/






























































































































Текст
А. П. ГРИГОРЬЕВ, О. Я. ФЕДОТОВА
ЛАБОРАТОРНЫЙ ПРДКТИКУМ
ПО ТЕХНОЛОГИИ ПЛАСТИЧЕСКИХ МАСС
В ДВУХ ЧАСТЯХ
под РЕДАКЦИЕЙ
АКАД. В. В. КОРШАКА
ИЗДАНИЕ ВТОРОЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Допущено Министерством высшего и среднего специального
образования СССР в квчестве учебного пособия для
студентов химико-технологических специальностей вузов
А. П. ГРИГОРЬЕВ, О. Я. ФЕДОТОВА
ЛАБОРАТОРНЫЙ ПРАКТИКУМ
М ТЕХНОЛОГИИ ПЛАСТИЧЕСКИХ МАСС
ЧАСТЬ I
П0ЛИМЕРИЗДЦИ0ННЫЕ
ПЛАСТИЧЕСКИЕ МАССЫ
МОСКВА
«ВЫСШАЯ ШКОЛА»
1977
6П7.55
Г83
УДК 678 (075)
Рецензент — проф. Г. П. Григорьев (Ленинградский ин-т целлю-
лозно-бумажной промышленности)
Григорьев А. П. и Федотова О. Я.
Г83 Лабораторный практикум по технологии пластических
масс. В двух частях. Ч. I. Полимеризационные пласти-
ческие массы. Учеб, пособие для химико-технол. вузов.
Изд. 2-е, перераб. и доп. М., «Высш, школа», 1977.
248 с. с нл.
Первая часть практикума содержит описания работ по полимеризационным
высокомолекулярным соединениям н пластмассам иа их основе. Каждому раз-
делу предпослано теоретическое введение. При описании синтезов приводятся
методы анализа исходных веществ. В новое издание внесены существенные
дополнения, вызванные появлением новых перспективных полимеров и новых
методов анализа.
Г
20505—241
001(01)—77
50—77
6П7.55
© Издательство «Высшая школа», 1977 г.
ПРЕДИСЛОВИЕ
В пятилетних планах развития народного хозяйства постоянно уделяет-
ся особое внимание увеличению производства полимеров и пластических
масс на их основе. На XXV съезде КПСС принято решение о дальней-
шем увеличении производства пластмасс и доведении их выпуска к
1980 г. до 5396—5964 тыс. тонн.
Такие грандиозные масштабы развития отрасли потребуют интен-
сификации научных исследований в этой области, а также подготовки
большого числа квалифицированных инженерно-технических и научных
работников.
Важнейшим звеном учебного процесса являются лабораторные за-
нятия, в значительной степени определяющие теоретическую и практи-
ческую подготовку будущего специалиста.
Настоящее учебное пособие объединяет в nepepa6offaHHOM и допол-
ненном виде два изданных ранее учебных пособия: А. П. Григорьев.
Практикум по технологии полимеризационных пластических масс. М.,
«Высшая школа», 1964, и А. П. Григорьев и О. Я. Федотова. Лаборатор-
ный практикум по технологии поликонденсационных пластических масс.
М., «Высшая школа», 1971.
Пособие состоит из двух частей: ч. I. Лабораторный практикум, по
технологии полимеризационных пластических масс и ч. II. Лаборатор-
ный практикум по технологии поликонденсационных и химически моди-
фицированных пластических масс. Содержание пособия увязано с учеб-
ником «Технология пластических масс» под ред. Коршака В. В. М., «Хи-
мия», 1976.
В руководстве собраны описания многочисленных' лабораторных
работ, достаточно полно охватывающих основные разделы химической
технологии полимеров и пластических масс на их основе. Первая часть
пособия посвящена полимерам полимеризационного типа и пластиче-
ским массам на их основе, вторая — поликонденсационным и химически
модифицированным полимерам и пластическим массам на их основе.
При описании каждого синтеза приводятся методы анализа исход-
ных мономеров, а в некоторых случаях методы их синтеза; даются под-
робные указания по проведению работы и по технике безопасности.
Работы не останавливаются на стадии химического синтеза полиме-
ра или олигомера, а продолжаются до получения изделий, подвергаю-
щихся механическим и иным испытаниям. Методики проведения испыта-
ний и свойства ряда основных промышленных полимеров приведены во
второй части учебного пособия.
Для изготовления многих пластических масс, пленочных материалов
широко используют различные пластификаторы (эфиры фталевой, себа-
циновой и других карбоновых и дикарбоновых кислот), стабилизаторы
(стеараты кальция, кадмия), которые также производят на заводах пла-
стических масс. Поэтому в практикуме приведены работы по получению
некоторых из перечисленных веществ.
5
Наряду с известными пластическими массами в пособии уделено
внимание перспективным новым классам полимеров, а также новым
представителям уже известных классов.
Лабораторный практикум предназначается в качестве учебного по-
собия для студентов химико-технологических вузов. Он может быть ис-
пользован студентами техникумов соответствующих специальностей, а
также работниками научно-исследовательских учреждений и централь-
ных заводских лабораторий.
Автор выражает глубокую признательность сотрудникам кафедры
химической технологии пластических масс МХТИ им. Д. И. Менделеева
К. П. Каменской, В. А. Быкову, И. А. Донецкому и А. В. Трезвову за
помощь, оказанную при написании учебного пособия.
Автор
ВВЕДЕНИЕ
Основой разнообразных пластических масс, имеющих огромное зна-
чение для развития народного хозяйства, новой техники, производства
предметов народного потребления, являются высокомолекулярные соеди-
нения (полимеры). Все высокомолекулярные соединения разделяются
на полимеризационные и поликонденсационные по типу реакций, лежа-
щих в основе их синтеза. Большое значение имеет и третий способ полу-
чения полимеров — химические превращения (модификация) олигоме-
ров и полимеров. Если в первых двух случаях исходными являются низ-
комолекулярные вещества, то третий способ основан на использовании
полимеров в качестве исходных веществ для синтеза новых типов этих
соединений.
Мономеры, имеющие ненасыщенный характер или структуру не-
устойчивого цикла, полимеризуются и сополимеризуются с образованием
высокомолекулярных соединений полимеризационного типа.
В этом случае полимеризация протекает с раскрытием двойной (трой-
ной) связи или неустойчивого цикла и образованием полимера, элемент-
ный состав которого не отличается от элементного состава мономера.
Полимеры поликонденсационного типа получаются в ре-
зультате поликонденсации мономеров за счет взаимодействия содержа-
щихся в них функциональных групп или атомов, причем образование
полимера сопровождается выделением низкомолекулярного продукта и
элементный состав полимера не совпадает с элементным составом исход-
ных веществ. Наряду с мономерами для синтеза полимеров широко ис-
пользуют олигомеры.
Химические превращения полимеров, приводящие к
образованию новых соединений этого класса, могут протекать как без
существенного изменения молекулярного веса и химического строения
основной цепи только за счет замены активных атомов основной цепи
или химического изменения боковых функциональных групп (химическая
модификация), так и с изменением молекулярного веса и химического
строения. В первом случае это реакция полимераналоговых превращений
и прививочная сополимеризация, во втором — блок-сополимеризация.
К высокомолекулярным соединениям полимеризационного типа от-
носятся полиэтилен, полипропилен, поливинилхлорид и др. К поликон-
денсационным — полиэфиры, полиамиды, полиметиленфенолы, полиме-
тиленмочевины и др.
Реакциями полимераналоговых превращений получают поливинило-
вый спирт, поливинилацетали, большой ассортимент ионообменных по-
лимеров на основе сополимеров стирола и дивинилбензола, производные
целлюлозы.
Прививочная сополимеризация лежит в основе способов получения
ряда важнейших полимерных материалов, таких, как ударопрочный по-
листирол, одним из способов получения которого является прививка сти-
7
рола на бутадиенстирольный каучук, и др. Прививочной сополимериза-
цией, сопровождающейся структурированием, получают обширный
ассортимент полиэфирстирольных связующих для изготовления стекло-
пластиков.
При реакции двух или более олигомеров различного строения полу-
чают блок-сополимер с новыми свойствами. Блок-сополимеризация дает
возможность из двух полимеров, например сополимера стирола с нитри-
лом акриловой кислоты я бутадиеннитрильного каучука, получить новый
полимер со специфическими свойствами — ударопрочный полистирол.
В зависимости от поведения полимеров при повышенных температу-
рах они разделяются на термопластичные и термореактивные. Термопла-
стичные полимеры как полимеризационного, так и поликонденсационно-
го типа способны к пластическому течению при повышенной температуре.
При охлаждении они затвердевают, сохраняя заданную форму. Такой
цикл превращений может повторяться неоднократно. Термореактивные
полимеры, также обоих типов, имеют стадию пластического течения при
повышенной температуре, при этом протекает структурирование (от-
верждение) и они переходят в неплавкое состояние. Эти твердые неплав-
кие и нерастворимые материалы не способны к повторному формованию.
Технология переработки полимеров в изделия базируется на этой клас-
сификации, которая определяет выбор метода переработки.
Пластические массы, изготовляемые на основе полимеров, имеют
более или менее сложный состав. Они могут быть однофазными (гомо-
генными). Это простые композиции, состоящие в основном из полимера
с добавкой небольших количеств стабилизатора, красителя и т. д. Более
сложные пластические массы — композиционные, состоящие из полиме-
ра и значительных количеств пластификаторов, наполнителей, стабили-
заторов, красителей. Они могут быть гомогенными и гетерогенными.
Количество пластификатора иногда может достигать 50—70 вес. ч. на
100 вес. ч. полимера (изделия из поливинилхлорида), а наполнителей —
50—100 вес. ч. на 100 вес. ч. полимера (изделия из сложных композиций
феноло-формальдегидных, амидо-формальдегидных олигомеров, поли-
изобутилена и др.). В этих случаях пластификаторы и наполнители из-
меняют технологические и эксплуатационные свойства пластических масс
настолько, что их уже нельзя назвать вспомогательными материалами.
В настоящее время число описанных в литературе полимеров, на
основе которых можно изготовлять пластические массы, волокна, клеи,
пленки, достигает нескольких сотен тысяч. Однако промышленное произ-
водство полимеров и пластических масс на их основе ограничивается
относительно небольшим ассортиментом. Это объясняется многими при-
чинами: комплексом наиболее ценных свойств, доступностью сырья, эко-
номическими преимуществами и др.
За последнее десятилетие наблюдается наибольший рост производ-
ства полиолефинов, полистирола, поливинилхлорида. Медленнее разви-
вается производство поликонденсационных материалов, таких, как фе-
нопласты, карбамидные полимеры и др.
Ведущее положение термопластичных полимеров: полиолефинов,
поливинилхлорида, полистирола и продуктов их модификации — сохра-
нится еще на долгие годы. Это объясняется тем, что комплекс свойств,
8
присущий полиолефинам, полистиролу и поливинилхлориду, а также де-
шевизна и доступность сырья определяют возможность их широкого
применения и такого многообразия изделий, какого невозможно достиг-
нуть в других случаях.
Возможности дешевой сырьевой базы для названных полимеров не-
ограниченны. Это разнообразное углеводородное нефтегазовое сырье, ко-
торое с возрастанием числа и мощности нефтеперерабатывающих й неф-
техимических заводов является стабильным -и надежным сырьем для
производства полимерных материалов.
Новые предприятия, предназначенные для производства пластиче-
ских масс на базе нефтяного сырья, проектируются и строятся в составе
комплексных нефтехимических комбинатов (удаленных от мест добычи
нефти и получающих ее по нефтепроводам), в которых предусматривают-
ся заводы по производству и очистке большого числа мономеров. На
этих же заводах из нефтяного сырья получают и другие органические
продукты, применяемые и синтезе мономеров.
Области применения пластических масс из года в год расширяются.
Главными потребителями являются автомобильная, авиационная, элект-
ро- и радиотехническая, строительная, судостроительная промышленно-
сти, железнодорожный транспорт, машиностроение, легкая и пищевая
промышленность, медицина, сельское хозяйство и многие другие.
Теоретические основы химической технологии
пластических масс
Теоретической основой технологии пластических масс является
прежде всего химия высокомолекулярных соединений, рассматривающая
механизмы и кинетику образования полимеров, а также методы их по-
лучения.
Физические и физико-механические свойства полимеров, способ пе-
реработки и применение пластических масс на их основе в значительной
степени определяются структурой макромолекул.
Вне зависимости от типа реакций образования полимеров (полиме-
ризация, поликонденсация) можно получать макромолекулы различного
строения: линейного, разветвленного, сетчатого и трехмерного. Форма
молекул предопределяется химическим строением исходных веществ.
Бифункциональные мономеры или их смеси обычно дают полимеры ли-
нейного или разветвленного строения. Основными факторами, влияющи-
ми на свойства таких полимеров, являются длина и гибкость цепи. Гиб-
кость цепи в свою очередь определяется химическим строением макро-
молекул, интенсивностью межмолекулярного взаимодействия,
надмолекулярной структурой полимера. При повышении хотя бы в одном
из мономеров числа реагирующих групп или атомов образуются полиме-
ры сетчатого или пространственного строения.
В зависимости от молекулярного веса полимеры одного и того же
химического строения имеют различную прочность. При увеличении мо-
лекулярного веса прочность растет, так как усиливается межмолекуляр-
ное взаимодействие, величина которого достигает определенного значе-
ния, после чего возрастание прочности прекращается.
• 9 ''
Высокомолекулярные соединения сетчатой структуры образуются
в результате полимеризации или поликонденсации полифункциональных
мономеров, олигомеров или смесей бифункциональных мономеров с по-
лифункциональными (мостикообразующими). Как правило, увеличение
длины «сшивающих мостиков» и размеров заместителей, расположенных
в боковых ответвлениях (до известного предела) приводит к повышению
деформируемости и, соответственно, снижению хрупкости продукта.
Полимеры сетчатого или трехмерного строения отличаются от линей-
ных полимеров, полученных из подобных по химическому строению мо-
номеров, по теплофизическим и физико-механическим свойствам. Они
обладают повышенной устойчивостью к термическим, химическим и меха-
ническим воздействиям и в зависимости от количества «сшивающих мо-
стиков» не растворяются в растворителях или набухают в них в большей
или меньшей степени. Поэтому такие полимеры наиболее пригодны для
использования в качестве поверхностных покрытий, изготовления склеи-
вающих материалов, для переработки прессованием и т. д.
Основными типами полимеризационных процессов являются цепная
и ступенчатая полимеризации.
В качестве исходных продуктов для получения полимеров использу-
ются низкомолекулярные вещества (мономеры), имеющие не менее двух
реакционноспособных групп (двойные связи, активные атомы водорода,
неустойчивые циклы и т. п.).
Цепная полимеризация
Одним из наиболее распространенных методов синтеза полимеров
является цепная полимеризация. По цепному механизму могут полиме-
ризоваться соединения, содержащие в молекуле одну, две и более крат-
ных связей.
В результате полимеризации соединений с С = С-связями, например
нёпредельных углеводородов, образуются карбоцепные полимеры. Поли-
меризация соединений со связями углерод — гетероатом (кислород, азот,
сера) приводит к образованию гетероцепных полимеров.
Молекулярный вес полимеров, получающихся при цепной полимери-
зации, обычно очень велик и может достигать нескольких тысяч и даже
миллионов. Полимеризация протекает с разрывом одних связей и обра-
зованием других. В зависимости от условий реакции (характера воздей-
ствия), природы мономера (полярный, поляризующийся) может проис-
ходить гетеролитический и гомолитический разрыв л-связей. В результа-
те гетеролитического разрыва образуются ионы, в результате
гомолитического — свободные радикалы.
В соответствии с характером активных центров различают ради-
кальную и ионную полимеризацию.
Радикальная полимеризация. При радикальной полимеризации ак-
тивным центром является свободный радикал, возникший тем или иным
способом: из молекулы мономера под влиянием какого-либо вида энер-
гии или при распаде специально введенного инициатора. Свободный ра-
дикал вступает в реакцию с различными мономерами, имеющими нена-
сыщенные связи, разобщает л-электроны: притягивает один из них, при
10
этом образуется пара (о-связь), другой, с противоположным спином,
остается неспаренным на внешней орбите второго углеродного атома и
определяет свободнорадикальную структуру образовавшегося соедине-
ния:
R 4- СН2=СН — RCH2—СН
Новый радикал взаимодействует с молекулой мономера по тому же
механизму:
RGH2-CH + СН2=СН —> RCH2-CH-CH2-CH
II II
R R R R
Эта стадия называется ростом цепи, причем растущая цепь остается
свободным радикалом, пока не произойдет ее обрыв в результате взаи-
модействия двух радикалов (рекомбинационный обрыв), диспропорцио-
нирования или реакции передачи цепи, которая протекает обычно при
взаимодействии активных центров или растущих макрорадикалов с
мономером, примесями, растворителем и т. п.
Так как образование свободного радикала протекает эндотермиче-
ски, молекуле мономера должно быть сообщено значительное количе-
ство энергии (тепловой, световой или какой-либо другой). Соответствен-
но, по способу инициирования различают термическую, фотохимическую
и радиационную полимеризацию. Инициирование мономера при воздей-
ствии свободного радикала, образовавшегося в результате распада ини-
циатора (вещества, дающего при распаде свободные радикалы), назы-
вают химическим инициированием, а полимеризацию — инициированной.
В производственных условиях применяются главным образом терми-
ческий и инициированный вид полимеризации. В качестве инициаторов
используются различные органические и неорганические перекиси, гидро-
перекиси, некоторые азо- и диазосоединения, которые легко распадаются
на свободные радикалы при повышении температуры. Количество при-
меняемого при полимеризации инициатора обычно невелико и колеблет-
ся в пределах от 0,1 до 1 % от массы мономера.
Для ускорения распада инициатора широко используют добавки
активаторов, снижающих энергию распада в результате протекания
окислительно-восстановительных реакций. Энергия активации иницииро-
вания снижается в последнем случае до 33,6 кДж/моль (8 ккал/моль),
что позволяет проводить полимеризацию при невысоких температурах
(20—30° С).
Как и всякий цепной процесс, инициированная полимеризация (в
мономере) протекает через три основные стадии:
1) инициирование
H->2R
скорость которого va определяется уравнением
®И=2АЛ< [И],
где kB— константа скорости распада инициатора; FB — коэффициент
эффективности инициатора, введение которого вызвано тем, что часть
И
радикалов, возникающих при распаде инициатора, рекомбинируется и,
следовательно, не участвует в реакции инициирования;
2) рост цепи
R-J-M — RM
RM 4- «М — RMn+1
скорость которой vv описывается уравнением
3) обрыв цепи реакцией рекомбинации
RM„+1-J-RMn-RM2n+IR
скорость которой
®o=MRF-
Общая скорость реакции определяется наиболее медленной стади-
ей — инициированием — и может быть рассчитана исходя из частных
уравнений, характеризующих основные стадии полимеризации:
Ч>6Щ=£[МПИ]°Л
Средняя степень полимеризации равна P=vp/vo, а следовательно,
Р=[М]МИ].
Наряду с этими основными реакциями в зависимости от условий и
природы мономера, инициатора, наличия растворителя и других добавок
может иметь место передача цепи на мономер, полимер, растворитель,
инициатор и т. п. В процессе передачи цепи материальная цепь обры-
вается (ее рост прекращается), а кинетическая продолжает сущест-
вовать:
RM„M+АВ — RM„MA 4- В
поэтому можно написать
Р = ^р/^пер'
В этом случае реакция передачи цепи может быть приравнена к реакции
обрыва цепи. От реакционной способности свободного радикала В* за-
висит, будет ли продолжаться кинетическая цепь. Если свободный ради-
кал В» способен при взаимодействии с мономером образовывать актив-
ный центр, то он дает начало новой материальной и кинетической цепи.
В противном случае В "может лишь рекомбинироваться (В"+В'-*-ВВ) и
тогда реакция растущей цепи с АВ будет, актом ингибирования и про-
изойдет обрыв цепи. (Поэтому ингибирование возникающих активных
центров применяют для предотвращения спонтанной полимеризации мо-
номеров при хранении.)
Передача цепи на полимер используется в случае прививочной по-
лимеризации и образования привитых сополимеров (см. ч. II, стр. 208).
Ионная полимеризация протекает в присутствии катализаторов, ко-
торые в отличие от инициаторов не расходуются в процессе полимериза-
ции и входят лишь в непрочные соединения, регенерируясь в конце ре-
акции.
12
к
В отличие от радикальной полимеризации, протекающей путем пе-
редачи по цепи неспаренного электрона, ионная полимеризация идет с
образованием при гетеролитическом разрыве связи положительного или
отрицательного заряда и сопровождается передачей его по цепи. В зави-
симости от характера катализатора и в соответствии с зарядом образую-
щегося иона различают катионную и анионную полимеризацию.
Катионную полимеризацию вызывают катализаторы Фриделя_________
Крафтса (апротонные кислоты А1С13, FeCl3, BF3, TiCl4 и др.) и кислоты
(серная, солдная и др.). Сокатализаторами являются вода и другие со-
единения, дающие протон. Катионы, инициирующие полимеризацию, об-
разуются в результате диссоциации комплексов — из апротонной кисло-
ты и сокатализатора. Таким образом, собственно катализатором являет-
ся комплекс, а не апротонная кислота.
Анионную полимеризацию вызывают катализаторы другого типа:
щелочные металлы, металлорганические соединения, окислы поливалент-
ных металлов.
Сущность действия катализаторов заключается в снижении энергии
активации процесса полимеризации.
Характерная особенность ионной полимеризации — очень высокая
скорость реакции в связи с низкой энергией ее активации. В некоторых
•случаях процесс ионной полимеризации протекает при низкой темпера-
туре (от —50 до —130° С).
В зависимости от природы растворителя, в частности его электро-
нодонорной или электроноакцепторной способности, ионная полимериза-
ция может протекать с участием свободных ионов или ионных пар, т. е.
по чисто ионному или ионно-координационному механизму. В первом
случае растворитель, сольватируя противоион, например положительный
в анионном процессе, освобождает карбанион, реагирующий свободно с
молекулой мономера, поляризуя ее таким образом, что образуется одна
ковалентная связь и регенерируется отрицательный заряд на конце ра-
стущей цепи.
Подбор растворителей для осуществлений ионной полимеризации
основан не только на учете их нуклеофильных и электрофильных свойств,
но и на диэлектрической проницаемости, сольватирующей способности
[3]. Природа растворителя в значительной степени определяет тонкий ме-
ханизм реакции, порядок реакции, а также строение макромолекулы.
В некоторых случаях растворитель может быть сокатализатором. Осо-
бое значение приобретает природа растворителя в процессе ионно-коор-
динационной полимеризации в присутствии катализаторов Циглера —
Натта, применяющихся для синтеза стереорегулярных полимеров изо-
тактического полипропилена, изотактического полистирола и др. Синтез
стереорегулярных полимеров успешно осуществляется в присутствии
таких катализаторов, как смеси металлорганцческих соединений (три-
алкилалюминия) с хлоридами металлов переменной валентности (TiCl3
и TiCU), т. е. элементов с незаполненной промежуточной электронной
оболочкой. Реакция протекает по анионно-координационному механизму.
Применение катализаторов дало возможность получить полиолефи-
ны и полидиены с ценными технологическими свойствами. В настоящее
•время описано много каталитических систем, определяющих ионно-ко-
13
/
ординационный механизм полимеризации, однако в промышленное1/! ис-
пользуют главным образом хлориды титана и алкилы алюминия./'
Полимеризация олефинов в присутствии катализаторов Циг/iepa —
Натта TiCl4 и А1(С2Н5)3 протекает по следующей схеме: переход TiCl4
в TiCl3 и TiCl2 облегчает хемосорбцию органических соединений сильно
, электроположительных металлов, таких, как Al, Mg, Be, обладающих
малыми ионными радиусами. Хемосорбция приводит к образованию
электрононедостаточных комплексов титана и другого металла:
TiCl3+А1(С2Н5)3 —Х>а/СН2СН’
° ^'Cllf' 'СН2СН3
Анз
Молекула мономера образует с таким комплексом л-комплекс, образова-
ние которого приводит к диссоциации связи Ti---CH2CH3 (I). После чего
происходит образование нового комплекса с участием мономера (II):
П
Образовавшийся комплекс стабилизируется, переходит в форму,
способную к образованию л-комплекса с новой молекулой мономера и
повторению описанного цикла превращений, приводящих к росту цепи:
)Ti<C x>ai\
сн2
снксн2сн3
Кинетика катионной и анионной полимеризации очень сходна. Ско-
рость образования активного центра с положительным зарядом иона
14
карбрния определяется следующими уравнениями:
' йакт
Кат 4-М Кат М+ (I)
®акт=*акг [М1 [Кат+],
где [Кат+]— концентрация Йонов карбония или протонов по схеме I.
Скорость роста цепи
\ Кат М+4-М —Кат ММ+
описывается уравнением
®Р=^Р[М][Кат М+],
где [Кат+] — концентрация растущих ионов карбония.
Для ионных процессов характерен мономолекулярный обрыв
Кат М+ -4-Кат М„
•скорость которого описывается уравнением и0=МКат М+]. Средний ко-
эффициент полимеризации образующегося полимера
— Vp vp
vo vaKt
или после преобразования
где k'=kplk0, а суммарная скорость полимеризации
®=£[Кат+ЦМ]2.
Совершенно аналогичные зависимости имеют место при анионной
полимеризации
Р=Л'[М]; -г?=/г[А-][М]2.
Сополимеризация. Совместная полимеризация двух или трех мономе-
ров является важнейшим методом синтеза многих полимеров. Этим ме-
тодом удается получать полимеры с новыми свойствами, изменять свой-
ства полимеров в желаемом направлении: увеличивать или уменьшать
эластичность, повышать или понижать температуру размягчения, изме-
нять термостойкость, растворимость и т. п. Однако свойства сополиме-
ров не являются простой суммой свойств полимеров, образуемых каж-
дым из данных мономеров в отдельности, так как изменение свойств,
вызываемое введением в полимер звеньев второго компонента, непропор-
ционально его количеству.
Метод сополимеризации позволяет также расширить круг мономе-
ров, так как значительное количество соединений, не способных полиме-
ризоваться, легко сополимеризуется с другими веществами. В качестве
примера могут быть названы стильбен, двуокись серы, простые винило-
вые эфиры (по радикальному механизму), диамины, гликоли и др.
Не все соединения, полимеризующиеся отдельно, могут образовы-
вать сополимеры. Так, при полимеризации стирола и винилацетата/обра-
зуется только полистирол, а полимеризация винилацетата задерживает-
ся. Некоторые мономеры полимеризуются совместно друг с другйм толь-
ко в определенных условиях. Например, бутадиен и акриловые /фиры не
образуют сополимеров при блочной сополимеризации, а в эмульсии сопо-
лимер легко получается. /
В случае отсутствия способности двух мономеров к сополимериза-
ции вводят третий компонент, например сополимеризаций стирола с
винилацетатом протекает легко в присутствии метилметакрилата. В этом
случае рост полимерной цепи происходит таким образом/что звенья не-
сополимеризующихся мономеров разделены третьим компонентом.
В присутствии ацетилацетоната марганца винилацетат вступает в
сополимеризацию со стиролом, при этом мономеры в- сополимере регу-
лярно чередуются.
Процесс совместной полимеризации двух и более мономеров, как и
полимеризация одного мономера, .зависит от реакционной способности
мономеров, определяющейся строением мономеров, полярностью их мо-
лекул, а также от способа инициирования. Следует отметить, что при
сополимеризации влияние полярности имеет более сложный характер.
Это определяется прежде всего взаимным влиянием мономеров в соот-
ветствии с направлением и характером поляризации каждого из них.
Состав сополимеров, т. е. количественное соотношение основных звеньев
в сополимере, как правило, не равно количественному соотношению мо-
номеров в исходной смеси.
Состав сополимера определяется реакционными способностями мо-
номеров и радикалов или ионов, образующихся при присоединении моле-
кул мономеров к растущим полимерным молекулам. При гомополимери-
зации соотношение активностей мономеров и концевых звеньев растущих
цепей остается всегда постоянным.
В ходе сополимеризации происходит постоянная смена одного моно-
мера или радикала другими, имеющими различную активность по отно-
шению друг к другу. Для характеристики процесса сополимеризации на-
чиная от возможности его осуществления до получения сополимера оп-
ределенного состава обычно измеряют так называемые константы
сополимеризации и и г2 (для бинарных систем). Эти константы называ-
ют оптимальными активностями. Они показывают отношение констант
скорости реакций присоединения радикала (иона) к «своему» и «чужо-
му» мономерам.
Дифференциальное уравнение состава сополимера можно написать
так:
d[M,] [MJ г,[М,] + [М2]
d[M2] [М2] ’ Г2[М2] + [М!] ’
где rf[Mi] и d[M2] — количество мономеров Mj и М2, расходуемых на об-
разевание сополимера. Следовательно, соотношение ~ .... , соответствует
d [м21
молекулярному соотношению основных звеньев в сополимере. Для ма-
16
лых Степеней превращения уравнение упрощается:
\ т\ _ [Mi] п [Mi] + [М2]
\ т2 [М2] гг[М2] + [М]]
где гп\ и щ2— содержание основных звеньев мономеров М! и М2.
Определение констант сополимеризации имеет исключительное прак-
тическое значение, так как оно предопределяет состав сополимера,
а следовательно, и заранее заданные свойства сополимеров и, соот-
ветственно, способы их переработки и применения. В табл. 1 при-
ведены значения Г\ и г% для ряда технически ценных мономеров.
При сополимеризации
двух мономеров возможны ТАБЛИЦА I
следующие варианты соот-
ношений Г\ и г$.
Константы сополимеризации некоторых мономеров
а) /1<1, г2>1; это зна- чит, что радикалы Mi и М2 будут легче реагировать с м, м. ft г.
мономером М2, что приводит к обогащению сополимера Стирол Акрилонитрил Бутадиен » 0,78 0,05 1,39 0,35
звеньями /Пг; Винилхлорид Винилацетат 1,68 0,23
б) Г]>1, г2<1; сополи- Стирол Метилметакрилат 0,52 0,46
мер обогащается звеньями Винилиденхлорид Акрилонитрил Винилхлорид Стирол 4,50 0,05 0,20 0,40
в) /1<1, /г<1; радикал » Винилхлорид 3,30 0,04
Метилметакрилат Метакрилонитрил 0,67 0,65
М 1 легче реагирует с моно-
мером М2, а радикал Mi —
с мономером Mi;
г) Г1=Г2=1; радикалы Mi и М2 одинаково легко реагируют с обои-
ми мономерами и состав сополимера соответствует составу исходной
смеси мономеров, что наблюдается крайне редко, это так называемые
азеотропные смеси;
д) Г1>1 и г2>1; радикалы легче реагируют со своими мономерами,
чем с чужими, и сополимер не образуется;
е) г 1/2=0; при сополимеризации двух мономеров, один из которых
не способен к гомополимеризации и константа сополимеризации которо-
го равна нулю, произведение Г]Г2 также равно нулю; в этом случае обра-
зующийся сополимер имеет наиболее регулярную структуру М4М2М1М2,
и чем ближе значение Г\Г% к нулю, тем более упорядочена структура об-
разующегося сополимера.
Важным фактором при сополимеризации является полярность двойт
ной связи мономеров. В связи с тем .что полярность двойной связи зави-
сит от природы заместителя (его нуклеофильности или электрофильно-
сти), установлено, что чем больше различаются мономеры по знаку по-
лярности, тем легче они сополимеризуются. На этом основании Алфрей
и Прайс разработали полуколичественную схему Q — е. Здесь Q —
удельная активность мономера, характеризующая резонансную стабили-
зацию, а е — величина, характеризующая полярность молекул мономе-
ров и образующихся из них радикалов, при этом делается упрощающее
расчеты допущение, что значение е для мономеров и радикалов одина-
17
ково. Для многих веществ Q и е определены, причем в качестве /стан-
дартного вещества взят стирол, для которого значение Q = 1 взятЬ про-
извольно, а е=—0,8 (см. табл. 2).
По схеме Алфрея и Прайса значения а и Гг определяются 'уравне-
ниями
n=-^-exp[-ei (eie2)];
<?2
- Г2=~ехр[ — е2(е2 — е1')]. /
Qi
Расчеты Q и е по этим уравнениям полностью подтверждают выска-
занные ранее закономерности сополимеризации, связанные с актив-
«остыо мономеров и полученных из них радикалов.
ТАБЛИЦА 2 Значения Q и е для некоторых мономеров Следует подчеркнуть, что актив- ность мономеров при ионной полиме- ризации резко отличается от их ак- тивности при радикальной сополиме-
Мономер Q е ризации ,в связи с различной способ- ностью мономеров поляризоваться под, влиянием анионных и катионных воз- "
Винилхлорид Вннилиденхлорид Акрилонитрил Метилметакрилат п-Хлорстирол N-Винилпиридин 0,02 0,20 0,44 0,74 0,88 1,07 +0,2 +0,6 + 1,2 +0,4 -0,3 -0,6 будителей полимеризации. Так, напри-’ мер, если стирол с почти одинаковым успехом образует карбкатион и карба- нион, то метилметакрилат, винилхло- рид, винилацетат и другие образуют только карбанион.
Ступенчатая полимеризация
и сополимеризация
При ступенчатой полимеризации реакция присоединения молекул
мономеров протекает постепенно и носит ступенчатый характер. Это
означает, что нарастание молекулярного веса происходит также посте-
пенно, причем процесс может быть прерван на любой стадии. Исходны-
ми веществами могут быть как ненасыщенные мономеры, так и цикли-
ческие. Одним из важных примеров ступенчатой полимеризации являет-
ся сополимеризация диизоцианатов с гликолями (полиуретаны), с
диаминами (поликарбамиды), диангидридов тетракарбоновых кислот с
диаминами (полиамидокислоты), окисей олефинов с дифенолами, диами-
нами, дикарбоновыми кислотами и т. д.
Так как сополимеризация протекает через миграцию атома водоро-
да от одного исходного соединения к другому, ее называют миграцион-
ной сополимеризацией или полиприсоединением в отличие от цепной по-
лимеризации, где перемещения атомов не происходит. В этих реакциях
участвуют два компонента, реагирующие с участием функциональных
групп, поэтому большое влияние на молекулярный вес полимера оказы-
вает их соотношение. Наибольший молекулярный вес получается при
эквимолекулярном соотношении исходных веществ. Избыток одного из
компонентов приводит к блокировке растущей молекулы полимера оди-
18
наковыми концевыми группами, что, естественно, вызывает прекращение
роста макромолекулы.
Аналогичное влияние на прекращение роста цепи оказывают добав-
ки низкомолекулярных монофункциональных веществ, таких, как спир-
ты, изоцианаты, амины и др.
Полимеризация гетероциклических соединений протекает также по
ступенчатому механизму под влиянием активаторов реакции, вызываю-
щих гидролиз гетеросвязи, предшествующий миграционной полимериза-
ции, или под влиянием катализаторов, вызывающих полимеризацию по
ионному механизму (см. стр. 211).
Способы проведения полимеризации
Радикальная полимеризация может быть осуществлена
различными способами: в среде мономера (в блоке), в водной среде —
эмульсии, суспензии, в газовой и твердой фазах, а также в растворе.
Ионную полимеризацию чаще всего проводят в мономере, растворителе
или в суспензии в органической среде.
Полимеризация в блоке протекает в условиях, когда реак-
ционной средой служит сам мономер. Блок образуется в случае раство-
римости полимера в мономере.
Полимеризация в мономере, приводящая к образованию
полимера, нерастворимого в мономере, протекает практически в диспер-
сии полимера в мономере. При инициированной полимеризации в моно-
мер вводится инициатор, а также различные добавки (стабилизатор,
пластификатор и др.).
Так как процесс роста цепи — экзотермическая реакция, необходим
отвод тепла', который по мере повышения вязкости реакционной среды и
снижения ее теплопроводности становится все более затруднительным.
В результате в образующемся блоке могут возникнуть пузырьки, вклю-
чения мономера и другие дефекты, что является недостатком этого спо-
соба полимеризации в случае периодического процесса.
Вместе с тем местные перегревы приводят к образованию полимера
с большой полимолекулярностыо (полидисперсностыо по молекулярно-
му весу), что значительно влияет на физико-механические свойства.
Вследствие более высокой температуры и затруднительного отвода тепла
во внутренних слоях блока молекулярный вес ниже, чем во внешних сло-
ях. Соответственно и механические свойства блочного полимера ухудша-
ются от периферии к центру.
Достоинством блочного метода полимеризации по сравнению с су-
спензионным и эмульсионным является получение полимера более высо-
кой степени чистоты, а также возможность изготовления блоков с высо-
кими оптическими свойствами.
Полимеризация в блоке может осуществляться в промышленном
масштабе как периодическим, так и непрерывным методами. При перио-
дическом методе полимеризации полимер приобретает форму реакцион-
ного сосуда, а при непрерывном он принимает форму и размеры отвер-
стия, через которое полимер выпускается из аппарата (ленты, стержни
и т. п.).
19
При полимеризации в среде мономера, не растворяющего полймер,
'последний выпадает из мономера, в виде либо порошка, либо кодокооб-
разной массы. /
Наиболее распространенный метод получения полимеров в Промыш-
ленных условиях — полимеризация в водных суспензиях и эмульсиях.
Суспензионная (капельная) гранульная йолиме-
,р и з а ц и я протекает в двухфазной системе мономер —вода/ однако по
механизму процесс является разновидностью блочной полимеризации.
Мономер диспергируется энергичным перемешиванием мещалкой в объ-
еме водной фазы до образования капель диаметром 0,1—1 мм. В водной
фазе присутствуют стабилизаторы. Капельки мономера Обволакиваются
слоем поверхностно-активного вещества — стабилизатора. В качестве
стабилизатора применяют слабые эмульгаторы: поливиниловый спирт,
-крахмал, желатин, метилцеллюлозу, тальк, бентонит, окислы метал-
лов и др.
Дисперсионной средой может быть раствор соли (если мономер рас-
творим в воде), глицерин, гликоли и другие вязкие, обладающие боль-
шой плотностью жидкости. Инициаторы, используемые в суспензионной
полимеризации, как правило, должны растворяться только в мономере.
Полимеризация от начала до конца протекает в капле, как в мини-
атюрном блоке. Отвод тепла осуществляется с помощью дисперсионной
среды. Кинетика реакции аналогична блочному процессу. Большим до-
стоинством этого метода является отсутствие необходимости осаждать
полимер в отличие от эмульсионной полимеризации. Однако образую-
щиеся гранулы необходимо промыть и высушить.
Диэлектрические свойства суспензионного полимера понижены
•вследствие примеси эмульгатора, который трудно полностью отмыть. Ко-
нец реакции суспензионной полимеризации определяют по падению гра-
нул на дно сосуда в связи с увеличением их плотности.
Суспензионные (гранульные) полимеры используют для литья под
давлением и экструзии, а также (например, сополимеры стирола с диви-
нилбензолом) для последующих химических превращений с целью по-
лучения ионитов.
Эмульсионная полимеризация также проходит в водной
среде, но отличается от суспензионной тем, что в реакционную среду
вводят растворимый в воде инициатор, а вместо защитных коллоидов и
нерастворимых в воде порошков применяют поверхностно-активные ве-
щества— сильные эмульгаторы (мыла, сульфокислоты и т. п.). Эмульга-
торы снижают поверхностное натяжение между фазами и способствуют
получению- устойчивой эмульсии в процессе полимеризации, образуют
солюбилизированные растворы мономера. Большая часть мономеров
нерастворима в воде. Растворы мыл в воде имеют мицеллярное
строение.
При наличии мицелл часть молекул мономера диффундирует во
внутреннюю часть мицелл и эту часть можно считать растворенной. Рас-
твор мыла может растворять мономер в некоторых случаях с образова-
нием 7—9%-ного коллоидного раствора. Концентрация раствора повы-
шается с увеличением концентрации мыла. Поэтому скорость реакции
и молекулярный вес образующегося полимера в значительной степени
20
зависят от концентрации мыла в растворе: чем она выше, тем'больше
скорость реакции.
Полимеризация начинается в мицеллах под воздействием свободных
радикалов, образовавшихся в водной среде в результате распада ини-
циатора. По мере увеличения объема образующихся частиц полимера
они перестают удерживаться ,в мицелле и выпадают, унося на поверхно-
сти слой эмульгатора. В такую частицу, имеющую свободный макрора-
дикал, диффундирует мономер из водной фазы, и в ней продолжается
радикальная полимеризация наряду с полимеризацией в мицелле. Обрыв
цепи осуществляется рекомбинацией со свободным радикалом, проник-
шим в полимерно-мономерную частицу.
Средний коэффициент полимеризации полимера, образующегося при
эмульсионной полимеризации, обратно пропорционален скорости обра-
зования радикала из инициатора (п). Так как только половина обра-
зовавшихся радикалов расходуется на инициирование, а вторая поло-
вина— на обрыв цепи, то скорость реакции и средний коэффициент по-
лимеризации равны
’ 2 2/Й ₽
где У— число частиц; Р — скорость образования радикалов из инициа-
тора.
При постоянном числе частиц (это наблюдается в пределах 15—
60%-ной конверсии) средний коэффициент полимеризации остается
постоянным. Увеличение числа частиц влечет за собой повышение моле-
кулярного веса. Увеличение концентрации мономера в частицах приво-
дит к тому же эффекту. Увеличение скорости реакции приводит к увели-
чению Р. Эта закономерность, обусловленная физико-химическими осо-
бенностями процесса эмульсионной полимеризации, и составляет ее
отличительную особенность по сравнению с блочной полимеризацией. При
блочной полимеризации, как известно, наблюдается обратная зависи-
мость.
После достижения 60%-ной конверсии концентрация мономера в ча-
стицах снижается, уменьшается число частиц за счет слипания и, как
следствие этих факторов, начинает снижаться молекулярный вес поли-
мера.
Известный способ активации распада инициаторов путем добавки
восстановителей широко используется для осуществления эмульсионной
полимеризации в промышленном масштабе. Так называемые окисли-
тельно-восстановительные системы состоят из инициатора и восстанови-
теля (перекиси водорода или персульфата калия, соли двухвалентного
железа); например: перекись водорода — диметиланилин;'гидроперекись
изопропилбензола — соль железа и т. д.
В качестве примера можно привести реакции, протекающие в сле-
дующей системе:
(С6Н5СОО)2 + C6H5N (СНзЪ ->'с6Н5СОб + С6НБСОО“ + [C6H5N (СН3)2]+
21
Высокие скорости образования радикалов при низких температурах
определяют большое практическое значение окислительно-восстанови-
тельного инициирования, так как позволяют провести процесс полиме-
ризации с меньшими энергетическими затратами.
Таким образом, достоинствами эмульсионного метода являются: вы-
сокая скорость, хороший отвод тепла, высокий молекулярный вес и воз-
можность получения полимера в виде тонкого порошка.
Недостаток этого метода заключается в загрязнении полимера
коагулянтами и другими примесями, что приводит к ухудшению его
диэлектрических свойств.
Полимеризация в растворителе, как показывает назва-
ние, протекает в среде вещества, растворяющего мономер и полимер. До-
стоинство этого метода — легкость отвода тепла, выделяющегося при ре-
акции, что приводит к получению полимера со сравнительно высокой
однородностью по молекулярному весу. Недостатки этого метода — не-
удобство работы с легко летучими растворителями, необходимость их
рекуперации, а также трудность удаления остатков растворителя из по-
лимера.
Молекулярный вес получающегося полимера в значительной степени
зависит от природы растворителя. Активные растворители (СС14, СНС1а
и т. д.), способные участвовать в процессе полимеризации, могут вызы-
вать преждевременный обрыв цепи вследствие реакции передачи цепи,,
что отрицательно сказывается на молекулярном весе (см. стр. 12).
Полимеризацию в растворителе обычно применяют в тех случаях,
когда необходимо получить лаки, клеи или когда нельзя использовать
другой метод полимеризации, например для прядения из растворов не-
которых видов полимеров.
Разновидностью этого метода является полимеризация в таком ве-
ществе (разбавитель), которое, растворяя мономер, не растворяет поли-
мер. Образующийся полимер в процессе реакции выпадает из раствора
и может быть легко отделен от растворителя. Условия теплообмена в
этом случае такие же благоприятные, как и при использовании раство-
рителя, а выпадение полимера в осадок облегчает его выделение, что яв-
ляется преимуществом этого метода.
Полимеризация в разбавителе применяется в промышленности для
производства полиэтилена высокой плотности, полипропилена и их со-
полимеров.
Изготовление изделий на основе лолимеризационных
пластических масс и образцов для испытания их свойств
Получение полимеров — только первая стадия изготовления техни-
ческих продуктов (изделий), обладающих необходимыми качествами.
С помощью различных добавок—пластификаторов, стабилизаторов,
наполнителей, красителей, отвердителей и других веществ — свойства по-
лимеров могут быть в значительной степени улучшены в соответствии с
требованиями, предъявляемыми различными отраслями техники. Мате-
риалы на основе полимеров, совмещенных с пластификаторами, напол-
нителями, стабилизаторами и т. д., называют пластмассами.
22
Не меньшее влияние, чем добавки, на свойства пластических масс
оказывает (правильный выбор метода и технологического режима пере-
работки полимера с учетом его физических свойств и структурных осо-
бенностей.
Выбор способа переработки определяется поведением полимера под
действием высокой температуры. Термопластичные полимеры
способны под влиянием повышенной температуры размягчаться, пере-
ходить в пластическое состояние, принимать желаемую форму, сохранять
ее при охлаждении и снова размягчаться при нагревании. К ним отно-
сятся полиолефины, их производные, полпакрилаты, полиамиды, поли-
эфиры линейного строения, поликарбамиды и т. д.
Жидкие олигомеры, а также полимеры, размягчающиеся при нагре-
вании и способные при этом необратимо переходить в неплавкое и не-
растворимое состояние, вследствие химических реакций, протекающих
под влиянием высокой температуры или катализаторов, относятся к типу
термореактивн ы х.
Наиболее прогрессивными способами переработки термопластичных
материалов являются литье под давлением, экструзия.
Для термореактивных полимеров широко используются различные
-способы прессования в присутствии разнообразных наполнителей, а так-
же литье под давлением и экструзия на червячных машинах.
Выбор метода переработки определяется также конфигурацией из-
делия и условиями его применения. Температурные условия переработки
полимера устанавливаются на основании данных термомеханических
кривых (кривых зависимости деформации от температуры), индекса
расплава, скорости отверждения.
В процессе переработки пластиков в изделия разнообразными ме-
тодами получают мелкие и крупногабаритные детали различного назна-
чения, профильные изделия, пленочные материалы и т. п. Применение
тех или иных методов переработки определяется физико-химическими
свойствами полимерного материала или композиции на его основе, фор-
мой изделия и областью его использования. Метод переработки должен
обеспечить максимальную реализацию полезных свойств полимера.
Технические детали и предметы широкого потребления из полиме-
>ризационных полимеров изготавливаются главным образом литьем под
давлением, реже прессованием. Профильные изделия технического на-
значения изготовляются экструзией. Пленочные и листовые материалы
можно получать различными методами: комбинированием экструзии с
последующей механической и пневматической вытяжкой, вальцеванием,
каландрованием и т. п. Изделия из листовых и пленочных материалов
обычно изготовляются путем штамповки, механической вытяжки, ваку-
умного и пневматического формования.
Необходимо отметить огромную роль наполнителей в модификации
свойств термопластичных материалов как полимеризационного, так и по-
ликонденсационного типа. Стекловолокно, стекловата, кварцевый песок,
рубленое стекловолокно все чаще используют для изготовления изделии
с повышенными физико-механическими свойствами.
Особое значение имеют газообразные наполнители для образования
поро- и пенопластов.
23
Одним из перспективных методов регулирования структуры поли-
меров является введение в них искусственных центров кристаллизации,,
чаще всего мелкодисперсных соединений тугоплавких металлов (окис-
лов, нитридов и т. д.).
Для оценки физико-механических свойств полимерных материалов
разработано много разнообразных методов испытаний как в статических,,
так и в динамических условиях.
Таким образом, получение конечного продукта с заданными свой-
ствами определяется всеми стадиями процесса, начиная с синтеза моно-
мера и кончая изготовлением изделий и их испытанием.
Интересным свойством многих полимеров, таких как полиэтилен,,
полиизобутилен, полистирол, полиметилметакрилат, а также многие со-
полимеры этих мономеров, является способность деполимеризоваться,,
превращаясь частично или полностью в мономеры. Некоторые из них
после очистки вновь способны к полимеризации с образованием полно-
ценных продуктов. Это позволяет использовать отходы, получающиеся
при переработке.
Другие полимеры, например поливинилацетат, поливинилхлорид, по-
ливиниловый спирт, при нагревании до высокой температуры не депо-
лимеризуются до мономера, а деструктируются с образованием различ-
ных жидких, твердых и газообразных продуктов.
ГЛАВА I. ПОЛИМЕРЫ НЕПРЕДЕЛЬНЫХ АЛИФАТИЧЕСКИХ
УГЛЕВОДОРОДОВ И ПЛАСТИЧЕСКИЕ
/МАССЫ НА ИХ ОСНОВЕ
Полиэтилен. Полипропилен
Многие исследователи пытались получить высокомолекулярные техни-
чески ценные продукты полимеризацией олефинов, однако получали
лишь олигомеры. Только в 1938 г. было осуществлено промышленное
производство полиэтилена (—СНа—СНг—) под высоким давлением
(около 500 атм) и 180—200° С в присутствии следрв кислорода по ра-
дикальному механизму. Полиэтилен, получаемый этим способом, назы-
вают полиэтиленом низкой плотности (высокого давления) в отличие
от полиэтилена высокой плотности (низкого давления), который полу-
чают по ионному механизму — на катализаторах Циглера — Натта.
Исключительно важным этапом в развитии производства полиоле-
финов явилось открытие Циглером катализаторов — комплексов алкил-
алюминия и хлоридов титана, — которые вызывали полимеризацию эти-
лена, пропилена и других олефинов при атмосферном давлении. Коли-
чество таких катализаторов в настоящее время значительно увеличилось.
Они представляют собой комплексы, состоящие из металлорганическйх
соединений Al, Be, Mg, Zn, Cd, Ba, Na и хлоридов металлов IV, V, VI и
VIII групп, т. е. элементов с незаполненной промежуточной электронной
оболочкой. Чаще всего используют хлориды титана TiCU и Т1С1з, причем
TiCl4 при взаимодействии с металлалкилами, в частности АЦСаЬЭДз,
восстанавливается до соединений более низкой валентности. В зависи-
мости от природы компонентов катализаторов, а также числа замести-
телей в олефине можно получать стереорегулярные полиолефины раз-
личной пространственной конфигурации: изотактические, синдиотактиче-
ские, диизотактические и т. п. (см. стр. 14).
Различная степень кристалличности и характер структуры опреде-
ляют комплекс ценных физико-механических свойств полиолефинов, по-
лученных на катализаторах Циглера.
Впоследствии был предложен другой способ полимеризации олефи-
нов (метод Филлипса): при давлении 35—70 атм и 130—170°С в среде
инертного углеводорода в присутствии катализатора, состоящего из окис-
лов металлов переменной валентности, например окислов хрома, нане-
сенных на алюмосиликат. В настоящее; время известно несколько моди-
фикаций этого метода, носящих общее название «полимеризации при'
среднем давлении».
Все три способа имеют промышленное значение для производства
полиэтилена, так как они позволяют получать полиэтилен с различными
свойствами.
25
Тепловой эффект полимеризации этилена составляет около
4200 кДж/кг (1000 ккал/кг). В это тепло входит теплота полимеризации
этилена, рассчитанная по энергиям связи и равная 3653 кДж/кг
(872 ккал/кг), теплота, выделяющаяся при переходе газообразного про-
дукта (этилена) в твердый продукт (полиэтилен), составляющая при
давлении 1 атм 481 кДж/кг (115 ккал/кг), а также теплота растворения
этилена в жидком углеводороде (в случае полимеризации при низком
давлении).
По методу свободнорадикальной полимеризации (1200—1500 атм
при 200° С в присутствии кислорода и других инициаторов) полиэтилен
может сополимеризоваться со многими мономерами, например с пропи-
леном, бутиленом, винил- и винилиденхлоридом, винилацетатом, метил-
метакрилатом, акрилонитрилом, тетрафторэтиленом и др.
Полимер этилена представляет собой твердый продукт. В зависи-
мости от метода получения он обладает различными свойствами и может
быть двух типов: 1) полиэтилен низкой плотности (получаемый при вы-
соком давлении) и 2) полиэтилен высокой плотности (получаемый при.
низком и среднем давлениях).
Первый имеет более низкую плотность (0,92 г/см3) и молекулярный
вес (18000—35 000), а также более низкую степень кристалличности (до
70%) по сравнению с полиэтиленом высокой плотности. Второй отли-
чается более высоким показателем плотности (0,94—0,96 г/см3), моле-
кулярным весом (до 3 000 000) и большим содержанием кристалличе-
ской фазы (до 93%).
От этих показателей (главным образом от структуры полиэтилена
и от величины молекулярного веса) зависят как физико-химические, так и
механические свойства полимера. Полиэтилен низкой плотности обладает
большей эластичностью, меньшей хрупкостью, более низкой тем-
пературой размягчения (108—120° С) по сравнению с полиэтиленом вы-
сокой плотности. Полиэтилен с молекулярным весом около 3-106 обла-
дает исключительно высокой прочностью, что очень ценно в производст-
ве как волокна, так и пластических масс.
Полиэтилен при комнатной температуре нерастворим ни в одном из
известных растворителей и только при температуре 80° С и выше он за-
метно начинает растворяться в четыреххлористом углероде, трихлорэти-
лене, бензоле, толуоле, ксилоле. При охлаждении этих растворов поли-
мер вновь выпадает в осадок.
Полиэтилен обладает высокой водостойкостью и химической стойко-
стью. При температурах до 60—80° С он устойчив к действию всех кис-
лот, включая фтористоводородную, а также щелочей. К действию концен-
трированной азотной кислоты полиэтилен неустойчив.'
При нагревании полиэтилена в присутствии воздуха уже при 120° С
начинается его окисление, которое ведет к поперечной сшивке линейных
макромолекул и образованию нерастворимых полимеров.
При температуре выше 290° С полиэтилен деструктируется с обра-
зованием жидких маслянистых и газообразных продуктов, в том числе
небольшого количества (около 3%) мономера.
При воздействии ультрафиолетовых лучей, кислорода воздуха и теп-
ла в процессе переработки и эксплуатации полиэтилен стареет, что
26
проявляется в ухудшении его физико-механических и диэлектрических
•свойств.
Что касается производства полимеров пропилена и других олефинов,
то обычно предпочитают пользоваться методами Циглера и Филлипса.
Твердый полимер пропилена
Г СНз
I
L-ch2-ch—
получается при
Л
анионно-координационной полимеризации и низком (2—6 атм) или сред-
нем (35—70 атм) давлении в присутствии гетерогенных катализаторов
{смеси алкилалюминия и хлоридов титана, окислов переменной валент-
ности Cr, Mo, V). При этом образуется значительное количество стерео-
регулярного (изотактического) полипропилена. Пропилен способен со-
полимеризоваться с этиленом и другими олефинами. Полипропилен вы-
пускается в виде порошка белого цвета или гранул.
Полипропилен стереорегулярного строения представляет собой твер-
дый кристаллический полимер, обладающий высокими показателями хи-
мических, физико-химических и диэлектрических свойств. Стереорегу-
лярный полимер может иметь изотактическую и синдиотактическую
структуру. Наряду со стереорегулярной структурой имеется и атактиче-
ская часть, и стереоблочный полимер, макромолекула которого содер-
жит как изотактические, так и атактические участки. Наиболее ценным
материалом является полимер с низким содержанием атактической и
стереоблочной структуры.
При комнатной температуре стереорегулярный полипропилен устой-
чив к действию органических растворителей. При 80° С и выше он рас-
творяется в бензине, толуоле, хлорированных углеводородах. Полипро-
пилен устойчив к действию кислот и оснований даже при повышенной
температуре.
При воздействии тепла и света полипропилен, как и полиэтилен,
стареет, но более интенсивно, поэтому полипропилен всегда стабилизи-
руют антиоксидантами. Полипропилен более чувствителен к действию
кислорода, особенно при повышенных температурах, что объясняется
наличием третичных атомов углерода. Однако полипропилен лучше про-
тивостоит агрессивным средам. Он водостоек: водопоглощение за 6 ме-
сяцев менее 0,5%.
Технология получения полиолефинов в присутствии
катализаторов Циглера — Натта
Технологические процессы получения полиолефинов, полиэтилена и
их сополимеров при низком давлении в основном сходны и состоят из
нескольких стадий: приготовления катализатора (комплекса), полиме-
ризации олефина, промывки и сушки полимера.
Катализатор — комплекс АЦСгИбЦО/ПСК приготавливается пу-
тем смешения раствора TiCh в бензине (уайт-спирите, н-гептане) с рас-
твором АЦСгНзЦ. Суспензия катализатора (концентрацией 1 г/л) по-
ступает в полимеризатор, куда затем подается этилен. Его количество
регулируется давлением в полимеризаторе. Отвод тепла полимеризации
осуществляется циркуляцией паро-газовой смеси олефин — бензин. Кон-
денсат бензина подается через холодильник обратно в полимеризатор.
27
В случае полимеризации пропилена аппарат охлаждается водой. При
получении полипропилена из пропан-пропиленовой фракции пропан яв-
ляется охлаждающим агентом. Отделение полимера от разбавителя про-
изводится на центрифугах.
Полимеризация олефинов осуществляется по следующим режимам:
Этилен Пропилен]
Т,°С . . ... .L.J. . -i.t.j. 70—80 65—70
Давление, атм . (в1.c.j.LBj. г.1.1 * 1,5—2 10—12
Степень конверсии, % • • 98 98
Полиэтилен низкого давления (высокой плотности) получается в
виде порошка, который затем гранулируется.
Полиэтилен высокого давления (низкой плотности) производится в
виде роговидных гранул, которые могут быть натурального (белого)
цвета и окрашенными в различные цвета. Для окрашивания полиэтиле-
на применяют органические красители и пигменты, 'например пигмент
алый Н, тиоиндиго, красно-коричневый Ж, пигмент желтый Ж, кубовый
ярко-фиолетовый К, пигмент синий антрахиноновый и др. Наряду с этим
применяются минеральные пигменты: крон свинцово-желтый, кадмий
лимонный, кадмий красный, двуокись титана, алюминиевая пудра и др.
Полиэтилен ВД выпускается по ГОСТ 16337—70 нескольких марок.
В основу классификации положен «индекс расплава»—величина, зави-
сящая от молекулярного веса полимера и плотности. Название марки
состоит из слова полиэтилен и восьми цифр, обозначающих способ по-,
лучения (высокое или низкое давление), аппарат, в котором получен
полимер (автоклав, трубчатый реактор), степень гомогенизации (усред-
нение холодным смешением, в расплаве). Например, полиэтилен
11802—070 расшифровывается так: полиэтилен высокого давления (1),
номер марки —18, усредненный холодным смешением (02), плотностью
0,910 (0) и с показателем текучести расплава 7 г/10 мин (070).
Полипропилен выпускается по МРТУ 6-05-1105—67 различных ма-
рок: 01П10/002 — для переработки литьем под давлением конструкцион-
ных деталей; 02П10/003, ОЗП10/005 — для переработки экструзией и т. д.
Комплекс полезных свойств приобретает сополимер этилена с про-
пиленом. По мере увеличения содержания пропилена в сополимере уве-
личивается гибкость, снижается кристалличность. Сополимер с 20 мол. %,
содержанием пропилена имеет ценные свойства и получается как по ме-
тоду Циглера — Натта при низком давлении (СЭП низкого давления),
так и при 35—40 атм с применением окислов металлов в качестве ка-
тализатора (СЭП среднего давления). При соотношении два звена эти-
лена на одно звено пропилена можно получить эластомер СЭП низкого
давления со средним молекулярным весом 80 000—500 000. Степень кри-
сталличности 58—75%. По сравнению с полиэтиленом низкого давления
СЭП отличается повышенным сопротивлением растрескиванию под дей-
ствием длительных нагрузок. Оба сополимера обладают высокой проч-
ностью, морозостойкостью и хорошими диэлектрическими свойствами
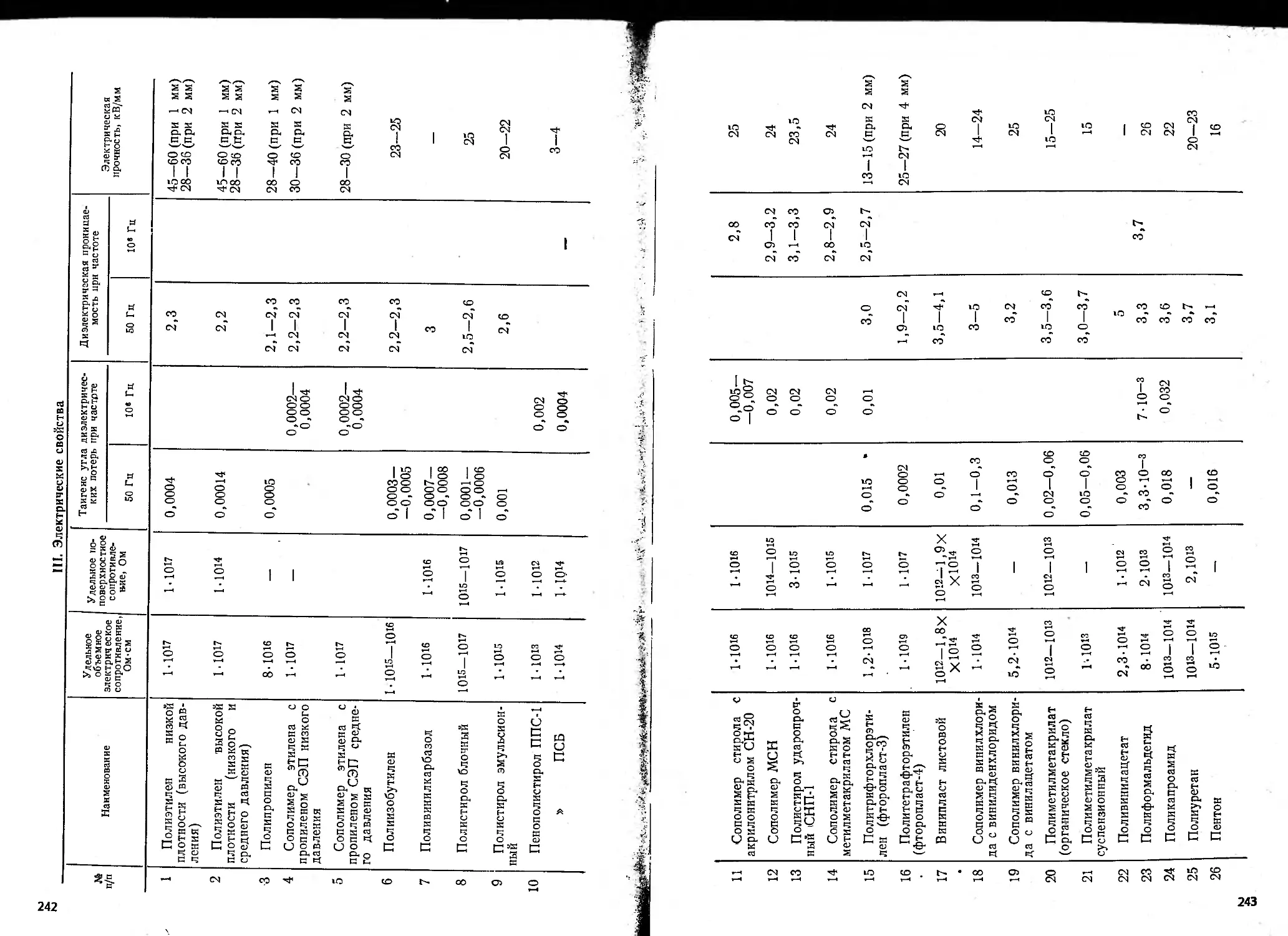
(см. приложение), не уступающими свойствам гомополимеров.
Полиолефины применяются в различных областях народного хозяй-
ства. Наиболее широко применяется полиэтилен ВД для изготовления
28
ЬПз’
I
—СН2—С— с высоким молекулярным весом
CH3J„
присутствии в качестве катализаторов различ-
При этом олефин полимеризуется по катионно-
пленок, листов, труб, шлангов, бутылей, бочек, ведер, флаконов и других,
изделий. Он применяется в кабельной промышленности, радиотехнике,
химической промышленности, сельском хозяйстве, для облицовки кана-
лов, в строительстве. Полиэтилен НД, а также его сополимеры с про-
пиленом весьма успешно применяются в строительстве для изготовления-
труб и санитарно-технических изделий. Полиэтилен представляет собой
неэластичный полимер, плавящийся и приобретающий свойства эласто-
мера только при 130° С.
На основе полиолефинов можно получать композиции с наполни-
телями (канальной сажей),.бутилкаучуком, отличающиеся высокой ус-
тойчивостью к растрескиванию.
Полипропилен с успехом можно применять для изготовления пред-
метов домашнего обихода н ухода за больными, требующих стерилиза-
ции. Он находит применение в текстильном машиностроении.
Полиизобутилен
I.
Полиизобутилен
был получен впервые в
ных апротонных кислот,
му механизму.
Полиизобутилен — аморфное вещество с молекулярным весом около-
200000, химически устойчив: выдерживает воздействие кислот (концен-
трированной азотной кислоты) и щелочей. Однако он уступает полиэти-
лену и политетрафторэтилену как по химической стойкости, так и ди-
электрическим свойствам.
Недостатком этого -полимера является его хладотекучесть под дей-
ствием собственного веса. Отличительная особенность поли-изобутилена
состоит в возможности совмещения с большим (до 150%) количеством
наполнителей, например -сажи, графита, что улучшает его физико-меха-
нические свойства и стабилизирует от воздействия УФ света.
Изобутилен легко сополимеризуется с различными мономерами:
стиролом, этиленом, пропиленом, акрилонитрилом, диенами и др. При-
чем в зависимости от природы мономеров и их соотношения, а также
условий полимеризации -можно получить сополимеры с разнообразными
свойствами: пластомеры, эластомеры и каучукоподобные продукты.
Полиизобутилены занимают промежуточное положение между пла-
стиками и каучуками. Однако введение этих полимеров в лабораторный
практикум по технологии пластических масс более целесообразно, -чем
его исключение.
Наряду с сополимерами со статистическим распределением звеньев
получают также блоксополимеры полиизобутилена с полиэтиленом, по-
листиролом, натуральным каучуком путем совмещения на вальцах
(см. ч. II).
2*
Химизм полимеризации изобутилена
Полипзобутилен получают в промышленном масштабе катионной
полимеризацией изобутилена в присутствии BF3 в жидком этилене. Про-
цесс идет в несколько стадий:
Р взаимодействие BF3 с основанием Льюиса:
BF3 + HR->H[BF3R]-
2) образование иона карбония, который вместе с противоионом об-
разует активный центр:
СН3 СН3
+ I I
Н [BF3R]“ + СН2=С -> СН3-С+ • • - [BF3R]~
I I
СН3 СН3
3) цепная реакция роста, в процессе которой сохраняется структура
активного центра на конце растущей макромолекулы:
СН3 СНз СН3г СНз-
I. Ill
СН3-С+ • • • [BF3R]“ 4- /гСН2=С -> СН3-С- -СН2-С-
I III
СНз СНз CH3L CH3J
CHg
- СН2— с+. • -[BF3R]“
I
п-1 СНз
4) обрыв цепи может происходить различными путями:
СНз СН3 СН3 СН3
а) ~ СН2-С+- • .[BF3R]~ + СН2=С -> ~ СН2-С=СН2 + СН3-С4 . • • [BF3R]~
СН3 СН3 СНз
СН3 СНз СНз
I , I I
б) ~ СН2-С+ • • • [BF3R]- + СН2=С -> ~ СН2—С=СН2 + BF3 + HR
I I
CH3 CH3
СН3 СН3
в) ~ CH2-i+. • .[BF3R]~-> ~ CH2-C-R + BF3
I I
CH3 СНз
Молекулярный вес полиизобутилена повышается с понижением тем-
пературы, так как в этих условиях снижаются константы скорости об-
рыва и передачи цепи. При комнатной температуре образуются только
олигомеры.
Ингибиторами полимеризации изобутилена являются сера, серово-
дород, меркаптаны, фтористый и хлористый водород. В присутствии
даже следов этих веществ резко падает выход полимера.
о На катализаторах Циглера — Натта можно получить стереорегуляр-
ный полиизобутилен.
зо
Технология полиизобутилена
В промышленности для полимеризации изобутилена чаще всего в
качестве катализатора применяют трехфтористый бор. Реакция осущест-
вляется в растворе в жидком этилене. Она очень экзотермична. С этиле-
ном испаряется также незаполимеризовавшийся изобутилен. Во избежа-
ние взрыва реакцию проводят для сохранения постоянства температуры
в среде растворителей, поглощающих при испарении за счет дросселиро-
вания в холодильном цикле выделяющееся тепло. В качестве такого рас-
творителя часто используют жидкий этилен, имеющий достаточно низ-
кую температуру кипения (—104° С) и не полимеризующийся при обыч-
ных условиях получения полиизобутилена.
Этилен смешивается с изобутиленом в соотношении 2,5 :1, смесь ох-
лаждается до —95° С и непрерывно подается в полимеризатор, пред-
ставляющий собой металлический короб, в котором движется бесконеч-
ная стальная лента (желоб), натянутая на барабан. На ленту непре-
рывно подается катализатор. Реакция завершается мгновенно при
смешении с катализатором. Этилен с незаполимеризовавшимся изобути-
леном подается на ректификацию и затем вновь возвращается в цикл.
Для разрушения оставшегося в полимере катализатора на ленту по-
лимеризатора непрерывно загружают стабилизатор.
Полиизобутилен из полимеризатора попадает в смеситель, нагрева-
ется до 100° С и нарезается затем специальным ножом на куски, ко-
торые охлаждаются.
Выпускаются марки: П-200, П-155, П-118, П-85. Индекс указывает
значение молекулярного веса в тысячах углеродных единиц. Низкомоле-
кулярный полиизобутилен выпускается следующих марок: П-50, П-ЗО>
П-20, П-5.
Важнейшей характеристикой полиизобутилена является молекуляр-
ный вес. Полимеры, у которых он ниже 50000,— жидкости. С увеличе-
нием молекулярного веса растет не только прочность и относительное
удлинение при растяжении, но и температура текучести.
Полиизобутилен каучукоподобен, легко окисляется кислородом воз-
духа при НО—130° С. При 180—200° С он легко формуется, а пр»
350—400° С разлагается с образованием маслянистых и газообразных
продуктов.
Наибольшее значение приобрело применение листового полиизобу-
тилена П-200, наполненного сажей и графитом (ПСГ), для футеровки
химической аппаратуры, бетонных и строительных конструкций.
Полиизобутилен ПСГ при 100° С устойчив к действию воды, 70 %-ной
серной кислоты, 1%-ной азотной, 25%-ной фтористоводородной, 100%-
ной уксусной, 17%-ного раствора соды и 25%-ного раствора аммиака.
При комнатной температуре ПСГ устойчив к действию 50%-ной азотной
и 40 %-ной фтористоводородной кислот.
Из наполненного полиизобутилена получают шланги, трубы и обо-
лочки. Его широко используют для изготовления уплотняющих прокла-
док, применяя в качестве наполнителей асбест, тальк, каолин и др. На
основе П-200 и графита при соотношении 1:1 получают материал марки
ПГ, который используют для изготовления обкладочных пластин. Для
31
этих целей может быть использован также материал, где наполнителями -
служат графит и асбест. К металлу, дереву и бетону обкладочный ма-
териал крепится клеями.
Полиизобутиленовые пластики могут/применяться лишь в пределах
40—100° С. Допустимая нагрузка при сжатии составляет 3 кг/см2.
В электронике используются для изоляции проводов и кабелей смеси
изобутилена с этиленом низкой плотности.
Исходное сырье для получения полиолефинов
Этилен СН2=СН2 простейший непредельный углеводород. При нор-
мальных условиях представляет собой бесцветный газ со слабым слад-
коватым запахом. Т. кип. —103,8° С; т. пл. —169,2° С; плотность при
т. кип. р=0,570 г/см3; показатель преломления п о100 =1,363; критиче-
ская температура 9,7° С; критическое давление 50,9 атм; молекулярный
вес 28,05.
Этилен устойчив до 350—400° С; выше этой температуры он начина-
ет разлагаться с образованием метана и ацетилена. При более высоких
температурах разложение идет до образования углерода и водорода.
Этилен может быть получен дегидратацией этилового спирта при
300—400° С в присутствии катализаторов (окиси алюминия, фосфорной
кислоты и др.). В промышленных масштабах этилен получают из газов
крекинга и пиролиза нефтяных углеводородов, а также из природных и
промышленных газов.
Пропилен СН3—СН = СН2 непредельный углеводород, при нормаль-
ных условиях газ. Т. кип. —47,7° С, т. пл. —185° С; плотность при
т. кип. р=0,610 г/см3; показатель преломления Пр47’7= 1,3623; критиче-
ская температура 92,0° С; критическое давление 45,4 атм; молекулярный
вес 42,08.
Пропилен может быть получен из пропилового или •изопропилового
спирта пропусканием их паров над окисью алюминия при 280—400° С.
В промышленном масштабе его получают из газов крекинга и пиролиза
нефтяных углеводородов.
Изобутилен — ненасыщенный углеводород, представляющий собой
при нормальных условиях газ. Т. кип. — 6,9° С; т. пл.— 148,35° С. Плот-
ность при т. кип. р = 0,5942 г/см3; показатель преломления пЪ25= 1,3814.
В промышленности изобутилен получают главным образом следую-
щими способами:
I) дегидратацией изобутилового спирта в присутствии А120з в ка-
честве катализатора при 360—370° С:
[АЦО,]
CH-j—СН—СН2ОН СН3-С=СН2 + Н2О
СН3 СН3
2) из бутан-бутиленовой фракции ректификацией, экстрактивной
или азеотропной дистилляцией, абсорбцией или экстракцией;
32
3) изомеризацией н-бутиленов в присутствии катализатора (фосфи-
новая кислота на носителе: шамоте, окиси алюминия и др.);
СН3
I
СН3 СН3 СН=СН
II I
СН3-СН2-СН=СН2 СН=СН СНз
а-бутилен р-н-бутилен-цис р-н-брилен-/пранс
Техника безопасности при полимеризации этилена,
пропилена и изобутилена при низком давлении
Этилен — горючий газ, горит светящимся пламенем. С кислородом
и воздухом образует взрывчатые смеси. Взрывоопасные концентрации с
воздухом: нижняя концентрация — 2,5 объемн. %; верхняя —
34,0 объемн. %.
Пропилен — горючий газ, образующий с воздухом взрывчатые сме-
си. Взрывоопасные концентрации лежат в пределах 2,0—11,0 объемн. %.
Особую осторожность необходимо соблюдать при обращении с ка-
тализаторами— трпэтилалюминием и диэтилалюминийхлоридом. Это
чрезвычайно горючие и взрывоопасные продукты, самовоспламеняющие-
ся на воздухе; с водой бурно реагируют, причем реакция идет со взры-
вом. Поэтому при хранении этих веществ должна быть обеспечена пол-
ная и надежная изоляция их от воздуха и влаги.
Концентрированные растворы триэтилалюминия и диэтил ал юминий-
хлорида в углеводородах при соприкосновении с воздухом также само-
воспламеняются, растворы меньших концентраций (до 20%) сильно ды-
мят, а при повышенных температурах способны к воспламенению.
Катализаторы бурно реагируют со спиртами, с четыреххлористым
углеродом реакция проходит со взрывом. Очень энергично, с большим
выделением тепла проходит реакция их с кислотами.
В очень небольших количествах (несколько граммов) их можно
хранить в запаянных ампулах из прочного стекла; в больших количе-
ствах они должны храниться в виде разбавленных растворов в каком-
либо углеводороде (например, бензине из парафинистых нефтей прямой
гонки, совершенно не содержащем влаги и тетраэтилсвинца). Для по-
лимеризации эти вещества применяются в виде растворов. Оба вещества
токсичны, при вдыхании их паров возможно заболевание легких. Жи-
вотные, находящиеся в атмосфере триэтилалюминия или диэтилалюми-
нийхлорида с концентрацией паров 50 г/м3 в течение часа, гибнут от кро-
воизлияния в легкие.
Необходимо остерегаться попадания катализаторов на кожу, так
как при этом образуются очень болезненные ожоги. Сильно действуют
на кожу и их концентрированные растворы (выше 40%); менее концент-
рированные растворы (в пределах 5—20%) болезненных ожогов не вы-
зывают.
2—1453 33
Применяемый для полимеризации этилен для предотвращения взры-
ва не должен содержать влаги и кислорода, а также примеси ацетилена.
По этим же причинам бензин, используемый в процессе полимериза-
ции как растворитель, должен быть совершенно сухим.
Применяемый в качестве сокатализатора реакции полимеризации че-
тыреххлористый титан представляет собой бесцветную жидкость с
т. кип. 136° С, дымящую на воздухе и разлагающуюся в присутствии вла-
ги. Поэтому четыреххлористый титан не должен соприкасаться с возду-
хом и водой, его следует хранить в герметически закрытой таре. Для про-
цесса полимеризации он, так же как и триэтилалюминий, применяется в
виде раствора в углеводороде (например, в бензине), не содержащем
влаги.
Реакционный сосуд перед полимеризацией должен быть тщательно
продут азотом, очищенным от кислорода.
Изготовление изделий из полиолефинов
Изготовление изделий методом литья под давлением. Одним из ос-
новных методов изготовления изделий из полиолефинов (полиэтилена
ВД, НД, полипропилена и их сополимеров) является литье под дав-
лением.
Литье под давлением — это процесс переработки термопластичных
материалов, при котором полимер нагревается до размягчения (до вяз-
Рис. 1. Инжекционная пресс-форма для
литья изделий под давлением:
1 — нижняя соединительная плита; 2—верхняя
соединительная плита; 3 — выталкиватель; 4 —
подвижная часть; 5 — линия разъема; 6 — не-
подвижная часть; 7 — направляющие колонки;
8 — выпускные каналы; 9 — кольцо; 10 — лит-
никовая втулка; 11— литниковый канал; 1% —
разводящий канал; 13 —- каналы для охлажде-
ния; 14 — плиты матрицы; /5 — выталкиватель
литника; 16 —- цапфа
котекучего состояния) в матери-
альном цилиндре и затем под дей-
ствием давления, которое созда-
ется поршнем или червяком лить-
евой машины, впрыскивается че-
рез узкое отверстие (сопло) по
литниковым каналам через впуск
в холодную или подогретую фор-
му, заполняет ее и затвердевает.
Готовое изделие извлекается при
разъеме формы с помощью вы-
талкивателей. Формы могут быть
различной степени сложности в
зависимости от конфигурации из-
делия и числа гнезд (рис. 1).
Полиэтилен хорошо перера-
батывается в изделия на обычных
литьевых машинах (рис. 2).
Высокая текучесть материа-
ла обеспечивает хорошее заполне-
ние формы, небольшую длитель-
ность цикла литья и позволяет работать при относительно низких удель-
ных давлениях (порядка 800—1000 кг/см2) с впускными каналами не-
большого сечения. Температура литья зависит от вязкости (молекуляр-
ного веса) применяемого материала и размеров литниковых каналов.
34
Для предотвращения чрезмерного затекания материала в зазоры не-
обходимо, чтобы обе половинки формы были тщательно подогнаны друг
к другу, а давление замыкания формы было больше давления литья.
При охлаждении
полиэтилена усадка со-
ставляет 3%, поэтому
пресс-форма во время
охлаждения должна по-
стоянно пополняться
материалом для ком-
пенсации уменьшающе-
гося объема *. Это до-
стигается непрерывным
давлением, оказывае-
мым на материал до
окончания цикла литья.
