/































































































































































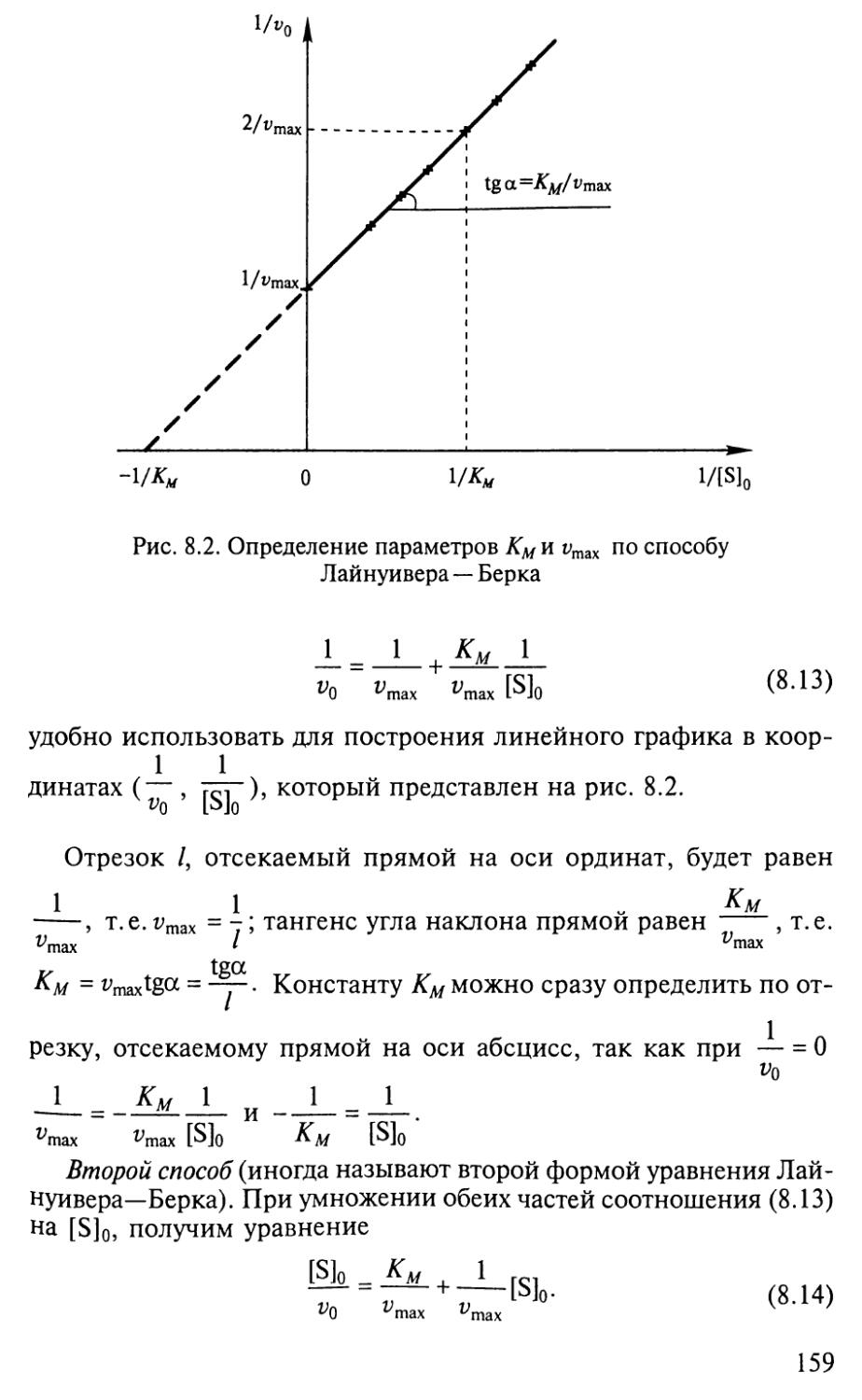


































































































Текст
ВЫСШЕЕ ОБРАЗОВАНИЕ
В. М. БАЙРАМОВ
основы
ХИМИЧЕСКОЙ
КИНЕТИКИ
И КАТАЛИЗА
Под редакцией академика РАН В. В. ЛУНИНА
Допущено
Советом по химии У МО по классическому университетскому образованию
в качестве учебного пособия для студентов
химических факультетов университетов, обучающихся
по специальности 011000 «Химия» и направлению 510500 «Химия»
Москва
ACADEMIA
2003
УДК 541.124
ББК 24.54я73
Б187
Рецензенты:
д-р хим. наук, профессор О. М. Полторак,
д-р хим. наук, профессор М. Я. Мельников (Московский государственный
университет им. М. В. Ломоносова)
Байрамов В.М.
Б187 Основы химической кинетики и катализа: Учеб. пособие
для студ. высш. учеб. заведений / Вадим Михайлович Байра¬
мов. — М.: Издательский центр «Академия», 2003. — 256 с.
ISBN 5-7695-1297-0
В пособии изложены теоретические основы различных направлений
химической кинетики (феноменологическая кинетика, сложные, цепные
и фотохимические реакции, теории химической кинетики), а также гомо¬
генного, ферментативного и гетерогенного катализа. Каждая глава посо¬
бия завершается вопросами и заданиями для самоконтроля.
Для студентов химических факультетов университетов и химических
специальностей вузов.
УДК 541.124
ББК 24.54я73
ISBN 5-7695-1297-0
© Байрамов В.М., 2003
© Издательский центр «Академия», 2003
Памяти моего сына Александра
ПРЕДИСЛОВИЕ
Данное учебное пособие написано на основе многолетнего опыта
преподавания автором физической химии в МГУ им. М. В. Ломо¬
носова в соответствии с утвержденной программой курса физиче¬
ской химии для химических факультетов университетов, а также
работы за рубежом, и посвящено разделам «Химическая кинети¬
ка» и «Катализ».
Цель учебного пособия — дать представление о теоретических
основах и современном состоянии различных направлений хими¬
ческой кинетики, таких, как феноменологическая кинетика, слож¬
ные, цепные и фотохимические реакции, теории химической
кинетики, а также гомогенного, ферментативного и гетерогенно¬
го катализа. Для облегчения усвоения материала при самостоя¬
тельном изучении каждая глава пособия завершается вопросами
для самоконтроля.
В ряде глав, например во 2, 4, 7 и 10-й, представленный мате¬
риал, возможно, превышает объем действующей университетской
программы, но отражает современный уровень исследований и
несомненно полезен для повышения знаний студентов. В пособии
не рассматриваются реакции в потоке, так как этот вопрос вхо¬
дит в программу курса химической технологии. Пособие предназ¬
начено для студентов химических факультетов университетов;
может быть полезно для аспирантов и молодых преподавателей,
ведущих семинарские занятия. Список рекомендуемой и допол¬
нительной отечественной и иностранной литературы приведен в
конце пособия.
Автор выражает глубокую признательность профессорам хими¬
ческого факультета МГУ О. М. Полтораку и М. Я. Мельникову за
внимательное прочтение рукописи и высказанные ими ценные
критические замечания, а также канд. хим. наук О. В. Давыдовой
за большую помощь при оформлении и подготовке рукописи к
печати.
Автор
ЧАСТЬ 1. ХИМИЧЕСКАЯ КИНЕТИКА
Это великая книга, на полстолетия
вперед наметившая путь развития хи¬
мической кинетики.
Н. Н. Семенов
(о книге Я.Х. Вант-Гоффа «Очерки хи¬
мической динамики», 1884)
ГЛАВА 1. ФЕНОМЕНОЛОГИЧЕСКАЯ КИНЕТИКА
1.1. Основные понятия и определения
Химическая кинетика — раздел физической химии, посвящен¬
ный изучению закономерностей протекания химических процес¬
сов во времени.
Слово «кинетика» (от греч. kinetikos — движение) означает из¬
менение. Химики используют этот термин для описания скорости
реакции и ее изменения в зависимости от различных парамет¬
ров.
Кинетическое исследование на первом этапе, традиционно на¬
зываемом формальной кинетикой, может включать в себя:
полный химический анализ реакционной смеси, позволяющий
написать уравнение баланса реакции;
измерение через определенные промежутки времени химиче¬
ского состава реакционной системы;
количественное изучение влияния каждого параметра, способ¬
ного изменить скорость реакции (температуры, давления, кон¬
центраций, природы растворителя, катализатора и т.д.);
наблюдение за особенностями протекания реакции (ускорени¬
ем, замедлением, изменением окраски и т.п.).
На базе полученных экспериментальных данных находят соот¬
ношения между различными измеряемыми величинами: концен¬
трациями и временем, скоростями и концентрациями, скоростя¬
ми и различными параметрами. Этот этап исследований приводит
к установлению кинетических закономерностей, которые харак¬
теризуют данную реакцию.
Рассмотрим химическую реакцию в общем виде
v 1В! + ... —» V, В ( + ..., (1.1)
где В], В,... — соответственно исходные вещества и продукты; vb
v(... — стехиометрические коэффициенты.
4
Для упрощения записи используем следующую форму уравне¬
ния (1.1):
o-»v;b; + ...-v,b, + ... = 2v,b,., (1.2)
/■
где коэффициент V,- считается алгебраически положительным
(V/>0), если компонент В,- относится к продуктам, и алгебраиче¬
ски отрицательным (v,<0), если В,- относится к исходным веще¬
ствам.
Изменение в процессе реакции количества вещества В,-, моль,
связано с химической переменной известным соотношением
сЦ = v,-dJ;, (1.3)
размерность £, в системе СИ — [моль]. Интегрирование формулы
(1.3) в интервале времени от t\ до t2 позволяет получить для пря¬
мой реакции, протекающей слева направо, следующее соотношение:
Л2-/11 =v(§2-$i),
где £2-£i>0.
В начальный момент времени tx = 0 реакция еще не протекает,
£, = 0 и соотношение (1.4) переходит в
n,-n0 = vt>l. (1-5)
Это уравнение позволяет вычислить значение глубины проте¬
кания реакции ко времени t на основании измерения количе¬
ства п только одного компонента, присутствующего в реакцион¬
ной смеси. Очевидно, на опыте выбирают компонент, наиболее
легко определяемый аналитически (одно из исходных веществ либо
один из продуктов).
Важным кинетическим параметром при изучении химических
реакций является скорость. Современное определение скорости ре¬
акции: число элементарных актов или изменение количества ве¬
щества (исходного или продукта) в единицу времени в единице
объема (V), рассчитанное на единицу его стехиометрического ко¬
эффициента (V,), дает не зависящее от природы компонента, ис¬
пользуемого для нахождения скорости, общее выражение
1
* v,V
d/i,-
“d7
(1-6)
Например, для химических реакций, протекающих соответ¬
ственно в газовой и жидкой фазах
2N205—»2N204+ 02
BrOj + 5Br" + 6Н+ -> 3Br2 + 3H20
скорости запишутся следующим образом:
1 d/?N2o5 _ 1 dflN2Q4 _ J_ d^o2_
2V dt 2V dt V dt
_ 1 d»BiQ3- 1 dnB[- _ 1 AnH+ _ 1 6nBt2
V At 5V At 6V At 3V At
Скорость по определению является положительной величиной,
поскольку Ащ> 0. Знак минус в ряде вышеприведенных выраже¬
ний указывает на расходование исходных веществ в течение реак¬
ции. Соотношение (1.6) с учетом формулы (1.3) примет вид
•4S-
Это уравнение наглядно показывает, что значение скорости v
не зависит от природы вещества В„ выбранного для определения
скорости. Определение скорости через соотношения (1.6) и (1.7)
справедливо для любого объема, но если объем постоянен, то его
можно ввести под знак дифференциала в соотношение (1.6) и
получить другое, наиболее часто используемое выражение
1 Ап, 1 ( Ап/
V = '-I'
V/F At V,-1 VAt
1 dC' (1.8)
V,- At
где С, ~~у~ конйентРайия компонента В„ выраженная в моль • л-1
или молек • см-3.
Отметим, что при переменном объеме скорость реакции не
dC
равна производной концентрации по времени, v * —— :
d/
dC A(n/V) 1 An n AV
At ~ At ~V At V2 At :
(1.9)
здесь первый член выражения —— указывает на изменение кон-
V At
центрации в ходе химической реакции, второй — на изменение
концентрации при изменении объема системы. Наглядным при¬
мером изменения концентрации без протекания химической ре¬
акции является движение газа в цилиндре с поршнем.
В дальнейшем рассматриваются в основном реакционные си¬
стемы при постоянном объеме, поэтому чаще используется со¬
отношение (1.8). Скорость реакции очевидно имеет размерность
[С-Г1], т.е. [моль л-1 с-1], [моль-см_3-с-1] или [молек-см-3• с'1].
6
Для реакций в газовой фазе при постоянном объеме стремле¬
ние выразить скорость реакции через парциальные давления р
требует аккуратности. Для идеального газа
p,V = tijRT, или Pi = HiRT/V= CjRT.
Если Т = const, то Pi прямо пропорционально С„ следовательно,
1 dp, 1 d С,
V,- d t v,- d t
(1.10)
Эти соотношения принимают другой вид, если в газовой фазе
р = const. Экспериментальным путем для многих реакций, проте¬
кающих при постоянной температуре, было показано, что ско¬
рость реакции в каждый момент времени пропорциональна про¬
изведению текущих концентраций взаимодействующих веществ,
возведенных в определенные степени.
Этот кинетический закон, получивший название основного по¬
стулата химической кинетики, был сформулирован впервые
К.Гульдбергом и П.Вааге в 1879 г. в виде общего выражения ки¬
нетической формы закона действующих масс. Для реакции (1.1)
запишем кинетическое уравнение
где к — коэффициент пропорциональности, который не зависит
от концентраций реагирующих веществ, но сильно зависит от тем¬
пературы, С, — концентрации реагентов, а, — экспериментально
определяемые показатели степени при концентрации /-го компо¬
нента.
По предложению Я. Вант-Гоффа к называют константой ско¬
рости реакции. В 1887 г. С. Аррениус предложил для £ другое назва¬
ние — «удельная скорость реакции», мотивируя это тем, что при
С; = 1, согласно формуле (1.11), fc = v, т.е. А:численно равна скоро¬
сти реакции при концентрациях реагентов, равных единице. Од¬
нако общепринятым названием к является предложенный Вант-
Гоффом термин «константа скорости реакции».
Коэффициент а, называют порядком реакции по веществу В,,
i = n — порядок реакции в целом.
Важно подчеркнуть, что ^а„ как правило, отличается от сум¬
мы стехиометрических коэффициентов v, уравнения (1.1) и толь¬
ко для простейших (элементарных) реакций Xa< = /I = Xv<-
Размерность константы скорости [/:] = [С]'“"[г]'1.
Часто используют термин «псевдо-» или «наблюдаемый поря¬
док» реакции, если в ходе ее концентрации одного или несколь¬
7
ких реагентов остаются практически постоянными. Например, клас¬
сическая реакция инверсии тростникового сахара
С|2Н220„ + Н20 н?°* > 2С6Н|206,
катализируемая (ускоряемая) в присутствии сильных кислот, дол¬
жна иметь, согласно основному постулату, третий порядок
v = ^[Ci2H220n][H20][H30+].
Однако, благодаря избытку концентрации воды в водном ра¬
створе по сравнению с концентрациями других реагентов и по¬
стоянству концентрации ионов гидроксония (катализатора), фак¬
тически со временем изменяется только концентрация сахара,
т. е. реакция имеет наблюдаемый первый порядок
» = *1С12Н22Оп],
где *' = *[Н20][Н30+].
Наряду с термином «порядок реакции», Я.Вант-Гофф ввел по¬
нятие «молекулярность»: «Ход химического превращения характе¬
ризуется исключительно числом молекул, при взаимодействии ко¬
торых происходит превращение... Именно с этим числом должна
быть связана естественная классификация реакций, которые я пред¬
полагаю называть моно-, би- и тримолекулярными реакциями».
Говоря современным языком, молекулярность реакции — число
молекул, участвующих в одном элементарном реакционном акте.
Различие важных в химической кинетике понятий «порядок» и
«молекулярность» заключается в следующем:
молекулярность реакции имеет ясный физический смысл, ука¬
зывая на конкретное число частиц (атомов и/или молекул), уча¬
ствующих в элементарной реакции. Она принимает целочисленные
значения, М= 1, 2, 3, поскольку вероятность одновременного стол¬
кновения четырех и более молекул близка к нулю. Наконец, поня¬
тие «молекулярность» неприменимо к любой сложной реакции;
порядок реакции — математическое число, формальная вели¬
чина, принимающая любые значения, в гомогенной среде обыч¬
но изменяется от 0,5 до 4; этот термин используется при описа¬
нии простых и сложных реакций. Порядок реакции зависит, как
правило, от механизма сложной реакции и, следовательно, от
влияющих на него параметров (концентрации, давления, темпе¬
ратуры, катализатора).
Механизмом химической реакции называют совокупность эле¬
ментарных стадий, составляющих процесс превращения исход¬
ных реагентов в конечные продукты. При изучении сложных реак¬
ций в кинетике решают две задачи.
Прямая задача: на основании известного механизма и кон¬
стант скорости каждой элементарной стадии с помощью кине¬
тического закона действующих масс составляют систему диффе¬
ренциальных уравнений, решение которой (в квадратурах или
путем численного интегрирования) позволяет получить кинети¬
ческое уравнение, выраженное через концентрации исходных
веществ и продуктов реакции.
Обратная задача: на основании экспериментально полученно¬
го набора кинетических кривых проверяют предполагаемый меха¬
низм реакции, определяют порядок и константу скорости каждой
из ее стадий. Эта задача часто оказывается достаточно сложной и
неоднозначной.
Вопросы и задания для самоконтроля
1. Что означает термин «кинетика»? Что составляет предмет химиче¬
ской кинетики?
2. Какие этапы включает в себя кинетическое исследование, тради¬
ционно называемое формальной кинетикой?
3. Какие кинетические закономерности может получить исследова¬
тель, используя экспериментальные данные?
4. Каким соотношением связаны изменение количества реагирующего
вещества и химическая переменная? Проинтегрируйте это соотношение и
укажите, какой важный практический вывод оно позволяет сделать.
5. Дайте современное определение скорости химической реакции. От¬
вет проиллюстрируйте на конкретном примере.
6. Может ли скорость реакции быть отрицательной величиной? От¬
вет мотивируйте.
7. Приведите выражение скорости реакции через химическую пере¬
менную. Какой вывод оно позволяет сделать?
8. Получите выражение скорости химической реакции через кон¬
центрации реагирующих веществ. При каком условии оно справедливо?
Укажите размерность скорости реакции для различных единиц измере¬
ния концентрации и времени.
9. Докажите, что при переменном объеме скорость реакции не рав¬
на производной концентрации по времени. Приведите пример измене¬
ния концентрации без протекания химической реакции.
10. Сформулируйте основной постулат химической кинетики и при¬
ведите его математическое выражение. Охарактеризуйте каждую из вхо¬
дящих в него величин.
11. Дайте определение понятий «порядок реакции по веществу» и
«порядок реакции в целом».
12. Связаны ли порядки реакции по отдельным веществам со стехио¬
метрическими коэффициентами брутто-реакции? Ответ мотивируйте и
поясните на примере.
13. Что означает термин «наблюдаемый порядок»? Ответ поясните на
примере.
14. Объясните термин «молекулярность» реакции.
15. В чем заключается различие понятий «порядок» и «молекулярность»
химической реакции? В каком случае эти величины могут быть равны?
9
16. Каков физический смысл константы скорости реакции? Какова
ее размерность?
17. От каких параметров зависит константа скорости реакции?
18. Что означает термин «механизм» химической реакции?
19. Сформулируйте прямую задачу химической кинетики.
20. В чем состоит обратная задача химической кинетики?
1.2. Кинетика односторонних реакций разных порядков
Для нахождения кинетического закона изучаемой реакции при
постоянной температуре необходимо записать выражения (1.6)
или (1.8) в случае закрытой системы для скорости реакции и вы¬
ражение (1.11) для основного постулата химической кинетики.
Приравнивая оба выражения, получаем дифференциальное урав¬
нение, связывающее переменные С,- и t
Интегрирование выражения (1.12) после разделения перемен¬
ных при известных значениях а,- и у, (напомним, что порядок ре¬
акции по веществу и в целом определяется экспериментальным
путем) приводит к математическому соотношению между кон¬
центрацией и временем, что позволяет сравнивать рассчитанные
по этому уравнению значения с экспериментальными кривыми
зависимости С, от /.
Реакции первого порядка. К ним относятся реакции изомериза¬
ции, разложения соединений в газовой фазе, например, (С2Н5)20,
CH3N = NCH3, а также процессы радиоактивного распада.
Рассмотрим схему реакции согласно уравнению
(1.12)
А L +
количество вещества (моль) / = 0 п0 0 [А]о - -тт - а
количество вещества
где А — вещество; L — продукт.
Уравнение (1.12) в этом случае принимает вид
(1.13)
10
Разделяя переменные х и 7, получим
— = kxdt. (1.14)
а-х
Интегрирование формулы (1.14) в пределах соответственно от
О до х и от 0 до Г дает - \п(а-х) = А:, Г + const. С учетом начальных
условий при 7=0, х = 0, const = -1пй. Следовательно, после под¬
становки получаем искомое кинетическое уравнение
In а - 1п(а - х) = In а = kxt. (1.15)
а-х
Потенцирование выражения (1.15) позволяет получить зависи¬
мость концентрации исходного вещества и продукта от времени
[А] = а-х-aexp(-kit) и [L] =х= я[1 -ехр(-А:17)]. (1.16)
Из выражения (1.15) находим выражение для константы ско¬
рости реакции первого порядка
к1=-1п — • (1.17)
7 а-х
По определению, данному выше, размерность [Arj] = [с]|-л[7]-1 =
= [7]-1, что видно и из выражения (1.17). Обычно [А:,] = с“] или мин-1.
Обратим внимание на то, что в выражении (1.17) отношение на¬
чальной и текущей концентраций (или количеств) реагента мо¬
жет быть заменено на отношение любых величин, пропорцио¬
нальных числу молей.
В кинетике для характеристики реакционных процессов широ¬
ко используют понятие время полупревращения, 71/2 — время,
необходимое для превращения половины исходного количества
реагирующего вещества.
Для реакции первого порядка после подстановки в формулу
(1.17) значения х = ^ при 7 = 71/2 получим
'1/2=Т- = (1Л8>
Как видно из формулы (1.18), время полупревращения в этом
случае зависит только от значения константы скорости реакции
первого порядка, к, и не зависит от начальной концентрации реа¬
гента. Благодаря этому свойству величина времени полупревраще¬
ния применяется в радиохимии в качестве основной характеристи¬
ки процесса радиоактивного распада (ее называют периодом по¬
лураспада). Например, для 2^Fr /,/2 = 4,8 мин. Согласно выражению
(1.18), вычислим константу скорости этого процесса
, _ 0,693 _ 2 41. in-3 c-i
4,8 -60 ~ С-
11
Введем еще один термин — среднюю продолжительность жиз¬
ни молекул реагирующего вещества, т. Используя математическое
понятие средней величины, запишем
x = ^-ltdN, (1.19)
Hot
где t — время жизни отдельной молекулы; N0 — общее число мо¬
лекул, N— число молекул в момент времени t\ dN — число моле¬
кул, прореагировавших за время dt.
С учетом принятых выше обозначений и уравнения (1.16) по¬
лучаем
ч оо 1 °° °°
т = — J td{a - х) = - J tk\at~k{,dt = к\ J te~kl'dt. (1.20)
а о а о о
После интегрирования по частям, принимая t = и и e~kl'dt = dv,
приходим к искомому результату
т = 1 /*,. (1.21)
Выражение (1.21) позволяет понять физический смысл кон¬
станты скорости реакции первого порядка: ее обратная величина
равна средней продолжительности жизни молекул реагирующего
вещества. Отметим, что для реакций первого порядка т> /|/2.
Реакции второго порядка. К ним относятся многие гомогенные
реакции в газовой и жидкой фазах, например, реакции разложе¬
ния и образования йодоводорода, омыления этилацетата щелочью,
димеризации циклопентадиена, а также большая группа бимоле¬
кулярных реакций с участием атомов и свободных радикалов.
При рассмотрении реакций второго порядка положим, что ре¬
акция имеет первый порядок по каждому из двух реагентов и оба
вещества реагируют в эквивалентных соотношениях.
Используя принятые ранее обозначения для концентраций, по
аналогии получим
А + В -> L+M
t=0 а b
t (а-х) (Ь- х)
В этом случае дифференциальное уравнение (1.12) примет вид
= ^7 = кЛа " Х)(Ь " х)’ 0 '22)
где к2 — константа скорости реакции второго порядка.
Разделение переменных приводит к уравнению вида
TZ -777 г = М'- (1.23)
(а - х)(Ь - х)
12
Рассмотрим два случая.
1. Концентрации реагентов А и В равны, т.е. а = Ь. Выражение
(1.23) преобразуется в дифференциальное уравнение
<U4>
которое легко интегрируется с учетом начальных условий (при
/=0 х = 0) и принимает вид искомого кинетического уравнения
—— -- = k2t. (1.25)
а-х а
Как видно из соотношения (1.25), величина, обратная теку¬
щей концентрации (а - х), линейно возрастает со временем. Зави¬
симость концентрации образующегося продукта, х, от времени
выражается из (1.25) формулой
* = (1.26)
1 + ak2t
Размерность константы скорости реакции второго порядка
[Лг2] = [с]|_л[/]_1= [с]-'[/]-1, т.е. константа выражается в моль_|-л-с_1
или моль-1-см3-с-1, причем численное значение константы зави¬
сит от выбора единиц концентрации. Понятие времени полупрев¬
ращения /1/2 однозначно применимо только для случаев, когда
одно вещество превращается по второму порядку или два соеди¬
нения присутствуют в одинаковых концентрациях. В данном слу¬
чае преобразуем выражение (1.25) к виду
к2=\ —. (1.27)
t а{а - X)
Подставляя в формулу (1.27) значение а-а/2 для t = t]/2, по¬
лучим
/,/2=-L (1.28)
ак2
Для реакции второго порядка (СА= Св) время полупревраще¬
ния обратно пропорционально константе скорости и начальной
концентрации реагирующего вещества.
Определим среднюю продолжительность жизни молекул реаги¬
рующего вещества для реакции второго порядка. Выразив (а-х)
из формулы (1.25) и подставив в формулу (1.20), получим
dt_ 1
a }0k2t +1 / а ак2
' ( 1 л
In
к21 + —
о
Стремление т —»со указывает на то, что для реакции второго
порядка скорость расходования исходного вещества замедляется
13
со временем в еще большей степени, чем для реакции первого
порядка.
2. Концентрации [А] и [В] первоначально различны, т.е. аФ Ь.
Интегрирование уравнения (1.23) проводят методом неопре¬
деленных коэффициентов. Для этого левую часть уравнения разла¬
гают на дроби
находят значения констант D=(b- а)~1 и Е-{а- Ь)~'. Затем урав¬
нение (1.23) интегрируют (левую часть — от 0 до х и правую часть
от 0 до О
С учетом начальных условий приходим к искомому кинетиче¬
скому уравнению
Интересен случай, когда концентрация b » а. Поскольку х ни¬
когда не может быть больше а, можно считать, что в соотношении
(1.32) (b-а) и (b-x) ~ b и уравнение существенно упрощается
Идентичность выражений (1.33) и (1.15) указывает на то, что
в данном случае речь идет о кажущейся реакции первого порядка
с константой Ат, = Ьк2. Это означает, что если реакция, имеющая
кинетический первый порядок по каждому из реагентов, осуще¬
ствляется в присутствии избытка одного из них, концентрация
второго реагента будет убывать по экспоненциальному закону со
скоростью, пропорциональной концентрации реагента, взятого в
избытке. В рассмотренном случае, когда а Ф Ь, нельзя говорить о
времени полупревращения реакции в целом, но можно приме¬
нять этот термин в отношении каждого из реагентов, учитывая,
что /|/2(А,) зависит от начальной концентрации обоих реагентов.
Реакции rt-го порядка. В этом случае дифференциальное и интег¬
ральное кинетические уравнения легко получить, если все реа¬
генты присутствуют в равных количествах и взаимодействуют в
эквивалентных соотношениях. Дифференциальное уравнение ско¬
рости (1.12) запишется так:
(а - х){Ь - х) а-х b-х’
(1.30)
(1.31)
(1.32)
(1.33)
d {а - х) _ dx
d t d/
—- = kn(a - x)n.
(1.34)
14
Разделяя переменные и интегрируя в пределах от 0 до х и соот¬
ветственно от 0 до /, для п ф 1 получим кинетическое уравнение
/ 1\ / \й-1 п-1 ~ ^п^ (1.35)
(л-1) (а-х)" 1 а" _
или
к =- 1 1 . (1.36)
л ^ (л -1) а" 1 (д - х)" 1
Размерность константы скорости реакции я-го порядка: [кон¬
центрация]1_л- [время]-1.
Общее выражение для времени полупревращения реакции по¬
лучим из уравнения (1.35) или (1.36) путем подстановки х=а/2
при t= tl/2:
1 1 2"-1 -1
<1J7>
Как видно из формулы (1.37), для любой реакции, порядок
которой отличен от единицы, время полупревращения реакции
обратно пропорционально начальной концентрации реагента, воз¬
веденной в степень (л - 1).
Рассмотрим частный случай реакций нулевого порядка, кото¬
рые встречаются в гетерогенных и фотохимических процессах. Под¬
ставляя значение п- 0 в кинетическое уравнение (1.35), после пре¬
образований получим выражение
а-х = a- kat, (1-38)
где ко — константа скорости реакции нулевого порядка. Размер¬
ность [А^] = [с][/]-1, например, моль-л-1-с-1.
Очевидно, что текущая концентрация исходного вещества в
этом случае линейно уменьшается со временем.
Время полупревращения реакции нулевого порядка находим
путем подстановки значения л = 0 в уравнение (1.37):
'./2=^- (1.39)
2ко
т. е. время полупревращения обратно пропорционально константе
скорости и прямо пропорционально начальной концентрации ре¬
агента. Оценим среднюю продолжительность жизни молекул реа¬
гирующего вещества для реакции нулевого порядка. Выражение
(а-х) из (1.38) подставляем в формулу (1.20) и находим
1 “ 1 а
х = -\(a-kot)dt = - \ (a-k0t)dt - —(1.40)
а о a q Zkq
15
Таким образом, т = /1/2, т.е. для реакции нулевого порядка
(в отличие от других) средняя продолжительность жизни моле¬
кул реагирующего вещества равна времени полупревращения.
К числу реакций третьего порядка относятся реакции оксида
азота(П) с галогенами, водородом и кислородом, например:
2NO + С12—»2NOC1,
а также большая группа процессов рекомбинации свободных ато¬
мов и радикалов при участии третьей частицы.
Рассмотрим вначале реакции типа ЗА—» продукты и А+В +
+ D —> продукты при условии, что начальные концентрации ис¬
ходных веществ равны: [А]0 = [В]0 = [D]0 = С0. Дифференциальное
кинетическое уравнение примет вид
-^- = кС\ (1.41)
at
Его интегрирование после разделения переменных приводит
к соотношению
u_L_J_"
2 С2 С02
= kt.
(1.42)
Из уравнения (1.42) после замены t на /1/2 и Сна С0/2 получим
выражение для времени полупревращения
3
/,/2 2кСп '
(1.43)
•2,-1
Размерность константы скорости реакции третьего порядка C~2t
и, как правило, приводится в моль-2-л2-с-1 или моль-2-см6-с-1.
Для реакции типа 2А + В —» продукты, если [А]0*[В]0 или
[А]0 Ф 2[В]0, дифференциальное кинетическое уравнение запишется так:
(1-44)
После интегрирования конечное выражение принимает вид
m-[au
[а]Да]
[AUB]
= kt. (1.45)
Для реакции типа А + В -» продукты, если [А]0 Ф [В]0 и приме¬
ним кинетический закон (1.44), получим интегральное кинети¬
ческое уравнение
1
([В],-[А],)([А]Э-[А]) [B^IA]
[аЩа] +1П[А]0[В]
= to. (1.46)
16
а-х
О
Рис. 1.1. Кинетические кривые реакций разных порядков п
для одинаковых концентраций исходных веществ
Для реакции A + B + D продукты, при условии, что [А]0*
Ф [В]0 Ф [D]0, дифференциальное уравнение запишется так:
На рис. 1.1 показано влияние порядка реакции (от 0 до 2) на
уменьшение текущей концентрации исходного вещества в зави¬
симости от времени.
1. Получите в общем виде дифференциальное уравнение, связываю¬
щее концентрацию и время. Какие сведения для этого необходимы?
2. Выведите дифференциальное и интегральное кинетические уравне¬
ния для односторонних реакций первого порядка. Приведите примеры
таких реакций.
3. Напишите выражение для константы скорости реакции первого
Порядка, используя начальную и текущую концентрации реагирующего
2-Зак. 159с. 17
(1.47)
Интегральное кинетическое уравнение имеет вид
in ([a]/[a]q ) чтдвь)
(W, -[ВЩО], -[AJ,)' ([AJ, -[вЩв], -Р1,)
■"([рури
(1.48)
Вопросы и задания для самоконтроля
вещества. Какова размерность константы скорости реакции первого по¬
рядка? Можно ли вместо концентраций использовать другие величины?
4. Что означает термин «время полупревращения», tX/{l От каких ве¬
личин зависит /1/2 реакции первого порядка?
5. Получите аналитическим путем выражение для средней продол¬
жительности жизни молекул т в случае реакции первого порядка. Какая
из величин больше — т или /1/2?
6. Приведите примеры химических реакций второго порядка. Явля¬
ются ли они бимолекулярными? Ответ поясните.
7. Выведите дифференциальное и интегральное кинетические урав¬
нения для реакций второго порядка А + В —> L + М при равных исходных
концентрациях реагентов.
8. Получите выражение для константы скорости реакции второго по¬
рядка, если начальные концентрации реагирующих веществ одинаковы.
Укажите размерность константы скорости.
9. Выведите выражение для времени полупревращения /1/2 реакции
второго порядка при одинаковых исходных концентрациях реагентов. От
каких величин зависит /1/2 в этом случае?
10. Получите выражение для средней продолжительности жизни мо¬
лекул т в случае реакций второго порядка ([А]0 = [В]0). Какой вывод мож¬
но сделать из полученного результата?
11. Выведите интегральное кинетическое уравнение для реакции вто¬
рого порядка при условии, что [А]0^[В]0.
12. Какой вид примет кинетический закон для реакции второго по¬
рядка при условии, что [А]0«с[В]0? Проанализируйте полученный ре¬
зультат.
13. Можно ли применять понятие о времени полупревращения для
реакций второго порядка в целом, когда [А]0^[В]0? Получите аналити¬
ческие выражения для времени полупревращения по каждому веществу
^ 1/2(А) и ^1/2(В)*
14. Приведите примеры реакций нулевого порядка. Выведите диффе¬
ренциальное и интегральное кинетические уравнения для реакции нуле¬
вого порядка. Какова размерность константы скорости в этом случае?
15. Получите выражение для времени полупревращения tl/2 реакции
нулевого порядка. От каких величин зависит /1/2 в этом случае?
16. Выведите выражение для средней продолжительности жизни т мо¬
лекул реагирующего вещества в случае реакции нулевого порядка. Срав¬
ните между собой значения т и /1/2 для реакций нулевого порядка.
17. Приведите примеры реакций третьего порядка. В чем заключается
особенность их протекания в ряде случаев?
18. Выведите дифференциальное и интегральное кинетические урав¬
нения для реакций третьего порядка при одинаковых концентрациях ре¬
агирующих веществ.
19. Получите выражение для времени полупревращения в реакции тре¬
тьего порядка при условии равенства исходных концентраций реагентов.
20. Выведите дифференциальное и интегральное кинетические урав¬
нения для реакций п-то порядка.
21. Приведите выражение для времени полупревращения в реакции
л-го порядка при равных начальных концентрациях реагирующих веществ.
18
22. Приведите размерности констант скорости реакций разных по-
рядков: а) 0; б) 1; в) 2; г) 3; д) п.
23. Получите уравнения зависимости концентрации образующегося
продукта X от времени для реакций: а) 0-го; б) 1-го; в) 2-го ([А]0 = [В]0)
порядков.
24. Укажите графический способ определения значений констант ско¬
рости для реакций целочисленных порядков: а) 0; б) 1; в) 2; г) 3. При¬
нять, что для в) [А]0 = [В]0 и г) [А]0 = [В]0 = [С]0.
25. Представьте графически зависимость текущей концентрации реаги¬
рующего вещества от времени для реакций различных порядков: а) 0; б) 1;
в) 2; г) 3. Какой вывод о скорости протекания реакций можно сделать?
1.3. Способы определения порядка реакций
из опытных данных
В начале рассмотрим способы, которые используют для нахож¬
дения общего порядка реакции. Экспериментальный метод иссле¬
дования в случае, если в реакции участвуют два и более реагента,
состоит в следующем: все реагирующие вещества берутся в рав¬
ных количествах (концентрациях), т.е. справедливы уравнения,
приведенные в подразд. 1.2 (1.25, 1.27, 1.35, 1.36).
Методы обработки опытных данных разделяются на две груп¬
пы в зависимости от того, используют ли они интегральное кине¬
тическое уравнение.
Интегральные методы определения порядков реакций. Они ос¬
нованы на применении интегрального кинетического уравнения
для обработки первичных экспериментальных результатов концен¬
трация-время. Эта обработка может осуществляться следующи¬
ми способами.
1. Способ подстановки позволяет установить соответствие опыт¬
ных данных одному из интегральных кинетических уравнений, вы¬
веденных в подразд. 1.2 для целочисленных порядков (от 0 до 3).
На практике используют алгебраический и графический варианты
способа. Первый вариант заключается в проверке справедливости
того или иного кинетического уравнения для объяснения массива
экспериментальных данных: проводится расчет константы скоро¬
сти последовательно для реакций разных порядков (от 0 до 3).
Если, например, для реакции нулевого порядка рассчитанные зна¬
чения константы скорости монотонно возрастают или убывают,
делают вывод о непригодности данного кинетического уравнения
и переходят к проверке следующего. В случае, когда при подста¬
новке опытных значений концентрации и времени в очередное
кинетическое уравнение рассчитанные значения константы ско¬
рости колеблются около постоянной величины (с учетом ошибки
определения самих экспериментальных величин), считают, что
19
Рис. 1.2. Графический способ определения порядка реакции:
а — при л = 0; б — при п= 1; в — при л = 2; г — при л = 3
изучаемая реакция в целом имеет искомый порядок. Если ни одно
из кинетических уравнений не дает удовлетворительного резуль¬
тата, можно сделать заключение о сложном характере реакции,
порядок которой может быть дробным и отрицательным. В этом
случае необходимо использовать другие способы определения по¬
рядка реакции, указанные ниже. В целом способ подстановки эк¬
спериментальных данных (С,-, г,) дает надежные результаты для
реакций, степень превращения в которых достигает 70 — 90 %. Гра¬
фический вариант этого способа определения порядка реакции
заключается в построении для реакций 0 —3-го порядков зависи¬
мостей (а-х), 1п(а-х), (а-х)~1 и (а-х)'2 от времени: если экс¬
периментальные данные линеаризуются в соответствующих коор¬
динатах, можно считать, что данная реакция отвечает искомому
порядку. Дополнительно в этих случаях рассчитывают численные
значения констант скорости по тангенсу угла наклона каждой пря¬
мой, как это показано на рис. 1.2.
2. Способ определения порядка реакции по времени полупре¬
вращения. Для реакции л-го порядка (л Ф 1) время полупревраще¬
ния выражается соотношением (1.37)
t 2”-' -1
1/2 к„(п-\)ап-1'
20
Произведение времени полупревращения на кинетически ак¬
тивную начальную концентрацию исходного вещества а, возве¬
денную в степень (л - 1), является постоянной величиной
txllan-x = ^——- = const. (1.49)
(л -1 )кп
Следовательно, для двух опытов, отличающихся начальной кон¬
центрацией исходного вещества, можно записать
(?i/2)iai" ' = (^/2)2^2" '• (1-50)
Логарифмирование выражения (1.50) позволяет алгебраическим
путем вычислить порядок реакции л, согласно уравнению
Я = 1_ЬМ2/М. (151)
\п(а2/щ)
Отметим, что этот способ позволяет найти любые значения
порядка реакции, включая дробные и отрицательные величины.
Широко применяют графическую разновидность указанного
способа. Для этого, логарифмируя выражение (1.37), получают
уравнение
1п?,/2 = 1п2'"';1 - (л - 1)1па. (1.52)
(л -1 )к„
Строя зависимость In /(/2 от 1па, находят порядок реакции по
угловому коэффициенту прямой (л= 1-tga) и константу скоро¬
сти по отрезку, отсекаемому построенной прямой на оси ординат
(рис. 1.3).
Напомним, что время полу¬
превращения реакции первого по¬
рядка не зависит от начальной
концентрации исходного вещества.
Следовательно, если на основании
опытных данных t{/2 не зависит от
начальной концентрации реагента,
реакция имеет первый порядок.
3. Разновидностью предыду¬
щего является способ определе¬
ния порядка реакции на осно¬
вании зависимости времени пре¬
вращения исходного вещества на
определенную долю tl/x от началь¬
ной концентрации (его часто на¬
зывают способом Оствальда —
Нойеса). Использование интег-
Рис. 1.3. Зависимость логарифма
времени полупревращения от лога¬
рифма начальной концентрации
21
рального кинетического уравнения реакции п-то порядка (1.37)
позволяет в этом случае получить новое соотношение
[х/(х - 1)Г‘ -1
6/х ='
(1.53)
(л - \)кап-х ’
В табл. 1.1 представлены значения времени превращения исход¬
ного вещества на определенную долю для различных порядков
реакции.
Таблица 1.1
Порядок
Время превращения 1\/х
6/2
6/3
*1/4
0
а/2к
а/Ък
а/Ак
1
1п2/к
1п(3/2)Д
1п(4/3)Д
2
1/ка
1/(2 ка)
1/(3 ка)
3
3/(2 ка2)
5/(8ка2)
7/(18&д2)
п
2"~1 -1
(3/2)*"1 -1
(4/3)«-1 _1
(л - 1)Ао"_|
(,п - \)капА
(.п - \)капА
Если известен большой массив данных одного опыта, можно
оценить время бд> соответствующее различным значениям х (х = 2,
3, 4...). Численные значения отношений /1/2А/4, Н/т/Н/г, 6/3/6/4
также зависят от порядка реакции, о чем свидетельствует табл. 1.2.
Таблица 1.2
Порядок
Отношение времен превращения
h/i! '1/4
6/2/6/3
6/з/6/4
6 ixl h/y
0
2,00
1,50
1,33
у/х
1
2,41
1,71
1,41
1п[х/(х-1)]
НуКу -1)]
2
3,00
2,00
1,50
(У-!)/(*-0
3
3,86
2,40
1,61
[х/(х -1)]2 - 1
[у/{у- I)]2 -1
п
2я-1 _ 1
2я-1 -1
1
т
[*/(*-1)Г‘-1
(4/3)"-1 -1
(3/2)»-' _ 1
(4/3)"-1 _1
\уКу -1)]"'1 -1
22
Преимущество интегральных методов связано с тем, что они
позволяют использовать первичные данные (концентрация — вре¬
мя), полученные непосредственно из опыта. Однако они приво¬
дят к расчету среднего значения (от 0 до t) константы скорости и,
следовательно, «сглаживают» ее изменения.
Дифференциальный метод определения порядка реакций. Пред¬
ложенный впервые Я. Вант-Гоффом для простых реакций, этот
метод состоит в следующем: для реакции (1.1), в которой началь¬
ные концентрации исходных веществ равны, применяется основ¬
ной постулат химической кинетики г> = /с[В]л в логарифмической
форме
1пц=1пЛ + л1п[В]. (1.54)
Порядок реакции находят либо алгебраическим путем, зная
минимум два значения пар скорость—концентрация (vx, [В], и
V2, [В]2),
ln(i>, /v2)
п =
1п([В],/[В]2)’
(1-55)
либо графически, строя зависимость \nv от 1п[В] — по танген¬
су угла наклона прямой к оси абсцисс (/j = tga), (рис. 1.4).
Экспериментальные результаты (значения v и [В]) могут быть
получены как из начальных участков кинетических кривых, так и
из полной кинетической кривой ([В] — /). Дифференциальный
метод Вант-Гоффа привлекателен своей простотой и теоретиче¬
ски позволяет найти любое значение п. Однако необходимые пары
данных скорость—концентрация экспериментально значитель¬
но труднее получить, чем пары значений концентрация —время.
Поэтому надежные результаты по определению порядка реакций
(с учетом неизбежных ошибок эксперимента) получают для на¬
чальных скоростей и соответствующим им начальных концентра¬
ций реагента.
Метод изолирования Оствальда.
Для определения порядка реакции
по каждому из веществ наиболее
распространенным является ме¬
тод изолирования Оствальда, назы¬
ваемый также методом понижения
порядка реакции.
Intb
Вис. 1.4. Зависимость lntfo от 1п[В]0
в дифференциальном методе
Вант-Гоффа
1п[В]0
23
Если реакция А + В —> С протекает при различных начальных
концентрациях исходных веществ, кинетический закон действу¬
ющих масс примет вид
v=yt[A]“[B]P. (1.56)
Суть метода состоит в том, что проводят изучение зависимости
скорости реакции от начальной концентрации одного из исход¬
ных веществ, например А, поскольку остальные вещества берутся
в большом (20 и более раз) избытке. В этом случае кинетический
закон упрощается
^=^каж[А]“, (1.57)
если [В] з> [А]; ккаж = £[Вр — величина кажущейся константы ско-
роста зависит от концентрации вещества В. Порядок реакции а по
веществу А находят одним из рассмотренных выше способов. Оп¬
ределяя затем при постоянной температуре численные значения
ккаж для различных значений [В]0 ([В]0»[А]0), можно алгебраи¬
ческим или графическим путем рассчитать порядок реакции (3 по
веществу В и константу скорости к по уравнению
1П£каж = 1п£+(3[В]. (1.58)
Зная порядки реакции по отдельным веществам, находят и об¬
щий порядок реакции (п = а + (3).
Вопросы и задания для самоконтроля
1. Какие экспериментальные методы исследований применяют для
определения порядка реакции: а) в целом; б) по отдельному веществу?
2. Какие опытные данные необходимы для определения порядка ре¬
акции: а) в интегральных; б) в дифференциальных методах исследова¬
ний?
3. В чем заключается различие интегральных и дифференциальных
методов определения порядков реакций?
4. Приведите описание алгебраического способа подстановки для оп¬
ределения порядка реакции.
5. Покажите, что для целочисленных порядков реакций (0 — 3) удо¬
бен графический вариант указанного выше способа. Какую дополнитель¬
ную информацию получают в этом случае?
6. Покажите возможности способа определения порядка реакции по
времени полупревращения а) в алгебраическом; б) графическом вари¬
антах.
7. Можно ли определить порядок реакции, зная зависимость времени
превращения исходного вещества на определенную долю /1/л. от его на¬
чальной концентрации?
8. Получите выражения для времени превращения исходного веще¬
ства на треть, Г|/3, в реакциях разных порядков: а) 0; б) 1; в) 2; г) 3.
9. Выведите выражения для времени превращения на четверть, Г1/4,
в реакциях разных порядков: а) 0; б) 1; в) 2; г) 3.
24
10. Вычислите значения отношений Л/2/Л/4» h/i/hp, U/i/t\/4 Для реак¬
ций нулевого порядка.
11. Определите численные значения отношений /1/2Д1/4, /1/2Л1/3» *\/з/*\/4
для реакций первого порядка.
12. Охарактеризуйте достоинства и недостатки интегральных методов
определения порядка реакции.
13. В чем заключается дифференциальный метод определения поряд¬
ка реакций? Каковы его разновидности? Укажите достоинства и недо¬
статки метода.
14. Покажите, каким образом получают пару значений скорость —
концентрация в дифференциальном методе: а) из начальных участков
кинетических кривых; б) из полной кинетической кривой? В каком слу¬
чае получают более надежные результаты?
15. В чем заключается метод изолирования Оствальда, или пониже¬
ния порядка реакции? В каком случае он применяется?
1.4. Зависимость скорости реакции от температуры
Первые количественные измерения температурной зависимос¬
ти скорости химической реакции были проведены JI. Вильгельми
в 1850 г. Позднее Я.Вант-Гофф на основании исследования мно¬
гих реакций в растворах при невысоких температурах сформули¬
ровал эмпирическое правило: при увеличении температуры на 10 °С
скорость гомогенной реакции возрастает в 2 —4 раза, т. е. темпера¬
турный коэффициент скорости реакции
У _ ^г+1о_ -2-4. (1.59)
кт
Следовательно, зависимость константы скорости от темпера¬
туры можно представить следующим образом:
кт ЗдЗ.
= Y 10 • (1.60)
Кт,
С. Аррениус в 1889 г. опубликовал статью, в которой проана¬
лизировал опытные данные восьми различных исследований
(табл. 1.3)*, посвященных температурной зависимости скорости
Реакции, и эмпирические уравнения, предлагавшиеся авторами
Для описания эксперимента.
На основании предложенного им уравнения
Ео
k = Aq rt, (1.61)
гДе А, Еа — не зависящие от температуры параметры уравнения;
— энергия активации реакции; R — универсальная газовая по-
* Штиллер В. Уравнение Аррениуса и неравновесная кинетика. — М.: Мир,
2000.
25
s
X
2
CD
s
s
CL.
a
+
+
Г4 м **+
as 5
OJ ® D,
4 S o.
5 &<
a*
0>'
£ *
« 8
O. 5
<u
03
in
,
СП
оо
СП
СП
СП
^з-
о
V—I
т—t
см
о
о
о
СП
1
СП
1
СП
1
СП
1
СП
1
1
1
СП
I
1
СП
1
Г"
1
X}-
1
оо
1
СП
1
СП
1
СП
1
ОО
ОО
г-
г-
as
Г'-
in
Tf
оо
см
см
см
см
см
СП
СП
см
§ §
g I
S rQ
сз ^
O.
E
cn
o\
о
о
in
II
P
I
in'
CH
<n
У 4
+
m
r-T
in
as
so
^ Г
чо к
о ^
P8
c-
in
I
II
-V
oo
I
in
CH
r-
CM
I
о
ОЙ
о ^
и 5\
^ in
J2P I
I
in
CH
t--
CM
I
CM
О S
f я
II о
чсГ
Jtf) I
Cs
s
Cf
X
03
5°
o3 Я
00 w
ё
5 о
* и
S s
X g
s
IS
5 «
Д <L>
О et
о
<
о
ш
СО
s
§
Cu
E*
3S g
° 5
X к
у g
о 5
i=; л
3s
о
a
О
s
X
H
о
с
Cl,
H
о
a,
<D
и
5S
о
X
о
CS
Q,
CS
X
CS
о
<D
5
Й
t=:
s
H
<£>
7^ 03
S a
>* s
Is
5 s
* о
<L> PQ
S gg
К H
o 3
* Й
° 2
S 5
i3 s
Oh *
in
^t
оо
as
^3-
оо
оо
оо
оо
оо
00
оо
оо
оо
оо
оо
оо
’■"н
1
1—1
Я
О
03
Z
+
С
о
и
и
гм
я
и
оз
Z
о
к
оз
a
2 3S
О, С
£ a.
о g
Он t
Ю к
о 5
a, £
et О
s а,
U VO
£ s
EJ с*
§
оо
оо
Tf
оо
оо
О.
О
Ё
<
S
х
О-
О
а
03
СО
X
<D
О,
>>
°< s 8
о н &
с * Е
В £3
•&
•е
•&
•е
•&
Uh cs
£ ^
Uh 03
U
ю
03
(1 а
х 3
03 ^
' о
к В
CS ^
1
н
X
03
CQ
в
CQ s
OQ S
CQ
S
26
стоянная. С. Аррениус рассматривал для каждой реакции отноше¬
ние констант скоростей при двух различных температурах, т.е. со¬
отношение
(1.62)
Это позволило ему в каждом случае (для данной реакции) оп¬
ределить константу Си затем вычислить по уравнению (1.62) зна¬
чения констант скорости к(Т) для всех температур, по которым
имелись экспериментальные данные. Оказалось, что в большин¬
стве случаев рассчитанные значения к(Т) совпадали с опытными
данными лучше, чем при использовании авторских эмпиричес¬
ких уравнений.
Выбор С.Аррениусом экспоненциальной формы уравнения
(1.61), по-видимому, связан с новейшими научными данными
того времени: во-первых, удобство дифференциальной формы
записи уравнения, предложенного Я. Вант-Гоффом (1884); во-вто¬
рых, высказанная в литературе идея о температурной функции
константы скорости «экспоненциального вида»; в-третьих, нали¬
чие энергетических соотношений, имеющих экспоненциальную
форму типа больцмановского множителя. Заслугой С. Аррениуса
является введение в уравнение (1.61) вместо известной в литера¬
туре и определяемой из опыта постоянной С: новой величины,
названной им энергией активации, Ea-CR. Уже к началу XX в.
уравнение Аррениуса стало общепринятым в кинетических ис¬
следованиях феноменологическим уравнением, достаточно точ¬
ным для многих реакций.
Термодинамическое обоснование температурной зависимости
константы скорости впервые было дано Я. Вант-Гоффом. Выве¬
денное им уравнение изохоры в современных обозначениях имеет
вид
где Кс — константа равновесия элементарной реакции, AU0 —
стандартное изменение внутренней энергии.
Учитывая, что по кинетическому закону действующих масс кон¬
станта равновесия элементарной реакции равна отношению кон¬
стант скорости прямой и обратной реакций, Кс= kx/k.x, уравне¬
ние (1.63) можно записать следующим образом:
din Л, din Л, А U0
(1.64)
d Т dT RT2
27
Полученное соотношение (1.64) позволяет «расщепить» ука¬
занную зависимость и записать выражения для каждой реакции
отдельно в виде
dInAtj _ Еа(i) f/nr\ lT dlnfc-i ^(-l) f(T\ (]
dr ~~dT~~'RT^' (L65)
где AC/0 = Ea(X)- Ea(_X),f(T) — произвольная функция температуры.
Полагая, что для выбранного небольшого интервала температур
величины Еа{Х) и Еа(.Х) не зависят от температуры и функция /(Г)
равна нулю, можно записать в общем виде
din к _ Еа
dТ ~ RT2'
Полученное уравнение называют дифференциальной формой
уравнения Аррениуса.
Отметим, что не существует особой формы внутренней энергии,
связанной с энергией активации. По Аррениусу, энергия актива¬
ции Еа для каждой реакции указывает на тот избыток внутренней
энергии (по сравнению со средней энергией всех молекул), кото¬
рым должны обладать молекулы для протекания реакции. Если ре¬
акция экзотермическая и ДС7°<0, то Еа{Х)<Еа(_Х) и энергия актива¬
ции прямой реакции меньше обратной. При недостаточном отводе
выделяющейся теплоты из реактора доля энергетически активных
молекул в реакционной смеси с ростом температуры возрастает,
что приводит к увеличению скорости реакции. Если реакция эндо¬
термическая и АС7°>0, то Еа(Х)>Еа(.х) и энергия активации прямой
реакции больше обратной. В этом случае должна быть больше Д IP.
Интегрирование уравнения (1.66) с учетом указанной выше
независимости Еа от температуры в небольшом интервале либо
логарифмирование уравнения (1.61) приводят к идентичному вы¬
ражению
1п£ = 1пЛ-^1, (1.67)
используемому для расчета «аррениусовских» параметров — энер¬
гии активации Еа и предэкспоненциального множителя А — по
опытным данным. Если экспериментально найденные значения
константы скорости в координатах In А:, 1 /Т ложатся на прямую
линию в небольшом интервале температур (Тх< Т< Т2), то можно
сделать вывод о справедливости уравнения Аррениуса (1.61) для
изучаемой реакции и графически определить параметры А и Еа,
как это показано на рис. 1.5.
Полезно учитывать, что интерполяция по мере удаления от ис¬
ходного температурного интервала может привести к искажению
результатов, так как возрастает вероятность изменения порядка ре¬
акции.
28
1 /Т2 1/Г, 1/Т
Рис. 1.5. Определение параметров Аррениуса в координатах lnfc, 1/7’
Предэкспоненциальный множитель и энергию активации можно
вычислить аналитическим путем, если известны значения к(Т)
при различных температурах. В этом случае из уравнения (1.67)
получим
In —= ■
R
_L__L
т, ъ
(1.68)
откуда
Ев =
Rm Лпь_
(г2-г,) ь
и с учетом формулы (1.67)
In А = 1пА: + — —.
R Т
(1.69)
(1.70)
В ряде случаев используют приближенные формы уравнения
(1.61)
ЧТ) = л ■ .
Е„/ 20т
1000
, Еа (кДж/моль),
-Е U 6—
~А Л0 7 ’ юоо> Еа (ккал/моль).
(1.71)
29
Для уравнений (1.61) и (1.71) из соотношения (1.59), при ус¬
ловии у = ■■7410 = 2, получают выражение
кт
Еа (кДж-моль-1) з>6-10"4(Г2 + ЮГ), (1-72)
которое показывает, что справедливость приближенного правила
Вант-Гоффа для у = 2 зависит от соотношения величин Еаи Т:
Г, К 200 300 500
Еа, кДж-моль4 25 56 153
С другой стороны, если известна для изучаемой реакции энер¬
гия активации Еа, уравнение (1.59) для у = 2 позволяет получить
следующую зависимость температуры от Еа:
Т{ К) = -5 +
7 + 104£о +25
6
V/2
(1.73)
здесь Еа измеряется в кДж • моль-1.
Как было указано выше, уравнение Аррениуса (1.61) позво¬
ляет дать удовлетворительное описание большого круга реакций
в достаточно узком температурном интервале. Однако в 60-х гг.
XX в. благодаря развитию новых кинетических методов исследова¬
ния (флеш-фотолиз, импульсный радиолиз, струйные методики,
реакции в ударных трубах) появились реальные возможности для
существенного увеличения температурных интервалов измерений
до сотен градусов.
Практическое использование этих методов в широком темпера¬
турном диапазоне показало, что зависимость 1п& от 1/Г перестает
быть линейной, т.е. параметры А и Еа зависят от температуры.
На феноменологическом уровне отклонения от классического
уравнения Аррениуса наблюдаются для нескольких классов реак¬
ций по ряду причин, к которым относятся: изменение механизма
реакции; квантовые эффекты (туннелирование, влияние внутрен¬
них степеней свободы); эффекты, обусловленные неравновеснос-
тью реакционной системы.
В настоящее время применяют модифицированное уравнение
Аррениуса
k(T) = AT"exp[-Ea/(RT)l (1.74)
которое включает три параметра: А, Еа и п. К преимуществам ис¬
пользования уравнения (1.74) относятся:
простая, но достаточно эффективная математическая структу¬
ра уравнения;
возможность описания «конкуренции» между кинетической
энергией RT поступательных степеней свободы и энергией «хи¬
мического сопротивления» Еа\
30
разнообразие форм получаемых зависимостей к от Т.
Предложенное эмпирическим путем уравнение Аррениуса по¬
тужило важным «мостиком» между физикой и химией, эффек¬
тность которого проявляется, по крайней мере, в следующих
направлениях:
уравнение позволяет достаточно просто и точно обрабатывать
кинетические данные, относящиеся к реакциям различных систем;
попытки обоснования уравнения Аррениуса и выявления физи¬
ческой сущности входящих в него параметров явились важным сти¬
мулом развития теоретических основ химической кинетики.
В ряде случаев для кинетических исследований кроме уравне¬
ния Аррениуса используют другие уравнения с тремя и более па¬
раметрами, например В и С. Одно из них
к = АТсе~в/т или lnk = lnA + ClnT-j; (1.75)
широко используется для реакций, протекающих в растворах.
Другое уравнение
к = Ае~втС или 1пк = 1пА- ВТС (1-76)
более адекватно, чем уравнение Аррениуса, описывает опытные
данные для дробных значений постоянной С, существенно от¬
личных от -1.
Вопросы и задания для самоконтроля
1. Приведите уравнение Аррениуса в интегральной и дифференци¬
альной формах. Какая из них является более точной?
2. Покажите, каким образом С. Аррениус продемонстрировал пре¬
имущества использования предложенного им уравнения при анализе эк¬
спериментальных результатов.
3. Какие аргументы можно привести в пользу экспоненциальной фор¬
мы уравнения Аррениуса?
4. Приведите термодинамическое обоснование температурной зави¬
симости константы скорости, данное Я. Вант-Гоффом.
5. Какие величины называют «аррениусовскими» параметрами? Ка¬
ков физический смысл термина «энергия активации»?
