/


















































































































































































































































Текст
ТРЕТЯК Н. М.
ГЕМАТОЛОГІЯ
Н. М. Третяк
ГЕМАТОЛОГІЯ
Рекомендовано Міністерством освіти і науки України
як навчальний посібник для студентів вищих навчальних закладів
(Рішення № 14/18.21657від 12.07.2005р.)
Київ — Зовнішня торгівля — 2005
УДК 616.15+612.11
Т66
Рекомендовано Вченою медичною радою Інституту гематології
та трансфузіології АМН України (протокол №6 від 29.06.04);
Рекомендовано Проблемною комісією “Гематологія та транс-
фузіологія ” АМН та МОЗ України (протокол №3 від 23.06.04);
Ангори:
ТретякН.М. — розд. “Кровотворення", “Анемії", “Гемобластози”, “Ле-
йкемоїдні реакції”, “Агранулоцитоз”, “Цитостатична хвороба”, “Лейко-
пенії”, “Еритроцитози”
Третяк Н.М., Калініна С.Ю. — розд. “Негоджкінські лімфоми ”, “Лім-
фогранулематоз”, “Геморагічні діатези ”
Третяк Н.М., Горяїнова Н.В. — розд. “Мієаодиспластичний синдром ”
Третяк Н.М., Перехрестенко Т.П. — розділ “Хронічна лімфоідна лей-
кемія”
Третяк Н.М., Кисельова ОА — розд. “Лімфоплазмоиртарналімфома”
Третяк Н.М.
Т 66 Гематологія: Навч. посібник. — К.: Зовнішня торгівля, 2005. — С. 240.
І8ВІМ 966-8517-07-5
У посібнику з урахуванням сучасних досягнень гематологічної науки
представлено дані про етіологію, патогенез, діагностику, диференційну
діагностику, клінічний перебіг та лікування таких захворювань системи
крові як анемії, гемобластози, геморагічні хвороби. Наведено критерії
диференційної діагностики захворювань терапевтичного, онкологічного,
хірургічного та інших профілів медицини. При написанні розділів терапії
враховано сучасні досягнення країн Європи та США в гематології.
Окремий розділ присвячено гематологічній патології, яка
супроводжує деякі внутрішні хвороби.
Навчальний посібник розраховано на студентів ВНЗ, але буде
корисним і спеціалістам (гематологам, гінекологам, терапевтам,
онкологам, хірургам, фахівцям інших медичних профілів).
УДК 616.15+612.11
І8ВК 966-8517-07-5
© Третяк Н.М.. 2005
© "Зовнішня торгівля”.
КРОВОТВОРЕННЯ
Кровотворення, або гемопоез — це тонко регульований процес
послідовних диференціювань родопочаткових клітин, що призво-
дить до утворення зрілих клітин крові всіх 8 ліній:
мієлоїдні: еритроцити, гранулоцити — базофільні, нейтрофіль-
ні, еозинофільні, мегакаріоцити, моноцити; макрофаги;
лімфоїдні: Т-лімфоцити,
В-лімфоцити.
Утворення клітин крові в кістковому мозку вперше незалежно
було показано Нейманном і Біццоцеро у 1868 р. Опрацьований
Ерліхом метод диференційного забарвлення клітин сприяв швид-
кому накопиченню знань про морфологію клітин крові. У 1898 р.
Паппенгейм використав методи забарвлення Романовського Д. Л.,
що дозволило йому описати перехідні форми клітин кісткового
мозку. Ці морфологічні описання лишаються класичними й на
сьогодні. Проте вивчення забарвлених клітин не давало змоги
встановити їх походження, оскільки наймолодші клітини кожної
лінії були дуже схожі.
На підставі клінічної різниці між мієлоїдними та лімфоїдними
лейкеміями Негелі дійшов висновку, що існують дві незалежні ро-
допочаткові кровотворні клітини. Вивчаючи гістогенез ембріональ-
ної кровотворної тканини, Максимов О. О. ще в 1910 р. обстоював
ідею моНофілитичного походження клітин крові із єдиного родо-
початкового попередника, і тільки розвиток таких сучасних мето-
дів дослідження, як радіаційна гематологія, цитогенетика, молеку-
лярна біологія, культура тканин, дозволив довести правоту
Максимова.
Вирішальну роль у підтвердженні ідеї існування єдиної кліти-
ни — попередниці кровотворення відіграли дослідження Лоренца,
Форда, Тіла та Мак-Кулоха, Меткалфа, Мул-лігана, Лемишки,
Мінца та інших. Зусиллями цих дослідників вдалося чітко встано-
вити послідовність диференціювання в кровотворній системі полі-
потентної стовбурової клітини.
Кровотворення у всіх вищих хребетних та людини відбуваєть-
ся в порожнинах трубчастих та плоских кісток, у просторі між си-
3
Н.М. Третяк | Гематологія
нусами, в так званому кровотворному мікрооточенні. Мікроото-
чення та строму складають клітинні та неклітинні елементи.
До клітинного мікрооточення належать ендотеліальні клітини,
адвентиціальні клітини, ретикулярні клітини (фібробласти кістко-
вого мозку), макрофаги, жирові клітини, остеокласти, остеобласти,
остеоцити тощо.
Позаклітинний матрикс складається із нерозчинних білків
(глюкозоаміноглікани, протеоглікани, фібронектин, ламінін, глі-
копротеїни та ін.), колагенових, еластинових волокон, у мережі
котрих розташовуються тяжі кровотворних клітин та основна речо-
вина кістки. У морфологічній організації кісткового мозку важли-
ву роль відіграє судинна система. Артеріальне кровозабезпечення
кісткового мозку здійснюється із двох джерел, головним із яких є
живильні артерії.
Здатність кровотворних клітин упізнавати клітини строми кіс-
ткового мозку та розташовуватись у ньому (хоммінг) зумовлена мо-
лекулами клітинної адгезії інтегринами та безпосередніми міжклі-
тинними контактами. Спеціалізовані молекули на поверхні
кровотворних клітин визначають їх специфічний хоммінг. Ця здат-
ність кровотворних клітин чітко проявляється при довенній транс-
плантації кісткового мозку: 85% введених клітин потрапляють
у кістковий мозок, котрий становить лишень 6% від маси тіла, 15%
клітин, що залишилися, розподіляються між печінкою, легенями,
селезінкою та іншими органами.
Родопочаткові стовбурові кровотворні клітини локалізуються
у кістковому мозку. Попередники Т- та В-лімфоцитів також утво-
рюються в кістковому мозку, проте їх остаточне диференціювання
відбувається у тимусі Т-к^ітини, в селезінці, лімфатичних вузлах та
пейєрових бляшках (В-клітини).
В онтогенезі відбувається поступове заміщення червоного кіс-
ткового мозку жировою тканиною. Зазвичай інтенсивність кровот-
ворення обернено пропорційна кількості жирових клітин. Проте
жировий кістковий мозок перетворюється на кровотворний в умо-
вах надзвичайного гемопоетичного стресу, наприклад при анемії.
Жирові клітини кісткового мозку не є жировим депо для організ-
му, оскільки при голодуванні вміст жиру в них не зменшується. Ці
клітини виконують якісь пов’язані із кровотворенням функції і є
його необхідним елементом.
4
| Кровотворення
КІСТКОВОМОЗКОВЕ КРОВОТВОРЕННЯ
Центральними органами гемопоезу є кістковий мозок та під-
грудинна залоза (тимус). Кістковий мозок містить пул стовбуро-
вих клітин, котрий безперервно поповнюється та є джерелом про-
дукування всіх гемопоетичних клітин; у ньому відбуваються
проліферація і дозрівання елементів мієлопоезу та початкові стадії
диференціювання В-лімфоцитів. У підгрудинній залозі дозрівають
Т-лімфоцити. До периферичних органів гемопоезу належать селе-
зінка, де дозрівають різні клітини крові, в тому числі й ретикуло-
цити та В-лімфоцити, а також лімфатичні вузли, мукоз — асоці-
йована лімфоїдна тканина (МАЬТ), периферична кров, котра
вміщує стовбурові клітини та імунологічно компетентний пул В- і
Т-лімфоцитів.
Кістковий мозок розташований на балках плоских та трубчас-
тих кісток. До складу кісткового мозку входять власне гемопоетич-
ні елементи всіх рівнів диференціювання та стромальні клітини.
Кровотворні клітини розташовані острівцями на підстилці із клі-
тин ендосту та епітеліальних клітин, обплутані та пронизані стро-
мальними елементами, такими як ендотеліальні та адвентиціальні
клітини, аципоцити, фібробласти й остеобласти (так звані елемен-
ти мікрооточення). Разом зі стромальними елементами активно
співпрацюють гемопоетичні клітини моноцитарно-макрофагаль-
ного ряду.
У функціонуванні системи мікрооточення важливу роль відіг-
рає позаклітинний матрикс, котрий є продуктом життєдіяльності
та розпаду клітин і складається із ламиніну, фібронекгину, гемо-
нектину, колагену, тромбосподину, глікозоаміногліканів. Матрикс
сприяє прикріпленню гемопоетичних клітин до елементів кістко-
вомозкового бар’єру та утримує їх до моменту остаточного дозрі-
вання. Різні складові частини матриксу взаємодіють із клітинами
різних ліній. Приміром, фібронектин зв’язується з елементами гра-
нулоцитарного ряду-', а гемонекгин — з еритроїдними клітинами.
Система мікрооточення забезпечує підтримання клітин кістко-
вого мозку, зберігаючи його структуру в кровотоку, індукує пролі-
ферацію та диференціювання стовбурових клітин, передає інфор-
мацію про потребу організму на периферії, продукує різноманітні
ростові фактори.
Система гемопоезу — це комплекс клітин, що забезпечує різно-
манітні функції і в той же час постійно регенерує. Згідно із сучас-
5
Н.М. Третяк | Гематологія
ною схемою кровотворення, існують попередники, котрі продуку-
ють різні типи клітинних форм. Ця схема пояснює взаємовідносини
гемопоетичних клітин у нормі та при патологічних станах, у тому
числі й при гемобластозах. Крім того, відомо, що частина ранніх по-
передників гемопоезу лишаються розсіяними в ретикулоендотелі-
альній системі різних органів і зберігають здатність до проліферації
та утворення екстрамедулярних вогнищ кровотворення протягом
усього життя людини.
Дослідженнями останніх років встановлено, що справжніми
родопочатковими клітинами гемопоезу є стовбурові клітини, котрі
закладаються в жовточному мішку ще в період формування плоду.
Кількість поліпотентних стовбурових клітин (ПСК) не велика і
в людини становить приблизно 40 000. З допомогою моноклональ-
них антитіл встановлено, що першим на поверхні ПСК з’являєть-
ся антиген стовбурових клітин (СО34), потім послідовно — анти-
ген гістосумісності II класу (НЬА-ВК), а також фермент
термінальна дезоксинуклеотидінтрансфераза (ТсІТ). ПСК не мають
ознак лінійного диференціювання.
Проліферацію стовбурових клітин індукує система мікроото-
чення, а також низка ростових факторів, що продукуються стро-
мальними елементами, циклони, рестриктини, адизини.
Пул поліпотентних попередників становить дуже малу (0,01%)
фракцію всіх ядровмісних елементів кісткового мозку, проте його
проліферативної активності вистачає, щоб забезпечити клітинни-
ми елементами кровотворну систему. У кістковому мозку людини
щодня продукується 2-Ю11 клітин, що становить 300 г. Цю потре-
бу забезпечує 5% ПСК, що перебувають у мітотичному циклі, в той
же час усі інші ПСК перебувають у стані спокою — Оо-фазі. ПСК
забезпечують постійність вихідного пула та вихід із нього частини
клітин у дозріваючу фракцію.
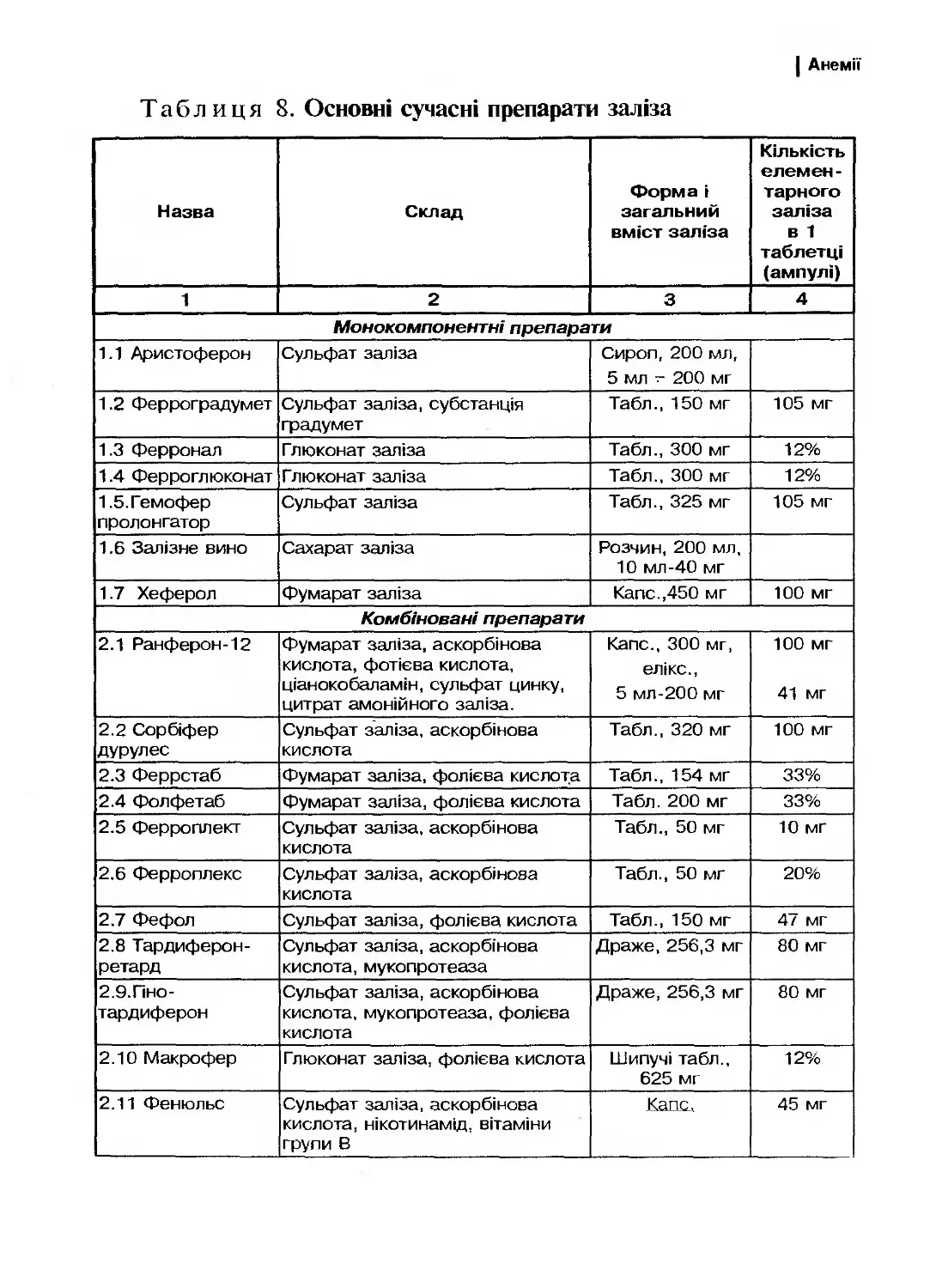
Подальший розвиток мієлоїдного та лімфоїдного паростків
кровотворення відбувається із окремого для кожної лінії мієло- або
лімфопопередника. Клітини мієлоїдного паростка проліферують та
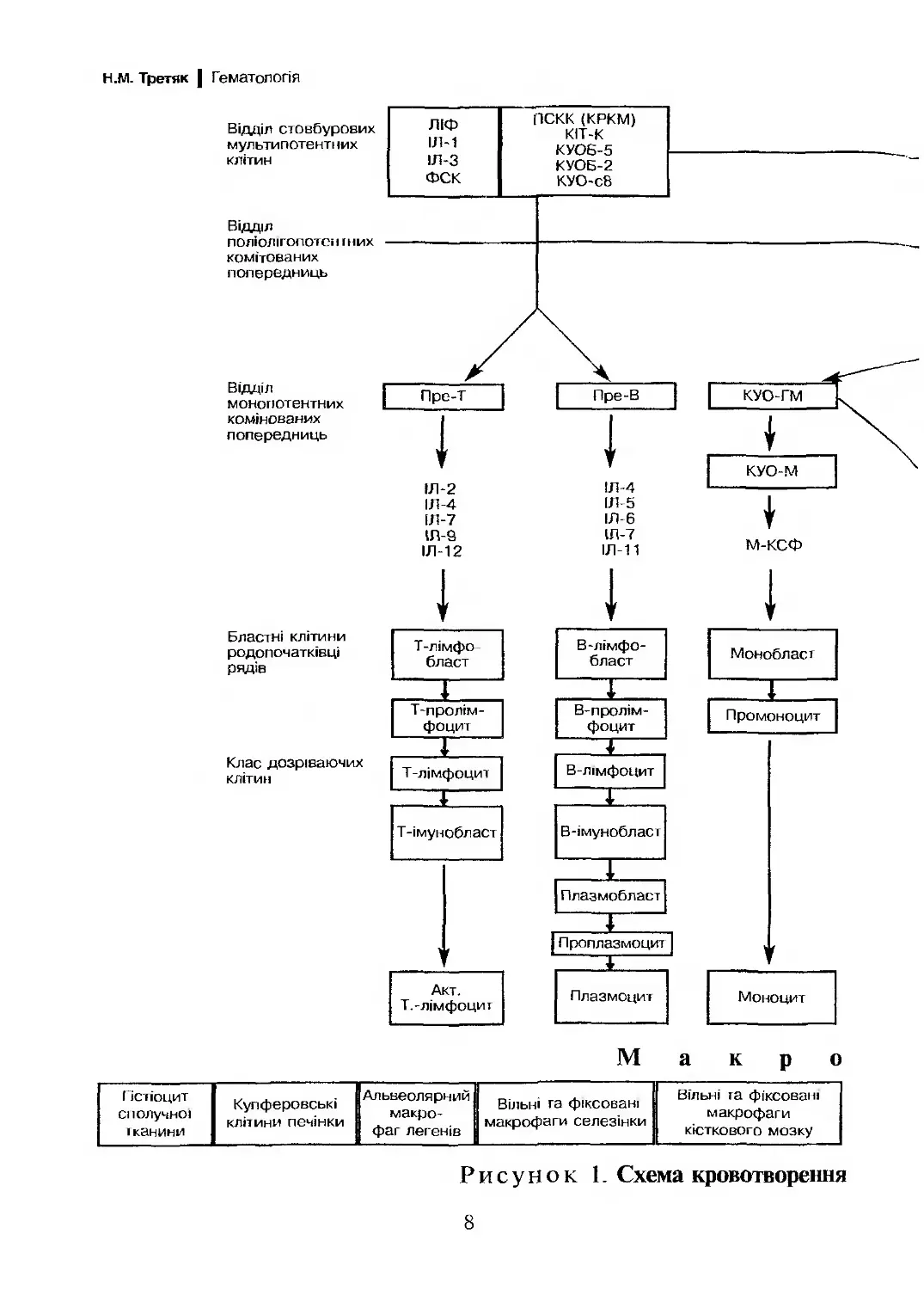
диференціюють у кістковому мозку до зрілих форм (рис. 1).
Загальним попередником мієлопоезу є поліпотентна колоніє-
утворююча одиниця гранулоцитів, еритроцитів, моноцитів/макро-
фагів, мегакаріоцитів. На ній експресуються макромолекули, спе-
цифічні для мієлоїдних клітин —- СІ2>33.
Індукція диференціювання всіх клітин-попередниць відбува-
ється під впливом ростових факторів (колонієстимулюючі фактори
6
| Кровотворення
(КСФ) та інтерлейкіни ІЛ), котрі специфічні для кожного парос-
тка. Більшість КСФ та ІЛ продукуються стромальними та гемопо-
етичними клітинами (фібробласти, ендотеліальні, епітеліальні клі-
тини, Т-лімфоцити. моноцити, макрофаги). Винятком є
еритропоетин та тромбопоетин. котрі екскретуються клітинами
нирки та являють собою глікопротеіни з низькою молекулярною
масою. Ростові фактори впливають на клітини-попередниці через
спеціальні мембранні рецептори, зумовлюючи їх активацію і про-
ліферацію.
Вирізняються ростові фактори, що активують родопочаткові
клітини, — ранодіючі фактори і ростові фактори, котрі реагують із
більш диференційованими клітинами — пізнодіючі фактори. До
ранодіючих факторів відносять стовбурово-клітинний фактор Сті-
ла (ВСЕ), ІЛ-1, ІЛ-3, ІЛ-6 та туморнекротичний фактор (ТКР).
ІЛ-1 та ТИР індукують вироблення факторів росту гранулоцитів
(Г-КСФ), гранулоцитів-макрофагів (ГМ-КСФ), еозинофілів
(ІЛ-5), В-клітин (ІЛ-6), Т-клітин (ІЛ-4 та ІЛ-2), а також беруть
участь у інших регуляторних механізмах. Встановлено, що ІЛ-1 не
лише здатен стимулювати клітини гемопоезу, а й підсилює актив-
ність власних стромальних клітин. Туморнекротичний фактор різ-
нобічно впливає на гемопоетичні клітини, індукуючи гранулоцито-
поез та пригнічуючи ранні попередники, а також попередники
еритропоезу та лімфоцитів. Особливістю ранніх цитокінів є їх ду-
же висока біологічна активність, взаємодія з клітинами різних па-
ростків на різних етапах диференціювання та висока кооперація з
іншими факторами росту.
Для кожного гемопоетичного паростка існує складна система
регуляції пізнодіючими факторами. Всі етапи диференціювання
клітин гранулоцитарного, еритроїдного і мегакаріопитарного па-
ростків представлені у кістковому мозку морфологічно ідентифіко-
ваними формами. У кожному паростку найбільш раннім елемен-
том є бласт. Загалом вони становлять 0,5-3% клітин кісткового
мозку.
Гранулоцитарний паросток складається з трьох типів клітин —
нейтрофіли, еозинофіли та базофіли.
Мієлобласти — це ранні форми гранулоцитів, які становлять
0,5-2% мієлокаріоцитів. Морфологічно це клітини середнього діа-
метра (10-15 мкм) з колоподібним ядром, дисперсною структурою
хроматину та 2-3 нуклеолами. Цитоплазма їхня забарвлюється
У світло-блакитний колір. При цитохімічному дослідженні у міє-
Н.М. Третяк | Гематологія
Відділ
монопотентних
комінованих
попередниць
Бластні клітини
родопочатківці
рядів
Відділ стовбурових
мультипотенті ІИХ
клітин
ВІДДІЛ
ПОЛІОЛІГОПОТЄі і і НИХ
комітованих
попередниць
КУО-М
М-КСФ
ІЛ-2
ІЛ-4
ІЛ-7
ІЛ-9
ІЛ-12
ІЛ-4
ІЛ-5
ІЛ-6
ІЛ-7
ІЛ-11
Клас дозріваючих
клітин
М а к р о
Пстіоцит сполучної тканини Купферовські клітини печінки Альвеолярнийі макро- фаг легенів Вільні га фіксовані макрофаги селезінки Вільні га фіксовані макрофаги кісткового мозку
Рисунок 1. Схема кровотворення
8
| Кровотворення
ф а Г И
Вільні та фіксовані макрофаги лімфатичних вузлів Перитонеальні макрофаги Плевральні макрофаі и Остсо- класти Клітини мікроглії нервової системи Дендритні клітини
(А. І. Боробйов, 1995 р.)
9
Н.М. Третяк | Гематологія
лобластах нейтрофільного й еозинофільного рядів визначається пе-
роксидаза, ліпіди у реакції із Суданом чорним В та ШИК-позитив-
на речовина, розташована в цитоплазмі дифузно. Наявність перок-
сидази та ліпідів є характерною цитохіміяною ознакою клітин
гранулоцитарного ряд)', що зберігається в них у період повної зрі-
лості — сегментоядерних форм.
Фенотип мієлобластів характеризується наявністю антигенів,
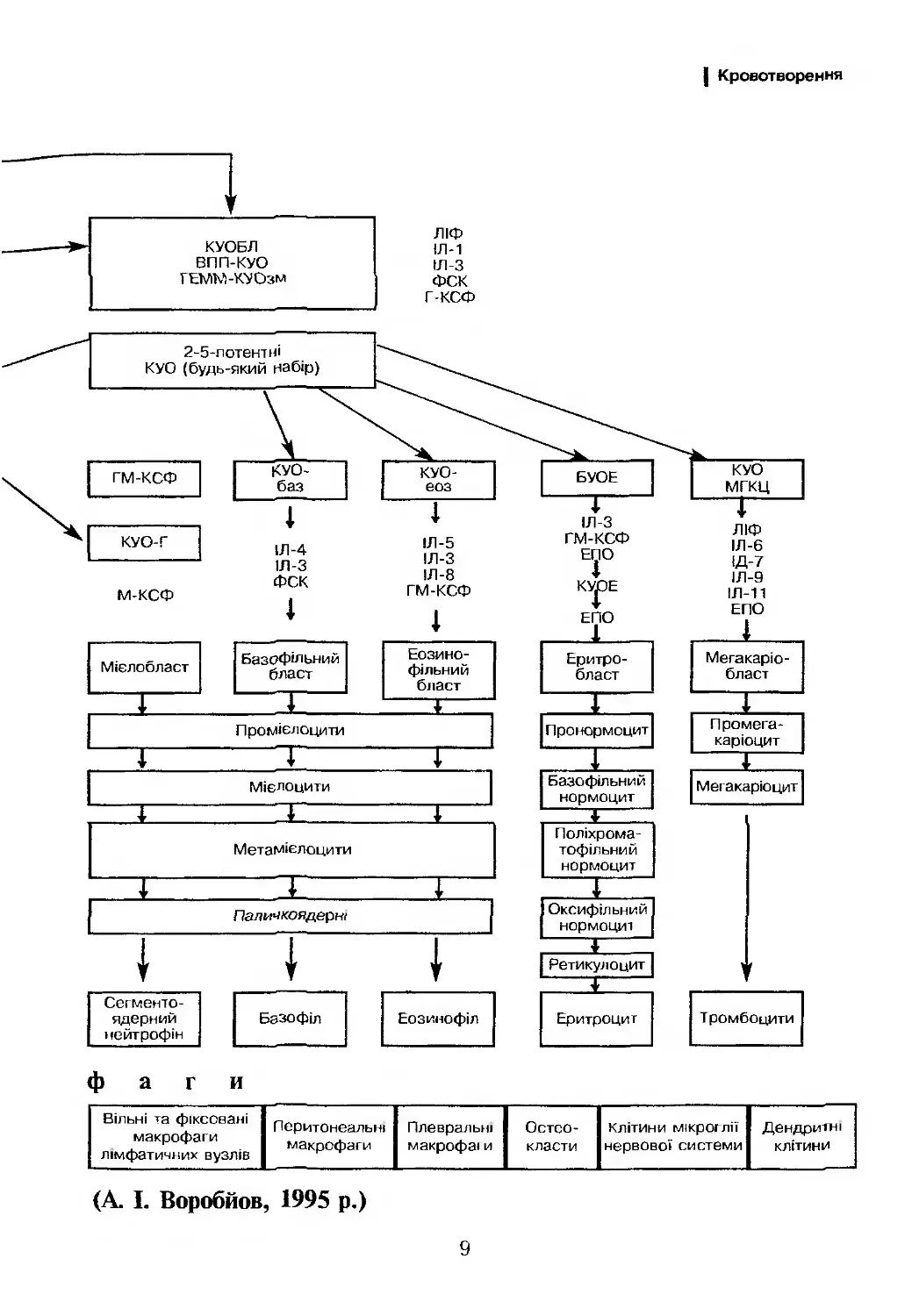
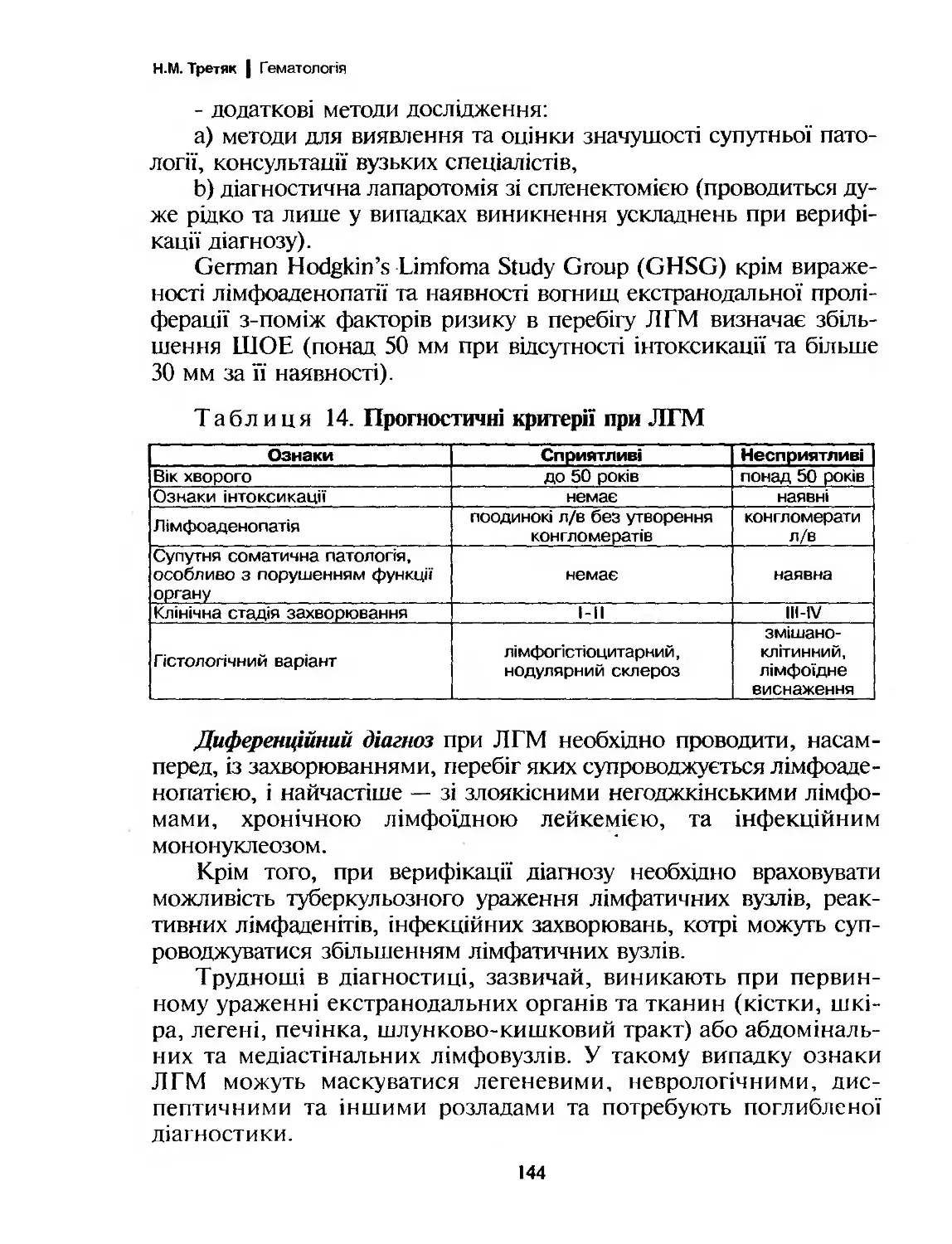
котрі виявляються з допомогою МКА СИ13, СО33 (рис. 2). Трива-
лість життя мієлобластів становить 18 годин, за цей час вони мо-
жуть здійснити 1-2 мітози.
Промієлоцити нейтрофільного ряду становлять 2-4% мієлока-
ріоцитів. Це великі клітини діаметром 13-18 мкм. Ядра в них роз-
ташовуються ексцентрично й містять тонкодисперсний хроматин
та 1-2 великі нуклеоли. Цитоплазма широка, слабко базофільна,
у ній синтезуються первинні лізосомальні гранули, що забарвлю-
ються азуром. У первинних гранулах міститься велика кількість
ферменту пероксидази та ліпідів, котрі є маркерами нейтрофілів.
Фенотипично характеризується антигенами СС]3. Сі533, СВ34,
НГА-1Ж.
Мієлоцити нейтрофільні становлять 13% мієлокаріоцитів, діа-
метр клітини — від 10 до 15 мкм, тривалість життя — до 104 год.,
за цей час вони спроможні зробити 3-4 мітотичні цикли. Це остан-
ня стадія розвитку нейтрофілу, на котрій клітина здатна до ділен-
ня. Мієлоцит містить ядро з дещо грубою структурою хроматину та
поодинокими дрібними нуклеолами. У цитоплазмі поряд із пер-
винними з’являються численні вторинні гранули. Наявність їх є
специфічною ознакою нейтрофільних мієлоцитів. У цих гранулах
містяться глікопротеіни (лактоферин, білок, зв’язуючий вітамін
В12, НАДФ-Н-оксидазу, цитохроми).
Метамієлоцити, паличкоядерні та сегментоядерні нейтрофіли
являють собою пул клітин, не здатних до ділення. Дозрівання їх
у кістковому' мозку триває 96-144 год. За цей час ядро набуває сег-
ментованої форми, збільшується ядерно-цитоплазматичне відно-
шення. Зрілі гранулоцити затримуються у кістковому мозку 3-4
дні, вони складають “кістковомозковий гранулоцитарний резерв”,
їхня кількість у 2-3 рази перевищує кількість молодих гранулоци-
тів і майже у 10-30 разів — кількість циркулюючих нейтрофілів.
У судинному руслі є два пули клітин — постійний (маргінальний,
капілярний) та пул циркулюючих гранулоцитів. У нормальних
умовах відбувається постійний обмін між обома пулами. У фізіоло-
10
| Кровотворення
Мієлоїдна стовбурова клітина
СО34
тат
НЇ-А-РР
СР10
Стовбурова клітина
СР34 (Тс1Т( (СР7) ।
Мієлоїдна стовбурова клітина
СРЗЗ
СР34
НЬА-РР
Про-В-клітина Ранній тимоцит Мієлобласт Монобласт Мегакаріобласт Еритробласт
НЬА-ОР СР2 НЬА-РР НЬА-ОР НЬА-РР НЬА-ОР
СОЮ (СРЗ) СО13 СО11с СО41 СО71
СР19 СО5 СРЗЗ СР13 СО42Ь глікофорін А
(СР22) СО7 (МРО) СО14 СО61 глікофорін С
тат тат СРЗЗ
(СР88)
(МРО)
Пре-В-клітина
НЬА-РР
(СР9)
СОЮ
СР19
СР20
С022
(СР24)
Суїоріавтік Ід ТсІТ
тат
Проміжний
тимоцит
СОІа
СР2
СРЗ
СО4
СР5
СР8
СР45РВ
Промієлоцит
СР13
СРЗЗ
(МРО)
Промоноцит
НЬА-РР
СО11Ь
С011с
СР13
СР14
СРЗЗ
(СР68)
(МРО)
Нормобласт
СР71
глікофорін А
глікофорін С
Незріла В-клітина
НЬА-РЯ
СОЮ
СР19
СР20
СР22
СР24
поверхневі Ід
Зрілий
тимоцит
С02
СРЗ
СО4/СР8
С05
СОЄ
СР7
Мієлоцит
СОЇ 1с
СР13
СР15
СРЗЗ
(МРО)
Мегакаріоцит
СР41
СО42Ь
СР61
В-лімфоцит Т-лімфоцит Нейтрофіл Моноцит Тромбоцит Еритроцит
НЦА-ОР СР2 (СОНЬ) НЬА-РР СО41 глікофорін А
СО19 СРЗ (СО11с) СО1ІСІ СО42Ь глікофорін С
СО20 С05 СР13 С011с СО61
(СО21) СОЄ СО15 СО13
СО22 С07 СР16 С014
СО23 СО4/СО8 СО24 СО35
(активовані СО25 СРЗЗ (СО68)
В-клітини) (активовані СО35 (МРО)
СО24 Т-клітини) (МРО)
С025
(активовані
В-клітини)
СР37
поверхневі Ід
СР43
СР45РО
НЬА-РР
(активовані
Т-клітини)
ОРР4
Рисунок 2. Поверхневі антигени клітини гемопоезу
11
Н.М. Третяк | Гематологія
гічних умовах нейтрофіли, що потрапили в тканини, не поверта-
ються у судинне русло: вони депонуються в капілярній сітці леге-
нів та меншою мірою в паренхімі печінки та селезінки.
За потреби спостерігається посилений вихід гранулоцитів
у тканини. Вони здатні рухатися в напрямку вогнища інфекції,
поглинати та перетравлювати бактеріальні мікроорганізми з допо-
могою складного процесу фагоцитозу.
Еозинофіли в процесі дозрівання під впливом еозинофільного
диференціювального фактору, котрий є лімфокіном і продукуєть-
ся Т-клітинами, проходять стадію промієлоцита, мієлоцита, мета-
мієлоцита, паличкоядерної та сегментоядерної форм. Дозрівання
еозинофілів у кістковому мозку триває 2-6 днів.
На мембрані зрілих еозинофілів визначаються рецептори до
І§О, І§Е, І§Е, СзЬ-компонента компліменту та рецептори, що бе-
руть участь у реакціях на різні хемотаксичні стимули у вогнищах
запалення.
Фізіологічна активність еозинофілів, як і інших кістковомозко-
вих клітин, залежить від впливу регуляторних цитокінів, таких як
ГМ-КСФ, Ш-5, ІЛ-3. Специфічним та найефективнішим для ут-
ворення еозинофільних колоній є ІЛ-5.
Еозинофілам властива імунозахисна функція проти гельмінтів,
бактерій, вірусів, грибів і, можливо, проти злоякісних пухлин. Во-
ни є модуляторами реакції гіперчутливості. Еозинофіли беруть
участь у реіуляції системи згортання, активуючи фактор ХП (Хаге-
мана) та плазминоген.
Базофіли, що містяться у кістковому мозку та периферичній
крові, й тучні клітини у тканинах становлять загальну популяцію
гістамінвміснмх елементів.
Базофільного бласту в кістковому7 мозку не виявлено. Базо-
фільний промієлоцит спостерігається дуже рідко.
Тучні клітини в нормі в периферичній крові не виявляються.
Зустрічаються поодинокі клітини в кістковому мозку. Зазвичай
тучні клітини містяться у сполучній тканині біля епітеліальних клі-
тин, в оточенні кровоносних судин, у серозних порожнинах. Сти-
муляторами базофілів є ІЛ-1, ІЛ-3 та колонієстимулюючий фактор
базофілів ГМ-Баз-КСФ.
У базофілів на відміну від еозинофілів та нейтрофілів відсутня
здатність до фагоцитозу.
Базофіли й тучні клітини секретують гістамін, кінін, специфіч-
ні естерази, трипсин, хемотрипсин.
12
| Кровотворення
На поверховій мембрані базофілів експресується рецептор до
І§Е, сполучення з котрим призводить до анафілактичної деграну-
ляцїї клітин. Таку ж реакцію можуть провокувати й деякі лікар-
ські препарати, гормони, тривале переохолодження. Під час цієї
реакції із гранул вивільнюються такі біологічно активні речови-
ни, як гістамін та метаболінін арахідонової кислоти. Гістамін бе-
ре участь у негайній відповіді гіперчутливості та є потужним хе-
моатрактантом еозинофілів, фіксуючи їх у місці дегрануляції
базофілів при астмі, уртикаріях, алергічних ринітах та анафілак-
сії. Водночас у еозинофілах міститься речовина, що інактивує
вміст гранул базофілів.
Базофіли разом із еозинофілами є медіаторами позаклітинно-
го запалення. При співдії з такими чинниками імунної системи, як
моноцити, макрофаги, лімфоцити, вони беруть участь у нормаль-
ній імунологічній відповіді на інфекцію, пухлину або паразитарну
інвазію.
Моноцити. Перша морфологічно розрізнювана клітина моно-
цитарного ряду в кістковому мозку — монобласт. Проходячи два
мітотичних цикли, він через фазу промоноцита утворює чотири
моноцити.
Зрілі моноцити кісткового мозку та периферичної крові явля-
ють собою великі — діаметром 15-20 мкм — клітини, що мають по-
ліморфне ядро частіше лопасної, бобоподібної або підковоподібної
форми. Цитоплазма їх забарвлюється у сіро-блакитний колір та
містить незначну пилоподібну зернистість.
У клітинах моноцитарно-макрофагального ряду на всіх етапах
диференціювання виявляється неспецифічна естераза, що пригні-
чується фторидом натрію, кисла фосфатаза, що пригнічується тар-
тратбм, ШИК-позитивна речовина в дифузній формі. У моноци-
тах міститься невелика кількість пероксидази та ліпідів.
Неспецифічна естераза є цитохімічним маркером моноцитів.
У нормі не всі попередники моноцитів проліферують до зрілих
форм. У кістковому мозку зберігається резерв, котрий використо-
вується організмом у разі виникнення запалень. У такому випадку
молоді моноцити із кісткового мозку потрапляють у периферичну
кров, де циркулюють від 36 до 104 годин, водночас формуючи
пристінчастий та циркулюючий пули.
В нормі основна частина життя моноцитів проходить у ткани-
нах, суттєво видозмінюючись морфологічно. Клітина збільшується
та набуває неправильної форми, в кістках утворюються поліплоїд-
13
Н.М. Третяк | Гематологія
ні багатоядерні форми, котрі називаються остеокластами. Перебу-
ваючи у тканинах, макрофаги утворюють дві великі підгрупи клі-
тин — альвеолярні макрофаги, клітини Купфера, остеокласти, по-
пуляцію фагоцитуючих форм та антигенпрезентуючих форм.
Фагоцитуючі клітини здатні до хемотаксису, фагоцитозу та
бактерицидної активності. Ці властивості реалізуються з допомо-
гою Рс- або СзЬ-рецепторів та мембрани клітини протеазами, що
знаходяться в гранулах, та системою окислювачів (перекис водню,
йодиди, броміди калію та натрію). Макрофаги можуть фагоцитува-
ти більшість мікобактерій, вірусів, грибів та найпростіших, але по-
рівняно з нейтрофілами їхня антимікробна функція значно слаб-
ша. Макрофаги поглинають та перетравлюють ушкоджені та
віджилі клітини. Макрофаги селезінки секвеструють дефектні та
застарілі еритроцити, повертаючи залізо до еритроцитарного пула.
Макрофаги легенів спроможні захоплювати сторонні частки, на-
віть азбест.
До антигенпрезентуючих форм макрофагів належать клітини
Лангерганса шкіри, інтердигітуючі клітини підгрудинної залози та
фолікулярні дендритичні клітини зародкових центрів лімфатичних
вузлів, котрі не здатні до фагоцитозу. Функція цих макрофагів по-
лягає в презентації антигенів Т-хелперам, котрі, в свою чергу, вза-
ємодіють із В-лімфоцитами, стимулюючи їх до продукції імуногло-
булінів.
Моноцитарно-макрофагальна система відіграє важливу роль у
імунологічному протипухлинному захисті організму7 при онкопро-
цесах. Не менш важливою є здатність моноцитарно-макрофагаль-
них клітин синтезувати та секретувати біологічно активні речови-
ни, які відносять до розряду цитокінів. До цих білків і пептидів
належать інтерферони, інтерлейкіни, лізоцим, туморнекротичний
фактор, фактор активації нейтрофілів, колонієстимулюючі — гра-
нулоцитарний та гранулоцитарно-макрофагальний фактори, рос-
товий трансформуючий фактор, макрофагальні загальні білки.
Клітини моноцитарно-макрофагального ряду продукують та-
кож такі гідролітичні фактори, як протеази (плазміноген, коллаге-
наза, еластаза), фактори згортання крові V, VII, IX, X та протром-
бін. Багато з цих факторів, комплексно взаємодіючи, беруть участь
у регуляції росту гемопоетичних клітин, у гострій фазі запалення,
метаболічних процесах печінки, в механізмі розвитку кахексії та
анемій, пов’язаних із хронічним запальним процесом, здійснюють
продукцію простагландинів.
14
| Кровотворення
Еритробласти кісткового мозку' мають вигляд великих клітин
правильної форми з округлим ядром, розташованим у центрі. Хро-
матин ядра має ніжносітчасту структуру, виявляється одна велика
нуклеола. Цитоплазма інтенсивно базофільна. Еритробласт експре-
сує антиген СО% та глікофарин А (рис. 2).
Здійснивши 4-5 мітозів, еритробласт переходить у категорію
нормобластів. При дозріванні забарвлення цитоплазми із базофіль-
ного поступово змінюється на поліхроматофільне, а потім на окси-
фільне внаслідок накопичення в ній специфічного білка (гемогло-
бін). У поліхроматофільній стадії у нормобласту зникає мітотична
активність. В період оксифільної стадії нормобласт втрачає ядро і
переходить до категорії ретикулоцитів.
Ретикулоцит набуває вигляду двоввігнутого диска діаметром 8-
9 мкм та протягом 1-2 діб дозріває у кістковому мозку. В цей час,
втрачаючи рецептори до трансферину та зменшуючись, цитоплазма
його стає еластичною. Набувши нових якостей, ретикулоцит із кіс-
ткового мозку переходить у периферичну кров і, потрапляючи в се-
лезінку, остаточно дозріває, переходячи в категорію еритроцита.
Специфічною цитохімічною реакцією для нормобластів та
еритроцитів є реакція на сідерофільні гранули. Збільшення вмісту
цих гранул у клітинах свідчить про неефективний еритропоез у кіс-
тковому мозку, зменшення гранул в еритроцитах — ознака дефіци-
ту заліза в організмі.
Регуляція еритропоезу відбувається під впливом цитокінів.
Найвідомішим із них є еритропоетин, котрий продукується в ос-
новному нирками і незначною мірою — клітинами моноцитарно-
макрофагального паростка.
Тривалість життя еритроцитів — 120 діб. Щодня продукується
в кістковому мозку та руйнується в системі циркуляції та селезін-
ці 2 • 10і 1 клітин. Еритроцити не мають кістковомозкового резерву,
деяка частина їх депонується в судинах шкіри та селезінки.
Мегакаріоцити розвиваються із недиференційованої стовбуро-
вої клітини і становлять надзвичайно малу частку серед ядровміс-
них клітин кісткового мозку. Весь життєвий цикл мегакаріоцитів
проходить у кістковому мозку.
Попередниками мегакаріоцитів у кістковому мозку, що мор-
фологічно ідентифікуються, є мегакаріобласти. На поверхневій
мембрані мегакаріобластів експресуються антигени СО( |.
Зрілі мегакаріоцити — найбільші кістковомозкові клітини
в кістковому мозку, до 40-50 мкм. Одна клітина містить 32-64 яд-
15
Н.М. Третяк | Гематологія
ра та пишну цитоплазму рожевого кольору, де формуються тром-
боцити.
Імунологічний фенотип клітин мегакаріоцитарного паростка
визначається експресією антигенів до пластинчатих факторів тром-
боцитів 4 та 8 (Р4 та 8), глікопротеїнів (ОР) II, 16 і ІІІа (СВ41,
со42, со42ь, СЕ>\¥49 та СО61) (рис. 2).
Регуляція мегакаріоцитопоезу відбувається за участі цитокінів
ІЛ-3, ГМ-КСФ та Мег-КСФ, що стимулюють його ранні етапи та
продукуються клітинами стромального оточення, нирок, лімфоци-
тами й моноцитами. Тромбопоетин, ІЛ-6, мегакаріоцитарний сти-
мулюючий фактор індукують диференціювання переважно зрілих
мегакаріоцитів, проте не виключений їх вплив на ранні форми.
До інгібіторів тромбоцитопоезу належать трансформуючий
ростовий фактор, тромбоцитарний ростовий фактор IV. Обидва ці
цитокіни посилюють мітотичну активність фібробластів, а тран-
сформуючий ростовий фактор стимулює синтез колагену в кістко-
вому7 мозку.
Кількість тромбоцитів у периферичній крові в нормі колива-
ється від 150 до 400 • 109/л; такі широкі межі свідчать, що їхня кіль-
кість регулюється не жорстко. Тромбоцити різні за розміром, ві-
ком, щільністю та функціональною активністю. Об’єм (МСУ)
тромбоцитів обернено пропорційний їхній кількості, тому7 в люди-
ни загальний об’єм тромбоцитної маси є постійною величиною
Тривалість життя тромбоцитів — 10 діб. Щодня тромбоцитів про-
дукується до 40 • 109/л.
Лімфоїдний паросток гемопоезу складається із В-, Т- та МК-
клітинних ліній. Клітина-попередник має морфологічну характе-
ристику бластів. Ідентифікація лінійної належності та етапів дифе-
ренціювання лімфоїдних елементів можлива лише за умови
імунофенотипового дослідження.
В-лімфоїдні попередники в кістковому мозку — центрально-
му органі В-лімфопоезу — проходять антигеннезалежну стадію
дозрівання. На найбільш ранніх про-В-клітинах зберігаються ан-
тигени стовбурових елементів (СО34 та СО38) і з’являються специ-
фічні макромолекули пан-В-СВ19, а потім цитоплазматичний
СВ22. Ці антигени характерні для всіх клітин В-лімфоїдного ряду.
Згодом на пре-пре-В-клітинах зменшується кількість стовбурово-
клітинних та з’являються інші В-антигени — СЦ10, СО2() та СО24.
На стадії пре-В-бластів у цитоплазмі клітин з’являються ланцюги
І§. На останньому етапі В-диферегщіювання на мембрані лімфо-
16
| Кровотворення
їдних клітин експресується повна молекула (легкі та великі лан-
цюги) І§М.
Морфологічно зрілі, але імунологічно “наївні” В-лімфоцити
потрапляють у кров та в периферичні органи імунітету — лімфа-
тичні вузли та селезінку. Після проходження в зародкових центрах
вторинних лімфоїдних фолікулів антигензалежної стадії диферен-
ціювання формується пул зрілих В-лімфоцитів та плазматичних
клітин, здатних синтезувати та продукувати імуноглобуліни різних
класів та брати участь у гуморальній відповіді імунної системи.
Попередники Т-клітин у кістковому7 мозку експресують стов-
бурово-клітинні (СП,4, НЕА-Ог) та (СП7, цитоплазматичний
СР3+) Т-антигени. Вийшовши із кісткового мозку, Т-попередни-
ки мігрують у тимус — центральний орган Т-лімфопоезу, після чо-
го надходять у периферичні лімфоїдні органи, де відбувається ан-
тигензалежна стадія диференціювання. У підгрудинній залозі
здійснюється перебудова генів, котрі кодують Т-клітинний рецеп-
тор — і він з’являється на поверхневій мембрані.
У період дозрівання в тимусі на лімфоїдних елементах дещо
змінюється комплекс мембранних антигенів. На ранніх тимоцитах
експресуються макромолекули ТЬуІ, СП5 та цитоплазматичний
СР3. Антиген СР3 є імуноспецифічною ознакою всіх Т-клітин. На
кортикальних тимоцитах визначаються антигени СОр СР4 та СР8.
На останньому етапі медулярні тимоцити розділяються на дві
фракції клітин, що експресують СО4 або СР8 Зрілі Т-лімфоцити
перебувають у периферичній крові, в тимусзалежних ділянках се-
лезінки, лімфатичних вузлів, мигдаликів, нейферових бляшок.
У периферичній крові Т-лімфоцитарний пул існує у вигляді двох
фракцій — хелпери/ефектори (СР4+) та супресори/цитотоксичні
клітини (СР8+). Т-лімфоцити СИ4+ поділяються на два субтипи:
Т-хелпери-1 і Т-хелпери-2. Т-хелпери-1 сприяють підсиленню
синтезу та продукції В-клітинами 1§; Т-хелпери-2 допомагають ін-
дукувати антигенспецифічну активність Т-супресорів.
Т-лімфоїдні клітини не синтезують і не секретують імуногло-
буліни. Вони продукують цитокіни, котрі регулюють проліферацію
та диференціювання інших клітин і беруть участь у клітинній імун-
ній відповіді.
Природні (натуральні) кілери (?4К-клітини) мають незалежну
лінію диференціювання. У периферичній крові морфологічно во-
ни визначаються як великі гранульовані лімфоцити, що мають яд-
ро з невеликою виїмкою та великими азурофільними гранулами.
17
Н.М. Третяк | Гематологія
Вони експресують СП3-. СВ16+, СП56+ антигени та мають на мем-
брані рецептор до СВ2 (С34), вірусу Епштейна—Барра, Ес-рецеп-
тор — для 1§. Натуральні кілери можуть відповідати за спонтанну
клітинну цитотоксичність (рис. 2).
МЕТОДИ ОБСТЕЖЕННЯ ГЕМАТОЛОГІЧНИХ ХВОРИХ
Основою для проведення диференційного діагнозу при захво-
рюваннях системи крові є грамотно виконаний та прочитаний ана-
ліз периферичної крові, тому що суттєва частина діагностики гема-
тологічної патології базується на морфологічних дослідженнях із
урахуванням як кількісних, так і якісних параметрів.
Загальний аналіз крові включає визначення концентрації ге-
моглобіну (НЬ), підрахунок кількості еритроцитів, лейкоцитів,
тромбоцитів у 1 л крові за міжнародною системою (СІ), визначен-
ня швидкості зсідання еритроцитів (ШЗЕ), колірного показника та
вивчення морфологічних особливостей клітин у забарвлених за ме-
тодом Романовського, Паппенгейма препаратах із підрахунком
лейкограми.
Кількісний склад та морфологія клітин крові в нормі характе-
ризуються високою стабільністю, що забезпечується чіткою робо-
тою механізмів внутрішнього гомеостазу людини.
Проте слід зазначити, що на показники крові можуть вплива-
ти фізичне або емоційне навантаження, зміна кліматичних, сезон-
них та метеорологічних умов, період доби, прийом їжі. Крім того,
оперативне втручання, медикаментозне лікування, іонізуюче опро-
мінення, фізіотерапевтичні процедури, деякі діагностичні заходи
можуть також змінювати показники периферичної крові.
При захворюваннях системи крові результати дослідження пе-
риферичної крові є основою для обрання схеми лікування та спос-
тереження за динамикою процесу.
Гемоглобін
Найточнішим методом визначення концентрації гемоглобіну є
ціанметгемоглобіновий метод, в основі котрого лежить перетво-
рення складових форм (оксигемоглобіну, карбоксигемоглобіну,
метгемоглобіну та ін.) гемоглобіну, що міститься в еритроцитах, на
ціангемоглобін.
У нормі показник концентрації гемоглобіну у чоловіків стано-
вить 132-1б4г/л, у жінок — 115-145г/л.
Зростання концентрації гемоглобіну в крові відбувається при
симптоматичних еритроцитозах та справжній еритремії.
18
| Кровотворення
Зниження вмісту гемоглобіну визначається при всіх формах
анемій, хронічних запальних захворюваннях та онкологічній і он-
когематологічній патології.
Еритроцити
Підрахунок еритроцитів може проводитись мануальним мето-
дом та електронними лічильниками. Автоматичний підрахунок є
більш точним методом визначення кількості клітин.
У нормі кількість еритроцитів у крові становить: у чоловіків
4,5-5,5 • Ю12/л, у жінок 3,5-4,5 • 1012/л.
Зростання кількості еритроцитів у крові відбувається при
справжній еритремії та симптоматичних еритроцитозах.
Зменшення вмісту еритроцитів спостерігається при апластич-
ній, гемолітичній, різних формах спадкових анемій. При залізоде-
фіцитній анемії кількість еритроцитів коливається близько ниж-
ньої межі норми.
Колірний показник
Вміст гемоглобіну в еритроциті не однаковий при різних
формах анемії. Серед індексів, що характеризують еритроцит,
найпоширенішим є колірний показник (КП), котрий показує се-
редній вміст гемоглобіну в еритроциті. В нормі КП становить
0,85-1,05. КП, нижчий за 0,8, характерний для залізодефіцитної
анемії. КП, вищий за 1,05, характерний для анемій, зумовлених
дефіцитом вітаміну В]2 та фолієвої кислоти. Нормальний вміст
гемоглобіну в еритроциті спостерігається при апластичній та ге-
молітичній анеміях, гемобластозах, анеміях, зумовлених трива-
лою інтоксикацією.
Для отримання повної і точної характеристики еритроцита
визначають їх середній об’єм, розміри, морфологічні особливості.
При дослідженні зазначених показників на геманалізаторах
нормальні величини їх становлять:
МСУ — середній об’єм еритроцита — 75-95 мкм3;
КЛАУ — ширина розподілення еритроцита за об’ємом —
11,5-14,5%;
МСН — середній вміст гемоглобіну в еритроциті — 27-31 п5;
МСНС — середня концентрація гемоглобіну в еритроцитах —
30-37 г/дл.
Важливе значення для діагностики має форма еритроцитів.
У мазку крові здорової людини еритроцити завжди приблизно од-
накові за розміром та круглі за формою. Мають рівномірне забар-
влення з невеличким просвітленням у центрі. При анеміях спосте-
19 -
Н.М. Третяк | Гематологія
рігаються еритроцити різні за розміром (анізоцитоз), за формою
(пойкілоцитоз), за забарвленням (анізохромія).
Для діагностики при різних формах анемій мають значення
морфологічні ознаки еритроцитів. Так, для В р-дефіцитної анемії
характерні макроцитоз та інтенсивне забарвлення (гіперхромія), для
залізодефіцитних анемій — мікроцитоз та бліде забарвлення зі знач-
ним просвітленням у центрі. При спадковій патології еритроїдного
паростка кровотворення спостерігається мікро-, сфероцитоз — при
хворобі Мінковського і Шоффара, при серпоподібній клітинній
анемії — серпоподібні еритроцити, при спадковому овалоцитозі —
овалоцити тощо.
Ретикулоцити
Ретикулоцити — це молоді форми еритроцитів, у цитоплазмі
яких після втрати ядра лишились агреговані рибосоми та міто-
хондрії у вигляді зернисто-нитчастої субінстанції. У здорової лю-
дини кількість ретикулоцитів становить 2-12%. Зростає кількість
ретикулоцитів при гемолітичному кризі, при великій крововтра-
ті. Знижена кількість ретикулоцитів спостерігається при аплас-
тичній анемії.
Лейкоцити
Одним із важливих показників гемограми є оцінка кількості
лейкоцитів. При мануальному методі дослідження підрахунок лей-
коцитів проводять у камері Горяєва. Нормальний вміст лейкоцитів
становить 4,5-9,0 • 109/л.
Збільшення вмісту лейкоцитів (понад 9,0 • 109/л) розцінюється
як лейкоцитоз і може спостерігатися при онкогематологічних, он-
кологічних, інфекційних захворюваннях та лейкемоїдних реакціях.
Сучасні технології дають змогу швидко та з високою точністю
підраховувати лейкоцити в автоматичних лічильниках з одночас-
ним визначенням гранулоцитів, лімфоцитів, моноцитів:
\¥ВС — кількість лейкоцитів (4,0-9,0 • 109/л);
ОКА — гранулоцити, 56-74%, абсолютний вміст 2,3-6,4;
1_¥С — лімфоцити, 25-40%. абсолютний вміст 1-3,6;
МОИ — моноцити, 2-6%, абсолютний вміст 0,08-0.54.
Зниження кількості лейкоцитів менш ніж 4 • 109/л розцінюєть-
ся як лейкопенія, причиною котрої може бути зменшення продук-
ції нейтрофільних гранулоцитів кістковим мозком — при захворю-
ваннях системи крові (апластична анемія, ідіопатичний
мієлофіброз). Крім того, існують лейкопенії, зумовлені зменшен-
ням тривалості циркуляції гранулоцитів у судинному руслі та зат-
20
| Кровотворення
римкою виходу їх із кісткового мозку, а також впливом інфекцій-
них, хімічних та фізичних факторів.
Підрахунок лейкограми (лейкоцитарна формула) має важливе
диференційно-діагностичне значення при встановленні діагнозу ба-
гатьох захворювань, особливо при діагностуванні таких захворювань
системи крові, як лейкемії гострі та хронічні, мієлоїдні та лімфоїдні.
Порушення в лейкограмі, так звані лейкемоїдні реакції, спос-
терігаються при гострих запальних, гнійно-септичних процесах,
онкопатології та інфекційних захворюваннях.
Нейтрофільний лейкоцитоз може мати місце при таких фізіо-
логічних станах, як вагітність та фізичне перевантаження.
Тромбоцита
Кількість тромбоцитів — важливий показник при захворюван-
нях системи крові, а також диференційно-діагностична ознака при
онкологічних, інфекційних захворюваннях та при патології систе-
ми гемостазу.
Підрахунок тромбоцитів проводять у камері Горяєва із вико-
ристанням хлориду кокаїну або оксалату амонію. Більш точним є
підрахунок тромбоцитів з допомогою кондукгометричного методу.
Цей метод одночасно визначає й морфологічні ознаки тромбоцита.
Межі коливань показників тромбоцитів наступні:
РЬТ — кількість тромбоцитів — 150,0-400,0 • 109/л;
МРУ — середній об’єм тромбоцитів — 7,4-10,4фл;
РП\У — ширина розподілення тромбоцитів за об’ємом —
10-20%;
РСТ — тромбокрит — 0,15-0.40%.
Визначення швидкості зсідання еритроцитів (ШЗЕ) прово-
диться з допомогою капіляра Панченкова та вимірюється у мілі-
метрах на 1 годину. Норма ШЗЕ становить для жінок 2-15 мм/год;
для чоловіків 1-10 мм/год.
Прискорення зсідання еритроцитів можливе при сепсисі, ін-
фекційних та онкологічних захворюваннях, а також при збільшен-
ні вмісту глобулінів, холестерину, фібриногену. ШЗЕ може упо-
вільнюватися при підвищеному вмісті еритроцитів, альбумінів,
жовчних кислот у крові та посиленій в'язкості крові.
Нормальні показники ШЗЕ — не завжди незаперечна ознака
відсутності патологічного процесу.
Дослідження кісткового мозку
Цитологічне дослідження кісткового мозку відіграє вирішаль-
ну роль у діагностиці захворювань системи крові та є одним із важ-
21
Н.М. Третяк | Гематологія
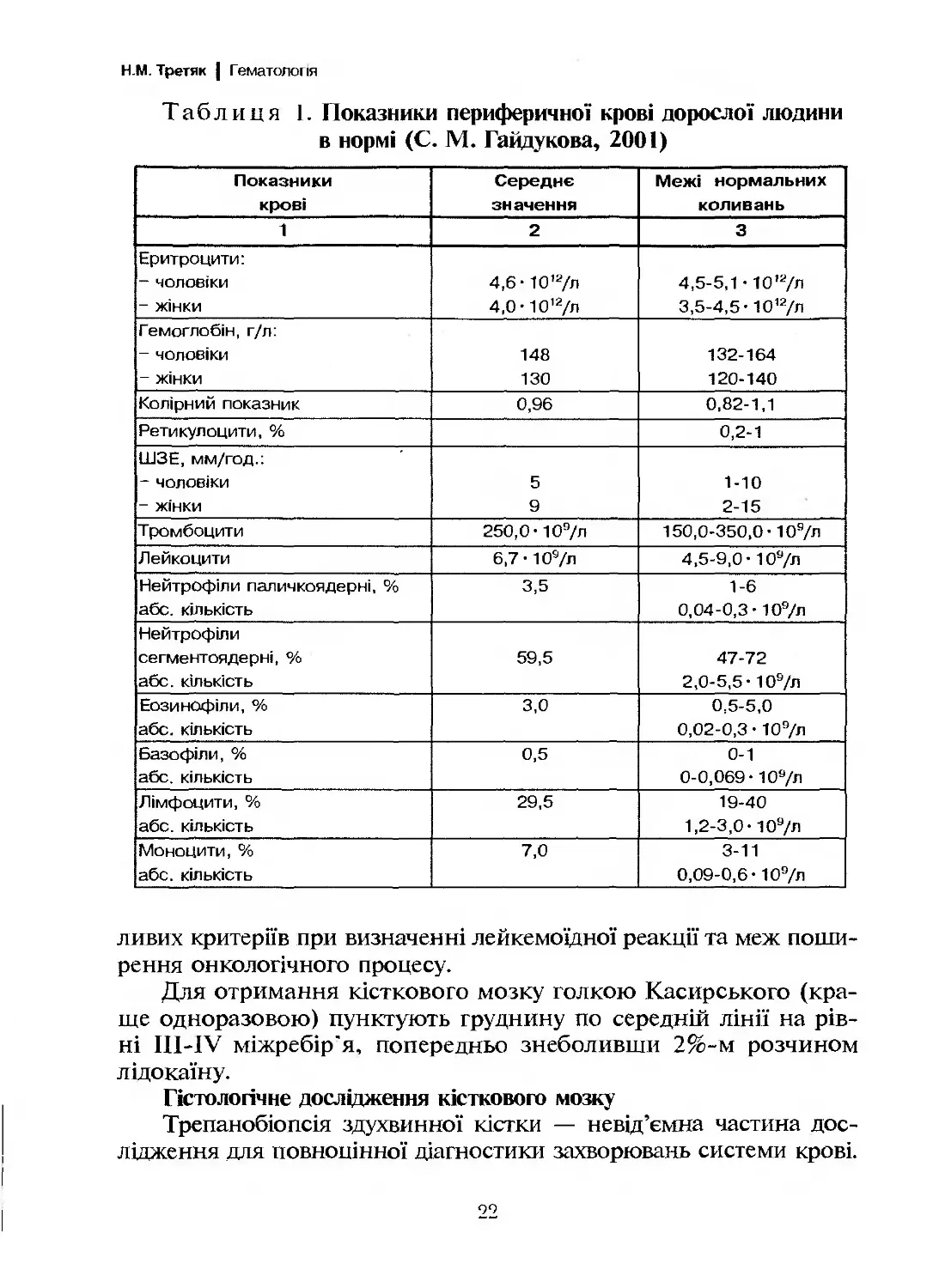
Таблиця 1. Показники периферичної крові дорослої людини
в нормі (С. М. Гайдукова, 2001)
Показники крові Середнє значення Межі нормальних коливань
1 2 3
Еритроцити: - чоловіки - жінки 4,6- 10'7л 4,0- 10'7л 4,5-5,1 • 10’7л 3,5-4,5- 10’7л
Гемоглобін, г/л: - чоловіки - жінки 148 130 132-164 120-140
Колірний показник 0,96 0,82-1,1
Ретикулоцити, % 0,2-1
ШЗЕ, мм/год.: - чоловіки - жінки 5 9 1-10 2-15
Тромбоцити 250,0- 109/л 150,0-350,0- 107л
Лейкоцити 6,7- 109/л 4,5-9,0- 107л
Нейтрофіли паличкоядерні, % або. кількість 3,5 1-6 0,04-0,3 • 109/л
Нейтрофіли сегментоядерні, % абс. кількість 59,5 47-72 2,0-5,5- 109/л
Еозинофіли, % абс. кількість 3,0 0,5-5,0 0,02-0,3 • 109/л
Базофіли, % абс. кількість 0,5 0-1 0-0,069- 109/л
Лімфоцити, % абс. кількість 29,5 19-40 1,2-3,0- 107л
Моноцити, % абс. кількість 7,0 3-11 0,09-0,6- 109/л
ливих критеріїв при визначенні лейкемоїдної реакції та меж поши-
рення онкологічного процесу.
Для отримання кісткового мозку голкою Касирського (кра-
ще одноразовою) пунктують груднину по середній лінії на рів-
ні ІП-ІУ міжребір'я, попередньо знеболивши 2%-м розчином
лідокаїну.
Гістологічне дослідження кісткового мозку
Трепанобіопсія здухвинної кістки — невід’ємна частина дос-
лідження для повноцінної діагностики захворювань системи крові.
| Кровотворення
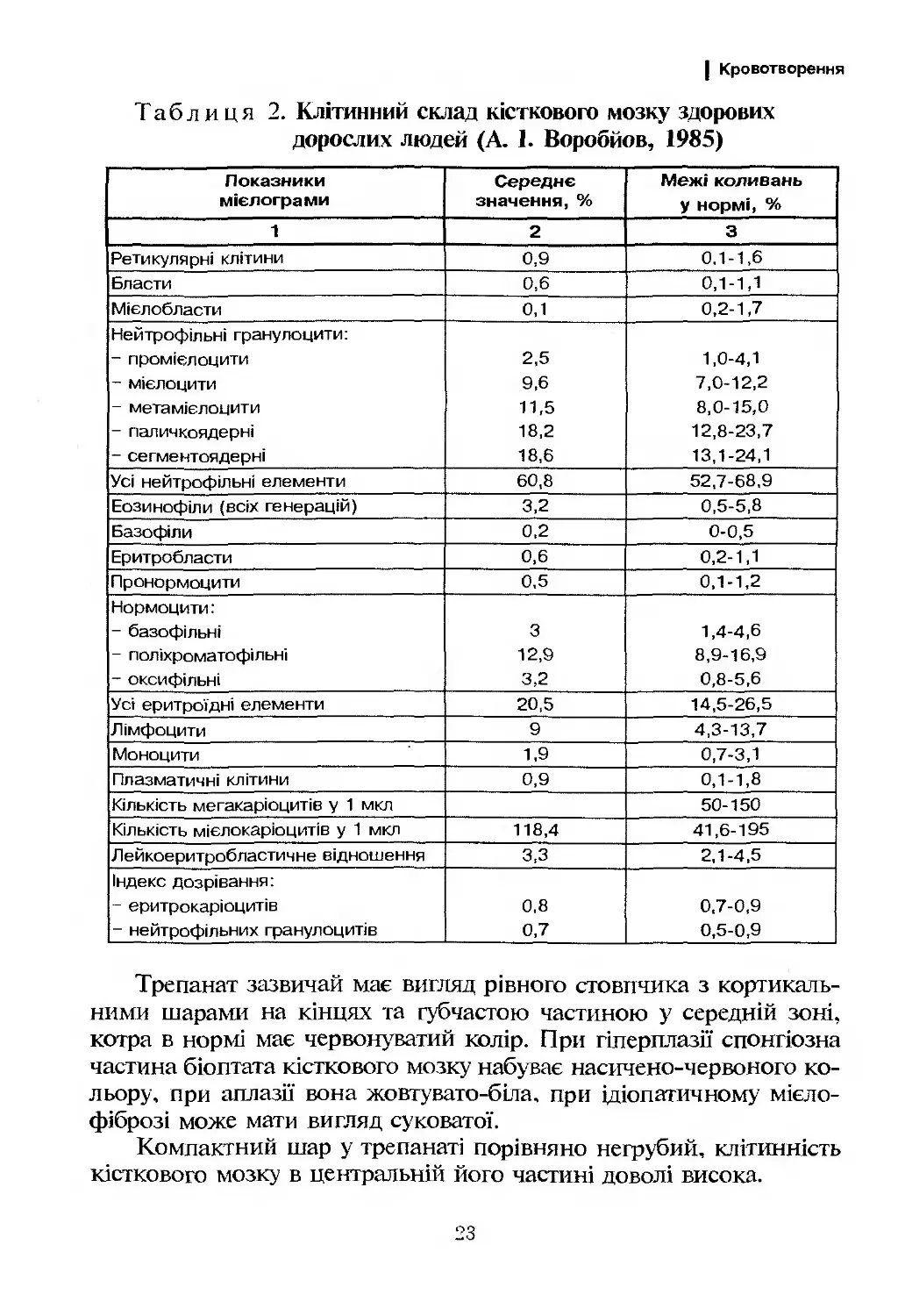
Таблиця 2. Клітинний склад кісткового мозку здорових
дорослих людей (А. 1. Воробйов, 1985)
Показники мієлограми Середнє значення, % Межі коливань у нормі, %
1 2 3
Ретикулярні клітини 0,9 0,1-1,6
Бласти 0,6 0,1-1,1
Мієлобласти 0,1 0,2-1,7
Нейтрофільні гранулоцити: ~ промієлоцити - МІЄЛОЦИТИ - мета мієлоцити - паличкоядерні - сегментоя дерні 2,5 9,6 11,5 18,2 18,6 1,0-4,1 7,0-12,2 8,0-15,0 12,8-23,7 13,1-24,1
Усі нейтрофільні елементи 60,8 52,7-68,9
Еозинофіли (всіх генерацій) 3,2 0,5-5,8
Базофіли 0,2 0-0,5
Еритробласти 0,6 0,2-1,1
ГІронормоцити 0,5 0,1-1,2
Нормоцити: - базофільні - поліхроматофільні - оксифільні 3 12,9 3,2 1,4-4,6 8,9-16,9 0,8-5,6
Усі еритроїдні елементи 20,5 14,5-26,5
Лімфоцити 9 4,3-13,7
Моноцити 1,9 0,7-3,1
Плазматичні клітини 0,9 0,1-1,8
Кількість мегакаріоцитів у 1 мкл 50-150
Кількість мієлокаріоцитів у 1 мкл 118,4 41,6-195
Лейкоеритробластичне відношення 3,3 2,1-4,5
Індекс дозрівання: - еритрокаріоцитів - нейтрофільних гранулоцитів 0,8 0,7 0,7-0,9 0,5-0,9
Трепанат зазвичай має вигляд рівного стовпчика з кортикаль-
ними шарами на кінцях та губчастою частиною у середній зоні,
котра в нормі має червонуватий колір. При гіперплазії спонгіозна
частина біоптата кісткового мозку набуває насичено-червоного ко-
льору, при аплазії вона жовтувато-біла, при ідіопатичному мієло-
фіброзі може мати вигляд суковатої.
Компактний шар у трепанаті порівняно негрубий, клітинність
кісткового мозку в центральний його частині доволі висока.
23
Н.М. Третяк | Гематологія
У губчастій частині трепаната існують численні неушкоджені
кісткові балки звивистої форми із включеними в їхні комірки кіс-
тковомозковими клітинами, гранулоцитарнсї, еритроїдної, мегака-
ріоцитарної та лімфоїдної ланок кровотворення.
У нормі співвідношення кровотворних клітин та жирового кіс-
ткового мозку' — 1:1. Дослідження трепану дозволяє скласти уяв-
лення про стан кістковомозкової тканини, її взаємозв’язок із мік-
рооточенням, про наявність резорбцій або утворення монореї
кісткової тканини (колагенові волокна) чи фіброзних вогнищ.
У трепанаті добре розрізняються мегакаріоцити, еозинофіли, плаз-
матичні клітини.