В случае образова-
ния в изделиях воздуш-
ных пузырей, а также
различных дефектов,
вызванных усадочными
явлениями, следует
увеличить .выдержку
при охлаждении, повы-
сить температуру литья
и удельное давление, а
материал впрыскивать
в пресс-форму, нагре-
тую до 50—60° С.
Для полиэтилена
НД, так же как и для
полиэтилена ВД, метод
литья под давлением
является одним из ос-
новных методов пере-
работки его в изделия.
Кроме литья под
давлением большое
применение нашел ме-
тод изготовления изде-
лий экструзией.
Наиболее важным
Рис. 2. Схема процесса литья под давлением на порш-
невой машине:
а — исходное положение формы; б — впрыск; в — разъем
формы; / — цилиндр; 2 — форма; 3— загрузочный бункер;
4 — плунжер
показателем, позволяющим отнести полиэтилен
к той или иной марке, является характеристическая вязкость, опреде-
* Усадка полиэтилена при охлаждении его, например, от 11'5 до 20° С достигает
15—16%. Во время литья под давлением под влиянием температуры материал расши-
ряется, а при одновременном действии давления сжимается. Окончательное оформление
изделия происходит под влиянием обоих факторов. Чем выше давление, тем больше
сжатие материала и тем больше конечный вес изделия. Когда давление снимается, то
материал вновь увеличивается в объеме; чем до некоторой степени компенсируется
большая усадка полиэтилена.
2*
35
ТАБЛИЦА 3
Зависимость
характеристической вязкости
от значения средневесового
молекулярного веса
полиэтилена низкого давления
Характеристи- ческая вязкость Средневесовой молекулярный вес
1,0 599000
2,0 180000
3,0 340 000
4,0 540 000
5,0 774000
ляемая в растворе декалина при 135° С ® среде азота, свободного от
примесей.
Величине характеристической вязкости соответствуют определенные
значения средневесового молекулярного веса полиэтилена низкого дав-
ления (табл. 3).
Важной характеристикой является индекс расплава (см. ч. II).
Изготовление изделий из полиолефинов
методом экструзии. Экструзией называется
процесс переработки термопластичных ма-
териалов, при котором полимер размягча-
ется, гомогенизируется, уплотняется и не-
прерывно выдавливается через оформляю-
щую головку экструзионной машины, ко-
торая придает изделию нужный профиль.
В зависимости от сечения формующего от-
верстия можно получать изделия в виде лис-
тов, пленок, стержней, труб, уголков и т. п.
Экструзионная машина (рис. 3) пред-
ставляет собой горизонтальную цилиндри-
ческую камеру с загрузочным бункером,
внутри которой вращается шнек. В конце
цилиндрической камеры устанавливают обо-
греваемую головку, в которой выдавливаемый шнеком материал оформ-
ляется в изделие нужного профиля. .
Экструзионные машины отличаются числом шнеков, их длиной и
диаметром, глубиной и шагом нарезки, заходностью, конструкцией го-
ловки.
Рис. 3. Схема экструзии пленочного или листового материала:
I — мотор; 2, 3 — передача; 4 — загрузочный бункер; 5 — цилиндр; 6 — шнек; 7 — головка
щелевая прямоточная; 8—гладильный каландр; 9— приемные охлаждающие валки; 10—
охлаждающий и разбраковочный стол; // — намотка готового материала
Увеличение числа шнеков от одного до трех позволяет увеличить
производительность и улучшить гомогенизацию композиции. Длина
шнеков, в таких машинах неодинакова, так же как и характер нарезки.
Наиболее удачным является такое'расположение шнеков, когда два из
них осуществляют смешение, гомогенизацию и подачу на третий шнек,
осуществляющий сжатие и выдавливание в головку.
36
Рис. 4. Головка к экструдеру прямо-
точная плоскощелевая:
1 — мундштук; 2 — поток расплава; 3 —
дорн; 4 — щель; 5 — регулировочные болты
Сжатие характеризуют коэффициентом сжатия К, который пред-
ставляет собой отношение количества материала, заключенного
между двумя нарезками в начале (а) и в конце шнека (6): K=alb.
Уменьшение глубины нарезки
или шага шнека усиливает сжатие
и увеличивает уплотнение материа-
ла на выходе, что обеспечивает вы-
сокое качество изделия.
Головки экструзионных машин
по направлению потока расплава
делятся на прямоточные и угловые,
а по форме щели — на кольцевые и
плоскощелевые (рис. 4, 5, 6, 7, 8).
Размеры кольцевого отверстия
регулируются изменением положе-
ния дорна относительно мундштука
в направлении потока. Размеры
кольцевого отверстия можно регули-
ровать также формующими кольца-
ми дорна и мундштука или одним
лишь кольцом мундштука.
В плоскощелевых головках (рис.
4 и 6) размер щели между подвиж-
ной и неподвижной губками (рис. 4)
либо между двумя подвижными губками (рис. 6, 1) изменяется с по-
мощью регулировочных болтов. Головка, изображенная на рис. 6, угло-
вая, материал направляется в щель из канала 2.
Головки с кольцевой щелью применяют для изготовления труб,
шлангов и рукавов (рис. 5, 7, 8), из которых путем раздувания получают
Рис. 5. Головка к экструдеру для изготовления труб
кольцевая прямоточная:
1 — шнек; 2 — цилиндр машины; 3 — мундштук; 4 — дорн
Рис. 6. Головка к экстру-
деру угловая плоскОЩеле-
вая:
1 — формующие губки; 2 —
канал; 3 — болт регулировоч-
ный
37 ‘
Рис. 7. Головка к экструдеру для изготовления ру-
кавов и труб:
1 — мундштук; 2 — кольцо мундштука; 3 — Дорн; 4 —
дорнодержатель; 5 — формующая часть дорна; 6 — регу-
лировочный болт; 7 — болт дорнодержателя; 8 — цилиндр
машины; 9 — сетки и решетки
Рис. 8. Головка к экструдеру
угловая кольцевая:
/ — мундштук; 2 — дорнодержатель;
3 — дорн; 4 — регулируемое кольцо
мундштука; 5 —рукав
Рис. 9. Схема получения пленки экструзией с пневматической вытяж-
кой (выдуванием):
/ экструдер; 2 головка; 3 — рукав; 4 — сплющивающие и охлаждающие
щеки; 5 — герметизирующие валки
1
Рис. 10. Угловая головка для об-
лицовки проводов:
1 —* голый провод; 2 — дори; 3 — мунд-
штук; 4 — облицованный провод
пленочный материал (рис. 9). Головки с плоской щелью используют для
изготовления пленок и листов.
Стержни изготавливают с помощью головки с отверстием в дорне.
Облицовку проводов производят на угловых головках (провод пропу-
скают в отверстие в дорне (рис. 10)).
В цилиндре должен быть дифференцированный по зонам обогрев
(электрический или жидким теплоносителем). В зоне загрузки необхо-
димо охлаждение. Зона перехода нагре-
вается. Червяк не следует ни нагре-
вать, ни охлаждать. Его длина должна
составлять 15—20£> (здесь D — диа-
метр шнека).
Рекомендуются однозаходные шне-
ки с уменьшающейся глубиной нарез-
ки, постоянным шагом и максимальным
коэффициентом сжатия 4:1. Между
червяком и головкой ставят много-
слойный фильтр (набор сеток) для
удаления пузырьков воздуха и нерас-
плавившихся частиц, а также для пре-
вращения вращательного движения в поступательное.
Температура должна повышаться от бункера к головке, причем
температура головки должна быть выше, чем температура зоны сжатия
цилиндра.
Экструзия, так же как и литье под давлением, широко применяется
для изготовления изделий из полиэтилена, полипропилена и других тер-
мопластов. Кроме изготовления изделий экструзионный метод исполь-
зуется также для гранулирования термопластичных материалов (поли-
этилена, полипропилена, полистирола, поливинилхлорида и др.).
Профилированные изделия могут быть получены непрерывным или
периодическим методами. В первом случае экструзия производится на
шнековых, а во втором случае — на поршневых экструзионных машинах.
Для изготовления профилированных изделий из полиэтилена применя-
ются главным образом шнековые экструзионные машины непрерывцого
действия (шнек-машины).
Методом экструзии можно перерабатывать полипропилен, полиэти-
лен как высокого, так и низкого давления и их сополимеры. Однако
надо иметь в виду, что при экструзии в отличие от литья под давлением
Рис. 11. Получение пленок с охлаждением в ваннах с понижающейся темпера-
турой:
1, 2 — ванны; 3 — приемные валки; 4 — намоточное устройство
39
и прессования оформленное .изделие отвердевает вне камеры формова-
ния. В результате этого форма изделия после выдавливания из мунд-
штука экструзионной машины при остывании на воздухе может подвер-
гаться значительным изменениям. Это обстоятельство определяет спо-
соб приема изделий, который зависит от его конфигурации. Прием
изделия осуществляется различными способами: охлаждением на ка-
ландрах (пленка, лиут, см. рис. 3), иа охлаждаемом столе, в насадке
сжатым воздухом (трубы), в охлаждающей ванне, в нескольких ваннах
с последовательно понижающейся температурой (прозрачные пленки
и т. д., рис. 11).
РАБОТА 1. ПОЛУЧЕНИЕ ПОЛИЭТИЛЕНА ВЫСОКОЙ ПЛОТНОСТИ
(НИЗКОГО ДАВЛЕНИЯ]
Исходные продукты: этилен; триэтилалюминий (раствор в бензине); четырех-
хлористый титан (раствор в бензине); бензин (т. кип. 80°С)*; метиловый спирт.
Оборудование: стальной автоклав, рассчитанный на давление не менее 8 атм,
снабженный манометром, рубашкой для обогрева и охлаждения, мешалкой и термо-
метром; трехгорлая колба, снабженная мешалкой с герметическим затвором, обрат-
ным холодильником и термометром; воронка Бюхнера; плоскодонные колбы на 250 мл
с притертой пробкой — 2 шт.; водяная баня с элект.ронагревом.
Автоклав предварительно продувают азотом **, загружают вначале
триэтилалюминий (1%-ный раствор в бензине), затем под давлением
2 атм этилен и, наконец, при интенсивном перемешивании четыреххло-
ристый титан (1%-ный раствор в бензине).
Катализатор вводится в количестве 0,5% от массы загруженного
этилена. Четыреххлористый титан добавляют исходя из расчета: на
1 моль триэтилалюминия 1 моль TiCl^.
Реакция полимеризации начинается с момента введения четырех-
хлористого титана и идет с выделением большогб количества тепла.
В это время необходимо охлаждать автоклав, пуская холодную воду в
рубашку аппарата. В течение всего процесса полимеризации температу-
ру в автоклаве следует поддерживать равной 50—60° С, а давление не
выше 2 атм. Полимеризация продолжается около 4 ч. О конце реакции
можно судить по снижению давления в автоклаве. По истечении этого
времени спускают оставшееся давление, открывают крышку аппарата и
полученную суспензию полимера переносят в трехгорлую колбу, снаб-
женную мешалкой с герметическим затвором, обратным холодильником
и термометром. В колбе полимер промывают от продуктов разложения
катализатора и сокатализатора.
Сначалд полимер промывают метиловым спиртом’ при нагревании
на водяной бане до 60—65° С и перемешивании в течение 15—20 мин.
После этого содержимое колбы отфильтровывают на воронке Бюхнера,
* Бензин для растворения катализатора и . сокатализатора должен быть тща-
тельно высушен над сернокислым натрием и перегнан при 80° С.
** Азот, применяемый для продувания аппаратуры, не должен содержать приме-
сей кислорода, ацетилена и влаги.
40
порошкообразный полимер возвращают обратно в колбу и вновь про-
мывают метиловым спиртом при перемешивании в течение нескольких
минут без нагревания, Промыйку при комнатной температуре с после-
дующей фильтрацией проводят трижды. Промытый спиртом полимер
промывают один раз дистиллированной водой на воронке Бюхнера, от-
жимают на листе фильтровальной бумаги и сушат при 40—50° С.
Полученный полиэтилен представляет собой порошкообразный про-
дукт белого или сероватого цвета.
Определяют характеристическую вязкость полиэтилена и рассчи-
тывают его молекулярный вес по табл. 3. Определяют индекс расплава
(см. ч. II).
РАБОТА 2. ПОЛУЧЕНИЕ СТЕРЕОРЕГУЛЯРНОГО
(ИЗОТАКТИЧЕСКОГО) ПОЛИПРОПИЛЕНА
При полимеризации пропилена в присутствии смеси алкилалюминия
с хлоридами титана получаются полимеры, содержащие наряду с изо-
тактичёским (стереорегулярным) значительное количество атактическо-
го (аморфного) полипропилена. Последний извлекают из смеси поли-
меров с помощью растворителей: эфира или гептана.
После извлечения атактического полипропилена остается нераст-
воримый изотактический (кристаллический) полипропилен. *
Исходные продукты: пропилен — 100 г; диэтилалюминийхлорид (в пересчете на
100%-ный продукт)—2,5 г; треххлористый титан (в пересчете на 100%-иый про-
дукт)— 1,2 г; бензин (т. кип. 80°С); метиловый спирт (абсолютированный).
Оборудование: стальной автоклав, рассчитанный на давление не ниже 15 атм,
снабженный рубашкой для обогрева и охлаждения, мешалкой, термометром и мано-
метром; колбы на 250 мл с притертой пробкой — 2 шт.; плоскодонные колбы на 1 л —
2 шт.
Стальной автоклав предварительно вакуумируют и продувают азо-
том, освобожденным от кислорода и влаги. Затем загружают в него
диэтилалюминийхлорид в виде 1%-ного раствора в бензине. После за-
грузки катализатора в автоклав под давлением вводят пропилен, не
содержащий ацетилена, кислорода и влаги, и, наконец, тонко измель-
ченную суспензию треххлористого титана в бензине, содержащую 10 г
сухого вещества в 1 л.
Процесс полимеризации проводят при 65—70° С и давлении 10—
12 атм в течение 5—6 ч при непрерывном перемешивании.
Тепло, выделяющееся в процессе полимеризации, отводят, пропу-
ская холодную воду через рубашку автоклава. Количество тепла, выде-
ляющегося при полимеризации пропилена, составляет около
58 кДж/моль (~14 ккал/моль), поэтому охлаж/гение автоклава водой
вполне достаточно и не представляет затруднений.
Количество загружаемых в аппарат компонентов рассчитывают
так, чтобы концентрация полимера в жидкой фазе (в бензине) к концу
Реакции составляла не более 300 г/л. При такой концентрации масса в
автоклаве достаточно подвижна и поддается перемешиванию механиче-
ской мешалкой, однако выгрузка ее затруднена. Для облегчения вы-
41
грузки массы из автоклава ее необходимо разбавить бензином в два
раза, т. е. довести концентрацию полимера до 150—160 г/л.
По окончании процесса полимеризации оставшееся в автоклаве
давление спускают, открывают крышку аппарата и выгружают полу-
ченный полимер, который затем подвергают очистке от продуктов раз-
ложения каталитического комплекса промывкой его метанолом точно
так же, как очищается: от загрязнений полиэтилен после полимеризации
при низком давлении (стр. 40).
Готовый полипропилен представляет собой порошкообразный про-
дукт белого или сероватого цвета с содержанием изотактического поли-
мера до 85%.
Определяют плотность и индекс расплава (см. ч. II).
РАБОТА 3. ПОЛУЧЕНИЕ ПОЛИИЗОБУТИЛЕНА В ПРИСУТСТВИИ
ЭФИРАТА ТРЕХФТОРИСТОГО БОРА
Исходные продукты: изобутилен, эфират трехфтористого бора BF3(C2H5)2O —
0,05 г; жидкий азот.
Оборудование: мерная ампула на 50 мл; сосуд Дьюара; прибор для перегонки
изобутилена (рис. 12). *
В изобутилене, находящемся в баллонах под давлением, содержит-
ся влага и некоторое количество полимера. Для очистки от этих приме-
сей его перегоняют. Изобутилен из баллона через вентиль Гоффера
пропускают в склянки 1 и 2, охлаждаемые льдом до 0°С. Влага вымо-
Рис. 12. Прибор для перегонки изобутилена:
I, 2 — склянки, помещенные в охлаждающую смесь; 3 — пустая склян-
ка; 4 — ампула; 5 — сосуд Дьюара
раживается на стенках склянок; изобутилен, проходя через пустую
склянку 3, нагреваемую водой до 30° С, перегоняется в мерные ампулы
4 емкостью 50 мл, помещенные в сосуды Дьюара 5 и охлаждаемые до
—20° С смесью сухого льда с ацетоном или спиртом. Заполненную до
метки ампулу с изобутиленом переносят в другой сосуд Дьюара, напол-
ненный жидким азотом, и охлаждают до —80° С. При этой температуре
в ампулу вносят 0,05 г (0,1%) эфирата трехфтористого бора — катализа-
тора катионной полимеризации и, не запаивая ампулу, выдерживают
42
изобутилен до образования каучукоподобного полимера. Так как BF3_
активный акцептор электронов, полимеризация протекает почти со
взрывной скоростью и заканчивается за несколько секунд. Полимер ис-
пользуют для работы 132, ч. II.
РАБОТА 4. ПОЛУЧЕНИЕ ПОЛИИЗОБУТИЛЕНА
В ПРИСУТСТВИИ TiCI4
Исходные продукты: изобутилен перегнанный — 20 г; TiCl4 — 0,1 г; метиловый
спирт— 100 мл, бензол — 100 мл.
Оборудование: сосуд Дьюара; пробирка диаметром 40 мм и высотой 300 мм; про.-
пеллерная мешалка (30—40 об/мин); вакуум-сушилка.
Изобутилен из баллона перегоняют, как описано в работе 3. Про-
бирку с перегнанным мономером помещают при —40° С в сосуд Дьюара,
наполненный жидким азотом и ацетоном. Устанавливают в пробирку
мешалку с механическим приводом и, перемешивая, охлаждают содер-
жимое пробирки до —80° С. При этой температуре и при перемешива-
нии добавляют 0,1 г ПС14 — катализатора катионной полимеризации.
В отличие от BF3 четыреххлористый титан менее активен и реагирует
лишь в присутствии влаги как комплексная кислота строения
НЦТЮчОН]-. Поэтому через реакционную смесь в течение 10 мин про-
дувают воздух, погружая в пробирку стеклянную трубочку, соединенную
с газометром.
Полученный полимер растворяют в бензоле и осаждают метиловым
спиртом, декантацией сливают растворитель. Осадок промывают спир-
том и высушивают в вакуум-сушилке при 50 °C и 400—500 мм рт. ст. .
Определяют предел прочности при растяжении * и относительное
удлинение стандартного образца (см. ч. II). Стандартный образец по-
лучают под прессом с помощью штампа из листа, изготовленного на
вальцах при 120—130° С.
РАБОТА 5. ИЗГОТОВЛЕНИЕ ИЗДЕЛИИ ИЗ ПОЛИЭТИЛЕНА
ВЫСОКОГО ДАВЛЕНИЯ (НИЗКОЙ ПЛОТНОСТИ)
ЛИТЬЕМ ПОД ДАВЛЕНИЕМ
Исходный продукт: полиэтилен высокого давления (молекулярный вес 1(8 000—
20 000) в виде гранул или крошки.
Оборудование: литьевая машина лабораторного типа с производительностью за
1 цикл 5—7 г.
Литье полиэтилена производят на обычных литьевых машинах лабо-
раторного типа. Схема процесса литья полиэтилена под давлением в
литьевой машине представлена на рис. 13.
В бункер машины загружают полиэтилен. Перед началом литья
обе половины формы (желательно иметь пресс-форму на стандартные
образцы) смыкают, в каналы пускают горячую воду (90—95°С). После
* Предел прочности или прочность при растяжении, сжатии, изгибе характери-
зуется разрушающим напряжением при соответствующих деформациях.
43
того как пресс-форма нагреется до 50—60° С, а материальный цилиндр
до 150—250° (температура литья) *, пускают в ход литьевую машину и
начинают литье.
После пуска литьевой машины обогрев форм прекращают, горячую
воду отключают и включают холодную для охлаждения формы, так как
в процессе литья форма нагревается за счет поступающего в нее нагре-
Рис. 13. Схема лабораторной машины для литья под давлением:
/ — цилиндр; 2 — плунжер; 3 — дозирующее устройство; 4 загрузочный
бункер; 5 — электрообогрев; 6 — гильза терморегулятора; 7 — сопло; 8 —
кронштейн; 9 — пружина; 10 — подвижная плита для крепления полумат-
рицы; 11— неподвижная полуматрица; 12—подвижная полуматрица; 13 —
плита’для крепления подвижной полуматрицы; 14— траверза; 15 — вытал-
киватель; 16 — пружина; 17 — упор
того материала. Во время литья температура загрузочной части машины
не должна превышать 70° С.
При правильно нагретой пресс-форме происходит медленное охлаж-
дение материала, способствующее образованию гладкой, глянцевитой по-
верхности изделия. Полученные стандартные образцы испытывают на
предел прочности при ударе и изгибе, диэлектрические свойства (см.
ч. II).
РАБОТА 6. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ПОЛИЭТИЛЕНА
НИЗКОГО ДАВЛЕНИЯ (ВЫСОКОЙ ПЛОТНОСТИ)
ЛИТЬЕМ ПОД ДАВЛЕНИЕМ
Исходный продукт: полиэтилен НД гранулированный.
Оборудование: литьевая машина лабораторного типа производительдостыо 5—7 г.
* Оптимальная температура литья устанавливается в зависимости от текучести рас-
плава полиэтилена, так называемого индекса расплава. Так, 'например, при индексе рас-
плава 7 г/10 мин температура литья составляет 190—200° С; при индексе 20 г/10 мин
155—1'70°С. У полиэтилена высокой плотности индексы расплава 0,2—0,5 г/10 мин.
У полимеров кристаллической структуры индекс расплава невелик. Литье под давле-
нием таких полимеров требует специального запорного устройства в сопле для устра-
нения вытекания расплава полимера из сопла при его отходе от литникового канала
пресс-формы. Температуру пресс-формы обычно поддерживают на 10—20° С ниже тем-
пературы плавления полимера. Несмотря на высокое содержание кристаллической
фазы, литье под давлением полиэтилена высокой плотности не требует специальных
устройств.
44
Полиэтилен с характеристической вязкостью в пределах 1,0—2,3
обладает наилучшими литьевыми свойствами. Полиэтилен с характери-
стической вязкостью более 3,3 может перерабатываться на литьевых
машинах при температуре материального цилиндра 270—380° С, однако
при этих температурах создается опасность разложения полимера, по-
этому трудно получить изделия с хорошей поверхностью. Хорошие литье-
вые свойства полиэтилена зависят также от его полимолекулярности.
Большое количество высокомолекулярных
фракций требует значительного повышения таблица*
температуры материала при литье.
Литье полиэтилена НД производится
аналогично литью полиэтилена ВД на ла-
бораторной литьевой машине лишь с неко-
торой разницей в температурном режиме
литья. Температура литья устанавливается
в пределах 200—270° С в зависимости от
молекулярного веса (характеристической
вязкости) полиэтилена и размеров литнико-
вых каналов (см. табл. 4).
На внешний вид изделия большое влия-
Зависимость температуры литья
от характеристической вязкости
Характеристи-
ческая вязкость
Температура литья
под давлением, °C
1,3
1,7
2,8
3,7
170—180
200-205
210—220
310
ние оказывает температура формы. Понижение температуры формы уве-
личивает усадочные дефекты и ухудшает внешний вид изделия. Опти-
мальная температура формы каждый раз должна устанавливаться экс-
периментально.
Для полиэтилена с характеристической вязкостью 1,2—2,2 и с цик-
лом литья от 40 до 90 сек температура пресс-формы должна поддержи-
ваться в пределах 50—70° С в зависимости от конфигурации и толщины
стенок изделия, сечения и формы литниковых каналов.
Удельное давление при литье полиэтилена низкого давления не
превышает 1000 кг/см2, что вполне обеспечивается литьевыми машинами
обычного типа.
Для литья полиэтилена высокой плотности должен применяться
только гранулированный полиэтилен, так как при этом достигается .точ-
ность дозировки на автоматических литьевых машинах и получаются
изделия высокого качества. В случае применения порошкообразного
полиэтилена переработка его затрудняется из-за попадания большого
количества воздуха.
РАБОТА 7. ИЗГОТОВЛЕНИЕ ИЗДЕЛИИ ИЗ ПОЛИЭТИЛЕНА
НИЗКОЙ ПЛОТНОСТИ ЭКСТРУЗИЕЙ
Исходный продукт: полиэтилен низкой плотности (гранулированный) — 100 г.
Оборудование: лабораторная экструзионная машина, диаметр шнека 50 мм, длина
шнека 15 D.
Схема экструзионной шнек-машины представлена на рис. 3. Поли-
этилен в виде гранул загружается в бункер 4, оттуда он непрерывно по-
ступает в обогреваемый цилиндр 5, в котором вращается винт шнека 6.
45
Шнек уплотняет и подает материал к головке машины 7, откуда и вы-
давливается профилированное изделие (пленка, лист).
При экструзии полиэтилена высокого давления температура мате-
риала в машине по мере его продвижения от загрузочного отверстия к
головке должна постепенно повышаться. В загрузочной части цилиндра
машины температура должна быть не выше 70° С, а в цилиндре у голов-
ки около 120—130° С в зависимости от вязкости полимера. Такое распре-
деление температур приводит к постепенному снижению вязкости и
созданию нужного давления материала в головке машины. При повыше-
нии температуры в цилиндре у загрузочного отверстия выше допустимой
происходит размягчение или расплавление полиэтилена и образование
сводов, что ведет к прекращению питания машины.
При температуре в головке ниже 120° С экструзия затрудняется,,
так как материал становится недостаточно мягким и гомогенным. В этом
случае изделия будут получаться плохого качества, а для выдавливания
потребуются очень большие усилия. Давление в головке машины при
экструзии полиэтилена низкой плотности составляет 80—100 кг/см2.
Между шнеком и мундштуком укрепляется решетка с набором ме-
таллических сеток.
Подача гранулированного полиэтилена при изготовлении изделий
должна быть непрерывной и равномерной, что обеспечивается установ-
кой питательного бункера с мешалкой или шнеком.
Важное значение имеет однородность применяемого материала ПО'
молекулярному весу. Применение полиэтилена с большой полидисперс-
ностью по молекулярному весу приводит к образованию неровной, ше
роховатой поверхности изделия.
На внешний вид изделия влияет также длина оформляющей части
мундштука. Чем она длиннее, тем более гладкой и глянцевитой получа-
ется поверхность изделия.
Полиэтилен имеет очень высокие коэффициенты термического рас-
ширения, что вызывает большую усадку при охлаждении изделий. На-
пример, усадка при снижении температуры от 115 до 20° С достигает
15—16% от первоначального объема.
В толстостенных изделиях наружные слои охлаждаются быстрее,
чем внутренние, которые остаются долгое время размягченными. В ре-
зультате этого’ происходит неравномерная усадка, возникают внутрен-
ние напряжения, что приводит к появлению трещин в готовых изделиях
в процессе их эксплуатации. Этот недостаток можно свести к миниму-
му, применяя медленное охлаждение изделия после экструзии.
РАБОТА 8. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ПОЛИЭТИЛЕНА
ВЫСОКОЙ ПЛОТНОСТИ ИЛИ СОПОЛИМЕРА ЭТИЛЕНА
С ПРОПИЛЕНОМ ЭКСТРУЗИЕЙ
Исходный продукт: сополимер этилена с пропиленом — 1'00 г (или полиэтилен вы-
сокой плотности).
Оборудование: экструзионная машина лабораторная с плоскощелевой головкой,,
выдающей ленту или пленку.
46
Для экструзии, так же как и для литья под давлением, полиолефинь!
и их полимеры должны применяться только в гранулированном виде.
Применение гранулированных полимеров обеспечивает равномерность
подачи его в шнек экструзионной машины, что гарантирует высокое ка-
чество изготовляемых изделий и более высокую производительность
агрегата.
Изделия из полиэтилена или его сополимера с пропиленом низкого
молекулярного веса с характеристической вязкостью до 1,8 плохо со-
храняют приданную им форму
после выхода из шнек-машины.
Такой материал мало пригоден
для изготовления изделий экстру-
зией.
Для полиэтилена или сопо-
лимера этилена с пропиленом вы-
сокого молекулярного веса, име-
ющего характеристическую вяз-
кость более 3,6, при экструзии
требуется высокая температура
(порядка 280—350°С), что созда-
ет опасность разложения полиме-
ра. Для экструзии рекомендуется
ТАБЛИЦА 5
Зависимость режима экструзии
от характеристической вязкости.полимера-
Характе- ристичес- кая вяз- кость т емпература по зонам, °C
I зона II зона III зона (мундштук)
1,9 50—60 130-140 180-200
2,2 50-60 140—150 200—210
3,2 60-70 160-170 220-230
3,7 80—90 190—200 250-260
4,0 80-90 200-220 280
применять эти материалы с ха-
рактеристической вязкостью в пределах 1,8—3,5.
На основании опытных данных установлен температурный режим
экструзии шлангов и труб из полиэтилена высокой плотности и его со-
полимера с различной характеристической вязкостью. Эти данные, ко-
торыми можно руководствоваться при выполнении работы *, приведены
,в табл. 5.
Высокая температура, особенно в III зоне, при экструзии полиэти-
лена и его сополимера может вызвать деструкцию полимера. Для пред-
отвращения разложения материала при переработке вводят тепловые
стабилизаторы. Эффективными стабилизаторами против теплового ста-
рения полиэтилена, полипропилена и их сополимеров являются амины
(например, N, N'-дифенил-и-фенилендиамин), дикрезилолпропан и др.
Стабилизаторы вводят в полимер при его грануляции. Для этого поли-
мер предварительно смешивают со стабилизатором в шаровой мельни-
це, а затем порошок подвергают гранулированию. Количество стабили-
затора составляет обычно 0,2—0,3% от массы полимера. Полипропилен,
подвергаемый повторной переработке, должен быть обязательно стаби-
лизирован.
Образец 5x2 см, вырезанный из экструдированной ленты, испыты-
вают на водостойкость при комнатной температуре (см. ч. II) и при
100° С в течение 1 ч. На стандартных образцах определяют разрушаю-
щее напряжение при разрыве, относительное удлинение и модуль эла-
стичности, диэлектрические свойства (см. ч. II).
* Данные относятся к работе экструзионной машины со скоростью вращения шне-
ка 9 об/мин.
47
ГЛАВА II. ПОЛИМЕРЫ НЕПРЕДЕЛЬНЫХ АРОМАТИЧЕСКИХ
УГЛЕВОДОРОДОВ И ПЛАСТМАССЫ
НА ИХ ОСНОВЕ
Полистирол
— продукт полимеризации ненасыщен-
ного углеводорода винилбензола (стирола). Это один из давно извест-
ных полимеров, впервые полученный еще в 1839 г. в результате само-
произвольной полимеризации стирола.
В настоящее время полистирол производится в больших количествах
почти во всех промышленно развитых странах, причем производство его
непрерывно растет. Стирол применяется также в производстве синтети-
ческих каучуков, где он является одним из наиболее важных сомономе-
ров бутадиена. Изменяя состав исходной смеси, можно получить раз-
личные сополимеры, свойства которых изменяются в широких пределах:
от каучуков, т. е. эластомеров, до эластопластических и даже пластиче-
ских масс. Стирол используется также в лакокрасочной, фармацевтиче-
ской и других отраслях промышленности.
Полимеризация стирола
Стирол может полимеризоваться как по свободнорадикальному ме-
ханизму с образованием полимеров высокого молекулярного веса, при-
годных для технических целей, так и по ионному (анионному, катионно-
му и ионно-координационному) механизму. Полимер, получаемый в ре-
зультате полимеризации по свободнорадикальному механизму,—
аморфный, атактического строения; полимер, получаемый в результате
ионной полимеризации, — кристаллический, имеет стереорегулярное
строение.
Аморфный полистирол может быть получен разными способами!: в
блоке, в эмульсии, в суспензии и в растворе в присутствии инициаторов
или без них.
В настоящее время в промышленности производят полистирол,
имеющий аморфное, строение, путем свободнорадикальной полимери-
зации.
Из возможных способов полимеризации стирола (в блоке, в эмуль-
сии, в суспензии, в растворе) используют главным образом первые три.
Полимеризация стирола в блоке может быть проведена как в при-
сутствии инициатора, так и без него. Инициаторами полимеризации
являются перекисные соединения, растворимые в мономере, но нераст-
48
воримые в воде, например перекись бензоила, динмтрил азаизомасляной
кислоты и др.
Полимеризация инициируется свободными радикалами, и рост маю
ромолекулы начинается в результате присоединения молекулы мономера-
к инициирующему радикалу (см. стр. 11). Обрыв цепи в случае по-
лимеризации стирола осуществляется, как показали результаты иссле-
дований, главным образом путем рекомбинации:
R + СН2=СН->- RCH2-CH
Продукты распада инициатора остаются в полимере и загрязняют
его. Поэтому полистирол, получаемый в присутствии инициатора, не
отличается высокой чистотой, а это сказывается в первую очередь на
ухудшении его диэлектрических свойств.
Полимеризация стирола в блоке в присутствии инициатора ускоря-
ется с повышением давления. Повышение давления в процессе полиме-
ризации приводит также к возрастанию молекулярного веса полистиро-
ла. Ускорение полимеризации достигается также увеличением количе-
ства инициатора, причем уменьшается молекулярный вес полимера. '
Для изготовления полистирола высокой степени чистоты, обладаю-
щего наиболее высокими диэлектрическими показателями, полимериза-
цию ведут без инициатора, вызывая образование свободных радикалов
нагреванием мономера до высокой температуры (термическая полиме-
ризация). При термической полимеризации процесс инициируется теп-
ловой энергией, в результате разрушается л-связь и образуются бира-
дикалы:
СН=СН2 нагрева^ СН-СН2-СН2-СН
I । ।
X/ X/ X/
Рост же цепи протекает по монорадикальному механизму.
Термическая полимеризация протекает очень медленно и сильно
зависит от температуры. В частности, скорость образования свободных
49
радикалов возрастает при повышении температуры, в результате увели-
чивается скорость полимеризации. При повышении температуры увели-
чивается также скорость реакции обрыва цепи. В связи с тем, что ско-
рость роста описывается уравнением vp=£P[R • ] [М], а скорость обрыва
t'o=^olR,]2> то при повышенных температурах, увеличивающих скорость
роста, еще более увеличивается скорость обрыва.
Присутствие кислорода воздуха при повышенных температурах спо-
собствует еще более резкому увеличению скорости полимеризации за
счет образования перекисей и их распада на свободные радикалы:
СН2=СН + О2 -> СН2-СН-С6Н5
I I I
о—о
Одновременно с этим ускоряются реакции, приводящие в обрыву цепи,
в результате молекулярный вес образующихся макромолекул снижается.
При значительном повышении температуры (до 300—350° С) образовав-
шийся полистирол деполимеризуется до мономера.
При достижении 90%-ной степени превращения мономера в поли-
мер реакция замедляется, а 98—99%-ной — почти совершенно прекра-
щается. Таким образом, даже при длительном нагревании невозможно
получить блочный полимер, совершенно не содержащий мономера, а это
-сказывается на качестве полистирола: изготовленные из него изделия
при эксплуатации с течением времени растрескиваются.
Полистирол с минимальным содержанием мономера можно полу-
чить при непрерывном методе производства, применяя ступенчатое по-
вышение температуры полимеризации, начиная с 100—110° С и кончая
220—230° С. На последних стадиях при повышенных температурах за
счет увеличения скорости реакции и снижения вязкости расплава уда-
ется достичь максимальной (99,5%-ной) степени превращения мономера
в полимер, в результате содержание мономера в полистироле снижается
до 0,5%. Поддержание высокой температуры на конечной стадии поли-
меризации необходимо также для того, чтобы облегчить вытекание рас-
плава полимера из аппарата.
В промышленности блочный полистирол изготовляют в настоящее
время термической полимеризацией непрерывным методом.
Термическая полимеризация стирола в мономере проводится с уче-
том всех указанных особенностей образования полистирола.
Полимеризация стирола в мономере непрерывным способом. Про-
изводство полистирола блочным методом осуществляется в двух основ-
ных аппаратах: 1) в форполимеризаторе— цилиндрическом реакторе со
сферическим днищем, снабженном мешалкой и рубашкой для нагрева
и охлаждения; 2) в полимеризационной колонне из хромоникелевой
стали, состоящей из шести царг с индивидуальным обогревом и корпус-
ного днища. В форполимеризаторах происходит предварительная поли-
меризация стирола до содержания в нем полимера около 30%. Процесс
протекает с выделением тепла, которое отводится циркулирующей в ру-
50
башке водой, в постоянном токе инертного газа. Температура смеси в:
течение всего процесса поддерживается в пределах 75—85° С.
После того как содержание полимера в форполимеризаторе достиг-
нет 30%, начинается слив вязкого раствора в полимеризационную ко-
лонну. Одновременно с непрерывным поступлением форполимера в ко-
лонну производят непрерывную подачу такого же количества стирола в
форполимеризаторы. Окончательная полимеризация происходит в поли-
меризационной колонне. Колонна снабжена шнеком для выгрузки рас-
плавленного полистирола.
Полистирол непрерывно вытекает из нижней конической части ко-
лонны и с помощью шнека выдавливается в виде прутков в охлаждае-
мую проточной водой ванну, затем в грануляторе режется на кусочки
цилиндрической формы (гранулы). К моменту выгрузки массы из ко-
лонны полимеризация заканчивается и содержание мономера в полиме-
ре составляет не более 0,5%.
Блочный полистирол выпускают в соответствии с ГОСТ 9440—60
в виде бесцветного (марка Д) для электроизоляционных изделий и окра-
шенного в различные цвета (марка Т) для технических изделий и това-
ров широкого потребления. Первый применяется как высококачествен-
ный диэлектрик (перерабатывается экструзией на пленку), второй—•
для изготовления различных изделий технического назначения (литьем
под давлением).
Производственные методы полимеризации стирола в эмульсии.
В производственных условиях полимеризация стирола в эмульсии мо-
жет быть осуществлена как периодическим, так и непрерывным ме-
тодами.
Технологический процесс производства полистирола периодическим
методом состоит из полимеризации стирола в водной среде в присутст-
вии эмульгатора олеата натрия (0,5—1% от веса стирола) и инициато-
ров: перекиси бензоила ,или персульфатов калия, аммония (0,25—0,5%)-
Процесс ведут в эмалированном реакторе, снабженном мешалкой тур-
бинного типа. Воду применяют умягченную, освобожденную от солей,
дистиллированную или деминерализованную. Последнюю получают про-
пусканием обычной водопроводной воды через колонны, заполненные
ионообменными смолами.
Полимеризация идет при 95—96° С в течение 7—8 ч до содержания
мономера в полимере около 0,5%.
Для коагуляции полученного латекса применяют электролиты: сер-
нокислый алюминий, алюмокалиевые квасцы A1iK(SO4)2, кислоты (сер-
ную, соляную или муравьиную).
Сушка эмульсионного полистирола опасна в пожарном отношении.
Поэтому она производится в сушилках с кипящим слоем с применением
инертного теплоносителя или сильно увлажненного воздуха, а также в
пневмосушилках в виде трубы с винтовой насадкой. Сушат полимер до
остаточной влажности не более 0,5%). Высушенный полистирол просеи-
вают через сито и упаковывают в мешки.
Полистирол выпускают в виде гранул. Целью гранулирования явля-
ется улучшение теплопроводности, транспортабельности полийера, сни-
жение молекулярного веса за счет частичной деструкции для облегчения
5Г.'
переработки полистирола литьем под давлением. Выпускаемые грану-
лы— цилиндрической формы, их диаметр 3 мм, высота 5 мм.
Непрерывным методом эмульсионный полистирол получают в кас-
каде аппаратов «идеального смешения» в окислительно-восстановитель-
ной среде.
Эмульсионный полистирол выпускается по ГОСТ 9440—60 в виде
высокодисперсного порошка или гранул двух марок: марка А — для тех-
нических целей и изделий широкого потребления, марка Б — для полу-
чения пенопластов.
Производство полистирола суспензионным (гранульным) способом.
Гранульный (суспензионный) полистирол применяют для изготовления
ионитов и суспензионного полистирола (стиропора). Он выпускается по
МРТУ 6-05-957—68 двух марок: ПС-С — полистирол суспензионный
обыкновенный и ПС-СП — полистирол суспензионный пластифициро-
ванный.
Полимеризация в суспензии, так же как и эмульсионная, проходит
в двухфазной системе, состоящей из дисперсионной среды (воды) и дис-
персной фазы. Полимеры, полученные этими двумя методами, разли-
чаются величиной частиц и молекулярным весом (см. стр. 20).
Структурированные полимеры стирола, предназначенные для изго-
товления ионитов, получают исключительно полимеризацией в суспензии
(гранульной полимеризацией), так как физическая форма такого поли-
мера (шарообразные гранулы) чрезвычайно удобна для дальнейшего
превращения его в ионообменные полимерные материалы и использова-
ния полученного ионита (см. ч. 11, работа 173).
Полистирол при обычных температурных условиях представляет
собой твердый продукт, пл. 1,05 г/см3. Блочный полистирол прозрачен,
бесцветен; светопрозрачность его достигает 90%. Он имеет высокий
коэффициент преломления (1,60), что позволяет использовать его для из-
готовления оптических стекол.
Полимер растворим в мономере (стироле), в ароматических и хло-
рированных углеводородах, в сложных эфирах, например в бутилаце-
тате, этиленгликольдиацетате и др. Он нерастворим в спиртах и в бен-
зине.
Один из лучших диэлектриков, полистирол широко применяется в
качестве электроизоляционного материала для высокочастотной техни-
ки, так как частота поля и температура почти не влияют на его диэлект-
рические показатели.
Полистирол обладает довольно хорошей химической стойкостью
к кислотам и щелочам, а также очень высокой водостойкостью.
Механическая прочность полистирола невысока. Особенно мала
ударная вязкость (около 16—20 кг-см/см2), что не позволяет применять
полистирол в качестве конструкционного материала для деталей, испы-
тывающих более или менее значительные механические нагрузки. По-
листирол обладает хрупкостью, которая возрастает с течением времени
в процессе эксплуатации изделий из него в результате старения поли-
мера.
При нагревании до высоких температур (300—400° С) полистирол
деполимеризуется с образованием мономера (до 42%)- Это свойство
52
используют для получения мономерного продукта из отходов полисти-
рола. Деполимеризацию следует проводить в инертной среде, не до-
пуская окисления.
Полистирол довольно легко подвергается окислению, сульфирова-
нию, нитрованию, галогенированию в ароматическом ядре. При дейст-
вии химических агентов изменяются свойства полистирола, например,
сульфированный полистирол становится растворимым в воде; при окис-
лении происходит пожелтение полимера и частичная деструкция.
Полистирол, получаемый по свободнорадикальному механизму, име-
ет аморфную структуру. Он характеризуется значительной разветвлен-
ностью макромолекул и полимолекулярностью. При растяжении полиме-
ра происходит ориентация его макромолекул, что приводит к значитель-
ному увеличению прочности в направлении вытяжки. Это обстоятельство
используется в технике для изготовления пленок толщиной 100—10 мкм
(0,1—0,01 мм), получаемых растягиванием полистирола в двух перпен-
дикулярных направлениях. Такие пленки, отличающиеся большой проч-
ностью, применяются как электроизоляционный материал для изготов-
ления конденсаторов, где они сочетаются с металлическими пластин-
ками.
Обладая высокими диэлектрическими свойствами, полистирол нахо-
дит большое применение в электротехнике. Он используется также в
качестве конструкционного материала для деталей, не испытывающих
механических нагрузок, для изготовления изделий широкого потребле-
ния (различной посуды, главным образом имитирующей хрусталь).
В значительных количествах полистирол (эмульсионный и гранульный)
применяется для изготовления пенопластов — легких материалов с
очень малым объемным весом.
Полистирол перерабатывается в изделия всеми способами, приме-
няемыми для переработки термопластичных материалов (литьем под
давлением, экструзией, вакуумным и пневматическим формованием
листов и др.).
К недостаткам полистирола следует отнести его небольшую меха-
ническую прочность (хрупкость) и сравнительно низкую теплостой-
кость (около 80°).
Эти недостатки полистирола частично или полностью устраняются
в полимерах производных стирола (хлорстирола, метилстирола, винйл-
толуола и др.), а также при сополимеризации стирола или полистирола
•с другими мономерами.
Исходное сырье для получения
полистирола
Чистый стирол С6Н5СН = СН2 представляет собой бесцветную про-
зрачную жидкость; т. кип. 145,2°С (при 760 мм рт. ст.), d <=0,9012,
показатель преломления «о =1,5439; молекулярный вес 104,14.
В технике стирол получают каталитическим дегидрированием этил-
бензола. Дегидрирование — обратимая реакция:
С6Н5СН2СН3 С6Н5СН=СН2 + Н2
53
Равновесие ее определяется уравнением kF=/\H—ГАЗ=20,720—31,1 Т„
Оптимальные условия: 600—630°С; 0,1 атм. Выход при этом равен
70—80%.
В небольших количествах стирол содержится в каменноугольном
дегте и продуктах пиролиза нефти, откуда может быть выделен. Моно-
мерный стирол очень хорошо растворим во многих органических раство-
рителях. Он смешивается в любом соотношении с метиловым и этиловым
спиртом, ацетоном, ароматическими углеводородами, уксусной кислотой,
этилацетатом и т. д. Растворимость стирола в многоатомных спиртах
ограничена. Стирол сам является хорошим растворителем различных
органических веществ. В нем растворяются полистирол и другие высо-
комолекулярные вещества.
В воде стирол растворим незначительно: при 20° С в 100 г воды
растворяется 0,0125 г стирола; в свою очередь растворимость воды в-
стироле при 25° С составляет 0,066%.
Стирол может быть стабилизирован гидрохиноном, п-трет-бутилпи-
рокатехином, серой, фенолом, алюминиевым порошком и другими ве-
ществами. Гидрохинон служит ингибитором полимеризации стирола
только в присутствии кислорода воздуха; при отсутствии кислорода
гидрохинон не задерживает процесс полимеризации; кроме того, дейст-
вие ингибитора связано с температурой. Так, гидрохинон стабилизирует
стирол лишь при температурах до 100°С. При хранении стирол обычно
стабилизируют гидрохиноном или и-трет-бутилпирокатехином.
Подготовка продукта для полимеризации
Перед полимеризацией стирол необходимо очистить, так как он
может содержать в качестве примесей ингибиторы (стабилизаторы),
вводимые в мономер для предохранения его от полимеризации во вре-
мя хранения. Кроме ингибиторов примеси обычно содержат небольшие
количества этилбензола, хлоридов, воды, серы, альдегидов, а также по-
лимера.
Примеси, содержащиеся в мономерном стироле, в значительной сте-
пени влияют на качество получаемого из него полимера. Так, например,
небольшое количество этилбензола, испаряясь из полистирола, может
вызывать его растрескивание или потускнение. Большое количество
этилбензола понижает молекулярный вес полистирола. Вода, растворен-
ная в мономере, способна вызвать помутнение полимера и ухудшить
его диэлектрические свойства. Альдегиды понижают молекулярный вес
полимера, ухудшают диэлектрические свойства. Альдегиды и перекиси
образуются в результате окисления стирола при действии на него кис-
лорода воздуха.
Пузырьки в блочном полистироле часто являются следствием при-
сутствия какого-нибудь газа, например двуокиси углерода, образую-
щейся при разложении перекисей при повышенных температурах. Сера
понижает молекулярный вес и ухудшает светостойкость полученного
полимера. Присутствие небольшого количества полимера, образующе-
гося в стироле при продолжительном хранении, несколько повышает его
вязкость, но не мешает дальнейшей полимеризации. Содержание поли-
стирола может оказать вредное влияние в том случае, когда мономер
применяют для получения сополимеров.
При очистке стирола следует избегать или свести к минимуму дей-
ствие па него меди или ее сплавов, следов кислот, воздуха и нагревания
(за исключением тех случаев, когда в стироле содержатся ингибиторы).
Мономер обычно очищают перегонкой под вакуумом, так как вследствие
сравнительно высокой температуры кипения стирола (145° С) во время
разгонки при нормальном давлении продукт полимеризуется. Перегонка
ведется в колбе Кляйзена при нагревании на водяной бане или электри-
ческой плитке.
Для предотвращения полимеризации стирола во время его перегон-
ки в колбу необходимо добавить вещества, препятствующие полимери-
зации (ингибиторы). Обычно вводят гидрохинон в количестве около
0,2% от веса перегоняемого мономера, а также серный цвет, алюминие-
вый порошок, фенол и некоторые другие вещества. Чрезвычайно эф-
' фективным стабилизатором стирола является п-трет-бутилпирокатехин,
введение 0,01 % (от веса стирола) которого вполне достаточно для ста-
билизации стирола, предназначенного для хранения.
Перегонка под вакуумом должна проводиться при температуре не
выше 100°С (давление 50—100 мм рт. ст.).
Дистиллят собирают в охлаждаемый льдом чистый сухой сосуд.
Для предотвращения окисления стирола во время перегонки ее ведут в
среде инертного газа, для чего через капилляр пропускают азот. Это
предохраняет также от толчков во время перегонки. Для уменьшения
толчков можно ввести в колбу кусочки необожженной глины.
Перегоняющийся стирол собирают по фракциям. Первую фракцию,
не имеющую постоянной температуры кипения при установленном не
меняющемся разрежении или мутную от присутствия влаги, отбрасыва-
ют; вторую фракцию, кипящую при постоянной температуре при уста-
новленном разрежении, собирают как чистый продукт; третью .фракцию,
тоже не имеющую постоянной температуры кипения, часто содержащую
возгоняющийся ингибитор, также отбрасывают.
Вторую фракцию, собранную в чистую сухую посуду, после анализа
употребляют для полимеризации.
Если в стироле не имеется других загрязнений, кроме стабилизато-
ра (гидрохинона или п-трет-бутилпирокатехина), их можно удалить про-
мывкой щелочью без последующей перегонки. В этом случае стирол
сначала промывают в делительной воронке несколько раз 5—10%-ным
водным раствором щелочи до прекращения окрашивания щелочного рас-
твора, затем для удаления щелочи его промывают дистиллированной
водой до нейтральной реакции промывной воды. Поглощенная стиролом
вода удаляется вымораживанием при температуре до —15° С или же
высушиванием безводными сернокислым или хлористым кальцием или
силикагелем. Другие осушители применять не рекомендуется во избе-
жание введения в стирол нежелательных загрязнений.
Стирол, предназначенный для полимеризации в эмульсии, после
промывки водой можно не осушать.'
55
Чтобы предохранить очищенный мономер от воздействия воздуха,
его запаивают в ампулы. Хранить стирол следует в темноте и при низ-
кой температуре для предотвращения полимеризации.
Качественное определение стирола
и полистирола
Качественное определение стирола в реактиве можно осуществить
путем фракционной перегонки и установления точки кипения или опре-
делением показателя преломления.
Идентификацию стирола можно проводить также окислением его
до n-нитробензойной кислоты и восстановлением последней до п-амино-
бензойной кислоты. В результате диазотирования п-аминобензойнон
кислоты с р-нафтолом получают краситель ярко-красного цвета (мето-
дику см. ниже).
Для качественного обнаружения полистирола производят предва-
рительный пиролиз или деполимеризацию. Полученные продукты ана-
лизируют названными выше методами. Деполимеризации подвергают не
только полимеры, но и сополимеры стирола с другими мономерами.
В колбу, снабженную шариковым холодильником, помещают 0,1—
0,2 г деполимеризата, вливают 5 мл концентрированной азотной кисло-
ты и нагревают при кипении в течение 1 ч. К раствору приливают 20 мл
воды, переносят его в делительную воронку и экстрагируют трижды по
10 мл серным эфиром. Экстракты соединяют, два-три раза промывают
водой, промывные воды отбрасывают. Эфирные вытяжки три раза обра-
батывают 5%-ным раствором NaOH порциями по 5 мл.
Эфирные слои отбрасывают, щелочные экстракты и промывную
воду соединяют, нейтрализуют концентрированной НС1 и добавляют
еще 5 мл НС1. Раствор нагревают на водяной бане и вносят 1 г грану-
лированного цинка для восстановления нитробензойной кислоты. После
растворения цинка жидкость нейтрализуют 20%-ным раствором NaOH,
прибавляют избыток его для растворения гидроокиси цинка и экстраги-
руют эфиром; эфирный слой отбрасывают.
Щелочной раствор подкисляют концентрированной НС1, охлаждают
до комнатной температуры и приливают 1 мл 0,5 н. раствора NaNO2.
Полученную смесь вливают в избыток насыщенного раствора 0-нафтола
в 5%-ном растворе NaOH. Появление ярко-красного окрашивания, уси-
ливающегося от прибавления 20%-ного раствора NaOH, указывает на
присутствие стирола.
Количественный анализ стирола
Определение содержания стирола реактивом Кауфмана. В две ко-
нические колбы с пришлифованными пробками емкостью по 250 мл от-
вешивают на аналитических весах по 0,2 г стирола, приливают из бю-
ретки по 30—35 мл реактива Кауфмана, взбалтывают и оставляют
стоять в течение 5—10 мин. После этого в каждую колбу из мерного
цилиндра прибавляют по 10—15 мл 10%-ного раствора йодистого ка-
56
лия, разбавляют равным количеством воды, перемешивают и через
10—15 мин оттитровывают выделившийся иод 0,1 н. гипосульфитом, при-
бавив в качестве индикатора несколько капель раствора крахмала.
Параллельно ставят контрольный опыт.
Содержание стирола в процентах (х) вычисляют по формуле
х (д— *) *0,0052-100
g
тц.е а — объем 0,1 н. гипосульфита, израсходованного на титрование
контрольной пробы, мл; Ь — объем 0,1 н. гипосульфита, израсходованно-
го на титрование пробы с навеской, мл; £ —поправка к титру 0,1 и.
гипосульфита; g— навеска стирола, г; 0,0052 — количество стирола, со-
ответствующее 1 мл 0,1 н. гипосульфита,-г.
Приготовление реактива Кауфмана. Берут 13—14 вес. ч. бромистого калия, пред-
варительно высушенного при 130° С, и растворяют в 100 вес. ч. перегнаииого метило-
вого спирта (выполнять в присутствии руководителя). Раствор фильтруют, затем к нему
прибавляют 5,1 мл брома на 1 л раствора и оставляют стоять 12—15 дней, после чего
устанавливают его титр. Титр устанавливают следуюшим образом: к 20 мл бромного
раствора прибавляют несколько капель концентрированной соляной кислоты и 20 мл
10%-ного йодистого калия. Смесь разбавляют равным количеством воды и оттитровы-
вают выделившийся иод 0,1 н. гипосульфитом. После того как титр реактива Кауфмана
практически перестанет изменяться, его используют для анализа.
Определение содержания стирола по методу Вийса. При определе-
нии стирола по методу Вийса метиловый спирт заменяется четыреххло-
ристым углеродом. В основе метода лежит реакция двойных связей с
хлоридом по схеме
С6Нг,СН==СН2 + IC1 -> С6Н5СН1-СН2С1
IC1 + KI=I2 + КС1
I2 -|- 2Na2S2O3=2NaI 4- Na2S4Og
Навеску стирола около 1 г (или около 10 г полистирола, если не-
обходимо определить в нем свободный мономер), взвешенную с точ-
ностью до 0,0002 г (во втором случае 0,01 г), вносят в мерную колбу,
приливают 150—200 мл СС14 и при. помешивании растворяют мономер
(или полимер). Затем добавляют СС14 до метки и взбалтывают. В две
конические колбы наливают по 10 мл (во втором случае 50 мл) приго-
товленного раствора и по 10 мл раствора Вийса. Колбы закрывают
пробкой, смоченной раствором KI, и помещают в темное место на
15 мин, затем добавляют 15 мл раствора KI и 100 мл дистиллированной
воды. Содержимое колбы тщательно перемешивают и титруют раствором
Na2S2O3, добавляя к концу титрования раствор крахмала.
В аналогичных условиях титруют контрольную пробу.
Содержание стирола (в %) вычисляют по формуле
(а — Ь) *-0,0052-100-250
Л"- ———
50g
где а и b — количество 0,1 н. раствора Na2S20a, пошедшего на титро-
вание контрольной и анализируемой проб, мл; k — поправка к титру
0,1 н. раствора Na2S2O3; 0,0052количество стирола, соответствующее
1 мл точно 0,1 н. раствора Na2S2O3, г; g— навеска полимера, г.
57
Приготовление раствора Вийса. В мерной колбе емкостью 1 л растворяют 8 г трех-
хлористого или однохлористого иода и 9 г металлического иода в 300 мл СС14 и 700 млг
ледяной уксусной кислоты. Раствор фильтруют в склянку из темного стекла и хранят-
в темном месте. Реактивом можно пользоваться через три дня после его приготовления.
Все вещества для приготовления реактива должны быть химически чистые.
Определение стирола в дисперсионной среде методом некомпенса-
ционного потенциометрического титрования. В стакан для титрования,
помещенный на магнитную мешалку, вливают 50 мл водно-спиртовой
смеси и из капельницы по каплям вносят 1—2 г дисперсионной жидко-
сти, взвешенной с точностью до 0,001 г, после чего титруют 0,1 н. раст-
вором брома в ледяной уксусной кислоте. В аналогичных условиях
титруют контрольную пробу (описание метода см. [5]).
Определение воды. Определение воды в мономере можно осущест-
вить с помощью реактива Фишера на ручной или автоматической элект-
рометрической установке.
Рис. 14. Колба для титрования воды Рис. 15. Колба для
реактивом Фишера: отбора проб
1 — платиновые электроды; 2 — микробю-
ретка; 3 — хлоркальциевая трубка; 4 —
пробка; 5 — магнитная мешалка
В колбу для титрования (рис. 14), снабженную магнитной мешал-
кой, наливают абсолютированный метанол или смесь метанола с мети-
ленхлоридом (1:1 по объему) в количестве, необходимом для погруже-
ния платиновых электродов. Реактивом Фишера оттитровывают воду,
содержащуюся в растворителе, и воду, адсорбированную стенками со-
суда и электродами. Сначала реактив приливают быстро (1 каплю в
секунду), при этом стрелка микроамперметра очень слабо отклоняется
от нулевого положения, при дальнейшем добавлении реактива стрелка
амперметра начинает сильно колебаться. В это время подачу реактива
производят медленно (1 каплю в 6—7 с). Титрование продолжают до
тех пор, пока стрелка амперметра не установится на определенном деле-
58
нии шкалы и останется в таком положении в течение 1 мин. Количество
реактива, пошедшее на титрование, не фиксируют.
Для определения воды в мономере в колбу с оттитрованным раст-
ворителем вносят через отверстие в пробке 4 (рис. 14) навеску анали-
зируемого вещества, которую передавливают сухим инертным газом из
колбы (рис. 15) и оттптровывают воду реактивом Фишера по описан-
ному выше методу.
Вычисление водного эквивалента реактива К (г/мл) и содержание
воды х (вес. %) в анализируемой пробе производят по формулам:
,, Ь аКЮО
К ——; х------------,
а Ь\
Здесь Ь и Ь} — навески воды (в контрольном опыте) и анализируемой
пробы, г; a, at — количество реактива, затраченное на титрование навес-
ки воды и анализируемой пробы.
Приготовление реактива Фишера. В склянку из темного стекла вносят 85 г метал-
лического иода (предварительно измельченного и высушенного в эксикаторе над СаСЬ
в течение 48 ч), 265 мл абсолютированного пиридина. Склянку закрывают пробкой,
оставляют при комнатной температуре и периодически взбалтывают до полного раство-
рения иода. Затем в раствор вносят 665 мл абсолютированного метанола, закрывают
пробкой и взбалтывают.
После перемешивания пробку из склянки вынимают и склянку быстро закрывают
пробкой с двумя стеклянными трубками (предварительно тщательно высушенными),
соединенными предварительно высушенным резиновым шлангом (рис. 15).
Склянку с содержимым помещают в холодную воду со льдом и реактив при
охлаждении насыщают SO2, сняв при этом резиновый шланг с наружных концов стек-
лянных трубок. SO2 вводят в склянку через длинную трубку до тех пор, пока вес склян-
ки с содержимым не увеличится на 65—75%.
Определение чистоты стирола криоскопическим методом. Метод ос-
нован на зависимости температуры замерзания стирола от содержания
в нем примесей и прежде всего воды и этилбензола.
Смесь из ацетона и четыреххлористого углерода в соотношении
1 : 1 помещают в сосуд Дьюара и охлаждают сухим льдом до —42° С.
Стирол (10 мл) заливают в чистую и сухую пробирку, помещенную в
охлаждающую смесь (рис. 16). В пробирку опускают термометр и сле-
дят за понижением температуры. Когда температура понизится до
—30° С, начинают перемешивание и фиксируют понижение температу-
ры с точностью до 0,01° С через каждые 30 с. При охлаждении темпера-
тура падает до минимума, затем быстро поднимается и снова начинает
опускаться. Фиксируют несколько точек при падении температуры. В
координатах время — температура строят кривую зависимости темпера-
туры от времени охлаждения. Температуру замерзания стирола находят
по кривым, приведенным на рис. 17. Для этого продолжают кривую
охлаждения твердого вещества до пересечения с кривой охлаждения
жидкости. Точка пересечения является температурой замерзания.
Содержание стирола х (%) вычисляют по формуле (5] .
л= 100+2,47[-/ + ( -30,63)],
59
где t — температура замерзания анализируемого стирола; —30,63 —
температура замерзания 100%-ного стирола; 2,47 — количество приме-
си этилбензола, понижающее температуру замерзания стирола на 1°, %.
Определение дивинилбензола в стироле спектрофотометрическим ме-
тодом. Метод основан на различном поглощении света дивинилбензолом
и стиролом в ультрафиолетовой области спектра при Х=313 нм* [5].
Для построения градуировочных графиков приготовляют ряд эта-
лонных растворов стирола с содержанием дивинилбензола 0,001; 0,003;
0,004; 0,005; 0,006; 0,008; 0,01 %.
Рис. 16. Прибор
для определения
температуры за-
мерзания стирола
Рис. 17. Зависимость температуры замерза-
ния стирола от времени охлаждения:
1 — кривая охлаждения жидкости; 2 — кривая пе-
реохлаждения жидкости; 3 — кривая охлаждения
твердого вещества
Оптическая плотность чистого стирола До=0,215 при Х=313 мкм
для толщины слоя 5 мм. Измерения Do производят по отношению к од-
ному кварцевому окошку.
Значения оптической плотности D для дивинилбензола в зависимо-
сти от его содержания в эталонных растворах {5] приведены ниже:
Содержание дивинилбензола в стиро-
ле, % ............ ...... . 0,001 0,003 0,004 0,005 0,006 0,008 0,010
Оптическая плотность ..... . . 0,180 0,500 0,660 0,820 0,9801,3061,603
На основании этих данных строят градуировочный график, отклады-
вая по оси абсцисс содержание дивинилбензола (в %), а по оси орди-
нат — оптическую плотность D, измеренную по отношению к чистому
стиролу при Х=313 нм для толщины слоя 5 мм.
Определение гидрохинона. Стирол не должен содержать гидрохино-
на. Для проведения пробы к 5 мл стирола добавляют 5 мл раст-
вора NaOH и смесь взбалтывают. При отсутствии гидрохинона нижний
|Слой не дает окраски. Если появляется желтая окраска, то стирол от-
-------——
* 1 нм (нанометр) = Г ммк (миллимикрон) = 10-9 м.
Ю
мывают от гидрохинона раствором NaOH, как указано выше. При этом
гидрохинон кислородом воздуха окисляется в хинон:
2С6Н4 (ОН)2 + О2 -> 2С6Н4О2 + 2Н2О
Техника безопасности при работе
со стиролом и полистиролом
Стирол — горючий углероводород, поэтому при работе с ним и при-
хранении его должны соблюдаться такие же меры предосторожности,,
как при работе с другими легковоспламеняющимися жидкостями (бен-
золом, толуолом и др.). Взрывоопасные концентрации стирола в смеси
с воздухом при атмосферном давлении и температуре 29,3—65,3° С на-
ходятся в пределах от 1,1 до 6,1 объемн. %. При работе со стиролом
следует избегать открытого огня, трения и статического электричества.
Пары стирола умеренно токсичны при вдыхании. Сладковатый
запах мономера и неприятный резкий запах, обусловленный присутст-
вием альдегидов, легко распознаются задолго до достижения токсичных
концентраций паров мономера. Раздражение слизистых оболочек глаз и.
носа также ощущается до достижения опасных концентраций, уже при
6,0—6,3 мг/л.
Опасны концентрации паров стирола в воздухе 10—12 мг/л, дейст-
вующие в течение 8 ч, или концентрация 46 мг/л в течение 30—60 мин.
Такие концентрации вызывали смерть у животных вследствие действия
на центральную нервную систему или пневмонии, вызванной раздраже-
нием легких.
Концентрация паров стирола 2 мг/л является максимально допусти-
мой при действии на человека в течение 8 ч.
Соприкосновения кожи с жидким стиролом следует избегать, так как:
повторное или продолжительное действие его вызывает раздражение.
Прием стирола внутрь организма опасен. Если он случайно попадает
через рот, то необходимо немедленно принять рвотное, которое не долж-
но содержать спирта, и вызвать врача.
При хранении стирола следует принять меры, предотвращающие
самопроизвольную полимеризацию, для чего в мономер добавляют ка-
кой-либо ингибитор. Образование полимера во время хранения нежела-
тельно не только потому, что это может отрицательно сказаться на
последующей работе, но и вследствие выделения тепла во время этого-
процесса, которое становится опасным, особенно в закрытом резервуа-
ре, где повышение давления паров мономера может создать опасность-
взрыв а.
Применяемый для полимеризации инициатор — перекись бензоила —
взрывоопасное вещество, взрывающееся в сухом виде при ударах, тре-
нии, нагревании и т. п. Поэтому при работе с ней нужно соблюдать
осторожность.
Перекись бензоила должна храниться в закрытых стеклянных или
жестяных банках во влажном состоянии (залита водой или спиртом)..
В сухом виде перекись бензоила выдается работающему в небольшом:
количестве, необходимом только для выполнения текущей работы.
61.
Переработка полистирольных пластиков в изделия осуществляется
при 150—240° С. В этих условиях при механическом воздействии может
иметь место частичная термомеханическая и термоокислительная дест-
рукция с выделением стирола, бензальдегида, ацетофенона и др. По-
этому в помещении необходимо иметь приточно-вытяжную вентиляцию.
Полистирольная пыль легко электризуется; в смеси с воздухом об-
разует взрывчатые смеси, .нижний предел взрываемости которых состав-
ляет 25—27,5 г/м3. Полистирол физиологически безвреден.
РАБОТА 9. СИНТЕЗ СТИРОЛА
ИЗ а-ХЛОРЭТИЛБЕНЗОЛА
Для осуществления синтеза стирола в условиях учебных лаборато-
рий предлагаются методы, не требующие специального оборудования и
сравнительно легко осуществимые.
Синтез стирола может быть осуществлен в лабораторных условиях
из а-хлорэтилбензола через метилфенилкарбинол. Реакция протекает в
три стадии:
1) а-Хлорэтилбензол получают хлорированием этилбензола без на-
гревания в присутствии катализатора — пятихлористого фосфора. В этом
случае хлор замещает водород в основном в боковой цепи в «-положе-
нии по схеме
РС15
1) С6Н5СН2СН3 + С12--► С6Н5СНС1СН3 4- НС1
2) Омыление а-хлорэтилбензола и получение метилфенилкарби-
нола:
+ Н2О
С6Н5СНС1СН3----> С6Н5СН (ОН) СН3 4- НС1
3) Дегидратация метилфенилкарбинола и получение стирола:
—то
С6Н5СН (ОН) СН3--> С6Н5СН=СН2
Получение а-хлорэтилбензола.
Исходные продукты: этилбензол (400 г); пятихлористый фосфор—(1% от веса
этилбензола) — 4 г; хлор (газ) в баллоне.
Оборудование: прибор (рис. 18); сосуд для раствора щелочи; склянки Тищенко
или Дрекселя — 2 шт.; прибор для перегонки в вакууме.
В цилиндрический сосуд диаметром 65 мм и высотой 300 мм, изго-
товленный из прочного стекла и снабженный обратным холодильником,
термометром и барботером для пропускания хлор'а (рис. 18), загружают
этилбензол и пятихлористый фосфор. Для улавливания избыточного
хлора и выделяющегося при реакции газообразного хлористого водорода
верхний конец обратного холодильника соединяют каучуковым шлангом
с отрезком стеклянной трубки, которую опускают в сосуд, наполненный
водным раствором щелочи.
Прибор собирают и начинают пропускать хлор в жидкость из бал-
лона через барботер. Предварительно хлор должен проходить через
62
склянки (Тищенко или Дрекселя): одну, заполненную концентрирован-
ной серной кислотой, вторую — пустую. Газ пропускают с такой ско-
ростью, чтобы температура смеси, повышающаяся за счет выделяюще-
гося при реакции тепла, поддерживалась около 70° С.
Реакцию ведут в течение 2 ч (увеличение в весе реакционной мас-
сы должно составлять около 60% от теоретического).
По окончании реакции полученный продукт хлорирования переносят
из реакционного сосуда в колбу
Кляйзена и отгоняют под вакуу-
мом не вступивший в реакцию
этилбензол — фракция, кипящая
до 45° С при 25 мм рт. ст.
После отгонки этилбензола в
колбе остается продукт, состоя-
щий из а-хлорэтилбензола и не-
большого количества высших
продуктов хлорирования.
Полученный продукт можно
не подвергать дополнительной
очистке и применять непосредст-
венно для синтеза стирола.
Получение метилфенилкар-
бинола.
Исходные продукты: а-хлорэтилбен-
зол — 200 г; сода кальцинированная —
97 г; бисульфат калия; сульфат натрия.
Оборудование: трехгорлая колба
емкостью 2 л, снабженная мешалкой с
герметическим затвором, обратным хо-
лодильником и термометром; баня во-
дяная; делительная воронка; установка
для перегонки с вакуумом; колба Вюрца с обратным холодильником и приемником;.
баня со сплавом Вуда или полиэтилсилоксановой жидкостью.
В трехгорлой колбе прибора (рис. 19) при перемешивании раство-
ряют кальцинированную соду в 873 мл дистиллированной воды, а затем
вводят а-хлорэтилбензол. Не выключая мешалки, содержимое колбы на-
гревают на бане. Реакция начинается при 85—90° С и заканчивается при
кипении смеси. Выделяющаяся при этом двуокись углерода, проходя че-
рез обратный холодильник, отводится в атмосферу.
Реакция продолжается приблизительно 2—2,5 ч до прекращения
выделения СО2. После окончания реакции содержимое колбы, состоящее
из двух слоев, переливают в делительную воронку, где верхний масля-
нистый слой отделяется от нижнего водного. Верхний слой, представля-
ющий собой метилфенилкарбинол, очищают перегонкой иод вакуумом.
Фракцию, кипящую при 98—100° С и давлении 20—22 мм рт. ст., отби-
рают как чистый продукт.
Получение стирола. Очищенный перегонкой под вакуумом метил-
фенилкарбинол загружают в колбу Вюрца и вводят туда же гидросуль-
фат калия KHSO4 (3—5% от веса метилфенилкарбинола).
43;
Колбу соединяют с прямым холодильником и приемной склянкой
для сбора стирола и нагревают содержимое колбы в бане со сплавом
Вуда вначале при 120° С, затем температуру поднимают до 200° С и
заканчивают реакцию при этой температуре.
Стирол по мере образования отгоняется вместе с получающейся при
реакции водой так, что в приемнике образуются два слоя. Верхний
-слой, представляющий собой стирол, отделяют в делительной воронке,
-сушат сульфатом натрия и перегоняют в вакууме в среде азота при
55° С и давлении 30 мм рт. ст.
Выход стирола составляет около 75% от теоретического.
РАБОТАЮ. СИНТЕЗ СТИРОЛА
ИЗ КОРИЧНОЙ КИСЛОТЫ
Удобным методом синтеза стирола в лабораторных условиях яв-
ляется также термическое декарбоксилирование' коричной кислоты по
схеме
нагревание
С6Н5СН=СНСООН-------> СёН5СН=СН2 + со2
Исходные продукты: коричная кислота—100 г; гидрохинон — 2,0 г.
Оборудование: прибор для перегонки с дефлегматором.
В круглодонную колбу емкостью 0,5 л, снабженную дефлегматором
высотой 24 см, прямым холодильником и приемной склянкой, помеща-
ют смесь из 100 г порошкообразной коричной кислоты и 1,5 г гидро-
хинона.
В приемную склянку вводят 0,5 г гидрохинона, а в колбу с коричной
кислотой добавляют несколько кусочков необожженной глины. Содер-
жимое колбы быстро нагревают пламенем горелки на асбестовой сетке
или воронке Бабо до начала отгонки стирола. Во время отгонки нагре-
вание регулируют таким образом, чтобы температура отходящих паров
все время была не выше 120° С.
Реакция продолжается в течение 3,5—5 ч и заканчивается тогда,
когда стирол уже больше не перегоняется и температура в верхней час-
ти дефлегматора начинает быстро возрастать.
Дистиллят состоит из маслянистого продукта, окрашенного в соло-
менно-желтый цвет, и небольшого количества воды; в реакционной колбе
остается смолистый остаток темного цвета. К дистилляту прибавляют
около 70 мл воды и водную смесь перегоняют еще раз. Стирол легко
перегоняется с водяным паром и отслаивается в виде бесцветного про-
дукта, который отделяют от воды в делительной воронке, затем сушат
небольшим количеством хлористого кальция и перегоняют под умень-
шенным давлением, охлаждая холодильник и приемник льдом или ледя-
ной вОдой до 5° С. Чистый -стирол собирают при 44—46° С (40 мм рт. ст.)
или при 60—63° С (60 мм рт. ст.).
Выход стирола — около 40% от теоретического.
-64
РАБОТА 11. ПОЛУЧЕНИЕ ПОЛИСТИРОЛА ТЕРМИЧЕСКОЙ
ПОЛИМЕРИЗАЦИЕЙ СТИРОЛА В БЛОКЕ
Исходный продукт: стирол (свежеперегнанный) — 10 мл.
Оборудование: толстостенная ампула из прочного стекла на 50 мл; пипетка; термо-
шкаф; прибор для определения термомеханпческой кривой; прибор для определения
температуры размягчения.
В толстостенную ампулу на 50 мл вводят с помощью градуирован-
ной пипетки 10 мл стирола. Ампулу запаивают на паяльной горелке,
помещают в термошкаф, нагретый до 170° С, и нагревают в течение
3—4 ч. Затем дают ампуле охладиться и осторожно вскрывают ее, на-
гревая в пламени горелки запаянный конец. Затем на расширенной час-
ти делают надрез напильником. Завернув ампулу в ткань, отламывают
надрезанный конец, освобождая блок полимера. Получается твердый
прозрачный бесцветный полистирол с выходом 98% от веса стирола.
Определяют зависимость деформации полистирола от температуры
(термомеханическую кривую) на приборе Журкова илн консистометре
Хеплера [7] и индекс расплава (см. ч. II, стр. 233).
РАБОТА 12. ПОЛУЧЕНИЕ ИЗДЕЛИЙ ИЗ БЛОЧНОГО
ПОЛИСТИРОЛА ЛИТЬЕМ ПОД ДАВЛЕНИЕМ
Блочный полистирол применяется для литья под давлением в виде
крупнозернистого материала без какой-либо предварительной подго-
товки.
Полистирол хорошо отливается в широком интервале температур
и большом диапазоне удельного давления.
Исходный продукт: полистирол гранулированный — 1Ю0 г.
Оборудование: лабораторная литьевая машина.
Учитывая высокую вязкость расплава, литье обычно ведут при наи-
большей температуре — в пределах 190—215° С. Применяемое удельное
давление колеблется в пределах 800—1500 кг/см2. Для .небольших изде-
лий цикл литья составляет 30—60 с при выдержке в форме в течение
нескольких секунд. Режим литья устанавливается опытным путем в
зависимости от молекулярного веса полистирола, поперечного сечения
литников, величины и конфигурации пресс-формы и т. п.
Пресс-форму литьевой машины охлаждают водой до 25—40° С, но
для уменьшения внутренних напряжений отлитых изделий можно повы-
сить температуру формы, чтобы изделия остывали медленнее.
Чтобы уменьшить внутренние напряжения, необходимо поддержи-
вать одинаковую температуру во всех частях внутренней поверхности
формы. Разность температур в различных частях пресс-формы, а также
поступающей и выходящей охлаждающей жидкости не должна пре-
вышать 3-—6°.
При литье полистирола возможно заклинивание изделий в пресс-
форме, что затрудняет их извлечение. Одной из основных причин закли-
3—1453 65
нивания является высокое давление при литье. Надо иметь в виду, что
во время процесса литья полистирола объем его под действием давления
значительно уменьшается по сравнению с исходным. Так, например, при
температуре литья 232° С и удельном давлении 1120 кг/см2 объем поли-
стирола уменьшается на 6,8% по сравнению с первоначальным объемом.
Когда же давление снимается и материал охлаждается, он снова уве-
личивается в объеме, следствием чего является иногда заклинивание из-
делия в форме.
Поэтому в процессе литья под давлением необходимо постоянно
учитывать такие важные факторы, как температура и давление: при на-
гревании материал расширяется, а под воздействием применяемого дав-
ления сжимается. В результате одновременного' действия обоих этих
факторов и происходит окончательное формование изделия.
Если увеличивать удельное давление при литье, то увеличивается
и вес изделия, вследствие чего окончательный объем материала в форме
будет больше, нежели при меньшем удельном давлении. Таким образом,
с увеличением удельного давления при литье размеры охлажденных из-
делий будут приближаться к размерам внутренней полости формы и
поэтому, с одной стороны, уменьшается усадка изделий, а с другой —
затрудняется их выемка.
Для получения наиболее чистых изделий из полистирола необходи-
мо тщательно предохранять исходный продукт от загрязнений. Во вре-
мя пересыпания, встряхивания и других операций частицы полистирола
особенно легко заряжаются статическим электричеством вследствие
взаимного трения отдельных крупинок при их перемещении. Заряжен-
ные же частицы полимера притягивают к себе пылинки из воздуха, что
ведет к загрязнению продукта. Поэтому во время работы литьевой ма-
шины загрузочный бункер должен быть обязательно закрыт крышкой,
а для хранения полистирола следует применять пыленепроницаемые
мешки из полиэтилена или бумаги.
Определяют удельную ударную вязкость, диэлектрические свойства
и водостойкость образцов (см. ч. II).
РАБОТА 13. ПОЛУЧЕНИЕ ПОЛИСТИРОЛА ПОЛИМЕРИЗАЦИЕЙ
СТИРОЛА В ЭМУЛЬСИИ
Исходные продукты: стирол (свежеперегнаиный) — 30 г; вода (дистиллирован-
ная)— ЮО мл; олеиновая кислота—1 г, щелочь — 0,5 г; персульфат калия или аммо-
ния— 0,3 г.
Оборудование: трехгорлая колба емкостью 0,5 л, снабженная обратным холодиль-
ником, мешалкой, герметическим затвором и термометром (см. рис. 19); капельная во-
ронка; стакан емкостью 100 мл; водяная баня; фарфоровый стакан емкостью 1 л;
воронка Бюхнера.
В трехгорлую колбу прибора' заливают дистиллированную воду,
нагретую до 50° С, и при работающей мешалке прибавляют олеиновую
кислоту и щелочь.
После 5—10 мин перемешивания, не выключая мешалки, через ка-
пельную воронку вносят стирол, а затем персульфат калия (или аммо-
66
ния), предварительно растворенный в 5 мл дистиллированной воды.
Смесь нагревают на водяной бане 6—8 ч при 80—85° С. Определяют
содержание мономера в латексе методом некомпенсационного потенцио-
метрического титрования [5].