6. В каком случае можно говорить о применимости уравнения Арре¬
ниуса при обработке экспериментальных данных?
7. Приведите способы определения параметров уравнения Аррениу-
Са: а) графический; б) аналитический.
8. Какой минимум опытных данных необходим для расчета энергии
Утивации реакции?
9. Какими причинами можно объяснить отклонение зависимости
°пытных данных в координатах \пк, 1/Т от линейного характера?
Ю. Какую величину называют температурным коэффициентом ско¬
рости реакции? Какие значения принимает у при невысоких температу¬
ру для определенного класса реакций?
31
11. Каковы возможные причины отклонения от уравнения Appcs
ниуса?
12. Почему экспериментальная зависимость In А: от 1/Т перестает быть
линейной в широком температурном интервале?
13. Приведите современную форму уравнения Аррениуса. Какие па>
раметры оно включает? В чем заключается преимущество его использо.
вания?
14. Какова роль уравнения Аррениуса в развитии химической кине-
тики?
15. Приведите другие уравнения с тремя параметрами, используе-
мые в кинетических исследованиях.
ГЛАВА 2. СЛОЖНЫЕ РЕАКЦИИ
Это было невероятно счастливой
случайностью, что мы занялись имен¬
но этой реакцией.
М. Боденштейн
(...о реакции разложения HI)
Сложными называют реакции, протекающие в несколько ста¬
дий, которые определенным образом связаны между собой через
исходные вещества и промежуточные соединения.
Кинетическое изучение сложных реакций проводят с использо¬
ванием, кроме основного, других постулатов химической кинетики.
Важную роль при кинетическом описании таких реакций игра¬
ет принцип независимости: если в системе протекает несколько эле¬
ментарных реакций, то каждая из них протекает независимо от
других, подчиняясь основному постулату химической кинетики.
Этот принцип имеет ограничения, в частности, он не применим
к сопряженным реакциям.
Принцип лимитирующей стадии: если процесс состоит из не¬
скольких последовательных стадий, то стадия, константа скорос¬
ти которой наименьшая, является лимитирующей, т.е. определя¬
ет скорость всего процесса.
Важное значение при изучении механизма сложных реакций
имеет принцип микрообратимости или детального равновесия, если
в сложном процессе устанавливается химическое равновесие, то
скорости прямой и обратной реакций должны быть равны для
каждой из элементарных стадий.
К характерным признакам сложных реакций, отличающих их
от элементарных стадий, относятся:
несовпадение порядка реакции и стехиометрических коэффи¬
циентов;
32
возможное изменение состава продуктов в зависимости от тем¬
пературы, начальных концентраций и других условий;
ускорение либо замедление процесса при добавлении в реак¬
ционную смесь небольших количеств веществ;
влияние материала и размеров реакционного сосуда на ско¬
рость реакции;
существенное влияние на скорость процесса незначительного
изменения условий проведения эксперимента: реакция либо ос¬
танавливается, либо протекает со взрывом.
2.1. Обратимые реакции
Реакции, в которых превращение реагентов в продукты сопро¬
вождается обратным процессом превращения продуктов в исход¬
ные вещества, называются обратимыми. С течением времени в та¬
ких реакциях устанавливается химическое равновесие.
1. Рассмотрим простейший процесс, в котором прямая и обрат¬
ная реакции имеют первый порядок. Примерами служат реакции
мутаротации глюкозы (а ^ (3), енодизации кетонов (R|CH2COR2^
^ R|CHC(OH)R2), превращения тиоционата в тиомочевину
(NH4NCS^(NH2)2CS).
Составим материальный баланс для простейшей схемы:
к-1
(2.1)
Количество вещества
в момент времени t= 0 п0 т0
Концентрации для / = О [А]0 = у* = а [В]0 = = b
Количество вещества
в момент времени t л0-£
Концентрации для / [А] = (п0-Q/V [В] = (/и0 + Q/V
[А] = а-х [В] = £ + х
x=$/V
Запишем выражение для скорости обратимой реакции
» = = (2.2)
а/ а/
или с учетом введенных обозначений
Цг-— = 4~ = ki(a - х) - k_t(b + х). (2-3)
d Г dr
После разделения переменных получим дифференциальное
Уравнение
Hr
— + х(&,+£_,) = (2.4)
Зак. 15%
33
Интегрирование этого уравнения с учетом начальных условий
приводит к соотношению
* = (2.5)
кх + £-i J
Преобразование выражения (2.5) позволяет получить важное
уравнение для определения суммы констант кх + к_х:
к\ + к_х =-\п— . (2.6)
t кха - k_\b - х(кх + k.x)
Теперь надо учесть условия равновесия, при достижении кото¬
рого можно записать
„-М.о,
следовательно,
кх[А]„= k_x[BU, (2.8)
где [А]„, [В]„ — равновесные концентрации, или
1®к = А. = ^ (2.9)
[AL
Отметим, что речь идет именно о концентрационной констан¬
те Кс, которая обычно отличается от термодинамической констан¬
ты Л'0, не имеющей размерности. В данном случае Кс также безраз¬
мерная величина, но для реакций типа А + В ^ С Кс имеет раз¬
мерность.
При достижении равновесия (г—> °°) х стремится к равновесно¬
му значению Соотношение (2.8) можно записать через а, Ь, х„:
kx(a-x„) = k_x(b + x„), (2.10)
после преобразований получим
kxa-k_xb = x„(kx+к.х), (2.11)
откуда
(2.12)
kx + к..
Подстановка выражения (2.11) в формулу (2.4) позволяет после
простых преобразований получить дифференциальное уравнение
dx
— = (kx + к.,)(х„ -x) = ккаж(х„ - x), (2.13)
которое формально записывается так же, как и для односторон¬
ней реакции первого порядка, где наблюдаемая, или кажущаяся
константа кКЗЖ = к] + к. х.
34
Интегрирование формулы (2.13) с учетом начальных условий
х = О при t- О дает кинетическое уравнение для определения сум¬
мы констант:
+*_, = -In——. (2.14)
t х„-х
Уравнение (2.14) можно представить в ином виде
ir \ /г — ^ In ~ р 1
к\ +к_х-- In [А] _ •
где [А]0 = а; [А]те= а-х * 0; [А] - [A]e. = fl-x^-(a-x„) = x«-x.
На основании уравнений (2.9) и (2.14) можно найти аналити¬
ческие выражения для констант кх и
= <tv C п1п —> <2Л6>
t(Kc + 1) X.. - х
1 -ln-Ь—. (2.17)
t(Kc +1) х„ - х
В случае, когда состояние равновесия в системе (2.1) не дости¬
гается, для определения суммы констант кх и к_х уравнение (2.3)
преобразуют к виду
^ = №+£_,)(М-х), (2.18)
где
М = к'а к-{Ь (2.19)
к\ + к_ 1
или путем деления числителя и знаменателя на к_
M-TTF' <220>
где К= kx/k_x.
После разделения переменных и интегрирования уравнения
(2.18) получают кинетическое уравнение
kl+k_x=- \n~P~. (2.21)
t М - х
Для нахождения численных значений каждой из констант ско¬
рости в этом случае требуется дополнительное знание константы
равновесия изучаемой системы при температуре опыта.
35
2. Рассмотрим обратимую реакцию, в которой обе реакции про,
текают по второму порядку. Типичный пример — реакция омыле>
ния сложного эфира
СН3СООС2Н5+ Н20 ^ СНзСООН + С2Н5ОН
Упростим задачу, представив схему реакции в виде
А+В ч *' -L + М. (2.22)
Скорость реакции запишется так:
~^ = к\ ([А]0 - х)([В]0 - х) - Аг_, ([L]0 + х)([М]0 + х). (2.23)
Положим, что [А]0 = [В]0 = a; [L]0 = [М]0 = 0, т. е. продукты реак¬
ции не присутствуют в начальный момент времени. Кинетическое
уравнение (2.23) примет вид
dx
— = кх{а-х)2 -к_хх2. (2.24)
В состоянии равновесия должно выполняться условие
кл х^
Кс = — = —^=-5-. (2.25)
Подставив в (2.24) выражение для /с_ь полученное из (2.25)
приходят к дифференциальному уравнению
>-2
dx ,
d<
(2.26)
XL
Полином допускает наличие двух корней квадратного уравнения
ах„
хх =*« и х2 = , его записывают через произведение вида
2*00 - а
т{х-хх)(х-х2). В конечном итоге из уравнения (2.26) получают
дифференциальное уравнение
dx _ к±а _ х^
d t xi
2х„ - а
у
(2.27)
Интегральная форма этого уравнения имеет вид
ki 2а(а - х„) _ j- 1п ах„ - х(2х, - а)
t а(хж - х)
Константу скорости прямой реакции кх можно определить, зная
кажущуюся константу скорости
V -L. Ма-х„)
которую находят экспериментальным путем при условии зна¬
ния Константу скорости обратной реакции к_х рассчитывают
по уравнению, аналогичному (2.9).
Возможны и другие случаи обратимых реакций, в которых одна
реакция — первого, а другая — второго порядка. Трудности, воз¬
никающие при рассмотрении обратимых реакций, в основном
носят математический характер и связаны с решением диффе¬
ренциальных уравнений.
3. Изучение кинетики быстрых обратимых реакций проводят с
помощью так называемого метода релаксации. Суть метода: на си¬
стему, находящуюся в состоянии равновесия, в течение коротко¬
го времени оказывается резкое воздействие путем изменения па¬
раметров (к ним относятся: температура, давление, напряжен¬
ность электрического поля и т.д.), в результате которого система
выходит из равновесного состояния. После прекращения возму¬
щающего систему воздействия она постепенно возвращается в
новое состояние равновесия. Процесс возвращения (релаксации)
системы в новое равновесное состояние, точнее, скорость релак¬
сации, изучают с помощью физических методов. При небольших
возмущениях процесс релаксации отвечает кинетике реакции пер¬
вого порядка и для обратимых реакций с порядком пф 1. Важной
характеристикой метода является время релаксации — время умень¬
шения измеряемой характеристики отклонения от равновесного
состояния в е раз.
В качестве примера рассмотрим обратимую реакцию типа
А + В \ С <2-30)
Концентрации [А] = ар + -*0 [В] = Ьр + Хо [С] = ср- х0
после возмущения
Концентрации [А]р = ар + х [В]р = Ьр + х [С]р = ср - х, х —> О
в новом состоянии
равновесия
Обозначают через др, 6Р, ср концентрации А, В, С при дости¬
жении системой нового равновесия. Величина х$ характеризует вы¬
званное возмущение системы с течением времени х -э 0.
Скорость возвращения в состояние равновесия запишется так:
V = = = + +Х)~к-'(СР “*>• О2-31)
Поскольку х0 а и jc0by величиной х2, как бесконечно малой
второго порядка, в соотношении (2.31) можно пренебречь:
djt
v = = кхарЬр + кхх{ар + Ьр) - к_хср + к_хх. (2.32)
37
Зная, что в новом равновесии выполняется соотношение
Jq_ = [С]р = _Ср_
*-i [А]Р[В]Р аХ
выражение (2.32) после подстановки кх из соотношения (2.33)
упрощается:
dr
v = -— = x[kx(ap + bp) + k_x], (2.34)
т.е. получаем кинетическое уравнение первого порядка, в кото¬
ром выражение в скобках не зависит от времени, его можно счи¬
тать константой скорости процесса возвращения системы в рав¬
новесное состояние.
Решение уравнения (2.34) имеет вид
х = х0е~'/т, (2.35)
где т — время релаксации, величина, обратная константе скоро¬
сти реакции первого порядка:
т-1 = кх(ар + bp) + Lx. (2.36)
Измерение т для различных значений ар и Ьр позволяет опре¬
делить константы к\ и к_и строя график в координатах (т-1, (яр+ Ьр)):
отрезок, отсекаемый прямой на оси ординат, равен к_и угловой
коэффициент прямой tga= кх.
Вопросы и задания для самоконтроля
1. Дайте определение сложных реакций и приведите примеры.
2. Перечислите характерные признаки, отличающие простую реак¬
цию от сложной.
3. Сформулируйте постулаты, которые используют при кинетиче¬
ском описании сложных реакций.
4. Какие реакции называются обратимыми? Приведите примеры.
5. Получите дифференциальное уравнение для скорости обратимой
реакции первого порядка А ^ В при условии, что [В]0 -Ъф0.
6. О какой константе идет речь при достижении равновесия в обра¬
тимой реакции В? Ответ поясните.
7. Получите интегральное уравнение для определения констант ско¬
рости кх и к_ь используя величины: а) х, х^; б) [А]0, [А] и [А]«.
8. Каким путем можно найти численные значения констант кх и к_{?
9. Получите интегральное уравнение для расчета констант скорости
рассматриваемой обратимой реакции А^В в отсутствие равновесия.
Какие дополнительные сведения необходимы в этом случае, чтобы
найти численные значения констант кх и к_х1
10*. Получите дифференциальное уравнение для скорости обратимой
реакции, в которой обе реакции протекают по второму порядку.
38
11*. Приведите решение полученного в вопросе 10 дифференциаль¬
ного уравнения. Каким путем находят значения констант кх и к_х в этом
случае?
12. Сопоставьте кинетические уравнения скорости обратимой реак¬
ции первого или второго порядка в обоих направлениях с уравнениями
скорости односторонней реакции того же порядка. Какой вывод можно
сделать?
13*. Получите дифференциальное и интегральное кинетические урав¬
нения для обратимой реакции, в которой прямая реакция имеет второй
порядок, а обратная — первый.
14*. Получите дифференциальное и интегральное кинетические урав¬
нения для обратимой реакции, в которой прямая реакция имеет первый
порядок, а обратная — второй.
15. Какой метод используют при изучении кинетики быстрых обрати¬
мых реакций? Сформулируйте его сущность. Назовите основную харак¬
теристику метода и поясните ее смысл.
16. Выведите уравнение, связывающее время релаксации и константы
скорости обратимой реакции первого порядка.
17*. Получите аналитическое выражение, связывающее время релак¬
сации и константы скорости обратимой реакции второго порядка в обо¬
их направлениях.
18*. Покажите, каким образом, зная времена релаксации, можно оп¬
ределить константы скорости кх и к_х для обратимой реакции второго
порядка.
19*. Получите аналитическое выражение для времени релаксации об¬
ратимой реакции (прямая реакция первого порядка, обратная — второго).
20*. Получите аналитическое выражение для времени релаксации об¬
ратимой реакции (прямая реакция второго порядка, обратная — первого).
2.2. Параллельные реакции
Реакции, в которых исходные вещества превращаются одно¬
временно по нескольким направлениям с образованием различ¬
ных продуктов, называются параллельными.
Вначале рассмотрим случаи, когда один реагент участвует в
двух реакциях. Например, превращение изопропенилциклобутана
идет по двум направлениям: с образованием 1-метилциклогексе-
на и по другой реакции — эквимолярных количеств этилена и
изопрена. Гидролиз 1-хлорпропан-2-ола в воде при -100 °С проте¬
кает с образованием по одной реакции соляной кислоты и ацето¬
на, по другой — пропан-1,2-диола.
1. Положим, что оба процесса являются необратимыми реак¬
циями первого порядка. В таком случае схема имеет вид
А У L
А <2 (2.37)
м
39
Обозначим через а начальную концентрацию вещества А; (а-х) —
текущая концентрация А в момент времени Г; х — убыль концен¬
трации А в ходе процесса по обеим реакциям, х = х1 + х2. Запишем,
согласно основному постулату химической кинетики и прин¬
ципу независимости, кинетическое уравнение в дифференциаль¬
ной форме
d[A] _ d(a - х) _ dx _
~~dГ" dr " dr “ (2.38)
= kx(a - x) + k2(a - x) = (kx + k2)(a- x).
После разделения переменных и интегрирования уравнение
(2.38) принимает вид
|ln —— = (*, + к2) = ккяж. (2.39)
Г а-х
Кажущаяся константа ккяж равна сумме констант скорости от¬
дельных реакций, kx + k2.
После потенцирования уравнения (2.39) получают экспоненци¬
альную зависимость убыли текущей концентрации вещества А со
временем
а-х = яехр[-(£| + k2)t\. (2.40)
Общая концентрация продуктов реакции будет равна
х = а[\ -е~(<Г|+*2)']. (2.41)
Пусть i;, и ^2 — степени превращения веществ по каждой из
параллельных реакций.
Положим Xi = £|/V, х2= £2/V, где V — постоянный объем сосуда.
Запишем выражения для текущих концентраций веществ A, L и М:
а-х=а-х 1-х2, [L] = [L]0+x, и [М] = [М]0+х2.
Превращения вещества А по обеим реакциям не являются неза¬
висимыми: отношение их скоростей равно отношению констант
скорости
-’¥- = Т- <2.«)
v2 dx2 k2
Если полагают, что в начале реакции нет следовых количеств
продуктов L и М, соотношение (2.42) интегрируют от 0 до X) и от
0 до х2:
Х| - *1 О >114
Т,~Т2' <2'43>
т.е. отношение концентраций (или количеств) продуктов посто¬
янно и не зависит от времени.
40
Используя соотношения (2.43) и (2.39), легко найти числен¬
ные значения констант скорости кх и к2. Получим аналитические
выражения для текущих концентраций:
а-х- а-хх{\ + к2/кх) - а- (хх/кх)(кх + к2)\ (2.44)
Х| =аг7г(1_е_(*'+*2),Нь]; (2-45)
х2 = 0-2—(1 - = [Ml; (2.46)
К\ + К2
Как видно из соотношений (2.45) и (2.46), концентрации про¬
дуктов возрастают со временем по экспоненциальному закону.
Если реагент А участвует одновременно в / реакциях первого
порядка по отношению к А, получают следующее уравнение:
[L] = a^(l-e-^>)'), (2.47)
где =
Для опытной проверки схемы (2.37) фиксируют концентра¬
ции обоих продуктов в зависимости от времени.
Если отношение текущих концентраций образующихся про¬
дуктов L и М не зависит от времени, убеждаются в том, что па¬
раллельные реакции имеют одинаковый порядок и выполняется
соотношение (2.43). Если желают получить чистый продукт L, не¬
обходимо подобрать экспериментальные условия таким образом,
чтобы выполнялось неравенство
kx»k2. (2.48)
В общем случае, если реагент расходуется в / параллельных ре¬
акциях, то
кх»кых. (2.49)
Контроль за чистотой продукта L осуществляют путем провер¬
ки для предложенной схемы равенства
d[Ll = _d[A] ^2 50^
dt dt
или
[L] = [А]0 — [А] =х. (2.51)
В противном случае получают
_d[L]/dU_A_
d[A]/d/ kl+k2 ( }
41
Следует обратить внимание, что для многих реакций по ме
накопления в реакционной среде продуктов возможно протек
ние обратных реакций с заметной скоростью. В этом случае ре
гент А частично регенерируется и устанавливается химичесю
равновесие между реагентом и продуктами
Ml = А и IMk = А. (2 5
[А]„ к.х [А]. к_2 ’ К ‘
а также между продуктами
[Ч„ _ кх к_2
(2.5-
[М]_ k_x к2
Если к\» к2, то происходит в основном образование продукта ]
но его концентрация останется преобладающей при достижени
химического равновесия только при выполнении дополнительнс
го условия
(2.5!
кг к-2
В случае, если
^ (2.5(
кг к. 2
в конце реакции, когда устанавливается химическое равновесие
концентрация продукта L становится меньше концентрации про
дукта М.
2. Рассмотрим случай, когда обе параллельные реакции подчи
няются кинетике второго или более высокого порядков. К этом
типу относятся многие реакции в органической химии, наприме
, R-CH2-CH2-0H + C1"
У
R-CH2-CH2-C1 + ОН’
r-ch=ch2 + сг + Н20
Если общие порядки обеих реакций равны, кинетическое опи
сание существенно упрощается. В общем случае схема реакци:
имеет вид
ус
А + В к2 (2.57
D
42
Составим таблицу (табл. 2.1).
Таблица 2.1
Показатель
А
В
С
D
Количество
вещества, t- 0
«0
щ
0
0
Концентрации,
/= 0
[А]0 =f = a
14 = ^ = »
[С]„=0
о
II
о
Q
Количество ве¬
щества в момент
времени t
ио-«, + ^)
ff»o-(5i + y
4,
Концентрации в
момент времени /
[А] = а-х
(x = y + z)
[В] = 6-х
(x = y + z)
II
II
Ч:
п
и
N
На основании данных табл. 2.1 запишем кинетические диффе¬
ренциальные уравнения
d[C] _ dy
dr dr
= kx{a-x)p{b-x)q,
Щ^ = ^ = к2{а-хУ{Ь-хГ,
at at
(2.58)
(2.59)
где p и q соответственно порядки реакции по каждому веществу.
Суммируя выражения (2.58) и (2.59), получим
% = % + % = № + к2)(а - хПЬ - x)q.
at at at
(2.60)
Разделив (2.59) на (2.58), находим для продуктов С и D:
d z _ к2
dу кх
или после интегрирования этого уравнения в пределах от 0 до z и
соответственно от 0 до у имеем
z к2 к2
- = -f и z = -*-y.
У К кх
(2.61)
Отношение концентрации продуктов, как и в первом случае,
не зависит от времени и определяется отношением констант ско¬
рости кх и к2. В случае, когда общий порядок реакции равен 2
(p = q- 1), уравнение (2.60) принимает вид
^ = (кх+к2)(а-х)(Ь-х).
at
(2.62)
43
После интегрирования методом неопределенных коэффициен¬
тов получим
*■ +к2 =Т~Цг1п^а~^- (2-63)
t(a- b) а(Ъ -х)
Соотношения (2.61) и (2.63) позволяют из опытных данных
определить значения кх и к2.
3. Рассмотрим параллельные (конкурирующие) реакции раз¬
ных порядков. Согласно схеме
А~~*L (2.64)
А + В—
Кинетические уравнения для убыли реагента А и образования
продукта L в этом случае имеют вид
= ^- = кх(а-х) + к2(а - х)(Ь -х) =
d/ dt (2.65)
= [kl+k2(b-x)](a-x);
^tl = A:l(fl-x). (2.66)
Отношение концентраций продукта L и реагента А в каждый
момент времени равно
d[L] _ кх
d[A] к\+к2(Ь-х)
или
(2.67)
гт 1 . к,+ к2Ь
L-тМп, 12—(2.68)
к2 k\+k2(b-x) v ’
Интегрирование уравнения (2.65) приводит в конечном итоге
к выражению
, 1. ffl[*i+*20-x)]l
(269>
в котором (а-х) и (b-х) взаимосвязаны.
Преобразовав уравнение (2.69) для извлечения х, получим
_ а(кх + k2b)(tk>' -1).
кхек< + k2b(ek'’ -1) ’ (2,70)
а-х _ кх(е*1' -1)
а ~ кхек" +к2Ь(ек" -\У (2,71)
44
Следует отметить, что для достаточно высоких концентраций
реагентов, когда [А] > к]/к2 (при условии, что [А] = [В]), протека¬
ет только реакция второго порядка
-М1 = £2[А]2 = £2(д-х)2. (2.72)
Напротив, для малых концентраций вещества А, когда [А]«:
<g.kjk2., в основном протекает реакция первого порядка
-Ml £*,[А] = *,(<*-*). (2.73)
4. Рассмотрим конкурирующие реакции одинакового порядка.
Положим, что продукт Р образуется одновременно в двух ре¬
акциях первого порядка по схеме
Ач
?Р (2.74)
В
Концентрации каждого из реагентов уменьшаются со време¬
нем по экспоненциальному закону
[A] = а — Х\ - aexp(-V); (2.75)
[B] = b - х2 = Ь ехр(-к2(). (2.76)
Текущие концентрации А, В и Р могут быть выражены через
х, = б,;/ К, например, для продукта Р получим
[Р] = [Р]0 + х, + х2 = [Р]0 + о(1 - *-*■') + 6(1 - е~к*). (2.77)
В случае, когда а = Ь и [Р]0= 0, т. е. продукт отсутствует в начале
реакции, уравнение (2.77) переходит в
[Р] = а(2 - е"*1' - е~*2'). (2.78)
Проверка справедливости такой схемы требует измерения кон¬
центраций обоих реагентов А и В в зависимости от времени.
Вопросы и задания для самоконтроля
1. Какие реакции называются параллельными? Приведите примеры.
2. Какие кинетические постулаты используют при изучении парал¬
лельных реакций?
3. Получите кинетическое уравнение для параллельных реакций типа
45
4. Приведите выражения для текущей концентрации реагента А и
продуктов в зависимости от времени.
5. Чему равно отношение концентраций (или количеств) продук¬
тов для указанной выше реакции? Зависит ли это отношение от вре¬
мени?
6. Получите выражение для зависимости концентрации продукта В
от времени в случае, если реагент А принимает участие одновременно
в i реакциях первого порядка.
7. Каким образом проводят опытную проверку рассматриваемой схе¬
мы параллельных реакций? При каких условиях будет получаться чистый
продукт С?
8. Получите соотношения между концентрациями реагента А и
продуктов В и С после установления химического равновесия в сис¬
теме.
9. Укажите условия, при которых будет образовываться в основном
продукт С и при достижении химического равновесия.
10*. Для параллельных реакций первого порядка получите количествен¬
ные соотношения
1^в
А^с
для определения суммы констант скоростей к{ + к2 + к3 и их отношения
к{: к2: к3.
11*. Получите дифференциальное и интегральное кинетические урав¬
нения параллельных реакций второго порядка.
12. Каким путем можно определить численные значения констант ско¬
рости к\К к2 параллельных реакций: а) первого порядка; б) второго по¬
рядка?
13*. Получите дифференциальное и интегральное кинетические урав¬
нения для конкурирующих реакций разных порядков согласно схеме
А—1—>L
А + В—>М
14*. Сформулируйте условия, при которых в рассматриваемой системе
(вопрос 13) протекает в основном: а) реакция первого порядка; б) реак¬
ция второго порядка.
15. Для конкурирующих реакций первого порядка
А ,
выразите текущие концентрации [А], [В] и [Р] через степень превраще¬
ния хг
46
2.3. Последовательные реакции. Принцип
квазистационарных концентраций
Реакции, которые протекают в две или более стадий таким
образом, что продукт, появляющийся в одной стадии, служит
реагентом в последующей, называются последовательными. При¬
мерами таких реакций являются, в частности, радиоактивные пре¬
вращения веществ и нуклеофильные замещения в ароматических
соединениях.
Для описания кинетики последовательных реакций необходи¬
мо решить систему дифференциальных уравнений. В общем случае
решение уравнений находят методами численного интегрирова¬
ния, что затрудняет их практическое использование. Точное ре¬
шение в аналитическом виде возможно лишь в случае двух после¬
довательных односторонних реакций первого порядка.
Составим таблицу для реакций: А—-—> В —-—> С (табл. 2.2).
Таблица 2.2
Показатель
А
в
С
Количество вещества
в момент времени
/=0
По
0
0
Концентрации в мо¬
мент времени /=0
>
о
II
II
а
о
II
о
CQ
о
о
II
о
Количество вещества
к моменту времени t
1-V
5'
Концентрации в мо¬
мент времени t
[А] = («„-£)/К
[А] -а-х
x=yv
[В]=(4-50/^
[В)=х-у
у =£,'/¥
[С]=57 v
[С]=у
Используя основной постулат кинетики и принцип независи¬
мости, запишем дифференциальные уравнения для первой стадии
= £i[A] или ^ = к\(а - х), (2.79)
для второй стадии
^51 = &[[А] - &2[В] = ki(a-x)-k2(x-y), (2.80)
*£1 = к2[Щ = к2(х-у).
at
(2.81)
47
Решение этой системы линейных дифференциальных уравне¬
ний с постоянными коэффициентами не представляет трудно¬
стей. Интегрирование уравнения (2.79) приводит к уравнению
[А] = [А]0е-*'' или а-х = ое"*1', (2.82)
выражение для х имеет вид
х = а{\- е"*1'). (2.83)
Подстановка уравнения (2.82) в (2.80) после преобразований
позволяет получить
М + Л2[В] = А:1[А]ое-*«'. (2.84)
ш
Общее решение дифференциального уравнения (2.84) записы¬
вается в виде
[В] = ае“^+Ре-^. (2.85)
Для определения (3 подставляют частное решение неоднород¬
ного уравнения ([В] = (Зе_/:|/) в дифференциальное уравнение (2.84)
-ЦЗе"*1' + к2[Зе-*1' = ^[Afoe-*'', (2.86)
откуда, если к2Ф ки
0 к\ [А]0
<2-87>
Для нахождения а используют в уравнении (2.85) начальные
условия: [В] = 0 при /=0
0 к\ [ А]о
<2-88>
Подставляя полученные значения а и Р в уравнение (2.85), по¬
лучают выражение для зависимости концентрации промежуточ¬
ного продукта В от времени
[В} = х-у = [АЬ- А-[е-*>' -е-*2']. (2.89)
К2 К\
Из уравнения материального баланса [А]0 = [А] + [В] + [С] либо
после подстановки выражения (2.89) в (2.81) и интегрирования
легко находят концентрацию продукта С:
[С] = [А]0
1 —— е'*1' н——— е~*2'
к-> - к, к-) - к,
(2.90)
На рис. 2.1 представлены зависимости концентраций А, В, С и
скоростей от времени для к2 = 1,5^,.
48
[А], [В], [С]
v
‘max
а
б
Рис. 2.1. Изменение концентраций и скоростей в зависимости
от времени, если к2 = 1,5^:
а — зависимость концентраций А, В, С; б — зависимость скорости
Как видно из рис. 2.1, концентрация реагента А уменьшается
со временем по кинетическому закону первого порядка. Этот про¬
цесс не зависит от последующих реакций. Скорость образования А,
очевидно, всегда отрицательна. Согласно выражению (2.89), кон¬
центрация промежуточного продукта В в начальный момент вре¬
мени, t = 0, и при t —» равна нулю, следовательно, можно счи¬
тать, что [В] проходит через максимум.
Рассмотрим подробнее частные случаи уравнения (2.89). При
кх = к2 = к возникает неопределенность, которая после раскрытия
по правилу Лопиталя приводит к простому выражению для кон¬
центрации промежуточного соединения В
В начальный момент времени [В] увеличивается пропорцио¬
нально времени, так как экспонента -а 1; при длительном проте¬
кании процесса экспонента -а 0, следовательно, [В]->0. Время
достижения максимальной концентрации промежуточного про¬
дукта определяется соотношением
В общем случае время достижения максимальной концентра¬
ций В находят следующим образом: уравнение (2.89) дифферен¬
цируют по времени и полученную производную, согласно требо¬
ванию экстремума, приравнивают к нулю; затем находят соотно¬
шение, отвечающее этому условию, его логарифмируют и, решая
относительно 1, приходят к искомому результату
[В] = акы*.
(2.91)
к
(2.92)
, _ In ki/ki
*max , ,
к2-кх
max
(2.93)
4. Зак. 1596
49
Подстановка соотношения (2.93) в выражение (2.89) позволя¬
ет оценить значения [В]тах в зависимости от соотношения кон¬
стант к2 и кх на основании полученного уравнения
1
[В]щах ^
1-*,/Аг2
(2.94)
В случае кх» к2 скорость образования промежуточного соеди¬
нения В существенно превосходит скорость его расходования, сле¬
довательно, с течением времени при /-><», как следует из выра¬
жения (2.94), [В]тах—> [А]0= а■ В случае к2 » кь когда скорость рас¬
ходования В существенно больше скорости его образования, при
»°° [В]тах—>0.
Рассмотрим полезные соотношения между концентрациями В,
С и А.
Если к2> ки то после длительного промежутка времени выра¬
жение (2.89) упрощается и переходит в
[В ] = а_£_е-*>'. (2.95)
К2 гС|
После деления формулы (2.95) на формулу (2.82) получают
выражение для так называемого переходного равновесия
[В] _
[А] к2 - кх
(2.96)
т. е. в стационарном состоянии отношение указанных концентрации
постоянно. В этом случае отношение концентрации вещества С к
начальной концентрации вещества А с учетом упрощения (2.90)
примет вид
й-‘-~ а97>
Если к2»киъ соотношении (2.96) можно пренебречь в знаме¬
нателе величиной кх, в итоге получают выражение, характеризу¬
ющее так называемое вековое равновесие
[В] _ kj _ Т2 _ 6/2(2) о
г А 1 7 4- ’ \Z.70)
[А] к2 Л/2(1)
где Tj, т2 — средние времена жизни; /1/20 >, /1/2(2) — периоды полу¬
превращения соответственно веществ А и В.
гС1
Очевидно в этом случае при / -> °° -L-L » 1.
[А]0
Как показано на рис. 2.1, кривая зависимости концентрации
продукта С от времени имеет такую форму, которая указывает на
50
наличие, во-первых, периода индукции, в течение которого ско¬
рость образования конечного продукта становится измеряемой ис¬
пользуемыми способами анализа; во-вторых, точки перегиба, вре¬
мя достижения которой совпадает со временем достижения мак¬
симальной концентрации промежуточного соединения В:
ВД. = Л. fd[Br
df2
d t
= o,
углах
(2.99)
In k2 / kx
k2 - kx
^nep = ^max = , • (2-100)
Таким образом, скорость образования продукта С проходит че¬
рез максимум, соответствующий точке перегиба и времени до¬
стижения [В]тах.
На рис. 2.2 представлены зависимости концентраций веществ А,
В, С и скоростей от времени для к2 = 20кх.
Сравнение графиков на рис. 2.1 и 2.2 с учетом полученных ра¬
нее уравнений позволяет сделать следующие выводы.
1. Значение [В)тах снижается с увеличением отношения к2/кх.
2. Время достижения [В]тах, ?тах, уменьшается с ростом отно¬
шения к2/кх, т.е. период индукции становится малым.
3. Скорость образования промежуточного соединения = 0
ДЛЯ t У /щах-
4. Отношение скоростей ~ и ~ стремится к минимуму для
^ ^шах-
а б
Рис. 2.2. Изменение концентраций веществ А, В, С (а) и скоростей (б)
в зависимости от времени; к2 = 20кх
Иллюстрацией этих выводов служат полученные численные зна¬
чения величин:
20
0,32
0,19
5
50
0,13
0,085
2
100 1000
0,09 0,012
0,063 0,009
1 0,1
На основании полученных выше данных сформулируем прин¬
цип квазистационарных концентраций (М. Боденштейн, 1913 г.): если
к2^>к{и при t> tmax концентрация промежуточного продукта В ос¬
тается очень малой и постоянной (квазистационарной) в течение
всего времени протекания реакции, то можно считать, что вы¬
полняются условия
В этом случае скорость образования продукта С будет практи¬
чески равна скорости расходования реагента А
Таким образом, можно сделать вывод, что первая стадия явля¬
ется лимитирующей, т.е. кинетически определяющей стадией всего
процесса. Этот принцип является приближенным, так как, строго
d[B] п
говоря, = и только в точке максимума, t = /тах; его нельзя
применять при значениях/:, > к2 и t< /тах.
Принцип квазистационарных концентраций имеет большое зна¬
чение для составления кинетических уравнений при описании слож¬
ных реакций, протекающих в три и более стадий. Применение это¬
го принципа позволяет путем замены части дифференциальных
уравнений алгебраическими выразить концентрации неустойчи¬
вых промежуточных соединений через концентрации аналитиче¬
ски определяемых исходных веществ и конечных продуктов.
Часто используют в кинетике и принцип квазиравновесных кон¬
центраций, называемый квазиравновесным приближением. Суть
принципа: если в сложной реакции одна или несколько стадий
протекают быстро и обратимо таким образом, что между реаген¬
тами и промежуточными продуктами устанавливается термоди¬
намическое равновесие, можно использовать соотношение К =
Рассмотрим применение этого принципа на примере класси¬
ческой реакции образования йодоводорода из простых веществ.
[В]=[В]тахи-^ = 0
(2.101)
= к,/к.,
52
М.Боденштейн (1894) на основании экспериментально установ¬
ленного кинетического уравнения
-^1 = №][!;] (2.103)
СП
сделал вывод, что эта реакция протекает по элементарному би¬
молекулярному механизму
Н2 + I2 = 2HI.
Позже (1967 г.) Дж. Салливэн показал, что рассматриваемая
реакция протекает по более сложному механизму, например:
1
12^=Ь^1 + 1
I + H2 r-^IHz
I + IH2 —^HI + HI J
(2.104)
Согласно принципу квазиравновесных концентраций получим
выражения для концентраций промежуточных веществ:
[I] = ^,1/2[12]1/2; (2.105)
[1Н2] = *2[ЩН2] = К\1гКг[ H2][I2F (2.106)
Уравнение скорости реакции, записанное по расходованию Н2
или через образование HI с учетом выражений (2.105) и (2.106),
примет вид
= -®г = *з[1Н2][1] = ММН2][12]. (2.107)
Полученное выражение отвечает кинетике реакции второго по¬
рядка и совпадает с опытным уравнением (2.103), считая к = къКхК2.
Вопросы и задания для самоконтроля
1. Дайте определение и приведите примеры последовательных реакций.
2. Для двух последовательных односторонних реакций первого порядка
А—!—>В—>С
составьте систему дифференциальных уравнений зависимости концент¬
рации участвующих веществ от времени.
3. Получите в аналитическом виде выражения для текущих концент¬
раций А, В и С.
4. Представьте графически зависимости концентраций А, В, С от вре¬
мени для к2 > кх. По какому закону меняется концентрация реагента А со
временем?
5. Представьте графически зависимости скоростей изменения [А], [В],
[С] от времени для к2> кх.
6. Получите аналитическим путем выражение для зависимости кон¬
центрации В от времени в случае кх = к2 и его проанализируйте.
7. Чему равно время достижения максимальной концентрации про¬
межуточного соединения В для кх - к{!
8*. Получите выражение для времени достижения [В]тах в общем слу¬
чае на основании точного решения (см. вопрос 3).
9. Выведите выражение для оценки значения [В]тах в зависимости от
соотношения констант кх и к2. Какие значения принимает [В]тах при /-»«>
в случае: а) кх^> к2\ б) к2^> кх1
10. Какое состояние называют переходным равновесием? При каком
условии оно достигается?
11. Чему равно отношение концентрации продукта С к начальной кон¬
центрации А при условии, что к2> кх1
12. Какое состояние системы называют вековым равновесием? При
каком условии оно устанавливается?
13. Какую информацию можно получить, анализируя кривую зависи¬
мости концентрации продукта С от времени?
14. Покажите аналитическим путем, что точка перегиба на кривой
зависимости [С] от времени совпадает со временем достижения [В]тах.
15. Представьте графически зависимости концентраций А, В, С от
времени для к2^>кх.
16. Представьте графически зависимости скоростей изменения А, В,
С от времени для &2» кх.
17. Каким образом с увеличением отношения к2/кх изменяется:
а) высота максимума для [В]тах;
б) время достижения [В]тах, /тах;
ч u l^R VTl . n
в) отношение скоростей — и — для t > /тах?
Va VC
18. Что такое период индукции? Как изменяется его величина с рос¬
том отношения к2/ к{!
19. Сформулируйте принцип квазистационарных концентраций. По¬
чему он является приближенным? Укажите границы его применения.
20. В чем заключается принцип квазиравновесных концентраций? Рас¬
смотрите его применение на примере классической реакции образова¬
ния йодоводорода из простых веществ. Приведите другие примеры.
ГЛАВА 3. ЦЕПНЫЕ РЕАКЦИИ
3.1. Основные понятия и стадии цепных реакций
Цепными реакциями называются сложные превращения ис¬
ходных веществ в продукты, осуществляемые при регулярном чере¬
довании нескольких реакций с участием активных центров и со¬
хранении свободной валентности.
54
Активными центрами могут быть атомы и свободные радикалы
с высокой реакционной способностью, обусловленной присут¬
ствием на их внешней орбитали неспаренных электронов, а также
ионы и возбужденные молекулы.
Различают реакции с энергетическими цепями, протекающие
с участием энергетически возбужденной молекулы (термин вве¬
ден М. Боденштейном в 1916 г.), и реакции с материальными це¬
пями, протекающие с участием атомов и свободных радикалов
(первая схема цепного процесса предложена В. Нернстом в 1918 г.
для описания реакции Н2 + С12 = 2НС1).
Примерами цепных радикальных реакций являются: окисле¬
ние органических веществ молекулярным кислородом, взаимо¬
действие водорода и углеводородов с хлором и бромом, терми¬
ческое разложение озона и кислородных соединений хлора, кре¬
кинг нефтепродуктов, полимеризация и поликонденсация, а также
ядерные реакции разложения урана или плутония в атомном ре¬
акторе.
Каждая цепная реакция состоит из трех основных стадий: за¬
рождения (инициирования), развития и обрыва цепи.
Стадия зарождения цепи, наиболее энергоемкая по сравнению
с другими, приводит к образованию активных центров из валент¬
но-насыщенных молекул. Возможные пути возникновения свобод¬
ных радикалов и атомов: мономолекулярное превращение (С2Н6 —>
—» 2’СН3), бимолекулярное взаимодействие (СН3СНО + 02 —»
—» СН3С*0 + Н02), гетерогенная реакция на стенке сосуда (С12 +
+ S —» СГ + С1адС) и воздействие физических агентов на реакцион¬
ную систему. В этом случае стадия зарождения цепи называется
инициированием. Оно происходит под воздействием света (С12 +
+ hv —» 2С1), либо при участии фотосенсибилизатора (Hg + hv —>
—» Hg*, Hg* + C3H8 —» ’C3H7 + Н* + Hg), либо в присутствии
специальных веществ, называемых инициаторами, которые образу¬
ют свободные радикалы. Такими веществами являются пероксид
бензоила С6Н5—С(О)—О—О—С(О)—С6Н5, азоизобутиронитрил
(CH3)2C(CN)-N=N-(CN)C(CH3)2, пары металлов (Na, Hg) и
многие неорганические соединения.
Стадия развития может включать в себя реакции продолжения
и разветвления цепи. Элементарные стадии, протекающие с обра¬
зованием свободной валентности и сопровождающиеся расходо¬
ванием реагентов и (или) образованием продуктов, называют
реакциями продолжения цепей. Для таких реакций характерны
небольшие энергии активации, к ним относятся:
взаимодействие атома или свободного радикала с молекулой
реагента, в результате которого возникает новый свободный ра¬
дикал
С2Н4 + СГ -» *С2Н4С1 R* + 02 -> RO*2
55
взаимодействие атома или свободного радикала с молекулой
реагента, сопровождающееся образованием нового радикала и мо¬
лекулы продукта:
СН4 + СГ -СНз + НС1 RH + СГ -> НС1 + R-
мономолекулярное превращение одного радикала в другой
сн3оо* -> сн2оон RO-2 -> R'o2h
мономолекулярный распад свободного радикала, приводящий
к образованию нового свободного радикала и молекулы продукта
реакции
СН3СО* -»’СН3 +СО СНО*->СО + Н*
взаимодействие двух свободных радикалов, приводящее к об¬
разованию свободного радикала (атома) и молекулы продукта
FO* + FO* -> 02 + 2F*
Реакциями разветвления цепей называют элементарные ста¬
дии, приводящие к образованию из одного активного центра не¬
скольких активных центров, например:
Н*+02 -> *ОН+*Оф
Элементарные стадии, приводящие к гибели свободных ради¬
калов или атомов, т.е. к исчезновению свободной валентности,
называются реакциями обрыва цепи. Обрыв цепи может происхо¬
дить (особенно при низких давлениях) на стенках сосуда в ре¬
зультате адсорбции активных центров поверхностью S:
W+S кf >HMC Cl'+S к> )Claje
с последующей рекомбинацией двух активных центров в обычную
молекулу. Скорость реакции обрыва цепи гу в этом случае имеет
первый порядок по концентрации активных центров, например
vf = kfS[М‘] и vf = ЗДСГ],
где S — площадь поверхности сосуда.
Принято называть реакции обрыва указанного типа линейным
обрывом цепей.
При больших давлениях обрыв цепи происходит в объеме в
результате тройного столкновения активных центров с другой мо¬
лекулой (М) газовой фазы:
Вг* + Вг* + М—^—>Br2 + М
F* + F* + F20—>F2 + F20
56
Скорость реакции обрыва цепи vf будет иметь второй порядок
по концентрации активных центров:
Vf = kf [M][Brf и vf= kf[M][F-}\ [M] = [F20]
В данном случае реакции обрыва называют квадратичным об¬
рывом цепей. В газовых реакциях чаще происходит линейный об¬
рыв цепи, а в растворах — квадратичный.
Любую цепную реакцию в общем виде можно представить сле¬
дующим образом:
А + аХ -> Р + (3Y,
где А — реагент; Р — продукт; X, Y — активные центры (Y может
быть идентичен X); а, (3 — целые числа, > 0.
В зависимости от значений а и (3 различают следующие стадии
реакции:
а = 0 (3*0 зарождение цепи (1)
а = (3 продолжение цепи (2)
а < Р разветвление (3)
а * 0 Р = 0 обрыв цепи (4)
3.2. Неразветвленные цепные реакции
По определению, неразветвленная цепная реакция включает в
себя стадии зарождения, продолжения и обрыва цепи.
Заслуга в разработке теории неразветвленных цепных реакций,
по оценке Н.Н. Семенова*, принадлежит школе Боденштейна
(1913 — 1929). Классическим примером реакций этого типа явля¬
ется синтез НС1 из водорода и хлора. Схема процесса предложена
В.Нернстом, теоретический анализ кинетики реакции проведен
в основном М. Боденштейном и его учениками и подробно изла¬
гается в учебной литературе.
Для характеристики неразветвленных цепных реакций исполь¬
зуют понятия звено и длина цепи. Началом звена цепи, как пра¬
вило, считают реакцию продолжения с участием того из актив¬
ных центров, который первым образуется в реакции зарождения
цепи.
Звено цепи — это совокупность последовательных элементар¬
ных реакций продолжения цепи, которая приводит к регенера¬
ции активного центра, уже участвовавшего в реакции. Например,
для реакции
С2Н6 + С12 -> С2Н5С1 + НС1
* Н.Н. Семенов — академик, лауреат Нобелевской премии, основатель ка¬
федры химической кинетики в МГУ.
57
звено цепи состоит из двух элементарных реакций
С2Н6 + СГ -» *С2Н5 + НС1 и *С2Н5 + С12 -> С2Н5С1 + СГ
Средняя длина цепи — это число полных звеньев, приходя¬
щихся в среднем на каждый активный центр, образовавшийся по
реакции зарождения цепи.
Средняя длина цепи v зависит от соотношения скоростей ре¬
акций продолжения цепи и ее обрыва и равна
где 3 — вероятность обрыва на каждом звене; а =1 - 3 — вероят¬
ность продолжения цепи; vR — скорость продолжения; iy— ско¬
рость обрыва цепи.
Если 3^1»
Длина цепи зависит от природы цепной реакции и условий ее
протекания: концентрации и чистоты реагентов, интенсивности
света, температуры, материала реакционного сосуда, его размера
и т.п.
Необходимым условием стационарного режима протекания не-
разветвленной цепной реакции является равенство скоростей за¬
рождения и обрыва цепей
Скорость неразветвленной цепной реакции (определяемая по
убыли концентрации исходных веществ или по накоплению ко¬
нечных продуктов) равна
К кинетическим особенностям неразветвленной цепной реак¬
ции относятся: рост скорости под воздействием на реакционную
смесь физических агентов (света и т.п.) и небольших количеств
инициаторов; зависимость скорости от размеров сосуда, материа¬
ла стенок и их состояния; быстрое уменьшение скорости при до¬
бавлении в реакционную смесь небольших количеств ингибито¬
ров. Последнее обстоятельство является характерным признаком
цепного механизма реакции.
Если цепная реакция начинается в момент времени t = 0, то
скорость ее не сразу достигнет некоторой постоянной величины
v=v0v, поскольку для развития цепи необходимо время.
(3.2)
v0 = Vf.
(3.3)
(3.4)
58
Дифференциальное уравнение для скорости изменения кон¬
центрации активных центров в ходе неразветвленной цепной ре¬
акции с линейным обрывом цепей запишется так:
— = v0-gn, (3.5)
где g — удельная скорость реакции обрыва.
Для решения уравнения (3.5) его интегрируют при начальных
условиях (п = 0 при /=0), считая i>0 и g постоянными, и получают
п = iJ“(i-e-*'). (3.6)
8
Зависимость скорости реакции от времени примет вид
v = — (1-е-*), (3.7)
8
где / — удельная скорость реакции продолжения цепи, ведущей
к образованию продукта.
Как видно из соотношения (3.7), при t —» «>, экспонента
exp[-g/] —> 0, а скорость реакции — к постоянному значению
— = v0v; таким образом, устанавливается стационарный режим
8
неразветвленной цепной реакции.
Теория обрыва цепей на стенках разработана Н. Н. Семеновым.
Различают диффузионную и кинетическую области протекания
реакции обрыва. В кинетической области скорость обрыва зависит
от скорости взаимодействия свободных радикалов или атомов со
стенкой; она пропорциональна отношению площади поверхно¬
сти сосуда к его объему (S/V), также зависит от величины в —
вероятности захвата стенкой свободных радикалов (в = 1 —10“5). Ос¬
новное уравнение константы скорости обрыва цепей на стенках
для цилиндрического сосуда имеет вид
кf =
d2 ( d vl
32 D tv
j
(3.8)
где D — коэффициент диффузии; d — диаметр реакционного со¬
суда; v — средняя арифметическая скорость.
В диффузионной области протекания реакции обрыва, т. е. при
d2 d
малых значениях D и выполнении условия -гт~- » —, константа
32Д ev
скорости реакции обрыва цепи равна
kf = ™- (3-9)
59
В кинетической области протекания реакции обрыва цепей,
d d2
т. е. при выполнении условия — » , константа скорости ре¬
ву 32 D
акции обрыва равна
kf=ev/d. (3.10)
3.3. Разветвленные цепные реакции
Цепная реакция, включающая в себя стадии зарождения, про¬
должения, разветвления и обрыва цепи, называется разветвлен¬
ной. Примерами таких процессов являются реакции окисления кис¬
лородом фосфора, фосфина, водорода, монооксида углерода(И)
и др.
Теория разветвленных цепных реакций разработана главным
образом Н. Н. Семеновым и его школой, определенный вклад в ее
развитие внес С.Н.Хиншельвуд.
Н.Н. Семенов показал, что при общем описании закономер¬
ностей развития разветвленных цепных реакций система кинети¬
ческих уравнений для активных центров может быть сведена к
уравнению для активных центров одного вида.
Дифференциальное уравнение для скорости изменения кон¬
центрации активных центров в этом случае по аналогии с уравне¬
нием (3.5) примет вид
^- = v0-gn + fn = v0-(g-f)n = vo+ ф«, (3.11)
ш
где gn — скорость гибели активных центров; fn — скорость обра¬
зования активных центров в результате реакций разветвления;
f-g=ф-
Интегрирование (3.11) при начальных условиях и = 0 для /=0,
принимая/и g постоянными и пренебрегая расходованием исход¬
ных веществ, приводит к уравнению вида
л = ^(е</'‘>'-1) = ^(е''-1)- (3.12)
Скорость реакции, определяемая приростом активных центров
в момент времени t, выразится следующим образом:
г; = /'1 = 7Г^(еФ'"1), (3.13)
где / — удельная скорость реакции продолжения цепи.
Анализ уравнений (3.12) и (3.13) позволяет оценить предель¬
ные значения пи v:
60
1. g >f При t —» 0
n = v0t и v = lv0t, (3.14)
т.е. в начальный период реакции п, скорость г;линейно зависит от
времени.
При t—>°°
v0 , v0
и ' F7' (315)
т. е. с течением времени в реакционной смеси устанавливается ква-
зистационарный режим, характерный для неразветвленной цеп¬
ной реакции.
2-f>g, f - g = Ф > 0. При f —> оо находим, что
п =
= — еф' и v = ln = — еф'. (3.16)
ф ф 4 ’
Иными словами, концентрация активных центров и соответ¬
ственно скорость процесса неограниченно возрастают со време¬
нем по экспоненциальному закону, и реакция после некоторого
периода индукции заканчивается взрывом даже при постоянной
температуре. Тепловыделение в этом случае принципиально несу¬
щественно, поскольку причина воспламенения заключается в
постоянном увеличении числа активных центров, что приводит к
спонтанному росту скорости реакции.
3. / = g, Ф = / - g = 0. В уравнениях (3.12), (3.13) возникает нео¬
пределенность, раскрывая которую по правилу Лопиталя полу¬
чим
v = lv0t, (3.17)
т. е. скорость реакции возрастает пропорционально времени, при
этом реакция протекает без воспламенения, во многих случаях с
чрезвычайно малой скоростью.
Уравнение (3.11) является модельным уравнением любой раз¬
ветвленной цепной реакции, а равенство ф = f - g = 0 — крити¬
ческим условием перехода от стационарного к нестационарному
взрывному режиму протекания цепного процесса.
Н.Н.Семенов показал, что уравнения типа (3.11) легко полу¬
чить при изучении кинетики конкретных разветвленных цепных
реакций, используя сформулированный им принцип частично-ста¬
ционарных концентраций', концентрации всех активных центров,
обладающих высокой реакционной способностью, можно считать
квазистационарными, кроме того активного центра, чья концен¬
трация существенно возрастает в ходе процесса.
61
Применяя принцип частично-стационарных концентраций к
реакции окисления водорода с учетом общепринятого механизма
ее протекания, можно считать, что
Я=,®=омЯ(0. (3.18)
d t at at
Это позволяет заменить часть дифференциальных уравнений
на алгебраические и получить кинетическое уравнение типа (3.11).
Полученные при анализе уравнений (3.12), (3.13) соотноше¬
ния (3.15), (3.16) позволили объяснить удивительные явления,
наблюдавшиеся при изучении реакций окисления фосфора и во¬
дорода кислородом. Особенность реакции окисления фосфора, изу¬
чение которой привело к открытию разветвленных цепных реак¬
ций, заключается в следующем: процесс идет быстро и сопро¬
вождается пламенем, если давление кислорода лежит в пределах
Р\ < Рог < Pi, и совсем прекращается, если р < р{ и р> р2; при¬
чем переход от фактического отсутствия реакции к быстрой реак¬
ции происходит очень резко при достижении критических давле¬
ний pi и р2, называемых пределами воспламенения. Нижний пре¬
дел области воспламенения фосфора достигается при давлении
кислорода, ро2 = 1(Г2 мм рт. ст., верхний предел имеет место при
Ро2 ~ 1 атм, давление же фосфора равно всего 1СГ2 мм рт. ст.
Согласно теории, при малых давлениях кислорода, р < ри цепи
не развиваются из-за обрыва на стенках, и реакция окисления
переходит на стационарный режим, т.е. выполняются условие
g >f и уравнение (3.15). Нижний предел воспламенения зависит
от состава смеси, диаметра сосуда, материала его стенок и незна¬
чительно от температуры. Для давлений кислорода в области вос¬
пламенения Р\< р < Рг разветвление цепей преобладает над их об¬
рывом, т.е. выполняются условие g>f и уравнение (3.16). При
давлении р > р2 обрыв цепей в объеме (в результате тройных столк¬
новений с частицами загрязнений), скорость которого пропор¬
циональна р2, начинает преобладать над разветвлением, скорость
которого пропорциональна р, и реакция снова переходит на ста¬
ционарный режим: g > /, уравнение (3.15). Верхний предел вос¬
пламенения зависит от температуры, природы и количества при¬
меси и мало зависит от формы сосуда, его диаметра и состояния
стенок.
Трудности изучения реакции окисления фосфора связаны с
отсутствием сведений о механизме развития цепи, но для реак¬
ций окисления Н2 и СО (в присутствии добавок Н2 или Н20)
такие механизмы можно считать установленными.
На рис. 3.1 в координатах (р, Т) приведены данные, характе¬
ризующие область воспламенения смеси Н2 и 02, получившую
название «полуостров воспламенения».
62
Рис. 3.1. Пределы воспламенения для стехиометрической смеси Н2 и 02
в сосуде диаметром 7,4 см, покрытом КС1*; ри р2, р3 — соответственно
первый, второй и третий пределы давления
Воспламенение гремучей смеси происходит только при усло¬
виях, соответствующих точкам внутри полуострова воспламене¬
ния. Из рис. 3.1 видно, что при температуре 773 К кроме нижнего
(первого, р{ = 1,5 мм рт.ст.) и верхнего (второго, р2) пределов
цепного воспламенения при давлении, равном 3000 мм рт. ст.,
наблюдается и третий предел (р3) воспламенения. Этот предел,
по-видимому, имеет тепловую природу, хотя при высоких давле¬
ниях не исключается и цепное разветвление в результате увеличе¬
ния вклада реакции
Н02 + Н2 -> Н202 + Н
Для лучшего усвоения теории цепных разветвленных реакций
рассмотрим подробно следующую задачу.
Пример 3.1. Пределы воспламенения смеси Н2+ 02. Для реакции окис¬
ления водорода в определенных условиях принимают следующую упро¬
щенную схему процесса:
* Lewis В. et von Elbe G. Combustion, Flames and Explosion of Gases. N. Y.: Academic
Press, 1953. P. 29.
63
Н2 + 02 1—>20Н зарождение (инициирование)
ОН + Н2 —^—»Н20 + Н продолжение
Н + 02 —-—> ОН + О разветвление
О + Н2 —-—> ОН + Н разветвление
Н + стенка—^—> у2Н2 гетерогенный обрыв (первый предел)
Н + 02 + М —^—> HOJ + М гомогенный обрыв (второй предел)
Эта схема позволяет объяснить экспериментальные результаты, каса¬
ющиеся первого и второго пределов в области воспламенения водорода,
которым соответствуют общие давления рх и р2:
А ,
C\*RT п г
Р\ =-L^—, Pi =Qe RT>
где d — диаметр сосуда; Еь Е2 — величины, идентичные энергии, С, —
постоянная.
1. Получите аналитическим путем выражение для фактора разветвле¬
ния ф в случае первого (нижнего) и второго (верхнего) пределов вос¬
пламенения.
Покажите, каким путем, зная ф, можно получить выражения для дав¬
лений, характеризующих оба предела, указанные в условии.
D
Принять, что к5 - —р-р > где D — коэффициент диффузии.
2. В области медленного протекания реакции концентрация радика¬
лов Н может достигать стационарного состояния. Найдите выражение для
этой концентрации и покажите, что из него можно получить условия
пределов воспламенения.
Решение. Пределы воспламенения смеси Н2 + 02 представлены на
рис. 3.1.
Предел воспламенения означает переход от стационарного режима к
взрывному. Известно, что критическое условие воспламенения можно
выразить двумя способами:
Ф = 0 или [Н]стац -> со.
1. Выражение для общего фактора разветвления, ф, находим из ос¬
новного уравнения разветвленных цепных реакций (3.11):
+ (1)
а) Для первого предела воспламенения, применив принцип частич¬
но-стационарных концентраций Н.Н.Семенова к радикалам Н, О, ОН,
получим
d[H]
—= v2 - v3 + v4 - v5. (2)
64
d[°] n
= u> следовательно,
v 3 = V4
d[OH]
dt
Подставляя (3) и (4) в (2), находим
d[H]
(3)
= 2v{ -v2+v3+v4=0. (4)
откуда
^ = 2v\ + 2v3 - v5 = 2v\ + [H](2fc3[02] - ks), (5)
<p = 2k3[02)-k5. (6)
При выполнении критического условия воспламенения ср = 0 полу¬
чаем
2к3[02] = к5. (7)
Выразив концентрацию 02 через давление
(8)
где pi — общее давление в смеси, p0l — парциальное давление, N0l —
мольная доля, и подставив (8) в (7) с учетом уравнения Аррениуса и
соотношения для к5 в задании 1, приходим к выражению
2A^T£JT-Wr (9)
откуда находим рх:
Р\ =
DRT
2N02A3
1/2 А-
2RT eRT
= (10)
d ' d
где С, =
DRT
Л ,r — фактически постоянная, поскольку Т1/2 изменя¬
ло, 4 ^
ется очень слабо по сравнению с экспонентой, Ех = Е3/2.
Как видно из формулы (10), полученный результат совпадает с при¬
веденным соотношением в условии задачи и показывает, что с увеличе¬
нием температуры и диаметра сосуда давление нижнего предела воспла¬
менения уменьшается.
б) Для второго (верхнего) предела воспламенения, применив снова
принцип частично-стационарных концентраций Н.Н. Семенова к ради-
s. Зак.1596
65
калам Н, О, ОН с учетом в механизме стадии (6) вместо (5), после
несложных преобразований получим
Ф = 2*3[02]-*«[02][М1; (11)
при ф = О
2*з=*в[М], (12)
где М — определенная молекула, концентрация которой равна
= (13)
р2 — давление на втором (верхнем) пределе воспламенения.
После подстановки [М] в (12) последнее в развернутом виде примет вид
2 Агъ~к? = jfc, (14)
откуда выразим р2.
_ AzA. _А
р2 = С2е лг =с2елг? (15)
полагая, что £3 - Ев = Е2> 0, так как £3 > Е6.
Как видно из выражения (15), полученная формула совпадает с ука¬
занной в условии задачи и показывает, что с ростом температуры наблю¬
дается и увеличение давления второго (верхнего) предела воспламенения
водорода.
2. Выразим стационарную концентрацию радикалов Н.
а) При рассмотрении реакции гетерогенного обрыва (стадия 5):
® = 2*,[02][Н2] + 2&3[Н][02] - Л3[Н] = 0, (16)
= 2Ц02][Н2]
I Jcrau lC5_2lCl[02]- (1?)
