотой 440 кГц. Результаты испытаний этого макета показали следующее.
Диаметр контрагированного разряда определяется в основном родом рабо-
чего газа и рабочей частотой генератора. Вкладываемая в разряд мощность
линейно растет с ростом расхода газа, и при изменении последнего в широ-
ких пределах среднемассовая температура остается практически постоян-
ной. Потери энергии на стенках разрядной камеры составляют пренебрежи-
мо малую долю вкладываемой в разряд мощности. Возможность использова-
ния меньших расходов газа при неизменном уровне вкладываемой в разряд
мощности позволяет получать плазму с высокой среднемассовой температу-
рой (среднемассовая энтальпия в 3—6 раз выше, чем в установках с вихре-
вой стабилизацией). Следовательно, генерируемая в таком ВЧИ-плазмо-
троне плазменная струя будет характеризоваться более'высокими значениями
технологических параметров, чем в случае ВЧИ-плазмотронов с вихревой
стабилизацией.
Рассмотрим некоторые результаты экспериментальных и теоретических
исследований ВЧИ-плазмотронов, которые могут представить интерес при
организации того или иного технологического процесса. Анализ результатов
измерений температуры аргоновой плазмы ВЧ-разряда показывает, что в
широком диапазоне изменения давления (0,5—3 ата), рабочей частоты
(0,3—30 МГц), расхода аргона (3—300 л! мин), диаметра трубки (1,2—12 ему
и вкладываемой мощности (1,5—20 кВт) как распределение температуры по
радиусу плазмы, так и ее максимальные значения изменяются очень мало.
Приблизительно 95% всех результатов измерения максимальной тем-
1.7
пературы укладывается в диапазон (9—11)-103° К, а характеры радиальных
распределений температуры отличаются лишь в периферийных зонах разря-
да (что можно объяснить возрастанием погрешности измерений температу-
ры в этих зонах) [13, 21, 31]. Уровень максимальной температуры, достига-
емый в плазме ВЧ-разряда в молекулярных газах при тех же уровнях мощ-
ности, что и в случае ВЧ-разряда в аргоне, несколько ниже.
В качестве иллюстрации на рис. 1.18 приведены радиальные распределе-
ния температуры в плазме ВЧ-разряда в кислороде, воздухе и азоте, а на
рис. 1.19 — распределение температур в осевом сечении плазмы и плазмен-
ной струи ВЧ-плазмотрона при работе на аргоне и кислороде [13]. Зависи-
мости средней по радиусу температуры плазмы в аргоне, кислороде, азоте
и воздухе от вкладываемой мощности при различных расходах плазмообра-
вующих газов приведены на рис. 1.20 [13]. В отличие от дуговых плазмотро-
нов скорость истечения плазмы из трубок ВЧИ-плазмотронов сравнительно
невысока и составляет, например, на оси потока аргоновой плазмы от — 7 до
— 60 м/сек при изменении мощности от — 9 до — 30 кВт и расходе от 3 до
3U л/мин [32]. Интересно отметить, что характер распределения массового
расхода газа по сечению трубки ВЧИ-плазмотрона существенно отличается
от такового для дуговых плазмотронов: не наблюдается относительного
уменьшения расхода плазмы на оси потока (рис. 1.21). На рис. 1.21 показаны
распределения скорости и массового расхода в аргоновой и кислородной плаз-
менной струях на выходе ВЧИ-плазмотрона [13].
Плазма, генерируемая ВЧИ-плазмотронами, при повышенных давлениях
может служить источником мощного излучения для проведения фотохими-
ческих реакций. Так, по измерениям авторов работ [33, 34] доля излучаемой
Рис. 1.18. Радиальные распределения температуры в плазме ВЧИ-разряда в кислороде
,(7), воздухе (2) и азоте (3)
Частоты 10 и 13 МГц, вкладываемая мощность 8—12 кВт, тангенциальный ввод газов с расходом
60 л/мин, удельная мощность 0,2—0,4 кВт/см*
Рис. 1.19. Изотермы плазмы ВЧИ-разряда с вихревой газовой стабилизацией в аргоне и
кислороде
Вкладываемая мощность для аргоновой плазмы — 12 кВт, для кислородной — 8 кВт, частота 10 МГц*
расход газа 30 л/мин-, числа у кривых — значения температуры, 103 °К
18
Рис. 1.20. Зависимость от вкладываемой мощности средней по радиусу температуры плаз-
мы ВЧИ-разряда с вихревой стабилизацией в аргоне (1, 2, 3), кислороде (4, 5, 6), азо-
те (7, 3, 9) и воздухе (10, 11)
Расходы газа (в л/мин): 1, 4, 7, 10 — 270; 2,5,8 — 140; 3, 6, 9, 11 — 70; диаметр разрядной камеры
60 мм, частота 5,28 МГц, индуктор — 4 витка диаметром 80 мм
Рис. 1.21. Радиальные распределения скорости и массового расхода pv в струе плазмы
на срезе ВЧИ-плазмотрона с вихревой стабилизацией разряда аргоном (1) и кислородом (2)
Распределения получены на плазмотроне, схема которого представлена на рис. 1.19 в режимах, ука-
занных в подписи к этому же рисунку
из разряда мощности Т^зл составляет для ксенона
Лгзл/Рр- 0,38 + 0,251g р, (1.8)
Где Рр — мощность, вкладываемая в разряд; р — давление, Н1м\
На рис. 1.22 эта зависимость представлена графически. Поскольку в ра-_
ботах [35, 36] показано, что ВЧИ-плазмотроны могут работать до давлений
— 40 ата, следует ожидать, что при давлении 40 ата доля излучаемой из раз-
ряда мощности составит — 80% (в ксеноне).
Рассмотрение результатов теоретических исследований ВЧИ-плазмо-
тронов показывает, что они [31, 37—43], как правило, удовлетворительно со-
гласуются с результатами экспериментов. Исключение составляют лишь дан-
ные экспериментов, свидетельствующие о наличии отклонений состояния
плазмы ВЧ-разрядов с протоком газа от модели ЛТР, закладываемой в рас-
чет. Эти данные [13, 44, 45] получают обычно сравнением результатов изме-
рений температуры электронов спектрально-оптическими методами с ре-
зультатами измерений температуры тяжелых частиц калориметрическими
(зондовыми) методами. Применение последних всегда связано с риском вне-
сения неконтролируемых возмущений в исследуемый поток плазмы. Тем не
менее при формулировании теоретической модели следовало бы, по нашему
мнению, учесть возможность отличия температур электронов и тяжелых
частиц плазмы, которое может явиться следствием наличия в плазме элект-
рических полей высокой напряженности и значительной скорости диффузии
электронов из столба плазмы [46, 47].
Для плазмохимической технологии теоретические исследования ВЧИ-
ллазмотронов представляют интерес прежде всего с точки зрения обеспечи-
ваемой ими возможности математического моделирования плазмохимиче-
ских процессов, организованных на базе использования ВЧ-плазмы. С этой
точки важны такие исследования, направленные на разработку критериев
19
подобия индукционных разрядов [48] и моделей, описывающих систему ге-
нератор — плазма [30]. В работе [48] предлагается использовать в качестве
критерия подобия эффективное волновое число определяемое из соот-
ношения
Кэ = Вэ / со|1бэ, (1.9)
где Rq — эффективный радиус разряда, со — круговая частота ВЧ-поля,
р — магнитная проницаемость и 0*3 — эффективная проводимость плазмы
разряда. При этом моделироваться будет ВЧ-плазмотрон, в котором увели-
чение мощности разряда сопровождается увеличением расхода плазмообра-
зующего газа и диаметра разрядной камеры при постоянной среднемассовой
Рис. 1.22. Зависимость доли
мощности, излучаемой плазмой
ВЧИ-разряда в ксеноне, от
давления газа в разрядной
камере
Абсолютные значения мощности из-
лучения составляют (в кВт):
1 — 1; 2 — 2; 3 — 3; 4—4; 5 — 5
температуре разряда (ВЧИ-плазмотрон с вихревой стабилизацией). Соглас-
но представлениям теории индукционного нагрева, для оптимальной пере-
дачи энергии в проводящий цилиндр достаточно обеспечить Кэ = 3,5. С ис-
пользованием этого критерия подобия в [48] были рассчитаны диаметр раз-
ряда и рабочая частота генератора на мощность от 50 до 106 кВт для случаев
использования в качестве плазмообразующих газов водорода, кислорода,
воздуха и аргона. Так, например, водородный ВЧИ-плазмотрон на мощность
1 МВт имеет диаметр камеры — 50 мм и рабочую частоту 630 кГц. Сниже-
ние рабочей частоты с увеличением мощности плазмотрона является весьма
полезным эффектом с точки зрения экономичности ВЧИ-плазмотронов [21,49].
Мы не рассматриваем здесь ВЧ факельных плазмотронов. С точки зре-
ния плазмохимической технологии они утрачивают основное преимущество
ВЧ-плазмотронов — способность генерации плазмы, не загрязненной мате-
риалом эрозии электродов. Поскольку достигнутые мощности таких плазмо-
тронов сравнительно малы, возможно, что их использование ограничится
лабораторными исследованиями.
[ 3. СВЧ-плазмотроны]
Физические процессы, протекающие в плазме ВЧ- и СВЧ-плазмотронов,
Принципиально ничем друг от друга не отличаются. Однако коэффициент
поглощения в плазме электромагнитной энергии на сверхвысоких частотах
сильно возрастает. Это позволяет эффективно передавать энергию в плазму
уже при температуре последней ~ 4000° К.
С точки зрения плазмохимической технологии СВЧ-плазмотроны интерес-
ны прежде всего тем, что их использование позволяет в принципе получать
резко неравновесную плазму при давлениях, близких к атмосферному.
В этой плазме температура тяжелых частиц не превышает — 1000° К,
а средняя энергия электронов приближается к 5—6 эВ при их концентрации
1012—1014 см* в воздухе, азоте и гелии при давлениях — 0,3 ата [50].
Увеличение частоты электромагнитного поля при переходе из ВЧ- в
СВЧ-диапазон приводит к принципиально отличному конструктивному
оформлению СВЧ-плазмотронов. В настоящее время применяются СВЧ-
20
плазмотроны трех основных конструкций: резонаторная, коаксиальная й
волноводная [51]. Резонаторный СВЧ-плазмотрон представляет собой замк-
нутую металлическую камеру в виде тороида или цилиндра с торцами [52].
Энергия вводится в камеру через отверстие в стенке камеры.
В принципе резонаторная конструкция обеспечивает поглощение основ-
ной доли энергии в плазме при условии, что электропроводность последней
не превышает электропроводности стенок резонатора. СВЧ-плазмотроны ко-
аксиальной конструкции аналогичны ВЧ факельным плазмотронам. Однако
^вследствие использования сверхвысоких частот между торцом центрального
электрода и факелом имеется значительно более широкий зазор, что сильно
снижает скорость эрозии электрода. Кроме того, СВЧ-плазмотроны коак-
сиального типа имеют более высокий КПД и обеспечивают значительно более
высокую плотность энергии в плазме [52, 53], чем ВЧ факельные плазмотроны.
Конструкции СВЧ-плазмотронов волноводного типа основываются на
взаимодействии бегущей электромагнитной волны с плазмой, возникающей
под ее действием в некоторой области волновода. На рис. 1.23 представлены
схема и один из вариантов конструкции плазмотрона этого типа, работаю-
щего на волне типа Н10. В этом плазмотроне разрядная трубка из диэлектри-
ческого материала вставляется в отверстие в широкой стенке волновода
таким образом, что ее ось располагается вдоль силовых линий электриче-
ского поля бегущей волны.
В другой конструкции СВЧ-плазмотрона волноводного типа используется
волна типа E1G; схема и один из возможных вариантов конструктивного
оформления этого плазмотрона представлены на рис. 1.24. В этом плазмо-
троне плазма образуется в отрезке круглого металлического волновода с
вставленной в него коаксиальной диэлектрической трубкой или без таковой
150]. Плазмотроны этого типа обеспечивают использование до 95% СВЧ-
энергии для нагрева плазмы. Плазмотрон на волне типа Н10 позволяет обес-
печить удельный вклад энергии в плазму, сравнимый с максимальным удель-
ным энерговкладом в дуговых плазмотронах, в то время как в плазмотронах
на волне 2Г10 удельный энерговклад быстро снижается с ростом температуры
плазмы, так что плазмотроны этого типа становятся неэффективными при
температурах ~7000° К. Для получения более высоких температур следует
использовать либо плазмотроны на волне Н10, либо резонаторные СВЧ-
плазмотроны [51]. Следует отметить, что в плазмотронах всех перечисленных
Рис. 1.23. Схема ((й) и один из вари-
антов (б) конструктивного оформле-
ния СВЧ-плазмотрона на волне типа
JTjo
1 — устройство запуска разряда.;
2 — корпус;
3 — кварцевая трубка;
4 — прямоугольный волновод;
5 — фланец для присоединения к тракту
подвода энергии;
6 — штуцер для ввода исследуемого газа;
7 — сопло;
jg — штуцер для ввода плазмообразующего
газа
21
выше конструкций стабилизация разряда осуществляется с помощью вихре-
вого обдува плазмообразующим газом.
В серийно выпускаемой отечественной установке «Фиалка» применен
плазмотрон на волне Н10 [51] с максимальной полезной мощностью 5 кВт и
рабочей частотой 2375 МГц. Плазмотрон позволяет получать столб плазмы
диаметром от 2 до 40 мм в воздухе, азоте, кислороде, хлоре, СО2 и аргоне
при расходе газов 5—100 л!мин. При атмосферном давлении минимальная
Рис. 1.24. Схема (а) и один из вариантов (б) конструктивного оформления СВЧ-плазмот-
рона на волне типа Ею
1 — стандартный волновод прямоугольного сечения; 2 — разрядная камера; 3 — отрезок круглого
волновода; 4 — короткозамыкающий поршень; 5 — перегородка из тефлона или керамики; 6 — теф-
лоновая шайба; 7 — дроссельный фланец; 8 — трубки для ввода защитного газа в зазор между пе-
регородками 5 и 6; 9 — отверстия для тангенциального ввода стабилизирующего газа; 10 — отверстие
в шайбе 6; 11, 13, 14 — патрубки для ввода и вывода охлаждающей воды; 12 — сопло плазмотрона
мощность, необходимая для поддержания разряда, составляет 0,3—0,5 кВт.
Температура газа в столбе плазмы увеличивается с уменьшением его диа-
метра и составляет 3000—7000° К.
Принципиальных ограничений мощности СВЧ-плазмотронов, по-види-
мому, не существует. Возникающая при увеличении мощности нестабиль-
ность разряда легко подавляется путем увеличения давления в разрядной
камере. Так, в камере СВЧ-плазмотрона мощностью 100 кВт на частоте
2375 Мгц при работе на воздухе должно поддерживаться давление не ниже
5 ата. а при работе на аргоне — не менее 25—30штш[51]. КПД современных
СВЧ-плазмотронов не превышает 50%. Существует, однако, мнение [55],
что при уровнях мощности в несколько сотен киловатт КПД этих плазмо-
тронов составит 70—80%.
Анализ результатов теоретических и экспериментальных исследований
СВЧ-разряда показывает, что, как и в ВЧ-разрядах, основными эффектами,
влияющими на параметры разряда, являются скин-эффект и теплоперенос.
Плазма СВЧ-разряда в аргоне с добавкой водорода (5% по объему) при ат-
мосферном давлении неравновесна: температура газа составляет в ней 4500° К»
температура заселения уровней аргона и водорода — 7000° К и концентра-
ция электронов — 1-1014 см~^ [56] при изменении мощности разряда в пре-
делах 800—2100 Вт. Плазма такого же разряда в среде азота, по-видимому,
находится в состоянии ЛТР [57]. Расчеты параметров азотной плазмы в ди-
электрической трубке импульсного СВЧ-плазмотрона на волне Я10 при дав-
лении 50 торр и мощности 1 кВт показали, что в указанных условиях мож-
но получить высокую степень ионизации газа при весьма незначительном его
нагреве [58].
22
4. Другие виды электрических разрядов
как источники низкотемпературной плазмы
Рассматривая плазмохимические процессы, в которых используются ду-
говые, ВЧ- и СВЧ-плазмотроны, нетрудно заметить, что результаты, полу-
ченные в этих процессах, не имеют, за редким исключением, никаких осо-
бенностей, связанных со спецификой плазмы. Однако в последнее время
появляется все больше данных (как правило, полученных спектрально-
оптическими методами), которые свидетельствуют о существовании в плазме
«странных» частиц и новых, ранее неизвестных соединений [59]. Одна из при-
чин того, что эти соединения не удается сохранить при выведении из плазмы,
«состоит, по-видимому, в невозможности обеспечить необходимую скорость
их закалки при охлаждении плотной «квазиравновесной» плазмы, созда-
ваемой указанными генераторами. Решение проблемы закалки можно об-
легчить путем проведения химических реакций в условиях неравновесной
плазмы. В такой плазме средняя энергия тяжелых частиц значительно мэнь-
тпе энергии электронов, под действием ударов которых и происходят ос-
новные химические превращения.
Следовательно, подбирая реакции, конечные продукты которых более
устойчивы к электронному удару, чем исходные реагенты, можно в принципе
найти условия, когда процесс будет селективен в отношении некоторого це-
левого продукта. При этом не требуется специальных мер для вывода избы-
точной энергии теплового движения молекул из системы, поскольку тяжелые
частицы в такой плазме практически «холодны». Примерами неравновесных
плазменных систем могут служить весьма короткие электрические разряды
(как, например, уже упоминавшийся выше импульсный СВЧ-разряд) или
стационарные тлеющий и коронный разряды.
Коронный разряд наблюдается в резко неоднородных электрических
полях. Ионизация и возбуждение молекул газа в коронном разряде имеет
место только в области наибольшей напряженности поля, вблизи наиболее
искривленных участков электродов. Перенос заряда в остальной части раз-
рядного промежутка осуществляется за счет дрейфа электронов или ионов,
образовавшихся в области короны. Коронный разряд наиболее подробно
изучен для цилиндрической системы электродов, представляющей обычно
металлическую трубку с расположенной на ее оси тонкой цилиндрической
проволокой. Возникновение коронного разряда вблизи поверхности ци-
линдрического электрода наблюдается в полях, напряженность которых мож-
но оценить, пользуясь полуэмпирической формулой [60]
Ео = 31/7 (1 + 0,308//^), кВ/см, (1.10)
где р — давление газа, ата; т\ — радиус коронирующего цилиндра, см.
Напряженности пробоя для положительной и отрицательной короны
различаются между собой. Разность потенциалов, соответствующую этой
напряженности электрического поля, можно рассчитать по формуле цилинд-
рического конденсатора
U = -Erjn (г2/гг), (1.10а)
где г2 — радиус внешнего электрода.
Интересной особенностью коронного разряда в цилиндрической системе
электродов является наличие очень широкой области с постоянной напря-
женностью поля и мощным пространственным зарядом. Вольт-амперная
характеристика коронного разряда описывается выражением [61]
I^U~U.)U0-—, (1.11)
In (r2/ri)
где Uo = Eorr In (г2/Г]) — потенциал начала коронирования (ибо 1 = 0 при
U < U0), р — подвижность ионов.
23
Это выражение удовлетворительно согласуется с экспериментом до не*
слишком высоких значений U, когда I ~ U (U — Uo) [61].
Тлеющий разряд характеризуется наличием нескольких диффузных све-
тящихся или темных зон и постоянной разностью потенциалов между элект-
родами в некотором диапазоне изменения тока. Относительный размер зон
определяется величинами давления и расстояния между электродами. Па-
дение напряжения на прикатодных зонах часто составляет основную часть,
всего приложенного напряжения. Пространственный заряд сосредоточен
в основном в прикатодной области. Следует отметить также, что концентра-
ция заряженных частиц в прикатодной области (зона отрицательного столба)
на один-два порядка выше, чем в области положительного столба разряда
[60]. Наиболее полно в настоящее время исследована зона положительного
столба. Теоретическое рассмотрение положительного столба разряда в ци-
линдрической трубке из диэлектрика позволило получить радиальное рас-
пределение концентрации электронов [60]
Ne W = Ne (О)/о (2,4 r/R), (1.12)
где Ne (О) — концентрация электронов на оси разряда, /0 — функция Бес-
селя нулевого порядка, R — радиус трубки.
Это выражение было получено при следующих предположениях:
1) в любой точке Ne = ^Nik (условие квазинейтральности);
к
2) заряженные частицы рекомбинируют на стенках, объемная рекомби-
нация не имеет места;
3) концентрации заряженных частиц на стенке равны нулю;
4) ионизация происходит только под электронным ударом;
5) отрицательных ионов не имеется;
6) радиус разряда много больше средней длины свободного пробега
электронов.
Предполагая, что электроны имеют максвелловское распределение по>
скоростям и сечение ионизации пропорционально температуре электронов
можно получить следующее соотношение [60]:
(еУг/кТе)~'1гехр 1еУ{/кТе] = i,2-iO^pR, (1.13)
где Vt — потенциал ионизации; р — давление, mopp\ R — радиус трубки
см\ [3 — константа для данного газа, которая приведена ниже:
Газ Не Ne Ar Hg N2 Н2 О2
3 4.IO-3 6.IO-3 4.10-2 7-10-2 4-10-2 1-10-2 2,9-10-2
Выражение (1.13) позволяет оценить электронную температуру в тлею-
щем разряде. Следует заметить, что в случае молекулярных газов предпо-
ложение о том, что электроны имеют максвелловское распределение по энер-
гии, не подтверждается результатами эксперимента [62—64].
5. Ударные трубы
и установки адиабатического сжатия
Использование ударных труб в качестве генераторов плазмы представ-
ляет, казалось бы, интерес с чисто научной точки зрения. Однако исследо-
вания на ударных трубах имеют с точки зрения плазмохимии и плазмохими-
ческой технологии и большое практическое значение, поскольку позволяют
определять механизмы и коэффициенты скоростей многих плазмохимических
реакций, свойства плазмы при различных температурах и давлениях и т. д.
Существует большое разнообразие установок для получения ударных волн,
но все они строятся по единому принципу: ударная волна образуется при
движении того или иного поршня в трубе, наполненной исследуемым газом..
24
Природа и свойства такого поршня меняются в зависимости от типа ударной
трубы (продукты взрыва, сгусток плазмы, сжатый газ и т. п.). В простейшем
варианте ударной трубы ударные волны образуются в трубе постоянного
сечения после разрыва диафрагмы, разделяющей камеру низкого давления,
наполненную исследуемым газом при пониженном давлении (несколько
торр), и камеру высокого давления, наполненную сжатым газом (несколько
атмосфер). Ударная волна распространяется по исследуемому газу. Одно-
временно в камере высокого давления в противоположную сторону распро-
страняется волна разрежения. Сжатый ударной волной газ нагревается до
высокой температуры. Принимая ряд допущений, перечисленных в [65],
можно получить выражение для отношения давлений на фронте ударной
волны
.11 = -_2l£ . ур _ Т1.— 1 /1
pi Ti+1 Ti + 1 ’ >
тде = cpJcVi — отношение удельных теплоемкостей для исследуемого
газа, М—число Маха ударной волны, равное отношению скорости распро-
странения фронта волны к скорости звука в газе перед фронтом ударной
волны.
Основным уравнением упрощенной теории ударной трубы является сле-
дующее [65]:
\Т1-Ь1 Y1 + 1 / L Т1 + 1 \ M/J ' 7
где р4 — давление толкающего газа, у4 — отношение теплоемкостей для
толкающего газа, с4 и — скорость звука в толкающем и рабочем газе со-
ответственно. Число М ударной волны возрастает с уменьшением отношения
молекулярных весов толкающего и рабочего газов и увеличением отношения
их темцератур. Изменение удельной энтальпии газа во фронте ударной вол-
ны находится из уравнения, называемого ударной адиабатой или адиабатой
Гюгонио [66]:
+ , (1.16)
\ pi р2 /
тде Hi — энтальпия газа, pz — плотность. Температура газа во фронте
ударной волны определяется выражением
Z2 = Т\ (Р2Р1/Р1Р2Л (1.17)
Ударные трубы позволяют без особого труда достигать температуры
~ 2-104°К и выше. Они широко используются для исследования физиче-
ских свойств плазмы, релаксационных процессов и химических реакций в га-
зах при высоких температурах [65—69].
В установках адиабатического сжатия нагрев газа происходит при сжатии
его поршнем в закрытой трубе. В отличие от ударных труб время сжатия в этих
установках относительно велико (^ 10~3 сек). С помощью таких установок
можно одновременно сжать газ до давления ~ 104 ста и нагреть до — 104° К.
На установках адиабатического сжатия были изучены многие химические
реакции, например, окисление азота, окисление метана, образование синиль-
ной кислоты и др. [70—73].
Принцип действия одноимпульсной установки адиабатического сжатия
«состоит в следующем (рис. 1.25, а). В исходном положении цилиндр разделен
поршнем на две полости — рабочую, где находится исследуемый газ (обыч-
но при атмосферном давлении), и полость толкающего газа, который нахо-
дится под давлением в десятки атмосфер. После освобождения поршня он
разгоняется до скорости в несколько десятков метров в секунду и сжимает
исследуемый газ. Поршень в цилиндре движется практически без трения,
но со столь малым зазором (несколько микрон), что перетекание газа из од-
ной полости в другую отсутствует. В тот момент движения поршня, когда
работа расширения толкающего газа становится равной работе сжатия ис-
25
следуемого газа, поршень останавливается. Температура исследуемого газа
в этот момент может достигать — 104 ° К и более, а давление во много раз
превышает давление толкающего газа. Поэтому поршень начинает двигаться
в обратном направлении к исходному положению. Если сжатие газа не со-
провождается каким-либо необратимым процессом, то все параметры описы-
ваемой системы изменяются во времени симметрично относительно момента
максимального сжатия. После возвращения поршня в исходное положение
специальное устройство его останавливает. Поршень в цилиндре движется
со скоростью, много меньшей скорости звука, и поэтому поле давлений и
температур в сжимаемом газе почти однородно [74, 75]. Весь цикл движения
Рис. 1.25. Схема (а) движения’
поршня в трубе установки
адиабатического сжатия и за
висимость температуры и дав-
ления сжимаемого газа от вре-
мени (б)
поршня длится, как правило, — 5-10“2 сек. На рис. 25, б показана типич-
ная зависимость температуры исследуемого газа от времени в течение цикла
работы установки. Видно, что газ находится при температуре, близкой к
максимальной, в течение 10“4—10“5 сек. Скорость изменения температуры
достигает 107 град/сек как при нагревании, так и при охлаждении. Аналогич-
ный вид имеет и зависимость давления от времени. Движение поршня в уста-
новке адиабатического сжатия описывается уравнением
d?x___ S , •.
(1.18)
где S — площадь торца поршня, М — масса поршня, р1ч р2 — соответствен-
но давления толкающего и исследуемого газов.
Это уравнение рассматривается совместно с уравнениями, описывающими
адиабатические изменения давления и температуры исследуемого газа при
переменном показателе адиабаты у (Т7) [76]:
£р2 Y w (1.19]
dt * х \ х / dt ’ ‘ '
(1.20)
Нетрудно оценить параметры газа в момент максимального сжатия, счи
тая, что в точке остановки поршня энергия его переходит во внутреннюю'
энергию сжатого газа: Ми2/2 ж суДТ7, где и — скорость поршня. Напри-
мер, при М = 200 г, и = 100 м/сек, cv — 3 кал/молъ, ДЕ = 300 см3, у = 5/3;
имеем АТ7 ж 6000°, pjp^ = 2,2-103.
Установки адиабатического сжатия в настоящее время применяются для
исследования кинетики химических реакций при высоких температурах
[71—76]. Однако после создания установок непрерывного действия они зай-
мут достойное место среди аппаратов промышленной плазмохимии.
26
ДИАГНОСТИКА НИЗКОТЕМПЕРАТУРНОЙ плазмы
’В задачу диагностики низкотемпературной плазмы входит определение
концентраций частиц и установление формы распределения энергии между
этими частицами. Поскольку лабораторная плазма неоднородна, возникает
необходимость исследовать также и распределения измеряемых величин
в пространстве. В случае нестационарной плазмы к этому добавляется за-
дача наблюдения за изменениями измеряемых величин во времени. В задачи
диагностики потоков плазмы входит определение газодинамических харак-
теристик этих потоков.
1. Простейшая диагностика
Для того чтобы перевести некоторое количество вещества, занимающего
определенный объем, в состояние плазмы, всегда приходится увеличивать
количество энергии, содержащейся в такой системе. При этом все существую-
щие способы увеличения энергии системы характеризуются коэффициентом
полезного действия ц, меньшим единицы. Под простейшей диагностикой
^десь подразумевается определение величины энергии Япл, которую удает-
ся ввести данным способом в систему в расчете на единицу массы вещества
системы. Наряду с исходным составом эта величина, называемая обычно
среднемассовой энтальпией, является одной из важнейших характеристик
данной системы; ее знание позволяет рассчитать равновесный состав систе-
мы. Таким образом, Я11Л = (ТГц/ттг), где Е — энергия, поступающая от
источника; т — масса вещества. Часто удобно пользоваться другой вели-
чиной, характеризующей уровень вкладываемой в систему энергии, относя
величину полезной энергии (£ц) к объему системы (например, при описании
ВЧ-разряда без протока газа). В том случае, когда вклад энергии произво-
дится в поток вещества (плазмы) с расходом т, среднемассовая энтальпия
определяется как отношение Hnsi = где Р — мощность генератора
плазмы.
Измерение энтальпии плазмы с помощью калориметрического зонда. Ло-
кальное значение энтальпии плазмы можно определить калориметрическим
способом. Для этого в плазму вводится калориметрический зонд, через
который производят отбор пробы газа из плазмы. Зонд интенсивно при-
нудительно охлаждается. По разности температур охлаждающей зонд
жидкости при наличии протока отбираемого газа и в отсутствие такового
нетрудно найти энтальпию плазмы в месте отбора пробы, если скорость про-
тока газа при отбора пробы известна [13, 77]. В отношении этого метода сле-
дует сделать одно замечание, хотя, по-видимому, стало уже банальностью
говорить о том, что идеальные методы диагностики плазмы предполагают
минимальное взаимодействие между датчиком и плазмой. Дело в том, что
применение зондов, вносящих неизвестные возмущения в плазму с неиз-
вестными и подлежащими определению свойствами, зачастую может только
запутать изучаемую проблему. Поэтому следует всегда тщательно оцени-
вать возмущения, которые может внести зонд в исследуемую плазму.
Ниже очень кратко изложены методы диагностики низкотемпературной
плазмы. Краткость изложения, сводящаяся в иных местах просто к перечи-
слению методов, обусловлена не только недостатком места, но и тем, что под-
робное обсуждение перечисляемых методов привело бы в сущности к повто-
рению материала, очень подробно изложенного в существующих в настоя-
щее время монографиях и сборниках [77, 80—82, 89, 91, 99] по методам
диагностики низкотемпературной плазмы.
2. Установление формы распределения энергии в плазме
Измерение температуры частиц плазмы. Введенная в плазму энергия
распределяется некоторым образом между частицами. Если бы плазма пред-
ставляла собой замкнутую систему, то распределение энергии со временем
27
приближалось бы к равновесному максвел л-больцмановск ому распределе-
нию. Лабораторная плазма всегда представляет собой систему незамкнутую,,
и поэтому некоторое неравновесное распределение энергии в ней может быть
стационарным. Часто обмен энергией внутри системы путем соударения
частиц идет по одной или нескольким степеням свободы системы очень бы-
стро, и для этих степеней свободы могут наблюдаться квазибольцмановские
распределения частиц по энергиям
Ni = 7Voexp [(е0 — ^)/кТ], (1.21)
где Nt — концентрация частиц в Z-м энергетическом состоянии, — энер-
гия i-ro состояния, Т — температура. Такое распределение устанавливает-
ся быстрее всего для поступательной и вращательной степеней свободы.
Поступательную температуру атомов (или молекул) можно измерить пу-
тем определения полуширины 6Х излучаемой этими атомами спектральной
линии, уширенной вследствие эффекта Доплера [78, 79]:
7,17-10“7 кУТ/М, (1.22)
где X — длина волны спектральной линий, М — атомный вес.
Из (1.22) получаем
Т = 1,95-1012 М (6Ш)2, °К. (1.22а>
Погрешность этого метода относительно невысока при использовании
спектральных линий, излучаемых легкими атомами при высоких темпера-
турах. Следует также учитывать влияние других возможных механизмов
уширения линий — штарковского, резонансного и др. [79, 81].
Вращательную температуру молекул определяют путем исследования
распределения интенсивности вращательных линий в молекулярных поло-
сах двухатомных молекул. Условиями применения метода являются макс-
велловское распределение скоростей молекул и больцмановское распределе-
ние заселенностей вращательных и колебательных уровней молекулы. Счи-
тается, что вращательная температура совпадает с температурой газа [65,
82]. Температура определяется по наклону прямой, являющейся графиком
зависимости
1п^г = -£вр/*7’, (1.23)
где Ij’j" — интенсивность вращательных линий, отн. ед.; j" — квантовые
числа верхнего и нижнего вращательных уровней соответственно; —
факторы интенсивности Хёнля — Лондона; v — частота излучения; 2ГВр —
= Bhcj' (j' + 1) — энергия верхнего вращательного уровня; В — постоянаяя
для данного электронно-колебательного состояния величина.
Приемлемая точность метода может быть достигнута при использовании
спектральных приборов с высокой разрешающей силой (обратная диспер-
сия не менее 1—2 А/мм). Можно, однако, пользоваться приборами с очень
низкой разрешающей способностью, вращательные линии на выходе кото-
рых сливаются и образуют непрерывное распределение интенсивности
[78, 83].
Колебательная температура молекул определяется по относительной ин-
тенсивности отдельных полос двухатомных молекул, принадлежащих одной
системе полос [82, 84], из наклона прямой, являющейся графиком зависи-
мости
g (?') fee
кТ
(1.24)
где — интенсивность полос (интегральная), r', v" — квантовые числа
соответственно верхнего и нижнего колебательных уровней перехода г'
28
-> v" , v — частота излучения, g (v') — колебательная постоянная для дан-
ного электронного перехода.
Температура свободных электронов плазмы. Обычно считают, что элект-
роны в плазме имеют максвелловское распределение по скоростям; это, по-
видимому, не вызывает сомнений, если концентрация электронов достаточно*
велика. Для определения в этом случае температуры электронов можно вос-
пользоваться результатами измерения интенсивности непрерывного излуче-
ния плазмы. Коэффициент излучения континуума плазмы определяется по*
следующей формуле [79]:
N N. Г h (v — v J1
ev = const £ (v, 7е)exp-------(v>vg), (1.25?
(ki e)/2 L e J
N N.
ev= const (v<vg), (1.25а/
e)
где Ne, Nt — концентрации электронов и ионов соответственно, Те — тем-
пература электронов, Vg — некоторая граничная частота в наблюдаемом
спектре, % (v, Те) — фактор, рассчитанный в [85] для благородных газов..
Температура электронов определяется по наклону прямой, являющейся гра-
фиком зависимости In [sv/| (v, Ге)] от частоты излучения v. Об особенно-
стях метода говорится в [78, 89].
Температуру свободных электронов можно измерить с помощью зондов:
Лэнгмюра [86—88], если электроны имеют максвелловское распределение
по энергиям. Поскольку по виду вольт-амперной характеристики зонда не?
всегда удается установить, существует ли максвелловское распределением
электронов в плазме, предпочтительнее производить непосредственное из-
мерение функции распределения [86, 87]. В настоящее время имеется много
данных, которые свидетельствуют о том, что в плазме тлеющего и других
разрядов при низких давлениях (несколько тпорр) в молекулярных газах
функция распределения свободных электронов по энергии не является макс-
велловской [62—64].
Среднюю энергию электронов (или температуру электронов при наличии
максвелловского распределения) можно оценить по излучению плазмы в;
микроволновой области спектра [89]. Для этого необходимо измерить мощ-
ность Р излучения плазмы, принимаемого согласованным волноводным?
приемником:
Р = ATr\f , (1.26>
где А — коэффициент поглощения плазмы, А/ — полоса пропускания прием-
ника, Тг — эффективная радиационная температура; при максвелловском^
распределении скоростей электронов Тг — Те, а при других распределениях:
Тг связана со средней энергией электронов [89, 90].
Температура электронов может быть найдена из измерений полуширины
линии рассеяния лазерного излучения в плазме в пределе томсоновского рас-
сеяния излучения на свободных электронах [78, 91, 92]. В этом случае пара-
метр теории рассеяния а 1 [93, 94], и при максвелловском распределении
скоростей электронов контур линии рассеяния имеет гауссову форму с по-
лушириной
AX = 4/^|}/^-sin4 , (1.27>
где X — длина волны зондирующего излучения, тп — масса электрона, 0 —
угол рассеяния. Этот метод в принципе позволяет найти функцию распре-
деления электронов по скоростям, однако экспериментов в этом направлении
пока не проводилось.
Температуру электронов плазмы, описываемой так называемой корональ-
ной моделью [78, 79, 95], можно найти по относительной интенсивности двух
29
спектральных линий одного и того же элемента. Для этого необходимо выби-
рать линии, функции возбуждения которых сильно отличаются (например,
комбинацию из синглетной и триплетной линий) [95].
Температура плазмы, находящейся в состоянии локального термиче-
ского равновесия (ЛТР), может быть определена путем измерения абсолютной
интенсивности I спектральной линии, испускаемой оптически тонким слоем
^однородной плазмы:
&0
где Ап — вероятность перехода; Л’о — концентрация частиц в основном со-
« (г)
стоянии; gm — статистический вес состояния ттг; g0 — статистическая сумма
-по всем состояниям излучающего атома; Ет — энергия верхнего уровня;
v — частота излучения; h — постоянная Планка; I — толщина излучаю-
щего слоя в направлении наблюдения.
В качестве эталонного излучателя используются вольфрамовые ленточ-
ные лампы и анодный кратер дуги постоянного тока между чистыми графи-
ковыми электродами. При диагностике неоднородной плазмы для определе-
ния коэффициента излучения спектральной линии или континуума следует
пользоваться интегральным1 преобразованием Абеля [78]. Вероятности
переходов А™ можно найти в [96].
Другим способом измерения температуры ЛТР-плазмы может служить
определение относительной интенсивности линий, излучаемых данным атомом.
Для этих линий должны быть известны относительные вероятности переходов
и энергии верхних уровней. Из (1.28) можно получить следующее выражение:
I. Ет- 1
In ---г---=-----const. (1.28а)
По наклону прямой, характеризующей зависимость левой части (1.28а)
от определяется температура Т. Очевидно, погрешность этого метода
тем меньше, чем более широкий диапазон изменения £’т. охватывается в
измерениях.
Наконец, для оценки температуры ЛТР-плазмы можно использовать ме-
тод Ларенца, основанный на том, что зависимость от температуры коэффи-
циента излучения спектральной линии при постоянном давлении приобре
тает максимум при некоторой температуре [78, 79].
3. Измерение концентраций частиц в плазме
Концентрации частиц плазмы, находящейся в состоянии ЛТР, могут быть
найдены в результате расчета с использованием системы уравнений, вклю-
чающей уравнение Саха, уравнение Дальтона, условие квазинейтральности
и уравнение сохранения начального состава [79, 96]. Для того чтобы убе-
диться, что в исследуемой плазме условия ЛТР выполняются, следует опре-
делить температуры и концентрации различных компонент плазмы. Концент-
рации частиц на возбужденных уровнях определяются по абсолютным ин-
тенсивностям соответствующих спектральных линий (см. уравнение (1.28)).
Концентрация частиц в основном состоянии может быть определена либо пу-
тем измерения поглощения излучения внешнего источника этими части-
цами, либо интерферометрическими методами. К сожалению, при заранее
неизвестном компонентном составе плазмы последний метод позволяет най-
ти лишь суммарную концентрацию тяжелых частиц в плазме [79]. Приме-
нение метода поглощения ограничено чисто техническими трудностями,
поскольку большинство резонансных линий атомов и молекул, обычно пред-
ставляющих интерес для исследователя плазмы, находится в области ваку-
умного ультрафиолета [78, 80].
30
Методы измерения концентрации свободных электронов в плазме более
многочисленны и разнообразны, чем методы измерения концентраций час-
тиц в основном состоянии.
Прежде всего необходимо назвать метод, основанный на измерении уши-
рения водородных линий. Уширение водородных линий в плазме обуслов-
лено линейным штарк-эффектом в микрополях заряженных частиц. Теория
уширения водородных линий развита в работах Грима [81]. Наиболее точ-
ным способом определения концентрации электронов по уширению водород-
ных линий является сравнение экспериментально полученного контура с
набором теоретических [97]. С несколько меньшей точностью можно поль-
зоваться зависимостью концентрации электронов от полуширины линии.-
Например, для линии [97]
In Ne = 1,452 1g АХ + 14,565, (1.29)
где Ne — концентрация электронов, см~3] Ак — полуширина линии Де, А.
Соответствующие выражения для других водородных линий приведены в
[81, 96].
Для определения концентрации электронов можно воспользоваться ре-
зультатами измерения ширины или сдвига изолированных спектральных ли-
ний, обусловленных квадратичным эффектом Штарка. К сожалению, здесь
нельзя дать общей формулы как в случае водородных линий, однако в на-
стоящее время в литературе можно найти расчеты указанных параметров
линий для атомов различных элементов [81, 98].
Концентрацию электронов можно рассчитать методом последовательных
приближений по температуре из выражения (1.25а) для коэффициента непре-
рывного излучения плазмы. Для этого необходимо измерить абсолютную
интенсивность этого излучения. Основной трудностью при проведении таких
измерений является учет вклада в интенсивность со стороны непрерывного
излучения примесных частиц в плазме [78, 79].
Оценку концентрации электронов в плазме можно произвести по правилу
Инглиза — Теллера, согласно которому главное квантовое число mmax по-
следней бальмеровской линии, которую можно наблюдать в спектре, связано
с концентрацией электронов следующим соотношением:
lg Ne = 23,26—7,51g rnmax. (1.30)
Погрешность такой оценки не превышает, по-видимому, одного порядка
величины [78, 81].
В случае исследования плазмы, находящейся в состоянии ЛТР, концен-
трацию электронов можно определить по относительной интенсивности линий,
принадлежащих атомам различных ступеней ионизации (например, атома
и первого иона):
Io _ v0 No Aogog(^ Ei — Ео
vi2V1/llglg«eXP И’
(1.31)
(в обозначениях, использованных в выражении (1.28)).
В сочетании с уравнениями Саха, Дальтона и условием квазинейтраль-
ности выражение (1.31) позволяет рассчитать температуру и концентрацию
электронов.
Удобным способом измерения концентрации электронов является интер-
ферометрия. Как известно, интерферометрия позволяет определять коэф-
фициент преломления п, который для свободных электронов находится из-
выражения
п2^!-/2//2, (1.32)
где /р = 8,98-IO3 сект1 — плазменная частота и / — частота зонди-
рующего излучения.
31
Используя в качестве источников зондирующего излучения СВЧ-гене-
раторы и оптические излучатели (в частности, лазеры), удается измерить Ne
в довольно широком диапазоне (от 1011 до 1017 см~8) [92, 99].Использование
лазеров в качестве источников света в интерферометрах упрощает конструк-
цию последних и позволяет увеличить их чувствительность [91, 92].
Концентрацию электронов измеряют по абсолютной интенсивности лазер-
ного излучения, рассеянного свободными электронами плазмы [91, 92]. Для
калибровки аппаратуры используется эффект релеевского рассеяния на ней-
тральных молекулах или атомах. Поскольку отношение сечений томсонов-
ского (на свободных электронах) и релеевского рассеяния не зависит от кон-
центраций рассеивающих частиц и может быть рассчитано с достаточной
Рис. 1.26. Схема калориметрического зонда
Твх, Гвых, тг — соответственно термопары для измерения температуры
охлаждающей зонд воды на входе и выходе из зонда и температуры
газа на выходе зонда
точностью, отношение интенсивности рассеянного плазмой света к интенсив-
ности света, рассеянного молекулами некоторого газа в тех же эксперимен-
тальных условиях, определяется выражением
I е/^м — N е^т/А^ m0r, (1.33)
где Ze, ZM — интенсивности света, рассеянного соответственно электронами
и молекулами газа; Ne4 Агм — концентрации соответственно электронов и
•молекул и пт, hr — сечения соответственно томсоновского и релеевского
рассеяния. Из (1.33) по измеренной величине отношения Ze/ZMj рассчитывают
концентрацию электронов Ne.
Для измерения небольших концентраций электронов (^1013 см”1) при-
меняют СВЧ-резонаторы [89]. Этот метод позволяет также определить эф-
фективную частоту столкновений электронов с тяжелыми частицами.
4. Измерение газодинамических параметров
потоков плазмы
Бесконтактные методы измерения скорости плазмы в сущности не отли-
чаются от обычных газодинамических методов [100]. Применение же зондо-
вых методов сопряжено с необходимостью использования либо хорошо ох-
лаждаемых датчиков, либо датчиков, время контакта которых с потоком
^существенно меньше времени их разрушения. Один из вариантов охлаждае-
мой трубки Пито представлен на рис. 1.26. Давление р, измеряемое трубкой
Пито, представляет, как известно, сумму статического pQ и динамического
.давлений в потоке:
Р = Ро + (рг2/2), . (1.34)
где v — скорость течения. Для определения скорости течения необходимо
знать плотность газа р в потоке. Поэтому обычно трубку Пито приспосаб-
ливают ДЛЯ' измерения местной энтальпии потока [77], калориметрируя
32
охлаждающую ее воду, и состава газа в потолке. Далее, в предположении
локального термического равновесия рассчитывают температуру и скорость
плазмы. При использовании неохлаждаемых трубок Пито для измерения
динамического напора плазменной струи эти трубки быстро проносят через
струю плазмы с помощью, например, маятникового устройства [13]. При этом
требуется, чтобы элемент, реагирующий на изменение давления в трубке,
обладал довольно высоким быстродействием (желательно, чтобы его ампли-
тудно-частотная характеристика простиралась без завалов до нескольких
килогерц). Потоки плазмы характеризуются обычно довольно высоким
значением вязкости и значительными градиентами скорости в радиальном
направлении. Влияние этих особенностей на результаты измерения с по-
мощью зондов газодинамических параметров потоков плазмы обсуждается
в [13].
С точки зрения плазмохимической технологии часто важно знать также
характеристики турбулентности плазменных струй. Поскольку степень
ионизации в струях довольно велика, флуктуации температуры струи могут
быть изучены путем измерения флуктуаций концентрации электронов.
В условиях ЛТР Ne ~ ехр (—Е^кТ), где Ег — энергия ионизации,
Ne — концентрация электронов и Т — температура. Следовательно, сравни-
тельно небольшие флуктуации температуры будут вызывать значительные
флуктуации концентрации электронов [101]. Флуктуации концентрации
электронов могут быть измерены с помощью охлаждаемых электростати-
ческих зондов [101, 102].
Исследование флуктуаций скорости струи можно производить с по-
мощью трубки Пито с соответствующим датчиком. Однако при этом возни-
кают значительные трудности при получении равномерной частотной харак-
теристики датчиков в широком интервале частот (до 10—20 кГц) [13].
По-видимому, наиболее удобным инструментом для измерения флуктуаций
скорости в плазменных струях в настоящее время является лазерный доп-
леровский измеритель скорости [103]. Этот прибор позволяет измерять сред-
нюю скорость и флуктуации скорости, обеспечивает высокое пространствен-
ное разрешение (измерительный объем ~ 1 мм3) и в принципе^ позволяет
исследовать даже флуктуации вектора скорости. Кроме того, совершенно
очевидно его преимущество как прибора для безконтактных измерений.
В ряде плазмохимических процессов требуется измерять скорость и тем-
пературу мелкодисперсных частиц, находящихся в потоке плазмы. В этом
случае лазерный доплеровский измеритель скорости [103] имеет значитель-
ные преимущества по сравнению с другими способами измерения скорости
(например, способом измерения скорости по трекам частиц). Такого же на-
дежного способа измерения температуры частиц в настоящее время не суще-
ствует. Обычно для нахождения температуры частицы в потоке плазмы при-
ходится решать систему уравнений движения и теплового баланса при нали-
чии многочисленных допущений о режиме движения и нагревания частицы
{13, 104]. В связи с этим следует отметить появление интересной экспери-
ментальной работы по определению температуры частиц в плазме [105].
Авторы [105] предложили использовать для измерения температуры частиц
метод многоцветовой пирометрии, позволяющий не делать каких-либо пред-
положений о коэффициентах черноты материала частиц.
2 Теоретич. и прикладная плазмохимия
2
ОСНОВНЫЕ ПОЛОЖЕНИЯ
НЕРАВНОВЕСНОЙ ХИМИЧЕСКОЙ КИНЕТИКИ.
ПЛАЗМОХИМИЧЕСКАЯ КИНЕТИКА
НЕРАВНОВЕСНАЯ СТАТИСТИЧЕСКАЯ ТЕРМОДИНАМИКА
И НЕРАВНОВЕСНАЯ ХИМИЧЕСКАЯ КИНЕТИКА
В не слишком далеком прошлом теория неравновесных явлений в термо-
динамике и неравновесные эффекты в кинетике рассматривались многими
как аналоги того, что делали древние географы, относя все ускользающее
от их знания к краям карты Земли и обозначая на ее полях «Болото мрака»,
«Далее безводные пески и дикие звери». Однако за последние 20—25 лет
ситуация кардинально изменилась. Развитие теории неравновесных (и/или
необратимых) процессов хотя еще далеко от завершения, но достигнутые
успехи позволяют говорить о создании новых научных областей неравновес-
ной статистической механики, неравновесной статистической термодинамики,
неравновесной термодинамики, неравновесной химической кинетики. Вместе
с тем происходит и процесс слияния некоторых из них, своеобразный про-
цесс интеграции.
Одной из характерных черт современной физики является изучение про-
цессов эволюции макроскопических систем. Возникающий при этом сложный
комплекс проблем рассматривается с различных точек зрения в перечислен-
ных выше новых областях знания. Эти исследования изменили и наш взгляд
на природу равновесных состояний, глубже раскрыв их связь с состояниями
неравновесными, и принципиальное значение флуктуационных явлений
[1-8, И].
Центральной проблемой неравновесной теории — теории эволюции —
является, в отличие от равновесной термодинамики, в которой время не вхо-
дит в число независимых переменных, введение времени в явном виде, что
в свою очередь стирает грань между неравновесной статистической термоди-
намикой и неравновесной кинетикой, в частности кинетикой химической.
Вообще говоря, химическая кинетика является разделом статистической
физики. В ней рассматривается временная эволюция систем, в которых,
кроме переноса энергии и импульса и гидродинамического и диффузионного
переноса частиц, происходит еще и специфический химический перенос и
перераспределение частиц. Химическая кинетика по самому существу ре-
шает многочастичную и многоканальную задачу.
В отличие от аррениусовой кинетики, где объектом исследования являет-
ся поведение молекулы при наличии одного канала химической реакции,
в неравновесной химической кинетике рассматривается многоканальная
задача, а объектом изучения является молекула в данном (определенном)
квантовом состоянии (динамическом и энергетическом).
Многие авторы считают, что химическая кинетика и термодинамика
имеют очень мало общего. Такое утверждение неверно. Термодинамика или,
точнее говоря, термохимические свойства веществ (AG, АЯ, Д£) наклады-
34
вают весьма жесткие ограничения на кинетические параметры, исполь-
зуемые для описания систем, изменяющихся во времени. Но дело не только
в этом. В число аксиом, лежащих в основе химической кинетики, которые
(насколько нам известно) в явной форме формулируются крайне редко [20],
входит следующее утверждение. Решение системы кинетических уравнений
таково, что при оо (или t достаточно большом) существует состояние,
для которого все макроскопически наблюдаемые скорости химических реак-
ций обращаются в нуль. Если считать, что никаких других градиентов и
потоков в реагирующей системе нет, то такое состояние будет равновесным
и устойчивым. Только в этой (строго говоря, асимптотически достигаемой)
точке кинетика совпадает с равновесной термодинамикой. В неравновесной
же термодинамике, в которой время — равноправная и явная независимая
переменная, уже нет различия между кинетическим и специфически термоди-
намическим подходом. Вся проблема сводится к разумному выбору и обосно-
ванию уравнений, описывающих временную эволюцию системы, а остальные
расхождения становятся только терминологическими.
Итак, фундаментальной проблемой современной феноменологической
неравновесной термодинамики является описание процесса, т. е. определение
термодинамических величин в неравновесном состоянии и включение вре-
мени в теорию в качестве независимой переменной при выполнении законов
сохранения и закона возрастания энтропии.
Попытки кинетического обобщения термодинамики делались с начала
XX в. Начиная с работ Онсагера (1931), можно уже говорить о системати-
ческом построении новой термодинамики необратимых процессов, развивае-
мой в дальнейшем главным образом бельгийской школой И. Пригожина.
Основными постулатами этой теории, применимой лишь к небольшим от-
клонениям от равновесия, являются: 1) утверждение о линейной зависимо-
сти обобщенных термодинамических потоков от обобщенных потенциалов;
2) соотношение Онсагера, выражающее равенство перекрестных коэффициен-
тов этой зависимости; 3) теорема Пригожина о минимальности произ-
водства энтропии. Значение этой теории состоит не столько в расширении
области исследования по сравнению с классической термодинамикой, сколь-
ко в попытке в общем виде построить аппарат феноменологической термо-
динамики так, чтобы в него с самого начала время входило равноправно
с прочими переменными. Однако в химической кинетике (особенно в области
химии высоких энергий) имеют место большие отклонения от равновесия,
что лишает возможности непосредственно использовать указанную термо-
динамическую теорию необратимых процессов [8].
Теоретическую основу неравновесной термодинамики составляет нерав-
новесная статистическая термодинамика аналогично тому, как обычная
статистическая термодинамика — основу равновесной термодинамики. Она
исследует процессы эволюции макроскопически физических систем (газы,
жидкости, твердые тела, плазма) и в первую очередь процессы переноса
энергии, импульса, частиц на основе статистической механики.
Обычная линейная феноменологическая неравновесная термодинамика
применима к любой системе при условии, что система слабо неравновесна,
т. е. находится вблизи состояния полного статистического равновесия.
В ней не проводится единая последовательная макроскопическая точка
зрения. Наряду с аксиоматическим термодинамическим методом она сущест-
венно использует аргументацию на микроскопическом уровне, а именно то
обстоятельство, что частицы подчиняются уравнениям движения механики
(например, так выводятся соотношения Онсагера из инвариантности урав-
нений движения относительно обращения времени). Однако используется
лишь существование уравнений движения, а не конкретный вид гамильто-
ниана. В неравновесной статистической термодинамике, которая, в отличие
от равновесной, еще находится в процессе развития и далека от своего за-
вершения, вводится с самого начала описание системы с определенным га-
мильтонианом и используются явно уравнения движения. Поэтому здесь
35
2*
отчетливо выступает несколько завуалированное в обычной статистической
термодинамике противоречие между обратимостью уравнений движения
отдельных частиц и необратимостью поведения макросистемы.
Следовательно, основной проблемой теории необратимых процессов яв-
ляется необходимость примирить необратимое поведение макросистем с обра-
тимостью основных микроскопических уравнений движения. Под обрати-
мостью микроскопических уравнений мы подразумеваем их инвариант-
ность относительно операции обращения времени (£-> — t). Возможны три
способа описания процессов в макроскопических системах: 1) использова-
ние уравнений движения микроскопических компонент системы (атомов,
молекул, электронов или других микрочастиц), что дает полное описание
макросистемы; все уравнения такого типа обратимы; 2) использование
обычных феноменологических соотношений (например, уравнений гидро-
динамики достаточно для правильного описания неравновесного поведения
многих жидкостей); уравнения такого типа неинвариантны относительно
обращения времени; 3) использование кинетических уравнений различного
типа, примерами которых являются кинетические уравнение Больцмана и
«управляющее» уравнение Паули (1928). Важным для нас свойством послед-
них уравнений является то, что они предсказывают стремление неравновес-
ной системы к равновесию при весьма общих начальных условиях. Однако
лишь в простейших случаях можно сказать, каким образом происходит уста-
новление равновесия в системе. Например, для разреженного газа (слабое
взаимодействие) уравнение Больцмана предсказывает монотонное прибли-
жение к равновесному состоянию, что отнюдь не обязательно для других
систем (в особенности для систем с сильным взаимодействием).
Как хорошо известно, классическое уравнение Лиувилля, позволяющее
изучать эволюцию функции распределения во времени, имеет вид
(2.1)
где L — линейный, эрмитовый оператор Лиувилля.
Это уравнение формально аналогично уравнению Шредингера
i^ = H¥. (2.2)
eft
Формальное решение уравнения (2.1) можно записать, если известно Л=о*
Тогда
/ (Р, 5» 0 = eiLtf (р, q, 0), (2.3)
где L не зависит явно от времени.
Получим теперь уравнение движения для динамической переменной
Л (р, q*, t), где последний аргумент показывает явную зависимость от времени.
Оно имеет вид
(2.4)
подобный уравнению Лиувилля (2.1).
Если известны Л;=о и L у= L (£), то уравнение (2.4) допускает формальное
решение
A (t) = e~iLtA (0). (2.5)
Оператор e~iLt называется оператором эволюции. При действии на про-
извольную функцию <р (р (0), q (0)) он переводит ее в <р(р (t), q (t)), т. е.
e-iL'<p(/)(O),g(O)) = <р(р (0,^(0). (2.6)
Рассмотрим теперь эволюцию изолированной квантовой системы, которая
в момент времени t = 0 была в состоянии | <р0>- Состояние системы в любой
36
последующий момент времени определяется уравнением
г/<р(> = Н | ф(>, (2.7)
где выбрана такая система единиц, что И = 1, Н Н (t). Формальное инте-
грирование дает
|<р,> = е-да|ф0>- (2.8)
Математическое ожидание произвольного оператора А в любой момент
может быть найдено из уравнений движения, которые называются основ-
ными кинетическими уравнениями (или уравнениями Паули) и которые были
получены из уравнения Шредингера или эквивалентного уравнения
ip - [Я, р], (2.9)
где р — оператор плотности [8].
ИСТОРИЧЕСКИЕ ЗАМЕЧАНИЯ
Химическая кинетика рассматривает поведение ансамбля и его подсистем,
она принадлежит к области статистической физики и к ней полностью отно-
сятся слова Льюиса Кэррола: «Тут не было «раз, два, три — и вперед».
Каждый начинал бежать, когда хотел, и останавливался тоже, когда хотел.
Таким образом, узнать, окончен ли бег, было нелегко».
Естественно начать рассмотрение основных положений химической ки-
нетики с исторических замечаний об аррениусовой кинетике. Хорошо из-
вестно, что центральное для этой кинетики выражение зависимости так на-
зываемой константы скорости химической реакции от температуры было по-
лучено Вант-Гоффом в 1883 г. из термодинамических соображений и изложено
в его книге «Очерки по химической динамике» [12], а в 1889 г. эта же
зависимость установлена Аррениусом, исходя из остроумной гипотезы
о существовании в реагирующей системе выделенного подкласса активных
молекул, и изложена в «Zeitschrift fur Physikalische Chemie» [13]. Весьма
подробный обзор истории аррениусовой химической кинетики можно найти
в [14]. Однако, к сожалению, ни в одном курсе химической кинетики мы не
находим упоминания о том, что, кроме термодинамического (Вант-Гофф) и
молекулярно-кинетического (Аррениус) решения, имеется еще независимый
экспериментальный источник (Худ) так называемой аррениусовой экспо-
ненты. Вопрос о наиболее правильном выражении зависимости скорости
химической реакции от температуры еще в 70—80-х годах прошлого века
пытались исследовать экспериментально Вардер, Миле, Макий, Урех,
Меншуткин, Лемуан, но только Худу, хотя и не сразу, удалось решить эту
задачу. Все указанные авторы, изучавшие различные реагирующие системы;
получали соответственно t2, а (6 + ct)/(d t), at. В своей первой работе
[15], увидевшей свет в 1878 г., Худ, изучавший реакцию жидкофазного
окисления сернокислого железа хлорноватокислым калием, получил так-
же t2 (такова была тогда точность эксперимента). Однако, исходя из требо-
ваний эксперимента, Худ отметил основные особенности тех реакций, кото-
рые описываются тогда еще неизвестной аррениусовой кинетикой: реакции
должны быть достаточно медленными, происходить с химически известными
молекулами и описываться обыкновенными дифференциальными нелиней-
ными уравнениями первого порядка. В следующей своей работе, опублико-
ванной через семь лет, в 1885 г. [16], Худ продолжил исследование той же
реакции и показал, что ранее он ошибся и что температурная зависимость яв-
ляется экспоненциальной. Эти первые кинетические кривые, обработка
которых позволила верно выразить температурную зависимость, показаны
на рис. 2.1.
Не рассматривая дальнейшее развитие аррениусовой кинетики в работах
Боденштейна, Касселя, Слэтера, Семенова и других многочисленных иссле-
37
дователей, можно сказать, несколько модернизуя ситуацию, что основы этой
кинетики, заложенные Худом, Вант-Гоффом и Аррениусом, таковы: объект
изучения — молекулы (например, N2); рассматриваются одноканальные си-
стемы; предполагается определимость и измеримость Т во все время реакции,
что имеет место, если поступательные и внутренние степени свободы равно-
весны или почти равновесны все время; = f (rij, Т) — вид уравнения хи-
мической кинетики; существует такое к, что к =f= к (щ, /), а к = к (Z). Тогда
из равновесной термодинамики или из статистической физики в случаях:
1) достаточно медленных реакций; 2) небольших плотностей; 3) достаточно
короткодействующих сил, чтоб не было корреляций; 4) достаточности модели
Рис. 2.1. Зависимость от вре-
мени концентрации сернокисло-
го железа в ходе его окисления
хлорноватокислым калием при
различных температурах [16]
бинарных соударений молекул, рассматриваемых как бесструктурные (твер-
дые) шары, получаем
к = А (Т) exp (~Еа1кТ).
Отказ, по крайней мере, от некоторых из перечисленных положений при-
водит к обобщенной (неравновесной) химической кинетике.
Таким образом, исторически первоначально изучались медленные хими-
ческие реакции, проходящие в сравнительно мягких условиях, для которых
возможно исследовать их скорости, предполагая закончившимися смешение,
диффузию, установление термического равновесия. Обычно это бимолеку-
лярные реакции с Еа кТ или реакции типа рекомбинации, когда плот-
ность активных частиц в пространстве мала. В силу этого огромное количе-
ство данных, полученных в химической кинетике, относится в основном
к системам, близким к равновесию по всем параметрам, кроме самой хими-
ческой реакции, поэтому все выражения скоростей описывают квазиравно-
весную ситуацию 117—21].
Физически квазиравновесное состояние реализуется, если времена уста-
новления термического равновесия в подсистеме возбуждений достаточно
малы по сравнению с временами рождения и уничтожения возбуждений,
так что между возбуждениями успевает устанавливаться статистическое
равновесие.
ОСНОВНЫЕ ПОЛОЖЕНИЯ ХИМИЧЕСКОЙ КИНЕТИКИ
Для выяснения основ следует прежде всего дать четкое определение
тех понятий (величин) и постулатов, которые необходимы (но вполне могут
быть недостаточны или даже избыточны) для ее построения [20].
В дальнейшем предполагается, что рассматриваемые химические реакции
не находятся под термодинамическим запретом и удовлетворяют всем требо-
ваниям теории валентности.
Назовем элементарным актом химической реакции химическое превраще-
ние, происходящее за время, меньшее времени между двумя соударениями.
38
Химическая реакция есть какая-либо последовательность элементарных
актов, переводящая
(2.Ю)
г 3
где Мь М?- — молекулы разных видов, причем под молекулой поднимается
система ядер и электронов, обладающая одной минимальной потенциальной
энергией 3. Тем самым химическая реакция определяется здесь как процесс,
приводящий к появлению «новых» молекул — частиц с иным, отличным от
исходного, минимумом потенциальной энергии. Заметим, что при любом
виде возбуждения молекулы значение минимума потенциальной энергии
остается неизменным. Это отличает собственно химическую кинетику от
тесно с ней связанной физической кинетики.
Рассмотрим теперь два основных понятия химической кинетики — кон-
центрацию вещества q и время t. Последнее является обычным физическим
временем, а концентрация определяется как число частиц nt данной компо-
ненты массы mi и в данном энергетическом состоянии q в некоторой единице
объема (который предполагается содержащим достаточно большое количество
частиц для макроскопического усреднения элементарных процессов). Таким
образом, по своему существу ct является статистической величиной. Пола-
гаем, что ct = Ci(t) непрерывна и по крайней мере дважды дифференцируе-
ма, Ci — скаляр.
Определим область существования и изменения q:
0 q ос,
т. е. Ci — существенно неотрицательная величина. Если рассматривать ее как
обобщенную координату системы, то она отличается от обобщенных коорди-
нат механики qt тем, что для последних имеем
— ос ос.
Постулируем выполнение в системе, в которой происходят химические
реакции, закона сохранения масс. Этот закон по существу первый постулат
химической кинетики. Введем следующие обозначения: mt — масса f-й ком-
поненты реагирующей системы, щ — число молекул i-ro сорта в этой системе,
тогда закон сохранения массы запишется так:
2 mini f,onst(j = l,2,. . ,)2. (2.11)
Так как nJY = Ci, где V — объем системы, то
V 2 mici — const (2.11а)
и при постоянном объеме
2 тгСг = const. (2.116)
Вводя в рассмотрение число атомов /-го сорта Nj в системе, можем на-
писать
2 Nj = const (2.11 в)
3
1 Превращение (2.10) может произойти только при возникновении некоторой определенной
конфигурации системы, для которой характерен специальный тип взаимодействия,
обычно представляемый в виде той или иной модели.
2 Закон сохранения массы (2.11) нужен прежде всего для разумного определения с,-. Как
и все законы сохранения, он выполняется в любой момент времени в замкнутой реаги-
рующей системе. В случае открытой системы с учетом потоковых членов обобщение
достигается без труда.
39
‘— закон сохранения числа атомов каждого сорта в химически реагирующей
системе. В записи (2.11в) мы имеем дискретный закон сохранения, формально
аналогичный дискретным законам сохранения в квантовой физике (и теории
элементарных частиц). Это обстоятельство еще не принято во внимание и не
использовано при анализе основ химической кинетики.
Уравнения (2.11а)—(2.Ив) определяют инвариант группы преобразо-
ваний1 процесса 2 М{—>2 Му, т. е. последовательности химических пре-
i з
вращений. Заметим, что такой инвариант не обязателен для механической
системы (для которой достаточны законы сохранения энергии, импульса,
момента и движения центра масс системы). Скорость реакции определим как
причем будем учитывать только то изменение которое обусловлено хи-
мической реакцией.
При постоянном объеме системы получим w = с^. Определим
Ci = fi (с1> С2ч • • •> Сп)> (2.12)
причем функциинепрерывны, имеют непрерывные производные и содержат
некоторые коэффициенты к} (коэффициенты скорости химической реакции) 2,
но не содержат времени явно. Такие системы в теории вероятностей называ-
ются марковскими [3—5]. Именно с таких позиций проблемы химической
кинетики рассматриваются, например, в недавно опубликованной работе [22].
Уравнение (2.12) устанавливает основную в химической кинетике связь
между t, с^. Для того чтобы придать этой формальной связи физический
смысл, необходимо принять некоторые предварительные утверждения (аксио-
матического характера).
1. В химических реакциях кроме выполнения закона сохранения масс
(уравнение (2.11)) выполняются также законы сохранения энергии, импульса
и момента 3.
2. Для протекания химической реакции необходимо столкнование по
крайней мере двух частиц.
3. Частицы, участвующие в химической реакции, являются статистически
независимыми.
4. Химическая реакция является вероятностным процессом, одним из ка-
налов перераспределения энергии в системе, приводящего ее в конечном
счете к состоянию с минимумом потенциальной энергии.
5. Для того чтобы химическая реакция стала возможной, система стал-
кивающихся частиц должна обладать некоторой минимальной энергией
(которую мы условимся называть пороговой)1 и подходящей ориентацией.
6. Существует такой состав 2 сг> который отвечает равновесному со-
стоянию, не изменяющемуся со временем, так что в этой равновесной точке
все Ci = 0.
В дальнейшем для наглядности и простоты изложения мы будем рассма-
тривать химическую реакцию в газовой фазе (считая газ мало отклоняющимся
от идеального). Пусть имеется реакция
A(f) + B(7)-C(Z) + DH,
1 Рассмотрение теоретико-группового аспекта химической и физической кинетики выхо-
дит за пределы настоящей книги.
2 Обсуждение физического смысла величины k-L и связанных с ней величин к и к(Т) см.
ниже стр. 66 и сл.
3 Мы не будем останавливаться на осложнениях, возникающих в связи с законом со-
хранения импульса при неупругих соударениях. Рассмотрение этого и связанных с
ним вопросов см. [23].
40
где i, 7, Z, m характеризуют энергетическое состояние молекул. Обобщение
на случай более сложных химических реакций непринципиально и не со-
ставляет труда.
Имеем на основании уравнения (2.12) и постулатов 1—3 (если не рассма-
тривать энергетическое состояние реагирующих молекул и продуктов
реакции)
— — ЛавСа^в + ^cd^ccd> (2.13)
где Лдв, ЛСГ) — коэффициенты скорости химических реакций не зависят
от концентраций и не зависят явно от времени.
Аналогичные уравнения 1 2 могут быть написаны для св, сс и cd- Решение
полученной системы дает ct = ct (t) при заданных начальных условиях
сД=„ = <0)2.
Сделаем несколько замечаний по поводу введенных постулатов.
Упростим уравнение (2.13) еще больше, допустив, что обратные реакции
отсутствуют, т. е.
с а === Лав^а^в (2.13а)
для реакции
А + В —> продукты.
Приведем некоторые соображения о постулате 2 на примере уравне-
ния (2.13а).
В первую очередь отметим, что в столкновении могут участвовать как
молекулы, атомы, радикалы, ионы, так и частицы типа нейтрона, электрона,
а-частицы и т. п., а также частицы, не имеющие массы покоя,— фотоны.
Во всех случаях последнего рода в соответствующем кинетическом уравне-
нии вместо концентрации одного из сортов частиц фигурирует плотность
потока (иногда интенсивность, мощность дозы). Именно принятие второго
постулата позволяет написать уравнение в форме (2.12) на основании теории
столкновений. Однако лишь принятие третьего постулата позволяет утвер-
ждать, что увеличение концентрации одного из компонентов, например
вдвое, приведет к увеличению скорости в два раза.
В хорошо разработанной в статистической физике теории столкновений
существуют различные формулы, где также имеется не зависящий от концен-
трации коэффициент z, который может входить в состав коэффициента Л,
но, конечно, не исчерпывает его. Поскольку, как известно, не все неупругие
соударения приводят к химической реакции и, кроме того, неупругие со-
1 Для химика уравнение (2.13) является тривиальной записью так называемого закона
действующих масс, и для него введение различных, приведенных выше постулатов
может показаться излишним. Однако за законом действующих масс стоит термодинамика
и ее статистическое истолкование, что в конечном счете приведет к явно выписанным
нами утверждениям. Заметим, что закон действующих масс был сформулирован еще в
1864—1867 гг. Гульдбергом и Вааге [25] как эмпирическое соотношение. Только позднее
он получил статистическо-термодинамическое обоснование [1, 12, 24]. Это обоснование
строго только для условий химического равновесия или для бесконечно близких к нему
состояний.
2 В общем виде уравнения химической кинетики для изотермических гомогенных газо-
вых реакций (в закрытых системах) можно записать в следующем виде:
п т
С г ~ Zd ^‘iinp^rn^p 4“ zL ^p£p/(Z),
т=г р^т р=1
i = 1, 2, . . ., n,
где — безразмерные концентрации, связанные с размерными множителями TJVqT;
I (z) — интенсивность потока частиц; к[тР, klP — коэффициенты скорости реакций вто-
рого и первого порядка; z0— время начала реакции; O^ct^l; к^ симметричны!
по отношению к перестановкам индексов.
41
(2-14)
ударения в системе не могут иметь места без существования упругих соуда-
рений, то это приводит к введенному выше постулату 4.
Мы здесь не будем останавливаться на специфике процесса соударения
и происходящих при этом явлений (эти вопросы, строго говоря, не рассма-
триваются пока в рамках химической кинетики). Тем не менее некоторые
замечания сделать необходимо.
При «столкновении» фотона с атомом или молекулой он может либо рас-
сеяться, либо поглотиться. Именно в последнем случае может произойти
химическая реакция. Специфика этого процесса состоит, в частности, в том,
что один из его участников перестает существовать вовсе.
Заметим, что в этом случае, так же как при соударении тяжелых частиц,
все законы сохранения (постулат 1) полностью выполняются С Так, напри-
мер, исчезнувший (поглощенный) фотон переводит молекулу (атом) в воз-
бужденное состояние, полная энергия которого отличается от исходного на
величину Av. Энергия молекулы складывается, как известно, из энергии
внутренних степеней свободы и энергии поступательного движения 2:
А ПОЛИ = ^ПОСТ + ^электр + ^колеб 4 А'вращ
В процессе соударения может происходить перераспределение энергии
путем, например, поступательно-колебательных, колебательно-вращатель-
ных и т. п. переходов. С этим связан многоканальный характер процесса,
что и выражается постулатом 4.
Для полноты картины необходимо отметить, что, хотя в выражение для
энергии поступательного движения входит квадрат относительной скорости
сталкивающихся частиц, их взаимная ориентация в пространстве отнюдь не
является безразличной для протекания химического процесса. Поэтому
только в случае использования грубой модели «точечных» частиц можно не
учитывать их взаимную ориентацию (обычно в химии говорят о «стерическом
факторе», «стерических препятствиях» и т. п.).
Пороговая энергия тЕ’пор» введенная постулатом 5, представляет собой
параметр, характерный для данной химической реакции и не зависящий от
состояния реагирующих частиц 1 2 3. Эта величина по своей природе — дина-
мическая (на это часто не обращают внимания), которая не может быть полу-
чена из обычного (классического) кинетического эксперимента [26].
Статистической величиной является энергия активации химической
реакции Еа, которая может быть определена из такого эксперимента с по-
мощью общеизвестной логарифмической анаморфозы.
Напомним, что энергией активации Еа называется разность между сред-
ней энергией реагирующих частиц и средней энергией всех частиц 4. Это опре-
деление энергии активации было строго получено еще Толменом [27] пять-
десят лет тому назад. Для его вывода существенно допущение максвелл-
больцмановского распределения. При таком допущении, дифференцируя
1 При изложенном здесь «расширенном» толковании понятия столкновения частиц (М +
+М, М + R, М + М -, М + hv и т. д.) второй постулат определяет конкретные условия»
обеспечивающие выполнение первого постулата и в первую очередь закона сохранения
энергии.
2 Что касается поступательной энергии, подчеркнем, что речь идет об относительной
энергии в системе центра масс сталкивающихся частиц, а не в лабораторной системе
координат. Мы не рассматриваем здесь вопрос о системах отсчета для других слагаемых
уравнения (2.14). Дальнейшее обсуждение не выходит за пределы требования, чтобы
все слагаемые полной энергии были представимы в виде квадратичных форм.
3 В связи с постулатом 5 укажем, что если первые четыре постулата определяют условия,
необходимые для протекания химической реакции, то этот постулат является одним из
достаточных условий, если его рассматривать совместно с постулатом 4.
4 Из определения Е.л отнюдь не следует, что эта разность обязательно положительна и
не равна нулю. Кроме того, здесь не предполагается, что эта разность не зависит от
температуры. Заметим, что хотя в законе Аррениуса утверждается, что Е 4= Е^(Г), од-
нако в его собственных экспериментах дело обстояло не так. Это показано тщательным
анализом, выполненным в [28].
логарифмическую форму выражения для к (Т7), найдем для £а:
= <£> - <#>, (2.15)
где <!?> — средняя энергия столкновений, приводящих к химической реак-
ции; {Еу = 3/2kT — средняя энергия всех соударений в системе независимо
от того, приводят они к реакции или нет. Из рассмотрения соотноше-
ния (2.15) следует, что в силу быстрого уменьшения числа частиц в высоко-
энергетической части максвеллова распределения по мере возрастания энер-
гии основной вклад в реакцию вносят только столкновения, энергия которых
близка к пороговой. С возрастанием температуры {Е} растет, вообще говоря,
слабее, чем <£>, а следовательно, Е& зависит от температуры. Эта темпера-
турная зависимость слабая, поскольку ^пор или {Е} велика по сравнению
с кТ, как это обычно имеет место. Последнее объясняет, почему аррениусова
схема, в которой Е<А =f= Е& (Т7), с успехом применяется в очень многих
случаях.
Однако в других случаях этой зависимостью «2Г> = ф (t)) пренебречь
нельзя и аррениусова кинетика оказывается недостаточно точной даже для
квазиравновесных систем.
При рассмотрении химических реакций в неравновесных системах не-
обходимо пользоваться, по-видимому, /Гпор, так как при немаксвелловом
распределении введение Е& по соотношению (2.15) связано с рядом трудно-
стей.
Связь между /Гпор и Е& может быть записана в виде какого-либо матема-
тического соотношения лишь для конкретного вида функции распределения.
В самом деле, пороговая энергия не зависит от функции распределения,
а энергия активации зависит от нее. Так как в принципе функция распреде-
ления может быть любой, то общего соотношения между -Мюр и быть не
может.
Сделаем лишь одно дополнение. Выполнение основных положений не
обеспечивает само по себе практически, т. е. при данном уровне экспери-
ментальной техники, измеримой скорости реакции.
Постулат 6, строго говоря, никогда не выполняется, если в системе про-
текают химические реакции, так как всякий процесс, протекающий с конеч-
ной скоростью, нарушает равновесие даже в системе, равновесной в началь-
ный момент времени.
ТЕОРИЯ АБСОЛЮТНЫХ СКОРОСТЕЙ РЕАКЦИЙ
Последнее положение предыдущего параграфа определяет место и роль
так называемой теории абсолютных скоростей реакций в системе химической
кинетики. Напомним, что в основе теории абсолютных скоростей реакции
лежат следующие предпосылки: 1) координата реакции рассматривается как
обычная декартова координата [9, стр. 187]; 2) концентрация исходных ве-
ществ всегда находится в равновесии с концентрацией активированных
комплексов [9, стр. 187]; 3) скорости движения активированных комплексов
вдоль координаты реакции распределены по Максвеллу [9, стр. 188]; 4) акти-
вированные комплексы всегда находятся в термическом равновесии с исход-
ными реагирующими компонентами [9. стр. 187]; 5) только часть активиро-
ванных комплексов, дошедших до вершины энергетического барьера, рас-
падается на продукты реакции, для учета этого обстоятельства вводится
«трансмиссионный множитель» [9, стр. 191]; 6) все реагирующие вещества
сохраняют единое максвелл-больцмановское распределение; 7) все активи-
рованные комплексы, достигшие вершины энергетического барьера, имеют
положительную скорость, двигаясь в направлении образования продуктов
реакции (обратный процесс не существует); 8) активированному комплексу
на вершине энергетического барьера соответствует не точка, а потенциальныц
ящик произвольной длины [9, стр. 187].
43
Легко видеть, что пункты 3 и 5, 4 и 5, 2 иЗит. д. несовместимы и протг»
воречат друг другу. Теория абсолютных скоростей реакций внутренне про-
тиворечива. Трудности, стоящие перед ней, видели уже ее создатели [10].
Во всяком случае из изложенного очевидно, что теория абсолютных
скоростей реакций неприменима для изучения кинетики быстрых неравно-
весных химических реакций и реакций, протекающих в условиях, далеких
от квазиравновесия.
МЕТОД СТАЦИОНАРНОГО СОСТОЯНИЯ
Метод стационарного состояния не зависит от того, равновесна систе-
ма или нет; он широко используется в химической кинетике. Рассмотрим его,
следуя [29], на простом примере, имея в виду прежде всего определение усло-
вий его применимости.
Имеем последовательные реакции:
аЛ2Мп М^сДр+М,,
ks kt (2.16)
М2 4-АР + Мх, МХ + МХ->А.
В сумме имеем стехиометрическое уравнение
А +С~>2Р, (2.17)
где А, С — реагирующие компоненты, Р — молекулы-продукты, Мх, М2 —
активные промежуточные частицы. Легко написать следующие кинетические
уравнения:
— А - АХА + &3М2А — *4МХ; — С = Л2МХС,
Р = Л2МХС + &3М2А, (2.18)
Мх - 2АХА + /с3М2А - *2МХС - 2А4МХ; М2 = *2МХС - А3М2Аа
причем число израсходованных молей А будет Ао — А = (Р/2) + (Мх/2),
а молей С будет Со — С = (Р/2) + (М2/2), при t = 0 имеем Р, Мх, М2 = 0.
Пусть для любого t Мх, М2 <<! А, С, Р, тогда
Ао — А = Р/2, Со - С = Р/2 (2.19)
или
-А = Р/2, - С - Р/2. (2.20)
Уравнения (2.20) означают, что нет временного отставания, или оно
по крайней мере очень мало, между исчезнованием исходных веществ А, С
и появлением продуктов Р. В этом случае М1? М2 практически равны нулю,
и уравнения для них в (2.18) становятся алгебраическими и разрешимыми
относительно Мх, М2.
Из последнего уравнения (2.18) имеем тогда
Л2МХС - *3М2А. (2.21)
Использовав предпоследнее уравнение в (2.18), получим
Мх = (к^к^АУ*, М2 - (к2/к3) (kJk^C/A^) (2.21а)
или
Мх/А = (£Х/Л4А)Ч М2/С = к2/к3 (Л^А)1/*. (2.216)
Эти уравнения позволяют количественно рассмотреть условия, необхо-
димые для применения метода стационарного состояния Мх, М2 А, С.
Для того чтобы ошибка в концентрациях не превышала 1%, необходимо,
чтобы Мх/А 0,01, М2/С 0,01. Это имеет место для Мх/А только тогда,
когда кг 10"4-А4А, а для М2/С тогда, когда Мх/А 0,01, к2 к3. Эти не-
равенства позволяют проверить принятый механизм.
44
ЗАМЕЧАНИЯ О КОНСТАНТЕ СКОРОСТИ
ХИМИЧЕСКОЙ РЕАКЦИИ
С ТОЧКИ ЗРЕНИЯ СТОЛКНОВИТЕЛЬНОЙ МОДЕЛИ
Рассматривая к элементарной реакции вида А + В С + D с класси-
ческой точки зрения, имеем: *
1. Число молекул, реагирующих в единицу времени, в большинстве слу-
чаев мало по сравнению с числом А — В соударений в единицу времени.
2. В то время как число нереактивных соударений изменяется как Г1/8,
число реактивных соударений очень часто показывает более сильную тем-
пературную зависимость, которая может быть аппроксимирована аррениу-
совым выражением ехр (—EJkT).
Рис. 2.2. Концентрации аммиа-
ка (7), цианистого водорода (2)
и альдегидов (3) в СЛ-образной
трубке прибора Миллера и
аминокислоты (4) в малой кол-
бе при воздействии искры на
смесь метана, аммиака, воды
и водорода [122]
Если предположить равновесное распределение (хотя бы как хорошее
приближение), то можно применить термодинамические соотношения.
Кажущаяся энергия активации может быть определена как
А’а = 1?7,2^р. (2.22а)
Здесь отношение константы кг прямой реакции и к2 обратной реакции
In kr - In к2 - In К = —\GIRT, (2.226)
где К — константа равновесия, G — свободная гиббсова энергия.
Все эти величины имеют характер термодинамических свойств и вследствие
этого дают весьма мало информации о механизме микроскопических реакций.
В газовой столкновительной модели (решительное преобладание бинар-
ных соударений, прямолинейное и равномерное движение частиц между
столкновениями, ускорение внешними полями, если оно имеется, просто
описывается в терминах классической механики) предполагается, что плот-
ность достаточно мала, силы достаточно короткодействующие, чтобы не было
корреляции, и бинарные соударения достаточны для описания элементар-
ных процессов. Кроме того, предполагается, что существует к =/= к t),
к — к (Т), тогда из равновесной термодинамики следует
к = A (Z) ехр;(-Еа/Л:Т). (2.23)
Это неприменимо, например, если в реакции участвуют моноэнергетиче-
ские электроны, фотоны, в реакциях для которых скорости реакции больше
45
скорости установления равновесия, а также в плазмохимии, в реакциях
за ударным фронтом, в соплах расширения, дугах, баллистических следах
и т. д.1 Т
Осложнения возникают также в том случае, когда система изменяется
в пространстве и времени, так как в аррениусовой кинетике неявно постули-
рована однородность пространства. Например, в плазменных струях и моле-
кулярных пучках распределение скоростей изменяется не только в простран-
стве скоростей, но и в обычном пространстве. Тогда / = / (г, v, t).
Известно, что для обычных реакций Е^кТ 1, поэтому в аррениусовом
выражении (2.23) определяющее значение имеет экспоненциальный множи-
тель, так как А (Г) слабо зависит от температуры — для простейшей газо-
кинетической модели А(Т) = аТ1!*. Когда же Т велико и EJkT 1,
ехр в (2.23) может быть разложена в ряд и основным членом будет А (Т).
Если А (Т) зависит от Т7 с малым положительным показателем степени,
к (Т) ведет себя подобно коэффициентам переноса, которые изменяются
— Т^2 (вязкость, теплопроводность) и Т3^2 (диффузия). Такой тип реакции
характерен для высокотемпературных реакций (атомы, радикалы), для ко-
торых Е% мала или равна нулю. В промежуточной области, где Е^кТ — 1
д In [Л (Т)]
или где —д , предэкспоненциальныи и экспоненциальный мно-
жители конкурируют между собой и имеет место сложная зависимость от
температуры.
НАБЛЮДАЕМАЯ (КАЖУЩАЯСЯ) ЭНЕРГИЯ АКТИВАЦИИ
В работе [30] рассмотрен вопрос о кажущейся энергии активации при
высоких температурах и показано, что наклон аррениусовой прямой в этом
случае не соответствует какой-либо разности энергий атомных или молеку-
лярных состояний. В предположении максвелл-больцмановского распреде-
ления существенное значение для теоретического расчета коэффициента
скорости химической реакции имеет вид функции ст (е) вблизи порога реакции.
Поведение этой функции вблизи порога может быть представлено в виде
простой степенной функции (Е — E1-lGp)m, где jE’nop — пороговая энергия.
Для ионизации при соударениях тяжелых частиц 2 тп 3 [31, 32], а для
электронного удара 1 тп 2 [33, 34].
Вычислив к в этих предположениях, получим для двух предельных
случаев следующие выражения:
/ 8 \
— \лрЗ / б°еХР( —Репор) (Р^пор4^ 1)>
(2.24)
где р — приведенная масса; [3 = ЦкТ\ <у0 — постоянная часть сечения o'.
Согласно этим выражениям, кажущаяся энергия активации, т. е. отри-
цательный наклон аррениусова графика, для указанных предельных слу-
чаев будет
ёпор 4 о (Р8пор^> !)•
d In к / Р
&пор 4“ 2р (Р^ПОр !)•
(2.25)
1 Это неприменимо и в процессах пр ед био логической химии, которая является сущест-
венно неравновесной (рис. 2.2).
46
Вблизи температур — 300° К, р ~40з2?-1и при 8пор — 10 эВ (ионизация)
этими поправками можно пренебречь: в этом случае наклон аррениусовой
прямой дает величину энергии активации. При Т = 104 °К и для типичного
значения ш — 2 поправочный член для той же реакции — 1,5 эВ. и его не-
обходимо учитывать, так же как и при 8Пор ~ 0,2 эВ (химическая реакция)
и той же температуре, когда поправка составляет даже 0,5 эВ.
Как показано в [11], наклон аррениусовой прямой отнюдь не обязательно
определяет энергию активации; иначе говоря, энергия активации высокотем-
пературных реакций не может быть получена на основе только скоростей ре-
акции без знания вида функции ст (е) даже при максвелл-больцмановском
распределении в системе [48].
Из изложенного видно, что даже в простейших случаях неравновесных
систем аррениусово описание скоростей химических реакций может привести
к принципиальным ошибкам как в величине к и в ее зависимости от параме-
тров системы, так и в понимании механизма реакции. Возникают три задачи:
1) рассчитать к для различных более реальных моделей строения и взаимо-
действия молекул при равновесных и неравновесных распределениях кине-
тической энергии и заселенностей уровней; 2) найти метод различения в ки-
нетическом химическом эксперименте реакций, протекающих в неравновес-
ной области, от «аррениусовых» реакций; 3) найти способ определения
из экспериментальных данных о А3ксп коэффициентов скоростей как функ-
ции энергий и заселенностей уровней подсистем единой системы, а также
определить (~Еа).
Для расчета kt и к необходимо знание сечений а (е), функций распределе-
ния / (е) и заселенностей уровней. На основании экспериментальных данных
Ci = ct (Z), выражения (2.24) и в условиях медленно протекающей реакции
(/ = /м (1 + ?), где V 1, /м — максвеллово распределение) можно найти а
или / с помощью рассмотрения интегрального уравнения Фредгольма первого
рода [35]. Для этого может быть использовано выражение (2.24). Запишем
задачу для простейшего частного случая, когда в силу относительно низкой
температуры скорости химической реакции малы, распределение очень слабо
отклоняется от максвелловского, а реакция практически идет с пер-
вого колебательного уровня основного электронного состояния. В левой
части выражения (2.24) имеем тогда определяемую экспериментально в
обычном химическом кинетическом эксперименте величину (к + Ек) ==
Г (K±AK)1
~(&о±А&о)ехр----------гй---- » где А&, АА0, А2Га означают соответствую-
щие ошибки эксперимента. Следовательно,
/г(7’) + Д/с= б1(е)/ё/м(8)й£, (2.26)
^пор
где /м (е) — максвеллово распределение, ТГдор — порог реакции, — энер-
гия, на которой обрезается распределение справа, так как вклад в реакцию
молекул с Е Ег делается пренебрежимо малым из-за их незначительного
(и убывающего) числа [38—47].
Уравнение (2.26) может быть разрешено относительно ог1 (s) при помощи
одного из способов решения такого рода так называемых некорректных за-
дач [49—51]. После нахождения Oj (s) для первого колебательного уровня ос-
новного электронного состояния можно найти сечения для более высоких
колебательных уровней, воспользовавшись экспериментальными данными
о значениях к для более высоких температур и уравнением Паули (см.
ниже). Предлагаемый метод рассмотрен в [35] на примере реакции
I + СН31 -> СН3 4- 12; 12 21. (2.27)
Как это принято в обычной химической кинетике, распределение частиц
по скоростям считается максвелловым, заселенность уровней — больцма-
новой, а возмущение, вносимое химической реакцией, пренебрежимо малым.
47
Если относительные скорости поступательного движения реагирующих
частиц распределены по Максвеллу /м (7\ и), то коэффициент скорости при-
нимает вид [11]
к(Т) = § fM(T,v)(5(v)vdv, (2.28)
£пор
т. е. берется его среднее значение по распределению /м (Т, г). Вводя энер-
гию 8, напишем
Л(Г) = 5 /м(Г,е)б(8)]/7й8. (2.29)
£пор
Согласно уравнению (2.23), в случае максвелл-больцмановского распре-
деления имеем
2£»(эксп) _ _ d In к(Т) __ ^2 dink (Т) gg^
Откуда [52]
оо
б (б) 82 ехр (— &]кТ) d&
£(аэксп)= _^Р------------------------f fcT. (2.31)
с (е) е ехр (— ъ/кТ) d&
£пор
Интеграл в знаменателе этого уравнения пропорционален распределению
соударений, приводящих к реакции, по энергии.
В общем случае даже для прямой бимолекулярной реакции А + В —>
-> С + D (так называемый случай «два в два», очень удобный для математи-
ческого рассмотрения) вероятность реакции имеет сложную функциональную
зависимость от сечений различных неупругих процессов, вероятности излу-
чательных переходов и т. п. функций распределения реагентов и продук-
тов по скоростям, заселенности внутренних степеней свободы и т. д.
Следовательно, записывая /м (Т7, 8) в явном виде, получим из (2.29)
оо
*(Л = А ^-б(8)еХр(-8/АЛЙ8, 4 =
Но
(2.32)
Решение интегрального уравнения (2.32) находим методом минимизации
некоторого сглаживающего функционала Ма, имеющего вид
Ма = \ [Л 0(e) ехр (—г]кТ) de — к (71)] * dT + aQ, (2.33)
Т, Е0
где а — параметр регуляризации, a Q было выбрано нами в виде
So
(2.34)
(регуляризация первого порядка). В этом случае задача приводится к ин-
тегрально-дифференциальному уравнению Эйлера
“de2 ( (e + t)2 \ kl\ + 1J
So
di (2.35>
к7\
48
страничными условиями o' (е0) = 0 и 6-1 _ =0; множитель А = ]Л8/лр,
внесен в о (s); t — переменная интегрирования. Уравнение (2.35) корректное
в том смысле, что при заданных граничных условиях и а -> 0 его решение
становится устойчивым. Параметр регуляризации а выбирали в виде
а = 0,Г1, где п — 10, 11, . . ., 18. Обычно а подбирается методом проб и
ошибок в ходе поиска устойчивого решения уравнения (2.35) на ЭВМ.
Выше мы отмечали, что нижним пределом при интегрировании правой
части уравнения (2.29) должна быть величина 8ПОр- Если она известна из
каких-либо независимых данных, то никаких трудностей не возникает.
Если же 8Пор неизвестна, то метод, развитый в предыдущем параграфе,
позволяет найти 8П0р вместе с в (е). Взяв несколько значений 80 в некоторой
разумной (определяемой из общих соображений) окрестности сп), находят
для этих значений 80 устойчивые решения уравнения (2.29). Затем по найден-
ным о* (е) для различных 80 определяется средняя квадратичная ошибка
отклонений б восстановленной константы к (Г) от экспериментальной вели-
чины к(Т):
т2
62 = тАт\ \lk^~k (2-36)
после чего строится кривая б (80). Минимум на этой кривой и определяет
значение 8 пор»
Предлагаемый метод определения значения 8Пор обладает общностью в том
смысле, что при любом, отличном от нуля, интервале (Z2, Tt) изменения
независимого переменного Т найдется такое п, для которого зависимость
б2п =
Ь -----
1 С ГЛ (ж) dk(x)l*
Ъ-а \ L ‘
(2.37)
от 80 обладает достаточно резким минимумом при 8 = 8п0р, чтобы из условия
^”1 1= о
д£° 1£0“еПОр
(2.38)
значение 8Пор можно было бы определить с достаточной точностью.
В общем случае надо из эксперимента найти и функции распределения.
Если экспериментальные методы нахождения функции распределения заря-
женных частиц в газовой фазе имеют уже солидную историю, то до самого
последнего времени не было эффективных способов найти реальную функцию
распределения нейтральных реагирующих частиц по электронным, колеба-
тельным и вращательным степеням свободы. Правда, в стационарных усло-
виях определение электронно-возбужденных состояний не представляет
труда. В этих же условиях в отдельных случаях [36] удавалось получить
данные и о заселенностях части колебательных уровней возбужденных элек-
тронных состояний.
Лишь недавно, в связи с развитием спектральной техники и в первую
очередь методов фотоэлектрической регистрации, появилась возможность
исследовать образование возбужденных колебательных состояний по
ИК-флуоресценции реагирующих молекул и продуктов реакции. Исследо-
вание спектров флуоресценции является общим методом исследования засе-
ленностей различных состояний реагирующих веществ и продуктов реак-
ции. В качестве частного метода, характеризующего некоторые стороны
отдельных процессов, могут быть использованы так называемые химические
лазеры [37].
49
УРАВНЕНИЕ ПАУЛИ
У равнение Паули записывается для крупноструктурной матрицы плот-
ности вероятности р (n, t) нахождения слабо взаимодействующих частиц
в совокупности Ап близко расположенных состояний (такая совокупность
является аналогом фазовой ячейки в классической механике) в виде
Эр g”’ - = 2 vnPrm'p (п', г<) — 2 Vn-Pn'nP (п, t), (2.39)
п' п'
где vn — число состояний в Ап; Рпп' — вероятности переходов в единицу
времени из состояния, принадлежащего п', в п. Для случая мономолекуляр-
ной реакции, протекающей в термостате инертного газа (концентрация ко-
торого является постоянной), уравнение Паули записывается в виде
[57 Л 58]
5 П • у’ —« «
gf = 2 (!) - 2 (0 - (0 + Ri (*)> (2-40)
FJ j
где ni — концентрация реагирующих молекул в i-м энергетическом состоя-
нии в момент времени Z; — вероятность (рассчитанная на одно столкнове-
ние) того, что при столкновении реагирующей молекулы с молекулой тер-
мостата она перейдет из /-го в z'-е энергетическое состояние; Рц — то же для
перехода из состояния i в состояние /; — коэффициент скорости химиче-
ской реакции для z’-ro энергетического состояния; Rt — скорость воз-
буждения i-ro уровня («накачка»); со — частота столкновений. Кроме чисто
математических преимуществ, которые имеет уравнение Паули с точки зре-
ния численных методов расчета, надо отметить следующие существенные об-
стоятельства. Строго говоря, это уравнение баланса, т. е. при правильной
записи оно всегда верно как всякое балансовое соотношение. Оно позволяет
единообразно объединить переходы между уровнями и собственно химиче-
ские переходы [56].
Существеннее значение имеет и то, что это уравнение, в известном смысле,
является прямым обобщением уравнений химической кинетики (и кинетики
заселенностей уровней и т. п.); если не учитывать переходы между уровнями,
оно сводится к уравнениям обычной химической кинетики.
Очевидно, что многие из методов решения кинетических задач, разрабо-
танных в классической химической кинетике (в частности, метод квазиста-
ционарных концентраций), найдут широкое применение в обобщенной хими-
ческой кинетике. Надо иметь в виду, что на определенных конечных интер-
валах изменения температуры (энергии) и концентраций реагентов и продук-
тов реакций можно будет, с учетом точности экспериментальных данных,
рассматривать скорости химических реакций с классической точки зрения.
Для того чтобы перейти от общей теории кинетики химических реакций,
охватывающей как неравновесные, так и (квази)равновесные процессы,
к частному и более простому случаю описания только последних (равно-
весных) реакций, необходимо допустить следующее:
1) сечение (вероятность) химической реакции не зависит (или слабо за-
висит) от энергии частиц при EnQ$ Е и равно нулю при Е^ > Е\
2) в реагирующей системе всегда сохраняется максвеллово распределение,
т. е. определена одна и только одна температура системы Т. Допущение
первое связано с упрощенной моделью взаимодействия «твердых шаров»,
в том или ином элементарном или несколько усложненном виде используе-
мой в обычной химической кинетике. Конечно, эта модель далека от своего
реального прообраза — реагирующих соударяющихся молекул. Допущение
второе, строго говоря, никогда не выполняется, если в системе происходит
химическая реакция, так как всякий процесс, протекающий с конечной
скоростью, нарушает равновесие даже в системе, равновесной в начальный
момент времени. Однако во многих случаях, характеризующихся неравен-
50
ством кТ (большие энергии активации, низкие температуры), нару-
шения максвелл-больцмановского распределения малы во всяком случае
потому, что касаются относительно небольшой группы частиц. Напротив,
при Е& ~ кТ распределение нарушено существенно.
Единая температура не существует также в целом ряде открытых систем,
где имеется «внешняя» неравновесность, создаваемая каким-либо источни-
ком: тем или иным видом излучения, разрядом и т. п. В этих случаях часто
бывает, что в системе в целом температура отсутствует, хотя для той или
иной подсистемы максвелл-больцмановское распределение может иметь место.
В огромном числе случаев в химических реакциях Тпост Ф Укол и т. п.
Например, мономолекулярные реакции почти всегда протекают через воз-
бужденные колебательные состояния, причем происходит нарушение равно-
весного распределения по колебательным степеням свободы, а тКОл Тдост?
где т — время релаксации.
Большое и зачастую определяющее значение имеет возбуждение молекул
и их фрагментов в экзотермических химических реакциях [249—251], реак-
циях, идущих через электронно-возбужденные состояния в тлеющем разря-
де, в газовых лазерах, в химических реакциях в ударных трубах, турбу-
лентных и сверхзвуковых потоках и т. п.
Не вызывает, таким образом, сомнений, что обычная кинетика является
существенно приближенной теорией, применимость которой к анализу
какого-либо конкретного процесса должна быть исследована заранее во
избежание сглаженного и просто неверного описания.
Подчеркнем то обстоятельство, что уравнение Паули имеет место для
ансамбля многих слабо взаимодействующих частиц (например, разрежен-
ные газы, очень слабоионизированная плазма и т. п.). Поэтому оно, строго
говоря, неприменимо при наличии в системе сильных взаимодействий,
например, для химических реакций на поверхности твердых тел, в сильно
неидеальных газах и плазме, высокоионизированной плазме ит. п. Пределы
применимости уравнения Паули в конкретных задачах такого типа должны
быть каждый раз исследованы отдельно.
Во всех такого рода выводах предполагается статистическая независи-
мость молекул до столкновения. Если а — радиус взаимодействия, к — длина
свободного пробега, N — полная плотность молекул, то условие статисти-
ческой независимости можно записать так :
а!к^ Ж3<1. (2.41)
При обычном обосновании уравнения Паули, впервые данном самим Пау-
ли [57], подразумевается, что приближение к равновесию вызывается воз-
мущающим членом Нг в гамильтониане системы, причем Н1 настолько малт
что вероятности перехода Ptj можно вычислять в первом приближении не-
стационарной теории возмущений. При этом вывод уравнения Паули опи-
рается на статистическую гипотезу, что фазы волновых функций, принадле-
жащих различным собственным значениям Н, распределены беспорядочно,
т. е. что матрица плотности считается диагональной в представлении невоз-
мущенного гамильтониана. Эта гипотеза беспорядочных фаз относится не
только к начальному состоянию, но многократно используется после каждого
из таких интервалов времени, для которых невозмущенная энергия Н при
переходе сохраняется. Аналогичная (и глубоко неудовлетворительная)
ситуация имеет место при допущении молекулярного хаоса в выводе кинети-
ческого уравнения Больцмана. Этот вопрос связан с тем, что надо получить
необратимость во времени, хотя исходные уравнения динамики обратимы.
Ван-Хов [59] показал, что повторное применение гипотезы беспорядоч-
ных фаз излишне при выводе квантовомеханического кинетического уравне-
ния, если при выводе его принять во внимание некоторое специфическое
свойство возмущения, обусловливающее существование эффектов переноса.
Мы не будем останавливаться на этом выводе, так как для неравновесной
химической кинетики (при не слишком больших отклонениях от равновесия)
51
допущения, закладываемые в вывод уравнения Паули, не ограничивают его
применимости.
Различные формы квантового уравнения Паули были получены начиная
с 1957 г. в работах Ван-Хова [61], Свенсона [69], Накаджимы [63], Цванцига
[64, 65], Резибуа [66, 68], Пригожина и Резибуа [67], Монтрола [69, 70].
В работе [71] Цванцдг показал, что все виды уравнения Паули (кроме [61])
полностью эквивалентны.
Выведем уравнение Паули при некоторых достаточно приемлемых допу-
щениях.
Рассмотрим систему, вероятность i-ro состояния которой в момент вре-
мени t равна Pt (t). В момент времени t -Т kt при Az -> 0 имеем Pt (t + Аг).
Величину Pi можно при довольно общих предположениях описать с по-
мощью уравнения Паули.
Введем следующее предположение:
А(* + Д0 = ЗЛЛ(^ (2-42)
к
где Aik — вероятности переходов, нормированные так:
= (2-43)
к
3 Агк — 3 Акг == 1 • (2.44)
г к
Используя (2.42), (2.43), (2.44), найдем
л(< + ^)-л(о (2-45)
к ' к
Введем вероятности переходов в единицу времени
Aik = (M)aik. (2.46)
В пределе Аг -> 0 уравнение (2.45) принимает вид
^ = У.^кРк~ак^). (2.47)
к
Это и есть уравнение Паули.
В силу (2.42) уравнение (2.47) содержит необратимость, что нетрудно по-
казать. Тем не менее его можно получить из обратимой динамики, что пред-
ставляет собой важную задачу статистической механики. Вывод самого
Паули [57] был основан на предположении, что фазы собственных функций
в каждый момент времени случайны (что аналогично допущению «молеку-
лярного хаоса»). В 1955 г. Ван-Хов [59] вывел уравнение Паули без этого
предположения, при весьма общих допущениях путем решения уравнения
Шредингера в пределе слабой связи [59, 60].
Пусть система описывается гамильтонианом Н:
н = Н.+ Hv (2.48)
где Но — гамильтониан невозмущенного состояния, II v — гамильтониан
возмущения.
Тогда для классической системы полная функция распределения / опре-
деляется из уравнения Лиувилля
g + [/, Н] = 0, (2.49)
где [/, Н] — скобки Пуассона.
52
Для квантовомеханической системы функцию распределения нужно за-
менить матрицей плотности р (£), удовлетворяющей уравнению
^+^[р,Я] = 0, (2.50)
Для Н вида (2.48) это уравнение имеет формальное решение
t
р(О = ро — ехр[— iH0(t — t')/h] [А, р] exp [£Я0(£ — t')/h]F (t')dt',
—oo
(2.51)
где p (— oo) = p0.
Легко показать, что уравнение (2.51) имеет смысл, только если рассма-
тривать достаточно «крупнозернистые» начальные распределения. Это еще
одна трудность, связанная с обоснованием и истолкованием кинетического
уравнения Паули.
НЕРАВНОВЕСНЫЕ ТЕОРИИ
И РАСЧЕТЫ ДИССОЦИАЦИИ - РЕКОМБИНАЦИИ
Необходимость неравновесной теории диссоциации—рекомбинации сле-
дует из экспериментальных данных, полученных с помощью ударных труб,
флеш-фотолиза, плазмохимии. Так, например, равновесная теория атомной
рекомбинации предсказывает, что Лрек не зависит от температуры. В то же
время эксперимент показывает, что &pgK уменьшается при низких темпе-
ратурах, как Г"1, и, видимо, как 7“2, при высоких температурах. Простые
неравновесные теории сочетают явно скорость реакции и скорость колеба-
тельной релаксации и приводят к правильной зависимости к — Т'1 при
низких температурах [72, 73].
В [74] эта проблема рассмотрена с помощью уравнения Паули. Рассмотрим
сначала равновесную задачу в термостате инертных молекул М, в который
помещены атомы и молекулы. Макроскопическое уравнение имеет вид
[А2] = А?рек [М] [А]2 &дис [М] [А2], (2.52)
где клис — коэффициент скорости диссоциации, &рек — то же рекомбинации.
Микроскопическое описание, естественно, требует учета колебательных
(или вращательных) переходов в возбужденной молекуле
[A2i] = &рек i [М] [А]2 - Лгдис i [М] [A2i] + [М] 2 kni [Аа„] - [М] 2 [А«],
i^n
(2.53)
где первый член — прямое заселение уровня i рекомбинацией А + А + М -+
—> А2 + М, второй — противоположный процесс, третий и четвертый —
переходы между различными внутренними степенями свободы молекулы.
Положим
N
[А2] = 2 [А2*Ь (2.54)
1=0
где N — число уровней энергии. Можно показать, используя (2.52)—(2.54),
что
N N
[А2] - [MJ [А]2 2 &рек г - [М] 2 йцис i [A2i]. (2.55).
i=0 i=0
53
Если распределение по внутренним степеням свободы двухатомной моле-
кулы равновесно, то
. ехр(-^Т)[А2]
[A2i] = --------------=5 Yi [А].
2 ехр(— Еп/кТ)
п=0
Тогда уравнение (2.52) принимает вид
^0 — [А2] = [М] [А]2 2 ^рек! — [М] [А2] 2 ^дисгТг-
1=0 г—0
(2.56)
(2.57)
Сопоставляя (2.52)
N
^рек “ 2 ^рек!»
1=0
и (2.57), найдем
N
^дис = 2 ^Дис iTi-
1=0
(2.58)
При [А2] 0 предположение о равновесном распределении по внутренним
степеням свободы не выполняется из-за возмущения его химической реакци-
ей, и обратно: это неравновесное распределение влияет на скорость химиче-
ской реакции.
Вывод кинетического уравнения для неравновесной теории рекомбина-
ции—диссоциации выполняется так же, как вывод его в случае равновесной
теории; необходимо только включить в схему, зависящую от времени, функ-
цию распределения по внутренним степеням свободы.
Не останавливаясь на выводе этого уравнения, укажем лишь, что оно
решается в основном для малых возмущений методом, аналогичным методу
Энскога. Окончательно уравнение неравновесной химической кинетики
рекомбинации—диссоциации двухатомной молекулы имеет следующий вид:
N N
[А2] = Vq — [М] [А2| Vq 2 ^рек1Т1/1 = Ц) [1 — [М] [А2] 2 ^рек lYi/d =
1=0 1=0
= (2.59)
здесь fi — функция распределения.
Таким образом, связь химической и колебательной релаксации представ-
лена функцией 1 (Т), которая есть первый член в разложении в ряд скорости
неравновесного процесса
V = v0 [1 - I (П - Р (П + . . . + (-ipr (Z)] = ср (T)v0. (2.60)
Макроскопическое уравнение будет иметь вид уравнения (2.52) с той
только разницей, что в неравновесном случае
N N
^рек — ф(^) 2 ^рек 1? ^дис = ф (^) 2 Т1^дис1- (2.61)
1=0 1=о
Задача рассматривается в рамках теории возмущения вблизи равновесия,
и поэтому при приближении скорости реакции к скорости колебательной
релаксации может оказаться необходимым учитывать члены более высокого
порядка.
Для модели переходов «ближайшего соседа» (одноквантовые переходы)
функция ф(Т) имеет вид
ф(П =
1 + Т/У^ДИС N
(2.62)
Расчет показывает, что /срек действительно убывает с ростом температуры
как Т~2 при достаточно высоких температурах. Надо отметить, что хотя
54
этот вывод получен для специального случая, однако основные найденные
выше уравнения имеют общий характер и могут быть использованы для изу-
чения и более реальной модели.
Запишем еще раз уравнение Паули для случая диссоциации и рекомби-
нации двухатомной молекулы в инертном разбавителе [75]. Пусть число мо-
лекул в i-м состоянии в единице объема в момент времени t будет (t), тогда
концентрация молекул
= (2-63)
г
полное же число атомов, сохраняющееся в системе (в предположении, что
атомы А, на которые распадается молекула А2, не имеет внутренних состоя-
ний),
Ад (0 + 2 2 ni(f) — const. (2.64)
Возможные процессы:
А2 (0 + М А2 (/) + М, (2.65)
А2 (0 + М А + А + М. (2.66)
Обозначим Р^ — вероятности возбуждения в прямой и обратной
реакции (2.66) и Pic, Pci — вероятности диссоциации и рекомбинации (все
эти вероятности не зависят от времени). Тогда нелинейное уравнение Паули
определит й щ при условии выполнения закона сохранения (2.64):
dn. v-,
•7Г = S (^Лч - пгРц) + м Pci - (2.67)
3
где i ~ 1, 2, . . ..
В силу нелинейности члена, описывающего рекомбинацию, нельзя полу-
чить точное решение (2.67) для задачи внутренней релаксации и одновремен-
ной диссоциации и рекомбинации.
Однако, используя метод возмущений, можно получить приближенное
решение для случая, когда характеристическое время внутренних релакса-
ционных процессов мало по сравнению с временем диссоциации и рекомби-
нации. Решение получается в виде разложения в ряд по степеням параметра
0фел/^дисс)1/2 1, причем первое собственное значение матрицы, описываю-
щей внутреннюю релаксацию и диссоциацию (но не рекомбинацию), равнэ по
порядку величины/€диТн) коэффициенту скорости, соответствующему равнове-
сию внутренних степеней свободы. Никаких конкретных предположений
о природе внутренних степеней свободы делать нет необходимости.
В работе [76] рассмотрено влияние (у—0-процессов на скорость диссоциа-
ции двухатомных молекул.
Уравнения Паули для одноквантовых переходов (в случае к ф- 1 колеба-
тельных уровней):
Л 0 = Р107V L — PqjN о,
~ ^л-i, ф- Pj-i, jN— Рj, j+lNj — Pj, j,
(2.68)
— Рф, ,Ф2У ф- PkN— PфNk — Pkl k.
Здесь Nj — концентрация частиц, находящихся в состоянии /; N — [А2];
Ф — [А]2//сравн А; Р — коэффициенты скорости перехода.
Если (у—0-переходы имеют место, коэффициенты скорости являются
функциями Nj и уравнения Паули нелинейны.
55
Не останавливаясь на деталях проведенного расчета, отметим только
полученные результаты. При низких температурах (у — ^-процессы слабо
увеличивают скорость диссоциации, а при высоких — скорость диссоциации
сильно уменьшается за их счет. Сочетание этих эффектов вызывает уменьше-
ние кажущейся энергии активации, т. е. уменьшение кажущейся энергии
активации есть общее следствие (у — 1?)-процессов в диссоциации двухатом-
ных молекул. Это значит, что (у — ^-процессы вносят значительный вклад
в термическую диссоциацию этих молекул, что подтверждается также экспе-
риментально.
Решению кинетических уравнений Паули для определения скоростей дис-
социации и связи ее с колебательно-поступательно-вращательной релакса-
цией двухатомного молекулярного газа (разбавленного инертным газом)
посвящено большое количество работ, выполненных Притчардом и соавто-
рами [74—86]. Решение системы обыкновенных дифференциальных уравне-
ний большой размерности связано с серьезными трудностями при численном
счете на ЭВМ.
Дело в том, что переменные и характерные времена их изменения отли-
чаются между собой по величине на несколько порядков. Поэтому, по мнению
авторов [77, 78], обычные методы численного интегрирования типа Рунге-
Кутта или прогноза и коррекции являются непродуктивными, так как тре-
буют существенно малого шага интегрирования и, следовательно, большого
времени решения кинетической задачи на ЭВМ. По этой причине в работах
[77, 80] был разработан метод решения, использующий самосогласованную
матричную процедуру при трех основных предположениях: 1) время — не-
прерывная переменная; 2) продолжительность столкновений, которые при-
водят к переходу исходных молекул в другие состояния, мала по сравнению
с рассматриваемым характерным временем; 3) начальное распределение
траекторий частиц должно быть таким, чтобы распределение времен первых
столкновений было случайным.
Данный метод позволил увеличить шаг интегрирования по сравнению
с методом Рунге-Кутта на несколько порядков величины с сохранением
необходимой точности решения.
Авторы рассматривают идеализированный эксперимент, в котором удар-
ная волна проходит через смесь двухатомных молекул А2 и молекул инерт-
ного газаМ, находящихся вначале при температуре TQ ([А2] <<: [М]). Нагрев
предполагается мгновенным, так что, когда Гпост = внутренние степени
свободы имеют при этом 7ВН = Установление равновесия между посту-
пательными и внутренними степенями свободы достигается при некото-
рой равновесной температуре Zp. Это происходит за счет процессов вида
М + А2 (г, J) М + А2 (у', J'), (2.69)
так как из-за условия [А2] [М] процессами вида
А2 (v, со) + А2 (у, J) А2 (v', со') + А2 (к', J') (2.70)
можно пренебречь.
Для описания диссоциации в [78] предположено, что она происходит
через образование скрытых пар, концентрация которых всегда находится
в равновесии с мгновенными концентрациями атомов, т. е.
А2 А 4-А с [А2] — X [А] [А]; (2 71)
МА*^±М4-А с [МА*]-6[М][А],
где % и б — константы равновесия. Тогда последовательность реакции диссо-
циации—рекомбинации можно представить так:
M + A2(p,J)t±M + a;^M+А +А, (2.72)
М +A2(p,Z)z±MA* + Aj±M + А+А; (2.73)
56
процессом
A2(v, со) + А2 (у, J) А2 (v', со') + А2 А2 (v', со') + А + Aj
пренебрегаем. В реакциях (2.72) и (2.73) трехчастичная рекомбинация опи-
сывается двумя последовательными двухчастичными реакциями (первая дает
скрытую пару, вторая — связанное состояние). В силу допущения 2 трудно-
стей при такой замене не возникает. Предположим далее, что давление мало
и ^-частичные реакции отсутствуют; пренебрегаем также радиационными
процессами.
Тогда, вводя обозначения: nt — заселенности i-x связанных вращатель-
но-колебательных состояний А2 (I = 0, 1, . . ., п — 1); пп, пп+1, —
то же для заселенностей А2, А, МА* соответственно; — вероятность,
рассчитанная на единицу времени и концентрации, перехода из состояния J
в состояние i; Wjt — то же из состояния i в J; Win — то же для процесса
(2.72), 1И(П+2)1 — то же для процесса (2.73), причем все W усреднены
по максвелл-больцмановскому распределению М при Т = Zo, а все TF —
по масквелловскому распределению энергий атомов А, получим
du. Г ул __ -
= [М] lWi^j — Wrfbt] + \ Winnn — PFninJ +
г___ и2 ______ р
+ pF {(п+2) у [од2] 2 — n+2inij j • (2.75)
В выражении (2.75) член под знаком суммы описывает все процессы (2.69),
а второй и третий члены в квадратных скобках представляют процессы, опи-
санные (2.72) и (2.73) соответственно.