По окончании полимеризации в колбу с латексом прибавляют 25—
30 мл концентрированной муравьиной или соляной кислоты для разру-
шения эмульсии и коагуляции полимера. Эмульсию разрушают в -тече-
ние 30—40 мин при перемешивании, после чего смесь оставляют на
3—4 ч.
В случае плохой коагуляции содержимое колбы переносят в литро-
вый стакан, разбавляют 200—300 мл воды, добавляют 20 мл кислоты
или метанола и кипятят 30—40 мин, после чего оставляют стоять на
2—3 ч для осаждения полимера.
После осаждения порошка полимера его отделяют от жидкой фа-
зы, промывают на воронке Бюхнера несколько раз 10—15%-ным раст-
вором щелочи, а затем 4—5 раз дистиллированной водой, нагретой до
50—60° С, и сушат до постоянного веса в сушильном шкафу или вакуум-
сушилке при 60—65° С.
Чтобы подготовить эмульсионный полистирол (порошкообразный)
для переработки в изделия методом литья под давлением, его предва-
рительно подвергают вальцеванию при 70—80° С с последующим охлаж-
дением и дроблением продукта или обработке на шнек-машинах. В ре-
зультате вальцевания и шнекования снижается молекулярный вес по-
лимера вследствие термомеханической деструкции. Это влечет за собой
снижение температуры перехода полимера в текучее состояние, сниже-
ние вязкости расплава, а следовательно, улучшение литьевых свойств
его. Кроме того, оба вида специальной подготовки дают возможность
получить полистирол в гранулированном виде.
В некоторых случаях эмульсионный полистирол сначала подверга-
ют холодному таблетированию с целью уменьшения его насыпного веса
и повышения теплопроводности, а затем термообработке при 160—
180° С в течение 30—40 мин.
Подготовленный таким образом эмульсионный полистирол в виде
крупных зерен, гранул или таблеток диаметром 5 мм и весом около 0,5 г
можно перерабатывать в изделия литьем под давлением (см. работу 12).
РАБОТА 14. ИЗГОТОВЛЕНИЕ ГАЗОНАПОЛНЕННОГО МАТЕРИАЛА
{ПЕНОПЛАСТА] ИЗ ЭМУЛЬСИОННОГО ПОЛИСТИРОЛА
Эмульсионный полистирол в значительных количествах применяется
для изготовления газонаполненных материалов (пенопластов), облада-
ющих очень малым объемным весом. Объемный вес некоторых марок
пенополистирола составляет 0,04—0,06 г/см3.
Пенополистирол широко применяется в строительстве; холодильном
машиностроении, на транспорте и в других отраслях народного хозяйст-
ва в качестве термо- и звукоизоляционного материала.
Получение пенополистирола возможно двумя методами: прессовым
(марки ПС-1 и ПС-4) и беспреосовым (марка ПС-Б).
3* «7
Технологический процесс производства полистирольных пенопластов
с применением внешнего давления состоит из трех основных стадий:
а) смешение полистирола с газообразователем (порофором); б) прес-
сование композиции; в) вспенивание отпрессованной заготовки в специ-
альной форме.
Исходные продукты: полистирол эмульсионный; порообразователь: карбонат аммо-
ния или порофор ЧХЗ.
Оборудование: шаровая лабораторная мельница (можно использовать ступку);
герметическая пресс-форма; пресс; термошкаф; ванна с горячей водой.
Порошкообразный эмульсионный полистирол (молекулярный вес
до 130000) смешивают с 4% просеянного карбоната аммония в шаровой’
мельнице в течение 12—24 ч. Вместо минерального порообразователя
можно применять органический, например динитрил азоизомасляной
кислоты (порофор ЧХЗ) в количестве 15% (на 85 г полистирола берут
15 г порообразователя).
После перемешивания в шаровой мельнице массу помещают в гер-
метическую пресс-форму и прессуют при удельном давлении 100—
150 кг/см2 и 150° С с выдержкой при этой температуре под давлением
в течение 1,5—2 мин на 1 мм толщины заготовки. При этом порофор
разлагается и газ равномерно распределяется по всей заготовке. Внеш-
нее давление при прессовании должно быть на 10—15% выше противо-
давления газов прессуемой заготовки.
Охлажденный под давлением до 40—50° С запрессованный образец
вынимают из пресс-формы и помещают в термокамеру, нагретую до
130—140° С для вспенивания. Процесс вспенивания происходит за счет
расширения газов, образовавшихся при разложении порообразователя
во время запрессовки заготовки. При разложении карбоната аммония
выделяются СО2 и NH3, а в случае употребления порофора ЧХЗ выде-
ляется азот. Вспенивание можно проводить также, опустив запрессо-
ванный образец в горячую воду, нагретую до 95—98° С. После вспени-
вания и увеличения в объеме образец всплывает на поверхность воды.
Зависимость длительности процесса вспенивания от толщины за-
прессованного образца приведена ниже:
Толщина, мм......................... 5 12 20 30
Продолжительность вспенивания, мин ... 20 30 60 60
При введении 15% динитрила азоизомасляной кислоты объемный
вес материала составляет 0,06 г/см3. При употреблении карбоната ам-
мония объемный вес меняется в зависимости от количества порообра-
зователя следующим образом:
Количество (МН4)2СОз, %.............2 4 5
Объемный вес, г/см3 ................ 0,1 0,07 0,042
Если при вспенивании образец пенополистирола покоробился, то
его выпрямляют. Выпрямление ведут при 80° С в прессе под давле-
нием до 5 кг/см2. Определяют объемный вес полученного материала
(см. ч. II).
68
РАБОТА 15. ПОЛУЧЕНИЕ ПОЛИСТИРОЛА ПОЛИМЕРИЗАЦИЕЙ
С ПЕРСУЛЬФАТОМ АММОНИЯ
Исходные продукты: стирол (свежеперегнанный) — 30 г; вода дистиллирован-
ная— 100 мл; персульфат аммония — 0,5 г.
Оборудование: прибор (см. рис. 19); стакан емкостью! л в качестве водяной ба-
ни, фарфоровый стакан.
Персульфат аммония растворяют в воде и загружают в трехгорлую
колбу емкостью 0,3 л. Затем вносят стирол, включают мешалку, пуска-
ют воду в холодильник и нагревают колбу на водяной бане до 80° С.
Через некоторое время после начала нагревания смесь в колбе приоб-
ретает молочно-белый цвет. При 80° С реакцию проводят 6—7 ч, после
чего содержимое колбы выливают в стакан, добавляют 10 мл концен-
трированной соляной кислоты и при нагревании и перемешивании про-
водят коагуляцию полимера. Полученный полистирол отфильтровывают
на воронке Бюхнера, промывают водой до нейтральной реакции про-
мывных вод и сушат до постоянного веса в термошкафу или вакуум-
сушилке при 60—65° С.
Рассчитывают выход полимера в процентах по отношению к взя-
тому стиролу. Определяют индекс расплава полимера ,и перерабатывают
литьем под давлением (см. стр. 65). Образцы испытывают на ударную
вязкость (см. ч. II).
РАБОТА 16. ПОЛУЧЕНИЕ ПОЛИСТИРОЛА ПОЛИМЕРИЗАЦИЕЙ
СТИРОЛА В СУСПЕНЗИИ
Исходные продукты: стирол (свежеперегнанный) — 1Ю г; перекись бензоила —
0,2 г; поливиниловый спирт — 0,34 г; вода (дистиллированная) — 60 мл. .,
Оборудование: пробирка диаметром 45 мм и высотой 1<90 мм, имеющая боковой
отвод и снабженная герметическим затвором, механической мешалкой и обратным холо-
дильником (рис. 20); химический стакан емкостью 1' л; воронка Бюхнера; вакуум,-су-
шильный шкаф.
В пробирке прибора для гранульной полимеризации (рис. 20) раст-
воряют в дистиллированной воде при нагревании до 60° С поливинило-
вый спирт. Затем отдельно в стироле растворяют перекись бензоила.
После растворения перекиси бензоила стирол с инициатором зали-
вают в пробирку, в охлажденный до комнатной температуры водный
раствор поливинилового спирта, включают мешалку, пускают воду в
холодильник и пробирку нагревают на водяной бане * до 80° С. Скорость
мешалки регулируют с таким расчетом, чтобы стирол разбивался на
отдельные шарики (гранулы) небольшой величины, не соединяясь в
общую массу. Установленную постоянную скорость мешалки необхо-
димо поддерживать в течение всего процесса полимеризации, не допу-
ская перерыва в работе и остановки мешалки во избежание слипания
шариков и образования бесформенного комка полимера.
* В качестве водяной бани применяют химический стакан, через прозрачные стенки
которого удобно наблюдать за образованием гранул в процессе полимеризации.
69
Температуру в бане рекомендуется поддерживать точно 80° С, не
допуская перегрева. Процесс полимеризации обычно заканчивается че-
рез 3—4 ч. Контролем окончания реакции может служить опускание
шариков полистирола на дно пробирки вследствие увеличения их плот-
ности. Если при остановке мешалки гранулы не
опускаются, то реакцию продолжают.
Готовый продукт извлекают из реакционного
сосуда, отфильтровывают, промывают теплой водой,
высушивают и взвешивают. Определяют выход по-
лимера в процентах по отношению к взятому для
полимеризации стиролу. В фильтрате определяют
содержание непрореагировавшего мономера, (стр.
58) и рассчитывают степень конверсии.
Полученный полимер используют для выполне-
ния работы 12.
Примечание. Для получения окрашенного продукта краси-
тель (судаковый красный, судановый оранжевый и др.) пред-
варительно растворяют в мономере и затем ведут полимериза-
цию по описанной методике.
Рис. 20. Прибор для
получения полимеров
в гранулах
РАБОТА 17. ПОЛУЧЕНИЕ СОПОЛИМЕРА СТИРОЛА
С ДИВИНИЛБЕНЗОЛОМ
ПОЛИМЕРИЗАЦИЕЙ В СУСПЕНЗИИ
Из сополимеров стирола особое значение имеют
сополимеры с дивинилбензолом, имеющие прост-
ранственное строение, устойчивые к воздействию
высокой температуры, растворителей. Путем химических превращений
из этих сополимеров можно получать один из важнейших классов поли-
мерных соединений — ионообменные материалы (см. ч. II).
Исходные Продукты: стирол (свежеперегнанный) — 10 г; дивинилбензол— 0,2 г;
перекись бензоила — 0,2 г; поливиниловый спирт — 0,34 г; вода (дистиллированная) —
’ 60 мл.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); химический
стакан емкостью 1 л; воронка Бюхнера; термошкаф.
В дистиллированной воде при нагоевании до 60° С паствоояют
поливиниловый спирт. Затем отдельно в стироле растворяют пепекись
бензоила и после полного растворения последней туда же добавляют
дивинилбензол. Смесь мономеров с инициатором заливают в пробирку,
в охлажденный до комнатной температуры водный раствор поливинило-
вого спирта. Далее работу проводят так же и в тех же условиях, как
описано в работе 16.
Определяют выход сополимера в процентах По отношению к взятой
для полимеризации сумме мономерных продуктов, растворимость сопо-
лимера и концентрацию непрореагировавших мономеров в водной фазе.
Сополимер используют для изготовления ионообменных материалов
(см. часть II).
70,
РАБОТА 18. ПОЛУЧЕНИЕ СОПОЛИМЕРА СТИРОЛА С НИТРИЛОМ
АКРИЛОВОЙ кислоты сню СУСПЕНЗИОННЫМ МЕТОДОМ
Сополимеры стирола с нитрилом акриловой кислоты получ;
преимущественно суспензионным методом, окрашенные и неокрашенн
с различным соотношением исходных мономеров: CH-10, CH-15, СН
СН-28 и СН-28В (число показывает долю нитрила акриловой кисл<
в сополимере). Сополимеры СН более теплостойки и механически пр
нее, чем полистирол. Применяются для тех же целей, что и полистир
Реакция стирола с нитрилом акриловой кислоты протекает по сх
лСН2=СН + тСН2=СН — Г— СН2-СН- 1 Г—СН2—СН— 1»
II I I
С6Н5 CN L C6H5J- L CN J-
Исходные продукты: стирол свежеперегиаииый — 8,0 мл; нитрил акриловой
лоты — 2,0 мл; перекись бензоила — 0,2 г; поливиниловый спирт — 0,34 г; вода (дне
лированная) — 60 мл.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); химиче
стакан емкостью 2 л; воронка Бюхнера с колбой Буизена.
Навеску поливинилового спирта помещают в реакционную проб
ку, растворяют при нагревании до 60° С в дистиллированной воде. За
отдельно в смеси мономеров растворяют перекись бензоила.
После полного растворения перекиси бензоила мономеры с иниг
тором заливают в пробирку, в охлажденный до комнатной температ;
водный раствор поливинилового спирта, включают мешалку, пуск;
воду в холодильник и пробирку нагревают на водяной бане до 80° (
Далее процесс ведут так же и в тех же условиях, как в работе
Определяют выход сополимера в процентах по отношению к взя'
для полимеризации мономерам и индекс расплава сополимера.
Полученный сополимер перерабатывают литьём под давлением
200—210° С в цилиндре и 60° С в форме, с выдержкой от 8 до 35
зависимости от конфигурации и размера изделия. Если возможно и;
товление стандартных образцов, их испытывают на ударную вязко
сопротивление изгибу и сравнивают результаты с показателями для
мополистирола.
Примечание. Для получения окрашенного продукта краситель (судаковый к
ный, судаковый оранжевый и др.) предварительно растворяют в мономерах и з.
ведут полимеризацию.
* В литературе не существует способа написания схемы реакции сополимер
ции, что связано с неоднозначностью коэффициентов при мономерах и степенью и
меризации их при статистическом распределении звеньев. В данном руководстве
сделали попытку условного написания схемы
nM.i+mM2—>(—Mi—)- (—М2—)-,
где п и т— средняя доля мономеров М, я М2 в сополимере со среднестатистичен
распределением звеньев.
РАБОТА 19. ПОЛУЧЕНИЕ СОПОЛИМЕРА СТИРОЛА С МЕТИЛМЕТАКРИЛАТОМ
В ГРАНУЛАХ (СУСПЕНЗИОННЫМ МЕТОДОМ)
Сополимер стирола с метилметакрилатом можно получать эмульси-
онным и суспензионным способами. Он хорошо растворим в метиленхло-
риде, дихлорэтане, бензоле; обладает стойкостью к бензину и смазочным
маслам. Применяется для изготовления деталей в автомобилестроении.
Благодаря высокой прозрачности сополимер стирола с метилметакри-
латом применяется для изготовления приборных щитков, а также часо-
вых стекол, галантерейных товаров и пр.
Реакция протекает по схеме
сн3
nCH2=CH + тСН2=С
I I
С6Н5 СООСНз
-сн2-сн—
I
c6H5 J
СН3 п
I
—сн2-с-
I
COOCH3J
п
Эти мономеры имеют близкие константы сополимеризации (у сти-
рола 0,52 и у метилметакрилата 0,46). Сополимер имеет среднестатис-
тическое расположение звеньев.
Исходные продукты: стирол свежеперегнанный — 40 г; метилметакрилат свеже-
перегнанный — 5,0 г; перекись бензоила — 0,25 г; поливиниловый спирт — 0,5 г; дистил-
лированная вода — 70 мл.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); химический
стакан емкостью 50 мл и 2 л; воронка Бюхнера с колбой Бунзена; термошкаф.
Навеску поливинилового спирта помещают в реакционную пробир-
ку, растворяют в 70 мл дистиллированной воды. Затем отдельно раст-
воряют перекись бензоила в смеси мономеров и после полного раство-
рения заливают раствор в пробирку, в охлажденный до комнатной тем-
пературы водный раствор поливинилового спирта. Далее процесс
проводят так же, как в работе 16. Температуру в бане следует поддер-
живать точно 80° С в течение 3—4 ч, а затем 90—95° С в течение 1 ч.
Реакция обычно заканчивается за 4—5 ч. Контролем окончания реакции
может служить опускание гранул сополимера на дно пробирки вследст-
вие увеличения их плотности.
Готовый продукт извлекают из реакционного сосуда, промывают,
теплой водой на воронке Бюхнера, высушивают и взвешивают.
Определяют выход сополимера в процентах к весу мономеров, взя-
тых для сополимеризации.
Сополимер стирола с метилметакрилатом перерабатывают литьем
под давлением 800—1000 кг/см2 при 200—220° С и температуре формы
40—45° С. Продолжительность выдержки от 8 до 35 с в зависимости от
|размеров и конфигурации изделия. Экструдируется сополимер при 130° С
в загрузочной зоне машины, 180—190° С в остальных зонах и 200—210° С
в головке. Определяют плотность и ударную вязкость, масло- и бензо-
стойкость (см. ч. II).
РАБОТА 20. ПОЛУЧЕНИЕ ТРОЙНОГО СОПОЛИМЕРА ТИПА МСН
СУСПЕНЗИОННЫМ МЕТОДОМ
Реакция стирола с нитрилом акриловой кислоты и метилметакрила-
том протекает с образованием сополимера со .среднестатистическим рас-
пределением звеньев мономеров:
СН3
I
иСН2=СН + тС[[2=СН + рСН2=С----->
I I I
С6Н5 CN СООСНз
—СН2—СН—
I
c6H5J
-сн2-сн-
I
CN
СН3 ..
I
—сн2-с-
I
СООСНз
Тройной сополимер имеет хорошую текучесть и применяется для
изготовления корпусов приборов, телефонных аппаратов, авторучек
и т. п., а также для изготовления бытовых изделий. Он обладает повы-
шенной по сравнению с полистиролом прочностью порядка 1.100—
1200 кг/см2.
Исходные продукты: свежеперегнанные мономеры: стирол — 5 г, метилметакри-
лат— 3 г, акрилонитрил — 2 г; сольвар*— 0,2 г или поливиниловый спирт — 0,34 г;
перекись бензоила — 0,2 г; дистиллированная вода — 70 мл.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); водяная баня
(стакан емкостью 1—2 л); воронка Бюхнера; колба Бунзена; стакан емкостью 50 мл.
В пробирке прибора растворяют в воде поливиниловый спирт или .
сольвар и добавляют к нему раствор инициатора в смеси мономеров;
пробирку соединяют с обратным холодильником, в который пускают
воду, включают мешалку и нагревают реакционную смесь при 80° С в
течение 4—5 ч на водяной бане (см. работу 16).'
Определяют выход полимера в процентах по отношению к смеси
мономеров и индекс расплава.
Сополимер перерабатывают в изделия (например, стандартный бру-
сок) литьем под давлением 800 кг/см2 при температуре в цилиндре
190—220° С и температуре формы 50—60° С. Выдержка 8—35 с, в зави-
симости от толщины и конфигурации изделия. Влажность гранул не
должна превышать 0,1% (см. стр. 58). Наличие влаги в сополимере
приводит к получению изделий с пониженной прочностью и поверхност-
ными дефектами: серебро, раковины, пузыри.
Из сополимера можно отпрессовать таблетки диаметром 3—4 мм
при давлении 250—300 кг/см2 и температуре формы 160—165° С. Вы-
держка под давлением 3 мин на 1 мм толщины изделия. Форма раскры-
вается только после охлаждения до 40—45° С. Таблетки испытывают на
хим стойкость.
Если необходимо изготовить стандартные образцы для проведения
физико-механических испытаний, загрузку увеличивают в 3—5 раз или
берут промышленный сополимер.
* Сольвар получают частичным омылением спирта до содержания ацетатных
групп 18—20%.
73
ГЛАВА III. ПОЛИМЕРЫ ГАЛОГЕНОПРОИЗВОДНЫХ НЕПРЕДЕЛЬНЫХ
УГЛЕВОДОРОДОВ И ПЛАСТИЧЕСКИЕ МАССЫ
НА ИХ ОСНОВЕ
Из большого числа галогенпроизводных полиуглеводородов лишь хлор-
и фторпроизводные имеют большое практическое значение и нашли ши-
-рокое применение в различных отраслях промышленности.
В качестве исходных мономеров для получения галогенсодержащих
полиуглеводородов используют винилхлорид, винилиденхлорид, тетра-
фторэтилен, трифторхлорэтилен, винилфторид, винилиденфторид.
А. ПОЛИМЕРЫ ХЛОРПРОИЗВОДНЫХ НЕПРЕДЕЛЬНЫХ
УГЛЕВОДОРОДОВ И ПЛАСТИЧЕСКИЕ МАССЫ
НА ИХ ОСНОВЕ
На основе поливинилхлорида получают широкий ассортимент плас-
тических масс с разнообразными свойствами: для легкой промышлен-
ности (плащи, занавески, скатерти, упаковочные материалы, галанте-
рейные товары, линолеум, сапоги, перчатки и т. д.); электро-
технической промышленности (кабельные оболочки); химической про-
мышленности (аппаратура, мерники, сборники, ванны, колонны, трубы,
шланги, покрытия по бумаге; защитные лаки и т. д.); для строительной
промышленности: пенопласты (блоки, листы, прокладки, набивки).
Поливинилхлорид имеет один существенный недостаток: темпера-
тура его разложения (130—140° С) значительно ниже температуры
течения. Таким образом, переработка в изделия чистого поливинил-
хлорида невозможна без стабилизации. Так как процесс распада его
происходит с выделением НС1 и образованием полиеновой структуры,
легко окисляющейся под влиянием УФ-лучей, то идеальным стабилиза-
тором должно было бы быть вещество, связывающее HCli и ингибирую-
щее окисление. Однако эти качества трудно соединить в одном вещест-
ве, поэтому поливинилхлорид целесообразно стабилизировать смесью
кислотоакцепторных стабилизаторов и антиоксидантов. В связи с тем
что НС1 является катализатором окисления двойных связей, в большин-
стве случаев связывание НС1 уже ингибирует процесс окисления. В со-
временных рецептурах широко используются такие кмслотоакцепторные
стабилизаторы, как мыла (соли высших жирных кислот и металлов Са,
Ba, Pb, Cd, Sn и др.), неорганические соли и окислы (окись свинца,
свинцовые белила, литопон, кадмиевый литопон, силикагель и др.). Не-
которые из них, являясь пигментами, поглотают УФ-лучи и предотвра-
щают окисление.
Наряду с указанными стабилизаторами используют амиды кислот:
мочевину, меламин и др., а также аренсульфамиды общей формулы
74
R'—Аг—SO2NR2, где Аг может иметь различное строение, R'— алии,
a R — водород или любой алкильный или арильный радикал. В завис!
мости от количества заместителей R и их строения кислотоакцепторнь
свойства этих соединений изменяются в широких пределах.
Большое практическое значение как стабилизаторы цоливинилхлс
рида имеют эпоксидные олигомеры, способные количественно связыват
НС1.
Для создания изделий из поливинилхлорида с высокой степень!
прозрачности используют оловоорганические соединения общей фо{
мулы (RCOO)2SnR2—диалкилдиацилолово, где R — радикал одно- ил
двухосновной кислоты, насыщенной или ненасыщенной, a R'— алкилт
ный радикал, содержащий не менее четырех атомов углерода. Смес
этих соединений и эпоксиолигомеров позволяют получить термостабил!
-ные композиции с хорошим внешним видом.
Повышают стабильность к окислению так называемые адсорбер!
УФ-света, принадлежащие к производным бензофенона и в зависимост
от строения поглощающие тот или иной диапазон волн, предотвраща
возможность старения (разложения) поливинилхлорида под воздей
ствием облучения.
Огромное разнообразие изделий из поливинилхлорида объясняет
ся также его способностью пластифицироваться различными пластифи
кагорами, что определяет возможность получения пластических масс
пленочных материалов, каучукоподобных масс и т. п. с комплексе!
разнообразных ценных свойств.
Пластификаторами могут быть вещества низкомолекулярные (угле
водороды, спирты, простые и сложные эфиры, амиды, эпоксиды и т. д.)
олигомеры (полиэпоксиды) и полимеры — органические и элементорга
нические. Из перечисленных соединений широко применяются сложны!
эфиры двухосновных кислот. Среди них наибольшее значение имею'
диэфиры фталевой, себациновой кислот и спиртов с радикалом, содер
жащим от 4 до 1’2 атомов углерода, а также эфиры о-фосфорной кисло
ты и фенолов.
Основными свойствами пластификаторов являются: совмещаемост!
с полимером в широком интервале температур, низкая летучесть, химй
ческая инертность. В зависимости от специального назначения изделиг
пластификаторы должны обладать также специфическими свойствами
морозостойкостью, диэлектрическими свойствами, негорючестью, тер-
мостойкостью. В каждом отдельном случае необходимо учитывать вза-
имное влияние и характер взаимодействия системы полимер — пласти-
фикатор.
Механизм пластификации заключается в образовании сольватов,
сопровождающемся нарушением полярных, водородных, дисперсионных
и других связей. Введение пластификатора, как правило, снижает тем-
пературу стеклования и течения полимера, а также вязкость его распла-
ва, что облегчает переработку полимера в изделие.
На физико-механических свойствах пластификация сказывается
следующим образом: при введении пластификатора резко снижается
прочность на разрыв, увеличивается относительное удлинение, проч-
ность на изгиб.
75
Для окраски изделий из поливинилхлорида используют красители,
растворимые в массе полимера (основные красители), и пигменты раз-
личных цветов и оттенков. Титановые и свинцовые белила являются
одновременно и стабилизаторами и красителями, окрашивающими в бе-
лый (непрозрачный) цвет.
Химизм образования хлорсодержащих
полиуглеводородов
Винилхлорид полимеризуется под влиянием УФ-лучей, .инициаторов
радикальной полимеризации, а также инициирующих излучений по схеме
пСН2=СНС1 —
-CH2-CH-I-
I
Cl Jn
Присоединение молекул протекает «голова к хвосту» (1,2-присоедине-
ние).
Винилхлорид — активный мономер, легко полимеризуется в присут-
ствии инициаторов как в среде мономера, так и в растворе, эмульсии и
суспензии.
Высокая активность макрорадикалов определяет возможность пе-
редачи цепи на растворитель, инициатор, примеси. Обрыв цепи в про-
цессах инициированной полимеризации происходит в одинаковой степе-
ни как путем рекомбинации, так и диспропорционирования, поэтому
поливинилхлорид имеет около 60% концевых ненасыщенных звеньев, на-
личие которых снижает устойчивость полимера к термоокислительной
деструкции. Благодаря легкости протекания передачи цепи на полимер
поливинилхлорид имеет одно ответвление на 75 звеньев.
Снижение температуры образования полимера в значительной сте-
пени уменьшает роль передачи цепи. Полимеризация винилхлорида со-
провождается реакцией дегидрохлорирования, которая, приобретает
существенное значение при повышении температуры. Так как реакция
протекает автокаталитически, то поливинилхлорид обязательно содер-
жит в центральных звеньях ненасыщенные связи.
Полярность концевого звена макрорадикала достаточно велика,
поэтому можно ожидать образования стереорегулярных поливинилхло-
ридов как синдио-, так и изотактического строения. Понижение темпе-
ратуры реакции (что можно осуществить, проводя полимеризацию в
окислительно-восстановительной системе) способствует образованию по-
лимера с большей степенью стереорегулярности, которая при —60° С
достигает 85%, причем температура стеклования повышается с 70 до
110° С.
Винилиденхлорид так же, как и винилхлорид, легко полимеризует-
ся на свету, поэтому поливинилиденхлоршд стал известен еще со времени
синтеза мономера, однако волокна из него были получены лишь через
несколько десятков лет.
Винилиденхлорид легко полимеризуется по радикальному механиз-
му как в блоке, так и в суспензии и эмульсии:
76
Cl г Cl ч
I I
пСН2=С—>• —CH2-C—
Cl L Cl J"
Полимеризация в блоке в присутствии перекиси бензоила протекает с
возрастающей скоростью до 30%-ной конверсии, что объясняют реак-
цией передачи цепи и гель-эффектом.
Эмульсионная и суспензионная полимеризация винилиденхлорида
подчиняется общим закономерностям этого процесса. Полимер винили-
денхлорида кристалличен, строения «голова к хвосту». Период иден-
тичности 4,67 А. Кристаллический поливинилиденхлорид плавится при
185—200° С. При 150° С полимер разлагается с выделением НС1, таким
образом в этом отношении он подобен поливинилхлориду и его нельзя
перерабатывать в отсутствие стабилизаторов. Стабилизаторы поливинил-
хлорида вполне пригодны для стабилизации поливпнилиденхлорида.
Технология производства хлорсодержащих
полиуглеводородов
Основными способами производства поливинилхлорида, поливини-
лиденхлорида и их сополимеров являются суспензионный и эмульсион-
ный. Полимеризация винилхлорида в массе связана с трудностью отво-
да тепла и нерастворимостью полимера в мономере. Однако этот способ
находит применение в виде ступенчатого двухстадийного процесса. На
первой стадии образуется форполимер в обычном автоклаве, а заканчи-
вается полимеризация в горизонтальном цилиндрическом аппарате,
обеспечивающем интенсивное измельчение полимера и отвод тепла.
Инициирование осуществляется инициаторами, растворимыми в моно-
мере.
Полимеризация в суспензии протекает как обычно, в водной среде
в присутствии инициаторов (перекиси лаурила и капроила, пероксиди-
карбонатов, динитрила азоизомасляной кислоты и др.).
Средняя величина частиц суспензии от 75—150 до 600 мкм. Важ-
нейшими параметрами в производстве поливинилхлорида всеми спосо-
бами является температура, определяющая степень разветвленности,
термостабильность полимера, молекулярновесовое распределение. Важ-
ное влияние на светостойкость, термостойкость оказывают природа^ини-
циатора, эмульгатора, чистота исходных веществ. Молекулярный вес
суспензионного поливинилхлорида колеблется в пределах от 12000 до
120000.
Для получения суспейзионного поливинилхлорида используется
умягченная вода. Процесс осуществляется в автоклаве с рубашкой и
мешалкой при 40—50°, давлении 5—14 атм в течение 20—30 ч до степе-
ни конверсии 80—90%. Окончание процесса сопровождается падением
давления в автоклаве. Гранулы отделяют от водной фазы, промывают и
сушат.
Эмульсионная полимеризация осуществляется в автоклавах, вра-
щающихся со скоростью 120 об/мин, вертикальных или горизонтальных,
77
снабженных мешалкой и рубашкой для обогрева и охлаждения. Темпе-
ратура реакции 50° С, продолжительность 15—20 ч, степень конверсии
87—88%. Инициирование осуществляется гидроперекисями, персульфа-
тами, перекисью водорода и другими водорастворимыми инициаторами.
Размер частиц латекса 0,05—0,5 мкм. Высушивание осуществляется в
распылительных сушилках. Если полимер выделяется коагуляцией, то
его промывают, центрифугируют и высушивают.
В промышленности поливинилхлорид выпускается различных марок,
например: суспензионный (ГОСТ 14332—69), ПВХ-С70 и ПВХ-С74 для
кабельного и медицинского пластиката и пленок различного назначения;
ПВХ-С63Ж для винипласта; ПВХ-С61 и ПВХ-С58 для винипласта,
литьевых изделий и грампластинок; ПВХ-6 70Т для искусственных по-
дошв, линолеума и т. д.
Эмульсионный поливинилхлорид (ГОСТ 14039—68) марок
ПВХ-Е74П, ПВХ-Е70, Е70П, Е66, Е62, Е54, Е58 и других предназначает-
ся для изготовления прочных пластикатов, пленок, искусственной кожи,
технических паст, формо- и пенопластов, а также лаков.
Поливинилхлорид массовый ПВХ-М (ТУ-6-01-678—72) выпускается
следующих марок: ПВХ-М70, М64, М67, М59, М56 и др. Номер марки
означает молекулярный вес, выраженный константой Фикентчера (Л),
которая является основной характеристикой той или иной марки поли-
винилхлорида (см. табл. 6). Буквы после цифры означают: Т — термо-
стабилизированный; М— мягкий; П — пастообразующий. Буквы перед
цифрой означают способ получения: С — суспензионный; Е — эмульсион-
ный и М — массовый.
ТАБЛИЦА 6
Зависимость между относительной вязкостью 0,5%-ного раствора
поливинилхлорида в циклогексаноне и величиной К
%тн К ^ОТН К
1,2 40,2 1,65 71,3
1,3 50,0 1,70 73,7
1,4 57,2 1,75 75,9
1,44 59,8 1,80 78,2
1,50 63,5 1,85 80,1
1,55 66,5 1,90 82,1
1,60 69,0
Большое практическое значение имеют эмульсионные и суспензион-
ные сополимеры винилхлорида с другими мономерами: винилацетатом —
«винилит» (ТУ 6-01-650—71) для изготовления грампластинок,, для
рельефных карт и других пленочных изделий; метилакрилатом — «хло-
винит». Хловинит МА-20 (ТУ 6-01-130—67) получают при соотношении
мономеров 80:20 для изготовления листового материала и прокладоч-
ных жгутов. На основе МА-20 выпускают листовой материал «вини-
проз» матированный и прозрачный (МРТУ 6-05-1222—69), для защиты
фотосхем, светокопировальных работ, картографии и т. п.
78
Создание сополимеров винилхлорида с винилиденхлоридом (ВХВД)
ГОСТ 6-02-18—72 и (СВХ-1) МРТУ 6-02-762—69 и других было вызвано
тем, что полнвинилиденхлорид плохо растворим в растворителях. Сопо-
лимеры же винилиденхлорида с винилхлоридом (а также бутилмета-
крилатом, акрилонитрилом) оказались особенно ценны и нашли боль-
шое распространение. Сополимеры ВХВД выпускаются с высоким
(более 70%), средним (30—60%) и малым (20%) содержанием винили-
денхлорида.
При содержании винилиденхлорида более 70% сополимеры с винил-
хлоридом кристалличны. Кристаллические сополимеры имеют сравни-
тельно низкую вязкость расплава и легко перерабатываются в изделия.
Полимеры и сополимеры винилиденхлорида в виде порошков, ла-
тексов применяются в основном для производства труб, стойких к дей-
ствию агрессивных сред, разнообразных деталей химической аппарату-
ры, пропиточных составов, грампластинок. Пленки применяются для
упаковки химических товаров и пищевых продуктов. Моноволокно, из-
готовляемое экструзией, применяется для изготовления фильтроваль-
ных тканей, рыболовных сетей, щетины, декоративных и обивочных тка-
ней. Сополимеры, как и гомополимер, необходимо стабилизировать кис-
лотоакцепторными стабилизаторами. Сополимеры винилиденхлорида с
акрилонитрилом обладают повышенной термостабильностью, легко
пластифицируются и растворяются в циклогексаноне, метилэтилкето-
не и др. Растворы используют в качестве лаков для нанесения покрытий
на металл и бумагу.
Исходное сырье для получения поливинилхлорида
и поливинилиденхлорида
Винилхлорид СН2=СНС1 при комнатной температуре и атмосфер-
ном давлении представляет собой бесцветный газ с приятным эфирным
запахом. Т. кип. — 13,9° С; т. зам.— 159,7° С; плотность жидкого винил-
хлорида d Г15 =0,9730; показатель преломления для жидкого вещества
п о15 = 1,38; молекулярный вес 62,50.
Винилхлорид растворим в обычных органических растворителях:
этиловом и метиловом спиртах, дихлорэтане, трихлорэтане, хлороформе,
сольвентнафте, керосине, соляровом масле, диэтиловом (серном) эфи-
ре, диоксане, бензоле, толуоле, ацетоне. Растворим в воде и соляной
кислоте. Мономер может быть стабилизирован гидрохиноном и п-трет-
бутилпирокалехином: 0,005% -ная концентрация последнего вполне до-
статочна для стабилизации винилхлорида, предназначенного для хране-
ния или перевозки.
В промышленности винилхлорид получают из ацетилена, действуя
на него хлористым водородом:
CHsCH + НС1->СН2=СНС1
Однако это очень дорогой способ, втрое дороже, чем синтез из олефино-
вых углеводородов. Винилхлорид может быть получен не только из эти-
лена, но и непосредственно из этана. Получение винилхлорида из этана
дает большой экономический эффект.
В лабораторных условиях винилхлорид наиболее удобно получать
действием спиртового раствора щелочи на дихлорэтан:
СН2С1-СН2С1 4- NaOH -> СН2=СНС1 + NaCl + Н2О
Винилиденхлорид (1,1-дихлорэтилен) СН2=СС12 представляет со-
бой бесцветную жидкость со слабым запахом, напоминающим запах
хлороформа. Т. кип. 31,7°С (при 760 мм рт. ст.), т. пл. — 122,5°С; плот-
ность &4°—1,2122; показатель преломления «о = 1,4246; молекулярный
вес 96,95.
Мономер, свободный от кислорода, полимеризуется очень медлен-
но, технический продукт полимеризуется относительно быстро. В каче-
стве инициаторов полимеризации в технике применяют главным обра-
зом перекись бензоила, перекись водорода и другие обычные инициато-
ры радикальной полимеризации. Полимеризацию ведут в растворителях
и в водной эмульсии в автоклавах под давлением.
В качестве стабилизатора для мономера чаще всего применяют гид-
рохинон. Ингибирующими свойствами обладают также фенолы, спирты,
амины (в частности, три метил а мин).
Поливинилиденхлорид представляет собой белый порошкообразный
продукт, нерастворимый в мономере. Он негорюч, хорошо противостоит
действию химических реагентов и плохо растворяется в горячем тетра-
хлорэтане, декалине, тетралине; частично растворим в бензоле, хлоро-
форме, сероуглероде; не растворяется в эфирах, спиртах и большинстве
других органических растворителей.
Полимер винилиденхлорида размягчается при 185—200° С, при на-
гревании до 150° С начинает разлагаться с отщеплением хлористого во-
дорода.
Подготовка продукта для полимеризации
Очистка винилхлорида. Сжиженный винилхлорид перегоняют по-
вторно для полного освобождения от дихлорэтана и метилового спирта,
которые могли попасть в приемную колбу (см. работу 21). При этом
используют ту же аппаратуру, для чего трехгорлую реакционную колбу
отъединяют от обратного холодильника и заменяют колбой, содержащей
жидкий винилхлорид. Повторную перегонку винилхлорида проводят,
переводя газообразный мономер в чистый сосуд, находящийся в бане с
сухим льдом. Для этого колбу, содержащую перегоняемый жидкий мо-
номер, нагревают до комнатной температуры за счет окружающего воз-
духа. Необходимости в дополнительном подогреве нет. Перегнанный
винилхлорид должен быть охлажден ниже температуры кипения и ис-
пользован возможно быстрее после его получения.
Очистка винилидеихлорида. Винилиденхлорид очищают перегонкой
его в атмосфере азота, освобожденного от примеси кислорода, так как
в присутствии даже следов кислорода образуются перекиси, а затем
происходит самопроизвольная полимеризация.
Мономер имеет низкую температуру кипения, поэтому перегонку
проводят при атмосферном давлении в круглодонной колбе с дефлегма-
тором высотой не менее 40 см. Холодильник для конденсации паров мо-
ей
номера должен хорошо охлаждаться водой с температурой 5—10° С.
Приемник для чистого продукта следует продуть азотом и поместить в
баню с водой и льдом. Приемником может служить колба Вюрца, плот-
но соединенная с форштоссом холодильника. К отводной трубке кол-
бы прикрепляют хлоркальциевую трубку, заполненную рыхлым сло-
ем ваты.
Перегонную колбу нагревают все время очень осторожно, так как
продукт легко воспламеняется. Ее погружают либо в водяную баню,
нагретую до температуры не выше 50—60° С, от которой предваритель-
но отнята горелка, либо в водяную баню, обогреваемую электрической
плиткой. Ни в коем случае не разрешается применение открытого огня.
Перед началом перегонки в винилиденхлорид в качестве ингибитора не-
обходимо добавить 0,1—0,2% гидрохинона.
Очищенный мономер следует тотчас использовать для работы, так
как без ингибитора он легко полимеризуется. В присутствии кислорода
воздуха чистый винилиденхлорид уже при комнатной температуре че-
рез несколько часов становится мутным от образующегося полимера,
который затем выпадает в осадок.
В качестве ингибитора полимеризации при перегонке винилиденхло-
рида можно применять также этиловый спирт в количестве около 1%.
Мономер, стабилизированный спиртом, можно перегонять в лаборатор-
ных условиях без применения инертного газа. Стабилизированный спир-
том и охлажденный до 0° С мономер не окисляется и не полимеризуется
в течение суток.
Для обнаружения гидрохинона в мономере может служить качест-
венная реакция, дающая возможность открыть 0,00001 % гидрохинона
(см. стр. 60).
Анализ винилхлорида и винилиденхлорида
Хотя полученный вышеописанным методом, а также продажный
винилхлорид (винилиденхлорид) — довольно чистые продукты, все же
их необходимо подвергнуть анализу для установления степени чистоты
и поведения при полимеризации. Винилхлорид промышленный мо-
жет быть загрязнен ацетиленом, метиловым спиртом и ацетальдегидом.
Анализ винилхлорида на эти примеси проводят методом газовой хрома-
тографии.
Определение плотности. Плотность жидкого винилхлорида (винил-
иденхлорида) определяют на весах Мора —- Вестфаля.
Определение хлористого водорода. Во взвешенную на аналитиче-
ских весах круглодонную колбу емкостью 200 мл, снабженную пробкой
и отводной трубкой с зажимом, помещают около 20 г жидкого винил-
хлорида. Колбу закрывают пробкой и погружают в баню с жидким ви-
нилхлоридом. С. помощью бани температура в колбе поддерживается
около —13° С и в ней не создается давления выше атмосферного. Колбу
с винилхлоридом быстро взвешивают на аналитических весах с точностью
до 1 г, для этого ее вынимают из Охлаждающей бани на очень короткое
время. Отводную трубку колбы соединяют с промывной склянкой, в ко-
торой содержится дистиллированная вода, затем открывают зажим и
81
колбу вынимают из бани. За счет тепла помещения колба постепенно
нагревается и испаряющийся винилхлорид барботирует через воду.
Когда колба освободится от жидкости, осторожным нагреванием
удаляют из нее оставшиеся пары. Если вода в промывной склянке име-
ет кислую реакцию, что указывает на присутствие хлористого водорода,
последний оттитровывают 0,1 н. едким натром, применяя в качестве ин-
дикатора фенолфталеин.
Содержание хлористого водорода в процентах (х) рассчитывают по
формуле
х=
а/<0,0036-100
g
где а — количество 0,1 н. NaOH, израсходованное на титрование, мл;
К — коэффициент нормальности 0,1 н. NaOH; g— навеска винилхлори-
да, г; 0,0036 — количество хлористого водорода, соответствующее 1 мл
0,1 н. NaOH, г.
Определение железа. Важное значение имеет определение железа в
винилхлориде, так как оно является инициатором его полимеризации.
Определение железа в винилхлориде осуществляется колориметрически.
Метод основан на взаимодействии железа с сульфосалициловой кисло-
той с последующим колориметрическим определением образующегося
в аммиачной среде комплексного соединения железа, окрашенного в
желтый цвет.
Испаряют 100 мл анализируемого винилхлорида, остаток растворя-
ют в 5 мл НС1 (х. ч.), приливают 5 мл дистиллированной воды, 3 капли
пергидроля и раствор кипятят 2—3 мин. После охлаждения раствор
переносят количественно в мерную колбу, вливают 10 мл раствора суль-
фосалициловой кислоты, по каплям раствор NH4OH до перехода окра-
ски в желтый цвет и после этого еще 2 мл NH4OH. Раствор доводят до
метки добавлением дистиллированной воды. Колориметрирование про-
водят на фотоэлектроколориметре.
Для этой цели вначале строят градуировочный график. Готовят
стандартный раствор: 0,8640 г сульфата железа-аммония (х. ч.), раст-
воряют в 15—20 мл 5%-ного раствора соляной кислоты и доводят объем
раствора соляной кислотой до 1 л. В 1 мл раствора содержится 0,1 мг
железа. В мерные колбы емкостью 50 мл вливают из микробюретки от
0,1 до 1 мл стандартного раствора с интервалом в 0,1 мл. Прибавляют
в каждую колбу по 5 мл соляной кислоты, 10 мл раствора сульфосали-
циловой кислоты и раствор аммиака до перехода окраски раствора в
желтый цвет, а затем еще 2 мл аммиака (избыток) разбавляют раствор
водой до метки и перемешивают.
Одновременно готовят исследуемый раствор. Приготовленные раст-
воры колориметрируют в фотоэлектроколориметре, применяя синий све-
тофильтр № 3, в кювете с толщиной слоя 50 мм. Колориметрирование
проводят согласно инструкции. Измерив оптическую плотность всех
растворов, строят градуировочный график, откладывая по оси абсцисс
количество железа (мг), а по оси ординат соответствующие значения
оптической плотности [5].
82
Качественное определение хлорсодержащмх
полимеров и сополимеров
Общей реакцией на присутствие любого галогена является реакция
Бейльштейна, которая заключается в сплавлении анализируемого ве-
щества в ушке тщательно прокаленной медной проволоки. В присутст-
вии галогена пламя горелки окрашивается в зеленый цвет.
Для обнаружения хлора в полимере существует несколько качест-
венных реакций.
1. Реакция с AgNO3. Небольшой кусочек анализируемого полимера
помещают в пробирку с кристалликом AgNO3, приливают к нему 1—
2 мл концентрированной HNO3 и одну каплю концентрированной H2SO4.
Смесь нагревают до кипения. После разложения вещества в пробирку
наливают 4—5 мл дистиллированной воды.
В присутствии галогенов образуется осадок серебряных солей, при-
чем AgCl растворим в аммиаке, другие галогениды серебра нераство-
римы.
2. Продукты пиролиза можно анализировать хроматографическим
методом.
3. Реакция с фениллитием. К раствору полимера в тетрагидрофура-
не прибавляют молярный раствор фениллития в тетрагидрофуране, при
этом выпадают окрашенные осадки. Реакцию проводят в атмосфере
азота. Если в растворе присутствует поливинилхлорид, образуется же-
леобразный осадок красного цвета, имеющий желтые и оранжевые
вкрапления. Полиперхлорвинил и сополимеры хлористого винила в
этих условиях образуют красный осадок, однако в отличие от поливинил-
хлорида после фильтрования этого осадка фильтрат окрашен в красный
цвет.
Эта реакция дает результат и в присутствии пластификаторов.
4. Реакция «Либермана—Шторха. Небольшое количество полимера
помещают на фарфоровую чашку, смачивают несколькими каплями
уксусного ангидрида и прибавляют одну каплю крепкой H2SO4 так,
чтобы она попала в жидкость. В присутствии поливинилхлорида поли-
мер медленно окрашивается в синий или зеленый цвет.
5. Реакция Винтершейдта. В пробирке на слабом пламени горелки
нагревают такое количество монохлоруксусной кислоты, чтобы получить
5 мл расплава, затем в расплав на кончике ножа вносят тонкоизмель-
ченный полимер и нагревают на умеренном пламени. После 2 мин на-
гревания появляется синяя окраска. Полиперхлорвинил окраски не дает.
Сополимеры с винилацетатом окрашиваются в красно-фиолетовый цвет.
Сополимеры с эфирами акриловой и метакриловой кислот дают слабую
красную окраску, а затем выпадает сине-фиолетовый осадок.
6. Качественная реакция на поливинилиденхлорид. (Реакция Век-
слера.) К 5 мл 0,1%-ного раствора полимера в пиридине добавляют
0,5 мл метилового спирта, содержащего 2% едкого натра, при этом по-
является коричневая окраска, переходящая в черную.
Описана также другая методика проведения этой реакции: 20 мг
тонкоизмельченного полимера или сополимера нагревают с 1 мл свеже
перегнанного пиридина; после охлаждения раствора добавляют 0,5 mj
8:
насыщенного раствора КОН в метиловом спирте и наблюдают окраши-
вание.
Количественный анализ хлорсодержащих
, полимеров и сополимеров
1. Количественное определение хлора, а) По методу Эшка. Тща-
тельно измельченную точную навеску полимера (0,1—0,2 г) смешивают
в фарфоровом тигле (высотой 25 мм, диаметром 30 мм) с 2—3 г смеси
Эшка. Затем тигель медленно нагревают до сплавления массы, поме-
щают в муфельную печь при 650—700° С и выдерживают там тигель до
полного разложения органического вещества. Полученный сплав выще-
лачивают водой, подкисляют азотной кислотой и определяют в растворе
ион С1~ титрованием 0,1 н. раствором нитрата серебра в присутствии
железо-аммонийных квасцов.
Смесь Эшка приготовляют смешением 60 вес. ч. окиси магния (х. ч.)
и 40 вес. ч. безводного углекислого натрия (х. ч.).
б) Быстрый метод определения хлора. В стальной тигель с навин-
чивающейся крышкой помещают 10 капель этиленгликоля и добавляют
2,5—3 г безводного углекислого натрия.
Отвешивают в ампуле на аналитических весах испытуемый полимер
и опускают ее открытым концом в тигель. Затем в тигель засыпают
2,5—3 г перекиси натрия, закрывают его навинчивающейся крышкой,
ставят на кольцо штатива в вытяжном шкафу и нагревают на неболь-
шом огне, пока не произойдет вспышка (1,5 мин). После вспышки на-
гревают еще 1,5 мин. Затем тигель охлаждают, открывают, ставят в
химический стакан и осторожно (особенно первую каплю) добавляют
воду. Хорошо вымывают содержимое тигля, помешивая стеклянной па-
лочкой с резиновым наконечником. После этого в стакан добавляют
концентрированной HNO3, пока не посинеет индикаторная бумага-
конго. Чтобы разложить оставшуюся перекись натрия, можно не-
много подогреть раствор или добавить немного 10%-ного раствора
Na2CO3.
Затем в стакан добавляют 20—25 мл 0,05 н. раствора AgNO3, рас-
твор квасцов и титруют 0,05 н. раствором роданистого аммония. Выпа-
дает осадок, а раствор приобретает бледно-розовую окраску, которая
уоиливается при добавлении лишней капли реактива.
Расчет содержания хлора к (%) производят по формуле
— 0,001778-100
g
где а — объем раствора AgNO3, мл; К, К' — поправки к титру раство-
ров; b — объем раствора роданистого аммония, мл; 0,001778 — количе-
ство хлора, соответствующее 1 мл 0,05 н. раствора, г; g— навеска поли-
мера, г.
П риготовление железо-аммонийных квасцов: 50 г квасцов растворяют в 100 г теп-
лой кипяченой воды. После растворения добавляют концентрированной HNO3 до пере-
хода коричневой окраски в желто-зеленую.
84
Определение количества полимера в латексе. Метод основан на оп-
ределении количества сухого остатка в латексе, полученного коагуля-
цией латекса алюмокалиевыми квасцами.
В коническую колбу вносят пипеткой 20 мл латекса, разбавляют
дистиллированной водой, добавляют 4 мл насыщенного раствора алю-
мокалиевых квасцов (ч. д. а.) и 10 мл раствора соды. Содержимое кол-
бы нагревают на водяной бане в течение 1 ч.
Выпавший осадок переносят на воронку Бюхнера с тканевым фильт-
ром и промывают дистиллированной водой до отрицательной реакции
фильтрата на ион SO4~ (проба с BaCla). Осадок переносят в чашку Пет-
ри и высушивают в сушильном шкафу при 50° С.
Содержание полимера х (в %) вычисляют по формуле ,
20р
где а — вес сухого остатка, г; .р — плотность латекса, г/см3 (определяют
ареометром).
Определение состава сополимеров винилхлорида с винилацетатом
или метилакрилатом. Определение связанного хлора (винилхлорида)
производится по описанной выше методике (см. стр. 84). Определение
связанного винилацетата основано на щелочном омылении сополимера
и последующем разложении соляной кислотой натриевой соли уксусной
кислоты, образующейся в результате омыления.
Выделяющуюся уксусную кислоту и избыточное количество соляной
кислоты титруют кондуктометрически раствором щелочи. В случае ана-
лиза сополимера винилхлорида с метилакрилатом выделяется акрило-
вая кислота и тогда титруют смесь акриловой и соляной кислот.
В этих условиях идет незначительное омыление хлора в звеньях
сополимера.
В колбу емкостью 250 мл, снабженную обратным холодильником
(на шлифе), вносят навеску сополимера 0,1—0,3 г, взвешенную с точ-
ностью до 2 мг, и 25 мл ацетона. Колбу нагревают на водяной бане до
растворения навески. В горячий раствор вносят пипеткой 5—20 мл (в
зависимости от навески сополимера) спиртового раствора NaOH. Колбу
соединяют с обратным холодильником, помещают в кипящую' водяную
баню и кипятят 5 мин (от начала кипения).
Затем колбу снимают, в горячий раствор вносят пипеткой 15—20 мл
раствора НС1, тщательно перемешивают и охлаждают. К раствору при-
бавляют 50—75 мл дистиллированной воды и фильтруют для освобож-
дения от смолистого осадка через вату в плоскодонную широкогорлую
колбу.
Колбу с воронкой и осадком тщательно промывают дистиллирован-
ной водой, присоединяя раствор к основному фильтрату, и титруют кон-
дуктометрически 0,1 н. раствором NaOH (кондуктометрический метод*
описан [5]).
Содержание винилацетата х (в %) вычисляют по формуле
аКО, 0086 -100
х=------1------,
g
' — 1 — •
* Кондуктометрический метод позволяет раздельно определять органические и ми-
неральные кислоты при их совместном присутствии.
85
где а — количество 0,1 н. раствора NaOH, затраченное на титрований
уксусной кислоты, мл; К — поправочный коэффициент 0,1 н. раство-
ра NaOH; 0,0086—количество винилацетата, соответствующее 1 мл
точно 0,1 н. раствора NaOH, г; g — навеска анализируемого сопо-
лимера, г.
В случае анализа сополимера винилхлорида с метилакрилатом в
расчетной формуле сохраняется тот же коэффициент — 0,0086.
Определение термостабильности
поливинилхлорида
Принцип определения температуры разложения основан на измене-
нии цвета реактивной бумаги конго красного под действием выделя-
ющегося хлористого водорода при температуре разложения поли-
винилхлорида (порошкообразного или пластифицированного плас-
тиката).
Испытание проводят в пробирках из тонкостенного стекла с внут-
ренним диаметром 12—13 мм и длиной 9,5 см, снабженных двумя круго-
выми метками: нижней, на расстоянии 3 см от дна пробирки, и верхней,
на расстоянии 7 см от дна пробирки.
Пробирки закрывают пробками и вставляют в крышку масляной
или глицериновой бани. Баня представляет собой химический стакан,
закрытый корковой пробкой или крышкой, имеющей в центре отверстие
для термометра, шесть равномерно расположенных отверстий для про-
бирок и одно отверстие для трубки, через которую проходит кольцевая
мешалка. Пробирки с испытуемыми образцами вставляют в отверстия
крышки так, чтобы дно пробирки отстояло от дна бани на 3 см, а верх-
няя метка на пробирке находилась на расстоянии не менее 0,5 см от
нижней поверхности крышки.
В пробирки помещают поливинилхлорид (порошок высотой слоя
3 см или кружки пластиката диаметром 11—12 мм, вырезанные про-
бочным сверлом соответствующего диаметра, чтобы они при уплотнении
палочкой образовали слой высотой 3 см, т. е. были на уровне нижней
метки). Затем в пробирки помещают полоски бумаги конго красного
шириной 0,5 см так, чтобы нижний конец полоски находился у верхней
метки, после чего пробирки закрывают пробкой.
Установив пробирки с испытуемыми образцами, отмечают началь-
ную температуру бани и нагревают ее так, чтобы температура равномер-
но повышалась на 2° в 1 мин. Температуру, при которой на нижнем крае
кольца бумаги конго красного появляется синее окрашивание, прини-
мают за температуру разложения испытуемого материала.
Термостабильность поливинилхлорида можно определять с помо-
щью комбинированного дифференциального термо- и газового анализа.
Термостабильность поливинилхлорида можно оценивать временем
от начала нагревания при 165° С до появления синего окрашивания ин-
дикатора.
Испытание проводят дважды; в случае расхождения результатов
двух последовательных или параллельных определений более чем на 3°
определение повторяют.
86
Техника безопасности при работе с винилхлоридом
и винилиденхлоридом и их полимерами и сополимерами
Винилхлорид при нормальных условиях (комнатной температуре и
атмосферном давлении) газ. Поэтому при хранении и во время работы
с ним должны быть созданы условия, предотвращающие утечку и поте-
рю продукта и обеспечивающие безопасность. Поэтому винилхлорид
должен храниться при низкой температуре, при которой он находится в^
жидком состоянии .(—14°С), и в герметически закрытых сосудах, вы-
держивающих более или менее значительное давление (баллоны, авто-
клавы; в лабораторных условиях — запаянные ампулы из прочного
стекла). Если жидкий винилхлорид хранится в запаянных ампулах, они
должны быть завернуты в металлическую сетку и помещены в сосуд
Дьюара, наполненный сухим льдом.
При низких температурах в пределах от —40 до —50° С винилхло-
рид может храниться без ингибитора в стальных резервуарах. При ком-
натной температуре без ингибиторов его хранят в атмосфере азота под
давлением. При перевозке же обычно применяют ингибиторы даже не-
смотря на то, что мономер, из которого удален ингибитор, полимеризу-
ется труднее, чем мономер, в который его не вводили. Сосуды, в которых
хранится или перевозится винилхлорид, не должны заполняться более
чем на 84% их полной емкости.
Винилхлорид обладает токсичностью средней силы. Симптомы, вы-
званные его действием, в основном сходны с симптомами действия нар-
котических веществ. Отравление при вдыхании винилхлорида в неболь-
ших концентрациях сопровождается потерей равновесия, координации
движений и неполным наркозом. При больших концентрациях наступает
потеря сознания, сопровождаемая конвульсиями, и, наконец, глубокий
наркотический сон с полной неподвижностью. Дыхание изменяется от
быстрого прерывистого в начале наркоза до медленного неглубокого.
При 20—40 %-ной концентрации винилхлорида в воздухе морские
свинки погружаются в состояние глубокого наркоза, оканчивающегося
смертью. Максимально допустимая концентрация винилхлорида в воз-
духе рабочих помещений — 0,5 %.
Газообразный мономер при низкой концентрации имеет приятный
запах. При вдыхании несмертельных доз его через несколько минут
наступает головокружение, теряется ориентация относительно располо-
жения и величины окружающих предметов, ощущается жжение в по-
дошвах ног. При равных концентрациях и при однократном воздействии
винилхлорид менее опасен, чем бензол, хлороформ и четыреххлористый
углерод. Концентрации от 5% и ниже вызывают головокружение и поте-
рю ориентации без вреда для организма.
Раздражающий запах винилхлорида проявляется лишь при высо-
ких концентрациях в воздухе, поэтому не служит достаточным предо-
стережением о его присутствии. При высоких концентрациях наркоти-
ческое действие винилхлорида проявляется очень быстро, поэтому чело-
век часто не замечает начальных предостерегающих симптомов, так как
потеря сознания и беспомощность могут наступить раньше, чем будет
обнаружено его присутствие.
87
Небольшое количество винилхлорида в воздухе, появляющееся в
результате утечки газа, не может вызвать сколько-нибудь сильного от-
равления. Оно более опасно из-за возможности взрыва, так как нижний
предел взрывоопасной концентрации его в воздухе невелик (4,0—
21,7 объемн. %)•• «
Поливинилхлорид с физиологической точки зрения безвреден. При
нагревании его выше 140° С выделяется хлористый водород; в процессе
переработки поливинилхлорида в изделия улетучиваются пластифика-
торы, поэтому перерабатывающие машины (вальцы, каландры) необ-
ходимо снабжать вакуум-зонтом для удаления летучих.
Поливинилхлорид не поддерживает горения (самозатухающий).
Пластифицированные изделия горят с большим количеством дыма. При
неполном сгорании поливинилхлорида выделяются ядовитые газы: НС1,
СО, хлор, фосген и др. В лаборатории необходимо иметь углекислотный
огнетушитель, противогаз, жидкий аммиак и работы проводить в вы-
тяжном шкафу с хорошим обменом воздуха.
Винилиденхлорид обладает сравнительно слабым физиологическим
действием. Пределом раздражающего действия на слизистые оболочки
является концентрация около 0,1 мг/л. Запах винилиденхлорида ощуща-
ется при содержании его в воздухе 0,2 мг/л. По сравнению с дихлорэта-
ном винилиденхлорид оказывает более слабое наркотическое влияние
на организм. Смертельная концентрация паров винилиденхлорида при
испытании на мышах составляет 15 мг/л, что примерно равно токсично-
сти хлорбензола. При нанесении мономера на кожу кроликов наблюда-
лось некоторое ее раздражение.
Винилиденхлорид сильно летуч и вдыхание его паров при открытом
испарении мономера вызывает острое отравление, поэтому работы с
ним следует проводить в вытяжном шкафу.
Так как винилиденхлорид легковоспламеняющееся вещество с очень
низкой температурой вспышки (—11°С), то при работе с ним необхо-
димо избегать открытого огня. Нагревание винилиденхлорида при пере-
гонке следует проводить в предварительно нагретой водяной бане, от
которой отнята горелка, или в бане, нагреваемой электрической плиткой.
Ни в коем случае не допускается непосредственный нагрев винилиден-
хлорида пламенем горелки.
Пары винилиденхлорида образуют с воздухом взрывчатую смесь.
Взрывоопасные концентрации лежат в пределах от 7 до 16 объемн.
При хранении чистого мономера без ингибиторов и в присутствии
кислорода воздуха наряду с полимерным продуктом образуется неболь-
шое количество перекисных соединений, которые в сухом состоянии под
действием кислорода воздуха способны разлагаться, иногда со взрывом.
Поэтому мономерный винилиденхлорид следует хранить с добавлением
ингибиторов (гидрохинона, фенола, спиртов, аминов), которые значи-
тельно замедляют образование перекисных соединений и последующую
полимеризацию, а также предохраняют мономер от воздействия кисло-
рода воздуха. Герметичную тару, предназначенную для хранения моно-
мера, необходимо предварительно продуть инертным газом (азотом,
двуокисью углерода).
Поливинилиденхлорид не горюч.
88
РАБОТА 21. ПОЛУЧЕНИЕ ВИНИЛХЛОРИДА
ИЗ ДИХЛОРЭТАНА
Исходные продукты: метиловый спирт—1'00 мл; едкий натр (химически чистый)—
80 г; дихлорэтан — 215 г; ацетон; сухой лед.
Оборудование: трехгорлая колба емкостью 750 мл, снабженная мешалкой, ртутным
затвором, термометром, капельной воронкой, обратным холодильником; колба емкостью
250 мл; термометр; стакан.
Реакцию ведут в приборе, изображенном на рис. 21. В верхней час-
ти обратного холодильника укрепляют второй термометр и присоединя-
ют нисходящий холодильник, нижний конец которого опускают в колбу,
погруженную в смесь ацетона с су*
хим льдом. Очень важно, чтобы
приемная колба была охлаждена
смесью ацетона с сухим льдом до
проведения опыта, в противном
случае вследствие неполноты кон-
денсации неизбежны потери винил-
хлорида.
В колбу помещают 100 мл мети-
лового спирта и 80 г химически чи-
стого едкого натра; при необходи-
мости добавляют еще некоторое ко-
личество метилового спирта, так что-
бы он покрывал едкий натр. Вклю-
чают мешалку и нагревают колбу
приблизительно до 50° С на водя-
ной бане, от которой отнята го-
релка, или с помощью электрона-
гревателя.
Для нагрева нельзя применять
открытый огонь, так как хлористый
винил очень легко воспламеняется
и взрывается. Необходимо обеспе-
чить бесперебойную подачу холод-
ной воды в оба холодильника. Ра-
бота обязательно проводится под
тягой (метиловый спирт ядовит). Рис. 21. Прибор для синтеза вииихлор»-
К раствору едкого натра в ме- да
тиловом спирте прибавляют из ка-
пельной воронки 215 г перегнанного дихлорэтана со скоростью около
100 капель в минуту. Когда температура в реакционной колбе достигнет
приблизительно 70° С, нагрев прекращают, так как для поддержания
необходимой температуры достаточно одной теплоты реакции.
Если температура понизится, то колбу снова немного нагревают,
примерно до 70° С. Во время проведения реакции метиловый спирт и
дихлорэтан, испаряясь, конденсируются в обратном холодильнике и
возвращаются в реакционную колбу, а винилхлорид поступает в охлаж-
89
даемый приемник. Если на термометре, укрепленном на обратном ход
дильнике, температура поднимется выше комнатной, необходим
снизить температуру в реакционной колбе или уменьшить скорость пр;
бавления дихлорэтана. В течение всего времени реакции переметив,
ние ведут с умеренной скоростью.
РАБОТА 22. ПОЛУЧЕНИЕ ВИНИЛИДЕНХЛОРИДА ДЕЙСТВИЕМ
ЕДКОГО НАТРА НА ТРИХЛОРЭТАН
Винилиденхлорид в лабораторных условиях получают из 1,1,2-тр:
хлорэтана, который дегидрохлорируют действием едких щелочей. Реа
ция идет по схеме
СН2С1-СНС12 Ч-NaOH СН2=СС12 4- NaCl + Н2О
Исходные продукты: 1,1,2-трихлорэтан—134 г; едкий натр — 45 г; дистиллирован-
ная вода — 300 мл; лед; гидрохинон.
Оборудование: прибор, изображенный на рис. 22 (емкость колбы 1 л); прямой'
холодильник; приемник; колба Вюрца; баня; склянка с притертой пробкой; холодиль-1
ник для хранения винилиденхлорида.
В дистиллированной воде растворяют едкий натр и раствор поме-
щают в колбу прибора. К дефлегматору присоединяют прямой холо-
дильник, а к последнему приемник для сбора продукта. Затем колбу с
раствором щелочи нагревают на водяной
бане до 75—78° С, включают мешалку, пу-
скают воду в холодильник и начинают мед-
ленно, в течение 3,5 ч, приливать из капель-
ной воронки трихлорэтан. Сразу же после
прибавления первых порций трихлорэтана
начинает отгоняться образующийся винили-
денхлорид, который собирают в приемник,
охлаждаемый водой со льдом. В приемник
предварительно вводят небольшое количе-
ство гидрохинона (около 0,1—0,2% -от веса
мономера). В качестве приемника ставят
колбу Вюрца, помещенную в баню со
льдом.
После загрузки всего трихлорэтана на-
гревание при 75— 78° С продолжают еще в
Рис. 22. Прибор для синтеза течение 20 мин. Температуру паров на вы-
винилиденхлорида ходе из дефлегматора поддерживают в ин-
тервале 31,5—33° С; что достигается ре-
гулированием скорости подачи трихлорэтана в реакционный сосуд.
Выход винилиденхлорида составляет 90—93% от теоретического.
Стабилизированный гидрохиноном технический винилиденхлорид, не
соприкасающийся с воздухом, можно хранить длительное время.
90
РАБОТА 23. ПОЛУЧЕНИЕ ВИНИЛИДЕНХЛОРИДА ДЕЙСТВИЕМ
ИЗВЕСТКОВОГО МОЛОКА НА ТРИХЛОРЭТАН
Винилиденхлорид может быть получен из 1,1,2-трихлорэтана дей-
ствием на него водной гидроокиси кальция (известкового молока). При
этом отщепляется хлористый водород. Реакция идет по схеме
2СН2С1-СНС12 4- Са (OHfc 2СН2=СС12 + СаС12 + 2Н2О
Исходные продукты: 1,1,2-трихлорэтан—267 г; известковое молоко (пл. 1,20 г/см3)—
343 мл *.
Оборудование: автоклав емкостью 3 л, снабженный мешалкой; колба Вюрца, хо-
лодильник и приемники; колба с елочным дефлегматором; приемники 2—3 шт.
В автоклав загружают 1,1,2-трихлорэтан и 'известковое молоко.
Реакционную смесь перемешивают при комнатной температуре в тече-
ние 3—4 ч. Затем содержимое автоклава медленно нагревают до 70—
80° С и перемешивают при этой температуре в течение 1—2 ч. После ох-
лаждения реакционной массы ее переносят из автоклава в колбу Вюрца
и отгоняют сырой винилиденхлорид при 40—60° С.
Для получения чистого продукта сырой винилиденхлорид загружа-
ют в круглодонную колбу, снабженную елочным дефлегматором высотой
40—45 см, термометром, прямым холодильником, и ведут перегонку при
нагревании на водяной бане. Фракцию продукта, кипящую в пределах
30—33° С, собирают как чистый продукт и анализируют (определяют
плотность и показатель преломления).
Выход чистого винилиденхлорида составляет около 90% от теорети-
ческого.
РАБОТА 24. ПОЛУЧЕНИЕ ПОЛИВИНИЛХЛОРИДА ПОЛИМЕРИЗАЦИЕЙ
ВИНИЛХЛОРИДА В РАСТВОРЕ
Исходные продукты: винилхлорид —100 г; ацетон —100 г; перекись бензоила —1 г.
Оборудование: автоклав емкостью 1 л, рассчитанный на давление не менее 20 атм,
освинцованный внутри, снабженный манометром, мешалкой и термометром; баллон
винилхлорида; фарфоровый стакан емкостью 250 мл.
В автоклав загружают из баллона при охлаждении до —20° С ви-
нилхлорид, ацетон и перекись бензоила. Закрыв крышку, включают ме-
шалку и при непрерывном перемешивании нагревают содержимое авто-
клава в течение 60 ч при температуре 40° С.
В процессе нагревания давление в автоклаве вначале повышается
и достигает 8—12 атм, но по мере протекания реакции полимеризации
оно постепенно падает и к концу реакции достигает почти атмосферного.
По окончании полимеризации автоклав охлаждают до комнатной тем-
пературы, спускают остаточное давление, соединив автоклав с атмосфе-
рой, и только после этого открывают крышку и выгружают полимер.
* Известковое молоко плотностью 1,20 г/см3 содержит 242 г/л гидроокиси кальция.
Са(ОН)2; 343 мл известкового молока соответствуют 93 г Са(ОН)2.
91
Полученный светло-желтый продукт растворим в хлорбензоле и ди4
хлорэтане.
Определяют относительную вязкость в вискозиметре Оствальда—:
Пинкевича, находят по табл. 6 значение К. Производят качественный
анализ на хлор. Полимер используют в виде лака для покрытия бумаги..
РАБОТА 25. ПОЛУЧЕНИЕ ПОЛИВИНИЛХЛОРИДА ПОЛИМЕРИЗАЦИЕЙ
ВИНИЛХЛОРИДА В ЭМУЛЬСИИ
[ЭМУЛЬСИОННЫЙ ПОЛИВИНИЛХЛОРИД]
Исходные продукты: винилхлорид— 100 г; перекись водорода (30 %-ной концентра- ‘
ции)—0,11 г; эмульгатор МК (натриевая соль мепазинсульфокислоты)—3,0 г; вода;
дистиллированная— 114 г; ортофосфорная кислота — 0,18 г; едкий натр (сухой) —0,2 г. <
Оборудование: автоклав, рассчитанный на рабочее давление до Г5 атм, снабжен-
ный рубашкой для нагрева и охлаждения, мешалкой и манометром; фарфоровый ста- j
кан емкостью 500 мл; колба емкостью 250 мл; воронка; 2—3 стакана химических емко- -
стью 200 мл.
Эмульгатор МК растворяют в дистиллированной воде до получения 1
раствора 20%-ной концентрации и фильтруют (должен получиться со-
вершенно прозрачный раствор).
В автоклав загружают приготовленный раствор эмульгатора, а за-
тем вводят при перемешивании указанные в рецептуре количества воды,
перекиси водорода, ортофосфорной кислоты и едкого натра.
После тщательного перемешивания смеси определяют pH среды,
который должен быть равен 6,5—7,0. При таком значении pH обеспечи-
вается нужная скорость распада перекиси водорода на свободные ради-
калы, а следовательно, и необходимая скорость реакции. Если pH не
соответствует указанной величине, то добавлением либо ортофосфорной
кислоты, либо едкого натра его доводят до нужного значения. После
такой подготовки водной фазы и установления pH автоклав гермети-
зируют и начинают загрузку винилхлорида.
По окончании загрузки винилхлорида включают мешалку, вращаю-
щуюся со скоростью 50—70 об/мин. Реакционную массу подогревают,
пропуская горячую воду через рубашку автоклава. После того как внут- .
ри аппарата температура поднимется до 50°С, подогрев прекращают, j
Если в результате экзотермической реакции температура начнет повы-
шаться, реакционную массу охлаждают, пропуская холодную воду через
рубашку автоклава.
Отвод тепла регулируют с таким расчетом, чтобы давление в авто-
клаве поддерживалось строго в пределах 7—7,5 атм, а температура бы-
ла равна 45—55° С. Окончание процесса полимеризации определяют по
падению давления в автоклаве. После этого реакционную массу охлаж-
дают до 25—30° С.
Процесс полимеризации с момента начала нагревания продолжает-
ся 38—40 ч. По окончании реакции остаточное давление в автоклаве ’’
(около 3 атм), создающееся за счет использования непрореагировавше- "
го винилхлорида, спускают и полученный в виде латекса поливинилхло-
92
рид выгружают. Определяют количество полимера в латексе (см.
стр. 85).
Сушку ведут распылением латекса в сушильных камерах. При от-
сутствии соответствующего оборудования латекс коагулируют, вводя
в него концентрированную соляную кислоту при нагревании и перемеши-
вании. Порошок полимера фильтруют на воронке Бюхнера, промывают
водой до исчезновения в промывных водах реакции на ион С1~ (с раст-
вором AgNO3) и сушат при 45° С. Полученный полимер перерабатывают
в изделия (см. работы 37, 38, 39).
Примечание. Для стабилизации поливинилхлорида в латекс вводят 5%-ный вод-
ный раствор кальцинированной соды до получения щелочности продукта, равной 0,35—
0,40%. Полимер стабилизируют при перемешивании в течение 10—15 мин без нагре-
вания.
РАБОТА 26. ПОЛУЧЕНИЕ ПОЛИВИНИЛХЛОРИДА ПОЛИМЕРИЗАЦИЕЙ
ВИНИЛХЛОРИДА В СУСПЕНЗИИ
Исходные продукты: винилхлорид—100 г; вода дистиллированная—120 мл; жела-
тин— 0,57 г; динитрил азоизомасляной кислоты (порофор ЧЗХ 100%-ный)—0,15 г;
едкий натр (сухой) — 0,7 г.
Оборудование: химический стакан емкостью 250 мл; сита № 30 и № 40, автоклав
емкостью 1 л, рассчитанный на рабочее давление не менее 15 атм, снабженный рубаш-
кой для обогрева и охлаждения, мешалкой, термометром и манометром; фарфоровый
стакан емкостью 4. л; воронка Бюхнера; термошкаф.
В отдельном сосуде готовят раствор желатина в воде. Его фильт-
руют через сито № 40 и загружают в автоклав. Затем при работающей
мешалке в аппарат вводят инициатор — динитрил азоизомасляной кис-
лоты.
Автоклав охлаждают до —20° С, пропуская через рубашку хлад-
агент, после чего вакуумируют и загружают в него имеющий ту же
температуру винилхлорид. Затем автоклав герметизируют, включают
мешалку и подогревают реакционную массу до 48° С, пропуская через
рубашку автоклава горячую воду. После окончания индукционного пе-
риода и начала реакции полимеризации, что определяется по повыше-
нию температуры внутри аппарата вследствие экзотермичности процес-
са, обогрев прекращают и охлаждают реакционную массу, пропуская
через рубашку аппарата холодную воду, а затем охлаждающий рассол
с температурой не выше 0° С. Отвод тепла регулируют с таким расчетом,
чтобы давление внутри автоклава поддерживалось в пределах 8—
8,5 атм, а температура — около 50° С.
Окончание процесса полимеризации определяют по понижению дав-
ления в автоклаве при некотором повышении температуры. В это время
охлаждение выключают и температура вследствие экзотермичности про-
цесса поднимается приблизительно до 70° С.
Когда давление в автоклаве снизится дс 3,5—3,0 атм, процесс за-
канчивают. Открыв кран, спускают давление, выгружают полимер и
подвергают его щелочной обработке и осаждению.
Щелочную обработку и высаждение полимера проводят в отдель-
ном сосуде 1 % -ным водным раствором едкого натра в течение 2 ч при
93
непрерывном перемешивании и нагревании острым паром до 94—96°С. |
Цель щелочной обработки состоит в том, чтобы разрушить эмульгатор, <
разложить и удалить адсорбированный на полимере винилхлорид.
После щелочной обработки реакционную массу отфильтровывают;
на воронке Бюхнера, промывают водой до нейтральной реакции про-
мывных вод (определяемой с фенолфталеином), сушат при 45—50°С и.
просеивают через сито № 30.
Полученный продукт подвергают анализу на содержание хлора
(стр. 84), определяют температуру разложения и используют для вы-
полнения работ № 27, 28, 30, 34 и др.
Примечание. Поливинилхлорид, предназначенный для получения хлорированного
поливинилхлорида (перхлорвинила), щелочью не обрабатывают, так как ее присутствие
в полимере замедляет процесс хлорирования.
РАБОТА 27. ИЗГОТОВЛЕНИЕ ЛИСТОВОГО
ВИНИПЛАСТА ПРЕССОВАНИЕМ
Винипласт — твердый пластический материал, получаемый термо-
механической пластикацией поливинилхлорида и содержащий стабили-
затор и смазку. Для его изготовления используются соответствующие
марки поливинилхлорида: ПВХ-С63Ж (суспензионный), блочный ПВХ
Б62-66.
Винипласт выпускается в виде пленки, листов, труб, стержней и
других профилей, а также сварочного прутка для сварки винипласта.
Винипласт устойчив к воде, агрессивным средам и растворителям. При
длительно действующих нагрузках прочность винипласта уменьшается
и через 3000 ч достигает 190 кг/см2 (долговременная прочность), после
чего меняется незначительно.
Винипластовая пленка каландрованная (ГОСТ 16398—70) являет-
ся полуфабрикатом для изготовления винипласта.
Из винипластовой пленки изготавливают на гофрировальных и пер-
форационных станках перфорированные и гофрированные пленки
(ГОСТ 15976—70) для электротехнической промышленности, прочные и
стойкие к действию кислот и щелочей.
Винипластовые листы (ГОСТ 9639—61) изготавливают прессова-
нием «пакетов» пленки определенного размера на многоэтажных прес-
сах. Трубы, стержни, профильные изделия из винипласта (ТУ 6-01-801—73
и 6-01-812—73) изготавливают экструзией или на гидравлических прес-
сах (штранг-прессах). Сварочный пруток изготавливают экструзией из
частично пластифицированного поливинилхлорида.
Изготовление листового винипласта складывается из следующих
операций: 1) приготовление композиции; 2) вальцевание композиции;
3) каландрование массы; 4) прессование.
Исходные продукты: поливинилхлорид суспензионный — 100 г; меламин — 2 г; сте-
арин — 1 г; трансформаторное масло — 2 г.
Оборудование: краскотерочная плита (или фарфоровая ступка); шаровая мель-
ница; вальцы лабораторные с фрикцией (рис. 23); каландр лабораторный, диаметр вал-
94
ков 150 мм (рис. 24); ножницы; лабораторный этажный пресс (рис. 25); полированный
стальной лист.
Меламин тщательно растирают на краскотерочной плите или в фар-
форовой ступке с трансформаторным маслом для получения однородной
Рнс. 23. Смесительные вальцы:
1 — валок с низкой температурой;
2 — валок с высокой температурой;
3 — лоток; 4 — вытяжная вентиля-
ция
Рис. 24. Лабораторный трехвалковый ка-
ландр с нижним выносным валком:
1 — композиция; 2 — верхний валок; 3 — сред-
ний валок; 4— н'нжний валок; 5 — выносной
валок; 6 ~ прижимной валок; 7 — приемные -
валки; 8 — готовый листовой материал
пастообразной массы. Затем порошок поливинилхлорида, измельчен-
ный стеарин и приготовленную массу загружают в шаровую мельницу
и перемешивают при комнатной температуре в течение 3 ч также до по-
лучения совершенно однородной смеси.
Рис. 25. Этажмый пресс:
1 — цилиндр; 2 — илиты; 3 — стол; 4 — тележка
95
Приготовленную смесь (композицию) вальцуют на вальцах, имею-
щих фрикцию 1,17, при 170° С на переднем валке и 165° С на заднем до
получения гомогенной пластичной массы.