На первом пределе воспламенения [Н]стац -> <», это возможно, если
к5 - 2Аг3[02] = 0 или 2к3[02] = к5
т.е. приходим к известному уже соотношению (7), из которого получаем
выражение (10) для давления первого предела воспламенения.
б) При рассмотрении реакции гомогенного обрыва (стадия 6):
ffl = 2*,[02][Н2] + 2*3[Н][02] - Аг6[М][02][Н], (18)
гН] __ 2*j[Q2][H2]
JcTau Л^[М](02] - 2Лг3[02] ' (19)
66
На втором пределе воспламенения [Н]стац -> °°, это возможно при
условии, что
^б[м][Ог] = 2£3[02] или 2кг = £6[М],
т.е. приходим снова к известному соотношению (12), из которого полу¬
чаем выражение (15) для давления второго (верхнего) предела воспла¬
менения водорода.
Примером разветвленной цепной реакции является также ре¬
акция деления ядер урана, используемая в атомной энергетике:
10п+ 2^и-э 3gSr + *gXe + 3l0n,
в результате которой происходит не только выделение огромного
количества энергии порядка 200 МэВ (1,93 -1013 Дж/моль), но и
образование трех (в среднем 2,5) нейтронов. Число нейтронов,
играющих роль активных центров, возрастает по ходу реакции в
геометрической прогрессии, что, в свою очередь, приводит к ла¬
винообразному возрастанию числа делящихся атомов и количе¬
ства выделяющейся энергии, т.е. к взрыву.
В рассмотренном примере пределы воспламенения смеси Н2 + 02
не зависят от значения скорости зарождения цепей. Такой резуль¬
тат связан прежде всего с тем, что реакции разветвления и обры¬
ва цепей рассматриваются как линейные относительно концент¬
рации свободных атомов (и радикалов), а другие процессы — квад¬
ратичные по концентрации активных центров, взаимодействия
последних друг с другом не учитываются.
Однако эксперимент показал, что увеличение скорости зарож¬
дения цепей, например под действием электрического разряда
различной мощности, приводит к значительному расширению об¬
ласти воспламенения гремучей смеси и, следовательно, к ускоре¬
нию процесса разветвления. В таком случае считают, что наблюда¬
ется положительное взаимодействие цепей.
Дифференциальное уравнение для скорости изменения кон¬
центрации активных центров в ходе разветвленной цепной реак¬
ции с положительным взаимодействием цепей запишется следу¬
ющим образом:
^■ = v0+(f-g)n + cn2, (3.19)
здесь слагаемое сп2 выражает квадратичное разветвление цепей.
При увеличении п условием воспламенения служит неравен¬
ство — >0. Для / > g это очевидно. В случае g > /> поскольку
dt
минимум производной — наблюдается при значении п = -——,
dt 2 с
условие воспламенения примет вид
67
Сg - /)2 | (s - ff ; Q
2c 4c
(3.20)
или
g < / + J4cv0.
(3.21)
Таким образом, смесь, находящаяся при условии g > / вне
области воспламенения, благодаря выполнению соотношения
(3.18) может попасть внутрь ее, что указывает на расширение об¬
ласти воспламенения с увеличением v0.
Реакции образования свободных радикалов, протекающие в
основном в результате превращений промежуточных продуктов,
называют реакциями «вырожденного взрыва» или цепными ре¬
акциями с вырожденным разветвлением цепей. Эти реакции прин¬
ципиально отличаются от рассмотренных выше разветвленных
цепных процессов, в которых зарождение новых цепей осуще¬
ствляется при участии активных центров: для них не наблюдает¬
ся переход в режим самовоспламенения или взрыва из-за малого
значения ф, кроме того, скорости реакций после достижения
некоторой максимальной величины уменьшаются. Характерное
время их ускоренного протекания не доли секунд, а десятки ми¬
нут и часов.
Примером реакций с вырожденным разветвлением служит про¬
цесс окисления кислородом ряда углеводородов. При низкотем¬
пературном окислении образующийся на одной из стадий про¬
должения цепи гидропероксид
может стать источником свободных радикалов по реакциям
ROOH -» RO*+*OH и ROOH -> R0*+R0*2+H20,
что приводит к возникновению новых цепей.
При высокотемпературном окислении образующийся на од¬
ной из стадий продолжения цепи формальдегид
•СН2ООН -» СН20 + 'ОН
может стать при взаимодействии с кислородом источником но¬
вых радикалов по реакции
сн2о + о2 -> но; + нс*о
На начальной стадии реакции, когда степень превращения ре¬
агентов невелика и расходованием промежуточных продуктов мож¬
RO; +RH -» ROOH + R
68
но пренебречь, система дифференциальных уравнений, описыва¬
ющих кинетику реакции с вырожденным разветвлением и линей¬
ным обрывом цепей, имеет вид
d« ^ dp 1
-jj = vo+fP~gn; = (3-22)
где р — концентрация промежуточного продукта, например
ROOH; / — удельная скорость продолжения цепи.
Решение этой системы уравнений можно получить либо путем
сведения к одному дифференциальному уравнению второго по¬
рядка, либо используя принцип квазистационарных концентра¬
ций, полагая <\п / dr <s. vQ + fp. В последнем случае
v0+fp-dn/dt v0+fp
П = ' — ■» ■■■■■ (3.23)
8 8
dp / If
~т~ = vq + — p. (3.24)
dr g g v '
Интегрирование (3.24) с учетом начального условия (р = 0 при
t = 0) приводит (при малой скорости разветвления) к конечному
результату
i'-l
(3.25)
В случае реакции с вырожденным разветвлением и квадратич¬
ным обрывом цепей для описания ее кинетики необходимо ре¬
шить следующую систему дифференциальных уравнений
77 = v0 + fp - sn2; 77 = (3.26)
ш at
Отметим, опуская решение, которое удобнее проводить с по¬
мощью безразмерных переменных и параметров, что накопление
промежуточного продукта идет по параболическому закону, т.е.
автоускорение выражено менее резко, чем для линейного обрыва
цепей.
Предельные явления для реакций с вырожденным разветвле¬
нием цепей наблюдаются, но выражены они не столь ярко, как
для разветвленных. Переход от медленно протекающей реакции к
медленно ускоряющейся не столь заметен, как переход от прак¬
тического отсутствия реакции к вспышке (воспламенению) в раз¬
ветвленных реакциях.
В заключение рассмотрим реакции с энергетическими цепями,
предсказанные М. Боденштейном и через 50 лет открытые и экс¬
периментально изученные в работах школы Н. Н. Семенова. Отли¬
69
чие этих реакций от рассмотренных выше материальных цепей
заключается в том, что энергия экзотермических реакций про¬
должения цепи переходит не сразу в химическую энергию атомов
и радикалов, а предварительно в энергию колебательного воз¬
буждения конечных или промежуточных продуктов реакции. Так,
для реакции между фтором и водородом, протекающей по цепно¬
му механизму, на стадии взаимодействия Н* с F2 часть энергии
(~50 кДж/моль, что составляет ~ 12 % от всего количества выделя¬
ющейся энергии) переходит в энергию возбуждения HF. Посколь¬
ку энергии колебательного возбуждения HF и Н2 близки (coHF =
= 4048 см_|,сон2 = 4280 см-1), происходит быстрый перенос энер¬
гии с HF* на Н2. Затем образующаяся колебательно-возбужденная
молекула Н* быстро взаимодействует с F2. Перечисленные про¬
цессы можно выразить схемой
Н* + F2 -> F* + HF*
HF* + Н2 -> HF + Hf
Щ + F2 —> 2HF + F* + H*
Суммарное уравнение приведенных выше реакций цикла про¬
должения цепи
2F2 + Н2 -» 2HF + 2F*
указывает на образование не только продукта реакции, но и ак¬
тивных центров — атомов F*.
Реакции с энергетическим разветвлением цепей могут проис¬
ходить и без передачи колебательной энергии на реагент. Напри¬
мер, при фторировании углеводородов по реакции
CHF2 +F2 ->F+CHF?
образуется колебательно-возбужденная частица CHF*, которая
быстро распадается на HF и CF2. Взаимодействие CF2 с фтором
приводит к рождению новых активных центров:
CF2 + F2 -» CF3* + Р
Таким образом осуществляется разветвленная цепная реакция.
Вопросы и задания для самоконтроля
1. Дайте определение цепных реакций.
2. Поясните термин «активный центр». Какие частицы можно отнести
к этому понятию?
70
3. В чем различие между материальными и энергетическими цепя¬
ми? Приведите примеры.
4. Перечислите стадии цепной реакции на конкретном примере.
5. Каковы возможные пути возникновения свободных радикалов и
атомов?
6. Какие вещества называются инициаторами? Приведите примеры.
7. Укажите различные типы реакций продолжения цепи.
8. Сформулируйте принцип неуничтожимости свободной валентно¬
сти, введенный Н. Н.Семеновым.
9. Поясните на примерах термины: а) «линейный обрыв»; б) «квад¬
ратичный обрыв».
10. Какие превращения являются неразветвленными цепными реак¬
циями?
И. Поясните термины: «звено цепи», средняя «длина цепи».Приве¬
дите математическое определение средней длины цепи.
12. Покажите на конкретном примере, что при стационарном проте¬
кании неразветвленной цепной реакции скорость зарождения цепи рав¬
на скорости обрыва, v0 = ty.
13. Приведите выражение для определения скорости неразветвлен¬
ной цепной реакции с учетом длины цепи.
14. Перечислите кинетические особенности неразветвленной цепной
реакции.
15. Приведите вывод кинетического уравнения зависимости скорос¬
ти неразветвленной цепной реакции от времени.
16. В чем заключается различие диффузионной и кинетической обла¬
стей протекания реакций обрыва цепей?
17. Приведите уравнение константы скорости реакции обрыва цепей
на стенках для цилиндрического сосуда.
18. Покажите, какой вид принимает уравнение константы скорости
реакции обрыва, если реакция протекает: а) в диффузионной области;
б) в кинетической области.
19. Какие реакции называются разветвленными? Приведите примеры.
20. Выведите уравнение для зависимости скорости разветвленной
цепной реакции от времени.
21. Проанализируйте кинетическое уравнение (вопрос 20) в случаях:
a) g > / ; б) / > g ; в) / = g.
22. Сформулируйте принцип частично-стационарных концентраций
Н. Н. Семенова.
23. Выведите кинетическое уравнение для скорости реакции окисле¬
ния водорода с учетом общепринятого механизма, используя принцип
частично-стационарных концентраций. Полученный результат сравните
с уравнением из вопроса 20.
24. Какие типы самовоспламенения возможны в химических реакци¬
ях? В чем существенное различие между ними?
25. Поясните термин: «предел воспламенения». Объясните с помощью
теории Н.Н. Семенова появление первого (нижнего) и второго (верхне¬
го) пределов воспламенения на примере реакции окисления фосфора.
26. От каких параметров зависит положение первого и второго преде¬
лов воспламенения?
71
27. Представьте в координатах (р, Т) область воспламенения грему¬
чей смеси (Н2 и 02). Какое название получила эта область? Укажите по¬
ложение первого и второго пределов воспламенения. Какой характер имеет
их зависимость от температуры?
28. Какова природа третьего предела воспламенения: в общем случае;
для реакции окисления водорода?
29. Каким путем можно определить выражение для точки мыса полу¬
острова воспламенения?
30. Перечислите кинетические особенности разветвленных цепных
реакций.
31*. В чем различие понятий: отрицательное и положительное взаимо¬
действие цепей?
32*. Покажите в случае положительного взаимодействия цепей, поче¬
му смесь может воспламеняться при условии /< g?
33*. Какие процессы называют реакциями «вырожденного взрыва» или
цепными реакциями с вырожденным разветвлением цепей? Приведите
примеры.
34*. Выведите кинетическое уравнение для начальной стадии реакции
с вырожденным разветвлением и линейным обрывом цепей.
35*. Получите кинетическое уравнение для реакции с вырожденным
разветвлением и квадратичным обрывом цепей.
ГЛАВА 4. ФОТОХИМИЯ
4.1. Основные законы и квантовый выход
Фотохимия — раздел науки, посвященный закономерностям
реакций, протекающих под действием света. Современная фото¬
химия находится на стыке физической химии, квантовой хи¬
мии, химической динамики, синтетической и аналитической хи¬
мии.
Фотохимические реакции могут протекать в газообразной, жид¬
кой и твердой средах, они осуществляются под действием види¬
мого и ультрафиолетового излучений с длинами волн в интервале
от 170 до 750 нм. Поглощение молекулой кванта энергии перево¬
дит ее в возбужденное состояние с отличными от исходного фи¬
зическими и химическими свойствами. Этим объясняется возмож¬
ность протекания различных химических и физических процес¬
сов, невозможных при термическом воздействии на реакционную
систему.
К фотохимическим реакциям, которых насчитывается множе¬
ство, относятся, например:
зрение — основу процесса составляет реакция фотоизомериза¬
ции ретиналя;
72
фотосинтез — сложная реакция ассимиляции углерода расте¬
ниями, протекающая под действием солнечной энергии с помо¬
щью хлорофилла, который является фотосенсибилизатором:
6С02 + 6Н20—2^->С6Н1206 + 602
фотография, в основе которой лежит способность галогенидов
серебра разлагаться под действием света с выделением металли¬
ческого серебра:
AgBr ———> Ag + Вг
образование витамина D из холестерола и эргостерола под дей¬
ствием: УФ (этот витамин способствует лучшему усвоению каль¬
ция в организме и предотвращает рахит).
Общие закономерности фотохимических реакций объясняют с
помощью следующих законов.
1. Закон Буге (1729) — Ламберта (1760) устанавливает неза¬
висимость поглощательной способности среды от энергии потока
и выполняется, как показали измерения в XX в., в широком ин¬
тервале потоков энергии (10~18 — 10 Дж-см'2-с-1).
Его математическая формулировка
где I — интенсивность светового потока, падающего на слой сре¬
ды (Е-см~2-с-1). Е — внесистемная единица Эйнштейн; dx — бес¬
конечно тонкий слой однородной поглощающей среды; L — по¬
стоянная.
2. Закон Бера (1852 г.) устанавливает связь между поглотитель¬
ной способностью среды (L) и концентрацией (и) поглощающих
молекул в этом слое:
L = кп, (4.2)
где к — молекулярный коэффициент поглощения или, говоря со¬
временным языком, мера поглотительной способности молекул
при данной длине волны.
Объединенный закон Буге—Ламберта—Бера на основании ра¬
венств (4.1) и (4.2) после интегрирования / в пределах от / до 1Х,
х от 0 до / (/ — общая толщина поглощающего слоя) и потенциро¬
вания примет вид
I, = /0е-ш, (4.3)
где /0 — начальная интенсивность светового потока; I, — интен¬
сивность светового потока после его прохождения через слой ве¬
щества толщиной /.
При переходе к мольному коэффициенту поглощения, е, со¬
гласно соотношению
кп - ес, (4.4)
объединенный закон Буге—Ламберта —Бера запишется так:
Woe-*, (4.5)
где с — концентрация поглощающего свет вещества.
Отношение /,//0 в литературе часто называют пропусканием, а
выражение [-lg(/,//0)] — поглощением среды или оптической
плотностью D.
Необходимо отметить, что объединенный закон применим только
к системам, в которых растворенное вещество не диссоциирует, не
ассоциирует, не взаимодействует и не реагирует с растворителем.
Поглощенная энергия 1а при прохождении светового потока че¬
рез слой толщиной / равна
/fl = /o-/,=/0(l-e-Ed). (4.6)
Это соотношение справедливо только для очень узкого интер¬
вала частот.
3. Закон Гротгуса (1817 г.) — Дрепера (1844 г.): только погло¬
щенный средой свет может произвести ее химическое изменение
(I закон фотохимии). Для осуществления фотохимических реак¬
ций это условие необходимое, но не достаточное.
4. Закон Вант-Гоффа (1904 г.): количество фотохимически из¬
мененного вещества пропорционально количеству поглощенной
световой энергии. Его математическая формулировка для скорос¬
ти фотохимической реакции иф х с учетом (4.6)
= c°nst/0 = const/0(l - е"£С/). (4.7)
Рассмотрим два предельных случая. Если ес/» 1, т. е. большой
коэффициент поглощения, тогда ехр[-ес/] —> 0 и уравнение (4.7)
переходит в
~^ = const/0, (4.7а)
т.е. скорость реакции пропорциональна световому потоку и не за¬
висит от концентрации поглощающего вещества (нулевой поря¬
док). Если ес/<к 1, поглощающий слой очень мал, тогда экспонен¬
ту можно разложить в ряд, ограничиваясь двумя членами,
ехр[-ес/] = 1 - ес/и уравнение (4.7) переходит в
— = const'С, (4.76)
т. е. скорость реакции имеет первый порядок по концентрации реа¬
гирующего вещества, const' = const • е//0.
5. Закон фотохимической эквивалентности Эйнштейна—Штарка
(1912 г.) в современной трактовке: каждый поглощенный в пер¬
74
вичном акте квант света вызывает превращение одной молекулы
(II закон фотохимии).
Для характеристики фотохимических процессов используют по¬
нятие квантового выхода, различая первичный, вторичный и об¬
щий квантовые выходы.
Первичный квантовый выход ф| равен отношению числа про¬
реагировавших возбужденных молекул в первичном акте к числу
поглощенных квантов. В соответствии со II законом фотохимии ф,
всегда < 1.
Общий квантовый выход Ф равен отношению числа образовав¬
шихся в процессе молекул продукта (или числа прореагировав¬
ших молекул реагента) к числу поглощенных квантов.
Для реакций, протекающих в растворах, Ф < 1; при значениях
Ф » 1 есть основания считать, что рассматриваемый фотохими¬
ческий процесс имеет цепной характер. Интервал возможных из¬
менений Ф: 10'3< Ф < 106.
Вторичный квантовый выход ф2 является мерой развития вто¬
ричных реакций и равен отношению числа образовавшихся в про¬
цессе молекул продукта к числу молекул, прореагировавших в
первичном акте. По определению, Ф = ф,ф2.
Количество энергии Ет, поглощаемое одним молем вещества,
по закону фотохимической эквивалентности равно
Ет = NAhv = NAhc(x> = Nk^-, (4.8)
где Na — число Авогадро, 6,02-1023 моль"1; А — постоянная План¬
ка, 6,626-10"34 Дж-с; с — скорость света, 3-108 м-с"1; v — часто¬
та (с-1); со — волновое число (м-1); X — длина волны излучения (м).
Величина Ет принимается равной 1 Эйнштейну.
В видимой части спектра энергия фотонов в фиолетовой обла¬
сти (Д. = 400 нм, Ет = 299 кДж-моль-1) выше, чем в красной (X =
= 700 нм, Ет = 171 кДж-моль'1).
Число моль вещества, которое в единицу времени и в единице
объема переходит в возбужденное состояние под действием излу¬
чения и может участвовать в первичном фотохимическом акте,
легко рассчитать
(4-9>
Подставляя в формулу (4.9) значение 1а из (4.6) и учитывая
первичный квантовый выход фь получим уравнение для скорости
фотохимической реакции
<4-Ю)
75
4.2. Физические и химические фотопроцессы
Любая фотохимическая реакция является сложным многоста¬
дийным процессом. Первичный процесс состоит из стадии погло¬
щения кванта энергии и сразу следующей за ней стадии превра¬
щения возбужденной молекулы. Начальная стадия поглощения
света
АВ + hv —» АВ*
приводит к образованию электронно-возбужденной молекулы, при
этом энергия электромагнитных колебаний трансформируется в
энергию движения электронов, которые переходят на более высо¬
кий энергетический уровень. Эта стадия с химической точки зрения
протекает мгновенно и заканчивается за время т = 10~15—10~17 с.
На следующей стадии электронно-возбужденная молекула пре¬
терпевает превращение по одному из возможных направлений пер¬
вичных процессов:
Превращение
Тип процесса
АВ*
—
А+В
диссоциация
—*->
ВА
изомеризация
>
AD + B
химическая реакция
+ D
АВ + hv
излучение (флуоресценция или
фосфоресценция)
—-—>
АВ
внутримолекулярный переход
энергии
—-—>
АВ + CD*
дезактивация при соударении
+ CD
или на стенке
Направления 1, 2, 3 — первичные химические процессы; 4, 5, 6 —
фотофизические процессы. По направлению (1) возможно проте¬
кание нескольких процессов:
диссоциация на свободные атомы и радикалы:
12—*л—-»Г + Г
фотоионизация:
Fe3+Cl“ —*^-> (Fe2+Cl*) -> Fe2+ + СГ
или распад на молекулы (и атомы):
NO* -» N0 + 0
По направлению (2) происходит изомеризация многоатомных
(в основном органических) молекул, например
цис СбН5СН = СНСбН? -> транс С6Н5СН = СНС6Н5
76
Химическая реакция (направление 3) может протекать либо с
участием другого вещества, например
Н* + HI -> Н2 +1*
либо внутримолекулярно:
С3Н7СОСЩ -> *С3Н6С(ОН)СН3 -> С2Н4 +СН3СОСН3
Рассмотрим подробнее фотофизические процессы. Для харак¬
теристики этектронных состояний молекул используют величину
мультиплетности, которая указывает на число вырожденных уров¬
ней, соответствующих данному электронному состоянию, и рав¬
на 2j + 1, где s — суммарный электронный спин молекулы. Муль-
типлетность обычно обозначают значком слева вверху. Как пока¬
зано на рис. 4.1*, исходные молекулы находятся в основном синг-
летном состоянии, обозначаемом символом S0, мультиплетность
которого равна 1. Поглощение света вызывает электронный пере¬
ход внутри молекулы АВ, который приводит к возбужденному
синглетному состоянию S„.
Время жизни возбужденного синглета 1СГ9 < xs < КГ6 с. Однако
возможны и переходы, когда спин электрона становится парал¬
лельным партнеру, тогда мультиплетность равна трем. Такое со¬
стояние системы называют триплетным, обозначая символом Т.
Время жизни триплета всегда больше, чем время жизни синглета:
КГ* <тг <10 с.
С электронно-возбужденной молекулой могут происходить бе-
зызлучательные и излучательные процессы. К первым относятся
колебательная релаксация, внутренняя конверсия и интеркомби¬
национный переход.
Колебательная релаксация, когда возбужденная молекула (на¬
правление 6) в результате столкновений может потерять свою из¬
быточную колебательную энергию и достичь состояния S\ (г>=0):
Hg* + Т1 Hg + Т1* и О* + N2 О + N2
В жидкой фазе этот процесс протекает очень быстро, т= 10-13—
10-11 с;
Внутренняя конверсия — переходы между состояниями одинако¬
вой мультиплетности (направление 5), когда молекула постоянно
переходит из одного колебательного состояния в другое до более
низкого электронного состояния: —> S0: С6Н6(5'1) —> С6Н6(50).
Энергия, выделяющаяся при этом, передается другим молекулам
среды. Длительность этого процесса т = 10~" — 10~10 с.
* Панченков Г. М., Лебедев В. П. Химическая кинетика и катализ. — М.: Хи¬
мия, 1985.
77
Синглетные
состояния
Триплетные
состояния
состояние
Рис. 4.1. Энергетические уровни безызлучательных (7, 2)
и излучательных (3, 4) процессов:
1 — внутренняя конверсия; 2 — интеркомбинационный переход; 3 — флуорес¬
ценция; 4 — фосфоресценция; волнистые стрелки — колебательная релаксация
Интеркомбинационный переход или конверсия (из состояния с
одной мультиплетностью в состояние с другой мультиплетнос-
тью, направление 5) обычно происходят с одного колебательно¬
го уровня синглета S{ на колебательно-возбужденный уровень трип-
летного состояния, например N(S{) -> N(T{) где N — нафталин.
Вероятность таких переходов в 103—106 раз меньше, чем внутрен¬
няя конверсия. Электронное распределение в состоянии Т{ может
привести к тому, что триплетное состояние становится более ре¬
акционноспособным, чем основное синглетное состояние £о, в
этом случае константы скорости интеркомбинационной деграда¬
ции 7j -» S0 изменяются в пределах 10-1—10 с-1.
78
Например, бензофенол, растворенный в 2-пропаноле, прак¬
тически не взаимодействует с последним. Однако при пропуска¬
нии через раствор УФ лучей, образование триплетного состояния
позволяет ему оторвать атом водорода у 2-пропанола, что приво¬
дит к образованию двух свободных радикалов:
PhCOPh (SQ) --hv > PhCOPh (У,)
PhCOPh (Sj) интеркомбинационный переход ^ PhCOPh (7])
PhCOPh(7J) + Me2CHOH -> Ph2C*OH + Me2C*OH
Следует отметить, что синглет-синглетный и триплет-синглет-
ный переносы энергии в основном происходят по индуктивному
механизму, т. е. в результате диполь-дипольного взаимодействия
возбужденной молекулы с невозбужденной, которое проявляется
на расстоянии до 10'8 м и позволяет достаточно эффективно осу¬
ществлять перенос энергии даже при малых концентрациях ак¬
цептора энергии (до 10"* моль/л) в жидких и твердых растворах.
Напротив, триплет-триплетный и синглет-триплетный переносы
энергии в жидких растворах протекают по обменному механизму.
К. излучательным процессам (направление 4) относятся флуо¬
ресценция и фосфоресценция.
Флуоресценция — излучательный переход между синглетными
состояниями, когда молекула, находящаяся на самом низком ко¬
лебательном уровне возбужденного синглетного состояния Su мо¬
жет вернуться в основное синглетное состояние ^0:
Si -*S0+hv'; Na* -> Na + hv',
испускаемая частота v' обычно близка к поглощаемой частоте v
для атомов, но для молекул характерно наличие стоксова сдвига,
иногда весьма значительного. Флуоресценция наблюдается редко,
так как другие переходы (направления 5 и 6) протекают более
быстро, т.е. более вероятны. Флуоресценция характерна для ли¬
нейных молекул со многими сопряженными я-связями и зависит
от соотношения констант скорости испускания и безызлучатель-
ных процессов.
Если испускание (наряду с внутренней и интеркомбинацион¬
ной конверсией) является единственным способом дезактивации
электронно-возбужденной молекулы, то величина, обратная кон¬
станте скорости высвечивания (реакция первого порядка), назы¬
вается естественным временем жизни т° возбужденного состоя¬
ния. Эта величина для флуоресценции находится в интервале от
10~9 до 10~6 с.
Фосфоресценция — излучательный переход между состояниями
разной мультиплетности, когда молекула, находящаяся на самом
79
низком колебательном уровне триплета Ть излучает квант энер¬
гии и переходит в основное синглетное состояние:
7] -> S0 + hv"; N(Ti) -> N(S0) + hv", N — нафталин
Фосфоресценцию обычно изучают в твердой фазе, поскольку
многие твердые соединения продолжают испускать свет в течение
достаточно длительного времени после прекращения их облуче¬
ния; в жидких растворах (кроме диацетила и дибензила) присут¬
ствующие примеси заметно снижают (тушат) фосфоресценцию.
Триплетные состояния являются метастабильными, излучатель-
ный переход при фосфоресценции может длиться от 10_3 до 10 с,
таким образом константа скорости испускания фосфоресценции
находится в пределах 10-1— 103 с-1.
Если разница в энергиях S и Тсостояний невелика, то в хими¬
ческих реакциях возбужденные Svi Т молекулы могут вести себя
одинаково, т.е. с одинаковой реакционной способностью, хотя
по физическим свойствам молекулы будут отличаться: молекулы
в триплетном состоянии — парамагнитны, например кислород,
оксид азота, в синглетном — диамагнитны.
К первичным фотохимическим процессам близко стоит фото¬
сенсибилизация: превращение молекул вещества, нечувствитель¬
ных к излучению данной частоты, но получающих энергию не¬
посредственно от поглощающих ее молекул. Примером фотосен¬
сибилизации является диссоциация водорода в присутствии паров
ртути, атомы которой поглощают свет, соответствующий резо¬
нансной линии ртути с длиной волны X = 253,67 нм. Схема про¬
цесса:
Образующиеся в этом процессе очень реакционноспособные
атомы водорода легко восстанавливают оксиды металлов, СО, N0,
Возбужденные атомы ртути разлагают также аммиак, метан и
другие органические соединения. Сенсибилизаторами являются так¬
же ионы железа, галогены, хлорофилл и др.
Количество излучения часто измеряют с помощью актиномет¬
ра, в котором протекает реакция фотохимического разложения
щавелевой кислоты, сенсибилизированной ионом уранила. Свет
поглощается в пределах длин волн от 254 до 435 нм окрашенным
ионом уранила. Полученная энергия передается бесцветной щаве¬
левой кислоте, которая разлагается (с квантовым выходом 0,57 в
растворе, содержащем 10'2 М U02S04 и 5 -10-2 М Н2С204). Ура-
нил-ион остается неизменным и в качестве сенсибилизатора мо¬
жет применяться неограниченно.
Hg + hv —^ Hg*,
Н2 + Hg* —> 2Н + Hg,
£=4,9 эВ
Dh2 = 4,5 эВ
С2Н4.
80
К вторичным процессам, протекающим без участия света от¬
носятся:
реакции появившихся в результате первичного процесса ато¬
мов и свободных радикалов с молекулами реагента, например,
для фотохимической реакции образования НВг из простых ве¬
ществ вторая стадия
Вг* + Н2 -э НВг + Вг*
или для фотолиза соединения Сг(СО)5+ СО -» Сг(СО)6;
реакции дезактивации образовавшихся в первичном акте мо¬
лекул;
реакции рекомбинации свободных атомов и радикалов, напри¬
мер
Вг* + Вг* + М —» Вг2 + М и F* + F* + F20 —» F2 + F20
4.3. Кинетическая схема Штерна —Фольмера
Важные характеристики фотохимического процесса: время жиз¬
ни возбужденных молекул, число и природа возбужденных состо¬
яний, первичный квантовый выход — определяются при количе¬
ственном изучении флуоресценции и фосфоресценции.
В подразделе 4.2 были рассмотрены шесть возможных направ¬
лений первичных процессов. Однако в практических исследовани¬
ях при изучении кинетики используют упрощенные схемы, к числу
которых относится механизм Штерна—Фольмера. Этот механизм
включает только три первичных процесса (флуоресценцию или
фосфоресценцию, дезактивацию или обмен энергией и диссоци¬
ацию), следующие сразу за начальной стадией поглощения кван¬
та энергии, согласно схеме
Поглощение АВ + hv >АВ* v\ = 1а (4.11)
Флуоресценция ... АВ* —^—> АВ + hv v2 = /фл = к2[АВ*] (4.12)
Дезактивация АВ* + АВ—^—> АВ + АВ уъ = &3[АВ*][АВ] (4.13)
Диссоциация АВ* —^—> А + В vA = &4[АВ*], (4.14)
где 1а — интенсивность поглощенного света, т. е. число квантов, по¬
глощенных в единицу времени в единице объема, \Ia] = Е • см-3 - с-1;
/фл — число квантов, испускаемых флуоресценцией в единицу
времени из единицы объема.
Применяя принцип квазистационарных концентраций к [АВ*],
находим
= 1а - £2[АВ*] - &3[АВ*][АВ] - fc,[AB*] = 0, (4.15)
6. 3.1К. 1 У>1-
81
откуда
г АВ*1 = ^ . (4.16)
1 J >t2 + Ar3[AB] + >t4 v 7
Уравнение для скорости диссоциации (4.14) с учетом (4.16)
примет вид
v. - = /с. Is = Is (4 17)
dt к2 + £3[AB] + fc4 1 + ^. + ^.[AB]'
k4 k4
Из соотношений (4.17) и (4.11) легко найти выражение для
первичного квантового выхода
Щ 1
ф! = — = —т—ъ • (4.18)
Если условия стационарности (4.15) применимы к рассматри¬
ваемому объему, можно написать
h = /ф* + *3[АВ*][АВ] + fc4[AB*]. (4.19)
С учетом (4.12) путем несложных преобразований получают
уравнение
-т2- -1 = + т^-[АВ]. (4.20)
^фл к2 к-2
Отношение числа поглощенных квантов к числу испускаемых
является мерой тушения флуоресценции и называется просто ту¬
шением. Экспериментальное определение тушения в зависимос¬
ти от концентрации АВ позволяет получить по уравнению (4.20)
в координатах
'f-
*фл
, [АВ] прямую линию. Угловой коэффи¬
циент прямой tga = k3/k2, отрезок, отсекаемый прямой на оси
ординат, равен к4/к2.
Таким образом, схема Штерна—1Фольмера позволяет найти от¬
ношения констант к3/к2, к4/к2, а также к3/к4. Подстановка этих ве¬
личин в уравнение (4.18) дает возможность определить первичный
квантовый выход. Соотношение (4.18) можно представить в виде
В координатах ( фг1 , [АВ]) получают также прямую линию, уг¬
ловой коэффициент которой tg$-к3/к4, отрезок, отсекаемый
прямой на оси ординат, равен (1 + к2/к4). Определив независи-
82
мым путем значение к2 для флуоресценции, можно рассчитать
величины констант к3 и к4.
Механизм Штерна —Фольмера, объединяя стадии процесса из
фотофизики и фотохимии, является наиболее популярным среди
исследователей.
4.4. Зависимость квантовых выходов
от различных условий*
В современной фотохимии известны три типа режимов прове¬
дения процессов в зависимости от характера возбуждения:
стационарное, когда система облучается непрерывным пото¬
ком света постоянной интенсивности;
импульсное — одиночными или повторяющимися импульсами
света от импульсных ламп (10_6— 10_3 с), лазеров (10~п — 10~3с)
или искровых источников света (10~9— 10-6 с);
модулированное возбуждение светом, интенсивность которого пе¬
риодически варьируется с помощью специальных источников света.
В настоящее время импульсные методы широко применяются
при изучении механизмов фотопроцессов, исследовании приро¬
ды возбужденных состояний и промежуточных продуктов, опре¬
делении их времен жизни и констант скорости реакции.
Кинетика гибели возбужденных молекул в большинстве случа¬
ев отвечает реакции первого или псевдопервого порядка
[N] = [N]0e-'/\ (4.22)
где [N]0, [N] — концентрации возбужденных молекул при t = 0 и
в момент времени t; т= 1 — время жизни возбужденных мо-
/
лекул.
Если гибель возбужденных молекул происходит в результате
спонтанных процессов (колебательная релаксация, внутренняя и
интеркомбинационная конверсия, испускание), величину т на¬
зывают естественным временем жизни и обозначают т°. Если же
гибель происходит в результате процесса тушения, т. е. взаимодей¬
ствия (физического или химического) с другой молекулой типа
N + X -> Р, (4.23)
время жизни (при сохранении первого порядка реакции по N)
уменьшается и может быть найдено по уравнению Штерна — Фоль¬
мера
* В этом разделе использованы материалы из работ профессора МГУ М. Г. Кузь¬
мина в кн.: Химическая и биологическая кинетика / Под ред. Н. М. Эмануэля,
И.В.Березина, С.Д. Варфоломеева. — М.: Изд-во МГУ, 1983. С. 47—113.
83
т°/т = 1 + Л/[Х],
(4.24)
где т° и т — времена жизни в отсутствие и в присутствии X; кх —
константа скорости взаимодействия N с X.
Для бимолекулярного процесса триплет-триплетной анниги¬
ляции
если он является определяющим по сравнению с другими, теку¬
щая концентрация 3М находится по гиперболическому закону
Для определения констант скорости приведенных выше реак¬
ций первого и второго порядка достаточно представить экспери¬
ментальные данные соответственно в полулогарифмических и об¬
ратных координатах:
где к — суммарные константы скорости; множитель 2 появляется
в результате гибели двух молекул при их взаимодействии в реак¬
ции второго порядка.
При импульсных измерениях часто применяют абсорбционные
или эмиссионные спектральные методы для измерения концент¬
рации, поэтому вместо концентрации используют оптическую
плотность D или интенсивность / испускаемого излучения.
Квантовый выход фотопроцессов при стационарном возбужде¬
нии зависит от многих факторов: величины констант скорости
фотохимических и фотофизических процессов, концентраций ис¬
ходных веществ, тушителей, сенсибилизаторов, растворителя, тем¬
пературы и иногда от интенсивности света.
1. Кинетические схемы мономолекулярных реакций. Выше была
рассмотрена простейшая кинетическая схема фотопроцесса Штер¬
на—Фольмера. Если стадию дезактивации рассматривать как мо-
номолекулярный обмен энергией за счет внутренней и интерком¬
бинационной конверсии, то для квантовых выходов флуоресцен¬
ции и реакции получим следующие соотношения:
3М+3М^ ‘М+‘М*,
(4.25)
(4.27)
(4.28)
(4.29)
84
(pi = kA ts (4.30)
где — время жизни синглетного возбужденного состояния.
Следовательно, константы скорости флуоресценции и реак¬
ции могут быть вычислены, если известны опытные квантовые
выходы и время жизни возбужденного состояния.
Рассмотрим более сложную кинетическую схему процесса, когда
реакция протекает из триплетного состояния:
Скорость
Поглощение
V\
(4.31)
Флуоресценция
1М*->1М + hv'
(4.32)
Внутренняя конверсия ...
^в.к['М*]
(4.33)
Интеркомбинационная ...
1Ы*-^3Ы
*и.к['М*]
(4.34)
конверсия
Фосфоресценция
3M->lM + hv"
кф[гМ]
(4.35)
Интеркомбинационная ...
А:„к.д[3М]
(4.36)
деградация
Реакция
3М—>N
КЛгМ]
(4.37)
В стационарном состоянии выполняются соотношения:
- [‘М*](^фл + квл + кКК) = 0; (4.38)
РР = КА1т - [3М](&Ф + кккл + krT) = 0. (4.39)
Квантовый выход всех триплетных состояний с учетом выра¬
жения (4.38) равен
ЛикРМ*] , о
Ф г = = К.А (4.40)
и может быть определен экспериментально методом счета трип¬
летов, использующим перенос энергии на фосфоресцирующий в
растворе акцептор, например диацетил.
Квантовые выходы фосфоресценции и реакции с учетом выра¬
жений (4.38) и (4.39) соответственно равны
_ *ф[3М] , о,
Фф - ^ K.k^sK^ti (4.41)
^г[3М] , о,
Фы = — = К.кЪКт*т, (4.42)
V\ v '
где К = (кфп + кВК + ки к)~' и тг = (кф + кшл + кгТУ' — времена жиз¬
ни возбужденных синглетного и триплетного состояний.
85
Квантовый выход реакции, протекающей одновременно из син¬
гл етного и триплетного состояний, равен
А^рМЧ + А^М] ,, , , ч
<pN = -М ———1 = (к„ Ts + kK'KkrTxT)xs. (4.43)
v\
В случае образования по реакции продукта в возбужденном со¬
стоянии может осуществляться и обратная реакция
y=A:_r[N*] (4.44)
с установлением равновесия в возбужденном состоянии
М* ^ N*. (4.45)
Для стационарного состояния (и превращений из синглетного
состояния) соотношения для скорости накопления концентра¬
ции возбужденных частиц принимают вид
d[M*i
dt
d[N*]
= vi- [M*] (кфп + kd + kr) + к., [N*] = 0, (4.46)
= *r[M*] - [N*](^ + k'd + k_r) = 0, (4.47)
dt
где kd — константа скорости дезактивации (за счет внутренней и
интеркомбинационной конверсии).
Квантовые выходы флуоресценции М(ф) и N(<p') и их отноше¬
ние в этом случае равны
Ф = — , (4.48)
кфл + к(/+кг/[\ + к.г/(к'(1 + к'фл)Г
Ф' = , (4.49)
(£фл +kd+ кг)(к’фл +k'd+ к_г) - кгк_г
Ф _ к Г
Ф £фл(£фл +k'd +к-гУ
(4.50)
Фотостационарное состояние будет приближаться к равновес¬
ному при условии к_г » (&фл + k'd):
£ = (4.51)
Ф кфлк_г
2. Зависимость от температуры. Влияние температуры на кван¬
товые выходы фотофизических и фотохимических процессов в це¬
лом невелико, поскольку указанные процессы характеризуются
небольшой энергией активации. Для излучательных процессов кон¬
станты скорости не зависят от температуры при сохранении неиз¬
менной химической структуры или конформации молекул.
86
Константы скорости безызлучательных процессов и реакций
возбужденных молекул можно выразить с помощью уравнения
Аррениуса (1.61)
к, = ^ exp [-Ej /(RT)].
Выражения для температурной зависимости времен жизни воз¬
бужденных состояний и квантовых выходов триплетов рассмот¬
ренной ранее схемы процесса (4.31) —(4.36) принимают с учетом
(1.61) вид
т, =
^"Фл -^в.к ехр
RT
+ л.К ехр
RT
тт =
кФ + Лк.д ехр
RT
-1
(4.52)
(4.53)
Фт- = Ал к ехр
RT
т..
(4.54)
Для достаточно широкого температурного интервала удобно ис¬
пользовать приближенные соотношения
xj1 = (xj1 )0 + Л ехр
RT
Ффл “ ‘
^фл
(xj'^ + Aexp
RT
(4.55)
(4.56)
Учитывая, что при понижении температуры время затуха¬
ния и квантовый выход флуоресценции увеличиваются, стре¬
мясь соответственно к пределам (тД и £фл(хД, можно оце¬
нить эффективную энергию активации безызлучательных про¬
цессов Eds графическим путем, строя зависимости ln[xj* - (tj1 ) ]
И 1п[1/ффл -(1/ффл)о] ОТ \/Т.
Для грубой оценки параметров можно использовать формулы
is ~ — ехр
ЕлЛ
RT
Ч /
кфл
Ффл - “Г" ехр
KRT,
\ j
1 (Ет
Tr^expUf
(4.57)
(4.58)
(4.59)
' 87
определяя энергию активации каждого процесса в координатах
Аррениуса Inт, 1/Т.
Учет протекания односторонней реакции из синглетного со¬
стояния приводит к следующим выражениям для времени жизни
возбужденной молекулы
^фл А.к
RT
+ А.к ехр
^и.к
RT
+ Д. ехр
IT'
RT
-i
(4.60)
и с учетом соотношения (4.30) для квантового выхода реакции
Е
<pN = Аг ехр
RT
т,.
(4.61)
Энергию активации реакции при условии, что Er » Eds, ив
области не очень высоких температур, когда kr т;1, находят из
линейной зависимости In <pN от обратной температуры.
Для равновесия в возбужденном состоянии (4.45) зависимость
квантовых выходов флуоресценции реагента <р и продукта реак¬
ции ф' от температуры может различаться для разных диапазонов
температур, в этом можно убедиться на основании приближен¬
ной оценки уравнений (4.48) —(4.50). Наибольший интерес пред¬
ставляют следующие зависимости:
In (ф'/ф) ~ In
при k.r /(£фл +k'd) « 1 и
In (ф'/ф) = In
кфлА
' кфЛ )
^фл А_г
E~E'd
RT
AH'
RT
(4.62)
(4.63)
при к.г/(к'фл +kd) » 1.
Строя график в координатах 1п(ф'/ф), l/Т, по тангенсу угла
наклона можно оценить энтальпию реакции в возбужденном со¬
стоянии для высоких температур и разность энергий активации
прямой реакции и дезактивации продукта в области низких тем¬
ператур.
3. Тушение. Наблюдаемое в присутствии добавок уменьшение
скорости фотопроцессов происходит за счет так называемого ди¬
намического тушения вследствие дезактивации возбужденных
молекул либо за счет статического тушения в результате образо¬
вания в основном состоянии несветящихся соединений. Приме¬
няя тушители различных типов, можно избирательно дезактиви¬
ровать триплетные или синглетные возбужденные состояния. При
выборе тушителя триплетов необходимо, чтобы энергия синг¬
летного возбужденного и триплетного состояний реагента была
88
выше триплетного и ниже возбужденного синглетного состоя¬
ний тушителя. К таким тушителям относятся сопряженные по-
лиены и ароматические углеводороды, а также парамагнитные
молекулы.
В качестве тушителей синглетных возбужденных состояний (пу¬
тем их конверсии в триплетное состояние) часто используют со¬
единения, содержащие тяжелые атомы, и вещества с электроно-
донорно-акцепторным взаимодействием. В последнем случае про¬
исходит конверсия реагента в состояние Тх либо в SQ, однако может
наблюдаться нежелательное образование радикалов или ион-ра¬
дикалов в полярных средах.
Вернемся вновь к простейшей кинетической схеме в обозначе¬
ниях данной главы:
Скорость
Возбуждение М + /*v-> М* vx
Флуоресценция М*—>M + /zv' ^ФлРМ*]
Дезактивация М* —> М(3М) &<у[М*]
(обмен энергией)
Реакция M*->N £гДМ*]>
добавив к ней процесс динамического тушения веществом Q
М* + Q -» М + Q*,
v = kqs[U*][Q]. (4.64)
По аналогии с рассмотренными ранее схемами несложно по¬
лучить интересующие нас соотношения для стационарного со¬
стояния:
vi = [М*](А:фл + kd+krs + kqs[ Q]), (4.65)
Ффл = ^фл /(^фл kd + kn + kqs [Q]), (4.66)
Фм = k„ /(кфл + kd + k„ + fc„[Q]) (4.67)
или представить их в следующем виде:
Ффл / Ффл = 1 + W [Q1 = 1 + ^т [Q1, (4.68)
Фм /Фм = 1 + W[Q] = 1 + MQ1. (4.69)
где т° = 1 /(k^ +kd+ кп), Ффл = кфлт? , = кп— время жизни и
квантовые выходы в отсутствие тушителя.
Уравнения типа (4.68) и (4.69) принято называть уравнениями
Штерна —Фольмера, а величину кт = kqsx°s — константой тушения
по Штерну—Фольмеру.
Значения кт определяют по тангенсу угла наклона прямых, по¬
строенных на основании экспериментальных данных в координа¬
тах (ффл/ффл, [Q]) или (фы/Фи, [Q])-
В случае избирательного действия тушителя по дезактивации
триплетных состояний получим аналогичное формуле (10.69) урав¬
нение
Фыг / Фиг = Фф / Фф = 1 + Лдт'ФЮ]» (4-70)
где ф| = £ф£и.кт?т£; ф^г = krTk„.KT0sT°T; = 1 /(к^КЛ+кф+кгТ).
Для тушителя, дезактивирующего совместно синглетное и трип-
летное состояния, справедливо следующее соотношение:
ФNT /ФЫГ = Фф /Фф = (1 + [QDO + (4.71)
и общий квантовый выход реакции будет равен
, „ _ Ф№ + Фиг /(1 + ^?rTr[Q])
Ф«=ФЫ, + Ч>ХГ = 1 + W[Q] • (4.72)
Наконец, в случае индуцированной тушителем интеркомбина¬
ционной конверсии Si в Г по схеме
>М* + Q ->3М + Q,
v = W‘M*][Q], (4-73)
выражения для квантовых выходов принимают вид
Фг = ^ + к“К'= (Фг + ^ИКД /%)<Рфл/ффл + *икл/kgs', (4.74)
1 + JCqSTs [К1\
<Pn. + Фкг(ф"г + [QB/0 + k,A [Q]
Ф(' = IT^iQi ’ <4-75>
где kqs = к„Кд + kd (за счет внутренней конверсии).
4. Фотосенсибилизация. В заключение рассмотрим фотосенси¬
билизацию, которую часто применяют для проведения фотопро¬
цесса при возбуждении светом большей длины, чем поглощаемой
реагентом. При ее осуществлении используют процессы безызлу-
чательного переноса энергии по индуктивному или обменному
механизму, соблюдая условие: энергия возбужденного состояния
акцептора должна быть меньше, чем у донора. У некоторых ве¬
ществ (например, родаминовых красителей) для осуществления
реакций из триплетного состояния из-за малого квантового выхо¬
да интеркомбинационной конверсии необходимо применять трип-
летные сенсибилизаторы, в качестве которых часто используют
кетоны и хиноны, обладающие близким к единице квантовым
выходом триплетов. Известны случаи, когда при взаимодействии
возбужденной молекулы сенсибилизатора с исходным веществом
90
происходит обратимый перенос электрона, индуцирующий хи¬
мическую реакцию в реагенте (возможно, через образование эк-
сиплекса), при этом процессе энергия возбуждения сенсибилиза¬
тора может быть значительно меньше энергии возбуждения ис¬
ходного вещества.
Составим кинетическую схему процесса фотосенсибилизации:
Зависимость квантового выхода испускания ф„сп (флуоресцен¬
ции или фосфоресценции) сенсибилизатора от концентрации ак¬
цептора энергии реагента М выражается также уравнением Штер¬
на—Фольмера
к — эффективная константа скорости переноса энергии.
Квантовые выходы испускания реагента — акцептора энергии —
и реакции имеют следующий вид:
где фисп = кпспт и фн = кгх — квантовые выходы испускания и ре¬
акции при прямом возбуждении реагента М; г| — квантовый вы¬
ход образования возбужденного состояния сенсибилизатора (ri =
= 1 для синглетных состояний и г| = фг для триплетных состоя¬
ний).
Соотношения (4.78), (4.79) можно представить в удобном для
линеаризации виде
(4.76)
s
м
Фисп = Ф°испЛ Мт*)°[М]/(1 + W)°[M]), (4.78)
Фн = Ф°нЛ *,(т*)°[М]/(1 + Л,(т*)°[М]), (4.79)
1/фисп=(1 + 1/№)°[М])/ф°спТ1,
l/Фм =(1 + 1/*,<т')0[М])/ф&л
(4.80)
(4.81)
или
[М] _ [М] + 1/*,(т*)°
Фисп ФиспЛ
(4.82)
91
[М] _ [М] + 1 / А:Дт5)0 ^ g^
cpN ф^л
Значения констант скорости переноса энергии к5 и предельных
квантовых выходов ф£сп и фм можно определить после построения
в соответствующих координатах по опытным данным прямых, из
тангенса угла наклона последних и отрезков, отсекаемых прямы¬
ми на оси ординат.
Вопросы и задания для самоконтроля
1. Дайте определение термина «фотохимия» и укажите предмет ис¬
следований.
2. Объясните, в чем заключается причина протекания фотохими¬
ческих реакций, часто невозможных при термическом воздействии на ту
же реакционную систему?
3. Приведите примеры известных фотохимических процессов.
4. Сформулируйте закон Буге —Ламберта и приведите его матема¬
тическую формулировку.
5. В чем заключается закон Бера?
6. Сформулируйте объединенный закон Буге —Ламберта—Бера и
приведите его математическую формулировку.
7. В чем заключается первый закон фотохимии?
8. Приведите математическую формулировку закона Вант-Гоффа.
9. Проанализируйте случаи, когда фотохимическая реакция имеет
нулевой или первый порядок по концентрации поглощающего свет ве¬
щества.
10. Сформулируйте закон Эйнштейна — Штарка. В чем заключается его
современная трактовка?
11. Что называют общим квантовым выходом Ф фотохимической ре¬
акции? Укажите интервал принимаемых Ф значений, ответ поясните.
12. Дайте определение первичного квантового выхода ф1# Может ли
он принимать значения больше единицы?
13. Что называют вторичным квантовым выходом ф2? Какая суще¬
ствует связь между ф2, Ф и ф!?
14. Какая величина носит название 1 Эйнштейн? Зависит ли она от
природы излучения?
15. Расположите в порядке возрастания энергетической активности
лучи разных участков спектра: зеленые, красные, ультрафиолетовые,
желтые, инфракрасные.
16. В чем заключается начальная стадия поглощения света? Какова ее
длительность?
17. Перечислите шесть возможных направлений второй стадии пер¬
вичных процессов.
18. Охарактеризуйте направления, на которых происходят первичные
химические процессы. Приведите примеры.
19. Какое состояние молекулы называется синглетным? Укажите вре¬
менной интервал жизни возбужденного синглета.
92
20. Какие возможны другие состояния системы? Укажите временной
интервал жизни возбужденного триплета.
21. Перечислите процессы, которые относятся к безызлучательным.
В чем заключается процесс колебательной релаксации и какова его дли¬
тельность?
22. Охарактеризуйте процессы внутренней конверсии и интеркомби¬
национного перехода. Какова их длительность? Может ли триплетное
состояние быть более реакционноспособным, чем основное синглетное?
Ответ мотивируйте.
23. Какой процесс называют флуоресценцией? Для каких молекул
она характерна? Каков временной интервал жизни для флуоресцен¬
ции?
24. Какой процесс называют фосфоресценцией? Укажите временной
интервал жизни для фосфоресценции.
25. Поясните термин «фотосенсибилизация». Приведите примеры.
26. Какие реакции относят к вторичным процессам?
27. Какие характеристики фотохимических реакций определяют при
кинетических исследованиях флуоресценции и фосфоресценции?
28. В чем заключается механизм Штерна— Фольмера? Какие первич¬
ные процессы учитываются в этом механизме?
29. Какую величину называют тушением? Покажите графически, на
основании схемы Штерна — Фольмера, какие величины могут быть оп¬
ределены при экспериментальном исследовании?
30. Каким образом, с помощью схемы Штерна — Фольмера, можно
рассчитать первичный квантовый выход?
31. Какие существуют пути возбуждения фотопроцессов?
32*. Выведите уравнения для квантовых выходов флуоресценции и
реакции из синглетного возбужденного состояния простейшей кинети¬
ческой схемы.
33*. Получите соотношения для квантовых выходов фосфоресценции
и реакции из триплетного возбужденного состояния полной кинетиче¬
ской схемы.
34*. Какие существуют пути для оценки эффективной энергии акти¬
вации безызлучательных процессов?
35. Каким образом находят энергию активации односторонней реак¬
ции из синглетного состояния?
36*. Каким способом (и при каких условиях) можно оценить энталь¬
пию реакции в возбужденном состоянии?
37. Поясните термины «динамическое тушение» и «статическое туше¬
ние». Каковы критерии выбора тушителей синглетных и триплетных воз¬
бужденных состояний?
38*. Получите для простейшей схемы с учетом процесса тушения син¬
глетного возбужденного состояния уравнение Штерна —Фольмера. Ка¬
кую величину называют константой тушения?
39*. Получите уравнение Штерна—Фольмера для кинетической схе¬
мы процесса фотосенсибилизации.
40. Возможно ли протекание фотопроцесса, если энергия возбужде¬
ния сенсибилизатора значительно ниже энергии возбуждения реагента?
Ответ поясните.
93
ГЛАВА 5. ТЕОРИИ ХИМИЧЕСКОЙ КИНЕТИКИ
В основе любой физической теории
лежат не формулы, а идеи и мысли.
А. Эйнштейн
5.1. Элементы молекулярно-кинетической теории газов
В основу этой теории положена модель идеального газа, кото¬
рую характеризуют следующие положения:
1. Размеры молекул исчезающе малы по сравнению с расстоя¬
ниями между ними, и их собственным объемом можно пренеб¬
речь по сравнению с объемом сосуда, в котором находится газ.
Этим объясняется, что все частицы могут свободно перемещаться
в доступном для них пространстве.
2. Между молекулами нет сил взаимодействия (ни сил притя¬
жения, ни отталкивания), поэтому, находясь постоянно в беспо¬
рядочном движении, молекулы между столкновениями движутся
прямолинейно.
3. Молекулы ведут себя при столкновениях (между собой и со
стенками сосуда) как упругие твердые сферы, этим объясняется
сохранение их энергии и количества движения при столкнове¬
ниях.
4. Средняя кинетическая энергия поступательного движения
молекул пропорциональна абсолютной температуре системы.
5. Число молекул, образующих газ, достаточно велико, чтобы
можно было осуществлять статистические усреднения.
При небольших давлениях и не очень низких температурах ре¬
альные газы близки к идеальному газу. При высоких давлениях
молекулы газа настолько сближаются, что пренебречь их собствен¬
ным объемом нельзя и между ними возникают заметные силы
притяжения.
При низких температурах кинетическая энергия уменьшается
и становится сравнимой с потенциальной энергией, которой уже
пренебречь нельзя.
Ниже приводится ряд формул и уравнений молекулярно-ки¬
нетической теории идеальных газов, которые используются в курсе
физической химии.
Основное уравнение молекулярно-кинетической теории свя¬
зывает между собой параметры состояния идеального газа и ха¬
рактеристики движения его молекул
P = ^nm(vc р.кв)2, (5.1)
94
или
или
pV = - jyrw^cP-KBji] = -Е,
3 1 2 3
pV = ^Nm(vcp,KB)2 = |wr( fcp.Ke)2,
(5.2)
(5.3)
где n — концентрация молекул; m — масса одной молекулы; тг =
= Nm — масса газа; N — число молекул в объеме газа ^ср.кв
средняя квадратичная скорость молекул; Е — суммарная средняя
кинетическая энергия поступательного движения всех молекул газа.
Средняя кинетическая энергия поступательного движения од¬
ной молекулы идеального газа равна
_ 1 — 3
£кин =-mv2 = 2къТ’
для 1 моль идеального газа
e = nas = 1rt,
(5.4)
(5.5)
где кБ — постоянная Больцмана; R — универсальная газовая по¬
стоянная; Na — постоянная Авогадро.
Скорости движения молекул:
наиболее вероятная скорость хаотического движения молекул
и» =
2 кБТ
m
1/2
средняя арифметическая скорость
vl+v2+... + vN _
N
&кБТ Л'/2
пт ,
rt^2
пМ
(5.6)
(5.7)
средняя квадратичная скорость
2,2, , 2
1>1 +V2 + ... + VA
3 кБТ
m
1/2
N
r3RT\1/2
М
(5.8)
Закон Максвелла для распределения молекул идеального газа
по скоростям при одномерном движении имеет вид
\1/2
dN
N А dvx
m
2 nkRT
*-mvl/(2kT)
(5.9)
95
при трехмерном движении
/
т
^ \3/2 mv-
f(v) = 1—- = 4nv2
Na dv
InkrT
e 2kT, (5.10)
где f(vx), или/(t>), — функция распределения молекул по скоро¬
стям — характеризует относительное число молекул из общего
числа Na молекул, скорости которых лежат в интервале от v до
v + df (или dvx).
Среднее значение любой функции h(v) находится по формуле
Л = f h(v)f(v)dv, (5.11)
о
где /(f)df = dN/NA.
Барометрическая формула
ph = potxpl-Mgih-^/iRT)), (5.12)
где ph и р0 — соответственно давление газа на высоте h и Л0; g —
ускорение силы тяжести.
Распределение Больцмана во внешнем потенциальном поле
п = «о ехр\-Mghl(RT)\ = щ ехр[--^] = щ ехР[~^]> (5-13)
где п, п0 — концентрация молекул на высоте h и h = 0; П = mgh —
потенциальная энергия молекулы в поле тяготения.
Согласно закону Больцмана, в случае, когда энергия системы
выражается двумя квадратичными членами (например, при стол¬
кновении двух молекул учитывается лишь относительная кинети¬
ческая энергия движения вдоль линии, соединяющей их центры),
доля молекул, энергия которых лежит в пределах от в до в + dB,
равна
dN 1
jr-W' (5л4>
После интегрирования (5.14) по е от е до °° получаем извест¬
ное соотношение для доли молекул, энергия которых > в,:
N
или
N,=Ne*r, (5.16)
или
Е
N, = Nt*T, (5.17)
96
Вопросы и задания для самоконтроля
1. Сформулируйте основные положения модели идеального газа.
2. Проанализируйте поведение молекул реального газа при высоких
давлениях и низких температурах.
3. Приведите разные формы основного уравнения молекулярно-ки¬
нетической теории газов.
4. Покажите, чему равна средняя кинетическая энергия поступа¬
тельного движения: а) для одной молекулы; б) для 1 моль идеального
газа?
5. Получите аналитическим путем выражения: а) для средней ариф¬
метической скорости; б) наиболее вероятной скорости движения моле¬
кул; в) средней квадратичной скорости.
6. Приведите уравнение закона Максвелла для распределения моле¬
кул идеального газа по скоростям: а) при одномерном движении; б) при
трехмерном движении.
7. Какой вид имеет распределение Больцмана во внешнем потенци¬
альном поле?
8. В каких случаях используется барометрическая формула? Приве¬
дите ее в аналитическом виде.
9. Напишите выражение закона Больцмана для случая, когда энер¬
гия системы выражается двумя квадратичными членами.
10. Приведите интегральное выражение закона Больцмана для доли
молекул, энергия которых ^ £/.
5.2. Параметры столкновений
В рамках молекулярно-кинетической теории были получены со¬
отношения для различных случаев столкновений между молеку¬
лами газа, перечислим некоторые из них.
Частота двойных столкновений, т.е. число столкновений, про¬
исходящих между двумя молекулами газа в единице объема за еди¬
ницу времени:
Zq = nd\2v = nd\2
'Ш'*2
7l[i
= nd\i
8 RT
Ml +M2
1/2
(5.18)
где dn — эффективный диаметр столкновений молекул разных
видов; р — приведенная масса; v — средняя скорость молекул. Раз¬
мерность z0 в системе СИ [z0\= м3/(молекулы • с) или м3-с-1, в
-1
системе СГС [z0] = см3/(молекулы • с) или см3 - с
Число столкновений одной молекулы с остальными молекула¬
ми одного вида
Z] 1 = 2|/2и,я^2,
8 кТлУ2
nnti
(5.19)
7. Зак. 1М('
97
и число столкновении одной молекулы со всеми молекулами вто¬
рого вида
Zl2 = H2nd?2
яц
(5.20)
где пи я2 — число молекул, содержащихся в единице объема газа
(в 1 м3 или в 1 см3), или молекулярная плотность газов; [zu] =
= [zl2] = с1.