Аналогично запишем
= [М] 2 - Wjnnn] + [^п(п+1)п*г+1) - W^1)nnnY (2.76)
э
dnn+J _____. ___ ______________ _______________,
d~j_ = [W(п+1)п^п Wn(n+l)^+1] 4~ [W(п+1) (п+2)^п+2 И (n+2) (п+1) [М] ^(п+2)1?
(2.77)
^Пп+2 Г —- 1
--— = W(n+2) (п+1)М/2(п+1) — TF(n+i) (п+2)^(п+2)1 +
+ [М] 2 [- W+2) • (2.78)
Легко видеть, что сумма уравнений (2.75)—(2.78) представляет собой усло-
вия сохранения числа частиц
п+2 п+2 7
1 dn •
= 2^ = 0- (2-79)
j—0 7=0
Строгое выполнение этих условий сохранения является решающим об-
стоятельством при численных расчетах системы (2.75) — (2.78).
Для решения задачи кинетики диссоциации — рекомбинации в рассматри-
ваемом случае система (2.75) — (2.78) приводится к системе неоднородных,
нелинейных интегральных уравнений Вольтерра второго рода. Для решения
ее предлагается удобная самосогласованная матричная итерационная про-
цедура. Эта процедура позволила произвести ряд конкретных расче-
тов для Н2 и D2 в атмосфере инертных газов [78, 80—86] и сделать ряд
интересных выводов. Например, в работе [82] показано, что более тесное
расположение колебательных уровней в D2 по сравнению с Н2 приводит к
увеличению вероятностей (у — ^-обмена и как следствие этого к увеличе-
нию скоростей диссоциации и рекомбинации, а также к выводу, что положе-
57
ние «узкого места», определяющее скорость диссоциации, не совпадает с
уровнем, с которого возможна прямая диссоциация.
Сравнение скоростей диссоциации при 1500 и 2000° К показывает, что
аррениусовы температурные коэффициенты диссоциации для D2 состав-
ляют 103,0 ккал/молъ, для Н2 — 104,4 ккал!молъ, т. е. в обоих случаях при-
мерно на 2 ккал!моль меньше, чем спектроскопическая энергия диссоциации.
Авторам не удалось найти простой связи между положением «узкого места»
в расположении колебательных уровней и аррениусовым температурным
коэффициентом диссоциации. Исследованию реакций типа Х2 + У2 2XY
для системы Н2 + D2 2HD посвящена работа [83]. Авторы считают, что
более полное описание диссоциации и рекомбинации разбавленного гомоядер-
ного двухатомного газа может быть достигнуто путем рассмотрения их сов-
местно с колебательно-поступательным обменом. Оказалось, что на време-
нах, превышающих характерное время колебательной релаксации, все фе-
номенологические коэффициенты скоростей или постоянны, или зависят от
времени как £2, хотя распределение заселенностей по колебательным уровням
существенно отлично от больцмановского. Авторы предполагают, что для
более детального описания этих процессов необходимо использовать больше
информации о внутреннем строении изучаемых молекул. В [84] исследова-
лась связь диссоциации и колебательной релаксации при истечении газа в
расширяющееся сопло, Н + Н + М^Н2 + М, без учета (г’ — г;)-обменаг
с одновременным изменением температуры газа, плотности, скорости газоди-
намического потока и распределения заселенностей колебательных уровней.
Начальное условие — полное термодинамическое равновесие при 3900° К.
Предполагалось, что скорость изменения температуры порядка (1—4)-109°К
в 1 сек. Диапазон изменения температуры 1500—3900° К. Хотя (v — г;)-обмен
не учитывался, распределение заселенностей небольцмановское и монотон-
но изменяется от одного уровня к другому. По мнению авторов, в некоторых
случаях возможно получение и абсолютной перезаселенности отдельных
колебательных уровней относительно друг друга. В результате расчетов ока-
залось, что коэффициент скорости рекомбинации соответствует обычному
феноменологическому коэффициенту, характерное время колебательной
релаксации изменяется на несколько порядков величины в зависимости от
вида аппроксимации, используемой для удовлетворительного описания за-
висимости колебательной энергии от времени. В ряде конкретных случаев
характерное время колебательной релаксации можно получить из экспери-
ментальных данных по колебательной релаксации в ударных трубах. Авторы
пытались также найти корреляцию между некоторым собственным значением
вычисляемой в процессе решения релаксационной матрицы и характерным
временем колебательной релаксации. Возможно также, что простое уравне-
ние, описывающее релаксацию колебательной энергии, dzldt = (в — ё)/тКОл,
имеет ограниченную область применимости. В следующей работе [85] учиты-
вался вклад вращения в диссоциацию и рекомбинацию. Детально изучались
диссоциация Н2 с J = 21 и рекомбинация атомов в это состояние. Враща-
тельный уровень J = 21 имеет два связанных квазисостояния: одно — дол-
гоживущее, второе — короткоживущее. Коэффициент скорости диссоциации
или рекомбинации с этого или на этот уровень фактически определяется
эффектами «узкого места», имеющими место ниже по колебательной
«лестнице».
Менее подробное изучение вклада в диссоциацию других вращательных
уровней показывает, что эти заключения являются сбшими для всех враща-
тельных уровней, за исключением тех, для которых нет сильного влияния
эффекта «узкого места», т. е. для очень высоких вращательных уровней
J > 30. Показано, что в предположении полного вращательного равновесия
сумма коэффициентов скоростей реакций с отдельных уровней ./, взвешенная
ио всем J, приводит к такому чрезмерному увеличению коэффициента скоро-
сти диссоциации, что можно предположить отсутствие вращательного равно-
весия на верхних вращательных уровнях. Вычисления показали, что с уче-
58
том только (г? — Т)- или только (R — 7)-процессов, в диапазоне температур
1500—5000° К верхние вращательные уровни сильно обеднены и аррениусов-
ский температурный коэффициент в коэффициенте скорости диссоциации
равен приблизительно 92—94 ккал!моль в зависимости от выбора вероятно-
стей для вращательных переходов. По мнению авторов, стадия, предшест-
вующая диссоциации в ударных волнах, т. е. стадия колебательной релак-
сации существенно зависит от величины вероятностей вращательных пере-
ходов и релаксация типа (vR— Т) более вероятна, чем (г?—/^-релаксация.
Проделаны также вычисления для D2 в диапазоне температур 700—5000° К
по этой модели, учитывающей связь между вращением, колебаниями и дис-
социацией. На основании этих вычислений можно предсказать, что возможен
обратный изотопический эффект для диссоциации и рекомбинации Н2 и
D2 : 1) если Не используется как третье тело, D2 может диссоциировать бы-
стрее, чем Н2, при высоких температурах, но при Т ниже 2000° К диссоциа-
ция D2 замедляется; 2) если в качестве третьего тела используется Аг, то
дейтерий рекомбинирует быстрее, чем водород, при высоких /, но ниже
1000° К рекомбинация дейтерия замедляется; 3) коэффициент скорости
рекомбинации водорода на водороде и дейтерия на дейтерии будет, видимо,
заметен в области температур около 1000 К, что указывает на необходимость
эксперимента в этой области. В последней известной нам работе [86]
изучаются характерные времена вращательно-колебательной релаксации и
связь некоторых собственных значений релаксационной матрицы с
вероятностями вращательно-колебательных переходов. Прямая пропорцио-
нальность между некоторыми вероятностями и некоторыми собственными
значениями матрицы, которая была получена ранее при учете связи
колебаний и диссоциации, в этом случае для ортоводорода не наблюдается.
Подтверждается ранее высказанное предположение, что процесс, пред-
шествующий диссоциации в ударных волнах, необходимо скорее отно-
сить к временам колебательно-вращательной релаксации, чем к временам
колебательной релаксации. Вычисления подтверждают недавно полученный
экспериментальный факт, что время вращательной релаксации сначала
уменьшается, а потом увеличивается с увеличением температуры. Таким
образом, чем выше температура, тем более сложной становится (vR — ^-вра-
щательно-колебательная релаксация. Кроме того, времена релаксации
для одних и тех же температур, измеренные ультразвуковой техникой
и в ударных волнах, не согласуются между собой.
Результаты данных работ представляют несомненно большой интерес.
Однако даже при разбавлении двухатомного молекулярного газа инертным
в соотношении 1 : 1000 роль (г — ^-обмена может быть существенной, осо-
бенно при низких газовых температурах. Тем более, что в работе [79], где
авторы пытались учесть (у — г?)-обмен, показано, что при эффективных
(у —г)-переходах решение затруднено, а (у — г?)-обмен не является малой по-
правкой в коэффициенте скорости диссоциации. Коэффициент этот определен
не совсем однозначно, скорость диссоциации записывается в виде kd [М]х- [Н2]?;,
где х 1 у. Порядки реакции х и у будут построены только в области
предельной концентрации, и общее число ш — (х + у) всегда меньше 2.
РАСЧЕТ КОЛЕБАТЕЛЬНОЙ РЕЛАКСАЦИИ МОЛЕКУЛ
ПРИ НАЛИЧИИ fv — T)-. (v-v)-. (е - ^-ПРОЦЕССОВ.
ВЗАИМНОЕ ВЛИЯНИЕ КОЛЕБАТЕЛЬНОЙ РЕЛАКСАЦИИ
И ДИССОЦИАЦИИ
Для расчета колебательной релаксации с учетом процессов возбуждения,
дезактивации и обмена колебательной энергией при соударениях частиц,
диссоциации молекул и рекомбинации атомов необходимо решать систему
кинетических уравнений Паули [11]. В настоящее время показано, что
59
например, в условиях электрических разрядов в молекулярных газах ста-
ционарное распределение заселенностей молекул по колебательным уровням
в основном электронном состоянии отлично от равновесного [87—90].
В качестве примера рассмотрим расчет кинетики релаксации распределе-
ния заселенностей по колебательным уровням на примере основного электрон-
ного состояния XxSg молекулы N2. Исследуется влияние степени ионизации
а на вид этого распределения и время релаксации (или на время достижения
стационарного распределения) [90].
При решении данной задачи были сделаны следующие основные предпо-
ложения: 1) среда однородна и изотропна; 2) распределение всех частиц по
энергиям — максвелловское, температура газа Тг и температура электро-
нов Те не изменяются во времени, при этом Тт =/= Те\ 3) энергия колебатель-
ных квантовых уровней состояния XxSg рассчитывается по модели ангармо-
нического осциллятора; 4) учитываются только следующие процессы: одно-
квантовые колебательно-колебательные (г — г) и колебательно-поступатель-
ные (у — ^-процессы, многоквантовые (Лг? = ± 1, +2 ,...,+ 51) (е — у)-
процессы; 5) в некоторых из выполненных расчетов учитывались диссоциа-
ция молекул и гетерогенная рекомбинация атомов на стенках разрядной
трубки. Предполагается, что поток, заселяющий последний колебательный
уровень г?* =51, равен потоку диссоциирующих молекул, т. е. тг51(г) = 0.
Считаем, что рекомбинация атомов происходит на стенках мгновенно и об-
разовавшиеся молекулы мгновенно распределяются по колебательным уров-
ням пропорционально их заселенностям. Фактически это означает, что мо-
лекулы образуются главным образом колебательно-невозбужденные или
слабовозбужденные.
Для нахождения распределения заселенностей по колебательным уров-
ням необходимо решать систему дифференциальных уравнений вида
= {[^r+1, V^V + l v+lnvl
V* V*
+ <^> |Г 2 Pv+L, V^s-lnv+l — 2 ^иД+Х^з71®! +
s=l S=1 J
V*— 1 V*—1 V* V*
+ Г 2 Pv^,v^s+lnv-l — 2 Л1 nsnr JJ + e I 2 vns — 2 (2.80)
L S=O s=0 1 s=o s=0
с начальным условием при t = 0
nv (0) “ /(*>)> v ~ 0,1 , . . ., Г*.
(2.80a)
Здесь nv — концентрация молекул на г-м колебательном уровне, г;* — гра-
ничный колебательный уровень (ближайший к пределу диссоциации, в на-
шем случае у* = 51); N — концентрация молекул; Ne — концентрация
V*
электронов, Ne = aTV, причем а не изменяется во времени; N = 2! п»(0 —
1?=0
закон сохранения числа молекул, он имеет место, естественно, при от-
сутствии диссоциации и рекомбинации и выполняется в данной модели дис-
социации с мгновенной рекомбинацией на стенках. В первой фигурной скоб-
ке записан баланс (г — 7)-процессов, переходы с уровней у ± 1 Z2 у\ во
второй — баланс (у — г?)-обмена, переходы с уровней s + 1 $, г? +1 у]
в третьей — баланс (е — г?)-процессов. <бг?) — усредненное газокинети-
ческое сечение соударений молекул, рассчитанное по формуле —
1/2
. При этом принималось 5n2_n2 = 3,714 A, |xn«-n2 =
= 14тпи PSv+i^v — безразмерные вероятности (г? — г?)-обмена — рассчитывались
в соответствии с теорией Герцфельда [91], с учетом экспериментальных дан-
ных [92] по СО — N2 (у — г?)-обмену; Pv+i,v — безразмерные вероятности
2 (SkT? \
3n2-n2
60
У Z7 Zf 25 25 55 55 25 25 5 /5 /5 25 25 55 55 252555и
!______I ~ I_________I_____J----- I------i------1------'-----—I------1
5 2 2 5 8 5 2 2 5 8 ЕитзВ
Рис. 2.3. Зависимость относигельных заселенностей колебательных уровней состояния
молекулы N2 от номера уровня v при N = 1016 см~3, Тг = 750° К, Те = 1 9В,
а = 10~3 без учета предположения 5 (а) и с учетом предположения 5 (б) на разных време-
нах релаксации
1 — t = ю-7 сек; 2 — 10~5; 3 — 3 -Ю"6; 4 — 2-10~4
(г?—^-процессов, рассчитывались по формулам работы [87], полученным на
основании той же теории; ks.v — коэффициенты скоростей возбуждения и
дезактивации колебательных уровней электронным ударом (в см3-сек~1).
Формулы для расчета Pr+ii и ks,v приведены в работе [90]. Вероятности пря-
мых и обратных процессов в любой момент времени связаны соотношениями
детального равновесия.
Выражение для расчета энергии колебательных уровней взято из рабо-
ты [93].
Расчеты проводились на ЭВМ БЭСМ-6. Получены решения при степенях
ионизации а = 10'3 Ю"6, при Те = 1 эВ. Тг = 750°К, N = 1016 см~3.
При этом начальное распределение пг(0) соответствовало больцмановскому
с Тг = Гг. Время расчета варианта с а = 10-6 составляло около 50 мин
[94]. Все распределения нормированы на N = 1. На рис. 2. 3, а представ
лены результаты расчета распределения заселенностей по колебательным
уровням при а = 10"3 без учета предположения 5 на разных временах ре-
лаксации. При t 2=2 10“7 сек несколько первых уровней имеют Tv = Zr.
Уровни v > 3 -т- 5 заселены с колебательной температурой, которая по
своему значению порядка электронной. С увеличением времени засе-
ленность уровня v = 0 уменьшается, а заселенности остальных увеличива-
ются. При t > (3—5)-10-5 сек распределение заселенностей на уровнях
v > 45 перестает быть квазибольцмановским вследствие процессов (у — Т)-
дезактивации. При t «2-10“4 сек распределение заселенностей стремится
к стационарному. При этом заселенности первых уровней хорошо описы-
ваются распределением Больцмана с Tv = Те. Заселенности уровней от
v ~ 20 до ~ 45 имеют колебательную температуру выше электронной,
что является следствием протекания процессов (у — г)-обмена, так как
решение в предположении отсутствия (у — г?)-обмена дает для этой части
распределения Tv = Те. Заселенности уровней v > 45 обеднены вследствие
(у — ^-дезактивации. Вблизи dnjdt = 0 решение совпадает со стационар-
61
рис. 2.4. То же, что на рис. 2.3, при а — 10-4 без учета предположения 5 (а) и с учетом
предположения 5 (б) на разных временах релаксации I
1 — t = Ю-7 сек; 2 — 10-5; 3 — 2-10-*; 4 — 4-10~4; 5 — 8-10-*
ним с относительной погрешностью 2—3% (погрешность решения стационар-
ной задачи равна 0,01%) для всех уровней. Следует отметить, что ни на
каких временах процесс релаксации нельзя описать распределением Больц-
мана с одинаковой для всех уровней колебательной температурой. На
рис. 2.3, 6 приведены результаты расчета для а = 10-3 с учетом диссоциации и
рекомбинации. Процесс релаксации в этом случае протекает так же, как и
при отсутствии диссоциации для всех уровней, кроме самых верхних. Обед-
нение на них происходит более интенсивно. Стационарное распределение
заселенностей, рассчитанное с учетом предположения 5, согласуется с не-
стационарным при t 2-Ю-4 сек с относительной точностью 1—3%.
На рис. 2.4, а представлены результаты расчета для а = 10-4 без учета
диссоциации и рекомбинации. При малых временах t 10-7 сек заселенно-
сти первых уровней имеют Tv = Тг. Но по сравнению с и = 10-3 на тех же
временах остальные уровни заселены на порядок величины меньше, так как
уменьшились скорости (е — г?)-процессов, а скорости (у — г?)- и (у — ТУ
процессов, видимо, еще невелики. С увеличением времени, например при
t х 2-10“4 сек, влияние (у — Г)-процессов проявляется сильнее, чем при
а = 10”3 (см. рис. 2.3); заселенности уровней v > 45 обеднены (по отноше-
нию к v = 30) сильнее, чем при о = 10“3, причем процесс релаксации про-
исходит медленнее. При Z>2-10“4 сек заселенности уровней v > 45 начинают
аномально увеличиваться. При £ ж 4-10-4 заселенность уровня v = 50
приближается к заселенности уровня v = 45 и при t = (6—8)-10~4 сек (т. е.
вблизи dnjdt = 0) становится «инверсной». Стационарное решение совпа-
дает в этом случае с нестационарным в пределах 20—30%, что значительно
меньше величины «перезаселенности» верхних уровней. Следовательно,
«перезаселенность» верхних уровней не является следствием накопления
ошибок при численном интегрировании.
62
На рис. 2.4, б представлены результаты расчета для того же режима,
но с учетом предположения 5. Релаксация распределения заселенностей
имеет в этом случае несколько иной характер: верхние уровни сильно обед-
нены вследствие процессов диссоциации и дезактивации. Вид распределения
на уровнях до v 30 н-35 существенно не меняется по сравнению с рис. 2.4, а.
Решение стационарной задачи совпадает с решением нестационарной с точ-
ностью 10—15%. Становится понятным происхождение «инверсной засе-
ленности», полученной без учета диссоциации при а = 10~4, и отсутствии ее
при а = 10-3. При а = 10"3 основной вклад в формирование распределе-
ния вносят (е — ^-процессы. В предельном случае наличия только (е — v)-
процессов стационарное распределение при t -> оо должно быть распределе-
нием Больцмана с Tv = Те. С другой стороны, (г — Т^-процессы стремятся
превратить распределение в больцмановское с Tv = Тг. Известно, что
(г? — г?)-обмен характерен тем, что при заданной Tv на первых уровнях г
которая в случае высокой степени ионизации определяется (е — ^-процес-
сами, дает сильный поток на заселение верхних уровней [87]. При а = 10~3
дезактивация колебательных уровней (е — v)- и (г — ^-процессами умень-
шает поток, заселяющий верхние уровни вследствие (г — г?)-обмена, даже
при отсутствии диссоциации. При а = 10-4 скорости (е — г)-процессов
меньше по сравнению с а = 10-3, а (г — ^-дезактивация не обеспечивает
компенсацию потоков, заселяющих верхние уровни вследствие (v — ^-об-
мена. Учет диссоциации ликвидирует перезаселение верхних уровней при
а = 10-4 за счет стока по этому каналу.
Эти результаты объясняют ранее непонятный факт обеднения верхних
уровней при расчете стационарной задачи для г?* = 25 [90]. Фактически рас-
чет там проводился с учетом предположения 5.
Таким образом, в зависимости от способа учета потоков, приходящих
на последний уровень и уходящих с него, вид распределения заселенностей
на верхних уровнях может существенно изменяться.
На рис. 2.5 представлены результаты расчета для степеней ионизации
а = 10“5 — 10“6 с учетом предположения 5. Вследствие процессов (г? — Т)~
дезактивации рост заселенностей верхних уровней происходит очень мед-
ленно по сравнению с уровнями от v 10 до v 35 до того момента, пока не
станет существенным (г — г?)-обмен. На рис. 2.6 изображена зависимость
заселенности предпоследнего уровня v = 50 от времени при различных сте-
пенях ионизации с учетом диссоциации. Для а = 10~6 имеем уменьшение
скорости заселения последнего уровня в некотором интервале времени (см.
рис. 2.6, кривая ^). Решение системы (2.80) для а = 10“6 в предположении
отсутствия (г? — ^-процессов не приводит к такому резкому уменьшению
скорости заселения последнего уровня (см. рис. 2.6, кривая <5). Это озна-
чает, что потоки, заселяющие верхние уровни (вследствие (е — г)-процессов
с нижних уровней), приблизительно равны дезактивирующим потокам при
10-5 сек <Z t < 4-10~3 сек для а = 10-6. Возможность существования по-
добного квазистационарного участка с релаксационной кривой подтвержда-
ется также грубыми оценочными расчетами. Тенденция к появлению такого
участка видна также из релаксационных кривых при других степенях ио-
низации. При t > 4-10~3 сек. т. е. когда (v — г)-процессы становятся значи-
тельными, заселенности верхних уровней резко увеличиваются.
Подобный вид релаксации верхних колебательных уровней может суще-
ственно повлиять на диссоциацию N2 под действием ударов тяжелых частиц.
Если предположить, что заселенности верхних колебательных уровней про-
порциональны количеству диссоциирующих молекул из под действием
ударов тяжелых частиц, то приращение концентрации атомов A[7V] вследст-
вие этого процесса должно иметь подобный вид при низких степенях ио-
низации а.
Этот результат еще раз подтверждает необходимость решения кинети-
ческих уравнений Паули для получения детальной информации о процессе
63
Рис. 2.5. То же, что на рис. 2.3, а —
— 10-5 (а), а = 10“6 (б) с учетом пред-
положения 5, на разных временах ре-
лаксации
1 — t = 10-’ сек; 2 — 10-8; 3 — 10-J;
4 _ ю-з; 5 _ 5-Ю-3;
6—8 -10-3; 7 — 2-Ю-2
Рис. 2.6. Зависимость относительной
заселенности колебательного уровня
v = 50 от времени при N = 1016 сл-3,
Тг = 750° К, Те — 1 эВ и различных
степенях ионизации а
1 — ю-3; 2 — 10~4; 3 — 10-5; 4 — 10~6;
5 — 10“3 (без (v — Т)-процессов)
релаксации отдельных уровней и подчеркивает невозможность описания та-
кой системы обычным релаксационным уравнением.
Время выхода на стационарный режим для а = 10“5 равно 5-10“3 сек,
для а = 10“6 — 2 -10“2сеи. Решение в этих случаях совпадает с решением ста-
ционарной задачи с точностью 10—20%.
В настоящее время нет полной уверенности в надежности данных по ве-
роятностям (е — г;)-, (г? — v)- и (г> — Т) -процессов, особенно для верхних
уровней. Поэтому исследовалась зависимость решения от величины вероят-
ностей переходов. Изменение вероятностей процессов на порядок величины
изменяет время достижения стационарного распределения в 3—5 раз, а за-
селенности верхних уровней изменяются при этом в 7—9 раз. Неопределен-
64
ности в значениях вероятностей переходов могут изменить время достижения
стационарного распределения и повлиять на относительный вклад тех или
иных процессов. Но качественно процесс релаксации, по-видимому, не
изменится.
Для того чтобы оценить возможное влияние диффузии колебательно-воз-
бужденных молекул и дезактивации их на стенках разрядной трубки на рас-
пределение заселенностей колебательных уровней состояния Х1^, были
проведены соответствующие расчеты стационарной задачи (dn^ldt = 0).
В уравнении (2.80) в этом случае появляется дополнительный член.
= 0 = (р=^0),
(2.81)
где у = 10"2 — 10"3—коэффициент аккомодации, Л = Дгруб (сл*)/2,4,
л п (°к)\1/2/2,69-1019 \
D \~300~ / N ) ’
Z)o(760 мм, 300° К) = 0,231 смЧсек.
Расчеты показали, что в интересующем нас диапазоне температур газа
300—3000° К и степеней ионизации а ~ 10~3 — 10~7 влиянием диффузии
можно пренебречь при N > 1016 см~\ если учитываются диссоциация и
мгновенная рекомбинация (если же учитывать диффузию и не учитывать
диссоциацию и рекомбинацию, то изменение в распределении заселеностей
верхних уровней может быть существенным и составлять порядки величины).
В ряде расчетов для а = 10“6 были использованы вероятности (е — г?)-
процессов, усредненные по экспериментальной функции распределения
электронов по энергиям, со средней энергией 8 ~ 3 эВ. При этом распределе-
ние заселенностей молекул по колебательным уровням изменялось не очень
существенно (на нижних уровнях до 10%, на верхних уровнях до 50—70%).
В данных расчетах не учитывались многоквантовый (v — г?)-обмен, коле-
бательно-колебательный обмен между различными электронными состоя-
ниями молекулы N2 и ряд других явлений и процессов, которые могут по-
влиять на результаты расчета. Поэтому влияние этих факторов необходимо
учитывать при сравнении результатов расчета с экспериментом. Несмотря
на это, в стационарном случае сравнение экспериментально определенной
в работе [95] колебательной температуры состояния с расчетной дало
удовлетворительные результаты '90] при а = 10“7 — 10“6 и Тг = 750° К.
В этом случае Tv на первых уровнях меньше Те и увеличивается при уве-
личении а. Для случая а = 10“3 и Тг ~ 5000° К расчетная колебательная
температура состояния в смеси Аг и N2 приблизительно была равна
температуре электронов, что подтвердилось экспериментально [96]. Удов-
летворительное согласие расчетов стационарной задачи с эксперименталь-
ными данными позволяет надеяться на качественно правильный характер
результатов нестационарного расчета в рамках данной модели.
На основании проделанных расчетов показано следующее:
1. Релаксация распределения заселенностей по колебательным уровням
основного электронного состояния X^g молекулы] N2 во времени при на-
личии (е — г>)-процессов не протекает через последовательность квазибольц-
мановских распределений. Это объясняется тем, что роль (е — v)-, (v — v)-,
(v — Т)-процессов при постоянных Те и Тг зависит от а и меняется
с течением времени в процессе релаксации.
2. Рассчитать кинетику "релаксации верхних колебательных уровней и
диссоциации молекул с учетом (е — v)~, (v — v)- (v — 7)-процессов можно
только путем непосредственного решения системы кинетических уравнений.
3 Теоретич. и прикладная плазмохимия
65
Таблица 2.1
Сравнение экспериментальных и расчетных данных параметров тлеющего разряда
в N2 особой чистоты
Р, торр г, ма No, W16 см~3 Ne, 10ю см~3 a-NjN0, 1и-в ЕЭЛ, Эв тг, °к грЭКСП 1 V" °К трасч v" ’ °К
0,7 75 1,13 3,40 3,00 3,10 560 8000 6500
30 1,35 1,30 0,97 2,65 470 6100 5400
10 1,47 0,40 0,27 2,90 430 4580
2,0 75 3,20 3,90 1,22 2,36 610 7900 5320
30 3,75 1,66 0,44 2,5 520 6200 5250
10 4,15 0,58 0,14 2,6 470 4800 4180
4,0 75 5,14 4,14 0,80 2,25 740 7600 5250
30 6,80 1,72 0,25 2,25 560 6100 4440
10 7,75 0,60 0,08 2,5 490 3800 4000
3. Показано, что процессы диссоциации и колебательной релаксации
связаны между собой. Диссоциация может существенно влиять на распре-
деление заселенностей верхних колебательных уровней молекул.
Данные табл. 2.1 иллюстрируют изменение параметров тлеющего разряда
в N2 особой чистоты. 7^сп — колебательная температура основного сос-
тояния определенная из экспериментально измеренных заселенностей
колебательных уровней состояния С3Пи [95]. Для определения
рассчитывалось стационарное распределение заселенностей колебательных
уровней состояния по методике, предложенной в [90].
В ряде случаев сравнение дает удовлетворительное согласие между Т^сп
трасч
И 1 ъ» .
КОЭФФИЦИЕНТ СКОРОСТИ ХИМИЧЕСКОЙ РЕАКЦИИ
Определим теперь статистические коэффициенты (константы!) скорости
химической реакции к как средние скорости (на единицу плотности реагиру-
ющих компонент) по всем доступным (динамически и энергетически) каналам
столкновений. Усреднение должно проводиться по скоростям и внутренним
квантовым состояниям реагирующих компонент. Это значит, что в выраже-
ние к должна входить в явном виде функция распределения реагентов и
продуктов, что позволяет оценить к для неравновесных реакций.
В простейшем случае диссоциации двухатомной молекулы, являющейся
малой добавкой в инертном газовом термостате при учете только колеба-
тельных степеней, имеем для i-го колебательного уровня
оо
К = § Oi (8)]/e/i(e)de. (2.82}
^пор
Для того чтобы отличить ki от к и к (Т), можно бы называть ki уровне-
вым коэффициентом скорости химической реакции.
Суммарный коэффициент скорости химической реакции Л, дающий «свод-
ную» характеристику диссоциации из всех (возбужденных и основного)
уровней, имеет вид
к = 2 «Л» (2.83)
i
где аг- — заселенности соответствующих квантовые уровней.
66
Так как элементарной стадией (элементарным актом) химической реак-
ции является процесс, происходящий с данной молекулой в данном энерге-
тическом состоянии, то только ki является коэффициентом скорости элемен-
тарной стадии химической реакции. Коэффициент Л, определяемый в хими-
ческом кинетическом эксперименте выражением (2.23), всегда является слож-
ной характеристикой, зависящей не только от сг (е), / (с), но и от и лишь
в очень частных случаях может совпадать с кг. Этот случай, вероятнее всего,
реализуется при относительно низких температурах и медленных (предпоч-
тительно термонейтральных) реакциях. В общем же случае к есть, как мы
видим, функция заселенностей отдельных уровней или относительных кон-
центраций молекул в данных энергетических состояниях. Эти концентрации
изменяются в весьма широких пределах. Именно поэтому аррениусова об-
работка экспериментальных данных в таких случаях, как разветвление
цепи через возбужденные молекулы, сильно экзотермические реакции и т. п.,
является физически неправильной, даже если сохраняется 7Пост = ?"кол.
Неправильными являются также выводы об изменении механизма реакции,
осли они основаны только на наличии изломов в аррениусовой зависимо-
сти [11].
Входящие в выражения для к и ki сечение можно во многих случаях
рассчитать или найти из пучковых экспериментов, а функцию распределения
и заселенности уровней также можно определить из эксперимента или для
медленных реакций принять их максвелловыми и больцмановыми. Тогда
можно, зная пороговую энергию реакции, рассчитать к, а затем сравнить с
этим значением величину А, найденную из химического кинетического экс-
перимента. Заметим, что в обычной полуэмпирической химической кинетике
теоретическое и экспериментальное значения к практически никогда не со-
поставляются. Естественно, что как возможные для молекулы химические
реакции, так и их скорости определяются ее строением (а для немономоле-
кулярных реакций также и строением других участников реакции). Понятие
отроения молекулы может быть сформулировано различным образом. Для
наших целей его лучше всего выразить так: строение молекулы, состоящей
из некоторых атомов,— это система ее квантовых уровней и пространствен-
ного распределения составляющих ее частиц.
В классической химической кинетике часто ставилась как одна из важ-
нейших следующая задача: связать «реакционную способность со строением
молекулы». Однако в рамках модели молекулы, применяемой в этой кинети-
ке, эта задача не могла быть решена. Она автоматически решается в нерав-
новесной химической кинетике, так как этого требует характер описания реак-
ций в ней. Это справедливо, конечно, при условии, если неясный термин «ре-
акционная способность» заменить на к той или иной реакции. Развитие
экспериментальной и электронно-вычислительной техники и новых обла-
стей математической физики сделало возможным исследование кинетики
неравновесных химических реакций при рассмотрении и учете реальной
сложной энергетической и пространственной структуры реагирующих мо-
лекул и динамики неупругих соударений.
Остановимся кратко (рассмотрим вопрос только качественно) на роли
многочастичных взаимодействий и внешних полей. Как указывалось выше,
выражение для kt (2.82) справедливо при учете лишь бинарных соударений,
а сечение реакций соответствует полностью некоррелированным парным со-
ударениям. Для плотных нейтральных газов (Na3 ~ 1) и в случае плазмы с
дальнодействующим кулоновым взаимодействием предположение о некор-
релированных парных соударениях непригодно. В этих случаях расчет по
формуле (2.82) может привести к неправильным результатам.
Присутствие внешних полей — электрического, магнитного или грави-
тационного — может влиять на молекулярную структуру и, следовательно,
на характер молекулярных сил сталкивающихся частиц. Это может приве сти
к изменению сечений и функций распределения реагентов и продуктов.
67 3*
Рис. 2.7. ki = /(1/ГП0СТ) для диссоциации О2 при 6-распределениях молекул по колебатель-
ным уровням (1—6), к для Тпост = Ткол (кривая 7)
Рис. 2.8. Зависимость 1g к от 1/Гв; 3 = ^0^
В случае слабых полей сечения меняются слабо, поскольку гамильтониан
взаимодействия остается почти неизменным, зато функция распределения
по поступательным степеням свободы, например для электронов или ионов,
может сильно измениться во внешнем электрическом поле.
Влияние сильных электрических полей более существенно. Если произ-
ведение напряженности поля на характерный молекулярный размер порядка
молекулярной энергии, то поле может существенно изменить электронные
волновые функции молекул и, следовательно, распределение электронов
в молекулах реагентов, молекулярную структуру реагентов и межмолеку-
лярные потенциалы. Поскольку размер молекул измеряется единицами ангст-
рема, а молекулярная энергия соответствует ~ 1 эВ, то для возникновения
подобных эффектов необходимы поля — 108 В/см. Такие большие электри-
ческие поля могут реализоваться в космохимии, в условиях космических
полетов, в пучке лазера большой интенсивности и т. п. При этом сечения
изменяются из-за значительного изменения гамильтониана взаимодействия,
а функции распределения существенно отклоняются от равновесной формы.
Использование уравнений (2.82) и (2.83) может приводить в указанных
случаях к существенным ошибкам, однако недостаток расчетных или экспе-
риментальных данных не позволяет дать количественных оценок указанных
эффектов.
Восстановление к через (рис. 2.7, кривая 7) 1 приводит не к аррениу-
совой прямой, а к более сложной зависимости. При низких температурах,
когда все определяется самым нижним уровнем, кривая 7 в правой части асим-
птотически совпадает с прямой для первого колебательного уровня, а при
более высоких температурах переходит через промежуточный участок к
прямой с энергиями активации соответственно 1,2; 2,05; 1,15 и изменением
значения к приблизительно на порядок. Именно поэтому аррениусова об-
работка экспериментальных данных в таких случаях, как сильно экзотерми-
ческие реакции, разветвление цепи через возбужденные молекулы и т. п.,
является физически неправильной, даже если и сохраняется ТПОст — ^кол
(рис. 2.8) [55].
Покажем теперь, что вид кривой 7 на рис. 2.7 не является следствием
выбранной нами модели диссоциации или расположения уровней. Легко
1 Для расчета кривой 7 (рис. 2.7) мы воспользовались моделью, предложенной в [97^ 98],
называемой часто моделью с преимущественной диссоциацией с верхних уровней.
68
показать [99, 100], что суммарный коэффициент к = 2°4&iHe будет прямой в
i
аррениусовых координатах, даже если все к; в отдельности подчиняются
уравнению Аррениуса, и в системе существует равновесное максвелл-больц-
мановское распределение. Действительно, пусть сечения процессов i дис-
социация таковы, что для коэффициентов кг для каждого уровня (рассмат-
ривая частицы в разных состояниях как частицы разных сортов) можно
написать
ki = bi exp (— г^кТ), (2.84)
где sai- — энергия активации соответствующего уровня. Тогда при больцма-
новской заселенности
У Ь. exp (— ea./fcT) exp (— 8./fcT)
к = 2 ’---v—<—--------------- (2-85)
i уехр(-е{/ЛТ)
i
Обозначим
У Ь.ехр [-(eai + е.)/]
У е. exp (— st) у b. (eai + 8.) exp [ — (eai + 8.)] t
г _ 1 dk
Уехр(—e.t)___________У l>. exp[— (8ai + 8{)] t_* dt
i i
Для того чтобы показать, что выражение (2.84) не будет представляться
прямой в аррениусовых координатах, рассмотрим асимптотики Ea(t). Най-
дем lim Еа (Z), т. е. при кТ 0. Предел первого члена правой части выра-
t—»оо
жения (2.87) при Z—> оо, как легко видеть, равен нулю, так как все члены
обеих сумм стремятся к нулю, кроме первого члена в сумме в знаменателе,
равного единице в силу того, что 8Х = 0.
Предел второго члена правой части выражения (2.87) равен еа1, так как
первые члены в суммах, у которых ех = 0, намного больше остальных.
Физически полученный результат совершенно естествен, так* как при
кТ -> 0 поведение системы определяется только* первым^уровнем.
Найдем lim 7?a(Z), т. е. при кТоо. Легко видеть, что
t->0
26i 2M6ai+ei)
lira Ea(0 = V “ —V7---------• (2-88)
['-и* 1 2 bi
i
В простейшем и весьма грубом допущении, что все одного порядка, имеем
2е«г
• (2.89)
1
Таким образом, показано, что асимптоты Еа (t) при t -> 0 и t оо различны.
В общем случае надо писать выражение для к с учетом наличия двух
температур 2"кол и 7\юст- Обозначим l//cZn0CT = tn. ИкТКОЛ = tk, Igk =
= R(Tn, Th). Тогда выражение (2.85) приводит к соотношению
# (Тп, тк) = 1g к = lg^—1——У bi exp (— eaitn) exp (— е^й). (2.90)
69
Так как Н (Тп, Тк) есть функция двух переменных, можно построить
поверхность И (рис. 2.9), однозначно определяемую уравнением. Равно-
весному случаю Тпост = ^кол соответствует сечение этой поверхности плос-
костью.
* Обычно для определения Еа находят производную d 1g к/d (1/Т). Для
(2.90) такая операция не является однозначной. Для скалярной функции
Рис. 2.9. Схема поверхности
Е 1g МЛюст» Гкол)
R (TniTk)B любой точке определены’производные по направлению s в плос-
кости, касательной к поверхности в данной точке:
dR dR , dR . /о пл \
- = —C0Sa4- ^sma. (2.91)
Обозначим частные производные dR/dtn и dR!dtt. через и Е*. Имеем
У е. ехр (- е.у J Ь. ехр (- eai у ехр (- е.у е.
Е* = 2___________________2______________________________,
У ехр (-8. у У Ъ. ехр (- eai у ехр (- е. у
г г
(2.92)
— = cos a 4- Еа sin a
(2.93)
Рассматривая a как параметр, можно получить семейство поверхностей
fslgk(tn, tk).
В классической (квази)равновесной химической кинетике обычно посту-
лируется: 1) Д7\ = Д7\; 2) Тп = Тк.
В 1951 г. Ван-Чанг и Уленбек [101] разработали формальную кинети-
ческую теорию для многоатомных молекул. В этой теории молекулы в раз-
личных внутренних квантовых состояниях рассматриваются как различные
частицы, а поэтому учитываются переходы между этими состояниями. Эта
теория была развита для расчета переносных свойств. Однако ясно, что пе-
реходы между электронными и колебательными уровями в молекуле можно
рассматривать как простейшие химические реакции. Поэтому теорию [101]
можно распространить на расчет скоростей химических реакций, для кото-
рых молекулы в различных квантовых состояниях не только «различные
частицы», но и обладают различной реакционной способностью [53].
Для бимолекулярной реакции
A(O + B(/)^C(A:) + D(Z)
имеем
-fe) =^(ПгеА|Пвп
' at / ,, it, i
(2-94)
(2.95)
70
где ki- — коэффициент скорости химической реакции частицы в i-м состоя-
нии или, суммируя для всех начальных состояний I, j и всех конечных /c,Z,
получаем
«А = SS^nAinB3-. (2.96)
ij kl
Число соударений в единице объема и за единицу времени, согласно ки-
нетической теории газов,
ПмПъз ЙЙ /а1/в;О« (Е, 0, ф) I Vy | sin QdQ dcp dvAi dvBj, (2.97)
где углы 0 и ф измеряются в системе координат центра масс сталкивающихся
молекул А и В; | | — величина начальной относительной скорости; vAi,
Vbj — векторы скорости молекул А и В. Предполагается, что функции рас-
пределения /Аь /в; удовлетворяют каждая своему уравнению Больцмана.
Интегрируя — дифференциальное сечение по углам 0 и ф, получаем
полное сечение
ан (Е) = g (Е, 0, ф) sin 0 de dtp, (2.98)
(Ji- назовем сечением реакции. Тогда из
= $Й /Аг/вг б (Е, 0, ф) I Vtf I sin 0 <70 dtp c7vA{ dv Bj (2.99)
получим
< = Й (Я) | vi} I (7vAi(7vBj. (2.100)
Покажем, что в случае, когда для реагентов имеет место максвелл-больц-
мановское распределение, выражение (2.100) перейдет в обычную аррениу-
сову константу скорости химической реакции к (Z).
Нормированное максвелл-больцмановское распределение имеет вид [102]
Ar’v’z)=(25r)/2exp(-S-) • <2-101)
Если молекулы А и В имеют распределение скоростей, не зависящее друг
от друга, то
$ = [^w] й ех₽ [~ 2КТ I vii Idv^ dy ^-
(2.102)
С помощью несложных подстановок и перехода к скорости центра масс двух-
молекулярной системы имеем согласно следующим формулам:
М = тл. + тв, И = mAmB/M,
иУ = -jf VAi + ЗГ VBj’ = VAi + Vb;’
где \iij — скорость центра масс, vZj- — относительная скорость. Поскольку
сечение реакции есть функция только кинетической энергии Е = р,г^, то
интегрирование по и по угловой части уг-7- можно отбросить. Тогда
4?(П = 4л(^)3/йехр(-И1;2.да0М(Е)|ру|М1;ф (2.103)
или, переходя к кинетической энергии,
4? (Г) = (яр)-*/* (^)S/’5 (Е) exp (- EdE. (2.104)
О
71
Легко видеть, что интеграл в выражении (2.104) есть преобразование
Лапласа величины Ей™ (Е). Преобразование Лапласа функции <р(£) опреде-
ляется как
оо
g (р) = § ехр (— pt) <р (() dt. (2.105)
о
Следовательно, в выражении (2.104) 1/кТ играет роль Р, а Е — роль t.
Это может быть полезно как [для получения к (Z) по известным (Е), так
и для нахождения (Е), исходя из известного к(!Г) — обратное преобразо-
вание Лапласа.
До сих пор нами не делалось никаких предположений о распределении
реагирующих молекул по внутренним степеням свободы. Допустим теперь,
что имеет место равновесное распределение по этим степеням свободы моле-
кул. Это распределение представляет собой сумму по энергетическим состоя-
ниям eAi, определяемую как
z™ = 3 ехр (- eAi/kE). (2.106)
г
Выражение для числовой плотности частиц в f-м состоянии будет иметь
вид
MAi = (wa/z^1) ехр (— ЕАг/кТ). (2.107)
Подставив последнее выражение в
- «а = 3 S nAinBjk^ (Т), (2.108)
ij kl
получим
3/ 00
- пА = пАпв ! JStAntSSехР(~gAi + евЖ) \с$(£)ехр (- jjW,
I W) zA ZB ij kl S )
(2.109)
где множитель при пАпв есть, очевидно, просто к — коэффициент (констан-
та) скорости химической реакции в предположении термического равнове-
сия для поступательных степеней свободы и для внутренних степеней свобо-
ды реагирующих молекул.
Покажем теперь на простом примере, как, зная (Е), найти /с(Т) с по-
мощью преобразования Лапласа. Предположим, что реагирующие молеку-
лы не имеют внутренней структуры (модель «твердых шаров») или, по край-
ней мере, что внутреннее состояние молекулы не влияет на вероятность
реакции. Тогда выражение (2.104) переходит в выражение
з/ 00
к(Т) = (лц)-* (А) с(£) tfexp (- ^) dE. (2.110)
о
Пусть реакция происходит только, если относительная кинетическая
энергия Е больше некоторого порогового значения ЕПОр и параметр удара
меньше диаметра соударения d. Тогда
|0 для £'<£,ПОр, (2 1111
(nd2 для Е>Еаор. { '
В терминах преобразования Лапласа это означает, что
7 (£')=/ ° ДЛЯ Е < Епор»
( nd2 для Е Епор.
Преобразование такой функции имеет вид
g(l/kT) = л^(ЛТ)2 ехр (-ЯПОРВД1 + (Япор/Щ], (2-112)
72
а следовательно,
к (Г) = 2^(2пкТ1^ exp (- E^IkT) [1 + (£пор//сТ)]. (2.113)
Выражение (2.113) хорошо известно и имеет место в том случае, когда вся
относительная энергия затрачивается в реакции.
НЕКОТОРЫЕ ПРОСТЕЙШИЕ ЧАСТНЫЕ СЛУЧАИ
КОЭФФИЦИЕНТА СКОРОСТИ ХИМИЧЕСКОЙ РЕАКЦИИ
1. Максвелл-больцмановское распределение
В работе [53] показано, что в этом случае к есть лапласово преобразо-
вание Е<з(Е). В рассматриваемом случае /д(г) (va) и (v)B имеют максвел-
лову форму и не зависят от внутренних энергетических состояний. Тогда
/ т* \3/2 / тА?Л\
и
к (Г) = ^Рб(г)/А (vA)/B (vb)<Zva <1vb. (2.115)
Пусть Та = Тв = Т, тогда переход от скоростей в лабораторной системе
координат к относительной скорости и скоростям в системе центра масс осу-
ществляется согласно выражениям:
v=vA—-vb; (mA + mB) V = TnAvA + mBvB, (2.116)
где v — относительная скорость, a V — скорость в системе центра масс,
dvA<ivB = dVdv,
У mA-VA + у msVB = Y И1-’2 + у (mA + тев) V2, (2.117)
где р, = mA/nB/(mA + тв) — приведенная масса. Использовав выражения
(2.114) — (2.117) и проинтегрировав по V, получим
(2^т)/2$с(г;)1?еХР (“S') dV’ (2Л18)
Используя то, что: 1) относительная кинетическая энергия Е =
2) в сферических координатах dV — 4niAZv, имеем
оо
ад Е°(£) ир dE- <2Л16>
О
Введем теперь в рассмотрение принятую в аррениусовой кинетике модель
взаимодействующих твердых сфер. Для этой модели имеем (пороговая энер-
гия реакции 2?Пор)’*
о (Е) = 0 для Е jEnopi
о (Е) = nd2 (1 — Еаор/Е) для Е * Й’пор- (2.120)
Подставив (2.120) в (2.119), получим
оо 1 / 8 \ */2 (* / ад=^Ш \ Н1- (2.121)
^пор
ИЛИ
tOT = «<pQ'’«P( -») > (2.122)
Это аррениусово выражение, в котором предэкспонент имеет вид
[54].
73
2. Химические реакции
при различных температурах Тг и Т2 реагентов
Равновесные распределения реагирующих частиц по скоростям в этом
случае имеют вид:
j ( \ \3/г [ miv2i\
h - (глЛЛ) ехР ’ (2.123)
, / \ / 7П22?2 \8/« / Ш22?22\
^ = (^1 exPb^)-
Для реакций с одного основного квантового уровня имеем
к = $ va f <vi) / (va) dvid v2’ (2.124)
где v = | Vi — v2 | — относительная скорость.
После ряда преобразований и интегрирования имеем
(\8/2О° / г \
-------.у1 * г Р3б(у) exp I-----I dv, (2.125)
Л I * 1 * 2 \ / J X \ *1 * 2 \ /
2л/с — +---) J о \ 2/c -4-----) /
\mi 1 m2 / \ mi 1 m2;
если ввести приведенную массу
ц = m1m2/(ml + m2)
и эффективную «температуру»
^Эф = (^1^2 4“ ^2)»
то получим выражение, аналогичное выражению для однотемпературной сис-
темы, в которое войдет ТЭф вместо настоящей равновесной Т:
<х>
ММ - (- dv. (2.126)
Недостатком формул (2.125), (2.126) является то, что в них не учитывается
необходимое для реакции смешение реагирующих компонент на молекуляр-
ном уровне (кстати, однородность пространства концентраций предположена
во всех приводимых выводах). Процесс смешения газов при различных тем-
пературах, очевидно, происходит при изменении во^времени 7\ и 7\, т. е. в
зависимости от конкретных условий надо учитывать, что за время реакции в
нее вносят вклад процессы при различных Т\ и Т2 и различных TJT2. Это
имеет место при разумно близких тп1 и тп2 и и Т2. Если это не наблюда-
ется, то с достаточным приближением можно вводить в рассмотрение только
температуру высокоэнергетической компоненты; это приближение дает
вполне удовлетворительные результаты при тп2 (как, например, в
задачах о химических реакциях под действием электронного удара, когда
обмен энергий затруднен).
3. Ионизация атомов электронным ударом
В случае низкотемпературной плазмы имеем: Те — температура электро-
нов, Тп — температура нейтральных атомов, причем Те > Тн, а потенциал
ионизации / кТР. Возьмем сечение первой ионизации в пороговой форме:
= 0 для pv2/2 < /, (2.127)
сг(г?) = а [(цг;2/2) — I] для цг;2/2 > /, (2.128)
где Ц = memH/(zne + mH) те, а — параметр сечения, имеющий размер-
ность см2/эрг, тогда из (2.125) получим
к (Те) = 2а (2kT(Jm^I exp (- ///с7е). (2.129)
1.3 за экспоненциального множителя к обычно мал.
74
4. Ионно-молекулярные реакции А+ + В —> С+ + D
Как показано в [103, 104], если принять дальнодействующий потенциал
для радиального движения иона и молекулы в виде
V (г) = - (ae2/2r*) + (ЕЬ2/г2), (2.130)
где а — поляризуемость нейтральной частицы, е — заряд электрона, Е —
начальная относительная кинетическая энергия, b — параметр удара, г —
расстояние ион — нейтральная молекула, тогда
bmax
0(р) = 2л = = (2.131)
•) v \ г /
о
где &тах = (4ае2/цг;2)1/4, а множитель g берется равным единице для экзо-
термической реакции и равным нулю для эндотермической. Согласно выра-
жению (2.131) va(y) есть постоянная величина, а следовательно, к не зави-
сит от температуры. Для к имеем
со
к = 4я(гаУ/! $р36v>ех₽ (-dv=2ng(tF (2Л32)
о
Таким образом: 1) ионно-молекулярные реакции происходят без энер-
гии активации; 2) к не зависит от температуры. Эти реакции имеют обычно
большие сечения (^ 10~14 см2).
5. Колебательно-поступательный обмен энергией
В наиболее простом варианте [105—108] теории колебательно-поступа-
тельного обмена энергией рассматривается гармонический осциллятор \ на
который действует зависящее от времени возмущение, представляющее ре-
зультат соударения с атомом или молекулой. В первом порядке теории та-
кого рода возмущений вероятность возбуждения осциллятора из основного
состояния на первый колебательный уровень имеет вид
оо
Ро^1 (0 = I <Ч>;| 0|<ро>ехр(—гсоО10 di|2, (2.133)
1 —оо
где (Ooi = (Ег — E0)/h; q?i(^) — волновая функция г-го уровня гармониче-
ского осциллятора; V(x, t) — взаимодействие, зависящее от времени, а сле-
довательно, от скорости. Ограничиваясь линейными членами в координатах
гармонического осциллятора и одномерной классической траекторией, оп-
ределяемой экспоненциальным отталкивательным потенциалом [110], полу-
чаем
б0->1 (у) = v2 ехр (— ^L/v), (2.134)
где L — параметр потенциала, v — относительная скорость сталкивающихся
частиц.
Для к^± имеем при трехмерном максвелловом распределении
7 /ГГ\ ( ^toLkT \ък ! 7tkT Г 3 / 16uwoi£ У'/з”] лоел
^1(П = (—р—) (-ejr) ехр[—] (2.135)
ИЛИ
*о->1 (Л - А Т2 ехр (- В/Т'к). (2.136)
Основная зависимость 7b0->i от температуры определяется экспоненциаль-
ным членом, причем имеет место экспоненциальная зависимость от Т^9
многократно и в широком интервале температур подтвержденная экспери-
ментально.
J- Учет ангармоничности см., например, [109].
75
Влияние различного вида неравновесных распределений на к рассмот-
рено, например, в [11]. Как мы неоднократно подчеркивали, неравновесный
характер химических реакций может создавать энергетически обогащенные
(возбужденные и «горячие») и в силу этого особенно реакционноспособные
молекулы, а также позволяет управлять кинетикой реакций (в частности,
в химических и газовых лазерах).
Наконец, в заключение отметим некоторые важные химические процес-
сы (кроме диссоциации), происходящие при взаимодействии тяжелых частиц:
1) ассоциативная ионизация:
N(2D) + О(3Р) -> NO+(XxSg) + е;
2) адиабатическая реакция, при которой энергия связи переходит в ко-
лебательную энергию:
Н]+С12-> HG1* (кол) + С1;
3) трехчастичная рекомбинация:
О (3Р) + О(3Р) + М -> О2(А32*; v = 9, 10) + М
(электронное и колебательное возбуждение).
ПРИНЦИП МИКРОСКОПИЧЕСКОЙ ОБРАТИМОСТИ
И ПРИНЦИП ДЕТАЛЬНОГО РАВНОВЕСИЯ
Как известно, принцип микроскопической обратимости непосредственно
вытекает из симметрии уравнения Шредингера (или классического уравне-
ния Лиувилля) по отношению к обращению времени. Этот принцип связы-
вает сечения прямой и обратной реакций. Принцип детального равновесия
устанавливает статистическое соотношение между константами скорости пря-
мого и обратного процессов в равновесии. Принцип детального равновесия
для коэффициентов скоростей прямой и обратной реакций может быть по-
лучен как следствие равенства скоростей прямой и обратной реакций в рав-
новесии, так и из соотношений микроскопической обратимости с использо-
ванием равновесного максвелл-больцмановского распределения по скоростям
и внутренней энергии.
Опуская математические выкладки, за которыми отсылаем к [111], запи-
шем общие выражения.
1. Принцип микроскопической обратимости:
а) в случае, когда молекулы не обладают внутренним моментом им-
пульса (спиновым, орбитальным или вращательным),
Рнач^ (1Цт1, Рнач» й) = Ркон^ (^V0*» ^кон? ^)> (2.137)
б) в случае, когда молекулы обладают внутренним моментом импульса
при отсутствии внешних электрических или магнитных полей:
Рнач^ (j'H']ml9 ^нач» £i) = Ркон^ А 7* » ^кон» ), (2.138)
где рнач» Ркон — начальный и конечный относительные импульсы; ml —
начальное, ij — конечное состояния; г?нач» ^кон — начальная и конечная от-
носительные скорости; Q — телесный угол, в который рассеивается частица.
Квантовые состояния, отмеченные штрихом, отличаются от состояний, не
отмеченных штрихом, знаком проекции момента импульса на ось z. Заметим,
что квантовомеханические вероятности переходов
®(ijlml\ v, Q) == ^f{mllij\ vf, Q') (2.139)
относятся к определенному начальному и конечному квантовым состоянием,
а сечения выражают долю начального потока частиц, рассеянного в телес-
ном угле AQ. В^силу этого для получения сечений из вероятностей перехода
76
необходимо разделить со на начальный поток и умножить результат на число
конечных состояний в заданном энергетическом интервале и в заданном
диапазоне AQ.
При анализе экспериментальных данных полезно соотношение между
усредненными сечениями, описывающими переход от одного набора вырож-
денных уровней к другому:
РначёгёР ^нач? — Рконётё1® ^кон> (2.140)
которое применимо только при отсутствии внешних полей, когда наборы
состояний I, ] вырождены.
2. Принцип детального равновесия:
к(Т)/к(Т)^К(Т)9
где К (Т) — константа равновесия реакции вида
А + В С + D.
Принцип детального равновесия представляет собой макроскопическое
выражение принципа микроскопической обратимости.
Использование этих принципов основано на том, что:
1) если определено сечение сг, то можно из принципа микроскопической
обратимости найти сечение о» обратной реакции;
2) если известно а и, кроме того, известно, что функции распределения
по поступательным и внутренним степеням свободы являются равновесны-
ми, то можно рассчитать к(Г);
3) рассчитав к(Т), можно по принципу детального равновесия найти
Вд.
Итак, принцип микроскопической обратимости может быть сформулиро-
ван следующим образом: для каждого элементарного химического процесса
существует обратный процесс. Эти два процесса в совокупности образуют
обратимую элементарную химическую реакцию, которая реализует хими-
ческое равновесие между реагентами и продуктами. При равновесии эле-
ментарная реакция должна самосбалансироваться так, что средняя скорость
прямого процесса равна средней скорости обратного процесса.
Микроскопическая обратимость есть основная черта элементарных мо-
лекулярных химических реакций. На молекулярном уровне все химические
взаимодействия и превращения обратимы. Поэтому обратимость вплетена в
наше описание элементарных химических процессов. В то же время в макро-
скопическое описание тех же процессов, если в них одновременно принимает
участие большое число молекул, неотделимо включена необратимость. Знак
t не имеет значения для элементарного химического процесса, но имеет су-
щественное значение в описании эволюции системы в направлении равнове-
сия. Время есть вектор, указывающий направление к равновесию для изо-
лированной многочастичной системы.
Рассмотрим квантовомеханический вывод принципа микроскопической
обратимости [112].
Пусть в системе имеются некоторые энергетические уровни со стацио-
нарными волновыми функциями Тг. При наблюдении этой системы, пренеб-
регая некоторыми взаимодействиями, найдем соответствующую волновую
функцию Tj. Любая группа функций (например, Фг и Ф2), переходы между
которыми мы исследуем из Tj и Tj, образуют полную ортогональную груп-
пу. Энергия, соответствующая Т/, будет е/, а полные волновые (зависящие
от времени) функции, считая для упрощения, что они вещественны и мни-
мость входит только во временной множитель (обобщение не составляет тру-
да), будут ехр(—2nieTtlh). Используя ортогональность волновых функций
7Т
Ф/ и Tj, напишем:
Tn = 2«ivA- (2.141)
J I
Умножая (2.141) соответственно на и TY и интегрируя по всему про-
странству, найдем, предполагая функции Ф нормированными:
&NK — Л^Фк^т, Ьмь~ ТьФ^йт. (2.142)
Заменяя М на К и L на А, получаем
bKN = $ Т^Фкйт = aNK. (2.143)
Рассмотрим систему в момент времени t = 0, находящуюся в состоянии
Ф. Волновая функция состояния этой системы в любой момент времени будет
ур = ^Ф! -р с2Ф2 + + • • • » (2.144)
где сг = 1, с2 = с3 = . . . = 0 при t — 0. Следовательно, при t = 0 имеем
также
Т = Фх = + 612Т2 + . . . (2.145)
Функция ¥ = Т (t) и может быть в любой момент времени выражена
как линейная функция волновых функций вида
Т=ЬиТ1еХр(-^^-) + Ь12Т2еХр(-^^)+ (2.146)
Это выражение переходит в (2.145) при t = 0. В момент времени t значение
с2 будет
с2 = [Ь11Т, ехр (- -^) + 612 Т2ехр (----+ • • • ]dr =
= felxa12 ехр ( - -^) + fe12a22 ехр ( - -^) + • • •, (2.147)
согласно уравнению (2.144). Подобным же образом, если мы начнем при
t = 0 с с2 = 1, с± = с3 = . . . = 0, то для t > 0 получим
С1 = Ь12«1г ехр (-) + ьма21 ехр ( — . (2.148)
Сравнивая (2.147) и (2.148) и приняв во внимание (2.143), видим, что эти
два выражения равны. Другими словами, если мы начнем с состояния Фо
придем к состоянию Ф2 с той же скоростью, с какой придем к состоянию Ф1Г
начав с Ф2, что и требовалось доказать»
ОПРЕДЕЛЕНИЕ ЭНТРОПИИ В НЕРАВНОВЕСНЫХ СОСТОЯНИЯХ
И КИНЕТИЧЕСКОЕ УРАВНЕНИЕ
Макроскопическая теория термодинамически равновесных систем осно-
вана на статистической механике. Однако эта теория может быть построена
чисто макроскопически, без применения микроскопических понятий. Пос-
ледние нужны для более глубокого понимания закономерностей термодина-
мического равновесия, но отнюдь не являются необходимыми для логически
замкнутого развития макроскопической теории.
В термодинамике необратимых процессов дело обстоит существенно ина-
че. В этой теории некоторые интенсивные переменные могут быть определены
для неравновесных состояний, если, по крайней мере, имеет место локальное
термодинамическое равновесие (ЛТР). Однако ЛТР может быть рассмотрено
78
лишь в терминах микроскопического подхода, что означает прежде всего
следующее: для каждого достаточно малого, хотя все еще макроскопического,
объема фазового пространства отклонение распределения вероятностей мик-
росостояний от гиббсова распределения пренебрежимо мало. Там, где это
не выполняется, возможен только статистический подход. Поэтому чисто
макроскопическая теория необратимых процессов, по-видимому, не может
быть построена. Одна из причин этого в том, что, в отличие от механики, обоб-
щение идет от равновесия к динамике, а не наоборот.
В работах [112—114] сделана попытка использовать общую теорию дви-
жения [115] для получения кинетических уравнений, описывающих нерав-
новесную систему. Заметим, что макроскопическую теорию необратимых
процессов можно строить, либо отказавшись вообще от понятия энтропии,
либо распространив это понятие на состояния, произвольно далекие от рав-
новесия. Трудности второго пути, который представляется физически есте-
ственным, возникают прежде всего из того, что такие величины, как темпе-
ратура и химические потенциалы, не определены в сильнонеравновесных
системах (без ЛТР).
В дальнейшем для простоты рассматривается однородная многокомпо-
нентная изолированная система, например химически реагирующая или
релаксирующая, причем имеющие в ней место изменения не возмущают
однородности. Состояние этой системы может быть представлено точкой М
в (п + 2)-мерном^ фазовом пространстве (число частиц п компонент 2
п
Е — внутренняя энергия, v — объем). Все эти переменные определяются
без использования термодинамики: число частиц — счетом (в принципе)
молекул каждого компонента, энергия — сложением всех энергий частиц.
Кинетическое уравнение не предполагает ЛТР и не образует какую бы то
ни было часть макроскопической теории необратимых процессов. Рассмат-
ривается система без гистерезиса (без памяти), которая эволюционирует
к равновесию так, что время не входит явно в кинетические уравнения, ко-
торые инвариантны относительно переносов времени.
Кинетические уравнения будут иметь вид
nt(Nn, N*,. . ., Eq, г0) 0 (i = 1.2, . . . ), (2.149)
причем Ео, Vo строго постоянны в изолированной системе и играют роль
параметров. Так как потоки масс через границу системы не существуют по
предположению, то Mq — const.
Тогда система (2.149) вместе с условием сохранения массы дает уравнения,
определенные в тг-мерном фазовом пространстве:
nt = , . . ., X ; Eq, Vq, M9). (2.150)
Введем координаты относительного отклонения от равновесия —
— H^/vq. Допустим, что система (2.150) имеет асимптотически равновесное
решение, т. е. что каждое решение a (t), начиная с некоторой области вблизи
<z (область притяжения), стремится к а с возрастанием времени
2(°Ч —аД2—>0 при (2.151)
Это предположение физически означает, что уравнение (2.150) описывает
необратимые процессы (или, по крайней мере, процессы, в которых есть не-
обратимая часть), т. е. исключены траектории, которые целиком инвариан-
тны относительно обращения времени. Опуская доказательство существова-
ния такого асимптотически устойчивого равновесного решения уравнения
(2.150), укажем важнейшее следствие из него. Пусть тривиальное решение
уравнения (2.150) асимптотически устойчиво и пусть функция обладает свой-
79
ствами, обеспечивающими существование решения для t 0. Тогда сущест-
вует положительно определенная функция ср:
. 0 < ср (аг) < оо, (2.152)
определенная по всем пространстве, производная по времени которой, взя-
тая вдоль пути реакции, отрицательно определена
71
(2-153)
г=1 i
так что равенство имеет место только в точке = 0. Функция ф на-
зывается функцией Ляпунова кинетического уравнения (2.150).
Введем теперь в качестве координат относительные отклонения коор-
динат от их равновесного значения, тогда по крайней мере вблизи равнове-
сия разность энтропии (равновесно определенной) s = S — So есть отри-
цательно определенная функция переменных at:
5(^) = 5-50<0. (2.154)
Причем равенство имеет место только в точке = 0. Для изолированной
системы второе начало термодинамики требует, чтобы производство энтро-
пии для системы, движущейся к положению равновесия, было положительно
или равно нулю (в равновесии), т. е. положительно определено:
* = (2-155)
Сравнение (2.153) и (2.154) показывает, что энтропия s, насколько она
определена, удовлетворяет дифференциальному уравнению функции Ляпу-
нова для рассматриваемого кинетического уравнения. Следовательно, энтро-
пия есть функция Ляпунова и, согласно свойствам этой функции, определе-
на во всем пространстве для произвольных неравновесных состояний там,
где применимо макроскопическое кинетическое уравнение.
Таким образом, устанавливается существование некоторой функции —
обобщенной энтропии, которая определена во всем пространстве неравновес-
ных состояний и совпадает с обычной энтропией вблизи равновесия; эта функ-
ция отрицательно определена, имеет положительно определенные производ-
ные вдоль пути реакции и может быть выбрана так, чтобы удовлетворялась
теорема Гленсдорфа — Пригожина [116].
Однако, чтобы выбрать функцию энтропии однозначно, движение долж-
но быть определено кинетическим уравнением, так как второе начало термо-
динамики имеет форму неравенства и не может поэтому определить конкрет-
ный вид движения.
Заметим, наконец, что кинетическое уравнение включает только перемен-
ные, определенные за пределами термодинамического равновесия. Построив
подходящую функцию энтропии (функцию Ляпунова), можно, образовав ее
производные, найти все термодинамические силы, включая температуру и
химические потенциалы. Тем самым окажется возможным полное описа-
ние химической реакции исходя из кинетического уравнения.
УРАВНЕНИЕ БОЛЬЦМАНА
Как мы видели, любая химическая реакция представляет собой один из
каналов перераспределения энергии частиц при их взаимодействии. Как и
другие каналы, она имеет статистическую природу и может быть охарактери-
зована своим дифференциальным сечением, что позволяет применить к
80
описанию ее результатов систему уравнений типа больцмановского 1
: ' Ч I' r I!; </'Г - /П <Г. (2 И6>
Обозначения здесь общепринятые (см., например, [117, 118]). Искомая
функция / имеет смысл плотности вероятности нахождения данного чис-
ла частиц в элементе объема dvdx фазового пространства (одночастич-
ного). Поэтому, зная ее, легко вычислить такие характеристики среды, как
те или иные концентрации, плотности потоков частиц и т. д.
Использование кинетических уравнений открывает один из путей пост-
роения микроскопической теории химических превращений. Тем не менее
следует иметь в виду, что уравнение (2.156) обладает рядом важных недос-
татков. Например, оно не описывает, по-видимому, очень быстрые процессы
[119]. Хорошо известно также, что не существует вполне безупречного вывода
его даже в случае одних только упругих столкновений (см., например, [4]);
при этом необходимо сделать предположение о независимости сечения рас-
сеяния частиц от внешнего поля, которое в действительности может оказать-
ся слишком грубым. Кроме того, уравнение Больцмана никак не отражает
возможности тройных соударений; наконец, по существу своему оно явля-
ется классическим, а описываемые им объекты — квантовыми. Тем не менее
система уравнений вида
п
v । - л/да^-; (2.157)
г L г 3=1
с успехом применяется для описания газов, молекулы которых могут хими-
чески реагировать [102, 120]. Обычно с ее помощью получают обобщен-
ные уравнения гидродинамики и кинетические коэффициенты (как в [ 121,122]),
а иногда уравнения типа тех, что применяются в химической кинетике [123].
Система (2.157) позволяет описать реакции класса «две в две», т. е.
Ах -|- А2 Вх -|- В2. (2.158}
Для реакций
Ах + А2 Вх + В2 + В3 (2.159)
необходимо уравнение, содержащее интеграл тройных соударений. Такое
обобщение известно [124—126] 2.
Как было сказано, уравнения (2.156) и (2.157) — классические или, во
всяком случае, «квазиклассические» (если используются квантовые диффе-
ренциальные сечения). Их квантовые аналоги существуют и для одноатомных
[127], и для многоатомных молекул [128]; в последней работе рассмотрено
влияние внешнего поля на процесс соударения частиц. Способ построения
1 Уравнению Больцмана посвящено огромное количество работ (см., например, [117])
Поэтому здесь будет сделано только несколько замечаний, относящихся к проблеме
неравновесной химической кинетики. При выводе уравнения Больцмана предполага-
ется статистическая независимость реагирующих компонент. Без этого предположения
или эквивалентного ему скорость реакции не является прямо пропорциональной произ-
ведению плотностей (концентраций) реагирующих компонент, но включает корреля-
ционную функцию, которая описывает отклонение от статистической независимости.
Нужна или нет корреляционная функция, зависит от начальных условий и отношений
сферы взаимодействия к среднему расстоянию между реагирующими компонентами.
В случае нейтральных частиц в разреженном газе сфера взаимодействия мала по срав-
нению с расстоянием между молекулами, и предположение о статистической независи-
мости хорошо применимо к макроскопической системе. Если обозначить сферу взаимо-
действия через а, среднюю длину свободного пробега через к и числовую (полную)
плотность через /V, то сформулированное выше условие статистической независимости
запишется так:
а/Х Na3 1.
2 Однако оно не используется при решении типичных конкретных задач из-за отсутствия
необходимых данных о распределении импульсов в такой системе после соударения.
81
квантовых кинетических уравнений, описывающих множественные столк-
новения, указан в [129, 130].
К сожалению, ценность такой теории существенно ограничивается сле-
дующими обстоятельствами. Вследствие целого ряда причин решение даже
уравнений (2.156) и (2.157), не говоря уже о более сложных, сопряжено с
громадными трудностями. Чаще всего оно ведется методами Энскога—Чемпе-
на [131, 132] или Трэда [133], разработанными для уравнения (2.157). Пер-
вый из них применим в некоторой «малой» окрестности равновесного распре-
деления (как мы видели, химическая реакция может сильно нарушать его),
второй обладает, возможно, большей общностью, но ни тот, ни другой не
позволяют получить сколько-нибудь высоких приближений из-за быстро
растущего объема вычислений.
Кроме того, в методе Энскога — Чемпена нет критерия, позволяющего
определить, насколько с его помощью можно отойти от равновесного состоя-
ния Ч Поэтому при решении каждой конкретной задачи необходим экспе-
римент, определяющий совпадение расчета с опытными данными, т. е. при-
менимость теории. Это — слабость метода. Неприменимыми в ряде случаев
оказываются и методы линеаризации (см., например, [134]), поскольку взаи-
модействие частиц одного и того же «сорта» — эффект нелинейный.
Все это, естественно, приводит к мысли об использовании вычислитель-
ных машин, посредством которых можно было бы получить те или иные част-
ные зависимости. Но уравнения так сложны, что построить удовлетворитель-
ную схему вычислений оказывается далеко не просто, и в этом направлении
осуществлены лишь отдельные попытки [135, 136], причем химические реак-
ции не рассматривались. Последний пробел можно восполнить, но необхо-
димое для этого время счета окажется, вероятно, весьма большим даже для
самых упрощенных моделей. Возможность использования численных мето-
дов может быть, в частности, обоснована теоремой, доказанной в [159] и
гласящей, что на некотором интервале времени, величину которого можно
оценить, уравнение (2.156) имеет единственное решение и последовательность
приближений, построенная определенным образом, равномерно сходится к
нему.
Однако до настоящего времени в литературе нет решений конкретных
задач химической неравновесной кинетики с помощью уравнения Больцма-
на. Оно использовалось неоднократно лишь для анализа общих особенностей
неравновесных реакций и для получения общих выражений соответствующих
членов в кинетических уравнениях и вида коэффициента скорости химиче-
ских реакций.
Рассмотрим некоторые полезные результаты такого анализа, имеющиеся
в научной литературе.
В работе [139] рассмотрено решение уравнения больцмановского вида
для гомогенных, бимолекулярных, газофазных, изотермических химических
реакций. Уравнение решалось сначала для системы частиц без внутренних
степеней свободы по схеме теории возмущений, аналогичной методу Энскога.
Основанием для этого было допущение, что время релаксации импульса мало
по сравнению с временем релаксации химической реакции. Если ограничить-
ся членом нулевого порядка в разложении в ряд Энскога, то получается
обычное макроскопическое выражение для скорости химической реакции с
коэффициентом скорости, не зависящим от времени и химических особен-
ностей реагентов. Учет членов высших порядков приводит к зависимости
коэффициента скорости от времени и химических параметров системы, а за-
кон действующих масс нарушается.
Такие же результаты были получены для реагирующей системы частиц
с внутренними степенями свободы.
1 В связи с этим попытка Б. В. Алексеева [138] применить метод Энскога вдали от рав-
новесия представляется чрезвычайно интересной.
82
Остановимся кратко на некоторых вопросах, поставленных в работе*
[139], и на развитом в ней математическом формализме. Рассматривается
реакция А + В С + D. Имеем
= 3 Яам + Ла (2.160).
м
и аналогичные уравнения для других частиц. Здесь
Eavl == пАпм \ • • • \ -£- <5ам (р, £1) [/а (Ра) /м (Рм) — /а (Ра) /м (Pm)] dp№dQ.,
' ‘ " (2.161)
Ра = \ " \ сав (р, £1) [«c^d/g (Pc) /d (р') — иа^в/а (Ра) /в (Рв)]
(2.162),
•Еам — интеграл упругих столкновений; Z?a — интеграл столкновений,
приводящих к химической реакции; т?м (t) — зависящая от времени плотность
частиц М; /м(рм, t) — одночастичная функция распределения по импульсам,
нормированная на единицу; сам (р, £2) — дифференциальное сечение упру-
гого рассеяния; р = | Ра — Рм | — | Ра — Рм | — инвариантный модуль от-
носительного импульса; блв (р, £2) — дифференциальное сечение химической
реакции как функция р = | Ра — Рв | и телесного угла рассеяния Q; р, —
приведенная масса сталкивающихся частиц.
В случае наличия внутренних степеней свободы имеем
=Z^AM(i,j)+ 3 1ам(ВД) + 2^а(Ш (2.163),
jM W
где
• • \ -£-<5ам (ij; Р, &) 1/iA/jM — Аа/3м] (2.164)
J г
6am (Jd/ij; Pt. О) [пкА^гм/лгА/гм —Иам«зм/{аЛм]^Рм^»-
(2.165)
Да (kllij) = V • • • \ -P- g* (klliy, P, £1) [пцсПтМт — niAnj^fiAf^] dpsd£l.
(2.166)
Здесь EAm (if) — интеграл столкновений для упругих соударений между
молекулами А в квантовом состоянии i и молекулами М в квантовом
состоянии /; /Ам (kl/if) — то же для неупругих соударений двух молекул А
и М с переходом внутренних состояний (if) и (кГ); R& (kl/if) — то же для со-
ударений, приводящих к химическим реакциям
A(Z) + В(у) С(к) + D(Z). (2.167)
При известных допущениях эти выражения могут быть преобразованы и
проинтегрированы. Полученные результаты в общем малоперспективны,
введение в [139] величины «сродства» мало привлекательно. Эту работу на-
до рассматривать лишь как подготовительную к более интересной работе
[140]. Как уже было отмечено выше, химическая реакция возмущает распре-
деления как по внутренним, так и по поступательным степеням свободы (см.,
например, [141—150]). Развитию этого физически разумного положения и
посвящена работа [140]. Рассматривается реакция А + В С + D или ее
частный случай — изомеризация в присутствии большого избытка инерт-
ного газа M:A + M^tB+M.
Уравнение для А в этой реакции имеет вид
dT~ “ ^f,tnAnB ^r,tncn^ (2.168)
^АМ (Z? j) — *
7am(W) = V”
J Jr
83
где kr^t — коэффициенты скорости химической реакции, представляющие
собой сечения химических реакций, усредненные по неравновесной функции
распределения. В общем kflt и krit зависят от состава реагирующей смеси, и
только при химическом равновесии отношение этих коэффициентов равно
константе равновесия К. Коэффициенты скорости, полученные при исполь-
зовании равновесной функции распределения, к®\ к^ и для них К ~ к^/к^»
В обычных кинетических экспериментах имеем
^ПА
- ~dT = kfnAnB - ктПСПЫ <2-169)
где /с/, кт не зависят от состава и времени; очевидно, что К = к^/кг.
Райс [151] отметил, что для реакции диссоциации и рекомбинации
kr^i^> кг. (2.170)
Для рассматриваемой реакции уравнение (2.170) может быть получено
из уравнения (2.169) и k^t, кг^ переходят в kf и кг, только если один из ком-
понентов реакции имеется в достаточно большом избытке или если скорость
реакции достаточно мала и реакция вызывает только пренебрежимо малые
возмущения распределения по внутренним степеням свободы.
Для расчета коэффициентов скоростей, вообще говоря, требуется знание
сечений химических реакций и неупругих соударений для реагентов и про-
дуктов.
Рассмотрим гомогенную изотермическую смесь идеальных газов, на ко-
торую не действуют внешние силы. Каждый из реагентов и продуктов А, В,
С, D может быть во внутренних квантовых состояниях /, /, к, I.
Эволюция такой системы во времени описывается уравнением (2.163).
Если предположить максвеллово распределение /м по поступательным
степеням свободы вместо /zm и т. д. и проинтегрировать по рд, то получим
С? 72. д жд
—= 2j [^ам (kl/ij) пКАП1№ — &АМ (ij/kl) niAnj№] +
+ 2 кf (ij/kl) niAnjB], (2.171)
где
&am (ij/kl) = Cam (ij/кк, Рам, ^/М^йраФм, (2.172)
kr (kl/ij) = Щ ^-ccd (kl/ij’, PCD, Q) /Ьо) dQ. dpc dpP, (2.173)
kf (ij/kl) = Щ c’ab (ij/kl; Рав, О) /(в0) d£l dpA dpB. (2.174)
Введем которые после некоторого начального периода не
зависят явно от времени, но зависят от времени через гад, ^в, ^d-
Просуммировав по А уравнение (2.172), получим уравнение (2.169),
где теперь будем иметь
kf,t = 2 XiAXjBkf(ij/kl), (2.175)
/Cr,t= 2 XKCXipkr (kl/ij). (2.176)
Эти коэффициенты не зависят явно от времени, но зависят от концентраций.
В том случае, когда имеет место обычное уравнение кинетики (2.170),
эти коэффициенты переходят в kf и кг. Отметим три случая, когда это осу-
ществляется:
84
1) реакция А + В С + D, когда один из компонентов имеется в боль-
шом избытке;
2) реакция А + В С + D, происходящая достаточно медленно, чтобы
пренебрежимо мало возмущать распределение по внутренним степеням
свободы молекул А, В, С, D;
3) реакция А + М В + М, происходящая при большом избытке инерт-
ных молекул М.
В общем же случае экспериментальное определение k^t и kr,t весьма
трудно. Оно требует измерения сечений химических реакций для индиви-
дуальных состояний, а также измерений неравновесных распределений по
внутренним состояниям во время реакции. Некоторый прогресс в измерении
этих величин в различных системах имеется. Однако в настоящее время
разумно точные измерения теоретических коэффициентов скорости пока
осуществимы только для систем, где эти коэффициенты сводятся к кц и к$-
Расчет других коэффициентов скоростей (или констант скоростей) тре-
бует знания и сечений химических реакций, и сечений неупругих соударе-
ний. В^принципе можно получить эти сечения либо из высокоточных экспе-
риментов по рассеянию, либо из теоретических расчетов рассеяния. Если
сечения известны, они могут быть использованы для определения неравно-
весного внутреннего распределения, полученного из решения соответству-
ющего уравнения переноса. Затем можно определить kfit и krtt при соответ-
ственной процедуре усреднения. Знание сечений реакций и равновесного
внутреннего распределения достаточно для расчета к$9 к$-
Итак, даже феноменологические константы скорости зависят от неупру-
гих сечений. Следовательно, изменения этих констант с температурой не обя-
зательно определяются только сечениями химических реакций. Этот факт
очень важен в теории скоростей диссоциации и рекомбинации двухатомных
молекул, где оказалось необходимым рассматривать зависимость коэффи-
циентов (констант) скорости химических реакций от неупругих сечений для
того, чтобы объяснить их аномальную температурную зависимость.
В настоящее время нельзя указать сколько-нибудь сложную химическую
реакцию, кинетика которой была бы достаточно подробно рассчитана с по-
мощью уравнения Больцмана. Некоторые результаты (хотя тоже для про-
стейших или даже переупрощенных случаев) получены при рассмотрении
влияния химической реакции на релаксационные процессы. Этот вопрос
представляет интерес для химической кинетики. В самом деле, при больших
областях реакции необходимо принимать во внимание как влияние релак-
сации на химическую реакцию, так и влияние реакции на релаксационный
процесс (см. стр. 59). Исследование таких принципиальных для кинетики
вопросов при посредстве уравнения Больцмана только начинается.
РЕЛАКСАЦИЯ РЕАГИРУЮЩЕГО ГАЗА
Рассмотрим задачу релаксации реагирующего газа [162]. Для упроще-
ния последующих вычислений рассмотрим газовый термостат, в котором
равномерно распределены молекулы другого газа, которые могут распа-
даться, если при ^соударении энергия относительного движения частиц
превышает некоторое критическое значение £*Пор- (Величины, относящиеся
к термостату, отмечены индексом 1, а те, что характеризуют реагирующий
газ, индексом 2.) Газовая смесь из двух компонентов описывается системой
уравнений Больцмана [162]
dh/dt - Z(/u Л) + I (Л, /2); dfjdt = I (/2,Л) + I (f2, f2), (2.177)
в которой / (/а, /р) (а, Р = 1, 2) представляют собой интегралы столкновений
вида
I (fa, ft) = Gap I Va — Vp | (Д/р — /a/p) dQ. dvp, (2.178)
85
где /а, — нормированные на числовые плотности функции распределения
частиц со скоростями va, vp; /а, — функции распределения после соуда-
рения этих частиц; ба^ — дифференциальные сечения рассеяния; dQ —
элемент телесного угла в пространстве скоростей; dvp = dv\dv^dv$', интегри-
рование распространено на все значения йй и dvp.
Пусть тл = т^т, т2 ~ тсн4, Тг = 1,5-104°К, п± = 101всш~3 и в началь-
ный момент времени Т2 = 1,5-103°К, п2 = 1015 ел*-3*. Так как п2<^п19
пренебрегаем изменением температуры 7\ и столкновениями молекул СН4
между собой, а распад метана считаем необратимым:
СН4 + Аг ->СН3 + Н + Аг. (2.179)
Следовательно, функция /j не меняется со временем, I (Д, /4) = I
/2) = I (/2> /2) = 0, а для /2 получаем уравнение
df2/dt = I (f2, /х). (2.180)
Отметим, что линеаризация проведена только для сокращения объема
вычислений; разработанные методы [158, 159] позволяют интегрировать,
нелинейные системы типа (2.117) при условии конечности полных сечений
рассеяния [153—155].
Интеграл I (/2, /4) описывает как упругие, так и неупругие процессы
столкновения. Для них примем следующую схему: пусть упругое рассеяние
характеризуется моделью максвелловских частиц (ограниченного радиуса),
а неупругое — моделью твердых шаров (см., например, [156]). Для того
чтобы эффект реакции оказался заметным, полные сечения обоих каналов,
считаем одинаковым (—10~15 см2). Тогда
I (j2, = $ K+f^dQ. dvr - J K-f^dil dvx, (2.181)'
где A""— ядро максвелловского оператора столкновений [152], если 1/21ы | v2 —
— vi |2 ^пор или ядро оператора столкновений для модели упругих шаров;
[153], если V2p, | v2 — vx |2 -Епор; К+ — ядро максвелловского оператора
столкновений, если 1/2jjl | v2 — v4 |2 7?пор; К+ = 0, если V2p, | v2 — vT |2 >
^пор (И — приведенная масса молекул СН4 и Аг); пренебрегаем также*
продуктами реакции.
Поскольку
/1 = С2 ехр {—рг I vx I2}, /2|(=о = с2ехр{—p2|v2|2},
Са = Па (Ра/л)3/2, ра = ma/2kTa,
целесообразно провести замену неизвестной функции (см. также [158])
/2 = gexp{— Ъ\ v2|2}, b = (2.183)
Получаем уравнения относительно g***
dgfdt = § Ktg'aSl dvx — g^K^d£ldv19 (2.184)
g|(=0 = g0^/2|(=oexp{b|v2|2}, ^scx^expt-Pilvx]2}. (2.185)
* Обозначения общепринятые: — масса частицы, na — числовая плотность, Та —
температура,
** Следовательно, использована обычная в химической кинетике упрощенная схема про-
цесса рассеяния [157]. В действительности сечение упругого процесса отлично от нуля
и при V2 ц 1222 — г?11 2 > Е , ио если в эт°й области оно существенно меньше сечения
реакции, им можно пренебречь. Кроме того, интересно проследить влияние отдельных
факторов (в данном случае — наличия неупругого рассеяния) на форму функции рас-
пределения /2. Точные оценки потребуют усложнения модели (введения квантовых
уровней и т. п.).
*** Нормировка функций /х и /2 обычная: fidvi = m (i), fzdvz = п2 (t). Вспомогательная
функция g может быть ненормируемой.
86
Задаче (2.184) — (2.185) соответствует интегральное уравнение [117]
t
g = Ag = ехр {— a (v2) (t — т)} K*eg'd£l dvx^dx + g0 ехр {— a (v2) t},
о
a (v2) = K^d£l c/V], (2.186)
в котором оператор Ь_т производит замену аргумента t параметром
интегрирования т. Методами функционального анализа можно показать, что
оно 1 при любом t оо имеет единственное непрерывное решение, к которому
равномерно сходится последовательность
Кч==А^}’ Л: = 0, 1, 2, . . . (2.186а)
Легко оценить, в каких пределах ук отличается от искомой функции
(158].
Рис. 2.10. Функция распре-
деления молекул CEU
по энергиям
1 — t = 0;
2 — t = 1 «IO-10 сек;
з 2*1O-10;
4 — 4-10-10;
5 — 5-Ю-10;
6 _ 6-Ю-10;
7 — 1 -10-9;
<6 — максвелловская функция
с температурой термостата
Чтобы построить решение уравнения (2.186), использована последова-
тельность (2.186а), достаточно высокий член ее отождествлен с функцией g.
За нулевое приближение взято
Уо = Щ (р (0/л)^ ехр {—р (0| v2 | 2},
Р (0 = р2 + (р' - рг) t/t0, р's mi/2kT1,
причем t0 = 10~9 сек, что примерно равно длительности быстрой стадии
процесса релаксации. Вычисление интегралов производилось методом Монте-
Карло [160] с точностью ±2%. Радиус сферы, в которой проведено интегри-
рование по компонентам скорости vx, выбран равным 1,2-106 см!сек, шаг —
4-104 см!сек\ аппроксимация функций — линейная. Вычислено 18 при-
ближений, поскольку последние три почти не отличаются друг от друга,
18-е принято за решение уравнения (2.186). Общее время счета на машине
БЭСМ-6 составило —40 мин.
На рис. 2.10 представлена временная эволюция функции Ф (t, v2) =
== 4 л | v2 |2 /2 (С v2). Пунктиром показано максвелловское распределение
с температурой термостата, к которому релаксировала бы функция Ф (t, v2)
в отсутствие канала неупругого рассеяния. Отчетливо различаются две
стадии процесса: быстрая (заканчивается к моменту —6-10~10 сек), во время
которой СН4 нагрелся, и медленная, которая закончится полным разложе-
* Вернее, аналогичное уравнение, у которого ядро К~ переходит в К+ непрерывным
образом; если этот переход осуществляется в достаточно малой области V2 р | v2 — vx |2
— ^пор> такая подмена уравнения не будет ощущаться при дискретном счете на элект-
ронной машине.
87
нием метана. Влияние реакции, как и следовало ожидать, сказывается в зна-
чительном уменьшении функции Ф (t, v2) при больших ] v2 | по сравнению
с максвелловской.
Расчет показывает, что на быстром этапе реакции разложению подвер-
гается около 10% первоначального количества метана. Отличие функции
распределения /2 от максвелловской на длинном этапе влечет за собой силь-
ное отклонение коэффициента скорости реакции от равновесного значения
его [162].
КОЭФФИЦИЕНТЫ СКОРОСТИ ХИМИЧЕСКОЙ РЕАКЦИИ
В РЕЛАКСИРУЮЩЕМ ГАЗЕ
Важной характеристикой химической реакции является его коэффициент
скорости к [И, 161]. Для реакции
А + В -> продукты (2-187)
его можно записать как
кС <3АВ I VA — Vb I WvAJvB.
(2.188)
Здесь va, vb — скорости поступательного движения частиц компонент А
и В, Fa и Fb — нормированные на единицу функции распределения; <зАв —
полное сечение реакции, которое принято равным
<5ав =
0’ V^AB I VA VB |2 ^пор’
i’ V^abI VA- 'вГ>Епор
(2.189)
(Иав — приведенная масса частиц). Величина оав* а следовательно и к, вы-
ражена в относительных единицах.
Рис. 2.11. Функции распреде-
ления веществ А и В [12] в
момент времени t = 0
1 — функции Фа;
2 — Фв; К — энергия поступатель-
ного движения частиц
Рис. 2.12. Зависимость от вре-
мени коэффициента к при раз-
личных законах релаксации
распределения (см. рис. 2.11, а)
и начальных условиях
Выясним зависимость кс от времени, полагая, что функции Fa и Fb изме-
няются по заданному закону. Рассмотрим [162] следующие варианты:
1. FA представляет собой максвелловскую функцию со средней энергией
Еа — ОДбЕпор, массы молекул тд и тпв равны, а функция Fb |ь=о соответствует
двухтемпературной смеси, т. е. задана как сумма максвелловских распреде-
лений со средними энергиями Е±в я Е%в, причем + ^2В = Еа (рис.
88
2.11, a); E±B, E^r — значения средних энергий при t = 0. Величины кс при
различных EiB и Е%в следующие:
Е°1В 0,10 0,075 0,05 0,04 0,03
Е°2в 0,20 0,225 0,25 0,26 0,27
кс 1,20 1,41 1,58 1,72 2,02
Допустим, что Fa. не меняется во времени, a Fb релаксирует по закону
Fb (0 = ^ове’1/Тр + Л (1 — ^'/Тр) (2.190)
ftpвремя релаксации; Fob Fb |г==0; А — максвелловское распределение
с Еа=£в). Значения кс, вычисленные для моментов времени t=0, х/20 тр,...
1тр, показаны на рис. 2.12 сплошной линией. Пунктиром на том же
рисунке дано изменение кс, когда средние энергии компонент Fb меняются
согласно соотношениям
£хв (0 = Е°хв + (Еа + ЯхвУ/Тр (х = 1, 2). (2.191)
2. Foa = Fa]i=o, Ров = Fb |/=о — максвелловские функции с Еа =
= 0,075i?nop, Ев = 0,2251?пор (рис. 2.11, б). Равновесию такой системы
(без химических реакций) соответствует, как и в первом случае, Еа = Ев =
= 0,15 £пор. Предположим, что релаксационный процесс идет так:
F. (t) = Foie~r‘^ + Л (1 - <Г1/тр) (i = А, В) (2.192)
или описывается соотношением (2.191). Результаты расчетов показаны на
рис. 2.12 (пунктирная кривая соответствует ра-
венствам (2.191), сплошная—равенству (2.192)).
3. Соотношение масс частиц возьмем
тпа/шв = 1-103, FA — максвелловское рас-
пределение, Fb — 6-образное, релаксирующее
по закону (2.190). Коэффициенты кс вычис-
лены для Ев = 0,8Епор, 1,2£пор, 2jEnop
(рис. 2.13).
На рис. 2.13 показано изменение со вре-
менем коэффициента скорости реакции
(2.179), которой соответствует вычисленная
выше функция Ф (£, v2).
Закономерности поведения кс показы-
вают, что данная химическая реакция
идет с существенно различной скоростью в
зависимости от того, как далеко от равно-
весия состояние системы и каким образом
оно меняется со временем по степеням сво-
боды, не связанным с химическими реакция-
ми. По мере приближения к равновесию зна-
чения кс могут и падать, и возраставшем.,
например, рис. 2.13, б). Предложенные мо-
дельные законы изменения функций распре-
деления качественно описывают условия
протекания химического процесса (ср. кри-
вую 1, рис. 2.13, б и рис. 2.13, в).
Рис. 2.13. Эволюция коэф фициента кс при различ-
ных законах релаксации распределений (см. рис.
2.11, б) (а); для газа, состоящего из двух видов ча-
стиц сильно разнящихся масс (б); изменение коэф-
фициента скорости ре акции (3) (в)
ц-кв|= 0,8 -L1.2 Е^-> К- 2Епор;[1б2]
89
РАДИКАЛЬНЫЕ И ЦЕПНЫЕ РЕАКЦИИ
ПРИ ВЫСОКИХ ЭНЕРГИЯХ (ТЕМПЕРАТУРАХ)
С ростом температуры длина цепи падает [163] и цепная реакция пере-
ходит в конце концов при плазмохимических температурах в свободноради-
кальную. Концентрация атомов и радикалов растет с температурой быстрее,
чем это можно было бы ожидать исходя из больцмановского множителя
ехр (—D/kT), где D — энергия диссоциации. Так, например, степень дис-
социации О2 при 3000° К составляет 1,4%, в то время как ехр (—D/kT) —
— 10~9 и 15% при 4000° К, когда ехр (—D/kT)— 10~6. При неравновесных
условиях концентрации атомов и радикалов могут значительно превосхо-
дить их равновесные концентрации. Так, например, аномальное возбуждение
ОН (2S+, v' = 2) в кислородно-водородных пламенах указывает на присут-
ствии О и (или) Н атомов в количестве, превышающем их равновесные кон-
центрации [164—166]. Такие сверхравновесные концентрации атомов и (или)
радикалов приводят к реакциям, не имеющим место в обычном пиролизе или
окислении. Так может возрастать вероятность реакций рекомбинации атом—
атом, радикал—радикал, хотя этот эффект ослабляется тем, что равновесие
при высоких энергиях смещено в сторону диссоциации. Большое значение
могут иметь, однако, реакции обмена радикалов и атомов и радикалов вида
А* + ВС —> АВ + С, например, [167]:
СН + ОН ->СО + Н,
С2 + ОН ->СН + СО,
NH + NH ->N2 + Н2,
NH + ОН ->NO + Н2.
УЧЕТ ВНУТРЕННИХ СТЕПЕНЕЙ СВОБОДЫ МОЛЕКУЛЫ
Одним из наиболее очевидных недостатков упрощенной теории столкно-
вений, применяемой в химической кинетике, является рассмотрение соуда-
ряющихся частиц как твердых сфер. С этой точки зрения лучше обстоит дело
в теории переходного состояния [9], однако эта теория предполагает некое
равновесие между реагентами и активированным комплексом. Это допущение
не всегда верно в равновесной химической кинетике и почти всегда неверно
в кинетике неравновесной. Поэтому в условиях неравновесной кинетики
надо пользоваться подходом теории столкновений, усложнив, однако, модель
молекулы так, чтобы можно было учесть внутренние степени свободы.
Впервые Видом и Бауер в 1953 г. [168] развили теорию процессов пере-
носа энергии при соударении колебательной-возбужденной молекулы и моле-
кулы в основном состоянии (на примере СО2 и Н2О), причем нашли, что часть
колебательной энергии в этом случае переходит в поступательную. Дальней-
шие исследования в этом направлении распадаются на две группы: в одной
не рассматриваются потенциальные поверхности, а в другой исследуются
кинематические условия реакций на гипотетической поверхности. В работе
[169] Элиасон и Гиршфельдер показали, что если в начальный момент вре-
мени имеет место максвелл-больцмановское распределение для реагирую-
щих молекул, то уравнение для скорости реакции в равновесной кинетике
получается обычного типа, даже если учитывать эффекты внутренних сте-
пеней свободы. Они показали также, что выражение для скорости химиче-
ской реакции, полученное Эйрингом [170] из теории переходного состояния,
может быть выведено из теории столкновений, хотя и при введении дополни-
тельных допущений.
Кинематические расчеты могут быть как классическими, так и квантово-
механическими при рассмотрении движения на потенциальной поверхности.
Классическое приближение применимо там, где можно пренебречь туннели-
90
рованием, т. е., например, при высоких температурах (энергиях), а кванто-
вомеханические подходы необходимы при низких энергиях и узких потен-
циальных барьерах для реакций. Кинематические расчеты во многих
случаях дают ясное понимание распределения энергии между продуктами
реакции.
Первыми, кто применил для таких расчетов ЭВМ, были Уолл, Хиллер
и Мазур [171, 172]. Одними из наиболее интересных расчетов, выполненных
с помощью ЭВМ, являются расчеты Карплюса, Портера, Шармы [173] для
реакции Н 4- Н2. Ими систематически исследовано влияние на полное се-
чение реакции относительной скорости сближения реагентов, колебательных
и вращательных квантовых уровней молекулы водорода. Они показали, что
сечение реакции монотонно возрастает с ростом относительной скорости за
порогом реакции и возрастает также с ростом колебательного и вращатель-
ного квантового числа [181].
ЭЛЕМЕНТАРНЫЕ РЕАКЦИИ ВОЗБУЖДЕННЫХ ЧАСТИЦ
Следующие типы элементарных реакций возбужденных частиц подробно
изучались экспериментально и теоретически (см. гл. 3, стр. 113):
1. Спонтанная диссоциация
АВ* -^А + В,
тде А, В — атомы, радикалы, молекулы. К этому типу реакций относится
предиссоциация, например диссоциация электронно-возбужденньгхрлефинов:
С2н; С2Н2 + Н2; Сзн; С3Н5 + Н.
2. Спонтанная изомеризация
АВ* -^ВА.
Некоторые такого типа реакции вызываются фотохимическими или тер-
мическими воздействиями через начальное электронное возбуждение.
Рассмотренные реакции исчерпывают возможные типы мономолекулярных
реакций. Следующие ниже элементарные реакции — бимолекулярные.
3. Перенос электронной энергии
А* + В -^А + В*.
К этому типу относится сенсибилизированная флуоресценция, напримерв
Hg m + Ti е₽1/2) ->Hges0) + ti*.i
4. Тушение соударением без какой-либо реакции
А* + В -^А + В,
например, тушение водородом электронно-возбужденного натрия
Na (2Р) + H2fS+) - Na fS) + Н2 (^).
5. Тушение, сопровождающееся диссоциацией молекулы и свободными
фрагментами реакции:
А* + ВС -^А + В + С,
например,
Hg(3Px) + н2 +H2es0) + н + н.
6. Тушение с диссоциацией и образованием новой молекулы
А* + ВС —>-АВ + С,
91
например,
Cd^PJ + На ->CdH(2S+) + Н.
7. Тушение с диссоциацией молекулы, с которой снимается возбуждение.
АВ* + С ->А + В + С,
например, индуцированная предиссоциация
OH(2S+) + Н2 ->О(3Р) + H(2S) + н2.
8. Тушение с изомеризацией молекулы, с которой снимается возбуждение,
АВ* + С ->ВА + С,
например,
1-С4Н8* + 1—С4Н8 ->2—С4Н8 + 1 - С4Н8.
9. Ассоциация возбужденной молекулы с соударяющейся при наличии
или отсутствии третьего тела
А* + В(+М) ->АВ(+М),
например,
O0D) + CO0SJ) + М ->CO2(xSg) + М.
МОНОМОЛЕКУЛЯРНЫЕ РЕАКЦИИ ВОЗБУЖДЕННЫХ ЧАСТИЦ
Для того чтобы мономолекулярная реакция произошла, молекула дол-
жна обладать энергией, достаточной для перестройки химических связей.
Эта энергия может быть получена за счет фотохимических или радиационно-
химических процессов, за счет электронного удара или соударений с другими
частицами (молекулами, атомами, радикалами) или за счет избытка энергии,
выделяющейся при экзотермических химических реакциях (химическая
активация). Линдеманн [174] предположил, что существенное значение для
мономолекулярных реакций, описываемых кинетическим уравнением пер-
вого порядка
—[А] = к [А], (2.193)
имеют соударения, а Хиншельвуд [175] показал, что если молекулярные
соударения могут активировать или дезактивировать реагирующую моле-
кулу, то такая активация может быть согласована с кинетикой по первому
порядку. Последующие теории мономолекулярных реакций позволили
учесть внутреннее возбуждение реагирующей молекулы и выявить влияние
его на скорость мономолекулярной реакции. Кассель [176] и Райс и Рамспер-
гер [177] распространили теорию Хиншельвуда на мономолекулярные реак-
ции возбужденных молекул, предположив, что энергия может свободно
переходить между несколькими или всеми нормальными модами возбужде-
ний в молекуле (теория РРК). Напротив, Слетер [178] предположил, что
потоки энергии внутри активированной молекулы не имеют места, а скорость
мономолекулярной реакции определяется наложением амплитуд строго
гармонических осцилляторов, составляющих молекулу, причем осуществле-
ние реакции зависит от фазовых соотношений. В теории переходного ком-
плекса при рассмотрении мономолекулярных реакций встречаются следу-
ющие трудности: 1) неясна возможность допущения даже приближенного
равновесия при низких давлениях; 2) неясно, что такое переходное состояние
в случае мономолекулярной реакции. Однако Маркусу [179, 180] удалось
все же развить довольно сложную квантовую теорию мономолекулярной
реакции, использовав теорию переходного состояния для обобщения РРК-
метода.
92
Напомним кратко основные выражения теории Линдеманна — Хиншель-
вуда [174, 175]:
fci *
А + А —> А + А активация,
* *2
А 4- А —> А + А дезактивация,
* #3
А —>продукты мономолекулярная реакция.
Решение стационарной задачи для А* дает
[А*] = ЛДАЖ [А] + к3 (2.194)
—[А] = к3 [А*] = ВДАР/Ш! + къ, (2.195>
при высоких давлениях к2 [А] к3 и из (2.195) получим
—[А] = кгк3 [A]/fc2, (2.196)
т. е. кинетический первый порядок реакций; при низких давлениях к2 [A]
<й3 и
—[А] = &JA]2, (2.197)
т. е. кинетический второй порядок.
Эксперимент удовлетворительно согласуется с этими выводами.
Пусть теперь к3 = к3 (Е) и существует такое активное состояние А^
которое имеет не равную нулю вероятность превратиться при соударении
в реакционноспособное состояние А$. Тогда образование А* опишется так:
[А*] = кс [A] [A1J, (2.198)
где кс = zaa3 [А][Аа1 = 4з2 (кТ/пр)1!* и называется коэффициентом скорости
соударения. Пусть, далее, сечение для активации и дезактивации А* одина-
ково, тогда скорость дезактивации
[Аг] дезакт == кс [А$] [А], (2.199)
а скорость мономолекулярного распада
-[А^реак = *1 [All, (2.200)
где к3 расщепилось на кг3 для каждого уровня энергии Et молекулы А^«
Решение стационарного уравнения для [AiJ дает
[AJ = кс [А] [А1]/(Агс [А] + Tel),
и, суммируя, получим
(2-202)
г i *с [А] + кг3
[Ас] может быть рассчитана исходя из распределения Максвелла — Больц-
мана для случая термического равновесия и равна
ГАЬ= Щ ^ехр(-£{/ЯГ)
5\.ехр(-£.//?:г) •
3
Для высоких давлений кг3 кс [А] получим из уравнения (2.202), приняв
во внимание (2.193)
ул|^ехр(-Е./Я7)
- [А] = [А] -4=-------------- ,
3
(2.201)
(2.203)
(2.204)
93
<а при низких давлениях &СА имеем
— [А] = /сс [А]2
7\ехр (-E-IRT)
г______________
yg.^V(-~E.IRT)
3
(2.205)
Таким образом, и при учете внутренних возбуждений и потоков по кван-
товым уровням мы получили в обоих случаях тот же кинетический поря-
док, что и в простой теории Линдемана — Хиншельвуда.
В заключение заметим, что слабость изложенных выше рассуждений за-
ключается в том, что предположение термического равновесия, которое
может быть оправдано при высоких давлениях, не выполняется при низких
давлениях. В самом деле, при этих условиях концентрация определяется
мономолекулярным распадом, а не столкновительной дезактивацией, и
если Аз тем больше, чем выше возбуждение молекулы, то скорость убирания
высокоэнергетических молекул может оказаться больше, чем скорость их
генерации, необходимой для поддержания равновесной концентрации.
НЕОБХОДИМЫЕ ЗАМЕЧАНИЯ
О СТОЛКНОВИТЕЛЬНОЙ ДЕЗАКТИВАЦИИ
Ввиду важности дезактивирующих соударений вида А + В и влияния
этого процесса на собственно химическую кинетику приведем некоторые
общие соображения.
Уточним, при каких условиях происходит неадиабатический перенос
энергии при соударении. Это будет иметь место в том случае, если возмущение
квантовых состояний соударяющихся систем, как это следует из соотно-
шения неопределенности, того же порядка, что и величина разности энергий
е между двумя соседними состояниями системы. Эффективное девозбуждение,
следовательно, будет иметь место, если 8тс0уД ~ где тСОуД — время со-
ударения. Оно обычно полагается равным a/v, где а — характеристическое
расстояние взаимодействия, v — средняя скорость столкновения. Итак,
га | vh ж 1. (2.206)
При обычных условиях (комнатная температура, а 1А) это означает, что
е должна быть меньше, чем (10—20) см"1 для эффективного переноса энергии.
В общем, следовательно, электронно-поступательный обмен энергией
ж 10 000 слГ1), колебательно-поступательный перенос энергии (8 ж
» 200—3000 оГ1) неэффективны, в то время как вращательно-поступатель-
ный перенос (Н2 : 8 ж 60 см~\ 12: 8 х 0,04 см'1) или резонансный колеба-
тельно-колебательный перенос очень эффективны.
Для оценки эффективности дезактивирующих столкновений введем вели-
чину и10 — среднее число соударений, необходимое чтобы дезактивировать
первый колебательный уровень. Тогда для z10 имеем: 1) z10 возрастает
с ростом энергии осциллятора; 2) и10 уменьшается с возрастанием темпера-
туры; 3) более легкие партнеры, вообще говоря, более эффективны, чем
более тяжелые; 4) обычно трехатомные молекулы релаксируют быстрее, чем
двухатомные; нелинейные трехатомные релаксируют быстрее, чем линейные,
что и показано ниже:
Соединение ............................ СО2 COS
Наинизший колебательный квант, смт1 672 527
zio (при комнатной температуре) . . • 5-104 104
SO2 Н2О
519 1595
5-102 102
5) большие молекулы релаксируют вообще быстрее. В гомологическом ряду
CnH2n z10 уменьшается с возрастанием п\ 6) примеси могут иметь большое
94
влияние, уменьшая время релаксации, например, Н20, спирт и т. п.
Для иллюстрации влияния примесей на процесс переноса энергии в со-
ударении молекул приведем некоторые данные об этом процессе в метане
при отсутствии и наличии примесей. В работе [182] рассмотрен перенос
энергии в метане, который возбуждался по моде¥3 (2497,9 см"1) с помощью
Не—Ne-лазера. Часть возбуждения быстро передавалась на v4 и измеря-
лась инфракрасная флуоресценция v3 и v4. Так как время жизни радиа-
ционных переходов (0,04 и 0,4 сек) больше, чем время жизни нерадиацион-
ных переходов, интенсивность флуоресценции пропорциональна модули-
рованным заселенностям возбужденных колебательных состояний.
Рис. 2.14. Диаграмма уров-
ней энергии СН4 и О2
Кроме v — ^-переноса энергии в СН4, авторы [182] изучили обмен энер-
гией между О2 и СН4, что представляет хороший пример влияния малых
добавок многоатомных молекул на времена релаксации. На рис. 2.14 при-
ведена диаграмма уровней энергии СН4 и О2. Обмен v — v для СН4—СН4
происходит быстро. Переход v3 v4 требует около 60 соударений. Около
440 соударений требуется для переноса одного кванта колебательного воз-
буждения от О2 к СН4. Для того чтобы перенести один колебательный квант
при v — i-переходе в чистом О2 при комнатной температуре, требуется ^>107
соударений. Тот же процесс в СН4 происходит быстрее — за ~104 соударе-
ний. Все это объясняет сильное влияние СН4 на релаксацию О2.
Имеются и другие вещества еще более эффективные, чем СН4 в О2, как
это видно из приведенных ниже данных, показывающих времена колебатель-
ной дезактивации СО2 различными партнерами по соударению М (а — эф-
фективность по отношению к соударению СО2—СО2):
М СО2 Не Н2 N2 СО Н2О СНзОН С6Н5СНз
а 1 20 200 40 223 103 2-103 3,4-103
РЕАКЦИИ РЕКОМБИНАЦИИ
ТРЕТЬЕГО КИНЕТИЧЕСКОГО ПОРЯДКА
Рассмотрим реакции рекомбинации вида
А+В+М->АВ+М (2.207)
или в частном случае
А + А + М А2 + М. (2.207а)
В случае рекомбинации двух атомов или радикалов возникают частицы,
обладающие всей энергией образовавшейся новой связи и называемые по-
95
этому «химически активированными», они могут спонтанно мономолекуляр*
но диссоциировать еще до стабилизации. Диссоциация происходит за одно
колебание, пока энергия остается только на одной связи. Стабилизация
колебательно-возбужденной молекулы колебательным излучательным про-
цессом крайне маловероятна, а в случае гомоядерной двухатомной молекулы
невозможна. Даже в том.случае, если молекула образовалась в электронно-
колебательном возбужденном состоянии, процесс электронного излучения
не может помочь стабилизации молекулы, так как электронные времена
жизни по отношению к излучению ~10~8 сек, а колебательный период
~1(Г13 сек. Поэтому наиболее вероятным процессом оказывается стабилизация
столкновением.
Однако если в процессе рекомбинации образуется много новых связей,
то колебательная энергия может легко создавать потоки по различным коле-
бательным степеням свободы многоатомной молекулы, так что энергия,
сконцентрированная вначале на вновь образованной связи, может распре-
делиться по нескольким связям в течение периода колебания. Тогда время
жизни молекулы по отношению к диссоциации возрастает, хотя вероятность
ее и не обращается в нуль. В этом случае лимитирующим процессом будет
только рекомбинация, а не рекомбинация и дезактивация, так как молекула
может претерпеть много дезактивирующих соударений. Поэтому кинетика
такой рекомбинации будет второго порядка. Однако при достаточно низких
давлениях (если исключить, конечно, дезактивацию на стенке) такой про-
цесс с необходимостью перейдет в процесс третьего кинетического порядка.
Реакции рекомбинации частиц, больших, чем, например, этильный ра-
дикал, хорошо описываются при всех экспериментальных условиях бимоле-
кулярной схемой, а рекомбинация атомов — только тримолекулярной. Ме-
тильный радикал обнаруживает промежуточное поведение.
Требование, предъявляемое к третьему телу в ходе рекомбинации, со-
стоит в том, чтобы оно могло эффективно убирать при столкновении колеба-
тельную энергию вновь возникшей молекулы. Эффективность этого процесса
тем больше, чем меньше разность энергий обменивающихся состояний, а
вероятность дезактивации при соударении тем больше, чем больше колеба-
тельных степеней свободы в дезактивирующей молекуле. Примером этого
могут служить данные, приведенные ниже, которые содержат коэффициенты
скорости третьего порядка для реакции О 4-О2 + М О3 + М:
Третье тело Коэффициент ско- рости Х1034, см—6 • мол ь—2 • сек—1 Третье тело Коэффициент ско-1 ростих Ю34, см—” • моль-2 • сек—1 Третье тело| Коэффициент ско- рости х 103*, см—6 • мол ь~2 -сек-1
Не 4 Ог 6,5 CF4 16
Аг 4 СО2 15 SF6 34
]\т2 5,6 N2O 15 Н2О 60
^ОСНОВНЫЕ ОСОБЕННОСТИ ПЛАЗМОХИМИЧЕСКИХ РЕАКЦИЙ
Прежде всего необходимо определить специфические отличия и законо-
мерности плазмохимических реакций.
Каковы особенности условий динамики химических реакций в области
высоких энергий? Следует отметить, что изучение соударений, приводящих
к химической реакции, хотя и тесно связано с кинетикой, но уже теперь есть
все основания выделить его в отдельную научную область, назвав ее, на-
пример, «квантовой динамикой химических реакций», изучающей собственно
так называемый «элементарный акт», в отличие от кинетики, которая явля-
ется частью статистической физики [И].
Динамика химических реакций определяется закономерностями столкно-
вений реагирующих частиц и полями, в области действия которых они нахо-
дятся. Суммарный (зачастую сильно сглаженный) результат изучения ди-
намики реакций может быть представлен в виде функции a (ijlkl-, е).
96
Условия, в которых развертывается «элементарный акт» (т. е. динамика
соударения) химической реакции в «классической» аррениусовой химии,
таковы: средняя энергия относительного движения соударяющихся частиц
(молекул) не больше ~0,1 эВ; заселенность основного колебательного уровня
первого электронного состояния решительно преобладает над всеми осталь-
ными; реакции типа е 4- М, М± 4-М, е + М~ и т. д. практически отсутст-
вуют; продукты реакции, как правило, не возбуждены; упругие столкнове-
ния решительно преобладают над неупругими. Поэтому для большинства
случаев можно представить молекулы моделью твердых упругих шаров.
В области плазмохимических реакций ситуация иная: средние энергии
молекул ^0,1 эВ; соударения часто испытывают возбужденные (по раз-
личным уровням) молекулы, а продукты химических реакций, происходящих
при соударении, оказываются в возбужденных электронно-колебательных
состояниях; соударения молекул с электронами и ионами (и последних
между собой) весьма существенны, причем, как правило, имеет место значи-
тельное различие скорости и энергии поступательного движения (в лабора-
торной системе координат) заряженных частиц малой массы (~1—50 эВ)
и тяжелых частиц (~ 0,03—2,3 эБ), а также колебательной энергии последних
(~0,2—0,8 эВ); поэтому пользоваться моделью упругих шаров уже нельзя
[183].
Кинетика химической реакции описывает определенный тип поведения
ансамбля молекул, а потому характер этого описания статистический. Следо-
вательно, кинетическое описание поведения ансамбля тех или иных частиц
определяется, предполагая динамику соударений (например, бинарных)
известной заселенностью квантовых уровней молекул, функцией распреде-
ления частиц по энергии поступательного движения и соотношением вероят-
ностей потоков по различным каналам.
В классической химии это обстоятельство не проявляется в явном виде,
так как в ней предполагается наличие в начальный момент максвеллова рас-
пределения, которое в дальнейшем не (или очень слабо) нарушается химиче-
ской реакцией, а заселенность считается больцмановой с решительным пре-
обладанием (на многие порядки) заселения основных уровней; переходами
вида М (i) (/) поэтому пренебрегают; все реагирующие частицы имеют
одну и ту же среднюю энергию (температуру) единого максвеллова распре-
деления [11, 183].
В плазмохимии ситуация иная, а именно: начальное распределение может
быть немаксвелловым, а заселенность — небольцмановой; даже если на-
чальное распределение максвеллово, оно нарушается химической реакцией,
причем вновь возникшее распределение в свою очередь влияет на скорость
реакции; различные компоненты реакции могут иметь различные по форме
и средней энергии функции распределения, которые, кроме того, могут
меняться с течением времени с различными временами релаксации; пре-
небрегать переходами вида М (?) (у) в описании химических реакций
уже нельзя; частота неупругих соударений не может считаться малой по
сравнению с упругими [11].
На рис. 2.15, взятом из [184], показано изменение с температурой вре-
мени колебательной релаксации и времени установления диссоциационного
равновесия молекулы О2, приводящее к их сближению и совпадению.
На рис. 2.16 приведена взятая из работы [185] зависимость коэффициента
скорости химической реакции от колебательной температуры (при замо-
роженной поступательной температуре) для реакции O+4-N2->NO + + N.
Отметим, что при изменении TK0Jl с 103 до 6-103 К коэффициент скорости
реакции возрастает примерно в 40 раз, а такое же изменение ZnocT привело
бы, согласно аррениусовой кинетике, к изменению в 60 раз. Эффекты вполне
сравнимые. Плазмохимические реакции могут быть неравновесными и (ква-
зи)равновесными. В табл. 2.2 показаны типы плазмохимических реакций.