Дослідження лімфатичних вузлів
Цитологічне й гістологічне дослідження лімфатичного вузла ві-
діграє важливу роль у диференційній діагностиці таких хронічних
лімфопроліферативних захворювань, як негоджкінська злоякісна
лімфома, лімфогранулематоз, хронічна лімфоїдна лейкемія, метас-
тази рака в лімфатичні вузли. Важливе значення має морфологіч-
не дослідження лімфатичного вузла при гострих та хронічних нес-
пецифічних лімфаденітах, туберкульозному лімфаденіті, саркоїдозі
Беньє—Бека—Шаумана.
Таблиця 3. Цитограма лімфатичного вузла в нормі
(за даними М. І. Абрамова, 1974)
Лімфобласти 0,1-0,5%
Пролімфоцити 65-80%
Лімфоцити 20-35%
Лімфоїдні ретикулярні клітини 0-0,9%
Плазматичні клітини 0,2-0,6%
Макрофаги 0,1-0,5%
Тучні, тканеві клітини 0-01%
Субстрат лімфатичного вузла в нормі складається, переважно,
із пролімфоцитів та лімфоцитів. Лімфоцити лімфатичного вузла
майже не відрізняються від лімфоцитів периферичної крові. Про-
лімфоцити мають ядро грубопухкої структури, іноді з ядерцем,
світлу ніжнобазофільну цитоплазму. Лімфобласти — клітини дещо
більшого розміру, ніж лімфоцити, з ніжною хроматиновою струк-
турою, ядра містять 1-2 ядерця, вузький обідок базофільної цитоп-
лазми з перинуклеарним просвітленням. Крім того, до клітинного
24
| Кровотворення
складу лімфатичного вузла входять стромальні клітини, макрофа-
ги, плазмоциди, лаброцити та зрілі клітини периферичної крові,
всього до 2%.
Гістологічна картина лімфатичного вузла представлена сполуч-
но-тканинною капсулою, корковою та мозковою речовинами.
У корковому шарі, котрий складається, в основному, із лімфатич-
них фолікулів, утворюються лімфоцити, які переміщуються в моз-
кову речовину.
Морфологічна картина лімфатичного вузла представлена зрі-
лими лімфоцитами, поодинокими лімфобластами, нейтрофілами
та макрофагами.
АНЕМІЇ
Анемія, або недокрів’я — це патологічний стан, котрий харак-
теризується зменшенням загального вмісту гемоглобін}7, що най-
частіше проявляється у зниженні його концентрації в одиниці
об’єму крові.
У більшості випадків, за винятком залізодефіцитних станів та
таласемії, анемія супроводжується зменшенням вмісту еритроцитів
у одиниці об’єм}' крові.
За етіологією, патогенезом та клініко-гематологічною симпто-
матикою анемії дуже різноманітні, що зумовило створення декіль-
кох класифікацій, у тому числі І. А. Касирського, Г. О. Олексієва
(1970 р.) та Л. І. Ідельсона (1979 р.).
На наш погляд, класифікацію Л. І. Ідельсона доцільно вико-
ристовувати фахівцям-гематологам.
Класифікація І. А. Касирського, побудована за патогенетич-
ним принципом та з урахуванням етіологічних і найважливіших
клініко-морфологічних ознак, зручна дчя використання в медичній
практиці лікарів загального профілю.
Класифікація анемій (І. А. Касирський, Г. О. Олексісв, 1970)
1. Анемії постгеморагічні:
— гостра постгеморагічна анемія;
— хронічна постгеморагічна анемія.
2. Анемії, пов ’язані з порушенням процесу кровотворення:
— залізодефіцитні анемії;
— анемії, пов’язані з порушенням синтезу порфіринів;
— анемії, пов’язані з дефіцитом мікроелементів;
— анемії, пов’язані з порушенням синтезу ДНК та РНК (мега-
лобластні анемії);
— апластичні (спадкові та набуті), що пов’язані з пригнічен-
ням проліферації
клітин кісткового мозку.
3. Анемії, зумовлені прискореним кроворуйнуванням (гемолітичні
анемії):
— спадкові, зумовлені порушенням структури мембрани ерит-
роцитів;
26
| Анемії
— спадкові, зумовлені порушенням активності ферментів
еритроцитів;
— набуті гемолітичні анемії.
Визначають три ступені тяжкості анемії, залежно від вмісту ге-
моглобіну в крові:
— анемія легкого ступеня — вміст НЬ 110-90 г/л,
— анемія середнього ступеня — вміст НЬ 89-70 г/л,
— анемія великого ступеня — вміст НЬ 69 г/л та менше.
В залежності від величини еритроцитів та їх насиченості гемог-
лобіном (за даними колірного показника) виділяють три морфоло-
гічних типи анемій:
— нормоцитарні нормохромні анемії, при котрих забарвлення
еритроцитів нормальне й колірний показник дорівнює 0,86-1,1;
— мікроцитарні анемії, котрі можуть бути гіпо- або нормох-
ромними, їх характеризує мікроцитоз, анізопойкілоцитоз, гіпохро-
мія, колірний показник, менший за 0,7;
— макроцитарні анемії, що характеризуються збільшенням
розмірів еритроцитів — макро’цитозом, мегалоцитозом та збіль-
шенням колірного показника (понад 1,1).
ГОСТРА ПОСТГЕМОРАГІЧНА АНЕМІЯ
Гостра постгеморагічна анемія розвивається внаслідок швидкої
втрати значної кількості крові. Гостра крововтрата може бути зу-
мовлена як внутрішніми, так і зовнішніми травмами, крововтрата-
ми під час оперативних втручань та пологів, а також кровотечами
при виразковій хворобі шлунка та кишечника, виразковому коліті,
дизентерії та черевному тифі, некротизованих судинах при пухли-
нах, туберкульозі, бронхоектатичній хворобі, позаматковій вагіт-
ності, менометрорагіях тощо.
У патогенезі основних клінічних проявів гострої крововтрати
провідну роль відіграє швидке зменшення загального об’єму крові,
і найперше її плазмової частини. Зменшення об’єму циркулюючих
еритроцитів призводить до гострої гіпоксії, що проявляється за-
дишкою та серцебиттям. Під час крововтрати та відразу після неї
відбувається посилена продукція наднирковими залозами катехо-
ламінів, що супроводжується спазмом периферичних судин, та мо-
білізація крові із депо, завдяки чому нормалізується гемодинаміка.
Водночас включається механізм аутогемоділюції, котрий мобілізує
власну міжтканинну рідину та спрямовує її в судинне русло. Акти-
Н.М. Третяк | Гематологія
вуються гуморальні механізми, які сприяють утриманню в організ-
мі води та іонів натрію. Такий механізм саморегуляції може ком-
пенсувати до 10-15% об’єму циркулюючої крові, що достатньо при
невеликих та повільних крововтратах.
Швидка втрата 33% маси циркулюючої крові та повільна кро-
вовтрата 50% крові без надання хворому відповідної допомоги мо-
же призвести до смерті. На фоні тканинної гіпоксії розвивається
картина геморагічного шоку з порушенням геодинаміки, внаслідок
патологічного депонування крові, та ексудацією її рідкою частини
в тканини. При цьому розвивається ацидоз, вихід іонів калію із
клітин, посилений катаболізм білків, дистрофічні зміни в паренхі-
матозних органах із недостатністю функції міокарда, центральної
нервової системи та ін.
Внаслідок гіпоксії, пов’язаної з крововтратою, підвищується
концентрація еритропоетину, що призводить до підсилення пролі-
ферації еритропоетинчутливих клітин зі збільшенням еритрокаріо-
цитів та вмісту ретикулоцитів у периферичній крові.
Клініка. Гостра постгеморагічна анемія зумовлює, насамперед,
симптоми колапса, а саме: значну блідість, спричинену спазмою
судин, запаморочення, задишку, непритомність, шум у вухах, ми-
готіння перед очима, холодний піт, блювання, а в деяких випадках
— судоми та втрату притомності. Об’єктивно — тахикардія, нитко-
подібний пульс, зниження артеріального тиску.
У периферичній крові в цей час виявляється гіпертромбоцитоз
(до 1000 • 109/л), нейтрофільний лейкоцитоз зі зрушенням до міє-
лоцитів, вміст еритроцитів та гемоглобіну — в межах норми.
На 2-3 день загальний стан хворого покращується внаслідок
поновлення об’єму циркулюючої крові та плазми. Водночас спос-
терігається зниження рівня гемоглобіну7 та кількості еритроцитів —
розвивається постгеморагічна нормохромна анемія, що є показни-
ком високої компенсаторної здатності організму Якщо крововтра-
ти не повторюються, а еритроцитопоез не порушений, то за 2-3
тижні кров повністю відновлюється.
Діагноз масивної кровотечі встановлюється на підставі клініч-
них та лабораторних даних і не є складним. Труднощі з діагнозом
виникають при внутрішніх кровотечах та захворюваннях, що суп-
роводжуються ознаками шоку та колапсу. У цих випадках самим
найбільш раннім гематологічним симптомом є гіпертромбоцитоз.
Проте цього симптому може не бути при розвиненні ДВЗ-синдро-
му із підвищеним спожиттям тромбоцитів.
28
| Анемії
Лікування починається з припинення кровотечі та протишоко-
вих заходів, що включають усунення гемодинамічних розладів та
поліпшення реологічних якостей крові, із застосуванням альбумі-
ну, реополіглюкіну, поліглюкіну, желатинолю. Еритропитну масу
(30-40% крововтрати) призначають тільки після поновлення кро-
вообігу через гемоділюцію.
Поповнення всієї витраченої крові тільки кров’ю не припусти-
ме, це може спричинити “синдром масивних трансфузій”.
ЗАЛІЗОДЕФІЦИТНА АНЕМІЯ
Залізодефіцитна анемія — це захворювання системи крові, зу-
мовлене дефіцитом заліза в організмі; супроводжується змінами
параметрів його метаболізму, зменшенням концентрації гемогло-
біну в еритроцитах, кількісними та якісними їх змінами, клінічни-
ми проявами анемічної гіпоксії, сидеропенії та метаболічної ін-
токсикації.
Історія вивчення залізодефіцитної анемії бере свій початок із
XVII століття. Саме тоді Варандал зробив опис “блідого знесилен-
ня” у дівчат пубертатного періоду, яке він назвав хлорозом внаслі-
док зеленкувато-блідого кольору обличчя. Колір обличчя порівню-
вали із кольором нестиглих зелених олив. Наприкінці XIX століття
поряд із уже описаним раннім хлорозом було описано пізній хло-
роз. У працях Фабера вперше було показано роль порушення зас-
воєння заліза та шлункової ахілії як етіологічних чинників у гене-
зі ахлоргідричної, або гастрогенної анемії. У 1933 р. Дамешек
сформулював поняття “есенціальна залізодефіцитна анемія”. По-
дальший процес вивчення залізодефіцитної анемії як захворюван-
ня пов’язаний із поглибленням знань про біологічну роль заліза,
його участь у синтезі гемоглобіну, у функціонуванні ензимів, меха-
нізмів порушення обміну, а також його вплив на формування ане-
мічних станів. Було, зокрема, встановлено, що ахлоргідрія не відіг-
рає суттєвої ролі в порушенні абсорбції заліза і може бути не
причиною, а наслідком його дефіциту.
Епідеміологія. За даними ВООЗ (1987 р.) від дефіциту заліза по-
терпав кожен п’ятий мешканець земної кулі. Тому ВООЗ розроби-
ла та висунула програму гемоглобінового оздоровлення населення
Землі до 2000 р., але, як свідчать дані сьогодення, проблема дефі-
циту заліза залишається актуальною як для високорозвинених дер-
жав, так і для таких, що розвиваються. Поширеність цього захво-
29
Н.М. Третяк | Гематологія
рювання в різних країнах значно відрізняється, що, очевидно, зу-
мовлено різним ступенем їх економічного розвитку, етнічними
традиціями, геохімічними особливостями місцевості проживання,
укладом охорони здоров’я.
У Центральній та Східній Європі 10-12% жінок та 3-8% чоло-
віків страждають на залізодефіцитну анемію Серед осіб молодого
(ювенільний період) віку 50% мають латентний дефіцит заліза чи
залізодефіцитну анемію, а серед жінок дітородного віку у 30% де-
фіцит заліза.
За даними МОЗ України поширеність та захворюваність на за-
лізодефіцитну анемію становить 88% усіх анемій.
Таблиця 4. Поширеність залізодефіцитних анемій в Україні
Все населення Дорослі Діти
Абс. число на 100 000 Абс. число на 100 000 Абс. число на 100 000
580 224 1163,9 247 823 610,2 332 401 3598,6
Захворюваність на залізодефіцитні анемії в Україні
201 659 | 404,5 [ 64 964 160 136 695 | 1479,9 |
Особливо високою є поширеність та захворюваність на залі-
зодефіцитну анемію (як серед дітей, так і серед дорослих) у Тер-
нопільський, Івано-Франківський, Черкаській та Вінницькій
областях. У Києві поширеність залізодефіцитної анемії серед до-
рослих становить 219 на 100 тис. населення, а серед дітей —
1538. Захворюваність на залізодефіцитну анемію серед дорослих
м. Києва становить 163,7, а серед дітей — 741,9 на 100 тис. на-
селення.
Залізодефіцитна анемія та латентний залізодефіцит для бага-
тьох країн є соціально-медичною проблемою, оскільки призводить
до порушення якості життя хворого, зменшення працездатності,
спричиняє функціональні розлади в органах і системах організму,
погіршує перебіг існуючих хронічних захворювань.
Проте, незважаючи на давню історію вивчення проблеми де-
фіциту заліза в організмі людини та залізодефіцитної анемії, відо-
мий етіологічний чинник, до сьогодні недостатньо вивчені проце-
си формування таких ускладнень дефіциту заліза, як анемічний,
сидеропенічний синдроми, механізми ушкодження органів та ці-
лих систем у організмі людини внаслідок гіпоксії та сидеропенії.
Тільки останнім часом з’явилися дані щодо синдрому метаболіч-
30
| Анемії
ної інтоксикації, котрий виникає внаслідок значного розбалансу-
вання обміну речовин і накопичення в тканинах сполук, що
справляють патологічний вплив та порушують функціонування
усіх органів і систем.
Патогенетичне усунення дефіциту заліза та корекція вторин-
них порушень метаболізму, шо супроводжують це захворювання,
залишаються актуальною проблемою для установ практичної лан-
ки охорони здоров’я.
Численними дослідженнями встановлено, що залізо є облі-
гатним біоматеріалом, котрий відіграє суттєву роль у забезпечен-
ні нормального функціонування клітин у всіх біологічних систе-
мах. Біологічна значущість заліза в організмі визначається його
здатністю зворотно окислюватися і відновлюватися. Ця власти-
вість забезпечує участь даного елемента у тканинному диханні,
що є обов’язковою умовою існування будь-якої клітини на всіх
етапах еволюції. Залізо як простетична група в комплексі з пор-
фіринами входить до складу білків-хромопротеїнів, а в складі ге-
ма — до структури гемоглобіну та міоглобіну. Дослідження остан-
ніх років показали участь заліза у забезпеченні таких важливих
процесів, як поділ клітин, клітинний та гуморальний імунітет, бі-
осинтетичні процеси, метаболізм фізіологічно активних сполук та
інше. Залізо відіграє визначальну роль у енергетичному обміні;
залізо необхідне для формування в клітинах мозку рецепторів до-
фаміну, що в багатьох випадках проявляється аномалією поведін-
ки людини і психічними порушеннями. Зазначені факти свідчать
про глобальність негативних наслідків порушень метаболізму за-
ліза у людини.
В організмі здорової людини міститься від 2,0 до 5,5 г заліза
(50 мг/кг у чоловіків, 35-40 мг/кг у жінок, 70 мг/кг у новонарод-
жених).
Баланс заліза в організмі людини визначається трьома факторами:
1) кількістю, що споживається з їжею та засвоюється у травно-
му тракті;
2) потребами — для забезпечення синтезу залізомістких сполук
та їх діяльності, насамперед гемоглобіну;
3) втратами заліза, що можуть бути зумовлені як фізіологічни-
ми, так і патологічними процесами.
Патогенетичним фактором дефіциту заліза є його від’ємний
баланс, зумовлений невідповідністю між вживанням, всмоктуван-
ням та засвоєнням або підвищеними втратами.
31
Н.М. Третяк | Гематологія
Таблиця 5.Розподілення заліза в організмі людини (чоловіки —
маса тіла 70 кг, жінки — 50 кг)
Розподілення заліза в організмі людини. Фракції заліза Абсолютна величина, МГ Відносна величина, %
чоловіки жінки чоловіки жінки
Гемоглобін 2670 1500 69,6 73,0
Феритин і гемосидерин 800 320 20,9 15,8
Міоглобін 350 220 9,10 10,6
Гемінові ензими 8 7 0,2 0,3
Трансферин 6 5 0,2 0,3
Разом 3834 2052 100 100
Залізо, що міститься в організмі, поділяється на:
1) функціональне — у склдці гемоглобіну, міоглобіну, ензимів і
коферментів;
2) транспортне — трансфери н, мобілферин;
3) депоноване — феритин, гемосидерин;
4) залізо, що утворює вільний пул.
У зв’язку зі статевими відмінностями в метаболізмі заліза, слід
зауважити, що менструальні кровотечі у жінок можуть легко стати
чинником залізодефіцита в організмі.
Добова потреба дорослої людини в залізі у стані фізіологічної рів-
новаги становить 1,0-1,5 мг, зростаючи у жінок під час місячних до
2,5-3,5 мг. Відомо, що об’єм середньої крововтрати при фізіологічних
місячних становить близько 40 мл за цикл. Близько 10% жінок втра-
чають до 80 мл крові за цикл, що еквівалентно втраті 3 мг заліза. Ос-
кільки в середньому абсорбується 1 мг заліза за добу з 10-20 мг залі-
за, що має міститися в їжі, то цілком очевидно, що в жінок із рясними
та тривалими місячними баланс заліза в організмі є досить нестійким.
На формування тканин плоду організм матері витрачає до 700 мг
заліза. Витрати його на формування плаценти становлять 250-300 мг.
Під час фізіологічних пологів втрачається близько 50 мг заліза з кро-
вотечею. Лактація при вигодовуванні немовляти супроводжується
втратами з лактоферином 1 мг заліза на кожний літр молока.
Всі ці витрати заліза зумовлюють потребу його у жінок під час
вагітності та вигодовування від 3 до 6 мг на добу.
Тобто жіноча стать є дуже вразливою щодо виникнення як ла-
тентного дефіциту заліза, так і залізодефіцитної анемії.
32
| Анемії
Усмоктування заліза, в основному, відбувається у верхніх від-
ділах тонкого кишечника. Шлунок, клубова і товста кишки в цьо-
му процесі беруть участь меншою мірою. Засвоєння заліза в орга-
нізмі визначається рівнем його абсорбції, переважно
у дванадцятипалій кишці та проксимальних відділах тонкої кишки.
У кишечнику дорослої людини всмоктується приблизно 1-2 мг за-
ліза на добу. Із їжі рослинного походження всмоктується заліза від
\% до 3%, тваринного походження — до 10-15%. У харчових про-
дуктах залізо міститься в окисній та закисній формах. Найкраще
всмоктується і засвоюється залізо в складі геміна і продуктів тва-
ринного походження. Тобто харчування продуктами, що містять
м’ясо, зводить до мінімуму імовірність виникнення дефіциту залі-
за. У рослинних, особливо зернових, продуктах переважна кіль-
кість заліза перебуває у важкозасвоюваній формі, зв’язана із фіти-
новою кислотою. Для засвоєння окисне (фері-) негемінове залізо,
в тому числі зі складу мінеральних компонентів дієти, повинне ви-
вільнитись із органічних комплексів і перетворитися на закисну
(фер-) форму. Тому такі багаті, на перший погляд, на залізо про-
дукти, як печінка, нирки, легені, не можуть бути рекомендовані
у дієтичному харчуванні при дефіциті заліза, оскільки містять залі-
зо у вигляді депонованих (фері-) форм.
Ступінь абсорбції заліза залежить як від його кількості в спо-
живаній їжі, так і від біодоступності. Цей процес регулюється особ-
ливими рецепторами слизової оболонки травного тракту, відпові-
дальними за накопичення заліза в організмі. Коли потреба в ньому
збільшується, внаслідок виснаження запасів при швидкому рості,
вагітності, менструальних або патологічних кровотечах, ефектив-
ність абсорбції збільшується на 10-20%.
Навпаки, при перевантаженні депо заліза, його абсорбція в ки-
шечнику істотно зменшується.
Різні патологічні процеси в тонкому кишечнику можуть приз-
водити до порушення усмоктування заліза.
Одним із факторів, що зумовлюють виникнення дефіциту за-
ліза (за умови достатньої його кількості у їжі), є ахілія, атрофічні
зміни слизової оболонки травного тракту, агастральні та аненте-
ральні стани, ентерити, які супроводжуються прискореним про-
ходженням хімусу в порожнині кишечника. Абсорбція заліза
в травному тракті є обернено пропорційною його запасам і підви-
щується при дефіциті. При вагітності абсорбція заліза підвищуєть-
ся до 4 мг на добу.
33
Н.М. Третяк | Гематологія
У нормі в тонкому кишечнику за добу всмоктується від 1-1,5
мг до 2,5 мг заліза, і приблизно стільки ж виводиться.
У плазмі крові здорової людини міститься 4-7 мг заліза, а йо-
го концентрація протягом доби може значно варіювати — від 12,5
до 30,4 мкмоль/л. Концентрація заліза в крові найвища вранці —
від 7 до 10 години, а найнижча увечері — між 20 та 22 годинами.
Зменшення вмісту заліза в плазмі крові спостерігається при
гострих і хронічних запальних процесах, пухлинах, гострому інфар-
кті міокарда. Пропасниця та гострі стадії інфекційних захворювань
завжди супроводжуються гіпоферимією, що настає як наслідок
компенсаторно-пристосовної реакції.
Зменшуючи постачання заліза до тканин, організм досягає
у такий спосіб гальмування розмноження бактерій, ділення кот-
рих залежить від наявності заліза та зменшення інтенсивності
альтернативних аутоокислювальних процесів у них. За виразного
дефіциту заліза, клінічним проявом якого є залізодефіцитна ане-
мія, вміст його у сироватці зменшується. Нормальні значення
вмісту заліза в сироватці не виключають наявності латентного де-
фіциту заліза.
У наш час встановлено взаємозв’язок обміну заліза з метабо-
лізмом інших мікроелементів. Біологічна ефективність викорис-
тання мікроелементів у організмі визначається рівнем збалансова-
ності раціонів поживними та біологічно активними речовинами,
ступенем засвоєння та депонування мікроелементів, їх взаємодією
між собою та іншими речовинами в процесі травлення, всмокту-
вання в травному тракті, транспортування, проміжного обміну та
засвоєння, екскреції, діяльності регуляторних систем, а також фі-
зіологічним станом організму, статтю, віком тощо.
Суттєве значення має не тільки абсолютний вміст у їжі мікро-
елементів, а і їхню засвоюваність.
У травному тракті утилізація мікроелементів залежить від
впливу багатьох сполук, значна частина яких здатні викликати ін-
гібіцію абсорбції. Так, теїни, що містяться в чаї, здатні зменшува-
ти всмоктування заліза, а фітати сприяють усмоктуванню цинку.
Утворюючи комплексні сполуки з органічними речовинами, мік-
роелемент може краще засвоюватись. Так, залізо, мідь та цинк
здатні утворювати комплекси з амінокислотами, що підвищує їхню
біодоступність. Численними дослідженнями доведено, що в орга-
нізмі людини міститься 50 мінеральних елементів. Є фактом і те,
що 26 із 50 елементів необхідні для забезпечення життєдіяльності.
34
| Анемії
Мікроелементами називають 14 елементів, концентрація яких
у організмі людини не перевищує 0,01%. До них належать залізо,
цинк, мідь, марганець, кобальт, олово, селен, нікель, хром, моліб-
ден, йод, ванадій, фтор, кремній.
Сьогодні доведено: метаболізм заліза дуже тісно пов’язаний із
обміном цих мікроелементів у організмі людини, і цілком очевидно,
що залізодефіцитну анемію слід розглядати як полімікроелементоз.
Етіологія. Загальновідомо, що основним етіологічним чинни-
ком залізодефіцитної анемії є дефіцит заліза. Розглянемо найвідо-
міші причини, що можуть спонукати виникнення залізодефіциту
в організмі.
Основні етіологічні чинники розвитку залізодефіцитної анемії
Захворювання травного тракту, які супроводжуються хронічни-
ми крововтратами:
виразкова хвороба, неспецифічний виразковий коліт, ерозивний
гастрит, хронічний тромбофлебіт гемороїдальних вен, тріщини прямої
кишки, пухлини, цироз печінки з явищами портальної гіпертензії.
Захворювання травного тракту, які супроводжуються порушен-
ням усмоктування заліза: ,
анентеральні стани (резекції), синдром мальабсорбцїї, хроніч-
ний ентерит, амілоїдоз кишечника.
Захворювання сечостатевої системи, ускладнені мікро- і макро-
гематурією:
гломеруло- і пієлонефрит, сечокам’яна хвороба, полікістоз ни-
рок, пухлини нирок та сечового міхура, поліпоз, фіброміома, ендо-
метріоз, тривалі та рясні місячні.
Захворювання ендокринної системи:
мікседема, хронічна недостатність надниркових залоз, пухлини
гіпофіза.
Захворювання серцево-судинної системи:
гіпертонічна хвороба з частими носовими кровотечами, атерос-
клероз мезентеріальних судин, портальна гіпертензія з кровотечами із
варикозно розширених вен стравоходу, кардіального відділу шлунка.
Захворювання дихальної системи:
легеневий гемосидероз, рак легенів і бронхів, бронхоектатична
хвороба.
Захворювання системи крові:
гемобластози із геморагічним синдромом, тромбоцитопенії, ге-
морагічні діатези і гемофілія, гіпо- та апластичні анемії, перебіг
яких ускладнений кровотечами.
35
Н.М. Третяк | Гематологія
Паразитарні та глистові інвазії.
Захворювання, що супроводжуються перерозподілом заліза:
гнійно-септичні стани, туберкульоз, гострі й хронічні інфекції,
ревматоїдний артрит, злоякісні пухлини, остеомієліт.
Геморагічні васкуліти та вторинні геморагічні синдроми.
Регулярна неконтрольована участь у донорстві.
Вагітність, лактація.
Патогенез виникнення дефіциту заліза відображає прогресую-
че використання та збіднення його депо, а також порушення фун-
кцій залізозалежних ензимів, білків, рецепторів тощо. Формування
та розвиток дефіциту заліза можна умовно представити у вигляді
трьох послідовних стадій.
Перша стадія — збіднення депо заліза — найбільш рання і мо-
же бути діагностована на підставі зменшення показників запасних
фондів заліза за нормальних значень його концентрації в сироват-
ці та гемоглобіну, підвищення параметрів абсорбції цього металу
в кишечнику (за даними радіологічних досліджень).
Друга стадія — її називають залізодефіцитом без анемії — ха-
рактеризується значним зниженням або відсутністю заліза в депо,
низьким його вмістом у плазмі, зменшенням насичення трансфе-
рину залізом за нормальних параметрів концентрації гемоглобіну
та кількості еритроцитів у периферичній крові.
Третя стадія розвитку залізодефіциту характеризується найсут-
тєвішими порушеннями параметрів заліза в крові та наявністю гі-
похромної мікроцитарної анемії.
На сьогодні не існує уніфікованої клінічної класифікації залі-
зодефіцитних анемій, котра відображала б механізм її розвитку' та
клінічні особливості перебігу7, полегшувала діагностику та вибір лі-
кувальної тактики.
Слід наголосити, що залізодефіцитна анемія завжди виникає
внаслідок дії якогось етіологічного чинника чи поєднання декіль-
кох причин, і тому, як правило, буває вторинною.
Есенціачьна, або первинна, залізодефіцитна анемія в практич-
ній роботі зустрічається дуже рідко.
Найзручнішою, на нашу думку, для використання в клінічній
практиці є класифікація, наведена нижче.
Клінічна юшсифікація залізодефіцитної анемії
1. Доклінічна стадія залізодефіцитної анемії'.
а) прелатентний дефіцит заліза;
16
| Анемії
Ь) латентний дефіцит заліза (крововтрати, нерегульована участь
у донорстві, підвищена потреба у залізі тощо).
2. Клінічна стадія залізодефіцитної анемії:
а) неускладнена форма, обумовлена:
— хронічними крововтратами (хронічна постгеморагічна фор-
ма залізодефіцитної анемії);
— підвищеною потребою у залізі (вагітність, лактація, період
росту і дозрівання, у спортсменів);
— недостатнім початковим рівнем заліза в організмі;
— недостатньою резорбцією заліза у травному тракті (агас-
тральні, пострезекційні стани);
— порушенням транспорту й утилізації заліза (гіпо- та атрас-
феринемія, ензимопатії, аутоімунні пронеси тощо);
— недостатнім вмістом заліза в їжі;
Ь) ускладнена форма, обумовлена причинами (див. п.2,1):
— анемічною гіпоксією (прояви: легкі, середні, тяжкі, тяжкі
з дистрофічними змінами органів — гіпоксична міокардідістрофія,
енцефалопатія тощо);
— сидеропенічним синдромом (прояви: неврологічні розлади,
а також емоційної сфери, смаку, нюху; дизурічні явища; зміни сли-
зової травного тракту, шкіри та її придатків, склер);
— метаболічною ендогенною інтоксикацією (за даними ком-
плексного лабораторного біохімічного дослідження вмісту середніх
молекулярних пептидів, молочної, піровиноградної кислот, вільно-
го гістаміну, вільного серотоніну, вільного гепарину в плазмі кро-
ві тощо).
3. Змішані форми залізодефіцитної анемії:
а) диформна анемія (поєднання залізодефіцитної анемії із віта-
мін В12 дефіцитною анемією);
Ь) поєднання залізодефіцитної анемії із дефіцитом вітамінів В,
Є, С;
с) поєднання залізодефіцитної анемії із дефіцитом мікроеле-
ментів;
сі) полідефіцитна анемія.
Ступінь тяжкості перебігу залізодефіцитних анемій рекоменду-
ється визначати за результатами комплексної оцінки таких показ-
ників (табл. 6).
Визначення ступеня тяжкості перебігу7 залізодефіцитної анемії
у клінічній практиці за показником концентрації гемоглобіну7 є до-
сить умовним і часто не відображає клінічного стану хворого.
37
Н.М. Третяк | Гематологія
Таблиця 6. Основні критерії оцінки ступеня тяжкості
перебігу залізодефіцитної анемії
Показник, одиниця виміру Ступінь тяжкості перебігу залізодефіцитної анемії
Легкий Середній Тяжкий Надтяжкий
Концентрація гемоглобіну, г/л 110-91 90-71 70-51 50 і менше
Ступінь гіпохромії еритроцитів 6-15% клітин 16-30% клітин 31-45% клітин гіпохромія понад 45%
Величина МСНС, пг (середня концентрація Нв у еритроциті) 30,0-31,5 29,0-30,5 28,5-29,0 28,5 і менше
Ступінь анізоцитозу 6-15% клітин 16-30% клітин 31-45% клітин гіпохромія понад 45%
Величина МСУ, мкм3 (середній вміст Нв у еритроциті) 76-80 66-75 56-65 55 і менше
Ступінь пойкілоцитозу 6-15% клітин 16-30% клітин 31-45% клітин гіпохромія понад 45%
Ступінь лоліхромазії, % 1,6-2,5 2,6-3,5 3,6-4,5 понад 4,6
Залізо сироватки, мкмоль/л 10,5-12,5 8,5-10,4 6,5-8,4 менше 6,4
Ферритин, мкг/л 10-12 7-9 4-6 3 і менше
Молочна кислота крові, ммоль/л 1,96±0,09 2,89+ + +
Вільний гістамін плазми, нмоль/л 2,7Г3,86 3,87-4,25 4,26-4,99 понад 5,0
Вільний серотонін плазми, нмоль/л 1,51±0,06 1,69±0,05 1,69±0,07 2,59±0,06
Молекули середньої маси сироватки крові, О.Д./Л 0,26±0,001 0,321±0,002 0.3381±0,002 понад 0,40
Повсякденна лікарська практика свідчить, що частіше тяжкість
стану хворого визначає не стільки ступінь зменшення концентра-
ції гемоглобіну, скільки термін, за який відбулося зменшення йо-
го рівня. Жінки суб’єктивно легше переносять зниження концен-
трації гемоглобіну, ніж чоловіки.
Клінічні прояви залізодефіцитної анемії (ЗДА) складаються із
загальних симптомів анемії, зумовлених гемічною гіпоксією, ознак
тканинного дефіциту заліза (сидеропенічний синдром) та метабо-
лічних порушень (синдром ендогенної метаболічної інтоксикації).
Симптоми клінічного поліморфізму ЗДА
1. Симптоми анемічної гіпоксії:
задишка при фізичних навантаженнях, тахікардія, артеріальна
38
| Анемії
гіпотонія, запаморочення, біль у ділянці серця, парестезії у кінців-
ках, набряки кінцівок, блідість шкіри;
2. Симптоми сидеропенічного синдрому:
втомлюваність, зниження пам’яті, м’язева слабкість, головний
біль, спотворення смаку, випадіння волосся, ламкість нігтів, “заї-
ди”, спотворення нюху, “блакитні склери”, гіпо- або анацидний
гастрит, сухість шкіри, нічний енурез, нетримання сечі, утруднен-
ня ковтання, щеміння язика;
3. Симптоми синдрому метаболічної інтоксикації:
втомлюваність, зниження пам'яті, головний біль, субфебрилі-
тет, тахікардія.
Основними скаргами та клінічними ознаками у хворих із лег-
ким перебігом залізодефіцитної анемії є втомлюваність, зниження
пам’яті, м’язева слабкість, біль у ділянці серця, запаморочення,
блідість шкіри, артеріальна гіпотонія.
У хворих на ЗДА із середнім ступенем тяжкості спостерігаєть-
ся задишка при фізичних навантаженнях, тахікардія, блідість шкі-
ри та інші симптоми анемічної гіпоксії. Більше виражені ознаки
сидеропенїї та метаболічної інтоксикації.
У хворих із тяжким перебігом залізодефіцитної анемії окрім
скарг і ознак, що зумовлені гіпоксією, спостерігається широкий
спектр симптомів сидеропенічного синдрому на фоні виражених
проявів метаболічної інтоксикації.
Щодо клінічної характеристики ЗДА слід також зазначити,
що загальними симптомами анемії є запаморочення, слабкість,
головний біль, задишка, відчуття серцебиття, схильність до неп-
ритомності, особливо в задушливих помешканнях, іноді — ми-
готіння “мушок” перед очима, часто спостерігається помірне
підвищення температури тіла, сонливість удень та погане заси-
нання вночі.
Іноді відзначається відчуття важкості в епігастральній ділянці
живота, погіршення апетиту, диспептичні симптоми, нудота, мете-
оризм, затвердіння або схильність до діареї. Внаслідок поганого
кровопостачання шкіри хворі гіперчутливі до холоду. Виразність
цих скарг залежить від адаптації до анемії. Кращій адаптації спри-
яє повільний темп анемізації.
Діагностика залізодефіцитної анемії є одночасно і простою, і
складною. Простою тоді, коли лікар володіє достатнім досвідом і
знанням класичного та атипових варіантів клінічного перебігу цьо-
го захворювання, сучасними методами діагностики.
39
Н.М. Третяк | Гематологія
Складною у випадках, коли залізодефіцитна анемія протікає
під маскою інших захворювань чи є проявом інших захворювань.
Лабораторна діагностика ЗДА базується на дослідженні карти-
ни периферичної крові з вивченням морфології' еритроцитів, виз-
наченні показників метаболізму заліза та ряду інших параметрів.
Основними критеріями лабораторної діагностики залізодефі-
цитної анемії є визначення таких параметрів (табл. 7).
Диференційна діагностика залізодефіцитної анемії — кропітка
справа, і передусім лікар повинен провести диференційний діагноз
із мієлодиспластичним синдромом, до якого сьогодні включають
гетерогенну групу захворювань, котрі також можуть супроводжува-
тися гіпохромією та макроцитозом із вторинними порушеннями
метаболізму заліза. Діагноз у таких випадках може бути встановле-
ний лише з допомогою морфологічного, цитохімічного та цитоге-
нетичного дослідження кісткового мозку.
Лікування хворих на ЗДА має бути патогенетично обґрунтова-
ним, комплексним і становити цілісну лікувальну програму, котра
повинна ретельно виконуватись пацієнтом та контролюватись лі-
карем. Лікування залізодефіцитної анемії має бути спрямоване не
тільки на усунення анемії як симптому, а й на ліквідацію дефіци-
ту заліза та поповнення його запасів у організмі. Програма лікуван-
ня повинна включати: усунення етіологічних чинників, раціональ-
не лікувальне харчування, патогенетичне лікування препаратами
заліза, профілактичні заходи з упередження її рецидиву.
Із харчових продуктів усмоктування заліза не перевищує
10-20% від загального вмісту, а тому дефіцит заліза в організмі не
можна поповнити лише дієтою. Дієта дорослого хворого на ЗДА
повинна включати 130 г білків, 90 г жирів, 350 г вуглеводів, 40 мг
заліза, 5 мг міді, 7 мг марганцю, 15 мг цинку, 15 мкг кобальту,
2 г метіоніну, 4 г холіну, вітаміни групи С, В.
Хворому рекомендують такі продукти: язик яловичий, м’ясо
кролика, індика, курки, яловичину, білі гриби, гречану та вівсяну
крупи, бобові, шоколад, какао, яйця, зелень, персики, абрикоси,
родзинки, чорнослив, яблука, фруктові соки, мед.
Слід пам’ятати, що залізо із продуктів тваринного походження
всмоктується в кишечнику в значно більшій кількості, ніж із рос-
линних продуктів.
Найкраще залізо засвоюється із телятини та іншого м’яса —
11-22%, із яєць, бобових, фруктів — до 3%, із рису, шпинату, ку-
курудзи — 1%.
40
| Анемії
Таблиця 7. Основні критерії лабораторної діагностики
залізодефіцитної анемії
Лабораторний показник Норма Зміни при ЗДА
1 Морфологічні зміни еритроцитів Нормоцити - 68% Мікроцити - 15,2% Макроцити - 16,8% Макроцитоз у поєднанні з анізоцитозом, пой кіл оцитозом
2 Колірний показник 0,86-1,05 Гіпохромія, показник менше 0,86
3 Рівень гемоглобіну Жінки - не менше 120 г/л Чоловіки - не менше 130 г/л
4. МСН (середній вміст Нв у еритроциті) 27-31 пг менше 27 пг
5. МСНС (середня концентрація Нв у еритроциті) 33-37% менше 33%
6. МС\/ (середній об’єм еритроцитів) 80-100 фл зменшений
7. НО\Л/ (показник анізоцитозу еритроцитів) 11,5-12,5% відхилений
8. Середній діаметр еритроцитів 7,55+0,009 зменшений
9. Кількість ретикулоцитів 2-10:1000 не змінена
10. Коефіцієнт ефективного еритропоезу 0,06-0,08 • 10’гл/добу не змінений або зменшений
11. Залізо сироватки Жінки -12-25 мкмоль/л Чоловіки - 13-30 мкмоль/л зменшене
12. Загальна залізозв’язуюча здатність сироватки 30-85 мкмоль/л підвищена
13. Латентна залізозв’язуюча здатність сироватки менше 47 мкмоль/л вище 47 мкмоль/л
14. Насичення трансферину залізом 16-50% зменшене
15. Дисфераловий тест 0,8-2 зменшене
16. Вміст протопорфіринів у еритроцитах 18-89 мкмоль/л підвищений
17. Забарвлення на залізо У кістковому мозку є сидеробласти зникнення сидеробластів у пунктаті
18. Рівень феритину 15-150 мкг/л зменшення І
41
Н.М. Третяк | Гематологія
Додільно призначати хворим на ЗДА фрукти, особливо яблука,
морси та соки, котрі багаті на аскорбінову, янтарну кислоти, оскіль-
ки вони значно підсилюють усмоктування заліза в кишечнику.
Здавна як у народній, так і в науковій медицині для лікування
залізодефіцитної анемії використовували мед. Він містить 40-60%
фруктози, яка сприяє всмоктуванню заліза в кишечнику. Доцільні-
ше рекомендувати темні сорти меду, оскільки вони містять заліза в
4 рази, марганцю — в 14 разів, міді — у 2 рази більше, ніж світлий
мед. За відсутності протипоказань хворим слід вживати 100 г меду
в 3-4 прийоми.
Таким чином, лікувальне харчування є складовою частиною
комплексного лікування хворих на ЗДА. Але дефіцит заліза не
можна відновити самою лише дієтою: обов’язковим є лікування
препаратами заліза.
Відомості про використання заліза ж лікарського засобу відо-
мі ще з часів Давнього Єгипту та Риму, але справжня ера застосу-
вання препаратів заліза для лікування залізодефіцитних станів нас-
тала в 1832 р., коли Ріегг Віапсі встановив ефективність сульфату
заліза при хлорозі. Сучасна медицина має в своєму арсеналі бага-
то препаратів заліза як для внутрішнього прийому, так і для парен-
терального введення.
Лікування залізодефіцитних станів базується на принципі від-
новлення дефіциту заліза в організмі та усуненні можливих причин
його виникнення.
Лікування залізодефіцитної анемії починають тільки після ве-
рифікації діагнозу та встановлення причини, яка зумовила її ви-
никнення. Комплекс заходів при лікуванні залізодефіцитної анемії
повинен включати усунення причин дефіциту заліза та патогене-
тично обґрунтовану терапію його препаратами.
При лікуванні ЗДА перевагу надають високодозним препара-
там заліза, добова доза двохвалентного заліза повинна становити не
менше 2 мг/кг маси тіла у дорослих, що в середньому становить на
добу 100-300 мг елементарного заліза.
Підбір препарату, що максимально добре переноситься хво-
рим, здійснюється лікарем емпірично, з урахуванням особливостей
перебігу' залізодефіцитної анемії' та супутніх захворювань.
Гемотрансфузії не є патогенетично обґрунтованим методом лі-
кування залізодефіцитної анемії.
Трансфузії еритроцитної маси, відмитих та розморожених
еритроцитів у комплексному лікуванні залізодефіцитної анемії мо-
42
| Анемії
Таблиця 8. Основні сучасні препарати заліза
Назва Склад Форма і загальний вміст заліза Кількість елемен- тарного заліза в 1 таблетці (ампулі)
1 2 3 4
Монокомпонентні препарати
1.1 Аристоферон Сульфат заліза Сироп, 200 мл, 5 мл - 200 мг
1.2 Ферроградумет Сульфат заліза, субстанція градумет Табл., 150 мг 105 мг
1.3 Ферронал Глюконат заліза Табл., 300 мг 12%
1.4 Ферроглюконат Глюконат заліза Табл., 300 мг 12%
1 .б.Гемофер пролонгатор Сульфат заліза Табл., 325 мг 105 мг
1.6 Залізне вино Сахарат заліза Розчин, 200 мл, 10 мл-40 мг
1.7 Хеферол Фумарат заліза Капе.,450 мг 100 мг
Комбіновані препарати
2.1 Ранферон-12 Фумарат заліза, аскорбінова кислота, фотієва кислота, ціанокобаламін, сульфат цинку, цитрат амонійного заліза. Капе., 300 мг, елікс., 5 мл-200 мг 100 мг 41 мг
2.2 Сорбіфер дурулес Сульфат заліза, аскорбінова кислота Табл., 320 мг 100 мг
2.3 Феррстаб Фумарат заліза, фолієва кислота Табл., 154 мг 33%
2.4 Фолфетаб Фумарат заліза, фолієва кислота Табл. 200 мг 33%
2.5 Ферроплект Сульфат заліза, аскорбінова кислота Табл., 50 мг 10 мг
2.6 Ферроплекс Сульфат заліза, аскорбінова кислота Табл., 50 мг 20%
2.7 Фефол Сульфат заліза, фолієва кислота Табл., 150 мг 47 мг
2.8 Тардиферон- ретард Сульфат заліза, аскорбінова кислота, мукопротеаза Драже, 256,3 мг 80 мг
2.9.Пно- тардиферон Сульфат заліза, аскорбінова кислота, мукопротеаза, фолієва кислота Драже, 256,3 мг 80 мг
2.10 Макрофер Глюконат заліза, фолієва кислота Шипучі табл., 625 мг 12%
2.11 Фенюльс Сульфат заліза, аскорбінова кислота, нікотинамід, вітаміни групи В Капе. 45 мг
Н.М. Третяк | Гематологія
Продовження табл. 8
1 2 3 4
2.12 Іровіт Сульфат заліза, аскорбінова кислота, фолієва кислота, ціанокобаламін, лізин моно гідро-хлорид Капе., 300 мг 100 мг
2.13 Актиферин Сульфат заліза, 0, Ь-серин. Сульфат заліза. Р. І_-серин . глюкоза, фруктоза, калія сорбаг Капе., 0,11385 г, краплі, 1 мл - 0,0472 0, 0345 г 0,0098 г
2.14 Тотема Глюконат заліза, марганцю глюконат, міді глюконат Ампули з розчином для пиття 50 мг
2.15 Глобірон Фумарат заліза, фолієва кислота, ціанокоболаїн, піридоксин, докузат натрію Капе., 300 мг 100 мг
Для парентерального введення
3.1 Фербітол Хелатні сполуки заліза Амп. 1 мл в/м 50 мг
3.2 Ферлецит Хелатні сполуки заліза, натрій- глюконатний комплекс Амп. 1 мл в/м Амп. 5 мл в/в 50 мг 100 мг
3.3 Ферковен ГІолінуклеарні гідроксильні комплекси заліза, сахарат заліза, кобальта глюконат Амп. 1 мл в/в 20 мг
3.4 Жектофер ГІолінуклеарні гідроксильні комплекси заліза, сорбітол, лимонна кислота Амп. 2 мл в/м 100 мг
3.5 Феррум-лек ГІолінуклеарні гідроксильні комплекси заліза, полі- мальтозний комплекс Амп. 2 мл в/м Амп. 5 мл, в/в 100 мг 100 мг
3.6 Мальтофер - // - Амп. 2 мл в/м 100 мг
3.7 Венофер - // - Амп. 5 мл в/в 100 мг
жуть застосовуватися лише в разі виникнення станів, що загрожу-
ють життю хворого.
Показання до гемокомпонентної терапії:
1) надтяжкий перебіг залізодефіцитної анемії — концентрація
гемоглобіну 50 г/л і менше, що супроводжується глибоким пору-
шенням перебігу метаболічних процесів та розвитком виразної
анемічної гіпоксії;
2) очікування пологів у вагітних, які хворіють на залізодефіцит-
ну анемію та мають показник концентрації гемоглобіну 70—80 г/л
і менше;
44
| Анемії
3) необхідність термінового оперативного втручання у хворих
на залізодефіцитну анемію, що мають показник концентрації ге-
моглобіну 70—80 г/л і менше;
4) посилення частоти нападів стенокардії у хворих на ішеміч-
ну хворобу серця, перебіг якої поєднується із залізодефіцитною
анемією;
5) наявність соматичного захворювання, тяжкість перебігу яко-
го прогресує через тяжку форму залізодефіцитної анемії;
6) виникнення гострої кровотечі на фоні існуючої залізодефі-
цитної анемії .
Показання до трансфузійної терапії при залізодефіцитній ане-
мії повинні бути максимально обмежені та чітко обґрунтовані. Зас-
тосування гемокомпонентної терапії як методу лікування може
супроводжуватись ускладненнями як імунологічної природи, так і
інфекційної, оскільки біологічна безпека компонентів крові не га-
рантована, а залізо із донорських еритроцитів реутилізується в ор-
ганізмі реципієнта повільно.
Первинну профілактику залізодефіцитних анемій проводять
вагітним, жінкам у період лактації, дівчаткам-підліткам, особливо
з рясними місячними, жінкам із тривалими рясними місячними.
Для первинної профілактики застосовують лікарські препара-
ти заліза для перорального вживання. Препарати для парентераль-
ного введення застосовувати не рекомендується.
Первинна профілактика проводиться шляхом щомісячного
призначення протягом 7-10 днів після місячних 30-40 мг елемен-
тарного заліза, або протягом року призначають 2 курси лікування
тривалістю 6 тижнів із прийомом 30-40 мг заліза щодня.
Вторинна профілактика залізодефіцитної анемії проводиться
у пацієнтів, які раніше хворіли на цю недугу та вилікувались від
неї, але в даний час є загроза рецидиву. Таким пацієнтам призна-
чають профілактичні курси пероральної феротерапїї тривалістю 4-
6 тижнів 2 рази на рік.
Усі хворі, які мають ризик виникнення чи рецидиву залізоде-
фіцитної анемії, повинні перебувати під диспансерним наглядом
у терапевта. Обов’язковим обстеженням для них є загальний ана-
ліз крові та дослідження сироваткового заліза (у разі потреби) дві-
чі На рік.
Н.М. Третяк | Гематологія
АНЕМІЇ, ЗУМОВЛЕНІ ПОРУШЕННЯМИ СИНТЕЗУ
ПОРФІРИНІВ
Анемії, що виникають унаслідок порушення синтезу порфіри-
нів, бувають спадковими (еритропоетична порфірия, еритропоетич-
на протопорфірия) та набутими — анемія, зумовлена свинцевою
інтоксикацією.
Анемії, що виникають внаслідок дефіциту піридоксину, бува-
ють також спадковими (вроджена піридоксиндефіцитна анемія) та
набутими — ідіопатична анемія, пов’язана з порушенням синтезу
гема; медикаментозно-індукована піридоксиндефіцитна анемія.
Еритропоетична порфірія успадковується за аутосомно-рецесив-
ним типом. Зустрічається надзвичайно рідко (описано 200 випад-
ків) і виникає внаслідок повної відсутності активності уропорфіно-
гену Ш-косинтетази та збільшення продукції в еритроїдних
попередниках кісткового мозку уропорфірину та копропорфірину,
переважно ізомеру І типу.
Клінічна картина характеризується гіперфоточутливістю шкіри
уже з перших днів після народження. Утворюються субендодер-
мальні відкладення порфірину з подальшим виникненням ерозій.
Наявність порфірину в дентині зубів зумовлює червоне, коричне-
ве та жовте забарвлення зубів, котрі червоно флюоресціюють при
ультрафіолетовому опроміненні. Можливий розвиток гемолітичної
анемії зі спленомегалією та порфіриновими конкрементами в жов-
чному міхурі. Інколи спостерігаються патологічні переломи кісток.
У периферичній крові виявляється нормоцитарна та нормох-
ромна анемія різного ступеня тяжкості, базофільна пунктація
еритроцитів, анізоцитоз, пойкілоцитоз, поліхромазія. У кістково-
му мозку спостерігається еритроїдна гіперплазія із вираженим ді-
зеритропоезом.
Діагноз встановлюється на підставі підвищення (у 20-60 разів)
вмісту порфірину в сечі та калі. Крім того, амніотична рідина у ва-
гітних та сеча у немовлят мають коричнево-рожеве забарвлення за
рахунок підвищеної концентрації порфірину.
Лікування обмежене лише попередженням сонячного опромі-
нення та застосуванням кремів із сонцезахисними фільтрами.
Спленектомія та трансфузії еритроцитарної маси на короткий час
покращують стан хворого.
Еритропоетична протопорфірія — спадкове захворювання, що
розвивається за аутосомно-домінантним типом та характеризується
46
| Анемії
зниженням активності ферохелатази. Виявляється в ранньому ди-
тинстві на підставі підвищеної фоточутливості шкіри, особливо об-
личчя та рук, а також утворення конкрементів у жовчному міхурі.
Діагноз. При лабораторному дослідженні спостерігається
збільшення вмісту протопорфірина в еритроцитах, плазмі та калі,
середнього ступеня тяжкості анемія, гіпохромія та мікроцитоз
еритроцитів.
Лікування обмежується запобіганням впливу ультрафіолетового
опромінення, вживанням ^-каротину. Трансфузії еритроцитарної
маси необхідні для супресії еритропоезу. Зменшує вміст протопор-
фірину застосування холестираміну по 4,0 г 3 рази на добу.
АНЕМІЯ, ЗУМОВЛЕНА СВИНЦЕВОЮ ІНТОКСИКАЦІЄЮ
Отруєння свинцем може виникати в осіб, що працюють на під-
приємствах, де виробничий процес пов’язаний із використанням
свинцю, а також у побуті, при користуванні неякісним посудом, та
у маленьких дітей — від іграшок, у яких застосовано фарби з до-
мішками свинцю.
Патогенез. Потрапляючи в організм, до 95% свинцю накопи-
чується в кістках у вигляді трьохосновного фосфату. Свинцева ін-
токсикація розвивається внаслідок того, що свинець блокує тіоло-
ві групи різних ферментів, у тому числі й тих,що беруть участь
у синтезі порфіринів.
Клінічна картина захворювання полісимптомна, оскільки вра-
жаються всі органи та системи. Ураження нервової системи супро-
воджується астеновегетативним синдромом. У складніших ситуаці-
ях може розвиватися енцефалопатія. Тривалі контакти зі свинцем
можуть супроводжуватися поліневритами. Ураження травного
тракту може проявлятися диспептичним синдромом. Характерною
є “свинцева кайма” на шийці зубів, що утворюється сульфідом
свинцю. Тяжким ускладненням свинцевого отруєння є свинцева
коліка, котра триває від декількох годин до кількох днів. При рен-
тгенологічному обстеженні виявляються спастико-атонічні зміни
у товстій кишці.
Клінічну картину свинцевого отруєння може доповнювати
токсичний гепатит із мінімальною симптоматикою. Лише присут-
ність жовтяниці із підвищенням вмісту непрямого білірубіну в си-
роватці крові може бути основною клінічною ознакою свинцевого
ураження печінки.
47
Н.М- Третяк | Гематологія
Свинцева інтоксикація серцево-судинної системи призводить
до токсичного міокардиту або спастико-атонічних змін у перифе-
ричних судинах, шо згодом може спричиняти виникнення артері-
альної гіпертензії, раннього склерозу та склерозу судин нижніх
кінцівок.
Свинцеве ураження кровотворної системи спричиняє повільне
розвинення гіпохромної анемії, котра набуває тяжких ступенів ли-
ше при сильному отруєнні. Підвищується кількість ретикулоцитів.
Рівень лейкоцитів і ШЗЕ — в межах норми. Еритроцити набува-
ють мішенеподібної форми з базофільною пунктацією.
У кістковому мозку виявляється підвищений вміст еритрокарі-
оцитів, у яких забарвлення залізом ідентифікує велику кількість
гранул, шо оточують ядро. Концентрація заліза в сироватці крові,
а також загальна та насичена залізозв’язуюча здатність сироватки
крові при отруєнні свинцем підвищені.
Для діагностики свинцевого отруєння важливим є виявлення
високого рівня вмісту свинця у крові — 0,5-0,6 мг/л (нормальне
значення 0,05-0,3 мг/л) та сечі — більш ніж 0,05 мг/л. Характер-
ним є підвищення концентрації у сечі <5-амінолевулинової кисло-
ти, яка іноді в десятки разів перевищує норму (0,5-1,5 мг/г), — до
40-100 мг/г креатиніну.
Лікування включає детоксикацію з допомогою комплексанів
(речовин, шо зв’язують свинець та виводять його з організму). Зок-
рема застосовують тетацин-кальцій (натрій-кальцієва сіль етіленді-
амінтетраоцтової кислоти). Вводять внутрішньовенно крапельно по
25 мг/кг на 500 мл 5%-го розчину глюкози. 3-5 триденних курси
з перервою у 3 дні із контролем вмісту свинця та <5-амінолевулино-
вої кислоти у сечі. Достатньо ефективний і інший комплексан —
пентацин.
Профілактикою є попередження потрапляння свинцю до орга-
нізму через дотримання заходів техніки безпеки на підприємствах
та норм гігієни в побуті.
Спадкові пірцдоксиндефіцитні анемії передаються за аутосомно-
домінантним, аутосомно-рецесивним типом та бувають х-зчепле-
ними.
До них належать ізольована сідеробластна анемія та синдром
Пірсона. Захворювання виявляються одразу після народження, по-
лісимптомні.
Спостерігається нормопитарна анемія зі зниженням вмісту ре-
тикулоцитів. Відзначається гіпохромія, мікроцитоз еритроцитів.
48
| Анемії
анизо-, пойкілоцитоз та мішенеподібні еритроцити. Підвищується
вміст фетального гемоглобіну, можлива лейкопенія, тромбоцито-
пенія. Кістковий мозок нормо- або гіперклітинний.
Лікування. З незначним позитивним ефектом застосування пі-
ридоксину (6% розчин) по 5-8 мл/добу внутрішньом’язово або пі-
ридоксальфосфату — 30-40 мг/добу внутрішньом’язово та 80-120
мг/добу внутрішньо.
Для виведення заліза — десферал по 500 мл/добу — 3-6 разів
на тиждень.
Набута піридоксиндефіцитна анемія може бути трактована як
медикаментозно-індуковане захворювання, етіологія котрого
пов’язана із використанням таких протитуберкульозних препара-
тів, як ізоніазид, фтівазид, салюзид, метазид. Вживання цих меди-
каментів призводить до підвищеної потреби піридоксину, котрий,
взаємодіючи із ізоніазидом, утворює нову сполуку — гідразид-три-
доксаль, що виводиться із сечею. Підвищена потреба у піридокси-
ні при цій формі анемії зумовлена тим, що він входить до складу
кофермента, котрий бере участь в утворенні 5-амінолевулинової
кислоти, необхідної для синтезу порфіринів.
Клінічна картина піридоксиндефіцитної анемії проявляється
гіпохромною анемією без інших ознак, характерних для анемій із
порушенням синтезу порфіринів.
При дослідженні крові виявляється помірна гіпохромна анемія
з мікроцитозом, анізо- та пойкілоцитозом, базофільною пунктаці-
єю гіпохромних еритроцитів.
Рівень заліза в сироватці крові та залізозв’язуюча здатність си-
роватки підвищені. Вміст заліза у тканинах нелікованого хворого
підвищений. У кістковому мозку посилений еритропоез та збіль-
шена кількість кільцевих сідеробластів. Концентрація вітаміну В6
у сироватці — на нижній межі норми, зменшується утворення та
виділення 5-амінолевулинової кислоти.
Лікування. Стан хворого покращується після зменшення дози
протитуберкульозних препаратів та призначення піридоксину
(100-200 мл щодня) до нормалізації показників крові.
МЕГАЛОБЛАСТНІ АНЕМІЇ
Мегалобластні анемії виникають унаслідок порушення синтезу
ДНК та РНК у еритроїдних клітинах та характеризуються наявніс-
тю в кістковому мозку мегалобластів.
49
Н.М. Третяк | Гематологія
Класифікація мегалобластних анемій Л. І. Ідельсона, 1979 р.
1. Анемії, зумовлені дефіцитом вітаміну В^.
— дефіцитні анемії, зумовлені порушенням секреції внутріш-
нього фактора;
— зумовлені атрофією слизової шлунку, що виникла внаслідок
дії ушкоджуючих_факторів;
— автоімунна форма, зумовлена експресією антитіл до паріє-
тальних клітин слизової оболонки шлунку або до внутрішнього
фактора;
— в осіб, що перенесли гастректомію;
— автосомно-рецесивне успадковане порушення синтезу внут-
рішнього фактора;
— В12-дефіцитна анемія, пов’язана з порушенням кишкового
всмоктування вітаміну В|2;
— В12-дефіцитна анемія, пов’язана з конкурентними витрата-
ми вітаміну В|2 у кишківнику;
— інвазія широким лентецем;
— при утворенні “сліпої петлі” внаслідок резекції кишківника
“бік у бік” або “кінець у бік”;
— при виникненні множинних дивертикулів тонкого кишків-
ника;
— В]2-дефіцитна анемія, зумовлена порушенням транспорту
вітаміну В12 унаслідок спадкового дефіциту транскобаламіну 11
(ТСП).
2. Анемії, зумовлені дефіцитом фолієвої кислоти:
— пов’язані з недостатнім вмістом фолієвої кислоти в їжі;
— унаслідок алкоголізму;
— при вживанні протисудомних засобів;
— при синдромі “сліпої петлі”;
— внаслідок спадкового порушення всмоктування фолієвої ки-
лоти;
— при підвищеній потребі у фолієвій кислоті;
— при вагітності на фоні початкового дефіциту або посилено-
го еритропоезу;
— унаслідок посиленої активації еритропоезу.
Етіологія та патогенез. В,-дефіцитна анемія відома з середи-
ни XIX ст. під назвою прогресуюча злоякісна анемія, або анемія
Аддісона—Бірмера за прізвищами авторів, котрі вперше описали
це захворювання (Аддісон у 1849 р., Бірмер у 1879 р.). Цікавим є
факт, що метод лікування цього захворювання був винайдений ше
50
| Анемії
до розкриття механізмів розвинення хвороби. 1926 р. 1 Міпеї та
Мигріїу вперше застосували сиру печінку для лікування В12-дефі-
цитної анемії та отримали вражаючий позитивний ефект. 1 тільки
у 1929 р. Кастл припустив, шо у м’ясі г якийсь зовнішній фактор,
а у шлунковому соку — внутрішній, котрі, відкладаючись у печін-
ці у вигляді протианемічного фактора, сприяють нормалізації кро-
вотворення, що пояснює ефективність лікування сирою печінкою.
Гіпотеза Кастла проіснувала до 1948 р., коли був синтезований ві-
тамін Вр. Згодом з’ясувалося, що він і є зовнішнім фактором,
а внутрішнім фактором є білок гастромукопротеїн, котрий утворю-
ється парієтальними клітинами шлунка. Потрапляючи із їжею
в організм, вітамін В|2 у шлунку поєднується з білком (К-протеї-
ном). У 12-палій кишці під впливом трипсину В-протеїн відокрем-
люється від вітаміну В|2та поєднується з гастромукопротеїном, зав-
дяки чому відбувається всмоктування вітаміну В12. Усмоктування
відбувається здебільшого у 12-палій та порожній кишках. У крові
вітамін Вр поєднується з транспортним білком транскобаламіном і
потрапляє в тканини та органи. Добова потреба вітаміну В12 стано-
вить 3,5 мкг, тоді як його вміст у печінці може сягати 3,5мг.
Виникнення В12-дефіцитної анемії зумовлене фізіологічною
властивістю його коферментних форм. Існують дві коферментні
форми вітаміну В12 — метил- та дезоксиаденозилкобаламін. Ме-
тилкобаламін каталізує перехід фолієвої кислоти в її активну фор-
му — 5,10-метилентетрагідрофолієву килоту. Ця форма бере участь
в утворенні тимідіну, що необхідний для синтезу ДНК із уридіно-
монофосфату. При недостатності ДНК порушується процес поділу
клітини, вона збільшується в розмірі внаслідок подвоєння кількос-
ті хромосом, а розділення не відбувається. Клітини еритроїдного
паростка значно збільшуються, нитки хроматику набухають, ста-
ють грубими. Ці клітини мають назву “мегалобласти”. Поряд із ме-
галобластами в кістковому мозку зберігаються і нормальні еритро-
їдні клітини, внаслідок чого при введенні вітаміну В|2 швидко
відтворюється нормальний гемопоез.
Дезоксиаденозилкобаламін бере участь у синтезі та розпаді
жирних кислот. При порушенні розпалу жирних кислот утворю-
ються пропіонова та метилмалонова кислоти, токсичні для нерво-
вої системи, внаслідок чого ушкоджуються бокові та задні стовпи
спинного мозку. Крім того, порушення синтезу жирних кислот зу-
мовлює дефекти при утворенні меланіну і з часом призводить до
пошкодження аксонів.
51
Н.М. Третяк | Гематологія
Клініка. При анемії середнього ступеня тяжкості хворі скар-
жаться на слабкість, запаморочення, задишку та серцебиття при
незначних фізичних навантаженнях, миготіння “мушок” перед
очима, печіння кінчика язика, особливо після солоної або гострої
їжі, відрижку та важкість у епігастральній ділянці. При тяжких
формах анемії з’являється відчуття повзання мурашок або поколю-
вання та відсутність упевненості в ногах, непевність ходи. Такий
симптомокомплекс називається фунікулярним мієлозом. Невроло-
гічні ураження можуть проявлятися поліневритом із подальшим
приєднанням порушень спинного мозку (насамперед страждають
нижні кінцівки). При невірно встановленому діагнозі та відсутнос-
ті лікування процес прогресує. Можуть порушуватися тактильна та
больова чутливість, здатність відрізняти холодне від гарячого, зни-
кати смакові відчуття, нюх та слух.
При об’єктивному обстеженні спостерігається бліда шкіра
з дешо кавовим відтінком, іктеричні склери. Маса тіла не знижу-
ється. Язик інтенсивно рожевий, сосочки згладжені — полірова-
ний, або гунтерівський язик (такий вигляд язика вперше описав
Гунтер). Іноді спостерігається незначне збільшення селезінки
та/або печінки.
У периферичній крові виявляється нормо- або гіперхромна
анемія. Зустрічаються великого розміру еритроцити, в багатьох із
них виявляються залишки ядра (тільця Жоллі, кільця Кебота). Як-
що діаметр клітини перевищує 8,5 мкм, МСУ більше 100-110 фл.,
такі клітини називають макроцитами. Мегалоцити мають середній
діаметр, більший за 11-12 мкм, та відсутнє просвітлення в центрі
клітини. Кількість ретикулоцитів знижена, може спостерігатися
лейкопенія та тромбоцитопенія, характерною є полісегментація
ядер нейтрофілів.
Кістковий мозок гіперклітинний, характерне переважання клі-
тин еритроїлного ряду, здебільшого базофільних його елементів,
тобто відбувається затримка дозрівання на стадії базофільно- та по-
ліхроматофільних клітин — “синій кістковий мозок”. Зустрічають-
ся мегалобласти — великі клітини з пухким хроматином. Існуван-
ня мегалобластів підтверджує порушення еритропоезу внаслідок
дефіциту вітаміну В|2. У сироватці крові вміст заліза дешо підвище-
ний, іноді нормальний, помірна гіпербілірубінемія за рахунок неп-
рямої фракції.
Діагноз встановлюється на підставі дослідження периферичної
крові — гіперхромна анемія, макро- та мегалоцитоз, гіперссгмен-
52
| Анемії
тація ядер нейтрофілів, тільця Жоллі, кільця Кебота, зменшена
кількість ретикулоцитів. “Кістковий мозок — Синій кістковий мо-
зок”, спотворення еритропоезу за мегалобластним типом. Врахову-
ються дані анамнезу та об’єктивного обстеження.
Диференційний діагноз проводиться із апластичною анемією,
фолієводефіцитною анемією, симптомокомплексом В|2-дефіциту
при злоякісних захворюваннях і, в першу чергу, шлунково-кишко-
вого тракту.
Лікування. В|7-дефіцитна анемія успішно лікується цианокоба-
ламіном по 100, 200 або 500 мкг/мл щодня. Існують аналоги — ок-
сикобаламін та аденозилкобаламін. Гемотрансфузії проводять за
життєвими показами, якщо хворий перебуває у надзвичайно тяж-
кому стані (анемічна кома). Діагноз та адекватність терапії підтвер-
джує ретикулоцитарна криза, котра спостерігається на 5-6 день лі-
кування. Повне одужання настає через 4-5 тижнів. У випадках
фунікулярного мієлозу дозу вітаміну В|2 підвищують до 1000
мкг/мл на добу.
Досягнуту ремісію закріплюють введенням вітаміну В|2 щотиж-
ня протягом двох місяців, а потім двічі на місяць по 400-500 мкг
протягом 6 місяців.
Для попередження загострень, із метою профілактики щороку
протягом 3 тижнів через день вводять вітамін В12 по 400 мкг.
Прогноз сприятливий.
ФОЛІЄВОДЕФІЦИТНА АНЕМІЯ
Дефіцит фолієвої кислоти може зумовлювати розвиток анемії,
що характеризується появою мегалобластів та руйнуванням ерит-
роцитів у кістковому мозку, іноді з випадками психічних розладів.
Етіологія та патогенез. Захворюваність на фолієводефіцитну
анемію значно менша, ніж на В12-дефіцитну патологію, і зустріча-
ється при порушенні всмоктування фолієвої кислоти при тривалих
проносах, інфекційних захворюваннях кишківника, резекції тонкої
кишки, синдромі сліпої кишки, при алкоголізмі та вживанні меди-
каментів, шо є аналогами або антагоністами фолієвої кислоти. Це
такі препарати, як протисудомні — дифеніл, гексамідін, фенобар-
бітал, аміноптерін. Дефіцит фолієвої кислоти може виникати внас-
лідок підвищеної потреби організму в період вагітності або прис-
кореного росту. У немовлят дефіцит фолієвої кислоти виникає при
вигодовуванні порошковим або козиним молоком.
53
Н.М. Третяк | Гематологія
Внаслідок того, що фолати в достатній кількості містяться
в печінці, дріжджах, шпинаті, м’ясі та інших продуктах, при зба-
лансованому харчуванні загроза виникнення захворювання обме-
жена. Загальна кількість кислоти в їжі при повноцінній дієті ста-
новить 400-600 мкг/добу.
Фолієвої кислоти в організмі людини міститься 5-10 мг у виг-
ляді глютамінової, або парамінобензойної кислоти, або птеридино-
вого кільця. Всмоктування відбувається у верхньому відрізку тон-
кої кишки, всмоктувальна здатність котрої може перевищувати
добову потребу.
Клінічна картина. На фолівоєдефіцитну анемію найчастіше
хворіють молоді люди, вагітні жінки. Основні скарги — загальна
слабкість, запаморочення, подеколи біль у язиці. Шкіра субікте-
рична, склери іктеричні. Спостерігається невиражена спленомега-
лія. Атрофічні зміни слизової оболонки рота та язика відсутні або
незначні. Атрофічного гастриту немає, проте часто буває знижена
шлункова секреція. Неврологічна симптоматика, як при В12-дефі-
цитній анемії, не спостерігається. У осіб, що страждають на епілеп-
сію, дефіцит фолієвої кислоти призводить до погіршення перебігу
основного захворювання.
Діагноз встановлюється на підставі дослідження периферичної
крові та кісткового мозку. В периферичній крові спостерігається
макроцитоз, гіперхромна анемія з анізопитозом, знижена кількість
ретикулопитів, тромбоцитів та лейкоцитів. У кістковому мозку ви-
являються мегалобласти та посилене руйнування клітин еритроід-
ного ряду. Крім того, для підтвердження діагнозу використовуєть-
ся мікробіологічний метод визначення вмісту фолієвої кислоти
в сироватці та в еритроцитах, котрий у нормі становить від 3 до
25 нг/мл у сироватці та від 100 до 425 нг/мл в еритроцитах. Вико-
ристовується також метод гістідінового навантаження: хворому да-
ють 15 г гістідіну, котрий, утворюючи глутамінову кислоту, виво-
диться із сечею у вигляді формімінглутамінової кислоти (норма —
1 — 18 мг. При дефіциті фолієвої кислоти за 8 годин виділяється від
20 до 1500 мг.
З метою диференціювання фолієводефіцитної анемії та В|2-де-
фіцитної анемії використовують забарвлення кісткового мозку алі-
зарином червоним, котрий зафарбовує тільки мегалобласти, що ут-
ворюються при дефіциті вітаміну В|2.
Лікування. Призначається фолієва кислота до 1,5 мг/добу про-
тягом 4-6 тижнів, шо достатньо для отримання клініко-гематоло-
54
| Анемії
гічної ремісії. При вагітності або вигодовуванні немовляти грудьми
призначається така ж доза препарату до нормалізації показників
периферичної крові. Надалі слід рекомендувати фолієву кислоту
у підтримуючій дозі (1 мг на добу) до завершення лактації.
Прогноз сприятливий.
Профілактику фолієводефіцитної анемії необхідно проводити
вагітним та жінкам у період лактації, а також особам, що стражда-
ють на різні форми гемолітичної анемії.
АПЛАСТИЧНА АНЕМІЯ
Для апластичної анемії (АА) характерним є глибоке пригнічен-
ня кістково-мозкового кровотворення (аплазія або гіпоплазія),
зниження швидкості проліферації та затримка дозрівання форме-
них елементів крові, порушення функції мікрооточення, наслідком
чого є розвинення анемії, лейкопенії та тромбоцитопенії, котрі зу-
мовлюють основні ознаки захворювання.
Епідеміологія. Апластична анемія вперше була описана у
1988 р. П. Ерліхом під назвою “панмієлофтіз”. Захворюваність не
висока — 1,5-2 на 100 тис. населення.
Етіологія. Причини виникнення захворювання остаточно не
встановлені, проте вирізняються набуті та вроджені форми. До на-
бутих АА відносять захворювання, виникнення котрих може бути
пов’язане з впливом таких хімічних або фізичних факторів, як лі-
карські засоби (левоміцетин, стрептоміцин, сульфаніламіди, мер-
казоліл, метилтіоурацил, цитостатики), мієлотоксичні отрути (бен-
зол та його похідні, миш’як, важкі метали — ртуть, вісмут), фізичні
фактори — іонізуюче випромінення та вплив променів ультрависо-
ких частот.
Значний внесок у захворюваність на АА належить вірусній ін-
фекції (вірус гепатиту А, В, С, Епштейна—Барра, інфекційного
мононуклеозу, грипу, ВІЧ-інфекції).
До вроджених форм АА належить анемія Фанконі, що успад-
ковується за аутосБмно-репесивним типом та характеризується гі-
перчутливістю до ДНК-пошкоджуючих впливів, прогресуючим
ураженням кісткового мозку і підвищеною схильністю до виник-
нення онкозахворювань.
Патогенез. Механізм розвинення АА до теперішнього часу ос-
таточно не Гясований. Проте існують експериментальні дослід-
ження, згідно з якими підґрунтям патогенезу АА можуть бути по-
55
Н.М. Третяк | Гематологія
рушення функції мікрооточення чи автоімунні реакції, що спричи-
няють порушення проліферації та диференціації стовбурової кліти-
ни або призводять до зменшення кількості стовбурових клітин.
Крім того, однією із ланок механізму виникнення АА може бути
порушення в системі регуляції апоптозу, що призводить до прис-
кореної загибелі клітин-попередниць.
Припускається, що розвинення АА можливе в разі поєднання
описаних ланок патогенезу.
Класифікація. Виділяється дві основні групи АА: вроджені
(конституційні) та набуті (первинна та вторинна) різної етіології.
Серед набутих АА виділяють форми захворювання із визначеною
етіологією та ідіопатичну — форму з невстановленою причиною
виникнення.
В залежності від глибини ураження кістково-мозкового кро-
вотворення виділяють трьохпаросткову АА та з переважною депре-
сією еритропоезу при відносно збереженому лейко- та тромбоци-
топоезі — парціальна червоноклітинна аплазія (ПЧА). Існування
ПЧА підтверджує можливість порушення кровотворення на рівні
комітируваної клітини.
Клінічна картина. Клінічний перебіг АА може розвиватися гос-
тро або повільно.
При повільному розвиненні захворювання переважають за-
гальноанемічні симптоми: слабкість, запаморочення, миготіння
“мушок” перед очима, задишка та серцебиття при невеликих фі-
зичних навантаженнях. Згодом приєднуються часті інфекційні зах-
ворювання та посилена кровоточивість ясен, менорагії, крововили-
ви та висипка на шкірі. Така форма АА спостерігається частіше
в осіб молодого та середнього віку. Характерних об’єктивних ознак
для цієї форми АА не існує. Діагноз встановлюється за лаборатор-
ними критеріями.