Общее число двойных столкновений между молекулами одно¬
го сорта
Z\\ =
n?nd?t(SkT\l/2 „ , „ (пкТ^12
21/2
птх
= 2л,2</,2,
= 2«M2i
.N1/2
щ
\
и между молекулами разного вида
nRT
М{
Zn = ti\ti2nd\2
^8 кТл1/2
ЯЦ
= Я1М12
8я RT
_1_ _1_
Mi + м2
\
Размерность Z,, и Z,2 в системе СИ [м_3-с-1], в системе СГС
[см-3 - с-1]. Величину яd2 называют эффективным сечением столк¬
новений.
Число ударов молекул о стенку площадью S в единицу време¬
ни (с)
1/2
(5.21)
(5.22)
ZSj = п
кТ
2я/и J
S.
(5.23)
Число ударов молекул в 1 с в 1 см2 плоской поверхности выра¬
жается уравнением Герца (1882 г.)
Z =
агл'/2
sfht
(2яткТУ2 ‘
(5.24)
Это уравнение широко используется при изучении процессов
адсорбции, конденсации и испарения.
В методе Кнудсена для измерения давления пара твердых тел
при его истечении (эффузии) через малое отверстие в стенке со¬
суда используется уравнение
p = v
2nRT\
М
1/2
Am
дIs
2я RT
М
1/2
(5.25)
где р — давление, [Па] или [дин/см2]; v — скорость истечения из
отверстия, в системе СИ [кг-м‘2-с'1]> в системе СГС [г-см-2-с-1];
98
Am — изменение массы вещества, в СИ [кг], в СГС [г]; At — время
эффузии, с; S — площадь сечения отверстия, м2 или см2.
Закон эффузии Грехема: скорость потоков двух газов через ма¬
лое отверстие обратно пропорциональна корню квадратному из
их молекулярных масс,
Средняя длина свободного пробега /, т.е. расстояние между
двумя столкновениями молекулы А определяется по формуле
где d — эффективный диаметр столкновений с учетом взаимодей¬
ствия. _
Среднее время между столкновениями t определяют либо по
формуле
v
либо как обратную величину числа столкновений одной молеку¬
лы в секунду (гп или z12), например
Надежное определение значений эффективных диаметров мо¬
лекул одного сорта связано с использованием эксперименталь¬
ных данных таких свойств газа, как вязкость, теплопроводность и
диффузия.
Молекулярно-кинетическая теория позволяет связать эти ве¬
личины с эффективным диаметром столкновений.
Для вязкости получены следующие соотношения:
где г) — динамическая вязкость, размерность в СИ [Па с] = [кгх
хм4-с4]; р — плотность газа [кг/м3]; jj — средняя скорость [м-с-1];
(5.26)
(5.27)
t
I
(5.28)
(5.29)
(5.30)
или
_ 5vm _ 5 (пткТ)'/2
Л “ 2‘/2 - 32^/2 “16 iid2 ’
(5.31)
или
11 = 32 nvm1.
(5.32)
99
/—средняя длина свободного пробега [м]; d — эффективный
диаметр столкновений, [м]; «—концентрация молекул газа в 1 м3;
т — абсолютная масса молекулы [кг].
Для коэффициента теплопроводности кТ [Дж-м_| с"1-К'1] полу¬
ченные уравнения имеют следующий вид:
где с^т) — молярная теплоемкость газа, Дж К_1-моль-1; с„ — теп¬
лоемкость молекулы, с„= cv(m)/NK.
Для коэффициента диффузии D, м2-с-1, получены уравнения
связи
Наиболее точные значения эффективных диаметров молекул
получены на основании экспериментального определения вязко¬
сти.
1. Дайте определение термина «частота двойных столкновений», z0,
и получите его выражение в аналитическом виде.
а) СИ; б) СГС?
3. Укажите размерность частоты двойных столкновений z0 в систе¬
ме: а) СИ; б) СГС.
4. Приведите выражения для числа столкновений одной молекулы:
а) с остальными молекулами одного сорта zM; б) со всеми молекулами
другого сорта z)2. Укажите размерность Zj i и z12.
(5.33)
или
, _25( кТ^'2 cv{m) _ 25 (пшкТ)'/2
Кг --ZT — »г .9 - ЗГГ —7Г~
Ъ2\пт) NAd2 32 nd2m
(5.34)
или
(5.35)
3 р
(5.36)
ИЛИ
п 3-21/2яг_ 3 (RT^2 1
D = ———lv-—77т —— —
64 8я1/2 М J nd2
(5.37)
Вопросы и задания для самоконтроля
2. Какова размерность выражения
8RT Г 1 1 V11/2
в системе:
п М\ М2
V 1 L J
100
5. Напишите выражения для общего числа двойных столкновений:
а) между молекулами одного сорта Zn; б) молекулами разного сорта Z12.
Укажите размерность Zlb Z12 в системе: а) СИ; б) СГС.
6. Какие выражения используют для расчета числа ударов о стенку сосу¬
да в единицу времени? Для каких процессов применяют уравнение Герца?
7. Дайте характеристику метода Кнудсена. Каким образом определя¬
ют давление в этом случае?
8. Сформулируйте закон эффузии Грехема.
9. Приведите выражение для определения средней длины свободно¬
го пробега, /.
10. Как находят среднее время между двумя столкновениями?
11. Укажите способы определения эффективных диаметров молекул.
В каком случае получают наиболее точные значения этой величины?
12. Приведите выражение, связывающее вязкость с «эффективным»
диаметром столкновений.
13. Какое соотношение существует между коэффициентом теплопро¬
водности и эффективным диаметром столкновений?
14. Каким образом связаны между собой коэффициент диффузии и
эффективный диаметр столкновений?
15. Приведите уравнение связи между коэффициентом диффузии и
вязкостью.
5.3. Теория активных столкновений (ТАС).
Бимолекулярные реакции
Рассмотрим элементарную бимолекулярную реакцию
А + В —^ Р| + Рз*
Уже на рубеже XIX и XX вв. стало ясно, что очевидное предпо¬
ложение о том, что химическая реакция протекает только между
сталкивающимися частицами, оказалось необходимым условием,
но явно недостаточным.
Простые вычисления, проведенные в рамках молекулярно-ки¬
нетической теории, показали, что опытное значение скорости ре¬
акции на много порядков меньше, чем рассчитанное по форму¬
лам (5.21, 5.22) общее число двойных столкновений
^оп ^ ^12-
. Достижением теории активных столкновений стала гипотеза о
том, что к химическому превращению молекул (рассматриваемых
как упругие твердые сферы) приводят только те столкновения,
при которых энергия реагирующих молекул равна или превышает
определенный критический уровень, называемый энергией акти¬
вации 6* (в расчете на моль, Е* = NKе*).
Доля активных столкновений при условии, что энергия выра¬
жается двумя любыми квадратичными членами, равна, как пока¬
зано в формуле (5.15), больцмановскому множителю:
101
(5.38)
или
£♦
Za = ZnQ RT.
(5.39)
Для ряда реакций было получено хорошее согласие между эк¬
спериментальным значением скорости и расчетной величиной
числа активных столкновений
Однако для большинства реакций von «: Za, поэтому для согла¬
сования расчетных и опытных данных Дж.Христиансен (1924 г.)
ввел поправочный множитель, названный стерическим фактором Р.
Первоначально полагали, что стерический фактор связан с необ¬
ходимой ориентацией сталкивающихся молекул и характеризует
вероятность определенной геометрической конфигурации частиц
при столкновении.
Оказалось, что, кроме соответствующей ориентации молекул,
существует еще ряд причин, приводящих к расхождению резуль¬
татов теории и опыта, поэтому целесообразно дать Р иную трак¬
товку: в общем случае этот множитель характеризует вероятность
протекания реакции между частицами, энергия которых больше
или равна Е*, по определению Р < 1.
Таким образом, теория активных столкновений дает следую¬
щее выражение для скорости указанной выше бимолекулярной
реакции:
С другой стороны, скорость данной реакции, согласно основ¬
ному постулату химической кинетики, выражается уравнением
где — константа скорости бимолекулярной реакции, лА и ив —
число молекул в 1 м3. Сопоставление выражений (5.41) и (5.42)
позволяет выразить константу скорости рассматриваемой элемен¬
тарной реакции
Подставляя значения частоты столкновений z0 (5.18) в уравне¬
ние (5.43), получим следующие соотношения:
(5.40)
= PnAnBz0e*T.
(5.41)
v = к,,пАп,
ПпАпВ>
(5.42)
£*
ки = Pz0e *т
(5.43)
102
-L+-L
мА мв
I/2 Е*
iRT, (5.44)
а) для молекул разного вида
кц = Pdi2 |8пЛГ
б) для идентичных молекул
И-гОТЛ1/2 .11
е (5.45)
/
М
\
где ^ — эффективный диаметр столкновений, т. е. наименьшее рас¬
стояние между центрами сталкивающихся молекул с поправкой
на взаимодействие.
Существуют различные способы расчета d, наиболее надеж¬
ный из них основан на экспериментальном определении вязко¬
сти, теплопроводности и диффузии (см. подразд. 5.2).
Эффективный диаметр зависит от температуры. Согласно по-
луэмпирической формуле Сезерленда, эта зависимость имеет вид
<#=<efi+£], (5.46)
\ /
где cL — эффективный диаметр молекул одного сорта при высо¬
кой температуре; С — постоянная Сезерленда, зависящая от при¬
роды газа и определяемая экспериментально, например, по зави¬
симости вязкости от температуры.
Сравнение выражения (5.43) с уравнением Аррениуса (1.61)
Ь.
к = Ае RT
указывает на очевидное их сходство по форме, что подтверждает
достоинства теории активных столкновений, предложенной
М.Траутцем (1916 г.) и В. Мак-Льюисом (1918 г.).
Эта теория не предлагает методов теоретического расчета энер¬
гии активации Е*, но позволяет установить количественное соот¬
ношение между Е* и опытной энергией активации Еа.
Для этого достаточно записать уравнение (5.43) с учетом (5.44)
или (5.45) в виде
ка = А'Г'Ч^. (5.47)
Последовательное логарифмирование и дифференцирование по
температуре уравнений (1.61) и (5.47) приводит к искомому со¬
отношению
Еа = E*+^RT (5.48)
ИЛИ
E* = Ea-~RT. (5.49)
103
Теория активных столкновений, вводя понятие о стерическом
множителе Р, не дает способов теоретического расчета его вели¬
чины. Оценку значения Р обычно проводят путем сопоставления
предэкспоненциальных множителей в уравнениях (5.43) и (1.61)
D А
Р = ~- (5.50)
Z0
Следует обратить внимание, что расчет Рпо выражению (5.50),
строго говоря, возможен в предположении об отсутствии разли¬
чия между опытной энергией активации Еа и энергией актива¬
ции £*, вводимой в теории активных столкновений (ТАС). Пра¬
вильнее поступать таким образом: уравнение (5.43) с учетом (5.49)
трансформируется в
_Еа_
кп = Pz0e1/2e RT, (5.51)
значение Р находят путем сопоставления предэкспоненциальных
множителей (при одинаковых экспонентах!) уравнений (5.51) и
(1.61)
D А
P = 77oJ- (5.52)
z0e
Предлагая ясную физическую модель процесса, ТАС из-за сво¬
ей схематичности не смогла объяснить влияние растворителя,
добавок инертных газов и других факторов на скорость реакции.
Представляется спорным положение теории о том, что процесс
превращения исходных веществ в продукты происходит мгновен¬
но в момент столкновения активных молекул, поскольку очевид¬
но, что химические превращения с учетом структуры молекул
представляют собой не единичный акт, а сложный процесс пере¬
распределения энергии между химическими связями. Кроме того,
возможно протекание химической реакции в несколько стадий,
что совершенно не учитывается теорией активных столкновений.
Вопросы и задания для самоконтроля
1. Сформулируйте основные положения теории активных столкно¬
вений.
2. Поясните термин «квадратичный член» в выражении для энер¬
гии.
3. Как выражается доля активных столкновений, если: а) энергия
выражается двумя квадратичными членами; б) энергия выражается б’-квад-
ратичными членами?
4. Дайте определение термина «стерический множитель» Р. Какие
причины обусловили его введение?
5. Приведите основное уравнение теории активных столкновений
для скорости бимолекулярной реакции.
104
6. Используя основной постулат химической кинетики и предыду¬
щее уравнение, получите выражение для константы скорости бимолеку¬
лярной реакции ки.
7. Какой вид принимает уравнение константы скорости реакции:
а) для молекул разного вида; б) для идентичных молекул?
8. Поясните термины: эффективный диаметр и эффективное сече¬
ние столкновений.
9. Каким образом эффективный диаметр столкновений зависит от
температуры?
10. Сравните уравнение константы скорости бимолекулярной реак¬
ции, полученное в теории активных столкновений, с уравнением Арре¬
ниуса. В чем заключается их сходство и различие?
11. Дайте определение энергии активации Е* в рамках теории актив¬
ных столкновений.
12. Получите количественное соотношение между энергиями актива¬
ции: опытной Еа и Е*.
13. Каким путем оценивают значение стерического множителя в рам¬
ках теории активных столкновений?
14. Можно ли объяснить с позиций теории активных столкновений
термины: «нормальные», «быстрые» и «медленные» реакции?
15. Сформулируйте достоинства и недостатки теории активных стол¬
кновений.
5.4, Теория активных столкновений. Мономолекулярные
реакции
По определению, мономолекулярная реакция — простейший тип
элементарной реакции, в которой молекула исходного вещества
превращается в продукты.
Мономолекулярные превращения, как правило, характерны для
молекул сложного строения или состоящих из большого числа
атомов, к ним относятся: реакции изомеризации, пиролиз угле¬
водородов, алкилгалогенидов и эфиров, термический распад кис¬
лородных соединений азота и галогенов, реакции образования
свободных радикалов и их распада.
При экспериментальном изучении мономолекулярных реак¬
ций был обнаружен ряд явлений, требующих логического объяс¬
нения.
В частности, понимание этих явлений требует ответа на следу¬
ющие вопросы: каков механизм активации реагирующей молеку¬
лы? Как объяснить наблюдаемый при низких давлениях переход
от кинетики первого порядка к кинетике второго порядка? Поче¬
му «инертные» газы часто увеличивают скорость мономолекуляр¬
ных реакций при низких давлениях? В чем причина высоких по
сравнению с «нормальными» реакциями значений предэкспонен¬
циальных множителей (до 1021) в ряде случаев?
105
Теория Линдемана (1922 г.). В основу теории положены следу¬
ющие идеи:
реагирующая частица А не может претерпевать мономолеку-
лярное превращение до тех пор, пока она не приобретет путем
столкновения избыток энергии, достаточный для ее возбуждения,
т. е. дестабилизации. Из нового, возбужденного состояния А* она
может либо вернуться в исходное состояние путем дезактивации,
либо перейти в конечные продукты;
продолжительность жизни возбужденной частицы должна быть
достаточной для протекания мономолекулярной реакции, т.е. су¬
ществует временная задержка между процессом активации и мо-
номолекулярным превращением.
Полагая, что среднее время жизни между двумя столкновени¬
ями обратно пропорционально давлению газа, Ф.Линдеман счи¬
тал, что выше определенного давления столкновения происходят
достаточно часто, чтобы поддерживать равновесную концентра¬
цию возбужденных молекул. Однако при низких давлениях время
между двумя столкновениями возрастает настолько, что большин¬
ство возбужденных молекул успевают прореагировать, т.е. пре¬
вратиться в продукты, до нового столкновения. В результате кон¬
центрация возбужденных молекул становится значительно мень¬
ше равновесного значения, поэтому при низких давлениях
константа скорости первого порядка будет существенно меньше,
чем при высоких давлениях.
Теорию Линдемана обычно иллюстрируют следующей схемой:
А+Ач - А*+А
А*—-—» В
(5.53)
где А и В два изомера одного соединения; А* — возбужденная
молекула. Процесс активации по стадии (1) происходит при би¬
молекулярном столкновении, константа скорости кх рассчитыва¬
ется из теории активных столкновений (см. подразд. 5.3)
кх = z01 ехр
Е* '
RT
(5.54)
Обратный первой стадии процесс дезактивации происходит при
каждом столкновении А*, поэтому полагают, что константа ско¬
рости к_{ не зависит от энергии, ее находят по формулам ТАС,
т.е. k.x~zQ(rX). Константу скорости собственно мономолекулярного
превращения, к2, полагают также не зависящей от энергии, со¬
средоточенной в А*.
Применив принцип квазистационарных концентраций к воз¬
бужденной частице А*
106
d[A*]
dt
= к1[Ар-к_1[А*ЦА]-к21А*] = 0,
находят
[A*] =
A:, [A]2
(5.55)
(5.56)
+ ^-llA]
что позволяет получить выражение для скорости образования про¬
дукта в мономолекулярной реакции:
2[А]2 (к\к2 / А:_,)[А]
\ + к2/{к_х[А)У
= d[Bl = = кМ
dt l[ ] к2+к_{[ А]
(5.57)
Из выражения (5.57) видно, что формально порядок реакции
является промежуточным между первым и вторым.
При высоких давлениях к_{ [А]» к2, поэтому соотношение (5.57)
переходит в уравнение кинетики первого порядка.
где кт =
к\к2
к-1
» = ^[A] = UA],
к. 1
— постоянная, не зависящая от давления;
(5.58)
= к2 ехр
Е*
RT
(5.59)
Таким образом, теория Линдемана подтверждает эксперимен¬
тальные данные о том, что при высоких давлениях наблюдается
кинетика первого порядка и отсутствует влияние добавок «инерт¬
ных» газов, поскольку достигнута и поддерживается равновесная
концентрация активных молекул.
При низких давлениях Jfc_i[A]«: к2, поэтому уравнение (5.57)
переходит в уравнение второго порядка относительно концентра¬
ции А:
v = k{[Af, (5.60)
что подтверждает опытные данные.
Для характеристики мономолекулярных реакций часто исполь¬
зуют следующие понятия. Запишем выражение для скорости (5.57)
в виде
v - Ач[А],
используя уравнения (5.57) и (5.58), находим к{:
к = — = —
1 [А] [А]
d[A]) кМА] _ А:,[А] к„
' dt J At2+Al,[A] { к_х[A] l + A^/^A])’
к2
(5.61)
(5.62)
107
где кх — эффективная константа скорости, зависящая от концен¬
трации или давления газа.
Относительная константа скорости, кх /к„ характеризует сте¬
пень уменьшения кх:
кх 1
к„ 1 + к2/(к_х[А])
(5.63)
Так называемое давление перехода рх/2, при котором кх/к^=\/2,
определяется из условия к2 = к_х [А]. Заменяя [А] полным давле¬
нием, получают формулу для определения рх/2 (при этом размер¬
ность и численное значение к2 изменяются):
к2 к„
р'12 = к=Т- <5-б4>
При этом давлении
*1=у- (5-65)
Из формулы (5.63) видно, что добавление «инертных» газов в
области перехода (ниже предела высоких давлений) приводит к
увеличению константы скорости кх до ее значения при высоких
давлениях, т.е. способствует достижению равновесной концент¬
рации активных молекул.
В целом теория Линдемана, хотя и дала ответ на ряд вопросов,
поставленных выше, оказалась неудовлетворительной. К ее недо¬
статкам относятся:
1) значительное расхождение между рассчитанными и опыт¬
ными значениями скорости реакции
^оп ^расч> (5.66)
т.е. Р»1, что по определению стерического множителя противо¬
речит физическому смыслу;
2) несоответствие между опытными значениями давления пе¬
рехода рхп и рассчитанными по формуле (5.64);
1 1
3) отклонение экспериментальной кривой в координатах ~г > —
кх р
для области малых давлений от линейной зависимости.
Современная трактовка схемы Линдемана состоит в следую¬
щем.
Основное дополнение: реагирующая молекула А может ак¬
тивироваться при столкновениях не только с идентичной молеку¬
лой, но и с любой другой. Поэтому необходимо добавить к схеме
(5.53) этап столкновения А с молекулой В:
А+В^==±А* + В (5.67)
108
Применяя принцип квазистационарных концентраций к А* для
схем (5.53) и (5.67), получают
^р = МА]2+<МА][В]-
-*_,[А][А*] - *_3[В][А*] - Лг[А*] = 0, (5.68)
откуда находят
[А*] -- ^СДА]2 + ^з[А][В]
" к_х [А] + А:_3 [В] + к2 ’
Обычно в кинетике обозначают концентрацию А в момент вре¬
мени /через (а-х) и В через х, в этом случае из формулы (5.69)
получают
[А*] = (*1 (а-х) + к3х}(а-х) (5 щ
к_\(а - х) + к_гх + кг
Скорость образования продукта по стадии 2 схема (5.53) запи¬
шется так:
Щ = dx = = к2{кх(а-х) + кгх}(а-х)
d/ dt 2 k_i(a - х) + к_3х + к2
Поскольку продукт реакции В является изомером А, обе моле¬
кулы должны иметь одинаковую молекулярную массу и похожие
размеры. Допускают, что константы скорости для процессов ак¬
тивации А и дезактивации А* приблизительно равны, т. е. к\ ~ к3 и
£_1 = &_з, тогда уравнение (5.71) переходит в
d / к_{а + к2
Таким образом, для суммарной схемы (5.53) и (5.67) реакция
изомеризации всегда подчиняется кинетике первого порядка. Со¬
гласно уравнению (5.72), константу скорости первого порядка к'
можно выразить следующим образом:
, (5.73)
^-i[A]o + к2
где [А] о — начальная концентрация А.
При высоких давлениях, к_х[К]^къ соотношение (5.73) пе¬
реходит в
, f к2к\ .
k~=Y~= 2 и (5-74)
где К\ — константа равновесия этапа 1.
109
При очень низких давлениях, к2 » Аг_, [А]0, уравнение (5.73)
переходит в
к'р-*о = к\ [ А]0, (5.75)
т.е. теория Линдемана предсказывает, что при низких давлениях
константа скорости реакции первого порядка уменьшается по срав¬
нению с ее значением при высоких давлениях и становится про¬
порциональной начальному давлению газа.
Иными словами, уравнение (5.74) применяется при условии,
когда в обратимых реакциях 1/-1 и 3/-3 устанавливается равнове¬
сие, так же как достигается и равновесная концентрация А* в
системе. В этом случае определяющей является константа скорос¬
ти стадии 2. Напротив, уравнение (5.75) относится к случаю, ког¬
да концентрация А* намного меньше равновесной: константа ско¬
рости стадии 2 теряет свою роль, так как скорость реакции конт¬
ролируется стадиями 1 и 3. В некотором смысле реакция не является
уже «мономолекулярной», поскольку стадии 1 и 3 — бимолеку¬
лярные процессы.
Для описания влияния добавок «инертного» газа необходимо
рассматривать начальную схему (5.53) + (5.67) с учетом обрати¬
мого соотношения:
А+М ^==ь А* + М, (5.76)
где М — молекула инертного газа. В этом случае уравнение (5.72)
принимает вид
<5-77>
dt *_,[А]о+*-4[М] + А2
Уравнение (5.77) показывает, что и в этом случае реакция под¬
чиняется кинетике первого порядка.
Кажущаяся константа скорости к" равна
к» _ *2(*,[A]q + ,» -о.
к-\ [А]0 + & ДМ] + к2 ;
При высоких давлениях, когда &^[М] » къ если [М] [А]0,
получаем
kZ = k2^. (5.79)
к4
Отношение ~г~ равно отношению равновесных концентраций
С К А
А* *-4
——, так же как
(5 80)
т.е. соотношение (5.79) идентично (5.74).
110
Следовательно, предельное значение константы к' при высо¬
ких давлениях достигается независимо от присутствия «инертно¬
го» газа М.
Теория Хиншельвуда (1927 г.). Основная причина указанных
выше недостатков теории Линдемана связана с выбранным спо¬
собом расчета константы скорости процесса активации (стадия 1)
по формуле (5.54). В рамках простой теории столкновений учиты¬
вается только относительная кинетическая энергия вдоль линии,
соединяющей центры сталкивающихся молекул, при условии, что
две классические степени свободы имеют энергию > Е*.
С. Хиншельвуд был первым, кто обратил внимание на необ¬
ходимость учета в процессе активации внутренней энергии мо¬
лекул.
Согласно его теории, молекула рассматривается как совокуп¬
ность определенного числа гармонических осцилляторов, между
которыми разрешен свободный обмен энергией. Накопление не¬
обходимой для активационного процесса энергии Е* происхо¬
дит при участии как поступательных, так и внутренних (в ос¬
новном колебательных) степеней свободы реагирующей моле¬
кулы.
Необходимым и достаточным условием для осуществления мо-
номолекулярной реакции является достижение внутренней энер¬
гией молекулы некоторой пороговой величины е* (или в расчете
на моль Е*). Теория не учитывает ни способ накопления энергии,
ни вероятность ее сосредоточения на отдельном осцилляторе, ни
наличие избытка в молекуле по сравнению с е*.
Учет внутренних степеней свободы приводит к увеличению чис¬
ла квадратичных членов, с помощью которых можно выразить
энергию участвующей в активационном процессе молекулы. С. Хин¬
шельвуд показал, что в этом случае доля молекул с энергией боль¬
ше или равной Е* (аналогично и доля активных столкновений) с
увеличением числа учитываемых внутренних степеней свободы
резко возрастает и при условии E*^>RT может быть найдена по
формуле
где s — число квадратичных членов; s = 2f+2,f — число участвую¬
щих в процессе активации колебательных степеней свободы.
Очевидно, что для s = 2 (или/=0) соотношение (5.81) пере¬
ходит в известное выражение для больцмановского множителя
(5.38). Выражения для констант скорости в модифицированной
теории Хиншельвуда—Линдемана с учетом формулы (5.81) при¬
мут следующий вид:
е АГ, (5.81)
111
при высоких давлениях
к] к*
■1*2
V-
7* \
RT
1
Е*
~RT •
(5.82)
/
при низких давлениях (если &-1 = го(-1)яго(о)
k\=7i
60(|)
■1
Е*
Jf
W-I
£*
" RT
(5.83)
Зависимость эффективной константы скорости к{ от давления
путем комбинирования уравнений (5.62) и (5.83) в этом случае
принимает вид
к,=
1
-5-1
7*
RT
II
" RT
(5.84)
На основании уравнений (5.82), (5.83) и (1.61) можно устано¬
вить связь между теоретической энергией активации Е* и ее опыт¬
ным, аррениусовским значением при различных давлениях.
При больших давлениях
Еа = Е*~
Is-1
RT = Е*-fRT.
При низких давлениях
Е„ = Е*~
— 5 — — \RT = E* +
2 2
RT.
(5.85)
(5.86)
Исходя из формул (5.85), (5.86), энергия активации фактичес¬
ки не зависит от давления, однако на опыте для многих мономо-
лекулярных реакций наблюдается заметное уменьшение энергии
активации с понижением давления. Объяснение такого явления
дается более поздними теориями мономолекулярных реакций.
Представления С. Хиншельвуда способствовали созданию бо¬
лее реальной, чем в теории Линдемана, модели процесса актива¬
ции мономолекулярной реакции.
По С. Хиншельвуду, учитывается энергетическая зависимость
константы скорости к, и принимается, что остальные константы
скорости к.\ и к2 не зависят от энергии; показано, что к2 умень¬
шается с увеличением числа колебательных степеней свободы /,
участвующих в активации молекулы. Последнее объяснимо на ка¬
чественном уровне: время жизни активной частицы (т = 1 /к2) все¬
гда возрастает с усложнением ее структуры, т. е. с ростом числа/
112
Основным недостатком теории Хиншельвуда является отсут¬
ствие метода теоретического определения необходимого числа
квадратичных членов s энергии (или колебательных степеней сво¬
боды /), участвующих в конкретной мономолекулярной реакции.
Вопросы и задания для самоконтроля
1. Какие реакции называют мономолекулярными? Приведите при¬
меры мономолекулярных превращений.
2. Перечислите группу вопросов, возникших при эксперименталь¬
ном изучении мономолекулярных реакций.
3. Каким образом пытались объяснить механизм активации моно¬
молекулярных реакций до Линдемана?
4. Сформулируйте основные положения теории Линдемана.
5. Почему, согласно аргументации Линдемана, константа скорости
реакции первого порядка при низких давлениях существенно ниже, чем
при высоких?
6. Приведите простейшую схему Линдемана и поясните, зависят ли
константы скорости входящих в нее реакций от энергии.
7. Получите с помощью принципа квазистационарных концентра¬
ций выражение для скорости образования продукта мономолекулярной
реакции по схеме Линдемана.
8. Какой вид принимает полученное ранее (вопрос 7) уравнение:
а) при высоких давлениях; б) при низких давлениях?
9. Какие величины называют эффективной и относительной кон¬
стантами скорости? От каких параметров они зависят?
10. Поясните термин «давление перехода», /?1/2. Каким путем его оп¬
ределяют теоретически?
11. Объясните роль добавок «инертных» газов в области перехода.
12. Постройте графически теоретическую кривую Линдемана в коор¬
динатах: а) 1 /kh 1 /р\ б) lgкь 1 gp. Согласуются ли с этой зависимостью
экспериментальные данные?
13. Перечислите недостатки теории Линдемана.
14. В чем заключается современная трактовка схемы Линдемана? Рас¬
смотрите ее на примере реакции изомеризации.
15. Приведите полную схему Линдемана с учетом добавления «инер¬
тного» газа. Какой порядок имеет реакция в этом случае?
16. Укажите основную причину недостатков теории Линдемана.
17. Сформулируйте основные положения теории Хиншельвуда. Ка¬
кие факторы не учитываются в этой теории?
18. Приведите выражение для константы скорости реакции в теории
Хиншельвуда—Линдемана: а) при высоких давлениях; б) при низких
давлениях.
19*. Установите соотношение между значением энергии активации
по Хиншельвуду и ее опытной величиной: а) при больших давлениях;
б) при малых давлениях.
20*. Укажите энергетическую зависимость констант скорости в схеме
Линдемана по Хиншельвуду. Как изменяется к2 при увеличении числа
8. Зак. 1596
113
колебательных степеней свободы в реагирующей молекуле? Ответ пояс¬
ните.
21. В чем заключается основной недостаток теории Хиншельвуда?
22*. Укажите отличия последующих теорий мономолекулярных реак¬
ций от теории Хиншельвуда—Линдемана.
5.5. Статистический аспект теории активированного
комплекса
Теория активированного комплекса была предложена в 1935 г.
независимо Г.Эйрингом, а также М.Поляни и М. Эвансом. Идея
теории состоит в том, что при протекании химической реакции,
например
XY+Z—>X+YZ (5.87)
конечная конфигурация атомов продуктов достигается из началь¬
ной конфигурации исходных веществ при непрерывном измене¬
нии межатомных расстояний, и ryz. Для каждой реакции суще¬
ствует так называемая критическая конфигурация X-Y-Z, при
достижении которой существенно возрастает вероятность завер¬
шения реакции. Эта критическая конфигурация, соответствующая
высшей точке наиболее выгодного пути реакции на поверхности
потенциальной энергии, называется активированным комплек¬
сом или переходным состоянием.
Отметим, что сходные идеи высказывались и другими автора¬
ми (Р. Марселей, О. Райс и Г. Гершинович), но никто из них не
использовал свойств поверхности потенциальной энергии, опре¬
деляющей природу активированного комплекса, поэтому они не
смогли указать правильный путь расчета абсолютной скорости ре¬
акции.
Активированный комплекс следует рассматривать как обык¬
новенную молекулу с обычными термодинамическими свойства¬
ми при одном допущении: движение вдоль координаты реакции
приводит к его распаду на части с определенной скоростью.
При выводе основного уравнения теории активированного ком¬
плекса используются следующие положения:
распределение молекул по состояниям в ходе химической ре¬
акции соответствует распределению Максвелла —Больцмана;
элементарный акт реакции происходит адиабатически, т. е. ядра
движутся независимо от движения электронов. Это позволяет счи¬
тать, что потенциальная энергия при движении ядер изменяется
непрерывно и является функцией межьядерных расстояний гху и ryz;
аппарат классической механики применим для описания дви¬
жения ядер в адиабатических условиях на вершине (или вблизи
ее) потенциального барьера;
114
исходные вещества всегда находятся в равновесии с активиро¬
ванными комплексами, которые распадаются с конечной скоро¬
стью. Это означает, что реакция не изменяет существенным обра¬
зом их равновесную концентрацию.
Представим основное уравнение, подученное теорией для кон¬
станты скорости реакции, в следующей форме:
к = эг^-К*, (5.88)
где аг — трансмиссионный коэффициент, или коэффициент про¬
хождения, указывает на долю активированных комплексов, пере¬
ходящих в продукты, как правило аг < 1; къ — постоянная Больц¬
мана; h — постоянная Планка; К* — константа химического рав¬
новесия между активированным комплексом и исходными веще¬
ствами с учетом его одномерного движения вдоль координаты
реакции.
Множитель , имеющий размерность частоты (с-1), назы¬
вают вслед за Эйрингом «эффективной скоростью перехода акти¬
вированных комплексов через энергетический барьер», он явля¬
ется функцией только температуры.
Отметим, что уравнение (5.88) справедливо для любого выбо¬
ра стандартного состояния при условии, что к и К* выражены в
одинаковых единицах.
В рамках статистической термодинамики константу равнове¬
сия К* можно выразить через суммы по состояниям, в этом слу¬
чае для реакции (5.87) получаем наиболее распространенную
форму основного уравнения теории активированного комплекса
кБТ Q* -#
<5-89>
где Qi — молекулярные суммы по состояниям, отнесенные к еди¬
нице объема активированного комплекса и реагирующих веществ:
Q* — имеет на одну колебательную степень свободы меньше обыч¬
ной молекулы; Е0 — истинная энергия активации, равная разно¬
сти нулевых энергий активированного комплекса и исходных ве¬
ществ, она идентична энтальпии или опытной энергии актива¬
ции при абсолютном нуле.
Уравнение (5.89) играет важную роль в химической кинетике,
так как позволяет рассчитывать константу скорости бимолекуляр¬
ных реакций (чаще — ее предэкспоненциальный множитель) на
основании сведений о размерах, геометрической конфигурации,
основных частотах колебаний реагентов.
Отметим, что теоретических путей расчета аг пока нет. Для боль¬
шинства реакций (в случае адиабатических процессов) ае = 1.
115
В практических расчетах принимают, что сумму по состояниям
можно выразить произведением определенного числа множите¬
лей — по одному для каждого вида энергии
Q = fnfU*
rt fr fv
п^вр^к >
где /„, /вр, /к — вклад в статистическую сумму одной степени
свободы каждого вида энергии — поступательной, вращательной,
колебательной; t,r,v — числа соответствующих степеней свободы.
Полагают, что значения /п, /вр) Л примерно равны для исход¬
ных веществ и активированного комплекса, и в ряде случаев Е0 -
С учетом этих допущений приведенное выше уравнение (5.89)
дает возможность оценить температурную зависимость предэкс-
поненциального множителя и константы скорости для различно¬
го типа реакций, а также установить количественную связь между
опытной Еа и истинной Е0 энергиями активации. В последнем слу¬
чае осуществляют стандартную операцию: последовательное ло¬
гарифмирование и дифференцирование по температуре уравне¬
ния Аррениуса и полученной в рамках теории активированного
комплекса зависимости к от Т, которая приводит к простому со¬
отношению
где п — показатель степени температуры в предэкспоненциаль-
ном множителе уравнения (5.89).
Теория активированного комплекса в статистическом аспекте
позволяет дать качественную интерпретацию стерического мно¬
жителя, вводимого теорией активных столкновений.
Для реакции
с участием частиц различной сложности при условии, что в ис¬
ходных веществах и активированном комплексе АВ54 отсутствуют
внутренние вращения, стерический множитель можно оценить с
помощью формулы
гдefK и /вр — колебательная и вращательная суммы по состояниям
реагирующих молекул, приходящиеся на одну степень свободы;
г = гК+ гв — суммарное число вращательных степеней свободы ис¬
ходных веществ; г* — число вращательных степеней свободы акти¬
вированного комплекса.
К достижениям теории активированного комплекса относится
успешная трактовка мономолекулярных реакций, в частности удов-
Еа= Е0 + nRT,
(5.90)
А + В
^ В*->Р
\г-г*+2
(5.91)
116
летворительное объяснение опытных значений предэкспоненци-
ального множителя для большинства реакций типа
A А* —> В + С
Учитывая указанные выше допущения, полагают также, что
структура и другие свойства активированного комплекса мало от¬
личаются от характеристик исходной молекулы. Поэтому в при¬
ближении равенства поступательных, вращательных сумм по со¬
стояниям и всех частот колебаний, за исключением частоты коле¬
бания вдоль разрываемой связи (которое при выводе основного
уравнения заменено в комплексе поступательным движением вдоль
координаты реакции), уравнение (5.89) упрощается и принимает
вид
к = аг —-——^е RT = аг-
h Qa
f _hv_
1 - t kT
A
RT
(5.92)
где v — частота колебания (с-1), присутствующего в реагирующей
молекуле и совершающегося вдоль рвущейся связи.
Для низких и умеренных температур, когда hv»kT, в уравне¬
нии (5.92) ехр (-Av / АГ) —»0, получают с учетом (5.90) прибли¬
женное выражение для константы скорости мономолекулярной
реакции
I rp Ер I rp Eg
к = RT = аг^-ее *т\ (5.93)
h h
При высоких температурах, когда hv <zkT, разлагая экспонен¬
ту в ряд и ограничиваясь двумя первыми слагаемыми, получают
следующее уравнение:
к = a. (5.94)
Основными недостатками теории активированного комплекса
являются: невозможность определения для большинства реакций
энергии активации неэмпирическим путем; отсутствие надежных
экспериментальных данных о строении и структуре активирован¬
ного комплекса и способов их оценки.
Вопросы и задания для самоконтроля
1. В чем заключается основная идея теории переходного состояния?
Есть ли различие в понятиях «переходное состояние» и «активирован¬
ный комплекс»?
2. Отличается ли активированный комплекс от обыкновенной моле¬
кулы? Ответ мотивируйте.
3. Какие положения используют при выводе основного уравнения те¬
ории активированного комплекса?
117
4. Приведите основное уравнение теории для константы скорости
реакции и поясните смысл каждой из входящих в него величин. К како¬
му стандартному состоянию относится это уравнение?
5. Поясните термин «трансмиссионный коэффициент», ае. Какие зна¬
чения принимает а?, существуют ли теоретические пути его нахожде¬
ния?
кТ
6. Как называют множитель — ? Укажите его размерность и чис-
h
ленное значение при 298 К.
7. Какой вид принимает основное уравнение в статистическом ас¬
пекте теории активированного комплекса?
8. Перечислите задачи, которые решают в химической кинетике с
помощью уравнения, рассматриваемого в вопросе 7.
9. Что понимают под истинной энергией активации Е0 в теории ак¬
тивированного комплекса?
10. Установите количественное соотношение между опытной Еа и ис¬
тинной Е0 энергиями активации.
11. Какова интерпретация стерического множителя в рамках статис¬
тического аспекта теории активированного комплекса?
12. Дайте трактовку мономолекулярных реакций с помощью данной
теории. Перечислите допущения, возможные в этом случае.
13. Какой вид принимает уравнение константы скорости мономолеку¬
лярной реакции: а) для низких температур; б) при высоких температурах?
14. Каким образом объясняет аномальную температурную зависимость
констант скорости тримолекулярных реакций теория активированного
комплекса?
15. Укажите основные недостатки теории активированного комп¬
лекса.
5.6. Термодинамический аспект теории активированного
комплекса
Теория активированного комплекса в отличие от уравнения
Аррениуса и теории активных столкновений установила количе¬
ственную связь между константой скорости реакции и констан¬
той равновесия К* процесса образования активированного комп¬
лекса из реагентов (см. подраздел. 5.5):
И
Применив к величине К* известные соотношения из термоди¬
намики, связывающие константу равновесия с энергиями Гиббса
и Гельмгольца, можно выразить константу скорости через изме¬
нения термодинамических функций при образовании активиро¬
ванного комплекса.
118
Если константа скорости выражается в единицах концентра¬
ции, например в моль-1 • см3-с-1, и все вещества находятся в стан¬
дартном состоянии (с°=1 моль см-3), используя выражения
Д F* =-RT\n К*
Д F* = AU*-TAS*
(5.95)
(5.96)
получают для реакции, протекающей при постоянном объеме,
где ДF* — стандартная энергия активации Гельмгольца; AS* —
энтропия активации при стандартном состоянии (с,°= 1 моль-см-3);
A U* — внутренняя энергия активации.
Множитель (с°)ду* необходим для получения в обеих частях
уравнения (5.97) одинаковой размерности, его значение зави¬
сит от выбора стандартного состояния и молекулярности реак¬
ции; Av* — изменение числа молекул при образовании активи¬
рованного комплекса; Av*= 1 - п, где п — число вступающих в
реакцию молекул.
Уравнение (5.97) показывает, что в зависимости скорости (и
константы к) реакции от температуры определяющую роль игра¬
ет энергия активации Гельмгольца. Напомним, что, по Аррениу¬
су, эту роль выполняет изменение внутренней энергии при обра¬
зовании активных молекул.
Для сравнения расчетных и опытных данных в соотношении
(5.97) заменяют неопределяемую на опыте величину AU* на экс¬
периментальное значение энергии активации Еа. Путем сравне¬
ния результатов последовательного логарифмирования и диффе¬
ренцирования по температуре уравнений Аррениуса и (5.88) или
(5.97) получают соотношение
Подстановка формулы (5.98) в (5.97) позволяет получить одну
из форм основного уравнения термодинамического аспекта тео¬
рии активированного комплекса
(5.97)
Еа = AU* +RT.
(5.98)
fr Т Д?£.
где множитель^ = ^-Д-ее R равен фактору частоты Аоп уравне-
h
ния Аррениуса, а энтропийный множитель ехр —- по смыслу
R
по смыслу
119
= аг-^-е * е~ RT (р°) . (5.100)
соответствует стерическому фактору Р, вводимому теорией ак¬
тивных столкновений.
Если реакция протекает в газовой фазе, в термодинамике обыч¬
но используют в качестве стандартного состояние газа при р°= 1 атм
(=1 бар). В этом случае использование формул (5.88), (5.95) и (5.96)
при замене AF* на Д G* приводит к уравнению
I ГТ-Т Д G*
кр = (p°fv‘ =
квТ
h
Отметим, что AS* зависит от выбора стандартного состояния в
отличие от AU* и АН*, которые для идеальных систем от него не
зависят.
Замена в уравнении (5.100), аналогично указанному выше спо¬
собу, энтальпии активации на опытную энергию активации
Еа = АН* + nRT (5.101)
с учетом уравнения (5.101) позволяет получить другую форму ос¬
новного уравнения
, гр as; Еа
кр =эе-^-е"е R е RT (/>°) , (5.102)
где п — число участвующих в образовании активированного ком¬
плекса молекул.
На практике константа скорости обычно выражается в едини¬
цах концентрации, поэтому с учетом термодинамического соот¬
ношения
Кр = Kc(RT)Av* (5.103)
формула (5.102) трансформируется в наиболее распространенную
форму основного уравнения
^ кБТ
я-1 Д St £.
е"е R е RT. (5.104)
Связь между энтропией активации при разных стандартных со¬
стояниях легко установить из соотношений термодинамики, ис¬
пользуя зависимость изотермического изменения энтропии иде¬
ального газа от давления S2 - Si = R In PJ P2, или путем сопостав¬
ления уравнений (5.99) и (5.104)
где AS* — изменение энтропии в процессе активации, когда ис¬
ходные вещества и активированный комплекс находятся при дав¬
лении р°- 1 атм.
Для практических расчетов по уравнению (5.104) удобно ис¬
пользовать следующие обозначения:
В =
'RT'*-'
Р° ,
v у
bS*p
А = 2?е"е R .
(5.106)
(5.107)
Множитель А из (5.107) равен фактору частоты в уравнении
Аррениуса. В итоге получают удобное выражение для вычисления
A S*
ASt = Л In.
р [Веп
= R
In
В
- п
(5.108)
Для мономолекулярных реакций (п = 1) уравнение (5.100) пе¬
реходит в (5.99); согласно (5.105) AS* =АSt ,B =
к*Т
и энтро¬
пию активации рассчитывают по формуле
AS* =R
In
-1
= R
In
Ah
-1
= R
In
>- \
AhNA
RT
-1
. (5.109)
Для бимолекулярных реакций (и = 2) уравнение (5.104) при¬
нимает вид
Ir Т ВТ
кс=хЩ^-^Цг&2е R е *т,
h ри
Энтропию активации вычисляют по формуле
(5.110)
ДА; = R
(5.111)
Для тримолекулярных реакций (и = 3) формула для вычисле¬
ния энтропии активации имеет вид
(5.112)
Во всех случаях расчет энтальпии активации АН* проводят по
формуле (5.101).
= я
In
(А)
-3
_
В
\ /
121
Вопросы и задания для самоконтроля
1. Выведите основное уравнение константы скорости, выраженной
в единицах концентрации, в рамках термодинамического аспекта тео¬
рии активированного комплекса.
2. Для каких целей вводят в это уравнение множитель (С°)ЛУ*?
3. Согласно термодинамическому аспекту теории, какая величина
играет главную роль в зависимости константы скорости реакции от тем¬
пературы?
4. Существует ли количественная связь между опытной энергией ак¬
тивации и внутренней энергией активации?
5. Получите основное уравнение для константы скорости кс, выра¬
женной в единицах концентрации через энтропию и опытную энергию
активации.
6. Приведите уравнение, связывающее константу скорости кр, вы¬
раженную в единицах давления, с энтропией и опытной энергией акти¬
вации.
7. Какое соотношение связывает энтропию активации при разных
стандартных условиях, ASC и A Spl
8. Найдите разность ДА/-ДА/ при 400 К для моно- би- и тримоле-
кулярных реакций и разных стандартных состояний концентрации:
а) С°= 1 моль-л-1; б) С°= 1 моль-см-3.
9. Приведите уравнение, связывающее константу скорости кс с энт¬
ропией AS* и опытной Еа энергией активации.
10. Получите соотношение, удобное для расчета AS*.
11. Зависят ли для идеальных систем значения величин AU*, АН* и
AS* от выбора стандартного состояния? Ответ поясните.
12. Каким образом оценивают значение стерического множителя в рам¬
ках термодинамического аспекта теории активированного комплекса?
13. Для реакций в газовой фазе при постоянном давлении между дву¬
мя реагентами А и В получите с помощью теории активированного ком¬
плекса: 1) соотношение между стандартной энтальпией активации ДН£
и опытной энергией активации Еа\ 2) выражение для частотного факто¬
ра уравнения Аррениуса.
14. Гидролиз метана при температуре 1200 К отвечает кинетике реак¬
ции первого порядка. Используя опытные значения энергии активации
Еа= 335 КДж- моль-1 и предэкспоненциального множителя lg Лоп = 12,2,
вычислите при этой температуре энтальпию и энтропию активации ре¬
акции. Оцените значение стерического множителя.
15. При изучении элементарной бимолекулярной реакции в газовой
фазе С1 + Н2 НС1 + Н получены опытные значения энергии актива¬
ции, Еа = 23 КДж • моль-1 и предэкспоненциального множителя константы
скорости, А = 9,5 • 1013 моль-1 • см3 • с-1. Рассчитывая энтальпию и энтро¬
пию активации, AS* и AS* при температуре 300 К.
16. Для бимолекулярной реакции в газовой фазе Вг + Н2 -» НВг + Н
экспериментальное значение предэкспоенциального множителя констан¬
ты скорости равно 6,9- 10,3моль-1* см3 • с-1. Вычислите при температуре
500 К энтропии активации AS* и. AS* и оцените значение вероятност¬
ного множителя.
122
17. Для бимолекулярной реакции СЮ2 + 0,5F2 -> FC102, протекающей
при пониженных температурах, опытное значение предэкспоненциаль-
ного множителя константы скорости равно 4- 1010моль-1- см3 • с-1. Вы¬
числите значения энтропии активации AА/ и ДА* при температуре 240 К
и оцените величину стерического множителя.
18. При изучении газофазной реакции Н2 + С2Н4 -> С2Н6 опытное
уравнение зависимости константы скорости от температуры имеет вид
21650
к = 6,3 • 1012е т ;[/:] = моль • см-3 • с”1; [Еа] = Дж • моль-1. Оцените зна¬
чения энтальпии, энтропии активации ДА/ и ДА/ при температуре 800К,
а также стерического множителя.
19. Бимолекулярная реакция 03 + NO -» N02 + 02, протекающая при
пониженных температурах, Т = 200 К, характеризуется следующими опыт¬
ными данными: предэкспоненциальный множитель константы скорости
равен 7,94 • 108 л • моль-1 • с-1, энергия активации равна 10,5 КДж • моль-1.
Оцените, используя теорию активированного комплекса, значения эн¬
тальпии, энтропии активации (ДА/ и ДА/) и стерического множителя.
20. При изучении кинетики газофазной реакции Н + НС1 -> Н2 + С1
для температур выше 20 °С получены экспериментальным путем зна¬
чения предэкспоненциального множителя константы скорости, А =
= 5-1013 см3 • моль-1- с-1 и энергии активации, Еа= 15 КДж-моль-1. Рас¬
считайте при температуре 300 К значения энтальпии, энтропии (ДА/ и
ДА*) и энергии Гиббса активации. Оцените значение константы равно¬
весия К*.
ЧАСТЬ 2. КАТАЛИЗ
Катализ — это явление химическое
Г. К. Боресков
ГЛАВА 6. ОСНОВНЫЕ ПОЛОЖЕНИЯ КАТАЛИЗА
6.1. Понятия и определения
Катализ имеет огромное значение в природе и современной
жизни: каталитическими являются практически все биохимиче¬
ские процессы, большинство реакций, изучаемых в лаборатории,
и технологических процессов, реализуемых в промышленности.
Перечислим некоторые примеры каталитических процессов: глу¬
бокая переработка нефти с помощью крекинга, гидроочистки,
гидрокрекинга и риформинга; синтез и окисление аммиака; про¬
изводство серной кислоты и метанола; гидролиз сложных эфи¬
ров, гидрирование олефинов; инверсия сахарозы и мутаротация
глюкозы; синтез белков и обмен веществ в биологических систе¬
мах и т.д. В настоящее время около 80% всей химической продук¬
ции получается с помощью каталитических процессов.
Учитывая сказанное выше, следует признать, что современ¬
ный катализ по праву занимает самостоятельное место в физичес¬
кой химии наряду с разделами термодинамики, кинетики и элек¬
трохимии.
По существу, катализ является связующим звеном, «мостом»
между теоретическими положениями в химии и практическим осу¬
ществлением большинства химических процессов.
В начале XIX в. было установлено, что ряд химических реакций
протекает в присутствии веществ, не принимающих в этих реак¬
циях видимого участия.
Были открыты, например, следующие превращения:
окисление диоксида серы в производстве серной кислоты в при¬
сутствии оксидов азота;
трансформация крахмала в сахар при добавлении разбавлен¬
ных кислот;
окисление паров спирта и эфира при обычной температуре в
присутствии платины.
124
Для объяснения указанных выше процессов Й.Берцеллиус
(1835 г.) впервые использовал термин «катализ» и дал его опре¬
деление: катализ — это ускорение скорости реакции в присут¬
ствии некоторых веществ, которые выделяются химически неиз¬
менными в конце реакции.
Катализатором по определению В. Оствальда (1905 г.) является
вещество, которое своим присутствием изменяет скорость реак¬
ции, не изменяя ее суммарного энергетического баланса, и оста¬
ется химически неизменным в конце процесса.
Таким образом, уже из классических определений ясно, что
каталитическая реакция отличается от обычной по меньшей мере
тем, что в ней всегда участвует как минимум один дополнитель¬
ный компонент—катализатор.
Обратим внимание на то, что катализатор — это вещество,
поэтому теплота и свет, хотя и влияют на скорость протекания
реакции, не являются катализаторами.
Современное определение катализа, сформулированное в ра¬
ботах академика Г. К. Борескова, гласит: катализ — это изменение
скорости химических реакций под влиянием веществ-катализато¬
ров, многократно вступающих в промежуточное химическое вза¬
имодействие с участниками реакции и восстанавливающих после
каждого цикла свой химический состав.
Существенно, что в определении катализа указывается на его
химическую сущность, чем проводится различие между явлением
катализа и изменением скорости химического процесса под вли¬
янием, например, физических параметров.
С другой стороны, подчеркивается стехиометрическое неучас¬
тие катализатора в реакции, т. е. он не расходуется в процессе ка¬
тализа. Из этого не следует, что в ходе реакции с катализатором
не происходит никаких изменений. Важно отметить, что возмож¬
ные изменения представляют собой побочные явления, не свя¬
занные с его каталитическим действием.
Причина каталитического действия различных веществ заклю¬
чается в изменении механизма реакции в присутствии катализа¬
тора, т.е. возникновении нового или нескольких протекающих
быстро путей реакции. Под механизмом реакции понимается со¬
вокупность элементарных химических стадий, которые имеют место
в реакционной системе при переходе от исходных веществ к про¬
дуктам.
Различают положительный и отрицательный катализ: в первом
случае скорость реакции в присутствии катализатора увеличива¬
ется, во втором — уменьшается. Под ускорением реакции следует
понимать также возможность протекания реакции, не идущей без
катализатора.
Объяснение этих явлений заключается в следующем. Если при
положительном катализе промежуточное химическое взаимодей¬
125
ствие катализатора с исходными веществами приводит к благо¬
приятному, энергетически доступному пути реакции, то при от¬
рицательном катализе такое взаимодействие оставляет менее дос¬
тупные реакционные пути, что вызывает уменьшение скорости
реакции или даже ее прекращение.
В настоящее время ИЮПАК предложила заменить термин «от¬
рицательный катализ» на «ингибирование», включая в последний
различные виды замедления химических реакций.
Классификацию каталитических процессов можно проводить
по разным признакам.
По фазовому составу компонентов каталитические реакции
обычно относят к гомогенному катализу (если исходные веще¬
ства, продукты реакции и катализатор находятся в одной фазе,
например, инверсия растворов сахарозы в присутствии кислот)
или к гетерогенному катализу (реакционная смесь и катализатор
находятся в разных фазах). Как правило, используются твердые
катализаторы, например, жидкофазное гидрирование непредель¬
ных соединений на переходных металлах (Ni, Pd), нанесенных на
силикагель.
Ферментативный катализ, занимающий промежуточное поло¬
жение между гомогенным и гетерогенным катализом, часто отно¬
сят к микрогетерогенному.
Недостаток такой классификации заключается в том, что в ней
не учитывается основная причина каталитического действия: осо¬
бенности взаимодействия катализатора с реагирующими вещества¬
ми, приводящие к изменению механизма реакции в присутствии
катализатора.
Разделение каталитических реакций на основные группы мож¬
но, например, провести на основе классификации, принятой в
химической кинетике, а именно:
гемолитический катализ, при котором промежуточное хи¬
мическое взаимодействие катализатора с реагирующими веще¬
ствами протекает по гомолитическому механизму, т.е. сопро¬
вождается разрывом одних и образованием других электронных
пар;
гетеролитический катализ, когда природа промежуточного хи¬
мического взаимодействия гетеролитическая, т. е. протекает без раз¬
рушения и образования электронных пар.
При таком подходе более очевидно, что природа промежуточ¬
ного взаимодействия указывает на свойства, которыми должен
обладать катализатор.
По гомолитическому механизму осуществляются реакции син¬
теза аммиака, спиртов из СО и Н2; гидрирования кратных связей
бензола, фенола, анилина; окисления S02 в S03, NH3 до NO,
метанола в формальдегид и т.д. Катализаторами таких реакций
являются вещества, способные отдавать неспаренные электроны
126
для образования новых электронных пар. К ним относятся прежде
всего переходные элементы с незамещенными d- или /-оболочка¬
ми в виде металлов или их соединений.
По гетеролитическому механизму протекают реакции крекин-
га углеводородов, дегидратации спиртов, гидратации олефинов
и т. п. В этом случае катализаторы должны проявлять способность к
образованию координационной связи, например А1203, А1С13 и
другие катализаторы реакции Фриделя —Крафтса.
Конечно, и эта классификация в связи с недостатком надеж¬
ных данных о глубоком механизме каталитических реакций не
является строгой. Однако хотя бы в общей форме она отражает
особенности химической природы катализа.
Катализ — явление специфическое, поэтому каталитическую
активность вещества можно оценивать только по отношению к
конкретной реакции, хотя ряд катализаторов проявляют актив¬
ность по отношению к группам реакций, например, соединения
на основе Сг203 катализируют различные реакции дегидрирова¬
ния (бутана в бутилен, бутилена в дивинил, этилбензола в сти¬
рол); катализаторы на основе А1203 ускоряют реакции дегидрата¬
ции спиртов, крекинга углеводородов.
В целом состав и химическое строение катализаторов очень раз¬
нообразны. Катализатор может быть индивидуальным веществом
(металлы, активированный уголь), соединением (оксид, соль),
комплексом металла с органическими лигандами (или сложным
соединением белковой природы, например ферментами).
Не существует стехиометрических соотношений между коли¬
чествами катализатора и превращаемых в продукты исходных ве¬
ществ. Например, в производстве азотной кислоты окислением
аммиака одна массовая часть катализатора вызывает превращение
106 массовых частей реагирующих веществ.
В гомогенном катализе даже очень малых количеств катализа¬
тора (от 10-13 М) достаточно для ускорения превращения боль¬
шого количества реагентов в продукты, так как катализатор мо¬
жет использоваться многократно. Экспериментально установлено,
что скорость гомогенной химической реакции прямо пропорцио¬
нальна концентрации катализатора. Этот факт является прямым под¬
тверждением того, что катализатор действительно участвует в ре¬
акции, образуя неустойчивые промежуточные соединения, и оп¬
ределяет ее течение по другому, чем в его отсутствие, направлению.
Конкретных механизмов катализа очень много, в качестве при¬
меров перечислим несколько основных.
«Переносный» катализ. В этом случае катализатором осуществ¬
ляется перенос атома, молекулы, электрона, фрагмента. Напри¬
мер, реакция
2S02 +02 -> 2S03
127
практически не протекает в отсутствие катализатора. Напротив,
присутствие современного катализатора (сульфованадата на по¬
верхности носителя при температурах 450 — 550 °С в виде жидкой
пленки) позволяет осуществить процесс в две стадии:
«Активационный» катализ. Взаимодействие с катализатором пе¬
реводит малореакционноспособное вещество в активную частицу.
Речь идет не о возбужденном состоянии исходного реагента, а об
образовании нестойкого в условиях реакции нового химического
соединения.
1. Активация водорода в процессе гидрирования на поверхно¬
сти переходных металлов протекает благодаря диссоциативному
взаимодействию реагента с катализатором. В результате этого про¬
исходит гемолитический разрыв связей в молекуле водорода
по реакции
Н2 -> 2Н, АГН = 430 кДж/моль
и замена одной прочной связи Н—Н на две менее прочные связи
в соединении Н —К (АН = 250 кДж/моль).
Термодинамическая осуществимость процесса активации (он
является благодаря замене связей экзотермичным) сочетается с
его кинетической эффективностью, поскольку каждое из проме¬
жуточных соединений является более реакционноспособным и
далее взаимодействует независимо.
2. Активация олефинов по А. А. Баландину с использованием
современных физических методов:
Rh, 77К Rh, 200К Rh, 300К Rh, 600К
3. Активация субстратов кислотами и основаниями (см. гл. 9).
Причина заключается в высокой реакционной способности мно¬
гих ионов органических соединений при малой активности нейт¬
ральных молекул.
«Координационный» катализ. Каталитическое превращение осу¬
ществляется во внутренней сфере комплексов металлов IV—VIII
групп, где происходит сближение реагентов, изменение их до-
норно-акцепторных свойств, снятие запретов по симметрии и мно¬
S02 + V205 —!—>S03 + V204 и V204 +у202 —г—>V205
н-н
Pt Pt
н н
Pt Pt
СНз
СН3
полимер 2С
128
гоэлектронные процессы. Наглядным примером является «Вакер-
процесс» — промышленный способ окисления этилена до аце-
тальдегида в присутствии двух катализаторов — ионов Pd2+ и Си2+.
Реакция
С2Н4 + ^02 ^СН3СНО
в отсутствие катализатора не идет при низких температурах, а при
высоких — окисление является неселективным. При каталитичес¬
ком осуществлении процесс идет со 100 %-й селективностью при
Т = 400 К и Р = 3 бар по схеме
PdCl2 + Н20 + С2Н4 -> Pd° + СН3СНО + 2НС1
Ионы Pd2+ окисляют этилен, восстанавливаясь до Pd°, затем
ионы Си2+ регенерируют Pd2+ по реакциям
2Cu2+ + Pd° -» 2Cu+ + Pd2+
2CuCl + 2HC1 + уг 02 -> 2CuC12 + H20
Реальным окислителем этилена является Pd2+, так как кисло¬
род не «соприкасается» с молекулой этилена. Предполагается, что
процесс протекает через несколько стадий с образованием коор¬
динационных соединений и заканчивается превращением
crpd2+ ОН-
сг С2Н4
СГ pd0 [ ]
СГ []
2-
+ СН3СНО + н+
Избирательность действия катализатора, т.е. его способность
ускорять конкретную реакцию из некоторого числа термодина¬
мически возможных направлений, имеет важное значение, так
как позволяет проводить процесс в сторону образования целевых
продуктов.