Из таблицы видно, что кинетика плазмохимических реакций представ-
ляет собой частный случай неравновесной химической кинетики.
4 Теоретич. и прикладная плазмохимия
97
Для плазмохимических газофазных реакций (которые только и будут
рассматриваться далее) характерно следующее.
1. Неупругие столкновения являются новой отличительной чертой газа
при высокой температуре (или высоких энергиях) по сравнению с «обычными»
газами (одноатомными), свойства которых хорошо описываются кинетиче-
ской теорией, основанной на учете упругих столкновений. Кинетическая теория
Рис. 2.16. Экспериментальная зависимость/^ от номера колебательного уровня для реак-
ции 0+ + N2 -» N0+ + N
Рис. 2.15. Время установления диссоциационного равновесия (2) и время колебательной
релаксации (7) для столкновений О2 — О2
объясняет некоторые особенности поведения многоатомных и реагирующих
газов в предположении, что неупругие соударения редки; такое прибли-
жение тем хуже, чем выше энергия (температура) газа.
Таблица 2.2
Типы плазмохимических реакций
Реакции Процессы в системе Характеристика системы Тип процесса
Неравно- весные Химические реакции в не- равновесных системах * Не равновесность системы, обусловленная химической реакцией Различие колебательной и поступательной температур Две подсистемы с различ- ными температурами Одна подсистема немаксвел- лова, другая максвеллова Стационарный Релаксационный
Квазирав- новесные Обе подсистемы немаксвел- ловы (и небольцмановы)
* Характерным для неравновесного состояния газовой системы или низкотемпературной плазмы
является, в общем случае, суммарный перенос массы, импульса и энергии или хотя бы какой-либо
одной из этих величин; этот перенос осуществляется через любой малый элемент поверхности,
ориентированный некоторым образом и движущийся со скоростью потока (или массовой скоростью).
2. Характерные времена различных физических и химических процес-
сов сближаются, и различные процессы нельзя уже разделить, как это до-
пускается в классической равновесной химической кинетике. Для иллюстра-
ции приведем некоторые данные о характерных временах (в сек) различных
процессов в интервале температур 3-103—1,5-104 °К:
98
Среднее время молекулярного взаимодействия..........—10~14
Среднее время свободного пробега молекул, совпадающее по поряд-
ку величины с временем максвеллоизации..............^10~9
Время вращательной релаксации молекул...............~10~9
Время колебательной релаксации молекул..............^10~7
Время релаксации для диссоциации Ог...................~ 10~7
Среднее время ожидания эффективного столкновения с пороговой
энергией 60 ккал!моль ~2,5 эв .................. ~ 10~5
3. В интересующей нас области энергий уже нельзя отделить физиче-
скую кинетику 1 от кинетики химической. Если скорость химической реак-
ции превышает частоту обычных столкновений с обменом энергией, проис-
ходит отклонение от равновесных условий. Это может привести к искажению
равновесного максвелл-больцмановского распределения по энергиям и
уменьшению доли его высокоэнергетической части из-за происходящих
реакций. Этот «хвост» распределения не может быть восполнен за счет столк-
новений с обменом энергией, поскольку такие столкновения слишком редки.
При этом принципы, лежащие в основе простой кинетической теории хими-
ческих реакций и теории абсолютных скоростей реакций, будут нарушаться.
Общее решение вопроса о том, насколько сильно химическая реакция иска-
жает функцию распределения, еще не получено. Однако в некоторых кон-
кретных случаях вопрос этот исследовался. Так, в [152] методом Монте-
Карло изучено влияние быстрой химической реакции на максвелл-больц-
мановское распределение и обратное влияние возникшего распределения на
скорость реакции.
Известны случаи, когда необходимо различать поступательную, колеба-
тельную, вращательную температуры, когда различные по вещественному
составу компоненты системы (например, электроны, ионы, нейтральные мо-
лекулы) имеют сильно различающиеся температуры, когда вообще системе
нельзя приписать какой-либо температуры (неравновесные, стационарные
и релаксирующие системы) 2. Во всех этих случаях использование аррениу-
совой кинетики, строго говоря, неправомочно, а обычное выражение для
константы скорости химической реакции не имеет смысла.
4. В плазмохимической кинетике необходимо рассматривать многока-
нальные процессы. С квантовомеханической точки зрения это означает
(для упрощения изложения будем говорить о двух каналах), что в некотором
интервале энергий возможно существование двух пар частиц Аг + Вг и
А2 4- В2, т. е. имеются две независимые и удовлетворяющие граничным усло-
виям волновые функции рассматриваемой системы. Как известно, для одно-
канальных процессов 5-матрица содержит в себе всю информацию о свойствах
взаимодействия системы, для многоканальных же процессов подобная задача
не решена [26]. Кроме того, чтобы восстановить гамильтониан по данным
рассеяния, необходимо знать все элементы 5-матрицы при всех энергиях
(не говоря уже о больших чисто математических трудностях).
5. Пороговый характер реакций (существование одного и только одного
порога, с которого начинается реакция), столь типичный для классической
аррениусовой кинетики, может не иметь места в плазмохимических реак-
циях. В них необходимо учитывать, что химические процессы происходят
из разных квантовых состояний системы и что вся система или по крайней
мере одна из ее подсистем могут в целом иметь надпороговую энергию.
6. Перечисленные пять особенностей плазмохимических реакций пред-
ставляют собой по существу характерные черты неравновесных химических
1 Основы физической кинетики впервые были сформулированы в классической книге
Л. Э. Гуревича «Основы физической кинетики». Л.— М., ГТТЛ, 1940.
2 Строго говоря, дело не только в том, что для неравновесной системы понятие
одной единой температуры не имеет смысла. Вопрос состоит также в том, что возникают
новые понятия, без которых сколько-нибудь полное описание становится невозмож-
ным. К ним относятся прежде всего заселенность уровней (числа заполнения), релакса-
ции по различным степеням свободы, изменение функции распределения во времени и
пространстве.
99
4*
процессов, частным (хотя и очень важным) случаем которых являются реак-
ции в низкотемпературной плазме. В настоящем пункте излагаются некото-
рые специфические черты именно плазмохимических реакций.
Наиболее важным критерием изменения состава плазмы является сте-
пень ее ионизации. В равновесном случае при заданных температуре и дав-
лении она определяется уравнением Саха, которое является следствием
закона действующих масс. Процесс ионизации (и неразрывно связанный
с ним процесс рекомбинации) обусловлен столкновениями тяжелых частиц
при высоких температурах (энергиях), фотоионизацией, столкновениями
с электронами, ион-молекулярными реакциями и т. д. Описание кинетики
всех этих процессов с микроскопической точки зрения требует обобщения
кинетической теории газов на случай плазмы. Это обобщение находится еще
в стадии разработки. В случае низкотемпературной плазмы, которая со-
стоит как из нейтральных, так и из заряженных частиц, кроме характерных
для обычного молекулярного газа соударений, происходящих в области
малых расстояний, имеются еще далекие соударения, обусловленные элек-
тромагнитным взаимодействием между заряженными частицами. При этом
надо принять во внимание очень малые углы рассеивания и, следовательно,
большое количество актов взаимодействия, при которых происходит весьма
незначительный перенос импульса; надо учесть также взаимодействие между
заряженными частицами и электромагнитными полями [186—187].
Когда электронный, ионный газы и газ нейтральных частиц не находятся
в тепловом равновесии, то существует обмен энергией между электронами
и нейтральными частицами и между электронами и ионами. Последний проис-
ходит путем кулоновских соударений. Оба процесса могут приводить непо-
средственно или через стадии возбуждения к химическим реакциям. Меха-
низм и вероятности этих реакций и надо изучить в плазмохимической кине-
тике на основе конкретного анализа всех взаимодействий в системе и ос-
новных принципов неравновесной химической кинетики.
В настоящее время представляется очевидной ограниченность аррениу-
совой кинетики, которая имеет смысл только вблизи равновесия (т. е.,
строго говоря, при малых возмущениях, когда система еще может считаться
квазиравновесной), другими словами, когда для системы может быть опре-
делена одна температура (рассматриваемая как параметр максвелл-бол ьцма-
новского распределения).
Неравновесное распределение реагентов имеет место, например, при им-
пульсном введении энергии в первоначально термодинамически и химиче-
ски равновесную систему при условии тимп трел.хим (импульсный электри-
ческий разряд, ударные трубы, флеш-фотолиз и т. п.).
ВЛИЯНИЕ НЕУПРУГИХ ПРОЦЕССОВ
НА РАСПРЕДЕЛЕНИЕ ЭЛЕКТРОНОВ ПО ЭНЕРГИЯМ
В ТЛЕЮЩЕМ РАЗРЯДЕ В N2, N2 + О2 И СО2
В [189] измерено энергетическое распределение / (е) свободных электро-
нов в положительном столбе тлеющего разряда в N2, N2 + О2 и СО2 при
давлении 2 торр, плотности разрядного тока 4,3 — 11 мА/см2 и различных
временах пребывания газа в зоне разряда (для СО2). Измерены также отно-
сительная величина концентрации электронов пе9 их средняя энергия ё,
напряженность продольного электрического поля Е, температура газа Тг
и колебательная температура Тj (для N2 и N2 4- О2), а также степень дис-
социации а (для СО2). Для измерения / (е), пе и Е использована зондовая
техника с двойным электрическим дифференцированием вольт-амперной
характеристики [188]. Температуру газа и колебательную температуру
определяли по неразрешенной вращательной структуре второй положитель-
ной системы полос N2. Величину а определяли методом хроматографического
анализа. Погрешности измерений составили: ±5% для Е, ±3% для а,
100
Рис. 2.17. Результаты измерений / (е)
а: 1 — в N2, е = 2,9 эВ; 2 — в смеси N2: О2 = 8 : 2, 3,7 эВ; 3 — то же, 4 : 6, 5,3 эВ; давление 2 тиорр;
ток разряда 30 мА; 4 и 5 — распределение Дрювестейна и Максвелла соответственно для е = 2,9 эВ;
б: 1 — в N2, Т = 0,23 сек, 8 = 2,9 эВ; 2 — в СО2, 0,23 сек, 3,2 эВ; 3 — то же, 0,4 сек, 4,2 эВ; 4—1,5 сек,
4,8 эВ; для 1—4 ток разряда равен 30 мА; 5 — 0,23 сек, 4,4 эВ; ток разряда 75 мА
±10—15% для Тг и Tv, Измеренные значения / (е) занижены примерно
на 40% вблизи максимума / (е). Эта погрешность уменьшается до 5—10%
в «хвосте» распределения. Случайные погрешности в измерении / (е) не пре-
восходят 5—7 %.
Результаты измерений показывают (рис. 2.17), что / (е) во всех упомяну-
тых условиях отличаются от максвелловского и дрювестейновского распре-
делений, что объясняется передачей энергии электронов на электронно-
колебательные уровни молекул. Распределение / (с) становится богаче быст-
рыми электронами по мере увеличения содержания О2 в смеси N2 ± О2,
что приводит к росту ё от 2,9 до 5,3 эВ (чистый N2 и N2 : О2 = 4 : 6). Это
вызвано, по-видимому, прилипанием медленных электронов к электроотри-
цательным частицам О2 и О. В то же время с увеличением содержания О2
в смеси происходит уменьшение Tv от 7000 (чистый N2) до 5700° К
(N2 : О2 = 4 : 6 по объему). Температура газа в этих экспериментах прак-
тически постоянна и составляет 460—500° К [189].
Распределение в СО2 при временах пребывания т менее 0,23 сек незначи-
тельно отличается от распределения в чистом N2 при том же токе. С увели-
чением т /(е) деформируется, причем е и пе увеличиваются, что показано
ниже:
/, мА ... 30 75 30 75 30 75
т, сек • • • 0,23 0,23 0,4 0,4 1,5 1,5
а, % . . . 9 20 14 28 23 43
пе, усл. ед. . . . . . . 180 600 400 1200 700 —
8, эВ . . . 3,2 4,4 4,2 4,7 4,8 —
Это связано с изменением химического состава газа в результате диссо-
циации СО2, что приводит к изменению сечений столкновений электронов
с тяжелыми частицами. Диссоциация СО2 на О и СО может происходить или
по реакции СО* + ё ->СО2 (3В2) — ё ->СО (XS) ± О (3Р) ± ё, где СО* —
молекула с энергией колебательного возбуждения 1—2 эВ, или через воз-
буждение молекулы СО2 из основного состояния однократным электронным
ударом с последующим распадом электронно-возбужденной молекулы.
Выделить один из этих механизмов пока не представляется возможным.
Таким образом, в тлеющем разряде в молекулярных газах N2, N2 ±
± О2 и СО2 вид / (е) в значительной мере связан с такими неупругими про-
цессами, как возбуждение электронно-колебательных уровней молекул,
прилипание медленных электронов с образованием отрицательных ионов и,
наконец, химическая реакция диссоциации молекул, приводящая к изме-
нению состава газа в разрядной трубке [190].
101
ВЛИЯНИЕ ФУНКЦИИ РАСПРЕДЕЛЕНИЯ ЭЛЕКТРОНОВ
ПО ЭНЕРГИИ НА КОЭФФИЦИЕНТ СКОРОСТИ
ХИМИЧЕСКОЙ РЕАКЦИИ
Важнейшими характеристиками плазмохимического процесса являются
его производительность, чистота полученного продукта и его стоимость.
Поэтому исследование кинетики и механизма процесса имеет целью главным
образом выяснить условия (давление, температуру и т. д.), при которых
реализуется наибольшая скорость образования целевого продукта при не-
обходимой селективности и экономичности. Каким образом эти параметры
процесса связаны с энергетическим распределением свободных электронов
в плазме реагирующих газовых компонент?
В плазмохимических системах при атмосферном или повышенном давле-
нии и больших степенях ионизации 2^>1О~2 этот вопрос имеет значение при
наличии очень быстрых процессов, в частности, химических реакций, диф-
фузии к стенкам и т. д., которые могут приводить к отрыву температуры
электронов от газовой (поступательной или колебательной) температуры.
В частности, в дуге постоянного тока [191] отрыв Те и 7V может быть суще-
ственным и заметно сказаться на скоростях физико-химических процессов,
например диссоциации молекул. Тем или иным способом воздействуя на
величину (Те—7Г), можно влиять на селективность и экономичность плаз-
мохимических процессов в этих условиях. Возможность существенного
отклонения функции распределения электронов по энергиям от максвеллов-
ской (равновесной) в этих условиях маловероятна. Однако в плазме с малой
степенью ионизации <Д0~6 отклонение вполне возможно и при атмосферном,
и при повышенном давлениях.
В плазме пониженного давления (например, в тлеющем, ВЧ- и СВЧ-
разрядах при давлении менее 10—20 торр) реализуется не только очень
большой разрыв средних энергий поступательного движения электронов
и тяжелых частиц, но и большое отклонение функции распределения элек-
тронов по энергиям (ФРЭЭ) от максвелловской. Это открывает большие воз-
можности оптимизации плазмохимических процессов в этих условиях по их
селективности, скорости и экономичности. Особенности ФРЭЭ сказываются
прежде всего на «динамике» элементарного акта химической реакции, про-
исходящего при столкновении электрона с молекулой. Усредненная по ФРЭЭ
скорость этой стадии
оо ___
™ = у ^-6(-^-,e)/(e)de (2.208)
КПор
зависит от энергетического распределения электронов — явно через ФРЭЭ
f (е) и неявно через эффективное (усредненное по начальным квантовым со-
стояниям ки I молекул М) сечение б (kl/ij, е). Распределение частиц сорта
М по квантовым состояниям может сильно зависеть от концентрации и ФРЭЭ.
Так, в тлеющем разряде при давлении —1 торр возбуждение колебатель-
ной степени свободы молекул N2 происходит при электронном ударе [192].
Заселение электронно-возбужденных состояний молекулы азота в этих усло-
виях также происходит в основном при однократном электронном ударе
И 93]. В то же время ФРЭЭ в тлеющем разряде в азоте существенно отлича-
ется от максвелловской [194], что показано на рис. 2.18. Таким образом, если
рассмотреть реакцию диссоциации азота в тлеющем разряде, то ее скорость
(она складывается из скоростей диссоциации «классическим путем» через
•основное электронное состояние молекулы и посредством предиссоциации
через электронно-возбужденные состояния молекулы [195]) будет опреде-
ляться концентрацией и энергетическим распределением электронов. Сход-
ная картина наблюдается в тлеющем разряде в СО2. Диссоциация молекул
102
Рис. 2.18. Функция распределения
электронов по энергиям в положи-
тельном столбе тлеющего разряда
в азоте [211]
1 — E/No = 11,4-io-1® В-см2\ 2 — 8,6-
• 10-16; з _ 7,3-10-16
Рис. 2.19. Функция распределения
электронов по энергиям в положи-
тельном столбе тлеющего разряда
в СО2 [215]
1 — ток разряда 30 мА', 2 — то же 75 мА.
Диаметр трубки 32 мм, время пребывания
газа в разряде 0,1 сек
Рис. 2.20. Функция распределения
электронов по энергиям в положи-
тельном столбце тлеющего разряда
в смесях N2 + Ог
1 — чистый азот; 2 — N2 : О2 = 4,1; 3 —
то же 2 : 3; 4 — распределение Максвелла
СО2 включает стадию электронного возбуждения молекулы при столкно-
вении со свободным электроном [196, 197]. Так же как и в разряде в N2,
колебательное возбуждение молекул СО2 в основном электронном состоянии
в значительной степени зависит от концентрации и ФРЭЭ [199], а ФРЭЭ
существенно немаксвеллова [190, 298—200] (рис. 2.19).
Если же нас интересует стадия реакции, происходящая при столкнове-
нии тяжелых частиц друг с другом, то и здесь роль ФРЭЭ может быть су-
щественной. Это видно из того, что исходные (до столкновения) состояния
реагирующих тяжелых частиц, т. е. распределения реагирующих частиц
по квантовым состояниям, в плазме пониженного давления сильно связаны
с параметрами электронной компоненты плазмы — концентрацией и ФРЭЭ.
Мы уже говорили о распределении по колебательным и электронно-возбуж-
денным состояниям молекул. В некоторых стадиях реакций основное значе-
ние имеют положительные ионы, и, следовательно, скорость таких реакций
будет определяться скоростью образования ионов. Эксперименты показы-
вают, что в некоторых условиях, например в тлеющем разряде в N2 [199],
ионизация происходит в результате однократных столкновений электронов,
с молекулами в основном электронном состоянии. ФРЭЭ в высокоэнергети-
ческом «хвосте», ответственном за ионизацию, может быть на порядок вели-
чины (азот [194, 200]) или даже на восемь-девять (инертные газы, разряд
в полом катоде [201]) богаче максвелловского распределения с той же сред-
ней энергией. На скорость образования отрицательных ионов, также игра-
ющих важную роль в некоторых химических реакциях, будет сильно влиять
особенность поведения ФРЭЭ в низкоэнергетическом интервале спектра
[202]. В этом интервале ФРЭЭ могут быть беднее максвелловской ФРЭЭ,
как это показывают, например, эксперименты в тлеющем разряде в смесях
103
N2 + О2 [189] (рис. 2.20). . Номера кривых, показанных на рис. 2.20, при-
ведены ниже:
№ п/п n2, % о2, % Ту, ° К Tv, °к е, эВ
1 100 0 500+35 7000±ЗО0 2,85
2 80 20 510+35 6600+700 3,70
3 40 60 460+30 5700+600 5,34
4 Максвел. 3,00
5 Максвел. 5,30
КИНЕТИКА РАЗЛОЖЕНИЯ УГЛЕКИСЛОГО ГАЗА
В ТЛЕЮЩЕМ РАЗРЯДЕ
Протекание химических реакций в условиях тлеющего разряда при пони-
женных давлениях давно привлекает внимание исследователей [200—214].
Высокие энергии электронов (порядка нескольких электронвольт) при от-
клонении функции их энергетического распределения от максвелловской и
низкие температуры тяжелых частиц, характерные для таких разрядов,
позволяют раздельно изучать влияние температуры газа, средней энергии
электронов и функции их энергетического распределения на протекание
реакций, что важно для создания теории неравновесных химических реак-
ций. Особое внимание в последнее время уделяется исследованию химических
реакций в тлеющем разряде в углекислом газе, поскольку имеются прямые
доказательства влияния химических реакций на длительность работы отпа-
янных лазеров на СО2 [215].
Однако до сих пор остаются невыясненными вопросы, связанные с меха-
низмом разложения СО2 в тлеющем разряде. Большой экспериментальный
материал, приведенный в работе [212], не может быть использован для вы-
яснения этого механизма по двум причинам. Во-первых, как правило, в этой
работе исследовали смеси СО2 с гелием и азотом, что сильно усложняет
анализ механизма реакции, и, во-вторых, не измерены истинные времена
пребывания газа в зоне разряда, без чего возможны ошибки даже при каче-
ственном истолковании результатов. Работа [214] непосредственно посвящена
изучению механизма разложения СО2 электронным ударом в предположении
о максвелловском распределении электронов по энергиям в тлеющем разряде
и малости вклада отрицательных ионов в ток разряда. Результаты работ
[216—218] опровергают справедливость* первого из этих предположений,
а второе требует экспериментальной проверки. Кроме того, работа [214]
проведена при малых плотностях тока, что далеко от условий разрядов,
применяемых в лазерах на СО2. В [217] исследованы начальные стадии раз-
ложения СО2 в тлеющем разряде при давлении газа 2 мм рт. ст. и плотностях
разрядного тока 0,6—12 мА/см2, близких к рабочим для лазеров на СО2.
Особое внимание при этом уделено измерению параметров, влияющих на
кинетику реакции.
Для начальных стадий разложения СО2 в тлеющем разряде с точностью
до членов порядка а/оср, где а — степень разложения, ар — степень разло-
жения, соответствующая динамическому равновесию в данных условиях,
пренебрегая изменением температуры газа со временем, можно записать
dNrn dNrn _
----+ Л,, (2.209)
где пе, Nqq2, Nqq — концентрации электронов, молекул СО2 и СО соответ-
ственно.
Первое слагаемое в правой части выражения (2.209) описывает разложе-
ние СО2 в результате однократного электронного удара, второе — разложе-
ние в результате ступенчатых процессов и реакций с участием частиц, пред-
варительно возбужденных (или ионизованных) электронным ударом Коэф-
104
фициенты скорости реакции и к2 зависят от энергетического распределения
электронов и механизма реакции. Вводя степень разложения а = TVco/
/(TVco + Мзог), равенство (2.209) можно переписать в виде
- /(1rf7a) = ^Пе+ (! - «)• (2-210)
Изменяя в широких пределах концентрации электронов и их энергети-
ческое распределение и регистрируя эти параметры, можно исследовать
механизм реакции и определить абсолютные значения коэффициентов ско-
ростей этой реакции.
Зависимость степени разложения СО2 от времени его пребывания в раз-
ряде при различных разрядных токах и давлении 2 мм рт. ст. приведена на
Рис. 2.21. Зависимость степени раз-
ложения СО2 от времени пребывания
в разряде при различных токах раз-
ряда
1 — 100 мА; 2 — 75; 3 — 30; 4 — 10; 5 — 5
Рис. 2.22. Зависимость концентра-
ции электронов от силы разрядного
тока
1 — зондовые измерения; 2 — расчет по
средней плотности тока и скорости дрейфа
электронов
Рис. 2.23. Зависимость величины
In (1 — а) от [концентрации элект
ронов
рис. 2.21. Точки, соответствующие различным длинам положительного
столба разряда (31,5 и 57 сж), с разбросом в пределах 10% ложатся на одну
кривую для данного тока разряда. Это показывает, что вклад приэлектрод-
ных областей разряда в измеряемые степени разложения при данной гео-
метрии разрядной трубки не превышает 10%. Давления на входе и вы-
ходе из разрядной трубки во всех опытах совпадали в пределах ошибки
измерения, т. е. градиенты давления вдоль трубки были пренебрежимо малы.
Для обработки полученных кривых по уравнению (2.210) необходимо
знать зависимость концентрации электронов от величины разрядного тока.
Эта зависимость приведена на рис. 2.22, на котором кривая 1 построена по
результатам обработки ионных ветвей зондовых характеристик по способу,
предложенному в [219] в предположении, что а — njne 2, где п_ — кон-
центрация отрицательных ионов, а также по результатам измерения пло-
щадей под кривыми функции распределения электронов по энергиям. Кри-
вая 2 (рис. 2.22) рассчитана по измеренной средней плотности тока разряда
и скоростям дрейфа, взятым из [220], по полученным значениям Е/N, При
токах ниже 50—70 мА обе кривые дают линейное увеличение пе с ростом
105
разрядного тока. Результаты измерения концентрации электронов СВЧ-
методом, полученные при давлении 2 жжрт. ст., токах 10—60 мА и диаметре
разрядной трубки 2 см в работе [221], также свидетельствуют о линейной
зависимости концентрации электронов от тока до 60 мА.
Различие абсолютных значений концентраций электронов, определенных
различными методами (кривые 1 и 2), может быть в основном следствием
завышения результатов зондовых измерений из-за присутствия отрицатель-
ных ионов в разряде. Если приписать все различие между кривыми влиянию
отрицательных ионов, то значение а = njne будет не более 15—20. Вклад
отрицательных ионов в разрядный ток при этом значении а будет составлять
не более 40%, и, следовательно, абсолютные значения концентрации, соот-
ветствующие кривой 2, завышены не более чем на 40%.
На рис. 2.23 изображена зависимость степени разложения СО2 от кон-
центрации электронов тге, вычисленной по плотности тока, для времени пре-
бывания газа в зоне разряда 0,1 сек. На оси абсцисс отложена концентрация
электронов, а на оси ординат — величина — In (1 — а). В результате инте-
грирования уравнения (2.210) получается связь последней величины с ко-
эффициентами скоростей реакции
— In (1 — а) — (к1пе + к2п^) т. (2.211)
Поскольку кажущийся порядок реакции разложения СО2 по концентра-
ции электронов (см. рис. 2.23) превышает единицу, но меньше двух, ясно,
что существенное значение имеют процессы, приводящие к разложению
СО2 в результате однократного электронного удара. Такими процессами
могут быть образование отрицательного иона СО2, возбужденной молекулы
СО2 или иона COJ с последующим распадом их без участия электронов и час-
тиц, предварительно возбужденных электронным ударом (под которыми
следует понимать также и ионы). Коэффициент (константа) скорости прили-
пания электрона к молекуле СО2 согласно данным работы [220] на два поряд-
ка величины ниже коэффициента скорости реакции разложения СО2, полу-
ченного в данной работе (см. табл. 2.3). Следовательно, процессами с участием
отрицательных ионов можно пренебречь и считать, что разложение идет
через образование СО2, т. е. реакция должна иметь порог не ниже энергии
разрыва связи СО—О (—5,45 эВ) и в реакции должны принимать участие
электроны только с энергиями выше 5,45 эВ. Как видно из рис. 2.19, на ко-
тором приведены функции распределения электронов по энергиям, измерен-
ные в [215], при увеличении тока разряда наблюдается увеличение доли
электронов с энергиями, превышающими пороговую энергию 8П0р- Это
должно привести к увеличению коэффициента скорости
оо оо
&i = с (е) ре/(е) de М / (е) d/, (2.212)
£пор > 0
где б (е) — сечение возбуждения электронным ударом молекулы СО2, при-
водящего к последующему ее разложению.
Учет зависимости коэффициента скорости от вида функции энергетического
распределения электронов приводит к уменьшению кажущегося порядка
реакции по концентрации электронов и приближает его к единице. В табл. 2.3
приведены значения коэффициентов скоростей реакции, вычисленные по
экспериментальным данным (см. рис. 2.23) в предположении первого по-
рядка реакции по концентрации электронов (/с1ЭКСп), а также коэффициенты
скорости, рассчитанные по формуле (2.12) с использованием измеренных
функций распределения и сечения, заимствованного из работы [214] (АДрасч)-
Удовлетворительное совпадение /с11ЭКСп и /с1расч показывает, что реакция
имеет первый порядок по концентрации электронов вплоть до токов в 75 мА
(j = 9 мА/см2), а это означает, что разложение происходит путем возбужде-
ния молекул СО2 однократным электронным ударом с последующим ее рас-
падом. Полученный вывод о механизме реакции при данной форме функций
106
Таблица 2.3
^Коэффициенты скорости реакции
Ток разряда, мА п • 10», см~3 тг, °к Е N-Ю-i», В-см3 fcl ЭКСП*10 10’ см3- сек-1 fcl расч'1и 10’ см3 • сек-1 3
5 0,33 370 5,25 1,5
10 0,66 400 5,25 1,8
30 2,00 510 5,23 2,3 1,9 0,140
75 4,80 680 6,45 3,4 3,3 0,220
100 6,10 780 7,25 3,7
распределения электронов по энергиям (см. рис. 2.24), по-видимому, мало
зависит от выбора величины сечения возбуждения молекулы и формы его
зависимости от энергии.
Выше показано, какое большое значение в величине коэффициента ско-
рости имеют реакции функции распределения электронов по энергиям f (е).
Изотермическая химическая реакция в рассматриваемом диапазоне энергий
(—1ч-3 эВ) является пороговым процессом, что отличает к от других коэф-
фициентов переноса: теплопроводности, диффузии и вязкости. В стационар-
ных условиях, когда f (е) не изменяется со временем, основной вклад в зна-
чение к (и, следовательно, в скорость реакции) вносит высокоэнергетическая
часть f (е), которая лежит выше значения 8П0р- Независимо от того, каким
путем восполняется убыль высокоэнергетических электронов (8 > еПор)>
происходящая за счет химической реакции, для расчета к существен
только вклад этой части / (е). Аналогичная ситуация наблюдается и в обыч-
ной аррениусовой кинетике, где в выражении для к основной вклад дает
множитель ехр {—г/кТ}. Здесь температура Т входит как измеряемая ве-
личина, однозначно определяющая функцию распределения, в том числе
ее высокоэнергетическую (химически активную) часть 8 > 8ПОр- В нерав-
новесном случае нельзя ввести температуру системы, поэтому в стационар-
ном случае в качестве наблюдаемой величины должна рассматриваться
вся функция распределения. Для грубой оценки изменения значения к
можно ввести величину
ос оо
Р = / (£) М / (8) ^8’ (2.213)
епор ' 0
которая определяет относительную долю частиц с 8 > 8П0р при заданной
/ (е) и, следовательно, число актов, приводящих к реакции, в единицу вре-
мени. Как видно из данных, приведенных в табл. 2.3, коэффициенты скорости
реакции приблизительно пропорциональны введенной величине р [222].
ОЦЕНКА КОЭФФИЦИЕНТОВ СКОРОСТИ РЕКОМБИНАЦИИ
ВОЗБУЖДЕННЫХ АТОМОВ АЗОТА
Известно [11, 223, 224], что коэффициенты скоростей реакций возбужден-
ных атомов могут значительно превышать константы скоростей реакций
невозбужденных атомов. Однако коэффициенты скоростей реакций реком-
бинации с участием возбужденных атомов мало изучены. Данные работы
[225] позволяют оценить величину такого коэффициента для реакции [137]
N (2D) + N (45) + Кг(Хг2Й —- N2 (С3П„, v = 4) + N2 (X4S+). (2.214)
В этой работе приведены осциллограммы послесвечения азота в проточной
системе в сечении, расположенном на выходе из разрядной трубки, из ко-
107
торых автором [225] получены квазистационарные концентрации [N2(A8Sj,
0, 1)], [N (45)] и [N (2Р)] при давлениях от 20 до 760 торр. По этим ос-
циллограммам можно оценить верхнюю границу отношения интенсивностей
излучения:
Л {N2 у" 4) N2 (BWg, у = 8)}
J2{N2(X32^^-0}-.N2(XiS+^' = 7)} ’ '
Л = А4_8 (С3Пи) [N2 (СШп, 4)],? (2,216)
J2 = Ао_7 (А3^) [N2 (A3S:, v - 0)], (2.217)
гдеЛ4_8 (С3Пи) иЛ0-7 G43Su) — вероятности соответствующих радиационных
переходов.
Максимально возможное значение кг можно определить, если предполо-
жить, что заселение уровня v = 4 состояния N2 (С3П..(,) происходит в резуль-
тате реакции (2.215), а дезактивация — за счет излучения и тушения тяже-
лыми частицами. Тогда
Av=4 IN,(С3Пи> V = 4)1 = MWHNpilNil , (2.218)
1 । vvl'=4/ ?;=4^
где vr=4 — частота тушения уровня v = 4 N2 (С3Г1Н) тяжелыми частицами,
а Аг==4 — суммарная вероятность радиационного распада этого уровня.
Образование N(2D) в условиях [225] происходит за счет излучательного
перехода N(2P) — N (2Р) с вероятностью Ар, дезактивация же этого уровня —
в результате диффузии с коэффициентом кд с последующим тушением на
стенке с вероятностью —1 [225, 226] и реакций (2.214) и (2.219):
N (2D) + N2 (XXS+) — N2 (X4£) + N. (2.219)
Квазистационарная концентрация1 [N (2D)] определяется так:
(N(2P)]= ------------Р.[.^-(2/>)1------ (2.220)
*i[N (45)][N2] + AnN2(X12+) ’
Таблица 2.4
Величины концентраций и параметров
Сорт частиц Концентра- ция, CM~S Литератур- ная ссылка Параметр Величина, сек-1 Литератур- ная ссылка
[N2A32+ [N(*P)] [NO] N(4S)] [N2] 2,6-Ю8 2.101° 2-103 1,5- IO13 6,2-1017 225,228 225 225,231 225,232 «. co “ II II brt © Д £ ft 8 4°^ <0,5 -1-102 0,154 0,173-Ю7 2,l-107 3,3-107 <6.10_i5 см31 сек 0,102 225 228 229,230 229,230 229 223 223
1 Квазистационарная концентрация [N (2D)] установится через
т = [N C5)J + /зд2 [N2 (Х%)]Г ^0,1,
где
T = Ci, t).
Наблюдения же длились часами [227].
(2.221)
(2.222)
108
Из уравнений (2.215) — (2.220) получим
|1 А4_8(СТ1ц)
[N(4<)J [а А0_7(Д32;)
N(2P)
[N2(A3S+,z>= 0)]
(2.223)
Все величины \ использованные в уравнении (2.221), приведены в табл.
2.4. Расчет кг при Р = 20 тпорр дает/q 1-10-28 см('/сек. Заметим, что коэф-
фициент скорости реакции
N (45) + N (45) + N2 — 2N2,
kt = 2-10-32—3.Ю-3» см6/сек.
(2.224)
(2.225)
РАВНОВЕСНЫЕ И КВАЗИРАВНОВЕСНЫЕ РЕАКЦИИ
В ПЛАЗМОХИМИЧЕСКОЙ КИНЕТИКЕ
Математическое описание таких реакций естественно может быть в пре-
делах точности экспериментальных кинетических данных выполнено в тер-
минах классической химической кинетики. Основная трудность состоит
в нахождении уверенных констант скоростей химических реакций, которые
для плазмохимических температур обычно отсутствуют (и их очень трудно
экспериментально определить). Экстраполяция констант скоростей, получен-
ных в интервалах (обычно узких) относительно низких температур, чревата
многочисленными источниками ошибок. Поэтому в таких случаях необходим
тщательный анализ возможных статистически-распределенных и системати-
ческих ошибок, ошибок аррениусовой аппроксимации и т. п.
Как правило, в плазмохимической кинетике приходится решать задачи
для плазменных струй, в которых необходимо совместное рассмотрение урав-
нений гидродинамики и химической кинетики, а также для зон разрядов,
для которых необходимо сочетание системы уравнений гидродинамики и
химической кинетики.
Необходимые указания о методах, расчетах, экспериментальных мето-
диках можно найти в [233, 234] и в разделе 5 настоящей книги.
СМЕШЕНИЕ РЕАГЕНТОВ
В ПЛАЗМОХИМИЧЕСКОЙ КИНЕТИКЕ
Проведение химических реакций при высоких энергиях (или температу-
рах) возможно тремя путями: 1) подогрев заранее приготовленной смеси
холодных реагентов (например, в электрической дуге и т. п.); 2) смешение
холодной компоненты с горячей (например, в плазменной струе); 3) смешение
двух горячих реагирующих газов. Второй и третий пути включают в каче-
стве необходимого этапа процесс смешения реагирующих компонент. Этот
процесс протекает за конечное время, в течение которого химическая реак-
ция может происходить в неоднородной, неизотермической системе со значи-
тельной переменной во времени и пространстве скоростью.
Известно, что процесс установления равновесия в пространстве скоростей
существенно отличен от установления равновесия в пространстве конфигу-
раций.
Распределение по скоростям в любом элементе координатного простран-
ства монотонно и весьма быстро (~10~9 сек — порядка среднего времени
между столкновениями при давлениях ~ 1 апгм) стремится к локальному
распределению Максвелла. Это распределение, конечно, никогда строго не
достигается, так как внешние силы и наличие потоков препятствуют этому.
1 Все величины концентраций пересчитаны по измеренным в [225] интенсивностям излу-
чения с использованием новых данных по вероятностям переходов [228, 231] и зависи-
мости интенсивности 1+ системы N2 в послесвечении от концентрации [Крб1)] [232].
109
Что же касается установления равновесия в координатном пространстве, то
оно вообще происходит немонотонно и требует значительно большего вре-
мени [235]. Это, конечно, не означает, что установление равновесия проис-
ходит в два последовательных резко отделенных этапа: а) максвеллиза-
ция; б) установление распределения в координатном пространстве вида
ехр (—pt7 (г)), где Р — постоянная, a U — потенциал внешнего поля. На самом
деле оба процесса взаимосвязаны и распределение по скоростям становится
строго максвелловским только при достижении полного равновесия [235].
Если же принять во внимание и химическую реакцию, то процесс уста-
новления равновесия еще более усложняется за счет эндо- и экзотермичности
реакции и размножения (рождения и соединения) частиц в каждом элементе
объема. Так, например, по данным, полученным спектральными, зондовыми
и теневыми [236] методами, в случае перемешивания плазменной струи аргона
(Гнач = 3000 К) и метана (Т^ач = 300° К) время перемешивания тпер
JjlO-5 сек, а время образования ацетилена из распадающегося метана треак —
—10~4 сек. Ясно, что некоторая (подлежащая специальному исследованию
и определению) часть реакции происходит в весьма сложных неравновесных
условиях (в отличие от обычной химии, где тпер <С гхимр)-
Поэтому проблема описания смешения реагирующих газов входит со-
ставной частью в плазмохимическую кинетику. Для этого необходимо ис-
следовать эту задачу экспериментально в условиях различных плазменных
струй и рассмотреть систему уравнений гидродинамики и химической кине-
тики, воспользовавшись также методами теории подобия [237].
Скорость газофазных химических реакций определяется, как известно,
эффективной частотой столкновений между молекулами реагентов. В том
случае, когда смесь реагентов не является гомогенной1, скорость химиче-
ской реакции зависит, вообще говоря, как от степени неоднородности данной
смеси, так и от скорости релаксации ее характеристик к характеристикам
гомогенной смеси. Таким образом, в этом случае может в значительной мере
проявиться связь физической и химической кинетики, представлениями ко-
торых обычно пользуются для описания поведения релаксирующих систем.
В качестве сравнительно простого примера системы, являющейся крайней
противоположностью гомогенной химически реагирующей смеси, можно
представить себе совокупность полубесконечных объемов двух веществ, где
в изотермических условиях наряду с взаимной диффузией (перемешиванием)
веществ происходит бимолекулярная химическая реакция. Скорость хими-
ческой реакции в такой системе тем сильнее отличается от скорости той же
реакции в гомогенной смеси тех же реагентов, чем ближе характерное время
Треак химической реакции к характерному времени тпер процесса диффузии.
В пределе треак <С тпер скорость химической реакции определяется, оче-
видно, скоростью физического процесса перемешивания реагентов («очень
быстрая» химическая реакция).
Для ряда плазмохимических процессов, целевой продукт которых полу-
чается в так называемой кинетической области, крайне важно решение про-
блемы организации перемешивания плазмы и реагентов [233]. Сходная с этой
проблема имеет место также при расчете процессов горения в турбулентных
потоках и определении параметров баллистических следов, остающихся за
телами, летящими с большой скоростью в газах и жидкостях.
Рассмотрение общего случая протекания химической реакции одновре-
менно с перемешиванием двух (или большего числа) турбулентных потоков
(один из которых может представлять собой, как это часто бывает в приклад-
ной плазмохимии, запыленную струю) является настолько сложной зада-
чей, что ее решение до сих пор не найдено. Это обусловлено не только
известными трудностями описания механизма турбулентности [237], нелиней-
1 Реагенты считаются гомогенно перемешанными, если для любого сколь угодно мало-
го (но достаточно для статистического описания) объема смеси отношение средних чи-
сел молекул реагентов в отсутствие реакции равно отношению исходных количеств
реагентов, взятых для приготовления смеси.
110
ностью кинетических уравнений, но и необходимостью точного учета простран-
ственной неоднородности полей температур и концентраций инертных и
реагирующих компонент.
Эта проблема подробно изложена в разделе 4 настоящей книги.
ХИМИЧЕСКИЕ РЕАКЦИИ В НЕИДЕАЛЬНОЙ ПЛАЗМЕ
Высокие плотности, при которых реализуется неидеальная плазма, де-
лают весьма своеобразным характер химических реакций в ней. Отдельные
особенности уже обсуждались в некоторых работах. Дадим краткое изложе-
ние проблемы, следуя [252].
Увеличение плотности приводит к тому, что в заметных концентрациях
могут образовываться многоатомные молекулы и даже весьма сложные конг-
ломераты частиц, содержащие — 10—102 атомов,— так называемые класте-
ры. Высокие температуры смещают равновесие в сторону образования ионов,
поэтому большее значение могут иметь не нейтральные комплексы, а ионы
многоатомных молекул и заряженные кластеры — положительные [238] и
отрицательные [239]. В связи с этим весьма важно установить номенклатуру
возможных многоатомных химических соединений, нейтральных и заряжен-
ных. Обзор ионных кластеров приводится в [240]. Были проведены оценки
состава литиевой плазмы с учетом положительных многоатомных ионов и
рассмотрено влияние на электропроводность изменения расчетного состава,
обусловленного учетом ионов [238]. Весьма интересна кинетика образования
и распада сложных многоатомных комплексов — скорости этих процессов
могут быть достаточно малыми, что может быть использовано для вывода
сложных соединений из плазмы. В работах [241—243] образование заряжен-
ных кластеров связывается с предпереходными явлениями, предшествующими
фазовым переходам.
Выше рассматривались химически устойчивые соединения с положитель-
ной энергией связи. Эти соединения могут быть выделены из плазмы; при от-
сутствии разрушающих столкновений и внешних воздействий они могут су-
ществовать сколь угодно долго. Кроме стабильных соединений, могут иметь
существенное значение и нестабильные. Наряду с флуктуационными и мета-
стабильными соединениями здесь можно указать на резонансы. Резонансы —
короткоживущие соединения с отрицательной энергией связи. Резонансы
спонтанно распадаются в результате автоионизации или автораспада. Авто-
ионизационные состояния хорошо известны для атомов, ионов и простых
молекул. Такие состояния должны также существовать для многоатомных
молекул и молекулярных ионов и кластеров. Резонансные кластеры могут
быть нейтральными и положительными, а также и отрицательно заряженны-
ми. Резонансы, как правило, живут недостаточно долго, чтобы их можно бы-
ло вывести из плазмы и наблюдать в изолированном виде, однако срок их
жизни достаточен для того, чтобы они могли вносить определенный вклад
в различные свойства неидеальной плазмы. Нестабильные кластеры могут
также определять предпереходные явления.
Влияние неидеальности сказывается в том, что меняется сам подход к
расчету химических равновесий. В неидеальной плазме в результате взаимо-
действия частиц изменяются потенциалы ионизации и диссоциации, межъ-
ядерные расстояния и т. п.: необходимо также уточнение понятия стати-
стического веса. Влияние неидеальности на диссоциацию CsCl рассматрива-
лось в [241]; если в разреженной плазме CsCl распадается преимущественно
на атомы Cs и С1, то в неидеальной плазме в силу снижения потенциала дис-
социации распад происходит преимущественно на Cs+ и С1~.
Уменьшение статистического веса может быть связано с микрополями и
конечностью объема, занятого окружающими частицами [242, 243]. Возмо-
жен и предельный случай, когда статистический вес с увеличением плотнос-
ти обращается в нуль и данный сорт частиц вообще не образуется в плазме
111
в таких условиях. Обращение статистического веса в нуль рассматривалось
для отрицательных ионов в [242], для атомов — в [243] и для молекул CsCl —
в [244]. Такой подход позволил в [244] объяснить, почему экспериментально
наблюдаемые спектры излучения азотной плазмы при 1 атм содержат спектр
фотоприлипания N“, а при 102 атм этот спектр пропадает.
Невозможность существования некоторых компонент в сильнонеидеаль-
ной плазме приводит к тому, что химические реакции идут не по всем кана-
лам. Приведем пример, рассмотренный в [241]. В обычных условиях концен-
трации зарядов в плазме CsCl определяются реакции Cs = Cs+ + е и С1~ —
= Cl + е. В сильнонеидеальном случае может возникнуть такая ситуация,
когда атомы Cs не образуются, поскольку их статистический вес обратился
в нуль, и остается только одна реакция С1~ = С1 + е, равновесие которой
следует рассчитывать с учетом того, что она происходит в сильнонеидеаль-
ной системе зарядов Cs+, С1~ и е.
Наличие взаимодействия частиц приводит к тому, что необходимо рассмат-
ривать термодинамическую устойчивость химического равновесия. Устой-
чивость равновесия CsCl = Cs+ + С1“ в сильнонеидеальном случае рассмат-
ривалась в [241]. Нарушение устойчивости было истолковано как указа-
ние на фазовый переход первого рода. Две фазы отличаются друг от друга
концентрацией ионов.
В целом характер изменения химического состава плазмы при переходе
от идеальногазовой к сильнонеидеальной плазме можно представить следую-
щим образом. Вначале происходит увеличение числа различных соединений,
существующих в плазме. Увеличивается доля стабильных и резонансных
многоатомных молекул и кластеров: с ростом температуры преобладают за-
ряженные соединения; увеличению доли заряженных многоатомных частиц
способствует уменьшение потенциалов ионизации. С дальнейшим ростом
плотности начинает связываться уменьшение статистического веса. Таким
образом, концентрация каждой компоненты, исключая простейшие, с ростом
плотности проходит через максимум [246]; очевидно, что для разных ком-
понент эти максимумы соответствуют разным условиям.
Доля сложных соединений данного размера (в качестве размера можно
принимать количество ядер, содержащихся в кластере) уменьшается в поль-
зу образования кластеров больших размеров. В последних возникает упо-
рядоченная структура [238, 240, 245]. При этом произведение доли новых
кластеров на их размер растет, хотя сама доля вновь образовавшихся клас-
теров уменьшается. В пределе она стремится к единице, и плазма переходит
в состояние, напоминающее жидкометаллическое, в состав которого входят
все ионы и электроны системы.
Весь этот переход от газовой к жидкометаллической плазме не обязатель-
но происходит плавно: ионизационное и химическое равновесия могут терять
устойчивость, т. е., вообще говоря, возможны фазовые переходы [252].
3
ОСОБЕННОСТИ МЕХАНИЗМОВ
ПЛАЗМОХИМИЧЕСКИХ РЕАКЦИЙ.8
РОЛЬ ЗАРЯЖЕННЫХ И ВОЗБУЖДЕННЫХ ЧАСТИП
Одно из основных отличий механизмов и кинетики химических реакций
в низкотемпературной плазме от механизмов и кинетики реакций в обычных
химических системах состоит в наличии заряженных и возбужденных час-
тиц, что связано с более высоким уровнем температур и способами получе-
ния плазмы (см. стр. 6—26) [1]. Эти особенности присущи не только плазмохи-
мическим системам. Они проявляются и в других областях химии высоких
энергий — радиолизе, фотолизе, лазерной фотохимии, ударных волнах, а
также существенны при анализе процессов, протекающих в верхних слоях
атмосферы, и т. д. Скорости реакций с участием возбужденных частиц, ионов
и радикалов, как правило, значительно превышают скорости образования
этих частиц [2]. Поэтому суммарная скорость химических превращений будет
определяться в основном скоростями образования и гибели таких частиц,
т. е. скоростями возбуждения, диссоциации, ионизации и рекомбинации.
При таких высоких уровнях температур, когда становится существенным
влияние возбужденных частиц на реакции, необходимо учитывать всю слож-
ную структуру квантовых уровней молекул, наличие конкуренции каналов,
приводящих к химическим превращениям и к физической релаксации —
процессам переходов между различными уровнями (см. стр. 50). Внешние
силы, действующие в типичных плазмохимических системах, электромагнит-
ные поля, поля излучений и т. п., а также интенсивный массо- и энергооб-
мен с окружающей средой приводят к нарушению максвелл-больцмановских
равновесных распределений частиц по поступательным и внутренним степе-
ням свободы. К отклонениям от равновесия может приводить и протекание
быстрых химических реакций. При этом часто реализуются такие случаи,
когда нельзя ввести единого параметра — температуры, и для описания си-
стемы необходимо использовать функции распределения частиц. Наиболее
часто реализуются в плазме такие случаи неравновесности, когда средняя
энергия электронов отличается от средней энергии тяжелых частиц (см. стр.
102) и распределения по внутренним степеням свободы также являются не-
равновесными (см. стр. 59). Иногда удается описать такие системы, вводя
различные температуры заселения уровней для каждой из внутренних сте-
пеней свободы: вращательного (7^), колебательного (Tv) и электронного
возбуждения (Гэл)-
В данной главе коротко изложены сведения о механизмах, сечениях и
коэффициентах скоростей возбуждения различных степеней свободы моле-
кул. Подчеркивается роль электронного удара в возбуждении. Затем рас-
смотрены процессы, приводящие к диссоциации и ионизации молекул под
действием электронного удара, и произведено сравнение эффективности раз-
личных процессов в инициировании химических реакций. Приведены сведе-
ния о ступенчатой диссоциации и ионизации молекул и атомов. Обсуж-
дены вопросы рекомбинации тяжелых частиц (ударно-радиационная ре-
113
комбинация), а также гетерогенная рекомбинация на поверхности раздела
фаз. Проведено теоретическое рассмотрение процессов диссоциативной ре-
комбинации молекулярных ионов с электронами и обратного ему процесса
ассоциативной ионизации, имеющих большое значение в установлении иони-
зационного равновесия.
Анализ всех этих процессов завершается рассмотрением вопросов диссо-
циационного и ионизационного равновесия в низкотемпературной плазме и
установлением причин, вызывающих отклонения от равновесия.
При изложении материала широко используется принцип микроскопиче-
ской обратимости (см. стр. 76) для связи сечений прямых и обратных про-
цессов, а также принцип квазистационарной заселенности короткоживущих
промежуточных состояний (см. стр. 44). Задача обобщенной химической ки-
нетики состоит в определении механизмов и скоростей химических превраще-
ний в системах с неравновесным распределением частиц по энергиям и уров-
ням исходя из заданной схемы уровней и вероятностей переходов между ни-
ми (прямая задача).
Расчет вероятностей переходов выходит за рамки данной книги, и в
соответствующих местах приводятся ссылки на литературу, посвященную
этим вопросам.
В ряде случаев полученное решение прямой задачи используется для на-
хождения по измеренным экспериментально коэффициентам скоростей про-
цессов вероятностей переходов (обратная задача) (см. стр. 159, 160, 183).
СТРУКТУРА УРОВНЕЙ И ПОТЕНЦИАЛЬНЫЕ КРИВЫЕ
ЭЛЕКТРОННЫХ СОСТОЯНИЙ МОЛЕКУЛ
Энергия молекулы в возбужденном состоянии Г ~ (i, /) G — набор
квантовых чисел, характеризующих электронное состояние молекулы; —
колебательное, /7 — вращательное квантовые числа) может быть представ-
лена в виде суммы [3, 4]
-Е.+ £7+ М, (3.1)
составляющие которой соответствуют энергии электронного, колебательного
и вращательного возбуждения при R = Re, где Re — равновесное межъ-
ядерное расстояние в молекуле. В общем случае энергия электронного взаи-
модействия зависит от межъядерного расстояния; будем обозначать ее
Ui (R). Часть из них имеет минимум в определенной области межъядерных
расстояний R, такие состояния мы будем называть стабильными; если же
кривые электронной энергии не имеют минимума ни при каких R — неста-
бильными. Заметим, что стабильные электронные состояния имеют минимум
энергии лишь в ограниченной области межъядерных расстояний. При ма-
лых расстояниях их энергия превышает энергию диссоциации, т. е. распада
на фрагменты. Эту часть потенциальной кривой стабильного состояния усло-
вимся называть отталкивательной ветвью кривой.
Энергия колебательного возбуждения молекул в данном электронном
состоянии представляется в виде ряда
= (Ое + “g") + ®еУе (17 + ~2") — ' ’ ' ’ (^*^)
где о)е — величина первого колебательного кванта; ®еУге и т. д.— па-
раметры, характеризующие ангармоничность колебаний молекулы в элект-
ронном состоянии I.
Энергия вращательного возбуждения также описывается рядом
= Blj (/ + 1) - Di/2 (7 + I)2 + • • •, (3.3)
114
где
Bv= Ве --- (у + х/2) + а2 (р + 1/2)2 - • • • ч
о о Л • (3.4)
Dv = De — рг (у + 1/2) + р2 (Р + ’’’Л)2 — • • •»
В\ = h/8myR2e. (3.5)
В этих формулах Ве — равновесное межъядерное расстояние в i состоянии,
|л — приведенная масса молекулы, с — скорость света, h — постоянная
Планка.
Для коэффициентов в формулах (3.3) — (3.4) выполняются соотношения
Bv^> Dv^>> • • •, о(2 , De^> pi^> р2* • •, (3-5)
ввиду чего при не слишком больших v и J в формулах (3.1) — (3.3) можно
ограничиться первыми членами разложения.
В настоящее время наиболее точным методом построения потенциальных
кривых Ut (R) является восстановление их из спектроскопических данных
[3—5]. Таким образом можно построить только ту часть кривой, которой
соответствуют энергии ниже соответствующего потенциала диссоциации
(связанные состояния). Отталкивательные ветви кривых стабильных состоя-
ний и потенциальные кривые нестабильных состояний можно определить
расчетным путем. Однако точно квантовомеханическая задача расчета энер-
гии в настоящее время решена только для атома водорода. А уже в случае
молекулы водорода, а тем более для других молекул используются различ-
ные приближенные методы [6—8]. В расчетах такого рода наблюдается замет-
ный прогресс в смысле улучшения точности и приближения результатов рас-
чета к результатам эксперимента [6]. Отталкивательные ветви кривых
можно определить несколько точнее, чем кривые потенциальной энергии не-
стабильных состояний, поскольку в этом случае возможна экстраполяция
потенциала взаимодействия, определенного спектроскопическим путем [3, 9].
Вместе с тем необходимо отметить, что потенциальные кривые нестабильных
и отталкивательные ветви стабильных электронных состояний играют боль-
шую роль в физике и химии молекул. Они являются причиной таких явле-
ний, как предиссоциация (диссоциация молекул из стабильного электрон-
ного состояния ниже соответствующего ему предела диссоциации), обратного
ей процесса излучательной (радиационной) и ударно-радиационной реком-
бинации атомов и фрагментов молекул в более сложные молекулы (см. стр.
153), диссоциативная рекомбинация ионов с электронами и ассоциативная
ионизация (стр. 168).
Для расчета вероятностей и коэффициентов скоростей всех перечисленных
процессов необходимо иметь точно построенные потенциальные кривые не-
стабильных состояний. Следовательно, прогресс в этой области будет возмо-
жен, по-видимому, только при увеличении точности расчета потенциальных
кривых. Однако в случае отталкивательных кривых возможно решение об-
ратной задачи — построение потенциальных кривых по измеренным вероят-
ностям предиссоциации [3, 10—12].
ВОЗБУЖДЕНИЕ ВРАЩАТЕЛЬНЫХ УРОВНЕЙ МОЛЕКУЛ
1. Возбуждение электронным ударом
При соударении электронов с молекулами прямое возбуждение вращатель-
ных переходов, связанное с передачей энергии электрона непосредственно
на вращение, малоэффективно ввиду малости массы электрона.
Теоретические расчеты для сечения вращательного возбуждения гомо-
ядерных двухатомных молекул электронами дают с учетом правила отбора
115
(&j = +2) следующее выражение [13]:
,i (Р\ - 8яао (/ + 2)(/ + 1) 9о Г, (4/ + 6)В’ j*
г;^+2( е) 15 (2/ + 3)(2/ + 1) А* |/
Для сечения обратного процесса [14]
.(Е ) = 8яа"
(3.7)
(3.8)
В этих формулах а0 — радиус Бора; q0 — квадрупольный момент молеку-
лы; е, те, Ее — заряд, масса и энергия электрона.
Методов прямого экспериментального измерения сечений вращательного
возбуждения в настоящее время не существует. Сравнение расчетов по (3.7)
с величинами потерь энергии электронов на вращательное возбуждение мо-
лекул в электронных роях [15] показывает, что они удовлетворительно опи-
сывают экспериментальные данные (N2, Н2, D2, О2). Величины сечений
вращательного возбуждения в области энергий от порога до 10~2—10-1 эВ
достигают для этих молекул нескольких единиц на 10~17 см2. В случае
негомоядерных молекул, взаимодействие которых с электронами обуслов-
лено дипольным моментом (осо), сечение больше и, согласно [16], равно
(^е)|—
^те / cto \2
ЗЕо \ еао /
СТ 1 Jn
2/ + 1
1——
(3-9)
Сравнение расчетов по этой формуле с результатами экспериментов в
роях для молекулы СО показывает, что расчет дает значение, завышенное
более чем на порядок величины. Из экспериментов следует, что сечение вра-
щательного возбуждения молекул электронами и в этом случае не превыша-
ет нескольких единиц на 10~16 см2 [15].
Кроме того, вращательное возбуждение молекул может происходить в
результате резонансного процесса образования и распада промежуточного
нестабильного молекулярного отрицательного иона. В этом случае сечения
вращательного возбуждения также могут достигать значений порядка не-
скольких единиц на 10~16 см2 [17—20]. Однако резонансные процессы имеют
место при больших энергиях электронов (несколько электронвольт), при ко-
торых более существенным при передаче энергии от электронов к молекулам
становится процесс колебательного возбуждения (см. стр. 118). Кроме того,
максимумы сечений резонансного возбуждения не сильно превышают сече-
ния прямого возбуждения, и ввиду малой ширины резонансов такие про-
цессы, по-видимому, не будут сильно влиять на скорость вращательного воз-
буждения молекул электронами.
2. Вращательное возбуждение
при соударении с тяжелыми частицами
Возбуждение вращательных уровней молекул при соударениях с тяже-
лыми частицами протекает более эффективно, чем при соударениях с элект-
ронами, поскольку в этом случае при любом соударении в направлении,
не совпадающем с осью молекулы, возможна передача большой доли посту-
пательной энергии тяжелых частиц на вращение.
Число соударений = Сч/т/)'1 (где tj — время вращательной, а т0 —
поступательной релаксации), необходимое для полного обмена энергией
между поступательной и вращательной степенями свободы тяжелых частиц
116
(Мг и M2), описывается выражением
у __ 3 (Л/1 + Л/2) (1 + Ь)2 /о ллч
- Т Mi ъ • (5ли)
В выражении (3.10) Ъ = 4//[xd2, |л = M1M2J(M1 + Af2), d = dr + d2;
Mt, dt — массы и диаметры сталкивающихся частиц; I — момент инерции
относительно оси, проходящей через центр масс.
Число соударений Zj, необходимых для установления равновесия между вра-
щательной и поступательной степенями свободы в чистых газах при Т =
— 300° К [21], показано ниже:
Газ N2 Ог Н2 d2 сн4 cd4
Zj 4—13 2-30 350—520 200 14—17 12
Газ cf4 С(СНз)4 SFe ecu SiBr4 SiH4
Zj 6 7 7 6 5 28
В чистом двухатомном газе
„ 3 (1 + 2bi)2 - 8 bi (3.11)
тде b1 = 21/^d^ Результаты расчетов по этим формулам удовлетворительно
согласуются с экспериментальными данными, полученными ультраакусти-
ческими методами или из коэффициентов переноса — диффузии, вязко-
сти, теплопроводности (табл. 3.1).
Таблица 3.1
Число соударений Z-, необходимых для вращательной релаксации при соударении
молекул Mi с одноатомными газами [21]. Т -- . 300° К
Ml M2 I Mi m2
He Ne Ar Kr Xe He Ne Ar Kr Xe
CH4 8 34 77 169 302 SFe 2 2 3 6 10
cd4 4 18 39 29 159 CC14 2 2 3 5 7
C(CH3)4 2 3 5 11 17 cf4 2 2 4 8 15
Для всех молекулярных газов Zj колеблется от нескольких единиц до
нескольких десятков. Увеличение массы партнера по соударению (Не
->Хе в табл. 3.1) приводит к увеличению Zj, т. е. замедлению вращательной
релаксации, что связано с меньшей эффективностью передачи энергии при
соударении частиц разной массы (6упр = 2М1/М^).
С ростом температуры число соударений Zj увеличивается:
(3-12)
чще Т* = &/к — величина, характеризующая глубину потенциальной ямы
Фхежмолекулярного потенциала [22].
Исключением является вращательная релаксация в водороде и дейтерии,
для которых существенны квантовые эффекты, что связано с большой вели-
чиной вращательного кванта для этих молекул. Для них квантово-механи-
ческим путем рассчитаны вероятности вращательных переходов [21]. Для
остальных молекул таких расчетов практически не имеется. Однако можно
сделать вывод, что вращательная релаксация протекает не через однокван-
«товые переходы Д/ — +1, а путем многоквантовых переходов.
117
3. Роль электронного удара во вращательной релаксации
Влияние электронного удара на заселенность вращательных уровней и
вращательную релаксацию можно оценить, сравнивая скорости возбуждения
молекул электронами и тяжелыми частицами.
С учетом приведенных выше данных имеем
wje) <фе> Ne 7 г м
w<№ 10~16 ’ ' те е ’
где хе = Ne/Nм — степень ионизации плазмы.
(3.13)
Отсюда следует, что при Те — Тт в азоте и кислороде заселение враща-
тельных уровней электронным ударом становится существенным при степе-
нях ионизации хе 7> 10“х, а в водороде, окиси углерода и, по-видимому, в
газе других линейных многоатомных молекул — при хе '>> 10“2.
Поэтому в низкотемпературной плазме с малой степенью ионизации
(хе 10“3) влиянием электронного удара на заселенности вращательных
уровней молекул можно пренебречь.
Этим обстоятельством пользуются для определения температуры посту-
пательного движения частиц в плазме спектральным методом относительных
заселенностей вращательных линий (см. стр. 30). Однако при использо-
вании этого метода следует иметь в виду, что приведенные выше оценки спра-
ведливы для основного электронного состояния молекулы и для таких воз-
бужденных электронных состояний, в которых время жизни достаточно ве-
лико по сравнению с временем вращательной релаксации.
Тем не менее вращательную температуру часто измеряют по спектрам
испускания молекул. В этом случае верхнее излучающее состояние имеет,
как правило, время жизни, малое по сравнению с временем вращательной
релаксации. Тогда для обоснования соотношения Тj = Т? необходимо спе-
циальное исследование механизма возбуждения верхнего излучающего со-
стояния. Анализ показывает, что это соотношение остается справедливым
при возбуждении прямым электронным ударом из основного состояния. Та-
кой механизм часто реализуется в разрядах с малой степенью ионизации
при пониженных давлениях (тлеющий, высоко- и сверхвысокочастотный),
но он не является единственно возможным даже в условиях этих разрядов
[23—26]. В частности, если разряды зажигаются в смесях молекулярных
газов с инертными, возможно заселение короткоживущих возбужденных со-
стояний молекул в результате процессов пеннинговской ионизации или пе-
редачи возбуждения, которые приводят к существенному отличию заселен-
ностей вращательных уровней от больцмановского закона с Тj = TY [26].
ВОЗБУЖДЕНИЕ
КОЛЕБАТЕЛЬНЫХ УРОВНЕЙ МОЛЕКУЛ
1. Колебательное возбуждение электронным ударом
Процесс прямого колебательного возбуждения молекул электронным
ударом также является малоэффективным. Соответствующие ему сечения
достигают величин порядка — 10“17—10“16 см2 [15, 16]. Они ниже значений,
наблюдаемых экспериментально для ряда двух- и многоатомных молекул.
Кроме того, в сечениях колебательного возбуждения большинства молекул
обнаружены резонансы, которые объясняются образованием нестабильного
промежуточного отрицательного иона, наличие которого подтверждается и
угловыми распределениями неупруго рассеянных электронов [27, 28]:
Ги(г'|/£'; Ее) ГИ(/Иг';Е')
АВ (Г) + е ;......... АВ" (А') 7........АВ (^). (3.14)
rW;Ke)
118
В настоящее время резонансы в сечениях колебательного возбуждения
электронами обнаружены для всех исследованных молекул: Н2, N2, О2, NO,
СО, СО2, Н2О, N2O и др. [14, 15, 17, 18, 19, 27, 29, 30-40]. В максимумах
сечения таких процессов достигают 10“16—10~15 см2 (рис. 3.1—3.3).
Сечение возбуждения колебательных уровней можно связать с вероят-
ностями автоионизации промежуточных отрицательных ионов, если вос-
пользоваться принципом микроскопического детального баланса. В квази-
стационарном приближении сечение возбуждения колебательного уровня
состояния i с уровня vio этого же состояния через колебательный уровень
промежуточного состояния vk отрицательного иона равно
/ I I . ЛЛЧ Г I Zc') г (q I/е')
° (vii | vk I Viv, — 2meEe (Ee — ЬЕ.,к,¥!№ + £ (Л')2/4 ’ ' ’ ’
здесь Г (f | k') — вероятность перехода в сект1, h — постоянная Планка.
Из выражения (3.15) следует, что ширина резонансных пиков в сечении
колебательного возбуждения равна величине ЛГ (к'), где Г (к') — полная
-вероятность распада уровня к' по всем каналам. После интегрирования
(3.15) по энергии электронов получаем выражение для полной площади пика
Г я2?;3 £к' Г (in I к') Г (L |/с')
\ б (Рй I vk I Ее) Ее dEe = —-----------0 г + - - - — . (3.16)
\ ч I л I ьо’ е/ е е g g., Г (kj ' '
Полное сечение возбуждения уровня vir с уровня vio, получается сумми-
рованием (3.15), (3.16) по всем промежуточным уровням vk. Оно имеет вид
отдельных резонансов, соответствующих образованию отрицательных ионов
на отдельных колебательных уровнях vk. Если время жизни промежуточ-
ного состояния отрицательного иона больше характерного времени колеба-
ния ядер, то вероятность распада можно записать в следующем виде:
Г (А') = 2 Ге (/, ) q^, (3-17)
= (3-18)
где — среднее межъядерное расстояние, Ге — вероятность электрон-
ного перехода. Согласно данным [40], такой случай реализуется для всех
молекул, кроме водорода. В случае азота и окиси углерода (см. ниже) эти
времена сравнимы по величине, и приближение (3.17), (3.18) применимо при
больших межъядерных расстояниях, но неприменимо при малых. Ниже при-
ведена зависимость Ге от межъядерного расстояния (R — Re) для иона азо-
та Ns (Х2Щ) [40]:
R — Re, ат. ед. —0,1 +0 +0,1 +0,2 +0,3 +0,4 +0,5 +0,6
Ге, 1014 сек-1 10,8 8,6 5,8 3,6 2,1 1,2 0,61 0,18
При отсутствии зависимости вероятности электронного перехода Ге от
межъядерного расстояния величины сечений в отдельных пиках определяют-
ся только взаимным расположением потенциальных кривых молекулы и со-
ответствующего отрицательного иона (<7*.^).
Выражения (3.15) — (3.18) можно использовать для нахождения сечений
резонансного колебательного возбуждения молекул с возбужденных коле-
бательных уровней (yi0 0) по измеренным сечениям колебательного воз-
буждения молекул из основного состояния (vi0 = 0). Параметры, характе-
ризующие потенциальные кривые ряда отрицательных ионов, и зависимости
вероятностей автоионизации от межъядерных расстояний приведены в рабо-
те [40]. Там же имеется библиография по расчету сечений колебательного
возбуждения ряда молекул.
119
б, етн.ед.
б, етн. ед.
Рис. 3.1. Сечения возбуждения колебательных уровней vi = 1—8 при столкновении
молекулы NgCX1!^ с электронами [35]
При Е е = 2,3 эВ сумма сечений равна 5*10~15 см2 [281
Рис. 3.2. То же, что на^рис. 3.1, для С0(Хх2^, v = 0)
При Ее — 3,8 эВ сумма сечений равнаУ8,3’10-1€ см2 [40]
fl
В заключение этого раздела необходимо отметить, что ни теоретического,
ни экспериментального исследования зависимости величины Ге от номера
вращательного уровня не проводилось до настоящего времени ни для одной
из молекул. Такая зависимость в принципе могла бы иметь место, если взаи-
модействие, приводящее к автоионизации, зависит от вращательного состоя-
120
ния иона (ср. с вероятностями автоионизации молекул, стр. 178). В то же
время наличие такой зависимости может существенно повлиять на сечение
колебательного возбуждения при повышении температуры заселения вра-
щательных уровней (например, в плазме различных разрядов).
В тех случаях, когда переход происходит на отталкивательные ветви
кривых иона АВ~ (&')> возникает конкурирующий процесс — распад иона
Рис. 3.3. То же, что на рис. 3.1,
для Vi = 1,2, v = 0)
1 — = 1; 2 — V. = 2J
АВ~ с образованием осколочного отрицательного иона. В этом случае резкой
колебательной структуры в сечениях наблюдаться не будет, а сечения ко-
лебательного возбуждения с участием иона АВ~ (к') должны уменьшаться.
Подробнее процесс диссоциативного прилипания электрона будет рассмот-
рен ниже (см. стр. 146).
Резонансное возбуждение колебательных уровней молекул электронами
приводит к большой вероятности обмена энергий между ними [15, 41].
Колебательное возбуждение
при соударениях тяжелых частиц
Возбуждение колебательных уровней молекул при соударениях с тяже-
лыми частицами исследовалось теоретически и экспериментально в большом
количестве работ (см., например, [21, 42, 43] и ссылки в цитируемых рабо-
тах).
Теоретические расчеты в большинстве случаев позволяют получить тем-
пературную зависимость вероятностей переходов, а абсолютные значения
Рис. 3.4. Зависимость вероят-
ностей V — У-пер входов
(PV, v+1 — кривые 1—6) и
V — 7-переходов (Р’Л’+1 —кри-
вые 7—12) при соударении моле-
кул азота от номера колебатель-
ного уровня при разных тем-
пературах [54]
I, 7 — Т = 3340°К;
2, 8 — 1500°;
3, 9 — 1000°;
4, 10 — 750°;
5, 11 — 500°;
6, 12 — 334°
необходимо уточнять путем сравнения результатов с экспериментальными
данными.
До недавнего времени единственным методом экспериментального иссле-
дования колебательной релаксации являлись ударные трубы [21, 44—47].
121
Однако с созданием лазеров появилась возможность исследования перехо-
дов между отдельными уровнями методом лазерной флуоресценции [48—53].
При исследовании колебательной релаксации в чистых многоатомных га-
зах или смесях наряду с поступательно-колебательными V — ^-перехода-
ми необходимо учитывать процесс обмена колебательными квантами (У —
У-процесс). Типичная зависимость вероятностей У — Т- и У—У-процессов
от номера колебательного квантового уровня при соударении с невозбужден-
ными молекулами для молекулы азота изображена на рис. 3.4.
3. Влияние электронного удара
на колебательное возбуждение молекул
Несмотря на то что сечения колебательного возбуждения молекул элект-
ронами не очень сильно превышают сечения вращательного возбуждения,
роль электронного удара в колебательном возбуждении оказывается более
существенной, чем в случае вращательного возбуждения (см. стр. 118). Это
обусловлено малой вероятностью колебательных переходов при соударе-
ниях тяжелых частиц.
В табл. 3.2 приведены результаты расчета вероятностей возбуждения
первого колебательного уровня молекулы азота электронами [55] и столк-
новениями с другими молекулами при Те = Тг [44]. В последнем столбце
указаны значения степени ионизации х^ при которой скорости обоих про-
цессов сравниваются.
Решение задачи совместного влияния возбуждения молекул электронным
ударом (е — V), колебательно-поступательных (У — ^-переходов и обмена
колебательными квантами (V — V) (см. стр. 59) [56, 57], выполненное нами
Таблица 3.2
Вероятности колебательного возбуждения N2 электронами и тяжелыми частицами
Т, Юз °к 4е?. см3 -сек-1 см3-сек-1 хе Т, Юз °к и,(е) и01 ’ с-мз-сек—1 см3-сек-1 хк хе
1 2-W-13 2,9. ю-18 1,4-10-5 8 1,3-ю-9 2,9-10-13 2,3-10-1
1,5 5,6-10-12 5,9-10-17 1,0-10~5 10 1,6-10-9 0,7-10-12 4,3-10-«
3 1,6-10-1» 3,3-10-15 2,0-10-5 15 2,3-10-9 2,6-Ю-12 1,1-Ю-3
5 6,0-10-1» 4,1-10-14 7,0-10-5
для молекул N2, в случае существенного отрыва температуры электронов
от температуры тяжелых частиц Те Тг показало следующее:
1. Распределение заселенностей колебательных уровней в общем случае
отлично от больцмановского и не может быть описано единым значением
колебательной температуры Tv (см. рис. 2.3—2.5) и в процессе релаксации
существенно нестационарно.
2. При степенях ионизации хе 10 ~4 распределение приближается к
больцмановскому с Те = Tv.
Полученные результаты являются следствием учета ангармоничности
молекулы и влияния колебательного обмена при достаточно эффективном
возбуждении колебательных уровней электронным ударом. Поэтому каче-
ственные черты процесса релаксации молекул при совместном влиянии
е — V-, V — Т-и V — У-процессов должны сохраниться и для других моле-
кул. В действительности численные расчеты, выполненные для молекулы СО
с учетом е — У-процессов [58, 59], а также без учета е — У-процессов, но в
предположении мощного потока на заселение первого колебательного уров-
ня Тг), что имитирует в некоторых чертах влияние электронного уда-
ра [58, 60], также дают картину стационарной заселенности колебательных
122
уровней, качественно совпадающую с результатами расчета для N2 |[56,
57, 61].
В заключение следует отметить, что колебательное возбуждение моле-
кул, обусловленное электронным ударом, может существенно повлиять на
ш скорости процессов возбуждения электронных уровней, диссоциации, иони-
зации и других процессов, инициируемых электронами в низкотемператур-
ной плазме.
ВОЗБУЖДЕНИЕ ЭЛЕКТРОННЫХ УРОВНЕЙ
1. Возбуждение электронным ударом
Задача расчета сечений возбуждения атомов и молекул электронным уда-
ром сводится к квантовомеханической задаче нахождения волновых функ-
ций системы, состоящей из налетающего электрона и возбуждаемой частицы.
Точное решение такой задачи в настоящее время невозможно, поэтому
расчет сечений возбуждения электронных уровней проводится в различных
приближениях.
Применимость используемых допущений, как правило, ухудшается по
мере приближения энергии электронов к порогу возбуждения рассматривае-
мого перехода, а в низкотемпературной плазме наибольший вклад в скорости
возбуждения дают именно эти низкоэнергетические области сечений. Поэто-
му особое значение приобретает эксперимент, который при сравнении с тео-
рией позволяет уточнить теоретические формулы и предложить различные
полу эмпирические методы расчета.
В частности, в приближении Борна — Оппенгеймера для сечения возбуж-
дения уровня к с уровня i, если оптические переходы между ними разрешены,
получается следующее выражение:
oiR (£'е) = 4n^/ifc (-Д-)2 In -Д- , (3.19)
\ е/ и/г/с ДЕг/. у , \ /
где а0 — радиус Бора; — сила осциллятора для данного оптического пе-
рехода гRy—потенциал ионизации атома водорода; &Eik = Ek —
-Et.
Выражение (3.19) является точным при больших энергиях электронов
(Ее/кЕ™) > 1.
Сравнение результатов расчета сечений по этой формуле с результатами
экспериментов по возбуждению атомов показывает, что по мере приближе-
ния энергии электронов к порогу возбуждения перехода расчетные сечения,
как правило, в 2—3 раза превышают экспериментальные [62]. Поэтому в
различных работах были предприняты попытки получения полуэмпириче-
ских формул для расчета сечений возбуждения разрешенных переходов в
атомах. Например, в работе [14] предложено выражение
(Ее) = 2л (-Д-Г nalf.k lnJ9d£ + O>9) t (3.20)
а в [63]
(Z?e) = 4л4 (-Д Г In (ЗД, (2.21)
где х = EJ&E1*.
В работе [62] приведены численные расчеты сечений возбуждения различ-
ных типов переходов в атомах и ионах, а также сечений ионизации в различ-
ных приближениях (Борна; Борна — Оппенгеймера с учетом обменного вза-
123
имодействия по методу Очкура в случае интеркомбинационных переходов, а
также в представлении парциальных волн) и результаты представлены в виде
таблиц параметров, с помощью которых можно рассчитать сечения конкрет-
ных переходов и ионизации и вероятности этих переходов, усредненные по
максвелловской функции распределения электронов по скоростям.
В случае столкновений молекул с электронами для сечения перехода с
колебательного уровня электронного состояния i на уровень ик электрон-
ного состояния к в приближениях Борна — Оппенгеймера и 7?-центроиды
имеем [64]
<^>к (Ее) = | Ме (£Е'.\, Ее) |2 (3.22)
где Ме (ДЕ^ 9 Ее) — матричный элемент перехода.
Это выражение отличается от соответствующих формул для возбуждения
атомов только зависимостью матричного элемента перехода от среднего межъ-
ядерного расстояния в молекуле и наличием фактора Франка — Кондо-
на.
В целях удобства сравнения результатов расчетов с экспериментальны-
ми данными выражение (3.22) можно представить в виде
°vivk “ I I2 VVk
\ V-V,. )
x Tn'
(3.23)
где величина | Ме | 2 определяет величину сечения в максимуме функции, а
<р (£e/A£^fe) описывает относительную зависимость сечения от энергии
налетающих электронов. В частности, для оптически разрешенных перехо-
дов в приближении Бете — Борна имеем
\ /
(3.24)
для возбуждения стабильных уровней и
|АГ?(ДЕ)|2 = —
(ЛЕ)
d(\E)
\2
ЬЕ* К „
(3.25)
для возбуждения нестабильных уровней, где е — основание натуральных
логарифмов, (Д2?) — сила осциллятора данного электронного перехода.
Функция ф (х) в этом случае имеет вид
Ы).
(3.26)
Все имеющиеся в настоящее время расчеты сечений возбуждения элек-
тронных переходов в молекулах в результате соударений с электронами вы-
полнены в приближении Борна — Оппенгеймера с различными варианта-
ми учета обменного взаимодействия [64] и 7?-центроиды (Я = 7?е). Экспе-
риментально исследованы только сечения возбуждения с нулевого колеба-
тельного уровня основного электронного состояния (рис. 3.5).
Сравнение выражения (3.23) с экспериментальными данными [64, 65]
показывает, что в случае возбуждения разрешенных переходов функция
<р (х) оказывается универсальной, т. е. в пределах погрешностей она не за-
висит от типа состояний Z, к и сорта молекулы. При этом наблюдается быст-
рый и плавный рост сечения к максимуму, который достигается при х ~ 4,
и медленный спад при дальнейшем увеличении х (рис. 3.6).
Универсальность функции ср (х) была обнаружена также и для иониза-
ции с получением ионов в определенном электронном состоянии (рис. 3.7).
В случае ионизации молекул вид функции ср (х) меняется от молекулы к мо-
124
Рис. 3.5. Сечения возбуждения
различных электронных состоя -
ний молекулы N2 (Х1^, v =0)
электронами
1 ~ а' ПАГ
2 — & Пи (^ = 0—4);
3 — переходы с ХЕ1^ = 12,96 эВ;'
4 — В3 П •
g
5 — С3 П ;
и’'
6 — a"^g;
7 - А3?*;
8 — E3Z+g (AEik = 11,87аВ)
Рис. 3.6. Функция ф(я) для возбуждения оптически разрешенных переходов электро-
нами
1 — Н(18 — 2Р); 2 — — Ш£*); 3 “ H2(X‘Sg — Cinw); 4 — H2(JT1S^ — D‘iy;
5 — СО (AAXg — АЧ1); 6 — N2(X1Zg — vk ~ 7 — расчет по формуле (3.20); 8 — среднее
из кривых 1—6; 9 — расчет по формуле (3.26)
Рис. 3.7. Функция <р(я) для ионизации молекул электронами с образованием иона в оп-
ределенном электронном состоянии
1 — N2(X»Zg, = 0) — (В22*, vk = 0); 2 — CO(X^g, = 0) — СО+(А2П, = 0); 3 —
COU'1^, = 0) — CO+(B2E, vk = 0); 4 — CO2(X‘Sg, vi = 0) — CO2 (a2jj); 5 — CO2(X‘Sg, r = 0) —
—CO2 (B2S); 6 — средняя кривая для полных сечений ионизации
лекуле более существенно (рис. 3.8). Это объясняется тем, что ионизация про-
исходит с образованием ионов в различных электронных состояниях. Кроме
того, большой вклад в сечение ионизации может давать автоионизация из
сверхвозбужденных (автоионизационных) состояний молекул. Все эти про-
цессы имеют различные пороги возбуждения, вследствие чего трудно ожидать
универсальности функции <р (х) для полного сечения ионизации молекулы
(см. стр. 135).
В случае возбуждения уровня к, оптические переходы с которого на уро-
вень i запрещены правилами отбора [3], вид функции qik (х) сильно изменя-
ется для разных переходов даже в одной и той же молекуле (рис. 3.9). Это
связано, по-видимому, с большим вкладом обменного взаимодействия (в слу-
чае разрешенных переходов численные расчеты показывают, что вклад об-
менного взаимодействия в сечение мал) (см., например, [66]), на которое
сильнее влияют детали строения электронных оболочек молекул. В целом
125
Рис. 3.8. Функция ф(ж) для полных сечений ионизации молекул электронами
1 — N,: 2 — СО: з — О2; 4 — Н2; 5 — СО2; 6 — СН4; 7 — С2Н4; 8 — N2O; 9 — средняя из 1—8; 10 —
расчет по формуле Гризинского
Рис. 3.9. Функции (jpifc (х) для возбуждения оптически запрещенных переходов электро-
нами
1 - N2(X‘S^ — С3Пп); 2 — N2(X‘Sg — ашр; 3 — N2(X‘Sg — bTJ; 4 — N2(X‘S* — BsHg); 5 —
CO(X12g — а3П). Кривая 3 приведена для сравнения
сечения возбуждения запрещенных переходов достигают максимума ближе
к порогу перехода, чем в случае разрешенных переходов ^тах = 1,25—1,6.
Значения же сечений в максимумах не уступают соответствующим значени-
ям для разрешенных переходов.
Величины сечений возбуждения оптически разрешенных переходов в
максимумах (Л/е), рассчитанные по формулам (3.24), (3.25), в случае моле-
кул лучше совпадают с результатами экспериментов, чем это имеет место в
том же самом приближении (3.19) для атомов (табл. 3.3).
Использование экспериментальных значений сил осцилляторов в форму-
лах (3.24), (3.25), позволяет избежать ошибки расчета, связанной с выбором
волновых функций, которая может быть довольна велика [66].
Необходимо отметить, что на сечениях возбуждения ряда электронных
состояний N2 (£32g), N2 [38, 68] (рис. 3.5), N2 (А32^), N2 (В3Щ)
[69] и СО (632+) наблюдалась резонансная структура.
Аналогичная структура была зарегистрирована и в сечениях упругого
рассеяния электронов на молекулах Н2, D2, N2, О2, СО, СО2, NO2, H2S, N2O,
С2Н4, СН4, С6Н6 [36, 70]. Эти резонансы обусловлены образованием промежу-
точных отрицательных молекулярных ионов в различных электронно-воз-
бужденных состояниях. В работе [71] теоретически было показано, что кон-
фигурация таких ионов соответствует остову положительного иона, в поле
которого находится два слабосвязанных ридберговских электрона. Поэтому
такие резонансы должны иметь место только при возбуждении уровней,
энергии которых близки к энергиям ридберговских состояний нейтральной
молекулы. Однако расчеты электронных термов отрицательных ионов (на-
пример, [7, 72, 73] ) показывают наличие у отрицательных ионов низколе-
жащих валентных состояний. С ними также могут быть связаны резонансы
в сечении более низколежащих электронных уровней молекул. Такие резо-
нансы наблюдались в дифференциальных сечениях возбуждения N2(A3Sj,
53Щ) [691.
При расчете сечений резонансные процессы не учитываются. Кроме то-
го, из экспериментальных данных для ряда переходов следует, что величи-
на | Ме |2 существенно меняется в зависимости от [64]. Это
обстоятельство также не отражено в расчетах сечений.
126
Таблица 3.3
Сравнение результатов расчета значений М2е для разрешенных переходов
с экспериментальными [67]
Исходное состояние молекулы Возбужденное состояние ДЕ Ry /(е) М%, ТШ&
Формула (3.24) Расчет в прибли- жении Борна Экспери- мент
H2(.¥iSg+, В^и+ 0,725 0,4 0,775 0,55
щ = 0) С’ПИ 0,905 0,3 0,55 0,40 —
1,05 0,05 0,067 0,034 —
N2(jnSg+ 61nw Vk = i 0,92 0,047 0,08 0,28—0,34 0,16
Vi = 0) 0,91 0,28 0,50 1,4—1,7 0,76
Ридберговские 0,92 0,253 0,45 0,9
расстояния
0,96 0,08 0,13 — 0,26
0,98 0,156 0,24 — 0,27
1,01 0,163 0,24 — 0,34
1,04 0,113 0,155 — 0,33
1,06 0,058 0,076 — 0,14
1,07 0,034 0,044 — 0,08
1,10 0,126 0,154 — 0,132
C0(Xi2g+, 0,59 0,1—0,2 0,4-0,8 — 0,51
Vi = 0) В^и+ 0,785 0,007—0,085 0,024—0,054 — 0,041
H(15) 2Р 0,75 0,416 1,1 — 0,28
Cs(6251/2) 62Ч 0,107 0,214 105 — 28
62Д /2 0,102 0,394 56 — 16
Поэтому для расчета сечений и вероятностей переходов между электрон-
но-колебательными уровнями молекул под действием электронного удара в
настоящее время наилучшие результаты могут быть получены при исполь-
зовании экспериментальных данных, представленных в виде (3.23). Такое
представление позволяет экстраполировать экспериментальные результаты,
полученные для возбуждения молекул на нулевом колебательном уровне
основного электронного состояния, на переходы с возбужденных колебатель-
ных уровней. При этом в большинстве случаев погрешность не должна пре-
вышать 20 %.
Для возбуждения разрешенных переходов можно рекомендовать исполь-
зование формул (3.24) — (3.26). Значения сил осцилляторов могут быть
получены из оптических спектров и спектров потерь энергии электронов
при больших энергиях налетающих электронов, которые исследованы для
большого набора молекул [74]. Погрешности расчета при этом могут дости-
гать 100% (см. табл. 3.3). В случае запрещенных переходов можно восполь-
зоваться тем обстоятельством, что сечения по порядку величины не отлича-
ются от сечений возбуждения разрешенных переходов [64]. При этом необ-
ходимо принять во внимание существование приближенных правил отбора
для переходов под действием электронного удара, вытекающих из теории
симметрии. Согласно анализу [75], сечения переходов между уровнями
<-> П„, Ag, «--► Ф„; (3.27)
ng &g, Ilg Ф„, Ag Фй;
П„^А„, Пи~Фв, Д„^Ф„
127
должны быть малы пэ сравнению с сечениями других эквивалентных пере-
ходов, не запрещенных правилами (3.27). Под эквивалентными переходами
имеются в виду переходы между состояниями с одинаковой конфигурацией
электронных оболочек.
Наличие этих правил отбора подтверждается экспериментальными дан-
ными, полученными для возбуждения молекул N2 и СО.
Для получения вероятностей переходов между различными электронно-
колебательными уровнями сечения возбуждения должны быть усреднены
Рис. 3.10. Зависимость CiR (те)
для |возбуждения электронным
ударом различных переходов
1 — оптически разрешенные пере-
ходы;
2 — N2(X‘S^ —
3 -N2(X‘2g~ С3Пр;
4 — N2(X^* - ВШ^);
5 — N2(AriSg — А3Е^)
по функции распределения электронов по энергиям. При этом резонансы,
наблюдаемые в сечениях возбуждения электронных уровней (см. стр. 126)
и имеющие малую ширину (вероятности автоионизации отрицательного ио-
на ~ 1,5-IO14 сект1 соответствуют ширине уровня — 0,1 эВ), будут давать,
как правило, малый вклад в вероятность перехода.
Однако из этого правила возможны исключения, если величина сече-
ния в резонансе больше сечения прямого возбуждения [см., например,
N2 (B3S+ а"1^) (см. рис. 3.5 )].
При максвелловской функции распределения электронов по энергиям
получаем
w(e) (i, Vi I к, vk- T e) = Cik (те) (Л^Д/21 ме 12 exp (- I kTe),
(3.28)
где те = (3/2)(kTe/EElk).
Зависимость величины Cik (те) от относительной средней энергии элек-
тронов приведена для ряда переходов на рис. 3.10. Из данных этого рисун-
ка следует, что при одинаковых значениях сечений в максимуме ве-
роятность возбуждения запрещенных переходов превышает в 1,5—2 раза
вероятность возбуждения разрешенных переходов при малых значениях
параметра те (т. е. при большом расстоянии между уровнями) и уменьшается
по мере роста те, в то время как вероятность возбуждения разрешенных пе-
реходов не меняется.
Эта картина должна обусловливать роль возбуждения запрещенных пе-
реходов в кинетике ступенчатого заселения электронных уровней молекул.
Их роль оказывается существенной в возбуждении низколежащих состояний
(переходы в нижние электронно-возбужденные состояния), но при перехо-
дах между возбужденными уровнями (ДЕг/с кТ^ они будут давать малый
вклад по сравнению с разрешенными переходами.
Для вероятности возбуждения состояния к из состояния i, ввиду малости
изменения коэффициента Сгк (АЕгк)1/2 по сравнению с экспонентой, имеем
приближенное выражение в случае М2 Ф / (А#):
128
_____________ 2
(i | к; Te) Cils (те) (ДЕ VI Me (i I к) p X ----------, (3.29)
2d \
где черта^обозначает усреднение по энергии данного перехода, 2VJ — засе-
ленности колебательных уровней состояния i. Если они заселены по больц-
мановскому закону, характеризуемому колебательной температурой Tv, то
(i I к; Te,Tv) = C’VeWTl Me (i | к) p exp (- Д£** / kTe) X
х[<?ж)г s ^Аехр(-^-)ехрГ-4-(4—4-)l- (3-30>
e ' L \ v e J
где Q\ (Tv) — У exp (—Evt/kTv) — колебательная статистическая сумма для
V
состояния i. Определение энергий Е\, Е% дано в формуле (3.1).
При равенстве температур Те — Tv вследствие правила сумм для факто-
ров Франка — Кондона из (3.30) следует
w(e) (i | к-, Те = Tv) = Cil£ (те) (ДД^‘)1/21 ме (I I к) Р х
X [Qi (Te)JQi О ехр (- Д^ I кТе). (3.31)
Это выражение 1 * * * 5 справедливо и для возбуждения нестабильных состояний.
В таком случае
Qi (П = \ 2 \ ехр (- / кТе) dE°k. (3.32)
гЕ*
Вероятности обратных переходов можно найти, используя принцип мик-
роскопического детального баланса для сечений, следствием которого при
максвелловском распределении электронов по энергиям является соотно-
шение
W(e) (к, rK I t, Vi, Те) = w(e) (i, Vi I к, Г,; Te)4^- exp (ДЕ^/ kTe), (3.33)
где gi>J1, g^1 — электронные статистические веса уровней. Тогда для вероят-
ности перехода к —> г, усредненной по колебательной структуре, в тех же
предположениях, что и (3.30), будем иметь
Ге)= ^-(7* (те) (Д£’*к)1/г| Me(i J А:)р. (3.34)
Соотношение макроскопического детального баланса для усредненных
по колебательной структуре вероятностей переходов i -> к и к -> i в общем
случае не выполняется, а имеет место только в случае Tv = Те:
(i | Л; Те)
&/С " ef
(3.35)
1 Усреднение по вращательным уровням с учетом правил отбора по вращательному
квантовому числу не влияет на сечение перехода i, vj -» к, под действием электрон-
ного удара. В случае зависимости факторов Франка — Кондона от номера вращательного
уровня вероятность перехода (3.28) будет зависеть от температуры заселения враща-
тельных уровней. Внешний вид формул (3.10) — (3.32) при этом не изменится.
5 Теоретич. и прикладная плазмохимия
129
2. Возбуждение и дезактивация электронных уровней
при соударениях тяжелых частиц
Сечения электронного возбуждения атомов и молекул (Мг) вследствие
прямой передачи поступательной энергии в энергию возбуждения при столк-
новении с другой частицей (М2) можно оценить по формуле, полученной в
работе [63] методом Томсона;
ЧЕм,) = 4ла20 (-Д-V F, (3.36)
v 7 L1+ мг+k-^"1)]
где
Сечение таких переходов имеет максимум при энергиях относительного
движения частиц Ем2 (103—104) эВ. Расчет по формуле (3.36) удовлетво-
рительно согласуется с экспериментальными данными, полученными для со-
ударений невозбужденных атомов аргона и гелия в своем газе. Аналогичные
значения сечений были получены из экспериментов по ионизации в ударных
волнах, распространяющихся в инертных газах [76] (табл. 3.4).
Таблица 3.4
Наклон сечений возбуждения атомов инертных газов
при столкновениях с атомами того же сорта
Атом Ек~ Ег> вВ dawIz)ldEM2'10-19 cлt2•эB",
Эксперимент [76] Расчет по (3-36) при
Аг 11,5 1,2 1,3
Кг 9,91 1,4 0,9
Хе 8,32 1,8 0,8
Усреднение сечений (3.36) по максвелловскому распределению скоростей
частиц дает для вероятностей переходов
мХМ!) (i I к) = 32лао
2*Гг
ДЕЙ
т
__е
Mz
ДГЙ
Хехр’.-^-
(3.37)
для обратного процесса девозбуждения
(к | i) = 32лао
Si f Rv \2 i ( kT? \I/2 Zl
gk \ ДЕ«* / Jik \ЛМ2 ) Mi y1
2kTr
(3.38)
Как следует из выражения (3.38), коэффициент скорости тушения уровней
увеличивается обратно пропорционально квадрату расстояния между уров-
нями (ДЕ)-2. При стремлении ДЕ1/С к нулю (к | i) -* оо, что физически
неверно.
Квантовомеханические расчеты коэффициентов скоростей переходов в
атоме водорода
Н (15) + Н (n) Н (п + 1) + Н (15),
выполненные в работе [77], показали, что при Т — 300°К коэффициенты
возбуждения при п 10 (ДЕП»П+1 0,023 эВ} достигают 10-9 см3-сек"1.
Расчеты по формуле (3.37) дают для этого случая (п | п + 1) =
130
— 4-10 8 см? • сек Ч Из сравнения этих результатов следует, что формулы (3.36) —
(3.38) перестают работать, по-видимому, уже при < 300 см'1. Этот ре-
зультат можно считать справедливым и для других атомов, поскольку такие
сильно возбужденные уровни (п > 10) являются водородоподобными. На-
пример, при исследовании ассоциативной ионизации при столкновении не-
возбужденных атомов аргона с атомами, возбужденными УФ-излучением,
в работе [78] были получены коэффициенты скорости девозбуждения уровней
id I1/2]0 — 0,25-Ю-10 и 65 [3/2]° —0,5-10-10 см^-сек'1. Если даже считать,
что девозбуждение этих уровней обусловлено переходами с наименьшим
изменением энергии AEi/£ = 160 см'1 (id — 5//), то по формуле (3.38) для
этого случая опять получаем сильно завышенные значения ш(М) =
= 2• 10-8 см?-сек'1.
Рис. 3.11. Зависимость сечений
перехода между уровнями ато-
мов, соответствующими различ-
ным компонентам тонкой струк-
туры, при соударении с тяже-
лыми частицами от разности
энергий
1 — столкновения с атомами;
2 — столкновения с молекулами [79]
При малых разностях энергии между уровнями (компонентами тонкой
структуры) атомов начинают сказываться неадиабатические переходы между
ними, индуцированные столкновениями с невозбужденными частицами. Та-
кие переходы имеют место как в чистых одноатомных газах, так и в смесях
с другими атомарными и молекулярными газами. Наличие этих переходов
обусловлено пересечением и квазипересечением термов молекул, составлен-
ных из партнеров по соударению. Сечения резко уменьшаются по мере
роста разности энергий между компонентами тонкой структуры. При соуда-
рениях с молекулами сечения оказываются в среднем значительно выше,
чем при соударениях с атомами рис. 3.11 [79]. Теория качественно правиль-
но описывает результаты экспериментов при тушении атомов атомами [43].
В смесях различных газов сечения тушения возрастают по сравнению с
результатами расчета по формуле (3.38) даже при столкновениях атомов
инертных газов (табл. 3.5). Они становятся еще выше при столкновениях с
некоторыми молекулами. Природа переходов, сопровождающихся тушением
возбужденных атомов, в настоящее время недостаточно ясна. При этом мо-
гут давать вклад процессы пеннинговской ионизации (если энергия возбуж-
дения атома больше потенциала ионизации тушащей частицы), резонансной
и квазирезонансной передачи энергии возбуждения (например, Не (2г5,
235) с неоном [79]), а также передача энергии возбуждения на поступатель-
ную и колебательную (в случае молекул) степени свободы. Большие сечения
тушения уровней можно было бы объяснить неадиабатическими квазирезо-
нансными переходами с передачей энергии на автоионизационные уровни
тушащих атомов или молекул (при тушении высоковозбужденных атомов
гелия и аргона). Такие процессы в настоящее время исследованы недостаточ-
но, и выводы из их результатов делать преждевременно.
Ранее считалось, что процессы тушения атомов молекулами носят резо-
нансный характер, так что вся энергия электронного возбуждения перехо-
дит в колебательную энергию молекул. Прямые измерения колебательного
131
5*
Таблица 3.5
Коэффициенты скорости тушения (к/l), 10-11 см3 • сек-1) возбужденных
атомов (Mi) молекулами и атомами другого сорта (М2). Т = 300° К
м2 Возбужденные
He(2xS) [81] He(23S) [81] Аг(зР2) [82] Аг(3Р0) [82] Hg(3Pt) [79, 80]
Не
Ne 6,4 0,39 — — —
Аг * 22 7,04 4-10-5 4-10-5 —
Кг 36 9,9 0,63 0,23 —
Хе 46 12 18 30 —
N — — — — ——
Н2 4,9 3,2 6,6 7,8 10
N2 16,7 7 3,6 1,6 0,21
О2 58 21 21 24 6,7
со 9,9 3,2 1,4 1,3 0,65
NO 43 24 22 25 0,5-7,5
СО2 110 58 53 59 16
СН4 38 4 33 55 0,06
* Коэффициенты скорости тушения атомов Аг(3Р2,о) аргоном
рассчитаны по
формуле (3.38).
возбуждения молекул СО и NO в результате тушения ими атомов ртути [80]
опровергли эти представления: на колебательное возбуждение передается
около 25% энергии возбуждения, причем наблюдается равномерное заселение
колебательных уровней v СО от 0 до 8 (энергия уровня v = 10 соответству-
ет примерно половине энергии возбуждения атома). Отсутствие зависимости
сечения тушения атома натрия различными молекулами от дефекта резонан-
са также опровергает эти представления [79]. Еще более наглядно отсутствие
такой зависимости проявляется при тушении атомов N (2Z>) молекулами
азота и окиси углерода. Дефект резонанса при возбуждении уровня N2 (v =
= 8) составляет — 550 см"1, а СО (у ~ 10) — 530 см"1. В то же время сече-
ния тушения отличаются друг от друга почти на три порядка величины.
Большие колебания коэффициентов скоростей тушения атомов при из-
менении химической природы тушащих частиц (см. табл. 3.5) позволяют
сделать вывод, что оно, по-видимому, целиком обусловлено наличием пере-
сечений и квазипересечений потенциальных кривых молекул, составлен-
ных из партнеров по соударениям. Ввиду сложности задачи расчета таких
процессов и отсутствия надежных данных, касающихся термов этих моле-
кул, создание общей теории таких процессов в настоящее время вряд ли воз-
можно [43].
Аналогичная ситуация имеет место и для коэффициентов скоростей ту-
шения электронно-колебательных уровней молекул (табл. 3.6).
Поэтому единственным надежным источником сведений о сечениях туше-
ния электронных уровней являются эксперименты. Основной недостаток
большинства экспериментов состоит в том, что редко удается идентифици-
ровать возбуждение внутренних степеней свободы партнеров после акта ту-
шения. Кроме того, большинство экспериментов выполняется при комнат-
ной или близкой к ней температуре [79]. В целях экстраполяции этих данных
на условия, характерные для ряда плазмохимических систем, необходимо
знать температурную зависимость коэффициентов скорости тушения. Раз-
личные модели тушения, позволяющие исследовать температурную зависи-
мость теоретически, изложены в работах [43, 97]. Если тушение обусловле-
132
частицы М2
Hg(3P0) [79, 80] Na(32P) [79] N(2D) [83, 84] N(2P) [83, 85, 86] O(W) [87] O(«S) [87]
— — 2-10-5 — — 3-10-5
— — — — — —
— — 2-10~5 7.10-5 — 3.10-5
— — — — — —
— — — — — —
— — — 0,18 — —
0,03 88 0,5 ЗЛО"4 — 10-4
4,5Л0-6 17 0,0016 2.10-7 8 зло-5
0,045 35 0,6 0,46 5 5-10-2
0,013—0,045 10 0,6 — — —
0,02—0,5 22 7 3,4 — 55
5.10-3 — 0ъ005 6-10-2 0,3 3-10-2
0,004 — 0,3 — — —
Остальные данные — результаты эксперимента.
но дальнодействующим поляризационным взаимодействием, его сечение мож-
но описать соотношением [98]
(3.39)
пс1 г
где [i — приведенная масса частиц; а — потенциал ионизации и поляри-
зуемость молекулы-тушителя; 7?с — радиус соударения. Этой формулой
описывается тушение электронных уровней молекул I2, SO2, NO2, СН3ОН
Таблица 3.6
Коэффициенты скорости тушения ряда электронно-колебательных уровней
молекул (ww(f/fc), в 10-11 СЛ1з.сек-1). т = 300° К
M2 Возбужденные частицы Mi
W2(A*Zu+, V) [85, 88, 89] N2(B3Hg) v = 2-18 J90-91] N2(C3Hu) v = 0—3 [92] СО(а3П) [93, 94] NO(A2S) [95] NO(B2n) v = 0—4 [96]
v — 0 V — 1 v — 0' = 1—4
He <10-3
Ar — — 0,4—0,9 — — — — —
N 5,0 5,4 — — — — — —
N2 1,910-7 l,5-10-5 0,5-7,5 1—2 4 8 — —
NO — — 3,7—17 — 31 70 12,5 8—10
CO — — — — 12 28 — —
CO 2 — — — — 3,4 — — 1
n2o — — — — — — — 4,5
С2Нб 0,029 0,36 — — — — — —
C2H4 6,4 10,8 — — — — — —
133
различными молекулами. В случае неадиабатических переходов между тер-
мами различной симметрии в различных приближениях также получается
зависимость [43]
о(М) (Arjt; (3.40)
В случаях (3.39), (3.40) коэффициент скорости тушения оказывается не
зависящим от температуры.
В большинстве исследованных случаев коэффициенты скоростей тушения
слабо зависят от температуры. Если представить зависимость в виде фор-
мулы:
Ww (к |i; Тг) = w(M) (k\i; T= 300° К) (7’r/300)n, (3.40а)
то наибольшая отрицательная температурная зависимость была обнаруже-
на в случае тушения атомов Na (2D) различными молекулами от п = 0 до
п — —0,16 [97]. В остальных случаях температурная зависимость отсут-
ствовала [91,99] или была слабо положительной. В некоторых случаях наб-
людалось резкое увеличение коэффициента скорости тушения с температу-
рой. Например, тушение атомов О ^А) молекулами СО2 и О2 описывается
формулами:
^(с°2)(&| f; 7г) = 3,3-10“п ехр (---100_\ см3 • сек”1,
w<°2} (&| i; Тг) = 4,9 • 10”12ехр (-850 100 Л См3 - сек”1,
\ 1 г /
в то время как коэффициенты скорости тушения О (х5) атомами аргона и азо-
та не зависят от температуры [99]. Это можно, по-видимому, объяснить на-
личием эндотермических химических реакций атомов О (1А) с молекулами-
тушителями.
Различные модели тушения электронных уровней обсуждаются в работе
[97], где приведен обширный список работ, посвященных этому вопросу.
ИОНИЗАЦИЯ
И ДИССОЦИАТИВНАЯ ИОНИЗАЦИЯ МОЛЕКУЛ
1. Ионизация атомов
Для ионизации атомов электронным ударом предложена полуэмпириче-
ская формула [63]
2 jj _।
0(е)(ф; Ее) =4л4(-^) ЛЛ-г-г~In [1,25^], (3.41)
\ Ali / и
где Ui = EeI^Eie\ kEie = | Е™ —Ег \ — разность энергий возбуждения уров-
ня и ионизации; — число эквивалентных электронов в оболочке.
Детальные расчеты сечений ионизации с различных уровней атомов вы-
полнены в [62] и результаты затабулированы.
В случае ионизации тяжелыми частицами в классическом приближении [63]
/ /? \ 2 9m W __ 1
о(М)(fl е, Ем.) = 4л4 (—Л-\ &ieг-------, (3.42)
\ W и I д me / Ml эг'геГ 2пгр 12 ’ ' '
V д 7 1 + -Тй — (Wi — 1)
L 1 те г J
где Wt = EmJAEie; Emz = M2v}/2; — масса ионизуемого атома, М2 —
масса налетающей частицы.
Сравнение этой формулы с результатами расчетов [77], как и в случае
возбуждения (см. стр. 131), показывает, что она дает завышение сечений
ионизации при кЕге < 300 слГ1.
134
2. Ионизация молекул
Если представить сечение прямой ионизации молекул с уровня V элек-
тронным ударом с образованием иона в определенном состоянии к' в виде
(см. стр. 124)
£е)=|Мв(Д4Д)|аф (3.43)
\ vivk /
то из сравнения с экспериментальными данными следует, что функция
ср (Ее/АЕг^к) в этом случае является универсальной и не зависит от типа
перехода и сорта молекулы (см. рис. 3.7). Тогда полное сечение прямой иони-
зации молекулы будет равно
б<е> (i | е; Ее) = 2=(е( G' | к'-, е; Ее).
к'
(3-44)
В этом случае мы не получим единой универсальной зависимости сечения от
относительной энергии электронов, поскольку разным к' отвечают различ-
ные пороги ионизации (см. рис. 3.8).
Кроме того, следует иметь в виду, что ионизация возможна в результате
двухступенчатого процесса — возбуждения автоионизационных (сверхвоз-
бужденных) состояний с последующей автоионизацией. Сечение такого про-
цесса можно представить в виде
(f [ т j к', е\ Ее) — 2 (т' 1 &') (к | т'\ (3.45)
га'
где (f | т') — сечение возбуждения автоионизационного состояния т'\
F” (т'| А') = Г77
v 1 ' Г (т )
эффективность ионизации из автоионизационного
состояния т'\ Ги (т' | к') — вероятность автоионизации; Г (тп') — суммарная
вероятность дезактивации уровня т .
Автоионизационные состояния обнаружены в настоящее время практиче-
ски для всех исследованных методом фотоэлектронной спектроскопии моле-
кул [100—102]. Эффективность ионизации для большинства автоионизацион-
ных уровней отличается от единицы, что связано с возможным предиссоциа-
ционным распадом уровней. Тогда
Уи (тг | к')
Ги {т' [ к')
Гя (т') + Ги (т') ’
(3.46)
где Гд (тп') — вероятность спонтанного предиссоциационного распада мо-
лекул.
В случае запрещенных автоионизационных переходов (будем считать ав-
тоионизацию запрещенной, если Ги ~ 109 сек~г; для разрешенной — Ги ~
~ 1014 сек-1 [103, 104]) возможна конкуренция спонтанных и индуцирован-
ных столкновениями с другими частицами переходов. В этом случае
Уи (т' I к’) = —----------Ги(пг'Р9 + ^и (пг'|Н-----------, (3.47)
Гя (m') -f- Ги (m') + -Vй (т') -j- Л'д (mr) -j- v (in’) -J- A (m')
где Vй (тп'), Vя (m'), v (m) — частоты индуцированной автоионизации, пре-
диссоциации и тушения автоионизационных состояний.
Если в процессе прямой ионизации или ионизации через автоионизацион-
ние состояние образуется ион в нестабильном состоянии либо в стабильном
состоянии, из которого возможна предиссоциация, то такой процесс иониза-
ции приведет к образованию осколочных ионов. Этот процесс называется
процессом диссоциативной ионизации. Поскольку основные электронные
состояния ионов молекул являются стабильными, то процесс диссоциатив-
135
ной ионизации имеет порог, равный сумме порогов ионизации молекулы и
диссоциации молекулярного иона. При этом все газы дают существенное
число осколочных ионов с кинетической энергией 4—10 эВ, а СН4, N2O,
Н2 и О2 — выше 10 эВ [105]. Кроме того, сечение диссоциативной ионизации
не может превышать суммарное сечение ионизации, а максимум этого про-
цесса достигается при энергиях, превышающих энергию, соответствующую
максимумам сечений ионизации. Экспериментальные исследования показы-
вают, что по мере увеличения числа атомов в молекулах сечение диссоциатив-
ной ионизации увеличивается, достигая половины сечения ионизации. Тог-
да как для двухатомных молекул они составляют менее 10 % от сечений иони-
зации (табл. 3.7).
Таблица 3.7
Пороги появления осколочных ионов (Д22д* и) и сечения диссоциативной ионизации
(°тах)и полной ионизации (<5^ах) ряда молекул при энергиях электронов,
соответствующих максимумам этих сечений [105, 106]
Молекула ДЕД- и, эВ Еи, эВ Ета>« ОД-И max’ Ю-i6 СМ2 Етах’эВ ои , max’ 10-16 СМ2
Н2 30 15,42 110 6.10-2 70 1,0
N2 25 15,6 140 0,65 120 2,5
О2 20 12,2 150 1,0 110 2,6
ND 21 9,25 150 0,9 110 3,2
СО 22 14,01 140 0,65 110 3,0
СО2 24 13,8 200 1,0 125 3,3
СН4 20 13,0 100 1,55 80 3,8
N2O 19 12,7 150 1,5 120 3,8
Прямая ионизация молекул при соударении с тяжелыми частицами яв-
ляется не единственным процессом ионизации, поскольку при таких соуда-
рениях с большой вероятностью протекают энергетически более выгодные
процессы ассоциативной ионизации (см. стр. 185).
ДИССОЦИАЦИЯ МОЛЕКУЛ
ЧЕРЕЗ ЭЛЕКТРОННО-ВОЗБУЖДЕННЫЕ СОСТОЯНИЯ
Возбуждение электронных уровней молекул может приводить к их дис-
социации в случаях:
1) возбуждения нестабильных состояний или переходов на отталкиватель-
ные ветви потенциальных кривых стабильных состояний;
2) возбуждения стабильных состояний, из которых возможна предиссо-
циация в результате взаимодействия с нестабильными состояниями;
3) возбуждения в стабильное состояние с последующим каскадным пе-
реходом в состояние, принадлежащее к двум первым типам.
Время жизни молекул в нестабильных состояниях составляет порядка
10~13—10-14 сек, что превышает характерные времена процессов возбужде-
ния электронами (~ 10-16 сек) и фотонами (~ 10~18 сек). Времена жизни ста-
бильных состояний еще больше, чем нестабильных. Поэтому процесс диссо-
циации молекул из электронно-возбужденных состояний надо рассматривать
как двухступенчатый — возбуждение промежуточного состояния и его по-
следующий распад.
Поэтому сечение диссоциации молекул, находящихся в состоянии V, че-
рез возбужденное состояние к' равно произведению сечения возбуждения
136
состояния к' из V и вероятности диссоциативного распада с этого уровня
(Гд (Л')):
Сд (Г | к') = с (Г | Л') Уд (Л')> (3.48)
Уд (£') = [Гд (Л') + Vя (О г*,. (3.49)
где Гд (А') — вероятность спонтанной предиссоциации стабильного или дис-
социации нестабильного состояния; ¥д (к') — частота предиссоциационных
переходов, индуцированных столкновениями окружающих частиц: —
время жизни уровня к', определяемое выражением
(ГгГ1 = А (кг) + Гд (к') + гд (/£') 4- V (Л'), (3.50)
где А (к') — вероятность радиационного распада, v (&') — частота тушения
уровня столкновениями с окружающими частицами, не приводящего к дис-
социации молекулы. В случае автоионизационных состояний в правую часть
(3.50)) необходимо добавить вероятность автоионизации с уровня к' —
— Ги (к').
В случае возбуждения нестабильных состояний и состояний, из которых
возможна разрешенная спонтанная предиссоциация (Гд (Л') 1010 сек~г),
радиационные и индуцированные соударениями переходы не могут конку-
рировать с Гд (Л') и при всех давлениях для них
Y*(k')~ 1. (3.51)
В случае автоионизационных уровней выражение (3.51) заменяется сле-
дующим:
УД(А') = Гд (Л')/] Гд (/,:')! ГИ(Л')]. (3.52)
Вероятности предиссоциации и автоионизации выражаются формулами
[9, 107]:
Гд (Л' |т') = (2л /К) V (к' \m'\R,r |2,
Ги(Л'|ш') = (2л/Л)|У(^|Г; К, г)|2, 1 '
где V (к' | тп'; R, г) и И (к' | Г, R, г) — матричные элементы взаимодействияя
приводящего к соответствующим переходам между состояниями к' т*
и к’ — i'\R — координаты ядер, г — электронов. В приближении Борна —
Оппенгеймера можно записать:
Гд (к’ I т') = Гд (к I m; R; /) q* ™ ,
»кЕт (3.54)
Ги(Л'|Г) = Геи(А:Н,/?;7)^ ,
П ъ
— к т к г v
где В — некоторое среднее расстояние между ядрами, — факто-
ры Франка — Кондона для соответствующих переходов:
Полные вероятности определяются суммированием по всем возможным
переходам:
Гд(А/) = ЗгД(/с'|т'),
(3.54а)
ГИ(А/) =2ги(&'|У),
137
Для того чтобы предиссоциационный переход имел место, должны выпол
няться следующие правила отбора [107]:
А5 = 0;+1; А/= 0; ДА = 0;+1,
+ <->+; — g^g,
(3.56)
где S — спин, Л — проекция орбитального момента на ось молекулы,
остальные знаки обозначают симметрию или четность состояний.
В случае слабости взаимодействия, приводящего к предиссоциации (на-
пример, в случае АД = +1 взаимодействие слабее, чем при AS = 0), нару-
шения правил отбора (3.56) или малости факторов Франка — Кондона для
предиссоциационных переходов (3.55) вероятности предиссоциации могут
достигать величины Гд — 105—102 сект1 (см. стр. 160). В этом случае должна
проявляться конкуренция процессов радиационных переходов, тушения
и т. д., и вероятность диссоциации с уровняй'может зависеть от давления, со-
става газа и его температуры (3.49). Она будет постоянна и равна Гд (й')/
![А (к') + Гд (Л')] при низких давлениях и падать обратно пропорционально
давлению в случае тд (к') = 0 или после падения в области промежуточных
давлений достигать постоянного значения, равного [108]
Уд (kf) — v* (к') / [v (к') + Vй (й')1.
Зависимость вероятности диссоциации от температуры заселения враща-
тельных уровней обусловлена тем, что величина матричного элемента взаи-
модействия может зависеть от / (3.54), либо предиссоциация может наблю-
даться с некоторого минимального уровня jmin (так называемая предиссо-
циация вращением [108, 109]). Как видно из (3.49), (3.50), температура газа
также может повлиять на величину Уд (Л'), поскольку она влияет на часто-
ты индуцированных процессов.
Расчет вероятностей предиссоциации и автоионизации [9, 103, 107] за-
труднен отсутствием надежных данных о потенциальных кривых и типах не-
стабильных состояний, взаимодействие с которыми приводит к таким перехо-
дам.