Пластицированную массу в горячем состоянии переносят на каландр
для получения тонкой однородной пленки одинаковой толщины по всей
длине и ширине ее полотна.
Толщина выходящей из каландра пленки должна быть не более
1 мм (обычно 0,5—0,6 мм).
Во время процесса каландрования температура валков каландра
постепенно повышается от верхнего валка к нижнему. Это делается для
того, чтобы полотно винипласта не прилипало к валкам каландра и
легко отставало от менее нагретого валка при движении полотна. Тем-
пературу верхнего валка устанавливают в пределах 160—165° С, сред-
него 165—170° С и нижнего 170—175° С.
Пленку винипласта, выходящую из каландра, после охлаждения
разрезают на листы одинакового размера и собирают в пакеты. Пред-
варительно из пленки вырезают стандартные образцы для испытания
на предел прочности на разрыв, относительное удлинение и химическую
стойкость.
Пакет собирается из нескольких листов пленки в зависимости от
требуемой толщины винипластового листа (плиты). Для укладки пакета
вначале на стол кладут металлическую плиту, затем несколько слоев
фланели, полированный тонкий стальной лист, затем лист целлофана.
После этого кладут несколько листов винипластовой пленки, на него
снова целлофан, тонкий стальной полированный лист, фланель и метал-
лическую плиту.
Такой «пакет» закладывают в лабораторный этажный пресс и прес-
суют при 170—175° С и удельном давлении 20—30 кг/см2, выдерживая
под давлением в течение 2 мин на 1 мм толщины изделия.
Запрессованное изделие охлаждают под давлением в прессе до
50° С, после чего снимают давление и вынимают готовые листы (плиты)
винипласта.
Определяют химическую устойчивость винипласта.
РАБОТА 28. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ЛИСТОВОГО
ВИНИПЛАСТА МЕТОДОМ ПНЕВМАТИЧЕСКОГО ФОРМОВАНИЯ
Изготовление изделий из листового и пленочного винипласта мето-
дом пневматического формования (выдувания) широко распространено.
Сущность метода заключается в том, что разогретый до размягчения
лист винипласта подвергают вытяжке и оформлению в изделие в форме
с ограничивающей поверхностью под действием сжатого воздуха, а так-
же и без формоограничения в случае изготовления изделий сферической
формы. Формование производят в интервале температур, при которых
винипласт находится в высокоэластическом состоянии.
При изготовлении изделий выдуванием необходимо учитывать ха-
рактерную особенность термопластов, заключающуюся в том, что
96
подвергнутый вытяжке винипласт при повторном нагреве выше темпе-
ратуры вытяжки способен к восстановлению первоначальной формы,
причем чем выше была температура нагрева листа винипласта при фор-
мовании, тем меньше склонность изделия к восстановлению йервона-
чальной формы при повторном нагреве в условиях'эксплуатации.
От температуры формования зависит также величина вытяжки ви-
нипласта. Как видно из ipnc. 26, наибольшая вытяжка винипласта (до
350%) имеет место при температуре около 110° С, но изделия, форму-
Температура, °C
Рис. 26. Зависимость вытяж-
ки винипласта от темпера-
туры нагревания
Рис. 27. Приспособление с ограни-
чивающей поверхностью для пнев-
матического формования виниплас-
та:
1 — заготовка; 2 — плита; 3 — каркас;
4 — прижимное кольцо; 5 — зажим; 6
воздушный кран
емые при этой температуре, при повторном нагревании теряют придан-
ную форму. Изделия же, отформованные при температурах выше 110° С,
хорошо сохраняют приданную им форму, хотя способность винипласта к
вытяжке при этих температурах падает. Это объясняется тем, что выше
110° С наблюдается переход от высокоэластического состояния к вязко-
текучему, когда деформации становятся необратимыми.
Оптимальная температура' нагрева винипластовых листов при фор-
мовацНи лежит около 130° С. При этой температуре вытяжка винипласта
достаточно велика (до 100%), и, что особенно важно, .изготовленное из-
делие при повторном нагреве до температуры вытяжки в условиях экс-
плуатации почти не изменяет свою форму.
Исходные материалы: винипласт листовой толщиной 2—3 мм или винипласт пле-
ночный толщиной 1 мм.
Оборудование: приспособление для пневматического формования; баллон со сжа-
тым воздухом или инертным газом; термошкаф.
Изготовление изделий пневматическим формованием проводят в
приспособлении с ограничивающей оформляющей поверхностью в виде
каркаса (рис. 27).
Винипластовую заготовку определенного размера, и соответствую-
щей формы нагревают в термошкафу или на металлической плите до
130° С и укладывают в приспособление между плитой 2 и каркасом 3.
4—1453 97
Заготовку прижимают кольцом 4 и закрепляют зажимами 5. Через воз-
душный кран 6 быстро впускают сжатый воздух под давлением 5—
6 атм. Воздух предварительно не подогревается. Под давлением сжато-
го воздуха размягченная заготовка растягивается и принимает форму,
соответствующую пръфилю каркаса.
После окончания формования и полного охлаждения изделия подачу
воздуха прекращают, перекрыв воздушный кран, снимают прижимы и
зажимное кольцо и вынимают готовое охлажденное изделие.
РАБОТА 29. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ЛИСТОВОГО
И ПЛЕНОЧНОГО ВИНИПЛАСТА ВАКУУМ-ФОРМОВАНИЕМ
Изделия из листового и пленочного винипласта можно изготовить
также вакуумным формованием, которое, как и пневматическое формо-
вание, проводят в температурном интервале, в котором винипласт нахо-
дится в высокоэластическом состоянии (90—170° С). Оптимальной
является температура около 130° С.
Исходные материалы: винипласт листовой толщиной 2—3 мм или винипласт пле-
ночный толщиной 1 мм.
Оборудование: установка для вакуум-формования; вакуумный насос; обогреваю-
щая рама.
Вакуумное формование проводят на установке для формования,
схема которой представлена на рис. 28.
Установка состоит из камеры /, прижимной рамы 2 и обогревающей
рамы 5 с электрическими нагревательными элементами. Камера присо-
единена к вакуум-насосу через отверг
стие 7, расположенное в нижней части
камеры. Обогревающая рама прикрепле-
на над прижимной рамой так, что ее
можно передвигать в горизонтальном
направлении.
Внутрь вакуумной камеры помещают
деревянную, гипсовую или металличе-
скую форму 4, изготовленную в соответ-
ствии с профилем формуемого изделия.
При формовании заготовку виниплас-
та нужной формы и размера кладут на
верхнюю часть камеры, прижимают за-
жимной рамой и при помощи струбцин 6
плотно закрепляют так, чтобы не было
подсоса воздуха в камёру. Обогреваю-
Рис. 28. Схема установки для
вакуумного формования изде-
лий:
/ — камера; 2 — прижимная рама;
3 — Заготовка винипласта; 4 — фор-
ма; 5 — обогревающая рама; 6 —•
струбцина; 7 — отверстие
щую раму устанавливают непосредствен-
но над заготовкой винипласта и включают ток для обогрева. Как толь-
ко заготовка нагреется до 130° С, что происходит приблизительно через
40 50 с после начала нагревания, включают вакуум-насос. Под влия-
нием атмосферного давления лист винипласта равномерно растягивает-
98
ся, вдавливаясь внутрь камеры, и принимает форму, соответствую!^1®^
профилю формы. х
По окончании формования ток выключают, обогревающую раму от,(\
водят в сторону и отключают вакуум-насос. Отформованное изделие
нимают из установки только после полного охлаждения-. '
РАБОТА 30. ИЗГОТОВЛЕНИЕ ПРОФИЛЬНЫХ ИЗДЕЛИЙ
ИЗ ВИНИПЛАСТА МЕТОДОМ ЭКСТРУЗИИ
Профильные изделия из винипласта могут быть изготовлены м^т5К
дом экструзии на горизонтальных экструзионных машинах (см. рис- ,
или на гидравлических профильных прессах (рис. 29). В первом сл/1^ '
оформление изделия происходит в формующей головке.. Методом ;
рузии из винипласта изготовляют трубы различных диаметров, уго/к^х^
стержни и другие профилированные изделия. '
Рис. 29. Схема гидравлического профильного пресса для выдав-
ливания труб и стержней из термопластов:
I — ретурный плунжер: 2 — ретурный цнлнндр; 3 — главный цилиндр; 4 —
главный плунжер; 5 — материальный плунжер; 6 — материальный цилиндр;
7 — головка материального цилиндра; 8 — прессуемый материал; 9 — рубаш-
ка обогрева
Изготовление профильных изделий на экструдере складываете/*^^
следующих основных операций: 1) приготовление композиции (см^г^к
мне компонентов); 2) вальцевание композиции; 3) экструзия ма^
Исходные продукты: поливинилхлорид (латексный, стабилизированный) —
свинцовый глет—-2,5 г; стеарин — 2,0 г; трансформаторное масло—1 г. . (с
Оборудование: лабораторная экструзионная машина с трубной головкой х
рис. 7); лабораторный смеситель (рис. 30); лабораторные вальцы; сито № 20.
Поливинилхлорид, растертые в тонкий порошок и просеянные
мелкое сито свинцовый глет и стеарин, а также трансформаторное
ло загружают в смеситель лабораторного типа, представляющий сг/^Х^
стальной аппарат с корытообразным днищем, в котором вращаются
Z-образные лопасти (см. рис. 30). Перемешивание ведут при комнатЛ^
температуре в течение 1 ч до получения совершенно однородной су
(композиции). oq
Приготовленную смесь переносят на вальцы и вальцуют при
170° С в течение 30 мин при зазоре между валками 0,4—0,5 мм. В
4‘ Ч
К
зультате вальцевания масса должна стать совершенно однородной
(гомогенной), без непровальцованных включений и полупрозрачной на
вид. Затем температуру валков поднимают до 170—180° С и при этой
температуре провальцованную массу закатывают в рулон, который за-
кладывают в лабораторный экструдер.
Рис. 30. Смеситель лабора-
торный»
1 — крышка; 2 — корпус; 3 —
Экструзию ведут при следующем режи-
ме: температура в цилиндре 175—185° С;
температура головки 195—200 °C. Трубу
обрезают по выходе из экструдера до не-
обходимой длины.
РАБОТА 31. ИЗГОТОВЛЕНИЕ ИЗДЕЛИИ
ИЗ ВИНИПЛАСТА СВАРКОЙ
Наиболее простым методом сварки ви
Z-образные лопасти
нипласта является сварка нагретым возду-
хом, осуществляемая при помощи специаль-
ных горелок.
Сварка винипласта основана на его спо-
собности переходить в вязкотекучее состо-
яние при 200—220° С и поэтому такая температура применяется во всех
процессах сваривания этого материала. Во избежание разложения ви-
нипласта нагревание его в месте сварки должно быть кратковремен-
ным, так как температура, при которой происходит сваривание, очень
близка к температуре разложения. При сварке нагретым воздухом ви-
нипласт размягчается и приобретает способность прочно соединяться
при небольшом давлении.
Сваривание винипластовых изделий может осуществляться как при
помощи присадочного материала, так и без него. Наиболее часто при-
меняется сварка при помощи присадочного материала — сварочного
прутка, который изготовляется из пластифицированного поливинилхло-
рида.
При прутковой сварке винипласта прутки размягчаются и прочно
скрепляются с поверхностями свариваемых деталей. Редко применяется
беспрутковая сварка. При этом размягчение свариваемых поверхностей
производится при помощи струи горячего воздуха.
Рис. 31. Электрическая горелка для сваривания винипласта:
электрический обогреватель; 2 — подвод электрического тока; 3 — сжатый воздух; 4 — выход
нагретого воздуха
100
Исходные материалы: винипласт листовой толщиной 2—3 мм; сварочный прутов
диаметром 1—2 мм.
Оборудование: горелка электрическая (рис. 31) или газовая (рис. 32).
Существуют различные виды сварного шва для винипласта
(рис. 33): встык, внахлестку, .впритык, угловой' Наиболее прочным яв-
ляется соединение встык.
Рис. 32. Газовая горелка системы ГГП-1-56 для свари-
вания винипласта:
/ — ствол; 2 —канал камеры; 3 —штуцер; 4 —трубка; 5 —кор-
пус; 6 — мундштук; 1 — газовая камера
Поверхность свариваемого материала и сварочного прутка должна
быть чистой, ровной и обезжиренной. Прочность соединения материала
с глянцевой поверхностью значительно ниже, чем с шероховатой, поэто-
му в местах сварки глянец снимают рашпилем или наждачной шкуркой.
Раскрой и подготовка листов винипласта производится в зависимо-
сти от изготовляемого изделия и вида шва. При изготовлении, например,
стойкой к агрессивным средам
ванны из винипласта применяют
соединение впритык или угловое.
В соответствии с этим произво-
дят раскрой материала и подго-
товку его для сваривания. В слу-
чае углового соединения или со-
единения встык производят под-
готовку кромок, заключающуюся
в снятии фаски на одном или
обоих свариваемых листах (рис.
33, а).
Температура воздуха на по-
верхности свариваемого материа-
ла должна составлять 200—
220° С. Для обеспечения такого
нагревания воздух в горелке дол-
жен быть нагрет до 230—270° С,
так как при прохождении рассто-
яния от сопла горелки до свари-
ваемых поверхностей струя воз-
духа остывает.
При сварке пруток следует
подавать под углом 90° к поверх-
fl
г
Рис. 33. Виды сварного шва для вини-
пласта:
а — встык; б — внахлестку; в впритык; г —
угловой
101
ности шва (рис. 34), а угол а
между направлением струи нагре-
того воздуха и швом, так назы-
ваемый угол подвода наконечни-
ка горелки, для свариваемого
листа винипласта толщиной до
5 мм должен составлять 20—25°;
для винипласта толщиной более
5 мм угол подвода наконечника
горелки составляет 30—45°.
Рис. 34. Сварка деталей из винипласта
при помощи горелки
При сваривании материала горелку необходимо держать правой
рукой, а пруток левой. Давление на пруток должно быть по возможно-
сти постоянным. По окончании процесса сварки сварочный пруток об-
резают ножом так, чтобы он на 3—5 мм выходил за край шва, й изде-
лие оставляют на воздухе для медленного охлаждения. \
РАБОТА 32. ПОЛУЧЕНИЕ СТАБИЛИЗАТОРОВ — СОЛЕЙ \
СТЕАРИНОВОЙ КИСЛОТЫ И МЕТАЛЛОВ
(КАЛЬЦИЯ, КАДМИЯ, БАРИЯ, СВИНЦА]
Стеараты кальция, кадмия, бария и свинца являются важнейшими
представителями кислотоакцепторных стабилизаторов. Свойства стаби-
лизированных композиций в значительной мере зависят и от аниона и
от катиона стабилизатора этого типа.
Реакция протекает ото схеме
5Ci7H35COONa + СаС12-*(С17Н35СОО)2Са + 2NaCl
или
2C17H35COONa + Cd (NO3)2-> (C17H35COO)2Cd + 2NaNO3
Исходные продукты: стеариновая кислота—10 г; 15%-ный раствор NaOH или
КОН—15 г; 110%-ный раствор хлористого калышя (азотнокислого кадмия пли бария).
Оборудование; фарфоровый стакан емкостью 0,5 л; мешалка; воронка Бюхнера;
термошкаф; ступка; асбестовая сетка; термометр.
Навеску стеариновой кислоты помещают в фарфоровый стакан и
осторожно нагревают на газовой горелке через асбестовую сетку до
90—100° С. Добавляют около 20 мл дистиллированной воды и прилива-
ют небольшими порциями 15%-ный раствор щелочи в количестве, необ-
ходимом для омыления стеариновой кислоты. Каждую порцию добавля-
ют лишь после того, как предыдущая прореагировала. Контроль реак-
ции производят пробой на фенолфталеиновую бумажку, красная
окраска которой указывает на избыток щелочи. Полученный
раствор натриевого (калиевого) мыла разбавляют горячей водой и при
помешивании доводят температуру до 90—100° С, а затем, не прекращая
перемешивания, осаждают соль стеариновой кислоты избытком раство-
ра соответствующей минеральной соли. Реакция обменного разложения
102
происходит мгновенно. Добавляя по стенке сосуда раствор соли, уста
навливают полноту осаждения.
Выпавший осадок соли стеариновой кислоты отфильтровывают н;
воронке Бюхнера, промывают горячей водой до нейтральной реакции i
отсутствия реакции на анион исходной соли (Cl-, NO3_ и др.). Стеара:
высушивают при 90—100° С, растирают в порошок и анализируют, оп
ределяя кислотное число (путем титрования раствора эфирного экстрак
та в спирте 0,1 н. спиртовым раствором КОН) и температуру плавленш
в капилляре (температура плавления стеарата кальция 175° С, стеаратг
кадмия 104°С, стеарата бария 200° С).
РАБОТА 33. ПОЛУЧЕНИЕ ДИБУТИЛФТАЛАТА
Реакция протекает по схеме
О
11
+2С4Н,ОН; [H2SO.l C-OG4H9
4-... ~_± +н2о
L ||-с-ос4н9
и
о
Исходные продукты: фталевый ангидрид—148 г; бутиловый спирт—170 г; H2SOj
(пл. 1,84 г/см3) —3 г; 0,5 н. раствор NaOH (или 20%-ный раствор соды).
Оборудование: круглодонная колба с насадкой Дина — Старка и Обратным холо-
дильником; прибор для перегонки в вакууме; делительная воронка.
Фталевый ангидрид, бутиловый спирт и серную кислоту помещают
в реакционную колбу, собирают прибор (рис. 35), нагревают на сетке
до кипения смеси и отделения воды в ловушке. Конец реакции опреде-
ляют по прекращению выделения воды. Не вошедший
в реакцию ангидрид нейтрализуют раствором соды, п
эфир промывают водой до нейтрал ьной.реакции в дели- II
тельной воронке, высушивают безводным сульфатом Ч-А
натрия и перегоняют в вакууме, собирая фракцию QT
206°/10 мм рт. ст. или 182°/5 мм рт. ст. 0L
Физико-химические свойства дибутилфталата: IlX
d4S= 1,047—1,050; По= 1,490—1,493; т]20=25 сП; рас- К
творимость в воде при 20° С 0,1%; растворимость во- М
ды в дибутилфталате при 25° С 0,3—0,5%; летучесть I
0,22 мг/(см2-ч) при 100° С. (Летучесть является не-
достатком дибутилфталата.) 13?
По аналогичной методике можно получить диок- / 4
тилфталат — менее летучий пластификатор с показа- • /\
телями: Тпл=— 40° С, Ткип=340°С и 18575 мм рт. ст.; / ( V
п о = 1,482; т]2О=40 сП; летучесть 0,25% за 24 ч при Jr
105° С; нерастворим в воде; растворимость воды в ди-
октилфталате — 0,6%. Рис 35. Прибор
В присутствии бензолсульфокислоты в качестве для синтеза
катализатора получают ди-п-октилсебацинат: ТПц= сложного эфира
103
=___55° С; 7Кип=24874 мм рт. ст.; df° ==0,913; и^ = 1,4496; ц20= 19,95 сП;
^80=4,04 сП; растворимость в воде 0,025%.
Диоктилсебацинат один из лучших пластификаторов, обеспечиваю-
щих композиции морозоустойчивость.
РАБОТА 34. ИЗГОТОВЛЕНИЕ ПЛЕНОЧНОГО
ПЛАСТИКАТА
Пластикат представляет собой эластичный материал, получаемый
из поливинилхлорида и содержащий также пластификатор, стабилиза-
тор, иногда краситель и другие добавки. В настоящее время выпускает-
ся около 20 марок пластифицированного пластиката и пленок различно-
го назначения (И40-13; И40-14; И50-13; И50-14 и т. д.), в том числе
пластикат изоляционный, светотермостойкий (ГОСТ 5960—72) и др.
Из пластифицированного поливинилхлорида выпускают липкие
изоляционные ленты (ГОСТ 16214—70; 17617—72 и др.) предназначен-
ные для ремонта и сращивания кабельных оболочек, трубки для галет-
ных батарей, масло- и бензостойкие изделия и линолеумы как на ткане-
вой основе, так и на основе прочных наполненных пленок. Изделия из
пластифицированного поливинилхлорида обладают прочностью, эластич»
ностью как при обычной, так и пониженной температуре.
Изготовляют пленочный пластикат вальцеванием с последующим
каландрованием или экструзией. Первый метод полунепрерывный и тре-
бует затраты ручного труда, поэтому он постепенно уступает место вто-
рому. Однако изготовление пленочных пластикатов вальцеванием и
каландрованием еще не потеряло своего значения, так как этот метод
дает пленки высокого качества с точной калибровкой по толщине.
Изготовление пленочного пластиката методом каландрования скла-
дывается из следующих основных операций: приготовление композиции,
вальцевание массы, каландрование.
Исходные продукты: поливинилхлорид суспензионный — ЮО г; дибутилфталат —
45 г; стеарат кальция — 3 г; стеарин — 1 г.
Оборудование: лабораторный смеситель; лабораторные вальцы; лабораторный ка-
ландр.
Поливинилхлорид, стеарат кальция, стеарин и дибутилфталат загру-
жают на 2 ч в лопастной смеситель лабораторного типа при нагревании
до 75—80° С.
Приготовленную смесь вальцуют на лабораторных вальцах в две
стадии. На первой стадии вальцевание проводят на вальцах с зазором
между валками 3—4 мм. Температуру рабочего валка поддерживают в
пределах 145—155° С, температуру холостого валка— 140—150° С. Валь-
цевание ведут 15—20 мин, подрезая массу ножом.
На второй стадии вальцевания зазор между валками уменьшают до
1—2 мм, а температуру повышают. Температуру рабочего валка поддер-
живают в пределах 150—160° С, а температуру холостого валка — 145—
|155°С. Вальцевание на второй стадии продолжается 6—7 мин.
После вальцевания горячую массу переносят на каландр. Каландро-
вание производят при следующем температурном режиме: температура
104
верхнего валка 125±5°; среднего 135±5°; нижнего 145±5°. Толщина вы-
ходящей из каландра пленки должна быть 0,2—0,3 мм.
Из готовой пленки вырезают стандартные лопатки и испытывают на
прочность при разрыве и относительное удлинение (см. ч. II).
РАБОТА 35. ИЗГОТОВЛЕНИЕ ЭЛАСТИЧНОГО ГУБКОПОДОБНОГО
МАТЕРИАЛА ИЗ ПОЛИВИНИЛХЛОРИДА
Исходные продукты: поливинилхлорид — Ю0 г; пластификатор (дибутилфталат
или ВИСФ)*— 1<00 г;, карбонат аммония — 8 г; гидрокарбоиат натрия — 8 г; окись цин-
ка'— 4 г.
Оборудование: лабораторный смеситель; форма с двумя отверстиями и зажимным
винтом (рис. 36); термошкаф.
Порошкообразный поливинилхлорид тщательно смешивают с кар-
бонатом аммония, гидрокарбонатом натрия и окисью цинка. Затем в
смесь добавляют пластификатор и композицию перемешивают в лопаст-
ном смесителе в течение 1,5—2 ч до образования равномерно окрашенной
гомогенной .массы.
Рис. 36. Форма для «получения эластичного губкоподобиого поливинилхлорида:
1 — матрица; 2— пуансон; 3 — крышка; 4— пластина; 5 — зажимной винт; 6 — проклад-
ка; 7 — штифт; 8 — пружина; 9 — запорный шарик
Приготовленную массу загружают в специальную форму (см.
рис. 36) с двумя отверстиями и зажимают винтом для быстрого выпус-
ка газов, скапливающихся в форме после разложения газообразователя.
Форму заполняют на ’/з—V2 ее объема, герметично закрывают и
помещают в термошкаф, в котором поддерживается температура
165—175° С. Через 2,5 ч горячую форму извлекают из шкафа, запорный
винт немного отвинчивают, выпускают часть газа, затем быстро вновь
завинчивают, герметически закрывая выходные отверстия запорными
шариками. Герметизированную форму охлаждают проточной водой до
20° С (охлаждение обычно продолжается 20—25 мин), затем раскрыва-
ют и извлекают из нее пористую массу, подобную губке.
* ВИСФ — фталаты высших изоспиртов С7—Сю-
105
Эластичный поливинилхлоридный поропласт имеет равномерную
пористую структуру и отличается высокими звуко- и теплоизоляционны-
ми свойствами. Объемная масса материала составляет 0,1-—0,2 г/см3
(см. часть II, стр. 225).
РАБОТА 36. ИЗГОТОВЛЕНИЕ ЖЕСТКОГО ПЕНОПЛАСТА
ИЗ ПОЛИВИНИЛХЛОРИДА
Исходные продукты: поливинилхлорид эмульсионный марки ПВХ-Е54— 100 г; ме-
тилметакрилат — 311 г; динитрил азоизомасляной кислоты — 0,5 г; карбонат аммония —
12 г; гидрокарбонат натрия — 8 г.
Оборудование: фарфоровая ступка; шаровая мельница; сито; пресс-форма с герме-
тизирующим пуансоном; лабораторный пресс; термошкаф или ванна с горячей водой.
В метилметакрилате растворяют динитрил азоизомасляной кисло-
ты. Карбонат аммония и гидрокарбонат натрия каждый отдельно рас-
тирают в фарфоровой ступке и просеивают.
В шаровую мельницу загружают порошкообразный поливинилхло-
рид, просеянные карбонат аммония и гидрокарбонат натрия и затем
метилметакрилат С растворенным в нем порофором. Измельчение и пе-
ремешивание всех 1компонентов в шаровой мельнице производят в тече-
ние 4 ч при температуре не выше 45° С. В случае повышения температу-
ры смесь охлаждают.
После смешения композицию просеивают через сито, затем загру-
жают в специальную пресс-форму, имеющую герметизирующий пуансон,
и подвергают 1прессованию при 160—170° С и удельном давлении
150—180 кг/см2. Выдержка должна составлять 2 мин на 1 мм толщины
заготовки.
Затем запрессованную заготовку охлаждают в пресс-форме, не сни-
мая давления, до 20—25° С, после чего извлекают ее из пресс-фор-
мы. Вспенивание полученной монолитной запрессовки производят,
нагревая ее в термошкафу при 105—110° С или же в горячей воде при
95—98° С.
Получается жесткий пенопласт с равномерной микроячеистой струк-
турой. Определяют удельный объем пенопласта (см. ч. II).
РАБОТА 37. ИЗГОТОВЛЕНИЕ ИСКУССТВЕННОЙ
КОЖИ — ТЕКСТОВИНИТА
Поливинилхлоридные пасты представляют собой дисперсии поли-
мера в жидком пластификаторе — пластизоли. В их состав входят кроме
пластификатора (диоктилфталата, диоктилсебацината, трикрезилфос-
фата и др.) стабилизаторы, пигменты, красители.
Пасты обладают специфическим свойством: их вязкость при нагре-
вании обратимо понижается до определенной температуры, после чего
начинает резко и необратимо увеличиваться. При критической темпера-
туре совмещения полимера с пластификатором масса превращается в
106
прочный гель (рис. 37). Участок АВ — обратимое снижение вязкости
пасты; ВС — необратимое увеличение вязкости —желатинизация; в
точке С — гель.
Нанося пасту на ткань через фильеру или с помощью ракли, полу-
чают искусственную кожу. В термокамере происходит желатинизация
пасты при 180—200° С. Затем уплотне-
5Z
г_
Рис. 37. Зависимость вязкости
пасты от температуры
ние на валках, после чего на тиснильных
валках наносят рисунок, имитирующий
рисунок кожи.
Для изготовления паст пригоден толь-
ко эмульсионный пастообразующий поли-
винилхлорид марок Е74П; Е70П; Е66П
или смесь 60% пастующегося и 40% сус-
пензионного со средним значением моле-
кулярного веса (/() и размерами частиц
1—2 мкм. Способность полимера образо-
вывать пасты, кроме размера частиц,
определяется также молекулярновесовым
распределением и связанной с ним рас-
творимостью в пластификаторе отдельных фракций полимера. Важное
значение имеет сольватирующая способность пластификатора на холо-
ду и влияние на нее добавок, составляющих композицию. Пасты обна-
руживают более или менее ясно выраженную тиксотропию. Хорошая
паста не должна расслаиваться при хранении.
Исходные продукты: поливинилхлорид пасто-
образующий— 100 г; хлористый калий—100 г;
диоктилфталат— 250 г; стеарат бария или каль-
ция—0,3 г; охра — 0,5 г; ткань (бязь, капрон,
трикотаж) — 0,25 м.
Вакуум
Рис. 39. Вакуум-выпарная чаша:
I — крышка; 2 — чаша; 3 — фланец с
герметизирующей прокладкой; 4 — слив-
ной штуцер; 5 — термометр
Рис. 38. Лабораторный гидравли-
ческий пресс с нижним давлением:
/ — основание; 2 — цилиндр; 3 — пор-
шень; 4 — плиты пресса; 5 — емкость
для масла; 6 — иасос
Оборудование: доска мраморная и пестик; пресс лабораторный (рис. 38); две
стальные полированные пластины; йаниа с проточной водой; шаровая мельница; смеси-
тель (пли ступка); лабораторный каландр; фарфоровая чашка; вакуум-чаша (рис. 39);
прибор для определения паропроницаемости; эксикатор.
407
Поливинилхлорид смешивают с хлористым калием в шаровой мель-
нице в течение 1 ч, а затем смесь растирают на мраморной доске с пла-
стификатором. Часть пластификатора (25%) заранее отделяют и на
доске затирают в пасту пигмент и стабилизатор, пасту переносят в сме-
ситель (ступку) и смешивают все компоненты с полимером до образо-
вания однородной массы, которая созревает при комнатной температуре
24 ч. После созревания пасту помещают в фарфоровой чашке в вакуум-
выпарную чашу и вакуумируют в течение 5 ч при 50—60° С (эту опе-
рацию при выполнении лабораторной работы можно не делать).
С помощью ракли слои пасты намазывают на материал, помещают на
подложку между плитами пресса, нагретыми до 180—200° С, и прессуют
5—10 мин. Полученный материал помещают на 5—7 дней в ванну с
проточной водой для вымывания соли, которое приводит к образованию
пор за счет разрыва пленочных оболочек вокруг каждого кристалла
соли вследствие возникающего осмотического давления. Готовый мате-
риал подсушивают и испытывают на паропроницаемость — важнейший
показатель для искусственной кожи.
Определение паропроницаемости. Простейшей методикой определе-
ния паропроницаемости является следующая: в алюминиевый стаканчик
с навинчивающейся крышкой, имеющей отверстие диаметром 2 см, нали-
вают воду, вкладывают в крышку кусок полученной искусственной кожи,
завинчивают ее, взвешивают на аналитических весах и ставят в эксика-
тор с концентрированной серной кислотой. Стаканчик взвешивают через
24 -и 48 ч. Потеря в весе, выраженная в процентах, дает представление о
паропроницаемости.
РАБОТА 38. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ
ИЗ ПОЛИВИНИЛХЛОРИДНЫХ ПАСТ
Исходные продукты: поливинилхлорид пастообразующпй — 40 г; дибутилфта-
лат— 60 г.
Оборудование: смеситель (ступка); краскотерка; фарфоровый стакан; пестик;
вав.уум-выпарная чаша; термошкаф; металлические формы.
Изготовление пасты. Поливинилхлорид и пластификатор тщательно
смешивают в лопастном лабораторном смесителе емкостью 1 л при 30° С
в течение 30 мин. По окончании смешения массу оставляют для набуха-
ния стоять («вызревать») 24 ч при 40° С; а затем при этой же температу-
ре ее перемешивают в смесителе в течение 2 ч. Полученную массу далее
обрабатывают на лабораторной пятивалковой краскотерке, валки кото-
рой охлаждаются водой, а затем вакуумируют для удаления пузырьков
воздуха.
Изготовление изделий методом окунания. Металлическую форму, на-
ружная поверхность которой соответствует профилю изготовляемого из-
делия, погружают в пасту. Затем форму извлекают, дают стечь избыточ-
ному количеству пасты и помещают в термошкаф, нагретый до 160° С,
где выдерживают до образования на поверхности формы прочной плен-
ки. После этого форму вынимают из термошкафа, охлаждают и получен-
ное изделие снимают.
108
Методом окунания из поливинилхлоридных паст изготовляют пер-
чатки, сапоги, калоши, мешки для упаковки и другие подобные изделия.
Изготовление изделий методом отливки в формах. Пасту заливают
в металлическую форму, внутренняя поверхность которой соответствует
профилю изготовляемого изделия, и нагревают в шкафу при 180° С до
образования прочного геля. После охлаждения изделие извлекают из
формы.
РАБОТА 39. ИЗГОТОВЛЕНИЕ ИЗ ПЛАСТИФИЦИРОВАННОГО
ПОЛИВИНИЛХЛОРИДА ФОРМ ДЛЯ ГИПСОВЫХ отливок
Исходные продукты: поливинилхлорид пастообразующий (или смесь пастообра-
эующего и суспензионного в соотношении 60 : 40) — 100 г; диоктилфталат или дибу-
тилфталат — 250 г; гипс.
Оборудование: фарфоровая ступка; термошкаф; баня, наполненная кремиийоргани-
ческой жидкостью; фарфоровый стакан — 2 шт.; фигурка гипсовая; нож; изоляционная
поливинилхлоридная лента.
Поливинилхлорид тщательно смешивают в фарфоровой ступке с
пластификатором и смесь выдерживают 1 ч в термошкафу при 70—80° С
для набухания. После этого ее переносят в фарфоровый стакан и нагре-
вают на масляной бане при 160—170° С, непрерывно перемешивая до
образования прозрачной очень вязкой массы. Для удаления пузырьков
воздуха, образующихся при перемешивании, массу выдерживают при
170° С без перемешивания в течение 30 мин.
Приготовленную массу заливают в стакан, в который предваритель-
но помещена фигурка из фарфора, гипса или другого термостойкого
материала, так, чтобы от стенок стакана до фигурки было расстояние
не менее 5—10 м,м.
После охлаждения эластичную болванку с находящейся в ней фи-
гуркой вынимают из стакана, надрезают острым ножом с одной сторо-
ны боковую»часть болванки так, чтобы из нее можно было вынуть
находящуюся в ней фигурку. Фигурку .вынимают и, сомкнув надрез, за-
крепляют изоляционной поливинилхлоридной лентой полученную фор-
му— негатив. В форме вырезают небольшое отверстие для заливки
гипсовой массы.
В фарфоровом стакане замешивают 60 вес. ч. гипса и 40 вес. ч. во-
ды и после нескольких минут тщательного перемешивания заливают
начинающую .загустевать сметанообразную гипсовую массу в отверстие
приготовленной формы. Через 1-—2 ч полностью затвердевшую гипсо-
вую фигуру вынимают из эластичной формы и, дав .вначале просохнуть
на воздухе в течение суток, окончательно высушивают в термостате при
50—60° С.
РАБОТА 40. ПОЛУЧЕНИЕ СОПОЛИМЕРА ВИНИЛХЛОРИДА
С ВИНИЛАЦЕТАТОМ (ВИНИЛИТА) В СУСПЕНЗИИ
Поливинилхлорид имеет ряд существенных недостатков, основными
из них являются недостаточная растворимость в органических раство-
109
рителях, слабая адгезия к металлу и другим материалам, .а также труд-
ность переработки в изделия литьем под давлением вследствие низкой
текучести расплава. Эти недостатки устраняются сополимеризацией
винилхлорида с другими мономерами.
Техническое значение имеют сополимеры винилхлорида с винилаце-
татом, так называемые винилиты, у которых указанные недостатки
отсутствуют. .Оба мономера легко образуют совместные полимеры.
Винилацетатное звено в макромолекуле сополимера нарушает
симметричность в расположении диполей, а следовательно, снижает
межмолекулярные силы взаимодействия. С этой точки зрения такую
сополимеризацию можно рассматривать как процесс структурной
(внутренней) пластификации.
В начале процесса сополимеризации реакция между мономерными
частицами винилхлорида идет несколько быстрее, чем между винилхло-
ридом и винилацетатом. К концу же реакции состав исходной смеси и
состав полимера становятся примерно одинаковыми. Таким образом,
полученный сополимер содержит макромолекулы с различным соотно-
шением исходных компонентов и потому полидисперсен как по величине
молекул, так и по структуре.
Техническое применение получили сополимеры с преобладающим
содержанием винилхлорида. Однако чем больше в сополимере ацетиль-
ных групп, тем лучше его растворимость и эластичность и тем ниже тем-
пература стеклования.
Варьируя соотношение компонентов и степень полимеризации,
можно получать сополимеры с разнообразными свойствами, что опреде-
ляет их применение.
Сополимеры винилхлорида выгодно отличаются от гомополимера
возможностью переработки их в изделия методом литья под давлением.
Процесс сополимеризации может проводиться как в растворителе, так
и в суспензии. В первом случае полимеризацию ведут в таких раствори-
телях, в которых растворяются оба мономера, но не растворяется полу-
чающийся сополимер. Процесс полимеризации можно проводить также
в ацетоне, в котором растворяются как оба мономера, так и сополимер.
Из ацетонового раствора сополимер осаждают спиртом. В случае поли-
меризации в суспензии процесс ведут в водной среде в присутствии ка-
кого-либо эмульгатора, например желатина.
Сополимер под названием «винилит» представляет собой продукт,
полученный совместной полимеризацией 80 вес. ч. винилхлорида и
20 вес. ч. винилацетата в присутствии инициатора (перекиси бензоила)
и эмульгатора (желатина).
По внешнему виду сополимер представляет собой однородный по-
рошок от белого до желтоватого цвета.
Реакция протекает по схеме
nCH2=CH + тСН2=СН----> Г-СН2—СН—
I I I
С1 ООССНз L CI
'-СН2-СН—
I
ООССНз
т
Исходные продукты: винилхлорид—80 г; винилацетат—20 г; перекись бензоила —
О.и г; желатин — 0,75 г; вода дистиллированная — 25 мл; раствор 1%-ной щелочи.
110
Оборудование: стакан емкостью 0,5 л; фильтр-сетка № 40; автоклав емкостью 1 л
(см. работу 25 и др.); колба плоскодонная емкостью 100 мл; колба трехгорлая емкостью
0.5 л с механической мешалкой; воронка Бюхнера.
Желатин предварительно заливают водой и оставляют набухать на
3—4 ч. Затем его нагревают при 65—70° С до растворения. Раствор
фильтруют через сетку № 40. Охлажденный до комнатной температуры
раствор желатина загружают в автоклав. Затем в винилацетате раство-
ряют перекись бензоила и после полного растворения инициатора моно-
мер при работающей мешалке заливают в автоклав. Впуская хлада-
гент* в рубашку автоклава, его охлаждают до —20° С, после чего в
него вводят имеющий ту же температуру винилхлорид.
По окончании загрузки винилхлорида автоклав герметизируют,
включают мешалку и реакционную массу нагревают до 48° С, пропуская
горячую воду через рубашку аппарата. Обогрев прекращают после того,
как температура внутри автоклава станет повышаться вследствие вы-
деления тепла за счет экзотермической реакции, и реакционную массу
начинают охлаждать, пропуская через рубашку аппарата холодную
воду.
Отвод тепла регулируют так, чтобы давление в автоклаве поддер-
живалось в пределах 10—12 атм, а температура на начальной стадии
полимеризации была 60—70° С и на конечной 75—95° С.
Окончание процесса полимеризации определяют по понижению
давления внутри автоклава. В этот период реакционную массу охлаж-
дают с таким расчетом, чтобы температура оставалась на прежнем
уровне до тех пор, пока давление не снизится до 3,5 атм. При этом
давлении полимеризацию заканчивают и автоклав охлаждают до 30—;
40° С. Открыв кран, спускают оставшееся давление, реакционную массу
выгружают и сополимер обрабатывают щелочью **.
Щелочную обработку производят в отдельном сосуде 1%-ным вод-
ным раствором едкого натра при непрерывном перемешивании и нагре-
вании до 40° С в течение 1—1,5 ч. В конце обработки щелочность реак-
ционной массы доводят до 0,15—0,2%.
После щелочной обработки сополимер отфильтровывают на ворон-
ке Бюхнера, промывают несколько раз теплой водой до нейтральной
реакции промывных вод (по фенолфталеину), сушат при 45—50°С и
просеивают через сито № 38 (900 отв/см2).
Определяют индекс расплава полученного сополимера и перераба-
тывают его литьем под давлением при температуре в цилиндре 160—
170° С, в форме 35—40° С на два стандартных бруска. Полученные об-
разцы испытывают на прочность на удар.
Определяют содержание винилацетата в водной фазе (стр. 57) [5].
* В качестве хладагента можно применять охлаждающий рассол (охлажденный
водный раствор СаС12).
** Щелочная обработка проводится для разложения частично непрореагировав-
шего инициатора, а также перевода бензойной кислоты в растворимую натриевую соль.
Реакции протекают по уравнениям:
2 (С6Н5СО)2О2 + 4NaOH -> 4C6H5COONa 4- 2Н2О 4- Ог
С6НБСООН 4- NaOH C6HsCOONa 4- Н2О
111
На основе сополимера изготовляют пасту, как было описано на
стр. 108 для поливинилхлорида, снизив лишь количество пластифика-
тора на 50%.
РАБОТА 41. ПОЛУЧЕНИЕ СОПОЛИМЕРА ВИНИЛХЛОРИДА
С МЕТИЛМЕТАКРИЛАТОМ
Реакция протекает по схеме
СН3
I
«СН2=СН + тСН2=С -------->
I I
С1 СООСНз
-сн2-сн—
I
С1
СНз
-сн2-с-
СООСНЗ- т
Исходные продукты: винилхлорид — 80 г; метилметакрилат — 20 г; персульфат
калия — 2 г; вода дистиллированная — 100 мл.
Оборудование: стакан емкостью 0,5 л; автоклав из нержавеющей стали емкостью
1 л, рассчитанный на рабочее давление 15 атм (см. работу 25); колба плоскодонная
емкостью ЮО мл; воронка Бюхнера; сито № 20; ступка фарфоровая.
В автоклав загружают персульфат калия и воду, после растворе-
ния соли добавляют метилметакрилат, затем автоклав охлаждают до
—20° С и вводят охлажденный до той же температуры винилхлорид.
После этого автоклав герметизируют, включают мешалку и нагревают
реакционную массу до 50° С, впуская горячую воду в рубашку аппара-
та. После того как'температура внутри автоклава станет повышаться
за счет экзотермической реакции, обогрев прекращают. Отвод тепла
регулируют за счет подачи в рубашку холодной воды так, чтобы давле-
ние в автоклаве поддерживалось в пределах 10—12 атм, а температура
на начальной стадии полимеризации была 60—70° С и на конечной
75—95° С.
Окончание реакции определяют по снижению давления в автоклаве.
Температуру продолжают поддерживать на прежнем уровне до тех пор,
пока давление снизится до 3,5 атм.
После этого автоклав охлаждают, открывают кран и спускают ос-
таточное давление. Реакционную массу выгружают, отфильтровывают
на воронке Бюхнера, промывают несколько раз горячей водой (в вы-
тяжном шкафу) и высушивают при 50—60° С. Определяют состав сопо-
лимера (см. стр. 85).
Полученный сополимер смешивают в фарфоровой ступке с мелами-
ном (2% от веса сополимера) и дибутил фтал атом (5—10% от веса со-
полимера). Количество пластификатора зависит от желаемой степени
эластичности, характеризуемой относительным удлинением при разры-
ве. Смесь тщательно растирают и полученную массу выдерживают в
термошкафу при 60° С в течение 1 ч, после чего помещают между двумя
пластинками из нержавеющей стали, насыпая ровным слоем в преде-
лах ограничительной рамки, и запрессовывают в прессе при 160—165° С
и удельном давлении 20 кг/см2 в течение 2 мин на 1 мм толщины листа.
Запрессованный лист охлаждают под давлением до 60—80° С, затем
распрессовывают и вынимают изделие — пластину.
112
Из пластины вырубают стандартные образцы — лопатки и опреде-
ляют сопротивление разрыву (предел прочности при растяжении) и от-
носительное удлинение; определяют температуру разложения исходного
сополимера и стабилизированного и пластифицированного (см. ч. II).
РАБОТА 42. ПОЛУЧЕНИЕ СОПОЛИМЕРА ВИНИЛХЛОРИДА
С МЕТИЛАКРИЛАТОМ (МА-20)
При получении сополимера винилхлорид и метилакрилат берут в
соотношении 80:20. Сополимер обладает большей термостойкостью по
сравнению с поливинилхлоридом и «винилитом»: температура разложе-
ния сополимера 160° С, винилхлорида 135° С, «винилита» 145° С,
Сополимер винилхлорида с метилакрилатом применяется для изго-
товления гибкого текстолита, прокладочных жгутов и т. п.
Реакция протекает по схеме
пСН2=СН + тСН2=СН
I I
С1 (СООСНз
СН2—CH—1—Г-СНг-СН-
С1 J- [ СООСНз
Исходные продукты: винилхлорид—80 г; метилакрилат (свежеперегнанный) —20 г;
персульфат аммония — 2 г; олеат калия — 2 г (или сульфанол); ткань (бязь) — 100 г.
Оборудование: автоклав емкостью 1 л (см. работу 25); колба плоскодонная на
100 мл; установка для пропитки и сушки ткани (рис. 40); пресс гидравлический лабо-
раторный; пластинки из нержавеющей стали; целлофан.
Рис. 40. Схема горизонтальной пропиточио-сушильиой машины:
/ — рулон материала; 2, 3, 4, 5, 9, 10 — направляющие валики; 6, 1 — отжимные ва-
лики; 8 — пропиточная ваниа; 11 — сушильная камера; 12 — ведущие валики; 13 — под-
держивающий валик; 14, 15, 16 — валики намоточного устройства; 17, 18 — вентилято-
ры; 19 — паровые змеевики
В автоклаве растворяют персульфат аммония в 80 мл воды. В от-
дельном сосуде приготовляют раствор олеата калия в 20 мл воды и
вливают в автоклав, туда же добавляют метилакрилат. При работаю-
щей мешалке автоклав охлаждают, впуская хладагент в рубашку до
—20° С и вливают в него имеющий ту же температуру винилхлорид.
По окончании загрузки винилхлорида автоклав герметизируют,
включают мешалку, нагревают до 50° С, пропуская горячую воду через
рубашку аппарата. После того как температура внутри автоклава ста-
113
нет повышаться вследствие экзотермичности реакции, обогрев прекра- -
щают и реакционную массу начинают охлаждать. Отвод тепла регули-
руют так, чтобы давление в автоклаве поддерживалось в пределах
10—12 атм; а температура на начальной стадии была 50—55° С, а на ко-
нечной 75—85° С.
Окончание реакции определяют по падению давления внутри авто-
клава. С этого момента реакционную массу охлаждают с таким расче-
том, чтобы температура оставалась на прежнем уровне до тех пор, пока
давление не снизится до 3,5 атм. При этом давлении процесс заканчива-
ют и автоклав охлаждают до 30—40° С. Открыв кран, спускают остаточ-
ное давление и полученную эмульсию выгружают в ванну лабораторной
пропиточной установки (см. рис. 40). Взвешенную и нарезанную на
ленты ткань пропитывают эмульсией и высушивают при 80—90° С.
Затем ткань взвешивают, если окажется, что привес меньше 50%, про-
питывают и просушивают еще раз. Пропитанные ленты нарезают на
куски, соответствующие размеру пластины; собирают пакет, закладыва-
ют между плитами пресса, завернув в целлофан, и прессуют при 160—
170° С под давлением 200—250 кг/см2, выдержка в прессе 2—3 мин на
1 мм толщины пластины. Ткань в данном Случае является наполните-
лем, упрочняющим полимерную основу. Материалы на тканевой основе
называют текстолитами.
Из полученных образцов вырезают стандартные лопатки и бруски
для испытания на предел прочности при разрыве, ударе и относительное
удлинение, которые в значительной степени зависят от сорта ткани (см.
ч. II). Определяют состав сополимера (см. стр. 85).
РАБОТА 43. ПОЛУЧЕНИЕ СОПОЛИМЕРА ВИНИЛХЛОРИДА
С ВИНИЛИДЕНХЛОРИДОМ СУСПЕНЗИОННЫМ МЕТОДОМ
Реакция протекает по схеме
С1
nCH2=CH + тСН2=С -> Г—СН2—СН—'
С1
С1
С1
-СН2-СС12-
п
Исходные продукты: 'винилхлорид (свежеперегнанный) — 75 г; винилиденхлорид
(свежеперегнаиный)—25 г; перекись бензоила — 0,3 г; желатин — 0,75 г; вода дистил-
лированная— 125 г; азот (баллон).
Оборудование: автоклав из нержавеющей стали емкостью 1 л (см. работу 26);
колба емкостью 200 мл; термометр; пластинка из белой жести; сетка № 40.
В отдельном сосуде при нагревании до 65° С растворяют в воде же-
латин. Раствор фильтруют через сетку № 40 и загружают в автоклав.
Перекись бензоила растворяют в винилиденхлориде и после полного
растворения инициатора в мономере при работающей .мешалке вносят
в автоклав. Впуская хладагент .в рубашку автоклава, его охлаждают до
20° С, затем вакуумируют и вводят имеющий ту же температуру ви-
нилхлорид. После этого автоклав продувают азотом, герметизируют,
114
включают мешалку и, впуская горячую воду в рубашку аппарата, на*
гревают реакционную массу до 48° С.
Обогрев прекращают, как только температура внутри автоклава
станет повышаться вследствие выделения тепла за счет экзотермиче-
ской реакции, после чего реакционную массу начинают охлаждать,
пуская хладагент в рубашку аппарата. Отвод тепла регулируют так,
чтобы температура внутри автоклава поддерживалась в пределах 48—
50° С, а давление не превышало 10 атм.
Окончание процесса полимеризации определяют по начинающему-
ся понижению давления в автоклаве. В это время охлаждение прекра-
щают и, когда давление достигнет 3,5—3 атм, процесс заканчивают. Ав-
токлав охлаждают до 30—40° С и, открыв кран, спускают остаточное
давление.
Полученный сополимер обрабатывают щелочью. Обработку прово-
дят в отдельном сосуде 1 %-ным водным раствором едкого натра при не-
прерывном перемешивании и нагревании до 40° С в течение 1,5 ч. После
этого сополимер отфильтровывают на воронке Бюхнера, промывают не-
сколько раз теплой водой до нейтральной реакции промывных вод (про-
ба с фенолфталеином) и высушивают при 45—50° С.
Сополимер растворяется в различных органических растворителях
(бензоле, ацетоне, бутилацетате и др.).
Готовят 25%-ный раствор сополимера в бутилацетате и наносят ки-
сточкой на предварительно обезжиренную растворителями поверхность
пластинки из белой жести. Подсушивают покрытие на воздухе и наносят
еще слой, после чего высушивают сначала на воздухе, а затем при
50-—60° С в термошкафу. Испытывают эластичность покрытия на шкале -
НИИЛКа.
РАБОТА 44. ПОЛУЧЕНИЕ ПОЛИВИНИЛИДЕНХЛОРИДА
ПОЛИМЕРИЗАЦИЕЙ СУСПЕНЗИОННЫМ МЕТОДОМ
Исходные продукты: винилиденхлорид свежеперегнанный — 100 г; вода дистиллн-
рованиная — 120 г; желатин — 0,57 г; перекись бензоила — 0,3 г.
Оборудование: химические стаканы емкостью 100 мл — 2—3 шт.; сетка № 40;
автоклав емкостью d л (см. работу 26); воронка Бюхнера; термошкаф.
Перекись бензоила растворяют в винилиденхлориде и загружают в
автоклав при перемешивании. После этого автоклав продувают азотом,
герметизируют, включают мешалку и нагревают до .50° С.
Начавшийся процесс полимеризации определяют по повышению
давления и температуры в автоклаве. В это время нагрев прекращают и,
пропуская холодную воду через рубашку аппарата, поддерживают тем-
пературу в течение всего времени реакции около 50° С. По окончании
реакции полимеризации, что определяют по падению давления внутри
автоклава, реакционную массу охлаждают до комнатной температуры,
спускают оставшееся давление й открывают автоклав.
Полученный полимер отфильтровывают на воронке Бюхнера, затем
415
несколько раз промывают теплой водой и сушат при 40—45° С. Готовый
продукт представляет собой порошок белого или желтого цвета.
Определяют индекс расплава и термостойкость по ВИКА (см. ч. И).
РАБОТА 45. ПОЛУЧЕНИЕ СОПОЛИМЕРА ВИНИЛИДЕНХЛОРИДА
С АКРИЛОНИТРИЛОМ И ПЛЕНКИ НА ЕГО ОСНОВЕ
Сополимеризация винилиденхлорида'с акрилонитрилом (5—15%)
позволяет получать сополимер с лучшей растворимостью и совмести-
мостью с пластификаторами (10—30%) и другими полимерами, нежели
чистый поливинилиденхлорид. Сополимеры растворимы в метилэтилке-
тоне, циклогексаноне и других кетонах. Сополимеры используют для по-
крытий на металле и бумаге, а также для получения пленок экструзией
с последующей пневматической вытяжкой.
Исходные продукты: винилиденхлорид (свежеперегиаииый)—76 г; акрилони-
трил— 25 г; персульфат калия — 2 г; дистиллированная вода — 200 мл.
Оборудование: автоклав емкостью 1 л (см. работу 25); воронка Бюхнера; термо-
шкаф; химические стаканы на 100 мл — 2 шт. /
Персульфат калия и дистиллированную воду загружают в автоклав
и растворяют соль при перемешивании. Затем заливают смесь мономе-
ров, автоклав продувают инертным газом (азот, аргон), герметизируют,
включают мешалку и нагревают до 60° С.
Начало процесса полимеризации определяют по повышению давле-
ния и температуры внутри автоклава. В это время нагрев пре-
кращают и, впуская холодную воду, поддерживают температуру внутри
автоклава около 50—60° С. Окончание реакции определяют по падению
давления внутри автоклава. Реакционную массу охлаждают до ком-
натной температуры, спускают остаточное давление и открывают
автоклав.
Полученный сополимер отфильтровывают на воронке Бюхнера, про-
мывают несколько раз теплой водой и высушивают при 40—45° С.
Определяют выход сополимера в процентах к общему количеству
смеси мономеров. Определяют температуру разложения сополимера и
сравнивают ее с температурой разложения поливинилхлорида.
Получают пленку на основе лака сополимера по следующей ме-
тодике.
10 г сополимера и 3 г диоктилфталата растворяют в 50 мл циклогек-
санона до образования однородного лака.
Металлическую пластинку (10x4 см) промывают растворителем,
высушивают и погружают в стакан с лаком так, чтобы полоска пластин-
ки шириной 3 см осталась непокрытой лаком. Пластину вынимают, дают
стечь избытку лака и ставят ее под углом 45°, прислонив к стойке шта-
тива. Пленке дают подсохнуть. После подсушивания пластинку окунают
вторично и снова подсушивают до исчезновения отлипа.
Покрытие испытывают на твердость по маятнику и эластичность
[8]. Сравнивают свойства сополимера и поливинилхлорида.
116
Б. ФТОРПРОИЗВОДНЫЕ ПОЛИУГЛЕВОДОРОДЫ
И ПЛАСТМАССЫ НА ИХ ОСНОВЕ
Исходным сырьем для получения фторпроизводных полиуглеводоро-
дов являются тетрафторэтилен, трифторхлорэтилен, винилфторид, вини-
лиденфторид и др. Эти полимеры обладают комплексом ценных свойств,
в частности повышенной термостойкостью (до 260°С), прочностью, хи-
мической стойкостью (даже к окислителям). Специфические свойства
фторопластов определяются в основном высокой степенью кристаллич-
ности, а также высокой прочностью связи С—F. Эти полимеры принадле-
жат к числу медленно кристаллизующихся, поэтому степень их
кристалличности можно изменять путем термообработки в различных
условиях.
Все перечисленные выше мономеры обладают высокой активностью
в радикальном процессе. Полимеризация протекает с большим экзотер-
мическим эффектом, поэтому ведут ее в водных средах: в эмульсии или
суспензии. Все названные фторсодержащие мономеры газообразны, в
связи с чем полимеризацию проводят под высоким давлением.
Применение фторсодержащих полимеров очень разнообразно: элект-
ротехнические изделия, антифрикционные, химически устойчивые и др.
Особенности переработки фторсодержащих полимеров определяют-
ся количеством атомов фтора в молекуле мономера. Так, например, по-
литетрафторэтилен (фторопласт-4, или фторлон-4) не плавится и отфор-
мовать его можно лишь на стадии вынужденной эластичности спеканием
при высокой температуре (360—380° С) предварительно отпрессованных
таблеток. Полимер трифторхлорэтилена уже можно перерабатывать
обычными методами, однако при очень жестких режимах. Полимеры ви-
нийфторида перерабатываются более легко. Поливинилфторид, подобно
поливинилхлориду, перерабатывается лишь в присутствии стабилизато-
ров. Поливинилиденфторид более Термостоек, чем поливинилфторид (при
260° С 12 ч).
Таким образом, как технология получения фторсодержащих полиме-
ров, так и их переработка в некоторых случаях достаточно сложны,
однако ценный комплекс свойств определяет необходимость их произ-
водства.
Химизм образования фторсодержащих
полимеров
Фторсодержащие мономеры полимеризуются при нагревании под
давлением в присутствии инициаторов, а в некоторых случаях без них
(тетрафторэтилен). Полимеризацию можно осуществить в блоке, в рас-
творителе и в водной среде (эмульсии и суспензии). При полимеризации
тетрафторэтилена выделяется 196 кДж/моль (47 ккал/моль) и в случае
полимеризации его в блоке возможен взрыв, связанный с местными пе-
регревами. В растворе, как обычно, получаются более низкомолекуляр-
ные полимеры. Проводя полимеризацию в присутствии СНС1з или ССЦ,
за счет передачи цепи получают олигомеры, применяющиеся в промыш-
117
ленности как пластификаторы и смазки:
R' + nCF2=CFCl -> R (CF2--CFC1)„
r (-cf2-cfci); + chci3 -+ r (cf2-cfci)„h + cci;
cci3 + nCF2=CFci -> cci3 (cf2-cfcd;
Высокомолекулярные полимеры образуются в эмульсии и суспен-
зии. Для снижения энергии активации полимеризации широко пользу-
ются окислительно-восстановительными системами, причем увеличивает-
ся скорость и снижается оптимальная температура полимеризации. Для
поддержания определенного значения pH среды (2,5—3) вводят буфер-
ные вещества.
Технология производства фторопластов
Полимеризация фторсодержащих мономеров проводится под давле-
нием в автоклавах из высокопрочных сталей, выдерживающих давление'
не менее 100 атм, с якорной мешалкой, снабженных рубашкой для на-
грева и охлаждения. Полимеризация тетрафторэтилена осуществляется
в водной среде (вода должна быть освобождена от кислорода) в присут-
ствии персульфата калия (0,2% от водной фазы). Перед вводом моно-
мера автоклав охлаждают и вакуумируют. После ввода мономера (30%
от водной фазы) поднимают температуру до 70—80° С, а давление до
40—100 атм. Конец реакции определяют по падению давления. За 1 час
образуется 86% полимера (от массы мономера). Автоклав охлаждают,
реакционную массу центрифугируют, полимер фильтруют, промывают
горячей водой и высушивают.
Политетрафторэтилен — фторопласт-4 — рыхлый порошок (ГОСТ
10007—72) кристаллической структуры с температурой плавления кри-
сталлитов 327° С. Выпускается трех сортов, отличающихся по физико-
механическим и диэлектрическим свойствам. Фторопласт-4 применяют в
авиационной, химической, радиотехнической, пищевой промышленности,
в виде деталей для химической аппаратуры, пленок, труб, волокна, поро-
пластов, антифрикционных изделий, антикоррозионных покрытий, на ко-
торые не действует даже царская водка и фтористоводородная кислота.
Разновидностью фторопласта-4 является фторопласт-4Д, отличаю-
щийся размером частиц, их формой и молекулярным весом. Фторо-
пласт-4Д перерабатывают обычными методами.
Политрифторхлорэтилен — фторопласт-3 (МРТУ 6-05-946—65) вы-
пускается трех марок: I — для получения смазок; II — для суспензий,
экструзии, прессования; III — для прессования изделий различного на-
значения.
Высокомолекулярный политрифторхлорэтилен обладает кристалли-
ческой структурой и содержит некоторое количество аморфной фазы.
Температура плавления кристаллитов 208—210° С. Степень кристаллич-
ности зависит от скорости охлаждения нагретого полимера: при медлен-
ном охлаждении степень кристалличности достигает 85—90%; пл. 2,15—
2,16 г/см3. При быстром охлаждении от 230° С до комнатной температу-
ры шэлитрифторхлорэтилен получается со степенью кристалличности
118
Политрифторхлорэтилен стоек к действию обычных органических
растворителей, но растворяется в некоторых фтор- и фторхлорорганиче-
ских соединениях. Пластифицируется низкомолекулярными полимерами
трифторхлорэтилена.
Исключительно устойчив к разбавленным и концентрированным ми-
неральным и органическим кислотам, в том числе к дымящей азотной,
фтористоводородной и другим кислотам, к царской водке, а также к
щелочам.
Разновидностью фторопласта-3 являются марки фторопласт-30 и
30 Л, растворимые в кетонах и сложных эфирах, дающие высококачест-
венные покрытия.
Политрифторхлорэтилен обладает высокой термической стойкостью,
выдерживая нагревание до 270—300° С. Выше этой температуры (напри-
мер, пиролиз при 400—650° С) полимер разлагается с образованием
низкомолекулярных веществ, в том числе значительного количества мо-
номера.
Политрифторхлорэтилен отличается высокими диэлектрическими'
свойствами, однако диэлектрическая постоянная и тангенс угла диэлек-
трических потерь до некоторой степени зависят от частоты поля и тем-
пературы вследствие несимметричности основного звена в цепи молекул
и присутствия атома хлора.
Благодаря сравнительно хорошей текучести политрифторхлорэтилен
перерабатывается в изделия обычными для термопластичных материа-
лов методами, в том числе экструзией, литьем под давлением и др.
Большое значение имеют неводные суспензии, изготовленные из по-
литрифторхлорэтилена, которые применяются в производстве пленок,
для нанесения полимера на металлические поверхности с целью защиты
их от коррозии и т. п. Низкомолекулярные полимеры трифторхлорэтиле-
на получают в среде хлороформа в присутствии перекиси бензоила при
150° С в течение 15 мин. Реакцию проводят в автоклаве. Образуются
масла, обладающие рядом ценных свойств.
Поливинилфторид и поливинилиденфторид получают в эмульсии,
суспензии и в растворе. Примеси углеводородов действуют ингибирующе
на полимеризацию этих мономеров, поэтому необходима их тщательная
очистка. Полимеризацию в растворе проводят в ацетоне, этиловом и "изо-
пропиловом спирте.
Поливинилфторид — фторопласт-1 — получают в виде порошка. Его
используют главным образом для получения эластичной пленки, отли-
чающейся хорошей погодоустойчивостью, химической стойкостью и вы-
сокой прочностью. Находят также применение сополимеры винилфтори-
да с другими мономерами.
Поливинилиденфторид выпускается в виде гранул и порошков, рас-
творимых в амидных растворителях, под названием фторопласт-2 и 2М.
Поливинилфторид и поливинилиденфторид перерабатывают экстру-
зией в присутствии высококипящих растворителей в стержни, пленки, ли-
сты; литьем под давлением — в фасонные детали; из всех фторопластов
можно изготавливать волокно; изделия можно сваривать прутком, под-
вергать стерилизации.
Общее число марок фторопластов превышает 25 наименований.
- 119
Исходное сырье для получения
фторопластов
Тетрафторэтилен CF2=CF2— бесцветный газ без запаха, нетоксич-
ный; т. кип.— 76,3°С; т. пл. 142,5°С; пл. 1,519 г/см3 (при—76,3°С).
Нерастворим в воде, растворим в органических растворителях. Легко
вступает в реакцию присоединения с хлором и бромом, а также в реак-
ции замещения галогенов и полимеризации.
В технике тетрафторэтилен получают из дифторхлорметана (фрео-
на-22) :
600-700° С
CHF2C1 —->2CF2:->CF2=CF2
•—rlCl
Трифторхлорэтилен CF2—CFC1 в нормальных условиях представля-
ет собой газ, конденсирующийся в жидкость при —26,8° С. Мол. вес
116,48; т. пл. — 157,2° С; плотность при /КИп 1,38° С.
При хранении мономер в присутствии кислорода воздуха способен
полимеризоваться при комнатной температуре.
Трифторхлорэтилен может быть получен из 1,2,2-трихлор-1,1,2-три-
фторэтана действием на него цинковой пыли в спиртовой среде
[Zn]
C1F2C-CC12F F2C=CFC1 + ZnCl2
[CaHgOri]
Применяется трифторхлорэтилен для бомополимеризации и сополи-
меризации с другими веществами. Он сополимеризуется с винил- и ви-
нилиденфторидом, винилхлоридом, винилацетатом, стиролом, метилме-
такрилатом и некоторыми другими мономерами. С винилиденхлоридом и
акрилонитрилом трифторхлорэтилен не сополимеризуется. Качественные
реакции и количественные определения хлора и фтора см. ниже.
Винилфторид CH2=CHF — бесцветный газ со специфическим запа-
хом. Т. кип. — 72,2° С; т. пл. 160,5° С; нерастворим в воде. Плотность
при —72° С 0,853 г/см3.
Промышленным методом получения винилфторида является гидро-
фторирование ацетилена в присутствии катализатора HgCl2, нанесенно-
го на уголь:
[HgCI2]
CHsCH + HF------->CH2=CHF
Самопроизвольно винилфторид не полимеризуется, он менее акти-
вен, чем винилхлорид. Полимер его плавится при 198° С и разлагается
при 300° С, растворяется при температуре выше 110° С в амидных рас-
творителях, тетраметиленсульфоне и др.
Винилиденфторид CH2=CF2—бесцветный газ с т. кип. —84° С;
т. пл. — 117° С; плотность при —26° С 1,03 г/см3. Растворим в спирте и
хлороформе, нерастворим в воде. Устойчив даже на солнечном свету.
Промышленным методом получения винилиденфторида является реак-
ция дифтордихлорэтана с цинковой пылью и Nal в растворе ацетамида
или 2-этилгексанола при 145° С:
[Zn]
CH2CI-CC1F2----> CH2=CF2 + ZnCl2
120
или пиролиз дифторхлорэтана или трифторэтана по следующей схеме:
870° с
СН3—CC1F2---> CH2=CF2 + НС1
820° с
СН3- CF3--->CH2=CF2 +HF
Винилиденфторид менее реакционноспособен, чем хлористый вини-
лиден. Для его полимеризации необходимы кроме инициаторов (переки-
си бензоила, перекиси ацетила, персульфатов) высокая температура и
давление (30—100 атм). Чаще всего полимеризацию проводят в раство-
ре. Практическое значение имеют сополимеры вннилиденфторида с эти-
леном, фтористым винилом и особенно с тетрафторэтиленом и трифтор-
хлорэтиленом.
Поливинилиденфторид и его сополимеры — аморфные, бесцветные
полимеры, устойчивые к действию минеральны?; кислот и щелочей; рас-
творяются в ароматических углеводородах, метаноле, хлороформе и че-
тыреххлористом углероде, диметилацетамиде и др.
Качественное определение фтора
в полимере
В пробирку вносят кусочек металлического натрия величиной с
горошину и прибавляют немного испытуемого вещества. Пробирку укреп-
ляют на штативе и нагревают ее нижнюю часть на горелке до тех пор,
пока не расплавится натрий. Пробирку охлаждают, добавляют еще не-
много вещества и снова нагревают дно пробирки до красного накала.
Пробирке дают остыть и приливают 1 мл этилового спирта для раство-
рения избытка металлического натрия. Затем ее вновь нагревают, спирт
испаряется, горячую пробирку опускают в стакан с 20 мл дистиллиро-
ванной воды и разбивают стеклянной палочкой. Содержимое пробирки
растворяется в воде, раствор нагревают до кипения и фильтруют. Около
4 мл фильтрата подкисляют уксусной кислотой, кипятят, охлаждают и
•прибавляют 2—5 капель насыщенного раствора хлористого кальция.
В присутствии фтора через несколько часов образуется студнеобразный
осадок фторида кальция.
Одну каплю раствора переносят на полоску реактивной циркониево-
ализариновой бумаги. В присутствии фторсодержащего соединения на
красной бумаге образуется желтое пятно.
Количественный анализ
фторсодержащих полимеров
Принцип метода основан на сжигании органического вещества в
колбе, наполненной кислородом, в присутствии платинового катализато-
ра. После поглощения продуктов сгорания водой образующуюся фтори-
стоводородную кислоту титруют азотнокислым торием р присутствии
натриевой соли ализаринсульфокислоты (ализариновый красный S) в
качестве индикатора.
121
Рис. 41. Колба для сжигания
по Шенигеру:
а — колба; б — складывание «пакети-
ка» ; в — ампулы
хивают в течение 5—10 мин
Содержание фтора определяют по специальному градуировочному}
графику, построенному после сжигания различных навесок заведомо из-
вестного фторорганического вещества.
В основе аналитического определения лежат реакции
Os
[—CF2—CF2—]„ — HF + лСО2 + mH/)
„ . HF + NaOH=NaF + Н2О
4NaF + Th (NO3)4=ThF4 + 4NaNO3
Навеску вещества 8—10 мг берут по разности из бюкса для взвеши-
вания, высыпают в середину фильтра (рис. 41, б) и аккуратно заверты-
вают в пакетик. Пакетик зажимают в платиновую сетку, причем узкий
длинный конец фильтровальной бумаги
должен быть обращен вверх. Платино-
вую сетку внизу загибают, благодаря
чему образец надежно удерживается и
обеспечивается полное сжигание.
В колбу (рис. 41, а) наливают 10 мл
дистиллированной воды, 1 мл 2 н. рас-
твора КОН, 3 капли пергидроля и напол-
няют кислородом. Выступающий конец
«пакетика» поджигают и быстро закры-
вают колбу пробкой. Сжигание продол-
жается 15—20 с, причем пробку необхо-
димо укрепить, чтобы ее не вытолкнуло
газами. (Сжигать в защитной маске из
оргстекла!). Содержимое колбы встря-
о полного исчезновения тумана в колбе,
после чего ее открывают, пробку и платиновую сетку тщательно про-
мывают дистиллированной водой над колбой. Раствор количественно
переносят в мерную колбу емкостью 100 мл и доводят дистиллирован-
ной водой до метки.
Для построения градуировочного графика проводят сжигание серии
навесок фторорганического вещества с таким расчетом, чтобы содержа-
ние фтора в каждой навеске последовательно увеличивалось от 0,3 до
1,2 мг. Для построения графика необходимо не менее 6—8 точек.
Аликвотную часть раствора отбирают в коническую кварцевую кол-
бу емкостью 100 мл и доводят объем водой до 50 мл (по предварительно
сделанной метке), добавляют 0,3 мл раствора ализаринового красного S
(0,05 %-ный раствор) и нейтрализуют 0,05 н. раствором NaOH до появ-
ления розового окрашивания. Затем добавляют 1—2 капли 0,05 н. рас-
твора НС1 до перехода окраски раствора в желтую. К нейтральному
раствору добавляют 0,5 мл буферного раствора и титруют 0,05 н. раство-
ром азотнокислого тория до розового окрашивания.
По полученным данным строят градуировочный график, откладывая
на оси абсцисс количество фтора, содержащееся в титруемой навеске
(мг), а по оси ординат — количество 0,05 н. раствора азотнокислого
тория, затраченное на титрование (мл).
122
Для каждого вновь приготовленного 0,05 н. раствора азотнокислого
тория градуировочный график строится заново.
Навеску вещества берут с таким расчетом, чтобы в титруемом рас-
творе содержалось 0,5—0,7 мг фтора. Сжигание и титрование проводят
в тех же условиях, что и при построении градуировочного графика.
Содержание фтора х (%) вычисляют по формуле х=~ 100, где а —
Ьс
количество фтора в титруемой пробе, найденное по градуировочному
графику; Ъ — количество раствора, взятое для титрования, мл; с — на-
веска анализируемого вещества, мг.
Определению мешают только фосфор и бор.
Анализ мономеров и полимеров, содержащих одновременно хлор и
фтор, производят по приведенной выше методике (см. стр. 84).
,Техника безопасности при работе
с фторсодержащими мономерами и полимерами
Фторсодержащие мономеры — очень токсичные вещества, поэтому
все работы с ними обязательно проводятся в вытяжном шкафу с эффек-
тивной вентиляцией. Токсичность тетрафторэтилена и трифторхлорэти-
лена объясняется их способностью при смешивании с воздухом окислять-
ся с образованием нестойких перекисных соединений, легко разлагаю-
щихся и образующих фторфосген, который обладает сильным
отравляющим действием, сходным с действием фосгена. Окисление мо-
номера протекает тем быстрее, чем выше температура помещения. Реаги-
руя с влагой воздуха, фторфосген гидролизуется с образованием двуоки-
си углерода и фтористого водорода; последний также весьма ядовит.
Отравляющее действие фторфосгена состоит в его способности легко
гидролизоваться при попадании на слизистые оболочки дыхательных
путей и легких, в результате чего выделяется фтористый водород, вызы-
вающий сильный химический ожог легких. При этом безразлично, про-
изошел ли гидролиз фторфосгена в воздухе или непосредственно на сли-
зистой оболочке ткани, — действие его на организм одинаково опасно.
Полимеры трифторхлорэтилена при нагревании до температуры вы-
ше 200° С начинают, хотя и очень медленно, разлагаться. Разложение их
сопровождается выделением газообразных ненасыщенных соединений,
многие из которых очень ядовиты. С повышением температуры до 270° С
разложение идет значительно быстрее. При этом выделяется до 0,04%
(от веса полимера) газообразных продуктов в час. При пиролизе же
фторполимеров при более высоких температурах в присутствии воздуха,
так же как и при окислении мономера, образуются ядовитые фторфос-
гены. Политетрафторэтилен разлагается при 500—550° С с выделением
фторфосгена.
При испытании отравляющего действия продуктов пиролиза поли-
трифторхлорэтилена (образующихся при 400° С) на животных последние
погибали при явлении нарастающего отека легких. Опасна концентрация
продуктов разложения полимера, равная 0,001 %.
Таким образом, как мономеры фторхлорпроизводных этилена, так
и продукты разложения полимеров (фторопластов), выделяющиеся при
123
термической их переработке или при других случаях нагревания, сильно •
токсичны и весьма опасны для здоровья. Опасность этих продуктов за-
ключается в том, что они не имеют запаха и бесцветны, поэтому отрав-
ление ими происходит незаметно.
Сильная степень отравления людей проявляется в раздражении
верхних дыхательных путей, а также в заболевании, известном под на-
званием «фтористой лихорадки», которое сопровождается высокой тем-
пературой и некоторыми другими симптомами, сходными с симптомами
гриппа. Такие заболевания часто неправильно диагностируются врачами,
поэтому вовремя не выявляются.
Большую опасность представляют собой пожары в помещениях, где
находятся фторполимеры или изделия из них. Подвергаясь во время
пожара действию высоких температур, в результате термического раз-
ложения они выделяют особо вредные газообразные вещества, в том
числе окись углерода.
Очень опасна содержащаяся в воздухе помещений пыль фторполи-
меров. Такая пыль при горящих газовых горелках, а также при курении
попадает на открытый огонь, разлагается, и продукты пиролиза, выде-
ляясь в воздух, отравляют его или попадают непосредственно в легкие
курящих людей.
Сами по себе полимеры как фтор-, так и фторхлорпроизводных эти-
лена (фторопласты) вследствие химической инертности совершенно
безвредны. Поэтому хранение их или использование изделий из них при
невысоких температурах никакой опасности для здоровья людей не пред-
ставляют.
РАБОТА 46. ПОЛУЧЕНИЕ ПОЛИМЕРА ФТОРОПЛАСТА-4 ПОЛИМЕРИЗАЦИЕЙ
ТЕТРАФТОРЭТИЛЕНА СУСПЕНЗИОННЫМ МЕТОДОМ
Исходные продукты: тетрафторэтилен — 30 г; вода, предварительно освобожден-
ная от растворенного кислорода,— 100 мл; персульфат калня — 0,2 г; бура—0,5 г;
инертный газ (баллон).
Оборудование: антоклав емкостью 1 л, рассчитанный на данление 100 атм, снаб-
женный якорной мешалкой и рубашкой; воронка Бюхнера; фарфоровая чашка; термо-
шкаф; муфельная печь; пресс гидравлический; пресс-форма на таблетку.
В автоклав, предварительно продутый инертным газом (аргоном,
азотом, освобожденным от кислорода), заливают воду и растворяют в
ней персульфат калия и буру. Затем впускают в рубашку охлаждающий
агент (рассол, жидкий азот, воздух), вакуумируют автоклав, вводят мо-
номер, герметизируют автоклав и поднимают температуру до 70—80° С.
Давление, поднявшееся в начале реакции, затем падает, что свидетель-
ствует об окончании реакции.
Автоклав охлаждают, газообразный мономер продувают инертным
газом, а реакционную массу фильтруют на воронке Бюхнера, промывают
несколько раз водой и высушивают в термошкафу при 80—90° С. Опре-
деляют выход по отношению к загруженному мономеру.
Из полученного продукта отпрессовывают таблетку при удельном
давлении 300—350 кг/см2, продолжительность выдержки в прессе опре-
124
деляется толщиной таблетки. Давление поднимают медленно, в течение
5—10 мин.
Полученные таблетки запекают в муфельной печи с терморегулято-
ром при 370° С в течение 2—10 ч в зависимости от размеров заготовки
(1 ч на 3 мм толщины).
Охлаждение изделия должно быть медленное (лучше всего вместе
с печью). При быстром охлаждении происходит закалка, степень кри-
сталличности в этом случае не выше 50%. При медленном охлаждении
степень кристалличности выше 60%. Определяют химическую устойчи-
вость изделия (см. ч. II, стр. 226).
РАБОТА 47. ПОЛУЧЕНИЕ ФТОРОПЛАСТА-3 ПОЛИМЕРИЗАЦИЕЙ
ТРИФТОРХЛОРЭТИЛЕНА В РАСТВОРЕ
Исходные продукты: трифторхлорэтилен (снежеперегнанный)—30 г; хлороформ
(счищенный от спирта)—270 г; перекись бензоила—1-,5 г; пятиокись фосфора — 5 г.
Оборудование: стальной автоклав емкостью Г л; 2 колбы Вюрца с водяным холо-
дильником; колба-приемник, охлаждаемая сухим льдом; баня с полиэтилсилоксановой
жидкостью.
Применяемый для работы хлороформ предварительно очищают от
спирта и перегоняют над пятиокисью фосфора. Трифторхлорэтилен пе-
регоняют непосредственно перед полимеризацией и растворяют в 220 г
хлороформа при охлаждении сухим льдом до —27° С. Перекись бензоила
растворяют в 50 г хлороформа и вводят в смесь в виде раствора.
В небольшой стальной автоклав загружают смесь исходных продук-
тов. Затем автоклав герметизируют и нагревают на бане при 100° С
1 ч 45 мин. После этого его охлаждают до комнатной температуры, спу-
скают оставшееся давление и полученный продукт переносят в колбу
Вюрца для отгонки растворителя.
После отгонки растворителя остается полимер в виде порошка бе-
лого цвета. Продукт используют в качестве пластификатора для высо-
комолекулярных фторопластов и определяют разрушающее напряжение
при ударе и растяжении пластифицированного образца и сравнивают эти
показатели с показателями образца без пластификатора.
РАБОТА 48. ПОЛУЧЕНИЕ ФТОРОПЛАСТА-3 ПОЛИМЕРИЗАЦИЕЙ
ТРИФТОРХЛОРЭТИЛЕНА СУСПЕНЗИОННЫМ МЕТОДОМ
Исходные продукты: трифторхлорэтилен — 50 г; вода — 75 мл, перекись бензои-
ла— 0,25 г; фосфат железа (II)—0,25 г; бисульфат натрия —0,26 г; инертный таз —
баллон.
Оборудование: автоклав емкостью 1 л (см. работу 46); воронка Бюхнера; фарфо-
ровая чашка; термошкаф.