Гострий початок захворювання спостерігається у 10-15% хво-
рих, переважно в осіб молодого віку, і є характерним для тяжкої
аплазії кровотворення.
Клінічний перебіг гострої форми АА може супроводжуватися
сильною лихоманкою, некротичними змінами слизової рота, но-
са, статевих органів, некротичною ангіною та іншими інфекцій-
но-запальними ускладненнями, тяжкими носовими, ясневими,
шлунково-кишковими, нирковими, матковими кровотечами. Ряс-
ними крововиливами у шкіру та слизові оболонки, очне дно, сіт-
ківку ока.
56
| Анемії
Об’єктивні дані залежать від тяжкості інфекційно-запального
або геморагічного синдромів. Специфічних для АА клінічних ознак
не існує.
Діагноз встановлюється на підставі показників периферичної
крові та кісткового мозку.
Аналіз периферичної крові характеризується панцитопенією:
нормохромна анемія (гіперхромна в окремих випадках), тромбоци-
топенія, лейкопенія за рахунок гранулоцитів, відносний лімфоци-
тоз, кількість ретикулоцитів зменшена, ШЗЕ прискорена.
Кістковомозковий пунктат бідний на клітинні елементи, змен-
шений відсоток вмісту клітин гранулоцитарного ряду, особливо
його молодих форм, порушені процеси проліферації та диференці-
ювання клітин еритроїдного ряду, спостерігається редукція мегака-
ріоцитарного паростка.
При тяжких формах АА кістковий мозок спустошений, вияв-
ляються лише поодинокі клітинні елементи, в основному лімфо-
цити, плазматичні клітини та еритробласти.
Гістологічні препарати трепанобіоптата клубової кістки харак-
теризуються зменшенням вогнищ активного мієлопоезу та замі-
щенням їх жировою тканиною, мегакаріоцити відсутні або їхня
кількість значно зменшена. Можуть зустрічатися залізовмісні піг-
ментні брилки та вогнища крововиливів.
Характерний підвищений вміст заліза у сироватці крові, а та-
кож у клітинах печінки та селезінки, що є ознакою гемосидерозу.
Критерії визначення тяжкості АА (Сатіііа еі. аі., 1979)
1. Тяжка форма:
а ) клітинність кісткового мозку <25%;
Ь ) наявність двох із зазначених трьох критеріїв:
— кількість нейтрофілів < 0,5 • 109/л,
— кількість тромбоцитів < 20 • 109/л,
— кількість ретикулоцитів < 20 • 109/л.
2. Дуже тяжка форма — те ж саме, що й тяжка, але кількість
Нейтрофілів < 0,2 • 109/л.
3. Легка форма — стан пацієнта не відповідає критеріям тяжкої
та дуже тяжкоїапластичної анемії.
Позитивними ознаками є подразнення еритроїдного паростка
та ретикулоцитоз, що дає надію на досягнення ремісії.
Диференційний діагноз проводиться з гострою лейкемією, міє-
лодиспластичним синдромом, злоякісними новоутвореннями (час-
тіше шлунково-кишкового тракту), ідіопатичною тромбоцитопе-
57
Н.М. Третяк | Гематологія
нічною пурпурою, В|2- та фолієводефіцитною анемією, туберку-
льозом.
Лікування. Для хворих, молодших за 40 років із важкою фор-
мою АА, шо мають родинного донора, сумісного за системою
НЬА, оптимальною є трансплантація кісткового мозку. За відсут-
ності такого донора проводиться імуносупресивна терапія:
— антитимоцитарний глобулін (АТГ) у дозі 0,75 мг/кг на добу
тривалою внутрішньовенною інфузією протягом 8-10 днів;
— антилімфоцитарний глобулін (Атгам) у дозі 10-20 мг/кг на
добу внутрішньовенно протягом 5-10 днів;
— циклоспорин А (неорал) 10-12 мг/кг на добу до отримання
ефекту, після чого продовжується терапія в підтримуючих дозах 3-
5 мг/кг маси на добу. Курс лікування — 6 місяців і більше;
— метил преднізолон у дозі 1-2 мг/кг на добу 2-4 тижні з пос-
туповим зниженням дози;
— гранулопитарні колонієстимулюючі фактори (нейпоген, гра-
ноцит) по 5 мкг/кг ваги тіла з 1 до 28 діб.
Протипоказане застосування імуносупресивної терапії при гі-
перчутливості до білкових препаратів, інфекційно-запальних та ге-
морагічних ускладненнях, котрі необхідно ліквідувати перед почат-
ком основної програми.
При неефектувності або при нестійкому позитивному резуль-
таті лікування АТГ (АЛГ) курс терапії можна повторити через 3-6
місяців.
При АА середньої важкості та легкій формі можливе лікуван-
ня циклоспорином А (Сандимун-Неорал). Тривалість лікування
становить 6-12 місяців по 6-10 мг/кг на добу до отримання ремісії
(покращання), з переходом на підтримуючі дози — 3-5 мг/кг на до-
бу. Для підтримання постійної концентрації в сироватці препарат
приймається в однакових дозах двічі на добу.
За відсутності позитивного ефекту протягом 10-12 тижнів пре-
парат поступово відміняється.
У таких випадках вирішується питання про призначення кур-
су АТГ (АЛГ), циклоспорину А, метилпреднізолону або виконан-
ня спленектомії.
Спленектомія в теперішній час використовується як допоміж-
ний метод лікування при частковому поновленні гемопоезу без
стійкої ремісії після курсу імуносупресивної терапії або при наяв-
ності автоантитіл до клітин крові. Операцію виконують у спеціалі-
зованих відділеннях хірургічної гематології.
58
| Анемії
Кортикостероїдні гормони як самостійний метод лікування АА
сьогодні не застосовуються.
Циклофосфамид у дозі 6-8 г на курс не дає стійкої ремісії. Ток-
сичний та імуносупресивний вплив високих доз не виправдовує їх
застосування. Використання окремо ростових факторів Г-КСФ,
ГМ-КСФ, Епо покращує стан хворих, проте не призводить до ре-
місії.
Супровідна терапія застосовується для лікування геморагічних
та інфекційних ускладнень.
Критерієм оцінки ефективності лікування АА є отримання
повної або часткової (неповної) ремісії.
Критерії оцінки ефективності лікування АА
1. Повна ремісія — відсутність клінічних проявів захворювання,
нормалізація рівня гемоглобіну, підвищення кількості гранулоци-
тів — більше 1,0 • 109/л, тромбоцитів — понад 100,0 • 109/л.
2. Часткова (неповна) ремісія — покращання кістковомозково-
го кровотворення, що дозволяє хворому не залежати від гемотран-
сфузій, кількість гранулоцитів більш ніж 0,5 • 109/л, що знижує ри-
зик важких інфекційних ускладнень.
При анемії Фанконі та АА, індукованій хімічними речовинами
або фізичними факторами, імуносупресивна терапія малоефективна.
Прогноз залежить від ступеня пригнічення кістковомозкового
кровотворення. Застосування сучасних методів терапії забезпечує
виживання до 80% хворих протягом року.
Профілактика АА включає заходи з попередження мієлоток-
сичного впливу хімічних та фізичних агентів, адекватне лікування
вірусних захворювань.
ГЕМОЛІТИЧНІ АНЕМІЇ
Гемолітичні анемії складають велику групу захворювань, що ха-
рактеризуються переважанням руйнування еритроїдних елементів
над кровотворенням, скороченням тривалості життя еритроцитів.
Гемоліз може відбуватись як внутрішньоклітинно, так і внутріш-
ньосудинно.
Розрізняють спадкові (вроджені) та набуті гемолітичні анемії.
Кіасифікація гемолітичних анемій (Ідельсон Л. І., 1974)
І. Спадкові гемолітичні анемії:
а) пов’язані з порушенням мембрани еритроцитів (мікросфе-
роцитоз, овалоцитоз, стоматоцитоз);
59
Н.М. Третяк | Гематологія
Ь) пов’язані з порушенням активності ферментів у еритроци-
тах (глюкозо-6-фосфатдегідрогеназа, піруваткіназа, глютаміонре-
дуктаза та ін.);
с) пов’язані із порушенням структури або синтезу ланцюгів ге-
моглобіну (таласемія. серпоподібно-клітинна анемія, носійство
аномального гемоглобіну).
2. Набуті гемолітичні анемії, пов 'язані з впливом таких факто-
рів, що спричиняють руйнування еритроцитів:
а) пов’язані з впливом до антитіл еритроцитів та еритроїдних
клітин кісткового мозку;
Ь) зумовлені змінами структури мембрани еритроцитів (хворо-
ба Маркіафави-Мікелі);
с) пов’язані з пошкодженням оболонки еритроцитів (штучні
клапани серця),
б) спричинені хронічним впливом на еритроцити гемолітичних
отрут, важких металів та свинцю, органічних кислот;
е) пов’язані з нестачею вітаміну Є;
0 зумовлені руйнуванням еритроцитів паразитами (малярія,
токсоплазмоз).
Спадкові гемолітичні анемії, зумовлені змінами мембра-
ни еритроцитів
До цієї групи належать захворювання, що успадковуються за
автосомно-домінантним типом і характеризуються скороченням
терміну життя еритроцитів унаслідок дефекту білків або ліпідної
оболонки клітини. У клінічній практиці найчастіше зустрічається
спадковий мікросфероцитоз (хвороба Мінковського—Шоффара)
та спадковий овалоцитоз.
Спадковий мікросфероцитоз — хвороба Мінковського—Шоффара
Захворювання успадковується за автосомно-домінантним ти-
пом та пов’язане із дефектом або відсутністю спектрину мембра-
ни еритроцитів, унаслідок чого порушується її щільність, відбува-
ється надмірне проникнення іонів калію в клітини. У результаті
цих процесів еритроцити нагрубають. втрачаючи здатність до де-
формації, шо утруднює їх проходження у вузьких місцях кровото-
ку. Такі еритроцити частково елімінуються, частково руйнуються
в селезінці.
Захворювання описане у І87І р. Уапіаіе, Махіи$. наприкінці
XIX сі. встановлено, шо патологія перелається спадково.
60
| Анемії
Клінічна картина. Захворювання характеризується переважно
внутрішньоклітинним руйнуванням еритроцитів, що зумовлює
розвиток анемії, жовтяниці, спленомегалії, схильність до утворен-
ня каменів у жовчному міхурі. Крім того, можуть спостерігатися
конституційні деформації скелету — баштоподібний череп, високе
піднебіння, широке перенісся, мікофтальмія, змінене розташуван-
ня зубів. У деяких хворих укорочені мізинці.
Клінічні ознаки захворювання, особливо анемія, виявляються
в підлітковому віці, рідше у дорослих. Тривалий час тільки легка
жовтяниця може свідчити про захворювання. Причиною загострен-
ня можуть бути інфекційні процеси, переохолодження, вагітність.
Ступінь анемії різниться в залежності від співвідношення між швид-
кістю руйнування еритроцитів та активністю регенераторної функції
еритропоезу та характеризується загальноанемічними симптомами.
Внаслідок руйнування еритроцитів розвивається жовтяниця
з підвищенням вмісту непрямого білірубіну в сироватці крові.
Жовтяниця та анемія зумовлюють лимонно-жовте забарвлення
шкіри. Кожне загострення захворювання посилює жовтяницю.
Захворювання часто ускладнюється жовчно-кам’яною хворобою
з утворенням каменів у жовчному міхурі та жовчних протоках і
розвиненням механічної жовтяниці. Крім того, через застій жов-
чі розвиваються біліарний гепатит та цироз печінки.
Важливим симптомом сфероцитозу є спленомегалія, шо зу-
мовлена гіперплазією фагоцитуючих клітин. Проте збільшення се-
лезінки не набуває великих розмірів, найчастіше вона на 2-4 см
виступає з-під края реберної дуги.
У період гемолітичної кризи (загострення) спостерігається різ-
ке погіршення загального стану, підвищення температури тіла,
поглиблення анемії та зростання концентрації білірубіну в сиро-
ватці крові; в сечі підвищується вміст уробіліну, а в калі — стерко-
біліну.
Тяжким ускладненням мікросфероцитозу є тимчасова недос-
татність еритропоезу — гіпопластична криза, шо триває 7-15 діб.
У таких випадках необхідна інтенсивна терапія.
Значно тяжчий перебіг мікросфероцитозу спостерігається у ді-
тей раннього віку та навіть у новонароджених. У цих хворих пору-
шується розумовий, фізичний та ендокринний розвиток.
Діагноз встановлюється на підставі даних прискіпливо зібрано-
го сімейного анамнезу та обстеження родичів, що може виявити
ознаки мікросфсроцитозу.
61
Н.М. Третяк | Гематологія
У периферичній крові більшості хворих анемія легкого та серед-
нього ступенів тяжкості — 90-100 г/л. У період гемолітичної кризи
вміст гемоглобіну іноді сягає критичних позначок — 20-30 г/л.
У крові серед еритроцитів переважають сфероцити — інтенсивно за-
барвлені, такі, що не мають просвітлення в центрі. Ці еритроцити
мають знижену осмотичну стійкість і можуть гемолізувати уже при
концентрації хлориду 0,60-0,70% замість 0,44-0,40% у нормі. Водно-
час підвищений автогемоліз та подовжена кислотна еритрограма.
Кількість ретикулоцитів, що відображають ступінь компенсаторного
кровотворення в кістковому мозку, помірно зростає. У кістковому
мозку спостерігається значна гіперплазія еритроїдного паростка.
Кількість еритроїдних клітин може сягати 30-50% при нормі 15-25%.
Тривалість життя мічених еритроцитів хворих мікросфероци-
тозом у власному кровообігу та в крові здорових донорів скороче-
на. Проба Кумбса негативна.
Диференційний діагноз необхідно проводити, в першу чергу, із
вірусним гепатитом, хронічним гепатитом, цирозом печінки, доб-
роякісною гіпербілірубінемією Жильбера, автоімунною гемолітич-
ною анемією.
Лікування. Основним методом лікування спадкового мікросфе-
роцитозу є спленектомія. Проте при стабільному перебігу захворю-
вання, відсутності ознак анемії, частих гемолітичних кризів, що
супроводжуються жовчно-кам’яною хворобою або інфарктом селе-
зінки, спленектомія не показана.
Прямими показаннями до спленектомії є часіі гемолітичні або
гіпопластичні кризи; виражена стабільна (навіть середнього ступе-
ня тяжкості) анемія у дітей, що негативно впливає на їхній фізич-
ний та розумовий розвиток; анемія, ускладнена жовчно-кам’яною
хворобою з нападами печінкової коліки або холестатичним гепати-
том. Такі випадки є показанням до виконання спленектомії та хо-
лецистектомії одночасно. У вагітних при спокійному перебігу мік-
росфероцитозу вагітність можна зберегти.
В результаті спленектомії зникають жовтяниця, анемія, змен-
шується кількість ретикулоцитів, поліпшується функція печінки,
нормалізується вміст білірубіну. Проте залишається мікросфероци-
тоз та знижена осмотична стійкість еритроцитів. Покращання до-
сягається шляхом видалення органу, в якому відбувався інтенсив-
ний гемоліз еритроцитів.
Трансфузії еритроцитарної маси показані лише у випадких
тяжкої гемолітичної або гіпопластичної кризи.
62
| Анемії
Глюкокортикоїдні гормони іноді показані лише при тяжких гі-
попластичних кризах.
Прогноз сприятливий, особливо при вчасно виконаній спле-
нектомії.
Спадковий еліптоцитоз (овалоцитоз)
Захворювання, шо передається за автосомно-домінантним ти-
пом і, як вважається в даний час, пов’язане зі зменшенням вмісту
АТФ та 2-3-ДФГ у мембрані еритроцитів.
Клінічна картина. Захворювання зустрічається так само час-
то, як і мікросфероцитоз. Оскільки в більшості випадків клініч-
ні симптоми не проявляються, діагноз встановлюється випадко-
во при дослідженні крові. В аналізі крові спостерігається:
зниження вмісту гемоглобіну та еритроцитів, ретикулоцитоз,
збільшення концентрації білірубіну за рахунок непрямої фракції.
Еритроцити еліптоїдної форми, анізо- та пойкілоцитоз. Осмо-
тична резистентність еритроцитів знижена, скорочений термін
життя і секвестрація їх переважної кількості відбувається в селе-
зінці. У кістковому мозку спостерігається подразнення еритроїд-
ного паростка.
Захворювання має спокійний, хронічний перебіг і лише в ок-
ремих випадках з невідомих причин набуває характеру гемолітич-
них криз із внутрішньоклітинним гемолізом. Розвивається анемія,
жовтяниця, спленомегалія.
Диференційний діагноз проводиться з ідіопатичним мієлофібро-
зом, цианкобаламіндефіцитною анемією, таласемією.
Лікування. Спленектомія є основним методом лікування, що
веде до одужання та запобігає виникненню ускладнень.
Прогноз сприятливий, особливо при вчасно виконаній спле-
нектомії.
Спадкові гемолітичні анемії, зумовлені порушенням
синтезу гемоглобіну та змінами активності ферментів
До цієї групи належать анемії, зумовлені порушенням рівнова-
ги синтезу однієї або декілької поліпептидних ланок гемоглобіну
(таласемія). Вирізняють а- та /?-таласемію, а також таласемію, вик-
ликану структурними порушеннями гемоглобіну. Ланка гемоглобі-
ну, котра надмірно продукується, відкладається в еритроцитах, що
призволить до ушкодження їхньої мембрани та прискореного руй-
нування клітин.
63
Н.М. Третяк | Гематологія
До спадкових анемій, захворюваність на які досить висока, на-
лежить також гемолітична анемія, в патогенезі котрої є дефіцит ак-
тивності глюкозо-6-фосфатдегідрогенази.
Зазначені форми спадкових гемолітичних анемій зустрічають-
ся, в основному, у корінних жителів країн Середземномор’я, Цен-
тральної та Східної Африки, Близького та Середнього Сходу, Пів-
денної та Південно-Східної Середньої Азії, Північного Кавказу,
Закавказзя, в американських негрів. Спорадичні випадки фіксу-
ються серед народів усієї планети.
Набуті гемолітичні анемії. Імунні гемолітичні анемії
Імунні гемолітичні анемії являють собою велику гетерогенну
групу захворювань, у механізмі розвитку котрих беруть участь ан-
титіла або сенсибілізовані лімфоцити, фо зумовлюють руйнування
власних еритроцитів хворого. В залежності від типу імунного про-
цесу анемії поділяють на ізоімунні, трансімунні, гетероімунні та ав-
тоімунні.
Ізоімунні (алоімунні) гемолітичні анемії розвиваються під
впливом антитіл, що-потрапляють у організм ззовні. Така ситуація
складається при гемолітичній хворобі плоду або новонародженого,
коли антитіла через плаценту потрапляють в організм дитини від
матері, організм якої продукує антитіла.
Ізоімунні антитіла потрапляють при трансфузіях еритроцитів,
не сумісних за системою АВО або резус-фактором.
Трансімунні гемолітичні анемії виникають у випадках, коли
антитіла тропні до загальноеритроцитарного антигена матері й ди-
тини від матері, що хворіє на гемолітичну анемію, через плаценту
потрапляють до організму плоду, зумовлюючи гемолітичну анемію
у дитини.
Гетероімунні (гаптенові) анемії розвиваються внаслідок фікса-
ції на мембрані еритроцита гаптенового комплексу (антитіло + хі-
мічна речовина або віруси, бактерії-), що спричиняє руйнування
еритроцита.
У механізмі розвитку автоімунної гемолітичної анемії беруть
участь антитіла, що продукуються проти антигена власних ерит-
роцитів.
Гемолітична хвороба новонароджених
Гемолітична хвороба новонароджених є наслідком існування ан-
титіл у організмі матері, шо утворилися при попередніх вагітностях
64
| Анемії
у відповідь на антиген батька. Крім того, такі антитіла можуть бу-
ти наслідком трансфузій несумісної за Кй-фактором крові або
у негативних за Кіі-фактором матерів при КЬ-позитивному плоді.
Клінічна картина. Тяжкість клінічних проявів захворювання
залежить від кількості антитіл, гцо потрапили через плаценту в ор-
ганізм плоду. Дитина народжується з анемією, набряками, серце-
вою недостатністю, збільшенням розмірів серця, печінки та селе-
зінки. Жовтяниця розвивається у першу добу.
Ядерна жовтяниця є тяжким наслідком інтоксикації нервової
системи, що виникає на фоні високого рівня непрямого білірубіну
— до 340 мкмоль/л та вище і проявляється гіпертонусом, ністаг-
мом, судомами.
Діагноз. У аналізі крові — нормохромна анемія різного сту-
пеня в залежності від тяжкості захворювання, ретикулонитоз,
еритрокаріонити, лейкоцитоз, нейтрофільоз зі зрушенням вліво.
Вміст непрямого білірубіну відповідний до тяжкості гемолізу.
Позитивна пряма проба Кумбса у дитини та непряма проба Кум-
бса у матері. Можуть виявлятись імунні теплові ізогемаглютині-
ни у матері.
У ВИ-негативних вагітних, шо мають КЬ-позитивних чоловіків,
проводиться антенатальне діагнозування гемолітичної хвороби но-
вонароджених на підставі динамічного спостереження за титром
анти- В1і0-антитіл.
Диференційний діагноз гемолітичної хвороби новонароджених
проводиться з постгеморагічною анемією, спадковими гемолі-
тичними анеміями (гемоглобінопатії, глюкозо-6-фосфатдегідро-
геназа).
Лікування. Лікувальні заходи спрямовані на усунення анемічної
гіпоксії, попередження інфекційних ускладнень, корекцію метабо-
лічних порушень, виведення з організму антитіл та нормалізацію
рівня непрямого білірубіну.
Замінне переливання крові в цьому сенсі — ефективний метод,
проте застосовувати його слід дуже зважено.
Абсолютним показанням до замінного переливання крові у доно-
шених дітей є:
— рівень непрямого білірубіну у пуповинній крові — 51,5
мкмоль/л та більше при народженні;
— на кінець І доби — 171 мкмоль/л;
— на кінець II доби — 220-250 мкмоль/л;
— на кінець 111 доби — 307 мкмоль/л.
65
Н.М. Третяк | Гематологія
У недоношених дітей абсолютним показанням до замінного
переливання крові є рівень непрямого білірубіну 256,5 мкмоль/л.
Крім того, для визначення необхідності прямого переливання
крові враховується погодинне підвищення концентрації непрямого
білірубіну: якщо вона перевищує 5,1 мкмоль/л/год. — виконується
операція.
Замінне переливання крові проводиться в асептичних умовах,
дитина має бути добре зігріта. Спочатку переливають 20 мл альбу-
міну для зв’язування білірубіну, після чого багаторазово випуска-
ють по 20 мл крові новонародженого та повільно вводять по 20 мл
свіжої одногрупної КИ-негативної донорської крові, всього 500-600
мл крові донора. Після введення кожних 100 мл крові донора вво-
дять 1-2 мл 10%-го глюконату кальцію.
Консервативний метод лікування включає введення 5-10%-го
розчину глюкози, 5%-го розчину альбуміну. Застосовується фено-
барбітал, вітаміни Є, С, групи В, фототерапія.
Після замінного переливання дітей вигодовують молоком до-
нора 2-3 тижні. За відсутності антитіл у молоці матері дозволяєть-
ся грудне вигодовування.
Профілактика. Попередженням резус-сенсибілізації жінок
з резус-негативним типом крові є введення 200-300 мл анти-КИ‘-
анти-П-імуноглобуліну відразу ж після пологів, та не пізніше 48 го-
дин після народження дитини, батько котрої був ВІї-позитивним.
Імунопрофілактика повинна виконуватись усім вагітним з Юі-
негативною кров’ю.
Прогноз залежить від категорії тяжкості анемії, вчасної діагнос-
тики та адекватно проведеної терапії.
Гемолітична анемія, зумовлена інфузією несумісної крові
Вливання не сумісної за групою або резус-фактором крові зу-
мовлює відразу або трохи згодом розвиток гемотрансфузійного шо-
ку, котрий характеризується болем у попереку, різким зниженням
артеріального тиску, тахікардією, порушенням дихання.
При проведенні адекватних реанімаційних заходів (замісне пе-
реливання одногрупної крові, протишокові заходи) настає другий
період — оліго- або анурія, тобто гостра ниркова недостатність. На
цій стадії доцільно підключати гемодіаліз.
У разі досягнення позитивного ефекту поновлюється діурез,
а з часом настає повне одужання хворого.
66
| Анемії
Токсична гемолітична анемія
Гостра токсична гемолітична анемія із внугрішньосудинним ге-
молізом виникає внаслідок отруєння біологічними токсинами (от-
рута змії, грибів, риби), хімічними речовинами (фенілгідразин,
миш’як та інші важкі метали, фенол, толуол, бензол, анілін, трих-
лоретилен, лізол, сірковий водень, оцтова та інші кислоти, мідь,
пестициди тощо). Гострий внугрішньосудинний гемоліз може роз-
винутися при акушерському сепсисі, руйнуванні еритроцитів пара-
зитами (малярія, токлоплазмоз) та внаслідок руйнування еритро-
цитів при протезуванні клапанів та перегородки серця.
Клінічна картина. Характеризується розвитком гемолітичної
кризи: анемія, тахікардія, гарячка, біль у попереку та животі, блю-
вота, пронос. Специфічним є виділення чорної або темно-бурої се-
чі. У важких випадках розвивається оліго- або анурія, гостра нир-
кова недостатність, порушення коагуляційного гомеостазу,
ДВЗ-синдром. Тяжкість процесу залежить від кількості отрути, що
потрапила в організм.
Лікування залежить від характеру отруєння.
Прогноз залежить від вчасного та адекватного надання допомоги.
Автоімунні гемолітичні анемії
Автоімунні гемолітичні анемії (АІГА) за своїм походженням по-
діляються на ідіопатичні (етіологію встановити неможливо) та сим-
птоматичні.
Симптоматичні АІГА розвиваються як автоімунне ускладнен-
ня захворювань із ушкодженням лімфоїдної системи. До них нале-
жать хронічна лімфоїдна лейкемія, лімфома Годжкіна та негод-
жкінська, множинна мієлома, гостра лімфобластна лейкемія,
хвороба Вальденстрема, а також системні захворювання сполучної
тканини (ревматоїдні артрити, системний червоний вовчак та ін.),
хронічні інфекції (сепсис), злоякісні пухлини.
За патогенезом АІГА поділяються на 5 типів:
— автоімунна гемолітична анемія з тепловими агглютинінами;
— автоімунна гемолітична анемія з тепловими гемолізинами;
— автоімунна гемолітична анемія з неповними тепловими агг-
лютинінами;
— автоімунна гемолітична анемія з повними холодовими агг-
лютинінами;
— автоімунна гемолітична анемія з двохфазовими гемолізинами.
67
Н.М. Третяк | Гематологія
Патогенез. Неповні теплові агглютиніни фіксуються на мем-
брані еритроцитів, змінюючи її проникність для іонів натрію, внас-
лідок чого розвивається сфероцитоз. Сфероцити руйнуються в се-
лезінці, при цьому гемоліз є внутрішньоклітинним.
Теплові гемолізини, двохфазові холодові гемолізини та
повні холодові агглютиніни зумовлюють руйнування еритро--
цитів у судинному руслі, наслідком чого є внутрішньосудин-
ний гемоліз.
Клінічна картина. А1ГА, незалежно від типу антитіл, шо зумов-
люють їх виникнення, може розвиватись як поступово, так і гос-
тро. Хворі скаржаться на слабкість, задишку, запаморочення, сер-
цебиття. При об’єктивному обстеженні виявляється жовтяниця,
збільшення селезінки.
Особливостями клінічного перебігу АІГА з холодовими агглю-
тинінами є непереносимість низьких температур, за яких відбуваєть-
ся аггл юти нація еритроцитів у капілярах пальців на руках і ногах.
У периферичній крові виявляється, різного ступеня тяжкості,
ретикулоцитоз, підвищення вмісту білірубіну за рахунок непрямої
фракції.
При АІГА з тепловими гемолізинами можуть спостерігатися
болі в животі, пов’язані з тромбозом мезентеріальних судин, а та-
кож тромбози периферичних вен.
Характерним є виділення чорної сечі, виявлення гемосидери-
ну, підвищеного вмісту уробіліну, лейкоцитоз зі зрушенням у фор-
мулі вліво.
Діагноз. Підтверджується позитивною прямою пробою Кумбса.
При низькому титрі антитіл або на висоті гемолізу проба Кумбса
може бути негативною, що не є ознакою відсутності АІГА.
Лікування. Основним методом лікування всіх форм АІГА є
призначення глюкокортикоїдних гормонів, доза котрих залежить
від тяжкості процесу (1-2 мг на 1 кг маси тіла). Знижувати дозу
гормонів рекомендується поступово, після досягнення ремісії.
При неефективності стероїдних гормонів використовуються такі
імуносупресивні засоби, як циклоспорин А (сандимун), імуногло-
булін, імуран та цитостатичні препарати (циклофосфан, вінкрис-
тин, метотрексат).
В окремих випадках доцільною є спленектомія.
Трансфузії відмитих або розморожених еритроцитів проводять-
ся лише за життєвими показаннями.
68
| Анемії
ПАРОКСИЗМАЛЬНА НІЧНА ГЕМОГЛОБІНУРІЯ
(ХВОРОБА МАРКІАФАВИ—МІКЕЛІ)
Пароксизмальна нічна гемоглобінурія (ПНГ) — це рідкісна фор-
ма набутих гемолітичних анемій, спричинених змінами структури
мембрани еритроцитів.
Патогенез. В основі механізму виникнення ПНГ лежить де-
фект у гені РІС, розташованому на Х-хромосомі. Внаслідок цієї
генетичної аномалії порушується структура мембрани еритроци-
та, посилюється чутливість клітини до комплементу. Характер-
ною особливістю цих еритроцитів є підвищення чутливості до
зрушення рН у кислу сторону, прокомулянтів пропердину та
комплементу. Процес гемолізу відбувається постійно, особливо
прискорюючись у нічний період, коли активуються прокоагу-
лянти та зрушується рН у кислу сторону. Дефект структури мем-
брани спостерігається не тільки в еритроцитів, а й нейтрофілів та
тромбоцитів, що наводить багатьох дослідників на думку про ге-
нетичне ушкодження стовбурової клітини. Патологічні зміни
нейтрофілів негативно впливають на їхню функціональну здат-
ність, унаслідок чого хворі на ПНГ схильні до інфекційних уск-
ладнень.
Клінічна картина. У більшості хворих захворювання почина-
ється поступово. З’являються слабкість, запаморочення, головний
біль, блідість одночасно із субіктеричністю склер та шкіри. Такий
латентний період може тривати декілька років до виникнення ге-
молітичної кризи.
Гемолітична криза проявляється різким погіршенням загаль-
ного стану, блювотою, гарячкою, болем у животі та попереку, що
може бути наслідком тромбозу мезентеріальних судин та закупор-
ки ниркових канальців у період протеїнурії на висоті кризи. Тром-
боз мезентеріальних судин виникає, незважаючи на знижену кіль-
кість тромбоцитів.
Гіперкоагуляційний синдром зумовлює еритроцитарний тром-
бопластин, шо у великій кількості потрапляє в кровотік при руй-
нуванні еритроцитів.
У період гемолітичної кризи рівень гемоглобіну може знижува-
тись до 50-30 г/л. Кількість лейкоцитів та тромбоцитів у перифе-
ричній крові знижена. Вміст ретикулоцитів трохи підвищений. Ви-
діляється чорна сеча, шо містить гемоглобін та гемосидерин.
У сироватці крові виявляється підвищений вміст білірубіну за ра-
69
Н.М. Третяк | Гематологія
хунок непрямої фракції, при посиленні гемолізу з’являється віль-
ний гемоглобін.
У кістковому мозку спостерігається подразнення червоного па-
ростка, зниження кількості мегакаріоцитів. У термінальних стадіях
захворювання може виявлятись гіпоплазія кровотворення.
Загострення змінюються періодами спокійного перебігу хворо-
би. Проте низький рівень гемоглобіну (не вище 100 г/л), гіпербілі-
рубінемія за рахунок непрямої фракції свідчать про постійне руй-
нування патологічного клону еритроцитів. З часом, унаслідок
постійного гемолізу, збільшуються розміри печінки та селезінки,
можуть розвинутись жовчокам’яна хвороба та трофічні виразки на
гомілках.
Діагноз. Хвороба Маркіафави—Мікелі встановлюється з допо-
могою проби Хема (кислотна проба) та сахарозної проби (додаван-
ня сахарози). Крім того, можна використовувати тест градієнтного
центрифугування, котрий дає змогу виявляти фракцію атипічно
легких еритроцитів, характерних для ПНГ.
Диференційний діагноз проводять з АІГА з тепловими гемолізи-
нами та АА, що ускладнена синдромом ПНГ.
Лікування. Патогенетичного методу лікування ПНГ не існує.
Застосування глюкокортикоїдних гормонів суттєво не впливає на
патологічний процес. Використовується симптоматична терапія —
трансфузії відмитих (не менше 6 разів) або розморожених еритро-
цитів. Інфузії свіжозаготовлених еритроцитів можуть спровокувати
гемолітичну кризу. Тривале застосування гемотрансфузій призво-
дить до утворення ізоантиеритроцитарних антитіл. У разі такого
ускладнення кров або розморожені еритроцити підбираються з до-
помогою проби Кумбса, відмиваються перед введенням 5-6 разів.
Останніми роками пропонується виконання трансплантації
алогенного кісткового мозку. Як симптоматична терапія застосову-
ються анаболічні гормони, антиоксидантні засоби (вітаміни Є, А),
за потреби — обережно препарати заліза.
Прогноз несприятливий.
70
МІЄЛОДИСПЛАСТИЧНИЙ СИНДРОМ
Мієлодиспластичний синдром (МДС) поєднує в собі гетерогенну
групу захворювань, які характеризуються клональними порушен-
нями кістковомозкового кровотворення, що призводить до нее-
фективного гемопоезу. Неефективність ця спричиняється затрим-
кою дозрівання та загибеллю прекурсорів у кістковому мозку. При
МДС спостерігається моно-, бі- або, частіше, панцитопенія в пе-
риферичній крові та гіперцелюлярний кістковий мозок, а також
підвищений ризик трансформації в гостру лейкемію. Морфологіч-
на характеристика диспластичного кісткового мозку проявляється
аномалією будови еритроцитів, що призводить до порушення їх
функції та скорочення терміну життя.
Раніше поняття МДС ототожнювали з передлейкемією. Термін
цей було впроваджено Віоск у 1953 р. для узагальнення стану, який
характеризувався двома основними рисами — анемією незрозуміло-
го походження та схильністю до розвитку маніфестних проявів гос-
трої лейкемії. З огляду на факт, що не в усіх пацієнтів із передлейке-
мічним синдромом розвивається гостра лейкемія, в 1982 р.
франко-американо-британською кооперативною групою було введе-
но поняття “мієлодиспластичний синдром” та запропоновано його
класифікацію.
Класифікація типів МДС
1. Рефрактерна анемія (РА) — 30-40% усіх хворих.
2. Рефрактерна анемія з кільцевими сидеробластами (РАКС) —
15-25%.
3. Рефрактерна анемія з надлишком бластів (РАНБ) — 15-25%.
4. Рефрактерна анемія з надлишком бластів у стадії трансфор-
мації (РАНБ-Т) — 10-15%.
5. Хронічна мієломоноцитарна лейкемія (ХММЛ) — 15%.
До нової класифікації ВООЗ увійшли наступні форми МДС:
РА, РАКС, РАНБ, рефрактерна цитопенія з мультилінійною дис-
плазією, 5ц-синдром, МДС, що не класифікується. ХММЛ було
перенесено в групу МДС/МПЗ, а РАНБ-Т виключено зовсім, то-
му що експерти ВООЗ дійшли висновку, що при вмісті бластів
у кістковому мозку 20% повинна діагностуватися ГМЛ.
71
Н.М. Третяк | Гематологія
Патологія стовбурової клітини при МДС може виникати сіє по-
уо і бути наслідком впливу хімічних канцерогенних речовин, раді-
ації, цитотоксичних медикаментів і спадкових генетичних пору-
шень (синдром Дауна, трисомія 8, моносомія 7 тощо). Клональні
цитогенетичні зміни, що виявляються у 50% випадків <іе поуо
МДС і більш ніж 80% випадків “наведеного” МДС, найчастіше
стосуються генів, локалізованих на довгому плечі хромосом 5 і 7, і
відповідають за продукцію колонієстимулюючих факторів (КСФ)
та інтерлейкинів (ІЛ-3, ІЛ-5), котрі беруть активну участь у регуля-
ції кровотворення. Порушення функції білків, які регулюють кро-
вотворення, призводять до порушення дозрівання клітин гемопое-
зу зі зміною їхніх морфологічних особливостей (дисплазією) та
функціональних властивостей. Крім того, виявилося, що до 75%
клітин кісткового мозку при МДС виявляють СЙ95 — маркер
програмованої клітинної загибелі — апоптозу. Результатом дії цих
патологічних механізмів є передчасне руйнування кровотворних
клітин у кістковому мозку (неефективний гемопоез) і різного роду
цитопенії в периферичній крові при гіпер- або нормоклітинному
кістковому мозку. Одним із можливих механізмів розвитку захво-
рювання є дефект мікрооточення, підтвердженням чого слугують
виявлені при МДС якісні та кількісні зміни клітин строми кістко-
вого мозку з порушенням продукції цитокінів.
Епідеміологія. Захворюваність на МДС становить 4,1 випадку
на 100 тис. населення. 80% випадків МДС припадає на осіб, стар-
ших 60 років. У європейських країнах серед осіб 50—69 років ре-
єструється 40 нових випадків МДС на 1 млн населення, а серед
осіб 70 років і старших — 150 нових випадків на 1 млн. населення.
Етіологія. Незважаючи на численні дослідження, причини роз-
винення МДС багато в чому залишаються неясними. У трупі етіоло-
гічних факторів розглядаються такі, що здатні викликати мутації клі-
тин і тим самим призводити до розвитку пухлини — віруси, іонізуюче
випромінювання, хімічні агенти. На сьогоднішній день яких-небудь
етіологічних факторів, специфічних для МДС, не встановлено. У ря-
ді випадків розвиткові МДС передує хіміотерапія солідних пухлин.
Патогенез. Початковим фактором у розвитку МДС є мутація
стовбурової клітини крові. Нащадки клітини, де відбулася мутація,
здобувають біологічну перевагу перед нормальними гемопоетични-
ми клітинами, що дозволяє їм цілком колонізувати кістковий мо-
зок, витісняючи нормальні гемопоетичні клітини. Особливістю му-
тації стовбурової клітини крові при МДС є часткове збереження її
72
| Міслодиспластичний синдром
нащадками здатності дозрівання до зрілих клітин крові. Однак
процес дозрівання носить неефективний характер, що призводить
до зменшення кількості зрілих клітин у периферичній крові. Крім
кількісних змін у складі клітин периферичної крові має місце і
зниження їхньої функціональної активності.
Значну роль у розвитку патологічного клону гемопоетичних
клітин відіграє стромальне мікрооточення, проте конкретні меха-
нізми залучення стромальної тканини в патологічний процес при
МДС вивчені ще недостатньо.
Неефективний характер гемопоезу (дисплазія кровотворення)
має чітко виражений морфологічний еквівалент — змінений як
морфологічних ознак гемопоетичних клітин, так і їхнього розташу-
вання у кістковомозкової стромі (зміна архітектоніки). Важливою
складовою морфологічних ознак дисплазії кровотворення є зміни
стромальної тканини.
У неопластичному клоні можуть відбуватися вторинні мутації
клітин. У ряді випадків це призводить до розвитку в кістковому моз-
ку клону гемопоетичних клітин, що втратили здатність до дозріван-
ня більшою мірою, ніж гемопоетичні клітини з попереднього клону.
Морфологічним еквівалентом цієї події є збільшення в кістковому
мозку кількості незрілих клітин — бласгів. Наявність понад 20%
бластних клітин у цитологічному препараті кісткового мозку — оз-
нака трансформації в гостру лейкемію. При цьому варто розуміти,
що дана ситуація не означає розвиток нового (другого) захворюван-
ня, а є закономірним продовженням перебігу даної нозологічної
форми мієлодиспластичного синдрому, що підкоряється закону пух-
линної прогресії. Трансформація в гостру лейкемію при МДС екві-
валентна розвиткові бластної кризи при хронічній мієлолейкемїї.
Клінічна картина МДС не має специфічних особливостей. Ос-
новні симптоми захворювання залежать від сполучення і глибини
ураження паростків кровотворення. Першою ознакою хвороби най-
частіше буває анемічний синдром, що спостерігається у 90% хворих
і виявляється зростаючою слабкістю, підвищеною втомлюваністю та
іншими ознаками, властивими анемії. У хворих на МДС із лейкопе-
нією на перший план виступають інфекційні ускладнення (бронхі-
ти, пневмонії та ін.), хоча у частини хворих причина лихоманки за-
лишається незрозумілою. Геморагічний синдром спостерігається у
10-32% хворих із тромбоцитопенією. Інтенсивність геморагічних
проявів дуже варіабельна, але найчастіше наявні крововиливи на
шкірі та слизових оболонках, іноді одночасно з кровоточивістю ясен
73
Н.М. Третяк І Гематологія
і слизової оболонки носа. Якої-небуць характерної органної патоло-
гії при МДС немає. Периферичні лімфовузли зазвичай не збільше-
ні. Іноді спостерігається незначне збільшення селезінки та печінки.
Діагноз. При дослідженні периферичної крові у більшості хво-
рих виявляється анемія, що в 30-40% випадків поєднується з лей-
ко- та/або тромбоцитопенією. У 19-28% хворих спостерігається
панцитопенія. Анемія при МДС завжди носить макроцитарний ха-
рактер за рахунок переважання серед еритроцитів макро- та овало-
цитів, нерідко спостерігається в еритроцитах базофільна пункгація
та тільця Жоллі. У крові можуть бути присутні клітини червоного
ряду, які містять ядра. Однією з важливих ознак МДС є стійка лей-
копенія з нейтропенією, причому для гранулоцитів характерна на-
явність псевдопельгеровської аномалії (лейкоцити з двохдольчасти-
ми ядрами) та дегрануляції цитоплазми (слабо виражена зернистість
чи її повне зникнення). У деяких випадках при МДС у крові спос-
терігається підвищений вміст моноцитів, що нерідко мають ядерну
атипію. Тромбоцитопенія зустрічається у 50% хворих із МДС. Се-
ред тромбоцитів зустрічаються гігантські та дегранульовані форми.
Кістковий мозок при МДС частіше гіперкпітинний, із ознака-
ми дисплазії одного чи декількох паростків кровотворення. Най-
більш чітко мієлодисплазія виявляється морфологічними аномалі-
ями в клітинах еритропоезу у вигляді мегалобластоїдних змін,
патології ядер і цитоплазми — еритрокаріоцитів (між’ядерні міс-
тки, багатоядерність, фрагментація і пікноз ядер, базофільна пун-
ктація та вакуолізація цитоплазми, збільшення кількості сидероб-
ластів із появою кільцевих форм). Дисплазія мієломоноцитарного
паростка кровотворення проявляється затримкою дозрівання гра-
нулоцитів на рівні мієлоцитів, порушенням процесу грануляції ци-
топлазми та підвищенням вмісту моноцитів. Дизмегакаріоцитопо-
ез характеризується перевагою мікроформ та гіпогрануляцією
цитоплазми клітин. При деяких формах МДС відзначається підви-
щений вміст у кістковому мозку бластних клітин (від 5% до 30%).
Хоча у більшості хворих на МДС — нормальна чи підвищена клі-
тинність кісткового мозку, у 8-28% хворих виявляється кістково-
мозкова гіпоплазія. При гістологічному дослідженні кісткового
мозку методом трепанобіопсії підвищене утворення ретикулінових
волокон виявляється майже у 50% хворих із МДС, причому різко
виражений мієлофіброз спостерігається у 10-15% хворих із МДС.
Спленомегалія зустрічається у 17% таких хворих, гепатомегалія —
у 13%, а лейкеміди — у 10%.
74
| Міслодиспластичний синдром
Рефрактерна анемія (РА) діагностується за наявності макроци-
тарної анемії, стійкої до терапії вітаміном В12 і фолієвою кислотою,
з ретикулоцитопенією, різними ознаками дизеритропоезу та рідше
— дизіранулоцитопоезу. Кістковий мозок гіперклітинний із вира-
женою гіперплазією червоного паростка й ознаками дизеритропо-
езу. Гранулоцитарний і мегакаріоцитарний паростки частіше нор-
мальні, вміст бластів не перевищує 5%.
РА з кільцевими сидеробластами (РАКС), або ідіопатична сиде-
робластна анемія. Основною відмінністю від попередньої форми є
наявність, крім ознак дизеритропоезу, понад 15% кільцевих сиде-
робластів з-поміж еритрокаріоцитів кісткового мозку.
РА з надлишком бластів (РАНБ). У всіх випадках анемія поєд-
нується з лейко- та/або тромбоцитопенією, завжди існують ознаки
дизгранулоцитопоезу й нерідко — гігантські тромбоцити. У крові
може бути присутня невелика кількість бластів (більше 5%). Кіс-
тковий мозок гіперклітинний з ознаками дисплазії всіх паростків
кровотворення і підвищеним вмістом бластів — від 5% до 20%.
РАНБ у трансформації (РАНБ-Т). Крім диспластичних змін
у периферичній крові й кістковому мозку, що спостерігаються при
РАНБ, виявляється більш високий вміст бластних клітин у пери-
феричній крові (понад 5%) та кістковому мозку (від 20% до 30%).
Хронічна мієломоноцитарна лейкемія (ХММЛ) діагностується на
підставі абсолютного моноцитозу (більше 1 • 109/л) у крові, що час-
то супроводжується збільшенням кількості зрілих гранулоцитів, не-
рідко з ознаками дисплазії: гіпогрануляція та пельгеровська форма
ядра. В кістковому мозку при ХММЛ дисплазія 3-х паростків кро-
вотворення поєднується з підвищеним вмістом моноцитів та їхніх
попередників, а також бластних клітин.
Як допоміжний метод діагностики може бути використане цито-
генетичне дослідження каріотипу гемопоетичних клітин. Різні хромо-
сомні поломки виявляються у 48% хворих на МДС. Частота аномалій
каріотипу варіює в залежності від нозологічного варіанту МДС. Так,
у хворих із РА хромосомні поломки виявляються у 30% випадків,
а у хворих із РАНБ-Т — у 60%. Виявлення хромосомних аномалій має
велике значення для визначення прогнозу перебігу захворювання.
Змінами каріотипу у хворих на РА, що зустрічаються найчасті-
ше, є делеція частини довгого плеча п’ятої хромосоми (5ц-). Озна-
чена аномалія частіше виявляється у жінок (співвідношення чоло-
віків і жінок серед хворих — 1:5). Для хворих із такою
хромосомною поломкою характерні яскраво виражені морфологіч-
75
Н.М. Третяк | Гематологія
ні аномалії мегакаріоцитів (мікромегакаріоцити), тромбоцитоз
у периферичній крові та досить сприятливий прогноз перебігу зах-
ворювання з низькою частотою трансформації в гостру лейкемію.
Диференційний діагноз. РА найчастіше доводиться диференцію-
вати від В|2-дефіцитної анемії, за якої також існує дисплазія кро-
вотворних клітин у вигляді мегалобластозу кісткового мозку й не-
ефективного еритропоезу. Швидка клінічна та гематологічна
відповідь на терапію вітаміном Вр вказує на причинний взає-
мозв’язок між анемією і дефіцитом цього вітаміну. Лікування РА
вітаміном Вр і фолієвою кислотою завжди неефективне. Значні
труднощі викликає диференційна діагностика РА, перебіг котрої
супроводжується гіпоплазією кісткового мозку, та апластичної ане-
мії. На користь діагнозу МДС свідчить наявність виразних ознак
дисплазії гемопоезу та цитогенетичних аномалій, що мають кло-
новий характер. Додатковою диференнійно-діагностичною озна-
кою МДС і апластичної анемії може слугувати низький рівень луж-
ної фосфатази нейтрофілів, що зустрічається у деяких хворих із
МДС, тоді як при апластичній анемії цей показник підвищений.
У 10-15% випадків РА має місце виражений фіброз кісткового
мозку. Наявність панцитопенії з трьохпаростковою мієлодисплазі-
єю, відсутність спленомегалії та швидке прогресування захворю-
вання дає змогу відрізнити цей варіант МДС від ідіопатичного мі-
єлофіброзу. Крім того, для ідіопатичного мієлофіброзу характерне
поєднання ділянок фіброзу кісткового мозку з вогнищами гіпер-
клітинного кісткового мозку з гіперплазією мегакаріопитарного
паростка, відсутність диспластичних змін клітин кісткового мозку
та крові, лейкоцитоз і тромбоцитоз, шо не властиво МДС.
РА з кільцевими сидеробластами (РАКС) варто диференціюва-
ти з іншими сидеробластними анеміями, зумовленими спадковими
порушеннями порфіринового обміну і свинцевою інтоксикацією.
При спадковому порушенні синтезу порфіринів анемія завжди ви-
являється в дитинстві та носить гіпохромний характер при високо-
му вмісті заліза в крові і тканинах. При дослідженні вмісту порфі-
ринів в еритроцитах у більшості хворих виявляється зниження
вмісту протопорфіринів і підвищення копропорфіринів. Для свин-
цевої інтоксикації, що зумовлює набуте порушення синтезу порфі-
ринів, характерна наявність базофільної пунктації еритроцитів,
збільшення вмісту сидеробластів у кістковому мозку, а також наяв-
ність ознак ураження нервової системи, біль у животі.
Складним іноді буває диференційний діагноз між РА (РАКС) і
76
| Мієлодиспластичний синдром
еритромієлозом, оскільки при обох захворюваннях спостерігається
значна гіпер- та дисплазія червоного паростка кровотворення. На
користь червоноклітинного варіанту лейкемії (еритромієлозу) свід-
чить підвищений вміст у кістковому мозку бластних клітин і вияв-
лення еритробластів з допомогою РАБ-реакції глікогену, а також
присутність у периферичній крові нормобластів, шо практично не
спостерігаються при РА і РАКС.
Необхідність диференційного діагнозу з пароксизмальною ніч-
ною гемоглобінурією зумовлена спільною для обох захворювань
панцитопенією при гіперклітинному кістковому мозку. Відмінною
ознакою ПНГ є ретикулоцитоз крові, позитивні проби Хема та са-
харозна, виявлення гемосидерину в сечі та вільного гемоглобіну
в сироватці крові.
РАНБ і РАНБ-Т, власне кажучи, є гострими малопроцентни-
ми лейкеміями й належать до МДС лише за ознакою дисплазії
кровотворних клітин та тривалого перебігу. Формально від гострої
лейкемії ці дві форми МДС відрізняються кількістю бластів у кіс-
тковому мозку — менше 20%.
ХММЛ також являє собою один із варіантів лейкемії і віднесе-
ний до МДС лише на підставі диспластичних змін кровотворення.
Диференційну діагностику ХММЛ іноді доводиться здійснювати
з хронічною мієлолейкемією (ХМЛ), перебіг якої супроводжується
високим вмістом у крові моноцитів. На користь ХМЛ із супутнім
моноцитозом свідчить наявність Філадельфійської хромосоми ((
(9;22)) і відсутність ферменту лужної фосфатази в гранулоцитах.
Лікування МДС багато в чому залежить від форми захворюван-
ня. Терапія РА і РАКС, особливо у людей похилого віку, здебільшо-
го носить симптоматичний характер: трансфузії еригроцитної маси
при вираженій анемії, тромбоконцентрату — при тромбоцитопенії
з геморагічним синдромом, введення антибактеріальних препаратів
при інфекційних ускладненнях. В останні роки виявлений позитив-
ний ефект при РА і РАКС імуномодулюючого препарату циклоспо-
рину А (Сандимун, Сандимун-Неорал), шо пригнічує функцію при-
родних кіллерів і Т-супресорів. Так, у хворих із РА і РАКС, особливо
у яких спостерігалися гіпоплазія кровотворення або лімфо'їдні скуп-
чення за даними гістологічного дослідження грепанобіоптатів, було
отримано позитивний ефект — від повної ремісії до зникнення пот-
реби в гемогрансфузіях — у 67% хворих при застосуванні циклоспо-
рину А в дозі 5 мг/кг на добу протягом 6-12 місяців.
Наявність при МДС двох-трьохпаросткової цитопенії стала під-
77
Н.М. Третяк | Гематологія
ставою для застосування комбінацій факторів росту: еритропоетину
(епрекс, рекормон), грунулоцитарних колонієстимулюючих .факто-
рів (граноцит, нейпоген). При поєднаному використанні цих цито-
кінів позитивний ефект спостерігається приблизно у 52% хворих.
Для лікування РАН Б і РАНБ-Т у осіб, молодших 50 років, зас-
тосовується програма ЕБАС, у осіб, старших 50 років, частіше зас-
тосовуються малі дози цитозару — 10 мг/м2 ( до 20 мг) двічі на день
підшкірно 14 днів щомісяця, або алкеран — 2 мг/добу до сумарної
дози 100-150 мг. Для лікування РАНБ-Т також застосовується хі-
міотерапія за програмами, шо використовуються в терапії гострих
лейкемій (7+3, КАСОР). У лікуванні ХММЛ найефективнішим є
застосування гідроксісечовини (Гідреа), що дає позитивний ефект
у 60% хворих. Крім того, нині в програми лікування МДС вклю-
чають антитимоцитарний глобулін, похідні ретиноєвої кислоти
(АТКА, весаноїд). Єдиним радикальним методом лікування МДС
є алогенна трансплантація кісткового мозку (ТКМ), що показана
хворим, молодшим 40 років, при кількості бластних клітин у кіс-
тковому мозку менш ніж 5%, тобто при ранніх варіантах із корот-
кими термінами від початку захворювання до ТКМ і нормальним
каріотипом клітин кісткового мозку.
Прогноз при МДС украй несприятливий. Середня тривалість
хвороби коливається в межах від 7,5 до 35,1 місяця. При адекватній
трансфузійній терапії хворі з РА і РАКС досить тривалий період збе-
рігають непогану форму і можуть бути працездатні. Прогноз у хво-
рих ХММЛ дешо гірший, а у хворих із РАНБ і РАНБ-Т — поганий.
Тривалість виживання хворих при трансформації кожної з нозоло-
гічних форм МДС у гостру лейкемію обмежується 4-6 місяцями.
Причинами летального результату можуть бути інфекційні та
геморагічні ускладнення внаслідок гранулоцитопенії та тромбоци-
топенії, однак у більшості випадків захворювання закінчується
трансформацією в гостру лейкемію, що дозволяє розглядати МДС
як передлейкемічний стан. Про несприятливий перебіг МДС свід-
чать такі ознаки: підвищення клітинності кісткового мозку, почат-
кова нейтропенія та тромбоцитопенія, а також підвищення вмісту
бластних клітин (понад 5%) у кістковому мозку. Таким чином,
найчастіше трансформація в гостру лейкемію відбувається у хворих
із РАНБ- і, особливо, РАНБ-Т-варіантами МДС. Прогностично
несприятливим є також порівняно немолодий вік хворих (понад 50
років) та вторинний характер МДС (унаслідок впливу радіаційних,
хімічних та медикаментозних агентів).
78
ГЕМОБЛАСТОЗИ
Гемобластозами називається група пухлин, що походять із од-
нієї мутованої кровотворної клітини, проліферація якої не підпа-
дає під контроль організму.
Гемобластози, при яких пухлинні клітини щільно заселяють
кістковий мозок, називаються гемобластозами кістково-мозкового
походження.
Класифікація гемобластозів
1. Гемобластози кістково-мозкового походження:
а) гострі лейкемії:
— гострі лімфобластні лейкемії (ГЛЛ),
— гострі нелімфобластні лейкемії (ГНЛ);
Ь) хронічні лейкемії:
— хронічна мієлоїдна лейкемія,
— хронічна лімфоідна лейкемія,
— ідіопатичний мієлофіброз,
— еритремія;
с) парапротеїнемічні гемобластози:
— множинна мієлома,
— хвороба Вальденстрема.
2. Гемобластози позакістково-мозкового походження:
а) лімфогранулематоз (лімфома Годжкіна);
Ь) негоджкінські злоякісні лімфоми.
ГОСТРІ ЛЕЙКЕМІЇ
Гостра лейкемія — це злоякісне захворювання системи крові,
що характеризується ураженням кісткового мозку морфологічно
незрілими, бластними клітинами — відповідними родопочатківця-
ми одного із паростків кровотворення.
Гострі лейкемії (ГЛ) посідають провідне місце в структурі зах-
ворюваності на гемобластози, становлячи майже 1/3 від їхньої за-
гальної кількості. Захворюваність на гострі лейкемії коливається
від 3,5 до 5 випадків на 100 тис. наседення на рік.
Гострі лімфобластні лейкемії спостерігаються, в основному.
79
Н.М. Третяк | Гематологія
в дитячому віці — до 80-90% випадків, у осіб після 40 років діаг-
ностується до 80% гострих нелімфобластних лейкемій.
Етіологія. Існуючі на сьогодні теорії виникнення лейкемій
свідчать про поліетіологічність їх походження.
Відомо, що на фоні генетичних порушень, таких як хвороба
Дауна, анемія Фанконі, синдроми Блума, Клайнфельтера, Віскот-
та-Олдрича, спостерігається підвищена захворюваність на ГЛ. Епі-
деміологічними дослідженнями встановлене виникнення “сімейної
ГЛ” у однояйцевих близнюків. Проте генетичні порушення розгля-
даються як підгрунтя для лейкемій, що розвиваються під впливом
хімічних факторів, опромінення або вірусу.
Причиною ГЛ може бути неконтрольваний вплив іонізуючої
радіації, що підтверджується зростанням захворюваності серед на-
селення після атомного бомбардування японських міст Хіросіми та
Нагасакі. На забруднених територіях унаслідок випробування ядер-
ної зброї, в промисловості та медицині, де використовується атом-
на енергетика, з’явилися випадки “радіаційних лейкемій” із харак-
терними для них хромосомними абераціями, а також розвинення
вторинних лейкемій після застосування променевої терапії при лі-
куванні хвороби Годжкіна, спондилітів тощо.
Еспериментальні та клінічні дослідження доводять, що ГЛ мо-
же спричинити тривале неконтрольоване використання бутадіону,
левоміцетину, цитостатичних препаратів, а також контакт із хлоро-
ваними фарбами, метілетілкетоном, бензолом, пестицидами.
Вірусна теорія походження лейкемій у птахів та гризунів дове-
дена виділенням вірусу Гросса мишей та вірусу Рауса курей, котрі
здатні ініціювати пухлинний процес не тільки в їхніх носіїв, але
й у мавп, та трансформувати гемопоетичні клітини людини в куль-
турі тканини. Науково підтверджено роль вірусу у виникненні
Т-клітинної лейкемії та лімфоми Беркитта в людини, при пору-
шенні імунітету і, особливо, функції протипухлинного нагляду.
Вірусна теорія припускає, що в геномі кожної клітини закла-
дена інформація у вигляді ДНК-провірусу, тотожна інформації
в геномі онковірусу. В нормі ДНК-провірус (онкоген) перебуває
в стані спокою і передається у спадок. Лише під впливом канцеро-
генних факторів при порушенні імунного, генетичного та гормо-
нального гомеостазу організму онкоген активується і призводить
до трансформації клітини.
Патогенез. На сьогодні абсолютно доведено клональне поход-
ження пухлини, тобто розвиток її із однієї мутованої клітини, кот-
80
| Гемобластози
рі на початковому етапі є однорідними — моноклонова (доброякіс-
на) стадія. Проте нащадки клітин цього клону мають високу здат-
ність до мутації, утворюючи таким чином субклони. Поява субкло-
нів свідчить про перехід пухлини в злоякісну стадію.
Перехід від доброякісної до злоякісної стадії при різних фор-
мах гемобластозів відбувається з неоднаковою частотою та інтерва-
лом. У мутованій стовбуровій клітині порушується механізм пролі-
ферації та диференціювання, внаслідок чого відбувається
неконтрольована проліферація лейкемічної клітини, котра здобу-
ває перевагу над нормальним клоном гемопоезу. При досягненні
лейкемічним клоном певної маси відбувається гальмування дифе-
ренціювання нормального клону стовбурових клітин за принци-
пом зворотного зв’язку. Наслідком такого гальмування є пригні-
чення кровотворення з розвиненням анемії, нейтропенії,
тромбоцитопенії. Так, для утворення 1 кг злоякісного клону клітин
потрібно 3,5 року, що підтверджується епідеміологічними спосте-
реженнями. Перший пік захворюваності на лейкемії у дітей фіксу-
ється у 2-4 роки.
В патогенезі лейкемій важливу роль відіграють процеси метас-
тазування мутованих стовбурових клітин, котрі здатні до переносу
та імплантації на стадії як доброякісного, так і злоякісного розвит-
ку. Оселяючись у різних органах і тканинах, мутовані клітини зу-
мовлюють формування екстрамедулярного вогнища кровотворен-
ня, що клінічно проявляється гепато- та спленомегалією,
лімфоаденопатією, лейкемідами на шкірі, нейтролейкеміями та ін.
Класифікація гострих лейкемій грунтується на особливостях
морфологічних, цитохімічних та імунофенотипових ознак бластних
клітин. У 1976 р. гематологами Франції, США, Великої Британії
з урахуванням морфологічних ознак була розроблена РАВ-класи-
фікація. Згодом, у 1980 р., на основі використання нових діагнос-
тичних методів були внесені уточнення.
За РАВ-класифікацією ГЛ поділяються на дві великі групи:
гострі лімфобластні лейкемії та гострі мієлобластні лейкемії, всере-
дині яких виділяють варіанти за цитохімічними та імунофенотипо-
вими ознаками (табл. 9, 10).
При цитохімічному дослідженні у всіх варіантах ГЛЛ виявля-
ється негативна реакція на мієлопероксидазу, судан чорний В (ре-
акція на ліпіди), хлорацетат естеразу, альфа-нафтилацетераза може
бузи позитивною у Т-лімфобластах. Найхарактернішою для ГЛЛ є
позитивна реакція на глікоген (РА8-реакція) у глибчастій або гра-
81
і
Н.М. Третяк | Гематологія
Таблиця 9. Гострі лімфобластні лейкемії. ЕАВ-класифікація
гострих лімфобластних лейкемій (ГЛЛ)
Варіант Морфологічні ознаки Частота виявлення
Ц Мікролімфобластний варіант: лімфобласти маленького розміру, правильної (іноді розщепленої) форми ядра з дрібними, погано візуалізованими нуклеолами або без них, з високим ядерно-цитоплазматичним співвідношенням 20-25% випадків у дітей
ц Лімфобласти найчастіше великого діаметра, ядро неправильної форми (може бути розщеплене). Нуклеоли < 1, чітко визначені, цитоплазма бліда в достатній кількості 70% випадків у дорослих
*-з Лімфобласти великого розміру, нагадують клітини при лімфомі Беркитта, ядра правильної форми (коло чи овал). Одна (або більше) велика, чітка нуклеола, виражені базофілія та вакуолізація цитоплазми > 5% випадків
Таблиця 10. Гострі мієлобластні лейкемії. РАВ-класифікація
гострих мієлобластних лейкемій (ГМЛ)
Варіант Морфологічні та цитохімічні ознаки Частота виявлення
1 2 3
Гостра мієлобластна недиферен- ційована лейкемія - Мо Мієлобласти великі, агранулярні, іноді схожі на лімфобласти, варіант і_2. Забарвлення на мієлопероксидазу та Судан чорний В негативне, але ультра структурним дослідженням виявляються пероксидазнопозитивні гранули. Експресують маркери (або один із них) мієло їдного спрямування (СВ33, СО13, С014). 2-3%
ГМЛ без дозрівання - М( Мієлобласти великі, низькодиференційо Вані, містять 1 або більше нуклеол. Від сутність або невелика кількість азурофільних гранул. 3% клітин забарвлюються позитивно на мієлопероксидазу. 20%
ГМЛ із дозріванням - М2 М2-баз. Від 30% до 89% мієлобластів із широкою цитоплазмою, містять багато нуклеол, азурофільних гранул та паличок Ауера. Характерним є вміст близько 3% промієлоцитів та понад 10% клітин еритроцитарної маси. У випадках виявлення великої кількості базофілів - варіант визначається як М2-баз. 25-30%
82
| Гемобластози
Продовження табл. 10
1 2 3
Гостра промієлоцитарна лейкемія - М3 м3- гіпогранулярний Більш як 30% клітин є аномальними тромієлоцитами, що містять великі азурофільні гранули, палички Ауера; розмір ядра заріює, може бути двохлопастеве. Активно забарвлюються на мієлопероксидазу. Промієлоцити схожі на моноцити з бобоподібним ядром та незначною зернистістю в цитоплазмі. 8-15%
Гостра мієломонобластна лейкемія - М4 м4 - еозинофільний Мієлобласти, промієлоцити, мієлоцити становлять < 30% нееритроїдних клітин, але не перевищують 80%, може виявлятися < 20% промоноцитів та моноцитів у кістковому мозку. Дають позитивну реакцію на неспецифічну естеразу, хлорацетатестеразу. Може підвищуватися вміст промоноцитів та моноцитів у периферичній крові. У випадках виявлення понад 5% аномальних попередників еозинофілів діагностується М4е.
Гостра монобластна лейкемія - М5 Підваріант М5а - гостра монобластна лейкемія без дозрівання Підваріант Ми - Гостра монобластна лейкемія із дозріванням Більш ніж 80% клітин кісткового мозку - це монобласти, промоноцити та моноцити. Понад 80% клітин кісткового мозку є монобластами, близько 3% - із ознаками диференціювання. Клітини кісткового мозку у 80% - зрілі форми промоноцитів. Реакція на неспецифічну естеразу позитивна, інгі-бується №Е. Відсоток моноцитів у периферичній крові більший, ніж у кістковому мозку. 5-6%
Гостра еритробластна лейкемія (еритромієлоз) - М6 У кістковому мозку більш ніж 50% ана- плазованих еритробластів та понад 30% мієлобластів. Еритробласти зазвичай дають високо позитивну реакцію на глікоген (РА8- реакція). 4-5%
Гостра мегакаріоцитарна лейкемія - М7 У кістковому мозку понад 30% великих та малих мегакаріобластів із високим ядерно- цитоплазматичним співвідношенням. Цитоплазма бліда, гіпогранулярна, у ній можна побачити вакуолі. Зрідка на периферії бластів можна побачити тромбоцити. У периферичній крові бластів мало, іноді зустрічаються уламки мегакаріоцитів. 1-2%
83
Н.М. Третяк | Гематолопя
нулярній формі. Майже у 90% хворих на ГЛЛ бластні клітини міс-
тять фермент ТсІТ (термінальна дезоксинуклеотиділтрансфераза).
Цінним набутком у диференційній діагностиці різних варіантів
гострих лейкемій є використання імунофенотипування та цитоге-
нетичного дослідження бластних клітин кісткового мозку і пери-
феричної крові. Як уже зазначалося, європейською групою гемато-
логів створено імунологічну класифікацію гострих лімфобластних і
мієлоїдних лейкемій.
За пропозиціями першої Європейської групи з імунологічної
класифікації лейкемій (ЕСІГ) виділені 5 основних форм ГЛ та їх
субваріанти.
Основні імунологічні форми гострої лейкемії та їх субваріанти
І. В-лінійні:
а) В-І (про-В) ГЛЛ — виявляються у 70% випадків ГЛЛ дітей
та у 50% дорослих,
Ь) В-1І (соттоп) ГЛЛ,
с) В-1П (пре-В) ГЛЛ — близько 25% ГЛЛ у дітей,
сі) В-ІУ (зрілий-В) ГЛЛ — діагностується у 2-5% випадків усіх
ГЛЛ;
2. Т-лінійнї.
а) Т-І (про-Т) ГЛЛ діагностуються у 15% випадків у дітей та у
20-25% у дорослих,
Ь) Т-И (пре-Т) ГЛЛ,
с) Т-П1 (кортикальний) ГЛЛ,
б) Т-ІУ (зрілий-Т) ГЛЛ,
е) Г-а/В і у/8,
()ГЛЛ із експресією 1 або 2 мієлоїдних маркерів (мієло+ГЛЛ);
3. Гострі мієлоїдні лейкемії (ГМЛ):
а) мієломоноцитарний,
Ь) еритроїдний (ранній), незрілий і зрілий,
с) мегакаріоцитарний,
сі) низькодифереційований мієлоїдний (ГМЛ-М0),
е) тат+гмл,
і) ГМЛ із експресією 1 або 2 лімфоїдних маркерів
(лімф+ГМЛ);
4. Біфенотипові гострі лейкемії;
5. Недиференційовані гострі лейкемії.
Імунофенотипування бластних клітин при ГЛ використовуєть-
ся з метою підтвердження діагнозу, встановлення діагнозу, якщо
цитоморфологічне дослідження не дало остаточного діагнозу, для
84
| Гемобластози
визначення біфенотипових варіантів гострих лейкемій, визначення
прогностичних груп.
Особливо важливого значення набуває імунофенотипування
при ГЛЛ, оскільки діагностування імунологічних субваріантів є ос-
новною складовою призначення адекватної, з урахуванням факто-
рів ризику, хіміотерапії та досягнення позитивних результатів —
отримання у 85-95% дітей та 75-80% дорослих повної ремісії.
Крім того, диференціювання таких мієлоїдних варіантів лей-
кемій, як Мо, Мр М6 та М7 проводиться за визначенням на по-
верхні бластних клітин таких антигенів, як СО33, СБ13, СО14, кот-
рі є маркерами мієлоїдної та моноцитарної ліній; СО4І, СІ)42Ь є
тромбоцитарними маркерами; глікофорин-А — еритроїдним мар-
кером.
Завдяки вивченню цитогенетичних аберацій пухлинних клі-
тин стало можливо створити класифікацію та прогностичні кри-
терії гострих лейкемій на основі хромосомних порушень, що ко-
релюють із морфологічним варіантом та клінічною картиною
захворювання.
Клони клітин із хромосомними аномаліями виявляються у 60-
80% випадків ГНЛ, найчастіше це такі характерні зміни: і(8;21),
1(5; 17), іпу(16), -5/5д-, -7/7я~; +8; +21, причому в більшості випад-
ків спостерігається по дві та більше змін каріотипу.
Транслокація ї(5; 17) (я22; д21) специфічна для гострої проміє-
лоцитарної лейкемії — М3, за якої вона спостерігається майже у
90-95% випадків.
Для вторинних лейкемій характерна значна перебудова каріо-
типу. Кількість хромосом у більшості випадків зменшена; спосте-
рігаються гіподиплоїдні клони з множинними хромосомними мар-
керами. Виявляються як зникнення або делеція довгого плеча
хромосоми 5 чи 7, трисомія 8, так і транслокацїї С(3;21) (ч26; я22),
і(1;7) (д!0; рІО), ї(11;16) (д23; рІЗ) та і( 16;21) (р12; ц22), котрі при
первинних лейкеміях майже не зустрічаються.
При ГЛЛ у 60-70% випадків виявляються клони клітин зі змі-
нами каріотипу та широким (понад 30) спектром хромосомних
аномалій. Найхарактернішими для ГЛЛ є транслокацїї 1(9;22) (д34;
411), 1(4; 11) (ч21; ч23), і(1;19) (ч23; рІЗ).
Однак, незважаючи на те, що хромосомний аналіз має велике
значення як для прогнозу, так і для моніторингу лікування, він не
є самостійним діагностичним критерієм. І повинен оцінюватися
в комплексі з усіма клініко-лабораторними даними.
85
Н.М. Третяк | Гематологія
Класифікація гострих лейкемій за стадіями перебігу патологіч-
ного процесу
І. Початкова стадія має прихований та компенсований пере-
біг, без чіткої клініко-гематологічної картини.
2. Розгорнута стадія характеризується значним пригніченням
нормального кровотворення, високим вмістом бластних клітин
у кістковому мозку.
3. Повна ремісія характеризується вмістом бластних клітин
у кістковому мозку, меншим 5%, або загальною кількістю лімфо-
їдних елементів, меншою 30%. Показники периферичної крові
близькі або нормальні протягом не менше одного місяця. Екстра-
медулярних лейкемічних інфільтратів не існує.
4. Одужання — повна ремісія без будь-яких подій протягом 5
років (безрецидивне 5-річне виживання).
5. Рецидив — повернення захворювання до показників 1 стадії
внаслідок активації залишкового лейкемічного пула. У кістковому
мозку — бластоз, у периферичній крові — частіше цитопенія.
6. Термінальна стадія — повне виснаження нормального кро-
вотворення та резистентність пухлини до цитостатичної терапії.
Справжній момент початку захворювання встановити у жодно-
му випадку неможливо. Хворі зазвичай почуваються добре, і ком-
пенсаторні механізми організму спрацьовують до моменту повсюд-
ного розселення пухлинних клітин (бластів) у кровотворній системі.
Діагноз гострої лейкемії встановлюється тільки при виявленні
понад 30% бластних клітин у кістковому мозку, при цьому у пери-
феричній крові бластні клітини іноді не визначаються.
Розгорнута стадія гострої лейкемії характеризується п’ятьма
основними синдромами.
Основні клінічні синдроми гострої лейкемії:
1) гіперпластичний,
2) геморагічний,
3) анемічний,
4) інтоксикаційний,
5) синдром інфекційних ускладнень.
Основними проявами гіперпластичного синдрому є помірне та
безболісне збільшення лімфатичних вузлів, печінки та селезінки.
Лейкеміди — лейкемічна інфільтрація шкіри у вигляді червонувато-
синіх, світло-коричневих або кольору шкіри утворень, шо можуть
виступати над поверхнею або бути на рівні шкіри, біль у трубчас-
тих кістках, гіперплазія ясен. У термінальній стадії або при рециди-
86
| Гемобластози
ві може спостерігатися лейкемічна інфільтрація легенів (лейкеміч-
ний пульмоніт), мозкових оболонок (нейролейкемія), яєчок.
Геморагічний та анемічний синдроми розвиваються внаслідок
лейкемічної гіперплазії та інфільтрації кісткового мозку злоякісни-
ми клітинами, що пригнічує нормальне кровотворення.
Анемічний синдром зумовлює такі симптоми, як адинамія,
блідість шкіри та слизових оболонок-, тахікардія, запаморочення,
болі в ділянці серця.
Геморагічний синдром може проявлятися окремими кровови-
ливами в шкіру або петехіальною висипкою. Іноді це помірні або
профузні носові, маткові кровотечі, кровоточивість ясен та гемора-
гії на слизових оболонках рота, склер. Найтяжчим ускладненням є
крововилив у речовину головного мозку.
Інтоксикаційний та інфекційний синдроми зумовлюють гаряч-
ку, болі в кістках. Інфекційні ускладнення спостерігаються у 80-
85% хворих на ГЛ і можуть бути спровоковані бактеріальними, ві-
русними та грибковими чинниками.