Для экспериментального измерения вероятностей применяются два ме-
тода: уширения линий и стационарных концентраций.
Полное время жизни уровня связано с его шириной (Av) соотношением,
вытекающим из соотношения неопределенности:
(т^)"1 - Г (к') - 1,88 • 10nAv (слГ1). (3.57)
Из этого соотношения следует, что данным методом можно надежно опреде-
лить время жизни уровня только в том случае, если Г (Л') превышает 1010 сек-1
(из-за влияния аппаратной функции прибора и допплеровского уширения
линий). Однако в этом случае конкуренция радиационных и индуцированных
столкновениями процессов несущественна и выполняется соотношение
(3.51). Таким образом, сам факт наличия уширения в спектрах (оптических—
диффузность полос [3], электронных потерь) свидетельствует о выполнимо-
сти (3.51) во всех случаях, если уровень не является автоионизационным. Для
автоионизационных уширенных уровней справедливо (3.52). В этом случае
для определения Гд {к') необходимо измерение эффективности ионизации
Уи (й') = Ги (й') / [Ги (к') + Гд (й')]. (3.58)
Такие измерения проведены для большого числа молекул методом фотоиони-
зации.
В работах [9, 107] указывается, что более надежным методом определе-
ния величины матричного элемента взаимодействия может служить сдвиг
линий, примыкающих к границе предиссоциации, поскольку этот сдвиг
больше уширения, да и само уширение не должно влиять на результаты из-
мерений. Такие измерения описаны в [107].
138
Если вероятности предиссоциации ниже указанного предела 109 сект1),
их измерение возможно методом стационарных заселенностей уровней в га-
зовых разрядах, электронных пучках, при фотовозбуждении и т. д. [108].
Частным случаем его является метод обрыва полос [3].
При известной скорости заселения и измеренной стационарной концент-
рации из (3.48) определяется Уд (Л'), а затем с использованием независимо
измеренных констант A, v — и Гд (к'). Кроме того, для оценки вероятнос-
тей предиссоциации можно использовать следующий факт: если уровень в
поглощении является не диффузным, а в спектре излучения он отсутствует
(при достаточной эффективности возбуждения), то
Ю10 сек-1 > Гд (к') > А (к') + v (к'). (3.59)
Определение вероятностей и типа предиссоциационных переходов воз-
можно также из измеренных коэффициентов радиационной рекомбинации
атомов и фрагментов молекул (см. стр. 154).
В заключение этого краткого обзора необходимо подчеркнуть, что знание
вероятностей диссоциации с данного электронно-колебательного уровня по-
зволяет рассчитывать сечение диссоциации молекул при любом известном
способе ее возбуждения — фотовозбуждения при фотолизе, возбуждения
электронами или тяжелыми частицами при радиолизе, в газовых разрядах,
плазмохимических системах и других случаях низкотемпературной плазмы.
Полное сечение диссоциации с уровня V молекулы определяется сумми-
рованием (3.48) по всем электронно-колебательным уровням, с которых воз-
можна предиссоциация, и интегрированием по энергии колебательного воз-
буждения нестабильных состояний
Од(г; Е) = 2 YR(k,vk)^---------------- +
к, vk 2J 1NV
vi
oo
. 0 /о
где Л1 — заселенность колебательного уровня исходного состояния i.
Коэффициенты скорости получаются из (3.60) усреднением по функциям
распределения частиц.
1. Диссоциация невозбужденных молекул
электронным ударом
через электронно-возбужденные состояния
Рассмотрим расчет сечцний диссоциации невозбужденных молекул (г^ =
— 0, i — основное электронное состояние) электронным ударом.
При расчете сечений будем исходить из схемы потенциальных кривых
(см. стр. 114), сечений возбуждения различных уровней электронным уда-
ром (стр. 123) и приведенных ранее формул (см. гл. 3, п. 6).
Водород. Из схемы уровней молекулы водорода [110] следует, что нижнее
триплетное состояние Ь3^и является нестабильным. Поэтому возбуждение
этого уровня, а также a32g и с3Пи, с которых наблюдаются каскадные пере-
ходы на 632и, приводит к диссоциации. Кроме того, наблюдается диссоциа-
ция, сопровождающаяся появлением возбужденных атомов, в результате
переходов на отталкивательные ветви потенциальных кривых состояний
В'^и, С1ПИ и B^t [ИИ, переходов в состояние с последующей спон-
танной предиссоциацией [112, 113], а также распад ряда ридберговских со-
139
стояний, в том числе и автоионизационных [101—103, 114]. Относительные
вероятности диссоциации со всех этих уровней, за исключением автоиони-
зационных, при р < 0,1 торр близки к единице.
Парциальные сечения диссоциации водорода через состояния й32^,
a3Sg и с3Пи были рассчитаны нами по дифференциальным сечениям возбуж-
дения, измеренным в работе [115], и рассчитанным в работах [115—117]
(рис. 3.12). На этом же рисунке изображено также сечение диссоциации с
образованием возбужденных атомов водорода 2Р и 2S [118, 119]. Образова-
ние атомов на уровнях п 3 можно не учитывать. Сечение образования ато-
мов 2Р и 2S по крайней мере наполовину обусловлено переходами на оттал-
кивательные ветви кривых СЧ1и, как следует из данных [111]
и формулы (3.25). Полное сечение диссоциации получается суммированием
этих парциальных сечений.
Рис. 3.12. Полное и парциаль-
ные сечения диссоциации элек-
тронным ударом через элект-
ронно-возбужденные состояния
молекулы H2(X1S^, v = 0)
1 — полное сечение [67, 120];
+
2 — через
+
3 — a3^g',
4 — с образованием атомов Н(2Р),
H(2S);
5 — сумма парциальных сечений
(2—4)
На рис. 3.12 приведено также полное сечение диссоциации водорода че-
рез нейтральное возбуждение молекулы, полученное из данных [120].
В области энергий Ее >25 эВ, где измерено сечение возбуждения
fe3S*, кривые 1 и 5 удовлетворительно совпадают, что свидетельствует о том,
что все основные каналы диссоциации учтены при построении кривой 5.
Из данных рис. 3.12 следует, что основной вклад в диссоциацию водоро-
да при низких энергиях электронов дает возбуждение триплетных уровней.
Однако при больших энергиях Ее 50 эВ определяющим становится пар-
циальное сечение диссоциации с образованием возбужденных атомов. Раз-
личие результатов расчета сечения 5 с данными [120] обусловлено в этой
области энергий погрешностью результатов измерений [120].
При увеличении давления относительные вероятности диссоциации че-
рез уровни fl3Sg и с3Пп, а также через ряд ридберговских состояний должны
уменьшаться. Например, возбуждение состояния a3Sj будет целиком при-
водить к диссоциации только при р < 1 мм рт. ст., а при больших давлени-
ях начнется конкуренция радиационных переходов с тушением [121]. В нас-
тоящее время неизвестно (как и в большинстве других случаев тушения),
на какие уровни совершаются переходы в результате этого процесса. Поэто-
му нельзя определить, насколько существенно изменится вклад парциально-
го сечения через a32g в полное сечение диссоциации водорода при повыше-
нии давления.
Азот. Из кривых потенциальной энергии молекулы азота [5] и анализа
сечений его возбуждения следует, что* все уровни, эффективно возбуждае-
мые электронным ударом из основного состояния, стабильны. Однако для
ряда электронно-колебательных уровней наблюдается предиссоциация [108].
Вероятность предиссоциации с уровней vk = 13—18 7?3Щ и > 7 a4Ig
пропорциональна температуре заселения вращательных уровней и при Тj =
= 300°К составляет (4—0,2)-107 и 4,2 -105 сек'1 соответственно. Сечения ту-
шения для них изменяются в пределах (1,1—2)-10~15 см2. Вероятности пре-
диссоциации уровней С3Па превышают 108 сек'1 [108]. Оценки вероятностей
140
Рис. 3.14. Сечение диссоциации электронным ударом через электронно-возбужденные со-
стояния молекулы O2(X3Sg, v = 0)
1 — полное сечение (сумма 2—9); 2 — через 4—[134]; 3-Д.Е*Й ~ 6,1аВ; 4 — 10,29; 5 — 9,9; 6 —
15,3; 7 — 16,8; 8 — 12,9; 9 — 19,7 i
спонтанной предиссоциации и радиационных переходов для уровней 6гПй
и всех, лежащих выше 12,4 эВ, были сделаны на основании сил осциллято-
ров, измеренных в поглощении в работе [122], а также данных [123—128]
по уширению полос и наблюдению излучения при возбуждении этих состоя-
ний. Если линия не наблюдалась в излучении, но уширение не было зарегист-
рировано, полагалось, что для таких уровней справедливо соотношение
(3.59). Относительные вероятности диссоциации для этих групп уровней
приведены ниже.
&Eik, эВ .... 12,4—13,05 13,06—13,30 13,31—13,80 13,81—15,58> 15,58
Уя(/с, vk) .... 0,63 0,33 0,55 1 0,4
Для автоионизационных уровней (АЕгк 15,58 эВ) вероятность диссо-
циации определялась по данным работы [126].
Сечение диссоциации молекулы азота с уровня — 0 основного состоя-
ния, рассчитанное по формуле (3.60), а также парциальные сечения диссо-
циации через ряд электронных состояний приведены на рис. 3.13. Использо-
вались сечения возбуждения уровней по данным [64] (кривая 2) и по [129] —
кривая 2а. Полное сечение диссоциации путем возбуждения нейтральной
молекулы азота, рассчитанное из данных [130] и относительных данных [131],
нормированных по [130] при Ее = 45 эВ, изображено на рис. 3.13 в виде
точек и кривой 1. При построении кривых 2 и 2а было также учтено сечение
диссоциации с образованием возбужденных атомов азота [132],половина ко-
торого обусловлена возбуждением уровней нейтральной молекулы [133].
Некоторое расхождение рассчитанных нами сечений диссоциации (кри-
вые 2 и 2 а) с данными [130, 131] обусловлено погрешностями в сечениях воз-
буждения уровней с КЕ 12,4 эВ. Тем не менее из этих данных следует, что
основные каналы диссоциации молекулы азота электронным ударом учтены
правильно.
Диссоциация молекулы азота целиком определяется возбуждением ста-
бильных электронных состояний с последующей предиссоциацией.
141
Кислород. При соударении электронов с невозбужденными молекулами
кислорода диссоциация может происходить в результате переходов на от-
талкивательные ветви потенциальных кривых всех состояний, сходящихся
к первому и второму потенциалам диссоциации (5,1 и 7,08 эВ): с1 A3Sj,
3Щ, 5Щ, 5ПП и В 32й (потенциальные кривые см. в [5]). Кроме того, все
возбужденные уровни кислорода, лежащие выше второго потенциала дис-
социации, в излучении не наблюдаются, а в поглощении соответствующие ли-
нии уширены. Уширены и линии поглощения с переходом на уровни vk 3
связанного состояния 7?35й, что соответствует вероятности спонтанной пре-
диссоциации Гд 1011 сект1 [127].
Парциальное сечение диссоциации через уровни состояния Б3 2^ (рис. 3.14)
было рассчитано по формуле (3.25) и <р (х) (см. рис. 3.6) в приближении Бе-
те — Борна. Результаты расчета (кривая 2) удовлетворительно согласуются
с экспериментальными точками [134]. С использованием сил осцилляторов из
[74] были рассчитаны парциальные сечения диссоциации для остальных
разрешенных переходов. В случае запрещенных переходов использовались
сечения [134] и формы кривых <р (х) (см. рис. 3.9).
На рис. 4.14 изображено также полное сечение диссоциации кислорода
через возбужденные уровни. Для всех автоионизационных состояний при-
нималось согласно (3.52)
Уд (Ze') = 1 — бфи/бфп, (3.61)
где (уФи, огФн — сечения фотопоглощения и фотоионизации, поскольку све-
чение, наблюдавшееся при поглощении в кислороде вакуумного ультрафио-
летового излучения, обусловлено высвечиванием возбужденных продуктов
диссоциации [128, 135, 136]. Для состояний, лежащих ниже границы иониза-
ции, предполагалось выполнение соотношения (3.51).
Основной вклад в полное сечение диссоциации кислорода через возбуж-
дение электронно-колебательных уровней электронным ударом дает возбуж-
дение континуума состояния В35^ (как и при фотодиссоциации). Однако вбли-
зи порога диссоциации существенным оказывается возбуждение запрещен-
ных переходов с порогом 6,1 эВ. Соответствующие уровни могут дать решаю-
щий вклад в условиях газовых разрядов. Автоионизационные уровни дают
вклад в полное сечение, достигающий 50% при больших энергиях электро-
нов Ее 25 эВ.
Окись азота. Предиссоциация окиси азота наблюдалась с уровней vh 4
A2S+, vh 1 Z)2S+, vk 7 Б2П [137]. Все соответствующие полосы погло-
щения не уширины, однако в излучении не наблюдаются при давлениях
вплоть до атмосферного. Квантовый выход фотодиссоциации при энергии
квантов hv 6,77 эВ близок к единице [138]. Все это позволяет сделать
вывод, что вероятности диссоциации с уровней, расположенных выше 6,77 эВ,
превышают 109 сек"1, но меньше 1011 сек"1. Следовательно, при всех давле-
ниях вплоть до атмосферного для них справедливо (3.51). Сечение фотодис-
социации через возбуждение автоионизационных уровней окиси азота из-
мерено в работах [139, 140] в предположении, что оно равно разности пол-
ного сечения поглощения и фотоионизации. Такое предположение оправды-
вается тем, что люминесценция молекул при фотовозбуждении наблюдалась
только в области AEili 18,3 эВ и связана с высвечиванием возбужденных
продуктов диссоциации [139, 141]. Сечение диссоциации электронным уда-
ром рассчитывалось нами с использованием этих данных для автоионизаци-
онных уровней по формуле (3.25) и ср (х) (см. рис. 3.6; рис. 3.15, кривая 4).
Парциальные сечения диссоциации через уровни АЕ*К > 6,77 эВ были рас-
считаны по силам осцилляторов, полученным из электронных спектров при
Ее = 59 эВ, 0 — 0° [74] путем нормировки по сечению возбуждения уровней
vh ~ 0—3 состояния А25+ (Л/g — 8,4-10~18 см2 [141]). Для расчета сечений
использовалась формула (3.24), поскольку все переходы являются раз-
142
Рис. 3.15. Сечение диссоциации электронным ударом через электронно-возбужденные
состояния молекулы NO(X2 П, v = 0)
1 — полное сечение по [142]; 2 — сумма парциальных сечений; 3 —через АЕг^ = 6,93—9,2 эВ; 4 —
то же, 9,2—17,3; 5 — 6,93—7,3; 6 — 8,5—9,2; 7 — 8,0—8,5; 8 — 7,6—8,0
Рис. 3.16. Сечение диссоциации электронным ударом через электронно-возбужденные
состояния молекулы С0(Хх2^ v = 0)
1 — полное сечение по [142]; 2 — сумма парциальных сечений; 3 — через АЕгй =13—14 эВ; 4—12,5—
13; 5 — AJE > 14,01
решенными. Полное сечение диссоциации через электронно-возбужденные
состояния получалось суммированием парциальных сечений (рис. 3.15). На
этом же рисунке изображено сечение диссоциации окиси азота, полученное по
данным [142] путем вычитания из них сечения диссоциативной ионизации.
Удовлетворительное согласие кривых 1 и 2 наблюдается при Ее < 40 эВ, что
свидетельствует о надежности данных в этой области энергий. Однако сильное
различие кривых при увеличении энергии электронов приводит к выводу,
что в результатах [142] возможна большая систематическая погрешность,
повышающаяся по мере увеличения энергии электронов. В противном слу-
чае пришлось бы предположить необычную зависимость сечений возбужде-
ния разрешенных переходов <р (х) от энергии налетающих электронов.
Из данных рис. 3.15 видно, что в полное сечение диссоциации молекулы
окиси азота дает вклад большое число электронно-колебательных уровней,
расположенных более или менее равномерно между первым потенциалом дис-
социации и А Егк 18 эВ. Вблизи порога диссоциации основной вклад
дает предиссоциация состояний Л22+, В2П и Z)2S+.
Окись углерода. В спектре излучения полос окиси углерода наблюдается
предиссоциация с уровней В1И+ ~ 0, / > 38; vk = 1, / ^> 18) (обрыв
полос), а уровни vk > 2 C'1S+ (vk 1), Е1И + 1) вообще не наб-
людаются в излучении, так же как и все уровни с потенциалами возбуждения
выше 12,5 эВ. В поглощении света и при электронном ударе переходы на эти
уровни наблюдаются [143—148]. Обрыв полос с уровня Вг^ + неполный, а ин-
тенсивность при р = 5 мм рт. ст. уменьшается примерно вдвое. Это соответ-
ствует вероятности предиссоциации порядка 108 сек"1. Для остальных полос
обрыв полный, но диффузности спектра поглощения не наблюдается, что сви-
детельствует о справедливости соотношения (3.59). Используя сечение
возбуждения полос Л1!! (о(е) = 20-10-18 см2 при Ее = 200 эВ) и спектр по-
терь энергии электронов при Ее =? 200 эВ 0 = 0° [145], получаем сечение
возбуждения полос с A Eik = 12,5—13 эВ — 7,8-10-18 см2 и полос с =
= 13—14,2 эВ — 22-10~18 см2 при этой энергии электронов. С помощью фор-
мулы (3.24) и<р(^) (см. рис. 3.6) находим сечение возбуждения этих состояний
и парциальные сечения диссоциации через них (рис. 3.16). Сечение диссоциа-
143
ции через автоионизационные уровни находим из спектров фотопоглощения
и фотоионизации [149—151]. На этом рисунке вместе с полным сечением дис-
социации через возбужденные состояния, полученным суммированием пар-
циальных сечений, изображено сечение диссоциации через возбуждение ней-
тральной молекулы, полученное из данных работы [142] вычитанием сече-
ния диссоциативной ионизации [105]. Различие кривых 1 и 2 вблизи порога
диссоциации частично можно объяснить неполным учетом всех уровней,
которые могут давать вклад в диссоциацию. В частности, плохо изучена пре-
диссоциация и сечения возбуждения триплетных состояний молекулы СО,
и эти уровни не учтены при нахождении полного сечения диссоциации (кри-
вая 2). Однако при больших энергиях электронов наблюдается систематиче-
ское расхождение кривых 1 и 2. которое может свидетельствовать о наличии
Рис. 3.17. Сечение диссоциации
электронным ударом невозбуж-
денной молекулы метана
1 — полное сечение диссоциации,
эксперимент [157, 158];
2 — сечение диссоциативной иони-
зации [106];
3 -- полное сечение диссоциации
через электронно-возбужденные
состояния;
4 — путем возбуждения оптически
разрешенных переходов;
5 — запрещенных переходов
систематических погрешностей в данных [142] при больших энергиях элек-
тронов, как и в случае молекулы окиси азота (см. стр. 143).
Метан и другие насыщенные углеводороды. При возбуждении электрон-
ным ударом и вакуумным ультрафиолетовым излучением метана и других
насыщенных углеводородов наблюдаются интенсивные переходы на элек-
тронно-колебательные возбужденные уровни, часть которых лежит ниже
потенциала ионизации, а часть являются автоионизационными [152—157].
Спектры полных потерь энергии электронов и потерь на ионизацию показы-
вают, что к ионизации молекул приводит лишь часть переходов в автоиони-
зационные состояния. Высвечивание возбужденных состояний неэффектив-
но (квантовый выход менее 10~3) [152], несмотря на то, что вероятности опти-
ческих переходов в поглощении превышают 108 сек"1. Фосфоресценция мо-
лекул связана с излучением возбужденных продуктов диссоциации [156].
Поскольку эти измерения проводились при малых давлениях (р < 10~3 мм
рт. ст.), тушение возбужденных уровней при соударениях с другими моле-
кулами маловероятно. Единственное объяснение отсутствия излучения из
возбужденных состояний молекул состоит в том, что все они либо нестабиль-
ны, либо стабильны, но вероятности предиссоциации из них велики по срав
нению с вероятностями радиационных переходов Гд (&') > 103-Л (к').
В спектрах поглощения света и спектрах потерь энергии электронов не удает-
ся разрешить ни вращательной, ни колебательной структуры (исключение
составляет этан, у которого наблюдалась колебательная структура в спектре)
[152]. В работе [153] было высказано предположение, что возбужденные со-
стояния являются нестабильными, и оценена вероятность распада из ниж-
него триплетного состояния — Гд ~ 3-10-15 сек-1. Поскольку это значение
является, по-видимому, завышенным, отсутствие структуры в спектрах может
быть связано только с большой плотностью состояний [154].
Высказанное выше предположение о роли распада в девозбуждении элек-
тронных уровней нашло непосредственное экспериментальное подтверждение
в случае метана. Полное сечение диссоциации этой молекулы, восстановлен-
ное нами [67] по данным [157,158], приведено на рис. 3.17. Вычитание из этой
кривой сечения диссоциативной ионизации [106] дает сечение распада моле-
144
кулы через электронно-возбужденные состояния (кривая 3). Кривая 4 пред-
ставляет собой сечение диссоциации через возбуждение разрешенных пере-
ходов и рассчитана по формуле (3.25) с использованием средних сечений пог-
лощения света и фотоионизации [153, 159]. Разность кривых 3 и 4 дает сече-
ние диссоциации путем возбуждения запрещенных переходов.
Из этих данных следует, что все акты возбуждения молекулы метана, не
приводящие к ее ионизации, приводят к диссоциации. При этом вблизи по-
рога возбуждения определяющим оказывается вклад парциальных сечений
диссоциации через возбуждение запрещенных оптических переходов. Ана-
логичные выводы можно сделать на основании данных но спектрам потерь
и для других насыщенных углеводородов.
О диссоциации других молекул. Полученные выше сечения диссоциации
ряда молекул электронным ударом через возбужденные электронно-колеба-
тельные уровни позволяют сделать ряд общих замечаний о закономерностях
их изменения. Прежде всего, процесс диссоциации является многоканальным.
За редким исключением нельзя указать одно состояние, дающее основной
вклад. При больших энергиях налетающих электронов основной вклад
дают многочисленные разрешенные переходы. При этом велика роль рид-
берговских состояний, лежащих ниже потенциала ионизации, а также авто-
ионизационных. Практически для всех исследованных молекул из ридбергов-
ских состояний, лежащих ниже границы ионизации, наблюдается настолько
сильная предиссоциация, что они, как правило, не наблюдаются в излуче-
нии. По мере увеличения числа атомов в молекулах максимум сечений воз-
буждения этих уровней сдвигается в область автоионизационных состояний
[152]. При этом увеличивается и роль диссоциативной ионизации в полных
сечениях диссоциации при больших энергиях электронов. Сечения диссоциа-
ции с образованием возбужденных продуктов (электронное возбуждение)
в большинстве случаев малы по сравнению с полными сечениями диссоциа-
ции.
Если молекула имеет низкорасположенные валентные нестабильные (водо-
род, метан) или стабильные состояния, из которых наблюдается предиссо-
циация (азот), оптические переходы в которые запрещены, то вблизи порогов
диссоциации основной вклад в сечения диссоциации молекул электронным
ударом будет давать возбуждение именно этих уровней. Пороги возбуж-
дения таких переходов лежат ниже порогов возбуждения разрешенных пере-
ходов, а максимумы сечений достигаются при меньших энергиях налетаю-
щих электронов. Значения же в максимумах сечений не уступают значениям
для разрешенных переходов. Это и обеспечивает преимущественную роль
возбуждения таких переходов (если они приводят к диссоциации) в диссоциа-
ции молекул в электрических разрядах в газе и в электронных роях.
По мере увеличения числа атомов в молекулах правила отбора для пре-
диссоциационных переходов между стабильными и нестабильными элек-
тронными состояниями должны смягчаться, что должно привести, как мы
видели выше на примере метана и других насыщенных углеводородов, к эф-
фективной диссоциации с большинства возбужденных электронно-колеба-
тельных уровней.
Однако при этом надо иметь в виду, что с увеличением числа атомов в мо-
лекуле при девозбуждении электронных состояний могут начать играть
роль и внутримолекулярные безрадиационные переходы, не приводящие к
распаду молекул,— изомеризация, интеркомбинационная конверсия (при на-
личии стабильных триплетных состояний) [138].
В действительности многочисленные фотохимические исследования мно-
гоатомных молекул показали, что поглощение света (а следовательно, и воз-
буждение уровней электронным ударом) приводит, как правило, к эффектив-
ной диссоциации молекул. Квантовые выходы фотодиссоциации большого
числа исследованных молекул — альдегидов, кетонов, органических кислот,
азот- и серосодержащих органических соединений, органических галоге-
нидов, гипохлоритов, олефинов, ацетиленовых углеводородов и т. д.— до-
145
стигают величины 0,5—1 [138]. Исключение в этом списке составляют моле-
кулы бензола, ароматических углеводородов и аминов, для которых наблю-
даются высокие выходы люминесценции, интеркомбинационной конверсии
и изомеризации.
ДИССОЦИАТИВНОЕ ПРИЛИПАНИЕ ЭЛЕКТРОНОВ
К МОЛЕКУЛАМ
Диссоциативное прилипание электронов к молекулам с образованием от-
рицательного осколочного иона происходит через образование нестабильного
(относительно диссоциации) отрицательного молекулярного иона:
:ГИ (г' | т')
е Ц- АВ (;.')? >АВ~(т').
Ги(т'|Г) (3.62)
АВ~ (т’) A’ (Z) + В (тг).
ГД (In | т')
Схема этого процесса аналогична схеме процесса диссоциативной реком-
бинации через нестабильное состояние. Поэтому для расчета скорости этого
процесса применимы все формулы, полученные для диссоциативной реком-
бинации (стр. 174—178). Процесс (3.62) возможен только в том случае,
если отрицательный ион A~(Z) стабилен относительно автоионизации (отщеп-
ления электрона).
Наибольший интерес для низкотемпературной плазмы представляет
прилипание с образованием иона в основном состоянии АВ- (т')
(табл. 3.8) группы 1, III. Процесс через образование ионов АВ- в воз-
бужденных электронных состояниях менее существен, поскольку сечения
этого процесса малы по сравнению с сечениями диссоциации через возбужден-
ные состояния нейтральных молекул (см. стр. 136).
В тех случаях, когда потенциальные кривые нестабильных состояний отри-
цательных ионов пересекают терм основного состояния молекулы АВ в обла-
сти Франка — Кондона для нулевого колебательного уровня, зависимость
сечения и коэффициента скорости диссоциативного прилипания от темпера-
тур (Те, ТГ, Tv) должна соответствовать температурным зависимостям, на-
блюдаемым для коэффициента диссоциативной рекомбинации (3.158),
(3.160) — (3.163). Эта зависимость имеет место для пиков в группе I, т. е.
соответствующие коэффициенты скорости должны падать с ростом темпера-
туры. Если же мы имеем дело с состояниями отрицательного иона, потенци-
альные кривые которых имеют минимум в некоторой области межъядерных
расстояний, и если отталкивательная ветвь потенциала отрицательного иона
лежит на краю или даже вне области Франка — Кондона нулевого колеба-
тельного уровня молекулы АВ, то с ростом колебательной температуры мо-
лекул будет наблюдаться значительное увеличение скорости диссоциативно-
го прилипания, а также могут появиться новые пики диссоциативного при-
липания.
Подтверждением этого вывода служит наблюдавшееся экспериментально
для молекулы N2O появление пика вблизи нулевой энергии электронов с ро-
стом колебательного возбуждения [161]. Этот пик не проявляется при комнат-
ной температуре, поскольку ион N2O- (пг) изогнут, а молекула N2O — ли-
нейная (переход за пределами области Франка — Кондона). Возбуждение
изгибных колебаний с увеличением температуры от 300 до 1000° К привело
к увеличению сечения соответствующего процесса в 104 раз. В то же время
сечение перехода, имеющего максимум при Ее = 2,2 эВ, не изменилось, так
как состояние иона N2O~ отвечающего этому переходу, также линейно
(при комнатной температуре переход осуществляется в области Франка —
Кондона). По этой причине не наблюдается и существенных изменений сече-
ний диссоциативного прилипания к молекуле СО2. Для двухатомных моле-
146
Таблица 3.8
Сечения диссоциативного прилипания электронов к ряду молекул [159, 160]
Группа Молекула Ион Emax-eB /71max\ од. n(Ke ), IO"!6 СЛ12 ^од. n(Ee}dEe, Ю-i6 см^-эБ Полуширина пика, эВ
HJ J- 0,0 230 3,7 —
DJ J- 0,0 130 2,6 —
J2 J- 0,3 30 — —
1 НВг Br- 0,28 2,7 0,74 0,24
DBr Br- 0,28 1,87 0,51 0,23
НС1 ci- 0,81 0,2 0,074 0,3
DC1 ci- 0,84 0,16 0,062 0,28
Н2О H- 6,45 6,94.10-2 6,6-10-2 1,0
D2O D- 6,5 5,2-10-2 3,9-10-2 0,8
О2 o- 6,7 1,43-10-2 6,6-10-2 1,0
СО2 0- 8,03 4,8.10-3 5,3-Ю-з 1,1
D2O D- 8,6 6-10-3 9-Ю-з 1,6
Н2о o- 8,6 1,3-10-2 2,5-10-2 2,1
11 NO o- 8,6 1,1-10-2 2,86-10-2 2,3
Н2 H- 10,1 l,2.10-4 4,84-10-4 3,6
HD H-, D- 10,3 6,2-10-5 2,5-10-4 3,2-4
d2 D- 10,6 2,6-10“5 8,8.10-5 3,0
n2o o- 11,0 9,2-10-3 — 3,6
d2 D- 14,0 1,1-IO”4 — 1,6
со o- 10,3 2,2-10-3 2,76-Ю-з 1,4
N2O o- 2,2 0,98-10-2 1,05-10-2 1,1
n2o o- 0 IO-3 — —
III н2 H- 3,75 1,75-IO-5 1,0-10-5 0,4
HD H-, D- 3,75 l,0-10-6 — —
d2 D~ 3,75 8-10-8 — —
CO2 o- 4,35 1,6-10-3 1,3-Ю-з 0,8
кул О2, Н2 нагрев до 1000° К, осуществляемый в экспериментах [162, 163],
недостаточен для колебательного возбуждения, и увеличения сечений не мог-
ло наблюдаться. В случае водорода, судя по положению потенциальных кри-
вых молекулы и отрицательного иона [73, НО], увеличения скорости диссоци-
ативного прилипания с колебательной температурой ожидать нельзя. Дру-
гая ситуация, по-видимому, имеет место для NO и О2 [5]. Отсутствие сильной
зависимости сечений диссоциативного прилипания от температуры газа
позволяет сделать вывод, что в этих случаях гамильтониан взаимодействия,
ответственного за прилипание электрона, не зависит от номера вращатель-
ного уровня.
О РОЛИ РАЗЛИЧНЫХ ПРОЦЕССОВ
ИНИЦИИРОВАНИЯ ХИМИЧЕСКИХ РЕАКЦИЙ
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
Сравнение сечений рассмотренных выше процессов при соударении элек-
тронов с молекулами, приводящих к диссоциации последних, позволяет
сделать ряд общих выводов о роли того или иного процесса в различных
плазмохимических системах, при радиолизе и других случаях использова-
147
ния низкотемпературной плазмы. Эта роль существенно определяется функ-
цией распределения электронов по энергиям (см. стр. 102). В различных
случаях генерации плазмы с помощью газовых разрядов средние энергии
электронов не превышают, как правило, 5—7 эВ. Поэтому преобладающими
процессами диссоциации молекул в этих условиях будут диссоциация через
электронно-возбужденные состояния1 и для ряда молекул (группа I в
в табл. 3.8) — диссоциативное прилипание электрона к молекулам [165].
Диссоциативная ионизация, а также вторичные процессы с участием
ионов (диссоциативная рекомбинация, ионно-молекулярные реакции) в этом
случае не могут давать существенного вклада. Это объясняется тем, что
каждый такой акт должен приводить к гибели исходного иона, и скорость
реакции будет лимитироваться скоростью ионизации, которая существенно
ниже скоростей диссоциации через электронное возбуждение нейтральной
молекулы. Исключение могут составлять различные смеси газов, когда ис-
следуемая молекула является малой примесью в газе с потенциалом иониза-
ции, равным или даже большим потенциала ионизации исследуемой моле-
кулы. Другим исключением из этого правила могут быть цепные ионно-мо-
лекулярные реакции с промежуточным возбуждением ионов, например:
А2 + е -» (А:/; (А2+)* + А2 а: + А; (3.63)
А3+ + ^->(Аз)*; (А:)* + А2-> а: + А. (3.63')
В этом случае скорость суммарной реакции будет определяться скоростями
образования возбужденных ионов (см., например, [166]).
Если энергия электронов в низкотемпературной плазме велика (напри-
мер, электронные пучки, радиолиз, верхние слои атмосферы и т. п.), т. е.
существенная доля электронов имеет энергию до 100 эВ, то в этих случаях
скорости образования радикалов через возбуждение нейтральных молекул
становятся сравнимыми со скоростями ионизации и диссоциативной иониза-
ции (особенно для сложных многоатомных молекул). В этом случае все эти
процессы могут конкурировать друг с другом. Поскольку при таких
энергиях электронов основное значение в диссоциации молекул на нейтраль-
ные продукты имеет возбуждение оптически разрешенных переходов, эти се-
чения и скорости соответствующих процессов могут быть рассчитаны по фор-
мулам (3.23) — (3.26).
Увеличение заселенностей возбужденных колебательных и вращательных
уровней приведет к еще большему увеличению роли процесса диссоциации
через электронно-колебательные уровни по сравнению с другими процес-
сами. Это связано с тем, что коэффициенты скорости диссоциации резко зави-
сят от номера колебательного уровня исходного состояния, а также могут
зависеть от вращательной температуры молекул. Экспериментальные ис-
следования процессов возбуждения, ионизации и диссоциации молекул
в электрических разрядах при пониженных давлениях описаны в рабо-
тах [23—25, 91, 110, 166—175]. Как следует из результатов, изложенных
в предыдущем параграфе, в некоторых газах может сильно возрасти роль дис-
социативного прилипания электронов в диссоциации молекул.
По мере повышения степени ионизации плазмы и снижения средней энер-
гии электронов (это, как правило, происходит при увеличении давления
газа, в котором генерируется плазма, до атмосферного и более высоких давле-
ний) возрастает роль ступенчатых процессов возбуждения колебательных,
электронно-колебательных уровней и ступенчатой ионизации. Эти про-
цессы будут рассмотрены в следующих параграфах.
1 Процесс прямой диссоциации молекул в основном электронном состоянии при соударе-
нии с электроном является маловероятным. Его сечение мало по сравнению с сечением
диссоциации через электронно-возбужденные состояния [ПО, 164].
148
СТУПЕНЧАТАЯ ДИССОЦИАЦИЯ МОЛЕКУЛ
ЭЛЕКТРОННЫМ УДАРОМ
1. Диссоциация через основное электронное состояние
Диссоциация молекул происходит в результате соударений с верхних
колебательных уровней. Положение узкого места» определяющего скорость
диссоциации, зависит от соотношения скоростей диссоциации с верхних
уровней и колебательного возбуждения и может находиться на более низко
лежащих колебательных уровнях (например, [21, 176, 177], а также см.
стр. 53). Поэтому возбуждение колебательных уровней молекул электрон-
ным ударом должно приводить к увеличению скорости диссоциации.
Такое предположение неоднократно высказывалось на основании оценок
[55] и расчетов колебательной релаксации молекул с учетом е — F-, V —
V- и V — Т- процессов [56, 57, 60] (см. стр. 122).
Действительно, такие расчеты [56—61, 178] показали, что совместное
влияние возбуждения е — V и обмена колебательными квантами V — V
может приводить к сильному перезаселению верхних колебательных уров-
ней, а следовательно, и к увеличению диссоциации молекул даже при низких
температурах газа (Т ~ 300° К) на много порядков величины по сравнению
с термической диссоциацией. Были предприняты специальные расчеты ско-
рости диссоциации с учетом колебательного возбуждения электронами
в азоте [57], которые показали, что этот процесс в принципе может превы-
шать скорость диссоциации молекул через электронно-возбужденные состоя-
ния в условиях тлеющего и других разрядов при пониженном давлении в азоте.
Полученный результат существенно зависит от величины вероятностей
V — V- и V — Z-переходов между верхними колебательными уровнями.
Существенное влияние может оказывать и гетерогенная релаксация коле-
бательно-возбужденных молекул, которая не была учтена в работах [57, 61].
Поэтому величина скорости диссоциации, полученная в этих работах, требует
уточнения. Тем не менее качественно такая возможность подтвердилась.
Существенным является вопрос, насколько общим может быть такой про-
цесс для других молекул. Расчеты для молекулы СО также подтвердили его
возможность, однако в этом случае наличие дипольных радиационных пере-
ходов существенно снижает заселенности верхних уровней по сравнению с
теми, которые получены в азоте [58, 59]. Эксперименты [173] не подтвердили
существования диссоциации СО в результате такого процесса.
На основании этих расчетов можно утверждать, что для других гомоядерных
двухатомных молекул этот процесс должен быть также эффективным. Для
негомоядерных молекул вопрос остается открытым. Открытым он является
и в случае многоатомных молекул ввиду их быстрой колебательной релак-
сации, что препятствует созданию больших концентраций колебательно-воз-
бужденных молекул. Аналогичный вопрос дебатируется в научной литера-
туре и для лазерного возбуждения нижних колебательных уровней. Задачи
расчета в этих случаях сходны, хотя имеется специфика, которая состоит
во влиянии электронного удара на переходы между возбужденными уров-
нями [179—181].
В заключение еще раз подчеркнем, что влияние электронного удара на
диссоциацию молекул через основное электронное состояние должно быть
и в случае термической диссоциации при достаточно высоких степенях иони-
зации.
2. Ступенчатая диссоциация
через электронно-возбужденные состояния
При достаточно высоких степенях ионизации наряду с рассмотренным
выше процессом возможно протекание диссоциации молекул посредством
ступенчатого возбуждения электронами их электронно-колебательных уров-
ней.
149
Для нахождения скорости этого процесса в общем случае необходимо ре-
шить систему уравнений Паули для всех электронно-колебательных уров-
ней с учетом возбуждения электронами и тяжелыми частицами, переходов
между электронно-колебательными уровнями, чисто колебательных пере-
ходов в пределах каждого электронного состояния, а также диссоциации
(как спонтанной, так и индуцированной) и ионизации с этих уровней.
В настоящее время решение такой задачи невозможно не столько вслед-
ствие недостаточности знаний о вероятностях различных процессов,
сколько из-за ограничений, связанных с объемом памяти и быстродействия
современных ЭВМ. Задача существенно упрощается, если просуммировать
уравнения по колебательным уровням (тем самым не рассматривая коле-
бательную релаксацию электронно-возбужденных состояний, в том числе
и обмен колебательными квантами между различными электронными состо-
яниями). Такое предположение может оказаться приемлемым для боль-
шого числа электронно-возбужденных состояний ввиду того, что скорости их
тушения оказываются больше, чем скорости колебательной релаксации (см.
стр. 121, 130). Колебательная релаксация в возбужденных состояниях
вследствие электронного удара также в большинстве случаев несущественна,
так как процесс будет являться нерезонансным, а сечения таких процессов
малы. Кроме того, вероятности переходов между различными электронно-
возбужденными уровнями превышают эти значения.
С другой стороны, распределение по колебательным уровням в основном
состоянии определяется главным образом взаимодействием е — F-, V — V-
и V — Z-процессов, а влияние переходов между электронными уровнями
(возбуждение и девозбуждение в основное состояние) оказывается, как пра-
вило, несущественным. Таким образом, задача расчета ступенчатого возбуж-
дения разбивается на две отдельные задачи, которые можно решить последо-
вательно: колебательная релаксация в основном состоянии (см. стр. 121) и
возбуждение электронных уровней без учета колебательной релаксации
в электронно-возбужденных состояниях. Распределение по колебательным
уровням в основном состоянии будет влиять на скорости возбуждения уров-
ней (см. стр. 129) и поэтому должно учитываться в решении второй части за-
дачи. Тогда система уравнений Паули принимает следующий вид:
dMdfW1- = 2 (Ь I к, V,- Te)Ne + 2 (i, V. I к, ps; Гг) x
i, Vi \ m
X [AB (i, — 2 [AB (&> vk) { 2 иА) (^’ Vk I vi', Te) Ne +
vi
+ 2 w(m) (A;, | j, Vi) ATm} - 2 [AB (A> vk)l {гд (к. V, I A. В) +
m
+ 2 vk | А, В) ЛЦ + [A] |В] |гц (A, В | k, Гг) +
m L
+ 2 <’> <A’ BI A’ Zr) Nn] . (3 64)
m
В этом уравнении квадратными скобками обозначены концентрации компо-
нент; Ne — концентрация электронов; Nm — концентрация всех тяжелых
частиц сорта — вероятность предиссоциации, индуцированной
столкновениями с частицами сорта гп.
Последние два члена в правой части отражают процессы предиссоциации
из данного электронного состояния и обратный ему процесс рекомбинации
частиц.
При приближении к равновесию оба процесса взаимно компенсируются,
а вдали от равновесия преобладает один из них: процесс диссоциации в слу-
чае низких по сравнению с равновесными концентрациями фрагментов и ре-
комбинации в случае высоких концентраций фрагментов (этот процесс под-
150
робно освещен ниже (см. стр. 153), и здесь мы на нем останавливаться не
будем. Этим членом будем пренебрегать). Вероятности переходов обсужда-
лись на стр. 123—134.
Наиболее просто задача ступенчатой диссоциации и ионизации решается
для молекулы водорода. В этом случае нижний возбужденный уровень не-
стабилен, а более высокие расположены на значительном удалении от него,
а кроме того, имеют малые энергии связи или предиссоциируют, поэтому
переходы в них с большой вероятностью приводят к диссоциации (см. стр. 139).
Поэтому для диссоциации водорода должно быть применимо прибли-
жение мгновенной диссоциации, и основной вклад будет давать возбуж-
дение первого уровня. Расчет возможен непосредственно с помощью формул
(3.31), (3.32).
Соударения с тяжелыми частицами в этом случае для расчета скорости
диссоциации электронным ударом несущественны при любой степени иони-
зации. Это справедливо и для других молекул, низшие возбужденные со-
стояния которых нестабильны (гидриды, галогены) [3], а также и для боль-
шинства многоатомных молекул, предиссоциация в спектрах которых настолько
сильна, что возбужденные уровни в излучении не наблюдаются (см. стр. 145).
Решения системы (3.64) во всех этих случаях не требуется, что значитель-
но облегчает расчет скорости ступенчатой диссоциации электронным ударом.
Иное положение наблюдается для двухатомных молекул, нижние возбужден-
ные электронные состояния которых стабильны (N2, NO, О2 и т. д.).
Рассмотрим более подробно процесс ступенчатой диссоциации в азоте.
Влиянием столкновений с тяжелыми частицами на тушение электронных
уровней можно пренебречь (см. стр. 133) при
7Ve/[N2] > 2• 10-6, JVe/[N] > 10-2,
поскольку тушение молекулами состояния А3 2^ мало, а состояние 53llg ту-
шится целиком с образованием N2 (Л3 ££)•
На рис. 3.18 приведены результаты расчета коэффициента скорости сту-
пенчатой диссоциации молекулы азота в приближении мгновенной дис-
социации (следующей за возбуждением) при Tv = Те (кривая 7). Для срав-
нения изображена зависимость коэффициента скорости прямой диссоциа-
ции через электронно-возбужденные состояния при Те = Tv (кривая 5).
Как видно из этих данных, ступенчатая диссоциация происходит значитель-
но быстрее прямой во всей области температур электронов. На этом же рисун-
ке точками нанесены результаты измерения коэффициента скорости диссо-
циации азота в слаботочной электрической
дуге в аргоне с примесью 0,25—1% азота
при атмосферном давлении [182]. При этом
Ne = (2—5)-1015 см~\ [N] = (2-8) ЛО15
€ж~3, [N2] - (1-5) • 1014 см~3, Тг = 4500 —
5000° К, а Те = 8300-8600° К. Как пока-
зывают оценки, тушением уровней тяже-
лыми частицами в этих условиях можно
было пренебречь.
Колебательная температура молекул на
нижних уровнях, существенных для воз-
буждения состояний Л32^ и В3Щ, была
близка к электронной. На верхних коле-
бательных уровнях, существенных для
Рис. 3.18. Зависимость коэффициентов скорости
диссоциации молекулы азота электронным уда-
ром от температуры электронов
1 — через ступенчатое возбуждение электронных состоя-
ний; 2 — путем прямого возбуждения электронных со-
стояний из основного состояния. Точки — эксперимент
151
диссоциации через основное состояние, она была близка к Тт, в результате
чего коэффициент скорости диссоциации через основное состояние оказался
ниже экспериментально измеренного значения. Как видно из рис. 3.18г
результаты экспериментов удовлетворительно совпадают с расчетом в при-
ближении мгновенной диссоциации.
Следовательно, ступенчатое возбуждение электронно-колебательных
уровней молекул приводит в основном к диссоциации (в отсутствие диссоциа-
ционного равновесия). Поэтому процессы ступенчатой ионизации могут ока-
заться существенно ослабленными из-за конкуренции с процессом диссоци-
ации и зависящими от установления диссоциационного равновесия. Диссоци-
ация молекул влияет на ионизационные процессы и через другой процесс
ионизации — ассоциативную ионизацию (стр. 185).
3. Влияние электронного удара
на степень диссоциации в низкотемпературной плазме
Влияние электронного удара на степень разложения молекул в низкотем-
пературной неравновесной плазме (Те Тг) при пониженных давлениях не
вызывает сомнения, поскольку в таких условиях протекание всех эндотерми-
ческих реакций инициируется электронным ударом. Поэтому в данном раз-
деле мы рассмотрим влияние электронного удара в условиях, близких к тер-
мическим (Те = Гг), а именно, в электрических дугах при атмосферном дав-
лении, горящих в чистом азоте.
В этом случае в работе [183] наблюдалось существенное отклонение отно-
шения интенсивности излучения молекулярного азота к интенсивности
атомарных линий, что свидетельствует об отклонении от термодинамичес-
кого равновесия при токах I = 13,5—70 А и диаметре канала плазмотрона
0,48 см.
Нами было высказано предположение, что наблюдаемая неравновесность
обусловлена диффузией атомов с оси дуги [184]. Экспериментально измерен-
ные величины yN — [N]/[N]° и z/n2 = [N2]/[N2]°, где нулями отмечены равно-
весные значения, приведены в табл. 3.9.
Расчеты отклонений от равновесия проводились с использованием соот-
ношения
4 ^4г = 2 [N2] - S 49) [N]2 + ад- Р [Ng ] Ne - Ги [N]2 -
Q q
— = о, (3.65)
TD
где — коэффициенты скорости диссоциации и рекомбинации (для
тяжелых частиц брались из работы [44]); ад р, |За и — коэффициенты диссо-
циативной рекомбинации и ассоциативной ионизации; тр — время диффузии
атомов с оси дуги.
Как показывают оценки, ионизационное равновесие в этом случае обус-
ловлено процессом ассоциативной ионизации, характерные времена которой
существенно больше времени амбиполярной диффузии. Вместе с тем основной
канал образования [N2] — ассоциативная ионизация, поэтому диссоциатив-
ная рекомбинация не может восполнять убыль атомов из дуги (перезарядка
неэффективна по сравнению с ассоциативной ионизацией). Следовательно,
причина отклонения от равновесия — диффузия атомов, поскольку при
увеличении радиуса [185] неравновесность пропадает. Однако расчет от-
клонения от равновесия с учетом только диссоциации тяжелыми частицами
дает более сильные отклонения, чем наблюдалось экспериментально (см.
табл. 3.9), причем расхождение расчета и эксперимента систематически уве-
личивается с ростом температуры.
152
Таблица 3.9
-Степень отклонения от равновесия в электрической дуге в азоте при атмосферном
давлении
Т, 10» °к хе Яд, см тр, 10—4 сек VN2/VN
эксперимент расчет (/£д=0) расчет (/г® ^0)
8 2-10~3 0,24 0,9 1,7—2 7,0 4,0
9 0,9.10-2 0,24 0,7 2,5 5,5 2,9
10 2,4-10-2 0,24 0,6 1,8—2,6 8,6 2,5
11 5,6-10-2 0,24 0,5 1,2—2 12 2,0
7 7-10~4 1,5 37 1 1,10 1,1
Учет ступенчатой диссоциации азота электронным ударом (см. рис. 3.18)
приводит к удовлетворительному согласию расчета с экспериментом (см.
последний столбец табл. 3.9).
Таким образом, диссоциация молекул электронным ударом существенно
влияет на установление диссоциационного равновесия и в термических усло-
виях при степенях ионизации хе 10“3 [182, 185].
РЕКОМБИНАЦИЯ ТЯЖЕЛЫХ ЧАСТИЦ
В МОЛЕКУЛУ
Рекомбинация тяжелых частиц является процессом, обратным диссоциа-
ции молекул. Она наряду с диссоциацией определяет стационарный уровень
концентрации радикалов в химических системах, в том числе и в плазмохими-
ческих:
- т = ГABJ [М] - [Ml [А] [ВЬ (3-66)
где Ад и Ар — коэффициенты скорости диссоциации и рекомбинации; А, В —
фрагменты молекулы АВ.
Теоретическому и экспериментальному исследованию этого процесса пос-
вящено большое число работ (см. стр. 95). Характерной особенностью проте-
кания процессов гомогенной рекомбинации является образование электронно-
возбужденных молекул, зарегистрированное практически во всех исследо-
ванных случаях. Однако существующие теории рекомбинации (см., например.
[43, 186, 187] и библиографию в этих работах) не учитывают возможные пе-
реходы между различными электронными состояниями в процессе рекомби-
нации. Поэтому, пытаясь восполнить этот пробел, мы подробно остановим-
ся на рекомбинации, обусловленной неадиабатическими переходами между
различными электронными состояниями молекул. При низких давлениях
такие переходы, обусловленные процессом обратной предиссоциации, приво-
дят к радиационной рекомбинации.
Расчету процесса радиационной рекомбинации атомов посвящена работа
[188], некоторые попытки расчетов предпринимались и ранее [189—192],
однако полная схема процессов, протекающих при этом, до сих пор не рас-
сматривалась.
Во второй части этого параграфа мы коротко коснемся результатов ис-
следования гетерогенной рекомбинации атомов, поскольку в низкотемпера-
турной плазме газовых разрядов при пониженных давлениях она может
играть определяющую роль.
153
1. Ударно-реакционная рекомбинация
тяжелых частиц
В общем случае схема процесса имеет следующий вид
ki(Tnnii')
А (пг) + В (п) < АВ (?), ЛГ1(г'| mn) (3.67)
Г«(г'|/С')
АВ (£'), (3.68)
ГД(Нг')
х '4(1'1 fc')
АВ (Г) + М lAB(A'), (3.69)
X
АВ (Г)^АВ(р'), (3.70)
— АВ(р') (3-71)
АВ (к') + М Р--Ав(4, (3.72)
-*АВ(Л, j). (3.73)
Здесь А (иг) и В (и) — рекомбинирующие частицы (в частном случае — ато-
мы); АВ (?) — нестабильное электронное состояние молекулы, характери-
зующееся энергией Е и моментом вращения /(? = £, Е, I) и вызываю-
щее предиссоциацию стабильного состояния АВ (к') (к' — к, vk где
fk — номера колебательного и вращательного уровней; v (Л/) — газоки-
нетическая частота соударений молекул АВ (к') с частицами М; Ре, Pvv',
Р™ — вероятности электронного тушения, колебательных и вращательных
переходов с уровня к', рассчитанные на одно соударение с частицами М.
Поскольку нас не интересуют нестационарные процессы заселения уров-
ней V и к', протекающие, как правило, за время, существенно меньшее ха-
рактерных времен рекомбинации, то мы можем провести квазистационарный
анализ системы уравнений, описывающих кинетику процесса (3.67) — (3.73).
Из выражения для квазистационарной концентрации молекул АВ (?) сле-
дует, что она может отличаться от квазиравновесного значения
[АВ (?)] = tA(m)l [B(n)]/ci(mn|fz) = [A(m)] [В (п)]
' ' ki (i' | m, n) Кir (mn | i'; T) ' ‘ '
только в случае
кг (i' \mn)~v (к') Pe + A (к') ~ Гд (к' | ?) + (k' | ? ). (3.75)
Ввиду того что величина коэффициента скорости кг (i'/mri) определяется
временем разлета частиц А и В после предиссоциации, то
к± (i' | тпп) ~ v^/a, (3.76)
где а — расстояние между частицами А и В в момент предиссоциационного
перехода; — относительная скорость этих частиц в системе их центра масс,
типичные значения кг (i | тпп) ~ 1012 — 1013 сек-1.
Следовательно, условие (3.75) может быть выполнено только при кон-
центрации молекул — 1022 — 1023 или Ne 1020 см-3. Такой случай не
1 В сложных многоатомных молекулах наряду с процессами (3.67) — (3.73) возможны
безрадиационные переходы между различными электронными состояниями с передачей
части энергии возбуждения на другие степени свободы (колебательное и вращательное
возбуждение). Учет таких переходов в рамках данной работы не представлял бы труда,
но несколько загромоздил бы изложение. Поэтому такие процессы не включены в схему.
Однако их учет мог бы, по-видимому, объяснить большие скорости и второй порядок
ряда реакций соединения сложных молекул с радикалами [2].
154
представляет практического интереса, поэтому можно считать, что во
всех случаях концентрация молекул в нестабильном состоянии АВ (?') дол-
жна находиться в квазиравновесии с концентрациями частиц А и В, т. е.
справедливо соотношение (3.74). Константа равновесия вычисляется по фор-
муле
Ktr (j/ | тп;
1 г)_ gv Л3 ехр
(3.77)
\ кТг /
где gm, gn, gi' — статистические веса частиц в соответствующих состояниях;
ц — приведенная масса частиц А и В; к и h — постоянные Больцмана и
Планка; Et, Ея — энергия молекулы в состоянии (£') и энергия диссоциа-
ции на частицы А и В соответственно (отсчитанные от основного состояния
молекулы АВ). В таком виде уравнение (3.77) справедливо при любом рас-
пределении молекул по внутренним степеням свободы частиц и следует из
принципа микроскопического детального баланса (см. стр. 76).
В общем случае решение системы кинетических уравнений, описывающих
процесс (3.67) — (3.73), даже в квазистационарном приближении возможно
только с помощью ЭВМ. Поэтому рассмотрим некоторые важные частные
случаи, в которых можно получить аналитические решения.
Случай 1
Гд (Г | Л:') 4- Vя (Г///) > А (Л:') + v(A').
(3.78)
Такое соотношение выполняется в случае разрешенной предиссоциации
Гд > 1010 — 1012 сек'1 [107] при всех давлениях вплоть до атмосферного и
концентрациях электронов Ne < 1019 см~3, а также при наличии индуциро-
ванной предиссоциации, если
V* (Г Iк') > V (к') (Ре + Pvv').
При выполнении неравенства (3.78) квазиравновесие успевает установить-
ся не только для реакции (3.67), но и для реакции (3.68), так что имеем
1 4 Ktr (к I шп\ тг)
(3.79)
гдеА^г (к’ | ттг, Тг) — константа равновесия, определяемая по формуле (3.77)
с заменой индексов (/' -> к'). Если записывать рекомбинационный поток в виде
(3.66), то суммарный коэффициент скорости рекомбинации вследствие
процесса (3.67) — (3.73) равен
Лр= 2 |А1а!вГ''
п ш, к' i J I 1
где
/1ф(тп|/,:')~4г|Х
oo oo Jmin~1
A(fc')+v(fc') (ре+ 2 2 pVV,+ 2 2 p"j
pr=pirun v'—o
X Ktr (к' | m/г; Tf)
(3.80)
J min — минимальный номер вращательного уровня, с которого возможна
предиссоциация на уровне г; rmin — минимальный номер предиссоциирую-
щего колебательного уровня. В выражении (3.80) не учтены обратные потоки
с уровней, лежащих ниже границы предиссоциации, на предиссоциирующие
155
уровни за счет колебательной и вращательной релаксации, которые будут
несколько уменьшать коэффициент скорости рекомбинации. Для нахождения
точного значения коэффициента скорости рекомбинации в этом случае не-
обходимо решать систему уравнений для v' < rmjn, /' < 7 min [с учетом (3.80)
для уровней V> rmin, / > 7minJ:
оо оо
2 2 [АВ (к, vk, Я)] V (к, vk, Я) (Р‘ + Р™’ + Р”') - [АВ (к, vk, $)] х
r=rmin 7=imin
оо оо
X Га (Л, vk, ivk) + V (к, vk, fk) (ре + 2' Р^ + 2' Р”’)].
V—о 7=0
(3.81)
Коэффициент скорости хемилюминесценции в результате рекомбинации
определяется соотношением [188—192]
/хл(тп|А:')= к*л(тп\к') [A(m)] [В (п)] = 2 S [АВ (//)] А (к') +
^='Ут1п ^—Лтп
rmin“1 ^min—1
+ 22 [ABOA(A'). (3.82)
Ъ>=0 j=0
Первая сумма в правой части этого выражения описывает поток хемилюми-
несценции через предиссоциирующие уровни. Как следует из формулы (3.79)
величина этого потока не зависит от давления. Это справедливо и для сум-
марного коэффициента хемилюминесценции в случае /тт = 0, rmin = 0.
Однако в общем случае /min =# 0, rmin =# 0 при увеличении давления вслед-
ствие колебательной и вращательной релаксации будут заселяться и уровни,
лежащие ниже границы предиссоциации (второй член в правой части (3.82).
Поэтому полный поток хемилюминесценции через данное электронное со-
стояние будет увеличиваться по мере увеличения давления. Для его
нахождения необходимо решать систему уравнений (3.81).
Случай 2
ГД(?|Л') +vB(f|«>/)<A(iHv(4 (3.83)
Такое соотношение может выполняться при запрещенной спонтанной, а так-
же при индуцированной предиссоциации при малых давлениях, когда А.
При выполнении неравенства (3.83) заселенности уровня АВ (к') будут су-
щественно ниже, чем в случае равновесия с атомами (3.79), и обратными по-
токами за счет предиссоциации АВ (&') -> АВ (?) можно пренебречь. Поэтому
для коэффициента скорости рекомбинации в этом случае имеем
% (тп | к') = 2 ГДУ1^ + УЛ(^1Л) • (3.84)
I / [М] KtT(ir \ тп\ тг) ' 7
В этом случае процессы колебательной и вращательной релаксации, а также
тушение данного электронного состояния не влияют на величину коэффици-
ента скорости рекомбинации. Но они будут существенно изменять распре-
деление заселенностей уровней, лежащих как ниже, так и выше границы
предиссоциации.
Для нахождения заселенностей снова необходимо решать систему урав-
нений, аналогичную (3.81). Если пренебречь вращательной и колебательной
релаксацией, то при выполнении условия (3.83)
[АВ (Л')] = [А (т)[ [В (п)] 2 — (3’85)
Г Ktr(i'\тп-,Тг) [A(/c') + v(fc')P ]
156
Коэффициент скорости хемилюминесценции тогда равен
{i + ^}~‘. (з.»в>
77 KtT (l I nUl' гг) 1 А J
В этом случае он будет падать с ростом давления (обратно пропорционально
последнему) только при
Гд(Гр')>тд(1"|/с') и v(fc'>A(£'). (3.87)
При преобладании индуцированной предиссоциации
Vя (Г j Л/)>ГД (Г | Л') (3.88)
и v (к') A (к') кхя должно расти с увеличением давления. Постепенно этот
рост будет замедляться, иприт !>* А зависимость от давления должна исчез-
нуть. Поэтому исследование зависимости кхл от давления в этом случае мо-
жет не только дать возможность определить коэффициент скорости рекомби-
нации, но и установить, является ли она спонтанной или индуцированной.
Случай 3
Гд (Г | к') + Vя (Г | к’) ~ А (к') + V (к') ( Ре + 2 р'”” + 3 • (3-89)
V У
Этот случай может реализоваться при индуцированной и запрещенной спон-
танной предиссоциации. Общие выражения для коэффициента рекомбинации
в аналитическом виде получить в этом случае не удается. В частном случае
преобладания тушения электронного состояния Ре Р7/, Pvv'
к (тп Ш - 1 У Г” V Ife') + Vя G' | fc')
кр(тп\к) [M] Zj Ktr(i’\mn; Tr) x
x __________[A(fc') + v(fc'He]_____ (3 90)
[ A (fc') + v (fc')Pe + Гд(£' |fc') + Vя (i' | fc')J '
И
к™(mn I к') - 2____________1ГД (Р|^)+_^1ЮМ(У)-------------------.
i' Ktr(V\mn- Tr)[A(k’)+v(k')Pe+ + | Л')1
(3.91)
Как следует из этих соотношений, коэффициент скорости рекомбинации либо
растет, либо не меняется с ростом давления, а коэффициент скорости хемилю-
минесценции падает медленнее, чем обратно пропорционально давлению, при
наличии предиссоциации любого типа.
Случай 4
Радиационная рекомбинация: Гд (Р | к') !>* тд (Z' | &'), А (к') !>> v (к').
В этом случае при любом соотношении вероятностей предиссоциации и ради-
ационных переходов имеем
кр (тп I &') = -L ...- Г.Д (/с' । Г) Л (fc,) —— 1-—. . (3.92)
PV 1 [М] [Гд(/с'1 £') + А (к')] Ktr(k lmn’7 r) '
Выражение (3.92) по существу не отличается от соответствующего выраже-
ния, полученного для коэффициента скорости радиационной рекомбинации
в работе [188] при рассмотрении процесса (3.67), (3.68), (3.70) как процесса
резонансного поглощения света на молекуле АВ. Отличие состоит в том,
что в работе [188] рассматривалась рекомбинация только через один колеба-
тельный уровень АВ (к'), и поэтому суммирование в формуле для kv прово-
дилось по I = /. На самом деле, как будет видно на рассмотренном ниже
примере радиационной рекомбинации двух атомов азота и кислорода с водо-
родом, рекомбинация через два или несколько колебательных уровней мо-
жет дать существенный вклад даже при кТ <<! сое.
157
В предельных случаях из (3.92) легко получаются соответствующие более
простые формулы:
Zcp (к' | тп; Тг) [М] = Лхл (к' | тп) =
А Г? A (k'\
(к' | m/г; Гг) ' \ ' )’
= +г^Х'г,
(3.93)
(3.94)
Сравнение с результатами экспериментов. Полученные выше формулы
для расчета коэффициентов скорости рекомбинации и сопровождающей ее
люминесценции (3.67) — (3.73) могут быть использованы для анализа раз-
личных экспериментально исследованных при комнатной температуре случа-
ев двухчастичной рекомбинации: двух атомов азота [191, 193], азота и кис-
лорода [192], серы [189], кислорода и водорода [190].
Пример 1
2N(4S)->N2 (3Sg) —
/ > N2 (В3 Щ, vk — 12, Jniin — 34; vk 13, /min — 0).
\—> N2 (бЙП^. Vk — 6, /min — 14, Vk 7, /min = 0).
Для этих случаев предиссоциации [194] Гд (к' | i') ^>А (к') [108]. Для радиа-
ционной рекомбинации справедлива формула (3.93). Результаты расчета
парциальных и полных коэффициентов скорости радиационной рекомбина-
ции через эти состояния вместе с экспериментально измеренными значения-
ми [193] приведены в табл. 3.10. В расчетах использовали следующие
значения вероятностей переходов 4(В3Щ, vk — 12, 13) — 2,2 • 10б сект1,
А (агЩ, vk = 6) = (0,7~1,0)-104 сек”1 [195, 196] и Ея - 78714 ±
± 40 см~г [194]. Экспериментальное значение коэффициента скорости радиа-
ционной рекомбинации через уровень a4Ig, v = 6, j 13 определялся нами
по данным работы [193] с помощью приближенной формулы
*XJI(a1ng,p = 6, />13)~
_________________________fcXJI (a^g, 2/fc = 6)____________________________
1 + v (/с') 2 ( v (&') 2 P”’ + ч(к’)Ре + A (к‘)
д' д'
которая следует из решения системы уравнений (3.81). Для этого уровня при
р = 6-Ю-3 торр vPe = 1,8-104 [108] и v (к’) У Р33' = 1,5-10* сек"1 [21] (см.
стр. 116).
Как следует из результатов расчета, основной вклад в рекомбинацию че-
рез состояние В3 Щ дает колебательный уровень vk = 13, однако вкладом
уровня vk = 12 пренебречь нельзя. Соотношение Лхл (vk = 12)/кхл (vk = 13)
Таблица 3.10
Сравнение расчетных и экспериментальных значений коэффициентов радиационной
рекомбинации
Ю-20 см3'сек-1
Состояние „ЭЛ V д° • 'пип Kjj.z, см"1 расчет эксперимент
6 12 13 12+13 34 0 79423 79 130 1,6±0,5 9,4±2,4 11,1±3 16±7
2 6 14 78 849 0,5 (io’L) (0,3-0,5)±40%
158
согласуется с экспериментальными данными [193]. В обоих случаях резуль-
таты расчета в пределах погрешностей согласуются с результатами экспери-
мента.
Пример 2
2 S (3Р) -> S2* -> S2 (В32^, vh > 10).
В этом случае также справедлива формула (3.93) для коэффициента скорости
радиационной рекомбинации, так как уровни vh > 10 в поглощении диффуз-
ны. Используя значения gs = 5,5, g™2 — 3 и АЕ = 0—140 слГ1 [189], Л10 =
= 3 • 107 сек"1 [189], получаем
АХЛ(В32~) = (O^-l^’lO-16^3.^-1. (3.95)
Это значение на порядок величины превышает экспериментальное [189]
^хл (в32“) = 0,64-10’17 см^сек"1. (3.96)
Такое расхождение обусловлено, по-видимому, неточностью оценки вероят-
ности радиационных переходов по данным [189]. Косвенным подтвержде-
нием этого является большое значение сечения тушения уровня vk =
= 10 B3Sg атомами аргона, которое получается из экспериментально изме-
ренного в работе [189] отношения A (vk)/v (vk) Ре — б(Аг) (B3SJ, v = 10) =
= 3,7-10-15 см2. Если использовать для определения вероятности радиацион-
ного перехода измеренное значение коэффициента скорости хемилюминес-
ценции (рекомбинации) серы [189], то получаем А (В32^, vh = 10) = (1,5—
—3)-106 сек"1. При этом сечение тушения данного уровня атомами аргона
составит б<Аг)) (А') = (1,8—3,7).10~16 см2, что удовлетворительно согласует-
ся с сечениями тушения аргоном других электронных состояний молекул
(например, для тушения состояния N2 (В3 Щ) аргоном с(Аг> (А/) = (2—6) X
Х10~16 см2 [193]).
Пример 3
О (3Р) + N (4Д) -> NO (а4П) NO (С2П, vh = 0).
Из измерений квантового выхода фотодиссоциации окиси азота [197] сле-
дует, что
ГД(С2П, vk = 0 | а4П) = (14-33) А (С2П, vk = 0),
и опять справедлива формула (3.93). В этом случае коэффициент скорости
хемилюминесценции не должен зависеть от давления в полном согласии
с экспериментальными данными [197]. Расчетные значения коэффициента
скорости люминесценции при Т = 300° К (ДЕ = Ед — Е (С2П, vk — 0) =-
-0):
&ХЛ(С2Ш— f ^-Ю 17сл13-с^ 1 при А(С2П) = 2,4-107с^ 1 [198],
“ I 3,8-Ю-17^3.^’1 при А(С2П) = 5,1-107 сек-1 [197].
Наиболее точное экспериментальное значение [192] при Т = 300°К
(С2П) = (1?5 ± о,3).1О-17 см3-сек"1.
Результаты расчета удовлетворительно согласуются с экспериментом,
если использовать первое из значений вероятностей переходов [198]. Это поз-
воляет нам с учетом полученных выше результатов сделать вывод, что зна-
чение вероятности радиационного перехода [198], по-видимому, является
более правильным.
Пример 4
О (3Р) + Н (2S) -> ОН (22~) ОН (A2S+, vk = 0,1).
В работе [190] в результате этого процесса наблюдалась хемилюминес-
ценция с уровней vk = 0 и vk = 1 при давлении р = 0,4—2 торр. Сростом
давления интенсивность хемилюминесценции Лхл падала.
159
При давлении р = 2 торр вероятность электронного тушения уровня
Л22+ — v (Л') Ре — 3,4-Ю5 сек-1, что сравнимо с вероятностью радиацион-
ного перехода А (Л22+) = 7,1-105 сек1 [190]. Зависимость интенсивности
хемилюминесценции от давления и концентраций атомов описывается про-
цессами (3.67), (3.71) — (3.73), если принять
7схл(Л22+) = (3±1)-10-21 см^сек'1, (3.97)
[kXJl(vk = 1)/Лхл(vk— 0)] — 1,15, Ахл(Л22+) — р”1.
Энергии колебательно-вращательных уровней состояния ОН (Л22+), рас-
считанные нами по данным [199], приведены в табл. 3.11, где указаны также
высоты вращательных барьеров для предиссоциации с этих уровней (Ед =
= 35400+Ю5 см'1 [199]).
Таблица 3.11
Параметры, характеризующие хемилюминесценцию молекулы
ОН(Л22+)
V E(A2L+, v^, fy, см—1 Ьд + ^В.б’ СМ-1 K(r(A*S+, vk, % 1 | О, Н; Тг), 10« слг->
и 13 35 487 36 180
14 35 962 36 290 —
15 36 470 36 420 37
16 37 013 36 550 530
1 0 35 391 35 400
1 35 423 35 408 2,5
2 35488 35424 2,3
3 35 714 35448 3,3
4 35 876 35 591 6,0
Из данных табл. 3.11 с учетом ошибки в значении Ед следует, что предис-
социации с уровня vk = 0 возможна только при /> 15—16, а с уровня vk =
= 1 — при / 1—3. (Вероятность прохождения частиц под барьером исче-
зающе мала.)
Из проведенного выше анализа следует, что зависимость коэффициентов
от давления (3.97), наблюдаемая в эксперименте, возможна только при соб-
людении условий (3.87), (3.93), т. е. если предиссоциация является спонтан-
ной, а частота тушения больше вероятности радиационных переходов. По-
скольку рассматриваемая предиссоциация протекает без изменения спина
и проекции орбитального момента, то вероятность предиссоциации не долж-
на зависеть от номера вращательного уровня [107]. Кроме того, ГД(£'|А/) =
= Гд (&'/£'), и из правил отбора следует I = j. Тогда согласно (3.94)
оо
Лхл(422+,р)£) = Гд(Л22+>рк) У ---------1. (3.98)
'min
С использованием экспериментальных данных (3.97) из этой формулы была
рассчитана вероятность предиссоциации для уровня vk = 1: ГД(Л22+, vh =
— 1) = (0,6—2)-103 сек"1. Для уровня vh = 0, если предиссоциация с j = 15
возможна, то ГД(Л') = Если предиссоциация начинается на уров-
не / = 16, то ГД(Л22+, vk ~ 0) = 7-105 сек-1. Оба последних значения опре-
делены с точностью до коэффициента 2. Следует отметить, что значение 7-105,
по-видимому, менее вероятно, поскольку в этом случае зависимость &хл от
160
давления должна была бы быть значительно слабее, так как условие (3.83)
при этом не выполняется, и если вращательную релаксацию учесть прибли-
женно, получаем
_____________гд (Л^+, VR) А (Л2£+, z?fe)_
1 Гд rk) + А (Л22+, vk) + v (Л22+, /Ре + Р"') '
' j'
Вращательной релаксацией пренебрегать в данном случае нельзя, по-
скольку согласно оценкам Ре Р™'. Это подтверждается наблюдавшимся
в экспериментах перераспределением заселенностей вращательных уровней
vk = 0. Влияние вращательной релаксации, не учтенное авторами [190]
при расчете значений коэффициентов скоростей (3.97), объясняет отмечав-
шееся ими некоторое расхождение значений Лхл при определении его при раз-
ных давлениях.
Низкие значения полученных нами вероятностей предиссоциации объ-
ясняются, с одной стороны, тем, что предиссоциационный переход запрещен
правилами отбора (+ А —) [107]. Различие же между значениями Гд для
уровней vk — 0 и 1 связано с влиянием факторов Франка — Кондона. Ка-
чественно оно согласуется с видом предполагаемого пересечения потенциаль-
ных кривых состояний Л22 + и 22~ и может быть использовано для более
точного построения кривой для состояния 22~.
Температурная зависимость коэффициентов скорости радиационной ре-
комбинации атомов. При выполнении условия
2Вг(7тт+1)<^,
что имеет место для молекул, составленных из тяжелых атомов, при темпе-
ратурах выше комнатной, суммирование по вращательным уровням в фор-
мулах (3.77), (3.93), (3.94) можно заменить интегрированием по вращатель-
ному квантовому числу. Тогда, например, из формулы (3.93) получаем (если
Л (А')^/(/))
к™ (к, vk) = A(k, vk)
gВ 9/ &еър[-(Ек,~Е^1 кТг]
ёАёв Вг(2лр)3/2 (А:ТгГ0’5
(3.99)
В случае радиационной рекомбинации атомов азота через состояние
N2 (В3Щ, vk = 12, / > 34; vk = 13) изменение температуры газа с 295 до
463° К привело к увеличению коэффициента скорости радиационной реком-
бинации в 2,2+0,4 раза [193]. По формуле (3.99) для этого случая получаем
изменение в 1,6+0,2 раза. Согласно данным той же работы [193], коэффици-
ент скорости радиационной рекомбинации двух атомов азота через состояние
N2 (бЛЩ, vk = 6, j 14) в том же диапазоне температур не изменялся. При
расчете по формуле (3.99) это изменение должно было составлять менее 3%,
и, естественно, не было обнаружено в эксперименте.
Коэффициент скорости радиационной рекомбинации атомов О (3Р) и
N (4S) через состояние NO (С2П) при изменении температуры с 600 до 2500° К
увеличивался в 1,65+0,35 раза [200]. Согласно расчету по формуле (3.99)
это изменение должно было составлять 100%, если не учитывать возможную
зависимость А (к') = f (/).
Таким образом, температурная зависимость коэффициентов скорости ра-
диационной рекомбинации атомов также в пределах погрешности описы-
вается полученными выше формулами.
Ударно-радиационная рекомбинация О (3Р) + СО (Х1^)
Хемилюминесценция молекулы СО2, сопровождающая рекомбинацию
нормального атома кислорода с молекулой СО, исследована в ряде работ
[201—203]. Однако до сих пор нет ясного представления о природе этой лю-
минесценции и ее связи с механизмом рекомбинации атома кислорода О (3Р)
с невозбужденной молекулой СО (XxSj) [201, 203—205]. Это связано отчасти
6 Теоретич. и прикладная плазмохимия
161
с большими систематическими погрешностями измерений разных авторов
в близких условиях.
Исследование спектра хемилюминесценции с большим разрешением поз-
волило сделать однозначный вывод о том, что излучающим состоянием яв-
ляется состояние СО2 (1В2). Равновесные длины связи О—С в этом состоя-
нии Re = 1,246 А, а угол между атомами кислорода составляет 122+2° 1206].
В спектрах поглощения вакуумного ультрафиолетового излучения переход
СО2 (Х1^^ — JB2) наблюдается с максимумом на длине волны 1450 А [4],
т. е. на уровень с = 8,45 эВ. Это объясняется тем, что при оптическом
переходе геометрия молекулы не меняется, следовательно, переход совершает-
ся в состояние 1В2 с линейной конфигурацией, что, согласно нашему расчету,
соответствует энергии колебательного возбуждения —2 эВ. Сила осциллятора
для перехода Х1^ — 1В2, определенная по коэффициенту поглощения СО2
[207], составляет /(е> = 10“2. Тогда радиационное время жизни уровней 1В2ч
наблюдаемых в результате рекомбинации атомов кислорода с молекулой СО,
должно составлять
трад (ХВ2) ~ 2,3.10"7 сек. (3.100)
Несмотря на то что переходы на уровень гВ2 наблюдаются в спектрах по-
глощения света и потерь электронов [208], излучение с него отсутствует. Это
свидетельствует о дополнительном канале дезактивации, которым может быть
(при низких давлениях р < 10-3 торр [208]) только спонтанная предиссо-
циация уровня ХВ2, вызываемая взаимодействием с отталкивательной ветвью
состояния 3В2. С учетом этих данных попытаемся проанализировать резуль-
таты работ [201—204] по хемилюминесценции СО2.
Во всех работах отмечалось, что коэффициент скорости хемилюминес-
ценции не зависит от давления. Однако в работе [201] были получены дан-
ные, свидетельствующие о зависимости Лхл от природы третьего тела, при
р = 0,6—2 торр. Поэтому был предложен следующий механизм хемилюми-
несценции:
О (3Р) + СО (Х1^) + М СО2 №) + М,
СО2 №) + М -> СО2 (Xx2j) + М, (3.101)
СО2 СВ2) -> СО2 (Х^) + hv.
Для того чтобы этот механизм описывал экспериментальные данные при ра-
диационном времени жизни трад (х52) — 2,3-10“7 сек, коэффициент скорости
тушения этого состояния аргоном должен составлять
хРе = w<a>-)(152)> = 2-10-1°сек, (3.102)
что представляется абсолютно невероятным, тем более что в работе [203]
отсутствие зависимости Ахл от давления было обнаружено при р =
= 0,2' торр.
Следовательно, предложенный ранее механизм хемилюминесценции про-
тиворечит экспериментальным данным о малом радиационном времени жизни
состояния СО2 (ХВ2), а также экспериментальному факту наличия спонтанной
предиссоциации молекулы СО2 (ХВ2).
Тогда нами было сделано предположение, что наблюдаемая хемилюминес-
ценция СО2 обусловлена ударно-радиационной рекомбинацией в результате
спонтанного предиссоциационного перехода:
А
GO (X12+) + О (3Р) СО2 (3В2),
к~
1
ГД(зВ2|Ш2)
С02(352)^=^С02(’В2),
ГД (>В2рВ2)
162
со2 (>z?2) —Д С02 (Xx2j) + hv, (3.103)
СО2 ев2) + М^СО2(У^ + м,
а зависимость Лхл от природы третьего тела [201] объясняется неучтенными
погрешностями эксперимента (это возможно, поскольку изменение не пре-
вышает 50% от средней величины). Коэффициент скорости хемилюминесцен-
ции рассчитывался с помощью формулы, полученной из (3.80):
к №1 О, СО; Тг) 1 + [v (1В21 X) ре]/[гд (1Д21 зв2)] ' (3.104)
Для расчета статистической суммы молекулы СО2 (1В2) использовались
формулы [199]
g(W = ^r.0<2>K.p, (З.Ю5)
где Щв2 = 2, так как состояние ГВ2 соответствует состоянию XAU линейной
молекулы [4, 206]:
<?Г. о = П -п----Г---ГГгГГ » (3.106)
<2ж. р = 4 (3.107)
тде Av = 5,9 см~\ Bv = Cv = 0,426 см~г — вращательные постоянные мо-
лекулы в состоянии гВ2; = 1225 слт1, со2 = 600 см~\ со3 = 2300 см~1 [4].
Тот факт, что коэффициент скорости хемилюминесценции не зависит от
давления вплоть до р = 0,5—1,5 атпм [209], позволяет нам сделать вывод, что
Гд (Ч321 3Я2) > v (ХВ2 | X) ~ 108 сек-1. Тогда
&расч№) - 1,8-10"18ехр(— АЕ / RT),
Лхл (ХВ2) = l,6-10“18exp(—2500/jRZ) [204], 335 <77<1500°К,
k^B2) = 10"17 ехр (-3700/ RT) [201], 300<Т<500°К.
Экспериментальные значения удовлетворительно согласуются друг с дру-
гом (погрешность их составляет около 100% [202]) в исследованном диапазоне
температур. Если предположить, что нижний уровень состояния гВ2 лежит
на 2,5 ккал/моль выше предела диссоциации на нормальные атом и молекулу
СО, то результаты расчета Арасч также удовлетворительно согласуются с
экспериментальными значениями. (Заметим, что полученное в работе [206]
значение АЕ = 6+2 ккал/молъ может быть завышено ввиду сложности ана-
лиза и интерпретации полученного спектра.) Таким образом, предложенный
нами механизм хемилюминесценции СО2 объясняет все имеющиеся экспери-
ментальные факты (за исключением отмеченного в [201] небольшого влия-
ния третьего тела на &хл), а теоретический расчет дает близкое к эксперимен-
тальному значение Ахл во всем исследованном экспериментально диапазоне
температур, если принять, что нижний уровень состояния ХВ2 лежит на
2,5 ккал!моль выше предела диссоциации СО2 на невозбужденные продукты.
Ввиду отмеченной выше несостоятельности механизма (3.101) представ-
ляется, что механизм (3.103) является единственно возможным механизмом
хемилюминесценции СО2.
Полученные выше результаты позволяют уяснить и механизм рекомби-
нации атомов О (3Р) и молекул СО (XT2j). Коэффициент скорости рекомби-
нации через состояние СО2 (ХВ2) можно записать в виде [Гд (гВ2) А (ХВ2) +
ч- v (^2 I X)]
4р(ХЯ2) [М] = 4ХЛ(\В2) . (3.108)
- I ( L>4)
163
6*
| Из теоретического анализа сечений тушения электронно-колебательных
уровней молекул [43, 98] следует, что сечения тушения состояния СО2 (ТВ2)
атомами аргона, молекулами СО и кислорода близки друг другу и, по-види-
мому, близки к газокинетическим, а коэффициенты скорости тушения не
должны зависеть от температуры (см. стр. 130—133)
v (ХВ2 |Х)Ре - 3-10"10 см3• сек-1 =f= f (Т). (3.109)
Из выражения (3.109) следует, что процесс рекомбинации может иметь второй
порядок по концентрациям частиц, если она является чисто радиационной
и Ар (ХВ2) [М] = Лхл Однако такое предположение противоречит экспе-
риментальным результатам, поскольку во всех без исключения работах
[201—205] было получено
Лр [М] Ахл.
(3.110)
Отсюда следует, что процесс рекомбинации протекает обязательно с участием
третьего тела. Это подтвердилось экспериментально результатами последней
по времени работы [210], где специально изучался вопрос о порядке реакции.
В работах [203—205] была обнаружена различная температурная зави-
симость коэффициента скорости рекомбинации. Положительная темпера-
турная зависимость (рост Ар с увеличением Т) может быть обусловлена только
рекомбинацией через состояние СО2 0В2), поскольку в настоящее время на-
дежно установлено, что рекомбинация через уровни, лежащие ниже границы
диссоциации, приводит к отрицательной температурной зависимости коэф-
фициента скорости.
Результаты расчета отношения коэффициента скорости рекомбинации
через состояние СО2 (ХВ2) к полному коэффициенту рекомбинации, измерен-
ному в [205, 203] соответственно, приведены в табл. 3.12.
Таблица 3.12
Значения Р = kp(1B2)/k^:)КСЦ при разных температурах. М = А, СО;
v(iB2 \Х )Ре
ТГ,°К 3 по данным [205] 0 по данным [203] тг, °к 0 по данным [205] 0 по данным [203]
300 IO"2 2-10-3 500 0,5 6-10~4
400 ю-1 Ю-з 600 1 5-10-4
Из данных табл. 3.12 следует, что положительная температурная зави-
симость коэффициента скорости рекомбинации [203] при Т < 500° К при та-
ких абсолютных значениях fcp, 8KCn на практике реализоваться не может, по-
скольку через состояние СО2(3В2) в этом случае может протекать лишь ма-
лый поток (Р < 10-1) рекомбинации. Основной поток рекомбинации при
этих температурах может идти через уровни, лежащие ниже границы диссо-
циации, т. е. через связанное состояние СО2(-3£?2). Следовательно, коэффици-
ент скорости рекомбинации должен уменьшаться по мере увеличения темпе-
ратуры [205]. Положительная температурная зависимость коэффициента
скорости рекомбинации при Т < 600° К [203] является следствием погреш-
ностей измерений.
При дальнейшем увеличении температуры (Т 600° К) коэффициент ре-
комбинации через уровень СО2 (ХВ2) начинает превосходить экстраполиро-
ванные значения коэффициента рекомбинации через СО2 (3В2) (см. табл. 3.12).
Это должно привести к смене температурной зависимости коэффициента ско-
рости рекомбинации.
На рис. 3.19 приведены результаты расчета коэффициента скорости удар-
но-радиационной рекомбинации этих частиц по формуле (3.108), а также име-
164
ющиеся наиболее достоверные экспериментальные данные, включая и] ре-
зультаты работы [211], в которой была предпринята более тщательная очи-
стка газов от примесей. Данные [210] были экстраполированы нами на об-
ласть высоких температур с учетом результатов [205].
Непонятным является наблюдаемое резкое различие результатов работ
[210, 211], выполненных при одинаковых условиях. Чистота газа вряд ли
могла сильно изменить коэффициент рекомбинации, поскольку он не так
сильно зависит от рода третьего тела. Тем не менее можно высказать предпо-
ложение, что это различие связано с тем, что в случае [210] имеются частицы,
приводящие к тушению состояния (3В2), а в [211] таких частиц нет. Тогда ре-
комбинация протекает целиком через состояние (ХВ2) и при низких темпера-
турах, а ход расчетной кривой качественно описывает наблюдаемую зави-
симость коэффициента рекомбинации от температуры.
Рис. 3.19. Зависимость коэф-
фициента скорости объемной
рекомбинации атомов кислоро-
да и молекул окиси углерода
от температуры
1 — расчет через состояние 1В2',
2 — через 3В2 согласно данным
[205, 210];
з — сумма (1) и (2); М = Аг, СО;
4 — по данным [211];
5 — экспериментальные точки [209],
М = Аг;
6 — [211], М = СО;
7 — [211], М == Аг
Из сравнения результатов, изображенных на рис. 3.19, можно сделать
только один вывод — рекомбинация атомов кислорода и молекул СО по край-
ней мере при температуре Т 1000° К целиком обусловлена ударно-радиа-
ционной рекомбинацией (3.67) — (3.73), т. е. связана с неадиабатическими
переходами.
О роли электронных переходов в рекомбинации частиц. Выше на примере
реакции рекомбинации невозбужденных атомов кислорода и молекулы СО
мы показали, что роль ударно-радиационной рекомбинации повышается по
мере увеличения температуры. При Т 1000° К она является определяющей,
что может привести к смене температурной зависимости коэффициента ско-
рости рекомбинации. Ниже приведены результаты оценки вклада такого
механизма в рекомбинацию двух атомов азота.
В табл. 3.13 приведены результаты расчета отношения коэффициента
скорости ударно-радиационной рекомбинации атомов азота через уровни
Таблица 3.13
Отношение коэффициента скорости ударно-радиационной
рекомбинации атомов азота через состояние №(В3Пд)
к коэффициенту скорости рекомбинации через состояния
N2(X1S+),N2(JSS+) [187]
т, °к р, торр
10 10* Юз 10*
• 300 зло-3 10-3 1,2-10-4 1,2-10“5
3 000 0,1 0,07 0,05 0,01
10 000 0,2 0,2 0,18 0,11
165
N2 (B3IIg, > 12) к суммарной скорости рекомбинации, рассчитанной в
работе [187] без учета электронных переходов при М=Аг. При этом были ис-
пользованы результаты работы [91] по вероятностям предиссоциации и туше-
ния уровней N2(B3ng) атомами Аг [212].
В табл. 3.14 сведены результаты аналогичных расчетов при наличии ту-
шения уровня N2 (В3Щ) электронным ударом (здесь хе — степень иониза-
ции) [64] (см. стр. 125).
Таблица 3.14
Отношение коэффициента скорости ударно-радиационной
рекомбинации атомов азота через N2(B3IIg) при наличии элек-
тронов к коэффициенту скорости рекомбинации через состояния
N2(.43S+)h N2(XxS+) [187], M = Ar, Тг = Те = 10* ° К
хе р, торр
10 102 Юз 104
1 40 4 3 0,5
10-1 4,0 3 1,6 0,5
ю-2 0,4 0,4 0,3 0,2
ю-3 0,3 0,3 0,2 0,1
Из приведенных расчетов для рекомбинации двух атомов азота и атома
кислорода с молекулой СО следует, что при повышении температуры и сте-
пени ионизации газа роль процесса ударно-радиационной рекомбинации
(т. е. рекомбинации путем переходов между различными электронными со-
стояниями образующейся молекулы) увеличивается и может стать опре-
деляющей в суммарной кинетике рекомбинации тяжелых частиц.
2. Гетерогенная рекомбинация
Наряду с объемной рекомбинацией атомов и радикалов в плазмохимиче-
ских системах большое значение имеет и гетерогенная рекомбинация — ре-
комбинация на стенках, ограничивающих плазму, или на поверхности твер-
дых частиц в ней.
Такое влияние было обнаружено при получении свободных радикалов
в электрических разрядах при пониженных давлениях [213], а также при ис-
следовании процессов рекомбинации атомов в послесвечении [214]. При этом
наблюдался как первый (водород, кислород), так и второй порядок реакции
относительно концентрации атомов, а также переход от первого ко второму
порядку. Уравнение для гетерогенной гибели атомов имеет в общем случае
следующий вид:
= + (3.111)
где V — полный объем системы; S — площадь поверхности раздела газовой
и твердой фазы; [А] — объемная концентрация атомов, — коэффициент
скорости рекомбинации Z-го порядка. (Для цилиндрической трубки (*S7V) =
2/7?w.)
Исследование гетерогенной рекомбинации показало, что величины коэффи-
циентов существенно зависят от типа поверхности (металл, диэлектрик), ее
чистоты, способа обработки. Основная часть исследований проведена в по-
слесвечении электрических разрядов на поверхности стекла и кварца. Ока-
залось, что гетерогенные процессы определяют гибель атомов при давлениях
ниже 1 торр [213, 214]. Покрытие поверхности различными веществами (фос-
166
форная и борная кислоты, полимеры и т. д.) приводит к снижению коэффици-
ентов гетерогенной рекомбинации.
Из этих результатов следует, что коэффициенты скорости рекомбинации
на стенке в самих электрических разрядах, подвергающейся бомбардировке
заряженных и возбужденных частиц, могут отличаться от измеренных в по-
слесвечении. Специальное исследование коэффициента гетерогенной реком-
бинации атомов азота на поверхности молибденового стекла непосредственно
в тлеющем и высокочастотном разрядах при давлении 1—10 торр и после
его выключения [215] подтвердило это предположение. Оказалось, что в раз-
ряде и непосредственно после его выключения коэффициент скорости гете-
рогенной рекомбинации атомов азота достигает величины, на порядок превы-
шающей коэффициенты гетерогенной рекомбинации на поверхности стекла,
не подвергавшегося воздействию разряда. Влияние разряда резко ограни-
чено зоной горения. Атомы, выходящие из разряда, в проточной системе не
оказывают такого влияния на стенку. Воздействие разряда значительно ста-
билизирует получаемые значения коэффициента рекомбинации: разброс при
постоянных давлении и режиме разряда не превышает ошибки измерения
(~15%), тогда как на стенке, не обработанной разрядом, он достигает 100—
200%. После выключения разряда коэффициент гетерогенной рекомбинации
монотонно падает со временем и через 20—30 мин достигает того же значе-
ния, которое получается на стенке, не подвергавшейся воздействию разряда.
Второй порядок реакции по концентрации атомов наблюдается на любой
стенке. Экспериментальные значения коэффициента рекомбинации в разряде
и непосредственно после его выключения можно представить эмпирической
формулой
= 4-1СГ14р-пС, см*-сек'1, (3.112)
где р — полное давление, торр} п = (1,24=0,4); m<Z 1 [215]. Они не зави-
сят от параметров разряда (концентрации заряженных частиц). Была сдела-
на попытка уменьшить коэффициент рекомбинации путем покрытия стенки
фосфорной кислотой. В первый момент после включения разряда действитель-
но наблюдалось снижение ко через несколько минут горения разряда
значение его увеличилось до той же величины, что и до покрытия.
Наблюдаемые зависимости коэффициента скорости гетерогенной реком-
бинации от параметров (3.112), покрытий и восстановление свойств стенки
после выключения разряда позволяют предположить, что механизм гетеро-
генной рекомбинации состоит в рекомбинации при соударении атомов из га-
зовой фазы с атомами, адсорбированными на поверхности. Покрытие стен-
ки различными веществами уменьшает число свободных центров адсорбции,
то же самое происходит и при повышении давления: свободные центры за-
нимают адсорбированные молекулы азота. Влияние разряда сводится к очи-
стке поверхности и освобождению центров адсорбции. После выключения раз-
ряда они снова занимаются молекулами примесей. Аналогия, наблюдаемая
в поведении коэффициента скорости рекомбинации и гетерогенной релак-
сации колебательной энергии молекул азота [216, 217], подтверждает такой
механизм и позволяет сделать вывод, что основное значение в этом процессе
имеют центры химической адсорбции. На поверхности стекла, не обработан-
ной разрядом, наблюдалось также монотонное падение коэффициента ре-
комбинации при увеличении давления, однако оно маскируется большим раз-
бросом экспериментальных данных. По-видимому, это обстоятельство, а так-
же способ определения коэффициентов гетерогенной рекомбинации [213, 214]
не позволяли заметить этот спад в проводившихся ранее исследованиях.
Вследствие резкого увеличения коэффициента при воздействии разряда
гетерогенная рекомбинация оказывает решающее влияние на стационарные
концентрации атомов азота в разрядах при давлениях до 10 торр и выше.
При дальнейшем повышении давления и увеличении размеров разрядных
трубок возможен случай, когда лимитирующей стадией станет не коэффици-
167
ёнт скорости гетерогенной рекомбинации, а диффузия. Тогда для нахож-
дения потока атомов, рекомбинирующих на стенке, необходимо решать урав-
нение диффузии с граничным условием
dr ]r=Rn Kw
(3.113)
где D — коэффициент диффузии атомов; — молярная доля атомов в объ-
еме [166].
ДИССОЦИАТИВНАЯ РЕКОМБИНАЦИЯ
МОЛЕКУЛЯРНЫХ ИОНОВ С ЭЛЕКТРОНАМИ
И АССОЦИАТИВНАЯ ИОНИЗАЦИЯ
В молекулярной плазме основным процессом гибели заряженных частиц
является диссоциативная рекомбинация молекулярных ионов с электронами.
Это связано с тем, что для протекания процесса не требуется столкновений
с третьей частицей, поскольку энергия, выделяющаяся при рекомбинации,
передается фрагментам, образующимся в результате диссоциации промежу-
точной возбужденной молекулы. Баланс заряженных частиц определяется
тогда разностью скоростей диссоциативной рекомбинации и обратного ему
процесса ассоциативной ионизации
- = «Д- ₽ [АВ-] Ne - Г и [А] [В],
(3.114)
где ссд р, |За и — коэффициенты диссоциативной рекомбинации и ассоциатив-
ной ионизации; [АВ+], [А], [В] — концентрация молекулярных ионов и обра-
зующихся фрагментов (в частном случае атомов, если молекула двухатомная).
Кроме того, экспериментальные исследования показали, что процессы
диссоциативной рекомбинации и ассоциативной ионизации цмеют решающее
значение и в плазме атомарных (например, инертных) газов при достаточно
низких температурах тяжелых частиц ввиду образования молекулярных
ионов.
В молекулярных газах при низких температурах могут образовываться
более сложные комплексные ионы (кластеры). Экспериментально показано,
что величина коэффициента диссоциативной рекомбинации растет по мере
увеличения числа частиц в молекулярном ионе. Поэтому образование таких
ионов может существенно влиять на кинетику ионизации.
1. Образование и гибель сложных ионов
Молекулярные ионы и кластеры могут образовываться из атомарных или
более простых молекулярных ионов в результате процесса ионной конверсии
на третьей частице С:
feCi
А++ В +CZ^AB+4-C.
/fc2
(3.115)
В гибели и образовании этих ионов могут принимать участие и процессы
(3.114).
Для случая образования молекулярных ионов из атомарных (А, В и С —
атомы) при комнатных температурах, где основную роль в захвате атомарного
иона атомом играет поляризационное взаимодействие, коэффициент скоро-
сти конверсии (в см6-сек-1) равен [104]:
/сС1 = 10-2У
1/2 /Ра \зд
( т ) ’
А = В - С,
168
в = С=£А,
А = В=#С,
А = С^В,
(3.116)
где Mi, рг — массы и поляризуемость частиц сорта i, ат. ед. (табл. 3.15),
я Т — температура, °К.
Таблица 3.15
Поляризуемость атомов в основном состоянии [104]
Атом м 3 Атом м 3 Атом м
Не 4 1,39 н 1 4,5 С 12 14
Ne 20 2,76 Li 7 165 N 14 7,6
Аг 40 П,1 Na 23 166 О 16 5,2
Кг 84 16,8 К 89 250 F 19 27
Хе 131 27,2 Rb 85 290 Са 40 140
Hg 201 37 J Cs 133 330 Ва 137 300
Сравнение результатов расчета коэффициентов конверсии атомарных ио-
нов инертных газов в молекулярные по формуле (3.116) с эксперименталь-
ными данными приведено в табл. 3.16.
Таблица 3.16
Расчетные и экспериментальные значения коэффициентов
конверсии [104]
Процесс kCi, 10-3i слг6-сек-i
расчет (3.116) эксперимент
Не+ + 2Не - -»He2+ + He 1,06 0,35-1,15
Ne+ + 2Ne"- Ne2+ + Ne 1,06 0,16—0,79
Ar+ + 2Ar - > Ar2+ + Ar 4,5 0,8—3,9
Если характерное время образования ионов путем конверсии и гибели
за счет обратного процесса (3.115) много меньше характерного времени диссо-
циативной рекомбинации и ассоциативной ионизации (3.114), то концентра-
ции молекулярных и кластерных ионов можно определить из термодинами-
ческого соотношения
[А+] [В]
[АВ+]
к
-Г = КзТ)-
(3.117)
Константа равновесия реакции (3.115)
#з(Л =
Qa+ Qb (Wa+b^),/2
^^АВ+ №
exp
#ab+ \
kT /
(3.118)
где Qa+, Qb, Qab+ — статистические суммы частиц; /с, h — постоянные Больц-
мана и Планка; ца+в — приведенная масса иона А+ и частицы В; £дв+ —
энергия диссоциации иона АВ+ (табл. 3.17).
169
Таблица ЗЛ7
Энергии диссоциации ряда молекулярных и кластерных ионов [104, 218, 219]
Ион еав+- эВ (эксперимент) Ион E«B+, эВ (теория)
Ht!2+ 2,24 Н4+ 0,54
Ne2+ 1,40 н5+ 0,47
АГ2+ 1,05 Нз+ —
КГ2+ 1,0—1,13 Нез+ 0,57—0,2
Хе2+ 0,98 НеН2+ 0,19
N4+ 0,5 (Не)2Н+ 0,96
№з+ 3,26 H2Li+ 0,59—0,1
1Л2+ 1,27—1,55 (H2)2Li+ 0,50
Na2+ 0,97—1,01 (Н2)зЫ+ 0,44
К2+ 0,75 (H2)4Li+ 0,36
Rb2+ 0,72—1,23 (H2)5Li+ 0,22
Cs2+ 0,69—1,17 (H2)&Li+ 0,30
При этом следует иметь в виду, что соотношения (3.117), (3.118) справед-
ливы только в случае полного термодинамического равновесия (Zr = Т^п. —
= Tj = TV = Те).
В общем случае для нахождения концентрации комплексных ионов в мо-
лекулярной плазме и молекулярных ионов в плазме инертных газов необхо-
димо совместное решение системы уравнений, описывающих процессы (3.114),
(3.115).
2. Экспериментальные исследования
диссоциативной рекомбинации и ассоциативной ионизации
Основная часть экспериментальных данных получена при исследовании
рекомбинации молекулярных ионов двухатомных молекул при температу-
рах электронов, близких к комнатной (Т — 200—700° К). Электроны и ионы
получались при этом в импульсном высокочастотном разряде. Чтобы исклю-
чить влияние диффузии на гибель заряженных частиц (Ne = 108 —1010 см~3),
исследуемый газ разбавлялся инертным (чаще всего неоном) при давле-
нии последнего (рые > 10 -т- 20 торр), концентрации исследуемого газа
1 — 10~4 торр.
Электроны после прекращения разряда охлаждались в результате соуда-
рений с тяжелыми частицами до температуры газа. Изменение температуры
газа достигалось помещением всей измерительной ячейки в термо зтат. При
таком способе получения ионов и электронов можно считать, что успевают
установиться равновесные распределения частиц по поступательным (мак-
свелловские распределения, Те ~ Тг) и по внутренним степеням свободы
(Гэл = Tv = Tj = Те). Большой разброс значений коэффициентов диссо-
циативной рекомбинации, измеренных в ранних работах, как показали по-
следующие измерения, был обусловлен переменным составом ионов. Поэтому
все последующие работы были выполнены при обязательном контроле соста-
ва ионов масс-спектральным методом [220—227]. Использование независимо-
го подогрева электронов с помощью постоянного слабого высокочастотного
поля позволяет менять температуру электронов (Те = 300—11 000 ° К) при
неизменной температуре тяжелых частиц (Тг = 300° К) [228—231]. При
этом температура заселения вращательных уровней остается равной темпера-
туре тяжелых частиц (Tj = Тг), а заселение колебательных и электронных
уровней обусловлено совместным влиянием возбуждения электронным уда-
ром и девозбуждения при соударении с тяжелыми частицами, а также излу-
170
Гением (см. стр. 122). Строгого решения задачи нахождения этих температур
в таких условиях до настоящего времени не имеется. Оценки показывают,
что в случае молекулярного иона азота [230] колебательная температура мо-
жет достигать —' 700—1000° К. Колебательное возбуждение при этом не дол-
жно оказаться существенным.
Поэтому изменение коэффициентов рекомбинации в этом случае в основ-
ном соответствует изменению температуры электронов. Результаты измере-
ний приведены в табл. 3.18. Погрешность этих измерений 10% для двухатом-
ных и ниже 20% для многоатомных ионов. При этом экспериментальные
результаты представлены в виде
ая- р (Те, Тг) = ад- р (Те = Тг = 300° К) Т^еТ^г. (3.119)
В ходе этих исследований было замечено, что результаты измерения коэф-
фициентов рекомбинации иона Nt зависят от способа нагрева (Те = Тг =
= Tj = Tv — Тэл и Те >> Тт = Т f, Tv, Тэл). Изменение температуры газа
от 200 до 500° К в первом случае не приводит к изменению ая*р, в то время
как нагрев электронов дает уменьшение коэффициента диссоциативной реком-
бинации.
В работах [234, 248, 249] была сделана попытка измерить коэффициенты
диссоциативной рекомбинации в ударной трубе, где, как полагают авторы
этих работ, Те = Тг = Ту — Т}- — В инертных газах ионы и электро-
ны получались в отсеке низкого давления с помощью импульсного высоко-
частотного разряда. Затем на эту область набегала ударная волна, нагревав-
шая газ до температуры 700—3500° К. Измерение скорости спада концентра-
ции ионов проводилось через ~20 мксек после прихода фронта ударной вол-
ны (полное давление за фронтом р 20 тпорр) методом тока насыщения, иду-
щего на двойной зонд. Оказалось, что измеренные таким образом коэффициен-
ты диссоциативной рекомбинации при низких температурах (Т < 900° К
для Net и Т < 670° К для Art) в пределах погрешности совпадают с измере-
ниями в печи с высокочастотным нагревом электронов (см. табл. 3.18), а при
больших температурах спадают более резко, чем при изменении только
Однако при этих измерениях были допущены две систематические погреш-
ности, которые существенно снижают точность измерений. Первая из них —
использование формулы Ленгмюра для тока насыщения на зонд, которая не-
применима в использованном диапазоне давлений [250]. Вторая — ионный
состав плазмы не контролировался. Вместе с тем при столь высоких темпера-
турах (~3500° К) равновесная концентрация молекулярных ионов должна
быть существенно ниже концентрации атомарных (стр. 169). Для Net при
р = 20 тпорр (сое = 0,1 эВ, Ё* + = 1,35 эВ) ниже приведено изменение рав-
JNe2
новесного ионного состава плазмы неона в зависимости от ^температуры:
[Ne2+]/[Ne+]'. . . 2-1017 7,0 2,0 0,35 1,5.10-4 4-10-6
Т, ° К......... 300 900 1000 1100 2100 3500
Как следует из этих результатов, равновесный состав ионов изменяется
в сторону образования атомарного иона неона. Времени, прошедшего от на-
грева до начала измерений — t ~ 20 мксек, согласно оценкам с использовани-
ем формул (3.116) — (3.118) при температурах (Т ~ 2000—3500°), оказы-
вается достаточно для того, чтобы исходные ионы Net успели продиссоци-
ировать к моменту начала измерений. Существенное изменение состава ионов
неона должно начинаться при Т ~ 900° К, иона Art — при ~700° К, а ксе-
нона — при ~ 800° К. Таким образом, наблюдавшиеся экспериментально
в работах [234, 248, 249] изменения температурной зависимости коэффициента
диссоциативной рекомбинации могут объясняться изменением состава ионов.
171
Таблица 3.18
Результаты измерений коэффициентов диссоциативной рекомбинации
Ион аД-Р, 10-7 см3-сек-1 те, °к тг, °к Ye Ye + Yr Литератур- ная ссылка
Нв2+ 0,09 300—1500 300 1,5 232
0,12 300 300 — — 233
Nea+ 1,7-0,4 300—11 000 300 0,43 — 228
1,8—0,4 300-4600 300 0,49 — 229
1,8—1,4 300-500 300-500 — 0,42 220
1,4-1,2 450—900 450-900 — 0,5 234
Al2+ 8-0,9 300—10 000 300 0,67 — 231
4,5-4,0 450—670 450—670 — 0,5 234
Хе2+ 14±2 300 300 — — 235
КГ2+ 12 300 300 — — 236
Н2+ <0,3 300 300 — — 237
Нз+ 2,9—2,0 205-450 205-450 — 0,5 226
Н5+ 36 300 300 — — 226
Ог+ 2 300—12 000 300 0,7 — 230
-0,3 1200-5000 300 0,56 —
3,0-1,0 205-690 205-690 — 1 234
N2+ 1,8-0,6 300—3000 300 0,39 — 230
2,7 205-480 250-480 — 0,02 221
NO+ 7,0—3,0 200—450 200-450 — 1,0 222
7,0—3,0 196-358 196—358 — 1,2 223
3,4 300 300 — — 227
СО+ 6,8—3,9 300-775 300 0,57 — 238
n4+ 20 300 300 — — 239, 240
10 300 300 — — 241
17±4 300 300 — — 222
СО2+ 3,8±0,5 300 300 — — 242
3,4±1,2 300 300 — — 227
Hg2+ 5,5—15 400 400 — — 247
35 470 470 — — 244
Cs2+ 1,7 600 600 — — 244
0,1 1250—1450 1250—1450 — — 245
5,4 1100 1100 — — 246
НСО+ 3,3—2,0 205—300 205-300 — 1,3 226
Н+(Н2О) 10 540 540 — — 247
Н+(Н2О)2 20 415—540 415—540 — — То же
Н+(Н2О)з 40-38 300—540 300-540 — — »
Н+(Н2О)4 49 ЗОЙ 300 — — »
Н+(Н2О)5 60 205 205 — — »
Н+(Н2О)б 75 205 205 — — »
Н+(Н2О)7 100 205 205 — — »
Прямое доказательство этого факта получено в экспериментах [251], где из-
мерение коэффициентов диссоциативной рекомбинации было выполнено в ста-
ционарном разряде методом тока насыщения на зонд с учетом диффузии
ионов [250]. При этом было зарегистрировано изменение коэффициента реком-
бинации на несколько порядков величины в соответствии с соотношениями
172
(3.117), (3.118). В измерениях коэффициентов диссоциативной рекомбинации
молекулярных ионов — NO+[252, 253] и N^, О2 [249] также возможны боль-
шие систематические погрешности, связанные как с неизвестным составом
ионов, так и со способом измерений и обработки результатов. В частности,
использование соотношения (3.117) является незаконным, поскольку в удар-
ной волне трудно ожидать установления термодинамического равновесия
[252, 254]. В [249] температурная зависимость не использовалась, а предпо-
лагалось ад р ~ Т”1’5, а после производилась обработка полученных резуль-
татов. Тем не менее в целом данные, полученные в ударных трубках для n£,
О2 [249] и NO+[252, 253], лежат ниже, чем результаты измерений в печи с на-
гревом электронов (см. табл. 3.18).
Таким образом, надежными являются только данные, полученные в печи
и в печи с нагревом электронов (см. табл. 3.18).
Энергия, выделяющаяся при диссоциативной рекомбинации молекуляр-
ных ионов с электронами, частично переходит в энергию возбуждения внут-
ренних степеней свободы (электронное возбуждение атомов и молекул, а
также, возможно, колебательное и вращательное возбуждения), а частично в
кинетическую энергию образующихся фрагментов. В работе [255] путем иссле-
дования доплеровского уширения линий атомов неона и аргона в послесвече-
нии разряда при пониженных давлениях было показано, что в результате
диссоциативной рекомбинации ионов NeJ и ArJ образуются атомы Ne с энер-
гиями возбуждения Ег = 18,3—18,92 эВ и Аг — с Et = 13,2—13,4 эВ. При
этом рекомбинация происходит по крайней мере с десяти колебательных
уровней основного электронного состояния и с трех — возбужденного элек-
тронного состояния иона NeJ* Возбуждение одного и того же состояния ато-
ма связано с рекомбинацией с нескольких колебательных уровней, и вклад
их примерно одинаков. Рекомбинация иона HeJ [256] приводит к образова-
нию атомов Не (335) и Не (23Р), а иона OJ — атомов О (3Р), О (ЧУ) и О (г5)
в отношении 1 : 0,9 ; 0,21 [257]. Из энергетических соотношений следует, что
в результате диссоциативной рекомбинации Hj могут образоваться только
атомы Н (п = 2) и Н (п = 1). Значительная доля энергии рекомбинации во
всех этих случаях расходуется на поступательное движение атомов
(табл. 3.19).
Таблица 3.19
Распределение энергии (в эВ) рекомбинации в продуктах
Ион Энергия ионизации атома Энергия связи иона El Допоет
Н2+ 13,6 2,65 10,2 0,93
Нб2+ 24,59 2,24 21—22,7 0—1,36
Ne2+ 21,56 1,35 18,3—18,9 1,26—1,86
Аг2+ 15,76 1,05 13,3-13,4 1,31-1,41
О2+ 13,61 6,65 1,97-4,2 2,76—4,99
Состояния, в которых могут образовываться атомы и фрагменты молекул
в результате диссоциативной рекомбинации, необходимо знать для оценки
роли этого процесса в возбуждении молекул, построения лазеров на реком-
бинации и т. д. Энергия, идущая на поступательные степени свободы, опре-
деляет баланс процессов ассоциативной ионизации и диссоциативной реком-
бинации в неизотермической плазме (Те =/=- Тг) (см. стр. 194).
Ассоциативная ионизация исследуется масс-спектрометрически по зави-
симости появления молекулярных ионов при возбуждении атомов монохро-
173
Таблица 3.20
Коэффициенты скорости ассоциативной ионизации
с участием возбужденных атомов (Т = 300° К)
Партнеры по столкновению
За-И (I, п), 10-1° см3-сек~1
Не (16) + Не (З1^)
Не (16) +Не (31D)
Не (16) +Не (33Р)
Не (16) + Не (3W)
Аг (36) + Ar (4P1Z)
Аг (36)+Аг (663/2)
Cs (626) + Cs (82Р)
Cs (62Р) + Cs (62Р)
8,35 [260]; 0,55±0,18 [261]
88 [260]; 3,5±0,7 [261]
0,35 [260]; 0,28+0,02 [261]
3,5 [260]; 0,8±0,09 [261]
0,2 [258]
0,5 [258]
0,012+0,003 [259]
0,42 ±0,13 [259]
Таблица 3.21
Коэффициенты скорости ассоциативной ионизации,
измеренные в ударных волнах
Процесс
Ра* и, см3 • сек-1
Литера-
турная
ссылка
N +О-*NO+ + e
N + N —N2+ + е
N + N _ N2+ + е
5 • 10-11 T-°>5 * * ехр (-32 500/Т)
9 • 10-11 Т-°>5 (1+1,3-10-4 Т +
+3,3-10-8 * * * Г2 — 10-12 Г3) ехр X
Х(—67 300/Т)
2,7.10-n ехр (—67 300/Т)
252
252, 262
263
матическим пучком электронов или фотонов от их энергии [258, 259] при ком-
натной или близкой к ней температуре. Потенциалы появления молекуляр-
ных ионов оказываются, как правило, несколько выше суммы энергии
возбуждения и энергии связи молекулярного иона, т. е. часть энергии расхо-
дуется на внутренние степени свободы иона и поступательное движение элек-
трона (см. табл. 3.19). Это свидетельствует о том, что наблюдаемая иониза-
ция может протекать не через те каналы, через которые идет диссоциативная
рекомбинация при комнатной температуре (табл. 3.20, 3.21).
Процесс ассоциативной ионизации, детально обратный диссоциативной
рекомбинации, при комнатной температуре не идет из-за недостатка энергии
поступательных степеней свободы атомов.
3. Теория диссоциативной рекомбинации
и ассоциативной ионизации
Процесс диссоциативной рекомбинации молекулярных ионов с электро-
нами и обратный процесс ассоциативной ионизации протекают через образо-
вание промежуточных автоионизационных состояний, из которых возможна
диссоциация (рис. 3.20) [9, 264—266].
В общем случае их схему можно записать в виде
ри
е АВ+ (Г) ZZZZt АВ (&'),
ГИ(/£'|Г)
Ги(г'|т')
е + АВ+ (i') , АВ (т'),
Ги(т'|г')
(3.120)
(3 121)
174
AB (7/)Z12AB (m'), (3.122)
ГД(т'|/Г)
Гд (m'\ln)
AB (m') <__12 A (Z) + В (n). (3.123)
ГД(?п|т')
Здесь AB (к') — связанное автоионизационное состояние; АВ (т') — несвя-
занное электронное состояние, распадающееся на фрагменты в состояниях
I и п (одно из них или оба могут быть возбужденными состояниями). Диссо-
циация из связанного состояния к' обусловлена взаимодействием с состоя-
нием т', приводящим к предиссоциации; АВ+ (/') — рекомбинирующий мо-
лекулярный ион в состоянии I'. Индексы со штрихом обозначают набор
Рис. 3.20. Схема потенциаль-
ных кривых состояний, участ-
вующих в диссоциативной ре-
комбинации
квантовых чисел, характеризующих электронный, колебательный и враща-
тельный уровни соответствующего состояния: V = (i, ЯД к' — (к, vk,
ЯД т' — (^, ]т), где в случае отталкивательного уровня 7™ —момент
вращения фрагментов А (/), В (п) относительно их центра тяжести. Кроме
того, на схеме реакций указаны коэффициенты скоростей прямых и обратных
процессов. Вероятности соответствующих прямых и обратных переходов
связаны между собой соотношениями микроскопического детального балан-
са (см. стр. 76). Поскольку мы рассматриваем переходы между отдельными
вращательными уровнями состояний к', т', I' , то независимо от распределе-
ния частиц по уровням, соответствующим внутренним степеням свободы,
в отсутствие внешних полей имеем [266]
g.£T« (А/ I m') = (m' j к’), (3.124)
^nocWrH(i'|^; £е)6(^-Ь2 + ^) = ^-Ги(4'Иг (3-125)
g^gig^ (In I т’- Е») 8 (£и - Ет. + Ег + Еп) = (т' |1, п), (3.126)
где ge, gi, gn — статистические веса электрона и фрагментов в соответствую-
щих состояниях; gf0CT, g|?0CT — плотности состояний в непрерывном спектре
поступательного движения электрона и иона и фрагментов A (Z) и В (п)
в системе их центра масс (ц — их приведенная масса), gy = gi + 1);
Ev
gk' = gk (2/fc + 1)» gm' = gm (tym™ + 1)
{gt — электронные статистические веса состояний; Я — вращательные кван-
товые числа); Et — энергии частиц в соответствующих состояниях, отсчиты-
ваемые от основного состояния молекулы АВ; 6 (х) — символ Кронекера.
1 В работе [266] принята другая система записи: вначале пишутся квантовые числа ко-
нечного, а затем — начального состояния.
175
Кроме того, для спонтанных автоионизационных и предиссоциационных
переходов существуют правила отбора, следующие из закона сохранения
момента количества движения, полного спина, симметрии и четности состоя-
ний (в случае гомоядерных двухатомных молекул). При переходе АВ (тп') ->
—> АВ+ (£') + е для двухатомных молекул они имеют вид в случае связи Гун-
да а [102, 107, 267]
AS = ± 1, А/ = ± (1 + ДА).
(3.127)
Здесь AS = Sm — St — изменение спина; А/ = у™ — /Г — изменение вра-
щательного квантового числа; ДА = Ат —— изменение проекции момен-
та орбитального движения электронов на ось молекулы. Правила отбора
для предиссоциационных переходов обсуждались на стр. 138 и в работе [4]
Соотношения (3.125), (3.126) отличаются от соответствующих выражений
для сечения захвата электрона ионом, полученных в работах [9, 264, 265]у
наличием статистических весов вращательных уровней.