Например, реакция термического разложения диэтилового эфи¬
ра в газовой фазе без катализатора идет по направлению
С2Н5ОС2Н5—»2СН4+ у2 С2Н4+СО
В присутствии же катализатора — паров иода — брутто-реакция
(гомогенный катализ) идет с ускорением в другом направлении:
С2Н5ОС2Н5 +12-> С2Н6+ СН3СНО +12
В гетерогенном катализе классическим примером избиратель¬
ного действия катализаторов служат многочисленные превраще¬
ния этанола, из которого в зависимости от подбора катализатора
и условий (Т, Р) можно получить различные продукты. Напри¬
мер, укажем на некоторые пути реакций:
9. Эак. 1596
129
^ >CH3CHO+H2 T = 473—523 К
>(С2Н5)20+Н20 Г=523 К
С2Н5ОН л, о
аьоз >с2Н4 + Н20 7= 623 К
—^^^СН2=СНСН=СН2 + Н2 + Н20 Г= 673—723 К
Для физико-химического объяснения избирательности и дру¬
гих свойств катализатора исключительно плодотворным оказалось
введение понятия каталитического или активного центра. Соглас¬
но этой идее, в химической реакции участвует не вся масса (объем,
поверхность) катализатора, а только небольшая ее часть, состав¬
ляющая активный центр.
В зависимости от типа катализа активный центр представляет
собой:
группу атомов или ионов кристаллической решетки, располо¬
женных на поверхности твердого тела в гетерогенном катализе;
определенный участок в молекуле белка для ферментативного
катализа.
Подробнее об активных центрах речь пойдет ниже (гл. 8, 10).
Избирательность действия различных катализаторов обуслов¬
лена химическим составом, строением и структурой их актив¬
ных центров. Именно активный центр катализатора участвует в
образовании с исходным веществом (или веществами) специ¬
фической химической связи (ковалентной, донорно-акцептор-
ной или водородной), которая приводит к появлению проме¬
жуточного соединения. В этом случае суммарная каталитическая
реакция может представлять собой несколько последователь¬
ных, параллельных или сопряженных реакций, приводящих к
образованию продукта и выделению катализатора в неизмен¬
ном виде.
Для оценки эффективности катализатора используют следую¬
щие величины.
В гомогенном ферментативном катализе мерой каталитической
активности является число оборотов реакции (turnover number),
т.е. количество молекул, превращающихся за единицу времени на
одном активном центре, которое равно
”к ~ Y' ’
'-''кат
где пк— число оборотов реакции, с-1; v — скорость реакции,
моль-л_1с_1; Скат — концентрация катализатора, моль-л-1.
Число оборотов принимает различные значения в зависимости
от типа катализа: 10“7—10~2 с-1 для катализаторов кислотно-ос¬
новного характера pi 1Q2 — 105 с-1 — для ферментов, причем у ката-
лазы достигает значения (2,5-5)-106 с-1.
130
Следует различать понятие цикла в каталитических и цепных
реакциях. Если активный центр катализатора может неоднократ¬
но (106 и более раз) сам участвовать в реакции с молекулами
исходных веществ, оставаясь неизменным после каждого цикла,
то в цепном механизме в каждом цикле, состоящем из несколь¬
ких стадий, участвует вновь образованная активная частица.
В гетерогенном катализе также было предложено использовать
термин «число оборотов» для характеристики активности реак¬
ции. Однако в гетерогенном катализе число оборотов не удается
определить, поскольку часто неизвестными остаются природа и
число активных центров на поверхности катализатора. Поэтому
вводят понятия: А, активность — величина, симбатная скорости
реакции, выражаемая количеством перерабатываемого реагента
или получаемого целевого продукта с единицы массы катализато¬
ра в единицу времени; а, удельная активность — та же величина,
А г ^
отнесенная к единице поверхности катализатора: а = — ~ .
J J
Удельная активность, конечно, более правильно отражает ис¬
тинную активность катализатора, однако на практике существен¬
ной может оказаться активность на единицу массы. Например,
для цеолитных и полимерных катализаторов понятие реакцион¬
ной поверхности теряет строгость.
По Г. К. Борескову, правильнее говорить о каталитической ак¬
тивности, характерной для всей системы, включающей катализа¬
тор и реакционную смесь данного состава, поскольку при изме¬
нении состава последней катализатор может изменять свой состав
и свойства.
Поэтому величина активности катализатора, как и скорости
реакции, подразумевает указание температурных и других усло¬
вий ее протекания, в том числе состава реакционной смеси, что
необходимо для сравнения различных образцов и типов катализа¬
торов.
Такое определение каталитической активности подразумевает
стационарное состояние катализатора в отношении взаимодей¬
ствия с реакционной средой, которое выполняется для большин¬
ства промышленных каталитических процессов.
Эффективность катализаторов оценивают также по избиратель¬
ности (селективности), различая:
интегральную избирательность, или селективность, — отноше¬
ние количества получаемого целевого продукта к теоретически
возможному из взятого количества исходного вещества, т.е. сте¬
пень превращения реагента в целевой продукт. Эта величина по¬
лезна для характеристики всего каталитического процесса и зави¬
сит от температуры, состава реакционной смеси и конечной сте¬
пени превращения;
131
дифференциальную селективность — отношение скорости об¬
разования целевого продукта к сумме скоростей реакций по всем
направлениям превращения реагентов. Эта величина, измеренная
в широком интервале составов реакционной смеси, используется
для оценки интегральной селективности и должна рассматриваться
в качестве основной характеристики селективного действия ката¬
лизатора.
Самой высокой селективностью обладают ферменты (95 —100 %)
и ряд гомогенных катализаторов. Для гетерогенного катализа эта
величина обычно ниже, порядка 70%.
6.2. Теория промежуточных соединений в катализе
Классическая теория промежуточных соединений в гомоген¬
ном катализе была сформулирована Е. И. Шпитальским (1926 г.)
на основе изучения реакции каталитического разложения перок¬
сида водорода в присутствии Сг2072" и в дальнейшем развита
Н. И. Кобозевым.
Основные ее положения таковы.
1. Катализатор образует с исходным веществом лабильное (не¬
устойчивое) промежуточное соединение.
2. Образование промежуточного соединения протекает обрати¬
мо и относительно быстро.
3. Неустойчивое промежуточное соединение относительно мед¬
ленно претерпевает дальнейшие превращения в направлении об¬
разования конечного продукта и выделения катализатора.
4. Общая скорость каталитической реакции пропорциональна
концентрации промежуточного соединения, но не реагента.
5. Один и тот же катализатор может взаимодействовать с реаген¬
том, образуя одновременно несколько промежуточных соединений.
Современная трактовка этой теории заключается в примене¬
нии кинетического закона действующих масс к обсуждаемой в
каждом конкретном случае схеме реакции.
Б качестве иллюстрации теории рассмотрим четыре механизма
каталитических реакций, получивших экспериментальное подтвер¬
ждение:
а) реакция с одним реагентом в отсутствие катализатора: А —> Р
протекает в присутствии катализатора по наиболее распростра¬
ненной схеме с образованием одного промежуточного соедине¬
ния АК:
A f Кх~—± АК—*2_>Р + К (6.1)
Примером такого механизма является реакция мутаротации глю¬
козы;
132
б) реакция с двумя реагентами имеет общий вид:
А+ В -+Р,
(6.2)
но в присутствии катализатора протекает в две стадии:
А + К—L_>АК и АК + В—-—>Р + К
Примером такого «переносного» механизма является реакция
окисления оксида серы(1У) кислородом в присутствии N0.
Брутто-реакция S02 + /202 = S03 протекает через образование
катализатором вместе с одним из исходных веществ промежуточ¬
ного соединения
no+;/2o2->no2
которое во второй стадии взаимодействует со вторым реагентом,
выделяя продукт реакции и регенерируя N0:
S02 + N02->S03 + N0
в) реакция с двумя реагентами протекает с образованием двух
продуктов по схеме
А+В —^ Р[ + Р2
В присутствии катализатора механизм этой реакции также со¬
стоит из двух стадий:
А + К——>Pi + КХ и КХ + В—^Р2+К (6.3)
Примером такого механизма реакции является гидролиз вод¬
ного раствора бромметана в присутствии иона I-.
Суммарная реакция
СН3Вг+Н20 = СН3ОН + НВг
практически протекает в две стадии, на первой образуется проме¬
жуточное соединение типа КХ и один из продуктов, на второй
стадии выделяется второй продукт и катализатор:
СН3Вг+Г—СН31 + ВГиСН31 + Н20—2—» СН3ОН + Г+Н+
г) реакция с двумя реагентами имеет вид А + В —> Pj + Р2, но в
присутствии катализатора механизм этой реакции, протекающей
также в две стадии, совершенно иной по сравнению с (в): на
первой стадии происходит обратимое образование промежуточ¬
ного соединения катализатора с молекулами исходных веществ,
на второй — его распад на продукты и регенерация активного цен¬
тра катализатора:
А + В + К^==±АКВ и АКВ—эР,+Р2 + К (6.4)
К. 1
Примером такого механизма является гидролиз простых эфи¬
ров в присутствии фермента.
133
Установление механизмов сложных каталитических реакций яв¬
ляется трудоемкой задачей и требует использования современных
спектральных, электрохимических и термохимических методов ис¬
следования для определения химического состава и строения про¬
межуточных соединений.
Широко известные механизмы таких гетерогенно-каталити¬
ческих реакций, как синтез аммиака, гидрирование, изомериза¬
ция и окисление олефинов, установлены с помощью изотопных
методов.
В отсутствие точных сведений о природе и структуре промежу¬
точных соединений, скорости их образования, используемые для
объяснения брутто-реакций механизмы часто являются гипотети¬
ческими. Аргумент о том, что опытные данные подтверждаются
расчетами по кинетическим уравнениям, выведенным для пред¬
ложенного механизма, является совершенно недостаточным. Нельзя
исключить, что при использовании другого механизма реакции
могут быть получены аналогичные кинетические уравнения.
6.3. Термодинамические и кинетические аспекты
в катализе
При определенной температуре, независимо от того, является
ли реакция каталитической или нет, она всегда характеризуется
теми же термодинамическими величинами: ArG°, АГН°, АГА°, не
зависящими от присутствия катализатора.
Если реакция в прямом направлении термодинамически не¬
возможна в данных условиях, т. е. ArG> 0, введение катализатора в
реакционную систему не приводит к ее протеканию, но может
ускорить обратную реакцию.
Если же реакция возможна (AG<0) и приводит к состоянию
равновесия, характеризуемого термодинамической константой К0,
присутствие катализатора позволяет только быстрее достичь со¬
стояния равновесия. При этом катализатор одинаково ускоряет
скорости прямой и обратной реакций, так как константа равно¬
весия К0 равна отношению констант скоростей прямой и обрат¬
ной реакций, но он не смещает положение равновесия — это сле¬
дует из известного соотношения AG°= -RTlnK0.
Допущение возможности смещения катализатором положения
равновесия равносильно совершению системой работы за счет хи¬
мической реакции при отсутствии разности температур, что оз¬
начало бы возможность существования вечного двигателя второго
рода, т.е. противоречит второму закону термодинамики.
Отметим, что гомогенный катализатор может изменять свой¬
ства среды, что приводит к изменению выхода продукта, при этом
меняется Кс, но не К°. Наглядный пример — гидролиз крахмала.
134
Если катализатор не влияет на термодинамические условия ре¬
акции, каким образом можно объяснить его ускоряющее воздей¬
ствие на химический процесс?
Химический процесс, как правило, сопровождается разрывом
связей в исходных веществах и образованием новых химических
связей в продуктах. Это становится возможным при переходе по
пути реакции через энергетический барьер, характеризуемый энер¬
гией активации.
Установлено для ряда изученных реакций, что энергия актива¬
ции существенно меньше суммы энергий разрываемых связей за счет
компенсации энергией, выделяемой при образовании новых связей.
Для характеристики способности веществ вступать в реакцию
Г.К.Боресков ввел понятие степени компенсации т|:
п ’ (6-5)
где Е— энергия активации; ХА — сУмма энергий разрываемых
связей. При £—г) —э 0, т.е. компенсации нет; при £—> О,
г) -» 1, т.е. степень компенсации 100 %.
Показано, что для реакций с участием радикалов степень
компенсации может превышать 95% (Н + С12-э НС1 + С1, Е =
= 8,4 кДж/моль), но для реакций между насыщенными молеку¬
лами компенсация значительно меньше, около 70% (Н2+С2Н4—»
—>С2Н6, £' = 180 кДж/моль).
Видно, что для реакций между молекулами необходимо пре¬
одолеть высокий активационный барьер, что и объясняет их ма¬
лую скорость.
Само по себе наличие энергетических барьеров стабилизирует
окружающий нас мир, поскольку если бы компенсация стала пол¬
ной и все энергетические барьеры исчезли, то это привело бы к
катастрофическим последствиям: быстро осуществились бы все
возможные реакции, органические вещества превратились бы в
воду и углекислый газ, земля стала бы пустыней, омываемой оке¬
аном слабой азотной кислоты.
Катализ играет большую роль именно в решении задачи пре¬
одоления энергетических барьеров, регулирования пути химичес¬
кого превращения, благодаря тому, что катализатор, взаимодей¬
ствуя с исходными реагирующими веществами и непосредствен¬
но участвуя в образовании активированного комплекса, повышает
степень компенсации, тем самым уменьшает величину энергети¬
ческого барьера — энергии активации и увеличивает скорость хи¬
мической реакции.
Отметим, что каталитическое воздействие на реакционный про¬
цесс может осуществляться в различных формах.
135
Большинство химических реакций протекает без нарушения рав¬
новесного распределения энергии в системе, т. е. соблюдается за¬
кон Максвелла—Больцмана: вероятность состояния с энергией,
Г Е
большей или равной Е, пропорциональна множителю ехр
RT
и быстро убывает с увеличением Е. Поэтому действие катализато"
ра сводится к тому, что из всего многообразия различных путей
реакции, характеризуемых энергетическими барьерами разной вы¬
соты, он открывает реакционный путь с наименьшей высотой
активационного барьера, т.е. обеспечивает наиболее вероятное
протекание химического превращения.
Для ряда химических процессов (цепных реакций) наблюдает¬
ся нарушение равновесного распределения энергии, обусловлен¬
ное существенным изменением свободной энергии реакции, и
образование частиц, способных аккумулировать энергию реакции
и использовать ее для продолжения и разветвления цепи.
В некоторых случаях действие катализатора можно объяснить
возбуждением цепного механизма в результате промежуточного
взаимодействия с катализатором. Например, реакция монооксида
углерода(П) с кислородом резко ускоряется в присутствии паров
воды, благодаря появлению богатых энергией частиц и возникно¬
вению нового цепного пути.
Другая форма каталитического действия — ускорение первич¬
ного образования активных частиц.
Например, реакция разложения растворов пероксида водорода
в отсутствие катализаторов характеризуется малой скоростью за¬
рождения активных частиц и соответственно самой реакции. При¬
сутствие катализатора, например солей железа, приводит в ре¬
зультате его взаимодействия с Н202 к образованию свободных ра¬
дикалов:
Fe2+ + Н202 -> Fe3+ + ОН' + ОН*
Fe3+ + Н202 -» Fe2+ + Н02 + Н+
В итоге стационарная концентрация активных частиц возраста¬
ет, что и объясняет резкое увеличение скорости разложения Н202.
В цепных реакциях встречается и явление каталитического ин¬
гибирования, когда в результате взаимодействия катализатора с
активными частицами происходит их рекомбинация и соответ¬
ственно уменьшается вероятность продолжения или разветвления
этой реакционной цепи.
Отличие каталитического ингибирования от химического свя¬
зано с тем, что катализатор многократно вступает во взаимодей¬
ствие с активными частицами, восстанавливая свой состав после
каждого акта обрыва цепи.
136
Остановимся подробнее на реакциях, характеризуемых равно¬
весным распределением энергии в изучаемой системе.
Ускорение реакции в присутствии катализатора, как было ука¬
зано выше, происходит за счет ее протекания по пути, характе¬
ризуемому минимальной высотой энергетического барьера. Состо¬
яние реакционной системы, отвечающее вершине этого барьера,
называется активированным комплексом, который образуется из
исходных реагирующих веществ с участием катализатора. Этот энер¬
гетически выгодный реакционный путь, как правило, сопровож¬
дается образованием стабильных промежуточных соединений с ка¬
тализатором и характеризуется более низкими значениями сво¬
бодной энергии образования активированных комплексов на всех
стадиях. Отметим, что рассматриваемая сама по себе стадийность
процесса не вызывает роста скорости реакции, если не происхо¬
дит снижения высот всех энергетических барьеров, отсчитывае¬
мых от энергетического уровня исходных веществ.
Для лучшего понимания причин, объясняющих действие ката¬
лизаторов, обратимся к термодинамическому аспекту теории ак¬
тивированного комплекса. Основное уравнение для константы ско¬
рости реакции (5.88)
к = х^К*
п
с учетом термодинамического соотношения (5.95)
AF° = -RT In Кс
примет следующий вид (5.97):
I гг-1 Д F* / rp AS* A U*
к = ае —е с0(1_х) = ас!^-еГТе~~кт с0(1-х),
h h
где ае — трансмиссионный коэффициент, указывающий на веро¬
ятность прохождения реагирующей системой — активированным
комплексом — через вершину энергетического барьера в сторону
образования продуктов, где — эффективная скорость про-
h
хождения активированного комплекса через вершину энергети¬
ческого барьера, с-1; AF* — энергия Гельмгольца или свободная
энергия, AS* — энтропия, A U* — внутренняя энергия процесса
активации при переходе исходных веществ в активированный ком¬
плекс в стандартных условиях. Множитель c0(1_jr) необходим для
соблюдения размерности, где {с0} = моль/л или моль/см3; х —
число молекул, участвующих в образовании активированного
комплекса. Легко в этом убедиться на примере бимолекулярной
реакции {к} = л-с_|моль-1.
Для реакций, протекающих в газовой фазе, удобнее применять
соотношение
137
cO(l-x)
р°,
v ^ у
(6.6)
Используя уравнение Аррениуса (1.61)
Еа_
к = At лг,
нетрудно преобразовать уравнение (5.97) к удобному для даль¬
нейшего анализа виду (5.99)
ь т — Е°
к = аг-^—-ее R е"*Гс0(|"1),
h
где Еа — так называемая «опытная» или «аррениусовская» энергия
активации (Ea=AU*+RT).
Как видно из уравнений (5.97) и (5.99), константа скорости
реакции зависит от свободной энергии активации и определяется
величиной энтропийного и энергетического множителей.
Энтропия активации при образовании активированного комп¬
лекса из исходных веществ с участием катализатора, как прави¬
ло, уменьшается по сравнению с энтропией активации активиро¬
ванного комплекса в отсутствие катализатора, поскольку присут¬
ствие катализатора требует большей упорядоченности в строении
активированного комплекса.
Присутствие катализатора способствует сближению и созданию
благоприятной взаимной ориентации молекул реагентов при об¬
разовании активированного комплекса.
Уменьшение энтропии активации при протекании реакции по
каталитическому пути приводит к существенному уменьшению
энтропийного множителя в уравнении (5.99), в результате этого
снижение скорости реакции (при одинаковой энергии активации)
может достигать 107—1010 раз.
Но из двух факторов, определяющих скорость реакции (5.99),
9энергетический множитель может изменяться в более широких
пределах. Нетрудно показать, что при комнатной температуре
уменьшение энергии активации (АЕ) примерно на 60 кДж уве¬
личивает скорость реакции в 10й раз. Поэтому даже небольшое
дальнейшее снижение энергии активации вызывает значительное
увеличение скорости реакции, таким образом проявляется ката¬
литический эффект. Реакций, протекающих в присутствии ката¬
лизаторов с уменьшением активационного барьера, много. Напри¬
мер, разложение йодистого водорода в присутствии платины со¬
провождается снижением энергии активации на Ais=120 кДж/моль.
Положительные значения AS* наблюдаются в ряде случаев для
реакций, в которых образование промежуточного соединения ак¬
тивного центра катализатора с исходным веществом сопровожда¬
ется ослаблением или разрывом связей реагента до формирова-
138
ния активированного комплекса. Это означает, что активирован¬
ный комплекс в каталитической реакции более «рыхлый», т.е. рас¬
положение атомов в нем более благоприятно, чем в активирован¬
ном комплексе некаталитической реакции.
Выразим предэкспоненциальный множитель А уравнения Ар¬
рениуса через параметры термодинамического аспекта теории ак¬
тивированного комплекса следующим образом:
А = аД <6'7>
или в логарифмической форме
\gA = B + $AS*, (6.8)
где А0, В, (3 — постоянные, R — универсальная газовая постоянная.
Наблюдаемый в ряде каталитических реакций рост предэкспо¬
ненциального множителя А указывает, как это видно из выраже¬
ния (6.8), на увеличение энтропии активации. Например, реак¬
ция гидролиза аденозинтрифосфорной кислоты (АТФ) в присут¬
ствии фермента (миозина) ускоряется по сравнению с действием
ионов Н30+в 1014 раз. Энергия активации при этом фактически не
изменяется, а результат достигается за счет увеличения предэкс¬
поненциального (т.е. энтропийного) множителя.
При изучении каталитических реакций было обнаружено, что
между параметрами уравнения Аррениуса Ак Е существует взаи¬
мосвязь, выражаемая уравнением логарифмического вида
1 ёА = С + уЕ, (6.9)
где С, у — константы. Сопоставление уравнений (6.8) и (6.9) по¬
зволяет получить интересное корреляционное соотношение меж¬
ду энтропией и энергией активации процесса
AS* = const + а Е, (6.10)
где const, а — постоянные. О других подобных соотношениях будет
сказано ниже (см. гл. 9).
Сложная связь между величиной энергии активации и скоро¬
стью каталитической реакции может быть следствием так называ¬
емого компенсационного эффекта, который наблюдается в ката¬
лизе. Для его объяснения удобно использовать уравнения (6.9) и
(6.10). Суть компенсационного эффекта заключается в следующем:
в ряде реакций даже значительное уменьшение энергии актива¬
ции в присутствии катализатора не сопровождается адекватным
увеличением скорости процесса, так как одновременно происхо¬
дит уменьшение AS* согласно (6.10), что приводит к снижению
Предэкспоненциального множителя в уравнении Аррениуса и ско¬
рости реакции в целом. Энергетический и энтропийный факторы
Действуют по существу в противоположных направлениях.
139
Компенсационный эффект наблюдается не всегда и может иметь
другие причины.
Сравнительная оценка каталитического и некаталитического
процессов (с одинаковыми исходными веществами) требует на¬
учно обоснованного и взвешенного подхода.
До сих пор в ряде изданий отечественной и зарубежной лите¬
ратуры сравнения каталитических и некаталитических реакций про¬
водятся путем сопоставления энергий активации обоих процес¬
сов, построения энергетических профилей обеих реакций в коор¬
динатах (потенциальная энергия £/, путь реакции), иллюстри¬
рующих снижение энергии активации при каталитическом про¬
цессе, и т.п.
Такой подход не представляется достаточно корректным. Срав¬
нение скоростей каталитической и некаталитической реакций, на¬
пример, в виде
kKSLT (АЕ
кат = ехр
к
^некат
RT
(6.11)
где АЕ = ЕКат- £Некат корректно только при сопоставлении однотип¬
ных, одностадийных процессов, для которых значения предэкспо¬
ненциальных множителей в уравнении Аррениуса очень близки. Та¬
кие процессы маловероятны, т. е. фактически не наблюдаются.
Если элементарные стадии для некаталитической и каталити¬
ческой реакций совершенно разные, то сравнение только значе¬
ний энергии активации служит лишь констатацией факта ускоре¬
ния каталитического процесса в целом (как и приведенный выше
пример разложения йодистого водорода в присутствии Pt).
В большинстве случаев речь идет о сложных реакциях, поэтому
прежде всего надо сравнивать их механизмы с разным числом эле¬
ментарных стадий. К тому же для сложной реакции со многими
стадиями неправомерно изображать потенциальные кривые на
плоскости, говорить о «координате реакции».
Как было сказано выше, увеличение скорости химической ре¬
акции при катализе в основном связано с повышением степени
компенсации благодаря участию катализатора в составе активи¬
рованного комплекса.
В заключение рассмотрим два основных механизма каталити¬
ческих превращений: слитный и стадийный.
При слитном (или синхронном) механизме на реакционном
пути сохраняется лишь один максимальный энергетический барь¬
ер. Слитный механизм широко распространен в гомогенном ката¬
лизе (мутаротация глюкозы, гидратация альдегидов). Этот меха¬
низм с сопряжением через катализатор является характерным для
ферментативного катализа, с ним связывают, как правило, высо¬
кую активность и специфичность ферментов.
140
В работах О. М. Полторака показано, что активные центры фер¬
ментов и реагирующих веществ образуют цепочки или «цепи пе¬
рераспределения связей», по которым в результате перемещения
электронов и протонов (последние лишь смещаются на доли дли¬
ны межатомных связей при гетеролитическом разрыве и возник¬
новении связей атомов водорода) синхронно происходит измене¬
ние кратности связей, что и приводит к высокой компенсации
энергии разрыва старых связей, позволяя осуществлять фермен¬
тативные реакции при невысоких (и даже малых) значениях энер¬
гии активации.
Формирование в ферментах цепей перераспределения связей,
действие которых управляется кислотно-основными центрами, яв¬
ляется, вероятно, наиболее общей чертой ферментативного ката¬
лиза и позволяет говорить о едином кислотно-основном механиз¬
ме большинства ферментативных реакций*.
При стадийном механизме этапы каталитического процесса про¬
текают раздельно и на реакционном пути имеется несколько энер¬
гетических барьеров с максимумами и минимумами. По стадий¬
ному механизму протекают многие реакции гомогенного катали¬
за и большинство гетерогенных процессов промышленного ката¬
лиза при повышенных температурах (600 — 800 К).
Стадийный механизм катализа при уменьшении молекулярно-
сти стадий позволяет существенно понизить энергию активации,
что может быть связано с уменьшением на всех стадиях суммы
энергий разрываемых связей.
Если молекулярность стадий не уменьшается, то возрастание
скорости каталитической реакции происходит лишь при повыше¬
нии степени компенсации энергии разрыва связей на стадии с
максимальной суммой энергий разрываемых связей.
Опытные данные показывают, что при стадийном механизме
энергии активации каталитических процессов сохраняют доста¬
точно высокие значения, что, по-видимому, связано с невысо¬
кой степенью компенсации.
Вопросы и задания для самоконтроля
1. Какое место занимает катализ в современной науке?
2. Дайте классические определения терминов «катализ» и «катали¬
затор». Являются ли теплота и свет катализаторами?
3. Сформулируйте современное определение катализа. В чем заклю¬
чается причина каталитического действия?
4. Каким образом различают каталитические реакции по фазовому
составу участников процесса9 Приведите примеры. В чем недостаток та¬
кой классификации?
* Полторак О.М., Чухрай Е. С. Физико-химические основы ферментативного
катализа. — М.: Высш. шк., 1971.
141
5. Существует ли иная классификация каталитических процессов?
Ответ поясните.
6. Существует ли стехиометрическое соотношение между количества¬
ми используемого катализатора и исходных веществ? Зависит ли ско¬
рость гомогенной химической реакции от концентрации катализатора?
Если да, то какой эта зависимость имеет вид?
7. Приведите примеры, характеризующие основные типы конкрет¬
ных механизмов катализа.
8. Что понимают под избирательностью действия катализатора? При¬
ведите примеры для различных катализаторов в гомогенном и гетероген¬
ном катализе.
9. Как вы понимаете термин «активный центр» катализатора? Что
представляет собой активный центр в ферментативном и гетерогенном
катализе?
10. Что является мерой каталитической активности в гомогенном и
ферментативном катализе? Каков порядок этой величины в каждом из
типов катализа?
11. Какие величины используют для оценки эффективности катали¬
затора в гетерогенном катализе? Какая из величин более точно отражает
реальную активность катализатора? Какую величину следует использо¬
вать для цеолитных и полимерных катализаторов?
12. В чем различие терминов: «интегральная» и «дифференциальная»
селективность? Ответ поясните.
13. Сформулируйте основные положения теории Е. И. Шпитальского.
Какая роль отводится в этой теории образованию промежуточного со¬
единения?
14. Приведите различные механизмы каталитических реакций, если
в отсутствие катализатора реакции протекают по схемам: а) А-»Р;
б) А+ В—>Р; в) А+В—>Р1 + Р2-
15. В каком случае предлагаемые для объяснения брутто-реакции ме¬
ханизмы являются гипотетическими? Почему?
16. Оказывает ли катализатор влияние на термодинамические усло¬
вия процесса? Смещает ли он положение равновесия? Может ли катали¬
затор изменить выход продукта? Ответ поясните.
17. Что означает термин «степень компенсации»? Каков порядок ве¬
личин степени компенсации и энергии активации для реакций: а) с
участием радикалов; б) между насыщенными молекулами.
18. Каким образом можно объяснить роль катализатора в увеличении
скорости химической реакции?
19. В каких формах может проявляться каталитическое действие в ре¬
акционном процессе?
20. Какова роль энтропии активации в каталитической реакции? В ка¬
ких случаях наблюдают положительные или отрицательные значения
энтропии активации в катализе?
21. Существует ли корреляционное соотношение между энтропией и
энергией активации? Если да, приведите его.
22. Каким образом может измениться скорость химического процес¬
са при уменьшении активационного барьера в присутствии катализато¬
ра: а) на 4 кДж/моль: б) на 40 кДж/моль?
142
23. В чем заключается явление компенсационного эффекта, наблю¬
даемого в гомогенном и гетерогенном катализе?
24. Какие каталитические реакции протекают по слитному механиз¬
му? В чем его особенности?
25. Приведите примеры стадийного механизма каталитических реак¬
ций. В каком из двух механизмов энергии активации каталитических про¬
цессов имеют, как правило, более высокие значения? Ответ поясните.
ГЛАВА 7. АВТОКАТАЛИЗ
Автокатализом принято называть ускорение процесса под дей¬
ствием одного из образующихся в ходе реакции продуктов.
Кинетические кривые автокаталитических реакций характери¬
зуются наличием областей индукции, когда в начальный момент
времени концентрация образующегося продукта-катализатора еще
мала и скорость реакции незначительна; самоускорения, когда
скорость реакции возрастает, и уменьшения скорости реакции в
результате существенного расходования реагирующих веществ.
По-видимому, первым примером автокаталитической реакции
явилось исследование Ф. Гаркуром (Оксфорд, 1866 г.) окисления
щавелевой кислоты перманганатом в кислой среде. Оказалось, что
реакция ускоряется, т.е. катализируется, одним из продуктов ре¬
акции ионом Мп2+.
Согласно современным представлениям, эта автокаталитиче-
ская реакция протекает в несколько стадий, самой медленной из
которых, т.е. лимитирующей весь процесс, является стадия обра¬
зования иона Мп3+
МПО4+ Мп2+—> МПО4Ч- Мп3+
Затем ион Мп3+ взаимодействует с щавелевой кислотой с об¬
разованием после нескольких стадий продуктов и регенерацией
иона Мп2+
Н2С204+ 2Мп3+—»2Мп2++ Н2+ 2С02
Большую, роль в изучении и понимании явления автокатализа
сыграли работы российского ученого Н.А. Меншуткина в 80-х гг.
XIX в. на примере реакции разложения третичного амилацетата с
образованием ацетанилида и уксусной кислоты. В настоящее вре¬
мя используются два подхода для описания кинетики автокатали¬
тических реакций.
1. В общем виде для реакции
А-эВ + С (7.1)
самая простая автокаталитическая реакция имеет вид
А + (В)—1—> В + (В) + С (7.2)
143
Обозначая начальную концентрацию А через а, текущую кон¬
центрацию А через (а-х), х — изменение концентрации А ко вре¬
мени t, равное концентрации образующегося вещества В, выраже¬
ние для скорости реакции (7.2) запишется следующим образом:
Щ^к1(а-х)х. (7.3)
at
Однако если продукт В первоначально отсутствует, то х = 0 и
начальная скорость также равна нулю, т.е. реакция не будет про¬
текать в отсутствие продукта В. Напротив, добавление даже не¬
большого количества вещества В к исходному веществу А позволяет
реакции протекать с возрастающей до некоторого момента скоро¬
стью. В этом случае уравнение скорости реакции имеет вид
л] у
v = - = kl(a-x)(b-x), (7.4)
где 6=[В]0 и Ь<ка.
Это уравнение для автокатализа впервые было получено В. Ост¬
вальдом (1883 г.) при изучении гидролиза уксуснометилового эфи¬
ра в присутствии добавленной до начала опыта уксусной кислоты.
Разделяя переменные в уравнении (7.4), разлагая дробь на эле¬
ментарные и интегрируя, получим выражение
J*‘d/ = /zrr
dx
1
г dx
•* а-х
■ dx
b + х
(7.5)
(а - x)(b + х) a + b
которое после простых преобразований сводится к кинетическо¬
му уравнению
1
а + Ь
-In
a(b + х)
Ь(а - х)
= kit.
(7.6)
Рис. 7.1. Изменение концентрации А в течение времени согласно
механизму (7.1) и уравнению (7.6) для 7: Ь= 10-3 моль-л-1;
2: Ь = 1(И моль-л-1; 3: b= 10-5 моль-л-1, при а= 10-1 моль-л-1
и кх = 1 л • моль-1 - с-1
144
Рис. 7.2. Изменение концентрации продукта автокаталитической
реакции (7.2) в координатах (x/b, ut) (для г = 0,1)
На рис. 7.1 представлены кривые, полученные из (7.6) при за¬
данных значениях а и ки для различных значений Ь. Из него вид¬
но, что выбор значения параметра b заметно влияет на время,
необходимое для достижения максимальной скорости, и незна¬
чительно изменяет форму сигмоидной кривой.
Из уравнения (7.6) можно получить выражение для х в зависи¬
мости от времени
На рис. 7.2 представлена зависимость концентрации образую¬
щегося продукта, отнесенной к его начальной концентрации в
смеси, от параметра ut = (а + b)kxt.
(7.7)
которое удобно представить в виде
х _ е"' -1
b 1+ rtm ’
(7.8)
где и = (а + />)£,; г = —
а
Ю. Зак. 1596
145
Как видно из рис. 7.2, кривая имеет сигмоидный вид с индук¬
ционным периодом, выпуклостью вниз, характерной для самоус-
корения, и точкой перегиба.
Нетрудно убедиться, что в точке перегиба количество прореа¬
гировавшего вещества равно максимальному
Xmax =±Y~ = \ (7-9)
при условии, что b «: а.
Скорость реакции достигает максимума
/ _ . ,\2
^тах — ^1
или при условии, что Ь «а,
а + Ь
(7.10)
/
„ ~к*
^тах — 1 ^ *
Время достижения максимальной скорости (для b <&а) опре¬
деляется из (7.8) соотношением
tnep = tl/2=--±-lnr. (7.11)
Уравнение (7.6) после преобразований запишем в виде
In -Х- = In — + (а + b)k{t. (7.12)
а-х а
При условии, что b <za, а количество прореагировавшего
вещества х достаточно велико, т.е. b<szx<a, можно допустить,
что Ь+х-х; а + Ь~а.
В этом случае уравнение (7.12) принимает вид
In * = In - + ak,t. (7.13)
а-х а
Видно, что, строя график в координатах (In ,t), можно по
а-х
тангенсу угла наклона прямой при известном значении а рассчи¬
тать константу скорости к\ автокаталитической реакции.
Уравнение (7.13) при условии х = ^> т е- в точке перегиба
(6iep=*i/2) преобразуется и примет вид
-In — = ak{tu2- (7.14)
а
Обозначив через а = — долю вещества А, превращенного за вре-
а
мя /, путем комбинирования уравнений (7.13) и (7.14), получим
146
новое соотношение
In
1 - а
а
= aki(ti/2-t),
(7.15)
из которого можно рассчитать долю прореагировавшего за время t
вещества А.
Время /и, в течение которого автокаталитическая реакция прак¬
тически не идет, называется периодом индукции. Для определе¬
ния этой величины удобно преобразовать (7.15) в
'и =*|/2 - -у- In
ак{
1-а„
0СИ
= /V2+4~ln
акх
а„
1-аи
(7.16)
где аи = —, tK — период индукции, хи — количество прореагиро-
а
вавшего вещества А ко времени tK.
2. Другой способ избежать трудностей, связанных с использо¬
ванием уравнения (7.3), состоит в следующем. Предположим су¬
ществование двух различных путей реакции: первая реакция —
некаталитическая первого порядка и вторая — автокаталитическая.
-эВ + С
А-
А + В-
Уравнение для скорости реакции в этом случае примет вид
dx
d t
при условии кх < к2 или
= ку (а - х) + к2х(а - х)
(7.17)
(7.18)
dx
— = к2
dt
к\
I +х
к2
(а-х).
(7.19)
Полученное уравнение (7.19) формально совпадает с уравне¬
нием Оствальда (7.4), выведенным для случая присутствия не¬
большого количества катализатора в исходной смеси при условии
кх
= Ь-
Разделяя переменные х и t в (7.18), затем разлагая дробь на
элементарные, получим соотношение
1 —f{JL
к, + к-,а: \а -
■+-А
х кх + к2х
dx = Jd/,
(7.20)
которое после интегрирования, используя начальное условие
х = 0 при t = 0, преобразуется в
147
ln(^+^£= (721)
(a - x)kx
Если k2 —> 0, то соотношение (7.21) очевидно переходит в урав¬
нение реакции первого порядка
In ——— = к\ t. (7.22)
а-х
В общем случае форма уравнения (7.21) такова, что значения
кх и к2 с трудом определяются из экспериментальных данных.
Из соотношения (7.21) получим выражение для зависимости
количества прореагировавшего вещества от времени
к] [е<*'+*^)' -1]
к2а + £,е('
Х а Ь- Л I /, ъ(к\ +kia)l ■ (7.23)
В предельных случаях при /=0 х=0; при /—><», пренебрегая сла¬
гаемыми 1 и к2а по сравнению с резко возрастающими показате¬
лями функций, х~а.
В общем случае, согласно формуле (7.23), кривая должна иметь
точку перегиба. Время ее достижения находим путем дифферен¬
цирования х дважды по времени и приравнивая производную нулю
in^)
кх + к2а
Время /пер зависит от соотношения констант скоростей и уве¬
личивается при уменьшении константы скорости к2 каталитиче¬
ской реакции.
Значение величины х в точке перегиба можно найти либо пу¬
тем подстановки выражения (7.24) в (7.23), либо путем диффе¬
ренцирования по х непосредственно скорости в уравнении (7.19)
а-к,/к2 а
^пер — -^шах — 2 ~ 2* (^‘^)
поскольку с учетом начального условия введения к} (к\«: к2), можно
tr
пренебречь членом L .
2к2
Полезно отметить общую закономерность автокаталитических
реакций: для двух разных механизмов (7.2) и (7.17), несмотря на
различие кинетических уравнений (7.3) и (7.19), скорость прохо¬
дит через максимум при значении х~а/2.
Зависимость скорости реакции v от времени получают путем
дифференцирования х в (7.23) по 1
148
dx
d7
*1 + 1
k, a
(k[ + k2a) exp[№ +Mf]
— (7.26)
j^- + -exp[(£i +A:2fl)?]
IA a
Учитывая, что по условию к2а ^>кь и при /->0 можно пренеб¬
речь вторым членом в знаменателе, в итоге получим в простой
форме зависимость скорости автокаталитической реакции в на¬
чальный период от времени
г> = ДефГ, (7.27)
где постоянные величины
= {к2/кх +l/a)(k{ + к2а) (J т
(k2/ki)2
И
у = к{ + к2а. (7.29)
Уравнение (7.27) аналогично полученному для скорости раз¬
ветвленной цепной реакции без учета расходования исходного ве¬
щества, т.е. также для начального периода времени.
Кинетика автокаталитических и цепных разветвленных реак¬
ций бывает очень похожей, что затрудняет их идентификацию при
исследовании, несмотря на различную природу.
Вопросы и задания для самоконтроля
1. Какое явление называется автокатализом? Какой вид имеет кине¬
тическая кривая автокаталитической реакции? Приведите примеры.
2. Какой механизм для объяснения автокатализа предложил В. Ост¬
вальд? Какой вид имеет уравнение скорости реакции в этом случае?
3. Получите методом неопределенных коэффициентов уравнение для
константы скорости автокаталитической реакции.
4. Из соотношения, полученного в вопросе 3, найдите выражение
для х — изменения концентрации вещества А ко времени t.
5. Для точки перегиба автокаталитической реакции определите:
а) максимальную концентрацию образующегося вещества х; б) скорость
реакции; в) время достижения максимальной скорости.
6. В чем заключается другой механизм автокатализа? Можно ли по¬
лучить в этом случае уравнение для скорости реакции, аналогичное урав¬
нению Оствальда?
7. Получите интегральное кинетическое уравнение для второго ме¬
ханизма автокаталитической реакции.
8. Получите аналитическим путем соотношение для времени дости¬
жения точки перегиба по механизму, обсуждаемому в вопросе 6.
9. Определите концентрацию образующегося вещества в точке переги¬
ба для автокаталитической реакции, протекающей по второму механизму.
149
10. В чем сходство и различие между автокаталитическими и разветв¬
ленными цепными реакциями?
ГЛАВА 8. ФЕРМЕНТАТИВНЫЙ КАТАЛИЗ
8.1. Общие определения и понятия
Ферментативные реакции подчиняются общим законам ката¬
лиза, но вследствие сложности состава и строения ферментов об ¬
разуют специфическую и весьма обширную область каталитичес¬
ких реакций.
Биологические катализаторы белковой природы, ускоряющие
каталитические реакции в живых системах, называют в отечествен¬
ной литературе ферментами, в англоязычной — энзимами.
Не каждый белок является ферментом, но в составе практи¬
чески каждого белка находится определенный набор групп, опре¬
деляющих активность фермента.
Академик И. П. Павлов называл ферменты «возбудителями жиз¬
ни». Так, при пищеварении ферменты амилазы превращают крах¬
мал в сахар, протеазы расщепляют белки до аминокислот. В при¬
роде ферментативные процессы протекают в крупных масштабах.
Например, объем мирового промышленного производства свя¬
занного азота составляет лишь несколько процентов от количества
азота, связываемого в природе с помощью фермента нитрогеназы.
Ферменты представляют собой компактные молекулы с моле¬
кулярной массой от 104 и диаметром от 20 А и выше. Входящие в
их состав глобулярные белки состоят из расположенных в опреде¬
ленной последовательности а-аминокислотных остатков, соеди¬
ненных пептидными связями (—СО—NH—), причем установле¬
но, что в построении большинства ферментных белков участвуют
20 или 21 аминокислот.
Внутреннее строение ферментов обычно характеризуют в био¬
химии наличием четырех типов структур:
первичная определяется только генетическим кодом и указы¬
вает последовательность аминокислот для каждого белка;
вторичная определяет характер свертывания в спираль, или
спирализации, цепи;
третичная структура характеризует способ пространственной
укладки частично или полностью свернутой в спираль полипеп-
тидной цепи;
четвертичная — взаимное расположение и характер объедине¬
ния структурно независимых глобул в активный олигомерный фер¬
мент.
150
Строго говоря, такие представления являются идеализацией,
поскольку не существует независимых вторичной и третичной
структур: в процессе пространственной укладки полипептидной
цепи происходит как скручивание, обусловленное силами притя¬
жения и отталкивания различных ее элементов, так и их укладка в
компактную глобулу фермента.
Тем не менее использование указанных выше терминов, хотя
и относится к приближенным способам описания белковых моле¬
кул, является полезным при качественном анализе общих струк¬
турных проблем в той области, которая соответствует разреше¬
нию ферментной глобулы примерно 6 —8 А.
Следует отметить, что именно третичная структура играет боль¬
шую роль в создании специфических отличий ферментов от ос¬
тальных катализаторов: при ее возникновении в глобуле фермен¬
та образуется в виде «щели» адсорбционный участок активного
центра.
Понятие четвертичной структуры приобретает ясный физиче¬
ский смысл, если объединение в глобулу не изменяет строения
объединяющихся частей. В действительности отдельные субъеди¬
ницы ферментов изменяют свои конформации при ассоциации,
поэтому понятие четвертичной структуры является еще менее стро¬
гим, чем двух предыдущих. По-видимому, речь идет о зависимос¬
ти пространственного строения белковой глобулы от всех межмо-
лекулярных взаимодействий в системе. В результате на каждом этапе
происходят конформационные изменения, что и делает нестро¬
гим понятие вторичной, третичной и четвертичной структур.
Каталитическое действие ферментов обусловлено наличием на
их поверхности активных центров, в состав которых входят кис¬
лотно-основные и простетические группы (небелковой природы)
при строго определенном их расположении друг относительно друга.
Например, в активный центр а-химотрипсина входят три важные
группы:
карбоксил-ион —COJ аспарагиновой кислоты;
гидроксил-ион серина;
имидазол гистидина, HIS57
Активный центр фермента можно представить (но не всегда)
в виде «щели», в которой расположены каталитические и адсорб¬
ционные участки. Адсорбционный участок осуществляет образо¬
вание комплекса субстрата с ферментом и ответственен за селек¬
цию субстрата и специфичность фермента. Каталитический учас¬
ток представляет собой совокупность функциональных групп,
осуществляющих перераспределение электронных плотностей и
перенос групп непосредственно в химическом акте катализа.
Основной структурной особенностью активного центра явля¬
ется большая вариация сорбционных участков, благодаря разно¬
образию способов укладки полипептидных цепей при построении
151
самой глобулы белка, и высокая консервативность каталитиче¬
ских участков, т. е. однотипное строение их активных групп.
Каталитические участки способны по своей химической при¬
роде взаимодействовать с большим числом субстратов, превра¬
щая их в продукты. Однако адсорбционные участки, выполняя
роль селектора, предоставляют возможность контакта с каталити¬
ческим центром только немногим из субстратов.
Оптимальные свойства ферментов достигаются при необходимой
для катализа геометрии активного центра и благоприятном распо¬
ложении адсорбционного участка относительно каталитического.
Такое двухслойное строение активного центра, раздельное на¬
хождение адсорбционных и каталитических участков обеспечива¬
ет резкое увеличение эффективности катализа в глобуле фермен¬
та и указывает исследователям путь построения серии катализато¬
ров с различными свойствами при одинаковом химическом составе
и близком строении.
Простетическая группа, входящая в состав активного центра,
может быть каким-либо небелковым органическим соединением
или ионом металла в форме комплекса. Часто в ферментах присут¬
ствуют соединения железа, цинка, меди, марганца, кальция, ко¬
бальта. Например, фермент карбангидраза с молекулярной мас¬
сой 3 • 104, содержащий один атом цинка на молекулу, находится
в красных кровяных тельцах и катализирует дегидратацию бикар-
бонат-ионов и гидратацию С02. Такие ионы, входящие на посто¬
янной основе в структуру некоторых ферментов, в классической
биохимии получили название «кофакторов», хотя с точки зрения
химической классификации этот термин является неточным. К ко¬
факторам можно отнести:
катионы металлов, необходимые для протекания ферментатив¬
ной реакции, но не принимающие непосредственного участия в акте
каталитического превращения субстрата. Такую роль играют ионы
металла в металлсодержащих пептидазах (концевых аминокислот);
ионы металлов, необходимые для ферментативных реакций пе¬
реноса фосфорильных групп (например, Mg2+), когда избыточ¬
ный отрицательный заряд на фосфате препятствует нуклеофиль¬
ной атаке каталитической группы;
ионы металлов, с помощью которых иногда осуществляется
построение необходимой для катализа четвертичной структуры
ферментов. Например, четыре иона цинка стабилизируют алко-
гольдегидрогеназные ассоциаты из четырех субъединиц.
В целом для осуществления функций кофактора может подхо¬
дить по свойствам большое число ионов, но белковая глобула фер¬
мента активируется избирательно лишь немногими ионами.
Во многих случаях функции иона металла-кофактора с доста¬
точной определенностью можно отличить в механизме реакции
от функций иона металла-катализатора.
152
Кофактор может измениться при построении фермент-субстрат-
ного комплекса, но он не претерпевает никаких изменений (в от¬
личие от катализатора) в элементарных актах каталитического пре¬
вращения фермент-субстратного комплекса в комплекс субстрат-
продукт.
Принципиальное отличие ферментов от катализаторов других
типов связано прежде всего с высокой специфичностью действия,
которая является их биологически необходимым свойством, что
позволяет проводить ферментативные процессы с выходом целе¬
вого продукта, близким к 100%.
Различают четыре типа специфичности действия ферментов.
1. Абсолютная специфичность, когда фермент ускоряет пре¬
вращение только одного вещества или одной пары субстратов (для
бимолекулярной реакции) в соответствующие продукты. Без аб¬
солютной специфичности большинства ферментов существующая
форма жизни оказалась бы невозможной, поскольку в клетке су¬
ществуют различные функционально близкие субстраты, кото¬
рые претерпевают совершенно различные типы превращений.
2. Абсолютно-групповая специфичность, когда фермент ката¬
лизирует превращение определенной группы веществ.
3. Относительная групповая специфичность, когда каталитичес¬
кое действие фермента проявляется не к группе веществ, а только
к ее части. Например, пероксидазы специфичны только к перокси¬
ду водорода, но не очень чувствительны к природе окисляемых
субстратов, если они являются ароматическими полифенолами.
4. Стереохимическая активность, когда фермент способен от¬
личать свой субстрат от его оптического изомера. Если название
фермента содержит буквы D- или L-, то всегда речь идет об абсо¬
лютной стереоспецифичности. Это означает, что адсорбционный
участок такого фермента строго комплементарен избранному суб¬
страту. Важно отметить, что в таких случаях второй изомер либо
вообще не способен соединяться с ферментом, либо, если такое
связывание происходит, он является конкурентным ингибитором.
Принятая Международным биохимическим союзом в 1961 г.
первая систематическая номенклатура ферментов, хотя еще не
совершенна, но отвечает современному состоянию дел в этой
области. Это связано прежде всего с тем, что большинство изу¬
ченных ферментативных реакций протекают на активных цент¬
рах, состоящих из ограниченного числа каталитических систем,
химический состав которых меняется незначительно.
В то же время полученные на основании физико-химических
Данных сведения о составе и строении активных центров в насто¬
ящее время имеются лишь для небольшой группы используемых
ферментов (-20 %). Поэтому последовательное физико-химичес¬
кое описание ферментов, как это принято для гомогенных ката¬
лизаторов, пока нельзя положить в основу их систематизации.
153
Учитывая все возрастающее число открываемых ферментов,
(в настоящее время их насчитывается более 2000), принято их клас¬
сифицировать не по типу химического строения, а по характеру
осуществляемых ими каталитических реакций и виду субстрата,
на шесть больших классов (табл. 8.1).
Таблица 8.1
Класс
Катализируемые реакции
Оксидоредуктазы
Трансферазы
Гидролазы
Лиазы
Изомеразы
Лигазы
Окисления — восстановления
Переноса функциональных радикалов
Гидролиза
Присоединения к двойным связям
Изомеризации
Образования связей (конденсации)
с использованием АТФ
Например, оксидаза аскорбиновой кислоты (молекулярная
масса 1,4 • 105, 8 атомов Си), широко распространенная в расте¬
ниях и микроорганизмах, катализирует окисление витамина С до
дегидроаскорбиновой кислоты.
В настоящее время встает вопрос о промышленном использо¬
вании ферментов, выделенных из биологических систем. В каче¬
стве примера можно привести производство сиропов для конди¬
терской промышленности из крахмала. Благодаря ферментам
осуществляется превращение крахмала в глюкозу, затем — изомери¬
зация глюкозы во фруктозу (сладкий сахар).
Однако для большинства ферментов их использование в широ¬
комасштабном производстве сопряжено с решением ряда проблем,
к числу которых относится нестабильность ферментов, короткое
время их существования. Перспективным путем решения этой про¬
блемы является иммобилизация ферментов, т.е. их закрепление в
каких- либо устойчивых и проницаемых для реагирующих веществ
матрицах.
8.2.. Кинетика ферментативной реакции с одним субстратом
Рассмотрим самую простую реакцию превращения субстрата (S)
в продукт (Р) при участии фермента (Е): S —> Р.
Уже в ранних работах было обнаружено, что начальная ско¬
рость ферментативной реакции при фиксированной концентра¬
ции фермента и малых концентрациях субстрата изменяется ли¬
154
нейно с концентрацией субстрата, но для высоких концентраций
субстрата скорость реакции уже не зависит от его концентрации.
Л.Анри (1903 г.) первым предположил, что фермент образует
промежуточное соединение ES с субстратом. JI. Михаэлис предло¬
жил для количественного описания кинетики этой реакции меха¬
низм, состоящий из двух стадий (первая — обратимая и быстрая,
вторая — необратимая и медленная):
E + S^-Д ' ES—*2—нР + Е (8.1)
Задача не имеет точного аналитического решения, но решает¬
ся методами квазиравновесных и стационарных концентраций.
Первый метод был использован для получения кинетического
уравнения Анри Л.Михаэлисом и М.Ментен (1913 г.)*, второй
метод, предложенный Г. Бриггсом и Дж.Холдейном** в 1925 г., стал
общепринятым в кинетике ферментативных реакций.
Для данного механизма скорость реакции определяется скоро¬
стью образования конечного продукта
» = “ = №]' (8.2)
а
В свою очередь, скорость образования промежуточного соеди¬
нения ES, согласно закону действующих масс, можно записать:
= к\ [S] [Е] - [ES] - к2 [ES]. (8.3)
at
Уравнения материального баланса для фермента и субстрата
имеют вид
[Е]0 = [Е] + [ES] (8.4)
и
[S]0 = [SJ + [ES] + [Р], (8.5)
где [Е]0, [S]0 — начальные концентрации фермента и субстрата;
[Е], [S] — их текущие концентрации; [ES], [Р] — концентрации
промежуточного соединения и продукта.
Решение системы из трех уравнений (8.3 — 8.5) находим при
выполнении следующих условий:
1) для /->0, [Р] —>0, следовательно, [S]0= [S] + [ES];
2) обычно [S]0»[E]0, поэтому с учетом условия (1) [S]0=[S];
3) концентрация ES стационарна; поэтому принимают в (8.3),
d[ES] „
что ——- = 0-
dt
* Michaelis L„ Menten M.L. Biochem. Z., 1913. V. 49. P. 333.
** Briggs G.E., Holdane J.B.S. Biochem. J., 1925. V. 19. P. 338.
155
Подставляя значение [Е] из (8.4) в уравнение (8.3) и решая
его относительно [ES], получаем выражение для
[Е s] = _AtiMSJo (8.6)
к\ [S]o + к-\ + к2
которое после деления числителя и знаменателя на кх принимает
вид
[ESI = [E]o[S]° • (8.7)
Км+[ S]o
к + к
Величину Км = —- принято называть по решению ИЮПАК
к\
константой Михаэлиса.
Подставляя выражение (8.7) в (8.2), получаем уравнение для
скорости образования продукта в начальные моменты времени
.. _ ВДоИо (й
Анализ уравнения (8.8) позволяет объяснить наблюдаемые на
опыте закономерности.
Если [^]0<к тогда
t,o=Mk[S]0, (8.9)
т.е. начальная скорость линейно зависит от концентрации суб¬
страта, что подтверждает наблюдаемый на опыте первый порядок
реакции по субстрату.
Если [iS’]0»A'A/, тогда из уравнения (8.8) получаем
v0 = k2[E)0, (8.10)
т. е. начальная скорость не зависит (как и в опыте) от концентра¬
ции субстрата, следовательно, порядок реакции по субстрату бу¬
дет нулевой, по ферменту, как и в предыдущем случае, — пер¬
вый, а сама скорость достигает предельного значения, fmax:
Щ-■= k2[E]0 = vmax. (8.11)
При достижении v0 значения vmiX (кроме предельной, ее назы¬
вают еще максимальной скоростью) концентрация фермент-суб-
стратного комплекса [ES] —»[ES]max, это означает, что весь фер¬
мент переходит в состав промежуточного соединения, т.е. фер¬
мент насыщен субстратом. Именно в этих условиях удобно
сравнивать скорости различных реакций, поэтому запишем урав¬
нение (8.8) в виде
v Jw[Sjo_ (8 12)
° *m+[S]0
156
где Км и i>max — параметры ставшего классическим уравнения Ми-
хаэлиса — Ментен.
Следует отметить, что уравнение типа (8.12) характерно для
кинетики гомогенных, ферментативных и гетерогенных катали¬
тических реакций, в частности, оно идентично (по форме) урав¬
нению Ленгмюра — Хиншельвуда (см. подразд. 10.6).
Физический смысл константы Михаэлиса можно пояснить,
исходя из уравнения (8.12): она численно равна концентрации
субстрата (Км = [S]0), при которой активность фермента составля¬
ет половину максимальной, т. е. v0 = vmax/ 2. Это наглядно видно из
графика в координатах (i>0, [S]0), представленного на рис. 8.1.
Км и [S]0 имеют одинаковую размерность концентрации М
(моль-л-1). Численные значения Км обычно лежат в пределах
10~2 —10~8 М, легко воспроизводятся и не зависят от концент¬
рации фермента. В то же время Км является функцией темпера¬
туры, pH среды, зависит от присутствия других веществ, игра¬
ющих роль ингибитора или активатора.
Если для рассматриваемого механизма реакции выполняется
следующее условие: » к2, тогда
г _k-i+k2 к_х_г
м ~ к ~к~ s'
/Vj /V|
где Ks — субстратная константа, т. е. константа диссоциации фер-
мент-субстратного комплекса на фермент и субстрат, характеризу¬
ющая меру связывания фермента с субстратом. В этом случае мож¬
но рассматривать Ки как величину, характеризующую сродство фер¬
мента к субстрату. В общем случае Км > Ks, причем эксперименталь¬
ное значение Км представляет собой максимальное значение Ks.
Рис. 8.1. Зависимость скорости ферментативной реакции
от концентрации субстрата
157
Иная картина наблюдается только для ферментов разложения
пероксида водорода, каталазы и пероксидазы, для которых к2~»кл.
В этих случаях константа Михаэлиса, Км~к2/кх сильно отличается
от субстратной константы К5.
Величина t!max, определяемая из экспериментальных данных по
уравнению (8.12), для ферментов с неизвестной молекулярной
массой и неочищенных препаратов является просто кинетическим
параметром. Но для ферментов с известной молекулярной мас¬
сой, т.е. таких, для которых можно рассчитать начальную концен¬
трацию [Е]0, эта величина становится фундаментальным парамет¬
ром, так как позволяет определить из соотношения (8.11) значе¬
ние константы к2:
I _ ^шах _ МОЛЬ • Л _ 1
К2 - —- - - мин ,
[Е]о (л • мин) • моль
характеризующей скорость распада фермент-субстратного комп¬
лекса на продукт и фермент.
Величину к2 называют также числом оборотов фермента, так
как она указывает на число моль субстрата, превращенных в про¬
дукт одним молем фермента в единицу времени.
В некоторых случаях (например, для амилазы) к2 легче рассчи¬
тать по числу моль продукта, образующегося в одну минуту. Одна¬
ко если фермент содержит не один, а несколько активных цент¬
ров, определение числа оборотов к2 приводит к неопределенному
результату.
При анализе простых ферментативных реакций полезно исполь-
к2
зовать соотношение -тг~, которое, если выполняется условие
к2»к_х, равно константе скорости к{ прямой реакции первой ста¬
дии механизма. Для сложных реакций константу к2 в уравнении
Михаэлиса—Ментен обычно заменяют на ккат — каталитическую
константу с той же размерностью [Г]-1.
8.3. Способы определения кинетических параметров
Для определения параметров Км и vmm используют несколь¬
ко способов линеаризации уравнения Михаэлиса —Ментен, ко¬
торые позволяют выразить экспериментальные данные в удобной
для анализа линейной графической форме.
Первый способ (в литературе носит название способа Лайнуиве-
ра —Берка)*.
После записи уравнения (8.12) в виде обратных величин, по¬
лученное соотношение
* Lineweaver Н., Burk D. J. Am. Chem. Soc., 1934. V. 56. P. 658.
158
Рис. 8.2. Определение параметров Кмк vmax по способу
JI айнуивера — Берка
vo vmax vm3X [S]0 (8.13)
удобно использовать для построения линейного графика в коор-
1 1
динатах (— , тггг-), который представлен на рис. 8.2.
Щ L^Jo
Отрезок /, отсекаемый прямой на оси ординат, будет равен
11 „ км
■ , т.е. vmm = тангенс угла наклона прямой равен , т.е.
1>тах »
t20C
Км = umaxtga = -у • Константу Км можно сразу определить по от¬
резку, отсекаемому прямой на оси абсцисс, так как при — = О
Щ
\ ^ Км \ 1 _ 1
^шах ^шах [S]0 Ки [S]0
Второй способ (иногда называют второй формой уравнения Лай-
нуивера—Берка). При умножении обеих частей соотношения (8.13)
на [S]0, получим уравнение
[S]0 _ Км , 1 ГС1
1T~V~ 7~[S]°- (8Л4)
v0 шах vma\
159
Рис. 8.3. Определение параметров Км и fmaxno второй форме
уравнения Лайнуивера — Берка
roi
Графическая зависимость ^ от [S]0 представляет собой, как
Ч
следует из рис. 8.3, прямую линию.