Клініко-гематологів на картина ГЛЛ
ГЛЛ становить приблизно 10-15% від усіх ГЛ, що виникають
у дорослих. У дітей ця форма лейкемії діагностується у 85-90% ви-
падків захворювання. До встановлення діагнозу хвороба може
з незначними проявами тривати місяцями, проте в деяких випад-
ках початок її може бути гострим.
Клініка. Перші ознаки захворювання — слабкість, сонливість,
субфебрильна температура (не пов’язана з інфекцією), болі в кіс-
тках, суглобах. У окремих випадках першими скаргами є болі в кіс-
тках та хребті без лімфоаденопатії, органомегалії та змін у аналізі
крові. Діагноз ГЛ ставиться в таких випадках зі значним запізнен-
ням. У 1% випадків ГЛЛ починається із головного болю, нудоти,
блювоти, втрати зору (власне спостереження), шо є наслідком за-
лучення до патологічного процесу, в першу чергу, центральної нер-
вової системи.
При об’єктивному обстеженні можуть спостерігатися блідість
шкіри, петехіальна висипка, крововиливи у шкіру різного розміру,
кровоточивість ясен, гарячка, лімфоаденопатія зі збільшенням
мигдаликів, гепато- та спленомегалія. У 5-10% випадків при рен-
тгенологічному дослідженні грудної клітки виявляється збільшен-
ня розмірів середостіння, що можливо як за рахунок залучення до
процесу ти мусу, так і внутрішньогрудних лімфатичних вузлів. У ра-
зі виконання діагностичної люмбальної пункції у 3-5% хворих ви-
87
Н.М. Третяк | Гематологія
являються в лікворі бластні клітини, що свідчить про ініціальне за-
лучення до лейкемічного процесу центральної нервової системи.
Діагноз. На початкових стадіях захворювання в аналізі крові
можуть бути еритрокаріоцити, мієлоцити, промієлоцити. У 60-70%
випадків спостерігається лейкоцитоз від 10 • 10-ч/л та вище, у 10%
хворих лейкоцитоз сягає 100 • 1бч/л та вище, що є негативною
прогностичною ознакою. Тромбоцитопенія від 50 • 109/л та менше
виявляється майже у 50% хворих. Анемія середнього ступеня тяж-
кості. В лейкоірамі кількість бластних клітин — від 35% до 99%,
у кістковому мозку — від 30% до 99%. Діагноз верифікується ци-
тологічним, цитохімічним, гістологічним, імунофенотипічним та
цитогенетичним дослідженням бластних клітин кісткового мозку
та периферичної крові.
Клініко-гематологічна картина ГНЛ
Група гострих нелімфобластних лейкемій (ГНЛ), або гострі мі-
єлоїдні лейкемії (ГМЛ) об’єднує лейкемії, що походять із клітин-
попередниць мієлоїдного або моноцитарного паростків кровотво-
рення. Проте вони мають морфологічні, цитохімічні,
імунофенотипові, цитогенетичні відмінності. Близько 10% ГМЛ
мають еритроїдне або мегакаріоцитарне походження.
Епідеміологія. ГМЛ діагностуються у будь-якому віці, але най-
частіше вони виникають у літніх людей — 60—65 років. Захворю-
ваність на ГМЛ становить 2,5-3,0 на 100 тис. населення на рік. Усі
ГНЛ у дорослих становлять близько 85%, у дітей, навпаки, 85%
становлять ГЛЛ.
Клініка. Спільними симптомами для всіх ГМЛ є слабкість, нез-
дужання, що можуть спостерігатися за декілька місяців до встанов-
лення діагнозу. У 15-20% хворих на початку захворювання спосте-
рігається пітливість та лихоманка, не пов’язана з інфекційним
процесом. Анемічний синдром — запаморочення, головний біль,
блідість шкіри та слизових оболонок. Дебютом захворювання мо-
жуть бути ангіни, пневмонії та інші інфекційно-запальні процеси.
Часто першим симптомом ГМЛ є геморагічний синдром. Петехі-
альна висипка на шкірі може спостерігатись у 50% хворих. Єдиною
першою характерною ознакою, особливо для гострої промієлоци-
тарної лейкемії (ГПмЛ), є кровотеча зі шлунково-кишкового трак-
ту, маткові, носові, ясневі та ниркові кровотечі. У половини хво-
рих відмічається схуднення, майже у 20% спостерігаються осалгії.
У 50% випадків виявляється лімфоаденопатія, збільшення розмірів
печінки та селезінки. Характерною для хворих з мієломоноблас-
88
| Гемобластози
тною і, особливо, з монобластною лейкемією, є гіперплазія ясен.
У 10% хворих спостерігається специфічна інфільтрація шкіри —
лейкеміди.
Діагностика. При лабораторному обстеженні майже у полови-
ни хворих виявляється лейкоцитоз, проте більше 100 • 109/л спосте-
рігається лише у 20% пацієнтів. У лейкограмі кількість бластних
клітин — від 5% до 80-90%. У кістковому мозку вміст бластів > 30%.
Частими є помірна анемія, тромбоцитопенія, кількість тромбоци-
тів < 100 • 109/л, іноді < 20 • Ю9/л.
За клініко-гематологічними ознаками з-поміж ГНЛ особливо
вирізняються гостра промієлоцитарна лейкемія (ГПмЛ) та гостра
еритробластна лейкемія (ГЕЛ), або хвороба Ді Гульєльмо.
Гостра промієлоцитарна лейкемія
ГПмЛ як самостійна форма лейкемії була описана в 1957 р.
НіПезіад. Захворювання характеризується важким геморагічним
синдромом, швидким перебігом та особливою морфологією блас-
тних клітин. ГПмЛ становить 10% усіх ГМЛ дорослих, у дітей зус-
трічається надзвичайно рідко.
Особливістю клінічної картини ГПмЛ є вираженість геморагіч-
ного синдрому (маткові, носові, ясневі кровотечі, геморагії в міс-
цях незначних травм) на фоні помірної тромбоцитопенії
(20-30 • 109/л), а іноді й при вмісті тромбоцитів понад 100 • 109/л.
Геморагічний синдром може бути першою ознакою захворювання
і з’являтися за декілька місяців до діагностування захворювання.
Далі розвивається важкий ДВЗ-синдром.
Ознаки пухлинної інтоксикації, слабкість, пітливість нароста-
ють повільно. Маса пухлини довго лишається відносно невеликою.
Вміст бластних клітин у кістковому мозку навіть у термінальній ста-
дії захворювання не перевищує 70-85%. Інфільтрація бластними
клітинами трубчастих кісток спостерігається не завжди, лімфатичні
вузли не збільшуються, гепато- та спленомегалія виникають рідко.
У патогенезі важкого геморагічного синдрому, ДВЗ-синдрому
та тяжкої інтоксикації головну роль відіграють лейкемічні клітини,
котрі мають на поверхні та в цитоплазматичних гранулах надлишок
тромбопластину. Руйнування цих клітин із вивільненням тканин-
ного тромбопластину та мезосомальних протеаз провокує ДВЗ.
Картина крові при ГПмЛ вирізняється з-поміж інших ГНЛ ви-
раженим поліморфізмом бластних клітин. Перші прояви захворю-
вання супроводжуються нормальними або дещо зниженими показ-
никами еритроцитів та гемоглобіну — нормохромною анемією
89
Н.М. Третяк | Гематологія
легкого ступеня. Вміст тромбоцитів та лейкоцитів у переважної
частини хворих зменшений.
За особливостями морфології лейкемічних клітин виділяють
два варіанти ГПмЛ. Перший — типовий макрогранулярний, коли
основну масу лейкемічних клітин складають бласти, що мають зер-
нистість, схожу за розмірами на зернистість базофілів та тучних
клітин, і зовсім малий відсоток — клітини з дрібного або пилково-
подібною азурофільною зернистістю. Другий — мікрогранулярний
варіант, що становить приблизно 20% від усіх ГПмЛ, коли перева-
жають бласти з дрібного та пилковоподібною азурофільною зер-
нистістю. Крім того, для них характерні дрібні з неправильними
контурами та потворні ядра, часто двох- та багатодольчасті. При
цьому варіанті частіше спостерігаються лейкоцитоз та вихід блас-
тних клітин у периферичну кров.
Імунофенотипові характеристики ГПмЛ тотожні маркерам
ГНЛ, проте на клітинах ГПмЛ не виявляється експресія ранніх клі-
тинних маркерів СП34, НЬА-ПК.
Майже в усіх випадках ГПмЛ виявляється транслокація —
і(15;17) (ц22; ці2-21), крім цих хромосомних змін описано ще
близько 17 варіантів транслокацій.
ГПмЛ може бути встановлено вже при вивченні морфології
бластних клітин. Цитохімічне дослідження підтверджує діагноз.
Гостра еритробластна лейкемія
Вперше описана у 1917 р. Ді Гульєльмо й становить лише 5%
від усіх форм гострих лейкемій у дорослих та 0,6% у дітей. У
10-20% випадків еритромієлоз має вторинний характер і розвива-
ється після хіміо- або променевої терапії у хворих на лімфограну-
лематоз, мієломну хворобу, еритремію.
Клінічна картина розвивається мляво. Тривалий час може
спостерігатися рефрактерна, частіше гіперхромна, анемія (не зу-
мовлена дефіцитом заліза або вітаміну В12). У гемограмі можуть ви-
являтись еритрокаріоцити, помірна лейкопенія або/та тромбоцито-
пенія, ретикулоцити не більше 1-3%. У міру прогресування
патологічного процесу розвивається лейкемізація: у периферичну
кров виходять еритрокаріоцити, бласти. Підвищується білірубін за
рахунок непрямої фракції, може бути гіпергамаглобулінемія, пози-
тивна проба Кумбса. ревматоїдний фактор. Хворий скаржиться на
болі в кістках, суглобах.
При дослідженні кісткового мозку виявляється значне зростан-
ня вмісту агипичних, багатоядерних клітин червоного паростка,
90
| Гемоблгстози
бластні елементи, за морфологічними ознаками яких еритромієлоз
поділяється на два варіанти — власне еритромієлоз, субстратом
котрого є еритрокаріоцити та недиференційовані бласти, та ерит-
ромієлоз, де поряд із еритрокаріоцитами виявляється велика кіль-
кість мієлобластів.
Підтверджується цитохімічним дослідженням клітин кістково-
го мозку і периферичної крові.
Характерними імунофенотипічними маркерами є експресія
СЮ[3, СЮ33, СІ)41, СС71, глікофорину А.
Цитогенетичні дослідження найчастіше виявляють моносомію
5 та/або 7, делецію 5у або 7ц. При гострих еритроїдних лейкеміях
часто зустрічаються структурні зміни хромосом (кільцеві хромосо-
ми, дицентрики).
Лікування гострих лейкемій
Лікування гострих лейкемій ґрунтується на використанні ци-
тостатичних препаратів, головним завданням яких є максимальне
викорінення пула пухлинних клітин.
Програма цитостатичної терапії складається з таких етапів:
— індукція ремісії;
— консолідація ремісії;
— профілактика нейролейкемії;
— підгримуюча терапія в період ремісії;
— лікування рецидиву;
— лікування резистентних форм.
Програми цитостатичної терапії розроблені з урахуванням клі-
тинної кінетики при ГЛ. Виходячи з цих позицій усі цитостатичні
засоби поділяються на 2 групи. До першої належать хімічні засоби,
що специфічно впливають на життєвий цикл клітини. До другої —
речовини, що діють незалежно від клітинного циклу.
Для лікування як ГЛЛ, так і ГНЛ розроблені стандарти ПХТ,
котрі дещо відрізняються, з огляду на клініко-гематологічні відмін-
ності, але ідеологія проведення програмної цитостатичної терапії
витримується в усіх випадках.
Лікування ГНЛ
Якщо не визначена група прогнозу за результатами цитогене-
тичного дослідження, індукція ремісії проводиться за схемою
“7+3”: у хворих віком до 60 років даунорубіцин — 45 мг/м2 на до-
бу, або ідарубіцин — 12 мг/м2 на добу, або мітоксантрон — 10 мг/м2
на добу в/в 15—30-хвилинною інфузією у 1—3-й дні та цитозар —
100 мг/м2 на добу тривалою в/в інфузією або 100 мг/м2 двічі на до-
91
Н.М. Третяк | Гематологія
бу у вигляді 1-годинних інфузій у 1—7-й дні. У хворих віком понад
60 років при задовільному загальному стані — даунорубіцин
ЗО мг/м2 на добу, або ідарубіцин 12 мг/м2 на добу, або мітоксантрон
10 мг/м2 на добу в/в 15—30-хвилинною інфузією у 1—3-й дні та ци-
тозар — 100 мг/м2 на добу в тривалій в/в інфузії або 100 мг/м2 дві-
чі на добу у вигляді 1-годинних інфузій на 1—7-й дні.
Якщо після першого курсу ремісії не досягнуто, з інтервалом
3-4 тижні проводиться другий аналогічний індукційний курс, але
доцільно застосовувати одночасне введення нейпогену 5 мг/кг п/ш
з 0 дня до 7-го дня курсу.
При досягненні ремісії проводиться її консолідація чотирма
курсами цитозару у високих (3000 мг/м2) дозах, а у хворих віком
понад 60 років у доброму загальному стані та з нормальною фун-
кцією нирок і при клиренсі креатиніну більше 70 мл/хв. — по
1000—1500 мл/м2 3-годинною інфузією щодванадцять годин двічі
на добу 1-й, 3-й та 5-й дні курсу. Курси проводяться кожного 29-го
дня від початку попереднього.
Після проведння курсів індукції та консолідації призначається
аміфостин по 500 мг в/в протягом 50 хв. тричі на тиждень до мо-
менту відновлення кровотворення.
Особам похилого віку після закінчення кусу хіміотерапії, а та-
кож тим, у кого розвинулись інфеційні ускладнення, призначаєть-
ся нейпоген по 5 мг/м2 на добу до досягнення рівня нейтрофілів
1000/мкл.
Пацієнтам, у котрих ремісії не досягнуто, після першого індук-
ційного курсу в якості підгримуючої терапії можливе проведення
після курсів консолідації ще чотирьох курсів за схемою “5+1”: да-
унорубіцин — 45 мг/м2 на добу для хворих віком до 60 років та
30 мг/м2 на добу для пацієнтів віком понад 60 років в/в протягом
15 хв. у 1-й день, та цитозар — 100 мг/м2 на добу в тривалій в/в ін-
фузії або 100 мг/м2 двічі на добу протягом 1-годинної інфузії у
1—5-й дні.
У разі виявлення цитогенетичних або молекулярно-біологічних
змін у пухлинному клоні використовуються наступні алгоритми:
після досягнення ремісії хворим із транслокацією і (8:21), або ї
(16:16), або інверсією 16-ї хромосоми (група сприятливого прогно-
зу перебігу захворювання) проводити чотири курси цитозару у ви-
соких дозах (цитозар 3000 мг/м2, а у хворих, старших 60 років,
у доброму загальному стані та з нормальною функцією нирок і клі-
ренсом креатиніну понад 70 мл/хв. — 100—1500 мг/м2 3-годинною
92
| Гемобластози
інфузією кожні 12 год. двічі на добу в 1-й, 3-й та 5-й дні курсу.
Курси проводяться кожного 29-го дня після попереднього. Потім
хіміотерапія припиняється, і за хворими ведеться спостереження.
Пацієнтам із проміжним ступенем ризику, в яких виявляється
транслокація 1(9:11), +8 або нормальний каріотип, після досягнен-
ня ремісії рекомендовано проведення алогенної трансплантацій
кісткового мозку від сумісного спорідненого донора або аутологіч-
ної трансплантації кісткового мозку. У разі неможливості здійснен-
ня трансплантації проводиться 4 курси консолідації ремісії, як і
в групах сприятливого прогнозу.
Пацієнтам високого ризику, у котрих визначаються —7, —5,
7 ц—, 5ц— аномалії Пц23 за винятком транслокації і (9:11) та і
(9:22), або комплексний каріотип, після досягнення ремісії реко-
мендується алогенна трансплантація кісткового мозку від сумісно-
го спорідненого донора чи альтернативного донора.
У разі неможливості проведення трансплантації пацієнт вклю-
чається до стандартних протоколів лікування.
Лікування рефрактерних та рецидивів гострих мієлобластних
лейкемій проводиться за окремими протоколами із застосуванням
високих дох цитозару та флударабіну (Р1АО: флударабін 25 мл/м2
на добу в/в протягом 2-х год. з 1-го до 5 дня через 2 год., цитозар
200 мт/м2 в/в протягом 4-х год. з 1-го до 5 дня, нейпоген ЗООмг п/ш
з 0 до 5-го дня.
Хворому може бути проведено алогенну трансплантацію або
введення стовбурових клітин периферичної крові.
Лікування ГПмЛ
Індукція ремісії: даунорубіцин — 50-60 мг/м2 на добу для хво-
рих віком до 60 років та 30 мг/м2 на добу для хворих віком понад
60 років, або ідарубіцин — 12 мт/м2 на добу в/в протягом 15 хв. З
дні, та весаноїд 45 мг/м2 на добу до досягення повної ремісії. При
ініціальній лейкопенії рекомендується починати лікування в моно-
режимі для мінімалізації коагуологічних розладів, а хіміопрепарати
вводити з 3-4-го дня індукційного лікування.
Консолідація ремісії може проводитися даунорубіцином — по
50-60 мг/м2 на добу для хворих до 60 років та по 30 мг/м2 на добу
для хворих після 60 років, або ідарубіцином — по 12 мг/м2 на до-
бу в/в протягом 15 хв. З дні, або за схемою “7+3” два курси.
Лікування ГЛЛ
Лікування хворих групи стандартного ризику (ГЛЛ соттоп-ти-
пу, пре-В- варіант, Т-ГЛЛ, при кількості лейкоцитів менш ніж
93
Н.М. Третяк | Гематологія
ЗО • 109/л, відсутності транслокацій і (9:22), і (4:11). Фаза циторедук-
ції (при гіперлейкоцитозі > 100,0 ’ 109/л) протягом 7-ми днів до по-
чатку лікування: преднізолон — по 40 мл/м2 на добу рег 08 за три
прийоми в 1—7-й дні фази циторедукцїї, вінкристин — 2 мл/добу
в/в, струмінно у 1-й день фази циторедукції, аллопуринол — 500
мл/м2 рег оз у перші три дні, по 200 мл/м2 у наступні 4 дні. Індукція
ремісії (перша фаза) з 1-го до 28 дня: вінкристин — 2 мл/добу в/в,
струмінно у 1-й, 8-й, 15-й, 22-й дні; даунорубіцин — 45 мг/м2 на до-
бу (у хворих віком понад 35 років — 40 мг/м2 на добу в/в 15-хвилин-
ною інфузією у 1-й, 8-й, 15-й, 22-й дні); преднізолон — по 60 мл/м2
на добу рег оз за три прийоми в 1—28-й дні, з 29-го дня доза змен-
шується протягом тижня до повної відміни; Ь-аспарагіназа 5000
ОД/м2 на добу в/в 60-хвилинною в/в ін фузією, 7 введень кожного
другого дня з 15-го дня, або ПЕГ-аспарагіназа у дозі 1000 ОД/м2 на
15-й день одноразово; метотрексат 15 мл інтратекально в 1-й день.
Індукція ремісії (друга фаза) з 29-го до 56-го дня: циклофосфа-
мід — 650 мг/м2 на добу в/в 60-хвилинною інфузією у 29-й, 43-й і
57-й дні, цитозар — 75 мл/м2 на добу в/в у 32—35-й, 39—42-й, 46—
49-й та 53—56-й дні, метотрексат 15 мг + цитозар 40 мг + декса-
метазон 4 мг інтратекально у 29-й, 36-й, 43-й, 50-й дні. Профілак-
тика нейролейкемії при неможливості інтратекального введення
цитостатиків на 9-12-й тиждень: опромінення голови в дозі 24 Гр.
Опромінення середостіння на 9-12-й тиждень (при його ураженні
у хворих Т—ГЛЛ) у дозі 24 Гр.
Консолідація ремісії на 13-й та 17—й тижні: етопозид —
100 мг/м2 на добу в/в 60-хвилинною інфузією та цитозар — 75 мг/м2
на добу в/в у 1—5-й дні, дексаметазон — 4 мг інтратекально у 1-й
день тижня. Реіндукція ремісії (перша фаза) на 21-24 тиждень: він-
кристин — 2 мг/м2 на добу в/в, струмінно у 1-й, 8-й, 15-й, 22-й дні
курсу, доксорубіцин — 25 мг/м2 на добу (у хворих, старших 35 ро-
ків, — 20 мг/м2 на добу) в/в 30-хвилинною інфузією у 1-й, 8-й,
15-й, 22-й дні курсу, преднізолон — 60 мг/м2 на добу рег 08 за
З прийоми з 1-го до 28-го днів курсу, з 29-го дня доза препарату
зменшується протягом тижня до повної відміни, метотрексат
15 мг+ цитозар 40 мг + дексаметазон 4 мг інтратекально у 1-й та
28-й дні курсу. Реіндукція ремісії (друга фаза) на 25-26 тиждень:
циклофосфамід — 650 мг/м2 на добу в/в 60-хвилинною інфузією у
29-й день курсу, цитозар — 75 мг/м2 на добу в/в 60-хвилинною ін-
фузією у 31—34-й, 38—41-й дні курсу, тіогуанін — 60 мг/м2 на до-
бу рег 08 з 29-го до 42-го дня.
94
| Гемобластози
Консолідація ремісії на 31 та 35 тижні: етопозид — 100 мг/м2
на добу в/в 60-хвилинною інфузією у 1—5-й дні, цитозар —
75 мг/м2 на добу в/в 60-хвилинною інфузією у 1—5-й дні, метот-
рексат 15 мг + цитозар 40 мг + дексаметазон 4 мг інтратекально у
1-й день тижня. Підтримка ремісії (до 142 тижнів від початку ліку-
вання): 6-меркаптопурин — 60 мг/м2 на добу рег 08 щодня, метот-
рексат — 20 мг/м2 на добу в/в або рег оз 1 раз на тиждень. Профі-
лактика нейролейкемїї: метотрексат 15 мг + цитозар 40 мг +
дексаметазон 4 мг інтратекально кожні 2 місяці.
Повна ремісія консолідується при нормальному співвідношен-
ні всіх паростків кровотворення, відсутності екстрамедулярих ура-
жень та при вмісті 5% і менше бластних клітин у аспіраті кістко-
вого мозку; кількості нейтрофілів у периферичній крові 1,5 • 109/л,
а тромбоцитів — 100,0 • 109/л та більше.
Рефракторна форма захворювання діагностується за відсутнос-
ті повної ремісії після двох фаз індукційної терапії.
Рецвдив діагностується при вмісті у кісткового мозку більш
ніж 5% бластних клітин та/або появі вогнища екстрамедулярного
росту.
У лікуванні хворих групи високого ризику (ранній пре-В-варі-
ант, білінійна ГЛЛ, ранні Т—ГЛЛ, при кількості лейкоцитів біль-
ше 30 • 109/л, наявності транслокації і (9:22), і (4:11), резистентних
форм та ранніх (до 6 місяців) рецидивів захворювання використо-
вують високі дози цитозару та метотрексату чи етопозиду або ви-
сокі дози метотрексату і аспарагіназу.
До таких програм належить Нурег-СУАВ (перша фаза): цикло-
фосфамід — 300 мг/м2 двічі на добу в/в протягом 3-х годин у 1—3-й
дні курсу; доксорубіцин — 25 мг/м2 на добу в/в протягом 24 год., че-
рез 12 год. після останнього введення циклофосфану (у 4—5-й дні
курсу); вінкристин — 1,4 мг/м2 (не більше 2 мг) в/в, струмінно в 4-й
та 11-й дні курсу; дексаметазон — 40 мг/день у 1—4-й та 11—14-й
дні курсу; цитозар — 75 мг/м2 на добу в/в протягом 60 хв. у 31—34-й,
38—41-й дні курсу.
Друга фаза проводиться через 21 день після першої: метотрек-
сат — 1000 мг/м2 на добу в/в протягом 24 год. у 1—й день; цито-
зар — 3000 мг/м2 протягом двох годин, кожні 12 год. у 2-й та 3-й
дні; лейковорин — 50 мл рег оз після зкінчення введення метотрек-
сату. Надалі лейковорин — по 25 мг рег оз кожні 6 год. протягом
48 год. Усього проводиться 4 курси з двох фаз.
95
Н.М. Третяк | Гематологія
При виявленні транслокації і (9:22) до лікування включають
глівек - по 400 мг/добу протягом 15 днів із кожною фазою програ-
ми Нурсг-СУАЕ).
Профілактика нейролейкемії проводиться при всіх формах
ГНЛ з ініціальним лейкоцитозом понад ЗО - 109/л, а також при мі-
єломонобластній, монобластній лейкеміях та гострій промієлоци-
тарній лейкемії, шо лікується препаратами ретиноєвої кислоти.
Перша діагностична пункція виконується до початку курсу ін-
дукції або в перші його дні до розвитку цитопенії. При неможли-
вості зробити пункцію в ці строки у зв’язку з глибокою цитопені-
єю (лейкоцитів < 1,0 • 109/л, тромбоцитів < 20-109/л), вона
виконується після курсу терапії та нормалізації показників перифе-
ричної крові.
При появі менінгеальних симптомів або виявленні у лікворі
бластного цитозу (більш ніж 10) діагностується нейролейкемія та
проводяться люмбальні пункції із введенням цитарабіну, метотрек-
сату, преднізолону. Після нормалізації цитозу потрібно зробити
щонайменше три пункції та надалі проводити профілактичні вве-
дення препаратів 1 раз на 2 місяці протягом усього часу лікування.
У разі появи ознак прогресування нейролейкемії або, якщо
після 8-10 люмбальних пункцій зберігається цитоз, необхідне про-
ведення краніального опромінення, котре може здійснюватись і
при нейтропенії (лейкоцити 1-3,0- 109/л).
Профілактика нейролейкемії у хворих на ГЛЛ — необхідний
захід у лікуванні. При невиконанні профілактики у 40-60% хворих
розвивається ураження центральної нервової системи.
Перша діагностична люмбальна пункція проводиться в пер-
ший день терапії, і зазвичай вводиться один метотрексат у дозі
12,5мг/м2 для уникнення розвитку менінгізму. Згодом вводяться
три препарати — метотрексат, цитарабін, преднізолон.
У період першої фази індукції ремісії виконується 5 люмбаль-
них пункцій з огляду на те, шо рання профілактика нейролейкемії
(в перший місяць лікування) є найефективнішою, та й імовірність
глибокої цитопенії менша, ніж у другій фазі лікування.
Профілактичне проведення люмбальних пункцій із викорис-
танням трьох препаратів надалі здійснюється раз на 3 місяці про-
тягом усього періоду лікування (до трьох років).
Супровідна терапія гострих лейкемій
У досягненні та утриманні позитивних результатів лікуван-
ня ГЛ важливу роль віліірає вчасно застосована та адекватна
96
| Гемобластози
супровідна терапія, шо включає боротьбу з інфекційними та ге-
морагічними ускладненнями, трансфузійну та детоксикаційну
терапію.
Інфекційні ускладнення у хворих на ГЛ на висоті цитостатич-
ної мієлосупресїї можуть бути спровоковані як бактеріальними, так
і мікотичними та вірусними чинниками. Для лікування бактеріаль-
них ускладнень необхідно використовувати антибіотики широкого
спектру дії: цефепім (максипім), лейкоплакін (таргоцид), амікоцин
(амікін), цефтазидім (фортум), уназин (сультаміцилін) з урахуван-
ням чутливості мікрофлори до препаратів.
Лікування вірусної інфекції проводиться відповідно до існую-
чих стандартів із використанням таких препаратів, як ацикловір
(зовіракс), валоцикловір, ганцикловір (цимевен).
Мікотичні ураження слизових оболонок або генералізовані мі-
котичні процеси, аж до виникнення сепсису, слід лікувати флюко-
назолом (дифлюканом), амфотеріцином В.
У боротьбі з геморагічним синдромом (кількість тромбоцитів <
ЗО • 109/л) застосовують трансфузії тромбоконцентрату, рекомбі-
нантний активований препарат фактор VII (ІЧоуосєуєп) (викорис-
товується при кровотечах, що загрожують життю хворого), е-амі-
нокапронову кислоту, діцинон. У випадку розвинення
ДВЗ-синдрому, особливо при ГПмЛ, одночасно із хіміотерапією,
препаратами ретиноєвої кислоти призначають гепарин — по 1,5-
3,0 мг/кг на добу, та свіжозаморожену плазму.
Замісна трансфузійна терапія включає трансфузії еритроцитів
при зниженні показників гемоглобіну < 80 г/л.
Найефективнішими дезінтоксикаційними засобами є неодез,
реополіглюкін, розчин альбуміну, 20%-й розчин сорбиту, 15%-й
розчин маніту.
Прогноз. Використання сучасних стандартних програм хіміоте-
рапії та адекватного супровідного лікування забезпечує, за останні-
ми даними, у 30-40% хворих на ГЛЛ та у 15-20% хворих на ГНЛ
п’ятирічне безрецидивне виживання.
ХРОНІЧНІ МІЄЛОПРОЛІФЕРАТИВНІ ЗАХВОРЮВАННЯ
До групи хронічних мієлопроліферативних лейкемій належать
захворювання, шо виникають на рівні ранніх попередників мієло-
поезу, всі нащадки котрих (гранулоцитарний ряд) походять із пух-
линного клону.
97
Н.М. Третяк | Гематологія
Основними нозологічними формами лейкемій цієї групи є
наступні: хронічна мієлоєдна лейкемія, ідіопатичний мієлофіброз
(сублейкемічна мієлолейкемія, остеомієлофіброз), еритремія
справжня (поліцитемія), хронічна моноцитарна лейкемія, есенці-
альна тромбоцитемія.
Хронічна мієлоїдна лейкемія
Хронічна мієлоїдна лейкемія (ХМЛ) — це мієлопроліферативне
захворювання, що виникає в результаті лейкемічної трансформації
поліпотентної стовбурової клітини. ХМЛ — єдине захворювання
системи крові, при котрому встановлена специфічна хромосомна
аномалія (філадельфійська хромосома) — транслокація 9;22. Унас-
лідок транслокацїї утворюється химерний ген Ьсг-аЬІ, що кодує
специфічний білок р-210, який має тирозинкиназну активність. У
95% випадків ХМЛ виявляється цей химерний ген, котрий, як вва-
жається на сьогодні, разом із білком р-210 відіграє провідну роль
у патогенезі захворювання.
Епідеміологія. Захворюваність на ХМЛ — 1-1,5 випадки на
100 тис. населення та становить від 7% до 15% серед усіх лейкемій
дорослих. Чоловіки хворіють частіше за жінок. Пік захворюванос-
ті припадає на вік 40-50 років. Особи, молодші за 20 років, хворі-
ють дуже рідко.
Патогенез. Захворювання проходить дві фази — доброякісну
(моноклонову) та термінальну злоякісну (поліклонову).
За клінічним перебігом ХМЛ виділяються:
— стадія хронічна: рання, пізня;
— стадія акселерації;
— стадія термінальна (бластна криза).
Рання хронічна стадія ХМЛ виявляється надзвичайно рідко
і випадково, іноді при дослідженні периферичної крові з іншої
причини, за умови уважного ставлення лікаря-терапевта, шо звер-
тає увагу на найменші зрушення в аналізі крові. (Власне спостере-
ження — діагноз верифікований лише цитогенетичним досліджен-
ням).
Пізня хронічна стадія ХМЛ — це період розгорнутих клініко-ге-
матологічних проявів.
Фаза акселерації — перехід у термінальну.
Термінальна стадія ХМЛ (стадія бластної кризи) — поліклоно-
ва трансформація.
98
| Гемобластози
Хронічна стадія є моноклоновою й характеризується тирозин-
киназною активністю, котра призводить до вираженої гіперпролі-
ферації гранулоцитів із порушенням їх дозрівання та активацією
проліферації мегакаріоцитарної ланки кровотворення. Тривалість
моноклонової стадії дуже варіабельна і під впливом інтерфероно-
терапії може сягати 12-15 років.
Фаза акселерації характеризується утворенням нових хромо-
сомних аберацій у стовбурових клітинах. Крім транслокації 9;22
виявляються трисомія 8, 19, ізохромосома 17, унаслідок чого роз-
вивається блок диференціювання бластних клітин та зростання їх-
ньої кількості. Такі цитогенетичні зрушення свідчать, що захворю-
вання перейшло в поліклонову стадію та набуло термінального
злоякісного характеру.
Клініко-гематологічна картина. Захворювання на ранній хро-
нічній стадії виявити практично ніколи не вдається.
Першими клінічними ознаками хронічної стадії ХМЛ є відчуття
тяжкості та іноді періодичний біль у лівому підребер’ї; пітливість,
нездужання, втомлюваність. При об’єктивному обстеженні привертає
увагу спленомегалія різного ступеня вираженості. Іноді збільшення
селезінки виявляється лише з допомогою УЗД або комп’ютерної то-
мографії і протягом багатьох місяців пальпаторно воно не визнача-
ється. Спленомегалію зумовлює мієлоїдна метаплазія в селезінці.
Для периферичної крові в цей період захворювання характер-
ний нейтрофільний лейкоцитоз зі зрушенням до поодиноких міє-
лоцитів та промієлоцитів. В окремих випадках спостерігаються по-
одинокі бластні клітини, але без атипових ознак. Вміст еритроцитів
та концентрація гемоглобіну тривалий час залишаються майже
незмінними. У більшості хворих кількість тромбоцитів утримуєть-
ся в межах норми, в 10-15% пацієнтів спостерігається тромбоцито-
пенія, а в 20-30% випадків виявляється тромбоцитоз та гіпертром-
боцитоз, котрий може сягати 1500-2000 • 109/л та більше.
Кістковий мозок у хронічній стадії багатий на клітинні елемен-
ти, але морфологія клітин у цей період захворювання не відрізняєть-
ся від норми. Проте при підрахунку формули крові виявляється омо-
лодження гранулоцитарного ряду, часто спостерігається підвищений
відсоток базофілів або еозинофілів та “базофільно-еозинофільна
асоціація” (одночасне збільшення вмісту базофілів і еозинофілів).
Патологічними ознаками гранулоцитів кісткового мозку в цей
період захворювання є порушення кількості або відсутність азуро-
фільних та специфічних гранул, зниження вмісту мієлопероксидази.
99
Н.М. Третяк | Гематологія
Специфічною ознакою для ХМЛ є зниження, аж до повної
відсутності, лужної фосфатази в зрілих нейтрофілах, що може ви-
користовуватися при диференційному діагнозі з іншими мієлопро-
ліферативними захворюваннями (ІМ, ЕТ) та лейкемоїдною реакці-
єю нейтрофільного типу.
У хронічній стадії ХМЛ при питогенетичному дослідженні
в кістковому мозку виявляється 85-95% Рй-позитивних клітин.
При гістологічному дослідженні кісткового мозку виявляється
майже повне витіснення жиру гранулоцитарними клітинами, акти-
візація мегакаріоцитарного паростка.
Астенічний синдром у період хронічної стадії ХМЛ зумовле-
ний посиленим розпадом клітин, котрий іноді супроводжується
підвищеним вмістом сечової кислоти в крові (гіперурікемія) з ут-
воренням каменів у нирках та печінці або гістамінемією (продуку-
ється гранулоцитами).
Якщо не проводиться або проводиться неадекватна терапія,
патологічний процес прогресує й переходить у проміжну стадію ак-
селерації. Ця стадія характеризується поступовим збільшенням се-
лезінки, зростанням кількості лейкоцитів, підвищенням відсотка
бластів, промієлоцитів і мієлоцитів у лейкограмі. Цитостатичні
препарати, що використовувались, втрачають ефективність. Зміна
терапевтичної тактики може на короткий час повернути перебіг
хвороби в хронічну стадію.
При цитогенетичному дослідженні виявляється, що Рй-гюзи-
тивні клітини набувають анеуплоїдних ознак, утворюючи 2-3 одна-
кових типи анеуплоїдії; пухлина набуває поліклонового злоякісно-
го характеру, і захворювання переходить у термінальну фазу —
стадію бластної кризи.
У периферичній крові в цій стадії спостерігається нормоцитар-
на анемія, поява у значній кількості ядерних еритроїдних клітин,
наростання лейкоцитозу, тромбоцитопенія. У лейкограмі — поява
більш як 5% бластних клітин, понад 20% базофілів. У кістковому
мозку — подальша гіперплазія гранулоцитарного та мегакаріоци-
тарного паростків, зростання кількості бластних клітин, звуження
еритроідного паростка.
Клінічно бластна криза проявляється немотивованою гаряч-
кою, болями у трубчастих кістках, швидким ростом розмірів селе-
зінки (часто з інфарктами в ній) та печінки, утворенням лейкемі-
дів у шкірі. В окремих випадках розвиваються вогнища саркомного
росту в лімфатичних вузлах. Ці прояви хвороби пов’язані з виник-
100
| Гемобластози
ненням нових мутантних субклонів, шо не здатні до диференцію-
вання, а лише активно проліферують, витісняючи попередньо іс-
нуючий клон клітин.
Останніми роками, внаслідок досить високої ефективності те-
рапії, що застосовується, та подовження термінів життя хворих
у термінальній стадії, почастішали випадки нейролейкемії, котра за
ознаками подібна до нейролейкемії при гострих лейкеміях.
У стадії бластної кризи в периферичній крові поглиблюються
анемія та тромбоцитопенія, зростає лейкоцитоз із підвищенням
відсотка бластних клітин та промієлоцитів і зниженням кількості
дозрілих гранулоцитів.
У кістковому мозку вміст, бластних клітин та мієлоцитів пере-
вищує 50% усіх ядерних елементів. За морфологічними ознаками
бласти в стадії бластної кризи відрізняються від клітин попередніх
стадій — з’являються атипові форми із широкою цитоплазмою та
неправильними контурами ядра й цитоплазми.
Цитохімічне дослідження дає змогу ідентифікувати бластні клі-
тини, котрі формують бластну кризу: переважно це мієлобласти
або лімфобласти, але можуть бути мієломонобласти, монобласти,
еритробласти, мегакаріобласти. В залежності від морфологічної та
цитохімічної ідентифікації бластів визначається форма бластної
кризи, шо впливає на тактику лікування.
У теперішній час програма лікування хворих на ХМЛ визнача-
ється з урахуванням критеріїв прогнозу та факторів ризику.
Як критерій прогнозу використовують модель Н.Капгаціап
(1990 р.), відповідно до якої пацієнти поділяються на три групи ри-
зику трансформації захворювання у прогресуючу чи термінальну
(бластну) стадії.
Ознаки несприятливого прогнозу:
— вік 60 років та більше;
— бласти в периферичній крові — 3% та більше;
— бласти в кістковому мозку — 5% та більше;
— базофіли в периферичній крові — 7% та більше або в кіс-
тковому мозку — 3% та більше;
— тромбоцити в периферичній крові — 700- 109/лта більше;
— спленомегалія (селезінка виступає на 10 см та більше з-під
реберної дуги).
І група — низького ризику, має 0-1 фактор ризику;
II група — проміжного ризику, має два фактори ризику;
НІ група - високого ризику, має три та більше факторів ризику.
101
Н.М. Третяк | Гематологія
Залежно від належності хворого до відповідної групи ризику
визначається тактика лікування. Пацієнти групи високого ризику
підлягають трансплантації кісткового мозку або периферичних
стовбурових клітин, при неможливості виконання цих заходів не-
обхідна активна хіміотерапія.
Глибоке вивчення патогенезу ХМЛ зумовило розробку нових
підходів до медикаментозного впливу на патологічний процес.
Опрацьовані та загальновизнані критерії визначення стадії
ХМЛ (Сиііііоі Е., 1999), застосування яких у клінічній практиці оп-
тимізує результати лікування.
Прогресуюча фаза:
— персистуюча або прогресуюча спленомегалія;
— резистентність до попередньої терапії (підвищення кількос-
ті лейкоцитів);
— активація фіброзоутворення в кістковому мозку;
— додаткові цитогенетичні аномалії;
— анемія та/або тромбоцитопенія, не пов’язані з попередньою
терапією;
— виражений тромбоцитоз;
— персистуюча гарячка;
— базофіли в периферичній крові > 20%;
— бласти та промієлоцити в периферичній крові > 30%;
— бласти в периферичній крові > 15%.
Бластна криза (термінальна стадія):
— бласти в периферичній крові > 20%;
— бласти в кістковому мозку > 30%;
— екстрамедулярні інфільтрати, що складаються із бластних
клітин.
Крім того, в гематологічних клініках світу для визначення прог-
ностичного індексу послуговуються формулою за 8ока1 (8окаі-іпс1ех,
81) та шкалою, запропонованою Комітетом з прогностичних факторів
при ХМЛ, котрі рівноцінно об’єктивно відображають стан хворого.
Формула Бокаї-ііиіех (51)
81 = ехр [0,0116(вік—43,4)4-0,0345 (РС-7,51 )4-0,188[(Тр:700)2-
О,563|4-О,887(БПК-2,1О)|.
РС — розміри селезінки (на скільки см виступає з-під краю ре-
берної дуги);
БПК — бласти в периферичній крові, у %;
ехр — стабільне число 2,718, яке підноситься до ступеня числа
у фігурних дужках.
102
| Гемобластози
За результатами підрахунку за цією формулою виділяють З
прогностичні групи:
1) зі сприятливим прогнозом (81 < 0,8);
2) із проміжним прогнозом (81 у межах 0,8-1,2);
3) з несприятливим прогнозом (81 >1,2).
Прогностична модель Комітету з прогностичних факторів при
ХМЛ
(0,6666 - ІВ + 0,0420 • РС + 0,0584 • БК + 0,0413 • ЕК + 0,2039 • ІБ +
+ 1,0956- ІТ) • 100
ІВ — індекс віку = 0 — при < 50, 1 > 50;
РС — розміри селезінки, в см, нижче реберної дуги;
БК — відсоток бластів у крові;
ЕК — відсоток еозинофілів у крові;
ІБ — індекс базофілів = 0 при < 3% і 1 — у інших випадках;
ІТ — індекс тромбоцитів = 0 при Гр. < 1500 г/л і 1 — у інших
випадках.
< 780 — сприятливий прогноз;
< 1480 — проміжний прогноз;
> 1480 — несприятливий прогноз.
Діагноз та диференційний діагноз. Діагноз ХМЛ ставиться на
підставі клінічних (гепато- та спленомегалія) і гематологічних да-
них. У периферичній крові виявляється нейтрофільний лейкоцитоз
зі зрушенням вліво, до мієлоцитів, промієлоцитів, базофільно-ео-
зинофільна асоціація, анемія, гіпертромбоцитоз або тромбоцитопе-
нія. Цитохімічне дослідження клітин периферичної крові та кістко-
вого мозку підтверджує діагноз (зниження або відсутність лужної
фосфатази). Імунофенотипування відіграє визначальну роль при
диференціюванні типу бластної кризи ХМЛ. Наявність клітин із
РЬ-хромосомою, виявлених при цитогенетичному дослідженні пе-
риферичної крові та кісткового мозку, є незаперечним аргументом
на користь ХМЛ. Захворювання слід диференціювати з лейкемоїд-
ними реакціями мієлоїдного типу, котрі можуть бути при інфек-
ційних захворюваннях (пневмонія, сепсіс, туберкульоз та ін.), зло-
якісних пухлинах різної локалізації.
Лікування. Тактика лікування ХМЛ на сьогодні має два нап-
рямки — паліативна терапія (мієлосан, гідроксисечовина), завдан-
ням котрої є контроль мієлоїдної гіперплазії, відстрочення розвит-
ку бластної кризи і, водночас, зменшення клінічних проявів
захворювання з поліпшенням якості життя пацієнта, та радикаль-
на терапія (інтерферон, ало- та автотрансплантація кісткового моз-
103
Н.М. Третяк | Гематологія
ку або стовбурових гемопоетичних клітин, іматініб), метою якої є
повна ерадикація лейкемічного клону та досягнення повного оду-
жання хворого.
Вибір методу лікування залежить від стадії захворювання, віку
пацієнта, прогностичної групи, наявності гістосумісного родинно-
го донора.
Лікування ранньої хронічної стадії ХМЛ починається відразу
після встановлення діагнозу. За наявності гістосумісного родин-
ного донора хворому пропонується алогенна трансплантація кіс-
ткового мозку, котра забезпечує 5-річну виживаність у 60-70% ви-
падків.
При відсутності донора курс лікування пацієнтів, молодших за
50 років, починають гідроксисечовиною, котра впливає на пролі-
феруючі мієлоїдні клітини, з розрахунку 40 мг/кг на добу та редук-
цією дози наполовину при зниженні рівня лейкоцитів до 20,09/л і
нижче. Доза препарату індивідуалізується для підтримання лейко-
цитів у межах норми.
Інтерферони — це, в основному, препарати генно-інженерно-
го походження, які випускаються різними фармакологічними фір-
мами під торговими назвами: інтрон-А, роферон-А, реаферон, ла-
ферон, еберон та ін.
Механізм дії препаратів інтерферону остаточно ше не вивче-
ний, але клінічна ефективність їх при онкозахворюваннях пов’язу-
ється із:
— антипроліферативною дією внаслідок пригнічення ростових
генів у злоякісних клітинах;
— індукцією клітинного диференціювання;
— імунною модуляцією шляхом активування макрофагів, при-
родних кілерів та цитотоксичних ефективних клітин;
— посиленням експресії поверхневих антигенів клітинних
мембран;
— посиленням регуляторного впливу кістковомозкового мік-
рооточення, оскільки інтерферон-а поновлює нормальний пригні-
чуючий вплив стромальних клітин на проліферацію клітин-попе-
редниць мієлоїдного ряду та посилює адгезію їх до стромальних
клітин.
Застосування інтерферону дає змогу досягти не тільки клініко-
гематологічної, а й цитогенетичної ремісії.
При неможливості застосування гідроксисечовини та інтерфе-
рону призначають лікування мієлосаном — Росія (бусульфан —
104
| Гемобластози
США, мілеран — Великобританія). Цей препарат при лікуванні
хронічної стадії ХМЛ використовується вже понад ЗО років і довів
свою досить високу ефективність (до 98% клініко-гематологічних
ремісій) та зручність для амбулаторного використання. Мієлосан
впливає, головним чином, на стовбурові кровотворні клітини,
з меншою активністю — на клітини-попередниці окремих парос-
тків кровотворення та майже зовсім не діє на швидко проліферу-
ючі клітини. Наслідком такого механізму дії є підвищення кількос-
ті лейкоцитів, іноді в 2-3 рази, через 10-12 днів вживання хворим
загальновизнаної терапевтичної дози мієлосану — 4-6 мг/добу.
Зниження вмісту лейкоцитів (в основному поступове, рідко швид-
ке) відбувається за 3-4 тижні безперервного прийому мієлосану.
Мієлосан призначається внутрішньо, доза його визначається
кількістю лейкоцитів та розмірами селезінки:
— при лейкоцитозі 110-150 • 1()9/л — 8 мг/добу;
— при лейкоцитозі 90-100 • 109/л — 8-6 мг/добу;
— при лейкоцитозі 60-80 • 109/л — 6 мг/добу;
— при лейкоцитозі 30-50 • 109/л — 4 мг/добу;
— при лейкоцитозі 15-20 • 109/ — 2 мг/добу;
— при лейкоцитозі 15-10- 109/л — 2 мг/добу 2-3 рази на тиж-
день.
По досягненні клініко-гематологічної ремісії проводять під-
тримуюче лікування (переривчасто-підтримуюче) — 2-4 мг 1-3 ра-
зи на 7-10 днів.
Курсова доза становить 250-300 мг. Клініко-гематологічна ре-
місія при лікуванні мієлосаном триває в середньому 33-35 місяців.
Цитогенетичної ремісії досягти не вдається.
Х’юстонські критерії ремісії
Ггматологічна ремісія:
— стабілізація кількості лейкоцитів периферичної крові на рів-
ні, меншому за 10 • 109/л, та нормалізація лейкоцитарної формули
— відсутність бластів та промієлоцитів.
— кількість тромбоцитів менше 450 • 109/л.
— відсутність усіх клінічних ознак захворювання, в тому числі
й спленомегалії.
Цшпогенетична ремісія:
— відсутність ремісії — РЬ+-хромосома визначається у всіх (не
менше 20) метафазах;
— мінімальна ремісія — Різ-позитивні метафази спостерігають-
ся більш ніж у 35%, але менш ніж у 100%;
105
Н.М. Третяк | Гематологія
— парциальна ремісія — Різ-позитивні метафази знайдені
у менш ніж 35% клітин, шо діляться;
— повна ремісія — повна елімінація клітин із Р1і+-хромосо-
мою;
— велика ремісія — повна або часткова відсутність Рїґ-нега-
тивнх клітин > 65%.
Найсерйознішим ускладненням мієлосанотерапії є розвиток
аплазії кровотворення. При застосуванні доз, вищих від терапев-
тичних (до 14 мг/добу), описані смертельні випадки. При тривало-
му використанні мієлосану у 70-100% жінок розвивається амено-
рея. Пігментація шкіри з’являється майже у 1/3 хворих на другий
рік лікування мієлосаном. Курсове та підтримуюче лікування міє-
лосаном призводить до ушкодження клітин епітелію більшості ор-
ганів, особливо страждають бронхи, легеневі альвеоли, внаслідок
чого розвивається фіброз легеневої тканини. Клінічно це проявля-
ється сухим кашлем, субфебрильною температурою, надалі — ци-
анозом та задишкою. При аускультації вислуховуються ділянки
крепітації, рентгенологічна картина подібна до дрібновогнишевої
пневмонії. Діагноз уточнюється лише при морфологічному дослід-
женні легеневої тканини.
Монотерапія у період хронічної фази ХМЛ досить швидко
призводить до клініко-гематологічної ремісії та сприяє її утриман-
ню. При лікуванні мієлосаном ремісія зі збереженням хворими
працездатності триває в середньому до 33 місяців, при лікуванні
гідроксисечовиною — до 41-56 місяців. При лікуванні із застосу-
ванням препаратів інтерферону медіана виживання хворих стано-
вить 61-89 місяців. Проте, незважаючи на оптимістичні результа-
ти, що досягаються при лікуванні хронічної стадії ХМЛ, це не
запобігає прогресуванню патологічного процесу і переходить
у прогресуючу фазу захворювання. Основною ознакою прогресую-
чої стадії (фази) є втрата чутливості до попередньої терапії, зрос-
тання кількості лейкоцитів, бластних клітин, еозинофілів, базофі-
лів у периферичній крові, збільшення розмірів селезінки у частини
хворих, підвищення температури тіла.
Метою лікування фази акселерації (прогресуючої стадії) ХМЛ
є віддалення моменту переходу захворювання в стадію бластної
кризи.
При резистентності до мієлосану та гідроксисечовини застосо-
вується 6-меркаптопурин - по 100 мг щодня внутрішньо, або тіо-
гуанін — по 80 мг щодня внутрішньо. Ці препарати ефективні
106
| Гемобластози
в поєднанні з мієлосаном або гідроксисечовиною. У цей період
захворювання ефективним є лікування малими дозами цитозару
(ара-С) — по 10-15 мг/м2 двічі на добу під шкіру протягом 10 днів
щомісяця, 4-6 місяців або протягом 20 днів з перервою у 20 днів —
2 курси. За потреби такі курси повторюються.
Лікування ХМЛ у стадії бластної кризи принципово відрізня-
ється від терапії попередніх стадій захворювання і розраховане на
знищення бластних клітин, кількість котрих швидко зростає. Ста-
дія бластної кризи подібна до гострої лейкемії з відповідним цито-
хімічним спрямуванням, тому й хіміопрепарати для лікування ви-
користовуються ідентичні.
Найчастіше для лікування бластної кризи мієлоїдного варіанту
використовують традиційні комбінації антрациклінових антибіоти-
ків та цитозин-арабінозиду за схемою “5+2”: доксорубіцин — 50
мг/м2 у перші дві доби лікування та цитозин-арабінозид — 150
мг/м2 двічі на лобу протягом п’яти днів. При недостатній ефектив-
ності цієї програми проводять лікування цими ж препаратами за
схемою “7+3”. Може бути виконана програма “7+3” із заміною
доксорубіцину етопозидом, мітбксантроном, ідарубіцином. З висо-
ким позитивним ефектом використовується програма ХТ, що
включає карбоплатин: 1—3-й день — карбоплатин по 150 мг/м2 на
добу безперервною інфузією, етопозид по 100 мг/м2 на добу одно-
годинною інфузією, цитозар по 500 мг/м2 кожні 12 год. одногодин-
ною інфузією. Такий курс повторюється на 8-10-й дні. На 14-й
день за кістковим мозком оцінюється стан гемопоезу, й надалі
проводиться ало- або автотрансплантація кісткового мозку. Така
програма забезпечує досягнення 70% ремісії.
Доволі ефективними при лікуванні лімфобластної бластної
кризи є застосування таких програм, як СОР+КиЬ (циклофосфан
— 400 мг/м2 в/в 1—5 день; вінкристин — 1,4 мг/м2 в/в 1 день, ру-
боміцин — 40 мг/м2 в/в 1 день, преднізолон — 40 мг/м2 р. о. 1—5
день); КАР (цитозар — 100 мг/м2 в/в 1—8 день, рубоміпин — 40
мг/м2 в/в 1—3 день, преднізолон — 40 мг/м2 р. о. 1—8 день); УАР
(вінкристин — 2 мг/м2 в/в 1, 8-й день, цитозар — 100 мг/м2 в/в 1 —
8 день, преднізолон — 40 мг/м2 р. о., в/м, в/в 1—8 день); УКР (він-
кристин — 2 мг/м2 в/в 1, 8-й день, рубоміпин — 40 мг/м2 в/в 2—4
день, преднізолон — 40 мг/м2 р. о., в/м, в/в 1—8 день).
У лікуванні лімфобластної бластної кризи ХМЛ можуть бути
використані й більш активні 15-денні курси ХТ. Найефективніши-
ми із них є:
107
Н.М. Третяк | Гематологія
УКР (15-денний варіант):
вінкристин — 2 мг в/в, струмінно у І, 8, 15-й дні;
рубоміцин — 40 мг/м2 в/в 3, 4, 5-й дні;
преднізолон — 40 мг/м2 р. о. 1 —15-й дні.
УРР + Ь-аспарагіназа:
вінкристин — 2 мг в/в, струмінно у 1, 8, 15-й дні;
рубоміцин — 40 мг/м2 в/в, крапельно 3, 4, 5-й дні;
Б-аспарагіназа — 5000 од/м2 в/в, крапельно 1 —15-й дні;
преднізолон — 40 мг/м2 р. о. 1 — 15-й дні.
СОАР:
циклофосфан — 50 мг/м2 кожні 8 год. в/в 4 дні;
вінкристин — 2 мг в/в 1-й день;
цитозар — 50 мг/м2 струмінно кожні 8 год.;
преднізолон — 60 мг/м2 р. о. 1—4-й дні.
СНОР:
циклофосфан — 750 мг/м2 в/в 1-й день;
вінкристин — 2 мг в/в 1-й день;
рубоміцин — 50 мг/м2 в/в, крапельно кожні 8 год.;
преднізолон — 60 мг/м2 р. о. 1—4-й дні.
Позитивна відповідь на ХТ при лікуванні бластної кризи мож-
лива за умови, шо воно розпочато з перших днів появи її ознак кри-
зи. Прагнення досягти ремісії моноХТ значно погіршує результати
подальшої ПХТ. Неефективність такої ПХТ зумовлена розвинен-
ням опірності пухлинних клітин до цитостатичних препаратів.
Нова ера в лікуванні ХМЛ почалася з винаходом та розробкою
програм застосування в клінічній практиці такого препарату, як
глівек (іматініб, мезилат, 8ТІ571 — §І£па1 ІгапзШсіог іп^іЬіїот — ін-
гібітор шляхів передачі сигналів). Механізм дії глівеку ґрунтується
на його сполученні з активними центрами ВСВ-АВІ. тирозинки-
нази (білка р210). Це порушує процеси взаємодії субстратів усере-
дині клітин, котрі містять білок р210 (переважно Ріі-позитивні клі-
тини), що призводить до загибелі останніх.
Багатоцентрові міжнародні дослідження довели ефективність
глівеку у пацієнтів у хронічній стадії ХМЛ, що резистентні до те-
рапії інтерфероном. Повні клініко-гематологічні ремісії через 2-4
тижні від початку лікування досягнуті у 91% хворих, котрі отриму-
вали глівек по 400 мл на добу. Велику цитогенетичну відповідь
(Рїґ-клітин у кістковому мозку < 35%) та повну цитогенетичну
відповідь (Р1і+-клітин у кістковому мозку — 0%) через 6 місяців лі-
кування було досягнуто у 61% та 21% хворих відповідно.
108
| Гемобластози
Клініко-гематологічної ремісії досягнуто у 44% пацієнтів у фа-
зі акселерації, велика цитогенетична відповідь констатована у 20%
хворих та у 14% повна цитогенетична відповідь — цитогенетична
ремісія.
Результати лікування глівеком хворих у бластній кризі значно
гірші. Клініко-гематологічного покращання вдалося досягти у 51%,
воно зберігалося лише 4 тижні. Цитогенетична відповідь зафіксо-
вана у 26% пацієнтів, із них велика — у 13,5%, повна — у 5%.
Хворі з мієлоїдним варіантом бластної кризи позитивну кліні-
ко-гематологічну відповідь дають у 50% випадків.
Набутий у світі досвід лікування глівеком ХМЛ на різних ста-
діях перебігу свідчить про збільшення частоти як клініко-гематоло-
гічної, так і цитогенетичної відповіді, а також про значне гальму-
вання прогресування захворювання. Це означає, що на сьогодні
глівек є препаратом першої лінії в лікуванні ХМЛ.
Для лікування хворих у хронічній стадії ХМЛ стандартна доза
глівеку становить 400 мг на добу в 1-й прийом їжі, запиваючи
1 склянкою води. Доза глівеку 300 мг та менше вважається субте-
рапевтичною й не може бути рекомендована до застосування. За
потреби (зниження показників периферичної крові) доцільніше
робити перерву в лікуванні, ніж постійно використовувати низькі
дози препарату.
У фазі акселерації ХМЛ пропонується проводити лікування
глівеком у дозі 600 мл на добу.
Лікування глівеком можна починати за будь-якої кількості
лейкоцитів. При гіперлейкоцитозі рекомендується збільшити вжи-
вання рідини та призначити аллопуринол. На фоні лікування глі-
веком можуть виникати нудота, судоми у м’язах, біль у кістках та
суглобах, висипка на шкірі, надмірна втомлюваність, збільшення
маси тіла, діарея. Такі побічні явища усуваються симптоматични-
ми засобами.
Прогноз. Захворювання хронічне, при застосуванні сучасних
методів терапії можливе одужання.
Ідіопатичний мієлофіброз
Ідіопатичний мієлофіброз (ЇМ) (синоніми: сублейкемічний міє-
лоз, мієлосклероз із мієлоїдною метаплазією, агноненна мієлоїдна
метаплазія, мієлоїдна спленомсгалія, алейкемічний мієлоз). Захво-
рювання належить до гемобластозів, котрі характеризуються мієлоп-
109
Н.М. Третяк | Гематологія
роліферацією у вигляді панмієлозу або мієломегакаріоцитарного мі-
єлозу, розвитком вторинного мієлофіброзу, остеомієлосклерозу,
трьохпаростковою мієлоїдною метаплазією селезінки, лейкоцитозом
та омолодженням гранулоцитарного паростка в крові.
Епідеміологія. Захворюваність на ІМ коливається від 1,0-1,5 на
100 тис. населення; діагностується в осіб середнього та старшого
(50-70 років) віку, проте останніми роками, у зв’язку з поліпшен-
ням діагностики, захворювання виявляється в осіб 40-45 років.
Патогенез. В основі патогенезу ІМ — фіброзування кісткового
мозку, що відбувається внаслідок патологічної проліферації мієло-
їдного паростка кровотворення. Згідно із сучасними уявленнями
припускається, що клональні порушення відбуваються на рівні по-
ліпотентної стовбурової клітини. Надмірна проліферація мієлоїд-
ного паростка призводить до гіперпродукції таких стимуляторів
росту, як фактор росту, шо продукується тромбоцитами, IV тром-
боцитарний фактор, котрий трансформує фактор р та р -тромбо-
глобулін. Під впливом цих факторів фібробласти надмірно проду-
кують колаген (колаген І, III, V та VI типів), а також фібронектин
і вітронектин, котрі відкладаються в кістковомозкових лакунах,
звужуючи таким чином плацдарм кровотворення.
На підставі уявлень про патогенез ІМ можна припустити такі
стадії механізму розвитку захворювання.
Стадії механізму розвитку ІМ:
І — проліферативна;
II — склеротична;
ПІ — бластна криза.
Проліферативна стадія характеризується надмірною проліфе-
ративною активністю мієлоїдного паростка, що зумовлює лейко-
цитоз, гіпертромбоцитоз та еритроцитоз у периферичній крові.
Склеротична стадія розвивається внаслідок надмірної продук-
ції колагену та відкладання його в лакуни кісткового мозку, що
звужує, вже виснажений за рахунок порушення проліферації плац-
дарм кровотворення, та зростаючого гіперспленізму.
Стадія бластної кризи. Захворювання набуває злоякісності, пе-
ребіг цієї стадії подібний до гострої лейкемії. Розвивається у 15-
25% хворих.
Клінічна картина відповідає морфологічним стадіям перебігу
захворювання. У першій проліферативній стадії хворі іноді скар-
жаться на слабкість, пітливість. При об’єктивному обстеженні або
на УЗД виявляється незначна спленомегалія. В аналізі периферич-
но
| Гемобластози
ної крові часто виявляється різного ступеня вираженості еритроци-
тоз, тромбоцитоз — до 1000-1500- 109/л та більше, незначний лей-
коцитоз (часто 15-20- 109/л), іноді в лейкограмі — поодинокі міє-
лоцити. У мієлограмі виявляється омолодження гранулоцитарного
паростка та збільшення кількості клітин —попередниць гранулоци-
топоезу. При гістологічному дослідженні кісткового мозку спосте-
рігається гіперпроліферація гемопоетичної тканини за рахунок гра-
нулоцитарного та мегакаріоцитарного паростків кровотворення
з витисканням жирової тканини, виявляється значний мегакаріо-
цитоз із гігантськими, мікро- та неправильної форми клітинами.
У такому гіперпластичному кістковому мозку вже є ретикуліновий
мієлофіброз, котрий виявляється імпрегнацією нітратом срібла.
У другій, склеротичній стадії скарги хворих набувають більш
характерних ознак: це пітливість, втрата ваги тіла, субфебрильна
температура, слабкість, запаморочення, серцебиття, важкість у лі-
вому підребер’ї або лівій половині живота.
При огляді пацієнтів виявляється спленомегалія різного ступе-
ня вираженості, шкіра бліда з іктеричним відтінком, нерідко збіль-
шена печінка.
Виникнення гіперспленізму у хворих у період склеротичної
стадії захворювання пояснюється утворенням компенсаторних по-
закістковомозкових вогнищ кровотворення на тлі недостатньої
функції кісткового мозку та внаслідок розвитку фіброзу. Для ІМ
у другій стадії типовим є синдром гіперспленізму на фоні вираже-
ної спленомегалії. Відбувається внутрішньоселезінкове руйнування
формених елементів крові та, насамперед, еритроцитів, чим пояс-
нюється наявність ознак гемолізу (жовтяниця меншого або біль-
шого ступеня вираженості, ретикулоцитоз та нормобласти в пери-
феричній крові, зростання кількості еритроїдних клітин
у кістковому мозку, гіпербілірубінемія за рахунок непрямої фрак-
ції білірубіну, темна сеча). Гепато-спленомегалія ускладнюється
розвитком портальної гіпертензії, шо утворюється внаслідок тром-
бозу селезінкової вени та портального шляху екстрамедулярними
вогнищами кровотворення і підвищеним наповненням їх кров’ю.
Портальна гіпертензія призводить до розвиток асциту та кровотеч
із варикозно розширених вен стравоходу. Наслідком порушення
функції тромбоцитів є виникнення внутрішньошкірних гематом та
можливість кровотеч зі слизової шлунково-кишкового тракту.
У період склеротичної стадії захворювання спостерігається
урикемія та урикозурія з ускладненнями у вигляді пієлонефриту,
НІ
Н.М. Третяк | Гематологія
нефросклерозу, артеріальної нирково-паренхиматозної гіпертонії.
Утворенню патології нирок сприяє їх зміщення збільшеними селе-
зінкою та печінкою.
Діагноз. У склеротичній стадії захворювання в периферичній
крові спостерігається анемія, часто лейкопенія, гіпертромбоцитоз
може змінюватися тромбоцитопенією. В лейкограмі виявляється
зрушення до мієлоцитів, промієлоцитів та поодиноких бластів, мо-
же зростати відсоток еозинофілів та/або базофілів — еозинофіль-
но-базофільна асоціація, анізо- та пойкілоцитоз еритроцитів.
Дослідження кісткового мозку в цій стадії пов’язане з трудноща-
ми отримання повноцінного аспірату, так званий “сухий пунктат”
унаслідок його прогресуючого фіброзування. В мієлограмі спостері-
гається подальше омолодження гранулоцитарного паростка.
При гістологічному дослідженні кісткового мозку виявляється
виражений фіброз із розростанням ретикулінових та колагенових
волокон, витісненням мієлоїдної та жирової тканин. Гемопоетичні
клітини залишаються у вигляді острівців, оточених фіброзною тка-
ниною
Прогресування захворювання призводить до остеосклерозу —
загрубіння балок губчастої частини кістки та вростання їх у кістко-
во-мозкові простори. Залишки кровотворної тканини можуть бути
представлені бластними клітинами. У периферичній крові в період
бластної кризи — анемія, тромбоцитопенія, лейкопенія зі зрушен-
ням у лейкограмі до бластів, але відсоток їх не буває таким висо-
ким, як при гострій лейкемії.
При цитохімічному дослідженні клітин периферичної крові ак-
тивність лужної фосфатази в нейтрофілах при ЇМ, на відміну від
ХМЛ, значно (до 300-400 у.о.) підвищена. Специфічних цитогене-
тичних змін не встановлено, проте у 30-70% випадків є хромосом-
ні аберації, що доводить клональне походження ІМ та є несприят-
ливою прогностичною ознакою.
Диференційний діагноз необхідно проводити з еритремією, хро-
нічною мієлоїдною лейкемією, хронічною мегакаріоцитарною лей-
кемією, есенціальною тромбоцитемією, волосатоклітинною лейке-
мією, мієлокарциноматозом.
Лікування. Терапевтична тактика залежить від стадії захворю-
вання. В першій проліферативній стадії традиційним є застосуван-
ня цитостатичних засобів. Мієлосан по 1-2 мг на добу в період ін-
дукції та по 2-6 мг на тиждень — підтримуюча терапія в період
ремісії. Гідроксисечовина по 0,5-1 г на добу в період індукції та по
112
| Гемобластози
1-3 г на тиждень — підтримуюча терапія при досягненні ремісії.
Ефективним є використання препаратів «-інтерферону (ІНФ-«) —
від 3 до 5 млн. МО/м2 тричі на тиждень — спостерігається змен-
шення розмірів селезінки, ліквідація гіпертромбоцитозу. Адекват-
но підібрана терапія на початковій стадії здатна загальмувати роз-
виток мієлофіброзу.
Як симптоматичні заходи використовуються еритроцитаферез
або гемоексфузїї, тромбоцитаферез.
У склеротичній стадії ІМ показані трансфузії еритроцитної ма-
си та призначення преднізолону по 20-50 мг на добу. При ознаках
прогресування захворювання — збільшення розмірів селезінки, га-
рячка, зрушення в лейкограмі до бластів та підвищення відсотка
мієлоцитів, промієлоцитів — призначаються цитостатичні засоби
(гідроксисечовина чи мієлосан) у тих же дозах, шо й у першій ста-
дії. За умови значних зрушень у лейкограмі використовується пу-
рінетол (6-меркаптопурин, 6-тіогуанін) та метатрексат, доза яких
визначається з урахуванням показників периферичної крові та за-
гального стану хворого.
Стадія бластної кризи, котра спостерігається лише у 15-25%
хворих на ІМ, супроводжується анемією, в основному із автоімун-
ним компонентом, тромбоцитопенією та підвищенням відсотка
бластних клітин у периферичній крові. Такі гематологічні показни-
ки потребують застосування цитостатичних засобів у поєднанні із
глюкокортико’їдними гормонами. Преднізолон — від 20 до 50 мг на
добу, 6-меркаптопурин — 50-100 мг на добу або цитозар — 12,5 мг
підшкірно двічі на добу до 21 дня. Препарати інтерферону у цій
стадії не ефективні.
Симптоматична терапія включає трансфузії еритроцитарної
маси, гепатопротектори, аллопуринол, комплекс вітамінів.
Прогноз. Застосування адекватних доз препаратів інтерферону
а на ранніх стадіях дає обнадійливі результати.
Справжня поліцитемія
Справжня поліцитемія (СП) (еритремія, хвороба Вакеза) — це
хронічний гемобластоз, що походить із ушкодженої клітини — по-
передниці мієлопоезу, здатної до необмеженого диференціювання
по 4 паростках, переважно еритроїдному.
Епідеміологія. Захворювання діагностується у 0,5-1,5 випадків
на 100 тис. населення. Хворіють, головним чином, особи, старші
113
Н.М. Третяк | Гематологія
за 60 років, переважно чоловіки, проте зустрічаються випадки СП
у молодих людей 25-30 років.
Патогенез. Клональна природа мієлоїдної проліферації при
СП доведена дослідженням 5-6-ФД у хворих мулаток, гетерозігот-
них за цим ферментом (існують А- і В-типи ферменту). В еритро-
цитах, гранулоцитах та тромбоцитах виявлений лише один тип із
цих ферментів, тоді як у фібробластах шкіри, кісткового мозку —
обидва типи цього ферменту.
Специфічних цитогенетичних аномалій при СП не виявле-
но. Зустрічаються спорадичні хромосомні аберації в мієлоїдних
клітинах, але їх присутність не є негативною прогностичною
ознакою.
Характерними патологоанатомічними змінами при СП є пере-
повнення всіх органів і тканин кров’ю, тромби в судинах. У ерит-
ремічній стадії в кістковому мозку спостерігається тотальна гіпер-
плазія трьох паростків із повним витісненням жиру, але можуть
бути такі варіанти, як гіперплазія тільки еритро'їдного та мегакарі-
оцитарного паростків, гіперплазія еритроїдного та гранулоцитар-
ного паростків або гіперплазія переважно еритроцитарного парос-
тка. Селезінка переповнена кров’ю з вогнищами інфарктів різних
строків давності, мають місце помірні чи значні ознаки мієлоїдної
метаплазії із локалізацією в синусах.
В анемічній стадії захворювання спостерігається посилена міє-
лоїдна метаплазія селезінки, печінки та гепатоспленомегалія. Кіс-
тковий мозок здебільшого фіброзований, при цьому мієлоїдна тка-
нина може бути як гіперплазована, так і редукована, зростає
кількість морфологічно змінених судин. Відбувається неефектив-
ний еритропоез.
Тривалість життя еритроцитів і тромбоцитів скорочується про-
порційно до збільшення селезінки.
Клінічна картина. Захворювання починається поступово,
й першими ознаками його можуть бути почервоніння шкіри об-
личчя та рук, слабкість, важкість у голові, тривалі кровотечі з ясен
після екстрації зубів.
\Уа88Єппап та СііЬегї у 1966 р. поділили симптоми клінічного
перебігу СП на три стадії.
Для І стадії характерна плетора. В аналізі крові — помірний
еритроцитоз. При гістологічному дослідженні кісткового мозку ви-
являються ознаки панмієлозу, в окремих випадках картина трепа-
ну може бути нормальною. Ця початкова стадія малосимптомна,
114
| Гемобластози
тому діагностувати СП у цей період складно. Проте тромботичні
ускладнення в І стадії СП уже спостерігаються.
II А-стадія СП — це еритремічний, або період розгорнутих
проявів захворювання з характерною метаплазією селезінки. Ця
стадія може тривати 10-15 та більше років. У цей період захво-
рювання хворих уже непокоїть слабкість, пітливість, свербіння
шкіри після миття, прояви еритромелалгії (гострий біль та по-
червоніння кінчиків пальців). При об’єктивному обстеженні ви-
являється почервоніння з пианотичним відтінком шкіри, особ-
ливо обличчя та кінцівок, слизових оболонок, ін’єкція склер.
Сплено- та гепатомегалія. Тромбо-геморагічні ускладнення ви-
никають частіше, ніж на 1 стадії захворювання. В аналізі крові
— еритроцитоз, вміст гемоглобіну перевищує норму, тромбоци-
тоз, лейкоцити в межах норми або дещо більше, в лейкограмі
нейтрофільоз зі збільшеним відсотком паличкоядерних клітин
та базофілів. У кістковому мозку — тотальна трьохпаросткова гі-
перплазія зі значним мегакаріоцитозом. При гістологічному дос-
лідженні кісткового мозку спостерігається повне витіснення жи-
рового кісткового мозку гіперплазованою гемопоетичною
тканиною з високим вмістом мегакаріоцитів. Фіброз майже
у всіх випадках відсутній, можливий ретикуліновий та вогнище-
вий, колагеновий мієлофіброз.
II В-стадія характеризується тими ж клінічними симптомами,
що й І стадія, але значно вираженішими. Ця стадія еритремічна,
але вже з мієлоїдною метаплазією селезінки. Спостерігається вис-
наження хворого, втрата маси тіла, частішають тромботичні та ге-
морагічні ускладнення. В периферичній крові виявляється підви-
щений вміст гемоглобіну (до 200-220 г/л), еритроцитоз,
тромбоцитоз та лейкоцитоз до 15-20 • 109/л зі зрушенням у форму-
лі до мієлоцитів, з’являються поодинокі еритрокаріоцити. Вміст
лужної фосфатази в нейтрофілах підвищений. При цитологічному
дослідженні кісткового мозку, як і в II А-стадії, виявляється пан-
мієлоз, але вже з перевагою гіперплазованого гранулоцитарного
паростка. В трепанаті спостерігається ретикуліновий та вогнище-
вий колагеновий мієлофіброз.
III стадія СП — анемічна. У периферичній крові виявляється
зниження кількості еритроцитів та вмісту гемоглобіну, тромбоци-
те- та лейкопенія. У збільшених селезінці та печінці відбувається
мієлоїдна метаплазія. У кістковому мозку виражений мієлофіброз,
вогнищевий мієлопоез почасти збережений.
115
Н.М. Третяк | Гематологія
Діагноз СП встановлюється на підставі даних цитологічного та
гістологічного дослідження кісткового мозку з урахуванням змін
у периферичній крові та клінічного обстеження. Виявлення в пе-
риферичній крові понад 170 г/л гемоглобіну та еритроцитів
6,0 • 109/л у чоловіків і 150 г/л гемоглобіну та еритроцитів 5,5 • 109/л
у жінок має бути поштовхом для подальшого обстеження пацієнта
з метою диференціювання СП та симптоматичного еритроцитозу.
Стандартні критерії діагностики СП, запропоновані Роїусуіке-
тіа Уегх Вішіу Сгоир (РУ8С):
Категорія А.