Усреднение соотношений (3.125), (3.126) по энергетическому распределе-
нию электронов и тяжелых частиц позволяет получить связь между макро-
скопическими средними значениями коэффициентов скоростей прямых и об-
ратных процессов (3.119) — (3.123). В частности, при максвелловских за-
конах распределения (Те =f= Тт) получим 1
Ги (г ю
Ги(Г|г';Т)
2gr (2лткТ)^
—--------— ----ехр
gr’ h3
где г' = к' или тп' и
^^) =^(г'Н',Ге),
(3.128)
Гя (т' | In)
ГД(Ч1 т'; Тг)
glgn / Ет,-Е1-Еп^
gm’ W Л
— Ktr (тг | In, Тг).
(3.129)
Здесь те — масса электрона и Ktr (т'Ип, Тг) nKtr Те) — парциаль-
ные константы трансляционного равновесия соответствующих процессов
(равновесия по внутренним степеням свободы при этом не предполагается).
Ниже для конкретности будем всюду предполагать максвелловское распре-
деление по скоростям и квазибольцмановские распределения по вращатель-
ным Tj, колебательным Tv и электронным уровням Тэл. В общем случае
будем полагать 7ЭЛ =/= Tv =f= Tj,
Квазистационарный анализ системы уравнений (см. стр. 44), описываю-
щих кинетику процесса (3.120) — (3.123), в общем случае громоздок. По-
этому мы ограничимся отдельным рассмотрением двух частных случаев —
нестабильного 2 и стабильного автоионизационных состояний, а затем пока-
жем, что взаимное влияние этих каналов рекомбинации несущественно.
Нестабильное автоионизационное состояние. В этом случае схема процес-
са описывается уравнениями (3.122) — (3.123), причем задание I, п неодно-
значно определяет пг. Поток частиц через уровень АВ (тп') после установле-
ния квазистационарности равен
j(О = 21АВ+ О Ги (Г I m', Те) -r77J/re) ~ 1А(01 [В (П)]•
1 V'*’ )
Гд (In | пг', Тг) Ги (?пг)
Г (т')
(3.130>
где [АВ+], [A(Z)1, [В (п)] — концентрации соответствующих компонент;
Ги (тп') = 2 Ги (тп' 1I'), Г (тп') = Гд (т') + Ги (пг').
1 Для стабильного состояния к это соотношение имеет место при Г(к') 2кТе.
2 Нестабильными состояниями условимся называть состояния, не имеющие минимума
потенциальной кривой в той области /?, где возможен автоионизационный переход.
176
Из сравнения выражений (3.130) и (3.114) следует
«нр = 2 2 j-B+ (Г)1 ги а' \т' т) гД(т'|,п)
[АВ+] Г(т<) ,
па.И—V [А (01 [В (л)] VI гд(/п|т'; Гг)Ги(т')
! Й 1АНВ] £ П-')
(3.131>
(3.132).
Введем парциальные коэффициенты скорости рекомбинации и ионизации
а5 Р (т' 1I', Те) = Г” (Г | т', Те) , (3.133),
оа.иП„ 1тЧ;Т} т’’ Гг) ГИ (m' | i')
₽н (ln\m\i,TT) =------------------------ . (3.134).
1 (/и )
Тогда для отношения парциальных коэффициентов с учетом (3.128), (3.129)>
имеем
₽-H(Zzz|m'|r,Tr) /Q4Qrx
с#** (Г | тп', Те) Ktr (m' | In. Tj *
Оно равно константе равновесия реакции А + В АВ+ + е только в слу-
чае Те = Тг (распределение по внутренним степеням свободы не имеет зна-
чения). Соотношение для полных коэффициентов скорости
Й’иК-р=А (А + В^АВ+ + е)
будет выполняться строго только при Те = TY = Tj = Tv =
Отношение
S (тп' | In) = Гд (тп' | In) / Г (тп')
(3.136)
(3.137)
есть вероятность того, что образующееся автоионизационное состояние не-
успеет снова автоионизоваться до полной диссоциации. При классическом
рассмотрении движения частиц А и В оно равно [104, 264, 268]
S (тп' | In) = ехр Ги(т')<й] , (3.138)
где Rc — расстояние между частицами А и В в момент образования АВ (тп')'.
Rs соответствует точке пересечения термов АВ (тп') и АВ+ (i'). Ввиду того
что наклон отталкивательных ветвей потенциальных кривых (dUldR)(rn').
состояний тп'. как правило, достигает 10—25 эВ1к, частицы А, В успевают
выйти из той зоны, где возможна автоионизация, за время меньше 4 • 10“15 сек,
что при реальных вероятностях автоионизации молекул Ги (пг')<С 1014 сект1
соответствует соотношению
5 (тп' [ In) ~ 1. (3.139)
Более точное квантовомеханическое рассмотрение задачи приводит к вы-
ражению, отличающемуся от (3.138), однако ввиду выполнения соотношения
(3.139) это отличие несущественно [9, 265].
Из (3.133), (3.134) с учетом (3.138) получаем выражения для парциальных
коэффициентов
Е
Д-Р / ' I •/ т \ О/ ' I 7 \ I О
<Хн Н 1 7 е) = —---S (тп In)--—!—ехр
v ‘ к 1 7 (2ятекТер
р (2 ] -4- 1) h3
РГ (In I m’ I Г; Гг) = Л1 Т, 3/2 5 (m' | | (') ехр
Ет--Ег'\ .
kTe ) ’
(3.140)
Em’-El-En\
kTr ) ‘
(3.141>
177
Из формул (3.127), (3.128), (3.140) и (3.141) следует, что парциальные
и суммарные коэффициенты диссоциативной рекомбинации зависят от рас-
пределения ионов по внутренним степеням свободы и температуры электро-
нов, а суммарный коэффициент ассоциативной ионизации — от распределе-
ния атомов по возбужденным уровням и температуры газа.
Стабильное автоионизационное состояние. В этом случае схема процессов
имеет вид (3.120), (3.122), (3.123). Поскольку вероятности спонтанной пре-
диссоциации Гд (к' /т') 1012—1013 сек-1 [107], то
Гд (т' | &')/Гд (m' | /п)< 1. (3.142)
Тогда из квазистационарного анализа системы кинетических уравнений,
описывающих процессы (3.120), (3.122), (3.123), для парциальных коэффи-
циентов получаем с учетом (3.128), (3.129):
с£ р (m' I к’ I i'; Те) =
(2/R1) h?
(27? + 1) (2ПтекТе)3^
Ги (к11 /') ГД (к'/т1) _ ( Ек, -
ПЙ кТе
ра-и (ln\m’\k'\i'-,TT) =
+ ги (fc' I Г) Гд (fc' I m')
glgn(2^kT^ W
(3.143)
fcTr J •
(3.144)
Как следует из сравнения формул (3.140) — (3.144), выражения для пар-
циальных коэффициентов диссоциативной рекомбинации и ассоциативной
ионизации при протекании их через стабильные автоионизационные состоя-
ния не отличаются от соответственных выражений в случае нестабильного
автоионизационного состояния.
Из полученных формул также следует, что влиянием индуцированных
столкновениями предиссоциационных и автоионизационных переходов (эти
процессы не отмечены в схеме реакций), а также запрещенными предиссоци-
ационными переходами при наличии спонтанных разрешенных процессов
можно пренебречь.
Кроме того, ввиду выполнения соотношений (3.139), (3.142) каналы дис-
социативной рекомбинации и ассоциативной ионизации через стабильные
и нестабильные автоионизационные состояния можно считать независимыми
даже в том случае, если предиссоциация стабильного состояния к' вызывает-
ся нестабильным т', с которого возможна автоионизация.
Вероятности автоионизации и предиссоциации. Квантовомеханические
выражения для вероятностей автоионизации и предиссоциации имеют вид
[107, 265]:
ги (rn' | П = -^15 Xi Л) Vin (г, R, ft, j£) (R, j£) dR |2 ,
ГИ (k’ | О | Xi (R, 7?) V* (r, R, ft, ft) Xk (Я, ft), dR |2 ,
(3.145)
W' | m') = | %m (R, %) V*m (r, R, ft, j£) (R, ft) dR |2 ,
Vlk (r, R, ft, /7) = ^i (r, R) Hils (r, R, ft, ft) (r, R) dr.
Здесь X; (7?, Я), (7?, Х^(7?,Я) — колебательно-вращательные, (г, 7?) —
электронные волновые функции соответствующих состояний; Hik — пол-
ные гамильтонианы взаимодействий, приводящих к автоионизации или
предиссоциации; г, 7? — координаты электронов и ядер.
178
В приближении Борна — Оппенгеймера, используя теорему о среднем,,
получим
Ги (т' |Г)=^| (Л, fo |2(Я) X» (й) dR |2 = Г* (m, | i-& В) q^
Ги (к' I i') = Г? (к, ft I i, ft- R) q*v., (3.146)
r^k'\m')=T*(k,ft\i,ft, R)q*™,
где д — факторы Франка -- Кондона для соответствующих переходов или
их плотности, если одно из состояний нестабильно.
Следовательно, расчет коэффициентов диссоциативной рекомбинации
и ассоциативной ионизации сводится к определению автоионизационных со-
стояний, из которых возможен спонтанный диссоциативный распад, и расчету
вероятностей автоионизации и предиссоциации из автоионизационных состо-
яний. Кроме того, необходимо знать и потенциальные кривые для молеку-
лярных ионов. Их нахождение вызывает затруднение в случае молекулярных
ионов инертных газов и многоатомных молекулярных ионов, поскольку по-
тенциальные кривые ионов стабильных двух- и трехатомных молекул могут
быть определены методами оптической и фотоэлектронной спектроскопии
[3, 4, 100]. Таким образом могут быть найдены и потенциальные кривые,
стабильных автоионизационных состояний.
Наибольшие затруднения возникают при нахождении потенциальных
кривых нестабильных автоионизационных состояний (см. стр. 115).
Прежде всего такие состояния должны удовлетворять правилам отбора
для автоионизации (3.127). Кроме того, факторы Франка — Кондона для
таких переходов должны быть отличны от нуля. Как показывает исследова-
ние оптических спектров и теоретическое рассмотрение вопроса их расчета,
неплохим приближением является представление колебательной волновой
функции нестабильного состояния в виде 6-функции в классической точке
поворота [9, 107, 269]. Тогда имеем
dU (m) I ПРИ >
<Гг a-dR~\R=Rc (3.147),
viEm
I 0 при R~i Rc <f Rt,
где dU(m)ldR — наклон потенциальной кривой нестабильного состояния;
а = Ri — Ri — амплитуда колебаний ядер в стабильном состоянии fz; R^,
Rt — классические точки поворота; т. е. фактор Франка — Кондона отли-
чен от нуля только в том случае, если классическая точка поворота отталки-
вательной кривой находится в области Франка — Кондона для соответству-
ющего связанного состояния i . Поэтому автоионизационные переходы воз-
можны только при пересечении потенциальных кривых т и i (см. рис. 3.20),
а автоионизация с отталкивательных ветвей потенциальных кривых других
электронных состояний, не пересекающих Ut (/?), несущественна. Расчеты
[270, 271] являются некорректными, поскольку в них не были учтены факто-
ры Франка — Кондона для переходов на такие непересекающиеся ветви
по-^енциальных кривых.
Одним из видов взаимодействия, приводящего к автоионизации и предис-
социации, а также возмущениям в оптических спектрах, является конфигу-.
рационное взаимодействие состояний i и т [9, 107, 265]. В этом случае авто-
ионизация возможна только из состояний с двумя возбужденными элек-
тронами. Это также сужает круг возможных автоионизационных состояний.
Однако кроме конфигурационного взаимодействия к автоионизации и предис-
социации могут приводить и другие типы — взаимодействие электронного,
и колебательного или вращательного движения ядер [103, 107, 1 14]. Поэтому
при выборе возможных автоионизационных состояний должны быть учтены
и эти взаимодействия.
179
Нахождение потенциальных кривых нестабильных состояний несколько
облегчается, если они имеют минимум при больших/?. Тогда возможна экстра-
поляция на малые R потенциала, определенного из оптических спектров [9].
Тем не менее в настоящее время задача отыскания автоионизационных со-
стояний не решена полностью ни для одной из молекул.
Прогресс здесь может быть достигнут, по-видимому, только путем теоре-
тического расчета соответствующих потенциальных кривых (см. стр. 115),
«а также дальнейшего экспериментального и теоретического исследования раз-
личных типов автоионизационных переходов.
Расчеты вероятностей автоионизации в настоящее время выполнены толь-
ко для ридберговских уровней водорода [103, 114], с ряда которых наблю-
дается и предиссоциация. При этом учтено только электронно-колебательное
взаимодействие. Сравнение с экспериментальными данными показывает [102],
что наблюдаются предиссоциационные и автоионизационные переходы Н2,
обусловленные и другими типами взаимодействия.
Полуэмпирический метод расчета вероятностей предложен в работе [9].
Он основан на предположении о том, что автоионизация и предиссоциация
(или возмущение состояния к состоянием т) автоионизационных состояний
вызываются одним и тем же типом взаимодействия, т. е. в выражениях (3.146)
можно положить = Нк™. Если считать, что волновые функции ридбер-
говских состояний подобны волновым функциям ионов, то отсюда следует
С (т | Г) ~ Ге (т' | £'), (3.148)
где лг* — эффективное главное квантовое число.
В работе [9] таким образом были определены вероятности автоионизации
состояний 52П и 6'2А молекул NO с использованием матричного элемента
взаимодействия этих состояний с ридберговскими, определенного из оптиче-
ских спектров. Очевидно, что этот метод можно применить и для нахождения
Ги (m'/i') по вероятности предиссоциации ридберговских состояний. Послед-
ние могут быть измерены экспериментально по ширинам автоионизационных
пиков в спектрах фотоионизации и эффективности фотоионизации (Уи (/с'))
Г(^)=Г“(П+Г«(Л'); (3.149)
1 (к )
В заключение этого раздела подчеркнем, что вероятности переходов мо-
гут зависеть от номера вращательного уровня [107, ИЗ] вплоть до зависимо-
сти типа
^\к;ц;Ь)~]{] + '1). (3.150)
Усреднение парциальных коэффициентов диссоциативной рекомбинации
и ассоциативной ионизации по вращательным уровням. Усреднение парциаль-
ных коэффициентов (3.140), (3.141), (3.143), (3.144) по вращательным уров-
ням с учетом правил отбора ((3.127), см. стр. 176) приводит к следующим вы-
ражениям:
а™ (т, Evm |i, v-, Те, Т-) = Ги (m, Evm | i, v-,
(2лтекТеУ1Л
xexp[— (Ет-Е^Еът-Е1)1кТе\, (3.151)
РГ (I, n I m I i, Vi', Tr) = g(mgW)3^- Г“ IX
glgn (2лтекТеУ*
xexp[-(£m + Е^-Ег-Е^/кТ^, (3.152)
Д-Р / г, | • . т т \ к3 (к’ । j)
О? (к, V]; I, Vi, Ге, Tj) = -----------л-т?-------— X
v 1 1 77 2g. (2nmekTe )3/2 F(fc,
X Гя (к, vk | i, v-, 7 j) exp [- (Ek - \ - £?) / kTe\, (3.153)
180
<Гг)
(2л^кТг)312
Ги (Л, vk | i, v^, Tj) X
Wn
г д (k 7? I m Ev • T.)
X t, ’ — ex₽ Ь + El-E>- En)IkTr], (3.154)
rA(fc, vh, T--)
/ Eiv \
где введены обозначения: Q\ (T) = 2 (2/ + 1) exp I-----j — вращательная
i \ /
статистическая сумма для уровня vt, i иона,
E^' = Eti + El -I- El, Ei' = Ei~\- El + E\, Em> — Em 4~ Em -}- E3m,
2 (2/ + 1) exp (- El I кТ,) Ги (/' / i')
Г (Z, к, [ i, v^, Tj) = ------------------j—----------------------
(3.155)
В том случае, если вероятности переходов Ги (пТ | /'), Ги (Л'| f), Гд (к' | т')
не зависят от номера вращательного уровня, зависимость средней вероятно-
сти перехода от Tj исчезает. Однако наличие зависимости вероятностей пере-
ходов от / должно приводить к росту парциальных коэффициентов диссо-
циативной рекомбинации и ассоциативной ионизации с ростом Tj, Например,
в случае зависимости (3.150) ад р (га'), |Заи (гп) растут прямо пропорционально
Tj, зависимость осд р и |За и может быть слабее [266].
Случай Те = Тг — Т$ = Tv = Тэл = Т. В этом случае, как следует из
формул (3.140), (3.143) и (3.131), полный коэффициент диссоциативной реком-
бинации
ад.р дз (Т)
(2ПткТ)3^ £ 2gAB+ Q7ab+ (Г) q» в+ (Г)
О3 (Т)
—— -------Ги (Л, Т) ехр
(Л<?АВ+(Л
(у/и \ р° / Fv ! Е \
\ Ги (т, У) ехр (— — )dEm’, (3.156)
Л-t /J \ /
Em(Ks)
ад-р = ______у ёк_____________
С (2Л«е7сГ)3/2 2?ав+ <Лв+
х Г г(^У\Г) ехР I- + Е^кТ^ <3’157)
1 {К, vk)
где (>ав+ (?) — колебательная часть статистической суммы иона, т. е. значе-
ния полного коэффициента диссоциативной рекомбинации определяются в ос-
новном свойствами автоионизационных состояний. Отличие Р от ос? Р состоит
только в замене суммирования по vh интегрированием по dEvm и видом мно-
жителя S (тп | In),
4. Сравнение теории с экспериментом
Зависимость коэффициента рекомбинации от температуры электронов
(Тг = Tj = 300° К, Те <; ое). Этот случай реализуется в экспериментах
для ионов N^, Ot и Art, Net, Het в части исследованного интервала темпера-
тур электронов (см. табл. 3.18). В этом случае возбуждением внутренних сте-
пеней свободы ионов можно пренебречь, и парциальные коэффициенты ре-
комбинации через одно электронное состояние равны:
оо
.а«‘р (т | Z, vr, Tj, Те) = - \ Г" (m, Evm | i, v-, Tj) X
(2лт кТ 2g. J
v ее' ьг де •
тг
X s (т, Em I In) ехр [— (Ет + Е„ — Et — El)/kTe] dEvm,
(3.158)
181
где
Д£ _ । Ёт + (7?s) ~ Ei ~ П₽И (7?s) + Ет > + Е^’
'mi ~ I 0 при Evm (Rs) + Ет < Е, + ЕГ„
Rs — точка пересечения кривых I, т и
*”<*।<*’*• Т>'г-> = (2„Л 2г“<*•*।7->х
6 6 Г/£
рЛ/д. 1) \тгг Т \
X T(fc,^) ех₽ I- + Е°* -Е>- Е^!кТе]. (3.159))
Полные коэффициенты рекомбинации получаются суммированием по
всем автоионизационным состояниям, через которые может идти процесс.
Если пренебречь изменением электронных моментов переходов в области
Франка — Кондона (это можно сделать ввиду малости изменения 2?), то из
(3.158) с учетом (3.147) получаем
а«'р (m | i, v-, ТЬ Te) = - -^Л(ш\ i, B, Tj) S (m, В \ln) X
(2nmekTey^
\ Л ехр(-^/ЛГе)йК.
mi
(3.160)
При вычислении интеграла в последнем выражении возможны два случая:
а) крутизна кривой dU (jn)/dR настолько велика, что изменением
|Х (7?с) |2 в области dU (ш) ~ кТе можно пренебречь. Тогда
«5’Р (тп | i, V-, Tj, Те) = — Г“(тп | i, В', Tj)S(m | In, В) X
y^TtfTL Ki j bj
кТ / \е . X
X —777ГТ-ТП-----ехР (----• (3.161а>
du (m) \ \ кГ ) \ е
а ЛГ |и=вс
Это выражение совпадает с полученным в [9, 265];
б) крутизна кривой dU (m)/dR настолько мала, что кривая U (ш) пере-
секает всю область Франка — Кондона кривой U (I) (рис. 3.21). Тогда полу-
чаем
(тп I i, V-, 2’-, Те) = - Геи (m I i; В, S (m | In, В) х
(2Лтек1 еЕ‘
/ ХЕ . \
X ехр[-^^) . (3.161б>
В реальном случае зависимость парциального коэффициента диссоциа-
тивной рекомбинации от температуры электронов должна быть между этими
крайними случаями. Так, при пересечении кривых т и i ниже vt — 0 kEmi —
= 0, и зависимость будет описываться степенной функцией (3.119) с 0,5 <
< Те < 1Д Численные расчеты [272] для NO+ с учетом двух возможных со-
стояний т дали 0,2 < Те < 0,8.
В случае инертных газов связанных автоионизационных состояний обна-
ружено не было [8, 273]. Поэтому диссоциативная рекомбинация их ионов
должна быть обусловлена нестабильными автоионизационными состояниями^
причем из результатов [255] следует, что в рекомбинации принимает участие
большое число автоионизационных состояний (10 для NeJ и 4 для ArJ). Срав-
нение измерений зависимости коэффициентов рекомбинации этих ионов от
Те (Те — 0,4—0,5) позволяет сделать вывод, что эти кривые, по-видимому,,
пересекают кривую иона i вблизи минимума потенциальной кривой и с боль-
182
Рис. 3.21. Квадраты амплитуды волновых функ-
ций и различные варианты пересечения потен-
циальных кривых иона и нестабильного авто-
ионизационного состояния (1—3)
1пим наклоном. В случае гелия возмож-
ных состояний значительно меньше (см.
табл. 3.19), и они, видимо, пересекают
черм i с малым наклоном.
Теоретическое сравнение вклада неста-
бильных и связанных автоионизационных
состояний в случае других ионов хотя и
указывает на малый вклад последних [265],
однако этот вывод нуждается в проверке.
Поэтому нами был рассчитан максимально
возможный вклад связанных состояний
в коэффициент диссоциативной рекомби-
нации иона Nj. Для этого использовал-
ся спектр автоионизационных состояний,
эффективности фотоионизации и ширины
тиков фотопоглощения, определенных из
экспериментов [122, 126]. При
расчете ширин пиков не учитывалось уширение за счет вращательной струк-
туры полос, но оно, по-видимому, несущественно, так как в спектре наблю-
даются и полосы поглощения с ширинами, значительно меньшими, чем основ-
ная масса полос.
Аналогичные расчеты с использованием экспериментальных данных [102,
274] были проведены и для иона (табл. 3.22) [266].
Таблица 3.22
Сравнение коэффициентов рекомбинации через стабильные
состояния с экспериментальными значениями
(Те = Тг = 300° К)
Ион а^’ Р, см3-сек~1 аэксп’
Ns+ 340-8 (1,8—2,7)-10-7
Н2+ 240-8 < io-8
Зависимость вклада связанных состояний от температуры электронов
щля Ng приведена на рис. 3.22 вместе с результатами измерений. Сравнение
показывает, что вклад рекомбинации через связанные состояния мал во всем
исследованном диапазоне температур. Однако наблюдается аналогичная тем-
пературная зависимость czc'p и аэксп- Выводы [265], что 06? р должно падать
с температурой, как Т^2, неверны. Обусловлено это тем, что не был принят
во внимание вклад уровней, лежащих выше предела ионизации (они располо-
жены примерно равномерно по энергии).
В случае иона Н2 вклад рекомбинации через связанные состояния оказы-
вается определяющим. К сожалению, надежные измерения коэффициента
рекомбинации в этом случае отсутствуют.
Величина уе и ее изменение с температурой электронов (например,
О2, см. табл. 3.19) могут зависеть от наличия вклада состояний (как
стабильных, так и нестабильных), пересекающих потенциальную кривую
иона выше его нулевого уровня.
На основании имеющихся экспериментальных данных (см. табл. 3.18)
можно высказать предположение, что увеличение коэффициента скорости
183
рекомбинации при переходе от ионов N2, 0^, N0+ к Ar2, Ne2, Хе2, Кг2 и мно-
гоатомным ионам в значительной мере обусловлено увеличением числа авто-
ионизационных состояний, дающих вклад в рекомбинацию. Это предположе-
ние подтверждается и тем, что вероятность автоионизации в случае N0+,
N2 приближается к максимальной для атомов [104].
Влияние вращательного и колебательного возбуждения ионов. Как уже
отмечалось выше (см. стр. 181), увеличение температуры заселения враща-
тельных уровней может привести только к увеличению коэффициентов диссо-
циативной рекомбинации. Это связано с возможной зависимостью величины
вероятностей автоионизации и предиссоциации от номера вращательного
Рис. 3.22. Зависимость коэф-
фициента диссоциативной ре-
комбинации ионов азота с
электронами от температуры
электронов
1 — эксперимент [230]; Те > Тг=
= 300°К;
2 — расчет через стабильные авто-
ионизационные состояния Т
= Те’
3 — эксперимент [211], Тг — Те
уровня. Этим можно было бы объяснить наблюдавшееся расхождение зави-
симостей коэффициентов от температуры для N2 [221, 230] (см. табл. 3.18,
стр. 171).
Теперь рассмотрим влияние колебательного возбуждения. Если не учи-
тывать вклада рекомбинации с возбужденных колебательных уровней ионов,
то из (3.131) следует (ср. с [268])
а»-Р (7е, Tv, Tj)— аД-Р (Ге, Tv = 300° К) [1 - ехр (- <ое / ЛТ,)]. (3.162)
В действительности вкладом возбужденных колебательных уровней пре-
небречь нельзя. Так, если по-прежнему считать, что If (i|m) не зависит от
В, то в самом неблагоприятном случае расположения кривых ш и i (кривая 1
на рис. 3.21) получаем
аД-Р (Те, Т Т„) ~ аД-Р (Те, Tv = 300° К) 1 ~ ехр (~ z ferP) . . (3 .163)
1 — -у- ехр (— 2cofi lkTv}
Численные расчеты [272] для NO+ в области 1000 < Tv < 7000° К пока-
зали, что
аД-Р(Ге, Tv) = a^Te)T~\
где yv < 0,5.
В том случае, если в рекомбинацию дает вклад большое число автоиони-
зационных состояний, зависимость от Tv должна еще больше сглаживаться.
Например, в случае Ne2 и Аг2 расстояние между кривыми m составляет 0,5—
1 эВ, и они, по-видимому, целиком перекрывают область Франка — Кондона
Vi = 0. Вклад рекомбинации с различных уровней должен быть одинаков,
что и подтверждается экспериментально [255]. Численные расчеты коэффи-
циента рекомбинации через связанные автоионизационные состояния для
N2 и Я2 также показали, что зависимость от Tv практически отсутствует.
В случае Те = Tv из теоретических выражений (3.156) — (3.157) следу-
ет, что коэффициент рекомбинации вообще не должен зависеть от распределе-
ния ионов по внутренним степеням свободы, поскольку он в этом случае це-
ликом определяется параметрами автоионизационных состояний.
184
Таким образом, различие коэффициентов диссоциативной рекомбинации
монов, измеренных в ударных трубах и в печах, не может быть объяснено
о точки зрения теории. Оно может быть только следствием погрешностей
и допущений, использованных при получении этих данных. Выше (см. стр. 171)
мы уже показали, что такие ошибки и неоправданные допущения имели мес-
то для ионов инертных газов. В случае ионов Oj, Nj и NO+ они могут быть
связаны либо с неравновесным заселением внутренних степеней свободы, ли-
бо с влиянием обратного процесса — ассоциативной ионизации — и будут
обсуждаться ниже (стр. 187) [266].
5. Ассоциативная ионизация
Для парциальных коэффициентов ассоциативной ионизации через неста-
бильные автоионизационные состояния, пренебрегая зависимостью Ге (m'|/f)
ют /?, получаем
S(m\ln, Л)Г”(т|£; R,Tr) х
(2лрАиг) ь ion
dU(m)
а dR
(3.164)
(3.165а)
X \ q™\ ехр[—(Ет + Em — Ее — En)/kTr]dEm-
mi
В тех же предельных случаях, что и (3.161а), (3.1616), имеем
(Т ) g
।т । Г = ^~п Г‘ <т ।' S 1х
i----ехР I— {Ет + Em(R^) — — Е-^/кТг],
lR=Rs
₽ани(/п|т|£;Гг)= R) X
(ЗлцкТ^ glgn
X exp [- (Em + Evm(R,) — Et — Еп)/кТГ]. (3.1656)
Отношение парциальных коэффициентов ассоциативной ионизации и дис-
социативной рекомбинации через состояние т равно
Гг) /п
v.-T-,Te} ~ \ I
J 5 (т, Е”т | In) Ги (т, | i; v.- ТГ) ехр [- (Ет + Evm - Е. - Е?)1кТг] dE”m
^гпг
з/2 е. (У {Т )
) ехр [__(£.+£”_ Е^Е^кТД X
\ S (т, Е»т | In) Ги (m, Е»т | i, v(, Tj) ехр {-(Ет +Е°т-Е.- Ef)l кТе] dE”m
(3.166)
Если положить Тj = Тг или Ги =/= / (Tj), то в случае
dU(m)
‘а dR
кТе, кТг
(ЗЛ67)
Р^(т Н,У.;Тг) / 1 1 УК ,
«нр (m I i,Tj, Те) gign \ Р / \ТГ / [ к Те)\
X ехр I- (^ + Е[ - Е, - En)/kTF}. (3.168)
185
В случае
а-^^|в=К8<^’АГг ’ (3-169)
имеем
ЗнИ("Ф, »Хг) = MX) /XX 3/2 ех Г _ +
a^(m\i,vi-,Tj,Te) gtgn \ Mr / Р Х
(3.170)
Поскольку при Те — Тг = 300° К рекомбинация идет через уровни тп,
для которых ДДт =0, то с помощью (3.166) в этом случае можно рас-
считать величину коэффициентов ассоциативной ионизации через те
же уровни (табл. 3.23), воспользовавшись измеренными коэффициентами
диссоциативной рекомбинации (см. табл. 3.18) и значением энергии Л7Г1ЮСТ.
идущей на кинетическую энергию осколков после рекомбинации
(см. табл. 3.19).
Таблица 3.23
Расчетные значения коэффициентов ассоциативной ионизации при Т е — Тг = 300° К
Ион Число уровней д^пост> эВ А (!) + В (п) IO-7 см3-сек~1 ^a. и смз.сек-1
при АКПОСТУ= =#> при AEdoct = = 0
1 Ne2+ i 10 1,25 Ne (215) + Ne (3p'[i/2]o) 0,2 1,3-Ю-33 1,3-ю-11
1,81 Ne (2'5) + Ne (3p[i/2]i) 0,2 4,5-10~42 4,5-10~12
АГ2+ 4 1,34 Ar (3i5) + Ar (4p'fVeil) 2 1,3-10-33 3,5-Ю-11
1,55 Ar (3’5) + Ar (4р'18Л]г) 2 2,1-Ю-37 2,1-Ю-11
NO+ 2 0,27 N (2D) -J- О («Р) ' 2,5 l,3.10-17 4,7-10-13 ‘
N2+ 2 2,26 N (45) + N (2P) 1,0 6-10~51 6-Ю-13
3,46 N (45) N (2D) 1,0 3,6-10-71 3,6.10-13:
Ог+ 3 5,10 О (3P) + 0 (iD) 1,5 l,2.10-98 1,25-Ю-12
3,13 20 (iD) 0,18 5,0-10-66 2,0.10-13
2,89 0 (3P) + 0 (15) 0,4 2.10-61 1,7-Ю-12
Из результатов расчета следует, что вклад атомов на уровнях, которые
заселяются при диссоциативной рекомбинации при комнатной температуре^
в ассоциативную ионизацию при той же температуре пренебрежимо мал.
Если же на поступательные степени свободы атомов расходуется энергия
Лапост ~ кТ, то коэффициенты ассоциативной ионизации через такие кана-
лы могут достигать величины ~ 10-11—10-13 еж3-сек-1, что по порядку вели-
чины согласуется с экспериментальными данными, полученными для ассо-
циативной ионизации аргона (см. табл. 3.20, 3.23).
Сравнение результатов расчета с экспериментом показывает, что термы
т, через которые идет ассоциативная ионизация при комнатной температу-
ре, должны пересекать термы молекулярных ионов на уровне, соответствую-
щем энергии возбуждения атомов, т. е. Лапост ~ кТе. Отсюда следует, что
эти термы не могут принимать участия в диссоциативной рекомбинации ио-
нов с электронами при комнатной температуре. Исключение могут состав-
лять лишь нижние термы, лежащие на уровне дна потенциальной ямы иона.
Интересно отметить, что ассоциативная ионизация наблюдается при
столкновении невозбужденных атомов с возбужденными с интервалом энер-
гий последних ~ 1100—1200 см~\ 600 см'1 у Ar I, ~500, ~ 1000 и 1900 слх1
у Кг ]; ~300, 600, 700, 1200 см-1 у Хе I. Это, по-видимому, близко к рассто-
янию между последующими колебательными уровнями иона (ср. для Ne2.
186
<ое = 800 см-1). Следовательно, вероятнее всего, энергия, выделяющаяся
при ионизации в результате столкновения возбужденных атомов с потенци-
ал ахми выше дна терма AJ, передается на колебательное возбуждение иона,
Поэтому рекомбинация на эти уровни в экспериментах с увеличением Те
и Тг “ 300° К не должна происходить. Эти каналы должны увеличивать
вклад рекомбинации колебательно-возбужденных ионов. Поэтому с повы-
шением температуры Tv для инертных газов следует ожидать увеличения вкла-
да рекомбинации через эти уровни, в результате чего значения коэффициен-
та рекомбинации при высоких температурах должны быть больше, чем
<хд-р (Ге, Tv), рассчитанные по (3.162), (3.163) (см. стр. 184 и работу [266]).
Что касается ассоциативной ионизации в ударных волнах, то на резуль-
таты этих измерений могут повлиять неравновесные заселенности различ-
ных уровней, что требует особого анализа. Например, в работе [252] коэффи-
циент ассоциативной ионизации N (45) + О (3Р) -+ NO+ + е измерялся
в фронте ударной волны. Время полной релаксации ионизации ~2-10_6 сек.
Реакция эта идет, как следует из данных [9, 265], через атомы N (2D)
(см. табл. 3.23). Нами была проведена оценка характерного времени нараста-
ния концентрации N (2Н) в ударной волне с использованием коэффициента
скорости дезактивации этого состояния (см. стр. 133). Оказалось, что засе-
ленность N (27?) к концу ионизационной релаксации может отличаться от
равновесной в четыре раза. Следовательно, измеренная скорость ассоциатив-
ной ионизации может быть занижена во столько же раз. Этим может объяс-
няться наблюдаемое отличие экспериментальных данных [252, 253] от рас-
четов [9, 265, 272]. Аналогичные систематические погрешности возможны
и при расчетах течения в экспериментах [249, 262, 263].
ОТКЛОНЕНИЯ ОТ ИОНИЗАЦИОННОГО РАВНОВЕСИЯ
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
Ниже будет рассмотрена кинетика процессов ионизации и рекомбинации,
приводящих к установлению ионизационного равновесия, и проанализиро-
ваны основные причины, приводящие к отклонению от ионизационного рав-
новесия.
При этом под равновесными будем понимать такие концентрации возбуж-
денных атомов и ионов, которые можно рассчитать по уравнениям Саха и
Больцмана с температурой, равной температуре электронов.
В общем случае кинетика процессов ионизации — рекомбинации заря-
женных частиц определяется большим числом элементарных процессов столк-
новения заряженных и тяжелых частиц, часть которых мы рассмотрели в пре-
дыдущих параграфах данной главы. Поскольку наибольшими сечениями
ионизации (см. стр. 134) обладают возбужденные атомы, их роль в кинетике
ионизации — рекомбинации будет, как правило, преобладающей. При этом
наряду с ионизацией электронным ударом может иметь значение и иониза-
ция ударами тяжелых частиц. Роль возбужденных молекул в процессе ио-
низации, ио-видимому, существенно ниже из-за конкуренции процессов
диссоциации (см. стр. 152), а роль диссоциативной рекомбинации будет
велика вследствие больших величин коэффициентов. В процессах ассо-
циативной ионизации и диссоциативной рекомбинации образуются или гиб-
нут возбужденные атомы, поэтому эти процессы должны также быть вклю-
чены в общую кинетическую схему процесса.
Ввиду сложности решения общей задачи мы ограничимся анализом ряда
важных случаев. Вначале будет рассмотрена кинетика ударно-радиационной
ионизации атомов под действием электронного удара. Затем рассмотрим влия-
ние выхода излучения, возбуждения и девозбуждения атомным ударом. Пос-
ле этого включим в рассмотрение ионно-молекулярные реакции (перезаряд-
ки и ионной конверсии), а также диссоциативной рекомбинации, ассоциатив-
ной ионизации и оценим влияние этих реакций на ионизационное равновесие.
187
1. Ударно-радиационная ионизация атомов
Рассмотрим вначале пространственно-однородную плазму, состоящую*
из тяжелых частиц (атомов и ионов одного сорта) и электронов с концентра-
циями N, Nt и Ne соответственно. Концентрацию атомов на возбужденном
уровне к обозначим Nк. Кинетика ионизации в ней определяется балансны-
ми уравнениями [275]
4г = 2 (МХ? - Ш?) = - «ад, (3.171}
где q обозначает сорт частиц, вызывающих переходы (электроны, тяжелые
частицы, фотоны). Поскольку максимальную вероятность ионизации, как
правило, дают соударения с электронами, то феноменологическое выражение
для скорости ионизации—рекомбинации цмеет вид, записанный в правой
части этого уравнения. Это уравнение записано без учета процессов диссоци-
ативной рекомбинации и ассоциативной ионизации. Для решения систе-
мы (3.171) необходимо определить концентрации возбужденных атомов. По-
скольку времена жизни атомов, как правило, существенно меньше харак-
терных времен ионизации — рекомбинации, можно воспользоваться ква-
зистационарным приближением j
2 =о. (3.172).
n,q
Следуя разработанному в работе [275] так называемому модифицирован-
ному диффузионному приближению, введем понятие потока электронов
в энергетическом пространстве связанных состояний атомов Jfe, пропорцио-
нального числу частиц, проходящих в единицу времени через сечение, про-
веденное между двумя уровнями к и к +1:
ь= 2 2 (ад^-ад^п). (з.17з>
п^к т>к
Тогда уравнение (3.172) приобретает вид
7 к — 7k+i = ^7fc = 0- (3.174)
Выражения для потоков имеют вид:
h=2<имд.%, д/к=д (2 4%адл), (з-175>
<2 Q
где z^k+1 — эффективные вероятности переходов между уровнями к -> к + 1
при соударениях с частицами сорта q, и введены относительные заселенности
уровней yk = Nk/Nk, характеризующие отклонение заселенности А-го
уровня от равновесного (А°). Аналогично ниже введем уе — Ne/Ne
и Уа+= Na+/Na+ ит. д.; kyk = yk — ук+1.
Учитывая (3.174), (3.175) и (3.171), имеем следующее решение для коэф-
фициентов скорости ступенчатой ионизации — рекомбинации и заселенно-
стей по возбужденным уровням:
рст = (Аде1е)-\ аст = [Ne да 51ер, (3.176)
Ук — (У1$ке + УвУх^Гк) (^le) S (3.177)
Sk = (Mz/г.шГ1, Sik = 2 St- (3.178)
1=к
Полные коэффициенты скорости включают член, описывающий ионизацию
невозбужденных атомов и рекомбинацию с образованием невозбужденных
188
атомов:
Р ~ Зет “Т Рш °C ~ #ст “Т
(3.179)
где
Зп = 2 ОСп = 2
<з а
В случае максвелловского распределения электронов по энергиям (Те)
для эффективных вероятностей перехода получены следующие выражения
[2751:
еХР\ Г ’
(е) 4(2jr)172Aie«7Ve / Д£’12 ч
212 ~ (те/сГе)1/2Д£12 еХР(— кТе )’
(3.180)
(3.181)'
гдеЛ^ — кулоновский логарифм, зависимость которого от отношения kTJkE
приведена на рис. 3.23. При расчете (3.180) и (3.181) использовалось при-
ближение Бете —Борна для сечений разрешенных переходов, а запрещенные
Рис. 3.23. Зависимость ку-
лоновского логарифма для
связанных состояний от от-
носительной энергии пере-
хода кТе!&Е
переходы не учитывались. В величинах z/3+i одноквантовые переходы к ->
-> к + 1 учтены точно, а многоквантовые — приближенно. Такое прибли-
жение вряд ли ухудшает точность расчетов, поскольку сами величины сече-
ний могут содержать значительные погрешности (см. стр. 123). В случае необ-
ходимости величины Zkj{+1 могут быть вычислены при любом заданном наборе
сечений и функции распределения. Аналогичные расчеты для случая соуда-
рений с тяжелыми частицами могут быть получены аналогичным путем [275].
При учете радиационных переходов между уровнями и радиационной ре-
комбинации выражения (3.176) — (3.178) необходимо заменить на
Зет = (адчад-1, аст = [1 + 2 4+1 да 5fc] х
X [Afe да ^еГ1, «V =
где
Щ — [1 4" (ttn+lNп / 2+1^71+1)1?
ап= 2 (М/С) =
/с>п Кк 1<п
(3.182)*
(3.183)'
— множители, отражающие влияние излучения (Ahl — вероятности спон-
танных переходов, ае1 — коэффициенты радиационной рекомбинации).
Заметим, что в ряде работ система уравнений (3.171), (3.172) решалась
численно [63, 276]. При этом использовались другие значения вероятностей
переходов. Тем не менее сравнение результатов численных расчетов с резуль-^
татами расчетов в изложенном выше модифицированном диффузионном при-
189
ближении показывает, что различие, если таковое и наблюдается, обусловле-
но больше выбором вероятностей переходов и заложенными до расчета пред-
положениями, чем расчетом кинетики процессов (сравнение результатов рас-
четов друг с другом и с экспериментом см. в [63, 275]).
Использование вместо вероятностей переходов (3.180), (3.181) экспери-
ментально измеренных сечений или рассчитанных согласно [62] не дает су-
щественных различий в результатах.
Мы выбрали для изложения данное приближение, потому что оно позво-
ляет представить решение аналитически. Это удобно для исследования зако-
номерностей, наблюдаемых в заселении возбужденных уровней, кинетике
ионизации и рекомбинации. Кроме того, в этом приближении сравнительно
несложно проследить качественно влияние различных факторов на отклоне-
ние от ионизационного равновесия.
В частности, в случае пространственно однородной^изотермической (Те =
= Тг) плазмы единственным источником неравновесного распределения ато-
мов по уровням и неравновесной степени ионизации является излучение. Вы-
ход излучения с Л-уровня становится несущественным при [275]:
2Ve>10^(ln'A.) (3.184)
для оптически тонкой плазмы. Если излучение реабсорбировано, критерий
смягчается. Выход излучения приводит к уменьшению заселенностей уров-
ней, что вызывает увеличение коэффициента рекомбинации и уменьшение
коэффициента скорости ионизации. Влияние атомного удара на коэффициен-
ты рекомбинации и ионизации в водородной плазме, согласно расчетам [63],
в которых использовалась формула (3.36) для сечений возбуждения атома-
ми, начинается при степени ионизации хе < 10-S в случае оптически тонкой
плазмы и хе < 10-4, если излучение серии Лаймана заперто. Согласно оцен-
кам [275], они становятся существенными только при хе < 10~5. В той же ра-
боте [275] приводятся выражения для расчета а, |3 и заселенностей уровней
в случае преобладания атомного удара.
Если плазма пространственно неоднородна, причиной отклонения от ио-
низационного равновесия может быть диффузия заряженных частиц. В этом
’Случае [275]
Уе = = ]/Др < 1,
(3.185)
где TD — характерное время диффузии заряженных частиц. Отклонение от
равновесной степени ионизации приводит к обеднению заселенностей уров-
ней (ук < 1) (3.177). Заметим, что в обоих рассмотренных случаях наруше-
ния равновесия при Tg = Тг влияние атомного удара приводит к уменьше-
нию отклонений от равновесия.
В случае неизотермической плазмы (Те 7"г) ситуация меняется. Влия-
ние атомного удара будет приводить к увеличению отклонения от равновесия
с Т — Тс. В предельном случае преобладания атомного удара степени иони-
зации и заселенности уровней можно рассчитать из уравнений Саха и Больц-
мана с Т = TY,
Следует отметить, что приведенные выше оценки степени ионизации, при
которых начинается влияние атомного удара, получены при использовании
формулы (3.36), которая справедлива только при соударениях атомов инерт-
ных газов одного сорта (см. стр. 130—132). Для водорода применимость этой
формулы не доказана. В случае атомов азота эта формула также неприме-
нима. При столкновениях атомов разного сорта, например аргона с ксено-
ном, сечения девозбуждения также существенно возрастают (см. табл. 3.5),
что связано с влиянием неадиабатических переходов. Рост сечений
должен приводить к значительно более существенному влиянию атомного
удара в таких смесях. Кроме того, при рассмотрении процессов ступенчатой
ионизации и рекомбинации мы не учли влияние ионно-молекулярных реакций.
190
[2. Влияние ионно-молекулярных реакций
на ионизационное равновесие
В целях упрощения соответствующих выражений рассмотрим влияние ион-
но-молекулярных реакций на кинетику ионизации и рекомбинации при пре-
обладании электронного удара в возбуждении и девозбуждении уровней и пе-
реходах между континуумом и уровнями при соударениях в пространствен-
но-однородной стационарной плазме [277, 278].
Поскольку в результате ионно-молекулярных реакций могут образовы-
ваться или гибнуть возбужденные атомы, то уравнение (3.174) в этом случае
становится неприменимым и его необходимо заменить следующим:
, (3.186)
где $ — поток атомов, образующихся на уровне к или уходящих с него за
счет протекания ионно-молекулярных реакций.
Бинарная смесь одноатомных газов. В случае смеси двух атомарных газов
при условии, что влиянием конверсии атомарных ионов в молекулярные,
а также квазирезонансным процессом ^передачи энергии возбуждения при
соударениях атомов разного сорта можно пренебречь (эти случаи будут рас-
смотрены ниже на конкретном примере — см. стр. 192—193), ионизация
будет определяться следующей схемой реакций:
Ра
А+ cZZZA+ + 2с, (3.187)’
аА
Рв
В+ eZZ±B++ 2с, (3.188)
ав
Yi
А+ + В^ГА + В++ ЪЕ, (3.189)
Y2
где 8Е Ед — Е& — разность потенциалов ионизации невозбужденных ато-
мов. В стационарном случае решение системы (3.187) — (3.189) имеет вид
([A+]/2Ve) + тХ) (тХ + чМ + осаЛХ,
(3.190)
(1В+Ж) = (₽iX + YtX) (тХ + + ХЧ
Как правило, в низкотемпературной плазме скорости перезарядки значи-
тельно превышают скорости ударно-радиационной рекомбинации. При вы-
полнении условия
«вМ ~ рХ, «аМ ~ ₽ATVe < ТX, 1Х (3.191)
из (3.189) следует
(Ув+/уа+) = [Г(Тг, Те)/Р(Те)Г, (3.192)
где
г (Тг, те) = [Т1 (Тг, те)/ъ (Тг, те)], хв = XX, (3.193)
-|Гд~ 1 f И + Г^в) (1 + Г°ЖВ ав/ал) Т |/ (1 + Г°жв) (1 + ГЯвав/аА) (3.194)
Используя квантовомеханический принцип микроскопической обрати-
мости при максвелловском распределении частиц по скоростям, получаем
Г = 4S44 = ех₽ (W4). (3.195).
г> SA+5B
191
Это выражение справедливо, если в перезарядке не принимают участия воз-
бужденные частицы. В некоторых случаях при большой разнице энергий
ЪЕ процесс может идти с образованием возбужденных частиц В или А+ и мо-
жет оказаться близким к резонансному. В таких случаях надо рассматривать
и кинетику возбуждения ионов. Однако для оценки можно просто использо-
вать в качестве величины 8Е не разность потенциалов ионизации, а дефект
резонанса. Из выражения (3.194) следует, что равновесная степень ионизации
смеси уе — 1 может быть получена только при Гг = Те (этот случай тривиа-
лен) и при а а. = «в- Но даже в последнем случае отношение концентраций
ионов разного сорта (Ад =f= ^в) должно отличаться от равновесного, что при-
ведет к неравновесному заселению уровней. Из (3.177) получим
У К = (?4 + yeyl+ 'Sllk) (-SL)-1, (3.196)
где I " А или В.
На рис. 3.24 для примера приведены результаты расчета отклонений от
равновесия в смеси атомов аргона и водорода при Те = 8300° К, Тг — 3300° К,
-хн = 0,025. Эти параметры близки к параметрам в слаботочной дуге при
атмосферном давлении [182, 279]. В этом случае, как показано в работе [280],
•атомным ударом и выходом излучения можно пренебречь. Тогда из (3.191) —
(3.195) получаем уе = 0,4, y^z+ = 3,1-10“2, ун+ = 4,5.
Результаты расчета отклонений от равновесных концентраций атомов во-
дорода и аргона для этого случая по формуле (3.196) приведены на рис. 3.24
(кривые 1,6). На этом же рисунке для сравнения нанесены эксперименталь-
ные значения у^ (точки 7) и укг (8), полученные в работе [182], и результаты
расчета по формулам (3.174), (3.185) без учета влияния ионно-молекулярных
реакций (кривые 2 и 4). Как видно из сравнения результатов расчетов и экс-
периментов, неучет перезарядки (кривые 2,4) дает обогащение концентраций
возбужденных атомов и ионов аргона по сравнению с водородом, что проти-
воречит экспериментальным данным. Учет перезарядки (3.188) позволяет по-
лучить качественно верный результат (кривые 1,5): обогащение заселенно-
стей возбужденных атомов и ионов водорода по сравнению с аргоном, кото-
рое наблюдалось экспериментально, обусловлено перезарядкой. Однако
количественного согласия с результатами экспериментов такой расчет не дает.
Объяснить это можно тем, что не были учтены другие ионно-молекулярные
реакции, протекающие в данной смеси газов [281]:
Аг+ + Н2 ZT HAr+ + Н, (3.197)
Y4
Д-Р
ак
НАг+ + с “Аг (Л = 1) + Н (к = 2), (3.198)
а также процессы квазирезонансной передачи энергии возбуждения от ато-
мов водорода (Ек = 105 291—109 191 см-1) к атомам аргона с возбуждением
последних (Ек = 105 462—108 722 см-1) [79]
Y5
Н(& — 5 — 15) 4-Аг (Л; - 1)ГГН(Л- 1)4-Аг(^-З) (Д£ = 56 -400 см'1).
(3.199)
Остальными ионно-молекулярными реакциями, приводящими к образова-
нию и гибели молекулярных ионов, в данном случае можно пренебречь ввиду
большой скорости реакции (3.197) — у3 = 1,5*10-9 см^-сек"1 [281]. Молеку-
лярные ионы, образующиеся в результате данной реакции, гибнут практи-
чески целиком в результате диссоциативной рекомбинации (3.198), если ее
коэффициент превышает ~ 10-11 см?* сек-1, что не вызывает сомнения (см.
стр. 172).
192
Рис. 3.24. Степень отклонения от равновесных концентраций электронов, ионов и воз-
бужденных атомов аргона (2, 5, 6, 8) и водорода (1, 3, 4, 7) в смеси при Те = 8300° К,
Гг = 3300° К, *н = 0,025
I, 6 —расчет по формуле (3.196) с учетом перезарядки (3.188); 2, 4—расчет без учета перезарядки
и других ионно-молекулярных реакций; 3, 5 — с учетом реакций (3.197) — (3.199); 7, 8 — экспери-
ментальные данные [182]. Энергия возбуждения атомов отсчитывается от границы ионизации. Зна-
I -I п Н Аг
чения у к при Ек/Е1 = 0 соответствуют ук = уеуц+; ук = УеУАт+
Рис. 3.25. Схематическое изображение уровней атомов аргона и водорода и потоков
между ними
Стрелками указаны направления, цифры над стрелками — величины потоков (см-3 ’сек-1). обуслов-
ленные реакциями (3.197), (3,198) — !, (3.199)—II, (3.188) — III и диффузией — IV. Остальные
переходы обусловлены электронным ударом
Реакция (3.197) является одной из причин диссоциации молекул водоро-
да в разряде. Если предположить, что диссоциация водорода целиком обу-
словлена этой реакцией, то
Т3 [Ar+][HJ = [Н]/2т?, (3.200)
где td — характерное время диффузии атомов водорода из дуги. Соотноше-
ние (3.200) позволяет оценить скорость этой реакции, не зная концентрации
молекул водорода.
Результаты расчета потоков между различными уровнями возбужденных
атомов и ионов разного сорта, обусловленные протеканием реакций (3.197)—
(3.199), приведены на рис. 3.25. Отклонения от равновесных концентраций
возбужденных атомов и ионов аргона и водорода, возникающие вследствие
протекания этих реакций, изображены на рис. 3.24 в виде кривых 3 и 5. Они
удовлетворительно описывают экспериментальные данные (точки 7, 8).
Таким образом, отклонения от равновесных концентраций возбужденных
атомов и ионов в неизотермической плазме (Те Ф Т^) в рассмотренном слу-
чае практически целиком обусловлены протеканием ионно-молекулярных
реакций. Потоки между уровнями атомов значительно превышают те, кото-
рые могли бы быть вызваны диффузией ионов из дуги (рис. 3.25). Атом водо-
рода при этом не участвует в ионизации смеси, поскольку поток с уровня к = 2
направлен вниз, т. е. он работает в рекомбинационном режиме.
Необходимо отметить, что ранее результаты аналогичных экспериментов
[279] были объяснены автором работы [280] только диффузионными потеря-
ми ионов из дуги, вызывающими отклонения от равновесной концентрации
7 Теоретич. и прикладная плазмохимия
193
заряженных частиц и возбужденных атомов; переходы между уровнями и в
ионизационный континуум при этом обусловлены только электронным уда-
ром, и справедливы уравнения (3.174). Однако при интерпретации резуль-
татов принималось, что отношение концентраций ионов водорода и аргона
в дуге соответствует равновесному с Т = Те, и не учитывалось обеднение
дугового газа атомами водорода вследствие диффузии последних [282—284].
Такие предположения, как следует из изложенного выше, не обоснованны.
Экспериментальные результаты [279] качественно не отличаются от исполь-
зованных для сравнения с расчетами результатов работы [182]. Однако
детальный анализ данных [279] затруднен возможным влиянием на резуль-
таты более легко ионизируемых примесей (С, циан) в дуговом газе.
Влияние ионной конверсии, диссоциативной рекомбинации и ассоциатив-
ной ионизации [277]. Рассмотрим однокомпонентный одноатомный газ, т. е.
такой газ, концентрация атомов в котором существенно превышает концен-
трацию молекул [А] ^>[А2].
На ионизационное равновесие в таком газе будут влиять, кроме реакции
(3.187), следующие реакции:
^С1
А+ + 2A^Z2 А2 + А + 6Е2, (3.201)
/;с2
А2 + ------А + Afe + §Екз.
(3.202)
Здесь 8Е2 и 8Екз — энергия, переходящая с внутренних степеней свободы
атомов и молекул на поступательную степень свободы (см. стр. 173). Поток
атомов, образующихся на к возбужденном уровне за счет реакции (3.202):
= а?-₽[АП^е-рГад, (3.203)
где [А2] — концентрация молекулярного иона,
MA+1 +
_________к_______
*сЛ+2а?'рЧ
к
(3.204)
Подстановка последнего выражения в (3.203) приводит к громоздким фор-
мулам. Поэтому рассмотрим два важных частных случая. При выполнении
условия
(3.205)
к
имеем
адр
(3-206>
к
где
кП,т, 2члх (2
2W 2 ’ <3'207)
к к
(Z) — константа равновесия реакции (3.201). Следствием условий [А2] <<:
[А] и (3.205) является соотношение
[At]<[A+J. (3.208)
194
Как показывают оценки, условие (3.205) выполняется для таких газов,
у которых потенциал диссоциации молекулярного иона больше половины по-
тенциала ионизации атома (азот, кислород и др.) при < 3*1019 мг3 прак-
тически при любых температурах. Кроме того, это условие выполняется
и при обратном соотношении потенциалов ионизации и диссоциации иона,
если равновесие реакции (3.201) сдвинуто в сторону образования молекуляр-
ных ионов. Например, при давлениях, близких к атмосферному, для водоро-
да это имеет место при < 2-103 °К, а для аргона — при Tr <С 103 °К.
В противоположном предельном случае
(3.209)
к
получаем
Л _ „д-р /V (А+1м h _ Л yj^i \ ,о олпч
1к к е к^\тт) ( к увУа.+ ) ’ (3.210)
где
^)(ГГ) (аД-Р)0
к ' К^(Те) (₽-И)0 »*•₽
и концентрация молекулярных ионов
[АП = [А+]ЛгЖр2)(7’г). (3.212)
С учетом этих потоков уравнение (3.174) в стационарном случае примет вид
(3.186).
В изотермической плазме Те -- Тг потоки = 0, и имеем случай, рас-
смотренный в первой части данного параграфа (см. стр. 188). В неизотерми-
ческой пространственно-однородной плазме (Тг =j= ?е) равновесие реакций
(3.201), (3.202) сдвигается, что приводит к отличному от нуля потоку $
и отклонению от равновесных степеней ионизации и концентрации возбуж-
денных частиц. Отклонение от равновесия описывается системой уравнений
(3.186) с учетом выражений для потоков (3.206), (3.210), а состав ионов —
формулами (3.208), (3.212). При этом необходимо, конечно, учесть квазиней-
тральность плазмы
[А2+] + [А+] = ЛГе. (3.213)
В наиболее простом случае, когда в результате диссоциативной рекомби-
нации возбужденные атомы образуются только на одном уровне (в частном
случае оба атома могут оказаться невозбужденными, к — 1), можно написать
•следующую систему уравнений:
(.Ук — УеУА+) (Site)-1 = Уг = У1 При I < к, (3.214)
У г = (j/l^ie + УеУА+ Skd^) (S^)-1 При I > к,
где величины Ski определяются по формуле (3.178). При выполнении условия
(3.205) из (3.214) получаем
У1 — Уе — ^1 (1 — fr/i/z/e), Дг — kCtNiSkeN е,
(3.215)
а величина б определяется формулой (3.207). Используя формулы (3.168),
(3.170), описывающие температурную зависимость коэффициентов ассоциа-
тивной ионизации и диссоциативной рекомбинации, а также (3.116) — (3.118)
для коэффициентов скорости конверсии, оценим отклонение от равновесия,
вызванное сдвигом равновесия ионно-молекулярных реакций, для азота.
В этом случае Л-1 (см. стр. 183), и при Те = 104° К, ТГ = 5500° К имеем б
— 10-6, кС1 - 4,8-10-32 см6-сек-1.
195
7*
Результаты расчета и уе по (3.125) при разных концентрациях атомов
приведены в табл. 3.24.
Таблица 3.24
Отклонения от равновесной степени
ионизации в атомарном азоте
ЛГ1, слг-3 Д1 Щ । Ni, ел-® Д1 Уе
1,0-1017 2,5-1017 0,34 0,84 0,81 0,4 3,0-101’ 1,0.1018 1,05 3,4 4,5-Ю-з 1,2.10-8
Ниже для примера приведены рассчитанные величины отклонения
от равновесной степени ионизации для атомов с водородоподобной схемой
уровней (водород, инертные газы, азот [275]) при Те — 104° К, и таких Гг,
что выполняется условие (3.205), если поток атомов поступает на другие уров-
ни к при постоянном и N е.
к Д1 уе
1 2,1 1,4.10-3
2 0,12 0,94
3 0,01 0,995
Из приведенных данных следует, что отклонение от равновесия значи-
тельно уменьшается, если к > 2.
В противоположном случае (3.209) из (3.214) получаем
(У1 — УеУА+) = A2Z/A+ fl — ,
\ УеУА.+ /
(3.216)
r/Д-Р (Т т ) __
Д2 =-----1..г’ е— /K1N31,2S]ceNe
К<Я(ТГ) r 1 1 е
И
J/a+ = уе И + NJK.% (Тг)1 [1 + NjKf (Те)]"\
где Кт — константа равновесия ионизации атома при 71 = Те, a опреде-
ляется формулой (3.211).
Например, для водорода при Те — 104° К, Тт = 3300° К и Nr = 2-
•1017 см~3 получаем б ~ 10-4. При ад-Р (300° К) — 2-10-8 см6-сек-1
(см. табл. 3.18, 3.22) и к = 2 из (3.216) получаем существенное отклонение
от равновесия: уе 0,2. Если принять ад р — 2-10”9 см6-сек~1, то уе <
<0,78.
При этих оценках использовалась формула (3.162) для зависимости ко-
эффициента диссоциативной рекомбинации от температуры газа. Реальная
зависимость будет слабее (см. стр. 184), что должно привести к увеличению
отклонений от равновесия.
Таким образом, как следует из анализа формул (3.194), (3.196), (3.210).
подкрепленного приведенными выше оценками, в реальных плазменных сис-
темах отклонения от равновесных концентраций электронов и заселенностей
уровней, обусловленные неизотермичностью плазмы (Те Ф Тг), могут быть
достаточно велики. При этом максимальное отклонение от равновесия наб-
людается при образовании в результате диссоциативной рекомбинации не-
возбужденных атомов, и оно уменьшается по мере роста номера уровня при
постоянном отношении ]^/Ne. Поэтому в тех случаях, когда в результате дис-
социативной рекомбинации атомы могут более или менее равномерно обра-
196
зовываться на различных возбужденных уровнях, наибольшее отклонение
от равновесия будет вызывать поток, поступающий на самый нижний уро-
вень из всех возможных.
При необходимости учет радиационных процессов, а также отклонения
функции распределения электронов по энергиям от максвелловской может
быть проведен так же, как в работе [275], но с использованием выра-
жения (3.186) вместо (3.174). В общем случае учет этих факторов дол-
жен привести только к увеличению возможных отклонений от равновесия
из-за влияния ионно-молекулярных реакций в неизотермической плазме
(Те =£= 77г) ввиду уменьшения соответствующих эффективных вероятностей
переходов между уровнями и величин Ske.
Не вызывает принципиальных затруднений и совместный учет влияния
неизотермичности (Те TY) и неоднородности плазмы (амбиполярной диф-
фузии ионов и электронов) на отклонения от равновесных концентраций.
Возможное уточнение значений и температурных зависимостей коэффи-
циентов скоростей ассоциативной ионизации, диссоциативной рекомбинации,
ионной конверсии и возбуждения атомов электронным ударом, а также учет
образования более сложных молекулярных ионов (Аз, . . .) не изменят ос-
новного вывода: в двухтемпературной плазме (Те Ф Тт) существенное
отклонение концентраций электронов, ионов и возбужденных атомов от равно-
весных, рассчитываемых по уравнениям Саха и Больцмана с Т — Те, воз-
можно даже в том случае, если ступенчатые переходы между возбужденны-
ми уровнями и возбужденными уровнями и континуумом происходят только
в результате электронных ударов, а диффузией заряженных частиц можно
пренебречь. Отклонение от равновесия в этом случае обусловлено влия-
нием ионно-молекулярных реакций, приводящих к дополнительному образо-
ванию или гибели возбужденных атомов.
4
ХИМИЧЕСКИЕ РЕАКЦИИ
В ПЛАЗМЕННЫХ СТРУЯХ
Современные плазмохимические процессы (и, в частности, ряд многотон-
нажных промышленных процессов) организуются, как правило, таким обра-
зом, что потоки плазмы и сырья вводятся в плазмохимический реактор раз-
дельно [1—3]. Для того чтобы достигнуть желаемого результата — провести
в реакторе химическую реакцию, необходимо прежде всего перемешать сырье
с плазмой. При этом по самой сущности химической реакции требуется, что-
бы молекулы реагентов находились в непосредственном контакте. Это озна-
чает, что сырье должно быть перемешано с плазмой до молекулярных масшта-
бов независимо от того, является ли плазма реагентом или только энергоно-
сителем; поскольку перенос энергии от частиц плазмы к молекулам реагента
происходит также на молекулярном уровне. Одной из характерных особен-
ностей плазмохимической технологии является использование весьма высо-
ких температур — от ~ 2-103 до (10—15)-103°К. При таких температу-
рах скорости химических реакций возрастают настолько, что характерные
времена этих реакций становятся сравнимыми с характерными временами
процессов переноса. Поэтому в процессе перемешивания реагента с плазмой,
характерное время которого становится сравнимым по величине с характер-
ным временем химических реакций, реагент может испытывать значительное
превращение. Для сокращения времени перемешивания последнее произво-
дят в условиях интенсивной турбулентности. Однако и в этих условиях вре-
мя перемешивания остается еще достаточно большим для того, чтобы реагент
в процессе перемешивания испытывал заметное превращение. При расчетах,
моделировании и оптимизации плазмохимического реактора необходимо учи-
тывать степень этого превращения, которая определяется геометрическими
и гидродинамическими особенностями реактора-смесителя. Следовательно,
возникает необходимость рассчитывать степень превращения данного реаген-
та в процессе его турбулентного перемешивания с плазмой в условиях, когда
характерные времена химического превращения и физического процесса тур-
булентного перемешивания сравнимы по величине между собой. Эта задача
неновая и возникает всякий раз, когда приходится иметь дело с «быстрыми»
и «очень быстрыми» химическими реакциями, например при расчете процес-
сов горения в турбулентных потоках, определении параметров баллистиче-
ских следов, остающихся за телами, перемещающимися с большими скоростя-
ми в газах и жидкостях, и определении констант скоростей биохимических
реакций в растворах [4, 5].
Рассмотрение общего случая протекания химической реакции одновре-
менно с перемешиванием турбулентных потоков в неизотермических услови-
ях представляет настолько сложную задачу, что ее решение до сих пор полу-
чить не удалось. Это обусловлено не только хорошо известными трудностя-
ми описания механизма турбулентности [6—9], но и необходимостью точного
учета пространственной неоднородности полей температуры и концентраций
компонент.
198
ХИМИЧЕСКИЕ РЕАКЦИИ]
В УСЛОВИЯХ ТУРБУЛЕНТНОСТИ
Задачей теории турбулентности является, как известно, предсказание
средних свойств турбулентного потока и их зависимости от граничных усло-
вий. При рассмотрении химических реакций, происходящих в условиях тур-
булентности, проблема может быть сформулирована следующим образом.
Требуется найти законы изменения во времени и пространстве средних вели-
чин и среднеквадратичных флуктуаций концентраций реагентов в процессе
турбулентного перемешивания последних в неизотермических условиях при
произвольном соотношении характерных времен реакции и процесса переме-
шивания.
Упрощенный подход. Кинетику химической реакции можно, очевидно,
рассматривать независимо от процессов переноса в том случае, когда харак-
терное время тх реакции много больше характерного времени тп процессов
переноса («очень медленные» химические реакции). Другой предельный слу-
чай —«очень быстрых» химических реакций, происходящих в условиях, ког-
да тх<<тп, довольно подробно проанализирован теоретически [10—24]. При
этом задача оказалась настолько сложной, что пришлось вводить ряд упро-
щающих предположений. Поэтому полученные результаты можно рассматри-
вать в основном в качественном аспекте. Предположения эти сводятся к сле-
дующему. Прежде всего рассматриваются изотермические условия. Турбу-
лентность считается однородной и изотропной [6], так что градиент средней
скорости турбулентного потока равен нулю. Рассматривается необратимая
термонейтральная химическая реакция типа А + пВ -> продукты, причем
реагенты А и В настолько разбавлены, что взаимной их диффузией можно
пренебречь.
Протекание химической реакции не влияет на гидродинамические харак-
теристики системы. Коэффициенты диффузии обоих реагентов считаются
одинаковыми и постоянными. В этих предположениях задача сводится к реше-
нию системы уравнений типа
П (х, tyTi (х, f) = — кТ\ (х, t), (4.1)
где
П (х, t) = + v (х, t)V — DV2, (4.1а)
Гi (я, t) — концентрация г-го реагента, v (х, t) — скорость, D — коэффици-
ент диффузии, к — коэффициент скорости рассматриваемой химической реак-
ции. Задача становится стохастической, если начальные условия для концен-
траций реагентов и скорости задаются в виде случайных функций. Поскольку
в условиях турбулентности Г\ — + у,, где — средняя по времени кон-
центрация и — флуктуация концентрации, то система уравнений (4.1)
не является замкнутой. Большинство существующих моделей турбулентных
потоков основано на использовании различных систем уравнений для момен-
тов, получаемых из уравнения Навье — Стокса [6], в которых всегда боль-
ше неизвестных, чем уравнений. Для нахождения решения требуется делать
какие-то предположения о свойствах моментов более высоких порядков.
Обычно систему (4.1) замыкают с помощью соотношений Буссинеска [6], ко-
торые позволяют связать напряжения Рейнольдса и коэффициенты турбулент-
ного переноса реагентов с местными градиентами средней скорости и средними
концентрациями реагентов. При этом новыми моментами, появляющимися
в результате учета химической реакции, пренебрегают (часто по физи-
чески не совсем ясно обоснованным причинам) [13, 23]. Иногда для удобства
вместо двух реагентов А и В рассматривают один; в этом случае считается,
что реакция идет по схеме А + А -> продукты [10, 11, 13, 15].
199
Основные результаты, полученные при таком весьма упрощенном подхо-
де к решению указанной проблемы, заключаются в следующем. Удается по-
казать, что дифференциальное уравнение (4.1), описывающее происходящие
одновременно массоперенос и химическую реакцию, можно преобразовать
в уравнение, описывающее только перенос «вещества» с концентрацией Г =
= Га — пГв [12]. Представляют интерес асимптотические зависимости от
времени средней концентрации реагента Г и его среднеквадратичных пуль-
саций Y2, которые удается получить, рассматривая последний этап вырожде-
ния турбулентности. Так, по данным [22, 23], при t ->- оо
ГА(0 = Гв(0~Г°’75, Та(0 = Тв(С~(О0-1’5. (4.2)
Если в дополнение к перечисленным выше ограничениям предположить,
что очень быстрая химическая реакция происходит в турбулентном потоке
в цилиндрическом реакторе, причем реагенты перемешаны в «грубом» мас-
штабе (т. е. на уровне глобул) в некотором начальном сечении реактора,
а флуктуации концентраций этих реагентов в каждой точке любого сечения
реактора распределены вблизи средней концентрации по нормальному закону
(в отсутствие реакции), то можно показать [12], что
F (z) = ц (z), (4.3)
где F (и) = 1 — ГА (^)/Га (0) представляет собой степень превращения и
ц (z) = 1 — [yl (z)/y& (0)]1/г. Равенство (4.3) справедливо при условии, что
реагенты вводятся в реактор в стехиометрическом соотношении, т. е. при 0 =
= Гв (0)/пГа(0) = 1. Если же 0 =[= 1, то
F (z) = 1 + (₽ - 1) {1 + g (z) [(? (0))V2 (1 - ц (z))]}, (4.4)
где g (z) = {z^y 2 ) ierfc (1/z У 2 ) и ierfc (1/z У2 ) — первый интеграл функции
ошибок.
Выводы упрощенной теории, сформулированные в виде выражений (4.3)
и (4.4), подтверждаются рядом экспериментальных данных [25—29]. Наибо-
лее обстоятельная проверка проводилась в работе [28]. При этом условия
эксперимента специально выбирались таким образом, чтобы выполнялись
многочисленные допущения, принятые в теоретическом анализе. Исследова-
лось протекание химических реакций, константы скоростей которых варьи-
ровались от 5-101 до 1 *10п л/моль-сек, в цилиндрическом реакторе в протоке
жидких, сильно разбавленных реагентов в изотермических условиях. Спе-
циальные меры были приняты для того, чтобы обеспечить режим течения,
близкий к однородной изотропной турбулентности (за исключением очень
небольших пристеночных участков). Полученные в этом эксперименте зави-
симости концентрации реагентов от расстояния по оси реактора хорошо сог-
ласовались с зависимостями, определяемыми по (4.3) и (4.4), в пределах из-
менения стехиометрического коэффициента 0 = 1 — 10.
Этот результат позволяет сделать следующий важный с практической точ-
ки зрения вывод: исследуя протекание очень быстрой химической реакции
и определяя зависимость от времени средних величин концентраций реа-
гентов в описанных выше условиях, можно находить характеристику данного
реактора как турбулентного смесителя. Затем можно использовать получен-
ную характеристику для расчета степени конверсии в реакторе в случае
произвольного соотношения скоростей реакции и процесса перемешивания
при условии, что гидродинамические характеристики реактора остаются
неизменными.
Стохастические модели. Упрощенный подход имеет одно важное преиму-
щество — он позволяет получать решение в аналитическом виде (хотя и
весьма приближенное в большинстве случаев). Однако для описания с по-
мощью полученного решения результатов эксперимента возникает необхо-
200
димость использовать эмпирические характеристики реальных турбулент-
ных полей [22, 24], что снижает, естественно, ценность полученных резуль-
татов. В связи с этим, а также благодаря быстрому развитию вычислитель-
ной техники в настоящее время появились подходы к решению обсуждаемой
проблемы, которые основаны на непосредственном использовании эмпири-
ческих данных [30—34]. В частности, в работе [32] предложена расчетная
модель химической реакции в турбулентном изотермическом потоке в ци-
линдрическом реакторе. Согласно этой модели, жидкость, входящая в реак-
тор, делится на некоторое постоянное число N одинаковых по размеру эле
ментов. Далее принимаются следующие допущения: 1) внутри каждого эле-
мента свойства жидкости постоянны; 2) элементы жидкости, расположенные
в одном и том же сечении реактора, сливаются попарно друг с другом слу-
чайным образом, после чего свойства жидкости в получившемся элементе
усредняются, вносятся поправки к величинам концентраций реагентов,
обусловленные протеканием химической реакции, а затем вновь образованный
элемент распадается на два идентичных; 3) скорость слияния и распада эле-
ментов жидкости является только функцией расстояния z по оси реактора;
4) однородность распределения по сечению реактора элементов, содержащих
жидкости-реагенты, достигается в непосредственной близости к входу реак-
тора; 5) аксиальной диффузией пренебрегают; 6) химическая реакция не
влияет на гидродинамические параметры потока. Авторы [32] вводят следую-
щую характеристику процесса смешения:
т] (z) = п (z)/F!\ (4.5)
где п (z) — число слияний и распадов, испытываемых N элементами жид-
кости за время прохождения расстояния z от входа в реактор. По смыслу эта
величина представляет собой половину среднего числа слияний и распадов
элементов, испытанных к моменту прихода в точку z (в расчете на один эле-
мент). Величину ц (z) можно измерить: измеряя величину среднеквадратич-
ных флуктуаций концентрации пассивной примеси, вводимой в поток, или
определяя степень конверсии (в функции z) исходного реагента, участвую-
щего в реакции, скорость которой контролируется перемешиванием (т. е.
в пределе тх тп). Выбор числа элементов N производился эксперименталь-
но в соответствии с требованием об инвариантности выводов, следующих из
предложенной модели, при увеличении N сверх некоторого Amin- С по-
мощью такой модели путем расчета на ЭВМ была получена зависимость от
величины ц (z) степени конверсии F одного из исходных реагентов в необра-
тимой бимолекулярной реакции (изотермические условия, несжимаемые
жидкости). Далее авторы [32] заимствовали результаты экспериментального
исследования [28] в виде зависимости от z степени конверсии F реагента в
реакции, контролируемой процессом перемешивания (т. е. в пределе тх <<!
тп). Из сопоставления расчетной F (ц (z)) и экспериментальной F (z) за-
висимостей была получена зависимость ц (z) для реальных условий экспери-
мента [28]. Получив эту характеристику реактора-смесителя, авторы [32]
рассчитали степень конверсии того же реагента в условиях, когда характер-
ные времена реакции и процесса перемешивания находились между собой в
произвольном соотношении. Результаты этих расчетов хорошо согласовались
с результатами соответствующих экспериментов [28]. Несмотря на значи-
тельную упрощенность обсуждаемой модели, из расчетов с ее использова-
нием следует, что перемешивание начинается с некоторым запаздыванием,
причем длина пути запаздывания очень близка к той же величине, предска-
зываемой теорией свободных струй [35].
В работах [30, 31, 33, 34] предлагаются по существу сходные модели. Ин-
тересно отметить, что в теории горения довольно давно пользуются так на-
зываемой микрообъемной моделью горения [36—40]. ничем по существу не
отличающейся от моделей, предлагаемых в [30—34]. Несмотря на то что в
расчетах скорости турбулентного горения газов с использованием микро-
объемной модели не учитывается реальный спектр скоростей и размеров эле-
201
ментов (глобул), пренебрегается аксиальной диффузией и делается допуще-
ние о мгновенности процесса молекулярного смешения в пределах глобул,
получаемые результаты в большинстве случаев хорошо согласуются с ре-
зультатами экспериментов — по меньшей мере в качественном аспекте [40].
Крайняя упрощенность моделей [30—34] вызвала появление ряда доволь-
но серьезных критических замечаний [41, 42]. В частности, указывалось, что
в этих моделях не учитывается влияние молекулярной диффузии [41]. Дейст-
вительно, в явном виде это влияние в модели [32] не учитывается. Однако та
операция усреднения свойств смеси реагентов, которая проводится в [32]
после каждого случайного слияния элементов жидкости, представляет со-
бой в сущности процесс мгновенного перемешивания до молекулярных мас-
штабов, т. е. такая операция позволяет учесть наличие молекулярной диф-
фузии.
В общем виде вопрос сводится к тому, какую стадию процесса «турбу-
лентного перемешивания до молекулярных масштабов» следует считать ли-
митирующей — стадию собственно турбулентного перемешивания или ста-
дию молекулярной диффузии. Будем рассматривать процесс турбулентного
перемешивания исходя из современной концепции локально-изотропной тур-
булентности, базирующейся на работах А. Н. Колмогорова [33], А. М. Обу-
хова и С. Корсина [43—48].
Итак, в условиях турбулентности при достаточно больших числах Рей-
нольдса происходит постепенное уменьшение размера турбулентных вихрей,
начиная с некоторого Zmax ~ d (где d — характерный размер течения) и
кончая размером порядка масштаба Колмогорова Zk ~ 10Zmax/Re3^.
Нетрудно убедиться, что время молекулярной диффузии в масштабах
~ 1к значительно меньше времени турбулентного дробления вихрей (от
Zmax До ^к), составляющего по оценке rT ~ Zmax/DT, где DT ~ Llv' —
коэффициент турбулентной£диффузии (определяемый по аналогии с коэф-
фициентом молекулярной диффузии — в соответствии с известной гипотезой
Прандтля); Ll — лагранжев масштаб турбулентности; vf — среднеквадра-
тичная скорость вихрей [6]. Положив, например, Zmax = 1 DT =
= 102 см21сек, получим Тт ~ 10~2 сек. Время молекулярной диффузии в мас-
штабе ZK составит при DM = 0,2 смЧсек, Re = 104 величину тм ~ Zr/Dm =
= 5-10“4 сек. Следовательно, несмотря на то что скорость турбулентного
перемешивания значительно выше скорости молекулярной диффузии (в
характерных масштабах течения), лимитирующей стадией суммарного про-
цесса является стадия турбулентного перемешивания.
Таким образом, при анализе стохастических моделей типа [30—34] сле-
дует прежде всего обращать внимание на достоверность описания моделью
стадии турбулентного перемешивания. С этой точки зрения указанные мо-
дели часто представляются неудовлетворительными. Так, порядок скорости
смешения по времени, согласно, например, [32, 33], равен единице, в то вре-
мя как по результатам [22] он составляет (— 1,5) при t-+ оо, а по данным
[13, 16] изменяется по мере затухания турбулентности.
Серьезным недостатком обсуждаемых моделей является также зависимость
следующих из них результатов от вида начальных распределений турбу-
лентных глобул [42]. Предлагаемая в [41] модель чисто диффузионного пе-
ремешивания глобул при наличии химической реакции по существу эквива-
лентна моделям /30—34] на стадии затухания турбулентности, и вопрос здесь
вовсе не в том, что «химическая реакция нечувствительна к деталям процесса
перемешивания» [41]. Действительно, при пользовании той или иной моделью
основным является вопрос, в какой мере м;одель отражает существенные фи-
зические черты описываемого с ее помощью явления. Характерным для мо-
делей [30 —34 и 41] является разбиение жидкости на элементы. Сохраняя
эту существенную черту процесса турбулентного перемешивания, можно, по
нашему мнению, предложить неограниченное число расчетных моделей и
описать ими результаты практически любого эксперимента, если произво-
202
дить подгонку результатов расчета к результатам эксперимента, проведен-
ного в условиях, когда тх <С тп-
В общем случае следует добиваться того, чтобы предлагаемая модель была
подобна описываемому явлению с геометрической, динамической и химичес-
кой точки зрения. От стохастических моделей процесса перемешивания с хи-
мическими реакциями требуется прежде всего, чтобы скорость смешения
изменялась по мере изменения интенсивности турбулентности и в пределе
очень быстрых реакций соблюдалось соотношение (4.3) для соответствующих
условий эксперимента.
Некоторые фундаментальные подходы. Как видно из предыдущего, ре-
зультаты, полученные при использовании упрощенных подходов и стохасти-
ческих моделей, позволяют описывать в лучшем случае весьма узкий круг
специально поставленных экспериментов. Анализ же практически интерес-
ных случаев — горения в турбулентном потоке, процессов, происходящих в
баллистических следах, плазмохимических реакций, осуществляемых в тур-
булентных плазменных струях,— требует разработки более фундаменталь-
ных подходов к решению задачи описания химических реакций в условиях
турбулентности. К сожалению, общей теории турбулентности в настоящее
время не существует. Поэтому в любом, даже весьма фундаментальном под-
ходе приходится пользоваться эмпирическими данными, касающимися ха-
рактеристик турбулентности. Естественно, при использовании этих подхо-
дов для решения указанной задачи в случаях, отличающихся от тех, для
которых эти подходы были предложены, приходится не только учитывать из-
менение характеристик турбулентности, но и уточнять отдельные положения
предложенных моделей.
Один из наиболее удачных приближенных методов расчета скоростей хи-
мических реакций и скоростей диссипации среднеквадратичных флуктуа-
ций концентраций реагентов в турбулентных потоках был предложен Ли-
нем [49] и получил название бимодального приближения. Основные ограни-
чения этого метода сводятся к следующему: а) коэффициенты скоростей
всех химических реакций заданы как функции температуры; б) скорости
химических реакций таковы, что для расчета процесса перемешивания прост-
ранственных неоднородностей в потоке можно использовать теорию пере-
мешивания в нереагирующих турбулентных потоках (например, используя
подходы Корсина [10, 11]); в) внешнее давление постоянно, химические реак-
ции не вызывают изменения давления в потоке; г) число Рейнольдса доста-
точно велико для того, чтобы обеспечивать наличие хорошо развитого тур-
булентного течения; д) число Маха мало настолько, что пульсациями кон-
центраций реагентов, обусловленными сжимаемостью, можно пренебречь
по сравнению с пульсациями, обусловленными турбулентной конвекцией ис-
ходных неоднородностей и химическими реакциями. Основная идея метода
базируется в сущности на представлениях о турбулентной конвекции, раз-
витых в [43—48].
Математическая формулировки модели основана на следующей схеме.
Для того чтобы использовать закономерности обычной химической кинети-
ки при исследовании макроскопически неоднородного поля течения, вся
жидкость в исходной «невязкой ламинарной области» делится на небольшие
объемы, в пределах которых химический состав считается постоянным. Пос-
ледующее движение этих элементов в турбулентном потоке описывается по
существу методом Лагранжа, причем влияние турбулентной конвекции и
молекулярной диффузии учитывается раздельно. Неупорядоченность траек-
торий рассматриваемых элементов, обусловленная турбулентной конвекцией,
учитывается путем усреднения по ансамблю'функций Лагранжа для тур-
булентного переноса вещества в физическом пространстве. Влияние же мо-
лекулярной диффузии описывается посредством деления каждого элемента
жидкости на две следующие отдельные объемные доли: Т, на которую не
влияет процесс молекулярной диффузии, и (1 — Т), целиком подверженную
влиянию последней. Далее предполагается, что динамические характерис-
203
тики поля турбулентности известны. Это позволяет отыскать вид функций
Sl. Наконец, из/геории турбулентного перемешивания нереагирующего по-
тока заимствуются характеристики пульсаций компонент и скорости диффу-
зионного смешения, что позволяет найти вид зависимости величины Т* от
времени.
В результате автору [49] удалось получить интегрально-дифференциаль-
ное уравнение, связывающее величину Т и концентрации перемешанных до
молекулярных масштабов компонентов в условиях осесимметричного турбу-
лентного следа. Как и следовало ожидать, полученное уравнение сравни-
тельно просто решается для реакции первого порядка. При учете реакций
более высокого порядка в многокомпонентной смеси возникает ряд труд-
ностей, которые в принципе можно преодолеть.
В качестве примера в бимодальном приближении было рассчитано поле
течения в квазиодномерном турбулентном следе [50]. Форма турбулентного
фронта следа, макромасштаб турбулентности и средняя скорость дисси-
пации турбулентной кинетической энергии заимствовались из результатов
соответствующих экспериментов. Были найдены величины турбулентных
пульсаций концентраций электронов и химически активных компонентов
следа. Результаты расчетов хорошо согласовались с результатами экспери-
ментов (по крайней мере, в качественном аспекте). В дальнейшем при исполь-
зовании бимодального приближения для описания турбулентных следов
[51, 52] обнаружилось, что для лучшего согласия результатов расчета и
эксперимента следует учитывать флуктуации температуры, сильно влияю-
щие на пульсации скоростей химических реакций (в основном уравнении
бимодальной модели [49] член, учитывающий пульсации скоростей химичес-
ких реакций, считался пренебрежимо малым и отбрасывался). В частности, в
работе^[52] флуктуации температуры в турбулентном баллистическом следе
исследовались путем наблюдения за флуктуациями интенсивности излуче-
ния молекулярного кислорода в ультрафиолетовой области спектра (молеку-
ла кислорода образуется в результате быстрой реакции рекомбинации двух
атомов кислорода).
Согласно современным представлениям явления переноса в турбулент-
ном потоке связаны в основном с турбулентным движением больших вихрей,
которые существуют сравнительно долго [6, 9]. Перенос различных субстан-
ций этими вихрями в некоторой мере аналогичен молекулярному переносу в
разреженном газе. Именно такую аналогию имел в виду Чанг [53, 54], ког-
да предложил рассматривать процесс турбулентного перемешивания с пози-
ций обобщенной теории броуновского движения [55]. При этом предполага-
лось, что переносимая субстанция является пассивной скалярной примесью
(т. е. влияние этой примеси на турбулентность пренебрежимо мало).
Основываясь на известных концепциях турбулентного переноса [6,9],
Чанг считает, что в турбулентном потоке существуют одновременно вихри
различных размеров. Статистические свойства элементов жидкости опреде-
ляются совместным влиянием этих вихрей. При достаточно больших числах
Рейнольдса в турбулентном потоке существует статистическое разделение
между статистически неравновесными низкими волновыми числами, представ-
ляющими вихри большего размера, и равновесными высшими волновыми чис-
лами, представляющими вихри меньших размеров. Большая часть наблюдае-
мых свойств турбулентного потока обусловлена поведением больших вихрей.
Кроме того, скорость молекулярного рассеяния, определяемая поведением
мелких вихрей, фактически контролируется скоростью вырождения крупных
вихрей.
Такую структуру поля турбулентности при высоких числах Рейнольдса
можно описать с помощью стохастического уравнения Ланжевена с соответ-
ствующей интерпретацией различных членов [53, 54]. Чангу удалось вывести,
используя уравнение Ланжевена и некоторые дополнительные упрощающие
предположения, уравнение типа Фоккера — Планка для функции распре-
деления эл l.j тов жидкости в фазовом пространстве. Помимо того, что это
204
уравнение корректно описывает поле турбулентности, оно является стати-
стически замкнутым; следовательно, все уравнения для моментов, получае-
мые из этого уравнения, также замкнуты. Более того, отпадает необходимость
отыскивать различные моменты как независимые неизвестные функции
(см. стр. 199), поскольку моменты всех порядков теперь связаны через функ-
цию распределения элементов жидкости. Следовательно, уравнения для мо-
ментов в теории Чанга имеют несколько другой смысл по сравнению с клас-
сическими статистическими теориями или с обычными феноменологическими
теориями турбулентного переноса. Эти уравнения нужны лишь для того,
чтобы облегчить решение уравнения Фоккера — Планка по аналогии с ме-
тодами, используемыми для решения уравнения Больцмана в кинетической
теории газов.
Как известно, в классических статистических теориях турбулентности
поле турбулентности описывается не самим уравнением Навье — Стокса, а
набором уравнений для моментов различных порядков. Чангу удалось по-
казать, что полученные из сконструированного им уравнения Фоккера —
Планка уравнения для моментов являются существенно теми же самыми, что
и полученные из уравнения Навье — Стокса (по меньшей мере вплоть до мо-
ментов второго порядка).
Для проверки предложенной теории рассчитывалось турбулентное тече-
ние Куэтта в канале при отсутствии химических реакций [53, 57]. Необходи-
мые для расчетов данные по скорости диссипации турбулентной энергии
заимствовались из эксперимента. Полученные результаты удовлетворитель-
но согласуются с результатами соответствующих экспериментов. Весьма ин-
тересные результаты были получены при анализе горения заранее непере-
мешанных горючего и окислителя в турбулентном потоке со сдвигом и пос-
тоянным градиентом осредненной скорости (гомологичный поток) [56].
Прежде всего следует отметить очень хорошее качественное согласие с ре-
зультатами эксперимента. Далее, в отличие от выводов феноменологических
теорий горения, из результатов, полученных Чангом, следует, что ширина
зоны пламени в пределе высоких значений числа Дамкелера (т. е. в пределе
очень быстрых химических реакций) равна по порядку величины локальному
интегральному масштабу турбулентности. Несколько неожиданным резуль-
татом является вывод, что перенос тепла в некоторых областях пламени мо-
жет иметь место в направлении, противоположном направлению местного
градиента средней температуры.
В заключение следует отметить, что современное состояние теории хими-
ческих реакций в условиях турбулентности не позволяет предсказывать ре-
зультаты соответствующих экспериментов с достаточной точностью. Поэто-
му, с одной стороны, желательны попытки построения более точной теории, а
с другой — необходимо производить более широкие экспериментальные ис-
следования с целью получения результатов, позволяющих ясно представить
структуру турбулентного движения и механизм перемешивания при нали-
чии химических реакций. Важно отметить также, что в ряде частных случаев
проблема может быть частично решена в результате исследования химиче-
ских реакций, степень превращения реагента в которых определяется харак-
теристиками процесса турбулентного перемешивания.
ИССЛЕДОВАНИЯ ТУРБУЛЕНТНОГО ПЕРЕМЕШИВАНИЯ
В ДОЗВУКОВЫХ ПЛАЗМЕННЫХ ПОТОКАХ
Потоки плазмы отличаются от «обычных» газовых течений высокой темпе-
ратурой газа (до 2-104° К и выше), наличием значительной диссоциации и
заметной ионизации, а также очень большими градиентами параметров. Так,
например, в плазменной струе аргона, генерируемой дуговым плазмотроном
постоянного тока и истекающей в атмосферу из сопла круглого сечения диа-
метром 3 мм, радиальные составляющие градиента температуры достигают
205
5-103 град/мм, составляющие в осевом направлении около 400 град/лии (см.
стр. 12).
Свободные плазменные струи. В настоящее время существует сравни-
тельно небольшое число работ, в которых исследовались характеристики сво-
бодных плазменных струй с теоретических позиций [58, 59, 62] и эксперимен-
тально [60—65]. Большинство результатов, полученных в этих исследованиях,
полностью согласуется с известными результатами исследований сво-
бодных турбулентных струй [35, 66, 67]. Отличия свободных плазменных
струй от струй газа сравнительно невысокой температуры сводятся к сле-
дующему: 1) плазменная струя рассеивается значительно быстрее; 2) длина
ее потенциального ядра меньше; 3) переход от ламинарного режима тече-
ния в струе к турбулентному существенно ускоряется по мере увеличения
скорости коаксиального струйного потока газа в весьма незначительных пре-
делах (что противоречит выводам, следующим из теории пограничного слоя
[68]). Такой переход осуществляется при весьма небольших значениях числа
Рейнольдса, составляющих, согласно данным [61], ~ 300 и 630—850 —
по данным [65].
Таким образом, результаты перечисленных выше исследований показы-
вают, что процессы ионизации, излучения и диссоциации, протекающие в
плазменных струях, оказывают в пределах погрешности экспериментов и рас-
четов пренебрежимо малое влияние на скорость перемешивания струи с ок-
ружающим газом. Полное перемешивание струй достигается на расстояниях,
равных 3—4 калибрам сопла генератора плазмы. Если, например, в качест-
ве диффузионной характеристики, соответствующей определенной степени
смешения с окружающим воздухом, выбрать длину £я светящегося участка
плазменной струи аргона, истекающей в атмосферу из сопел диаметром 2—6 жж,
то критериальное соотношение, связывающее £я, диаметр сопла d, средне-
массовую скорость струи гсм и ее среднемассовую температуру Тем, будет
иметь вид [64]
где g — ускорение силы тяжести, То — начальная температура газа на^вхо-
де в плазмотрон. Основные параметры плазменной струи можно рассчиты-
вать с помощью существующих теорий струй при учете диссоциации и осе-
вого изменения коэффициента турбулентного обмена, являющегося функцией
числа Маха [63]. При этом, как и в свободных турбулентных течениях газа
при невысоких температурах, скорость турбулентного переноса массыТвыше
скорости переноса энергии, которая в свою очередь выше скорости переноса
количества движения [66, 67]. На основном участке струй радиальные про-
фили основных параметров хорошо описываются’кривой Гаусса. Диапазоны
исследованных параметров составляют: по температуре — до 13*103°К, по
скорости (0,1—0,8)М.
Потоки плазмы в каналах. При движении плазменной струи в цилиндри-
ческом канале происходит перемешивание плазмы со сравнительно холод-
ным газом, распространяющимся в периферийных зонах струи. Характерис-
тики этого процесса перемешивания не отличаются от таковых для свободной
плазменной струи вплоть до расстояния, равного трем’калибрам канала,
где процесс перемешивания практически завершается [62]. Здесь и в преды-
дущем параграфе речь идет о турбулентном перемешивании, т. е. процессе,
приводящем к выравниванию радиальных профилей параметров струи. Если
в качестве критерия степени перемешивания выбрать,^как это^принято в ра-
боте [64], длину £яТсветящегося ядра плазменной струи и рассматривать
распространение плазменной струи в спутном потоке различных газов, то
оказывается, что £я|сильно зависит от рода газа в'спутном потоке и несколь-
ко слабее — от скорости последнего. Так, подданным [69], величина Ln!d =
= 3,5 при обдуве аргоновой плазменной струи азотом и Lnld = 2 при обду-
ве водородом и метаном (отношение диаметра канала реактора D к диаметру
206
сопла плазмотрона d составляет Did ~ 2,5, диапазон изменения среднемас-
совой скорости струи 100—300 м/сек, скорости спутного потока 10—100 м/сек,
диапазон изменения среднемассовой температуры плазменной струи
4000—5000° К). Ускорение процесса смешения спутных потоков при исполь-
зовании химически реагирующих (диссоциирующих) газов может быть объяс-
нено влиянием химических реакций на процесс турбулентного перемешива-
ния. Это влияние, согласно теоретическим исследованиям Корсина [76],
оказывается очень сильным только в том случае, когда велика скорость мо-
лекулярной диффузии. Поэтому представляется совершенно естественным
сокращение длины высокотемпературного, светящегося, почти ламинарного
ядра струи, в области которого коэффициент молекулярной диффузии может
превышать коэффициент турбулентного переноса. Однако ядро плазмен-
ного потока занимает обычно весьма незначительную часть (^30%) сече-
ния последнего, и основным процессом, приводящим к выравниванию про-
филей параметров потока в канале, следует считать процесс турбулентного
перемешивания. Из результатов работы [70], где проводилось исследование
перемешивания аргоновой плазменной струи со спутным потоком азота в
цилиндрическом канале (D/d = 2,45), следует, что возникающая в канале
рециркуляция газа приводит к очень быстрому завершению перемешивания.
Полученные в этой работе с помощью калориметрического зонда [71] профи-
ли концентрации азота устанавливаются на расстоянии около 3D. Решение
весьма упрощенной системы уравнений, описывающей процесс перемешива-
ния в указанной системе [70], показало, что для объяснения полученных
экспериментальных результатов необходимо положить коэффициент диффу-
зии равным — 200 см2/сек. Это свидетельствует о том, что основным механиз-
мом перемешивания является турбулентный перенос.
Таким образом, даже сравнительно слабое воздействие на поток плазмы со
стороны спутного потока приводит к существенному ускорению перемеши-
вания [62, 60, 70]. Естественно, при более сильном взаимодействии смеши-
вающихся потоков скорость перемешивания возрастает в еще большей сте-
пени. Так, например, в уже упоминавшейся выше (см. стр. 8) камере
смешения многодугового подогревателя [72] интенсивность перемешивания
в системе трех сталкивающихся плазменных струй настолько высока, что
уже на расстоянии около двух калибров канала камеры смешения от места
ввода струй профиль температуры можно считать установившимся (и весьма
равномерным) в пределах погрешности эксперимента.
Аналогичные результаты были получены в [73] при вводе в плазменный
поток холодных газовых струй через отверстия, расположенные в плоскости
сечения канала, перпендикулярной его оси. При длине зоны перемешивания,
равной двум калибрам канала, максимальная неравномерность профиля тем-
пературы составляет 5—7% на диаметрах, равных 0,75D. Поскольку стен-
ки канала интенсивно охлаждаются снаружи, профиль температуры может
все более отклоняться от прямолинейного вследствие теплообмена газа со
стенкой. Профиль температуры тем ближе к прямолинейному, чем меньше
диаметр отверстий ввода холодного газа и чем больше расход холодного газа.
Так, при уменьшении площади отверстий примерно в два раза при сохране-
нии числа отверстий и величины расхода холодного газа максимальная не-
равномерность температурного профиля снижается почти в три раза [73].
По-видимому, на скорость установления профилей параметров в потоке сме-
си оказывает влияние величина отношения динамических напоров смешивае-
мых струй (pi^i/p2^, где р1? р2 — плотности газов в струях, v19 г?2 — скорости).
Об этом свидетельствуют результаты экспериментов со сравнительно слабо-
подогретыми струями [77, 78], а также результаты работ, в которых иссле-
дуются траектории холодных струй в поперечных плазменных потоках
[75, 79].
Модель перемешивания газовых потоков в цилиндрических каналах. На
основе анализа результатов перечисленных выше теоретических и экспери-
ментальных работ, а также принимая во внимание результаты собственных
207
исследований [74, 75, 80] и представления теории турбулентных струй [66],
можно предложить следующую приближенную модель процесса перемеши-
вания газовых потоков в цилиндрических каналах. Пусть в цилиндрическом
канале диаметром D распространяется поток газа, в который вдувается
газ из отверстия диаметром d<D, расположенного на оси потока (рис.
4.1, а).
Предположим, что степень турбулентности в струе вдуваемого газа доста-
точно высока для того, чтобы распространение этой струи можно было опи-
сать с помощью полуэмпирических соотношений, предложенных в [66] для
описания свободных затопленных турбулентных струй. Вдуваемую струю
можно считать свободной до тех пор, пока ее пограничный слой не сопри-
касается со стенками канала. Справедливость этого подтверждается резуль-
татами экспериментов [80, 81] в пределах изменения отношения Did от 1,4
Рис. 4.1. Схема перемешивания турбулентной струи с потоком газа в канале
а — симметричное заполнение сечения канала пограничным слоем струи; б — несимметричное запол-
нение; в — схема распространения струи при поперечном вдуве
до 13. Очевидно, профиль концентрации вдуваемого газа в канале можно счи-
тать установившимся (в грубом масштабе), когда внешние границы вдувае-
мой струи пересекутся со стенками канала, при условии, что расстояние £п,
на котором происходит заполнение сечения канала пограничным слоем вду-
ваемой струи, не меньше длины начального участка этой струи.
Оценим величину Ln исходя из представлений полуэмпирической теории
свободных турбулентных струй [66]. Согласно этим представлениям, тангенс
угла наклона внешней границы свободной турбулентной струи к ее оси со-
ставляет tg а/2 = 0,22—0,3 в широком диапазоне изменения значений числа
Рейнольдса, скоростей и температуры турбулентных струй. Следовательно,
длина зоны установления профиля концентрации вводимого в поток газа
составит
Ln = l)/2tg -^ = (1.7- 2,3) D. (4.7)
В крайнем случае, когда вдуваемая струя смещена с оси канала и распо-
лагается у стенки канала (рис. 4.1, б), величина £п составит, очевидно,
(3,4—4,6)2). В случае поперечного ввода струи газа в поток (рис. 4.1, б) до-
биться строго симметричного заполнения сечения реактора пограничным слоем
холодного газа практически невозможно, поскольку форма сечения вводимой
струи в сносящем потоке существенно деформируется [66], будет сказываться
влияние величины отношения динамических напоров струй и потока, профи-
лей параметров струй и потока и других факторов. Поэтому, для того чтобы
обеспечить достижение минимального значения Ln в условиях радиального
ввода струй, необходимо пользоваться результатами исследования траек-
торий струй в сносящих потоках [69, 75, 79, 80, 82—84]. Например, траекто-
рии струй холодных газов в сносящем плазменном потоке аргона в канале
описываются с погрешностью не более 12% следующим выражением:
y/d = q°^8(x/d)G^ (4.8)
208
где у, х — координаты точки траектории (оси струи), отсчитываемые соот-
ветственно нормально и параллельно оси плазменного потока, мм; q =
= Pi^i/pa^ — отношение динамического напора струй холодного газа к
среднему по сечению динамическому напору плазменной струи; р2 и
v2 — плотности и скорости холодного и нагретого газов соответственно;
d — диаметр отверстия для ввода холодного газа, мм.
Следует отметить, что соотношение (4.8) получено в диапазоне изменения
q = 0,25—4,35 при сохранении постоянным динамического напора плазмен-
ной струи, среднемассовая температура которой поддерживалась равной
3000 + 350° К при расходе аргона 10—15 л/мин через сопло диаметром 4 мм.
В качестве «холодных» газов использовались аргон, азот, кислород и метан;
газы вводились радиально в плазменный[поток через одно отверстие, диаметр
которого варьировался в пределах 0,4—1,3 мм.
Поскольку диапазон исследованных параметров потоков [75] сравнитель-
но узок, а полученные результаты согласуются с погрешностью не более
40% с результатами расчета по формуле, предложенной^ в [83] и проверенной
в значительно более широком диапазоне изменения параметров струй, мож-
но рекомендовать пользоваться этой формулой для оценок формы траекто-
рии струй холодных газов в сносящих плазменных потоках
ax/d = 195q”1»3 (ay/d)3, (4.9)
где а — коэффициент структуры струи газа, вводимой в поперечный поток
[83]. Дальнобойность h струи плазмы, вводимой в поток холодного газа, т. е.
в наших обозначениях расстояние по оси у от стенки канала до оси вводимой
струи после разворота ее в направлении сносящего потока, можно оценить
по формуле, предложенной в [69]:
hid = 2,48 1(р3^)/(р1^)]°,2в, (4.10)
где использованы те же обозначения, что и в формуле (4.8). Соотношение
(4.10) справедливо для vx — 10 — 50 м/сек. v2 — 400—700м/сек и среднемас-
совой температуры струи (3—4)-103°К*
Поскольку угол раскрытия се плазменных струй больше угла раскрытия
струй холодного газа [85], длина £п зоны установления профиля концентрации
газа плазменной струи, вводимой в поперечный поток холодного газа, будет
меньше величины определяемой соотношением (4.7). С точки зрения
обеспечения минимальной длины зоны установления профиля концентрации
примени в потоке ввод плазменных струй в поперечный поток представ-
ляет некоторые преимущества по сравнению со способом ввода струй хо-
лодного газа в поток плазмы [86, 87]. Однако с позиций организации плаз-
мохимических процессов, для которых важно обеспечить минимальное время
турбулентного перемешивания реагентов и плазмы в канале реактора, пос-
ледний способ преимуществ, по-видимому, не имеет. Действительно, для
уменьшения времени перемешивания следует увеличивать скорость транс-
портирующего потока в канале реактора. Этого легче добиться, очевидно,
используя в качестве транспортирующего потока струю плазмы.
Таким образом, согласно предлагаемой приближенной модели турбулент-
ного перемешивания газовых потоков в канале, длина £п канала от места вво-
да турбулентной струи до сечения, в котором профиль концентрации вводи-
мого газа можно считать установившимся (в грубом масштабе), сохраняется
постоянной для данного канала диаметром D и равно!! 2D с погрешностью
+ 15% (см. формулу (4.7)). Очевидно, для сокращения длины смесителя сле-
дует уменьшать диаметр канала, учитывая при этом условие, приведенное вы-
ше при формулировке обсуждаемой модели и касающееся соотношения дли-
ны начального участка турбулентной струи и величины Ln. Действительно,
сокращение Ln путем уменьшения диаметра канала D целесообразно прово-
дить до тех пор, пока Ln не станет сравнимой с длиной начального участка
турбулентной струи /0. Согласно экспериментальным измерениям [80] и
209
теоретическим оценкам [35, 66], выполненным с учетом того, чтопривдуве
струи в поток газа меньшей плотности величина Zo несколько увеличивается,
имеем 10 = (5—7)d, где d — диаметр отверстия для вдува.
Следовательно, минимальная величина диаметра смесителя составит
Dmin (2,5—3,5)d, что не противоречит одному из основных положений мо-
дели — о свободном характере распространения струи холодного газа в ка-
нале реактора [80].
Время установления профиля концентрации введенного в реактор газа
составит, очевидно, тп = LJv. где v — линейная скорость движения сме-
си в канале. По оценкам [80], в канале диаметром D — 3,5 мм величина тп
составляет 20—30 мксек.
Перемешивание реагентов и плазмы до молекулярного уровня. До сих
пор речь шла о перемешивании плазменных потоков со струями газов-реа-
гентов на макроскопическом уровне, т. е. на уровне отдельных глобул газа,
каждая из которых может в принципе содержать молекулы только плазмы или
газа-реагента. Как уже подчеркивалось выше (см. стр. 198), для проведения в
этих условиях химической реакции необходимо осуществить перемешивание
плазмы и реагентов на молекулярном уровне независимо от того, является
плазма одним из реагентов или только выполняет роль теплоносителя. Од-
нако ни один из используемых в газодинамике методов изучения процессов
перемешивания (в том числе использующие зонды — пробоотборники) не
позволяет установить, что перемешивание произошло именно на молекуляр-
ном уровне. Объясняется это не столько грубостью самих методов, сколько
тем, что для решения соответствующих задач газодинамики вполне достаточ-
но исследовать процесс перемешивания в некотором макроскопическом мас-
штабе, меньшем, например, характерного масштаба исследуемого течения.
При анализе процесса турбулентного перемешивания струй плазмы и реа-
гента до молекулярных масштабов будем исходить из известных концепций
теории турбулентного переноса [6,9, 43—48] и считать, что в турбулентном по-
токе существуют глобулы различных размеров. С течением времени проис-
ходит дробление глобул вплоть до момента, когда их размер сравнивается
по порядку величины с масштабом Колмогорова /к — 10d/Re3/4, где d — харак-
терный размер течения и Re — число Рейнольдса. Выше показано (см. стр.
202), что характерное время молекулярной диффузии в масштабе /к намного
меньше характерного времени процесса, приводящего к постепенному умень-
шению размеров глобул в турбулентном потоке. Следовательно, скорость мо-
лекулярной диффузии в масштабе d, т. е. процесса, приводящего к переме-
шиванию на молекулярном уровне, фактически будет определяться скоро-
стью уменьшения размеров крупных глобул. Для того чтобы проследить
за процессом турбулентного перемешивания плазмы и реагентов до молеку-
лярного уровня, авторы предложили использовать очень быструю химичес-
кую реакцию [88]. В отличие от других работ, где использовался метод быст-
рой химической реакции [25—29], в данном случае исследуемая система яв-
ляется неизотермической. Фактически здесь необходимо с помощью метода
быстрой химической реакции проследить за процессом нагревания холодно-
го газа, вводимого в поток плазмы. Подбор химической реакции производил-
ся с учетом следующих требований. Во-первых, характерное время реакции
должно быть значительно меньше характерного времени процесса турбу-
лентного перемешивания, которое можно оценить для конкретных условий
эксперимента. Пусть характерный размер турбулентного течения равен диа-
метру канала реактора d — 5-10-1 см\ коэффициент турбулентной диффузии
в сходных условиях составляет, по оценке [80], величину DT^1-103 см21 /сек.
Таким образом, характерное время турбулентного перемешивания со-
ставит тт ~ d2/DT = 2,5-Ю’4 сея. Во-вторых, механизм реакции должен быть
достаточно простым для того, чтобы в исследуемых условиях его можно бы-
ло в хорошем приближении описать схемой типа А + М -> продукты, где
А — молекула исходного реагента, М — молекула инертного газа плазмен-
ной струи. Если в качестве реагента выбрать закись азота N2O, то механизм
210
термического разложения молекулы N2O может быть описан следующей схе-
мой:
N2O + М -> N2 + О + М, (1)
N2O + О N2 + О2, (2)
N2O + О 2NO, (3)
N2O + N2O 2N2 + О2, (4)
N2O + N2O -> 2NO + N2, (5)
O+O+M->O2+M, (6)
О + NO + M -> NO2 + M, (7)
О + NO2 NO + O2, (8)
NO + N2O -> NO2 + N2. ' (9)
Для того чтобы пренебречь всеми вторичными реакциями (2)—(9), в экс-
перименте используется закись азота, разбавленная аргоном так, что содер-
жание закиси азота в исходной смеси составляет 15%. Такое разбавление
позволяет также исключить влияние химической реакции на гидродинами-
ческие характеристики смешивающихся потоков. Расход аргоновой плазмы,
подаваемой в реактор, выбирается таким образом, чтобы концентрация за-
киси азота в расчете на полную смесь газов равнялась ~ 5—7%. Характер-
ное время реакции (1) составляет (по убыли концентрации N2O в е раз) ве-
личину тх ~ 1-10-5 сек при температуре реакции 3000° К и концентрации
N2O в аргоне, равной 5% (при атмосферном давлении) [88]. Таким образом,
для реакции термического разложения N2O требование тх тп = тт
выполняется.
Для отбора пробы из потока смешиваемых плазмы и холодного газа ис-
пользовался калориметрический водоохлаждаемый зонд. Методика экспе-
римента подробно описана в [80, 89], и здесь мы на ней останавливаться не
будем. Следует отметить только, что скорость охлаждения (закалки) пробы
газа в зонде должна быть достаточно высокой для того, чтобы степень превра-
щения реагента в канале зонда была незначительной. Элементарные оценки
показали следующее. Если отбираемая зондом проба состоит частично из
холодного, содержащего закись азота аргона и частично из аргона при тем-
пературе ~ 3000° К, скорее происходит охлаждение горячей части пробы,
чем перемешивание в канале зонда холодного и горячего газов (при ламинар-
ном режиме течения в канале зонда). В наихудшем случае, когда проба газа
содержит закись азота при температуре 3000° К, степень превращения пос-
ледней в канале зонда составит не более 12%. Скорость закалки на на-
чальном участке (длиной менее одного калибра канала зонда) достигает
2*108 град/сек при температуре 3000° К и диаметре входного отверстия
0,5 мм [90].
С помощью метода быстрой химической реакции термического разложения
закиси азота исследовалось перемешивание^аргоновой плазмы с холодным ар-
гоном (примесь N2O составляет 15%) в плазмохимическом реакторе двух кон-
фигураций: с подачей холодного газа в реактор в виде спутного потока не-
большой скорости (Re ~ 103) и с радиальным вводом холодного газа в плаз-
менный поток через одно или три отверстия диаметром 0,3—1,5 мм (Re ~
~ 104). На рис. 4.2 представлены схемы этих реакторов-смесителей. Реак-
тор со спутным вводом реагента исследовался также в режиме с предвари-
тельной турбулизацией плазменной струи, осуществляемой путем ввода в по-
ток плазмы на выходе плазмотрона высоконапорной струи аргона. На рис. 4.3
приведены зависимости средней по сечению реактора степени конверсии
F закиси азота от длины реактора-смесителя. Величина F определялась в
211
нормальных сечениях реактора по формуле
F (z) = 1 — [Гм2о (z)/rN2o (оо)],
где
R R
Гя2о = Ги2о (z/r) г dr I г dr,
(4.11)
(4.11а)
Гтед (z, г) — осредненная по времени концентрация N2O в пробе газа,
отобранной зондом в точке (z, г) цилиндрического реактора; R — радиус ка-
нала реактора; rNeo (°°) — концентрация N2O в расчете на полную смесь
Рис. 4.2. Схемы конструкций плазмохимических рёакторов, в которых исследовался про-
цесс турбулентного перемешивания потока плазмы со струями холодных газов
а — реактор со спутным вводом холодного газа; б — реактор со спутным вводом холодного газа и
предварительной турбулизацией потока плазмы; в — реактор с радиальным вводом струй холодного
газа
газов (т. е. концентрация N2O после завершения перемешивания в отсутст-
вие химической реакции). В условиях радиального ввода холодного газа-
реагента в поток плазмы в начальных сечениях смесителя (до z ~ 2D, где
D = 2R) имеет место резкая несимметричность профилей концентрации
Рис. 4.3. Зависимости от времени и расстояния вдоль оси реактора степени превращения
закиси азота в быстрой реакции ее термического разложения, происходящей в процессе
перемешивания струй реагента с потоком плазмы в канале плазмохимического реактора
1 __ радиальный ввод реагента через одно или три отверстия диаметром 0,3—1,5 мм; 2 — спутный
ввод реагента; 3 — спутный ввод с предварительной турбулизацией потока плазмы
Tn2o (z, г). Вследствие этого приходилось измерять rN2o (z, г) в N (до 8)
азимутальных направлениях в каждом начальном сечении канала смесителя
и определять величину
N R R
Fn2o (z) = 2 Fn2o(z, г, <pi)rdr / rdr. (4.116)
i=1 о о
Погрешность определения степени конверсии в начальных сечениях была
поэтому существенно выше, чем в последующих (при z>;2D). Этим объяс-
няется значительный разброс точек на графиках зависимости F (z) для сме-
212
сителя с радиальным вводом реагента (при z < 2/)) и спутным вводом без
предварительной' турбулизации.
Кривые F (2), представленные на рис. 4.3 выражают зависимости от z
доли реагента, перемешавшегося с плазмой до молекулярных масштабов к
данному сечению z канала смесителя. Следовательно, наилучшие условия сме-
шения реагента и плазмы обеспечивают реактор с радиальным вводом реа-
гента (см. рис. 4.3, кривая I), наихудшие (среди исследованных) — реактор
со спутным вводом реагента без предварительной турбулизации плазменного
потока (кривая 5). Реактор со спутным вводом реагента и предварительной
турбулизацией плазменного потока занимает промежуточное положение
между двумя остальными (кривая 2). Таким образом, метод быстрой хими-
ческой реакции позволяет показать, что изменение интенсивности турбулент-
ности в канале смесителя приводит к изменению скорости процесса переме-
шивания. Кроме того, этот метод дает возможность оценивать характерное
время процесса перемешивания до молекулярных масштабов в реакторах-
смесителях различных конфигураций. В частности, величина времени пол-
ного перемешивания для реактора с радиальным вводом реагента состав-
ляет ~ 50 жтссекпри диаметре канала реактора 3,5 жж и скорости потока сме-
си в канале ~ 300 м/сек. Таким образом, средняя скорость нагревания реа-
гента до температуры 3000° К составит в этом реакторе ~ 6-107 град/сек.
Очевидно, в реакторе с радиальным вводом можно получить и среднюю ско-
рость закалки (охлаждения) реагирующей смеси того же порядка при введе-
нии в реактор достаточного количества холодного газа.
В заключение отметим, что длина зоны полного перемешивания £Пм
холодного газа с плазмой до молекулярных масштабов значительно превы-
шает £п — длину зоны установления профиля концентрации вводимого газа
в канале смесителя (например, приблизительно в 2,3 раза в случае реактора
с радиальным вводом [89]). По-видимому, между величинами £п и £Пм долж-
на существовать определенная связь, характер которой должен определяться
особенностями турбулентности в исследуемых условиях. Существование та-
кой связи позволило бы пользоваться для определения величины £Пм ре-
зультатами более грубых и менее трудоемких измерений величины £п. Для
проверки этого предположения необходимо, очевидно, проведение соответ-
ствующих экспериментов в широком диапазоне изменения характеристик
турбулентных потоков.
