/












































































































































































































































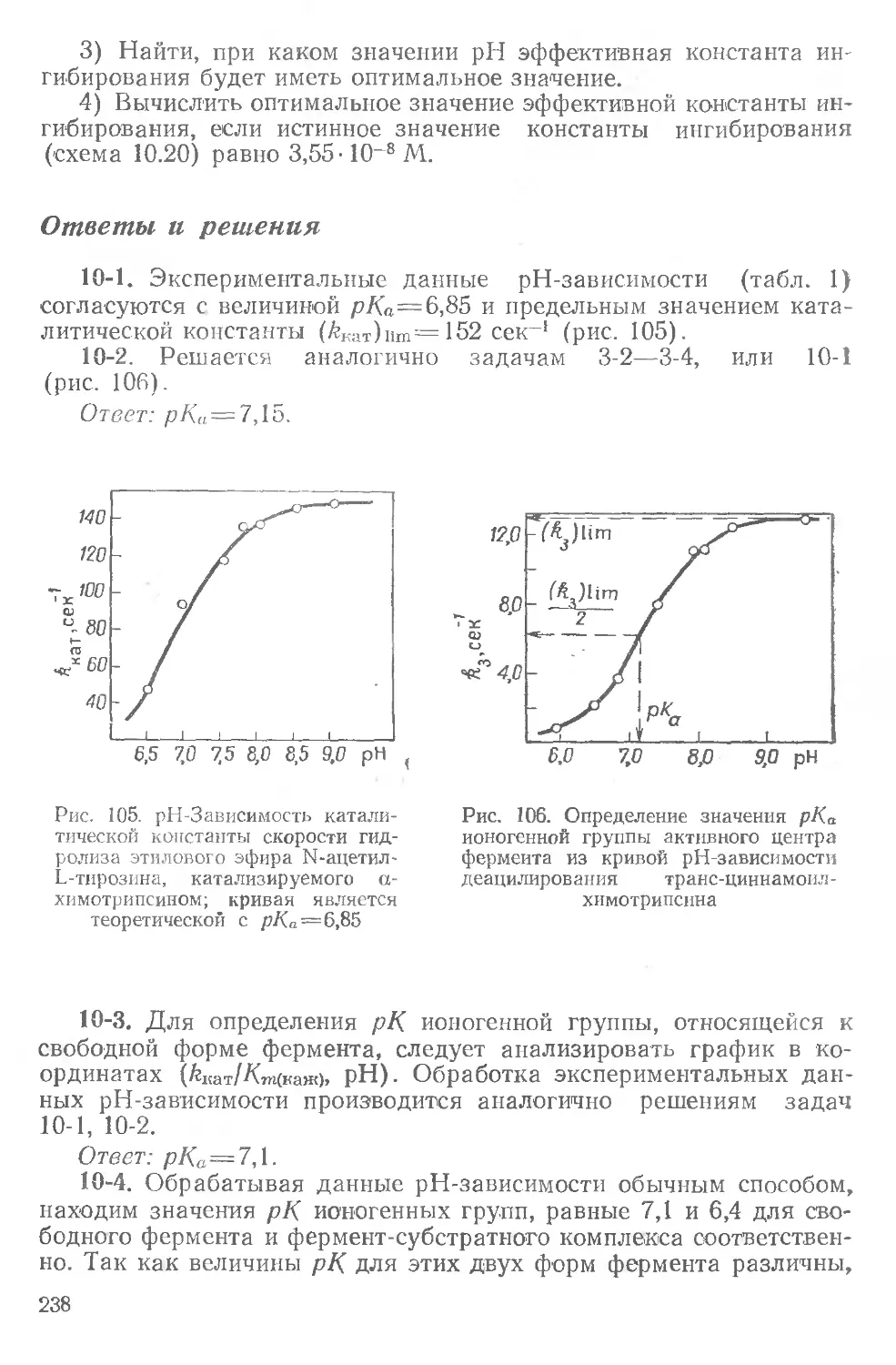























































































Текст
И. В. БЕРЕЗИН, А. А. К ЛЕСОВ
ПРАКТИЧЕСКИЙ
КУРС
ХИМИЧЕСКОЙ
И ФЕРМЕНТАТИВНОЙ
КИНЕТИКИ
V*-
И. В. БЕРЕЗИН, А.А. КЛЁСОВ
ПРАКТИЧЕСКИЙ
КУРС
ХИМИЧЕСКОЙ
И ФЕРМЕНТАТИВНОЙ
КИНЕТИНИ
Допущено Министерством
высшего и среднего специального образования СССР
в качестве учебного пособия для студентов
химических специальностей университетов
ИЗДАТЕЛЬСТВО
МОСКОВСКОГО УНИВЕРСИТЕТА 1976
УДК 541.1
Настоящее пособие — первое в мировой литературе учебное ру
ководетво по анализу и обработке кинетических данных
ферментативных реакций. В первой части курса изложены методы анализа
кинетических закономерностей простых химических реакций,
изучение их необходимо для дальнейшего понимания кинетики и
механизма действия ферментов. Во второй части книги рассмотрены
методы обработки кинетических данных ферментативных реакций.
Особое внимание здесь уделено новым подходам, не нашедшим до
последнего времени отражения в учебной литературе (новые
методы нахождения элементарных констант, влияние диффузии на
кинетику действия иммобилизованных ферментов, использование
интегральных форм кинетических уравнений и др.). Каждая глава
сопровождается оригинальными задачами с подробными решениями.
Книга предназначена для студентов, аспирантов н
преподавателей химических и биологических факультетов университетов, а
также для научных и научно-технических работников всех
специальностей, имеющих отношение к химической и биологической кинетике.
Рецензенты:
кафедра физической химии Новосибирского университета;
проф. А. Е. Шилов
Илья Васильевич БЕРЕЗИН, Анатолий Алексеевич КЛЁСОВ
ПРАКТИЧЕСКИЙ КУРС ХИМИЧЕСКОЙ
И ФЕРМЕНТАТИВНОЙ КИНЕТИКИ
Тематический план 1975 г. № 138
Редактор Л. И. Чиркова. Художник В. С. Казакевич. Художественный редактор
М. Ф. Евстафьева. Технический редактор В. В. Лебедева. Корректоры Я. А. Большакова,
И. С. Хлыстова, И. И. Коновалова, С. Ф. Будаева.
Сдаио в набор 25/111 1975 г. Подписано к печати 14/XI 1975 г. Л-23010 Формат 60Х90'/Ч
Бумага тип. № 1 Физ. печ. л. 20,0 Уч.-изд. л. 18,70 Изд. № 2428 Зак. 158 Тираж 6800 экз.
Цена в переплете № 7 — 89 коп. Цена в переплете № 5—76 коп.
Издательство Московского университета. Москва, К-9, ул. Герцена, 5/7.
Московская типография № 13 Союзполиграфпрома при Государственном комитете
Совета Министров СССР по делам издательств, полиграфии и книжной торговли.
107005, Москва, Б-5, Денисовский пер., д. 30
Отпечатано с готового набора в типографии Лзд-ва МГУ. Москва, Ленгоры. Зак. 370-
(С) Издательство Московского университета, 1976 г.
20503—157 Q
Б 077(02)—76 llj8~7o
ПРЕДИСЛОВИЕ
До настоящего времени базис химической кинетики остается
наименее строгим среди всех разделов физической химии. Однако
значение кинетических методов в современной химии и биохимии
неуклонно растет. В первую очередь это связано с тем, что хотя
теория элементарного акта далека от совершенства,
математический аппарат формальной кинетики и макрокинетики вполне строг
и приводит к однозначным результатам. Не менее важно и то
обстоятельство, что результаты кинетического эксперимента часто
удается поставить в четкое соответствие с другими
физико-химическими параметрами реагирующих систем и таким образом
получить законченное описание химического или биохимического
процесса.
С экспериментальной точки зрения нахождение константы
скорости элементарного процесса является одной из самых
фундаментальных задач. Эта задача решается без труда в случае простых
реакций и неизмеримо усложняется в случае быстрых
многостадийных процессов. За последние годы достигнут существенный
прогресс в этом направлении, и исследование кинетики даже сложного
процесса сегодня перестало быть камнем преткновения для
исследователя.
Практический курс, предлагаемый вниманию читателя, может
рассматриваться как руководство по обработке экспериментальных
данных ферментативной кинетики. В основу данной книги
положены лекции и практические занятия по кинетике ферментативных
реакций. Так как понимание кинетических закономерностей и
механизма действия ферментов невозможно без знания кинетических
законов простых химических реакций, последним в книге также
уделено существенное внимание.
С точки зрения химика фермент — это прежде всего
катализатор. Поэтому многие закономерности, которые наблюдаются в
ферментативных процессах, почти наверняка найдут себе аналогии в
гомогенном химическом катализе и наоборот. По существу нет
резких границ между всеми известными нам видами химического и
биологического катализа, и противопоставление их друг другу
требует скорее классификация, чем сущность явления катализа.
Практическое руководство построено с таким расчетом, что
изучение его может начать человек, неискушенный в химической
кинетике, а тем более в ее экспериментальных тонкостях. Мы
надеемся, что подробное рассмотрение методов обработки кинетиче-
3
ских кривых простых реакций, а также основных закономерностей,
связанных с влиянием на кинетику температуры и концентрации
ионов водорода, создаст хорошую основу для дальнейшего
восприятия материала по кинетике ферментативных процессов.
Во второй части книги, посвященной ферментам,
рассматриваются как традиционные вопросы биокинетики (уравнение Миха-
элиса — Ментен, различные виды ингибирования, влияние рН на
скорость ферментативных реакций и т. д.), так и новые, не
нашедшие пока отражения в учебной литературе (новые методы
нахождения элементарных констант из данных стационарной кинетики,
влияние диффузии на кинетику действия иммобилизованных
ферментов, использование интегральных форм кинетических
уравнений, кинетический анализ систем со взаимным истощением, анализ
нетривиальных типов ингибирования, применение теории графов
в ферментативной кинетике и др.). Требования систематизации
курса способствовали созданию новых методов обработки
кинетических данных ферментативных реакций, описываемых в
главах 5—8, 10, 11.
Каждая глава курса сопровождается серией задач с
подробными решениями. Эти задачи, материал для которых почерпнут из
научной периодики и собственных экспериментальных
исследований, и их решения являются по существу примерами анализа дан-
пых оригинальных работ и позволяют читателю более глубоко
вникнуть в некоторые особенности обработки данных
кинетического эксперимента.
Мы надеемся, что книга окажется полезной особенно сейчас,
когда значение кинетических методов исследования чрезвычайно
возросло. Проработать систематически весь этот курс смогут в
основном только те студенты, аспиранты и исследователи, которые
специализируются в области химической или биологической
кинетики, однако можно надеяться, что книга, снабженная подробным
предметным указателем, в любом случае будет удобным
справочником по методам обработки кинетических данных химических и
ферментативных реакций.
Авторы выражают благодарность К. Мартинеку, А. М. Клиба-
нову и А. К. Яцимирскому за участие в составлении ряда задач
настоящего курса. Глава 12 написана А. М. Клибановым.
Часть I
КИНЕТИКА ГОМОГЕННЫХ
ХИМИЧЕСКИХ РЕАКЦИЙ
Глава 1
ПОРЯДОК РЕАКЦИИ И ЕГО ОПРЕДЕЛЕНИЕ
При определенных внешних условиях (температура, давление,
реакционная среда и т. д.) скорость химической реакции обычно
является функцией концентраций реагирующих веществ:
« = M*i]"«'...[cft]n*, (1.1)
где Сг — концентрация i-го реагирующего вещества. В таком случае
величины Пг называют порядком реакции по веществу и Величины
tii в общем случае могут принимать как целые, так и дробные
значения. В случае простых реакций п,- оказываются целыми
положительными числами, совпадающими по величине с молекулярностью
реакции. Сумму порядков реакции по всем реагирующим
веществам называют общим порядком реакции. Наиболее важное
значение в кинетике простых реакций имеют реакции первого, второго
и третьего порядков (соответственно, моно-, би- и тримолекуляр-
ные реакции). Простые реакции более высокого порядка
практически неосуществимы из-за чрезвычайно малой вероятности
одновременного соударения четырех или более молекул.
Основным свойством уравнения (1.1) является то, что
константа скорости реакции k и порядки реакции по ее компонентам
должны оставаться постоянными в течение всей реакции, а также
при вариации начальных концентраций реагентов (при
неизменных условиях эксперимента).
При определении порядка реакции на практике можно
использовать либо начальные участки серии кинетических кривых, либо
одну полную кинетическую кривую.
5
§ 1. Анализ начальных скоростей
реакций
Скорость расходования вещества в реакции «-го порядка в
начальный момент времени можно записать следующим образом:
«о - - ^ = **"»• (1-2)
Логарифмируя уравнение (1.2), получим
log v0 = log k + n log cn. (1.3)
Из выражения (1.3) очевидно, что порядок реакции можно
определить при анализе начальных скоростей реакции (v0),
соответствующих различным начальным концентрациям реагента (со). В этом
случае зависимость в координатах (logt>0, logc0) должна иметь
вид прямой линии с тангенсом угла наклона, численно равным
порядку реакции п.
Порядок реакции можно приблизительно определить по
значениям начальных скоростей реакций (г^ и ^(02))5 которые
соответствуют двум начальным концентрациям субстрата (с^ и с(20)),
используя соотношение (1.3):
logv™=logk-\-nlogcm; (1.4)
log vM = log k + n log C&K (1.5)
Вычитая (1.5) из (1.4), получим
log tfv - log г£> = n (log J? - log cW)
или
Л1)
rm ■
0
c0
(1.6)
§ 2. Анализ полной кинетической кривой
1. Скорость расходования вещества в реакции п-го порядка
записывается следующим образом:
Очевидно, что порядок реакции можно определить из единственной
кинетической кривой, логарифмируя уравнение (1.7):
logi^ = log k -f- /г log с. (1.8)
Из полученного уравнения видно, что зависимость в координатах
(log v, log с) в случае простых реакций должна быть прямолиней-
6
ной с тангенсом угла наклона, равным п. Следует отметить, что
в случае сложных реакций (см. выражение (1.9)) прямолинейная
зависимость в координатах (log v, log с) может не выполняться:
—§ = kc + k'c*. (1.9)
При использовании соотношения (1.8) на практике скорость
реакции при данной текущей концентрации субстрата находят обычно
графическим дифференцированием кинетической кривой (см.,
например, рис. 2).
2. Если Gi — доля исходного-вещества, не вступившая в
химическую реакцию к моменту времени ti(ai = cJco), и йг — доля
исходного вещества, не вступившая в реакцию к моменту времени
Ыа2—Сг/со), причем a.2=a2i, то порядок реакции можно найти из
следующего выражения [1]:
п=\- '"«('«ft-1). (1.10)
Вывод. Если а — доля исходного вещества, не вступившая в
химическую реакцию к моменту времени t, то а=с/с0 и с=асо.
Подставляя выражение для с в уравнение
получаем
dt
-% = ka»c*-\ (1.12)
Разделяя переменные и интегрируя уравнение (1.12) в пределах
(1, а), (0,/), получим
-й^(-^-1)-Лс*"1'- (1ЛЗ>
(Очевидно, что соотношение (1.13) не применимо для случая я=1.)
Для определения двух неизвестных величин Jfc и п запишем два
уравнения:
а
1 / I
4=kc%~42. (1.15)
■1. ,2
Делением уравнения (1.15) на (1.14) приходим к соотношению
1 .1
,л—1
1
1 Г =1Г (Ы6)
«?_I
Решение его можно упростить, если выбрать на кинетической
кривой два значения а, связанных соотношением а2=а2\. В этом сл>-
7
чае получаем выражение, логарифмирование которого приводит
ч уравнению (1.10):
(1П7)
а
1
я—1
1 t„
3. Если имеется несколько кинетических кривых, полученных
при различных начальных концентрациях субстрата, то порядок
реакции можно определить, используя преобразованное
выражение (1.13):
log^ = logl7^|^-l]-(/t-l)logCo. (1.18)
Здесь а — доля исходного вещества, не вступившая в реакцию
к моменту времени t (а=с/са). Если для всех кинетических кривых
реакционной серии выбрать одинаковое значение а (постоянное
для всех Со), то выражение (1.18) можно записать в виде
log t = const — (п — 1) log c0. (1-19)
Очевидно, что зависимость в координатах (\ogt, logc0) в случае
простой реакции должна быть прямолинейной с тангенсом угла
наклона, равным —(п—1).
4. Если на двух кинетических кривых, соответствующих двум
различным значениям начальной концентрации субстрата (с^ и
с(02)), выбрать два значения времени (t\ и U), соответствующие
достижению одинаковой доли превращения субстрата, то порядок
реакции может быть вычислен по формуле
t-l+,'tf'ir t1-20)
log4 /"о
Формула (1.20) может быть легко получена делением друг на
друга двух уравнений типа (1.13), соответствующих двум начальным
концентрациям субстрата и двум временам достижения
определенной доли превращения субстрата.
Задачи
1-1. В таблице 1 приведены значения начальных скоростей
реакций 2-фенил-4,4-диметил-2-оксазолин-5-она с этиловым эфиром
аланина, соответствующие начальным концентрациям реагентов
[2]. Найти порядок реакции по каждому компоненту.
1-2. Кинетика термического распада хлористого гидразония
в смеси его с гидразином при 185° С описывается общим
уравнением a=£[N2H4]a[N2H5Cl]6. Используя данные табл. 2, найти
константу скорости реакции и порядок реакции по гидразину и
хлористому гидразонию.
8
Таблица 1
Таблица 2
Начальные скорости реакции
2-фенил-4,4-диметил-2-оксазолин-
-5-она (А) с этиловым эфиром
аланина (В).
Условия реакции: четыреххлористый
углерод; 20° С
Vo-10',
миль/л-сек
4,06
4,20
12,10
36,80
|А]„-10»,
моль/л
моль /л
2,15
10,0
6,38
10,0
15,1
3,36
15,1
29,4
Зависимость скорости
термического распада хлористого
гидразония в смеси его с гидразином
от концентраций реагентов [3]
1-3. В таблице 3
приведены кинетические данные
обесцвечивания красителя проф-
лавипа (3,6-диамипоакридина)
под действием
ультрафиолетового света. Определить
порядок реакции и значение константы
сителя.
tf-10*,
лголь/л-сек
27,7
27,4
26,1
21,9
10,5
9,86
6,48
3,57
3,34
3,30
2,58
2,29
2,24
2,19
[NjH*],
моль/л
12,65
12,50
11,90
10,00
4,80
4,51
2,96
1,71
1,71
1,71
1,71
1,71
1,71
1.71
[N5H5CI1,
моль/л
17,8
17,8
17.8
17,8
17.8
17,8
17,8
17,0
15,9
15,7
12,25
10,9
10,55
10.4
скорости обесцвечивания кра-
Таблица 3
Кинетика обесцвечивания профлавина под действием
ультрафиолетового света. Условия опыта: рН 6,9; 25° С
Время, мин
Г) *
0
0,63
2
0,56
4
0,51
7
0,44
п
0,38
15
0,32
20
0,25
* Оптическая плотность раствора профлавина при алиие
волиы X = 444 нм.
1-4. Определить константу скорости и порядок реакции по
данным табл. 4.
1-5. В таблице 5 приведены кинетические данные
расщепления диэтилового эфира изобутил-литием [4]. Определить
порядок реакции и найти значение константы скорости.
1-6. В таблице 6 приведены данные эксперимента по
изучению кинетики денатурации ДНК в водном растворе
формальдегида [5]. Найти порядок реакции и константу скорости реакции
денатурации.
9
Таблица 4
Кинетика щелочного гидролиза метилового эфира
N-ацетилглицина. Условия опыта: рН 11,8; 25е С
Время, мин
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
С-105, моль/л
8,80
7.S6
7,23
6,52
5,87
5,35
4,85
4,36
4 00
Время, мин
0.9
1,0
1,1
1,2
1,3
1,4
1,5
1,6
С-101, моль/л
3,55
3,20
2,85
2,60
2,40
2,20
1,95
1.75
Таблица 5
Кинетика реакции диэтилового эфира
с изобутил-литием. Условия опыта: 30° С;
начальные концентрации диэтилового эфира
и изобутил-лития равны 4,5 моль/л и 0,15 моль/л
соответственно
Время реакции, часы
0
1
2
3
4
5
6
{Изобутил-литий!, моль/л
0,150
0,132
0,120
0,108
0,097
0,090
0.075
Таблица 6
Кинетика денатурации ДНК. Условия опыта:
рН 9,1; 34,2° С; 37% формальдегида;
2-10-2 мг/мл ДНК (молекулярный вес 2,810е)
Лоля натнвной ДНК, %
100 -
94,4
74,4
61,0
48,1
37,5
29,4
22,5
17,5
12,5
Время, мин
0
1
3
5
7
10
12
14
17
20
1-7. Под действием ультразвука происходит необратимая
инактивация фермента сс-химотрипсина. В табл. 7 приведены
значения ферментативной активности при различных временах
озвучивания раствора фермента. Найти порядок реакции и константу
скорости инактивации.
Таблица 7
Кинетика необратимой инактивации
а-химотрипсина под действием ультразвука.
Условия опыта: [Е]0 = 1,3-10-7М; интенсивность
излучения 2 вт/см2; температура 15° С
Время озвучивания, мин
0
15
20
40
50
Активность фермента,
условн. ед.
1,80
1,44
1,30
0,99
0,85
1-8. В таблице 8 приведены результаты кинетического
изучения необратимой денатурации бычьего оксигемоглобина [6].
Определить порядок реакции денатурации.
Таблица 8
Кинетика необратимой денатурации
бычьего оксигемоглобина. Условия опыта:
рН 11,9; температура 2° С
Время, часы
Доля денатурированного
белка, %
1
2
6
18
21
26
31
39
43
47
7
13
30
53
58
62
69
78
80
82
1-9. Реакция распада двуокиси азота при достаточно малых
давлениях и больших температурах практически необратима.
Исходя из данных табл. 9, определить порядок этой реакции.
1-10. Начальная концентрация реагирующего вещества
уменьшилась за 10 минут вдвое. Начальная концентрация того же
вещества, в 5 раз большая, уменьшилась вдвое за 24 секунды.
Определить порядок реакции.
11
1-11. При изучении реакции термического распада диоксана
было найдено, что при двух начальных коицентрациях его, равных
800 и 400 мм рт. ст., времена полупревращения составляют 13,9 и
19,0 мин соответственно. Определить порядок реакции.
Таблица 9
Временная зависимость термического распада
двуокиси азота
Время, сек
0
20
40
60
80
100
[NOil-10", моль/л
17,8
10,6
7,1
5,4
4,6
4,0
Таблица 10
Значения начальных скоростей реакций декаборана
с к-бутиловым спиртом в бензоле (скорость реакции
определялась по кинетике расходования декаборана).
Условия опыта: 25° С
v0- 10s, моль/л сек
2,00
4,02
8,05
12,02
16,10
20,10
67,10
53,40
40,20
26,70
13,50
6,70
IB,0HuIo, моль/л
0,01
0,02
0,04
0,06
0,08
0,10
1,00
1,00
1,00
1,00
1,00
1,00
[С4Н»ОН]0, моль/л
3,0
3,0
3,0
3,0
3,0
3,0
1,0
0,8
0,6
0,4
0,2
0,1
1-12. В работе [7] было найдено, что реакция декаборана с
алифатическими спиртами описывается следующим стехиометри-
ческим уравнением:
30 ROH + В10Н14 = 10 В (OR)3 + 22Н2.
Используя данные табл. 10, найти порядок реакции декаборапа
с н-бутанолом по каждому компоненту.
12
1-13. Реакцию этерификации лауриновой кислоты лаурило-
вым спиртом изучали в среде продукта реакции, лауриллаурата
[8]. Кинетика этерификации приведена в табл. 11. Найти общий
порядок реакции.
Таблица 11
Кинетика этерификации лауриновой кислоты
(0.200М) лауриловым спиртом (0,200М).
Условия опыта: температура 195° С
Время, мин
0
15
30
60
120
180
240
300
360
420
480
555
600
660
780
900
1080
1320
1620
Доля превращении,-^%
0
6,20
8,50
18,5
33,1
40,7
44,7
49,7
53,6
57,0
59,7
61,6
62,9
64,6
66,9
69,2
71,6
74,1
76,6
Ответы и решения
1-1. Используя формулу (1.6) для определения порядка
реакции по двум начальным скоростям, соответствующим двум
различным начальным концентрациям реагента, получаем
_ log 12,1/4,06 _ 1
/Z(A) "" log6,38/2,15 ~~ '•
_ log 36,8/4,2 1
(В) log 29,4/3,36
Таким образом, реакция имеет первый порядок по обоим
реагентам.
1-2. Логарифмируя общее уравнение реакции, получаем
logv = logk + alog [N2H2] + blog [N2H5C1].
В координатах (log v, log [N2H4]) при постоянном значении
концентрации хлористого гидразония получаем прямолинейную за-
13
висимость (рис. 1), тангенс угла наклона которой равен а, и
отрезок, отсекаемый на оси ординат, равен log k-{-b log [N2H5CI].
Аналогично, в координатах (logo, log [N2H5CI]) при
постоянном значении концентрации гидразина получаем прямую (рис. 1)/
тангенс угла наклона которой равен Ъ, и отрезок на оси ординат
равен log к-\-а log [N2H4]. Используя вычисленные таким образом
величины порядков реакции по отдельным компонентам
(равные в обоих случаях единице), из значений отрезков на осях
ординат можно найти величину константы скорости реакции, равную
1,23-10 5 л/моль-сек.
0,5 10 1,5-
log[NH] log[MHCLj
2 4J
0,7
0,6
0,5
0,4
0,3
0,2
0,1
->
-"
0
1
5
\
10
1 1
15 20
Время,мин
Рис. 1. Определение константы
скорости и порядка реакции термического
распада хлористого гидразония
Рис. 2. Графическое
дифференцирование кинетической кривой
обесцвечивания профлавина под
действием ультрафиолетового света
1-3. Графически дифференцируя кинетическую кривую (рис. 2),
можно найти значения скоростей реакции, соответствующие
определенным концентрациям красителя. Порядок
реакции,'определенный в координатах (log v, log D444), приблизительно равен единице.
Значение константы скорости реакции обесцвечивания,
определенное в координатах (In c0/c, t), равно 4,7- Ю-2 мин-1 (рис. 3).
Исходя из времени полупревращения реакции, равного приблизительно
15 мин, можно также найти значение константы скорости реакции,
равное 4,6- Ю-2 мин-1..
1-4. Значение константы скорости реакции, найденное
линеаризацией экспериментальных данных в координатах (In Со/с, t),
равно 0,017 сек-1.
1-5. Из графика в координатах (In Со/с, 0 можно найти, что
реакция расщепления диэтилового эфира имеет первый порядок по
изобутил-литию, с константой скорости, равной 3,06-Ю-5 сек-1.
14
1-6. Линеаризацией данных табл. 6 в координатах первого
порядка (рис. 4) находим величину константы скорости реакции.
Ответ: реакция денатурации ДНК протекает по первому
порядку; константа скорости реакции равна 0,103 мин-1.
10 15 20
Время,мин
Рис. 3. Определение константы
скорости первого порядка
обесцвечивания профлавина
10 15 20
Время,мин
Рис. 4. Определение константы
скорости денатурации ДНК в
водном растворе формальдегида
1-7. Ответ: реакция первого
тивации равна 0,15 мин-1.
1-8. Порядок реакции по
одной кинетической кривой
удобно находить с помощью
формулы (1.10). Так, для
/i=l час а.=0,93. Тогда
02=0,865 и /2=2 часа.
Окончательно получаем
порядка; константа скорости инак-
и= 1
log 1
log 0,93
= 1.
Для проверки следует
взять еще одну точку на
кинетической кривой, например
*i = 2I час, Gi=0,42. В этом
случае а2=0,177 и £2=46
часов,
'0,46
1,23.
п
1
log 0,42
Таким образом, порядок
реакции денатурации
приблизительно равен единице.
60 100
Время,сек
Рис. 5. Определение порядка реакции
распада двуокиси азота с
использованием одной кинетической кривой
15
1-9. а) Выберем на кинетической кривой (рис. 5) две точки,
удовлетворяющие условию а2=а2ь где а.\ и а2 — доли пепрореаги-
ровавшего вещества. Этому условию удовлетворяют, например,
точки (*i=20 сек, ai = 0,595) и (f2=50 сек, 02=0,354). Порядок
реакции можно определить из формулы (1.10):
„--\ !£I_Li_==i8
log 0,595
б) Графически дифференцируя кинетическую кривую (см.
рис. 5), можно найти значения скоростей реакции,
соответствующие определенным концентрациям исходного вещества.
Линеаризуя полученные значения в координатах (logy, log с), получим, чт©
тангенс угла наклона прямой, численно равный порядку реакции,
равен двум.
Ответ: порядок реакции равен 2; уравнение реакции,
следовательно, можно записать в виде
2N02 - 2NO + 02.
1-10. Используя формулу (1.20), найдем, что порядок
реакции равен трем.
1-М. Применяя формулу (1.20), получим м=1,46.
1-12. Реакция имеет первый порядок как по декаборану, так
и по н-бутанолу.
1-13. Используя формулу (1.10), можно найти, что порядок
реакции равен трем.
ЛИТЕРАТУРА
1. Эмануэль Н. Ж., Кнорре Д. Г. Курс химической кинетики. М., „Вые.
шая школа", 1962, стр. 182.
2. R о d r i g u e z Н., С h u a q u i С, A I a 1 a S., M a r q u e z A. .Tetrahedron",
27, 2425, 1971.
3. Рубцов Ю. И., Ma не лис Г. Б. ЖФХ, 46, 627, 1972.
4. Барышников Ю. Н., Весиовская Г. И., Мушкаринова В. Н.
ДАН СССР, 205, 1353, 1972.
5. U tly am a H., D о I у P. „Biochemistry", 10, 1254, 1971.
6. Haurowltz F., Hardin R. L., Dicks M. „J. Phys. Chem.", 58,103, 1954.
7. Beacheli H. C, Meeker T. R. .J. Amer. Chem. Soc", 78, 1796, 1956.
8. H a m a n n S. D., Solomon D. H., Swift J. D. „J. Macromol. Sci.", A2(l),
153, 1968.
Глава 2
ОПРЕДЕЛЕНИЕ КОНСТАНТ СКОРОСТЕЙ
РЕАКЦИИ ИЗ ЭКСПЕРИМЕНТАЛЬНЫХ
ДАННЫХ
§ 1. Общие методы обработки
кинетических кривых
1. В случае односторонней реакции первого порядка
дифференциальное уравнение, описывающее зависимость скорости
химической реакции от концентрации реагента, имеет вид
—U-kc. (2.1)
Интегрирование уравнения (2.1) при начальном условии с = с0
при £=0 приводит к соотношению
с = c0e-kt (2.2)
или
In с- lnc0 — kt. (2.3)
Из выражения (2.3) следует, что для реакций первого порядка
график в координатах (In с, t) имеет вид прямой линии, тангенс
угла наклона которой равен —к (так называемая
полулогарифмическая анаморфоза). Аналогично график в координатах (\nc0/c,t)
имеет вид прямой линии, проходящей через начало координат
с тангенсом угла наклона, равным к.
а) Если в распоряжении экспериментатора имеется
кинетическая кривая реакции первого порядка, начальный период которой
не был зафиксирован, то константу скорости реакции удобно
находить графически из следующих выражений:
1п([Р|м-[Р1) = 1п[Р]00-^1 (2.4)
[р1-
~[Р] — [pi " С---"3)
где [Р]—концентрация продукта реакции в момент времени t;
[Р]оо — концентрация продукта после завершения реакции.
Наконец, константу скорости реакции первого порядка часто
приблизительно рассчитывают, используя формулы (2.6) или (2.7),
вытекающие из соотношений (2.2) и (2.5) соответственно:
£ = ^log-^. (2.6)
, 2,303 , 1Р]„ ,0 „.
* = -г-1ое-р]—[рГ (2-7)
Из выражения (2.2) — (2.7) видно, что в случае реакций первого
порядка абсолютная величина константы скорости не зависит от
17
того, в каких единицах выражаются концентрации реагентов или
продуктов реакции. Поэтому для расчета константы скорости
реакции можно использовать любые физические величины,
пропорциональные концентрациям (оптическая плотность, угол вращения
плоскости поляризации света, объем добавленной в реакционный
сосуд щелочи или кислоты и т. д.).
б) Время, в течение которого реакция первого порядка
пройдет на 50% (время полупревращения), определяется соотношением
, In 2 0,693 ,9R.
Отсюда, зная время полупревращения реакции, можно легко
рассчитать численное значение константы скорости реакции.
2. Так как уравнение скорости реакции п-го порядка можно
записать в виде
v = kcn, (2.9)
то
log v = log k + п log с. (2.10)
Отсюда график в координатах (logw, log с) должен иметь вид
прямой линии, из тангенса угла наклона которой можно определить
порядок реакции, а из точки пересечения с осью ординат —
значение константы скорости реакции. Значения скоростей реакции
при разных концентрациях субстрата можно найти графическим
дифференцированием кинетической кривой (см. рис. 2).
3. Для реакции второго порядка
k
А + В —» продукты (2.П)
кинетическое уравнение можно записать в виде
log [$Ещ = log Щ>0 + 0.434 k ([А]0 - (В]0) t, (2.12)
где [Р] — концентрация продукта реакции. Отсюда для реакции
Л [А1о—Р] Л
второго порядка зависимость в координатах (*°g mi _ г pi * w имеет
вид прямой линии, из угла наклона которой можно найти
значение константы скорости реакции.
а) При эквимолярном соотношении реагентов ([А]0=[В]0=
= Со) кинетическое уравнение реакции второго порядка
приобретает вид
— = — + kt. (2.13)
Отсюда тангенс угла наклона зависимости (2.13) в координатах
(1/с, t) численно равен k.
б) В реакции второго порядка при эквимолекулярном
соотношении реагентов время полупревращения определяется
выражением
*!„-£. (2.14)
18
Таким образом, для реакции второго порядка в отличие от реакции
первого порядка при определении константы скорости реакции из
времени полупревращения необходимо знать начальную
концентрацию реагента (или реагентов).
4. Реакции третьего порядка типа
к
А + В + С — продукты (2.15)
встречаются чрезвычайно редко, и кинетическое уравнение их
может быть записано в следующем виде [1]:
in W 1д]° \«в1в~'С1оУ Е!» V,c,0—IA,0> v
\\ [А].-[Р] ) Ub]o-[P] ) Х
Х ( [С]|С1°[Р1 У'"'0"16'0'} = k ([А]0 - [ВЫ ([Во] - [С]0) ([С]0 - [ А]0) t.
(2.16)
а) В случае, если начальные концентрации двух веществ в
реакции (2.15) совпадают, кинетическое уравнение записывается
в виде
■*P- = k ([А]0 - [Р]) ([В]0 - [P]f (2.17)
или
, 1„ Ио [В]о — [А]о /с, 10Л
+ 1п[аь-—Ш>—• (2Л8)
В этом случае из тангенса угла наклона прямой в кординатах
/1п [В1.-РЧ [В].-[А]. \
\ [А]0-[Р] [В],-[Р] ' Г
равного k([A]0—[В]о)2, можно найти численное значение
константы скорости реакции третьего порядка.
б) Если концентрации всех трех реагентов в реакции (2.15)
равны друг другу ([Л]о=|[£]о= [С]0=с0), то кинетическое
уравнение можно записать в виде
l/c2=\/cl + 2kt. (2.19)
Очевидно, что тангенс угла наклона зависимости (2.19) в
координатах (1/2 с2, t) численно равен k.
с) В реакции третьего порядка при эквимолекулярном
соотношении реагентов время полупревращения равно
'"2="г--^г- <2-20>
Отсюда, определив экспериментально время полупревращения
реагента в реакции третьего порядка и зная начальную концентрацию
реагента, из формулы (2.20) можно вычислить константу скорости
реакции.
19
§ 2. Специальные методы обработки
кинетических кривых
Нередко встречаются такие условия проведения эксперимента,
в которых нельзя зафиксировать ни начало, ни конец кинетической
кривой. Это обычно равносильно тому, что в ходе реакции
неизвестны текущие концентрации реагента или продукта реакции, или
неизвестно начальное время реакции, что делает невозможным
применение для кинетических расчетов уравнений предыдущего
раздела. В подобных случаях используются специальные методы
нахождения констант скоростей реакций.
Нахождение констант скоростей первого
(псевдопервого) порядка
1. Метод Гуггенгейма [2]. Если кинетика реакции
регистрируется по изменению подходящего физического свойства системы Ф
(оптическая плотность, электропроводность, количество
добавленного титранта и т. д.), то уравнение (2.2) может быть записано
в виде
_(Ф,-Фсо) = (Ф0 —Фсо)е-«. (2.21)
Если зафиксировать времена реакции ti и ^i+А, где А —
произвольный интервал времени, то на основании выражения (2.21)
можно записать следующие уравнения:
(Ф! — Фсо) = (Ф0 - Фсо) е~*Ч (2.22)
(Ф1 - Ф~) - (Ф0 - Фот) е~" <«•+»>. (2.23)
Здесь Ф; и Ф\ — физические свойства системы во времени tx
и t\ = tx -f Д соответственно (рис. 6). Подобные соотношения можно
записать также для t2 и t'2 = t2 + А И т. д. Вычитая (2.23) из (2.22),
получим
ф, - ф', = (Ф0 - Фи) е-»« (1 - е-«). (2.24)
Для времени t2 и t'2 = t2 + Д аналогично получим
ф2 _ ф'2 = (ф0 — фм) е-«» (1 — е-*4) (2.25)
или в общем случае при Д = const
Ф. — Ф;=Ае-«/, (2.26)
где постоянная величина А равна
А = (Ф0 - Фсо) (1 - е~*д). (2.27)
Логарифмируя выражение (2.26), получим
In (Ф; — Ф'.) -= const — ktt. (2.28)
20
Из уравнения (2.28) видно, что для реакций первого порядка
график в координатах [In (Ф, — Ф,), tt] будет иметь вид прямой
линии, тангенс угла наклона которой равен — k.
В оптимальном случае интервал Д должен соответствовать
двум-трем временам полупревращения исследуемой реакции. Для
практического выбора величины Д изучаемую кинетическую
кривую удобно разделить на две части, ф
каждая из которых содержит
одинаковое число измерений Ф и
определить А как временной интервал
между первыми измерениями в
каждой группе (см. рис. 6).
Очевидно, что в этом случае каждому
значению Ф первой группы
соответствует значение Ф' второй
группы и ни одно измерение не
используется в расчетах дважды.
2. Метод Мангельсдорфа [3].
Мангельсдорф также предложил
метод нахождения констант
скоростей первого порядка, основываясь
на уравнениях типа (2.22), (2.23).
Если разделить попарно левые и
правые части этих уравнений, то
новое уравнение, в
Время
Рис. 6. Обработка
кинетической кривой первого порядка
согласно методу Гуггенгейма.
Ф-подходящее физическое
свойство системы
мы получим
(ф0_фоо)е-«.
или
котором отсутствует член
(Ф;-Фоо) = (Ф,-Фсо)е
-ы
(2.29)
Ф
Ф^ + В,
(2.30)
где постоянная В равна Фте(1 — е-*4). Дая второй пары точек,
разделенных отрезком времени Л, мэжнэ также записать
ф; = Ф2е-^4-В (2.31)
или в общем случае
ф; = ф|в-м + В. (2.32)
Из полученного уравнения видно, что график в координатах (Ф/, Ф/)
должен иметь вид прямой линии, тангенс угла наклона которой
равен е~
-ьь.
и отрезок, отсекаемый прямой на оси ординат, равен
Ф=»(1 — е-*4). Из уравнения (2.32) также видно, что точка
пересечения прямой в координатах (ф|, Ф^) с прямой линией Ф/ = Ф/
будет иметь координаты (Фоо, Фоо).
Выбор величины Д при обработке кинетических кривых
первого порядка методом Мангельсдорфа производится таким же
образом, как и в методе Гуггенгейма. Статистический анализ
показывает [3], что методы Гуггенгейма и Мангельсдорфа дают опти-
21
мальные результаты при обработке кинетических кривых,
полученных на протяжении приблизительно трех-четырех времен
полупревращения реакции.
3. Для обработки кривых первого порядка существует
вариант метода Гуггенгейма, который заключается в следующем. Если
зафиксировать условное начало отсчета на кинетической кривой
и принять шаг времени постоянным, равным А, то приращение
переменного физического свойства системы Ф (рис. 7) на n-ном шаге
будет равно
Ф,,+в4 - Ф,,+ (в_1) л = (Ф« - Ф0) (1 - е~* <"+"А> - 1 + е- " с+ (»- D д>)
ИЛИ
Ф', + „л -Ф^(„_1)д = (Фм-Ф0)е-«'-е-й("-1'д(1~е-Л- (2-33)
Логарифмируя уравнение (2.33), получаем
1п(Ф</ + яЛ — Фц +(„-1) л) = const — k (л — 1)Д. (2.34)
Таким образом, графике координатах [1п(Ф«/+/гд —Ф^+(П-1)д. {и— 1)А]
для реакции первого порядка должен быть прямолинейным с
тангенсом угла наклона, равным —k.
Нахождение констант скоростей второго
порядка
Метод .нахождения констант скоростей второго порядка из
одной кинетической кривой, начало и конец которой не
зафиксированы, был предложен Розвери [2]. Этот метод распространяется на
Ф,
-/!д| i
/i Н i
f f+пд Время
Рис. 7. Обработка кинетической
кривой первого порядка с
помощью модифицированного метода
Гуггенгейма
t t t Время
Рис. 8. Обработка
кинетической кривой второго порядка
методом Розвери
кинетические уравнения типа (2.35) или (2.36) при условии
эквивалентности концентраций реагентов [А] и [В] во втором случае:
v = k [A]2, (2.35)
<у = £[А]-[В]. (2.36)
22
Согласно методу Розвери, константа скорости'реакции второго
порядка определяется выражением
и [(Ф2-Ф.)-№-Ф2)]2
2Д (Фг-Ф,) (Ф„—Ф.) (Ф,—Ф„)
(2.37)
где Фь Фг и Фз — значения подходящего физического свойства
системы, по изменению которого производится регистрация кинетики
реакции при временах t\, t% и t3, разделенных постоянным
интервалом времени Д (рис. 8).
Следует отметить, что для расчета констант скоростей второго
порядка, в отличие от констант скоростей первого порядка,
необходимо знать связь между регистрируемым физическим
параметром Ф и абсолютными концентрациями реагентов (или
продуктов). В противном случае константа скорости реакции k будет
иметь размерность Ф/сек.
Задачи
2-1. Обесцвечивание красителя профлавина (3,6-диаминоак-
ридина) под действием ультразвука (частота 880 кгц,
интенсивность 2 вт/см2) подчиняется кинетике первого порядка (табл. 1).
Определить значение константы скорости реакции.
Таблица 1
Кинетика обесцвечивания профлавина
под действием ультразвука. Условия опыта:
15° С; концентрация профлавина 1,87-10_6М
Время, мин
0
10
20
30
40
50
60
80
100
120
140
160
180
240
Оптическая плотность
раствора (Л=444 нм)
0,635
0,525
0,460
0,410
0,365
0,325
0,295
0,240
0,190
0,160
0,150
0,140
0,130
0,125
2-2. За процессом денатурации пепсина следили при помощи
калориметрического метода (табл. 2). Используя данные
таблицы, найти константу скорости денатурации, если известно, что по-
23
Таблица 2
Кинетика денатурации пепсина. Условия опыта: 15е С, рН 6,9
(фосфатный буфер); концентрация фермента 5 мг/мл
Время,
. мин
19
20
21
22
23
24
25
26
27
28
Начальная точка отсчета
(базовая линия), усл. ед*
3,23
3,17
3,32
3,26
3,30
3,34
3,34
3,33
3.23
3,40
'Конечная точка отсчета,
соответствующая
окончанию реакции, усл. ед. *
8,19
8,11
8,18
8,14
8,18
8,18
8,14
8,14
8,10
8,19
Текущие значения
поглощения тепла, усл. ед.
4,14
4,32
4,57
4,75
4,92
5,09
5,25
5,40
5,52
5,еб
* Без поправки на дрейф.
глощение тепла при инактивации пепсина следует кинетике
первого порядка [4].
2-3. Обмен 5-Н-пиримидинового кольца на дейтерий при
обработке 2/,3'-0-изопропилиденуридина смесью CPbONa и CH3OD
проходит по кинетике первого порядка, со временем
полупревращения 2,84 часа [5]. Найти константу скорости реакции.
HOFLC о
2-4. Кинетику кислотного гидролиза снмм-ди(2-карбокснфе-
нокси)-диметилового эфира регистрировали спектрофотометриче-
ски по выделению метилсалицилата (табл. 3). Найти константу
скорости первого порядка реакции гидролиза.
2-5. В таблице 4 приведены результаты кинетического
изучения гидролиза н-нитрофекилацетата, катализируемого имидазолом
[7]. Найти константу скорости второго порядка реакции
гидролиза, если каталитически активной формой является нейтральная
молекула имидазола, и рКа имидазола в водном растворе
равно 7,23.
24
Таблица 3
Кинетика гидролиза
симм-ди(2-карбоксифенокси)-диметилового эфира [6].
Условия опыта: рН 3,49; температура 30" С;
ионная сила 0,1; концентрация эфира 1,0-10_4М
Время, дни
0
0,4
0.8
1.8
2,9
4,0
4,6
5,8
6,7
7.5
8,6
9,6
11.7
15,5
18,4
22,7
25,5
29,6
Оптическая плотность
(Х=300 нм)
0,129
0,137
0,141
0,154
0,162
0,171
0,181
0,187
0,200
0,204
0,213
0,221
0,229
0,245
0,258
0,267
0,275
0,280
Таблица 4
Кинетика гидролиза /г-нитрофенилацетата,
катализируемого имидазолом. Условия опыта: рН 6,20;
температура 20° С; концентрация п-нитрофенилацетата
1,8-J0-4M; концентрация имидазола 0.569М;
ионная сила 0,15М(КС1)
10
20
30
40
60
80
100
120
140
оо
Оптическая плотность продукта
реакции и-нитрофенола (Х=348 нм)
0,23
0,42
0,58
0,72
0,93
1.08
1,18
1,26
1,31
1,44
2-6. Реакция между 2-фенил-4,4-диметил-2-оксазолил-5-оном и
этиловым эфиром DL-аланина в четыреххлористом углероде имеет
первый порядок по каждому из реагентов [8]. Найти константу
скорости реакции, используя кинетические данные образования
25
продукта реакции, этилового эфира N- (N'-бензоил-а-аминоизобути-
рил)-БЬ-аланина (табл.5).
О • О °jH*
^-/ У-'х У +NH2—CH—СООС2Н6—>
СН3
сн3 сн3
*# Ч—С—NH—С — С—NH—СН—СООС2Н5
х=/ II I II
О СН, О
Таблица 5
Кинетика образования этилового эфира
М-(1Ч'-бензоил-а-аминоизобутирил)-1)1,-аланина
из оксазолона и этилового эфира аланина.
Условия опыта: 20е С; концентрации оксазолона
и этилового эфира аланина равны 9,96- 103М
и 2,22-10-2М соответственно
Время, сек
700
1700
2700
3700
4700
5700
6700
7700
8700
9700
Концентрация продукта
реакции, моль/л-Ю3
1,59
3,30
4,55
5,52
6,27
6,88
7,36
7,76
8,10
8,36
2-7. Реакция этерификации лауриковой кислоты лауриловым
спиртом имеет общий третий порядок [9]. Используя данные
табл. 6, определить константу скорости реакции.
2-8. Реакция между динитрофторбензолом и бензимидазолом
в водном растворе протекает по следующей схеме:
Н
N
/ \/\ /
OaN— <^ Ч>— F+/ | J —> OaN—<^ ^>—N N+HF
NO, N NO. ^
\
Кинетику этой реакции регистрировали по увеличению оптической
плотности продукта при длине волны 330 нм в условиях, когда
концентрация одного реагента значительно превышала концентрацию
26
Таблица 6
Кинетика этерификации лауриновой кислоты
лауриловым спиртом в лауриллаурате. Условия опыта:
температура 163° С; концентрация лауриновой кислоты
0,200М; концентрация лаурилового спирта 0.200М
Время, мин
0
30
60
120
180
240
300
360
420
480
540
600
720
840
960
1С80
1200
1380
1560
Степень протекания реакции,
0
5,48
9,82
18,1
23,8
27,1
32,4
35,2
38,0
40,7
42,3
44,6
48,0
50,1
53,2
54,8
57,5
59,8
61,7
%
Таблица 7
Кинетика реакции
динитрофторбензола
с бензимидазолом. Условия опыта:
концентрация динитрофторбензола
5-Ю-4 М; концентрация
бензимидазола 7,5-Ю-3 М
второго (табл. 7). Определить константу скорости реакции
псевдопервого порядка, используя метод Гуггенгейма.
2-9. Кислотно-катализируемый гидролиз метилового эфира
2,4-дихлорфенил-1М-метилфосфорамида имеет первый порядок по
эфиру [Ю]. Определить
константу скорости реакции гидролиза,
используя данные табл. 8.
2-10. Этерификация
лауриновой кислоты лауриловым
спиртом, катализируемая п-толуол-
сульфоновой кислотой, имеет
первый порядок как по кислоте,
так и по спирту [9]. Исходя из
данных табл. 9, рассчитать
константу скорости реакции.
2-11. При изучении кинетики
реакции в четыреххлористом
углероде между 2-фенил-4,4-диме-
тил-2-оксазолин-5-оном (А) и
этиловым эфиром DL-аланина (В),
катализируемой уксусной
кислотой, было найдено, что константа
скорости реакции второго порядка
Время, мин
1
2
3
4
5
6
7
8
■^330
0,005
0,220
0,400
0,550
0,660
0,750
0,815
0,860
27
уменьшается при увеличении концентрации сложного эфира
(табл. 10). При дальнейшем изучении реакционной системы нашлн,
что уксусная кислота в среде четыреххлористого углерода образует
Таблица 8
Кинетика гидролиза метилового
эфира 2,4-дихлорфенил-М-метилфос-
форамида. Условия опыта: 25%
диоксана; 0,19 N НС1; 25° С
Время, мин
8
10
12
14
16
18
20
22
24
26
28
30
32
34
36
п п*
t (/+10 мин)
8.3
7,5
7,0
6,1
5,8
5,2
4,8
4,2
4,1
3.8
3,4
3,0
2,8
2,6
2,5
* Разность оптических плотностей
(в усл. ед.).
Таблица 10
Влияние концентрации этилового эфира DL-аланина
на кинетику реакции А с В, катализируемой уксусной
кислотой. Условия опыта: концентрация А равна
1-10-2М; концентрация уксусной кислоты 16,85-10-4М
Концентрация этилового эфи-
jpa DL-аланина, моль/л-10'
1.463
1,679
1,811
2,221
2,940
3,079
Значение константы скорости
реакции ft, л/моль сек-103
14,81
13,61
13,23
11,65
10,04
9,33
свои димеры с константой равновесия /G = 2252 М-1, а также
ассоциируется с этиловым эфиром DL-аланина (с образованием ионной
пары) с константой равновесия 7(2=228 М-1. Наконец, было
найдено, что скорость изучаемой реакции описывается соотношением
V— (k0+k'[M]) [А] [В], где [М]—концентрация мономерной
формы уксусной кислоты в СС14, k0 — константа скорости иекатализи-
Таблица 9
Кинетика этерификации лауриновой
кислоты (0.200М) лауриловым
спиртом (0.200М) в лауриллаурате
(0.800М), катализируемой
я-толуолсулвфоновой кислотой
(0,004М). Условия опыта:
температура 163° С
Время, мин
0
5
10
15
20
25
35
44
50
60
70
Доля превращения, %
0
36,7
59.7
69,4
74,3
78,0
83,5
86,5
88,0
89,8
90,9
28
руемой реакции, k' — константа скорости катализируемой .реакции
[8]. Пользуясь данными табл. 10 и значениями констант
равновесия К\ и Кь определить значения констант скоростей k0 и k'.
Ответы и решения
2-1. Из рис. 9 следует, что глубина реакции обесцвечивания
профлавина ограничена значением .D444~0,I25 и не падает до нуля
в конце озвучивания. Поэтому для расчета константы скорости
100 150
Время, мин
Рис. 9. Кинетическая кривая
обесцвечивания профлавина под
действием ультразвука
50 ЮС 15С
Время.мим
Рис. 10. Определение
константы скорости первого порядка
обесцвечивания профлавина под
действием ультразвука
реакции удобно использовать выражение (2.5) и линеаризовать
данные табл. 1 в координатах Пп^ _£>, /I (рис. 10).
Ответ: /г=2,14-10-2 мин-1.
2-2. Применительно к данным табл. 2 выражение (2.2)
можно записать в виде
/■с*, — г = (/%» — r0)e~kt,
где Го — начальная точка отсчета (базовая линия) при измерениях
поглощения тепла калориметрическим методом; г<„ — конечная
точка отсчета; г — текущие значения поглощения тепла; k —
константа скорости реакции денатурации. Тогда, линеаризуя данные
можно найти значение кон-
табл. 2 в координатах ( 'n r _г . Ч,
станты скорости реакции.
Ответ: k—0,051 мин-1.
2-3. Используя формулу (2.8), найдем &=6,8-10~~5 сек-1.
29
2-4. Кинетическую кривую изучаемой реакции удобно
обрабатывать методом Гуггенгейма. Для этого разделим кривую
условно на две части, соответствующие интервалу Д, равному 16
дней, так что первыми измерениями в каждой группе точек будут
являться £=0 и £'=16 дней. Далее, логарифмируя разности
значений оптических плотностей, соответствующих парам значений
времени, разделенных интервалом Д, и откладывая полученные
величины в координатах уравнения (2.28), найдем величину k
(рис. И).
Ответ: £=0,0075 дней-'=8,7 • 10~7 сек-1.
2-5. Линеаризуя данные табл. 4 в координатах
\ [Р]„-[Р]' /
находим значение константы скорости реакции псевдопервого
порядка, fe1=l,73-I0~2 сек-1. Для нахождения концентрации
нейтральной формы имидазола в условиях эксперимента (рН 6,20)
воспользуемся соотношениями
Im + H+- - ImH+. / (2.38)
[1т]общ = [1т] + [1тН+], (2.39)
= рт1[Н+] 2 0
А" [Im H+] ' ^ V)
из которых находим
fIml = -E^j-. (2.41)
Из условия задачи [1т]общ = 0,569 М, Ка = 5,89-10~8 М, [Н+] =
= 6,3-10-7М. Из формулы (2.41) находим [1т] = 4,86- 10~2М.
Из соотношения
k> = k" [Im],
где k1 — константа скорости реакции псевдопервого порядка
реакции гидролиза, катализируемого имидазолом; kn — константа
скорости второго порядка, находим &11|:=0,356 л/м-сек.
2-6. Интегрируя дифференциальное кинетическое уравнение
для скорости реакции второго порядка
*/[Р]/Л = *((А]0-[Р])([В]0-[Р]),
где [А] о и [В] о — начальные концентрации реагентов,
[Р]—концентрация продукта реакции в момент времени t, получаем
следующее выражение:
Ы - 2'303 log [в1о([А1о-[р1) (2 42)
М~ [A1„-[B1. 10g[A]0([B]0-[P])- VAZ>
30
Учитывая, что в нашем случае [А]0— [В]0=1,22-10-2 М, запишем
выражение (2.42) в виде
ДВ]„-([А]0-[Р])
! ifjo-u"jo-f-j; a=S3- 1П_з.ы
(2.43)
Далее с помощью линеаризации экспериментальных данных в
координатах уравнения (2.43) (рис. 12) находим численное значение
константы скорости реакции.
Ответ: £=1,13-Ю-2 л/м-сек.
2.0
1.6
<=Г
"сП?
с
*
**
0.8
l i i
5 Ю 15
Время.дни
Рис. 11. Определение
константы скорости реакции первого
порядка с помощью метода
Гуггенгейма
2
,'.-°
1—1
э
0,6
Q4
0,2
,\
/ 1
/ 1
/i i i i i
2 4 б 8 10
Время, 10\сек
Рис. 12. Определение константы
скорости реакции второго порядка
2-7. Для решения задачи запишем >равнение (2.19) в виде
(С.-Р?- d +^f'
(2.44)
где Со — начальная концентрация реагентов, Р — концентрация
продукта реакции. Уравнение (2.44) легко преобразовать к виду
= 1 + 2сШ.
Константу скорости реакции третьего порядка можно найти из
тангенса угла наклона прямой в координатах [(1/1—a)2, t], где
а=Р/с0 —степень протекания реакции (рис. 13).
Ответ: ft=4,71 • 10~2 л2/м2-мин.
2-8. Используя уравнение (2.34), запишем выражение для
линеаризации экспериментальных данных
In (Д£>„, „_!) = const — k (n — 1) Д.
31
Принимая в нашем случае Д = 1 мин, построим табл. 11 для
применения модифицированного метода Гуггенгейма
Таблица 11
ri
1
•1
3
4
5
6
7
й Dn, п-1
0,215
0,180
0,150
0,110
0,090
0,065
0,045
/ j
4+1п(ДОя„_1)
2,463
2,285
2, ЮЗ
1,793
1,592
1,267
0,899
Из рис. 14 видно, что зависимость в координатах [In (Л/?п, n-i),
(п—1)Л] является прямолинейной с тангенсом угла наклона,
равным 0,26 мин-1.
Ответ: £=0,26 мин-1.
2-9. Решение аналогично
решению задачи 2-8.
400
В00 1200 W00
Время,мин
Рис. 13. Определение константы
скорости реакции третьего порядка эте-
рифнкации лауриновой кислоты лау-
риловьш спиртом
Ответ: /г=4,35-10-2
МИН"
/23456
Рис. 14. Определение константы
скорости реакции псевдопервого
порядка с помощью
модифицированного метода Гуггенгейма
2-10. Исходя из того, что доля превращения в реакции равна
отношению [Р] /со, где [Р] — концентрация продукта реакции, с0 —
начальная концентрация реагента, перестроим табл. 9 в виде
табл. 12.
Для нахождения константы скорости реакции "второго порядка
воспользуемся методом Розвери
а) [РЬ = 0,109; [Р|2 = 0,167; [Р]3 = 0,180; Д = 25 мин;
k = 0,76 л/м-мин;
б) |Р],= 0,073; |Р]2 = 0,149; [Р]3 = 0,167; А = 15 мин;
k = 0,77 л/м-мин;
32
Таблица 12
Бремя, мин
0
5
10
15
20
25
35
44
50
60
70
Концентрация цродукта
реакции, М
0
0,0734
0,109
0,139
0,149
0,156
0,167
0,173
0,176
0,180
0,192
в) [РЬ =0,149; [PJ2 = 0,167; (Pj3 = 0,176; А -15 мин;
к = 0,д2 л/м-мин.
Ответ: №етя« = 0,72 л/м-мин.
2-11. На основании условия задачи запишем
Кг
[D]
и Ко
[ИП].(1+/С,-[М])
где [D] — концентрация димерной формы уксусной кислоты;
[ИП]—концентрация ионных пар между мономерпой формой
уксусной кислоты [М]; [В] о — начальная концентрация этилового
эфира аланина. Подставляя выражения для [D] и [ИП] в
уравнение материального баланса
[АсОН] = 2 [DJ + (ИП] + [М],
получим кубическое уравнение относительно концентрации
мономерной формы уксусной кислоты [АсОН]:
2Ki - К* г»*is . 1+K.-IB1, —K.IAcOH] 1ЖЙ1 [АсОН]
Ш13 I ffM-^Ks Гд/[12 _i_ l+Kg-lBlo — Кг {АсОН] m
Iм! +-2КЖГ lMJ + 2кЖ, [
Ж Ж,
= 0.
Решая полученное уравнение обычным путем [11], получаем
таблицу значений концентраций мономера [М], соответствующих
начальным концентрациям сложного эфира [В]0 (табл. 13).
Таблица 13
IBlo-10", мочь/л
1,463
1,679
1,811
2,221
2,940
3,079
[MJ-lO4, моль/т
3,02
2,86
2,75
2,44
2,03
1,97
/МО3, л/моль-сек
14,81
13,61
13,23
11,65
10,04
9,33
Заказ 158
33
В координатах (k, [M]) получаем прямолинейную
зависимость, тангенс угла наклона которой равен k', а отрезок,
отсекаемый на оси ординат, равен k0.
Ответ: А'=48,4 М^-сек"1; /г0=0.
ЛИТЕРАТУРА
1. Эмануэль Н. М., Кнорре Д. Г. Курс Химической кинетики. М.т
„Высшая школа", 1962, стр. 179.
2. Frost A. A., Pearson R. О. Kinetics and-Mechanism, 2-nd edn. N. Y.,
Wiley, 1961, p. 49.
3. Margerison D. In „Comprehensive Chemical Kinetics", ed. by Bamford
C. H. and Tipper С F. H. v. 1. Elsev. publ. сотр., Amsterdam, 1969, p. 343.
4. BuzzeN A., Sturtevant J. M. „J. Amer. Chem. Soc", 73, 2454, 1951.
5. Santi D. V., Brewer С F. „J. Amer. Chem. Soc", 90, 6236, 1968.
6. Dunn В. М., Bruice Т. С. „J. Amer. Chem. Soc", 93, 5725, 1971.
7. Martin С J., Oza N. В., Marini M. A. „Eur. J. Biochem.", 20, 276,
1971.
8. Rodriguez H., ChuaquiC, Atala S., Marguez A.
"Tetrahedron", 27, 2425, 1971.
9. Ham an n S. D.,. Solomon'D. H., Swift J. D. „J. Macromol. Sci.\
A2(l), 153, 1968.
10. Garrison A. W., Boozer С. Е. „J. Amer. Chem. Soc", 90, 3486, 1968.
11. Бронштейн И. Н., С е м е н д я е в К. А. Справочник по математике. М.,
„Наука", 1964, стр. 138.
Глава 3
ВЛИЯНИЕ рН НА СКОРОСТЬ ХИМИЧЕСКИХ
РЕАКЦИЙ
Влияние рН на скорость реакции в большинстве случаев
обусловлено двумя причинами. Во-первых, протон или гидроксильный
анион могут сами принимать непосредственное участие в реакции,
например:
А + Н+ ► продукты, (3.1)
или
В + ОН- продукты. (3.2)
В таком случае выражение для скоростей реакций можно
записать соответственно в виде
v~k[h]-[H+] (3.3)
или
v = k[B]-[OH-]. (3.4)
Из выражений (3.3) и (3.4) очевидно, что в зависимости от
типа реакции логарифм скорости реакции линейно уменьшается или
растет при увеличении рН с тангенсом угла наклона, численно
равным —1,0 (3.1) или +1,0 (3.2). В том случае, когда катализатором
реакции является ион водорода (гидроксония), такой катализ на-
34
зывают специфическим кислотным катализом (или в общем
случае специфическим катализом ионами лиония). Катализ при
помощи ионов гидроксила называют специфическим основным
катализом или в общем случае специфическим катализом лиат-ионами
(например, катализ ионами этилата в спиртовом растворе).
Помимо этого влияние рН на скорость реакции часто
выражается в том, что реакционная способность реагентов обычно
изменяется при отщеплении или присоединении протона.
Рассмотрим, например, реакцию, в которой нейтральная молекула А
взаимодействует с анионом В~
к
А + В- » продукты, (3.5)
Ка
В- + Н+ + ВН. (3.6)
В этом случае общая скорость реакции равна
г»=А[Л]-[В~].
Так как [В-НМ|1, тс ,^Ка™. (3.7)
Из уравнения материального баланса
[В]„-[В-] + [ВН|
находим
[вн] = [В]0-^И
и далее
[ВН] - [В1° . (3.8)
1 4- а
1 + [Н+]
Подставляя (3.8) в (3.7), получаем окончательное выражение для
скорости реакции
*=*,фф[А]. [В]0,
где
*9*Ф= Ка + [°H+J • ^'^
Из выражения (3.9) можно найти, что в кислой области рН,
где [Н+]^>/Сй, логарифм эффективной константы скорости реакции
равен
log /гэфф = log k - рКа + рН
и график в логарифмических координатах (logfe3(Mb рН) в кислой
области рН имеет вид прямой линии с тангенсом угла наклона
+ 1,0. В щелочной области рН, где выполняется соотношение
[Н+]<сКа, эффективная константа скорости реакции не зависит
от рН
logA8(M,-logfc. (3.10)
2* 35
, , "эфф1" 'J /о 1 1\
«эфф=« й \РЛЧ
Таким образом, рН-профиль константы скорости реакции в
логарифмических координатах в целом должен иметь сигмоидную
форму, и из точки пересечения касательных к обеим ветвям кривой
можно найти значение рКа диссоциации реагента (см., например,
рис. 15).
Одним из методов определения констант k и Ка (3.5—3.6)
является построение данных кинетического эксперимента в
координатах (1//гЭфф, [Н+]). Эта зависимость, вытекающая из (3.9),
должна иметь вид прямой, отсекающей на осях ординат и абсцисс
величины l/k и —Ка соответственно. Аналогично зависимость (3.9)
можно записать в виде
*эфф [Н+]
и анализировать в координатах (&Эфф, £эфф • [Н+]), где точка
пересечения прямой с осью ординат есть k, а тангенс угла наклона
прямой равен —l/Ка- В случае, если активной формой
реагирующего вещества является его кислотная форма
k
А + ВН ► продукты, (3.12)
B- + H+ZZZZ2BH, (3.13)
эффективная константа скорости равна
«эфф = k [Н+] • (3-14)
Из соотношения (3.14) видно, что значения k и Ка в этом случае
можно найти построением экспериментальных данных в
координатах (&Эфф, &эфф/[Н+]). Если реакция катализируется молекулами
веществ, способными при диссоциации давать протоны (кислотами
Бренстеда (3—12)), такой катализ называют общим кислотным
катализом. Если каталитическое действие на реакцию оказывают
молекулы веществ, способные присоединять протон (основания
Бренстеда (3—5), такой катализ называют общим основным
катализом.
В сложных реакциях, в которых принимают участие способные
к ионизации вещества, полное уравнение скорости должно
включать скорости реакции для каждой ионной формы реагента. При
вычислении доли реагента в данной ионной форме иногда удобно
пользоваться уравнением Хендерсона — Хассельбалха
,, г, , , [основание!
pH = pKa + \og [кислота/
или
РН =/>/<■„ +log-г^-,
где а — доля реагента в основной форме.
В общем случае в зависимости от механизма реакции и числа
ионогенных групп реагентов рН-профиль константы скорости реак-
36
ции может иметь различный вид — «колокола» или
«перевернутого колокола», иметь острый или широкий максимум, или иметь
ступенчатый вид. Теория подобных рН-зависимостей подробно
изложена в ряде монографий (например, [1—3]).
Задачи
3-1. Образование лактама при циклизации 1Ч-(о-аминофенокси-
ацетил)-глицилглицина проходит согласно следующей схеме [4]:
СН, О
О
S\/
\
С
NH +
С— N Н— СН,—CON Н -СН,—СООН-
-н+
V
сн2
о \
s\
-NH
/
С=0 -f г лицил-глицин
В таблице 1 приведена зависимость эффективной константы
скорости реакции первого порядка от рН. Вычислить значение рКа
анилиний-иона и истинное значение константы скорости
циклизации, не зависящее от рН среды.
Таблица 1
Влияние рН на константу скорости
внутримолекулярного аминолиза
1^-(о-амикофеноксиацетил)-глицнлглнцнна.
Условия опыта: температура 52° С; ионная сила
1,0М(КС1); значения констант скоростей являются
экстраполированными к нулевой концентрации
буферного раствора
рН
2,0
2,8
3,2
3,6
3,8
4,1
4,3
4,4
4,7
5,0
*эФф-105-сек '
15,50
14,50
13,60
12,10
9,60
6,50
5,04
4,18
2,70
1,42
37
3-2. При изучении реакции N-ацетилциклосерина (4-ацетамино-
3-изоксазолидона) с я-нитрофенилацетатом (3.15) было найдено,
что при избытке первого реагента ацилирование протекает
согласно псевдопервому порядку. В табл. 2 приведены результаты
исследования зависимости эффективной константы скорости реакции от
рН [5]. Найти значение истинной константы скорости второго
порядка реакции (3.15) и величину рКа N-ацетилциклосерина.
сн3
с=о
NH О
1 ^ k
1 +СНа-С-0-<^ ^>-N02
NH I ==
\ /
О
сн3
1
с=о
1
н
N—С—СН3+НО— <f Ч—N(
\ / I! Х==
О О
Таблица
(3.15)
рН-Зависимость константы скорости реакции
N-ацетилциклосерина с п-нитрофеннлацетатом.
Условия опыта: 28° С; 0,2М ацетатный
(рН 5,0—5,9) и 0,2М фосфатный (рН 6,4—7,0)
буфер. Концентрации N-ацетнлциклосерина
и п-нитрофенилацетата равны 5-10-2М и 2-10_6М
соответственно
рН
5,00
5,35
5,90
6,40
7,00
Лэфф-Ю'. сек '
1,45
2,50
4,33
5,33
6,00
3-3. Реакция гидролиза я-нитрофенилацетата, катализируемая
N-метилимидазолом, имеет первый порядок по каждому из
реагентов. Используя данные табл. 3, вычислить рКа N-метилимида-
38
зола и константу скорости второго порядка гидролиза, если
известно, что каталитической активностью обладает лишь непротони-
рованная форма N-метилимидазола [6].
Таблица 3
рН-Зависимость эффективной константы скорости
псевдопервого порядка реакции гидролиза
гс-нитрофенилацетата, катализируемого
N-метилимидазолом. Условия опыта: 20° С;
ионная сила 0,15 (КС1); концентрации
N-метилимидазола и и-нитрофенилацетата равны
0,4 Ш и 1,8-1 (ММ соответственно
рН
6,80
7,21
7,60
8,00
8,40
8,60
8,80
9,00
Лэфф-Ю2, сек '
0,678
1,47
2,94
5,26
7,44
8,71
8,57
9,14
3-4. Рацемизация L-валина (а-аминоизовалериановой
кислоты), катализируемая ионами гидроксила, протекает по следующей
схеме [71:
СН3 СН3 СН, СН3
\ / \ /
— г\-~
Ка
~* NH+-
NH3+
сн
I
СН—СООН.
СН
-СН—СОО
■+н+.
(3.16)
D-изомер
Используя данные табл. 4, определить величину рКа
карбоксильной группы L-валина при 135° С.
Таблица 4
рН-Зависимость эффективной константы скорости
рацемизации L-валина в начальный период
реакции при 135° С
Рн
0,48
0,89
1,01
1,44
1,78
2,20
3,51
Лэфф-Ю7, сек '
0,3
0,7
1,0
1,7
2,4
3,2
3,9
39
3-5. При изучении профиля рН-зависимости эффективной
константы скорости гидролиза метилового эфира тиолмуравьинои
кислоты
НгО+Н—С—SCH3-f-H+ -^ HCOOH+CH3SH-f-H+
о
авторами работы [8] было высказано предположение, что
простейшим механизмом реакции может являться параллельное
протекание кислотно-катализируемого и водного гидролиза тиолового
эфира. Проверить это предложение, исходя из данных табл. 5.
Таблица 5
Зависимость эффективной константы скорости
гидролиза метилтиолформата от кислотности
раствора. Условия опыта: 30° С; ионная сила
0,97М (LiCl)
- log [HCI]
*эфф-|0!'ынн"
0,013
0,110
0,208
0,314
0,411
0,509
0,615
0,712
0,848
1,01
1,11
1,26
1,30
1,47
1,61
1,81
2,01
2,21
2,41
2,51
2,61
2,80
3,00
4,47
3,70
2,88
2,36
1,94
1,59
1,34
1,12
0,791
0,569
0,476
0,383
0,330
0,270
0,198
0,135
0,095
0,067
0,051
0,045
0,043
0,033
0,029
3-6. Циклизация N-(о-карбоксифенил) мочевины в водном
растворе с образованием 2,4-дигидроксихиназолина может
проходить согласно одному из четырех вероятных механизмов [9]:
40
II
III
NH—C=0
"^e^— nh2
■ i
NH—C=0
H°\ ^— NH
NH—C=0
IV
"°\c^ NH
NH— C=0
На основании данных рН-зависимости реакции (табл. 6)
определить, по какому из механизмов (I—IV) проходит циклизация.
Таблица 6
Зависимость констант скоростей реакции
циклизации от рН. Условия опыта: 30° С;
ионная сила 1,0 М (КС1)
Рн*
йэфф-НЛ сек
13,70
13,01
12,61
11,34
410
85
34
1,7
* Все значения рН намного превышают рКа
карбоксильной группы N-(о-карбокснфенил)
мочевины.
•3-7. Реакция образования фенилозазона глюкозы при
взаимодействии фенилгидразона глюкозы с избытком фенилгидразина
41
в уксуснокислом буферном растворе протекает по механизму
общего кислотного катализа [10].
кр
Фенилгидразон.г|-СНзСООН +_ *
X
фенилгидразин
фенилозазон+СНзСОО-.
где X — промежуточное соединение; k — константа скорости
лимитирующей стадии реакции. Используя данные рН-зависимости
реакции (табл. 7), определить величину константы диссоциации
уксусной кислоты в условиях опыта.
Таблица 7
рН-Зависимость эффективной константы скорости реакции
образования фенилозазона из фенилгидразона глюкозы
и фенилгидразина в буферной системе
(CH3COOH+CH3COONa).
Условия опыта: 30° С; 50% водный этанол
рН
3,25
3,35
3,87
4,19
*3<b<b''°5' л/молЬ'Сек
4,33
4,00
3,25
2,65
рН
4,21
4,60
4,75
5.05
*эфф105' -'/«оль-сек
2,35
1,28
1,13
0,63
3-8. Кинетику гидролиза циклического эфира 2-(2-карбоксифе-
нил) -1,3,2-диоксафосфоринан-2-оксида регистрировали спектро-
фотометрически по образованию салициловой кислоты при длине
волны 298,5 нм (изобестическая точка для кислоты и ее аниона)
ОО О О
S\/ \|| ,0 ч ка ^\/ \| О
■ "' II р< >
р\
ч/\
О"
\
/-
соон
^/\
чг
+н+
COO-
IS. 17)
он
^\/
° о f\.
+но-<0^> + \ II
он
соон
ч/\
соо-
Было показано [11], что эта реакция имеет общий псевдопервыи
порядок, причем эффективная константа скорости реакции зависит
от рН среды (табл. 8). Найти значения констант скоростей k0 и k\,
если из независимых экспериментов было найдено, что величина
рКа карбоксильной группы циклического эфира равна 3,15.
42
рН-Зависимость константы скорости гидролиза
циклического эфира при 39° С и ионной силе
1,0М (КС1)
Таблица 8
рН
1.00
2.00
2.10
2,22
2,40
2,70
ЗЛО
4,08
5,30
*эФф-105- сек~'
4,29
5,12
5,23
5,52
5,84
7,02
8,75
12,90
13,50
3-9. рН-Зависимость эффективной константы скорости
гидролиза этилового эфира 2-окси-5-нитрофенилкарбоната приведена
в табл. 11 [14].