В автоклав, предварительно продутый инертным газом (аргоном,
азотом, освобожденным от кислорода), заливают воду и растворяют в
ней фосфат железа, бисульфат натрия. Затем автоклав вакуумируют и
охлаждают (рассолом, жидким азотом). В мономере растворяют инициа-
тор и засасывают с помощью вакуума *в автоклав, который вновь герме-
тизируют и при температуре не выше 35° С полимеризуют мономер до
125
конверсии 90%. Падение давления свидетельствует об окончании реак-
. ции.
Автоклав открывают, содержимое отфильтровывают на воронке
Бюхнера, промывают несколько раз горячей водой и высушивают в тер-1
мошкафу или на воздухе (в эксикаторе над хлористым кальцием).
РАБОТА 49. ИЗГОТОВЛЕНИЕ ПЛЕНКИ НА ОСНОВЕ
СУСПЕНЗИОННОГО ПОЛИТРИФТОРХЛОРЭТИЛЕНА
При промышленном производстве пленки суспензию наливают на
бесконечную металлическую ленту, которая проходит через сушильное
отделение и отделение спекания. Несущая полоса покрыта слоем поли-
тетрафторэтилена, что облегчает отделение готовой пленки. Этот слой
служит также защитой от каталитического действия металла ленты.
В лабораторном методе в качестве основы используют алюминиевую
фольгу.
Исходные продукты: политрифторхлорэтилен высокомолекулярный—15 г; поли-
трифторхлорэтилен низкомолекулярный — 4,5 г (см. работу 47); спирто-ксилольная
смесь (или хлорированный дифенил —сонол) —50 мл.
Оборудование: колба плоскодонная емкостью 250 мл; коллоидная мельница; лис-
товая алюминиевая фольга; вакуум-эксикатор; термошкаф с регулировкой темпера-
туры.
Политрифторхлорэтилен (см. работу 47) смешивают в коллоидной
мельнице с растворителем и пластификатором (низкомолекулярный по-
литрифторхлорэтилен) до образования частиц размером 1—2 мкм. Дис-
персию наливают ровным тонким слоем на алюминиевую фольгу и высу- !
шивают вначале при комнатной температуре в вакуум-эксикаторе, затем
при 90° С в термошкафу, после чего образовавшийся порошкообразный
слой запекают в том же термошкафу при 260—275° С. Продолжитель-
ность спекания 20—40 мин. Для увеличения эластичности горячую плен-
ку вместе с пластинкой опускают в холодную воду: происходит закалка.
Чтобы получить покрытие достаточной толщины и без дефектов, по-
лив, сушку и запекание повторяют до закалки несколько раз.
Для снятия пленки подложку (фольгу) растворяют в концентриро-
ванном растворе щелочи. Пленку испытывают на прочность при разрыве
и относительное удлинение при различных температурах.
Можно изготовить пленку из дисперсии в соволе (хлорированном
дифениле). В этом случае ее сушат сразу при 260—270° С. Следует пом-
нить, что качество покрытия зависит от однородности частиц полимера
по величине, равномерности высыхания слоя и от дефектов (трещин),
образующихся при высыхании и запекании слоев.
Определяют диэлектрические свойства и химическую устойчивость
материала.
РАБОТА 50. ПОЛУЧЕНИЕ СОПОЛИМЕРА ТРИФТОРХЛОРЭТИЛЕНА
С ВИНИЛИДЕНФТОРИДОМ
Сополимеры трифторхлорэтилена с винилиденфторидом эластичны
(относительное удлинение 600—800%) и аморфны, хотя и склонны к
кристаллизации при вытяжке. Состав сополимеров колеблется от 30 до
126
80% винилиденфторида. Сополимеры можно отверждать аминами, пере-
кисями и изоцианатами.
Отвержденный сополимер устойчив к тепловому старению, действию
концентрированных кислот, щелочей. По свойствам он подобен полиси-
локсановым каучукам. Перерабатывается экструзией, каландрованием;
является основой для клея. Из сополимеров изготавливают кислотостой-
кие изделия: обувь, перчатки, прокладочные кольца и уплотнения, об-
кладки для баков.
Реакция протекает по схеме
nCF2-CFCl + /nCH2=CF2—>[—CF2—CFC1—СН2—CF2—]—
Исходные продукты: трифторхлорэтилен — 25 г; винилиденфторид — 25 г; вода
дистиллированная — 120 мл; персульфат калия — 0,5 г; тиосульфат калия — 0,3 г; инерт-
ный газ.
Оборудование: автоклав емкостью 1 л (см. работу 47); воронка Бюхнера; химиче-
ский стакан емкостью 200 мл.
В автоклав, предварительно продутый инертным газом (аргоном,
очищенным от кислорода азотом), заливают воду, растворяют в ней пер-
сульфат и тиосульфат калия. Затем впускают в рубашку охлаждающий
агент (рассол, жидкий азот или воздух), вакуумируют автоклав и вво-
дят мономер. Затем автоклав герметизируют и поднимают температуру
до 80—90° С. Давление, поднявшееся в начале реакции, через некоторое
время падает вследствие окончания реакции сополимеризации.
Автоклав охлаждают, газообразный мономер отдувают . инертным
газом, а реакционную массу фильтруют на воронке Бюхнера, промывают
несколько раз горячей водой и высушивают в термошкафу при 80—
90° С.
Определяют выход сополимера по отношению к исходной смеси мо-
номеров, температуру течения (или плавления) сополимера, предел проч-
ности и относительное удлинение отпрессованного стандартного образца
(см. ч. II). Прессование производят при 110° С и давлении. 25 кг/см2.
РАБОТА 51. ПОЛУЧЕНИЕ ОТВЕРЖДЕННОГО СОПОЛИМЕРА
ТРИФТОРХЛОРЭТИЛЕНА И ВИНИЛИДЕНФТОРИДА
Исходные продукты: сополимер — 20 г; гексаметилендиамин — 1‘ г.
Оборудование: вальцы лабораторные; пресс гидравлический; плиты из нержавею-
щей стали — 2 шт; ограничительная рамка—1 шт.; пресс-форма изделия любой кон-
фигурации; термошкаф.
Сополимер смешивают на вальцах с гексаметилендиамином при
100° С в течение 30 мин; затем переносят продукт в форму или помеща-
ют между плитами в ограничительную рамку и прессуют при 110° С и
давлении 25 кг/см2 в течение 1 ч. Для улучшения свойств образец под-
вергают термообработке при 135° С в течение 0,5—1 ч.
Отвержденный продукт испытывают на те же показатели, что и ис-
ходный сополимер (см. работу 50}.
127
РАБОТА 52. ИЗГОТОВЛЕНИЕ ПЛЕНКИ ИЗ РАСТВОРА
НА ОСНОВЕ СОПОЛИМЕРА ТРИФТОРХЛОРЭТИЛЕНА
И ВИНИЛИДЕНФТОРИДА
Исходные продукты: сополимер — 10 г; метилэтилкетон — 40 мл; перекись бензо-
ила —0,3 г.
Оборудование: термошкаф; колба емкостью 250 мл; обратный холодильник; нодя-
ная баня; вакуум-эксикатор; алюминиевая фольга.
Перекись бензоила растворяют в метилэтилкетоне и добавляют со-
полимер. Реакционную смесь нагревают в колбе с обратным-холодильни-
ком на водяной бане до полного растворения сополимера. Раствор охла-
ждают и покрывают им алюминиевую фольгу. Растворитель испаряют
при комнатной температуре в вакуум-эксикаторе. После подсушки, если
необходимо увеличить толщину пленки, наносят второй и даже третий
слои. После каждого полива следует подсушка в вакуум-эксикаторе. За-
тем пленку отверждают при 150° С в течение 5 ч.
Полученную эластичную пленку испытывают на прочность и относи-
тельное удлинение при разрыве, а также на устойчивость к концентри-
рованной серной кислоте.
РАБОТА 53. ПОЛУЧЕНИЕ ПОЛИМЕРА ВИНИЛФТОРИДА
ПОЛИМЕРИЗАЦИЕЙ суспензионным методом
Исходные продукты: винилфторид — 50 г; вода, предварительно освобожденная
от кислорода, — 150 мл; персульфат калия —0,5 г; тиосульфат калия — 0,5 г; инертный
газ — баллон.
Оборудование: автоклав (см. работу 46); воронка Бюхнера; фарфоровая чашка;
термошкаф.
В автоклав, предварительно продутый инертным газом, заливают
воду и растворяют в ней персульфат и тиосульфат калия. Впускают в
рубашку охлаждающий агент (жидкий азот, воздух), вакуумируют ав-
токлав и вводят мономер. Затем автоклав герметизируют и поднимают
температуру до 70° С. Давление вначале поднимается, но по мере про-
текания реакции падает. При давлении 3—4 атм автоклав начинают
охлаждать, оставшийся мономер продувают инертным газом. Реакцион-
ную массу фильтруют на воронке Бюхнера, промывают несколько раз
водой и высушивают в термошкафу при 90° С.
Определяют выход полимера (в процентах по отношению к мономе-
ру). Определяют индекс расплава, температуру плавления в запаянном
с обоих концов капилляре.
При температуре, превышающей температуру плавления на 20°,
вальцуют полимер, добавив в него 2—3% (от веса полимера) минераль-
ного масла. Полученную пленку напрессовывают на винипластовую плен-
ку в лабораторном прессе при 160—170° С и удельном давлении 100—
150 кг/см2 в течение 10 мин, затем постепенно охлаждают материал в
прессе. Из дублированной пленки вырубают лопатки — образцы для
испытания на предел прочности и относительное удлинение при разрыве.
Определяют диэлектрические свойства и устойчивость слоя из поливи-
нилфторида к УФ-облучению.
128
ГЛАВА IV. ПОЛИВИНИЛАЦЕТАТ, ЕГО СОПОЛИМЕРЫ
И ПЛАСТИЧЕСКИЕ МАССЫ НА ИХ ОСНОВЕ
Поливинилацетат
Г—СН2—СН—
обычно рассматривают как исходный
COCH3J„
продукт для производства поливинилового спирта и его производных.
Однако он имеет довольно значительное самостоятельное применение.
Свойства поливинилацетата зависят от способа получения, который
определяет значение молекулярного веса, полимолекулярность, а также
технологию дальнейшей переработки. В настоящее время поливинилаце-
тат, полученный в эмульсии и суспензии, широко применяется в самых
разнообразных отраслях техники и строительства в виде компонента
лаков, красок и клеев, в производстве кожзаменителей, искусственной
замши, в полиграфии и других отраслях промышленности.
Химизм образования поливинилацетата
Винилацетат легко полимеризуется под воздействием УФ-лучей,
тепла и инициаторов. В качестве инициаторов применяются как органи-
ческие, так и неорганические перекиси, а также другие вещества, спо-
собные возбуждать радикальную полимеризацию.
Винилацетат полимеризуется с большой скоростью. Скорость поли-
меризации зависит как от реакционной способности мономера, так и от
активности образующегося из него свободного радикала. Известно, что
активность свободных радикалов зависит от их строения. Если неспа-
ренный электрон находится в сопряжении с другими связями, то плот-
ность электронного облака у атома углерода с нечетным числом элект-
ронов становится наиболее однородной и активность радикала в этих
условиях невелика. В случае же полимеризации винилацетата, малоак-
тивного мономера, образуется свободный радикал, в котором эффект
сопряжения неспаренного электрона с другими связями близок к нулю:
~ сн2-сн
I
ОСОСНз
Поэтому такой радикал очень активен. Скорость же реакции роста за-
висит в основном от активности радикала.
В процессе радикальной полимеризации винилацетата образуется
полимер главным образом строения «^голова к хвосту», однако имеет
место и присоединение «голова к голове». Характерной особенностью
полимеризации винилацетата является ярко выраженная склонность к
передаче цепи. При проведении полимеризации в CCI4 образуются лишь
низкомолекулярные черные маслообразные продукты.
5—1453 >29
В процессе полимеризации винилацетата происходит также переда.
ча цепи на мономер, которая заметно снижается, если проводить поли-
меризацию в инертном растворителе.
Винилацетат сополимеризуется с другими сложными эфирами, на-
пример, эфирами фумаровой и малеиновой кислот, а также винилхлори-
дом, образуя ценный пластик «винилит» (см. стр. ПО).
Поливинилацетат растворим в мономере; при нагревании до высо-
кой температуры (250—300° С) он деструктируется с выделением уксус-
ной кислоты и других низкомолекулярных продуктов, не образуя моно-
мера.
Поливинилацетат растворяется в спирте, -бензоле, этил ацетате и
других растворителях.
Технология производства
поливинилацетата
В зависимости от назначения поливинилацетата полимеризацию осу-
ществляют различными методами: блочным, лаковым, суспензионным
и эмульсионным.
Блочная полимеризация проводится в аппаратах с мешалками якор-
ного типа и обратными холодильниками с большой поверхностью охлаж-
дения. Реакцию проводят при 80—90° С, причем выделяется тепло:
89 кДж/моль (21,3 ккал/моль). По окончании экзотермической реакции
массу нагревают до ПО—120° С и направляют на грануляцию. Блочный
поливинилацетат выпускают в виде гранул.
При полимеризации в растворителях в качестве инициаторов приме-
няют перекись бензоила, динитрил азоизомасляной кислоты. Выбор рас-
творителя определяется назначением поливинилацетата: для склеивания
и лакирования кожи поливинилацетат получают в этилацетате; для скле-
ивания бумаги, ткани, стекла — в среде этилового спирта. Поливинил-
ацетат в виде спиртового раствора (ТУ 6-10-1081—70) выпускают трех
марок, отличающихся по вязкости мольных растворов (в сантипуазах):
С-4 (3—6 сП), С-8 (6—10 сП); С-12 (10—14 сП). Концентрация лаков —
45—55% полимера; содержание мономера не более 1,2%.
Суспензионная полимеризация винилацетата проводится в водной
среде, где поверхностно-активным веществом является поливиниловый
спирт с содержанием 10—15% ацетатных групп, а инициатором — пере-
кись бензоила или динитрил азоизомасляной кислоты. Суспензионный
поливинилацетат (ТУ 6-05-1593—72) выпускают семи марок, отличаю-
щихся вязкостью мольного раствора: 15, 25, 50, 75, 100, К, Ц. Марка К
близка к № 50, а марка Ц — к № 15. Номер марки соответствует вяз-
кости мольного раствора в сантипуазах. Высокомолекулярный поливи-
нилащетат марок 75 и 100 в спиртах не растворяется.
Эмульсионной полимеризацией получают поливинилацетатные су-
спензии (ГОСТ 18992—73). Их выпускают 22 марки, отличающиеся по
вязкости, содержанию пластификатора, размеру частиц и по физико-
механическим свойствам.
Пленки и покрытия из эмульсионного поливинилацетата обладают
хорошей адгезией и светостойкостью.
130
Исходное сырье для получения
поливинилацетата
Винилацетат СН2=СНОСОСН3 (представляет собой бесцветную про-
зрачную жидкость с эфирным запахом. Т. кип. 73° С при 760 адм рт. ст.,
20° С при 90 мм рт. ст. и 0°С при 30 мм рт. ст.; ^4°=0,9568, d420=0,9342,
«и20 =1,3958; мол. вес 86,05.
Винилацетат легко растворим в ароматических углеводородах, а
также в спирте, этилацетате .и других органических растворителях.
В воде при 20° С растворяется 2,5% винилацетата; растворимость
воды в винилацетате при этой же температуре составляет 0,1%. Винил-
ацетат устойчив при нагревании до 400° С.
Ингибитором, обычно применяемым для стабилизации мономера,
служит резинат меди *, придающий винилацетату синевато-зеленоватую
окраску. Исчезновение окраски после продолжительного хранения пока-
зывает, что медь высадилась из раствора и ее стабилизирующее действие
прекратилось. Добавление 0,05—0,2% резината меди вполне достаточно
для стабилизации мономера, предназначенного для хранения и пере-
возок.
В качестве ингибитора иногда применяют и дифениламин. Его пре-
имущество перед солями меди состоит в том, что он замедляет появле-
ние нежелательной кислотности даже при продолжительном хранении
винилацетата в стальных резервуарах. Введение дифениламина в коли-
чествах 0,01—0,1% уже в значительной степени замедляет полимериза-
цию. При перевозках мономерный винилацетат рекомендуется стабили-
зировать 0,01—0,02% дифениламина и для большей гарантии добавить
еще 0,01% резината или ацетата меди. Стабилизированный таким обра-
зом мономер может храниться более двух лет, не полимеризуясь и за-
метно не изменяя своих качеств.
В качестве ингибиторов можно применять также серу и гидрохинон.
Резинат меди и сера — нелетучие ингибиторы, поэтому они предпочти-
тельнее (особенно если мономер очищается перегонкой) по сравнению
с летучим гидрохиноном.
Нестабилизированный мономерный винилацетат нельзя хранить бо-
лее 24 ч. Дальнейшее хранение, особенно в больших количествах и в
плотно закрытых сосудах, может привести к взрыву.
Мономерный винилацетат, применяемый в промышленности пласти-
ческих масс, должен быть прозрачной бесцветной жидкостью, не содер-
жащей взвешенных частиц. Недопустимо содержание уксусной кислоты
более 0,02%. В интервале 71,8—73э С (при 760 мм рт. ст.) должно пере-
гоняться не менее 97% винилацетата. В перегнанном винилацетате не
должны присутствовать альдегиды и гидрохинон.
Представляет большой интерес разработанный в СССР метод полу-
чения винилацетата из этилена в растворе уксусной кислоты в присут-
ствии хлористого палладия:
СН2=СН2 + СН3СООН + PdCl2 -> СН3СООСН=СН2 + 2НС1 + Pd
* Резинат меди — смесь медных солей кислот, входящих в состав каиифоли.
5’ 131.
Благодаря присутствию в реакционной среде веществ, способствую-
щих протеканию окислительно-восстановительной реакции (СиС12 и
CuCl), металлический палладий снова превращается в хлористый:
2CuC12 + Pd ->2CuCl + PdCl2
2CuCl + 2HC1 + 0,5O2 -> 2CuCl2 + H2O
Реакция протекает при 180° С и 12 атм в колонных аппаратах, за-
полненных раствором солей палладия и меди ® уксусной кислоте, через
который пропускают смесь этилена и кислорода. Побочными продукта-
ми в этом процессе являются этилендиацетат и ацетальдегид. Наряду
с хлоридом палладия и хлоридами меди в этом процессе можно исполь-
зовать и другие каталитические системы, содержащие палладий.
Винилацетат в промышленных масштабах получают также винили-
рованием уксусной кислоты:
СНз-СООН + СНнСН -> СН3СООСН=СН2
Реакция протекает как в жидкой фазе, так и в паровой. Парофаз-
ный метод имеет ряд преимуществ перед жидкофазным, так как дает
более высокую степень превращения и меньший выход побочного про-
дукта —этилидендиацетата.
Жидкофазный метод состоит в пропускании ацетилена через уксус-
ную кислоту при 60—65° С в присутствии катализатора — ртутной соли
ацетилсерной кислоты. В качестве побочного продукта получается эти-
лидендиацетат:
СНз-СООч
СНзСООН + СН3-СООСН=СН2 -> >СН-СН3
СНз-СОО7
Винилацетат можно получать из уксусного ангидрида и уксусного
альдегида в две стадии:
СН3СО. Feci, СН3СООХ
1) >О + СН3СНО-------> >СН-СН3
СН3ССХ СНзСОО/
СН3СООЧ ArSo,H
2) >СН-СНз——>СНзСООСН=СН2 +СНзСООН
СНзСОО7 135 с
Подготовка продукта к полимеризации
Наиболее удобным методом отделения винилацетата от загрязне-
ний и остатков веществ, применявшихся при синтезе, а также от боль-
шинства ингибиторов является перегонка. Обычно перегонка ведется
при атмосферном давлении, поскольку винилацетат при температуре
кипения (73° С) полимеризуется с небольшой скоростью. Для разложе-
ния некоторых примесей рекомендуется, чтобы источник тепла имел тем-
пературу около 110° С.
Перегонку можно вести также в вакууме или с водяным паром.
Если винилацетат был стабилизирован только резинатом меди, то по-
следний можно полностью удалить промывкой 10%-ным раствором ук-
суснокислого натрия или соды. В этом случае при последующей про-
132
мывке мономера водой и сушке его безводным сульфатом натрия винил-
ацетат полимеризуют, не подвергая перегонке.
Винилацетат очищают перегонкой обычным методом, нагревая его
в круглодонной колбе, снабженной дефлегматором и прямым холодиль-
ником. Вследствие низкой температуры кипения и легкой воспламеняе-
мости мономера нагревать его рекомендуется на электрической плитке,
прикрытой листом асбеста, или в горячей водяной бане, от которой от-
нята горелка. Ни в коем случае не допускается нагревание открытым
пламенем горелки.
Для предотвращения полимеризации винилацетата во время пере-
гонки в колбу необходимо добавить 0,2% гидрохинона от веса перего-
няемого мономера. Для работы отбирают фракцию, кипящую в преде-
лах 71—73° С. Фракции, кипящие ниже 71° С, а также выше 73° С, от-
брасывают. Очищенный винилацетат анализируют и употребляют для
полимеризации.
Анализ винилацетата
Определение содержания винилацетата. Содержание винилацетата
в образце можно определить двумя методами: йодометрическим титро-
ванием и некомпенсационным потенциометрическим титрованием [5].
Метод титрования основан на присоединении брома по месту двой-
ной связи винилацетата, избыток брома определяется иодометрически:
СН2=СНОСОСН3 + Вг2 —> СН2Вг—СНВг—ОСОСНз
Вг2 + 2KI—2КВг + 12
I2 + 2Na2S2O3=2NaI + Na2S40g
В мерную колбу, содержащую 50 мл раствора уксусной кислоты
и взвешенную на аналитических весах, вносят 0,4 мл винилацетата и
снова взвешивают. Мерную колбу заполняют до метки ледяной уксусной
кислотой и после перемешивания переносят пипеткой 10 мл раствора в
колбу со 100 мл охлажденной до 5° С дистиллированной воды. К полу-
ченному раствору приливают по каплям 0,1 н. раствор брома в ледяной
уксусной кислоте до слабо-желтого окрашивания, а затем еще 0,5—1 мл
реактива с таким расчетом, чтобы расход составил целое число милли-
литров (для удобства расчетов). Прибавляют 10 мл раствора KI и че-
рез 5—10 мин титруют выделившийся иод 0,1 н. раствором Na2S2O3 в
присутствии нескольких капель раствора крахмала, периодически встря-
хивая колбу. Содержание винилацетата х (в %) рассчитывают по фор-
муле
Y (а/<1 ~ ^2)0,0043-100-100
£10
где а — количество 0,1 н. раствора брома, пошедшее на титрование на-
вески винилацетата, мл; b — количество 0,1 н. раствора Na2S2O3, пошед-
шее на титрование избытка брома, млц Ki —поправка к 0,1 н. раствору
брома; /<2— поправка к 0,1 н. раствору Na2S2O3j 0,0043 — количество
винилацетата, соответствующее '1 мл точно 0,1 н. раствора Na2S2O3, г;
g— навеска анализируемого винилацетата, г.
133
Определение активности винилацетата. Активность винилацетата
можно определить дилатометрическим методом, т. е. по изменению объ-
ема мономера, помещенного в дилатометр, при нагревании в течение 1 ч
при 72—72,5° С.
В колбу вносят 20 г винилацетата, взвешенного с точностью до
0,01 г, и добавляют 0,02 г перекиси бензоила (с точностью до 0,0002 г).
После растворения инициатора в колбу добавляют
4 мл бензола, перемешивают и 20 мл смеси заливают
в дилатометр (рис. 42), который погружают в стакан
с предварительно нагретой до 72—72,5° С водой; че-
рез 5 мин после погружения дилатометра в воду от-
мечают точно объем смеси (в мл).
Сокращение объема смеси (в мл) в течение 1 ч
при 72—72,5° С характеризует полимеризационную
активность винилацетата.
Определение содержания уксусной кислоты в ви-
нилацетате. В коническую колбу с притертой проб-
кой наливают около 50 мл нейтрального этилового
спирта, затем вносят навеску 9—10 г винилацетата,
взятую с точностью до 0,01 г, взбалтывают и быстро
титруют 0,1 н. едким натром в присутствии фенол-
фталеина.
Содержание уксусной кислоты %i (в %) рассчи-
тывают по формуле
akO,006-100
*1 =---------- ,
g
где а — количество 0,1 н. едкого натра, израсходован-
ное на титрование, мл; k — коэффициент нормально-
сти 0,1 н. едкого натра; g— навеска винилацетата, г;
0,006 — количество уксусной кислоты, соответствую-
щее 1 мл 0,1 н. едкого натра, г.
Качественное определение ацетальдегида в винил-
ацетате. Сначала готовят реактив: в 1 л 95%-ного
этилового спирта растворяют 5 г солянокислого гид-
роксиламина, затем прибавляют 5 мл 0,02%-ного раствора метилового
оранжевого. Цвет реактива доводят до оранжевого, добавляя по каплям
раствор соляной кислоты. Реактив может храниться длительное время.
В пробирку помещают 5 мл реактива и прибавляют к нему 1 мл
испытуемого винилацетата. Пробирку встряхивают и через 1 мин срав-
нивают окраску реактива до и после прибавления винилацетата. При
отсутствии ацетальдегида изменение цвета не происходит. При содер-
жании в пробе уже 0,05% альдегида появляется красное окрашивание.
Определение содержания гидрохинона в винилацетате. В пробирку
наливают 3—4 мл винилацетата, добавляют 1 мл 0,5 н. едкого натра и
хорошо встряхивают. Водный слой раствора щелочи должен оставаться
бесцветным. В присутствии гидрохинона раствор щелочи окрашивается
в желтый цвет, причем интенсивность окраски зависит от содержания
134
Рис. 42. Дилато-
метр:
1 — воздушный холо-
дильник высотой
45 см; 2 — шлиф; 3 —
градуированная часть
пробирки диаметром
10—11 мм, высотой
20 см, с ценой деле-
ния 0,1 мл; 4 — рас-
ширенная часть про-
бирки (диаметром
18—20 мм, высотой
50 мм, емкостью —>•
25—27 мл)
сутствии 0,0005% гидрохинона.
Определение содержания резината меди в винилацетате. В совер-
шенно чистую и сухую фарфоровую чашку, взвешенную на аналитиче-
ских весах, помещают 20 мл анализируемого винилацетата и выпари-
вают на песчаной бане. Когда вся жидкость выпарится, остаток озоляют
в муфельной печи до постоянного веса. Золу растворяют в 2 мл концент-
рированной азотной кислоты и 3 мл концентрированной соляной кисло-
ты и осторожно нагревают до полного прекращения выделения паров
окиси азота, затем прибавляют несколько капель концентрированной
серной кислоты и выпаривают до появления паров серного ангидрида.
После охлаждения остаток в чашке разбавляют 10 мл дистиллиро-
ванной воды и нейтрализуют избытком водного раствора аммиака. Оса-
док гидроокиси железа отфильтровывают и фильтрат разбавляют ди-
стиллированной водой до объема 100 мл в трубке для колометрических
определений.
Окраску раствора сравнивают с эталонами, содержащими известные
количества меди в эталонных трубках. Эталоны готовят растворением
1,964 г кристаллической сернокислой меди в 1000 мл дистиллированной
воды: 1 мл такого раствора содержит 0,50 мг меди.
Количество металлической меди Xj (в %) рассчитывают по формуле
100-0,0005-100
,
1 20р
где 0,0005 — количество металлической меди, содержащееся в 1 мл рас-
твора, г; р — плотность винилацетата.
Содержание резината меди х2 (в %) равно x2=10xi.
Анализ поливинилацетата
Качественное определение поливинилацетата. Поливинилацетат —
прозрачный бесцветный полимер, его плотность 1,191 г/см3; nD20= 1,4665.
1. С монохлоруксусной кислотой поливинилацетат дает красно-
фиолетовую окраску; с дихлоруксусной кислотой — сине-фиолетовую.
2. При смешении раствора поливинилацетата в тетрагидрофуране
с мольным раствором фениллития в том же растворителе появляется
коричневый осадок.
3. При обработке спиртового раствора поливинилацетата спиртовым
раствором щелочи появляется белый осадок поливинилового спирта.
Количественное определение суспензионного поливинилацетата, ла-
ка и поливинилацетатной эмульсии. В суспензионном поливинилацетате
определяют остаточный мономер, летучие вещества, вязкость, термоста-
бильность, молекулярный вес; в поливинилацетатной эмульсии опредё-.
ляют остаточный винилацетат, дибутилфталат, сухой остаток, величину
частиц.
Количество винилацетата в полимере, в отроне, растворе, эмульсии
или суспензии можно определить обычным титрованием. В колбу емко-
стью 250 мл, содержащую 20 мл раствора уксусной кислоты, вносят
0,6—0,8 г полимера, взвешенного с точностью до 0,0002 г (5—10 г лака
. - »35
или эмульсии, взвешенных с точностью до 0,01 г). Колбу помещают в
охлаждающую смесь (вода со льдом), приливают 100 мл дистиллиро-
ванной воды и после охлаждения до 5—7° С титруют 0,1 н. раствором
брома в ледяной уксусной кислоте.
Содержание винилацетата х (в %) рассчитывают по формуле
(а — <>)/<. о, 0043-100
g
где а и b — количества 0,1 н. раствора брома, затраченные на титро-
вание соответственно анализируемой и контрольной проб, мл; К — по-
правка к 0,1 н. раствору брома; 0,0043 — количество винилацетата, со-
ответствующее 1 мл точно 0,1 н. раствора брома, г; g — навеска анали-
зируемого .вещества, г.
Определение летучих веществ в бисерном поливинилацетате произ-
водят сушкой под инфракрасной лампой. Навеску 1 г высушивают под
лампой в течение 2 мин, взвешивают; высушивают еще 2 раза по 1 мин
до постоянного веса. Потерю в весе относят в % к исходному весу ве-
щества. Высушивание можно производить в шкафу при 150° С в тече-
ние 2 ч.
Молекулярный вес определяют по характеристической вязкости при
20° С раствора суспензионного поливинилацетата в высушенном и пере-
гнанном ацетоне (ч. д. а.).
Величина частиц (гранул) эмульсии (в мкм) определяется при
25-кратном разбавлении испытуемой пробы дистиллированной водой с
помощью оптического микроскопа типа МБП-6 или МИМ-7 при 400-крат-
ном оптическом увеличении, с последующим двукратным фотографиче-
ским увеличением.
Определение вязкости полимера. 1. Полимеризация винилацетата.
В круглодонную колбу емкостью 100 мл, имеющую тубус для термо-
метра и соединенную при помощи шлифа с холодильником, через кото-
рый пропущена мешалка, вводят 15 г .винилацетата и 6 г абсолютного
этилацетата (что соответствует 40 вес. ч. этилацетата на 100 вес. ч. ви-
нилацетата), затем прибавляют свежеперекристаллизованной перекиси
бензоила 0,1% от веса винилацетата. Колбу помещают в водяную баню
и нагревают при температуре кипения (72—78° С) и перемешивании в
течение 8 ч. Легкое кипение начинается при 72° Сив ходе процесса
температура постепенно повышается до 78° С.
Если реакционная масса раньше указанного времени загустеет на-
столько, что мешалка не может больше вращаться, и температура до-
стигнет 78° С, процесс заканчивают. Колбу охлаждают до 15—20° С и
через тубус берут две пробы: 2—3 г для определения вязкости полимера
и 4—5 г для определения термостабильности.
2. Определение вязкости. Пробу, взятую для определения вязкости,
помещают .в бюкс диаметром 40—50 мм и растворяют в 10—15 мл этил-
ацетата. Бюкс с раствором ставят б вакуум-термостат и высушивают до
постоянного веса. Начинают сушку при комнатной температуре, посте-
пенно повышая вакуум. К концу сушки бюкс нагревают до 100° С.
После охлаждения высушенного полимера берут навеску 0,925 г,
помещают в колбу емкостью 20—30 мл, добавляют 10 мл бензола, закры-
136
вают колбу корковой пробкой и взвешивают с точностью до 0,01 г. Кол-
бу с обратным холодильником нагревают на водяной бане до полного
растворения полимера, после чего вновь взвешивают и, если нужно, до-
бавляют бензол до первоначального веса.
Перед определением вязкости находят плотность полученного раство-
ра при помощи пикнометра.
Для определения вязкости раствор заливают в вискозиметр Остваль-
да, который погружают в сосуд с водой, где поддерживают температуру
20±1°С. После 20-минутной выдержки вискозиметра в сосуде при по-
мощи секундомера определяют время истечения раствора. Для расчета
берут среднее из трех последовательных определений.
Предварительно устанавливают константу вискозиметра по 60 %-но-
му раствору сахара (60 г сахара в 40 г воды). Плотность раствора сахара
1,286 г/см3.
Константу вискозиметра К определяют по формуле
56,5 __43,94
1,286и и ’
где 56,5—абсолютная вязкость 60%-него раствора сахара; т — время
истечения раствора сахара, с.
Вязкость полимера ц (в сантипуазах) рассчитывают по формуле
»]=K/ip, где К — константа вискозиметра; t\—время истечения испыту-
емого раствора, с; р — плотность испытуемого раствора.
Техника безопасности при работе
с винилацетатом
Винилацетат представляет собой легковоспламеняющуюся жидкость
с температурой вспышки (в открытом сосуде)—5° С. Поэтому при работе
с ним следует соблюдать необходимые меры противопожарной безопас-
ности. Все работы с винилацетатом проводятся вдали' от источников
открытого огня.
Винилацетат перегоняют на предварительно нагретой водяной бане,
от которой отнята горелка, или в специальном электрическом колбона-
гревателе. Ни в коем случае не допускается нагревание колбы с моно-
мером открытым пламенем горелки.
Вследствие экзотермичности реакции полимеризации, приводящей к
выделению большого количества тепла и сильному саморазогреванию,
не допускается хранение сколько-нибудь значительных количеств винил-
ацетата без стабилизатора более одних суток. Особенно недопустимо
хранение его без стабилизатора в плотно закрытых сосудах, так как при
начавшейся полимеризации сильный разогрев массы может привести к
взрыву.
РАБОТА 54. ПОЛУЧЕНИЕ ВИНИЛАЦЕТАТА
Удобным лабораторным методом получения винилацетата является
жидкофазный метод, в основе которого лежит реакция ацетилена с ук-
сусной кислотой, протекающая в«жидкой фазе:
СН = СН + СН3СООН-^-СН2=СНОСОСН3
137
Исходные продукты: уксусная кислота—1000 г; уксусный ангидрид—150 г;
мышьяковая кислота — 3 г; фтористоводородная кислота (плавиковая) 40%-ной кон-
центрации — 9 г; окись ртути — 2,8 г; ацетилен — 235 г; уксуснокислый натрий — 5 г.
Оборудование: установка (рис. 43); колбы плоскодонные емкостью 250 мл — 2 шт.;
прибор для перегонки в вакууме; водяная баня с термометром.
Установка для получения винилацетата в лабораторных условиях
(рис. 43) состоит из трехгорлой колбы, помещенной в водяную баню.
Колба снабжена мощной мешалкой с герметическим затвором, и двумя
трубками для ввода и вывода газа. Перед поступлением в реакционный
Рис. 43. Установка для синтеза винилацетата:
1— моностат; 2— баллон с ацетиленом; 3— промывная склянка с водой; 4 — склянка
с хромовой кислотой; 5 — осушительные колонки с хлористым кальцием; 6 — реометр?
7 — осушительные колонки с хлористым кальцием и силикагелем; 8 — реакционная
колба; S — обратный клапан; 10— промывная склянка; // — резервуар с водой для ре-
гулирования давления
сосуд ацетилен промывают водой и концентрированным раствором хро-
мовой кислоты. Промытый таз проходит затем через реометр для изме-
рения его подачи и через несколько осушительных склянок, наполненных
прокаленным гранулированным хлористым кальцием н силикагелем.
Трубка для выхода газа из реакционной колбы соединена с колонкой,
наполненной хлористым кальцием, и через обратный клапан — с пере-
вернутой промывной склянкой, которая, в свою очередь, соединена с ре-
зервуаром для воды. Повышая или понижая уровень воды в резервуаре,
можно менять давление в реакционной колбе. Во время реакции избы-
точное давление равно около 10 см водяного столба. По изменению
уровня воды наблюдают за изменением скорости поглощения ацетилена
в реакционном сосуде.
Реакционную смесь, состоящую из уксусного ангидрида, уксусной,
мышьяковой и плавиковой кислот, а также окиси ртути, готовят в от-
дельном сосуде, затем переносят в реакционную колбу. Уксусный ангид-
рид соединяется с водой, содержащейся в смеси (вода понижает эффек-
тивность каталитического действия мышьяковой и фтористоводородной
кислот). Предполагается, что в отсутствие воды образуется более реак-
ционноспособное фтористое соединение.
138
Во время пропускания ацетилена температуру в реакционной колбе
поддерживают между 15 и 20° С. Скорость поглощения ацетилена не
должна изменяться в продолжение приблизительно 8 ч. По истечении
этого времени прибавляют сухой уксуснокислый натрий для нейтрали-
зации смешанного кислого катализатора и перемешивание продолжают
еще 15—30 мин. В этих условиях выход винилацетата составляет около
88%, считая на количество поглощенного ацетилена.
Винилацетат рекомендуется немедленно отделить от неорганических
соединений и поддерживать низкую температуру для предотвращения
дальнейшей реакции винилацетата с кислотой. Чтобы добиться макси-
мальной отгонки этилидендиацетата и других высококипящих соедине-
ний, реакционную смесь перегоняют под уменьшенным давлением. В на-
чале перегонки давление поддерживают около 100—150 мм рт.ст., в
конце 10—20 мм рт. ст. Весь дистиллят конденсируют, охлаждают до
—10 или —20° С, после чего подвергают фракционной разгонке в колбе,
снабженной дефлегматором.
Первую фракцию собирают при температуре до 71° С. Вторая фрак-
ция, отгоняющаяся при 71—75° С, в основном состоит из винилацетата.
Следующая фракция, кипящая при 75—115° С, является смешанным ди-
стиллятом. Во фракции, перегоняющейся при 115—120° С, преобладает
уксусная кислота.
Продукт, перегоняющийся выше 120° С, состоит из этилидендиаце-
тата, некоторого количества уксусной кислоты и уксусного ангидрида.
РАБОТА 55. ПОЛУЧЕНИЕ ПОЛИВИНИЛАЦЕТАТА ПОЛИМЕРИЗАЦИЕЙ
ВИНИЛАЦЕТАТА В БЛОКЕ
Исходные продукты: винилацетат — 30 г; перекись бензоила — 0,03 г.
Оборудование: круглодонная колба, снабженная обратным холодильником; водя-
ная баня.
В кругло донную колбу, снабженную обратным холодильником, за-
гружают винилацетат и перекись бензоила и перемешивают жидкость
до полного растворения инициатора. Затем колбу помещают в водяную
баню, пускают воду в холодильник и нагревают до 70—75° С. Эту тем-
пературу поддерживают в течение 2 ч до образования в колбе прозрач-
ной густой неперетекающей массы.
Ход реакции полимеризации контролируют по изменению вязкости
и показателя преломления. Показатель преломления .готового полимера
должен быть равен около 1,4665. 1 >
Полимер используют для получения лака и поливинилового спирта.
Примечание. Показатель преломления следует определять через каждые 15 мин
после начала реакции полимеризации.
РАБОТА 56. ПОЛУЧЕНИЕ ПОЛИВИНИЛАЦЕТАТА ПОЛИМЕРИЗАЦИЕЙ
ВИНИЛАЦЕТАТА В ЭГМУЛЬСИИ И ИЗГОТОВЛЕНИЕ
ЭЛАСТИЧНЫХ ПЛЕНОК И ПОКРЫТИЙ НА ЕГО ОСНОВЕ
Исходные продукты: винилацетат — 50 г; поливиниловый спирт — 3,3 г; уксусная
кислота — 0,63 г; перекись водорода-— 0,8 г; сульфат железа — 0,0058 г; вода дистил-
лированная — 50,5 мл; аммиачная вода ('25%-ная).
Оборудование: прибор (рис. 44); водяная баня; .рН-метр.
139
Поливиниловый спирт растворяют в дистиллированной воде в колбе
прибора при 60° С и перемешивании. После полного растворения спирта
в колбу вводят сначала уксусную кислоту (содержание ее в растворе
должно составлять 1,45—1,55%), затем приливают 7,5%-ный водный
раствор сульфата железа. Смесь хорошо
Рис. 44. Прибор для проведения
реакции в токе инертного газа
с перемешинанием и капельной во-
ронкой для введения реагентов
перемешивают и при работающей ме-
шалке добавляют терекись водорода.
После этого включают холодильник, про-
пускают в колбу газообразный азот и
при быстро работающей мешалке из ка-
пельной воронки начинают вводить ви-
нилацетат. Температуру во время поли-
меризации поддерживают 65—85° С. Ре-
акция продолжается 4—5 ч.
По окончании реакции содержимое
колбы охлаждают до 30° С и полученный
продукт 'нейтрализуют аммиачной водой
до pH 2,5—3,5. Нейтрализацию ведут
при тщательном перемешивании.
По внешнему виду полученный про-
дукт представляет собой вязкую смета-
нообразную массу белого цвета. Опреде-
ляют содержание винилацетата и сухо-
го остатка в эмульсии (см. стр. 135).
Полученной эмульсией склеивают бума-
гу, ткань, бумагу с тканью и т. д.
Для получения эластичной пленки с высокой морозоустойчивостью
в поливинил ацетатные эмульсии вводят до 35% дибутилфталата.
Для более равномерного распределения дибутилфталата в массе
эмульсии его предварительно эмульгируют поверхностно-активными
веществами: ОП-Ю или сольваром (поливиниловый спирт с 15%-ным
содержанием ацетатных групп). Для этого 0,3 г эмульгатора ОП-10 рас-
творяют в 8 мл воды и к раствору добавляют небольшими порциями
при очень интенсивном перемешивании 10 г дибутилфталата. Эмульги-
рованный дибутилфталат вводят при энергичном перемешивании в по-
ливинилацетатную эмульсию, после чего она готова к употреблению.
Содержание пластификатора в эмульсии с содержанием сухого остатка
52,7% составляет 9,3 г (15%). Полученную эмульсию используют для
изготовления искусственной замши, моющихся обоев. В первом случае
эмульсией покрывают ткань, подсушивают и прокатывают подогретым
до 60°С валиком, имеющим «ворсистую» поверхность (рисунок замши).
Во втором случае эмульсией покрывают обои, подсушивают, покрывают
еще раз и подсушивают окончательно. Полученный материал прокаты-
вают между подогретыми валками вальцев.
РАБОТА 57. ПОЛУЧЕНИЕ СУСПЕНЗИОННОГО
ПОЛИВИНИЛАЦЕТАТА
Исходные продукты: винилацетат—10 г; дистиллированная вода—60 г; динитрил
.азоизомасляной кислоты — 0,1 г; сольвар — 0,2 г или поливиниловый спирт — 0,34 г.
Оборудование: прибор для суспензионной полимеризации (см. рис. 20); стаканы
химические на 25 мл и 1 л.
В широкой пробирке диаметром 45 мм и высотой 190 мм, имеющей
боковой отвод и снабженной мешалкой с герметическим затвором и об-
ратным холодильником, при нагревании до 50—60° С растворяют соль-
вар или поливиниловый спирт в дистиллированной воде. Затем готовят
раствор инициатора в мономере и заливают в реакционный сосуд с ох-
лажденным до комнатной температуры водным раствором сольвара (или
поливинилового спирта).
Включают мешалку, пускают воду в холодильник и нагревают ре-
акционную смесь на водяной бане до 65° С в начале процесса и до 95° С
•в конце. Скорость вращения мешалки регулируют так, чтобы образова-
лись капельки мономера. Установленную скорость поддерживают в те-
чение 3—4 ч, пока шарики (бисер) не начнут стремиться осесть на дне
пробирки в результате увеличившейся плотности. Во время реакции под-
держивают pH 3—4, добавляя уксусную кислоту или бикарбонат натрия
в зависимости от pH раствора.
Определяют выход полученного полимера по отношению к количе-
ству взятого мономера, а также количество непрореагировавшего моно-
мера (см. стр. 136).
РАБОТА 58. ПОЛУЧЕНИЕ СОПОЛИМЕРА АКРИЛОНИТРИЛА
С ВИНИЛАЦЕТАТОМ
чСН2=СН + тСН2=СН > Г—СН2—СН—
I I I
CN ОСОСНз L CN
-СН2—СН—
ОСОСНз]-
Исходные продукты: нитрил акриловой кислоты—10 г; винилацетат—15 г;
.динитрил азоизомасляной кислоты — 0,05 г; диметилформамид— 100 г.
Оборудование: прибор (см. рис. 19); стаканы емкостью 100 мл — 2; нодяная баня;
кювета или чашки Петри.
Инициатор растворяют в одном из мономеров, добавляют второй
мономер. В колбу прибора вливают диметилформамид и приготовлен-
ную смесь мономеров с инициатором. Раствор нагревают в течение 2 ч
-при перемешивании на водяной бане при 60° С до образования вязкого
сиропа.
Раствор сополимера используют для изготовления пленки. Кювету
.для полива пленки (или чашку Петри) тщательно отмывают, высуши-
вают и протирают мягкой тканью. Затем ее устанавливают по уровню
в вытяжном шкафу и наливают раствор сополимера слоем толщиной
4—5 мм.
Растворитель испаряют до образования пленки, зат,ем помещают
кювету в термостат и выдерживают при 80° С до образования прочной
•пленки. Пленку аккуратно снимают с подложки, вырезают стандартные
лопатки (см. ч. II) и определяют предел прочности при растяжении.
ГЛАВА V. ПОЛИМЕРЫ АКРИЛОВОЙ И МЕТАКРИЛОВОЙ КИСЛОТ.
ПОЛИМЕРЫ ПРОИЗВОДНЫХ АКРИЛОВОЙ
И МЕТАКРИЛОВОЙ КИСЛОТ И ПЛАСТИЧЕСКИЕ
МАССЫ НА ИХ ОСНОВЕ
К классу полиакрилатов и полиметакрилатов относят полимеры и сопо-
лимеры акриловой и метакриловой кислот, их сложных эфиров, амидов,
нитрилов, а также эфиры хлоракриловой и цианакриловой кислот. Осо-
бое значение приобрели эфиры этих кислот, на основе которых произ-
водят органические стекла различного назначения, прессованные и лить-
евые пластические массы, а также нитрилы для изготовления синтети-
ческих волокон, литьевых и прессовочных материалов.
Полиэфиры акриловой и метакриловой кислот и высших спиртов
имеют различную консистенцию в зависимости от строения радикала
спирта. Чем больше число атомов углерода в спиртовом радикале, тем
ниже температура размягчения; начиная с октилового эфира полимеры
представляют собой вязкие жидкости или полужидкие массы. Поли-
меры эфиров метакриловой кислоты более теплостойки, чем полимеры
эфиров акриловой кислоты. Так, температуры размягчения полиэфиров
акриловой кислоты: полиметилакрилата +3° С, полиэтилакрилата
—25° С, полибутилакрилата —45° С, а соответствующих полиэфиров ме-
такриловой кислоты +100°С, +65° С и +33° С.
Растворимость акрилатов и метакрилатов также зависит от строе-
ния спиртового радикала: с увеличением его длины растворимость улуч-
шается.
Различие между метакриловыми и акриловыми полимерами прояв-
ляется и в их химической и термической устойчивости. Полиметакрила-
ты более водостойки, чем полиакрилаты. При температуре разложения
первые деполимеризуются с образованием мономера, а вторые разру-
шаются с образованием около 1 % мономера, довольно больших фраг-
ментов цепей и продуктов более глубокого распада: кислот, спиртов.
Химизм полимеризации акрилатов
и метакрилатов
Особенности полимеризации ионогенных мономеров (акриловой и
метакриловой кислот) заключаются в их склонности к изменению ре-
акционной способности под влиянием среды.
При изучении кинетики полимеризации акриловой и метакриловой
кислот в водных растворах при различных pH показано, что с возраста-
142
нием pH до 6—7 скорость реакции резко падает, что связано с v •
чением в системе числа малореакционноспособных анионов кисло^ и
дальнейшем увеличении pH скорость полимеризации*и молекул '
веса полимеров значительно возрастают при pH 7—12 для метакр^*Рн
кислоты и pH 7—11 для акриловой. По-видимому, возрастание ckq °Е
полимеризации является результатом увеличения локальной коцц>е^о<
ции ионов Na+ вблизи ионизованных концевых радикалов, что доНт1
привести к увеличению скорости роста цепи. Особый интерес 'ВЫд?™
использование в этой реакции аминов в качестве нейтрализующие*1®'
тов, основность и строение которых можно широко изменять. аг'
Так, в щелочной области в присутствии изобутиламина скород
димеризации метакриловой кислоты с ростом pH возрастает бол^Ь 1
ко, чем в присутствии NaOH. ₽
При pH>7,5, когда макрорадикалы в основном ионизовдц
концах растущих цепей образуются ионные пары с участием Кар§ ’
дат-анионов макрорадикалов и органических катионов: ок'
СН3 СН3
СН2—С’---СН2=С
I -+ I
COONH3 соо~
I
R
Полимеры акриловойГ—СН2
метакриловой
-сн2-£
п
что, как и в случае ионов Na+, облегчает рост цепи.
Образующиеся ионные пары оказывают влияние и на структур^
тущей цепи. В этих условиях можно получить до 85% синдиотактц^^
го полимера. Присоединение мономеров происходит главным образ
по типу «голова к хвосту».
-СН—
I
соон
кислот и их производных: эфиров, нитрилов и амидов — можно noj^*
всеми известными методами: блочным, эмульсионным, суспензионном
в растворителях.
Полимеризация тщательно очищенных мономеров под влинни
тепла и УФ-лучей протекает лишь при высоких температурах. Эти лО]
меры могут полимеризоваться как по радикальному, так и по аниоино
механизму. Однако в технике принято полимеризовать их по Радика>
ному механизму в блоке, суспензии и эмульсии.
Обрыв цепи происходит в зависимости от условий диспропорц^
рованием или рекомбинацией.
При блочной полимеризации метилметакрилата вь>Дедяе1
56,8 кДж/моль (13,6 ккал/моль) тепла, что при плохой теплопроВод]
сти может приводить к образованию пузырей. Это вызывает Heo6XOj
мость тщательного регулирования температуры в процессе блочц0д ,
димеризации.
Эмульсионная полимеризация эфиров акриловой и метакридов
кислот и их нитрилов протекает в присутствии растворимых в воде j
рекисей и персульфатов, а также окислительно-восстановительных ини-
циирующих систем. Трудно получить стабильную эмульсию высших
эфиров (выше С4) в связи с их низкой температурой размягчения. Это
относится и к суспензионной (гранульной) полимеризации.
г — СН2— СН— '
I
CN
Полимеризация нитрилов акриловой
и метакри-
СН3п
I
-сн2-с-
I
CN
При увеличении концентрации инициатора ингибирующее действие сни-
жается. Другие эфиры акриловой и метакриловой кислот нечувстви-
тельны к действию кислорода и даже, напротив, его присутствие ска-
зывается положительно.
Акриламид Г—СН2—СН—
ловой
Jn
кислот ингибируется присутствием кислорода.
самопроизвольно не полимеризуется,
и
L CONH2J„
но в присутствии перекисей в атмосфере инертного газа полимеризация
протекает с высокой скоростью. Полиакриламид имеет разветвленную и
частично сшитую структуру, что связано со способностью амидной груп-
пы к химическим превращениям. Проведение реакции в растворителе
снижает возможность образования разветвлений. В присутствии метал-
лического натрия имеет место миграционная полимеризация (полипри-
соединение), в результате чего образуется поли-.р-аланин:
CH2=CHCONH2+ nCH2=CH—conh2->
: t
-> CH2=CHCONHCH2CH2CONH2 -> СН2=СН (-CONHCH2CH2-)„CONH2
Полимеризация в водной среде при 90° С приводит, к омылению
амидной группы. Более низкие температуры (ниже 50° С) обеспечива-
ют образование линейного полимера с высоким молекулярным весом.
Степень полимеризации можно регулировать добавлением изопропило-
вого спирта.
Технология производства акрилатов
и метакрилатов
Акриловую и метакриловую кислоту полимеризуют в водных рас-
творах в присутствии кислорода и перекисей или персульфатов. Мета-
криловая кислота менее реакционноспособна, чем акриловая, в связи с
наличием в a-положении метильной группы. Полимеризацию осуществ-
ляют в реакторах с мешалкой. Полимеры растворимы в воде и нераство-
римы в органических растворителях. Полимерные кислоты широко ис-
пользуют в качестве эмульгаторов и диспергаторов при водноэмульсион-
ной полимеризации. Их растворы обладают свойствами электролитов.
Соли поликислот используют в качестве загустителей.
Наибольшее количество марок полимеров, выпускаемых промыш-
ленностью на основе эфиров метакриловой кислоты, представляют собой
144
листовые материалы, предназначенные для остекления и приборострое-
ния. Получают листовые материалы блочной полимеризацией между
силикатными стеклами. Листовой материал может быть получен также
экструзией через плоскощелевую головку, однако прозрачность этих
листов, их оптические свойства хуже, чем у блочного.
Блочный полиметилметакрилат выпускают около 20 марок: стекло
органическое листовое для остекления (СО-95; СОЛ; СО-120; СТ-1
и т. д.); стекло органическое часовое СОП-4; стекло органическое Т-2-55.
Выпускается много марок пластифицированного дибутилфталатом поли-
метилметакрилата СОП. Опаловое стекло — пластифицированный и за-
мутненный полистиролом полиметилметакрилат. Перламутровое стек-
ло— полиметилметакрилат, наполненный патом и эмульсионным поли-
стиролом (МРТУ 6-01-412—69).
Стекло органическое светотехническое (ГОСТ 9784—61) представ-
ляет собой непластифицированный полиметилметакрилат или его сопо-
лимеры, замутненные различными добавками, и предназначается для
изготовления рассеивателей, светильников с люминесцентными лампами
накаливания.
Сополимеризацией в массе метилметакрилата с 4% бутилметакри-
лата получают дакрил-4Б (ТУ 6-01-742—72) для медицинских целей; с
2% метилакрилата — дакрил-2МО — общего назначения (ТУ 6-01-544—
70). Их перерабатывают литьем под давлением и экструзией. Эти же
сополимеры выпускают замутненными полистиролом.
Пластифицированный полибутилметакрилат, полученный в массе,
применяют для изготовления пленки методом строжки блока, использу-
емой для прокладок.
Эмульсионный полиметилметакрилат получают в присутствии ини-
циаторов, растворимых в воде. Эмульгаторы — мыла, соли органических
сульфокислот. Процесс можно осуществлять как периодическим, так и
непрерывным способом в аппаратах с мешалкой. Латекс при необходи-
мости можно коагулировать электролитом, центрифугировать, промыть
и высушить.
Суспензионный полиметилметакрилат получают методом суспензи-
онной полимеризации в присутствии инициаторов, растворимых в моно-
мере (перекиси бензоила, динитрила азоизомасляной кислоты), а также
персульфатов калия или натрия в водной среде, содержащей стабилиза-
тор суспензии (желатин, поливиниловый спирт, метилцеллюлозу или
соли акриловой или метакриловой кислот, карбонат магния и т. п.). По-
лимеризацию проводят в эмалированном реакторе с мешалкой лопаст-
ной или турбинной при 70—85° С. Гранулы отделяют и высушивают в
гребковых сушилках при 100° С.
Полимер выпускается по ТУ 6-01-67—72 марки ЛСОМ и ЛСОМ
4Б — сополимер метилметакрилата с 4% бутилметакрилата; Л-1—по-
лимер метилметакрилата пластифицированный.
Полимеры и сополимеры перерабатываются в изделия литьем под
давлением, экструзией, прессованием; хорошо вальцуются и смешива-
ются с наполнителями. Они применяются для изготовления различных
технических изделий сложной конфигурации, радиодеталей, деталей, со-
прикасающихся с бензином и маслами.
145
Полибутилметакрилат суспензионный (ТУ 6-01-252—68) получают
суспензионной полимеризацией бутилметакрилата в тех же условиях,
что и полиметилметакрилат. Он широко используется в производстве
художественных красок н полимеризующейся мастики.
Производство полиакрилонитрила можно осуществить периодиче-
ским и непрерывным способами в аппарате с мешалкой в водной среде
в присутствии персульфата и тиосульфата калия при 30—40° С. Реакция
продолжается 10—20 мин. Выход 95—98%. Полимер выпадает из реак-
ционной смеси, центрифугируется, промывается и высушивается. В свя-
зи с высокой скоростью реакции легко осуществляется непрерывный
.процесс по принципу идеального вытеснения в одном реакторе.
Полиакрилонитрил растворяется лишь в диметилформамиде, тетра-
метиленсульфоне, в концентрированных водных растворах некоторых
-солей, например бромистого лития, роданистого натрия или кальция,
хлористого цинка.
Полиакрилонитрил из растворов дает волокно «нитрон» («орлон»).
Формование волокон осуществляется по сухому и мокрому способам.
В случае мокрого прядения берут 15—20%-ный раствор полимера в ди-
метилформамиде, а в качестве осадителя—воду или разбавленный ди-
метилформамид.
Полиакриламид получают в водно-спиртовой среде в присутствии
церсульфата калия в реакторе с мешалкой при 65—80° С. Образуется
прозрачный раствор полиакриламида с вязкостью 35 П (при 25° С).
Полиакриламид применяется в качестве стабилизатора латексов,
составной части клеев, коагулянта суспензии фосфоритов, структурооб-
разователя почвы. При сополимеризации метакриламида с бутилмета-
крилатом в изопропиловом спирте образуется сополимер — смола АС
(ТУ 6-01-370—69), применяющаяся для изготовления лаков и эмалей.
Большой практический интерес представляют сополимеры мономе-
ров акрилового и метакрилового ряда с другими мономерами (стиролом,
винилхлоридом, винилиденхлоридом), сополимеры отдельных предста-
вителей этих групп, например метилметакрилата с акриловой и метакри-
.ловой кислотами (эмульгатор), метилметакрилата с бутилметакрилатом
(мастики, краски, оргстекло), метилметакрилата с акрилонитрилом и
стиролом (литьевой материал). Многие из этих сополимеров уже опи-
саны в соответствующих разделах. Следует отметить также практиче-
ское значение сополимеров метилметакрилата со сшивающими агента-
ми, такими как диметакрилаты диэтилен- и триэтиленгликолей, диаллил-
малеинат, диаллилметакрилат и диаллилфталат. Сополимеры этого типа
неплавки и нерастворимы, имеют повышенную твердость и теплостой-
кость. Применение их для изготовления теплостойких стекол имеет боль-
шое значение.
Исходное сырье для получения полиакрилатов
и полиметакрилатов
Акриловая кислота СН2=СНСООН — прозрачная бесцветная жид-
кость с резким запахом, напоминающим уксусную кислоту. Т. пл. 12,3° С;
т. кип. 141° С при 760 мм рт. ст., 71° С при 50 мм рт. ст. и 52—53° С при
146
22 мм рт. ст.; di6= 1,062, d-4 =1,051; nD —1,4224; мол. вес 72,06. Хоро-
шо растворяется в спирте, эфире и смешивается с водой в любых соот-
ношениях.
Акриловую кислоту получают нагреванием этиленциангидрина с
разбавленной серной кислотой через оксиакриловую кислоту:
СН2ОН +2H,o+h,so. СН2—ОН —hso
| ~ - ~„п > | > СН2=СНСООН
CH2-CN -nh‘[iso‘ сн2-соон
В промышленности акриловую кислоту получают реакцией окиси
углерода с ацетиленом в присутствии воды или спирта. Источником СО
является карбонил никеля Ni(CO)4. Реакция протекает в среде мине-
ральных или органических кислот: соляной, уксусной, фосфорной (в ви-
де водных растворов) по суммарному уравнению:
4СН=СН 4- 4Н2О + Ni (СО)4 4- 2НС1 -> 4СН2=СНСООН 4- NiCl2 4- Н2
В присутствии спиртов образуются эфиры акриловой кислоты.
Метакриловая кислота СН2=С(СН3)СООН— бесцветная жидкость
с острым запахом, напоминающим уксусную кислоту. Т. кип. 161° С при
760 мм рт. ст. и 60—63°С при 12 мм рт: ст.; т. пл. 15°С; d4°=l,015; пр =
= 1,4314; мол. вес 86,09. Хорошо растворяется в воде, со спиртом и эфи-
ром смешивается в любых соотношениях.
Метакриловую кислоту получают 'взаимодействием ацетонциангид-
рина с разбавленной серной кислотой, при этом образуется оксиизомас-
ляная кислота, которая при последующей дегидратации дает метакрило-
вую кислоту:
СНз
/ОН +н,о /ОН ]
(СНз)2С/ ---->(СН3)2С< —>сн2=с
XCN ^СООН ~н“° |
соон
Метакриловую кислоту можно получить из метакролеина с 96 % -
ным выходом при его окислении под давлением 4 атм в присутствии аце-
татов меди и никеля при комнатной температуре:
СНз СН3
СН2=С—СНО 4- 0,5О2 -> СН2=С—СООН
Метакриловую кислоту и ее нитрил можно получить исходя из изо-
бутилена без применения синильной кислоты:
СНз СН3 СНз
I I NaOH * |
СН2=С-СН3 4- С12 ——> СН2=С-СН2С1-> СН2=С—СН2ОН ->
СН3 СНз
—% СН2=С-СНО сн2=с—соон
СН3 СН3 СНз
I +NH, '| +о, I
- СН2=С—СН2С1 —СН2=С—CH2'NH2 — ^>СН2=С— CN
147
Метиловый эфир акриловой кислоты (метилакрилат)
€Н2=СНСООСНз представляет собой прозрачную жидкость с резким
запахом. Т. кип. 80,3° С при 760 мм рт. ст., 50° С при 270 мм рт. ст. и
20° С при 72 мм рт. ст.; d|° =0,9735; df =0,9558; n£б) * 8 =1,4117; п™ =
= 1,404; ид = 1,401; мол. вес 86,09.
Метилакрилат легко растворяется в обычных растворителях; в воде
растворимость его составляет 5,2 вес. %.
Мономер может быть стабилизирован гидрохиноном, серой, метал-
лической медью, п-трет-бутилпирокатехином, пикриновой (кислотой и не-
которыми другими веществами. В отличие от гидрохинона пикриновая
кислота обладает хорошим ингибирующим действием и в отсутствие
кислорода.
Теплота полимеризации метилакрилата при 25° С равна 86,9±
±4,1 кДж/моль (20,8 ±0,98 ккал/моль).
Температура кипения технически пригодного продукта 79—82° С,
кислотность, считая на акриловую кислоту, 0,02%.
Метилакрилат можно получать по той же схеме, как и акриловую
кислоту, из ацетилена, окиси углерода и спирта (вместо воды) в присут-
ствии карбонила никеля.
В промышленности метилакрилат (а также этилакрилат и н-бутил-
;акрилат) тоже можно получать по измененной схеме из окиси углерода,
ацетилена и хлористого водорода в присутствии спирта и карбонила
никеля (см. стр. 147). В спирте растворяют хлорид никеля и этим рас-
твором орошают насадочный реактор колонного типа. Противотоком в
реактор пропускают под давлением при 150—180° С ацетилен и окись
углерода:
сн=сн + со + roh->ch2=ch-coor
В лабораторных условиях метиловый эфир акриловой кислоты мож-
но получать взаимодействием этиленциангидрина с метиловым спиртом
в присутствии серной кислоты.
Реакция идет в две стадии по схеме:
а) гидролиз этиленциангидрина
СН2ОН СН2ОН
I +2H2O±H2SO4-> | +NH4HSO4
ch2cn сн2соон
б) этерификация оксиакриловой кислоты и последующая дегидра-
тация
СН2ОН СН2ОН СН2
I + СН3ОН -> | -> || + Н2О
СН2СООН СН2СООСН3 СНСООСНз
Метилметакрилат СН2=С—СООСН3. Чистый метиловый эфир мета-
СИ3
криловой кислоты представляет собой бесцветную жидкость с приятным
эфирным запахом. Т. кип. 100,3°С; di°=0,936; Пд = 1,413; мол. вес 100,11.
Метилметакрилат легко смешивается с серным эфиром и спиртом
и хорошо растворяется во всех обычных органических растворителях.
М 48
Растворимость его в воде при комнатной температуре составляет
1,5 вес. %.
Мономер стабилизируют гидрохиноном, пирогаллолом, фенолом, ре-
зинатом меди и другими веществами. Гидрохинон может быть ингибито-
ром только в присутствии растворенного кислорода. При хранении и пе-
ревозках мономер обычно стабилизируют гидрохиноном.
В технике метилметакрилат получают синтетическим путем из аце-
тона и синильной кислоты через ацетонциангидрин.
Продажный метиловый эфир метакриловой кислоты, применяемый в
промышленности пластмасс, должен содержать метилового эфира мета-
криловой кислоты не менее 99,7%, метакриловой кислоты не более 0,2%.
Метиловый эфир метакриловой кислоты в лабораторных условиях
можно получить из ацетонциангидрина. Реакция протекает в одну ста-
дию:
ОН
СН3. ,ОН +h2so* СН3. | zNH —HjO
)С<--------*- >c-c<f ---------->
CH3Z XCN CH3Z XOSO3H
CH3
>ДН +CH.OH I
-* >C—Cf-------------->CH2=C
H3CZ 4>SO3H -<NH*hS0< ]
C00CH3
н-Бутиловый эфир метакриловой кислоты (бутилметакрилат)
СН3 о
| || представляет собой про-
СН2=С—С-О-СН2-СН2—СН2-СН3
зрачную бесцветную жидкость. Т. кип. 163—164° С при 760 мм рт. ст.,
73° С при 30 мм рт. ст., 51—52°С при 11 мм рт. ст., dl's’e =0,889; с?4° =
=0,894, dи =0,895, иИ’е = 1,426; nf=1,421, мол. вес 142,19.
Бутилметакрилат хорошо растворяется в различных органических
растворителях и практически не растворяется в воде. Он легко полиме-
ризуется при нагревании в присутствии инициаторов.
В технике бутилметакрилат получают из ацетонциангидрина и
«-бутилового спирта в условиях, аналогичных получению метилметакри-
лата в одну стадию.
Аллиловый эфир метакриловой кислоты (аллил метакрил ат)
СН2=С—С—ОСН2—СН=СН2 представляет собой бесцветную
I
СН3
прозрачную жидкость с т. кип. 67° С при 50 мм рт. ст., 62—63° С при
43 мм рт. ст., 55° С при 30 мм рт. ст. и 42° С при 15 мм рт. ст.;
<4° =0,9335, djo =0,9342; пц’7 =1,4392, пр =1,4385, Пд =1,4358;
мол. вес 126,15.
Этиленгликолевый эфир метакриловой кислоты
СН3 О О СН3
.1 II II I
СН2=С—С-О-СН2-СН2-О-С-С=СН2
149
предотавляет собой прозрачную бесцветную жидкость. Т. кип. 83—85° С ’
при 1 мм рт. ст., 100—102° С при 7,5—8 мм рт. ст., 122—126° С при
15 мм рт. ст. и 125—130°С при 20 мм рт. ст.; с?4° = 1,0468; Пд=1,4545;'
мол. вес 198,2. ;
Обладая четырьмя функциональными группами, диметакрилат эти-
ленгликоля при полимеризации образует макромолекулы пространствен-
ного строения.
Мономер может применяться как для гомополимеризации, так и для
сополимеризации с метилметакрилатом и другими .мономерами с целыо
получения неплавких и нерастворимых продуктов.
Полимер диметакрилата этиленгликоля представляет собой твердый
стекловидный продукт с плотностью р= 1,2323 г/см3, не размягчающийся
при нагревании до 200—220° С. Деполимеризация полимера начинается
выше 220° С, при этом образуется до 70% жидких продуктов, из кото-
рых около 40% составляет исходный мономер.
Диэтиленгликолевый эфир метакриловой кислоты (диметакрилат
.диэтиленгликоля) СН2= С С—О—СН2—СН2—О—СН2—СН2—О—С—С=СН2
СН3 О О СН3
представляет собой прозрачную бесцветную жидкость. Т. кип. 150—
152°С (при 7—8 мм рт. ст.); df = 1,0821; Пд =1,4685; бромное число
132,07; мол. вес 242,26.
Диметакрилат диэтиленгликоля получают по методике, описанной
для диметакрилата этиленгликоля.
Диметакрилат диэтиленгликоля может применяться для сополиме-
ризации с метилметакрилатом и другими мономерами с
ния неплавких и нерастворимых продуктов.
Триэтиленгликолевый эфир метакриловой кислоты (диметакрилаг
СН2=С—С-(-ОСН2-СН2-)зО—с—с=сн2
триэтиленгликоля) I II II I
СН3 О О СН3
представляет собой прозрачную бесцветную жидкость. Т. кип. 160° С
при 5 мм рт. ст., dl0 =1,062; пд =1,457; бромное число 171,0; мол.
вес 286.
Диметакрилат триэтиленгликоля применяется для сополимеризации
с метилметакрилатом и другими мономерами с целью получения неплав-
ких и нерастворимых теплостойких полимеров, а также в производстве
ненасыщенных полиэфиров.
Получается ,как и диметакрилат диэтиленгликоля переэтерификаци-
ей метилметакрилата.
Акрилонитрил (нитрил акриловой кислоты) СНг=СН—CN — проз-
рачная бесцветная жидкость. Т. кип. 17,3° С; d2t =0,8060, dl5 =0,8004;
«d = 1,3911, п д = 1,3884; мол. вес 53,06.
Он в любых соотношениях смешивается с большинством полярных
и неполярных растворителей, например ацетоном, этиловым и метило-
вым спиртами, эфиром, этилацетатом, диоксаном, ароматическими угле-
водородами, четыреххлористым углеродом, петролейным эфиром, керо-
сином. В воде при 20° С растворяется 7,3 вес. % акрилонитрила. Рас-
творимость воды в акрилонитриле при 20° С составляет 3,1 вес. %.
150
целью получе-
.ОН
(СНзЪС/^
Акрилонитрил может продолжительное время храниться в металли-
ческой таре или стеклянных сосудах со стеклянными пробками в при-
сутствии небольшого количества ингибитора (гидрохинона, пирогаллола,
^-нафтиламина и др.). При температуре около 0°С образование полиме-
ра практически прекращается.
Метакрилонитрил (нитрил метакриловой кислоты) СН2=С—CN
СН3
— бесцветная прозрачная жидкость со специфическим неприятным запа-
хом. Т. кип. 90° С при 760 мм рт. ст.; dl° =0,786; пд =1,3780; мол. вес
52,21. Во всех отношениях смешивается с большинством полярных и не-
полярных растворителей. Нерастворим в воде.
Метакрилонитрил получают в промышленности дегидратацией аце-
тонциангидрина в присутствии пятихлористого фосфора
СН3
I
+ РС15 -> CH2=C-CN + РОС13 + 2НС1
или из изобутилена (см.стр. 147).
Акриламид (амид акриловой кислоты) СН2 = СН—CONH2 — белый
хлопьевидный порошок с т. пл. 84,5° С; d%5= 1,127; мол. вес 71. Хоро-
шо растворим в воде, метиловом и этиловом спиртах, ацетоне и слабее —
в этилацетате, хлороформе и дихлорэтане. Перекристаллизовывается из
-бензольного раствора.
В щелочной среде реагирует с формальдегидом, образуя метилол-
акриламид.
Акриламид получают различными методами. Наиболее удобным яв-
ляется гидролиз, акрилонитрила концентрированной серной кислотой:
+н2о
CH2=CHCN----> СН2=СН—CONH2
Реакция протекает 30—45 мин.
Акриламид можно получать в присутствии порошкообразной меди и
-серной кислоты (выход 95%):
100° С: HjSO»
CH2=CHCN----—-----> CH2=CH-CONH2
Этим же способом получают метакриламид:
СНз 100» С; HjSOt СНз
CH2=C-CN icu] CH2=O-CONH2
Качественный анализ полимеров акриловой
и метакриловой кислот и их производных
1. Окрашивание пламени. Полиакрилаты, внесенные в пламя горел-
ки, окрашивают его в желтый цвет. Полиметакрилаты окрашивают пла-
мя в синий цвет.
2. Деполимеризация. При деполимеризации акрилатов образуется
смесь мономеров и продуктов более глубокого разложения, -состоящих
151
из кислот и производных спиртов. При деполимеризации полиметакри-
латов образуются главным образом мономеры.
Для идентификации продуктов пиролиза можно использовать ме-
тод Тиниуса; бромировать их бромом в среде четыреххлористого угле-
рода. В случае акрилатов образуются 2,3-дибромпропионовые эфиры;
в случае метакрилатов — 2,3-дибромизомасляные эфиры. Их идентисЬи-
цируют по температуре кипения. Так, у метилового эфира дибромпропи-
оновой кислоты т. кип. 206 ° С, у этилового эфира — 211° С; метиловый
эфир дибромизомасляной кислоты кипит при 200° С.
3. Реакция с фенилгидразином: 0,5—1 г полимера смешивают &
колбе Вюрца или Кляйзена с сухим песком, осторожно разлагают обра-
зец и собирают дистиллят в охлаждаемый снегом приемник. Дистиллят
высушивают безводным хлористым кальцием и перегоняют: в колбу
вносят 4—5 капель дистиллята, 4—5 капель свежеперегнанного фе-
нилгидразина и 5 мл толуола и кипятят с обратным холодильником
30 мин.
Около 1 мл этого раствора несколько минут встряхивают с 5 мл
85%-ной муравьиной кислоты и 1 каплей 30%-ной перекиси водорода
(можно немного нагреть).
В присутствии акриловой кислоты или акрилатов постепенно появ-
ляется темно-зеленая окраска, не исчезающая несколько часов. Чистая
мономерная акриловая кислота дает голубую окраску.
Метакриловая кислота и ее эфиры не дают окрашивания.
Сополимер метилакрилата с метилметакрилатом (1:1), полиэтила-
крилат, сополимер акриловой кислоты с метилметакрилатом (1:1) дают-
очень слабую положительную реакцию.
4. Реакция с фениллитием. К 1 г образца, растворенного в 35 мл аб-
солютного тетрагидрофурана (тетрагидрофуран высушивают над без-
водным сульфатом натрия, затем над металлическим натрием и перего-
няют над натрием), приливают 10 мл 1 м раствора фенчллития, пропу-
ская через жидкость ток сухого инертного газа.
В присутствии акрилатов или метакрилатов появляется оранжевая
окраска. В избытке фениллития 'полиметилметакрилат дает сначала-
желто-зеленую окраску, которая при стоянии переходит в темно-крас-
ную. Полиакрилаты дают оранжевую окраску, не изменяющуюся при
стоянии.
5. Гидролиз полиакрилатов. 1 г полимера растворяют в колбе с
обратным холодильником в ацетоне или тетрагидрофуране, нерастворив-
шуюся часть отфильтровывают. Добавляют 50 мл 1 н. спиртового рас-
твора КОН и кипятят с обратным холодильником в течение 30 мин. По-
лиакрилат калия выпадает в осадок в виде белой массы. Жидкость де-
кантируют, ос-адок промывают один или два раза ацетоном и высуши-
вают в чашечке на водяной бане. Затем кипятят осадок с водой в тече-
ние короткого промежутка времени для его растворения. Раствор фильт-
руют, охлаждают и нейтрализуют. Если в исходном полимере присут-
ствовали акрилаты, раствор будет давать следующие реакции:
а) при добавлении к раствору разбавленной минеральной кислоты
выпадает осадок акриловой кислоты, который при добавлении аммиака
снова растворяется;
152
б) при добавлении гидроокиси бария или хлористого бария раство]
мутнеет. При добавлении кислоты выпадает осадок полиакрилатг
бария, который при стоянии превращается в каучукоподобную массу
Сульфат алюминия, азотнокислое серебро вызывают образование
осадка. Хлорное железо дает осадок от оранжево-желтого до корич
невого цвета.
С хлорной медью и NH4C1 образуется голубой осадок, который рас
творяется в аммиаке, давая голубой раствор. Хлорид кобальта и NH4C
дает розовый осадок, который после высушивания становится фиолето-
вым. Сульфат никеля с NH4C1 дают зеленый осадок, при растворении
которого в аммиаке появляется зеленовато-голубая окраска.
6. Проба на азот. Полиакрилонитрил и полиакриламид можно ка-
чественно обнаружить пробой на азот: в сухой пробирке сплавляют на
голом огне равные количества полимера и металлического натрия (не
более 0,5 г) до получения однородной массы. При сплавлении полимер
разлагается и образуется цианистый калий.
Горячую пробирку осторожно опускают в стакан с небольшим коли-
чеством воды. (Осторожно, может быть вспышка, работать в вытяжном
шкафу с закрытым стеклом!) Пробирка растрескивается, масса раство-
ряется в воде.
Полученный водный раствор смешивают с несколькими каплями
растворов FeSO4 и FeCl3, смесь сильно взбалтывают и. прибавляют 10%-
ную кислоту до слабокислой реакции на лакмус. При наличии KCN в
растворе (и, следовательно, азота в полимере) сначала появляется синее
окрашивание, а затем выпадает осадок берлинской лазури. Реакция про-
текает по схеме
2KCN + FeSO4=Fe (CN)2 + K2SO4
Fe (CN)2 4-4KCN=K4 [Fe (CN)6J
ЗК4 [Fe (CN)6J + FeCl3=Fe4 [Fe (CN)6]3 + 12KC1
Пиролизат полиакрилонитрила имеет запах анилина. При сплавле-
нии полимера с содой или поташом выделяются белые пары и появ-
ляется запах анилина.
7. Цветные реакции на полиакрилонитрил.
а) Если растворить полиакрилонитрил в горячем N.N-диметилфор-
мамиде и к полученному раствору прилить раствор NaOH до сильноще-
лочной реакции, после нагревания появляется яркое'оранжево-красное
окрашивание.
б) При нагревании полиакрилонитрила с 10%-ным раствором NaOH
образец окрашивается в красно-коричневый цвет.
Количественный анализ мономеров акрилового
и метакрилового рядов
Определение содержания акриловой кислоты. В две конические кол-
бы с притертыми пробками емкостью 200—250 мл отвешивают на ана-
литических весах с точностью до 0,0001 г приблизительно по 1 г акрило-
153
вой кислоты, быстро приливают в каждую по 15 мл нейтрализованного
этилового спирта и после растворения навесок, добавив в качестве ин-
дикатора несколько капель фенолфталеина, содержимое колб титруют
0,5 н. едким кали до появления слабо-розового окрашивания. Содержа-
ние акриловой кислоты х (в %) рассчитывают по формуле
с/<0,036-100
«
g
Здесь a — объем 0,5 н. щелочи, израсходованной на титрование, мл;.
К — коэффициент нормальности 0,5 н. щелочи; g— навеска акриловой
кислоты, г; 0,036 — количество акриловой кислоты, соответствующее
1 мл 0,5 н. раствора щелочи, г.
Определение содержания метакриловой кислоты проводится по этой
же методике. Однако в расчетную формулу следует внести изменение:
количество метакриловой кислоты, соответствующее 1 мл 0,5 н. раствора*
щелочи, равно 0,043.
Содержание акриловой и метакриловой кислот можно определить-
методом бромирования с помощью бромид-броматной смеси (аналогично
определению содержания метилметакрилата, см. ниже).
Определение содержания метилового эфира метакриловой кислоты.
В две конические колбы емкостью по 250 мл, снабженные холодильни-
ками, отвешивают на аналитических весах по 1 г эфира, добавляют по
10—15 мл нейтрализованного этилового спирта и оттитровывают содер-
жащуюся в эфире метакриловую кислоту 0,1 н. КОН в присутствии-
фенолфталеина до появления неисчезающего слабо-розового окрашива-
ния. Затем в каждую колбу из бюретки приливают по 40 мл 0,5'н. спир-
тового раствора едкого кали, присоединяют колбы к обратным холодиль-
никам и нагревают на кипящей водяной бане в течение 3 ч. Верхние кон-
цы холодильников закрывают трубками, наполненными натронной
известью. Одновременно ставят контрольный опыт. По окончании
нагревания колбы охлаждают и содержимое их оттитровывают 0,5 н. со-
ляной кислотой.