Отрезок, отсекаемый прямой на оси ординат, равен ——, а на
^шах
оси абсцисс равен -Км. Тангенс угла наклона прямой равен .
Гщах
Третий способ (в некоторых странах называют способом Эди —
Хофсти)*. После умножения обеих частей уравнения (8.13) на
vm&xvo и несложных преобразований получим
v0 = vmax ~ Км (8.15)
Vo
Построение графика в координатах (щ, ттгг), как видно из
L^Jo
рис. 8.4, дает прямую линию, тангенс угла наклона которой tga =
= ~КМ (tgP = Км), а отрезок, отсекаемый на оси ординат, соответ¬
ствует нтах. Кроме того, отрезок, отсекаемый на оси абсцисс, дол¬
жен быть равен , так как при v0—»0 fmax ~ Км и
Км г 0 [S]0 Км [S]0
Во всех способах обработки результатов, кроме графических,
широко используется метод наименьших квадратов.
Указанные способы линеаризации основного уравнения (8.12),
учитывая вид используемых линейных соотношений, охватывают
различные области концентраций субстрата, в которых возможны
отклонения от линейности. Поэтому полезно значения Км и нтах,
полученные разными способами, сравнивать между собой.
Важно помнить, что если обработка опытных данных, напри¬
мер по способу Лайнуивера —Берка в координатах (i\f\ [S]0-1)»
указывает на линейную зависимость, это еще не означает, что
‘ Eadie G.S. J. Biol. Chem. 1942. V. 146. P. 85.
160
Рис. 8.4. Определение параметров Kmvl vm способом Эди—Ховсти
реакция протекает по простому механизму, предложенному Ми-
хаэлисом и Ментен.
Нетрудно показать, что аналогичная (8.12) форма уравнения
может быть получена и для более сложных механизмов реакций.
Например, субстрат взаимодействует с ферментом с образова¬
нием через два промежуточных соединения двух продуктов X и Y
по схеме
E + S^
— ES—-—»ES' + X—-—»Е +Y
Выражение для начальной скорости примет вид
к2%[ E]q[S]q
Vn =
^jrX + Plo
(8.16)
где % =
. Это уравнение аналогично по форме уравнению
к2 + к3
Михаэлиса—Ментен (8.12), оно также легко линеаризуется в ко¬
ординатах (v0_l, [S](f')-
Возможно протекание более сложных ферментативных реак¬
ций с участием двух субстратов.
8.4. Ингибирование ферментативных реакций
Как сказано выше, вещества, присутствие которых в системе
понижает активность фермента, т.е. уменьшает скорость реакции,
называются ингибиторами.
Ч. Зак. 1596
161
Если для ферментативного катализа необходимы предваритель¬
ная адсорбция субстрата и его строгая ориентация относительно
активных групп каталитического центра, то для ингибирования
достаточно не только взаимодействия со всем адсорбционным уча¬
стком, но и простого связывания ингибитора с отдельными уча¬
стками адсорбционного центра.
Соединения проявляют свойства ингибиторов либо благодаря
возможности образовывать с каталитическим центром прочные
комплексы (H2S, цианиды), либо из-за воздействия на карбониль¬
ную группу фермента, либо благодаря своей способности денату¬
рировать белки (например, соли Ag, Си, Hg).
Эффект ингибирования может иметь место по разным причи¬
нам, но выяснение природы этого явления часто дает необходи¬
мые сведения о строении и свойствах самих ферментов. Среди воз¬
можных типов обратимого ингибирования остановимся на трех.
L. Ингибитор I конкурирует с субстратом за активный центр,
так как по строению близок к нему и является псевдосубстратом,
образуя с ферментом неактивный комплекс (EI). Если при увели¬
чении концентрации субстрата в растворе активность фермента вос¬
станавливается, такое ингибирование называется конкурентным.
2. Ингибитор не конкурирует с субстратом за активный центр,
так как присоединяется к другой части молекулы белка с образова¬
нием неактивных комплексов (EI и ESI), что приводит к сниже¬
нию активности фермента, но не изменяет его сродство к субстра¬
ту. Если достигается обратимость ингибирования, т.е. восстановле¬
ние активности фермента под действием отличных от субстрата
веществ, такое ингибирование называется неконкурентным.
3.Если при ингибировании по типу 2 образуется только один
неактивный комплекс ESI (фермент—субстрат—ингибитор), име¬
ет место бесконкурентное ингибирование.
Необратимое ингибирование, приводящее к полной потере ак¬
тивности фермента, наблюдается при участии в реакции катали¬
тических ядов, например солей ртути, свинца, меди, образую¬
щих прочные комплексы на поверхности фермента.
Рассмотрим подробнее реакцию конкурентного ингибирования,
протекающую по следующему механизму:
называют константой ингибирования или константой диссоциа¬
ции комплекса EI.
E+S *L- 1ES—Ь—»Е + Р
Л-1
(8.17)
Е +1 Е‘ (неактивный комплекс)
Константу
(8.18)
162
Для математического решения задачи используем следующие
допущения:
при /->0 [Р]-э0, [S]0=[S] + [ES];
[S]0»[E]0, поэтому [S]0= [S];
rnci d[ES] n.
[ES] — стационарна, т.е. = 0;
[I]0>> [E]0, т.е. [I] = [I]0 — начальная концентрация ингибитора.
Скорость реакции определяется скоростью образования про¬
дукта (8.2)
d[P]
v = ■
d t
= к2[ ES],
В присутствии ингибитора уравнение материального баланса
для фермента примет вид
[Е]0= [Е] + [ES] + [Е1]. (8.19)
Заменяя [EI] на Kj из формулы (8.18), откуда [Е1]=
К,
, и
подставляя в формулу (8.19), получим
[Е]0 = [ES] + [Е]
1 +
Що
К,
(8.20)
Для нахождения концентрации [ES] используем принцип ста¬
ционарных концентраций
d[ES]
At
:*,[S][E]-*_,[ES]-*2[ES] = 0,
откуда
^[E][S]0 _ [E][S]0
*_,+*2 Ku •
(8.21)
Из соотношения (8.21) выражаем концентрацию фермента
[esja:*,
[Е] =
[Slo
(8.22)
полученное выражение подставляем в уравнение (8.20), которое
решаем относительно [ES]:
[Е]0 = [ES] +
[ES ]КМ
[S]o
1 +
Що
К,
-*[E]0[S]o = [ES]
[S]0 + Км
(8.23)
163
->[ES] =
[E]0[S]0
[S]0 + К
м
1 +
Що
К,
Знание концентрации ES позволяет получить на основании фор¬
мулы (8.2) выражение для начальной скорости реакции, ускоря¬
емой ферментом в присутствии ингибитора
d[P] , ft;ci
v01 - —7— = ^[ES] -
at
^2[E]0[S]0
JS]0
[S]0 + Km
1 +
Що
К,
эф
+ [S]0
(8.24)
где
К
М эф
= К
м
1+!Ь
(8,25)
эффективная константа, зависящая от [1]0 и К{.
Сравнение уравнений Михаэлиса — Ментен (3.13) и (3.24) по¬
казывает, что начальная скорость реакции в присутствии ингиби¬
тора всегда меньше скорости реакции в его отсутствие. Предель¬
ные значения скорости в обоих случаях для больших концентра¬
ций субстрата одинаковы и равны (8.9)
*>max = к2[Е] 0.
Если [1]о=А> и [S]0= Км, скорость реакции ингибирования бу¬
дет равна —р-.
Выражение для константы ингибирования получим из (8.25):
[1]о
К, =
Км эф/Км ~ 1
(8.26)
Таким образом, для определения Кь кроме знания начальной
концентрации ингибитора [1]0, необходимо найти эффективную
константу реакции в присутствии ингибитора и константу Миха¬
элиса в его отсутствие.
Графическую обработку результатов опытов в отсутствие ин¬
гибитора или в его присутствии удобно проводить одним из ука¬
занных ранее способов, например в координатах Лайнуивера—
Берка
1 —I
у*\Щ
Это позволяет, как видно из рис. 8.5, найти
также, зная Шо, К,
Км, Км Эф, а
164
Рис. 8.5. Конкурентное ингибирование:
1 — в отсутствие ингибитора, [1]0 = 0; 2 — в его присутствии, [1]0> 0
Константу ингибирования Kj можно также найти непосредствен¬
но из графика, так как отношение тангенсов угла наклона пря-
'i+ra*
Для неконкурентного ингибирования рассмотрим простейший ме¬
ханизм процесса:
мых в точке пересечения с осью ординат равно
а
ES
кг
Р + Е
E + I
ES +1 т-
с учетом, что
EI
- ESI
(8.27)
[E][I]
[EI] ’
_ [ES] [I]
греи ' (8.28)
[ESI]
Если оба комплекса неактивны, т.е. не дают продукта, а кон¬
станты их диссоциации равны между собой: К[ - К', то это равно¬
сильно дополнительному условию: ингибитор не мешает взаимо¬
действию субстрата с ферментом, но уменьшает скорость реак-
165
иии из-за неактивности образующихся комплексов на поверхнос¬
ти фермента.
Решая задачу аналогично предыдущей, получаем выражение
для начальной скорости в следующем виде:
_ ^тахэфИо /0
<8 9)
где
^ахэф=-!%г- (8.30)
1+ff
Из выражения (8.29) видно, что в этом случае изменяется
предельная скорость процесса, но константа Михаэлиса остает¬
ся постоянной. Если [1]0 = К{ и = [S]0, скорость реакции инги-
V
бирования будет равна .
Выражение для константы ингибирования, полученное из фор¬
мулы (8.30),
К,= [1]о _ , (8.31)
^шах/^шахэф
показывает, что Kj легко вычислить при известной [1]0, измеряя
скорости реакции в отсутствие и в присутствии ингибитора.
Обработка экспериментальных данных для неконкурентного ин-
1 1 }
позволяет получить урав-
гибирования в координатах
нение
_1_ J_
v0’ [S]„
-U—L_ + (8.32)
Щ ^ max эф ^ max эф l lo
которое позволяет определить Км, vmax, vmm эф и Kj, зная [1]0, как
это показано на рис. 8.6.
При бесконкурентном ингибировании механизм реакции имеет
вид
ES +1 ESI
(8.33)
166
-1 /Км
О
Рис. 8.6. Неконкурентное ингибирование:
1 — в отсутствие ингибитора, [1]0=0; 2 — в его присутствии, [1]0 > О
В этом случае образуется один неактивный комплекс ESI за
счет присоединения ингибитора к промежуточному соединению ES
с константой равновесия
К -[ES][I] /о 341
К' ' [ESI] ‘ (04)
Решение задачи аналогично разобранным ранее примерам и
приводит к следующему выражению скорости реакции в началь¬
ный период времени:
Рис. 8.7. Бесконкурентное ингибирование:
/ — в отсутствие ингибитора, [1]0 = 0; 2 — в его присутствии, [1]0 > 0
167
Vo I =
^max эф[$]р
Км эф + [S]0
(8.35)
где
К
М эф
Км
(8.36)
В этом случае эффективная константа отличается от значения
для конкурентного ингибирования, но эффективная скорость вы¬
ражается уравнением, аналогичным для неконкурентного инги¬
бирования.
1
1
Обработка опытных данных в координатах ,
[S]o,
ставленная на рис. 8.7, позволяет рассчитать значения ц
К,
М эф>
^тах эф>
и К,, зная [1]0.
пред-
ах> Км>
8.5. Активность ферментов
Активность фермента определяется прежде всего химическим
строением активного центра, зависит от концентрации, pH сре¬
ды и температуры.
Зависимость скорости реакции (и, следовательно, активности
фермента) от концентрации субстрата определяется уравнением
(8.8); в начальный период времени, согласно (8.9), эта зависи¬
мость имеет линейный характер.
Установлено, что активные центры ферментов с изученным ме¬
ханизмом действия имеют в своем составе функциональные груп¬
пы, способные присоединять или отщеплять протон. Константы иони¬
зации этих групп лежат в пределах изменения рК (= —lgAT) от 3 до 12.
Причем активность проявляют только кислотно-основные груп¬
пы фермента в необходимом для катализа состоянии ионизации.
Активность фермента сильно зависит от pH раствора, причем для
каждого фермента существует свой интервал значений pH, внут¬
ри которого активность фермента максимальная. Обычно оптиму-
мы pH лежат в нейтральных или слабощелочных областях, хотя
возможны и отклонения (для пепсина оптимум при pH = 2, для
аргиназы — при pH =10,2). Интервал оптимума активности, как
правило, находится в пределах 0,2—0,4 единицы pH, но возмож¬
ны отклонения, например, глюкозоредуктаза сохраняет высокую
активность в интервале pH от 6 до 9.
От pH раствора зависят и кинетические параметры фермента¬
тивной реакции: предельная скорость t>max и константа Михаэли-
са Км, причем во втором случае зависимость более сложная.
168
Появление оптимума pH объясняют разными причинами, но,
по-видимому, главная из них связана с прохождением через мак¬
симум эффективной скорости распада фермент-субстратного ком¬
плекса.
Влияние температуры на скорость ферментативной реакции имеет
сложный характер, связанный в том числе с проблемой термола¬
бильности фермента, поскольку положение максимума pH среды
существенно зависит от температуры. Кроме того, при повышении
температуры происходит изменение третичной структуры фермен¬
та, изменяющее геометрические параметры активного центра.
В результате повышения температуры наблюдается увеличение
активности (скорости реакции) фермента до достижения макси¬
мального значения, обусловленное тепловой активацией хими¬
ческих процессов и температурной зависимостью концентраций
активных форм фермента. При последующем увеличении темпе¬
ратуры активность фермента убывает (скорость реакции уменьша¬
ется) в результате инактивации фермента, т.е. его тепловой дена¬
турации, что приводит к уменьшению доли реально участвующих
в реакции глобул фермента.
Температурный оптимум ферментативных реакций, как пра¬
вило, имеет значение 50 — 60 °С, однако возможны и отклонения
в обе стороны: для а-амилазы он достигает 100 °С, в то время как
для люциферазы находится при 25 — 30 °С.
Эффективные энергии активации для большинства ферментатив¬
ных процессов не превышают, как правило, значений 50 кДж/моль.
Высокая активность ферментов по сравнению с неорганичес¬
кими катализаторами объясняется рядом причин, в том числе су¬
щественным снижением в их присутствии энергии активации био¬
химической реакции. Например, реакция гидролиза мочевины ус¬
коряется ферментом уреаза в 1014 раз по сравнению с обычным
кислотным катализом.
Возникает вопрос, каким образом можно оценить активность
фермента, эффективность его действия? Полный ответ на него
возможен, если удастся рассчитать константы скоростей и энер¬
гии активации всех основных стадий изучаемого процесса. При
отсутствии сведений о механизме реакции невозможно оценить
константы скоростей элементарных стадий на основании «эффек¬
тивных» кинетических параметров, определенных из опыта.
Чаще всего ограничиваются сравнением «эффективных» кон¬
стант, входящих в кинетическое уравнение каталитической реак¬
ции. За меру активности фермента принимают значение констан¬
ты к2 (или ккат), характеризующей скорость распада фермент-суб-
стратного комплекса.
Однако для многих ферментов молекулярная масса неизвест¬
на, поэтому для них нельзя рассчитать начальную концентрацию
и, следовательно, согласно формуле (3.12), невозможно опреде¬
169
лить значение к2. Поэтому Комиссия по ферментам ИЮПАК сфор¬
мулировала следующие рекомендации для характеристики актив¬
ности ферментов: за единицу количества фермента Е принимают
такое количество, которое катализирует превращение одного мик¬
ромоля субстрата за 1 мин в стандартных условиях (298 К, опти¬
мум pH, субстратное насыщение).
Введены в употребление новые термины:
удельная активность (а): количество фермента в единицах Е на
1 мг белка;
молекулярная активность (Л): число молекул субстрата, пре¬
вращаемых в продукты за 1 мин под действием 1 молекулы фер¬
мента в условиях субстратного насыщения. Отметим, что величи¬
на А равна к2.
Обе величины связаны между собой соотношением
Л=10"3М7,
где а —- в ферментных единицах Е\М— молекулярная масса фер¬
мента, г/моль.
Вместо термина «число оборотов» теперь предпочитают назва¬
ние «молекулярная активность» фермента. Для иллюстрации при¬
веденных выше понятий рассмотрим следующий пример.
Пример 8.1. Неочищенный препарат содержит 20 мг белка на 1 мл.
10 мкл препарата катализируют превращение субстрата в 3 мкмоль про¬
дукта в объеме 0,5 мл за 1 мин при оптимальных условиях (pH, ионной
силы, концентрации и температуры). Требуется рассчитать: а) началь¬
ную скорость реакции, выраженную в моль-л-1 • мин-1; б) концентра¬
цию фермента в условных единицах Е, мл-1; в) удельную активность
фермента в единицах Е на 1 мг белка.
„ , dпр 3 10-М03 с ]П_з , , |N
Решение: a)v = —£■ = — — = 6-10 (моль-л-1 • мин-1);
Vat 0,5 • 1
б) из определения Е и условий примера следует, что 10 мкл фермен¬
та позволяют получить три ферментных единицы. Находим концентра¬
цию фермента:
3
Сф = = (единиц Е• мл-1);
в) из определения удельной активности и знания концентрации фер¬
мента (см. б) получим искомое значение а:
300 ед. Е • мл-1 _
° = 20 мг_мл-' = 15 (ед‘ Е на 1 мг белка)‘
Сравнительная оценка активности различных ферментов во
многих случаях затруднена, поскольку ферментативные реакции
могут протекать по более сложным, чем схема Михаэлиса, меха¬
низмам. Рассчитываемые на основании опытных значений мак¬
170
симальной скорости константы к2 в таких случаях являются слож¬
ными функциями констант скоростей элементарных стадий ре¬
акции.
Использование при сравнительной оценке эффективности фер¬
ментов только одного критерия — константы к2, характеризую¬
щей скорость превращения фермент-субстратного комплекса в про¬
дукт, может оказаться недостаточным из-за компенсационного
эффекта.
Установлено, что параметры уравнения Аррениуса (1.61)
Е
к = At**
А и Е могут изменяться симбатно при переходе от одного катали¬
затора к другому. Например, уменьшение константы скорости к в
связи с уменьшением предэкспоненциального множителя может
компенсироваться увеличением множителя ехр[~E/(RT)\, связан¬
ным с уменьшением энергии активации. Поэтому необходимо од¬
новременно учитывать влияние двух параметров: температуры и
энергии активации.
Большая осторожность требуется при сравнении активности
ферментов и катализаторов других типов, так как каталитические
реакции могут осуществляться по совершенно различным меха¬
низмам. Перспективным путем исследований представляется со¬
поставление функций, выполняемых отдельными группами в ак¬
тивных центрах ферментов и в растворе, а также сравнение меха¬
низмов ферментативного и гомогенного катализа.
Оказалось, что изомеразы, гидролазы, лиазы, трансферазы яв¬
ляются кислотно-основными катализаторами, и большинство фер¬
ментативных реакций протекают по механизмам общего кислот¬
но-основного катализа.
Вопросы и задания для самоконтроля
1. Какие соединения получили название «ферменты»? Какова их при¬
рода?
2. С помощью каких типов структур характеризуют внутреннее уст¬
ройство ферментов?
3. Какое понятие используют для объяснения каталитического дей¬
ствия ферментов? Охарактеризуйте его с точки зрения состава, строе¬
ния, размеров.
4. Какой смысл вкладывают в термин «специфичность» фермента?
Перечислите различные типы специфичности действия ферментов.
5. Дайте определения следующих величин: единица количества фер¬
мента; удельная активность; молекулярная активность фермента.
6. Какие факторы определяют активность фермента?
7. Каким образом pH среды влияет на активность фермента?
8. Зависит ли активность фермента от температуры? Ответ аргумен¬
тируйте.
171
9. В чем заключается причина более высокой активности ферментов
по сравнению с неорганическими катализаторами?
10. Перечислите основные классы, на которые подразделяют фер¬
менты. Почему их классифицируют не по типу химического строения?
11. Какой механизм был предложен Михаэлисом для объяснения
ферментативной реакции с одним субстратом?
12. Приведите аналитический вывод уравнения Михаэлиса—Ментен
методом квазиравновесного приближения.
13. Получите это же уравнение, используя принцип стационарных
концентраций.
14. Проанализируйте уравнение Михаэлиса—Ментен и объясните
наблюдаемую на опыте закономерность перехода порядка реакции по
субстрату от единицы до нуля.
15. Поясните физический смысл Км. Как называется эта константа и
при каком условии Км- Ks— субстратной константе? Характеризует ли
Км сродство фермента к субстрату?
16. В каком случае кинетический параметр vm2tx играет роль фундамен¬
тального параметра?
17. Какую величину называют числом оборотов фермента?
18. Для определения параметров Км и vmax охарактеризуйте три гра¬
фических способа:
а) б) [S]0; в) Vo, fg-.
V0 ь0 v0 IpJo
19. Почему значения Км и z;max, полученные разными способами, по¬
лезно сравнивать между собой?
20. Всегда ли линейная зависимость, например, в координатах (^о_1>
[S]0-1) означает, что реакция протекает по простейшему механизму, пред¬
ложенному Михаэлисом и Ментен? Ответ поясните.
21. Какие вещества называются ингибиторами? Перечислите возмож¬
ные типы обратимого ингибирования.
22. В каком случае ингибирование называется конкурентным? Ука¬
жите схематически механизм этого процесса, и выведите уравнение
для начальной скорости ферментативной реакции в присутствии инги¬
битора.
23. Для конкурентного ингибирования покажите:
а) каким образом эффективная константа КМэ§ связана с Км\
б) каким путем можно определить Kf — константу ингибирования;
в) можно ли графически определить величины Км, vmax, Км эф и Kf на
основании опытных данных в отсутствие и в присутствии ингибитора?
Ответ поясните.
24. Какое ингибирование считают неконкурентным? Укажите схема¬
тически механизм этого процесса и выведите уравнение для начальной
скорости ферментативной реакции в присутствии ингибитора.
25. Для неконкурентного ингибирования покажите:
а) какова связь между vmax и vmax эф — эффективной скоростью ре¬
акции;
б) выражение для определения константы ингибирования, Kj\
в) графический способ определения Км, vmax, vmax зф и К/.
172
26. Какое ингибирование называют бесконкурентным? Укажите ме¬
ханизм такого процесса и получите уравнение для начальной скорости
ферментативной реакции с участием ингибитора.
27. В случае бесконкурентного ингибирования покажите:
а) какова связь между Км и КМэф;
б) как связаны между собой vmax и vmax эф;
в) каким путем можно определить К7;
г) графический способ определения vmax, ^тахэф, Км, КМэф и Kj.
28. В каком случае ингибирование называется смешанным? Укажите
механизм этого процесса и выведите уравнение для начальной скорости
ферментативной реакции с участием ингибитора.
29. Для смешанного ингибирования покажите:
а) какова взаимосвязь между vmax эф и vmax;
б) как связаны между собой Км и КМэф\
в) графический способ определения vmax, i;max эф, Км.
30. Что представляет собой самоингибирование? Какой вид имеет в
этом случае зависимость начальной скорости реакции от концентрации
субстрата?
ГЛАВА 9. КИСЛОТНО ОСНОВНОЙ КАТАЛИЗ
9.1. Теории кислот и оснований
Реакции в растворах часто ускоряются в присутствии веществ,
относящихся к различным типам кислот и оснований: Аррениу¬
са, Бренстеда, Льюиса и Усановича.
По определению С. Аррениуса (1884 г.), кислота — это соеди¬
нение, образующее при диссоциации в водном растворе ионы во¬
дорода Н+; основание — это соединение, образующее при диссо¬
циации в водном растворе гидроксид-ионы ОН-.
Одним из наиболее важных доказательств соответствия модели
Аррениуса опыту оказались каталитические свойства кислот, ко¬
торые теория электролитической диссоциации связала с концен¬
трацией ионов водорода. К недостаткам теории Аррениуса следует
отнести ее ограниченность, поскольку она применима только к
водным растворам, исключая реакции, протекающие в органи¬
ческих растворителях и в газовой фазе.
По Дж.Бренстеду и Т. Лоури (1923 г.), кислота — это вещество,
являющееся донором протона, а основание — вещество, являющееся
акцептором протона. Кислота и основание, связанные уравнением
НА Н+ + А-
(9.1)
Кислота Протон Основание
173
называются сопряженными. Протон в растворе обычно соединяется
с молекулами растворителя, например в воде: Н++ Н20 -> Н3(Э+. Если
молекулы растворителя не способны ни присоединять, ни отда¬
вать протоны, то растворенное вещество не проявляет ни кислот¬
ных, ни основных свойств, а сам растворитель называется апро-
тонным.
Раствор кислоты в воде — наглядный пример системы, состо¬
ящей из двух пар сопряженных кислот и оснований:
НА + Н20 Н30+ + А"
(9.2)
Кислота Основание Кислота Основание
первая сопряженная пара — НА и А', вторая — Н30+ и Н20; кис¬
лотами и основаниями, как это видно из примера, могут быть не
только молекулы, но и ионы.
С позиций протонной теории определение понятия «основа¬
ние» сильно отличается от определения Аррениуса: не требуется
ни особого типа ионов, ни особого растворителя. Основаниями
являются не только вещества, содержащие ионы гидроксила, но
и такие, как пиридин, аммиак и др.
Отличие протонной концепции кислоты от теории Аррениуса
состоит в том, что, по Бренстеду, кислота сохраняет свои свой¬
ства и в присутствии, и в отсутствие растворителя. Таким обра¬
зом, протонная теория Бренстеда существенно расширила область
кислотно-основных реакций.
По Г.Льюису (1938 г.), кислота — это акцептор неподеленной
пары электронов, а основание — это вещество, являющееся до¬
нором электронной пары, которая может быть использована для
образования устойчивой электронной конфигурации другого ато¬
ма. На примере реакции
S03 + Н20 H2S04 (9.3)
видно, что Н20 является основанием, так как обладает свобод¬
ной парой электронов, a S03 — кислотой, поскольку использует
эту электронную пару.
Кислотами Льюиса являются такие соединения, как FeCl3,
А1С13, А1Вг3, BF3; основаниями — NH3, N2H4, NO5, С6Н6, C6H5N.
Было установлено, что кислотно-основные реакции Льюиса та¬
кого типа действительно могут быть проведены до конца в при¬
сутствии окрашенного индикатора так же, как титрование про¬
тонных кислот.
Теория Льюиса гораздо шире, чем предыдущие теории, одна¬
ко она имеет и ряд недостатков:
неубедительная трактовка протонных кислот, таких как H2S04
и НС1, поскольку они не могут присоединять электронную пару с
образованием ковалентных связей;
174
несостоятельность в решении вопроса о силе кислот, посколь¬
ку, по Льюису, это зависит от особенностей реакции.
Примером, в котором кислоты Льюиса являются лучшими ка¬
тализаторами, чем протонные кислоты, является реакция депо¬
лимеризации паральдегида в эфире.
При изучении гетеролитических реакций обычно используют
термины: «электрофильность» и «нуклеофильность» для характе¬
ристики реакционной способности веществ. Частица, являющая¬
ся донором пары электронов, называется нуклеофилом; другая
частица называется электрофилом, так как выступает в качестве
акцептора этой пары электронов.
По М. И.Усановичу (1939 г.), кислота — это вещество, спо¬
собное отдавать катионы, соединяющиеся с анионами или элек¬
тронами, или нейтрализующее основание с образованием соли.
Основание — вещество, способное отдавать анионы или электро¬
ны, соединяющиеся с катионами, или нейтрализующее кислоты
с образованием соли.
Можно считать, что теория Усановича является самой общей
из всех изложенных выше теорий, она включает также и окисли¬
тельно-восстановительные реакции как особый класс кислотно¬
основных реакций. Однако объединять кислотно-основные и окис¬
лительно-восстановительные процессы на практике мало продук¬
тивно, их лучше разделять.
В теории Усановича обращается большое внимание на степень
координационного насыщения центрального атома в соединении,
а также учитывается влияние его положения в Периодической
таблице Д. И. Менделеева. Например, центральным атомом в ди¬
оксиде серы является сера, которая координационно ненасыще-
на, поэтому способна присоединить анион, например О2', т.е.
проявляет кислотные свойства.
9.2. Типы кислотно-основного катализа
В зависимости от природы катализатора различают несколько
типов катализа.
Специфический кислотный катализ. В этом случае катализатора¬
ми являются кислоты Аррениуса, т.е. ионы Н30+, реакция проте¬
кает по схеме
S + H30+^=|^SH++H20—►Р+Н-.СР (9.4)
Первая стадия, связанная с внедрением протона в реагирую¬
щую часть молекулы субстрата (S), протекает быстро. Вторая ста¬
дия, в которой образовавшийся катион SH+ отщепляет протон с
образованием продукта (Р), протекает медленно и является ли¬
митирующей стадией всего процесса.
175
По этому механизму протекают реакции гидролиза сложных
эфиров и ацеталей, гидратации ненасыщенных альдегидов и ал-
кенов, дегидратации третичных спиртов, кето-енольной изоме¬
ризации, инверсии сахарозы.
Общий кислотный катализ. Его отличие от специфического кис¬
лотного катализа состоит в том, что катализаторами процесса яв¬
ляются кислоты Бренстеда (НА), т.е. донором протона не являет¬
ся ион Н30+; реакция в этом случае протекает по схеме
S + HA—> SH++A~—~^ Р+НА (9.5)
Причем медленной стадией является не распад иона SH+, а его
образование, т. е. лимитирующей является первая стадия процесса.
По этому механизму протекают реакции переноса протона
к атому углерода (разложение эфиров диазоуксусной кислоты, гид¬
ратация 4-метокси-а-метилстирола), отрыва протона от атома уг¬
лерода (кето-енольные переходы), присоединения к карбониль¬
ной и карбоксильной группе (образование сложных эфиров и ос¬
нований Шиффа).
Электрофильный катализ. В этом случае катализаторами явля¬
ются кислоты Льюиса. Объяснение роли кислот Льюиса как ката¬
лизаторов связывают с образованием ими за счет донорно-акцеп-
торной связи промежуточного соединения с одним реагентом,
которое более легко вступает в реакцию с молекулами второго
реагента благодаря наличию областей с повышенной или пони¬
женной электронной плотностью.
По этому механизму протекают реакции Фриделя—Крафтса
(алкилирования ароматического кольца в среде неполярного ра¬
створителя в присутствии А1С13), алкилирования бензола ал-
килгалогенами, хлорирования ароматического кольца, гидролиза
сложных эфиров аминокислот в присутствии катионов переходных
металлов.
Специфический основной катализ. Катализаторами являются ос¬
нования Аррениуса, т. е. гидроксид-ионы ОН'. Промежуточное со¬
единение является анионом и образуется либо присоединением
иона ОН", либо путем отрыва протона от молекулы воды или
другого реагента. Реакция протекает по схеме
SH + 01Г S~ + Н2° S* + 0Н~—+ 0Н ’ (9-6)
где S* — новое промежуточное соединение, причем первая ста¬
дия является быстрой, но вторая — медленной, т.е. лимитирую¬
щей весь процесс.
По этому механизму протекают реакции гидролиза сложных
эфиров, гидратации альдегидов, альдольной конденсации, клас¬
сические органические перегруппировки Кляйзена, Михаэля,
Перкина.
176
Общий основной катализ. Отличие его от специфического ос¬
новного катализа состоит в том, что катализаторами процесса вы¬
ступают основания Бренстеда (В), т.е. акцептором протона не яв¬
ляется ион ОН-. Другое отличие заключается в том, что медлен¬
ной стадией процесса является образование промежуточного
активного аниона по схеме
SH + B —^—> S'+BH+ > S*+B быстрая * Р+В (9-7)
По этому механизму протекают реакции гидролиза эфиров,
конденсации альдегидов, разложения нитрамида в водном растворе
карбоновых кислот.
Нуклеофильный катализ. В этом случае катализаторами являют¬
ся основания Льюиса, т.е. доноры электронных пар. Классичес¬
ким примером реакции данного типа является гидролиз сложного
эфира в присутствии имидазола. К нуклеофильным катализаторам
относятся также анионы CN", СЮ", N02", амины R3N, R2NH,
RNH2.
Принято обозначать нуклеофильное бимолекулярное замеще¬
ние символом yN2, а нуклеофильное замещение, протекающее
по типу мономолекулярной реакции, символом 5N1
Если в каталитической реакции одновременно участвуют:
кислота й основание Аррениуса, ее называют реакцией кис¬
лотно-основного катализа;
кислота и основание Бренстеда — это реакция общего кислот¬
но-основного катализа;
кислота и основание Льюиса — в этом случае имеем пример
реакции электрофильно-нуклеофильного катализа.
В особом случае, когда общий кислотно-основной или элект-
рофильно-нуклеофильный катализ осуществляется одновремен¬
но путем тримолекулярного столкновения, речь идет о механиз¬
ме, называемом пуш-пульным.
9.3. Кинетика реакций кислотно-основного катализа
Рассмотрим наиболее общий случай, когда реакция катализи¬
руется одновременно кислотами и основаниями Аррениуса (Н30+,
ОН") и Бренстеда (Н20).
Механизм такой реакции, состоящий из нескольких стадий,
можно представить следующим образом:
SH + Н30+ —HSH+ + Н20 (9.8)
SH + ОН" —» S" + Н20 (9.9)
SH + Н20 —HSH+ + ОН" (9.10)
12. Зак. 1596 J77
SH + H20 —£—» s- + н30+
(9.11)
Реагент SH участвует одновременно в четырех реакциях, отве¬
чающих разным типам катализа. Скорость расходования реагента
SH запишется так:
= (£3 + yfc4)[H20][SH] + yfc,[H30+][SH] + fc2[OH-][SH], (9.13)
Положим, что:
(к}+ £4)[Н20] =£0, где к0 — константа скорости реакции взаи¬
модействия реагента с водой (эту стадию часто рассматривают
как некаталитическую);
/:,[Н30+] = ^н+[Н+], где &н+ — константа скорости реакции, ка¬
тализируемой ионами водорода;
&2[ОН-] = &он-[ОН-], где кон— константа скорости реакции,
катализируемой ионами гидроксила.
Кажущаяся константа скорости каталитической реакции в итоге
примет вид
С учетом ионного произведения воды, [Н+] [ОН-] = А^о пре¬
дыдущее соотношение можно представить следующим образом:
где к'он-= к0Н-КН20( С0)2; С0 — стандартная концентрация.
Это общее уравнение позволяет рассмотреть все случаи ката¬
лиза ионами водорода и гидроксила. Различные возможности дан¬
ного типа реакций были подробно изучены давно (А.Скрабал,
1927 г.) и представлены на рис. 9.1.
Наиболее общий случай (а) представлен линией, указываю¬
щей на три различные области pH, характеризуемые прямыми
отрезками, наклон которых соответственно равен -1, 0 и 1. В за¬
висимости от значения pH наблюдается преобладание либо спе¬
цифического кислотного, либо основного катализа, в промежу¬
точной области преобладает каталитическое действие воды. По дан¬
ному типу катализа протекает реакция мутаротации глюкозы.
В случае (в) каталитическое действие воды происходит очень
медленно по сравнению с реакциями, катализируемыми ионами
VSH = VX + v2 + v2 + v4
(9.12)
или
^каж~ ^0+^H+ [Н+] + fcoH'[OH ].
(9.14)
In I
(9.15)
178
Рис. 9.1. Зависимость от pH логарифма кажущейся константы скорости
реакций (a—f), катализируемых кислотой и/или основанием
Н+ и ОН", горизонтальный участок кривой исчезает: существуют
только два отрезка прямой (кривая Ь), которые пересекаются в
точке, соответствующей минимальному значению кажущейся кон¬
станты скорости. Точке минимума отвечают значения
[Н+]
^0Н~^Н20
кн+
1/2
И pH = ilg
^он-^н2о
(9.16)
Этому типу катализа отвечает реакция галогенирования ацетона.
Кривая (с) относится к реакции, катализируемой только ионами
ОН" в растворах, когда pH имеет повышенные (> 7) значения.
Кривая, (d) представляет случай, когда реакция всегда катали¬
зируется ионами гидроксила (ОН").
Кривая (е) описывает случай реакции, катализируемой ионами
водорода в растворах, когда pH имеет достаточно малые значения.
Кривая (/) представляет случай, когда реакция всегда катали¬
зируется ионами водорода.
Рассмотрим классический пример реакции галогенирования
ацетона, детально исследованной В. Оствальдом в 80-х гг. XIX в.:
СН3СОСН3+Х2= СН3СОСН2Х + НХ (9.17)
Реакция в кислой среде. Экспериментально установлено, что ско¬
рость реакции в этом случае пропорциональна концентрации аце¬
тона и ионов гидроксония, но не зависит от концентрации гало¬
гена Х2, поэтому был сделан вывод: Х2 не участвует в промежу¬
точных стадиях процесса, в том числе и в самой медленной,
определяющей скорость реакции в целом. Ацетон существует в ке-
тонной и енольной формах, причем первая является преобладаю¬
щей. Процесс перехода кетонной формы в енольную, с которой
быстро взаимодействует Х2, осуществляется в два этапа. На пер¬
вой стадии происходит быстрое и обратимое присоединение про¬
тона к карбонильному кислороду с образованием промежуточно¬
го соединения (YH+) по схеме
ОН
СН3СОСН3 + Н30+ —4 СН3ССН3 + Н20 (9.18)
YH+
Запишем выражение для константы равновесия
^ [YH+]
1 [кетон][Н30+] (919)
На последующих стадиях образуется енольная форма путем от¬
рыва протона от одной из метальных групп промежуточного со¬
единения YH+ с помощью различных форм оснований, выступа¬
ющих в качестве катализатора:
ОН
, I
YH+ + ОН" —^ СН3С:СН2 + Н20
ОН
YH+ + Н20 —2—з сн3с:сн2 + Н20+ (9.20)
ОН
YH+ + А" —СН3С:СН2 + НА
Скорость медленного этапа образования енола определяется сум¬
мой скоростей трех стадий
d[C”0Jl] = £2[YH+] [ОН] + кг[ YH+] + к4[ YH+] [А-]. (9.21)
at
Подставляя в это соотношение концентрацию соединения YH+,
выраженную через Кх (см. 9.19), получим
d[C”0Jl]- = ^,[кетон][Н30+]{А:2[ОН-]+А:з+^4[А"]}. (9.22)
а;
Обозначив КНг0 = [Н30+][0Н_] и Ка[НА\ = [Н30+][А"], прихо¬
дим к конечному уравнению скорости для медленного этапа об¬
разования енольной формы
^^ = №етон]{А:2*Н20 +А:3[НзО+]+^[НА]}. (9.23)
180
Последний этап реакции — взаимодействие Х2 с енольной
формой
СН3С(ОН):СН2+ Х2—СН3СОСН2Х + НХ (9.24)
протекает очень быстро для Х2=Вг2 и 12, поэтому общая скорость
реакции галогенирования определяется лимитирующей стадией
образования енольной формы, т.е. уравнением (9.23)
d[CH3C°CH2X1 = [СН3СОСН3]{£0+ кн + [Н30+] + £на[НА] } (9.25)
где [кетон] = [ацетон], /с0=к2К1 KHl0 , kH, = k3K{ и k^-k^K^
Проанализируем полученное уравнение. Константа к0 относит¬
ся к некаталитической реакции, более точно она представляет
вклад растворителя в реакцию катализа, так как стадии 1 и 2 мо¬
гут протекать даже при pH = 7. Образование енола по стадии 2 не
зависит от pH, как это следует из уравнения (9.20).
Слагаемое &н+ [Н30+] представляет вклад стадии 3 механизма
образования енола и относится к специфическому кислотному ка¬
тализу, в то время как /сНа Г НА] характеризует общий кислотный
катализ и вклад стадии 4 с участием сопряженного основания А".
Опытное определение констант скорости ко, кн+ и кнА ослож¬
нено наличием среди продуктов реакции сильной кислоты НХ.
При отсутствии слабой кислоты, НА, кажущаяся константа ско¬
рости из (9.25) принимает вид
£каж=£о+ *нЛН3°+]. <9-26)
Строя график в координатах (ккаж, [Н30+]), по отрезку, отсека¬
емому на оси ординат, находят ко и по наклону прямой, кн+.
Если полагают, что катализатором является слабая кислота,
опыты проводят с буферными растворами НА/А" различной кон¬
центрации при постоянном значении pH. Зависимость опытной
(кажущейся) константы скорости от концентрации будет иметь
вид либо прямой, параллельной оси абсцисс, указывая этим на
отсутствие катализа, либо прямой, положительный наклон кото¬
рой равен &НА-
Реакция катализируется основаниями. Механизм катализа будет
иным, чем в кислотном катализе. Определяющим для скорости,
т. е. самым медленным, является этап отрыва протона от молеку¬
лы ацетона с участием различных форм оснований:
СН3СОСН3+ОН'—СН3СГ0СН2+Н20
СН3СОСН3+Н20 —7—> СН3С'ОСН2+Н30+ (9.27)
СН3СОСН3+ В" —»СН3СГОСН2+ вн
181
На втором этапе галоген быстро реагирует с образовавшимся
анионом
СН3С"ОСН2+Х2 СН3СОСН2Х + Х' (9.28)
Выражение для скорости процесса примет вид
г;= [СН3СОСН3]{&7+А;6[ОН~] + А:8[В"]}, (9.29)
где константы к7, к6, к% оцениваются способом, аналогичным опи¬
санному выше для идентификации констант скорости реакции,
катализируемой кислотами.
Кислотно-основной катализ. Если реакция катализируется толь¬
ко ионами Н+ и ОН-, используя уравнения (9.25) и (9.29), полу¬
чим единое выражение для кажущейся константы скорости ката¬
литической реакции
Лиж=Аь+ kH> [Н30+] + k0H- [ОН-] =
= *Ь+ V [Нз0+] + *ОН-*н2о [НзОТ1. (9.30)
Зависимость lg£Kax от pH на рис. 9.2 представляет собой кри¬
вую, аналогичную линии а на рис. 9.1.
При малых значениях pH в уравнении (9.30) преобладает вто¬
рой член, наклон участка прямой равен минус единице; при боль¬
ших pH, преобладает третий член, наклон прямолинейного участка
Рис. 9.2. Зависимость от pH логарифма кажущейся константы скорости.
Линейные участки соответствуют (слева направо) преобладанию каталитиче¬
ских механизмов под действием кислот, растворителя
и оснований
182
равен 1, между этими значениями pH доминирующим будет пер¬
вый член, в этом случае наклон прямой равен нулю, образуется
плато.
9.4. Солевые эффекты в катализе
Правило Оствальда (1884 г.): сила каталитического действия
кислот прямо пропорциональна их электропроводности. Этот вы¬
вод был сделан на основании установленной им корреляции между
возрастанием константы скорости реакции и увеличением кон¬
станты диссоциации слабой кислоты Ка. Причем все полностью дис¬
социирующие сильные кислоты обусловливали приблизительно оди¬
наковое возрастание константы скорости катализируемой реакции.
С. Аррениус открыл два новых эффекта.
Первичный солевой эффект: каталитическое действие кислоты
увеличивается при добавлении в раствор соли, не имеющей об¬
щего иона с этой кислотой.
Например, скорость инверсии тростникового сахара в присут¬
ствии уксусной кислоты существенно возрастает при добавлении
10% (молярная доля) хлорида натрия.
Объяснение этого явления заключается в том, что при добав¬
лении нейтральной соли увеличивается ионная сила раствора, со¬
ответственно и степень диссоциации электролита. Это приводит к
увеличению концентрации ионов гидроксония и константы ско¬
рости каталитической реакции.
Вторичный солевой эффект: каталитическое действие слабой кис¬
лоты также увеличивается при добавлении в раствор ее соли (со¬
держащей одноименный ион).
Первоначально считалось, что для слабой кислоты в качестве
катализатора прибавление соли с одноименным анионом должно
смещать равновесие реакции
НА+ Н20 А"+ Н30+
с константой равновесия
[НзО^[А1
[НА] ’
в сторону образования недиссоциированной кислоты и уменьшать
каталитическое действие.
Однако С.Аррениус (1893 г.) показал, что это уменьшение
существенно меньше ожидаемого по закону действующих масс.
Объяснение этого эффекта связано также с увеличением ионной
силы раствора при введении одноименного иона, что приводит к
росту скорости кислотного катализа.
183
Количественное объяснение солевых эффектов и зависимости
константы скорости реакции от ионной силы раствора связано с
использованием уравнений Бренстеда—Бьеррума и различных при¬
ближений теории Дебая—Хюккеля.
Рассмотрим реакцию в растворе между ионами, которая может
быть записана следующим образом:
А+В^=^АВ*——>Р, (9.32)
где А, В — ионы, АВ54 — активированный комплекс, Р — про¬
дукт.
Константа скорости этой реакции, согласно теории активиро¬
ванного комплекса, выражается уравнением
к = s^-K* = зг ^ . (9.33)
п п САСВ
В то же время термодинамическая константа равновесия акти¬
вированного комплекса и исходных веществ для неидеальных си¬
стем выражается через активности и имеет вид
К* =fABL = = Iabl; (9 34)
аАав САСВ YaYb YaYb
где dj — активность соответствующих частиц; С, — концентра¬
ции; у, — коэффициент активности.
Подставляя в формулу (9.33) выражение для К* из формулы
(9.34), получаем искомое уравнение Бренстеда—Бьеррума, кото¬
рое в наиболее общей форме показывает причины отличия реак¬
ций в газе и растворе:
= = , (9.35)
h Yab* W
где ко — константа скорости при бесконечном разбавлении, когда
отношение коэффициентов активности равно единице.
Прямые количественные расчеты по уравнению (9.34) прово¬
дить нельзя, так как из-за отсутствия сведений о структуре и свой¬
ствах активированного комплекса в растворах нет данных о его ко¬
эффициенте активности. Однако уравнение (9.34) позволяет уста¬
новить зависимость скорости реакции от различных свойств среды,
влияющих на коэффициенты активности (химической природы
частиц, их заряда, размеров и диэлектрической проницаемости).
Согласно первому приближению теории сильных электроли¬
тов Дебая —Хюккеля, уравнение для определения среднего коэф¬
фициента активности у± имеет вид
lgY± =-zAzBhJI, (9.36)
184
где zA, zB — заряды ионов; / = -^Cjzf — ионная сила раствора;
1,826 -106
С, — концентрации ионов; А — постоянная, равная ’—-—3^-- ;
ё — диэлектрическая проницаемость растворителя.
Для реакции (9.32) при взаимодействии между ионом А с за¬
рядом zA и ионом В с зарядом zB образуется активированный ком¬
плекс с зарядом zA+zB.
После логарифмирования уравнения (9.35) и подстановки в
него значений коэффициентов активности из (9.36), получим
Для водного раствора при 25 °С постоянная А равна 0,51, сле¬
довательно, выражение (9.37) примет вид
Таким образом, при реакции между ионами в разбавленных
растворах логарифм константы скорости является линейной фун-
(9.37)
lgfAl = l,02zAzBV7.
/Сл
V и У
(9.38)
lg*A0
1
ОД
о
-0,1
3
Рис. 9.3. Влияние заряда ионов на константу скорости:
1 — при zAzB = 4; 2 — при zAzB = 0; 3 — при zAzB = -1
185
кцией квадратного корня из ионной силы, как это показано на
рис. 9.3.
Сущность вторичного солевого эффекта заключается в том, что
посторонние электролиты, уменьшая коэффициенты активности
ионов, увеличивают степень диссоциации слабого электролита,
что приводит к росту концентрации иона, принимающего учас¬
тие в реакции.
При увеличении ионной силы, особенно в присутствии мно¬
гозарядных ионов, линейная зависимость lg(k/ko) от 41 наруша¬
ется, поэтому полезно использовать при расчетах второе и третье
приближения теории Дебая —Хюккеля.
9.5. Корреляционные соотношения в катализе
Уравнение Бренстеда. Для реакций общего кислотно-основного
катализа, катализируемых кислотами или основаниями одного го¬
мологического ряда, Дж.Бренстед и JI. Педерсон (1925 г.) первы¬
ми предложили корреляционные уравнения между константами
скорости и константами ионизации и основности соответствую¬
щих кислот и оснований в растворе.
где кНА и кь — константы скорости кислотного и основного ката¬
лиза; Са и а — константы, а обычно < 1, для гомологического ряда
кислот; Съ и Р — константы, (3 обычно < 1, для гомологической
серии оснований; Каи Кь — константы ионизации кислот и основ¬
ности оснований.
Соотношения (9.39, 9.40) могут быть представлены в логариф¬
мической форме:
Чтобы исключить из рассмотрения величину постоянной С,
обычно проводят сравнительный анализ, используя эталонное или
«отсчетное» состояние катализирующих процесс с одним субстра¬
том кислот или оснований.
Для серии различных кислот и оснований получим
кнА=СаК:
kb=CbKl
(9.39)
(9.40)
1пА:на= lnQ+alnA'o,
\nkb = lnQ+pinA),.
(9.41)
(9.42)
(9.43)
kx Кх
1 V 1 /
186
или
In — = a In
к\
(9.44)
Таким образом, зная рКа для данной кислоты и используя урав¬
нения (9.43) и (9.44), можно рассчитать константу скорости и
скорость каталитической реакции без проведения эксперимента.
Важная роль, которую уравнение Бренстеда сыграло в кинетике и
катализе, обусловлена следующими причинами:
это уравнение позволяет (хотя и приближенно) перейти к ана¬
лизу взаимосвязи энергий активации и тепл от реакций, что явля¬
ется важной проблемой теории элементарных актов химических
превращений;
по аналогии с этим уравнением, на его основе или независимо
от него предложены другие корреляционные соотношения в фи¬
зической органической химии (например, уравнение Гаммета).
Используя аппарат термодинамики и теории активированного
комплекса, нетрудно показать, что константа скорости и кон¬
станта ионизации кислоты связаны с соответствующей энергией
Гельмгольца (поскольку речь идет только о реакциях в конденси¬
рованных фазах, когда AG совпадает с AF):
где ДF* — энергия Гельмгольца активации; ДF0 — энергия Гельм¬
гольца диссоциации кислоты НА.
После последовательного логарифмирования уравнений (9.45),
(9.46), подстановки полученных выражений в (9.41) и выделения
постоянных величин в отдельное слагаемое получаем корреляци¬
онное соотношение
т.е. уравнение связи между энергией Гельмгольца активации ре¬
акции (ДF*) и энергией Гельмгольца (ДF0), 0 < а < 1.
Уравнение (9.47) является достаточно точным и иногда вы¬
полняется при изменении К: на 10 порядков в реакциях общего
кислотного и общего основного катализа.
Существенно менее точным является корреляционное соотно¬
шение между энергиями активации и тепловыми эффектами ре¬
акций общего кислотно-основного катализа, хотя первоначально
именно этот тип энергетических соотношений считался наиболее
(9.45)
и
(9.46)
AF*= const + аД F°;
(9.47)
187
важным. При постоянной энтропийной составляющей это урав¬
нение связи имеет вид (а< 1)
Еа-const + аАЯ; (9.48)
Аналогичные уравнению (9.48) корреляционные соотношения
были предложены и для других типов реакции. Д. Хориути и М. По-
ляни (1932 г.) при изучении замедленного разряда Н30+ на раз¬
ных катодах получили уравнение
£ = 0,5JFA<p = 0,5Q, (9.49)
где F — число Фарадея; Д<р — потенциал электрода; Q — теплота
процесса.
Н.Н.Семенов (1955 г.) после обобщения большой экспери¬
ментальной базы данных по взаимодействию радикалов, атомов
с валентно-насыщенными молекулами для реакций типа А + ВС
= = АВ + С предложил следующие приближенные соотношения
между энергией активации Е и теплотой реакции Q:
Е = 48,1-0,25(9 (9.50)
для экзотермической реакции, Е и Q в кДж/моль, и
Е= 48,1+0,750 (9.51)
для эндотермической реакции.
Следует отметить, что сама форма записи соотношения (9.50)
указывает на ограниченность области его применения: при Q>
> 192,5 кДж энергия активации становится отрицательной, что не
имеет физического смысла. Н. Н. Семенов подчеркивал фундамен¬
тальное значение для химической кинетики соотношений Брен-
стеда и Поляни.
Несмотря на указанную выше ограниченность корреляцион¬
ных соотношений, справедливых лишь в пределах отдельных групп
реакций, соотношение Бренстеда имеет большое значение для
явлений катализа, поскольку служит пока основным средством
предвидения изменения скорости реакции при вариации состава
и природы катализатора.
По Г. К. Борескову, это соотношение позволяет найти связь
между скоростью каталитической реакции и энергией промежу¬
точного взаимодействия при катализе и свести задачу предвиде¬
ния каталитического действия к поискам зависимости энергии
промежуточного взаимодействия от химического состава ката¬
лизатора.
Уравнение Гаммета. Для многих реакций, например, для реак¬
ции гидролиза
АгС02СН3+0Н'^АгС02+СН30Н (9.52)
возможно замещение отдельных атомных групп доноров или ак¬
цепторов электронов в различных участках ароматического коль¬
188
ца без нарушения реакционного процесса. Объяснение изменений
константы скорости в этих случаях связывают с эффектом влия¬
ния заместителей на изменение энергии активации реакции, ко¬
торое зависит от природы и положения атомов заместителей.
Обычно равновесные измерения проводят быстрее и более точ¬
но, чем кинетические. Поэтому более удобно оценить эффект вли¬
яния различных заместителей на константу диссоциации для ре¬
акции (9.52):
jQ^-C02H^^ jQ^-C02 + H+ (9.53)
X X
Л.Гаммет в 1935 г. сформулировал правило: эффект влияния
заместителя X на изменение энергии Гиббса реакции (9.53) ли¬
нейно связан с его эффектом влияния на энергию активации ре¬
акции (9.52); следовательно существует корреляция между рКа дис¬
социации кислоты и константой скорости реакции гидролиза.
Выразим параметр замещения следующим образом:
5* = lg К* - lg К° = РК2 - рЦ, (9.54)
где и Kg — константы ионизации незамещенной бензойной
кислоты и Л'-замещенной кислоты.
В этом случае уравнение Гаммета примет вид
Шх/к0) = р8х (9.55)
или
\gkx = \gkQ+p8x, (9.56)
где /cq — константа скорости реакции, протекающей с незаме¬
щенным соединением; кх — константа скорости реакции с X-за¬
мещенным соединением; р — постоянная, параметр реакции, ко¬
торый находят по наклону прямой, строя график зависимости
lgкх от Ьх. Смысл параметра р реакции можно лучше понять, ком¬
бинируя уравнения (9.54) и (9.55):
Шх/ко)
p~-wiiky <9-57)
Таким образом, р — это отношение эффекта влияния X на
уменьшение энергии активации реакции (9.52) к эффекту, про¬
изводимому в реакции (9.53) на понижение энергии Гиббса.
Значение и Знак констант 8Х и р характеризуют тип реакцион¬
ного превращения, позволяют оценить скорость протекания ре¬
акции. Например, рассчитав на основе известных для бензойных
кислот значений параметров замещения Зд-постоянную р для кон¬
кретной реакции, можно затем использовать это значение для
нахождения других 8* констант. Уравнение Гаммета применимо
только к ароматическим системам.
189
Уравнение Тафта. Корреляционное соотношение, внешне ана¬
логичное уравнению Гаммета, было предложено Р. Тафтом для
расчета констант скоростей реакций превращения алифатических
соединений в зависимости от индуктивного эффекта заместителя:
lg (кх/ко) = р*8|,
(9.58)
где ко — константа скорости для стандартного соединения, содер¬
жащего СН3 — группу; кх — константа скорости для Х-замещен-
ного соединения; р* — константа реакции; 8| — полярная кон¬
станта заместителя.
Зная величину 8$, которая является мерой электроноакцеп¬
торной способности заместителя, т. е. характеризует индуктивный
эффект, можно найти величину р* для различных серий реакций
по наклону прямой, построенной в координатах ( lg^, 8*).
Для нахождения значений 8* использовалась реакция основ¬
ного и кислотного гидролиза эфиров карбоновых кислот, так как
оказалось, что разница между отношениями lg(&rAo) ДЛЯ этих
типов гидролиза пропорциональна индуктивному эффекту, т. е. 8*.
Тафт для расчета константы 8* использовал соотношение
Ц =
2,5
lg
У / N
-lg
fk, '
уосн
h
Укисл.
(9.59)
На основании полученных значений 8| и р* и уравнения (9.59)
можно сравнивать скорости реакций для Х-замещенного соедине¬
ния реакционной серии и стандартного соединения.
9.6. Функция кислотности Гаммета
JI. Гаммет предложил и разработал метод сравнения сильных
протонных кислот в водных и неводных средах с помощью инди¬
каторной шкалы кислотности.
Запишем реакцию ионизации индикатора — основания В:
В + Н30+ ВНЧ Н20 (9.60)
для которой константа основности субстрата имеет вид
К„ = йвн'а»2° . (9.61)
аВаН30+
Выразим отношение концентраций протонированной и непро-
тонированной форм субстрата, используя соотношение (9.61):
[ВН+] „ °н3о+ VB „ ,
где множитель
= (9.63)
ан:о Ybh*
где h0 — это кислотность среды, которая характеризует ее способ¬
ность передавать протон реагенту В.
Величина
tf0=-lgA0 (9-64)
называется функцией кислотности Гаммета. Обычно используют
следующую форму записи этой функции:
<9-65>
где В — индикаторное основание, ВН+ — его протонированная
форма; рКвн+= -IgK для диссоциации ВН+.
[ВН+]
Соотношение ГР1 можно измерить спектрофотометрически.
|bj
Если применять основания с очень низкой основностью (отрица¬
тельные значения рК), то можно расширить шкалу Н0 в область
отрицательных значений.
В разбавленных растворах, когда Ув=Увн* = 1, %2о = 1 и аН}0+ =
= CHj0+, из соотношений (9.63) и (9.64) следует, что
К ~ 6^н3о+ и Я0 = pH.
Функция Гаммета является аналогом pH в области значений
О —14 и за пределами этого интервала характеризует протонирую-
щую способность среды. В концентрированных растворах кислот
значения Н0 могут существенно отличаться от pH, о чем свиде¬
тельствуют следующие примеры:
кислота ... H2SO4(50%) H2S04(97%) НР(жидк.) H2S207 HS03F
Н0 -3,4 -9,0 -11,0 -14,1 -15,0
Смесь HS03F и SbF5 в молярном соотношении 1:1 называют
магической кислотой. Самое высокое значение Щ получено для
HS03F при содержании 7% SbFs: Я0=-19,4.
Способность SbF5 повышать кислотность HS03F в основном
обусловлена равновесием
2HS03F+SbF5=H2S03F++ SbF5(S03F)“ (9.66)
Сверхкислоты используют для разных целей, чаще всего для
Протонирования молекул, которые в обычных условиях не являют¬
ся основаниями, например алифатических углеводородов, алканов.
191
Функция кислотности Гаммета Н0 и соответственно h0 могут
быть использованы при описании кинетики специфического кис¬
лотного катализа. В этом случае реакция протекает по схеме (9.4)
S + H30+^=!f=±SH++H20—k-эР +Н30+
Скорость реакции определяем по скорости образования про¬
дукта
v = ^p = *2[SH+]- (9.67)
Для нахождения концентрации промежуточного соединения SH+
используем соотношение (9.62) в виде [SH+] = XiA0[S] и уравнение
материального баланса по субстрату
[S]0= [S] + [SH+]. (9.68)
После подстановки [S] = [S]0- [SH+] в (9.62) и несложных пре¬
образований получим
[5Н.] = М!!а •
1 + АьПо
Выражение для скорости реакции (9.67) с учетом (9.69) при¬
нимает вид
_ k2Kbho[S]o _ , г™
"" Т7ТТ ” (9.70)
I + АьПц
где
, k2Kbh0 к2
\ + Kbh0~ \ + (КМ-] (9’71)
кажущаяся (опытная) константа скорости реакции.
Для определения констант к2 и Кь удобно использовать графи¬
ческий способ: соотношение (9.71) запишем в обратных величи¬
нах
1111
*каж k2+k2Kbh0• (9-72)
1 1
Строя зависимость (9.72) в координатах (т—>т“), находим
^каж "0
по величине отрезка, отсекаемого на оси ординат, значение ~г,
1 *2
а по тангенсу угла наклона прямой — значение , v , затем —
к2аь
численные значения каждой из констант к2 и Кь.
Рассмотрим два предельных случая для соотношения (9.71).
192
Если Ayi0« 1, тогда кклж=к2КьИ0 и, логарифмируя последнее
выражение, получим
1ё^каж= Ык2Кь) + lgh0=lg (k2Kb) -Н0. (9.73)
Графическое представление зависимости (9.73) в координатах
(lg^Ka*> Но) позволяет по величине отрезка, отсекаемого на оси
ординат, найти только произведение величин к2Кь, но не каждую
из констант в отдельности.
Если Kbh0» 1, в этом случае
ккаж=к2. (9.74)
Вопросы и задания для самоконтроля
1. Какие соединения называются кислотами и основаниями Арре¬
ниуса? Приведите примеры. Перечислите недостатки теории электроли¬
тической диссоциации.
2. Дайте определения кислот и оснований по Бренстеду. Какие со¬
единения называют сопряженными? В чем заключается отличие протон¬
ной теории от теории Аррениуса?