О О
II II
02N О—С—ОС2Н5 02N О—С—ОС2Н5
I
\^\/
4/V
он
%/\
о-
*0 |
о
02N ОН
\^\/ II
|| + НО—С—ОС2Н6 +
02N
+ Н+
к, (3.18)
ОН
%/\
он
I !
о-
Найти значение констант скоростей k0 и k\ и константы
диссоциации Ка-
Влияние рН на эффективную константу скорости гидролиза
этилового эфира 2-окси-5-нитрофенилкарбоната>
Условия опыта: 30° С; ионная сила 0,5М (КО)
Таблица 9
рН
1,0
2,0
2,6
2,9
3,6
4,0
*эфф ■ I05, сек *
0.16
0,16
0,16
0,16
0,25
0,44
рН
4,5
4.6
5,0
5,1
5,6
6,0
Йэфф-Ю5. сек-1
1.0
1,2
2,3
2,8
4,7
6,4
рН
6,2
6.6
7,0
7,6
8.0
Йэфф-ЮБ, сек-1
7,2
7,8
7,9
8,0
8,0
3-10. В таблице 10 приведена зависимость эффективной
константы скорости гидролиза фенилового эфира тиолмуравьинои
кислоты от рН [13].
н—с-
II
о
S—/ Ч —— ->• НС—ОН + HS— / Ч
о
На основании характера рН-зависимости предложить механизм
гидролиза и найти значения констант скоростей реакции
Таблица 16
рН-Зависимость гидролиза фенилтиолформата
при 25° С и ионной силе 1,0М(КС1)
рн
0,10
0,26
0,50
0,70
1,07
1,46
2,06
3,10
4,99
*эфф-10*, сек '
12,66
8,90
5,40
3,67
2,00
1,20
0,95
0,65
0,55
3-11. Гидролиз Ы-(п-нитрофенил)-2,2-диметилпропионогидра-
зонил бромида протекает по следующей схеме [12]:
СН.
i 8 ка
СН8—С — C=N—NH—/ Ч—NO, ; *
СН. Вг
сн8
:СН,-С- C=N— N— ^ Ч-NO, +
I I N /
Н +
сн8
СН„ —С—СО—NH—NH—^ Ч—NOa
СН3
Используя данные табл. 11, определить значения констант kQ,
h и Ка-
44
Таблица 11
рН-Зависимость констант скоростей псевдопервого
порядка реакции гидролиза гидразонил бромида.
Условия опыта: 25° С; 70% диоксан
рН
4,15
4,30
4,50
4,70
5,00
5,50
5,95
6,42
Йэфф-Ю3, сек '
3,52
3,69
4,04
4,51
6,02
11,0
22,0
38,0
3-12. Гидролиз 3,5-динитросалициловой кислоты протекает по
механизму внутримолекулярного общекислотного катализа
карбоксильной группой в кислой области рН и внутримолекулярного
катализа карбоксилат-анионом в нейтральной области рН [15]
О О
N02 || NO, ||
| О— С— СН, | О—С—СН,
f\/
/V\
N02
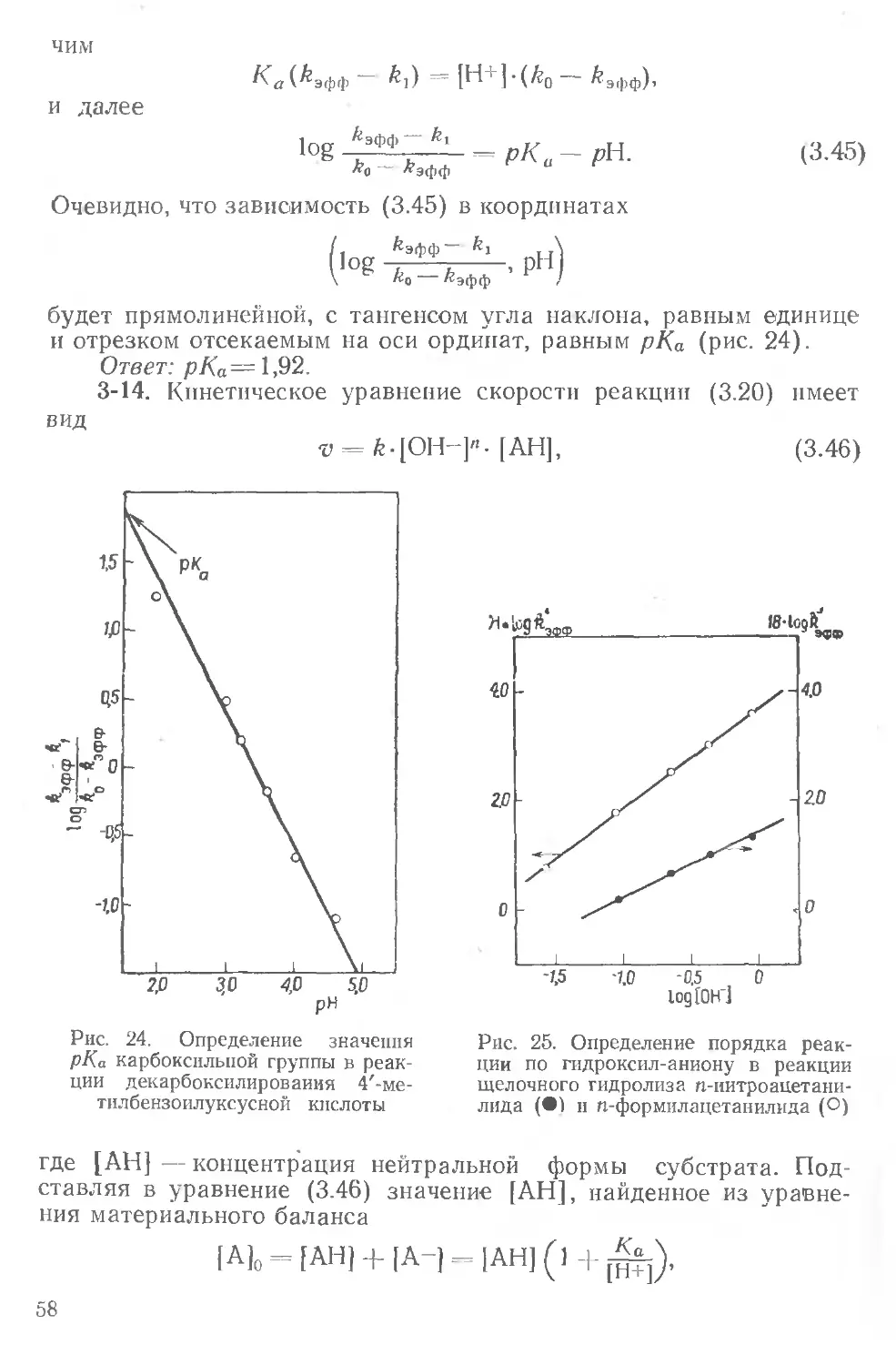
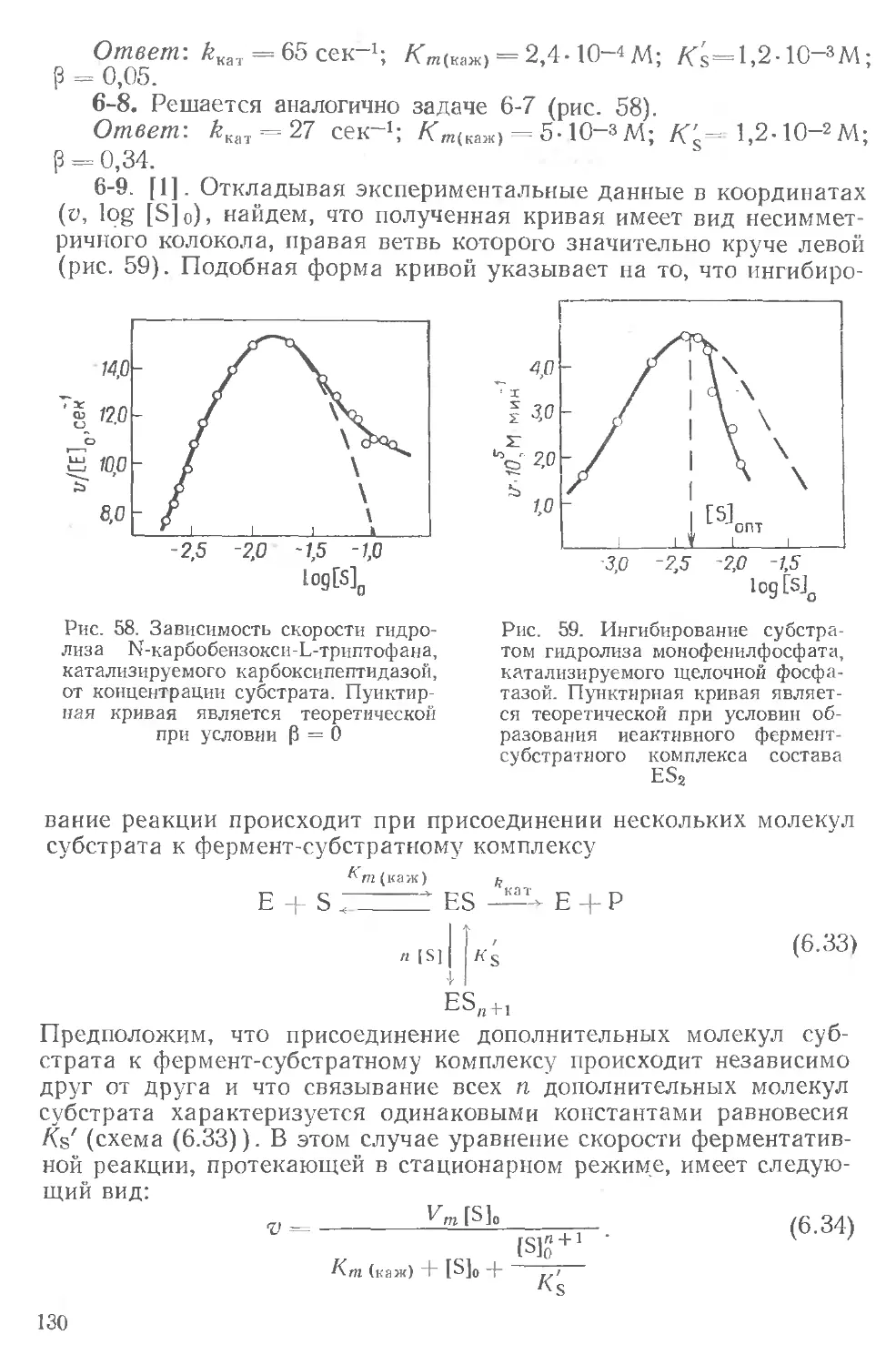
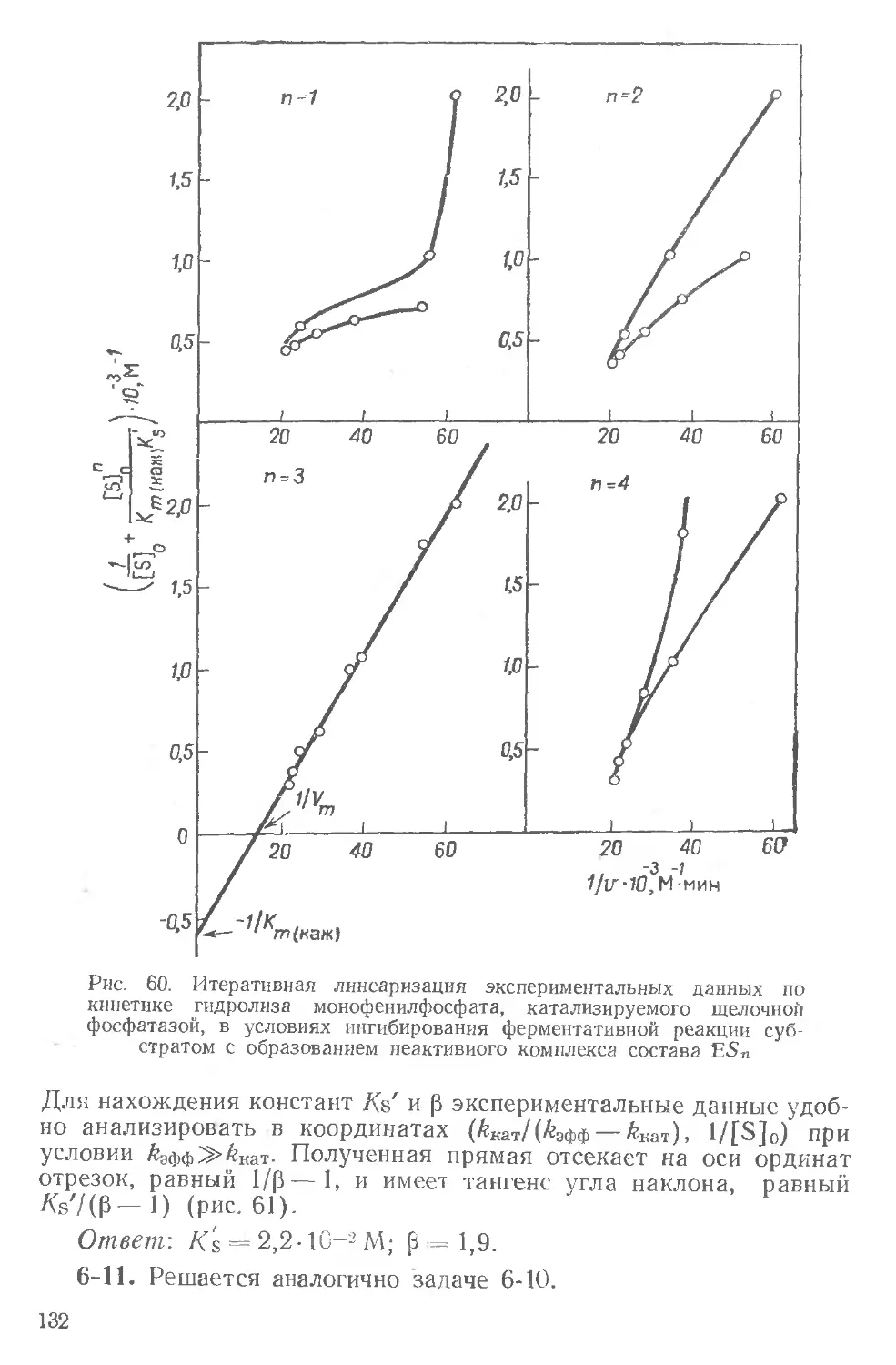
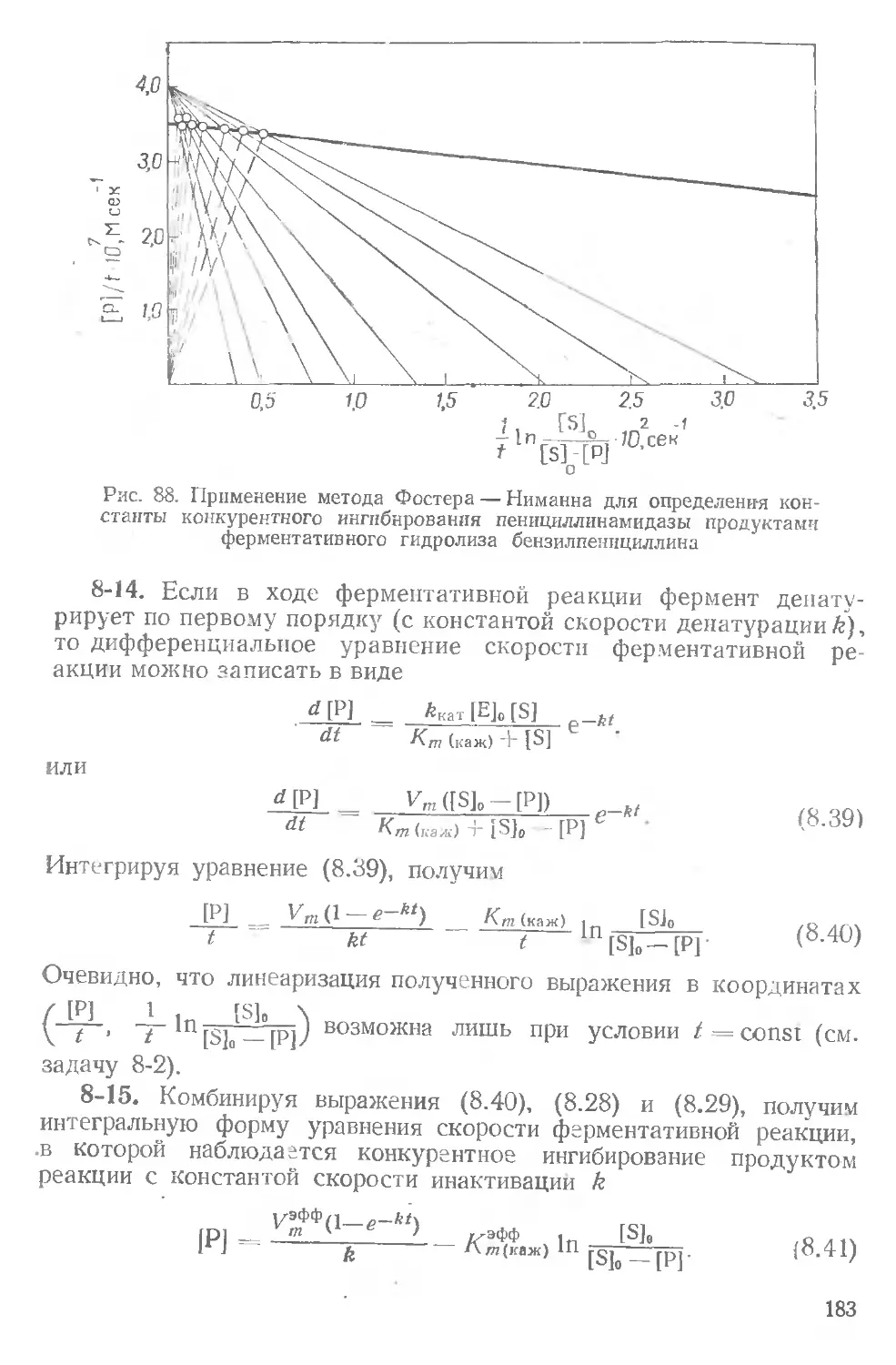
соон
: I I
/v\
NO, COO-
+н+
(3.19)
N02
I
OH
о
II '
+HO—С—СН3+
N02
I
OH
/V\
N02 COOH
NO. COO-
Исходя из данных рН-зависимости реакции, найти значение рКа
карбоксильной группы субстрата и величины констант k0 и k\.
Таблица 12
рН-Зависимость эффективной константы скорости гидролиза 3,5-динитроаспирина.
Условия опыта: 35° С; ионная сила 0,1.
рН
0
0,4
0,8
1,2
1,5
*эфф- мин-'
7,4
7,4
7,3
6,8
5,8
рН
1,6
1,8
2,1
2,4
2,6
*эфф' мин-'
5,4
4,7
3,4
2,2
1,6
рН
2,9
3,6
4,8
5,6
6,4
*эфф- мин '
1,0
0,4
0,3
0,3
0,3
45
3-10. В таблице 10 приведена зависимость эффективной
константы скорости гидролиза фенилового эфира тиолмуравьиной
кислоты от рН [13].
Н—С— S—<( Ч —— - НС—ОН -f HS— <f Ч
II \=/ II \=/
о о
На основании характера рН-зависимости предложить механизм
гидролиза и найти значения констант скоростей реакции
Таблица I©
рН-Зависимость гидролиза фенилтиолформата
при 25° С и ионной силе 1,0М(КС1)
рн
0,10
0,26
0,50
0,70
1,07
1,46
2,06
3,10
4,99
^эфф-Ю1, сек '
12,66
8,90
5,40
3,67
2,00
1,20
0,95
0,65
0,55
3-11. Гидролиз N- (n-нитрофенил) -2,2-диметилпропионогидра-
зонил бромида протекает по следующей схеме [12]:
СН.
i 8 ка
СН8—С — C=N—NH—<( Ч—NO, ~ "*•
СН„ Вг
сна
:сн
,-C--C=N—N— <( Ч-NO, + Н+
К
СН,
СН„—С—СО—NH—NH—<f V-N02
СН,
Используя данные табл. 11, определить значения констант k0,
h и Ка-
44
Таблица И
рН-Зависимость констант скоростей псевдопервого
порядка реакции гидролиза гидразонил бромида.
Условия опыта: 25° С; 70% дноксан
рН
4.15
4,30
4,50
4,70
5,00
5,50
5,95
6,42
Йэфф-Ю3, сек •
3,52
3,69
4,04
4,51
6,02
11,0
22,0
38,0
3-12. Гидролиз 3,5-динитросалициловой кислоты протекает по
механизму внутримолекулярного общекислотного катализа
карбоксильной группой в кислой области рН и внутримолекулярного
катализа карбоксилат-анионом в нейтральной области рН [15]
О О
NO, ll NO, и
| О—С—СН, | О С—СН,
fY K« fY
I II z=l I II +н+
/V\ /V\
N02 COOH N02 COO-
fto
NO,
I
I I
/V\
OH
о
II '
+ HO—С—CH3+
I ftl
*
N02
I
(3.19)
OH
NO, COO-
NO, " COOH
Исходя из данных рН-зависимости реакции, найти значение рКа
карбоксильной группы субстрата и величины констант k0 и k\.
Таблица 12
рН-Зависимость эффективной константы скорости гидролиза 3,5-динитроаспирина.
Условия опыта: 35е С; ионнаи сила 0,1.
рН
0
0,4
0,8
1.2
1,5
*эфф- мин-'
7.4
7,4
7,3
6,8
5,8
рН
1.6
1.8
2,1
2,4
2,6
*эфф- мнн-'
5,4
4,7
3,4
2,2
1,6
рН
2.9
3,6
4,8
5,6
6,4
*эфф- мнн '
1,0
0,4
0,3
0,3
0,3
45
3-13. В реакции декарбокснлирования одноосновных |3-кето-
кислот участвуют как свободная кислота, так и ее анион [19].
О
.О к.
Х^ \=
£* СУ'-
^
о
? - V-c—сн — с^ :==: <> >-с—сн— с'-
x-G^?=cH2+c°2
он
/3
с—сн„ + со,
II
о
быстро
г\
>1стро
УГ v=
с—сн,
о
Исходя из рН-зависимости декарбокснлирования 4'-метилбензо-
илуксусной кислоты (табл. 13), рассчитать значение рКа
карбоксильной группы субстрата.
Таблица 13
рН-Зависимость декарбокснлирования 4'-метилбензоилуксусной кислоты
в водном растворе при 50,3° С
рн
0,5
1,0
2,0
3,0
3,2
*эфф-101' «*"-•
9,7
9,7
9,2
7,4
6,1
рн
3,6
4,0
4,6
7,0
*эфф-10'- «*-'
4,1
2,0
1,0
0,36
рН
8,0
10,0
*эфф-,01.«к-
0,36
0,36
-1
3-14. Щелочной гидролиз n-нитро- и п-формил-ацетанилидов
проходит согласно общей схеме
О
СН3—С—NH—<Г y~X+nOW -
о
СН3—С—О""+ HN—/ ^>—Х+Н20, (3.20)
причем реакция имеет первый порядок по анилиду [16]. В табл. 14
приведены начальные скорости реакций гидролиза анилидов при
различных значениях концентраций гидроксильных ионов.
Начальные концентрации субстратов равны 7,27- Ю-5 М и 8,22- 10~5 М для
n-нитро- и п-форм'Илацетанилида. Определить порядки реакцпй по
гидроксил-анионам и значения константы скорости реакции k в
46
обоих случаях, если известно, что при ионизации анилида
образуется анион, не вступающий в реакцию гидролиза:
О о-
СН,
С—NH—(^ Ч -Х+ОН- * СН, - С=N-/ V-Х+НаО.
Величина рКа ионизации я-нитроацетанилида и я-формилацетани-
лида равны 13,8 и 14,3 соответственно.
Таблица 14
Зависимость начальных скоростей (vD) реакции гидролиза
анилидов от концентрации анионов гидроксила в растворе.
Условия опыта: 25е С; 0,99% диметилсульфоксида
X
N02
СНО
[OH-J-IO', М
9,59
24,0
47,1
95,9
96,7
2,80
9,27
23,7
47,1
94,0
t>0-lOs, М-сек— 1
8,34
22,9
38,4
51,8
50,3
0,0419
0,424
2,26
5,77
18,5
3-15. Рацемизация L-аспарагиновой кислоты, катализируемая
ионами гидроксила, протекает в водных растворах в начальный
период реакции согласно следующей схеме:
соон соон соо-
сн2
NHg1"—сн—соон;
снг
NH31"—сн—соо-;
сн2
1NH,— СН—СОО- (3.21)
ft, [он-]
ki [ОН-]
ft3 [он~]
D-изомер D-изомер D-изомер
Было найдено [7], что при 117° С значения кинетических
констант рацемизации соответствующих ионных форм аминокислоты
равныfei= 1,0■ Ю5М-> сек-1,А8=41 М-1 сек-', /г3=6,5- 10-3М-' сек"1.
Вычислить значения начальных скоростей рацемизации
L-аспарагиновой кислоты при значениях рН: 3,0, 5,0 и 7,0, если из
независимых опытов были найдены значения pKi = l,88 и p/G=3,65 и
начальная концентрация L-аспарагиновой кислоты в кинетических
экспериментах равнялась 0,1 моль/л.
47
3-16. Эффективная константа скорости псевдопервого порядка
гидразинолиза «-толуидида уксусной кислоты
СН3—/ \—О—С—CH„+NH2—NHS—>
О
v CH„-<f \-OH+NH2-NH-C-CH3
=== О
описывается выражением
*эфф = К [N2H4J + k2 [N2H4]2 + k3 [N2H4] - [N2H+]. (3.22)
Исходя из данных табл. 15, найти значения констант ky, k2 и kb,
если значение рКа гидразина равно 8,40.
Таблица 15
Зависимость эффективной константы скорости гидразинолиза
гс-толуидидацетата от рН среды и концентрации свободной
формы гидразина при 18° С
рН
8,72
8,72
8,73
8,74
8,39
8,40
8,40
8,40
8,41
8,41
8,20
8,21
8,21
8,21
8,00
8,00
8,00
8,00
IN,H,1, М
0,0614
0,123
0,185
0,256
0,060
0,120
0,180
0,180
0,250
0,250
0,0471
0,0941
0,141
0,196
0,0342
0,0684
0,103
0,143
*эфф- ыии~1
0,0408
0,141
0,320
0,613
0,0442
0,152
0,315
0,328
0,666
0,637
0,0304
0,105
0,239
0,452
0,0201
0,0705
0,142
0,286
3-17. Щелочной гидролиз трифторацетанилида протекает
согласно следующим схемам [20]:
О ..О
CF3—C-NH—<^ Ч~—"-* CF„ — С -N— <^ ^> + Н+
О
о-
CF3—С—N Н—^ ^+ОН- *- * CF„-C -NH—<^ ^>
(3.23)
(3.24)
ОН
О-
CF,
_(LNH-<;~y'+M0H I CF8-C-0- + H.N-^3 (3'25)
ОН
48
Исходя из зависимости эффективной константы скорости
гидролиза трифторацетанилида от рН (табл. 16), найти значения
констант k\, /г_1, k2 и k3, если из независимых экспериментов
найдены рКа=--9,50, К„=[Н+] -[ОН-] = 1,48-Ю-14 (при 30° С).
Таблица 16
рН-Зависимость гидролиза трифторацетанилида при 30° С
рН
13,23
12,83
12,75
12,53
12,32
12,13
12,01
^фф-103'1-"1»-1
71,5
62,5
58,9
50,0
38,5
31,3
25,7
рН
11,83
8,16
8,00
7,92
7,82
7,70
7,62
*эФф-103- ш,н—
19,2
0,125
0,0970
0,0800
0,0663
0,0508
0,0446
3-18. В таблице 17 приведена рН-зависимость образования
Ы-метил-2-беызоксазолинона при внутримолекулярной циклизации
фенилового эфира Ы-(2-оксифенил)-Ы-метилкарбамата [17]:
СН3 СН3
,N—С О { \
S\/1
\_
^\/N\
о-
i
о
\/\
\
S\
о-
\/\0/
/
>с=о+
"V
Определить значение константы диссоциации ионогенной группы
субстрата, контролирующей скорость реакции.
Таблица 17
рН-Зависимость образования циклического
карбаматного эфира в реакции
внутримолекулярной циклизации. Условия опыта:
25° С; ионная сила 0,5М (КС1)
рН
7,6
7,8
8,3
8,7
9,0
9,2
9,6
9,8
10,0
11,3
11,8
йэфф' сек-1
0,10
0,16
0,45
0,84
1,20
1,50
1,81
1,98
2,08
2,20
2,22
49
3-19. Сультон р-(2-окси-3,5-динитрофенил)этансульфоновой
кислоты образуется при циклизации соответствующего сульфо-
нилимидазольного производного [18]
NO.,
l
0,N.
+ll
0,N
v^^^^V^^R,—SO —N-^^NH
_ !-— i
(3.26)
0..N.
N*^NH
Эффективная константа скорости циклизации псевдопервого
порядка имеет колоколообразную зависимость от рН (табл. 18).
Используя данные таблицы, найти значения констант К\, Кг и k.
рН-Зависимость эффективной константы скорости образования
циклического сульфоната
Таблица 18
рН
0,8
1,4
1,4
1,5
1,6
2,0
йэфф' се,<-1
0,16
0,57
0,69
0,74
0,79
1,42
рН
2,0
2,4
2,8
3,1
3,1
3,6
*эфф- сек-1
1,52
2,48
2,82
2,52
2,80
1,60
рН
3,8
4,1
4,3
4,7
4,9
5,0
йэфф, сек-'
1,00
0,57
0,50
0,18
0,15
0,09
3-20. Реакцию образования хлорамина из аммиака и
хлорноватистой кислоты можно записать в виде
N1Ц+ + OCI- _ NH3+HOCI —-* NH2CI+H20. (3.27)
Реакция между гипохлорит-ионом и ионом аммония
обратима и по сравнению с образованием хлорамина протекает быстро.
Константы кислотности иона аммония и хлорноватистой кислоты,
найденные из независимых опытов, равны 6,1 • 10~10 М и 3,7- 10-8 М
50
соответственно. Константа скорости реакции k (не зависящая от
рН) равна 5,3 М-'-мин-1 [21].
а) Определить форму зависимости эффективной константы
скорости реакции от рН.
б) Найти, при каком значении рН эффективная константа
скорости реакции будет иметь максимальное значение.
в) Вычислить максимальное значение эффективной константы
скорости в рН-оптимуме реакции.
Ответы и решения
3-1. Обработку данных табл. 1 можно проводить в
координатах (log&, рН). В этом случае профиль рН-зависимости имеет вид
сигмоидальной кривой (рис. 15), в которой абсцисса точки
пересечения касательных с нулевым и единичным наклонами к ветвям
рН-зависимости равна рКа-
Эфф
/[Н^м'-сек'
Рис. 15. Определение значения
рКа анилиний-иона в реакции
внутримолекулярной
циклизации Ы-(0-аминофеноксиаце-
тил) -глицина
Рис. 16. Обработка рН-
зависимости
внутримолекулярного аминолиза,
при котором реакционно-
способной формой
реагирующего вещества
является его кислотная форма
Однако этот способ определения констант диссоциации ионо-
генных групп реагента из кинетических данных не является
оптимальным. Как видно из рис. 15, при таком способе из расчета
практически выпадают точки, соответствующие значениям рН в
районе рКа, и точность определяемой константы диссоциации за-
51
висит в основном от точности экспериментальных значений
кинетических параметров в экстремальных областях рН. Обычно для
увеличения точности линеаризуют всю кривую рН-зависимости в
подходящих координатах (см. теоретическую часть). Так как
в данном случае схема реакции формально описывается
уравнениями (3.12—3.13), то эффективную константу скорости можно
записать в виде
^эфф = Л pq+j •
Из рис. 16 можно найти значения констант Д'а=Ю~4М, и
k= 15,6-10"5 сек-1.
3-2. Обрабатывая данные табл. 2 в координатах (/гэфф, £Эфф[Н+]),
находим ^ = 6,ЬЮ-4сек-1; /Св — 3,12- ГО-6М; рКа =-5,5. Так как
константа скорости псевдопервого порядка kx равна произведению
константы скорости второго порядка ku на концентрацию N-аце-
тилциклосерина k\ = kn [N-ацетилциклосерин], то
*" = 5Л0^Ж = Х&- Ю-2Ж-1.сек->.
Ответ: № = 1,22- Ю-Mf-1-сек-1; рКа = 5,5.
3-3. Из условия задачи и табл. 3 очевидно, что кинетическая
схема реакции принадлежит к следующему типу:
*
А + В > Продукты, (3.28)
ка
В+ Н+ - ВН+. (3.29)
Обработка уравнений (3.28)—(3.29) приводит к выражениям (3.9)
и (3.11). Отсюда, линеаризуя данные табл. 3 в координатах
{£эфф, /2Эфф1Н+]), находим значения £' = 0,102 сек-1, Ка.= 1,08х
X Ю-8М. Наконец, из выражения
kx = /г" [N-метилимидазол]
находим значение константы скорости второго порядка kn.
Ответ: рКа = 7,97; Л» = 0,249 М-1 сек-1.
3-4. Кинетическая схема (3.16) принадлежит к общему типу
k
А- > Продукты, (3.30)
А- + Н+ АН. (3.31)
Обработка уравнений (3.30) — (3.31) приводит к следующему
выражению для эффективной константы скорости реакции:
, k
«эфф "- гН+1 ■ (3.32)
1 + т<7
52
Преобразовывая выражение (3.32), получаем
, , £эфф[Н+]
«эфф « ^ •
Линеаризуя данные табл. 4 в координатах (£Эфф, ^эфф[Н+]),
находим Ка=-2,72-Ю~2М.
Ответ: рКа=\,57.
3-5. При параллельном протекании кислотного и водного
гидролиза сложного эфира эффективная константа скорости
псевдопервого порядка должна определяться выражением
k3^-kH[H+} + kw, (3.33)
где /гн и kw — константы скоростей соответствующих реакций.
В этом случае в координатах (/гЭфф, [Н+]) должна наблюдаться
прямолинейная зависимость, из которой можно раздельно
определить &н и kw. Как видно из рис. 17, реакция гидролиза метил-
Рис. 17. Влияние концентрации во- Рис. 18. рН-Зависимость эффектив-
дородных ионов на эффективную ной константы скорости образова-
константу скорости гидролиза ме- нин фенилозазона глюкозы
тилтиолформата
тиолформата не описывается соотношением (3.33) и не
соответствует чистому случаю специфического кислотного катализа.
3-6. [9]. При значениях рН, намного превышающих рКа
карбоксильной группы реагента, механизмам I—IV должны
соответствовать следующие рН-зависимости:
I— с увеличением рН скорость реакции должна уменьшаться;
II и III (кинетически неразличимые механизмы)—скорость
реакции не должна зависеть от рН;
IV-—с увеличением рН скорость реакции должна
увеличиваться.
53
Как видно из следующей таблицы, константа скорости
реакции псевдопервого порядка линейно возрастает с уменьшением
[Н+], что соответствует механизму IV
Таблица 19
[Н + ]-10"М,
йэФф-106' сек~
2,0
9,8
24,6
457
410
85
34
1,7
3-7. Характер зависимости &Эфф от рН (рис. 18) показывает,
что каталитически активной формой уксусной кислоты является
ее недиссоциированная форма. Запишем выражения для скорости
реакции, констант равновесия (при условии быстрого
установления равновесия в системе), а также уравнение материального
баланса
v = k [X], (3.34)
^Р ~[ФГ] [СН3СООН] ' (3.35)
_ [сн3соо-][н+] п чс
к ° ~ [сНзСОбН] • (6-60)
[СН3СООН]0 = [СН3СОО-] + [СНдСООН], (3.37)
где [ФГ] - концентрация фенилгидразона. Подставив (3.35)—(3.37)
в (3.34), получим уравнение скорости реакции
■v- Аэфф[ФГ].[СН3СООН]0>
где
эфф /Са + [Н+] '
Преобразуя полученное выражение для эффективной константы
скорости реакции в виде
*Эфф Kg
"эфф "/ч р [W + ]
и линеаризуя его в координатах (&Эфф. *эфф/ [H+J), найдем
/Го = 6,03-10-5М.
Ответ: рКа = 4,22.
54
3-8. Реакция (3.17) принадлежит к общему кинетическому
THI1V
АН, А- + Н+
(3.38)
fco ft, |
Продукты
Отсюда скорость реакции (3.38) определяется выражением
г/ = А0[АН] + МА-].
Заменяя [АН] и [А-] через [А] о и Ка из уравнения
материального баланса и последующей формулы, получаем общее уравнение
для скорости реакции псевдопервого порядка
[А]0=[АН] + [А-], (3.39)
^ = *8фФ[А]о, (3-41)
где эффективная константа скорости реакции равна
у _ «о [H+J . ktKg /q ЛС\\
Яэ** Ка + [H+J + Ка + [H+J • ^' >
Выражение (3.42) можно записать в виде
КФФ{Ка + \)Л+]) = кй\\\+}^к1Ка.
Наконец, линеаризуя данные табл. 8 в координатах (£э<м>(^а+
+ [Н+]), [Н+], находим величины kc и k\ (рис. 19).
Ответ: /г0=4,53-10"5 сек-1, /г, = 1,37- 10~4 сек"1.
3-9. Ход решения является аналогичным решению задачи 3-8.
Однако в данном случае линеаризацию экспериментальных
данных в координатах (&эфф (Ка+[Н+]), [Н+]) необходимо
проводить, подбирая численное значение Ка (рис. 20).
Ответ: /г0=1,15-10-е сек-1, А, = 7,95- К)"5 сек"1, Ка=4,4-10-еМ.
3-10. Профиль рН-зависимости эффективной константы
скорости гидролиза (рис. 21) может быть следствием двух основных
механизмов реакции:
1. Гидролизу подвергаются как нейтральная, так и протониро-
ванная форма субстрата
«а
АН+ , А+Н+
(3.43)
+ +
Продукты
55
В этом случае эффективная константа скорости реакции
определяется следующим выражением:
*о [Н+] , k,Ka
^Эфф Ка + (H + j
+
Ка + [Н+]
(см. решение задачи 3-8).
2. Субстрат подвергается как специфическому кислотному, так
и водному гидролизу
А
k'0[H+] k[[U20\.
f +
Продукты
В этом случае £Эфф=£'о[Н+] +A'i[H20]. В условиях Ка
ханизмы (3.43) и (3.44) можно различить
(3.44)
[Н+] ме-
кинетически. При
4fl , SO
[н]-Я),м
Рис. 19. Обработка рН-зависимости
реакции, в которой реакционноспо-
собными формами являются как
нейтральная, так и ионизованная форма
субстрата (на примере гидролиза
циклического триэфира фосфорной
кислоты)
Рис. 20. Итеративная линеаризация
экспериментальных данных рН-зависимости
гидролиза этилового эфира 2-окиси-5-
нитрофенилкарбоната, принимая
значение рКа ионизации субстрата равным
(•) 3,2-Ю-6 М, (О) 5,0-10~6 М, (А)
4,4-Ю-6 М
7(а>[Н+] варианты 1 и 2 являются кинетически неразличимыми,
приводя в обоих случаях к прямолинейной зависимости в
координатах (£эфф, [Н+]), что и наблюдается в нашем случае (рис. 22).
Ответ: на основании экспериментальных данных (табл. 10)
можно предположить по меньшей мере два механизма реакции,
описываемые схемой (3.43) с константами k0/Ka=l,5lX
Х10-3 М-'-сек-1; /г,=6-1()-5 сек"1 и схемой (3.44) с константами
£'0=1,51 -Ю-3 М-1 -сек-1; ft/I = l,08-10"6 М-'-сек-1.
56
3-11. Решается аналогично задаче 3-9.
Ответ: К„=3,8-10"7 М, й0=3,16-10"3 сек"-', /г, = 7,37- Ю^сек"1.
3-12. Так как изучение реакции гидролиза проводилось в
достаточно широком интервале рН (табл. 12), значения констант k0
и k\ могут быть определены
непосредственно из профиля рН-зави-
симости эффективной константы
скорости реакции (рис. 23).
Величину рКа в этом случае удоб- щ
го
&
8-
СП
о
~ 08
in
0,4
^~~~""°—-— г..
I I I I I
1,0 2,0 3,0 4,0 5,0
рН
Рис. 21. рН-Зависимость
эффективной константы скорости
гидролиза фенилтиолформата
Ц4 t 0,6
[И ],М
Рис. 22. Обработка рН-зависимо-
сти гидролиза фенилтиолформата
но определять графически как значение
значению эффективной константы скорости
следует из выражения (3.42) при
Ка= [Н+].
Ответ: &о=7,4 мин-', ki =
= 0,3 мин^1. рКа=2,0.
3-13. Значения констант
&о и k\, найденные
непосредственно из рН-профиля
эффективной константы скорости реакции,
равны /2о=9,70- 10-4 сек-1, kx =
0,36-Ю-4 сек-1. Для определения
значения рКа можно
воспользоваться графическим методом,
использованным в решении
предыдущей задачи. Однако более
точным является следующий метод
обработки рН-зависимости.
Преобразуя выражение (3.42), полу-
рН,
сэфф
соответствующее
К + kx
= о как
Рис. 23. Определение значения рКа
из профиля рН-зависимости
эффективной константы скорости
гидролиза 3,5-дпнитроаспирина
57
чим
и далее
Ка{КФф - kj) = [Н+]-(/г0- /гэфф),
log.
'эфф "
*0~*3ФФ =^-^н- ^-45>
Очевидно, что зависимость (3.45) в координатах
Г^Г' рН
будет прямолинейной, с тангенсом угла наклона, равным единице
и отрезком отсекаемым на оси ординат, равным р/(а (рис. 24).
Ответ: рКа=1,92.
3-14. Кинетическое уравнение скорости реакции (3.20) имеет
вид
v = k-[OH-]».[AH], (3.46)
И.Ьодй,
ЭШФ
I8-looft
л ал
Рис. 24. Определение значения
рКа карбоксильной группы в
реакции декарбоксилироваиия 4'-ме-
тилбензоилуксусной кислоты
-7.0 -0.5 0
ЮдЮН"]
Рис. 25. Определение порядка
реакции по гидроксил-аниону в реакции
щелочного гидролиза п-иитроацетани-
лида (9) и n-формилацетанилида (О)
где [АН] — концентрация нейтральной формы субстрата.
Подставляя в уравнение (3.46) значение [АН], найденное из
уравнения материального баланса
[А)о = [АН] -f [A-] ~ [АН] (1 + *^),
58
получаем общее выражение для начальной скорости реакции
*> = *9фф[А10,
где [А]0 — общая начальная концентрация субстрата и
Обозначая ^фф - кэфф (Ка [ОН-] + Kw) и логарифмируя обе части
выражения (3.47), получим
log ^ФФ = log (kKw) + n log [OH-]. (3.48)
Очевидно, что зависимость (3.48) в координатах (logk'., log [ОН-])
будет прямолинейной с тангенсом угла наклона, равным п (рис. 25),
и отрезком, отсекаемым на оси ординат, равным (—14 -\- log k).
Ответ: для щелочного гидролиза n-нитроацетанилида л = 1,1,
£ = 2,09-10~3 М-'-сек-1; для щелочного гидролиза п-формилаце-
танилида и=1,9, /г = 3,98-10~4 М^-сек-1.
3-15. Из схемы (3.21) следует, что скорость рацемизации
аминокислоты описывается уравнением
v - А, [А,] • [ОН-] + k2 ]А2] • [ОН-] + k3 [А3] • [ОН-], (3.49)
где [Ai], [Аг] и [А3] —концентрации соответствующих ионных
форм субстрата. Используя уравнение материального баланса в
начальный период реакции
[A]0=[A1] + [A2J + [A3] (3.50)
и выражения
Кг-ЩРР-. (3-51)
[А2].[Н+]
[А,]
[А.НН+]
уравнение (3.49) можно преобразовать к киду
^=-^фф[А],-[ОН-], (3.53)
где
- [н+i -*■ [н+i' п 54,
1 ' [Н+] r [H+]2
Подставляя в соотношения (3.53) — (3.54) численные значения
кинетических и равновесных констант, а также значения [Н+] и
[ОН~] из условия задачи, найдем величины скоростей реакций
рацемизации.
Ответ: и = 5,9-10-9 м/л-сек (рН 3,0); и = 5,2- 10"10 м/л-сек
(рН 5,0); v = 2,6-Ю-10 м/л-сек (рН 7,0).
59
3-16. С учетом константы диссоциации протонированной
формы гидразина выражение (3.22) можно записать следующим
образом:
Аэфф = *i • [N2H4] + (А2 + k3 Ю) • [N2H4p. (3.55)
При постоянном значении рН выражение в круглых скобках
уравнения (3.55) является константой, так что
£9фф/[№Н4] Ai + A'fNaHJ- (3.56)
где
k' -k2 + k,-^. (3.57)
Из выражения (3.56) видно, что значения k\ и k' можно найти из
данных эксперимента при постоянном значении рН в координатах
(^эфф/ [N2H4], [N2H4]). Наконец, из зависимости k' от [Н+] (см.
уравнение 3,57) можно найти значения констант k2 и k3.
Ответ: k 1=0,13 М-1-мин-1; £2 = 7,9 М^-мииг1: £3=
=2,0 М-2-мин-1.
3-17. Запишем схему реакций (3.23) — (3.25) в сокращенном
виде:
АН-—^А~+Н+, (3.58)
АН + ОН- Z^ В, (3.5У)
й—1
fts+£3[0H—1
В ■ * Продукты. (3.60)
При условии стационарности концентрации промежуточного
соединения В в процессе реакции (при небольших глубинах
превращения) будет выполняться соотношение
4Цг - *i [АН]-[ОН-] - (А_х + А2+А3[ОН-])-[В] = 0.
Отсюда концентрация [В] будет равна
fR1_ МАННОН-]
' J '" ft_, J- k2 + ft, [OH-] ■
Из (3.60) видно, что уравнение скорости реакции имеет вид
v-(k2+k3[OH-])-[B]
или
„, (ft, + ft, [ОН-]) ft, [AHHOH-] nfin
^ ft_, + k2 + ft, [OH-] • \p.v4
С помощью уравнения материального баланса
[А]0 = [АН] + [А-] + [В] (3.62)
и выражения для константы равновесия (3.58) преобразуем
уравнение (3.61) и найдем выражение для эффективной константы ско-
60
роста реакции
_ fc.Kw (*, + *■ [ОН-])
*8фф - kiKw -+ (Ka + [н+]) (ft_, + k2 + k3 [ОН-])-
(3.63)
Из выражения (3.63) видно, что при значениях [Н+]3>/Са (при
малых концентрациях гидроксильных ионов) может выполняться
соотношение
'эфф
ktKw + (ft-, + k2) [H+]
или
1 +^Т^[Н+]
- * "Г и и V I" I- (3-64)
*Эфф «2 kjkiK-w l J
В этом случае экспериментальные данные при значениях [Н+] 3>/Са
(вторая серия данных табл. 16) должны линеаризоваться в
координатах (1/&эфф> [Н+]), что и наблюдается на опыте (рис. 26). При
2.0
15
W
1
0,5
-
/
>6
1.0
2,0
!н*]-ю!м
I
■^
i;
п"
5,0
40
ЯП
W
48
W
/
У
11111
2,0 1.0 6,0 S.C 10,0
[ОН"]
70. М
Рис. 26. Зависимость эффективной
константы скорости гидролиза
трифторацетанилида от
концентрации ионов водорода при
значениях [Н+] > Ка
Рис. 27. Зависимость эффективной
константы скорости гидролиза
трифторацетанилида от концентрации
гидроксильных ионов при значениях
[Н+] « Ка
высоких значениях рН (когда [Н+] <С/(а, первая серия данных
табл. 16) выражение (3.63) может быть преобразовано к линейной
зависимости от [ОН-] лишь в случае &3[ОН~] 3>k2. Тогда
^эфф
KKvih [OH-]
ftiKW + К a (ft-i + k, [ОН"])
или
'эфф
ktKW + k-xKg l
k,Kwk3 [он-
+
_Ка
кгК
W
(3.65)
61
Линеаризация экспериментальных данных в координатах
(1/Аэфф, 1/[ОН"]) (рис. 27) подтверждает правильность
сделанного предположения. Зная численные значения констант kx и k2,
найденные из точек пересечения зависимостей (3.64) и (3.65)
с осями ординат, из тангенса угла наклона зависимости (3.64)
можно рассчитать значение &_ь и далее из тангенса угла наклона
зависимости (3.65) —значение k3.
Ответ: £i = l,74-103 М-1-мин-1; fe_, = 8-10-3 мин-1: k2=
= 3,85- Ю-4 мин-1; £3=2,75 М^-мин"1.
3-18. Решается аналогично задаче 3-4.
Ответ: 7(о=1,2-10-9М.
• 3-19. Кинетическая схема (3.26) принадлежит к общему типу
реакций
АН ~ Продукты, (3.66)
АН2+ ZZZ1 АН + Н+, (3.67)
АН Z=^ А- + Н+, (3.68)
рН-зависимость которых имеет вид колоколообразной кривой.
Выражение для эффективной константы скорости реакции находят
обычным методом, комбинируя уравнение скорости реакции,
уравнение материального баланса и выражения для констант
диссоциации комплексов АН + и АН+
v-^k\hH] -Аэфф|А]п, (3.69)
[А]0 = [А-] + [АН] + [АН+], (3.70)
[АН]-[Н+]
Ка~ [АН+] ' {6Jl)
K^—lm1- (3-72)
Очевидно, что выражение описывает колоколообразную зависимость
эффективной константы скорости реакции от рН.
£Эфф = jjppj ^— (3.73)
Из рис. 28 видно, что экспериментальная зависимость £эфф от
рН имеет острый максимум. Это означает, что численные значения
Ка и Кь близки, так что раздельное определение этих констант
графическим методом не представляется возможным. Для
определения констант диссоциации Ка и Кь и истинного значения
константы скорости реакции k в случае острого максимума рН-зави-
симости обработку экспериментальных данных можно проводить
по следующей схеме:
62
1. Приблизительно определить значен'ия Л"0 и k из левой ветви
кривой рН-зависимости с помощью обычных методов (см.
решение задач 3-2—3-4).
2. Приблизительно определить значения Кь и k из правой
ветви кривой рН-зависимости (см. решение задачи 3-1 и 3-7).
3. Провести теоретическую кривую рН-зависимости с
использованием найденных значений Ка, Кь и k согласно выражению
(3.73).
4. Варьируя значения Ка, Кь и k, подобрать теоретическую
кривую, оптимально удовлетворяющую экспериментальным
данным (провести оптимизацию подбора значений Ка, Кь и k).
На рис. 29 показан профиль
реакции (3.26) в координатах
(log £Эфф, рН), на котором
приведены характеристические точки
рН-зависимости, найденные с
помощью описанного метода.
2,5
Гх2,0
ш
о
1,0
0,5
до
А
: /\
J \
■/ \
1,0 2,0 30 4,0 5,0
* ' Рн
Рис. 28. рН-Зависнмость эффективной
константы скорости реакции
образования циклического сульфоната.
Теоретическая кривая проведена с
использованием значений констант
Л=4,25 сек-', рК„ = 2,2 и рКь = 3,4
1,0 2,0 3,0 4,0 5,0
рн
Рис. 29. Профиль рН-зависимости
логарифма эффективной
константы скорости реакции образования
циклического сульфоната
Ответ: Ki = 4-W-4 М, /?К, = 3,4; К2 = 6-10-3 М, р/(2=2,2; k =
=4,25 сек-1.
3-20. Как следует из уравнения (3.27), скорость реакции
определяется выражением
*> = A[NH3].[HOCl]. (3.74)
Для нахождения выражения для эффективной константы скорости
реакции, зависящей от рН, запишем уравнения материального
баланса для аммиака и хлорноватистой кислоты
cN = [NH3] + [NH4+], (3.75)
Сс = [НОС1] + [ОС1-] (3.76)
63
и выражения для констант диссоциации иона аммония и
хлорноватистой кислоты
[NH,]-[H+]
ATn
/<с =
[NH+] '
[QCi-HH*-]
(3.77)
(3.78)
[НОС1] •
Комбинируя выражения (3.74) — (3.78), найдем уравнение
начальной скорости реакции
v = k
эфф
CNCc,
где
'эфф
1 +
[Н+]\
1 +
[H+l
(3.79)
Из выражения (3.79) видно, что зависимость &Эфф от [Н+] должна
проходить через максимум. Для нахождения координат
максимума рН-зависимости продифференцируем &Эфф по [Н+], и
полученное выражение приравняем нулю. Таким образом найдем, что
максимальное значение &Эфф соответствует значению [Н+], равному
[Н+] = К^Ж- (3.80)
Подставляя (3.80) в выражение (3.79), найдем, что максимальное
значение &Эфф в рН-оптимуме реакции равно следующей величине:
* (3.81)
эфф
('♦/§■'
Наконец, подставляя численные значения Ks и Л'с из условия
задачи в выражения (3.80) и (3.81), найдем рН
= 6,9-Ю-2М-1-мин-1.
= 8 32 &тах =
ОПТ V,^*, «Эфф
ЛИТЕРАТУРА
1. Гаммет Л. Основы физической органической химии. М., „Мир", 1972,
стр. 407.
2. Джен кс В. Катализ в химии и энзимологии. М., „Мир", 1972, стр. 430.
3. Ашмор П. Катализ и ингибирование химических реакций. М., „Мир",
1966, стр. 53.
4. Kirk К. L., Cohen L. A. „J. Amer. Chem. Soc.«, 91, 8142, 1972.
5. Whish W. J. D., V i s w a n a t h a T. „Can. J. Biochem.", 48, 218, 1970.
6. Martin C. J., Oza N. В., Marini M. A. „Eur. J. Biochem."
1971.
7. В a da J. L. „J. Amer. Chem. Soc", 94, 1371, 1972.
8. Hershfield R., Schmir G. L. „J. Amer. Chem. Soc", 94, 1263, 1972.
9. Hegarty A. F., В r u i с е Т. С „J. Amer. Chem. Soc",- 9i, 4924, 1969.
10. Афанасьеве. А., Стрельцова И. Ф. ЖФХ, 46, 2545, 1972.
11. Bromilow R. H., Khan S. A., Kirby A. J. „J. Chem. Soc", (B), 1971,
1971.
20, 276,
64
12. H e g a r t у Л. F, Cashman M. P., Scott F. L-. . J. Chem. Soc. Perkin", II,
44, 1972.
13. Barnett R, Jencks W. P. „J. Org. Chem.", 34, 2777, 1969.
14. Fife T. H., Hutchlns J. E. С „J- Amer. Chem. Soc", 94, 2837, 1972.
15. Fersht A. R., К i r by A. J. .J. Amer. Chem. Soc", 89, 5961, 1967.
16. Pollack R. M., Bender M. L. »J. Amer. Chem. Soc", 92, 7190, 1970.
17. Hutch ins J. E. C, Fife T. H. „J. Amer. Chem. Soc.*, 95, 2282, 1973.
18. Berg W., Campbell P., К a i s e г E. T. .J. Amer. Chem. Soc", 94,7933,
1972.
19. StraubT. S., Bender M. L. „J. Amer. Chem. Soc", 94, 8881,1972.
20. Stauf fer С E. „J- Amer. Chem. Soc", 91, 7887, 1972.
21. Weill I., Morris J. С „J. Amer. Chem. Soc", 71, 1664, 1949.
Глава 4
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА СКОРОСТЬ
ХИМИЧЕСКИХ РЕАКЦИИ
§ 1. Зависимость констант скоростей
реакций от температуры
Зависимость константы скорости реакции от температуры в
простейшем виде определяется уравнением Аррениуса
Е
k- k0e «\ (4.1)
где k0 — предэкспоненциальный множитель;. Е — энергия
активации; R — газовая постоянная (1,987 - ), Т — абсолютная
температура. Логарифмируя уравнение (4.1), получаем
In k = In k0- -^ . (4.2)
Из уравнения (4.2) видно, что графически зависимость в
координатах (In k, l/T) выражается прямой линией, тангенс угла наклона
которой равен — E/R.
Так как скорость реакции связана с константой скорости
соотношением
V = kc"
и концентрации веществ не зависят от температуры, то уравнение
типа (4.1) применимо и для зависимости скорости реакции от
температуры
е_
v = v0e RT
и далее
In v = In v0 ~ ^Y.
Величина энергии активации в случае простых химических
реакций, протекающих в одну стадию, показывает, какой избыточ-
3 Заказ 151 65
нои энергией по сравнению со средней энергией молекул при
данной температуре должны обладать молекулы, чтобы они могли
вступить в химическую реакцию. Уравнение Аррениуса
формально применимо и к температурным зависимостям сложных
многостадийных процессов. Однако в этих случаях энергия активации
обычно эффективная величина, не имеющая определенного
физического смысла.
Более строго зависимость константы скорости реакции от
температуры определяется в рамках теории абсолютных скоростей
реакций, одно из основных уравнений которой
k Т А/"^
к^ъ-^-е *г, (4.3)
или
кпТ
ДЛ'^ ШФ
А = *-2— е R e к? . (4.4)
Здесь к — трансмиссионный коэффициент, обычно принимаемый
равным единице (за исключением некоторых специальных
случаев); AF*— стандартная свободная энергия активации
(характеризующая изменение свободной энергии системы в стандартных
условиях при переходе ее из исходного состояния в
активированное), AS* и АН*— стандартная энтропия и стандартная
энтальпия активации, kB — постоянная Больцмана (1,38-1016 эрг/град) и
h — постоянная Планка (6,625-10-27 эрг-сек). Из уравнения (4.3)
видно, что константа скорости реакции определяется главным
образом изменением свободной энергии при переходе системы в
активированное состояние, так что любой внешний фактор,
уменьшающий свободную энергию активации, будет способствовать
ускорению химического процесса.
Из сопоставления уравнений (4.1) и (4.4) нетрудно получить
соотношение
ДН^ = E — RT,
из которого видно, что численные значения энтальпии активации
и эмпирической энергии активации отличаются на величину RT
(0,6 ккал/моль при 300° К).
§ 2. Изокинетические зависимости
При изучении влияния температуры на скорости химических
реакций довольно часто встречаются линейные зависимости
между изменениями стандартной энтальпии активации и стандартной
энтропии активации в реакционных сериях (отличающихся
строением реагентов или условиями проведения реакции). Обычно
подобные зависимости встречаются в тех реакционных сериях, когда
изменения в структуре реагентов не затрагивают их реакционные
центры. Соотношения типа
АН* = А + рД5^ (4.5)
66
называют «изокинетическои» или «^компенсационной»
зависимостью.
Для выяснения физического смысла постоянных А и |3 в
соотношении (4.5) запишем известное уравнение
М?Ф = &НФ - TbS^ (4.6)
в виде
Д# = bF* _|_ T&S*. (4.7)
Из выражения (4.7) видно, что если в какой-то реакционной
серии значения АРф при температуре Т совпадут для всех членов
ряда, то график в координатах (АНФ, AS^) будет иметь вид
прямой линии, тангенс угла наклона которой равен Т. Температура,
при которой значения свободных энергий активации (или
значения констант скоростей реакций) совпадают в пределах данной
реакционной серии, называется изокинетическои температурой
(I изок) ■
Задачи
4-1. Используя данные табл. 1, вычислить значение энергии
активации щелочного гидролиза карбометоксидигидроизокарбо-
стирила
О О
II I
сн2 с сн2 с
/\/ \ / \ s\/ \ / \
сн осн, * | II сн о-
\/
HN СН3
\ /
с
II
о
—СН30Н
V
HN СН3
\ /
с
II
о
Таблица 1
Температурная зависимость константы скорости
щелочного гидролиза
карбометоксидигидроизокарбостирила [1].
Условия опыта: рН 8,5; ионная сила 0,1 М (KCI);
4% ацетонитрила
Т°1<
298
303
307,5
312
321
330
к-10*. сек-'
0,40
0,75
0,95
1,48
3,03
7,72
67
4-2. В таблице 2 приведена зависимость константы скорости
реакции гидразина с диаминотрифенилметановым красителем —
малахитовым зеленым
сн3 сн3
\ /
N
I
Y
N
/ \
сн3 сн,
СН3 СН8
\ /
N
I
+ NH2NH2
V
/ Ч—С—NHNH2+H+.
I
N
/ \
сн, сна
Вычислить значения стандартных энтальпии и энтропии
активации данной реакции.
Таблица 2
Температурная зависимость константы скорости
реакции гидразина с малахитовым зеленым [2].
Условия опыта: рН 11,0; ионная сила 1,0М (KCI);
концентрация красителя 5-10-6М
/°с
7,0
14,8
23,8
30,0
38,4
k, M -мин *
1060
1580
2480
3750
4680
4-3. В таблице 3 приведена температурная зависимость
константы скорости щелочного гидролиза метилового эфира N-аце-
тил-Ь-фенилаланина
V
I
сн2
CH.CONH—СН—СООСН,
-Сн.он
V
I
сн2
I
-* CH.CONH—СН—СОО"
68
Вычислить значения стандартных энтальпии и энтропии
активации реакции.
Таблица 3
Зависимость константы скорости щелочного
гидролиза метилового эфира
К-ацетил-Ь-фенилаланина от температуры [1].
Условия опыта: рН 8,5; ионная сила 0,1М (КС1);
4% ацетонитрила
ГС
25
30
34,5
39
48
57
/S-105, сек-'
0,50
0,68
1,03
1,62
3,95
8,95
4-4. В таблице 4 приведена зависимость константы скорости
кислотного гидролиза бензоилимидазола от температуры [3].
_i_s~ // \_с_он н- Н№ Г-NH
\=/|| \=^
О
Вычислить значения активационных параметров гидролиза,
АН* и AS*.
Таблица 4
Температурная зависимость константы скорости
гидролиза бензоилимидазола в 32,5% -ной
серной кислоте
14
15,54
25,00
34,53
44,92
/г, мин 1
0.138
0,312
0,638
1,22
4-5. Константа скорости реакции равна 100 мин-1. Определить
значение стандартной свободной энергии активации реакции.
4-6. Показать, что энергия активации и энтальпии активации
химической реакции связаны соотношением E=AH*-\-RT.
4-7. В таблице 5 приведены температурные зависимости
констант скоростей реакций глицилглицина и гидразина с
малахитовым зеленым [2]. Можно ли достичь в условиях эксперимента
такой температуры, при которой значения констант скоростей этих
двух реакций будут одинаковыми.
69
Таблица Б
Зависимость констант скоростей реакций глицилглицииа
и гидразина с малахитовым зеленым от температуры.
Условия опыта: рН 11,0; ионная сила 1,0М (КС1);
концентрация малахитового зеленого 5-10-6М
Реагент
I идразин
Глнцилглицнн
/ с.
7,0
14,8
23,8
30,0
38,4
7,0
14,8
23,8
30,0
38,4
k, M—1-мин—1
1060
1580
2480
3750
4680
31,4
52.1
90,2
133
199
4-8. В работе [4] показана возможность обмена водорода на
тригий в химотрипсиногене после обратимого развертывания
молекулы белка
*■ , *.
Нативный белок ., Развернутый белок ■—* Обмен (4.8)
*-■
Зависимости констант скоростей отдельных стадий реакции (4.8)
от температуры приведены в табл. 6. На основании данных
таблицы вычислить величину эффективной энергии активации реакции.
Таблица 6
Температурная зависимость констант скоростей отдельных стадий
реакции (4.8) при рН 2
("С
25
30
35
40
ft, .10*, сек-'
0,0398
0,631
6,31
44,6
fc_,-10», сек-1
2,5
8,0
16,0
20,0
ft.-10', сек-1
1.5
2,6
5,1
8,8
4-9. Константы скоростей щелочной -денатурации (рН 11,9)
бычьего гемоглобина при 2 и 28° С равны, соответственно,
2,0-Ю-5 сек-1 и 1,5-Ю-4 сек-1 [5]. Рассчитать значения
стандартных энтальпии и энтропии активации реакции денатурации.
70
4-10. В таблице 7 приведена температурная зависимость
константы скорости термического разложения хлористого гидразония
3N2H5C1 — — 3NH4C1 + NH3 + N2.
Вычислить значения стандартных энтальпии и энтропии
активации реакции.
Таблица 7
Зависимость константы скорости термического разложения
хлористого гидразония от температуры [6]
i°C
183,6
191.5
192,0
198,6
198,6
199,4
207,8
ft-10«, сек-*
1,44
2,53
2,40
5,18
4,94
5,13
10,20
("С
221,0
221,0
221,5
221,9
235,0
235,0
239,5
239,5
ft-10«, сек-1
39,5
37.0
37,5
42,0
99,0
114,0
179,0
175,0
4-11. Найти значения энергии активации химического
процесса, если при переходе от 25 до 35° С константа скорости реакции
возрастает а) в 2 раза, б) в 3 раза, в) в 5 раз, г) в 10 раз.
4-12. В таблице 8 приведена температурная зависимость
скоростей тепловой денатурации р-лактоглобулина [7]. Найти
энергию активации денатурации, а также значения стандартных
энтальпии и энтропии активации реакции.
Таблица 8
Зависимость константы скорости тепловой
денатурации {5-лактоглобулина от температуры
i°C
64,3
67,2
69,8
72,0
74,6
ft-1С, сек"1
0,46
1,11
2,69
7,30
14,59
4-13. Щелочная денатурация белков, приведенных ниже,
характеризуется следующими значениями энтальпии и энтропии
активации:
71
Белок
ккал/ моль
AS^=, э. i
Оксигемоглобин человека
Окснгемоглобин крысы
Гемоглобин крысы . .
Окснгемоглобин кролика
Гемоглобин кролика .
Оксигемоглобин быка .
7,6
15,1
12.2-
18,5
16,9
21,0
—39
—17
—27
— 8
—13
— 6
Найти а) значения констант скоростей денатурации при 25° С;
б) при каком значении температуры эксперимента константы
скоростей реакций денатурации всех белков будут равны по величине.
4-14. В таблице 9 приведена температурная зависимость
константы скорости перегруппировки диэтил-2-гидрокси-1,1,1,3,3,3-гек-
сафторизопропилфосфоната до 1,1,1,3,3,3-гексафторизопропил-
диэтил фосфата (4.9) под действием пиридина в дихлорметане [8].
Вычислить значения стандартных энтальпии и энтропии активации
реакции.
k С2Н5Оч О
W /CF, (4.9)
л/ -ел -_сн<
XCF,
с2н„ох о
/К
с,нлу
с
I
он
<
CF3
F3
с,н5о'
нотабли ц а
Зависимость константы скорости
перегруппировки фосфоната в фосфат (4.9)
от температуры
t с
32,6
42,3
44,6
54,1
57,1
ft-10*. M—J сек—1
0,40
1,07
1,33
3,15
3,80
Ответы и решения
4-1. Ответ: £'=17,5 ккал/моль (рис. 30).
4-2. Уравнение (4.4) при х=1 можно записать в виде
или
А
т
1п-=- -- const
RT
RT
(4.10)
Из выражения (4.10) вицно, что зависимость в координатах (lnft/Г,
1/Г) должна быть прямолинейной, с тангенсом угла наклона,
равным —АНФ/Я (рис. 31). Далее найдя величину свободной энергии
активации реакции при 25° С по формуле, полученной
преобразованием выражения (4.3),
AF* = RT In kBT/kh = 0,593 (29,4 — In k), (4. 11)
7.0
-8.0
-9,0
Щ0
•up
4
\
N.
Цч> =
i i i
•
еЧ
i
3.0
31 3.2
3.3
I
T
3.1
W
Рис. 30. Определение энергии
активации щелочного гидролиза кар-
бометоксидигндроизокарбостирила
2,8
2,4
-а? |е- 2,0
с
16
»,?
Ао
"*- — 1 то.
г\
1 \
1 \
| 1*1 1 ~П
3,2 3,3 3,4 3,5 3,6
f*8
Рис. 31. Определение энтальпии
активации (ДН1^) и константы
скорости реакции (йст) в
стандартных условиях для
взаимодействия гидразина с
малахитовым зеленым
находим величину стандартной энтропии активации из выражения
AF* = АН* — TAS*, (4.12)
или
Ш* — hF*
AS* =
(4.13)
Примечание: При использовании формулы (4.11) для
вычисления AF* размерносхь константы скорости реакции должна быть
т1^е1Г^~ттГМ=:г- сек ' шля реакций первого или второго порядка
соответственно (так как при расчетах числовых коэффициентов в
формуле (4.11) постоянная Планка использовалась в
размерности эрг-сек).
Ответ: Д//^ = 7,96 ккал/моль; AS* = ~ 24,3 э. е.
4-3. Решается аналогично задаче 4-2.
Ответ: АН* = 16,4 ккал/моль; AS* = ~ 37 э. е.
4-4. Решается аналогично задаче 4-2.
Ответ: АН* = 13,2 ккал/моль; AS*=~ 24,8 э. е.
4-5. Решается с использованием формулы (4.11) (см.
примечание к задаче 4-2).
Ответ: AF* = 17,15 ккал/моль.
73
4-6. Логарифмируя выражения (4.1) и (4.4) и дифференцируя
их по температуре, получим соответственно
dink E
dT ~ ~W>~ '
dink 1_ АНФ
dT — т + RTz ■
Приравнивая друг другу полученные выражения, найдем
Е = Mi* + RT.
4-7. Обрабатывая данные табл. 5 в координатах (In k/T, 1/-7"),
найдем д//=£ и далее Д5^ (см. решение задачи 4-2) для реакций
гидразина и глицилглицина с малахитовым зеленым: uhf =
= 7,96 ккал/моль; ASf = 24,3 э. е.; ДТ/f = 9,95 ккал/моль; &St =
= —24,2 э. е. Так как полученные значения энтропии активации
двух реакций очень близки, то значение температуры, при
котором константы скоростей соответствующих реакций будут
одинаковыми, очень высоко. Легко также показать графически, что
равенство энтропии активации двух реакций приведет к
пересечению двух соответствующих прямых в координатах (In k/T, 1/7")
на оси ординат, т. е. при Т — со.
4-8. Запишем уравнение (4.8) в виде
*• ь
А ^12 В—-+Р, (4.14)
откуда следует, что общая скорость реакции определяется
выражением
v = ka[B]. (4.15)
Так как для реакции (4.14) k—l'^>k1 и k2^>kx (табл. 6), то можно
полагать, что концентрация вещества В при малых глубинах
превращения находится в стационарном состоянии, так что
1Ш. = к, [А] - (£_, + к2) [В] = 0. (4.16)
Из выражения (4.16) можно найти стационарную концентрацию
вещества В:
Из уравнения материального баланса
[А]0 = [А] + [В]
найдем
[А]-^ёУА1°- Г4Л8)
Комбинируя выражения (4.15), (4.17) и (4.18), получим
•о = £эфф [А]0,
74
где
^э*ф = л, + k-[ + k2 ■ (4-19)
Подставляя данные табл. 6 в выражение (4.19) и линеаризуя
полученную зависимость в координатах (1п£эфф, 1/^0 > найдем
эффективную энергию активации реакции.
Ответ: £'=81,5 ккал/моль.
4-9. Выражения для констант скоростей реакции при двух
различных температурах Т\ и Т% в рамках теории абсолютных
скоростей реакций можно записать следующим образом:
*i = -V- е R e RTl. (4-2°)
k2 = ^j-l-e ч е «■». (4.21)
кв7"2
Логарифмируя обе части выражения, полученного при делении (4.21)
на (4.20), найдем
' Л,/?". V Д№".(1/7\-1/Г2)
Hffi)
R
или
АНФ = ^ W*' . (4.22)
тх~ т
2
Подставив данные задачи в выражение (4.22), найдем численное
значение Д//=£. Далее, используя выражение (4.22) и найденную
величину Д//^% вычислим значение константы скорости реакции
в стандартных условиях (при 25° С). Наконец, используя формулы
(4.11) и (4.13), найдем значение стандартной энтропии активации
реакции.
Ответ: Д//^= 12 ккал/моль; AS=£= — 36,3 э. е.
4-10. Решается аналогично задаче 4-2.
Ответ: Д//=£ = 39,3 ккал/моль; ДЗ^^О.
4-11. Константы скоростей реакций при температурах Тх и 7\>
равны соответственно
_ Л.
k1=k0e wlf (4.23)
Е
*2=V *г' (4.24)
(см. выражение 4.1). Разделив (4.24) на (4.23), получим
|_Д(ЙГ-7Т)# (425)
75
Логарифмируя (4.25) и преобразуя полученное выражение, найдем
выражение для энергии активации
F R In kjkt
Ответ: Е = а) 12,5 ккал/моль; б) 19,9 ккал/моль; в) 29,1
ккал/моль; г) 41,6 ккал/моль.
4-12. Решается аналогично
задаче 4-2.
Ответ: Е = 86,5 ккал/моль;
Д//=£ = 85,9 ккал/моль; AS* =
= 176 э. е.
4-13. Для решения задачи
используются соотношения (4.4)
(при и=1) и (4.7), рис. 32.
Ответ: a) ft=4,97-10-2 сек-1
1,0-Ю-2 сек-1; 8,61-Ю"8 сек-1
3,02-Ю-3 сек-1; 3,31 -lO"8 сек"1
1,20-Ю-4 сек-1; б) ГИЗОк^100°С.
4-14. Решается аналогично
задаче 4-2.
Ответ: АН*= 17,7 ккал/моль;
bS±= — 20,8 э. е.
10 20 30
дб, кал/моль- град
Рис. 32. Определение величины изо-
кинетической температуры для
щелочной денатурации белков
ЛИТЕРАТУРА
1. Antonov V. К., Rumsh L. D. „FEBS Letters", 9, 67, 1970.
2. Dixon J. E., Br ul се Т. С. „J. A,mer. Chem. Soc", 93, 3428, 1971.
3. Smith C. R., Yates K. „J. Amer. Chem. Soc", 94, 8811, 1972.
4. Rosenberg A., Enberg J. .J. Biol. Chem.", 244, 6153, 1969.
5. H a u г о w i t z F., H a r d i n R. L., D i с k s M. „J. Phys. Chem.", 58, 103, 1954.
6. Рубцов Ю. И., Манелис Г. Б. ЖФХ, 46, 627, 1972.
7. Larson В. L., Jennes R. „J. Amer. Chem. Soc", 74, 3090, 1952.
8. Jansen A. F., Smyrl Т. G. .Can. J. Chem.", 50, 1205, 1972.
Часть II
КИНЕТИКА
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
Глава 5
КИНЕТИЧЕСКАЯ ОБРАБОТКА ПРОСТЫХ
ДВУХСТАДИЙНЫХ ФЕРМЕНТАТИВНЫХ
РЕАКЦИИ (АНАЛИЗ НАЧАЛЬНЫХ
СКОРОСТЕЙ)
§ 1. Кинетическое описание
двухстадийных ферментативных реакций
Простейшей схемой для описания кинетики ферментативных
реакций является так называемая двухстадийная схема
Е +S—'"F.SJS R + P, (5.1)
в которой Е — фермент; S — субстрат; Ks — константа
диссоциации фермент-субстратного комплекса
и kz —■ константа скорости первого порядка распада фермент-
субстратного комплекса ES с образованием продуктов реакции Р
и регенерацией фермента. В том случае, когда концентрация
субстрата намного превышает концентрацию фермента ([S0] ^> [Е]0),
что является обычным условием изучения кинетики
ферментативных реакций (за исключением случаев, описанных в главах 6 и 9),
уравнение материального баланса по ферменту можно записать
в виде
[E]0 = [E] + [ES]. (5.3)
Комбинируя выражения (5.2) и (5.3) с выражением для скорости
распада фермент-субстратного комплекса
v=k2[ES], (5.4)
получаем уравнение скорости ферментативной реакции
_ *,[EUS] ,5 5)
V - Ks + [S] ' V'6>
При изучении начальных скоростей реакции (когда расходом
субстрата можно пренебречь) можно полагать [S] = [S0] и
V ~ Ks + [S]. • V-b>
77
Уравнение (5.6) описывает зависимость скорости ферментативной
реакции, протекающей по схеме (5.1), от начальной концентрации
субстрата и позволяет определять на опыте значения констант ki
и Ks, являющихся важнейшими характеристиками ферментативной
реакции. В том случае, когда определяемые из эксперимента
константы k2 и Ks являются эффективными величинами (зависящими
от рН среды, присутствия в системе ингибиторов или активаторов,
наличия дополнительных стадий в схеме (5.1) и т. д.), их
называют соответственно «каталитической константой» (&кат) и
«кажущейся константой Михаэлиса» (/Ст(каш)) данной ферментативной
реакции. Тогда уравнение (5.6) выглядит следующим образом"-
«кат l^Jo l^Jo
^т (каж)'
„. «кат tcJo I'-'Jo /C7\
v— ~к _i_rsi ' \°-'f
это так называемое уравнение Михаэлиса —■ Ментен. Оно
иллюстрирует гиперболическую зависимость скорости ферментативной
реакции от начальной концентрации субстрата и линейную
зависимость — от концентрации фермента. Произведение &Кат [Е] 0,
имеющее размерность скорости реакции, обычно называют
максимальной скоростью реакции и обозначают Vm (из уравнения (5.7)
видно, что при [5]0>/Ст(каш) выполняется равенство v=Vm).