Содержание метилового эфира метакриловой кислоты в процентах
(х) рассчитывают по формуле
(a —fe)/<0,05-100
g
где а — объем 0,5 н. соляной, кислоты, израсходованный на титрование
контрольной пробы, мл; b — объем 0,5 н. соляной кислоты, израсходо-
ванный на титрование пробы с навеской, мл; К — коэффициент нор-
мальности 0,5 н. соляной кислоты; g— навеска эфира, г; 0,05 — количе-
ство метилового эфира метакриловой кислоты, соответствующее 1 мл
0,5 н. едкого кали, г.
Количественное определение метилового эфира акриловой кислоты
производят по этой же методике, только в расчетной формуле коэффи-
циент 0,05 заменяют на 0,043. При определении содержания этилмета-
крилата этот коэффициент равен 0,057, а бутил мет акр плата — 0,071.
Определение содержания свободной метакриловой кислоты в эфире.
В две конические колбы с пришлифованными пробками емкостью по
154
100—150 мл вводят по 10 мл нейтрализованного этилового спирта и взве-
шивают на аналитических весах с точностью до 0,01 г.
Затем туда же отвешивают по 10 г метилового эфира метакриловой
кислоты и содержимое колбы титруют 0,1 н. едким кали в присутствии
фенолфталеина до исчезновения в первый момент розового окрашивания.
Во время титрования сильно встряхивать колбу не рекомендуется.
Содержание метакриловой кислоты х (в %) находят по формуле
аКО, 0086-100
X -- “ ' у
g
где а — объем 0,1 н. едкого кали, израсходованный на титрование, мл;
К— коэффициент нормальности 0,1 н. едкого кали; g— навеска эфира, г;
0,0086 — количество метакриловой кислоты, соответствующее 1 мл 0,1 и.
едкого кали, г (для акриловой кислоты — 0,0072). « -
Определение степени чистоты метилметакрилата методом бромиро-
вания. Описание метода дано для метилметакрилата; однако он может
быть использован и для определения других мономеров акрилового и ме-
такрилового рядов; лишь в формуле для определения Хг теоретическое
бромное число изменяется в зависимости от строения мономера.
В бюкс на аналитических весах отвешивают около 2 г вещества, рас-
творяют навеску в небольшом количестве 50°/о-|ной уксусной кислоты и
через воронку переносят в мерную колбу емкостью 100 мл. Бюкс и во-
ронку несколько раз обмывают кислотой и промывную жидкость слива-
ют в колбу. Уксусной кислотой доводят уровень до метки и, закрыв
мерную колбу притертой пробкой, смесь тщательно перемешивают.
В две чистые сухие конические колбы с пришлифованными пробками
емкостью по 500 мл вводят пипеткой по 50 мл 0,1 н. бромид-броматного
раствора* и 10 мл приготовленного раствора метилметакрилата в уксус-
ной кислоте. Затем туда же мерным цилиндром быстро добавляют по
10 мл концентрированной соляной кислоты, закрывают колбы пробками
и оставляют на 20 мин на рассеянном дневном свету. После этого быстро
мерным цилиндром прибавляют по 10 мл 10%-ного раствора йодистого
калия, колбы встряхивают и выделившийся иод оттитровывают 0,1 н.
гипосульфитом натрия, добавив в качестве индикатора несколько капель
раствора крахмала. Параллельно ставят контрольный опыт.
Количество присоединившегося брома (в %) — бромное число Xj—•
рассчитывйют по формуле
(д — fe)7(0,00799-100-100 (а — 6)К-79,9
g
g
где а — объем 0,1 н. гипосульфита натрия, израсходованный на титрова-
ние контрольной пробы, мл; Ь—объем 0,1 н. гипосульфита натрия, из-
расходованный на титрование пробы с навеской, мл; К — коэффициент
нормальности 0,1 н. гипосульфита натрия; g— навеска вещества, г;
0,00799 — количество брома, соответствующее 1 мл 0,1 н. гипосульфита
натрия, г.
* Бромид-броматный раствор содержит 5,939 г КВг и 1,666 г КВгОз в 1 л воды.
155
Степень чистоты метилметакрилата в процентах (х2) определяют по> -1
формуле 1
xilOO
159,8
где Xi — найденное бромное число; 159,8 — теоретическое бромное число- '
метилметакрилата.
Определение содержания стабилизатора (гидрохинона). Метод ос- |
нован на взаимодействии гидрохинона с иодом, взятым в избытке, и на 1
последующем определении количества не вступившего в реакцию иода j
титрованием гипосульфитом натрия.
При анализе метилметакрилата, содержащего не более 0,005% гид- <
рохинона, в коническую колбу с притертой пробкой отвешивают 100 г '
испытуемого вещества, прибавляют 100 мл дистиллированной воды, точ-
но 5 мл. 0,5 н. раствора иода и около 5 мл 5%-ного раствора крахмала.
Колбу энергично встряхивают в течение 1 мин и содержимое ее титруют ,
0,1 н. раствором гипосульфита натрия. Параллельно ставят контрольный j
опыт с образцом метилметакрилата, не содержащим гидрохинона.
Содержание гидрохинона х (в %) рассчитывают по формуле
_ (а- Ь) КО,0055-100 1
•Я-— » 1
g ’
где а — объем 0,1 н. гипосульфита натрия, израсходованный на титро- -
вание контрольной пробы, мл; b — объем 0,1 н. гипосульфита натрия,. :
израсходованный на титрование пробы с навеской, мл; К—коэффици- 1
ент нормальности 0,1 н. гипосульфита натрия; g— навеска образца ме-
тилметакрилата, г; 0,0055 — количество гидрохинона, соответствующее 1
1 мл 0,1 н. гипосульфита натрия, г.
Этот же метод используют для количественного определения гидро-
хинона в других мономерах.
Определение содержания головной фракции. Головной фракцией ме-
тилметакрилата считается фракция, кипящая до 98° С. Количество ее оп-
ределяют отгонкой 100 г метилметакрилата в колбе с дефлегматором.
Нагревание ведут на глицериновой бане, при 130—135° С. ’
Фракцию, кипящую до 98° С, собирают в предварительно взвешен-
ный приемный сосуд. Как только температура достигнет 98° С, перегонку
прекращают и приемную колбу с отгоном взвешивают. Масса продукта •;
в граммах соответствует содержанию головной фракции <в процентах. ;
Если перегонка велась при барометрическом давлении ниже нормаль-
ного (ниже 760 мм рт. ст.), то процентное содержание этой фракции, ;
приведенное к нормальному давлению, находят в табл. 7.
Рассмотрим пример расчета. Предположим, что отгонка проводи-
лась при давлении 745 мм рт. ст.; при этом получено 4,3 г головной
фракции. После приведения к нормальному давлению (760 мм рт. ст.)
содержание этой фракции будет равно 2,4%.
Количественное определение акрилонитрила реакцией с сернисто-
кислым натрием. В основу метода положена реакция акрилонитрила с ;
сернистокислым натрием с последующим титрованием выделяющейся
156 * ’
ТАБЛИЦА Г
Зависимость содержания головной фракции от давления
Давление фактиче- ское, мм рт. ст.' Содержание головной фракции, %
760 1,2 1,6 2,0 2,4 3,0 3,5 4,0 4,6 5,0
755 1,4 1,8 2,3 2,75 3,4 4,0 4,5 5,1 5,5
750 1,8 2,4 2,8 3,4 4,0 4,7 5,6 5,8 6,3
745 2,7 3,2 3,7 4,3 5,0 5,7 6,4 7,0 7,6
740 3,7 4,0 4,7 5,2 6,0 6,7 7,6 8,4 9,4
735 4,8 5,7 6,3 7,0 8,0 8,9 9,8 10,7 12,0
щелочи раствором соляной кислоты обычным или потенциометрическим
титрованием.
В две конические колбы емкостью 250 мл вносят 25 мл 0,25 н. рас-
твора Na?SOs, навеску 0,3—0,35 г акрилонитрила (взвешивают с точно-
стью до 0,0002 г). Пробы выдерживают в течение 30 мин при комнатной
температуре и периодическом перемешивании и затем титруют 0,-1 н.
раствором НС1 в присутствии 3 капель тимолфталеина до исчезновения
синей окраски. В аналогичных условиях титруют контрольную пробу
с раствором Na2SOs. Содержание акрилонитрила х (в %) вычисляют по
формуле
v х (g —ft)/<0,0053-100
g
где а и b — объемы 0,1 н. раствора НС1, израсходованные на титрование-
контрольной пробы и пробы с навеской, мл; К — поправка к титру НС1;
0,0053 — количество акрилонитрила, соответствующее 1 мл точно 0,1 н.
раствора НС1, г; g— навеска, г.
Для потенциометрического титрования взвешивают в коническую-
колбу 0,1—0,15 г акрилонитрила (с точностью до 0,0002 г) и заливают
25 мл 0,25 н. свежеприготовленного раствора Na2SO3; колбы выдержи-
вают 30 мин при комнатной температуре, периодически помешивая со-
держимое.
Потенциометрическое титрование проводят на потенциометре со-
стеклянным электродом; перед титрованием необходимо измерить потен-
циал стеклянного электрода. Для этого в стакан вносят пипеткой 25 мл
раствора Na2SO3 и 150 мл дистиллированной воды. В раствор опускают
стеклянный и каломельный электроды, включают мешалку и замеряют
на потенциометре значение потенциала стеклянного электрода (в мВ).
Пробу с навеской переносят количественно в стакан, доливают водой
до 150 мл и погружают в раствор электроды. Выделившуюся щелочь от-
титровывают раствором НС1 (при перемешивании) до значения потен-
циала стеклянного электрода (в мВ) в контрольной пробе. Расчет про-
изводят по приведенной выше формуле.
157
круглодонную колбу емкостью 200—300
Рис. 45. Установка для отгонки аммиака
. с водяным паром по методу Кьельдаля:
J — парообразователь; 2 — конденсатор; 3 — загру-
зочная воронка; 4 — колба Кьельдаля; 5 — брызго-
улавливатель; 6 — холодильник; 7 — приемник
Определение содержания азота по Кьельдалю. Акрилонитрил можно
определить по содержанию азота в веществе (по методу Кьельдаля) или :
по процентному содержанию нитрильных групп. --:
Сущность определения азота по Кьельдалю состоит в том, что органи- '
ческое вещество при нагревании с концентрированной серной кислотой
разлагается до образования прозрачного бесцветного раствора. Углерод
при этом окисляется в углекислоту, а азот превращается в аммиак, ко-
торый остается в растворе в виде сернокислой соли. К разбавленному
кислому раствору прибавляют избыток щелочи и отгоняют аммиак, 1
улавливая его определенным количеством титрованной кислоты, оста-
ток которой оттитровывают щелочью.
Точную навеску анализируемого вещества (0,1—0,5 г) помещают в
мл с длинным горлом (колбу .
Кьельдаля). Прибавляют
3 мл концентрированной
серной кислоты так, чтобы
она смочила навеску, и не-
плотно закрывают колбу
небольшим стеклянным ша-
риком с короткой запаян-
ной трубкой. Затем колбу
закрепляют с помощью лап-
ки в слегка наклонном по-
ложении, ставят в вытяж-
ной шкаф и нагревают не-
большим пламенем микро-
горелки Бунзена.
Нагревание ведут посте-
пенно, так как смесь может
вспениваться, особенно при
больших навесках. Смесь в
течение 15 мин осторожно
доводят до кипения, затем в
колбу прибавляют 1 г сухо-
го порошкообразного суль-
фата калия и кристаллик
сульфата меди и продолжа-
ют нагревание до тех пор,
пока смесь не станет прозрачной и бесцветной или же слегка окрашен-
ной в зеленоватый цвет. После охлаждения жидкость разбавляют 30 мл
дистиллированной воды и присоединяют к установке для отгонки с па-
ром (рис. 45).
В колбу парообразователя наливают дистиллированную воду и на-
гревают. Конец дистилляционной трубки холодильника Либиха погру-
жают в колбу-приемник. Перед началом дистилляции в колбу, содержа-
щую навеску, через делительную воронку приливают 10 мл концентри-
рованного раствора едкого натра. Во избежание толчков в колбу вносят
3—4 г цинкового порошка (или чистой цинковой фольги), который одно-
временно разлагает амидные соединения, если они образуются.
458
Выделяющиеся пары, содержащие аммиак, отгоняют и последний
улавливают в приемной колбе, в которую налито 20 мл титрованного
раствора 0,1 н. H2SO4. В качестве индикатора применяют метиловый
оранжевый или метиловый красный. Для выделения всего аммиака до-
статочно отогнать 100 мл дистиллята.
Оттитровав 0,1 н. раствором щелочи (NaOH или КОН) избыток сер-
ной кислоты в приемной колбе, находят количество выделившегося ам-
миака и рассчитывают содержание азота в анализируемом продукте-
Содержание азота х (в %) рассчитывают по формуле
(аК— 6/<1)0,0014-100
X — — —————j
£
где а — объем 0,1 н. H2SO4, взятый для улавливания аммиака, мл; Ь —
объем 0,1 н. щелочи, израсходованной на титрование избытка серной
кислоты после улавливания аммиака, мл; К — коэффициент нормально-
сти 0,1 и. H2SO4; Ki — коэффициент нормальности 0,1 н. щелочи; g—•
навеска анализируемого вещества (акрилонитрила), г; 0,0014 — количе-
ство азота, соответствующее 1 мл 0,1 н. H2SO4, г.
На основе полученных данных рассчитывают процентное содержа-
ние акрилонитрила (х^ по формуле
х100
где х — содержание азота в анализируемом веществе, %; 26,41—расчет-
ный коэффициент для акрилонитрила.
Определение содержания азота омылением нитрильных групп. Сущ-
ность метода, состоит в том, что нитрильную группу гидролизуют кипя-
чением с избытком разбавленного раствора едкого натра и выделивший-
ся аммиак оттитровывают кислотой. Однако этот метод неприменим к
летучим нитрилам, в частности к акрилонитрилу, поэтому его видоизме-
няют следующим образом.
Вначале в колбе проводят реакцию 100 мл этилового спирта с не-
большим кусочком металлического натрия. На аналитических весах в-
маленькой ампуле взвешивают 0,5—1 г акрилонитрила. Ампулу, поме-
щают «в колбу и там разбивают. Колбу закрывают пробкой и оставляют
на час. Акрилонитрил вступает в реакцию, образуя высококипящий нит-
рил этоксипропионовой кислоты. Получ‘енпый продукт переносят в пере-
гонную колбу (такую же, как при определении азота по методу Кьельда-
ля) и, влив через делительную воронку избыток разбавленного раствора
едкого натра, кипятят до полного гидролиза нитрильной группы. Выде-
ляющийся аммиак улавливают титрованным 1 н. раствором H2SO4.
Дальнейший ход анализа и расчеты проводят так же, как при оп-
ределении содержания азота методом Кьельдаля.
Определение нитрильных групп в полимере полиакрилонитриле или
его сополимере можно определять этим же методом, внеся лишь изме-
нение в предварительную стадию. В случае полимера нет необходимости
в обработке металлическим натрием.
Определение акрилонитрила в "полимере, в дисперсиях, отгонах. Так
же как при определении акрилонитрила в реактиве, в основу метода по-
15>
ложена реакция акрилонитрила с сернистокислым натрием с последую-
щим титрованием выделяющейся щелочи раствором соляной кислоты
потенциометрическим методом или обычным титрованием в присутствии
тимолфталеина.
В две конические колбы вносят пипеткой 25 мл раствора Na2SO3 и
5—Ю г дисперсии (маточного раствора, отгона), взвешенной с точ-
ностью до 0,01 г. Смеси выдерживают 30 мин при комнатной темпера-
туре, периодически перемешивая. Титрование проб производят 0,1 н.
раствором НС1 в присутствии 2—3 капель тимолфталеина до исчезно-
вения синей окраски. В аналогичных условиях титруют контрольную
пробу. Содержание акрилонитрила х (в %) рассчитывают по формуле
д/<-0,005306-100
—-—1-----------
g
здесь а — объем 0,1 и. НС1, пошедший на титрование пробы с веществом,
мл; К— поправка к 0,1 н. раствору НС1; 0,005306 — количество акрило-
нитрила, соответствующее 1 мл точно 0,1 н. раствора НС1, г; g— навеска
пробы, г;
Проведение потенциометрического титрования см. стр. 157.
Это же определение можно осуществить и другими методами: коло-
риметрическим и полярографическим. Последний дает возможность раз-
дельно определить концентрацию нескольких мономеров, в то время как
потенциометрический не позволяет отделить акрилонитрил от других
производных акриловой и метакриловой кислот [5].
Определение примесей в акрилонитриле. В качестве примесей в ак-
рилонитриле всегда присутствуют альдегиды. Определить их количество
можно оксимированием по следующей реакции:
R—С^° + NH2OH-HC1 -> R-C^N°H 4- NCI + Н2О
ХН ХН
В две конические колбы емкостью 250 мл берут навески акрило-
нитрила (около 5 г), взвешенного с точностью 0,01 г, заливают их 20 мл
7%-нбго спиртового раствора солянокислого гидроксиламина, перемеши-
вают и оставляют на 1 ч. Затем приливают 100 мл воды, 4—5 капель рас-
твора метилового оранжевого и титруют образовавшуюся в результате
реакции соляную кислоту 0,1 н. раствором NaOH. Оттитровывают конт-
рольную пробу.
Содержание альдегидов х (в %) в пересчете на карбонильную груп-
пу рассчитывают по формуле
(а — fc)/<0,0028-100
g
где а и Ь — объемы 0,1 н. раствора NaOH, пошедшие на титрование конт-
рольной пробы и пробы с навеской, мл; К — поправочный коэффициент
0,1 н. раствора NaOH; 0,0028 — коэффициент пересчета на карбониль-
ную группу, соответствующий 1 мл точно 0,1 н. раствора NaOH; g—
навеска вещества, г.
Определение содержания амида метакриловой кислоты. В чистый
•сухой взвешенный бюкс емкостью 10 мл отбирают 5—6 г исследуемого
160
вещества. Бюкс с веществом охлаждают до комнатной температуры и
взвешивают на аналитических весах с точностью до 0,01 г. Содержание
бюкса через воронку переносят в мерную колбу емкостью 250 мл. Бюкс
и воронку тщательно ополаскивают дистиллированной водой и промыв-
ные воды сливают в ту же колбу, объем жидкости доводят до метки и
тщательно перемешивают содержимое колбы.
В две конические колбы с притертыми пробками емкостью по 250 мл
наливают из бюретки по 25 мл 0,1 н. бромид-броматной смеси (см.
стр. 155) пипеткой приливают по 10 мл испытуемого раствора, затем мер-
ным цилиндром в каждую колбу добавляют по 5 мл концентрированной
соляной кислоты. Быстро закрывают колбы пробками, хорошо взбалты-
вают и оставляют на 5 мин в темноте. Затем мерным цилиндром быстро
вливают по 5 мл 10%-ного раствора йодистого калия, приняв меры'пре-
досторожности против потерь брома. Выделившийся иод оттитровывают
0,1 н. раствором гипосульфита до обесцвечивания раствора, добавив- в
качестве индикатора несколько капель 2%-ного раствора крахмала.
Параллельно ставят контрольный опыт.
Содержание амида х (в %) рассчитывают по формуле
_(а — Ь) /<0,00425-250.100
gio
где а — объем 0,1 н. раствора гипосульфита, израсходованный на титро-
вание контрольной пробы, мл; b — объем 0,1 н. раствора гипосульфита,
израсходованный на титрование пробы с навеской, мл; К — поправка на
нормальность 0,1 н. раствора гипосульфита; g — навеска исследуемого
вещества, г; 0,00425 — количество амида, соответствующее 1 мл 0,1 н. ги-
посульфита, г.
Техника безопасности при работе
с акрилатами и метакрилатами
Метилакрилат. При вдыхании паров метилового эфира акриловой
кислоты разрушается слизистая оболочка дыхательных путей. Длитель-
ное воздействие метилакрилата на организм, вызывающее острое от-
равление, сопровождается разрушением нервной ткани, внутренним кро-
воизлиянием и патологическими изменениями внутренних органов.
Попадание жидкого мономера на кожу вызывает ее раздражение,
а попадание в глаза — раздражение конъюнктивы.
Содержание паров метилакрилата в воздухе рабочих помещений
свыше 25 мг/л вызывает обильное выделение мокроты, что уже служит
признаком недопустимых концентраций.
Учитывая сильное и опасное действие метилакрилата на организм*
а также острый неприятный запах, работы с метилакрилатом следует
проводить под тягой и с соблюдением соответствующих мер предосто-
рожности.
Мономерный метилакрилат хранят в присутствии ингибитора: гид-
рохинона (от 0,25 до 1,0% от веса мономера) или пикриновой кислоты —
наиболее эффективного ингибитора.
6—1453 16Т
Перегнанный и полностью очищенный от ингибитора мономер хра-
нят при температуре не выше 5° С, так как чистый эфир без ингибито-
ра при комнатной температуре способен быстро полимеризоваться.
Полимеризация в отсутствие следов кислорода иногда проходит со
взрывом.
Метилметакрилат — горючий, легко воспламеняющийся продукт с
очень низкой температурой вспышки (10°С), поэтому при работе с ним
необходимо тщательно соблюдать правила противопожарной безопасно-
сти. В рабочем помещении обеспечивается хорошая вентиляция, не дол-
жно быть открытого пламени и искрящего оборудования, так как с воз-
духом метилметакрилат образует взрывчатые смеси. Взрывоопасная
концентрация его в воздухе составляет 4,99—12,5 объемн. %.
Пары метилметакрилата менее токсичны, чем этилацетат, и более
токсичны, чем ацетон. Первый признак острого отравления — угнетенное .
состояние, после чего наступает нарушение дыхания, раздражение сли-
зистой оболочки глаз.
Местное действие жидкого мономера на кожу оказывает временное
слабое раздражение.
Высшие эфиры метакриловой кислоты (этил, аллил- и бутилмета-
крилаты) вызывают токсическое действие, сходное с действием метило-
вого эфира, но более слабое.
Акрилонитрил при нормальных условиях — жидкость, поэтому он мо-
жет безопасно храниться в сосудах, предназначенных для обычных го-
рючих веществ, например бензина, спирта и пр.
При хранении и работе с акрилонитрилом особенно необходимо
иметь в виду такие его свойства, как легкая воспламеняемость, неустой-
чивость в присутствии перекисей и токсичность. Максимально допусти-
мой концентрацией паров акрилонитрила в атмосфере рабочих помеще-
ний при ежедневном воздействии без вреда на организм считается 0,02%.
Однако следует избегать действия паров акрилонитрила на организм
даже при такой концентрации. Присутствие небольшого количества ак-
рилонитрила в воздухе обнаруживается по запаху, сходному с запахом
пиридина в разбавленном растворе. Необходимо избегать попадания
жидкого мономера на кожу.
При длительном хранении акрилонитрила без ингибиторов необхо-
димо иметь в виду его склонность к полимеризации, хотя она происхо-
дит только в присутствии перекисей, которые при действии кислорода
воздуха обычно не образуются, однако даже небольшое количество за-
грязнений в мономере может легко привести к их образованию. Образу-
ющийся полимер выпадает на дно сосуда, загрязняя продукт.
Низкая температура при хранении (около 0°) практически полно-
стью предотвращает полимеризацию.
Чтобы свести к минимуму образование перекисей вследствие воз-
можного окисления содержащихся в мономере загрязнений, сосуды, в
которых хранится акрилонитрил, следует заливать возможно полнее для
уменьшения свободного воздушного пространства. Во избежание взрыва
большие резервуары, предназначенные для хранения мономера, перед
наполнением должны быть вычищены, высушены и воздух из них вытес-
нен инертным газом.
162
Акрилонитрил образует с воздухом взрывчатые смеси. Нижний пр.
дел взрывоопасной смеси составляет 3,05 объемн. %, верхний *
17,5 объемн. %-
РАБОТА 59. ПОЛУЧЕНИЕ АКРИЛОВОЙ КИСЛОТЫ
ИЗ ЭТИЛЕНЦИАНГИДРИНА
Исходные продукты: этиленциангидрин— 71 г; серная кислота (р=1,84)—98
ацетон — 140 г; гидрохинон — 0.5 г; медная фольга — 2 г. Г1
Оборудование: четырехгорлая колба, снабженная мешалкой с герметическим
ТЕором, обратным холодильником, капельной воронкой и термометром (см. рис. 4^?*
баня с холодной водой; прибор для перегонки в вакууме, делительная воронка.
В четырехгорлую колбу загружают этиленциангидрин и около
медной фольги. При перемешивании и охлаждении колбы смесью ль г
с солью из капельной воронки в течение получаса прибавляют концецу'
рированную серную кислоту. После этого в колбу добавляют 18 мл вод^ы*
пускают воду в холодильник и смесь осторожно нагревают на водян^.’
бане. При 80° С начинается энергичная реакция, в результате котор.^
температура может повыситься до 130—140° С. Реакция идет 16 ч пу\„
95° С. Затем реакционную смесь охлаждают и из полутвердой мас^
кристаллов бисульфата аммония экстрагируют в делительной ворон^
акриловую кислоту ацетоном, содержащим 0,5 г гидрохинона. Ацет^н
отгоняют, а чистую акриловую кислоту выделяют фракционной пе^е_
гонкой под вакуумом. Получают продукт с т. кип. 132—141° С. Выход <уо_
ставляет 68% от теоретического.
РАБОТА 60. ПОЛУЧЕНИЕ АКРИЛОВОЙ КИСЛОТЫ
ИЗ МЕТИЛАКРИЛАТА
Наиболее удобным методом получения акриловой кислоты в лабо-
раторных условиях является взаимодействие муравьиной кислоты с ь^е_
типовым эфиром акриловой кислоты в присутствии серной кислоты в
честве катализатора.
Реакция идет по схеме
СН2=СНСООСН3 + НСООН -> СН2=СНСООН + НСООСНз
Исходные продукты: метиловый эфир акриловой кислоты — 516 г; муравьицдя
кислота — 92 г; гидрохинон—15 г; серная кислота (концентрированная)1 мл.
Оборудование: круглодонная колба емкостью 1 л, снабженная дефлегматором $
сотой 100 см с регулируемым отбором дистиллята.
В метиловом эфире акриловой кислоты растворяют гидрохинон и
раствор загружают в круглодонную колбу, снабженную дефлегматор^
с регулируемым отбором дистиллята. Затем туда же вводят муравьи-
ную кислоту и концентрированную серную кислоту (р=1,84 г/сму
Смесь кипятят до тех пор, пока температура в верхней части дефлегма-
тора не понизится до 32° С (температура кипения метилового эфира му-
равьиной кислоты). Образующийся метиловый эфир муравьиной кисл0_
6* Чз
ты отгоняют, на что требуется 8—12 ч. Когда образование эфира прекра-
тится, избыток метилового эфира акриловой кислоты отгоняют под
вакуумом (140 мм рт. ст.) при 35° С. Затем выделяют акриловую кисло-
ту, собирая фракцию, кипящую при 56° С и 25 мм рт. ст.
Выход продукта составляет около 88% от теоретического.
Очистка акриловой кислоты. Акриловая кислота очищается пере-
гонкой под вакуумом (см. табл. 8), которая ведется в колбе Кляйзена
в присутствии стабилизатора гидрохинона, добавляемого в акриловую
кислоту в количестве 5% от веса перегоняемого продукта.
ТАБЛИЦА 8
Зависимость температуры кипения чистой акриловой кислоты от давления
Давление, мм рт. ст, Т. кип., °C Давление, мм рт. ст. Т. кип., °C
760 141 15 48,5
753 140 14 42
249 100
50 71 12 39,5-40,2
30 70 7,7 20
РАБОТА 61. ПОЛУЧЕНИЕ МЕТАКРИЛОВОЙ КИСЛОТЫ
ИЗ АЦЕТОНЦИАНГИДРИНА
Реакция протекает по схеме
ОН ОН
I +hsso4 I --->
СНз-C-CN------->-СН3—С—C=NH -н.О
I I I
СН3 СН3 OSO3H
СН,=С—C=NH . +Hg% сн2=с—c-,nh2
I I -H.SO, 2 I ||
СН3 OSO3H сн3 о
+н,о
------> СН2=С—С-ONI ц —сн2=с—соон
I II -NHa I
сн3 о сн3
Исходные продукты: ацетонциангидрин — 200 мл; серная кислота (пл. 1,84) —
200 мл; гидрохинон—0,84 г; медная стружка.
Оборудование: четырехгорлая колба емкостью 1 л, снабженная мешалкой с герме-
тическим затвором, капельной воронкой, обратным холодильником и термометром; при-
бор для перегонки в вакууме.
Получение амида метакриловой кислоты (дегидратация ацетонци-
ангидрина). В колбу прибора (см. рис. 44) при 25° С загружают сер-
ную кислоту (моногидрат) и при работающей мешалке и охлаждении
медленно, по каплям, в течение 1 —1,5 ч через капельную воронку вводят
ацетонциангидрин. Подачу ацетонциангидрина регулируют в зависимо-
сти от температуры реакционной смеси, которая должна быть около 80—-
164
По окончании загрузки всего ацетонциангидрина охлаждение колбы
прекращают и начинают ее медленно, в течение 20 мин, нагревать до
90—110° С. При этой температуре смесь выдерживают 25 мин, после че-
го ее нагревают до 125—130° С и выдерживают при этой температуре
30 мин. Затем нагрев прекращают и берут первую пробу на амид. Если
содержание амида в этой пробе ниже 26%, то нагрев продолжают еще
15 мин и берут вторую пробу на амид.
. Независимо от результатов анализа второй пробы нагрев прекра-
щают и реакционную смесь в колбе охлаждают с помощью водяной бани
до 60—70° С. Содержание амида не ниже 27—29% указывает на пра*
вильное течение реакций дегидратации, при этом выход метакриловой
кислоты будет в пределах нормы.
Омыление амида. Амид метакриловой кислоты оМыляют водой в том
же сосуде, в котором дегидратировали ацетонциангидрин.
В колбу, подогретую до 80° С, при работающей мешалке и включен-
ном обратном холодильнике вводят 106 мл воды и добавляют 0,84 г
гидрохинона. Затем температуру смеси повышают до 100° С и при этой
температуре процесс омыления ведут в течение 3 ч.
Отгонка метакриловой кислоты. По окончании процесса омыления
реакционную смесь охлаждают до 70° С и образовавшуюся метакрило-
вую кислоту отгоняют под вакуумом при остаточном давлении 300—
350 мм рт. ст., в процессе отгонки температуру повышают до 125° С. От-
гоняется 180—200 мл метакриловой кислоты-сырца со средним содержа-
нием метакриловой кислоты около 50%.
Полученную сырую метакриловую кислоту хранят до ее ректифика-
ции в присутствии медных стружек (0,5—1,0 г).
Очистка сырой метакриловой кислоты. Окончательно метакриловую
кислоту-сырец очищают перегонкой под вакуумом.
В колбу Кляйзена с елочным дефлегматором загружают полученную
сырую метакриловую кислоту, добавляют в качестве ингибитора около
1 г медной стружки и отгоняют ее под вакуумом при остаточном давле-
нии не более 100 мм рт. ст.
Первая фракция (головная) может быть отогнана без вакуума при
температуре до 100° С. Ее собирают отдельно. Содержание метакриловой
кислоты в этой фракции составляет 15—20%-
Вторая фракция, представляющая собой чистую метакриловую кис-
лоту (90—100%-ную), отбирается под вакуумом. Чистую метакриловую
кислоту хранят при температуре не выше 10° С.
РАБОТА 62. ПОЛУЧЕНИЕ МЕТАКРИЛОВОЙ КИСЛОТЫ
ИЗ МЕТИЛМЕТАКРИЛАТА (ОМЫЛЕНИЕМ)
Исходные продукты: метилметакрилат —100 мл; раствор едкого иатра (40%-ной
концентрации) — 75 мл; спирт этиловый — 7—9 мл; вода — 60 мл; гидрохинон; пова-
ренная соль; соляная кислота (р=1,19 г/см3).
Оборудование: четырехгорлая колба емкостью 0,75 л, снабженная мешалкой с гер-
метическим затвором, капельной воронкой, обратным холодильником и термометром;
водяная баня; делительная воронка; прибор для перегонки в вакууме.
6*—1453 165
В реакционную колбу прибора загружают метилметакрилат, воду к
спирт. Включают мешалку, пускают воду в холодильник и при постоян-
ном перемешивании из капельной воронки тонкой струей вливают 75 мм.
40%-ного раствора едкого натра. Перемешивание не прекращают до-
окончания реакции.
Начало омыления эфира сопровождается повышением температуры-
смеси до 80—85° С. При повышении температуры выше 80° С смесь не-
обходимо охладить, опустив колбу в баню с холодной водой. Реакция
продолжается 2—3 ч. Окончание ее определяют по исчезновению запаха-
эфира и появлению запаха метилового спирта.
В полученный раствор натриевой соли метакриловой кислоты до-
бавляют 5—10 г поваренной соли и при продолжающемся перемешива-
нии и охлаждении колбы в бане с холодной водой в смесь вводят не-
большими порциями из капельной воронки 90 г концентрированной со-
ляной кислоты, в которой растворено 0,2 г гидрохинона.
При повышении температуры реакционной смеси выше 30—35° С
следует прекратить подачу соляной кислоты и охладить смесь до 10—
14° С, после чего можно снова добавлять соляную кислоту. Окончание
реакции контролируется индикатором конго красным.
После полного разложения соли метакриловой кислоты полученный,
продукт переносят в делительную воронку и отстаивают 20—30 мин.
Верхний слой, представляющий собой сырую метакриловую кислоту,,
осторожно сливают и фильтруют. Добавление поваренной соли способ-
ствует ускорению отделения метакриловой кислоты от раствора мине-
ральных солей.
Очистка метакриловой кислоты. Метакриловую кислоту-сырец очи-
щают перегонкой в колбе Кляйзена, снабженной дефлегматором и пря-
мым холодильником. Перед разгонкой в сырой продукт, содержащий
50—60% метакриловой кислоты, вводят медную стружку из расчета
0,5 г на 100 г сырца.
Продукт разгоняют на три фракции: первая, кипящая до 108° С при-
нормальном давлении, отбрасывается; вторая отгоняется при давлении;
не более 200 мм рт. ст. Третья фракция, отгоняющаяся при давлении
не более 12 мм рт. ст. и температуре 60—63° С, представляет собой 98—
100%-ную метакриловую кислоту.
Повторной разгонкой второй фракции можно выделить еще некото-
рое количество чистого продукта.
РАБОТА 63. ПОЛУЧЕНИЕ МЕТИЛОВОГО ЭФИРА
МЕТАКРИЛОВОЙ КИСЛОТЫ
Исходные продукты: ацетонциангидрин — 200 мл; сериал кислота (р=1,84 г/см8)—
195 мл; метиловый спирт—140 мл; гидрохинон — 2 г; безводный сульфат натрия—-
40 г.
Оборудование: четырехгорлая колба емкостью 1 л, снабженная мешалкой с герме-
тическим затвором, капельной воронкой, обратным холодильником и термометром.
К нагретому до 75° С в четырехгорлой колбе ацетонциангидрину?
при перемешивании прибавляют сначала серную кислоту, а затем мети--
166
ловый спирт и гидрохинон (в качестве ингибитора полимеризации). Пос-
ле этого температуру повышают до 80—100° С. К продуктам реакции
добавляют безводный сульфат натрия. Смесь подвергают перегонке в
приборе для перегонки и собирают эфир-сырец при 90—125° С.
Очистка эфира-сырца. Полученный эфир-сырец промывают в дели-
тельной воронке 200 мл дистиллированной воды, слегка подкисленной
серной кислотой. После двух промывок водой в сырой эфир добавляют
соду для нейтрализации и после фильтрования перегоняют в колбе с
дефлегматором. Для предотвращения полимеризации .во время перегон-
ки в колбу необходимо добавить гидрохинон или несколько кусочков
медной проволоки. Перегонную колбу нагревают в специальном'элект-
рическом колбонагревателе или на электрической плитке, закрытой ли-
стом асбеста.
Перегоняющийся метилметакрилат собирают по фракциям. Первую
фракцию, кипящую до 98° С, собирают отдельно. Вторую фракцию, ки-
пящую в интервале 98—100,5° С, собирают в тарированную чистую сухую
посуду. Третья фракция, кипящая при температуре выше 100,5° С, часто
содержит возгоняющийся стабилизатор.
Первую и третью фракции отбрасывают, а вторую фракцию, являю-
щуюся чистым продуктом, анализируют и используют для полимериза-
ции любым из описанных ниже методов.
РАБОТА 64. ПОЛУЧЕНИЕ БУТИЛМЕТАКРИЛАТА
ПЕРЕЭТЕРИФИКАЦИЕЙ МЕТИЛМЕТАКРИЛАТА
В лабораторных условиях бутиловый эфир метакриловой кислоты
удобно получать переэтерификацией метилового эфира .метакриловой
кислоты н-бутиловым спиртом по схеме
СН2=С—СООСНз + С4Н9ОН -> СН2=С—СООС4Н9 + СН3ОН
СНз СНз
Исходные продукты: метилметакрилат — 40Q г (4 моля); м-бутиловый спирт —
148,4 г (2 моля); гидрохинон — 8 г; серная кислота (х. ч., р=1,84 г/см3) —2,5 мл.
Оборудование: круглодонная колба емкостью 1—1,5 л, снабженная стеклянной ко-
лонкой типа елочного дефлегматора, насадкой, термометром, обратным шариковым
холодильником, прямым нисходящим холодильником и стеклянным краном между холо-
дильниками (рис. 46); баня, наполненная полиэтилсилоксановой жидкостью № 5; прибор
для фракционной вакуумной разгонки; склянки Тищенко или Дрекселя; скляики из тем-
ного стекла.
В круглодонную колбу прибора загружают перегнанный «-бутило-
вый спирт (фракция с т. кип. 114—118°), метиловый эфир метакриловой
кислоты, гидрохинон и серную кислоту.
Реакционную жидкость нагревают на полиэтилсилоксановой бане
при кипении 8—10 ч. Колонка работает с.закрытым краном и с полным
возвратом дистиллята до тех пор, пока температура в парах не снизится
до 63—65° С, т. е. до температуры кипения азеотропной смеси метиловый
спирт — метиловый эфир метакриловой кислоты.
g**
167
L
После этого кран открывают и образующуюся смесь отгоняют, при-
чем следят, чтобы температура в парах не поднималась выше 67—68° С.
После отгонки азеотропной смеси оставшийся в колбе продукт реакции
подвергают фракционной вакуум-разгонке в токе двуокиси углерода или
азота.
Очистка бутилметакрилата. Полученный технический бутиловый
эфир метакриловой кислоты (450—500 г) загружают в круглодонную
колбу, снабженную елочным дефлегматором высотой 50 см, прямым
Рис. 46. Прибор для синтеза
бутилметакрилата переэтери-
фикацией метилметакрилата
холодильником, термометром и приемни-
ком. В колбу вставляют также толстостен-
ный капилляр, через который во время
разгонки пропускают двуокись углерода
или азот. Инертный газ предварительно
должен проходить через две склянки Вуль-
фа или Тищенко с концентрированной сер-
ной кислотой для сушки и через пустую
предохранительную склянку.
Вначале отгоняют головную фракцию,
кипящую до 95° С при 300-—400 .мм рт. ст.
Затем собирают фракции с т. кип. 40—53° С
при 29 мм рт. ст. и с т. кип. 52—54° С при
12—.13 мм рт. ст.
Выход второй (основной) фракции —
около 131,5 г, что составляет 23,0% от тео-
ретического.
Разогнанный бутиловый эфир метакри-
ловой кислоты хранят на холоду в склян-
ках из темного стекла с притертыми проб-
ками, которые заливают парафином, или в
запаянных ампулах. Во избежание полиме-
ризации в эфир вносят несколько кристал-
ликов гидрохинона. Не вступивший в реак-
цию метиловый эфир метакриловой кисло-
ты может быть регенерирован из головной
фракции.
РАБОТА 65. ПОЛУЧЕНИЕ АЛЛИЛМЕТАКРИЛАТА
Аллиловый эфир метакриловой кислоты в лабораторных условиях
удобно получать путем переэтерификации метилового эфира метакрило-
вой кислоты аллиловым спиртом. Реакция идет по схеме
СН2=С—СООСНз + НОСН2-СН=СН2 -> СН2=С—СООСН2—СН=СН2 + СНзОН
СН3 СНз
Исходные продукты: метилметакрилат — 218 г; аллиловый спирт — 67 г; бензол —
225 г; гидрохинон— 112 г; серная кислота (р= 1,84 г/см3) —5 г.
Оборудование: круглодонная колба емкостью 1 л, снабженная елочным дефлег-
матором с термометром и прямым холодильником; делительная воронка; колба Вюрца
емкостью 0,5 л; колбы-приемники — 3 шт.
168
В круглодонную колбу прибора (см. рис. 46) загружают аллиловый
спирт, бензол и метилметакрилат, в котором предварительно растворен
гидрохинон. При взбалтывании и охлаждении в смесь небольшими пор-
циями вносят серную кислоту и нагревают ib течение часащри 50° С, пос-
ле этого температуру повышают до 60—65° С (температуру измеряют в
парах термометром, установленным в дефлегматоре). При этой темпе-
ратуре отгоняется образующийся в процессе переэтерификации метило-
вый спирт.
После отгонки всего метилового спирта температуру в парах по-
вышают до 80—85° С. При этом отгоняется в основном бензол, который
собирают в отдельный приемник. Оставшуюся в колбе смесь переносят
в делительную воронку и несколько раз обрабатывают раствором щелочи
для удаления серной кислоты и гидрохинона, затем раствором соды и,
наконец, промывают дистиллированной водой до нейтральной реакции
промывных вод. Образуется стойкая труднорасслаивающаяся эмульсия
коричневого цвета, для разделения которой требуется несколько часов
отстаивания.
Промытую нейтральную жидкость сушат безводным сульфатом нат-
рия, затем переносят в колбу Вюрца и перегоняют. Головная фракция,
кипящая ниже 120° С, содержащая остатки бензола и непрореагировав-
ший метилметакрилат, отбрасывается. Вторую фракцию, кипящую в ин-
тервале 120—144° С, собирают отдельно, еще раз высушивают с безвод-
ным сульфатом натрия и разгоняют под вакуумом.
Фракцию продукта, отбираемую при 59—66° С и 43 мм рт. ст.,
окончательно очищают повторной перегонкой при давлении 43 мм рт. ст.
и собирают продукт, кипящий при 62—63° С.
Полученный аллиловый эфир метакриловой кислоты должен иметь
следующие показатели: т. кип. (при 43 мм рт. ст.)— 62—63° С; dlo =
=0,9342; п.о —1,4385; бромное число 239,5 — 241,2; молекулярная реф-
ракция АГтеор — 35,245; молекулярная рефракция 7ИПракт— 34,81.
Степень чистоты аллилметакрилата определяют по относителвной
плотности, показателю преломления и бромному числу. Бромное число
определяют с бромид-броматным раствором (см. стр. 155).
РАБОТА 66. ПОЛУЧЕНИЕ ДИМЕТАКРИЛАТА ЭТИЛЕНГЛИКОЛЯ
Диметакрилат этиленгликоля получается при взаимодействии эти-
ленгликоля с метакриловой кислотой. Реакция идет по схеме
О СН3
О || |
СН2ОН || СН2-О-С—С=СН2
I +CH2=C-C-OH-> | +2Н2О
СН2ОН | сн2—о—с—с=сн2
Исходные продукты: метакриловая кислота—104,2 г (1,2 моля); этиленгликоль —
31,03 г(0,5 моля); серная кислота (р=1,84 г/см3)—2 г; гидрохинон — 25 г; бензол —
50 мл.
169
Оборудование: четырехгорлая колба с мешалкой, герметическим затвором, спе-
циальной ловушкой с краном и обратным холодильником, трубкой для подачи инерт-
ного газа (рис. 47).
В четырехгорлую колбу прибора загружают свежеперегнанную ме-
такриловую кислоту и перегнанный под вакуумом этиленгликоль. После
Рис. 47. Прибор для
проведения реакции с
перемешиванием в то-
ке инертного газа с ло-
вушкой Дина — Стар-
ка
нескольких минут перемешивания, не останавли-
вая мешалки, вводят в смесь концентрирован*
ную серную кислоту, гидрохинон и обезвожен-
ный бензол. После этого колбу нагревают с об-
ратным холодильником 2 ч при 97—103° С. Вы-
деляющаяся в результате взаимодействия эти-
ленгликоля с метакриловой кислотой вода не-
прерывно отгоняется вместе с бензолом и соби-
рается в специальной ловушке. Об окончании
реакции судят по прекращению отделения воды,
количество которой должно составлять около
17,5 мл. Реакция ведется при нерерывном про-
пускании в колбу тока сухого азота. По оконча-
нии реакции смесь охлаждают в токе азота, а
затем обрабатывают сухим углекислым натрием
для нейтрализации серной кислоты и фильтруют.
Фильтрат высушивают в течение суток над про-
каленным хлористым кальцием или безводным
сульфатом натрия и перегоняют в вакууме в то-
ке азота.
Фракцию, кипящую в интервале 98—100° С
(при 4—5 мм рт. ст.), собирают как чистый про-
дукт, выход которого составляет 55—60% в пе-
ресчете на исходный этиленгликоль.
РАБОТА 67. ПОЛУЧЕНИЕ ДИМЕТАКРИЛАТА ДИЭТИЛЕНГЛИКОЛЯ
Диметакрилат диэтиленгликоля в лабораторных условиях можно
получать из метилметакрилата и диэтиленгликоля методом переэтерифи-
кации. Реакция идет по схеме
НОСН2СН2-0-СН2СН2ОН + 2СН2=С—СООСНз ->
I
СНз
сн2=с—С—(—О—СН2СН2—)2—О—С—С=СН2 + 2СН3ОН
I II II I
СНз О О СН3
Исходные продукты: метилметакрилат 500 г (5 молей); диэтилеигликоль—106 г
(1 моль); серная кислота (р=1,84 г/см3)—3 мл; гидрохинон — 25 г; бикарбонат нат-
рия — 6 г.
Оборудование: круглодонная колба емкостью 1 л, снабженная елочным дефлегма-
тором длиной 1 м с термометром и прямым холодильником; приемник для эфира; во-
ронка Бюхнера; прибор для фракционной перегонки в вакууме.
В круглодонную колбу прибора (см. рис. 46) загружают указанные
количества метилметакрилата, диэтиленгликоля, серной кислоты и гид-
170
рохинона и после тщательного перемешивания нагревают 5-—6 ч при
118—120° С. Выделяющийся в результате взаимодействия диэтиленгли-
коля с метилметакрилатом метиловый спирт непрерывно отгоняется вме-
сте с избыточным метилметакрилатом в виде азеотропной смеси, ки-
пящей при 65° С (показатель преломления азеотропной смеси л ™ ==
= 1,3720).
После отгонки 120 мл азеотропной смеси синтез заканчивают. Кол-
ьбу с реакционной смесью охлаждают, нейтрализуют сухим гидрокарбо-
натом натрия, фильтруют на воронке Бюхнера, сушат прокаленным хло-
ристым кальцием и разгоняют под вакуумом. Фракцию диметакрилата
диэтиленгликоля, кипящую в интервале 150—152° С при 7—8 мм рт. ст.,
собирают как чистый продукт. Выход его составляет 145 г (60% от тео-
ретического) .
По этой же методике можно получить диметакрилаты этиленгликоля
и триэтиленгликоля. Этиленгликоля берут 62 г, а триэтиленгликоля —
150 г. . •
При получении диметакрилата этиленгликоля собирают фракцию,
кипящую в интервале 98—100° С при 4—5 мм рт. ст., а при получении
диметакрилата триэтиленгликоля — фракцию, кипящую в интервале
160—161° С при 5 мм рт. ст.
РАБОТА 68. ПОЛУЧЕНИЕ АКРИЛОНИТРИЛА
В промышленности акрилонитрил получают либо из окиси этилена
через этиленциангидрин, либо прямым синтезом из ацетилена и циани-
стого водорода:
СН=СН + HCN -> CH2=CHCN
В лабораторных условиях акрилонитрил удобнее получать дегидра-
тацией этиленциангидрина:
—н,о
НОСН2—CH2CN----> CH2=CHCN
Исходные продукты: этиленциангидрин — 50 г, порошкообразное олово — б г; про-
каленный хлористый кальций.
Оборудование: круглодонная колба емкостью 0,25 л с прямым холодильником;
водяная баня; прибор для перегонки; делительная воронка; колбы-приемники — 2 шт.
Этиленциангидрин дегидратируют нагреванием с порошкообразным
оловом *, которого берут 10%, от веса этиленциангидрина. Реакцию ве-
дут на водяной бане в круглодонной колбе с прямым холодильником
одновременно с отгонкой воды.
Отогнанный продукт, состоящий из акрилонитрила и воды, разде-
ляют в делительной воронке и высушивают над едким кали или хлори-
стым кальцием. Выход акрилонитрила составляет 75—85%.
* Порошкообразное олово получают действием на концентрированный водный рас-
твор двухлористого олова цинковой пылью. Осадок обрабатывают уксусной кислотой
для растворения непрореагировавшего цинка, отфильтровывают, промывают спиртом
и высушивают.
171
Для получения чистого продукта акрилонитрил очищают простой
перегонкой. Если необходима особо тщательная очистка с удалением,
следов ингибитора, то продукт вначале промывают разбавленной сер-
ной кислотой или щелочью (в зависимости от природы ингибитора), за-
тем сушат хлористым кальцием, после чего мономер перегоняют под
небольшим вакуумом или без него, собирая фракцию, кипящую при
77,3°С (±0,1°).
Во время промывки надо следить за хорошим расслаиванием про-
мывных вод и мономера, так как акрилонитрил растворим в воде (до
7%) и могут быть большие потери его с промывными водами.
РАБОТА 69. ПОЛУЧЕНИЕ ПОЛИМЕРА АКРИЛОВОЙ КИСЛОТЫ
ПОЛИМЕРИЗАЦИЕЙ В РАСТВОРИТЕЛЕ (В ВОДЕ]
•он
пСН2=СН--->
I
соон
—сн2-сн—
соон
Исходные продукты: акриловая кислота — 72 г (1 моль), 100%-ная перекись водо-
рода— 1 г (0,03 моля). (30%-ная перекись водорода — 3,4 г); дистиллированная
вода — 240 мл.
Оборудование: ампулы из тугоплавкого стекла; термошкаф.
В дистиллированной воде растворяют перекись водорода и акрило-
вую кислоту, заливают раствор в чистые сухие ампулы из прочного ту-
гоплавкого стекла, заполнив их на половину. Узкий конец ампул запаи-
вают на паяльной горелке и ставят их в термошкаф для полимеризации,
которую ведут 11 дней при 100° С. Затем термошкаф выключают, ампу-
лам дают охладиться в шкафу до комнатной температуры, после чего их
осторожно вскрывают. Полученная вязкая жидкость представляет собой
раствор полимера акриловой кислоты в воде.
Из полученных высоковязких растворов полимер может быть вы-
делен путем выпаривания воды в вакууме. Определяют кислотное число
полученного продукта и рассчитывают концентрацию полимера. Поли-
мер может быть использован в качестве эмульгатора или водораство-
римого ионита.
По этой же методике получают полимер метакриловой кислоты; оп-
ределяют его кислотное число и используют для тех же целей, что и по-
лиакриловую кислоту.
РАБОТА 70. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
ПОЛИМЕРИЗАЦИЕЙ В БЛОКЕ
СНз
I [С.ЩСОО*]
пСН2=С ---------------»
I [C.Hg]
СООСНз
СН3 Л
I
—СН2—С—
I
СООСНз-1я
Исходные продукты: метилметакрилат — 1*00 г; перекись бензоила — 0,2 г; дибу-
тилфталат— 10 г.
172
Оборудование: трехгорлая колба, снабженная мешалкой с герметическим Затво-
ром, обратным холодильником и Термометром; водяная баня; стеклянные формы; термо-
шкаф «ли водяной термостат.
В трехгорлую колбу загружают метилметакрилат, в котором пред-
варительно растворены перекись бензоила и дибутилфталат. Включают
мешалку, пускают воду в холодильник и нагревают содержимое колбы
на водяной бане при 80—85° С до образования густого сиропообразного
продукта.
Полученный форполимер охлаждают, разливают в стеклянные фор-
мы и окончательно полимеризуют в термошкафу или водяном термоста-
те при 55—65° С до образования однородных, твердых, прозрачных об-
разцов органического стекла.
После окончания полимеризации формы размачивают в теплой во-
де (40° С) и вынимают готовые листы органического стекла.
Изготовление форм. Изготовляют 2—3 стеклянные формы размером 13X18 см
с величиной просвета 1—3 мм.
На хорошо очищенную, промытую спиртом сухую стеклянную пластинку уклады-
вают по углам четыре кусочка поливинилхлоридного пластиката размером 10X15 мм
соответствующей толщины, обернутые в целлофан или кальку. На эту пластинку сверху
кладут вторую чистую сухую стеклянную пластинку такого же размера так, чтобы края
обеих пластин совпали. Торцевые части пластинок оклеивают полосками кальки, про-
мазанной крахмальным или казеиновым клеем. Изготовленную стеклянную форму высу-
шивают в сушильном шкафу при температуре не выше 50° С, во избежание коробления
наклеенной кальки. Чтобы придать склеенной форме большую прочность и избежать
утечки мономера, после сушки на кальку наклеивают слой плотной (кабельной) бума-
ги и вновь высушивают форму при 50р С.
Проделав в форме небольшое отверстие, заливают в нее приготовленный для поли-
меризации сиропообразный полимер так, чтобы ои не доходил до верхнего края формы
на 1—1,5 см.
Примечание. Для получения окрашенного органического стекла перед началом на-
гревания в мономере растворяют около 0,07 г красителя (судановый красный или суда-
ковый оранжевый). Блочный полиметилметакрилат хорошо поддается поверхностному
окрашиванию спиртовыми растворами органических красителей при нагревании.
Так как блочный полиметилметакрилат хорошо склеивается, из него
можно изготовить различные изделия (цилиндры, трубы, коробки .ит.п.)
методом склеивания. Склеенные изделия имеют такую же прочность и
прозрачность, как и монолитный материал.
Для склеивания применяется полиметилметакрилатный клей, пред-
ставляющий собой 5%-ный раствор полиметилметакрилата в раствори-
теле (ледяной уксусной кислоте, дихлорэтане, муравьиной кислоте
и т. д.). Для склейки может применяться также один из растворителей —
дихлорэтан или ледяная уксусная кислота. Растворитель вызывает на-
бухание полимера и склеивание происходит в результате диффузии мак-
ромолекул, расположенных на склеиваемых поверхностях.
РАБОТА 71. ИЗГОТОВЛЕНИЕ НЕПРОЗРАЧНОГО ОРГАНИЧЕСКОГО
СТЕКЛА МОЛОЧНО-БЕЛОГО ЦВЕТА
Исходные продукты: метилметакрилат — 100 г; перекись бензоила — 0,5 г; поли-
стирол (эмульсионный) — 3,5 г.
173
Оборудование: водяная баня; колба емкостью 0,25 л; трехгорлая колба, снабжен-
ная мешалкой с герметическим затвором, обратным холодильником, термометром; фор-
мы стеклянные.
При слабом нагревании на водяной бане в метилметакрилате раст-
воряют полистирол, затем в раствор вводят перекись бензоила и загру-
жают приготовленный раствор в трехгорлую колбу и нагревают содер-
жимое колбы на водяной бане с обратным холодильником, перемеши-
вая непрерывно при 70—80° С до образования сиропообразного про-
дукта.
Полученный продукт охлаждают, разливают в формы (приготовле-
ние форм см. работу 70) и окончательно полимеризуют в термостате
при постепенном повышении температуры с 55 до 100° С в течение 8—
10 ч до получения твердых образцов органического стекла молочно-
белого цвета. Для извлечения органического стекла формы размачива-
ют в воде при 70° С в течение 0,5—1 ч и разнимают.
РАБОТА 72. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ЛИСТОВОГО
ОРГАНИЧЕСКОГО СТЕКЛА ВАКУУМ-ФОРМОВАНИЕМ
Листовое органическое стекло хорошо перерабатывается в изделия
•методом вакуумного формования. Этим методом можно изготовлять
изделия различной конфигурации, главным образом полусферы, гипер-
болоиды вращения и пр. Нагретый до размягчения лист органического
-стекла под действием вакуума подвергается вытяжке — формуется, при-
обретая конфигурацию, соответствующую профилю формы (см. стр. 96).
Вакуум-формование, можно производить и без формы (шаблона).
В этом случае получают изделия (полусферы или гиперболоиды враще-
ния) с гладкой поверхностью.
Установка для формования органического стекла при помощи ва-
куума (рис. 48) представляет собой цилиндрическую камеру, соединен-
ную с вакуум-насосом. Верхние
края камеры имеют фланец, на ко*
торый кладут лист размягченного
органического стекла и закрепляют
по краям прижимным кольцом.
В камере при помощи вакуум-насо*
са должен создаваться вакуум до
80—100 мм рт. ст.
Исходный продукт: листовое органиче-
ское стекло толщиной 2—3 мм.
Оборудование: вакуум-формовочная
установка.
Лист органического стекла опре-
деленного размера и соответству-
ющей формы нагревают в термо*
шкафу или с помощью ИК-ламп до
120—130° С. Размягченную заготов-
ку кладут на протяжное кольцо, по-
ли
•Рис. 48. Схема установки для вакуум-
формования листовых материалов:
J — вакуум-камера; 2 — световой луч: 3 фо-
тоэлемент; 4 — протяжное кольцо; 5 — прижим-
ное кольцо; 6 —• заготовка; 7 — ИК-лампы
Вакуум
-174
мешенное на фланец вакуум-формовочной установки; сверху’ наклады-
вают зажимное кольцо и заготовку закрепляют зажимами на бортах
цилиндрической камеры как можно плотнее, чтобы не было подсоса
воздуха после создания вакуума в камере.
После этого включают вакуум-насос и создают вакуум в камере
формовочной установки. Под действием атмосферного давления лист
органического стекла начинает вытягиваться и принимает сферическую
форму. Внутри камеры на определенной высоте устанавливают фото-
элемент. Как только формуемая деталь пересечет пучок света, срабаты-
вает фотореле и выключается вакуум-насос. Вследствие повышения дав-
ления в камере в связи с неполной герметичностью заготовка будет
подниматься за счет релаксации напряжений и луч снова попадет на
фотоэлемент, который, в свою очередь, включит вакуум-насос. Таким
образом удается поддерживать заданную глубину вытяжки с точностью
± 1 мм. Обычно к моменту полного оформления изделия лист органиче-
ского стекла остывает настолько, что теряет способность к дальнейшей
деформации. После остывания изделия вакуум выключают, камеру ва-
куум-формовочной установки соединяют с атмосферой и отформованное
изделие вынимают.
Для получения качественного изделия цикл формования должен
занимать не более 2 мин.
РАБОТА 73. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
В БЛОКЕ БЕЗ НАГРЕВАНИЯ
Для получения толстостенных блоков, совершенно свободных от
пузырей, трещин и других дефектов, а также при консервации различ-
ных предметов органического и минерального происхождения для со-
хранения их цвета и формы, полимеризацию метилметакрилата блоч-
ным методом проводят без нагревания.
Исходные продукты: метилметакрилат — 150 г; полиметилметакрилат (суспензион-
ный чистый)—20 г; перекись бензоила — 0,1 г; 2%-ный водный раствор щелочи —
500 мл; сульфат натрия безводный.
Оборудование: делительная воронка; прибор для перегонки в вакууме; трехгорлая
колба с мешалкой с герметическим затвором и обратным холодильником; водяная баия;
стеклянные формы любой конфигурации; термошкаф.
Очистка мономера. Для полимеризации без нагревания мономер
необходимо особенно тщательно очистить от содержащегося в нем ин-
гибитора. Перегонкой метилметакрилата при нормальном давлении не
всегда удается получить достаточно чистый продукт, так как возможно
попадание в дистиллят гидрохинона, применяемого обычно в качестве
ингибитора. Чтобы получить возможно более чистый мономер, перегон-
ку следует вести под вакуумом, а для полного удаления гидрохинона мо-
номер предварительно промывают 2—3 раза слабым раствором щелочи
(1—2%-ным). После этого продукт промывают дистиллированной водой
до нейтральной реакции промывных вод (по фенолфталеину). Затем
мономер высушивают безводным сульфатом натрия и перегоняют под
175
вакуумом. Перегонку ведут в колбе Кляйзена, снабженной елочными
дефлегматором, при нагревании на водяной бане.
Так как мономер под вакуумом перегоняется при низкой температу-
ре (т.кип.46°С при 100ммрт.ст. и 61°С при 200ммрт.ст.),а при боль-
шом разрежении перегонка может вестись даже при комнатной темпе-
ратуре, ингибитор не вводят. Первую фракцию отгона, не имеющую по-
стоянной температуры кипения при установленном не меняющемся раз-
режении или мутную от присутствия влаги, отбрасывают. Вторую фрак-
цию, кипящую при постоянной температуре при установленном разре-
жении, собирают в сухую чистую колбу, как чистый продукт, годный для
полимеризации. Остаток в колбе также отбрасывают.
У чистого мономера определяют показатель преломления. Доста-
точно чистый продукт может быть использован для блочной полимериза-
ции на холоду, в противном случае очистку необходимо повторить.
Полимеризация метилметакрилата. В 100 г очищенного мономера
вносят перекись бензоила и после его полного растворения отдельными
порциями добавляют 20 г полиметилмстакрилата, который растворяют
при перемешивании на холоду или при слабом нагревании на водяной
бане.
Полученный вязкий раствор после выделения пузырьков воздуха-
заливают в стеклянные формы и оставляют для полимеризации на рас-
сеянном дневном свету (на окне), поддерживая температуру около
20—25° С.
При этой температуре полимеризация продолжается 2—3 недели
до образования твердых блоков. Полученные изделия дополнительно*
выдерживают в термошкафу при 100° С в течение 1 ч.
Примечания. 1. Чтобы избежать усиления реакции, ведущей к образованию-
пузырьков в полимере, формы во время полимеризации следует предохранять от пря-
мого солнечного света, закрыв их листом белой бумаги.
2. Для получения окрашенных изделий в мономер до растворения в нем полимера
вводят краситель (судаковый красный, судаковый оранжевый и др.).
3. Для консервации в формы можно поместить высушенные цветы, листья, различ-
ных мелких насекомых (жуков, бабочек, предварительно обработанных спиртом) и дру-
гие предметы и, залив их приготовленным вязким раствором, провести полимеризацию.
4. Если при полимеризации предметы, подлежащие консервации, обесцвечиваются
вследствие воздействия на пигменты перекиси бензоила, ее не вводят в мономер И полу-
ченные блоки не подвергают дополнительному нагреванию в шкафу. В этом случае
полимеризацию на холоду проводят более длительное время.
РАБОТА 74. ПОЛУЧЕНИЕ ПОЛИЭТИЛМЕТАКРИЛАТА
ПОЛИМЕРИЗАЦИЕЙ В БЛОКЕ
СНз
I [С,Н5СОО"1
пСН2=С----------------->
I [С.Щ)
СООС2Н5
СН3 Я
I
-сн2-с—
I
COOC2H5J„
Исходные продукты: этилметакрилат—100 г; перекись бензола — 0,2 г; дибутил-
фталат — 5 г.
Оборудование: трехгорлая колба с обратным холодильником, снабженная механи-
ческой мешалкой и термометром.
176
В этилметакрилате растворяют перекись бензоила и дибутилфталат.
Раствор загружают в трехгорлую колбу прибора, включают мешалку"
•пускают воду в холодильник ,и нагревают содержимое колбы на водяной
бане при 80—85° С до образования густого сиропообразного продукта.
После охлаждения полученный продукт (форполимер) разливают в
стеклянные формы (см. работу 70) и окончательно полимеризуют в тер-
мостате при 55—65° С до образования твердого прозрачного полимера.
После окончания полимеризации формы с полимером погружают в
нагретую до 70° С воду для размачивания бумажной окантовки, затем
осторожно разнимают и вынимают готовые листы полиэтилметакрилат^.
Определяют остаточное содержание мономера путем анализа ма-
точного раствора, полученного после переосаждения из подобранных
растворителя и осадителя навесок, взятых сверху и снизу пластины.
iP А Б О Т А 75. ПОЛУЧЕНИЕ ПОЛИБУТИЛМЕТАКРИЛАТА
В БЛОКЕ
СН3
I [C.HsCOO’J
СН2=С------------------>
I [С.Н,]
СООС4Н9
СН3 1
I
-СН2-С-
I
COOC4H9J
Исходные продукты: бутилметакрилат — 30 г; перекись бензоила — 0,03 г.
Оборудование: колба емкостью 100 мл; водяная баня; ампулы из тугоплавкого
•стекла — 2 шт.; термошкаф; вакуум-сушилка.
Перекись бензоила растворяют в бутилметакрилате при слабом на-
гревании на водяной бане и вносят в ампулы, заполняя их на 7г объема
(отверстие ампул протирают комочком ваты для удаления следов мо-
номера) . Ампулы охлаждают, помещая их в сухой лед, а затем осторож-
но запаивают их на паяльной горелке и помещают в термошкаф при
70° С для полимеризации продукта.
Через 5—6 ч термошкаф выключают, дают ампулам охладиться в
шкафу, после чего осторожно вскрывают и извлекают полимер.
Для определения непрореагировавшего бутилметакрилата в блоке
пользуются методом, описанным на стр. 155, или выдерживают навеску
в воздушной или вакуумной сушке при 40—50° С до постоянного веса.
РАБОТА 76. ПОЛУЧЕНИЕ ПОЛИАЛЛИЛМЕТАКРИЛАТА
В БЛОКЕ
Органическое стекло из полиметилметакрилата имеет невысокую
теплостойкость и недостаточную поверхностную твердость. Стекла из
полифункциональных мономеров, способные отверждаться с образова-
нием полимеров пространственной структуры, лишены этих недостат-
ков. К таким мономерам относятся аллилметакрилат, полиэтиленгли-
кольметакрилаты. Интересным свойством аллилметакрилата является
образование линейного полимера под влиянием УФ-облучения, когда
1/7
реагирует лишь одна двойная связь. Аллильная группа, как мало реак-
ционноспособная, в этих условиях не образует свободных радикалов.
Теплостойкость органических стекол из полимеров пространствен-
ного строения достаточно высока (около 250°С). Недостатком их явля-
ется повышенная хрупкость. Более оптимальными свойствами обладают
сополимеры метилметакрилата с аллилметакрилатами и полиалкилен-
гликольметакрилатами.
Исходные продукты: аллилметакрилат—10,0 г; перекись бензоила — 0,051 г.
Оборудование: ампулы из тугоплавкого стекла; металлическая сетка; термостат
водяной.
При слабом нагревании на водяной бане в аллилметакрилате раст-
воряют перекись бензоила, и заливают раствор в две чистые сухие ам-
пулы, заполняя их не более чем на */г объема. Ампулы осторожно запаи-
вают, завертывают в металлическую сетку и помещают для полимери-
зации в термостат при 75° С.
Через 5 ч термостат выключают и дают ампулам охладиться в нем
до комнатной температуры. Охлажденные ампулы вынимают из тер-
мостата, осторожно вскрывают и извлекают полученный продукт.
Полимер аллилового эфира метакриловой кислоты представляет
собой бесцветный прозрачный хрупкий продукт, не размягчающийся
при нагревании до 250° С и нерастворимый в органических раствори-
телях.
РАБОТА 77. ПОЛУЧЕНИЕ ПОЛИАЛКИЛЕНГЛИКОЛЬДИМЕТАКРИЛАТА В БЛОКЕ
Условная схема реакции:
СН3 СНз СНз
I I [С.ЩСОО’1 I ?
иСН2=С— COO-R—ООС—С=СН2--------->~CH2-C-COO-R—ООС—С-СН2
(С.нй I |
СНз
где R —это —СН2СН2О —, (—СН2СН2О—)2 и (—СН2СН2О—)3.
Исходные продукты: диметакрилат этиленгликоля (диэтиленгликоля или триэти-
ленгликоля)— 15 г; перекись бензоила (1% от веса мономера)—0,15 г.
Оборудование: химический стакан емкостью 100 мл; ампулы — 3 шт.; термостат.
В диметакрилате этиленгликоля при слабом нагревании на водяной
бане растворяют перекись бензоила. Приготовленный мономер с ини-
циатором разливают поровну в три чистые сухие ампулы, заполняя их
на ’/г—*/з объема. Запаянные ампулы помещают в термостат .и нагре-
вают при 80° С до образования совершенно твердого полимера. Череа
1,5—2 ч весь мономер переходит через гелеобразное состояние в стекло-
образный продукт. По окончании полимеризации шкаф выключают и
ампулы оставляют в нем до полного охлаждения. Охлажденные ампулы
вскрывают и извлекают полученный полимер.
Полиэтиленгликольдиметакрилат исследуют на растворимость в ор-
ганических растворителях, твердость и термостойкость.
178
РАБОТА 78. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
С АЛЛИЛМЕТАКРИЛАТОМ В БЛОКЕ
Условная схема реакции:
СН3 СН3 снз СНз
I I [С,Н,СОО'1 I |
пСН2=С 4- тСН2=С -------> ~СН2-С-СН2-С ~
I I [c.Hii | |
СООСНз СООСН2СН=СН2 СООСНз СООСН2СНСН2
г
Исходные продукты: метилметакрилат — 8 г; аллилметакрилат — 2 г; перекись,
бензоила — 0,05 г.
Оборудование: колба емкостью 150 мл; водяная баня; ампулы — 2 шт.; термо-
стат.
В метилметакрилате при слабом нагревании на водяной бане раст-
воряют перекись бензоила, затем добавляют аллилметакрилат. Приго-
товленную смесь заливают в две чистые сухие ампулы на ’/г объема.
Узкий конец ампул осторожно запаивают и помещают их в термостат
при температуре 65° для полимеризации.
Через 8 ч термошкаф выключают и дают ампулам охладиться до
комнатной температуры. Затем их вынимают из термошкафа, осторожно*
вскрывают и извлекают продукт.
Сополимер представляет собой бесцветный прозрачный продукт, от-
личающийся высокой температурой размягчения (около 240° С) и боль-
шой устойчивостью к органическим растворителям.
Определяют растворимость в органических растворителях и тепло-
стойкость.
РАБОТА 79. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
С ДИМЕТАКРИЛАТОМ ЭТИЛЕНГЛИКОЛЯ В БЛОКЕ
СНз СН3 СНз
I R- I I
иСН2=С + тСН2=СН СН=СН2-> ~СН2-С-СН2-С~ СН3
III lil
СООСНз COOROOC соосн3 coorooc-c-ch2 -
1
Исходные продукты: метилметакрилат—10,0 г; диметакрилат этиленгликоля —
0,1 г; перекись бензоила — 0,1 г.
Оборудование: колба; водяная баня; стеклянные формы. г
В метилметакрилате при слабом нагревании на водяной бане раст-
воряют перекись бензоила. Затем в полученный раствор добавляют ди-
метакрилат этиленгликоля. Смесь мономеров с инициатором заливают
в стеклянные формы (см. работу 70) и ставят в термошкаф при 60° С
для полимеризации. Через 2—3 ч образуется твердый стеклообразный
сополимер трехмерного строения, не размягчающийся при нагревании
до 200° С и не растворяющийся в органических растворителях. Полиме-
ризация проходит без заметного нарастания вязкости мономера, с боль-
шим периодом индукции через образование нерастворимого геля.
179
По окончании полимеризации формы вынимают из термошкафа,
размачивают в теплой воде (около 70° С) бумажную окантовку и из-
влекают твердые образцы органического стекла.
РАБОТА 80. ПОЛУЧЕНИЕ ПОЛИАКРИЛОНИТРИЛА
В МОНОМЕРЕ
Схема реакции:
иСН2=СН
CN
—сн2-сн-
I
CN
Исходные продукты: акрилонитрил — 10 г; перекись бензоила — 0,01 г.
Оборудование: ампулы — 2 шт.; водяные термостаты — 2 шт.; колба емкостью
50 мл; бюкс — 1 шт.
Перекись бензоила растворяют в акрилонитриле и жидкость разли-
вают в две ампулы, заполняя их на ’/г объема. Ампулы осторожно за-
паивают и ставят для полимеризации в термостаты.
Одну ампулу нагревают при 100° С .и наблюдают за ходом полиме-
ризации и образованием полимера. Приблизительно через 2 мин начи-
нается очень энергичная реакция и образующийся полимер вследствие
нерастворимости его в мономере выпадает в виде порошка на дно ампу-
лы, поэтому блочный способ полимеризации акрилонитрила называют
полимеризацией в мономере. Через небольшой промежуток времени
реакция прекращается, так как инициатор быстро расходуется.
Вторую ампулу нагревают в термостате при 70° С. При этой тем-
пературе индукционный период продолжается около 40 мин, но реакция
протекает так же, как и при 100° С.
По окончании реакций ампулы охлаждают, осторожно вскрывают
и извлекают полимеры.
Определяют выход полимеров и их растворимость в органических
растворителях.
Примечания. 1>. Следует избегать более высоких, чем указано в рецептуре, концент-
раций перекиси бензоила, так как во время полимеризации при концентрации инициа-
тора около 1 % часто происходят взрывы.
2. При запаивании ампулы с акрилонитрилом, а также во время наблюдения за
ходом полимеризации при открывании термостата необходимо предохранять глаза и
руки, обязательно надевая защитные очки, а на руки — толстые резиновые перчатки
или обвернув руки полотенцем.
РАБОТА 81. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА,
АКРИЛОНИТРИЛА И ДИМЕТАКРИЛАТА
ТРИЭТИЛЕНГЛИКОЛЯ В БЛОКЕ
Условная схема реакции:
СН3
I R*
лСН2=С . + /иСН2=СН + дСН2=СН СН=СН2->
I I II'
СООСНз CN COOROOC
180
СН3
I г »
~ СН2—С---СН2 —СН—СН2—СН СН—сн2 ~
СООСНз CN COOROOC
Исходные продукты: метилметакрилат—1Ю г; акрилонитрил — 5,3 г; диметакрилат
триэтиленгликоля—0,198 г; перекись бензоила — 0,0775 г.
Оборудование: водяная баня; колба плоскодонная емкостью 50 мл; ампулы_
3 шт.; термостат.
В метилметакрилате при слабом нагревании на водяной бане раст-
воряют перекись бензоила, затем в полученный раствор вводят акрило-
нитрил и димстакрилат триэтиленгликоля. Смесь мономеров с инициа-
тором разливают поровну в три чистые сухие ампулы, заполняя йх на
72 объема. Ампулы запаивают на паяльной горелке, предварительно
надев защитные очки и приняв меры предосторожности против вос-
пламенения продукта, и помещают в термостат для полимеризации
при 60° С.
Через 7 ч нагревания, надев защитные очки и резиновые перчатки,
осторожно вынимают из шкафа одну ампулу (контрольную) и после
полного охлаждения вскрывают ее. Если полимеризация закончилась и
полимер представляет собой совершенно твердый прозрачный бесцвет-
ный продукт, шкаф выключают, ампулам дают охладиться в шкафу и
после охлаждения осторожно вскрывают. В противном случае нагрева-
ние при той же температуре продолжают еще 2 ч и после этого вскры-
вают вторую ампулу и т. д.
Извлеченный из ампулы готовый сополимер исследуют на раство-
римость в органических растворителях (ацетоне и диметилформамиде)
и на теплостойкость.
РАБОТА 82. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
СО СТИРОЛОМ В БЛОКЕ
Выше были описаны сополимеры стирола с метилметакрилатом,
в которых преобладал стирол. Введение в сополимеризацию небольших
количеств стирола модифицирует свойства полиметилметакрилата, при-
давая повышенную термостойкость органическому стеклу на его основе
и специфическую растворимость в ароматических углеводородах.
Условная схема реакции:
СН3 СНз СН3
I R- I I
пСН2=С + тСН2=СН -+ ~ СН2-С-СН2-С—СН2-СН ~
I I III
СООСНз С6Н5 СООСНз СООСНз С6Н5
Исходные продукты: метилметакрилат—100 г; стирол — 5 г; перекись бен-
зоила — 0,5 г.
Оборудование: трехгорлая колба емкостью 0,5 л, снабженная мешалкой с герме-
тическим затвором, обратным холодильником и термометром; водяная баня; стеклян-
ные формы (см. работу 70); термостат; стакан емкостью 150 мл.
В смеси мономеров растворяют перекись бензоила, приготовленный
раствор загружают в трехгорлую колбу прибора. Включают мешалку,
•tw
пускают воду в холодильник и нагревают содержимое колбы на водя-
ной бане при 70—80° С до образования сиропообразного продукта. По-
лученный продукт охлаждают, разливают в формы и окончательно по-
лимеризуют в термостате при постепенном повышении температуры от
55° до 110° С в течение 8—10 ч до получения твердых образцов прозрач-
ного органического стекла.
Для извлечения готовых изделий формы помещают на 30—60 мин.
в нагретую до 70° С воду для размачивания бумажной окантовки. По-
лученные листы органического стекла обрезают по краям.
Определяют термостойкость и растворимость в бензоле.
РАБОТА 83. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
С ПЕРСУЛЬФАТОМ АММОНИЯ
Схема реакции:
СНз г снз 1
I [ОН] |
пСН2=С --------> —СН2—С—
I I
СООСНз L COOCH3J„
Исходные продукты: метилметакрилат — 30 г; вода дистиллированная — 100 г; пер-
сульфат аммония — 0,5 г.
Оборудование: трехгорлая колба, снабженная мешалкой с герметическим затво-
ром, обратным холодильником и термометром; водяная баня; воронка Бюхнера, колба-
плоскодонная емкостью 200 мл; термостат.
Персульфат аммония растворяют в воде и помещают в трехгорлую-
колбу. Туда же загружают метилметакрилат, включают мешалку, пу-
скают воду в холодильник и нагревают колбу на водяной бане при
80° С. При быстром вращении мешалки через 10—15 мин содержимое
колбы приобретает молочно-белый цвет.
При 80° С реакция полимеризации продолжается 2—2,5 ч. После
этого в колбу с суспензией добавляют 10—15 мл концентрированной
соляной кислоты (пл. 1,19) и при нагревании перемешивают до коагуля-
ции полимера и расслоения смеси.
Осадок полиметилметакрилата отфильтровывают на воронке Бюх-
нера и быстро промывают вначале небольшим количеством спирта, за-
тем водой до нейтральной реакции промывных вод (проба с метиловым
оранжевым). Промытый продукт, представляющий собой белый тонкий
порошок, высушивают в термостате при 65—70° С, взвешивают и опре-
деляют выход полимера по отношению к взятому мономеру. Определяют
количество не вступившего в реакцию мономера;
Полимер перерабатывают экструзией при температуре в цилиндре
170° С, в головке 200° С. Стандартные образцы испытывают на удар и
разрушающее напряжение при изгибе (см. ч. II).
182
«РАБОТА 84. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
В ГРАНУЛАХ
Исходные продукты: метилметакрилат—10 г; перекись бензоила — 0,1 г; вода
дистиллированная — 60 г; поливиниловый спирт — 0,34 г.
Оборудование: пробирка диаметром 45 мл и высотой 190 мм, имеющая боковой
•отвод и снабженная мешалкой с герметическим затвором и обратным холодильником
(см. рис. 20); химический стакан (в качестве водяной бани) емкостью 1 л; стакан ем-
костью 50 мл — 2 шт.; воронка Бюхнера; чашка Петри.
В пробирке прибора для гранульной полимеризации (см. рис. 20)
при нагревании до 60° С растворяют поливиниловый спирт в дистилли-
рованной воде. Затем отдельно .в метилметакрилате растворяют перекись
бензоила и заливают полученный раствор в пробирку с охлажденным
до комнатной температуры водным раствором поливинилового спирта,
включают мешалку, пускают воду в холодильник и пробирку нагревают
на водяной бане до 80° С.