А.1. Збільшення маси циркулюючих еритроцитів: для чоловіків
— більше 36 мл/кг, для жінок — понад 32 мл/кг.
А.2. Нормальне насичення артеріальної крові киснем (більше
92%).
А.З. Спленометалія.
Категорія В.
В.1. Лейкоцитоз понад 12- 109/л (за відсутності інфекції та ін-
токсикації).
В.2. Тромбоцитоз більше 400 • 109/л (за відсутності кровотечі).
В.З. Підвищення вмісту лужної фосфатази нейтрофілів (за від-
сутності інфекції та інтоксикації).
В.4. Підвищення ненасиченої вітамін ВІ2-зв’язуючої здатності
сироватки крові (понад 220 пг/мл).
Існування двох позитивних ознак категорії А та В або всіх
трьох ознак категорії А підтверджує діагноз СП.
Диференційний діагноз слід проводити між СП та вторинними
абсолютними еригроцитозами.
Вторинні еритроцитози можуть виникати при генералізованій
тканинній гіпоксії — хронічні обструктивні захворювання легенів,
вроджені “сині” пороки серця, артеріовенозні співустя, ожиріння
— синдром Піквіка, надмірне паління (карбоксигемоглобінемія),
гемоглобінопатії з підвищеним спорідненням до кисню, дефіцит
2,3-дифосфоглицерату в еритроцитах.
При пухлинах — гіпернефроїдний рак нирок, гемангіобласто-
ма мозочка, гепатома, міома матки, пухлини (або гіперплазії) кор-
кового та мозкового шарів наднирників, аденома та киста гипофі-
зу, маскулінізуючі пухлини яєчників.
При локальній ішемії нирок — кисти нирок (солітарні, мно-
жинні), гідронефроз, стеноз ниркової артерії, відторгнення нирко-
вого трансплантату.
116
| Гемобластози
Лікування СП включає механічне видалення надмірної кількос-
ті еритроцитів на перших стадіях захворювання. Подальший роз-
виток мієлоїдної метаплазії зумовлює необхідність використання
цитостатичних засобів (мієлосан, гідроксисечовина).
Крововидалення — ефективний метод терапії, використовую-
чи котрий можна досягти нормалізації рівня гематокриту < 0,45%.
За умови проведення надалі підтримуючої терапії кровопускання-
ми або препаратами а-2в-інтерферону — по 3 млн. МО підшкірно
тричі на тиждень.
Крововидалення здійснюють на фоні застосування антикоагулян-
тів — гепарин 5 тис. од. підшкірно в передню черевну стінку. Обов’яз-
ковим є проведення гемодилюції сольовими розчинами або низько-
молекулярними декстранами, котрі поліпшують мікроциркуляцію.
Для попередження тромботичних ускладнень призначають дезагрега-
ційні засоби — трентал, курантил, тіклід, аспірин.
У другій А, В-стадії використовують цитостатичні препарати —
мієлосан по 2-6 мг на добу протягом 4-6 тижнів. Короткі курси лі-
кування зумовлені вираженим мієлосупресивним впливом препа-
рату. Гідроксисечовина призначається по 1,0-1,5 г на добу. Особ-
ливо ефективний цей препарат при лікуванні хворих із
гіпертромбоцитозом. При досягненні клініко-гематологічної ком-
пенсації (стабілізації кількості тромбоцитів) підтримуючу терапію
доцільно проводити препаратами а-2в-інтерферону, застосування
якого запобігає розвиненню мієлофіброзу.
У III стадії захворювання проводиться, в основному, симпто-
матична терапія.
Прогноз. Вчасне застосування адекватної терапії нині сприяє
виживанню хворих до 10-20 років при достатньо високій якості
життя.
ХРОНІЧНІ ЛІМФОПРОЛІФЕРАТИВНІ ЗАХВОРЮВАННЯ
Хронічна лімфоїдна лейкемія
Хронічна лімфоїдна лейкемія (ХЛЛ) належить до групи хронічних
лімфопроліферативних захворювань та характеризується накопи-
ченням непроліферуючих морфологічно зрілих лімфоідних клітин
у периферичній крові, кістковому мозку, лімфатичних вузлах та се-
лезінці з відносно доброякісним перебігом та повільним прогресу-
ванням.
117
Н.М. Третяк | Гематологія
Епідеміологія. Хронічна лімфоїдна лейкемія (ХЛЛ) — найпо-
ширеніший вид гемобластозів, що становить майже 30% усіх лей-
кемій та 9% усіх злоякісних новоутворень. Щорічно захворюва-
ність на ХЛЛ дорівнює 2,5-3 на 100 тис. населення, а для осіб віком
понад 60 років — до 20 випадків. Чоловіки хворіють частіше за жі-
нок у співвідношенні 2:1. Приблизно у 70% пацієнтів захворюван-
ня діагностується в 70 років, у 10-15% хвороба виникає до 40 ро-
ків. Середній вік пацієнтів становить 55 років.
З епідеміологічних досліджень відомо, шо ризик розвитку ХЛЛ
у родичів хворих у 3-6 разів вищий, ніж у загальній популяції. При
цьому в багатьох випадках спостерігається вертикальна передача,
що вказує на аутосомно-домінантний характер успадкування.
В наступному поколінні хвороба починається раніше й перебігає
тяжче.
Етіологія, патогенез. Численні дослідження до цього часу не
змогли оцінити ролі будь-яких мутагенних факторів (радіація, хі-
мічні агенти тощо) у виникненні ХЛЛ. У той же час встановлено,
що невипадкові хромосомні аберації, котрі з’являються під впли-
вом мутагенів, спостерігаються в більшості хворих на ХЛЛ. Такі
хромосомні аномалії, як трисомія 12 хромосоми, делеції 11ц, 6ц,
17р, крім делеції 13ц, є прогностично несприятливою ознакою при
ХЛЛ.
Останніми роками при ХЛЛ виявлені деякі молекулярні зміни,
що відіграють значну роль у виникненні та перебігу злоякісних но-
воутворень. Так, ген р53, що в нормальному стані є супресором
пухлинного росту, у 10-15% хворих на ХЛЛ являє собою мутан-
тний тип і асоціюється з прогресуванням хвороби та резистентніс-
тю до терапії. Давні уявлення про те, що при ХЛЛ відбувається
поступове накопичення довго живучих лімфоцитів, в останні роки
дістали підтвердження та пояснення. Виявлено, що в більшості
хворих спостерігається підвищення рівня гену ВСЬ-2, якому нале-
жить головна роль у запобіганні апоптозу — програмованій клітин-
ній смерті.
Приблизно 95-98% хворих на В-ХЛЛ мають В-клітинний імуно-
фенотип з експресією на поверхні лейкемічних лімфоцитів антиге-
нів диференціації. таких як НЕА-ОВ, СО|9, СГ)2(р СГ)24, СГ)5, СО23.
Клінічна картина. Хронічна лімфоїдна лейкемія починається
поступово (особливо у хворих похилого та старечого віку) і в біль-
шості випадків у 0-1 ст. за Ваі та А, В ст. за Віпеї прогресує по-
вільно. З розвитком захворювання поступово зростає лейкоцитоз.
118
| Гемобластози
Таблиця 11. Класифікація ХЛЛ.
Класифікація ХЛЛ за Каі (1975)
Стадія Характеристика Прогноз Медіана виживаності, роки
0 Лімфоцитоз більше 15,0- 109/л у крові, понад 40% у кістковому мозку сприятли- вий Така ж, як у загальній популяції
1 Лімфоцитоз + збільшення лімфовузлів проміжний 9
II Лімфоцитоз + спленомегалія та/або гепатомегалія незалежно від збільшення лімфатичних вузлів проміжний 6
III Лімфоцитоз + вміст гемоглобіну нижче 110 г/л незалежно від збільшення лімфатичних вузлів та органів несприят- ливий 1,5
IV Лімфоцитоз + вміст тромбоцитів менше 100,0- 109/л незалежно від збільшення лімфатичних вузлів та органів несприят- ливий 1,5
Класифікащя ХЛЛ за Віпеї (1981)
Стадія Характеристика Медіана виживаємості, роки
А Вміст гемоглобіну вище 100 г/л, тромбоцитів більше 100,0- 109/л, збільшення лімфатичних вузлів у 1-2 зонах Така ж , як у загальній популяції
В Вміст гемоглобіну більше 100,0- Ю9/л, тромбоцитів - понад 100,0- 109/л, збільшення лімфатичних вузлів у 3-х та більше зонах 7
С Вміст гемоглобіну менше 100,0- 109/л, тромбоцитів менше 100,0- 109/л незалежно від кількості збільшених зон 2
котрий з роками без лікування інколи досягає величезних цифр
(500-1000,0- 109 /л). Одночасно в лейкоцитарній формулі збіль-
шується кількість лімфоцитів — до 75-99%. Переважають зрілі
форми, але зустрічаються приблизно 5-10% пролімфоцитів та не-
рідко 1-2% лімфобластів. Вміст еритроцитів, гемоглобіну, тром-
боцитів у периферичній крові на початкових стадіях захворюван-
ня зберігається нормальним. При прогресуванні захворювання
відбувається скорочення плацдарму еритро- та тромбонитопоезу
за рахунок неопластичної проліферації кісткового мозку лейке-
мічними клітинами, що витісняють нормальні паростки кровот-
ворення, з розвитком анемії за тромбоцитопенії. При досліджен-
119
Н.М. Третяк | Гематологія
ні кістковомозкового пунктату вже на ранніх стадіях хвороби
спостерігається збільшення кількості лімфоцитів до 40-50-60%.
З часом, без лікування, при прогресуванні ХЛЛ, вміст цих клітин
може досягати 99%.
У більшості хворих виявляється повільне генералізоване збіль-
шення шийних, пахвових, підпахвових лімфатичних вузлів тістопо-
дібної консистенції, безболісних при пальпації. При рентгенологіч-
ному дослідженні визначається збільшення лімфатичних вузлів
середостіння, а при ультразвуковому дослідженні — збільшення
абдомінальних та зачеревних вузлів. Розміри лімфатичних вузлів
можуть коливатися в широких межах — від 1,5-2 см до 10-15 см
у діаметрі. Характерним для ХЛЛ є збільшення розмірів селезінки,
яка в деяких хворих може набувати величезних розмірів і займати
всю ліву половину живота. Також при ХЛЛ може збільшуватися
печінка. Не відмічається стійкої кореляції ступеня лімфоідної ін-
фільтрації кісткового мозку та розмірів лімфатичних вузлів, селе-
зінки, печінки.
Темпи розвитку хвороби, швидкість збільшення вмісту лейко-
цитів, розмірів лімфатичних вузлів та селезінки при ХЛЛ колива-
ються в широких межах. У частини хворих захворювання постійно
прогресує і, незважаючи на лікування, тривалість життя становить
2-3 роки. Водночас майже у 20% пацієнтів клінічні та гематологіч-
ні ознаки захворювання протягом 10-15 років, а інколи 20-30 ро-
ків, залишаються стабільними, спостерігається лише незначне
збільшення кількості лейкоцитів — 10-20 • Ю9/л, процент лімфоци-
тів у крові становить 60-70%, у кістковому мозку — 45-55%, кіль-
кість еритроцитів, гемоглобіну, тромбоцитів зберігається в межах
нормальних значень, відсутні лімфаденопатія та спленомегалія..Та-
ку форму ХЛЛ називають доброякісною. Е. Мопїзсггаі та співавто-
ри також виділили окрему форму ХЛЛ із тривалим, спокійним пе-
ребігом, для якої використовують термін „«тоИегіп^” (тліюча
ХЛЛ). Для таких пацієнтів у всьому світі прийнята тактика “очікуй
та спостерігай”. При цій формі ХЛЛ тривалість життя може взага-
лі не залежати від наявності даного захворювання.
При ХЛЛ у розвитку клінічних проявів, окрім лейкемічної
лімфоідної проліферації, важлива роль належить якісним змінам
як патологічних, так і нормальних лімфоцитів. В-лімфопити ви-
робляють знижену кількість нормальних імуноглобулінів та анти-
тіл у відповідь на бактеріальну інфекцію, внаслідок чого у пацієн-
тів виникають інфекційні ускладнення (бронхіти, пневмонії,
120
| Гемобластози
абсцеси, сепсис, інфекції сечовивідних шляхів тощо), котрі стають
частою причиною смерті хворих. Крім того, при ХЛЛ у хворих із
нормальним вмістом Т-лімфоцитів та натуральних кілерів (Т\'К-
клітин) їхня функція значно знижена, що пояснює характерну для
цього захворювання схильність до повторної інфекції. Частота ви-
никнення інфекційних ускладнень за даними різних авторів ста-
новить 60-80%. Нерідко у хворих на ХЛЛ виникає Ііегрек хокіег,
який може набувати генералізованих форм з ураженням внутріш-
ніх органів.
У 10-25% хворих на ХЛЛ виникає ще одне серйозне усклад-
нення — симптоматична автоімунна гемолітична анемія (АІГА).
Автоімунний конфлікт розвивається в будь-який період хвороби
й часто не відображає тяжкості проявів лейкемічного процесу.
У багатьох випадках (але необов’язково) збільшується вміст рети-
кулоцитів у крові, однак він ніколи не досягає високих значень, як
при ідіопатичній гемолітичній анемії. У сироватці крові виявляєть-
ся підвищення рівня непрямого білірубіну. Діагноз автоімунного
гемолізу підтверджується, зазвичай, позитивною прямою пробою
Кумбса. У більшості хворих з АІГА на еритроцитах виявляються
антитіла класу фО, лише у 10-15% — класу І§М. Наявність анти-
тіл класу ІеМ асоціюється з несприятливим перебігом ХЛЛ. Виді-
ляють форму гемолітичної анемії з антитілами проти еритрокаріо-
цитів кісткового мозку.
Автоімунна тромбоцитопенія зустрічається значно рідше,
приблизно у 2-3% хворих. Поєднання АІГА та автоімунної тром-
боцитопенії носить назву синдрому Фішера-Івенса.
Діагностика. Міжнародною робочою групою запропоновані
наступні критерії діагностики ХЛЛ: 1) абсолютний лімфопитоз
крові в межах 10,0 • 109/л, більшості лімфоцитів притаманна мор-
фологія зрілих клітин; 2) у кістковому мозку лімфоцити становлять
не менш ніж 30% усіх ядерних клітин; 3) більшість лімфоцитів пе-
риферичної крові мають В-клітинні маркери.
Лікування. Неоднорідність клінічного перебігу В-ХЛЛ потребує
різних підходів до лікування хворих. Приблизно третина хворих не
потребують лікування та помирають від причин, не пов’язаних із
ХЛЛ; ше в третини хворих після початкової фази настає стадія
прогресування; у частини пацієнтів виявляється агресивний пере-
біг захворювання віл самого початку, і вони потребують негайного
121
Н.М. Третяк | Гематологія
лікування. При появі будь-яких симптомів прогресування хвороби
треба невідкладно розпочинати лікування.
Показання для початку специфічної терапії хворих на ХЛЛ:
1) поява лихоманки неінфекційного генезу, пітливості, втрата
маси тіла;
2) анемія чи тромбоцитопенія, зумовлені інфільтрацією кістко-
вого мозку лейкемічним клоном;
3) автоімунні анемії чи тромбоцитопенія;
4) подвоєння кількості лімфоцитів менш ніж за!2 міс.;
5) гіперлейкоцитоз периферичної крові (понад 150,0- 109/л);
6) масивна лімфаденопатія або спленомегалія;
7) підвищена схильність до інфекційних ускладнень;
8) масивна лімфоцитарна інфільтрація кісткового мозку (біль-
ше 80% лімфоцитів у мієлограмі);
9) наявність комплексних хромосомних аберацій.
Упродовж багатьох років найпоширенішим лікувальним засо-
бом ХЛЛ був хлорамбуцил (хлорбутин, лейкеран}, який належить
до алкілюючих речовин. Цей препарат переважно застосовується
в якості первинно-стримуючої терапії. Механізм дії цього цитоста-
тичного засобу полягає в тому, що він зв’язується з ДНК патоло-
гічної клітини та призводить до порушення її синтезу. Найчастіше
його призначають у малих дозах: по 0,1 мг/кг щоденно до досяг-
нення максимальної відповіді або по 0,4-1 мг/кг кожні 4 тижні (К.аі
К. В.., 1993, Воробйов А. 1., 1995). Застосування хлорамбуцилу ре-
комендоване, насамперед, хворим похилого віку з вираженою су-
путньою соматичною патологією. Комбінована терапія хлорамбу-
цилу з глюкокортико'їдними гормонами показана при виникненні
автоімунних ускладнень.
Циклофосфамід також належить до групи алкілюючих цитос-
татиків. Ефективність цього препарату виявилась аналогічною
ефективності хлорамбуцилу. Він найчастіше застосовується при
первинній резистентності до хлорамбуцилу, а також при неперено-
симості останнього. Призначають по 50-100 мг/добу щодня до до-
сягнення позитивного терапевтичного ефекту або по 500-600-700
мг/м2 шодня кожні 2-3-4 тижні.
Найпоширеніші програми поліхіміотерапїї при лікуванні ХЛЛ
1. Схема СОР: циклофосфан — 300-400 мг/м2 в/в у 1—5-у до-
бу, вінкристин — 1,4 мг/м2 в/в у 1-у добу (але не більше 2 мг).
122
| Гемобластози
преднізолон — 60 мг/м2 у таб. в 1 —5-у добу.
2. Схема СНОР: циклофосфан — 750 мг/м2 в/в у 1-у добу, він-
кристин — 1,4 мг/м2 в/в у 1-й день, доксорубіцин — 50 мг/м2 в/в
у 1-у добу, преднізолон — 40-60 мг/м2 у таб. в 1-5-у добу.
3. Схема САР: циклофосфан — 500-750 мг/м2 в/в у 1-у добу,
доксорубіцин — 50 мг/м2 в/в у 1-у добу, преднізолон — 60 мг/м2 в
1-5-у добу.
Інтервали між циклами становлять 3-4 тижні в залежності від
показників крові. Дози окремих препаратів у цих схемах можуть
змінюватися. Проводять у середньому від 6 до 12 циклів терапії.
Окрім запропонованих схем, інколи застосовується схема М-2
з двома алкілюючими препаратами — циклофосфаном і мелфала-
ном та ЬУРР, у якій алкілюючим препаратом є хлорамбунил (лей-
керан).
Табл иця 12. Критерії ефективності терапії ХЛЛ
Результат терапії Міжнародна робоча група Національний інститут раку
Повна ремісія Ознак хвороби немає. Кількість лімфоцитів менш ніж 4,0- 109/л, гранулоцитів більш ніж 1,5- 109/л, тромбоцитів понад 100,0- 109/л, нормальний клітинний склад кісткового мозку, можливі нодулярні скопичення лімфоцитів Ознак хвороби немає. Рівень гемоглобіну більш ніж 110 г/л без гемотрансфузій. Усі показники зберігаються не менше 2 місяців
Часткова ремісія Повернення від стадії С до А чи В або від В до А Вираженість усіх ознак хвороби, котрі спостерігалися до лікування, зменшується на 50% та більше
Стабілізація Змін у стадіях хвороби не відбулося Повної чи часткової ремісії не досягнуто, але захворювання не прогресує
Прогресування Перехід від стадії А до В чи С або В до С Збільшення на 50% та більше проявів будь-якої з присутніх ознак хвороби чи поява інших. Трансформація в пролімфоцитарну ХЛЛ чи синдром Ріхтера
Новим етапом у лікуванні хронічних лімфопроліферативних
захворювань, зокрема В-ХЛЛ, стало застосування аналогів пурино-
вого нуклеозиду, яким притаманна висока протипухлинна актив-
ність. До препаратів цієї групи належать флударабін, клалрибін,
123
Н.М. Третяк | Гематологія
пентостатін. Найбільш поширеним у лікуванні ХЛЛ став флудара-
бін. Він пригнічує ряд ферментів, що беруть участь у синтезі ДНК.
Крім того, флударабін інгібує синтез РНК, що є його унікальною
властивістю. У результаті припинення синтезу ДНК та РНК відбу-
вається загибель клітини за механізмом апоптозу (Габеєва, Півник,
2000). Уже перші клінічні випробування цього препарату показали
його високу ефективність: ремісії вдалося досягти у 45% хворих на
ХЛЛ, шо були резистентні до всіх попередніх схем терапії. Ще
більш переконливі результати отримані при лікуванні первинних
нелікованих хворих. У залежності від стадії захворювання лікуваль-
ний ефект вдається отримати, за даними різних авторів, у 67-85%
хворих, при цьому у 50-73% — повні ремісії. Флударабін високое-
фективний і при повторному застосуванні при рецидиві ХЛЛ, що
виник після першого застосування флударабіна у хворих, які були
рефрактерні до всієї попередньої терапії. У багатьох проведених
рандомізованих дослідженнях було доведено безумовну перевагу
застосування флударабіну над поширеними схемами моно- та по-
ліхіміотерапії, на підставі чого зроблено висновок, шо саме флуда-
рабін повинен стати препаратом першої лінії терапії ХЛЛ.
Флударабін зазвичай призначається по 25 мг/м2 внутрішньо-
венно в 1—5 добу кожні 28 днів. Кількість курсів лікування зале-
жить від досягнення повних чи часткових ремісій (мінімально —
3-4 курси, максимально — 6). Після досягнення ремісії проводять
2 курси консолідації. При порушенні функції нирок та зниженні
кліренсу креатиніну від 60 до 30 мл/хв. проводиться редукція до-
зи на 50%.
У теперішній час рекомендується застосування комбінованої
терапії флударабіну з такими препаратами, як циклофосфамід
(флударабін — 25 мг/м2 в/в у 1—3 добу, циклофосфамід — 250-300
мг/м2 в/в у 1—3 добу), мітоксантрон, ідарубіцин та інші.
Використовується також інша лікарська форма — флударабін
у таблетках. Доза препарату 40 мг/м2 відповідає дозі флударабіну 25
мг/м2 для внутрішньовенного застосування.
Альтернативним препаратом у лікуванні ХЛЛ є моноклональ-
ні антитіла (мкАТ) проти антигенів, які експресуються на пухлин-
них клітинах із метою прицільного ураження клітин злоякісного
клону та зведення до мінімуму ушкодження здорових тканин орга-
нізму. Для імунотерапії було відібрано окремі ХЛЛ-асонійовані ан-
тигени, такі як СІ)., СЕ)|9, СО20, СО5,. Сьогодні для лікування
ХЛЛ застосовують антитіла, спрямовані проти антигена СГ)52
124
| Гемобластози
(Маб Кампат, Алемтузумаб) та СО?() (Ритуксімаб). При ХЛЛ мкАт
до СТ)20 виявився менш ефективним, шо зумовлено слабкою екс-
пресією СО20 на лейкемічних лімфоцитах. З іншого боку, Маб
Кампат визнано високоефективним засобом.
Маб Кампат вводиться у дозах, що зростають протягом 3-7
днів: 3 мг—ІОмг—ЗО мг. Останню дозу вводять тричі на тиждень,
максимально до 12 тижнів.
Маб Кампат показаний для лікування хворих на ХЛЛ, що не
дали повної чи часткової ремісії після лікування алкілюючими пре-
паратами, або котрі були рефрактерними до попередньої терапії
флударабіном.
Лікування автоімунних ускладнень при ХЛЛ включає застосу-
вання імуносупресорної терапії — кортикостероїдних гормонів або
циклоспорину А (середня добова доза — 3 мг/кг). При повільному
розвитку гемолітичної кризи достатньо призначати 40-50 мг пред-
нізолону на добу. При гострих гемолітичних кризах дози збільшу-
ють до 70-100-150 мг на добу. У випадках різко вираженого гемо-
лізу, особливо при поєднанні АІГА з автоімунною
тромбоцитопенією та за відсутності ефекту від гормонів протягом
2-3 тижнів, показана спленектомія.
Трансплантація гемопоетичних стовбурових клітин не розгля-
дається як стандартний метод лікування ХЛЛ. Він показаний хво-
рим віком до 55 років, головним чином при резистентності до те-
рапії флударабіном. Частіше застосовується автологічна
трансплантація — в зв’язку з високою летальністю після проведен-
ня алогенної трансплантації кісткового мозку.
Рідкісні форми ХЛЛ
В-клітинна пролімфоцитарна хронічна лімфолейкемія
В-клітинна пролімфоцитарна хронічна лімфолейкемія — варіант
ХЛЛ, при якому в крові та кістковомозковому пунктаті переважа-
ють пролімфонити. Імунофенотипічне дослідження показує, шо
ці клітини не експресують типовий для В-ХЛЛ антиген СЕ)5. Па-
цієнти з цим варіантом захворювання, в середньому, старші за ві-
ком. Характерний високий лейкоцитоз та спленомегалія. Ця фор-
ма захворювання відрізняється від типового ХЛЛ агресивнішим
перебігом, короткою тривалістю життя (приблизно 3 роки). Тера-
пія пролімфонитарної лімфолейкемії малоефективна. Деякий
ефект спостерігається при опроміненні селезінки, шо зменшує
масу пухлини. Найбільш ефективні аналоги пурину — флудара-
бін, пентостатін.
125
Н.М. Третяк | Гематологія
Волосато-клітинна лейкемія (ВКЛ)
Для неї характерна дифузна інфільтрація кісткового мозку. Ци-
топлазма лейкемічних клітин має нерівні вінця, в типових випад-
ках обривчаста та ворсинчаста. Специфічною цитохімічною озна-
кою є яскрава дифузна реакція на кислу фосфатазу в лімфоцитах,
котра не інгібується тартратом натрію. Найхарактернішим клініч-
ним симптомом ВКЛ є спленомегалія. При лікуванні застосовують
інтерферонотерапію, аналоги пурину (кладрібін, пентостатин),
спленектомію.
Т-клітинний варіант ХЛЛ
Зустрічається у 2-5% хворих. За цієї форми лейкемії експресу-
ються антигени СО4 та майже не експресуються СП8. Характерни-
ми є високий лейкоцитоз, спленогепатомегалія. Відзначається ре-
зистентність до алкілюючих препаратів. Ефект лікування значно
підвищується від застосування пуринових аналогів.
Іншою формою Т-ХЛЛ є лейкемія, за якої пухлинні клітини
представлені великими гранулярними лімфоцитами. Ця форма
підрозділяється на лейкемію з імунофенотипом Т-лімфоиитів та
лейкемію із клітин з імунофенотипом натуральних кілерів. Пер-
ший варіант характеризується невираженою симптоматикою та по-
вільним перебігом, хоча в зв’язку з нейтропенією часто виникають
інфекційні ускладнення. Другому варіанту притаманні високий
лейкоцитоз та масивна гепатоспленомегалія. Цей варіант має агре-
сивний перебіг із резистентністю до терапії.
Негоджкінські лімфоми
Негоджкінські злоякісні лімфоми (НЗЛ) — це злоякісні лімфоп-
роліферативні пухлини, гетерогенні за своїм походженням, біоло-
гічними якостями, морфологічною будовою, клінічними проява-
ми, відповіддю на терапію та прогнозом.
Епідеміологія. Захворюваність на НЗЛ має постійну тенденцію
до зростання й у розвинених країнах за останні 20 років вона під-
вищилась більш ніж на 50%. Чоловіки хворіють удвічі частіше за
жінок. Захворюваність на НЗЛ різниться в залежності від раси: бі-
лі хворіють значно частіше, ніж чорні, і зовсім рідко ця патологія
зустрічається серед представників жовтої раси.
Вікова категорія є найбільш значущим фактором ризику ви-
никнення НЗЛ: рівень захворюваності осіб у 15-20 років майже в
10 разів нижчий у порівнянні з таким у осіб, старших за 75 років.
126
| Гемобластози
Етіологія НЗЛ дотепер ше остаточно не з’ясована. Вважається
можливою участь у виникненні різного типу лімфом інфекційних
агентів. Вірус Епштейна—Барра є причиною розвитку так званої
класичної, або ендемічної, лімфоми Беркитта, котра поширена
в ендемічно малярійних регіонах Африки. Захворюваність дітей (до
15 років) у цих регіонах становить 4,0 на 100 тис. населення. Спо-
радична лімфома Беркитта зустрічається значно рідше, і захворю-
ваність становить 0,2 на 100 тис. населення.
Вірус імунодефіциту значно підвищує ризик виникнення НЗЛ.
Дослідженнями Національного ракового інституту США засвідче-
но, що ступінь ризику залежить від фактора часу: у хворих на СНІД
або ВІЛ-інфекції з клінічними проявами він становить 8% у перші
2 роки та 19% через 3 роки від початку антивірусної терапії.
Т-клітинний лімфоїдний вірус 1 типу людини (НТБУ-І) був ви-
ділений СаІІо як фактор, що спричиняє розвиток Т-клітинної лей-
кемії/лімфоми, та описаний на початку 70-х років Т. СсИіуата.
Встановлено, шо передача інфекції відбувається вертикально за на-
явності інфікованих лімфоцитів у материнському молоці та горизон-
тально при статевих контактах. Можлива передача з кров’ю донора.
Є повідомлення про участь у виникненні НЗЛ герпесвірусу
людини VI типу, проте ці свідчення ще вивчаються.
Певну роль у розвиненні НЗЛ можуть відігравати фактори зов-
нішнього середовища, такі як пестициди (гербіциди, інсектициди,
фунгіциди та ін.).
Первинний та вторинний імунодефіцити — важливі чинники
у формуванні НЗЛ. Виявляється чіткий зв’язок між імунною дис-
функцією та частотою розвитку НЗЛ при низці спадкових імуно-
дефіпитних синдромів: синдроми Віскотта—Олдрича, Чедіака—Хі-
гаші, Клайнфельтера атаксія-телеангіектазія.
Найбільший ризик виникнення НЗЛ становить застосування
імуносупресивної терапії при проведенні трансплантації органів.
У цих’випадках НЗЛ розвивається в 46 разів частіше, ніж у загаль-
ній популяції.
Встановлено чіткий взаємозв’язок між частотою НЗЛ та автоі-
мунними захворюваннями: при ревматоїдному артриті ризик зрос-
тає в 2-3 рази, при синдромі Фелті — в 2-13 разів.
Не доведено впливу на частоту виникнення НЗЛ іонізуючої
радіації, ультрафіолетового опромінення та родинного зв’язку.
Класифікація. Історично класифікація НЗЛ була побудована на
морфологічному принципі, проте досягнення в галузі імунології,
127
Н.М. Третяк | Гематологія
молекулярної біології та цитогенетики створили умови для глибо-
кого вивчення патогенезу захворювання, й на підставі цих дослід-
жень створено сучасні, більш уживані класифікації — КЕАЬ (1994)
та ВООЗ (1995).
ЯЕАЬ-класифікація лімфоїдних неоплазій
І. Пухлини із В-клітин
1. Лімфоми з клітин-попередниць:
— В-лімфобластна лімфома/лейкемія з клітин попередниць;
2. Лімфоми з “периферичним” фенотипом:
— В-клітинна хронічна лімфоцитарна лейкемія/пролімфоци-
тарналейкемія/лімфома з малих лімфоцитів;
— лімфоплазмоцитарна лімфома/імуноцитома;
— лімфома з клітин мантійної зони;
— лімфома з клітин фолікулярних центрів, фолікулярна: а) із
малих клітин,
б ) змішана з малих та великих клітин,
в ) із великих клітин;
3. В-клітинна лімфома маргінальної зони:
— екстранодальна (МАІТ-тип+/- моноцитоїдна);
— лімфома маргінальної зони селезінки (+/- ворсинчасті лім-
фоцити);
4. Волосатоклітинна лейкемія;
5. Плазмоцитома/плазмоклітинна мієлома;
6. Дифузна В-крупноклітинна лімфома:
— первинна медіастінальна В-клітинна лімфома (тимус);
7. Лімфома Беркитта:
— В-клітинна лімфома високого ступеня злоякісності, типу
лімфоми Беркитта.
II. Пухлини із Т-клітин та ПК-клітин
1. Лімфоми з Т-клітин-попередниць:
— Т-лімфобластна лімфома/лейкемія з клітин-попередниць;
• 2. Т-клітинні лімфоми із клітин з “периферичним” фенотипом
та ЬЖ-клітинні пухлини:
— Т-клітинна хронічна лейкемія/пролімфоцитарна лейкемія;
— лейкемія з великих грануловмісних лімфоцитів:
а) Т-клітинний тип,
б) ІЧК-клітинний тип;
3. Грибоподібний мікоз/синдром Сезарі;
4. Т-клітинна лімфома з “периферичним” фенотипом, неуточ-
нена:
128
| Гемобластози
— із клітин середнього розміру;
— змішана із середніх та великих клітин;
— із великих клітин;
— лімфоепітеліоїдно-клітинна;
5. Ангіоімунобластна Т-клітинна лімфома;
6. Ангіоцентрична лімфома;
7. Т-клітинна лімфома кишечника;
8. Т-клітинна лімфома/лейкемія дорослих;
9. Анапластична крупноклітинна лімфома.
III. Хвороба Годжкіна
1. Лімфоїдне переважання (лімфогістіоцитарний варіант);
2. Нодулярний склероз;
3. Змішаноклітинний варіант;
4. Лімфоїдне виснаження:
— класична хвороба Годжкіна з великою кількістю лімфоцитів.
Подальше удосконалення методів дослідженя в імунології
й молекулярній біології та виявлення нових інтимних механізмів
розвитку лімфом допомогли фахівцям міжнародної робочої групи
з виявлення лімфом сформулювати класифікацію ВООЗ.
Класифікація ВООЗ пухлин, що походять з лімфоїдної тканини
І. Пухлини із В-клітин
1. Лімфоми із попередниць В-клітин:
— В-лімфобластна лімфома/лейкемія із клітин-попередниць;
2. В-клітинні пухлини із периферичних (зрілих) клітин:
— В-клітинна хронічна лімфоцитарна лейкемія/лімфома із ма-
лих лімфоцитів;
— В-клітинна пролімфоцитарна лейкемія;
— лімфоплазмоцитарна лімфома;
— лімфома маргінальної зони селезінки (+/- ворсинчасті лім-
фоцити);
— волосатоклітинна лейкемія;
— плазмоклітинна мієлома/плазмоцитома;
— екстранодальна В-клітинна лімфома маргінальної зони
МАТТ-типу;
— фолікулярна лімфома;
— лімфома з клітин мантійної зони;
— дифузна В-крупноклітинна лімфома:
а) медіастинальна дифузна В-крупноклітинна лімфома,
б) лімфома, первинно асоційована з випотом;
— лімфома/лейкемія Беркитга.
12‘
Н.М. Третяк | Гематологія
II. Пухлини Т- і ИК-клітинні
1 . Лімфоми із Т-клітин-попередниць:
— Т-лімфобластна лімфома/лейкемія із клітин попередниць;
2 . Т-клітинні лімфоми із периферичних (зрілих) клітин:
— Т-клітинна пролімфоцитарна лейкемія;
— лейкемія з великих грануловмісних лімфоцитів;
— агресивна МК-клітинна лейкемія;
— Т-клітинна лімфома/лейкемія дорослих (НТІЛ1+);
— екстранодальна ІМК/Т-клітинна лімфома, назальний тип;
— Т-клітинна лімфома, асоційована з ентеропатією;
— гепатолієнальна у<5Т-клітинна лімфома;
— грибоподібний мікоз/синдром Сезарі;
— анапластична крупноклітинна лімфома;
— периферична Т-клітинна лімфома, неуточнена;
— ангіоімунобластна Т-клітинна лімфома.
III. Лімфома Годжкіна / хвороба Годжкіна
1. Нодулярний варіант хвороби Годжкіна з лімфоїдним перева-
жанням;
2. Класичний варіант хвороби Годжкіна:
— нодулярний склероз ХГ (градації 1 і 2);
— класична ХГ із великою кількістю лімфоцитів;
— змішаноклітинний варіант ХГ;
— лімфоїдне виснаження ХГ.
Попередньо розроблені класифікації лімфом, такі як Ьикеь—
Соїііпз (1976 р.), Кільська (1974 р.) із модифікаціями (1976, 1988,
1989 рр.)? Британська (1974 р.), класифікація ВООЗ (1966 р.) та ро-
боче формулювання (Л¥огкіп§ Рогтиіайоп, 1982 р.), у теперішній
час являють собою лише історичний інтерес.
З оглвду на те, що всім класифікаціям притаманні ті чи інші не-
доліки, розроблено зручну ддя використання в клінічній практиці
систему стадіювання лімфом Апп-АгЬог (модифікована в Сойлуоійз) та
міжнародний прогностичний індекс (ІРІ) для негоджкінських лімфом.
Система стадіювання лімфом Апп-АгЬог (1971) модифікована в
Соїхнюїдв, 1973)
І стадія — ураження однієї групи лімфатичних вузлів чи орга-
нів (селезінка, тимус, кільце Вальдеєра).
II стадія — ураження двох чи більше груп лімфатичних вузлів
по один бік діафрагми (середостіння та лімфатичні вузли воріт вва-
жаються окремими локусами). Кількість груп уражених органів ін-
дексується так — ІІ2.
130
| Гемобластози
III стадія — ураження групи лімфатичних вузлів по обидва бо-
ки діафрагми:
Пір з або без лімфатичних вузлів у воротах селезінки, печінки,
портальних чи черевних;
ІІІ2: з парааортальними, з духвинними та мезентеріальними
лімфатичними вузлами;
IV стадія — екстранодальні групи уражень, не визначені в п. Е:
А: без симптомів,
В: гарячка, профузне пітніння, прогресуюча втрата ваги,
X: “Виїку сіізеазе” :>1/3 поперечного розміру середостіння
або >10 см у діаметрі лімфатичного ураження,
Е: ураження поодинокого екстранодального локусу, дотичного
чи просимального до певного нодального локусу,
8: ураження селезінки.
Таблиця 13. Міжнародний прогностичний індекс (ІРІ) для
негоджкінських лімфом
Прогностичний чинник виживання Міжнародний прогностичний індекс
Критерій 0 балів 1 бал Категорія Кількість балів
Вік < 60 років > 60 років Низький (Ьо«) 0; 1
лдг < N > N
Загальний стан (згідно з ЕСОС) 0; 1 2; 3; 4 Низький-проміжний (ко^-іпіегтесііаіе) 2
Стадія (Апп-АгЬог) І/ІІ ІІІ/ІУ Високий-проміжний (НідГі-іпІептіесІіаІе) 3
Екстранодальні ураження <_1 > 1 Високий (НідП) 4;5
Прогностичні групи хворих на НГЛ, згідно з індексом ІРІ, роз-
поділяються таким чином:
— 0-1 пункт — низький ІРІ, прогностично найсприятливіший;
— 2 пункти — низький/проміжний ІРІ;
— З пункти — проміжний/високий ІРІ;
— 4-5 пунктів — високий ІРІ, прогностично найнесприятливі-
ший.
Один несприятливий пункт визначається за такими показни-
ками:
— вік хворого > 60 років;
— загальний стан > 1;
131
Н.М. Третяк | Гематологія
— стадія ПІ або IV;
— рівень ЛДГ > ІхМ;
— кількість екстранодальних уражень > 1.
Діагноз НЗЛ встановлюється на підставі результатів мофологіч-
ного дослідження пухлинного утворення. В амбулаторних умовах
можливе виконання цитологічного дослідження, котре є достатньо
інформативним. Проте для впевненості в діагнозі неодхідно прово-
дити гістологічне дослідження біоптата пухлини з гістохімічним та
імунофенотипічним дослідженням. Для виявлення можливого лей-
кемічного ураження кісткового мозку проводиться цитологічне,
гістологічне, цитохімічне, гістохімічне та імунофенотипічне дослід-
ження клітин кісткового мозку (пунктат і трепанобіоптат).
Застосування імунофенотипування дає змогу визначити В- або
Т-лінію імунної належності НЗЛ та рівня порушення диференцію-
вання.
Імунофенотипові особливості різних морфологічних варіантів
НЗЛ, які залежать віл рівня порушення диференціювання лімфоїд-
ної клітини, є високоінформативними для діагностики та прогно-
зу захворювання.
Найчастіше (майже у 90% випадків) НЗЛ має В-клітинне по-
ходження, експресуючи пан-В-клітинні антигени СО|3, СО2(), СО22
в поєднанні з НІА/ОК та молекулами поверхневих імуноглобулі-
нів. Присутність інших В-клітинних агтигенів (СП5, СП10, СО38,
СО23 та ін.) дозволяє з упевненістю встановити В-клітинний варі-
ант НЗЛ, що є вагомим критерієм вибору адекватної терапії.
Для Т-клітинних пухлин характерна присутність антигенів
со4, сі)7, со8.
Для виявлення меж поширеності патологічного процесу, тобто
клінічної стадії захворювання, обов’язкове проведення УЗД,
комп’ютерної томографії, ЯМР, сцинтіграфії печінки, селезінки та
кісток скелету. При особливих клінічних варіантах НЗЛ (ураження
оболонок головного та спинного мозку) застосовуються такі мето-
ди дослідження, як люмбальна пункція з визначенням клітиннос-
ті цереброспинальної рідини, її біохімічного та цитохімічного скла-
ду. Після встановлення діагнозу на підставі клінічних,
морфологічних та імунологічних ознак визначають поширеність
патологічного процесу відповідно до Апп-АгЬог-класифікації
(1971), модифікованої в Соі8\уоісі8 (1973).
Диференційний діагноз проводиться з хронічною лімфо'їдною
лейкемією, хворобою Годжкіна, інфекційним мононуклеозом,
132
| Гемобластози
лімфоаденопатією внаслідок туберкульозу, метастазами в лімфа-
тичні вузли.
Клінічна картина. НЗЛ починається з появи одного пухлинно-
го вузла та поширюється з часом лімфогенним та гематогенним
метастазуванням. Первинне пухлинне вогнише зазвичай локалізу-
ється в лімфатичному вузлі (недальнє ушкодження) — 45-50%, або
в інших органах та тканинах (селезінка — 30-40%; шлунково-киш-
ковий тракт — 10-24%; кістковий мозок — 30-40%) — екстрано-
дальне ураження.
На початку захворювання лімфатичні вузли щільні, безболісні,
не спаяні зі шкірою та оточуючими тканинами, при прогресуван-
ні патологічного процесу утворюються конгломерати. Клінічні
прояви захворювання зумовлені розташуванням пухлинних вог-
ниш. При ураженні лімфатичних вузлів шиї та середостіння може
спостерігатися стиснення стравоходу та трахеї, що утруднює про-
ходження їжі та спричиняє кашель. Стиснення великих судин
грудної порожнини (частіше в задньомедіастинальному просторі)
зумовлює застій у системі верхньої порожнистої вени. Спостеріга-
ється ціаноз та набряк верхньої половини тіла та обличчя з пору-
шенням дихання й тахікардією. Великі лімфатичні вузли черевної
порожнини, що розташовані у функціонально важливих зонах, мо-
жуть зумовлювати кишечну непрохідність, застій у поральній сис-
темі та порушення лімфовідтоку з нижньої половини тіла, шо
спричиняє розвинення асциту, набряків нижніх кінцівок та стате-
вих органів, крім того може виникати механічна жовтяниця, пору-
шення сечовиділення.
Основними клінічними проявами лімфопроліферативного
процесу в носоглотці є утруднення носового дихання та погіршен-
ня слуху. Лімфоїдні пухлини в носоглотці мають бугристий вигляд,
блідо-рожевого кольору, швидко ростуть, можуть проростати до
решітчастого лабіринту, у верхньощелепову пазуху, очну ямку,
спричинюючи екзофтальм. Глоточні мигдалики можуть значно
збільшуватись, іноді майже змикатися між собою, з подальшим ут-
воренням виразок.
Лімфома шкіри проявляється по-різному. Можливе утворення
солітарного пухлинного вузла в товщі шкіри та підшкірній жировій
клітковині або утворення маленького внутрішньошкірного вузлика
з подальшим їх розмноженням. Зазвичай ці лімфатичні вузли різні
за розміром, щільні, безболісні, можуть розташовуватись групами та
зливатися в щільні інфільтрати з бугристою поверхнею, схильні до
133
Н.М. Третяк | Гематологія
утворення виразок. Шкіра над ними має темно-червоне ціанотич-
не забарвлення. Перші симптоми лімфоми шкіри часто схожі на
дерматит, і хворі тривалий час лікуються в дерматолога.
Для пухлини шлунково-кишкового тракту специфічних, при-
таманних тільки НЗЛ клінічних ознак немає. Хворі скаржаться на
нудоту, погіршення апетиту, зменшення маси тіла. Такі скарги мо-
жуть бути-при будь-якій пухлині, розташованій у шлунково-киш-
ковому тракті. Вираженість клінічних проявів захворювання зумов-
лена локалізацією та формою росту пухлини.
Найчастіше зустрічається лімфома шлунка та тонкої кишки —
як при ізольованому, так і при поєднаному залученні їх у патоло-
гічний процес. У 2/3 хворих лімфома травного тракту є першою оз-
накою захворювання (первинне ураження). Виникнення пухлини
вторинного походження відбувається переважно метастатичним
шляхом майже у 90% випадків. Інфільтративна форма росту пухли-
ни зустрічається найчастіше — у 50% хворих, майже у 15% випад-
ків спостерігається виразкова форма пухлини, котра може усклад-
нюватися кровотечею, перфорацією, перитонітом та кишечною
непрохідністю. Прогноз за таких ускладнень несприятливий.
Ізольована, первинна лімфома ЦНС зустрічається у 10% ви-
падків. Клінічна картина може бути мало вираженою та залежить
від локалізації пухлини. Утворення поодинокого або множинних
пухлинних утворень у речовині головного мозку супроводжується
неврологічними симптомами, котрі можуть виникати при уражен-
ні тих чи інших відділів головного мозку. При залученні до пато-
логічного процесу оболонок головного мозку розвиваються озна-
ки, що нагадують менінгіт, — головний біль, нудота, блювота,
позитивні менінгеальні симптоми.
Прогноз первинної лімфоми ЦНС несприятливий. Злоякісний
перебіг мають також первинні лімфоми яєчка, яєчників, кісток,
молочної залози.
До прогностично сприятливих форм лімфоми з 5-річною ви-
живаністю у 60% хворих належать первинні НЗЛ шлунково-киш-
кового тракту, кільця Пирогова—Вальдейєра, орбіти, слинних за-
лоз. Терапевтична тактика в усіх цих випадках не однакова.
Лікування НЗЛ проводиться з використанням усіх видів проти-
пухлинної терапії. Основними факторами, шо впливають на виз-
начення терапевтичної тактики, є поширеність процесу (клінічна
стадія 1, II, ПІ, IV), морфологічний варіант пухлини, первинна або
переважна локалізація злоякісного процесу та фактори прогнозу.
134
| Гемобластози
Хірургічне лікування застосовується не часто. Головним пока-
занням до хірургічного втручання є первинні поодинокі вогнища
НЗЛ у шлунково-кишковому тракті. Спленектомію застосовують
рідко. Видалення селезінки проводять із метою корекції гематоло-
гічних показників при вираженому гіперспленізмі, що буває нас-
лідком злоякісного ураження селезінки.
Променева терапія є високоефективним методом лікування
НЗЛ і може використовуватись на всіх етапах захворювання, од-
нак її терапевтичний результат не завжди позитивний. Як самос-
тійний лікувальний захід променева терапія застосовується рід-
ко, значно більший ефект досягається при поєднанні хіміо- та
променевої терапії. Значного поширення з позитивним результа-
том набуло комбіноване хіміо-променеве лікування при І—Ш
стадіях НЗЛ.
У багатьох клінічних ситуаціях НЗЛ основним лікувальним за-
ходом є хіміотерапія — як самостійний метод та в поєднанні з ін-
шими терапевтичними підходами.
Поліхіміотерапію проводять, як правило, у вигляді коротких
(різних за інтенсивністю й тривалістю) курсів, дотримуючись виз-
наченого інтервалу. Лікування триває до повної ремісії або до того
часу, поки протипухлинний вплив від курсу до курсу наростає.
Проте якшо 2 курси хіміотерапії не дали позитивного ефекту, схе-
му хіміотерапії потрібно замінити іншою. Після досягнення повної
ремісії проводиться 2-3 курси консолідації ремісії, з використан-
ням курсів індукції ремісії.
Тривалість життя хворих на НЗЛ високого ступеня злоякіс-
ності перебуває в прямій залежності від результатів лікування:
5-річна виживаність при досягненні повної ремісії становить 50%,
при частковій ремісії — 15%. Такі показники зумовлюють необхід-
ність призначення інтенсивної терапії відразу після встановлення
діагнозу.
При лімфомах низького ступеня злоякісності залежність три-
валості життя від ефективності лікуванця виражена менше: 5-річ-
на виживаність перевищує 80%, незалежно від досягнення повних
або часткових ремісій.
Для лікування НЗЛ використовуються такі схеми хіміотерапії
• При 1-11 стадії захворювання терапією вибору є локальна
променева терапія в дозі 35-40 Гр.
• При ІІІ-ІУ стадії захворювання проводиться хіміотерапія за
наступними схемами:
135
Н.М. Третяк | Гематологія
СОР:
вінкристин — 1 мг/м2 в/в 1-й день;
циклофосфамід — 300 мг/м2 у таблетках або в/в 1—5-й дні;
преднізолон — 40 мг/м2 у табл. 1—5-й дні.
Курси повторюються кожні 28 днів.
СНОР:
вінкристин — 1,4 мг/м2 в/в 1-й день;
циклофосфамід — 750 мг/м2 в/в 1-й день;
доксорубіцин — 50 мг/м2 в/в 1-й день;
преднізолон — 50 мг/м2 у табл. 1—5-й день.
В останнє десятиріччя для лікування НЗЛ із високим сту-
пенем ефективності використовується флударабіна фосфат
(Флудара).
З метою лікування застосовуються моноклональні антитіла
проти антигена СО20, що є патогенетичним підходом до терапії
НЗЛ. Це такі препарати, як Мабтера (ритуксімаб) або Маб Кампат.
Монотерапія та в комплексі зі схемою СНОР дозволяє досягати
повних ремісій у 55% хворих. Стандартним є 4-годинне внутріш-
ньовенне введення ритуксімабу в дозі 375 мг/м2 один раз на тиж-
день 4 тижні поспіль.
Р:
Флударабін — 25 мг/м2 внутрішньовенно 1-5 дні. Курси повто-
рюються кожні 28 днів, усього 6-8 курсів. Або флударабін — 40
мг/м2 у таблетках 1 -5 дні кожні 28 днів, усього 6-8 курсів.
РС:
Флударабін — 25 мг/м2 внутрішньовенно 1-3 дні та циклофос-
фамід — 250 мг/м2 1-3 дні внутрішньовенно. Курси повторюються
кожні 28 днів, всього 6-8 курсів. Або флударабін — 30 мг/м2 у таб-
летках1-3 дні та циклофосфамід — 250 мг/м2 у таблетках 1-3 дні.
Курси повторюють кожні 28 днів, усього 6-8 курсів.
II):
флударабін — 25 мг/м2 внутрішньовенно 1-3 дні;
мітоксантрон — 10мг/м2 внутрішньовенно 1-й день.
РМВ:
флударабін 25 мг/м2 внутрішньовенно 1-3 дні;
мітоксантрон — 10 мг/м2 внутрішньовенно 1-й день;
дексаметазон — 20 мг/м2 у таблетках 1 -5 дні.
К:
ритуксімаб — 375 мг/м2 внутрішньовенно 1 раз на тиждень 4
тижні.
136
| Гемобластози
СНОР-К:
вінкристин — 1,4 мг/м2 внутрішньовенно 1-й день;
циклофосфамід — 750 мг/м2 внутрішньовенно 1-й день;
доксорубінин — 50 мг/м2 внутрішньовенно 1-й день;
ритуксімаб — 375 мг/м2 внутрішньовенно 1-й день;
преднізолон — 50 мг/м2 у таблетках 1-5 день.
Хворим молодого віку за наявності факторів несприятливо-
го прогнозу після стандартної ініціальної терапії показано прове-
дення високодозової хіміотерапії з наступною автологічною транс-
плантацією стовбурових клітин.
Зважаючи на те, шо проведення ПХТ, особливо високодозових
програм, призводить до пригнічення нормального кровотворення,
розвивається гранулоцитопенія і як наслідок — інфекційні усклад-
нення. Для скорочення тривалості гранулоцитопенїї в останні ро-
ки широко використовуються колонієстимулюючі фактори — Г-
КСФ, ГМ-КСФ. У разі виникнення тромбоцитопенії
внутрішньовенно вводиться тромбоконцентрат.
Нечутливість до загальноприйнятої терапії, рецидив після пер-
шої повної ремісії розцінюється як резистентний, у таких випадках
високодозова хіміотерапія та алотрансплантація кісткового мозку
можуть дати позитивний результат.
Курси хіміотерапії другої лінії
МАСОР-В:
доксорубіцин — 50 мг/м2 внутрішньовенно в 1, 3, 5, 7, 9, 11-й
тижні;
циклофосфамід — 350 мг/м2 внутрішньовенно в 1, 3, 5, 7,
9,11-й тижні;
вінкристин — 2 мг внутрішньовенно в 2, 4, 6,8,10,11-й тижні;
блеоміцин — 10 мг/м2 внутрішньовенно в 4, 8,12-й тижні;
преднізолон — 75 мг у таблетках щодня, доза знижується в ос-
танні 15 днів;
метотрексат — 400 мг/м2 у 2, 6, 10-й тижні;
кальціумфолінат — 15 мг у таблетках (8 введень) кожні 6 год., по-
чинаючи через 24 години після закінчення введення метотрексату.
т-ВАСОВ:
блеоміцин — 4 мг/м2 внутрішньовенно в 1-й день;
доксорубіцин — 45 мг/м2 внутрішньовенно 1-й день;
циклофосфамід — 600 мг/м2 внутрішньовенно в 1-й день;
вінкристин — 1 мг внутрішньовенно в 1-й день.
дексаметазон — 6 мг у таблетках 1-5 дні;
137
Н.М. Третяк | Гематологія
метотрексат — 200 мг/м2 внутрішньовенно 8,15-й дні;
кальціумфолінат — 10 мг внутрішньовенно або в таблетках
(8 введень) кожні 6 год., починаючи через 24 години після за-
кінчення введення метотрексату.
РгоМАОЕ/СуіаВОМ:
циклофосфамід — 650 мг/м2 внутрішньовенно в 1-й день;
доксорубіцин — 25 мг/м2 внутрішньовенно 1-й день;
етопозид — 120 мг/м2 внутрішньовенно в 1-й день;
преднізолон — 60 мг/м2 у таблетках 1-14 дні;
вінкристин — 2 мг внутрішньовенно 8-й день;
блеоміцин — 5 мг/м2 внутрішньовенно 8-й день;
цитозар — 300 мг/м2 внутрішньовенно 8-й день;
метотрексат — 120 мг/м2 внутрішньовенно 8,15-й дні;
кальціумфолінат — 15 мг у таблетках через 24, 30, 36, 42 год.
після закінчення введення метотрексату.
Курси проводяться кожні 4 тижні.
Курси хіміотерапії третьої лінії
т-ВАСО8:
метотрексат — 1000 мг/м2 внутрішньовенно в 10-й день (3-го-
динна інфузія);
кальціумфолінат — 15 мг внутрішньовенно в таблетках (8 вве-
день) кожні 6 год., починаючи через 24 години після закінчення
введення метотрексату;
доксорубіцин — 50 мг/м2 внутрішньовенно 1-й день;
вінкристин — 1,4 мг/м2 внутрішньовенно в 1-й день;
блеоміцин — 10 мг/м2 внутрішньовенно в 1-й день (за 15 хви-
лин);
циклофосфамід — 750 мг/м2 внутрішньовенно в 1-й день (за
15 хв.);
метілпреднізолон — 500 мг/м2 внутрішньовенно 1-3 дні (за
15 хв.).
Таким чином, провідну роль у лікуванні НЗЛ відіграє хіміоте-
рапія. Лише правильне використання всієї групи хіміопрепаратів та
засобів, що запобігають виникненню ускладнень, приводить до по-
зитивних наслідків.
Лімфогранулематоз (хвороба Годжкіна)
Лімфогранулематоз (ЛГМ) — це системний злоякісний лімфоп-
роліферативний процес позакістковомозкової локалізації, шо ура-
138
| Гемобластози
жає практично будь-який орган, котрий містить лімфоїдну ткани-
ну. Клітинним субстратом цієї пухлини є специфічна гігантська
клітина Березовського—Штернберга. У спектрі сучасної захворю-
ваності кровотворної тканини лімфогранулематоз (ЛГМ) посідає
одне з провідних місць.
Вперше увазі клініцистів повне, не описане раніше захворю-
вання представив англійський лікар Т. Годжкін у 1832 р., а звич-
ний сьогодні термін “ЛГМ” запропонував К. Штернберг. У сучас-
ній гематологи ЛГМ розглядається як одна з форм злоякісних
лімфом.
Епідеміологія. Вивчення епідеміології ЛГМ дозволило встано-
вити, що частіше на нього хворіють мешканці країн із низьким рів-
нем життя або ті, хто проживає в екстремальних географічних умо-
вах (геомагнітні зони, значна інсоляція, високий радіаційний,
температурний фон), а представники білої раси частіше, ніж мон-
голоїдної та негроїдної. В Україні поширеність цього захворюван-
ня становить близько 50 осіб на 100 тис. населення. Розподіл віко-
вої захворюваності за даними різних авторів має трипіковий
характер: в середньому 4—6 років, 20—36 років та понад 50 років.
Чоловіки хворіють дещо частіше за жінок.
Етіологія. Незважаючи на досягнуті успіхи у вивченні ЛГМ,
етіологія цього захворювання, як і більшості пухлинних процесів,
залишається остаточно не з’ясованою. З-поміж великої кількості
факторів, що можуть спровокувати пухлинну трансформацію, виз-
начають основні, найбільш суттєві.
До них належать:
- генетичні аномалії;
- вірусні інфекції (вірус Епштейна—Барра, РНК-ретровіруси);
- неадекватність імунного захисту;
- умови життя.
Однак і при цьому можна говорити лише про підвищення ри-
зику, більшу або меншу імовірність неоплазії, а не про абсолютну
значущість цих факторів у її розвитку.
Патогенез. В основу патогенезу ЛГіуі покладено пухлинну
трансформацію лімфатичної клітини, яка походить, переважно,
з лімфоцитів зародкового центру фолікула та лімфатичних вузлів.
В імунному фенотипі основними маркерами є експресовані СО15,
СО30, а в Т-клітинній лінії — СО4.
Під впливом етіологічного чинника В-лімфоиити втрачають
спроможність до основної функції — секреції І£ і, крім того, ста-
Н.М. Третяк | Гематологія
ють більш стійкими перед апоптозом (запрограмована загибель
клітини). Трансформовані таким чином, вони накопичуються
й руйнуються Т-лімфоцитами. На певному етапі пул Т-лімфоци-
тів виснажується, клітинна ланка імунітету не спроможна адек-
ватно контролювати аномальний клон, наслідком чого є прогре-
сивний рост пухлини. Саме тому деякими клініцистами ЛГМ
розглядається як імунне захворювання, оскільки початково в йо-
го основі лежить імунний конфлікт.
Надалі до патологічного процесу залучаються лімфатичні вуз-
ли, групи лімфатичних вузлів, потім — система груп, в тому числі
селезінка і тимус, пізніше метастазування поширюється на екстра-
нодальні (не лімфоідні) органи та тканини, тобто виходить за ме-
жі лімфатичної системи. Поширення пухлинних клітин із первин-
ного вогнища відбувається переважно лімфогенно та меншою
мірою гематогенно. Таке уявлення про патогенез ЛГМ було покла-
дено в основу розробки сучасних схем хіміо- та променевої тера-
пії, в котрих враховані зони регіонарного метастазування та розу-
міння необхідності знищення пухлинного клону не локально, а по
всьому пухлинному полю.
Патоморфологія. При дослідженні біоптату лімфатичного вуз-
ла визначаються порушення його загальної структури. В клітинно-
му складі серед морфологічно повноцінних клітин переважають
малі лімфоцити, зустрічаються еозинофіли, гранулоцити, плазма-
тичні клітини. Патологічний субстрат представлений клітинами
Березовського—Штернберга, котрі мають абсолютне діагностичне
значення, можуть визначатися їхні попередники — клітини Год-
жкіна. Фактично сам діагноз ЛГМ може вважатися остаточно ве-
рифікованим тільки за наявності цих клітин.
Класифікація. На сьогоднішний день існує дві основні класи-
фікації ЛГМ: гістологічна, в основу якої покладено клітинний
(морфологічний) склад пухлини, що утворює лімфоїдну гранульо-
му, та за стадіями клінічного перебігу, котра фактично відображає
анатомічну поширеність процесу.
Гістологічна класифікація ЛГМ (Міжнародна класифікація
ВООЗ, 2001)
І Лімфогістіоцитарний
II Нодулярний склероз
ПІ Змішаноклітинний
IV Лімфоїдне виснаження
І та II гістологічні варіанти захворювання зустрічаються часті-
мо
| Гемобластози
ше в молодому віці та мають найсприятливіший перебіг, ПІ та IV
варіанти менш сприятливі, діагностуються частіше в людей похи-
лого віку.
Клінічна класифікація (Апп-АгЬог, 1971)
І стадія — ураження лімфатичних вузлів однієї групи або од-
ного екстранодального (позалімфатичного) органу чи тканини
(ІЕ);
II стадія — ураження лімфатичних вузлів двох або більше ді-
лянок по один бік діафрагми (II,, П3) або, додатково до цього, ура-
ження одного екстранодального органу (НЕ) чи тканини (ІГО) по
той же бік діафрагми;
III стадія — ураження лімфатичних вузлів будь-яких ділянок
з обох боків діафрагми або, додатково до цього, ураження одного екс-
транодального органу чи тканини, а також/чи селезінки (ШЕ, 1118);
IV стадія — дисеміноване ураження одного чи більше екс-
транодального органу з ураженням лімфатичних вузлів або без
цього.
Позначки', ураження екстранодальне — Е, селезінки — 8, ле-
гень — Е, плеври — Р, шкіри — О, кісткового мозку — М, кісток
— О, наявність симптомів інтоксикації — В, без них — А.
До сьогодні залишається остаточно не з’ясованим питання: чи
різні стадії перебігу ЛЕМ є послідовними етапами пухлинної прог-
ресії, які поступово змінюють один одний, чи вони є різними фор-
мами одного захворювання.
Клінічна картина. ЛГМ, як злоякісному неопластичному зах-
ворюванню, притаманні основні пухлинні феномени: пухлинна
прогресія, проліферація та інтоксикація. Саме від ступеня їх вира-
женості, зрештою, залежить клінічна картина.
Симптоматика захворювання, передусім, визначається віком
пацієнта, розповсюдженням пухлинного клону в лімфатичній сис-
темі та глибиною ураження внутрішніх органів аж до значного по-
рушення їх функції. Крім того, суттєве значення може мати наяв-
ність супутньої патології, котра не пов’язана з основним
захворюванням, але спроможна негативно вплинути на клінічний
перебіг процесу.
На фоні досить варіабельної клінічної симптоматики можна
визначити основні, найбільш характерні прояви.
Основні клінічні прояви ЛГМ:
1) ураження лімфатичних вузлів;
2) ураження внутрішніх органів та тканин;
141
Н.М. Третяк | Гематологія
3) автоімунні зрушення;
4) прояви інтоксикації.
Лімфатичні вузли при ЛГМ уражаються найчастіше (до 90%),
особливо налдіафрагмальні — шийні, надключичні, пахвові, меді-
астінальні. Взагалі, можливе ураження вузла будь-якої локалізації.
При пальпації вузли можуть бути як безболісними, так і болючи-
ми, на початкових стадіях — м’якої консистенції з подальшим
ущільненням аж до кам’янистості. За умов залучення до патологіч-
ного процесу капсули, вузли зливаються в конгломерати, інколи
значних розмірів, зрощуються з навколишніми тканинами (м’яза-
ми, підшкірною клітковиною, шкірою), можуть тиснути на нерво-
ві скінчення та судини.
Селезінка залучається до гранулематозного процесу на різних ста-
діях розвитку майже в 50% випадків. Вона стає доступною для паль-
пації, інколи болюча. У деяких випадках можливі імунні розлади та
прояви гіперспленізму (гемоліз, тромбоцитопенія). Слід зазначити,
шо не завжди ступінь ураження селезінки відповідає ступеню сплено-
мегалп. інколи, навіть при дифузній пухлинній трансформації, спле-
номегалії може не бути зовсім, або вона може бути мінімальною.
Легенева локалізація ЛГМ майже така сама за частотою, як і
селезінкова. Процес може бути одно- і/або двостороннім, як пра-
вило, супроводжується збільшенням лімфатичних вузлів, часто —
залученням плеври. Хворі потерпають від кашлю, задухи, у важких
випадках — аж до легеневої недостатності, особливо за умов гідро-
тораксу. Перкуторна та аускультативна картина типова для інфіль-
тративного ураження легенів.
• Крім того, первинно або на певному етапі розвитку хвороби
можливе ураження структур кістяка, найчастіше грудини та попе-
рекового відділів, рідше грудного, ребер та інші локалізації. Клініч-
но це супроводжується появою болю за типом корінцевого або
проходить асимптоматично.
При ураженні печінки симптоматика може відповідати карти-
ні гепатиту з наявністю типово локалізованого болю та жовтяниці.
У важких випадках порушення функції печінки може вкрай нега-
тивно вплинути на перебіг захворювання і навіть спричинити
смерть хворого.
До патологічного процесу можуть залучатися інші органи, шо
супроводжується певними клінічними проявами. Тимус — слаб-
кість, підвищення температури тіла; шлунково-кишковий тракт —
болі, шлункова та кишкова диспепсія; шкіра — свербіння, мацера-
142
| Гемобластози
ція, інфільтративне ураження зі зміною кольору шкіри, можливий
розвиток псоріазу; кістковий мозок —анемія, геморагії; молочні за-
лози — набряк, збільшення розміру залози. Частота їх невелика,
приблизно 1-10%, і вкрай рідко є первинною локалізацією.
Крім того, для ЛГМ характерні ознаки інтоксикації. До них на-
лежать:
- слабкість;
- підвищення температури тіла без певної причини та добової
залежності;
- пітливість, інколи дуже значна, особливо в нічні години;
- зменшення маси тіла.
Діагностика ЛГМ складна та кропітка, оскільки необхідно не
тільки з’ясувати факт наявності ЛГМ, а й максимально точно вста-
новити анатомічну поширеність неоплазії в лімфатичній системі,
виявити екстранодальні ураження та їх ступінь, можливу супутню
патологію, котра може вплинути на оцінку прогнозу, а згодом — і
на вибір програми лікування.
Діагностичний пошук має включати об’єктивне обстеження
хворого та лабораторно-інструментальні методи дослідження, в то-
му числі інвазивні.
1. Об’єктивний огляд пацієнта:
- оцінка загального стану за органами та системами, наяв-
ність ознак інтоксикації, лімфоаденопатії (локалізація вузлів, іхні
розміри, консистенція, болючість, чи зв’язані вони з навколиш-
німи тканинами), спленомегалії, екстранодального ураження (ге-
патомегалія, прояви ураження кісток, молочних залоз, шкіри та
інші).
2. Лабораторно-інструментальна діагностика:
- загально-клінічні методи обстеження: загальний аналіз крові,
сечі, калу, біохімічне дослідження крові, ЕКГ;
- спеціальні методи дослідження:
а) гістологічне дослідження відбитка первинно збільшеного
лімфатичного вузла та за показаннями селезінки, шкіри, мо-
лочної залози, трепанату здухвинної кістки,
Ь) імунофенотипування,
с) УЗД, за потреби — комп’ютерна томографія органів грудної
та черевної порожнин, малого тазу, головного мозку, мамографія,
б) ФГДС із біопсією,
е) рентгенологічне дослідження легенів, кісток (за потреби —
сцинтіграфія);
143
Н.М. Третяк | Гематологія
- додаткові методи дослідження:
а) методи для виявлення та оцінки значущості супутньої пато-
логії, консультації вузьких спеціалістів,
Ь) діагностична лапаротомія зі спленектомією (проводиться ду-
же рідко та лише у випадках виникнення ускладнень при верифі-
кації діагнозу).
Сеппап НосІ£кіп’$ ЕітГота Зійду Сгоир (СН8С) крім вираже-
ності лімфоаденопатїї та наявності вогнищ екстранодальної пролі-
ферації з-поміж факторів ризику в перебігу ЛГМ визначає збіль-
шення ШОЕ (понад 50 мм при відсутності інтоксикації та більше
ЗО мм за її наявності).
Таблиця 14. Прогностичні критерії при ЛГМ
Ознаки Сприятливі Несприятливі
Вік хворого до 50 років понад 50 років
Ознаки інтоксикації немає наявні
Лімфоаденопатія поодинокі л/в без утворення конгломератів конгломерати л/в
Супутня соматична патологія, особливо з порушенням функції органу немає наявна
Клінічна стадія захворювання І-ІІ ІІІ-М
Гістологічний варіант лімфогістіоцитарний, нодулярний склероз змішано- клітинний, лімфоїдне виснаження
Диференційний діагноз при ЛГМ необхідно проводити, насам-
перед, із захворюваннями, перебіг яких супроводжується лімфоаде-
нопатією, і найчастіше — зі злоякісними негоджкінськими лімфо-
мами, хронічною лімфоїдною лейкемією, та інфекційним
мононуклеозом.
Крім того, при верифікації діагнозу необхідно враховувати
можливість туберкульозного ураження лімфатичних вузлів, реак-
тивних лімфаденітів, інфекційних захворювань, котрі можуть суп-
роводжуватися збільшенням лімфатичних вузлів.
Труднощі в діагностиці, зазвичай, виникають при первин-
ному ураженні екстранодальних органів та тканин (кістки, шкі-
ра, легені, печінка, шлунково-кишковий тракт) або абдоміналь-
них та медіастінальних лімфовузлів. У такому випадку ознаки
ЛГМ можуть маскуватися легеневими, неврологічними, дис-
пептичними та іншими розладами та потребують поглибленої
діагностики.
144
| Гемобластози
Таблиця 15. Диференційно-діагностичні критерії при ЛГМ
Критерій/ нозологія ЛГМ ХЛЛ НЗЛ Інф. мононуклеоз
Первинна локалізація лімфоїдні органи або екстранодальні органи кістковий мозок лімфоїдні органи або екстранодальні органи лімфатичні вузли
Пухлинний субстрат клітини Березовського- Штернберга зрілі лімфоцити зрілі лімфоцити пухлинного субстрату немає
Лімфоаденопатія наявна наявна наявна наявна
Консистенція лімфатичних вузлів від м’якої до дуже щільної еластично- м’які середньо- еластичні щільні
Конгломерати лімфатичних вузлів наявні, з’язані з оточуючими тканинами зрідка, не зв’язані з оточуючими тканинами зрідка, не зв’язані з оточуючими тканинами відсутні
Кістковий мозок специфічних змін немає значний лімфоцитоз специфічних змін немає незначний лімфоцитоз, моноцитоз
Периферична кров специфічних змін немає лейкоцитоз, лімфоцитоз специфічних змін немає лейкоцитоз, мононуклеари, можливі плазматичні клітини
Інтоксикація наявна можлива можлива наявна
Лікування будь-якого пухлинного захворювання до сьогодні є
надзвичайно складною та далеко не вирішеною проблемою. Успі-
хи фармакології, біологічної та молекулярної хімії дозволили син-
тезувати сполуки, які вкрай агресивно впливають на пухлинну клі-
тину. Завдяки розробці спеціальних програм, ЛГМ сьогодні вже не
вважається захворюванням з абсолютно несприятливим прогнозом
і переведений до розряду виліковних.
Наразі для лікування ЛГМ використовують променеву тера-
пію, хіміотерапію або їх комбінацію. Вважається встановленим
той факт, що ізольована променева терапія показана хворим пе-
реважно на початкових стадіях розвитку захворювання та є мало-
ефективною в усіх інших випадках. Самостійна поліхіміотерапія
без опромінення являє собою ефективний метод для індукції ре-
місії, але кількість рецидивів, у тому числі ранніх, при цьому дос-
татньо велика. Тому сучасні програми передбачають інтенсифіка-
цію лікування, тобто комбінацію променевої та хіміотерапії, шо
145
Н.М. Третяк | Гематологія
дозволяє досягти максимального ефекту — тривалої ремісії, по-
довження безрецидивних проміжків і, таким чином, загальної ви-
живаності пацієнтів аж до одужання.
При цьому слід пам’ятати про можливість розвитку індуко-
ваних терапією інших неопластичних захворювань, найчастіше
гострих лейкемій, тому підходи до її вибору мають бути макси-
мально ефективними щодо ерадикації пухлини, але мінімально
агресивними для пацієнта.
Програми першої лінії (індукція ремісії). При діагностуванні 11
ст. захворювання без несприятливих ознак найчастіше використо-
вуються МОРР, МУРР, СУРР, а з цими ознаками — АВУО. Хімі-
отерапія комбінується з опроміненням у невеликих дозах. Сумар-
но вона дорівнює 36-40 Гр, але за потреби може бути збільшена до
40 Гр. При цьому вплив на всі лімфоідні колектори, котрі знахо-
дяться вище діафрагми, є обов’язковим.
Друга лінія (лікування резистентних форм, ранніх та пізніх ре-
цидивів) —Г)еха-Г)ЕАМ, СЕР, РЕСС. Доза опромінення може не
збільшуватись.
Позитивними критеріями прогнозу на фоні лікування є вчас-
не досягнення ремісії згідно з протоколом та відсутність рецидивів
щонайменше 2 роки.
Несприятливими ознаками є несвоєчасне досягнення ремісії
або розвиток резистентної форми ЛГМ, ранні рецидиви, велика за-
лишкова маса пухлинної тканини та наявність ознак інтоксикації
після проведення основної терапії.
Третя лінія — високодозова хіміотерапія (ВЕАМ, СВУ) із вико-
ристанням колонієстимулюючих факторів та підсадок стовбурових
та ембріональних клітин.
Лімфоплазмоцитарна лімфома (макроглобулінемія
Вальденстрема)
У 1944 р. Лап Со$їа \Уа1сіеп$ігот описав двох пацієнтів із но-
совими кровотечами, лімфаденопатією, гепатоспленомегалією,
прискореною швидкістю зсідання еритроцитів, високою в’язкістю
крові, відсутністю патології на рентгенограмах кісток та інфільтра-
цією кісткового мозку лімфоцитами. Ця стаття стала першою
згадкою в медичній літературі широковідомого, проте досить рід-
кісного захворювання, котре дістало назву макроглобулінемії
Вальденстрема.
146
| Гемобластози
Лімфоплазмоцитарна лімфома (ЛЛ) належить до групи В-клі-
тинних парапротеїнемічних гемобластозів, морфологічно представ-
лених у кістковому мозку лімфоцитами, плазмоцитами та їхніми
перехідними формами, що мають здатність до гіперпродукцїї мо-
ноклонального імуноглобуліну М.
Відповідно до сучасних класифікацій, ЛЛ — це лімфоплазмоци-
тарна лімфома, котра характеризується дифузною лімфоїдно-плаз-
моклітинною інфільтрацією кісткового мозку з переважно інтратрабе-
кулярним ростом пухлини та супроводжується І§М-моноклональною
гамапатією.