Более строго уравнение (5.7) можно вывести, исходя из
предположения о стационарном режиме протекания ферментативной
реакции (5.1). В этом случае скорость образования
фермент-субстратного комплекса ES должна быть практически равна скорости
его расходования:
ft.
E + S^—-ES — -*Е + Р, (5.8)
^[EHSJ^^ + ^-OIES], (59)
так что концентрация [ES] должна оставаться постоянной
(стационарной) в ходе ферментативной реакции, во всяком случае
в начальный период реакции (при [S] ~ [S]0). Это предположение
является логичным, если принять во внимание высокую
реакционную способность фермент-субстратных промежуточных соединений
(что уже доказано в ряде случаев экспериментально, см.
например, [1]). Комбинируя выражения (5.3), (5.4) и (5.9), найдем
уравнение скорости двухстадийной ферментативной реакции (5.8),
протекающей в стационарном режиме:
*i
*a+[S].
Очевидно, что уравнение (5.10) является частным случаем
уравнения (5.7) при &кат = k2 И Кт(кат) = (k-i + k2)/ki. ТаКИМ обрЭЗОМ,
константа Михаэлиса даже в двухстадийных ферментативных
реакциях в общем случае является кинетической, а не равновесной
78
константой (переходя в константу равновесия лишь в случае
k-i > k2, что, впрочем, часто имеет место в реальных
системах, [2]). Поэтому при дальнейшем изложении константы/w
и Кт(кат) будут называться в общем случае кинетическими
параметрами ферментативных реакций.
§ 2. Определение кинетических
параметров ферментативных реакций из
экспериментальных данных
Существуют различные способы линеаризации зависимости
начальной скорости ферментативной реакции от концентрации
субстрата. Наиболее распространенным способом является
линеаризация экспериментальных данных в координатах (l/v, 1/[S]0),
называемых координатами Лайнуивера-Берка или координатами
«двойных обратных величин». Как
следует из выражения
1 1 | *^т (каж)
"5" ~~ VZ + Ут
[SJ. '
(5.11)
график зависимости (5.7) в
координатах Лайнуивера-Берка имеет вид
прямой линии, пересекающей ось
абсцисс и ординат в точках
—1/Кт(каж) и 1/Vm соответственно
(см. рис. 36).
Довольно часто для обработки
кинетических данных
ферментативных реакций используют так
называемое уравнение Иди (5.12),
которое также легко получить
преобразованием уравнения (5.7):
V = V т — Л'т(каж)
Рис. 33. Линеаризация
зависимости начальной скорости
ферментативной реакции от
концентрации субстрата в
координатах Иди
v
(5.12)
При графическом построении экспериментальных данных в
координатах Иди (рис. 33) полученная прямая линия пересекает ось
ординат в точке Vm и имеет тангенс угла наклона,
равный — Кт(каж)- Остальные четыре возможных способа
трансформации i равнения Михаэлиса (5.7) в линейную функцию не имеют
широкого применения [3].
§ 3. Влияние обратимых эффекторов
(ингибиторов и активаторов)
на кинетику ферментативных реакций
В общем случае влияние обратимо действующих эффекторов на
двухстадийную ферментативную реакцию может быть передано
схемой (5.13). В этой схеме предусмотрено взаимодействие эффек-
79
торов как со свободной формой фермента, так и с
фермент-субстратным комплексом
Е+ S^db ES —+Е + Р
(5-13)
ЭЕ+S ^ZlBES —-> ЭЕ + Р
aKs |
Можно показать, что кинетическая обработка схемы (5.13)
приводит к следующему выражению для начальной скорости
ферментативной реакции (при [Э] о 3> [Е] 0) '-
к2
°^э + Р [Э]
<*КЭ+[Э]
[E].[S].
v-—*±w • <5Л4>
аК*аКэ+[Э]+№»
В зависимости от численных значений множителей а и (3
эффектор Э может выступать в роли либо ингибитора, либо промотора
(активатора) ферментативной реакции. Полный кинетический
анализ и сводная таблица возможных частных случаев
ингибирования и активации фермента даны в работе [4]. Однако некоторые
частные случаи имеют особое значение и широко применяются для
описания кинетики ферментативных процессов. К их числу
относятся полное конкурентное ингибирование, полное неконкурентное
ингибирование, бесконкурентное ингибирование, простая
активация и некоторые типы смешанного ингибирования и активации.
1. Полное конкурентное ингибирование (а—»-оо, р не имеет
определенного смысла). В случае полного конкурентного
ингибирования начальная скорость ферментативной реакции
определяется выражением
Ks(i + 7^) + [S]„
и зависимость в координатах Лайнуивера-Берка имеет вид пучка
прямых, пересекающихся на оси ординат (см. рис. 38). Константу
конкурентного ингибирования Ki можно определить с помощью
выражения
Kmlmx) = Ks[l + -^-), (5.16)
откладывая экспериментальные данные в координатах (К-щквт), Щ)-
Для определения константы ингибирования распространен
также так называемый метод Диксона, согласно которому
экспериментальные данные откладывают в координатах (1/f, [I]) (см.
рис. 46).
80
2. Полное неконкурентное ингибирование (а=1, р = 0).
В случае полного неконкурентного ингибирования выражение для
начальной скорости ферментативной реакции имеет вид
~ПГШ7кГ[ US]o /t-17,
v = кГ+Ж ' {5Л7)
и зависимость в координатах Лайнуивера-Берка имеет вид пучка
прямых, пересекающихся на оси абсцисс (см. рис. 39). Константу
неконкурентного ингибирования можно определять с помощью
выражения
*«" = 1 + [l)IKl (5J8)
построением экспериментальных данных в координатах (1/Акат, [I])»
или с помощью метода Диксона (см. рис. 41).
3. Бесконкурентное ингибирование (а=р< 1). В случае
бесконкурентного типа ингибирования константы &кат и Кт(кв.ж)
ферментативной реакции уменьшаются в одинаковой степени (5.19),
так что график соответствующей зависимости в координатах
Лайнуивера-Берка имеет вид семейства параллельных прямых (см.
рис. 44):
*i + [II
"*■«•*, +[I][E]o[S].
v = -—кГШ ■ (5Л9)
«KsctK, + [I] + [S]«
Для анализа уравнения (5.19) выражение
*i +
«*i + [I]
АКат = <**2 „у , m (5-z0>
можно преобразовать к виду
1 L_ , ^j_ _L (ч 9п
kKaT/ks-\ - а-1 Т"«-1 " [I] ' ^'Zl;
из которого видно, что линеаризация экспериментальных данных
в координатах (\ —77^ ~ 1,1/[I] J позволяет раздельно найти
значения а и К\.
4. Неконкурентная активация (а = 1, |3 > 1). В случае
неконкурентной активации субстрат и активатор связываются
независимо с активным центром, образуя тройной комплекс (фермент-
субстрат-активатор) и приводя к увеличению скорости
образования продукта, так что начальная скорость ферментативной
реакции определяется выражением
Кк + №\
*2 *а + [А1 [EUS]°
v = кТ+Щ ' (5-zz)
81
и зависимость в координатах Лайнуивера-Берка имеет вид пучка
лрямых, пересекающихся на оси абсцисс (см. рис. 48). Анализ
уравнения (5.22) можно проводить с помощью выражения
^кат — %2 ^ I гд] > (5.2d)
преобразованного в виде
1 +^Г-Ш- (5-24)
^кат . Р— 1 ' Р— 1 [А]
Из выражения (5.24) видно, что построение графика в
координатах (1/—~ 1, 1/[А]) позволяет провести раздельное ОПреДе-
ление констант |3 и Ка-
5. Смешанные типы ингибирования и активации (а =7^1,
§ Ф 1). В случае смешанных типов ингибирования или активации
графики в координатах Лайнуивера-Берка имеют вид пучка
прямых, соответствующих различным концентрациям эффектора и
пересекающихся в общей точке в правом верхнем, левом верхнем
или левом нижнем квадранте (в зависимости от числовых
значений а и р и соотношения между ними). Координаты точки
пересечения во всех случаях являются следующими [4}:
[SIT = 1<~s'T=f* (5-25)
_L = 1 . IzA (5.26)
v Vm a — p v '
Из выражений (5.25) и (5.26) видно, что положение точки, в
которой происходит пересечение пучка прямых зависимости l/v от
1/ [S] о при различных концентрациях эффектора, определяется
лишь значениями констант а и |3 и не зависит от величины Ка
(см. 5.13). Таким образом, вид графика в координатах
Лайнуивера-Берка может быть использован для определения типа влияния
эффектора на ферментативную реакцию и для оценки интервалов
значений аир [4].
6. Применение метода Диксона к анализу нетривиальных типов
ингибирования. Как отмечалось выше, обработка данных по
влиянию ингибиторов на кинетику ферментативных реакций может
быть проведена в координатах Лайнуивера-Берка (l/v, 1/[S]0)
или, согласно методу Диксона, в координатах (1/и, [I]). Метод
Диксона обладает тем преимуществом, что он позволяет
определять значение константы ингибирования непосредственно из
кинетических данных, не прибегая к дополнительным построениям.
С другой стороны, вид графика в координатах Диксона не
позволяет отличить смешанный тин ингибирования от конкурентного
или неконкурентного ингибирования, как это обычно можно
сделать при построении в координатах Лайнуивера-Берка. Однако
«2
можно показать, что в ряде случаев ингибирования наиболее
эффективным методом обработки экспериментальных данных
является совместный анализ графиков в координатах Диксона и Лай-
нуивера-Берка [5].
Приведем уравнение (5.14) к виду, удобному для анализа
вариантов ингибирования в координатах (1/а, 1/[S]0) или (1/а, [I]):
• М'+Щ) "-(■+$)-я-'
Очевидно, что выражение (5.27) в общем виде линеаризуется
в координатах Лайнуивера-Берка и лишь в отдельных случаях
в координатах Диксона, давая тем самым дополнительную
возможность для подбора подходящего механизма, описывающего
изучаемую ферментативную реакцию. Рассмотрим некоторые частные
случаи схемы (5.13), приводящие к нетривиальным типам
ингибирования.
1) р = 0. В этом случае вид графика в координатах
Лайнуивера-Берка иллюстрирует классическое смешанное ингибирование
или (при а-*-<х>) полное конкурентное ингибирование.
Независимо от численного значения а абсцисса точки пересечения пучка
прямых в координатах Диксона равна — Ki (см. рис. 46) и
ордината равна (1 — \ 1а) IVт. Отсюда видно, что в зависимости от
величины а общая точка пересечения прямых в координатах
Диксона может находиться в левом верхнем квадранте (а>1,
смешанное или конкурентное ингибирование), на оси абсцисс (а = 1)
или в левом нижнем квадранте (а<1). Во всех этих случаях
величина Ki обычно определяется с хорошей точностью (как
абсцисса общей точки пересечения).
2) р = 1. Этот вид ингибирования называется
псевдоконкурентным, так как из следующего выражения видно, что характер
графика в координатах Лайнуивера-Берка идентичен случаю
классического конкурентного ингибирования:
1 1
К
(•+£) .
Так как выражение (5.28) не линеаризуется в координатах
Диксона, сопоставление обоих типов графиков позволяет отличить
псевдоконкурентное ингибирование от обычного конкурентного.
3) р = 0; Ki = °°. Этот тип ингибирования называется
антиконкурентным, так как ингибитор связывается только с фермент-
субстратным комплексом и не взаимодействует со свободным
ферментом. В этом случае график в координатах Лайнуивера-
Берка имеет вид семейства параллельных прямых и неотличим
от случая бесконкурентного ингибирования (где а = р, см. раз-
83
дел 3). Однако зависимости в координатах Диксона в случае
1
V
1 , PI
1+^г
—
v m
, ^s
Л
v m
1
[SU
(5.29)
антиконкурентного ингибнрования имеют вид параллельных
прямых (с тангенсом угла наклона, равным l/a/dVm), в отличие от
гиперболических кривых в случае бесконкурентного ингибнрования:
14-Ж
Vm[l+K1)
4) a = р. В случае бесконкурентного ингибнрования (5.30)
зависимости в координатах Диксона являются гиперболическими
и перестают зависеть от концентрации ингибитора при [I]//(i>
> 1 < [I]/a/Ci, приводя к лимитирующей величине l/v, равной
l/Vm(l/a+/Cs[5]0), откуда можно найти значение а.
Таким образом, совместное применение координат Лайнуивера-
Берка и Диксона позволяет выявить некоторые варианты
нетривиальных типов ингибирования.
§ 4. Кинетический метод
двухкомпонентного обратимого
ингибирования ферментативных реакций
Важную информацию о строении активного центра фермента
можно получить с помощью метода двухкомпонентного обратимого
ингибирования [6]. Для этого вводится понятие о
взаимонезависимых и взаимозависимых ингибиторах. По определению,
взаимонезависимые ингибиторы могут одновременно (и независимо)
связываться с активным центром фермента, в то время как для
взаимозависимых ингибиторов это исключено.
а. Влияние взаимозависимых конкурентных ингибиторов на
кинетические параметры ферментативной реакции. Схема двух-
стадийной ферментативной реакции в присутствии двух
взаимозависимых конкурентных ингибиторов выглядит следующим
образом:
Е12
t
+
к)
Е 4- S7_ES "Е 4- Р. (5.31)
Ml
EI,
84
Кинетическая обработка схемы (5.31), проводимая обычным
способом (по методу установившегося равновесия или по методу
стационарных концентраций), приводит к выражению (5.7) для
начальной скорости ферментативной реакции, где
"чсаг == "-2' (5.<32)
Km(Ka3K) = Ks(l + |j- + |]-). (5.33)
Графически зависимость (5.33) можно представить в координатах
(К-щкъж), [U]) прямой линией (при [I2] = const), тангенс угла
наклона которой равен отношению Ks/Ki, а отрезок, отсекаемый
на оси ординат, равен Ks(l + [I2]/AY) (рис. 34). Таким образом,
зная значение Ks из опытов, проведенных в отсутствие
ингибиторов, можно определить величины Ki и Ki'.
Рис. 34. Зависимость
кажущейся константы Михаэлиса от
концентрации ингибитора [I,]
(при [12] = const) в
присутствии двух взаимозависимых
конкурентных ингибиторов
Рис. 35. Зависимость
кажущейся константы Михаэлиса от
концентрации ингибитора [Ii]
(при [I2] = const) в
присутствии двух взаимонезавнсимых
конкурентных ннгабиторов
б. Влияние взаимонезависимых конкурентных ингибиторов на
кинетические параметры реакций. При наличии двух
взаимонезависимых конкурентных ингибиторов схему двухстадийной
ферментативной реакции можно записать в виде
Е1,1
E + S
-^ЕЗ-^Ет-Р
(5.34)
Кинетическая обработка схемы (5.34) приводит к выражениям для
кинетических параметров ферментативной реакции:
"-кат = «2» (5.do)
^m(Ka», = ^s(l+|J-)(l + |i). (5.36)
85
Значения констант Кг и Кг можно определить с помощью
построения зависимости (5.36) в координатах (/Ст(каж), [Ii]), как и в
случае анализа действия взаимозависимых конкурентных ингибиторов
(рис. 35). Однако на рис. 35 не только отрезок, отсекаемый на оси
ординат, но и тангенс угла наклона, равный Ks/Ki(l + [h]/Ki'),
зависят от концентрации второго ингибитора [12]. Значение Ks
может быть найдено из опытов, проведенных в отсутствие
ингибиторов, и тогда величины Кг и Кг могут быть найдены из отрезков,
отсекаемых на осях.
в. Влияние взаимозависимых неконкурентных ингибиторов на
кинетические параметры реакции. Кинетическая обработка схемы
EI2 + S^Z^1EI2S
«I I I Jfs I I *l
+ I * +1 ft.
E + S ZZZ. ES -E + P. (5.37)
I IKi
EI,+S-;—»EI,S
приводит к выражениям для кинетических параметров
ферментативной реакции в присутствии двух взаимозависимых
неконкурентных ингибиторов:
*2 (5.38)
! , [У. , М.
к
т(каж)
= к<
(5.39)
Графический анализ выражения (5.38) следует проводить в
координатах (1//гКат, [Ii]), подобно обработке зависимости (5.33).
В этом случае из графика, аналогичного рис. 34, можно найти
численные значения констант Кг и Кг'-
г. Влияние взаимонезависимых неконкурентных ингибиторов
на кинетические параметры реакции. В присутствии двух
взаимонезависимых неконкурентных ингибиторов схему двухстадиинои
ферментативной реакции можно представить в виде
я.
Е1,12
Е12 « . =+ ESI5
JtESI,!.,
i,
У
(5.40)
1 + *
«I.
*■,»№& loi.
Обрабатывая схему (5.40) обычным способом, найдем выражения
для кинетических параметров реакции:
k —
kt
1 +
ЭД(. + $)
К
т(каж)
= к5
(5.41)
(5.42)
Вычисление значений Кг и Кг в этом случае может быть проведено
анализом зависимости (5.41) в координатах (1/Акат, [Ii]), подобно
анализу зависимости (5.36) (см. рис. 35).
Задачи
/5-1.^Определить значения кинетических параметров (Акат и
Ктхкящ) гидролиза метилового эфира N-ацетил-Ь-валина,
катализируемого а-химотрипсином [7], исходя из данных табл. 1.
Таблица I
Зависимость начальной скорости ферментативной
реакции от концентрации субстрата
для гидролиза метилового эфира
N-ацетил-Ь-валина, катализируемого
а-химотрипсииом. Условия опыта: рН 7,8; 25° С;
ионная сила 0,1М (КС1), [Е]0 = 3.8-10-5М
[SI„, M
0,200
0,124
0,124
0,091
0,091
0,071
0,071
0,060
0,060
К.-10», М-сек-1
4,57
3,83
3,84
3,33
3,31
2,97
2,93
2,67
2,74
5-2. Определить значения kKaT и Кт(каж) Для гидролиза метило-
ього эфира Ы-бензоил-Ь-аминомасляной кислоты, катализируемого
«-химотрипсином [7], исходя из данных табл. 2.
5-3. Исходя из данных табл. 3, определить кинетические
параметры гидролиза метилового эфира Ы-ацетил-Ь-норвалина,
катализируемого а-химотрипсином [7].
\*~£s)Определить значения кинетических параметров гидролиза
метилового эфира К-ацетил-Ь-аланил-Ь-фенилаланина,
катализируемого а-химотрипсином [7], исходя из данных табл. 4.
5-5. Определить кинетические параметры гидролиза этилового
эфира М-транс-циннамоил-Ь-тирозина, катализируемого
а-химотрипсином, исходя из данных табл. 5.
87
Таблица 2
Зависимость начальной скорости ферментативной
реакции от концентрации субстрата для гидролиза
метилового эфира ^бензоил-Ь-аминомасляной
кислоты, катализируемого а-химотрипсином.
Условия опыта: рН 7,8; 25° С; ионная сила
0,Ш (КС1), [Е1о = 2,16.10-<М
[S1„-I0», M
2,24
2,24
1,49
1,49
1,12
1,12
0,90
0,90
0,75
0,75
V„-I07, М-сек~1
4,25
4,31
3,52
3,60
3,10
3,12
2,71
2,77
2,45
2,40
Таблица 3
Зависимость начальной скорости реакции
от концентрации субстрата для гидролиза
метилового эфира ^ацетил-Ь-норвалина,
катализируемого а-химотрипсином.
Условия опыта: рН 7,8; 25° С; ионная сила
0,1М (КС1); [Е]0 = 2,62.10-7М
ISlo-W, M
4,00
4,00
2,00
2,00
1,33
1,33
1.С0
1,00
0,80
0,80
IVI07, М-сек—'
9,70
10,0
7,77
7,85
6,51
6,41
5,50
5,51
4,80
4,76
5-6. Определить значения /гкат и Кт(кат) гидролиза
бактериальных клеток Мс. lysodeikticus, катализируемого лизоцимом, исходя
из данных табл. 6.
5-7. Определить кинетические параметры гидролиза
метилового эфира Ы-ацетил-Ь-фенилаланина, катализируемого трипсином,
исходя из данных табл. 7.
5-8. При изучении кинетики гидролиза метилового эфира
N-ацетил-глицина, катализируемого а-химотрипсином, наряду
Таблица 4
Зависимость начальной скорости гидролиза
метилового эфира
М-ацетил-Ь-аланил-Ь-фенилаланина,
катализируемого а-химотрипсином,
от концентрации субстрата. Условия опыта:
рН 7,8; 25° С; ионная сила 0,!М (КС1);
[Е]о = !,035 10-8М
[S]0-l№, М
1,530
1,530
0,765
0,765
0,510
0,510
0,383
0,383
0,306
0,306
Vo-10', М-сек '
4,94
4,98
4,20
4,30
3,74
3,70
3,02
2,99
3,32
3,28
Таблица 5
Зависимость начальной скорости гидролиза
этилового эфира М-транс-циннамоил-Ь-тирозина,
катализируемого а-химотрипсином,
от концентрации субстрата. Условия опыта:
рН 7,8; 25° С; ионная сила 0,!М (КС1);
2% ацетонитрила; |Е]0 = 3,ЫО-9М
[SJo-lC, М
3,6
1,8
1,2
0,90
0,72
1/о-107, М-сек-1
1,94
1,84
1,75
1,67
1,59
с ферментативным происходит также спонтанный (щелочной и
водный) гидролиз субстрата (табл. 8). Определить кинетические
параметры ферментативной реакции.
5-9. В работе [8] было показано, что а-кетоглутарат является
конкурентным ингибитором реакции окисления М-метил-Ь-глутама-
та, катализируемого N-метилглутамат-дегидрогеназой. Определить
константу диссоциации комплекса фермент-ингибитор, исходя из
данных табл. 9.
5-10. Бензоат 1,2,5-триметилпиперидол-4(р-изомер) является
ингибитором гидролиза бутирилхолина, катализируемого холин-
эстеразой [9]. Определить тип ингибирования и найти константу
89
Таблица 6
Зависимость начальной скорости гидролиза
бактериальных клеток, катализируемого
лизоцимом, от весовой концентрации суспензии
субстрата. Условия опыта: рН 4,9 (ацетатный
буфер); 25° С; [Е]0 = 5,7210-8М
[Slo-10'. мг/мл
l/-10s,
ед. опт. плотности/сек
1,17
1,33
1,55
1,87
2,33
3,08
3,73
5,60
6,53
7,46
8,40
9,33
11,2
15,5
0,380
0,396
0,480
0,516
0,650
0,770
0,800
0,914
1,02
1,10
1,11
Ы7
1,18
1,31
Таблица 7
Зависимость начальной скорости гидролиза
метилового эфира 1Ч-ацетил-Ь-фенилаланина,
катализируемого трипсином, от концентрации
субстрата. Условия опыта: рН 7,8; 25° С;
ионная сила 0,1М (КС1); [Е]0 = 1,4Ы0-6М
isi„-io*, м
1,00
1,00
0,80
0,80
0,67
0,55
0,55
0,50
W10", М-сек-'
3,25
3,41
2,81
2,79
2,45
2,15
2,13
2,01
диссоциации комплекса фермент-ингибитор, исходя из данных
табл. 10.
5-11. В некоторых работах за показатель ингибирующей
способности эффектора принимают такую концентрацию последнего,
которая вызывает уменьшение скорости ферментативной реакции
в два раза (1бо) - Найти связь между 150 и Ki для случаев
конкурентного и неконкурентного ингибирования.
90
Таблица 8
Зависимость начальных скоростей спонтанного и химотриптического гидролиза
метилацетурата от концентрации субстрата. Условия опыта: рН 7,8; 25° С;
ионная сила 0,Ш (KCI); 1Е]0 = 3,82-1 О^М
[S]0, М
1,00
0,67
0,50
0,50
0,40
0,33
0,33
Начальная скорость спонтанного
гидролиза сз'бстрата (в
отсутствие фермента), (М-сек *)-107
11,9
9,31
7,88
7,90
6,45
5,88
5,78
Суммарная начальная скорость
спонтанного и ферментативного
гидролиза, (М-сек—*)-107
34,2
27,3
23,05
23,10
19,70
17,50
17,30
Таблица 9
Влияние а-кетоглутарата на кинетику окисления N-Memn-L-глутамата,
катализируемого N-метил-глутамат-дегидрогеназой. Условия опыта: рН 7,4, 30° С,
фосфатный буфер; 3-10~6М 2,6-дихлориндофенола, [Е]0 = 6-10—2 мг/мл
1а-кетоглутарат]*103, М
0
0,6
[SJ„-10S М
ЬСО
0,625
0,500
0,417
0,264
1,67
1,00
0,625
0,500
0,330
IV 10е, М-мин *
1,67
1,43
1,33
1.25
1,00
1,67
1,43
1,18
1,04
0.83
3,0
5,00
1,67
1,00
0,667
0,500
1,56
1,00
0,77
0,57
0,45
(5-12_'В работе [10] изучали влияние н-бутанола на кинетику
реатщйи гидролиза сложных эфиров, катализируемых карбокси-
пептидазой В. На основании зависимости скорости
ферментативной реакции от начальной концентрации субстрата (табл. 11)
предложить кинетическую схему реакции (приняв двухстадийныи
механизм действия фермента) и определить константу ингибиро-
вания н-бутанолом.
5-13. В работе [11] было показано, что метйодид 2-пиридинкар-
бальдоксима является ингибитором гидролиза 1-нафтилацетата,
91
Таблица р[
Влияние fS-изомера бензоата 1,2,5-триметилпиперидола-4
на кинетику гидролиза бутирилхолина, катализируемого
холинэстеразой. Условия опыта: рН 7,4; 25° С; фосфатный буфер
(0.0ОШ)
[11-10=. М
0
0,5
1,0
2,0
3,0
[S]„-10«, М
10,00
2,50
0,91
0,50
10,00
2,50
0,91
0,50
10,00
2,50
0,91
0,50
10,00
2,50
0,91
0,50
10,00
2,50
0,91
0,50
vt усл. ед.
5,55
4,45
2,94
2,09
4,77
3,78
2,56
1,79
4,00
3,18
2,16
1,49
2,86
2,28
1,52
1.С6
2,38
1,85
1,24
0,87
катализируемого ацетилхолинэстеразой, причем прямые в
координатах Лайнуивера-Берка, соответствующие различным
концентрациям ингибитора, имеют вид семейства параллельных прямых.
Предложить схему ингибирования (приняв двухстадийный
механизм ферментативной реакции) и рассчитать константу
ингибирования.
5-14. Кофермент А (СоА) является ингибитором реакции,
катализируемой фосфат-ацетилтрансферазой [12]:
Ацетил — СоА + фосфат -> Ацетилфосфат + СоА. (5.43)
В табл. 13 приведены данные по влиянию СоА на начальную
скорость реакции (5.43) при вариации концентраций обоих
субстратов. Определить тип ингибирования коферментом по отношению
к нуклеотидному (Ацетил — СоА) и неорганическому субстрату
и вычислить соответствующие константы ингибирования.
92
Таблица 11
Влияние н-бутанола на кинетику гидролиза гиппурового
эфира L-аргинина, катализируемого карбоксипептидазой В.
Условия опыта: рН 8,0; 23° С; трис-ацетатный буфер (0.025М);
[Е]0 = 6,05-10-6 мг/мл
[н-бутанол], М [Slo-105, M
0
0,05
0,15
0,30
[0,0
5,00
3,33
2,50
2,00
1,67
10,0
5,00
3,33
2,50
2,00
1,67
10,0
5,00
3,33
2,50
2,00
1,67
10,0
5,00
3,33
2,50
2,00
1,67
v-W, М-мин— *
16,7
12,5
10,5
8,8
7,7
6,7
9,1
7.7
6,5
5,7
5.0
4,55
5,35
4,60
4,07
3,64
3,26
3,00
3,33
2,94
2,64
2,38
2,18
2,00
Таблица 12
Влияние метиодида 2-пиридинкарбальдоксима на кинетику
гидролиза 1-нафтилацетата, катализируемого
ацетилхолинэстеразой. Условия опыта: рН 6,85; 25° С;
ионная сила 0,1М (NaCl); 1,6% метанола
[11-10', М
0
1,04
2,56
lS]o-10', М
10,0
5,00
3,33
2,50
1,43
10,0
5,00
3,33
2,50
1,43
10,0
5,00
3,33
2,50
1,43
v, >сл. ед.
5,00
3,30
2,50
2,00
1,25
2,08
1,73
1,47
1,28
0,91
1,19
1,06
0,955
0,87
0.69
Таблица 13
Влияние СоА на начальную скорость реакции фосфотрансацетилирования,
катализируемую фосфат-ацетилтрансферазой. Условия опыта: рН 8,32; 25° С;
ионная сила 0,ЗМ; [Е]0=5-10_8М
[СоА]-10s, M
0
2,5
4,0
6,7
12,0
0
4,0
12,0
18,0
21,0
0
4,0
8,0
12,0
16,0
22,0
0
3,0
7,5
10,0
17,5
0
6,0
10,0
15,0
22,0
0
5,5
10,0
16,0
25,5
[Ас—СоА]. 10s, M
3,5
3,5
3,5
3,5
3,5
8,5
8,5
8,5
8,5
8,5
12,5
12,5
12,5
12,5
12,5
12,5
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
7,4
[фосфат] • 10", М
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
6,1
2,0
2,0
2,0
2,0
2,0
4,0
4,0
4,0
4,0
4,0
6,0
6,0
6,0
6,0
6,0
v-10s, М-мин
2,94
2,18
1,88
1,50
1,09
6,25
4,35
2,64
2,04
1,82
7,70
5,30
4,00
3,30
2,80
2,22
1,74
1,30
0,935
0,815
0,582
2,94
1,75
1,39
1,09
0,835
3,85
2,38
1,82
1,39
0,99
5-15. В работе [13] изучалось активирующее влияние
красителя профлавина на гидролиз этилового эфира М-бензоил-Ь-аргини-
на, катализируемый папаином (табл. 14). Исходя из двухстадий-
94
ного механизма ферментативной реакции, предложить
кинетическую схему активации и вычислить возможные кинетические и
равновесные параметры реакции.
Таблица 14
Влияние профлавина на кинетику гидролиза этилового эфира
^бензоил-Ь-аргинина, катализируемого папаином. Условия опыта: рН 6,6;
25° С; ионная сила 0,3M (KC1); 5 10-3М цистеина; 1-10-3М ЭДТА;
[Е]0= 1,2.16-6М
[профлавин]-10*1 М
0
0,49
3,08
[Slo-10*, M
5,0
2,0
1,25
0,8
0,5
0,4
5,0
2,0
1,25
0,8
0,5
0,4
5,0
2,0
1,25
0,8
0,5
0,4
г/-106, М-сек—'
1.62
1.24
0,995
0,770
0,565
0,470
1,77
1,42
1,20
0,86
0,72
0,61
1,85
1,60
1,42
1,20
0,96
0,84
5-16. В таблице 15 приведены данные по влиянию профлавина
на кинетические параметры гидролиза этилового эфира N-бензоил-
глицина, катализируемого папаином [13]. Исходя из
предположения о двухстадийном механизме ферментативной реакции,
предложить кинетическую схему реакции папаина в присутствии
профлавина и вычислить возможные кинетические и равновесные
параметры реакции.
Таблица 15
Влияние профлавина на гидролиз этилового эфира N-бензоилглицина,
катализируемый папаином. Условия опыта: рН- 6,6; 25° С;
ионная сила 0,3M (KC1); 5-10~3М цистеина; Ы0-8М ЭДТА, [Е]0 = 1,2-10-вМ
[профлавин]-1С*, М
0
0,492
3,080
"кат, сек
2,29
2,32
2,44
*т<Каж)-10Э- М
12,9
7,7
4.2
05
5-17. При изучении гидролиза п-нитроанилида L-лейцина,
катализируемого лейцинаминопептидазой, было найдено, что добавки
катионов Mg2+ приводят к возрастанию каталитической константы,
не влияя на величину кажущейся константы Михаэлиса [14]
(табл. 16). Предложить кинетическую схему ферментативной
реакции в присутствии катионов Mg2+ и вычислить возможные
значения кинетических и равновесных параметров реакции.
Таблица 16
Влияние катионов Mg+2 на кинетические параметры гидролиза п-нитроанилида
L-лейцина, катализируемого лейцинаминопептидазой.
Условия опыта: рН 8,5; 25° С; [Е]0 = 3,32-10-7М
[Mg2+]-lCP, М
0
0,1
0,5
10
100
*кат. сек *
0,572
0,745
1,37
6,31
10,3
к 1 )-103, М
1,01
0,98
1.06
0,97
1,00
5-18. В работе [15] было продемонстрировано активирующее
действие метирапона на реакцию превращения фенилэтиленоксида
в фенилэтиленгликоль, катализируемую эпоксид-гидразой. На
основании данных табл. 17 предложить кинетическую схему
активации ферментативной реакции и оценить значения кинетических
и равновесных параметров реакции.
Таблица 17
Влияние метирапона на гидратацию стирол-оксида,
катализируемую эпоксид-гидразой. Условия опыта: рН 9,0; 37° С
[метирапон]-103, М
0
0,25
2.0
[S]„ Ю*. М
2,5
2,0
1,5
1,0
2,5
2,0
1,5
1,0
2,5
2,0
1,5
1,0
v, усл. ед.
1,59
1,35
1.08
0,76
1,84
1,52
1,18
0,81
2.00
1,63
1,24
0,85
96
5-19. При изучении кинетики анаэробной реакции р-хлоралани-
на с оксидазой D-аминокислот было найдено, что цианид-анионы
ускоряют реакцию гидрирования фермента при окислении
субстрата [16]. Исходя из данных табл. 18, найти значение константы
диссоциации комплекса фермент-активатор и величину
максимальной скорости ферментативной реакции при избытке цианид-ионов.
Таблица 18
Влияние цианид-анионов на эффективную
константу скорости реакции первого порядка
гидрирования оксидазы D-аминокислот
в реакции окисления |3-хлораланина.
Условия опыта: рН 8,3; 25° С: 0,1М пирофосфата
натрия; [Щй = 1,4-10-6Л1; IS10 = 410-SM
[KCNl-lO*, M
0
1,0
2,5
5,0
7,5
10,0
*эфф, сек '
0
0,013
0,023
0,033
0,036
0,039
5-20. При изучении кинетики гидролиза пептидного субстрата,
катализируемого карбоксипептидазой В, был обнаружен
активирующий эффект добавок к-бутанола [10]. Исходя из данных
табл. 19, предложить кинетическую схему реакции в
предположении двухстадииного механизма действия фермента и рассчитать
константу активации н-бутанолом.
5-21. Известно [17], что неконкурентное ингибирование
ферментативной активности а-химотрипсина борной кислотой
обусловлено взаимодействием ингибитора с имидазольной группой остатка
гистидина-57 активного центра фермента. В табл. 20 приведены
результаты совместного воздействия борной и н-гексилборной
кислот на кинетику гидролиза анилидного субстрата, катализируемого
а-химотрипсином [18]. Определить, принимает ли гистидин-57
активного центра фермента участие в связывании н-гексилборной
кислоты.
5-22. Авторами [19] был предложен аналитический метод
определения параметров Кпцкащ и Vm ферментативной реакции,
согласно которому значение Кт(каж)1Ут, определяемое как
котангенс угла наклона касательной к экспериментальной кривой в
координатах (v, [S]o) (при малых концентрациях субстрата),
подставляется в выражение
1 1 Кт(каж) 1
V^~ v Vm "IS],
(5.44)
4 Заказ 158
97
Таблица 19
Влияние н-бутанола на кинетику гидролиза гиппурил-Ь-аргинина,
катализируемого карбоксипептидазой В. Условия опыта: рН 8,0;
23° С, трис-ацетатиый буфер (0.025М); |Е]0 = 1,21 Ю"4 мг/мл
[н-бутанол], М
0
0.025
0,04
0,1
0,33
[S)0-lO*. М
10,0
1,5
1.0
0,67
0,50
10.0
1,5
1.0
0,67
0,50
10,0
1,5
1,0
0,67
0,50
10,0
1.5
1,0
0,67
0,50
10,0
1,5
1.0
0,67
0,50
v-10s, М-мин
1,79
0,92
0,71
0,54
0.43
2,44
1,29
1,11
0,85
0,69
2,67
1,57
1,28
0,98
0,80
3,33
2.18
1,82
1,45
1,19
4,17
3,13
2,64
2,22
1,89
полученное преобразованием выражения (5.11). Находя таким
образом величину Vm для всех экспериментальных значений
начальной скорости реакции (v) при соответствующих значениях
[S]0 и зная величину Ктгцкаж)/Ут для изучаемой реакции, можно
ВЫЧИСЛИТЬ Значение Кг>Мкаж)-
В таблице 21 приведены значения максимальной скорости
гидролиза дихлориндофенилацетата, катализируемого холинэстеразой,
рассчитанные при помощи описанного выше аналитического
метода [19] при условии, что котангенс угла наклона касательной
к кривой в координатах (и, [S]0) при малых концентрациях суб-
98
страта равен 0,023 усл. ед.-'М-сек. Очевидно (табл. 21), что
вычисленная величина Vm последовательно возрастает с увеличением
[S]o- Найти с помощью алгебраических методов выражение для
систематической ошибки и определить истинные значения Vm
Таблица 20
Влияние борной и н-гексилборной кислот на кинетику гидролиза
n-нитроанилида Ы-(3-карбоксипропионил)-Ь-фенилаланина,
катализируемого а-химотрипсином. Условия опыта: рН 8,0; 30° С,
ионная сила 0,1 М (NaCl); [S]0 = 1,3-10-4М
[H3B03], М
0
0,21
|C,H13B(OH)j]-103, М
1,3
2,6
4.0
5.1
6.5
0
1,3
2,6
4,0
5,1
6,5
V
1,45
2,00
2.40
2,75
3,25
3,70
4,15
4,50
5,05
5,45
5,93
и Хт(каж) для данной реакции при условии, что начальные скорости
реакции (v) при данных значениях [S] 0 определены с высокой
степенью точности. Точность проведения касательной к кривой в
координатах (v, [S]o) при [S] о -> 0 невелика.
Таблица 21
Зависимость начальной скорости гидролиза дихлориндофенилацетата,
катализируемого холинэстеразой, от концентрации субстрата
и значения максимальной скорости реакции, рассчитанные
с помощью уравнения (5.44) при условии
/Ст(каж)/Ут = 0,023 усл. ед.-'М-сек. Условия опыта: рН 8,0;
25° С; 0,05 М фосфатный буфер; 1Е]0 = 2-lO-"M
IS]0-M«, М
0,2
0,6
1,0
1.5
2,0
ti-103, усл. ед./сек
0,77
1,90
2,70
3.30
3,85
^„•Ю", уел- ед./сек
3,0
5,2
5,9
6.0
6,2
99
Ответы и решения
5-1. Ответ: (см. рис. 36): Vт = 6,58- Ю-6 М сек-1; £ка
= 0,173 сек-ь /Гт(каж) = 8,77- Ю~2 М.
5-2. Ответ: £КЯТ = 0,32 сек-1; /^т(каж) = 1,4Ы0-з М.
5-3. Ответ: kKaT = 5,08 сек-1; /^т(каж)=- 1,43-Ю-2 М.
5-4. Ответ: £кат = 57,3 сек-1; Кт(ыЖ) = 2,96- Ю-4 М.
5-5. Ответ: £кат = 66,5 сек-1; /Ст(каж) = 2,13-10—5 М.
5-6. Ответ: (рис. 37): £кат = 2,92-104 ед-л/сек-моль;/Ст(каж)=
= 0,04 мг/мл.
£40
^ 30
i 2,0
-1/К """ ^
-*
III!
-/,0 -0,5 0 0,5 V '.5
V[s]n/oV
3.0
СО
и
v^ 2.0
' о
III!
-4,0 0 4.0
7[S] JO,
0
8.0
Рис. 36. Определение
максимальной скорости и кажущейся
константы Михаэлиса для гидролиза
метилового эфира К-ацетил-Ь-ва-
лина, катализируемого а-химо-
трипсином, в координатах Лай-
нунвера-Берка
Рис. 37. Определение кинетических
параметров гидролиза бактериальных
клеток Мс. lysodeikticus,
катализируемого лизоцимом
5-7. Ответ: &кат = 6,53 сек-1; Кт(каж)= 1,85-10—2 М.
5-8. Ответ: £кат = 0,109 сек-1; /^т(каЖ) = 0,862 М.
5-9. Обрабатывая данные табл. 9 в координатах Лайнуивера-
Берка (рис. 38) находим значение Кт (каж) при [I] - - 0 и значения
А'т(каж) при двух концентрациях ингибитора. Откладывая
найденные значения констант А4ихаэлиса в координатах (Кт(квж)/К°т(кажу
[I]), получим прямую линию, тангенс угла наклона которой равен \/К] '
Ответ: К\ = 6.2 • 10~4 М.
5-10. Из графика в координатах Лайнуивера-Берка (рис. 39)
видно, что ингибирование ферментативной активности имеет
неконкурентный характер, и прямая в координатах (v0lvi, [I])
указывает на полный неконкурентный тип ингибирования (рис.40).
Для определения величины Д'г можно использовать или
координаты (vo/vi, [I]), прямая линия в которых имеет тангенс угла
наклона, равный l/Ki (рис. 40) или координаты Диксона (рис. 41).
Ответ: Ki = 2,2-Ю-5 М.
100
5-11. Делением друг на друга выражений (5.6) и (5.15; можно
показать, что для конкурентного типа ингибирования выполняется
соотношение
V
К.
Ь+%)
+ [S].
или
= 1 +
Ks + [S].
^i(^s + [S]„) '
Если отношение скоростей реакций в отсутствие (v) и в
присутствии конкурентного ингибитора (vi) равно двум, то концентрация
ингибитора при этом равна
I
50"
■Ki
('+%)
(5.45)
2,0
т
т* Ь5
X
■> 1-°
^г
05
/е
— -ч, /
/ б
г J* a
- 9 rJr ^^^
ill
2 4
1/[S]010",V
Рис. 38. Конкурентное ингибирова-
ние а-кетоглутаратом реакции
окисления, катализируемой N-ме-
тилглутамат-дегидрогеназой.
Концентрации ингибитора (а) — 0;
(б) —6-Ю-4 М; (е) — 3-Ю-3 М
<=t ^cr
% 1,0
с_>
-у>
■51 05
1 1 1 1
-1J0
1,0 2,0
'/[s]orc,V
Рис. 39. Неконкурентное ннги-
бирование бензоаточ 1,2,5-три-
метилпиперидола-4 реакции
гидролиза бутирилхолина,
катализируемой холипэстеразой.
Из выражения (5.45) видно, что в случае конкурентного типа
ингибирования показатель 150 не может быть принят за меру инги-
бирующей способности эффектора при произвольной
концентрации субстрата.
Для неконкурентного типа ингибирования выполняется
отношение
v
V,
1 +
HL
/с,
101
В этом случае концентрация ингибитора, вызывающая уменьшение
скорости ферментативной реакции вдвое, численно равна константе
неконкурентного ингибирования
ho = К\ •
5-12 Ч Из графика в координатах Лайнуивера-Берка (рис. 42)
очевидно, что н-бутанол ингибирует ферментативную реакцию по
смешанному типу. Обрабатывая
экспериментальные данные с
помощью общего уравнения
скорости реакции (5.14), находим зна-
ZD5
Ш-/0.М
Рис. 40. Определение
константы неконкурентного
ингибирования гидролиза бутирилхоли-
на, катализируемого холин-
эстеразой
12
0,6
1
п/г
/ J^
1 j i
-3,0 -2,0 -1,0 0
W 20 3,0
ГН-я/Гм
Рис. 41. Определение в координатах
Диксона константы неконкурентного
ингибирования гидролиза бутирилхо-
лина, катализируемого холинэстера-
зой. Концентрации субстрата (а) —
1-Ю-3 М; (б)—2,5-Ю-4 М; (е)—
9,1-Ю-5 М; (г) — 5-IO-s M
чения параметров схемы (5.13) а, (3, /G (рис. 43). Найденные
значения кинетических и равновесных параметров ферментативной
реакции можно также проверить с помощью координат общей точки
пересечения семейства прямых на графике Лайнуивера-Берка
(см. выражения 5.25—5.26). Так как для данной ферментативной
реакции константа промотирования р = 0, то общую схему
реакции в присутствии н-бутанола можно представить в виде
^ S к
Е + STUPES—-> Е + Р.
«I
L J
ffi+s;
оЯс
IES
**i
(5.46)
1 В работе [10] количественная обработка ингибирования не проводилась.
102
Ответ: Ks = 4,17- Ю-5 М; Vm = 2,33- Ю-5 М-мин-'; <х =
==0,27; р = 0; Ki =0,15 М.
5-13. Как отмечалось в теоретическом введении к настоящей
главе, параллельность прямых в координатах Лайнуивера-Берка
(рис. 44) может указывать на бесконкурентный (схема 5.13,
а = р < 1) или антиконкурентный (схема 5.13, р = О, КЭ = °°)
характер ингибирования,
различить которые можно построением
в координатах Диксона (1/а,
[I]). При бесконкурентном типе
ингибирования линеаризация в Т
координатах Диксона не должна
наблюдаться (см. выражение
5.30). В случае антиконкурентно- ч
го типа ингибирования прямые в
координатах Диксона, соответст-
2,0
1,5
1,0
0,5
L
-- ^
1
а*
^^к.
<*&>-ii
i i i
i 1
■'о; ^
! 0,5
X
'± 0,4
ijo .
'S 0,3
■^^-2
-01
с/г
_r/P jf^b
-У^\у^6
r&cf\ra-s>'^a
у 1 1
2 4 6
- 1 -4 -i
^ &■"■"'
0
10 15
20
10 15 20
Рис. 42. Иллюстрация смешанного
типа ингибирования н-бутанолом
реакции гидролиза гиппурового эфира
L-аргинина, катализируемого карбо-
ксипептидазой В. Концентрации
ингибитора (а) — 0; (б) — 0,05 М; (е) —
0,15 М; (г)— 0,30 М
Рис. 43. Определение кинетических
и равновесных параметров
ингибирования н-бутанолом гидролиза
гиппурового эфира L-аргинина,
катализируемого карбоксипептида-
зой В, с помощью обработки
значений каталитических констант
(а) и констант Михаэлиса (б) в
координатах уравнения (5.14)
вующие различным концентрациям субстрата, должны быть
параллельными, что и наблюдается для данной ферментативной реакции
(рис. 45). Из тангенса угла наклона в координатах Диксона,
равного l//CiVm, можно найти константу ингибирования
фермент-субстратного комплекса (Ki). Итак, в данной ферментативной
реакции ингибитор связывается только с фермент-субстратным комп-
103
лексом и не связывается со свободным ферментом
Е +STATES—^Е + Р
IES
Ответ: К\ =4,05-Ю-8 М.
5-14. По отношению к нуклеотидному субстрату кофермент А
является конкурентным ингибитором с константой ингибирования,
2.0 4.0 6,0 8.0
?/[S]-!0,M
Рис. 44. Антиконкурентное
влияние метйодида 2-пиридиикарбаль-
доксима на кинетику гидролиза
1-нафтилацетата, катализируемого
ацетилхолинэстеразой.
Концентрации ингибитора (а) —0; (б) —
1,04-Ю-7 М; (е)—2,56-10-7 М
! '3,0 -2,0 -1,0 0 1,0 2,0 30
[1]ЮМ
Рис. 45. Обработка кинетических
данных, соответствующих
антиконкурентному типу ингибирования, в
координатах Диксона
равной 6,3- 10~5 М (рис. 46). По отношению к неорганическому
субстрату кофермент А является неконкурентным ингибитором
с константой ингибирования 9- Ю-5 М.
5-15'. Как видно из рис. 47, добавление активатора не влияет
на величину лкат, но уменьшает /Ст(каж) ферментативной реакции.
В применении к общему уравнению (5.14) это соответствует
случаю активации, которую можно назвать синергистическои, так как
связывание активатора с активным центром фермента увеличивает
сродство фермента к субстрату, и наоборот (а< 1, р = 1). Таким
образом, схему (5.13) в применении к данному случаю можно за-
1 В работе [13] количественная обработка экспериментальных данных
не проводилась.
104
писать в виде
E+SZ=I ES-^E + P
А I А
АЕ + S ;ZZ!AES > АЕ -f P
и уравнение скорости реакции примет вид
*» [Е]. [S].
•V
, * А + [А]
•sa/CA4 [Л]
(5.47)
/5 ?0
Рис. 46. Применение координат
Диксона для определения константы
конкурентного ингибирования кофер-
ментом А реакции фосфотрансацети-
лирования, катализируемой фосфат-
ацетилтрансферазой. Концентрации
нуклеотидного субстрата (Ацетил-
СоА): (а) 1,25-10-* М; (б)—8,5Х
X Ю-5 М; (е) —3,5-10-5 М
1/щюУ
Рис. 47. Синергистическая активация
профлавином реакции гидролиза
этилового эфира 1Ч-бензоил-Ь-аргинина,
катализируемой папаином. Концепт-
рации активатора (а) —0; (б) —
4,9-Ю-5 М; (е)—3,08-10-* М
Для определения параметров уравнения (5.47) проведем
преобразование
А т(каж) = аК s ..- , (0.4С)
К
т(каж)
' °Ка + [А] '
_j , aI<A _L
1 - а ~Т" 1 — а ' [А]
(5.49)
Откладывая значения констант Михаэлиса, найденные из рис. 47,
в координатах
^■т(каж) ' [А] |
V1" «* /
тг; \, найдем значения а и Ка-
105
Ответ: ^2=14,5 сек-1; Ks= 1,33- Ю-2 М; а=0,33; /СА=2-10~4 М.
5-16 :. Решается с использованием общей схемы (5.13) и
общего уравнения (5.14), аналогично задаче 5-15.
Ответ: k2 = 2,29 сек"1; Ks = 1,29-10-2 М; а = 0,23; р = 1,02;
Ка= 1,9 ■ Ю-4 М.
5-172. Из графика в координатах Лайнуивера-Берка (рис. 48)
видно, что влияние катионов Mg2+ на ферментативную реакцию
относится к неконкурентному типу
активации (см. § 3) и описывается
схемой
Е + S 7=^ ES —--> Е + Р
Кк
АЁ + S ~ AES -
(5.50)
PS, 1
АЕ + Р
где Р > 1. Уравнгние скорости реакции
в данном случае имеет вид
Ка + ИА]
0
VlS]oVm
Рис. 48. Неконкурентная
активация катионами Mg++
реакции гидролиза п-нитроанилида
L-лейцина, катализируемого
лейцинаминопептидазой.
Концентрации активатора (а) — 0;
(б)-Ы0-1.М; (е)—5-10-4М;
(г)— I -10—2 М;" (б)—ЫО-'М
v ■■
КА + [А]
[Е]о [S]„
a:s + [sj0
(5.51)
Для определения параметров р и К а ,
выражение для kKST в (5.51) можно
преобразозать к виду (5.52) и
анализировать последнее в координатах
Ч Акат/^2-1 ' ТА]/'
гкат/^2 1
1
[А]
1
Кд
^кат/^г
1
р-1
1
ж (5-52)
Ответ: /г2=0,572 сек-,; Ks=l-10~3 М; КА=5,6-10~3 М; р=18.
5-183. Из графика в координатах Лайнуивера-Берка (рис. 49)
видно, что активатор увеличивает в одинаковой степени значения
&кат и /Ст(каж), что соответствует случаю бесконкурентной
активации (а = р > 1, см. схему 5.13 и выражение 5.14). Таким образом,
данная ферментативная реакция описывается схемой
+ S '—* FS —--* Е + Р
К А
aKS
AE-f-S
i
AES
(5.53)
afc5
HAE-f P
1 В работе [13] количественная обработка экспериментальных данных
не проводилась.
2 В работе [14] количественная обработка явления активации не проводилась.
3 В работе [15] количественная обработка экспериментальных данных
не проводилась (прим. авторов).
106
и уравнение скорости реакции можно записать в виде
К к + ГА]
v = А + [ ' (5.54)
КА + [А] • v '
aKs о.кА + [А] + Is!»
Для определения значений констант а и /С а выражение для £кат
в уравнении (5.54) можно преобразовать к виду
I = _L_+^L _J_ (5.55)
и анализировать его в координатах ( -—-гт р, Тдт)-
\ «кат/«г — 1 [гЦ /
Ответ: Ks = 6,6.10-4М; /СА= 1,8-Ю-4М; а = 5.
5-19. Схему активации реакции гидрирования фермента
(проходящей по первому порядку) можно представить в виде
ES —-->- Продукты
*А
I ft,
AES * Продукты
Запишем выражение для скорости реакции
v = kx [ES] + k2 [AES], (5.56)
уравнение материального баланса
[ES]„ = [ES] + [AES] (5.57)
и выражение для гонстанты равновесия
К ь-Sffl-, (Б.58,
затем, подставив (5.57), (5.58) в (5.56), найдем ьыражение для
эффективной константы скорости реакции
kj + fc2
k
[А]
*А
ФФ [ES]0 ГА] •
+ *А
Согласно данным табл. 18, kl = 0. Отсюда
, k2 [A] _L_ = ±.^a J_
эФф КА + [А] *9фф k2 + k2 • [А]
107
Обрабатывая данные табл. 18 в координатах (1/&эфф, 1/[А]),
найдем значения /ег и Ка (рис. 50).
Ответ: k2 - - 0,05 сек-1; К а = 2,8- Ю-2 М.
0,25 0,50 0у75
'/[sJ-roV
Рис. 49. Бесконкурентный тип
активации метирапоном
ферментативной активности эпоксид-гидра-
зы в реакции гидратации стирол-
оксида. Концентрации активатора
(а)—0; (б)— 2,5-Ю-4 М; (е)—
2-Ю-3 М
100
е-
8-
У
у
-50
50 100
'/[KCNj.M
Рис. 50. Определение кинетических и
равновесных параметров активации
цианистым калием гидрирования окси-
дазы D-аминокислот в реакции с
Р-хлораланином
5-20.1 н-Бутанол является смешанным активатором
ферментативного гидролиза гиппурил-Ь-аргинина (рис. 51). Определение
кинетических и равновесных параметров ферментативной рекции (схема
5.13) можно провести по методу, аналогичному применявшемуся
в решении задачи 5-12 с помощью обработки экспериментальных
данных в координатах (^—р, -L) и (T_-L__ _^).
V,,
2,13-Ю-5 М; а = 0,2; р = 2,3;
Ответ- Ks = 2-10-4 М;
Ка = 0,15 М.
5-21. График в координатах (v/vi, [I]), где [I] —концентрация
н-гексилборной кислоты (рис. 52), указывает на несовместимость
1 В работе [10] количественная обработка активации ферментативной
реакции не проводилась (прим. авторов).
108
борной и н-гексилборной кислот при связывании их с ферментом
(см. § 4 настоящей главы). Таким образом, оба ингибитора
связываются с одной и той же группой активного центра фермента
(в данном случае — с остатком имидазола-57).
5-22 [19]. Из условия задачи можно заключить, что источник
систематической ошибки — недостаточная точность проведения
касательной к кривой в
координатах (у, [S]0) при [S]o-^0. Так
как котангенс угла наклона этой
касательной равен KmlVm, то
примем, что истинное значение
(Кт/У,т,)тт отличается от эксие-
* 0,20
'о
s>
^ 0,10
-~-~~~1^^Ф
Уа
/ Б
s ^/>* г
/sjr^0'— °
.1111
-1.0
V .4 $°
Рис. 51. Смешанная активация н-бу-
танолом гидролиза гнппурнл-L-apni-
нина, катализируемого карбоксипеп-
тидазой В. Концентрации активатора
(а)—0; (б)—2,5-Ю-2 М; (е)—
4-Ю-2 М; (г) —ЫО-'М; (д) —
3,3-Ю-1 М
6.0
[сбн13в(он)2] ю,3м
Рис. 52 Влияние двух
взаимозависимых ингибиторов — борной и
н-гексилборной кислот — на
скорость реакции гидролиза амидного
субстрата, катализируемого а-
химотрипсином: (а) — без
добавления НзВОз, (б) — концентрация
Н3ВОз равна 0,21 М. Пунктирная
прямая соответствует
альтернативному механизму взапмонсзави-
симого ингибнровапия
рименталыю
MKjVm),
найденной величины
fKm\ .._ (Кт\
\УтЛст \VmJb
(Am/ Vт)экс
на
величину
(5.59)
Найдем выражение для A (KmlVm).
Из выражения (5.44) следует, что для любых двух концентраций
субстрата [S]t и [S]2 будут справедливы следующие соотношения:
тЬ-£-(£)«-тзр (5-60)
(5.61)
109
_ 1 (Кт\
[S], •
_J
[Sb •
Вычитая (5.61) из (5.60), получаем
А fc) = т: - тг ~ (т^)9КсП [т§г ~ ikl (5'62)
С другой стороны,
_L__L = (4^) [4—^1. (5-63)
v, t>2 V Vm /ист L [S], [S]2 J ■ v '
Подставляя выражение (5.63) в (5.62), находим
ИЛИ
4 (0-4fc) [-иг-гаг]- (5М)
Среднюю величину систематической ошибки A(Km/Vm) можно
рассчитать из уравнения (5.64) при использовании комбинаций
экспериментальных значений [S]0 и v из табл. 21. Затем с помощью
уравнения (5.59) можно найти пределы изменения (KmlVm)vci,
варьируя в которых можно добиться постоянства величины lfVm,
рассчитываемой по уравнению (5.44). Наконец, зная истинные
значения величин /(т(каж)/Ут и Vm, можно вычислить значение
Лтп{каж) ■
Ответ: 1/т=(6,94±0,14)-10-3 усл. ед./сек; Ят(1<аж)=(1,59гЬ
±0,04) • Ю-4 М.
ЛИТЕРАТУРА
1. Hess G. P., Mc Conn J., Ku E., Мс С on key G. .Phil. Trans. Roy
Soc. Lond.", B257, 89, 1970.
2. Z erne г В., В e n d e г M. L. .J. Amer. Chem. Soc", 86, 3669, 1964.
3. Яковлев В. А. Кинетика ферментативного катализа. М., .Наука*, 1965,
стр. 42.
4. Берез н и И. В., М а рти н е к К. „Мол. биол.", 5, 347, 1971.
5. Butterworth P. J. „Biochim. Biophys. Acta", 289, 251, 1972.
6. Березнн И. В., Мартинек К. „Теор. эксп. химия", 3, 458, 1967.
7. Berezin I. V., К a z a n s k ay а N. F., Klyosov A. A. „FEBS Letters",
15, 121, 1971.
8. Н е г s h L. В., Stark M. J., Worthed S., F i е г о М. К. .Arch. Biochem.
Biophys.", 150, 219, 1972.
9. Унковский Б. В., Черкасова Е. М., Луцык А. И., Богат-
ков С. В., Соколова Т. Д., Малина Ю. Ф. .Биохимия", 37, 1156.
1972.
10. F о 1 k J. Е., Wolff Е. С. S с h i г m е г Е. W.. С о г n f i e 1 d J. „ J. Biol.
Chem.", 237, 3105, 1962.
11. Rosen berry T. L., Bernhard S. A. .Biochemistry", 11, 4308, 1972
1 Вследствие громоздкости расчетного аппарата данный метод представляет
лишь теоретический интерес и, не имея преимуществ по сравнению с
классическими методами определения кинетических параметров ферментативной реакции,
не может быть рекомендован в качестве альтернативного для обработки
реальных систем.
ПО
1'2. Ку г topoulo s S. A.. Satchell D. P. N. „Biochim. Biophys. Acta',
276, 383, 1972.
13. Hall P. L., Anderson С D., Crawford G. D. „Arch. Biochem.
Biophys.", 153, 162, 1972.
14. Lasch J., Kudernatsch W., Hanson H. „Eur. J. Biochem.", 34, 53,
1973.
15. Oesch F., Kaubisch N., Jerina D. M., D a 1 у J. W, „Biochemistry",
!0, 4858, 1971. .
16. Porler D. J. Т., Voet J. G., Bright H. J. „Biochem. Biophys. Res.
Commun. ", 49, 257, 1972.
17. Бе резин И. В., Коломнйцева Г. Я., Левашов А. В., М а р т и-
не к К. „Мол. биол.", 1, 425, 1967.
18. Антонов В. К., Иванина Т. В., Березин И. В., МартинекК.
„Мол. биол.", 4, 558, 1970.
19. В г est k in A. P., Rozengart Е. V., Samokish V. A., S о b о 11>
val. N. „Biochim. Biophys. Acta.", 191, 155, 1969.
Глава 6
КИНЕТИЧЕСКИЙ АНАЛИЗ ДВУХСТАДИИНЫХ
ФЕРМЕНТАТИВНЫХ РЕАКЦИИ,
НЕ ПОДЧИНЯЮЩИХСЯ УРАВНЕНИЮ
МИХАЭЛКСА — МЕНТЕН
§ 1. Ингибирование субстратом
Согласно уравнению Михаэлиса — Ментен (5.7), при
увеличении концентрации субстрата начальная скорость ферментативной
реакции гиперболически возрастает, стремясь к своему
предельному значению, называемому максимальной скоростью реакции.
Однако в ряде случаев при увеличении концентрации субстрата
начальная скорость ферментативной реакции проходит через
максимум и затем уменьшается. Обычно подобный тип
зависимости v от [S]0 можно количественно описать, исходя из
предположения об образовании тройного комплекса ES2, не обладающего
ферментативной активностью
*s
E+S7=^ES ЛЕ +P. (6.1)
+
ES2
Кинетическая обработка схемы (6.1) обычными способами (см. § 1,
гл. 5) приводит к уравнению скорости ферментативной реакции
MEUS].
I
Ks +[S]„+-
v = K'l"j0loJ° „ (6.2)
[Sf0
Анализ данного уравнения целесообразно проводить раздельно
при низких ([S]o'C/Gs/) и высоких ([S]o^>/Cs) концентрациях
Ш
субстрата. В первом случае уравнение (6.2) упрощается до
классического уравнения Михаэлиса — Ментен
v = -2[EjofS]o (6 3)
Ks + IS]. ' ^'°'
линеаризацией которого в подходящих координатах (см. гл. 5)
можно найти значения k2 и Ks,- При высоких концентрациях
субстрата уравнение (6.2) примет вид
v= *,[g? , (6.4)
линеаризация которого в координатах (\/v, fS]o) позволяет найти
значения ki и Ks'- Совпадение значений k2, найденных при
использовании уравнений (6.3) и (6.4), является доказательством
применимости схемы (6.1) для формального описания кинетики
изучаемой ферментативной реакции.
Ингибирование субстратом при высоких его концентрациях
будет также наблюдаться, если тройной комплекс ES2 будет
обладать активностью, но меньшей по сравнению с
фермент-субстратным комплексом (р < 1, схема 6.5).
E + SZ=^ES--E + P (6.5)
ES2—-ES + P.
В этом случае скорость ферментативной реакции будет
описываться уравнением
(,, + №) И.и.
' - " isi— (6'6)
A's + [S]„ -f ——
Для того чтобы определить на опыте, обладает ли
ферментативной активностью тройной комплекс ES2, следует построить
зависимость экспериментально найденных значений скорости в
координатах (v, log [S]0). Симметричный вид полученной колоколооб-
разной кривой указывает на отсутствие ферментативной
активности комплекса ES2. Если же правая ветвь графика (при больших
концентрациях субстрата) более пологая по сравнению с левой
ветвью, это указывает на величину р, отличную от нуля (0<р<
< 1, схема 6.5). В этом случае уравнение (6.6) при высоких кон-
112
центрациях субстрата можно записать в виде
>[S].
1
1
К
£9фф = ^2 [Sjo ■ (6-7)
где'^дфф = v/[E]0.r Преобразовывая выражение (6.7), получим
1 1 , Ks 1
Из полученного выражения видно, что зависимость в координатах
(1/(1 — &)фф/&г), l/[S]o) при высоких концентрациях субстрата
должна иметь вид прямой, из тангенса угла наклона которой и
ординаты точки перееечения можно найти значения |3 и Ks'.
Существуют виды субстратного ингибирования, когда к
фермент-субстратному комплексу присоединяется несколько молекул
субстрата, переводя его в неактивное состояние. Для анализа
подобных случаев разработан графический способ [1],
позволяющий определить число молекул субстрата в неактивном фермент-
субстратном комплексе. Если п — число дополнительных молекул
субстрата, присоединившихся к фермент-субстратному комплексу,
то, откладывая экспериментальные данные в координатах
(1/[S]" + [S] o/Ks-Ks', i/v), подбирают значение п. при котором
наблюдается линеаризация
_L . [S]" -Xhl А L /fi ол
Для применения данного метода необходимо предварительно
графически найти значение концентрации субстрата, при которой
скорость ферментативной реакции достигает оптимума ([S]oht).
Из соотношения
«+i
[S]onT=j/7Cs-/Cs//i (6.10)
можно найти произведения Ks-Ks', соответствующие различным
значениям п, и, подставляя их в выражение (6.9), произвести
обработку экспериментальных данных, последовательно варьируя п.
§ 2. Активация субстратом
Случаи активации субстратом ферментативных реакций были
обнаружены экспериментально лишь в последнее время (см.
задачи настоящей главы). Простейшая интерпретация полученных
кинетических данных может быть проведена в рамках схемы (6.5)
при условии р > 1. Обработка экспериментальных данных в этом
случае проводится так же, как и при анализе ингибирования
ферментативной реакции субстратом (6.6) — (6.8). Следует лишь отме-
113
тить, что при р>1 (схема 6.5) линеаризацию кинетических
данных, соответствующих высоким концентрациям субстрата, удобно
проводить в координатах
1
. Яэфф/«2 1
V[S]0Y
§ 3. Кинетика ферментативных реакций
при условии [EJo«i[Slo. Определение
абсолютной концентрации фермента из
кинетических данных
Все рассмотренные в предыдущих разделах методы обработки
кинетических данных относились к условиям избытка субстрата по
сравнению с ферментом ([S]0 3> ГЕ] о) - Рассмотрим теперь случай,
когда концентрация субстрата сравнима по величине с
концентрацией фермента ([S]0«[E]o), и выведем уравнение скорости
ферментативной реакции, протекающей по двухстадииному механизму
при условии быстрого установления равновесия на стадии
образования фермент-субстратного комплекса
Е + S ^ИГ ES —- Е + Р. (6.11)
Особенностью обработки схемы (6.11) в случае [EJo^ [S] о
является необходимость использования уравнений материального
баланса как по ферменту (6.12), так и по субстрату (6.13), отчего
подобные системы называют иногда «системами со взаимным
истощением» [2]
[Е]0 = [Е] + [ES], (6.12)
[S]o = [S]+[ES]. (6.13)
Подставляя в уравнение (6.12) значение [ES], найденное из
обычного выражения для константы равновесия
tfs = ^jj* (6Л4)
и производя несложные преобразования, получим
и-Ж (6Л5)
Комбинируя далее выражения (6.13)—(6.15), найдем
[S]o = [S]+^|j. (6-16)
Выражение (6.16) можно записать в виде квадратного уравнения
[S]2 + *[S]-/Cs[S[o^O, (6.17)
114
где
« = Ks + [Е]0 - [S]0. (6.18)
Рещая уравнение (6.17), получим
fC1 -a+V°* + 4Ks[S)0
Подставляя значение равновесной концентрации [S] в
выражение (6.15), найдем значение равновесной концентрации [Е]:
[Е]
KS[E]0
Ks +
V<** + 4Ks[S]o-<*
lsr 2
Найденные значения [S] и [Е] подставим в общее выражение
для скорости ферментативной реакции
*> = ft2/#s[E][S],
получим
2^S — a + )/a2 + 4A:s[S]„ '
где Ут = £2 [E]0. Приводя далее выражение (6.19) к виду
1 2#Ч 1
— S = г- + ~ (6.20)
v Vm(ya* + 4Ks[S]0-a) ^ Vm \
и вводя в него равенство
-/iqlfci* —-*■ (6.2!)
получим соотношение
хг-г—г = х- (6-22)
Vm/v — i v >
Из формулы (6.22) видно, что каждому значению скорости
реакции v соответствует определенное значение "к. Таким образом,
значения К соответствующие значениям [S]0, можно вычислить
непосредственно из кинетических данных, если предварительно на
опыте найти значение максимальной скорости реакции.
Записывая равенство (6.21) в виде
2X^s + a=/a2 + 4/^s[S]0,
возводя обе части его в квадрат и подставляя значение а из (6.18),
получим выражение
VKs + Ч/Cs + [Е]„ ~ fS]o) - f S]0 - 0. (6.23)
ЗаписыЕая выражение (6.23) в виде
*/Cs(l + X) + A[E0] = [S]0(l-r-X)
115
и разделив обе его части на X JE]0(1 -j- X), придем к окончательному
выражению
l + х Х[Е]. [Е]0 V0"4*'
Очевидно, что зависимость (6.24) в координатах (1/1 + X, [S]oA)
должна быть прямолинейной с абсциссой точки пересечения,
равной Ks, и тангенсом угла наклона, равным 1/[Е]о- Таким образом,
изучение зависимости скорости ферментативной реакции от
концентрации субстрата в условиях [Ejo^ [S]0 позволяет определить
абсолютную концентрацию фермента. Так как выражения (6.12) —
(6.24) являются симметричными относительно концентраций
субстрата и фермента, абсолютная концентрация субстрата может
быть определена обработкой экспериментальных данных в
координатах (1/1 + Я, [Е]0/л) при фиксированной концентрации
субстрата, принимая Vm = &2[S]o
1 + X - X[S]. [S]. ' ( '
§ 4. Кинетика ферментативных реакций
при избытке фермента ([Е]0^ [S]o)
В отдельных случаях кинетические исследования
ферментативных реакций удобно проводить в условиях избытка фермента по
сравнению с субстратом (например, при малой растворимости
субстрата в воде, или при высоком молекулярном весе субстрата).
Детальный кинетический анализ подобного рода систем
проводится в главе 9. Здесь отметим только, что уравнение скорости
ферментативной реакции, протекающей в режиме установившегося
равновесия в условиях [Е]0 ^ [S]o, является симметричным
классическому уравнению Михаэлиса — Ментен относительно
концентраций реагентов. Так, при избытке фермента скорость реакции
имеет первый порядок по концентрации субстрата, и смешанный —
по концентрации фермента
v = К [Е1о [S]0 -.g 26i
Ks+ [E]. • <D-/D>
Прочие типичные случаи отклонения кинетического поведения
ферментативных реакций от уравнения Михаэлиса—Ментен
(реакция двух субстратов с одним ферментом, реакция двух
ферментов с одним субстратом, влияние примесей в препарате
фермента на кинетику реакции т. д.) будут рассмотрены при решении
соответствующих задач.
Задачи
6-1. 3,4-Дихлорфенилэтаноламин является одновременно
субстратом и ингибитором реакции трансметилирования,
катализируемой фенилэтаноламин-Ы-метилтрансферазой [3]. На основании
116
данных табл. 1 предложить схему ферментативной- реакции и
определить кинетические и равновесные параметры реакции.
Таблица 1
Зависимость скорости трансметилироваиия, катализируемой
феиилэтаиоламин-^метилтрансферазой, от концентрации субстрата
(Sl0-10», M
1,00
1.14
1,33
1,60
2,00
2,67
5,00
п-Ю'о, М-мин-'
4,СО
4,25
4,55
4,76
5,00
5,26
5,45
[Sb-io«, м
1 0,0
25,0
50,0
75,0
100,0
125,0
150,0
v-W°, М-мин-1
5,16
3,85
2,82
2,33
1,79
1,47
1,28
6-2. Реакция гидролиза йодида Ы-метил-7-ацетоксихинолина,
катализируемого ацетилхолинэстеразой, ингибируется субстратом
[4]. Исходя из данных табл. 2, определить кинетические и
равновесные параметры ферментативной реакции.
Таблица 2
Влияние концентрации йодида М-метил-7-ацетоксихинолина на кинетику
гидролиза, катализируемого ацетилхолинэстеразой. Условия опыта: рН 6,85;
25° С; 0.05М фосфатный буфер; 1,6% метанола
IS]0-№, M
0,125
0,143
0,167
0,200
0,250
0,333
0,500
1,000
v, уел, ед.
0,174
0,195
0,220
0,254
0,300
0,364
0,465
0,625
[SJo-Ю5. М
2,0
5,0
10,0
20,0
30,0
40,0
50,0
60,0
71,0
v, усл. ед,
0,735
0,833
0,800
0,715
0,645
0,572
0,514
0,476
0,431
6-3. n-Нитроанилид Ы-бензоил-Ь-аргинина является
одновременно субстратом и ингибитором гидролиза, катализируемого трипси-
ноподобным ферментом микробного происхождения [5]. Исходя
из данных табл. 3, найти кинетические и равновесные параметры
■ферментативной реакции.
6-4. Реакция восстановления пирувата, катализируемая лактат-
дегидрогеназой, ингибируется высокими концентрациями субстрата
[6]. На основе данных табл. 4 определить значения кинетических
и равновесных параметров ферментативной реакции.
117
Таблица 3'
Кинетические данные для гидролиза и-нитроаиилида Г^-бензоил-Ь-аргинииа,
катализируемого трипсииоподобным ферментом из St. fradiae.
Условия опыта: рН 9,0, 25" С, 0.02М СаС12
[S]o-10B, M
1,98
2,50
3,00
4,00
6,00
8,00
10,0
г//[Е]„, сек '
4,15
4,65
5,13
5,89
6,63
7,15
7,15
[Slo-10s, M
20,0
50,0
100
200
300
396
»/[Е]0, сек
7,50
6,91
5,72
4,22
3,30
2,74
Таблица 4
Зависимость скорости реакции восстановления пирувата, катализируемого
лактатдегидрогеназой, от концентрации субстрата. Условия опыта: рН 6,80;
27,5° С; 0.05М фосфатный буфер; [NAD-Н] = 1,29-10-BM; [E]0 = 2-10-s мг/мл
[Slo-103, M
0,020
0,029
0,040
0,050
0,100
0,500
г»-10", М-мин
3,15
3,70
4,10
4,39
4,92
4,90"
[Slo-lCP, М
0,70
1,10
3,00
5,00
7,00
10.00
гМО6, М-мин
4,30
3,90
2,40
1,80
1,50
1,10
6-5. При изучении кинетики гидролиза ацетилхолина,
катализируемого ацетилхолинэстеразой, было показано, что
ферментативная реакция ингибируется субстратом с константой диссоциации
неактивного тройного комплекса ES2 (см. схему 6.1), равной
3,2-Ю-2 М [7]. Найти значение концентрации субстрата, при
которой скорость ферментативной реакции достигает максимального
значения в условиях эксперимента, если величина константы Ми-
хаэлиса, найденная при использовании низких концентраций
субстрата, равна 2,6-10~4 М.