3. Дайте определение и приведите примеры кислот и оснований Лью¬
иса. Каковы недостатки этой теории?
4. Сформулируйте определения кислот и оснований по Усановичу.
Приведите примеры.
5. Какой принцип классификации используют в кислотно-основном
катализе? Приведите механизмы катализа, характеризующие его различ¬
ные типы.
6. Какой вид имеет уравнение для кажущейся константы скорости
реакции кислотно-основного катализа? Проанализируйте различные слу¬
чаи катализа ионами гидроксония и гидроксила.
7. Представьте графически зависимость от pH логарифма кажущей¬
ся константы скорости для различных реакций, катализируемых кисло¬
той и/или основанием.
8. Укажите механизм процесса и выведите уравнение для общей ско¬
рости реакции галогенирования ацетона в кислой среде.
9. Укажите механизм процесса и выведите уравнение для общей ско¬
рости реакции иодирования ацетона, катализируемой основаниями.
10. В чем заключается правило Оствальда? На основании каких на¬
блюдений оно было установлено?
11. Дайте определение первичного солевого эффекта и его объяс¬
нение.
12. В чем заключается вторичный солевой эффект и объяснение этого
эффекта?
13. Выведите уравнение, описывающее зависимость константы ско¬
рости реакции от ионной силы раствора, используя уравнение Бренсте¬
да—Бьеррума и первое приближение теории Дебая —Хюккеля.
14. Приведите корреляционное уравнение Бренстеда и поясните смысл
входящих в него величин.
,3- Зак. 1596
193
15. Покажите, используя теорию активированного комплекса и ап¬
парат термодинамики, возможность получения различных «энергетичес¬
ких» корреляционных соотношений.
16. Приведите корреляционное уравнение Гаммета и поясните смысл
каждого из его членов.
17. Какой вид имеет уравнение Тафта? Поясните смысл каждой из
входящих в него величин и покажите, каким путем можно найти значе¬
ние константы реакции р*?
18. Дайте определения терминов: «кислотность среды» h0, «функция
кислотности Гаммета» Н0. Как связаны между собой hQ и [Н30+], Н0 и
pH? Какие значения может принимать величина Я0?
19. Выведите уравнение для описания кинетики специфического кис¬
лотного катализа, проанализируйте его и покажите, каким образом можно
определить значения входящих в это уравнение констант к2 и Кь.
20. Выведите уравнение для описания кинетики специфического ос¬
новного катализа, проанализируйте его и покажите, каким путем мож¬
но найти значения констант, входящих в это уравнение.
ГЛАВА 10. ГЕТЕРОГЕННЫЙ КАТАЛИЗ
10.1. Общие положения
Гетерогенными называются реакции, участники которых и ка¬
тализатор находятся в разных фазах.
В качестве катализаторов обычно используют твердые вещества
(металлы, оксиды, соли и т.п.) и изучают в основном системы
«газ—твердое тело» и «жидкость—твердое тело». Реже применя¬
ются жидкие катализаторы, которые представляют собой кисло¬
ты (H2S04 или Н3Р04), иногда растворы активных веществ в воде
или органических растворителях. Например, при синтезе S03 из
S02 и 02 в производстве серной кислоты используют в качестве
катализатора жидкий сульфованадат на поверхности носителя при
температуре 450 — 550 °С.
Каталитическое действие в гетерогенном катализе связывают с
наличием на поверхности твердых тел так называемых активных
центров, природа и число которых часто неизвестны.
Впервые этот термин ввел X. Тейлор (1925 г.), который, ана¬
лизируя явления отравления, указал, что не вся поверхность ка¬
тализатора однородна и каталитическая реакция протекает лишь
на определенных участках поверхности, названных им активными
центрами.
По X. Тейлору, активные центры образуются на тех местах по¬
верхности, где атомы слабее всего связаны с кристаллической ре¬
шеткой металла, т.е. обладают большей или меньшей степенью
координационной ненасыщенности.
194
В ряде случаев активные центры, в пересчете на единицу пло¬
щади, могут составлять только незначительную долю поверхнос¬
ти катализатора, например, 4% для некоторых алюмосиликатов.
Однако, как показано в работах Г. К. Борескова по каталитичес¬
кой активности платины в реакции окисления S02 и силикагеля в
реакции гидролиза хлорбензола, каталитическое действие может
быть присуще и всем атомам поверхности.
Понятие активного центра не относят к жидким катализато¬
рам, поскольку поверхность жидкости однородна и сами катали¬
заторы не столь чувствительны к действию ядов.
Согласно химическому подходу к сущности каталитического дей¬
ствия, Г. К. Боресков называет катализатором вещество, изменяю¬
щее скорость химической реакции своим участием в образовании
активированного комплекса одной или нескольких стадий хими¬
ческого превращения и не входящее в состав конечных продуктов.
При гетерогенном катализе механизм действия катализатора
аналогичен по своей сути его действию в гомогенной системе:
катализатор, благодаря промежуточному химическому взаимодей¬
ствию с реагирующими веществами, открывает новый энергети¬
чески выгодный реакционный путь с минимальным активацион¬
ным барьером. В отличие от гомогенного катализа при химичес¬
ком взаимодействии на поверхности твердого катализатора
происходит в силу ряда причин образование более сложных, час¬
то многоатомных, промежуточных соединений. К этим причинам
относятся:
физическая природа твердых катализаторов (они представляют
собой либо совокупность кристаллов — металлы, оксиды и т.д.,
либо макромолекулы — силикагель или органический полимер,
содержащие большое число атомов);
характер химического взаимодействия с реагирующими веще¬
ствами в результате которого не происходит полного разрыва свя¬
зей поверхностных атомов с остальными атомами катализатора;
неоднородность энергий связи при поверхностном взаимодей¬
ствии, обусловленная нарушениями регулярности в строении по¬
верхности, которые связаны с содержанием примесей или откло¬
нениями от стехиометрии основного состава катализатора.
Наблюдаемые на опыте изменения гетерогенных катализато¬
ров (их структуры и даже состава) вызваны взаимодействием с
примесями или с основными компонентами реагирующих веществ
и представляют собой побочные явления.
Очевидно, что эффективность гетерогенного катализатора оп¬
ределяется структурой и химическим составом поверхностного
слоя, соответствием между химическими свойствами катализато¬
ра и реагирующих веществ.
Отметим, что в первой половине XX в. наблюдалось отрица¬
тельное отношение к химическому подходу в катализе. В 20 — 30-е гг.
195
были предприняты попытки объяснить явления гетерогенного ка¬
тализа без учета химических свойств катализатора, главная роль
отводилась его различным физическим свойствам.
К числу этих представлений следует отнести так называемую
радиационную теорию катализа, электронную теорию, предло¬
женную JI. В. Писаржевским, взгляды X. Тейлора и Ф. Констебля
на неоднородность поверхности. Их работы получили широкое,
но одностороннее развитие, в результате чего появление катали¬
тических свойств стали связывать исключительно со структурны¬
ми нарушениями катализаторов.
Все эти взгляды, как и цепная теория катализа, предложенная
в 50-е гг. Н. Н. Семеновым и В. В. Воеводским, представляют в на¬
стоящий момент лишь исторический интерес. Подробнее их изло¬
жение и анализ различных теорий катализа приведены в моногра¬
фии Г. К. Борескова*.
В гетерогенном катализе за меру каталитической активности
системы А принимают изменение скорости химической реакции
при участии катализатора
Л^кат-г'оО-ф). (10.1)
где vKaT и v0 — скорости реакции соответственно в присутствии
катализатора и в его отсутствие; ср — доля объема системы, при¬
ходящаяся на катализатор и недоступная для реагирующих веществ.
В случаях, когда вторым слагаемым в (10.1) можно пренеб¬
речь, получаем
A=Vкат- (10.2)
Удельную активность гетерогенного катализатора а характери¬
зуют скоростью, отнесенной к единице его поверхности, S
a = (Ю.З)
Впервые на это было указано в работах Г. М. Панченкова и
К. В. Топчиевой, посвященных алюмосиликатным катализаторам.
Отметим, что активность связана прежде всего с величиной
удельной поверхности (м2/т) катализатора, но не с массой или
объемной концентрацией и возрастает, часто прямо пропорцио¬
нально поверхности, с его измельчением.
Для однотипных катализаторов при одинаковом механизме про¬
цессов отношение удельных активностей катализаторов конкрет¬
ной реакции можно принять равным отношению констант скоро¬
стей реакции.
В приближении равенства предэкспоненциальных множителей
в уравнении Аррениуса для однотипных реакций это отношение
принимает вид
* Боресков Г. К. Гетерогенный катализ. — М.: Наука, 1986.
196
^ ь
(Ех-Е2
RT
V
\
У
(Ю.4)
Важное место в теории и практике гетерогенного катализа за¬
нимает «правило Борескова» о приблизительном постоянстве
удельной каталитической активности веществ. Исследования пос¬
ледних десятилетий показали, что удельная каталитическая ак¬
тивность (для ряда однокомпонентных оксидных и металлических
катализаторов) не зависит от дисперсности активного компонен¬
та, степени его кристалличности и способа приготовления при
неизменном составе катализатора.
Правило Борескова, выполняющееся, как правило, для ста¬
дийного механизма при повышенных температурах, базируется
на представлениях о катализаторе и реагирующих веществах как
единой каталитической системе, в которой химические превра¬
щения претерпевают не только реагенты под влиянием катализа¬
тора, но и катализатор изменяет свой состав и свойства в резуль¬
тате взаимодействия с реагентами. В результате под воздействием
реакционной смеси устанавливается стационарный состав, опре¬
деляющий удельную каталитическую активность, которая не за¬
висит от исходного состояния поверхности катализатора, т.е. от
условий его приготовления и предварительной обработки. Так для
ряда металлов удельная каталитическая активность не изменяется
вплоть до размеров кристаллов больше 3 нм.
В случае многокомпонентных катализаторов каталитические
свойства часто обусловлены возникновением нового химического
соединения, поэтому правило постоянства удельной каталитичес¬
кой активности следует относить к поверхности этого вещества,
если оно образует отдельную фазу.
В последние десятилетия растет число исследований, связан¬
ных с созданием устойчивых каталитически активных соединений
путем закрепления на поверхности носителей комплексов опре¬
деленного состава, образования кластеров из нескольких одина¬
ковых или различных атомов переходных металлов. В связи с этим
представляет теоретический и практический интерес установле¬
ние зависимости атомной каталитической активности компонен¬
тов этих соединений от их состава, строения и координационной
ненасыщенности.
Хотя сущность гетерогенного катализа, химическая природа ка¬
талитического действия в основном определена и установлена,
задача предвидения каталитической активности остается актуаль¬
ной и наиболее сложной в теории катализа.
Перспективным для решения указанной задачи представляет¬
ся путь использования корреляционных соотношений Бренсте¬
да—Поляни (для однотипных катализаторов со сходным механиз¬
мом каталитических реакций) между изменением энергии акти-
197
вации и энергии определенных связей, характеризующих взаимо¬
действие катализатора и реагентов. Речь идет прежде всего о свя¬
зях, образующихся или разрывающихся при превращении акти¬
вированного комплекса.
Следует отметить, что часто используемые в корреляционных
соотношениях термодинамические характеристики катализатора
и реагирующих веществ служат лишь средством приближенной
оценки энергии этих связей.
10.2. Катализаторы в промышленных процессах
Промышленные катализаторы применяют обычно в форме гра¬
нул или цилиндров диаметром ~10-3 м, которые должны обладать
механической прочностью, пористостью и высокими значениями
удельной поверхности.
1. Переходные металлы (особенно VIII группы) способны ка¬
тализировать многие химические реакции гидрирования, дегид¬
рирования, окисления важнейших промышленных процессов, на¬
пример:
окисление NH3 до N0 на Pt в сплаве с Rh, Rd;
гидрирование СО до СН4 (Ni на Сг203, А1203);
получение водорода конверсией метана с водяным паром и
кислородом (Ni на носителях).
Ниже приведена принятая в литературе классификация пере¬
ходных металлов по каталитическим свойствам.
А
В
С
D
Ti
V
Сг
Mn
Fe
Co
Ni
Cu
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Ag
Hf
Та
W
Re
Os
Ir
Pt
Au
В группу А входят металлы, которые сильно хемосорбируют в
диссоциативной форме органические молекулы, а также такие газы
как N2 и СО, обладающие высокой энергией связи. Скорости де¬
сорбции молекул с этих металлов малы, вследствие чего они обыч¬
но являются плохими катализаторами.
Металлы группы В являются, в частности, катализаторами ре¬
акций Фишера —Тропша (СО + Н2) и синтеза аммиака.
Металлы группы С катализируют скелетные реакции углеводо¬
родов, а также гидрогенизационные процессы. Так, медь обладает
способностью гидрировать альдегиды, кетоны, органические кис¬
лоты.
198
Серебро является катализатором эпоксидирования этилена и
окисления метанола в формальдегид.
Однако ни один из металлов группы D не способен катализи¬
ровать химические реакции, требующие разрыва связей С—С или
более прочных.
Условность приведенной классификации связана с тем, что,
во-первых, границы между классами могут смещаться в зависи¬
мости от природы реагентов и условий проведения реакции; во-
вторых, каталитические свойства металлов сильно зависят от спо¬
соба приготовления катализаторов, влияющего на степень струк¬
турной неоднородности поверхности. Поэтому для металлических
катализаторов влияние неоднородности поверхности на катали¬
тические свойства исследовано наиболее подробно.
Укажем несколько путей приготовления катализаторов. Мно¬
гие из них получают нанесением растворов солей металлов (на¬
пример, Pt, Pd, Ni, Со) на пористые носители с высокоразвитой
поверхностью (например, силикагель, алюмогель, уголь, оксид
хрома(Ш) и т.п.), которые затем высушивают и обрабатывают
водородом при 250 — 500 °С. Восстанавливаемый металл покрыва¬
ет поверхность и поры носителя.
Аналогичным образом получают катализаторы, являющиеся по
своей природе органометаллическими комплексами — металл-
фталоцианины, которые используют в ряде реакций окисления в
жидкой и газовой фазах.
Широко используемые в жидкофазных процессах гидрирова¬
ния и дегидрирования олефинов катализаторы получают из спла¬
вов Ni, Со, Fe, Си с алюминием в соотношениях 1:1. Затем сплав
металла с алюминием измельчают до частиц размером до 10‘6 м и
обрабатывают раствором щелочи. Остающийся после растворения А1
металлический «скелет» обладает высокой механической прочно¬
стью.
Ряд катализаторов в форме оксидов А1203, Сг203, Fe203, ZnO
получают осаждением растворимых солей в виде гидроксидов с
последующим высушиванием и термической обработкой.
Рассмотрим подробнее несколько примеров промышленных ка¬
талитических процессов.
Синтез аммиака из азота и водорода в течение многих десятиле¬
тий осуществлялся на катализаторе Митташа — Fe304 + К20 +
+А1203 при температуре 450 —550 °С и давлении 200 — 500 атм.
В этом режиме катализатор работает несколько лет, но при пере¬
ходе к более высоким давлениям (до 1000 атм) — только 5 — 6
месяцев.
Современные достижения в этом процессе связаны не с хими¬
ей синтеза, а главным образом с механикой — созданием турбо¬
компрессоров, которые позволяют уменьшить затраты на сжатие
исходной смеси.
199
Реакция Фишера —Тропша
г-> метанол
СО + н,
-» смеси спиртов, кетоны, эфиры
-> метан и другие углеводороды
позволяет осуществлять синтез по разным направлениям в зави¬
симости от выбора катализатора и скорости осуществления про¬
цесса. Процесс синтеза метанола проводят в аналогичной по типу
аппаратуре, как и синтез аммиака, но изготовленной из нержаве¬
ющей стали, поскольку железо взаимодействует с монооксидом
углерода(И), образуя Fe(CO)5. Условия синтеза СН3ОН: 300—400 °С,
100 — 600 атм на катализаторах сложного «патентного» состава со¬
единений меди, хрома и цинка. Синтез углеводородов осуществ¬
ляют на сложных Со- и Ni-катализаторах.
Общий недостаток процесса заключается в невысокой селек¬
тивности, что снижает его привлекательность по соображениям
экологии.
2. Широкое применение в реакциях гидратации, дегидратации,
изомеризации находят катализаторы кислотно-основного типа.
К твердым кислым катализаторам относятся:
нелетучие минеральные кислоты (Н3Р04, Н3В03) на инертных
носителях — силикагеле, угле, кизельгуре и т.д.;
натуральные минералы (монтмориллонит, каолинит, бентонит);
смешанные оксиды (Al203 Si02, А1203 В203 и т.п.);
соли сильных кислот (А1С13, CuS04, NiS04, TiCl4 и др.).
Примером является гетеролитический процесс гидратации оле-
финов в присутствии Н3Р04 на носителе.
К твердым основным катализаторам относятся:
неорганические основания и амиды (NaOH, КОН, KNH2
и т. п.), нанесенные на инертные носители;
неорганические соли и оксиды основного характера (ВаО, СаО,
MgO, К2С03, СаС03 и т.д.).
Существенное отличие каталитических реакций с твердыми кис¬
лотами и основаниями от аналогичных в растворах состоит в том,
что если в растворе реакция протекает при одном значении фун¬
кции кислотности Гаммета Я0, то на поверхности твердых кислот
и оснований существуют центры с различными значениями Щ.
Поэтому активность катализатора по целевому продукту будет про¬
порциональна концентрации активных центров некоторой опти¬
мальной кислотности, как это наблюдается, например для реак¬
ции олигомеризации пропилена. В гетерогенном катализе, как и
при гомогенном кислотном, имеют место реакции, специфически
ускоряемые бренстедовскими или льюисовскими кислотами.
Некоторые реакции могут протекать по механизму бифункци¬
онального катализа, если на твердой поверхности существуют вбли¬
200
зи друг друга кислотные и основные центры. Такой механизм пред¬
ложил Д.Хориути для синтеза HCN из СО и NH3 с катализатором
на основе А1203- Th02.
К гетерогенным кислотно-основным катализаторам следует от¬
нести ионообменные смолы (иониты). Иониты — это сшитые по¬
лимеры, имеющие в своем составе функциональные ионогенные
группы, которые могут генерировать катионы и анионы. Различа¬
ют соответственно твердые кислоты — катиониты и основания —
аниониты. Ионы Н+ и ОН', нейтрализующие заряд ионогенных
групп, называют противоионами, они подвижны и сольватирова-
ны. Реакции, катализируемые ионитами, аналогичны реакциям,
ускоряемым растворимыми кислотами и основаниями. К недо¬
статкам большинства ионитных катализаторов следует отнести их
относительно низкую термическую стабильность, порядка 150 —
180 °С.
Особое место среди гетерогенных кислотных катализаторов зани¬
мают цеолиты, широко применяемые в процессах крекинга углево¬
дородов и гидрокрекинга и многих других кислотно-основных про¬
цессах. Природные цеолиты — алюмосиликаты (их около 40), содер¬
жащие в своем составе оксиды щелочных и щелочноземельных
металлов, отличающиеся строго регулярной структурой пор. Общая
химическая формула цеолитов Ме2/лО • AI203 xSi02 yH20, где
Me — катион металла, п — его валентность.
Обычно в состав цеолитов в качестве катионов входят натрий,
калий, кальций, реже барий, стронций и магний. Кристалличес¬
кая структура цеолитов образована тетраэдрами Si04 и А104. Кати¬
оны компенсируют избыточный отрицательный заряд анионной
части алюмосиликатного скелета цеолита. Пространственная ре¬
шетка цеолитов является пористой, причем адсорбционные по¬
лости соединяются друг с другом входами — «окнами» строго оп¬
ределенного размера (от 0,3 до 1,0 нм). Этим объясняется наличие
у цеолитов молекулярно-ситовых свойств, т. е. их способность вза¬
имодействовать только с проникающими через «окна» в глубь по¬
лостей молекулами. Кислотостойкие природные цеолиты, в пер¬
вую очередь высококремнистые формы типа морденита (отноше¬
ние Si02: А1203 колеблется в интервале 8,3 — 10,7) и клиноптило-
лита нашли широкое применение в процессах очистки отходящих
промышленных газов от NOx, S02, NH3, удаления соединений
аммония из сточных вод, извлечения цезия из радиоактивных от¬
ходов. Наряду с адсорбционными свойствами клиноптилолит об¬
ладает высокой каталитической активностью в процессах нефте¬
переработки и органического синтеза, например, при гидрирова¬
нии бензола в циклогексан, окислении этилена, деалкилирова-
нии толуола, гидрокрекинге.
Последние 50 лет широкое применение в адсорбции и катализе
получили синтетические цеолиты. Большая заслуга в решении этой
201
проблемы принадлежит английскому физико-химику Р. Барреру,
который осуществил синтез морденита, шабазита, фожазита и дру¬
гих цеолитов. Кислотные формы цеолитов впервые были получены
Р. Баррером (1949 г.) путем окисления их аммонийных форм. Поз¬
же их стали получать термическим разложением аммонийных форм
(после замещения катионов на группу NHJ) в вакууме. Например,
для получения Н-формы морденита достаточно прогреть цеолит в
течение одного часа при температуре 350 °С. «Ультрастабильные» це¬
олиты способны выдерживать температуру до 1000 °С, их получают
деалюминированием каркаса гидротермальной обработкой при по¬
вышенной температуре (-400 °С) исходных алюмо- и силикагелей.
Цеолиты в протонизированной форме и при замещении прото¬
на металлом функционируют как кислотные катализаторы, в де-
катионированной форме они являются льюисовскими кислотами.
Ангидридные формы кислотных форм цеолитов получают де¬
гидратацией поверхностных гидроксильных групп алюмокремни-
евых кислот при их нагревании (различном для разных видов це¬
олитов и для достижения определенной степени дегидратации).
В настоящее время цеолиты являются наиболее удобной для ката¬
лиза формой алюмосиликатов, на основе которых создано боль¬
шинство твердых кислотных катализаторов.
Например, одной из первых стадий переработки нефти явля¬
ется каталитический крекинг, при котором происходит разрыв
С—С связей в тяжелых углеводородах, их изомеризация, а также
разрушение N- и S-содержащих соединений, что позволяет уве¬
личить выход низкокипящих фракций нефти.
В этом процессе долгое время использовался катализатор Гуд-
ри (патент), в последние годы его заменили на современные ка¬
тализаторы (синтетический цеолит + аморфный алюмосиликат +
ионы РЗЭ), что позволило повысить выход полезных продуктов
на 30%.
Крекинг осуществляют при 450 — 500 °С в крайне неблагопри¬
ятных для катализатора условиях, препятствующих его длитель¬
ному использованию. Современные катализаторы выдерживают до
1000 циклов.
Гидрокрекинг — это одновременно протекающие процессы кре¬
кинга, гидроочистки, изомеризации, осуществляемые на суль¬
фидах (W—Ni) и (Со—Мо), нанесенных на твердые кислоты. Рань¬
ше использовали в качестве носителей кислотные формы монт¬
мориллонитов, сейчас — катализаторы на основе цеолитов с
добавками Pt, Pd, Ni, Co.
3. В промышленности катализаторы представляют собой, как
правило, многокомпонентные и многофазные системы. Напри¬
мер, при получении водорода из СО и Н20, а также при синтезе
спиртов из СО и Н2, используют оксидный медьцинкхромовый
катализатор (СиО —Сг2Оэ—ZnO).
202
Добавление к катализатору в небольших количествах веществ,
называемых промоторами, повышает его активность за счет уве¬
личения поверхности, избирательности и срока действия. Разли¬
чают структурообразующие и модифицирующие промоторы. Пер¬
вые препятствуют увеличению размеров кристаллов катализато¬
ра, стабилизируя его активную фазу от различных воздействий
(Т, Р и др.). Например, добавка 1 % А1203 к катализатору синтеза
аммиака увеличивает поверхность активного железа с 0,5 до 10 м2
и препятствует ее уменьшению за счет спекания, которое приво¬
дит к уменьшению удельной активности.
Модифицирующие промоторы влияют на строение и химичес¬
кий состав активной фазы катализатора. На примере реакции окис¬
ления метилпиридинов на ванадий-молибденовых оксидных ка¬
тализаторах были изучены различные механизмы действия про¬
моторов. Установлено, что оксиды Ag, Ni, Си перестраивают
решетку катализатора — твердого раствора основных компонен¬
тов; оксид кадмия за счет иона металла вызывает появление ак¬
цепторного дефекта; оксиды титана и никеля снижают прочность
связи V—О и стабилизируют соотношение ионов V^/V5*.
Среди многофазных катализаторов особый интерес представ¬
ляют полифункциональные катализаторы, на которых можно про¬
водить превращения, неосуществимые на исходных компонентах.
Например, в последние годы большую роль в процессе рифор-
минга бензиновых фракций играют бифункциональные Pt-ката¬
лизаторы, нанесенные на у- или ц-А1203. Механизм реакции де¬
гидроциклизации парафинов заключается в том, что олефины, об¬
разующиеся по реакции дегидрирования на платине, затем цикли-
зуются на кислотном катализаторе — оксиде алюминия. Добавляе¬
мый к катализаторам риформинга рений играет роль промотора.
Провести грань между катализаторами на носителях и много¬
фазными катализаторами непросто, так как носители, как прави¬
ло, не являются полностью инертными. Примером чисто инерт¬
ного носителя является пемза в серебряном катализаторе окисле¬
ния метанола в формальдегид.
Определенный интерес представляют каталитические системы,
получаемые путем механического смешения различных катализа¬
торов. Например, смесь А1203 и силикагеля при условиях, когда
взаимодействие между ними отсутствует, многократно увеличи¬
вает скорость реакции изомеризации окиси этилена по сравне¬
нию с действием каждого из компонентов.
Каталитические системы целесообразно применять к процес¬
сам, стадиями которых могут быть реакции, протекающие само¬
стоятельно. Так, процесс деалкилирования пиридинов водяным
паром на Ni-катализаторе протекает в одну брутто-стадию:
C5H4NCH3 + Н20—Ni )C5H5N + СО + 2Н2
203
При использовании каталитической системы V205—Мо03 —
— ТЮ2 + Ni в присутствии воздуха протекает двухстадийный про¬
цесс, существенно более селективный:
C5H4NCH3 + о2 —^^c5h4ncoh + н2о
C5H4NCOH—^C5H5N + CO
Катализаторы очень чувствительны к действию ядов — некото¬
рых веществ, присутствующих даже в малых количествах, кото¬
рые необратимо сорбируются на поверхности катализатора и за¬
трудняют доступ к ней реагирующих веществ. Это явление назы¬
вают отравлением катализаторов.
К типичным каталитическим ядам относятся соединения H2S,
CS2, тиофен, тиоспирты, синильная кислота, оксид углерода(Н),
соединения фосфора, мышьяка и др.
Каждый катализатор имеет свой набор ядов. Например, даже
малое содержание серы (0,01%) в железе заметно снижает его
каталитическую активность в синтезе аммиака, а увеличение ко¬
личества серы до 0,1 % приводит к полной потере железом ката¬
литических свойств.
Для оценки степени отравления катализатора обычно исполь¬
зуют кривые отравления, точнее их начальные участки, аппрок¬
симируемые по линейному закону, согласно эмпирическому урав¬
нению
Лотр.к/А=1-аС, (Ю.5)
где Аотр.к — активность отравленного катализатора; А0 — актив¬
ность катализатора до отравления; а — коэффициент отравления,
С — концентрация яда.
Снижение активности катализатора может происходить не толь¬
ко в результате действия ядов, но и из-за «старения» самого ката¬
лизатора, которое связано с рядом процессов: перекристаллиза¬
ции в поверхностном слое, наблюдаемой при высоких температу¬
рах; появления на поверхности катализатора других продуктов за
счет протекания побочных реакций; изменения структуры или хи¬
мического состава носителя и т. п.
Применение носителей в гетерогенном катализе способствует
улучшению характеристик процессов, так как позволяет умень¬
шить расходование дорогостоящих катализаторов (Pt, Pd, Au, Ag,
Os, Ir); увеличить в некоторой степени активность и избиратель¬
ность катализатора (небольшой промотирующий эффект); повы¬
сить устойчивость адсорбционных или нанесенных катализато¬
ров к температурным воздействиям (спекание) и к отравлению
ядами по сравнению с массивными металлическими катализато¬
рами.
204
10.3. Физическая адсорбция и хемосорбция
Исследования гетерогенного катализа показали, что каталити¬
ческая реакция протекает на поверхности катализатора, поэтому
для объяснения существа вопроса необходимо прежде всего по¬
нимание процесса адсорбции. Напомним, что адсорбция — это
процесс самопроизвольного изменения концентрации вещества
на границе раздела фаз, сопровождающийся уменьшением энер¬
гии Гиббса, Д(7<0.
В гетерогенном катализе адсорбентом является катализатор, ад-
сорбатом — молекула реагирующего вещества. Различают физи¬
ческую адсорбцию и хемосорбцию, хотя между ними нет четкой
границы. Физическая адсорбция протекает уже при низких темпе¬
ратурах достаточно быстро с малой энергией активации. По сути
она аналогична конденсации при сжижении газа, т. е. в этом слу¬
чае силы межмолекулярного взаимодействия слабы и аналогичны
силам Ван дер Ваальса.
Величина физической адсорбции слабо зависит от химической
природы адсорбата, но существенно возрастает при температу¬
рах, близких к температуре сжижения (или конденсации) адсор¬
бата. Однако температурная область протекания физической ад¬
сорбции не может существенно превышать температуру конден¬
сации адсорбата.
Физическая адсорбция, как правило, обратима, так как стан¬
дартная энтальпия адсорбции хотя и экзотермична, но невелика
и лежит в пределах —10, —50 кДж-моль-1, только в некоторых слу¬
чаях достигая —70 кДж-моль-1. Однако невысокие значения тепло¬
ты адсорбции не являются достаточным основанием, чтобы счи¬
тать адсорбцию физической, поскольку и для хемосорбции в ред¬
ких случаях наблюдаются малые величины теплоты адсорбции.
Необходимо пояснить, почему физическая адсорбция всегда
экзотермична, т. е. АН S 0. Это объясняется тем, что энтропия в
этом процессе уменьшается (AS^ 0), так как возрастает упорядо¬
ченность молекул в адсорбционном слое по сравнению с газовой
или жидкой фазами. Очевидно, что для уменьшения энергии Гиб¬
бса адсорбции: AG— АН— ГАДпри отрицательных значениях AS ве¬
личины ДЯдолжны быть экзотермичны, т. е. меньше 0 или термо¬
химические теплоты адсорбции, Q>0.
В подтверждение сказанного выше приведем термодинамиче¬
ские характеристики физической адсорбции ряда веществ на сзже.
для Аг при Т= 140К ДД° =-79,4 Дж • моль -К , АН -
=-8,9 кДж-моль'1; 0_
для С6Н6 при Г=291К ДД°=-97 Дж-моль-' К-', АН -
= -36,2 кДж-моль'1.
Физическая адсорбция характеризуется невысокой специфично¬
стью, так как ее величина слабо зависит от химического состава
205
поверхности катализатора. По этой причине удобно использовать фи¬
зическую адсорбцию, например азота или аргона, для определения
общей поверхности катализатора, формы и размера его частиц.
Температурная зависимость скорости физической адсорбции
проявляется слабо, но количество адсорбированного вещества с
ростом температуры быстро уменьшается (для р- const) в соот¬
ветствии с принципом подвижного равновесия.
Хемосорбция представляет собой поверхностную химическую
реакцию, которую можно рассматривать как химическое взаимо¬
действие газа или растворенного вещества с поверхностью твер¬
дого катализатора, не сопровождающееся образованием объем¬
ной фазы.
В этом случае силы, удерживающие молекулы адсорбата на по¬
верхности адсорбента, аналогичны химическим связям между ато¬
мами в молекуле, в результате чего происходит существенное пе¬
рераспределение электронной плотности в адсорбированном ком¬
плексе, приводящее к ослаблению или разрыву некоторых связей
в хемосорбированной молекуле.
Значения энтальпии хемосорбции, как и любой химической
реакции, лежат в широком интервале, они превышают в боль¬
шинстве случаев по абсолютной величине 100 кДжмоль-1, дости¬
гая, например, для хемосорбции 02 на молибдене значения АЯ°=
= -720 кДжмоль-1. Однако при хемосорбции энтропия адсорбции
не всегда меньше нуля, поэтому принципиально возможна хемо¬
сорбция с поглощением тепла (Д#>0). Подтверждением этого слу¬
жат надежные измерения для хемосорбции молекулы Н2 на пори¬
стом стекле: Д#°=30 кДж моль-1. Хемосорбция в отличие от физи¬
ческой адсорбции высоко специфична по отношению к химичес¬
кому составу адсорбента, поэтому очень чувствительна к чистоте
поверхности катализатора.
Хемосорбция может протекать не только при соприкоснове¬
нии молекул реактива из газовой фазы с поверхностью катализа¬
тора, но и через промежуточную стадию физической адсорбции.
В любом случае хемосорбция осуществляется через образование
промежуточного активированного комплекса, энергия которого
может существенно превышать энергию исходных веществ на ве¬
личину энергии активации.
Температурная область протекания хемосорбции не имеет огра¬
ничений, она может происходить как при низких, так и при вы¬
соких температурах. С ростом температуры количество хемосорби-
рованного вещества уменьшается, а скорость процесса возрастает
согласно уравнению Аррениуса. В связи с высокой экзотермично-
стью большинства процессов хемосорбции десорбция может иметь
место только при повышенных температурах.
Хемосорбция является обратимой, если при десорбции адсор-
бат переходит в газовую фазу в той же форме, какую он имел до
206
адсорбции. В ряде случаев хемосорбция необратима, поскольку при
десорбции может происходить разрыв прочных хемосорбционных
связей и переход в газовую фазу атомов твердого тела, связанных
с адсорбатом. Например, кислород, хемосорбированный на воль¬
фраме или молибдене, десорбируется при высоких температурах в
виде оксидов в результате реакции сублимации. Адсорбция водоро¬
да на платине при 200 К (с образованием разных связей Pt Н)
частично обратима, а частично — необратима.
В ряде случаев можно наблюдать оба типа адсорбции для одно¬
го и того же адсорбата: при пониженных температурах происхо¬
дит физическая адсорбция, при высоких хемосорбция. Проме¬
жуточная область характеризует процесс перехода от физической
адсорбции к хемосорбции, который обычно сопровождается рос¬
том количества адсорбированного вещества с повышением тем¬
пературы. Например, величина адсорбции водорода на меди умень¬
шается в интервале температур 70 —100 К, затем увеличивается с
ростом температуры до 450 К, выше этой температуры снова умень¬
шается. -л
Из вышесказанного следует, что отличие физическои адсорб¬
ции от хемосорбции можно установить с помощью ряда критери¬
ев, проводя сравнение энергий и энтальпий активации, темпера¬
турных интервалов процессов, зависимости скорости адсорбции
от температуры. Однако более однозначным критерием является
сравнение температурных зависимостей скорости десорбции для
обоих процессов. Поскольку энергия активации десорбции равна
сумме энергии активации адсорбции и теплоты адсорбции, оче¬
видно, что слабая зависимость скорости десорбции от температу¬
ры наблюдается только при небольших величинах энергии акти¬
вации и теплоты адсорбции. Это характерно для физической ад¬
сорбции.
Из двух процессов адсорбции только хемосорбция приводит к
заметному изменению энергетического состояния адсорбирован¬
ной молекулы и существенному увеличению ее реакционной спо¬
собности за счет снижения энергии активации, что и объясняет
каталитический эффект. Однако физическая адсорбция может пред¬
шествовать хемосорбции и являться стадией каталитического про
цесса.
10.4. Адсорбционная теория Лэнгмюра
Для количественного рассмотрения процесса хемосорбции удоб¬
но использовать модель адсорбционного слоя, впервые предло¬
женную Лэнгмюром. Основные ее допущения.
поверхность адсорбента состоит из определенного числа оди¬
наковых адсорбционных центров, вероятность адсорбции на всех
207
центрах одинакова (энергетически однородная поверхностей не¬
зависимость энергии хемосорбции от степени заполнения поверх¬
ности);
каждый адсорбционный центр может удерживать только одну
частицу (молекулу или атом), поэтому процесс адсорбции закан¬
чивается образованием на поверхности раздела фаз слоя толщи¬
ной в одну молекулу (мономолекулярный адсорбционный слой);
силами притяжения и отталкивания между молекулами или ато¬
мами, адсорбированными на соседних центрах, можно пренеб¬
речь.
На основании этих допущений должен выполняться следую¬
щий кинетический закон действующих поверхностей: скорость ад¬
сорбции пропорциональна числу столкновений молекул адсорба¬
та со свободными адсорбционными центрами на поверхности ад¬
сорбента; скорость десорбции пропорциональна числу занятых мест
на поверхности; скорость реакции между адсорбированными мо¬
лекулами пропорциональна произведению величин адсорбции
каждого из веществ.
Рассмотрим несколько механизмов адсорбции.
1. Процесс хемосорбции вещества А из газовой фазы схемати¬
чески можно представить следующим образом:
А(г)+8^ф^(А...8)адс, (10.6)
d
где s — свободный адсорбционный центр поверхности адсорбен¬
та; (А. . . s)MC — адсорбированная частица А или «заполненный»
центр адсорбции; ка — константа скорости адсорбции частицы А;
kd — константа скорости десорбции.
Для вывода кинетического уравнения этой реакции введем по¬
нятие степени заполнения поверхности или доли занятых адсор¬
бированными молекулами центров
0 = —, (10.7)
где а — величина адсорбции при заданном давлении; ат — пре¬
дельная величина адсорбции, равная емкости монослоя.
Согласно закону действующих поверхностей скорость адсорб¬
ции va пропорциональна доле свободной поверхности и давлению
газа
va=kap{ 1-0), (10.8)
где (1 -0) — доля свободной поверхности.
Скорость десорбции пропорциональна степени заполнения
vd=kdQ. (10.9)
В целом адсорбция протекает со скоростью
v = va-vd = kap(l-Q)-kde (10.10)
208
После установления равновесия между газовой и адсорбцион¬
ной фазами v=0, следовательно,
kap(\-Q) = kdQ. (10.11)
Решая соотношение (10.11) относительно 0 и вводя константу
адсорбционного равновесия
К = ^-, (10.12)
получаем известное уравнение изотермы Лэнгмюра:
0=:г=ттк' (10ЛЗ)
ат 1 + Кр
Часто вместо К используют введенную автором теории величи¬
ну Ь, которую называют адсорбционным коэффициентом веще¬
ства А. Для обратимой адсорбции b имеет физический смысл кон¬
станты равновесия между веществом на поверхности и в газовой
фазе
-ДСщк АЛдас — А//^с Q
К = Ь = с~кг~ = е~е~кт~ =ЬьеЯТ) (10.14)
где Ь0 — коэффициент, связанный с энтропией адсорбции; Д G^c —
энергия Гиббса адсорбции; АИ^С — энтальпия адсорбции; Q —
теплота адсорбции.
И.Лэнгмюр получил свое уравнение в 1918 г. кинетическим
путем, изложенным выше. Покажем, что оно может быть легко
выведено термодинамически.
Для реакции (10.6) условие адсорбционного равновесия выра¬
зим через химические потенциалы исходных и конечных компо¬
нентов
Ра + Ps = Pas-
Значения химических потенциалов имеют следующий вид:
для газовой фазы рА = р^ + RT ln-^-, где р — давление компо-
Р
нента А в газовой фазе; р° — стандартное давление, его можно
опустить, если давление измеряется в бар или атм; р°= 1:
для свободных адсорбционных центров ps = Ps + RT ln(l - 0)
для занятых адсорбционных центров Pas = Pas + ЛГ1п0.
Из условия адсорбционного равновесия получаем
Ра + RT lnp + ps + RT ln(l - 0) = p^s + RT In 0,
откуда путем перегруппировки слагаемых находим
•4. Зак. 1596
209
RT ln ТГТ^Г = ^ + ^ ^ = -A<%K,
(1 - @)p
где A<70MC — изменение энергии Гиббса при адсорбции.
Очевидно, что выражение под логарифмом в полученном урав¬
нении ■ ® имеет физический смысл адсорбционной констан-
(1 - в)р
ты равновесия (достаточно вспомнить, что Д G° = -RT In К0). Сле¬
довательно,
Q ..у
(1 -9)р ’
Решая полученное уравнение относительно величины 0, кото¬
рую нельзя непосредственно измерить, приходим к уравнению
Лэнгмюра
0 = ^L = _t
ат 1 + Кр
Если равновесное давление остается достаточно малым, т. е. вы¬
полняется условие Крс 1, тогда (10.13) переходит в
Q = Kp. (10.15)
В этом случае 0 растет пропорционально давлению и по накло¬
ну касательной к начальному участку прямой в координатах (0, р)
находят величину К.
Рис. 10.1. Изотерма адсорбции Лэнгмюра
210
Если равновесное давление достаточно велико, чтобы выпол¬
нялось условие Кр^> 1, получаем
0=1. (10.16)
Величина 0 перестает зависеть от давления, это означает, что
поверхность полностью покрыта мономолекулярным слоем, на¬
ступает состояние насыщения.
Как видно из рис. 10.1, при значении р = —, Кр= 1 и степень
К
заполнения 0 равна — ; причем касательная достигает значения
0 = 1 именно в точке р = .
К
Для нахождения параметров уравнения Лэнгмюра — величин
ат и К, обработку экспериментальных данных по зависимости ад¬
сорбции а от давления р можно проводить тремя способами:
а) записав уравнение (10.13) в обратных величинах - и -,по-
а р
лучим
11 11
-а-^*ТГ,Т <1017>
1 1
Из графика, построенного в координатах легко нахо-
/ - 1
дим ат по отрезку /, отсекаемому прямой на оси ординат ат - - ,
и А" — по тангенсу угла наклона прямой и указанному значению ат
tgoc - —— и К-- ,
Кат tga
б) преобразуем уравнение (10.15), умножая обе его части на р,
a=jk+ZP- (10Л8>
Строя линейную зависимость — = f(p), находим ат по танген-
а
су угла наклона прямой ат = —— и рассчитываем К, используя
tga
полученное значение ат и величину отрезка /, отсекаемого пря-
, - 1 , ^ tga
Мои на ординате = / и К = ;
Кат I
м. 211
в) уравнение Лэнгмюра, умножив обе части уравнения (10.17)
на ата, можно представить в виде
а = ат-\г-. (10.19)
К р
Строя график в координатах (о, —), находим ат по отрезку,
Р , г
отсекаемому прямой на оси ординат, ат = / и л по тангенсу угла
- г 1
наклона прямой К = .
tga
2. Рассмотрим случай, когда адсорбция молекулы А2 сопро¬
вождается ее диссоциацией на атомы. Схема этого процесса вы¬
глядит так:
А А
II* 11
А2+— s — s — у ‘ — — s —s— (10.20)
Запишем выражения для скорости адсорбции
va=kap( 1-0А)2 (10.21)
и скорости десорбции
vd=kdel. (10.22)
При равновесии kap(l-@A)2=kd@A. Решая это соотношение отно-
/с
сительно 0А, полагая К = — , получим
kd
= (Кр)уг (10.23)
ИЛИ
Q .(кр)у_2_ (ю 24)
А 1 + (Кр)У2' { }
В начальный момент времени, Кр«: 1, следовательно 0А~ р1/г.
В дальнейшем для нахождения параметров уравнения (10.24), удоб¬
но использовать линейную зависимость в виде
©а = 1 + (КрУ1/2 (10.25)
3. Рассмотрим случай, когда при одновременной адсорбции двух
газов происходит их конкуренция за адсорбционные центры. Пусть
0А и 0В — степени заполнения поверхности соответственно моле¬
кулами А и В (без их диссоциации), рА и рв — парциальные давле¬
212
ния этих газов. Выражения для скоростей адсорбции и десорбции
примут вид:
Va,\ = £о,а/>а(1-0а -0в); (10.26)
va,B = ^а,вРв(^ ~ ®А - 0в)> (10.27)
frf.A = kd,A®A> (10.28)
vd, в = kd,B®B- (10.29)
В условиях равновесия (va=vd), обозначив через Кк =
'V. '
/ \
Vd
и
получим
/А
и решая систему уравнений относительно ©А и 0В,
КаРа
©А =
0R =
1 + АдРл + КвРв
КвРв
1 + ^а Ра + КвРв
(10.30)
(10.31)
Если АТдРа и Квра<$:1, тогда 0А~рА и 0в~Рв •
Если один из газов сильно адсорбируется, например АдРа»
»1+А’врв, тогда 0А=1 и 0В - — , т.е. адсорбция вещества В за¬
медляется адсорбцией А.
Ра
10.5. Нелэнтюровские изотермы адсорбции
Эксперимент показывает, что во многих случаях реальные изо¬
термы не описываются уравнением Лэнгмюра. По-видимому, мо¬
дель «идеального адсорбированного слоя», в котором поверхность
адсорбента уподобляется шахматной доске, оказывается заметно
упрощенной.
Причинами, объясняющими эти отклонения в рамках модели
Лэнгмюра, являются энергетическая неоднородность поверхнос¬
ти реальных катализаторов, что приводит к зависимости теплот
адсорбции от степени заполнения поверхности адсорбента: умень¬
шению теплоты адсорбции по мере увеличения степени заполне¬
ния; наличие сил притяжения и отталкивания между адсорбиро¬
ванными молекулами.
213
Рассмотрение моделей адсорбции, учитывающих указанные
выше причины, выходит за рамки данного пособия, поэтому ог¬
раничимся упоминанием ряда полезных уравнений для описания
реальных изотерм адсорбции.
Изотерма Генри
9 = Кр (10.32)
в ряде случаев представляет собой начальный участок изотермы Лэнг-
мюра и выполняется в небольшом интервале степеней заполнения.
Изотерма Фрейндлиха
@ = Qp1/C2, (10.33)
где С| и С2 — постоянные, С2> 1; часто используется для описа¬
ния хемосорбции газов на металлах в области средних заполнений
(О,3<0<О,7).
Логарифмическая изотерма, получившая в иностранной лите¬
ратуре название изотермы Темкина:
0 = c!ln(c>) (10.34)
или
0= Q+ С21пр, (10.35)
где сь с' и Ch С2 — постоянные.
Полезно отметить, что эмпирические уравнения (10.33) и
(10.34) можно получить как в предположении неоднородности по¬
верхности, так и при учете сил межмолекулярного взаимодействия.
Несмотря на то что соотношения (10.33 и 10.34) являются толь¬
ко экстраполяционными формулами, так как а,—при р—
они часто используются в некотором интервале заполнения вбли¬
зи 0А~О,5 для описания медленного роста адсорбции при изме¬
нении парциального давления на несколько порядков.
При физической адсорбции, если энергии межмолекулярных
связей соизмеримы, после насыщения первого адсорбционного
слоя наступает полимолекулярная адсорбция с образованием вто¬
рого и последующих адсорбционных слоев. Полимолекулярная
адсорбция имеет место во многих процессах с участием органи¬
ческих соединений при повышенных давлениях.
Удобную модель для описания полимолекулярной адсорбции па¬
ров на мезо- и макропористых адсорбентах предложили С. Бруннауэр,
П. Эммет и Э. Теллер. Ее основные допущения состоят в следующем:
учитывается только минимальная совокупность парных взаи¬
модействий молекул, которая может привести к возникновению
полимолекулярного слоя;
при значительной разнице в теплотах адсорбции первого и пос¬
ледующих адсорбционных слоев принимается, что
к2 = к2 = к4 = ... = кК0ИД = Р;\ (Ю.36)
214
где К, — константа равновесного взаимодействия пары частиц во
втором и далее слое; Ку.онл — константа равновесия процесса кон¬
денсации; ps — давление насыщенного пара сорбата при темпера¬
туре опыта;
полагают, что Kj » ЛГК0НД, считая, что взаимодействие адсор¬
бент — адсорбат сильнее, чем взаимодействие адсорбат — адсор-
бат. Здесь Кх — константа адсорбционного равновесия в первом
«ленгмюровском» слое. Для удобства математического решения
задачи в качестве ее параметра используют соотношение
Kl/KKom = KlPs=C»l. (10.37)
Некорректность предложенной авторами модели привела к тер¬
модинамически некорректному уравнению, которое описывает
опытные данные в узком интервале изменения относительного
давления. Однако несмотря на эти недостатки, уравнение поли-
молекулярной адсорбции (БЭТ), предложенное авторами, в тече¬
ние полувека было единственным уравнением, позволяющим ко¬
личественно описывать полимолекулярную адсорбцию паров и
оценивать площадь поверхности адсорбентов. Обратим внимание
на то, что это уравнение относится к случаю, когда эксперимен¬
тально измеряемая избыточная адсорбция может быть приравне¬
на к абсолютной.
Уравнение полимолекулярной адсорбции БЭТ, полученное пу¬
тем несложных математических операций, имеет следующий вид:
а = а!”СШ.РЛ (10.38)
(1-р/Л)[1 + (С-1)(р/Л)]‘
Для определения параметров ат и С уравнение БЭТ приводят к
линейному виду
У = ~^Р!Р) + (Ю.39)
^(1 Р/Ps) O'irP' Ps
Строя графическую зависимость в координатах (у, —), нахо-
,А 1
дят величину отрезка, отсекаемого на оси ординат, / = ——, и
тангенс угла наклона прямой
атС
tga = —-гг = (С -1)1.
йтС
С помощью этих данных легко найти искомые параметры
am=(l + tga)-1 иС = 1 + у.
Знание ат, что является целью расчета, позволяет рассчитать
Доступную поверхность адсорбента S, если известна эффективная
215
площадка s, занимаемая молекулой адсорбата в плотном слое:
S=a„sNK.
Принято считать, что уравнение БЭТ можно применять для
определения поверхности с точностью 5—10%, если С > 40. В этих
условиях описание полимолекулярной адсорбции до значений
p/ps< 0,3—0,4 с помощью уравнения БЭТ является достаточно точ¬
ным.
Использование уравнения БЭТ оказало большое влияние на
развитие теории и практики катализа, позволило установить ко¬
личественную связь между скоростью каталитических процессов
и величиной поверхности катализаторов.
На макропористых адсорбентах, например в цеолитных ката¬
лизаторах (кристаллических алюмосиликатах), молекулы адсор¬
бата проникают в глубь кристаллической решетки, характеризую¬
щейся наличием полостей. Поэтому понятие поверхностной ад¬
сорбции здесь неприменимо. Оценку величины адсорбции проводят
с помощью уравнения из теории объемного заполнения микро-
пор М. М. Дубинина
п2
lg^ = lg| Щ-\-втг
(10.40)
где W — адсорбированный объем (в жидком состоянии); fV0 —
объем пор; V— мольный объем жидкого адсорбата; В — константа;
р — равновесное давление адсорбата при температуре Т, р5 — дав¬
ление насыщенных паров адсорбата при той же температуре.
Однако использование уравнения (10.40) в катализе ограниче¬
но достаточно низкими значениями —. Поэтому для описания
адсорбции на цеолитах часто пользуются эмпирическими форму¬
лами типа
а = г4^> (1041)
1 + Врт
где А, В, т — константы для данного типа адсорбата, 0<m< 1.
10.6. Кинетика гетерогенно-каталитических реакций
Гетерогенно-каталитическая реакция представляет собой слож¬
ный процесс, состоящий из следующих стадий:
1) диффузия исходных веществ из объема газовой или жидкой
фазы к внешней и внутренней поверхностям катализатора;
2) адсорбция реагирующих веществ на поверхности катализа¬
тора;
216
3) химическая реакция на поверхности катализатора;
4) десорбция продуктов реакции;
5) диффузия продуктов реакции от внутренней и внешней по¬
верхностей катализатора в объем.
Общая скорость такого сложного процесса определяется ско¬
ростью наиболее медленной стадии, часто называемой лимитиру¬
ющей стадией.
Обычно условия эксперимента выбирают таким образом, что¬
бы процессы транспорта (стадии 1 и 5) не являлись лимитирую¬
щими. Однако этого не всегда можно добиться, так как диффузия
в адсорбционные центры катализатора, которые могут находить¬
ся в микропорах молекулярных размеров, осуществляется очень
медленно.
Ограничимся далее рассмотрением процессов, для которых ско¬
рости адсорбции и десорбции велики по сравнению со скоростью
стадии 3: можно считать, что в любой момент времени каждая
частица находится в равновесии между адсорбированной и газо¬
вой фазами, т. е. адсорбционное равновесие не нарушается хими¬
ческой реакцией, протекающей на поверхности катализатора.
В этом случае скорость химической реакции является лимитиру¬
ющей стадией для всего процесса.
О сходстве механизмов гетерогенного и гомогенного катализа
можно судить по тому, что стадия хемосорбции в первом анало¬
гична стадии образования промежуточного соединения во втором.
Различие их состоит в том, что на поверхности гетерогенного ка¬
тализатора, благодаря наличию нескольких типов активных цент¬
ров, могут образовываться различные типы активированных ком¬
плексов, приводящих к появлению разных продуктов реакции.
В результате гетерогенные катализаторы характеризуются более
низкой селективностью, чем катализаторы в гомогенном ката¬
лизе.
1. Рассмотрим мономолекулярную каталитическую реакцию, когда
вещество А хемосорбируется на поверхности катализатора и про¬
дукт Р выделяется по схеме
A(r)+s ч (А... s)^ —P(r)+s (10.42)
По определению скорость гетерогенной химической реакции
равна количеству вещества, реагирующего в единицу времени на
единице площади поверхности катализатора:
'-55- (10-43>
где п — количество реагирующего вещества в момент времени /;
S — общая площадь поверхности катализатора, на которой идет
процесс.
217
С другой стороны, согласно основному постулату химической
кинетики, скорость этой реакции прямо пропорциональна по¬
верхностной концентрации веществ, которая, в свою очередь,
пропорциональна доле поверхности 0А, занятой молекулами ад¬
сорбированного вещества А, поэтому можно записать
(10.44)
Поскольку поверхность катализатора, как правило, постоян¬
на, величину общей поверхности катализатора можно ввести в
константу скорости k'S=k. Уравнение для скорости запишется в
более простом виде:
v = k&А. (10.45)
Используя уравнение Лэнгмюра для нахождения 0А, получим
уравнение скорости для данной реакции
кКрА
v = кО А =
(10.46)
А 1 + ^Рд’
где к — константа скорости химической реакции; К — константа
адсорбционного равновесия.
Анализ полученного уравнения (10.46) показывает, что для до¬
статочно высоких давлений, когда Кр »1, получаем v~ к, т.е. ре¬
акция имеет нулевой порядок по веществу А.
Рис. 10.2. Потенциальная кривая для гетерогенной каталитической
реакции:
1 — реагенты; 2 — адсорбированный комплекс; 3 — адсорбированные
реагенты; 4 — адсорбированные продукты реакции; 5 — конечные продукты
218
В этом случае константа скорости общей реакции фактически
равна константе скорости поверхностной химической реакции, и
кажущаяся энергия активации реакции Екаж равна энергии акти¬
вации поверхностной химической реакции Еа,ъ как показано на
рис. 10.2.
Напротив, при достаточно низких давлениях, если Кр«. 1, урав¬
нение (10.46) примет вид v=kKpA, т.е. реакция имеет первый по¬
рядок по веществу А.
В этих условиях кажущаяся константа скорости: &каж = кК. Ис¬
пользуя уравнения Вант-Гоффа (для нахождения энтальпии ад¬
сорбции АЯ^дс) и Аррениуса (для определения Еа к), находят ка¬
жущуюся энергию активации каталитической реакции
Учитывая, что адсорбция является экзотермическим процес¬
сом (АН%дС<0), выясняется, что кажущаяся энергия активации
имеет меньшее значение, чем энергия активации стадии, на ко¬
торой происходит поверхностная химическая реакция.
Примерами гетерогенно-каталитических процессов, для кото¬
рых характерно существенное снижение кажущейся энергии ак¬
тивации по сравнению с энергией аналогичных гомогенных нека¬
талитических реакций (Д£’=Ягом-Дсаж.гех) являются:
а) реакция окисления S02 в присутствии Pt (АЕ = 160 кДж/моль);
б) реакция синтеза аммиака: на W-катализаторе (АЕ= 163 кДж/
моль), на Fe-катализаторе (АЕ ~ 200 кДж/моль).
Обычно понижение энергии активации в гетерогенном ката¬
лизе приводит к значительному росту скорости реакций в 106 —
1016 раз.
В некоторых случаях продукты реакции могут также адсорбиро¬
ваться, уменьшая поверхность катализатора, доступную для ис¬
ходного вещества А, как показано на схеме:
A<r)+ s v (А... s)^ —к—> (В... s)№C ч В(г)+ s (10.48)
где к — константа скорости лимитирующей стадии.
Если обозначить через 0В — долю поверхности, покрытую про¬
дуктом В при парциальном давлении рв, доля поверхности, по¬
крытая А, имеет вид согласно формуле (10.30):
(10.47)
1+ КаРа + КвРв
Скорость реакции запишется так:
<а - каРа
UA ~ , , ъг _ ,
v = keA= -—~thAEA
1 + ^А+^В
(10.49)
219
Продукт В играет в этом случае роль ингибитора. Если веще¬
ство А очень сильно адсорбируется, т.е. выполняется соотноше¬
ние: КА рА^>1 + Кврв, тогда
т.е. реакция имеет нулевой порядок по веществу А и скорость ее
достигает максимального значения, так как вся поверхность ката¬
лизатора занята молекулами А.
Если напротив, оба вещества А и В слабо адсорбируются, т.е.
KaPa+KrPb <*с 1, реакция имеет первый порядок по А:
Если давление исходного вещества достаточно низкое, т.е. вы¬
полняется условие А'дрА«:1 + Кврв, и если к тому же продукт В очень
сильно адсорбируется (выполняется соотношение К^рв> 1), выра¬
жение для скорости (10.49) упрощается:
минус первый по веществу В, которое выступает в качестве инги¬
битора. Обозначив через АЛадс(В) энтальпию адсорбции продук¬
та В, аналогичным путем, как описано ранее, получают выраже¬
ние для кажущейся энергии активации каталитической реакции
Энергия активации суммарной реакции возрастает по срав¬
нению с реакцией (10.47), так как для десорбции продукта В с
поверхности катализатора требуются дополнительные затраты
энергии (-АЯадс(В)>0).
Все разобранные случаи наблюдаются в процессе разложения
аммиака, катализируемого металлами. Например, реакция, ката¬
лизируемая вольфрамом, имеет нулевой порядок при достаточно
высоких давлениях аммиака и первый порядок при очень низких.
2. В случае протекания бимолекулярных реакций А+ В -» Р,+Р2 обыч¬
но ограничиваются рассмотрением двух механизмов процесса.
Механизм Лэнгмюра —Хиншельвуда: реакция протекает между дву¬
мя адсорбированными веществами на соседних центрах по схеме
A^j.)+St (A...s)MC B(r)+s ^ (B...s)^c
(10.50)
v ~ kKfj}A.
(10.51)
(10.52)
где куж = кажущаяся константа скорости.
Реакция в !том случае имеет первый порядок по веществу А и
^каж- Л-Т/зд^д) АЯадс(В).
(10.53)
(А...8)адС+ (B...S)MC > (P.-.S)^
(10.54)
(P...S)MC P, +P2 + S
220
Если допустить, что оба вещества адсорбируются на одинако¬
вых центрах, т. е. конкурируют между собой, степени заполнения
поверхности А и В имеют вид согласно формулам (10.30) и (10.31):
К А р X
0. = А^А •
1 + %аРа + %вРв
0В =
Кврв
1 + %аРа + КвРв
При условии, что продукты реакции адсорбируются слабо, урав¬
нение для скорости реакции запишется в следующем виде:
Ьг\ гл кКкКъРхРв /1Л сс\
А В (1 + КаРа + КвРв)2 ( }
Выражение для скорости реакции упрощается при слабой ад¬
сорбции газов, т.е. при малых значениях рА и рв, если КрР/^1,
КвРв ^ 1 •
v = kKAKBpApB. (10.56)
Видно, что скорость пропорциональна рА и рв, реакция имеет
первый порядок по каждому веществу, общий порядок равен двум.
Такая зависимость наблюдается, например, при гидрировании эти¬
лена на поверхности меди.
Если вещество А адсорбируется сильно, В — слабо и выполня¬
ется условие Kxj)A»l + Кврв , тогда выражение для скорости при¬
мет вид
v = Мв £в = к^жРъРА ■ (10.57)
Ад Ра
Реакция имеет первый порядок по веществу В и минус пер¬
вый — по А, скорость ее уменьшается при увеличении рА за счет
вытеснения вещества В с поверхности.
Если вещество В адсорбируется сильно, А — слабо, при выпол¬
нении условия Кврв » 1 + КхрА, получаем
1сК а Да * -1
v = —^^ = kKa*pApB. (10.58)
Лв Рв
Реакция в этом случае имеет первый порядок по веществу А и
минус первый по В, т.е. В замедляет адсорбцию А, необходимую
для протекания реакции.
Скорость реакции, протекающей по данному механизму, со¬
гласно (10.55), проходит через максимум при значении
_ 1 + КвРв
^ А
Рл(тах) ^ (10.59)
221
и постоянном давлении рв, затем медленно убывает (приблизи¬
тельно на 10% при увеличении вдвое указанного значения рА).