ПЛАЗМОХИМИЧЕСКИЙ РЕАКТОР
На основании изложенного выше, представляется разумным рассматривать
плазмохимический реактор состоящим из двух частей: смесителя плазмы и
сырья и собственно реактора. Смеситель должен обеспечивать получение го-
могенной смеси реагентов на входе в реактор при некоторой температуре,
величина которой определяется исходя из кинетических и (или) термодина-
мических характеристик данного химического процесса. Одной из особен-
ностей плазмохимических процессов является то, что часть химических пре-
вращений происходит в процессе перемешивания сырья с плазмой. Наличие
этой особенности и диктует необходимость раздельного анализа и моделиро-
вания смесителя и реактора. На вход плазмохимического реактора поступает
из смесителя турбулентный поток гомогенно перемешанных реагента и плаз-
мы. В случае реактора цилиндрической формы характеристики такого реак-
тора будут близки к характеристикам идеального реактора вытеснения.
Прежде чем обсуждать возможные отклонения характеристик реального
плазмохимического реактора от характеристик идеального реактора и их
причины, представляется разумным напомнить об одном способе описания
потоков жидкости (газа) в химических реакторах, основанном на понятии о
функциях распределения жидкости (газа) по временам пребывания в объеме
реактора.
213
Функции распределения по времени пребывания. Рассмотрим поток жид-
кости (газа), проходящий через реактор в условиях неизменной плотности и
отсутствии химической реакции. Тогда среднее время пребывания жидкости
в объеме реактора составит [91]
т = V/G. (4.12)
где V — объем реактора, G — объемный расход жидкости. В дальнейшем но
мере необходимости будем пользоваться понятием безразмерного времени,
определяемого как
0 = Нт = GtIV. (4.13)
где t — натуральное время.
Различные элементы жидкости могут проходить через реактор различны-
ми путями, и, следовательно, периоды присутствия этих элементов в объеме
реактора будут неодинаковыми. Пусть каким-то способом удается зарегист-
рировать распределение I интервалов времени, в течение которых эти эле-
менты находятся в реакторе (считая от некоторого момента t = 0). Тогда
Idt составит долю элементов жидкости, для которых интервалы времени^при-
сутствия внутри реактора находятся в пределах от t до t + dt. Поскольку
сумма всех долей жидкости в реакторе равна единице, то
со
\ Idt--= i. (4.14)
о
Очевидно, доля содержащихся в реакторе элементов жидкости, для кото-
рых время присутствия меньше некоторого tr. составит^Idt\ доля находя-
о
щихся в реакторе элементов жидкости с временем присутствия больше
со 4,
будет § Idt = 1 — Idt. [Пусть теперь удалось зарегистрировать распреде-
41 О
ление Е интервалов времени, прошедших с момента попадания данного эле-
мента жидкости в объем реактора до момента появления этого же элемента
жидкости в потоке на выходе реактора. По аналогии с предыдущим Edt со-
ставит долю элементов жидкости, для которых время от момента их попадания
в объем реактора до появления в потоке на выходе реактора находится в диа-
пазоне от t до t + dt. и
J Edt = 1. (4.15)
о
Среднее время пребывания жидкости в реакторе составит, таким образом,
l = \tE(t)dt = V/G. (4.16)
О
Функция Е (t) представляет собой плотность вероятности пребывания жид-
кости в*1 реакторе и связана з функцией I (£) известным соотношением
[91, 92]*
<4Л7>
Для определения функций I (t) и Е (f) используют методы, основанные на
изучении отклика данной системы на возмущение. Возмущающее воздейст-
вие в рассматриваемом случае проточного реактора может заключаться во вве-
дении в поток какого-либо вещества (трассера), химически не реагирующего
с веществом потока. Сигналы возмущения могут в принципе иметь различ-
ную форму, однако чаще всего используют ступенчатое или импульсное воз-
214
мущение, поскольку это существенно упрощает анализ состояния исследуе-
мой системы. Действительно, можно показать [91, 92], что при внесении воз-
мущения в виде ступенчатой функции отклик F системы связан с функцией
распределения I жидкости по временам присутствия в объеме реактора весь-
ма простым соотношением
I - 1 — F, (4.18)
а при использовании возмущающего воздействия в виде импульса, длитель-
ность которого много меньше среднего времени пребывания жидкости в реак-
торе, исследование отклика С системы дает функцию Е распределения жид-
кости по времени пребывания, т. е. С = Е. (4.19)
На рис. 4.4 представлены функции распределения I и Е, а также кривые
отклика для реакторов различного типа при нанесении возмущений импульс-
ной и ступенчатой форм [91]. Видно, что функции распределения для некото-
рого реального реактора представляют собой нечто среднее между соответ-
ствующими характеристиками идеальных реакторов вытеснения и смешения.
При использовании безразмерного времени, определяемого в соответствии
с выражением (4.13), следует учитывать, что I (0) cl (£), Е (0) = хЕ (t)
со со
и Z?(0)d0= 1, /(0)d0= 1. Идеальным реактором вытеснения здесь на-
о о
зывается реактор, в котором в любом поперечном сечении, нормальном к на-
правлению движения потока жидкости, скорость и свойства жидкости (давле-
ние, температура и состав) распределены равномерно, а продольные диффу-
зионные потоки отсутствуют. Следовательно, в реакторе идеального вытес-
нения все элементы жидкости прохо-
дят через реактор за одинаковое вре-
мя, и последовательность изменения
давления, температуры и концентра-
ции в элементах одинакова для всех
элементов. Поскольку к тому же ?
диффузия реагентов и продуктов ре-
акции из одного «поперечного» эле-
мента в другой отсутствует, степень
превращения в каждом элементе оста-
ется одной и той же. Это позволяет f
рассматривать каждый такой элемент /
как реактор периодического действия
с длительностью цикла, равной вре-
мени прохождения элемента через
реактор. Реактор идеального переме-^=г
юшвания является по смыслу край-
ней противоположностью реактора
идеального вытеснения и определяет-
ся как реактор, в котором поступаю-
щая в него жидкость мгновенно пере-
мешивается с содержимым объема//''/
реактора, так что состав смеси в лю- г
бой точке объема реактора одинаков.
Как видно из рис. 4.4, функции рас-
пределения по времени пребывания в
Рис. 4.4. Функции [распределения времени^^
пребывания и кривые отклика двух идеаль-
ных реакторов ирреального реактора
1 — реактор с потоком ^идеального вытеснения;
2 — реактор* с потоком идеального'. перемешига-
ния; 3 — реальный ^реактор; 4 — максимальная
Производная; 5 — максимальная величина
215
идеальных реакторах вытеснения и перемешивания представляют собой соот-
ветственно 6-функцию и экспоненту e~Q.
Если ^-функция распределения для данного реактора определена, то при
известной кинетике изучаемой химической реакции степень превращения
Х-реагента в реакторе в случае реакций первого порядка определяется выра-
жением [91]
_ со
X = 1 — е~^Е (0 dt, (4.20)
о
где Г — средняя концентрация реагента на выходе реактора, Го — исходная
концентрация реагента, к — константа скорости реакции. Для реакций с
нелинейными кинетическими уравнениями знания Е (/) оказывается недоста-
точно для расчета степени превращения исходного реагента в реакторе.
Однако по уравнению (4.20) можно в этом случае оценить верхнюю или ниж-
нюю границу степени превращения соответственно для реакций с порядком
больше и меньше единицы [91].
Для предварительной оценки принадлежности данного химического реак-
тора к классу аппаратов идеального смешения или вытеснения следует со-
поставить скорости массообмена со скоростью химической реакции. Для
этого можно воспользоваться, например, «критерием перемешивания» Кпг
предложенным в [93]:
^п = (^,/Рэф)(Ж/Г0), (4.21)
где £р — длина реактора, — эффективный коэффициент массообмена,
W — скорость химической реакции, Го — исходная концентрация реагента.
При К к 1 режим потока в реакторе близок к режиму идеального реак-
тора перемешивания и при Кп 1 — к режиму идеального реактора вы-
теснения. Оценим величину критерия Ки для той части плазмохими-
ческого реактора, которая выше была названа собственно реактором, на
примере лабораторной установки пиролиза природного газа в струе водород-
ной плазмы [94]; для этого плазмохимического реактора Лр ж 10d = 6 см,
характерное время химической реакции (Го/И7) ~ 1-10~4 сек (при темпера-
туре реакции ~ 2000° К) и эффективный коэффициент турбулентной диф-
фузии в рассматриваемых условиях DQ$ ~ 103 смЧсек (исходя из результа-
тов измерения времени перемешивания до молекулярных масштабов, кото-
рые приводились выше).
Таким образом, критерий перемешивания для данного плазмохимического
реактора составит Кп 4 • 102 1.
Следовательно, можно ожидать, что характеристики расположенного
непосредственно за смесителем собственно реактора близки к характеристи-
кам идеального реактора вытеснения.
Характеристика реального реактора. Различие указанных характерис-
тик может быть обусловлено следующими причинами. Прежде всего, исполь-
зование реактора с интенсивно охлаждаемыми стенками приведет к тому, что
появятся радиальные градиенты температуры и связанные с ними радиаль-
ные градиенты скорости потока. Снижение степени превращения, обуслов-
ленное понижением температуры реагирующей смеси вблизи стенок реак-
тора, лишь в весьма незначительной степени скомпенсируется увеличением
времени пребывания более холодного газа в реакторе, обусловленным сни-
жением скорости потока вблизи охлаждаемых стенок. Для того чтобы в этих
условиях обеспечить расчетную степень превращения, необходимо увеличи-
вать длину реактора. Но это приведет к существенному снижению селектив-
ности процесса, целевой продукт которого является промежуточным в цепи
химических превращений, имеющих место в реакторе, поскольку скорость
превращения в высокотемпературном ядре потока остается весьма высокой.
Следовательно, плазмохимический реактор, предназначенный для про-
ведения процесса, целевой продукт которого является промежуточным.
216
должен иметь теплоизоляцию, позволяющую поддерживать температуру стен-
ки максимально близкой к температуре проведения реакции (естественно,
насколько позволяет материал, из которого реактор изготовлен).
Второй причиной, приводящей к отличию характеристик реального плаз-
мохимического реактора от характеристик идеального реактора вытеснения,
является влияние турбулентной диффузии в направлении вдоль оси реакто-
ра (продольная диффузия). Влияние продольной диффузии тем сильнее, чем
больше величина D^/vL^ = Ре"1, где Ре — критерий Пекле. Для реактора
упомянутой выше лабораторной установки пиролиза природного газа в плаз-
менной струе водорода имеем D^/vLv 3-10~3 (при средней скорости по-
тока ~ 500 м/сек). Таким образом, влиянием этого эффекта на функцию рас-
пределения времени пребывания газа в реакторе можно пренебречь. В тех
случаях, когда указанный параметр достаточно велик, для расчета харак-
теристик реактора можно воспользоваться результатами Левеншпиля [91].
Диффузия в радиальном направлении является эффектом положительным,
способствуя выравниванию радиальных распределений параметров потока
смеси и приближению характеристик реактора к характеристикам идеаль-
ного реактора вытеснения [95].
Наконец, еще одним эффектом, приводящим к появлению различий меж-
ду характеристиками реального и идеального реакторов вытеснения, являет-
ся наличие радиального профиля скорости потока, связанного с режимом те-
чения реагирующей смеси в канале реактора. При турбулентном режиме те-
чения профиль скорости потока в канале определяется выражением
г(г) = р(0)(1 (4.22)
где В — радиус канала реактора, и показатель степени т равен 5 для шеро-
ховатых стенок, 7 для гладких стенок при 2300 Re 105 и 8 при Re >
105. Если рассчитать время пребывания т (0) газа в реакторе, пользуясь
осевым значением скорости, т. е. т (0) = L^/v (0), то среднее время пребыва-
ния т газа в реакторе составит [96, 97]!
т ........ " " 8
г...........• 1,32т(0) 1,22т(0) 1,19т(0)
Интересно отметить, что для ламинарного режима течения т = 2т (0);
следовательно, необходимо отдать предпочтение турбулентному режиму те-
чения.
Эти данные позволяют уточнить объем реактора, рассчитанного на задан-
ное время пребывания в нем реагирующей смеси.
Способы учета влияния перечисленных выше эффектов на степень превра-
щения реагента в реальных реакторах вытеснения рассматриваются кроме
[91, 97] в книге [98].
Моделирование химических реакторов. Задачей моделирования является
нахождение условий, позволяющих рассчитывать аппарат заданного мас-
штаба исходя из результатов, полученных на аппарате другого масштаба,
например рассчитывать промышленный химический реактор на основе ре-
зультатов, полученных на модельной лабораторной (пилотной) установке.
При физическом моделировании пользуются критериями геометрического
и физического подобия, получаемыми либо из дифференциальных уравнений,
описывающих рассматриваемый процесс, либо на основании анализа размер-
ностей величин, определяющих протекание процесса. С целью выведения
критериев подобия для химических процессов формально приводят члены ос-
новных дифференциальных уравнений, описывающих сохранение материи
и энергии с учетом химических превращений, к безразмерным комплексам.
Это позволяет получить четыре критерия Дамкелера (Даг, Дан, Даш и
Дару), а для обратимых реакций — критерии контакта (Ко) и равновесности
(Ра) [92, 99]. По виду критериев химического подобия можно заключить, что
одновременно они все выполняться не могут, поскольку линейный размер I
217
входит в них в различных степенях. Эти критерии оказываются также несов-
местимыми с критериями физического подобия (например, с критерием Рей-
нольдса).
Следовательно, прямое моделирование химических процессов с использо-
ванием критериальных уравнений оказывается практически невозможным,
если требовать полного подобия модели и натуры — гидродинамического,
теплового, диффузионного, геометрического и реакционно-кинетического.
Моделирование химических реакторов в случаях частичного подобия подроб-
но рассмотрено в [99]. В частности, при рассмотрении гомогенной реакции в
трубчатом реакторе, коэффициент теплоотдачи в котором зависит от Re0’8,
при отказе от геометрического и гидродинамического подобия и соблюдения
теплового, диффузионного и реакционно-кинетического подобия отношения
размеров натуры и модели и скоростей потока в них составят [92, 99]
d2/dx = п0’2?6, L2/Lx =n0’428, v2/vx = г№\ (4.23)
где d, L — диаметр и длина реактора, п — коэффициент увеличения произ-
водительности реактора.
Таким образом, в настоящее время можно осуществлять лишь прибли-
женное моделирование химического реактора по критериальным уравнениям.
Прогресса в этом направлении следует, по-видимому, ожидать по мере раз-
вития методов математического моделирования физико-химических процес-
сов [92, 100-102].
Приближенное моделирование плазмохимического реактора. Рассмотрим
цилиндрический плазмохимический реактор. Формально его можно разде-
лить на смеситель плазмы и реагентов и собственно реактор. Характеристи-
кой смесителя является кривая распределения времени перемешивания (на-
гревания), получаемая, например, с помощью метода быстрой химической
реакции (примеры таких кривых представлены на рис. 4.3). При переходе к
реактору другого масштаба следует, очевидно, добиваться идентичности
распределений времени перемешивания в том и другом реакторе. Поскольку
скорость перемешивания определяется в основном характеристиками полей
турбулентности и температуры в смесителе, следует добиваться идентичности
указанных полей в модели и натуре.
Характеристики турбулентности в струях вводимого в модельный реактор
реагента легко воспроизвести и в натурном реакторе; характеристики же
турбулентности и поля температур плазменной струи могут сильно варьи-
роваться в зависимости от типов плазмотронов, используемых в модели и
натуре. Поэтому представляется необходимым производить тем или иным
способом предварительную турбулизацию плазменной струи, обеспечиваю-
щую получение более однородных полей турбулентности и температуры.
С этой точки зрения весьма перспективными представляются генераторы плаз-
мы, использующие многодуговые камеры смешения (см. стр. 8).
В условиях идентичности полей турбулентности и температуры в смеси-
телях модели и натуры можно предполагать, что длины £ПМ1 и £ПМ2 зон пол-
ного перемешивания (до молекулярных масштабов) будут связаны с диамет-
рами dx и d2 реакторов-смесителей соотношениями ЛПМ1 = Adj и £Пм2 =
-- Ad2, установленными на основании исследований процесса перемешивания
на модельной установке (см. стр. 212) для конкретной конфигурации смеси-
теля. Тогда среднее время перемешивания в модели и натуре составит соот-
ветственно тпм’ = LmsJi\ и тПм2 = где v± и v2 — средние скорости
движения смеси в канале реакторов (средние скорости потоков плазмы в мо-
дели и натуре при радиальном вводе струй реагента). Из условия сохранения
степени превращения реагента в смесителе натуры на уровне, достигаемом в
смесителе модельного реактора, следует, что тПМ1 — тПМ2 и поэтому
dx!vx = d2/z,2.! (4.24)
Пусть производительность натурного реактора выше в п раз производи-
тельности модельного реактора, тогда объемные расходы в модели и натуре
218
связаны соотношением пр1г;151 = р2^2» гДе Pi, и Рз, — соответственно
средняя плотность смеси и площадь сечения канала реактора в модели и на-
туре. Поскольку выше было поставлено условие идентичности полей тем-
ператур в смесителях модели и натуры, можно полагать, что средние плот-
ности в том и другом случае также будут одинаковыми, если давление в реак-
торах одинаково fa = р2).
Таким образом, получаем
nv^ = (4.25)
Из (4.24) и (4.25) находим следующие соотношения для расчета диаметра
натурного реактора-смесителя и скорости потока в нем:
d2 - (4.26)
v2 = VjjiVs. | (4.27)
Из смесителя на вход собственно реактора поступает гомогенная смесь
реагентов при 7Р, задаваемой кинетическими особенностями исследуемой
реакции. Для точного соответствия условий проведения процесса в модели и
натуре следует потребовать идентичности соответствующих распределений
времени пребывания смеси в реакторах модели и натуры. Выше было показа-
но, что при достаточной теплоизоляции плазмохимического реактора режим
потока в нем близок к режиму идеального реактора вытеснения. Поэтому для
приближенного моделирования можно потребовать равенства средних вре-
мен пребывания смеси в реакторе: тР1 = тР2. Отсюда следует, что
— ^рг/^25 (4.28)
и с учетом (4.27)
£Р2 - W/3« (4.29)
Приближенные условия подобия плазмохимических [реакторов
Параметр Условия подобия процессов в модели (индекс 1) и натуре (2)
Конфигурация смесителя Конфигурация смесителя — idem
Конфигурация реактора^— цилиндрическая тепло- изолированная труба Конфигурация реактора — idem
Производительность установки Q Q2 = nQi
Среднемассовая температура плазменного потока Тсм Т =Т СМ2 CM1
Давление в реакторе р Р2 —
Среднемассовая скорость плазменного потока г?см yCM2^
Среднее время перемешивания тпм ^ПМ2 ТПМ1
Диаметр смесителя d dz — din к
Длина смесителя £пм ^ПМ2 ' ^ПМ1П
Диаметр реактора с?р d = d ₽2 Р1
Температура проведения реакции 7р Т ==Т Р2 Р1
Среднее время контакта тр ТР2 = ТР1
Длина реактора Lp rn = Ln п/з Рг Pi
Расход плазмообразующего газа б?пл ^ПЛ2=/г^ПЛ1
Расход сырья G G2 = nGi
Температура внутренней поверхности [канала реак- тора Ля л ст2 л СТ1 р
Качество обработки внутренней поверхности канала реактора v \72 = V1
219
В таблице перечислены выведенные здесь приближенные условия подобия
плазмохимических реакторов. В качестве примера рассмотрим процесс полу-
чения ацетилена из метана в плазменной струе водорода.
Пусть лабораторная установка характеризуется следующими парамет-
рами: полезная мощность ~ 20 кВт, производительность по ацетилену
Q (СзЩ)! = 20 m/год, среднемассовая скорость потока плазмы г2СМ1 »
500 м/сек, конфигурация реактора (включая смеситель) — цилиндрическая
труба круглого сечения диаметром dL —- 0,6 см, конфигурация смесителя —
набор радиальных отверстий для ввода метана, расположенных в одном
сечении, нормальном оси реактора, среднемассовая температура водорода
T'cMi ~ 4000° К, давление в реакторе 1 ата, температура проведения
реакции 7Р1 ~ 2000° К, полная длина реактора (включая смеситель) состав-
ляет 10 см, расходы водорода и метана равны соответственно 0,1 и 1,8 г/сек.
Тогда некоторые параметры пилотной установки производительностью 300 т
ацетилена в год и полезной мощностью 300 кВт составят (п = 15): диаметр
реактора d2 « 1,5 см, общая длина Ь2 ж 25 см, скорость потока плазмы
v2 1200 м/сек, расход водорода 1,5 г/сек, метана — 27 г/сек. Остальные па-
раметры процесса должны в соответствии с таблицей сохраняться такими же,
как и в лабораторных условиях. Более производительные установки должны,
очевидно, рассчитываться исходя из результатов испытаний пилотной уста-
новки. Однако приблизительную оценку полупромышленного реактора про-
изводительностью 2000 т ацетилена в год (п — 102) можно сделать исходя из
данных лабораторных испытаний. В этом случае получаем: d2 ~ 2,8 см,
L2 х 45 см, v2 ж 2300 м/сек при полезной мощности установки ~ 2 МВт.
5
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
(ОРГАНИЧЕСКИЕ И НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ,
МАТЕРИАЛЫ)
Одним из важных промышленных применений низкотемпературной плаз-
мы является прикладная плазмохимия, охватывающая широкий круг про-
цессов, представляющих значительный интерес для различных областей
народного хозяйства: химической, металлургической, электронной, радио-
технической, электротехнической промышленности, промышленности стро-
ительных материалов и т. д.
Проведение химических процессов при температурах выше 1000—2000° К
(высоких с точки зрения физика-химика, химика-технолога) было вызвано
рядом обстоятельств, в частности тем, что:
а) для многих процессов, например типа получения ацетилена из угле-
водородов в органической химии или тугоплавких соединений в неоргани-
ческой, равновесие сдвинуто в сторону продуктов при высоких температурах;
б) с повышением температуры резко возрастают скорости реакций.
Появившиеся в основном в последние 20 лет источники концентрированной
энергии при высоких температурах — низкотемпературной плазмы, термоди-
намические и кинетические данные для многих соединений, эксперименталь-
ные и расчетные методики исследования химических реакций при высоких
температурах позволили осуществить химические реакции при температурах
1000—10 000° К, давлениях от 10-4 до 102 атм при временах контакта от
10-6 сек до нескольких секунд в равновесных и неравновесных условиях.
Для описания высокотемпературных химических процессов, явившихся раз-
витием классических химических процессов, используются методы, приме-
няемые в классической химии,— равновесной термодинамики и классиче-
ской химической кинетики. Применение этих методов, а также интенсивное
изучение проблемы закалки продуктов значительно ускорили исследование
и описание высокотемпературных химических процессов в низкотемператур-
ной плазме — равновесных и квазиравновесных плазмохимических процессов,
протекающих при температурах в несколько тысяч градусов и давлениях,
близких к атмосферному, и разработку научных основ технологии их. В ре-
зультате в настоящее время многие важные плазмохимические процессы
этого типа находятся на стадиях опытно-промышленной, промышленной про-
верки и промышленного внедрения.
Однако с повышением температуры физическая обстановка в низкотем-
пературной плазме изменяется, скорости реакций возрастают и могут стать
соизмеримыми со скоростями смешения, диффузии и даже установления тер-
мического равновесия. Кроме того, стационарная газоразрядная плазма при
пониженных давлениях и импульсная при нормальных часто существенно
неравновесны. Химические реакции, протекающие в ней, также неравновес-
ны. В этих условиях заселение квантовых уровней, химические реакции, ре-
лаксационные процессы, процессы смешения протекают и проявляются одно-
временно. Естественно, что классическая химическая кинетика уже не в
221
состоянии описать эту ситуацию. Теория химических процессов, протекающих
в неравновесных условиях, только создается [1,247]. Для описания их необ-
ходима огромная информация, включающая детальное строение молекулы,
сечения реакций, функции распределения реагирующих и образующихся
частиц и т. д. Эти вопросы изложены в главах 2 и 3.
Низкотемпературная плазма может быть использована как высокоэнталь- ’
пийный источник энергии, источник положительных и отрицательных ионов
для ионно-молекулярных реакций, мощный источник светового излучения для
фотохимических реакций. Низкотемпературная плазма может генерироваться
в генераторах плазмы с дугой высокой интенсивности, плазмотронах постоян-
ного тока, переменного тока промышленной частоты, высокочастотных и
сверхвысокочастотных, а также в тлеющем и коронном разрядах, установках
адиабатического сжатия, ударных трубах, с помощью мощных лазеров (см.
гл. 1).
Процессы в низкотемпературной плазме особенно перспективны для про-
мышленной реализации в следующих случаях [2,230]:
равновесие смещено в сторону высоких температур;
скорости резко возрастают с повышением температуры (времена контакта
~ Ю-3—10“5 сек), что обусловливает резкую миниатюризацию техники;
высокие выходы достигаются в существенно неравновесных условиях;
используется широкодоступное малоценное неустойчивое по составу
сырье (например, в плазмохимическом пиролизе природного газа примеси к
метану до 20—25% не влияют на выход целевых продуктов).
Значительные перспективы имеются в области получения чистых и высо-
кочистых, например полупроводниковых, материалов, так как в плазмохи-
мических процессах в ВЧ- и СВЧ-плазме чистота продуктов определяется
только чистотой исходного сырья и даже может быть повышена в ходе про-
цесса.
Большинство представляющих практический интерес плазмохимических
процессов с технологической точки зрения одностадийно, процессы хорошо
моделируются, оптимизируются и управляются. Управление низкотемпе-
ратурной плазмой может быть осуществлено газо- и электродинамическими
методами, применение которых позволяет уменьшить требования к конструк-
ционным материалам плазмотрона и реактора.
Важным достоинством плазмохимических процессов является общность
их технологической организации. Дело в том, что аппарат для осуществления
плазмохимического процесса состоит обычно из генератора низкотемператур-
ной плазмы (плазмотрона), собственно реактора и закалочного устройства.
В некоторых случаях плазмотрон и реактор совмещены.
□ПИСАНИЕ
ПЛАЗМОХИМИЧЕСКИХ ПРОЦЕССОВ
Протекание и описание плазмохимических процессов определяется усло-
виями, реализующимися в плазме. Низкотемпературная плазма может нахо-
диться в состоянии локального термодинамического равновесия (ЛТР),
и химические процессы в ней могут быть равновесными или квазиравновес-
ными; низкотемпературная плазма может быть и в неравновесном состоянии.
Химическая реакция, являющаяся в принципе неравновесным процес-
сом, скорость которой мала относительно скорости установления ЛТР,
не нарушает ЛТР, и для ее описания можно использовать методы равновес-
ной термодинамики и классической химической кинетики. Последняя пред-
полагает, кроме наличия ЛТР во все время протекания реакции, закончив-
шимися к ее началу процессы смешения, диффузии, малую плотность актив-
ных частиц, преобладающую заселенность основного колебательного уровня
первого электронного состояния, малый вклад реакций с участием возбу-
жденных и заряженных частиц, решительное преобладание упругих столкно-
222
вений над неупругими, среднюю энергию относительного движения сталки-
вающихся частиц не более 0,1 эВ [1, 3].
Однако с изменением условий (температуры и давления) некоторые из
перечисленных выше постулатов, четко сформулированных в [1, 3], не вы-
полняются. С повышением температуры даже при наличии ЛТР возрастает
концентрация возбужденных частиц, и роль реакций с их участием в меха-
низме и кинетике возрастает. Сближаются характерные времена их релакса-
ции и химических реакций. Кроме того, увеличивающиеся скорости хими-
ческих реакций становятся сравнимыми со скоростями процессов смешения.
Если же энергия активации химической реакции £а становится сравнимой со
средней энергией кТ тяжелых частиц (Е^/кТ ^5), то она нарушает равно-
весие, даже если оно и было в начальный момент времени; возникшая нерав-
новесность в свою очередь меняет скорость химической реакции.
С понижением давления до — 0,1 атпм газоразрядная плазма становится:
неравновесной в связи с уменьшением частоты столкновений и затруднен-
ностью обмена энергией между электронами и тяжелыми частицами из-за
соотношения масс. Эта неравновесность может проявляться в значительном
превышении средней энергии электронов над средней энергией тяжелых
частиц, в неравновесной функции распределения электронов, в значительно
превышающей равновесную (отнесенную к температуре газа) степень иони-
зации. Эти факторы приводят к большим концентрациям возбужденных час-
тиц в связи с относительно большими сечениями процессов возбуждения под.
действием электронного удара [1, 20, 231].
Таким образом, физическая обстановка, в которой протекают неравновес-
ные плазмохимические реакции и те квазиравновесные, когда часть постула-
тов химической кинетики не выполняется, существенно отличается от усло-
вий классической химии. Поэтому эти плазмохимические реакции необходимо
описывать с помощью разрабатываемой в настоящее время обобщенной (не-
равновесной) химической кинетики [1], излагаемой в гл. 2.
КВАЗИРАВНОВЕСНЫЕ ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
К процессам этого типа относятся такие высокотемпературные процессы,
как пиролиз углеводородов, хлоруглеводородов, фторуглеводородов в орга-
нической химии, получения окислов азота, восстановления элементов из руд,
окислов, хлоридов, получения тугоплавких соединений (карбидов, нитридов
окислов) в неорганической химии. Эти процессы проводят обычно при тем-
пературах 1000—5000° К, давлениях, близких к атмосферному.
Конечный результат и характер химических превращений, происходящих
при температурах порядка нескольких тысяч градусов, в значительной сте-
пени определяются термодинамическими свойствами веществ, участвующих
в той или иной из ее стадий. Но прежде чем перейти в равновесное состояние,
определяемое термодинамикой, система проходит через ряд промежуточных
состояний, скорость прохождения которых описывается кинетикой процес-
са: физической — для установления равновесного распределения энергии
по степеням свободы и химической — для установления равновесного хими-
ческого состава. Плазмохимические реакции в общем случае характеризу-
ются сильным взаимным влиянием факторов физической и химической кине-
тики, и скорости их могут сильно зависеть от скоростей таких физических
процессов, как молекулярная диффузия, турбулентный перенос, а также ма-
кроскопическое перемешивание реагентов.
Таким образом, исследование квазиравновесного плазмохимического
процесса предполагает в общем случае изучение элементарных актов соуда-
рений, термодинамики, физической и химической кинетики процесса, вопро-
сов газодинамики перемешивающихся потоков реагентов с учетом взаимо-
осложняющих воздействий всех этих факторов друг на друга. Кроме того
необходимо специально исследовать процесс закалки продуктов реакции,
имеющей решающее значение для большинства плазмохимических процессов
223
этого типа. Сложность такой задачи ясна. Поэтому правомерно принять
некоторое физически осмысленное упрощение отдельных сторон вопроса,
разграничение отдельных факторов и их влияний.
Термодинамика плазмохимических процессов. Для пояснения термоди-
намического подхода к равновесным плазмохимическим процессам рассмо-
трим следующие уравнения!
AG - АЯ - TAS, (5.1)
AG = - ВТ In К. (5.2)
Условие равновесия: AG — 0, где А(? — изменение изобарного потенциа-
ла; АЯ — изменение энтальпии; AS — изменение энтропии; К — констан-
та равновесия.
Легко видеть, что с повышением температуры возрастает роль энтропий-
ного фактора. Так как для многих реакций изменения АЯ и AS с температу-
рой невелики по сравнению с Т, то TAS и AG можно приближенно считать
линейными функциями Т, a In Я — линейной функцией ИТ, Поэтому при
высоких температурах и постоянном давлении наиболее существенными дол-
жны быть процессы диссоциации и разложения, что связано с возрастанием
роли энтропийного фактора.
Однако действительная картина сложнее. Наряду с процессами диссо-
циации сложных веществ, резким повышением реакционной способности и
возрастанием скоростей реакций осуществляются процессы, приводящие к
образованию соединений, не существующих при обычных температурах, а
также новых видов частиц (например, CaCl, А12О, АЮ, SO, С3, С9, Na2, Ва2О3
и др.). Тем не менее термодинамические расчеты дают большую и полезную
информацию о плазмохимических процессах.
Располагая надежными термодинамическими константами, можно, а в
большинстве случаев и необходимо, оценить оптимальные условия проведе-
ния реакций: диапазоны температур, давлений, соотношений реагентов,
величины ожидаемых выходов продуктов, как целевых, так и побочных, а
также энергетические показатели процесса.
Из анализа результатов сопоставления экспериментальных и рассчитанных
термодинамически равновесных и квазиравновесных составов можно выяс-
нить следующее: 1) реализуется ли в условиях экспериментов равновесие
или квазиравновесие; 2) оптимален ли закон закалки, имевший место в экс-
периментах; 3) какова роль в полученных экспериментальных выходах
продуктов тех или иных радикалов и промежуточных соединений, реагирую-
щих в процессе закалки.
Как показывают результаты экспериментов, газофазные плазмохимиче-
ские реакции протекают за время ~ 10~3—10~4 сек. Это время может быть
недостаточным для установления химического равновесия для всех реакций.
В частности, за это время может не успеть произойти образование конден-
сированной фазы, поскольку скорости газофазных реакций значительно вы-
ше скорости образования конденсированной фазы [4]. Кроме того, возможно,
не успеют образоваться многие сложные соединения, требующие тройных
столкновений каких-либо промежуточных соединений (например, бензола
при пиролизе углеводородов). Поэтому для описания газофазных плазмохими-
ческих процессов часто более правильным является рассмотрение не истинно
равновесного состава, а состава квазиравновесного, не учитывающего обра-
зования конденсированной фазы и каких-то (конкретных для данного про-
цесса) соединений. «
Методика расчета равновесного состава. Расчеты равновесных составов
заключаются в решении системы уравнений, состоящей из:
1) I уравнений вида
= KPj, (5.3)
224
где i — рассматриваемые соединения; Ра, Ръ, Рс, Pj — парциальные дав-
ления, ата, атомарных компонент и /-го компонента состава ambnct соот-
ветственно; Кр. — константа равновесия данного вещества с атомарными
продуктами разложения;
2) уравнения полного давления
Ра + РЪ+Ре+2 Р1 = Р'' (5-4)
3) уравнений элементарного баланса
л,+2"‘Л-
-----------= а, ------------= 3. (5.5)
.7==1
Если при рассматриваемых температурах возможно существование одного
из атомарных компонент в конденсированной фазе, то последнее уравнение
заменяется условием существования этой фазы (Р с индексами а, Ъ, с равно
равновесному давлению атомарного вещества над конденсированной фазой).
В последнем случае элементарный состав в газовой фазе не сохраняется, и
по его изменению можно определить количество твердого вещества. Метод
численного решения подобных систем уравнений предложен в работах [5, 6].
Кинетика плазмохимических процессов. Кинетические расчеты позволяют
определить временные, а для условий процессов в плазменной струе — прост-
ранственно-временные характеристики, включая режим закалки их продук-
тов (момент начала ее и закон снижения температуры). Процессы в плазмен-
ной струе описываются системой уравнений химической кинетики и гидро-
динамики [7, 8]
l(pV) = O, (5.6)
А(И.У) = ИЛ. (5.7)
ру^-+4^=°’ (5-8>
ру_^ + 1^0. + ру2^. = 0, (5.9)
r dz 1 dz 1 k dz \ /
P = nkT. (5.10)
Здесь p — плотность, V — скорость газового потока, nt — число молекул
Z-ro вида в единице объема газа. Р — давление, U — внутренняя энергия,
Т — температура, — скорости рассматриваемых химических реакций,
к — постоянная Больцмана.
Граничные условия задаются при z = 0: = ni0, Т = ТР = Р0,
V = Vo, р = р0. Обычно эти уравнения решаются в предположении мгно-
венного перемешивания и нагрева реагентов и изоэнтальпийности процесса,
т. е. они описывают ситуацию, когда характерное время перемешивания тп
намного меньше характерного времени химической реакции тх. Для ряда
химических процессов такая ситуация реализуется при проведении их в
лабораторных установках с реакторами, диаметры которых до 5—6 мм.
В работах [9, 10] сделаны попытки выяснить влияние процесса перемешива-
ния на кинетические зависимости. Для этого задавалась зависимость поступ-
ления реагента в плазменную струю от длины реактора z, который затем
«мгновенно» нагревался. При расчете стадии закалки задаются изменением
энтальпии или температуры во времени [7, 8]. Однако кинетические расчеты
выполнены для относительно небольшого числа плазмохимических процес-
8 Теоретич. и прикладная плазмохимия
225
сов: для процессов пиролиза и окисления углеводородов [7, 8], для образо-
вания окиси азота [7] и т. д. Дело в том, что для выполнения расчетов необ-
ходима информация о механизме химических реакций и кинетических кон-
стантах их при высоких температурах, однако такой информации в настоящее
время явно недостаточно. Ситуация в большинстве случаев подобна рассма-
триваемой (см. стр. 233). Возможно, это связано с тем, что для обеспечения
малых конверсий необходимо изучать многие реакции в интервале времен
контакта ~ 10~5 — 10-6 сек, что трудно технически осуществимо. Кроме
того, скорости закалки продуктов должны быть огромны. Например, если изу-
чать пиролиз углеводородов при ~ 3000° К при времени ~10“6 сек, скорость
закалки должна быть не менее 109—1010 град/сек, что в настоящее время
практически недостижимо.
Анализ результатов расчетов кинетики плазмохимических процессов по-
казывает, что они йротекают не в изотермических, а в изоэнтальпийных ус-
ловиях; максимальные концентрации целевых продуктов образуются за время
~ 10-3 сек\ величины их согласуются с экспериментальными данными. По-
казано также, что гидродинамические характеристики потока не оказывают
существенного влияния на характер протекания реакций, а лишь растяги-
вают их в пространстве. Химические же реакции существенно влияют на
динамику движения струи.
Закалка продуктов плазмохимических процессов. Специальное теорети
ческое и экспериментальное исследование проблемы закалки является одним
из основных вопросов плазмохимической технологии. Существуют два типа
плазмохимических реакций, при которых состав продуктов зависит от режима
закалки. В случае, когда необходимо зафиксировать промежуточные про-
дукты химических превращений (например, ацетилен при пиролизе угле-
водородов), существенны скорость закалки и момент начала ее. Опоздание
с началом закалки приблизительно на 2*10-3 сек в плазмохимическом пиро-
лизе метана приводит к падению концентрации С2Н2 с 15,5 до 10 об.% [11R
Во втором случае, когда получаемое вещество — конечный продукт реак-
ции, протекающей при высокой температуре,— достаточно устойчиво при
комнатной температуре, необходимо охладить продукты так, чтобы они не
успели разложиться в промежуточном интервале температур. К этому типу
реакций относится, например, термическое образование окиси азота в воз-
духе. Здесь важно обеспечить необходимый режим закалки (dT/dt) (7) и
не начать ее слишком рано, когда равновесие еще не установилось. Естест-
венно, что режим закалки определяется кинетикой процесса.
Необходимая скорость закалки продуктов плазмохимических процессов
колеблется от 103 до 105 град/сек, причем обычно в момент начала она должна
быть максимальной, а затем по мере охлаждения продуктов может снижаться.
В качестве иллюстрации рассмотрим закон закалки нитрозных газов, пред-
ставленный на рис. 5.1, заимствованном нами из работы [12]. Скорость за-
калки достигает максимального значения при температуре на 200—300°
ниже температуры начала закалки и резко падает. Нарушение этого закона
в каком-либо интервале температур нельзя компенсировать увеличением
скорости закалки в другом.
Основные способы закалки. Наиболее распространенным способом закалки
является охлаждение в теплообменниках, например в водоохлаждаемой труб-
ке [12, 13]. Скорость закалки в теплообменнике зависит от конструкции, ре-
жима течения газа, температуры и физических свойств потока. На начальном;
участке теплообменника она может достигать — 107 град/сек, в среднем она
составляет — 10е град/сек. Методы расчета скорости закалки в теплообмен-
никах описаны, например, в [14, 15].
Широко применяется метод закалки продуктов плазмохимического про-
цесса затапливанием струями жидкости (воды или реагента) или газа. Про-
цесс взаимодействия струй воды с потоком горячего газа рассмотрен в [7,
12, 16]. Скорость охлаждения при Т — 3000° К, давлении — 10 ата, соот-
ношении расходов газа и воды 1 : 1 достигает — 108 град/сек. Те же скорости
226
закалки осуществимы и в реакторе с кипящим слоем [17]. Практический ин-
терес представляет использование в качестве закаливающего агента того или
иного реагента, приводящее к улучшению процесса и иногда к экономически
эффективному изменению технологии.
Охлаждение горячих газов можно проводить в сопле Лаваля. При началь-
ной температуре газа — 4000° К и диаметре сопла 1 мм скорость закалки до-
стигает ~ 108 град/сек. Однако скорость охлаждения уменьшается с повы-
шением температуры, в то время как она должна быть в ряде случаев макси-
мальной вблизи Т0. Кроме того, так как при торможении потока газа его
темйература принимает начальное значение, сопло Лаваля необходимо при-
менять совместно с другими методами, позволяющими отводить тепло. Оцен-
ки эффективности различных способов закалки продуктов плазмохимических
процессов, выполненные в работе [18], согласуются с приведенными выше.
Рис. 5.1. Предельные законы
охлаждения нитрозных газов
1 — Гнач ~ 3000°К, Р = 1 сипа;
2 — 3300° К, 10 anta;
3 — 3300° К, 20 ата;
4 — 3300° К, 50 ата;
5 — 3100° К, 20 ата
Заметим, что часто экономическая эффективность процесса может быть
•обеспечена лишь в случае утилизации отводимого при охлаждении реагирую-
щей системы тепла.
От режима и способа закалки зависят свойства получаемых конденсиро-
ванных продуктов плазмохимического процесса (см. стр. 231).
Перемешивание реагентов с плазменной струей. Характерное время пере-
мешивания тп зависит кроме других факторов также и от характерного раз-
мера устройства, в котором происходит смешение, и с ростом размера уста-
новки тп растет. Соотношение тп <5 тх может не выполняться. Кроме того,
существует значительное количество быстрых химических реакций, для ко-
торых справедливо тх тп. Описание общего случая протекания химиче-
ской реакции одновременно с перемешиванием турбулентных потоков в
неизотермических условиях чрезвычайно сложно. Это связано как с труд-
ностью описания механизма турбулентности, так и с необходимостью учета
пространственных неоднородностей полей температуры и концентрации ком-
понент [19, 20]. Эти вопросы рассматриваются в гл. 4.
НЕРАВНОВЕСНЫЕ ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
Неравновесные плазмохимические процессы могут протекать в газораз-
рядной стационарной плазме пониженного давления (тлеющий разряд постоян-
ного и переменного тока промышленной частотой, тихий, коронный и другие
типы разрядов, высокочастотный и сверхвысокочастотный электродные и
безэлектродные разряды), импульсной плазме при среднем и нормальном
давлениях, а также в плазме, образованной ударными волнами, быстрым
адиабатическим сжатием, и под действием излучения лазера.
227
8*
Физическая обстановка в неравновесной газоразрядной плазме, реализу-
ющейся при давлениях до десятков торр в стационарном режиме ее генерации
и до атмосферного в импульсном режиме генерации, обычно такова:
степень ионизации достигает 10~4—10-1;
средняя энергия электронов, составляющая 1—20 эв, более чем на поря-
док превышает среднюю энергию поступательного движения тяжелых час-
тиц (7Г до (2—3)-103° К);
концентрация возбужденных по внутренним степеням свободы тяжелых
частиц значительно превышает равновесные (отнесенные к температуре газа),
и соответственно температуры заселения Z3 значительно выше газовой (773 ~
~ (5—10)-103° К). В ряде случаев распределение заселенностей по уровням
может быть небольцманово;
могут реализоваться значительные концентрации радикалов.
В таких условиях интенсивно протекают неупругие процессы первого и
второго рода и химические реакции, которые являются также одним из видов
неупругого процесса. Причем в энергообмене эти процессы значительно пре-
обладают над упругими.
Неравновесность газоразрядной плазмы может быть обусловлена зна-
чительными напряженностями электрических полей и химическими реакциями.
Напряженность электрического поля, например в СВЧ-плазме, колеблется
в пределах 101—104 В 1см. Химические реакции могут как искажать функцию
распределения частиц по энергиям поступательного движения, «съедая»
высокоэнергетическую, запороговую часть [8], так и вызывать нарушение
больцмановой заселенности колебательных уровней.
Описание неравновесных плазмохимических реакций. Константы скорос-
тей химических реакций К с участием возбужденных частиц могут быть боль-
шими и зависеть от того, в каком квантовом состоянии находится частица.
Каждая частица в том или ином квантовом состоянии должна рассматриваться
как самостоятельный «сорт частиц». Переходы между различными кванто-
выми уровнями, меняющие концентрации возбужденных частиц, влияют на
скорость химической реакции, которая в свою очередь влияет на эти
переходы.
Естественно, что реакции с участием возбужденных частиц могут вносить
существенный вклад в механизм и кинетику химических реакций в неравно-
весной плазме [20]. Не меньший, а часто определяющий вклад в механизм и
кинетику химических процессов и процессов возбуждения внутренних сте-
пеней свободы тяжелых частиц вносят реакции под действием электронного
удара [20].
С другой стороны, концентрации и средние энергии заряженных частиц
(в основном электронов) определяют электродинамические параметры плаз-
мы, влияющие на распространение электромагнитных волн в ней и часто ее
создающие.
Коэффициенты скоростей химических реакций зависят от сечений их,
функций распределения реагирующих и образующихся частиц по энергиям
поступательного движения. Для реакций с участием электронов для описан-
ных выше условий они определяются соответствующими сечениями и видом
энергетического распределения электронов /е (е). Поскольку химические ре-
акции и процессы возбуждения внутренних степеней свободы тяжелых час-
тиц электронным ударом в большинстве своем пороговые, необходимо знать
/е (е) особенно детально за порогом. Последние могут быть найдены из реше-
ния системы уравнений, состоящей из кинетического уравнения для электро-
нов (например, уравнения Больцмана), уравнений химической кинетики
для всех реакций с их участием и в общем случае уравнений электродина-
мики.
Решение такой задачи необычайно сложно и часто из-за отсутствия мно-
гих данных (сечений реакций и т. д.) практически невозможно. Поэтому в
зависимости от задачи, стоящей перед исследователем, упрощают ту или иную
сторону вопроса.
228
В плазмохимии основное внимание уделяется реакциям, протекающим в
плазме. В связи с этим более детально описывают кинетику реакций, а урав-
нения электродинамики в разумных пределах упрощают.
Математическая модель неравновесных плазмохимических реакций в од-
ном из важных типов разрядов — СВЧ-разряде, рассмотренная нами в [21],
состоит из двух частей Ч
1. Уравнения кинетики гомогенных химических реакций
dNjdt=sssww (5.Н)
г 3 к
где Л t — концентрации любых из участвующих в реакциях частиц: элек-
тронов, ионов, возбужденных частиц различных* типов. Коэффициенты а^
и Рг7ь1 2 являются коэффициентами скоростей различных процессов, которые
могут иметь неаррениусовский вид и зависеть от напряженности электриче-
ского поля из-за влияния последней на вид функции распределения элек-
тронов.
2. Уравнения электродинамики Максвелла, из которых при выполнении
неравенств
| ldt)(dEldt) К | е' (д*Е!д?) |, (5.12)
| (d^'ldt*) Е К | 8' (д*Е1дР) |
получается известное стационарное волновое уравнение
ЬЕ (г) — grad div Е (г) + (со2/с2) г'Е (г) = 0. (5.13)
Здесь 8' — комплексная диэлектрическая проницаемость, которая для
изотропной плазмы выражается следующим образом:
8' = 8 — i (4л/со) б, (5.14)
где 8 — диэлектрическая проницаемость, б — проводимость плазмы. Вели-
чины 8 и б в основном определяются концентрацией электронов
8 = 1 — [4ле27Уе/ш (со2 + \’эф)], б = e27Vev9$/[m (со2 + ¥2Ф)], (5.15)
где е и т — заряд и масса электрона; тэф — эффективная частота соударений
электронов с тяжелыми частицами с передачей импульса, зависящая от энер-
гии электронов и концентрации тяжелых частиц.
Система дифференциальных уравнений (5.11) и (5.13) является существен-
но нелинейной, так как коэффициенты скоростей и могут зависеть от
напряженности электрического поля, являющейся решением волнового урав-
нения (5.13), а величина 8', входящая в уравнение (5.13), в свою очередь за-
висит от изменяющейся во времени концентрации электронов, получаемой в
результате решения системы уравнения (5.11), и также от самого электриче-
ского поля, влияющего на величину Метод решения рассматриваемой
системы уравнений описан в [21].
Таким образом, для описания кинетики химических реакций в неравно-
весной плазме необходима обширная информация, включающая: 1) элек-
тронно-колебательные термы реагирующих и образующихся соединений;
2) сечения процессов возбуждения, дезактивации, химических реакций; 3) за-
селенности электронно-колебательных состояний рассматриваемых соедине-
ний, плотности электронов и их пространственно-временные распределения;
4) функции распределения электронов и их пространственно-временные распре-
деления; 5) напряженности переменного и постоянного электрических полей
и их пространственно-временные распределения.
1 В данной работе не рассматриваются и не решаются уравнение энергии для тяжелых
частиц и кинетическое уравнение для электронов.
2 Реакции нулевого и третьего порядка не рассматриваются, так как встречаются крайне
редко, а включение их в схему не представляет трудностей.
229
В связи с отсутствием многих сечений указанных выше процессов и ме-
тодов решения систем нелинейных дифференциальных и интегродифферен-
циальных уравнений вряд ли возможно детальное описание кинетики хими-
ческих реакций в неравновесной плазме.
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
С КОНДЕНСИРОВАННОЙ ФАЗОЙ
Значительную часть исследованных плазмохимических процессов состав-
ляют процессы, в которых либо используются реагенты в конденсированной
фазе, либо образуются целевые продукты в конденсированной фазе.
В случае проведения плазмохимических реакций с использованием в ка-
честве реагентов конденсированных веществ наблюдается сильное взаимное
влияние факторов, связанных с протеканием химических процессов и процес-
сов тепло- и массообмена. В [22] предложена математическая модель, опи-
сывающая поведение частиц, введенных в плазменную струю. При составле-
нии этой модели были приняты следующие основные допущения: порошок
по сечению струи распределен равномерно, частицы порошка имеют сфери-
ческую форму, температура по сечению частиц постоянна. Для получения
более общих представлений о поведении конденсированных частиц в плазмен-
ной струе были рассмотрены некоторые системы газ — материал, которые
представляют крайние случаи сочетания теплофизических свойств: аргон —
вольфрам, водород — трехокись вольфрама. Результаты расчетов позволили
исследовать динамику изменения температур частиц и газа, их скоростей,
коэффициента теплоотдачи, радиуса частиц и степени испарения в зависимо-
сти от начальной температуры струи, размера и расхода порошка, теплофизи-
ческих свойств плазмообразующего газа и реагента. Было показано, что на
степень перехода в газовую фазу в каждой рассматриваемой системе газ —
материал сильно влияют начальная температура потока плазмы и размер
частиц.
Экспериментальные исследования поведения твердых частиц в плазмен-
ной струе, выполненные, например, в работе [23], показали, что частицы и
газ движутся с различными скоростями. Наблюдается так называемый эф-
фект проскальзывания, т. е. обтекания частицы газом. Обтекание твердых
частиц потоком плазмы при атмосферном давлении может осуществляться
в режиме непрерывного течения, течения со скольжением и свободномоле-
кулярного движения в зависимости от значения числа Рейнольдса для пото-
ка плазмы. В работе [23] показано, что наличие порошка в плазме приводит
к снижению температуры газа и более равномерному распределению пара-
метров по сечению. Кроме того, порошок турбулизирует струю (в случае
ламинарного ее течения) и уменьшает турбулентность (в случае начального
ее турбулентного течения). Вопросы теплообмена в потоке плазмы с вве-
денными в него твердыми частицами рассмотрены также в работах [24, 25].
Используя опытные данные по значениям скоростей и температур частиц
и потока плазмы, можно оценить время, необходимое для расплавления ча-
стиц различных размеров из разных материалов. Так, для частиц воль-
фрама диаметром 5 мкм при температуре аргоновой плазмы в 5000° К
необходимо — 10“5 сек для их расплавления; увеличение диаметра частиц
до 100 мкм приводит к увеличению необходимого времени до единиц секунд.
Пренебрежение эффектом проскальзывания приводит к существенной
погрешности в оценке времени, необходимого для испарения частиц порошка.
Естественно, что степень испарения частиц зависит от теплофизических
свойств системы газ — материал. Так, при одинаковой начальной темпера-
туре потока аргона (8000° К) и радиусе частиц 2,5-10-4 см степень испарения
частиц углерода в три раза меньше, чем частиц вольфрама. При замене ар-
гона водородом, в связи с ростом коэффициента теплоотдачи и увеличением
времени пребывания частиц в зоне высоких температур, резко возрастает
степень испарения частиц [26].
230
По описанной в [22] методике были выполнены расчеты для процесса
восстановления трехокиси вольфрама. Оказалось, что время пребывания
частиц в зоне высоких температур ~ 10“4 сек недостаточно для развития
гетерогенного процесса восстановления. Напротив гомогенная кинетика
обеспечивает необходимые скорости реакций. Эти результаты согласуются с
экспериментальными данными, полученными при исследовании этого про-
цесса. Таким образом, плазмохимические процессы с использованием конден-
сированных веществ лимитируются скоростью перехода компонентов в газо-
вую фазу.
Свойства получаемых конденсированных продуктов процессов в плазмен-
ной струе зависят от способа и режима закалки.
Выбором той или иной скорости закалки в низкотемпературной плазме
можно получать вещества как предельного, стехиометрического, так и несте-
хиометрического составов, любых промежуточных образований равновесного
и неравновесного типов. Примеры таких соединений будут приведены при
описаний конкретных плазмохимических процессов. Образование подобных
соединений связано с тем, что в плазме, в отличие от традиционных источ-
ников энергии, применяемых для получения тугоплавких веществ, присутст-
вуют свободные электроны и электронно-возбужденные атомы и молекулы
реагентов [27]. Например, при восстановлении окислов и других соединений
до металлов и неметаллов, которое связано с приобретением остовом атома
(иона) металла или неметалла электронов взамен отданных атомам кислорода,
хлора, используются свободные электроны плазмы. Таким образом, в плазме
процесс восстановления ускоряется, что подтверждено экспериментально.
Использование различных режимов закалки, например в плазмохимических
процессах восстановления, позволяет получить металлы в виде порошков
различной дисперсности, нитевидных образований, слитков. Соответствую-
щим подбором парциального давления паров металла и степени пересыщения
(изменением расхода порошка и газа, а также температуры на входе в закалоч-
ное устройство) были получены ультрадисперсные порошки вольфрама
сферической формы, а подбор скорости закалки позволил ограничить их
размеры в пределах 400—500 А. В случае закалки в сопле Лаваля при усло-
вии, если среднемассовая температура струи на входе в сопло близка к
температуре начала конденсации продуктов, более вероятно образование
большого числа частиц с размерами, близкими к критическим. Частицы
крупных размеров можно получить, если конденсация их протекает при более
высоких температурах.
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
В ОРГАНИЧЕСКОЙ ХИМИИ
В настоящее время изучены сотни реакций углеводородов в низкотемпе-
ратурной плазме. Разрабатываются научные основы технологии плазмохими-
ческих процессов пиролиза углеводородов, окисления их, селективного син-
теза ценных соединений.
ПИРОЛИЗ УГЛЕВОДОРОДОВ В ПЛАЗМЕ Н2 и СН4
1. Квазиравновесные процессы
Термодинамика системы С—Н. Равновесная смесь при 1,0 ата (рис. 5.2)
и температурах до 2000° К состоит из Н2 и конденсированного углерода, от
2000 до 5000° К она содержит С1? С2, С3, Н, Н2, СН, С2Н, С3Н, С4Н, СН2,
С2Н2, С4Н2, С4Н4 [4, 29, 30]. До 3000° К основным углеводородом является
С2Н при более высоких температурах концентрации [С2Н], [С3Н], [С4Н]
превышают [С2Н2] и достигают максимума при 4000° К. Концентрации
[С2Н2] в газе после закалки равновесной смеси, в процессе которой постули-
231
Рис. 5.2. Равновесный состав продуктов
пиролиза углеводородов. Р — 1 ата
Рис. 5.3. Влияние изменения давления на
концентрацию ацетилена в квазиравновес-
ных составах
С : Н = 1 : 1,66; 1 — Р = 0,1 ата, 2 — 1,0;
«3 — 10
руется протекание следующих реак-
ций: н + с2н->с2н2, н + н->н2,
CnHx->C2H, С2Н2, Cs и Н2; Cn->CS
(Cs — твердый углерод), возрастают
с 12 до 42 об. % при увеличении С/Н
от 0,25 до 2,5. Затраты электроэнер-
гии превышают величины, характер-
ные для промышленных процессов, и
составляют 23 кВт-ч! нм? С2Н2 для
1,0 ата и Т ~ 3200° К; затраты мини-
мальны при 15 ата и 3400° К
(~ 13,8 кВт-ч! нм? С2Н2).
В квазиравновесных составах, рас-
считанных без учета конденсирован-
ного углерода Cs [31], до — 1600° К
основными углеводородами являются
метан и толуол, а при 1600° < Т <
2000° К — ацетилен. Выход толуо-
ла максимален при 1400° К и колеб-
лется от 80 до 35% в зависимости от
величины С/Н (от 1 : 2,6 до 1 :8,3).
При 1800—2000° К выход ацетилена
достигает максимальных значений и
не отличается от приведенных ниже.
В составах, рассчитанных без уче-
та Cs и ароматических соединений,
максимальные концентрации [С2Н2] и
[С2Н4] реализуются до 2000° К, кон-
центрации же соединений [С2], [С3],
[СН], [С2Н], [СН2], [С4Н] малы. При
1000-1200° К (1,0 ата С/Н - 1 : 3,2)
[32] в составах одновременно присут-
ствуют [С2Н2], [С2Н4], [С3Н6], причем
[С2Н4] в этой области проходит через
максимум (12,5 об. %). Максимальное
содержание С3Н6 имеет место при
700—800и К, а [С2Н2] максимальна
при 1800° К (29,5 об. %). [С2Н2] :
: [С2Н4] варьируется от 1:8 до
1 : 0,013. Увеличение С/Н от 1:5
до 1 : 2,4 [30] (именно в этих преде-
лах изменяется С/Н в технических
углеводородных смесях, представ-
ляющих практический интерес) при-
водит к значительному повышению
[С2Н2]. Как показано в [30], наиболь-
шее значение ее близко к максималь-
но возможному, рассчитанному в
предположении образования только
С2Н2 и Н2. Изменение давления от
0,1 до 10 ата при Z^IBOO0 К прак-
тически не влияет на [С2Н2], при
1000° < Т < 1800° К она резко падает
(рис. 5.3), возрастают минимально
необходимые затраты энергии на по-
лучение 1 нм? С2Н2 (а), и значения
а смещаются в сторону больших тем-
ператур. Заметим, что влияние уве-
232
личения давления на состав равносильно уменьшению С/Н. Значения «мини-
мальны при 1500—1800° К. Приводимые в [30] величины а из различных
углеводородов таковы: из СН4 — 7,8 кВт-ч!нм3', из С2Н6 — 6,4; из
С3Н8 — 5,9; из С4Н10 — 5,7; из С5Н12 — 5,5 кВт-ч1нм\
В [30] описана удобная связь [С2Н2], степени превращения в [С2Н2], а
с энергетическим критерием Аэн, представляющим отношение всей затра-
ченной на образование соединений при рассматриваемой температуре энер-
гии к энергии, необходимой для получения только С2Н2 и Н2 при стандарт-
ной температуре1. Использование зависимостей показателей процесса от
позволит получить заданный состав при изменении С/Н (состава сырья)
соответствующим изменением подводимой энергии.
Приведенные выше результаты расчетов позволили нам оценить величину
«химического» коэффициента полезного действия, представляющего отноше-
ние энергий, необходимых для образования продуктов при стандартной и рас-
четной температурах, и рассчитанного для каждого значения С/Н для соста-
ва, когда затраты энергии на его образование минимальны. Величина «хими-
ческого» КПД достигает 55% при С/Н = 1 :2,5, значительно превышая зна-
чение, характерное для условий окислительного пиролиза (30—35%).
Термодинамика системы С—Н—S—О—N. Для определения выходов воз-
можных соединений серы, кислорода, азота, присутствующих в технических
углеводородных смесях (которые необходимо знать для технологического
оформления процесса), в [34] выполнен расчет системы С : Н : S О : N =
— 1:3: 0,013 : 0,004 : 0,001, соответствующей составу нефти Ишимба-
евского месторождения с учетом разбавления ее водородом плазменной струи.
Основные примеси с увеличением Т от 1000 до 1800° К колеблются в пределах
(в об. %) : 0,53 > [СО] > 0,27; 0,85 > [CS2] > 0,01; 0,01 < [CS] < 0,85;
0,11 > [HCN] > 0,064; 0.034 < [H2S] < 0,247.
Анализ приведенных выше результатов термодинамических расчетов рав-
новесных и квазиравновесных составов для системы С—Н позволяет опреде-
лить оптимальные условия проведения следующих процессов:
получения ацетилена и технического водорода из природного газа;
получения ацетилена, этилена и технического водорода из любых углево-
дородов, кроме метана;
получения ацетилена из конденсированного углерода и водорода или ме-
тана;
получения конденсированного углерода и технического водорода.
Макрокинетика плазмохимического пиролиза углеводородов. Одноступен-
чатый процесс. Выяснению механизма и исследованию кинетики термического
разложения углеводородов при температурах до 1000° К посвящено много
работ (см. например, [35]). Развиваемые в них представления удовлетвори-
тельно согласуются с экспериментальными данными. Однако вопросы кинети-
ки при Т^> 1000° К разработаны недостаточно. Возможно, это связано с темт
что для обеспечения малых степеней превращения углеводородов с числом
углеродных атомов, большим 2, необходимо исследовать пиролиз в интерва-
ле времен контакта ~ 10“5—10~6 сек, что технически трудно осуществимо.
Кроме того, скорости закалки продуктов должны быть огромны. Например,
если изучать пиролиз при Т ~ 3000° К, при временах — 10“6 сек, скорость
закалки должна быть не менее 109—1010 град/сек, что в настоящее время
практически недостижимо.
Однако некоторые экспериментальные и расчетные работы позволяют уста-
новить качественно изменение состава продуктов пиролиза с ростом темпера-
туры и наметить брутто-стадии процесса. На основании анализа литератур-
ных данных в [36] можно сделать следующие выводы: крекинг углеводородов
при Т 1000° К является цепью последовательных реакций, причем не
предельные соединения — промежуточные продукты, а углерод в конден-
1 В [33] показано, что показатели процесса зависят не только от 2^эн, но и от отноше-
ния lid (I — длина реактора, d - его диаметр).
233
сированной фазе и водород — конечные продукты этих реакций. С ростом Т
уменьшается доля цепных процессов, увеличивается доля радикальных и
молекулярных и исчезает торможение разложения углеводородов продуктами
его. При температурах 1100—1300° К конденсированные продукты обра-
зуются по радикальному механизму, выше 1300° К сажа образуется молеку-
лярным путем на поверхности углерода. Скорость образования сажи умень-
шается при разбавлении углеводородов водородом. Начальные стадии раз-
ложения алканов, начиная с этана, непредельных (С3Н6, С4Н8), ароматических
соединений при температурах, реализуемых после смешения их с плазменной
струей, практически не влияют на процесс пиролиза, поскольку характерные»
времена их разложения на порядки меньше характерных времен распада
СН4, С2Н2, С2Н4. Поэтому, по-видимому, можно считать, что за малые (отно-
сительно продолжительности всего процесса разложения углеводородов)
времена образуется смесь СН4, С2Н4, Н2 и в случае присутствия в сырье аро-
матических соединений1 * * * *— С2Н2.
Далее метан разлагается по брутто-схеме, предложенной Л. Касселем
[37]; разложения этилена и ацетилена включены в нее в качестве отдельных ста-
дий, что подтверждено экспериментально. Этот механизм включает следующие
реакции:
сн4 -> сн2 + н2, сн2 +. СН4 -> С2Н6, С2Н6 -> С2Н4 + н2.
С2Н4 -> С2Н2 + Н2, С2Н2 -> 2CS + Н2.
При выборе отношения СН4 : С2Н4 : Н2 необходимо учитывать, что при
рассматриваемых температурах вероятности разрыва связей С—С и С—Н
в исходном углеводороде становятся равными. Начальные составы можно
получить в предположении различных мест разрыва связей С—С с образо-
ванием разных комбинаций С2Н6, С3Н8, С3Н6, С3Н4 (в случае разложения пре-
дельных и непредельных соединений), которые дают описанные в литературе
определенные отношения СН4 : С2Н4 : Н2. Например, при разложении
н-С5Н12 возможны такие крайние комбинации: 1СН4 + 2С2Н4 и ЗСН4 +
+ 1С2Н4 — 2Н2 (знак минус обозначает использование водорода плазменной
струи).
Используемые для пиролиза технические углеводородные смеси (бензины,
газоконденсаты, нефти) обычно содержат кроме алканов циклические предель-
ные углеводороды, ароматические, многоядерные ароматические соединения.
Исходя из характерных времен распада, результатов плазмохимического
пиролиза их (анализ этих работ выполнен в [36]), можно также предложить
начальные составы для расчета макрокинетики пиролиза. Например, в случае
разложения бензола начальный состав состоит из ЗС2Н2, а в случае разложе-
ния толуола — СН4 + ЗС2Н2 — Н2 и т. д. Величина отношения СН4 : С2Н4 :
: Н2 : (С2Н2) может быть уточнена при сопоставлении результатов расчетов
с экспериментальными кинетическими данными плазмохимического пиролиза
углеводородов. Заметим, что в экспериментах обычно углеводороды значи-
тельно разбавлены теплоносителем — водородом.
В [11,38, 39,232] описаны результаты расчетов макрокинетики плазмохими-
ческого разложения СН4, С3Н8; нами выполнены расчеты макрокинетики раз-
ложения газоконденсата и низкооктанового бензина в плазменной струе [36].
Решалась система уравнений химической кинетики и гидродинамики плаз-
менной струи (см. стр. 225). Предполагались мгновенный нагрев реагентов
с образованием состава, мгновенное перемешивание и изоэнтальпийность
процесса. В наших расчетах учитывались потери тепла в экспериментальном
реакторе и в закалочном устройстве.
1 Более вероятно разложение углеводородов в рассматриваемых условиях на различные
радикалы за времена ~ (10~8—10“7) сек. Радикалы с числом углеродных атомов
С 3 примерно за то же время распадаются с образованием С2Н4, С3Нв и т. д. Ради-
калы, содержащие С < 2, рекомбинируют с Н или друг с другом за времена ~ (10“б—
—10“7) сек, образуя СЩ, С2Не [35].
234
Кинетические зависимости концентраций продуктов пиролиза для рас-
смотренных углеводородных смесей и углеводородов подобны. Однако в слу-
чае пиролиза газоконденсата (рис. 5.4) и бензина максимальные значения
концентраций этилена и ацетилена смещены в сторону меньших времен и реа-
лизуются соответственно при 10-6 и 10“5—10“4 сек (при пиролизе метана —
10“6—10“5 и 10“4—10-3 сек). Это связано со значительно меньшим тепловым
эффектом реакции по сравнению с соответствующей величиной образования
ацетилена из метана (при тех же TQ), Метан расходуется практически пол-
ностью за 10-5 сек (пиролиз газоконденсата). Температура струи в течение
до — 10“4 сек уменьшается, что связано с эндотермическими реакциями
разложения СН4 и С2Н4, а затем после прохождения минимального значения
начинает повышаться, что связано с выделением тепла в процессе образо-
вания углерода из С2Н2. Поэтому через ~ 10~4 сек следует начинать прину-
дительную закалку со скоростью 5-106 град/сек. Изменением начальных усло-
вий и времени начала закалки можно варьировать отношение С2Н2 : С2Н4
в продуктах (от 1:1 до 1 : 0,15 при пиролизе газоконденсата).
Потери энергии, которые реализуются в охлаждаемых экспериментальных
реакторах, сильно влияют на кинетические зависимости начиная с ~ 10~5 сек:
концентрации [С2Н2] уменьшаются, а этилена увеличиваются по сравнению
с их значениями, полученными без учета потерь энергии. Начиная же с
~ 3-10-5 сек концентрации продуктов практически не меняются, т. е. в реак-
торе с потерями энергии происходит закалка продуктов пиролиза со ско-
ростью 107 — 106 град/сек, Вероятно, этим объясняются полученные многими
авторами данные, свидетельствующие
о незначительном влиянии на пока- к
затели процесса изменения длины 2666
реактора в пределах порядка вели-
чины начиная с некоторого ее зна-^/^
чения.
Экспериментальные данные по
плазмохимическому пиролизу газо- /666
конденсата были получены нами при
изменении длины реактора и закалке^
продуктов в теплообменнике. При7
выполнении макрокинетических рас-
четов на заданных длинах реактора/Ж7
«включалась» закалка со скоростью
(1—5) -106 град/сек. Эксперименталь-
ные данные хорошо совпадают с рас-
четными (пиролиз метана) и удовле-
творительно в случае пиролиза газо-
конденсата при временах 10“4—10“3
сек. при которых осуществим отбор
проб. Эти факты могут подтверждать
положенный в основу расчета брутто-
механизм Л. Касселя. Расхождения
в содержании метана в расчетных и
экспериментальных составах, наблю-
даемые при плазмохимическом пиро-
лизе газоконденсата и бензина
(табл. 5.1), вероятно, вызваны сле-
дующими причинами:
Рис. 5.4. Кинетика плазме химического
пиролиза газоконденсата
1 — без учета потерь энергии в реакторе;
2 — с учетом этих потерь
235
Таблица 5.1
Сопоставление экспериментальных и расчетных данных (вес. доли)
плазмохимического пиролиза газоконденсата *
Газ Длина реактора
1 см 2 см 3 см 4 см
Опыт ** Расчет *** Опыт Расчет Опыт Расчет Опыт Расчет
н2 0,18 0,202 (0,194) 0,18 0,193 (0,187) 0,18 0,(188 0,20 0,188>
СН4 0,066 0,01 (0,001) 0,07 0,001 (0,001) 0,08 0,001 0,08 0,001
С2Н2 0,53 0,627 (0,637) 0,60 0,639 (0,635) 0,54 0,634 0,55 0,634
С2Н4 0,12 0,026 (0,0841) 0,10 0,084 (0,128) 0,12 0,123 0,14 0,122
cs Не опре- делялось 0,144 (0,08) Не опре- делялось 0,083 (0,049) Не опре- делялось 0,054 Не опре- делялось 0,055
* Групповой состав газоконденсата: н-алканы (С8Н18) — 0,21; нафтено-изопарафины (С7Н14) — 0,58;
ароматика (C9Hi4) — 0,10; смолы, многоядерная ароматика (С12Н10) —0,11.
** Опыты проводились при мощности в плазме 8,5 кВт, расходе сырья 2 кг/ч, расходе водорода
3,0 м3/ч\ степень превращения в газообразные продукты — 96—99%.
**♦ Результаты расчета получены в предположении, что скорость закалки составляет 1-Ю6 град/сек,
а в скобках — 5-Ю6 град/сек.
1) в составах бензина и газоконденсата присутствуют изосоединения.
Начальные составы для расчета макрокинетики их разложения должны со-
держать больше метана и меньше этилена, чем в случае разложения н-алка-
нов;
2) для выполнения расчетов использовались константы скоростей реакций
разложения чистых углеводородов. Известно, что эти величины могут отли-
чаться от соответствующих значений в случае разложения их в смесях.
Макрокинетика двухступенчатого пиролиза углеводородов. Как следует
из приведенного выше анализа результатов расчетов макрокинетики пиро-
лиза углеводородов, для получения целевых продуктов в задаваемом соотно-
шении через ~ 10~4 сек, начиная с температур ~ 1800° К, необходимо
проводить их закалку со скоростью ~5 • 106 град!сек. Однако при этих темпе-
ратурах возможно проведение пиролиза дополнительно вводимых углево-
дородов. В [39, 40] был рассчитан такой двухступенчатый процесс пиролиза
углеводородов. Варьировали промежуток времени между началом процесса
и вводом реагента на вторую ступень А/, относительное количество вводимого
углеводорода т = GJGA (Gr и G2 — количества реагентов, подаваемых на
первую и вторую ступени), состав углеводородов. Предполагалось, что в
момент введения реагента на вторую ступень происходит мгновенное пере-
мешивание его с продуктами пиролиза первой ступени и распад его на СН4,
С2Н4, Н2 за времена, которые меньше времени образования С2Н2 на порядок
или более. Температура смеси понижается «мгновенно» за счет разбавления
подаваемым углеводородом и его развала. Количества метана, этилена и во-
дорода, образуемые при развале реагента, выбираются в соответствии с со-
ображениями, описанными на стр. 234. Расчеты выполнялись для следующих
соотношений СН4 : С2Н4 =- 1 : 1 и 3 ; 2, а также для случаев подачи на вто-
рую ступень метана и этилена, т меняли от 0 до 0,7. На первой ступени про-
исходил пиролиз СН4 и бензина, состав которого описан в [36]. Резуль-
таты расчетов двухступенчатого процесса сопоставляли с соответствующими
величинами для одноступенчатого пиролиза. В зависимости от момента на-
чала закалки (3,6-10-5, 3,7 -10~4 и 7,3-10~4 сек) меняется соотноше-
236
ше С2Н2 : С2Н4 в продуктах и т. Например, при закалке продук-
тов пиролиза метана пропаном при т — 0,5 С2Н2/С2Н4 меняется от 1,5 до
1,15 (в одноступенчатом процессе это чисто ацетиленовый режим). При за-
калке в момент 3,6 -10-3 сек С5Н12 при т = 0,7 наблюдаются лучшие показа-
тели процесса: 25 вес. % С2Н2, 46 вес. % С2Н4 — затраты энергии на полу-
чение суммы С?Н2 с С2Н4 снижаются в 2,5 раза (рис. 5.5). Если же начинать
закалку вблизи максимума ацетилена (3,7 -Ю-4 сек) при т = 0,5, то в продук-
тах будут одинаковые концентрации С2Н2 и С2Н4— по 32 вес.%, энергоза-
траты снижаются в два раза.
Результаты экспериментального изучения плазмохимических процессов
получения ацетилена и технического водорода и ацетилена, этилена и тех-
нического водорода. В настоящее время изучен плазмохимический пиролиз
многих углеводородов и их технических смесей, представляющих практи-
ческий интерес. Нами исследован пиролиз пропан-бутановой фракции [32],
^-гептана [32], бензинов с концом кипения 165 и 200° С [32, 34], газоконден-
сатов [41], газойля [34,42], нефти Ишимбаевского месторождения [34, 42]
и в литературе описаны пиролиз метана [И, 30, 43, 44, 228, 232] природного
газа различных составов [И, 45], пропана[30, 45], пропилена [45], ft-гептана
[48], бутадиена [46], циклогексана [46], бензинов [46—49], сырых нефтей
нескольких месторождений [45, 49—
51].
Наиболее развитым в промышлен-
ном отношении оказался процесс раз-
ложения природного газа непосред-
ственно в электрическом разряде
(так называемый электрокрекинг).
В настоящее время в ряде стран рабо-
тают промышленные установки мощ-
ностью (6—8)-103 кВт. Лучшие по-
казатели электрокрекинга природно-
го газа таковы [45]: общая степень
превращения ~ 70%, степень пре-
вращения в ацетилен 50%, содер-
жание ацетилена в продуктах до
14,5 об. % при затратах электроэнер-
гии 13,6 кВт-ч на 1 нм3 природного
газа. Однако в продуктах содержат-
ся значительные количества сажи и
гомологов ацетилена. Процесс плохо
управляется и оптимизируется. Серь-
езные недостатки, свойственные
электрокрекингу природного газа,
вызваны тем, что в реакторах, в кото-
рых протекает процесс, имеют место
огромные градиенты температур.
Ряд фирм США [52, 53] в резуль-
гате применения плазмотрона с маг
нитной стабилизацией дуги, образую-
щей как бы сплошной конус, вместо
цуги длиной [45] — 1 м создали свой
Рис. 5.5. Кинетика двухступенчатого про
цесса пиролиза углеводородов в плазмен
ной струе
[ ступень — пиролиз метана в плазменной струе
12; II ступень — пиролиз пропана при введении
то через 1,7 *10-4 сек (1), 3,7*10-* сек (2) и
\3*10~4сек (3) после начала процесса. Tq ][ —
=293» К, GG3Hs/GCH4 = 0,5
237
вариант электрокрекинга. В этом процессе степень конверсии метана в аце-
тилен достигает 80%, концентрация ацетилена в продуктах 20 об. % при
затратах электроэнергии 12,5—13,3кВт-ч на 1 кг ацетилена. Столь высо-
кая концентрация ацетилена в пиролизном газе создается благодаря прове-
дению процесса в две стадии (крекинг природного газа происходит в дугег
а в струе газов крекинга при закалке их тяжелыми углеводородами проте-
кает пиролиз последних). Приведенные показатели процесса получены на
промышленной установке производительностью 25 000 тп ацетилена в год.
В экспериментах, выполненных в плазменной струе, были определены
диапазоны температур 1 * * и времен проведения процессов, необходимых для
получения заданного отношения продуктов, исследовано влияние на показа-
тели процессов различных способов закалки, составов плазмообразующих
сред и сырья, условий подачи его в плазменную струю. Оказалось, что ре-
жимы с максимальным количеством ацетилена реализуются при температу-
рах 1800—2000° К (для СН4и С3Н8) и 1600—1880° К (для более тяжелых угле-
водородов) и временах контакта 10~5 — 10“4 сек (отношение С2Н2 : С?Н4 —
= 4:1). Составы продуктов плазмохимического пиролиза в этих режимах
(табл. 5.2) удовлетворительно согласуются с термодинамическими квази-
равновесными составами. Результаты кинетических расчетов также
согласуются с экспериментальными данными. В этих режимах выход аце-
тилена совместно с этиленом достигает 65—78,5 вес.%, удельные зат-
раты энергии на их получение колеблются от 4,6 кВт-ч/кг (в случае пиро-
лиза бензина — нефти) и до 9,5 кВтп-ч/кг (в случае пиролиза метана). При
температурах 1300—1600° К и примерно тех же временах контакта при
пиролизе углеводородов с числом углеродных атомов С, большим 3, обра-
зуются составы, содержащие значительные количества этилена (например^
при пиролизе бензина с концом кипения 165° С — до 13 об. %, а С2Н2 : С2Н4 =
= 1 : 1). С термодинамически рассчитанными согласуются лишь концентра-
ции Н2, СН4 и суммы непредельных соединений, в то время как [С2Н4] и
[С2Н2] значительно отличаются. Возможно, это связано с тем, что за указан-
ные времена не успевает устанавливаться квазиравновесный состав. Кинети-
ческий расчет подтверждает это.
При пиролизе углеводородов с числом углеродных атомов С 3 состав
сырья (алканов) слабо влияет на показатели процесса. Однако с увеличе-
нием высококипящих углеводородов в сырье выходы С2Н2 и С2Н4 снижаются
[45, 46, 50, 54]. Кроме того, увеличение содержания высших ароматических
соединений приводит к росту выхода сажи.
Повышение давления ухудшает показатели плазмохимического пиролиза.
Так, по данным [43, 44, 55, 232], при плазмохимическом пиролизе метана
с повышением давления с 1 до 6 ата уменьшается выход С2Н2 и возрастают
энергетические затраты на его получение.
Изучено влияние температуры на концентрации высших ненасыщенных
углеводородов (метил-, винил-, диацетилена, бутадиена, пропадиена и бути-
лена), являющихся нежелательными примесями в данном процессе [56].
С увеличением температуры с 1300 до 1700° К концентрация винилацетилена
вначале растет с 0,18 до 0,30 об.% при 1500° К, а затем падает до 0,24 об.%;
концентрация диацетилена не превышает 0,1 об.%, а остальных соединений
падает с 1,44 до 0,4 об.%.
При пиролизе нефти, содержащей в своем составе кислород, азот и серу,
образуются СО, H2S, HCN, концентрации которых хорошо согласуются с
термодинамически рассчитанными [34].
При проведении плазмохимического процесса в этилен-ацетиленовом
режиме степени превращения сырья в газообразные продукты могут быть от-
носительно небольшими (~ 60—80%). Жидкие продукты состоят из непрореа-
1 Протекание реакций в плазменной струе описывается не изотермическим, а изоэнталь-
пийным процессом, когда энергия, полученная сырьем после перемешивания с плаз-
менной струей, больше не пополняется. Поэтому экспериментально определяется (рас-
считывается) температура продуктов реакции перед закалкой.
238
Таблица 5.2
Показатели плазмохимического пиролиза углеводородов
Метан [И] Пропан [45] Бензин Бензол «сырой» [45] Газокон- денсат [41] Газойль [42] Нефть
«легкий» [45] с концом кипения 165° С [34]
Ишимбая [34] «легкая» [45] «сырая» [50]
Газ, об. %
С2Н2 15,50 13,70 14,80 17,00 18,10 16,50 16,20 15,95 15,00 14,5 0,31 0,30 0,38 6,51 1,12 0,17 0,23 0,33 0,20 0,38 6,04 0,26 0,12 0,22 0,26 68,5
С3Н4 0,17 0,23 0,34 0,30 0,14 0,56 0,27 0,28 0,35 0,39 0,41 6,43
СаН2 0,36 0,25 0,25 0,16 0,54 U,25 0,10 0,16
С4 Н4 0,09 0,32 0,36 — 0,35
С3Щ 0,50 5,83 7,47 7,03 0,96 7,05 6,52 7,00
СзНб — 0,74 1,13 0,40 0,09 0,10 0,69 0,70 1,00 0,18 0,22 0,026 0,16 0,25 6,26 0,23 0,05 0,02 0,15 68,20
Аллен —• 0,09 0,13 0,15 0,06 0,15 0,10 0,10
С4Н8 С4Н6 С5Ш СбНв — 0,13 0,23 0,11 0,17 0,24 0,32 0,11 0,13 0,04 0,08 0,84 0,68 — 0,12 0,22
СН4 С2Не СзН8 С4Н10 С5Н12 н2 со О2 5,80 76,00 8,38 0,36 2,87 0,05 66,40 7,92 0,32 0,03 64,10 7,50 67,31 2,9б 0,01 75,9it 5,37 69,72 6,11 69,89 5,82 69,37
— 0,03 — — — 0,26 0,33 0,15
n2 — 0,23 0,33 0,04
Углеводороды Cs — — 2,12
H2S HCN — — — — — — — 0,14 0,06 —
Показатели
Выход С2Н2 + С2Н4, вес. % Выход сажи, г/кВт»ч Энергозатраты, кВт*ч/кг СгН2 -f- + С2Н4 78,5 70,00 2,0 70,00 6,0 72,0 65,0 40,0 77,0 74,5 74,5 9,0 4,8 71,0 —
9,5 6,6 6,4 4,6 8,5 4,7 4,9 7,0 —
Мощность, кВт 30 (5—8) -103 (5—8) -103 30—200 (5—8) -103 30 30 30 (5—8).1О3 8,5.10s
гировавшего сырья и ароматических соединений. Например, в жидких про-
дуктах плазмохимического пиролиза парафино-нафтеновой фракции газо-
конденсата, не содержавшей ароматических соединений, их доля достигает
20%. Основными соединениями были толуол, бензол, ксилолы. Эти продукты
можно возвращать на плазмохимический пиролиз, или, если это будет оправ-
дано экономически, выделять. Экспериментальные исследования плазмохи-
мического пиролиза углеводородов проводились на установках различных
масштабов (с плазмотронами мощностью от 30 до 10 000 кВт). Составы газов,
имевшие место на установках промышленных масштабов (с плазмотронами
мощностью до 10 МВт), хорошо согласуются с составами, полученными на
лабораторных установках. Однако наблюдаются значительный выход сажи
и большие затраты энергии на получение целевых продуктов. ’Это связано,
по-видимому, с недостатками в организации смешения сырья с плазменной
струей.
Возможные пути улучшения показателей процесса. 1. Большинство экс-
периментов проводили в плазменной струе Н2. Последний выбран в качестве
теплоносителя, так как он является одним из продуктов пиролиза, эффекти-
вен как теплоноситель (при нагреве его приблизительно до 3700° К и про-1
ведении реакции примерно при 1300° К используются более 90% энергии)
и благоприятно влияет на процесс, подавляя образование сажи и диацетилена.
Технически оправдано использование в качестве теплоносителя смеси
80—90% Н2 с 20—10 об.% СН4, получаемой после выделения С2Н2 и С2Н4.
При этом увеличивается выход последних на 5—10% по сравнению с пироли-
зом в чистом Н2. Описаны пиролиз углеводородов в плазменных струях
природного газа в [43, 44, 232, 249] и водяного пара в [48]. В последнем случае
концентрации С2Н2 и С2Н4 значительно повышаются, но одновременно растут
и энергозатраты. Так, при пиролизе бензина [С2Н2] и [С2Н4], полученные на
неоптимальном режиме, возрастают с 5,6 и 9,4 об. % (пиролиз в плазменной
струе Н2) до 11 и 19% соответственно, а энергозатраты повышаются на 25%.
2. Закалка продуктов пиролиза сырьем также улучшает показатели про-
цесса. В [45] описаны результаты закалки газов электрокрекинга метана про-
паном и бензином. Во втором случае показатели процесса оказались несколько
лучше, однако в жидких продуктах значительно больше высококипящих
примесей, затрудняющих выделение С2Н2.
3. Предварительный подогрев углеводородов также улучшает показатели
процесса. По данным [43], подогрев природного газа до 500° С позволяет
увеличить выход С2Н2 и уменьшить удельные энергозатраты при пиролизе
природного газа в плазменных струях Н2 и СН4 примерно на 25%.
4. Рекуперация тепла газов пиролиза может улучшить энергетические
показатели процесса. Оказалось [46], что 60—70% энергии пиролизного га-
за можно использовать для получения технологического пара.
Некоторые технологические схемы получения химических продуктов,
включающие плазмохимические процессы получения ацетилена, этилена и
технического водорода. Как известно, ацетилен и этилен являются важней-
шими исходными продуктами для синтеза целого ряда мономеров (винил-
хлорида, хлорпрена, акрилонитрила и т. п.). Поэтому включение плазмо-
химических процессов получения ацетилена и этилена, а также технического
водорода в технологические схемы производства указанных продуктов при-
водит к существенному и эффективному усовершенствованию их, а иногда
к полному изменению технологии.
Технологические схемы производства винилхлорида, включающие плазмо-
химический пиролиз углеводородов. Необходимо, чтобы разрабатываемый
процесс был сбалансированным по хлору. Из анализа экспериментальных
данных,’ приведенных выше, следует, что пиролиз углеводородов, в частности
бензина с концом кипения 165° С, можно проводить в плазменных струях,
содержащих Н2, СН4. В зависимости от состава плазмообразующей смеси
в газе пиролиза содержатся ацетилен и этилен (до 25—28 об. %). В рассмат-
риваемой ниже схеме плазмохимический пиролиз совмещается с получением
240
винилхлорида гидрохлорированием ацетилена и хлорированием этилена без
их выделения из пиролизных газов с последующим пиролизом дихлорэтана.
Процесс гидрохлорирования разбавленного ацетилена описан в работе [57]
и реализован в промышленном способе производства винилхлорида, разра-
ботанном японской фирмой «Куреха» [58].
На рис. 5.6 приведена технологическая схема получения винилхлорида
[59, 60]. Пиролиз углеводородов осуществляется в плазменной струе, со-
держащей 90—80 об. % Н2 и 10—20 об. % СН4 (стадия 1). Закалка продук-
тов пиролиза производится затапливанием струями воды или бензина.
После отделения от закалочной жидкости состав газа таков (в об. %):
60,8-66,9 Н2, 12,1-20,0 С2Н2, 12,9-5,0 С2Н4; 0,3-0 С2Н6, 10,5-7,5 СН4,
1,9—0,1 С3Н6, высшие ненасыщенные (винилацетилен, метилацетилен, ди-
ацетилен, пропадиен, бутадиен) составляют 1,5—0,5 об.%.
На стадии 2 газообразные продукты плазмохимического пиролиза очи-
щаются от высших ненасыщенных углеводородов абсорбцией керосином
при температуре (—15' С) и давлении 6—10 ата. Затем они смешиваются с хло-
ристым водородом, расход которого подбирается так, чтобы его концентра-
Рис. 5.6. Технологическая схема про
изводства винилхлорида-мономера
1 — плазмо химический пиролиз;
2 — очистка от высших ненасыщенных
углеводородов;
з — гидрохлорирование ацетилена;
4 — хлорирование этилена;
5 — термический пиролиз дихлорэтана
ция была равной концентрации ацетилена. При этом используется НС1,
получаемый при пиролизе дихлорэтана и, если необходимо, с других хлор-
органических производств. Гидрохлорирование ацетилена протекает на ста-
дии 3 в присутствии сулемового катализатора, нанесенного на активирован-
ный уголь. Из газа, содержащего Н2 (63,1—67,3 об.%), С2Н3С1 (12,5—
20 об.%), С2Н4 (13,3-5,0 об.%), С2Н6 (0,3—0 об.%), СН4 (10,8-7,6 об.%),
выделяют винилхлорид абсорбцией дихлорэтаном при давлении до 10 ата.
Цалее, на стадии 4 после подачи в смесь С12 осуществляется хлорирование
этилена в жидком дихлорэтане в присутствии катализатора — хлорного
келеза при давлении 5 ата. После этой стадии часть газов, состоящих из
55,5—90 об. % Н2 и 14,5—10,0% СН4, подают в плазмотрон. После пред-
варительной очистки дихлорэтан подвергается термическому пиролизу
температура 450—550° С, 7 атм), в результате которого образуются винил-
хлорид и хлористый водород. Последний возвращается на стадию гидро-
хлорирования ацетилена. Технико-экономические показатели описанного
троцесса обсуждаются ниже.
Технологическая схема условного специального нефтехимического комплекса
НХК), перерабатывающего сырую нефть и включающая плазмохимический
шролиз [61]. Предлагаемый НХК может выпускать в различных соотноше-
шях моторное топливо и нефтехимическую продукцию (этилен, пропилен,
>утилен-дивинильную фракцию и др.).
Один из рассмотренных в [61] вариантов схемы включает следующие про-
цессы: атмосферно-вакуумную перегонку нефти, пиролиз газойля и бензина
5 трубчатых печах, плазмохимический пиролиз нефти, каталитический ри-
юрминг, непрерывное коксование, гидрокрекинг, гидроочистку дизельного
оплива, депарафинизацию дизельного топлива, гидроочистку бензина,
азофракционирование. В другом варианте схемы гидрокрекинг заменяется
каталитическим крекингом, а газойль, получаемый в процессах каталити-
ческого крекинга и непрерывного коксования, пиролизуется в трубчатых
чечах.
241
Для процессов гидроочистки и гидрокрекинга используется дешевый
водород, получаемый в достаточном количестве при плазмохимическом пиро-
лизе нефти вместе с ацетиленом, этиленом и пропиленом.
Экономические преимущества плазмохимического пиролиза углеводоро-
дов. Технико-экономические оценки плазмохимических процессов получения
ацетилена, этилена и технического водорода из различных видов углеводород-
ного сырья выполнены, например, в [61] по нашим данным, специалистами
фирмы МГД (США) — по данным американских исследователей [62]. Сопо-
ставление показателей плазмохимических процессов пиролиза с показателя-
ми промышленных процессов окислительного пиролиза свидетельствует о
значительных преимуществах плазмохимических процессов. В последних
возможна переработка любого вида углеводородного сырья, включая сырую
нефть, выход ацетилена и этилена достигает 70—80 вес.%, а при организа-
ции рецикла 85 вес.%, расход сырья на 1 кг ацетилена составляет 2—2,8
против 4,5—6 кг в термоокислительном пиролизе, используется до 45% под-
водимой энергии непосредственно на реакцию против 35% в окислительном
пиролизе, нет СО, СО2 и относительно малы концентрации побочных про-
дуктов — это приводит к значительному упрощению аппаратурного оформ-
ления процесса, продуктами их являются С2Н2, С4Н4 и технический водород,
который может быть использован в нефтехимии, концентрации ацетилена в
газах относительно велики и его можно применять без выделения, например
в процессе получения винилхлорида.
Существенным достоинством плазмохимических процессов является воз-
можность переработки отходов производств, что позволяет практически пол-
ностью устранить загрязнение атмосферы и сточных вод.
Себестоимость и приведенные затраты на 1 т ацетилена при плазмохими-
ческом пиролизе метана на 20—30% ниже, чем эти величины в термоокисли-
тельном пиролизе; в случае плазмохимического пиролиза нефти эти показа-
тели снижаются в два раза. Включение же плазмохимического пиролиза
в технологическую схему специального нефтехимического комплекса, пере-
рабатывающего сырую нефть, позволит выпускать рентабельную нефтехи-
мическую продукцию и моторное топливо в различных соотношениях, при-
чем с ростом доли нефтехимической продукции (~ 25% от перерабатывае-
мой нефти) рентабельность производства возрастает.
При использовании газов плазмохимического пиролиза углеводородов
для получения винилхлорида себестоимость последнего резко снизится по
сравнению с величиной, характерной для способа получения его из этилена,
даже несмотря на относительно низкую себестоимость этилена.
В [63] описаны результаты пиролиза углеводородов терпенового ряда
в плазменной струе азота. Реагенты вводили в жидком состоянии, закалка
продуктов осуществлялась в водоохлаждаемой камере. Конверсия возрас-
тала с увеличением мощности. При пиролизе fJ-пинена конверсия в мирцен
достигала 26%. В случае же пиролиза а-терпинена образуются а-пинен и
пара-цимол, но степени конверсии не превышают 4%.
2, Реакции углеводородов в неравновесной плазме
Реакции углеводородов исследовались в неравновесной плазме, генери-
руемой в плазмотронах тлеющего, ВЧ-, СВЧ- и коронного разрядов. Первые
работы были посвящены реакциям полученных в плазме атомов водорода,
азота, кислорода с углеводородами, обзоры их приводятся в [64—67,182]. За-
тем изучались процессы изомеризации, отрыва отдельных функциональных
групп — элиминирования, димеризации и полимеризации. Некоторые примеры
таких процессов приведены в табл. 5.3—5.6, заимствованных нами из [64].
Анализ показателей процессов и полученных продуктов (табл. 5.3, 5.4,
5.5, 5.6) свидетельствует о высоких степенях конверсии некоторых из них
и большей селективности. Это позволило автору [64] предложить использо-
вать неравновесную плазму для препаративной химии.
242
Таблица 5.3
Примеры реакции изомеризации углеводородов в неравновесной плазме
Реагент Конверсия, % Продукт; содержание, %
транс-Стильбен 20 tyiw-Стильбен — 95
Анизол 67 Крезол, орто — 48, пара — 25
Фенилэтанол 30 Этилфенол, орто — 41, пара — 29
н-П ропилфенилэфир 30 Пропилфенол, орто — 38, пара — 19 Этилфенол, орто —1, пара — 0,5 Крезол, орто — 4, пара — 7
1-Нафтилметилэфир 13 2 1Мстил1 нафтол — 48
2-Нафти лмети лэф ир 88 1-Метил-2-нафтол — 45
Дифенилэфир 40 2-Фенилфенол — 36 4-Фенилфенол —18 Дибензофуран — 9
М,М-Диметиланилин 15 N-Метил-о-толуидин — 28 N-Метил-п-толуидин —15
н-Метиланилин 6 Толуидин, орто — 28, пара — 5
Циклооктатетраен 80 Стирол — 40
Пирол 6 цис-, трянс-Кротоннитрил — 57
Таблица 5.4
Примеры реакций элиминирования углеводородов в неравновесной плазме
Реагент Конверсия, % Продукт; содержание, %
Бензоальдегид 63 Бензол — 81, дифенил —16
2-Пиридин формальдегид 25 Пиридин — 70; 2,2-дипиридил — 20
Бензофенол 63 Дифенил — 70
Дибензоил 16 Дифенил — 98
Циклогексанон 8 Циклопентан — 5
1-Нафтол 20 Инден — 76
2-Нафтол 24 Инден — 93
Фталевый ангидрид 70 Дифенилен — 60
Трифенилен — 27
Тетралей 50 Нафталин — 60
В неравновесной плазме изучали пиролиз углеводородов [65—69]. В плаз-
ме тлеющего разряда при давлениях 40—50 торр метан до 80% превращает-
ся в ацетилен, концентрация его достигала 9 об. %, энергозатраты составили
13 кВт-ч!.м? С2Н2 [65]. Однако процесс идет с большим выделением сажи.
В плазме тлеющего разряда при давлении до 100 торр подвергали разло-
жению пары ацетона и бензола [65]. Продуктами в первом случае были СО,
СО2, С2Н4, С2Н6, Н2, а во втором — Н2, СН4, С2Н4, С2Н2.
Однако механизмы этих процессов практически не исследовались. Сос-
тав продуктов их часто близок к составам, полученным в процессах терми-
ческого пиролиза, фотохимических процессах и в масс-спектрометрии. Наб-
людаются также реакции, не протекающие ни в одном из указанных типов
процессов.
Значительное число работ посвящено изучению процессов полимериза-
ции в неравновесной плазме, а также исследованию поведения полимеров
в ней. Эти исследования были вызваны главным образом необходимостью
243
Таблица 5.5
Примеры реакций образования углеводородов с шестичленными кольцами
в неравновесной плазме
получения полимерных диэлектрических пленок на различных подложках.
На закономерностях образования таких пленок мы остановимся ниже (см.
стр. 277). В неравновесной плазме ВЧ-электродного разряда, генерированной
на частоте 3,14 мГц при давлениях от 0,5 до 5 торр, плотности тока 0,1 —
3 мА/см2, напряженностях поля 150—400 в/см и температурах стенок разряд-
ной камеры от 0 до 50° С, в [70] из мономеров — стирола, этилена, тетра-
фторэтилена, винилхлорида, винилфторида, винилацетата, метилметакрила-
та, винилиденхлорида — были получены соответствующие полимеры. Их об-
разование наблюдалось в этой работе только на электродах и практически
отсутствовало на стенках разрядной камеры. Скорость осаждения их воз-
растала с уменьшением температуры. По мнению автора [70], механизм по-
лимеризации включает адсорбцию молекул на поверхности электродов, реак-
цию их с положительными ионами, образованными в плазме.
В [70, 71] и обзорной работе [73] приводятся результаты изучения про-
цессов полимеризации бензола, толуола, этилбензола и стирола в тлеющем
разряде в диапазоне температур от —115 до —130° С. Оказалось, что ско-
рость роста пленок экспоненциально уменьшается с температурой, что сви-
детельствует о гетерогенном характере процесса. Кажущаяся энергия акти-
вации определяется суммарным действием полимеризации и адсорбции. Про-
244
Таблица 5.6
Примеры реакций образования углеводородов с пятичлеиными кольцами
в неравновесной плазме
дуктами полимеризации этилбензола были 1,2- и 1,3-дифенил бутаны, а
полимеризации толуола — 1,2-дифенилэтан [72].
Широко исследуется в неравновесной плазме полимеризация кремний-
органических мономеров. Так, в [74] из тетраэтил-, триэтилвинил- и три-
этилэтинилсиланов в тлеющем разряде на поверхности твердого тела полу-
чены полимеры. Скорость образования пленок зависит от активности моно-
меров при полимеризации и минимальна при полимеризации Et4Si. Поли-
меризация октаметилциклотетрасилоксана в тлеющем разряде на стенках
разрядной трубки при температуре —78° С и давлении 0,3 торр описана
в [75]. Полученный полимер характеризуется заметной степенью кристаллич-
ности, плохо растворим в органических растворителях, но хорошо раство-
ряется в мономере.
Неравновесная плазма может вызывать и изменение структуры полимера,
находящегося в ней [76, 233]. Например, в пленке полистирола, помещен-
ной в зону плазмы воздуха, кислорода или азота, происходят процессы
деструкции и сшивания полимерных цепей (меняется молекулярный вес).
Образуется большое число кислородсодержащих продуктов [76].
245
3. Плазмохимический процесс получения
ацетилена из паров углерода и водорода или метана
Процесс образования ацетилена из паров углерода с водородом изучали
в установках с дугой высокой интенсивности в равновесных условиях. Ре-
зультаты его приведены в обзорной работе [29]. При давлении 1 атм и тем-
пературе сублимации графита в газообразных продуктах находилось
19,3 об. % С2Н2, в случае проведения процесса в смеси водорода (0,33 атм)
и гелия (0,67 атм) концентрация С2Н2 возросла до 24%. Эти величины хоро-
шо согласуются с термодинамически рассчитанными. Если же на той же
установке проводить реакцию метана с графитом, то выход ацетилена дос-
тигает 52%, причем концентрация ацетилена растет с увеличением расхода
газа при достаточном для реакции количестве паров углерода. И эти резуль-
таты хорошо согласуются с результатами термодинамических расчетов для
оптимальных условий. Однако затраты электроэнергии велики и значительно
больше величин, характеризующих используемые в настоящее время промыш-
ленные процессы.
4. Плазмохимический процесс образования углеводородов
из водорода и окиси углерода
Этот процесс исследовали в неравновесной плазме тлеющего разряда в.
смесях СО и Н2 при давлении 10 торр [65] и СВЧ-разряда при давлениях
10—50 торр и длительностях пребывания в разрядной зоне порядка несколь-
ких минут [66]. В первом случае наблюдались малые выходы ацетилена и
метана, в то время как во втором случае конверсия углерода в углеводороды
достигала 80%. Продуктами были метан, ацетилен и пары воды.
5. Плазмохимический процесс получения сажи
и водорода
Если при плазмохимическом пиролизе углеводородов не организовано-
принудительной закалки, то в результате можно получить сажу и техни-
ческий водород. Качества получаемых саж, которые являются высокодис-
персными и обладают развитой вторичной структурой, намного выше, чем
у сажи, получаемой в печах. В работе [77] предложена кинетическая модель
сажеобразования и приведены результаты расчетов и экспериментального
исследования процесса. Продполагалось, что процесс состоит из термического
разложения углеводородов до углеродного пара и углеводородных радикалов
с образованием зародышей новой фазы, роста и коагуляции частиц. Кроме
того, учитывалась возможность образования зародышей в объеме при
взаимодействии молекул углеводородов с сажевыми частицами.
Сажу получали при разложении метана в аргоновой плазменной струе.
Характеристики ее таковы: удельная поверхность 145—150 <м2/г, масляное
число 1,4—1,6 мл/г, pH = 8,25.
Результаты экспериментального исследования плазмохимического про-
цесса получения сажи и технического водорода пиролизом углеводородов
описаны в основном в патентной литературе [78—87]. Эксперименты прово-
дили в электродуговых и высокочастотных безэлектродных плазмотронах,
а также в установках с дугой высокой интенсивности. В качестве сырья
использовали метан, природный газ, пропан, бензин, специально изготов-
ленные смеси, содержавшие до 40% ароматических углеводородов (толуол,
ксилол, нафталин), ароматические и нафтеновые углеводороды. Процесс
проводили как непосредственно в дуге, так и в плазменных струях аргона,
азота, водорода при температурах от 1500 до (5—6) • 103 СК, временах пребы-
вания в реакционной зоне ~ 10~3—10~2 сек. Закалка осуществлялась за-
тапливанием струями холодного газа (иногда углеводородами) либо на ох-
лаждаемой поверхности. Как и следовало ожидать из кинетических сообра-
жений, максимальный выход сажи имел место при пиролизе ароматических
246
углеводородов и составлял от 45 до 70 вес. %, энергозатраты колебались от
7 до 13 кВт-ч!кг сажи. При пиролизе парафиновых и нафтеновых углеводо-
родов выход сажи снижался до 18 вес. %, а затраты возрастали до ~ 70 кВт-
-ч/кг. Масляное число, характеризующее структуру сажи, составляло 1,8—
4,0 смЧг, в то время как для наиболее высокоструктурной ацетиленовой
сажи оно не превышает 2,4 см?1г.
Высококачественную сажу с заданной формой кристаллов получают так-
же из дешевого углеродосодержащего материала (включающего до 90 вес. %
углерода в расчете на сухое вещество) или низкосортной сажи [80]. В плаз-
му сырье подается в таком количестве, чтобы большая часть углерода пере-
ходила в газовую фазу. В процессе закалки продуктов происходила конден-
сация углерода. Свойства полученной в плазме сажи как наполнителя нам-
ного превосходят свойства саж, полученных другими способами. Однако в
настоящее время не существует промышленного плазмохимического процес-
са получения сажи. Необходимы как кинетические исследования процесса,
так и изучение технологического оформления его.
Одновременно с образованием конденсированной фазы в объеме конденси-
рованные продукты осаждаются на поверхности реактора. При плазмохи-
мическом пиролизе углеводородов на поверхности образуется пирографит
в результате реакции твердого углерода непосредственно с углеводородами.
Этот процесс протекает как в равновесной плазме при температурах 1800—
2100° К и давлении ~ 1 атм [88, 89], так и в неравновесной плазме, напри-
мер коронного разряда, возбуждаемого на частотах 50 Гц — 50 кГц [90].
6. Плазмохимические процессы получения дициана
Термодинамика системы С—N. По данным [91, 92], максимальное значе-
ние концентрации C2N2 (~ 0,5 об. %) реализуется при Т ~ 3500° К, однако
в диапазоне 3500—5000° К концентрации радикала CN, из которого воз-
можно образование C2N2 в процессе
закалки, достигает 40—55% (С : N =
= 1:1, 1,0 ата; рис. 5.7). Мини-
мальные затраты энергии на полу-
чение 1 кг C2N2 в этом предположе-
нии составляют 10,2—11,2 кВт-ч.
Кинетика плазмохимического про-
цесса получения дициана. Механизм
образования дициана в плазме рас-
смотрен в [93]. Предполагается, что
реакции идут в газовой фазе, образо-
вание дициана осуществляется при
рекомбинации радикала GN. Автора-
ми [93] рассмотрены следующие ре-
акции: N2 4- М N + N 4-М, N4-
4- С 4- М CN 4- М, CN 4- CN 4-
+ М C2N2 + М.
Уравнения химической кинетики
для этих реакций были решены на
ЭВМ для процесса в реакторе (при
постоянной энтальпии) и в закалоч-
ном устройстве (при задаваемом вре-
менном снижении температуры).
Результаты расчетов приведены на
рис. 5.8. В реакторе при рассмотрен-
Рис. 5.7. Температурная зависимость со-
става системы С—N
Р « 1 ата, С : N = 1 : 1
247
Рис. 5.8. Кинетика образования дициана
Стрелкой отмечен момент начала закалки
ных условиях (температура 7000° К, N : С = 1 : 1) происходит диссоциация
азота и образование радикала CN, стационарная концентрация которого
достигается за ~ 5- 10~4с^к. Дициан образуется на стадии закалки (времен-
ной закон снижения температуры приведен на рис. 5.8), его концентрация
достигает ~ 12 об.%.
Экспериментальное изучение процесса получения дициана. Дициан обра-
зуется при взаимодействии испаренного с электрода углерода с плазмой
N2 [94] или с введенной в азотную плазму сажей при Т ~ 4000 К. Концен-
трация его не превышает 1 об. %, конверсия углерода достигает 15%. В по-
следних работах получены концентрации дициана порядка 18%, что хорошо
согласуется с расчетными [92].
ПИРОЛИЗ УГЛЕВОДОРОДОВ В ПЛАЗМЕ N2
1. Равновесные процессы
Термодинамика системы С—Н—N. По литературным данным [91, 95,
96], в равновесных составах максимальная концентрация HCN достигает
5—6 об. % при Т ~ 3000° К, а концентрация С2Н2 не превышает 1%
(Р = 1 ата, С : Н : N = 1 : 4 : 2). В квазиравновесных составах оптималь
ные условия образования С2Н2 и HCN реализуются при температурах 1500—
2500° К, отношениях C:H:N = 1:2:2h1:1:4hP = 1,0 ата, когда
[С2Н2] = 22,1 и [HCN] - 20,6 об.% и [С2Н2] = 13,9 и [HCN] - 12 об. %
соответственно, а энергетические затраты во втором случае составляют
4,8-5,3 кВт*ч!кг С2Н2 с HGN [96].
Кинетика образования цианистого водорода и ацетилена из метана в
азотной плазме. В предложенном в работе [93] механизме процесса цианис-
тый водород образуется при взаимодействии атомарного азота с продуктами
разложения метана, протекающего по схеме Л. Касселя. Он включает сле-
дующие реакции:
2СН4-^С2Н4 + Н2,
С2Н4 С2Н2 -j- Н2,
HCN + М CN + Н + М,
с2 + м = с + с + м,
248
С2Н2 -> 2С + Н2,
N + C2H4~>HCN + СНз,
N + С2Н2 — HCN + СН,
N + СНз -» HCN + Н2,
N2 + М N + N + М,
н2 + м —+ н + м,
CN + М С + N + М,
CN + H2z±HCN +Н,
с + Н2 — СН + н,
СНз + СНз С2Н4 4- Н2,
СН + СН — С2Н2,
Н + CN 4- М HCN 4- М,
Н 4- С + М СН 4- М.
Кинетические расчеты проводили для процесса в реакторе и на стадии
накалки при предположениях, описанных на стр. 234. В качестве началь-
ного состава азотной плазмы принимали равновесный состав, соответствую-
щий задаваемой температуре азотной плазмы. Результаты расчета для
Т^2 = 6500° К, отношения азота к метану, равного 3, приведены на рис.
5.9. Закалка включалась в момент максимальной концентрации HCN, сред-
няя скорость ее составляла ~ 106 град/сек. В рассматриваемых условиях
максимальные концентрации ацетилена и цианистого водорода разделены
во времени. Конечный состав продуктов определяют рекомбинационные
процессы, протекающие на стадии закалки.
Результаты экспериментального изучения плазмохимического процесса
получения смесей С2Н2 4- HCN. По данным [94, 97—99], реакционная смесь,
содержащая С2Н2 и HCN, может быть получена как пиролизом углеводородов
в плазменной струе азота, так и в результате реакции испаряющегося > ка-
тода плазматрона углерода с Н2 или NH3 в плазменной струе N2[94]. Во вто-
ром случае 50% испаренного углерода переходит в HCN, однако скорость
образования HCN мала и определяется скоростью испарения углерода.
При введении СН4 в плазменную струю N2 скорости образования HCN и
С2Н2 резко возрастают, 91% углерода переходит в целевые продукты. Луч-
шими были получены следующие показатели процесса [97]: в пиролизном
газе содержатся до 10,7 об. % HGN, 12,9 об. % С2Н2 и 74 об. % Н2, которые
хорошо согласуются с термодинамически рассчитанными. Примерно те же
показатели процесса приводятся в патентах [100, 101].
Рис. 5.9. Кинетика образования HCN
Стрелкой отмечен момент начала закалки
249
2. Получение С2Н2 и HCN в неравновесной плазме
Процесс исследовали в [65, 231] в плазме тлеющего разряда в смесях азота
и метана при давлении 10—15 торр. Конверсия зависит от плотности тока и
соотношения компонентов. Максимальной величины конверсия (~ 80%)
достигает при концентрации СН4 в смеси ~ 15 об.%. Одновременно с HCN
образуется и С2Н2. В зависимости от плотности тока меняется соотношение
этих продуктов.
ПИРОЛИЗ УГЛЕВОДОРОДОВ В ПЛАЗМЕННЫХ СТРУЯХ,
СОДЕРЖАЩИХ ХЛОР
1. Квазиравновесные плазмохимические процессы
получения реакционной смеси для производства винилхлорида
Термодинамика системы С—Н—С1. В квазиравновесных составах (без
учета Gs и ароматических углеводородов) при температурах 1200—1800° К
присутствуют только углеводороды С2Н4, С2Н2, СН4, Н2 и НС1 [60, 102, 103]
(см., например, рис. 5.10). Максимальная концентрация С2Н2 реализуется
при 1700° К, при меньших температурах на концентрацию С2Н2, выход аце-
тилена, затраты энергии на получение 1 кг смеси С2Н2 с НС1 сильно влияют
Рис. 5.10. Температурная за-
висимость квазиравновесного
состава системы С—Н—С1
Р = 1 ата,
С : Н : С1 = 1 : 4 : 0,435
величины отношений С/Н и С/С1. Уменьшение отношения С/С1 и увеличение'
С/Н увеличивают выход С2Н2. Изменение давления сильно влияет на составы
при 1200—1500° К. При 1500—1800° К при С/С1 > 2 в составах содержатся
равные [С2Н2] и [НС1] (до 20 об. %), выход ацетилена (по углероду) достигает
80—90 вес. %, а затраты энергии не превышают 2,0 кВт-ч/кг смеси С2Н2
с НС1.
Макрокинетика плазмохимического пиролиза хлоруглеводородов. Ки-
нетика разложения хлоруглеводородов при температурах выше 1000° К так-
же практически не исследована. Оценки характерных времен разложения их,
выполненные в [36] по описанным в литературе кинетическим данным,
экстраполированным до Т ~ 2000—3000° К, свидетельствуют об относитель-
но малых временах по сравнению с временами образования конечных про-
дуктов. Анализ же экспериментальных данных плазмохимического пиро-
лиза их [60, 102, 103] показывает, что при температурах реакции 1600—
2000° К основными продуктами являются НС1, С2Н2, С2Н4, Н2, СН4. При-
чем практически весь хлор, содержавшийся в исходном хлоруглеводороде,
переходит в НС1, а соотношение углеводородов характерно для случая
плазмохимического пиролиза углеводородного сырья примерно при тех же
температурах и отношениях С/Н. Поэтому можно предположить, что разло-
жение хлоруглеводородов протекает следующим образом: за относительно
250
малые времена (~ 10~6 сек) исходные соединения распадаются на НС1, СН4,
С2Н4, Н2 и в случае присутствия в сырье ароматических групп — С2Н2,
затем эти углеводороды распадаются по уравнениям схемы Л. Касселя для
разложения метана. Отношения СН4: С2Н4 : Н2 : [С2Н2] выбираются в соот-
ветствии с описанными (см. стр. 234) соображениями.
Нами были выполнены расчеты макрокинетики плазмохимического пиро-
лиза смеси хлоруглеводородов для условий пиролиза в плазменной струе
водорода [36]. Смесь описывается формулой С3,35Н8,35С1Ь45, средний молеку-
лярный вес 100. Решалась описанная выше система уравнений, закалка со
скоростью 5-106 град/сек «включалась» на различных длинах реактора. Ва-
рианты начальных условий подбирались по изложенным выше соображениям.
Экспериментальные и расчетные концентрации [С2Н2], [Н2] и [НС1] хорошо сог-
ласуются друг с другом; [СН4] в экспериментах больше расчетных, а [С2Н4] —
меньше (табл. 5.7). Однако характер зависимости содержания С2Н4
в пиролизном газе от длины реактора одинаков. Отличия в абсолютных
значениях могут быть сязаны с причинами, обсужденными выше на
стр 236.
Таблица 5.7
Сопоставление экспериментальных и расчетных данных (вес. доли)
плазмохимического пиролиза хлоруглеводородов
Газ Длина реактора
2,5 см 5 см 10 см 15 см
Опыт * Расчет Опыт Расчет Опыт Расчет Опыт Расчет
€Н4 0,06 0,038 0,06 0,036 0,06 0,035 0,06 0,034
С2Н4 0,06 0,138 0,05 0,106 0,04 0,072 0,04 0,052
С2Н2 0,24 0,239 0,29 0,263 0,29 0,283 0,28 0,291
€ Не опре- делялась 0,013 Не опре- делялась 0,020 Не опре- делялась 0,031 Не опре- делялась 0,042
Н2 0,07 0,067 0,07 0,070 0,07 0,074 0,07 0,076
НС1 0,51 1 0,501 1 0,51 0,501 0,51 0,501 0,50 1 0,501
Расход: 4 нлг3/ч Н2, 7,2 кг/ч сырья, мощность
в плазме 11,8 кВт; закалка в теплообменнике.
Плазмохичические процессы получения смесей С2Н2 с НС1. Реакционная
смесь с равными концентрациями С2Н2 и HG1, необходимая для производства
мономера винилхлорида, может быть получена пиролизом углеводородов в
плазменной струе, содержащей Н2, HG1, С12, СН4, либо пиролизом хлоругле-
водородов или их смесей, например, отходов хлорорганических производств,
в плазменной струе Н2 [60, 59].
Весьма подробно [59, 60] изучен пиролиз бензина с концом кипения 165° С
в плазменных струях смесей Н2—НС1, Н2—НС1—Cl2, Н2—Cl2—НС1—СН4
в интервале температур 1300—1700° К и времен контакта ~ 10-5—
10~4 сек. Отношение компонент в плазмообразующей смеси подбиралось
так, чтобы можно было создать сбалансированный процесс получения син-
тез-газа для винилхлорида с использованием образующихся газов после вы-
деления целевых продуктов.