6-6. В работе [8] было показано, что окисление тестостерона
в Д5-андростен-3,17-дион под действием р-оксистероид-дегидрогена-
зы при увеличении начальной концентрации субстрата проходит
через максимум, достигая 76% теоретической максимальной
скорости реакции, при [S]oht = 6-10_6 М. На основании полученных
данных рассчитать значение константы диссоциации неактивного
тройного комплекса ES2 (см.. схему 6.1).
6-7. Скорость реакции гидролиза 0-(бензоилглицин)-2-оксиизо-
валериановой кислоты, катализируемой карбоксипептидазой А,
118
уменьшается в присутствии избытка субстрата [9] Исходя из
данных табл. 5, предложить схему ферментативной реакции и
определить ее кинетические и равновесные параметры.
Таблица 5
Зависимость скорости реакции гидролиза
0-(бензоилглицин)-2-оксиизовалериановой кислоты, катализируемой
карбоксипептидазой А, от концентрации субстрата. Условия опыта: рН 7,5;
25° С; ионная сила 0,5М (NaQ); IElo = 6,Ы0-'° — 1,2-10-вМ
[Slo-lC, M
0,400
0,445
0,5С0
0,572
0,667
0,800
1,000
1,410
1,780
г;/[Е]0, сек '
9,57
10,50
11,50
12,80
14,50
16,70
19,60
25,00
28,00
[S]„10«, М
2,00
3,16
4,СО
5,62
10,00
10,60
16,80
17,80
24,00
сек '
29,2
33,8
35,6
36,2
34,7
33,4
29,2
28,4
24,8
[Slo-10*, М
31,6
44,7
56,2
74,1
100,0
141,0
178,0
316,0
562,0
г,/[Е]0, сек '
21,00
16,70
15,00
12,50
10,60
8,&ч
7,50
5,83
4,80
6-8. В работе [10] было показано, что при увеличении
концентрации субстрата в реакции гидролиза Ы-карбобензокси-Ь-трипто-
фана, катализируемого карбоксипептидазой, скорость
ферментативной реакции проходит через максимум, затем снижается и
достигает постоянного уровня (табл. 6). Предложить схему реакции
и определить значения ее равновесных и кинетических параметров.
Таблица 6
Зависимость скорости гидролиза 1Ч-карбобензокси-Ь-триптофана,
катализируемого карбоксипептидазой, от концентрации субстрата.
Условия опыта: рН 7,5; 5° С; 0.04М фосфатный буфер; 0,5М КО
[S)o-10\ M
2,00
2,22
2,50
2.86
3,33
4,00
5,00
6,67
10,00
vllE]0, сек-1
7,70
8,33
9,00
9,80
10,80
11,80
12,80
13,90
14,90
[Slo-103, М
20,0
39,0
50,0
70,0
76,0
98,0
101,0
130,0
150,0
v /[Е]0, сек"'
15,0
13,6
12.8
12,0
П,8
10,9
11,0
10,9
10,8
6-9. При изучении кинетики гидролиза монофенилфосфата,
катализируемого щелочной фосфатазой, было найдено, что субстрат
119
является одновременно и ингибитором ферментативной реакции
[1]. На основании данных табл. 7 предложить схему реакции и
определить кинетические и равновесные параметры
ферментативного гидролиза.
Таблица 7
Влияние концентрации субстрата на скорость
гидролиза монофенилфосфата, катализируемого
щелочной фосфатазой. Условия опыта:
рН 10,0; 37° С; [Е10—4Ю"3 мг/мл
[5]0-103, М
0,5
1,0
2,0
4,0
5,0
6,0
7,0
10
12
p-llV, М-мин '
1,60
2,80
4,10
4,65
4,64
4,36
3,45
2,60
1,84
6-10. В реакции гидролиза метилового эфира п-толуолсульфо-
нил-Б-аргинина, катализируемого трипсином, субстрат является
одновременно активатором фермента [11]. Исходя из данных
табл. 8, предложить схему ферментативной реакции и определить
ее кинетические и равновесные параметры, если из экспериментов,
проведенных при небольших концентрациях субстрата (0,32—
— 50-Ю-5 М), были найдены значения kKaT = 0,74 сек-1; Кт(кащ =
= 2,7-10-* М.
Таблица 8
Зависимость скорости гидролиза метилового эфира я-тозил-О-аргинина,
катализируемого трипсином, от концентрации субстрата.
Условия опыта: рН 8,0; 25° С; 0,2М КО и 0.05М СаС12
[S]o, M
0,001
0,005
0,010
0,012
0,015
0,020
0,022
0,025
г//[Е]0, сек '
0,61
0,84
0,93
0,97
1,02
1,08
1,09
1,10
[S]e. М
0,026
0,027
0,030
0,032
0,042
0,050
0,100
WlElo. сек
1,11
1,13
1,12
1.16
1,18
1,21
1,30
120
6-11. При изучении кинетики гидролиза /г-нитроанилида N-бен-
зоил-Ь-аргинина, катализируемого трипсином, было обнаружено,
что при высоких концентрациях субстрат является также и
активатором ферментативной реакции [12]. На основании данных
табл. 9 определить кинетические и равновесные параметры
реакции, если при изучении кинетики ферментативного гидролиза
в области низких концентраций субстрата были найдены значения
&кат = 1,2 сек-'; Кт(кан<) = 3-10~4 М.
Таблица 9
Влияние концентрации субстрата на скорость
гидролиза /г-нитроанилида 1Ч-бензоил-Ь-аргинина,
катализируемого трипсином. Условия опыта:
рН 8,2; 25° С; 0.05М трис-НС1-буфер; 0.02М
СаС12; 1% диметилформамида;
[Е1о = 2,58-10-7М
ISlo-10', М
0,80
1,00
2,00
3,20
5,00
6,15
10,00
WIE]0, сек *
1,6
1,8
2,5
3,2
3,9
4,3
5,9
6-12. В реакции гидролиза н-каприлового эфира ж-оксибензой-
ной кислоты, катализируемого карбоксилэстеразой, субстрат
является также активатором фермента [13]. На основании данных
табл. 10 определить кинетические и равновесные параметры фер-
Таблица 10
Зависимость скорости гидролиза
«-каприл-ж-оксибензоата, катализируемого
карбоксилэстеразой, от концентрации субстрата.
Условия опыта: рН 8,0; 30° С;
0,01 М трис-НС1-буфер
[S]0- l№, M
8,95
6,00
4,60
3,30
2,30
1,85
1,00
0,65
0,30
V, усл. ед.
7,70
6,47
5,68
4,83
3,95
3,54
2,51
1,96
1,07
121
ментативной реакции, если значения Vm и Кт(каж) реакции,
определенные при невысоких концентрациях субстрата, оказались
равными 2,4 усл. ед. и 4- Ю-4 М соответственно.
6-13. В работе [14] было показано, что метиловый эфир «-то-
луолсульфонил-Ь-аргинина является одновременно субстратом и
активатором реакции гидролиза, катализируемой трипсином.
Исходя из данных табл. 11, определить кинетические и равновесные
параметры ферментативного гидролиза, если из экспериментов,
проведенных при низких концентрациях субстрата, были найдены
значения Кт(каж) — 8- Ю-6 М; Vm = 10 усл. ед.
Таблица 11
Влияние концентрации метилового эфира
и-тозил-Ь-аргинина на скорость гидролиза,
катализируемого трипсином. Условия опыта:
рН 8,5; 25°С; [Е10 = 2-10-8М
[Sb-lCP, M
1,0
2,0
4.0
10,0
50,0
vt усл. ед.
11,8
13,3
16,0
21,6
32,7
6-14. При изучении кинетики гидролиза «-нитроанилида L-лизи-
на, катализируемого трипсином, было обнаружено, что субстрат
является также активатором ферментативной реакции [15]. При
низких концентрациях субстрата были найдены значения /Ст(каж>
и &кат, равные 5,1-Ю-4 М и 2,95-Ю-3 сек-1 соответственно. Исходя
из данных табл. 12, определить значения равновесных и
кинетических параметров промотирования ферментативного гидролиза.
Таблица 12
Зависимость скорости гидролиза /г-нитроанилида L-лизина, катализируемого
трипсином, от начальной концентрации субстрата.
Условия опыта: рН 8,53; трис-буфер; 25° С; ионная сила 0,1 (СаС12)
|S|o-10», М
0,210
0,274
0,325
0,520
0,750
1,140
1,670
»/[Е]о-10', сек-1
0,10
0,12
0,13
0,16
0,21
0,25
0,30
[S]o.103, М
2,19
3,64
7,74
10,00
12,60
16,00
16,50
W[E]0.1C*,
сек-1
0,35
0,40
0,58
0,69
0,73
0,80
0.91
[Slo-Ю3, М
21,4
33,3
34,0
39,1
57,0
100.0
122,0
WIE]„-10',
сек *
0,96
1,00
1,11
1,13
1,32
1,43
1,49
122
6-15. В таблице 13 приведена зависимость начальной скорости
гидролиза п-нитроанилида Ы-ацетил-Ь-тирозина, катализируемого
а-химотрипсином, от концентрации фермента [16]. Определить
значения кинетических параметров ферментативной реакции, если
концентрация субстрата во всех случаях равна 1 ■ Ю-4 М.
Таблица 13
Зависимость начальной скорости гидролиза л-нитроанилида
М-ацетил-Ь-тирозина, катализируемого а-химотрипсином,
от концентрации фермента. Условия опыта: рН 6,1; 25° С;
[Sl0== 1-10-«М
[Е]0.10', М
2,580
2,070
1,550
1,037
0,929
0,516
»■!()•, М-сек—*
2,060
1,840
1,710
1,320
1,320
0,898
[Е]0-103, М
0,3350
0,2070
0,2010
0,1205
0,1037
0,0723
»-10', М-сек *
0,698
0,501
0,508
0,337
0,328
0,222
6-16. Зависимость скорости гидролиза п-нитрофенилсульфата
от концентрации арилсульфатазы (неочищенного экстракта печени)
имела нелинейный характер [17]. После тщательного диализа
препарата фермента та же зависимость стала линейной (см. табл. 14).
Предполагая, что первоначальное отсутствие пропорциональности
между скоростью реакции и концентрацией фермента вызвано
наличием конкурентного ингибитора в препарате фермента, найти
отношение концентрации ингибитора к концентрации фермента в
неочищенном препарате арилсульфатазы.
Таблица 14
Влияние концентрации арилсульфатазы на скорость гидролиза
«-нитрофенилфосфата до и после диализа препарата фермента
Концентрация фермента,
усл. ед.
0,20
0.25
0,40
0,50
1,00
Начальная
скорость реакции,
катализируемой неочищенным
ферме
нтом, усл. ед.
3.8
4,6
7,0
8,3
13,5
Начальная
скорость реакции,
катализируемой очищенным
ферментом, усл. ед.
5.4
6,8
11,0
13,6
27.2
6-17. В работе [18] изучалась кинетика реакции двух
конкурирующих субстратов (5а-андростан-3,16-диона и 5а-андростан-3-она)
123
с одним ферментом — кортизонредуктазой. В ходе эксперимента
были определены значения кинетических параметров
ферментативной реакции восстановления 5а-андростан-3,16-диона в отсутствие
второго субстрата, а также реакции восстановления смеси обоих
субстратов при постоянном отношении их концентраций (табл. 15).
Определить значения Vm и Кт(кат) восстановления 5сс-андростан-3-
она, катализируемого кортизонредуктазой, в отсутствие первого
субстрата.
Таблица 15
Значения кинетических параметров восстановления смеси
5а-андростан-3,16-диона (А) и 5а-андростан-3-она (Б), катализируемого
кортизонредуктазой, при различных соотношениях концентраций субстратов.
Условия опыта: рН 5,2; 25° С; [NAD■ Н] = 1,45- 10-5М
Соотношение концентраций
субстратов в смеси (А + Б)
[Б]/
[Б]/
[Б] = 0
А] = 10
А] = 30
Vm-W, М.мин—!
25,3
2,25
1,44
*т(каж>-106.М
42,0
2,24
0,825
6-18. При изучении кинетики одновременного восстановления
смеси 5и-андростан-3,16-диона и 5а-андростан-3-она,
катализируемого кортизонредуктазой, концентрация первого субстрата
поддерживалась постоянной, а концентрация второго субстрата
варьировалась [18]. Используя данные предыдущей задачи, рассчитать,
при какой постоянной концентрации 5а-андростан-3,16-диона в
смеси субстратов начальная скорость ферментативной реакции
будет оставаться постоянной при изменении концентрации
5а-андростан-3-она в любых пределах.
6-19. При изучении кинетики гидролиза п-нитроанилида L-ала-
нина, катализируемого аминопептидазой М в стационарном режиме
протекания реакции было найдено, что зависимость начальной
скорости ферментативной реакции от концентрации субстрата
имеет вид 5-образной кривой при малых концентрациях и-нитро-
анилида [19]. Предполагая, что фермент в условиях реакции
претерпевает обратимую изомеризацию с образованием неактивного
конформера (Е*, схема 6.27), и исходя из данных табл. 16,
определить значения кинетических параметров ферментативной
реакции и константу равновесной обратимой изомеризации фермента:
(6.27)
E+S-
ES
-* Е + Р.
124
Таблица 16
Зависимость скорости гидролиза и-нитроанилида L-аланина,
катализируемого аминопептидазой М, от концентрации субстрата
[S]o.106, М
1,0
2,0
3,0
4,0
6,0
8,0
10,0
12,5
14,0
16,0
18,0
20,0
22,5
v-W*, М-мнн *
0,179
0,364
0,550
0,742
1,14
1,57
2,00
2,40
2,64
2,96
3,25
3,51
3,84
[Slo-105, М
26,0
28,0
30,0
32,0
34,0
36,0
38,0
39,5
42,0
44,0
46,0
48,0
v-Ш, М-мин
4,26
4,50
4,72
4,93
5,15
5,33
5,51
5,65
5,85
6,03
6,20
6,34
6-20. Известно, что в промышленном препарате а-химотрипсина
обычно содержится примесь трипсина, причем оба этих фермента
способны катализировать гидролиз этилового эфира N-бензоил-
аргинина. Исходя из данных табл. 17, определить процентное
содержание активного трипсина в препарате а-химотрипсина, если
в отдельном эксперименте найдено, что для гидролиза этилового
эфира Ы-бензол-Ь-аргинина, катализируемого трипсином, Кт(каж) =
= 4- Ю-6 М; kKaT = 29,6 сек-1.
Таблица 17
Зависимость скорости гидролиза этилового эфира
1Ч-бензоил-Ь-аргинина, катализируемого
препаратом а-химотрипсина, от концентрации
субстрата. Условия опыта: рН 7,8; ионная сила
0,1М (NaCI); 25° С; концентрация препарата
а-химотрипсина в растворе равна 8,5-10-вМ
[S]o.103, М
1,60
2,00
2,67
4,00
8,00
к-106, М-сек—'
0,802
0,929
1,10
1,51
2,41
6-21. В таблице 18 приведены кинетические данные по
изучению ингибирования профлавином гидролиза этилового эфира
Ы-ацетил-Ь-тирозина, катализируемого а-химотрипсином. Исходя
125
из двухстадиинои схемы ферментативной реакции, предложить
механизм ингибирования и рассчитать возможные кинетические и
равновесные параметры реакции.
Таблица 18
Влияние профлавина на кинетику гидролиза этилового эфира
1Ч-ацетил-Ь-тирозина, катализируемого а-химотрипсином ([Е]0 = 1,73-10~8М)
[I], М
0
1 ■ 10-5
3,3-Ю-5
6,2-Ю-5
[Slo-103, М
1.25
1,67
2,50
5,СО
1,43
1,82
2,50
4,00
6,67
1,25
1,67
2,50
5,00
10,00
1,25
2,СО
2,50
v-W, М-сек—1
2,18
2,39
2,65
2,96
1,97
2,18
2,38
2,70
2,92
1,32
1,56
1.89
2,41
2,80
0,90
1,24
1,42
П1, М
6,2-Ю-5
8,6-10-s
1,16-10-*
1,54-10-*
[Slo-101, М
5,00
10,0
1,43
2,00
2,78
5,00
10,00
2,00
2,50
3,33
5,00
10,00
2,50
3,33
5,00
10,00
v-№, M-cei(—1
2,00
2.50
0,725
0,935
1.19
1,67
2,21
0,69
0,83
1,01
1,32
1,89
0.С25
0,785
1,05
1.59
6-22. В таблице 19 приведены результаты кинетических
исследований гидролиза п-хлоранилида N-ацетил-Ь-тирозина, катализи-
Таблица 19
Зависимость начальной скорости гидролиза
и-хлоранилида 1Ч-ацетил-Ь-тирозина, катализируемого
а-химотрипсином, от концентрации субстрата.
Условия опыта: рН 8,0; 25° С; ионная сила 0,1М (КО);
5% диметилформамида
ISlo-W, М
5,0
6,6
10
12
15
20
Z'-IO7, М-еек-'
2,04
2,63
3,92
4,66
5.72
7,35
[S]o-J0s, М
30
40
50
60
68
80
г<-107, М-сек-'
10,3
12,8
15,1
17,0
18,7
20,2
126
руемого а-химотрипсином, при сравнимых по величине значениях
концентраций субстрата и фермента. Определить значения
кинетических параметров ферментативной реакции и вычислить величину
абсолютной концентрации а-химотрипсина в условиях
эксперимента, если значение Vm, определенное при той же концентрации
фермента и избытке субстрата, равно 4,1 ■ Ю-6 М-сек-1.
Ответы и решения
6-1. Откладывая данные табл. 1 в координатах (v, log [S]o),
получаем кривую в виде симметричного колокола (рис. 53).
Уменьшение скорости реакции при значительном увеличении
концентрации субстрата (правая ветвь
кривой) указывает на ингибирование
субстратом, а симметричность
полученного колокола свидетельствует
о том, что образующийся при
связывании дополнительной молекулы
субстрата тройной комплекс ES2
является неактивным (см.
теоретическую часть настоящей главы).
Таким образом, кинетические данные
соответствуют схеме реакции
E + S
*/л(каж)
ES
ES,
■fs
Е + Р
(6.28)
и скорость ферментативной реакции
определяется выражением
-50 -4,5
' log[S]
Рис. 53. Ингибирование
субстратом в реакции
трансметилирования, катализируемого фенилэтанол-
aMHH-N-мети лтр ансфер азой
V =
Km (каж) + [S]o +
(6.29)
Линеаризуя кинетические данные задачи в координатах (1/г1, 1/[S]0)
при низких концентрациях субстрата (рис. 54) и в координатах
(l/v, [S]0) при высоких концентрациях субстрата (рис. 55), найдем
значения Кт&аЖ), Vm и Ks-
Ответ: /Г1В(К.»)= 8-1С-7М; Vт = 7,15- 1С-" М-мин-1; Ks=*
= 3,25-Ю-5М.
6-2. Решается аналогично задаче 6-1.
Ответ: Кт{КЙЖ) = 6- 10-6М; Vm = 1.0 усл. ед.; K's= 5,3- 10~*М.
6-3. Решается аналогично задача 6-1.
Ответ: /Cm(Ka«) = 2,5-10-s M; £кат = 9,5 сек-1; Ks = 1,6- HHW.
127
6-4. Решается аналогично задаче 6-1.
Ответ: /Ст(каЖ)= 1,8- 10~SM; К1В=6-10-6М.мин-1; /Cs=2,4 X
X Ю-з М.
6-5. Дифференцируя уравнение скорости реакции (6.29) по
переменной начальной концентрации субстрата и приравнивая
полученное выражение нулю, найдем значение концентрации субстрата,
'0,75 -0,25 0 0,25 0,75
//[S]>,V
Рис. 54. Определение кинетических
параметров реакции
трансметилирования, катализируемой фенилэтанол-
амин-М-метилтрансферазой, в
условиях ингибирования высокими
концентрациями субстрата
1 8
Т.
^ 6
' о „
v A
'Ja
-А-^ о
i t^1
Г^^
1 1 1 1 1 1
-50 -25 0
50
100 150
Мо-го,м
Рис. 55. Определение константы
диссоциации неактивного тройного
комплекса ES2 в условиях
ингибирования ферментативной
реакции субстратом
при котором скорость ферментативной реакции достигает
максимума:
[Son-rl = V ^т(каж) Ks.
(6.30)
Ответ: [S]otlT = 2,9 • Ю-3 М.
6-6. Из уравнения (6.2) видно, что оптимальная скорость
реакции определяется выражением
Vm [Sic
*\т1каж) + 1ч1опт~Г
[S1L
(6.31)
/с
Подставляя соотношение (6.30) в (6.31) и преобразуя полученное
выражение, получим систему из двух уравнений с двумя
неизвестными:
[Ь]опт :
к,
т(каж)
Ks,
-^ 1 = 2]/ к
каж)
S
128
решая которую можно найти значения Л"„,(Каж) и АГ$-
*т 1 __ ^ t^JonT
v опт
1
(каж)
^опт [^JonT
Ответ: /<т(кгж) = 9,5- 10-'М; Ks = 3,8- Ю-5М.
6-7. Из графика в координатах (v, log [S]o) видно, что
полученная колоколообразная кривая не является полностью
симметричной (рис. 56), что указывает на наличие некоторой
ферментативной активности тройного комплекса ES2, причем 0 < |3 < 1
*т(каж) £
Е + S " ES —--* Е + Р
t
к s
(6.32
ES9
РК
ES + P
3,0 -2,0
1°дИ0
Рис. 56. Ипгибнрование субстратом
реакции гидролиза О-бензоилглицин-
2-оксиизовалериановой кислоты,
катализируемой карбоксипептидазой А.
Пунктирными линиями проведены ось
симметрии кривой и правая ветвь
теоретической кривой зависимости
скорости ферментативной реакции от
концентрации субстрата при условии
0 = 0
Рис. 57. Обработка кинетических
данных по неполному ингибироваиию
субстратом реакции гидролиза
О- (бензоилглицнн) -2-оксиизовалериа-
повой кислоты, катализируемой
карбоксипептидазой А
Обрабатывая данные эксперимента в координатах (1/&эфф. l/[S]o)
и в координатах (&Нат/(&кат — /гэфф), 1/[S]0), где &Эфф=а/[Е]о
(рис. 57), находим значения констант схемы (6.32).
5 Заказ 158
129
Ответ: kKaT = 65 сек-1; Кт{кяж) = 2,4-10-4м. /Cs=l,2-10-3M;
Р = 0,05.
6-8. Решается аналогично задаче 6-7 (рис. 58).
Ответ: £кат = 27 сек-1; /Ст(каж) = 5- 1(Н»М; /С'= 1,2-10-2 М;
Р = 0,34.
6-9. [1]. Откладывая экспериментальные данные в координатах
(v, log [S]o), найдем, что полученная кривая имеет вид
несимметричного колокола, правая ветвь которого значительно круче левой
(рис. 59). Подобная форма кривой указывает на то, что ингибиро-
2,0 -1,5 -1,0
1094
Рис. 58. Зависимость скорости
гидролиза М-карбобензокси-Ь-триптофана,
катализируемого карбоксипептидазой,
от концентрации субстрата.
Пунктирная кривая является теоретической
при условии Р = 0
4,0
X
г 3,0
W 2,0
Ь
1,0
/TV
/ I Л '\
/ I V \
. / | \ \
/ | \
I [S]
I опт
-3,0
-2,5 -2,0 -1,5
ЮдИо
Рис. 59. Ингибирование
субстратом гидролиза монофенилфосфата,
катализируемого щелочной фосфа-
тазой. Пунктирная кривая
является теоретической при условии
образования неактивного фермент-
субстратиого комплекса состава
ES2
вание реакции происходит при присоединении нескольких молекул
субстрата к фермент-субстратному комплексу
^т(каж) jj
Е + S : ES —- Е + Р
п [S]
(6.33)
Предположим, что присоединение дополнительных молекул
субстрата к фермент-субстратному комплексу происходит независимо
друг от друга и что связывание всех п дополнительных молекул
субстрата характеризуется одинаковыми константами равновесия
/Cs' (схема (6.33)). В этом случае уравнение скорости
ферментативной реакции, протекающей в стационарном режиме, имеет
следующий вид:
V =
+ [S]„ +
[S]0"
n + 1
(6.34)
К
130
Дифференцируя уравнение (6.34) по начальной концентрации
субстрата и приравнивая полученное выражение нулю, получим
соотношение, связывающее величину оптимальной концентрации
субстрата с параметрами схемы (6.33) (см. решение задачи 6-5):
п+\
Записывая далее уравнение (6.34) в виде
и производя несложные преобразования, получим
-±-+ [S]° , =-**— ^--j^—. (6.37)
Вычислив произведения Km (каноне с использованием значения [S]otIT=
= 4,3- 1СН3 М (см. рис. 59) и формулы (6.35) для величин п = 1, 2, 3
и т. д., отложим полученные данные в координатах (1/[S]0 +
+ [Щ"1 Кт(каж) Ks, 1/v). Из рис. 60 видно, что линейный характер
зависимости (6.37) соответствует лишь значению п = 3. Таким
образом, неактивный фермент-субстратный комплекс в данном
случае имеет состав ES4. Наконец, из отрезков, отсекаемых
полученной прямой (рис. 60) на координатных осях, можно определить
значения Кт(каж) и Vm ферментативной реакции.
Ответ: Кт(каж) = 1,8- 10-»М; Vm = 7,7- lO-sM-мин-1; K's =
= 5,7-10-7М.
6-10. Из данных табл. 8 видно, что при увеличении
концентрации субстрата скорость ферментативной реакции превышает
теоретическую максимальную скорость (Vm), рассчитанную при
использовании небольших концентраций субстрата. Подобная
зависимость v от [S]0 согласуется со следующей схемой
ферментативной реакции, где р>1:
"т (каж) k
IS _J^L* E + Р
*s I (6.38)
ES2 -^* ES + P
Уравнение скорости реакции (6.38) можно записать в виде
i + JSfMra.
^эфф = ~Щ^~ = ^кат " ^jT- (6-39)
Кт (каж) + [S]o + ~'
5*
131
20 40
7/1Г-70,Ммин
т(каж)
Рис. 60. Итеративная линеаризация экспериментальных данных по
кинетике гидролиза монофенилфосфата, катализируемого щелочной
фосфатазой, в условиях ингибирования ферментативной реакции
субстратом с образованием неактивного комплекса состава ESn
Для нахождения констант AV и |3 экспериментальные данные
удобно анализировать в координатах (кка-г1 (кЭфф — £Кат), 1/[S]0) при
условии £ЭфФ>£кат- Полученная прямая отсекает на оси ординат
отрезок, равный 1/(3—I, и имеет тангенс угла наклона, равный
/Cs'/(P— 1) (рис 61).
Ответ: K's = 2,2-10--? М; р = 1,9.
6-11. Решается аналогично "задаче 6-10.
132
Ответ: K's = 1,45- Ю-2М; р — 10.
6-121. Решается аналогично задаче 6-10.
Ответ: /Cs = 2-10-2M; р = 8.
6-13. Решается аналогично задаче 6-10.
Ответ: Ks = 1,6 • Ю-2 М; р = 4.
6-14. Решается аналогично задаче 6-Ю.
Ответ: K's = 3.1(Н*М; р = 6.
6-15. Так как из данных задачи следует, что в условиях
эксперимента концентрация фермента превышает концентрацию
;/[5] ,М
1 о
Рис. 61. Обработка кинетических
данных по активации субстратом
гидролиза метилового эфнра п-то-
зил-О-аргинина, катализируемого
трипсином
Л12-0
*| 8,0
1 >П
-1,0
&Т
~<Z% [S]
I 1 1 1
3 1.0 . . 2.0
УСЕ]-/о.У
0
Рис. 62. Определение
кинетических параметров гидролиза п-ни-
троанилида К-ацетил-Ь-тирозина,
катализируемого сс-химотрипси-
ном при избытке фермента
субстрата, для определения кинетических параметров реакции
следует воспользоваться уравнением (6.26), преобразовав его к виду
J_ 1 . *s 1
v
+
кг IS]0 ' k2 [S],
[Е]о
(6.40)
Значения кинетических параметров &2 и Ks можно найти из
графика в координатах (1/и, 1/[Е]0) (рис. 62).
Ответ: k2 = 3- KHceiH; Ks = 1,25- Ю-3 М.
6-16. Если в препарате фермента содержится примесь
конкурентного ингибитора, то схему ферментативной реакции при равно-
1 В работе [13] количественная обработка результатов эксперимента
не проводилась
133
весных условиях образования комплексов фермент-субстрат и
фермент-ингибитор можно записать в виде
Е + S TZZl ES Е + Р,
EI
где
к И [SJ
К,=
[Е] [I]
[EI]
(6.41)
(6.42)
(6.43)
Особенность кинетического анализа данного случая
заключается в том, что при сравнимых по величине значениях молярных
концентраций фермента и ингибитора необходимо использовать
соответствующие уравнения материального баланса
[1]0 - [I] + [EI], (6.44)
[E]0=[E] + [ESl + [EIi, (6.45)
где отношение концентраций [1]о/[Е]о—-постоянная величина
в условиях эксперимента при вариации [Е]0. Вводя равенство
[I]0/[E]0=R (6.46)
и используя соотношения (6.42) — (6.44), уравнение (6.45) можно
записать в виде
[Е]0 = [Е]
[E][S] R[E][E]„
+ /Ci + [Е] ■
Ks
(6.47)
Подставляя в выражение (6.47) вместо переменной концентрации
свободной формы фермента [Е] значение скорости ферментативной
реакции
(6.48)
« = £[E][SI
и приводя полученное соотношение к виду квадратного трехчлена,
получим уравнение
Ks (Ks + [S]0) tf + k2 [S]0 {/C, (Ks + [S]0) + Ks [E]0 (R - 1)} v -
-*|tfi[S]?[E]0 = 0. (6.49)
При постоянной величине [S]o это уравнение характеризует
зависимость между скоростью ферментативной реакции и начальной
концентрацией фермента, содержащего примесь конкурентного
ингибитора [20]. Разложим левую часть полученного уравнения
на множители и запишем его в виде
134
*i*. [S].) / MS].[E},(R
1) ■ ^г [S]0 /ClR
ks + [S], "•" ks (R -1)
*2[S]2*?R
K2S(R
I)2
(6.50)
График уравнения (6.50) представляет собой гиперболу (при R ф 1),
асимптоты которой равны
*i[S]»[E].(R—1) _ fea [S]0 Ki R
KS + IS]„ /CS(R —0 "
(6.52)
Приравнивая друг другу выражения (6.51) и (6.52), найдем
значения координат центра гиперболы:
Ki kz [S]„
ордината = щ^,
Ki(Ks + [S]0)(R+ 1)
абсцисса = -
/CS(R —1)2
Известно, что в случае равносторонней гиперболы, проходящей
через начало координат (v, [E]o) и имеющей асимптоты,
параллельные координатным осям (уравнение 6.53)
* = 1ятЬ (6-53)
координаты центра гиперболы определяются соотношениями
ордината = а, абсцисса = —Ь.
В этом случае зависимость начальной скорости ферментативной
реакции от концентрации фермента, содержащего примесь
конкурентного ингибитора, определяется уравнением
^s(R-i) [EUS]o ,, ы
P = *.(Ks+[S].)(R+l) • (6'54)
KS(R — l)2 +[E]o
Приводя уравнение (6.54) к виду, удобному для анализа в
координатах двойных обратных величин, получим
J_ (*s -HS].)(R + 1) 1 /cs(R—1)
v *i[S],(R 1) [E], + k2[S]0Ki ■ (6-55)
Из выражения (6.55) видно, что при наличии примеси
конкурентного ингибитора в препарате фермента тангенс угла наклона
зависимости в координатах (l/v, 1/[E]0) равен
, (Ks +[S].)(R+D /fiI-fi,
tgy'^ *.[S].(R-D • (6-56)
При отсутствии ингибитора в препарате фермента тангенс угла
наклона зависимости в координатах (\/v, 1/[E]0) равен
К* + [S]0
^'-ж'- <6-57>
135
Разделив (6.56) на (6.57), получим соотношение
tgfi = R + 1
tg9» R —1'
преобразуя которое, придем к окончательному выражению
tg¥,
R= tgVo
+ 1
tg?i
tg9o ""'
Подобным образом, обрабатывая данные табл. 14 в координатах
(l/v, 1/[Е]о), найдем значение R (рис. 63).
Ответ: [1]/[Е] о=8,3.
6-17. Реакцию двух конкурирующих субстратов с одним
ферментом (не образующих смешанный тройной комплекс) можно
записать в виде схемы
E + S^ES^E + P j (65g)
Е + S2 ^ZT ES2 —'■-* Е + Р '
В случае регистрации общего продукта при одновременном
протекании реакций (6.58) общая скорость ферментативной реакции
выражается уравнением
Ух Ve
^f[S,]+^-[S2]
^общ= [s,j 2 TsJ' (6'59)
1 + К. + Кг
где V'j = k1 [E]0 и V2 = k2 [E]0. При постоянном отношении
концентраций обоих субстратов [S2]/[S,] = а уравнение (6.59)
линеаризуется в координатах Лайнуивера-Берка:
1 J_ а
_!_ = fs'] + к\ к* (6.60)
v общ Vj_ oVj_ ■ v '
К\ Кг
Решая уравнение (6.60) совместно с (6.61), соответствующим
случаю а=0 (отсутствие второго субстрата в системе),
4-=w + ^7' (6-61)
найдем, что положение точки пересечения прямых в координатах
(l/v, \/[S\]) не зависит от величины а и определяется
следующими значениями абсциссы и ординаты:
1 _ Уг-Уг
[S,] - KtVz •
1 ^ 1
v ' v2 ■
136
Определив таким образом значение V<i при графическом
анализе данных табл. 15 (рис. 64), найдем далее значение /С2, приняв во
внимание, что величина общей максимальной скорости
ферментативной реакции двух субстратов с одним ферментом (при
oc=const) определяется выражением (см. уравнение 6.60)
V„
Кг + аК,
Из соотношения (6.62) легко получить величину К2:
К2 =
Уг-Ут
Ответ: V2 = 9,7- Ю-^М-мин-1; К2 = 2,5- 10-5М.
(6.62)
V[l\ ,услед.
Рис. 63. Зависимость обратной
скорости ферментативной
реакции от обратной концентрации
фермента для неочищенного
(•) и очищенного (°)
препарата арилсульфатазы
Рис. 64. График в координатах
Лайнуивера-Берка для реакции
двух конкурирующих субстратов
(5а-андростан-3-16-диона и 5а-аи-
дростан-3-она) с одним
ферментом — кортизонредуктазой.
Отношение концентрации второго
субстрата к первому равно: (а) — 0;
(б) —10; (е) — 30
6-18. Скорость ферментативной реакции будет оставаться
постоянной при изменении концентрации субстрата в любых
пределах (вплоть до очень высоких концентраций) только в том случае,
если она будет равна максимальной скорости ферментативной
реакции. Исходя из этого, запишем уравнение (6.59) в виде
V, ' У2
V, = Kl rc , Kz tc ,-■ (6.63)
1 +
[S.] , [S,]
Ki
+
K*
137
Упрощая соотношение (6.63), находим, что при постоянном значении
1/ К
[S\\ = \r s \r скорость ферментативного превращения смеси двух
V 1 V 2
субстратов равна постоянной величине (V2) при любом значении
[5г]. Используя данные предыдущей задачи, можно найти, что при
постоянной концентрации 5а-андростан-3,16-диона, равной 1,67Х
X10~6 М, скорость ферментативной реакции будет равна 9,7-10~7
М-мин-1 при любой концентрации 5а-андростан-3-она.
Ответ: [51]=1,67-10-6М.
6-19. Для вывода уравнения стационарной скорости
ферментативной реакции, в которой происходит обратимая изомеризация
фермента с образованием неактивного конформера (схема 6.27),
запишем уравнение материального баланса по ферменту (в случае
[S]o^>[E]0), а также выражения для скорости распада фермент-
субстратного комплекса и для константы диссоциации фермент-
субстратного комплекса:
[Е]0 -^ [Е*] + [Е] + [ES], (6.64)
v=-k2[ES], (6.65)
Ks = ff. (6.66)
Комбинируя уравнение материального баланса и соотношение
(6.66), можно показать, что концентрация активной формы
фермента зависит от начальной концентрации субстрата:
[Е]= ГС""^1 ■ (6.67)
Полагая, что концентрация неактивного конформера [Е*]
стационарна в условиях эксперимента, т. е.
^- = ft_1[E]-ftI[E*]=0, (6.68)
и подставляя соотношение (6.67) в выражение (6.68), найдем
стационарную концентрацию [Е*]:
[Е*] = *-'siE1° • (6-69)
Наконец, комбинируя выражения (6.65) — (6.69), получим
уравнение скорости ферментативной реакции
^ Vm [S]o 'I Ks ) /g 7Qv
^s+[S]o / [S],\ . ' ( )
где Vm = k2[E]0. При высоких концентрациях субстрата, при
которых выполняется неравенство kx 11 + У-°) > k-v уравнение (6.70)
138
упрощается до обычного уравнения Михаэлиса — Ментен
Vm [S].
Ks + Ю. '
линеаризируя которое в подходящих координатах (см. гл. 5) можно
найти значения Vт и Ks (рис. 65). В области низких концентраций
субстрата ([S]0<^ Ks) скорость ферментативной реакции должна
линейно зависеть от [S]0:
*i Vm [SJ0
V
kl+k^i
К,
или
i-0+-^-)-
Ks
Vm [S]„
(6.71)
Из выражения (6.71) видно, что график в координатах (1/о,
1/ [S] о) позволяет найти значение константы обратимой
изомеризации фермента £_i/£i (рис. 66).
Ответ: Vm=l,64-1(HM; Ks=7,5- 1(Н М; /Сравн=й1/Й_1 = 4,6.
0,3
J*
'е
^ о,/
Jf^
i i i 1 ..1
-007 0 007 002 ЦОЗ 0,04 0.05
Рис. 65. Определение кинетических
параметров гидролиза га-нитроанили-
да L-аланина, катализируемого ами-
нопептидазой М
0,2 0,4 0,6 0,8 1,0
Рис. 66. Обработка кинетических
данных, соответствующих
невысоким концентрациям субстрата, при
обратимой изомеризации
фермента, которая приводит к
образованию неактивного конформера
6-20. Схема превращения одного субстрата, катализируемого
двумя различными ферментами, может быть представлена в
следующем виде:
Ej + S
кт(каж)
.(1)
iEjS-^Ej + P,
ь<2)
кт(каж)
,<<>
E2 + SZ
E2S
Т.Е2+Р-
Общая скорость реакции в этом случае должна быть равна сумме
скоростей обеих ферментативных реакций
139
причем для рассматриваемого случая скорость гидролиза,
катализируемого трипсином, должна являться максимальной (так как
[!3]о»Ят(каж) для катализа трипсином). Отсюда суммарная
скорость ферментативной реакции может быть записана в виде
^.[ElotSb +/е[Т]о, (6-72)
^СбЩ
I'm (каж) "г [Мо
где [Е]0 — концентрация промышленного препарата к-химотрип-
сина в растворе; [Т] 0 — концентрация трипсина, присутствующего
в виде примеси в препарате а-химотрипсина. Из выражения (6.72)
15
ш ю
О
аГ
о
UU
5Р
. /
■ /
/ У
f JT
/ S
/ s _^*
/ <У ^Г
/ У^ ^г
£ /ъ^
/^ S>*^
J^
i i
6
Г
а
уу*~
1
-1Р О
2,0 4,0
6.0
1lmQio2M1
8 П
(i];o?m
Рис. 68. Влияние концентрации
ингибитора на величину
кажущейся константы Михаэлиса
ферментативной реакции в случае
связывания с активным центром
фермента двух молекул
конкурентного ингибитора
Рис. 67. Определение количества
трипсина в препарате а-химотрпп-
сииа с помощью специфического
субстрата трипсина — этилового
эфира Г\Г-бензоил-Ь-аргинина: а —
результаты эксперимента; бив —
значения скоростей
ферментативной реакции рассчитаны в
предположении, что в препарате
а-химотрипсина содержится 0,075 и
0,2% трипсина соответственно
очевидно, что зависимость в координатах (1/1>общ, l/[S]o) не
должна в общем случае быть линейной, что и наблюдается на опыте
(рис. 67). Для нахождения величины [Т]0 выражение (6.72)
можно записать в виде
*W - k(K{\ [Т]„ = vt - -щ ——
^т(каж) "г l°J»
и, произвольно варьируя возможное содержание трипсина в
препарате а-химотрипсина, откладывать полученные значения в
координатах (1Д>ь l/[S]o). Для рассматриваемого случая кривая
линеаризуется в предположении, что исходный препарат
а-химотрипсина содержит 0,075% активного трипсина (см. рис. 67).
140
6-21. Обработкой данных табл. 18 в координатах Лайнуивера-
Берка можно показать, что профлавин является конкурентным
ингибитором а-химотрипсина. Однако зависимость в координатах
(/(т(каж), [I]) в данном случае является нелинейной (рис. 68), в
отличие от простого конкурентного типа ингибирования. Одной из
возможных причин подобной зависимости может быть связывание
с активным центром фермента двух молекул конкурентного
ингибитора:
E + s3ES-^E + P (6.73)
| +
*il I
i I
EI
/ I
Е12
Для вывода уравнения скорости этой ферментативной реакции
запишем уравнение материального баланса по ферменту и
выражения для констант диссоциации фермент-субстратного и фер-
мент-ингибиторного комплексов:
[Е]0 = [Е] + [ES] + [EI] + [Е12], (6.74)
^, = ™, (6.76)
А, щ^ \У-ч)
Комбинируя выражения (6.74) —(6.77) с выражением для скорости
распада фермент-субстратного комплекса.
v = k2 [ES],
найдем уравнение скорости ферментативной реакции
V—~R л_rsi • (o./S)
Ат(каж) т l°Jo
где
АГ»(Мж) = АГ5(1 + -Ш-+ J||r). (6.79)
Преобразуя выражение (6.79), получим
IS
^т (каж)
*S _ J, _Ш_
[I] Ki + KiK[ '
141
/ /п(каж) \
Откладывая данные табл. 18 в координатах \ - , [I]/,
получаем прямолинейную зависимость (рис. 69). Таким образом,
схема (6.73) описывает результаты эксперимента с константами
ингибирования /Ci=2,2- Ю-5 М; К\= 1,2-10~4 М.
Ответ: /г2=193 сек-1; /Cs = 6,63-10-4 М; /Ci=2,2-10-5 М- К\ =
= 1,2-КИМ.
Рис. 69. Определение констант Рис. 70. Графический вариант оп-
ингибнрования ферментативной ределения констант ингибирова-
реакции в случае связывания с ак- ния ферментативной реакции в
тивным центром фермента двух случае связывания с активным
молекул конкурентного ингиби- центром фермента двух молекул
тора конкурентного ингибитора
(уравнение 6.80)
Примечание. Анализ кинетики ферментативной реакции, в
которой с активным центром фермента могут связываться две
молекулы конкурентного ингибитора, можно провести также следующим
образом. Из уравнений (6.78) — (6.79) можно легко получить
выражение
J L = _*s_n!L . W2 \
Vm [S]o V^i ^~ KxK\ I
и далее
1гГ(---иг-#^(1+-ЩЛ (6.80)
[I] \vi v j KiVm[S]0 \ K{ )
В случае справедливости схемы (6.73) обработка
экспериментальных данных в координатах (1/[I] (1/f,— 1/f), [I]) должна
приводить к пучку прямых, соответствующих различным начальным
концентрациям субстрата, пересекающихся на оси абсцисс в
точке— K'i и имеющих тангенс угла наклона, равный Ks/KiVm[S]0
(рис.70).
142
Наконец, выражение (6.80) можно записать в виде
JSjo/J 1\ = _^s /, , [I]
[I] \Vi v J KiV„
(1 + f>
(6.81)
1.0-
анализ которого в координатах (Цтг- ■ (— Л , [I] ) приводит ь
единственной прямой (рис. 71), пересекающей ось абсцисс в точке
— К и и ось ординат — в точке
KslKxVm.
6-22. Используя соотношения
(6.22) и (6.24), проведем
линеаризацию экспериментальных данных
в координатах (1/1 -f- X, [S]0/A), ogi
0,6-
0.4-
0.2
йо/Д-ИГ.М
Рис. 72. Определение абсолютной
концентрации фермента из
кинетических данных при условии [EJo^fSjo
Рис. 71. Графический вариант
определения констант ингибирова-
ния ферментативной реакции в
случае связывания с активным
центром фермента двух молекул
конкурентного ингибитора
(уравнение 6.81)
где X =
1
Vjn
v
Из абсциссы точки пересечения и тангенса угла
наклона полученной прямой (рис. 72) найдем значения k2, Ks и [Е]с.
Ответ: [Е]с = 3,6-10-' М; tfs = 6,2-10-4M; /г2=1,Нх
X Ю-2 сек-1.
ЛИТЕРАТУРА
1. БресткинА. П., Новикова Н. В., Прокофьева Е. Г., Ржехи-
на Н. И. „Биохимия", 26, 266, 1961.
2. Уэбб Л. Ингибиторы ферментов и метаболизма. М., „Мир", 1966 стр 80
3. Fuller R. W., Marsh M. M. „J. Med. Chem.", 15, 1068, 1972.
4. Rosenberry Т. L., Bernhard S. A. „Biochemistry", 11, 4308, 1972.
5. Nakata H., Yoshida N., Narahashl Y. Ishii S. „J Biochem " 71
1085, 1972.
143
6. Hakala M. Т., 01 aid A. J., S с h w e r t 0. W. „J. Biol. Chem.", 221,
191, 1956.
7. Wilson I. В.. Bergmann F. „J. Biol. Chem.", 186, 683, 1950.
8. Marcus P. 1., Talalay P. „Proc. Roy Soc", B144, 116, 1955.
9. Bunting J. W., Murphy J. „Biochem. Biophys. Res. Commun." 48, 1316,
1972.
10. L urn г у R., Smith E. L., О I a n t z R. R. „J. Amer. Chem. Soc.', 73,4330,
1951.
11. Trowbrige С. О., Krehbiel A., Laskowski M. „Biochemistry",
2, 843, 1963.
12. Nakata H., Ishii S. — I. „Biochem. Biophys. Res. Commun. 41, 393, 1970.
13. Hofstee В. Н. J. „Biochim. Biophys. Acta', 258, 446, 1972.
14. Bechet J. — J., Yon J. „Biochim. Biophys. Acta'. 89, 117, 1964.
15. Sevdoux F. „Eur. J. Biochem.", 17, 209, 1970.
16. I n a g a m i Т., S t u r t e v a n t J. M. „Biochemistry", 4, 1330, 1965.
17. Диксон М., Уэбб Э. Ферменты. М., „Мир", 1966, стр. 58.
18. Pock ling ton Т.. Jef f ery J. „Biochem. J.", 112, 331, 1969.
19. Femfert U., Cichocki P. „FEBS Letters", 27, 219, 1972.
20. Katsumata M. „J. Theoret. Biol.", 24, 294, 1969.
Глава 7
СТАЦИОНАРНАЯ КИНЕТИКА
ТРЕХСТАДИЙНЫХ ФЕРМЕНТАТИВНЫХ
РЕАКЦИЙ. ОПРЕДЕЛЕНИЕ ЗНАЧЕНИЙ
КОНСТАНТ СКОРОСТЕЙ ПРОМЕЖУТОЧНЫХ
СТАДИЙ
§ 1. Кинетическое описание
трехстадийных ферментативных реакций
Большое число ферментативных реакций (в особенности
реакций гидролитического типа) протекают по так называемому трех-
стадийному механизму
Ё + S 5==i: ES —4*- ЕА —^ Е + Р, (7.1)
где ЕА—ацилферментное промежуточное соединение,
образованное в результате ацилирования активного центра фермента
молекулой субстрата; Pi и Рг — продукты реакции, образованные на
стадиях ацилирования и деацилирования соответственно; k2 и k3 —
индивидуальные константы скоростей реакций ацилирования и
деацилирования. Если фермент-субстратный комплекс ES
находится в квазиравновесном состоянии с исходным субстратом и
ферментом, то Ks называется истинной константой Михаэлиса.
Определяемые на опыте параметры ферментативной реакции,
каталитическая константа &кат и кажущаяся константа Михаэлиса Кт(каж)
144
связаны со значениями индивидуальных констант схемы (7.1)
соотношениями
Кп (квж) = ^S Х+~£Г ' ^'^
входящими в простое уравнение Михаэлиса — Ментен
_. __ «кат [Ejo [SJq ,j a\
(каж)
Из уравнения (7.4) следует, что кинетика трехстадийных
ферментативных реакций формально описывается классической двухста-
дийной схемой (7.5), и, следовательно, к ним применимы все
основные методы кинетической обработки двухстадийных реакций,
рассмотренные в гл. 5:
Кщ (каж) д,
E + ST:=^ES^I^E + P. (7.5)
Довольно сложной проблемой в изучении кинетики
трехстадийных ферментативных реакций является определение значений
индивидуальных констант k2, k3 и Ks (схема 7.1). Как видно из
уравнения (7.4), кинетический анализ трехстадийнои реакции в
стационарном режиме ее протекания не позволяет в общем случае
раздельно определять значения индивидуальных констант. Однако в
ряде случаев значения k2, k3 и Ks можно определить путем
селективного воздействия каких-либо эффекторов на отдельные стадии
(ацилирования или деацилирования) стационарной
ферментативной реакции.
§ 2. Селективное влияние эффекторов
(ингибиторов или активаторов)
на стадию ацилирования
Если имеется возможность влиять на стадию ацилирования
трехстадийнои ферментативной реакции с помощью селективно
действующего эффектора, то при этом эффективные кинетические
параметры уравнения
*= ;-[EUS]° (7.6)
Km (каж) + tSlo
будут определяться выражениями
k* = . , (7.7)
кат k2 + k3 ' ( '
145
где индекс (*) указывает на величину, измененную в присутствии
эффектора. Решая совместно уравнения (7.4) и (7.6), нетрудно
показать, что в координатах Лайнуивера-Берка семейство прямых
(l/v, 1/[S]0), полученных при различных концентрациях
эффектора, пересекаются в общей точке, расположенной в левом верхнем
квадранте, с координатами
[S]7 = ~/V (7-9)
4--*к- - (7Л0)
Определив таким образом значения индивидуальных констант
&з и Ks, легко найти значение k2 из соотношения
-т--^ 1. (7-и)
Следует отметить, что положение точки пересечения в данной
координатной системе не зависит от характера функциональной
зависимости константы k<i от концентрации эффектора. Таким образом,
применение ингибитора или активатора, избирательно влияющего
на скорость стадии ацилирования, позволяет проводить раздельное
определение индивидуальных констант k2, k3 и /Cs независимо от
конкретного механизма действия эффектора.
§ 3. Влияние дополнительных
нуклеофильных агентов на стадию
деацилирования
В случае гидролитических ферментативных реакций,
протекающих по трехстадийному механизму, нуклеофильные агенты,
добавленные в реакционную систему, могут конкурировать с водой
на стадии деацилирования ацилфермента
^Е+Р2
к,
Е + S z£=*i ES -<*- ЕА ^^ (7 12)
Pi *^/^Е + Р9
где N — дополнительный нуклеофильный агент; Р3 — продукт
реакции переноса ацильной части субстрата на нуклеофил,
происходящей с константой скорости второго порядка k4; &3=&'3 [Н20].
Кинетический анализ схемы (7.12) для реакции, протекающей в
стационарном режиме, приводит к уравнению скорости (7.4), где
И
. ^ k2(k, '-Ъ[Щ) п ,,-.
146
при регистрации продукта Р^
k =
"■кат
при регистрации продукта Р2 и
йкат" *, + *,"+\[N] (7Л5>
ь k'ki [N] (7 16>
k, + k, + k,[N] k • '
при регистрации продукта Рз [1]. Если кинетика реакции (7.12)
регистрируется по выделению кислоты Р2, то, как можно показать
совместным решением двух уравнений (7.4) с учетом соотношений
(7.2) — (7.3) и (7.13), (7.15), в координатах Лайнуивера-Берка
семейство прямых (l/v, 1/[S]0), соответствующих различным
концентрациям [N], пересекаются в общей точке, расположенной в
верхнем левом квадранте, с координатами (7.9) и (7.10), откуда
можно определить значения индивидуальных констант k2, k3 и К&
[2]. Отношение констант скоростей сольволиза ацилфермента
добавленным нуклеофильным агентом и водой, ki/k3, можно
определить построением экспериментальных данных в координатах (l/v,
[N]). В этом случае прямые, соответствующие различным
начальным концентрациям субстрата, пересекаются в левом верхнем
квадранте в общей точке с абсциссой [N] =—k3/k4.
Задачи
7-1. Для реакций гидролиза ряда субстратов, производных N-
бензоил-Ь-тирозина, катализируемых а-химотрипоином, были
найдены значения /гКат (табл. 1). Вычислить значения индивидуальных
Таблица 1
Значения йкат реакций гидролиза ряда производных
1^-бензоил-Ь-тирозина, катализируемых а-химотрипсином
Производные N-бензоил-Ь-тирозина
Амид
Этиловый эфир
n-Нитрофениловый эфир
0,24
200
300
констант скоростей ферментативного гидролиза субстратов, если
в случае п-нитрофенилового эфира стадией, лимитирующей
скорость гидролиза, является деацилирование промежуточного
ацилфермента [3].
7-2. В таблице 2 приведены значения кинетических параметров
гидролиза ряда субстратов, сложных эфиров N-бензилоксикарбо-
нил-Ь-аланина, катализируемого стрептококковой протеиназой [4].
Оценить значения индивидуальных констант k2, k3 и Ks фермен-
147
тативного гидролиза, принимая, что истинная константа Михаэли-
са не зависит от структуры уходящей группы субстрата в данном
ряду.
Таблица 2
Кинетические параметры реакций гидролиза ряда субстратов,
катализируемых стрептококковой протеиназой.
Условия опыта: рН 5,5; 25е С; ионная сила 0,2 .(ацетатный
буфер); 3,17% ацетонитрила
Сложный эфир К-бензиюксикар-
боннл-Ь-аланина
я-Нитрофениловын
ж-Нигрофениловый
о-Нитрофениловый
Феииловьы
ут (каж)
1С< М
118,0
119,0
35,5
82,5
1.32
1,35
6,58
3,64
7-3. Ингибирование ионами Си++ реакций гидролиза сложно-
эфирных субстратов, катализируемых а-химотрипсином,
происходит согласно схеме
IE + S:
1ES
*■ Е + P.
(7.17)
В таблице 3 приведены данные по влиянию ионов Си++ на
кинетические параметры гидролиза этилового эфира Ы-бензоил-Ь-тирози-
на, катализируемого а-химотрипсином. На основании данных
таблицы и схемы (7.17) определить значения индивидуальных
констант ферментативного гидролиза.
Таблица 3
Влияние ионов Си++ на кинетические параметры
гидролиза этилового эфира N-бензоил-Ь-тирозина,
катализируемого а-химотрипсином. Условия опыта:
рН 6.0; 25° С; иоииая сила 0,Ш (KCI);
6% ацетонитрила; [Е]0 = 10~7М
(Cu+ + l-lf, M
0
1,0
2.0
3,0
5,0
8,0
W сек
86
69
56
46
34
26
кт (каж)-10', М
3,7
3,6
4,8
6,0
6,7
7,7
148
7-4. В реакциях гидролиза сложноэфирных субстратов,
катализируемых а-химотрипсином, изменение ионной силы раствора
оказывает влияние лишь на константу скорости ацилирования
фермента (&2), в ™ время как значения констант й3 и Ks (схема 7.1)'
остаются неизменными [6]. Исходя из данных табл. 4, найти
отношение констант скоростей k2jk3 и значение истинной константы
Михаэлиса Ks для гидролиза метилового эфира N-ацетил-Ь-вали-
на, катализируемого а-химотрипсином, при концентрациях КС1,
равных 0,1М и 2.7М.
Таблица 4
Влияние концентрации КО на кинетические параметры
гидролиза метилового эфира N-ацетил-Ь-валина,
катализируемого а-химотрипсином [7J.
Условия опыта: рН 7,8; 25° С
[KCI], М
0,1
0,3
0,5
0,8
1,0
1,5
2,0
2,7
W >"сл- с-!-
2,20
2,30
2,40
2,50
2,60
2,65
2,70
2,80
т (каж)
0, 108
0,081
0,073
0,052
0,044
0,035
0,025
0,017
7-5. В таблице 5 приведены кинетические данные для
гидролиза этилового эфира L-тирозина, катализируемого а-химотрипсином
в присутствии дополнительного нуклеофильного агента, 1,4-бутан-
диола. Определить значения индивидуальных констант k2, k3 и Ks
ферментативной реакции.
Таблица 5
Влияние 1,4-бутандиола (N) на кинетику гидролиза этилового
эфира L-тирозина, катализируемого а-химотрипсином.
Условия опыта: рН 6,5; 25е С: ионная сила 0,1М (КС1);
[EJ0= 1,6!0-7M
1,80
1,45
1,20
1,02
0,90
0,77
0,67
[N1 = 0
5,43
5, СО
4,57
4,27
4,00
3,64
3,34
г<-10в, М-сек '
|N] =0,11 М
3,78
3,37
3,05
2,76
2,54
2,28
2,08
N = 0,22 М
2,88
2,54
2,27
2,05
1,88
1,68
1,50
149
7-6. Определить значения индивидуальных констант k2, k3 и Ks
для гидролиза метилового эфира N-ацетил-Ь-валина,
катализируемого а-химотрипсином в присутствии дополнительного нуклеофиль-
ного агента, 1,4-бутандиола (табл. 6).
Таблица 6
Влияние 1,4-бутандиола (N) на кинетику гидролиза
метилового эфира 1М-ацетил-Ь-валина, катализируемого
а-химотрипсином. Условии опыта: рН 7,8; 25° С; ионная сила
0,!М (КС!); [Е]0 = 3,8 !0-5М
|SJ„, М
0.2С0
0,193
0,189
0,Ь4
0,091
0,071
0.Г6П
IN] = 0
4,57
—
—
3,83
3,32
2,95
2,70
г>-106, М-сек-1
N = 0,33 М
3,68
—
3,01
2,60
2,16
1,97
N = 0,55 М
—
3,03
2,47
2,03
1,68
1,53
7-7. Определить значения индивидуальных констант гидролиза
метилового эфира М-ацетил-Ь-фенилаланина, катализируемого
трипсином в присутствии метанола, являющегося дополнительным
нуклеофильным агентом (табл. 7).
Таблица 7
Влияние метанола на кинетику гидролиза метилового эфира
1>)-ацетил-Ь-фенилаланина, катализируемого трипсином.
Условия опыта: рН 7,8; 25° С; ионная сила 0,1М (КС1);
|Е]0 = !,4!10-6М
(S]„-103, М
10,0
8,00
6,67
5,55
5,00
1СН3ОН] = 0
3,26
2,80
2,47
2,16
2,00
V' 10е, М-сек *
[СН3ОН] = 0,5 М
1,95
1,67
1,47
1,28
1,18
1СН3ОН] = 1 М
1,39
1,19
1,05
0,915
0,843
7-8. В таблице 8 приведены кинетические данные для гидролиза
метилового эфира N-бензоилглйцина, катализируемого а-химотрип-
150
сином, в присутствии дополнительного нуклеофильного агента,
1,4-бутандиола [2]. Определить значения индивидуальных констант
k2, k3 и Ks ферментативной реакции.
Таблица 8
Влияние 1,4-бутандиола (N) на кинетику гидролиза
метилгиппурата, катализируемого а-химотрипсином.
Услович опыта: рН 7,8; 25° С; ионная сила 0,1М (NaCl);
[EJa = 3,87-!0-6M
[SIo.10!, М
4,00
2,00
1,33
1,00
0,80
v-107, М-сек '
[N] = 0
11,05
9,98
9,28
8,54
7,94
[N] = 0,1IM
6,72
6,05
5,50
5,05
4,65
[N] = 0,22 M
4,72
4,25
3,85
3,52
3,25
7-9. При изучении кинетики сольволиза метилгидроциннамата,
катализируемого а-химотрипсином в смеси воды с метанолом,
выступающим в качестве дополнительного нуклеофильного агента,
определяли зависимость максимальной скорости образования
гидрокоричной кислоты (продукт Рг на схеме 7.12) от концентрации
метанола [1]. Исходя из данных табл. 9, определить значения
констант скоростей k2, k3 и kt (см. схему 7.12), если для
ферментативного гидролиза метилгидроциннамата отношение й2/^з,
определенное из независимого эксперимента, равно 2,5.
Таблица 9
Влияние метанола на максимальную скорость
гидролиза метилгидроциннамата, катализируемого
а-химотрипсином. Условия опыта: рН 7,8; 25° С,•
1Е]0 = 8,810-7М
ICHjOH], М
0
0,12
0,50
1,0
2,0
3,0 |
1,67
1,43
0,80
0,53
0,30
0,22
7-10. Определить значения индивидуальных констант
гидролиза метилового эфира Ы-ацетил-Ь-норвалина, катализируемого
а-химотрипсином в присутствии метанола, выступающего в роли
дополнительного нуклеофильного агента (табл. 10).
151
Таблица 10
Влияние метанола на значения кинетических параметров
гидролиза метилового эфира Г>)-ацетил-Ь-норвалина,
катализируемого а-химотрипсином. Условия опыта: рН 7,8;
25°С; ионная сила 0,!М (КС1); [Е]0 = 2,7910-7М
[СН^ОНЬ
0
0,5
1,0
1,5
м
*кат> сек~'
7,97
7,36
6,60
6,14
Кт
(каж)-Ю',
1,43
2,22
2,94
3,45
м
7-11. В работе [8] изучали сольволиз ряда производных глута-
ровой кислоты, катализируемый со-амидазой в присутствии
дополнительного нуклеофильного агента, гидроксиламина. В табл. 11
приведены отношения скоростей гидроксиламинолиза к скорости
гидролиза для исследованного ряда субстратов при нескольких
концентрациях гидроксиламина. Показать, в пользу двух- или
трехстадийного механизма гидролиза свидетельствуют получен-
ные данные.
Та блица 11
Влияние концентрации гидроксиламина на отношение скоростей
гидроксиламинолиза и гидролиза для производных глутаровой
кислоты при катализе ш-амидазой. Условия опыта: рН 7,0; 30° С;
0,05М фосфатный буфер; [Е]с = 2,8-Ю-2 мг/мл
INH,OH].103, M
1,0
1,5
2,0
3,0
ыетилгл\'тарат
1,05
1,60
2,20
3,37
^NH.OH/^HjC
глутарамат
1,10
1,70
2,17
3,23
л-нитрофенилглутарат
1,17
1,65
2,25
3,30
7-12. Реакция гидролиза амида глутаровой кислоты,
катализируемого со-амидазой, проходит по трехстадийному механизму [8],
Исходя из данных табл. 12, определить, какая из двух стадий —
ацилирование или деацилирование — лимитирует скорость фермен*
тативной реакции, если кинетика гидролиза глутарамата регистра
ровалась по выделению аммиака (продукт Р| на схеме 7.12).
152
Таблица 12
Влияние гидроксиламина на каталитическую константу
превращения глутарамата, катализируемого ю-амидазой.
Условия опыта: рН 7,0; 30° С; 0,05М фосфатный буфер;
[Е1о = 2,8-!0-2 мг/мл
[NH,OH|-l(P. M
0
2,0
4,0
8.0
k -ДО6, ыоль/мин-ыг белка
5,55
7,15
9,10
12,50
7-13. При конкурентном гидролизе и этанолизе п-нитрофенил-
гиппурата под действием папаина конечными продуктами реакции
являются гиппуровая кислота и этилгиппурат [9]. Исходя из
соотношения между концентрациями этих продуктов в реакционной
смеси (табл. 13), вычислить отношение констант скоростей этано-
лиза и гидролиза kjk^.
Таблица 13
Зависимость концентрации гиппуровой кислоты
(ГК) в реакционной смеси от концентрации
этанола после окончания сольволиза
n-нитрофенилгиппурата, катализируемого
папаином. Условия опыта: рН 6,0; 40° С;
0,3M KC1; !,ЗМ ацетоиитрила; [Е]0 = 5,7Ю-7М
IC2HsOH], М
0
0,86
1,72
2,58
[ГК]-10*, М
3.58
2,87
2,49
2,29
7-14. Кинетику гидролиза n-нитрофенилфосфата,
катализируемого щелочной фосфатазой, изучали по образованию п-нитрофе-
нола и фосфат-иона (продукты Pi и Р2 схемы 7.1 соответственно).
Было найдено, что при проведении ферментативной реакции в 1М
трис-буфере (1М водный раствор трис-оксиметил-3-аминометана)
отношение концентраций продуктов Pi/P2 в процессе реакции
равнялось 2,1. Исходя из предположения, что трис-буфер принимает
участие в ферментативной реакции в качестве дополнительного
нуклеофильного агента [10], определить отношение констант k4/k3
(см. схему 7.12).
7-15. Краситель эриохром черный Т является неполным
неконкурентным ингибитором гидролиза этилового эфира N-ацетил-Ь-ти-
розина, катализируемого а-химотрипсином {Кт(каж)=7,58-10~4 М
153
для всех концентраций ингибитора в интервале [I] =0—1,35Х
Х10-4 М). Исходя из данных табл. 14, определить возможные
кинетические и равновесные параметры схемы (7.18), если
известно, что для исследуемой реакции выполняется соотношение
Таблица 14
Влияние эриохрома черного Т
на каталитическую константу гидролиза
этилового эфира 1М-ацетил-Ь-тирозина,
катализируемого а-химотринсином.
Условия опыта: рН 7,0; 25° С; ионная сила
0.1М (КС1); [Е]о = 4!0-8 М
Шо-Ю5. м
0
1,0
2,0
2,4
3,0
5,0
7,0
13,5
*кат- сек-1
135,0
92,5
80,0
71,2
68,7
60,0
58,8
55,0
кг^>кз и константа димеризации красителя, определенная из
независимого эксперимента, равна 2,5-Ю-4 М (димер красителя не
ингибирует ферментативную реакцию)
1E+S
Е + Р,
IE + Р,
(7.18)
7-16. При взаимодействии а-химотрипсина с Ы-ацетил-Ь-трип-
тофаном происходит быстрое образование равновесного комплекса
фермент-кислота, за которым следует реакция ацилирования
фермента свободной кислотой [11]. При низких значениях рН
([Н^] ^Ка, где Ко.—константа диссоциации Ы-ацетил-Ь-триптофа-
на) кинетическую схему этой реакции можно записать в виде
Е + Р ;z=! ЕР 7ZH ЕА + Н20,
(7.19)
где Р — протониров'анная форма Ы-ацетил-Ь-триптофана, КР —
константа диссоциации комплекса фермента со свободной
кислотой (ЕР).
154
1) Исходя из данных табл. 15,-найти константу скорости ацили-
рования фермента свободной кислотой (/г_3), если из независимых
экспериментов были найдены значения й3=10-2 сек-1 и Кр —
=з-ю-зм.
Таблица 15
Кинетика установления равновесия при взаимодействии
а-химотрипсина с N-ацетил-Ь-триптофаном. Условия
опыта: рН 2,3 (цитратный буфер); 25е С; ионная сила
0,0Ш; [Р]о = 5,94- 10~6М. Концентрация свободного
фермента определялась титрованием его
n-нитрофениловым эфиром N-ацетил-Ь-триптофана
Время, сек
0
10
20
30
40
70
4С0
Концентрация свободного
фермента. %
100
91,6
74,7
59,9
44,3
41,3
31,2
2) Предложить графический способ определения констант fe_3
и Кр для реакции (7.19), основанный на измерении эффективной
константы скорости установления равновесия в системе при
различных концентрациях свободной кислоты.
Ответы и решения
7-1. Так как все исследуемые субстраты а-химотрилсина
(табл. 1) имеют одинаковую ацильную часть, то константа
скорости деацилирования (/г3) имеет для них одинаковое значение,
равное 300 сек-1 (для п-нитрофенилового эфира &кат=^з, поскольку
для этого субстрата стадия деацилирования лимитирует скорость
ферментативного гидролиза). Используя соотношение (7.2) и
данные табл. 1, можно вычислить значения kz для ряда субстратов
(для п-нитрофенилового эфира можно указать лишь нижний
предел величины k2).
Ответ: для амида й2=0,24 сек-1; /г3=300 сек-1; для этилового
эфира /г2=600 сек-1; /г3=300 сек-1; для п-нитрофенилового эфира
й2>3000 сек-1; й3=300 сек-1.
7-2. Так как для п-нитрофенилового и ж-нитрофенилового эфи-
ров .Ы-бензилоксикарбонил-Ь-аланина значения каталитической
константы не зависят от структуры уходящей группы, можно
полагать, что для этих субстратов йкат = й3- Учитывая, что значение k3
постоянно для всего исследуемого ряда субстратов (см. решение
предыдущей задачи), находим значения k2 и Ks с помощью
соотношений (7.2), (7.3) и (7.11).
155
Ответ: (результаты округлены с учетом точности вычислений):
Для всех субстратов й3=120 сек-1; /Cs = 10—3 М; для п-нитрофе-
нилового эфира £2=800 сек-1; для ж-нитрофенилового эфира k2=
= 750 сек-1; для о-нитрофенйлового эфира /г2=50 сек-1; для фени-
лового эфира й2=270 сек-1.
7-3. Для вывода уравнения скорости, связывающего значения
кинетических параметров ферментативной реакции (7.17),
протекающей в стационарном режиме (при [S]03>[E]o), с концентрацией
добавленного ингибитора (ионов Си++) запишем уравнение
материального баланса по ферменту
[Е]0 = [Б] + [ES] + [ЕА] 4- [IE] + [IES], (7.20)
выражения для констант диссоциации фермент-субстратного и фер-
мент-ингибитсрного комплексов
выражение для скорости образования продукта Р2
v = k3 [EA] (7.23)
и условие стационарности концентрации ацилфермента в ходе
реакции
^ = k2 [ES] - къ [ЕА] = 0. (7.24)
Комбинируя выражения (7.20) - (7.24), получаем уравнение скорости
ферментативной реакции в присутствии ионов Си++
_, кат
[Е]„ [S].
Ч] ■ — :
^т(каж) т [S]0
где
k\ = -
КЗТ 1 , А , W
1 "г ь |
,f> (каж) ~ А S k „J •
Из выражения (7.25) видно, что тангенс угла наклона зависимости
в координатах (1/^ат' 14) Равен Vk2Kx (рис. 73):
kl k2 k3 k2K\
кат
Значение константы ингибирования (Ki) ферментативной
активности ионами меди можно найти линеаризацией экспериментальных
156
данных в координатах (Кт{клж) /&'кат, [Ц) (рис. 74). Определив
таким образом значение k2, из выражений (7.2) и (7.3) легко
можно найти значения констант й3 и Ks-
Ответ: /г2=320 сек1-, /г3=120 сек"1; Ks=9,7-10-5 М.
7-4. Так как изменение ионной силы раствора избирательно
влияет лишь на константу скорости ацилирования, для
раздельного определения индивидуальных констант ферментативной реакции
удобно применить метод, описанный в § 2 настоящей главы.
Согласно этому методу, в координатах Лайнуивера-Берка семейство
Рис. 73. Обработка результатов экс- Рис. 74. Определение констан-
перимепта по ингибированию ионами ты ингибирования а-химотрип-
меди гидролиза этилового эфира сипа ионами меди в реакции
N-6eH30iw-L-THpo3ima, катализируемо- гидролиза этилового эфира
го а-химотрипсином 1Ч-бензоил-Ь-тирозина
прямых (l/v, l/[S]o), полученных при различных значениях ионной
силы раствора, пересекается в левом верхнем квадранте с
абсциссой общей точки пересечения, равной l/[S]o=—l/i\s. Таким
образом, для нахождения величины Ks необходимо воссоздать ход
прямых зависимости \jv от l/[S]o, соответствующих различным
значениям ионной силы раствора. Это можно сделать, если учесть, что
в координатах Лайнуивера-Берка прямая ([E]o/f, l/[S]o) отсекает
на оси ординат и абсцисс отрезки 1//гкат и —\IKm{™m (рис. 75).
Определив таким образом величину Ks, из соотношения (7.11)
находим значения k2/ks.
Ответ: /(s=0,4M независимо от ионной силы раствора; при
[KCI] =0,1 М; k2/k3=2,7; при [KCI] =2,7 М; k2lk3=23.
7-5. Для нахождения значений индивидуальных констант
ферментативной реакции удобно применить метод, описанный § 3
настоящей главы. Согласно данному методу, в координатах
Лайнуивера-Берка прямые, соответствующие различным концентрациям
дополнительного нуклеофильного агента, пересекаются в левом
157
верхнем квадранте с абсциссой общей точки пересечения, равной
l/[S]o= — 1/As. Определив таким образом величину Ks (рис. 76),
из соотношений (7.2) и (7.3) можно найти значения констант k2
и k3.
Ответ: й2=260 сек"1; й3=68 сек"1; /(S=5-10-2M.
-6,0
-40
-2,0 ...
//[S]q10'!m
Рис. 75. Определение истинной
константы Михаэлиса в реакции
гидролиза метилового эфира N-
ацетил-Ь-валина, катализируемого
а-химотрипсином, при селективном
влиянии ионной силы раствора на
константу скорости ацилнровашш
фермента. Концентрации КО: а —
0,1 М; 6 — 0,3 М; в —0,5 М; г —
0,8 М; д— 1,0 М; е— 1,5 М; ж —
2.0 М; 3 — 2,7 М
х:
сь
Г° 8.0
Г
lO .
S 6,0
is
i> 4,0
1 ЛГ
X S*°
/ XT
i i i i i
-0,8
0,8 _ W
УЩ10М
у:
си
t_>
'г
'о „
' О
_^
6.0
4,0
2,0
Г *
/В
/ Л
р if
/ Cf j*^
i i i i
'0,8
-h
к
0,8 -2 ,16
Рис. 76. Определение
индивидуальных констант скоростей
гидролиза этилового эфира L-тнрозина,
катализируемого а-химотрипсн-
пом, в присутствии
дополнительного нуклеофильного агента, 1,4-бу-
тандиола. Концентрации 1,4-бутан-
дпола: с —0; б — 0,11 М; в — 22 М
Рнс. 77. Влияние дополнительного
нуклеофильного агента, метанола,
на кинетику гидролиза метилового
эфира Ы-ацетил-Ь-фенилаланина,
катализируемого трипсином.
Концентрации метанола: а — 0; б —
0,5 М; в— 1,0 М
7-6. Решается аналогично задаче 7-5.
Ответ: й2=0,98 сек"1; /г3=0,21 сек-1; /(S=0,5M.
7-7. Решается аналогично задаче 7-5 (рис. 77).
Ответ: /г2 = 6,4 сек-1; К&= 1,8- 10"2М; /г3>/г2.
158
7-8. Решается аналогично задаче 7-5.
Ответ: £2=0,42 сек"1; h=\,\7 сек"1; /(s=5,78- 10"3М.
7-9. Для вывода уравнения скорости образования продукта Р2
(схема 7.12) в присутствии дополнительного нуклеофильного
агента N запишем уравнение материального баланса по ферменту
(7.26), выражение для константы диссоциации
фермент-субстратного комплекса (7.27) и условие стационарности концентрации
ацилфермента (7.28) в ходе ферментативной реакции
[E]0=[E] + [ES] + [EA], (7.26)
^—тёЦЬ <7-27>
ЩР = k2 [ES] - {ks + kt [N]) [EA] = 0. (7.28)
Комбинируя уравнения (7.26) — (7.28) с выражением
^- = Й3[ЕА|, (7.29)
получаем уравнение скорости ферментативной реакции
k2k3
dP2 A2 + A, + ft4[N] [E]o [S]o
dt K k3 + k,[N]
s A. + fc. + A.lN] +l5Jo
(7.30)
Из уравнения (7.30) видно, что зависимость максимальной
скорости образования продукта Рг (при [S]o>Km(naH<),) от концентрации
добавленного нуклеофильного агента определяется выражением
*т ksk3 С7 ЯП
[Е]. _ ft2 + ft3 + ft4[N] • К ■ '
Линеаризуя выражение (7.31) в координатах ([E]0/Vm, [N]) и
учитывая, что для изучаемой реакции k2/k3=2,5, находим значения
индивидуальных констант скоростей ферментативного сольволиза.