Епідеміологія. Захворюваність на ЛЛ становить 1,7 випадку на
1 млн. населення на рік серед жінок та 3,4 — серед чоловіків, шо
приблизно дорівнює 2% від загальної захворюваності на гемоблас-
този або 10-20% від поширеності мієломної хвороби. Середній вік
пацієнтів становить 65 років. Захворюваність на ЛЛ зростає в за-
лежності від віку — від 0,1 на 1 млн. на рік серед осіб, молодших
за 45 років, до 36,3 серед тих, чий вік перевищує 75 років.
Клінічна картина. Приблизно в однієї третини пацієнтів із ді-
агнозом ЛЛ спостерігається лімфоденопатія, гепато- та спленоме-
галія. Для них характерна нічна пітливість, слабкість, зниження ва-
ги, періодично — субфебрильна температура тіла.
Макроглобулін, котрий продукується патологічно зміненими
лімфоїдними та плазмоцитарними елементами, може циркулювати
в плазмі, збільшуючи в’язкість крові, а також відкладатися у тка-
нинах. З-поміж симптомів, пов’язаних із циркуляцією патологіч-
ного І§М у плазмі, виділяють симптом гіперв’язкосД, кріоглобуті-
немії та холодової аглютинаційної хвороби. До симптомів,
пов’язаних із накопиченням його в різних тканинах, відносять
прояви периферичної полінейропатїї, параамілоїдозу внутрішніх
органів, ураження нирок та шкіри.
На момент встановлення діагнозу синдром гіперв’язкості спосте-
рігається у 10-30% хворих та клінічно характеризується кровотечами
з ясен та носу, слабкістю та ускладненнями з боку серцево-судинної
(серцева недостатність) та нервової систем (головний біль, запаморо-
чення, атаксія, енцефалопатія). Як правило, подібні симптоми з’яв-
ляються в тих хворих, у яких рівень І§М у плазмі перевищує 30 г/л.
Кріоглобуліни — це протеїни плазми чи комплекси протеїну,
котрі демонструють феномен преципітації при зниженні темпера-
тури тіла. Тип 1 кріоглобулінів складається із моноклонального
1§М і спостерігається у 10-20% хворих на ЛЛ. Клінічні прояви цьо-
147
Н.М. Третяк | Гематологія
го патологічного стану є у 5% пацієнтів. Вони характеризуються
синдромом тяжкого імунокомплексного васкуліту з ураженням
шкіри, центральної та периферичної нервової системи, синдромом
Рейно, гломерулонефритом.
Приблизно у 10% хворих на ЛЛ патологічний протеїн плазми
поводить себе як температурозалежне антитіло, котре не взаємодіє
з еритроцитами людини при температурі тіла, проте має високу
афінність до них при переохолодженні. У таких пацієнтів спосте-
рігаються явища акроцианозу, холодової кропивниці, синдрому
Рейно, а також може бути гостра чи хронічна гемолітична, анемія
різного ступеня тяжкості.
До симптомів, пов’язаних із відкладанням патологічного І§М-
протеїну в різних тканинах, належать прояви периферичної поліней-
ропатії, параамілоідозу внутрішніх органів, ураження нирок та шкіри.
Полінейропатія трапляється у 10% хворих на ЛЛ. Серед пато-
генетичних механізмів розвитку цього ускладнення виділяють ін-
фільтрацію плазматичними клітинами периферичних нервів, інду-
кування антитіл проти глікопротеїнів та гліколіпідів периферичних
нервів, а також відкладання параамілоїду. Клінічно при полінейро-
патїї спостерігаються симптоми чутливих та рухових порушень,
атаксія, парестезія, слабкість та атрофія дистальних м’язів кінцівок,
що супроводжується больовим синдромом.
Параамілоїдоз у хворих на ЛЛ у 76% випадків пов’язаний із
легкими ланцюгами А [§М має системний характер та вражає сер-
це, нирки, легені, печінку у приблизно 2% хворих на ЛЛ.
Для симптомів ураження шкіри при ЛЛ, котрі пов’язані із від-
кладанням патологічного І§М-протеїну в тканинах, характерні
тверді, напівпрозорі інтраепідермально розташовані папули або ур-
тикарна висипка (БсИпіїхІег’.ч зупсіготе).
Діагноз ЛЛ базується на трьох основних критеріях — виявлен-
ні інфільтрації кісткового мозку лімфоплазмоцитарними клітина-
ми при морфологічному дослідженні, визначенні патологічного
моноклонального 1§М у плазмі та характерному для цього захво-
рювання імунофенотипі лімфоїдних клітин.
Головним у діагностиці ЛЛ є інфільтрація кісткового мозку
лімфоплазмоцитами з імунофенотипом І§М+СП5+СП10-
СОі9+СО20+СВ2,+СО23-СП,5+СЕ),7+ЕМС7+СО103-СО138-. При
цитологічному дослідженні виявляються лімфоцити, котрі мають
ознаки плазматизанії (клітини з інтенсивно базофільною цитоплаз-
мою, але лімфоцитоподібним ядром), плазматичних клітин та їх
148
| Гемобластози
перехідні форми. В залежності від щільності інфільтрації кістково-
го мозку патологічними клітинами у пацієнтів в аналізі перифе-
ричної крові може спостерігатися тільки анемія різного ступеня
(від незначної до глибокої) або анемія в поєднанні з вираженою
цитопенією (лейкопенією, нейтропенією, тромбоцитопенією).
Наявність І§М-моноклонального протеїну, котрий секретуєть-
ся патологічно зміненими лімфоцитами, вважається надзвичайно
важливим критерієм у встановленні діагнозу ЛЛ. Кількість цього
протеїну в сироватці крові варіює у різних пацієнтів, але, як пра-
вило, перевищує нормальні величини (більш ніж 0,5 мг/мл).
Лікування. За пацієнтами із безсимптомною ЛЛ необхідно наг-
лядати, не призначаючи лікування, оскільки їхній стан залишаєть-
ся стабільним протягом багатьох років. Таким пацієнтам не слід
призначати лікування тільки тому, що в них виявляється підвище-
на концентрація моноклонального протеїну або знайдена патоло-
гічна інфільтрація кісткового мозку лімфоплазмоцитарними кліти-
нами.
Показанням до призначення хіміотерапії пацієнтам з ЛЛ є
зменшення гемоглобіну (<100 г/л) чи тромбоцитів (<100 • 109/л),
зумовлене патологічним процесом у кістковому мозку. Такі усклад-
нення хвороби, як синдром гіперв’язкості, сенсорно-моторна пе-
риферична нейропатія, системний параамілоїдоз, ниркова недос-
татність чи симптоматична кріоглобулінемія, також потребують
лікування.
Плазмафарез є допоміжним заходом, котрий може швидко
зменшити рівень в’язкості крові та запобігти виникненню серйоз-
них ускладнень, пов’язаних із надмірною гіперв’язкістю плазми.
Лікування хлорамбуцилом у режимі монотерапїї або в поєднан-
ні з преднізолоном протягом тривалого часу використовується як
терапія першої лінії у хворих на ЛЛ. Така терапія вважається ефек-
тивною приблизно у 50-60% хворих, проте повні ремісії досягають-
ся рідко. Тривалість життя хворих становить приблизно 5 років.
Відповідь на терапію хлорамбуцилом зазвичай повільна (в серед-
ньому 18 місяців). Хлорамбуцил у комбінації із плазмафарезом (як-
що є симптом гіперв’язкості) — адекватне лікування для хворих
похилого віку, котрі не мають серйозних ускладнень.
Монотерапія пуриновими аналогами (флударабін) призначається
в дозі 25-30 мг/м2 в/в протягом 5 днів, 6-8 курсів лікування, курси
повторюються кожні 4 тижні. Приблизно 80% хворих із уперше
встановленим діагнозом ЛЛ позитивно відповідають на лікування
149
Н.М. Третяк | Гематологія
флударабіном (повна або часткова ремісія). Флударабін також од-
ним із перших досить успішно почав застосовуватися у пацієнтів із
рецидивом захворювання та в тих, котрі виявилися рефрактерни-
ми до лікування алкілюючими агентами (хлорамбуцилом). Відпо-
відь на призначення препарату досить швидка, і, як правило, по-
зитивний результат досягається протягом перших двох циклів
лікування. Тому лікування флударабіном оптимальне для хворих із
серйозними ускладненнями, шо становлять загрозу для життя, як-
то синдром гіперв’язкості чи периферична нейропатія.
Ритуксімаб (Мабтера) призначається в дозі 375 мг/м2 один
раз на тиждень повільною внутрішньовенною інфузією протягом
4 тижнів як у пацієнтів із уперше встановленим діагнозом, так і
під час рецидиву захворювання. Препарат має досить високу
ефективність при лікуванні периферичної нейропатії, особливо
в пацієнтів, у яких значно виражені симптоми моторно-сенсор-
них порушень.
Курси поліхіміотерапії (СОР, СНОР, АУАМР, М2). У хворих із
уперше встановленим діагнозом ЛЛ СОР є ефективним у 65% ви-
падків, 11 % хворих із рефрактерною та рецидивною ЛЛ відповіда-
ють на лікування САР, середня тривалість відповіді становить З
місяці.
Автологічна трансплантація показана для порівняно молодих
пацієнтів із рефрактерною або рецидивною ЛЛ і є досить ефектив-
ною для більшості хворих, включаючи й тих, котрі є резистентни-
ми до аналогів пурину.
Кортикостероїдні гормони не мають самостійного значення
в лікуванні та використовуються, в основному, при імунній ге-
молітичні анемії, холодовій аглютинаційній хворобі та кріоглобу-
лінемії.
Призначення терапії хворим на ЛЛ може подовжити тривалість
життя хворих та позитивно вплинути на його якість. При досяг-
ненні повної ремісії (відсутність у плазмі та сечі хворого на ЛЛ мо-
ноклонального 1§М, злоякісних клітин при гістологічному дослід-
женні кісткового мозку, нормалізація розмірів лімфатичних вузлів,
печінки та селезінки, відсутність невмотивованої періодичної ли-
хоманки > 38,4°С, нічної пітливості, зменшення ваги більш ніж на
10%, синдромів гіперв’язкості та кріоглобулінемії) або часткової
ремісії (зменшення більш ніж на 50% рівня моноклонального І§М
у плазмі від того рівня, котрий був у пацієнта на початку лікуван-
ня, зменшення на 50% розмірів лімфовузлів, печінки та селезінки)
150
| Гемобластози
пацієнт повинен перебувати під ретельним наглядом гематолога та
перевіряти рівень І§М у плазмі кожні 3 місяці. При виникненні ре-
цидиву захворювання, в залежності від тривалості попередньої ре-
місії, або призначається аналогічна схема терапії, або пацієнт пе-
реводиться на лікування за більш складним протоколом.
Прогноз. Середня тривалість життя у хворих на ЛЛ становить 5
років, хоча як мінімум 20% із них живуть понад 10 років. Негатив-
ний вплив на тривалість життя справляють такі параметри: вік па-
цієнтів 65 або більше років на момент встановлення діагнозу, чо-
ловіча стать, рівень альбуміну < 40 г/л, рівень гемоглобіну менш
ніж 120 г/л, кількість тромбоцитів < 150- 109/л, кількість лейкоци-
тів менш ніж 4 • 109/л або глибока цитопенія, значна гепатомегалія.
При проведенні адекватної терапії тривалість життя пацієнтів
збільшується до 7-9 років.
Мієломна хвороба (множинна мієлома)
Мієломна хвороба (МХ) — це злоякісне новоутворення системи
крові, що належить до групи парапротеїнемічних гемобластозів і
виникає в результаті безконтрольної проліферації в кістковому
мозку плазматичних клітин, котрі мають здатність продукувати ве-
лику кількість моноклонального імуноглобуліну (І§С, І§А, і§Б,
І§Е) або легких ланцюгів каппа (к) чи лямбда (А).
Епідеміологія. Захворюваність на мієломну хворобу становить
приблизно 40 випадків на один мільйон населення. Середній вік
пацієнтів на момент встановлення діагнозу — 60-65 років, близько
2% хворих старші за 40 років. Чоловіки хворіють частіше за жінок.
Етіопатогенез. Етіологія мієломної хвороби не відома. До по-
тенційних факторів ризику відносять контакт із радіоактивними
речовинами, пестицидами, бензолом, деякими органічними роз-
чинниками.
Як правило, мієлома проявляється множинними літичними
пухлинами в кістках, остеопорозом та дифузним плазмоцитозом
у кістковому мозку. Значна кількість патологічного моноклональ-
ного імуноглобуліну (мієломні білки), котрий продукується плаз-
матичними клітинами, циркулює в плазмі, внаслідок чого може
збільшуватися її об’єм та загальна в’язкість крові. Мієломні білки
взаємодіють із факторами згортання крові та, огортаючи тромбо-
цити, впливають на їхню функцію й підсилюють кровоточивість.
Також патологічні імуноглобуліни можуть відкладатися в міокарді,
151
Н.М. Третяк | Гематологія
нервах (периферична полінейропатія), нирках (нефросклероз, нир-
кова недостатність), призводити до амілоїдозу внутрішніх органів.
Крім мієломного білка, плазматичні клітини мають здатність також
продукувати різні цитотоксичні фактори, наприклад фактор, кот-
рий активує функцію остеокластів. Остеокласти зумовлюють ре-
зорбцію кісткової тканини, в результаті чого з’являються болі в кіс-
тках, патологічні переломи та виникає гіперкальціємія. Наслідком
інфільтрації кісткового мозку плазматичними клітинами є анемія
різного ступеня тяжкості, лейкопенія та/або тромбоцитопенія.
Зменшення кількості лейкоцитів та порушення синтезу нормаль-
них імуноглобулінів може викликати депресію гуморального та,
меншою мірою, клітинного імунітету, шо робить таких хворих ду-
же чутливими до багатьох інфекцій (переважно бактеріальних).
Клінічна картина. Клінічні ознаки захворювання досить різно-
манітні. Хворі, як правило, скаржаться на постійний біль у кістках,
спині та попереку, шо поєднується зі зменшенням зросту та осте-
опорозом (особливо у чоловіків та в жінок до періоду менопаузи).
Можлива компресія спинного мозку та спиномозкових нервів із
виникненням відповідної неврологічної симптоматики (аж до
нижнього парапарезу та порушення функції тазових органів). Зви-
чайним явищем для пацієнтів є патологічні переломи.
Хворі на МХ потерпають від частих інфекційних ускладнень
(викликаних переважно Зїгерїососсш рпеишопіе, ЗіарЬуІососсих
аигеих, грам-негативними мікроорганізмами), відчувають слабкість,
задишку, запаморочення, шо пов’язані з анемією та пухлинною ін-
токсикацією, можлива наявність симптомів, характерних для нир-
кової недостатності. Наслідком коагулопатії та тромбоцитопенії
можуть бути прояви підвищеної кровоточивості. У частини пацієн-
тів спостерігається безсимптомний перебіг МХ, і захворювання ви-
являється при дослідженні аналізу крові, котре проводиться з ін-
шого приводу.
Діагностика. У загальному аналізі крові може відмічатись ерит-
роцитопенія, лейкопенія та тромбоцитопенія. Особливе діагнос-
тичне значення має збільшення ШОЕ.При дослідженні сироватки
крові нерідко привертає увагу виражена протеїнемія, котра вини-
кає за рахунок збільшеної продукції плазматичними клітинами мо-
ноклональних імуноглобулінів певного класу. У сечі хворих на МХ
можна виявити протеїнурію, часто позитивною є реакція на білок
Бенс-Джонса (зазначимо, що ця реакція не є патогномонічною для
МХ і може спостерігатися за інших патологічних станів).
152
| Гемобластози
Рентгенологічне дослідження кісток скелету (грудної КЛІТКИ,
тазу, черепа, плечової та стегнової кістки, хребта) дає змогу вияви-
ти в них вогнища резорбції кісткової тканини та остеопорозу. Про-
те слід пам’ятати, що специфічних рентгенологічних ознак, харак-
терних для МХ, не існує. Відсутність остеодеструкцій не виключає
захворювання, а їх наявність вважається недостатньою для підтвер-
дження діагнозу.
Для верифікації діагнозу мієломної хвороби необхідне прове-
дення стернальної пункції з подальшим підрахунком мієлограми,
що дає змогу встановити плазмоклітинну інфільтрацію кісткового
мозку. Проте при множинно-вогнищевих формах захворювання,
коли не спостерігається дифузного ураження кісткового мозку, мі-
єлограма може бути нормальною. За наявності інших ознак МХ
(остеодеструкнія, моноклональна імуноглобулінопатія) для під-
твердження діагнозу слід призначати повторні проколи грудини
в різних місцях, трепанобіопсію клубової кістки, проводити пун-
кції'в місцях кісткових пухлин, при спірних питаннях можливе
проведення резекції ураженої кістки (ребра, лопатки).
Діагноз мієломної хвороби вважається достовірним при сукуп-
ності наступних ознак.
1. Кількість плазматичних клітин у кістковому мозку >/= 10%
та/або встановлена присутність солітарної плазмоцитоми, котра
підтверджена з допомогою біопсії.
2. Виявлення моноклонального білка в плазмі та/або сечі (як-
що моноклональний протеїн не виявлено, то кількість плазматич-
них клітин у кістковому мозку має перевищувати 30%).
3. Рівень кальцію в крові — на верхній межі норми або вище,
ниркова недостатність (креатинін > 20 мкмоль/л), анемія (гемогло-
бін менше 100 г/л), вогнища деструкції в кістках або остеопороз
(якщо присутній тільки остеопороз, то для підтвердження діагнозу
необхідно мати більш ніж 30% плазматичних клітин у кістковому
мозку).
Лікування. Лікування пацієнтів із мієломною хворобою полягає
в проведенні хіміотерапії, спрямованої на боротьбу із пухлинним
клоном, та в корекції ускладнень, пов’язаних із перебігом хвороби
(синдром анемії, синдром резорбції кісткової тканини, ниркова не-
достатність та ін.).
Слід зазначити, шо стан пацієнтів, у яких відсутні клінічні
симптоми захворювання, нормальний рівень гемоглобіну та каль-
цію в плазмі, немає порушень у функції нирок та синдрому резор-
153
Н.М. Третяк | Гематологія
бції кісток, може лишатися стабільним протягом тривалого часу
(“тліюча”, або “лінива” мієлома) й не потребувати специфічного
лікування. Таким хворим, одначе, необхідно проходити ретельний
лікарський огляд та дослідження парапротеїну в плазмі та сечі кож-
ні 3 місяці.
Хворим, у котрих захворювання має виражені клінічні ознаки
або спостерігається прогресування захворювання, слід проводити
курси хіміотерапії.
Найчастіше на першому етапі лікування пацієнтам признача-
ється комбінація мелфалану із преднізолоном. Як правило, така лі-
кувальна схема прийнятна для пацієнтів похилого віку, котрим не
планується проведення високодозної терапії із автологічною транс-
плантацією кісткового мозку. Мелфалан застосовують у дозі 6-8
мг/м2 на добу разом із преднізолоном у дозі 40-60 мг/добу протя-
гом 4-7 днів із 4-6-тижневими інтервалами. Лікування триває до
досягнення максимальної відповіді на лікування (загалом 9-12 мі-
сяців). Така лікувальна схема дозволяє досягти зменшення рівня
парапротеїну на 50% у 50% хворих протягом кількох місяців. Три-
валість позитивного ефекту від лікування дорівнює 18-24 місяці,
середня тривалість життя пацієнтів, котрі лікуються за цією схе-
мою — 2-4 роки. Продовження хіміотерапії після досягнення ста-
більної фази захворювання (фаза плато) не впливає на тривалість
ремісії. Повні ремісії досягаються рідко. Слід враховувати, що мел-
фалан потрібно застосовувати з пересторогою у хворих із нирко-
вою недостатністю.
Досить широковживаним є протокол УАО. При лікуванні за
цим протоколом використовують вінкристин та адріаміцин протя-
гом 4 днів безперервною внутрішньовенною інфузією в поєднанні
з пероральним прийомом дексаметазону у великій дозі (40 мг/до-
бу). Таке лікування дозволяє досягти значного відсотка повних ре-
місій (60-70% за короткий проміжок часу). Протокол УАО має нез-
начний вплив на стовбурову клітину та є ідеальним режимом, шо
застосовується перед автологічною трансплантацією. У той же час
проведення такого лікування має значні незручності, пов’язані із
необхідністю використання для інфузії хіміопрепаратів централь-
ного венозного катетера, що може зумовити виникнення катетер-
асоційованої інфекції та коагуляційних ускладнень.
Пульсова терапія дексаметазоном у дозі 40 мг на день протя-
гом 4 днів із 4-денною перервою в лікуванні також вважається до-
сить ефективним методом лікування вперше виявленої мієломної
154
| Гемобластози
хвороби. Лікувальна відповідь становить 40-50% та більше, пози-
тивна відповідь спостерігається після декількох циклів. Проте
пульсова терапія дексаметазоном має багато побічних ефектів, ана-
логічних тим, котрі виникають при застосуванні високих доз глю-
кокортикостероїдних гормонів (порушення настрою, безсоння,
подразливість, порушення уваги, затримка рідини в організмі,
збільшення ваги, стероїдний діабет, гастроінтестинальні розлади,
інфекційні ускладнення, слабкість проксимальних м’язів, пору-
шення зору, включаючи розвиток катаракти).
У деяких випадках пацієнтам призначається комбінована полі-
хіміотерапія (протоколи М2, АВСМ та ін.). Як правило, викорис-
тання комбінації з декількох хіміопрепаратів не має значної пере-
ваги перед зазначеними вище схемами, але підвищує токсичний
вплив на організм пацієнтів.
Пацієнти, котрі не відповіли позитивно на терапію першої лі-
нії, розглядаються як потенційні кандидати на проведення автоло-
гічної трансплантації периферичної стовбурової клітини.
Високодозова хіміотерапія з подальшою антологічною транс-
плантацією (АТ) вважається одним із досить ефективних сучасних
методів лікування МХ (повних ремісій досягнуто у 24-75% хворих,
часткових ремісій — у 76-90%), однак АТ остаточно не виліковує
дану патологію (у більш ніж 90% хворих у майбутньому прогнозу-
ється виникнення рецидиву). Середня тривалість життя у хворих
після проведеної високодозової терапії становить 4-5 років, безре-
цедивний період триває приблизно 18-24 місяці. Смертність,
пов’язана із процедурою трансплантації, низька (близько 1%). Ав-
тологічна трансплантація не показана пацієнтам похилого віку
(старшим за 70 років) і тим, хто має серйозну супутню патологію
У наш час не існує чітких рекомендацій щодо призначення те-
рапії хворим, котрі досягли ремісії. У рамках дослідних протоколів
із цією метою найчастіше застосовують преднізолон, альфа-інтер-
ферон, ведеться вивчення ефективності використання талідоміду.
Для ефективного лікування кісткових уражень при мієломній
хворобі надзвичайно важливим є призначення з самого початку лі-
кування адекватної хіміотерапії та, за потреби, опромінення кісток.
Опромінення кісток призначається при компресії спинного мозку,
тривалому сильному больовому синдромі, для лікування та попе-
редження патологічних переломів.
З метою прискорення процесу репарації кісткової тканини ре-
комендується призначення біфосфонатів — памідронату (Аредіа),
155
Н.М. Третяк | Гематологія
золедронової кислоти (Зомета), клодронату (Бонефос). Аредіа зас-
тосовується внутрішньовенно один раз на 3-4 тижні у дозі 120 мг і
вважається найбезпечнішим препаратом для хворих із порушенням
функції нирок. Одним із найкращих препаратів серед представни-
ків цієї групи для корекції ііперкальціємії є зомета. Препарат приз-
начається у дозі 4 мг у вигляді 15-хвилинної інфузії один раз на 3-
4 тижні. Бонефос застосовують внутрішньовенною 2-годинною
інфузією в дозі 300 мг на день протягом 7 днів, як альтернативний
метод лікування може використовуватися пероральний прийом
препарату — 1600 мг на добу.
Новим методом лікування деструкцій хребта є застосування кі-
фопластики із введенням рідкого цементу, що дає змогу зміцнити
хребетний стовп та зменшити больовий синдром. Хворим, у кот-
рих не виявлено патологічних переломів, рекомендуються помірні
фізичні навантаження, особливо плавання та ходьба.
Для корекції синдрому анемії, котрий супроводжує мієломну
хворобу, застосовують препарати заліза, вітаміну В12 (у випадках
супутнього дефіциту цих елементів), проводять переливання
еритроцитарної маси. Також для лікування анемії патогенетично
обґрунтованим є призначення еритропоетину (Епрекс, Рекормон
та ін.). На першому етапі при лікуванні еритропоетинами відбу-
вається “навантаження дозою”, коли препарат вводять у дозі
40 000 одиниць на тиждень. Якщо після 4 тижнів прийому пре-
парату в зазначеній дозі не відбулося підвищення гемоглобіну,
доза еритропоетину підвищується до 60 000 одиниць. Терапія
триває до того часу, поки рівень гемоглобіну не зросте до
120 г/л, і припиняється при гемоглобіні 140 г/л. Підтримуюча те-
рапія еритропоетином проводиться тричі на тиждень у дозі
10 000 одиниць із підвищенням дози в разі потреби до 20 000 оди-
ниць.
Важливою складовою лікування пацієнтів із мієломною неф-
ропатією є достатня гідратація, переливання лужних розчинів та
плазмафарез. У пацієнтів із нирковою недостатністю слід, по мож-
ливості, обмежити використання нефротоксичних препаратів
(внутрішньовенні контрасти, аміноглікозиди, ванкоміцин, амфоте-
ріцин В, ацикловір, циклофосфамід, діуретики, біфосфонати — за
винятком аредіа).
При проведенні хіміотерапії у хворих із нирковою недостатніс-
тю перевагу в лікуванні слід надавати протоколу УАП та дексаме-
тазону в якості монотерапії, оскільки вони забезпечують швидкий
156
| Гемобластози
лікувальний ефект без додаткового токсичного впливу на нирки,
як це можна спостерігати у випадку з мелфаланом.
Причиною інфекційних ускладнень у хворих із уперше вста-
новленою МХ, як правило, є Зігеріососсиз рпеишопіае, НеторИі-
1п2 іпОтзе, Негрез /ойег. Встановлено, що інфекційні ускладнення
найчастіше трапляються в перші 3 місяці від початку лікування та
під час рецидиву захворювання. У. цей період хворим на МХ мож-
ливе призначення антибіотиків та сульфаніламідів (наприклад бі-
септолу) із метою профілактики інфекційних ускладнень. Лікуван-
ня бактеріальної інфекції проводиться за загальними принципами
раціональної антибіотикотерапії з урахуванням нефротоксичності
окремих препаратів.
Нині опрацьовуються нові технології лікування МХ із вико-
ристанням інгібіторів ангіонеогенезу (талідоміду) в режимі моноте-
рапії або в комбінації із дексаметезоном. Вивчається також роль ін-
терферонів для проведення підтримуючої терапії в період ремісії.
Прогноз. Сучасна терапія сприяє подовженню життя хворих на
МХ. Середня тривалість життя при адекватному лікуванні стано-
вить 50 місяців.
157
ЛЕЙКЕМОЇДНІ РЕАКЦІЇ
Лейкемоїдними реакціями називають патологічні реактивні
стани кровотворення, за яких картина крові подібна до лейкеміч-
них змін при інших пухлинах системи крові, проте вони не тран-
сформуються в ту пухлину, на яку схожі.
Етіопатогенез лейкемоїдних реакцій надзвичайно різноманіт-
ний. До виникнення лейкемоїдних реакцій молоть призводити такі
екзогенні фактори, як отруєння сульфаніламідними препаратами,
дигіталісом, адреналіном, фенацетином, оловом, ртуттю, миш’яком,
чадним газом, вплив іонізуючого опромінення та інші. До ендоген-
них факторів, що можуть зумовлювати лейкопенії, належать віруси,
токсини гельмінтів (трихінельоз, фасціольоз, опісторхоз, стронгілої-
доз, лямбліоз, токсоплазмоз), продукти лізису клітин крові внаслідок
гемолізу та масивного розпаду пухлинних клітин при онкопатології,
сепсис, системні захворювання сполучної тканини, гостра кровоте-
ча. Етіологічним фактором, котрим зумовлена лейкемоїдна реакція,
визначається й тип лейкемоїдної реакції. Так, наприклад, при злоя-
кісних пухлинах, сепсисі та захворюваннях сполучної тканини роз-
виваються лейкемоїдні реакції мієлоїдного типу. Інфекційні проце-
си, що супроводжуються потужною імунною відповіддю, призводять
до лейкемоїдних реакцій лімфоцитарного типу. Зазвичай при оду-
жанні від основного захворювання зникає й лейкемоїдна реакція.
Класифікація лейкемоїдних реакцій
І. Мієлоїдні:
1) псевдомієлобластного типу;
2) промієлоцитарного типу;
3) нейтрофільного типу.
II. Еозинофільні:
1) еозинофілія паразитарного генезу;
2) гіпереозинофілія як симптом злоякісного новоутворення;
2) еозинофільна реакція при колагенозах;
3) гіпереозинофільна реакція при захворюваннях легенів;
4) гіпереозинофільна реакція при захворюваннях системи крові;
5) родинний еозинофільний синдром.
III. Лімфоцитарні.
IV. Моноцитарно-макрофагальні.
158
| Лейкемоїдні реакції
МІЄЛОЇДНІ РЕАКЦІЇ
Лейкемоїдні реакції псевдобластного та промієлоцитарного ти-
пів спостерігаються перед виходом із імунного агранулоцитозу
(цей період триває від декількох годин до однієї доби). У пункта-
ті кісткового мозку виявляється високий відсоток клітин із гомо-
генним ядром, поодинокими нуклеолами, блакитною вузькою
цитоплазмою без зернистості. Іноді зустрічаються великі кліти-
ни, дуже схожі за формою, структурою ядра та величиною ци-
топлазми на промієлоцит, але з бідною зернистістю. Відсутність
типових бластів із ніжною мережею та рівномірністю розташу-
вання ниток хроматину дозволяє віддиференціювати лейкемоїд-
ну реакцію псевдобластного типу від гострої лейкемії. В наступ-
ні дні в пунктаті кісткового мозку виявляється величезна
кількість промієлоцитів з численною зернистістю. Таку картину
кісткового мозку можна сплутати зі змінами при гострій промі-
єлоцитарній лейкемії. Проте для гострої промієлоцитарної лей-
кемії характерні значне пригнічення мегакаріоцитарного парос-
тка, геморагічний синдром, анемія та тромбоцитопенія. Бластні
клітини при промієлоцитарній лейкемії мають усі ознаки атипії
(потворність форми ядра та цитоплазми, величезні бластні кліти-
ни, двоядерність, присутність у цитоплазмі поліморфної рясної
зернистості, яка дає позитивну реакцію на сульфатовані кислі
мукополісахариди). Ці ознаки відсутні при промієлоцитарній
лейкемоїдній реакції.
Надзвичайно високе промієлоцитарне зрушення в кістковому
мозку та поява промієлоцитів у периферичній крові спостерігаєть-
ся також у випадку тяжкої токсикоінфекції (сальмонельоз), при ін-
фекційних захворюваннях (черевний тиф), алергічному дерматиті
медикаментозного походження, злоякісних новоутвореннях, при
спадкових нейтропеніях. Проте характерною ознакою спадкової
нейтропенїї є еозинофілія та моноцитоз у кістковому мозку та пе-
риферичній крові. Цього ніколи не буває при гострій промієлоци-
тарній лейкемії.
Лейкемоїдна реакція нейтрофільного типу може бути реакцією
на найрізноманітніші фізіологічні та патологічні фактори, а також
Першим симптомом патології системи кровотворення. Одначе пер-
винні зміни в системі гемопоезу при захворюваннях крові є озна-
кою сталою, тоді як нейтрофільні лейкемоїдні реакції, що виника-
ють у відповідь на запальні та інфекційні захворювання, минущі.
159
Н.М. Третяк | Гематологія
Нейтрофільна лейкемоїдна реакція характеризується не тільки
відсотковим збільшенням нейтрофільних гранулоцитів, а й зрос-
танням їхньої абсолютної кількості.
Нейтрофільна лейкемоїдна реакція може бути зумовлена ба-
гатьма факторами.
По-перше, гострими запальними та інфекційними захворюван-
нями, збудником яких є кокова інфекція (стафілокок, стрептокок,
пневмокок та ін.), віруси, гриби, паразити.
Лейкоцитоз у периферичній крові в цих випадках може сягати
20,0-50,0 • 109/л. У лейкограмі кількість паличкоядерних нейтрофі-
лів збільшується до 20-50%, з’являються молоді форми нейтрофі-
лів — метамієлоцити та мієлоцити. При деяких захворюваннях
спостерігається токсична зернистість у цитоплазмі нейтрофілів,
а також дегенеративні зміни — вакуолізація ядра та цитоплазми.
По-друге, вираженою нейтрофільною лейкемоїдною реакцією
супроводжуються сепсис, шкарлатина, бешиха, крупозна пневмо-
нія, дифтерія, дизентерія, ПОЛІОМІЄЛІТ, ВИСИПНИЙ тиф, йегрез 2О8ІЄГ,
ревматизм, гнійні процеси.
По-третє, тривале вживання сульфаніламідних препаратів, ди-
гіталісу, адреналіну, фенацетину, а також отруєння хініном,
миш’яком, оловом, ртуттю, чадним газом може призвести до під-
вищення кількості лейкоцитів у периферичній крові (15,0-
40,0 • 109/л) із появою молодих форм нейтрофілів. Водночас у кіс-
тковому мозку збільшується відсоток мієлоцитів та промієлоцитів і
відповідно змінюється лейко-еритроїдне співвідношення (10:1; 20:1
та більше).
По-четверте, нейтрофільна лейкемоїдна реакція може бути зу-
мовлена впливом ендогенних факторів, котрі утворюються при
азотемічній уремії, діабетичному ацидозі тощо.
При злоякісних новоутвореннях нейтрофільна лейкемоїдна ре-
акція розвивається внаслідок подразнення мієлоїдного паростка
метастазами або в результаті токсичного впливу речовин розпаду
пухлини. Зазвичай при цьому відбувається подразнення й еритро-
їдного паростка, внаслідок чого в периферичній крові поряд із лей-
коцитозом та зрушенням вліво в лейкограмі з’являються ядерні
елементи еритропоезу. Помірна чи глибока анемія та присутність
еритрокаріоцитів у периферичній крові, що супроводжують ней-
трофільну лейкемоїдну реакцію, є переконливою ознакою злоякіс-
ного новоутворення. Підтверджують діагноз значне прискорення
ШОЕ (40-70 мм/год.) та гіпертромбоцитоз, що може сягати 600,0-
160
| Лейкемощні реакції
900,0 • 10^/л. Для лейкемоїцних реакцій нейтрофільного типу ха-
рактерне збільшення в кістковому мозку промієлоцитів, метамієло-
цитів та плазматичних клітин, іноді еозинофілів, а також мегакарі-
оцитів. Виявлення в кістковому мозку метастазів раку підтверджує
діагноз.
При лімфогранулематозі спостерігається нейтрофільна лейке-
моїдна реакція. У цій ситуації в лейкограмі, окрім зрушення вліво,
спостерігаються еозинофілія, моноцитоз та абсолютна лімфопенія.
При тривалому впливі радіаційного опромінення розвивається
лейкемоїдна реакція нейтрофільного типу, причому в лейкограмі
збільшується відсоток молодих нейтрофільних гранулоцитів та ео-
зинофілів. Такі зміни в периферичній крові можуть бути ознакою
гострої або хронічної променевої хвороби.
Лейкемоїдна реакція нейтрофільного типу може виникати при
великих крововтратах — особливо внутрішній кровотечі та лікуван-
ні кортикостероїдними гормонами. Характерним для обох випад-
ків є підвищення кількості лейкоцитів до 15,0-20,0 • 109/л та омо-
лодження лейкограми з появою метамієлоцитів і мієлоцитів. Після
відміни гормонів або припинення кровотечі показники крові нор-
малізуються.
Нейтрофільна лейкемоїдна реакція, як правило, виникає внас-
лідок оперативних втручань, переломів кісток, опіків, широкого
розчавлення тканин та шоку іншого генезу. Така ж реакція мієло-
їдного паростка характерна для інфаркту міокарда, можлива при
пароксизмальній тахікардії.
Досить часто спостерігається лейкоцитоз зі збільшенням від-
сотка нейтрофілів та зрушенням до поодиноких мієлоцитів у лей-
кограмі при вагітності. Крім того, існує фізіологічний нейтрофі-
льоз зі зрушенням вліво новонароджених і такий, що виникає
внаслідок тривалої фізичної напруги (пологи, тривалі судоми,
марш).
Диференційна діагностика лейкемоїцних реакцій нейтрофіль-
ного типу від мієлопроліферативних захворювань системи крові,
в першу чергу, проводиться на підставі таких ознак.
1. Для лейкемоїцних реакцій не характерна спленомегалія, хо-
ча існування збільшеної селезінки не виключає вторинного харак-
теру змін у гемограмі. Відомо, що при таких захворюваннях, як
сепсис або інші інфекційні захворювання, може спостерігатися по-
мірна спленомегалія. Однак селезінка в цих випадках має еластич-
ну консистенцію, а стан хворого виражено тяжкий і може супро-
161
Н.М. Третяк | Гематологія
воджуватися такими симптомами, як зменшення маси тіла, осал-
гії, тяжка лихоманка, тромбоцитопенія.
2. Виявлення значного лейкоцитозу зі зрушенням вліво в лей-
кограмі за відносно непоганого загального стану хворого найчасті-
ше зустрічається при захворюваннях системи крові. Присутність
збільшеної щільної селезінки тільки підтверджує існування хроніч-
ного мієлопроліферативного захворювання.
3. При морфологічному дослідженні клітин нейтрофільного
ряду виявляються токсична зернистість у цитоплазмі нейтрофіль-
них гранулоцитів, різноманітні дегенеративні зміни: такі, наприк-
лад, як вакуолізація ядра або цитоплазми, що підтверджує наяв-
ність лейкемоїдної реакції. При тяжких септичних станах може
спостерігатися некробіоз ядра майже в усіх клітинах крові.
Переконливим диференційно-діагностичним критерієм може
бути активність лужної фосфатази нейтрофілів периферичної кро-
ві. В разі існування лейкемоїдної реакції лужна фосфатаза нейтро-
філів периферичної крові залишається в межах норми.
При хронічній мієлоїдній лейкемії активність лужної фосфата-
зи нейтрофілів чітко знижується. При таких хронічних мієлопролі-
феративних захворюваннях, як ідіопатичний мієлофіброз або
справжня поліцитемія, активність лужної фосфатази нейтрофілів
підвищується. Крім того, на користь лейкемоїдної реакції нейтро-
фільного ряду свідчить нормальна загальна клітинність кісткового
мозку, незмінена кількість мегакаріоцитів та коливання вмісту ео-
зинофілів і базофілів у межах норми.
Диференційний діагноз лейкемоїдної реакції значно ускладню-
ється при злоякісних новоутвореннях та мієломній хворобі.
При дослідженні кісткового мозку у хворих з онкопатологією
може виявитись плазмоклітинна реакція з підвищеним вмістом
плазматичних клітин — до 20-30% та більше в мієлограмі. Найчас-
тіше такі реакції зустрічаються при гіпернефромах та пухлинах
шлунка. У таких випадках на користь множинної мієломи свідчить
наявність характерних деструкцій у плоских кістках, моноклонової
імуноглобулінопатії (патологічний білок у сироватці крові — М-
градієнт, білок Бенс-Джонса в сечі, збільшення вмісту імуноглобу-
лінів одного класу), морфологічно змінених (двоядерних, “по-
лум’яніючих”) та незрілих плазматичних клітин (плазмобластів,
плазмоцитів) у кістковому мозку.
На відміну від мієломної хвороби в кістковому мозку хворих
на онкопатологію плазматичний ряд складається зі зрілих плаз-
162
| Лейкемоїдні реакції
моцитів, можуть зустрічатися “атипові” (пухлинні) клітини, про-
те відсутність їх у препараті не дає підстав виключити онкозахво-
рювання.
ЛЕЙКЕМОЇДНІ РЕАКЦІЇ ЕОЗИНОФІЛЬНОГО ТИПУ
Лейкемоїдні реакції еозинофільного типу найчастіше пов’язані
з алергічними реакціями організму на будь-який агент, тобто чу-
жорідний білок або на білок власних тканин, що набув антигенних
властивостей.
Основними причинами еозинофіли' можуть бути такі патоло-
гічні процеси:
— інфекційні (еозинофілія паразитарного генезу, котра суп-
роводжує анкілостомоз, шистомоз, аскаридоз, трихінельоз, опіс-
торхоз, ехінококоз, особливо його тканинні стадії та форми, лям-
бліоз);
— алергічні (бронхіальна астма, сезонний риніт та ін.);
— автоімунні (еозинофільний фасциїт, ендокардит Лефлера,
хронічний гепатит, ревматоїдний артрит, вузликовий периартеріїт);
— тяжкі дерматози та дерматити;
— злоякісні новоутворення, в тому числі гемобластози (реци-
дивуючий гранулематозний дерматит — синдром Велля; аденокар-
цинома шлунка; рак піхви, щитовидної залози, носоглотки; злоя-
кісний гістіоцитоз; лімфогранулематоз; хронічна мієлоїдна
лейкемія);
— медикаментозні (пеніцилін, ампіцилін, цефалоспоріни, про-
титуберкульозні засоби, нітрофуранові сполуки, дифенін, каптоп-
ріл та ін.);
— ідіопатичні (еозинофільний синдром, легеневий еозино-
фільний інфільтрат, системний мастоцитоз із еозинофілією, а та-
кож родинно-спадкова еозинофілія).
Для лейкемоідної реакції еозинофільного типу характерне під-
вищення кількості лейкоцитів до 40-50 109/л із вмістом еозинофі-
лів до 60-90% за рахунок зрілих форм. При морфологічному дос-
лідженні в еозинофілах периферичної крові виявляється
гіперсегментація ядер і вакуолізація цитоплазми клітин. У кістко-
вому мозку на фоні нормальної клітинності спостерігається підви-
щення кількості зрілих еозинофілів.
При паразитарних захворюваннях еозинофілію спричиняють
не тільки гельмінти, коли паразити локалізуються в тканинах, а й
163
Н.М. Третяк | Гематологія
їхні личинки, а також міграція личинок аскарид, не адаптованих до
організму людини (іохосага сапіь та іохосага саііз). При паразитар-
них захворюваннях личинка або паразит, тісно стикаючись із тка-
нинами хворого, виділяють хемотаксичний фактор, у відповідь на
котрий збільшується продукція еозинофілів кістковим мозком.
Частина еозинофілів виходять із кістковомозкових синусів і мігру-
ють до місця інвазії паразита, де, оточуючи й прилипаючи до його
поверхні, виділяють ферменти, які призводять до загибелі шкідни-
ка. Перебіг інвазії найпростішими супроводжується еозинофілією
до 12-25%, а іноді й більшою.
Паразитарна кишкова інфекція у дітей не завжди супроводжу-
ється еозинофілією. Лікування спрямоване на усунення етіологіч-
ного фактора. При позитивному результаті лікування основного
захворювання показники крові нормалізуються.
Автоалергічні еозинофілії спостерігаються при різних формах
колагенових захворювань. Зокрема, це класична форма вузликово-
го периартеріїту, ревматоїдний артрит, системна склеродермія,
дерматоміозіт, ревматизм, недиференційований колагеноз. При цій
патології утворюються підшкірні вузлики, поширюючись на плев-
ру та перикард, уражаючи серце та легені.
У хворих на класичну форму ревматоїдного артриту еозинофілія
може сягати 20-90%. Активація ревматичного процесу може також
супроводжуватись високою еозинофілією, що є негативною озна-
кою перебігу хвороби. Гіпереозинофілія характерна для недиферен-
ційованих колагенозів, ускладнених пристінковим тромбозом ендо-
карда (пристінковий фібропластичний ендокардит). Ізольовані
ураження міокарда з еозинофілією в периферичній крові спостеріга-
ються у вигляді дифузних форм при фідлерівському міокардиті, а та-
кож при вогнищевих еозинофільних інфільтратах міокарда.
Найчастіше еозинофілія локалізується в легенях. Проте легене-
ва еозинофілія — це збірне поняття. Сюди входять летючі еозино-
фільні легеневі інфільтрати, еозинофільні пневмонії та гранульоми,
васкуліти різноманітної етіології (від паразитарної до медикамен-
тозної). Схильність до еозинофільної інфільтрації легенів пов’яза-
на з великим вмістом у легенях тучних клітин, котрі кооперують-
ся з еозинофілами при імунній відповіді. Крім того, важливо те, що
легені, як і шкіра, найчастіше стикаються з чужорідними антиге-
нами. Плевральна еозинофілія не є самостійною нозологічною
формою, вона спостерігається у хворих на плеврити різної етіоло-
гії, часто буває непостійною. Найчастіше плевральні еозинофілії
164
| Лейкемоїдні реакції
спостерігаються при злоякісних новоутвореннях. В ексудаті вміщу-
ється більш ніж 10% еозинофілів, водночас еозинофілія в перифе-
ричній крові спостерігається приблизно в половині випадків.
Підвищена кількість еозинофілів у периферичній крові може
спостерігатися при гістіоцитозі, хронічній мієлоїдній лейкемії на
ранній хронічній стадії, множинній мієломі, годжкінських та не-
годжкінських злоякісних лімфомах, гострій лейкемії, особливо
в період рецидиву; під час ремісії еозинофілія зникає.
Ураження шлунково-кишкового тракту з дифузною еозино-
фільною інфільтрацією органів та значною еозинофілією в пе-
риферичній крові зустрічається рідко. Іноді спостерігаються ео-
зинофільні гастрити та гепатити, крім того, можливі
еозинофільні інфільтрати у вигляді гранульом у різних ділянках
шлунково-кишкового тракту та підшлункової залози. Імовірно,
ця патологія пов’язана з паразитами, в тому числі з міграцією
в організмі людини личинок не адаптованих до нього паразитів,
або, в окремих випадках це може бути результатом алергії на ме-
дикаменти.
При алергічних проявах у відповідь на медикаментозний анти-
ген збільшується концентрація І§Е, який у фізіологічних дозах за-
безпечує нормальну функцію місцевого захисту. Надмірна кіль-
кість І§Е призводить до ушкодження тучних клітин, виділення
великої кількості гістаміну, простагландину Д та ін., що діють як
хемотаксичний фактор та призводять до зростання кількості еози-
нофілів у периферичній крові. Невелика кількість антигена приз-
водить лише до місцевої реакції. Якщо потрапляє велика кількість
антигена, процес стає генералізованим, що спричиняє ушкоджен-
ня оточуючих тканин. Еозинофіли фагоцитують імунні комплекси,
що вміщують І§Е, секретують гістаміназу, котра пригнічує гістамін,
і таким чином припиняють алергічний процес.
Бронхіальна астма зазвичай супроводжується помірною еози-
нофілією. Тривала значна еозинофілія при бронхіальній астмі час-
то буває передстадією вузликового періартерїїту. Останнім часом
виділяють астматичний варіант вузликового періартерїїту.
Еозинофілія часто спостерігається при імунодефіцитних син-
дромах із дефіцитом одного або декількох класів імуноглобулінів.
Еозинофілія виявляється при патології Т-лімфоцитів, тобто при
зменшенні їх кількості або зниженні їхньої функціональної актив-
ності. У таких випадках рівень еозинофілів у периферичній крові
може сягати 40-60%.
165
Н.М. Третяк | Гематологія
Синдром Віскогга-Олдрича, що характеризується екземою,
тромбоцитопенією та підвищеною схильністю до інфекцій, часто
супроводжується еозинофілією та підвищенням рівня І§Е.
Інший вид імунодефіциту, пов’язаний із дефектом хемотакси-
су нейтрофілів, зумовлює схильність до інфекції, часто супровод-
жується зростанням вмісту еозинофілів та концентрації І§Е.
Пневмоцистні інфекції, що виникають на фоні імунодефіцит-
ного стану, можуть призводити до еозинофілі! При імунодефіциті
високу еозинофілію в периферичній крові може спровокувати ста-
філококова інфекція, що міститься в різних органах, і навіть у гай-
морових пазухах.
Еозинофільні інфільтрати в судинах можуть спостерігатися при
так званих “системних васкулітах”, гранульомах у кістках та м’язах,
травному каналі, при гострій кропив’янці, бульозному дерматиті,
пфемфігусі, пфемфігоїді, універсальній екземі При цьому спостері-
гається відкладення С43-компонента компліменту на базальній
мембрані шкіри та підвищення рівня І§ у сироватці крові, що свід-
чить про можливість опосередкованого через хемотаксичного ге-
незу еозинофілі! Сполучення різних симптомів з ураженням шкіри
існує при поворотному гранулематозному дерматиті та еозинофіль-
ному фасциїті (синдром Шульмана), що характеризується гаряч-
кою, ознобом, міальгіями, артралгіями. У шкірі відбувається скле-
роподібний процес за рахунок накопичення в епідермісі І§Сі та
С43-компонента компліменту. Ушкодження фасції призводить до
стягування шкіри, контрактур, спазмів м'язів. При гістологічному
дослідженні шкіри виявляється великий відсоток еозинофілів, при-
чому в периферичній крові вміст еозинофілів може бути меншим.
Ураження шкіри бувають також вторинними при так званих гі-
переозинофільних синдромах і часто невизначеного генезу.
Гіпереозинофільний синдром. Це захворювання трактувалося ра-
ніше як “еозинофільна лейкемія”, “еозинофільний колагеноз”,
“пристінковий фібропластичний ендокардит Леффлера”. Всі ці син-
дроми мали декілька спільних ознак: спочатку — тривала висока ео-
зинофілія в периферичній крові, яка іноді поєднується з астматичним
синдромом, прогресуюча серцева недостатність та гепато-спленомега-
лія, з часом з’являється лейкоцитоз зі зрушенням у лейкограмі вліво,
до бластів. У мієлограмі спостерігається значне омолодження за раху-
нок недиференційованих клітин та молодих форм еозинофілів. При
патоморфологічному дослідженні в серці майже завжди спостерігаєть-
ся фібропластичний ендокардит із пристінковим тромбозом у порож-
166
| Лейкемоїдні реакції
нинах шлуночків (частіше лівого), а також еозинофільна інфільтрація
печінки, селезінки, енщо- та міокарда.
Прогноз не сприятливий, лікування не дає позитивного ре-
зультату.
Лікування всіх лейкемоїдних реакцій еозинофільного типу
спрямоване на усунення етіологічного фактора. У важких випадках
необхідно призначати десенсибілізуючі препарати, глюкокортико-
їдні гормони, котрі найефективніші при колагенозах та алергічних
реакціях. Кортикостероїдні гормони, які призначаються від 40 до
80 мг на добу, справляють еозинопенічний ефект, сприяють зни-
женню температури тіла та поліпшують загальний стан хворого.
Гіпереозинофільний синдром неясного генезу може бути спро-
вокований туберкульозом та стафілококовою інфекцією, тому
в сумнівних випадках доцільно призначати специфічну терапію ех
ІцуапііЬиз.
Псевдоеозинофілія. У деяких випадках нейтрофільна зернис-
тість забарвлюється еозином, і такі нейтрофіли можна помилково
сприйняти за еозинофіли. Такі клітини називаються псевдоеозино-
філами. З допомогою цитохімічного дослідження клітин, зокрема
реакції на хлорацетатестеразу, котра є характерною тільки для ней-
трофілів, та реакції на пероксидазу, що характерна й для нейтро-
філів, і для еозинофілів, при низькому вмісті ціанідів або Сіїїогазої
Газі ріпк можна визначити справжню природу клітин.
При неможливості встановити діагноз необхідне спостережен-
ня за хворим, щоби за найменших ознак прогресування захворю-
вання починати лікування десенсибілізуючими препаратами, глю-
кокоргикоїдними гормонами або навіть цито< іатичними засобами.
ЛЕЙКЕМОЇДНІ РЕАКЦІЇ ЛІМФОЦИТАРНОГО ТИПУ
Інфекційний мононуклеоз
Захворювання вперше описане Н. Ф. Філатовим у 1885 р. Си-
нонімічні назви: залозиста гарячка, моноцитарна ангіна, хвороба
Філатова—Пфайфера.
\\Ї5Іп£ у 1938 р. довів, що збудником інфекційного мононукле-
озу є вірус, що фільтрується, типу Негрек, який у 1964 р. був де-
тально описаний Епштейном та Барром. Хворіють на цю патоло-
гію переважно діти, частіше хлопчики, та особи молодого віку.
Захворювання передається повітряно-крапельним та пероральним
167
Н.М. Третяк | Гематологія
шляхом від хворої людини. Вірус лімфатичними шляхами потрап-
ляє в регіональні лімфатичні вузли, спричиняє гіперплазію тимус-
незалежних ділянок, і відбувається інтенсивна проліферація в кор-
тикальному шарі Т-клітин.
Клініка. Початок захворювання зазвичай гострий, але іноді від-
мічаються продромальні ознаки, такі як нездужання, біль у м’язах,
запаморочення. Температура підвищується зненацька і сягає
39-39,5°С за 1-2 дні. Проте іноді в перші 8-10 діб температура ут-
римується на субфебрильних цифрах і лише пізніше підвищується
до 39-40°С .
В окремих випадках температура утримується на рівні 38-39°С
до 2 тижнів, а надалі — на субфебрильних цифрах протягом 1-2 мі-
сяців. Підвищення температури часто передує ангіні та набряку
лімфатичних вузлів, проте буває навпаки. Часто всі три симптоми
виникають майже водночас.
Постійний і головний симптом захворювання — системне
збільшення периферичних лімфатичних вузлів. Найтиповішим є
збільшення лімфатичних вузлів, розташованих по задньому краю
грудинно-ключично-соскоподібного м’яза (у 95% випадків), але
разом із тим збільшуються також передні шийні, підщелепні, пах-
вові, пахвинні та стегнові лімфатичні вузли: вони стають розміром
від 1 до 3 см у діаметрі, щільні, рухливі, неспаяні між собою, по-
мірно болючі при пальпації. Трапляються випадки, коли лімфатич-
ні вузли не збільшуються, проте є болючими, шо підтверджує їхню
участь у патологічному процесі. Дуже рідко відбувається збільшен-
ня лімфатичних вузлів середостіння та черевної порожнини. Опи-
сані випадки захворювань, клінічна картина яких імітує гострий
апендицит.
Важливим симптомом є ангіна. Форми ангіни найрізноманіт-
ніші: від катаральної та фолікулярної до виразково-некротичної та
виразково-дифтеритичної,- Іноді ангіна супроводжується значною
гіперемією слизової зіва та носоглотки. В окремих випадках ангіна
може на 10-12 днів передувати характерним змінам у периферич-
ній крові. Дуже рідко спостерігаються петехіальні геморагічні ви-
сипання на нижніх кінцівках або носові кровотечі.
З боку внутрішніх органів характерне у 80-90% випадків збіль-
шення селезінки на 2-3 см нижче лівої реберної дуги, на дотик во-
на щільна, не болісна. Печінка збільшується у 65-70% хворих —
приблизно на 1-3 см нижче правої реберної дуги. Досить частим
симптомом (20-30% випадків) є гепатит із проявами жовтяниці.
168
| Лейкемоїдні реакції
Жовтяниця розвивається за рахунок прямого білірубіна, відзнача-
ється високий рівень трансамін^з, лактатдегідрогенази. Іноді роз-
вивається гепатаргія, яка може призвести до смерті хворого.
Крім того, інфекційний мононуклеоз супроводжується астені-
єю. Майже в усіх хворих спостерігається головний біль, порушен-
ня зору, міальгії, млявість, іноді болі в животі з нудотою та блюво-
тою. В окремих випадках може виникати фотофобія, біль у м’язах
очей, кашель, у разі ускладнень — еритема шкіри, тромбоцитопе-
нія, психоз, парез черепних нервів, радикулоневрити.
За особливостями клінічного перебігу інфекційний мононук-
леоз поділяють на три форми:
— субклінічна, тобто мононуклеари в периферичній крові ви-
являють тільки при випадковому обстеженні;
— з мінімальними симптомами за відсутності температури, ан-
гіни, лімфаденіту, спленомегалії;
— моносимптоматична, коли в клінічній картині домінує одна
з основних ознак — гіпертермія, ангіна, збільшення лімфатичних
вузлів, селезінки.
Деякі автори, на підставі домінуючих симптомів, виділяють
різні клінічні варіанти інфекційного мононуклеозу: ангінозний,
залозистий, гарячковий, кишковий, енцефалітний, ревматоїдний,
спленомегалічний, шкіряний, септичний, печінковий, з уражен-
ням слизових оболонок, стертий варіант, афебрильний, варіант
без ангіни, варіант, ускладнений автоімунною гемолітичною ане-
мією.
Крім гострої та підгострої стадії, у певної групи хворих захво-
рювання набуває рецидивуючого або хронічного характеру. Проте
прогноз інфекційного мононуклеозу сприятливий. Основні сим-
птоми захворювання зникають за 1-3 тижні, але астенізація збері-
гається досить довго. У разі ускладнення перебігу хвороби прогноз
залежить від складності патологічного процесу. Вмирають хворі
надзвичайно рідко.
Картина периферичної крові відзначається лейкоцитозом, що
сягає від 10,0- 109/л до 30,0-40,0- 109/л, в основному за рахунок
збільшення кількості лімфоцитів (до 50-70%) та моноцитів — від
10-12% на початку захворювання до 40-50% у розпалі хвороби. Іно-
ді ці коливання відбуваються протягом кількох днів.
При мікроскопічному дослідженні ядра в моноцитах мають губ-
часту структуру та більш базофільну, ніж звичайно, цитоплазму. Для
моноцитів при інфекційному мононуклеозі характерний феномен
169
Н.М. Третяк | Гематологія
відриву фрагмента ядра, котрий локалізується окремо в цитоплазмі
та є наслідком деструктивного впливу вірусу, шо проникає в ядро.
Мононуклеари мають широку цитоплазму із дешо бузковим
забарвленням та зоною перинуклеарного просвітлення. Ядро
клітин, що мають вакуолізовану або пінисту цитоплазму і відсут-
ність гранул, називають віроцитами. Крім того, для інфекційно-
го мононуклеозу характерна поява плазматичних клітин серед-
нього розміру, до 12-15 мкм. їхня кількість коливається від 4-5%
до 15-25%.
У важких випадках із самого початку захворювання кількість
плазматичних клітин у периферичній крові сягає 40%. Крім опи-
саних клітин, спостерігаються атипові мононуклеари, присутність
яких є патогномонічноЮ ознакою захворювання. Такі самі клітини
виявляються в мазках із зіва та у спинномозковій рідині.
Анемія не характерна для інфекційного мононуклеозу, про-
те в розпалі захворювання гемоглобін та кількість еритроцитів
дешо знижуються, й особливо це стосується випадків, коли ос-
новний процес ускладнюється автоімунною гемолітичною ане-
мією. Вміст тромбоцитів, як правило, залишається в межах нор-
ми, й лише в окремих випадках їхня кількість зменшується до
100,0-80,0 • 109/л, не знижуючись до критичних цифр. У пункта-
ті кісткового мозку виявляється помірне збільшення кількості
лімфоцитів, моноцитів та плазматичних клітин, 10% із них ста-
новлять атипові мононуклеари. Проте картина кісткового мозку
не має вирішального діагностичного значення.
Гістологічно в лімфатичних вузлах при інфекційному моно-
нуклеозі виявляються зміни, характерні для реактивного лімфаде-
ніту з активною проліферацією лімфоїдних клітин.
Патоморфологічні зміни в селезінці подібні до змін у лімфа-
тичних вузлах — рисунок стертий, у грабекулах великі судини опо-
виті муфтами з великих моноцитарних та плазматичних клітин.
Розростання таких самих клітин спостерігається під епітелієм сли-
зової оболонки кишечника, тут же можуть спостерігатися вогнища
некрозів. У печінці гістологічні зміни подібні до таких при хворо-
бі Боткіна: великі інтерстипійні ушкодження призводять до змен-
шення портальних просторів та скупчення значної кількості вели-
ких моноцитарних та лімфоїдних клітин.
Діагностика інфекційного мононуклеозу базується на дос-
лідженні периферичної крові та виявленні в ній підвищеної
кількості мононуклеарів у поєднанні із лімфоцитозом. Для ве-
170
| Лейкемоїдні реакції
рифікації діагнозу проводиться серологічна реакція Пауля—Бун-
неля (1932). Ця реакція заснована на тому, що в крові людини
при інфекційному мононуклеозі зростає рівень гемаглютинінів
проти еритроцитів барана, морської свинки та кролика. Пози-
тивною реакцією вважається титр, не нижчий 1:64. Найвищий
титр антитіл виявляється на 6-7-й день захворювання й утриму-
ється до 21-24-го дня. Титр антитіл зменшується, починаючи із
6-го тижня реконвалесценції, та має здатність підвищуватись
після будь-якої аденовірусної інфекції протягом тривалого часу.
Під час інфекційного мононуклеозу підвищується концентрація
в сироватці крові імуноглобулінів — І§М (на 100-300%), І&А (на
100-200%), І§С (на 50-70%). Антитілами в реакції Пауля—Бун-
неля є І§М із легкими ланцюгами того ж типу. Проте реакція не
є специфічною й може бути позитивною при вірусному гепати-
ті, гострій лейкемії, хронічних лімфо- та мієлолейкеміях, ревма-
тизмі, туберкульозі, сифілісі, лептоспірозі та в інших випадках,
але в нижчих титрах.
При імунологічному дослідженні в перший тиждень захворю-
вання спостерігається диспропорція у співвідношенні Т- та В-лім-
фоцитів. Кількість В-лімфоцитів зростає в перший тиждень і змен-
шується на 3-4-й тиждень. Вміст Т-лімфоцитів підвищується на
10-14-й день та нормалізується на 5-6-й тиждень. У розпалі захво-
рювання у хворих на інфекційний мононуклеоз можуть спостері-
гатися незначна альбумінурія, уробілінурія та невелика кількість
еритроцитів у сечі.
Диференційна діагностика інфекційного мононуклеозу прово-
диться із дифтерією, ангіною Венсана, агранулоцитозом, гострою
лейкемією, хронічною лімфоїдною лейкемією, негоджкінською
злоякісною лімфомою, туберкульозом та туляремією.
Лікування інфекційного мононуклеозу, котре застосовувалося
раніше (жарознижуючі препарати, вітаміни групи В та вітамін С,
а у важких випадках — антибіотики), тепер носить скоріше тради-
ційний характер.
Найефективнішим на сучасному етапі є використання пре-
паратів інтерферону рекомбінантного альфа-26 (це лаферон,
інтрон-А, роферон-А, реальдирон, еберон та ін.) по 3 млн. МО
щодня підшкірно до 15 днів. Крім того, хворого необхідно ізо-
лювати на 10-15 днів, у особливо складних випадках — постіль-
ний режим.
Протипоказані фізичні навантаження, заняття спортом.
171
Н.М. Третак | Гематологія
Інфекційний лімфоцитоз
Інфекційний лімфоцитоз — це самостійне захворювання (хворо-
ба Сміта, асимптоматичний доброякісний лімфоцитоз), на яке хво-
ріють переважно діти до 10 років, але зустрічаються випадки зах-
ворювання дорослих. Прогноз хвороби сприятливий.
Збудником є ентеровірус із групи Коксаки або аденовірус типу
12. Інфекційний лімфоцитоз — надзвичайно контагіозне захворю-
вання, іноді набуває епідемічних спалахів у дитячих колективах,
найчастіше уражає дітей 2—6 років. Шлях передачі — повітряно-
крапельний, інкубаційний період — 12-21 день.
Клінічна картина розвивається за 12-24 години після проник-
нення вірусу. Як правило, захворювання починається із грипопо-
дібних симптомів. У залежності від переважаючих ознак розрізня-
ють клінічні форми: тифоподібну, легеневу, шлунково-кишкову,
неврологічну, гангліозну, що характеризується збільшенням пери-
феричних та вісцеральних лімфатичних вузлів.
Гострий період триває 15-20 діб, підвищена температура, час-
тіше субфебрильна, зберігається до 7 днів. Характерними є слаб-
кість, астенізація, катаральні явища, іноді бувають нудота, блюво-
та, діарея до 4-7 разів на добу, болі в животі, симптоми ураження
верхніх дихальних шляхів, коро- або шкарлатиноподібний висип
на шкірі. Дуже рідко — неврологічні симптоми, міальгії, менінгіт,
енцефаліт. Периферичні лімфатичні вузли та селезінка не збільше-
ні. При ультразвуковому обстеженні можуть фіксуватися збільшен-
ня абдомінальних лімфатичних вузлів.
У периферичній крові виявляється лейкоцитоз — 30-80, іно-
ді до 100,0- 109/л. У лейкограмі переважають дозрілі лімфоцити
(80-90%), зустрічаються моноцити, лімфоцити з плазматизова-
ною цитоплазмою. Абсолютний лімфоцитоз, пов’язаний із під-
вищеним виходом лімфоцитів, зберігається до 15-30 днів, іноді
триває до 2-3 місяців. Можуть спостерігатися тіні Гумпрехта.
Крім того, у 30% випадків виявляється еозинофілія до 6-10% та
полісегментація ядер нейтрофільних гранулоцитів. Кількість
еритроцитів та ШОЕ коливається в межах норми.
У мієлограмі лімфоїдної метаплазії немає. Морфологічна кар-
тина пунктату лімфатичного вузла — в межах норми. У розпал зах-
ворювання підвищується концентрація альфа-2- та гама-глобулінів
у сироватці крові.
Диференційний діагноз проводиться з інфекційним мононукле-
озом, хронічною лімфоїдною лейкемією, гострою лімфоїдною
172
| Лейкемоїдні реакції
лейкемією, негоджкінською злоякісною лімфомою, коклюшем,
кором, скарлатиною, гострим апендицитом, фізіологічним лім-
фоцитозом у немовлят та лейкемоїдними реакціями лімфоцитар-
ного типу.
Лікування захворювання симптоматичне.
Прогноз: захворювання закінчується одужанням.
Хвороба котячої подряпини
Хвороба котячої подряпини — це гостре інфекційне захворюван-
ня, шо виникає після подряпини або укусу кішки. Синоніми —
доброякісний лімфоретикульоз, небактеріальний регіонарний лім-
фаденіт.
Збудник захворювання належить до групи орнітозу-хламіді-
озу. У місці проникнення збудник зумовлює запалення, після
чого лімфогенними шляхами потрапляє в найближчий регіонар-
ний лімфатичний вузол, призводячи до його запалення. При ди-
семінації інфекції розвивається екзантема та гепатолієнальний
синдром.
Найчастіше хворіють діти, особливо в зимовий період. У при-
роді резервуаром збудника є дрібні гризуни та птахи. Інкубаційний
період — 1 -2 тижні, рідко до 60 днів.
Клініка. Захворювання починається з утворення невеличкої па-
пули із червоним кружальцем навколо місця проникнення збудни-
ка. Згодом розвиваються виразка та гнійник при нормальному за-
гальному стані хворого. За 2-3 тижні після цього виникає
регіональний лімфаденіт, що є найхарактернішим симптомом зах-
ворювання. Збільшуються шийні та пахвові лімфатичні вузли, до 2-
5 см у діаметрі, болючі та рухливі при пальпації. У 50% випадків
уражені лімфатичні вузли нагноюються, виділяючи густий зелено-
жовтий гній. Клінічну картину доповнює слабкість, головний біль,
підвищена температура тіла, відсутність апетиту, збільшені печінка
й селезінка. Клінічні прояви зберігаються 2-3 тижні. В однієї тре-
тини хворих розвивається екзантема у вигляді краснухи або вузлу-
ватої еритеми, яка зникає за кілька днів. Лімфаденопатія може три-
вати від декількох місяців до років.
У периферичній крові спостерігається лейкопенія на початку
захворювання та помірний лейкоцитоз (до 15-20 • 109/л) із підвищен-
ням відсотка нейтрофільних гранулоцитів у лейкограмі, шо свідчить
про нагноєння лімфатичних вузлів. У окремих випадках може зус-
173
Н.М. Третяк | Гематологія
трічатися збільшення, до 40-60%, кількості лімфоцитів у лейкограмі,
причому серед них зустрічаються лімфоїдні форми, що нагадують
атипові мононуклеари при інфекційному мононуклеозі.
Діагноз хвороби котячої подряпини верифікується наявністю
позитивної проби з орнітозним антигеном або орнітозним алерге-
ном. Кістковий мозок не зазнає специфічних змін.
Лімфатичний вузол при гістологічному дослідженні має полі-
морфну структуру: виявляється гіперплазія гістіоцитарних елемен-
тів із присутністю плазматичних клітин та еозинофілів. При нагно-
єнні лімфатичного вузла з’являються нейтрофільні гранулоцити.
Рідко виявляються клітини, що за морфологією нагадують клітини
Березовського—Штернберга.
Диференційна діагностика проводиться з бактеріальним лімфа-
денітом, туберкульозним лімфаденітом, інфекційним мононуклео-
зом, лімфогранулематозом, сифілісом, туляремією, бруцельозом.
Прогноз сприятливий.
Лікування симптоматичне при легких формах. У разі важкого
перебігу захворювання призначають антибіотики та невеликі, до ЗО
мл на добу, дози глюкокортикоідних гормонів; інструментальне
видалення гною з лімфатичного вузла. В окремих випадках мож-
ливе використання фізіотерапевтичних процедур.
Ієрсиніоз
Ієрсиноз — це інфекційне захворювання, збудником котрого є
Уеіьіпіа епіегосоїйіса, що переноситься гризунами.
Клінічна картина дуже різноманітна. Першим симптомом мо-
же бути пронос, гарячка. Можливі помірна лімфаденопатія, збіль-
шення селезінки, короподібний сип на дистальних відділах кінці-
вок. Іноді в клінічній картині превалюють поліартралгії разом із
ознаками міокардиту: тахікардія, систолічний шум на верхівці, не-
гативні зубці Т у передніх грудних відведеннях.
У периферичній крові — лейкоцитоз до 15,0-20,0 • 109/л, вели-
кий відсоток лімфоцитів зі значною присутністю широкоплазмо-
вих із перинуклеарним просвітленням імунобластів та еозинофілів
у лейкограмі.
Діагноз підтверджується позитивною специфічною серологіч-
ною пробою.
Лікування проводиться левоміцетином (по 2 г на добу) або
фторхінолонами (цифран по 500 мг 2 р. на добу). Клінічні прояви
174
| Лейкемоїдні реакції
зникають за декілька днів. Селезінка залишається збільшеною три-
валий час.
Прогноз сприятливий.
Слід пам’ятати, що лейкемоїдні реакції лімфоцитарного типу,
крім зазначених вище, можуть бути симптомами таких інфекцій-
них захворювань, як черевний тиф, паратиф, вірусний гепатит А,
В, С, скарлатина, коклюш, кір, вітряна віспа, туберкульоз, малярія,
гострий поліомієліт, бруцельоз, орнітоз, туляремія, геморагічні ли-
хоманки, пароксизмальний рикетсіоз, вісцеральний лейшманіоз.
У всіх випадках лейкемоїдних реакцій діагноз повинен бути ве-
рифікований. Починати лікування хворого без точно встановлено-
го діагнозу неприпустимо.
МОНОЦИТАРНО-МАКРОФАГАЛЬНІ ЛЕЙКЕМОЇДНІ
РЕАКЦІЇ
Моноцитарно-макрофагальні лейкемоїдні реакції можуть вини-
кати при будь-якому інфекційному процесі. Найчастіше вони суп-
роводжують туберкульоз.
У периферичній крові в таких випадках спостерігається нез-
начний лейкоцитоз, від 10,0 до 15,0 • 109/л, та підвищений відсоток
лімфоцитів і моноцитів у лейкограмі. У кістковому мозку вміст мо-
ноцитів та промоноцитів збільшується до 2-4%. В уражених палич-
кою Коха тканинах утворюються моноцитарно-макрофагальні ін-
фільтрати (гранульоми). Основним морфологічним субстратом
туберкульозної гранульоми є макрофаг, який частіше називають
епітеліоїдною клітиною. Крім того, до морфологічного складу ту-
беркульозного горбика входять багатоядерні макрофагальні еле-
менти — клітини Лангерганса.
При тривалому застосуванні глюкокортикоїдних гормонів або
цитостатичних препаратів унаслідок порушення імунітету туберку-
льозні гранульоми не утворюються, і процес набуває міліарного ха-
рактеру із розвиненням некрозів у місцях проникнення туберку-
льозної інфекції.
Моноцитарно-макрофагальна лейкемоїдна реакція спосте-
рігається при патології імунних комплексів (ревматизм, сарко-
їдоз Бека, хвороба Вегенера, так звані хвороби гіперчутливос-
ті легенів).
У периферичній крові підвищується кількість моноцитів.
У тканинах утворюються макрофагально-лімфоцитарні гранульоми
175
Н.М. Третяк | Гематологія
(при ревматизмі — ашот-талалаєвські гранульоми). У легенях мак-
рофагальні гранульоми локалізуються переважно в міжальвеоляр-
них перетинках, призводячи до їх значного потовщення, зменшен-
ня розмірів альвеолярних порожнин, унаслідок чого виникає
гостра дихальна недостатність.
Клінічні прояви дуже бідні; аускультативно відмічається жорс-
тке або бронхіальне дихання, періодичні крепітуючі хрипи.
Гістіоцитоз синусів — це своєрідна макрофагальна реакція на
бактеріальну, вірусну або паразитарну інвазію.
Клінічна картина характеризується значним збільшенням ок-
ремих або всіх груп лімфатичних вузлів, котрі мають щільну кон-
систенцію; збільшеною, іноді болючою при пальпації селезінкою
та тривалою гіпертермією.
Діагноз встановлюється лише на підставі результатів гістологіч-
ного дослідження лімфатичного вузла або селезінки. У лімфатич-
ному вузлі спостерігаються розширені синуси, заповнені світлими
великими однотипними клітинами з великими ядрами без нукле-
олей. Маючи світлу широку цитоплазму, вони виділяються на фо-
ні темних пікнотичних лімфоцитів і стирають рисунок лімфатич-
ного вузла.
Гістіоцитоз синусів може спостерігатися й окремо в селезінці.
Селезінка збільшується внаслідок макрофагальної реакції. Утриму-
ється постійна, рецидивуюча гіпертермія, і в окремих випадках ли-
ше після спленекгомії може бути встановлений діагноз.
Описані випадки гістіоцитозу синусів, що локалізуються в шкі-
рі з масивними інфільтратами у дермі.
-Лікування гістіоцитозу синусів бактеріальної природи має по-
зитивний ефект при застосуванні антибактеріальних препаратів.
ГІСТІОЦИТОЗ X (ГІСТІОЦИТОЗ ІЗ КЛІТИН ЛАНГЕРГАНСА)
Це група захворювань, шо об’єднує еозинофільну гранульому
(хвороба Таратинова), хворобу Хенда—Шюллера—Крісчена та хво-
робу Летгерера—Сіве.
Діагноз встановлюється з допомогою морфологічного дослід-
ження біоптатів шкіри, селезінки, печінки.