В этом температурном диапазоне содержатся только углеводороды и HG1,
характер температурных зависимостей концентраций углеводородов такой
же, как и в случае пиролиза углеводородов с тем же С/Н в плазменной струе
Н2 или смеси Н2—СН4. Увеличение температуры в этом интервале приводит
к росту [С2Н2] с 10 до 20 об.%, максимальная [С2Н4] реализуется при 71 =
= 1400° К и достигает 14 об.%. Равные концентрации [С2Н2] и [НС1] (17—
20 об. %) (табл. 5.8) могут быть получены при Т ~ 1550— 1700° К и времени
251
Таблица 5.8
Пиролиз бензина в плазменной струе смеси Н2, НС1. С12, СНд
Режим
1 2 3 4
Газ, об. %
Водород 43,86 48,59 47,79 45,25
Метан 6,94 8,12 8,11 7,94
Хлористый водород 21,00 16,80 17,00 21,00
Ацетилен 21,70 16,73 17,00 21,00
Этилен 5,14 8,06 8,50 3,71
Этан — — 0,07 —
Пропилен 0,40 0,38 0,45 0,20
Пропадиен 0,15 0,17 — —
Аллен — 0,16 0,09
Метилацетилен 0,17 0,31 0,30 0,18
Диацетилен Следы Следы Следы Следы
Винилацетилен 0,27 0,24 0,25 0,13
Бутадиен 0,08 0,18 0,15 —
Бутилен — 0,02 —
Бутан —. —- — —
Пентан 0,30 0,42 0,20 0,20
Гексан —. — — —
Показатели
Мощность, кВт 27,1 20,4 13,1 14,8
Расход, нм21ч
Н2 2,4 3,6 2,6 2,2
СБ — — 0,45 0,73
НС1 3,6 2,4 0,9 0,7
СН4 — — 0,35
Расход бензина, кг/ч 7,2 6,0 4,4 4,0
т, °к 1650 1600 1550 1700
Время контакта, 10~4 сек 4,5 5,6 7,9 7,3
Количество продуктов, им?/ч 17,1 14,3 10,6 10,4
Конверсия, вес.% 98 97 99 96
Выход С2Н2, вес. % 62 48 49 67
Выход С2Н2 + С2Н4, вес. % 78 72 73 79
Энергозатраты, кВт-ч/кг (СгНг + СгН^ 5,0 4.7 4,2 4,7
контакта ~ (5—15)-10“5 сек. Степень превращения сырья и выходы С2Н2
и смеси С2Н2 с С2Н4 высоки. Энергозатраты 4,2—5,0 кВт-ч/кг С2Н2 +
+ С2Н4, причем меньшие значения соответствуют условиям пиролиза в
плазменной струе, содержащей С12, что связано с выделением энергии при
реакции образования HCL
Закалка продуктов реакции бензином позволяет получить дополнитель-
ное количество С2Н4 и снизить на 15—20% энергозатраты по сравнению с
соответствующими величинами, полученными при закалке в теплообменнике.
Оптимальные результаты получаются при закалке продуктов ацетиленового
режима и отношении расходов сырья, идущего на пиролиз и закалку, равно-
го I : 0,7.
Не менее подробно изучен плазмохимический пиролиз хлоруглеводоро-
дов, которые (и смеси которых) являются побочными продуктами хлорорга-
нических производств [102, 103]. Показатели пиролиза дихлорэтана, тре-
тичного бутилхлорида, нетоксичного изомера гексахлорана и смеси хлор-
252
Таблица 5.9
Плазмохимический пиролиз хлоруглеводородов
Дихлорэтан, 8 кг/ч Третичный бутилхлорид, 5,4 кг 1ч Гексахлоран * Смесь ** 7,2 кг/ч
режим 1 режим 2 режим 1 режим 2 режим 1 режим 2
Газ, об. %
Водород 47,46 41,82 56,24 56,49 56,31 52,34 52,90
Метан 0,68 0,64 7,80 6,53 1,45 6,72j 5,23
Хлористый водород 34,05 38,57 12,73 13,53 28,51 19,43 19,50
Ацетилен 12,35 17,00 13,08 20,50 12,30 14,73 19,53
Этилен 2,69 1,18 2,25 2,03 1,03 3,27 2,11
Пропилен — — 0,40 — — 0,40 0,10
Аллен — — 1,61 0,23 — 0,53 0,16
Метилацетилен 0,11 0,10 1,80 0,24 — 0,63 0,25
Диацетилен Следы Следы Следы Следы Следы Следы Следы
Винилацетилен 0,10 0,18 0,23 0,23 — 0,21 —
Изобутилен — — 1,51 0,08 — — —
Винилхлорид 2,56 0,61 0,15 — — 1,03 —
Бутилхлорид — — 2,20 0,14 — — —
Бутадиен — — — — — 0,44 —.
Хлор этил — — — — — 0,27 —
Окись углерода — — — — 0,40 — —
Показатели Мощность, кВт 6,9 9,8 9,5 13,4 10,0 11,6 14,3
Расход, нлР/ч
Н2 4 4 4 4 4 3,4 3,5
СН4 — — — — — 0,6 0,36-
С2Н4 — —. — — — — 0,14
т, °к 1400 1700 1400 1650 — 1400 1600
Количество продуктов, нмР/ч 7,8 9,1 8,6 9,5 — 10,2 11,2
Конверсия, % 85 99 85 99 — 90 99
Выход С2Н2, вес. % 67 87,5 46 75 — 64 88
Энергозатраты, кВт-ч/кг С2Н2 6,15 5,45 7,2 6,0 — 6,6 5,7
С2Н2 + НС1 1,26 1,30 3,0 3,1 — 2,3 2,3
* Плазменная струя вводилась в сосуд с сухим сырьем.
** 15 об.% трихлорэтана, 15 об.% дихлорэтана, 15£об.% третичного бутилхлорица, 10 об.% хлористого
этила, 5 об.% дихлоризобутанов, 5 об.% трихлоризобутиленов, 5 об.% монохлорбутенов, 30 об.%,
остальное.
углеводородов в плазменной струе при температурах от 1400 до 1700° К и
времени контакта ~ 10~4 сек приведены в табл. 5.9. С повышением Т возрас-
тает [С2Н2] и уменьшаются [С2Н4], [C3H6J, [^о-С4Н8] и [С2Н3С1]. Для образо-
вания составов, удовлетворительно согласующихся с термодинамическими,
необходимы времена контакта ~ 10~4 сек. Результаты кинетических расче-
тов удовлетворительно согласуются с экспериментальными. Закалка про-
дуктов пиролиза может быть организована с использованием бензина или
смеси хлоруглеводородов. В последнем случае достигается сохранение це-
левых продуктов без увеличения в газе пиролиза ненасыщенных углеводо-
родов (отношение расходов на пиролиз и закалку 1 : 2, хлоруглеводороды
с двумя углеродными атомами). Если же используется смесь хлоруглеводо-
родов с четырьмя углеродными атомами, в процессе закалки образуются
253
изобутилен, пропилен и аллен. Описанные выше процессы пиролиза бензи-
на в хлорсодержащих плазменных струях и плазмохимического пиролиза
хлоруглеводородов, позволяющие получить реакционную смесь для произ-
водства винилхлорида, также упрощают технологию и резко снижают его
себестоимость.
Себестоимость винилхлорида, получаемого из газов плазмохимического
пиролиза бензина и побочных продуктов хлорорганических производств,
соответственно в 1,6 и 2,3—2,5 раза ниже его себестоимости в процессах
фирмы «Куреха» и «сбалансированного» из этилена. Процесс получения винил-
хлорида, включающий плазмохимический пиролиз углеводородного сырья,
не столь жестко связан с нефтеперерабатывающей промышленностью, как
другие, что позволит развивать хлорную промышленность в наиболее опти-
мальных районах страны, не привязывая ее к нефтехимическим комплексам.
Стоимость пускового комплекса при одинаковой мощности производства
винилхлорида в семь-восемь раз ниже, чем для «сбалансированного» процес-
са из этилена.
Технологические схемы производства мономера винилхлорида (ВХМ).
В разработанном в [59, 60, 102, 103] способе получения ВХМ сырьем могут
служить любые дешевые технические углеводородные смеси, включая бензи-
ны, газойли, сырую нефть, хлор и (или) хлористый водород; выходы ВХМ
не ниже 70—80%, считая на углеводородное сырье; затраты энергии, холода,
растворителей значительно ниже, чем в известных способах; подготовка
реакционной смеси для синтеза ВХМ может происходить совместно с полу-
чением компонентов этой смеси. Процесс получения ВХМ включает сле-
дующие стадии: а) пиролиз углеводородного сырья в плазменной струе,
содержащей Н2, СН4, HG1 и (или) С12. Отношение компонент подбирается так,
чтобы в продуктах пиролиза были равные концентрации [С2Н2] и [HC1J;
б) закалку продуктов пиролиза водой или бензином; в) очистку газов пироли-
за от высших ненасыщенных углеводородов (абсорбцией керосином при
—15° С и 6—10 ата); г) гидрохлорирование ацетилена (в присутствии суле-
мового катализатора на активированном угле) и выделение ВХМ (абсорбцией
дихлорэтаном при 10 ата); д) смешение с хлором; е) хлорирование этилена в
жидком дихлорэтане в присутствии катализатора (хлорного железа при
5 ата); ж) пиролиз дихлорэтана (450—550° С), выделение ВХМ и использование
НС1 в составе плазмообразующего газа. Газы стадии хлорирования этилена
(смесь Н2 и СН4) используются частично в составе плазмообразующего газа,
топлива при пиролизе дихлорэтана, для предварительного подогрева угле-
водородного сырья или получения товарного водорода.
В качестве сырья предлагается использовать бензин с концом кипения
165° С или сырую нефть.
ВХМ может быть получен при плазмохимическом пиролизе смеси хлор-
углеводородов — побочных продуктов хлорорганических производств. Пи-
ролиз хлоруглеводородов проводится в плазменной струе смеси Н2, СН4 и
С2Н4, возвращаемой со стадии выделения ВХМ. Сырьем служат побочные
продукты отдельных или нескольких хлорорганических производств, под-
бираемые и в случае необходимости разбавляемые углеводородами так, что-
бы отношение С : Cl = 1 : 0,42; в этом случае в газе пиролиза — равные
концентрации [С2Н2] и [HC1J. Закалка продуктов производится затаплива-
нием струями жидких углеводородов или хлоруглеводородов; последние
не должны содержать соединения с числом С > 2. После очистки газов пи-
ролиза от высших ненасыщенных углеводородов проводят гидрохлорирова-
ние ацетилена. ВХМ выделяется, остальные газы используются в качестве
плазмообразующего, топлива для подогрева сырья или процессов гидриро-
вания.
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ ОКИСЛЕНИЯ
УГЛЕВОДОРОДОВ
1. Процессы окисления углеводородов в равновесной плазме
Термодинамика системы С—Н—О. В работе [104] выполнен термодинами-
ческий анализ процессов конверсии углеводородов (метана, этана, пропана
и углеводородов ряда С?1Н2П) водяным паром и двуокисью углерода при коэф-
фициенте расхода окислителя 1—1,5 при давлениях 1—30 ата и в диапазоне
температур 400—6000° К. Оказалось, что концентрации непредельных угле-
водородов, углеводородных радикалов, формальдегида в описанных выше
условиях не превышают 10-4 об.%. При отношении С : О = 1 : 1 при Р -
= 1 ата и температурах, больших 1200° К, в квазиравновесных составах
присутствуют только Н2 и СО; их соотношение зависит от величины G : Н.
При Т 2500° К начинается заметная диссоциация Н2; СО диссоциирует
при температурах выше 6000° К. Минимальное значение затрат энергии на
получение 1 нм3 смеси СО с Н2 при С : О = 1 реализуется при Т ~ 1000° К
и составляет ~ 1 кВт-ч. При температурах 1200—2500° К оно возрастает
незначительно в связи с тем, что состав практически не меняется.
Кинетика процессов окисления углеводородов. В работах [104—106]
описаны результаты расчетов кинетики окисления метана кислородом [105,
106] и двуокисью углерода и водяным паром [104]. В первом случае предпо-
лагали, что процесс протекает по радикально-цепному лгеханизму. Резуль-
таты расчета кинетики этого процесса (на ЭВМ) по этому механизму удов-
летворительно согласуются с экспериментальными данными. На рис. 5.11
показаны расчетные кривые относительного выхода формальдегида, расхода
метана, образования окиси углерода, а также экспериментальные точки.
В интервале 800—1700° К экспериментальные точки удовлетворительно ло-
жатся на расчетные кривые, и, следовательно, положенный в основу расчета
цепной механизм процесса имеет место в указанном интервале температур.
В [104] рассмотрен радикальный механизм окисления СН4 углекислым
газом или водяным паром. Однако окисление непредельных соединений и
ацетилена не включалось в механизм процесса. Анализ расчетов кинетики
процесса для одного из вариантов расчетов — изоэнтальпийных условий
(предполагалось, что в реакции с СН4 вступают имеющие квазиравновесный
состав СО2 при 3500° К) показывает, что превращение метана происходит за
времена ~ 10~4 сек, атомарный и молекулярный кислород — продукты
диссоциации СО2 — реагируют за времена 3*10-7 и 5-10~6 сек соответственно.
Рис. 5.11. Зависимость выхода формальдегида (1), окиси углерода (2), расхода метана (3)
от температуры реакции
Время контакта ~ 10-4 сек; 1—з — расчетные кривые; X, О, • — экспериментальные точки
255
Результаты экспериментального изучения плазмохимических процессов
окисления углеводородов. В работах [106, 107] изучался процесс окисления
метана в плазменной струе аргона. Экспериментально изучено влияние тем-
пературы реакции на выход продуктов (Н2, НСНО, СО, СО2, Н2О, Н2О2).
Максимальный выход формальдегида (3,5% на исходный метан) наблюдался
при 1600-1700° К.
В [104, 108] исследовали конверсию метана, природного и сжиженного
газов в плазменных струях СО2 и Н2О. В интервале температур плазменной
струи 2400—3600° К основными продуктами были Н2 и СО при ~ 100%
конверсии углеводородного сырья. Продукты термического пиролиза угле-
водородов — промежуточные в данном процессе — наблюдались при его
проведении при Т 2400° К.
2. Окисление углеводородов в неравновесной плазме
Весьма перспективными являются процессы получения окисей олефинов
и спиртов в неравновесной плазме. Окислы олефинов С2—С10 образуются в
результате окисления соответствующих олефинов СО2 [110] или смесями
О2 с N2, N2O [111], или Н2О [112]. Процесс протекает при температуре сте-
нок 15—100° С, давлениях от 10 торр до 3,5 ата и временах пребывания в
разрядной зоне от 30 до 100 сек в кольцевом зазоре ~ 1,5 мм между коакси-
альными цилиндрическими остекленными электродами. К последним при-
ложено переменное электрическое поле от 30 до 60 Гц, напряженность его
в плазме составляет от 30 до 120 тыс. В1см. Выход окислов олефинов дости-
гает 20—30% и составляет ~ 100 г!кВт-ч. При окислении этилена водяным
паром в озонаторе [112] газообразные продукты состоят из Н2, СО, СО2 и
следов углеводорода, жидкая фракция содержит ~ 75 различны^ углеводо-
родов от Сг до С14, в том числе парафины, олефины и спирты. Увеличение
соотношения Н2О: С2Н4, мощности и температуры стенок приводит к росту
конверсии С2Н4. Уменьшение времени пребывания за счет повышения расхо-
да реагентов снижает степень превращения этилена. Спирты были получены
на установке, описанной в [110], из парафиновых углеводородов и СО2
примерно в тех же условиях [ИЗ]. Парафиновые углеводороды избирательно
превращаются в спирты с тем же числом атомов углерода.
В рассматриваемых процессах окисления, вероятно, немаловажную роль
в механизме играют процессы на стенках разрядной камеры. Так, в [229]
показано, что с увеличением температуры стенок разрядной трубки с 25
до 75° С в системе СН4—СО2 (600 торр, СН4 : СО2 = 1 : 1, 12,6 мА) воз-
растают скорости образования и выходы карбонильных и карбоксильных
соединений и подавляется образование полимеров на стенках. В газовой
фазе при окислении углеводородов образуются СО и Н2 и в некоторых слу-
чаях С2И2. Так, в неравновесной плазме тлеющего разряда в смесях СН4 с
СО2 и СН4 с Н2О при 50 торр и больших плотностях тока в продуктах были
СО и Н2 при 100%-ной конверсии СН4, а при малых плотностях тока — СО,
Н2 и С2Н2 [65].
ПИРОЛИЗ СЛАНЦЕВ И УГЛЕЙ
Изучен также процесс окисления горючего сланца (состав органической
части: 69,4% С, 8,7% Н)парами воды в плазменной струе аргона [109]. В эк-
спериментах были получены газообразные продукты, содержавшие СО
(~ 29,3%). Удельные затраты энергии на 1 нм3 газа составляли ~0,5 кВт-ч.
Проводятся исследования пиролиза углей различных месторождений в
плазменных струях дугового электродного [234] и СВЧ [235] плазмотронов
при атмосферном давлении. Газообразные продукты состоят в основном из
ацетилена, метана, водорода и окиси углерода. В твердых продуктах при
пиролизе бурого угля Подмосковного месторождения содержится — 94% С
[234].
256
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ ПИРОЛИЗА
ФТОРУГЛЕВОДОРОДОВ
1. Пиролиз фторуглеводородов в равновесной плазме
Термодинамика системы С—F. В равновесных составах основным фтор-
углеродным соединением от 2000 до 4000° К при 1 ата является C2F2 [114]
(рис. 5.12). При температурах, больших 4500° К, продукты состоят из ато-
марных частиц. Концентрации C2F4 не превышают 10~б мол.%. Без учета
конденсированного углерода в квазиравновесных составах дифторацетилен
в больших концентрациях присутствует до 2000° К. Это соединение ос-
тается основным при изменении отношения С : F от 0,1 до 10 и давлений от
0,1 до 10 ата. Однако экспериментально соединение G2F2 не было обнаружено
в указанном диапазоне температур. Кроме того, отсутствуют термодинами-
ческие данные для соединений типа CJF, где х = 1, 2, 3, которые, возможно,
существенны вблизи температуры сублимации углерода. Поэтому, вероят-
но, в будущем необходимо будет изучать эту систему опять, после появле-
ния надежных термодинамических данных для фторуглеродных соединений.
Кинетика плазмохимических процессов пиролиза фторуглеводородов.
В литературе нет полных кинетических данных для образования и разложе-
ния фторуглеродных соединений. В [114] предполагается, что синтез C2F<
Рис. 5.12. Температурная зависимость квазиравновесного (а) и равновесного состава (б)
системы С—F
Тs — температура сублимации. При Т < Ts Р — 1 ата, при Т > Ts Р > 1 ата, С : F = 2
9 Теоретич. и прикладная плазмохимия
257
(получаемый экспериментально с относительно большими выходами) в плаз-
ме дуги высокой интенсивности с графитовыми электродами протекает сле-
дующим образом:
1. В дуге при температурах выше 2000° К
CF4 —> C2F2.
2. В процессе закалки C2F2 + F -> GoF3, C2F3 + F C9F4, 2CF2
C2F4, 2G2F2 C2F4 + 2GS.
CF4, присутствующий в продуктах пиролиза, может быть получен по реак-
циям С + F -> CF, CF + F -> CF2, CF2 + F -> CF3, CF3 + F CFr
Газообразные соединения углерода участвуют в этих реакциях или кон-
денсируются на стенках в процессе закалки.
Экспериментальное изучение пиролиза фторуглеродных соединений в
плазме. Процесс образования тетрафторэтилена из тетрафторметана в ус-
тановке с дугой высокой интенсивности с угольными электродами изучался
в работе [114]. В результате было установлено, что выход тетрафторэтилена
возрастает с увеличением мощности и снижением давления. Максимальный
выход (~ 75 мол. %) был достигнут при мощности 20 кВт и давлении 0,1 ата.
Основными компонентами в продуктах после закалки были CF4, G2F4, C2F6y
C3F8. Подробная библиография по разложению различных фторуглеродных
соединений приводится в [114].
ПИРОЛИЗ ФТОРУГЛЕРОДНЫХ СОЕДИНЕНИЙ
В ПЛАЗМЕ АЗОТА
Термодинамика системы С—N—F. В равновесных составах при 1 ата,
соотношении С : N : F —- 1 : 4 : 4 до 4000° К основными фторуглеродными
соединениями являются CF2 и C2F2, при более высоких температурах — GFr
между 2500 и 4500° К — соединения, содержащие азот, FCN и CN. Концентра-
ция FCN достигает максимума ~ 1 мол. % при 3500° К, a [GN] — 5 мол. %
при 4500° К [114].
Кинетические данные для соединений этой системы отсутствуют. Вероят-
но, что продукты образуются главным образом в процессе закалки, когда
происходят последовательное фторирование неустойчивых промежуточных
соединений и димеризация [114].
Экспериментальное изучение пиролиза. Экспериментально исследован
пиролиз CF4, CHF3 [115] и других углеводородов [114] в плазменной струе
азота, закалка производилась в зонде со скоростью ~ 106 град/сек. Основ-
ными продуктами, содержавшими связанный азот, были NF3, CF3NF2 и в
небольших количествах N2F4, N2F2. Максимальная конверсия азота во фто-
риды составляла 0,85%. Выходы их увеличиваются с увеличением мощности
в плазме и отношения F/N.
При пиролизе CHF3 [115] в продуктах были обнаружены 57,7% CF4r
18,2% C,F6, 12,9% C2F4, 7,8% CF3CN и 3,4% C3F8.
ПЛАЗМОХИМИЧЕСКИИ ПИРОЛИЗ
ХЛОРФТОРУГЛЕРОДНЫХ СОЕДИНЕНИЙ
Пиролиз ряда хлорфторуглеродных соединений в плазме приведен в
[114] в сводке изученных работ по исследованию реакций фтора (табл. 5.10)
и описан в [116, 117]. Хлорфторметаны подвергали пиролизу в плазме аргона
при 1 ата и мощностях 2—10 кВт. Продуктами были хлорфторэтаны и про-
изводные бензола [116], которые, по предположениям авторов [117], обра-
зуются по радикальному механизму.
258
Таблица 5.10
Сводка работ по изучению плазмохимического пиролиза фторуглеводородов
Реагент Давление, торр Мощность, кВт Расход реагентов, (ммоль/ /сек), их соотноше- ние Продукт Конструкция реактора
cf4 CsFe C3F6 CsFie f2 cf4 + Cl2 CF2C1CF2C1 c2f5ci cf2ci2 CF3C1 CF4 + CC14 cf4 4- C6H6 CF4 + CH4 CF4 + C2H2 CF4 + NO CF4 4- 02 CF3COOH /O^/CsF? k/ CF4, Ar CF4 + n2 COF2+ NO n2 ZCF4 N N 1 1 FC CF \n^ cf4 + C2N2 CF4 + HCN c2f4 4- Br2 C2F4 + C2F4Br2 C2F4Br2 35-760 32 40 10 2 55 45 45 45 45 760 20 20 20 20—65 20 8 250 760 28 60 20 20 100—500 luO—500 100—500 0,1-28,5 0,4 0,4 0,4 0,068 0,5 0,45 0,45 0,45 0,45 3,0 1,3 1,3 1,3 0,7— 2,0 0,75 0,42 0,33 0,8 20 2,3 1,7 1,4 1,4 0,4 0,4 0,4 0,02-3,7 0,98 1,4 0,01 Постоян- ный объем То же 0,1 0,1 0,007 0,08 9 : 1 3 : 1 3 : 1 0,04; 0,08 0,06; 0,06 0,007 Постоян- ный объем 0,02; 0,2 35; 18 0,17; 0,10 0,19 0,19 2 : 1 2 : 1 C2F4, CF4, C2F6, CsFe C2F4, CF4, CsFs, C2Fg, C3F6 C2F4, CF4, C3F6, C2Fe, CsFg c2f4, cf4 cf4, c2f4, CsF6 CF4, CF2C12, CF3CI, C2F6, cf2cicf2ci C2F4, CF4, CF3CI, cf2ci2 CF4j C2F4, C2Fe, CF3GI C2F4, CF4, CF3CI, CF2C1CF2C1 cf4, c2f4, cf2ci2 cci2f2 C2F4, CF4, C2F6, HF C2F4, CF4, C2F6, HF C2F4, CF4, C2F6, HF c2f4, cf4, c2f6, n2, co, COF2, NF3, NO, N2O C2F4j CF4, C2Fe, co c2f4, cf4, C2F61 C3Fs, HF, CO c2f4, cf4, cof2, c2f6, co2 Смола NF3, CF3NF2, N.2F4, C2F6, n2f2, c2f4 NF3, n2f2, cf4, co2, n2o, NO, COF2 FCN,C2F4,CF4,C2F6,C2N2, N = :C—C=C—C—N c2f4, cf4, fcn, CF3CN, c2n2, n2 HF, N2, C2F4, CF4 Перфтор-ct, (о-дибром-ал- каны, перфтора лканы, 1-бромперфторалканы 1 -Бр оМперфтор алканы CFBr3, C2F3Br, Br2(CF2)4, смеси олефинов Дуга высокой ин- тенсивности, закал- ка на холодной стенке Угольная дуга с за- калкой То же » » Угольная дуга с вводом С12 Угольная дуга с за- калкой То же » Индукционная ВЧ- плазма аргона Угольная дуга с за- калкой То же » » » » СВЧ-разряд, вне его — графит Плазменная струя N2 с вводом фторида Угольная дуга, за- калка на холодной’ поверхности То же » » »,
259
9’
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
В НЕОРГАНИЧЕСКОЙ ХИМИИ
ПРОЦЕСС ОКИСЛЕНИЯ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
1. Окисление азота
Первым промышленным плазмохимическим процессом явилось окисление
атмосферного азота в плазме электрической дуги, реализованное в 1900 г.
в Норвегии (процесс Биркеланда — Эйде [119]) и в 1902—1904 гг. в США
(процесс Брэдли — Лавджоя [119]). В этих процессах концентрация NO
составила 1,8—2,0 об.%. В связи с успехами плазмохимии и технологии этот
процесс вновь привлек к себе внимание в 60-х годах. В настоящее время ис-
следован на установках лабораторных и промышленных масштабов в равно-
весной [12, 91, 120—123] и неравновесной плазме.
Окисление азота в равновесной плазме
Термодинамика системы N—О. Максимальная концентрация NO зависит
от давления и отношения N : О. При 1 ата в воздухе ойа составляет 5,2 об. %
при 3500° К и возрастает до 8 об. % при 4200° К при 10,0 ата [91, 123]
NO], л? %
27.--------
Рис. 5.13. Зависимость равно
весной концентрации окиси
азота от температуры при раз-
личных давлениях
О2 : N2 = 1 : 4.
1 — Р — оо шпсг,
2 - 10;
3 - 1,0;
4 — 0,1
I — экспериментальные точки при
давлениях 5—10 ата
(рис. 5.13). При более высоких температурах концентрация NO уменьшается,
но возрастают концентрации атомарного кислорода и азота. Последние, по
мнению авторов [91], могут участвовать в образовании NO на стадии за-
калки.
Кинетика образования окислов азота в плазме. В [91, 123] рассматри-
вается образование NO в газовой фазе по цепному механизму, предложенному
в [124], включающему следующие реакции:
О2 + М О + О + М, О + N2 NO + N, N + О2 NO + О,
N2 + М N + N + М, NO + М N ч- О + М.
Кинетические характеристики образования и закалки окиси азота в
J123] рассчитывали по уравнению, полученному Зельдовичем [124]:
d(NO) Л Г лпз ехр (—86000//?Г) ПАТП12 / з
——— = 1,3.1Ш ———------------------{[NO]2 — (NO)2} моль/см? • сек
тде [NO] — равновесная концентрация при расчетной температуре для изо-
термических условий (3500° К), давления 1 ата и стехиометрической азотно-
кислородной смеси. В этих условиях равновесная концентрация [NO]
260
(7,8 об.%) устанавливалась за ~ 2-10~4 сек. Закалка со скоростью
108 град/сек сохраняет 95% равновесной концентрации в охлажденных про-
дуктах, причем практически закалка происходит при Т ~ 2500° К. При
скорости закалки 107 град/сек в продуктах остается 85% окиси азота от ее*
равновесного значения, при dT/dt ~ 106 град/сек — 63%.
Концентрация окислов азота в нитрозных газах зависит также от темпе-
ратуры Т0, с которой начинается закалка. Она максимальна при
~ 3500° К и с повышением 710 до 7000° К уменьшается до 2 об. % [91]. Вклю-
чение в кинетическую модель реакций рекомбинации на поверхности приво-
дит к незначительному повышению концентрации NO при 70 % 3500° К
(при скорости закалки ~ 107 град/сек). Если же обеспечить скорость охлажде-
ния, большую 1010 град/сек, то доминирующими в механизме будут реакции
рекомбинации на поверхности, и это может привести к получению сверх-
равновесных концентраций NO [91].
Экспериментальное изучение окисления азота в плазме. Образование
окислов азота исследовали в плазменной струе азота при введении кислорода,
водяного пара при 1 ата [12, 91, 123] и при 10 ата [12] на установке лабора-
торных масштабов мощностью до 30 кВт., а также в плазме воздуха на про
Рис. 5.14. Зависимость концентрации окислов азота в воздушной плазме от среднемас-
совой энтальпии
Р = 1 ата. Закалка на поверхности; 1,2 — экспериментальные точки, полученные на лабораторной к.
промышленной установках соответственно
мышленной установке мощностью 2500 кВт [120—122]. Закалка нитрозных
газов производилась либо в закалочном зонде [12, 123], либо затапливанием
струями газа [12, 123]. Экспериментальные данные хорошо согласуются с
термодинамическими [12] при проведении процесса до 4000° К (см. рис. 5.13).
На промышленной установке при закалке на поверхности получили те же
концентрации NO в воздушной плазме, что и на лабораторной установка
до средних энтальпий ~ 1200 кал/г (Т ~ 3100° К) [120—122] (рис. 5.14).
При проведении процессов в плазменной струе водяного пара [125] после его*
конденсации концентрация NO достигает 14 об. %, но энергозатраты резко
возрастают.
Возможная технологическая схема процесса. В [12] предложена техноло-
гическая схема образования окислов азота с равновесной концентрацией в
воздушной плазме с закалкой ранее охлажденными нитрозными газами.
В плазмотрон поступает воздух, предварительно подогретый в теплообменнике-
до температур 1600—1800° К. В плазмотроне воздух подогревается до темпе-
ратур ~ 3000—3300° К, далее воздушная плазма закаливается холодными
нитрозными газами со скоростью ~ 108 'град/сек до температур ~ 2000—
1800° К. При этих температурах скорость закалки продуктов реакции долж-
на составлять (2—5)-105 град/сек, что вполне может быть обеспечено в обыч-
261 '
ных теплообменниках, куда и направляют нитрозные газы. Продукты реак-
ции после подогрева в теплообменнике воздуха, поступающего в плазмотрон,
попадают в турбокомпрессор.
На основании оценок авторов [12] оптимальными условиями проведения
процесса являются следующие: диапазон давлений 20—30 ата, температур
3000—3500° К. Только в предположении 80% КПД плазмотрона с реакто-
ром и 30% КПД турбокомпрессора процесс окисления азота в плазменной
струе характеризуется затратами электроэнергии в размере 8 кВт-ч на
1 кг NO.
Окисление азота в неравновесной плазме
Процесс окисления азота исследовали в неравновесной плазме тлеющего
разряда (в области отрицательного свечения [65], положительного столба
1126], приэлектродных областях [127, 128] постоянного и переменного тока
промышленной частоты, высокочастотного электродного [127] на частоте
270 кГц, безэлектродного на частоте 27,5 мГц [129] и СВЧ [130] разрядов, а
также в имнульсных разрядах [131]. Разряды возбуждали в смесях азота
с кислородом либо в воздухе в широком диапазоне давлений от 0,7 до
500 торр. Полученные результаты довольно противоречивы.
В области отрицательного свечения тлеющего разряда при давлении
0,7—4 торр максимальный выход NO наблюдали при соотношении N2 : О2 =
= 68 : 32, а в приэлектродных областях — при соотношении 1:1.
В плазме электродного разряда на частоте 270 кГц при 180 торр и мощ-
ности в плазме 65 Вт концентрация NO достигала 5 об.%, что в три раза
больше, чем в низкочастотном разряде той же мощности. Однако с дальней-
шим повышением частоты до 27,5 мГц и мощности до 375 Вт концентрация
NO падает до 2%. В то же время в плазме тлеющего разряда в области поло-
жительного столба при давлениях 50—300 торр концентрация NO, линейно
зависящая от плотности тока в исследованном диапазоне ее изменения,
достигала 5—6%, а в прикатодной области — 8%.
В плазме же СВЧ-разряда в воздухе концентрация NO зависит от параметра
Р = Pz/E2x, где Р — давление, z — импеданс, Е — напряженность электри-
ческого поля в плазме, ат — длительность пребывания в зоне разряда.
Максимальный выход NO составляет 0,8 моль на 1 кВт-ч, В плазме импульс-
ного разряда в воздухе при давлениях 20—500 торр, мощности в импульсе
~ 20 МВт и его длительности 3 мксек содержание NO не превышало 2,5 об. %.
Ни в одной из этих работ механизм процесса не был изучен. Вероятно,
поэтому приведенные данные столь противоречивы.
Нами была предпринята попытка исследовать образование окислов азо-
та в неравновесной плазме импульсного СВЧ-разряда в воздухе [132]. Ана-
лиз результатов расчетов процесса ионизации молекулярного азота в СВЧ-
импульсе, описанных в [21], показывает, что за времена 10~7 сек происходит
быстрый рост концентрации заряженных частиц, скорость изменения кото-
рой далее резко уменьшается. В этот же период времени (10~7 сек) велика
скорость образования возбужденных по внутренним степеням свободы тя-
желых частиц, причем происходит практически только накопление. По-
скольку реакции с участием возбужденных частиц обычно вносят сущест-
венный вклад в механизм образования химических продуктов, изменение
длительности импульса и паузы, влияющие на концентрации этих частиц,
должны влиять и на выход продуктов. Нами были выполнены предваритель-
ные эксперименты по изучению образования окислов азота в плазме возду-
ха [132]. В опытах разряд возбуждался в воздухе при 400—500 торр, рас-
ход его составлял 10 нл/мин. При длительности СВЧ-импульсов (0,1 —
0,05)-10~6 сек, частоте их повторения 1000 Гц, мощности в импульсе 0,5—
1,5 МВт, времени пребывания в зоне разряда 0,1 сек концентрация окислов
азота достигала 3,5—3,0 об. %. Энергозатраты составляют 6—6,5 кВт-ч/кг NO.
Полученные нами показатели могут представлять и практический интерес.
262
Увеличение длительности импульсов ухудшает показатели процесса, о чем
свидетельствуют описанные в [130] результаты.
Определенную роль в кинетике и механизме образования окислов азота
в неравновесной газоразрядной плазме могут играть реакции диссоциатив-
ной рекомбинации молекулярных ионов N2 и О2, описанные в [133, 134].
Значительная доля возбужденных атомов в газовых разрядах может образо-
ваться по этим реакциям, а не возбуждаться из основного состояния. Если
время жизни возбужденных атомов порядка или больше характерного вре-
мени элементарного акта химической реакции, их роль в механизме и кине-
тике может быть существенной, поскольку коэффициенты скорости таких
реакций велики. Рассмотрим с этих позиций возможный механизм образо-
вания окислов азота. Пусть в плазму N2, в которой протекает диссоциатив-
ная рекомбинация, вводится О2. Тогда протекают экзотермические безак-
тивационные реакции
N (2D) + О2 -> NO (Х2л) + О (ЗР) + 3,31 эВ, К = (1-7)-Ю"12 смЧсек,
N (2D) + NO (Х2л) -> N2 (А'2) +0 (ЗР) 1,44 эВ, К = 1,8Л^смЧсек.
Квазистационарная концентрация NO с учетом этих реакций составляет
(0,6—4)-10~2 [О2]. Высокие концентрации NO в верхних слоях атмосферы
могут быть объяснены этими реакциями.
Анализ относительно малого количества данных о коэффициентах скорос-
тей, имеющихся в настоящее время, свидетельствует о необходимости учета
реакций с участием N (2D) в механизме образования N2H4, C2N2, HCN.
2. Окисление фосфора в воздушной плазме
В [135] описан процесс окисления фосфора в воздушной плазме с целью
получения окислов фосфора и азота, необходимых для производства комплекс-
ных фосфор-азотсодержащих удобрений.
Из анализа температурных зависимостей изменения изобарно-изотерми-
ческого потенциала для реакций окисления газообразного фосфора следует,
что при температурах от 1000 до 4000° К в равновесной смеси могут присут-
ствовать окислы фосфора (Р2О5, РО, Р2О3) и окись азота. До 2100° К основ-
ным окислом будет Р2О5, в интервале температур 2100—3250° К — смесь
Р2О3 и Р2О5, а в диапазоне 3250—4100° К — Р2О3 и РО. При температурах
выше 4100° К основным продуктом окисления фосфора будет РО [135].
Кинетический расчет для этого процесса не производился.
Экспериментальные исследования процесса проводили при мощности
плазменной струи 5—12 кВт, расходах воздуха и красного фосфора соот-
ветственно 1,4—1,9 г/сек и 2—10 г!мин. Продукты реакции закаливали затап-
ливанием струями воды или воздуха. Оказалось, что введение фосфора в
плазменную струю воздуха приводит к увеличению концентрации окислов
азота. Концентрация Р2О5 не превышала 0,3 г/л, а NO — 2,5 об.%. При
окислении фосфора выделяется энергия, составляющая ~ 30% энергии плаз-
мы. Судя по полученной концентрации NO в газах после закалки, последняя
недостаточно эффективна.
Авторы [135] предложили использовать в качестве сырья вместо элемен-
тарного фосфора печной газ фосфорного производства, содержащий ~ 25%
фосфора, 75% СО, без его охлаждения с температурой 500—700° С. В этом
случае используется энергия, выделяемая при окислении СО.
По мнению авторов [135], организация процесса совместного получения
окислов фосфора и азота сжиганием элементарного фосфора или печных
газов фосфорного производства в воздушной плазме позволит снизить энерго-
затраты на 20—30% в плазмохимическом процессе получения только окис-
лов азота из воздуха.
263
3. Окисление хлоридов металлов
Плазмохимический процесс получения ТЮ2 окислением его тетрахлорида.
Процесс получения двуокиси титана окислением тетрахлорида в плазме
промышленно реализован в ряде стран 1136]. Термодинамическая оценка
реакции TiCl4 + О2 -> ТЮ2 + 2С12 показывает, что равновесие в сторону
желательных продуктов сдвинуто уже при комнатной температуре, однако
скорость реакции ничтожно мала. При температурах — 1000—1500° К ско-
рость реакции резко возрастает, и она протекает за времена ~ 10“2—10”3 сек.
Один из возможных способов реализации этого процесса таков: в плаз-
менную струю кислорода, полученную в безэлектродном ВЧ-плазмотроне,
вводят пары тетрахлорида титана. Порошок двуокиси титана собирают в
фильтрах, смесь же газообразного хлора, образующегося в результате реак-
ции (~ 85 об.%) и остатка кислорода (~ 15 об. %), подвергают низкотем-
пературной перегонке [137, 248].
Процесс можно проводить в любом кислородсодержащем газе [138,
139]. Так, в [138] TiO2 получают по реакции его тетрахлорида с СО2.
Повышение температуры реагентов в результате их предварительного
подогрева приводит к возрастанию скорости реакции, вызывающее увели-
чение количества центров кристаллизации. Этот факт, а также уменьшение
времени взаимодействия позволяют получить тонкодисперсный продукт
с размером частиц менее 1 мкм. Изменением температурного и газового ре-
жима процесса можно получить рутильную или анатазную модификации
TiO2 [140].
Плазмохимическое получение окислов металлов окислением галогенидов
и металлов. Подобным же способом получают и окислы других металлов, и
смеси окислов. Так, в [141] описаны результаты исследования образования
смесей окислов хрома и алюминия, а также хрома и титана окислением их
галлоидов в ВЧ-плазме кислорода. О возможности получения окислов U,
Mo, V, Ti, Zr, Nb, Та окислением их хлоридов в плазме упоминается в [140].
Анализ результатов термодинамических расчетов процесса окисления тетра-
хлорсилана [142] свидетельствует о том, что двуокись кремния присутствует
в продуктах в значительных концентрациях при температуре — 2000° К; в ин-
тервалах температур от 3000 до 4000° К практически весь кремний содержит-
ся в моноокиси его (1 ата, Si : С1 : О = 1 : 4 : 2). Порошки смеси окислов
кремния были получены при окислении SiCl4 в ВЧ-плазме аргона с добав-
кой кислорода [142].
Окислы различных металлов могут быть получены также окислением их
порошков. Таким образом, окислением в ВЧ-плазме кислорода порошка
алюминия был получен тонкодисперсный порошок у-А12О3 [143, 144].
Процессы окисления металлов с образованием окисных пленок на их
поверхности мы рассмотрим на стр. 279.
4. Окисление хлористого водорода в плазме
Этот процесс исследовался в равновесных и неравновесных условиях.
Анализ результатов термодинамических расчетов реакции 2НС1 + 0,5О2 ->
Н2О + С12, приведенных в [145], свидетельствует о том, что равновесие
в сторону целевых продуктов сдвинуто уже при 300° К. Максимальный вы-
ход С12 реализуется при температурах 4000° К и отношении НС1/О2, равном
4, и достигает 85% при энергозатратах 5,5 кВт-ч/кг С12 (давление 1 ата).
Присутствие азота не влияет на выход4 хлора, в то время как в атмосфере во-
дяного пара выход хлора уменьшается на 20—30%. Минимальные энерго-
затраты 3,2кВт-ч на 1 кг С12 при 3000° К, когда конверсия НС1 в хлор 45%.
Экспериментальные исследования этого процесса проводили в плазмен-
ных струях воздуха или смеси воздуха с кислородом [145]. При температуре
реакции 4200° К конверсия составила 96%, но энергозатраты были больши-
ми — 14 кВт-ч1кг С12. Когда же процесс провели при Т ~ 3000° К, конвер-
264
сия составила 45% при затратах энергии — 3,4 кВт-ч/кг С12. Эти показатели
хорошо согласуются с термодинамически возможными* Состав продуктов
был таким (об. %): 18 — С12, 43 — НС1. 12,5 — О2, 26.5 — Н2. Окисление
хлористого водорода осуществляется также в плазменной струе водяного
пара при температурах 1500—3000° К (это предложено в [146]), конверсия
хлористого водорода составляет 45%.
Протекание реакции окисления НС1 кислородом в неравновесной плаз-
ме тихого разряда [147] при 1 ата характеризуется тем, что: а) максималь-
ная конверсия реализуется при соотношении О2 : НС1 = 1:4; б) конверсия
линейно возрастает с ростом напряженности электрического поля Е, что
связано с увеличением параметра E/N (N — плотность частиц, сл/3), от кото-
рого зависит средняя энергия электронов. Тем самым предполагается, что
в процессе существенное значение имеют реакции электронного удара.
В исследованных условиях конверсия хлористого водорода достигала 18%
при затратах энергии 10 кВт-ч/кг С12. По мнению автора [147], оптимальные
условия лежат в области пониженных давлений.
В неравновесной плазме СВЧ-разряда этот процесс изучали при давле-
ниях от 5 до 40 торр, мощностях в плазме 200—350 Вт при стехиометриче-
ском соотношении реагентов [148]. Максимальная конверсия (— 50%) реали-
зуется при давлениях, зависящих от мощности в плазме (для мощности
200 Вт составляет 15 торр, а для 340 Вт —20 торр). Затраты энергии
— 20 кВт-ч!кг С12. По мнению авторов [148], реакция идет по свободнора-
дикальному механизму с участием атомов О и Н.
5. Синтез озона в плазме
Этот процесс исследован в плазменной струе аргона при введении в нее
кислорода при атмосферном давлении [149] и в неравновесной плазме корон-
ного разряда в кислороде или воздухе, например в [150, 151].
В первом случае образование О3 происходило при мощностях в плазмен-
ной струе 350—850 Вт, закалка его осуществлялась в зонде, максимальная
концентрация О3 не превышала 2 об. %.
В разряде в озонаторе при атмосферном давлении концентрация озона
составила — 0,5 об.%. Однако в [151] описывается установка производи-
тельностью 454 г!сут. В [150] рассмотрен вероятный механизм образования
озона в неравновесной плазме тихого разряда. На основании следующих
экспериментальных данных установлено, что концентрация О3 и его выход
прямо пропорциональны количеству электронов за полпериода низкочастот-
ного поля (50—60 Гц) и напряжению и обратно пропорциональны расходу
газа. Кроме того, оказалось, что при подаче на электроды низкочастотного
поля в плазме генерируется быстрозатухающее высокочастотное поле час-
тотой 106 — 107 Гц. Автор работы [151] предполагает, что инициирование
процесса протекает в ВЧ-поле за время — 10“6—10~7 сек, когда молекулярный
кислород под электронным ударом диссоциирует на атомы. Последние в зо-
нах, свободных от разрядных стриммеров, реагируют с молекулярным
кислородом, образуя О3.
В литературе упоминаются и другие плазмохимические процессы окисле-
ния. В [148] сообщается о том, что в неравновесной плазме СВЧ-разряда
при 10 торр и мощности в,ней 280 Вт идет реакция окисления кислородом сер-
нистого ангидрида в серный. При этом температура газа не превышала
600° К. В неравновесной плазме тлеющего разряда могут протекать реакции
окисления водорода и окиси углерода [65].
6. Образование фторидов в плазме
Синтез фторидов азота в равновесной плазме. Анализ результатов термо-
динамических расчетов, приведенных в [114, 152], свидетельствует о том, что
при комнатной температуре при 1 атм термодинамически устойчивым со-
265
единением является NF3. Отсутствие же реакции между фтором и азотом при
этих условиях вызвано, вероятно, кинетическими причинами. В интервале
же температур 1500—6000° К концентрации соединений фтора малы, в
продуктах преобладают F2 и N2. Максимальная концентрация NF реализует-
ся при отношении N/F = 1, возрастает с увеличением давления, достигая
при 100 атм 0,1 мол. %.
Кинетические данные для образования и разложения фторидов азота
практически отсутствуют. Экспериментальные исследования синтеза фтори-
дов азота проводили в дуге с графитовыми электродами и в плазменной струе
азота [114]. В первом случае независимо от состава фторидного сырья полу-
чали от 1 до 2,5 мол. % фторидов азота. При проведении процесса в плазмен-
ной струе в качестве фторсодержащего сырья использовали CF4, SF6. Про-
дукты охлаждались со скоростью — 106 град/сек. Были получены NF3,
N2F2, N2F4. Максимальное превращение во фториды азота составляло
— 1 мол. %.
Синтез фторидов азота в неравновесной плазме. В плазме тлеющего раз-
ряда выход NF3 при синтезе его из азота и фтора при давлениях 10—40 торр.
токе 10—25 мА и температуре стенок от —150 до —196’ С зависит от расхода
реагентов и при расходе до 0,1 л/ч достигает 10—70%. Причем от соотношения
F/N степень превращения не зависит. Затраты энергии на единицу объема
реагирующих газов велики и составляют — 325 Вт-ч/л [152].
В плазме тихого разряда выход NF3 мал (1,5—2,0%) [152]. NF3 в тлею-
щем разряде при температуре стенок комнатной и выше легко распадается
на азот и фтор (конверсия 80%). В этом случае не образуется N2F4. Если же
при разложении NF3 тем или иным способом, например по реакции с парами
ртути, связывать образующийся атомарный фтор, то возможно получение
N2F4 и N2F2. Выход этих соединений в расчете на разложившийся NF3 со-
ставляет 50—67 и 12—16% соответственно [152]. Сам же тетрафторгидразин
может легко распадаться в тлеющем разряде с образованием NF3 и N2.
В плазме тлеющего разряда в смесях фтора и воздуха, фтора и окислов
азота, трифторида азота и дифторида кислорода при давлении 10—15 торр.
температуре стенок —196° С, токе 30—50 мА образуется F3NO с выходом
10—15% [152]. Механизм процесса неясен. Возможно, это соединение об-
разуется из продуктов распада NF3.
В неравновесной плазме тлеющего разряда были получены соединения
с ранее неизвестным катионом NF£. NF4AsFe получили в стеклянной труб-
ке тлеющего разряда, охлажденной до —78° С, при разряде в смеси NF3 +
+ AsF5 + F2 с отношением 1 : 1 : 2 и давлении 80 торр в циркуляционной
системе; 1 г этого вещества образовался за — 40 ч.
Синтез фторидов кислорода в плазме. Фториды кислорода с общей фор-
мулой OnF2 (п = 2—6) были получены главным образом в неравновесной плаз-
ме тлеющего разряда из фтора и кислорода при охлаждении стенок разряд-
ной трубки до температур —168—213° С при давлениях от 0,1 до 10 торр
[114, 152]. В зависимости от состава фторкислородной смеси и условий син-
теза образуются фториды кислорода разного состава. Высшие полиоксифто-
риды образуются при малой силе тока и мощности разряда, более низкой
температуре стенок реактора, малом расходе реагентов.
Синтез фторидов кислорода в рассматриваемых условиях идет через обра-
зование радикала O2F, существование которого установлено [152]. Вероятно,
эти соединения образуются по следующей схеме [152]:
F2—>2F; F+ 02~>02F,
—_O2F2
/ +OtF
O2f —-----> O4F2
\^O4F^O6F2.
266
Как показано в обзоре [152], возможно существование только соединений,
включенных в эту схему.
Синтез фторидов благородных газов в плазме. В настоящее время описа-
ны реакции получения фторидов ксенона и криптона в плазме тлеющего раз-
ряда [114, 152]. При синтезе фторидов ксенона стенки разрядной трубки
охлаждают до —78°, в то время как при синтезе фторидов криптона — до
—183 или —196°. Состав продукта зависит от состава смеси исходных реа-
гентов. Условия образования этих соединений приведены в табл. 5.11.
Таблица 5.11
Условия образования фторидов благородных газов
Условия опытов Реагент Продукт
2-15 торр- 10-30 мА- 1100-2800 В; 78° К Хе : F2 = 1 : 2 XeF4
2—15 торр- 10—30 мА-, 1100—2800 В; 78° К Хе : F2 - 1 : 3 XeF6
20 торр-, 0,05 кВт-, 90° К Kr : F2 = 52 : 48 KrF2
10 торр-, 0,05 кВт-, 86° К Kr : F2|= 1 : 2 KrF4
Синтез фторидов серы и хлора в плазме. Фториды серы (SF2, S2F2, SF6)
были получены в плазме СВЧ-разряда при давлении 2 торр и мощности
— 0,1 кВт из SF4 или смесей SF4 с N2 [114]. SF5C1 образуется из смесей SF4,
SF6 или S2F10 с С12 в тех же экспериментальных условиях [114]. Из смесей
SF6 и S2F10 с О2 образуется SF4O в СВЧ-плазме [114].
В обзоре [152] описаны условия получения соединений C1F, C1F3, C1F5
в плазме тлеющего разряда при 30 торр, температуре стенок —78° и кон-
денсации продуктов в ловушке при —196°, соотношении F2 : С12 = 10.
ПРОЦЕССЫ ВОССТАНОВЛЕНИЯ
НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
Низкотемпературная плазма применяется для проведения восстанови-
тельных процессов в металлургии и «вскрытия» рудных материалов [153,
154, 228, 230, 231, 236—239].
Восстановление окислов или хлоридов до элементов обычно проводят на
установках с дугой высокой интенсивности или с проникающей дугой типа
плазменных плавильных печей с использованием анода из спрессованного
с угольным порошком сырья либо в плазменной струе. В последнем случае
методика экспериментов была такой: реагент в виде газа или порошка по-
давался в плазмотрон в зону разряда или в плазменную струю. Закалка
продуктов производилась одним из известных методов (на охлаждаемой
металлической поверхности, затапливанием холодным газом, газодинамиче-
ским способом и другими способами). Целевые продукты могли быть в газо-
вой или конденсированной фазе.
1. Восстановление хлоридов в плазме
Плазмохимические процессы восстановления хлоридов металлов и метал-
лоидов в равновесных условиях. Подробный термодинамический анализ
выполнен для процессов разложения тетрахлоридов титана [155], германия
[155] и тетрахлорида кремния нами в [142] в нейтральной и восстановитель-
ной атмосфере. Разложение тетрахлоридов металлов до металлов протекает
с образованием промежуточных соединений МеС13, МеС12 и МеС1. При 1 ата
в продуктах разложения в газовой фазе появляются Ge при 2300° К, Si —
3000, Ti — 3500° К, и выходы их достигают практически 100% соответствен-
267
но при температурах 4000, 4500 и 5000° К. Уменьшение давления до 0,1 ата
снижает значения температур, необходимых для разложения хлоридов
— на 1000° К. Восстановление хлоридов водородом начинается при более
низких температурах (примерно на 300° меньших). На рис. 5.15 в качестве
примера приведена температурная зависимость выхода кремния при разло-
жении его тетрахлорида в нейтральной и восстановительной атмосфере.
Таким образом, получение металлов целесообразно проводить в восстанови-
тельной атмосфере при температурах —350СГ К (для Ge) и —4500° К для
Si и Ti.
Кинетика разложения рассматриваемых хлоридов изучалась в диапазоне
3000—6000° К [155, 156J. Начальная стадия разложения исследовалась на
Рис. 5.15. Температурная зависимость выхода кремния при разложении его хлорида в
нейтральной и восстановительной атмосфере
1 — Р = 1,0 атпа, Н : С1 = 0; 2 — 1,0; 1 : 1; 3 — 0,1, 1 : 2; 4 — 0,1, 1 : 1; 5 — 0,1, 2 : 1
Рис. 5.16. Температурная зависимость выхода кремния при восстановлении его тетра-
хлорида водородом
1 — экспериментальная кривая; 2 — расчетная
установке адиабатического сжатия и расширения, разложение же до метал-
лов изучалось в плазменной струе. Показано, что разложение тетрахлори-
дов происходит путем последовательного отрыва атомов хлора, причем ста-
бильными продуктами являются соединения состава МеС12 и Me.
Наиболее медленной стадией разложения МеС14 является стадия разло-
жения МеС12. Значения констант скоростей разложения хлоридов, проте-
кающего по первому порядку, приведены в табл. 5.12.
Таблица 5.12
Кинетические данные разложения хлоридов металлов
К, сек-1
Элемент
MeCJ4—>МеС12
МеС12 -> Me
Ge
Si
Ti
/ 82 000+5000
109exp (-
5-108exp (
RT )
88 000+5000 \
RT )
/ 83 000 + 5000
4,5-109exp (—-----------
7 -107exp
5-107exp
108exp
112 000+11000
"" RT
126 000+Ю000
~ RT
144 000+15 000
” RT
268
Экспериментальные исследования процессов получения Ge, Si и Ti из их
тетрахлоридов проводили в плазменных струях электродного и ВЧ безэлек-
тродного плазмотронов в восстановительной атмосфере. Были получены вы-
ходы металлов, близкие к термодинамически возможным. Например, тем-
пературная зависимость выхода Si при восстановлении SiCl4 приведена на
рис. 5.16. Чистота полученных металлов в безэлектродном плазмотроне за-
висит только от чистоты реагентов.
Выполнены экспериментальные исследования плазмохимических про-
цессов восстановления многих хлоридов металлов и металлоидов. Обзоры этих
работ приведены, например, в [67, 135, 157]. В [158, 240] описано получение
непирофорных металлов (Ti, Zr, Та, Nb, W, V, V, Hf, Mo, Th), восстановле-
ние их галогенидов в плазме Н2. Размеры полученных частиц 0,03—0,1 мкм
кубической, октаэдральной или сферической форм.
В [159] в плазме безэлектродного ВЧ-разряда при атмосферном давлении
получены некоторые тугоплавкие интерметаллические соединения на основе
ниобия и ванадия восстановлением их хлоридов. В экспериментах был до-
стигнут 100%-ный выход металлов из их хлоридов, состав твердой фазы пол-
ностью соответствовал составу газовой фазы. Продукт реакции кристалли-
зовался на подложке, нагретой до температуры —1000° G. Были получены
•образцы материалов, содержащих ванадий и кремний, ванадий и германий
различного состава, состоящие из отдельных кристаллов. Кроме того, в этой
же работе синтезированы кристаллы соединений V3Ge, Nb3Al, Nb3Ge,
а также кристаллы тройного соединения Nb3Al0,8Ge0,2.
Восстановление хлоридов в неравновесной плазме. В неравновесной плаз-
ме тлеющего разряда исследовались реакции водородного восстановления
галогенов титана, циркония и бора [160—163]. При восстановлении TiCl4
получили 90% Ti чистотой 99,6 и 10% низших его хлоридов [160, 161]. Низ-
шие галогениды циркония (ZrCl3, ZrBr3, ZrJ3 ZrF2) были получены в тлею-
щем разряде в смесях галогенидов с водородом при 3—4 торр [162]. Бор чис-
тотой 99,9% был получен в тлеющем разряде в смесях ВС13 : Н2 =- 1,5 при
давлении 30—200 торр [163].
2. Восстановление окислов в плазме
Эти процессы реализуют на установке с дугой высокой интенсивности
либо в плазменной струе. Подробно исследован плазмохимический процесс
получения моноокиси кремния восстановлением его двуокиси [142, 164,
165]. При термическом разложении SiO2 при 1 атм в интервале температур
3500—5000° К доля SiO в кремнийсодержащих продуктах составляет
45—100%). Быстрая закалка продуктов разложения SiO2 с температуры
—3500° К до температуры ниже начала диспропорционирования твердой
моноокиси кремния (— 670° К) позволит ее получить.
Экспериментальные результаты исследования процесса
Si + SiO22SiO
в ВЧ-плазме аргона приведены в [164, 165]. Порошок продукта собирали на
металлокерамических фильтрах. Полученная моноокись кремния светло-
коричневого цвета, имеет диэлектрическую проницаемость 5—6 при tg 6 <
< 1 °о и пробивном напряжении ^>2-106 В1см, она рентгеноаморфна.
В плазменной струе аргона или гелия при температурах 2000—8000° К
и времени контакта —10-4 сек изучен процесс восстановления окисла вольфра-
ма углеводородом (вводили в плазменную струю смесь сажи с окислом) [22,
28] и водородом [63]. Степень восстановления достигала 15 и 95% соответст-
венно. При использовании в качестве восстановителя метана степень восста-
новления 60%. Порошок вольфрама характеризовался удельной поверх-
ностью 6—8 ж2/г. Полученные данные в этом процессе были использованы
при разработке модели поведения конденсированных частиц в плазменной
269
струе [22], описанной на стр. 230. На основании этих данных были разрабо-
таны способы получения порошков металлов вольфрама, молибдена, титана,
ванадия, циркония восстановлением соответствующих окислов в плазменной
струе водорода, изучены параметры процессов на лабораторных установках,
спроектированы укрупненные и опытно-промышленные установки [166].
Кроме процессов восстановления окислов, приведенных выше, в литерату-
ре описаны восстановление А12О3 до А1 в ВЧ-плазме Аг + СО или Аг +
+ СН4 [167] или в плазменной струе Аг + Н2 электродугового плазмотрона
[63], реакции восстановления окислов железа (выход —100%), пятиокиси
тантала (—50%) в плазменной струе гелия с добавлением водорода [63].
В этих же условиях восстановления TiO2 и ZrO2 не наблюдалось.
Восстановление окислов металлов и металлоидов, температуры плавле-
ния которых ниже температуры кипения металла или металлоида, на уста-
новках типа плавильных печей [167, 168] и восстановление окислов железа,
кремния, магния на установках с дугой высокой интенсивности описаны
в обзорах [67, 135]. На основании результатов термодинамических расчетов
и экспериментальных исследований плазмохимических процессов восста-
новления окислов кремния и алюминия природным газом сконструированы
плазмохимические установки [169].
Восстановление карбонила никеля в плазменной струе Аг + Н2 описано
в [63]. В продуктах содержалось ~70% окисла и 30% металла.
3. Восстановление руд и минералов в плазме
В [135] подробно описаны результаты исследования восстановления фос-
фатов в плазме. Термодинамические оценки [135] свидетельствуют о том, что
при температурах 2500—3500° К равновесие сдвинуто в сторону образова-
ния фосфора, фтористого водорода, кальция и углекислого газа. Наиболее
приемлемыми восстановителями являются углерод и продукты разложения
природного газа.
Экспериментально изучены процессы восстановления трикальцийфосфа-
та углеродом в смеси с кремнеземом до элементарного фосфора в псевдоожи-
женном плазмой слое и фосфатов в плазменной струе природного газа. В пер-
вом случае выход фосфора достигал—100% [135, 170]. Во втором случае при
среднемассовой температуре плазмы 3000—4000° К, времени контакта 10”2—
5-10-2 сек, соотношении фосфат : плазмообразующий газ 1 : 10 степень вос-
становления составляла 55—7596. В газовой фазе присутствовали ацетилен
и некоторые предельные и непредельные углеводороды, РН3, СО, СО2; а в
твердой фазе — карбид и фосфид кальция, содержание каждого из кото-
рых к общему количеству конденсированной фазы достигало —20 вес. %
[135]. В этой же работе предложена технологическая схема опытно-промыш-
ленной установки восстановления фосфатов в плазме. Восстановление же-
лезной руды (с содержанием Fe—'66—67%) в плазменной струе водорода
описано в [171, 238, 239], а ильменитовой руды — в [172]. Степень восста-
новления выше при использовании более мелких порошков и колеблется
в пределах 33—69%. В работе [228] описаны результаты изучения восста-
новления ильменита в плазменной струе продуктов разложения СН4 на
полупромышленной установке мощностью 3000 кВт. Протекает реакция
FeOTiO2 + СН4-^ Fe + TiO2 + СО + 2Н2.
Порошок ильменита вводят в плазменную струю и направляют в футе-
рованную камеру. СО и Н2 отводят через верхнюю часть камеры, шлак,
содержащий TiO2, отводят через летку в средней части камеры, а расплав
железа собирают на дне камеры и отводят через специальную летку. Расход
энергии — 1100 кВт-ч на 1 т обогащенной руды.
270
ПРОЦЕССЫ ДИССОЦИАЦИИ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
1. Термическое разложение руд и минералов
Большинство таких процессов проводят в установках с дугами высокой
интенсивности. В качестве расходуемого анода используется специально
спрессованный штабик графита с рудой. При конденсации находящихся
в плазме паров элементов, входящих в состав сырья, и углерода образует-
ся смесь окислов. Их разделение осуществляют фракционной конденса-
цией.
Исследовано разложение следующих минералов: берилла, каолина, радо-
нита и циркона [173]. Например, при разложении радонита MnSiO3 получе-
ны окислы МпО и SiO2. Расход электроэнергии составляет 31 кВт-ч на 1 кг
Мп.
Рис. 5.17. Зависимость степени
диссоциации фосфатов К от
температуры при различных
затратах энергии, отнесенных
к 1 кг сырья
1 — 11 кВт-ч/кг',
2 — 16,5;
3 — 29;
4 — 39
В обзоре [135] сообщается о диссоциации окислов кремния, магния и ди-
сульфида молибдена [174] в аргоновой плазме.
Хлорирование руд в плазме используется для получения хлоридов, ко-
торые в связи с различием в температурах кипения позволяют провести се-
лективную конденсацию. В [175] исследовали процесс хлорирования бе-
рилла, каолина, буры. Однако, по данным [175], при получении А1 из као-
лина таким способом необходимо затратить 100 кВт-ч на 1 кг А1, что на поря-
док превышает затраты энергии в современных промышленных процессах
его получения.
Изучению разложения фосфатов в плазме посвящена значительная часть
книги [135]. Анализ термодинамических оценок свидетельствует о том, что
при температурах — 3500е К и выше могут протекать реакции диссоциации
трикальцийфосфата и фторапатита, основных компонентов фосфатов. В га-
зовой фазе при этом должны присутствовать окислы фосфора РО, Р2О3, Р2О5,
соотношение которых зависит от температуры. В присутствии азота возмож-
но образование окислов азота.
Экспериментальные исследования получения пятиокиси фосфора из фос-
фатов в азотной и воздушной плазме выполнялись на установках мощностью
50 кВт [135]. В опытах получали твердые продукты (шлак), сконденсирован-
ные из газовой фазы, и газообразные продукты. Шлак представляет собой
расплав, частично покрывающий стенки реактора и закалочной камеры. Это
кристаллическое вещество, в котором, вероятно, содержатся трикальций-
фосфат, силикат кальция, фторапатит, карбонаты Mg и Al и одно- и двухза-
мещенные фосфаты кальция. В сконденсированных продуктах присутствуют
фторидные и нитридные соединения кальция и СаО в свободном состоянии.
В составе газовой фазы при диссоциации фосфатов в плазме присутству-
ют азот, кислород, фтор, окислы углерода и кальция и пятиокись фос-
фора.
271
Изучено влияние температуры, времени контакта и расхода газа, пода-
ваемого на закалку продуктов, на степень диссоциации фосфатов. На рис. 5.17
приведена зависимость степени диссоциации фосфатов К от среднемассовой
температуры Т при различных удельных затратах энергии на сырье. С уве-
личением удельных затрат энергии степень диссоциации сырья возрастает.
В экспериментах степень диссоциации достигала 85%, условная концентра-
ция Р2О5 составляла 0,2—0,4%.
2. Разложение молекулярных и трехатомных газов
в равновесной и неравновесной плазме
Разложение молекулярных газов в низкотемпературной плазме при дав-
лениях, близких к атмосферному и более высоких, в большинстве случаев
является равновесным процессом [63]. Термодинамические и кинетические
особенности этих процессов хорошо изучены. Исключением является дис-
социация молекулярных газов в электрических дугах при атмосферном дав-
лении при малых токах и в пристеночных областях ВЧ-разряда [176], когда
нарушаются условия ЛТР.
Не менее подробно исследованы плазмохимические процессы диссоциации
трехатомных молекул при атмосферном давлении, являющиеся в большин-
стве случаев равновесными процессами.
Проведение процессов диссоциации молекулярных газов в неравновесной
плазме используется для получения атомов. Во многих обзорах описаны эти
реакции.
Молекулярный водород в плазме СВЧ-разряда при давлении 0,5 торр
и мощности 100 Вт диссоциирует на 90% [177]. Малые добавки (0,1—0,3%)
Н2О или О2 значительно увеличивают выход атомов Н. Возможно, это свя-
зано с уменьшением рекомбинации атомов Н на стенках, вызываемым этими
добавками.
Молекулярный кислород в СВЧ-разряде при давлениях 0,2—1.6 торр
и мощности до 800 Вт диссоциирует на 10—20% [177], и в этом случае при-
сутствие следов (0,01—0,05%) N2, N2O или NO увеличивает степень диссо-
циации.
Значительно меньшие степени диссоциаций N2 наблюдали примерно
в тех же условиях в СВЧ-разряде [177]. Описаны реакции разложения га-
логенов (Cl2, Br2, F2) и фосфора [177].
К диссоциации молекул, наиболее важной стадии инициирования многих
химических реакций, приводят следующие процессы под действием элек-
тронного удара, подробно рассматриваемые в гл. 3:
возбуждение молекул в электронные состояния, лежащие выше потен-
циала их диссоциации, с последующим распадом, если эти состояния неста-
бильны или из них возможны переходы в нестабильные состояния:
диссоциативное прилипание электронов к молекулам;
диссоциативная ионизация;
электронно-ионная диссоциативная рекомбинация.
Сечения и скорости диссоциации молекул по этим каналам, их отношения
могут меняться в зависимости от строения конкретной молекулы и от степени
возбуждения колебательных, вращательных и электронных уровней. Так,
сечение диссоциации Н2 определяется суммой сечений возбуждения неста-
бильных электронных уровней. Диссоциация N2 протекает через те электрон-
ные уровни, для которых наблюдается предиссоциация, обусловленная вза-
имодействием с нестабильными уровнями (максимальное сечение близко к се-
чению ионизации), при больших концентрациях электронов — в результате
электронно-ионной диссоциативной рекомбинации. Сечение диссоциативной
ионизации растет с увеличением числа атомов в молекуле и достигает зна-
чений сечения ионизации для многоатомных молекул. Сечения диссоциатив-
ного прилипания велики для молекул галогенов, галогеносодержащих сое-
динений, некоторых окислов.
272
Изменение заселенностей вращательных и колебательных уровней меняет
значения коэффициентов скоростей диссоциации. Перезаселение верхних
колебательных уровней, часто реализующееся в экспериментальных усло-
виях, может увеличить эти коэффициенты.
Процессы диссоциации N2, СО2 в тлеющем разряде подробно рассматри-
ваются в гл. 3, диссоциации молекулярного кислорода в плазме ВЧ-разря-
да — в [178], хлора — в [179], окиси углерода — в [241].
Диссоциация трехатомных молекул в неравновесной плазме тлеющего
и других типов разрядов описана в [67, 180, 181], в работе [170] — разложе-
ние SO2, а в [171] — разложение N2O.
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ ПОЛУЧЕНИЯ
ТУГОПЛАВКИХ СОЕДИНЕНИЙ
1. Получение нитридов тугоплавких металлов
Ряд процессов описан в обзорах [67, 135]. Феноменологически изучены
процессы образования нитридов титана, циркония, алюминия, вольфрама,
бора и магния в плазменных струях, генерируемых в электродных и без-
электродных плазмотронах. В этих исследованиях порошки металлов вводятся
в плазменную струю азота. Так, в работах [183—185] показано влияние вре-
мени пребывания порошка циркония и титана в струе плазмы N2 или Аг +
+ N2 на степень азотирования. При т — 10~4—10-3 сек (стадия 1) увеличение
степени азотирования происходит линейно в зависимости от т. Затем в интер-
вале т ж 10-3—10-2 сек — по параболе, что свидетельствует о том, что на
стадии 1 процесса на поверхности частиц образуется нитридная пленка, да-
лее образование нитрида лимитируется процессом диффузии атомов азота
через эту пленку.
Нитрид алюминия был получен в плазменной струе N2, истекавшей из
ВЧ электродного плазмотрона, куда вводился порошок алюминия 20—80 мкм
[186]. Оказалось, что процесс нитрования идет на поверхности частиц при
температуре газа до 2500° К. Лучшие показатели достигнуты в случае введе-
ния в плазменную струю паров хлоридов. Нитриды различных металлов
(алюминия, кремния, бора, циркония, титана, гафния, ниобия и тантала)
получают восстановлением их хлоридов в водородной плазме в присутствии
азота или NH3 [187—191, 242, 243]. Например, выход Si3N4, полученного
таким способом, составлял 80%.
Рассмотрим подробнее процесс получения нитрида титана, реализованный
в СВЧ-плазме азота при введении в нее смеси Н2 + TiCl4 и описанный
в [192—194]. Анализ результатов термодинамических расчетов равновесного
состава гетерогенной системы Ti — N—Н—С1 [194] (рис. 5.18) свидетель-
ствует о том, что максимальные концентрации нитрида титана реализуются
в диапазоне 1000—1700° К. Выход его существенным образом зависит от раз-
бавления TiCl4 азотом и водородом, и с увеличением отношений N/C1 и N/Ti
с 1 до 5 и с 1 до 100 он возрастает с 40 до 109%. Экспериментально была
реализована практически 100%-ная конверсия хлорида титана, получен
тонкодисперсный порошок нитрида титана состава TiN0,8. Энергозатраты
составили 18 кВт-ч!кг нитрида титана, что близко к термодинамически необ-
ходимым. Если же в СВЧ-плазму N2 подается смесь TiCl4 + ВС13 + Н2, то в
зависимости от соотношения реагентов образуются порошки составов TiN,
TiN + TiB2 и TiN + TiB2 + В [193].
Наряду с проведением указанных процессов в плазменных струях изу-
чаются процессы образования тугоплавких соединений на установках с про-
никающей дугой типа плазменных плавильных печей [195, 196]. В этих уста-
новках применяется электродуговой плазмотрон обычной конструкции
(~10 кВт), герметичный охлаждаемый реактор, в котором помещен медный
или графитовый анод, где находится металлический пруток или таблетка,
10 Теоретич. и прикладная плазмохимия
273
Рис. 5.18. Равновесный состав гетерогенной си-
стемы Ti—Н—N—С1
Р = 1,0 ата, Н : Cl = 1, Ti : N = 1, Ti : Cl = 0,25
спрессованная из окислов, смеси окислов
или силикатов с графитом. Продукты обра-
зуются на поверхности расплава, анода и
реактора. Обычно в работах [195—198] ана-
лизируется газовая фаза (спектральными
методами и отобранная проба — хромато-
графически), твердая фаза — температура
поверхности Т& определяется пирометри-
чески, состав ее — различными методами.
В экспериментах получены зависимости Ts
от параметров процесса (силы тока, рас-
хода газа), состава твердых продуктов,
времени опыта, соотношения реагентов и
конверсии реагентов в продукты от тех
же параметров. На основании этих зави-
симостей и термодинамических оценок
делается вывод о возможных брутто-реак-
циях в механизме процесса. Так, в работе
[195] на описанной установке исследованы процессы образования нитридов
из Al, Ti, Zr, Та, Mo, W в плазме азота. Основными стадиями процесса яв-
ляются следующие: плавление металла, растворение атомарного азота в рас-
Таблица 5.13
Условия образования нитридов и карбидов тугоплавких металлов в плазме
Реагенты и их Условия в плазме Литера-
соотношение Продукт Бутто-реакция турная ссылка
А1 N2, 9Ю0—94С00 К AIN N + Al = \1N 195
Ti N2. 9100—9400° К TiN N + Ti = TiN 195
Zr N2, 9100—9400° К ZrN N -j- Zr = ZrN 195
Та N2, 9100-9400 К TaN N 4- Ta = TaN 195
Мо N2, 9100—9400 К M02N N + 2Mo = Mo2N 195
W N2, 9100—9400° К w2n N -{- 2W - W2N 195
С' AI2O3 1-5,3: N2 AIN, CO A12O3 + 3C + 2N -2A1N + 195
+ 3CO
1-3 | TiC
С : ТЮ2 3—5,2 1 N2 TiN ТЮ2 + 2C + N = TiN +2CO 195
2,5 J TiN + TiC
1—2 1 ZrN
С: ZrO2 3—5,2 I N2 ZrC ZrO2 + 2C + N = ZrN + 2CO 195, 196
2,5 J ZrN + ZrC
1-3 ) ZrO2 + ZrN ZrSiOi = ZrO2 r
С: ZrSiCU 4 N2 ZrN + ZrC + SiO2; ZrO2 + SiO2 + 3C + 195, 196
5—5,3 J ZrC + SiC + N = ZrN + SiO + 3CO
С: (AI2O3: : SiO-z = 1:1) CO 1 1 СЛ CO СЛ 2 bS AIN SiC AI2O3 + SiO2 + 5C + 2N = ==2A1N 4-l/2SiO + + l/2SiC4-9/2CO 196
С ZrO2 3, Ar, 8400° K, Zr, ZrC, ZrO2 ZrO2 + 3C —ZrC -Ь 2CO 197
1 мин
Ar+H2, 9500° К, ZrC 197
10,5 мин
Ti Ar + O2, 8300° К TiO, Ti2O, Ti2O3, TisOs, 198
ТЮ2
274
плаве и его реакции. Лимитирующей стадией стал процесс диффузии атомов
азота. Оказалось, что выход продукта выше в случае образования стабиль-
ных нитридов. В табл. 5.13 приведены условия образования некоторых нит-
ридов.
2. Получение карбидов тугоплавких металлов
Плазмохимические процессы получения карбидов урана, кальция описа-
ны в обзорных работах [67, 135]. Карбиды различных металлов могут быть
получены восстановлением в плазме водорода их хлоридов в присутствии
галогеносодержащего углеводорода, например СС14 [187] или С2Н4С12 [199,
200]. Таким образом был получен практически чистый карбид титана стехио-
метрического состава. Этот порошок черного цвета с частицами 0,01 —
0,5 мкм можно использовать в лакокрасочной промышленности.
Описан также процесс получения карбида кремния из газообразных крем-
нииорганических соединений в водородной плазме в присутствии углеводо-
родов [201]. В качестве сырья использовали CH3SiCl3, CH3SiHCl2 и СН4.
(З-SiC применяют в качестве абразивного материала. Карбиды вольфрама
и тантала были получены в плазменной струе гелия при подаче в нее
порошков металлов или окислов в потоке метана [63]. Конверсия в кар-
биды вольфрама (W2C, WC) достигала 30—40%, а тантала до 70% (из метал-
лического порошка) и —20% из его окиси.
3. Получение интерметаллических соединений
в плазме
Некоторые химические соединения металлов друг с другом — интерме-
таллиды характеризуются тугоплавкостью и высоким сопротивлением к окис-
лению [206]. Они плавятся без разложения при более высокой температуре,
чем слагающие их металлы. Они особенно перспективны для защиты туго-
плавких металлов. Образуются эти соединения с выделением тепла. Синте-
зируются они в плазменной струе во время напыления, в которую вводятся
в виде смесей. Поскольку прогрев их объемный (за счет экзотермичности реак-
ции), частицы интерметаллидов попадают на покрываемую поверхность
жидкими и образуют плотное покрытие. Наиболее тугоплавкое соединение
этого типа ZrBe3 (1930° С), а наименее тугоплавкое из применяемых NbBe2
(1580 С). Некоторые свойства интерметаллидов, полученных в плазме, при-
ведены в табл. 5.14.
Таблица 5.14
Некоторые свойства интерметаллидов [206]
Интерметал- лид Температура плавления, °C Плотность, г!смА Увеличение веса после нагревания в воздухе при 1250° С в течение 100 час, мг/см2-ч
ZrBe3 1930 2,72 0,062
СгВе2 1840 4,34 0,023
ЬаВег 1810 2,57 0,020
ТаВе2 1830 9,79 0,039
МоВег 1715 3,03 0,026
NbBe2 1580 3,04 0,013
WBe5 1800 6,91 0,051
ТаА1з 1690 6,92 0,064
NbAl3 1660 4,57 0,022
Sn3Zr5 1985 6,61 —
NiAl 1640 4,82 —
275
ю*
ПОЛУЧЕНИЕ ГИДРИДОВ И БОРИДОВ
Бориды тугоплавких металлов могут быть получены при одновременном
восстановлении хлоридов бора и металла в ВЧ-плазме смеси Аг с Н2 [187, 242,
243]. О возможности синтеза гидридов некоторых металлов при атмосферном
давлении упомянуто в [202]. Получены же гидриды (фосфора, серы, мышьяка)
в неравновесной плазме тлеющего разряда в водороде в тех случаях, когда
стенки разрядной трубки были покрыты соответствующими элементами [66].
Гидриды бора (В2Н6, B10Hi6, В20Н16) с выходами до 40% образовались в тлею-
щем разряде при 20 торр в смесях Н2 : ВС13 = 12:1 [66]. Описаны условия
получения гидридов германия парафинового ряда из GeH4 (озонатор
300 торр, температура стенок —78° С) [177] и кремния на той же установке,
но при более низких давлениях. В последнем случае в продуктах наблю-
дались Si2H6 (66%), Si3H8 (23%), Si4H10 (11%). Возможно образование даже
высших силанов типа Si8Hi8. Если же разряд возбуждать в смесях гидридов,
возможно образование высокомолекулярных систем из трех элементов? Та-
ким образом, в плазме эквимолекулярной смеси GeH4 и РН3 наблюдалось
образование GeH3 РН2, Ge2PH7 и других высших гомологов [177]. В плазме
смеси GeH4 и AsH3 получили только один трехэлементный гидрид GeH3—
AsH2. С хорошими выходами образуется новое соединение H3GeSiH3 в плаз-
ме GeH4 и SiH4. В разряде же в смесях SiH4 и РН3 образуются Si2H5PH2
и (SiH3)2PH, а в смесях SiH4 и AsH3 — смешанные гидриды кремния и мышь-
яка [177].
ПОЛУЧЕНИЕ СЕРОВОДОРОДА В ПЛАЗМЕННОЙ СТРУЕ
Сероводород был получен в плазменной струе гелия при подаче в него
порошка серы в потоке водорода [63]. Максимальный выход H2S достигал
36%. Показатели процесса приведены на рис. 5.19.
Рис. 5.19. Зависимость конвер
сии и выхода H2S на 1 квт-ч
от затрат энергии на 1 г серы
ПЛАЗМОХИМИЧЕСКОЕ ПОЛУЧЕНИЕ ЦИАНАМИДА КАЛЬЦИЯ
Цианамид кальция был получен в плазменной струе азота при введении
в нее смеси порошков окислов кальция и сажи [203] по реакциям
СаО + 2С2 + N2-> CaCN2 + GO.
Эксперименты проводили в диапазоне температур 2500—3000° К и времени
контакта — 3-10“3 сек. Полученный продукт содержал до 20% связанного
азота. Энергетические затраты, по оценкам авторов [203], составляют
—5000 кВт-ч на 1 т связанного азота.
Этими же авторами был изучен двухстадийный процесс получения циа-
намида кальция. На первой стадии получали цианистый водород при пиро-
лизе углеводородов в плазменной струе азота (результаты его описаны hs
стр. 249). Газы пиролиза охлаждались до температур 600—1000° С и посту-
276
пали на синтез цианамида кальция в аппарат кипящего слоя, где реагиро-
вали с известью При пропускании через этот аппарат пиролизных газовг
содержащих HCN, через слой СаО в течение 30 мин получили цианамид каль-
ция с 25% связанного азота.
ПОЛУЧЕНИЕ ШПИНЕЛИ В ПЛАЗМЕ
Синтез MgAl2(J4 из окислов МцО и А12О3 в плазме рассмотрен в работах
[204, 205]. Использовали смесь окислов с отношением 1:1. Получен продукт,
содержащий кристаллы шпинели размером 110—160 А. По мнению авторов,
MgAl2O4 образуется при конденсации окислов алюминия (А12О, АЮ, А12О3)
и магния, присутствующих в плазме при температурах выше 2000 С.
ПОЛУЧЕНИЕ СФЕРИЧЕСКИХ
И ТОНКОДИСПЕРСНЫХ ПОРОШКОВ
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ
Способы получения сферических и тонкодисперсных порошков в плазме*
их свойства и области применения подробно описаны в книгах и многочис-
ленных обзорах, например в [135, 154, 207, 208] и патентах [209—215]. В ка-
честве примера приведем некоторые характеристики порошков диэлектриков
до и после обработки в плазме (табл. 5.15).
Таблица 5.15
Характеристики порошков диэлектриков до и после обработки
в плазме
Материал Удельная поверхность, ж2/з Размер частиц после обработки, мкм
до обработки после обработки
Si 4-SiO2-> S1O 0,1 75 0,04
А120з 0,7 25 0,06
SiO2 0,1 - 65 0,03
Однако во время пребывания частиц в плазме, кроме их округления, из-
менения гранулометрического состава, образования пор, происходят: а) про-
цессы окисления или нитридирования поверхности; б) рафинирование ма-
териала; примеси с высокой упругостью пара улетучиваются из материала,
например, содержание в вольфрамовом порошке кремния и цинка уменьша-
ется в несколько раз, а марганца и свинца — на порядок; в) фазовые пере-
ходы. Например, при плазменной обработке порошка окиси алюминия в слу-
чае его испарения при закалке с большой скоростью образуется а А12О3.
При обработке же карбида вольфрама WC в зависимости от мощности в плаз-
менной струе (температуры) с увеличением ее уменьшается содержание угле-
рода и образуются новые соединения WC-> W2C-> W [207].
ПОЛУЧЕНИЕ ПЛЕНОК
В НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЕ1
Широкое применение в промышленности, главным образом в микроэлек-
тронике, находят в настоящее время тонкие пленки, осажденные из низко-
температурной плазмы. Различные диэлектрические пленки используются
1 В настоящей главе не рассматриваются вопросы плазменного нанесения покрытия.
Они подробно описаны в книгах [154, 206] и обзорах [216—218].
277
для изготовления элементов тонкопленочных гибридных схем, пассивации
интегральных схем и в качестве диффузионных масок при изготовлении тран-
зисторов [73, 219,220].
Плазмохимические пленки могут быть кристаллическими или аморфными.
Необходимая толщина пленок колеблется от долей микрона до 100 мкм.
Скорость роста пленки должна быть такой, чтобы можно было управлять
отдельными стадиями процесса — вводить примеси, от которых зависят
электрические свойства пленок, создавать требуемые нарастающие друг
на друга слои.
Естественно, что кинетические закономерности химического процесса
влияют на образование пленок, их морфологию, характер и распределение
дефектов и в результате на электрофизические свойства пленок.
Для выращивания монокристаллических пленок необходимо, чтобы [221]:
1) скорости подвода вещества к подложке и роста затравочных кристаллов
были одинаковыми; 2) была эмпирически выбрана оптимальная темпера-
тура подложки; 3) для предотвращения срастания соседних кристаллов
зона кристаллизации была большой; 4) отсутствовали градиенты темпера-
туры в зоне кристаллизации для предотвращения испарения выросших кри-
сталлов.
Скорость роста зависит от: 1) кристаллографической ориентации под-
ложки. Эта зависимость связана с различием коэффициентов прилипания
на разных кристаллографических гранях. Медленнее всего кристаллы растут
в направлении 100 и быстрее всего — в 110; 2) от состояния поверхности
подложки.
В большинстве случаев пленки получают в плазме кислорода, омываю-
щей поверхность металла, в результате высокочастотного распыления
диэлектриков и в плазме, где протекают химические реакции, в результате
которых осаждаются те или иные пленки [73, 219].
Для плазмохимического осаждения пленок применяют установки с тлею-
щим или ВЧ-разрядом пониженного давления (10“х—10 торр).
1. Экспериментальные установки
для осаждения пленок из низкотемпературной плазмы
Схема установки для осаждения пленок в тлеющем разряде приведена
на рис. 5.20. На этой установке можно осаждать пленки при давлениях от
10"1 до 10 торр, плотности тока приблизительно нескольких мА/см,1. При
возбуждении плазмы постоянным током наблюдали рост пленки преиму-
щественно на катоде. Если же разряд возбуждается переменным полем час-
тотой 50 Гц или 2 мГц [73], пленка образуется на обоих электродах.
Схема установки для осаждения пленок в плазме ВЧ-разряда показана
на рис. 5.21.
Иногда разряд возбуждается не в парах смеси реагентов, а в од-
ном из них, например в кислороде [222], который поступает в камеру, где вы-
зывает разложение другого реагента и осаждение пленки.
Оптимальная область частот /опт возбуждения плазмы определяется сле-
дующим выражением:
0,5- 109/W < /опр < 1,5-Ю9 od2,
где о — электропроводность плазмы, d — диаметр разряда [223]. Критерием
оптимальности, по мнению авторов [73], служит величина температуры элект-
ронов.
Осаждение пленки из плазмы тлеющего разряда проводят при более
высоких давлениях. В связи с этим понижается воспроизводимость состава
пленок и повышается возможность загрязнения их материалом электродов
1731.
278
Рис. 5.20. Схема установки для осаждения пленок в тлеющем разряде
1 — подача сырья; 2 — держатели электродов; з — плоские электроды; 4 — защитные пластины для
ограничения объема плазмы; 5 — колпак рабочей камеры; 6 — к насосу; 7 — к манометру; 8 — токо-
подводы
Рис. 5.21. Схема установки для осаждения пленок в ВЧ-разряде
7 — к насосу; 2 — колпак; 3 — печь; 4 — вакууметр; 5 — подложка; 6 — кварцевые весы; 7 — ввод
ВЧ-мощности (волновод); 8 — подача реагента; 9 — ВЧ-генератор; 10 — реагент; 11 — термостат;
12 — подача кислорода
2. Образование пленок окислов
при окислении металлов в плазме
Вопросы образования окисных пленок материалов в кислородной плаз-
ме мало изучены, и в настоящее время не существует теории процесса. Ре-
зультаты работ довольно противоречивы.
Экспериментально установлено, что на электродах из алюминия, меди,,
железа, магния, цинка при разряде в кислороде образуется пленка окисла.
Если эти металлы использовались в качестве анодов, то при фиксированном
токе наблюдалась предельная глубина окисления. Рост тока приводил к про-
должению процесса. На катодах, изготовленных из этих металлов, толщина
пленки окисла не зависела от плотности тока (до 4 мА/см2), Обычно на аноде
образуются аморфные пленки, на катоде же — кристаллические. Характер
окисления катода для всех металлов почти одинаков. Строение окислов на
катоде такое же, как и у окислов, образованных на этих металлах при высо-
ких температурах. Возможно, это связано с тем, что окисление катода проис-
ходит в результате его локальных перегревов из-за бомбардировки кисло
родными ионами [73].
Толщина пленки окисла на поверхности металла, вносимого в ВЧ-плазму,
определяется температурой подложки, параметрами разряда, величиной
приложенного к образцу постоянного напряжения. Зависимость толщины
пленки от времени квадратичная при ее росте в ВЧ-плазме и линейная при
окислении в плазме тлеющего разряда постоянного тока. Эти закономерно-
сти установлены при окислении кремния, арсенида галлия, тантала.
3. Осаждение пленок
в результате химических реакций в плазме
В зависимости от требований, предъявляемых к пленке, выбг раются реаген-
ты и тип разряда. В качестве реагентов обычно используют металл органи-
ческие соединения, гидриды, хлориды, карбонильные соединения и углево-
дороды.
279
Пленки полимерных материалов. Пленки полимеров осаждаются из не-
равновесной плазмы при давлениях от 10~1 до 10 торр в различных углево-
дородах. В [224] они были получены из 57 углеводородов. Скорость осажде-
ния пленок проходит через максимальное значение при увеличении расхода
реагента. В газовой фазе полимер образуется только при высоких давле-
ниях и плотностях тока. Скорость роста пленки больше при низких темпе-
ратурах подложки.
Физические свойства пленок определяются режимом осаждения. Пленки,
образованные в плазме тлеющего разряда при высоком давлении и низкой
плотности тока, чистые и прозрачные, мягкие и клейкие, что свидетельст-
вует о неполной полимеризации. Пленки же, полученные в плазме низкого
давления (в пределах рассматриваемого диапазона) и высокой плотности
тока, прочные, желтого или коричневого цвета. В таких пленках сильно
развиты поперечные связи.
Описываемые пленки полимеров свободны от микропор даже при малых
толщинах. Они отличаются от напыленных термическим способом, в котором
пленка формируется в результате образования и роста зародышей. Отдель-
ные области срастаются, покрывая всю поверхность. Пленки растут в верти-
кальной и горизонтальной плоскостях одновременно, что приводит к разно-
толщинности их.
Свойства полимерных пленок, осажденных в плазме. Плазмохимические
полимерные пленки характеризуются низкой электропроводностью (—10“16—
10“14 (Ом-см)-1. малыми диэлектрическими потерями (е ~ 2 — 4, tg 6
~ 10~3—10~2) и высокими напряжениями пробоя — 106—107 В/см).
Энергия активации проводимости —1,4 jB. В электрических полях большой
напряженности пленки толщиной от 50 до 2500 А обладают нелинейной про-
водимостью. Сопротивление пленок в вакууме не меняется, а на воздухе воз-
растает. Фотопроводимость пленок сильно зависит от состава полимера.
Пленки неорганических материалов. Пленки неорганических материалов,
осажденные в низкотемпературной плазме, плотные, аморфные и характе-
ризуются значительной твердостью. Экспериментально получены следующие
пленки: а) кремния из плазмы в парах силана осаждением на холодную по-
верхность; б) окислов кремния из плазмы в парах силана и кислородосодер-
жащего газа (СО2, Н2О, NO) или разложением тетраэтоксисилана в кисло-
родной плазме; в) нитрида кремния из плазмы в смеси силана и аммиака,
SiCl4. NH3, N2 и N2O в различных соотношениях.
Описано также осаждение пленок карбида кремния из плазмы смеси си-
лана и этилена, двуокиси титана — из смеси TiCl 4 и СО2. Пленки металлов
были получены из их карбонилов.
Подробно изучено образование пленки из тетраэтоксисилана в ВЧ-плаз-
ме кислорода при давлениях 0,5—3,0 торр на частотах 0,5 и 40 МГц, мощ-
ностях до 400 Вт на подложках из различных материалов — NaCl, плавле-
ного кварца, платины, алюминия и окиси алюминия при температурах
подложки до 400°С [73]. Оказалось, что количество кислорода в молекуле
(C2H5O)4Si достаточно для полного окисления кремния. Скорость роста плен-
ки линейно зависит от давления, увеличиваясь с ростом его. Повышение
температуры подложки (от 4 до 50n С) приводит к уменьшению скорости роста
пленки. Химический состав пленки существенно зависит от давления. При
давлениях в разряде, больших 2,5 торр, осаждались пленки белого цвета,
содержавшие до 20% углерода. Предполагается, что это полимерные пленки.
При меньших давлениях в разряде осаждались пленки цвета кофе, аморфной
«структуры. Содержание в пленках SiO2 и углерода зависит от давления в раз-
ряде. Увеличение мощности в плазме приводит к росту содержания SiO2
и уменьшению содержания углерода в пленке. Скорость осаждения пленок из
плазмы, возбужденной на частоте 40 МГц, значительно ниже скорости роста
пленки из плазмы, генерированной на частоте 0,5 МГц. В первом случае
в пленках концентрация SiO2 составляла 6—15%, а С — 42—60%. В их
состав входят органические радикалы. Различие в скоростях роста пленок
280
и их составах в рассмотренных случаях связано не с влиянием частоты на
процесс, а с величиной поглощаемой разрядом мощности на данной частоте.
Не менее подробно исследовано плазмохимическое получение пленок нит-
рида кремния [219]. Установлено, что при постоянных мощности ВЧ-гене-
ратора, скорости подачи реагентов, температуре подложки скорость роста
пленок Si3N4 существенно зависит от концентрации SiH4. При увеличении
концентрации силана от 3 до 50 % скорость роста пленки возрастает линейно
от 35 до 180 А в 1 мин. Скорость роста пленки линейно зависит от ВЧ-мощ-
ности и увеличивается до 400 А в 1 мин с увеличением мощности от 100 до
700 Вт. Плотность пор зависит от предварительной обработки поверхности
подложки. Эффективной оказалась обработка поверхности подложки в тлею-
щем разряде в N2, О2 или Н2. Пленки, осажденные в оптимальных условиях,
имели менее 10 пор на 1 см2.
Термообработка сильно влияет на структурные свойства пленок Si3N4.
В них возникают структурные нарушения, обусловленные температурными
напряжениями и плохой адгезией пленки к подложке. По мере повышения
температуры подложки, при которой производили осаждение пленки, воз-
растала стабильность. Пленки, осажденные при комнатной температурег
отслаивались после отжига при температуре — 500° С, а в пленках, полу-
ченных при 300° С, возникали дефекты и трещины. Пленки же, выращенные
при температуре —600° С, сохраняются при нагреве до 1200° С.
Свойства пленок неорганических материалов. Регулированием условий
в плазме можно получать пленки неорганических материалов, электричес-
кие свойства которых лежат в широком диапазоне [73, 244, 245]. В качестве
примера рассмотрим свойства пленок нитрида кремния, выращенных в плаа-
ме разряда.
Диэлектрическая проницаемость в зависимости от концентрации SiH4
находится в пределах 4—11; диэлектрическая прочность 106—107 В/см,
tg 6 ~ (6—7)-10~4 (при 1 кВц), удельное сопротивление 5-Ю140ж-сж, пока-
затель преломления 1,95—2,1, пик поглощения в ПК-области спектра лежит в
интервале 1,4—12,2 мкм. Эти пленки пригодны для применения в качестве
пассивирующего покрытия, защитной маски и в структурах металлнит-
рид — окисел — полупроводник [219].
Вероятный механизм образования пленки. По мнению авторов [73], плен-
ки в плазме образуются по двум одновременно протекающим процессам:
в газовой фазе происходит возбуждение и диссоциация реагента с образова-
нием различных активных частиц: на подложке идет адсорбция промежуточ-
ных продуктов диссоциации и их взаимодействие с заряженными частицами
плазмы. Рассмотрим с этих позиций образование пленки SiO2 из тетраэток-
силана (C2H5O)4Si. По мнению авторов [73], наибольшую вероятность раз-
рыва имеют связи С—О и С—С. После распада этой молекулы на ионы и ра-
дикалы предполагаются реакции
Н ]- Н -> Н2? СХН^ + 0,5 у О + Я)2 ->rrGOs + 0,5 у Н2О.
Время пребывания адсорбированной молекулы определяется теплотой
адсорбции и температурой подложки. Продукты с малой теплотой адсорб-
ции быстро удаляются (откачка непрерывная).
За время пребывания на подложке радикалы реагируют друг с другом
либо с заряженными частицами из плазмы разряда. Но вблизи подложки^
(изолированной) в плазме возникает электрическое поле — она заряжена,
отрицательно из-за более высокой подвижности электронов. Напряженность
поля и толщина слоя, в котором оно существует, зависят от концентрации
заряженных частиц и температуры их. Поэтому адсорбированные радикалы
подвергаются бомбардировке быстрыми электронами, способными преодолеть
потенциальный барьер около подложки, и положительными ионами, уско-
ренными полем. Таким образом, химический состав пленки должен зависеть,
от концентрации заряженных частиц в плазме и их температуры. С увеличе-
нием давления газа растет концентрация заряженных частиц, увеличивается!
281
степень диссоциации реагента. С повышением концентрации заряженных
частиц усиливается интенсивность бомбардировки подложки. В результате
с повышением давления разложение более полное, увеличивается содержание
SiO2 в пленке и уменьшается содержание С. Увеличение мощности в плазме,
приводящее к увеличению концентрации электронов, приводит к тому же
эффекту. Постоянное электрическое поле, увеличивающее увод электронов на
стенки разрядной камеры и уменьшающее их концентрацию в объеме, при-
водит к уменьшению содержания SiO2 в плазме.
ИСПОЛЬЗОВАНИЕ ПЛАЗМЫ,
СОЗДАВАЕМОЙ ИЗЛУЧЕНИЕМ ЛАЗЕРА, И ИЗЛУЧЕНИЯ ПЛАЗМЫ
С появлением лазера многие исследователи попытались проводить хими-
ческие реакции под действием его излучения. Использовались либо мощные
лазеры, позволяющие создавать плотную плазму, либо, напротив, мало-
мощные, интенсивность излучения которых ослаблялась до исчезновения
пробоя в исследуемой среде. Подробный обзор работ по получению плазмы
с помощью лазера, ее свойства, взаимодействие лазерного излучения с по-
верхностью приведены в работе (225], а возможные применения лазера в фи-
зико-химических исследованиях описаны в обзоре [226].
Меняя состав газа и условия разряда, можно получить интенсивное
излучение в нужном для фотохимии диапазоне длин волн. В США вы-
пускается специальное плазменное оборудование для фотохимических
процессов [227]. При использовании аргона в качестве плазмообразу-
ющего газа наблюдается интенсивное излучение в ультрафиолетовой об-
ласти, при использовании неона — в инфракрасной. Смеси ксенона с ар-
гоном дают излучение, близкое по спектральному составу к солнечному.
Анализ показателей подобного плазменного генератора, работающего на
аргоне при давлении 17 атм, показывает, что —31 % от общей мощности
уходит в излучение, причем в ультрафиолетовой области —11%. В этой
же работе предлагается применить плазменную установку для изготовления
капролактама, получения акрилонитрила из пропилена и аммиака в присут-
ствии кислорода, для прямого окисления бензола в фенол.
НЕКОТОРЫЕ ДРУГИЕ ПРИМЕНЕНИЯ НЕРАВНОВЕСНОЙ ПЛАЗМЫ
В последние 5—10 лет неравновесная плазма нашла новые сферы при-
менения, в частности в химическом и физико-химическом анализах, в техно-
логии обработки поверхностей твердых тел, в микроэлектронике и при изго-
товлении полупроницаемых мембран [246].
В плазме реализованы основные стадии подготовки образцов для хими-
ческого и физико-химического анализов. В кислородной плазме при давлении
0,5—1,5 торр производят окисление органических и полимерных материа-
лов. Из-за относительно низкой температуры образца не происходит его
возгонки, плавления, что сохраняет начальный химический состав, мор-
фологию и тонную структуру. В плазме легко удаляется органиче-
ская часть, а неорганический остаток не меняется. Часто в плазме в резуль-
тате обработки материала получают газообразные продукты, которые воз-
можно анализировать хроматографически или масс-спектрально. Очистку
поверхности для восстановления ее оптических характеристик, осаждение
пленок для образования реплик также осуществляют в неравновесной
шлазме [246].
Плазмохимическая технология широко применяется в микроэлектронике
для удаления фоторезиса, травления, осаждения диэлектрических пле-
нок [246]. Обработка поверхностей твердых материалов в плазме позволяет
модифицировать их структуру и состав [246].
ЛИТЕРАТУРА
К главе 1
1. О. Левеншпилъ. Инженерное оформ-
ление химических процессов. М., «Хи-
мия», 1969.
2. Т. Peters. Naturwissenschaften, 41,
571 (1954).
3. Ионные, плазменные и дуговые ра-
кетные двигатели. М., Госатомиздат,
1961.
4. Исследования при высоких темпера-
турах. М., ИЛ, 1962.
5. Генераторы низкотемпературной плаз-
мы. Под ред. В. М. Иевлева. М.,
«Наука», 1969.
6. М. Ф. Жуков, В. Я. Смоляков,
Б. А. Урюков. Электродуговые нагре-
ватели газа (плазмотроны). М., «На-
ука», 1973.
7. Д. Т. Ильин, В. И. Сидоров,
Б. А. Урюков, А. Э. Фридберг,
Е. П. Чернавина. В сб.: Генераторы
низкотемпературной плазмы. М.,
«Энергия», 1969, стр. 296.
8. Обобщение характеристик электр о-
дуговиги подогревателя постоянного
тока двустороннего истечения с пе
ременным диаметром электродов.
Минск, ИТМО АН БССР, 1966.
9. Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. Под ред. Л. С. Полака. М.,
«Наука», 1965.
10. Обобщенные характеристики электро-
дугового нагревателя водорода по-
стоянного тока. Совместный отчет
ГИПХ и ИТПМ СО АН СССР,
№ 90—68, 1968.
11. А. И. Жидович, С. К. Кравченко,
О. И. Ясько. В сб.: Генераторы низко-
температурной плазмы. М., «Энергия»,
1969, стр. 218.
12. Применение плазмотрона в спектро-
скопии. Сборник. Фрунзе, «Илим»,
1970.
13. Физика и техника низкотемператур-
ной плазмы. Под ред. С. В. Дресвина.
М., Атомиздат, 1972.
14. М. L. Thorpe, L. W. Scammon. TAFA
Division Final Report, 1968, NAS 3,
9375.
15. H. H. Рыкалин, И. Д. Кулагин,
Л. М. Сорокин. В сб.: Физика, тех-
ника и применение низкотемператур-
ной плазмы. Алма-Ата, 1970, стр. 714.
16. Р. И. Dundas, М. L. Thorpe. Chem.
Engng., 78, N 14, 123 (1969).
17. Г. И. Бабаш. Вести, электропромыш-
ленности, вып. 3, 1 (1942).
18. Т. В. Reed. J. Appl. Phys., 32, 821
(1961); 32, 2534 (1961).
19. А. В. Донской, С. В. Дресеин,
Д. Г. Ратников. Теплофизика высо-
ких темп., 3, 922 (1965).
20. А. Миронер. Ракетная техн, косм.,
1, 225 (1963).
21. Н. U. Eckert. The Induction Arc.
A State-of-the-Art Review, Air Force
Report SAMSO-TR-72-227. Los Ange-
les 1972.
22. И. Г. Блинов, В. В. Гусев, Л. Н. За-
харов, С. А. Неустроев, Б. А. Сухо-
даев. В сб.: Генераторы низкотемпе-
ратурной плазмы. М., «Энергия», 1969,
стр. 318.
23. Ф. Б. Вурзель, Н. Н. Долгополов*
А. И. Максимов, Л. С. Полак, В. И.
Фридман. В сб.: Кинетика и термоди-
намика химических реакций в низко-
температурной плазме. М., «Наука»,.
1965, стр. 223.
24. J. ВеЪоих. Ingrs. et techniciens. N 166»
109 (1963).
25. Б. M. Дымшиц, Я. П. Корецкий^
Ж. техн, физ., 34, вып. 9, 1677 (1964).
26. В. И. Пименов. Электротермия, вып.
35, 25 (1964).
27. В. И. Пименов, В. Д. Сидоренко.
Электротермия, вып. 50, 32 (1966).
28. Б. М. Дымшиц, Я. П. Кореикий.
В сб.: Плазмохимия-71. М., ИНХС
АН СССР, 1971, стр. 46.
29. Б. М. Дымшиц. Исследование кон-
трагированного индукционного раз-
ряда. Автореф. канд. дисс. М., 1970.
30. Б. М. Дымшиц, Я. П. Кореикий.
В сб.: Моделирование и расчет физико-
химических процессов в низкотемпе-
ратурной плазме. М., «Наука», 1974,
стр. 230.
31. Ю. П. Райзер. Усп. физ. наук, 99,
№ 4, 545 (1969).
32. С. В. Дресвин, В. С. Клубникин. Тепло-
физика высоких темп., 9, 475 (1971)..
33. Р. Е. Ровинский, В. А. Груздев*
И. П. Широкова. Теплофизика высо-
ких темп., 4, 35 (1966).
34. В. А. Груздев, Р. Е. Ровинский,
И. П. Широкова. Теплофизика высо-
ких темп., 5, 557 (1967).
35. Р. В. Митин, К. К. Прядкин. Ж.
техн, физ., 35, 1205 (1965).
36. К. К. Прядкин, Р. В. Митин,
Н. Н. Климов. Ж. прикл. спектр.,
10, 721 (1969).
37. Р. Е. Ровинский, А. П. Соболев:.
Теплофизика высоких темп., 6, 216;
(1968); 7, 203 (1969).
283
38. В, А. Груздев, Р. Е. Ровинский,
А. П. Соболев. Ж. прикл. мех., техн,
физ., 1, 143 (1967).
39. В. Н. Сошчиков, Е. С. Грехов. Тепло-
физика высоких темп., 4, 166 (1966);
4, 324 (1966); 5, 522 (1967); 9, 488
(1971).
40. D. D. Holhster. Phys. Letters, 27А,
672 (1968).
41. Н. U. Eckert, D. С. Pridmore-Brown.
J. Appl. Phys., 42, 5051 (1971).
42. H. U. Eckert, J. Appl. Phys., 41, 1520
(1970); 41, 1529 (1970).
43. Б. E. Мейерович, JI. П. Питаевский.
ЖЭТФ, 61, 235, 1891 (1971).
44. J. Besombes-Vailhe. Rev. phys. appl.,
2, 225 (1967).
45. S. L. Leonard. J. Quant. Spectr.
Radiat. Transfer, 12, 619 (1972).
46. H. U. Eckert. J. Appl. Phys., 41, 3633
(1970).
Al. H. U. Eckert. Contrib. Papers, 10-th
Internal. Confer. Phenomena Ionized
Gases. Oxford, 1971, p. 244.
48. И. Д. Кулагин, H. H. Рыкалин,
Л. M. Сорокин. В сб.: Физика, тех-
ника и применение низкотемпера-
турной плазмы. Алма-Ата, 1970,
стр. 717.
49. И. U. Eckert. AIAA Journal, 9, 1452
(1971).
4)0. Л. М. Блинов, В. В. Володько, Г. Г.
Гонтарев, Г. В. Лысов, Л. С. Полак.
В сб.: Генераторы низкотемператур-
ной плазмы. М., «Энергия», 1969,
<стр. 345.
51. Г. В. Лысов. В сб.: Плазмохимия-71.
ИНХС АН СССР. М., 1971, стр. 53.
52. Г. Г. Подлесный, В. А. Жданов,
В. Ф. Игнатович. В сб.: Вопросы
физики низкотемпературной плазмы.
Минск, «Наука и техника»., 1970,
стр. 184.
53. S. Murayama. J. Appl. Phys., 39,
5478 (1968).
54. G. Seifert, R. Kappelt. Exper. Technik
Phys., 13, 194 (1965).
55. СВЧ-энергетика. Под ред. Э. Окресса.
М., «Мир», 1971.
56. Л. М. Балтин, В. М. Батенин.
В. Р. Гольдберг, Н. И. Цемко. В сб.:
Генераторы низкотемпературной плаз-
мы. М., «Энергия», 1969, стр. 438.
57. Л. М. Балтин, В. М. Батенин,
В. Р. Гольдберг, И. И. Девяткин,
Н. И. Цемко. В сб.: Физика, техника
и применение низкотемпературной
плазмы. Алма-Ата, 1970, стр. 673.
•58. Ф. Б. Вурзелъ, Г. В. Лысов, Л. С. По-
лак, Ю. Л. Хаит, Э. Н. Червочкин.
Химия высоких энергий, № 5, 105
(1971).
59. Ф. Б. Вурзелъ. В сб.: Очерки физики
и химии низкотемпературной плазмы.
Под ред. Л. С. Полака. М., «Наука»,
1971, стр. 411.
<60. A. von Engel. Ionized Gases. Oxford,
Clarendon Press, 1965.
*61. M. M. Shahin. Reactions under Plasma
Conditions. N. Y., Wiley-Interscience,
1971, p. 285.
,62. Ю. А. Иванов, Л. С. Полак, Д. И. Сло-
вецкий. Теплофизика высоких темп.,
9, 1151 (1971).
63. 10. А. Иванов, В. И Макаров, А. А.
Овсянников, Л. С. Полак. Химия вы-
соких энергий, 5, 159 (1971).
64. Ю. А. Иванов, А. А. Овсянников,
Л. С. Полач, Д. И. Словецкий. Энер-
гетическое распределение электронов
в тлеющем разряде. Препринт ИНХС
АН СССР, 1972, № 5.
65. Е. В. Ступоченко, С. А. Лосев,
А. И. Осипов. Релаксационные про-
цессы в ударных волнах. М., «Наука»,
1965.
66. Я. Б. Зельдович, Ю. II. Райзер. Физи-
ка ударных волн и высокотемператур-
ных гидродинамических явлении. М.,
«Наука», 1966.
67. A. G. Gaydon, I. R. Hurle. The Shock
Tube in High Temperature Chemical
Physics. N. Y., Reinhold, 1963.
68. E. F. Greene, J. P. Toennies. Chemical
Reactions in Shock Waves. London,
Edward Arnold, 1964.
69. Основные результаты экспериментов
на ударных грубах. Под ред. Ферри.
М., ИЛ, 1963.
70. Ю. Н. Рябинин. Газы при больших
плотностях и высоких температурах.
М., Физматгиз, 1959.
71. А. М. Маркевич, В. В. Азатян,
Н. А. Соколова. Кинетика и катализ,
3, 431 (1962).
72. И. Е. Волохонович, А. М. Маркевич,
И. Ф. Мастеровой, В. В. Азатян.
Докл. АН СССР, 146, 387 (1962).
73. Ф. Б. Вурзелъ, Л. С. Полак, В. С. Щи-
пачев. Кинетика и катализ, 7, 1068
(1966).
74. И. В. Игонина, Л. С. Полак, В. С. Щи-
пачев. Кинетика и катализ, 9, вып. 1,
15 (1968).
75. В. С. Щипачев. Особенности химиче-
ской кинетики в условиях быстрого
адиабатического сжатия. Препринт
ИНХС АН СССР, 1972, № 13.
76. С. Г. Гагарин, Л. С. Полак, В. С. Щи-
пачев. В сб.: Генераторы низкотемпе-
ратурной плазмы. М., «Энергия», 1969,
стр. 70.
77. Грей, Джекобс, Шерман. Приборы
научн. исслед., № 7, 29 (1962).
78. Методы исследования плазмы. Под
ред. В. Лохте-Хольтгревена. М.,
«Мир», 1971.
79. А. А. Овсянников. В сб.: Очерки физи-
ки и химии низкотемпературной плаз-
мы. Под ред. Л. С. Полака. М.,
«Наука», 1971, стр. 386.
80. Спектроскопия газоразрядной плаз-
мы. Под ред. С. Э. Фриша. Л., «Нау-
ка», 1970.
81. Г. Грим. Спектроскопия плазмы. М.,
Атомиздат, 1969.
82. Оптическая пирометрия плазмы. Под
ред. Н. Н. Соболева. М., ИЛ, 1960.
83. А. М. Губанов. Оптика и спектр., 30,
211 (1971).
84. J. в. Tatum. Astrophys. J. Suppl.,
14, N 124, 21 (1967).
85. D. Schldter. J. Quant. Spectr. Radiat.
Transfer, 5, 87 (1965).
284
•86. О. В. Козлов. Электрический зонд
в плазме. М., Атомиздат, 1969.
87. Ф. Чен. В сб.: Диагностика плазмы.
Под ред. Р. Хаддлстоуна и С. Лео-
нарда. М., «Мир», 1967.
88. L. Schott. In «Reactions under Plasma
Conditions». M. Venugopalan (Ed.).
N- Y., Wiley—Interscience, 1971,
p. 515.
89. В. E. Голант. Сверхвысокочастотные
методы исследования плазмы. М.,
«Наука», 1968.
90. G. Bekefi, S. С. Brown. Phys. Fluids,
4, 173 (1961).
91. Л. А. Душин, О. С. Павличенко.
Исследование плазмы с помощью ла-
зеров. М., Атомиздат, 1968.
92. A. A. Ovsyanikov. In «Reactions under
Plasma Conditions». M. Venugopalan
(Ed.). N. Y., Wiley—Interscience,
1971, p. 471.
93. E. E. Salpeter. Phys. Rev., 120, 1528
(1960); 122, 1663 (1961).
94. A. T. Ситенко. Электромагнитные
флуктуации в плазме. Харьк. гос.
ун-т, 1965.
95. Р. Макуиртер. В сб.: Диагностика
плазмы. М., «Мир», 1967, стр. 165.
96. В. М. Гольдфарб. В сб.: Очерки фи-
зики и химии низкотемпературной
плазмы. М., «Наука», 1971, стр. 169.
97. R. A. Hill. J. Quant. Spectr. Radiat.
Transfer, 7, 401 (1967).
98. В. Визе. В сб.: Диагностика плазмы.
М., «Мир», 1967, стр. 218.
99. Диагностика плазмы. Сборник под
ред. Р. Хаддлстоуна и С. Леонарда.
М., «Мир», 1967.
100. С. М. Горлин, И. И. Слезинеер.
Аэромеханические измерения. М.,
«Наука», 1964.
101. А. Деметриадес, Е. Доуман. Ракетная
техника и космонавтика, № 3, 88
(1966).
102. Дж. Грей, П. Джексбс. Ракетная
техника и космонавтика, № 1, 98
(1967).
103. Б. С. Ринкевичюс. Усп. физ. наук,
111, вып. 2, 305 (1973).
104. Л. С. Полак, Н. С. Суров. Физика и
химия обр. матер., № 2, 2 (1969).
105. В. Д. Шиманович, А. К. Шипай,
Б. М. Соловьев, А. Ф. Пузряков. В сб.:
Физика, техника и применение низко-
температурной плазмы. Алма-Ата,
1970, стр. 209.
К главе 2
1. С. де-Гроот, П. Мазур. Неравновес-
ная термодинамика. М., «Мир», 1964.
2. И. Пригожин. Неравновесная термо-
динамика. М., «Мир», 1965.
3. И. Пригожин. Неравновесная стати-
стическая механика. M.J «Мир», 1964.
4. М. Кац. Вероятность и смежные во-
просы в физике. М., «Мир», 1965.
5. М. Кац. Несколько вероятностных
задач физики и математики. М.,
«Наука», 1967.
6. Дж. Честер. Теория необратимых
процессов. М., «Наука», 1966.
7. С. Фудзита. Введение в неравновес”
ную квантовую статистическую ме“
ханику. М., «Мир», 1969.
8. Д. Н. Зубарев. Неравновесная стати-
стическая термодинамика. М., «Нау-
ка», 1971.
9. С. Гласстон, К. Лейдлер, Г. Эйринг.
Теория абсолютных скоростей реак-
ций. М., ИЛ, 1948.
10. В. J. Zwolinski, Н. Eyring. J. Amer.
Ghem. Soc., 69, 2702 (1947).
11. Л. С. Полак. Очерки физики и химии
низкотемпературной плазмы. М.,
«Наука», 1971.
12. Я. Г. Вант Гофф. Очерки по хими-
ческой динамике. М., ОНТИ, 1936.
13. S. Arrenius. Z.Phys. Chem., 4, 226 (1889).
14. В. Н. Кондратьев. В сб.: Вопросы
истории естествознания и техники,
1965, стр. 1.
15. J. J. Hood. Philos. Mag., 6, 371 (1878).
16. J. J. Hood. Philos. Mag., 20, 323 (1885).
17. Л. С. Кассель. Кинетика гомогенных
газовых реакций. ОНТИ, 1937.
18. N. В. Slater. Theory of Unimolecular
Reactions. N. Y., 1959.
19. С. Бенсон. Основы химической кине-
тики. М., «Мир», 1964.
20. Ю. А. Колбановский, Л. С. Полак.
Препринт ИНХС АН СССР, 1971, № 02.