Ответ: £2=0,665 сек-1; /г3=0,266 сек"1; £4=2,04 М-1-сек-1.
7-10. Из уравнения (7.30) находим
J L _i_ _L -I *« lNl
KKST Kg К2Кз
Рассматривая полученные выражения, видим, что линеаризация
кинетических параметров ферментативной реакции в координатах
(1/&кат, [N]) и (/Ст(каж)/&кат, [N]) позволяет провести раздельное
определение значений k2, k3, &4 и Ks (рис. 78).
Ответ: £2=55,8 сек-1; £3=9,30 сек-1; fe4= 12,8 М"!-сек '; Ks=
= 0,1 М.
159
1,0
1,5 0,5
[сн,он],м
Рис. 78. Определение индивидуальных констант гидролиза и метанолиза
метилового эфира Ы-ацетил-Ь-норвалина под действием а-химотрипсина
7-11. Из рис. 79 видно, что отношение скорости гидроксиламино-
лиза к скорости гидролиза одинаково для амида, метилового
эфира и n-нитрофенилового эфира глутаровой кислоты, что
свидетельствует в пользу существования общего ацилферментного
промежуточного соединения для всех изученных субстратов (схема 12).
7-12. Для вывода уравнения скорости образования продукта Pi
(см. схему 7.12) в присутствии дополнительного нуклеофильного
агента выражения (7.26) — (7.28) комбинируют с выражением
dPl *2[ES],
dt
получая в итоге уравнение
dP>
M^ + MN])
k2 + k3 + fe, [N]
[E]«[S]„
(7.32)
(7.33)
dt &3 + MN] •
Ks ке + к3 + к4[Щ +lbl»
Из полученного уравнения видно, что если ацилирование
(«2*С«з) лимитирует скорость ферментативной реакции, величина
«кат не должна зависеть от [N]. В случае «2>«з+«4 [N] величина
«кат линейно зависит от [N]. Наконец, при соизмеримых значениях
«2 и «з величина ккат не должна являться линейной функцией от
концентрации добавленного нуклеофильного агента.
Так как для гидролиза амида глутаровой кислоты зависимость
160
в координатах (£кат, [N]) линейная (рис. 80), для этой реакции
выполняется неравенство £2»£3, т. е. стадией, лимитирующей
скорость ферментативного гидролиза, является деацилирование.
1-16. из схемы (7.12) следует, что отношение концентраций
продуктов сольволиза (Р3) и гидролиза (Р2) равно отношению
скоростей их образования, т. е.
[Р3] К [Щ [ЕА]
[Р2] kt [ЕА] '
или
-^- = _L Ж. /7Ч/Ч
кг [N] ■ [Р2] • ('-Щ
Таким образом, зная отношение концентраций образующихся
продуктов Р3 и Р2, можно вычислить отношение констант скоростей
LNH 0Н]-70.М
Рис. 79. Отношение констант
скорости"! гидроксиламинолиза
и гидролиза для амида (О);
метилового эфира (А) и п-ни-
трофенилового эфира (•) глу-
таровой кислоты
[NH ОН] Л
Рис. 80. Обработка
кинетических данных по влиянию
дополнительного нуклеофильного
агента, гидроксиламина, на
гидролиз глутарамата,
катализируемый со-амидазой
ферментативного сольволиза и гидролиза kjk3. Пренебрегая
изменениями 'концентрации воды при добавлении этанола в
реакционную систему, получаем kjka=029 М"1 при [С2Н5ОН1 =0,86 М
kjk3=0,26 М-1 при [С2Н5ОН] = 1,72 М и kjk3=0,22 M"1 при
[С2Н5ОН]=2,58М.
Ответ: kjk3= (0,26+0,03) Mr1:
7-14. Из схемы (7.12) вытекает, что для ферментативной
реакции, протекающей в присутствии дополнительного нуклеофильного
агента, выполняется соотношение
[Pi] = [Р2] + [Р3],
или
[p.] [P8i L (7--*>)
6 Заказ 158
161
Подставляя соотношение (7.35) в (7.34), получаем
К 1 / [Р,
К
[N] V [PJ J '
Ответ: k4/k3=l,l М-1.
7-15. Для вывода уравнения скорости ферментативной реакции
(7.18), протекающей в стационарном режиме, запишем выражение
для скорости образования продукта
v -= k2 [ES] + -\k, [IES], (7.36)
выражения для констант диссоциации комплексов (при условии
быстрого установления равновесия в системе (7.18)):
^s ~ "[EST ' (7'37)
^, = °Щ]-, (7-38)
»*•—&' <7"39>
p^—w-- <7-40>
условие стационарности концентраций ацилферментных
промежуточных соединений ЕА и IEA
tf([EA]rf+[IEA]) = fe2 [ES] - k3 [ЕА] + т£2 [IES] - bk3 [IEA] = 0 (7.41)
и условие материального баланса по ферменту
[Е]0 = [Е] + [ES] + [ЕА] + [IE] + [IES] + [IEA]. (7.42)
Из выражения (7.41) следует, что
h [ES] + fk2 -№L = k3 [EA] + 6*3 -^Шг1
aKi 'ML—J . »"3 ^
или
1 + UL
[EA] = A ^-[ES]. (7.43)
Комбинируя выражения (7.37) — (7.43) и подставляя их в
выражение (7.36), получаем уравнение скорости ферментативной реакции
... "кат
(каш) + [SJo '
162
где
7 И
ъ "А1 у П 44)
1 ['] *» l1+«/ft A + p*i /
1+ а/С, + из , , »И
1+ PACi
/cm(Ka»)=/cs гТТ^ПлТТТЖТ"' (7"45)
1
1_ГР/С1
В выражениях, приведенных выше, концентрация [I]
соответствует мономерной форме красителя. Из схемы реакции
димеризации
I + I—D
и уравнения материального баланса по ингибитору
[I]o = [I] + 2[DJ,
где [1]0 — общая концентрация ингибитора; [D] — концентрация ди-
мера и /f d = [I]2/[Dj, можно найти соотношение
2[1р + ЛЪ[1]-/С0[1]о = 0.
Решая полученное квадратное уравнение, находим
[Ч -^*+/ж + -^[Чо. (7-46)
Зная численное значение константы димеризации, из выражения
(7.46) можно найти концентрацию мономера в растворе для всего
использованного интервала концентраций ингибитора (см.
табл. 14).
Из выражений (7.44) и (7.45) можно получить соотношение
^кат 7 [I]
1 + "^
Кт(каж) aR\ „ ._
Ks + К,
где отношение k2/Ks равно отношению ккаг/КпцКяж) в отсутствие
ингибитора. Преобразуя соотношение (7.47), получаем
Km (каж) 1 \ а . . аК\ _1
kj I 7 —а Т — а [I] '
*S /
6* 163
Так как для ингибирования <х-хи\.отрипсина эриохромом черным Т
величина Кт(шм) не изменяется в присутствии ингибитора, то
линеаризация экспериментальных данных в координатах
("кат I | 1
где [I] —концентрация мономера, рассчитанная с помощью
соотношения (7.46), позволяет найти значения Ki и у/а (рис. 81).
Наконец, из рассмотрения выражения
(7.45) можно заключить, что
величина Кт(Каж) не будет зависеть от
концентрации ингибитора только в
случае выполнения равенств у/а=
= б/р, а=1, р=1.
Ответ: а=1; Р=1; у=0,3; б =
= 0,3; Кг= 1.1 - 10-Б М; h =
135 сек-1.
7-16. Для вывода уравнения
скорости установления равновесия
в системе (7.19) запишем
выражение для скорости накопления ацил-
фермента
Рис. 81. Обработка кинетических
данных по неполному
неконкурентному ипгибированию
эриохромом черным Т гидролиза
этилового эфира Ы-ацетил-Ь-тирозина,
катализируемого а-химотрипсином
*Ш- = k„3 [ЕР] - k3 [EA], (7.48)
dt
уравнение материального баланса по
ферменту
[Е]0 = [Е] + [ЕР] + [ЕА] (7.49)
и выражение для константы равновесия в системе в начальный
период реакции
Kt
[Е] [Р]
(7.50)
' Р [ЕР]
Из соотношений (7.49), (7.50) найдем выражение для текущей
концентрации свободной формы фермента
(Е] =
[Е]„-[ЕА]
1 +
Kv
Дифференцируя выражение (7.51) по времени, получаем
dt
dt
(7.51)
(7.52)
Kv + [P]
Подставляя выражение (7.48) в (7.52) и комбинируя полученное
выражение с соотношениями (7.50), (7.51), получаем
«Р|В1._^+1^ЛЧ)|Е]. (7.53,
dm
dt
КР + [Р]
КР+ [Р]>
164
Интегрирование уравнения (7.53) при начальных условиях [Е] = [Е]0
при t = 0 приводит к соотношению
[Щ — [Е]рагн -kr,,t it кл\
■■ е 5ФФ , (/■54)
l^Jo ■ [t-Jpaiii
где эффективная константа скорости первого порядка,
характеризующая скорость установления равновесия, равна
^эфф = «з + ^р ^ pj (7.55)
и [Е]раВн — равновесная концентрация свободной формы фермента
(при t —» оо).
1) Из выражения (7.54) следует, что величину кэфф можно
найти линеаризацией экспериментальных данных в координатах
/i [E] —[Е]раон Л
(In fE| _fE1 . t\. Далее, используя соотношение (7.55) и
известные значения констант k3 и Кт>, можно нёйти величину k~z.
2) Линеаризация соотношения (7.55) в виде
1 =_i_ , Кр 1
ь "г
*эфф— k3 k-s r k-3 [P]-
позволяет определить значения констант #_3 и ^р из тангенса угла
наклона и точки пересечения прямой в координатах (1 1{кэфф -\- k3), 1 / [Р]).
Для использования этого метода необходимо предварительно
определить величину k3 из независимого эксперимента.
Ответ* £_3 = 2,6-Ю-2 сек-1.
ЛИТЕРАТУРА
1. В е n d е г М. L., С 1 е m e n t G. E., G u n t е г С. R., К е z d у F. J. . J. Amer.
Chem. Soc.", 86, 3697, 1964.
2. Berez in I. V., Kazanskay a N. F., Klyosov A. A. „FEBS Letters",
15, 121, 1971.
3. О и t f г е и n d H., Hammond B. R. „Biochem. J.", 73, 526, 1959.
4. Kortt A. A., Liu T.-Y. „Biochemistry", 12, 328, 1973.
5. Березин И. В., Билль X., Мартинек К., Яцимирский А. К.
ДАН СССР, 179, 214, 1968.
6. Б е резн н И. В., Мартинек К. В сб.: „Структура и функция
ферментов", вып. 1. Изд-во МГУ, 1972, стр. 20.
7. Martin R. В., Niemann С. „J. Amer. Chem. Soc", 80, 1481, 1958.
8. Hersh L. В. „Biochemistry", И, 2251, 1972.
9 H e n г у А. С, К i г s с h J. F. „Biochemistry", 6, 3536, 1967.
10 H i n b e r g I., L a i d I e г К. J. „Can. J. Biochem", 50, 1360, 1972.
11 KezdyF J., Clement G. E., В e n d e r M. L. „J. Amer. Chem. Sec",
86, 3690, 1964.
Глава 8
ПРИМЕНЕНИЕ ИНТЕГРАЛЬНОЙ ФОРМЫ
УРАВНЕНИЯ СКОРОСТИ ДЛЯ
КИНЕТИЧЕСКОГО АНАЛИЗА
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
§ 1. Интегральный анализ кинетики
действия ферментов
Обработка кинетических данных ферментативных реакций
может быть проведена как с использованием начальных скоростей
(см. гл. 5—7), так и с использованием временного хода реакции,
применяя интегральную форму кинетического уравнения скорости.
В своей дифференциальной форме уравнение Михаэлпеа-Мен-
тен (8.1) справедливо для двух- и многостадийных стационарных
ферментативных процессов при условии [S] 3> [Е] о
d[S] _ Vm[S] (R n
dt ' Km („») + [S] ■ ' '
Кинетические параметры: Vm — максимальная скорость и /\т(каж)—
кажущаяся константа Михаэлиеа ферментативной реакции —
функции констант скоростей индивидуальных стадий (см. соотношения
5.10, 7.2, 7.3). Для раздельного определения этих параметров в
общем случае нельзя использовать интегральные методы обычной
неферментативной кинетики вследствие смешанного порядка
ферментативных процессов. Например, метод Гуггенгейма (см. гл. 2)
пригоден для обработки кинетики ферментативных реакций только
в случае /Ст{каж)3> [S]0 или [E]o3>[S]0, т. е. только при наличии
кинетики первого порядка по субстрату или продукту.
Интегрирование уравнения (8.1) при начальных условиях
[S] = [S]o при 2=0 приводит к уравнению (8.2), определяющему
зависимость концентрации продукта реакции Р от времени
[Р] = VJ - /Ст(каж) In -тзйр]-. (8-2)
Линеаризация уравнения (8.2) может быть проведена различными
способами, более или менее пригодными для практической работы.
Например, широко известен метод Уокера—Шмидта [1], согласно
которому обработка экспериментальных данных проводится в
координатах ([PJ/*, \Ц In -jsjfzripj-)
[Р] I/ Km (каж) | Но /о о\
— - * т t Ш -[S]„-[P] {Ъ-6)
Из выражения (8.3) очевидно, что прямая в координатах Уокера-
Шмидта имеет тангенс угла наклона, равный — Кпцкаж), и отсекает
на оси ординат отрезок, равный Vm.
166
Если в распоряжении экспериментатора имеется полная
кинетическая кривая (рис. 82), более удобным является метод
обработки, в котором вместо абсолютных значений параметров реакции
(начальной концентрации субстрата, начального времени реакции,
абсолютной концентрации продукта реакции и т. д.) используются
относительные, что значительно повышает точность результатов
эксперимента. Рассмотрим в качестве примера кинетическую
кривую накопления продукта
ферментативной реакции (см. рис. О
82), удовлетворяющую уравне- ^
нию (8.2). Откладываемая на ?
оси ординат величина D может ^2~&,
соответствовать различным ха- п
рактеристикам системы, которые
удобны для прямого измерения
(величина оптической
плотности, количество щелочи, идущей
на титрование продукта
реакции, проводимость раствора
и т. д.). В общем случае [Р] =
= <z(D — D0), где а —
переходный коэффициент, a D0 и D —
значения, соответствующие
начальному и текущему моментам
реакции. Для двух любых
моментов времени, различающихся на величину Д, в соответствии
с выражением (8.2) справедливы следующие уравнения:
*iW, Н
Рис. 82. Полная кинетическая кривая
накопления продукта реакции. D—
некоторый физический параметр
системы, удобный для регистрации
кинетики
t = °(Dl ~ °°) J_ Кт (каж) In
£>
А,
V„
v„
А,
А, •
*i = *! + A<
a (D\ — D„ ) Km (каж) A
r? Г V ln
A„
Vr
D„
A,
(8.4)
(8.5)
Подобные уравнения должны быть верны для t2 и t2 и т. д.
Вычитая (8.4) из (8.5), имеем
a(D[~D1) КтЫаж) . А» ~ А
v„
А»-Я
или в более общем виде
D',-Dt =
Vm-&
Кт (каж) i_ _ °° l
D»
A
При построении результатов эксперимента в координатах
D[-Di, ln-
£>
D\
(8.6)
(8.7)
получаем прямую с тангенсом угла наклона
пересечения на оси ординат VmA/a [2].
-Кт(каш)/а И ТОЧКОЙ
167
§ 2. Ингибирование продуктом реакции
При интегральном анализе кинетики ферментативных реакций
особый интерес приобретает случай конкурентного ингибирования
продуктом реакции
т (каж) и
Е + S «. _JT ES —Ы+ Е + Р, (8.8)
Е + Р Kv , ЕР.
Кинетическая обработка схемы (8.8) приводит к зависимости,
концентрации продукта ферментативной реакции от времени
[S]„
к- Л . _[§!
*\ги (каж) I l ~l~ yv-
t __ Km (каж) Л Km (каж) \ [S]0 [Я] v
Одним из методов определения константы ингибирования
продуктом (Кт>) является метод Фостера — Ниманна [3]. Согласно этому
/[Р] 1 . [S]„ \
методу прямая, проведенная из начала координат (-г-, -j lnr§i _rpi)
с тангенсом угла наклона, численно равным [S]0, пересекает
прямую, описываемую уравнением (8.9), в точке, соответствующей
начальному (Времени реакции, /=0 (в которой, разумеется,
ингибирование продуктом реакции отсутствует). Имея ряд точек,
полученных подобным образом при различных начальных концентрациях
субстрата, можно определить все кинетические и равновесные
параметры схемы (8.8).
Величину константы ингибирования продуктов Kv можно
также определить, откладывая обратные значения отрезков,
отсекаемых на оси абсцисс прямыми в координатах Уокера-Шмидта, в
зависимости от [S]0 (см. выражение 8.9). В случае ингибирования
продуктом реакции подобная зависимость прямолинейна и отсекает
на осях абсцисс и ординат отрезки, равные —Kv и Кт(к&т)1Ут
соответственно [2].
Наконец, величину Kv можно также определить, если из
независимого эксперимента (при использовании начальных скоростей
реакции) найти значения Vm и /Ст(каш) ферментативной реакции и
сопоставить их со значениями эффективных кинетических
параметров, найденных из полной кинетической кривой при ингибирующем
действии продукта реакции.
§ 3. Инактивация фермента в ходе
реакции
При изучении кинетики ферментативных реакций, протекающих
до высоких степеней превращения субстрата, необходимо
принимать в расчет возможность инактивации фермента в ходе реак-
168
ции. В ряде случаев было найдено, что связывание субстрата с
ферментом ускоряет инактивацию последнего; иногда субстрат не
оказывает влияние на инактивацию фермента. Однако чаще всего
связывание субстрата предохраняет фермент от инактивации, т. е.
фермент-субстратный комплекс инактивируется медленнее, чем
свободный фермент.
Рассмотрим в качестве примера схему реакции, в которой
субстрат полностью предохраняет фермент от инактивации, в то время
как свободный фермент денатурирует с константой скорости
инактивации первого порядка /гин
E + S^ES^l+E + p. (8Л0)
Неактивный фермент
Если [Е*]0 — общая текущая концентрация активного фермента
в системе (8.10), то можно записать
[E*]0=[E] + [ES] (8.11)
и
-£gb = *„B<[E*]0-[ES]). (8.12)
Из схемы (8.10) следует, что скорость расходования субстрата
определяется выражением
или, учитывая соотношение
*s = i§f, (8.14)
имеем
- ^г = кг ([Е*]о ~[ES]) [S]- (8-15)
Разделив уравнение (8.12) на (8.15), получаем
d[E*]0 Kskm
d [S] k, [S]
или
/Cc kKn d [S]
Wb—itr-W- <8Л6)
Интегрируя уравнение (8.16) при начальных условиях [Е*]0=[Е]0
при [S] = [S]0,
lE*l° „ h [si
[Elo [ S]„
169
получаем
ДЕ%-[Е]Р-*^1п-^-. (8.17)
Из полученного выражения следует, что количество непрореагиро-
вавшего субстрата в системе после прекращения реакции
(вследствие полной инактивации фермента, при [Е*]0—0) равно
_ к, [Е]0
[S] = [S]„e *™*s. (8.18)
Если из независимого эксперимента (при использовании
начальных скоростей ферментативной реакции) определить значения
^2 и Ks, то, зная начальные концентрации субстрата и фермента
и долю субстрата, прореагировавшего до прекращения реакции, из
соотношения (8.18) можно рассчитать константу скорости
инактивации фермента в условиях опыта.
Комбинируя выражения (8.11), (8.13) и (8.14), получим
дифференциальное уравнение скорости ферментативной реакции
'tf[S] *,[E*].[S] /R1Q.
dt Ks +[S] • [p.iv)
Подставляя выражение (8.17) в (8.19), получим
d[S] *.[EUS] KslS]km« [S],
ЧГ = Ks f [SI ~ *s + [S] ln -\sf (8"2°)
Вводя вспомогательную переменную z, связанную с [S]
зависимостью
~ ' [si. e •
где
q __ Ъ [Е].
преобразуем уравнение (8.20) к более простому виду
dz Ks йич г In г
dt Ks+[S]0e-"z-
(8.21)
Интегрирование уравнения (8.21) "при начальных условиях z = е"
при t = 0 приводит к выражению
t = -±- In
1 [S]0e~" Г dz
л 1 [S] Ksk
4 [S]0
— \ -41-. (8.22)
f dz ,
где \ -: — табличный интеграл, равный
f dZ . . , . . (1П2)2 , (1П2)3
170
Подставляя в выражение (8.22) известные значения констант k2,
р<\\ъ, Ks и начальные концентрации [Е]о, [S]0, можно рассчитать,
какое количество субстрата превратится в продукт под действием
фермента за определенное время t. Из выражения (8.22) также
можно рассчитать время, за которое ферментативная реакция
пройдет до заданной степени превращения субстрата.
Задачи
8-1. Найти значения точек пересечения на координатных осях
и угловых коэффициентов прямых при линеаризации интегральной
формы уравнения скорости ферментативной реакции в координатах
*> t • t iU[S].-[P]'
6)-L _LIn_lsi._.
°' [Pj ■ [Pj Ш [S].
в)
[P] ■
[P]
In:
[S].
In i
[S].
^r-> [S]o-[P] ,U[S].-[P]
Гв-^/При изучении кинетики гидролиза амида N-ацетил-Ь-гек-
сагйдрофенилаланина, катализируемого ct-химотрипсином,
эксперимент ставили следующим образом. Меняя начальную
концентрацию субстрата, определяли концентрацию продукта,
образовавшегося через 1 ч после начала ферментативной реакции [4]. На
основании данных табл. 1 определить кинетические параметры (&кат и
Кт(кьш)) реакции.
Таблица 1
Зависимость концентрации продукта,
образовавшегося через 1 ч после начала
ферментативного гидролиза амида
К-ацетил-Ь-гексагидрофенилаланина,
от концентрации субстрата. Условия опыта:
рН 7,9; 23° С; начальная концентрация
а-химотрипсина 6,14-10-5М
[S]o-10>, M
1,00
1,24
1,25
1,43
1,50
1,67
2,00
2,22
2,55
3,00
3,50
4,01
IPleo мин ■«>■, м
1,85
2,26
2,35
2,44
2,58
2,84
3,13
3,46
3,62
3,90
4,15
4,51
171
\Jwp В таблице 2 приведены кинетические данные для
гидролиза метилового эфира Ы-ацетил-Ь-фенилаланил-Ь-лизина,
катализируемого трипсином в присутствии 1-метил-2-аминоиндола [2].
Определить значения кинетических параметров и константу инги-
бирования ферментативной реакции.
Таблица 2
Кинетика расходования субстрата в реакции гидролиза метилового эфира
К-ацетил-Ь-фенилаланил-С-лизина, катализируемого трипсином,
в присутствии 1-метил-2-аминоиндола. Концентрация субстрата выражена в см
диаграммного листа самописца, так что перемещение пера самописца на 1 см
соответствует изменению концентрации субстрата на 3,74-10-5М.
Условия опыта: рН 7,8; 25° С; ионная сила 0,Ш (СаС12); [Е]0 = 1,82-10-8М
Время, сек
0
30
60
90
120
150
180
195
210
225
240
255
270
285
300
315
330
345
360
390
[П = о
15,4
13,1
11.1
9,0
7,1
5,2
3,6
—
2,1
—
1,1
—
0,4
—
—
—
.—
—
—
—
[S
[1]= 1,46-10—3 М
16,9
14,9
13,0
11,2
9,4
7,7
6,1
—
4,6
—
3,4
—
2,4
—
1,6
—
1,0
—
0,5
—
, см
[1] = 1,84-10—3 М
16,8
15,2
13,2
11,2
9,4
7,6
—
5,6
—
4,6
—
3,3
—
2,2
—
1,5
—
0,9
—
—
[I] 3,25-10—3 М
18,0
16,2
14,5
12,8
11.2
9.7
8,2
—
6,8
—
5,6
—
4,5
—
3,5
—.
2,6
—
2,0
1,4
8-4. В таблице 3 приведены кинетические данные для
гидролиза этилового эфира Й-ацетил-Ь-тирозина, катализируемого сс-хи-
мотрипсином в присутствии конкурентного ингибитора, метилового
эфира М-ацетил-О-фенилаланил-Ъ-аланина. Определить значение
константы ингибирования фермента DD-дипептидом, если
начальная концентрация субстрата и начальное время реакции
неизвестны. Определение концентрации непрореагировавшего субстрата
в ходе ферментативной реакции проводилось в каждом случае
через равные промежутки времени.
8-5. Кинетика дезаминирования аденозина, катализируемого
аденозиндезаминазой, изучалась в работе [5] кондуктометриче-
ским методом (табл. 4). На основании данных табл. 4 определить
значения Vm и Кт(каж) ферментативной реакции.
172
Таблица 3
Зависимость концентраций субстрата (в усл. ед.), определяемых в процессе
ферментативной реакции через одинаковые промежутки времени,
от концентрации конкурентного ингибитора. Условия опыта: рН 7,8; 25° С;
ионная сила 0,1М (КС1)
[1] = 0
18,2
16,4
14,5
12,7
11,0
9,4
7,9
[I] = 2,8-10-3 М
16,9
15,2
13,4
11,7
10,1
8,7
5,9
[I] — 7,3-10—3 М
[8,9
17,2
15,5
13,9
12,3
10,9
9,5
[1]=0
6,5
5,2
4,0
3,0
2,1
1,4
1.0
[I] - 2.8-10-3 М
4,8
3,7
2.8
2,0
1,5
[I] - 7,3-10—3 М
8,1
6,8
5,7
4,6
3,5
2,9
2,3
Таблица 4
Кинетика накопления продукта дезаминирования
аденозина, катализируемого аденозиндезаминазой.
Условия опыта: рН 6,5; 37° С; [Sj0 = 4,95-1()-3М
Время, мин
0,4
0,9
1.3
1,8
2,7
3,3
3,9
4,6
5,2
5,8
6,3
7,0
7,7
оо
[ Р]-1С», М
0,28
0,67
0,96
1,34
1,85
2,36
2,74
3,22
3,54
3,92
4,30
4,58
4.85
4,95
8-6. В таблице 5 приведена кинетика накопления п-нитрофе-
нола в реакции гидролиза n-нитрофенилфосфата, катализируемого
щелочной фосфатазой [6]. Определить значения Vm и /(т(каж)
ферментативной реакции.
8-7. В таблице 6 приведены кинетические данные по изучению
реакции гидролиза мочевины, катализируемой уреазой, полученные
с помощью кондуктометрического метода [5] Определить
значения Vm и Кт(каж) ферментативной реакции.
173
Таблица 5
Кинетика накопления n-нитрофенола в реакции
гидролиза n-нитрофенилфосфата, катализируемого
щелочной фосфатазой. Условия опыта: рН 8,0;
25°С; 1М трис-буфер; |S]0 = 5,Ы0-3М
Время, мин.
5
10
20
30
40
50
60
80
100
120
150
180
оо
Р-103, М
0,171
0,466
1,16
1,78
2,36
2,90
3,31
3,95
4,36
4,66
4,88
5,00
5,10
Таблица 6
Зависимость изменения проводимости раствора
(До) от времени гидролиза мочевины,
катализируемого уреазой. Условия опыта: рН 8,0;
37° С; 0.01М трис-буфер; [Sj0 = 110-3M
Время, сек
10
20
30
60
90
120
150
180
оо
До-10е, ом '■ см '
40
62
77
ПО
130
140
147
149
150
8-8. Анализируя полную кинетическую кривую гидролиза гип-
пурилфенилглицина, катализируемого карбоксипептидазои, авторы
[7] получили отрицательные значения ккат и Кт(кат)- Дать
объяснение полученным результатам.
8-9. В таблице 7 приведены кинетические данные для
гидролиза бромацетил-ОЬ-фениллактата, катализируемого
карбоксипептидазои [7]. Найти значение константы конкурентного ингибирова-
ния продуктом реакции, если значения kKaT и Кт<ка-ж), определенные
из начальных скоростей ферментативной реакции, равны 57,4 сек-1
и 1,6 • 10-3 М соответственно.
174
Таблица 7
Кинетика накопления продукта гидролиза
бромацетил-БЬ-фениллактата, катализируемого
карбокснпептидазой. Условия опыта: рН 7,5;
25° С; [Slo = 7,3.10-3M; [EJ0 = 5,68-10-7М
Время, сек
96
144
228
324
624
834
1146
|Р]-103, М
2,63
3,29
4,29
5, СО
6,27
6,73
7,20
8-10. Предложить метод определения кинетических параметров
ферментативных реакций, основанный на определении площади под
кинетической кривой в координатах ([S], t).
8-11. Предложить аналитический метод определения константы
Михаэлиса, исходя из уравнения касательной к кинетической
кривой в начальный момент времени и интегральной формы уравнения
кинетической кривой ферментативной реакции.
8-12. Доказать, что прямая, проведенная из начала координат
(■^г, ~т~ 1" rsi —."гот) с угловым коэффициентом численно
равным [S]0, пересекает другую прямую, описываемую уравнением (8.3)
в точке, соответствующей начальному времени реакции.
8-13. Гидролиз бензилпенициллина, катализируемый пеницил-
линамидазой, ингибируется продуктом реакции, фенилуксусной
кислотой. В табл. 8 приведены значения констант Михаэлиса, рас-
Таблица 8
Зависимость кажущейся константы Михаэлиса
от начальной концентрации субстрата
для гидролиза бензилпенициллина,
катализируемого пенициллинамидазой
Условия опыта: рН 7,5; 25° С; ионная сила
0,1М (КС1); [Е|0 = 2,310-7М
[ S]0 -10*, М
0,70
0,90
1,20
2,30
3.50
4,70
7,00
9,40
11,70
*т(каЖ)-,05'М
1,26
1,49
1,99
3,43
4,80
5,30
11,10
11,60
15,50
175
считанные из анализа полной кинетической кривой, при различных
значениях начальной концентрации бензилпенициллина.
Определить значения кинетических параметров и константы ингибирова-
ния фермента фенилуксусной кислотой, если 1величина Vm,
определенная с помощью интегрального метода анализа, равна
4- 1(У~7 М-сек-1 при всех значениях [S]0.
8-14. Указать, как должен быть поставлен кинетический
эксперимент, чтобы была возможна линеаризация экспериментальных
данных в координатах МР]Д, \lt In .„,_' j в случае заметной
денатурации фермента в ходе реакции. Принять, что связывание
субстрата с ферментом не влияет "на скорость инактивации последнего.
8-15. Кинетические параметры гидролиза бензилпенициллина
до 6-аминопенициллановой кислоты (6-АПК) под действием
иммобилизованной пенициллинамидазы при 40° С равны йнат=15 сек"1,
^Ст(каж)=3,1 • 10~6 М. Константа инактивации пенициллинамидазы
в условиях проведения реакции равна Ю-5 сек-1, причем
связывание с ферментом субстрата или продуктов реакции не влияет на
скорость инактивации фермента. Рассчитать, -какое количество
6-АПК (мол. вес 217) можно получить с помощью
иммобилизованной пенициллинамидазы в периодически действующем реакторе
объемом 100 л (начальная концентрация активного фермента
равна 3-10~6М, начальная концентрация субстрата равна 1,0М), если
степень конверсии субстрата в каждом реакционном цикле должна
составлять 99%. В расчетах учесть, что константа конкурентного
ингибирования пенициллинамидазы вторым продуктом реакции,
фенилуксусной кислотой, равна 2,8-10~5 М.
8-16. Определить константу скорости инактивации фермента,
вызываемой субстратом (см. схему 8.23), если фермент инактиви-
руется практически полностью при прохождении реакции
превращения субстрата на 20%. Начальная концентрация фермента
равна 2-10~5М, начальная концентрация субстрата равна 0,8 М,
йкат=0,2 сек-1
т (каж) h
Е + S ;=TES—!^> Е + Р (8.23)
Неактивный фермент
8-17. В реакции гидролиза нитрокатехолсульфата,
катализируемого арилсульфатазой А, фермент инактивируется под действием
субстрата согласно схеме
*s *„
Е + S 7=^ ES Е + Р (8.24)
Неактивный фермент
176
где сс<С 1 [Й] ■ Исходя из данных табл. 9, найти значения k2 и ее,
если эксперимент проводился при условиях [S] ^Ks-
Таблица 9
Кинетика накопления пирокатехола в реакции
гидролиза нитрокатехолсульфата,
катализируемого арилсульфатазой А.
Условия опыта: рН 5,3; 37° С; 0,03М формиата
натрия; [Е]0 = 1,25-Ю-8 мг/мл; [S]0 = 5-10"3М
Время, мин
0,5
1,0
2,0
2,5
5,0
10,0
20,0
[Р]-10=, М
0,67
1,14
1,79
2,08
2,77
3,12
3,70
Ответы и решения
8-1. Преобразуя интегральную форму уравнения Михаэлиса
[Р] = ^т<- /Ст(ка») 1П
[So]
[S]„-[P]
(8.25)
и обозначая тангенс угла наклона прямой через а, отрезок,
отсекаемый прямой на оси ординат, через Ъ, и отрезок, отсекаемый на
оси абсцисс, через с, получаем
a) IE1 = ут - КгпЫж) ,п_ [S]o _
[S].-[P]'
а = ~К
т (каж)
*\ * £_ | Km (каж)
; [Р] ~ vm ^ vm
- Ь = Vn, C = VjKm (
каж) !
[S].
[P] [S]„-[P]'
a - /Ч('"Ж) , 6=1/^я, с = -1/Л-я(к.ж);
в)
In
[S].
[S]„[P]
a = \/Vm, b =
Km (к;
^m (кап
Vm
+
[P]
[S].
^'"[SL-iP]
С = A m (каж)-
8-2.)Описанный в задаче эксперимент — один из вариантов
интегрального анализа кинетики ферментативных реакций. Однако
в отличие от классического метода, в котором при данной началь-
177
ной концентрации 'субстрата определяется текущая концентрация
продукта, в данном случае варьируется начальная концентрация
субстрата при постоянной величине t в уравнении (8.25). Таким
образом, для линеаризации экспериментальных данных можно
применять все виды интегрального уравнения Михаэлиса—Ментен
см. например (рис. 83).
Ответ: /г1(ат=0,038 сек'1; Кт(каж)=3,1 • Ю~2 М.
20
15
10
5
\ w
- \
\
i
о\
I
чо
\
2 4 6 8
i,nr%_.fflfceK'
f [5J0-[P]
Рис. 83. Определение
кинетических параметров
гидролиза амида М-ацетил-Ь-гек-
сагидрофенилаланина,
катализируемого сс-химотрипси-
ном, с помощью
интегральной формы уравнения
Михаэлиса
.П
8,0
6,0
40
2.0
. _ т(каж1\ \\
1 1 \i \\! 1
N
2,0
4,0
6.0
In
Д»-Д,
Рис. 84. Определение кинетических
параметров гидролиза метилового
эфира &-ацетил-Ь-фенилаланил-Ь-ли-
зина, катализируемого трипсином, в
присутствии конкурентного
ингибитора, 1-метил-2-аминоиндола. а [I] =0,
Л = 150 сек; б [I] = 1,46- Ю~3 М,
А =180 сек; в [I] = 1,84-10-3 М.
А = 195 сек; г [I] = 3,25-Ю-3 М.
Д = 210 сек
8-^/Для обработки данных табл. 2 удобно использовать коор-
динаты типа ( Di — Dh In , где применительно к этой
£>
D
таблице (Di — Dt) — разница концентраций субстрата, выраженная
в см диаграммного листа через равные интервалы времени (А),
(Do» — Dt) и (Z)oo — D'i) — концентрации непрореагировавшего суб-
178
страта в моменты времени tt и t\ (см. рис. 82). Так как согласно
уравнению (8.7)
D't~Dt =
Vm A Km (каж)
In
D.
£)/
A — D
(8.26)
где а=3,74 10~5 М, то линеаризация в координатах уравнения
(8.26) позволяет определить значения Vm и /Ст(КаЖ) при различных
концентрациях ингибитора (рис. 84), и далее найти значение /Ст.
Ответ: Лт(каж)=7,5-Ю-5 М; £кат=170 сиг1; /Ci=l,8-10"3 M.
8-4. Решается аналогично задаче 8-3.
Ответ: /u=2-10-2 M.
8-5'. Решается аналогично задаче 8-3 (рис. 85)
Ответ: Кт=1,3-10-8М-сек-1; KmiKam)=2,9■ 10~4 М.
8-6. Решается аналогично задаче 8-3 (рис. 86)
Ответ: Ут^З,5-10-б М-сек"1; Кщкаш)=^7,9■ К)-3 М.
8-7'. Для обработки
полной кинетической кривой
(табл. 6) удобно использовать
интегральное уравнение
скорости ферментативной
реакции (8.26). Если принять, что
изменение проводимости в
ходе реакции пропорционально
концентрации продукта, то
значение переходного
коэффициента а в уравнении (8.26)
равно 6,67М-ом-см.
Ответ: Ут=4,9-10-5 MX
Хсек-1; Кгы**ж)= 1,8-10-3 М.
8-8. Если продукт
ферментативной реакции является
конкурентным ингибитором
фермента, то интегральная форма уравнения скорости определяется
выражением
к- , /и. [S]^
А,и (каж) | * +
Рис. 85. Определение кинетических
параметров дезаминирования адено-
зина, катализируемого аденозиндез-
аминазой, с помощью интегрального
уравнения (8.26) скорости (Д =
= 225 сек)
t
1
Vm
Km (каж)
ЛЧ
t \
('
к
m (каж]
In
[S].
[S1.-PT
(8.27)
КР '1' КР
Из выражения (8.27) видно, что эффективные отрицательные
значения максимальной скорости и константы Михаэлиса
ферментативной реакции ^соответствуют случаю К?<Кт{кыК), когда продукт
реакции имеет большее сродство к ферменту по сравнению с
исходным субстратом.
8-9. Определение кинетических параметров ферментативной
реакции с помощью интегральной формы уравнения Михаэлиса
1 В работах [5, 6] количественная обработка кинетической кривой не
проводилась.
179
(рис. 87) приводит к значениям Vm=—9,3-10~6 М-сек-1; Кпцкаж)—
=—9,7-10-3 М. Как указывалось в решении предыдущей задачи,
эффективные отрицательные значения Vm и К-щкат)
свидетельствуют о наличии значительного конкурентного ингибирования
продуктом реакции, причем ftp < Лакано- Из выражения (8.27)
следует, что
,эфФ _ Vm (g28)
1/ЭфФ
v т —
1
Km (каж)
iv-эфф
Л т(каж)
К
т (каж)
к*
i +
[SJ.
Am (каж)
Кг,
(8.29)
i" d - d;
Рис. 86. Обработка с помощью
уравнения (8.26) полной
кинетической кривой гидролиза
гс-нитрофенилфосфата под
действием щелочной фосфатазы
(Д = 1 час)
Рис. 87. Необычный ход прямой,
приводящий к отрицательным
значениям кинетических параметров
гидролиза бромацетнл-ОЬ-феннл-
лактата, катализируемого карбо-
кснпептидазой, полученный при
обработке полной кинетической
кривой с помощью интегральной
формы уравнения скорости
Подставляя в выражения (8.28), (8.29) значения Vm и Кт(кат),
определенные из начальных скоростей ферментативной реакции,
можно найти величину константы ингибирования фермента
продуктом реакции.
Ответ: /Ср=3,6-10-4М.
180
8-10 [9]. Интегрируя повторно интегральную форму уравнения
скорости ферментативной реакции (8.2) в пределах (0, [S]0),
имеем
IS1„ IS,„ [SI„
5 Vmtd[S]= \ ([S]0-[S])d[S]+ \ /Cffl(K.«)lnigj-d[S]. (8.30)
0 0 0
Интегрируя правую часть выражения (8.30), получаем
о
(Slo
Так как интеграл в левой части полученного выражения \ td [S]
о
есть площадь под кинетической кривой в координатах ([S], t), то,
обозначая его через А, приходим к выражению
ZA ^Кщ (каж) i l^Jo /fi oil
Очевидно, что в координатах (2A/[SJ0, [S]o) зависимость (8.31)
должна иметь вид прямой линии с тангенсом угла наклона \/Vm
и пересекающей ось ординат в точке 2Кт(кат)/Ут.
8-11 [10]. Уравнение касательной к кинетической кривой в
начальный момент времени можно представить в виде
v°= IC-r^trsT- (8'32)
Am (кал) т LJjo
Преобразуем выражение (8.32) следующим образом:
^о Ут
. [S]. „ . . Л , [S].
и далее
Л , [S], \
Am (каж) I '"г is , . j
\ Аи (каж) /
(8.33)
Ащ (каж) [^Jo
Vm ~ ^Л , [S]. \ ■
\ Кщ (каж) '
Запишем основное интегральное уравнение в виде
/ Am (каж) ( ["J i i_ _[£Jo^_\ /о од\
Комбинируя выражения (8.33) и (8.34), получим
t = [S]o / [Р] , ,_ [S]. \
ш (каж) 1 [S].- [P]J-
4 Am (каж) /
или
. V0t
[S]» 1 +
Am (каж
[S]. (/Cm (каж) +ln[S]c-[P])- (8'35>
181
Таким образом, при данных [S]0 и v0 каждому значению / может
соответствовать определенное значение правой части соотношения
(8.35), из которого легко определяется величина Кт(кв.ж)-
8-12. Тангенс угла наклона прямой, проведенной из начала
координат ([P]/t, 1// In[s]0 —'[Р])' равен
^°тЛг' (8-36)
* ln[S]o—[Р]
В начальный момент времени (при t —»0) имеем [Р] —> 0 и
In [SJ° --,0
[S].-[P]
Таким образом, для предела выражения (8.36) при £ —> U мы имеем
случай неопределенности типа 0/0. Пользуясь правилом Лопиталя,
числитель и знаменатель выражения (8.36) запишем в виде
}?Л-*&—Ф- <8-37>
||п !iS@,_ ' Ш.. (8.38)
Разделив уравнение (8.37) на (8.38), окончательно получаем при
t — 0
tg«P = [S]0.
8-13. Преобразуя выражение (8.29) к виду
^эфф _ ^Р | [S]o
Ат(каЖ)= ■ jr Т" К
Km (каж) Кт (каж)
и линеаризуя экспериментальные данные в координатах (/С?$аж),
[S]0), получаем прямолинейную зависимость, из тангенса угла наклона
которой и отрезков, отсекаемых на координатных осях, можно
определить значения /Ср и Кпцкату Далее, используя полученные
данные, из выражения (8.28) находим значение Vm.
Альтернативным способом обработки экспериментальных
данных является 'воссоздание вида прямых в координатах
([Р]/<, Vt ln[S1 _rpA пересекающих оси ординат и абсцисс в
точках 1/*фф и /СФ(Фкаж)/1^ФФ соответственно (рис. 88). Применив
затем метод Фостера — Ниманна (см. задачу 8.12), находим значения
Vт и Km (каж). Наконец, из соотношения (8.28) находим величину /Ср .
Ответ: ЛтскаЖ)=3,Ы0-6 М; им,—1,5 сек"1; Ар=2,8-10"5 М.
182
f ln[s]-TPJ
2 -1
Ю, сек
Рис. 88. Применение метода Фостера — Ниманна для определения
константы конкурентного ингибнрования пенициллинамидазы продуктами
ферментативного гидролиза бензилпенициллина
8-14. Если в ходе ферментативной реакции фермент
денатурирует по первому порядку (с константой скорости денатурации k),
то дифференциальное уравнение скорости ферментативной
реакции можно записать в виде
d[P)
[Е]о [S]
dt
или
d[p)
Km (каж) + IS]
Vm([S)0-[P])
-kt
[P]
dt Km (каж) т [SJo
Интегрируя уравнение (8.39), получим
[P] __ Vm (1 - e-kt) _ Km (каж) ln
o—kt
[SJ.
Г8.39)
(8.40)
t kt t "'[SJ,— [P]'
Очевидно, что линеаризация полученного выражения в координатах
(-у1-, —г- In.g. __mi) возможна лишь при условии / = const (см.
задачу 8-2).
8-15. Комбинируя выражения (8.40), (8.28) и (8.29), получим
интегральную форму уравнения скорости ферментативной реакции,
в которой наблюдается конкурентное ингибирование продуктом
реакции с константой скорости инактивации k
[PJ =
У»Ф*(1—e-«)
Кт(каж) 1П
[S].
[SJ»—[P]-
(8.41)
183
Значения АГтф$аж) и 1/„фф в уравнении (8.41) определяются
соотношениями (8.28) и (8.29) и могут быть легко рассчитаны из цифровых
данных задачи (Кв£&аЖ) = 0,124М; 1/^фф = 5,С6 • 10-s M • сек-1).
Из уравнения (8.41) можно рассчитать время, необходимое
для завершения каждого последующего реакционного цикла (до
99%-ной степени превращения субстрата, т. е. до образования 0,99М
продукта) при данной исходной концентрации активного
фермента. Так, время необходимое для завершения первого реакционного
цикла (при начальной концентрации активного фермента, равной
3-10_6М), равно 10,3 часа; при этом концентрация активного
фермента после завершения цикла составит 69% от первоначальной.
Для завершения второго цикла потребуется 16,3 часа, после
которых концентрация активного фермента составит 38%
первоначальной. Третий реакционный цикл пройдет за 45,5 часов, и
концентрация активного фермента к окончанию цикла понизится до 6,7%
от исходной. Практически полная инактивация фермента должна
произойти во время четвертого цикла реакции. Таким образом, за
72 часа непрерывной работы реактора можно осуществить три
полных реакционных цикла и получить при этом 300 л раствора 6-АПК
концентрации 0,99М. При количественном выделении продукта
выход 6-АПК составит 297 молей или 64,5 кг.
8-16. Если [Е*]0 — общая текущая концентрация активного
фермента, то из схемы (8.23) следует, что
-T-*™IES]. (8.42)
Так как скорость превращения субстрата определяется выражением
--^T- = A2[ES], (8-43)
то, разделив уравнение (8.42) на (8.43), получим
d [Е*]„ кт
d[S] k
или
d[E*]0«^5d[S]. (8.44)
Интегрируя выражение (8.44) с начальными условиями [S] - [S]0 при
[Е*]0 = [Ё]0, получим
[E*]-[E*l0=|f([S]0-[S]).
При полной инактивации фермента ([Е*]0 = 0)
_ fe. [EJ,
"н [S]e-[S]-
Ответ: kan = 2,5- Ю-5 сек-1.
1R1
8-17 [11]. Из схемы (8.24) следует, что скорость инактивации
фермент-субстратного комплекса равна
--^- = ^2[ЕБ]. (8.45)
Интегрируя выражение (8.45) с начальными условиями [ES] = [Е]0
при t = 0 (так как [S]>/Cs), получим
[ES] = [Е]0 *-«*•'. (8.46)
Рис. 89. Определение кинетических параметров
ферментативной реакции при наличии быстрой инактивации
фермент-субстратного комплекса (на примере гидролиза нитрокатехол-
сульфата, катализируемого арилсульфатазой А)
Исходя из схемы (8.24) и выражения (8.46), скорость образования
продукта реакции равна
*ip-~k2[E]0e-*'<. (8.47)
Интегрирование выражения (8.47) приводит к зависимости
концентрации продукта реакции от времени
[Е]„
[Р]
Ио c—ah,t_
(8.48)
Преобразуем выражение (8.48) к виду
_1 а_
[Р]
и далее
[Е]„(1
-ай^П
1
[Р]
а а
-ak?t
1
<>-«*> '
185
Так как а< 1, то, разлагая экспоненциальные члены в ряд Макло-
рена, получим
[р] = [1J7 + k2[E]0t ■ (8-49>
Очевидно, что зависимость (8.49) в координатах (1/[Р], 1//)
должна быть прямолинейной, с тангенсом угла наклона, равным
1/&2[Е]о, и отрезком, отсекаемым на оси ординат, равным а/[Е]0
(рис. 89).
Ответ: k2=l,27-lQr2 М-мл/мг-мин; а=30 мг/М-мл; kmi=
= 0,381 мин-1.
ЛИТЕРАТУРА
1. Walker А. С, Schmidt С. L. A. „Arch. Biochem.", 5, 445, 1944.
(25 К лесов А. А., Бе резин И. В. „Биохимия", 37, 170, 1972.
ЗГ Foster R. J, Niemann С. „Proc. Nat. Acad. Sci.", 39, 999, 1953.
<2B Jennings R. R., Niemann C. „J. Amer. Chem. Soc", 75, 4687, 1953.
5.'La wrence A. J., Moores O. R. „Eur. J. Biochem.", 24. 538, 1972.
ФН in berg I., Laidler K. J., ,Can. J. Biochem.", 50, 1360, 1972.
Snoke J. E., Neurath H. „J. Biol. Chem.", 181, 789, 1949.
Stinshoff K. „Biochim. Biophys. Acta", 276, 475, 1972.
9. Banfield J. E. „J. Chem. Soc", 383. 1964.
10. F lei she r O. A. „J. Amer. Chem. Soc". 75. 4487, 1953.
11. Roy A. B. „Biochim. Biophvs. Acta", 276, 488. 1972.
Глава 9
НЕСТАЦИОНАРНАЯ КИНЕТИКА
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
Превращение субстрата под действием фермента проходит
обычно через ряд короткоживущих промежуточных соединений,
изучение реакционной способности которых помогает выяснить
механизм ферментативного катализа. К настоящему времени
разработаны такие методы, которые позволяют определять значения
констант скоростей промежуточных стадий ферментативных
реакций при изучении кинетики их протекания в стационарном
режиме (см. гл. 7). Однако наиболее прямую и надежную
информацию о кинетике промежуточных стадий можно получить, изучая
ферментативные реакции в нестационарном режиме их протекания
(с применением методов изучения быстрых реакций в растворах).
Рассмотрим общую схему ферментативной реакции,
включающей образование п промежуточных соединений:
ft, *2 "i *j + ,
Е -f- S «_. Xj ., ...ч X,- ...
ft-(/+i)
...— Xn7=!E + P. (9.1)
186
Для каждого из п промежуточных соединений, а также для
фермента, субстрата и продукта можно на основе законов
химической кинетики записать дифференциальное уравнение,
описывающее изменение концентрации вещества во времени. С учетом
материального баланса по ферменту и субстрату, полное изменение
концентраций всех веществ во времени будет представлено
системой (n-j-1) дифференциальных уравнений с постоянными
коэффициентами и двух алгебраических уравнений:
^1 = К [Е] [S] + £_2 [Х2] - (А_, + k2) [X,],
Щ- = kt [X,-,] + k- (/+1) [Xl+I] - (*_, + kl+1) [X,],
(9.2)
^i = ^[X„-1] + *-(„+i)[E][P]-(^„ + ^+1)[X„],
i-^=*»+i[XJ-*-(e+i)[E][Pj,
[Е]0 = [Е] + [Х1]-1 .+[Х„], (9.3)
[S]0 = [S] + [X1]+ .+ [X„] + [P]. (9.4)
Так как система уравнений (9.2) содержит члены второго
порядка &i[E]- [S] и &_(m+i)[E] • [Р], она нелинейна и получить
аналитическое решение данной системы при произвольных
соотношениях констант скоростей реакций и концентраций реагентов не
представляется возможным. Решения подобных систем уравнений могут
быть найдены или путем численного интегрирования на цифровых
вычислительных машинах [1] или моделированием на аналоговых
вычислительных машинах [2]. Однако в некоторых частных
случаях систему уравнений (9.2) можно превратить в линейную
систему, которая может иметь аналитическое решение. В настоящее
время при анализе кинетики ферментативных реакций,
протекающих в нестационарном режиме, наибольшее развитие получили
два подхода, основанные па предпосылках, упрощающих
кинетическое рассмотрение:
а) предстационарная кинетика, или кинетика начальных
стадий ферментативной реакции;
б) релаксационная кинетика, или кинетика смещения
положения равновесия ферментативной реакции.
§ 1. Методы предстационарной кинетики
Одним' из вариантов упрощения системы дифференциальных
уравнений (9.2) является использование большого избытка одного
из реагентов (например, создание условий [S]0^>[E]0 или [Е]0>
187
^>[S]o) и изучение кинетики реакции в начальный (предстацио-
нарный) период времени на небольшой глубине реакции. В этом
случае накоплением концентрации продукта во времени можно
пренебречь ([Р] ?»0), и члены второго порядка становятся
зависимыми с достаточной степенью точности лишь от переменной
концентрации одного реагента. Полученная таким образом новая
система дифференциальных уравнений является линейной и имеет
аналитическое решение. Так как для обработки
экспериментальных данных имеется большое число вариантов применения методов
предстационарной кинетики, анализ конкретных типовых
ферментативных систем будет рассматриваться в решениях задач
настоящей главы.
Кинетика ферментативных реакций в предстационарной
режиме их протекания изучается в основном методами «остановленной
струи». Принципиальная особенность этих методов состоит в
быстром смешивании растворов реагирующих веществ в смесительной
камере специальной конструкции [3, 4], после чего смесь поступает
в измерительную кювету. Чаще всего в установках типа
«остановленной струи» используется спектрофотометрический метод
измерения с осциллографической регистрацией сигнала от
фотоумножителя. «Мертвое» время лучших современных установок подобного
типа составляет 0,1—0,3 мсек.
В последнее время работами Хесса с сотрудниками [5—7] на
примере а-химотрипсина был развит новый метод изучения
кинетики начальных стадий ферментативных реакций, получивший
название метода «вытеснения профлавина». Метод основан на том
факте, что краситель профлавин (3,6-диаминоакридин) при
связывании с а-химотрипсииом в водном растворе изменяет свой спектр
поглощения в ультрафиолетовой области. Величина разностного
спектра поглощения, имеющего максимальное значение при длине
волны 465 нм, пропорциональна концентрации комплекса фермент-
профлавин. Введение в систему фермент-профлавин субстрата,
конкурирующего с красителем за связывание на активном центре
а-химотрипсина, приводит к двум последовательным процессам
вытеснения профлавина. Первый, очень быстрый 'процесс,
заключается в обратимом вытеснении красителя из комплекса его с
ферментом за счет образования нековалентного фермент-субстратного
комплекса. Второй процесс, времена прохождения которого лежат
обычно в пределах разрешения установок типа «остановленной
струи», вызван химическим взаимодействием субстрата с
ферментом (например, образованием ацилферментного промежуточного
соединения), что приводит к дополнительному уменьшению
концентрации комплекса фермент-профлавин. Изучение кинетики
второго процесса при различных концентрациях субстрата в
дополнение к изучению кинетики ферментативной реакции в стационарном
режиме позволяет сделать заключения о стадийности изучаемой
реакции, а также найти значения констант скоростей
промежуточных стадий ферментативной реакции.
188
§ 2. Релаксационные методы
Если при изучении ферментативной системы, находящейся в
состоянии равновесия, резко изменить внешние условия
(температуру, давление), то равновесие в системе на некоторое время
нарушается. В этом случае члены второго порядка системы уравнений
(9.2)_fti[E]-[S] и £_(ИН)£Е] • [Р] принимают вид fti(_[E]+A[E])X
X([S]+AS) и *_(„+„ ([Е]+Д[Е])([Р]+Д[Р]), где [Е], [S] и [Р] -
равновесные концентрации фермента, субстрата и продукта при
исходном равновесии; Д[Е], A[S] и Д[Р] —переменные (зависящие
от времени) концентрации реагентов при переходе системы к
новому равновесию. Если сдвиг равновесия достаточно велик, то
уравнения, описывающие поведение ферментативной системы,
являются сложными, включая произведения переменных
концентраций фермента и субстрата и опять приводя к нелинейной системе
уравнений типа (9.2). Но при небольшом смещении равновесия эти
произведения становятся пренебрежимо малыми, и члены второго
порядка системы уравнений (9.2) приобретают вид
MEHS] + M"S]A[E] + ME]A[S]
и
£_с„+1) [Ё] • [Р] + *_(я+1) [Pj Д [EI + л_(я+1) [Ё] Д [Р].
Таким образом, бимолекулярные члены системы уравнений (9.2)
становятся псевдомономолекулярными относительно переменных
концентраций реагентов, и система становится линейной. Тогда
кинетическое уравнение реакции можно записать в виде
где Д [X] — отклонение концентрации реагента или продукта (по
изменению концентрации которого производится регистрация
кинетики) от равновесной; т — константа, характеризующая скорость
достижения нового положения равновесия и называемая временем
релаксации. Из выражения (9.5) очевидно, что время
.релаксации— величина, обратная константе скорости псевдопервого
порядка. Ее .связь с кинетическими и равновесными параметрами
изучаемой системы, а также с равновесными концентрациями
реагентов определяется характером конкретной системы.
Следует отметить, что изменение внешних условий должно
происходить так быстро, что скорость, с которой достигается новое
равновесие в системе, должна значительно превышать скорости
прямой и обратной реакций. На практике смещение равновесия
производят обычно с помощью так называемых методов
«температурного скачка» или «скачка давления». Например, пропустив
через раствор импульс тока высокого напряжения, можно повысить
температуру раствора на 10° менее чем за 10~е сек. Резкое
изменение давления в системе можно производить или скачкообразно,
189
или в периодической форме при использовании ультразвуковых
волн. Важное преимущество релаксационных методов по
сравнению со струевыми состоит в том, что для них не требуется
быстрого смешивания реагентов перед началом регистрации кинетики, а
это в свою очередь позволяет намного уменьшить «мертвое время»
установки. Применение релаксационных методов для изучения
ферментативных реакций позволило достичь разрешения во
времени порядка 0,01 мсек, что дает возможность еще более детально
описать механизмы действия ферментов [8].
Задачи
9-1. На рисунке 90 приведен пример типичной кинетической
кривой, полученной при изучении двухстадийкого ферментативного
процесса в начальный период времени
*' ft,
Е + S ^Zl ES —+ Е + Р.
(9.6)
Используя данные табл. 1, найти константы скоростей ku k_{ и k2.
[Pjl 1 D
Время
Рис. 90. Зависимость концентрации
продукта реакции от времени в
начальный (предстационарный) период
протекания двухстадийной
ферментативной реакции
Время
Рис. 91. Кинетическая кривая
накопления продукта Pi (схема
9.8) в предстацион арном (о) и
стационарном (б) периодах
ферментативной реакции. На оси
ординат — оптическая плотность
раствора
9-2. Кинетику реакции фицина с n-нитрофенилгиппуратом
изучали методом остановленной струи в условиях [EJo^fSJo- Было
найдено, что образование n-нитрофенола в начальный период
реакции подчиняется кинетике первого порядка, причем эффективная
константа скорости реакции зависит от концентрации фермента
(табл. 2) и не зависит от концентрации субстрата [9]. Значения
&кат и /Сгтцкаж), найденные при изучении этой реакции в
стационарном режиме, равны 5,0 сек-1 и 1,28-10~4 М соответственно. Прини-
190
Таблица 1
Зависимость кинетики двухстадийной ферментативной реакции
от начальной концентрации субстрата. [Е]0 во всех случаях
равно 1,0 10-6М
[Sol-Ю5, М
1,0
4,0
5,0
6,0
8,0
10,0
т*, сек '
0,91
0,72
0,67
0,63
0,56
0,50
г,.Ю?, ** М-сек~'
0,41
1,30
1,53
1,72
2,08
2,44
* Отрезок, отсекаемый на оси абсцисс продолжением
стационарного участка кинетической крчвой (см. рис. 90).
** Величина стационарной скорости ферментативной
реакции.
Таблица 2
Значения эффективных констант скоростей
первого порядка образования п-нитрофенола
в реакции п-нитрофенилгиппурата (5-10_6М)
с фицином. Условия опыта: рН 3,9; 25° С
*эфф,
[ГЬ-10*. М
0,98
1,22
1,61
2,38
3,45
0,29
0,36
0,54
0.S9
1,50
мая, что механизм изучаемой реакции согласуется с трехстадиинои
схемой, определить значения констант k2, k3 и Ks'.
Е+ S;
ES
ft.
EA
k.
E + P,
(9.7)
9-3. В таблице З приведены результаты кинетического
исследования ацшшрочания папаина лг-нитрофениловым эфиром карбо-
бензоксиглицина [10] в условиях [E]o^[S]o. При изучении этой
реакции в стационарном режиме было найдено, что величина
кажущейся константы Михаэлиса равна (1,89±0,17) ■ Ю-5 М, а
величина &катАг\т(кан<) равна (1,15+0,08) -105 М-1 сек-1. Принимая трех-
стадийную схему протекания ферментативной реакции (9.7), найти,
191
скорость которой из двух кинетических стадий — ацилирования или
деацшшрования — является лимитирующей.
Таблица 3
Кинетика реакции ацилирования папаина
(1,1Ы0-4М) лг-нитрофениловым эфиром
карбобензоксиглицина (1,0-10-6М). Условия
опыта: рН 6,8; 0,021 М фосфатный буфер; 25° С
Время, мсек
0
10
20
30
40
60
80
100
120
СО
Оптическая плотность
раствора в относит, ед.
0,00
0,18
0,29
0,37
0,42
0,55
0,66
0,73
0,78
1.00
9-4. а-Химотрипсин и N- (2-фурил) акрилоил-Ь-триптофан при
низких значениях рН находятся в равновесии с ацилферментным
соединением, N- (2-фурил) акрилоил-Ь-триптофанил-химотрипсином
При резком увеличении значения рН раствора (от 2,4 до 7,9) с
помощью метода остановленной струи (так называемый «рН-ска-
чок») наблюдается быстрое уменьшение оптической плотности
раствора при длине волны 340 нм, соответствующее необратимому
деацилированию ацилфермента (табл. 4). Определить значения
констант скоростей ацилирования (k2) и деацилирования (k3) в
Таблица 4
Кинетика деацилирования
1Ч-(2-фурил)акрилоил-1^-триптофанилхимотрипсина.
Условия опыта: рН 7,9 (трис-буфер 0,05М);
ионная сила 0,15; .25° С
Время, мсек
10
20
30
40
50
70
90
со
Оптическое пропускание
раствора, %
47,5
48,9
50,0
50,8
51,8
52,9
53,2
54,0
192
реакции гидролиза метилового эфира М-(2-фурил)акр.илоил-Ь-
триптофана, катализируемого а-химотрипсином (см. схему 9.7)
при рН 7,9, если при этом значении рН величина каталитической
константы скорости &кат, определенная в стационарных условиях,
равна 25 сек-1 [11].
9-5. Реакция гидролиза n-нитрофенилтриметилацетата,
катализируемого эластазой, протекает по следующей общей схеме:
E + S^ES~E + P2. (9.8)
Pi
На рисунке 91 приведен типичный пример кинетической кривой
выделения «-нитрофенола (продукт А) в начальный период
реакции. В табл. 5 приведена временная зависимость изменения разно-
Таблица 5
Кинетика ацилирования эластазы (1,86-10_6М)
п-нитрофенилтриметилацетатом (1,03- 10_4М)
при рН 7,9 (см. рис. 91)
Время, мсек
10
30
50
70
90
110
до
1,86-Ю-2
1,20-Ю-2
7,94-10~3
5,25-10-3
3,47-Ю-3
2,29-Ю-3
сти оптической плотности (AD) между наблюдаемой кривой и
экстраполированным участком прямой, отвечающей стационарному
режиму реакции в условиях [S]o^[E]o. Определить значение
бимолекулярной константы скорости ацилирования фермента (/гц),
если из кинетических экспериментов, проведенных в стационарном
режиме, найдено значение k3=2,6-10-3 сек-1 [12].
9-6. Гидролиз о-нитрофенил-а-мальтозида, катализируемый
а-амилазой, изучали с помощью метода остановленной струи.
В табл. 6 приведена зависимость концентрации образующегося в
реакции о-нитрофенола-от времени в начальный период реакции.
Найти, двух- или трехстадийной схеме ферментативной реакции
соответствуют данные табл. 6, если значения /Ст(каж) и &КаТ,
определенные в стационарном режиме протекания реакции, равны
8- Ю-4 М и 6,1 сек-1 соответственно [13].
7 Заказ 158
193
Таблица 6
Кинетика выделения о-нитрофенола в реакции
гидролиза о-нитрофенил-а-мальтозида,
катализируемого а-амилазой, в начальный период
времени. Условия опыта: рН 5,3; 17° С;
[S]0 = 1,8510-3М; [EJ0 = 1,710"4M
Время, мсек
10
20
30
40
50
60
70
80
90
IP.I-IO", М
0,725
1,450
2,180
2,900
3,630
4,350
5,070
5,800
6.520
9-7. При изучении начального периода гидролиза п-нитрофе-
нилового эфира триметилуксусной кислоты, катализируемого эла-
стазой (см. рис. 91), было найдено, что разность оптической
плотности (AD) между наблюдаемой кинетической кривой и
экстраполированным участком прямой, отвечающим стационарному
периоду реакции, уменьшается со временем согласно кинетике первого
порядка [12]. Было показано, что значение эффективной
константы скорости реакции первого порядка зависит от начальной
концентрации субстрата (табл. 7). Принимая, что ферментативная
реакция протекает по схеме (9.8), определить значения констант
kn и къ.
Таблица 7
Зависимость эффективной константы скорости
первого порядка образования п-нитрофенола
в реакции гидролиза
п-нитрофенилтриметилацетата, катализируемого
эластазой, от концентрации субстрата.
Условия опыта: рН 7,9; 25° С; [Е]0 = 1,86-10-6М
[SIo-105, M
3,00
5,81
8,57
10,28
*эфф-10"> eeK_I
0,98
1,40
1,87
2,12
9-8. Один из методов расчета концентрации активных центров
фермента основан на регистрации стехиометрического «выброса»
первого продукта реакции (схема 9.7), если образующееся при
194
этом производное фермента является практически неактивным и не
регенерирует фермент за время эксперимента (так называемое
«титрование активных центров», рис. 92). В реакции гидролиза
n-нитрофенилгиппурата, катализируемого фицином, образующийся
гиппурил-фицин не является
таким устойчивым соединением,
вступая в реакцию с водой [9].
Определить, какую долю
концентрации фермента составит
«выброс» «-нитрофенола в
реакции гидролиза п-нитрофенилгкп-
пурата (10_4М) при следующих
условиях эксперимента:
а) рН 5,9; начальная
концентрация фицина 1,55-10~5 М;
А2=44 сек-1; £3=8,5 сек-';
/Cs=2,8-10-4 M.
б) рН 3,9; начальная КОН- Рис. 92. Определение «выброса» я
центрация фицина 3,06-10-5 М; продукта в начальный период фер-
,г9 = 7,8 сек-1; /г3=13,9 сек"1: ментатавной реакции
9-9. В таблице 8 приведены численные значения «выброса»
(я, см. рис. 92) n-нитрофенола в начальный период реакции эла-
стазы с п-нитрофенилтриметилацетатом для ряда концентраций
субстрата при постоянной начальной концентрации фермента [12].
Таблица 8
Зависимость «выброса» «-нитрофенола
в начальный период ферментативной реакции
(см. рис. 92) от начальной концентрации
субстрата. Условия оыта: рН 7,9; 25е С
[S]„-10s, М
3,00
5,81
8,57
10,28
it-10», М
0,797
1,215
1,344
1,480
Принимая, что механизм реакции согласуется со схемой (9.8),
рассчитать начальную концентрацию активных центров фермента в
растворе.
9-10. Кинетику гидролиза этилового эфира М-ацетил-Ь-трипто-
фана, катализируемого а-химотрипсином, изучали в начальный
период реакции с помощью метода «вытеснения профлавина» [5].
Было найдено, что уменьшение оптической плотности раствора во
времени, соответствующее уменьшению концентрации комплекса
фермент-профлавин, описывается кинетикой первого порядка с эф-
7*
195
фективной константой скорости, зависящей от начальной
концентрации субстрата (табл. 9). При изучении ферментативного
гидролиза субстрата в стационарном режиме протекания реакции было
найдено, что йкат равна 2 сек^1. Из независимых экспериментов
было также найдено, что значение константы диссоциации комплекса
Таблица 9
Значения эффективных констант скоростей
первого порядка вытеснения профлавина
из комплекса фермент-краситель
при ацилировании фермента субстратом.
Условия опыта: рН 5,4; 28° С; начальная
концентрация фермента Ы0-5М; начальная
концентрация профлавина [F]c = 5-10_бМ
[S]0-1(F, М
0,50
0,61
0,75
0,90
1,45
1,77
2,25
2,70
*эфф > сек 1
13,0
14,5
17,0
19,9
28,0
32,0
38,0
42,0
фермент-профлавин при тех же условиях опыта равно 3,3-Ю-5 М.
Принимая, что ферментативная реакция протекает по трехстадий-
ной схеме, найти значения индивидуальных констант k2, kz и Ks,-
9-11. Кинетику реакции а-химотрнпсина с п-нитрофениловым
эфиром М-ацетил-Ь-триптофана изучали методом. остановленной
Таблица 10
Зависимость эффективной константы скорости
первого порядка выделения п-нитрофенола
в начальный период реакции от начальной
концентрации фермента [6|. Условия опыта:
рН 3,5; 26,5° С; 1,6% ацетонитрила; ионная сила
0,05М; начальная концентрация субстрата
2-10-6М
[Е]„.1№, М
1,38
2,13
3,00
4,40
5,15
5,95
8,50
11,60
16,60
*эфф' сек '
1,8
2,6
3,6
4,7
5,3
6,0
7,8
10,0
11,1
196
струи. Было найдено, что образование п-нитрофенола в начальный
период реакции в условиях [E]0>[.S]o подчиняется кинетике
первого порядка, причем эффективная константа скорости реакции
зависит от концентрации фермента (табл. 10). Принимая, что
изучаемая реакция проходит по трехстадийной схеме, найти константы
k2 и К&.
9-12. Гидролиз этилового эфира N-ацетил-Ь-тирозина,
катализируемый а-химотрипсином, изучали методом остановленной струи
в условиях [S]0^>[E]o по вытеснению профлавина из комплекса
его с ферментом [7] (константа диссоциации комплекса фермент-
профлавин в условиях эксперимента равна 4,4-Ю-5 М).
Уменьшение оптической плотности раствора (Я=465 нм) во времени
подчинялось кинетике первого порядка (табл. 11). Из кинетических
Таблица 11
Значения констант скоростей вытеснения
профлавина из комплекса его с а-химотрипсином
под действием субстрата. Условия опыта: рН 5,0;
25° С; [EJc = 10-5М; начальная концентрация
профлавина [F]0=5-10_5M; ионная сила 0,4М
[SJo-lC, M
4,5
6,9
10,2
14,7
18,7
21,6
*эфф- сек Х
12
16
21
28
31
32
экспериментов, проведенных в стационарном режиме, было
найдено, что значение каталитической константы ферментативной
реакции равно 3,0 сек-1. Найти величины индивидуальных констант
реакции k2, kz и Ks, если известно, что для изучаемой реакции
выполняется условие k2~>kz (см. схему 9.7).
9-13. Гидролиз метилового эфира М-ацетпл-Ь-фенилаланина,
катализируемый а-химотрипсином, изучали методом
остановленной струи, используя методику «вытеснения профлавина» из
комплекса его с ферментом [6]. Было найдено, что уменьшение
оптической плотности раствора при длине волны 465 нм
(соответствующей максимуму поглощения комплекса фермент-пр'офлавин)
описывается кинетикой первого порядка (табл. 12). Из независимых
экспериментов нашли, что константа диссоциации комплекса фер-
мент-профлавин при условиях опыта равна ЫО-4 М. Значение
каталитической константы, найденное в стационарном режиме
проведения реакции ферментативного гидролиза, равно 0,03 сек-1.
Найти значения индивидуальных констант k2,' k3 и Ks (схема 9.7)
при условии k2^>ks для изучаемой реакции.
197
Таблица 12
Кинетика вытеснения профлавина из комплекса
фермент-профлавин под действием субстрата.
Условия опыта: рН 3,1; 25° С; ионная сила 0,4М;
[Е]0 = 9,5-10"6М; начальная концентрация
профлавина [F]0 = 4,75-10~5М
[S]o-I08, М
5,80
8,65
11,20
18,00
22,10
*эфф
-105, сек-1
5,2
6,2
6,8
8,6
9,4
9-14. Кинетику гидролиза n-нитрофенилового эфира N-ацетил-
L-триптофана, катализируемого а-химотрипсином, изучали
методом остановленной струи по вытеснению профлавина из комплекса
фермент-профлавин в начальный период реакции (в условиях
[S]o^ [E]o). Уменьшение оптической плотности раствора при
длине волны 480 нм (соответствующей уменьшению концентрации
комплекса фермент-краситель) описывалось кинетикой первого
порядка (табл. 13). Из независимых экспериментов было найдено;
Таблица 13
Зависимость эффективной константы скорости
вытеснения профлавина из комплекса
фермент-профлавин от концентрации субстрата.
Условия опыта: рН 4,3; 26,5° С; 1,6%
ацетонитрила; [EJ0 = 5-10~6М; начальная
концентрация профлавина [F]0=5-10-6M
ISIo-W5, М
2,5
4,0
4,5
5,7
7,5
10,0
*эфф, сек '
23
40
44
58
75
97
что константа диссоциации комплекса фермент-профлавин в
условиях опыта равна 7,9-Ю-5 М. Вычислить значения
индивидуальных констант, если величина kKSLT, найденная в стационарных
условиях ферментативной реакции, равна 0,2 сек-1 [6].
9-15. Кинетику гидролиза этилового эфира N-фурил-акрилоил-
L-тирозина, катализируемого а-химотрипсином, изучали методом
остановленной струи в условиях [S]o^>[E]0 по вытеснению
профлавина из комплекса его с ферментом (константа диссоциации
198
комплекса фермент-краситель равна 4-10~s M в условиях
кинетического эксперимента). Скорость вытеснения профлавина
подчинялась кинетике первого порядка с эффективной константой
скорости, зависящей от начальной концентрации субстрата (табл. 14).
Значение AiKaT, найденное при изучении ферментативного гидролиза
субстрата в стационарном режиме протекания реакции, равно
1,5 сек-1 [6]. Найти значения индивидуальных констант k2, kz и
Ks, если известно, что для изучаемой реакции k2 ^> k3.
Таблица 14
Значения эффективных констант скоростей
вытеснения профлавина из комплекса
фермент-профлавин при ацилировании фермента
субстратом. Условия опыта: рН 5,0; 24,5° С;
3% ацетонитрила; JE]0 = 1,25-10-5М; начальная
концентрация профлавина [F]0 = 4,75-10~5М
IS]„.1C«, м
0,С8
0,49
0,77
1,00
1,47
2,00
3,04
4,67
*эфф- сек '
1,8
3,3
4,3
5,2
6,5
8,3
11,2
15,5
9-16. При изучении взаимодействия профлавина с трипсином
(схема 9.9) методом температурного скачка [14] было найдено
одно время релаксации, зависящее от концентраций реагентов
(табл. 15). Определить значения элементарных констант скоростей
реакции
E + DZ3ZTED.