Скорость мешалки регулируют так, чтобы метилметакрилат раз-
•бивался на отдельные шарики (гранулы) наибольшей величины, не со-
•единяясь в общую массу. Установленную постоянную скорость мешалки
.необходимо поддерживать в течение всего процесса полимеризации, не
допуская перерыва в работе во избежание слипания гранул и образова-
ния бесформенного комка полимера.
Температуру в бане рекомендуется поддерживать точно 80° С, не
допуская перегрева. Полимеризация обычно заканчивается через 3—4 ч.
•Об окончании реакции судят по опусканию шариков полиметилметакри-
лата на дно пробирки вследствие увеличения удельного веса полимера.
Готовый продукт извлекают из реакционного сосуда на воронку
Бюхнера, промывают теплой водой, высушивают и взвешивают. Выход
полимера определяют в процентах от в*зятого для полимеризации мо-
номера.
В маточном растворе определяют остаточный мономер (см. стр. 58,
136). В гранулах определяют количество летучих весовым методом (см.
стр. 136).
Примечание. Для получения окрашенного продукта краситель (судановый крас-
ный, судаковый оранжевый или другой) предварительно растворяют в мономере, а за-
тем ведут полимеризацию по описанной методике.
РАБОТА 85. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
В СУСПЕНЗИИ
СН3
I (CHs)2CCN
nCH2=C--------------->
I
СООСНз
СН3 -1
I
-сн2-с-
I
СООСНз J
Исходные продукты: метилметакрилат—10 г; дииитрил азоизомасляной кислоты —
0,02 г; поливиниловый спирт — 0,34 г.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); стаканы хи-
мические емкостью 50 мл—2 шт.; воройка Бюхнера; чашка Петри; водяной термостат
(стакан); термометр.
183
В пробирке прибора растворяют навеску поливинилового спирта в
60 мл, дистиллированной воды при нагревании. Затем отдельно в ме-
тилметакрилате растворяют динитрил азоизомасляной кислоты и прили-
вают к охлажденному до комнатной температуры раствору поливинило-
вого спирта. Включают мешалку, пускают воду в холодильник и нагре-
вают пробирку в стакане с водой до 80° С. Далее процесс проводят так,
как это описано в предыдущей работе.
Реакция заканчивается обычно за 2—2,5 ч. Контролем служит осе-
дание гранул на дно сосуда. Гранулы отфильтровывают на воронке
Бюхнера. В маточном растворе определяют содержание мономера бро-
мированием в ледяной уксусной кислоте (см. стр. 136). Определяют
содержание летучих весовым методом (см. ч. II).
Гранулы перерабатывают литьем под давлением при 190—235° и
удельном давлении 1200 кг/см2 или экструзией при 170—200° С. Опреде-
ляют прочность образца на удар, водостойкость (см. ч. II).
РАБОТА 86. ПОЛУЧЕНИЕ ПЛАСТИФИЦИРОВАННОГО
ПОЛИМЕТИЛМЕТАКРИЛАТА В СУСПЕНЗИИ
Исходные продукты: метилметакрилат — 30 г; перекись бензоила — 0,3 г; дибутил-
фталат (5% от массы метилметакрилата) — 1,5 г; вода дистиллированная — 180 г;
поливиниловый спирт — 1 г.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); колба емко-
стью 1Ю0 мл; воронка Бюхнера, сито № 30.
В пробирке прибора растворяют при нагревании до 60° С поливини-
ловый спирт в дистиллированной воде. Затем отдельно растворяют в
метилметакрилате перекись бензоила и после ее полного растворения
вводят дибутилфталат. Приготовленный раствор при непрерывном пе-
ремешивании (1200—1500 об/мин) загружают в пробирку с охлажден-
ным до комнатной температуры водным раствором поливинилового
спирта. Затем пускают воду в холодильник м содержимое колбы нагре-
вают 3—4 ч на водяной, бане при 80° С. Об окончании процесса полиме-
ризации можно судить по опусканию порошка полиметилметакрилата
на дно колбы вследствие увеличения удельного веса полимера.
Готовый продукт извлекают ,из реакционного сосуда, отфильтровы-
вают на воронке Бюхнера, промывают теплой водой, высушивают. Вы-
ход полимера определяют в процентах от суммы взятых для полимери-
зации мономера и дибутилфталата. Используют в работе 87.
Примечание. Для получения окрашенного продукта краситель (судановый крас-
ный, судаковый оранжевый или др.) предварительно растворяют в мономере, а затем
полимеризуют указанным выше методом.
РАБОТА 87. ИЗГОТОВЛЕНИЕ СВЕТЯЩИХСЯ ИЗДЕЛИЙ
ИЗ ПОЛИМЕТИЛМЕТАКРИЛАТА МЕТОДОМ ГОРЯЧЕГО
ПРЕССОВАНИЯ
Исходные продукты: полиметилметакрилат (суспензионный), содержащий 5—10%
пластификатора, — 10 г; светосостав (люминофор) — 1 г.
Оборудование: сито; металлическая форма; лабораторный гидравлический пресс.
Полиметилметакрилат, просеянный через сито № 30, тщательно
смешивают со светосоставом стеклянным или фарфоровым шпателем,
184
избегая трения смеси о стенки сосуда. Приготовленный порошок прес-
суют в изделие в металлической форме при 140—150° С и удельном дав-
лении 250—300 кг/см2.
Выдержка при нагреве под давлением составляет 2 мин на 1 мм
толщины изделия.
Не снимая давления, изделие охлаждают в прессе до 30° С, затем
вынимают из пресс-формы.
РАБОТА 88. ИЗГОТОВЛЕНИЕ НЕПРОЗРАЧНЫХ ОКРАШЕННЫХ ИЗДЕЛИИ
ИЗ ПОЛИМЕРИЗУЮЩЕЙСЯ МАСТИКИ НА ОСНОВЕ
СУСПЕНЗИОННОГО ПОЛИМЕТИЛМЕТАКРИЛАТА
Исходные продукты: полиметилметакрилат (суспензионный) — 70 г; метилметакри-
лат — 30 г; перекись бензоила — 0,03 г; пигмент — 5 г.
Оборудование: ступка фарфоровая; форма; термошкаф.
Полиметилметакрилат с пигментом (люминофором) тщательно пе-
ремешивают в ступке или фарфоровом стакане до получения однород-
ной смеси. Отдельно растворяют в мономере перекись бензоила и по-
степенно вливают мономер при постоянном перемешивании в смесь по-
лиметилметакрилата с пигментом.
Хорошо смешав массу, ее выдерживают 15—20 мин для набухания
полимера, закрыв стакан стеклянной пластинкой, чтобы избежать испа-
рения мономера. После этого продолжают перемешивание до образова-
ния тестообразной массы.
Полученную массу помещают в форму (металлическую, стеклянную
или гипсовую) и при давлении 0,5—1 кг/см2 уплотняют, равномерно рас-
пределив ее внутри формы. Форму ставят в термошкаф для полимери-
зации массы и выдерживают ее 1 ч при 55° С, затем 1 ч при 60° С, 2 ч
при 70° С и 2 ч при 80° С.
После этого нагревание продолжают при 100° С до получения со-
вершенно твердого изделия. Затем форму вынимают из шкафа, охлаж-
дают до комнатной температуры и вынимают изделие.
Примечании. В качестве пигмента можно применять тонко измельченные мумию,
двуокись титана, литопон, желтый крон, окись хрома и др. Вместо минеральных пиг-
ментов можно применять также органические красители (судановый красный, судано-
вый оранжевый и др.). В этом случае краситель предварительно растворяют в мономе-
ре, а затем вводят в порошок полиметилметакрилата.
РАБОТА 89. ПОЛУЧЕНИЕ ПОЛИБУТИЛМЕТАКРИЛАТА
В СУСПЕНЗИИ
сн3
I
сн2=с
СООС4Н9
[С.Н.СОО']
-------:—>
(С«н«)
СН3 -1
I
-сн2-с
I
COOC4H9J
Исходные продукты: бутилметакрилат свежеперегнанный —40 г; перекись бен-
зоила— 0,2—0,3 г; поливиниловый спирт — 0,34 г (или сольвар — 0,1' г); дистиллирован-
ная вода —. 60 мл.
7—1453
185
Оборудование: прибор для гранульной полимеризации (см. рис. 20); стеклянный
стакан емкостью 1 л; термометр; воронка Бюхнера; стаканы емкостью 50 мл — 2 шт.
В пробирке прибора растворяют при нагревании поливиниловый
спирт в дистиллированной воде. Отдельно в бутилметакрилате раство-
ряют перекись бензоила и вливают в охлажденный до комнатной тем-
пературы раствор поливинилового спирта. Собирают прибор и переме-
шивание ведут с такой скоростью, чтобы капли мономера равномерно
распределялись по всему объему жидкости.
Содержимое пробирки нагревают на водяной бане до 80° С и под-
держивают эту температуру 5—6 ч, пока гранулы образовавшегося по-
лимера не начнут опускаться на дно сосуда. Скорость вращения мешал-
ки должна быть постоянной и регулироваться с помощью реостата (см.
работу 84).
Полученные гранулы отфильтровывают на воронке Бюхнера, про-
мывают водой и высушивают на воздухе в чашке Петри.
Определяют удельную вязкость 1 %-ного раствора в дихлорэтане,
содержание влаги и летучих. Перед употреблением для изготовления
полимеризующейся мастики продукт высушивают в вакуум-сушилке.
Полимеризующуюся мастику готовят так, как описано в работе 88.
РАБОТА 90. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
С БУТИЛМЕТАКРИЛАТОМ СУСПЕНЗИОННЫМ МЕТОДОМ
Схема реакции:
СН3 СН3
I I ic.h.coqt
лСН2=С -}- Л1СН2—С
I I [C.Hil
СООСНз СООС4Н9
СН3 СН3 СНз
~ сн2-с-----СН2-С----СН2-С ~
I I I
СООСНз СООСНз СООС4Н9
Исходные продукты: метилметакрилат — 75 г; бутилметакрилат — 25 г; перекись
бензоила — 0,75 г; крахмал растворимый — 2,0 г; вода дистиллированная — 200 мл;
NaCl —1,5 г.
Оборудование: прибор для гранульной полимеризации (см. .рис. 20); химические
стаканы емкостью 10 мл — 2 шт.; воронка Бюхнера; енто № 30.
В смеси мономеров растворяют перекись бензоила, затем в отдель-
ном сосуде при нагревании растворяют в дистиллированной воде крах-
мал и NaCl. После охлаждения крахмальный раствор заливают в про-
бирку прибора. Включают мешалку и при энергичном перемешивании
в пробирку вводят подготовленную смесь мономеров и инициатора.
После 10 мин перемешивания определяют pH смеси. Если pH 6,8-?7,
то добавлением 20%-ного раствора щелочи его доводят до 7. Затем
включают холодильник и содержимое пробирки постепенно (около 4° за
10 мин) в течение 2 ч нагревают на водяной бане до 74—76° С при энер-
гичном и очень равномерном перемешивании. При 76° С полимеризацию
продолжают еще около 3,5—4 ч.
186
По окончании полимеризации сополимер переносят на воронку
Бюхнера и тщательно промывают водой до полного удаления крахмала
что определяется раствором иода по прекращению окрашивания про-
мывных вод в синий цвет. Промытый сополимер сушат при 55—60° С и
просеивают через сито № 30.
Продукт перерабатывают в изделия методом горячего прессования
•е последующим охлаждением или методом формования с добавкой мо-
номера (метилметакрилата) (см. работу 88).
РАБОТА 91. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
СО СТИРОЛОМ ПОЛИМЕРИЗАЦИЕЙ С ПЕРСУЛЬФАТОМ АММОНИЯ
Схема реакции:
СН3 СН3
I [*ОН] 1
пСН2=С + пСН2=СН -----> ~ СН2-С--СН2-СН ~
I I II
СООСНз С6Н5 СООСНз С6Н5
Исходные продукты: метилметакрилат —15 г; стирол—15 г; персульфат аммо-
ния — 0,5 г; вода дистиллированная — 100 мл.
Оборудование: трехгорлая колба емкостью 0,5 л, снабженная мешалкой с герме-
тическим затвором, обратным холодильником и термометром; водяная баня; воронка
Бюхиера. ч
В трехгорлую колбу вносят растворенный в воде персульфат ам-
мония, метилметакрилат и стирол, включают мешалку, пускают воду в
холодильник и смесь нагревают на водяной бане при 80° С. Через неко-
торое время содержимое колбы приобретает молочно-белый цвет. После
4—5 ч нагревания в колбу добавляют 10—15 мл концентрированной со-
ляной кислоты и продолжают нагревание при перемешивании до полной
коагуляции сополимера. Полученный порошкообразный продукт фильт-
руют на воронке Бюхнера, несколько раз промывают водой до нейт-
ральной реакции промывных вод, высушивают при 40—50° С и взвеши-
вают. Выход сополимера определяют по отношению к сумме взятых мо-
номеров.
Определяют теплостойкость по Мартенсу и сравнивают с тепло-
стойкостью гомополимеров.
РАБОТА 92. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
С МЕТИЛАКРИЛАТОМ В СУСПЕНЗИИ
СН3
тСН2=СН -Ь пСН2=С
I I
СООСНз СООСНз
[С,Н,СООЧ
---------->
[с.н;]
СН2—СН----
I
СООСНз
СНз
СН2-С----СН2-
СООСНз
СНз
СООСН.З
Исходные продукты: метилметакрилат — 75 г; метилакрилат — 25 г; перекись бен-
зоила — 0,715 г; дибутилфталат — 5 г; крахмал растворимый — 2,0 г; дистиллированная
вода — 200 мл.
7*
187
Оборудование: колба емкостью 200 мл; трехгорлая колба емкостью 0,5 л, снабжен-
ная мешалкой с герметическим затвором, обратным холодильником и термометром; во-
ронка Бюхнера; сито № 30.
Вначале перекись бензоила растворяют в смеси мономеров, затем
добавляют дибутилфталат. В отдельном сосуде при нагревании раство-
ряют в дистиллированной воде крахмал и после охлаждения крахмаль-
ный раствор помещают в колбу прибора. Включают мешалку и при энер-
гичном перемешивании в колбу вводят подготовленную смесь мономе-
ров. После 10 мин перемешивания определяют pH смеси. Если pH ниже
6,8—7, то добавлением небольшого количества 20%-ного раствора ще-
лочи его доводят до 7. Затем пускают воду в холодильник и температу-
ру смеси медленно (1,5—2 ч) доводят, нагревая колбу на водяной бане,,
до 74—76° С при продолжающемся энергичном перемешивании. После
этого при 76° С полимеризацию продолжают еще около 2—2,5 ч.
По окончании процесса полимеризации сополимер переносят на
воронку Бюхнера, маточный раствор анализируют на содержание моно-
меров (см. стр. 136). Осадок многократно промывают водой до полного
удаления крахмала, что определяют раствором иода по прекращению
окрашивания промывных вод в синий цвет. Промытый сополимер сушат
при 55—60° С и просеивают через сито № 30.
Выход продуктов составляет 80—90%. Сополимер перерабатывают
литьем под давлением при температуре цилиндра 170° С, температуре го-
ловки 200° С и температуре формы 40—45° С. Определяют твердость,
теплостойкость, предел прочности при разрыве. Сравнивают результаты
с показателями для гомополимеров.
РАБОТА 93. ПОЛУЧЕНИЕ СОПОЛИМЕРА МЕТИЛМЕТАКРИЛАТА
С ВИНИЛАЦЕТАТОМ СУСПЕНЗИОННЫМ МЕТОДОМ
Условная схема реакции:
СН3 СН3 СНз
I [(CH,),C-CN] I . |
nCH2=C + тСН2=СН --------------> ~ СН2-С-СН2-С—СН2-СН -
II III
СООСН3 ООССНз СООСНз СООСНз ООССНз
Исходные продукты: метилметакрилат—10 г; винилацетат — 3 г; кварцевая му-
ка— 5 г; динитрил азоизомасляной кислоты — 0,025 г; поливиниловый спирт — 0,34 г.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); стакан
емкостью 1 л; термометр; воронка Бюхнера; лабораторные вальцы; лабораторная шаро-
вая мельница (или медная ступка); пресс гидравлический.
В метилметакрилате растворяют инициатор и раствор смешивают
с винилацетатом. В реакционной пробирке растворяют при нагревании
навеску поливинилового спирта. Охлаждают раствор и в него вливают
смесь мономеров. Регулируют уровень лопастей мешалки и скорость
перемешивания так, чтобы масса мономеров разделилась на отдельные
шарики— 1—2 мл. Мешалка должна вращаться с постоянной скоростью
(регулируют реостатом), ее нельзя останавливать на длительный срок,
так как мелкие полимерно-мономерные частицы при этом слипаются в
188
комок. При слишком большой скорости образуется очень тонкая дис-
персия. Реакция длится 2—3 ч. Конец реакции определяют по осажде-
нию гранул на дно реакционной пробирки вследствие увеличившейся
плотности.
Гранулы отделяют от жидкости фильтрованием на воронке Бюхне-
ра, хорошо промывают водой (проМывные воды присоединяют к фильт-
рату) и высушивают на воздухе. В фильтрате определяют суммарное
содержание мономеров бромированием раствором брома в уксусной кис-
лоте (см. стр. 136).
На основе сополимера, высушенного в вакуум-сушилке до постоян-
ного веса, получают материал, наполненный кварцевой мукой. Для этой
цели 10 г сополимера смешивают на лабораторных вальцах с 5 г квар-
цевой муки при 70° С; измельчают полученный материал в шаровой
мельнице и прессуют стандартные бруски при 180° С и удельном давле-
нии 200—300 кг/см2, выдержка в форме 2 мин на 1 мм толщины изделия.
Охлаждают до 40° С в прессе под давлением. Определяют теплостой-
кость образцов, твердость и разрушающее напряжение при ударе.
РАБОТА 94. ПОЛУЧЕНИЕ ПОЛИМЕТИЛМЕТАКРИЛАТА
В РАСТВОРИТЕЛЕ
Исходные продукты: метилметакрилат —'50 г; бензол (чистый)—50 г; перекись
бензоила — 0,25 г.
Оборудование: трехгорлая колба емкостью 250 мл; снабженная мешалкой с герме-
тическим затвором, обратным холодильником и термометром; водяная баня; рефракто-
метр.
В трехгорлую колбу прибора помещают метилметакрилат, в кото-
ром предварительно растворена перекись бензоила, а затем вводят бен-
зол. Включают мешалку, пускают воду в холодильник и нагревают колбу
7—8 ч на водяной бане при 75—80° до получения сиропообразного про-
дукта.
Течение реакции контролируют по изменению вязкости и показате-
ля преломления раствора. В начале реакции их определяют через каж-
дые 30 мин (5—7 раз), затем через каждый час. К концу реакции поли-
меризации показатель преломления не должен изменяться в течение
часа. По полученным данным составляют таблицу и вычерчивают кри-
вые изменения этих показаний.
Продукт применяется в качестве лаков для покрытий и пропитки,
а также для склеивания полиметилметакрилата.
РАБОТА 95. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ОТХОДОВ
ПОЛИМЕТИЛМЕТАКРИЛАТА МЕТОДОМ ГОРЯЧЕГО ПРЕССОВАНИЯ
Исходные продукты: отходы органического стекла (опилки или стружки)—-.917 г;
краситель (нигрозин) — 3 г.
Оборудование: шаровая мельница; сито № 30; фарфоровая ступка; пресс-форма
стандартного бруска; гидравлический пресс.
Отходы органического стекла, не содержащие загрязнений в виде
примесей и минеральных масел, измельчают в шаровой мельнице в те-
чение 6—10 ч до стадии тонкого помола (время помола зависит от раз-
189
мера частиц отходов, поступающих на переработку). Измельченный про-
дукт просеивают через сито № 30, после чего в порошок добавляют ниг-
розин и тщательно перемешивают 0,5—1 ч в шаровой мельнице или в
фарфоровой ступке до получения однородной смеси.
Полученный порошок загружают в металлическую пресс-форму
«стандартный брусок» и прессуют при следующем режиме: температура
190—200° С; удельное давление 50—80 кг/см2; выдержка при нагреве под
давлением 2—3 мин на 1 мм толщины стенки изделия. Охлаждение до
40° С в прессе под давлением.
Определяют твердость, теплостойкость и ударную вязкость образ-
цов. Сравнивают результаты с данными, приведенными в приложении.
РАБОТА 96. ПОЛУЧЕНИЕ ПОЛИАКРИЛОНИТРИЛА
В ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЙ
ИНИЦИИРУЮЩЕЙ СИСТЕМЕ
Инициирование полимеризации акрилонитрила с помощью окисли-
тельно-восстановительной реакции в системе персульфат — тиосульфат
в водной среде приводит к образованию порошка полимера, который
после отмывки от солей удобно перерабатывать любым способом.
Окислительно-восстановительные реакции можно описать следую-
щей схемой:
S2OI 4- SjO2 —> SC)4 4- SO4 4- S2O3
SO^‘ 4- Н2О HSOJ- 4- ОН
ОН 4- СН2=СН -> НОСН2-СН 4- nCH2=CH -> HO-CH2-CHCN [-CH2-CHCN-]„
I I I
CN CN CN
Исходные продукты: нитрил .акриловой кислоты, перегнанный — 20 г; персульфат
натрия — 0,402 г; тиосульфат — 0,28 г.
Оборудование: трехгорлая колба емкостью 250 мл с обратным холодильником, ме-
шалкой и термометром; водяная баня; воронка Бюхнера.
Навески персульфата и тиосульфата натрия растворяют в колбе
прибора в 40 мл дистиллированной воды. К раствору приливают моно-
мер и при перемешивании мешалкой нагревают 1—2 ч на водяной бане
с обратным холодильником при 40—60° С. Выпавший осадок отфильтро-
вывают на воронке Бюхнера. Фильтрат анализируют на содержание не-
прореагировавшего мономера (см.стр. 159).
Осадок полимера промывают водой до отрицательной реакции на
сульфат-ион и высушивают при 60° С в термостате. Определяют процент-
ное содержание летучих весовым способом.
РАБОТА 97. ПОЛУЧЕНИЕ СОПОЛИМЕРА ПОЛИАКРИЛОНИТРИЛА
В ВОДНОМ РАСТВОРЕ РОДАНИСТОГО НАТРИЯ
Исходные продукты: роданистый натрий — 25 г; акрилонитрил — 22,5 г; метила-
крилат — 2 г; динитрил азоизомасляной кислоты — 0,025 г; итаконовая кислота — 0,2 г.
Оборудование: колба трехгорлая, снабженная механической мешалкой, обратным
холодильником и термометром; воронка Бюхнера; стаканчик емкостью 50 мл; термо-
стат; стеклянная ванночка для коагуляции (рис. 49).
190
Готовят 50%-ный раствор роданистого нат-
рия в воде и заливают его в реакционную колбу.
Растворяют инициатор в акрилонитриле, к раст-
вору добавляют другие мономеры и смесь влива-
ют в раствор роданистого натрия. Включают ме-
шалку, пускают воду в холодильник и нагревают
раствор при энергичном перемешивании при 80° С
в течение 2 ч. Образовавшийся раствор со-
полимера используют для получения волокна
или пленки методом высаживания в воду в спе-
циальном приборе (рис. 49). Полученную пленку
промывают водой и сушат при 60—70° С в тер-
мостате. Определяют разрушающее напряжение
при растяжении пленки (волокна).
Рис. 49. Ванночка для
коагуляции раствора по-
лимера на вращающемся
валике:
1 — фильера; 2 — ванна; 3 —
вращающийся валик; 4 —
осаждающая жидкость; 5 —
отжимные валки; 6 — пленка
РАБОТА 98. ПОЛУЧЕНИЕ ПОЛИАКРИЛАМИДА В РАСТВОРИТЕЛЕ
Схема реакции:
[R*]
пСН2=СН —->г-сн2-сн-
I I
CONH2 CONH2]n
Исходные продукты: акриламид — 10 г; изопропиловый спирт — 1,55; персульфат
калия — 0,02 г; двуокись углерода или инертный газ; этиленгликоль — 1г.
Оборудование: колба трехгорлая, снабженная обратным холодильником, механиче-
ской мешалкой с герметическим затвором и термометром; водяная баня.
80 мл дистиллированной воды заливают в колбу прибора, добавля-
ют изопропиловый спирт, персульфат калия. После растворения соли
вливают мономер и перемешивают раствор, нагревая его на водяной
бане при 60° С в течение 2 ч. Определяют вязкость полученного раствора
полимера и используют в качестве клея для бумаги.
РАБОТА 99. ПОЛУЧЕНИЕ СОПОЛИМЕРА АКРИЛАМИДА
С АКРИЛОВОЙ КИСЛОТОЙ В ВОДНОЙ СРЕДЕ
[•ОН]
пСН2=СН + тСН2=СН -----*- ~ СН2—СН-СН2-СН ~
II II
conh2 соон соон conh2
Исходные продукты: акриламид—10 г; акриловая кислота, 50%-ный водный рас-
твор — 20 г; персульфат калия — 0,4 г; тиосульфат калия — 0,4 г.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная обратным холо-
дильником; механической мешалкой с герметическим затвором и термометром; водяная
баня.
В реакционной колбе в 80 мл дистиллированной воды растворяют
персульфат и тиосульфат калия. Затем заливают мономеры и нагревают
с обратным холодильником при перемешивании на водяной бане при
60° С до образования водного раствора.
Определяют вязкость полученного полимера и используют его в ка-
честве покрытия ткани, которая, приобретает жесткость и несминаемость.
191
ГЛАВА VI. ПОЛИМЕРЫ НА ОСНОВЕ ПРОИЗВОДНЫХ ЭТИЛЕНА
СО СЛОЖНЫМИ ЗАМЕСТИТЕЛЯМИ
К производным этилена со сложными заместителями относят N-винил-
карбазол, N-винилпирролидон, 2-винилпиридин. N-Винилкарбазол —
дешевый мономер, основным поставщиком которого является коксохи-
мическая промышленность. Полимеры на основе этих мономеров доволь-
но термостойки и химически устойчивы. Особенную техническую цен-
ность представляют их сополимеры с различными винильными соедине-
ниями, применяющиеся в различных отраслях техники.
-СН-СН2-
I
N
У\/\/Ч.
Поливинилкарбазол
получают из N-винил-
— X/ — п
карбазола по радикальному и ионному механизмам при нагревании, в
присутствии инициаторов и катализаторов. Это белый полимер с плот-
ностью 1,2 г/см,3 теплостойкостью по Мартенсу 150° С, т. разлож. вы-
ше 300° С.
В связи с высокой температурой размягчения (211° С) поливинил-
карбазол трудно отливать и экструдировать. Его прессуют при 220—
230° С.
Поливинилкарбазол по свойствам подобен полистиролу, отличается
от него более высокой теплостойкостью и большей хрупкостью.
Поливинилпирролидон
-сн2-сн-
I
N
образуется из N-ви-
н,с со
I I
Н2С—сн2_
нил-а-пирролидона радикальной полимеризацией в блоке и в растворе в
присутствии перекиси водорода. Гомополимер обладает достаточной теп-
лостойкостью, высокой химической устойчивостью. Он применяется в ка-
честве заменителя плазмы крови.
Как пластические материалы в технике применяются сополимеры
N-винил-а-пирролидона с винильными мономерами, малеиновым анги-
дридом и др. Сополимеры образуют пленки и волокна с высокими физи-
ко-химическими показателями, особенно в ориентированном состоянии.
-СН2- СН-
Поливинилпиридины
образуются из 2-винил-
п
п
192
пиридина, а также из 4-винилпиридина в присутствии инициаторов ра-
дикальной полимеризации. По физико-химическим свойствам поливинил-
пиридины близки к полистиролу, однако в отличие от полистирола
растворимы в кислотах, что определяет их применение в качестве поли-
электролитов. Они растворимы в воде и лрименяются в качестве эмуль-
гаторов.
Технология производства полимеров N-винилкарбазола, N-винил-
пирролидона и винилпиридинов ничем не отличается от обычной поли-
меризации в присутствии радикальных инициаторов в растворе или в
суспензии.
Исходное сырье для получения полимеров
из производных этилена
___/ч
N-Винилкарбазол 4/\N/\Z представляет собой бесцветный твер-
I
сн=сн2
дый кристаллический продукт, темнеющий при хранении на свету, с т. пл.
от 61 до 67° С. Чистый продукт, перекристаллизованный из метилового
спирта, плавится при 67° С. Т. кип. при 1 мм рт. ст. 110° С; при 2 мм рт.
ст. 150—155° С; плотность расплавленного продукта при 70° С
1,094 г/см3; при 100° С 1,075 г/см3; мол. вес 193,24.
Винилкарбазол нерастворим в воде; он растворяется в метиловом и
этиловом спиртах, ацетоне, хлорбензоле, четыреххлористом углероде,
эфире, уксусной кислоте, хлороформе, хлористом метилене, тетралине,
гексане, циклогексане, диоксане, пентрне, тетрагидрофуране, бензоле,
лигроине.
Хранить мономер рекомендуется в темноте, в среде инертного газа.
Ингибиторами для мономера, предотвращающими потемнение про-
дукта при хранении на свету, являются вещества основного.характера
амины и амиды, например морфолин (тетрагидрооксазйн), формамид,
диметилформамид, а также некоторые спирты, например стеариловый и
октадециловый.
NH
н2с1/х1сн2
Морфолин и стеариловый спирт С47Н35СН2ОН —
* Н2С>^ Z'CH2
о
наилучшие ингибиторы для винилкарбазола. Однако они надежно ста-
билизируют мономер только при температурах до 100° С. Выше этой
температуры их действие заметно снижается, а выше 120° С совершенно
прекращается.
Полимеры винилкарбазола растворяются в ароматических углеводо-
родах, хлорбензоле, хлороформе, дихлорэтане, трихлорэтилене и нерас-
творимы в алифатических и ненасыщенных алициклических углеводоро-
дах, спиртах, простых эфирах, четыреххлористом углероде, смазочных
маслах.
193
Поливинилкарбаэол устойчив к действию разбавленных кислот и ос-
нований. При действии нагретых концентрированных серной и азотной
кислот он разрушается.
N-Винилкарбазол получают из карбазола и ацетилена при 180—
260° С и давлении 10 атм по реакции
С довольно высоким выходом он получается из карбазола и окиси
этилена:
КОН или
------>
NaOH
калиевая соль
карбазола
алкоголят
ок си этилкарбазол а
сн2сн2он сн=сн2
N-p-оксиэтилкарбазол N-винилкарбазол
N-Винилпирролидон — прозрачная жидкость, кипящая при 214—
215° С и 71—72° С при 3 мм рт. ст.; р= 1,036 г/см3, nD—1,513. Хорошо
растворяется в воде и органических растворителях.
Получают N-винилпирролидон винилированием а-пиррилидона в две
стадии: сначала получают пирролидон-калий, который затем под дав-
лением переводят в N-винилпирролидон:
СН2—СН2 +кон Н2С----СН2 +снвСН; +н,о Н2С--СН2
• ।---------* 1 । -----------—* 1 1
С112 с=о Н2С с=о ~кон Н2С с=о
^NH XrZ 4N- СН=СН2
2-Винилпиридин—бесцветная жидкость с острым запахом, т. кип.
159° С, р20=0,975 г/см3; «о =1,5490; растворим в органических раствори-
телях и слабо растворим в воде.
2-Винилпиридин получают из 2-метилпиридина в две стадии: взаи-
модействием с формальдегидом с образованием 2-этанолпиридина с по-
следующей дегидратацией в щелочной среде:
.о
нс\н /X
I I —-I I II
Х/\ перегонка
N СН3 N СН2СН2ОН N СН=СН2
194
Аналитические методы определения аналогичны методам определе-
ния других виниловых соединений.
Подготовка N-винйлкарбазола для полимеризации
Чистота винилкарбазола, применяемого для полимеризации, имеет
большое значение: чем чище мономер, тем легче вызывается его поли-
меризация.
Мономер может быть очищен тщательной перегонкой под уменьшен-
ным давлением в присутствии ингибиторов (например, щелочного метал-
ла или не слишком низкокипящего органического основания) для пре-
дотвращения полимеризации.
Чистый N-винилкарбазол, обладающий высокой склонностью к по-
лимеризации, получают повторной кристаллизацией. Кристаллический
N-винилкарбазол, полученный из растворителей, содержащих гидро-
ксильные группы (например, из метилового спирта), полимеризуется
медленнее, чем полученный из бензола или лигроина.
Анализ N-винилкарбазола
Содержание N-винилкарбазола определяют методом оксимирования
продукта его кислого гидролиза; элементным анализом на содержание
азота, углерода и водорода, а также определением молекулярного веса
и ненасыщенности (бромированием, см. стр. 58).
Определение N-винилкарбазола методом оксимирования продукта
гидролиза. N-Винилкарбазол гидролизуют раствором ортофосфорной ки-
слоты:
СН3С
СН=СН2
Выделившийся ацетальдегид определяют
тодом:
Ю .NOH
CH3Cf + NH2OH- НС1 -> СН3-С<
\н хн
гидроксиламлновым ме-
+ HCI + Н2о
В трехгорлую колбу емкостью 250 мл, снабженную форштоссом с
брызгоуловителем, капельной воронкой и тубусом для введения мономе-
ра (рис. 50), вливают 50 мл этилового спирта и точную навеску 0,3—0,4 г
N-винилкарбазола.
В колбу-приемник наливают 25 мл 7%-ного спиртового раствора со-
лянокислого гидроксиламина и помещают ее в охлаждающую баню со
снегом или льдом. В капельную воронку наливают 3—4 мл 10%-ноге
раствора ортофосфорной кислоты. о
Сначала нагревают реакционную колбу на водяной бане до 50—60° С
для растворения N-винилкарбазола в спирте. Затем приливают по кап-
195
лям ортофосфорную кислоту и на кипящей бане отгоняют образовав-
шийся ацетальдегид в приемник, где он реагирует с солянокислым гид-
роксиламином. Отгонка продолжается около 1 ч, после чего в приемник
вливают через фор штосс 100 мл воды,
Рис. 50. Прибор для определения
N-винилкарбазола методом окси-
мирования продукта гидролиза
закрывают его пробкой и оставляют
на 1 ч при 20° С.
В аналогичных условиях проводят
контрольный опыт, для чего в колбу
вливают 50 мл спирта, прибавляют из
капельной воронки 3—4 мл раствора
ортофосфорной кислоты и проводят
перегонку в течение ~1 ч, через фор-
штосс вливают 100 мл воды, закры-
вают пробкой и оставляют на 1 ч при
20° С.
Обе пробы титруют раствором 0,5 н.
NaOH из микробюретки в присутствии
метилового оранжевого в качестве ин-
дикатора.
Содержание N-винилкарбазола х
(в %) рассчитывают по формуле
Х=
(а — Ь) /<0,0966-100
g
где а и b — объемы 0,5 н. раствора NaOH, затраченные на титрова-
ние анализируемой и контрольной проб, мл; К. — поправка к титру
0,5 н. NaOH; 0,0966 — количество винилкарбазола, соответствующее 1 мл
точно 0,5 н. раствора NaOH, г; g — навеска мономера, г.
Техника безопасности при работе с .производными
этилена со сложными заместителями
N-Винилкарбазол — токсичное вещество, поэтому при работе с ним
необходимо соблюдать соответствующие меры предосторожности.
При действии этого вещества на кожу развивается дерматит, степень
заболевания зависит от продолжительности воздействия и от чувстви-
тельности к винилкарбазолу, которая у разных людей неодинакова.
При отравлении винплкарбазолом в начале заболевания наблюдает-
ся резкое повышение температуры. Необходимо избегать вдыхания паров
и пыли винилкарбазола и их попадания на кожу.
При работе с вини-лкарбазолом рекомендуется соблюдать следую-
щие меры предосторожности:
1) рабочее помещение должно иметь хорошую вентиляцию. Все ра-
боты -с винплкарбазолом проводить в вытяжном шкафу;
2) во время работы с винплкарбазолом необходимо надевать за-
щитную одежду, включая резиновые сапоги и резиновые перчатки;
3) для защиты лица, шеи и других открытых частей тела, подвер-
гающихся действию винилкарбазола, следует смазывать их защитной
мазью или вазелином.
196
I
N-винилпирролидон и 2-винилпиридин образуют в газообразном или
парообразном состоянии взрывоопасные смеси с кислородом воздуха.
Винилпиридины физиологически активны, что следует учитывать при
работе с этими веществами.
РАБОТА 100. ПОЛУЧЕНИЕ N-ВИНИЛКАРБАЗОЛА
Исходные продукты: очищенный карбазол—170 г; едкое кали — 56 г или едкий
натр — 40 г; ксилол — '500 г; окись этилена — 44 г.
Оборудование: железный тигель; трехгорлая колба с механической мешалкой и
капельной воронкой; установка для перегонки в вакууме; вакуум-эксикатор.
Карбазол 2—3 ч нагревают с едким кали при 240° С в железном тиг-
ле (при применении едкого натра нагревают при 340° С).
После испарения воды, которое сопровождается возгонкой некоторо-
го количества карбазола, остается твердая калиевая соль, окрашенная в
желтый цвет. Ее извлекают из тигля и охлаждают в вакуум-эксикаторе.
Выход соли около 200 г.
Калиевую соль карбазола измельчают и переносят в трехгорлуго
колбу, где находится 300 г ксилола. Из капельной воронки при перемеши-
вании прибавляют в течение около 2 ч раствор жидкой окиси этилена в
"200 г охлажденного ксилола. Температуру медленно повышают до 50° С
и продолжают перемешивание еще 3 ч. Затем содержимое колбы фильт-
руют, прибавляют немного воды и перемешивают, чтобы разложить
алкоголят оксиэтилкарбазола. Получают коричневую жидкость. После
удаления растворителя перегонкой при атмосферном давлении остав-
шуюся жидкость перегоняют под вакуумом.
N-р-Оксиэтилкарбазол перегоняется при давлении 10 мм рт. ст. и
230° С. Выход продукта около 190 г.
N-р-Оксиэтплкарбазол, перекристаллизованный из ксилола, пред-
ставляет собой бесцветные мелкие иглы с т. пл. 81—82° и т. кип. 234° С
при 10 мм рт. ст.
Дегидратацией N-р-оксиэтилкарбазола с помощью едкого калй полу-
чают N-винилкарбазол.
РАБОТА 101. ПОЛУЧЕНИЕ ПОЛИВИНИЛКАРБАЗОЛА
ПОЛИМЕРИЗАЦИЕЙ В ЭМУЛЬСИИ
Исходные продукты: N-винилкарбазол — 40 г; натриевая соль изобутилнафтаяин-
.-сульфокислоты (иекаль) —0,2 г; ализариновое масло—0,1 г; дистиллированная вода —
50 г; 20%-ный водный раствор аммиака; перекись водорода (30%-ная)—0,3 г; соляная
кислота (конц.).
Оборудование: трехгорлая колба, снабженная мешалкой с. герметическим затво-
ром, обратным холодильником и термометром; водяная баня; воронка Бюхнера.
В трехгорлой колбе прибора при перемешивании растворяют в воде
некаль и ализариновое масло. После их полного растворения при тща-
тельном перемешивании вводят растертый в тонкий порошок N-винил-
карбазол, затем добавляют несколько капель водного раствора аммиака
и интенсивным перемешиванием эмульгируют смесь.
Полученную эмульсию нагревают при перемешивании до 95—98° С
и медленно прибавляют перекись водорода. Во время полимеризации
необходимо следить за тем, чтобы раствор все время оставался слабо
щелочным, в противном случае добавляют еще небольшое количество
раствора аммиака.
Реакция продолжается 30 ч, после чего для коагуляции полимера
вводят небольшое количество соляной кислоты. Выпавший осадок от-
фильтровывают на воронке Бюхнера, промывают сначала метиловым
спиртом, затем водой до исчезновения реакции на ион С1~ и сушат при
40—45° С. Полимер прессуют при 220—230° С и давлении 200—
250 кг/см2.
Определяют теплостойкость образца, сравнивают с полистиролом.
РАБОТА 102. ПОЛУЧЕНИЕ СОПОЛИМЕРА N-ВИНИЛКАРБАЗОЛА
СО СТИРОЛОМ В СУСПЕНЗИИ
[R'l
nCH2=CH + тСН2=СН -> ~ СН2—СН—СН2-СН ~
II II
N С6Н5 С6Н5 N
/\/\/\ У\/\/Ч
fill 11 I I
Ч/—xz \/—\z
Исходные продукты: N-винилкарбазол — 10 г; стирол — 2 г; перекись бензоила —
0,1 г (или динитрил азоизомасляной кислоты—0,33 г); поливиниловый спирт—0,38 г.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); стаканчики
емкостью 50 мл; водяная баня (химический стакан емкостью 1 л); воронка Бюхнера;
термошкаф.
Рис. 51. Форма для
пресс-литья с гори-
зонтальной плоско-
стью разъема:
1 — тигель с плунже-
ром; 2 — литниковый
канал; 3 форма
В пробирке растворяют поливиниловый спирт в 60 мл дистиллиро-
ванной воды при нагревании на водяной бане. Затем охлаждают раствор
и вливают смесь мономеров, в которой предва-
рительно растворяют перекись бензоила. Переме-
шивание реакционной смеси стараются проводить,
так, чтобы капли мономеров находились во взве-
шенном состоянии во всем объеме жидкости.
Перемешивание должно быть равномерным и
постоянным во избежание коагуляции на стадии
образования полимерно-мономерных частиц. Реак-
цию проводят при 80° С в течение 6—7 ч, пока об-
разовавшиеся гранулы не начнут осаждаться на
дне пробирки.
Гранулы отфильтровывают на воронке Бюхне-
ра, высушивают в термошкафу. _
Определяют выход, температуру плавления, ин-
декс расплава. Перерабатывают пресс-литьем при
температуре расплава 180° С и температуре формы 30° С. Определяют
теплостойкость и термостойкость образцов (см. ч. II).
Пресс-литье или литьевое прессование является периодическим методом литья,
под давлением, которое осуществляется в специальных формах, состоящих из двух
частей (рис. 51'): для получения расплава и для формования изделия. Характерная
особенность литья под давлением в данном случае соблюдается: расплав полимера
через узкий канал (литниковый канал) поступает в замкнутую полость, образуемую»
двумя полуматрицами. Расположение формы может быть горизонтальным и верти-
кальным.
198
РАБОТА 103. ПОЛУЧЕНИЕ СОПОЛИМЕРА N-ВИНИЛКАРБАЗОЛА
С АКРИЛОНИТРИЛОМ В ПРИСУТСТВИИ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЙ ИНИЦИИРУЮЩЕЙ
СИСТЕМЫ
Исходные продукты: N-винилкарбазол — 16 г; нитрил акриловой кислоты — 4 г;
персульфат натрия — 0,4 г; тиосульфат натрия — 0,28 г.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная механической ме-
шалкой с герметическим затвором, обратным холодильником и термометром; стаканчики
емкостью 50 мл; водяная баня; термошкаф; воронка Бюхнера.
Навески персульфата и тиосульфата натрия растворяют в 40 мл
дистиллированной воды в круглодонной трехгорлой колбе прибора.
К раствору приливают мономеры и при непрерывном перемешивании
нагревают на водяной бане при 60° С в течение 2 ч. Выпавший осадок
промывают на воронке Бюхнера водой до отрицательной реакции на
сульф ат-ион.
Определяют температуру размягчения сополимера, индекс распла-
ва. Прессуют сополимер в пресс-форме на стандартный брусок при
180° С, удельном давлении 200 кг/см2; выдержка в прессе 2 мин на 1 мм
толщины изделия. Определяют твердость по Бринеллю и теплостойкость
по Мартенсу.
РАБОТА 104. ПОЛУЧЕНИЕ ПОЛИ-К-ВИНИЛПИРРОЛИДОНА
В ВОДНОМ РАСТВОРЕ
СН2—СН- -
I
N
н2с—сн2
Исходные продукты: N-винилпирролидон — 20 г; перекись водорода — 0,02 г; ам-
миак 25%-ный— 0,02 г; ацетон для высаждения полимера.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная механической ме-
шалкой с герметическим затвором, обратным холодильником и термометром; капельная
воронка; водяная баня.
В колбу прибора помещают 70 мл дистиллированной воды, перекись
водорода и аммиак; затем в раствор через капельную воронку прилива-
ют мономер и при энергичном перемешивании нагревают реакционный
m
раствор при 50° С в течение 2 ч. Полимер осаждают из раствора ацето-
ном (или раствором NaCl). Определяют содержание непрореагировав-
шего мономера бромированием (стр. 136).
РАБОТА 105. ПОЛУЧЕНИЕ СОПОЛИМЕРА N-ВИНИЛПИРРОЛИДОНА
С АКРИЛОНИТРИЛОМ
R"
СН2= СН + лСН2=СН ->
N CN
Н2С С=О
I I
н2с—сн2
сн2-сн-сн2-сн—сн2—сн,~
I I I
CN N N
H2d/'c=o н2с/Хс=о
Il II
НгС—СН2 НгС—СН2
Исходные продукты: N-винилпирролидон — 20 г; нитрил акриловой кислоты — 5 гр
динитрил азоизомасляной кислоты — 0,05 г; поливиниловый спирт — 0,76 г.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная механической ме-
шалкой с герметическим затвором, обратным холодильником и термометром; водяная
баия; воронка Бюхнера: стаканы емкостью 50 мл.
В трехгорлой колбе прибора при нагревании растворяют поливини-
ловый спирт в 150 мл дистиллированной воды. Инициатор растворяют в
акрилонитриле и к раствору добавляют N-винилпирролидон. Смесь,
мономеров вливают в охлажденный раствор поливинилового спирта, на-
гревают 2 ч при постоянном перемешивании с обратным холодильником
при 60—70° С. Образовавшийся сополимер высаживают добавлением'
кристаллической NaCl. Осадок отфильтровывают, промывают дистил-
лированной водой и высушивают в термошкафу, постепенно поднимая
температуру от 40 до 70° С.
Определяют выход сополимера и растворимость.
Изготавливают пленку поливом из раствора и определяют разру-
шающее напряжение и относительное удлинение при разрыве.
РАБОТА 106. ПОЛУЧЕНИЕ СОПОЛИМЕРА N-ВИНИЛПИРРОЛИДОНА
СО СТИРОЛОМ
тСН2=СН—N + «СН2=СН 1(СН‘),СС^
Н2с/\С=О
Н2С----сн2
СН2-СН-СН2—СН -
I I
N
н2с|/\с=° | II
н rl 1гн
112'-' С Г12
.Исходные продукты: N-винилпирролидон — 8 г; стирол — 2 г; динитрил азоизомас-
ляной кислоты — 0,1 г; поливиниловый спирт — 0,35 г; ацетон.
Оборудование: прибор для гранульной полимеризации (см. рис. 20); воронка Бюх-
нера; стаканы емкостью 50 мл; термошкаф; водяная баня.
В пробирке прибора растворяют при нагревании поливиниловый
спирт в 60 мл дистиллированной воды. В стаканчике растворяют инициа-
тор в стироле и к раствору добавляют N-винилпирролидон. Мономеры.
200
вливают в реакционную пробирку и 2—4 ч нагревают при 60—70° С при
энергичном перемешивании. Сополимер высаждают из раствора кри-
сталлическим NaCl. Осадок отфильтровывают, отмывают дистиллиро-
ванной водой от иона С1_ и высушивают сначала на воздухе, а затем в
термошкафу, постепенно поднимая температуру от 40 до 70° С.
Изготавливают пленку поливом из раствора и определяют разру-
шающее напряжение и относительное удлинение при разрыве.
РАБОТА 107. ПОЛУЧЕНИЕ ПОЛИВИНИЛПИРИДИНА
В ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЙ ИНИЦИИРУЮЩЕЙ
СИСТЕМЕ
Исходные продукты: 2-винилпиридин — 20 г; персульфат натрия — 0,45 г; тиосуль-
фат натрия — 0,30 г; хлористый натрий.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная мешалкой с герме-
тическим затвором, обратным холодильником и термометром; водяная баня; воронка
Бюхнера; термошкаф.
В реакционной колбе в 40 мл дистиллированной воды растворяют
соли и вливают мономер. После 2 ч нагревания с обратным холодильни-
ком на водяной бане при 60° С и постоянном перемешивании образовы-
вается полимер, его осаждают из раствора кристаллическим NaCl, от-
фильтровывают на воронке Бюхнера, промывают до отрицательно»
реакции на ионы SO|~ и С1~, высушивают в термошкафу при 60° С. Оп-
ределяют растворимость в разбавленных кислотах, температуру размяг-
чения. Сополимер используют в качестве эмульгатора и водораствори-
мого ионита.
РАБОТА 108. ПОЛУЧЕНИЕ СОПОЛИМЕРА 2-ВИНИЛПИРИДИНА
С АКРИЛОНИТРИЛОМ БЛОЧНЫМ МЕТОДОМ
[В’1
+ тСН2=СН----> - СН2—СН—СН—СН2
Исходные продукты: 2-винилпиридин — 10 г; акрилонитрил — 5 г; динитрил азоизо-
масляной кислоты — 0,15 г.
Оборудование: ампула из тугоплавкого стекла емкостью до 50 мл; термостат во-
дяной; термометр контактный.
В ампулу заливают 2-винилпиридин, акрилонитрил и инициатор,
тщательно протирают горлышко ампулы и запаивают ее на огне паяль-
ной горелки. Растворяют инициатор, взбалтывая содержимое ампулы, и
устанавливают ее в термостат при 60° С. Реакцию ведут до образования
полимера (неперетекающая масса). В полученном сополимере определя-
ют количество непрореагировавших мономеров бромированием (см-
стр. 136), растворимость его в воде, в разбавленных кислотах.
ГЛАВА VII. ГЕТЕРОЦЕПНЫЕ ПРОСТЫЕ ПОЛИЭФИРЫ
И ПЛАСТИЧЕСКИЕ МАССЫ НА ИХ ОСНОВЕ
Полиформальдегид (полиоксиметилен) (—СН2—О—)п — высокомоле-
кулярный гомолог полиоксиметиленов, полученных впервые Бутлеровым
методом постепенного концентрирования формалина в вакууме. Он
является первым представителем класса простых полиэфиров. Обладая
молекулярным весом 150 000, полиформальдегид, полученный в резуль-
тате ионной полимеризации формальдегида или его циклического три-
мера (триоксана), представляет собой ценный полимер, обладающий
высокой прочностью, устойчивый к действию органических растворите-
лей, с высокой износоустойчивостью. К недостаткам его следует отнести '
способность при 185—205° С деполимеризоваться до мономера. Стабили-
зация полиформальдегида блокировкой концевых групп дает возмож-
ность повысить предел его термической устойчивости и поставить его в
ряд таких технически ценных полимеров, как полиамиды, полиари-
латы и др.
Химизм образования полиформальдегида
Формальдегид очень реакционноспособен и полимеризуется спонтан-
но, причем этот процесс инициируется разнообразными полярными при-
месями. Характерной особенностью формальдегида является его способ-
ность к реакциям нуклеофильного и электрофильного характера в зави-
симости от природы катализирующих полимеризацию соединений.
Полимеризацию формальдегида вызывают амины и BF3, четвертич-
ные аммониевые основания и металл алкилы. Даже незначительные ко-
личества воды вызывают полимеризацию формальдегида; естественно,
что механизм полимеризации во всех случаях различен.
Вода, инициируя полимеризацию, участвует в передаче цепи и, сле-
довательно, снижает молекулярный вес:
Н (-ОСН2-)ЛО~ + Н2О -> Н (~ОСН2-)ЛОН + он
ОН -|- СН2О НОСН2О~
носн2о_ + СН2О -> НОСН2О (—СН2О-)Л-СН2—
Аналогично действует метанол. Катионная полимеризация в присутствии
+
комплекса H[BF3OH]~ протекает по следующей схеме:
4“ +СН3=О — + 4"CHj=O —Ь
H[BF3OH]~-----> BF3OHCH2-OCH2OH------> BF3OHCH2O-CH2O-CH2OH
202
Анионная полимеризация в присутствии аминов и следов воды, со-
держащихся в формальдегиде:
R3N + НОН -> [R3NH] ОН
ОН 4- СН2=О -* НОСН2-О~
НОСН2-О“ 4- СН2О -> НОСН2—ОСН2-О~
Полиформальдегид.можно получать в суспензии или в газовой фазе-
над катализатором.
Триоксан полимеризуется в любом агрегатном состоянии по катион-
ному механизму (катализаторы — кислоты Льюиса). Твердофазную по-
лимеризацию триоксана осуществляют при у-облучении. Максимальный
выход полимера при облучении достигается при температуре, близкой к
температуре плавления триоксана.
Деполимеризация полиформальдегида протекает по миграционному
механизму:
о 6 ой 6 он
X /X /X / -сн.о X /X/ -сн„о
СН2 СН2 СН2--------->- СН2 СН2------>- и т. д.
Замещение гидроксильных концевых групп на простые или сложно-
эфирные стабилизирует полимер. К стабилизации приводит также сопо-
лимеризация с 1,3-диоксоланом, окисью этилена, 1,3-диоксаном и др.
Т. пл. полимера 175—180° С, степень кристалличности около 75%;
р= 1,475 г/см3. Полимер белый, непрозрачный, легко окрашивается кра-
сителями. Сильные кислоты и щелочи разрушают его.
Технология производства полиформальдегида
Полимеризацию газообразного формальдегида осуществляют в реак-
торе, наполненном толуолом, ксилолом, пентаном или циклогексаном в
присутствии катализатора. Скорость подачи формальдегида должна со-
ответствовать скорости образования полимера. Полимер выделяется в
виде белого порошка. Температура реакции от —20 до +50° С, в зависи-
мости от природы катализатора.
Полимер отделяют от растворителя, отмывают водой и спиртом от
низкомолекулярных фракций и высушивают в вакуум-сушилке при 40—
60° С. Стабилизируют антиоксидантами в смеси с полиамидом 548/27.
Наибольший практический интерес представляют полимеры с моле-
кулярным весом 30 000—100 000.
Полиформальдегид выпускают по СТУ 36-13-8—64 двух марок: мар-
ка А — для изготовления литьевых изделий с индексом расплава не ниже
1,8; марка 13—-.для изготовления изделий литьем или экструзией с ин-
дексом расплава не ниже 1,2, термостабильность при 222° С не менее
20 мин.
Полиформальдегид превосходит полиэтилен по непроницаемости для
паров и спиртов, эфиров. Одиако проницаемость его для водяного пара
почти в 10 раз больше, чем у полиэтилена.
203
Сополимеризацию “‘триоксана с 1,3-диоксаланом, окисью этилена и
.другими мономерами производят в расплаве или в суспензии.
Для изготовления полимеров предложен ряд методов: сополимери-
зация на бесконечно движущейся ленте в камере под азотом с после-
дующим нагревом до 100° С в реакторе с конусным днищем (рис. 52);
сополимеризация во вращающемся барабане в камере в атмосфере азота
Рис. 54. Аппарат с
ложным днищем:
/ — обогреваемый до-
затор; 2 — разбрызги-
вающее устройство;
3 — обогревающая ка-
мера; 4 — распыли-
тельное устройство
для газа
Рис. 52. Производство полиформаль-
.дегида: бесконечная лента в камере
под инертным газом и реактор с ко-
нусным днищем:
1 — линия ввода триоксана и сомономера;
2—мешалка; 3— линия ввода инициатора;
4 — обогреваемый сосуд; 5 — вход горячего
• азота; 6 — подвижная лента; 7—продукт;
8 — выход горячего азота %
Рис. 53. Вращающий-
ся барабан для сопо-
лимеризации триокса-
на с диокса л а ном:
1 — обогреваемый доза-
тор; 2 — барабан; 3 —
продукт; 4 —нож
в присутствии BF3 при 100° С (рис. 53); в двушнековом смесителе «ко-
кнетере» при последовательно повышающейся температуре от 0 до
115°С (время реакции около 10 мин). В «ко-кнетере» реакционная мас-
са перемещается по спирали через цилиндр смесителя. Кроме того/
имеет место возвратно-поступательное движение шнека вдоль оси.
Сополимеризацию можно осуществить также в аппаратах с ложным
днищем, на которое помещен катализатор, а смесь мономеров поступает
через верхнее отверстие из бункера смесителя (рис. 54).
Сополимеры формальдегида представляют собой термопластичные
материалы с высокой степенью кристалличности, однако с более низкой .
температурой плавления по сравнению с гомополимером. Преимущест-
вом сополимера является повышенная термостабильность (90 мин) и
формуемость. Сополимеры устойчивы к действию концентрированных ;
щелочей и солей при 100° С. ;
До 60° С они устойчивы к действию практически всех растворителей.
Кислоты разрушают сополимеры так же, как и гомополимеры. !
204
В полиформальдегиде удачно сочетаются высокая механическая
^прочность с хорошими диэлектрическими свойствами (см. приложение).
Полиформальдегид имеет низкий коэффициент трения по стали, почти
не изменяющийся в интервале 20—120° С и при нагрузке до 175 кг/см2.
По сопротивлению истиранию и по усталостной прочности полиформаль-
дегид превосходит большинство известных термопластов. Эти свойства
определяют применение его в качестве заменителя стали, цветных метал-
лов, цемента, дерева и других материалов. Полиформальдегид прекрас-
но обрабатывается на станках. Его можно пилить, резать, забивать в не-
го гвозди, клепать, штамповать.
Из полиформальдегида изготавливают втулки, вкладыши подшипни-
•ков скольжения, сепараторы и кольца подшипников качения, тела каче-
ния, втулки роликовой цепи, бесшумные шестерни, зубчатые ролики в
•счетных устройствах и т. п., а также различные бытовые детали и при-
боры.
Из полиформальдегида можно получить волокно, а также прессован-
ные пленки.
Исходное сырье для получения полиформальдегида
и его сополимеров
а-Полиоксиметилен. Газообразный формальдегид получают из По-
лиоксиметилена, содержащего не менее 99,7—99,9% формальдегида.
«-Полиоксиметилен в виде белого порошка выделяют из обычного фор-
малина при его концентрировании в вакууме в присутствии щелочей.
Высушенный а-полиоксиметилен загружают в деструктор и нагрева-
ют до 160—180° С с целью деполимеризации до газообразного формаль-
дегида. Последний очищают от примесей и влаги вымораживанием
при —20° С.
О
Н2с/ 'ХСН2 20
Диоксан । । — жидкость ст. кип. ЮГ С; d4 =1,033; ядо-
Н2С СН2
вит. С водой образует азеотропную смесь с т. кип. 87,8° С, содержащую
81,6% диоксана. Диоксан получают перегонкой этиленгликоля с 4%-ной
H2SO4: о
НгС^
2СН2ОН-СН2ОН -- (L
о
Н2С СН2 _ ci
Триоксан I | — кристаллическое соединение с т. пл. or
ОО '
Хсн2
205
62° С и т. кип. 114—115° С, т. разл. 300° С. Растворим в воде и полярных
растворителях. Триоксан получают тримеризацией формальдегида в.
водном растворе в присутствии 2%-ной H2SO4:
СН2
ЗСН2О->- o' 'о
I I
Н2С сн2
oz
1,3-Диоксолан— жидкость с т. кип. 105—106° С ;с?4° = 1,034. Получа-
ют диоксолан реакцией окиси этилена с формальдегидом:
СН2-СН2 + СН2=О->-
сн2
с/ 'о
I I
н2с—сн2
Реакцией окиси этилена с ацетальдегидом образуется 2-метил-
1,3-диоксолан.
Качественное определение полиоксиметилена, диоксана, триоксана
и 1,3-диоксолана основано на деполимеризации и идентификации фор-
мальдегида (см. ч. II). Количественное определение формальдегида
см. ч. II.
РАБОТА 109. ПОЛИМЕРИЗАЦИЯ ФОРМАЛЬДЕГИДА
В ПАРОВОЙ ФАЗЕ
Схема реакции:
(С,Н5)=Р
пСН2О------>(-СН2О—)п
Исходные продукты: безводный параформальдегид — 1'00 г; пентан безводный —
600 мл; трифеннлфосфин — 0,2 г.
Оборудование: трехгорлая колба, снабженная механической мешалкой с гермети-
ческим затвором, обратным холодильником и трубкой для ввода формальдегида; ло-
вушки — 2 шт.; воронка Бюхнера; колба из тугоплавкого стекла для пиролиза пара-
фсрмальдегида; термометр, ванна для охлаждения с ацетоном н сухим льдом (рис. 55).
Безводный параформальдегид подвергают пиролизу в колбе при
180° С. Выделяющиеся пары проходят через ловушки, помещенные в
охлаждающую смесь с температурой —20° С. К ловушкам присоединя-
ют трехгорлую реакционную колбу, в которую заливают пентан и три-
фенилфосфин. Пары формальдегида проходят при энергичном переме-
шивании в реакционную среду, температура которой 25° С. По мере про-
пускания паров образуется белый порошкообразный продукт, который
используют для переработки в изделия (см. работу ПО).
206
Рис. 55. Прибор для получения полиформальдегида в паровой фазе
РАБОТА 110. ИЗГОТОВЛЕНИЕ ПЛЕНОК НА ОСНОВЕ
ПОЛИФОРМАЛЬДЕГИДА
Исходные продукты: полиформальдегид — 30 г; неозон Д — 0,05 г; стеарат каль-
ция — 1 г.
Оборудование: пластины из нержавеющей стали; пресс лабораторный; плиты на-
гревательные; ступка фарфоровая; ограничительные рамки разной толщины — 0,5—1
и 2—3 мм.
Полиформальдегид смешивают с неозоном Д и стеаратом кальция
в ступке и таблетируют на холоду между полированными пластинами с
ограничительной рамкой. Затем меняют ограничительную рамку на бо-
лее тонкую и прессуют на лабораторном прессе при 200° С, удельном
давлении 100 кг/см2 и выдержке 5 мин. Из отформованных пленок гото- ,
вят стандартные образцы (см. ч. II) и испытывают их на разрушающее
напряжение при разрыве, изгибе, относительное удлинение при разры-
ве и диэлектрические свойства.
РАБОТА 111. ПОЛУЧЕНИЕ СОПОЛИМЕРА ТРИОКСАНА С 1,3-ДИОКСАЛАНОМ
В РАСПЛАВЕ В ПРИСУТСТВИИ BF3
Схема реакции:
О
Н2С СН2 /О—СН2 вр„
т | | 4- пН2С< |-------> ~ СН2О—СН2О—СН2СН2О ~
О О Ч)—СН2
сн2
207
Исходные продукты: триоксан — 30 г; дпоксалан — 9 г; эфират трехфтористого*'2
бора (10-3 М раствор в гептане)—0,3—0,6 мл; 30%-ный раствор триэтаноламина в.
ацетоне; ацетон; дифенилолпропан — 0,26 г; дицнанднамии — 0,25 г.
Оборудование: пробирка диаметром 45—50 мм, высотой 150—170 мм с хлоркаль- .
циевой трубкой; ступка фарфоровая; экструдер; склянка для промывных растворов;,
баня с полиэтнлоксановой жидкостью; баня со сплавом Вуда; вакуум-установка.
В пробирку загружают триоксан и помещают в баню с полиэтил-
силоксановой жидкостью, нагретой до 70° С. После расплавления триок-
сана добавляют диоксалан и катализатор. При введении последнего-
происходит почти мгновенная полимеризация. После 30 мин выдержки
полимера при 70° С его извлекают из пробирки, дробят и промывают
горячей водой, 30%-ным триэтаноламином в ацетоне и, наконец, ацето- :
ном. Продукт высушивают и определяют индекс расплава при 190° С.
Для стабилизации полученного полиформальдегида добавляют
0,5% дифенилолпропана и 0,5% дициандиамида. Стабилизацию осуще-
ствляют, пропуская смесь полимера и стабилизаторов через экструдер.
Стабильность определяют по времени разложения на воздухе 1 % образ-
ца при 222° С. Для удаления нестабильной части полимер нагревают до-
250° С в бане со сплавом Вуда в вакууме (10—15 мм рт. ст.) в течение
30 мин.
РАБОТА 112. ПОЛУЧЕНИЕ СОПОЛИМЕРА ТРИОКСАНА
С 1,3-ДИОКСАЛАНОМ В РАСПЛАВЕ
Схема реакции:
О
^СНг
п I I
О О
,о-сн2
+ mH2C< |
ХО-СН2
СН2О—СН2О-СН2СН2О
Исходные продукты: триоксан—50 г; -1,3-диоксалан—15 г; фторборат п-иитро-
фенилдназония — 0,01 г; 30%-ный ацетоновый раствор триэтаноламина; ацетон; дифе-
нилолпропан — 0,25 г; дициандиамид — 0,25 г.
Оборудование: баня с полиэтилсилоксановой жидкостью; пробирка диаметром
45—50 мм, высотой 150—170 мм, снабженная хлоркальциевой трубкой на шлифе; ступка
фарфоровая; экструдер; склянка для промывных растворов.
В пробирку загружают триоксан и помещают ее в баню, нагретую
до 70° С. После расплавления триоксана добавляют диоксалан, катали-
затор и перемешивают.
После периода индукции (4 мин) масса быстро полимеризуется и
выдерживается 30 мин при 70° С. Затем продукт извлекают из пробирки,,
дробят в фарфоровой ступке, там же промывают горячей водой, 30%-ным
раствором триэтаноламина и ацетоном, сливая растворы для регенера-
ции.
Продукт высушивают на воздухе и определяют индекс расплава.
Для стабилизации полученного продукта поступают так же, как описано
в работе № 111. Определяют термостабильность.
208
РАБОТА 113. ПОЛУЧЕНИЕ СОПОЛИМЕРА ТРИОКСАНА
С 1.3-ДИОКСАЛАНОМ В СУСПЕНЗИИ
Схему реакции см. стр. 208.
Исходные Продукты: триоксан—10 г; диоксалан—3 г; BF3(C2H5)2O— О, В—0,3 мл
1С~3 М раствора в гептане; гептан — 30 мл; 30%-ный раствор триэтаноламина в ацето-
не; ацетон; стабилизаторы: дифенилолпропан —0,1 г, дициандиамид — 0,1- г.
Оборудование: пробирка диаметром 45—50 мм, высотой 150—170 мм, снабженная
-обратным холодильником с хлоркальциевой трубкой, мешалкой и тубусом для ввода
катализатора; баня с полиэтилсилоксановой жидкостью; термометр; фарфоровая ступ-
ка; экструдер; склянка для слива растворов; воронка Бюхнера.
В пробирку при перемешивании загружают триоксан, диоксалан,
гептан и нагревают до 70° С на бане, закрыв холодильник хлоркальцие-
вой трубкой, до образования суспензии. Затем вводят катализатор через
боковой тубус, который затем закрывают притертой пробкой, после че-
го смесь перемешивают еще 2—3 ч.
Образовавшийся продукт фильтруют, промывают и стабилизуют,
как описано в работе 111. Определяют индекс расплава и перерабатыва-
ют литьем или экструзией как описано в работе 114.
РАБОТА 114. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ ПОЛИФОРМАЛЬДЕГИДА
ЛИТЬЕМ ПОД ДАВЛЕНИЕМ И ЭКСТРУЗИЕЙ
Полиформальдегид перерабатывают'в изделия литьем под давле-
нием и экструзией.
Профилированные изделия (трубы, уголки, листы) экструдируют
через угловые (см. рис. 8) и прямоточные (см. рис. 4, 5) головки с обя-
зательным охлаждением в водяной ванне (см. рис. 11). Кабельную изо-
ляцию для проводов небольшого диаметра изготавливают экструзией
через кольцевые головки (см. рис. 10).
Переработку полиформальдегида экструзией или литьем под давле-
нием производят при температуре в цилиндре не выше 205° С, т. е. толь-
ко на 30° С выше температуры плавления полимера. Это вызвано тем,
что вязкость полиформальдегида при повышении температуры не изме-
няется и, следовательно, нецелесообразно перегревать материал во из-
бежание разложения.
Полиформальдегид как кристаллический полимер имеет довольно
четкую температуру плавления и быстро застывает в форме при неболь-
шом охлаждении, поэтому выдержка при формовании литьем и экстру-
зией незначительна. Целесообразно охлаждение экструдированных
изделий в ванне с подогретой водой для снятия внутренних напряжений.
Невысокая термостабильность расплава вызывает необходимость
точной регулировки температуры в цилиндрах машин. Экструдеры вы-
бирают с отношением длины к диаметру 20:1, а литье под давлением
желательно производить на червячных машинах или машинах с пред-
пластикаторами (рис. 56, 57), обеспечивающими более мягкие условия
нагрева материала.
209
Перед работой машина должна быть очищена от загрязнений. Для
этой цели лучше всего воспользоваться полиэтиленом, который разогре-
вают и формуют при температуре формования полиформальдегида.
Рис. 56. С/ем а шнековой машины для литья под
давлением:
/ — форма; 2— цилиндр; 3 — шиек; 4 — загрузочный бун-
кер; а — впрыск (форма заполнена частично); б — впрыск
(форма заполнена); в — разъем формы
Исходные продукты: ста-
билизированный полиформаль-
дегид— 100 г.
Оборудование: лаборатор-
ная литьевая машина или экст-
рудер с водяной ванной.
Предварительно разо-
гревают цилиндр очи-
щенной машины да
205° С, затем загружают
полиформальдегид в за-
грузочный бункер литье-
вой машины (экструде-
ра). Температура в фор-
ме 60—75° С, температу-
ра в головке экструдера
не выше 230° С.
Для снятия напряже-
ний и сохранения ста-
бильности размеров гото-
вое изделие подвергают
термообработке при
160° С в течение 10—20-
мин. На стандартных об-
разцах определяют раз-
рушающее напряжение
при растяжении и изгибе
(ч. II).
Рис. 57. Схема литья под давлением
на машинах с предпластикаторами
поршневым (а) и червячным (б):
1 — плунжер предпластикатора; 2 — плун-
жер литьевой машины; 3 — форма; 4 — за-
грузочный бункер; 5— шнек предпластика-
тора
ГЛАВА VIII. ПОЛИМЕРЫ, ПОЛУЧАЕМЫЕ ПОЛИМЕРИЗАЦИЕЙ
НЕУСТОЙЧИВЫХ гетероциклических соединении
Из полимеров на основе гетероциклических мономеров наибольшее зна-
чение имеют полимеры, получаемые из лактамов (е-капролактама, энан-
толактама). Значительно уступают им по значению полимеры окиси
этилена и окиси пропилена. В результате полимеризации лактамов об-
разуются полиамиды строения HI—HN—R—СО—]ОН, а из окисей ал-
киленов— полиалкиленоксиды строения Н[—О—R—]ПОН, т. е. простые
полиэфиры, вещества, имеющие одинаковую общую формулу с полиме-
рами альдегидов, получающихся методами ионной полимеризации.
Большой практический интерес представляет полиэфир — полиоксахлор-
метилциклобутан (пентапласт), получающийся полимеризацией соответ-
ствующего циклического соединения 3,3-бпс-(хлорметил) -оксациклобу-
тана и также содержащий простую эфирную связь.
Химизм образования полимеров
из гетероциклических мономеров
Раздельное описание полиамидов, получаемых методом полимери-
зации напряженных гетероциклов и методом поликонденсации, оправда-
но тем, что различные механизмы реакций определяют различную тех-
нологию. То же относится и к раздельному описанию процессов получе-
ния простых полиэфиров полимеризацией альдегидов и напряженных
гетероциклов.
Возможность образования полимеров из циклических мономеров
определяется термодинамической и кинетической устойчивостью цикла.
Полимеризация циклического .мономера возможна в том случае,
когда свободная энергия системы AF уменьшается при переходе к поли-
меру, т. е. с увеличением теплового эффекта реакции и с повышением
энтропии системы.
При превращении напряженного цикла в линейную молекулу не
возникает новых связей, поэтому энтальпия определяется напряженно-
стью цикла. Чем более напряжен цикл, тем выше тепловой эффект реак-
ции. Наиболее напряженные циклы — трех- и семичленные. Полимери-
зация трехчленных циклов происходит главным образом за счет измене-
ния энтальпии, а полимеризация циклов, имеющих семь и более членов,
возможна и за счет изменения энтропии, так как изменение энтальпии
при переходе таких соединений из циклической формы в линейную близ-
ко к нулю.
Кинетическая устойчивость цикла характеризуется подвижностью в
нем связей в условиях реакции. Поэтому наиболее кинетически неустой-
чивыми оказываются гетероциклы, содержащие связь С—гетероатом.
211
Однако для создания кинетической неустойчивости необходимо вводить- я
активаторы, чаще всего воду, кислоты, амины. Так, лактамы и окиси- I
алкиленов легко полимеризуются в присутствии воды при высокой тем- •ч
пературе (200—270°С). -J
Полимеризация циклических соединений в присутствии активаторов
протекает по механизму гидролитической или миграционной полимери- ;
зации, называемому полиприсоединением. Присоединение мономера к ;
растущей цепи происходит до тех пор, пока не установится равновесие:. ’
R A
—R—A—
Полимеризация циклических соединений—процесс экзотермиче-
ский, следовательно, константа равновесия с повышением температуры
уменьшается и реакция образования полимера затрудняется.
Процесс раскрытия цикла автокаталитический, проходит через ин-
Аукционный период. Катализатором является аминокапроновая кисло- •<
та, а в дальнейшем олигомеры, содержащие группы NH2 и СООН.
В случае полимеризации окисей алкиленов катализаторами являются
диоксипроизводные.
Однако в отсутствие воды аминокислоты и гликоли не катализиру- ч
ют полимеризацию, что указывает на ионный механизм каталитическо- <
го действия этих соединений. Катализирующим действием обладает их 4
ионная форма: NH3(CH2)5COO и НОСН2СН2ОН.
Равновесие между циклическим и линейным соединением определи- 1
ет выход полимера, а средний коэффициент полимеризации определяет- J
ся амидным равновесием. ?
В случае полимеризации трехчленных циклов в связи с их высокой ‘.
напряженостью реакция практически неравновесна.
Семи- и трехчленные циклы могут полимеризоваться не только по ri
ступенчатому гидролитическому механизму, но и по ионному цепному
(см. работы 119—121). Четырехчленный цикл 3,3'-бис-(хлорметил)
циклобутана полимеризуется только по ионному механизму и в присут-
ствии BF3 и воды.
H
'i
Технология производства полимеров
из гетероциклических соединений
Капролактам образует полимер — поликапроамид-6 в присутствии
воды и соли АГ (соль гексаметилендйамина и адипиновой кислоты) в
условиях как периодического, так и непрерывного процесса. Периодиче- :
ский процесс осуществляют в автоклавах при повышенном давлении, ;
Непрерывный процесс —в прямоточном колонном аппарате идеальнога j
вытеснения или в U-образной трубе — все больше внедряется в произ- -
водство. Полимер, образующийся в автоклаве или в другом аппарате,
при 270° С выдавливается в виде ленты, гранулируется и перерабатыва- i
ется литьем под давлением и экструзией. Поликапроамид — твердый ро-
212
говидный продукт с молекулярным весом 20000—40000, кристалличе-
ской структурой; т. пл. 215° С, р= 1,12—1,13 г/см3.
Ионная полимеризация е-капролактама протекает с высокой скоро-
стью. Поэтому ее используют для получения блоков заданного размера
и формы, из которых механической обработкой получают изделия нуж-
ного профиля.
Промышленность выпускает несколько марок поликапроамида и
смешанных полимеров с капролактамом. Капрон (поликапроамнд)
(ВТУ УХП 69—58) выпускается в виде гранул от белого до светло-жел-
того цвета трех марок: А, Б и В, отличающихся молекулярным весом..
Капрон марки Б — литьевой материал.
Капролон (МРТУ 6-05-988—66) —продукт полимеризации е-капро-
лактама в присутствии щелочных катализаторов. Выпускается в виде
блоков-заготовок. Марки В, Bi и Вг отличаются по монолитности, проч-
ности и молекулярному весу. Применяются в качестве электроизоляци-
онного и антифрикционного материала.
Капролит— полимер капролактама, получающийся анионной поли-
меризацией в присутствии многофункциональных активаторов (мета-
крилоилкапролактам); отличается высокой ударной прочностью (в три
раза выше, чем у капрона).
Важным применением капрона является изготовление материалов;
с различными наполнителями: тальком — КТ5 и КТ10; BaSO4 — К-Ва10;
графитом — КГ-10 (10% коллоидального графита). Они применяются1
для изготовления деталей антифрикционного и конструкционного назна-
чения (наполненные смешанные полимеры описаны в разделе «Полиа-
миды») .
Капрон, полученный в присутствии .воды и соли АГ, перерабатыва-
ют на волокно и пленку из расплава. Пленка ПК-4 применяется в строи-'
тельстве и на железнодорожном транспорте.
Полимеры окиси этилена Н—-[—ОСН2СН2—]п—ОН и окиси пропиле-
сн,
I
на Н—(—ОСН2—СН—]„—ОН, полученные гидролитической полимериза-
цией, представляют собой скорее олигомеры, чем полимеры, растворимы;
в воде, их молекулярный вес колеблется от 300 до 5000. Они применяют-
ся в качестве полигликолей при синтезе полиэфируретанов. Высокомо-
лекулярные полимеры получаются при анионо-координационной поли-
меризации окисей.
При ионной полимеризации 3,3/-быс-(хлорметил)оксациклобутана‘
в присутствии BF3 в среде растворителя (метиленхлорида, SO2 и др.)
образуется кристаллический полимер — полиоксахлорметилциклобута»
(пентапласт) с т. пл. 180° С, нерастворимый в хлорированных углеводо-
родах, степень кристалличности около 30%. Содержание хлора 45,5%.
Молекулярный вес достигает 250—400 тыс. Деструкция происходит выше-
285° С. Расплав имеет низкую вязкость, что определяет легкость перера-
ботки полимера. Пентапласт достаточно термостоек вплоть до 120° С.
Этот перспективный пластик выпускается по ТУ 6-05-1422—71 в„ широ-
ком диапазоне молекулярных весов, применяется в химической про-
мышленности, машиностроении, в виде изделий, полученных литьем под
давлением, экструзией, а также в виде пленок и защитных покрытий.
При введении в пентапласт минеральных наполнителей (графит, стек-
ловолокно, мелкоизмельченная слюда) увеличивается его прочность,
снижается усадка.
Исходное сырье для получения полимеров
из гетероциклических мономеров
I---------1
Е-Капролактам — лактам Е-аминокапроновой кислоты NH(CH2)sCO
по внешнему виду представляет собой белое кристаллическое вещество
в виде порошка или плоских оплавленных кусочков; т. пл. 70° С; т. кип.
258°С при 760 мм рт. ст. и 139°С при 12 мм рт. ст.; р= 1,02 (при 25°С);
я ™=1,4784, лЬ = 1,4768, Ид =1,4710.
Капролактам хорошо растворяется в воде. При обычной температу-
ре можно получить растворы, содержащие до 80% вещества. Чистый
капролактам растворим в органических растворителях (спирте, бензоле,
ацетоне, эфире, хлороформе и др.) и в растворах кислот и солей. Для
очистки с успехом применяется уксусноэтиловый эфир, из которого кап-
ролактам выкристаллизовывается в виде снежно-белых кристаллов.
Вследствие гигроскопичности капролактам рекомендуется хранить
в герметически закрытой таре.
Сырьем для промышленного получения капролактама служат фе-
нол, циклогексан и анилин. Капролактам, применяемый для изготовле-
ния капрона, должен иметь температуру затвердевания не менее 67,7° С,
влажность не более 0,2%; механических примесей не более 0,00075%.
Раствор его в уксусной кислоте должен быть стабильным по цвету в те-
чение 24 ч.
Наиболее перспективным является производство капролактама че-
рез циклогексанон, получаемый окислением циклогексана воздухом:
/\ /\/° /\ZON ,™2
II О, < । NHjOH-HjS04 I I перегруппировка Бекмана / \
hl I -------------Н I --------------------<f=o
Х// Ч'/ Н2С NH
I I
н2с—сн2
Капролактам очищается перекристаллизацией из уксусноэтилового
эфира. Степень чистоты устанавливают по температуре плавления про-
дукта.