Головною ознакою є присутність клітин Лангерганса в дослід-
ному матеріалі. Клітини Лангерганса містять овальне, ниркоподіб-
не або порізане ядро із ніжною структурою хроматину та широку
світлу цитоплазму, яка не містить зернистості або включень. Крім
176
| Лейкемоїдні реакції
того, у вогнищі ураження можуть бути еозинофіли, лімфоцити,
нейтрофільні гранулоцити та гігантські багатоядерні клітини.
Діагноз верифікується електронно-мікороскопічним дослід-
женням (виявлення гранул Бірбека) та імуноцитохімічним дослід-
женням — забарвлення клітин Лангерганса лектином арахісу і на-
явність моноклональних антитіл до білка 5-100.
Здатність до накопичення та зберігання ліпідів також властива
макрофагам, вона гіпертрофована при хворобі Хенда—Шюллера—
Крісчена та при еозинофільній гранульомі, у клітинах котрої також
є надлишок ліпідів.
При хворобі Леттерера—Сіве макрофаги не містять надлишку
ліпідів. Захворювання описано Ееііегег (1924), 8іу/є (1933). Має гос-
тру прогресуючу форму з важким перебігом та дуже часто — нес-
приятливий прогноз. Діагностується здебільшого у немовлят і
призводить до смерті хворого через 1 -2 роки.
У клінічній картині переважає проліферація макрофагів у внут-
рішніх органах — селезінці, печінці, нервовій системі та шкірі.
Внаслідок спленомегалїї розвивається гіперспленізм, а згодом
тромбоцитопенія та геморагічний синдром.
Диференційний діагноз необхідно проводити з гострою моноб-
ластною та лімфобластною лейкемією, негоджкінською злоякіс-
ною лімфомою з лейкемізапією.
Еозинофільна гранульома (хвороба Таратинова) характеризу-
ється майже виключно внутрішньокістковою проліферацією гісті-
оцитів із присутністю великої кількості еозинофілів.
Часто до еозинофільної гранульоми або до ксантоматозу Хен-
да—Шюллера—Крісчена відносять такі хронічні деструктивні про-
цеси в кістках, як фіброзна остеодистрофія Олбрайт чи гіперпара-
тіреоз, при яких у вогнищах деструкції виявляють ксантоматозні
клітини.
У випадках солітарних дефектів у кістках найефективнішим є
хірургічне видалення проліферату або його опромінення, менш
ефективне застосування глюкокортикостероідних гормонів.
177
АГРАНУЛОЦИТОЗ
Агранулоцитоз — це клініко-гематологічний синдром, що ха-
рактеризується повним або майже повним зникненням із перифе-
ричної крові нейтрофільних гранулоцитів.
У 1922 р. Вернер Шульц звернув увагу лікарів на синдром не-
відомої етіології, який спостерігався, в основному, у жінок серед-
нього віку та характеризувався тяжкою ангіною, виснаженням,
значним зменшенням чи й повним зникненням гранулоцитів із
периферичної крові, швидким розвитком сепсису, що призводив
до смерті хворого. Він назвав цей стан агранулоцитозом.
Перші випадки агранулоцитозу були описані в Європі та Спо-
лучених Штатах Америки, проте захворювання спостерігається
в усіх частинах світу, й причиною його виникнення був амідопірин.
Частота захворювань у жінок порівняно з чоловіками стано-
вить 2-3 до 1.
Етіологія. Найчастіше виникнення агранулоцитозу пов’язане із
вживанням медикаментів. Якщо при використанні цитостатичних
засобів виникнення нейтропенії є майже закономірним наслідком і
пов’язане з основним механізмом дії, то агранулоцитоз унаслідок
застосування амідопірину або аміназину має інший патогенез.
У літературі є багато повідомлень про виникнення агранулоци-
тозу у хворих на тиреотоксикоз, котрі використовували такі проти-
тиреоїдні засоби, як метилтіоурацил, рідше — мерказолил. Абсолют-
но доведено, шо нейтропенічний вплив виявляють сульфаніламіди,
в тому числі й протидіабетичні, а також такі медикаменти, як бута-
діон, фенілбутазон, та препарати важких металів (вісмут, ртуть,
миш’як, срібло, золото), антигістамінні засоби (дипразин), нейро-
лептичні препарати та транквілізатори (аміназин, триоксазин, елені-
ум), барбітурати, новокаїн та новокаїнамід, протисудомні засоби
(дифенін, триметин), хінін та його похідні. Агранулоцитарні реакції
можуть виникати при використанні більшості сучасних антибіотиків
та протитуберкульозних препаратів.
Проте існують випадки захворювання на агранулоцитоз, котрі
не можна чітко пов’язати із впливом будь-якого екзогенного фак-
тора. Висловлюється припущення про участь генетичних факторів
у патогенезі такої форми агранулоцитозу.
178
| Агранулоцитоз
Імунні агранулоцитози
Концепція патогенезу імунних (гаптенових) агранулоцитозів
була сформульована МоєбИііп еі. аі. на початку 50-х років. Автори
дійшли висновку, шо лейкопенії, які виникають при алергічних
станах, найчастіше бувають пов’язані з появою в організмі антитіл
(аглютинів, лізинів тошо), дія яких спрямована проти власних лей-
коцитів. Припускають, що медикаментозні препарати відіграють
роль гаптенів, які утворюють комплексні сполуки з циркулюючи-
ми в крові білками або з білками оболонки лейкоцитів. Такий ком-
плекс “чужорідний” для організму та є своєрідним антигеном. Ан-
титіла, що продукуються у відповідь на даний антиген, фіксуються
на поверхні лейкоцитів, крім того, можливе й безпосереднє поєд-
нання антигена в крові із подальшою адсорбцією циркулюючих
імунних комплексів на лейкоцитах.
Перебіг імунного агранулоцитозу супроводжується порушенням
гранулоцитопоезу, ступінь якого широко варіює. На висоті імунного
агранулоцитозу розвивається гіпоплазія кісткового мозку за рахунок
клітин мієлоїдного паростка. В період одужання кістковий мозок стає
гіперпластичним унаслідок проліферації великої кількості незрілих
клітин гранулоцитарного ряду (промієлоцитів та мієлоцитів).
Імунний агранулоцитоз може бути автоімунним (системний
червоний вовчак, ревматоїдний артрит та ін.) або пов’язаним із ут-
воренням антитіл до екзогенних антигенів — гаптенів. Імунний аг-
ранулоцитоз часто набуває циклічно редицивуючої форми, особли-
во якщо його провокує інфекція (частіше вірусна).
Мієлотоксичні агранулоцитози
Причиною мієлотоксичного агранулоцитозу є шкідливий
вплив медикаментозних препаратів, котрий призводить до пригні-
чення проліферативної активності клітин гранулоцитарного парос-
тка з подальшим розвитком гіпоплазії гранулоцитопоезу.
Особливістю мієлотоксичного агранулоцитозу є зв’язок між
можливістю його виникнення та сумарною дозою вжитого препара-
ту, причому в кожного хворого ця доза не перевищує терапевтичну.
Розвиток мієлотоксичного агранулоцитозу може бути зумовлений
порушенням метаболізму препаратів унаслідок ферментативних де-
фектів у печінкових або інших клітинах, що беруть участь у метабо-
лізмі лікарських сполук. Крім того, порушення клітинного метабо-
лізму пов’язане зі здатністю препаратів посилювати проникність
клітинних мембран, спричиняти преципітацію полінуклеотидів, що
може призводити до інактивації багатьох ензимів. Взаємодія препа-
179
Н.М. Третяк | Гематологія
рату з ензимами мітохондрій викликає роз’єднання окислювального
фосфорилювання, результатом якого є пригнічення проліфератив-
ної активності клітин та зупинка клітин у С-фазі. Оскільки фермен-
тативні системи гранулоцитарних елементів змінюються в процесі
диференціювання та дозрівання, то й чутливість до медикаментів
клітин, що перебувають на різних стадіях диференціювання, може
бути різною. При максимальному ушкодженні активності родона-
чальних клітин перебіг патологічного процесу буде важким, і апла-
зія гранулоцитарного паростка може бути незворотною. При мен-
шому ушкодженні пула стовбурових клітин можливе повне
відродження гранулоцитарного паростка та одужання хворого.
Клінічна картина медикаментозного агранулоцитозу характери-
зується гострим або підгострим початком через 7-14 днів після пер-
шого курсу прийому препаратів, до котрих сенсибілізований орга-
нізм, або через 1-2 дні при повторному вживанні цих ліків.
Захворювання починається ознобом та гіпертермією, артралгіями.
Пацієнт блідий, проте слизові оболонки зберігають нормальний ко-
лір, можуть бути цианотичними, з’являється ііегрез. Водночас розви-
вається гранулоцитопенія, а через 12-24 години настає повний агра-
нулоцитоз. На мигдаликах виявляється брудно-сірий наліт. Згодом
утворюються некротичні бляшки та виразки, що пов’язано з пору-
шенням міграції нейтрофілів через слизові поверхні. Поширюючись,
некротизація захоплює піднебінні дужки, язичок, ясна, м’яке та
тверде піднебіння. Поширюючись углиб, некротичний процес охоп-
лює слизову оболонку стравоходу, кишечника й набуває характеру
номи, зумовлюючи болі в животі, здуття, нудоту, блювоту, пронос
(некротична ентеропатія). При руйнуванні глибоко лежачих тканин
і судин може виникнути значна кровотеча. В тяжких випадках агра-
нулоцитозу може розвиватися жовтяниця внаслідок некротичних
процесів у печінці. Некротизація тканин може спостерігатись у нир-
ках (протеінурія, гематурія, циліндрурія), надниркових залозах, що
призводить до гострої надниркової недостатності, легенях (пневмо-
нії, абсцеси, гангрени). Іноді виразково-некротичний процес охоп-
лює сечовий міхур та жіночі статеві органи.
У серцево-судинній системі особливих змін не спостерігається,
за винятком гіпотонії, артеріальної й венозної, та анемічних серце-
вих і судинних шумів. Селезінка, зазвичай, не збільшена. Іноді
спостерігається незначне збільшення регіонарних лімфатичних
вузлів, головним чином шийних та підщелепних. При виникненні
геморагічного синдрому з’являється гематурія.
180
| Агранулоцитоз
Хворі апатичні, проте притомними лишаються до самої агонії.
Велику загрозу життю хворого становить септицемія. Важкість*
інфекційного ускладнення визначається тривалістю агранулоцитозу
(що довший період агранулоцитозу, то тяжчий перебіг захворюван-
ня та інфекційних ускладнень). Важливим елементом, що визначає
частоту та складність інфекції, є глибина агранулоцитозу. При імун-
ному й, особливо, спровокованому гаптенами агранулоцитозі
в крові, зазвичай, гранулоцитів немає. У цих випадках інфекційні
ускладнення обов’язково виникають уже в перші дні захворювання.
Часто спостерігається грам-негативний сепсис, причому джерелом
інфекції є сапрофітна флора — кишкова паличка, протей, синегній-
на паличка. Грам-негативний сепсис супроводжується тяжкою ін-
токсикацією, гіпертермією до 40-41°С, тенденцією до артеріальної
гіпотонії, гіперемії тіла. При синегнійному сепсисі можлива поява
дрібних метастатичних вогнищ інфекції на шкірі, які мають чорне
забарвлення й розмір котрих може збільшуватися протягом десяти
хвилин. Такі ж самі вогнища можна спостерігати в легенях, нирках
та в інших органах під час розтину. Ці вогнища складаються майже
із чистої культури синегнійної палички.
Діагностика імунного агранулоцитозу базується на характер-
них змінах у периферичній крові.
При гаптеновому агранулоцитозі із периферичної крові ізольо-
вано зникають гранулоцити та моноцити. Інші формені елементи
не зазнають суттєвих змін. При затяжному процесі іноді буває нез-
начна лейкопенія. Вихід із агранулоцитозу супроводжується поя-
вою в крові плазматичних клітин, поодиноких мієлоцитів, водно-
час із ними, а іноді й на день .раніше, виникає моноцитоз. Надалі
бурхливо відновлюється кількість дозрілих гранулоцитів. Протягом
тижня склад периферичної крові нормалізується. Ізольовані “нулі”
гранулоцитів — характерна ознака гаптенового агранулоцитозу.
Крім того, визначається пікноз та розпад ядер, токсична зернис-
тість та вакуолізація цитоплазми поодиноких гранулоцитів, що за-
лишилися. Базофілів немає, іноді зустрічаються еозинофіли, ШЗЕ
збільшена.
При автоімунному агранулоцитозі картина крові така ж сама,
але при ньому частіше зберігаються поодинокі гранулоцити.
Якщо при автоімунному агранулоцитозі кількість гранулоцитів
наближається до своєї верхньої позначки (750 клітин у 1 мкл), то ін-
фекційного ускладнення може не бути протягом 2-3 тижнів. Це доз-
воляє вважати критичними не 750, а 500 гранулоцитів у 1 мкл крові.
181
Н.М. Третяк | Гематологія
При обох формах агранулоцитозу кількість лейкоцитів знижу-
ється помірно до 1,5-3,0- 109/л за рахунок збереження лімфоцитів.
Автоімунний агранулоцитоз, котрий ускладнює основне захво-
рювання, зазвичай проявляється помірним зниженням кількості
гранулоцитів. Проте на гематологічну картину в таких випадках
впливає основне захворювання, яке може спровокувати, й автоі-
мунний тромбоцитоліз, й автоімунний лізис еритроцитів. Кістко-
вий мозок на висоті гаптенового агранулоцитозу не містить ніяких
клітин гранулоцитарного паростка. Разом із тим, у трепанаті кіс-
тковий мозок клітинний за рахунок гіперплазії плазматичних клі-
тин, до того ж, спостерігається мегакаріоцитоз.
При виході з агранулоцитозу в кістковому мозку з’являються
великі із бобовидним або круглим ядром клітини, котрі дещо пе-
ревищують за розміром промієлоцити та містять бідну пилоподіб-
ну зернистість (клітини виходу). Наступного дня з’являється бага-
то про мієлоцитів, і вже через день у кістковому мозку досить
велика кількість дозрілих гранулоцитів.
Диференційний діагноз проводиться між імунним агранулоцито-
зом та лейкопенічною й алейкемічною формами гострої лейкемії,
особливо це необхідно в разі підгострого або хронічного перебігу
імунного агранулоцитозу.
Диференційний діагноз ґрунтується на морфологічному дослід-
женні кісткового мозку: при агранулоцитозі немає бластозу, збере-
жені еритроїдний та мегакаріоцитарний паростки кровотворення.
Лікування. Терапевтична тактика при обох формах агранулоци-
тозу включає, насамперед, боротьбу з інфекційними ускладнення-
ми. При гаптеновому агранулоцитозі необхідно виключити вико-
ристання препарату, котрий є гаптеном чи може ним бути. Нині
при цій формі агранулоцитозу не призначаються глюкокортикоїд-
ні гормони, тому що їх застосування веде до ще більшого знижен-
ня протиінфекційного імунітету.
При автоімунному агранулоцитозі глюкокортикоїдні гормони,
безперечно, призначаються.
Профілактика'.
— хворий, що переніс медикаментозний агранулоцитоз, не по-
винен використовувати препарати, які вміщують цей гаптен;
— при призначенні препаратів, котрі потенційно можуть спри-
чинити агранулоцитоз, необхідно контролювати показники крові
не рідше, ніж один раз на 10 днів.
182
ЦИТОСТАТИЧНА ХВОРОБА
Цитостатична хвороба є багатосиндромним захворюванням,
котре спричиняється дією цитостатичних факторів та зумовлене
загибеллю клітин, які діляться. Це, в першу чергу, клітини кіс-
ткового мозку, епітелію оболонки травного тракту, шкіри та її до-
датків.
Визначення “цитостатичні фактори” об’єднує різні агенти. По-
перше, це цитостатичні препарати, які використовуються для ліку-
вання онкозахворювань і серед котрих є такі, що впливають на ок-
ремі фази клітинного циклу чи на клітини, що перебувають поза
цим (Со) циклом, або на клітини, що перебувають і в тому, і в ін-
шому стані. По-друге, це речовини, які не використовуються як
цитостатичні засоби, але мають цитостатичні властивості, що дове-
дено (Різсіопа, 1969). Крім того, такі препарати, як левоміцетин,
проявляють свої цитостатичні якості при лікуванні хворих, які ма-
ють індивідуальну чутливість до нього, тобто ці пацієнти мають ге-
нетичні особливості клітинних структур, чутливих до впливу хіміч-
них засобів (У/а§ао еі. аі., 1969). Цитостатичний ефект справляють
усі види іонізуючого опромінення, яке ушкоджує клітини у фазах
клітинного циклу.
Усі ці речовини поєднує одна спільна властивість — здатність
до ушкодження клітин, що призводить до їх загибелі та розвинен-
ня аплазії кісткового мозку. Найтяжчим, таким, що загрожує жит-
тю, проявом аплазії є агранулоцитоз. Агранулоцитозом вважається
зниження вмісту лейкоцитів менше 1,0 • 109/л або рівня гранулоци-
тів нижче 0,75 • 109/л .
Патогенез. Ушкодження всіх трьох паростків мієлопоезу озна-
чає, що цитостатичний препарат вплинув на клітину — поперед-
ницю мієлопоезу. Закономірно, що чим більш ранні клітини кро-
вотворення ушкоджує цитостатичний препарат, тим пізніше,
навіть при великих його дозах, з’являються зміни в периферичній
крові.
При цигостатичній хворобі картина крові характеризується
зменшенням кількості лейкоцитів і тромбоцитів. При глибших
ураженнях клітин-попередниць розвивається анемія.
183
Н.М. Третяк | Гематологія
Лейкопенія проявляється зникненням із периферичної крові
всіх гранулоцитів, моноцитів, лімфоцитів і може сягати дуже низь-
ких показників — 100 і навіть 50-25 клітин у 1 мкл., проте пооди-
нокі гранулоцити в крові зберігаються. Тромбоцитопенія також
може бути дуже глибокою — до 10-5 тисяч тромбоцитів у 1 мкл.
Критичним рівнем тромбоцитів вважається такий, за якого ше не
розвивається геморагічний синдром. Таким умовно критичним мо-
же вважатися рівень тромбоцитів 20,0 • 109/л, за якого геморагічний
синдром, зазвичай, ще не розвивається.
Разом із тим, різні цитостатичні препарати неоднаково пригні-
чують нормальне кровотворення. Відомо, шо циклофосфан і він-
кристин у терапевтичних дозах мало впливають на тромбоцитопо-
ез у протилежність рубоміцину та метотрексату. В період
найбільшої активності патологічного процесу тромбоцити можуть
знижуватись до нуля. Ретикулоцитів у периферичній крові в пері-
од мієлотоксичного агранулоцитозу не знаходять. У кістковому
мозку в цей період майже повністю зникають як гранулоцитарні
елементи, так і еритронормобласти й мегакаріоцити. Зберігаються
майже виключно лімфоїдні, ретикулярні та плазматичні клітини.
За 2-3 дні до виходу із мієлотоксичного агранулоцитозу в кіс-
тковому мозку з’являється велика кількість промієлоцитів та поо-
динокі нормобласти.
Вихід із цитостатичної мієлодепресії супроводжується зростан-
ням вмісту тромбоцитів та лейкоцитів у периферичній крові, поя-
вою молодих форм нейтрофільних гранулоцитів (мієлоцити та ме-
тамієлоцити), інколи виявляються плазматичні клітини та
моноцити, що є першою ознакою активації кровотворення.
При використанні терапевтичних доз цитостатичних препара-
тів мієлотоксичний агранулоцитоз спостерігається 1-2 тижні. Про-
те при передозуванні мієлосану, хлорбутину, а також застосуванні
звичайних доз алкерану у хворих із нирковою недостатністю гли-
бока цитопенія може тривати довго.
Найпостійнішою ознакою агранулоцитозу є ураження слизової
оболонки порожнини рота та глотки, тому що цитостатичні засо-
би, впливаючи на різні фази клітинного циклу, з однаковою агре-
сивністю ушкоджують клітини кісткового мозку та клітини слизо-
вої оболонки травного тракту, шкіри, волосяних головок.
Найнебезпечнішим ускладненням тривалої цитостатичної гра-
нулоцитопенії є ураження шлунково-кишкового тракту — некро-
тична ентеропатія.
184
| Цитостатична хвороба
Окремі гематологи (Атготіп, 1968) розрізняють 4 морфологіч-
них варіанти некротичного ентероколіту при лейкеміях.
Перший тип — ішемічний ентероколіт (або псевдомембраноз-
на ентеропатія) проявляється некрозом слизової оболонки, най-
частіше тонкої кишки. Некроз розвивається внаслідок ішемії, зу-
мовленої порушенням центральної геодинаміки, та
мікроциркуляторних розладів при шоку будь-якого генезу (після-
геморагічний, септичний, бактеріальний).
Другий тип — виразково-некротична ентеропатія тонкого та
товстого кишечника (найчастіше ураження дистального відрізка
тонкого, висхідного відрізка товстого кишечника та сліпої кишки).
Крім множинних поширених ерозій слизової оболонки, спостері-
гається значне потовщення всієї стінки кишечника внаслідок наб-
ряку (часто геморагічного) та поширення некрозу до серозного ша-
ру. Можливі перфорація та перитоніт. Особливістю цього типу
некротичної ентеропатїї є локалізація уражень у відрізках кишеч-
ника з розвинутою лімфатичною системою. При лейкеміях під-
ґрунтям ентеропатїї цього типу може бути лейкемічна проліфера-
ція в лімфатичній структурі кишечника.
Третій тип — геморагічна некротична ентеропатія, тобто кро-
вовиливи в стінку тонкого та товстого кишечника із вторинним ін-
фікуванням флорою кишечника й наступним розвиненням вираз-
ково-некротичних змін.
Четвертий тип — виразково-некротичні зміни порожнини ро-
та, глотки, стравоходу, прямої кишки, відхідника, піхви. Відомо,
що ці ділянки найчастіше вражаються мікотичною інфекцією.
Поширена ентеропатія, особливо другого та третього типів,
обов’язково призводить до розмноження та розселення кишкової
флори, котра у більшості хворих на мієлотоксичний агранулоцитоз
спричиняє грам-негативну септицемію з розвиненням ендотокси-
нового шоку.
Клінічна картина некротичної ентеропатїї при цитостатичному
агранулоцитозі має деякі особливості.
Одним із перших симптомів є гіпертермія, потім з’являється
пронос, кашкоподібні випорожнення або запор. Часто спостеріга-
ється помірне здуття живота й нападоподібний біль, частіше в іле-
оцекальній ділянці, який супроводжується напруженням черевної
стінки та симптомами подразнення очеревини.
Об’єктивно початковими ознаками некротичної ентеропатїї є
плеск, буркотіння та болючість пальпації ілеоцекальної ділянки.
185
Н.М. Третяк | Гематологія
Водночас язик у хворого обкладений та сухуватий. Наростання оз-
нак подразнення очеревини, зникнення перистальтики кишечни-
ка, виявлення випоту в черевну порожнину, сухість язика — все це
ознаки загрози проколювання або проколювання, що вже сталося.
Тривалість некротичної ентеропатії в умовах сучасної терапії
не перевищує 1-1,5 тижня та зазвичай закінчується одужанням.
Іншим загрозливим ускладненням цитостатичної хвороби є
гострий епітеліальний гепатит, котрий виникає після одужання від
агранулоцитозу, зумовленого цитостатичними препаратами. Різні
цитостатичні препарати мають неоднакову гепатотропність. Най-
небезпечніші — це циклофосфан, 6-меркаптопурин, метотрексат,
рубоміцин. Гепатит, спричинений циклофосфаном, найнебезпеч-
ніший.
Клінічною особливістю цитостатичного гепатиту є відсутність
будь-яких провісників хвороби, таких, наприклад, як підвищення
температури тіла, біль у суглобах, під правим ребром, ангіна.
Хвороба починається із жовтяниці при відносно задовільному
самопочутті хворого. Спостерігається помірне збільшення печінки,
АСТ, АЛТ різко підвищені, білірубін відносно невисокий за раху-
нок прямої фракції. Зникнення жовтяниці, нормалізація розмірів
печінки та трансаміназ свідчать про одужання хворого і можливість
продовжувати, за потреби, цитостатичну терапію.
Гепатит, зумовлений цитостатичними засобами, становить заг-
розу життю хворого.. Лікування цього ускладнення проводиться за
загальними правилами терапії гострого епітеліального гепатиту.
Відсутність гранулоцитів при цитостатичному агранулоцитозі
— патогенетична основа для розвитку різних інфекційних усклад-
нень, таких як септицемія з високою гарячкою, значною інтокси-
кацією, проливними потами без локальних вогнищ запалення. Од-
ним із найчастіших інфекційних ускладнень цитостатичного
агранулоцитозу є медіастеніт, пневмонія з дуже бідною симптома-
тикою: сухий кашель, задишка, цианоз, обмеженість ділянки брон-
хіального дихання, поодинокі сухі хрипи або крепітація в цьому
місці. В окремих випадках навіть при рентгенологічному дослід-
женні визначити вогнище запалення в легенях неможливо.
Збільшення кількості гранулоцитів у крові призводить до ос-
лаблення інтоксикації. Проте іноді посилюються фізікальні ознаки
пневмонії. Зазвичай клінічна розв’язка цитостатичного агрануло-
цитозу відбувається дещо раніше від нормалізації гематологічних
показників.
186
| Цитостатична хвороба
Лікування та профілактика. Для попередження інфекційних
ускладнень при цитостатичній хворобі зі зниженням у периферич-
ній крові вмісту гранулоцитів та тромбоцитів до критичного рівня
хворого одразу необхідно перевести до ізолятора. Під час перебу-
вання в ньому хворому протягом доби, окрім ночі, проводять оп-
ромінення ультрафіолетовими лампами, підвішеними на рівні 2
м від підлоги, причому лампа повинна бути екранована знизу. Під-
логу, стіни, устаткування палати щодня протирають дезінфікую-
чим розчином (діацид, рокал, 1%-й розчин хлораміну). Хворого
щодня обмивають або обтирають (якщо його загальний стан тяж-
кий) асептичним розчином, перевдягають у стерильну білизну, змі-
нюють простирадла. Медичний персонал повинен заходити до па-
лати в масках, бахілах, шапочках, мити руки асептичним
розчином. Такі заходи знижують частоту інфекційних запалень
верхніх дихальних шляхів майже у 10 разів.
Система протиінфекційних заходів включає не тільки бороть-
бу з екзогенною інфекцією, а й санацію шлунково-кишкового
тракту хворого від патогенної флори. З цією метою використову-
ється бісептол (по 3 г/добу), полімексин В (по 0,4 г/добу) та амфо-
терицин В (по 2 г/добу); налідіксова кислота (по 1000 мг/кг на
день), полімексин В (по 10 мг/кг на день) та амфотерицин В (по
1 г/добу).
Хворим на агранулоцитоз призначають дієту, що виключає
консервовані продукти, надлишок клітковини та страви, які рані-
ше призводили до диспептичних розладів. Для повного очищення
кишечника від флори страви стерилізують.
При підвищенні температури тіла до фебрильних цифр або ви-
явленні вогнищ інфекції необхідно зробити посів крові, сечі або
інших рідин організму для визначення флори та чутливості її до
антибіотиків, зробити рентгенограму органів грудної клітки й того
ж дня призначити антибіотики широкого спектру дії та ністатин —
не менше 6-10 млн. ОД/добу.
У період агранулоцитозу, як і під час глибокої тромбоцитопе-
нії, підшкірні та внутрішньом’язові ін’єкції не призначають. Усі
препарати вводять внутрішньовенно або всередину. При виявлен-
ні синегнійного сепсису використовують схему ступінчастої хіміо-
терапії: цифран — 200-400 мг в/в 2 р. на добу, після стабілізації ста-
ну хворого — орально 500-750 мг цифрану 2 р. на добу, залежно від
тяжкості перебігу' інфекції, або гентаміцин — до 240 мг/добу разом
із карбеніциліном (до 30 г/добу). Можна застосувати вібраміцин —
187
Н.М. Третяк | Гематологія
до 80 мл 2-3 р. на добу окремо або разом із карбеніциліном, крім
того, ефективним є амікацин — по 150 мг в/в 2-3 р. на добу.
При діагностуванні стафілококового сепсису застосовують це-
порин, лінкоміцин. При пневмококовому сепсисі призначають пе-
ніцилін у максимально високих дозах. Контрольні посіви крові
проводять щодня. Вогнище інфекції у м’яких тканинах лікується за
всіма правилами хірургічного лікування таких ускладнень.
При виникненні некротичної ентеропатїї призначається повне
голодування. Хворому дозволяється пити тільки кип’ячену воду.
Вода не повинна мати ніякого смаку, щоб не провокувати відділен-
ня секрету в шлунку, підшлунковій залозі та із жовчного міхура.
Забороняються мінеральні води, соки, чай. Холод та тепло на жи-
віт не використовуються. Всі лікувальні препарати вводяться тіль-
ки внутрішньовенно.
Термін голодування закінчується після зникнення всіх ознак
некротичної ентеропатїї але, зазвичай, не перевищує 7-10 діб. Ду-
же рідко голодування може тривати близько місяця.
Вихід із голодування триває приблизно стільки ж, як і сам
період голодування. Спершу збільшують кількість разів прийо-
му їжі. В перші дні дають 300-400 мл води, сироватку, можна
сир 50-100 г або вівсяну кашу, все це за 2-3 рази. Поступово ді-
єту розширюють за рахунок гречаної та манної каш, додають са-
лати із сирої капусти та моркви. Пізніше дозволяються страви із
м’яса (фрикадельки, парові котлети). Хліб дозволяють вживати
останнім.
У хворих на цитостатичний агранулоцитоз часто утворюються
некротичні виразки на слизових оболонках рота, глотки. У таких
випадках стоматолог або отоларинголог повинен взяти мазки на
посів та щодня проводити туалет слизових оболонок роту та глот-
ки, зрошувати розчином перекису водню, граміцидіном, розчином
білка з курячого яйця, змащувати виразки вітаміном А, обліпихо-
вою олією, прополісом; при появі молочниці на слизовій оболон-
ці рота проводять полоскання гідрокарбонатом натрію та левори-
ном, змазують слизову оболонку бурою з гліцерином та
ністатином.
Дуже часто при агранулоцитозі інфекція локалізується в піхві
або в ділянці відхідника. Лікування такого парапроктиту прово-
диться хірургом за всіма хірургічними правилами.
Дуже тяжкими ускладненнями цитостатичної хвороби є тром-
боцитопенія і пов’язаний із цим геморагічний синдром.
188
| Цитостатична хвороба
Показанням для трансфузії тромбоконцентрату є глибока, не
менш як 2,0 • 109/л, тромбоцитопенія, а також геморагії на шкірі,
слизових оболонках, склерах, кровотечі шлунково-кишкового
тракту, матки, сечовивідних шляхів.
Профілактичні трансфузії тромбоконцентрату необхідні при
проведенні курсів цитостатичної терапії в умовах глибокої тромбо-
цитопенії.
Одиниця тромбоконцентрату — це кількість тромбоцитів,
отримана із 100-5000 мл. донорської крові, зазвичай це
0,5-0,90 • 10п/л тромбоцитів. Лікувальну дозу становлять 4-4,5 ОД
тромбоконцентрату, отриманого від одного донора. Ефективність
лікування тромбоцитами визначається припиненням кровотечі та
підвищенням кількості тромбоцитів у реципієнта за 1-24 години
після переливання. Гемостатичний ефект перелитих тромбоцитів
може зберігатися від 2 до 7 днів, після чого рівень тромбоцитів мо-
же знижуватись.
При виникненні кровотеч, що загрожують життю хворого
внаслідок ДВЗ-синдрому та тромбоцитопенії, тромбоконцентрат
потрібно вводити на фоні терапії гепарином і контрикалом та вли-
вання свіжомороженої плазми під прискіпливим коагулологічним
контролем.
За потреби кількаразових переливань тромбоконцентрату слід
проводити підбір донора за НІА-антигенами. Таких донорів, найі-
мовірніше, можна знайти серед братів та сестер хворого. Для поо-
диноких трансфузій тромбоконцентрату можна обмежитися підбо-
ром донора за АВО-антигенними системами.
189
ЛЕЙКОПЕНІЇ
Лейкопенії — одна з актуальних проблем сучасної гематології.
Найчастіше розвиток лейкопенії пов’язаний зі зменшенням у кро-
ві абсолютної кількості нейтрофілів. Вміст нейтрофілів, менший за
4,0- 109/л, називається лейкопенією, при цьому кількість нейтро-
фільних гранулоцитів становить менш ніж 2,0* 109/л.
Основні причини лейкопенії (нейтропенїї)
1. Інфекції:-
— бактеріальні (тифи, паратифи, бруцельоз, рідко — туляре-
мія);
— вірусні (грип, інфекційний гепатит, кір, вітряна віспа, крас-
нуха, СНІД);
— рикетсіальні (везикулярний рикетсіоз, висипний тиф);
— гарячка скелястих гір;
— протозойні (малярія, кала-азар, кліщовий поворотний тиф).
2. Усі види генералізованих інфекцій:
— міліарний туберкульоз, септицемія, особливо у виснажених
пацієнтів із низькою резистентністю.
3. Фізичні та хімічні агенти:
— іонізуюча радіація, бензин, нітросполучення, уретан;
— цитостатики — антиметаболіти (антагоністи фолієвої кисло-
ти, аналоги пурину та піримідину), вінбластин, колхіцин, антра-
цикліни.
Зазначені хімічні та фізичні речовини призводять до мієлосуп-
ресії при застосуванні в досить високих дозах. Іноді ці фізичні та
хімічні агенти спричиняють лейкопенію внаслідок підвищеної чут-
ливості організму.
4. Окремі гематологічні захворювання та інші стани невідомої та
маловивченої етіології. Лейкопенія може розвиватися внаслідок не-
ефективного гемопоезу (перніциозна та апластична анемія). Лей-
копенія пов’язана з підвищеною утилізацією, деструкцією або сек-
вестрацією нейтрофілів, що спостерігається при цирозі печінки
спленомегалією, вовчаку червоному, синдромі Ееіїу, синдромі
Вапіу, хворобі Саисіїег, гемодіалізі.
5. Кахексія та ослаблений стан.
190
| Лейкопенії
6. Анафілактичний шок та ранні стадії реакції на чужорідний білок.
7. Рідкісні спадкові, вроджені та родинні патології (циклічна
нейтропенія, хронічна гіпопластична нейтропенія, дитячий гене-
тичний агранулоцитоз, первинна селезінкова нейтропенія).
Кінетична класифікація нейтропеній виділяє:
1) нейтропенії, зумовлені зменшенням продукції нейтрофілів
у кістковому мозку;
2) нейтропенії, зумовлені уповільненням виходу нейтрофілів із
кісткового мозку у кров;
3) нейтропенії, зумовлені скороченням терміну циркуляції
нейтрофілів у кровоносних судинах.
Зменшення продукції нейтрофільних гранулоцитів спостеріга-
ється, в основному, при захворюваннях системи крові, що доклад-
но висвітлено у відповідних розділах.
Спадкові та вроджені лейкопенії зустрічаються дуже рідко і яв-
ляють собою надзвичайно гетерогенну за патогенезом та клінічною
картиною групу захворювань, перебіг котрих супроводжується зни-
женням рівня нейтрофільних гранулоцитів у крові та кістковому
мозку.
До спадкових форм лейкопеній відносять таку нейтропенію, як
хвороба Костмана, що успадковується за автосомно-домінантним
типом. Характеризується ця хвороба важкою нейтропенією й роз-
вивається одразу ж після народження дитини. У перші дні життя
виникають фурункульоз, карбункули, сепсис, виразки та некрози
на слизових оболонках. У периферичній крові вміст нейтрофілів
менший за 0,5 • 109/л, але загальна кількість лейкоцитів утримуєть-
ся в межах норми за рахунок збільшеної кількості моноцитів.
Спостерігається помірна анемія.
У кістковому мозку переважають промієлоцити, зрілі грануло-
цитарні елементи відсутні. Зустрічаються зрілі еозинофіли та базо-
філи, атипічні еритроідні клітини.
Навіть за умови інтенсивної антибактеріальної терапії при пов-
торних рецидивах уже в перший-третій тижні дитина може загину-
ти.
Лікування стероїдними та анаболічними гормонами, як і спле-
нектомія, не ефективні. Доцільно рекомендувати трансплантацію
кісткового мозку.
Покращує прогноз застосування гранулоцитарно-макрофа-
гальних колонієстимулюючих факторів та адекватна протиінфек-
ційна терапія.
191
Н.М. Третяк | Гематологія
Спадкова нейтропенія автосомно-домінантного типу (хвороба
Сагазіепіа)
Основою патогенезу цього захворювання є зменшення продук-
ції нейтрофілів у кістковому мозку в поєднанні з перерозподілом
нейтрофілів у кровоносних судинах.
Захворювання проявляється у дітей уже в перші місяці життя
схильністю до запально-некротичних процесів, зумовлених ней-
тропенією.
При дорослішанні дитини, уже після 7 років, значно зменшу-
ється схильність до інфекційно-запальних захворювань, і це зумов-
лює сприятливий прогноз. Проте, незважаючи на нормалізацію
з віком кількості нейтрофілів у периферичній крові, у кістковому
мозку утримується знижена кількість гранулоцитів мітотичного пу-
ла та клітин — попередниць гранулоцитопоезу. Це свідчить, що де-
фект стовбурових клітин зберігається й у період “ремісії”.
Лікування під час загострення лейкопенії симптоматичне, крім
того, призначається літій, андрогени. При дорослішанні дитини
потреба в терапії зникає.
Родинна доброякісна нейтропенія автосомально-домінантного типу
Характеризується значним збільшенням кількості клітин — по-
передниць грануломоноцитопоезу в кістковому мозку хворих, про-
те дозрівання цих клітин порушено, внаслідок чого в периферич-
ній крові спостерігається суттєве зменшення нейтрофільних
гранулоцитів (до 20-30%). Клінічний перебіг захворювання подіб-
ний до картини при спадковій нейтропеніі автосомно-домінантно-
го типу.
Вроджена нейтропенія автосомно-домінантного типу
Характеризується стійкою гранулоцитопенією при нормально-
му співвідношенні в мієлограмі. Розвинення гранулоцитопенії мо-
же бути пов’язане з порушенням надходження нейтрофілів у кров.
Захворювання зустрічається тільки у єменських євреїв.
Перебіг хвороби доброякісний і не супроводжується знижен-
ням опору до інфекцій. Специфічного лікування хворі не потребу-
ють. Прогноз сприятливий.
Синдром “ледачих лейкоцитів”
Синдром “ледачих лейкоцитів” уперше описаний Хиеігег (1964)
і належить до вроджених із доброякісним, сприятливим для пацієн-
та перебігом нейтропеній. Проявляється зменшенням кількості доз-
рілих нейтрофільних гранулоцитів у периферичній крові в поєднан-
ні зі збільшенням їх у кістковому мозку. Відсоткове співвідношення
192
| Лейкопенії
між іншими гранулоцитарними елементами не порушене. Припус-
кається, що в основі патогенезу синдрому “ледачих лейкоцитів” ле-
жить дефект клітинної мембрани, що зумовлює порушення виходу
гранулоцитів із кісткового мозку в периферичну кров.
Клінічно синдром проявляється частими інфекційно-запаль-
ними захворюваннями. Особливо характерні для нього інфекційні
ускладнення з боку верхніх дихальних шляхів. Зазвичай селезінка
при синдромі “ледачих лейкоцитів” не збільшена, проте при інс-
трументальному обстеженні (УЗО, Ко-скопія) в деяких хворих від-
значається незначне її збільшення.
Патогенетичної терапії до цього часу не існує. Глюкокортико-
їдні гормони, спленектомія не дають бажаного ефекту. Застосову-
ється симптоматична терапія, Прогноз відносно сприятливий.
Циклічна нейтропенія
Це одна із рідкісних та незвичайних форм нейтропеній. Харак-
терною особливістю захворювання є циклічність його перебігу. Ре-
цидиви виникають, як правило, кожні 3-4 тижні та проявляються
типовими для агранулоцитозу клініко-гематологічними ознаками.
Частіше циклічний агранулоцитоз починається в ранньому дитячо-
му віці, проте в деяких випадках діагностується у літніх людей. Зах-
ворювання з однаковою частотою зустрічається у чоловіків і жінок.
Спадковий фактор відіграє не останню роль у генезі захворю-
вання.
Припускається три можливих механізми розвитку циклічної
нейтропенії: внутрішній дефект стовбурових гемопоетичних клі-
тин, порушення гуморальної регуляції гемопоезу, порушення ко-
роткодистанційних регуляторів гемопоезу.
Клінічна картина характеризується періодичним фазовим зни-
женням кількості нейтрофільних гранулоцитів у периферичній
крові. Рецидив захворювання за клініко-гематологічними проява-
ми подібний до гострого агранулоцитозу. Однак кількість грануло-
цитів починає значно знижуватися за 1-3 дні до появи клінічних
ознак. Фаза вираженої гранулоцитопенії триває близько 3-5 днів,
після чого вміст гранулоцитів починає зростати. В розпалі лейко-
пенії в мієлограмі виявляється картина так званого “гальмування
дозрівання” — з переважанням у мієлограмі промієлоцитів та
збільшенням відсотка мієлобластів.
У період ремісії склад периферичної крові та кісткового мозку
суттєво не відрізняється від норми. Рецидиви виникають через 3-4
тижні.
193
Н.М. Третяк | Гематологія
Лікування спрямоване на боротьбу з інфекційними ускладнен-
нями. В період агранулоцитозу призначають антибіотики, імуног-
лобулін, а також свіжу нативну плазму, котра містить колонієсти-
мулюючі фактори. Глюкокортикоїдні гормони та спленектомія не
ефективні.
Прогноз сприятливий, при дорослішанні хворого клініко-гема-
тологічна симптоматика пом’якшується.
Синдром Швахмана
Описаний у 1964 р., але причини виникнення цього виду ней-
тропенії не з’ясовані.
Родинний характер захворювання, співіснування в окремих
хворих нейтропеній із тромбоцитопенією та анемією дозволяють
припустити, що підґрунтям нейтропенії є ушкодження родона-
чальних клітин-попередниць.
Нейтропенія виявляється в ранньому дитинстві. У багатьох
дітей спостерігається відставання у фізичному розвитку, пору-
шення формування епіфізів, значно рідше виявляється розумове
недорозвинення, крім того, нейтропенія поєднується з кістозним
фіброзом підшлункової залози, порушенням її екзокринної фун-
кції та діареєю.
При значному зменшенні нейтрофілів у цих хворих значно
знижується резистентність до інфекцій. У частини хворих зберіга-
ється гранулоцитарна відповідь на запалення, але вона виражена
менше, ніж у гематологічно здорових людей.
Лікування інфекційних ускладнень зводиться до призначення
антибактеріальних препаратів та вітамінів. При вираженій грануло-
цитопенії показані трансфузії гранулоцитів. Доцільне призначення
андрогенів та літію.
Синдром Чедіака—Хігаші
Патогенез лейкопенії при цьому синдромі не з’ясовано, але
припускається, шо лейкоцити швидше руйнуються, ніж продуку-
ються кістковим мозком (неефективний грануломоноцитопоез або
скорочення тривалості життя).
Типовим для хворих є схильність до розвитку тяжких гнійних
ускладнень, зумовлених пригніченням фагоцитарної функції ней-
трофілів.
Патогенетична терапія цього захворювання не опрацьована.
Існують спостереження щодо позитивного впливу на перебіг зах-
ворювання та функцію нейтрофільних гранулоцитів великих доз
аскорбінової кислоти, застосування котрої призводить до знижен-
194
| Лейкопенії
ня в нейтрофілах активності цАМФ та до відновлення їхньої фаго-
цитарної активності.
При швидкому прогресуванні захворювання, коли спостеріга-
ється лімфогістіопитарна інфільтрація печінки, селезінки, нервової
системи тощо, деякі дослідники рекомендують застосування він-
кристину та преднізолону.
Синдром Цинсера—Коула—Енгмена
Синдром Цинсера—Коула—Енгмена описаний у 1910 р. 2іп-
88ег, у 1926—1932 рр. класифікований Соїе як вроджений дискера-
тоз, для якого характерна дискератотична дистрофія шкіри та сли-
зових оболонок. Ця патологія проявляється дистрофією нігтів,
гіпотрихозом вій, сльозотечею та закупоркою слізних протоків,
ураженням ендокринних залоз. Поряд із ураженням шкіри та сли-
зових оболонок у частини хворих спостерігаються зміни в системі
крові. Це може бути гіпоплазія з панцитопенією, окремо анемія,
тромбоцитопенія або нейтропенія. Зміни в системі кровотворення,
множинні вроджені ураження інших органів, особливо шкіри та її
фрагментів, а також слизових оболонок роблять це захворювання
подібним до анемії Фанконі. Проте в клінічній картині цих захво-
рювань є суттєві відмінності: при анемії Фанконі цитопенічний
синдром проявляється в перші десять років життя, тоді як при син-
дромі Цинсера—Коула—Енгмена зміни з боку системи крові з’яв-
ляються значно пізніше, у 20-30 років.
Патогенез цитопенії при вродженому дискератозі не вивчено,
але є дослідження, що свідчить про внутрішній дефект стовбуро-
вих клітин або патологію кровотворного мікрооточення.
НЕЙТРОПЕНІЯ ПРИ ПЕРВИННИХ ІМУНОДЕФІЦИТНИХ
СТАНАХ
Агамаглобулінемія Брутона
Агамаглобулінемія Брутона, яка передається рецесивно та
зчеплена з Х-хромосомою, — типовий представник імунодефіцит-
них станів із переважаючим дефектом з боку В-лімфоцитів.
Для хворих на В-імунодефіцит характерне зниження концентра-
ції імуноглобулінів усіх класів, стійка нейтропенія в периферичній
крові, шо поєднується з морфологічною картиною “гальмування
дозрівання” у кістковому мозку. Клінічний перебії характеризується
значним зниженням резистентності до інфекцій, рецидивуючими
виразками на слизових оболонках ротової та інших порожнин.
195
Н.М. Третяк | Гематологія
Телеангіектатична атаксія
Телеангіектатична атаксія з нейтропенією має подібні клініко-
гематологічні ознаки.
Синдром Ді-Джоржа
Характеризується імунодефіцитним станом із переважаючим
ушкодженням Т-лімфоцитів — синдром III та ІУ-фарингеальних
кишень і синдром II та III фарингеальних кишень. Ці синдроми
характеризуються тяжкими імунодефіцитними зрушеннями клі-
тинної ланки імунітету та нормальним функціонуванням гумораль-
ної ланки імунітету.
У хворих із цим синдромом часто виявляється патологія ендок-
ринних залоз та аорти, в багатьох випадках зустрічається нейтро-
пенія, патогенез котрої зовсім не вивчений. Хворі, як правило, по-
мирають від вірусних пневмоній.
Синдром Гуда
Це лімфопенічна агамаглобулінемія з тимомою, котра характери-
зується патологічним порушенням функції як В-, так і Т-лімфоцитів.
Синдром Віскотга-Олдрича
Це спадкова гамаглобулінемія з тромбоцитопенією та екземою,
пов’язана з дефектом Х-хромосоми.
При всіх вище зазначених агамаглобулінеміях може розвину-
тись тяжка нейтропенія, й особливо вона характерна для синдро-
му Віскотта-Олдрича.
Лікування хворих із первинно імунодефіцитними станами сим-
птоматичне. При інфекційно-запальних ускладненнях проводить-
ся активна антибактеріальна терапія, призначається імуноглобулін.
Глюкокортикоідні гормони та спленектомія позитивного результа-
ту не дають.
Описані випадки повного виліковування дітей з допомогою
трансплантації кісткового мозку або стовбурових клітин ембріо-
нальної печінки в поєднанні із трансплантацією тимусу.
НЕЙТРОПЕНІЯ НОВОНАРОДЖЕНИХ ІМУННОГО ГЕНЕЗУ
Проявляється відразу ж після народження дитини підвищеною
чутливістю до інфекцій, котра зумовлена глибокою гранулоцитопе-
нією. В основі патогенезу захворювання — присутність автоанти-
тіл у організмі дитини, мішенню для яких є нейтрофіли. Вважають,
що продукція цих антитіл пов’язана з антигенною несумісністю
гранулоцитів матері та плоду.
196
| Лейкопенії
Захворювання не відносять до вроджених, хоча гранулоцитопе-
нія й виявляється у дітей уже при народженні. Проте у більшості
новонароджених склад крові нормалізується вже за 5-7 тижнів піс-
ля народження.
Прогноз — як близький, так і віддалений — сприятливий.
ЛЕЙКОПЕНІЇ ПРИ СПЛЕНОМЕГАЛІЯХ
При захворюваннях, що супроводжуються спленомегалією,
у більшості хворих у периферичній крові спостерігається нитопе-
нія. В деяких випадках на перший план виступає анемія, в інших
— лейкопенія або тромбоцитопенія, часто панцитопенія.
Майже завжди лейкопенія спостерігається при так званих зас-
тійних спленомегаліях (порушення портального кровообігу при ци-
розах печінки, порушення кровообігу в системі воротної вени та ін.).
Лейкопенії при цирозі печінки
З-поміж різних форм цирозів печінки спленомегалія найчасті-
ше спостерігається при портальному цирозі. Механізм розвитку
спленомегалії при цирозах різний. Вирішальну роль у цьому проце-
сі відіграє підвищення тиску в системі воротної вени із подальшим
застоєм крові в селезінці. Тривалий застій крові сприяє розвитку
сполучної тканини зі збільшенням у ній вмісту фагоцитуючих мак-
рофагів.
Провідним механізмом “гіперспленізму” вважається руйнуван-
ня формених елементів крові на периферії. Проте далеко не в усіх
хворих на цироз печінки лейкопенія може бути пояснена тільки
з позицій “гіперспленізму”. Оскільки в патогенезі хронічних гепа-
титів та цирозів печінки суттєву роль відіграє імунологічний ком-
понент, не виключається розвинення й імунних цитопеній (ней-
тропеній). Одним із механізмів, що спричиняє нейтропенію, може
бути зниження дезінтоксикаційної функції печінки при порталь-
них цирозах.
Лейкопенія у цих хворих клінічно не проявляється. При об’єк-
тивному обстеженні печінка не збільшена, виявляється тільки
спленомегалія. Ступінь спленомегалії при портальних цирозах ши-
роко варіює. Лейкопенія має стійкий характер і не перевищує
2,0-109/л, проте прямої кореляції між значною лейкопенією та
спленомегалією не існує. У разі приєднання інфекційно-запальних
ускладнень кількість лейкоцитів збільшується, але меншою мірою,
ніж у здорової людини. Для хворих цієї групи не характерні зміни
197
Н.М. Третяк | Гематологія
в лейкоцитарній формулі. Іноді зменшується вміст сегментоядер-
них нейтрофілів та збільшується відсоток паличкоядерних форм.
В основному в усіх хворих кістковий мозок гіперцелюлярний. У мі-
єлограмі спостерігається зменшення відсотка сегментоядерних
нейтрофілів із деяким збільшенням відсотка молодших елементів
(промієлоцитів, мієлоцитів та метамієлоцитів) — так зване “галь-
мування дозрівання” гранулоцитів. Проте існує думка, шо лейко-
пенія у хворих на портальний цироз зумовлюється двома фактора-
ми — швидкістю видалення лейкоцитів із циркуляції та темпами
кровотворення. Більшість хворих на цироз печінки не потребують
корекції нейтропенії, оскільки вона ніяк не проявляється.
При виникненні інфекційно-запальних ускладнень застосову-
ється активна антибактеріальна терапія. Всі лікувальні заходи
спрямовуються на лікування основної патології, успіх якої призво-
дить до покращання показників периферичної крові.
Тромбофлебітична спленомегалія
Запальний процес у селезінковій вені, як правило, виникає
в осіб із частими гострими або ж хронічними запальними захворю-
ваннями. Флебіт найчастіше поєднується з патологічним процесом
портальної вени. Проте тромбофлебітична спленомегалія — доволі
рідкісний процес. Загострення захворювання проявляється дуже
сильними болями в черевній порожнині, лихоманкою, іноді озна-
ками динамічної непрохідності кишечника, збільшенням розмірів
селезінки та появою асциту. Незважаючи на лихоманку, характер-
на лейкопенія. У деяких хворих виникає геморагічний синдром,
пов’язаний із тромбоцитопенією. Прогресування тромбофлебіту
призводить до розвитку синдрому портальної гіпертензії з типови-
ми клінічними ознаками. Патогенез лейкопенії не відрізняється від
такого ж при цирозах печінки. Діагноз встановлюється на підставі
даних клінічного обстеження.
Терапія спрямована на корекцію тромбофлебіту. Виражена
спленомегалія в поєднанні з лейкопенією, а також тромбоцитопе-
нією може бути показанням до спленектомії.
Подібний патогенез нейтропенії спостерігається при хворобах
накопичення, ліпідозах, синдромі Фелті, хворобі Стилла—Шоффа-
ра, синдромі Лібмана—Сакса, червоному вовчаку, а також при ряді
інфекційних та паразитарних захворювань, що протікають зі спле-
номегалією (сифіліс, бруцельоз, малярія, лейшманіоз, гістоплазмоз).
Терапія нсйтропеній, зумовлених зменшенням циркуляції ней-
трофільних гранулоцитів у кровообігу, зводиться, передусім, до лі-
198
| Лейкопенії
кування основного захворювання. Позитивний результат лікування
основного захворювання покращує показники периферичної крові.
Глибока цитопенія може бути показанням до спленектомії.
Хронічна доброякісна лейкопенія
Найчастіше спостерігається у хворих із різними захворювання-
ми травного тракту, такими, наприклад, як хронічний гастрит із
зниженою секреторною активністю, хронічний холецистит, хро-
нічний коліт, хронічний панкреатит та ін. Перерозподіл нейтро-
фільних гранулоцитів у судинному руслі за цієї патології зумовле-
ний ваготонією. При парасимпатикотонїї більша за нормальну
кількість нейтрофільних гранулоцитів переходить із циркуляторно-
го до маргінального пула.
Клінічно хронічна доброякісна лейкопенія ніяк не проявляєть-
ся та може бути випадково виявлена при дослідженні периферич-
ної крові з іншої причини.
У крові визначається відносна та абсолютна- нейтропенія та
відносний лімфоцитоз. Функція нейтрофільних гранулоцитів не
змінена. Показники кістковомозкового кровотворення — в нормі.
Спеціальне лікування доброякісної лейкопенії не показане.
Необхідно активно лікувати основну патологію.
Прогноз сприятливий.
199
ЕРИТРОЦИТОЗИ
Еритроцитозом називається підвищення кількості еритроцитів
у поєднанні зі збільшенням концентрації гемоглобіну, показників
гематокриту та маси циркулюючих у периферичній крові еритро-
цитів, незалежно від фізіологічної норми.
Згідно із сучасними уявленнями еритроцитози можуть бути
класифіковані таким чином.
Класифікація еритроцитозів
1. Абсолютний еритроцитоз, котрий буває при таких захворю-
ваннях системи крові (первинний еритроцитоз), як полицитемія
справжня, еритроиитемічна фаза ідіопатичного мієлофіброзу, син-
дром Вєуоі.
2. Вторинний еритроцитоз (виникає внаслідок підвищеного ут-
ворення еритропоетинів):
— при генералізованій тканинній гіпоксії, зумовленій захворю-
ваннями легенів (хронічний обструктивний бронхіт, бронхіальна
астма, хронічні необструктивні захворювання легенів, облітерація
плевральних порожнин, ексудативний плеврит, пухлини легенів, ре-
зекція легенів, силікоз, антрактоз, азбестоз, склероз системи легене-
вої артерії (синдром Аехга-Аггі11а§), дифузний прогресуючий про-
міжний фіброз легенів (синдром Наттап-Кісй), значна деформація
грудної клітки (уроджені, набуті); патологією серцево-судинної сис-
теми — уроджені пороки “синього” типу (синдром Тоесиг—Бінга,
синдром Тоесиг (Таиззів), синдром Тоесиг—Стелла—Альберса (Таи$-
8І£-8пеІ1а—АІЬегз), синдром Ейзенменгера (Еікептсп^ег), тріада, тет-
рада, пентада Фалло, синдром Ебштейна (ЕЬяеіп), синдром Гайсбе-
ка (ОаікЬоск), дисциркуляторні еритроцитози при хронічній
серцево-судинній недостатності, серцево-легенева недостатність);
— при пухлинах надниркових залоз, нирок, гіпофізу, гонад
у чоловіків, яєчників, щитоподібної залози, гепатокарциномі, раку
підшлункової залози, пухлинах матки;
— при захворюваннях нирок — полікістоз, гідронефроз, урод-
жені та набуті аномалії ниркових артерій, стеноз, атеросклеротич-
ний стеноз, гіпоплазія, аневризма, паранефрит, стискання нирко-
вих артерій ззовні, посттрансплантаційний нирковий еритроцитоз;
200
| Еритроцитози
— при інших захворюваннях — дифузний токсичний зоб, змі-
шаний токсичний зоб, токсична аденома щитоподібної залози, кіс-
ти яєчників, туберкульоз яєчників, клімактеричний період у жінок,
виразкова хвороба пілоричного відділу шлунка та дванадцятипалої
кишки, туберкульоз селезінки, стеноз судин селезінки, неврологіч-
ні захворювання;
— “професійні” еритроцитози при обробці марганцевих руд,
вібраційній хворобі, у пілотів, робітників-кесонників.
3. Відносний “гемоконцентраційний ” еритроцитоз при:
— сильному потовиділенні;
— захворюваннях, що супроводжуються діареєю та блюван-
ням;
— діабетичному кетоацидозі та кетоацидотичній комі;
— захворюваннях, шо супроводжуються виходом рідкої части-
ни крові в тканини;
— опіковій хворобі;
— некротично-виразкових змінах шкіри та слизових оболонок;
— синдромі Лайела.
Із наведених даних ясно, що еритроцитоз може бути як гема-
тологічним проявом захворювання системи крові (поліцитемія
справжня або еритремічна стадія ідіопатичного мієлофіброзу), так
і, часто, вторинного походження, як наслідок негематологічного
захворювання, або мати відносний (при зменшенні об’єму плазми)
характер. Це означає, що еритроцитоз є як діагностичною, так і
диференційно-діагностичною проблемою.
При виявленні еритроцитозу в периферичній крові необхідно
простежити динаміку показників гемограми протягом тижня, приз-
начаючи три аналізи крові з інтервалом 2-3 дні. Це дозволить вик-
лючити або підтвердити наявність стрес-еритроцитозу. Якщо є впев-
неність у постійному характері еритроцитозу, слід провести ретельне
клініко-лабораторне обстеження з обов’язковим виміром об’єму
циркулюючої крові, маси циркулюючих еритроцитів та об’єму плаз-
ми. Цитологічне та гістологічне дослідження кісткового мозку та
визначення показника лужної фосфатази нейтрофільних гранулоци-
тів мають вирішальне значення для диференціювання еритроцитозу
та еритремії (поліцитемії справжньої та еритремічної стадії ідіопа-
тичного мієлофіброзу). В разі виявлення абсолютного еритроцитозу
необхідно встановити причину, що зумовила його розвиток.
Лікування спрямоване на усунення причини симптоматичного
еритроцитозу та основної патології.
201
ГЕМОРАГІЧНІ ДІАТЕЗИ
Гемостаз у судинах мікроциркуляції підтримується двома сис-
темами — прокоагулянтною (згортання) та антикоагулянтною
(протизгортання). Перебуваючи в динамічній рівновазі, ці системи
адекватно підтримують оптимальну реологію (текучість крові), що
запобігає тромбоутворенню, з одного боку, та кровотечам — з ін-
шого. Цей багатофакторний та складнорегульований процес забез-
печує, насамперед, локальний гемостаз, не спричиняючи його ге-
нералізацію по всьому судинному руслу.
Функціональна спроможність системи гемостазу забезпечуєть-
ся трьома основними компонентами: судинним, клітинним (пере-
важно тромбоцитарним) та плазмовим (коагуляційним). Усі скла-
дові мають одночасно, більшою чи меншою мірою, як про-, так і
антикоагулянтний потенціал. Патологічні стани, спричинені кож-
ною з цих складових, можна віднести відповідно до вазопатій (мік-
ротромбоваскуліти), тромбоцитопеній/патій, коагулопатій.
Процес прокоатуляції реалізується двома шляхами — зовнішнім та
внутрішнім. їх взаємодія забезпечує запуск каскадних, взаємозалежних
реакцій, кожний із компонентів яких є активатором для іншого.
Зовнішній механізм запускається при травмі судини потрап-
лянням у кров тканинного тромбопластину. Внутрішній реалізу-
ється через активацію фактора Хагемана, що є результатом контак-
ту крові зі сполучною тканиною судини внаслідок порушення їі
цілісності.
У процесах згортання крові судинний, клітинний та плазмовий
компоненти беруть участь взаємозалежно.
Судинний компонент забезпечує:
1) збереження крові в судинному руслі,
2) зменшення площі кровотечі через вазоспазм,
3) активацію тромбоцитів.
Тромбоцитарний компонент забезпечує:
1) виділення в кровоток вазоконстрикторів (тромбоксану,
простагландинів пресорної дії, катехоламінів, серотоніну);
2) утворення первинного тромбоцитарного тромбу через адге-
зію та агрегацію;
202
| Геморагічні діатези
3) активацію плазмової ланки гемостазу через виділення в кро-
воток специфічних тромбоцитарних факторів;
4) живлення судинної стінки.
Плазмовий гемостаз забезпечує дві основні фази багатоетапно-
го ферментативного процесу згортання крові:
1) активацію протромбіну з подальшим утворенням тромбіну,
2) перетворення фібриногену на фібрин.
У цій каскадно-комплексній системі коагуляційної ланки згор-
тання крові беруть участь наведені нижче фактори.
Таблиця 16. Плазмові фактори згортання крові
Позначка фактора Найменування фактора
1 Фібриноген
II Протромбін
III Тканинний тромбопластин
IV Іони кальцію
V Ас-глобулін, проакцелерин
VII Проконвертин
VIII Анти гем офільний глобулін
1 ІХ РТС-фактор, фактор Кристмаса
X Фактор Стюарта-Пауера
XI РТА-фактор
XII Фактор Хагемана, контактний фактор
XIII Фібрин-стабілізуючий фактор
- Плазмовий прекалікреїн, фактор Флетчера
- Високомолекулярний кініноген, фактор Фітцжеральда, фактор Фложак, фактор Вільсона
Унаслідок взаємодії судинного, тромбоцитарного та плазмово-
го компонентів гемостазу в місці ушкодження судинної стінки ут-
ворюється тромбін, котрий завершує процеси агрегації та адгезії
тромбоцитів. Надалі він активує плазмову ланку згортання, резуль-
татом чого є утворення фібрину (фібринмономерного комплексу),
шо є кінцевою фазою згортання крові.
Друга складова гемостазу — це система протизгортання. Вона
включає могутню систему фібринолізу та протизгортальні власти-
вості судин.
Фібриноліз — ферментна система, яка зумовлює поступове
руйнування (деградацію) фібриногену/фибрину. Головним компо-
нентом цієї системи є фермент плазмін (фібринолізин), котрий
циркулює в плазмі у вигляді плазміногену. Так само, як і в проко-
203
Н.М. Третяк | Гематологія
аіулянтній системі реалізація фібринолізу відбувається незалежни-
ми один від одного шляхами — внутрішнім та зовнішнім. Перший
реалізується також через активацію фактора Хагемана, а другий —
через специфічний активатор тканинного типу. Цей активатор ви-
вільняється з ендотелію судин при всіх видах тромбозів, та навіть
при стисканні судини. Крім того, він міститься в багатьох типах
тканин організму, а також у клітинах крові — еритроцитах, тром-
боцитах, лейкоцитах. У цілому фібриноліз адаптований для руйну-
вання фібрину як у сформованих тромбах, так і в розчинних ком-
плексах.
Антикоагулянтна функція судинної стінки реалізується через
синтез інгібітора агрегації тромбоцитів — простациклін, синтез
тканинного активатора фібринолізу, а також утворення та підтри-
мання антикоагулянтного потенціалу на межі кров—тканина.
Для підтримки рівноваги та запобігання надмірній активності
антикоагулянтної системи в плазмі крові циркулює найактивніший
інгібітор фібринолізу — антиплазмін, котрий належить до о^-гло-
булінів, а також інші, менш активні білкові сполуки.
Виходячи з цього, зрозуміло, що порушення в системі гемос-
тазу на будь-якому етапі може стати серйозним ускладненням, заг-
розою кровотечі чи тромбозу.
Не зупиняючись на рідкісних варіантах захворювань, які май-
же не зустрічаються в клінічній практиці, особливо неспеціалізова-
них відділень, головну увагу буде приділено основним нозологіч-
ним формам.
ВАЗОПАТІЇ
Вазопатія (мікротромбоваскуліт) — структурна патологія стін-
ки судини, яка врешті зумовлює порушення її функції, наслідком
чого є розвиток дисемінованого мікротромбоутворення (ДВЗ-син-
дром) та погіршення реологічних властивостей крові.
До захворювань, котрі можуть спричинити такі розлади, нале-
жать вроджені ангіопатії, а також інфекційні хвороби (геморагічні
лихоманки, тиф), бактеріальний ендокардит, васкуліти при сис-
темних захворюваннях сполучної тканини (вузликовий артерііт,
системний червоний вовчак, дерматоміозит), медикаментозні вас-
куліти, гемолітико-уремічний синдром та ін. Типовим представни-
ком вазопатії є васкуліт Шенлейн-Геноха.
204
| Геморагічні діатези
Васкуліт Шенлейн-Геноха (ВШГ)
вшг належить до групи мікротромбоваскулітів і виникає на
основі імунно-комплексного порушення структури стінок мікросу-
дин та асептичного запалення на фоні ДВЗ-синдрому.
Епідеміологія. Найчастіше захворювання зустрічається у дітей
віком до 14 років. Серед дорослого населення — переважно в мо-
лодому віці з поступовим зменшенням захворюваності. Жінки та
чоловіки хворіють приблизно однаково.
Етіологія. Етіологія захворювання до сьогодні остаточно не
з’ясована. Простежується зв’язок зі стрептококовою та вірусною
інфекціями. Провокуючими факторами можуть бути щеплення,
харчова або медикаментозна алергія, переохолодження, сенсибілі-
зація ендогенними білками тощо. У багатьох випадках визначити
провокуючий фактор не вдається.
Патогенез. Нині доведено належність ВШГ до імунокомплек-
сних захворювань із розвитком гіперсенситивних васкулітів.
1. Під впливом певного чинника, внаслідок реакції сенсибілі-
зації, в організмі починають продукуватися автоантитіла до анти-
гена епітеліоцитів судинної стінки. Утворення низькомолекуляр-
них фіксованих имунних комплексів спричиняє порушення її
структури та функції.
2. Порушення структурної цілісності та антикоагуляційного
потенціалу судинної стінки — основний чинник розвитку тромбо-
утворення і, як ускладнення, десимінованого внутрішньосудинно-
го згортання крові, основним проявом якого є геморагічний син-
дром.
3. Виснаження антикоагуляційних (через ДВЗ-синдром), а зго-
дом і прокоагулянтних (через надмірне споживання) механізмів ге-
мостазу підтримує й посилює геморагічний синдром.
4. Реологічні порушення: мікротромбози, ДВЗ-синдром.
Класифікація (3. С. Баркаган, 1988)
Клінічні форми:
1) шкірна,
2) суглобова,
3) абдомінальна,
4) ниркова,
5) змішана.
Варіанти перебігу:
1) блискавичний;
2) гострий;
205
Н.М. Третяк | Гематологія
3) хронічний:
— переметуючий,
— рецидивуючий (із частими або рідкими загостреннями).
Ступінь активності:
1) мінімальна,
2) помірна,
3) висока,
4) дуже висока.
Ускладнення:
1) кишкова непрохідність, перфорації, перитоніт;
2) ДВЗ-синдром з тромбоцитопенією потреби, кровотечі;
3) постгеморагічна анемія;
4) тромбози та інфаркти.
Клінічна картина. Оскільки при ВШГ до патологічного проце-
су можуть залучатися мікросудини будь-якої ділянки шкіри, сугло-
бових поверхонь або внутрішніх органів, у тому числі нирок,
шлунково-кишкового тракту, легенів, мозку чи його оболонок та
ін., клінічна картина буде зумовлена, передусім, анатомічною ло-
калізацією патологічного процесу.
У клінічному перебігу ВШГ залежно від форми виділяють ос-
новні клінічні синдроми: шкірний, суглобовий, абдомінальний,
нирковий, які за різного ступеня вираженості можуть комбінувати-
ся один з одним, а при рецидивуючому перебігу можливе перева-
жання одного з них.
Найчастіше в клініці зустрічається шкірна форма ВШГ, котра
характеризується появою симетричної папульозно-плямистої (вас-
кулітно-пурпурної) висипки, в першу чергу на кінцівках — через
високий периферичний опір у судинах, а потім на тулубі, сідницях,
украй рідко — на обличчі. Геморагії можуть бути поодинокими або
зливними, як правило вони мономорфні, інколи із запальною ос-
новою, та в багатьох випадках при пальпації визначаються як
ущільнення. При натисканні елементи висипки не зникають.
У важких випадках папули можуть некротизуватись, а також зали-
шати після себе тривалу пігментацію.
Суглобовий синдром виникає або окремо, або разом зі
шкірним. У великих суглобах відмічається летючий біль різної
інтенсивності, який через кілька діб зникає. Але при появі но-
вої висипки може виникати знову. В деяких випадках суглобове
ураження характеризується стійкістю та клінічно нагадує ревма-
тоїдний поліартрит.
206
| Геморагічні діатези
Абдомінальний синдром часто передує шкірним проявам, що
особливо ускладнює правильну діагностику. Його основною ознакою
є сильний біль у животі, постійний або нападоподібний, інколи над-
звичайно інтенсивний. Больовий синдром обумовлений крововили-
вами у стінку кишки або в брижу. У клініці можуть мати місце кри-
вава блювота, мелена або наявність свіжої крові в калі, імовірні як
проноси, так і закрепи. Із самого початку захворювання можлива ли-
хоманка, при значних кровотечах — розвиток колапсу та гострої пос-
тгеморагічної анемії. Діагностика абдомінального синдрому при
ВШГ надзвичайно складна, оскільки потребує верифікації з гострою
хірургічною патологією, такою як апендицит, панкреатит, кишкова
непрохідність та ін., або ургентними патологічними станами, за яких
васкуліт є основною причиною розвитку ускладнення.
Приблизно у половини хворих на ВШГ має місце нирковий
синдром, перебіг якого найчастіше відбувається за типом гострого
чи хронічного гломерулонефриту. Така форма супроводжується
мікро- або макрогематурією, протеїнурією та циліндрурією. Арте-
ріальна гіпертензія при цій формі нефриту визначається зрідка, але
можливий розвиток нефротичного синдрому. Ураження нирок
часто виникає через один-чотири тижні після початку захворюван-
ня, а перебіг може бути тривалим та хронічним, шо значно погір-
шує прогноз. У частини хворих відмічається швидкопрогресуюче
ураження нирок із переходом в уремію в перші два роки захворю-
вання. В цілому ураження нирок є потенціально загрозливим та
найнесприятливішим проявом ВШГ.
Діагностика. Важливе значення для оцінки ступеня тяжкості
перебігу процесу мають наступні дослідження.
1. Кількісне визначення вмісту фактора Віллєбранда у плазмі.
Його рівень при ГВ збільшується в 1,5-3 рази й більше, до того
ж ступінь збільшення відповідає тяжкості та поширеності уражен-
ня мікросудин.
2. Визначення вмісту в плазмі циркулюючих імуноглобулінів
6. При ВШГ їх рівень, як правило, підвищений, хоча не виявлено
відповідності між ступенем цього підвищення і тяжкістю хвороби.
3. Гіперфібриногенемія, підвищений вміст у плазмі альфа-2- та
гамма-глобулінів. Визначення цих показників важливе для оцінки
ефективності гепаринотерапії.
4. Визначення у сироватці крові кріоглобулінів.
5. Визначення антитромбіну-3 та ступеня гепаринорезистен-
тності плазми.
207
Н.М. Третяк | Гематологія
Активність запального процесу оцінюється за загальноприйня-
тими гострофазовими показниками: С-реактивний білок, фібрино-
ген, ступінь активації моноцитів, а також ШОЕ.
Лікування. В основу лікувальних програм ВШГ покладені па-
тогенетичні механізми, а їхня головна мета — розірвати ланцюго-
ву реакцію тромбоутворення.
Легкі форми ВШГ лікуються призначенням терапевтичних доз
антигістамінних препаратів (фенкарол, кетоф, кларітин та ін.). Хво-
рим не показані антибіотики, сульфаніламіди, акскорбінова кислота
та рутин. Доведено, шо призначення преднізолону виправдане тіль-
ки при значній активності процесу. Препарат не скорочує тривалість
ВШГ, не зменшує частоту рецидивів та тривалість ремісій, не запо-
бігає розвиткові нефриту й не полегшує його перебігу. Крім, того,
кортикостероїди можуть підвищувати згортання крові, інгібувати
фібриноліз, поглиблювати ДВЗ-синдром. Тому призначення препа-
ратів цієї групи виправдане тільки на короткий термін (3-5 діб), у не-
великих дозах (0,3-0,5 мг/кг на добу), під прикриттям достатніми до-
зами антикоагулянтів. Основними показаннями для призначення
преднізолону є сильно виражена запальна реакція з інфільтративни-
ми елементами висипки з некрозами, значні гіперфібриногенемії, на-
явність нефротичного синдрому при нирковій формі, високий вміст
альфа2-глобулінів у сироватці крові та ШОЕ, вища за 45 мм/год.
До загальноприйнятого стандарту лікування ВШГ входять:
— гепарин — 300-400 ОД/кг 4 рази на добу, під шкіру навко-
лопупкової ділянки. У важких випадках або для запобігання уск-
ладненням — по 10 тис. ОД/кг двічі на добу внутрішньовенно
(препарат вводиться під контролем аутокоагуляційного тесту);
— свіжозаморожена плазма — 300-400 мл/добу (у важких ви-
падках — до 800 мл/добу) протягом 3-4 діб;
— плазмаферез;
— дезагреганти: курантил, аспірин, трентал, тіклід, плавекс та ін.;
— нестеро’їдні протизапальні засоби: індометацин, вольтарен,
моваліс та ін.
Симптоматична терапія', знеболюючі засоби, за потреби —
місцева зупинка зовнішніх кровотеч, жарознижуючі засоби тощо.
При суглобовій формі використовують компреси (димексид,
гепарин, вольтарен, гідрокортизон), при нирковій — у схемі ліку-
вання враховуються особливості перебігу нефриту.
Крім ВШГ до групи захворювань із порушенням судинного ге-
мостазу належать спадкова геморагічна телеангіектазія (хвороба
208
| Геморагічні діатези
Рандю—Ослера), синдром Луї—Бар, хвороба Фабрі. Серед них час-
тіше зустрічається хвороба Рандю—Ослера, котра характеризується
вогнищевим витонченням стінок і розширенням судини, а також
неповноцінним локальним гемостазом через недостатній розвиток
субендотелію. Клінічно характеризується наявністю телеангіектазів
ще в дитячому віці та проявляється кровоточивістю з них.