21. L. S. Polak. In «Reactions under Plas-
ma Conditions», v. 2. Venugopalan
(Ed.). N. Y., Willey — Interscience,
1971, Ch. XIII.
22. ИС M. Gelbart, S. A. Rice, K. F. Freed.
J. Chem. Phys., 52, 5718 (1970).
23. Дж. Кларк, M. Макчесни. Динамика
реальных газов. М., «Мир», 1967.
24. Р. Фаулер, Э. Гуггенгейм. Статисти-
ческая термодинамика. М., ИЛ, 1949.
25. Leicester, Klickstein. Source Book in
Chemistry. N. Y., Me Graw-Hill
Book Co., 1952, p. 468.
26. A. И. Бязь, Я. Б. Зельдович, А. М. Пе-
реломов. Рассеяние, реакции и распа-
ды в нерелятивистской квантовой
механике. М., «Наука», 1966.
27. R. С. Tolman. J. Amer. Chem. Soc.,
42, 2506 (1920).
28. E. A. Moelvin-Hughes. Kinetics of
Reactions in Solution. N. Y., Oxford
Univ. Press, 1947.
29. О. K. Rice. Statistical Mechanicsand
Kinetics. N. Y., 1967.
30. C. F. Hansen. Phys. Fluids, 12, 5, 1127
(1969).
31. N. G. Utterback. Phys. Rev. Letters,
20, 1021 (1968).
32. N. G. Utterback, G. H. Miller. Phys.
Rev., 124, 1477 (1961).
33. L. I. Kiefter, G. H. Dunu. Rev. Mod.
Phys., 38, 1 (1966).
34. Л. Спитцер. Физика полностью иони-
зованного газа. М., «Мир», 1965.
35. А. М. Губанов, Е. Н. Ерощенков,
О. А. Малкин, Л. С. Полак. Кинетика
и катализ, 13, № 1, 33 (1972).
36. Ю. А. Иванов, Л. С. Полак, Д. И. Сло-
вецкий. Труды IV Всесоюзной кон-
ференции по физике и генераторам
низкотемпературной плазмы. Алма-
Ата, 1970.
285
37. М. С. Джиджиев, В. Т. Плашоненко,
Р. В. Хохлов. Усп. физ. наук, 100,
№ 4, 641 (1970).
38. М. Menzinger, R. Wolfgang. J. Chem.
Phys., 50, 2991 (1969).
39. E. F. Creene, J. Ross. Science, 159,
587 (1968).
40. A. Kuppermann, J. M. White. J. Chem.
Phys., 44, 4352 (1966).
41. R. Wolfgang. Amer. Rev. Phys. Chem.,
16, 15 (1965).
42. Ion-Molecules Reactions in the Gas
Phase. R. F. Could (Ed.). Amer. Chem.
Soc. PubL, N 58. Washington, 1966.
43. Л. С. Полак. Сборник ИНХС АН СССР,
М., 1974.
44. М. Karplus, R. N. Porter, R. D. Shar-
ma. J. Chem. Phys., 45, 387 (1966).
45. M. Menzinger, R. Wolfgang. Angew.
Chem., 8, 438 (1969).
46. L. Rodney Le Roy. J. Phys. Chem., 73,
4338 (1969).
47. G. H. Melton, T. L. Gordon. J. Chem.
Phys., 51, 5449 (1969).
48. Z. A. Polak. Pure Appl. Chem., 39,
N 3, 307 (1974).
49. A. H. Тихонов. Докл. АН СССР, 153,
49 (1963).
50. А. Н. Тихонов. В сб.: Вычислительные
методы и программирование, вып. 8.
Изд-во МГУ, 1967, стр. 3.
51. А. Н. Тихонов, Б. В. Гласко. Ж. вы-
числ. мат. и мат. физики, 4, 564 (1964).
52. R. I. Tolman. J. Amer. Chem. Soc.,
42, 2506 (1920).
53. M. Eliason, J. О. H irschf elder. J. Chem.
Phys., 30, 1426 (1959).
54. К. E. Shuler. Chemische Elementar-
prozesse. Springer-Verlag, 1968.
55. Л. С. Полак. Химия высоких энерг.,
6, 206 (1972).
56. Л. С. Полак. Основные положения
обобщенной неравновесной химиче-
ской кинетики. Препринт ИНХС АН
СССР, 1972.
57. W. Pauli. In«Probleme der modernen
Physik». Zum A. Sonmertled 60. Ge-
burtstage. Leipzig, 1928, S. 30.
58 J. Oppenheim. Advanc. Chem. Phys.,
1, 1 (1958).
59. L. Van-Hove. Physica, 21, 517 (1955).
60. L. Van-Hove. Physica, 23, 441 (1957).
62. R. J. Swenson. J. Math. Phys., 3, 1017
(1962).
63. A. Nakajima. Progr. Theoret. Phys.,
20, 948 (1958).
64. R. Zwanzig. J. Chem. Phys., 33, 1338
(1960).
65. R. Zwanzig. Lectures Theoret Phys.,
3, 106 (1960).
66. P. Resibois. Physica, 27, 541 (1961).
67. J. Prigogine. P. Resibois. Physica, 27,
629 (1961).
68. P. Resibois. Physica. 29, 721 (1963).
69. E. И. Montroll. In Fundamental
Problems in Statistical Mechanics,
1962, p. 230.
70. E. W. Montroll. Lectures Theoret.
Phys., 3, 221 (1960).
71. R. Zwanzig. Physica, 30, 1109 (1964).
72. S. W. Benson, T. Fueno. J. Chem.
Phys., 32, 1001 (1960).
73. E. В. Ступоченко, А. И. Осипов.
Ж. физ. химии, 33, 36 (1959).
74. М. J. Kolker. J. Chem. Phys., 44, 582
(1966).
75. C. A. Brau, J. C. Keck, G. F. Carrier.
Phys. Fluids, 9, 1885 (1966).
76. E. W. Montroll, K. Shuler. Advanc.
Chem. Phys., 1, 361 (1968).
77. V. A. LoDato, D. L. S. McElwain*
H. O. Pritchard. J. Amer. Chem. Soc.,
91, 7688 (1969).
78. D. L. S. McElwain, H. O. Pritchard.
J. Amer. Chem. Soc., 91, 7693 (1969).
79. D. L. S. McElwain, H. O. Pritchard.
J. Amer. Chem. Soc., 92, 5027 (1970).
80. D. L. S. McElwain, H. O. Pritchard.
13-th Sympos. (Internat). Combustion.
Combustion Inst., Pittsburgh, 1971,
p. 37.
81. E. Kamaratos, H. O. Pritchard. Canad.
J. Chem., 49, 2617 (1971).
82. D. L. S. M cElwain, H. O. Pritchard.
Canad. J. Chem., 49, 3915 (1971).
83. D. L. S. McElwain, H. O. Pritchard.
Canad. J. Chem., 50, 897 (1972).
84. N. I. Labib, D. L. S. McElwain,
H. O. Pritchard. Canad. J. Chem.,
50, 3832 (1972).
85. T. Ashton, D. L. S. McElwain, H. O.
Pritchard. Canad. J. Chem., 51, 237
(1973).
86. E. Kamaratos, H. O. Pritchard. Canad.
J. Chem., 51, 1923 (1973).
87. J. W. Rich. J. Appl. Phys., 42, 2791
(1973).
88. H. H. Соболев, В. В. Соковиков.
Препринт ФИ АН СССР, 1971, № 108.
89. G. Е. Caledonia, R. Е. Center. J. Chem.
Phys., 55, 552 (1971).
90. Л. С. Полак, П. А. Сергеев, Д. И. Сло-
вецкий. Химия высоких энергий, 7,
№ 5, 387 (1973).
91. К. Herzfeld, Т. A. Litovitz. Absorption
and Dispersion of Ultrasonic Waves
N. Y., Acad. Press, 1959, Chap. VII
92. G. Hancock, J. W. Shith. Appl. Opt.u
10, 1827 (1971).
93. Термодинамические свойства индиви-
дуальных веществ, т. 1. Под ред.
В. П. Глушко. М., 1962, стр. 42, 352.
94. Алгоритмы метода Монте-Карло и
численного интегрирования диффе-
ренциальных уравнений с автомати-
ческим выбором шага. М., ОНТИт 1970.
95. Л. С. Полак, Д. И. Словецкий, А. С. Со-
колов. Оптика и спектр., 32, 472 (1972).
96. Ю. Г. Аванесов, Л. С. Полак, Д. И.
Словецкий, А. С. Соколов. Тезисы до-
кладов на семинаре «Химическая ки-
нетика и вычислительная математика».
Препринт ИНХС АН СССР, 1971, №01.
97. Ch. Е. Treanor, F. W. Marrone. Phys.
Fluids, 5, 1022 (1962).
98. F. W. Marrone, Ch. E. Treanor. Phys.
Fluids, 6, 1215 (1963).
99. Л. С. Полак, A. H. Темчин, A. B. Xa-
чаян. Кинетика и катализ, 12, 813
(1971).
100. L. S. Polak, A. W. Chatchojan. Trans.
Faraday Soc., 67, 1980 (1971).
101. C. S. Wang-Chang, G. E. Uhlenbeck.
Transport Phenomena in Polyatomic
286
Molecules. Univ. Michigan Publ.,
CM-681, 1951.
102. Дж. Гиршфелъдер, Ч. Кертисс,
Р. Берд. Молекулярная теория газов
и жидкостей. М., ИЛ, 1961.
103. Н. Eyring, J. О. Нirsclrjelder, Н. S. Tay-
lor. J. Chem. Phys., 4, 479 (1936).
104. G. Gionmousis, D. P. Stevenson. J. Chem.
Phys., 29, 294 (1958).
105. L. D. Landau, E. Teller. Phys. Z.
Sowjetunion, 10, 34 (1936).
106. R. N. Schwarz, K. F. Herzfeld, Z. I.
Slawsky. J. Chem. Phys., 20, 1591
(1952).
107. R. N. Schwarz, K. F. Herzjeld. J.
Chem. Phys., 22, 767 (1954).
108. J. Ross, J. C. Light, К. E. Shuler.
In «Kinetic Processes in Gases and
Plasmas». A. R. Hochstim (Ed.). N. Y.,
Acad. Press, pt. VIII.
109. Б. В. Гордиец, A. И. Осипов, Л. A. Ше-
лепин. Препринт ФИАН СССР, 1970,
№ 17.
110. D. Rapp, Т. Е. Sharp. J. Chem. Phys.,
38, 2641 (1963).
ill. D. L. Bunker. Theory of Elementary
Gas Reaction Rates. N. Y., Perga-
mon-Press, 1966.
112. J. Wei. J. Chem. Phys., 36, 1578 (1962).
113. B. D. Coleman, Г. J. Mizel. Arch.
Rational Meeh. Analyses, 25, 243
(1967).
114. F. Hofflich. Z. Phys., 226, N 5, 395
(1969).
115. W. Hahn. Stability of Motion, Sprin-
ger, 1967.
116. П. Гленсдорф, И. Пригожин. Термо-
динамическая теория структуры, ус-
тойчивости и флуктуаций. М., «Мир»,
1973.
117. М. Н. Коган. Динамика разреженных
газов. М., «Наука», 1967.
118. В. П. Шидловский. Введение в дина-
мику разреженного газа. М., «Наука»,
1965.
119. К. Хуанг. Статистическая механика.
М., «Мир», 1966.
120. С. Чемпен, Т. Каулинг. Математиче-
ская теория неоднородных газов. М.,
ИЛ, 1960.
121. Дж. Бонд, У. Уотсон, Д. Уэлч. Фи-
зическая теория газовой динамики.
М., «Мир», 1968.
122. S. L. Miller. Biochim. Biophys. Acta,
23, 488 (1957).
123. К. E. Shuler. Phys. Fluids, 2, N 4, 442
(1959).
124. F. C. Andrews. J. Chem. Phys., 35,
N 3, 992 (1961).
125. E. G. Cohen. Physica, 28, N 10, 1025
(1962).
126. M. S. Green. Phys. Rev., 135, N 4A,
905 (1964).
127. E. Uehling, G. Ublenbeck. Phys. Rev.,
43, 522 (1933).
128. S. Hess. Z. Naturwiss., 22a, N 12, 1871
(1967).
129. M. H. Коган. Докл. АН СССР, 175,
785 (1967).
130. Н. Р. Сибгатулин. Докл. АН СССР,
180, 48 (1968).
131. J. Т. O'Toole, J. Dahles. J. Chem.
Phys., 39, N 12, 3214 (1963).
132. С. В. Волландер, И. А. Егорова,
M. А. Рыдалевская. Вести. ЛГУ, 7,
155 (1964).
133. Г. Грэд. В сб.: Некоторые вопросы
кинетической теории газов. М., «Мир»,
1965, стр. 7, 93.
134. R. F. Suider. J. Chem. Phys., 41, N 3,
591 (1964).
135. В. H. Рыков, Г. И. Чуканова. В сб.:
3-й Всесоюзный съезд по теоретиче-
ской и прикладной механике. 1968,
стр. 202.
136. М. Wachman, В. В. Hamel. Rarefied
Gas. Dynamics, v. 1. N. Y., Acad.
Press, 1967.
137. A. H. Темчин. Инж. физ. ж., 16, 5
(1969).
138, Б. В. Алексеев. Докл. АН СССР, 182,
№ 2, 288 (1968).
139. J. Ross, Р. Mazur. J. Chem. Phys.,
35, N 1, 19 (1961).
140. N. S. Snider, J. Ross. J. Chem. Phys.,
44, 1087 (1966).
141. E. F. Green, A. L. Moursund, J. Ross.
Advanc. Chem. Phys., 10, 135 (1966).
142. P. Hammering, J. D. Teare, B. Kivel.
Phys. Fluids, 2, 422 (1959).
113. M. Karplus, L. M. Raff. J. Chem.
Phys., 41, 1266 (1964).
144. К. E. Shuler, G. H. Weiss. J. Chem.
Phys., 38, 505 (1963).
145. C. W. Pyun, J. Ross. J. Chem. Phys.,
40, 2572 (1964).
146. J. T. O'Toole, J. S. Ross. J. Chem.
Phys., 39, 3214 (1963).
147. S. H. Bauer. Science, 141, 867 (1963).
L48. О. K. Rice. J. Chem. Phys., 9, 258
(1941).
149. A. G. Gaydon, J. R. Hurle. The Shock
Tube in High Temperature Chemical
Physics. London, 1963.
150. The Study of Fast Reactions. Discuss.
Faraday Soc., N 17, 1954.
151. О. K. Rice. J. Chem. Phys., 9, 258
(1941).
152. Применение вычислительной матема-
тики в химической и физической ки-
нетике. Под ред. Л. С. Полака. М.,
«Наука», 1969.
153. Л. С. Полак, А. Н. Темчин, А. В. Ха-
чоян. Кинетика и катализ, 11, 831
(1970).
154. В. М. Жданов. ЖЭТФ, 53, 6, 2099
(1967).
155. В. D. Present. J. Chem. Phys., 48,
11, 4875 (1968).
156. У. Yoshizawa. Rarefied Gas Dynamics,
v. 1. N. Y., Acad. Press, 1967.
157. С. А. Денисик, С. H. Лебедев, Ю. Г.
Малама, Л. С. Полак. Химия высоких
энергий, 2; 4, 297 (1968).
158. Л. С. Полак, А. Н. Темчин. В кн.:
Применение вычислительной мате-
матики в химической и физической
кинетике. Под ред. Л. С. Полака.
М., «Наука», 1968.
159. А. Н. Темчин. Инж. физ. ж., 17, 5,
900 (1969).
160. Н. П. Бусленко, Ю. А. Шрейдер.
Метод статистических испытаний (ме-
287
тод Монте-Карло). М., Физматгиз,
1961.
161. К. Е. Shuler. 5-th Sympos. Internal,
on Combustion. N. Y., 1955.
162. Л. С. Полак, A. H. Темчин, A. B. Xa-
чоян. Теплофизика высоких темп., 9,
№ 2, 230 (1971).
163. И. Н. Семенов. О некоторых пробле-
мах химической кинетики и реакци-
онной способности. М., Изд-во АН
СССР 1958.
164. II. Р. Brovda, К. Е. Shuler. J. Chem.
Phys., 20, 168 (1952).
165. К. Е. Shuler. J. Chem. Phys., 21, 624
(1953).
166. A. G. Gaydon, H. G. Woljhard. Proc.
Roy. Soc. London, A208, 63, (1951).
167. К. E. Shuler. Fifth Symposium (Inter-
nal) on Combustion. N. Y., 1955, p. 56.
168. B. Widom, S. H. Bauer. J. Chem.
Phys., 42, 1670 (1953).
169. R. V. Serauskas, E. W. Schlag. J.
Chem. Phys., 42, 3009 (1965).
170. H. Eyring. J. Chem. Phys., 3, 107
(1935).
171. F. T. Wall, L. A. Hiller, J. Mazur.
J. Chem. Phys., 29, 255 (1958).
172. F. T. Wall, L. A. Hiller, J. Mazur.
J. Chem. Phys., 35, 1284 (1961).
173. M. Karplus, R. N. Porter, R.D. Shar-
ma. J. Chem. Phys., 43, 3259 (1965).
174. F. A. Lindemann. Trans. Faraday Soc.,
17, 598 (1922).
175. G. N. Hinschelwood. Proc. Roy. Soc.
London, A133, 230 (1927).
176. L. S. Kassel. J. Phys. Chem., 32, 225
(1928).
177. О. K. Rice, H. C. Rampsperger. J.
Amer. Chem. Soc., 49, 1617 (1927).
178. N. B. Slater. Proc. Roy. Soc. London,
A194, 112 (1948).
179. R. A. Markus. J. Chem. Phys., 49,
2610 (1968).
180. R. A. Markus. J. Chem. Phys., 49,
2617 (1968).
181. В сб.: Возбужденные частицы в хи-
мической кинетике. Под ред. К. Б эм-
форд, К. Типпер. М., «Мир», 1973.
182. К. J. Laidler, J. С. Polanyi. Progr.
Reaction Kinetics, 3, 1 (1965).
183. Л. С. Полак. В сб.: Плазмохимия-71.
М., ИНХС АН СССР, 1971, стр. 1—15.
184. Е. В. Сшупоченко, А. И. Осипов,
С. А. Лосев. Релаксационные процес-
сы в ударных трубах. М., Физматгиз,
1966.
185. A. L. Schmeltekopf, Е. Е. Ferguson,
F. С. Rosenfeld J. Chem. Phys., 48,
2966 (1968).
186. А. Энгель. Ионизованные газы. М.,
Физматгиз, 1959.
187. Бай-Ши-И. Магнитная газодинамика
и динамика плазмы. М., «Мир», 1964.
188. Ю. М. Каган, В И. Перелъ. Усп. физ.
наук, 81, 409 (1963).
189. А. М. Губанов, Ю. А. Иванов, А. А.
Овсянников, Л. С. Полак. Ж. прикл.
спек р., 12, 8 (1970).
190. Л. С. Полак. В кн.: Применение вы-
числительной математики в физиче-
ской и химической кинетике. (Введе-
ние). М., «Наука», 1969.
191. L. S. Polak, D. I. Slovetsky. XI Inter-
nal. Confer. Phenomena Ionized Gases.
Prague, 1973, p. 51.
193. Ю. А. Иванов, И. А. Сергеев, Л. С. По-
лак, Д. И. Словецкий. А. С. Соколов,
Б. Б. Шварцман. Труды IV Вс союз-
ной конференции по физике и генера-
торам низкотемпературной плазмы.
Алма-А а, 1970, стр. 18.
194. Ю. А. Иванов, Л. С. Полак, Д. И. Сло-
вецкий. Теплофизика высоких темп.,
№ 6, 1151 (1971).
195. Л. С. Полак, Д. И. Словецкий, Е. Р.
Мамиконян. Химия высоких энергий,
6, № 6, 483 (1973).
196. К. К. Corvin, S. J. В. Corrigan. J.
Chem. Phys., 50, 2570 (1969).
197. Л. С. Полак, Д. И. Словецкий,
А. С. Соколов. Химия высоких энер-
гий, 6, № 6, 396 (1972).
198. Л. С. Полак. Химия высоких энер-
гий, 6, № 3, 266 (1972).
199. Ю. А. Иванов. Автореф. канд. дисс.,
ИНХС АН СССР, 1971.
200. Ю. А. Иванов, А. А. Овсянников,
Л. С. Полак, Д. И. Словецкий. До-
клад на семинаре «Обобщенная нерав-
новесная химическая кинетика».
Препринт ИНХС АН СССР, 1972, № 5.
201. А. Н. Солдатов. Оптика и спектр.,
31, вып. 2, 181 (1971).
202. И. С. Бучелъникова. Усп. физ. наук,
65, вып. 3 (1958).
203. В. И. Светцов, А. И. Максимов.
Г. П. Добр якова. Химия высоких
энергий, 1, 174 (1967).
204. . W. N. Kolesnikow, Е. В. Kuprifanowa,
N. N. Sobolew. J. Quant. Spectr.
Radiat. Transf., 9, 1025 (1969).
205. J. W. McConcey, F. R. Simpson. J.
Phys., B, 2, 923 (1969).
206. P. N. Stanton, R. M. S. John. J. Opt.
Soc. America, 59, 252 (1969).
207. Л. С. Полак, Д. И. Словецкий, А. С.
Соколов. Химия высоких энергий, 7,
№ 2, 183 (1972).
208. А. К. Brewer. J. Chem. Phys., 35,
1281, 1293 (1931).
209. Г. С. Ушакова, Е. Н. Еремин. Ж. физ.
химии, 31, 233 (1959).
210. С. К. N. Patel. Phys. Rev. Letters,
12, 588 (1954).
211. A. H. Мальцев, E. H. Еремин, В. Л.
Ивантер. Ж. физ. химии, 41, 1190
(1967).
212. Е. С. Гасилевич, В. А. Иванов,
Э. Н. Лоткова, В. Н. Очкин, Н. Н. Со-
болев, И. Г. Ярославский. Ж. техн,
физ., 39, 126 (1969).
213. J. N. Noon, Р. R. Blazuk, Е. Н. Holt
Appl. Phys., 39, 5518 (1968).
214. К. К. Corvin, S. J. В. Corrigan.
J. Chem. Phys., 50, 2570 (1969).
215. Э. H. Лоткова, В. И. Макаров,
Л. С. Полак, Н. Н. Соболев. Химия
высоких энергий, 2, 278 (1968).
216. М. 3. Новгородов, А. Г. Свиридов,
Н. И. Соболев. Письма в ЖЭТФ,
8, 341 (1968).
217. Ю. А. Иванов, В. И. Макаров, А. А.
Овсянников, Л. С. Полак. Химия вы-
соких энергий, 5, 159 (1971).
288
218. М. 3. Новгородов, В. Н. Очкин, Н. Н.
Соболев. Препринт ФИАН СССР, 1969,
№ 172.
219. R. L. F. Boyd, J. В. Thompson. Proc.
Roy. Soc. London, A252, 102 (1959).
220. R. D. Hake, A. V. Phelps. Phys. Rev.,
158, 70 (1967).
221. Л. M. Брусиловская, M. 3. Новгоро-
дов, А. Г. Свиридов, H. Н. Соболев.
Препринт ФИАН СССР, 1969, № 32.
222. Ю. А. Иванов, Л. С. Полак, Д. И. Сло-
вецкий. Химия высоких энергий, 5,
382 (1972).
223. R. J. Donovan, D. Husain. Chem. Rev.,
70, 489 (1970).
224. T. Carrington, D. Garvin. Comprehen-
sive Chemical Kinetics, Amsterdam -r-
London — N. Y., Elsevier Publ. Corp.,
1969.
225. J. F. Noxon. J. Chem. Phys., 36, 926
(1962).
226. S. N. Foner, R. L. Hudson. J. Chem.
Phys., 37, 1665 (1962).
227. A. N. Wright, C. A. Winder. Active
Nitrogen. N. Y.— London, Acad.
Press, 1968.
228. D. E. Shemansky. J. Chem. Phys., 51,
689 (1969).
229. M. Jeunehomme, A. B. F. Duncan.
J. Chem. Phys., 41, 1962 (1964).
230. R. N. Zare, F. O. Larsson, K. A. Berg.
J. Mol. Spectrosc., 15, 117 (1965).
231. А. А. Коньков, В. Я. Нейланд,
В. М. Николаев, Ю. А. Пластинин.
Теплофизика высоких темп. 7, 140
(1969).
232. В. A. Thrush. J. Chem. Phys., 47,
3691 (1967).
233. Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. Под ред. Л. С. Полака. М.,
«Наука», 1965.
234. L. S. Polak. Invited Papers 10-th In-
ternal Conf, on Phenomena Ionized
Gases. Oxford, 1971, p. 113.
235. Дж. Уленбек, Дж. Форд. Лекции по
статистической механике. М., «Мир»,
1965.
236. А. А. Овсянников, Л. С. Полак, М. Б.
Сафонова, М. С. Федорова. Теорет.
основы хим. технологии, 5, № 6, 891
(1971).
237. А. А. Овсянников, Л. С. Полак,
М. А. Куманцев. Химические реак-
ции в условиях турбулентности. Пре-
принт ИНХС АН СССР, 1972, № 16.
238. Н. В. Ермохин, Б. М. Ковалев, П. П.
Кулик, В. А. Рябый. Физика, тех-
ника и применение низкотемператур-
ной плазмы. Алма-Ата, 1970, стр. 93.
239. А. Г. Храпак, И. Т. Якубов. ЖЭТФ,
945, 1970.
240. R. Clampit. 10-th Internal. Conf, on
Phenomena Lonized Cases. Oxford, 1971.
241. В. А. Зейгарник, Г. А. Кобзев, Ю. К.
Куриленков, Г. Э. Норман. III Все-
союзная конференция по физике низ-
котемпературной плазмы. М., 1971,
стр. 104.
242. И. В. Авилова, Г. Э. Норман. Тепло-
физика высоких темп.., 2, 517 (1964).
243. Г. Э'. Норман. ЖЭТФ, 60, 1686 (1971).
244. Г. А. Кобзев. Оптика и спектр., 31,
37, 1971; ЖЭТФ 61, 582, 1971.
245. Е. J. Robbins, R. Е. Leckenby, Р. Wil-
lis. The properties of Liqzid metals.
Proc. Internal. Conf. Brookhaven Lab.,
Sept. 1966.
246. П. П. Кулик, Г. Э. Норман, Л. С. По-
лак. Всесоюзная конференция по хи-
мии высоких энергий. М., 1971.
247. J. Amdur, G. G. Hammes. Chemical
Kinetics: Principles and Selected To-
pics. N. Y., 1966.
248. В. И. Веденеев, А. В. Парийская.
Кинетика и катализ, 12, 21, 293, 543,
839 (1971); 14, 1116 (1973).
249. Б. А. Медведев, М. А. Тейтпелъбойм,
В. И. Веденеев. Кинетика и катализ,
13, 50 (1972).
250. В. И. Веденеев, Б. А. Медведев,
М. А. Тейтелъбойм, А. Е. Шилов,
Ж. физ. химии, 40, вып. 1, 2924
(1972).
251. И. Г. Федотов, В. И. Веденеев, О. Н.
Саркисов. Докл. АН СССР, 208, 401
(1973).
252. П. П. Кулик, Г. Э. Нопман, Л. С. По-
лак. Плазмохимия-71. ИНХС АН СССР.
М., 1971.
К главе 3
1. Очерки физики и химии низкотемпе-
ратурной плазмы. Под ред. Л. С. По-
лака. М., «Наука», 1971.
2. В. Н. Кондратьев. Константы скоро-
сти газофазных химических реакций.
Справочник. М., «Наука», 1970.
3. Г. Герцберг. Спектры и строение двух-
атомных молекул. Пер. с англ. Под
ред. В. Н. Кондратьева. М., ИЛ, 1949.
4. Г. Герцберг. Электронные спектры и
строение многоатомных молекул.
Пер. с англ. Под ред. В. М. Татев-
ского. М., «Мир», 1969.
5. F. R. Gilmore. J. Quant. Spectr.
Radiat. Transfer, 5, 369 (1965).
6. J. A. Pople, D. L. Beveridge. Appro-
ximate molecular orbital theory (ser.
Advances in chemistry). N. Y., Me Graw-
Hill, 1970.
7. M. Krauss, D. Neuman, A. C. Wahl,
G. Das, W. Zemke. Phys. Rev., A7,
69 (1973).
8. У. Tanaka, K. Yoshino. J. Chem.
Phys., 53, 2012 (1970).
9. J. N. Bardsley. J. Phys. В, 1, 349
(1968).
10. R. S. Mulliken. J. Chem. Phys., 55,
288 (1971).
11. P. K. Carrol. J. Chem. Phys., 37, 807
(1962).
12. J. N. Murrel, J. M. Taylor. Mol. Phys.,
16, 609 (1969).
13. E. Gerjouy, S. Stein. Phys. Rev., 97,
1671 (1955).
14. Б. M. Смирнов. Атомные столкнове-
ния и элементарные процессы в плаз-
ме. М., Атомиздат, 1968, гл. VI.
15. А. V. Phelps. Rev. Mod. Phys., 40, 399
(1968).
16. К. Takajanagi. Progr. Theoret. Phys.,
suppl., N 40, 216 (1967).
289
17. R. J. W. Henry, E. S. Chang. Phys.
Rev., A5, 276 (1972).
18. G. J oyer, J. Comer, F. H. Read. J.
Phys. B, 6, 2727 (1973).
19. D. G. Truhlar, J. R. Rice. J. Chem.
Phys., 52, 4480, 4532 (1970).
20. /V. Chandra, P. G. Burke. J. Phys. B,
6, 2355 (1973).
21. E. В. Ступоченко, С. А. Лосев,
А. И. Осипов. Релаксационные про-
цессы в ударных волнах. М., «Наука»,
1965, гл. 4.
22. Дж. Гиршфелъоер ,4. Кертисс, Р. Берд.
Молекулярная теория газов и жидкое
тей. Пер. с англ. Под ред. Е. В. Сту-
поченко. М., ИЛ, 1961, гл. 13.
23. А. С. Соколов, Т. В. Федосеева. В сб.:
Нефтехимический синтез и высоко-
молекулярные соединения. М., «Нау-
ка», 1973, стр. 92.
24. Л. С. Полак, Д. И. Словецкий, А. С.
Соколов. Возбуждение электронных
состояний и С3ли молекулы
азота в тлеющем разряде. Препринт
ИНХС АН СССР, 1974.
25. О. П. Бочкова, Н. В. Чернышева,
10. А. Толмачев. V Всесоюзная кон-
ференция по физике электронных и
атомных столкновений. Ужгород,
1972, стр. 175.
26. J. Н. Moore, J. Р. Doering. Phys.
Rev., 177, 218 (1969).
27. J. N. Bardsley, J. Mandle. Repts
Progr. Phys., 31, 1 (1968).
28. Л. С. Полак, Д. И. Словецкий. Тепло-
физика высоких темп., 10, 645 (1972).
29. D. G. Truhlar, S. Trajmar, W. Wil-
liams. J. Chem. Phys., 57, 3250, 3260
(1972).
30. A. Stomatovic, G. J. Schulz. Phys.
Rev., 188, 213 (1969).
31. I. W. Larkin, J. B. Hasted. Chem.
Phys. Letters, 5, 325 (1970).
32. M. J. W. Boness, G. J. Schulz. Phys.
Rev. Letters, 21, 1031 (1968).
33. D. Spence, G. J. Schulz. Phys. Rev.,
A2, 1802 (1970).
34. D. Spence, G. J. Schulz. Phys. Rev.,
A3, 1968 (1971).
35. G. J. Schulz. Phys. Rev., A135, 988
(1964).
36. L. Sanche, G. J. Schulz. Phys. Rev.,
A6, 69 (1972).
37. G. E. Chamberlain, H. G. M. Hyde-
mann, С. E. Kyatt. J. Chem. Phys.,
44, 355 (1966).
38. H. Ehrhardt, K. Willmann. Z. Phys.,
204, 462 (1967).
39. J. C. Y. Chen. Phys. Rev., 146, 61
(1966).
40. G. J. Schulz. Rev. Mod. Phys., 45,
423 (1973).
41. A. X. Мнацаканян, Л. M. Биберман.
Теплофизика высоких темп., 4, 491
(1966).
42. К. F. Herzjeld, Т. A. Litovitz. Absorp-
tion and Dispersion of Ultrasonic
Waves. N. Y., Acad. Press., 1959,
Ch. VII.
43. E. E. Никитин. Теория элементар-
ных атомно-молекулярных процессов
в газах. М., «Химия», 1970.
44. С. А. Лосев, М. С. Яловик. Химия
высоких энергий, 4, 202 (1970).
45. С. А. Лосев, О. П. Шаталов. Химия
высоких энергий, 4, 263 (1970).
46. R. L. Taples, S. Bitterman. Rev. Mod.
Phys., 41, 26 (1969).
47. J. D. Lambert. J. Chem. Soc. Faraday
Trans., 68, pt 2, 364 (1972).
48. J. C. Stephenson, С. B. Moore, G. Bra-
dley. J. Chem. Phys., 56, 1295, 4813
(1972).
49. G. Hancock, I. W. M. Smith. Chem.
Phys. Letters, 8, 41 (1971).
50. W. A. Rosser, J. R. D. Sharma,
E. T. Gerry. J. Chem. Phys., 54, 1196,
4131 (1971).
51. M. A. Kovacs. J. Chem. Phys., 58,
4704 1973).
52. D. J. Eckstrom. J. Chem. Phys., 59,
2787 (1973).
53. K. G. Anlauf, P. H. Dawson, J. A. Her-
man. J. Chem. Phys., 58, 5354 (1973).
54. С. E. Treanor, J. IF. Rich, R. G. Rem.
J. Chem. Phys., 48, 1798 (1968).
55. Ю. Г. Аванесов, Л. С. Полак, Д. И.
Словецкий, А. С. Соколов. Химическая
кинетика и вычислительная матема-
тика. Препринт ИНХС АН СССР,
1971, № 01, стр. 16.
56. Л. С. Полак, Д. И. Словецкий, П. А.
Сергеев. Химия высоких энергий, 7,
395 (1973); Экспериментальные и тео-
ретические исследования неравновес-
ных физико-химических процессов.
М., ИНХС АН СССР, 1974, стр. 392.
57. Л. С. Полак, В. А. Кузьмин, П .А. Сер-
геев, Д. И. Словецкий. Химия вы-
соких энергий, 8, 129 (1974).
58. Н. Н. Соболев, В. В. Соковиков. Обра-
зование инверсии в С0-лазере. Пре-
принт ФИАН СССР, 1971, № 108.
59. G. Е. Caledonia, R. Е. Center. J. Chem.
Phys., 55, 552 (1971).
60. Б. Ф. Гордиец, А. И. Осипов, Л. А. Ше-
лепин. Усп. физ. наук, 108, 655 (1972).
61. J. W. Rich. J. Appl. Phys., 42, 2719
(1971).
62. Л. А. Вайнштейн, И. И. Собелъман,
Е. А. Юков. Сечения возбуждения
атомов и ионов электронами. М.,
«Наука», 1973.
63. Н. W. Drawin. Z. Phys., 211, 404
(1968); 225, 470, 483 (1969).
64. Д. И. Словецкий. В сб.: Моделирование
и методы расчета физико-химических
и физических процессов в низкотем-
пературной плазме. Под ред. Л. С. По-
лака. М., «Наука», 1974, стр. 3.
65. Д. И. Словецкий. В сб.: Плазмохи-
мия-71. М., ИНХС АН СССР, 1971,
стр. 19.
66. S. Chung, Ch.-C. Lin. Phys. Rev.,
6A, 988 (1972).
67. Д. И. Словецкий. В сб.: Химия плаз-
мы. Под ред. Б. М. Смирнова. М.,
Атомиздат, 1974, стр. 283.
68. S. A. Lawton, F. М. J. Pichanick.
Phys. Rev., 7А, 1004 (1973).
69. J. Mazeau, F. Gresteau. R. I. Hall,
G. Joyer, J. Reinhardt. J. Phys. B, 6,
862, 873 (1973).
290
70. L. Sanche, G. J. Schulz. J. Chem.
Phys., 58, 479 (1973).
71. A. W. Weiss, M. Krauss. J. Chem.
Phys., 52, 4363 (1970).
72. Ch. R. Claydon, G. A. Segal, H. S. Tay-
lor. J. Chem. Phys., 52, 3387 (1970).
73. F. Linder, H. Schmidt, Z. Natur-
forsch., 26a, 1603 (1971).
74. E. N. Lassettre. Canad. J. Chem., 47,
1733 (1969).
75. W. A. Goddard HI, D. L. Huestis,
D. C. Cartwright. Chem. Phys. Let-
ters, 11, 329 (1971).
76. A. J. Kelly. J. Chem. Phys., 45, 1723,
1733 (1966).
77. M. R. Flannary. Ann. Phys., 61, 465
(1970).
78. R. E. Huffmann. D. H. Katyama. J.
Chem. Phys., 45, 138 (1966).
79. А. Каллир, Дж. Ламберт. В сб.:
Возбужденные частицы в химической
кинетике. Пер. с англ. Под ред.
А. А. Борисова. М., «Мир», 1973.
80. G. Karl, Р. Kruus, J. С. Polanyi.
J. Chem. Phys., 46, 224, 244 (1967).
81. A. L. Schmeltekopf, F. C. Fehsenfeld.
J. Chem. Phys., 53, 3173 (1970).
82. L. G. Piper, J. E. Velazco, D. W. Set-
ser. J. Chem. Phys., 59, 3323 (1973).
83. C.-L.-Lin, F. Kaufman. J. Chem.
Phys., 55, 3760 (1971).
84. R. J. Donovan, D. Husain. Chem. Rev.,
70, 489 (1970).
85. Л. С. Полак, Д. И. Словецкий, P. Д.
Тодесайте. Химия высоких энергий,
10, N 1 (1976).
86. D. Husain, L. J. Kirsch, J. R. Wiesen-
feld. Discuss. Faraday Soc., N 53, 201
(1972).
87. E. C. Zipf. Canad. J. Chem., 47, 1863
(1969).
88. J. A. Meyer, D. W. Setser, D. H. Sted-
man. J. Phys. Chem., 74, 2239 (1970).
89. J. W. Dreyer, D. Perner. J. Chem.
Phys., 58, 1195 (1973).
90. M. Jenehomme. J. Chem. Phys., 45,
1805 (1966).
91. Л. С. Полак, Д. И. Словецкий, А. С.
Соколов. Оптика и спектр., 32, 472.
(1972).
92. J. М. Calo, R. С. Axtmann. J. Chem.
Phys., 54, 1332 (1970).
93. R. A. Young, G. Volkenburgh. J.
Chem. Phys., 55, 2900 (1971).
94. T. S. Wauchop, H. P. Broida. J.
Chem. Phys., 56, 330 (1972).
95. L. A. Melton, W. Klemperer. J. Chem.
Phys., 59, 1099 (1973).
96. J. M. Cambell, B. A. Thrush. J. Quant.
Spectr. Radiat. Transfer., 8,1571 (1968).
97. B. L. Earl, R. R. Herm, Sh. M. Lin,
Ch. A. Mims. J. Chem. Phys., 56,
867 (1972).
98. J. E. Selvin, J. I. Steinfeld. Chem.
Phys. Letters, 4, 217 (1969).
99. R. Atkinson, К. H. Welge. J. Chem.
Phys., 57, 3689 (1972).
100. Ф. И. Вилесов, В. И. Клейменов,
Ю. В. Чижов. Успехи фотоники. Л.,
1971, стр. 3.
101. J. Berkowitz, W. A. Chupka. J. Chem.
Phys., 51, 2341 (1969).
102. W. A. Chupka, J. Berkowitz. J. Chem.
Phys., 51, 4244 (1969).
103. R. S. Berry, S. E. Nielsen. Phys.
Rev., 1A, 395 (1970).
104. Б. M. Смирнов. Атомные столкнове-
ния и элементарные процессы в плаз-
ме. М., Атомиздат, 1968, гл. 10.
105. Р. Englander-Golden, D. D. Briglia.
J. Chem. Phys., 42, 4081 (1965).
106. L. J. Kiefer. Atomic Data, 1, 133
(1969).
107. V. Kovacs. Rotational Structure in
the Spectra of Diatomic Molecules.
Budapest, Akad. Kiado, 1969, chap.
108. Л. С. Полак, Д. И. Словецкий,
А. С. Соколов. Химия высоких энер-
гий, 6, 396 (1972).
109. R. S. Mulliken. J. Chem. Phys., 33,
247 (1960).
110. Г. Месси, Е. Бархоп. Электронные и
ионные столкновения. Пер. с англ.
М., ИЛ., 1958, стр. 196.
111. А. С. Allison, A. Dalgamo. Atomic
Data, 1, 91 (1969).
112. Р. S. Julienne. Chem. Phys. Letters,
8, 27 (1971).
113. F. J. Comes, S. Schumpe. Z. Natur-
forsch., 26a, 538 (1971).
114. G. Herzberg, Ch. Junge. J. Mol. Spec-
trosc., 41, 425 (1972).
115. S. Trajmar, A. Kuppermann. J. Chem.
Phys., 49, 5464 (1968).
116. S. P. Khare. Phys. Rev., 149, 33
(1966); 157, 107 (1967).
117. R. Clampitt, A. S. Newton. J. Chem.
Phys., 50, 1997 (1969).
118. D. A. Vroom, E. J. De Heer. J. Chem.
Phys., 50, 580 (1969).
119. W. L. Fite, R. T. Brackmann. Phys.
Rev., 112, 1151 (1958).
120. S. J. B. Corrigan. J. Chem. Phys.,
43, 4381 (1965).
121. R. E. Center. J. Chem. Phys., 54,
3499 (1971).
122. V. L. Carter. J. Chem. Phys., 56,
4195 (1972).
123. K. Dressier. Canad. J. Phys., 47, 547
(1969).
124. P. K. Caroil, K. Yoshino. J. Phys.,
B, 5, 1614 (1972).
125. W. Sr oka. Z. Naturforsch., 24a, 398
(1969).
126. M. Ogawa, G. R. Cook. Canad. J
Phys., 43, 256 (1965).
127. R. D. Hudson, V. L. Carter. Canad.
J. Chem., 47, 1840 (1969).
128. G. R. Cook, P. H. Metzger. J. Chem.
Phys., 41, 321 (1964).
129. R. T. Brinkmann, S. Trafmar. Ann.
Geophys., 28, 201 (1970).
130. H. F. Winters. J. Chem. Phys., 44,
1472 (1966).
131. A. Niehaus. Z. Naturforsch., 22a,
640 (1967).
132. J. F. M. Aar's, F. J. De Heer. Physi-
ca, 52, 45 (1971).
133. J. M. Ajello. J. Chem. Phys., 53, 1156
(1970).
134. S. Trafmar, W. W. Williams, A. Kup-
permann. J. Chem. Phys., 56, 3759
(1972).
291
135. R. E. Huffman, Y. Tanaka, J. C. Lar-
rabee. J. Chem. Phys., 37, 159 (1964).
136. K. D. Bayer, К. H. Welge. J. Chem.
Phys., 51, 5j23 (1969).
- 137. A. Gaydon. Proc Phys. Soc., A.56,
95, (1944).
138. Дж. Калверте, Дж. Питтс. «Фото-
химия». Пер. с англ. Под. ред.
А. X. Багдасарьяна. М., «Мир»,
1968, гл. 5—7.
139. Р. Н. Metzger, G. R. Cook, М. Ogawa.
Canad. J. Phys., 45, 203 (1967).
140. К. Watanabe, F. M. Matsunaga,
dajitne Sakai,. Appl. Optics, 6, 391
(1967).
141. M. M. Повч, Г. Ю. Герзанич, В. В.
Скубенич. V Всесоюзная конферен-
ция по физике электронных и атомных
столкновений. Ужгород, 1972, сгр. 45.
142. А. И. Максимов, В. М. Светцов,
В. Ф. Соколов, Т. А. Беззубенко.
Химия высоких энерги i, 4, 543 (1970).
143. Y. Tanaka, A. S. Jursa, F. Le Blanc.
J. Chem. Phys., 26, 862 (1957).
144. R. Schmid, L. Gero. rL. Phys., 93, 656
(1935).
145. E. N. Lassettre, V. D. Meyer. J. Chem.
Phys., 43, 805 (1965).
146. E. N. Lassettre, A. Skerbele. J. Chem.
Phys., 54, 1597 (1971).
147. E. N. Lassettre, V. D. Meyer. J. Chem.
Phys., 51, 1608 (1971).
148. J. M. Afello. J. Chem. Phys., 55,
3156, 3158, 3169 (1970).
149. D. Coster, F. Brons. Physica, 1, 155
(1934).
150. G. R. Cook.C&naA. J. Phys., 43, 1703
(1965).
151. R. B. Cairns. J. Opt. Soc. America,
56, 526 (1966); J. Geophys. Res., 70,
99 (1965).
152. В. И. Макаров, Л. С. Полак. Химия
высоких энергий, 4, 3 (1970).
153. R. W. Ditchburn. Pror. Roy. Soc.
London, A229, 44 (1955).
154. D. R. Salahub, C. Sandorfy. Theoret.
chim. acta, 20, 227 (1971).
155. H. Ehrhardt, F. Linder. Z. Natur-
forsch., 22a, 444 (1967).
156. D. A. Vroom, F. J. De Heer. J. Chem.
Phys., 50, 573 (1969).
157. Ch. Melton, R. S. Rudolph. J. Chem.
Phys., 47, 1771 (1965).
158. J. E. Manton, A. W. Tickner. Canad. J.
Chem., 38, 858 (1960).
159. V. L. Tairose, G. V. Karachevtsev.
In: Reaction under Plasma Condi-
tions, v. 2. M. Venugopalan (Ed.).
N. Y., Wiley Interscience, 1971, p.
35.
160. L. G. Christopherou. Pros. 3-rd. Conf,
on Radiat. Chem. Tihany, Hungary,
1971.
161. P. J. Chantry. J. Chem. Phys., 51,
3369 (1969).
162. D. Spence, G. J. Schulz. Phys. Rev.,
188, 280 (1969).
163. D. Spence, G. J. Schulz. J. Chem.
Phy^., 54, 5424 (1972).
164. M. А. Бурнашев, В. Д. Объедков.
Докл. АН ТаджССР, 12, № 11, 12
(1969).
165. А. В. Горохов, А. И. Максимов,
В. Д. Сизов, Л. С. Степанова Ж.
гехн. физ., 42, 2176 (1972).
166. Д. И. Словецкий, Р. Д. Тодесайте.
Химия высоких энергий, 8, 135 (1974).
167. Ю. А. Иванов, Л. С. Полак, Д. И. Сло-
вецкий. Химия высоких энергий, 5,
382 (1971).
168. Е. С. Гасилевич, В. А. Иванов,
Э. Н. Лоткова, В. Н. Очкин, Н. Н. Со-
болев, И. Г. Ярославский. Ж. Гехн.
физ., 39, 126 (1969).
169. К. К. Corvin, S. J. В. Corrigan. J.
Chem. Phys., 50, 2570 (1969).
170. Л. С. Полак, Д. И. Словецкий,
А. С. Соколов. V Всесоюзная конфе-
ренция по физике электронных и
атомных столкновений. Ужгород,
1972, сгр. 186.
171. 10. А. Иванов, П. А. Сергеев, Л. С. По-
лак, Д. И. Словецкий, А. С. Соколов,
Б. Б. Шварцман. Труды IV Всесоюз-
ной конференции по физике и генера-
торам низкотемпературной плазмы.
Алма-Ата, 1970, сгр. 18.
172. Е. Р. Мамиконян, Л. С. Полак,
Д. И. Слов цкий. Химия высоких
энергий, 6, 483 (1972).
173. А. А. Микаберидзе, В. Н. Очкин,
Н. Н. Соболев. В сб.: Плазмохимия-
71. М., ИНХС АН СССР, 1971, стр.
112.
174. В. Н. Очкин, Н. А. Шубина. В сб.:
Плазмохимия-71.М., ИНХС АН СССР,
1971, сгр. 114.
175. Д. И. Словецкий, Р. Д. Тодесайте.
Химия высоких энергии, 7, 291 (1973).
176. Н. М. Кузнецов. Теорет. и эксп. хи-
мия, 7, 22 (1971).
177. D. L. S. Me Elwain, Н. О. Pritchard.
. Canad. J. Chem., 49, 3915 (1971).
17&. Б. Ф. Гордиец, А. И. Осипов, Л. А.
Шелепин. ЖЭТФ, 61, 562 (1971).
179. И. Д. Артамонова, В. Т. Платоненко,
Р. В. Хохлов. ЖЭТФ, 58, 2195 (1970).
180. В. С. Летохов, А. А. Макаров.
ЖЭТФ, 63, 2064 (1972).
181. Н. Г. Басов, Э. М. Беленое, Е. П. Мар-
кин, А. Н. Ораевский, А. В. Панкра-
тов. ЖЭТФ, 64, 485 (1973).
182. Л. С. Полак, Д. И. Словацкий. Теп-
лофизика высоких темп., 12, 926
(1974).
183. W. Н. Venable fr., J. В. Shumaker
jr. J. Quant. Spectr. Radiat. Tranfer,
9, 1215 (1969).
184. L. S. Polak, D. I. Slovetsky, R. D.
Todesaite. Contributed Papers, 11-th
Internal. Conf on Ion Phenomena in
Gases, Prague, 1973, p. 43.
185. В. H. Колесников, E. Б. Куприянова,
H. H. Соболев. J. Quant. Spectr. Rad.
Transf., 8, 1851 (1968).
186. J. C. Keck. J. Chem. Phys., 32, 1035
(1960).
187. Ven-H. Schui, J. P. Appleton,
J. C. Keck. J. Chem. Phys., 53, 2547
(1970).
188. T. Carrington. J. Chem. Phys., 57,
2033 (1972).
189. R. W. Fair, B. A. Thrush. Trans. Fa-
raday. Soc., 65, 1208 (1969).
292
190. S. Ticktin, G. Spidler, Я. I. Skiff.
Discuss. Faraday. Soc., N 44, 212 (1967).
191. К. H. Becker, A. Elser, W. Groth,
D. Kley. T. Natarforsch., 26Л, 929
(1971).
192. A. Mandelman, T. Carrington,
R. A. Eoung. J. Chem. Phys., 58,
84 (1973).
193. К. H. Becker, E. H. Fink, W. Groth,
W. Jud, D. Kley. Discuss., Faraday
Soc., N 53, 35 (1972).
194. P. H. Carrol. J. Chem. Phys., 37,
805 (1962).
195. D. E. Shemansky, A. L. Broadfoot.
J. Ouant. Spectr. Radiat. Transfer.,
11, 1335 (1971).
196. M. J. Pilling, A. M. Bass, W. Braun.
J. Quant. Spectr. Radiat. Transfer,
11, 1593 (1971).
197. A. B. Callear, M. J. Pilling. Trans.
Faraday Soc., 66, 1618 (1970).
198. L. Flias. J. Chem. Phys., 42, 4311
(1965).
199. Термодинамические функции инди-
видуальных веществ, т. 1. Под ред.
В. И. Глушко. М., Изд-во АН СССР,
1962.
200. R. W. Gross. N. Cohen. J. Chem. Phys.,
48, 2582 (1968).
201. M. A. Clyne, B. A. Trush. Proc. Roy.
Soc. London, A269, 404 (1962).
202. B. F. Meyers, E. R. Bartie. 3. Chem.
Phys., 47, 1783 (1967).
203. В. H. Mahan, R. B. Solo. J. Chem.
Phys., 37, 2669 (1962).
204. R. A. Hartunian, W. P. Thompson,
E. W. Hewitt. J. Chem. Phys., 44,
1765 (1966).
ш205. E, И. Интезарова, В. H. Кондратьев.
Кинетика и катализ, 9, 477 (1968).
206. R. N. Dixon. Proc. Roy. Soc. London,
A275, 431 (1963).
207. В. A. Thompson. J. Geophys. Res.,
68, 6431 (1963).
208. H. M. Brongersma, A. J. H. Boerboom,
J. Kistemaker. Physica, 444, 49 (1969).
209. T. A. Brabbs, F. E. Belles. 11-th Sym-
posium (Internat.) on Combustion. Com-
bustion Inst.. Pittsburg, 1967, p. 125.
210. В. В. Азатян, H. E. Андреева,
E. И. Интезарова, В. H. Кондратов?.
Кинетика и катализ, И, 290 (1970).
211. Т. G. Slunger. J. Chem. Phys., 57,
233 (1972).
212. N. Jonathan, R. Petty. J. Chem. Phys.,
50, 3804 (1969).
213. Г. П. Бройда. Стабилизация свобод-
ных радикалов. Пер. с англ. М.,
ИЛ, 1963.
214. A. N. Wright, С. A. Winkler. Active
Nitrogen. N. Y., 1968.
215. Д. И. Словецкий, P. Д. Тодесайте.
Химия высоких энергий, 7, 297 (1973).
216. Ю. М. Гершензон, В. И. Егоров,
В. Б. Розенштейн. Химия высоких
энергий, 7, 77 (1973).
217. V. J. Egorov, Ju. М. Gerschenson,
V. В. R&senshtein, S. Ja. Umansky.
Chem. Phys. Letters, 20, 77 (1973).
218. R. Clampitt. 10-th Internat. Conf, on
Phenomena Ionized Gases. Oxford,
1971, p. 231.
219. B. Brocklehurst, F. A. Downing. J.
Chem. Phys., 46, 2976 (1967).
220. W. H. Kasner. Phys. Rev., 167, 148
(1968).
221. W. H. Kasner. Phys. Rev., 164,
194 (1967).
222. O. S. Weller, M. A. Biondi. Phys.
Rev., 172, 198 (1968).
223. R. C. Gunton, T. M. Shaw. Phys.
Rev., 140, 44 (1965).
224. W. H. Kasner, M. A. Biondi. Phys.
Rev., 1374, 317 (1965); 174, 139 (1968).
225. M. H. Mentzoni. J. Appl. Phys., 36,
57 (1965).
226. M. T. Leu, M. A. Biondi, R. Johnsen.
Phys. Rev., A8, 413, 420 (1973).
227. M. B. Mahdavi, J. B. Hasted,
M. M. Nakshbandi. J. Phys. B., 4,
1726 (1971).
228. L. Frommhold, M. A. Biondi, F. J.
Mehr. Phys. Rev., A140, 778 (1965).
229. J. Philbrick, F. J. Mehr, M. A. Bion-
di. Phys. Rev., 181, 271 (1969).
230. F. J. Mehr, M. A. Biondi. Phys.
Rev., 181, 264 (1969).
231. F. J. Mehr, M. A. Biondi. Phys. Rev.,
185, 98 (1969).
232. C. L. Chen, С. C. Leiby, L. Goldstein.
Phys. Rev., 121, 1391 (1961).
233. A. W. Johnson, J. B. Gerardo. Phys.
Rev., A5, 1410 (1972); A7, 807, 1339
(1973).
234. A. J. Cunningham, R. M. Hobson.
Phys. Rev., 185, 98 (1969).
235. R. J. Chantry. In Atomic Collision
Processes. Amsterdam, 1964, p. 565.
236. H. J. Oskam, V. P. Mittelstadt. Phys.
Rev., 132, 1445 (1963).
237. К. B. Persson, S. C. Brown. Phys.
Rev., 100, 729 (1955).
238. M. H. Mentzoni, J. Donohoe. Phys.
Letters, 26A, 330 (1968).
239. W. H. Kasner, W. A. Rogers, M. A.
Biondi. Phys. Rev. Letters, 7, 321
(1961).
240. R. Hackam. Planet. Space Sci., 13,
667 (1965).
241. J. J. Gibbons. Abstr. DASA Reaction
Rate Conf. Bould., Colorado, 1961.
242. C. S. Weller, M. A. Biondi. Phys.
Rev. Letters, 19, 59 (1967).
243. M. A. Biondi. Phys. Rev., 90, 730
(1953).
244. С. B. Kretschmer, H. L. Peterson.
J. Appl. Phys., 34, 3209 (1963).
245. L. P. Harris. J. Appl. Phys., 36, 1543
(1965).
246. J. M. Hammer, J. J. Thomas, В. B.
Aubrey. Bull. Amer. Phys. Soc., 9,
184 (1964).
247. M. T. Leu, M. A. Biondi, R. Johnson.
Phys. Rev., 7A, 292 (1973).
248. A. J. Cunningham, R. M. Hobson.
J. Phys., B, 5, 1773 (1972).
249. Данн, Лорди. Ракетная техника и
космонавтика, 8, № 4, 183 (1970);
№ 4, 9 (1970).
250. К. Н. Ульянов. Ж. техн, физики, 40,
790 (1970).
251. К. Р. Novikova. Proc. 10-th Internat.
Conf, on Ionized Phenom. Gases.
Oxford, 1971, p. 9.
293
252. S. C. Lin, J. D. Tear. Phys. Fluids,
6, 355 (1963).
253. R. P. Stein, M. Scheibe, M. W. Syver-
son, T. M. Shaw, R. C. Gunton. Phys.
Fluids, 7, 1641 (1964).
254. M. Б. Железняк, A. X. Мнацаканян.
Теплофизика высоких темп., 6, 390
(1968).
255. L. Frommhold, M. A. Biondi. Phys.
Rev., 185, 244 (1969).
256. Г. H. Герасимов, Г. M. Старцев.
V Всесоюзная конференция по физике
электронных и атомных столкнове-
ний. Ужгород, 1972, стр. 182.
257. Е. С. Zipf. Bull. Amer. Phys. Soc.,
15, 418 (1970).
258. R. F. Huffman, D. H. Katayama.
J. Chem. Phys., 45, 138 (1966).
259. A. H. Ключарев, И. С. Рязанов. V
Всесоюзная конференция по физике
электронных и атомных соударений.
Ужгород, 1972, стр. 137.
260. W. Peterson, И. R. Steiger. J. Geophys.
Rev., 71, 2267 (1966).
261. Н. F. Wellenstein, W. W. Robertson.
J. Chem. Phys., 56, 1072 (1972).
262. T. Wilson. Phys. Fluids, 9, 1913 (1966).
263. С. А. Лосев, Г. Д. Смехов. Теплофи-
зика высоких темп., 6, 281 (1968);
7, 1015 (1969).
264. Д. Бейтс, А. Далгарно. В сб.: Атом-
ные и молекулярные процессы. Пер.
с англ. М., «Мир», 1964, стр. 238.
265. J. К. Bardsley, М. A. Biondi. Advanc.
Atomic and Mol. Phys., 6, 1 (1970).
266. Л. С. Полак, Д. И. Словецкий. В сб.:
Экспериментальные и теоретические
исследования неравновесных физико-
химических процессов. М., ИНХС
АН СССР, 1974, стр. 100.
267. Н. Beutler, Н. О. Junger. Z. Phys.,
100, 80 (1936).
268. Т. F. O'Malley. Phys. Rev., 150,
14(1966); 162, 98 (1967); 185, L01 (1969).
269. W. R. Jarmain, R. W. Nicholls.
Proc. Phys. Soc., 84, 417 (1964).
270. C. S. Warke. Phys. Rev., 144, 120
(1966).
271. F. T. Chan. J. Chem. Phys., 49, 2533
(1968).
272. J. N. Bardsley. Phys. Rev., A2,
1359 (1970).
273. Y. Tanaka, K. Yoshino. J. Chem. Phys.,
57, 2964 (1972).
274. J. Berkowitz, W. A. Chupka. J. Chem.
Phys., 51, 2341 (1972).
275. Л. M. Биберман. В. С. Воробьев,
И. Т. Якубов. Усп. физ. наук, 107,
393 (1970).
276. D. R. Bates, A. Kingston. Proc. Roy.
Soc. London, Л279, 1376 (1964).
277. Л. С. Полак, Д. И. Словецкий. Химия
высоких энергий, 8, 135 (1974).
278. L. S. Polak, D. I. Slovetsky. Contri-
buted Papers, 11-th Tnternat. Conf, on
Ionized Gases. Prague, 1973, p. 44.
279. В. H. Колесников. Труды ФИ АН
СССР, 30, 66 (1964).
280. В. С. Воробьев. ЖЭТФ, 51, 327 (1966).
281. F. С. Fehsenfeld, A. L. Schmeltekopf,
Е. Е. Fergusson. J. Chem. Phys.,
46. 2802 (1967).
282. Л. С. Полак, Д. И. Словецкий. VI
Всесоюзная конференция по генера-
торам низкотемпературной плазмы.
Тезисы докладов. Фрунзе, изд-во
«Илим», 1974, стр. 407.
283. W. Frie, Н. Ма cker. Z. Phys., 162
69 (1961).
284. G. L. Rogoff. Contributed Papers,
11-th Internat. Conf, on Phenomena
Ionized Gases, Prague, 1973, p. 206.
К главе 4
1. Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. Под ред. JI. С. Полака. М.,
«Наука», 1965.
2. Очерки физики и химии низкотемпе-
ратурной плазмы. Под ред. Л. С. По-
лака. М., «Наука», 1971.
3. Плазмохимия-71. М., ИНХС АН
СССР, 1971.
4. А. А. Овсянников, Л. С. Полак,
М. А. Куманцев. Химические реак-
ции в условиях турбулентности. Пре-
принт ИНХС АН СССР, 1972, № 16.
5. Progress in Reaction Kinetics, v. 4.
G. Porter (Ed.). Oxford, Pergamon,
1967.
6. И. О. Хинце. Турбулентность. M.,
Физматгиз, 1963.
7. А. А. Таунсенд. Структура турбулент-
ного потока с поперечным сдвигом.
М., ИЛ, 1959.
8. Дж. Бэтчелор. Теория однородной
турбулентности. М., ИЛ, 1955.
9. А. С. Монин, А. М. Пелом. Статис-
тическая гидромеханика, т. I, II.
М., «Наука», 1965.
10. A. Corrsin. Phys. Fluids, 1, 42 (1958).
11. A. Corrsin. .1. Fluid Meeh., 11, 407
(1961).
12. H. L. Toor. A. I. Ch. E. Journal, 8r
70 (1962).
13. S. Corrsin. Phys. Fluids, 7, 1156 (1964).
14. E. E. О Brien. Phys. Fluids, 9, 215
(1966).
15. E. E. О Brien. Phys. Fluids, 9, 1561
(1966).
16. J. Lee. Phys. Fluids, 9, 1753 (1966).
17. E. E. O'Brien. Phys. Fluids, 11, 1883
(1968).
18. E. E. O'Brien. Phys. Fluids, 11, 2087
(1968).
19. E. E. О Brien. Phys. Fluids, 12, 1999-
(1969).
20. E. E. O'Brien, R. M. Eng. Phys.
Fluids, 13, 1393 (1970).
21. J. C. Hill. Phys. Fluids, 13, 1395
(1970).
22. E. E. O'Brien. Phys. Fluids, 14,
1804, 1326 (1971).
23. E. E. O'Brien, С. H. Lin. Phys.
Fluids, 15, 931 (1972).
24. J. J. Riley. Phvs. Fluids, 16, 1160е
(1973).
25. D. Kristmanson, P. V. Danckwerts.
Chem. Engng. Sci., 16, 267 (1961).
26. F. J. W. Roughton, B. Chance. In:
Technique of Organic Chemistry. S.
L. Friess a. o. (Ed.),v. 7, pt 2. N. Y.r
Interscience, 1963, p. 703.
294
27. R. N. Keeler, E. E. Petersen, J. M.
Prausnitz. A. I. Ch. E. Journal, 11,
221 (1965).
28. G. Vassilatos, H. L. Toor. A. I. Ch. E.
Journal, 11, 666 (1965).
29. H. Kramers, jr. Internal. Ser. Monogr.
Chem. Engng, 1, 45 (1967).
30. R. L. Curl. A. I. Ch. E. Journal, 9,
175 (1963).
31. L. A. Spielman, O. Leveuspiel. Chem.
Engng Sci., 20, 247 (1965).
32. A. Kattan, R. J. Adler. A. I. Ch. E.
Journal, 13, 580 (1967).
33. 1. J. Harris, R. D. Srivastava. Canad.
J. Chem. Engng, 46, 66 (1968).
34. D. P. Rao, 7. /. Dunn. Chem. Engng
Sci., 25, 1275 (1970).
35. Бай-Ши-И. Теория струй. M., Физ-
матгиз, 1960.
36. Горение в турбулентном потоке. М.,
сборник. Изд-во АН СССР, 1959.
37. Д. Сполдинг. Основы теории горения.
М., Госэнергоиздат, 1959.
38. Б. В. Канторович. В сб.: Тепло- и
массоперенос, т. 1. Минск, Изд-во
АН БССР, 1962, сгр. 243.
39. Б. В. Канторович, Г. Н. Делягин.
Internal. J. Heat Mass Transfer, 5,
И (1962).
40. E. С, Щетинное. Физика горения га-
зов. М., «Наука», 1965.
41. К. W. Мао. Н. L. Toor. A. I. Ch. Е.
Journal, 16, 49 (1970,.
42. Р. W. Coldrey, R. D. Radford, 1. J.
Harris. A. I. Ch. E. Journal., 17,
1253 (1971).
43. A. H. Колмогоров. Докл. АН СССР,
30, 301 (1941).
44. А. Н. Колмогоров. Докл. АН СССР,
30, 453 (1941).
45. А. Н. Колмогоров, А. М. Обухов. Изв.
АН СССР, серия геогр. и геофиз.,
13, 58 (1949).
46. A. Corrsin.J. Appl. Phys., 22,469 (1961).
47. A. A. Kolmogorov. J. Fluid Meeh.,
13, 82 (1962).
48. A. M. Obukhov. J. Fluid Meeh., 13,
77 (1962).
49. S. C. Lin. Ракетная техника и кос-
монавтика, № 2, 16 (1966).
50. S. С. Lin. Ракетная техника и кос-
монавтика, № 2, 24 (1966).
51. G. W. Sutton. Ракетная техника и
космонавтика, № 12, 239 (1968).
52. G. W. Sutton, М. Сатае. Ракетная
техника и космонавтика, № 12, 199
(1968).
53. Р. М. Chung. Ракетная техника и кос-
монавтика, № 10, 165 (1969).
54. Р. М. Chung. Phys. Fluids, 13, 1153
(1970).
55. I. Prigogine. Non-equilibrium Sta-
tistical Mechanics. N. Y., Interscien-
ce, 1962.
56. P. M. Chung. Phys. Fluids, 15, 1735.
(1972).
57. P. M. Chung. Phys. Fluids, 16, 980
(1973).
58. П. П. Корявое. Ж. вычисл. мат. и
мат. физики, 1, № 5 (1961).
59. В. А. Голубев. В сб.: Исследование
турбулентных струй воздуха, плазмы
и реального газа. Под ред. Г. Н. Аб-
рамовича. М., 1967.
60. J. Grey, Р. F. Jacobs. Ракетная тех-
ника и космонавтика, № 3, 25
(1964).
61. F. Incroper a, G. Leppert. Ракетная
техника и космонавтика, № 6, 164
(1966).
62. J. Grey, М. Р. Sherman, Р. М. Wil-
liams, D. В. Fradkin. Ракетная тех-
ника и космонавтика, № 6, 364 (1966).
63. Т. J. O'Connor, Е. Н. Comfort,
L. A. Cass. Ракетная техника и кос-
монавтика, № 11, 181 (1966).
64. В. Н. Троицкий, А. И. Смородин,
С. Н. Шорин. В сб.: Явления перено-
са в низкотемпературной плазме.
Минск, «Наука и техника», 1969,
стр. 149.
65. Я. С. Суров. Теплофизика высоких
темп., 7, 304 (1969).
66. Г. Н. Абрамович. Теория турбулент-
ных струй. М., Физматгиз, 1960.
67. Л. А. Вулис, В. II. Кашкаров. Тео-
рия струй вязкой жидкости. М.,
«Наука», 1965.
68. Г. Шлихтинг. Теория пограничного
слоя. М., ИЛ, 1956.
69. Е. П. Марцевой, И. Н. Карп,
О. В. Каминская, К. А. Зубкова.
В сб.: Физика, техника и применение
низкотемпературной плазмы. Алма-
Ата, 1970, стр. 511; в сб.: Плазмо-
химия-71. М., ИНХС АН СССР,
стр. 184.
70. D. L. Smith, R. Н. Kadlec, S. W.
Churchill. A. I. Ch. Е. Journal, 17,
482 (1971).
71. J. Grey, P. F. Jacobs. Приборы научи,
исслед., № 7, 29 (1962).
72. M. Ф. Жуков, Ю. И. Сухинин,
Н. И. Воробьева. В сб.: Физика, тех-
ника и применение низкотемператур-
ной плазмы. Алма-Ата, 1970, стр. 518.
73. Г. И. Морцева, Б. А. Поздняков,
В. Я. Смоляков, В. К. Сторожук,
В. И. Ядроз. Там же, стр. 531.
74. А. А. Овсянников, Л. С. Полак,
М. Б. Сафонова. Там же, стр. 507.
75. А. А. Овсянников, Л. С. Полак,
М. С. Федорова. Там же, стр. 543.
76. S. Corr sin. Ракетная техника и кос-
монавтика, № 9, 229 (1968).
77. К. А. Зубкова, Г. Г. Васюкова,
О. В. Каминская, Е. П. Марцевой.
Хим. технол., № 6, 16 (1972).
78. А. М. Цирлин. Теорет. основы хим.
технол., 1, 522 (1967).
79. Э. К. Добринский, В. А. Любимов,
В. И. Сидоров, Б. А. Урюков,
В. В. Шумский, А. Э. Фридберг. В
сб.: Физика, техника и применение
низкотемпературной плазмы. Алма-
Ата, 1970, стр. 504.
80. А. А. Овсянников, Л. С. Полак,
М. Б. Сафонова, М. С. Федорова.
Теорет. основы хим. технол., 5, 891
(1971).
81. Б. В. Канторович, Б. А. Байрашев-
ский. В сб.: Тепло- и массоперенос,
т. 10. Минск, Изд во АН БССР,
1962, стр. 94.
295
82. M. A. Patrick. Trans. Inst. Chem.
Engr, 45 (1), T16—T31 (1967).
83. Ю. В. Иванов. Основы расчета и
проектирования газовых горелок. М.,
Гостоптехиздат, 1963.
84. 10. В. Иванов, В. А. Гендриксон.
В сб.: Тепло- и массоперенос,
т. 1. М., «Энергия», 1968, стр. 394.
85. В. Ф. Сивиркин. Автореф. канд. дисс.
Куйбышев, 1965.
86. В. Д. Пархоменко, И. И. Сорока,
С. И. Ганз. Теорет. основы хим.
технол., 8, 60 (1974).
87. В. Д. Пархоменко, П. И. Сорока,
С. Н. Ганз. Теорет. основы хим.
технол., 8, 307 (1974).
88. М. А. Куманцев, А. А. Овсянников,
Л. С. Полак, М. Б. Сазонова. В сб.:
Плазмохимия-71. М., ИНХС АН СССР,
1971, стр. 186.
89. М. А. Куманцев, А. А. Овсянников,
Л. С. Полак. Теорет. основы хим.
технол., 8, 872 (1974).
90. Р. R. Ammann, R. S. Timmins.
A. I. Ch. Е. Journal, 12, 956 (1966).
91. О. Левеншпилъ. Инженерное оформле-
ние химических процессов. М., «Хи-
мия», 1969.
92. В. В. Кафаров. Методы кибернетики
в химии и химической технологии.
М., «Химия», 1971.
93. Г. Н. Абаев. В сб.: Моделирование
химических процессов и реакторов,
т. 1. Новосибирск, «Наука», 1971,
стр. 65.
94. Г. В. Гуляев. Л. С. Полак. В сб.:
Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. М., «Наука», 1965, С'р. 72.
95. ТГ. Г. Денбиг. Теория химических
реакторов. М., «Наука», 1968.
96. R. С. L. Bosworth. Transport Proces-
ses in Applied Chemistry. N. Y.,
Wiley, 1956.
97. Я. M. Брайнес. Введение в теорию и
расчеты химических и нефтехимиче-
ских реакторов. М., «Химия», 1968.
98. X. Крамере, К. Вестертерп. Хими-
ческие реакторы, расчет и управление
ими. М., «Химия», 1967.
99. А. М. Брайнес. Подобие и моделиро-
вание в химической и нефтехимической
технологии. М., Гостоптехиздат, 1961.
100. М. Г. Слинъко. Моделирование хими-
ческих реакторов. Новосибирск, «Нау-
ка», 1968.
101. А. И. Бояринов, В. В. Кафаров. Ме-
тоды оптимизации в химической тех-
нологии. М., «Химия», 1969.
102. D. М. Himmelblau, К. В. Bischoff.
Process Analysis and Simulation.
N. Y.— London — Sydney, Wiley, 1968.
К главе 5
1. Л. С. Полак. В сб.: Очерки физики
и химии низкотемпературной плаз-
мы. Под ред. Л. С. Полака. М., «Нау-
ка», 1971; In: Reactions under Plasma
Conditions, v. 2. M. Venugopalan (Ed.)
Interscience. 1971, chap. XII; В сб.:
Плазмохимия-71. M., ИНХС АН СССР,
1971, стр. 1; Основные положения обоб-
щенной неравновесной химической
кинетики, Препринт ИНХС АН СССР,
1972, № 1.
2. Ф. Б. Вурзелъ, Л. С. Полак. Журнал
ВХО им. Д. И. Менделеева, № 3,
320 (1969); Industr. and Engng Chem.,
12. N 6, 8 (1970); In: Reactions under
Plasma Conditions, v. 2. M. Venugo-
palan (Ed.)., Interscience, 1971,
p. 299; Пластические массы, № 4, 68
(1970); Химическая технология, № 6,
9 (1972); в сб.: Плазменные процессы
в металлургии и технологии неорга-
нических материалов. М., «Наука»,
1973, стр. 197; Ф. Б. Вурзелъ. В сб.:
Очерки физики и химии низкотемпе-
ратурной плазмы. Под ред. Л. С. По-
лака. М., «Наука», 1971, стр. 411.
3. IO. А. Колбановский, Л. С. Полак.
Основные положения химической ки-
нетики. Препринт ИНХС АН СССР,
1971, № 2.
4. R. Е. Duff, S. Н. Bauer. J. Chem.
Phys., 36, 1754 (1962).
5. Г. Б. Синярев. Некоторые вопросы
механики. М., Оборонгиз, 1962.
6. Я. М. Буждан, Н. М. Акимутин.
Изв. СО АН СССР, серия хим. наук,
3, № И, 61 (1963).
7. Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. Сборник. Под ред. Л. С. По-
лака. М., «Наука», 1965.
8. Применение вычислительной матема-
тики в физической и химической ки-
нетике. Сборник. Под ред. Л. С. По-
лака. М., «Наука», 1969.
9. Е. П. Марцевой. Теорет. основы хим.
технол., 3, № 4 (1969).
10. В. Д. Пархоменко, Л. С. Полак и др..
Химия высоких энергий, 8, №5,
(1974).
11. Г. В. Гуляев, Л. С. Полак. В сб.:
Кинетика и термодинамика хими-
ческих реакций в низкотемператур-
ной плазме. Под ред. Л. С. Полака..
М., «Наука», 1965, стр. 72.
12. Л. С. Полак, В. С * Щипачев. Там же%
стр. 151.
13. J. F. Skrivan, W. Jaskowski. Industr.
and Engng Chem., 4, 371 (1963).
14. Д. Б. Сполдинг. В сб.: Современные
проблемы теплообмена. М., «Энергия»,
1966.
15. А. Л. Сурис. В сб.: Плазмохимия-71.
М., ИНХС АН СССР, 1971, стр. 171.
Химия высоких энергий, 7, 309 (1973).
16. С. И. Шорин. Теплопередача. М.,
«Высшая школа», 1964.
17. М. Bauerns, F. Fetting. Chem. Engng
Sci., 20, 273 (1964).
18. И. M. Андрианова, С. Н. Шорин*
В сб.: Плазмохимия-71. М., ИНХС
АН СССР, 1971, стр. 169.
19. А. А. Овсянников, Л. С. Полак,
М. А. Куманцев. Химические реак-
ции в условиях турбулентности. Преп-
ринт ИНХС АН СССР, 1972, № 16.
20. Плазмохимия-71. М., ИНХС АН СССР,
1971.
21. Ф. Б. Вурзелъ, Г. В. Лысов, Л. С. По-
лак, Ю. Л. Хаит, Э. Н. Червочкин,
296
Химия высоких энергий, 5, 105
(1971).
22. С. А. Панфилов, Ю. В. Цветков.
Теплофизика высоких темп., 5, 294
(1967); в сб.: Металлургия цветных
и редких металлов. М., «Наука»,
1967, стр. 128.
23. Н. С. Суров, Л. С. Полак. Физика и
химия обработки материалов, № 2, 19
(1969).
24. G. R. Kubanek, W. П. Gauvin, Р. Che-
valier. Canad. J. Chem. Engng, 46,
101 (1968).
25. G. R. Kubanek, W. H. Gauvin. Chem.
Engng, 222, 332 (1968).
26. Ю. В. Цветков. Исследование термо-
динамики и кинетики восстановления
некоторых окислов цветных ме аллов
в различных агрегатных состояниях.
Автореф. докт. дисс. М., ИМЕТ им.
А. А. Байкова, 1968.
27. Г. В. Самсонов. В сб.: Плазменные
процессы в металлургии и техноло-
гии неорганических материалов. М.,
«Наука», 1973, стр. 214.
28. Ю. В. Цветков, Д. М. Чижиков,
С. С. Дейнека, И. К. Тагиров,
И. Я. Басиева. В сб.: Порошковая
металлургия в новой технике. М.,«На-
ука», 1968, стр. 143.
29. Дж. Кларк. В сб.: Использование
плазмы в химических процессах. М.,
«Мир», 1970, Стр. 152.
20. А. Л. С] рис, С. И. Шорин. Химия
высоких энергий, 3, № 2 (1969).
21. Л. Е. Слынько, Ф. Б. Вурзелъ,
Ю. В. Валибеков, Б. Е. Гутман.
Докл. АН ТаджССР, № 12 (1973).
22. Ф. Б. Вурзелъ, Л. С. Полак. В сб.:
Кинетика и термодинамика химиче-
ских реакций в низкотемпературной
плазме. М., «Наука», 1965, сгр. 101.
23. О. С. Нурсултанов, Л. С. Полак,
В. Т. Попов. Химия высоких энергий,
8, № 5 (1974).
24. Н. Л. Володин, Ф. Б. Вурзелъ.
Л. С. Полак, П. И. Эндюсъкин.
В сб.: Некоторые вопросы н)фтехими-
ческого синтеза. Уфа, 1970, стр. 11;
Физика, техника и применение низ-
котемпературной плазмы. Сборник.
Алма-Ата, 1970, стр. 576.
25. А. Д. Степухович. Кинетика и меха-
низм термического крекинга алканов.
Изд-во Саратов, ун-та, 1965.
26. Ю. В. Валибеков, Н. Л. Володин,
Ф. Б. Вурзелъ, Б. Е. Гутман,
Л. С. Полак, П. Н. Эндюсъкин,
И. Л. Эпштейн. Химия высоких
энергий, 9, № 1 (1975).
27. L. S. Kassel. J. Amer. Chem. Soc.,
54, 3949 (1932).
28. А. Л. Сурис, С. H. Шорин. Тезисы
доклада на Всесоюзной конференции
по проблеме «Получение ацетилена
пиролизом природного газа». Севе-
родонецк, 1968.
29. С. Ш. Гершуни, А. Л. Сурис, С. Н. Шо-
рин, Н. В. Алексеев. Химия высоких
энергий, 9, № 1 (1975).
40. Ф. Б. Вурзелъ, Л. С. Полак, И. Л. Эп-
штейн. Химия высоких энергий, 9,
№ 2 (1975).
41. Ф. Б. Вурзелъ, Ю. В. Валибеков,
Б. Е. Гутман, Л. С. Полак. Химия
высоких энергий, 7, № 3 (1973).
42. Н. Л. Володин, Ф. Б. Вурзелъ,
В. Т. Дятлов, Л. С. Полак, Ю. И.
Шмыков, П. И. Эндюсъкин. Химия
высоких энергий, 5, 3, (1971).
43. Ю. Н. Кобзев, Г. И. Худяков. Элек-
тронная обработка материалов, 19,
22 (1961); в сб.: Плазмохимия-71.
М., ИНХС АН СССР, 1971, стр. 155.
44. Г. И. Козлов, Г. Н. Худяков,
IO. Н. Кобзев. Нефтехимия, 7, 224
(1967).
45. Н. Gladisch. Hydroc. Proc. Pet. Ref.,
41, N 6, 159 (1952); Chimie et In-
dustrie, 88, 471 (1962); Chem.-Ingr.-
Techn., N 4, 204 (1969).
46. K. Senneivald, E. Schallus, F. Pohl.
Chem.-Ingr.-Techn., 35, 1 (1963).
47. K. Senneivald. Patent BRD, N 1468159,
5.8. 1964; N 1468161, 11.8.1964.
48. Д. T. Ильин, E. H. Еремин. Ж.
физ. химии, 36, 1560, 2222 (1962);
Вести. МГУ, 6, 41 (1962); Ж. прикл.
химии, 38, 12 (1965); в сб.: Низко-
температурная плазма. М., «Мир»,
1967, стр. 576; в сб.: Современные
проблемы физической химии, т. 2. М.,
Изд-во МГУ, 1968, стр. 61.
49. Л. С. Полак,. 8-й Мировой нефтяной
конгресс. Дискуссионный симпозиум
18. «Производство олефинов и ацети-
лена как сырья для химической про-
мышленности». М., 1971, стр. 40.
50. К, Герман, X. Шмидт. Там же,
стр. 60.
51. А. Л. Моссэ, В. К. Забродин,
И. А. Крылова. В сб.: Физика, техника
и применение низкотемпературной
плазмы. Алма-Ата, 1970, стр. 578;
Инж.-физ. ж.,20,3 (1971); Химия высо-
ких энергий, 7, 491 (1973).
52. Chem. Week, 94, N 3, 65 (1964).
53. D. A. Maniero, P. F. Kienast, C. Hi-
rayima. Westinghouse Eng., 26, N 3,
66 (1966).
54. M. Mitscuio, K. Yasio. J. Chem. Soc.
Japan, Industr. Chem., 71. N 4, 492
(1968); 3, N 2, 306 (1970).
55. Л. С. Полак, Ю. В. Валибеков
Г. В. Гуляев, Г. М. Болотов. Изв.
АН ТаджССР, 1(31), 43 (1969).
56. Л. С. Полак, П. Н. Эндюсъкин,
В. Н. Углев, Н. Л. Володин. Химия
высоких энергий, 3, 2 (1969).
57. Sh. Gomi. Petrol Refiner, 43, N 11,
165 (1964).
58. Gr. Brit. Patent, N 1068793, 30.3.1965.
59. TV. L. Volodin, L. S. Polak, P. N. An-
diuskin, R. I. Levenson, S. M. Krugly,
I7. r.Dijtlov. Brit. Patent, N 1316668,
3.06. 1969.
60. H. Л. Володин, Ф. Б. Вурзелъ,
Л. С. Полак, П. Н. Эндюсъкин.
В сб.: Плазмохимия-71. М., ИНХС
АН СССР, 1971, стр. 157.
61. А. Л. Рабкина, П. А. Борисов,
В. Т. Пономарева. Хим. промышлен-
ность, № 1, 35 (1971); Экономика,
297
организация и управление в нефте-
перерабатывающей и нефтехимической
промышленности, № 2, 1 (1971); в сб.:
Плазмохимия-71. М., ИНХС АН СССР,
1971, стр. 44.
62. Chem. Engng, 71, N 3, 29 (1964).
63. С. S. Stokes. In: Reactions under
Plasma Conditions, v. 2. M., Venugo-
palan (Ed.). N. Y.— London — Syd-
ney — Toronto, J. Wiley, 1971, p. 259.
64. H. Suhr. Fortschr. chem. Forsch.,
36, 39 (1973).
65. А. Б. Illexmep. Химические реакции
в электрических разрядах. М.—Л.,
ОНТИ, 1935.
66. F. К. McTaggart. Plasma Chemistry
in Electrical Discharges. Amsterdam—
London — N. Y., Elsevier Publ. Co.,
1967.
67. Ф. Б. Вурзелъ. В сб.: Использование
плазмы в химических процессах. М.,
«Мир», 1970, гл. X.
68. J. Amouroux. Vide, 26, N 154, 164
(1971).
69. N. Friedman, II. II. Bovee, S. L. Miller.
J. Org. Chem., 36, N 19, 2894 (1971).
70. A. R. Westwood. Amer. Chem. Soc.
Polymer Preprints, 80, N 1, 433 (1969);
Europ. Polymer. J., 7, N 4, 363, 377
(1971).
71. А. Б. Гилъман, В. M. Колотыркин,
И. Н. Туницкий. Кинетика и катализ,
11, 1267 (1973).
72. A. N. Singh, I. S. Singh. lodian J.
Pure and Appl. Phys., 8, N 2, 119(1970).
73. К. А. Осипов, Г. Э. Фолманис. Осаж-
дение пленок из низкотемператур-
ной плазмы и ионных пучков. М.,
«Наука», 1973.
74. Б. В. Ткачук, В. М. Колотыркин,
Н. П. Сметанкина. Высокомолекул.
соединения, А12, 1458 (1970).
75. F. Denes, С. Ungurenasu, I. Haiduc.
Europ. Polymer. J., 6, N 8, 1155
(1970).
76. S. A. Acasov, M. A. Bogirov,
V. P. Malin. Kinetics and Meeh.
Polyreactions, 5, Preprints. Budapest,
1969, p. 317.
77. H. H. Рыкалин, Ю. В. Цветков,
В. А. Петруничев, И. К. Глушко.
В сб.: Физика, техника и применение
низкотемпературной плазмы. Алма-
Ата, 1970, стр. 602.
78. И. М. Артюхов, Э. С. Безмозгин,
X. Л. Винокуров, В. Л. Клименко,
Н. С. Пригожин. В сб.: Термоката-
литические методы переработки уг-
леводородного сырья. Л., «Химия»,
1969, стр. 101.
79. N. W. Ryan. Patent US, N 3420632,
18.11.1966.
80. Cabot. Corp., Brit. Patent, N 1202587,
10.11.1967.
81. D. Gardner. US Patent, N 2572851,
30.10.1951.
82. C. Sheer. US Patent, N 3009783,
21.11.1961.
83. П. Orbach. US Patent, V 3288696,
29.11.1966.
84. R. F. Baddour. US Patent, N 3333927,
1.8.1967.
85. B. Latham. US Patent, N 3344051,.
26.9.1967.
86. D. Bjomson. US Patent, N 34094U3.
5.11.1968.
87. Patent BRD, N 2015914, 4.4.1969.
88. П. А. Теснер, Б. H. Алыпшуллер.
Докл. АН СССР, 187, № 5, 1100
(1969); Газовая промышленность, №6,
4 (1969).
89. П. А. Теснер. Образование углерода
из углеводородов газовой фазы. М.,
«Химия», 1972.
90. Unone Kiyoshy. US Patent, N 3513014,.
1.3.1967.
91. P. Тиммине, P. Амман. В сб.: Ис-
пользо ание плазмы в химических
процессах, d., «Мир», 1970, стр. 116.
92. С. Н. Ганз, В. Д. Пархоменко,.
Ю. И. Краснокутский. В сб.: Генера-
торы низкотемпературной плазмы. М.,
«Энергия», 1969, стр, 529.
93. Л. С. Полак, В. Д. Пархоменко,
Б. И. Мельников, С. Н. Ганз. Химия
высоких энергий, 7, 303, 408 (1973).
94. Н. W. Leutner. Industr. and Engng
Chem. Process Design and Developm.
1, 166 (1962); 2, 315 (1963).
95. C. W. Marinov)ski, R. C. Phillips,.
N. K. Hiester. Industr. and Engng
Chem. Fundamentals, 1, 52 (1962).
96. С. H. Ганз, Ю. И. Краснокутский.
Тезисы докладов семинара «Приме-
нение низкотемпературной плазмы в
технологии неорганических веществ
и порошковой металлургии». Киев,
апрель 1968, стр. 18; Химическая тех-
нология, № 19. Харьк. гос. ун-т,
1971, стр. 18.
97. С. Н. Ганз, В.Д. Пархоменко, IO. И.
Краснокутский. Получение ацетиле-
на и цианистых соединений в плазме.
Киев. Укр. НИИНТИ, 1968; Хим.
промышленность, № 5, 356, 518 (1970).
98. Е. Schallus, A. Gotz. Patent BDR,
N 1012899, 1957; US Patent. N 2616534
1959; US Patent, N 25218, 1962.
99. К. K. Neuman. Erdol und Kohle, 17,
N 9, 708 (1964); US Patent, N 3114691,
1963.
100. C. Hiray irrru, D. A Maniero. US Pa-
tent, N 3460902, 2.8.1967.
101. T. Yasui, T. Akitani. Patent Japan,
N 02215/69, 4.2.1965.
102. H. Л. Володин, Ф. Б. Вурзелъ,
Л. С. Полак. П. И. Эндюсъкин. В
сб.: Некоторые вопросы нефтехими-
ческого синтеза. Уфа, НИИнефтехим,
1970, сгр. 24.
103. Н. Л. Володин, Ф. Б. Вурзелъ,
Л. С. Полак, П. Н. Эндюсъкин. В
сб.: Физика, техника и применение
низкотемпературной плазмы. Алма-
Ата, 1970, стр. 583.
104. Н. В. Алексеев, А. Л. Сурис, С. П. Шо-
рин. В сб.: Теория и практика сжига-
ния газа, вып. 5. Л., «Недра», 1972;
сб.: Низкотемпературная плазма в
технологии неорганических i еществ
Новосибирск, «Наука», 1971; сб.:
Плазмохимия-71. М., ИНХС АН СССР,
1971, стр. 149; сб.: Физика, техника
и применение низкотемпературной
298
плазмы. Алма-Ата, 1970, стр. 589,
592.
105. Ф. А. Бухман, В. Г. Меламед,
Л. С. Полак. В сб.: Применение вы-
числительной математики в физиче-
ской и химической кинетике. М.,
«Наука», 1969, стр. 39.
106. А. А. Овсянников, Л. С. Полак,
В. И. Юдин. Химия высоких энер-
гий, 2, 5 (1968).
107. А. А. Овсянников, Л. С. Полак. В
сб.: Кинетика и термодинамика хи-
мических реакций в низкотемператур-
ной плазме. Под ред. Л. С. Полака. М..
«Наука», 1965, стр. 118.
108. В. В. Михайлов, А. Б. Сулейменов.
В сб.: Плазменные процессы в ме-
таллургии и технологии неорганиче-
ских материалов. М., «Наука», 1973,
стр. 152.
109. О. С. Нурсултанов, Л. С. Полак,
В. Т. Попов. Химия высоких энергий,
8, № 4, 1974.
110. J. С. Burleson, ИС F. Jates. US Patent,
№ 3475308, 3.1.1967.
111. R. Weisbeek, D. Hullstrung. Chem.-
Ingr-Techn., 42, N 21, 1302, A1949,
A 1951 (1970).
112. Th. C. Ruppel, P. F. Mossbauer,
M. F. Ferrer, D. Beinstock. Industr.
and Engng Chem. Product. Res. and
Developm., 9, N 3, 369 (1970).
113. J. C. Burleson, W. F. Jates. US Pa-
tent, N 3497436, 3.1.1967.
114. Б. Бронфин. В сб.: Использование
плазмы в химических процессах. М.,
«Мир», 1970, стр. 182.
115. G. Bjornson. US Patent, N 3471546,
28.9.1966.
116. К. Бегин, Д. Изв’, Л. Салве.кини,
Д. Томпсон, Д. Вайскроц, Д. Мар-
грейв. В сб.: Использование плазмы
в химических процессах. М., «Мир»,
1970, стр. 49.
117. G. Bjornson. US Patent, N 3494974,
28.9.1966.
118. E. S. Lo, J. D. Roadio, S. W. Os-
born. J. Org. Chem., 38, N 5, 907
(1973).
119. Fixed Nitrogen H. A. Curtis (Ed.).
N. Y., Chemical Catalog Co., 1932,
chap. 4.
120. Получение технологического газа
(фиксация атмосферного азота). Сбор-
ник статей. М., ГИАП, 1968.
121. А. Я. Лыткин, Н. А. Зыричев,
В. Т. Пендраковский. В сб.: Физика,
техника и применение низкотемпера-
турной плазмы. Алма-Ата, 1970,
стр. 569.
122. А. Я. Лыткин, В. Т. Пендраковский,
В. Г. Варапаева. Азотная промышлен-
ность, № 7 (1969).
123. Г. В. Гуляев, Г. И. Козлов, Л. С.' По-
лак, Л. Н. Хитрин, Г. Н. Худяков,
В. С. Щипачев. В сб. Кинетика и тер-
модинамика химических реакций в
низкотемпературной плазме. Под ред.
Л. С. Полака. М., «Наука», 1965,
стр. 132.
124. Я. Б. Зельдович, П. Я. Садовников,
Д. А. Франк-Каменецкий. Окисление
азота при горении. М., Изд-во АН
СССР, 1947.
125. В. Д. Пархоменко, А. И Руденко,
С. Н. Ганз. Химия высоких энергий,
3, 274 (1969).
126. Е. Н. Еремин, А. Н. Мальцев. Ж.
физ. химии, 30, 1615 (1956).
127. Е. Н. Еремин, С. С. Васильев,
Н. И. Кобозев. Ж. физ. химии, 8,
814 (1936).
128. Л. М. Крыхтина, А. И. Мальцев,
Е. Н. Еремин. Ж. физ. химии, 40,
2784 (1966).
129. С. S. Stokes, J. J. Correa, L. A. Streng,
Н. W. Leutner. J. Amer. Inst. Chem.
Engr., 11, 370 (1965).
130. R. L. McCarthy. J. Chem. Phys., 22,
1360 (1954).
131. M. M. Богородская, E. H. Еиемин.
Ж. физ. химии, 38, 1849 (19б4).
132. Ф. Б. Вурзелъ, Г. Г. Гонтарев,
Г. В. Лысов, Л. С. Полак. III Все-
союзная конференция по физике низ-
котемпературной плазмы. Изд-во МГУ,
1971, стр. 279.
133. В. Ф. Бабалъянц, Ф. Б. Вурзелъ,
А. М. Губанов, Л. С. Полак. Оптика
и спектр., 32, 249 (1972).
134. Ф. Б. Вурзелъ. В сб.: Плазмохимия-71.
М., ИНХС АН СССР, 1971, стр. 88.
135. А. Л. Моссэ, В. В. Печковский. При-
менение низкотемпературной плазмы в
технологии неорганических веществ.
Минск, «Наука и техника», 1973.
136. Р. Н. Dundas, М. L. Thorpe. Chem.
Engng Progr., 66, N 10, 66 (1974).
137. H. В. Антипов, А. Б. Гугняк, С. H.
Дмитриев, И. Д. Кулагин, Я. М. Лип-
кес, А. Б. Логинов, С. В. Огурцов,
Н. Н. Рыкалин, Л. М. Сорокин,
Т. П. Сушко. Физика и химия обра-
ботки материалов, № 4 (1968).
138. Плазменные процессы в металлургии
и технологии неорганических мате-
риалов. Сборник. М., «Наука», 1973.
139. W, L. Wilson, F. Strain, Н. Н. Но-
ckje. US Patent, N 3443897, 7.4. 1965.
140. Д. M. Чижиков, С. С. Дейнека,
В. Н. Макарова. В сб.: Исследования
в области металлургии цветных и ред-
ких металлов. М., «Наука», 1969,
стр. 136.
141. Т. I. Barri, R. К. Bailiss, L. A. Lay.
J. Mater. Sci., 3 (3), 229, 239 (1968).
142. Ф. Б. Вурзелъ, Н. Н. Долгополов,
А. И. Максимов, Л. С. Полак,
В. И. Фридман. В сб.: Кинетика и
термодинамика химических реакций
в низкотемпературной плазме. Под
ред. Л. С. Полака. М., «Наука»,
1965, стр. 223.
143. С. А. Шевченко, С. М. Павлов. В
сб.: Низкотемпературная плазма в
технологии неорганических в ществ.
Новосибирск, «Наука», 1970, стр. 29.
144. Н. А. Синайский. Там же, сгр. 33.
145. П. Н. Эндюсъкин, Л. С. Полак,
Н. Л. Володин, В. Н. У глее. Химия
высоких энергий, 3, 202 (1969).
146. J. В. Margeloff. US Patent, N 3254958,
7.6.1966.
299
147. J. D. VanDrumpt. Industr. and Engng
Chem. Fundamentals, 11, N 4, 594
(1972).
148. P. Баддур, П. Дундас. В сб.: Ис-
пользование плазмы в химических
процессах. М., «Мир», 1970, стр.
103.
149. О. Е. Скадченко, В. П. Вендилло,
Ю. В. Филиппов. Вести. МГУ, № 5,
594 (1972).
150. Н. Sigimitsu. Bull. Chem. Soc. Japan,
43, 1927 (1970).
151. F. F. Lowther. Patent BRD, N 2026622,
4.6.1966; Patent France, N 2052492,
3.6,1970.
152. И. В. Никитин, В. Я. Росоловский.
Усп. химии, 34, 1161 (1970); 40, № 11,
1971.
153. Ю. В. Цветков, С. А. Панфилов. Плаз-
менные процессы в металлургии и
технологии неорганических материа-
лов. М., «Наука», 1973, стр. 207.
154. А. И. Краснов, В. Г. Зилъберберг,
С. Ю. Шаривкер. Низкотемператур-
ная плазма в металлургии. М., «Ме-
таллургия», 1970.
155. В. В. Крапухин, Э. А. Королев. Изв.
высш, учебн. завед. Цвет, мет., № 6
(1968).
156. Ф. Б. Вурзелъ, Л. С. Полак, В. С. Щи-
пачев. Химия высоких энергий, 1,
268 (1967).
157. F. К. McTaggart. Proc. Sympos.
Advances Extr. Melals, London, 1967,
Pub. 1968, p. 541: US Patent,
N 3533777, 2.1. 1965.
158. Brit. Patent Abstr., 1969, 10, N 8,
74.
159. Ю. А. Башкиров, С. А. Медведев.
Генераторы низкотемпературной плаз-
мы. М., «Энергия», 1969, стр. 501.
160. I. К. Ingraham, К. W. Downes,
Р. Marier. Canad. J. Chem., 35, 850
(1957).
161. К. Ishizuka. US Patent, N 2860094,
1958.
162. I. E. Newman, J. A. Watts. J. Amer.
Chem. Soc., 82, 2113 (1960).
163. Л. Ю. Марковский, В. И. Львова,
Ю. Д. Кондрашов. Труды конферен-
ции по химии бора и его соединений.
М., 1958, стр. 36.
164. И. Д. Кулагин, В. К. Любимов,
К. Г. Марин, Б. А. Сахаров,
Л. М. Сорокин. Физика и химия об-
работки материалов, № 2 (1967).
165. Б. А. Сахаров, К. Г. Марин, В. К. Лю-
бимов, Д. Л. Федорова, И. Д. Кулагин,
Л. М. Сорокин. Изв. АН СССР. Неор-
ган. материалы, № 11 (1968).
166. Д. М. Чижиков, С. С. Дейнека,
Ю. В. Цветков, В. И. Макаров. Тезисы
докладов I сёминара по применению
низкотемпературной плазмы в тех-
нологии неорганических веществ и
порошковой металлургии. Киев,
1968.
167. В. К. Rains, В. Н. Kadlek. Meta lurg.
Trans., 1, N 6. 1501 (1970).
168. R. L. Stewart. Patent US, N 3586613,
31.3.1967.
169. Д. А. Балакирев, Г. Б. Синярев,
А. С. Сахиев, В. Е. Медведев, Л. Е.
Слынъко, Г. П. Стельмах, П. А. Чес-
ноков. В сб.: Илазмохимия-71. М.,
ИНХС АН СССР, 1971, стр. 168.
170. J. Е. Longjield, D. Hyman, J. F. Skri-
van, J. D. Chase. Patent BRD,
N 2053206, 29.10.1969.
171. H. L. Gilles, C. W. Clump. Industr.
and Engng Chem., Process Leung
and Developm., 9, N 9, 194 (1969).
172. R. A. Brown. Canad. Met. Quart.,.
10, Nl, 47 (1971).
173. U. Harries, I. Holmgen, S. Korman,
G. Sheer. J. Electrochem. Soc., N L0,
L06 (1969).
174. P. A. Huska, C. W. Clump. Industr.
and Engng Chem. Process Desing and
Developm.. 6, A 2, 238 (1967).
175. J. Warren, H. Shimizu. Canad. Mining
and Metallurg. Bull., 58, N 6, 551
(1965).
176. С. В. Дресвин. А. В. Донской,
В. M. Гольдфарб, В. С. Клубникин.
Физика и техника низкотемператур-
ной плазмы. М., Атомиздат, 1972.
177. М. М. Shahin. In: Reactions under
Plasma Conditions, v. 2. M. Venu-
gopalan (Ed.). N. Y.— London — Syd-
ney — Toronto, J. Wiley, 1971,
p. 237.
178. A. T. Bell, K. Kwong. A. I. Ch. E.
Journal, 18, N 5, 990 (1972); Industr.
and Engng Chem. Fundamentals, 10,
N 3, 373 (1971).
179. А. В. Горохов, А. И. Максимов,
В. Д. Сизов, Л. С. Степанова. Ж.
техн, физ., 42, 2176 (1972).
180. D. G. Kuehn, L. М. Chanin. J. Appl.
Phys., 43, 339 (1972).
181. J. Prashad. Roczn. Chem., 46, 1629f
(1972).
182. I. P. Wightman. Proc. IEEE, 62, A 4
(1974).
183. У. А. Циелен, T. H. Миллер. В сб.:
Низкотемпературная плазма в тех-
нологии неорганических веществ. Но-
восибирск, «Наука», 1971.
184. Т. Н. Миллер. У. А. Циелен^
А. А. Шпате. В сб.: Плазмохимия-71,.
М., ИНХС АН СССР, 1971, стр.
140.
185. Я. П. Грабис, Т. Н. Миллер,.
Г. М. Хейдемане, М. Рацис. Там же,,
стр. 142.
186. А. А. Ашеулов, М. Г. Бардический,.
Г. В. Куртуков, А. А. Корнилов,.
В. В. Марусин. В сб.: Низкотемпера-
турная плазма в технологии неоргани-
ческих I ешеств. Новосибирск,.
«Наука», 1971, стр. 21.
187. Н. D. Murdoch, S. М. Hamblyn. Pa-
tent France, N 1582154, 3.7.1967.
188. Patent France, N 2003568, 9.03. 1968.
189. Brit. Patent, N 1199811, 4.02.1969.
190. H. Winterhager. K. Hanusch. Ber.
Dtsch. Keram. Ges., 46, N 4. 181
(1969).
191. E. Neuenschwander, K. Schnet, W.
Scheller. Patent, US N 342966,1,
2.12. 1965; N 3545922, 2.12.1965.
192. В. H. Троицкий, M. И. Айвазов,
В. M. Кузнецов, В. С. Корягин. В
300
сб.: Низкотемпературная плазма в
технологии неорганических веществ.
Новосибирск, «Наука», 1971, стр. 19.
193. В. Н. Троицкий, Б. М. Гребцов,
М. И. Айвазов. В сб.: Плазмохимия-71.
М., ИНХС АН СССР, 1971, стр.
145.
194. В. С. Корягин, А. Л. Сурис,
В. Н. Троицкий, С. Н. Шорин. Хи-
мия высоких энергий, 7, 215 (1973).
195. О. Matsumoto, У. Shirato, Y. Hayaka-
wa. J. Electrochem. Soc. Japan, 37,
151, 165 (1969).
196. O. Matsumoto, Y. Shirato. Denki
Kagaku, 38, 170, 39, 82 (1970).
197. O. Matsumoto, T. Miyazaki. Denki
Kagaku, 39, 388 (1971).
198. O. Matsumoto. J. Electrochem. Soc.
Japan, 37, 200 (1969); Denki Kagaku,
38, 827 (1970).
199. Brit. Patent, № 1241864, 5.6.1968.
200. L. R. Swaney. Patent, US
N 3485858, 26.9.1968; Patent France,
N 1571542, 5.6.1968.
201. J. Silbiger, C. Schnell. Patent Suisse,
N 472337, 10.6.1968.
202. Ю. А. Шмакин, В. В. Марусин.
Тезисы докладов II Всесоюзного се-
минара по применению низкотемпе-
ратурной плазмы в технологии неор-
ганических веществ и порошковой
металлургии. Новосибирск, 1970.
203. С. Н. Ганз, А. II. Мельник,
В. Д. Пархоменко. Получение свя-
занного азота в плазме. Киев,
Укр. НИИНТИ, 1967; Тезисы докла-
да на VIS Всесоюзной конференции
по технологии неорганических ве-
ществ и минеральным удобрениям.
Минск, 1970.
204. В. А. Вишневская, 3. А. Констант,
Т. Н. Миллер, А. Я. Вайвад. Изв.
АН ЛатвССР, серия хим., № 1 (1969).
205. Н. Mayer. Вег. Dtsch. Keram. Ges.,
39, N 2, 115 (1962).
206. Л. H. Усов, А. И. Борисенко. Приме-
нение плазмы для получения высоко-
температурных покрытий. М.— Л.,
«Наука», 1965.
207. Н. Н. Рыкалин, В. А. Петруничев,
И. Д. Кулагин, Л. М. Сорокин,
Е. Б. Королева, А. Б. Гугняк. В сб.:
Плазменные процессы в металлургии
и технологии неорганических мате-
риалов. М., «Наука», 1973, стр. 220.
208. В. Waldie. Trans. Inst. Chem. Engrs,
48, N 3, 90 (1973).
209. E. Neuenschwander. US Patent,
N 3592627, 7.6.1966; N 3545922,
2.12.1965.
210. A. A. Lonza. Patent France, N 2039121,
31.3.1970.
211. H. Houseman. Pat. BRD, N 1667188,
11.11.1967.
212. W. R. Barner, D. Barby. Patent
BRD, N 1667765, 7.12.1967; N 1667766,
7.12.1967.
213. T. Kugler, J. Silbiger. Patent BRD,
N 2008029, 31.3.1969.
214. Askoda, Kazuo, T. Takabatake. Pa-
tent Japan, N 40163/70, 14.2.1961.
215. Brit. Patent, № 1018874, 8.10.62.
216. Г. В. Самсонов, А. П. Эпик. Туго-
плавкие покрытия. M., «Металлур-
гия», 1973.
217. В. В. Кудинов. В сб.: Плазменные
процессы в металлургии и техно-
логии неорганических материалов. М.,
«Наука», 1973, стр. 158.
218. Н. Н. Рыкалин, М. X. Шоршоровг
В. В. Кудинов, Ю. А. Галкин. Там
же, стр. 187.
219. Электронная техника. ЦНИИэлек-
троника, № И, 56 (1973).
220. Chem. Age, 102, N 2705, 12 (1971).
221. Кристаллизация из газовой фазы.
М., «Мир», 1965.
222. D. R. Secrist, J. D. Mackensie. J.
Electrochem. Soc., 113, 914 (1966).
223. P. E. Ровинский, А. П. Соболев.
Теплофизика высоких температур, 6,
№ 2 (1968).
224. Е. Linder, A. Davis. J. Phys. Chem.,
35, 3649 (1931).
225. R. G. Meyerand. AIAA Journal, 5,
N 10, 3 (1967).
226. 1. 1 . Haught. Annual Rev. Phys.
Chem., 19, 342 (1968).
227. C. A. Papp. Industr. and Engng
Chem., 55, N 4, 48 (1963); Chem.
Engng Progr., 59, N 6, 51 (1963).
228. А. П. Ляхович. Электрическая про-
мышленность, серия электротермия,
вып. 8 (144), 21 (1974).
229. В. Б. Лукьянов, Е. Ф. Симонов,
А. М. Можайский. Ж. физ. химии,
47, 2838 (1973).
230. Р. Н. Wilks. Pure Appl. Chem.. 39.
415 (1974).
231. E. Molina ry. Pure Appl. Chem., 39,
343 (1974)?
232. Ю. H. Кобзев, Г. И. Козлов, Г. Н. Ху-
дяков. Плазмохимический процесс пе-
реработки природного газа в ацети-
лен, водород и сажу. Препринт ИПМ
АН СССР и ЭНИН им. Кржижанов-
ского. М.. 1974, № 48.
233. R. R. Sowell, N. J. DeLollis, Н. J. Gre-
gory, О. Montoya. Recent Adv. Adhes.,
Proc. Symp. Am. Chem. Soc., 1971,
Pub. 1973, L., Gordon a. Breach,
p. 77. C. A., 82. N 4. 17615 (1975).
234. Г. H. Разина, С. В. Кафтанов,
С. Д. Федосеев, В. В. Клейменов.
Труды МХТИ, 74, 69 (1973).
235. D. М. Bodily, С. L. Che, W. II. Wiser.
Am. Chem. Soc., Div. Fuel Chem.,
Preprints. 18 (2), 221 (1973). C. A.,
82, N 14, 88360 (1975).
236. F. W. Bach, F. Erdman-Jesnitzer. Me-
tall (Berlin), 28, 986, 1091 (1974).
237. Applied Mineralogy, v. 3. Arc Plasma
Technology in Material Science, Ed.
D. A. Gerdeman, N. L. Hecht, Sprin-
ger, N. Y., 1972.
238. F. J. Skriian. Patent US, N 3856918,
24.12.1974.
239. K. A. Akashi, R. Ishizuka. T. Mutobe.
Proc. 4-th Int. Conf. Vac. MetalL,
1973, Pub. 1974, p. 163. C. A., 82,
\ 6, 33641, 1975.
240. Ю. H. Туманов, H. П. Галкин. Атом-
ная энергия, 37, 340 (1974).
301
241. Y. Nishimura, T. Takenouchi, W. Sa-
kai. High Temp.— High Pressures,
6(2), 149 (1974).
242. I. A. DeVink. Silikates Ind., 35 (12),
308 (1970); 36 (1), 17 (1971), 36 (2),
49 (1971).
243. W. Dale. Patent US, N 3848068,
12 XI 1974.
244. D. V. McCaughan, R. A. Kushner,
Proc. IEEE, 62 (9), 1236 (1974).
245. A. Д. Сулимин, Л. П. Остаткам и
др. Физ. и хим обработки матер.,
3, № 2, 57 (1957).
246. Techniques and Applications of Plas-
ma Chemistry, Ed. J. R. Hollahan,
A. T. Bell. Wiley-Interscience, N. Y.
1974.
247. Экспериментальные и теоретические
исследования неравновесных физико-
химических процессов, т. 1—3. Ред.
Л. С. Полак. ИНХС АН СССР, М.,
1974.
248. Д. М. Чижиков, С. С. Дейнека, В. Н.
Макарова, Ю. В. Цветков. Авт. свпд.
№ 240691. Бюлл. изобрет., № 13, 22
(1969).
249. Г. И. Козлов, Г. Н. Худяков, Ю. И.
Кобозев. Авт. свид. № 256758. Бюлл.
изобрет., № 35, 24 (1969).
ОГЛАВЛЕНИЕ
Предисловие.......................................................... 2
1
ГЕНЕРАЦИЯ
И ДИАГНОСТИКА НИЗКОТЕМПЕРАТУРНОЙ ПЛАЗМЫ
Генераторы низкотемпературной плазмы................................. б
Диагностика низкотемпературной плазмы............ ............... 27"
2
ОСНОВНЫЕ ПОЛОЖЕНИЯ
НЕРАВНОВЕСНОЙ ХИМИЧЕСКОЙ КИНЕТИКИ.
ПЛАЗМОХИМИЧЕСКАЯ КИНЕТИКА
Неравновесная статистическая термодинамика и неравновесная химическая
кинетика........................................................... 34
Исторические замечания.............................................. 37
Теория абсолютных скоростей реакций................................. 43
Метод стационарного состояния....................................... 44
Замечания о константе скорости химической реакции с точки зрения
столкновительной модели............................................ 45
Наблюдаемая (кажущаяся) энергия активации......................... 46
Уравнение Паули..................................................... 50
Неравновесные теории и расчеты диссоциации — рекомбинации........... 53
Расчет колебательной релаксации молекул при наличии (v — Ту , (г? — &)-,
(е — ^-процессов. Взаимное влияние колебательной релаксации и диссо-
циации ............................................................ 59
Коэффициент скорости химической реакции............................. 66
Принцип микроскопической обратимости и принцип детального равнове-
сия ............................................................... 76
Определение энтропии в неравновесных состояниях и кинетическое уравне-
ние ............................................................... 78
Уравнение Больцмана................................................. 80
Релаксация реагирующего газа........................................ 85
Радикальные и цепные реакции при высоких энергиях (температурах) ... 90
Учет внутренних степеней свободы молекулы........................... 90
Элементарные реакции возбужденных частиц............................ 91
Мономолекулярные реакции возбужденных частиц........................ 92
Необходимые замечания о столкновительной дезактивации............... 94
Реакции рекомбинации третьего кинетического порядка................. 95
Основные особенности плазмохимических реакций....................... 96
Влияние неупругих процессов на распределение электронов по энергиям в
тлеющем разряде в N2, N2 + О2 и СО2.............................. 100
Влияние функции распределения электронов по энергии на коэффициент
скорости химической реакции....................................... 102
Кинетика разложения углекислого газа в тлеющем разряде............. 104
Оценка коэффициента скорости рекомбинации возбужденных атомов азота . 107
Равновесные и квазиравновесные реакции в плазмохимической кинетике . 109
Смешение реагентов в плазмохимической кинетике..................... 109
Химические реакции в неидеальной плазме.......................... 111
303
3
ОСОБЕННОСТИ МЕХАНИЗМОВ
ПЛАЗМОХИМИЧЕСКИХ РЕАКЦИЙ.
РОЛЬ ЗАРЯЖЕННЫХ И ВОЗБУЖДЕННЫХ ЧАСТИЦ
Структура уровней и потенциальные кривые электронных состояний моле-
кул ............................................................. 114
Возбуждение вращательных уровней молекул......................... 115
Возбуждение колебательных уровней молекул........................ 118
Возбуждение электронных уровней.................................. 123
Ионизация и диссоциативная ионизация молекул..................... 134
Диссоциация молекул через электронно-возбужденные состояния...... 136
Диссоциативное прилипание электронов к молекулам................. 146
О роли различных процессов инициирования химических реакций в низ-
котемпературной плазме........................................... 147
Ступенчатая диссоциация молекул электронным ударом............... 149
Рекомбинация тяжелых частиц в молекулу.......................... 153
Диссоциативная рекомбинация молекулярных ионов с электронами и ассо-
циативная ионизация.............................................. 168
Отклонения от ионизационного равновесия в низкотемпературной плазме . 187
4
ХИМИЧЕСКИЕ РЕАКЦИИ В ПЛАЗМЕННЫХ СТРУЯХ
Химические реакции в условиях турбулентности . ................. 199
Исследования турбулентного перемешивания в дозвуковых плазменных
потоках......................................................... 205
Плазмохимический реактор......................................... 213
5
ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
(органические и неорганические соединения, материалы)
Описание плазмохимических процессов.............................. 222
Плазмохимические процессы в органической химии................... 231
Плазмохимические процессы в неорганической химии................. 260
Получение сферических и тонко дисперсных порошков в низкотемператур-
ной плазме....................................................... 277
Получение пленок в низкотемпературной плазме..................... 277
Использование плазмы, создаваемой излучением лазера, и излучения плаз-
мы .............................................................. 282
Некоторые другие применения неравновесной плазмы................. 282
Литература....................................................... 283
ТЕОРЕТИЧЕСКАЯ И ПРИКЛАДНАЯ ПЛАЗМОХИМИЯ
Утверждено к печати Институтом нефтехимического синтеза им. А. В. Топчиева Академии наук СССР
Редактор издательства В. М. Медер. Художественный редактор Н. II.-Власик
Художник А. Г. Кобрин. Технический редактор Ю. В. Рылина
«Сдано в набор 3/VII 1975 г. Подписано к печати 31/Х 1975 г. Формат 70xl081/i6. Бумага № 1 Усл.
печ. л. 26,6. Уч.-и д. л. 28,4. Тираж 2000. Т-17011. Тип. зак. 2593. Цена 2 р. 10 к.
Издательство «Наука». 103717 ГСП, Москва. К-62, Подсосенский пер., 21
2-я типография издательства «Наука». 121099, Москва, Г-99, Шубинский пер., 10
СПИСОК ИСПРАВЛЕНИЙ
Стра- ница Строка Напечатано Должно быть
66 26 сн. I у экс и j. X)' Т эксп 1 у,,
66 25 сн. XVg+
84 3 сн. (2.170) (2.169)
107 12-13 св. скорости имеют реакции скорости реакции имеют
120 подпись к рис. 3.1 510~15 см2 510“1е см2
164 15 сн. СО2(3В2) СО2(Ч32)
192 19 св. У az* ^АГ*
276 9 сн. 2С2 2С
Теоретич. и прикладная плаамохимия