Окись этилена СН2—СН2 — простейший представитель эпок-
сидов; легко летучая, бесцветная жидкость со специфическим эфирным
запахом. Т. кип. 10,7° С; т. заст. —113,3° С; с водой образует гидрат с
-семью молекулами Н2О (т. пл. 12,8°С). Окись этилена смешивается с во-
дой во всех отношениях, с воздухом образует взрывчатые смеси (преде-
лы взрываемости 3,0—80 объемн. %). Окись этилена широко применяет-
ся в промышленности, поэтому производится в огромных количествах
каталитическим окислением этилена:
Ag
СН2=СН2 + 0,5О2 ->СН2— СН2
-214
„ сн2-сн-сн3 л
Окись пропилена \q/ — бесцветная жидкость с эфир-
ным запахом; т. кип. 33,9° С; т. заст. —104,4° С; горюча; с воздухом об-
разует взрывоопасные смеси. Растворимость в воде 40,5 вес. % при 20° С..
Растворимость воды в окиси пропилена 12,8 вес. %.
Окись пропилена получают из пропилена по схеме
нею
СН2=СН-СН3---->
|->-СН3-СНОН-СН2С1--. +Са(ОН)а
-----*
1->СН3-СНС1-СН2ОН—1
-* 2СН2—СН—СН3 + СаС12 + 2Н2О-
,СН2 ,СН2С1
3,3'-бис-(Хлорметил)-оксациклобутан °\сн z\qH о —
кристаллический порошок с т. кип. 101° С при 27 мм рт. ст., 42° С при
4 мм рт. ст.
Его получают из пентаэритрита в несколько стадий. Пентаэритрит
ацетилируют в присутствии катионообменной смолы.
Под действием хлористого тионила тетраацетат пентаэритрита дает-
моноацетат трихлоргидрина пентаэритрита, который омыляют этиловым
спиртом в присутствии п-тол.уолсульфокислоты в трихлоргидрин пента-
эритрита. Последний при обработке водной или спиртовой щелочью пре-
вращается в 3,3/-бпс-(хлорметил)-окса|циклобутан:
СН2ОН СН2ООССН3
I + (CH,CO)2O I [SOC1,
С(СН2ОН)2-------->СН3СООСН2-С—СН2ООССН3------->
СН2ОН СН2ООССН3
СН2С1 СН2С1
I I кон /СН2ч ,СН2С1
-+ С1СН2-С-СН2С1 С1СН2-С-СН2С1 ——о< >с<
I 1 2 | с’н‘он . ^сн/ ХСН2С1.
СН2ООССН3 СН2ОН
Качественный анализ капролактама
и поликапроамида
1. в-Капролактам в водном растворе в присутствии иодида висмута-
калия в кислой среде дает оранжево-красный осадок с характерной
кристаллической структурой. Цвет осадка зависит от кислотности рас-
твора и концентрации реагента. Этот метод позволяет обнаружить менее
0,3% .капролактама,
2. При кислотном гидролизе е-капролактама после нейтрализации
NaOH до pH 6,3—6,5 и добавления раствора нингидрина образуется
вещество фиолетового цвета, нерастворимое в эфире и четыреххлористом
углероде.
215-
Характерное окрашивание, зависящее от pH, получается в хлоро-
форме и бензоле.
Качественный анализ полимеров на основе
окиси этилена и окиси пропилена
1. К 1 капле водного раствора образца добавляют 0,02 г кодеина и
0,5 мл концентрироваиной H2SO4. В присутствии полиоксиэтилена появ-
ляется фиолетово-красное окрашивание.
2. 0,1 г образца растворяют в 2 мл воды. Добавляют 3 мл 5%-ного
раствора пирогаллола в H2SO4. Появление золотисто- или красно-корич-
невого окрашивания указывает на присутствие полиоксиэтилена.
3. Крупинку или каплю исследуемого образца нагревают с бисуль-
фитом калия. Полиоксиэтилен окрашивает в синий цвет фильтроваль-
ную бумагу, смоченную 1%-ным раствором нитропруссида натрия и пи-
перидина. При добавлении раствора NaOH синее окрашивание перехо-
дит в красное.
Полиоксипропилен дает оранжевое окрашивание, переходящее в
темно-коричневое.
Количественный анализ полимеров
из гетероциклических мономеров
Поликапроамид имеет концевые группы —NH2 и — СООН. Количе-
ственное определение концевых групп дает возможность рассчитать
молекулярный вес полимера.
Группу —NH2 можно определить потенциометрическим или ациди-
метрическим титрованием в неводной среде.
Определение аминного числа. Берут две точные навески (0,5—1,0 г)
полиамида в конические колбочки с притертыми пробками и заливают
точно отмеренным количеством (25 мл) 0,1 н. спиртового раствора НС1.
-Одновременно ставят контрольный опыт без навески. Через 3 ч пробы
отфильтровывают, отбирают 10 мл фильтрата и определяют в нем избы-
ток кислоты путем обратного титрования 0,1 н. спиртовым раствором
КОН в присутствии метилового оранжевого. Аминное число (Ам. ч.),
выраженное в милиграммах НС1 на 1 г вещества, рассчитывают по фор-
муле
Ам. ч,- №-^8)2,5-1000
g
где а — объем 0,1 н. раствора НС1, израсходованный в контрольном опы-
те, мл; b — объем 0,1 <н. раствора КОН, израсходованный на обратное
титрование, мл; Т\ — титр раствора НС1, г/мл; Т%— титр раствора КОН
ио НС1, г/мл; g — навеска полимера, г.
Аминные числа полимеров, растворимых в спирте, можно определять
прямым титрованием спиртового раствора полимера 0,1 н. спиртовым
раствором НС1.
216
Определение аминогрупп в полиамиде потенциометрическим мето-
дом, К 3 г полиамида прибавляют 75 мл свежеперегнанного ж-крезола,
перемешивают до полного растворения и титруют потенциометрически
0,5 н. раствором хлорной кислоты в метиловом спирте [4].
Определение кислотного числа. Две точные навески 1—2 г полиами-
да помещают в конические колбы емкостью 250 мл и добавляют точно
отмеренное количество (25 мл) 0,1 н. спиртового раствора КОН. Одновре-
менно ставят контрольный опыт без навески. Через 2 ч отфильтровыва-
ют пробы, берут 10 мл фильтрата и оттитровывают 0,1 н. раствором НС1
до исчезновения розовой окраски по фенолфталеину. Кислотное число
(К. ч.) рассчитывают по формуле
,, (а — О 72,5-1000
К. ч. =———-——— ,
g
где а — объем 0,1 н. раствора НС1, израсходованный в контрольном
опыте, мл; b — объем 0,1 н. раствора НС1, израсходованный в опыте с
навеской, мл; g— навеска полимера, г; Т — титр раствора НС1 по КОН,
г/мл.
Из двух определений принимают среднее значение кислотного числа.
Кислотное число полиамидов, растворимых в спирте, можно опре-
делять прямым титрование^ спиртового раствора полимера 0,1 н. спир-
товым раствором КОН.
Определение карбоксильных групп можно проводить с достаточной
степенью точности кондуктометрически. Однако аминогруппа определя-
ется этим методом со значительной погрешностью.
Определение концевых гидроксильных групп в полиалкиленокаидак
можно проводить по методу Вер лея.
Две точные навески полимера помещают в конические колбы с при-
тертыми пробками и приливают в каждую по 10—15 мл (в зависимости
от предполагаемого содержания гидроксильных групп) смеси, состоящей
из 12 вес. ч. уксусного ангидрида и 88 вес. ч. перегнанного над окисью
бария пиридина. Параллельно ставят контрольный опыт без навески.
Притертые пробки закрепляют, колбы помещают в водяную баню и на-
гревают 0,5—2 ч. (в зависимости от количества групп —ОН и раствори-
мости полимера) при 60° С. После охлаждения во все три колбы добав-
ляют равные объемы воды и титруют выделившуюся уксусную кислоту
0,5 н. раствором NaOH в присутствии фенолфталеина.
Уксусный ангидрид реагирует с гидроксильными группами по схеме
—R— + (СН3СО)2О -> -R- + СНзСООН
I I
ОН ООС—СН3
Образовавшаяся уксусная кислота образует с пиридином соль
СН3СООН \ , устойчивую в безводной среде и быстро разлагаю-
щуюся при добавлении воды. Связывая кислоту пиридином, устраняют
возможность гидролиза образовавшегося эфира.
8—1453 217
Количество ОН-групп х (в %) рассчитывают по формуле
(а— 6) КО, 0085 -100
Хг - ' у
g
где а — объем 0,5 и. раствора NaOH, израсходованный в контрольном
опыте, мл; Ь — объем 0,5 н. раствора NaOH, израсходованный на титро-
вание пробы с навеской, мл; 0,0085 —коэффициент пересчета на гидро-
ксильную группу, соответствующий 1 мл 0,5 н. раствора NaOH; g — на-
веска испытуемого вещества, г; К — поправка к титру раствора.
Определение гидроксильных групп этерификацией фталевым ангид-
ридом. Готовят раствор 4 г свежевозогнанного фталевого ангидрида в
20 г абсолютно сухого, перегнанного над окисью бария пиридина. В две
конические колбы емкостью 250 мл берут точные навески по 0,1—0,2 г
полимера, приливают в каждую колбу точно по 10 мл приготовленного
раствора фталевого ангидрида в пиридине. Параллельно ставят конт-
рольный опыт без навески.
Колбы соединяют с обратными холодильниками, снабженными
хлоркальциевыми трубками со свежепрокаленным СаСк, и 2—3 ч нагре-
вают микрогорелкой на асбестовой сетке. Затем разбавляют содержимое
каждой колбы 10 мл воды и титруют 0,5 и. раствором NaOH в присут-
ствии фенолфталеина до появления розовой окраски.
Химизм реакций, лежащих в основе метода, аналогичен приведен-
ному выше для метода Верлея.
Химический анализ пентапласта основан на определении хлора (см.
стр. 84).
Техника безопасности при работе
с гетероциклическими мономерами
и полимерами на их основе
е-Капролактам хорошо растворим в воде, имеет горький вкус, не-
приятный запах. При остром отравлении капролактамом наблюдаются
одышка, судороги и смерть от остановки дыхания.
Пыль поликапроамида действует на печень и почки. Смачивание
кожи раствором капролактама вызывает гиперемию и шелушение. Дли-
тельное воздействие капролактама при концентрации в воздухе 0,003—
0,009 мг/л вызывает тошноту, потерю аппетита, утомление, головную
боль.
Все работы с капролактамом и его полимером следует проводить в
вытяжном шкафу.
Окись этилена, окись пропилена и их полимеры. Эти соединения
мало токсичны и требуют при работе с ними соблюдения обычных пра-
вил гигиены.
Пентапласт. Вредны продукты термического распада- пентапласта,
в число которых входят хлорангидриды низших кислот, фосген, СО,
альдегиды, в том числе формальдегид, хлористый водород. Они вызыва-
ют раздражение слизистой оболочки глаз и верхних дыхательных путей.
Переработка пентапласта должна проводиться в условиях сильной при-
точно-вытяжной вентиляции.
218
РАБОТА 115. ПОЛУЧЕНИЕ ПОЛИКАПРОАМИДА ГИДРОЛИТИЧЕСКОЙ
ПОЛИМЕРИЗАЦИЕЙ е-КАПРОЛАКТАМА
В РАСПЛАВЕ
Образование полимера протекает по механизму ступенчатой поли-
меризации. Схематически реакция может быть представлена в следую-
щем виде:
+CO(CHa)sNH
+н2о !______!
ОС (CH2)5NH------> НООС (CH2)5NH2------------->
I I
CO(CH2)5NH
I______I
-> HOOC (CH2)sNHCO (СН2)5МН2------->
->HOOC(CH2)5NHCO(CH2)sNHCO(CH2)5NH2 h t. Д.
В общем виде уравнение этой равновесной реакции можно запи-
сать:
nHN (СН2)5СО [-HN (СН2)5СО-]П
I I
Исходные продукты: капролактам—100 г; соль АГ (гексаметилендиамииадипи-
нат) — 4 г.
Оборудование: четырехгорлая колба емкостью 0,5 л, снабженная механической
мешалкой с герметическим затвором, обратным холодильником, термометром и трубкой
для ввода инертного газа (рис. 58); стаканчик емкостью 50 мл; лист жести; воздушный
холодильник; воронка Бабо.
Рис. 58. Прибор
для проведения ре-
акции в токе
инертного газа
•Рис. 59. Схема ручной вытяжки во-
локна
В отдельном сосуде готовят 50%-ный водный раствор соли АГ, затем
в четырхгорлую колбу прибора помещают капролактам и вносят приго-
товленный водный раствор соли АГ. Включают мешалку, пускают воду
в холодильник, пропускают в колбу медленный ток азота и реакционную
смесь нагревают пламенем горелки на асбестовой сетке при 110 С.
219
Через 1 ч водяной холодильник меняют на воздушный, температуру
в колбе поднимают до 270° С и нагревают еще 2 часа на воздушной бане
(на воронке Бабо) до получения полимера, что определяется визуально
по образованию длинных нитей поликапролактама при вытягивании
расплавленного продукта с помощью стеклянной палочки. Если продукт
не вытягивается в длинную нить или она быстро обрывается, нагревание
следует продолжить еще полчаса и вновь взять пробу на образование
нити.
Полученный готовый полимер в расплавленном состоянии вылива-
ют на лист жести, где он и застывает в твердый продукт.
Определяют кислотное число, аминное число, индекс расплава и из-
готовляют методом литья под давлением изделие или образец для оп-
ределения разрушающего напряжения при растяжении (работа 116)
или ручной вытяжкой получают волокно (рис. 59).
РАБОТА 116. ИЗГОТОВЛЕНИЕ ИЗДЕЛИЙ ИЗ КАПРОНА
МЕТОДОМ ЛИТЬЯ ПОД ДАВЛЕНИЕМ
Изделия из поликапроамида (капрона) изготавливаются методом
литья под давлением.
У поликапроамида очень узкий температурный интервал перехода
из твердого состояния в жидкое-, поэтому литье необходимо вести при
строго определенной температуре. Она должна 'быть примерно на 10—
20° выше температуры плавления материала (для капрона т. пл. 215° С),
так как, с одной стороны, должно быть обеспечено полное расплавление
продукта, а с другой — он не должен разлагаться вследствие перегрева.
Оптимальной температурой литья является верхний предел размягчения
материала.
В расплавленном состоянии поликапроамид обладает высокой теку-
честью, поэтому из него можно готовить изделия сложной конфигурации,
однако это не позволяет использовать обычные незапирающиеся сопла,
так как в этом случае расплав вытекает из отверстия сопла даже тогда,
когда во время паузы в цикле литья в материальном цилиндре нет дав-
ления. Поэтому при литье под давлением всех полиамидов и, в частно-
сти, капрона следует применять самозапирающиеся сопла, которые
автоматически открываются только при соприкосновении с пресс-фор-
мой или под давлением материала в цилиндре. Эти сопла имеют инди-
видуальный обогрев; так как они длиннее обычных, то возникает
опасность застывания материала в канале сопла. В нерабочем положе-
нии сопло всегда закрыто. Оно открывается только тогда, когда проис-
ходит впрыск материала (рис. 60).
Исходный продукт: поликапроамид (капрон) гранулированный или в виде крошки.
Оборудование: литьевая лабораторная машина.
Капрон гигроскопичен, и при нормальных условиях содержание
влаги в нем составляет 1,5—3,5%. Влажность капрона влияет на процесс
литья и качество отлитых изделий, так как при плавлении материала во-
да испаряется и вспенивает расплав. Наряду с этим влага может вызвать
220
гидролитическую деструкцию полимера. Поэтому применяемый ДЛ1
литья капрон необходимо непосредственно перед литьем высушивать д<
содержания влаги 0,25—0,35%.
Чтобы ускорить процесс, а также предохранить материал от окисле
ния, сушку ведут под вакуумом при 80—85° С. При нормальной грануля
ции капрона, т. е. толщине гранул или крошки не более 3—3,5 мм4 сущю
под вакуумом ведут'2—4 ч. Высушенный материал, если он сразу hi
применяется для литья, следует хранить в герметически закрытой таре
Рис. 60. Схема литья поликапроамида под давлением
1 — загрузочный бункер; 2 — материальный цилиндр; 3 — пор-
шень; 4 — обогревательный цилиндр; 5 — электрообогрев;
6 — торпеда; 7 — сопло; 8 — форма
Литье капрона производят на литьевой машине (см. рис. 60). Высу
шенный капрон загружают в бункер литьевой машины, откуда он чере:
автоматический дозатор поступает в материальный цилиндр. Затеи
поршнем материал подается в обогревательный цилиндр, где нагревается
до температуры плавления. Температура его постепенно повышается О'
начальной (у загрузочного отверстия) до конечной (на выходе), уставов
ленной технологическим режимом.
Вследствие низкой теплопроводности .капрона расплавить его i
обычном материальном цилиндре быстро и равномерно по всей толщин»
невозможно. При плавлении в этих условиях в толще материала .могу'
оставаться нерасплавленные частицы, а у стенок цилиндра вследстви(
перегрева материала он может частично разлагаться. Чтобы обеспечит!
более равномерный подогрев материала за короткое время, в обогрева
тельный цилиндр устанавливают специальную деталь—торпеду. Онг
представляет собой цилиндр с коническими днищами. Материал) сопри
касаясь с нагретыми стенками торпеды, быстро и равномерно прогревает
ся и плавится.
На выходе обогревательного цилиндра установлено сопло, которо»
обеспечивает подачу материала в форму с необходимой скоростью, а так
же служит запирающим приспособлением обогревательного цилиндра
В момент литья пресс-форма должна быть плотно прижата к соплу
чтобы не допускать вытекания материала и обеспечить надлежащее дав
ление литья. Обогревательный цилиндр остается неподвижным, пресс
форма перемещается в горизонтальном направлении. Литье поликапро
амида производится при 225—235° С.
На качество изделий из.капрона сильно влияет температура пресс
формы. При литье в холодную пресс-форму изделия получаются более
22
эластичными. Если же материал отливается в горячую форму, то изделия
после охлаждения приобретают большую жесткость и твердость. Это
связано со структурными изменениями в полимере в момент перехода
его из жидкого состояния в твердое.
При быстром остывании расплавленного капрона в холодной форме
образуется аморфная структура. Наличие аморфной фазы в полимере
определяет его высокую ударную прочность, эластичность, вязкость.
При литье в горячую пресс-форму .происходит медленное остывание
полимера и его кристаллизация. Это обусловливает меньшую эластич-
ность деталей, однако повышает их износостойкость на истирание. Поэто-
му при литье изделий, предназначенных для работы на истирание, следу-
ет создавать условия, обеспечивающие преобладание кристалличе-
ской структуры. Одним из таких условий является литье в нагретую
форму. Так, например, литье подшипников, которые работают на истира-
ние, следует производить в пресс-формах, нагретых до 60—80° С.
Усадка поликапроамида достигает 1—2,5%. При увеличении давле-
ния литья она уменьшается. Для уменьшения усадки необходимо также,
чтобы материал в форме находился все время под давлением до полного
остывания изделия.
Давление при литье не имеет большого значения, так как вследст-
вие низкой вязкости расплава для его выдавливания требуются неболь-
шие усилия. Максимальное давление литья для поликапроамида не уста-
навливается и практически оно ограничивается только прочностью
пресс-формы и величиной давления замыкания формы. Поэтому литье
капрона может происходить в очень широком интервале удельных давле-
ний: примерно от 20 до 2000 кг/см2. Обычно удельное давление при литье
поликапроамида находится в пределах 150—800 кг/см2, а при малых
впускных каналах достигает 1200—1700 кг/см2.
Остывание в пресс-форме под давлением отлитых из капрона дета-
лей толщиной 1—2 мм должно продолжаться приблизительно 3 с.
Готовые изделия целесообразно подвергнуть термической обработке,
нагревая детали в минеральном масле или воде. При этом снимаются
внутренние напряжения, возникающие при остывании материала в пресс-
форме под давлением, и достигается равновесное состояние структуры
материала.
Рекомендуется следующий режим термообработки в масле для та-
ких изделий, как, например, подшипники: температура нагревания 150—
160° С; время выдержки 5—10 мин на 1 мм толщины стенки изделия.
Нагревание проводят в масляной ванне. После масляной обработки изде-
лия подвергают влагонасыщению на воздухе или в воде. Полученные
стандартные образцы испытывают на разрушающее напряжение при
ударе и растяжении (см. ч. II).
РАБОТА 117. ПОЛУЧЕНИЕ N-АЦЕТИЛКАПРОЛАКТАМА
Для получения в лабораторных условиях ацетилкапролактама при-
меняют е-капролактам и уксусный ангидрид. Реакция протекает по схеме
2HN (СН2)5СО + (СН3СО)2О -> 2CH3CON (СН2)5СО + Н2О
I_____I I I
222
Исходные продукты: е-капролактам (0,58 моля) — 65,7 г; уксусный ангндр
(1,95 моля) — 200 г.
Оборудование: трехгорлая колба емкостью 0,5 л, снабженная мешалкой с герме:
ческнм затвором, обратным холодильником с хлоркальциевой трубкой и термометре
колба Вюрца; холодильник Либиха; прибор для перегонки в вакууме.
В трехгорлую колбу прибора загружают е-капролактам и уксусш
ангидрид. Включают .мешалку, пускают воду в холодильник и нагрева!
содержимое колбы на масляной бане при 130° С. Через 4 ч реакционна
смесь переносят в колбу Вюрца и отгоняют избыток уксусного ангидри,
и уксусной кислоты. Остаток подвергают перегонке под вакуумом. Фра
цию, кипящую при 98—103° С и 2 мм рт. ст., собирают как чистый пр
дукт.
Выход составляет 60—68 г (около 67% от теоретического, считая j
взятый для реакции е-капролактам).
РАБОТА 118. ПОЛУЧЕНИЕ КАПРОЛОНА ЛИТЬЕМ
В ФОРМЫ
Изделия из поликапроамида, в особенности крупногабаритные, мо;
но изготовлять также отливкой расплава мономера в формы, в которг
и происходит полимеризация е-капролактама по анионному механизм
в присутствии металлического натрия и N-ацетилкапролактама:
Na
HN (СН2)5СО -> NaN (СН2)5СО -> “N (СН2)5СО ->
I I |I I!
CH,OCN(CH,),CO HN(CHa)5C0
!_____! _ !_____I
-------------> CH3CON (CH2)5CON (СН2)5СО-------->
I I
-> CH3CONH (CH2)5CONH (CH2)5CON (CH2)sCON (CH2)5CO -> и T.
I I
Исходные продукты: е-капролактам (2,21 моля) — 250 г; металлический натр:
(0,35 мол. %)—0,178 г; N-ацетилкапролактам свежеприготовленный (0,5 мол. %)
1,08 мл.
Оборудование: прибор для синтеза в токе инертного газа (см. рис. 58); металла
ская форма для отливки (сталь, алюминий); баня с полиэтилсилоксановой жидкость
В четырехгорлую колбу прибора загружают совершенно сухой св
жеперегнанный е-капролактам. Через трубку для ввода инертного газа
колбу пропускают азот, освобожденный от влаги и кислорода, и нагрев
ют капролактам на масляной бане до 90—95° С. При этой температуре
непрерывном перемешивании к расплаву мономера добавляют 0,178
металлического натрия.
После прекращения выделения водорода температуру масляной б,
ни повышают до 135° С и при этой температуре к расплаву мономера д<
бавляют 1,08 мл (0,35 мол. %) N-ацетилкалролактама.
Содержимое колбы перемешивают в течение 3 мин, затем быстр
выливают расплав в нагретую до 170° С металлическую форму, изгото:
ленную из стали или алюминия.
2
Форму с расплавом помещают .в термошкаф, где выдерживают
40—50 мин при 170—178° С.
В данном случае полимеризация мономера проходит при температу-
ре ниже температуры плавления полимера, и жидкий е-капролактам сра-
зу превращается в твердый поликапроамид — «капролон».
По истечении указанного времени обогрев термошкафа выключают
и дают полимеру медленно охладиться. Изделие извлекают из формы
после охлаждения до комнатной температуры.
Примечание: для получения совершенно сухого е-капролактама последний непо-
средственно перед полимеризацией перегоняют под вакуумом. Фракцию, кипящую при
температуре 130° и давлении 5 мм рт. ст., собирают как чистый сухой продукт, содер-
жащий минимальное количество влаги. Капролактам можно высушить также в вакуум-
сушилке, нагревая его при 40—60° в течение нескольких часов.
РАБОТА 119. ПОЛУЧЕНИЕ ПОЛИЭТИЛЕНОКСИДА
В ПРИСУТСТВИИ ЩЕЛОЧИ
Реакция протекает по схеме
сн2-сн2
NaOH -+ \>/ —+
СН2—СН2---> НОСН2—СН2—ONa------> HOCH2-CH2-OCH2-CH2-ONa и т. д.
Общая формула .полиэтиленоксида Н—(—ОСН2—СН2—)п—ОН.
Исходные продукты: окись этилена — 50 г; едкий натр—0,25 г; баллон с инерт-
ным газом.
Оборудование: автоклав с манометром, рубашкой для обогрева; мерная ампула;
лед для охлаждения.
Мерную ампулу охлаждают во льду и наливают в нее из баллона
50 г окиси этилена. В автоклав помещают едкий натр, 1 мл дистиллиро-
ванной воды, охлаждают до 5—10° С, затем вводят окись этилена; воз-
дух вытесняют азотом или другим инертным газом. Герметизируют
автоклав и начинают его нагревать до 100° С, пока давление не снизится
до нормального.
После вскрытия автоклава полимер выгружают и определяют его
молекулярный вес по концевым гидроксильным группам [7].
Полиэтиленоксид применяют для изготовления воска.
РАБОТА 120. ПОЛУЧЕНИЕ ПОЛИПРОПИЛЕНОКСИДА
В ПРИСУТСТВИИ ЩЕЛОЧИ
Реакция раскрытия цикла может протекать по двум направлениям:
по вторичному и по третичному атому углерода:
Р -Г —
₽ а NaOCH2—СН—СН
СН2-СН-СН3 4- NaOH- ।
ЧО/ l-Д- -СН2-СН-СН3
i“Na+
3
224
'СН—СН2— —он
I
R
Термодинамически более вероятна 0-схема. Однако практически э'
определяется условиями полимеризации.
Общая формула полипропиленоксида
Исходные продукты: окись пропилена — ЮО г; дипропиленгликоль — б” г; гидр
окись натрия — 0,75 г; баллон с инертным газом.
Оборудование: автоклав с манометром и рубашкой для обогрева н охлажден!
мерный цилиндр.
В автоклав помещают едкий натр и дипропиленгликоль, нагрева!!
их до растворения; затем охлаждают до комнатной температуры и пр
ливают окись пропилена. Автоклав герметизируют и постепенно за 1,5
2 ч нагревают до 120—160° С. Конец реакции определяют по паденг
давления. После этого автоклав открывают и извлекают, продукт реа
ции.
Определяют молекулярный вес полимера по концевым гидроксил
ным группам, введя поправку на щелочность [7]. Продукт использун
для получения полиэфируретанов (см. ч. II).
РАБОТА 121. ПОЛУЧЕНИЕ ПОЛИМЕРА
ИЗ 3,3'-БИС-(ХЛОРМЕТИЛ)-ОКСАЦИКЛОБУТАНА
Полимеризация З,3'-бпс-(хлорметил) -оксациклобутана протекает
раскрытием цикла по катионному механизму. Катализаторами являют
+
кислоты Льюиса и, в частности, комплексная кислота H[BF3OH]~
+
Н [BF3OH]~ + О
<сн„. ZCH,C1
)с<
СН2/ ХСН,С1
/СНг^с/СНгС!
'чсн2// ^сн2с1
СН2С1
I +
-> НОСН2-С-СН2 [BF3OH]~ ->
СН2С1
СН2С1 СН2С1
I I +
НОСН2—С-СН2-О-СН2—С—CH2[BF3OH]- и т.
I I
СН2С1 СН2С1
Исходные продукты: 3,3'-бис-(хлорметил)-оксациклобутан—10 г; трехфторист
бор — незначительное количество; метиленхлорнд — 50 мл.
Оборудование: трехгорлая колба емкостью 250 мл, снабженная мешалкой с гер»
тнческим затвором, обратным холодильником и тубусом для ввода реагентов; ба
охлаждающая с сухим льдом и ацетоном; воронка Бюхнера.
В трехгорлую колбу, помещенную в охлаждающую смесь с темпер
турой —30° С, вводят метиленхлорнд, катализатор и мономер. Реакц:
протекает очень быстро, причем выпадает белый осадок полимера. Пр
дукт отфильтровывают на воронке Бюхнера, промывают несколько р
метанолом для удаления следов катализатора, высушивают при 80° С
определяют индекс расплава.
Полимер перерабатывают на экструдере при температуре в цилин
ре 200° С, в головке 220° С. В зависимости от конструкции головки пол
чают пленки, трубки, шланги. На стандартных образцах определи!
прочность на растяжение и модуль упругости при этом испытании.
ГЛАВА IX. ПОЛИМЕРЫ, ПОЛУЧАЕМЫЕ ПО РЕАКЦИЯМ
СТУПЕНЧАТОЙ МИГРАЦИОННОЙ СОПОЛИМЕРИЗАЦИИ
ИЛИ ПОЛИПРИСОЕДИНЕНИЯ
Реакция ступенчатой миграционной сополимеризации лежит в основе
производства важной группы полимерных материалов — полиуретанов
(эфиров карбаминовой кислоты).
Полиуретаны представляют собой продукты совместной полимери-
зации диизоцианатов с соединениями, содержащими две или более гид-
роксильных группы. В качестве гидроксилосодержащих соединений ис-
пользуют гликоли, полигликоли, сложные эфиры с концевыми гидро-
ксильными группами. В последнем случае образуются блоксополимеры.
Полиуретаны в зависимости от строения исходных компонентов могут
быть твердыми, эластичными, монолитными или пористыми, как линей-
ного, так и трехмерного строения.
Полиуретаны находят широкое применение для изготовления син-
тетического волокна, пленок, щетины, пластических масс, клеев,
лаков.
Полимочевины (поликарбамиды) — полимеры, содержащие группу
I —NH—СО—NH—, могут быть получены различными путями: как по
реакции полимеризации, так и поликонденсации (ч. II). Ступенчатый
миграционный механизм реакции лежит в основе получения полимочевин
из диаминов и диизоцианатов. Реакция протекает со скоростью ионных
реакций и в простейшем оборудовании. Полимочевины алифатического
строения высокомолекулярны, обладают высокой прочностью, хорошей
адгезией, используются для изготовления волокна, пленок, клеев. Не-
достатком этих полимеров является невысокая термостойкость, которая,
однако, значительно выше, чем термостойкость полиуретанов. Близость
температур плавления и разложения затрудняет переработку по-
лимочевин, однако стабилизация дает возможность увеличить этот
интервал.
I
Химизм образования полиуретанов
и поликарбамидов
Диизоцианаты, имеющие в группе O = C = N— два неспаренных элек-
трона у атома азота, активно взаимодействуют с веществами, имеющи-
ми подвижный атом водорода.
Нуклеофильная атака диизоцианатом веществ, содержащих протон,
приводит к образованию переходного четырехчленного цикла:
226
—R—N— C=0
II.
H O—R—
полиуретан
•• +5 /-~ч —В
—R—N=C=O
—R—N—C=0 —R—N—C=0
; ! i —* I I ।
H—N—R— H NH—R—
H полимочевина
(поликарбамид)
Так .как реакция протекает при взаимодействии функционалы^
групп двух веществ: диизоцианата и диола или ди^йина, молекулярнь
вес полимеров зависит от соотношения этих соединений.
В случае реакции диамина с диизоцианатом взаимодействие прои
ходит с высокой скоростью, что приводит к мгновенному нарастанию вя
кости, затрудняющей диффузию реагирующих молекул, это определи
сравнительно невысокий молекулярный вес. Полимер выпадает из ра
твора на ранней стадиц реакции, что также вызывает снижение молек
лярного веса.
В случае реакции диизоцианатов с диолами и другими .веществам
содержащими группу ОН-, их растворимость в органических растворит
лях обеспечивает возможность получения достаточно высокомолекуля
ного полимера. Примеси родственных монофункциональных соединен!
ведут себя как блокирующие цепи, а при высоких температурах как ги
ролизующие агенты.
Добавление исходных веществ к полимеру может вызвать увелич
ние молекулярного веса, так как механизм реакций ступенчатый. Кат
лизаторами реакции могут служить щелочи и третичные амины.
Технология производства
полиуретанов и поликарбамидов
Процесс образования полиуретанов заключается в сополимеризащ
диолов или других гидроксилсодержащих соединений с диизоцианатаА
в расплаве или в растворе в хлорбензоле или смеси хлорбензола с д
хлорбензолом.
Получение полиуретанов или полиэфируретанов в расплаве осущ
ствляется в аппарате с быстроходной мешалкой в токе инертного та:
при 190—210° С. По достижении вязкости расплава 600—900 П
190° С реакцию считают законченной и расплав вакуумируют для удал
ния пузырьков газа, после чего выдавливают расплав из аппарата сж
тым газом в виде ленты, которую после охлаждения гранулируют.
Получение полиуретанов в растворителе осуществляют в реактор?
с мешалкой при температуре кипения реакционной массы 160—170° С
течение 4—5 ч. В зависимости от природы исходных компонентов пол!
мер либо выпадает в виде осадка, либо растворитель отгоняют остры
2
Качественный анализ исходного сырья
и полимеров
Качественное определение диизоцианата. В 5—10 мл ледяной уксус-
ной кислоты растворяют при комнатной температуре 0,5 г изоцианата и
к полученному раствору добавляют 0,1 г n-диметил аминобензальдегида.
Через 20—30 мин появляется интенсивная желтая окраска. Эту реакцию
дают и блокированные смолы из изоцианатов.
Качественная реакция на этиленгликоль. К нескольким миллилитрам
испытуемого раствора прибавляют 20 мл водного раствора кодеина и
4 капли концентрированной серной кислоты, после чего смесь осторожно
нагревают. В присутствии этиленгликоля она окрашивается в красно-
фиолетовый цвет.
Качественная реакция на глицерин. Испытуемый раствор или поли-
мер, смешивают в пробирке с безводным сульфатом магния. Пробирку
закрывают корковой пробкой, с отводной трубкой. Смесь сначала осто-
рожно нагревают, затем повышают температуру и собирают образую-
щийся дистиллят в пробирку с 1 мл воды. Наличие акролеина в продук-
тах разложения легко установить по их слезоточивому действию и по вос-
становлению аммиачного раствора нитрата серебра.
Качественное определение полиуретанов основано на определении
спиртов и диаминов в продуктах кислотного или щелочного гидролиза.
Спирты и амины определяют приведенными выше методами.
Количественный анализ исходного сырья
и полимеров
Определение изоцианатного числа. Для определения изоцианатного
числа используют обычно реакцию вещества, содержащего изоцианатные
группы (изоцианата, олигомера, полимера), с диалкиламином, приводя-
щую к образованию замещенной мочевины:
Rx Rx
>NH + R'—N=C=O-> >N—C—NH-R'
Rz Rz II
О
Изоцианатным числом называется'количество НС1, мг, соответствую-
щее количеству амина, пошедшего на взаимодействие с 1 г вещества, со-
держащего изоцианатные группы.
В две чистые сухие конические колбы с пришлифованными пробками
емкостью 250 мл отвешивают на аналитических весах точные навески
(0,5—1 г) вещества, содержащего изоцианатные группы (в зависимости
от предполагаемого содержания изоцианатных групп навеска может ко-
лебаться от 0,5 до 5 г), и приливают 0,2 н. раствора ди-п-бутиламина в
толуоле.
Полученные смеси взбалытывают. Через 30 мин добавляют в колбы
по 100 мл изопропилового 'Спирта и оттитровывают избыток дибутил ами-
на 0,1 н. раствором НС1 в присутствии индикатора бромфенолового сине-
го до появления желто-зеленой окраски. Одновременно ставят контроль-
ный опыт без навески.
230
Изоцианатное число (Из. ч.)-рассчитывают по формуле
„ (a- b) Т-1000
Из. 4.=i------------,
g
где а—количество 0,1 <н. НС1, пошедшее на титрование контрольной про-
бы, мл; b — количество 0,1 н. НС1, пошедшее на титрование пробы с на-
веской, мл; Т — титр раствора НС1; g— навеска, г.
Количественное определение этиленгликоля и глицерина в полиме-
рах или олигомерах (полиэфирах). Навеску 2—5 г испытуемого полиме-
ра (олигомера) омыляют 25 мл 2 н. водного раствора щелочи в кругло-
донной колбе с обратным холодильником в течение 3 ч на кипящей
водяной бане. После этого избыток щелочи нейтрализуют, содержимое
колбы количественно переносят в фарфоровую чашку и смешивают с
50—70 г прокаленного сульфата натрия.
Образовавшуюся кашицу переносят в патрон из асбеста или бума-
ги и помещают в аппарат Сокслета. В колбу аппарата заливают серный
эфир и нагревают на водяной бане с закрытым электрообогревом до тех
пор, пока проба эфира из аппарата не перестанет оставлять жирное пят-
но на фильтровальной бумаге. Экстракт переносят во взвешенную на
аналитических весах колбочку и отгоняют эфир.
По весу остатка в колбе рассчитывают процентное содержание гли-
коля или глицерина (в %) '
g210°
где gi — вес исследуемого продукта; g2 — вес экстракта.
Методы получения, качественное и количественное определение ди-
аминов описаны в ч. II.
Количественное определение олигомеров, содержащих простую и
сложноэфирную связи. Так как олигомеры простых и сложных эфиров,
используемые в производстве полиуретанов, содержат концевые гидро-
ксильные группы, их можно определять по способу Верлея (стр. 217) в
том случае, если они не содержат воды.
Определение состава полиэфируретанов и полимочевин. В состав
полиуретанов могут входить изоцианаты и полиэфиры, полученные из
двухосновных кислот и спиртов.
Для определения состава полиуретанов их подвергают гидролизу
щелочью или кислотой, а также аминолизу.
Для омыления полиуретанов существует следующая методика.
Навеску 2—3 г образца омыляют 1 н. спиртовым раствором щелочи
(КОН) при нагревании на водяной бане. В результате гидролиза двухос-
новные кислоты выделяются в виде калиевых солей, нерастворимых в
спирте. Их отфильтровывают и промывают спиртом, .высушивают и
взвешивают.
К щелочному раствору прибавляют 60%-ную серную кислоту до
нейтральной реакции, после чего добавляют 1 мл НС1 (конц.). Получен-
ный раствор экстрагируют смесью этилового эфира и ацетона (1:1);
экстракт сушат прокаленным'сульфатом натрия, фильтруют и отгоняют
растворитель.
231
Остаток высушивают при 60° С до постоянного веса в вакууме. По-
лучают спирты, которые идентифицируют по плотности, показателю
преломления и качественным реакциям (см. стр. 228—230).
Кислотный гидролиз полиуретана можно проводить нагрева-
нием полимера с 60%-ной серной или фосфорной кислотой, в течение
2—3 ч, при этом выделяется СОг, диамин и спирт. При гидролизе поли-
уретана из 1,4-бутандиола и 1,4-гексаметилендиизоцианата выделяются
СО2, гексаметилендиамин и бутандиол-1,4. Под действием минеральной
кислоты бутандиол превращается в тетрагидрофуран. Его константы:
б/420=0,888; nD20= 1,4060; т. кип. 55—66° С. Из продуктов гидролиза его
выделяют экстракцией изобутиловым спиртом с последующей разгонкой
экстракта.
Аминолиз полиуретанов проводят анилином. Полиуретаны типа
—R—NH—COOCH2R'— при кипячении с анилином разрушаются с об-
разованием дифенилмочевины, которую отфильтровывают, промывают
спиртом, высушивают и взвешивают. Диамины и спирты остаются в
фильтрате, из которого их можно извлечь (см. выше).
Полимочевины при нагревании до температуры деструкции деполи-
меризуются с образованием смеси диаминов, соответствующих диизоциа-
нату и исходному диамину. Образовавшиеся диамины идентифицируют
по температуре плавления и содержанию азота.
Техника безопасности при получении полиуретанов
и поликарбамидов
Из исходных продуктов, применяемых для изготовления полиурета-
нов, с точки зрения техники безопасности заслуживают внимания изо-
цианаты.
Толуилендиизоцианат обладает раздражающим действием, он пора-
жает слизистые оболочки глаз и органов дыхания. При поражении ды-
хательных путей появляется кашель и развивается общая форма брон-
хиальной астмы.
При попадании на кожу он вызывает ее слабое раздражение. Случа-
ев сильного поражения кожи у работающих с толуилендиизоцианатом
не наблюдалось.
Концентрация паров толуилендиизоцианата в воздухе, равная
0,1 мг/л, безопасна для работы и считается предельно допустимой в
рабочих помещениях. Содержание 0,4 мг/л — нижний предел концентра-
ции, при которой толуилендиизоцианат различается по запаху. При
больших концентрациях, порядка 0,5—0,8 мг/л, уже раздражается слизи-
стая оболочка дыхательных путей.
Степень токсичности различных видов диизоцианатов определяется
их летучестью: чем более летучи изоцианаты, тем более они опасны.
В помещениях, где работают с диизоцианатами, должна быть обес-
печена хорошая вентиляция. При взвешивании, переливании и загрузке
изоцианатов в сосуды необходимо пользоваться резиновыми перчатками,
защитными очками, предохраняющими руки и глаза от случайного по-
232
падания продукта. При попадании толуилендиизоцианата в глаза необ-
ходимо немедленно промыть их водой или растительным маслом.
При аварийных случаях проливания или утечки изоцианат следует
немедленно нейтрализовать, используя его растворимость в некоторых
веществах. Изоцианаты хорошо растворяются в спиртах, например в
изопропиловом спирте. Для смывания изоцианатов рекомендуется при-
менять водные растворы спиртов и мыла. В воде толуилендиизоцианат
растворяется незначительно, медленно с ней реагируя.
РАБОТА 122. ПОЛУЧЕНИЕ ПОЛИУРЕТАНА ИЗ ГЕКСАМЕТИЛЕНДИИЗОЦИАНАТА
И ДИЭТИЛЕНГЛИКОЛЯ В РАСТВОРЕ
Реакция протекает по схеме
nHOCH2CH2OCH2CH2OH + nO=C=N-(CH2)6--N=C=O ->
Г—OCH2CH2OCH2CH2OCNH (CH2)6NHC—
Исходные продукты: гексаметилендиизоцианат — 50 г; диэтиленгликоль — 16,9 г
(или этиленгликоль—10 г); хлорбензол, высушенный над гидридом кальция, — 330 г;
о-дихлорбензол, высушенный над гидридом кальция,— 50 г (можно взять 380 г хлор-
бензола).
Оборудование: трехгорлая колба емкостью 0,75 л, снабженная мешалкой с герме-
тическим затвором, обратным холодильником с хлоркальциевой трубкой, термометром;
фарфоровая чашка; термошкаф.
В трехгорлую колбу прибора при работающей мешалке последова-
тельно загружают смесь растворителей, гексаметилендиизоцианат и'
диэтиленгликоль. Затем включают обратный холодильник и нагревают
колбу пламенем горелки на асбестовой сетке при кипении реакционной
массы.
Через 5—6 ч реакционную смесь переносят в фарфоровую чашку,
охлаждают до комнатной температуры, при этом полимер выпадает из
раствора. Растворитель декантируют, полимер высушивают в термо-
шкафу при 80° С и определяют его выход и температуру размягчения.
Полимер используют для получения пленки вальцеванием или прес-
сованием между плитами при температуре, которая на 20° С выше
температуры размягчения. При вальцевании температуру переднего вал-
ка устанавливают на 5° С выше, чем заднего, чтобы формование пленки
происходило на переднем валке. Вальцевание заканчивают, когда обра-
зуется однородная масса; ее подрезают ножом вдоль валка, остановив
вальцы; затем аккуратно снимают пленку, вновь включив мотор, приво-
дящий валки в движение.
Прессование производят на обогреваемых плитах размером 200X
Х200 мм. На нижнюю плиту устанавливают металлическую рамку^ тол-
щина которой равна толщине прессуемого образца. Из полученной тем
или иным способом пленки вырубают стандартный образец для испыта-
ния на предел прочности при разрыве и tg6 (см. ч. II).
233
РАБОТА 123. ПОЛУЧЕНИЕ ПОЛИУРЕТАНА ИЗ ГЕКСАМЕТИЛЕНДИИЗОЦИАНАТА
И 1,4-БУТАНДИОЛА В РАСТВОРЕ
Реакция протекает по следующей схеме:
«НО (СН2)4ОН + nO=C=N(CH2)6N=C=O -> Г-О (CH2)4O-C-NH (CH2)6NH-C—'
Исходные продукты: 1,4-бута н диол— 30,4 г; 1,4-гексаметилендиизоцианат—50 г;
хлорбензол, осушенный гидридом кальция,— 320 г; о-дихлорбензол — 80 г; диметилфор-
мамид— 100 мл; метанол— 100 мл.
Оборудование: трехгорлая колба емкостью 0,5 л, снабженная мешалкой с герме-
тическим затвором, капельной воронкой, обратным холодильником с хлоркальциевой
трубкой; колба емкостью 0,5 л; воронка Бюхнера; вакуум-эксикатор.
В колбу прибора загружают раствор 1,4-бутандиола в 200 мл смеси
растворителей, присоединяют холодильник, нагревают смесь до кипения
на асбестовой сетке >и через капельную воронку быстро вливают поло-
вину раствора диизоцианата в 200 мл растворителей. Вторую половину
вливают по кашлям в течение 3 ч, после чего реакционную смесь нагре-
вают еще 1 ч, охлаждают раствор до комнатной температуры, причем
полимер высаждается.
Растворитель декантируют, а полимер растворяют в 100 мл горяче-
го диметилформамида. Не ожидая охлаждения раствора, в него влива-
ют 100 мл метанола. Прозрачный раствор переносят в плоскодонную
колбу и оставляют в холодильнике на 12 ч. Высадившийся полимер
отделяют фильтрованием и высушивают в вакуум-эксикаторе. Опреде-
ляют выход полимера и его температуру размягчения.
Полимер используют для изготовления пленки вальцеванием или
прессованием между плитами пресса при температуре, на 20—30° С
превышающей температуру размягчения. При вальцевании в полимер
можно добавить 3—5% пигмента (титановые белила, кадмиевый лито-
пон, цинковые белила и др.) для окраски пленки.
РАБОТА 124. ПОЛУЧЕНИЕ ПОЛИУРЕТАНА ИЗ ГЛИЦЕРИНА
И ТОЛУИЛЕНДИИЗОЦИАНАТА
Реакция протекает по схеме
иСН2-СН—СН2 +
I I I
ОН ОН ОН
y\/NC0
I II -
H3c/Y
NCO
-> O=C=N-
NHCOCH2CHCH2OC-NH~-
n
234
В полиуретане, полученном при соотношении глицерина и диизоци-
аната 1:1, сохраняются функциональные группы (вторичный гидро-
ксил), обладающие пониженной реакционной способностью, которые
можно использовать для структурирования образовавшегося полимера.
Эту реакцию применяют для составления клеевых композиций горячего
отверждения. Реакция полиуретана с диизоцианатом в присутствии тре-
тичного амина в качестве катализатора идет по схеме
2O=C=N-
NHCOCH2—СН-СН2—OC-NH-
•CH2-OC-NH-~
(CaH5)3N
---->-* O=C=N-
NH—СОСН2—СН
\У\/
ОСО—NH
—СН3
я
O=C=N-
^/V-NH— СОСН2—CH-CH2-OC-NH—
II
о
о
.НзС/^/
Л
Исходные продукты: глицерин безводный—23 г; толуилендиизоцианат — 87 г;
хлорбензол — 360 мл; о-дихлорбензол— 120 мл; триэтиламин — 3 г.
Оборудование: четырехгорлая колба, снабженная мешалкой с герметическим за-
твором, обратным холодильником с хлоркальциевой трубкой, капельной воронкой и тер-
мометром; фарфоровая ступка; колба для приготовления смеси растворителей емкостью
0,5 л; термошкаф.
В колбу прибора загружают глицерин и растворяют его в 200 мл
смеси растворителей. 43,5 г толуилендиизоцианата растворяют в 280 мл
смеси, заливают раствор в капельную воронку и по каплям вливают его
в реакционную колбу при работающих мешалке и обратном холодиль-
нике при 160° С. Реакция идет 3—4 ч.
Образовавшийся на первой стадии линейный, растворимый полиуре-
тан при охлаждении выпадает из раствора. Раствор охлаждают для
полного осаждения полимера до —5 или +5° С и декантируют смесь рас-
творителей, которую после перегонки снова используют для проведения
реакции.
Образовавшийся полимер высушивают в термошкафу и смешивают
в фарфоровой ступке с 43,5 г толуилендиизоцианата и триэтиламином
(в вытяжном шкафу). Полиуретан структурируется при 100°С в тече-
ние 30 мин на поверхности металла или дерева (см. ч. II).
235
РАБОТА 125. ПОЛУЧЕНИЕ ПОЛИКАРБАМИДА
ИЗ 2И-ТОЛУИЛЕНДИАМИНА И ГЕКСАМЕТИЛЕНДИИЗОЦИАНАТА
Реакция протекает по следующей схеме:
СН3
1 /NH2
+ OCN(CH2)6NCO
о
СН3
—NH-C-NH.
п
NH2
NH—С—NH (CH2)6—_„-NHCO—
Исходные продукты: 2,4-толуилендиамин—12,2 г; гексаметилендиизоцианат —
16,2 г; ацетон — 200 мл.
Оборудование: трехгорлая колба, снабженная механической мешалкой с гермети-
ческим затвором и двумя капельными воронками (рис. 61); колбы плоскодонные емко-
стью 150 мл; воронка Бюхнера; термошкаф.
Готовят отдельно растворы диизоцианата в
100 мл ацетона и диамина в таком же количестве
ацетона, заливают их в капельные воронки и сли-
вают при энергичном перемешивании в реакцион-
ную колбу. Почти мгновенно начинает выпадать
осадок полимочевины. После окончания реакции
осадок отфильтровывают на воронке Бюхнера, про-
мывают ацетоном и высушивают при 50—60° С в
термошкафу.
Определяют выход полимера, температуру плав-
ления в капилляре. Готовят образцы прессованием
при 235° С и удельном давлении 50 кг/см2. Стан-
дартные образцы испытывают на ударную вяз-
кость.
Рис. 61. Прибор для получения поликарбамндов
РАБОТА 126. ПОЛУЧЕНИЕ ПОЛИКАРБАМИДА
ИЗ N, Н'-ДИЭТИЛ-Д.Д'-ДИАМИНО-З.З'-ДИМЕТИЛДИФЕНИЛМЕТАНА
И ГЕКСАМЕТИЛЕНДИИЗОЦИАНАТА
СН3 СНз
I__ I
HN-/ V-СН2-^ NH + (п + 1) O=C=N-(CH2)6-N=C=O ->
I \==/ I
С2н5 С2Н5
СНз СНз о
I • __। ||
-N-Z S- СН2-£ Ч—N—С—NH—(CH2)g—NH—СО—
I х=/ Х=/ (
C2Hs С2Н5
236
Реакции, протекающие при обработке паром:
r-N=C=O + Н2О -> R—NH2 + СО2
R—NH2 + O=C=N-R -> R—NH—С—NH—R
II
О
где R— полимерная цепь.
Исходные продукты: N, Н'-диэтил-4,4'-диамино-3,3'-диметилдифенилметан — 9,2 г;
гексаметилендиизоцианат—П,2 г; ацетон, содержащий следы Н2О, — 100 мл.
Оборудование: трехгорлая колба, снабженная механической мешалкой с гермети-
ческим затвором и двумя капельными воронками (см. рис. 61); воронка Бюхнера; уста-
новка для перегонки с водяным паром; колбы для приготовления растворов емкостью
100 мл; термошкаф.
Готовят раствор диамина в 50 мл ацетона и раствор диизоцианата в
таком же количестве ацетона. Растворы заливают в капельные воронки
и при работающей мешалке одновременно по каплям сливают их в реак-
ционную колбу. Через 1 ч реакционную массу переносят в колбу установ-
ки для перегонки с водяным паром и пропускают острый пар в течение
1 ч.
Выпавший в виде желтоватой массы полимер отфильтровывают и
высушивают в термошкафу (при 70°С). Определяют выход. Реакции,
протекающие при обработке паром (см. выше), приводят к увеличению
молекулярного веса полимера.
Полимер исследуют, определяют температуру размягчения, термо-
стабильность, .вязкость расплава. Его можно использовать для вальцева-'
ния, переработки прессованием, пресс-литьем или экструзией. Вальцева-
ние проводят при НО—120° С; прессование в рамке при 130—140° С;
пресс-литье при 170° С (температура формы — 80—90° С). При перера-
ботке экструзией температура в цилиндре машины 160° С; в головке
140—150° С, температура в загрузочной воронке 30—35° С,
Стандартные образцы, полученные из пленок и листов, испытывают
на разрушающее напряжение при растяжении и изгибе и относительное,
удлинение (см. ч. II).
ЛИТЕРАТУРА
к Технология пластических масс, под ред. Коршака В. В. М, — Л., «Химия», 1976.
2. Николаев А. Ф. Синтетические полимеры и пластические массы на их осно-
ве. Изд. 2-е, М. — Л., «Химия», 1968.
3. Л о с е в И. П., Т р о с т я н с к а я Е. Б. Химия синтетических полимеров. Изд.
3-е, «Химия», 1071.
4. К р е ш к о в А. П., Быкова Л. Н., Казарян Н. А. Кислотно-основное
титрование в неводных средах. М. — Л., «Химия», 4967.
5. Баландина В. А. и др. Анализ полимеризационных пластмасс. М. — Л.,
«Химия», 1967.
6. К а с т е р и н а Т. Н., Калинина Л. С. Химические методы исследования
полимеров, синтетических смол и пластических масс. М., Госхимиздат, 1963.
7. Л о с е в И. П., Федотова О. Я- Практикум по химии высокополимерных
соединений. Изд. 2-е, М. — Л., Госхимиздат, 1962.
8. С о р о к и н М. Ф., Л я лю ш ко К. А. Практикум по синтетическим поли-
мерам для лаков. М., «Высшая школа», 1965.
238
ПРИЛОЖЕНИЕ
СВОЙСТВА ПОЛИМЕРОВ, ПОЛУЧАЕМЫХ ПО РЕАКЦИЯМ ПОЛИМЕРИЗАЦИИ, И ПЛАСТМАСС НА ИХ ОСНОВЕ
Условные обозначения: Н— теплостойкость по методу НИИПП; В— теплостойкость по методу ВИКА; С —стойкий мате-
риал; с — относительно стойкий; МИзг— модуль упругости при изгибе; Меж — модуль упругости при сжатии; А4растяж— мо-
дуль упругости при растяжении.
I. Теплофизические свойства
№ п/п Полимер Плотность, г/см3 Коэффи- циент ли- нейного расшире- ния, (1°С)Х10“в Коэффициент теплопровод- ности, Дж/(см-сх Х°С)Х10~* Удельная теплоемкость, Дж/(г-°С) Теплостой- кость по Мар- тенсу, °C Волопог лоще- ние за 24 ч,% Маслоиог ло- щение за 24 ч, % Бензинопо- глощение за 24 ч, %
1 Полиэтилен низкой плотности (высокого давления) 0,92—0,93 22-55 6-8 0,5-0,68 50-60 100-110(H) 0,01 Набухает Набухает
2 Полиэтилен высокой плотности (низкого и среднего давления) 0,94-0,96 10-18 9,6 0,55 100 120-128 (Н) 0,005 (2 месяца) Относитель- но стойкий Относитель- но стойкий
3 Полипропилен 0,9 1,1 3,3 0,46 105-110 (В) 0,03 — Растворяет- ся в бензоле выше 20 °C
4 Сополимер этилена с пропиленом СЭП (среднего давления) 0,94-0,95 —— — — 115-128(1-1) Стойкий Стойкий
5 Сополимер этилена с пропиленом СЭП низкого давления 0,925-0,937 6,2-6,8 (при 40 °C) 0,54-0,56 120-126 (Н) — — —
6 Полиизобутилен 0,91-0,93 0,45 Под нагруз- кой течет на холоду. Пла- вится при 80 °C Растворя- ется в угле- водородах
7 Поливинилкарба- зол 1,2 5,5 0,3 0,3 150-170 0,10 Стойкий Стойкий
8 Полистирол блоч- ный 1,05-1,08 6-10 1,9-3,2 0,32 80 0,20 Набухает
9 Полистирол эмуль- сионный 1,05-1,08 6-10 1,9 0,32 80 0,30 » »
10 Пенополистирол ПС-1 ПСБ 0,06-0,П 0,02-0,03 — 0,03-0,0332 0,03-0,038 — — 0,05-1,25 0,05-1,25 — —
11 Сополимер стирола 1,04 9,5 — — 85-90 — — —
с акрилонитрилом СН-20 113(B) 0,01-0,05 0,06
12 13 Сополимер МСН Полистирол ударо- 1,12 1,10 6,0-8,0 6-10 1,9 0,32 72-74 75 Стойкий Стойкий
14 прочный Сополимер стирола 1,20 7-8 — — 75 Не более 0,07 •
с метилметакрилатом МС *
0,00
15 Политрифторхлор- 2,11-2,16 6 1,4 0,22 70 — —
этилен (фторопласт-3) 0,22 200-250 0,00 0,00 0,00
16 По литетр а фторэти- 2,2-2,3 5,5-11 6 0,25
17 лен (фторопласт-4) .Винипласт листовой 1,35-1,4 6-7 4 0,24-0,51 65 0,001 Стойкий —
18 Сополимер винил- хлорида с винилиден- хлоридом 1,4 1,34 16 2,2 0,316 70-95 0,04 0,05
19 Сополимер винил- хлорида с винилацета- — -
20 том Полиметилметакри- 1,18-1,2 8-12 3,5-6 0,36-0,4 52-58 0,1-0,5 2 2
21 лат Полиметилметакри- 1,2 8-12 3,5-6 0,36-0,4 50 0,3-0,5 Стойкий —
22 лат суспензионный Поливинилацетат 1,18-1,19 8,6 3,8 0,39 244-50 3 я Стойкий
23 Поликапроамид 1,14-1,15 6,0 0,55 190—200(B) 55-60 3,5(1 ч.)
24 Полиуретан 1,2 10,6-13,5 7,5 — 55-60 0,2-0,4 0,00 0,00
25 Полиформальдегид 1,4 8,1 0,22 0,35 160—170(B) 0,2 0,01 — —
26 Пентон 1,4 7,8—8,0 — —
27 Сополимер триокса- на с 1,3-диоксаланом 1,4-1,41 — — — 150—155(B)
‘ Объемная масса; 2 В кДж/(м-ч-°С).
II, Механические свойства
№ ж/п Полимер Удельная ударная вязкость, кг «см/см2 Разрушающее напряжение при растяже- нии, кг/см2 Разрушающее напряжение при статичес- ком изгибе, кг/см’ Разрушающее напряжение при сжатии, кг/мм2 Твердость по Бринеллю, кг/мм2 Относительное удлинение при разрыве, % Модуль упругости, кг/см2
1 Полиэтилен низкой плотности (высокого дав- ления) Не лома- ется 120-160 120-170 125 1,4-2,5 150-600 1500-2500 (^растяж)
2 Полиэтилен высокой плотности (низкого и среднего давления) То же 220-400 200-300 — 4,5-5,8 200-900 5000-8000 (Мрастяж)
3 4 Полипропилен Сополимер этилена с пропиленом СЭП (сред- него давления) 80 300-350 140-220 700-800 660-760 6,3 400-800 150-800 8000-10 000 (Мрастяж) 3000-6000 (Мнзг)
5 Сополимер этилена с пропиленом СЭП (низ- кого давления) — 220-320 200-220 500-900 2000-3300 (Мн3г)
5 Полиизобутилен — 20-45 — — 35 (по Шору) 550-1000 2000
7 Поливинилкарбазол 8-10 250 500-900 — 10,0 — 30 000 (Мрастяж)
8 Полистирол блочный 12-16 350-400 950-1000 1000 14-15 1,5 12 000-32 000
9 Полистирол эмульсион- ный 15 350-400 Не менее 1000 1000 14-15 1,5 26 000 (Мизг)
10 Полистирол ПС 1 » ПСБ 0,6-1,1 0,1-0,3 1,8-4,5 13-14 2,25 — 550 (Мсж) 40-90
11 Сополимер стирола с нитрилом акриловой кис- лоты СН-20 18-20 490 1000 — 17 1,7 27 000 (AfH3roT)
12 Сополимер МСН 22 1100-1200 16-17
13 Полистирол ударопроч- ный 40-50 330 890 — 12 1,2-1,54 34 700 22 000 (М„зг)
14 Сополимер стирола с Не менее — 950-1000 — 16-17 2
метилметакрилатом МС 10 11 600-14500 (Л4ИЗГ)
15 Политрифторэтилен 20-160 350-460 600-800 500-570 10-13 Незакален- ный 20—40
(фторопласт-3) Закаленный
70—200
16 Политрифторэтилен (фторопласт-4) 100 Незакален- ный 140-250 Закаленный ' 110-140 • 120—200 3-4 250-500 4700-8800 (Л4изг)
160-315
17 Винипласт листовой 120 400-600 900-1200 800—1600 15-16 10-25 4000
18 * Сополимер винилхлори- да с винилиденхлоридом вхвд — 300-500 1000 500-600 - 4 2100
19 Сополимер винилхло- — 1000 — — —’ —
рида с винилацетатом
20 (Винилит) Полиметилметакрилат (органическое стекло) 8,5-12 400—700 600-650 700-850 7-16 3-4 19000-28 000 21 000-35 000
21 Полиметилметакрилат 6 420-700 910-1330 840—1330 12 1-15
22 суспензионный Поливинилацетат 5-8 150-250 300-400 — — 80-100 17000-20 000(Мизг)
23 Поликапроамид 140-160 500-840 700-1000 700-800 8-12 50-150 15 000
24 Полиуретан 40—55 500-850 700-850 800-850 8-12 100—150 25 000
25 Пентон — 420 . 775 — 100 (по Роквеллу) 35-100 11200
26 27 Полиформальдегид Сополимер триоксана с 75—130 80 700 580-620 800-1100 1300 29-25 20-40 12-30 42 000 25 000-30000
диоксаланом 1
III. Электрические свойства
№ Наименование Удельное объемное электрнческое сопротивление Ом-см Удельное не- поверхностное Тангенс угла диэлектричес- ких потерь при частоте Диэлектрическая проницае- мость при частоте Электрическая прочность, кВ/мм
п/п сопротивле- ние, Ом 50 Гц 10» Гц 50 Гц 10s Гц
1 Полиэтилен низкой плотности (высокого дав- ления) МОП 1 1017 0,0004 2,3 45—60 (при 1 мм) 28—36 (при 2 мм)
2 Полиэтилен высокой плотности (низкого и среднего давления) 1 1017 1-1014 0,00014 2,2 45—60 (при 1 мм) 28—36 (при 2 мм)
3 Полипропилен 8-1016 — 0,0005 2,1-2,3 28—40 (при 1 мм)
4 Сополимер этилена с пропиленом СЭП низкого давления 1-1017 — 0,0002— 0,0004 2,2-2,3 30—36 (при 2 мм)
5 Сополимер этилена с пропиленом СЭП средне- го давления 1-1017 0,0002— 0,0004 2,2-2,3 28—30 (при 2 мм)
6 Полиизобутилен 1-1015—1016 0,0003- -0,0005 2,2-2,3 23-25
7 Поливинилкарбазол 1-1016 1-1016 0,0007— -0,0008 3 —
8 Полистирол блочный 1015—1017 1015-1017 0,0001 — -0,0006 2,5-2,6 25
9 Полистирол эмульсион- ный 1-1015 1-1015 0,001 2,6 20-22
10 Пенополистирол ППС-1 1-1013 1-1012 0,002 3-4
» ПСБ 1-1014 1-1014 0,0004
И Сополимер стирола с 1-1016 1-1016 0,005— 2,8 25
акрилонитрилом СН-20 —0,007
12 Сополимер МСН 1-1016 1014-1015 0,02 2,9-3,2 24
13 Полистирол ударопроч- ный СНП-1 1-1016 3-1015 0,02 3,1-3,3 23,5
14 Сополимер стирола с метилметакрилатом МС 1-1016 1-1015 0,02 2,8-2,9 24
15 Политрифторхлорэти- лен (фторопласт-3) 1,2-1018 1-1017 0,015 • 0,01 3,0 2,5-2,7 13—15 (при 2 мм)
16 Политетрафторэтилен (фторопласт-4) 1-1019 1-1017 0,0002 1,9-2,2 25—27 (при 4 мм)
17 Винипласт листовой 1012-1,8х 1012-1,9х 0,01 3,5-4,1 20
ХЮ14 Х1014
18 Сополимер винилхлори- да с вииилиденхлоридом 1-1014 1013-1014 0,1-0,3 3-5 3,2 14-24
19 Сополимер винилхлори- да с винилацетатом 5,2-1014 — 0,013 25
20 Полиметилметакрилат (органическое стекло) 1012-1013 1012—1013 0,02-0,06 3,5-3,6 15-25
21 Полиметилметакрилат суспензионный Г-1013 — 0,05-0,06 3,0-3,7 15
22 Поливинилацетат 2,3-1014 1-1012 ' 0,003 5 3,7 —
23 Полиформальдегид 8-1014 2-1013 3,3-10-3 7-Ю-з 3,3 26
24 Поликапроамид 1013-Ю14 1013-1014 0,018 0,032 3,6 22
25 Полиуретан 1013-1014 2,1013 — 3,7 20-23
26 Пентон 5-1015 — 0,016 3,1 16
243
ОГЛАВЛЕНИЕ
Стр.
Предисловие ............................................................. 5
Введение.................................................................. 7
Глава I. Полимеры непредельных алифатических углеводородов и плас-
тические массы иа их основе................................ ).............25
Работа 1. Получение полиэтилена высокой плотности (низкого
давления).................................................40
Работа 2. Получение стереорегулярного (изотактического) по-
липропилена .............................................41
Работа 3. Получение полиизобутилена в присутствии эфирата
трехфтористого бора.......................................42
Работа 4. Получение полиизобутилена в присутствии TiClj . . 43
Работа 5. Изготовление изделий из полиэтилена высокого дав-
ления (низкой плотности) литьем под давлением.............43
Работа 6. Изготовление изделий из полиэтилена низкого дав-
ления (высокой плотности) литьем под давлением............44
Работа 7. Изготовление изделий из полиэтилена низкой плот-
ности экструзией..........................................45
Работа 8. Изготовление изделий из полиэтилена высокой плот-
ности или сополимера этилена с пропиленом экструзией . . 46
Глава II. Полимеры непредельных ароматических углеводородов и пласт-
массы на их основе ...................................................... 48
Работа 9. Синтез стирола из а-хлорэтилбензола............62
Работа 10. Синтез стирола из коричной кислоты............64
Работа 11. Получение полистирола термической полимериза-
цией стирола в блоке......................................65
Работа 12. Получение изделий из блочного полистирола литьем
под давлением.............................................65
Работа 13. Получение полистирола полимеризацией стирола в
эмульсии.............................................. 66
Работа 14. Изготовление газонаполненного материала (пено-
пласта) из эмульсионного полистирола......................67
Работа 16. Получение полистирола полимеризацией с персуль-
фатом аммония........................................... 69
Работа 16. Получение полистирола полимеризацией стирола в
суспензии.................................................69
Работа 17. Получение сополимера стирола с дивинилбензолом
полимеризацией в суспензии............................... 70
Работа 18. Получение сополимера стирола с нитрилом акрило-
вой кислоты СН-20 суспензионным методом...................71
Работа 19. Получение сополимера стирола с метилметакрила-
том в гранулах (суспензионным методом)....................72
Работа 20. Получение тройного сополимера типа МСН суспен-
зионным методом......................................... 73
Глава III. Полимеры галогенопроизводных непредельных углеводородов
и пластические массы на их основе........................................ 74
А. Полимеры хлорпроизводных непредельных углеводородов
и пластические массы на их основе........................74
Работа 21. Получение винилхлорида из дихлорэтана.........89
Работа 22. Получение винилиденхлорида действием едкого нат-
ра на трихлорэтан........................................ 90
244
Стр.
Работа 23. Получение винилиденхлорида действием известково-
го молока на трихлорэтан . .............................91
Работа 24. Получение поливинилхлорида полимеризацией ви-
нилхлорида в растворе...................................91
Работа 25. Получение поливинилхлорида полимеризацией ви-
нилхлорида в эмульсии (эмульсионный поливинилхлорид) 92
Работа 26. Получение поливинилхлорида полимеризацией ви-
нилхлорида в суспензии. ...._-....................... 93
Работа 27. Изготовление листового винипласта прессованием 94
Работа 28. Изготовление изделий из листового винипласта ме-
тодом пневматического формования............. 96
Работа 29. Изготовление изделий из листового и пленочного
винипласта вакуум-формованием.......................... 98
Работа 30. Изготовление профильных изделий из винипласта
методом экструзии..................................... 99
Работа 31. Изготовление изделий из винипласта сваркой . . . 100
Работа 32. Получение стабилизаторов — солей стеариновой
кислоты и металлов (кальция, кадмия, бария, свинца) . . . 102
Работа 33. Получение днбутилфталата................... 103
Работа 34. Изготовление пленочного пластиката...........104
Работа 35. Изготовление эластичного губкоподобного материа-
ла из поливинилхлорида ...............................Ю5
Работа 36. Изготовление жесткого пенопласта из поливинил-
хлорида ............................................. . .106
Работа 37. Изготовление искусственной кожи — текстовинита 106
Работа 38. Изготовление изделий из поливинилхлоридных паст 108
Работа 39. Изготовление из пластифицированного поливинил-
хлорида форм для гипсовых отливок.................... 109
Работа 40. Получение сополимера винилхлорида с винилаце-
татом (винилита) в суспензии...........................109
Работа 41. Получение сополимера винилхлорида с метилмета-
крилатом ............................................ Н2
Работа 42. Получение сополимера винилхлорида с метилакри-
латом (МА-20)...........................................ИЗ
Работа 43. Получение сополимера винилхлорида с винилиден-
хлоридом суспензионным методом.........................114
Работа 44. Получение поливинилиденхлорида полимеризацией
суспензионным методом...................................ИЗ
Работа 45. Получение сополимера винилиденхлорида с акрило-
нитрилом и пленки на его основе............... . . ... . Пб
Б. Фторпроизводные полиуглеводороды и пластмассы на их
основе...................................................
Работа 46. Получение полимера фторопласта-4 полимеризацией
тетрафторэтилена суспензионным методом...................
Работа 47. Получение фторопласта-3 полимеризацией трифтор-
хлорэтилена в растворе ..................................
Работа 48. Получение фтороплаета-3 полимеризацией трифтор-
хлорэтилена суспензионным методом...................... • •
Работа 49. Изготовление пленки на основе суспензионного по-
литрифторхлорэтилена.....................................
Работа 50. Получение сополимера трифторхлорэтилена с вини-
лиденфторидом ...........................................
Работа 51. Получение отвержденного сополимера трифторхлор-
этилена и винилиденфторида.......................... .
Работа 52. Изготовление пленки из раствора на основе сополи-
мера трифторхлорэтилена и винилиденфторида . .. ..... •
Работа 53. Получение полимера винилфторида полимеризацией
суспензионным 'методом...................................
124
125.
125
126
126
127
128
128
245
Глава IV.
Глава V.
Поливинилацетат, его сополимеры и пластические массы иа их
основе..................................................129
Работа 54. Получение винилацетата......................138
Работа 55. Получение поливинилацетата полимеризацией ви-
нилацетата в блоке 139
Работа 56. Получение поливинилацетата полимеризацией ви-
нилацетата в эмульсии и изготовление эластичных пленок и
покрытий на его основе...................................139
Работа 57. Получение суспензионного поливиннлацетата . . . 141
Работа 58. Получение сополимера акрнлоннтрила с винилаце-
татом .............................................. . . . 141
Полимеры акриловой и метакриловой кислот. Полимеры про-
изводных акриловой н метакриловой кислот и пластические
массы на их основе..................................... 142
Работа 59. Получение акриловой кислоты из этиленциангид-
рина ...................................................163
Работа 60. Получение акриловой кислоты из метилакрилата 163
Работа 61. Получение метакриловой кислоты нз ацетонциан-
гидрина ................................................164
Работа 62. Получение метакриловой кислоты из метилметакри-
лата (омылением)........................................165
Работа 63. Получение метилового эфира метакриловой кисло-
ты ................................................... 166
Работа 64. Получение бутилметакрилата переэтерификацией
метилметакрилата . . . .................................167
Работа 65. Получение аллилметакрилата...................168
Работа 66. Получение диметакрилата этиленгликоля.........169
Работа 67. Получение диметакрнлата днэтиленгликоля ... .170
Работа 68. Получение акрилонитрила.............,........171
Работа 69. Получение полимера акриловой кислоты полимери-
зацией в растворителе (в воде) ........................ 172
Работа 70. Получение полиметилметакрилата полимеризацией
в блоке.................................................172
Работа 71. Изготовление непрозрачного органического стекла
молочно-белого цвета .................................. 173
Работа 72. Изготовление изделий из листового органического
стекла вакуум-формованнем...............................174
Работа 73. Получение полиметилметакрилата в блоке без на-
гревания ...............................................175
Работа 74. Получение полиэтилметакрнлата полимеризацией в
блоке...................................................176
Работа 75. Получение полибутилмета крилата в блоке . . . .177
Работа 76. Получение полиаллнлметакрилата в блоке.......177
Работа 77. Получение полиалкнленгликольднметакрилата в
блоке...................................................178
Работа 78. Получение сополимера метилметакрилата с аллил-
метакрилатом в блоке....................................179
Работа 79. Получение сополимера метилметакрилата с днмета-
крнлатом этиленгликоля в блоке..........................179
Работа 80. Получение полиакрилонитрила в мономере . . . .180
Работа 81. Получение сополимера метилметакрилата, акрило-
нитрила и диметакрнлата трнэтилевгликоля в блоке .... 180
Работа 82. Получение сополимера метилметакрилата со стиро-
лом в блоке........................................... .181
Работа 83. Получение полнметилметакрнлата с персульфатом
аммония.................................................182
Работа 84. Получение полиметилметакрилата в гранулах . . . 183
246
Работа 85. Получение полиметилметакрилата в суспензии . . . 183
Работа 86. Получение пластифицированного полиметилметакри-
лата в суспензии ..... ............................... 184
Работа 87. Изготовление светящихся изделий из полиметилме-
такрилата методом горячего прессования.................184
Работа 88. Изготовление непрозрачных окрашенных изделий
из полимеризующейся мастики на основе суспензионного по-
лиметилметакрилата ....................................184
Работа 89. Получение полнбутилметакрилата в суспензии . . . 185
Работа 90. Получение сополимера метилметакрилата с бутил-
метакрилатом суспензионным методом.....................186
Работа 91. Получение сополимера метилметакрилата со стиро-
лом полимеризацией с персульфатом аммония..............187
Работа 92. Получение сополимера метилметакрилата с метила-
крилатом в суспензии...................................187
Работа 93. Получение сополимера метилметакрилата с винила-
цетатом суспензионным методом..........................188
Работа 94. Получение полиметилметакрилата в растворителе 189
Работа 95. Изготовление изделий из отходов полиметилмета-
крилата методом горячего прессования...................189
Работа 96. Получение полиакрилонитрила в окислительно-вос-
становительной инициирующей системе .......... . 190
Работа 97. Получение сополимера полиакрилонитрила в вод-
ном растворе роданистого натрия........................190
Работа 98. Получение полиакриламида в растворителе .... 191
Работа 99. Получение сополимера акриламида с акриловой
• кислотой в водной среде.............................. 191
Глава VI.
Полимеры на основе производных этилена со сложными заме-
стителями ................. . ...........................192
Работа 100. Получение N-винилкарбазола...................197
Работа 1'01. Получение полнвинилкарбазола полимеризацией в
эмульсии...............................................197
Работа 102. Получение сополимера N-винилкарбазола со сти-
ролом в суспензии......................................198
Работа 103. Получение сополимера N-винилкарбазола с акри-
понитрилом в присутствии окислительно-восстановительной
инициирующей системы ..................................199
Работа 104. Получение поли-№винилпирролидона в водном
растворе...............................................1®9
Работа 105. Получение сополимера N-винилпирролидона с акри-
лонитрилом ............................................200
Работа 106. Получение сополимера N-винилпирролидона со
стиролом ............................................. 200
Работа 107. Получение полнвинилпиридина в окислительно-вос-
становительной инициирующей системе....................201
Работа 108. Получение сополимера 2-винилпиридина с акрило-
нитрилом блочным методом...............................201
Глава VII.
Гетероцепные простые полиэфиры и пластические массы иа их
основе.......................................................202
Работа 109. Полимеризация формальдегида в паровой фазе. . 206
Работа НО. Изготовление пленок ла основе полиформальде-
гида.......................................................207
Работа 111. Получение сополимера триоксана с 1,3-диоксала-
ном в расплаве в присутствии BF3...........................207
Работа 112. Получение сополимера триоксана с 1,3-диоксала-
ном в расплаве.......................................... 200
Работа 113. Получение сополимера триоксана с 1,3-диоксала-
ном в суспензии.............................................2W
24
Работа 114. Изготовление изделий из полиформальдегида
литьем под давлением и экструзией . ... . ... . . 209
Г
Глава VIII. Полимеры, получаемые полимеризацией неустойчивых гетеро-
циклических соединений..................................................... 211
Работа 115. Получение поликапроамида гидролитической поли-
меризацией е-капролактама в расплаве.........................219
Работа 116. Изготовление изделий из капрона методом литья
под давлением................................................220
Работа 1117. Получение N-ацетнлкапролактама................222
Работа 118. Получение кашролона литьем в формы......223
Работа 119. Получение полиэтиленоксида в присутствии ще-
лочи ....................................................... 224
Работа 120. Получение полипроппленоксида в присутствии ще-
р лочи.................................................... 224
Работа 121. Получение полимера из 3,3'-бпс-(хлорметил)-окса-
циклобутана ............................................... 225
Глава IX. Полимеры, получаемые по реакциям ступенчатой миграционной
сополимеризации или полиприсоединения ......................................226
Работа 122. Получение полиуретана из гексаметилендинзоцна-
ната и диэтиленглнколя в растворе............................233
Работа 123. Получение полиуретана нз гексаметилендиизоциа-
ната и 1,4-бутапдиола в растворе......................'. . . 234
Работа 124. Получение полиуретана из глицерина и толунлен-
динзоцианата........................................... . 234
Работа 125. Получение поликарбамида из .м-толуиленднамина
и гексаметилендинзоцианата...................................236
Работа 126. Получение полнкарбамида .из N, М'-диэтил-4,4'-дн-
амино-З.З'-диметилднфенилметана и гекса метилен днизоциа-
ната.........................................................236
Литература...................................................................237
Приложение...................................................................238
Алексей Петрович Григорьев, Ольга Яковлевна Федотова
ЛАБОРАТОРНЫЙ ПРАКТИКУМ ПО ТЕХНОЛОГИИ ПЛАСТИЧЕСКИХ МАСС
Часть I
ПОЛИМЕРИЗАЦИОННЫЕ ПЛАСТИЧЕСКИЕ МАССЫ
Редактор А. В. Бородина
Художественный редактор В. П. Спирова
Оформление художника Ю. Д. Федичкине
Технический редактор 3. А. Муслимова
Корректор С. К. Марченко
ИБ № 637
Сдано в набор 16/1Х—76 г. Подп. к печати 11/1II—77 г.
Формат 70X90’/i6 Бум. тип. № 2. Объем 15,5 печ. л. Усл. п. л. 18,14. Уч.-изд. л. 18,38
Изд. № ХИМ-528 Тираж 15 000 экз. Цена 1 р. 02 к.
План выпуска литературы издательства
«Высшая школа» (вузы и техникумы) на 1977 г. Позиция № 50
Издательство «Высшая школа»,
Москаа, К-51, Неглинная ул., д. 29/14,
Московская типография № 8 Союзполиграфпрома при Государственном комитете Совета
Министров СССР по делам издательств, полиграфии и книжной торговли. Хохлоаский пер., 7.
Зак. 1490.
2