Лікування передбачає зрошування слизової оболонки, що кро-
воточить, тромбопластином, тромбіном, перекисом водню та Е-
АКК. Останнім часом використовують локальне лікування лазером.
КОАГУЛОПАТІЇ. ГЕМОФІЛІЇ
До групи коагулопатій належать як вроджені, так і набуті пору-
шення в плазмовій ланці гемостазу, котрі проявляються кількісною
або якісною недостатністю. Набуті форми найчастіше пов’язані
з окремими патологічними станами, перебіг яких супроводжується
сорбцією факторів (наприклад амілоїдом), підвищеною потребою
(ДВЗ-синдром) чи утворенням автоантитіл до них.
До вроджених геморагічних коагулопатій належать захворю-
вання, які зчеплені зі статтю та передаються через покоління. Най-
частіше серед них зустрічаються дефіцитні стани за фактором VIII
— гемофілія А та хвороба Віллєбранда, фактором IX — гемофілія
В (хвороба Кристмаса) та фактором XI — гемофілія С (хвороба Ро-
зенталя).
Епідеміологія. Частота гемофілій серед коагулопатій коливаєть-
ся, в середньому, від 6 до 18 на 100 тис. мешканців чоловічої ста-
ті. Із цієї кількості до 75% припадає на гемофілію А, 10-15% — на
хворобу Віллєбранда, до 10% — на гемофілію В, і дуже рідко, в по-
одиноких випадках, зустрічається гемофілія С — 1-2%.
Захворюваність на набуті форми коагулопатій залежить від по-
ширеності соматичних захворювань, перебіг яких вони можуть
ускладнювати.
Етіологія. Гемофілії передаються по материнській лінії. Хворі-
ють переважно хлопчики. Ген гемофілії, локалізований у Х-хромо-
сомі, передається як рецесивна ознака, крім гемофілії С, котра
спадкується аутосомно. У зв’язку з цим жінки не страждають на
кровоточивість. І хоча активність одного з факторів у них може бу-
ти зниженою майже вдвічі, вони залишаються лише кондукторами
цих захворювань. Прояви коагулопатії у жінок мають місце лише
при значних травмах чи оперативних втручаннях.
209
Н.М. Третяк | Гематологія
Патогенез. Зменшення в периферичній крові рівня будь-якого
з факторів плазмової коагуляції, незалежно від причини, порушує
каскадний, взаємозалежний механізм згортання крові, де один
фактор є активатором для іншого. Так, фактор VIII разом з ЇХ
у присутності іонів кальцію активує фактор X, а фактор XI вклю-
чає до прокоагуляції IX.
Унаслідок генетичного дефекту порушуються опосередковані
прокоагулянтними факторами VIII, IX чи XI послідовні реакції ко-
агуляції, результатом чого є порушення утворення фибринмоно-
мерного комплексу, а через не — формування кров’яного згустку
(тромбу) та зупинка кровотечі.
Класифікація. Гемофілії можна класифікувати за формою дефі-
цитного фактора — А, В та С, про шо ішлося вище, та за клініч-
ними формами.
Ступінь тяжкості клінічного перебігу % активності фактора
в плазмі крові)
1. Латентна форма > 25%
2. Легка форма 5—15%
3. Середньої тяжкості 2—5 %
4. Тяжка 1-2%
5. Украй тяжка 0-1 %
Клініка. Клінічні синдроми та тяжкість їх проявів при всіх фор-
мах гемофілії практично однакові. На виникнення кровотеч та їх
значущість впливає не сам факт дефіциту активності фактора,
а виключно ступінь цього дефіциту. За активності фактора понад
25% клінічні прояви гемофілії не відзначаються взагалі або мають
місце при значних травматичних ушкодженнях. При 10-15% актив-
ності геморагічний синдром більш значущий, але, як правило, ви-
никає при зовнішньому впливі. Наявність у сироватці крові менш
ніж 5% активності фактора супроводжується спонтанними крово-
течами, гематомами, гемартрозами.
Основні клінічні синдроми:
1) спонтанні та післятравматичні гемартрози (крововиливи
в суглоб) та гемофільні артропатії;
2) внутрішньо- та міжм’язеві гематоми;
3) профузні кровотечі, гостра та хронічна постгеморагічна анемія;
4) ниркова патологія аж до ниркової недостатності.
Кровотечі у хворого на гемофілію виникають не одразу після
травми, а через І—5 годин після неї. Швидкість виникнення кро-
вотечі залежить від тяжкості захворювання.
210
| Геморагічні діатези
Гемартрози — одна з найхарактерніших ознак гемофілії. Кро-
вотечі походять із синовіальної оболонки суглоба й супроводжу-
ються вираженим больовим синдромом, порушенням активних ру-
хів у суглобі і, нарешті, його анкілозуванням. Уражаються,
переважно, великі суглоби (колінний, ліктьовий, кульшо-стегно-
вий). Крім того, гемартроз певної локалізації, який мав місце впер-
ше, надалі буде рецидивувати. Зазвичай у гемофіліка уражається
від 1 до 5-8, й навіть більше, суглобів. При внутрішньосуглобово-
му крововиливі суглоб збільшений у розмірах, шкіра над ним гаря-
ча на дотик, рухи значно обмежені й супроводжуються різким бо-
лем. Наслідком рецидивуючого гемартрозу є поступові
деструктивні зміни в синовіальній оболонці та хрящі, узурація кіс-
ткових поверхонь, утворення епіфізарних кист, можливий розви-
ток внутрішньосуглобових переломів. З часом, унаслідок тривалої
сенсибілізації, у 10-30% хворих можливе приєднання автоімунного
запалення навіть у неушкоджених раніше суглобах із розвитком
вторинного ревматоїдного синдрому — гемофільної артропатїї
(синдром Баркагана—Єгорової).
Внутрішні та міжм’язеві гематоми будь-якої локалізації (під-
шкірні, міжфасціальні, зачеревні, в попереково-здухвинний м’яз та
ін.) — друге часте ускладнення перебігу гемофілії. Об’єм гематоми
може сягати 300-1000 мл, і навіть більше, й через стискання нер-
вових закінчень, як правило, супроводжується сильним болем. Ін-
коли, в залежності від локалізації гематоми (шлунок, кишковик,
брижа, сальник), больовий синдром може імітувати гостру хірур-
гічну патологію.
Тиск, котрий спричиняє гематома на оточуючі тканини, приз-
водить до порушення нервової та судинної трофіки з розвитком ат-
рофії, й навіть некрозів. Серйозного негативного впливу зазнає
й кісткова тканина, найчастіше верхніх та нижніх кінцівок і мис-
кової кістки, в яких розвиваються виражені деструктивни зміни.
У деяких випадках гематома може нагноюватися, кальцифікува-
тись або осифікуватися з утворенням нової кістки, що також є
причиною глибокого порушення рухової активності суглоба.
Загрозою для життя хворого можуть стати геморагічні інсуль-
ти, шлунково-кишкові кровотечі, особливо при анамнестичних ви-
разкових чи ерозивних процесах, а також ниркові на фоні прийо-
му медикаментів (анальгетиків), супутнього пієлонефриту чи
травми. Ці стани можуть спричинити постгеморагічну анемію, кот-
ра значною мірою погіршує стан пацієнта.
211
Н.М. Третяк | Гематологія
До сьогодні серйозною проблемою в перебігу захворювання є
розвиток інгібіторної форми гемофілії, яка є наслідком ало- та ав-
тоімунних зрушень і супроводжується появою в крові хворого у ви-
соких титрах специфічного інгібітора фактора згортання. Інгібітор-
ні форми мають украй важкий перебіг із сильними тривалими
кровотечами, оскільки традиційна зупинка геморагічного синдро-
му в таких випадках не можлива.
Діагностика. Діагностика гемофілії, як правило, проводиться
в період після новонародженості або в ранньому дитинстві на під-
ставі підвищеної спонтанної та посттравматичної кровоточивості.
У дорослому віці вперше діагноз гемофілії ставиться вкрай рідко.
У діагностичному пошуку використовується кількісне визна-
чення антигемофільних факторів VIII, IX та XI або метод із вико-
ристанням детромбінізованої плазми, котра містить один із факто-
рів. Крім того, досліджуються можливі ускладнення в перебігу
гемофілії: рентгенологічне дослідження суглобів, пункція суглобів,
оцінка функції нирок та мозкового кровотоку.
Лікування. До сьогодні найкращим методом лікування при ге-
мофіліях є введення хворому недостатнього антигемофільного
фактора VIII, IX чи XI. Для припинення незначних кровотеч або
проведення минімальних втручань (екстракція зуба) достатньою є
концентрація VIII фактора в плазмі крові на рівні 15-20%. Цього
можна досягти введенням 15 мл/кг на добу антигемофільної плаз-
ми або кріоприципітату. Дещо меншою ця доза може бути при ге-
мофілії С (приблизно 9-10 мл/кг) При більш серйозних кровови-
ливах присутність фактора має бути збільшена до 30-40%, а при
великих оперативних втручаннях та тяжких кровотечах — до 40-
60% і більше.
Розрахунок дози антигемофільного препарату проводять за
формулою:
де Д — доза кріопреципітату в одиницях активності,
А — маса тіла хворого в кілограмах,
В — необхідний рівень фактора у відсотках.
Кріопреципітат — універсальний препарат, який являє собою
концентрат фактора VIII, фібриногену, а також факторів XIII, IX,
XI й не містить альбуміну; Ново-Севен — рекомбінантний VПа-
фактор. При гемофілії В ефективний препарат РР8В, ЕЕ1ВА (кон-
212
| Геморагічні діатези
центрат тромбіну, проконвертину, фактора Стюарта та антигемо-
фільного фактора В). У якості симптоматичної терапії вводяться
опосередковані гемостатики: діцинон, Е-АКК, контрикал, гордокс,
трасилол. Особливе значення в тактиці ведення хворого на гемофі-
лію має лікувальна пункція ураженого суглоба, евакуація крові
з його порожнини та подальше введення кортикостероїдних гор-
монів (діпроспан, кеналог). Використовується також нетривала
імобілізація кінцівки (до 3-5 діб) із наступним поступовим збіль-
шенням об’єму рухів у суглобі. Можливе застосування фізіотера-
певтичних методів. Охолодження суглоба не використовують.
При лікуванні крововиливів у суглоб використовують також
синовектомію, котра запобігає подальшому розвитку гемартрозів та
важких остеоартрозів. Найефективніший цей метод на І-ІІ стадіях
розвитку артрозів.
Лікування інгібіторних форм гемофілії вимагає введення дуже
великих доз необхідного фактора та використання плазмаферезу,
а також РР8В, ЕЕІВА. Введення кортикостероїдих препаратів, за
даними більшості клініцистів, позитивного ефекту майже не дає.
У випадках розвитку постгеморагічної анемії показані препара-
ти заліза, а при масивних кровотечах — замісна терапія еритроци-
тарною масою. За умов розвитку значного больового синдрому
можливе призначення знеболювальних (анальгін, кетанов, барал-
гін) аж до наркотиків та транквілізуючих засобів (седуксен, імован,
радедорм).
Велике значення для хворих на гемофілію має режим зі зведен-
ням до мінімуму можливості порізів, забоїв, травматичного ушкод-
ження суглоба та якнайшвидше введення при цьому антигемофіль-
ного препарату.
Вивчаються можливості генної інженерії з використанням під-
садки специфічних генів.
Хвороба Віллебранда
Хвороба Вімебранда — форма спадкових коагулопатій, в осно-
ві якої лежить порушення синтезу одного з компонентів фактора
VIII — крупномолекулярного білка (УІП:ФВ), або так званого
фактора Віллебранда, шо є головною причиною кровотеч. Розріз-
няють два типи захворювання: І тип зі зниженням кількісного
синтезу компоненту фактора VIII, але без порушення мультимер-
ної структури та її активності, і кілька варіантів типу II (ПА, ПВ),
213
Н.М. Третяк | Гематологія
при яких мають місце якісні порушення в стуктурі фактора без
кількісних зсувів.
Епідеміологія. Серед спадкових геморагічних діатезів хвороба
Віллебранда за частотою посідає третє місце після тромбоцитопа-
тій та гемофілії А (близько 3 хворих на 100 тис. населення).
Етіологія. Захворювання належить до коагулопатій, котрі ус-
падковуються за аутосомно-домінантним типом. Надзвичайно рід-
ко в літературі згадується аутосомно-рецесивна форма, яка потре-
бує подальшого вивчення.
Патогенез. Притаманне хворобі Віллебранда порушення син-
тезу фрагмента УІП:АГ спричиняє порушення активного включен-
ня прокоагулянтного фактора VIII у каскадну реакцію коагуляції,
внаслідок чого і розвивається геморагічний синдром. Захворюван-
ня належить до патології коагуляційного гемостазу, але, крім того,
має риси ангіопатії, оскільки саме ендотелій судини синтезує фак-
торний фрагмент, а також риси тромбоцитопатії у зв’язку з тим,
що фактор Віллебранда забезпечує адгезивну та агрегаційну здат-
ність тромбоцитів. Таким чином, при хворобі Віллебранда можли-
ві порушення в плазмовій, судинній та клітинній ланках гемоста-
зу.
Клініка. В залежності від ступеня дефіциту фактора клінічний
перебіг може бути як легким, так і надзвичайно тяжким, із загроз-
ливими для життя пацієнта кровотечами. Можливі петехіально-
плямисті геморагії в шкіру, носові кровотечі, шлунково-кишкові,
ниркові, маткові. Не завжди прослідковується кореляція між сту-
пенем порушення синтезу УІІІ:АГ та ступенем кровотечі.
Діагностика. При верифікації діагнозу необхідно:
1) дослідити спадковий анамнез пацієнта;
2) визначити характер кровоточивості;
3) оцінити час кровоточивості, функцію тромбоцитів (адге-
зію й агрегацію) та можливість її відновлення під впливом фак-
тора VIII;
4) визначити вміст фактора Віллебранда в сироватці крові.
Лікування. Допомога хворому забезпечується введенням препа-
ратів, котрі містять фактор VIII, а разом із ним і фактор Віллебран-
да — кріоприпипітат, антигемофільну плазму, фактор VIII як та-
кий. Слід зазначити, що введення прокоагулянтного препарату при
хворобі Віллебранда дає набагато більший і триваліший ефект, ніж
при гемофілії А. Крім того, можна використовувати Е-АКК. Ліку-
вання з використанням тромбоконцентрату ефекту не дає.
214
| Геморагічні діатези
ТРОМБОЦИТОПЕНІЇ
Тромбоцитопенії — це патологічні стани, які супроводжуються
зменшенням у периферичній крові тромбоцитарних пластинок або
тромбоцитів, наслідком чого є розвиток геморагічного синдрому.
Класифікація тромбоцитопеній
1. Спадкові.
2. Набуті:
а) первинні:
— імунні (алоімунні, гетероімунні, трансімунні, автоімунні),
— неімунні;
б) вторинні (симптоматичні).
До того ж, тромбоцитопенія, як один із синдромів, може суп-
роводжувати перебіг деяких вроджених захворювань: анемії Фан-
коні, тромботичної тромбоцитопенія ної пурпури, хвороби Алпор-
та, синдрому Віскотта-Олдрича.
Спадкові ізольовані тромбоцитопенії — надзвичайно рідкісна
патологія, яка є наслідком генетичного дефекту в утворенні тром-
бопоетину або внутрішньотромбоцитарної ензимопатії.
До набутих імунних тромбоцитопеній у дорослих належать
стани, за яких надмірне руйнування тромбоцитів пов’язане із не-
сумісністю з донорськими тромбоцитами у випадках, коли тром-
боцити набувають антигенних властивостей або, навіть незміне-
ні, являють собою об’єкт агресії з боку власної імунної системи,
та при імунній реакції на чужорідний антиген, фіксований на
тромбоциті.
Ідіопатична тромбоцитопенічна пурпура (ІТП)
Найчастішою та найбільш значущою серед усіх тромбоцитопе-
ній є ідіопатична тромбоцитопенічна пурпура. Це захворювання є
результатом імунного конфлікту, внаслідок якого в периферичній
крові зменшується кількість тромбоцитів.
Епідеміологія. Зазвичай захворювання зустрічається в юному,
молодому та зрілому віці. У людей старечого й похилого віку воно
має місце надзвичайно рідко й потребує ретельного підтверджен-
ня. Переважна кількість випадків припадає на 15—30-річних осіб і
меншою мірою — на осіб після 45 років. Жінки білої раси хворі-
ють удвічі частіше за чоловіків.
Етіологія. Етіологічні фактори на сьогоднішній день не з’ясо-
вані, тому термін “ідіопатична пурпура” не втратив актуальності.
215
Н.М. Третяк | Гематологія
Патогенез. В основу патогенезу ІТП покладена спотворена ре-
акція на власні тромбоцити внаслідок порушення імунної толеран-
тності, тобто співвідношення між В-лімфоцитами та Т-супресора-
ми. Імунний конфлікт відбувається переважно в периферичній
крові й не. стосується кісткового мозку. Тільки в невеликому від-
сотку випадків, як правило при тривалому або агресивному пере-
бігу, автоантитіла можуть проникати в кістковий мозок і мати
афінність (тропність) до мегакаріоцитів.
Основні ланки патогенезу
1. Ідіопатична агресивна реакція імунної системи на власні
тромбоцити.
2. Утворення антитромбоцитарних антитіл (І§ С, субклас 3).
3. Фіксація автоантитіл на мембрані тромбоцитів.
4. Руйнування тромбоцитів.
5. Геморагічний синдром.
Клініка. Клінічна картина при ІТП може мати як гострий, рап-
товий початок, так і повільно-прогресуючий. Симптоматика зале-
жить виключно від рівня тромбоцитів у периферичній крові. Зва-
жаючи на це, геморагічний синдром, як основна ознака
захворювання, може мати факультативний (вміст тромбоцитів по-
над 30,0 • 109/л) та облігатний (вміст тромбоцитів менш ніж
30,0 • 109/л) перебіг.
Характер кровоточивості петехіально-плямистий, при значній
тромбоцитопенії можливі носові, маткові, шлунково-кишкові, рідко
— ренальні кровотечі. Крововиливи можуть локалізовуватися на будь-
якій ділянці тіла чи кінцівок хворого (передня поверхня тулуба, спи-
на, кінцівки), але частіше в місцях більшого механічного ушкоджен-
ня. Розташовані вони, як правило, асиметрично, можуть бути
поодинокими у вигляді екхимозів або множинними зливними синця-
ми. Несприятливою в прогностичному плані є наявність геморагій на
обличчі, твердому чи м’якому піднебінні, оскільки захворювання в та-
ких хворих частіше ускладнюється розвитком геморагічного інсульту.
При значних кровотечах у пацієнтів можливий розвиток хро-
нічної чи гострої постгеморагічної анемії з відповідною, притаман-
ною їй, клінічною картиною та змінами в периферічній крові.
Крім проявів геморагічного синдрому при об’єктивному обсте-
женні хворого можливе підвищення температури тіла, з боку внут-
рішніх органів змін, як правило, немає. Зрідка можлива незначна
лімфоаденопатія і в поодиноких випадках — мінімальна спленоме-
галія, як супровід імунної реакції.
216
| Геморагічні діатези
Діагностика. Зменшення вмісту тромбоцитів у периферичній
крові нижче за норму дає підстави запідозрити 1ТП. Діагностика
цієї патології передбачає встановлення факту тромбоцитопенії,
відсутності депресії кістково-мозкового кровотворення внаслідок
пухлинної, жирової чи склеротичної проліферації та виключення
симптоматичного характеру тромбонитарного дефіциту.
Об’єктивний огляд пацієнта:
— оцінка загального стану за органами та системами, наявність
ознак геморагічного синдрому (оцінка його характеру та пошире-
ності), лімфоаденопатії (локалізація вузлів, іхні розміри, консис-
тенція, болючість, чи зв’язані вони з навколишніми тканинами),
спленомегалії.
Лабораторно-інструментальна діагностика:
1) загальноклінічні методи обстеження: загальні аналізи крові,
сечі, калу (в тому числі на приховану кров чи тест Грегерсена), б/х
крові, ЕКГ, рентгенологічне дослідження органів грудної порож-
нини;
2) спеціальні:
а) дослідження кістково-мозкового пунктату з оцінкою мегака-
ріоцитарної ланки кровотворення,
в) імунологічне дослідження з визначенням титру специфічних
антитромбоцитарних антитіл,
с) УЗД органів черевної порожнини;
3) додаткові:
— методи, котрі дозволять виявити та оцініти значущість су-
путньої патології, консультації окремих спеціалістів (гастроентеро-
лога, гінеколога та інших).
Лікування. Сучасні погляди на лікування ІТП спираються на
патогенез захворювання й передбачають, у першу чергу, вплив на
агресивну імунну відповідь, а також активну симптоматичну те-
рапію.
Лікування хворих на ІТП включає дві основні групи заходів:
1. Медикаментозні:
— глюкокортикостероідні гормони;
— імуносупресивні засоби (сандимун, антитимоцитарний іму-
ноглобулін);
— цитостатики;
— вазапротектори;
— гемостатики.
217
Н.М. Третяк | Гематологія
2. Немедикамснтозні:
— спленектомія;
— плазмаферез.
Лікування хворого починається при встановленні прогресуючої
тромбоцитопенії. Зазвичай, у таких випадках кількість тромбоцитар-
них пластинок у периферичній крові становить менш ніж
100,0 • 109/л. В залежності від віку пацієнта, агресивності імунної від-
повіді та чутливості організму до лікування, воно може мати як абсо-
лютну ефективність, до повного одужання хворого, так і мінімальну.
Препаратами першої лінії є кортикостероїди (преднізолон,
медрол) у дозі 1 мг/кг ваги, до отримання стійкого позитивного ре-
зультату. Надалі доза препарату поступово зменшується, бажано до
повної відміни. При сприятливих формах вміст тромбоцитів почи-
нає збільшуватися вже через кілька днів.
За неможливості досягти результату за 2-3 тижні доза протягом
кількох діб збільшується до 2 мг/кг ваги, а в деяких випадках — до
З мг/кг. Загальний термін лікування кортикостероїдами не пови-
нен перебільшувати 6-7 місяців через значну кількість побічних
ефектів, особливо негативний вплив на систему гемостазу.
При неефективності лікування (нестабільний результат, недос-
татньо високий вміст тромбоцитів у крові) хворому рекомендована
спленектомія. Ефективність методу зумовлена видаленням органу,
в ретикуло-ендотеліальній системі котрого руйнуються тромбоцити
та який бере участь у імунній відповіді. Позитивний ефект спленек-
томії відмічається вже в перші години після операції, а за добу вміст
тромбоцитів збільшується в кілька разів і навіть може досягти норми.
У разі неефективності спленектомії можуть повторно призна-
чатися кортикостероїди, в дозі 1 мг/кг ваги, або циклоспорин А, по
5мг/кг, як активний імуносупресант.
Якщо ефекту не досягнуто або він незначний, у схему лікуван-
ня можна вводити імуносупресивні цитостатики (циклофосфан,
вінкристин, вінбластин) у середніх дозах.
З-поміж препаратів симптоматичної терапії активно викорис-
товуються гемостатики — діцинон (етамзилат), Е-АКК, гордокс,
трасилол, а також ангіопротектори — аскорбінова кислота, рутин.
Особливо обережно слід призначати тромбоконцентрат хворим
на ІТП, оскільки можливе потенціонування імунної реакції через
сенсибілізацію донорськими тромбоцитами. Показаннями для пе-
реливання донорських тромбоцитів є активна кровотеча або підго-
товка хворого до оперативного втручання.
218
| Геморагічні діатези
Тромботична тромбоцитопенічна пурпура
Тромботична тромбоцитопенічна пурпура (ТТП) — захворюван-
ня на основі множинного тромбоутворення, котре характеризуєть-
ся активною дисемінованою агрегацією тромбоцитів, утворенням
мікротромбів та розвитком гемолітичної анемії.
Епідеміологія. На ТЛІ хворіють люди будь-якого віку, без ві-
кових, географічних, расових та сезонних піків захворюваності;
жінки, в середньому, дещо частіше, ніж чоловіки.
Етіологія. Етіологічні чинники до сьогодні не з’ясовані. Від-
мічається певна залежність від попередньої вірусної чи бактері-
альної патології, але однозначно пов’язувати розвиток захворю-
вання з нею підстав немає. Незважаючи на деякі ознаки, не
знайшли підтвердження ні вірусна чи бактеріальна інфікованість,
ні імунна агресія.
Патогенез. Патологічні зміни в системі гемостазу починають-
ся зі спонтанного тромбоутворення, котре швидко набуває генера-
лізованого характеру й уражає дрібні судини артеріального типу.
Головним чинником, котрий запускає цей процес, є наявна в плаз-
мі хворих надзвичайно активна, без ферментних властивостей ре-
човина білкової природи, яка погано піддається інгібіції. В нормі
у здорових людей присутній її інгібітор як антифактор. Саме за
умов його відсутності й розвивається ТТП.
Отже, основні ланки патогенезу наступні:
1)білковий дефект (спонтанна відсутність специфічного інгібі-
тора мікротромбоутворення);
2) множинний дисемінований мікротромбоз;
3) тромбоцитопенія потреби;
4) геморагічний синдром;
5) гемолітичний синдром через порушення реології крові.
Клініка. Захворювання, як правило, починається гостро, має
швидко прогресуючий, агресивний перебіг. Зрідка можливі прод-
ромальні прояви (слабкість, нездужання, підвищення температури
тіла). Основною клінічною ознакою є геморагічний синдром різ-
ного ступеня вираженості — від петехіально-плямистої висипки до
метрорагій та шлунково-кишкових кровотеч. Крім геморагічних
проявів можливі підвищення температури тіла до фебрильних
цифр, шлункова та кишкова диспепсія (нудота, блювота, болі
в животі), мелена, гепато-спленомегалія, неврологічна симптома-
тика (судоми, порушення зору, мови), психічні розлади.
219
Н.М. Третяк | Гематологія
Суттєвим компонентом клінічної картини при ТТП є анеміч-
ний синдром, розвиток якого пов’язаний із приєднанням неімун-
ної гемолітичної анемії. До геморагічної висипки приєднуються
жовтяниця, гепато- та спленомегалія, ознаки порушення функції
нирок та гіпоксії внутрішніх органів.
ТРОМБОЦИТОПАТІЇ
Однією з функцій тромбоцитів у організмі людини є забезпе-
чення первинного гемостазу, провідна роль у якому належить їхній
адгезивно-агрегаційній здатності. У разі ушкодження судинної стін-
ки тромбоцити контактують із колагеном, котрий є головним сти-
мулятором адгезії (прилипання до судинної стінки). Активовані
тромбоцити набрякають, утворюють псевдоподії та адгезуються до
субендотеліальних структур. Важливими плазмовими кофакторами
цього процесу є іони кальцію та фактор Віллебранда. Одночасно
з адгезією відбувається й агрегація (склеювання між собою) тром-
боцитів, для якої потрібні колаген, тромбін, АДФ та адреналін.
Поняття тромбоцитопатії (ТП) об’єднує всі захворювання
з порушенням гемостазу, зумовленим якісною неповноцінністю
або дисфункцією тромбоцитів. Це досить поширена група хвороб
та синдромів, котрі часто проявляються так званими “малими”
формами кровоточивості (менорагії, носові та ясневі кровотечі, ге-
морагії, що легко виникають на шкірі, тривалі кровотечі після екс-
тракції зубів та незначних порізів). Поява такої кровоточивості на
фоні нормальної або дещо зниженої кількості тромбоцитів і відсут-
ності відхилень у показниках коагулограми може свідчити про
функціональну неповноцінність тромбоцитів.
Епідеміологія. Серед спадкових геморагічних діатезів ТП посі-
дають перше місце й становлять 36% від загальної кількості хворих.
При активному виявленні хворих із легкими формами ТП цей по-
казник сягає 60-65%.
Класифікація тромбоцитопатії (за С. МиеІІег-ЕсккагІЇ аі аі.,
1993)
І. Спадкові тромбоцитопатії:
1) розлади адгезії тромбоцитів:
— синдром Бернарда—Сульє;
2) розлади агрегації тромбоцитів:
— тромбастенія Гланцмана,
— тромбоцитарний синдром Віллебранда;
220
| Геморагічні діатези
3) розлади звільнення внутрішньотромбоцитарних субстанцій:
— дефіцит альфа-гранул,
— дефіцит сігма-гранул,
— інші дефекти секреції;
4) розлади прокоагуляційної активності;
5) розлади функції тромбоцитів, які спостерігаються при інших
вроджених дефектах.
//. Набуті тромбоцитопатії.
Причини набутої тромбоцитопатії:
а) медикаменти;
б) соматична патологія:
— хронічна ниркова недостатність,
— антитромбоцитарні антитіла,
— хронічні захворювання печінки;
в) гематологічні захворювання:
— мієлопроліферативні хвороби,
— мієлодисплазія та гостра лейкемія,
— моноклональна гамапатія,
— набутий синдром Віллебранда.
Спадкові тромбоцитопатії
Синдром Бернарда—Сульє — макроцитарна тромбодистрофія,
єдине, відоме на даний час захворювання з порушенням, переваж-
но, адгезії тромбоцитів. Успадковується за аутосомно-рецисивним
типом, проявляється вже в ранньому дитинстві й зустрічається ду-
же рідко. В основі патогенезу — первинна аномалія мегакаріоцитів
і тромбоцитів, що характеризується помірною тромбоцитопенією
та відсутністю в цитоплазматичній мембрані обох видів глікопро-
теїну 1, який взаємодіє з фактором VIII, V, IX. Порушення адгезії
тромбоцитів призводить до подовження часу кровотечі. Захворю-
вання має три форми перебігу: легку, середню та тяжку. Клінічни-
ми проявами захворювання є типові ознаки геморагічного синдро-
му з петехіально-плямистою висипкою, а також кровотечі з носа,
ясен, шлунково-кишкового тракту та ін.
Діагностика синдрому Бернарда—Сульє полягає у визначенні
мегатромбоцитів у мазку периферичної крові. Диференціально-ді-
агностичним тестом є відсутність нормалізації ристоцетин-агрега-
ції тромбоцитів у хворих при додаванні плазми здорової людини
або фактора Віллебранда.
221
Н.М. Третяк | Гематологія
Лікування симптоматичне. Зменшення кровотечі досягається
вливанням концентрату тромбоцитів. Спленектомія та глюкокор-
тикостероїди не ефективні.
Тромбастенія Гланцмана належить до дезагрегаційних ТП, пе-
редається за рецесивно-аутосомним або домінантним типами. Ха-
рактеризується подовженням часу кровотечі за Дюком, вираженим
порушенням ретракції кров’яного згустку, порушенням агрегації
тромбоцитів під дією АДФ при нормальній кількості тромбоцитів.
В основі патогенезу тромбастенії Гланцмана — спадковий де-
фіцит глікопротеїнів 11 та IV в оболонках тромбоцитів, що пору-
шує їхню агрегаційну активність. Захворювання проявляється
з раннього дитинства й характеризується кровоточивістю мікро-
циркуляторного типу (кровотечі зі слизових оболонок носа, ясен,
шлунково-кишкового тракту, нирок, матки).
Діагностика базується на виявленні порушення ретракції згус-
тку крові, відсутності агрегації тромбоцитів під дією АДФ.
Лікування симптоматичне. При значних кровотечах ефективне
вливання тромбоконцентрату. Кортикостероїдні гормони не ефек-
тивні.
Розлади звільнення внутрішньотромбоцитарних субстанцій є нас-
лідком спадкового дефіциту в тромбоцитах білкових альфа- або сіг-
ма-гранул, неспроможності пластинок накопичувати та виділяти
для здійснення гемостазу АТФ, серотонін, адреналін, кальцій тощо,
а отже, порушення агрегаційної функції тромбоцитів та тромбони-
тарного гемостазу. Основними симптомами цієї групи захворювань
є геморагії на слизових оболонках носа, ясен, петехіально-плямис-
ті крововиливи на шкірі, маткові кровотечі, тривалі кровотечі після
екстракції зубів. У частини хворих захворювання може ускладнюва-
тися розвитком мієлофіброзу без спленомегалії. Крім того, дефіцит
сігма-гранул може проявлятися формою без альбінізму та з таким
(синдром Хержманського—Пуддака).
Синдром Чедіака—Хігаші — родинна аномалія гранулоцитів та
тромбоцитів, що успадковується за аутосомно-рецисивним типом.
Характеризується не тільки кровоточивістю через дисфункцію
тромбоцитів (порушення звільнення внутрішньотромбоцитарних
субстанцій), а й помітно зниженою резистентністю до інфекцій,
нейтропенією зі значним зниженням хемотаксису та фагоцитарної
активності, диспігментацією шкіри та очним альбінізмом.
Клінічний перебіг проявляється схильністю до інфекцій (рес-
піраторні захворювання, отити, пневмонії та ін.), іноді спостеріга-
222
| Геморагічні діатези
ються генералізована лімфоаденопатія, гепатоспленомегалія, гіпох-
ромна анемія, фотофобія, ністагм. Геморагічний синдром проявля-
ється петехіями, екхімозами й носовими кровотечами; на пізніх
стадіях можливі профузні шлунково-кишкові кровотечі.
Діагноз встановлюється на підставі виявленого порушення збе-
рігання та вивільнення АТФ, АДФ, серотоніну. Спостерігається
подовжений час кровотечі.
Лікування симптоматичне. Прогноз не сприятливий. Хворі
вмирають до 10-річного віку.
Парціальні дезагрегаційні тромбоцитопатії зі збереженням реак-
ції вивільнення внутрішньотромбоцитарних субстанцій включають
аномалію Мея—Хегліна, аномалію тромбоцитів Персона—Стоба,
тромбоцитопатію з ізольованим порушенням колаген-агрегації або
з ізольованим порушенням АДФ та/чи тромбін-агрегацїї, вроджену
(спадкову) афібриногенемію. Усі ці форми ТП успадковуються за
аутосомно-домінантним типом.
Диференційна діагностика досить складна й можлива лише за
ретельного комплексного дослідження функції тромбоцитів. Ліку-
вання симптоматичне. При значних кровотечах позитивний ефект
досягається інфузіями тромбоконцентрату. Кортикостероїдна тера-
пія не ефективна.
Набуті (симптоматичні) тромбоцитопатії
Набуті (симптоматичні) ТП — це вторинні дисфункції тром-
боцитів, що можуть виникати при тяжких ураженнях печінки, ви-
раженій нирковій недостатності, колагенозах, системних захворю-
ваннях сполучної тканини, септичних процесах, ендогенній
інтоксикації, захворюваннях системи крові. Вторинні ТП розвива-
ються також у випадках дисемінованого внутрішньосудинного зсі-
дання крові, при тромбоемболіях, масивних гемотрансфузіях.
Діагностика та диференційна діагностика ТП базується на дос-
лідженні мікроциркуляторного гемостазу, адгезивно-агрегаційної
та коагуляційної функцій тромбоцитів, на оцінці вмісту в них
тромбоцитарних факторів і гранул та реакцій вивільнення, на виз-
наченні кількості, розмірів, морфології та інших якостей тромбо-
цитів.
Геморагічний синдром, що розвивається у хворих із гострою та
нирковою недостатністю, пов’язаний з якісною неповноцінністю
тромбоцитів та тромбоцитопенією. Найчастіше спостерігаються
223
Н.М, Третяк | Гематологія
кровотечі зі слизової оболонки носа, кишково-шлункового тракту
та урогенітальних органів. Вважається, шо вплив уремічних токси-
нів на тромбоцити є основною причиної їх функціональних розла-
дів. Основним методом патогенетичного лікування таких хворих є
гемодіаліз. При захворюваннях печінки, особливо ускладнених
синдромом портальної гіпертензії, часто відмічається не тільки ТП,
а й тромбоцитопенія, що зумовлено порушенням дозрівання мега-
каріоцитів та прискоренням секвестрації тромбоцитів у селезінці.
При тяжких гепатитах причиною геморагічного синдрому може бу-
ти ДВЗ крові. Життєво важливим лікуванням є інфузія концентра-
ту тромбоцитів. Попередження кровотеч досягається лише лікуван-
ням основного захворювання.
Синдром дисемінованого внутрішньосудинного згортання крові
(ДВЗ) суроводжується вираженою гіпофункцією тромбоцитів,
втратою повноцінного пула, дегрануляцією їх у зоні мікроциркуля-
ції та пригніченням адгезивно-агрегаційної функції кров’яних
пластинок продуктами ферментного розщеплення фібриноге-
ну/фібрину. Тобто при ДВЗ-синдромі порушення агрегації тромбо-
цитів поєднується з вираженими коагуляційними зрушеннями та
тромбоцитопенією витрати.
При захворюваннях системи крові основною причиною гемора-
гічного синдрому є тромбоцитопенія гіпорегенераторного типу, яка
поєднується з порушенням властивостей мегакаріоцитів і тромбоци-
тів. При обстеженні системи згортання крові у хворих на гостру лей-
кемію та хронічні мієлопроліферативні захворювання (остеомієло-
фіброз, поліцитемія, бластна криза хронічної мієлолейкемії)
спостерігаються як тромбоемболічні, так і геморагічні ускладнення,
які виникають унаслідок дисфункції тромбоцитів, обумовленої зло-
якісною трансформацією мегакаріоцитопоезу. Основні зрушення аг-
регаційної функції тромбоцитів, особливо зниження агрегації, відмі-
чаються у хворих після лікування цитостатичними препаратами.
У хворих на мієломну хворобу та хворобу Вальденстрема гемора-
гічний синдром є наслідком дисфункції тромбоцитів, що зумовле-
на “окутуванням” їх парапротеїнами з подальшим порушенням ак-
тивації та розладами адгезивності тромбоцитів. Встановлено, що
ступінь дисфункції тромбоцитів і клінічні прояви геморагічного
синдрому залежать від концентрації імуноглобулінів та їх класу.
Розлади колаген- та АДФ-агрегаиії починаються при концентрації
І&С та І§А близько 50 г/л, а І&М блокує ці функції у значно ниж-
чих концентраціях.
224
| Геморагічні діатези
Лікування полягає в призначенні цитостатичної терапії та про-
клейці плазмаферезу. При кровотечах, шо загрожують життю хво-
рого, показане введення тромбоконцентрату.
Тромбоцитопатії, спричинені медикаментами, залежно від доміну-
ючого впливу на властивості тромбоцитів поділяють на такі групи:
Таблиця 17. Диференційна діагностика при рідкісних формах
спадкових ТП
Показник синдром Бернарда- Сульє хвороба Віллєбранда Аномалія Мея-Хегліна
Розмір тромбоцитів мегатромбоцити норма норма
Кількість тромбоцитів норма або нижче норма частіше зниження
Аномалія гранулоцитів немає немає спостерігається
Адгезія до скла знижена знижена норма або знижена
Адгезія до колагену знижена знижена норма
Корекція порушень нормальною плазмою немає спостерігається немає
Рівень фактора VIII норма знижений норма
Антиген фактора VIII і фактор Віллєбранда норма знижений норма
Агрегація під впливом бичачого фактора VIII знижена норма норма
1) інгібітори утворення тромбоксану А, (високі дози глюкокор-
тикостероїдів, практично всі нестеро'ідні протизапальні засоби, імі-
дазол, простациклін та його синтетичні аналоги;
2) препарати, що підвищують у тромбоцитах вміст цАМФ
(простагландин Е, простациклін та його аналоги);
3) інгібітор фосфодіестерази (дипірідамол, пентоксифілін, па-
паверин, теофілін та ін.);
4) антагоністи кальцію (верапаміл, ніфедипін, ділтіазем та ін.,
високі дози фуросеміду);
5) антибіотики пеніцілінового ряду, цефалоспорини;
6) антикоагулянти прямої дії (гепарин);
7) декстрани (реополіглюкін);
8) цитостатичні засоби;
9) фенобарбітал.
Принципи лікування ТП
При дсзагрегаційних ТП. особливо есенніальній атромбії, пар-
ціальних дсзагрсгаційних формах, що перебігають як із нормаль-
225
Н.М. Третяк | Гематологія
ною, так і з порушеною реакцією вивільнення, при хворобі Віллеб-
ранда введення епсилон-амінокапронової кислоти (Е-АКК) по 8-
12 г на добу дорослому хворому значно зменшує кровоточивість
при носових кровотечах і менорагіях (крім дуже тяжких форм хво-
роби Гланцмана та Віллебранда). Для попередження маткових кро-
вотеч Е-АКК. призначають регулярно, разом з протизаплідними за-
собами, а в період кровотечі — в комплексі з препаратами, котрі
скорочують матку. Відмічають також позитивний ефект при вико-
ристанні АТФ, магнезії сульфату, рибоксину. При носових крово-
течах рекомендовані тампони з гемостатичною губкою або адрок-
соном. Слід уникати тугої тампонади носа, вишкрібання
порожнини матки та припікання слизових оболонок.
Лікування медикаментозних тромбоцитопатій, насамперед, по-
лягає у відміні провокуючого засобу. Для поліпшення гемостазу
використовують етамзилат натрію, Е-АКК, адроксон, при загрозі
життю — введення концентрату тромбоцитів.
226
ПЕРЕЛІК ПОСИЛАНЬ
1. Абдулкадьіров К. М., Бессемельцев С. С. Апластическая ане-
мия. — М.: Наука, СПб.: Ї\'Н, 1995. — 231 с.
2. Абдулкадьіров К. М., Бессемельцев С. С., Рукавицьін
О. А. Лечение хронического миелолейкоза. — М.: Наука, СПб.:
ЛЕКА, 1999. - 150 с.
3. Абдулкадьіров К. М., Рукавицьін О. А. и др. Гемолитические
синдроми в общей клинической практике: Справочник. — СПб:
ЗЛБИ, 1999. — 125 с.
4. Возианов А. Ф., Бутенко А. К., Зак К. Л. Цитокиньї. — К.:
Наукова думка, 1998. —317 с.
5. Воробьев А. И. Руководство по гематологии. В 2-х тт. — М.:
Ньюмед, 2002.
6. Гайдукова С. М., Видиборець С. В., Колесник І. В. Залізоде-
фіцитна анемія: Навч. посіб. для студентів. — К.: Науковий світ,
2001. —132 с.
7. Гематологія і трансфузіологія: Підручник / Під ред. проф.
С. М. Гайдукової. — К.: Три крапки, 2001. — 752 с.
8. Гусєва С. А., Вознюк В. П., Дубкова А. Г. Принципи діаг-
ностики та лікування: Довідник / Під ред. проф. С. А. Гусєвої. —
К.: Фахівець, 1999. — 289 с.
9. Глузман Д. Ф., Абраменко И. В. и др. Лабораторная диагнос-
тика гематологических заболеваний. — К.: Морион, 1998. — 333 с.
10. Дворецкий Л. И. Железодефицитньїе анемии. — М.: Нью-
диамед, 1998. — 211 с.
11. Исследование системи крови в клинической практике
/Под ред. Г. И. Козинца, В. А. Макаровой. — М.: Триада-Х, 1997.
— 480 с.
12. Иммуноцитохимия и моноклональньїе антитела в онкоге-
матологии І Под ред. В. Г. Пинчук, Д. Ф. Глузман. — К.: Наукова
думка, 1990. — 232 с.
13. Геморагічні захворювання / Під ред. Я. І. Виговської. —
Львів, 1999. — 235 с.
14. Идельсон Л. И. Гипохромньїе анемии. — М.: Медицина,
1981. — 121 с.
227
Н.М. Третяк | Гематологія
15. Идельсон Л. И. Методи лабораторной диагностики гемоли-
тических и мегалобластньїх анемий. -- М.: ЦОЛИУВ, 1990. — ЗО с.
16. Ковалева Л. Г. Острьіе лейкозьі. — М.: Медицина, 1990. —
271 с.
17. Классификация всемирной организации здравоохранения
опухолевьіх заболеваний кроветворной и лимфоидной ткани: Се-
минарьі по гематологии. — К., 2001. — 36 с.
18. Новик А. А. Классификация злокачественньїх лимфом: Ре-
комендации ВОЗ. — СПб.: ЗЛБИ, 2000. — 125 с.
19. Симбирцева Л. П., Холсти Л. Л. Лимфогранулематоз. — М.:
Медицина, 1985. — 302 с.
20. Справочник по гематологии / Под ред. А. Ф. Романовой. —
К.: Здоров’я, 1997. — 320 с.
21. Суховий М. В. Гемофилия как хирургическая проблема. —
К.: АДЕФ-Украина, 2001. — 270 с.
22. Флюдарабин в лечении хронического лимфолейкоза /
Н. Г. Габеева, А. В. Пивник, Т. Н. Мойсеева и соавт. // Тер. Архив.
- 2000. - Т.72. - №8. - С. 42-45.
23. Химиотерапия опухолевьіх заболеваний: Краткое руководс-
тво / Под ред. Н. И. Переводчиковой. — М.: Медицина, 2000. —
391 с.
Апластична анемія
1. Васі§аІііро А., Сйаріе М., Но\с8 8. еі аі. (1993). ТгеаїтепС оГ
аріазйс апетіа (АА) ауійі апйІутрИосуІе §1оЬи1іп (АБО) апсі теїйу-
Іргейпіхоїопе (МгесІ) МЙі ог суіЙіопі ап<1го§еп8: а гапсіотіхесі ігіаі
йот Йіе ЕВМТ 8АА \Уогкіп§ Рагіу. ВІН. 83,145—151.
2. Васі§а1иро А., ВгапсІ К.., Опеїо К., аі аі. (2000). Тгеаітепі оГас-
циігесі 5еуеге аріазйс апетіа: Ьопе тагголу Ігапзріапіайоп сопірагесі суіЙт
іттипокирргеззіуе Йіегару — Тйе Еигореап Сігоир Гог ВІоод апсі Маг-
гоу/ Тгапзріапіайоп ехрегіепсе. Зеті пак іп Нетаїоіо^у, 37, 69—80.
3. Васі§а1иро А., ЕосаіеИі Р., Зосіе О. еі аі. (2202). РІисІагаЬіпе,
сус1ор1іо8Ііаті<1е апсі АТО Гог аііетаііуе сіопог ггапзріапїаїіоп іп ар-
Іазйс апетіа — а герой оГ Фе 8АА \Уогкіп§ РагГу. Вопе Маггош
Тгапзріапіайоп, 29 (8ирр1. 2), 312а.
4. РгіскйоГеп ІЧ, Неітреї Н., Каїйуаззег І.Р. ей аі. (2003). Апййі-
утосуіе §1оЬи1 і п Мій ог МЙіоиі сус1о8рогіпе А: 11 уеаг Го11о\у-ир оГ а
гапйотіхесі Сгіаі сотрагіп§ їгеаїтепї8 оГ аріазйс апетіа. Віоой, 101,
1236-1242.
5. ОогсІоп-ЗтіФ Е.С. (1991). Асциігесі аріазїіс апетіа. Іп: Нета-
їо1о§у “Вазіс Ргіпсір1е8 апсі Ргасйсе” ( есі Ьу К. НоЬЬтап Е.І., Вепх
228
| Перелік посилань
8.І., 8йаііі1 В. Гигіе апйСоЬеп). Рр. 160—192. СЬигсЬіІІ. Ьіуіп§8іопє,
Ьопйоп.
6. Непйгу С.Ь., МагзЬ Оогйоп-8тіі1і Е.С., 8Іуа-Китагап
М. (2002)/ Кеіарке оГ зєуєгє аріазііс апетіа аГіег іпЯиепга іттипіга-
ііоп ВІН., 119, 283—284.
7. КіЛіск 8.В., МагзЬ І.С.ЛУ. (2002). Аріазііс апетіа: тапа§етепі.
Віоой Кєуієлуз, 14, 157—171.
8. Магзй І.8А¥. (2002). Нетаіороіеііс §гоуЛИ Гасіог іп Йіе раїіто-
§епезіз апй Гог Йіе ігеаітепі оГ аріазііс апетіа. 8етіпагз іп кетаіо-
іоеу, 37, 84-90.
9. Мс.Сапи 8.В., Разз\Уе§ І., 8іогЬ., Оеей Н.І., (2000). НЬАійеп-
іісаі зіЬ1іп§ Ьопе таггоу/ ігапзріапіаііоп іо ігеаі зєуєгє аріазііс апетіа.
Іп: Аріазііс апетіа, раіЬоіо§у апй ігеаітепі (ей. Ьу Н. ЗсИехептїІіег,
А. Васі^аіиро), рр. 230—257. СатЬгій§е Ііпіуегзіїу Ргезз, СатЬгій§е.
10. ЗсЬгегептеіег Н., Васі§а1иро А., А§1іеііа М. еі. аі. (2002).
Сопзепзиз йоситепі оГ а §гоир оГ іпіетаііопаї ехрегіз. Іп: Аріазііс
апетіа: раікорЬузіо1о§у апй ігеаітепі (ей. Ьу Н. ЗсЬгегешпеіег, А.
Васі§а1иро) рр. 308—315. СатЬгій^е Опіуегзііу Ргезз, СатЬгій^е.
Мієломна хвороба
1. Андреева Н. Я. Диагностика и лечение множественной мие-
ломьі. — М.: Ньюмед-А. О., 1998.
2. Клиническая онкогемагология / Под ред. М. А. Волковой. —
М.: Медицина, 2001.
3. Стандарти в гематології / Під ред. Я. І. Виготської, В. Л. Но-
вака. — Львів, 2002.
4. ХУіпігоЬе’з Сііпісаі Нетаіо1о§у, 10°“ ей. 1998, У2.
5. Нетаіоіо^іа Кііпісхпа у/ хагузіе рой. Кей. Т.КоЬака, Ьоойг. 1998.
6. Ьесгепіе гоггокіоу/усії сЬогоЬ ІіетаіоІо£Іс2пісІі рой гей. А.
Отозхупзкіе], у/уй. III, ЬиЬІіп, 2002.
7. Оигіе В.О.М., Заїтоп 8.8. А сііпісаі 8Іа§іп§ кузіет Гог тиііір-
іе туеіота/ Соггеїаііоп оГ теазигей туеіота сеіі тазз \уіЙі ргекеп-
ііп§ сііпісаі Геаіигек, гезропзе іо ігеаітепі апй кигуіуаі. Сапсег 1975;
36 (10): 842-854.
8. Сгііегіа Гог еуа1пайп§ йізеазе гезропзе апй рго§ге58Іоп іп раііепіз
Мій тиіііріе туеіота ігеаіей Ьу Ьі§й йозе іИегару апй Ьетороіеііс
зіет сеіі ігапзріапіаііоп. Вг І. оГНетаі.1998; 102: 1115—1123.
9. Оигіе Ь.О.М 8іа§іп§ апй кіпеіісз оГтиІіірІе туеіота. Сііп. На-
етаіоі. 1982: 11: 181—210.
10. Куіе В.А. Огеірр В.Р. Ріазта сеіі йізсгазіаз: сиггепі зізіиз.
Сегі. Кєу. Опсої. Нетаіоі 1988; 6: 93—152.
229
Н.М.Третяк | Гематологія
Гостра лімфобластна лейкемія
1. Наггіз N. Дайе Е, ОіеЬоїд 1. еі. Аі ХУНО СіаззіГісаІіоп оГ №-
оріазііс Візеазе оГ Нетаїороіеіід апд ЕутрИоід Тіззиез: Керогі оГ Йіе
СІіпісаІ Адуізоіу Соттіїїее МееІіп£ Аігііе Ноизе, Ущцпіа, КоуетЬег
1997,1. СІіп. Опсої. 1999; 17: 3835-3849.
2. Ноеіхег В., ОоекЬи^еІ М., ОИтапп О. еі аі. Асиїе ІутрИоЬІаз-
Ііс Іеикетіа. Нетаїоіо^у 2002; 162—192.
3. Ноеіхег В. Тгеаїтепі орііопз іп адиіі асиїе ІутрИоЬІазІіс Іеике-
тіа. Іп: Еигореап Нетаїоіо^у аззосіаііоп Едисаііоп Ьоок 2001: 49—53.
4. ЗЬіЬаиі А., Уотапі І.Р., Ве§оз Ь. е1.а1. Адиіі асиїе ІутрЬосуІіс
Іеикетіа зійду Іезііп§ сИетоІІіегару апсі аиіоіо^оиз апсі а11о§епіс Ігапз-
ріапіаііоп. А 1оІіои-ир герой оГ Йіе Егспсії ргоіосої ГАЬА 87. Нета-
Іо.ОпсоІ Сііп. Могіїї Ат. 2000: 14: 1353—66.
5. Аппіпо Ь., Уе§па М.Ь. Сатега А. еі аі Тгеаїтепі оГ адиіі аси-
їе ІутрИоЬІазІіс Іеикетіа (АІХ) Іоп^-Іегт Го11оу/-ир оГ Йіе СІМЕМА-
А1X0288 гапдотіхед зійду. Віоод. 2002: 99: 863-871.
6. Капдагуап М.У., О’Вгіеп 8., 8тііЬ .IX. еі. аі. Кезиііз оГ Ігеаі-
тепі оГ \уііИ курег СУАВ, а дозе іпієпзіує ге§ітеп, іп асиїе Іутркос-
уііс Іеикетіа. 1. СІіп. Опсої. 2000; 18: 547—561.
7. Аззеїіп ВХ. ТЬе іЬгее азрага^іпазез. Сотрагаїіуе рЬагтасо!о§у
апд оріітаї апд оріітаї изе іп сМдЬоод Іеикетіа. Аду. Мед. Віоі.
1999; 457: 621-629.
8. Оіеіззпег В., Оок1Ьи§еІ К., Вагігт С.К еі.аі. Ьеадіп§ рго§поз-
Ііс геїеуапсе оГ Йіе ВСК-АВЬ Ігапзіосаііоп іп адиіі асиїе Вітане
ІутрИоЬІазІіс Іеикетіа: а ргозресііуе зійду оГ Йіе Оегтап Миііісепіег
Тгіаі Огоир апд сопГігтед роїутегазе сИаіп геасііопз апаїузіз. Віоод.
2002; 99: 1536-1543.
Гостра мієлоїдна лейкемія
1. Наггіз Н, Іаїїе Е., ВіеЬоїд 1. еі аі. ХУНА Сіаззіїїсаііоп о£Ме-
оріазііс Візеазе оГ Наггіз N. -Іайс Е, ВіеЬоїд .1. еі. А1 \\'НО Сіаззійса-
Ііоп оГ №ор!азііс Візеазе оГ Нетаїороіеіід апд Ьутркоід Тіззиез: Ке-
рогі оГ Йте Сііпісаі Адуізогу Соттіїїее МееІіп§ Аігііе Ноизе, Уіг§іпіа,
^уетЬег 1997,1. СІіп. Опсої. 1999; 17: 3835—3849.
2. \¥еск І.К., Кореску К.Л., АрреІЬаит Р.К. еі. АІ. А гапдотіхед
Іпуе8іі§а1іоп оПті§Ь-дозе уегзиз зіапдагд-дозе суіозіпе агаЬіпозіде \УІІк
даипогиЬісіп іп раїіепіз \уііЬ ргеуіоизіу ипігеаіед асиїе туеіоід Іеике-
тіа: А зоиікіу/езі Опсоїоеу Огоир зійду. Віоод 1996; 88: 2841—2851.
3. Апдегзоп Л.Е., Коеску К.ї. ХУіІІтап С.Ь., еі аі. Оиісоте аГіег
іпдисііоп сИетоІІіегару Гог оїдег раїіепіз \уіЙі асиїе туеіоід Іеикетіа
із поі ітргоуед у/іііт тіїохапігопе апд еіорозіде сотрагед суіііт суіага-
230
| Перелік посилань
Ьіпе ап<1 даііпогиЬісіп: а Зоиіітсуезі опсоїо^у Огоир зійду. Віоод 2002;
100: 3869—3876.
4. ВізИор З.Р., Маііііелуз З.Р. ¥оип£ О.А., еі. аі. А гапдотіхед
зійду оГ Ьі§Ь-дозе суіогаЬіп іп іпдисііоп іп асиіе туеіоід Іеикетіа.
Віоод 1996; 87: 1710-1717.
5. Напп І.М., 8ієуєпз Е.Р., Соїдзіопе А.Н., аі. аі. А гапдотіжд сот-
рагізоп оГ ВАТ уегзиз АОЕ аз іпдисііоп сИетоИіегару іп сіііідгеп апсі уо-
игщег адиііз \уіИі асиіе туеіоід іеикетіа: Кезиііз оГ Йіе Медісаі ЕезеагсИ
Соипсії’з 10 Ні АМЬ ігіаі (МЕС АМЬ 10). Віоод 1997; 89: 2311-2318.
6. Витеіі АК., Ооідзіопе А.Н. 8ієуєпз Е.М.Р., еі. аі. А гапдо-
тіхед сотрагізоп оГ аддіііоп оГ аиіоіо^оиз Ьопе тагтосу ігапзріапіаіі-
оп іо іпіепзіуе сИетоІІіегару Гог асиіе туеіоід Іеикетіа іп Йгзі гетіз-
зіоп: гезиііз оГ МЕС АМЬ 10 ігаіі. Ьапсеі 1998; 351: 700—708.
7. Репаих Р., Сказіап§ С., СИєугєі 8., еі. аі. А гапдотігед сот-
рагізоп оГ аіі ігапзгеііпоіс асід (АТЕА) ріиз скетоЙїегару апсі іііе гііе
оГ таіпіепапсе іИегару іп песуіу діа^позед асиіе рготуеіосуііс іеике-
тіа. Віоосі 1999; 94: 1192—1200.
8. 8апх М.А. Магііп С., Еауоп с. еі. аі. А тодійед АЮА ргоіо-
сої \УІ11і апйігасусііпе — Ьазед сопзоіідаііоп гезиііз іп Иі^іі апіііеике-
тіс ейісасу апсі гедисед іохіїу іп пеу/Іу діа§позед РМЬ/ЕАЕ асиіе
рготуеіосуііс іеикетіа. РЕТНЕМА §гоир Віоосі 199; 94: 3015—3021.
Хронічна мієлоїдна лейкемія
1. Радегі 8., Таїрах М., Езіхоу 2. еі. аі. Сіігопіс туе1о§епиз Іеи-
кетіа Віріреу апсі іііегару. Апп Іпіет Мед. 1999; 131: 207— 219.
2. 8а\ууегз С.Ь. Сіігопіс туеіоід Іеикетіа. N. Еп§1. 3. Мед. 1999;
340: 1330-1340.
3. СІіГі Е.А., 8іогЬ Е: Маггоу/ ігапзріапіаііоп Гог СМЬ: Тке 8еаі-
ііе ехрегіепсе. Вопе Маггосу ігапзріапі 1996; 17 (8ирр1 3): 8 1—3.
4. Капіагуап Н.М. Согіез З.Е. О’Вгіеп 8. еі. аі. ІтаііпЬ тезуїаіе
іп пеМу діа§позед раііепіз \уіі1і РМадеІрИіа скготозоте — розіііуе
скгопіс туек>§епоиз Іеикетіа: кі§1і іпсідепсе оГ еагіу сотріеіе апд
та]ог суіо§епезіз гезропзез. Віоод 2003; 101: 97—100.
5. Капіагуап Н.М., 8а\ууегз С. НоскЬаиз А. еі. аі. НетаіоІо§іс
апд суіо§епеііс гезропзез іо ІтаііпЬ тезуїаіе іп сИгопіс туе1о§епоиз
Іеикетіа. N. Еп§1. 3. Мед. 2003; 348: 1048—1050.
6. Абдулкадьіров К. М., Бессмельцев С. С. Апластическая ане-
мия. — М.: Наука, СПб: NН, 1995. — 231 с.
7. Абдулкадьіров К. М., Бессмельцев С. С, Рукавицьш О.А. Ле-
чение хронического миелолейкоза. — М.: Наука, СПб: ЛЕКА,
1999. — 150 с.
231
Н.М. Третяк | Гематологія
8. Абдулкадьіров К. М, Рукавицьш О. А. и др. Гематологичес-
кие синдроми в общей клинической практике: Справочник. СПб:
ЗЛБИ, 1999. — 125 с.
9. Возианов А. Ф. Бугейко А. К. Зак К. Л. Цитокиньї. — К.:
Наукова думка. 1998. — 317 с.
10. Воробьев А. И. Руководство по гематологии: В 2-хтг. — М.:
Ньюдиамед— Москва, 2002.
11. Гайдукова С. М., Видиборець С. В. Колесник І. В. Залізоде-
фіцитна анемія: Навч. посібник для студентів. — К.: Науковий світ,
2001. - 132 с.
12. Гематологія та трансфузіологія: Підручник І Під ред. проф.
С. М. Гайдукової. — К.: ВПУ “Три крапки”, 2001. — 752 с.
13. Гусєва С. А., Вознкж В. П., Рубкова А. Г. Анемии: принци-
пи диагностики и лечения: Справочник І Под ред. проф. С. А. Гу-
севой. — К.: Фахівець, 1999. — 289 с.
14. Глузман Д. Ф., Абраменко И. В. и др. Лабораторная диагнос-
тика онкогематологических заболеваний. — К.: Морион, 1998. —
333 с.
15. Дворецкий Л. И. Железодефицитнне анемии. — М.: Нью-
диамед,1998. — 211 с.
16. Исследование системи крови в клинической практике І Под
ред. Г. И. Козинца, В. А. Макаровой. — М.: Триада-Х, 1997. — 480 с.
17. Иммуноцитохимия и моноклональнне антитела в онкоге-
матологии І Под ред. В. Г. Пинчук, Д. Ф. Глузман. — К.: Наукова
думка, 1990. — 232 с.
18. Геморагічні захворювання / Під ред. Я. І. Виготської. —
Львів, 1999. — 235 с.
19. Идельсон Л. И. Гипохромнме анемии. — М.: Медицина,
1981. — 121с.
20. Идельсон Л. И. Методи лабораторной диагностики гемоли-
тических и мегалобластньїх анемий. — М.: ЦОЛИУВ, 1990. — 30 с.
21. Ковалева Л. Г. Острьіе лейкози. — М.: Медицина, 1990. —
271 с.
22. Классификация Всемирной организации здравоохранения
опухолевьіх заболеваний кроветворной и лимфоидной ткани: Се-
минарн по гематологии. — К., 2001. — 36 с.
23. Новик А. А. Классификация злокачественних лимфом: Ре-
комендации ВООЗ. — СПб.: ЗЛБИ, 2000. — 125 с.
24. Симбирцева Л. П. Холости Л. Л. Лимфогранулематоз. —
М.: Медицина, 1985. — 302с.
232
| Перелік посилань
25. Справочник по гематологии / Под ред. А. Ф. Романовой. —
К.: Здоров’я, 1997. —320 с.
26. Суховий М. В. Гемофилия как хирургическая проблема. —
К.: АДЕФ—Украйна, 2001. — 220 с.
27. Флюдарабин в лечении хронического лимфолейкоза /
Н. Г. Габеева, А. В. Пивньж, Т. Н. Мойсеева и соавт. //Тер. Архив.
— 2000. — Т. 72. — №8. — С. 42-45.
28. Химиотерапия опухолевьіх заболеваний: Краткое руководс-
тво / Под ред. Н. И. Переводчиковой. — М.: Москва, 2000. — 391 с.
233
СПИСОК СКОРОЧЕНЬ
АА АІГА АЛГ АПТЧ АТГ БКХМЛ ВКЛ ВФ Г-6-ФДГ ГА ГЕС Г-КСФ ГЛ ГЛЛ апластична анемія автоімунна гемолітична анемія антилімфоцитарний глобулін активований парціальний тромбопластиновий час антитимоцитарний глобулін бластна криза хронічної мієлоїдної лейкемії волосковоклітинна лейкемія внутрішній фактор глюкозо-6-фосфатдегідрогеназа гемолітична анемія гіпереозинофільний синдром гранулоцитарний колонієстимулюючий фактор гостра лейкемія гостра лімфобластна лейкемія
ГМ-КСФ гранулоцитарно-макрофагальний колонієстиму-
люючий фактор
ГМЛ гостра мієлобластна лейкемія
гхн гемолітична хвороба новонароджених
ЕПО еритропоетин
ЗДА залізодефіцитна анемія
ІЛ інтерлейкін
їм інфекційний мононуклеоз
ІГІІ ідіопатична тромбоцитопенічна пурпура
ІФН інтерферон
їм ідіопатичний мієлофіброз
КП колірний показник
ЛГМ лімфогранулематоз
лдз латентний дефіцит заліза
ЛЛ лімфоплазмоцитарна лімфома
МДС мієлодиспластичний синдром
МХ мієломна хвороба
НЗЛ негоджкінські злоякісні лімфоми
СП справжня поліцитемія
ткм трансплантація кісткового мозку
ХЛЛ хронічна лімфоідна лейкемія
ХМЛ хронічна мієлоїдна лейкемія
ХММЛ хронічна мієломоноцитарна лейкемія
234
ЗМІСТ
КРОВОТВОРЕННЯ............................................ з
Кістковомозкове кровотворення.................... . 5
Методи обстеження гематологічних хворих...............18
АНЕМІЇ...................................................26
Гостра постгеморагічна анемія.........................27
Залізодефіцитна анемія... 29
Анемії, зумовлені порушеннями синтезу порфіринів .....46
Анемія, зумовлена свинцевою інтоксикацією ............47
Мегалобластні анемії ................................49,
Фолієводефіцитна анемія . 53
Апластична анемія 55
Гемолітичні анемії ...................................59
Спадкові гемолітичні анемії, зумовлені змінами
мембрани еритроцитів........................... 60
Спадкові гемолітичні анемії, зумовлені порушенням
синтезу гемоглобіну та змінами активності ферментів ... 63
Набуті гемолітичні анемії. Імунні гемолітичні анемії.64
Гемолітична хвороба новонароджених 65
Гемолітична анемія, зумовлена інфузією несумісної
крові .......................................... .66
Токсична гемолітична анемія ..................... 67
Автоімунні гемолітичні анемії.................... 67
Пароксизмальна нічна гемоглобінурія
(хвороба Маркіафави—Мікелі) ......................... 69
МІЄЛОДИСПЛАСТИЧНИЙ СИНДРОМ ..............................71
ГЕМОБЛАСТОЗИ.............................................79
Гострі лейкемії...... 79
Хронічні мієлопроліферативні захворювання . 97
Хронічна мієлоїдна лейкемія . ... 98
Ідіопатичний мієлофіброз 109
Справжня поліцитемія ... 113
Хронічні лімфопроліферативні захворювання .117
Хронічна лімфоїдна лейкемія . ... 117
Негоджкінські лімфоми ........................ .126
Лімфогранулематоз (хвороба Годжкіна)............138
Лімфоплазмоцитарна лімфома (макроглобулінемія
Вальденстрема)..................................146
Мієломна хвороба (множинна мієлома) . 151
ЛЕЙКЕМОЇДНІ РЕАКЦІЇ ...................................158
Мієлоїдні реакції 159
Лейкемоїдні реакції еозинофільного типу .... 163
Лейкемоїдні реакції лімфоцитарного типу . 167
Інфекційний мононуклеоз . 167
Інфекційний лімфоцитоз......................... 172
Хвороба котячої подряпини . .173
Ієрсиніоз 174
Монопитарно-макрофагальні лейкемоїдні реакції .. .175
Гістіоцитоз X (гістіоцитоз із клітин Лангерганса) . 176
АГРАНУЛОЦИТОЗ......................................... 178
ЦИТОСТАТИЧНА ХВОРОБА ................................. 183
ЛЕЙКОПЕНІЇ.............................................190
Нейтропенія при первинних імунодефіцитних станах 195
Нейтропенія новонароджених імунного генезу . 196
Лейкопенії при спленомегаліях 197
ЕРИТРОЦИТОЗИ...........................................200
ГЕМОРАГІЧНІ ДІАТЕЗИ ...................................202
Вазопатії..... ..... .204
Васкуліт Шенлейн-Геноха . . 205
Коагулопатії. Гемофілії .. . 209
Хвороба Віллєбранда . .213
Тромбоцитопенії 215
Ідіопатична тромбоцитопенічна пурпура (ІТП) . .215
Тромботична тромбоцитопенічна пурпура .219
Тромбоцитопатії............. . 220
Спадкові тромбоцитопатії ...................... 221
Набуті (симптоматичні) тромбоцитопатії. . 223
ПЕРЕЛІК ПОСИЛАНЬ........................................227
СКОРОЧЕННЯ .............................................234
До розділу "Мієломна хвороба (множинна мієлома)", с. 151-157.
Клініко-анатомічна класифікація ММ:
— дифузно-вогнищева;
— дифузна;
— множинно-вогнищева;
— склерозуюча;
— переважно вісцеральна.
Стадії ММ (В. С. М/ І)игіе, 8. Е. 8а1шоп, 1975)
Стадії Критерії Пухлинна маса, кг/мг
І Сукупність таких ознак: - рівень НЬ > 100 г/л; - нормальний рівень Са у сироватці; - відсутність остеолізису або солітарне кісткове вогнище; - низький рівень М-компонента (Ідб < 50 г/л, ІдА < ЗО г/л, білок Боне Джонса у сечі < 4 г/24 г. До 0,6 (мала)
II Показники середні між І та II стадіями 0,6-1,2
III Одна чи більше з наступних ознак: - рівень Нб < 85 г/л; - рівень Са сироватки вищий за норму; - виражений остеодеструктивний процес; - високий рівень М-компонента (ІдС > 70 г/л, ІдА > 50 г/л, білок Бенс Джонса у сечі > 2 г/24 і. Більше 1,2
Помічені помилки
На сторінці 11 у підписі до рисунка замість “клітини гемопоезу”
— “клітин гемопоезу”;
На сторінці 13 у шостому рядку першого абзацу замість “ метабо-
лії іін" — “метаболіти”;
На сторінці 15 у п'ятому рядку другого абзацу замість “нормоблас-
ту” — “нормобласті”;
На сторінці 27 в останньому рядку другого абзацу замість “вели-
кого ступеня” — “тяжкого ступеня”;
На сторінці 49 в першому рядку третього абзацу та третьому ряд-
ку восьмого абзацу замість “мл” — “мг”;
На сторінці 55 у другому рядку другого абзацу нового розділу за-
мість “1988” - “1888”;
На сторінці 68 у першому рядку першого абзацу замість “агглю-
тиніни” — “аглютиніни”.
ДЛЯ ЗАМІТОК
Навчальне видання
Третяк Н. М.
ГЕМАТОЛОГІЯ
навчальний посібник
Рецензенти:
Гайдукова С.М. — завідувач кафедри гематології та транс-
фузіології Київської медичної академії післядипломної освіти, заслужений
діяч науки, доктор медичних наук, професор;
Палієнко І.А. — Наиіональний медичний університет імені О.О.
Богомольця, кафедра пропедевтики внутрішніх хвороб № 2, доктор ме-
дичних наук;
Білько Н.М. — керівник центру молекулярних та клітинних до-
сліджень Національного університету "Києва-Могилянська академія ”
Міністерства освіти і науки України, доктор медичних наук, професор.
Відповідальний редактор: Гнатюк М.
Худоджник-дизайнер: Бєлов А.
Редактор: Книш /.
Верстальник:. Кириличев І.
Підписано до друку 7.06.2005 р. Формат 60x84/16.
Папір офсет. Друк офсетний. Гарнітура Тітез ЕТП.
Ум. друк. арк. 10,85. Обл.-вид. арк. 9,2.
Тираж 1000 прим. Зам. №296.
Видавництво "Зовнішня торгівля"
01042, м. Київ, вул. Раєвського, 36, оф. 408;
т./ф.: 528-10-02; т.: 528-85-83
е-таіі: го\1ог£@иаїї.ес1и.иа
Свідоцтво про внесення суб'єкта видавничої справи
до Державного реєстру видавців, виробників і розповсюджувачів
видавничої продукції: Серія ДК№ 1378 від 30.05.2003 р.
Надруковано ЗАТ «М кроні вська друкарня»
Свідоцтво про внесення суб'єкта видавничої справи
до Державного реєстру видавців, виготівників
і розповсюджувачів видавничої продукції
ДК №2101 від 16.02.2005 р.
м. Миронівка, Київської області
ТРАДИЦИИ СОЗДАНИЯ НОВЬІХ НАПРАВЛЕНИМ
ВосЬе
«Ф. ХОФФМАНН-ЛЯ РОШ Лтд.»
В ОНКОГЕМАТОЛОГИИ
Рекормон
зпозтин бета
Нейпоген
филграстим Г-КСФ
к ВЕСАНОИД
рофєрои-л
интерферон альфа-2а
Стимулятор зритропозза,
единственний в Украине
зритропозтин, разрешенньїй
для подкожного применения.
Лечение анемии почепного и
онкологического генеза,
анемии новорожденньїх.
Наиболее изученний и
предпочитаемий в мире
гранулоцитарний колоние-
стимулирующий фактор.
Лечение нейтропении.
- Индуктор дифференциации
клеток (системний ретиноид).
Единственний в мире препарат
для патогенетической -ерании
острой промиелоцитарной
лейкемии.
Рекомбинантний интерферон,
не содержащий сивороточного
альбумина человека.
Терапия хронических
гепатитов В и С, солидньїх
опухолей, новообразований
лимфатической системи.
ПРЕДСТАВИТЕЛЬСТВО КОМПАНИИ «ХОФФМАНН-ЛЯ РОЦІ Лтд.» в УКРАИНЕ
01001, г. Киев, пер. Музейньїй, 2-Б, 2-зтаж; тел: (044) 490-70-50: факс: (044) 490-70-29
мллм.госЬе.сот.иа мллгуу.госІіе-опсоІоду.сот.иа
Третяк Наталія Миколаївна
Доктор медичних наук, професор кафедри військової терапії Української військової ме-
дичної академії.
Завідувач відділення захворювань системи крові Інституту гематології та трансфузіології
АМН України.
Голова проблемної комісії "Гематологія та трансфузіологія" МОЗ та АМН України, член Єв-
ропейської асоціації гематологів, член Вченої ради та член Спеціалізованої ради Інституту ге-
матології та трансфузіології АМН України.
Закінчила Київський медичний інститут ім. О.О. Богомольця, кандидатську дисертацію
захистила у 32 роки, докторську — у 46 років.
Крім клінічної і наукової діяльності, займається викладацькою діяльністю (курс гемато-
логії у Військовій медичній академії). Також надає консультації у Центральному госпіталі
МВС, Військово-медичному управлінні СБУ, Центральному клінічному військовому госпіталі,
лікарні для вчених АМН України.
Наукові напрямки відділення захворювань системи крові, очолюваного Н. М. Третяк, сто-
суються вивчення патогенезу, особливостей клінічного перебігу, діагностики та опрацюван-
ня оптимальних програм лікування хронічних та гострих мієло - та лімфопроліферативних
захворювань, анемій. Н. М. Третяк має п'ять патентів на винаходи, автор 212 наукових пуб-
лікацій у вітчизняних і зарубіжних фахових виданнях, 6 монографій, підручників і навчаль-
них посібників. Зокрема: "Гематологія" (підручник); "Медикаментознеє лечение онкологичес-
ких больньїх" (наук, монографія); "Цитостатическая терапия злокачественньїх заболеваний"
(наук, монографія); "Онкология" (довідник).
* * *
Нагороджена орденом Дружби народів (за участь у ліквідації наслідків аварії на ЧАЕС),
знаком "Відмінник охорони здоров'я", медалями "1500 років Києва", Ветеран праці".