Примером реакции, протекающей по механизму Лэнгмюра—
Хиншельвуда, является окисление оксида углерода(И) молеку¬
лярным кислородом на кварце или платине. Скорость этой реак¬
ции выражается аналогичным (10.58) уравнением
v = ^тсаж РОг РсО •
Механизм Или—Ридиела. Реакция осуществляется между адсор¬
бированной молекулой и молекулой в газовой фазе по схеме:
A(r)+s^=^^(A...s)aac пплт
(A...s)MC+B->P+s <10'60>
В общем случае В может адсорбироваться, даже если никакой
роли не отводится его адсорбированному соединению. Уравнение
для скорости реакции в этом случае запишется так:
v = k&*PB = ]+tKfA+Pl и • (10-61>
1 + каР\ + квРв
Это уравнение показывает, что при постоянном рв скорость
монотонно возрастает с ростом рА. Наличие или отсутствие мак¬
симума для скорости в зависимости от давления одного из ис¬
ходных веществ при сохранении постоянным давления второго
компонента позволило в ряде случаев найти различие между ме¬
ханизмами Лэнгмюра—Хиншельвуда и Или—Ридиела. В некото¬
рых случаях реакция гидрирования этилена, катализируемая ме¬
таллами, может быть интерпретирована механизмом Или—Ри¬
диела.
3. Большинство исследований гетерогенного катализа посвящено
газофазным реакциям, поэтому следует обратить внимание на спе¬
цифику жидкофазных процессов.
Наблюдаемые на опыте отличия гетерогенно-каталитических
реакций, которые протекают в жидкой фазе от аналогичных в
газовой фазе: разные скорости протекания конкретной реак¬
ции на идентичном катализаторе; различный состав получае¬
мых продуктов; большая вероятность объемного продолжения
реакции.
Для объяснения указанных отличий следует учитывать, во-пер¬
вых, различное термодинамическое состояние системы; во-вто¬
рых, наличие сольватационного слоя над поверхностью катализа¬
тора для жидкофазных реакций; наконец, возможность возник¬
новения электрохимических механизмов при использовании даже
слабых электролитов.
Первый фактор, термодинамический, имеет в основном энт¬
ропийную природу. Изменение энтропии реакции происходит по
222
разным причинам. С одной стороны, тепловой эффект реакции в
жидкой фазе может существенно отличаться от теплового эффек¬
та в газовой фазе за счет разности теплот испарения и растворе¬
ния реагентов и продуктов, это приводит соответственно к замет¬
ному изменению энтропии реакции.
С другой стороны, в жидкости существует ближний порядок в
расположении молекул, т. е. определенная их ориентация и взаим¬
ное влияние. Поэтому при образовании активированного комп¬
лекса исходных веществ с катализатором происходит существен¬
ное уменьшение энтропии, что приводит к изменению скорости
реакции в целом.
Кроме того, образование сольватационного комплекса из мо¬
лекул может сопровождаться понижением энергии активации про¬
цесса, как это имеет место при интермолекулярных превращени¬
ях, что ускоряет реакцию в жидкой фазе.
Например, скорость восстановления ароматических нитросо¬
единений на близких по составу катализаторах в жидкой фазе
может быть на 3 — 5 порядков выше скорости реакции в газовой
фазе.
Примером получения различного состава продуктов реакции
при ее переносе из газовой в жидкую фазу служат результаты га¬
зофазного и жидкофазного окисления парафинов нормального
строения на оксидных катализаторах.
На V—W-м катализаторе при газофазном окислении н-гептана
не удается получить, кроме формальдегида, продукты неполного
окисления. Для той же реакции и катализатора в жидкой фазе
образуется с почти количественным выходом сумма кислот, аль¬
дегидов и кетонов С3—С7 с заметным содержанием энантовой
кислоты.
Особенность жидкофазных реакций проявляется в процессах,
связанных с передачей электронов (окисление, восстановление,
гидрирование и т.д.), так как в проводящей среде возникает элек¬
трохимический механизм катализа. Твердый катализатор, поме¬
щенный в электропроводящую жидкость, можно рассматривать
как электрод, на котором возникает электрохимический потен¬
циал. Благодаря последнему увеличивается адсорбция реагента
(окислителя или восстановителя), который принимает электроны
от катализатора или отдает их ему. Образующиеся в этом процессе
ионы обладают повышенной реакционной способностью.
В качестве иллюстрации электрохимического механизма ката¬
лиза можно привести сравнение результатов окисления на Pt-
черни водных растворов спиртов и абсолютного изопропилового
спирта. В первом случае реакция полностью гетерогенна с дока¬
занным механизмом электронного переноса через ионы ОН-
воды, во втором случае доля гетерогенной реакции составляет
только 30%.
223
10.7. Макрокинетика гетерогенно-каталитических
процессов
Предмет макрокинетики в курсе физической химии включает
в себя математический анализ одновременного протекания хими¬
ческой реакции и процессов переноса вещества на поверхности и
в зерне катализатора (без учета закономерностей теплопереноса).
Как показано в подразд. 10.6, гетерогенно-каталитический про¬
цесс осуществляется в несколько стадий, каждая из которых, яв¬
ляясь в определенных условиях лимитирующей, может опреде¬
лять общую скорость всего процесса. В зависимости от условий
проведения процесса различают четыре основные макрокинети-
ческие области в гетерогенном катализе.
Внешнекинетическая область: наиболее медленной стадией явля¬
ется химическая реакция на внешней поверхности зерна катали¬
затора. Подробное описание кинетики процесса в этой области
рассмотрено в подразд. 10.6. К отличительным особенностям гете¬
рогенно-каталитических реакций во внешнекинетической облас¬
ти относятся отсутствие зависимости скорости процесса от разме¬
ров зерен катализатора и его пористости, линейной скорости га¬
зового потока, условий перемешивания и быстрое увеличение
скорости процесса с ростом температуры.
Внешнедиффузионная область: скорость процесса лимитирует¬
ся диффузией реагентов из объема к внешней поверхности катали¬
затора или переносом продуктов реакции в обратном направлении.
Внутридиффузионная область: наиболее медленной стадией яв¬
ляется диффузия реагентов от внешней поверхности зерна ката¬
лизатора к его внутренней поверхности пор или диффузия про¬
дуктов реакции в обратном направлении. Отметим, что скорость
процесса в этой области, как будет показано ниже, контролиру¬
ется не только диффузией, но и кинетикой химической реакции.
Внутрикинетическая область: наиболее медленной стадией яв¬
ляется химическая реакция, протекающая на внутренней поверх¬
ности пор зерна катализатора. На практике четкой границы между
указанными выше областями действия катализатора не существу¬
ет, поэтому проводят анализ поведения реакционной каталити¬
ческой системы и в промежуточных (переходных) областях.
Внешняя диффузия. Рассмотрим гетерогенно-каталитическую ре¬
акцию, наиболее медленной стадией которой является перенос
реагента из объема к внешней поверхности зерна катализатора
или перенос продуктов в обратном направлении. Согласно теории
стационарной конвективной диффузии Нернста, вблизи поверх¬
ности твердого тела (в данном случае, зерна катализатора) обра¬
зуется диффузионный слой, толщина которого 5 существенно за¬
висит от скорости движения газового (или жидкого) потока от¬
224
носительно твердой поверхности. Полагают, что внутри диффузи¬
онного слоя конвекция отсутствует и происходит основное изме¬
нение концентрации между объемом и поверхностью.
В рамках так называемой одномерной задачи считают (полагая
зерна катализатора непористыми и линейные размеры поверхно¬
сти существенно превосходящими длину пути диффузии), что кон¬
центрация переносимых к/от поверхности реагентов или продук¬
тов изменяется в одном направлении — по нормали к поверхнос¬
ти. Излагаемый ниже подход к решению задачи был предложен
Д. А. Франк-Каменецким и получил название метода равнодос¬
тупной поверхности*.
При стационарном режиме (условием стационарности при диф-
d С
фузии является соотношение = const) концентрация реаген¬
та внутри диффузионного слоя меняется линейно с расстоянием
до поверхности катализатора, поэтому градиент концентрации ра¬
вен
dC _ АС _ Cs -Су (10.62)
dx ~ 5 6 ’
где Су— концентрация вещества у внешней поверхности катали¬
затора; Cv — концентрация в объеме газовой (жидкой) фазы.
Согласно первому закону Фика скорость диффузии, отнесен¬
ная к единице площади поверхности катализатора, пропорцио¬
нальна градиенту концентрации
dп „ dC
dx’ ( }
где D — коэффициент диффузии, м2/с; S — площадь поверхности
катализатора, на которой осуществляется диффузия, м2; С — кон¬
центрация, моль/м3. После подстановки соотношения (10.62) в
(10.63) получим
*диф =1§-(cs- Су) = р(Су - Cs). (10.64)
Здесь Р = ^ — коэффициент массопередачи или константа массо-
о
обмена между объемом газовой или жидкой фазы и внешней по¬
верхностью катализатора; Р зависит от размера зерен и толщины
диффузионного слоя, возрастая с их уменьшением.
Диффузия реагента к поверхности катализатора сопровождает¬
ся протеканием каталитической реакции, скорость которой для
* Франк-Каменецкий Д.А. Диффузия и теплопередача в химической кинети¬
ке. — М.: Наука, 1987. — 492 с.
15. Зак. 1596
225
реакции первого порядка прямо пропорциональна поверхност¬
ной концентрации вещества
(10.65)
При достижении стационарного состояния скорости диффу¬
зии и поверхностной химической реакции равны, следовательно
Выразив из формулы (10.66) поверхностную концентрацию ве¬
щества через объемную
и подставив выражение (10.67) в (10.65), получим общее выраже¬
ние для гетерогенно-каталитического процесса, отвечающее ки¬
нетике первого порядка
Таким образом, обратная величина Аэф (общего коэффициента
массопередачи) равна сумме обратных величин |3 и к, характери¬
зующих соответственно процессы диффузии и химической реак¬
ции.
Рассмотрим предельные случаи уравнения (10.68). Если
(диффузия к поверхности осуществляется быстрее химической ре¬
акции), то величиной к в знаменателе уравнения (10.68) можно
пренебречь, после чего оно переходит в уравнение первого по¬
рядка относительно Су
Сравнивая уравнения (10.70), (10.68) и (10.65), получаем к^-к
и Cs ~ Cv. Эту область протекания каталитического процесса на¬
зывают внешнекинетической, ее отличительные особенности ука¬
заны выше.
Если 3 «А: (химическая реакция протекает заметно быстрее диф¬
фузии), то уравнение (10.68) принимает вид
kCs=V(Cv-Cs).
(10.66)
(10.67)
(10.68)
Соотношение к^ =
-—- для анализа удобно представить в виде
к + Р
1 _ 1 1
£эф 3 к’
(10.69)
vx.p — kCv.
(10.70)
^диф — 3 Су .
(10.71)
226
Сравнивая уравнения (10.71), (10.68) и (10.64), получаем £Эф~Р
и Cs ~ — Cv -» 0 . В этом случае процесс происходит во внешне-
к
диффузионной области. Учитывая, что скорость диффузии пропор¬
циональна концентрации в первой степени, в указанной области
химическая реакция любого кинетического порядка будет проте¬
кать как реакция первого порядка и подчиняться уравнению (10.71).
Отличительными признаками протекания процесса во внешне¬
диффузионной области являются зависимость скорости диффу¬
зии от скорости газового потока и интенсивности перемешива¬
ния жидкой фазы, размера зерен катализатора; слабая зависи¬
мость скорости процесса от температуры; отсутствие зависимости
скорости диффузии от пористости катализатора и диффузионно¬
го сопротивления от времени. Кроме того, если процесс прервать
и возобновить через определенный период при первоначальных
условиях, то его кинетика будет характеризоваться теми же пара¬
метрами, что и до перерыва.
Если оба коэффициента Р и к величины одного порядка, обе
стадии оказывают влияние на скорость процесса (т. е. лимитирую¬
щей стадии нет) и необходимо использовать уравнение (10.68).
В этом случае считают, что процесс протекает в промежуточной
области.
Отметим, что для работы катализатора в трех указанных выше
областях (внешнедиффузионной, промежуточной и внешнекине¬
тической) важное значение имеет величина внешней поверхнос¬
ти катализатора, но не пористость его зерна, поэтому в этих обла¬
стях используют в качестве катализаторов непористые вещества.
Для процессов, протекающих во внешнедиффузионной (и про¬
межуточной) областях, увеличение линейной скорости газового
потока и интенсивности перемешивания, а также уменьшение раз¬
мера зерен вызывают рост скорости диффузии и переход процес¬
са во внешнекинетическую область.
Рассмотрим влияние температуры на скорость процесса в раз¬
ных областях. Увеличение температуры приводит к резкому увели¬
чению скорости во внешнекинетической области, так как зависи¬
мость константы скорости реакции к от температуры выражается
уравнением Аррениуса (1.61). Напомним, что энергия активации
Еа для поверхностных химических реакций может достигать сотен
кДж/моль. Напротив, во внешнедиффузионной области коэффи¬
циент массообмена (3 зависит от температуры в существенно мень¬
шей степени, чем константа скорости химической реакции. При¬
чина этого заключается в следующем. Коэффициент диффузии D
(пропорциональный р) изменяется с температурой по уравне¬
нию, аналогичному уравнению Аррениуса
Ed
Z) = A,e«\ (К).72)
15*
227
где D0 = -z гг; г — радиус зерна; ri0 — коэффициент вязкости,
6nrr\0Na
очень слабо зависящий от температуры; Ed — энергия активации
диффузионного процесса. Для газов и жидкостей эта величина су¬
щественно меньше энергии активации химической реакции и со¬
ставляет 4—16 кДж/моль, поэтому температурный коэффициент
скорости диффузии близок к единице. При значительном повы¬
шении температуры, когда к »|3, процесс переходит из внешне¬
кинетической во внешнедиффузионную область.
Внутренняя диффузия. Для большинства пористых катализато¬
ров, используемых в промышленности, суммарная площадь внут¬
ренней поверхности пор на несколько порядков больше площади
внешней поверхности катализатора. Поэтому гетерогенно-катали¬
тический процесс при определенных условиях может характери¬
зоваться равенством концентраций реагента на внешней поверх¬
ности (у входа в пору) катализатора и в объеме газовой (или жид¬
кой) фазы, Cv = Су, а также наличием внутренней диффузии
исходного вещества за счет разности концентраций на внешней
поверхности и внутри пор.
Характер массопереноса внутри пор отличается от внешней (мо¬
лекулярной) диффузии и зависит во многом от соотношения дли¬
ны свободного пробега молекул Т и диаметра поры d = 2г.
Для узких пор, если Т > d, существенно возрастает возмож¬
ность столкновения молекулы со стенкой поры, в этих условиях
эффективный коэффициент диффузии пропорционален диамет¬
ру поры (речь идет о кнудсеновском течении). Для широких пор,
если / < d, принимают, что массоперенос подчиняется закону
Фика для молекулярной диффузии в объеме, в этом случае коэф¬
фициент диффузии пропорционален длине свободного пробега
молекул /. Если внутри пор протекает гетерогенно-каталитическая
реакция, необходимо учитывать дополнительный поток вещества
внутри поры за счет разности давлений, внешнего и внутреннего,
в поре.
Кинетическая задача Зельдовича. Л. Д. Зельдович предложил рас¬
сматривать модель пористого зерна катализатора как сплошную
однородную неподвижную среду, в которую линейным массопе-
реносом доставляется с поверхности реагент, вступающий затем
на стенках поры в химическую реакцию, а продукты реакции диф¬
фундируют к поверхности.
Фактически речь идет о модельной поре, направленной вдоль
максимального градиента концентрации реагента от внешней по¬
верхности к центру зерна. Для сферического зерна модельная пора
является конической.
Адсорбция реагента на поверхности катализатора для упроще¬
ния не учитывается. В случае протекания внутри зерна каталити¬
228
ческой реакции первого порядка (В —»Р) второй закон Фика при
стационарной линейной диффузии запишется так:
Здесь первое слагаемое указывает на изменение количества ве¬
щества В в процессе диффузии, второе — за счет каталитической
реакции (знак — показывает на расходование вещества В). Дф —
эффективный коэффициент диффузии вещества В в поре зерна;
кзф — эффективная константа скорости реакции. Эти величины
должны учитывать особенности массопереноса в порах и отличие
упрощенной модели от реального зерна катализатора.
Уравнение (10.73) решается при различных краевых условиях,
выберем следующие:
а) С = Су при х = 0 (х — расстояние от поверхности зерна), где
Су — концентрация Св на поверхности зерна;
dC „ d .
б) — = 0 при х = —; d — диаметр зерна.
dx 2
Смысл второго условия заключается в том, что диффузия от
поверхности поры для х=0 и x=d к ее центру симметрична, по¬
этому касательная к кривой в координатах (С, х) прих = ^ гори¬
зонтальна.
Вначале запишем решение (10.73) в форме С = е*/х, где X —
некоторый коэффициент. Взятие второй производной от С, под¬
становка полученного выражения в (10.73) после сокращения на
е-*Л позволяет получить уравнение
откуда путем извлечения квадратного корня находим выражения
Для двух коэффициентов
Следовательно, имеем два частных решения уравнения (10.73)
(10.73)
(10.74)
(10.75)
с, = e'L И с2 = e~x/i
и общее решение принимает вид
С = Axe>L + A2rx'L ,
гДе А\ и А2 — коэффициенты.
(10.76)
(10.77)
229
Для нахождения коэффициентов Д, и Д2 используем краевые
условия. При первом условии из (10.77) находим С= Cs,
Cs = Л\ + Д2 и А2 = С^ ~ А\. (10.78)
При втором краевом условии из выражения (10.77) получим
^died/(2L) _ 2^2 e-d/(2L) _ Q ^ (10.79)
а а
Решая совместно уравнения (10.78) и (10.79), определяем Д,
и Д2
Л^Т^- <10'80)
После подстановки значений Ах и Д2 из уравнения (10.80) в
формулу (10.77) приходим к искомой зависимости концентрации
реагента от параметра х в интервале от входа в пору (х=0) до ее
/ d ч
центра (х = - = г)
<1(Ш>
Отметим, что, согласно выражению (10.75), L — параметр,
зависящий от эффективных значений коэффициента диффу¬
зии и константы скорости химической реакции внутри поры,
L — yj Дэф / ^эф •
Оценим из соотношения (10.81) отношение концентраций ре¬
агента В в центре (x=d/2) и на поверхности зерна, у входа в пору.
Эта величина характеризует степень использования внутренней
поверхности поры, обозначим ее через /1/2:
( С Л 2ed/W
•'HzH =ih^- (10-82)
)x=d/2 1 + e
Рассмотрим предельные случаи. Если выполняется соотноше¬
ние 1 > f\/2 £0,99 или 1 - /1/2 <0,01, концентрация реагента в цен¬
тре зерна фактически не отличается от его поверхностной кон¬
центрации, поэтому скорость процесса лимитируется не массопе-
реносом, а скоростью химической реакции на внутренней
поверхности зерна. Считают, что катализатор работает во внутри-
кинетической области. В этом случае параметр L превосходит диа¬
метр зерна в несколько раз. Действительно, преобразуя уравнение
(10.82) с учетом замены e‘//(2i) = у, получим квадратное уравнение
1 + у
230
Поскольку d и L всегда > 0, искомое значение у=1,15, d/(2L) =
= 1пу = 0,14, откуда L> 3,6d.
Во внутренней кинетической области скорость процесса не за¬
висит от размера зерен катализатора, но в реальных процессах
удобнее использовать крупные зерна для уменьшения гидравли¬
ческого сопротивления потоку газа или жидкости через слой ка¬
тализатора.
Во втором случае положим Л/2 ^ 0,01, т.е. концентрация в цен¬
тре зерна близка к нулю, что очевидно связано с трудностями
внутреннего массопереноса. Считают, что катализатор работает во
внутридиффузионной области. В этих условиях параметр L суще¬
ственно меньше диаметра зерна, поэтому, полагая exp(d/Z)»l,
уравнение (10.82) можно упростить и выразить через него L
f/2 = 2e~rf/(2i) или L =
2 In
2 v
Л/2
(10.83)
Для Л/2 — 0,01 из (10.83) L<0,ld. Поэтому действительно ed/i» 1.
На основании полученных выше математических соотношений
выделим отличительные признаки протекания процесса во внут¬
ридиффузионной области.
Количество вещества В, доставляемое на внутреннюю поверх¬
ность зерна, равно тому количеству, которое поступает за счет
внешней (линейной) диффузии на поверхность зерна у устья поры,
поэтому для определения скорости массопереноса внутрь зерна
используем первый закон Фика
^вндиф — ^эф
^dC, л
dx
(10.84)
Jx=0
Для нахождения производной в правой части соотношения
(10.84) воспользуемся уравнением (10.81), которое при малых L
после несложных операций приходит к виду
Св = Csg~x/l.
Производная равна
'dC, л
dx
= -9s_p-x!l
х=0 -
х=0
L
(10.85)
(10.86)
Выражение (10.84) для скорости внутренней диффузии с уче¬
том (10.86) и (10.75) запишется так:
D.
^вн-диф = —j^-SCs - Зу]ВэфкЭфС$.
(10.87)
231
Если отнести увнл,иф к единице объема пористого катализатора
Выясняется, что скорость процесса в этой области пропорцио-
Д ф), но и химической реакцией (через к^). В отличие от внешне¬
диффузионной области здесь отсутствует лимитирующая стадия.
Параметр L, как видно из выражений (10.85) и (10.87), оказы¬
вает существенное влияние на процесс в целом. Уменьшение L
приводит к уменьшению толщины работающего слоя зерна. При
значительном уменьшении L процесс может перейти во внешне¬
кинетическую область, поскольку сама каталитическая реакция
успевает осуществиться на внешней поверхности зерна. Объемная
скорость процесса, согласно уравнению (10.88), возрастает с умень¬
шением диаметра зерна, т. е. с измельчением катализатора.
Анализируя уравнение (10.88) и учитывая слабую зависимость
Дэф от температуры (см. формулу (10.72)), можно сделать вывод,
что наблюдаемая энергия активации во внутридиффузионной об¬
ласти примерно равна половине энергии активации реакции во
внутрикинетической области
Отметим, что в предложенной модели зерна влияние размера
пор на скорость внутридиффузионного процесса не рассматри¬
вается.
Кинетическая задача и параметр Тиле. Е.Тиле рассматривал в
качестве модели цилиндрическую пору диаметром 2г и длиной 21,
направленную вдоль максимального градиента концентраций веще¬
ства от внешней поверхности зерна к середине плоской пластины.
Запишем уравнение материального баланса для бесконечно ма¬
лого элемента поры dx. Скорость диффузионного накопления ве¬
щества, определяемая по разности диффузионных потоков через
сечение поры (я г2) внутрь ее и обратно, согласно закону Фика,
равна
где Д — истинный коэффициент диффузии реагента в порах.
' ^ ^ ™ з ' d
уравнение (10.87) примет окончательный вид
4 1 6
(для сферической формы зерна S = 4яг2 V = —яг и S/V = — ),
(10.88)
нальна ^Аф^ф > т-е- контролируется не только диффузией (через
(10.89)
^вндиф
(10.90)
232
Скорость химической реакции характеризует убыль вещества
на внутренней поверхности поры площадью 2пгдх и для реакции
первого порядка (выбор порядка реакции связан с удобством по¬
лучения аналитического решения задачи) равна
dt>x.p = -2пгкСйх. (10.91)
Из условия стационарного течения процесса (^ = 0), исполь¬
зуя формулы (10.90) и (10.91), получим
Air
itr2D-^-f dx = 2nrkCdx (10.92)
или
0 = (10.93)
Для решения уравнения (10.89) выбраны следующие краевые
d С Л
условия: С= Су при х=0 и — = 0 при х=1 (по условию симмет¬
рии), где / — половина толщины пластины. Вначале осуществим
переход к безразмерным переменным. Вводя безразмерную кон¬
центрацию % = С/Су, преобразуем уравнение (10.93) к виду
<|0-94>
Обозначив через характерную длину поры, равную
rD
V='2к) ’ (10'95)
введем безразмерную координату л = —
¥
г) = Мх. (10.96)
V rD
Теперь уравнение (10.94) в безразмерных координатах примет вид
бц
Решение уравнения (10.97) находим с учетом новых краевых
2 = 5- (10.97)
jt
условий 4=1 ПРИ Л =0 и -^ = 0 при максимальном значении л,
ал
233
где А — безразмерная величина, называемая параметром Тиле. Ана¬
литическое решение уравнения (10.97) имеет вид
С _ сЬ(Л-л)
С5 ch(A) ’
(10.99)
где гиперболический косинус
(10.100)
и г| изменяется в пределах 0 < т| < А.
Параметр А оказался удачным критерием для определения внут-
ридиффузионной и внутрикинетической областей протекания про¬
цесса. Как видно из формулы (10.98), А зависит от отношения
константы скорости реакции к коэффициенту диффузии, а также
радиуса, размера пор и (через D) от температуры. Анализ режима
работы гетерогенного катализатора удобно проводить с помощью
величины /, равной отношению опытной скорости реакции (с
учетом внутридиффузионного торможения) к максимальной, когда
отсутствует градиент концентрации вещества по длине поры.
Величину/в литературе называют по-разному: фактором диф¬
фузионного торможения, фактором эффективности, относитель¬
ной глубиной протекания реакции, степенью использования по¬
верхности.
Для нахождения скорости внутренней диффузии воспользуем¬
ся первым законом Фика, поскольку эта скорость должна быть
равна скорости переноса реагента через внешнюю поверхность
поры (для х=0)
Находим теперь значение производной в формуле (10.102)
von _ ^вндиф
(10.101)
^тах ^вн.х.р
2 п
^вндиф — D ,
)х=0
Дифференцируя £ из (10.95) по г), получим
(10.102)
(10.103)
(10.104)
234
Подставляя выражение (10.104) в формулу (10.102), приходим
к искомому выражению для скорости гетерогенного процесса с
учетом внутренней диффузии
vBH^ = nr2DCSjth(h). (10.105)
Для химической реакции внутри поры в отсутствие диффузи¬
онного торможения (реакция протекает по всей внутренней по¬
верхности поры) скорость для половины длины поры равна
tWp = 2nrlkCs. (10.106)
Получим теперь выражения для фактора диффузионного тор¬
можения с учетом (10.105) и (10.106)
f Ath(A) th(A) .. 1 е*-е~* ПО 1071
7 " (2k/rD)l2 h h eh + е_Л ’ 1 '
Для небольших значений h (<0,2), как видно из (10.107),
th(A) - h и / - 1. (10.108)
В отсутствие внутридиффузионного торможения процесс пере¬
ходит во внутреннюю кинетическую область, его скорость конт¬
ролируется скоростью химической реакции на внутренней поверх¬
ности пор с энергией активации Дн кин, определяемой по уравне¬
нию Аррениуса (1.61).
Для больших значений Л (> 2), пренебрегая в числителе и зна¬
менателе выражения (10.103) ехр(-А), получим
th(A) = 1 и / = }. (10.109)
п
В этом случае фактор эффективности/<0,5 указывает на суще¬
ственное уменьшение концентрации вещества внутри поры, т.е.
уменьшение степени использования поверхности и переход ката¬
литического процесса во внутридиффузионную область. Скорость
реакции, согласно выражениям (10.105) и (10.109) будет равна
л к \1/2
^внлиф ” nr2D€s[%) = я2'/2/'3/2(*7))1/2с^ = Vi. (10-110>
V /
где
k^=comt4kD, (10.111)
т.е. эффективная константа опытной скорости реакции пропор¬
циональна среднему геометрическому из константы скорости ка¬
талитической реакции и коэффициента диффузии.
Учитывая слабую зависимость коэффициента диффузии в га¬
зовой фазе от температуры
235
D = ^vl = const Tl/1,
(10.112)
где v — средняя арифметическая скорость (5.7); / — средняя
длина свободного пробега (5.27); можно считать, что определяе¬
мая во внутридиффузионной области энергия активации пример¬
но равна половине Ешлмн для внутренней кинетической области,
как показано в (10.89).
Отличительными признаками протекания процесса во внутри¬
диффузионной области служат: зависимость скорости от диаметра
зерна, радиуса и извилистости пор катализатора; рост диффузи¬
онного сопротивления со временем и независимость скорости ка¬
талитического процесса от скорости движения газа и интенсив¬
ности перемешивания жидкой фазы.
При очень больших значениях А уравнение (10.99) можно за¬
писать иначе:
4=е-\ 0<л<~. (10.113)
d С .
Появляется новое граничное условие для х= /: кроме = 0
оказывается и С = 0, что позволяет получить аналитические ре¬
шения для реакций л-порядка во внутридиффузионной области.
При значениях А в интервале 0,2<h<2 каталитический про¬
цесс смещается в промежуточную область, для которой характер¬
но изменение опытной энергии активации от /v.khh Д° ^£внкин.
При решении задачи с учетом протекания химической реак¬
ции л-ro порядка, полученные выше уравнения и соотношения
примут следующий вид:
(10.89) ->
(10.90) ->
(10.91) -*•
(10.92) -»
(10.93) -»
(10.94) ->
d 2С
dx2
-2kC-
rD
(10.114)
d2S
dx2
(10.115)
V =
I rD
(10.116)
pkQ-1
Л = 1
(10.117)
d4
dr|2
(10.118)
A = ,
ёС!"'
(10.119)
236
рис. 10.3. Зависимость констан- lg*
ты скорости гетерогенно-катали¬
тической реакции от температу¬
ры в координатах Аррениуса для
крупнопористых катализаторов:
1 — внугрикинетическая; 2 — внут-
ридиффузионная; 3 — внешнеки¬
нетическая; 4 — внешнедиффузион¬
ная; — промежуточная
область
0 1/Г
Во внутридиффузионной области скорость процесса выража¬
ется соотношением
увижф=п-2^г^(кОу/2С^2. (10.120)
Как видно из формулы (10.120) наблюдаемый на опыте поря¬
док реакции равен среднему арифметическому между порядком
ее во внутрикинетической области и единицей, отвечающей по¬
рядку в диффузионной области.
Для реакций, протекающих на внутренней поверхности пор с
«лэнгмюровской» кинетикой, при определенных допущениях
А. Я. Розовским получены аналитические решения, имеющие до¬
вольно сложный вид*. Поэтому ограничимся математическими со¬
отношениями, полученными в рамках приближенного решения,
для фактора эффективности
_L rD0±W (l012l)
J 72/ i к \ + Kp/2 (10Л21)
и определяемой на опыте скорости реакции во внутридиффузи¬
онной области
г'вн.диф = я • 2'/2г3/2р^Т^рД- (10.122)
Как и в предыдущих случаях, скорость реакции ~ -JkD, следо¬
вательно, наблюдаемая энергия активации равна ^
В заключение рассмотрим зависимость протекания каталитиче¬
ского процесса в различных макрокинетических областях от темпе¬
ратуры на примере кривой Зельдовича, представленной на рис. 10.3.
* Розовский А.Я. Гетерогенные химические реакции. Кинетика и макрокине¬
тика. — М.: Наука, 1980. — 323 с.
237
При достаточно низких температурах в отсутствие диффузион¬
ного торможения (Ed Ехр) каталитическая реакция протекает
во внутренней кинетической области (участок 7). При повыше¬
нии температуры процесс последовательно переходит, как пока¬
зано на рис. 10.3, во внутридиффузионную (участок 2), внешне¬
кинетическую (участок 3) и внешнедиффузионную (участок 4)
области. По тангенсу угла наклона прямолинейного участка мож¬
но оценить энергию активации процесса в каждой из рассмотрен¬
ных областей. Из рис. 10.3 видно, что наименьшая энергия актива¬
ции отвечает внешнедиффузионной области, она составляет, как
указывалось ранее, 4—16 кДж/моль. Таким образом, варьирова¬
ние температуры может служить «ключом» для перехода в ту или
иную макрокинетическую область каталитического процесса.
Вопросы и задания для самоконтроля
1. Какие реакции называются гетерогенными? Приведите примеры.
2. Какое понятие используют для объяснения каталитического дей¬
ствия в гетерогенном катализе? Охарактеризуйте его.
3. В чем заключается основа каталитического действия?
4. Какие факторы определяют активность гетерогенного катализатора?
5. Сформулируйте «правило Борескова» о приблизительном посто¬
янстве удельной каталитической активности веществ.
6. По какому принципу классифицируют гетерогенные катализато¬
ры? Какие реакции относятся к процессам: а) окисления —восстановле¬
ния; б) кислотно-основного типа? Какие катализаторы используются в
этих процессах?
7. В чем заключается отличие каталитических реакций с твердыми
кислотами от аналогичных реакций в растворе?
8. Дайте определение цеолитов и характеристику их свойств. Приве¬
дите примеры их использования в катализе.
9. Приведите примеры промышленных процессов с использовани¬
ем многокомпонентных и многофазных катализаторов.
10. Объясните механизм действия бифункционального катализатора
в процессе риформинга бензиновых фракций.
11. Какие вещества называют промоторами? Каким образом они ока¬
зывают влияние на каталитический процесс?
12. Какие вещества называются в катализе ядами? Приведите конк¬
ретные примеры воздействия ядов на каталитические процессы.
13. Каким образом проводят оценку степени отравления катализатора?
14. В чем заключается позитивная роль использования носителя в ге¬
терогенном катализе?
15. В чем сходство и различие между физической адсорбцией и хемо¬
сорбцией? Какой процесс приводит к каталитическому эффекту?
16. Сформулируйте основные положения адсорбционной теории Лэнг¬
мюра. В чем заключается кинетический закон действующих поверхностей?
17. Приведите кинетический вывод уравнения изотермы Лэнгмюра и
проанализируйте его.
238
18. Приведите термодинамический вывод уравнения Лэнгмюра.
19. При каком значении давления р степень заполнения поверхности 0
равна У2, а касательная к начальному участку кривой Лэнгмюра дости¬
гает значения 0=1?
20. Покажите, что для определения параметров уравнения Лэнгмюра
удобно использовать графические способы в следующих координатах:
а)
Р;
б)
в)
-,Р
а /
V х у ^ ' \ ж У
21. Получите выражение для степени заполнения в случае диссоциа¬
тивной адсорбции молекулы А2.
22. Получите аналитическим путем выражения для степеней запол¬
нения при одновременной адсорбции двух газов.
23. В чем причины отклонения реальных изотерм от модели «идеаль¬
ного адсорбированного слоя» Лэнгмюра?
24. Какой вид имеют уравнения для изотерм: а) Генри; б) Фрейнд¬
лиха; в) в логарифмической форме?
25. Сформулируйте основные допущения теории полимолекулярной
адсорбции, проанализируйте уравнение БЭТ, укажите его роль в ката¬
лизе и пределы применимости.
26. Укажите, из каких стадий состоит гетерогенно-каталитическая
реакция? Каким образом определяют общую скорость такого процесса?
27. Покажите механизм, по которому протекает мономолекулярная
гетерогенно-каталитическая реакция. Выведите уравнение для скорости
этой реакции и проанализируйте его.
28. Какая существует связь между кажущейся энергией активации и
истинной для гетерогенной каталитической реакции? Ответ поясните.
29. Какой вид имеет связь между кажущейся и истинной энергиями
активации в случае адсорбции исходного вещества и продукта реакции?
30. В чем заключается механизм Лэнгмюра—Хиншельвуда? Получите
уравнение для скорости реакции в этом случае и проанализируйте его.
Приведите пример реакции, протекающей по данному механизму.
31. В чем заключается механизм Или — Ридиела? Получите уравнение
для скорости реакции в этом случае. Приведите пример реакции, проте¬
кающей по данному механизму.
32. Какой прием можно использовать для установления четкого раз¬
личия между механизмами Лэнгмюра—Хиншельвуда и Или —Ридиела?
33. Перечислите возможные отличия гетерогенно-каталитических ре¬
акций, протекающих в жидкой фазе, от реакций в газовой фазе.
34. Какую роль играет термодинамический фактор в объяснении осо¬
бенностей протекания жидкофазных реакций?
35. В чем заключается электрохимический механизм катализа? Для
каких процессов он характерен?
36. ^Перечислите основные макрокинетические области в гетероген¬
ном катализе и охарактеризуйте каждую из них.
37. Укажите отличительные особенности каталитических процессов
во внешнекинетической области.
38. Сформулируйте в аналитическом виде первый и второй законы
Фика.
239
39. Какой параметр вводится теорией конвективной диффузии Нер-
нста и от чего он зависит?
40. Получите в аналитическом виде уравнение для скорости гетеро¬
генно-каталитического процесса с учетом внешней диффузии и хими¬
ческой реакции на поверхности катализатора.
41. Проанализируйте предельные случаи полученного в задании 40 урав¬
нения. * •
42. Перечислите отличительные признаки протекания процесса во
внешнедиффузионной области.
43. При каких условиях процесс протекает в промежуточной области?
44. Сравните влияние температуры на скорость процесса во внешне¬
кинетической и внутридиффузионной областях.
45. В чем заключается отличие внутренней диффузии от внешней?
46* Сформулируйте кинетическую задачу Зельдовича и приведите ее
аналитическое решение для реакции первого порядка.
47*. Проанализируйте полученное в вопросе 46 уравнение с учетом
степени использования внутренней поверхности /1/2 и параметра L.
48. Охарактеризуйте условия протекания процесса во внутренней ки¬
нетической области.
49*. Получите в аналитическом виде выражение для скорости процес¬
са во внутридиффузионной области.
50. Какая существует количественная связь между опытной энергией
активации во внутридиффузионной области и энергией активации во
внутренней кинетической области?
51. Какую роль играют во внутридиффузионной области параметр L и
диаметр зерна?
52*. Сформулируйте кинетическую задачу Тиле и приведите ее анали¬
тическое решение для реакции первого порядка.
53. От каких величин зависит параметр Тиле Л?
54. Какую величину называют фактором диффузионного торможения?
Получите ее аналитическое выражение для реакции первого порядка.
55. При каких значениях h каталитический процесс протекает во внут-
рикинетической и внутридиффузионной областях?
56. Какое соотношение между энергиями активации в указанных выше
внутренних областях получено в рамках задачи Тиле?
57. Перечислите особенности протекания процесса во внутридиффу¬
зионной области.
58. При каких значениях h появляется дополнительное краевое усло¬
вие для решения задачи Тиле?
59*. Приведите выражение для скорости процесса во внутридиффузи¬
онной области с учетом протекания химической реакции л-го порядка.
Чему будет равен наблюдаемый на опыте порядок реакции?
60*. Оцените значение опытной энергии активации процесса во внут¬
ридиффузионной области при наличии реакции с «лэнгмюровской» ки¬
нетикой.
61. Дайте характеристику метода равнодоступной поверхности.
62. Покажите, каким образом в задаче Зельдовича параметр L зависит
от значений коэффициента диффузии и константы скорости химичес¬
кой реакции.
240
63. Какую роль в задаче Тиле играет фактор диффузионного торможе¬
ния?
64. В какой области «работает» катализатор, если для степени исполь¬
зования его внутренней поверхности /,/2 в задаче Зельдовича выполняет¬
ся соотношение 1 - У1/2 < 0,01.
65. В какой области «работает» катализатор, если выполняется соотно¬
шение /i/2< 0,01. Перечислите отличительные признаки протекания про¬
цесса в этой области.
66. При каких значениях фактора эффективности /в задаче Тиле ката¬
литический процесс протекает во внутрикинетической и внутридиффу¬
зионной областях?
ЛИТЕРАТУРА
Рекомендуемая
Еремин Е.Н. Основы химической кинетики. — М.: Высш. шк., 1976.
Панченков Г. М., Лебедев В. П. Химическая кинетика и катализ. — М.:
Химия, 1985.
Семиохин И. А., Страхов Б. В., Осипов А. И. Кинетика химических ре¬
акций. — М.: Изд-во МГУ, 1995.
Эмануэль Н.М., Кнорре Д. Г. Курс химической кинетики. — М.: Высш.
шк., 1984.
Дополнительная
Бенсон С. Основы химической кинетики. — М.: Мир, 1964.
Боресков Г.К. Гетерогенный катализ. — М.: Наука, 1986.
Глесстон С., Лейдлер К., Эйринг Г. Теория абсолютных скоростей реак¬
ций. — М.: Иностранная литература, 1948.
Денисов Е. Т. Кинетика гомогенных химических реакций. — М.: Высш.
шк., 1988.
Иоффе И.И., Решетов В.А., Добротворский А.М. Гетерогенный ката¬
лиз. — Л.: Химия, 1985.
Катализ (Фундаментальные и прикладные исследования) / Под. ред.
О. А. Петрия и В. В.Лунина. — М.: Изд-во МГУ, 1987.
Киперман С.А. Основы химической кинетики в гетерогенном катали¬
зе. — М.: Химия, 1979.
Кондратьев Б.Н., Никитин Е.Е. Термические бимолекулярные реак¬
ции в газах. — М.: Наука, 1976.
Полторак О.М. Лекции по теории гетерогенного катализа. — М.: Высш.
шк., 1968.
Полторак О.М. Термодинамика в физической химии. — М.: Высш. шк.,
1991.
Полторак О. М. Чухрай Е.С. Физико-химические основы ферментатив¬
ного катализа. — М.: Высш. шк., 1971.
Робинсон Я, Холбрук К. Мономолекулярные реакции. — М.: Мир, 1975.
Семенов Н.Н. О некоторых проблемах химической кинетики и реакци¬
онной способности. — М.: Изд-во АН СССР, 1958.
Семенов Н.Н. Цепные реакции. — М., Л.: Госхимтехиздат, 1934.
242
Стромберг А.Г., Семненко Д.П. Физическая химия. — М.: Высш. шк.,
1999.
Физическая химия в вопросах и ответах / Под ред. К. В. Топчиевой,
Н.В.Федорович. — М.: Изд-во МГУ, 1981.
Физическая химия / Под ред. К.С. Краснова. — М.: Высш. шк., 1995.
Химическая и биологическая кинетика / Под ред. Н. М. Эмануэля,
И. В. Березина, С. Д. Варфоломеева. — М.: Изд-во МГУ, 1983.
Штиллер В. Уравнение Аррениуса и неравновесная кинетика. — М.:
Мир, 2000.
Atkins P. Physical Chemistry. — 5th Edit. — Oxford Univ. Press, 1994.
Bowker M. The Basis and Applications of Heterogeneous Catalysis. — Oxford
Univ. Press, 1998.
Laffitte M., Rouquerol F’ La reaction chimique. — Paris: Masson, 1990.
Logan S. Introduction a la cinetique chimique. — Paris: Dunod, 1998.
Mortimer R. Physical Chemistry. — California: The Benjamin, 1993.
Wayne C.E., Wayne R.P. Photochemistry. — Oxford Univ. Press, 1996.
Wright M.R. Fundamental Chemical Kinetics. — Horwood Publishing
Chichester, 1999.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автокатализ 143
Автокаталитические реакции 143
, кинетические уравнения
144, 147
, механизм кинетики 143, 147
, период индукции 147
Адсорбат 205
Адсорбент 205
Адсорбция 205
—, изотерма 209, 210, 213—215
—, константа (коэффициент) 209
—, механизм 208, 212
— физическая 205, 207
—, хемосорбция 205—207
—, энтальпия 209, 219
—, энтропия 209
Активированный комплекс 114
, основное уравнение 115
, статистический аспект 115
, степени свободы 116
, термодинамический аспект
119-120
, трансмиссионный коэффи¬
циент 115
, энергия активации 119
Активность
— катализатора 131, 196
, общая 131, 196
, удельная 131, 196
— ферментов 168
— —, молекулярная 170
, удельная 170
Активный центр 130
— — в гетерогенном ката¬
лизе 194
фермента 151
в цепных реакциях 54
Актинометр 80
Акцептор
—, по Бренстеду 173
—, по Льюису 174
Апротонный растворитель 174
Аррениуса
— уравнение 25
, дифференциальная
форма 28
, параметры 25, 28
, в современном виде 30
, энергия активации 25
Барометрическая формула 96
Бимолекулярные реакции 101
Боденштейна принцип 52
Больцмана
— постоянная 95
— множитель 101
— распределение по энергиям 96
— распределение во внешнем
потенциальном поле 96
Бренстеда
— энергетические соотношения
187, 188
— уравнение 186
Бренстеда—Бьеррума уравнение
185
Вакер-процесс 129
Вековое равновесие 50
, уравнение 27
Вероятностный фактор, см, Сте-
рический множитель
Верхний предел воспламенения 62
Взаимодействие цепей
положительное 67
Внешняя диффузия 224
Внутренняя
— диффузия 228
244
— конверсия 77
— энергия 34
Возбуждение
— модулированное 83
— импульсное 83
— стационарное 83
Время жизни
молекулы (среднее) 12
синглета 79
триплета 80
— полупревращения 11
— превращения на определенную
долю 21
— релаксации 37
Вырожденное разветвление
цепей 68
Вязкость 99
Гаммета
— уравнение 189
— функция кислотности 191
Генри изотерма адсорбции 214
Герца уравнение 98
Гетерогенный катализ 126, 194
Гетеролитический механизм 126
Гомогенный катализ 126
Гомолитический механизм 126
Грехема закон 99
Гудри катализатор 199
Давление перехода 108
Дезактивация 106
Десорбция 217
Двойные столкновения 97
, частота 97
, число 97
Диссоциация молекул
— —, в водном растворе 173
, под действием света 76
Диффузионный слой Нернста 224
Диффузия
— внешняя 224
— внутренняя 228
кнудсеновская 228
коэффициент 225
" линейная 228
скорость 225
стационарная 224
Длина цепи 57
Донор
протона 173
— электронной пары 174
Дубинина уравнение 216
Задача
— Зельдовича 228
— Тиле 232
— химической кинетики 11
, прямая 11
, обратная 12
Закон(ы)
— Больцмана 96
— Буге—Ламберта—Бера 73
— Вант-Гоффа 74
— Гротгуса—Дрепера 74
— Максвелла 95
— Фика 225, 229
— фотохимии 74
— эффузии Грехема 99
— Эйнштейна-Штарка 74
Зарождение цепей 55
Звено цепи 57
Зельдовича
— задача 228
— кривая 237
Изотерма адсорбции 209, 214
Генри 214
Лэнгмюра 209
Темкина 214
Фрейндлиха 214
Или — Ридиела механизм 222
Ингибирование
— бесконкурентное 162
— конкурентное 162
— неконкурентное 162
— смешанное 173
— субстратом 173
— ферментативных реакций 161
Инициирование, см. Зарождение
цепей
Ионная сила раствора 183
Ионообменные
смолы (иониты) 201
Интеркомбинационная
— конверсия 78
— деградация 85
Истинная энергия активации 115
Катализ 124, 125
— активационный 128
— гетерогенный 126
245
Катализ (продолжение)
— гетеролитический 126
— гомогенный 126
— гомолитический 126
— кислотно-основной 173
— координационный 128
— нуклеофильный 177
— общий кислотный 176
— общий основной 177
— переносный 127
— специфический кислотный 175
— специфический основной 176
— ферментативный 126
— электрофильный 176
—, механизм
—, — пуш-пульный 177
—, — слитный 140
—, — стадийный 141
Катал изатор(ы) 125, 194
—, активность 131, 196
—, активный центр 130, 194
— бифункциональные 203
— гетерогенные 195
— гомогенные 127, 129, 132
— кислотно-основные 200
, иониты 201
, цеолиты 201, 202
— металлические 198
— Митташа 202
— оксидные 199
—, отравление 204
—, приготовление 199
—, промотирование 203
—, поверхность 131, 195
—, селективность 131
—, спекание 204
—, старение 204
Каталитические
— системы 203
— яды 204
Квадратичный обрыв цепей 57, 69
Квантовый выход
вторичный 75
общий 75
первичный 75
флуоресценции 84, 86
фосфоресценции 85
Кинетика химических реакций
гетерогенно-каталити¬
ческих 216
гомогенных 177
обратимых 33
односторонних 10
параллельных 39
последовательных 47
фотохимических 81, 82
цепных 58
с вырожденным развет¬
влением 69
с положительным взаи¬
модействием
цепей 67
— неразветвленных 59
— разветвленных 60
Кинетическая
— кривая 23
— область 60
Кислота(ы)
—, по Аррениусу 173
—, по Бренстеду и Лоури 173
—, по Льюису 174
—, по Усановичу 175
—, каталитическое действие 182
Кислотность среды 191
Колебательная релаксация 77
Конверсия
— внутренняя 77
— интеркомбинационная 78
Константа
— Михаэлиса 156
— равновесия 34, 115, 184, 209
— тушения 89
Константа скорости реакции 7
активированного комп¬
лекса 115
бимолекулярной 102
кажущаяся 24, 34, 110,
178, 211
мономолекулярной 107,
109
обратимой 34
односторонней 11, 13,
16, 18
параллельной 40, 44
последовательной 47, 48
размерность И, 13, 16,
19
способы определения
20, 23
Координата реакции 115
Корреляционные соотношения 186
Бренстеда 187, 188
Поляни 188
Семенова 188
Кофакторы 152
Коэффициент(ы)
— адсорбционный 209
— активности 184, 190
— диффузии 100, 225
— массообмена (массопередачи)
225
— отравления 204
— прохождения (трансмиссион¬
ный) 115
— температурный 25
— теплопроводности 100
Крекинг 124, 202
—,гидрокрекинг 124, 202
Кривая Зельдовича 237
Лимитирующая стадия 32, 175,
181
Линейный обрыв цепей 56, 69
Линдемана теория 106
Лэнгмюра изотерма 209
Лэнгмюра—Хиншельвуда
механизм 220
Максвелла распределение
по скоростям 95
Макрокинетика 224
Метод(ы)
— определения порядков
реакций 19
дифференциальный 23
изолирования Ост¬
вальда 23
интегральный 19
— равнодоступной
поверхности 225
— релаксации 37
Михаэлиса константа 156
Михаэлиса—Ментен
уравнение 157
Механизм(ы)
— адсорбции 208, 212
— Или—Ридиела 220
— каталитических реакций 132,
134
— Лэнгмюра —Хиншельвуда 222
— пуш-пульный 177
— Штерна —Фольмера 81
Молекулярность реакции 8
Молекулярно-кинетическая
теория газов 94
Мономолекулярные реакции
105-114
Мыс полуострова
воспламенения 72
Неконкурентное ингибирование
162, 165
Неразветвленные цепные
реакции 57
Нижний предел воспламенения
62
Носители катализаторов 203,
204
Нуклеофил ьность 175
Область гетерогенного катализа
внешнедиффузионная
224, 227
внешнекинетическая
224
внутридиффузионная
224, 228
внутрикинетическая
224, 230
Обратимые реакции 33 — 39
Обрыв цепей 56
квадратичный 57
линейный 56
на стенках 59
Односторонние реакции 10
Основания 173, 175
Основной постулат химической
кинетики 7
Оствальда
— правило 183
— метод изолирования 23
Параметр Тиле 232, 234
Первичный солевой эффект 183
Переходное равновесие 50
Переходы
—, внутренняя конверсия 77
— излучательные 79
— интеркомбинационные 78
— электронные 76
Период
— индукции 51, 147
— полураспада 11
247
Поверхность
— катализатора 130, 194
— потенциальной энергии 114
Полуостров воспламенения 62
Порядок реакции 7 — 8
дробный 21
общий 7
псевдо 7
, способы определения 19—25
Правило
— Борескова 197
— Вант-Гоффа 25
— Оствальда 183
Пределы воспламенения
верхний 62
, зависимость от темпера¬
туры 62
нижний 62
третий 63
Принцип
— квазиравновесных концентра¬
ций 52
— квазистационарных
концентраций 47, 52
— лимитирующей стадии 32
— микрообратимости (детального
равновесия) 32
— независимости реакций 32
— неуничтожимости свободной
валентности 71
— частично-стационарных кон¬
центраций 61
Пробег, средняя длина 99
Продолжение цепей 55
Промоторы 203
Простетические группы 151, 152
Процесс(ы)
— адиабатический 114
— активации 106, 111
— Вакер- 129
— вторичные 81
— квазистационарный 61
— первичные 76, 80
— спонтанный (взрывной) 67
— стационарный 58, 62, 64
Равновесие
— адсорбционное 209
— вековое 50
— переходное 50
Радикалы свободные 55
Разветвление цепей 55, 56
Развитие цепи 59
Реакции
— авто каталитические 143
— бимолекулярные 101
— во внутридиффузионной
области 224, 231, 234
— второго порядка 12
— с вырожденным разветвлением
68, 69
— гетерогенные 194
— гетеролитические 126
— гомогенные 175
— гомолитические 126
— каталитические 126
— с «лэнгмюровской» кинетикой
237
—, механизм 8, 126, 132, 155
—, молекулярность 8
— мономолекулярные 105 — 114
— нулевого порядка 15
— обратимые 33 — 39
— обрыва цепи 55 — 57
— односторонние (необратимые)
10-19
— параллельные 39 — 46
— первого порядка 10 — 12
— поверхностные 206
—, порядок 7 — 8
—, —, способы
определения 19 — 25
— последовательные 47 — 52
— радикальные 55
— развития цепи 55
—, скорость 5
—, —, константа 7
— сложные 32, 33
— третьего порядка 16 — 17
— тримолекулярные 8, 118
— ферментативные 150, 154
— Фишера —Тропша 200
— фотохимические 72, 73
— цепные 54, 72
— — с вырожденым разветвлени¬
ем 68, 69
— — неразветвленные 57 — 60
— — разветвленные 60 — 68
— элементарные 7, 56
Рекомбинация атомов
и радикалов 19, 81
Релаксации метод 37, 38
248
Самоингибирование 173
Селективность катализатора 131
дифференциальная 132
интегральная 30
Сезерленда формула 103
Синглетное состояние 77, 79, 85
Скорость
— диффузии 225
— наиболее вероятная 95
— средняя арифметическая 95
— средняя квадратичная 95
— химической реакции 5
— — —, зависимость
от температуры 25
— — —, константа 7
Сложные реакции 32 — 54
— — обратимые 33 — 39
— — параллельные 39 — 46
— — последовательные 47 — 54
Солевой эффект
— вторичный 183
— первичный 183
Соотношения
— Бренстеда 187
— Поляни 188
— Семенова 188
Сопряженные кислоты
и основания 173
Способы определения порядка
реакции 19 — 25
Строение катализатора 127, 150,
151
Степень компенсации 135
Стерический множитель 102, 104,
116, 120
Столкновения
— активные 101
— двойные 208, 209
—, частота 208
—, число 208
Субстрат 151, 154
Тафта уравнение 190
Темкина уравнение 214
Температурный коэффициент
скорости 25
Теория(и)
активированного комплекса
114-122
^ активных столкновений
105-114
— Линдемана 106 — 110
— промежуточных соединений
132-134
— трех пределов воспламенения
63
— фотохимических реакций
73-75
— Хиншельвуда 111 — 114
— цепных реакций
57-62, 67-69
Тиле
— задача 232, 233
— параметр 234
Трансмиссионный коэффициент
115
Тримолекулярные реакции 8, 118
Триплетное состояние 78
Туннельный эффект 30
Тушение
— динамическое 88
—, константа
— статическое 88
Уравнение(я)
— Аррениуса 25
— Больцмана 96
— Бренстеда 186
— Бренстеда-Бьеррума 184, 185
— Бэт 214-216
— Гаммета 188, 189
— Генри адсорбционное 214
— Герца 98
— Дебая —Хюккеля 184, 185
— Дубинина 216
— Лэнгмюра 209
— Максвелла 95, 96
— Михаэлиса—Ментен 156
— Тафта 190
— Темкина адсорбционное 214
— Траутца—Льюиса 102, 103
— Фрейндлиха адсорбционное
214
— Штерна—Фольмера 89
Фактор диффузионного торможе¬
ния 234
Фермент(ы) 150
—, активность 168—171
—, активные центры 151, 152
—, специфичность 153
—, структура 150, 151
249
Ферментативные реакции 150,
154, 155
— —, ингибирование 161 — 168
— —, кинетические
параметры 158
— способы определения
158-161
— —, скорость 154
Фермент-субстратный
комплекс 153
Ферментная единица 170
Физическая адсорбция 205, 207
Фишера—Тропша реакция 200
Флуоресценция 76, 79
Фосфоресценция 76, 80
Фотосенсибилизация 80, 90
Фотосинтез 73
Фотохимические реакции 72
Фотохимия 72 — 93
Фрейндлиха уравнение 214
Функция кислотности Гаммета
190, 191
Хемосорбция 205, 207
Химическая
— кинетика 4
— —, основной постулат 7
— переменная 5
Химический акт (элементарный)
7, 104
Цеолиты 201, 202
— природные 201
— синтетические 202
Цепи
—, зарождение 55
— материальные 55
—, обрыв 55, 56
—, перераспределения связей 141
—, продолжение 55
—, развитие 55
—, разветвление 56
— энергетические 55, 69
Цепные реакции 54, 70
Частота столкновений 97
Число
— столкновений 97, 98
— — с поверхностью 98
— оборотов 130, 131, 158
Штерна — Фольмера
— механизм 81
— константа тушения 89
— уравнение 89
Электрофильный катализ 176
Энергия
— активации 25, 27, 28
— —, вычисление 28 — 30
— — истинная 115
— — кажущаяся 219, 220
— —, связь с предэкспоненци-
альным множителем 29
— Гельмгольца 119, 187
— Гиббса 120, 209
Энзимы, см. Ферменты 150
Энтальпия активации 120, 121
Энтропийный множитель, см.
Стерический множитель
Энтропия активации 119 — 121,
137
Эффект
— компенсационный 118
— солевой 183
Эффективный диаметр столкно¬
вений 97, 99
Яды каталитические 204
ОГЛАВЛЕНИЕ
Предисловие 3
ЧАСТЬ 1. ХИМИЧЕСКАЯ КИНЕТИКА
Глава 1. Феноменологическая кинетика 4
1.1. Основные понятия и определения 4
1.2. Кинетика односторонних реакций разных порядков 10
1.3. Способы определения порядка реакций
из опытных данных 19
1.4. Зависимость скорости реакции
от температуры 25
Глава 2. Сложные реакции 32
2.1. Обратимые реакции 33
2.2. Параллельные реакции 39
2.3. Последовательные реакции. Принцип
квазистационарных концентраций 47
Глава 3. Цепные реакции 54
3.1. Основные понятия и стадии цепных
реакций 54
3.2. Неразветвленные цепные реакции 57
3.3. Разветвленные цепные реакции 60
Глава 4. Фотохимия 72
4.1. Основные законы и квантовый выход 72
4.2. Физические и химические фотопроцессы 76
4.3. Кинетическая схема Штерна —Фольмера 81
4.4. Зависимость квантовых выходов от различных условий 83
Глава 5. Теории химической кинетики 94
5.1. Элементы молекулярно-кинетической теории газов 94
5.2. Параметры столкновений 97
5.3. Теория активных столкновений (ТАС). Бимолекулярные
реакции 101
5.4. Теория активных столкновений. Мономолекулярные
реакции 105
5.5. Статистический аспект теории активированного
комплекса 114
5.6. Термодинамический аспект теории активированного
комплекса 118
251
ЧАСТЬ 2. КАТАЛИЗ
Глава 6. Основные положения катализа 124
6.1. Понятия и определения 124
6.2. Теория промежуточных соединений в катализе 132
6.3. Термодинамические и кинетические аспекты
в катализе 134
Глава 7. Автокатализ 143
Глава 8. Ферментативный катализ 150
8.1. Общие определения и понятия 150
8.2. Кинетика ферментативной реакции с одним субстратом 154
8.3. Способы определения кинетических параметров 158
8.4. Ингибирование ферментативных реакций 161
8.5. Активность ферментов 168
Глава 9. Кислотно-основной катализ 173
9.1. Теории кислот и оснований 173
9.2. Типы кислотно-основного катализа 175
9.3. Кинетика реакций кислотно-основного катализа 177
9.4. Солевые эффекты в катализе 183
9.5. Корреляционные соотношения в катализе 186
9.6. Функция кислотности Гаммета 190
Глава 10. Гетерогенный катализ 194
10.1. Общие положения 194
10.2. Катализаторы в промышленных процессах 198
10.3. Физическая адсорбция и хемосорбция 205
10.4. Адсорбционная теория Лэнгмюра 207
10.5. Нелэнгмюровские изотермы адсорбции 213
10.6. Кинетика гетерогенно-каталитических реакций 216
10.7. Макрокинетика гетерогенно-каталитических процессов ....224
Литература 242
Предметный указатель 244
Учебное издание
Байрамов Вадим Михайлович
Основы химической кинетики и катализа
Учебное пособие
Редактор В. Е. Венюкова
Технический редактор О. С. Александрова
Компьютерная верстка: Н. В. Протасова
Корректоры М.Г.Дахнова, J1. П. Кравченко
Изд. № A-704-I/1. Подписано в печать 16.05.2003. Формат 60x90/16.
Бумага тип. № 2. Печать офсетная. Гарнитура «Таймс». Уст нем. 16,0.
Тираж 20 000 экз. (1-й завод 1-5100 жт). ’Заказ 159f\
Лицензия ИД № 02025 от 13.06.2000. Издательский центр «Академия».
Санитарно-эпидемиологическое заключение JS° 77.99.02.953.Д.002682.05.01 от 18.05.2001.
П7342, Москва, ул. Бутлерова, 17-Б, к. 223, Тел./факс: (095)334-8337, 330-1092.
Отпечатано с готовых диапозитивов
на ОАО «Альянс «Югполиграфиздат» ИПК «Офсет»
400001, г. Волгоград, ул. КИМ, 6.