" (9-9)
9-17. Скорость образования донорно-акцепторкого комплекса
между тетрацианоэтиленом и гексаметилоензолом (схемя 9.10) в
1-хлорбутане изучалась методом температурного скачка,
используя микроволновое нагревание [15]:
A + B^Z^AB. (9.10)
На основании данных табл. 16 определить значения констант кл и k-v
9-18. Кинетика взаимодействия рибонуклеазы с уридин-З'-мо-
нофосфатом (схема 9.11) изучалась в работе [16] методом темпе-
199
Таблица 15
Зависимость времени релаксации для связывания
профлавина с трипсином от суммы равновесных
концентраций реагентов. Условия опыта: рН 7,7;
17° С; 0.02М СаСЬ; 0,07М трис-буфера
(IF-I) + [PI-10', М
0,20
0,50
0,63
1,05
1,22
1,45
2,05
2,18
2,33
2,45
2,72
3,25
t-iO5, сек
9,25
7,27
7,02
5,72
4,94
4,87
3,70
3,88
3,60
3,60
3,33
2,94
Таблица 16
Зависимость времени релаксации системы,
содержащей тетрацианоэтилен [AJ, гексаметилбензол [BJ
и донорно-акцепторный комплекс от равновесных
концентраций реагентов. Условия опыта: температура
равна —83° С
IAJ-105, М
20,4
15,1
22,6
11,1
11,4
12,7
8,8
5,1
[В]-10', М
4,9
11,6
3,0
3,33
19,8
31,2
51,2
61,2
т-106, сек
18,6
17,0
18,3
23,3
14,4
11,8
9,6
8,4
ратурного скачка. Используя данные табл. 17, определить значения
элементарных констант скоростей реакции
ft.
E + P^ZZTEP. (9.11)
*->
9-19. Кинетику взаимодействия антител кролика и гаптена
(схема 9.10) изучали методом температурного скачка. Используя
данные табл. 18, определить значения констант k\ и k-\.
200
Таблица 17
Зависимость времени релаксации реакции (9-11)
от суммы равновесных концентраций
рибонуклеазы и уридин-З'-монофосфата. Условия
опыта: рН 5,5; 25° С; ионная сила 0,2М (КС!)
([EI+ [Р])-1С, М
0,55
0,72
0,90
1,20
1,60
1,82
2,00
т-Ю5, сек
6,72
6,10
5,75
5,00,
4,43
4,10
3,84
Таблица 18
Зависимость времени релаксации реакции (9.10)
от суммы равновесных концентраций антитела
и гаптена
([A]+[B])-K>5, M
0,62
0,90
1,28
Т'Ю3, сек
5,25
3,70
2,94
9-20. При изучении методом температурного скачка кинетики
связывания профлавина с активным центром а-химотрипсина было
обнаружено, что кинетическая кривая является суммой двух
экспоненциальных зависимостей. В табл. 19 приведены значения
времен релаксации для ряда равновесных концентраций фермента и
Таблица 19
Зависимость времен релаксации комплексообразования профлавина
с а-химотрипсином от суммы равновесных концентраций реагентов
([E1 + [D])105. M
■с,-10*, сек
т2-10*, сек
3,2
2,3
1,45
5,6
2,0
1,25
8,0
1,70
1,23
11,4
1,65
0,95
18,0
1,54
0,72
20,0
1,50
0,67
красителя. Предложить кинетическую схему
комплексообразования и найти значения элементарных констант скоростей реакции.
9-21. При изучении кинетики равновесного связывания KAD-H
с глутаматдегидрогеназои методом температурного скачка были
201
использованы одинаковые начальные концентрации фермента и
кофсрмепта [17]. Полагая, что механизм комплексообразования
описывается схемой (9.12), и исходя из данных табл. 20,
определить значения элементарных констант скоростей реакции
ft.
Е + N Г
EN.
(9.12)
Таблица 20
Зависимость времени релаксации
комплексообразования NAD • Н
с глутаматдегидрогеназой от суммы начальных
концентраций реагентов. Условия опыта: рН 7,94;
22,3° С; 0,5М NaCl; 0,1М трис-буфер; [Е]0 = ]N]0
([EI„ + [Nlo)-10«,
1,0
3.5
7,0
10
12
14
м
г-Ю3, сек
2.60
2,37
2,06
1,92
1.82
1,70
0-22. В работе [17] с использованием метода температурного
скачка изучалось равновесие в системе
ft, ft? [О]
E + N^C.ZZZTCj, (9.13)
где Е — глутаматдегидрогеназа; N—NAD-H; G — глутамат; Ci и
С2 — промежуточные комплексы. На основании данных
предыдущей задачи и табл. 21 определить значения элементарных
констант скоростей реакции (9.13).
Таблица 21
Зависимость произведения обратных времен
релаксации от суммы равновесных концентраций
фермента и кофермента (NAD-H). Условия
опыта: рН 7,94; 22,3° С; 0,5М NaCl;
0,1 М трис-буфер; концентрация глутамата во всех
случаях равна 1,6-10_2М
([Е] + [NI)-10«. M
3,0
3,6
4,7
5.8
6.8
8,3
I з г
-^—■10 3, сек г
т,т,
15.2
18,0
21,5
25.5
29,0
35,0
202
Ответы и решения
9-1 [18]. Для решения задачи необходимо найти
функциональную связь между величиной т (см. рис. 90) и начальной
концентрацией субстрата [S]0.
Исходя из схемы (9.6), запишем дифференциальные уравнения
для концентрации фермент-субстратного комплекса и продукта
реакции, а также уравнение материального баланса по ферменту
(так как в нашем случае [S]0^> [Е]о)
^l=ft1[E][S]0-(*_1 + *2)[ES], * (9.14)
-*gL = *2[ES], (9.15)
[E]0 = [E] + [ES]. (9.16)
Подставляя соотношение (9.16) в (9.14) и разделяя переменные,
получим
[ES] t
J *i[E].[S]Q- (ft1[S]Q + A_I + AJ[ES] J" K '
о о
Интегрируя уравнение (9.17), находим
rpci __ ki tEio [S]o [1 _ ,-№|Slo+*_i+*!)< 1
^^k^Slo + k^ + kt I1 e i
Подставляя выражение для [ES] в уравнение (9.15) и интегрируя
его в пределах (0, Р), (0, t), получаем уравнение, описывающее
зависимость концентрации продукта ферментативной реакции от
времени реакции:
rni = kxk2 [Е]о [S]o f kxk, [E]0 [S]0 r 1 _ _ (ft, [SJo+ft_i+ft!)q
1 ' *, [S], + k-t t k2 (ft, [S]„ + A_, + k2f I* J"
Как видно из полученного уравнения, при сравнительно
больших временах реакции зависимость концентрации продукта от
времени будет линейной
mi к\к2 \Е\0 [Ь\0 , к\Кг [Ь.\0 [bjo ,q <г>\
1 J~ "*. [SI.+ *-, + *, (A.ISJ. + A-. + AO* ' <- '
Из рассмотрения уравнения (9.18) очевидно, что продолжение
линейного участка зависимости в координатах ([Р], t) до
пересечения с осью времени будет определять характеристическое время
реакции т (см. рис. 90):
Т = *, [S]. + *_, + k2 ■ (9Л9)
Как следует из выражения (9.19), зависимость в координатах
(1/т, [S]o) должна быть прямолинейной, с тангенсом угла
наклона, численно равным k\, и ординатой точки пересечения, равной
k-i+k2 (рис. 93).
203
Значение константы k2 можно найти линеаризацией данных
табл. 1 для стационарного режима протекания ферментативной
реакции в подходящей системе координат (см. гл. 5).
Ответ: £i = l,0-104 М-'-сек-1; /г_1=0,5 сек-1; £2=0,5 сек-1.
9-2. В соответствии со схемой (9.7) кинетика образования
продукта Pi (в данном случае «-нитрофенола) описывается
уравнением
^J- = £2[ES]. (9.20)
Образование продукта Pi в начальный период реакции следует
кинетике первого порядка; это означает, что равновесие при
образовании фермент-субстратного
комплекса (ES) устанавливается
очень быстро, в пределах
«мертвого времени» прибора. Таким
образом, в изучаемом
временном интервале ферментативного
процесса действительно
соотношение
rajs]
Ks -
Поскольку в данном эксперименте
соблюдается условие [Е] ^ [Е]0
([Е]0 » [S]0), то уравнение (9.20)
можно записать в виде
*[Р.] =jb_[E]o[S]i (9_22)
2.0
1,5
1.0
0.5-
_
-
■1 2
' 1
£TtgcP'£,
i i i i
[ES] =
(9.21)
5 s7
Й-Ю.М
Рис. 93. Определение элементарных
констант двухстадийной
ферментативной реакции
dt
Комбинируя выражения (9.20) —
(9.22) с уравнением материального
баланса [S]0 = [S] + [ES] -f- [ЕА], получаем
dpi] k2 [E]Q([S].-[EA])
dt
Kc
1 +
[ЕЬ
К*
ИЛИ
<*[Р,1
dt
*i[E].[S].
Ks + [E].
*2 jE-Jo
KS + tElo
[EA].
(9.23)
Так как в случае [E]oS>[S]o соблюдается равенство [ЕА] =[Pi]
(см. схему 9.7), то, разделяя переменные и интегрируя уравнение
(9.23) в пределах (0, Pi) и (0, t), можно получить выражение для
зависимости концентрации продукции [Pi] от времени реакции
[Pi] = [SLVi—e
l!, [Е]о
Ks+l
Е]о I-
204
Очевидно, что накопление продукта Pi должно подчиняться
кинетике первого порядка с эффективной константой скорости
^эфф = к$ + [Е°]о • • (9-24)
Линеаризуя зависимость (9.24) в координатах (1/&эфФ, 1/[Е]о),
получаем прямую линию с угловым коэффициентом Ks/k2 и точкой
пересечения с осью ординат, равной 1/&2 (рис. 94).
Определив таким образом значения k2 и Ks и подставляя их в
выражение для /гкат или Кт<кат) (см. соотношения 7.2 и 7.3),
находим значение константы k3.
Ответ: £2=6,7 сек-1; /г3=19,7 сек-1; /Cs=l,7-10~4 М.
Рис. 94. Определение
индивидуальных констант &2 и Ks из
данных предстационарной кинетики
гидролиза п-нитрофенилгиппурата,
катализируемого фицином
2.0
1.5
W
О
с?
1=
0
-0,5
Xj
I
оЧ
i N
20 з 60
f-70. сек
Рис. 95. Определение константы
скорости деацилирования ацилфер-
9-3. Накопление продукта Pi ферментативной реакции,
протекающей по трехстадийной схеме (9.7), должно подчиняться
кинетике первого порядка с эффективной константой скорости
*2 [Ео] (9.25)
сэфф
к.
[Е].
в условиях [E]0^>[S]o (см. решение задачи 9-2).
Из данных табл. 3 можно найти, что реакция ацилирования
папаина л*-нитрофениловым эфиром карбобензоксиглицина (в
условиях [E]o^>[S]o) действительно описывается кинетической за-
205
висимостью первого порядка с константой скорости, равной
12,1 сек-1. Подставляя полученное ' значение &эфф в выражение
(9.25), получим &2/(/Cs+[E]o) = l,09-105 М-1-сек-1. Очевидно, что
эта величина совпадает в пределах ошибки эксперимента с
величиной ^кат//Ст(каж)=/г2//;С8= (1,15+0,08)-105 М-1-сек-1, определенной
в стационарном режиме протекания ферментативной реакции.
Таким образом, в данном случае соблюдается условие Ks^> [E]o, или
/Cs»10-4 М. Так как Л'„,(„аж, = Ks -j^tkT = (i,89±0- ,7> Ю~5 М'
то для изучаемой реакции k2lk3^>5, т. е. стадией, лимитирующей
скорость, является деацилирование.
9-4. Значение константы скорости деацилирования ацилфер-
мента, найденное из данных табл. 4 согласно кинетике первого
порядка (рис. 95), равно 30,9 сек-1. Зная численное значение
k к
^каг = k *,*k ■ равное 25 сек-1, и k3, равное 30,9 сек, можно
найти значение k2.
Ответ: £3=30,9 сек-1; /е2= 131 сек-1.
9-5. Кинетика накопления продукта Pi (схема 9.8) при
условии [S] о^ [Е] о определяется соотношением
^f = ka [E]. [S]0 = fcn ([E]0 - [ES]) ['S]0. (9.26)
Из схемы (9.8) следует также соотношение
^Р = ku ([Е]0 - [ES]) [S]0 - кг[ES], (9.27)
интегрируя которое в пределах (0, [ES]), (0, t) можно получить
следующее уравнение:
[ES] = *"rtgE1jBlS!bB [1- e-t*nlsb+*.)']. (9.28)
fell L^Jo ~г ^з
Подставляя (9.28) в соотношение (9.26), получаем уравнение
d[P,\ _ , rF, rci №i[So])2[E]0[-, _-(ftn[S]„+fc3)n
~аГ~ яп1Ь;о1Ь10- -ka[S]a^kt I1 e J'
интегрируя которое в пределах (О, [Р,]), (0, t) можно получить
выражение для зависимости концентрации продукта ферментативной
реакции от времени
m, *n*,[EUS]. , , №i[S]0)2[E]ori
,Pl] = ftnlSb + A, ' + (л^ГнУ1Ь~е J- (9'29)
При значительных временах реакции t кинетика образования
продукта Р, описывается выражением
lFlJ~*n[S]„ + ft3'r+ (ft„[S].-> ksy ■ V-6V>
Вычитая из (9.30) выражение (9.29), получим
д р = (^и[SJq)2 [EJ0 -(л,, [Sb+ft.) t
(fen [SJ„ -I- ktf
206
Таким образом, уменьшение со временем разности оптической
плотности между наблюдаемой прямой (AD, см. рис. 91)
подчиняется кинетике первого порядка с эффективной константой
скорости
*,ФФ = *ii lS]o +*3- (9-31)
Линеаризацией экспериментальных данных (табл. 5) в
координатах (InAD,if), находим величину &Эфф, равную 2,1-Ю-2 сек-1. Зная
из условия задачи значения k3 и [§]о, из выражения (9.31)
находим величину k\\.
Ответ: kn= 179 М-1 • сек-1.
9-6. Для решения задачи необходимо найти аналитическую
зависимость концентрации продукта [Pi] от времени реакции для
более общего случая трехстадийной схемы (9.7), так как двухста-
дийная схема (9.6) формально-кинетически эквивалентна
трехстадийной в частном случае k2<^k3.
В соответствии со схемой (9.7) кинетика образования продукта
Pi в случае [S]0S> [E]0 определяется уравнением
^L = ^-[E][S]0. (9.32)
Из уравнения материального баланса
[EJ0 = [Е] + [ES] + [ЕА] = [Е] ( 1 + Щ + [ЕА| (9.33)
получаем
[Е] = 1E]'~'EA]- (9.34)
Подставляя соотношение (9.34) в (9.32), имеем выражение
Ч[РЛ .___ *»[SJo([E].-[EA])
dt Ks + [S]0
(9.35)
При записи выражений (9.32) — (9.35) мы принимаем
установившимся равновесие на стадии образования фермент-субстратного
комплекса. Это следует из того факта, что зависимость в
координатах (I^i], t) не имеет лаг-периода (индукционного периода)
(рис. 96).
Из схемы (9.7) также следует уравнение
^ = A2IES]-*3|EA]. (9.36)
[я (9.33), (9,34) и (9.с6), получаем
■/fsTisjT ~ ( /cs + [sic + k*}[EA] •
dt
Комбинируй уравнения (9.33), (9,34) и (9.с6), получаем
d [ЕА] __ *,[EJ0[S]. I k2 [S]„
dt
207
Интегрируя полученное уравнение в пределах (О, [ЕА]), (О, t),
приходим к выражению
[EAJ. МШЯ [,_е-(-Йп^ + *')'].
подставляя которое в уравнение (9.36) и интегрируя последнее
в пределах (О, [Р,]), (U, t), получаем выражение для зависимости
концентрации продукта реакции [Р^ от времени:
IP.]-
Якат [ч!о [^Jo
К.
t +
[Е]„
(К,
т (каж)
+ [S],
т (каж) + [S].
( MS], у
о)2 \ k2 + k3J А
(9.37)
f /О.сек
И-ю.м
Рис. 96. Кинетика гидролиза о-ни-
трофенил-а-мальтозида,
катализируемого а-амилазой, в начальный
период реакции
Рис. 97. Обработка предстационарно-
го участка кинетической кривой
гидролиза и-нитрофенилтриметилацетата,
катализируемого эластазой
Из рис. 96 видно, что [Р,] линейно зависит от t, причем [Р2] - О
при t = 0. Это может быть лишь в случае
"(*,
,(Ka») + tS]o)S \Ъ+Л, ) L1 J
(см. 9.37) Из рассмотрения полученного равенства очевидно, что
оно будет выполняться лишь в случае &2-С&з, что формально
соответствует двухстадийной схеме протекания ферментативной
реакции.
9-7. Для изучаемой реакции &офф=&и[$]о+&.ч (см. решение
задачи 9-5), поэтому график зависимости &Эфф от [S]o должен иметь
вид прямой линии, из угла наклона и точки пересечения которой
208
с осью ординат можно найти численные значения кинетических
констант (рис. 97).
Ответ: ка=\Ъ7 М-1-сек-1; йз^б-Ю"3 сек"1.
9-8. В случае трехстадийной схемы ферментативной реакции
(9.7) зависимость концентрации продукта реакции [Pi] от
времени (в условиях [S]03>[E]o) определяется следующим
выражением (см. решение задачи 9-6):
гр 1 __ "кат [Е]0 [S]„ , , [EJ0 v.
*vra (каж) + 1^]о (^т(каж)
+ [S]o)2 ^
х-{-m-yi-A^■*-)■] ^
При больших временах t выражение (9.38) переходит в
гр 1 «2ft3 [tij0 [aj0 , | [c,J0 (ft2 l^Jo) /q oq\
1 " k,Ks + [S]. (*, + *,) "•" [k,Ks -, [S]. (*, + ft3)]! ' ^-oy'
так что отрезок, отсекаемый на оси ординат экстраполированным
участком прямой в координатах ([Р^, Л (см. рис. 92), определяется
выражением
k2 у
k2 + k3 ts3o
it =
(каж) + [S].
■[Е]0. (9.40)
В рассматриваемых нами случаях
а) Кт(каж)=4,54-10-5 М; k2=U сек"1; £3=8,5 сек-1; [S]0 =
= Ю-4 М;
б) Кт(каж)=1,28-10-4 М; *2=7,8 сек-1; кя= 13,9 сек-1; [S]0=
= Ю-4 М.
Подставляя эти значения в формулу (9.40), можно рассчитать,
какую долю концентрации фермента составит «выброс» л-продукта
реакции Р].
Ответ: а) 33%; б) 2,5%.
9-9. Рассматривая уравнение (9.30), видим, что
экстраполяция линейного участка кинетической кривой в координатах ([Pi],0
на ось ординат (см. рис. 92) отсекает отрезок, равный
(ftn[S]„ + ft3)2 • ( • '
Преобразовывая выражение (9.41), получаем
1 = 1 fe3 1
/iT /[Ё]"о + *"(SI° "Г[ЁЬ"
Отсюда график в координатах (1/)/тс, 1/[S]0) должен иметь ьид
прямой линии с отрезком на оси ординат, равным 1/)'[Е]0 (рис. 98).
Ответ: [Е]0= 1,97.1С-6 М.
9-10. Для решения задачи необходимо найти аналитическое
выражение для эффективной константы скорости первого порядка
209
вытеснения профлавина из комплекса с ферментом под действием
субстрата. Общую схему реакции (9.42—9.43), выражение для
констант диссоциации комплексов фермент-субстрат (9.44) и фер-
мент-профлавин (9.45), а также уравнение материального
баланса по ферменту (9.46) можно записать в следующем виде (при
условии [S]o>[E]0, [F]0>[E]o):
Е + S ~■*- ES 4=^ ЕЛ -%- е + р, ( 9.42)
2 )
^
E + F^=iEF' (9-43)
A'S==JW|-' <9"44)
к?=-Щг> (9-45)
[Е]0 = [Е] + [ES] + [ЕА] + [EF]. (9.46)
Кинетика ферментативной реакции в нашем случае регистрируется
по изменению концентрации комплекса фермент-краситель (EF),
поэтому необходимо концентрации всех веществ, принимающих
участие в реакции, выразить через концентрации [EF]:
КР [EF]
[Е]= F
[ESj =
[F]„ '
KF [EF] [S]„
Ks [F],
a „ ^f[S]o
.заменяя постоянный член r^r— через а, получим
Ks In»
[ES] = a[EF]. (9.47)
Из уравнения материального баланса (9.46) найдем
[EA] = [E]0-6[EF], (9.48)
КР
где & = 1 + о + тр!—• Подставляя выражения (9.47) и (9.48)
L* Jo
в уравнение скорости ферментативной реакции
^J = ft2[ES] - МЕА],
получаем
_ &£Ш = {k2a + kbb) [EF] - k3 [E]0. (9.49)
Из уравнения (9.48) следует, что при t = 0 величина [EF] равна
[Е]0/£. Отсюда, интегрируя уравнение (3.49) в пределах (0, t) и
210
[E]0/b, [EF]), получим
[E]„
ftja+ft3*
[EF] = w/^i* »м Kb J + k2ae
1 > b (k2a + k3b) ч J l l
Таким образом, уменьшение концентрации комплекса фермент-
профлавин (EF) во времени следует кинетике первого порядка
с эффективной константой скорости
*2 [S]0
"'эфф —
к,
■№)
Я-г.
[S].
(9.50;
юо
ео
т 60
о
е-
о
20
0
^
1
К
10
20
2,0 3,0
1/[s}wW
о
Рис. 98. Определение концентрации
активных центров фермента из
данных предстациоиариой кинетики
А
30
;3 -1 -1
L о
Рис. 99. Определение
индивидуальных констант скоростей
реакции гидролиза этилового
эфира М-ацстпл-Ь-триптофана,
катализируемого а-химотрипсн-
ном, с помощью метода
«вытеснения профлавина»
Выражение (9.50) можно преобразовать к следующему виду:
Ks(1+-kJt)(**M'-*')
(^эфф Я3) — К0
оче
эфф ~з; — «-2 ■ [S]o
Из выражения (9.51) очевидно, что в координатах
^эфф ~~ "з
(9.51)
*з).
[S].
-J
211
точка пересечения прямой с осью ординат даст величину k2, а
тангенс угла наклона прямой равгн /Cs (1 +\P]qIKf). Так как в нашем
случаг величина &кат для всех значений [S]0 значительно меньше
£эфф, то для изучаемой реакции kKaT^k3. Отсюда, откладывая
данные табл. 9 в координатах уравнения (9.51), найдем величины k2
и Ks (рис. 99).
Ответ: fe2=I04 сек-1; k3=2 сек"1; Ks=l,7-10"3 M.
9-11. Образование продукта Pi в трехстадийной
ферментативной реакции (см. схему 9.7) в условиях [E]o3>[S]o должно
подчиняться кинетике первого порядка с эффективной константой
скорости:
, МЕ]0
«эфф к$ + [Е]о
(см. решение задачи 9-2). Отсюда зависимость в координатах
(&эфф, &эфф/[Е]0) должна быть прямолинейной с отрезком,
отсекаемым на оси ординат, равным k2, и тангенсом угла наклона,
равным — Ks (рис. 100).
Ответ: £2=21,3 сек"1; Ks=l,5-10~4 М.
9-12. Решается аналогично задаче 9-10.
Ответ: /г2=85 сек-'; k3=3 сек-1; Ks= 1,8-10"2 М.
9-13. Решается аналогично задаче 9-10.
Ответ: £2=0,205 сек"1; £3=0,03 сек-1; /Cs=3,2-10~2 M.
9-14. Так как для изучаемой реакции величина £Кат Для всех
значений [S]0 значительно меньше &Эфф (см. табл. 13), то
справедливо соотношение k2^>k3 и, следовательно, &кат~&3 (см.
выражение 9.50). В этом случае выражение (9.50) приобретает вид
fe2[S]
^эфф -^ ', rpi ^ (У.52)
или
Ь и ^ £_1
лэфф к2 Г§1
В данном случае график в координатах (&Эфф, &эфф/[5]о) должен
иметь вид прямой линии, параллельной оси ординат, т. е. при всех
концентрациях [S]0 величина ^эфф/fSlo должна оставаться
постоянной. Это означает, что величина &эфф пропорциональна [S]0 или,
как следует из уравнения (9.52), Ks^>[S]o- Очевидно, в этом
случае невозможно найти раздельно значения индивидуальных
констант k2 и Ks, но из точки пересечения прямой с осью абсцисс
можно найти их отношение k2/Ks (рис. 101).
Ответ: £3=0,2 сек"1; k2/Ks=lfi-106 М-1-сек-1.
9-15. Решается аналогично задаче 9-10.
Ответ: £2=65 сек"1; k3= 1,5 сек"1; Ks=7,7-10"4 М.
212
9-16. При небольшом смещении равновесия (9.9) методом
температурного скачка текущие концентрации фермента, красителя и
комплекса фермент-краситель будут определяться соотношениями
[Е] = [Е] + Д [Е], [D! = [Б] + Д [D], [ED] = [Щ + Д [ED],
где [Ё], [Б] и [ED] —
концентрации реагентов в исходном
равновесии. Дифференциальное
уравнение, описывающее переход систе-
12.0
10,0
1
8,0
г£ 6,0
О
в-
&
■*?4,0
2.0
-
_
1
\
\ о
\
\
i 1 \
0,4 0.8 1.2 1.6
*эфф/И0Ю5.П сек'
Рис. 100. Определение
индивидуальных констант реакции
гидролиза «-нитрофенилового эфира
N-aцeтил-L-тpиптoфaнa,
катализируемого а-химотрипсином, из
данных предстационарной кинетики
Т^ 60
Рис. 101. Определение
константы скорости второго порядка
(k2/Ks) реакции гидролиза
и-нитрофенилового эфира N-
ацетил-С-триптофана,
катализируемого а-химотрипсииом, с
помощью метода «вытеснения
профлавина»
мы (9.9) к новому равновесию, имеет вид
^ = *,|B1[D|-*_,[ED1.
или
tf(A[ED])
-й = *i([E] + AfE])([D]-|-A[D])-^1([EDj-|-A[ED]). (9.53)
Уравнение материального баланса для рассматриваемой системы
213
можно записать в виде
A[E]-f-A[ED]=0,
A[D] + AjEDJ = 0.
(9.54)
(9.55)
Подставляя значения А [Е] и A [D] из соотношений (9.54) и (9.55)
в уравнение (9.53) и пренебрегая г, алым по величине произведением
A[£]A[D], получим
<*(А[ЕР])
dt
'■к
о
= const - [ft, ([Е] + 1D|) + k-x\ A [ED], (9.56)
где const ft, [EJ [D] - ft_, [ED].
Очевидно, уравнение (9.56)
является кинетическим уравнением
псевдопервого порядка по
концентрации A[EDJ с константой
скорости
1
= К (IE] + [D]) + к-
ь
'еИ.о
3,0
([ЁМР])--70?М
Рис. 102. Определение
элементарных констант скоростей комплек-
сообраэования трипсина с профла-
вином с помощью
релаксационного метода; т — времена
релаксации при различных значениях
равновесной концентрации реагентов
где
§ 2
вая
элементарных
■10?М-'-сек
-1.
т — время релаксации (см.
настоящей главы). Отклады-
экспериментальные данные
в координатах (7т, [Е] + [Т5]),
получим прямолинейную
зависимость (рис. 102), из тангенса
угла наклона которой и
ординаты точки пересечения можно
найти значения
констант.
Ответ: ft, = 7,6 •
ft_i=9500 сек-1.
9-17. Решается
задаче 9-16.
Ответ: ki = 1,45-108 М-'-сек"1; ft_, = 2-104 сек-'.
9-18. Решается аналогично задаче 9-16.
Ответ: ft,==7,79-107M-1-ceK-1; ft_,= l,05-10* сек-1.
9-19. Решается аналогично задаче 9-16.
Ответ: kx = 2,3-107 М-1-сек-1; ft_i = 50 сек-1.
9-20. Так как на экспериментальной кривой наблюдаются два
времени релаксации, кинетическая схема реакции должна
включать по меньшей мере одно промежуточное соединение
аналогично
E + DZ=^EDZ=^ED,
(9.57''
Тогда выражения для текущих концентраций реагирующих
веществ и промежуточных соединений можно записать в следующем
214
виде:
[Е] = [Ё] + Д [Е], [D] = [DJ + A [D],
[ED] = [ED] + Д [ED], [ED,] = [Щ] + A [EDJ.
Кинетика перехода системы (9.57) к новому равновесию полностью
описывается двумя дифференциальными уравнениями (см. решение
задачи 9-16)
^И = к, ([Ё] + Д [Е]) ([D] + Д [D,) + £_2 ([Щ] + A [EDJ) -
-(fc_, + *2)([ED] + A[ED]), (9.58)
tf (A5Dll> = ^([ЁЩ + A [ED]) - fe_2 ([Щ] + Д [ED,]) (9.59)
и двумя уравнениями материального баланса
A[E] + AIED] + A[ED1] = 0, (9.60)
A[D] + A[ED]+A[ED,] = 0. (9.61)
Раскрывая скобки в уравнении (9.58) и пренебрегая произведением
£ХД [Е] Д [D] (при условии незначительного смещения равновесия
в системе), получим
tf(A[ED]) =fei.tE][D] + fe1[E]A[Dl + fei[D]A[E] +
-f- £_2 (fEDiJ + A [EDx]) - (A_! + h) ([ED] + Д [EDJ). (9.62)
Исключая из уравнения (9.62) переменные Д [Е] и Д [D] с помощью
соотношений (9.60), (9.61), приходим к системе двух линейных
дифференциальных уравнений с постоянными коэффициентами
*£Ш = ki {щ {ц _ ki {[щ + pj) (Д [ED] +Д [ED,]) +
+ А_2 ([Щ] + Д [ED,]) - (к-г+ k2) ([ED] + Д [ED]),
И*Ш>_= кЛ[Щ + д [ED]) - £-2([Щ] + Д [ED,]).
Находя из второго уравнения величину A[ED] как функцию
переменных Д [EDj] и —* * '" , дифференцируем ее и подставляем
наиденные выражения для Д [EDJ и —- в первое уравнение.
Полученное при этом дифференциальное уравнение второго порядка
имеет вид
^(МЕВЛ + L iKAgDJi. + ш [EDi] = д (9 63)
где постоянные величины L, М и А определяются значениями
кинетических констант и равновесных концентраций системы (9.57).
215
Решение дифференциального уравнения (9.63) имеет вид
Д [ED,] = С,е-^- +'С2е- '/*.,
где т-1 и т-1 — корни характеристического уравнения
X2 + ZX 4 М = 0.
Воспользовавшись свойствами корней квадратного уравнения
+ Ч1
L,
•с-'.т-' =ЛГ
г-1
(9.64)
(9.65)
и подставляя в соотношения (9.64), (9.65) выражения для
постоянных L и М, найденные из (9.63), получим
тГ1 + тГ1 = К ([Ё] 4- [D]) 4 *-, + ft2 + *-a,
(тхт2)-' = kx (k2 4- ft-2)([E] 4 [D]) + k-,k-2.
Наконец, откладывая экспериментальные данные в координатах
(•сГ1 4'СГ1, [Е] 4- [D]) и (тГ'тГ1, [Е] 4- [D]), найдем значения
элементарных констант реакции (рис. ЮЗ).
24
"х 16
^ 8
+
-и-г"
-
-
I 1 1 1 1
-
У-_
S
J^
1 1 1 1 I
120
-80'
40
б /2 /6 20
4 8
(Гё]+[ц1)-ю?м
12 16 20
I
Рис. 103. Определение элементарных констант двухстаднйпой
реакции комплексообразования по двум временам релаксации
Ответ: Jfe1==6,2-107 М-1-сек-1; ft_i=-2,6-103 сек-1; k2= 50 сек-1;
k-a=6,55-103 сек"1.
9-21 [17]. Постановка задачи отличается от предыдущих
вариантов тем, что в расчетах должны быть использованы не
равновесные, а начальные концентрации реагентов. Выражение для
константы равновесия системы (9.12) можно записать в виде
к_ \ш\
Ten!
(9.66)
216
где [EJ, [NJ и [ENj — разновесные концентрации реагентов.
Учитывая уравнения материального баланса
[Е]0 = [Ё] + [EN], [NJ0 = [N] + [Щ,
соэтношеме (9.65) мэжчо представить в виде
К =
gE].-[EN])_(t_Nlo-[ENl)
[ENj
(9.67)
Приводя выражение (9.67) к виду квадратного уравнения
[EN]* - [ENJ ([E]0 + [NJ0 + K) + [E]0 [NJ0 = О,
находим соотношение, связывающее равновесную концентрацию
комплекса с начальными концентрациями реагентов
ГЩ = [Е]° + № + К± /([Е]„ + [N]0 + Kf -4 [Е], [N]„
Переходя в выражении
К ([EJ -f- [NJ) + ft-,
1ций исходных ре;
4- = ft1([E]0+[N]0-2[ENj) + ft_1
от равнозесных концентрации исходных реагентов к начальным
1
(9.68)
(9.69)
:м
(9.70)
Рис. 104. Определение
элементарных констант скоростей
связывания NAD-H с глутаматдегидро-
геназой с помощью
релаксационного метода при вариации
начальных концентраций реагентов
8,0 120
(см. решение задачи 9-16) и комбинируя соотношения (9.68) и (9.70),
получим
4" = *i (~ К ± /([E]0 + [Nlo+/C)2-4[E]0[N]0) + ft_,
или
— = kx V ([Е]0 + [N]0 + Kf - 4 [Е]0 [N]0.
Возводя обе части выражения (9.71) в квадрат, находим
-L = k\[([Е]0 - [Щ0? + 2К ([Е]0 -f [N]0) + К2].
(9.71)
(9.72)
217
Наконец, при условии [Е]0 = [N]0 выражение (9.72) упрощается
-i- = 2k,k-, ([E]0 + [N]0) + fti,. (9.73)
2
Откладывая экспериментальные данные в координатах (т-2, [Е]0 +
+ [N]0), найдем значения элементарных констант скоростей реакции
комплексообразования (рис. 104).
Ответ: fei=2-106 М~'-сек-1; &_i=364 сек-1.
9-22. Решается аналогично задаче 9-20.
Ответ: Jfe,==2-106 Mr1-сек-1; &_i=364 сек-1; Jfe2=l,09-104 М~'Х
Хсек-1; &_2=11 сек-1.
ЛИТЕРАТУРА
1. Сб. «Применение вычислительной математики в физической и химической
кинетике», под ред. Л. С. Полака. М., «Наука», 1969, стр. 12.
2. Сб. «Экспериментальные методы химической кинетики", под ред. Н.
М.Эмануэля. М., «Высшая школа», 1971, стр. 155.
3. Бернхард С. Структура и функция ферментов. М., «Мир», 1971,
стр. 127.
4. Уэстли Дж. Ферментативный катализ. М. «Мир», 1972, стр. 182.
5. В г a n d t К. О., Н i m о е А., Н е s s G. P. «J. Biol. Chem.», 242, 3973, 1967.
6. Н i m о е А., В г a n d t К. С, D e S a R. J., H e s s G. P. «J. Biol. Chem.»,
244, 3483, 1969.
7. Mc Conn J., Ku E., Himoe A., Brandt К. С Hess G. P.
«J. Biol. Chem.», 246, 2 18, 1971.
8. Chook P. B. «Biochimie», 53, 161, 1971.
9. Hoi I a way M. R., Antonini E., Brunori M. «Eur. J. Biochem.»,
24 332 1971
10. Hubbard C. D, Kirsch J. F. «Biochemistry», 7, 2f69, 1968.
11. Miller С G., Bender M. L. «J. Amer. Chem. Soc», 90, 68f0, 1968. „
12. Bender M. L., Marshall Т. Н. «J. Amer. Chem. Soc», 90, 201, 196$.
13. S u e l s u g u N.. H i г о m i К., О n о S. «J. Biochem.», 69. 421, 1971.
14. Guilaln F., Thus i us D «J. Amer. Chem. Sec», 92, 5534,1970.
15. С a 1 d I n E. F., С г о о k s L. E., O'D о n n e 11 D., S m i t h D, Toner S.
«J. Chem. Sec, Farrday Tr?ns I», pt. 5. 849, 1972.
16. Hammes G. G.. W a 1 z F. G., Jr. «J. Amer. Chem. Soc», 91, 7179, 1969.
17. Malcolm A. D. B. «Eur. J. Bicchem.», 27, 453, 1972.
18. Уолтер Ч. Кинетика ферментативных реакций. М., «Мир», 1969, стр. 48.
Глава 10
ВЛИЯНИЕ рН НА СКОРОСТЬ
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
Ионогенные группы имеют особенно важное значение для
ферментативного катализа. В активных центрах всех изученных до
настоящего времени ферментов обнаружены функциональные
группы, способные присоединять или отщеплять протоны в области рН,
оптимальной для проявления ферментативной активности. Исходя
из этого, естественно, что рН-эффекты используются для
выявления каталитически важных ионогенных групп фермента и
выяснения способов их участия в общем механизме ферментативного
катализа.
218
§ 1. рН-Зависимость двухстадийной
ферментативной реакции
Зависимость скорости ферментативной реакции от рН на
практике часто отвечает следующей схеме:
EH2 + S~
:eh2s
EH + S-
«s
EHS-
EH + P.
(10.1)
E + S:
ES
- Здесь необходимо указать, что символы Е, ЕН, ЕН2 и т. д.
описывают только состояние ионизации определенных групп
фермента, контролирующих ферментативную реакцию. Ионизация
остальных групп белковой глобулы здесь вообще не рассматривается.
Согласно схеме (10.1) активный центр фермента имеет две ионо-
генные группы, причем константы их диссоциации в свободном
ферменте и в фермент-субстратном комплексе являются
различными (в принципе, схема (10.1) может описывать и реакцию
фермента, активный центр которого содержит четыре ионогенные
группы, две функционируют в свободной форме фермента и две — в
фермент-субстратном комплексе).
Обработка схем рН-зависимости ферментативных реакций типа
(10.1) формально сводится к подсчету концентрации активной
формы фермента с помощью констант диссоциации ионогенных
групп его активного центра (или ионогенных групп фермента,
контролирующих состояние активного центра). Для проведения
обработки схемы (10.1) запишем, как обычно, уравнение скорости,
уравнение материального баланса по ферменту (в случае [S]0^
^> [Е] о) и выражения для констант равновесия:
■y = £2[EHS], (10.2)
[Е]о
[Е|
К,
А» =
[ЕН] + [ЕН,]
[ЕН]-[Н+]
[EHJ '
[Е]-[Н+]
[ЕН]
+ 1ES]
К' =
А% =
f [EHS] + [EH2Sj,
[EHS]-[H+]
[EH2S]
[ES]-[H+]
(10.3)
[EHS]
[EH]-[S]
[EHS]
Следует отметить, если рН влияет на ферментативную активность,
то допущение о равновесных условиях присоединения или
отщепления протона вполне допустимо, поскольку скорость ионных
реакций с участием протона достаточно высока.
Комбинируя выражения (10.2) и (10.3) с выражениями для
219
констант равновесия, получим общее уравнение скорости
ферментативной реакции
1) = ^кат [E]q-[S]0 , jq ^д
Кт (каж) + f Sl«
где
*кат = ~ J7r~, (Ю.5)
[Н+] Кь
1 + К'а + [Н+]
/Ст(каж) = /С5 ^ ^5^—• (Ю.6)
_[НЦ *»_
1 + 7<; + [н+]
Из выражений (10.5) — (10.6) видно, что анализ зависимости &Кат
от рН позволяет найти значения констант диссоциации групп
фермент-субстратного комплекса (К'а и К'ь), а анализ рН-зависимо-
сти константы скорости второго порядка kKSi?/Кщкаж) приводит к
значениям констант диссоциации ионогенных групп свободного
фермента Ка и Кь-
Если субстрат, связываясь с ферментом, не изменяет значений
констант диссоциации ионогенных групп активного центра (Ка=
— К'а, Къ=К'ъ на схеме 10.1), то в этом случае константа Миха-
элиса ферментативной реакции не зависит от рН
[Е]. [S]„
v-—"'"«s-Hsj; • (la7>
Практически это может означать, что ионогенные группы,
контролирующие ферментативную реакцию, входят в состав
каталитического, а не сорбционного участка активного центра.
Если активный центр свободного фермента имеет две
ионогенные группы, ионизация которых переводит фермент в неактивную
форму, не способную связывать субстрат (К'а = оо, К'ь = 0),
ЕН2
t
ЁН + S^Z^l EHS —--EH + P
то уравнение скорости ферментативной реакции приобретает
следующий вид:
V = &2 [E],[S]0 , 1Q g.
[S]„
220
Зависимость скорости ферментативной реакции от рН типа (10.8)
может соответствовать случаю, когда ионогенные группы
активного центра входят в состав сорбционного участка фермента, и не
принимают участие в последующей каталитической стадии.
Очевидно, что уравнения (10.7) и (10.8) описывают
симметричную колоколообразную кривую рН-зависимости скорости
ферментативной реакции (при небольшой концентрации субстрата в
случае 10.8). Однако на практике иногда наблюдаются колоколооб-
разные кривые зависимости скорости ферментативной реакции от
рН. Для их описания необходимо учитывать возможность
нескольких ступеней ионизации активной формы фермента в кислую или
щелочную сторону. Например, схеме (10.9) соответствует
уравнение (10.10) скорости ферментативной реакции, согласно которому
график в координатах (log&KaT, рН) или (log &Кат/Кт(каж), рН)
имеет вид соединенных плавными переходами отрезков с
тангенсами угла наклона +2, -f-1, 0, —1, так что общий график рН-зави-
симости имеет вид несимметричной колоколообразной кривой,
левая ветвь которой круче правой:
EH8 + S ^ZH EH3S
EH0 + S -» EH2S
EH + S
+ S "
► EI
Kb
ES
(10.9)
IS —*-* EH + P
•v = ■
[H+] [H+]«
l+—^r- + 77Г777 +
К'
Kh
KaK'b [H+]
[EJ.IS1.
[H+] [H+P Kc
1 + Kb + KaKb + [H+]
K<
!+■
[H+] [H+]2
K'h
+
- +
К
KaKb [H+]
+ [S].
(10.10)
§ 2. рН-Зависимости трехстадийной
ферментативной реакции
В общем виде схему рН-зависимости трехстадийной
ферментативной реакции с двумя ступенями ионизации можно записать
следующим образом:
221
EH2 + S
^EH2S-
-EH2A 6 > EH2 + P2
EHS-
*
*-EHA-
E + S:
ES
•#.
ЕЛ
EH + P„
EtP,
(10.11)
Полная обработка данной схемы приводит к довольно сложным
выражениям для кинетических параметров &кат и /Ст(каж)
ферментативной реакции. Однако на практике рН-зависимость
ферментативных реакций, протекающих по трехстадийному механизму,
имеет более простой вид. Как правило, ряд протонированных или
депротонированных состояний фермента или не обладают
способностью связывать субстрат, или не вступают в реакции ацилирова-
ния или деацилирования, что приводит к упрощениям
соответствующих схем рН-зависимости. Например, в реакциях гидролиза
специфических субстратов, катализируемых а-химотрипсином и
трипсином, схема (10.11) упрощается:
ЕН,
EH + S
*"s
EHjS
EHS
ЕН2А
ЕНА
ЕН + Р (10.12)
В этом случае уравнение скорости реакции имеет вид
k2 -[EMS].
•v =
к
1+ Ка +
Кь
[Н+]
(1+ к'а ) + *.( К )
■+[S].
(10.13)
и эффективные индивидуальные константы ферментативной
реакции, зависящие от рН tk2, k3 и A*s), определяются следующими
выражениями:
7, - k*
1 +
Ш1
"<
222
1 +
/Cs = /Cs-
1 +
[H+]
«a
[Н+]
1 +
(10.14)
/Си
[Н+]
[Н+
Из уравнения (10.13) видно, что рН-зависимость скорости
ферментативной реакции, протекающей по трехстадийной схеме, в общем
случае будет различной в зависимости от соотношения констант
скоростей стадий ацилирования и деацилирования (k2/k3). С
другой стороны, рН-зависимость константы скорости второго порядка
&кат/Кт(каж), как и для двухстадийной схемы (10.1), определяется
только константами диссоциации ионогенных групп активного
центра свободного фермента (Ка и Кь), контролирующих
дальнейшее превращение фермент-субстратного комплекса.
§ 3. Ионизация субстрата
В тех случаях, когда и фермент, и субстрат имеют в своем
составе ионогенные группы, протонирование или депротонирование
которых влияет на скорость ферментативной реакции, обработка
соответствующих схем рН-зависимости существенно усложняется.
Схема (10.15) иллюстрирует двухстадийную ферментативную
реакцию, в которой активный центр фермента имеет две ионогенные
группы (с константами диссоциации Ка и Кь в свободной форме
фермента), и две формы субстрата находятся в прототропном
равновесии друг с другом (с константой диссоциации Кс)
EH,SH=:
ESH
EH..S
К dK £f
S ^- » EHS < ^ ES
(10.15)
EH + P
Если из трех ионизационных состояний фермента лишь одно
является активным, и обе формы субстрата являются реакционноспо-
223
собными (схема 10.15), скорость ферментативной реакции
определяется обычным выражением
[E].IS].
•V
К
т (каж)
+ [S].
где
k2 +
k
"•кат
*2h*s [H+]
1 +
[Н+]
к'
+
[н+] /
+
Ks [Н+]
1 +
JH+]
+N-
«
[Н+]
/С:
m (каж)
= Ks
O + ^ + T&X' + T)
V /С; ' [Н+]/+ ^sh^c I
1 +
[Н+]
к:
■+
[Н+]/
Более часто встречается случай, когда из двух форм субстрата
лишь одна связывается с ферментом или вступает в
ферментативную реакцию, приводя к продукту. Например, если в
ферментативной системе, описываемой схемой (10.16), субстрат претерпевает
ионизацию, причем из двух форм субстрата — нейтральной и
отрицательно заряженной — лишь нейтральная форма связывается с
ферментом, то скорость ферментативной реакции будет
определяться выражением (10.18)
EH2+S
EH2S
EH + S
E + S
к*
4-
EHS
t
«b
■ EH + P
(10.16)
ES
:S- + h+,
i +
[H+] Kb t£b[S]0
V = ■
Ka + [H+]
/Cs(l + -THT]-j + [S]„
(10.17)
(10.18)
Вывод выражения (10.18) отличается от вывода (10.7) лишь
введением уравнения материального баланса по субстрату, [S] о=
= [S] + [S-]. Из сравнения выражений (10.7) и (10.18) очевидно,
что ионизация субстрата приводит к зависимости значения Кт(иат)
от рН, анализ которой позволяет найти константу диссоциации
субстрата Кс-
224
В том случае, если субстрат имеет две ступени ионизации
кс к'с
S-Z— S S+,
-н+ -н+
выражения для &Кат и Лт(каж) в уравнении скорости
ферментативной реакции, протекающей по схеме (10.16), будут иметь вид
*^ If Я Т
1 + Ка + [Н+]
(,. [Н+] Кб \Л , Kg [Н+]\
v ^ ' "^ WM1+W+"*r)
А ш (каж) -"As ЩЦ —^~
1 ' "Ж" ^ Тн+Г
§ 4. Нахождение значений рК по кривым
рН-зависимостей ферментативных
реакций
Нахождение значений рК ионогенных групп фермента (или
субстрата), контролирующих скорость реакции, по профилям
зависимости кинетических параметров ферментативной реакции от рН
производится по тем же правилам, что и при обработке рН-зави-
еимостей неферментативных реакций (см. гл. 3). Например,
логарифмируя левую и правую части выражения (10.14), получим
~ /' [Н+]
log k2 = log k2 — log 1 + -^7-
Очевидно, что при [Н+] <£ Ка значение log k2 не зависит от рН
и равно логарифму константы скорости ацилирования. Если же
[Н+] » К'а, то
log %2 = log k2 — рКа + рН,
т. е. логарифм эффективной константы скорости ацилирования
изменяется пропорционально рН. На основании подобных
представлений можно легко показать, что графики зависимостей
логарифмов кинетических параметров ферментативной реакции от рН
должны иметь вид прямолинейных отрезков, соединенных
плавными переходными участками (см. гл. 3). Экстраполяция линейных
участков до пересечения их друг с другом дает величины рК
ионогенных групп, контролирующих изменения данных кинетических
(или равновесных) параметров ферментативной реакции.
Основным способом проверки и уточнения найденных значений
рК (в случаях кривых рН-зависимости любой формы) является
8 Заказ 158
225
проведение теоретической кривой рН-зависимости и коррекция ее
вплоть до оптимального согласования с результатами
эксперимента.
Задачи
10-1. Гидролиз специфических сложноэфирных субстратов,
катализируемый а-химотрипсином, протекает при участии имидазоль-
ной группы остатка гистидина активного центра фермента,
выступающей в качестве общекислотно-общеосновного катализатора.
На основании данных табл. 1 определить величину рКа этой
группы [1].
Таблица 1
рН-Зависимость каталитической константы
скорости гидролиза этилового эфира
Г\1-ацетил-Ь-тирозина, катализируемого
а-химотрипсином. Условия опыта: 25° С;
ионная сила 0,1М (NaCl);
[Е]0 = (0,17-2,2) ■ 10-7М
рн
6,5
7,0
7,5
7,8
8,0
8,5
9,0
Лкат> сек '
48
95
120
137
139
148
150
10-2. В таблице 2 приведена зависимость константы скорости
деацилирования транс-циннамоил-химотрипсина от рН [2]. Опре-
Таблица 2
рН-Зависимость деацилирования
транс-циннамоил-химотрипсина. Условия опыта:
25° С; 1 % ацетонитрила
рН
5,91
6,48
6,82
7,39
7,94
8,06
6,49
9,54
10,95
11,80
Лз-Ю3, сек-*
0,73
2,02
3,65
7,82
11,1
11,2
12,5
12,4
12,4
12,4
226
делить величину рК ионогеннои группы активного центра
фермента, контролирующей реакцию деацилирования.
10-3. В таблице 3 приведена рН-зависимость кинетических
параметров гидролиза n-нитрофенилацетата, катализируемого
бактериальной протеазой Е3оШ [3]. Определить значение рК ионогеннои
группы активного центра свободной формы фермента.
Таблица 3
рН-Зависимость гидролиза я-нитрофенилацетата,
катализируемого бактериальной протеазой.
Условия опыта: 25° С; 1Е]0 = 1,52-10-6М;
[S]0= (0,32 —8,0)-10-4М
рн
5,92
6,47
6,82
7,01
7,42
7,74
8,00
9,04
*кат-Ю», сек-1
1,99
5,49
8,23
11,97
18,80
18,80
21,90
21,90
^ш(каж)-101. М
7,14
8,33
8,33
9,С9
9,С9
7,69
7,69
7,14
10-4. В таблице 4 приведена рН-зависимость гидролиза семи-
карбазида N-фopмил-L-фeнилaлaнlинa, катализируемого
а-химотрипсином [4]. Определить значения рК ионогенных групп актив-
Таблица 4
рН-Зависимость кинетических параметров гидролиза
семикарбазида N-формил-Ь-фенилаланина, катализируемого
а-химотрипсином. Условия опыта: 25° С; ионная сила
0,1 М (КС1)
рн
5,47
5,95
6,47
6,98
7,48
7,79
*кат-10», сек-.
0,32
0,96
1,92
2,44
3,06
2,88
*т(кажГ103' М
7,00
6,70
5,60
3,45
2,24
2,28
ного центра свободной формы фермента и фермент-субстратного
комплекса, и предложить схему рН-зависимости реакции.
10-5. В работе [5] изучали кинетику гидролиза этилового
эфира 1\[-ацетил-1--тр!иптофана, катализируемого а-химотрипсином, в
8*
227
окиси дейтерия. Используя данные табл. 5, рассчитать значение
рК иопогенной группы активного центра, контролирующей скорость
ферментативной реакции.
Таблица 5
Влияние D20 на реакцию гидролиза этилового
эфира 1Ч-ацетил-Ь-триг!тофана, катализируемого
а-химотрипсином. Условия опыта: 25° С; 0,81 %
ацетонитрила, [SJ0 = (0,23—1,55)-103М;
[Е]0= Ю-5—10"7М
pD
6,29
7,05
7,45
8,19
8,74
9,25
9,90
10,57
"кат' «К"1
1,59
5,50
8,76
14,14
13,45
13,5о
14,СО
13,90
10-6. В таблице 6 приведена рН-зависимость гидролиза фор-
милгидразида М-формил-Ь-фенилаланина, катализируемого
а-химотрипсином [4]. Определить значения рК ионогенных групп
свободной формы фермента и фермент-субстратного комплекса,
контролирующих реакцию, и предложить схему рН-зависимости
ферментативного гидролиза.
Таблица 6
Влияние рН на кинетические параметры гидролиза
формилгидразида 1Ч-формил-Ь-фенилаланина, катализируемого
а-химотрипсином. Условия опыта: 25° С; ионная сила
0.1М (КС1)
рН
4,48
4,74
4,98
5,24
5,47
5,98
6,48
6,73
6,98
7,47
7,78
*кат, сск-*
0,0097
0,022
0,029
0,048
0,065
0,143
0,256
0,306
0.3С6
0,348
0,362
*т(каж)-№. «
22
24
12,5
11,6
8,4
7,3
6,3
4,46
3,3
2,7
2,6
228
10-7. В таблице 7 приведена рН-зависимость каталитической
константы скорости гидролиза бензилового эфира N-бензилокси-
карбоиил-Ь-лизина, катализируемого папаином [6]. Определить
рК ионогенной группы фермент-субстратного комплекса,
контролирующей скорость реакции.
Таблица 7
рН-Зависимость гидролиза бензилового эфира К-бензилоксикарбонил-Ь-лизина,
катализируемого папаином. Условия опыта: 25° С; ионная сила 0,2М (буферные
растворы); 1,6% ацетонитрила; [Е|0 = (1,4— 14,6)-10~7M, [S]0 = 1,3910-3М
(значения йкат были найдены из анализа полной кинетической кривой)
рн
3,15
3,42
3,47
3,70
3,77
3,96
4,01
4,57
*кат' «к-1
11,9
21,7
22,2
27,4
29,3
33,2
36,9
37,2
рн
4,60
5,32
5,51
5,58
6,00
6,14
6,92
*кат- сек~'
40,9
40,1
43,5
44,0
43,2
44,4
44,5
10-8/ Скорость необратимого ингибирования папаина L-1-хлор-
3-тозиламидо-4-фенил-2-бутаноном зависит от рН среды [6]. На
основании данных табл. 8 определить рК ионогенной группы,
контролирующей инактивацию фермента, а также найти истинное
значение константы скорости инактивации.
Таблица 8
рН-Завнсимость константы скорости инактивации
папаина хлоркетоном. Условия опыта: 25° С;
ионная сила 0,1М (буферные растворы); 3,2%
ацетонитрила; [EJ0=2,9-10-6M
рн
6,56
7,05
7,55
8,08
8,30
8,61
9,02
9,60
k , М-'-сек—1
инакт'
20,0
67,9
176
448
554
755
981
1020
10-9. рН-Зависимость гидролиза n-нитроанилида Ы-бензоил-Ь-
аргинина, катализируемого папаином, имеет вид колоколообразной
229
кривой [7]. Исходя из данных табл. 9, вычислить значения рК
ионогенных групп свободного фермента, принимающих участие в
реакции.
Таблица 9
Влияние рН иа гидролиз гг-нитроанилида
1Ч-бензоил-Ь-аргинина, катализируемый папаином.
Условия опыта: 25° С; ионная сила 0.3JW;
[SJo = (0,40 — 2,00)- 10-3М;
[EJ0 = (4,13 —8,26)-10-7М
рн
3,98
4,50
4,99
5,49
5,96
6,49
7,01
7,48
7,65
8,28
8,51
8,82
8,91
9,25
*кат- сек~1
0,29
0,47
0,72
0,76
0,74
0,73
0,69
0,63
0,63
0,53
0,45
0,40
0,34
0,21
КтЫжУ^.Ы
4,07
3,23
3,29
3,17
2,86
2,95
3,00
3,18
3,59
5,06
4,39
5,47
5,76
6,06
10-10. В таблице 10 приведена рН-зависимость гидролиза
амида Ы-ацетил-Ь-фенилаланина, катализируемого а-химотрипсином
[5]. Найти значения рК ионогенных групп фермент-субстратного
комплекса, контролирующих ферментативную реакцию.
Таблица 10
Влияние рН на кинетику гидролиза амида
1М-ацетил-Ь-фенилалаиина, катализируемого
а-химотрипсином. Условия опыта: 25° С;
[S]0= (0,35—3,5) • 10"2М; [Е]0= (2,0—5,0) • 10~5М
рн
5,71
6,34
6,87
7,62
7,92
8,49
8,81
9,05
9,08
9,18
9,35
9,95
0,61
1,4
3,6
4,3
4,6
3,9
3,9
2,8
2,4
3,0
2,6
1,2
230
10-11. В таблице 11 приведена рН-зависимость максимальной
скорости гидролиза п-нитрофенилового эфира н-масляной кислоты,
катализируемого кишечной эстеразой [8]. Определить значения
рК ионогенных групп фермент-субстратного комплекса,
контролирующих скорость реакции.
Таблица 11
Влияние рН на максимальную скорость
гидролиза гс-нитрофенилбутирата, катализируемого
кишечной эстеразой
рн
4,5
5,2
5,5
6,0
6,5
7,0
7,3
7,5
8,0
8,5
9,0
V .10е. моль
ul" пин мг белка
24,2
73,3
126,5
166,2
235,6
281,9
257,2
230,1
160,6
61,3
21,6
10-12. В таблице 12 приведена рН-зависимость гидролиза
амида Ы-ацетил-Ь-триптофана, катализируемого а-химотрипсином [5].
Определить значения рК групп фермент-субстратного комплекса,
принимающих участие в катализе.
Таблица 12
Влияние рН на гидролиз амида
1М-ацетил-Ь-триптофана, катализируемого
а-химотрипсином. Условия опыта: 25е С;
[S]0 = (0,71- 12,7) -10-3М;
[EJ„= (1,0-2,5) -10-4М
рН
5,73
6,72
7,09
7,73
8,00
8,66
9,17
9,70
*кат.10>,сек-1
0,249
1,44
2,70
4,10
4,37
3,40
1,42
0,974
10-13. В таблице 13 приведена рН-зависимость кинетических
параметров гидролиза этилового эфира Ы-бензоил-Ь-аргинина, ка-
231
тализируемого клострипаином [9]. Определить величины рК ионо-
генных групп активного центра свободного фермента и фермент-
субстратного комплекса и предложить схему рН-зависимости
реакции.
Таблица 13
Влияние рН на гидролиз этилового эфира
]^-бензоил-Ь-аргинина, катализируемый клострипаином. Условия
опыта: 25° С; ионная сила 0,1М (КО);
[EJ0=(0,6—1,2)-10-7M; [Slo = (0,10—0,75)10-3М
рн
5,50
6,00
6,88
7,37
7,89
8,50
8,95
9,39
*кат- сек-1
83
79
89
90
80
47
15,7
6,6
^т(каж)-103'М
4,80
0,94
0,39
0,41
0,40
0,42
0,35
0,31
10-14. В таблице 14 приведена рН-зависимость константы
скорости реакции второго порядка ацилирования а-химотрипсина
п-нитрофенилацетатом [5].
Вычислить значения рК групп активного центра свободного
фермента, контролирующих скорость реакции.
Таблица 14
рН-Зависимость ацилирования а-химотрипсина п-нитрофенилацетатом.
Условия опыта: 25° С; 1,6% ацетонитрила
рн
4,18
5,08
5,38
5,91
6,47
6,58
6,74
6,83
6,97
7,09
ЫА'5 ' М—ьсек-i
14,6
78,5
186
487
1120
1350
1970
1850
2350
2490
рн
7,21
7,46
7,66
7,83
8,13
8,71
8,73
9,21
9,38
9,98
1г?1К$ , М—1-сек—1
2850
3190
3000
3410
3330
2660
1850
1810
1420
670
10-15. В таблице 15 приведена рН-зависимость гидролиза фе-
нилового эфира а-]М-бензилоксикарбонил-Ь-лизина, катализируемо-
232
го стрептококковой протеиназой [10]. Определить значения рК
ионогенных групп свободной формы фермента и
фермент-субстратного комплекса, контролирующих реакцию гидролиза.
Таблица 15
рН-Зависимость гидролиза фенилового эфира
а-]Ч-бензилоксикарбонил-Ь-лизина, катализируемого
стрептококковой протеиназой. Условия опыта: 25е С; ионная
сила 0,2 (буферные растворы); 3,17% ацетонитрила;
[Sl0= (0,293— 1,17)- 10-3М; [Е]0 = (0,641 — 1,80)- 10~8М
рн
3,42
3,71
4,04
4,33
4,55
4,74
5,00
5,53
6,21
6,73
6,99
7,25
*кат> сек'
45,4
60,8
94,1
114,5
142.6
150,5
163,2
176,1
182,4
178,0
189,5
173,7
К с \-10*. М
т \каж) •
15,20
11,76
12,38
9,49
8,33
8,02
7,01
6,67
6,64
6,05
6,37
5,88
10-16. В таблице 16 приведена рН-зависимость гидролиза
n-нитрофенил'тиолацетата, катализируемого а-химотрипсином [11].
Определить значения рК ионогенных групп свободной формы
фермента, принимающих участие в реакции.
Таблица 16
рН-Зависимость гидролиза
n-нитрофенилтиолацетата, катализируемого
а-химотрипсином. Условия опыта: 25е С; ионная
сила 0,005М (буферные растворы);
1,6% ацетонитрила; [S]0 = 2,60-10-5М;
[Е1о = 5,99-10-5М
рН
4,47
4,93
5,43
5,80
6,48
7,15
7,41
7,52
7,69
8,20
8,64
8,98
9,65
As/#S' М—ьсек—«
40
117
278
560
1360
2500
2340
2670
2970
3000
2570
2080
1140
233
10-17. В таблице 17 приведена рН-зависимость гидролиза
амида Ы-ацетил-Ь-фенилаланил-Ь-тирозина, катализируемого пепсином
[12]. Определить значения рК групп активного центра свободной
формы фермента, принимающих участие в реакции.
Таблица 17
рН-Зависимость гидролиза амида
К-ацетил-Ь-фенилаланил-Ь-тирозина, катализируемого
пепсином. Условия опыта: 35° С; 3% метаиола;
[SJ0 = (0,85 — 8,49)-10-4М; [Е]0 = (1,39 — 2,21)- 10-5М
рн
0,50
0,70
1,11
1,50
1,95
2,50
3,15
4,20
4,68
4,70
4,97
5,43
5,44
fcKaT-10', сек-1
0,99
1,64
3,48
5,05
7,80
5,59
7,02
4,70
1,82
2,07
3,59
0,76
0,24
*т(каж)'103-М
1,67
2,28
2,34
2,36
2,41
1,88
2,33
2,54
1,87
1,68
4,47
1,94
0,72
10-18. В таблице 18 приведена рН-завнсимость гидролиза фе-
нилового эфира Ы-бензилоксикарбонил-Ь-аланина,
катализируемого папаином [10]. Определить значения рК ионогенных групп
свободной формы фермента, контролирующих реакцию.
Таблица 18
Влияние рН на гидролиз фенилового эфира 1^-бензилоксикарбонил-Ь-аланина,
катализируемый папаином. Условия опыта: 25° С; ионная сила 0,2М;
3,17% ацетонитрила; [S]0 = 1,4310-5М; [Е]0 = 7,43-10-8М
рн
3,42
3,71
4,04
4,33
4,55
4,74
5,00
5,39
5,53
—— -ю6,
^т (каж)
М— 1-сек—1
1,33
3,10
5,35
7,34
8,09
8,90
9,12
9,73
9,65
рН
6,21
6,48
6,73
6,99
7,40
7,73
8,00
8,19
8,65
кат .10я
^т (каж)
М—1-сек—1
9,15
9,64
9,53
9,65
8,74
7,52
5,69
4,15
2,20
234
10-19. Гидролиз метилового эфира Ы-бензоил-Ь-аланииа,
катализируемый трипсином, протекает по трехстадийному механизму,
причем для этой реакции скорость-лимитирующей стадией
является ацилирование. На основании данных рН-зависимости реакции
гидролиза (табл. 19) вычислить значения рК ионогенных групп
свободного фермента и кинетически существенных промежуточных
соединений фермента и предложить схему рН-зависимости
реакции.
Таблица 19
рН-Зависимость кинетических параметров гидролиза
метилового эфира N-бензоил-Ь-аланина, катализируемого
трипсином [3J. Условия опыта: 25° С; иониая сила
0,Ш (СаС12 и NaCl); [SJ0 = (0,167 — 4,33)- 10~2М
рн
6,5
7,0
7,5
7,8
8,0
8,5
9,0
9,2
9,5
9,7
10,0
Ь — 1
"кат, сек
0,109
0,185
0,232
0,248
0,265
0,248
0,184
0,168
0,122
0,105
0,070
^т(каж), М
0,243
0,171
0,129
0,112
0,111
0,098
0,078
0,077
0,052
0,047
0,035
10-20. В таблице 20 приведена рН-зависимость гидролиза п-пи-
трофенилацетата, катализируемого папаином [13]. Определить
Таблица 20
Влияние рН на гидролиз гс-нитрофенилацетата,
катализируемый папаином. Условия опыта: 25е С;
ионная сила 0,Ш (буферные растворы);
ISb = (2,0— 10,0)- 10-2М; [EJ0 = 5,Ы0-6М
рн
4,35
5,12
5,50
5,90
6,00
6,35
7,00
7,30
7,90
8,00
8,15
кат'"т (каж)'
М-'-сек-*
1,41
4,16
4,69
6,45
6,50
6,74
6,62
6,21
4,10
3,05
3,11
235
значения рК ионогенных групп активного центра свободной формы
фермента, контролирующих реакцию гидролиза.
10-21. Профиль рН-зависимости кинетических параметров
гидролиза Ы-ацетил-Ь-фенилаланил-Ь-триптофана, катализируемого
пепсином, имеет колоколообразную форму, причем левая ветвь рН-
зависимости обусловлена протонированием карбоксильной группы
активного центра фермента, а правая — депротонированием
карбоксильной группы субстрата [12]. На основании данных табл. 21
Таблица 21
рН-Зависимость гидролиза
М-ацетил-Ь-фенилаланил-Ь-триптофана, катализируемого
пепсином. Условия опыта: 35° С; 3% метанола
рН
0,30
0,50
0,70
1,11
1,50
1,89
1,95
1,96
2,50
2,94
3,01
3,55
3,56
3,73
4,06
4,20
Йкат-1С5, сек-1
1,18
1,35
2,64
2,11
5,24
6,38
5,70
5,24
5,05
4,51
6,14
3,64
3,06
4,03
2,24
3,82
Кт (каж)-Ю', М
18,1
10,9
17,1
9,3
10,1
9,6
8,2
7,0
6,6
7,6
12,3
13,1
10,3
17,1
24,1
53,9
Таблица 22
рН-Зависимость гидролиза
К-трифторацетил-Ь-фенилаланина, катализируемого пепсином.
Условия опыта: 34° С; ионная сила 0,1М;
[Slo = (0,75 — 4,0)- 10-2М; [EJo = 510-«M
рН
1,70
2,25
2,80
3,05
3,35
3,60
4,05
4,50
4,65
5,40
*кат-Ю3, мин-'
2,4
6,0
14,3
26,8
34,1
31,7
21,7
15,8
13,1
5,5
^т(каж)-103. М
6,6
5,9
8,0
14,0
16,6
15,5
21,6
28,6
29,0
81,8
предложить схему рН-зависимости и вычислить значения рК ионо-
генкых групп свободного фермента и субстрата, контролирующих
ферментативную реакцию.
10-22. Реакция гидролиза М-трифторацетил-Ь-фенилаланина,
катализируемая пепсином, происходит только в том случае, если
карбоксильная группа активного центра фермента является прото-
нированной, а карбоксильная группа субстрата — депротонирован-
ной [14]. Исходя из данных рН-зависимости ферментативной
реакции (табл. 22), вычислить значения рК ионогенных групп
субстрата и фермента, принимающих участие в реакции.
10-23. В работе [15] была предложена следующая схема инги-
бирования а-химотрипсина 2-фенилэтанборной кислотой:
ЕН2 + I Z^- EI I, I
t
EH + I ;ZZ2 EH1 (10.19)
t I t
In il /
«\ \ I кь
'E4-I El
. ч
Были найдены значения pKa=G,4; рКь=83; рК'а<4,8; рК'ъ>
>10,3.
1) Найти выражение для эффективной константы ингибирова-
ния фермента.
2) Определить форму рН-зависимости эффективной константы
ингибирования.
3) Найти, при каком значении рН ингибирование
ферментативной активности будет максимальным, если изучение
ингибирования проводится в интервале рН 5,0—10,0 и активной формой
фермента является ЕН.
10-24. В работе [16] предложена следующая схема
ингибирования карбоангидразы цианид-анионами:
*i
ЕН+ + CN- 7Ш EHCN
it it
*е| +н+ *а | |-н+ (10.20)
Е HCN
Согласно этой схемы анион ингибитора (р/(а=9,21) связывается с
активным центром фермента, имеющим положительно заряженную
группу (р/Се=6,5).
1) Найти выражение для эффективной константы
ингибирования, зависящей от рН.
2) Определить форму рН-зависимости эффективной константы
ингибирования.
237
3) Найти, при каком значении рН эффективная константа ин-
гибирования будет иметь оптимальное значение.
4) Вычислить оптимальное значение эффективной константы ин-
гибирования, если истинное значение константы ингибирования
(схема 10.20) равно 3,55-10~8 М.
Ответы и решения
10-1. Экспериментальные данные рН-зависимости (табл. 1)
согласуются с величиной р/Со=6,85 и предельным значением
каталитической константы (&кат)ит= 152 сек-1 (рис. 105).
10-2. Решается аналогично задачам 3-2—3-4, или 10-1
(рис. 106).
Ответ: рКи = 7,15.
140
120
Т*100
QJ
и-80
ъ*60
W
о/
I ! I I I
6,5 7,0 7,5 8,0 8,5 9,0 рН
12.0
8,0-
че: 4,0
~(*3)[im
2
^~'~J
- J*
' 1
1
'p*w
1 a
6.0
7,0 8J0 9,0 pH
Рис. 105. рН-Зависимость
каталитической константы скорости
гидролиза этилового эфира N-ацетил-
L-тирозина, катализируемого о>
химотрипсином; кривая является
теоретической с р/и=6,85
Рис. 106. Определение значения рКа
ионогенной группы активного центра
фермента из кривой рН-зависимости
деацилирования транс-циннамоил-
химотрипсина
10-3. Для определения рК ионогенной группы, относящейся к
свободной форме фермента, следует анализировать график в
координатах (йкат/ТСтпскаж), рН). Обработка экспериментальных
данных рН-зависимости производится аналогично решениям задач
10-1, 10-2.
Ответ: рКа=7Л-
10-4. Обрабатывая данные рН-зависимости обычным способом,
находим значения рК ионогенных групп, равные 7,1 и 6,4 для
свободного фермента и фермент-субстратного комплекса
соответственно. Так как величины рК, для этих двух форм фермента различны,
238
то экспериментальные данные можно описать следующей
простейшей схемой рН-зависимости
ЕН2 + S * ЕН2 S
а ■ .
EH + S - EHS -t EH + P. (10.21)
Из анализа схемы (10.21) можно получить следующие выражения
для эффективных кинетических и равновесных параметров:
' ft -- "
'^
*«(»«,-*S1 + [H+]//c7.
Ответ: рКа=7,1\ рД"„=6,4; /г2=0,032 сек-1; k2IKs=l6A
М-1 -сек-1; Ks= 1,95- Ю-3 М.
10-5. Решается аналогично задаче 10-2.
Ответ: рКа—72-
10-6. Решается аналогично задаче 10-4.
Ответ: схема рН-зависимости идентична схеме (10.21); рКа=
= 7,0; рД"„=6,1.
10-7. Решается аналогично задаче 10-2.
Ответ: рКа=3,5.
10-8. Решается аналогично задаче 10-2.
Ответ: рКа=8,3; £инакт= 1110 М-1 • сек-'.
10-9. Данные табл. 9 можно описать следующим выражением
рН-зависимости константы скорости второго порядка
ферментативной реакции
"кат -' S
1+^^- +
Ка ^ [Н+]
где Ка и Кь — константы диссоциации ионогенных групп
свободного фермента, контролирующих реакцию.
Для определения значений Ка и Кь можно применить обычный
Метод обработки колоколообразных кривых рН-зависимости —
сначала построить профиль рН-зависимости в логарифмических
координатах (log ^кат/Д'т(каж), рН) и оценить приближенно значения
рКа, рКь и k2IKs (рис. 107), а затем провести коррекцию
найденных значений констант до оптимального согласования теоретиче-
239
ски рассчитанной кривой с экспериментальными результатами
(рис. 108).
Ответ: р/(„=4,3; р/Сь = 8,2.
10-10. Решается аналогично задаче 10-9.
Ответ: p#a=6,55; рКъ=9,23; (£Кат)пт=4,76-10~2 сек"1.
10-11. Решается аналогично задаче 10-9.
Ответ: рКа=5,8; рКь=8,0; (Vmax)iim=3,08- Ю~* моль/мин-мг
белка.
10-12. Решается аналогично задаче 10-9.
Ответ: рКа=7,16\ рКь = &,90; (&кат)ш„=5,08-10-2 сек-'.
6,0 7,0 6,0 9,0
РН
Рис. 107. Приблизительная оценка
значений констант диссоциации ноно-
генпых групп активного центра па-
папна в реакции гидролиза п-нитро-
анилнда 1\]-бензоил-Ь-арпшина
40 50 6,0 7,0 8,0 9,0
рН
Рнс. 108. Иллюстрация подбора
теоретической кривой рН-зависн-
мости, списывающей
экспериментальные данные по влиянию рН
на гидролиз, катализируемый па-
паином. Пунктирная кривая
рассчитана с использованием
значений рКа=4,5; р/Сь=8,4; k2/Ks =
=251 М-1-сек-1 (найденных из
рис. 107). Сплошная кривая
рассчитана с использованием
значений р/Со=4,3; рКь=8,2; kdKs =
= 251 М-1-сек-1
10-13. Значения констант ионизации можно определить
построением экспериментальных данных в логарифмических
координатах (log &кат//Ст(каяф рН); (log£KaT, рН) и проведением к ветвям
рН-зависимости касательных с угловыми коэффициентами,
равными + 1,0 и —1. Проекция точек пересечений касательных на ось
абсцисс даст значения рКа, рКь и рК'ъ (рис. 109). Форма кривых
рН-зависимости согласуется со следующими выражениями для
240
кинетических параметров реакции:
1 +
_Кь_
[Н+!
Km (каж) = A's-
"ка
+ Kg + [H+]
К
т(каж)
1 +
Кь
[Н+]
1 + JHiI+._^
Ка ^ [Н+]
(10.22)
(10.23)
(10.24)
Выражениям (10.22) - (10.24) соответствует следующая схема
рН-зависимости:
ЕН,
E + S
>
<—■ ■
<
EHS
I +
- EH + Р
ES
•<ь
Здесь рКа=Ъ,40-г р/(ь=8,35; рК'ь=8,2Б (см. рис. 109).
10-14. Решается аналогично задаче 10-9 (рис. ПО).
Ответ: рКа=6М; pKb = 9,04; (As//Cs)iim=3940 М-1-сек-1.
10-15. Решается аналогично задаче 10-4.
Ответ: /?/С«=4,46; (*иа1/^вкваяо)вт=3- Ю5 М-1-сек-1; рК'а=
=4,13; (^KaT)iim=192ceK-1.
10-16. Решается аналогично задаче 10-9.
Ответ: р/Са=6,89; рКь=9,01; (*2//Cs)ihii=3350 М-'-сек-1.
10-17. Решается аналогично задаче 10-9.
Ответ: рКа=1,17; рКь=4,ЗБ; (А;кат//Ст{Каж))пт=32,5 М-'-сек-1.
10-18. Решается аналогично задаче 10-9.
Ответ: рКа=3,56: рКь = 8,07; (kKa^/Km^m)hm=l07 М-1-сек-1.
10-19. Обрабатывая экспериментальные данные (табл. 19)
обычным способом, находим, что величина #кат зависит от
состояния двух ионогенных групп фермент-субстратного промежуточного
соединения с рК'а—€>,7 и рК'ь=:9,5 и &кат/^(т(каж) зависит от
состояния одной ионогенной группы свободного фермента с рКа=7,1.
Для данной ферментативной реакции стадией, лимитирующей
скорость гидролиза, является ацилирование (&2<§;&з), поэтому фор-
241
мально-кинетически реакцию можно рассматривать как двухста-
дийнук,и , и
£кат = ~ ттг~. (10.25)
И~^ + *'
Ка [Н+]
kJKs
К,
т(каж)
Л'т(каж) = AS
1 +
1 +
[Н+] '
Ка
[Н+]
Ка
1Н+] К'ь
к
[Н+]
(10.26)
(10.27)
3000
2000
v
Рис. 109. Определение констант
ионизации ионогепных групп активного
центра клострипаина,
контролирующих реакцию гидролиза
Выражениям (10.25) — (10.27)
рН-зависимости ферментативной
1000
4fl 5,0 6.0 7,0 8,0 9,0 10,0
РН
Рис. ПО. Профиль рН-зависимости аци-
лирования а-химотрипсина п-нитрофе-
нилацетатом; кривая является
теоретической для /t?/Ca=6,85; ^6=9,04;
(fe2/Ks)iim=3940 М-1-сек-1
соответствует следующая схема
реакции:
EH2S
*s
EH + S
4
EHS
^E + P
(10.28)
Kb
ES
242
Схема (10.28) может быть интерпретирована таким образом, что
субстрат при связывании индуцирует каталитически неактивное
конформационное состояние фермента, концентрация которого
регулируется ионогенной группой с константой диссоциации К'ъ-
10-20. Решается аналогично задаче 10-9.
Ответ: рКа=Ь,0; рКъ=&,0.
10-21. Согласно условию задачи, схема рН-зависимости
реакции должна включать два ионных состояния фермента —протони-
рованное (неактивное) и депротонированное (активное) и два
состояния субстрата — нейтральное (активное) и отрицательно
заряженное (неактивное). Условию задачи могут удовлетворять два
основных варианта реакции: (а) анион субстрата образует
нереакционный фермент-субстратный комплекс и (б) анион субстрата не
связывается с ферментом. Рассмотрим сначала более общий
случай (а)
EHSH
(10.29)
Е + Р
Кинетическая обработка схемы (10.29) проводится обычным
способом. Запишем уравнение скорости реакции, уравнения
материального баланса по ферменту и субстрату (принимая [Е]0<С
•С [S] о) и выражения для констант равновесия
(10.30)
(10.31)
(10.32)
(10.33)
v = k2[ESH],
[Е]0 = [Е] + [EH] + [ESH] + [EHSH] + [ES"
[S]0 = [S-] + [SH],
[Е] [SH] . „ [Е] [S-]
+ [EHS-],
Ks
Кп
К
[ESH]
[Е][Н+].
[ЕН] '
[ES-] [Н+]
[EHS~]
Ks--
а-: =
[ES-] '
[ESH] [H+]
[EHSH]
Кс =
[S-] [н+]
[SH]
(10.34)
(10.35)
Комбинируя выражения (10.31) — (10.35) друг с другом и
подставляя их в (10.30), получим уравнение скорости реакции в обычной
форме
^кат l^Jo [bjo
v--
К
m (каж)
+ [SJ«
243
где
h + ^rJ+^TH^i1 - < J
Для бо lee частного случая (б) Ks- = oo, K"a = оо, так что в этом
случае
1 -ь-Чт^
, .,>^)0 + т&г) 1103И
Ат(каж) = А5 jprpi (10.39)
1+ L
Ка
Очевидно, что независимо от вида выражений (10.36) — (10.37)
или (10.38) — (10.39), отношение &кат и Кт(к&т) равно
«кат "2/A s
'<„,„„» ~ Л , ШЧЧЛ , *« Л ' (10-4°)
f—- 0+J5£L)0+i&r)'
так что график в координатах (Ькат/Кт(кьж), рН) для
рассматриваемой схемы (10.29) имеет вид колокола. Левая ветвь рН-зависи-
мости (10.40) (для которой [Н+] ^>КС) соответствует выражению
■*^"7ГШ" (10-41)
Соответственно для правой ветви рН-зазисимэсти (для которой
[H+]«?/Q
*кат *»/*S /ЩДОЧ
-77 = ^ . (10.42)
л/л(каж) 1 , Ас
1+ [НН]
Оценив значения /(а и Д'с с использованием выражений (10.41) и
(10.42) обычными методами обработки рН-завмсимостей сигмоид-
ной формы (см. гл. 3), необходимо провести последующую
коррекцию найденных значений констант ионизации в координатах
(ктак/Кщкаж), рН) до оптимального соответствия теоретически
вычисленной кривой рН-зависимости с экспериментальными данными
(рис. 111).
Ответ: pKa=l,4; рКс=3,2.
244
10-22. Условию задачи удовлетворяет следующая схема рН-за-
зисимости реакции:
ESH
(10.43)
EHSH
Проводя кинетическую обработку схемы (10.43) аналогично
схеме (10.29) предыдущей задачи, получим выражение для рН-за-
висимой константы скорости
второго порядка реакции
ферментативного гидролиза 80
Йкят ■»-
к,
т(каж)
ЫК<
(, + 1^)(1 + -!^)
(10.44)
[Н+]
Значения констант
диссоциации Кь и Кс ионогенных групп
фермента и субстрата можно
найти с помощью метода,
изложенного в решении предыдущей
задачи.
Ответ: рКь=3,7; рКс=2,&.
10-23. Из уравнения
материального баланса по ферменту
[Е]0 = [Е] + [ЕН] + [ЕН2] +
+ [EI] + [EHI] + [EH2I]
и выражений для констант
равновесия схемы (10.19) можно
найти концентрацию активной
формы фермента в отсутствие
[Е]„
[ЕН]
1 +
[н+]
+ ■
Кь
(10.45)
Рис. 111. рН-Зависимость гидролиза
М-ацетил-Ь-фенилаланил-Ь-триптофа-
на, катализируемого пепсином.
Кривая теоретическая и рассчитана на
основании значений рКа = 1,4 для
ионизации фермента и рКс = 3,2 для
ионизации субстрата
Ка т [Н+]
и в присутствии ингибитора
[ЕН](1) =
[Е]0
0 +
[н+1
Ка
-4-
[н
+] J + Ki
1+-
[Н+1
к:
[Н+]
(10.46)
94Л
По своему физическому смыслу константа ингибирования
численно равна концентрации ингибитора, при содержании которой в
растворе концентрация активного фермеита уменьшается вдвое, т. е.
[ЕН]=2 [ЕН](1) при [1]=/С,эФФ. В этом случае из выражений
(10.45), (10.46) следует выражение
V + Ка + [Н+] J " V1 + Ка + [Н+] J +
д-эфф / [Н+] К'
К\ [1+ Ка ' [Н+]
[Н+] К'„ ' '
1 + — +
Ка [Н + ]
которое определяет зависимость эффективной константы
ингибирования от концентрации водородных ионов. Из выражения (10.47)
очевидно, что форма рН-зависимости эффективной константы
ингибирования загисит от соотношения констант диссоциации
ионогенных групп фермента (например, если Ка = К'а и Кь = Кь,
то /Cf** = К\ = const во всем интервале рН). Так как из простых
термодинамических соображений следует, что К\ К'а = КаК\\
К'ЬК\ = КЬК\ (см. схему 10.19), и из условия задачи К'а^>Ка
и К'ь <С Кь, то легко получить для нашего случая К{^> К1 и
Ki">Ki- Это означает, что как протонирование, так и депротони-
рование активной формы фермента ЕН приводит к ухудшению
константы ингибирования фермента, поэтому зависимость
эффективной константы ингибирования от рН имеет вид перевернутого
колокола.
При изучении ингибирования фермента в интервале рН 5,0—
10,0 будут соблюдаться соотношения
-Щ-«1 и^«1,
ка [н+] ^
так что
tff*=^1 + J^+wr)- {Ю'48)
Для нахождения рН-оптимума реакции ингибирования (положение
минимума кривой в координатах (К\фф, рН)) следует
продифференцировать выражение (10.48) по [Н+] и, приравняв
производную нулю, вычислить концентрацию водородных ионов, при
которой эффективная константа ингибирования достигает своего
минимального значения
'*!** к [1 *s_J\.= o
246
откуда
или
рКа + РКь
Р^опт _
Если рКа=е>,4; р/С6=8,8, то рН0пт = 7,6.
10-24. Из уравнения (10.20) следует, что истинная константа
ингибирования определяется выражением
„ [EH+][CN~]
Al [EHCN] "
Для нахождения эффективной (рН-зависимой) константы
ингибирования запишем уравнения материального баланса по
ферменту и синильной кислоте (пренебрегая во втором случае расходом
синильной кислоты на образование фермент-ингибиторного
комплекса, т. е. принимая [1]о^> [Е]0):
[Е]0 = [Е] + [ЕН+] + [EHCN],
[HCNJ0 = [CN-] + [HCN],
или
[Е]0 = [ЕН+] (l + ^ + Ш), (10.49)
[HCN]0 = fCN-](l+-^ii]. (Ю.50)
Подставляя (10.50) в (10.49), получим
[Е].
ГЕН+] = к
, , - , [HCN]0
1 + гн+1 +
Kl( + ТГ
В отсутствие ингибитора
[ЕН+] = -
[Е]„
К
1 +
[Н+]
и концентрация синильной кислоты, уменьшающая вдвое
концентрацию активной формы фермента и численно равная эффективной
константе ингибирования (см. решение предыдущей задачи),
определяется выражением
[HCN]0 = *;«* -Кг (l + -^j (l + '^). (Ю.51)
Из выражения (10.51) видно, что эффективная константа
ингибирования ухудшается при значениях [Н+]</Се и [Н+]>/Са,
приводя к рН-зависимости типа «перевернутого колокола».
247
Продифференцировав выражение (10.51) по [Н+] и приравняв
первую производную нулю, найдем концентрацию водородных
ионов, при которой ингибирование является оптимальным (константа
ингибирования имеет минимальное значение)
или
[Н+]опт = 1/Л'еЛ'а, (10.52)
РКЕ + РКА
Когда /?/(е=6,5 и р/Са=9,21, рН0пт=7,85. Наконец, подставив
выражение (10.52) в (10.51), найдем формулу для оптимального
значения константы ингибирования (в рН-оптимуме реакции)
^'-«^+]/Ш
Подставив в полученную формулу значения Ке, Д'а и К\, найдем
К\т = 1,98-10~5 М.
ЛИТЕРАТУРА
I. Березин И. В., Казанская Н. Ф., Клёсов А. А. „Биохимия,,
36, 108, 1971.
2. Bender M. L., Schonbaum О. R., Zerner В. „J. Amer. Chem. Soc."
84, 2562, 1962.
3. Генов Н., Велчева П., КараджоваМ. „Известия на отделението
за химически науки, Българска академия на науките", 4, 637, 1971.
4. Fersht A. R., Requena Y. „J. Amer. Chem. Soc", 93, 7079, 1971.
5. BenderM. L., С I e m e n t G. E., К e z d у F. J., Heck H. d" A. J. Amer.
Chem. Soc", 86, 3680, 1964.
6. Bender M. L., Brubacher L. J. „J. Amer. Chem. Soc", 88. 5880, 1966.
7. Mole J. E., H or ton H. R. „Biochemistry", 12, 816, 1973.
8. Malhotra O. P., Philip O. „Biochim. Biophys. Acta", 309, 97, 1973.
9. Cole P. W., Murakami K., I n a g a m i T. „Biochemistry". 10, 4246, 1971.
10. Kortt'A. A., Liu T.-Y. „Biochemistry", 12, 338, 1973.
II. FrankfaterA., KezdyF. J. „J. Amer. Chem. Soc", 93, 4029, 1971.
12. Denburg J. L., Nelson R., Silver M. S. „J. Amer. Chem. Soc",
90, 479, 1968.
13. Williams A, Lucas E. C, R i m m e r A. R. „ J. Chem. Soc. Perkin II",
621, 1972.
14. Hunk a pill er M. W., R i с h a r ds J. H. „Biochemistry", 11, 2829,1972,
15. Koehler K. A., Li en hard О. Е. „Biochemistry", 10, 2477, 1971.
16. Taylor P. W., Bur gen A. S. V. „Biochemistry", 10, 3859, 1971.
Глава 11
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА КИНЕТИКУ
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
§ 1. Зависимость кинетических
и равновесных параметров
ферментативных реакций от температуры
Влияние температуры на реакции, катализируемые ферментами,
по существу не отличается от влияния температуры на любые
другие химические реакции. Поэтому методы обработки
температурных зависимостей кинетических и равновесных параметров
ферментативных реакций также основываются на классических принципах
термодинамики и кинетики (см. гл. 4).
Согласно закону Вант-Гоффа, влияние температуры на
константу равновесия химической реакции выражается уравнениями
din К АЯ m п
dT ~ RT2 • К11Л)
или
й In К АЯ
7Щ-"*-- 1"'2)
так что график зависимости в координатах (In К, 1/Т) имеет вид
прямой с тангенсом угла наклона, равным —AHjR. Зная величину
энтальпии АН равновесного процесса и свободную энергию
процесса AF при данной температуре, найденную из соотношения
AF= -RTlnK, (11.3)
можно найти величину энтропии процесса
Д^АЯ-А^ (И4)
Таким образом можно обрабатывать температурные
зависимости констант Михаэлиса ферментативных реакций, констант инги-
бирования или констант диссоциации ионогенных групп активного
центра фермента. Однако при обработке зависимостей констант
равновесия ферментативной реакции от температуры следует
принимать во внимание, что они могут являться эффективными
величинами. Так, константы Михаэлиса в общем случае не являются
истинными константами равновесия даже при наличии двухстадий-
иого механизма ферментативной реакции (см. выражение 5.10).
Даже в том случае, если изучаемый процесс в действительности
равновесный, его константа равновесия может оказаться
эффективной величиной, зависящей, например, от рН среды (см. гл. 10).
249
В этом случае для корректной обработки температурной
зависимости реакции необходимо учитывать теплоты ионизации ионогенных
групп субстрата или активного центра фермента.
Все вышеприведенные рассуждения в равной мере относятся и
к анализу температурных зависимостей кинетических параметров
ферментативных реакций, которые зачастую представляют собой
комбинации констант скоростей «индивидуальных» процессов, в
свою очередь обычно зависящих от рН среды. Поэтому изучение
температурных зависимостей имеет действительную ценность в тех
случаях, когда известен механизм протекания реакции и
установлена его лимитирующая стадия.
На практике анализ зависимостей констант скоростей
ферментативных реакций от температуры проводится аналогично анализу
температурных зависимостей неферментативных реакций (см.
гл. 4). Однако зачастую для реакций, катализируемых
ферментами, график в координатах (In k, l/T) или (\nk/T, l/T) имеет вид
линии с изломами. Наличие подобных изломов на аррениусовской
зависимости можно объяснить как сменой лимитирующей стадии
реакции при изменении температуры, так и переходом активного
центра молекулы фермента в узком температурном интервале в
другое информационное состояние со сменой активационных
параметров реакции.
Особенностью ферментативных реакций является также
наличие колоколообразной зависимости скорости реакции от
температуры в достаточно широком интервале, которая характеризуется
«температурным оптимумом» реакции. Эта особенность
объясняется наложением двух эффектов — возрастанием скорости реакции
при увеличении температуры и ускорением тепловой денатурации
белковой молекулы, приводящей к инактивации фермента при
достаточно высоких температурах. При достаточно корректной
постановке эксперимента оба эти явления можно изучать раздельно.
§ 2. Изучение термодинамики
конформационных изменений активных
центров ферментов
Изучение термодинамики обратимых конформационных
переходов в белках можно проводить на основе уравнения Вант-Гоффа
(11.1) — (П-2), исследуя температурную зависимость константы
равновесия процесса. Однако существуют подходы, позволяющие
изучать термодинамику равновесных процессов с помощью
кинетических методов. Один из них основан на изучении влияния
температуры на кинетику инактивации ферментов под действием
ультразвука [1] Рассмотрим кинетическую схему (11.5) инактивации
двух молекул фермента, отличающихся конформацией активного
центра и находящихся друг с другом в равновесии. Естественно
полагать, что вследствие различной конформации активных центров
молекул фермента Е и Е' кинетика их инактивации также будет
250
различной, характеризуясь константами скоростей инактивации
k\ и k2: ' -.«—• - ^j-
F —" F/ (П.5)
+ I
Продукты инактивации
фермента
Рассмотрим, как с помощью изучения температурной зависимости
эффективной константы скорости инактивации можно рассчитать
термодинамические параметры АН и AS, характеризующие
обратимые конформационные изменения активных центров ферментов.
Общая скорость инактивации фермента, как следует из схемы
(11.5), определяется выражением
v = k1[E\ + k2[E'\.
На основании уравнения материального баланса
[Е]0 = [Е] + [Е']
и выражения для константы равновгсия процгсса можно записать
ft, [E], k2 [E]„K
v 1 + К+ \ + К •
или
v = йэфф [Е]0,
где
ks^ = k-^~f- (П.6)
Выражение (11.6) устанавливает связь между эффективной
константой скорости инактивации фермента и константой равновесия
(11.5), зависящей, согласно уравнению Вант-Гоффа, от
температуры (при АНФО). Дифференцируя обе части выражения (11.6) по
Т (считая k\ и k2 постоянными, так как конформационные
переходы активных центров ферментов происходят, как правило, в
довольно узком температурном интервале), получаем
^эфф k2 — k1 dK
dT ~~ (1 +KY ' dT'
Из уравнения (11.7) следует, что скорость изменения &Эфф с
температурой в точке К= 1 (при температуре Тс) равна
^зфф _ k2 — kt UK
dT ~ 4 ' dT'
Наконец, преобразуя уравнение Вант-Гоффа
(11.7)
(11.8)
л,, RT* dK m ш
AH = -K-df- (и-9>
251
и подставляя выражение (11-8) в (11.9), найдем выражение
для энтальпии конформащюнного перехода в точке Т=ТС
(рис. 112):
ДЯ=-
4RT;
kt
dk,
|фф
dT
(11.10)
Рис. 112. Иллюстрация к
формуле (11.10) для определения
энтальпии конформационного
перехода активного центра
фермента
Зная АН конформационного перехода, по формулам (П.З) и
(11.4) можно найти значения свободной энергии и энтропии
данного процесса при температуре Тс.
Задачи
П-1. При изучении влияния рН и температуры на
максимальную скорость прекращения D-глюкозо-б-фосфата в D-фруктозо-б-
фосфат под действием глюкозофосфатизомеразы [2] были
найдены значения рК ионогенной группы активного центра фермента,
равные 9,35+0,06 и 8,71±0,11 при температурах 30 и 40° С
соответственно. Оценить значение теплоты ионизации найденной
ионогенной группы и вычислить ошибку экспериментально
определенной величины Д#ион.
11-2. Эффективность нековалентного связывания аниона
гидрокоричной кислоты с а-химотрипси'ном зависит от ионизационного
состояния а-аминогруппы остатка изолейцина-16 активного центра
фермента [3]. Вычислить теплоту ионизации этой группы на
основании данных табл. 1.
11-3. При изучении кинетики гидролиза /г-нитроанилида N-бен-
зоил-ВЬ-аргинина, катализируемого трипсином, была определена
температурная зависимость константы диссоциации ионогенной
группы фермента, контролирующей реакцию (табл. 2). Вычислить
теплоту ионизации этой группы.
11-4. В таблице 3 приведена температурная зависимость
константы ассоциации лизоцима с N-ацетилхитотриозидом [4].
Рассчитать значения стандартных энтальпии и энтропии комплексо-
образования.
П-5. При нековалеитном связывании молекулы ингибитора
активный центр а-химотрипсина претерпевает медленное обратимое
252
Таблица 1
Таблица 2
Температурная зависимость
константы диссоциации группы,
контролирующей связывание
гидрокоричной кислоты
с а-химотрипсином. Условия опыта:
ионная сила 0,2М (NaCl); конц.
фермента 2-10-5М; конц.
гидрокоричной кислоты Ы0_2М
*»с
12
20
28
36
Р«а
8,81
8,41
7,90
7,52
Влияние температуры на константу
диссоциации ионогенной группы
трипсина, участвующей в реакции
гидролиза п-нитроанилида
1М-бензоил-ОЬ-аргинина. Условия
опыта: ионная сила
0,1 М (0.033М СаС12)
/»с
15
25
35
ркв
7,30
7,10
6,95
Таблица о
Влияние температуры на константу
ассоциации лизоцима с N-ацетилхитотриози-
дом. Условия опыта: рН 5,3; ионная сила 0,1 М
(•С
9,0
9,0
20,0
20,0
31,4
31,4
45,0
45,0
60,0
60,0
^„..ю—5, м~'
10,70
9,77
2,95
3,09
1,32
1,20
0,427
0,447
0,159
0,148
конформационное изменение [3]
E + I;
к */
—* EI Е'1.
(11.11)
На основании данных, приведенных в табл. 4, рассчитать значения
стандартных энтальпии и энтропии активации прямой и обратной
реакций (11.11), а также стандартных энтальпии и энтропии
равновесной изомеризации.
11-6. В таблице 5 приведена температурная зависимость
константы ассоциации диэтилглутарата с активным центром сс-химо-
трипсина [5]. Определить значения стандартных энтальпии и
энтропии комплексообразования.
253
Таблица 4
Влияние температуры на константы скоростей
прямой (kf) и обратной (kr) реакций изомеризации
активного центра а-химотрипсина при связывании
с анионом гидрокоричной кислоты. Условия опыта:
ионная сила 0,2М (NaCl); конц. а-химотрипсина
2-10^5М, конц. гидрокоричной кислоты 1-10~2М
t°c
20
28
36
kr, °еК '
/'
0,48
1,78
6,67
к сек •
Г
0,23
0,48
1,34
Таблица 5
Зависимость константы ассоциации
диэтилглутарата с а-химотрипсином
от температуры. Условия опыта: рН 6,9; ионная
сила 0,Ш (КС1)
ГС
15
20
25
30
35
40
45
^асс.Ю~4, м-1
7,24
3,00
2,10
1,05
0,599
0,525
0,290
11-7. В таблице 6 приведена зависимость константы скорости
необратимой инактивации аденозинтрифосфатазы от температуры
[6]. Вычислить значения стандартных энтальпии и энтропии
активации процесса.
Таблица 6
Влияние температуры на константу скорости
инактивации аденозинтрифосфатазы. Условия
опыта: рН 7,0; ионная сила 0,5М (КС1)
("С
15
20
25
30
35
ft -10s, сек-1
0,556
1,28
3,06
6,95
18,6
254
11-8. В таблице 7 приведена температурная зависимость
констант скоростей прямой и обратной реакций конформационного
изменения молекулы а-химотрипсина [7]. Вычислить значения
стандартных энтальпии и энтропии активации прямого (kf) и
обратного (kr) процесса.
Таблица 7
Зависимость кинетических параметров обратимого
конформационного перехода молекулы а-химотрипсина
в растворе. Условия опыта: рН 9,8; ионная сила 0,2М (NaCl);
конц. фермента 2-10_5М
ГС
12
20
28
36
к ., сек 1
0,242
0,703
1,973
5,129
ft , сек '
0,157
0,321
0,582
1,192
11-9. В работе [8] было показано, что нековалентный
комплекс ксантенового красителя родамина 6G с активным центром
а-химотрипсина представляет собой соединение «включения». На
основании данных табл. 8 определить значение энергии активации
реакции комплексообразования.
Таблица 8
Влияние температуры на константу скорости
образования комплекса родамина 6G с активным
центром а-химотрипсина. Условия опыта: рН 8,0;
ионная сила 0,1М (КС1)
* с
10
15
30
*.
м
—LceK-1
75
135
535
11-10. В таблице 9 приведена температурная зависимость
каталитической константы гидролиза этилового эфира М-бензоил-L-
аргинина, катализируемого трипсином, в 2%-ном геле агар-агара.
Вычислить значения стандартных энтальпии и энтропии активации
реакции.
11-11. При изучении термодинамики гидролиза ацил-химотрип-
синов, образующихся при реакции метиловых эфиров М-ацетил-L-
аминокислот с а-химотрипсином [9], были получены активацион-
ные параметры, приведенные в табл. 10. Найти изокинетическую
температуру для реакции деацилирования фермента.
Таблица 9
Влияние температуры на кинетику гидролиза
этилового эфира N-бензоил-Ь-аргинина,
катализируемого трипсином. Условия опыта:
2%-ный гель агар-агара; рН 5,4; ионная сила
0,075 (КО); [Е1о = 4-10-8М; [SJo = 10~2М
fC
10
15
20
25
30
35
43
*кат' сек 1
0,33
0,46
0,64
1,16
1,74
2,27
5,10
Таблица 10
Активационные параметры гидролиза ацил-химотрипсинов,
производных 1Ч-ацетил-Ь-аминокислот. Условия опыта: рН 8,0;
ионная сила 0,1 М (КС1); 5% диметилсульфоксида
L-ашшокислота
а-Аминомасляная
Норвалин
Норлейцин
Фенилаланин
Триптофан
Тирозин
к-Аынногептановая
з-Аминооктановая
4"^=' ккал/моль
16,9
15,6
13,7
11,3
12,3
10,3
12,5
14,9
S*¥=, э. е.
—0,7
—2,4
—6,8
—11,4
—10,0
—13,4
—9,4
—3,0
Таблица 11
Температурная зависимость константы скорости
инактивации а-химотрипсина под действием
ультразвука. Условия опыта: рН 8,1;
0.01М КН2Р04; [ЕЬ = 3-10-6М; интенсивность
ультразвука 2 Вт/см2
ГС
15
20
23
26
28
32
35
^фф-10'- ыин_1
2,20
1,85
1,50
4,10
5,90
8,30
8,20
Таблица 12
Температурная зависимость константы скорости
инактивации а-трипсина под действием
ультразвука. Условия опыта: рН 4,2; ионная сила
0.01М (буферный раствор); [Е]0 = 210-7М;
интенсивность ультразвука 2 Вт/см2
ГС
14,9
20,3
26,4
32,2
37,1
40,8
44,8
48,2
51,2
ft^.-lO1, мин-1
эфф
0,76
0,65
0,83
1,10
2,10
3,40
5,60
6,20
6,30
Таблица 13
Влияние температуры на эффективную константу скорости
инактивации (5- и ^-трипсина под действием ультразвука.
Условия опыта: рН 4,2; ионная сила 0,01 М (буферный
раствор); [Е]о = 2-10~7М; интенсивность ультразвука
2 Вт/см2
Фермент
Р-Трипсин
f-Трипсин
t с
14,9
20,3
26,5
31,4
36,6
40,9
44,2
48,4
15,2
20,8
25,8
31,3
36,3
40,7
43,5
48,2
*эфф-10'' мин_1
0,62
0,60
0,94
1,50
2,53
3,91
4,30
4,50
0,26
0,34
0,53
1,42
2,16
2,85
3,36
3,53
11-12. В таблице 11 приведена температурная зависимость
эффективной константы скорости инактивации а-химотрипсина под
Действием ультразвука [1]. Найти температуру конформационного
перехода (Тс) и рассчитать значения энтальпии и энтропии кон-
" Заказ 158
257
формационного перехода активного центра фермента в данном
температурном интервале.
11-13. В таблице 12 приведена температурная зависимость
эффективной константы скорости инактивации а-трипсина под
действием ультразвука. Найти температуру конформационного перехода
и рассчитать значения энтальпии и энтропии конформационного
перехода активного центра фермента.
11-14. В таблице 13 приведены температурные зависимости
инактивации р- и ^-трипсина под действием ультразвука. Найти
значения температуры конформационных переходов активных
центров обоих ферментов и вычислить значения энтальпии и энтропии
конформационных переходов.
Ответы и решения
11-1. Для нахождения теплоты ионизации ионогенных групп
удобно пользоваться уравнением Вант-Гоффа (11.2), преобразовав
его в виде
или
рк° = 1гЩ--г- <11Л2>
Если значения рКа определялись при двух значениях
температуры, то
^i-w-тг- (11ЛЗ)
рк*=%Ш-к <11Л4>
и, вычитая (11.14) из (П. 13), получим
Шяоп = Щ1рК1=1рЫ. (11Л5)
Для обработки выражения (11.15) с учетом ошибок измеряемых
на опыте величин рКа вычислим сначала ошибку разности (pKidz
dzApKi) —(рК2±АрКг)- Из теории ошибок известно, что квадрат
средней ошибки алгебраической суммы независимых слагаемых
равен сумме квадратов средних ошибок этих слагаемых [10], так
что ошибка разности (pKi—pKz) будет равна
^(pKi — pK2)~ l/С/162+ 0,115 = 0,13. (11.16)
С учетом найденной величины ошибки (11.16) выражение (11 15)
будет выглядеть следующим образом:
КИ 2,3^(0,64 + 0,13)
"•** игн 11"
Т7 —Т7
258
Из теории ошибок известно также [10], что средняя ошибка
произведения независимой переменной на постоянную величину равна
средней ошибке независимого переменного, помноженной на эту
постоянную величину. Исходя из этого, получаем
-^ион = (27,9 + 5,7) ккал/моль.
Ответ: А//1ЮН= (28±6) ккал/моль.
11-2. Из выражения (11.12) следует, что график зависимости
в координатах (рКа, 1/Т) должен иметь вид прямой, тангенс угла
наклона которой равен Д#Ион/2,3 R (рис. 113).
Ответ: Д#ИОн:=22,4 ккал/моль.
11-3. Решается аналогично задаче 11-2.
Ответ: ДЯИОн=7,3 ккал/моль.
11-4. Для решения задачи
воспользуемся уравнением Вант-Гоффа (11.1)
в виде
iog/Ca
дя
2,3/?
-у- + const.
(11.17)
Откладывая экспериментальные данные
(табл. 3) в координатах (log/(arc, 1/T),
найдем АН комплексообразования (рис.
114).Далее, определив по формуле (11.3)
величину стандартной свободной
энергии AF (при 25°С), найдем величину
стандартной энтропии
комплексообразования по формуле (11.4).
Ответ: Д//°=—15,1 ккал/моль; AS°=
= —26,2 э.е.
11-5. Для расчета активационных
параметров прямой и обратной реакций
(схема 11.11) следует провести анализ
зависимостей (табл. 4) в координатах
(\nkjT, 1/Т), тангенс угла наклона кото-
рых равен— —^—(см. гл. 4). Далее, найдя значения стандартных
свободных энергий активации прямой и обратной реакций,
рассчитываем величины AS* по формуле
Рис. 113. Определение
теплоты ионизации а-
аминогруппы активного
центра а-химотрипсини
AS*-
UH*—UF*
Так как константа равновесия изомеризации фермента равна отно"
шению k,\kr,
kbr
ля;
A"7
RT
Ь-Sf - Lsf
kT
= e
Ш
(11.18)
то из соотношения (11.18) находим Д//° и Д5°.
Ответ: b.Hf= 30,4 ккал/моль; \Ht = 19,9 ккал/моль;
= 43,6 э. е.; Д5? = 6,2 э. е.; Д#°= 10,5 ккал/моль; ДS°= 37,4 э. е.
Рис. 114. Определение
стандартных энтальпии и свободной
энергии комплексообразования лизоци-
ма с N-ацетилхитотриозидом
Рис. 115. Определение изокинети-
чеекой температуры для
гидролиза ряда ацил-химотрипсинов,
производных И-ацетил-Ь-аминокислот
11-6. Решается аналогично задаче 11-4.
Ответ: Д//° = —16,3 ккал/моль; AS0 =—34,9 э. е.
11-7. Решается аналогично задаче 11-5.
Ответ: Д//=* = 30,8 ккал/моль; Д5^ = 24,2 э. е.
11-8. Решается аналогично задаче 11-5.
Ответ: ДЯ^=21,7 ккал/моль; Д/zf = 13,9 ккал/моль; kSf =
= 15,1 э. е.; ASf=- 13,0 э. е.
11-9. Ответ: Е = 20,9 ккал/моль.
11-10. Решается аналогично задаче 11-5.
Ответ: Д//=£= 15,1 ккал/моль; 1S^=—7J э. е.
11-11. Решается"аналогично задаче 4-13 (см. также §2 гл.4)
(рис. 115).
Ответ: Тизок = 482° К.
11-12. Для решения задачи см. §2, гл. 11
можно найти k2— kl = 6,9- Ю-2 мин~!, tc = 27°C,
Из рис
116
= 9,3 X
260
X 1С-3 мин-1 -град-1 (в точке t - tc). Подставляя полученные значения
в выражение (11.10), найдем Д//=96,5 ккал/моль. Далее,
используя формулу (11.4) и полагая AF—0 при t=tc (в этой точке
константа равновесия равна единице), найдем Д5=320 э. е.
Ответ: ТС=300°К (27°С); Д#=96,5 ккал/моль; Д5=320 э. е.
Рис. 116. Определение температуры
конформационного перехода
активного центра а-химотрипсина по
температурной зависимости эффективной
константы скорости инактивации
фермента под действием ультразвука
Рис. 117. Определение температуры
конформационного перехода
активного центра а-трипсина по
температурной зависимости эффективной
константы скорости ультразвуковой
инактивации фермента
11-13. Решается аналогично задаче 11-12 (рис. 117).
Ответ: 7"С=313°К (40° С) ДЯ=61,9 ккал/моль; Д5=198 э. е.
11-14. Решается аналогично задаче 11-12.
Ответ: для р-тр:ипсина Д#=50,3 ккал/моль; Д5=163 э. с; Тс=
= 309° К (36°С); для -у-трипсина Д#=37,2 ккал/моль; Д5=
= 121 э. е.; ГС=308°К (35°С).
ЛИТЕРАТУРА
1. К л и б а н о в А.
878, 1974.
2. Dyson J. E. D.,
3. Kim Y., L um г у
4. Banerjee S. К
5. H v m es A. J. "
1969.
6. Takahashi К
M., Мартин ек К., Бе резин И. В. „Биохимия", 39,
Noltmann Е. A. „J. Biol. Chem.", 243. 1401, 1968.
R. ,J. Amer. Chem. Soc", 93, 5882, 1971.
Rupley J. A. „J. Biol. Chem.", 248, 2117, 1973.
■ ■ ~ ~ С a n a d у W. J. ~"
С u p p e t t С. С
Y
asui Т., Hashimoto Y..
Biochem. Biophys.", 99, 45, 1962.
„J. Biol. Chem.", 244, 637,
Tonomura Y. „Arch.
261
7. Kim Y. D., Lumry R. „J. Amer. Chem. Soc", 93, 1003, 1971.
8. Варфоломеев С. Д., Мартинек К., Березин И. В. „Мол. биол.",
7, 115, 1973.
9. Martinek К., Dorovska V. N., V а г i o-l о m е у е v S. D., В е г е-
zin I. V. „Biochim. Biophys. Acta", 271, 80, 1972.
10. Пирятин В. Д. Обработка результатов экспериментальных измерений
по способу наименьших квадратов, ч. 1. Изд-во Харьковского
университета, 1962, стр. 15.
Глава 12
ВЛИЯНИЕ ДИФФУЗИИ НА СКОРОСТЬ
ГОМОГЕННЫХ ХИМИЧЕСКИХ РЕАКЦИЙ
И РЕАКЦИЙ, КАТАЛИЗИРУЕМЫХ
ИММОБИЛИЗОВАННЫМИ ФЕРМЕНТАМИ
§ 1. Феноменологическое описание
диффузии
Диффузией называется переход молекул вещества из области с
более высокой его концентрацией в область с более низкой
концентрацией. Закономерности диффузии описываются законами Фика.
Первый закон Фика определяет величину переноса вещества б
процессе диффузии
Здесь dm/dt — количество вещества в молях, перенесенное в
единицу времени через поперечное сечение А (поток диффузии);
дс/дх— градиент концентрации диффундирующего вещества в
направлении х; D — коэффициент пропорциональности, называемый
коэффициентом диффузии. Отрицательный знак в правой части
уравнения показывает, что вещество диффундирует в направлении,
обратном увеличению концентрации. Из уравнения также видно,
что размерность коэффициента диффузии L2T~l (обычно см2/сек).
Согласно первому закону Фика коэффициент диффузии не зависит
от концентрации диффундирующего вещества. Тот факт, что в
реальных систерлах при высоких концентрациях вещества D начинает
зависеть от концентрации, связан с отклонением раствора от
идеального. В таком случае следует в первом законе Фика
использовать не концентрации, а активности веществ.
Уравнение (12.1) нельзя использовать непосредственно для
расчетов коэффициентов диффузии, так как в процессе диффузии
градиент концентрации — переменная величина. Если считать, что
количество молей диффундирующего вещества сохраняется в процессе
диффузии и что коэффициент диффузии является постоянным в
направлении диффузии, то из уравнения (12.1) можно получить
второй закон Фика
~^=D~. (12.2)
dt дх- v '
262
Аналитическая форма этого уравнения зависит от граничных
условий.
Другой подход к вычислению коэффициентов диффузии был
развит Эйнштейном, согласно которому для частицы произвольной
формы величина D равна
D = k«TB, (12.3)
где kB — константа Больцмана, Т — абсолютная температура; В —
коэффициент трения частицы при движении ее в сплошной среде.
Если частица имеет сферическую форму (что часто используется
в качестве приближения), то величина коэффициента трения
определяется известным уравнением Стокса
£=7rV. (12-4)
где R — радиус диффундирующей частицы; т] — вязкость среды.
Комбинируя уравнения (12.3) и (12.4), получим уравнение
Стокса — Эйнштейна
D = ■££-■ (12.5)
Зная радиус диффундирующей частицы и измеряя вязкость среды
при данной температуре, из уравнения (12.5) можно вычислить
значение коэффициента диффузии.
Из уравнения Стокса—Эйнштейна следует одна важная
особенность параметра D. Если молекулы представить в виде шариков
с постоянной плотностью d, то, как известно, масса каждой из них
(т) будет равна" 4/3 я R3d. Учитывая, что пг = M/N, где М —
молекулярный вес диффундирующего вещества; N— число Авогадро,
уравнение Стокса — Эйнштейна можно представить в виде
D -const M~~, (12-6>
i
где const— ^~\\dW) Из соотношения (12.6) видно, что
коэффициент диффузии должен не очень сильно зависеть от
молекулярного веса диффундирующего вещества. Так, увеличение
молекулярного веса в 100 раз должно приводить к уменьшению
величины коэффициента диффузии молекулы лишь в 4,6 раза.
Еще одной особенностью, характерной для коэффициента
диффузии, является слабая зависимость его от температуры. Как
следует из уравнения (12.5), температурный ход коэффициента
диффузии определяется параметром Г/т]. Так как изменением
температуры в обычно используемом диапазоне (270—320° К) можно
пренебречь, коэффициент диффузии должен изменяться с
температурой обратно пропорционально изменению вязкости среды.
Поскольку вязкость не очень чувствительна к изменению температуры
(несколько уменьшается при увеличении Г), то и величина коэффи-
263
циента диффузии имеет лишь небольшой температурный
коэффициент. Если D представить в виде
_ Е
D = D0e w (12.7)
где постоянная D0 не зависит от температуры, то при диффузии в
водной среде величина энергии активации процесса диффузии (Е)
равна, например: 4,5 ккал/моль для мочевины; 4,8 икал/моль для
аланина; 5,0 ккал/моль для бычьего альбумина (белка с
молекулярным весом 68000). Как видно из приведенных данных,
величина энергии активации процесса диффузии практически не зависит
от молекулярного веса диффундирующего вещества.
Действительно, для большинства органических растворителей величина Е
определяется исключительно свойствами диффузионной среды и
лежит в интервале 1—5 ккал/моль.
Следует отметить, что для описания свойств реальных систем
приведенные выше уравнения являются приближенными,
поскольку они не учитывают межмолекулярного взаимодействия и
истинной формы молекул. Наличие межмолекулярных или ионных
взаимодействий фактически означает, что потенциальная энергия (U)
частицы -в растворе есть функция ее расположения. В таком случае
первый закон Ф'ика принимает вид
ж--"*[& + -& ш\ <>2-8>
Если известен конкретный вид взаимодействия, или, другими
словами, если задана функция U(x), уравнение (12.8) может быть
получено в аналитической форме.
§ 2. Влияние диффузии на кинетику
гомогенных химических реакций
Для химической кинетики одним из наиболее важных является
вопрос о влиянии диффузионных факторов на константы
скоростей химических реакций. Эта проблема рассматривалась в
работах Смолуховского [1], Дебая [2], Нойеса [3]. Если диффузия
частиц, участвующих в химической реакции, протекает медленнее
по сравнению со скоростью самой реакции, то взаимное
пространственное расположение реагирующих частиц не будет
одинаковым, что приводит к различиям в скоростях реакции. Таким
образом, уже из самого общего рассмотрения очевидна важная
роль диффузии в химических реакциях. Ясно, что для протекания
мономолекулярных реакций диффузия не имеет существенного
значения. Тримолекулярные реакции маловероятны и факт их
протекания в растворах не доказан окончательно. Следовательно,
наибольший интерес представляет анализ роли диффузии в кинетике
бимолекулярных процессов.
264
Для количественного описания бимолекулярной реакции будем
считать, что молекулы представляют собой шарики, движущиеся
друг относительно друга, причем сближение двух молекул на
расстояние а (для нейтральных молекул а равна сумме радиусов
реагирующих частиц) приводит к химической реакции
А + В ——-* Продукты. (12.9)
Здесь k — истинная константа скорости химической реакции. Если
зафиксировать молекулу А, то, используя уравнения для обычной
(фиковской) диффузии, можно найти концентрацию молекул как
функцию расстояния от А. Если при отсутствии диффузионных
затруднений скорость реакции (12.9) равна
г»=£[А][В], (12.10)
то в рамках принятой модели, когда роль диффузии существенна,
можно получить выражение для эффективной константы скорости
химического процесса
£эфф = Ц— (12-И)
1 + 4tcDo
где D=Da-\-Db. o=Ra-\-Rb, и размерность констант скоростей
равна см3/сек. Как видно из соотношения (12.11), диффузия не
влияет на скорость, химической реакции (&Эфф=&) в случае
/г/4я£>а<С1. Если же /г/4я/)а^>1, то эффективная константа
скорости реакции не зависит от величины истинной константы скорости
и пропорциональна коэффициенту диффузии
*9фф = 4*Яо. (12.12)
Химические реакции такого типа называются диффузионно-контро-
лируемыми. Из соотношения (12.11) также видно, что переход
реакции в диффузионно-контролируемую область может
осуществляться не только из-за уменьшения коэффициента диффузии, а
также из-за увеличения истинной константы скорости реакции.
Рассмотрим некоторые характерные особенности диффузионно-
контролируемых реакций. Во-первых, реакции, лимитируемые
диффузией, характеризуются очень высокими значениями констант
скоростей. Коэффициенты диффузии небольших молекул в
обычных растворителях при обычных условиях имеют порядок
10~6 см2-сек-1. В этом случае при а—5 А из соотношения (12.12)
можно вычислить &Эфф=4-109 М-1-сек-1. Действительно,
экспериментальные значения констант скоростей реакций, лимитируемых
диффузией, часто лежат в интервале 109—1010 М-1-сек-1.
Во-вторых, диффузионно-контролируемые реакции обычно
характеризуются малыми значениями энергии активации (1—5 ккал/моль),
поскольку, как следует из соотношения (12.12), температурный ход
эффективной константы скорости реакции определяется
температурным ходом коэффициента диффузии. В-третьих, как следует из
265
соотношений (12.5) и (12.12), эффективная константа скорости
диффузионно-контролируемой реакции изменяется обратно
пропорционально изменению вязкости среды.
При рассмотрении влияния диффузии на скорость химической
реакции мы исходили из предположения о фиковском характере
диффузии. В том случае, когда межмолекулярными
взаимодействиями пренебречь нельзя (например, в реакциях ионного типа),
вместо уравнения (12.11) необходимо использовать уравнение
Дебая
£9фф = -г^ . (12.13)
k
\ е drjr-
1 +
4т; D
Если предположить, что молекулы не взаимодействуют друг с
другом, когда расстояния между ними превышают о (т. е. (7=0 при
г>о), то уравнение Дебая переходит в уравнение (12.12), которое,
следовательно,— частный случай уравнения (12.13). Как
показывает уравнение Дебая, значение £эфф зависит от конкретного вида
функции U—U(r). В реакции двух ионов взаимодействие между
ними можно считать кулоновским и тогда
U=^L, (12.14)
где 2а и Zb — заряды взаимодействующих ионов; е — заряд
электрона; е — диэлектрическая проницаемость среды; г — межионное
расстояние (г>а). В этом случае из уравнения (12.13) нетрудно
получить выражение, описывающее влияние диффузии на
эффективную скорость реакции между двумя ионами:
, __ &__
зФФ kzkB Г {[ехр (*А гв *Ve°*B J')] - 1}
! + ■
4nDzA zB e*
Если реакция диффузионно-контролируемая, то
4r.D^A zB e2
**ФФ = zkB Т {[ехр (*А zB e*l*ok„ 1)] - 1} • <12Л5>
Выражение (12.15) можно записать в виде
■ ^^^Dc-^, (12.16)
'—Г
где
ейв7"о
(12.17)
Если хотя бы один из двух реагентов не является ионом, то
2ж2в=0, 6 = 0 и выражение (12.16) переходит в (12.12),
справедливое для незаряженных реагентов.
266
В заключение отметим, что для некоторых реакций переноса
протона экспериментальные значения &Эфф значительно превышают
теоретически предсказанные. Это можно объяснить наличием
туннельного эффекта. Поскольку, согласно де Бройлю, частица с
массой т является в то же время волной длины hjmv, где v —
скорость частицы, то представление о движении частицы,
преодолевающей энергетический барьер, можно заменить представлением о
волне, падающей на энергетический барьер. Из квантовомеханиче-
ских расчетов следует, что даже для системы, энергия которой
меньше величины энергетического барьера, имеется конечная
вероятность проникновения через барьер. Эта вероятность тем больше,
чем меньше масса частицы и ее энергия. При обычных
температурах'туннельный эффект имеет место, по-видимому, лишь в
некоторых окислительно-восстановительных реакциях (туннелиро-
вание электрона) и в некоторых реакциях переноса протона или
дейтерия. Можно полагать, что роль туннельного эффекта в
кинетике химических реакций является значительной лишь при
сверхнизких температурах, когда энергия частиц мала.
§ 3. Роль диффузии в реакциях,
катализируемых иммобилизованными
ферментами
Иммобилизованными ферментами называются ферменты,
переведенные в нерастворимое состояние с сохранением (частичным
или полным) их каталитической активности. Для иммобилизации
ферментов применяют следующие методы:
1. Ковалентное присоединение фермента к водонерастворимому
носителю (целлюлоза, стекло, бумага, ткань, пластмассы и т. д.).
2. Захват фермента в сетку геля или полимера.
3. Адсорбция фермента на твердой поверхности.
4. Ковалентная сшивка молекул фермента друг с другом при
помощи какого-либо полифункционального реагента (например,
глутарового альдегида).
5. Микрокапсулирование фермента (помещение раствора
фермента в полупроницаемые капсулы размером 5-—300 мкм).
Рассмотрим гранулу иммобилизованного фермента, помещенную
в раствор субстрата. Для осуществления ферментативной реакции
субстрату необходимо, во-первых, подойти к грануле. Это
перемещение молекулы субстрата происходит обычно не за счет
молекулярной диффузии, а за счет конвективного движения, скорость
которого намного выше скорости диффузии. Во-вторых, молекуле
субстрата необходимо продиффундировать через неперемешиваю-
щийся слой жидкости (слой Нернста), прилегающий к
поверхности в любой гетерогенной системе. В этом слое происходит лишь
молекулярная диффузия при отсутствии конвективного движения,
что значительно замедляет общий процесс. Толщина слоя Нернста
267
(несколько долей миллиметра в водных растворах) уменьшается
при перемешивании. Наконец, молекулам субстрата необходимо
продиффундировать через поры гетерогенного носителя (или
через пространственную сетку геля) для подхода непосредственно
к молекулам фермента.
Рассмотрение первых двух процессов входит в круг вопросов,
решаемых не физической химией, а гидродинамикой. Мы
проанализируем здесь третью проблему, а именно: влияние диффузии
субстрата в гранулу иммобилизованного фермента на
кинетические параметры ферментативной реакции.
Рассмотрим следующую модель. Имеется плоская мембрана,
толщина которой равна / и площадь поверхности А, содержащая
иммобилизованный фермент с концентрацией в мембране [Е]0.
Мембрана погружена в раствор субстрата, концентрация
которого равна [S]o. Коэффициент распределения субстрата между
раствором и мембраной равен Р. Требуется найти зависимость между
скоростью появления продукта в растворе и кинетическими
параметрами (йКат и Кт(ке.ж)) ферментативной реакции. Подробный
анализ этой модели приводится в работах [4, 5]. Если скорость
ферментативной реакции мала по сравнению со скоростью диффузии
и концентрация фермента мала по сравнению с концентрацией
субстрата, начальная скорость ферментативной реакции на
единицу объема мембраны будет равна
«кат [Ер] [S].
V ■■
(каж) , г„,
р + L3Jo
Для простоты расчетов в дальнейшем будем полагать, что
матрица, в которой находится фермент, гидрофильна и что между
матрицей и субстратом нет электростатических взаимодействий.
В этом случае Р~1.
В соответствии со вторым законом Фика скорость диффузии
субстрата в мембрану (в направлении х, перпендикулярном ее по-
/ д2 \S] \
верхности) равна D ( дх2 ). Скорость ферментативной реакции
в мембране определяется уравнением Михаэлиса-Ментен
= _*кат [Е]0 [S]
Km (каж) + [S] "
В стационарном состоянии 1 системы скорости этих двух процессов
1 В некоторых случаях стационарное состояние в системе устанавливается
довольно долго. Если в мембрану диффундирует субстрат, то при отсутствии
химической реакции в мембране время т установления стационарного состояния
в системе (с ошибкой не более 10%) равно [5]
1г
■c = 0,257-g-,
268
должны быть одинаковыми
V дх* ) Кт (каж) + [SJ
При рассмотрении стационарного случая частные производные в
уравнении (12.18) можно заменить на обычные (при граничных
условиях [S] = [S]0 при х=0, и [S]=[S]0 при х = 1). Отметим,
что для известного вида функции 5=5 (х) можно легко найти с
помощью первого закона Фика скорость расхода субстрата через
каждую поверхность
■и
= -ОА(*ш =£>л(4Э-) • о2-19)
Ч dx А=о \ dx Jx=i
При анализе уравнения (12.18) возможны три случая:
1. [S]>'/Cm(Kam)- Тогда
d2 [S] -feKaT [E]0
X12.20)
dx2 D
Решение уравнения (12.20) дает выражение для скорости
ферментативной реакции на единицу объема матрицы
Очевидно, что в этом случае диффузия не злияет на скорость
ферментативной реакции.
2. [S]C/Cm(Ka>K). Тогда
^, = «2[S], (12.21)
где
а = ]/ йкат[Е]0~ (12.22)
Дифференциальное уравнение (12.21) легко решается с помощью
метода Эйлера (подстановкой [S]=efa). Учитывая граничные
условия и уравнение (12.19), суммарная скорость ферментативной
реакции на единицу объема носителя равна
^ £кат [Е]о [S]p 2 COSh al — 1 П2 23)
К«(каж) al Sinha/ ' \ • )
Рассматривая полученное уравнение, видим, что вклад диффузии
учитывается фактором
р== 2 COSh a/-1
al SinhaZ v '
Если ввести обозначение -9~ = Т> то выражение (12.24) можно
записать в виде
т '
269
На рис. 118 приведен график зависимости F от а/. При
а/^1 F отличается от единицы не более, чем на 10%, т. е.
влиянием диффузии на кинетику ферментативной реакции практически
можно пренебречь. При а/5>1 величина F з'аметно меньше
единицы. Исходя из этого, отличие произведения а/ от единицы (в ту
или иную сторону) может быть критерием влияния диффузии на
скорость реакции, катализируемой
иммобилизованными ферментами.
3. [S] « Кт(каж). Тогда
d* [S] Aw [E]„ [S]
dx* /Ст(каж)+ [S]
1
D
Решение этого дифференциального
уравнения с учетом выражений
(12.22) и (12.24) приводит к
выражению
Рис. 118. Зависимость между
параметрами, характеризующими
ферментативную реакцию
V ■■
V-
^кат 1ч|0
а =
«кат
[E].[S].
^чп (каж)
Kmb.*)Dj- и диффузией-
ным фактором F (см. выражения
12.23—12.24)
где
Am (каж)-
Am (каж)
Таким образом, роль диффузии в кинетике реакций,
катализируемых иммобилизованными ферментами, сводится к увеличению
кажущейся константы Михаэлиса1.
До сих пор в качестве модели иммобилизованного фермента
мы рассматривали мембрану, помещенную в раствор субстрата.
Разумеется носитель фермента может иметь любую другую
форму: шарика, цилиндра, эллипсоида и т. д. Рассмотрим общий
критерий, позволяющий оценить вклад диффузии в любой
гетерогенный процесс [6—8].
Пусть для осуществления химической реакции веществу
необходимо продиффундировать в некоторую частицу, которой может
быть гранула с иммобилизованным ферментом, клетка
растительного или животного происхождения, клеточная органелла, зерно
гетерогенного катализатора и т. д. В этом случае эффективная
скорость химической реакции равна произведению истинной скорости
на диффузионный фактор ц. В свою очередь ц есть функция
модуля Тиле (Ф), который определяется соотношением
Ф
*Vh
(12.25)
«ЕслиРчМ то А-Эф* - Кт (каж)
если f=jt=i, то Аш (каж) — р^-
270
где R — размер частицы (параметр, аналогичный радиусу в случае
сферической частицы); k — константа скорости превращения
вещества. Соотношение (12.25) можно преобразовать так, чтобы в него
входили лишь непосредственно измеряемые на опыте параметры.
Если с — концентрация реагирующего вещества, a dm/dt—
скорость его каталитического превращения, то
Ф
*V% 75- <12'26>
Зависимость ц от Ф несколько различается для случаев
реакции нулевого, первого, второго или смешанного порядков, но в
общем эта зависимость почти аналогична зависимости F от а/ (см.
рис. 118). Если Ф<1, диффузия практически не влияет на общую
скорость реакции; если Ф>1, диффузия оказывает существенное
влияние на скорость процесса.
Следует отметить, что а/ (см. выражение 12.22) явтается
частным случаем модуля Тиле. Действительно, если в уравнении (12.26)
заменить ~ на к"1 "^ '" и с на [S]0, то
"* Аш (каж)
' Лт(кяж)«
КтЫаж)<2
Случай [S] > Кт(Каж) соответствует Ф<4.
Задачи
12-1. Концентрации раствора в двух его различных точках
различны. Оценить время выравнивания концентраций.
!2-2. Оценить радиус глобулярного белка цлтохрома с
(молекулярный вес 13000), если его коэффициент диффузии в воде при
20еС равен 1,01-Ю-6 см2/сек. Вязкость воды в условиях опыта
равна 0,89 спз.
12-3. Вычислить, во сколько раз будут различаться
коэффициенты диффузии глицина (мол. вес 75) и глобулярного белка уреа-
зы (мол. вес 480 000) в воде.
12-4. Коэффициент диффузии некоторого вещества в пиридине
(■п —0,95 спз) при 20е С равен Ы0~5 см2/сек. Оценить значения
коэффициентов диффузии этого вещества в ацетоне (т] = 0,33 спз),
этиленгликоле (т]=16,9 спз), глицерине (т)== 1500 спз).
Предполагается, что молекулы диффузанта не взаимодействуют с
растворителем.
12-5. Вычислить, какой по величине должна быть константа
скорости реакции двух близких по размеру молекул в воде при
25° С, чтобы:
а) можно было не учитывать скорости диффузии;
б) можно было считать реакцию диффузионно-контролируемой.
271
12-6. Решить предыдущую задачу при условии, что реакция
проходит при 20°С в ацетоне (т] = 0,33 спз), глицерине (ц =
1500 спз), патоке (т] = 27000 спз).
12-7. В работе [9] исследован перенос протона в воде при 25е С
в реакции
Н30+ + NH3 -> Н20 + NH4+,
причем было найдено значение k, равное 4,3-1010 М-1-сек-1.
Считая, что скорость реакции контролируется диффузией, оценить
величину межмолекулярного расстояния при взаимодействии частиц,
если коэффициенты диффузии иона гидрония и молекулы
аммиака равны 0,94-Ю~4 см2/сек и 0,24-10~4 см2/сек соответственно.
12-8. В работе [10] с помощью метода импульсного фотолиза
исследована лимитируемая диффузией реакция рекомбинации
атомов йода
I + I - 12,
причем величина эффективной константы скорости реакции
оказалась равна 1,31 • 1010 М-1-сек-1 при 25°С. Определить вязкость
растворителя, в котором проходила рекомбинация.
12-9. В работе [11] методом ЯМР исследована реакция
Н20 + Н80+ 2 Н80+ + Н80,
скорость которой является диффузионно-контролируемои. Реакция
изучалась при 25°С в воде, содержащей 0,8% 170. Оценить
значение эффективной константы скорости реакции, если вязкость воды
в условиях опыта равна 1 спз.
12-10. В работе [12] исследована лимитируемая диффузией
реакция тушения кислородом триплетного возбужденного состояния
антрацена. Вычислить эффективную константу скорости реакции в
бензоле (т] = 0,65 спз) при 20° С, если молекулярные радиусы
антрацена и кислорода равны 3,3 и 1,7 А соответственно.
12-11. Определить величину константы скорости k-\
диссоциации протонированной формы акридина в воде (т] = 0,89 спз), если
этот процесс контролируется диффузией.
н
Значение рКа акридина при 25° С равно 5,5, радиус молекулы
акридина 3,4 А.
12-12. Расстояние максимального сближения двух заряженных
частиц в реакции, контролируемой диффузией, равно 5 А. В каких
пределах будет изменяться значение эффективной константы
скорости реакции в воде при 25° С, если, не меняя максимального
сближения, варьировать заряд каждой из частиц от —2 до +2.
272
12-13. В работе [13] методом электрического импульса
исследовано взаимодействие n-нитрофенолят-иона с протоном
OeN—<С ^>-Р- + Н80+ ;—'-* OsN—(^ у~ОН + Н20.
Считая, что эта реакция лимитируется диффузией, оценить
значение k-i в воде при 25°С, если рКа п-нитрофенола равно 7,0; сумма
коэффициентов диффузии аниона и катиона равна 10~4 см2/сек и
расстояние минимального сближения при взаимодействии ионов
равно 5 А.
12-14. Согласно Эйгену [14] перенос протона в реакции
нейтрализации
Н30+ + ОН- - . 2Н20 (12.27)
происходит следующим образом. Ионы гидрония и гидроксил-ионы
сближаются на расстояние, равное примерно 7,5 А, что
соответствует трем водородным связям. После этого вдоль образованной
системы водородных связей происходит быстрое перемещение
протона. Очевидно, что согласно этому механизму реакция
переноса протона лимитируется диффузией. Вычислить эффективную
константу скорости реакции (12.27), если коэффициенты
диффузии ионов гидрония и гидроксила равны 0,94-10~4 см2/сек и
0,54- Ю-4 см2/сек соответственно.
12-15. В таблице 1 приведены значения констант скоростей
реакции переноса протона
Н30++ (CH3)3N - Н20 + (СН3)з NH+. (12.28)
Показать, является ли данная реакция диффузионно-контролируе-
мой. Сумма коэффициентов диффузии иона гидрония и триметила-
мина в воде равна 1 • 10~4 см2/сек, величина межмолекулярного
расстояния при взаимодействии равна 5 А.
Таблица 1
Влияние температуры на константу скорости
переноса протона между ионом гидрония
и триметиламином в воде [15]
t°c
ft-1010, М-'-сек-i
35
3,15
50
3,58
65
4,18
80
5,07
12-16. Реакцию димеризации органической молекулы изучали
в двух растворителях одинаковой полярности — гексане (ц =
= 0,22 спз) и парафиновом масле (т] = 13,3 спз). Определить
истинную константу скорости реакции димеризации, если при 20° С
величина эффективной константы скорости второго порядка в
гексане в 10 раз больше, чем в парафиновом масле.
273
12-17. В работе [16] приведены данные по взаимодействию
ферменга фумаразы (мол. вес 204000), имеющего в своем составе
шесть активных центров, с анионом фумаровой кислоты (мол. вес
115) в воде при 25° С. Предполагается, что субстрат
взаимодействует с одной положительно заряженной группой в каждом
активном центре фермента. Считая, что реакция образования
фермент-субстратного комплекса лимитируется диффузией, оценить
величину эффективной константы скорости второго порядка
взаимодействия субстрата с ферментом, если коэффициент диффузии
фумарат-иона равен 9,3 -10-6 см2/сек; расстояние максимального
сближения субстрата с активным центром фермента в момент
взаимодействия принять равным 5 А.
12-18. Химический реактор состоит из колонки, заполненной
гранулами иммобилизованного фермента. Исследование показало,
что осуществляемый ферментативный процесс лимитируется
диффузией. Что в общем случае более предпочтительно для
увеличения эффективности ферментативного процесса — измельчение
гранул иммобилизованного фермента или уменьшение начальной
концентрации фермента? Ответ пояснить.
12-19. В работе [17] исследовали гидролиз о-нитрофенилового
эфира pD-галактопиранозида под действием (З-галактозидазы
(мол. вес 540000), иммобилизованной в 15%-ном геле полиакрила-
мида. Эксперимент проводили следующим образом: в раствор
субстрата ([S]0=l,66-10~3 М) помещали пластинку определенной
толщины, вырезанную из куска геля, с равномерно
распределенным в ней ферментом, и регистрировали реакцию по образованию
в растворе о-нитрофенолят-иона. Ферментативная реакция,
катализируемая ферментом в гомогенном водном растворе,
характеризуется значениями &кат=273 сек-1; /(т(каж)= 1,73-10~4 М.
Коэффициент распределения субстрата между водой и гелем равен
единице, коэффициент диффузии субстрата в геле равен 1 • 10~6 см2/сек.
а) Концентрация фермента в геле равна 0,25 мг/мл. При какой
толщине пластинки геля диффузионные затруднения приведут к
уменьшению начальной скорости реакции в 1,5 раза?
б) Толщина пластинки геля равна 1 мм. Какова должна быть
концентрация фермента в геле, чтобы с ошибкой не более 20%
можно было считать, что эффективное значение константы Миха-
злиса пренебрежимо мало по сравнению с начальной
концентрацией субстрата.
12-20. Кинетика гидролиза этилового эфира 1Ч-ацетил-Ь-тиро-
зина, катализируемого а-химотрипсином, иммобилизованным в
геле агар-агара, изучалась согласно методике, описанной в
предыдущей задаче. Концентрация фермента в геле равна 1-Ю-7 М,
концентрация субстрата в растворе равна ЫО-2 М. Коэффициент
диффузии субстрата в геле агар-агара в условиях опыта равен
2-10-6 см2/сек. Найти, при какой толщине пластинки геля реакция
будет полностью (с ошибкой не более 5%) контролироваться
диффузией, если кинетика ферментативргого гидролиза в гомогенном
274
водном растворе характеризуется значениями kKaT—l90 сек г,
/(т(каж)=8- 10"4 М.
12-21. Имеется сосуд, разделенный надвое полимерной
мембраной, содержащей иммобилизованный фермент. Найти выражение
для общей скорости образования продукта, если субстрат
находится только по одну сторону мембраны и начальная концентрация
субстрата меньше константы Михаэлиса ферментативной реакции.
12-22. Энергия, необходимая для работы мышцы, выделяется
в результате ферментативного гидролиза АТФ под действием
мышечного белка миозина. Удельный вес мышцы, содержащей 10%
миозина (мол. вес 2-Ю7), приблизительно равен единице,
коэффициент диффузии АТФ в мышце равен Ю-6 см2/сек. Реакция
гидролиза АТФ под действием миозина характеризуется значениями
&кат=Ю0 сек-1, /(т(каж)=10_5 М. Оценить, при какой толщине
мышечного волокна (моделируя его пластинкой) работа мышцы
начнет лимитироваться диффузией, если начальная концентрация АТФ
равна 1-10"3 М.
12-23. Имеется раствор фермента с молекулярным весом 100000.
Плотность фермента в кристаллическом состоянии равна 1,25 г/см3.
С помощью критерия Тиле оценить, при каких значениях
&кат/^т(каж) реакция фермента с нейтральной молекулой субстрата
небольшого размера начнет зависеть от диффузии, если
коэффициент диффузии субстрата в растворе равен 3-10~6 см2/сек. В
расчетах учесть, что в растворе объем молекулы белка за счет
гидратации увеличивается примерно на 30%.
12-24. Рабочей частью химического реактора является колонка,
заполненная гранулами фермента, иммобилизованного в геле по-
лиакриламида ([Ё]о=1-10-5 М). Через колонку пропускают
раствор субстрата в концентрации Ь Ю-2 М. Ферментативная реакция
в растворе характеризуется параметрами &кат=10 сек-1, Кщк&ж)=
= 0,1 М. Каков должен быть размер гранул иммобилизованного
фермента, чтобы диффузия не играла существенной роли в
ферментативной реакции, если коэффициент диффузии субстрата в геле
равен 10~6 см2/сек.
12-25. Энергия, необходимая для движения сперматозоида
морского ежа, вырабатывается в результате реакции
ферментативного гидролиза АТФ, которая характеризуется нулевым порядком
по АТФ и скоростью гидролиза, равной 6-Ю-18
моль/сек-сперматозоид [18]. Физиология сперматозоида такова, что для
осуществления ферментативной реакции молекуле АТФ необходимо продиф-
фундировать через хвостик сперматозоида длиной 4-Ю-3 см,
причем коэффициент диффузии АТФ, измеренный в модельных
условиях, равен 2,5-Ю-6 см2/сек. Оценить наименьшую концентрацию
АТФ в сперматозоиде, при которой движение его еще не будет
лимитироваться диффузией субстрата.
12-26. В работе [19] молекулы папаина (мол. вес 25000) иммо-
билизовывали «сшиванием» их друг с другом с помощью глута-
275.
рового альдегида. Кристаллы иммобилизованного фермента
(средний размер кристаллов равен 8 мк) суспендировали в растворе
субстрата — этилового эфира ацетилглицина. Было найдено, что
константа скорости второго порядка (£кат/'Кпцкащ)
ферментативного гидролиза, катализируемого иммобилизованным ферментом
([Е]о—I мг/мл), в 5 раз меньше константы скорости реакции под
действием нативного фермента (в последнем случае Акат=1 сек-1,
^т(каж)=:0,45 М). Вычислить, влияет ли диффузия на кинетику
реакции, катализируемой иммобилизованным папаином, если
коэффициент диффузии субстрата принять равным 10_6 см2/сек.
12-27. В процессе некоторого производства, для устранения из
реакционной смеси перекиси водорода, раствор пропускают через
слой гетерогенного катализатора реакции разложения перекиси,
включенного в зерна инертного носителя. Концентрация перекиси
водорода в растворе составляет Ю-4 М, скорость ее разложения
под действием катализатора равна 10~5 М/сек. Рассчитать, каков
должен быть размер зерен катализатора, чтобы диффузия
заведомо не влияла на скорость каталитического процесса, если
коэффициент диффузии молекул перекиси равен 1(Н см2/сек.
Ответы и решения
12-1 [20]. Отметим, что время выравнивания концентрации т
не должно зависеть от величин самих концентраций, так как при
изменении их значений в несколько раз во столько же раз
изменится и диффузионный поток dm/dt, оставляя время т, прежним.
Единственными величинами, от которых может зависеть т, являются
коэффициент диффузии D и размеры области L, где концентрации
различны. Из соображений размерности очевидно, что существует
лишь одна возможная комбинация т, D и L, а именно
L"
•; — -£-.
12-2. Считая белковую глобулу сферой, из уравнения Стокса —
Эйнштейна имеем
Численные значения параметров уравнения (2.29) в системе СИ
равны: kB= 1,38-10-23 н-м/град, Г=293°К, tj = 8,9-10~4-^^-
м '
D = 1,01 • Ю-10 м2/сек. Подставляя эти значения в уравнение (12.29),
получим /? = 0,24- Ю-8 м.
Ответ: # = 24А.
12-3. Согласно уравнению Стокса — Эйнштейна
глицина
А,
-уреазы &щр.
уреазы
276
Отсюда
*\уреазы
^уреазы *^гли
Считая молекулы глицина и уреазы шариками с одинаковой
плотностью, получим (см. соотношение 12.6)
з
^глицина F Муреазы
~п 3
^уреазы r-jTj
V ■'"глицина
Ответ:1 ^.глицииа = 19.
^уреазы
12-4. Из уравнения Стокса — Эйнштейна следует, что для
данного диффузанта при данной температуре отношение
коэффициентов диффузии обратно пропорционально отношению вязкости
растворителя
Dj т
где индексы i и / соответствуют различным растворителям.
Ответ: £>ац=2,9-10-5 см2/сек; £>ет = 5,6- К)-7 см2/сек; £>гл =
= 6,3-10-9см2/сек.
12-5. Величина эффективной константы скорости
бимолекулярной реакции определяется следующим выражением (см. § 2
настоящей главы):
ь fe
геэфф k
Так как согласно условию задачи ргагируют двэ близкие по
размеру молекулы, то Dt ~ D2 и ^~ /?2. Используя уравнение
Стокса — Эйнштейна, получим
**фф=—4sr- (12-3°)
i + —-
SkBT
Подставляя в выражение (12.30) численные значения параметров,
лолучим
^ФФ = ^—Ж-iceK-i.
1 + 7.5-109
а) Если скоростью диффузии можно пренебречь, выполняется
неравенство 1 > 7 " или k <^ 7,5-109 М-1 сек-1.
1 Экспериментально определенная величина (D глицина/^уреазы = 27)
отличается от теоретически рассчитанной менее, чем в 1,5 раза, что обусловлено
лесферической формой молекул.
277
б) Если же реакция диффузионно-контролируемая, -ыполняется,
неравенство 1 <С 7 5 ю9 ' 1"и ^ >^,5-WM-1 сек-1.
12-6. Решается аналогично предыдущей задаче.
Ответ: а) /г<2-1010; 4,3-106; 2,4-105М"1 -сек-1;
б) /г»2-1010; 4,3-106; 2,4-105 М-'-смт"1.
12-7. По условию задачи скорость реакции лимитируется
диффузией; поэтому эффективная константа скорости равна
^эфф = 4 ТС (-°HsO+ + ^HN3 ) О-
Ответ: с = 4,8 А.
12-8. Эффективная константа скорости реакции определяется
выражением
Авфф = 4« (Д + А) (/?, + /?,). (12.31)
Используя уравнение Стокса — Эйнштейна, выражение (12.31) можно
преобразовать к виду
*эФф=^- -|-. (12.32)
откуда
8_ kBT'
1 3 &эфф
Ответ: iq = = 0,51 спз.
12-9. Поскольку одна из принимающих участие в реакции
частиц нейтральна, можно воспользоваться следующим
соотношением:
^эфф = 4л(^)н3о++ ^н,о) (^?н3о++ ^н3о).
Для целей приближенной оценки вполне можно считать, что
^НзО4 — £*н,о, и RH 0+ ~ /?н2о. В этом случае, используя
соотношение (12.32) можно легко рассчитать значение £эфф.
Ответ:1 /гэфф = 7,5-109 М-1-сек-1.
12-10. Эффективная константа скорости реакции равна
^эфф = 4л (Дштр + А).) (^?аитр"+ Ro,)-
Используя уравнение Стокса — Эйнштейна, получим
э*ф 3 • ц ' Ла„трЛ0, •
Ответ: /гэфф = 1,1-1010М-1-сек-1.
12-11. Oreer: /e_i=0,89- 10s сек"1.
12-12. Если в случае бимолекулярной реакции хотя бы одна из
частиц незаряжена, то реакция протекает согласно законам фиков-
1 Экспериментально найденное значение йэфф равно H-lO'M—'-ceKi]" [~Ч
278
ской диффузии, характеризуясь константой скорости k0. Если же
обе частицы приобретут заряд, то, согласно уравнению Дебая,
, , s
«эфф «о еЪ _ 1 ,
где
s- ztz2e2
tkBTo'
Учитывая, что для воды е =78, можно рассчитать отношение £Эфф/£0
для возможных комбинаций произведения zxz2.
Ответ: zxz2 =„+4, £эфф/£0 = 0,083; zxz2 = + 2, кэфф//г0 = 0,17;
zxz2 = + 1, /гэфф//г0 = 0,45; zxz2 = 0, /гэфф//гс = 1; z\z-i = — L
^эфф/^о= 1.9; 2^2 = — 2, йэфф/^о = 3,0; zxz2 = — 4, кэфф/1г0 = 4,0.
12-13. Значение константы £, при взаимодействии катиона
и аниона равно
/ej = 4ти (£>+ + £>_) с-
где —g—— = 1,9 (см. решение предыдущей задачи).
е«-1
Находя значение &, и используя соотношение
К0 = k-i/kly
рассчитываем величину константы &_,.
Ответ: k~x = 7,8- lCPceK-1.
12-14. Решается аналогично предыдущей задаче.
Ответ1: 4вфф=1,3-1011 М^-сек-'.
12-15. Если считать, что реакция (12.28) контролируется
диффузией, то теоретическое значение эффективной константы скорости,
вычисленное с помощью соотношения
£ЭФФ =41,:(£)ИзО+ +Асн„)3м)е,
равно 3,7-1010 М-1-сек-1. Из данных табл. 1 видно, что
теоретическое значение £Эфф находится в интервале экспериментальных
значений констант скоростей реакции.
Помимо этого, значение энергии активации реакции,
определенное из данных табл. 1, равно 2 ккал/моль, что также
указывает на диффузионный характер реакции.
12-16. Так как в реакции димеризации принимают участие две
одинаковые молекулы, то величина £Эфф определяется выражением
k
k
1 + 8 kBT
1 Экспериментально найденное значение £Эфф равно (1,4+0,2) ■ 101* М— '-сек-
279
(см. решение задачи 12-5). Зная отношение эффективных констант
скоростей реакций в двух растворителях, можно рассчитать
численное значение k.
Ответ: /г=5,25-109 М-1-сек-1.
12-17. Величина эффективной константы скорости
взаимодействия двух заряженных частиц (в данном случае субстрата и
активного центра фермента) равна
А9фф = 4те(А + Л2)о.-г?-г (12.33)
(ом. решения задач 12-12, 12-13). Коэффициент диффузии фумара-
зы можно рассчитать с помощью соотношения (12.6), и далее, с
помощью уравнения (12.33), найти значение &Эфф для
взаимодействия молекулы субстрата с одним активным центром фермента,
равное 7,2-109 М-1-сек-1. Так как молекула фумаразы содержит
шесть активных центров, то теоретическое значение &эфф должно
увеличиться в шесть раз.
Ответ: 1 /гэфф=4,3-1010 М-1 -сек-1.
12-18. Влияние диффузии на скорость ферментативной реакции
определяется диффузионным фактором F (см. § 3 настоящей
главы), для уменьшения которого необходимо уменьшить величину at
(см. рис. 118), где
V-
-Щг- (12.34)
Лиг (каж) ^)
и / — размер гранул иммобилизованного фермента. Практически
для уменьшения величины al можно уменьшить или размер гранул
катализатора, или начальную концентрацию фермента. Однако
параметр al зависит от / в первой степени и от [Е]0 в степени 7г-
Таким образом, для достижения одинаковой эффективности
работы реактора можно, например, или уменьшить размер частиц в
10 раз при неизменной величине [Е]0, или уменьшить
концентрацию фермента в 100 раз (резко замедлив этим общую скорость
процесса) при неизменном размере гранул иммобилизованного
фермента. В общем случае более предпочтительным, по-видимому,
оказывается первый вариант.
12-19. Начальная скорость ферментативной реакции в водном
растворе и в геле определяется уравнениями Михаэлиса — Ментен
1) = ^кат [ч!о [SJo /12 451
Аш (каж) ~г [ч1о
[E]„[SJ„ /1QQR4
ь^гель i го 1 • \ *° /
Ат(каж) "Г Poi
где Ктл£ж) = Kmi™*)lF (см. § 3 настоящей главы)
а) Разделив (12.35) на (12.36), получаем
V ^т (каж)
^гель is 1 гс] • (1Z..OIJ}
v Ат(каж) + laJ
1 Экспериментально найденное значение &Эфф :> 3-10'°М—'-сек—'.
280
Из соотношения (12.37) и исходных данных задачи, можно найти
значение Кт\каж) и далее величину диффузионного фактора F,
равную 0,16. Так как
F tht
if '
где т=а//2, то отсюда можно найти, что уменьшению скорости
реакции в 1,5 раза соответствует величина а.1 = 12,5 или толщина
пластинки / = 0,147 мм.
б) Для того чтобы с ошибкой не более 20% можно было
пренебречь величиной /Ст(каж> по сравнению с начальной
концентрацией субстрата, необходимо выполнение неравенства
l^Jo <^- ЭДя (каж)
или
[S]0>5 К"'р™\ (12.38)
Подставляя численные значения параметров в полученное
неравенство, получаем F^0,52. Соответствующие значения а.1,
найденные из рис. 118, должны удовлетворять неравенству а/^3,65.
Наконец, используя соотношение (12.34), можно рассчитать искомое
значение начальной концентрации фермента.
Ответ: [Е]о=£=8,44-1010 М=4,55-10"4 мг/мл.
12-20. Решается аналогично предыдущей задаче.
Ответ: 1^4,Б см.
12-21. Применяя подход, аналогичный рассмотренному в § 3
настоящей главы, для данной модели имеем
dx
где
^gi=a2[S], (12.39)
-VI
[EJ„
i (каж) l)
и граничные условия [S] = [S0] при х = 0, и [S] -- 0 при х = /.
Применяя метод Эйлера для решения дифференциальных уравнений
типа (12.39) (подстановкой [S] = ekx), приходим к выражению
[S] = Схеах -f- C2e~ax
Учитывая граничные условия, получаем
„а(1—х) „—а (1-х)
tSl-tSol ,,,"'1
е" — е-
или, переходя к гиперболическим функциям,
[S] ^ [SJ0 s,;hg/ >. (12.40)
281
Дифференцируя выражение (12.40) по х, получаем
d [S] [С, cosh a (Z— х)
- — —a pj0
dx ' 1V sinhaZ
Применяя первый закон Фика
dt dx '
где А — площадь мембраны, можно определить количество молей
продукта, выходящего из мембраны
п , гс, cosh а (I — х)
v = aDA S n ~--,——.
1 J0 Sinha/
12-22. [17] Диффузия начинает'оказывать существенное влияние
на скорость ферментативной реакции при условиях КЭт^аж) ~^> [Sj0,
где Кэт$гж) = Km(каж)/^, т. е. в данном случае при F .: Ю-2. Из
соотношения (12.24) нетрудно получить, что при достаточно
высоких величинах а/ (ео всяком случае выше значения а/ = 5, что
соответствует значению F^0,4) выполняется равенство
F = 2/и/.
Таким образом, для рассматриваемого случая должно выполняться
неравенство
VKaT[E1n->200- (12-41>
Подставляя численные значения параметров в неравенство
(12.41), находим /^0,28 мм. Итак, при толщине мышечного
волокна 0,28 мм диффузия АТФ начнет сказываться на работе
мышцы.
12-23. Для решения задачи удобно рассматривать белковую
глобулу как сферическую матрицу, содержащую один активный
центр. Объем одной молекулы фермента в кристаллическом
состоянии, рассчитанный из данных условия задачи, равен
1,33-10-19 см3. С учетом гидратации белковой глобулы, объем
молекулы фермента в растворе составляет 1,73-10~1Я см3, что
соответствует радиусу молекулы, равному 35 А. Для решения
поставленной задачи необходимо найти, при каком значении &кат/Кт(каж)
величина модуля Тиле
'У
Ф ^ /? ]/"-*к" [Ео]п (12.421
' Km (каж)^
будет больше или равна единице. Величина [Е]0 в выражении
(12.42) отвечает концентрации активных центров фермента в
единице объема матрицы. В нашем случае, зная объем молекулы
фермента в растворе, легко рассчитать концентрацию активных
центров, приходящуюся на единицу объема белка, [Е]о=
=9,6-10~3 М. Наконец, принимая, что диффузия субстрата в бел-
282
ковой глобуле не отличается существенно от диффузии в растворе,
из соотношения (12.42) находим £кат/Лт(каж) ^ 2,6■ 109 М-1-сек-1.
12-24. Для того чтобы диффузия не играла существенной роли
в ферментативной реакции, необходимо, чтобы величина модуля
Тиле (12.43) не превышала единицу
*=«V^>> о2-43)
где
г. "ч;ат [fcjolbjn
Km (каж) + [S]o '
Так как ° рассматриваемом случае выполняется соотношение
А гп (каж)
> [Sj0, ТО
Ответ: R < 0,32 мм.
12-25. Для решения задачи воспользуемся критерием Тиле.
Для того чтобы диффузия АТФ не лимитировала движение
сперматозоида, необходимо, чтобы величина модуля Гиле (12.43) не
превышала единицу. При Ф=1 будет выполняться соотношение
[S] = -^-. (12.44)
Подставляя значения параметров соотношения (12.44), находим
величину [S] =38- Ю-18 моль/сперматозоид'.
12-26. Воспользуемся критерием Тиле (12.43). Так как
концентрация субстрата неизвестна, примем, что отношение a/[S]o равно
^кат [Е] о//(т(каж) (преувеличивая этим влияние диффузии на
скорость ферментативной реакции). Подставляя численные значения
параметров в соотношение (12.43), находим Ф=0,0035. Так как
рассчитанная величина модуля Тиле является намного меньшей
единицы, то диффузия практически не оказывает влияния на
изучаемый ферментативный процесс.
12-27. Чтобы диффузия заведомо не влияла на скорость
химической реакции, величина модуля Тиле не должна превышать
единицу.
*-*/£ -f •-*<••
Ответ: /?<;0,1 мм.
1 Любопытно, что значение концентрации АТФ, найденное при прямом
эксперименте, оказалось равным 30-10—,8 моль/сперматозоид, что совпадает с
теоретически вычисленным значением. Отметим, что поскольку АТФ расходуется
в реакции по нулевому порядку, то в целях его экономии сперматозоид должен
вырабатывать минимальные количества этого вещества. Однако при слишком
малых концентрациях АТФ процесс передвижения сперматозоида будет
лимитироваться диффузией субстрата. Таким образом, в процессе естественного отбора
морские ежи приспособились вырабатывать оптимальное количество АТФ.
283
ЛИТЕРАТУРА
1. Smoluchowski М. „Z. Phys. Chem.", 92, 129, 1917.
2. Debye P. „Trans. Amer. Electrochem. Soc", 82, 265, 1942.
3. Noyes R. M., in „Progress in Reaction Kinetics", ed. G. Porter, v. 1, 1961,
p. 128.
4. Goldman R., Kedem O., Katchalski E. „Biochemistry", 7,4518,
1968.
5. L a i d 1 e r K. J., S u n d a r a m P. V. In „Chemistry of cell interface", p. A,
ch. V., N. Y., Acad. Press. 1971, p. 255.
6. Thiele E. W. „Ind. Eng. Chem.", 31, 916, 1939.
7. Weisz P. В., Hicks J. S. „Chem. Eng.^Sci.", 17, 265, 1962.
8. Weisz P. B. „Science", 17Э, 433,' 1973.
9. EmersonM. T, G r u n vv a I d E., Kromhout R. A. „J. Chem. Phys.",
33, 547, 1960.
10. Rabinovi tch E., W ood B. „Trans. Faraday Soc", 32, 547, 1936.
11. Meiboom S. „J. Chem. Phys.", 34, 375, 1961.
12. Boven E. J. „Trans. Faraday Soc", 50, 97, 1954.
13. Eigen M., Kustin K. „J. Amer. Chem. Soc", 82, 5952, 1960.
14. Eigen M., De Maeyer M. In „Structure of Electrolytic Solutions",
ed. Hamer. Chapman and Hall, 1959.
15. Grunvvald E. „J. Phys. Chem.", 67, 2208, 1963.
16. Albert у R. A., Hames G. G. „J. Phys. Chem.", 62, 154, 1958.
17. Bu ntin'g P. S., Laidler K. J. „Biochemistry", 11, 4477, 1972.
18. Nevo A. C, Rikmenspoel R. „J. Theoret. Biol.", 26, 11, 1970.
19. Sluyterman L. A. E., De Graaf M. J. M. „Biochim. Biophys. Acta",
171, 277, 1969.
20. Ландау Л. Д., Ахиезер А. И., Лифшиц Е. М. Курс общей физики.
М., „Наука", 1969, гл. 14.
Глава 13
ПРИМЕНЕНИЕ ТЕОРИИ ГРАФОВ
ДЛЯ СОСТАВЛЕНИЯ УРАВНЕНИЙ
КИНЕТИКИ ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
Большая часть проблем ферментативной кинетики сводится к
анализу предполагаемых схем ферментативных реакций, выводу
уравнений скорости, соответствующих этим схемам, и
сопоставлению полученных зависимостей с данными эксперимента. Когда
мы рассматривали простые кинетические схемы ферментативных
реакций (двух- и трехстадийные механизмы действия ферментов,
двухстадийные ферментативные реакции в присутствии
простейших эффекторов — ингибиторов и активаторов и т. п.), т. е. когда
мы имели систему из двух-трех алгебраических уравнений, ее
можно было легко решить обычным путем, не прибегая к
существенным упрощениям.
Однако при усложнении системы, с увеличением числа
промежуточных соединений быстро возрастает число кинетических
констант, характеризующих ферментативный процесс, так что даже
стационарное решение становится слишком громоздким, не
говоря уже о нестационарном. Далее нахождение уравнения скорости
еще более усложняется, если молекула фермента имеет несколько
284
активных центров по отношению к субстратам и эффекторам.
В качестве примера можно привести следующую схему
ферментативной реакции, которая рассматривается в известной монографии
М. Диксона и Э. Уэбба «Ферменты», стр. 89:
(13.1)
Выражение для скорости реакции (13.1) имеет вид
дробно-рационального выражения, числитель которого содержит 8
слагаемых, а знаменатель — 32 слагаемых. Обработка подобной системы
(кстати, далеко не самой сложной для реакций, катализируемых
ферментами) по методу стационарных концентраций — довольно
трудная задача. Значительное упрощение таких схем может быть
достигнуто с помощью теории графов, применение которой к
анализу кинетики ферментативных реакций было разработано
главным образом в работах М. В. Волькенштейна [1—8], а также в
работах Кинга и Альтмана [9—10], Фромма [11], Орси [12],
Келети [13].
§ 1. Элементы теории графов
Теория графов является одной из ветвей топологии и
отличается геометрическим подходом к изучению объектов Основное
понятие теории, граф-—система линий, соединяющих заданные
точки. В дорожном деле — это дороги, соединяющие населенные
пункты, в электротехнике — проводники, соединяющие различные
детали схемы; в химической кинетике при изображении
кинетических схем реакций точками могут быть представлены химические
соединения (исходные или промежуточные), а линиями — стрелки,
указывающие направление протекания реакции. В общем случае
линии графа могут быть прямыми, кривыми или извилистыми
в зависимости от конкретной задачи.
Линии графа, соединяющие заданные точки, обычно называют
ветвями. Ветви соединяют вершины графа. Обычно граф можно
изображать по-разному: заменять прямолинейные ветви на
криволинейные, располагать произвольно вершины его на плоскости.
Графы, имеющие принципиально одно и то же строение,
называются изоморфными. Если ветви графа пересекаются только в
вершинах, такой граф называется плоским. Графы химических
реакций обычно плоские.
Путь графа — непрерывная последовательность ветвей в
каком-либо одном направлении, в котором ни одна из вершин не
285
встречается более одного раза. Если путь замкнут, т. е. начинается
и заканчивается в одной и той же вершине, то это называется
циклом, а соответствующий граф — циклическим.
Граф называется связным, если каждую его вершину можно
соединить с любой другой вершиной некоторым путем. Очевидно,
что граф любой химической реакции является связным. Однако
если в системе идет несколько независимых химических реакций,
то они образуют несвязный граф, состоящий из нескольких
связных. В этом случае общий граф распадается на связные
компоненты. Если же в системе присутствует соединение, не
вступающее в реакцию вообще, то ему соответствует так называемая
изолированная вершина, или нуль-граф. Идеальный растворитель
должен был бы являться таким соединением.
Каждой ветви графа можно придать определенное численное
значение. Например, в кинетике химических реакций каждая
стрелка кинетической схемы характеризуется константой скорости
данного превращения. Граф, на котором указано направление
каждой его ветви, называется ориентированным (или направленным).
Граф, на котором имеются как ориентированные, так и
неориентированные ветви, называется смешанным.
Можно выбрать одну из вершин графа и считать ее базовой
вершиной, или базой. Тогда базовым деревом называется
совокупность всех ветвей, проходящих через все вершины графа и
направленных к базе. Ветви базового дерева ие образуют циклов. Сумма
величин всех базовых деревьев, направленных к данной базе,
называется базовым определителем графа. Практически решение
графа, т. е. в нашем случае нахождение уравнения скорости
ферментативной реакции, сводится к нахождению зсех базовых
определителей (определителей всех состояний фермента). Решение этой
задачи существенно упрощается благодаря следующим свойствам
графа:
I. Величина пути графа равна произведению величин всех
ветвей этого пути. Гак, в случае графа
ъ
о - d
А В
путь (A-^D) =abc.
2. Параллельные ветви графа складываются
ь
<£+ 2 Дэ = о* о
ABA В
Таким образом можно уменьшить число деревьев графа.
286
3. Ветви, направленные к одной базе, перемножаются
о >-о-« о
ABC
4. Пользуясь симметрией графа, можно сливать некоторые его
ветви (см. пример 3, § 2).
§ 2. Нахождение уравнений скорости
стационарных ферментативных реакций
методом графов
Для анализа кинетических схем ферментативных реакций мы
будем рассматривать направленные графы, причем каждой
вершине и каждой ветви графа будут отвечать определенные величины
(концентрации реагентов и константы скоростей реакции). Общая
формула для скорости ферментативной реакции записывается в
виде
г' = —„ 1Е!о. (13-2>
5-1
где Ds — базовые определители всех состояний фермента; Dr —
определители тех состояний фермента, из которых образуется
конечный продукт ферментативной реакции; kr — константы
скоростей стадий, приводящих к образованию продуктов. В случае
графа с одним выходом (т. е. при отсутствии параллельного
образования продуктов на нескольких стадиях ферментативной реакции)
выражение (13.2) для скорости записывается в виде
ч> = -т^-\Щ*- О3-3)
.9^1
Как видно, для нахождения уравнения скорости ферментативной
реакции достаточно найти выражения для базовых определителей
графа.
Пример 1. Рассмотрим простую схему лвухстадийиой
ферментативной реакции
Е + S 7ZZT ES —-* Е + Р,
287
которую для дальнейших операций удобно представить в виде
графа
Е ! - ES
(13.4)
Уравнение скорости ферментативной реакции записывается
согласно формуле (13.3) в виде
и т\
(13.5)
#2DES
— BTFEr,'^
Так как параллельные ветви графа складываются (см. свойство 2
графов), то граф (13.4) можно записать в виде
*-E.S-
— Р
откуда
DE = £_, + k2, DES -= kx [Sj.
Подставляя полученные выражения для определителей в
формулу (13.5), получим
„,_ ft. кг [Е]. [S]
или
*i [E], [S]
Л_1 + fes
' + [S]
что совпадает с выражением (5.10), полученным с помощью
алгебраического вывода.
Пример 2. Конкурентног ингибирозание двухстадийной
ферментативной реакции
v =
feaDES
DE +DES +
L'Fl
'El
(13.6)
288
Так как ветви, направленные к одной базе, перемножаются
(правило 3) и параллельные ветв'и складываются (правило 2), то
Df> = k-3(k-1 + k2). (13.7)
Так как величина пути графа равна произведению величин ветвей
этого пути (правило 1), то
DES = *-3MS], (13.8)
DEi = (k^ + k2)k3[l]. О3-9)
Подставляя выражения для определителей (13.7)—(13.9) в
формулу (13.6), приходим к уравнению скорости реакции
*А£-з [Е]„ [S]
V ■■
или
где
k-3{k-t + k2) + £_3*i [S] + k, (й_, + k2) [I] '
k2 [E]0 [S]
V =
Kmo+s+ra'
IV- k — x -\- k2 Tj- k—з
Пример 3. Схема трехстадийной реакции с участием ацилфер-
ментного промежуточного соединения (см. гл. 7)
Е + S ^Г^ ES <|> ЕА 3 > Е + Р2 (13.10)
Представим схему (13.10) в виде графа
ЕА
Скорость образования второго продукта реакции будет
определяться выражением
d [Р2] £зЕ>еа
dt DE + DES + DEA
[E]0.
Проведя построение всех возможных деревьев или путей, ведущих
к данной базе и не образующих циклов, выпишем значения
базовых определителей
DE = k-y k3 + к2 k3, DES = къ kx [S], DEA = kx [S] k2.
19 Заказ 158 289
Отсюда
dt
ktk3
k2 + k3
[E]„-[S]
i + k2
ft.
k2 + k3
+ [S]
Пример 4 [1]. Рассмотрим фермент, содержащий два
эквивалентных активных центра (или две активные субъединицы),
взаимодействующие друг с другом (13.11). Этот процесс кооперативеи:
если оба активных центра заняты, реакция превращения субстрата
в продукт идет со скоростью, отличающейся от скоростей реакции
при связывании субстрата одним активным центром
(13.11)
Складываем параллельные ветви и, пользуясь симметрией
графа (13.11), сливаем некоторые его ветви, получая таким образом
упрощенный граф
Согласно формуле (13.2), уравнение скорости реакции можно
записать в виде
AeDes + 2й* DSES
■» = -
DE + DES + D,
^SES
[E]o,
(13.12)
где
DE = 2 (vfc-g + k4) (£_, + k2), DES = 2ky [S] 2 (£_3 + £4),
Dses = 2A1[S]*3[S].
Подставляя выражения для базовых определителей в формулу (13.12)
и преобразуя выражения, получаем
<v =
KmKm+2K,m[S] + lSY
![E]o,
290
где
Кп
Kn
+ kt
Как видно, метод графов позволяет избежать прямого решения
алгебраических уравнений. Для рассматриваемых в настоящем
разделе случаев это не столь существенно, однако по мере
усложнения кинетических схем преимущества данного метода становятся
все более значительными.
Обработка схем сложных стационарных ферментативных
реакций в модификации Фромма.
Фромм [11] предложил упрощенный вариант обработки кине»
тических схем сложных стационарных ферментативных реакций с
помощью метода графов, позволяющий избегать утомительной
процедуры подсчета всех многочисленных деревьев сложного графа.
Рассмотрим в качестве примера двухсубстратную ферментативную
реакцию с неупорядоченным присоединением субстратов
ьДа]
*«[А]
*2Св]
(13.13)
ЕАВ
Как и в рассмотренных выше случаях, скорость реакции (13.13)
определяется по формуле (13.3), однако вычисление базовых
определителей согласно методу Фромма производится следующим
образом:
а) выписываются величины всех ветвей, направленных к базе
и соединяющих только два состояния фермента. Например, для
свободного фермента на схеме (13.13) подобными ветвями будут
являться ЕА->-Е, ЕВ—>-Е, EAB-vE, численно равные k-\, &_з и k^,
соответственно;
б) каждая из этих ветвей умножается на сумму величин ветвей,
выходящих из всех остальных состояний фермента. Например, для
рассматриваемого случая ветвь ЕА—*-Е умножается на сумму
величин ветвей, выходящих из вершин ЕВиЕАВ, &_i (&_2+&-4+&s) X
X (&_з+&4[А]); ветвь ЕАВ—>-Е умножается на сумму величин
ветвей, выходящих из вершин ЕА и ЕВ, и т. д.;
в) для нахождения величины базового определителя
полученные произведения суммируются. Например, величина определителя
свободного фермента равна
DE = k-i {k-2 + &_4 + ks) (k-3 + К [А]) + &_з {k-x + k2 [В]) (k-2 +
+ *-4 + h) + К (A_! +k2 [B]) (A_3 + k4 [A]); (13.14)
19*
291
г) из полученного выражения для базового определителя
исключаются члены, содержащие произведения констант скоростей
прямой и обратной реакций (например, k^ki [A], k-2k2[B] и т. п.).
Помимо этого, из повторяющихся членов в выражении для
определителя следует оставить лишь один.
Применяя данные правила к рассматриваемой системе,
выражение (13.14) можно упростить до (13.15)
DE = &_, £_3 (£_2 + £_4 + k5) + k-YkA [A] (£_2 + k5) +
+ k2 k-z [В] (£_4 + k5) + k2k,k5 [А] [В]. (13.15)
Подобным образом находим значения остальных определителей
DEA= ^_з[А] (£_2 + £_4 + ks) + k,kA [A]2(k-2 +k5) +
+ ^^8*4 [А] [В],
DEB== ft_, £3 И (А-2 + А-4 + АБ) + Мз И2 X
X(A-4+AB) + *iM-4[A][B],
Daeb = (AiM-в + *-iW [А] [В] + A,A2A4 [A]2 + k2\kA [A] [B]2.
При подстановке найденных значений определителей в
формулу (13.16) можно легко получить уравнен„г скорости
v = В + D "5+в"" + D 1Е1о (13-16)
UE + UEA + UEB + UEAB
двухсубстратной ферментативной реакции (13.13) в стационарном
режиме ее протекания.
§ 3. Нахождение уравнений скорости
ферментативных реакций, включающих
равновесные стадии
В целях упрощения обработки кинетики квазиравновесных
ферментативных реакций с помощью метода графов, М. В. Воль-
кенштейн с сотрудниками разработали так называемый
диаграммный метод анализа ферментативной кинетики [5—8]. Согласно
данному методу, дальнейшее упрощение анализа графов
достигается тем, что для обратимых стадий ферментативного процесса
выписываются не константы скорости, а константы равновесия.
В этом случае линии, Соединяющие вершины графа, называют
дугами (аналоги ветвей в графах стационарных реакций).
Величина дуги равна отношению констант скоростей прямой и обратной
реакции (по отношению к ориентации дуги). Дуги ориентируются
от входа, за который обычно принимают состояние свободного
фермента. Наконец, выходом диаграммы называют вершину, из
которой получается продукт ферментативной реакции и свободный
фермент. В этом случае из выхода ведет не дуга, а ветвь,
величина которой равна константе скорости стадии образования
продукта.
292
Если произведение величин дуг пути, ведущего из входа /в
вершину s обозначить символом М (i—>-s), причем М (£—>-i_) = l, то
скорость ферментативной реакции будет определяться выражением
v -
YkrM{l-+r)
2 м (' ■*■s)
[Е]о,
(13.17)
где М (i—*г) — произведение величин дуг путей, ведущих из
входа i в выход г; kr — константа скорости стадии образования
продукта на выходе г, и суммирование в знаменателе (13.17)
производится по всем п вершинам диаграммы. В случае ферментативных
реакций с одним выходом выражение для скорости записывается
в виде
kr M (/ -з- г)
v = ■
[Ео
(13.18)
2М(^«)
5=1
Пример 1. Рассмотрим схему двухсубстратной необратимой
ферментативной реакции с упорядоченным присоединением
субстратов, в которой ингибитор конкурентно вытесняет второй
субстрат В из комплекса с ферментом:
кк «в ч
Е 7=Z^ ЕА 7=^ ЕАВ —'-■* Е + Р
А'А
IE-
(13.19)
IEA
Согласно принципу микроскопической обратимости, изменение
свободной энергии в системе при равновесном переходе от Е к IEA
не зависит от пути перехода, так что имеет место соотношение
КкК'^КуК'к- (13.20)
Таким образом, любые три константы равновесия соотношения
(13.20) однозначно определяют четвертую константу равновесия,
которая не является, следовательно, независимой. Отсюда видно,
что схема реакции (13.19) эквивалентна, в частности, схеме
Е ZIZT ЕА —
ЕАВ
Е + Р
(13.21)
IE
IEA
Руководствуясь аналогичными соображениями, циклический граф
типа (13.19), в котором направления дуг в цикле при данном
293
входе задаются неоднозначно, можно легко привести к
ациклическому графу типа (13.21) с однозначной ориентацией дуг.
Запишем далее схему реакции (13.21) в виде диаграммы,
выбрав в качестве входа вершину Е
1А] [В]
—- ЕА — > ЕАВ —
ш
(13.22)
IE
IEA
Следует отметить, что, согласно правилам построения диаграмм,
если направление дуги соответствует присоединению реагента X,
то ее величина равна Х/К; если же направление дуги
соответствует отщеплению реагента, ее величина равна К/Х. Величины
путей из входа Е в различные вершины диаграммы (13.22) равны
произведению соответствующих дуг
М(Е —Е) = 1,
М (Е - ЕА) = [А]1Ка ,
М(Е-ЕАВ)= £]'*В] ,
М (Е-* IE) = [1]//Гь
М (Е — IEA)
[А] • [I]
КАК1
Подставляя полученные значения М в формулу (13.18), получим
выражение для скорости ферментативной реакции
£з[Е]0
v =
[А] [В]
Ка*в
1+ КА r КАКВ + Кх + КА К,
или
v =
Ъ [Е]„ [А] [В]
KaKb(i+1§-) + [A]{[B] + /<b(i + ^)} '
Задачи
13-1. С помощью метода графов найти выражение для скорости
двухстадийной ферментативной реакции в присутствии неконку-
294
рентного ингибитора, протекающей в стационарном режиме
ЙзШ
IE
MS)
■*-■
*-з *-
ft, IS] ^
k,
ES _^1^E + P
IES
(13.23)
13-2. Вывести уравнение скорости ферментативной реакции
(13.24) при условии ингибирования субстратом
Е + S 7=Г ES
Аз
Е + Р
(13.24)
ES9
*s
13-3. Вывести уравнение рН-зависимости скорости
ферментативной реакции для случая, когда и фермент, и субстрат
претерпевают ионизацию (см. схему 10.15).
13-4. Найти выражение для скорости стационарной
ферментативной реакции, протекающей по механизму Теорелла-Чанса [14]
*,[а]
:ЕА
*,[в]
э-EY
X
-*-Е + Y
(13.25)
где X и Y — продукты реакции.
13-5. Найти уравнение скорости трехстадийной ферментативной
реакции, протекающей в стационарном режиме, в которой помимо
продуктивного фермент-субстратного комплекса ES образуется
также непродуктивный комплекс ES* [15]
Е « *• ES
fc,'[s] l*-i
К-1
ЕА
Р,
-*-Е + Рг
ES
13-6. Найти выражение для скорости двухсубстратной
ферментативной реакции, в которой ингибитор Ii является конкурентным
ингибитором субстрата S2, а ингибитор 1г конкурентным
ингибитором субстрата Si [6]
295
t
к,
ES,
t
Кг
EI,"
t
E + P
/f«
к,
ES0
EIj
#3
I2^
1EL
r,S2l2
13-7. Кинетика восстановления молекулярного азота до
аммиака под действием нитрогеназы бактериального происхождения [16]
описывается, в частности, следующей схемой ферментативной
реакции:
ft, [M] fe,[M] ft3[S]
Е ~1 ЕМ ZH ЕМ, —
EM2S
Е + Р,
где [М] —'концентрация модификатора (железосодержащего
белка); [S] ■—концентрация молекулярного азота. Найти выражение
для скорости ферментативной реакции, протекающей в
стационарном режиме.
13-8. Гидролиз 0-фруктозо-1,6-дифосфата, катализируемый
фруктозодифосфатазой [17], описывается схемой
E + S
/г.
ES
SES
ki
Е + Р, + Р2
SE + P, + P2
(13.26)
где Pi — D-фруктозо-б-фосфат, и Р2 — ортофосфат. Найти
выражение для начальной скорости ферментативной реакции.
13-9. В работе [18] был найден следующий порядок
присоединения субстратов в реакции, катализируемой триптофан: тРНК-
лигазой
к,
ES,
r,0[b2 ■
EP + Z,
где Si —АТФ, S2 — триптофан, Р — аминоациладенилат, Z — пи-
рофосфат. Найти выражение для начальной скорости образования
пирофосфата в данной реакции.
13-10. Найти выражение для скорости трехсубстратной
ферментативной реакции со случайным присоединением
субстратов [6]
296
ES,S3 « "2 > ES S2S3—^—* E + P
vli; ^
(13.27)
13-11. В работе [19] было показано, что образование
непродуктивного тройного комплекса лактатдегидрогеназа (ЛДГ) — НАД-
пируват происходит по следующей схеме:
*"n ^nx ft,
Е ZZZZ EN7ZZI ENX ZZZT ТС, (13.28)
*-i
SZZZZX,
где Е — связующий центр в молекуле ЛДГ (изофермент М4), N —
NAD, ENX — промежуточный тройной комплекс, ТС —
непродуктивный тройной комплекс, S и X — две формы пирувата
(предположительно кето- и енольная), так что Кх = Lf ~ \~. Найти вы-
[C3J [C3J0
ражение для начальной скорости образования непродуктивного
тройного комплекса.
13-12. Гидролиз глюкозо-6-фосфата до глюкозы и ортофосфата,
катализируемый глюкозо-6-фосфатазой, описывается следующей
кинетической схемой [20]:
Е + S
ES
к.,
ft-2[Pi]
ЕР
Е + Р.
I
(13.29)
где Pi — глюкоза, Рг — ортофосфат. Найти уравнение скорости,
описывающее гидролиз глюкозо-6-фосфата в присутствии
добавленной глюкозы, если кинетика ферментативной реакции
регистрируется по образованию ортофосфата.
13-13. Схема (13.30) иллюстрирует кинетический механизм
реакции |3-элиминирования р-хлоро-Ь-аланина, катализируемой ас-
партатаминотрансферазой [21]
Е + S~*~ES:
к..
ES,
:ES,
CI
:es.
■e + p
(13.30)
297
Найти выражение для начальной скорости ферментативной
реакции, протекающей в буферном растворе в присутствии
добавленных хлорид-ионов.
13-14. «Пинг-понг» — механизм двухсубстратных
ферментативных реакций заключается в том, что реакция фермента со вторым
субстратом происходит лишь после образования одного из
продуктов ферментативного превращения. Согласно этому механизму
проходит, например, реакция гс-нитроацетанилида с
ароматическими аминами, катализируемая ариламин-ацетилтрансферазой [22]
»■ . ft,
Е + Sj ^ZZl ES, -^— EA + Plf
*-i
ft3 . (13.31)
EA + S27=i EAS2 —-^ E -f P2.
ft-3
Найти выражение для начальной скорости ферментативной
реакции (13.31), протекающей в стационарном режиме.
13-15. В работе [23] было показано, что реакция окисления
р-гидроксибутирата до ацетоацетата, катализируемая D-p-гидрок-
сибутират-дегидрогеназой, происходит по так называемому
«упорядоченному Биби»-механизму, при котором NAD является первым
субстратом, связывающимся с ферментом, и NAD-H — последним
продуктом, уходящим с фермента
*|[N] ft,[S] к к
Е „ '^EN^: "ENS-^-»EWH—^-«'Ё + NH (13.32)
*-i к-г Чр
где N — NAD, NH — NAD-H, S-p-гидроксибутират, Р-ацетоацетат.
Найти выражение для скорости ферментативной реакции (13.32)
в стационарном режиме.
13-16. Перенос ацетильной группы от ацетил-СоА на изопро-
пил-р-О-тиогалактопиранозид, катализируемый тиогалактози-
трансацетилазой, описывается «упорядоченным Биби»-механизмом
[24] и представлен на следующей кинетической схеме:
fc,[A] k2[B] *, к к
*-i *-г *-i >р
где А — ацетил-СоА; В — изопропил-р-О-тиогалактопиранозид;
Р-изопропил ацетил-р-О-тиогалактопиранозид; Q — СоА. Найти
выражение для начальной скорости реакции трансацетилирования,
протекающей в стационарном режиме.
13-17. Гидролиз 2-фосфо-ГЗ-глицерата до фосфоенолпирувата,
катализируемый фосфопируват-гидратазои (енолазой), протекает
в соответствии со следующей схемой [25]:
298
ун+ >он-
k\ fto-X k3 _ ki/' ft,
г + s =^:es z^ns-^ пр-он rf?: ep-^-e+p (13.зз>
*., it.2[Hl] ft., *-<Гон-1
Найти выражение для начальной скорости реакции, протекающей
при постоянном значении рН среды.
13-18. Окисление D-аланина под действием оксидазы D-амино-
кислот'в присутствии акцептора электронов — феназинметасульфа-
та (ФМС) проходит согласно следующей реакционной схеме [26]:
*> *» ft3 fte [ФМС]
E + S^^TES ■ E'S' >ЕН„ >Е + Р
1».,ф«с] " С13-34)
ЕР > Е + Р,
где Е — окисленная форма фермента; ЕН2 — восстановленная
форма фермента; E'S' — промежуточное соединение («пурпурный ин-
термедиат»). Найти выражение для начальной скорости реакции,
протекающей в стационарном режиме.
13-19. Окисление уридиндифосфатглюкозы (УДФГ) до уридин-
дифосфатглюкороновой кислоты (УДФГлур) под действием NAD,
катализируемое УДФГ — дегидрогеназой, проходит по
кинетическому механизму типа «гекса уни пинг-понг» [27] и описывается
следующей схемой:
*|ГЧ *, *эГв], к. к, к6[в] к
Е, < * Е,^» Е *• Е, —'-*Е -^-Ей^ * Е,—*-*- Е + Р,
*■-! " X k-i ' ' %. к-Ч 4
Pi P,
где [А]—концентрация УДФГ, [В]—концентрация NAD, Pi —
восстановленная форма NAD, P2 — УДФГлур. Найти уравнение
скорости ферментативной реакции, протекающей в стационарном
режиме.
13-20. В работе [28] рассмотрена кинетическая модель двухсуб-
стратной ферментативной реакции, в которой фермент,
реагирующий по «пинг-понг»-механизму, претерпевает в ходе реакции
обратимый аллостерический переход:
ЕЛ ^-»-Е С
е" V (13-35)
ED-« Е'В
299
На схеме (13.35) Е и Е' соответствуют двум устойчивым
состояниям фермента. Найти выражение для начальной скорости
реакции, протекающей в стационарном режиме.
13-21. В реакции окисления p-D-глюкозы до D-глюконо-б-лак-
тона и перекиси водорода, катализируемой глюкозооксидазой,
субстрат взаимодействует только с депротонированнои формой
окисленного фермента [29]. На схеме (13.36), иллюстрирующей эту
реакцию, Е и Ej— окисленные формы фермента (активная и
неактивная), Ел — восстановленная форма фермента.
Н+Е
к., si к.
ES-
^Er^F
=tE„
fc,[0,]
,[fl4
к.Л
H202
Б;
кЛя+1
*>,] (13.36)
н,о2
Найти выражения для образования б-лактона в стационарном
режиме протекания ферментативной реакции.
Ответы и решения
13-1. Переходя от циклического графа (13.23) к ациклическому
и выписывая значения базовых определителей, получаем
*-Р
ft,[i] fc3[i]
\к-з
IE
IES
DE = &_3 (£_, + k2) k-3, DES = £_., kx [S] k-3,
Die = k-3 (A_x + *2) £3 [I], DIES = k-zkx [S] k3 [I].
Подставляя значения базовых определителей в формулу скорости
реакции
•v ■
£2DES
DF n D
D
получим
V =
ES + DIE
* А*13 [S] [E]0
IES
■[E]o,
*l3(*-i + **) + *-s*3 [I] (*-i + *s) + *3*i [SJ + *i*-A HI [S]
300
или
„ 1 + [I]/Ki[Е]о [S]o ,
Кт + [S]0
где
IS k—i + Кг jr
Ain = % , Ai =
13-2. Записывая схему (13.24) в виде диаграммы
[S]
E-^ES-**P
Ls
к
ES.
*s
и выбирая в качестве входа свободное состояние фермента Е,
найдем величины путей, ведущих от входа:
М(Е — Е)=1, (13.37)
M(E-*ES) = -P- (13.38)
М (Е — ES2) = -Р- • Щ-. (13.39)
11 Ks К
s
Подставляя полученные значения (13.37) — (13.39) в формулу (13.18),
найдем уравнение скорости реакции
* [S]
v = -
2 Ks [Е]„
, , [S] [S]2
или
ft» LE]„ IS]
г/ =
* s + [S] + -р"
что совпадает с выражением (6.2).
13-3. Расцикливая граф (10.15) и выбирая в качестве входа
ациклической диаграммы свободное состояние фермента, получим
диаграмму с двумя выходами
•км
EH^S
EH2SH
к"
EH.
[H_]
M
1
K„
]*2B £b_
EHSH
[SH]
-*■ ESH
-«
EHS -
- EH
—;—*~
-*■ E
ft.
DO
ES
Находя величины путей, направленных от в: эда диаграммы ко
всем состояниям фермента, и используя формулу (13.17), получаем
Кь [ *2 К* '
V =
*s +km KSH
j [E]o
1+Ш
Кь
(■^(■^+w)+d,+^+^)+«!)'
Полученное уравнение можно привести к обычному виду (см. § 3
гл. 10), если учесть ионизацию субстрата
А'С
SH ^ZZ^ S- + Н+
и уравнение материального баланса по субстрату
[S]. = [SH] + [S-].
13-4. Записывая схему (13.25) в виде графа
EY X
найдем значения базовых определителей
DE = k^k3 + k2 [В] k3, DEa-=Mi[A], Dey = ^[A]^2[B].
Подставляя полученные значения в формулу
v
£3DEY
DE + DEA + DEY
получим уравнение скорости реакции
fe,ktk, [А] [В] [Е],
[Е]о,
v =
k,k2 [А] I В] + k3 (Л, [А] + k-t + k2 [В])
302
13-5.
o-ES
EA P,
Применяя метод Фромма нахождения базовых определителей (см.
теоретическую часть), получаем
DE = £!_! (*-! + k2) къ + k^kifa + къ¥_х (£_! + k-A),
DES = К [S] klfa,
Dea = ^11(^i[SJ + ^[SJ),
Вычеркивая из определителей DE и Dea слагаемые, содержащие
произведения k*_xk*v и повторяющиеся слагаемые (см. § 2
настоящей главы), и подставляя значения определителей в формулу
получим
. V =-
fe2DES
DE + DES + DEA + DES,
^ v [E]„[S]
■[E]o,
w =
Ks
£2 /<s
1 + -r- + —r
+ [S]
где
/Cs =
i + fts
, /r;=-
13-6.
ЕЭЛ
'tis,]
E,
ES,
[s,i
IS.]
Ki
ES^a—P
is,]
ES9
[i.
EI,I2
IU1
EI,
[Ы
tf4
i-fOijlo
303
Выпишем значения путей, ведущих из входа диаграммы
М(Е^Е)=1' м^ебаз-.Ж.РД,
M(E->ES2I2) = ^---M,
МСЕ-.ЕБД)»^-.!^-.
Подставляя полученные значения путей в формулу (13.18),
найдем выражение для скорости реакции
MSJISJIE]. ,
К,К2 + Кг [S,] (l + [j£) + Кг [S,l (1 + j^) + [S,l [S2] +
*. [Si] [S2] [E]B
~* . Ж ■ Ж.В«ИЫ'
+ Кг + K* + Кз/С4
M(E —ESX) =
M (E — ES2) =
M (E — EI,) -
M (E — EIX) =
[Si]
K,
[SJ
Кг
(У
[I.]
V =
13-7.
EM
EM,
EM,S
V =
**^EMSS
[E]0-
(13.40)
DE + DEM + DEM, + DEM3S
Применяя метод Фромма, находим значения базовых определителей
DE = А_х (А-2 -f h [S]) (*4 + Л-8) + *4 (*-i + *2 [М]) (£_2 + k3 [S]),
DEM = *г [М] (£_2 + kz [S]) (A4 + £_3) + £_2 (*4 + £_3) *i [M],
Dem, = К [M] (*4 + &_3) k, [M] + Л^ [M] {к-, + k2 [M]),
DEM,s = *3 [S] *! [M] (A_x + £2 [M]).
Вычеркивая лишние слагаемые определителей и подставляя
полученные значения в формулу (13.40), найдем выражение для
скорости ферментативной реакции
ft,&2&3&4 [S] х
v =
<*«, + k-i) <Л_гЙ_2 + Й!*-, [М] + fe A [M]2) +
X [Ml2 ГЕ1
+ [SI {A-!*,** + *,fc4 [М] (йг + /г2) + *!*„*, [М]2}
304
13-8. Схему реакции (13.26) удобно анализировать в виде
диаграммы (13.41) с двумя выходами
IS]
или
13-9.
или
/с.
ES -> Е + Р
[S1
1<1
»»
SES >SE + P
[S]
[S] [S]
•v =
1 ._13.,Ж Is!
+ Кг + Кг ' Kt
|Е]о,
IS,]
fe3/C2 [S] + k, [SI*
К./С2 + К2 [S] + [S]:
ESi
■[E]0.
is,]
*■ ESjS2
[Si] [SJ
EP + Z
/fi
K2
, _,_ [S.] , [SJ [SJ
1 + ~~i} T
■[E]o,
г> ^
К, ' ЯЛ,
fe3[E]0[S,][S2]
(13.41)
KiK* + Кг [SJ + [S,] [S2] •
13-10. Расцикливая граф (13.27), представим его в виде
ациклической диаграммы
к
ES,
А
ts3]
ES,S3
А
ES,
[SJ
ES,S,
[SJ
ES,SiS3
"[SJ
ES2S3
-^es;
[SJ
E + P
в которой дуги ориентированы однозначно, и направлены от входа
Е. Находя значения путей диаграммы от входа ко всем вершинам
и подставляя их в формулу (13.18), получим выражение для
скорости реакции
v =
k [S.][SJ[S,]
8 КЖгКг l Jo
, , JSJ [sj , [sj [sj[sj [sj[s,;
+ K, + K2 + Ks + K3K, + K2Ki
+
[SJ [SJ [SJ '
К\КгКз
305
или v = К [Е]„ [S,] [S2] [S3]
*,*,«, (1+'f )(. + f-)(.+^) '
13-11. В начальный момент времени реакции обратимостью ее
последней стадии можно пренебречь, так что схему (13.28) можно
представить в виде диаграммы
Ш JIL
Кц ft" NX к,
Е > EN . ENX — — ТС .
11ринимая во внимание, что [X] ~ /Сх [Sj0 и используя формулу
•X13.18), найдем выражение для начальной скорости реакции
„ [NJ Kx [S].
кг —irT [E]o
V =
[N] [N]/CX[S]. '
+ ^n + ^nKnx
ИЛИ v = ^г [Е]о [S]o
_^NX_
#Х V ^ [N]
V rNl /
+ [S]o
13-12. Записав схему (13.29) в виде циклического графа
вычислим величины базовых определителей
DE = k-2 [PJ k-x + &2&з + k-xkz,
DES = ft1[SlA-2[PI] + Mi[S],
DEp = kx [S] &2.
Подставляя полученные значения определителей в формулу
^DEP[E]0
V = "DE + DES + DEP •
найдем уравнение скорости
*i*A [Е]. [S]
-у =
£3 (*_i + А,) + А_,А_, [Р,] + ft, [S] (А, + А, + А-, [P.D
306
13-13. Записаз схему (13.30) в виде диаграммы, дуги которой
ориентированы от входа Е
[S1
Е —% ES -^ ESX -Щ ES2—I ES3~ E + Р,
и применяя формулу (13.18), найдем выражение для начальной
скорости ферментативной реакции
[SJ
1
К~---гг-
К,
К
v
К
Кг ' [Н + ] ' [С1-]
±f[E]o
1 +
[S] /. 1
[S] /
или
V =
+ Кг V + [Н+]
ЬКгКш [Е], [S]
1 +
Кг
[С1-
графа
KiKs [Н+] [С1-] + [S] (К2/С3 + [H+J [CI-] + К2 [С1-] + Кг [Н+] [CI-]) •
13-14. Представим схему реакции (13.31) в виде циклического
«4is,3
k-i
*»ts,3
ES,
ft,
К,
Применяя метод Фромма, находим значения базовых определителей
DE = k4 (k-L + k2) къ [S2] + k-x (k4 + k-z) kz [S2],
Des, = К fsi] (*4 + k-s) h [S2],
Dea = Mi [Sj] (£4 + А-з),
;Deas, = *3[S2](*-i + *2)MSi],
после сокращения которых получаем
De = /г3&4 [S2] (^-i + k2), DESl = /e^^ [Sj] [S2],
DE„ = Лгй2 [Sj] (k-з + £4), DE*s5 = kxk2kz [S,\ [S2].
Подставляя значения определителей в формулу
fe*DEASs Ио
DE + DESl + Dea + DEASs '
находим выражение для начальной скорости реакции
*^[E].[SJ[SJ
v =
Л—г + fe2 *-
^ ki [S2] Н—
+ Й4
*»[S,] + (*, + *i)[S,][SJ
307
13-15.
NH
k3D
г1 =
ENS
[El.
DEN + DENS + D
'ENH
(13.42)
Вычисляя значения базовых определителей
DE"= k-ok-ik^ -f- k2 [S] k3kt -f- k-xk^k^
DEN = k^ky [N] -f k^k^ [N],
DENs = Mi[NJ*2[S],
Denh = *i [N] £2 [SJ £3
и подставляя их в формулу (13.42), найдем выражение для
скорости ферментативной реакции
v =
fe.^MN] [S] [Е].
ft_,£4(ft-2 - ft,) + /гА [N](ft_2 + A3) -f ft2ftsft4[S] H- ft,ft2[N][S](ft, + ft,)'
13-16.
ЕА:
ьЛВ'З
А,[А]
ЕЛ В
4
Ь-э
(13.43)
EQ
EPQ
Применяя метод Фромма, найдем значения базовых определителей
графа (13.43), которые после сокращений имеют следующий вид
DE =- k-tk- {k-2k-3 + /г_2/г4 + k3k4) + k2hk*ko [ВЬ
DEA = kxk5 [A] (k-2k-z + k-2ki + k3kt),
DEab = Л A*e [AJ [B] (£_3 + kt),
DEPQ = ^2M5[A|[B],
DEQ = k,k2k3kA [A] [B].
Подставляя полученные значения в формулу
V = ■
DF
^sDeq [E]q
DEA + DEAB + DEPQ
+ D
EQ
308
можно найти выражение для начальной скорости ферментативной
реакции.
13-17. Обработку кинетической схемы (13.33) можно
провести с помощью метода графов, представляя ее в виде циклического
графа и подставляя значения определителей состояний фермента
в формулу
v =
5 ЕР I Jo
Dp
D
ES
D + D
ES- EP-OH
D
EP
Однако значительно проще, рассматривая промежуточные стадии
реакции как квазиравновесные, представить схему (13.33) в виде
диаграммы
[S] К, 1 Ki
*' Гн+1 ^з _ Гон-] *,
Е ч-ES—-ES" .ЕР-ОН i—-IeP—->E + P
и с помощью формулы (13.18) найти выражение для начальной
скорости ферментативной реакции
[SJ
К.
1
/с*
v = ■
Кг ' [Н + ] " /Сз
[он-
[Е]0
1 +
[SJY
Кг \
1+-
К,
■ + ■
Кг
кж*
или
v ■
[н+] "г" [н+]/Сз ■ [н+]/сг[он
ft5/C2/C4[£]o [S]
=r)'
KiKyf+'lSUKiiKw + KilOH-])- КЛК* [ОН-])} •
13-18, Представив схему реакции (13.34) в виде циклического
графа
запишем выражения для базовых определителей
DE = k2k3k6 [ФМС] k5 + k2k4 [ФМС] /г5/г6 [ФМС] +
+ /г-^б [ФМС] k-0 + /г_,£4 [ФМС] ft5ft6 [ФМС],
DES = ksk6 [ФМС] &5£, [S ] + k4 [ФМС] ft5ft6 [ФМС] ft, [S],
DE'S' = &6 [ФМС] ftx [S] ft2&5,
DEP - k6 [ФМС] ft, [S] k2k4 [ФМС],
Deh, = k5kx [S] /г2/г3.
309
Подставляя полученные величины определителей в формулу
ft6 [ФМС] DEH2
v -
ft5DEP
DF
DE
[E]o,
JE "Г ^ES + DE'S' + DEP + DEH2
найдем выражение для начальной скорости реакции
МА&6 [ФМС] [S] [Е], х
v ■
ksk6 [фмс] (ft_, + k2) {k3 + ft4 [фмс]) ; maisji*mc]x
X (ft, + К [ФМС])
X (ft2 + ft3 + ft4 [ФМС]) 4- ft,ft2ft3ft5 [S] + ft,ft2ftA[S] [ФМС]"
13-19,
V :
7
V
S-=l
(1
Dc
D, = k-fa [B]2 kjibk&k1 + /e2£3 [B]2 k4k,k6k7,
iV- = MB]2MAMi[A],
D3 = M5*6Mi [A] [B] &2 + k-sksk6k7ki [A] £2 [B],
D4 = MeMi [A] AAJB]2,
D5 = M7*i|A]ft2ft8[B]2ft4,
D6 = k7kx [А] &2&з [B] &4&5 + k-&kx [A] &2&з [B] Ms.
D7 = ^[A]M3[B]2^A-
Подставляя значения определителей в формулу (13.44), по.
уравнение скорости ферментативной реакции.
13-20. Найдем величины базовых определителей графа
DE = k2k3k4 [В] ksk6 4- kzkA [В] k5k6k^,
Dea=*s*4P]Wi[A].
Efe'c = MB] Me*i [A] *2,
DE' — k5k6kt [A] k2kz 4- &_4£6&i [A] /г2/г3,
DE'B=Ml[A]M8*4[B],
DED=^i[A]^2^4[B]^5-
310
Подставляя значения определителей в формулу
fe6DED tElo
V ~~ DE + DEA + DE,C + DE, + DE,B + DED'
получим выражение для скорости ферментативной реакции
v ' М, [А] [В] [Е]0
13-21. Используя метод Фромма, находим значения базовых
определителей графа (13.36). Подставляя затем значения
определителей в формулу
fe2DES [E].
V DE+DEH+ + DEs+DER+DE--DEr+DE, '
можно найти уравнение скорости стационарной ферментативной
реакции
ЛИТЕРАТУРА
1. Волькенштейн М. В. Физика ферментов. М., „Наука", 1967, стр. 155.
2. V ol kens t ein M. V., Goldstein В. N. „Biochim. Biophys. Acta",
115, 471, 1966.
3. Волькенштейн М. В., Гольдштейн Б. Н., Стефанов В. Е.
„Мол. биол.\ 1, 52, 1967.
4. Гольдштейн Б. Н., Магаршак Ю. Б., Волькенштейн М. В.
ДАН СССР, 170, 1172, 1970.
5. Волькенштейн М. В., Магаршак Ю. Б. „Биофизика", 15,777, 1970.
6. Волькенштейн М. В., Магаршак Ю. Б. „Биофизика", 15, 949,1970.
7. Волькенштейн М. В., Магаршак Ю. Б. ДАН СССР, 192, 665,1970.
8. Волькенштейн М В., Магаршак Ю. Б., Стефанов В. Е.
„Биофизика". 17, 379, 972.
9. King Е. L., Altman С. A. „J. Phys. Chem.", 60, 1375, 1956.
10. King E. L. „J. Phys. Chem.", 60, 1378, 1956.
11. Fromm H. J. „Biochem. Biophys. Res. Commun.", 40, 692, 1970.
12. Orsi B. A. „Biochim. Biophys. Acta", 258, 4, 1972.
13. Keleti T. „Acta Biochim. et Biophys. Acad. Sci. Hung.", 3, 247, 1968.
14. Hijazi N. H., Laidler K. J. „Biochim. Biophys. Acta", 315, 209, 1973.
15. Березин И. В., Казанская Н. Ф., Клесов А. А. „Биохимия", 36,
227, 1971.
16. Bergersen F- J, Turner G. L. „Biochem. J.", 131, 61, 1973.
17. Coo per man B. S., Buc H. „Eur. J Biochem.", 27, 503, 1972.
18. Зиновьев В. В., Киселев Л. Л, К н о р ре Д. Г., К о ч к и н а Л. Л.,
Малыгин Э. Г., Фа во ров а О. О. „Биохимия", 37, 443, 1972.
19. Су г роб о в а Н. П., Курганов Б. И., Я к о в л е в В. А. „Мол. биол.",
7, 481, 1973.
20. Arion W. J., Wallin В. К. „J. Biol. Chem", 248, 2372, 1973.
21. John R. A., Tudball N. „Eur. J. Biochem.", 31, 135, 1972.
22. Riddle В., Jencks W. P. „J. Biol. Chem.", 246, 3250. 1971.
23. Nielsen N. C, Za tiler W. L., F 1 e i s с h e r S. „J. Biol. Chem.", 248,
2556, 1973.
24. Musso R. E, Zabin I. „Biochemistry", 12, 553, 1973.
25. D inovo E. C, Boy er P. D. „J. Biol. Chem.", 246, 4586, 1971.
26. RaoN. A., Nlshlklml M., Yagi K. „Biochim. Biophys. Acta", 267,
350, 1972.
27. Шибаев В. Н., Дружинина Т. Н. „Мол. биол.", 6, 471, 1972.
28. Sumi Т., Ui M. „Biochim. Biophys. Acta", 276, 12, 1972.
29. Weibel M. К., В r i g h t H. J. „J. Biol. Chem.", 246. 2734, 1971.
311
УКАЗАТЕЛЬ ИСПОЛЬЗУЕМЫХ НАИМЕНОВАНИЙ ФЕРМЕНТОВ
(в скобках даны номера ферментов согласно классификации)
Аденозиндезаминаза (3.5.4.4) 172
Аденозинтрифосфатаза (3.6.1.3) 254
а-Амилаза (3.2.1.1) 193
со-Амидаза (3.5.1.1) 152, 153
Аминопептидаза М 124
Ариламин-ацетилтрансфераза (2.3.1.5.) 298
Арилсульфатаза (3.1.6.1) 123, 176
Аспартатаминотрансфераза (2.6.1.1) 297
Ацетилхолинэстераза (3.1.1.7) 92, 117, 118
Бактериальная протеаза Е3оШ 227
Р-Галактозидаза (3.2.1.23) 274
D-p-гидроксибутират-дегидрогеназа (1.1.1.30) 298
Глутаматдегидрогеназа (1.4.1.2) 201, 202
Глюкозооксидаза (1.1.3.4.) 300
Глюкозо-6-фосфатаза (3.1.3.9) 297
Глюкозофосфатизомераза (5.3.1.9) 252
Енолаза (4.2.1.11) 298
Карбоангидраза (4.2.1.1) 237
Карбоксилэстераза (3.1.1.1) 121
Карбоксипептидаза А (3.4.2.1) 118, 119, 174
Карбоксипептидаза В (3.4.2.2.) 91, 97
Клострипаин 355, 232
Лактатдегидрогеназа (1.1.1.27) 117, 297
Лейцинаминопептидаза (3.4.1.1) 96
Лизоцим (3.2.1.17) 88, 252
N-Метилглутамат-дегидрогеназа (.1.5.3.2) 89
Нитрогеназа 296
Оксидаза D-аминокислот (1.4.3.3) 97, 299
Р-Окснстероид-дегидрогеназа (1.1.1.51) 118
Папаин (3.4.4.10) 94, 95, 153, 191, 229, 230, 234, 235, 275
Пенициллинамидаза (3.5.1.11) 175, )76
Пепсин (3.4.4.1) 23, 234, 236, 237
Рибонуклеаза (2.7.7.16) 199
Стрептококковая протеиназа 147, 233
Тиогалактозид трансацетилаза (2.3.1.18) 298
Трипсин (3.4.4.4) 88, 120, 122, 125, 150, 172, 199, 235, 252, 255, 258
Трипснноподобный фермент микробного происхождения 117
Триптофан: тРНК-лигаза 296
Уреаза (3.5.1.5) 174
УДФГ-дегидрогеназа (1.1.1.22) 299
Фенилэтаноламин-1Ч-метилтрансфераза 116
Фицин (3.4.4.12) 190, 195
Фосфат-ацетилтрансфераза (2.3.1.8) 92
Фосфопируват-гидратаза (4.2.1.11) 298
Фруктозодифосфатаза (3.1.3.11) 296
Фумараза (4.2.1.2) 274
Химотрипсин (3.4.4.5) 11, 87, 88, 97, 123, 125, 126, 147, 148, 149, 150, 151,
153, 154, 171, 172, 188, 192, 195, 196, 197, 198, 201, 226, 227, 228, 230,
231, 232, 233, 237, 252, 253, 255, 256, 274
Холинэстераза (3.1.1.8) 89, 98
Щелочная фосфатаза (3.1.3.1) 119, 153, 173
Эластаза (3.4.4.7) 193, 194, 195
Эпоксид-гидраза 96
Эстераза кишечная 231
312
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютная концентрация фермента, определение 114, 19
Адсорбция ферментов на носителях 267
Активация бесконкурентная 106
неконкурентная (простая) 81, 106
простая (неконкурентная) 81, 106
синергистическая 104
смешанная 82, 108
субстратом 113
Активных центров фермента титрование 195, 209
Аллостерический переход фермента в ходе реакции 299
Анаморфоза полулогарифмическая 17
Антиконкурентное ингибирование 83, 103
Аррениуса уравнение 65
Ациклический граф 294
Ацилирования константа скорости 144
Ацилирования фермента стадия 144
Ацилферментное промежуточное соединение 144, 188
Базовая вершина (база) графа 286
Базовое дерево 286
Базовые определители графа 286
Бесконкурентная активация 106
Бесконкурентное ингибирование 81, 83, 84, 103
Бимолекулярная реакция 5
Больцмана постоянная 66
Бренстеда кислоты 36
основания 36
Вант-Гоффа закон 249, 251, 258
Вершины графа 285
Ветви графа 285
Взаимозависимые конкурентные ингибиторы 84, 85
неконкурентные ингибиторы 86
Взаимонезависимые конкурентные ингибиторы 85
неконкурентные ингибиторы 86, 87
Влияние рН на скорость химических реакций 34 ел.
Влияние температуры на константу равновесия 249
Внутримолекулярный общекислотный катализ 45
Вре\1я полупревращения 18
в реакции первого порядка 18
в реакции второго порядка 18
в реакции третьего порядка 19
Время релаксации 189, 199, 214
Второго порядка реакции 21 ел.
Второй закон Фика 262
«Вытеснения профлавина» метод 188, 195, 209—211
Выход графа 287
Вязкость, влияние на диффузию 263 ел.
влияние на скорость реакции 265
Газовая постоянная 65
«Гекса уни пинг-понг» механизм ферментативной реакции 299
Гомогенных химических реакций кинетика 5 ел.
Граф, определение 285
свойства 286
Графическое дифференцирование кинетической кривой 7, 14, 16, 18
Графов теория 284 ел.
Гуггенгейма метод определения констант скоростей 20 ел., 30, 32
«Двойных обратных величин» координаты 79
Двух конкурирующих субстратов реакция с ферментом 136—138
Двух молекул ингибитора связывание с ферментом 141, 142
Двух ферментов действие на один субстрат 139, 140
Двухкомпонентного обратимого ингибирования метод 84 ел.
Двустадийные ферментативные реакции 77 ел.
рН-зависимость 219 ел.
Двухсубстратные ферментативные реакции 291
Деацилирования фермента стадия 144
Деацилирования константа скорости 144
Дебая уравнение 266, 279
Диаграммный метод анализа ферментативной кинетики 292 ел.
Диксона метод определения констант ингибирования 80, 82 ел.
Димеризация ингибитора 163, 164
Дифференцирование графическое кинетической кривой 7, 14, 16, 18
Диффузионно-контролируемые реакции 265, 278
Диффузионные факторы 269—271
Диффузия
влияние на константу Мпхаэлиса ферментативной реакции 270
влияние на скорость бимолекулярных реакций 264 ел.
влияние на скорость ионных реакций 266, 267
влияние на скорость ферментативных реакций 267 ел,
коэффициент 262
феноменологическое описание 262 ел.
Дополнительных нуклеофильиых агентов влияние на ферментативну
146, 147, 158
Закон Вант-Гоффа 249, 251, 258
Законы диффузии Фика 262, 263, 264
Иди координаты 79
Иди уравнение 79
Изокинетические зависимости 66, 255
Изолированная вершина (нуль-граф) 286
Изомеризация обратимая фермента в ходе реакции 124, 138
Изоморфные графы 285
Иммобилизация ферментов, методы 267
Иммобилизованные ферменты 176, 267 ел.
влияние диффузии на кинетику 267 ел.
Инактивация фермента в ходе реакции, кинетика 168 ел., 176, 182 ел.
под действием субстрата 169, 176, 184—186
под действием ультразвука 11, 250 ел., 257
Ингибирование
антиконкурентное 83, 103
бесконкурентное 81, 83, 84, 103
двухкомпонентное 84 ел.
конкурентное 80, 83, 101
неконкурентное 81, 97, 101, 102, 153, 295
необратимое 229
обратимое 79 ел.
продуктом реакции 167, 168, 179, 180
псевдоконкурентное 83
смешанное 82 ел.
314
субстратом 111 ел., 127 ел., 295
субстратом неполное 112 ел.
Ингибиторы взаимозависимые 84 ел.
взаимонезависимые (совместимые) 84 ел.
Индивидуальные константы скоростей ферментативной реакции 145
определение 145 ел., 156 ел.
Интегральная форма уравнения Михаэлиса-Ментен 166, 177
Ионизация субстрата, влияние на рН-профиль ферментативной реакции 223—225,
236, 243 ел.
Ионной силы влияние на кинетику действия ферментов 149, 157
Истинная константа Михаэлиса 144
Кажущаяся константа Михаэлиса 78, 144
Каталитическая константа 78, 144
зависимость от рН 220 ел.
Кинетические параметры ферментативной реакции 79
определение из площади под кинетической кривой 181
Кислотный общий катализ 36
специфический катализ 35
Кислоты Бренстеда 36
Ковалентная иммобилизация ферментов 267
Колоколообразная форма рН-зависимости 37, 50, 62 ел., 221 ел.
Компенсационные зависимости 67
Конвективное движение в растворе 267
Конкурентное ингибирование 80, 83, 100, 101
Константа диссоциации ионогеиных групп свободного фермента, определение 220,
238 ел.
Константа ингибирования 80
продуктом, определение 168
Константа диссоциации ионогениых групп фермент-субстратного комплекса
220 ел.
определение
Константа Михаэлиса
зависимость от рН 220 ел.
истинная 144
кажущаяся 78, 144
Константа скорости реакции 5
определение 17 ел.
Конформационный переход активного центра фермента 251
Кооперативный ферментативный процесс 290
Коордчиаты Иди 79
Лайнуивера-Берка 79, 83
Коэффициент диффузии 262 ел.
вычисление 262, 263
зависимость от температуры 263
зависимость от молекулярного веса диффузанта 263
размерность 262
Критерий Тиле 271, 283
Лайнуивера-Берка координаты 79, 83
Лиат-ионами специфический катализ 35
Лиония ионами специфический катализ 35
Максимальная скорость ферментативной реакции 78
зависимость от ионной силы 149, 156, 157
зависимость от рН 220 ел.
Мангельсдорфа метод определения констант скоростей 21
Метод двухкомпонентного обратимого ингибирования 84 ел.
Гуггенгейма 20 ел., 30, 32, 166
графов 287
Диксона 80, 82 ел.
Мангельсдорфа 21
315
Розвери 22, 23, 32
Фостера-Ннманна 168, 182, 183
Фромма обработки кинетических схем 291, 292
Микрокапсулирование фермента 267
Микроскопической обратимости принцип 293
Михаэлиса константа
истинная 144
кажущаяся 78, 144
Михаэлиса-Ментен уравнение 78
Модуль Тиле 270, 271
Молекулярность реакции 5
Мономолекулярные реакции 5
Направленный граф 286
Начальных скоростей химических реакций анализ 5 ел.
Неконкурентная (простая) активация 81, 106
Неконкурентное" ингибирование неполное 153, 162—165
полное 81, 101, 295
Необратимое ингибирование 229
Непродуктивный фермент-субстратный комплекс 295
Нернста слой 267
Несвязный граф 286
Несимметричные колоколообразные кривые рН-зависимости 221
Несовместимые ингибиторы 108
Нестационарная кинетика ферментативных реакций 186 ел.
Нетривиальные типы ингибирования ферментативной реакции 82 ел.
Неупорядоченное присоединение субстратов к ферменту 291
Нуль-граф (изолированная вершина) 286
Общий кислотный катализ 36, 42, 45
основной катализ 36
Общий порядок реакции 5
Ориентированный граф 286
Основания Бренстеда 36
Основной специфический катализ 35
«Остановленной струи» методы 188
Отрицательные значения кинетических параметров, физический смысл 1<
Ошибка средняя алгебраической суммы, вычисление 258
произведения, вычисление 259
рК диссоциации реагента, определение 36 ел., 51 ел.
рК ионогенных групп активного центра, определение 225 ел.
рН-профиль константы скорости реакции 35, 36, 57
«рН-скачок», применение для определения констант скоростей быстрых
ферментативных реакций 192
Первого порядка реакции 17 ел.
Первый закон Фика 262, 264
«Пинг-понг» механизм ферментативной реакции 298, 299
Планка постоянная 66, 73
Плоский граф 285
Полное конкурентное ингибирование 80
Полное неконкурентное ингибирование 81, 100
Полной кинетической кривой анализ 6
Полной кинетической кривой ферментативной реакции анализ 166 ел.
Полулогарифмическая анаморфоза 17
Полупревращения время
в реакции первого порядка 18
в реакции второго порядка 18
в реакции третьего порядка 19
Порядок реакции 5 ел.
316
Поток диффузии 262
Предстационарная кинетика 187 ел.
Предэкспоненциальный множитель в уравнении Аррениуса 65
Примеси ингибитора в препарате фермента, влияние на кинетику 133—136
Принцип микроскопической обратимости 293
Продуктом ферментативной реакции ингибирование 168, 179
Промоторы ферментативной реакции 80
Простая (неконкурентная) активация ферментативной реакции 81, 106
Простые химические реакции 5
Прототропное равновесие 223
Псевдоконкурентное ингибирование 83
Путь графа 285
Равновесная ферментативная реакция 155, 164, 165
Расцикливание графа 301, 306
Релаксации время 189, 213, 214
Релаксационная кинетика 187, 189 ел., 212—214
Розвери метод определения констант скоростей 22, 23, 32
Свободная энергия активации 66 ел.
Свободная энергия равновесного процесса 249
Связный граф 286
Селективное воздействие эффекторов на отдельные стадии ферментатиг
реакции 145 ел.
Сигмоидная форма рН-профиля 35, 51
Синергистическая активация 104
Системы со взаимным истощением 114
«Скачка давления» метод 189
Скорость реакции 5, 6
Слой Нернста 267
Смешанная активация ферментативной реакции 82, 108
Смешанное ингибирование ферментативной реакции 82 ел.
Смешанный граф 286
Сольволиз ацилфермента нуклеофильными агентами 146 ел.
Специфический катализ ионами лиония 35
Специфический кислотный катализ 35
основной катализ 35
Средняя ошибка алгебраической суммы, вычисление 258
произведения, вычисление 259
Стандартная свободная энергия активации 66, 259
энтальпия активации 66, 259
энтропия активации 66, 259
Стационарное состояние при диффузии, время установления 268
Стационарности условие 78
Стационарный режим ферментативной реакции 78
Стационарных концентраций метод 85
Стокса уравнение 263
Стокса-Эйнштейна уравнение 263, 277
«Температурного скачка» метод 189, 199
Тиле критерий 271, 283
модуль 270, 271
Теорелла-Чанса механизм ферментативной реакции 295
Теория графов 284 ел.
определение, терминология 285, 286
применение для анализа кинетических уравнений 286 ел.
Температура конформационного перехода 251
Температуры влияние на кинетику ферментативных реакций 249 ел.
на константу равновесия 249, 250
на скорость 249, 250
«Температурный оптимум» ферментативной реакции 250
Теплота ионизации ионогенных групп активного центра 252
Термодинамика обратимых конформационпых переходов активного центра 2
Титрование активных центров фермента 195, 208, 209
Трансмиссионный коэффициент 66
Третьего порядка реакции 19 ел.
Трехстадийная ферментативная реакция 144
рН-зависимость 221—223
Грехсубстратная ферментативная реакция 296
Тримолекулярные реакции 5
Трис-буфера влияние на кинетику действия ферментов 153
Туннелирование электрона 267
Туннельный эффект 266, 267
Ультразвуковая инактивация ферментов, кинетика 250—252
Уокера-Шмидта метод определения кинетических параметров ферментат
реакции 166
«Упорядоченный Биби»-механизм ферментативной реакции 298
Уравнение Аррениуса 65
Вант-Гоффа 249, 251, 258, 259
Дебая 266, 279
Михаэлиса-Ментен 78
Стокса 263
Стокса-Эйнштейна 263, 277
Хендерсона-Хассельбалха 36
Установившегося равновесия метод 85, 207
Факторы диффузии 269—271
Фермент-субстратный комплекс 77 ел., 188
Фика законы диффузии 262, 263, 264
Фостера-Ниманна метод 168, 182, 183
Фромма вариант метода графов 291 ел.
Хендерсона-Хассельбалха уравнение 36
Циклический граф 286
Элементарные константы скоростей реакций комплексообразования, опреде.'
199 ел., 217
Энергия активации 65
Энергия активации диффузии 264
Энтальпия активации 66, 259
конформационного перехода активного центра 251
равновесного процесса 249
Энтропия активации 66, 74, 259
равновесного процесса 249
Эффективная энергия активации 66
Эффекторы обратимые, влияние на кинетику действия ферментов 79 ел.
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ ... . . 3
Часть I. Кинетика гомогенных химических реакций
Глава 1. Порядок реакции н его определение . 5
§ 1. Анализ начальных скоростей реакций 6
§ 2. Анализ полной кинетической кривой 6
Задачи.. . . 8
Ответы и решения . 13
Литература 16
Глава 2. Определение констант скоростей реакций из
экспериментальных данных . .17
§ 1. Общие методы обработки кинетических кривых 17
§ 2. Специальные методы обработки кинетических кривых 20
Задачи.. .23
Ответы и решения . 29
Литература. . 34
Глава 3. Влияние рН на скорость химических реакций 34
Задачи.. • 37
Ответы и решения 51
Литература ... 64
Глава 4. Влияние температуры на скорость химических реакций 65
§ 1. Зависимость констант скоростей реакций от температуры 65
§ 2. Изокинетические зависимости 66
Задачи. 67
Ответы и решения 72
Литература 76
Часть II. Кинетика ферментативных реакций
Глава 5. Кинетическая обработка простых двухстадииных
ферментативных реакций (анализ начальных скоростей) . .... 77
§ 1. Кинетическое описание двухстадииных ферментативных реакций 77
§ 2. Определение кинетических параметров ферментативных реакций
из экспериментальных данных . . . .... 79 ■
§ 3. Влияние обратимых эффекторов (ингибиторов и активаторов) на
кинетику ферментативных реакций ...... 79
§ 4. Кинетический метод двухкомпонентного обратимого ингибиро-
вания ферментативных реакций 84
Задачи. 87
Ответы и решения. 100
Литература 110
Глава 6. Кинетический анализ двухстадииных ферментативных реакций,
не подчиняющихся уравнению Михаэлиса — Ментен 111
§ 1. Ингибирование субстратом 111
§ 2. Активация субстратом . . .113
§ 3. Кинетика ферментативных реакций при условии [E]o~[S]0.
Определение абсолютной концентрации фермента из кинетических данных 114
§ 4. Кинетика ферментативных реакций при избытке фермента
([E]0>[S]o) " 46
Задачи. 116
Ответы и решения 127
Литература . 143
Глава 7. Стационарная кинетика трехстадийных ферментативных
реакций. Определение значений констант скоростей промежуточных
стадий 144
§ 1. Кинетическое описание трехстадийных ферментативных реакций 144
§ 2. Селективное влияние эффекторов (ингибиторов или активаторов)
на стадию ацилирования 145
319
§ 3. Влияние дополнительных нуклеофильных агентов на стадию
деацилирования
Задачи .
Ответы и решения
Литератур,а
Глава 8. Применение интегральной формы уравнения скорости для
кинетического анализа ферментативных реакций
§ 1. Интегральный анализ кинетики действия ферментов
§ 2. Ингибирование продуктом реакции .
§ 3. Инактив.ация фермента в ходе реакции
Задачи .
Ответы и решения
Литература
Глава 9. Нестационарная кинетика ферментативных реакций
§ 1. Методы предстационарной кинетики
§ 2. Релаксационные методы
Задачи
Ответы и решения
Литература
Глава 10. Влияние рН на скорость ферментативных реакций
§ 1. рН-Зависимость двухстадийной ферментативной реакции
§ 2. рН-Зависимости трехстадийной ферментативной реакции
§ 3. Ионизация субстрата
§ 4. Нахождение значений рК по кривым рН-зависимостей
ферментативных реакций
Задачи .
Ответы и решения
Литература
Глава 11. Влияние температуры на кинетику ферментативных реакций
§ 1. Зависимость кинетических и равновесных параметров
ферментативных реакций от температуры
§ 2. Изучение термодинамики конформ.ационных изменений активных
центров ферментов
Задачи .
Ответы и решения
Литература
Глава 12. Влияние диффузии на скорость гомогенных химических
реакций и реакций, катализируемых иммобилизованными ферментами
§ 1. Феноменологическое описание диффузии
§ 2. Влияние диффузии на кинетику гомогенных химических реакций
§ 3. Роль диффузии в реакциях, катализируемых
иммобилизованными ферментами
Задачи .
Ответы и решения
Литература
Глава 13. Применение теории графов для составления уравнений
кинетики ферментативных реакций
§ 1. Элементы теории графов
§ 2. Нахождение уравнений скорости стационарных ферментативных
реакций методом графов . ...
§ 3. Нахождение уравнений скорости ферментативных реакций,
включающих равновесные стадии
3 ,а д а ч и
Ответы и решения
Литература
Указатель используемых наименований ферментов
Предметный указатель
320
СПИСОК ЗАМЕЧЕННЫХ ОПЕЧАТОК
Страница
Строка
Напечатано
Следует читать
14 сверху
3 снизу
уравнение ( )
10 сверху
11 сверху
14 сверху
5 сверху
уравнение (9.37)
уравнение (9.73)
8 сверху
17 сверху
сноска
19 сверху
2 снизу
с(0)
a log {N%Hv\
Ф,- — Ф^
ASf = 24,3 э. е.
l/a + /Cs[S]0
Кр _ 1
k-s [Р]
/г_(ин)[Е]-[Р]
/ *j»[s]B у
\ Ь + Ь I
1
t2
Наблюдаются колоколо-
образные кривые
Ф=Я
/
[Е]0
К d
Ат(каж)
11 • ЮвМ^сек^- [-1]
[Е]0 < 8,44 • 1010 М
М
с0
alog[W2tf4]
ф,-—ф;
ASf = — 24,3 э. е.
l/a + /Cs/[S]e
Кр 1
*-. ' [Р]
*_(„+1) [Е]-[Р]
/ fe2[S]o у
1
Наблюдаются
несимметричные колоколообраз-
ные кривые
*=*Yi
■ IE],
т(каж)^
11- 10вМ-1сек-1[П]
[Е]0<8,44 • 10-1° М
k
-\-
-S
Зак. 370